/










































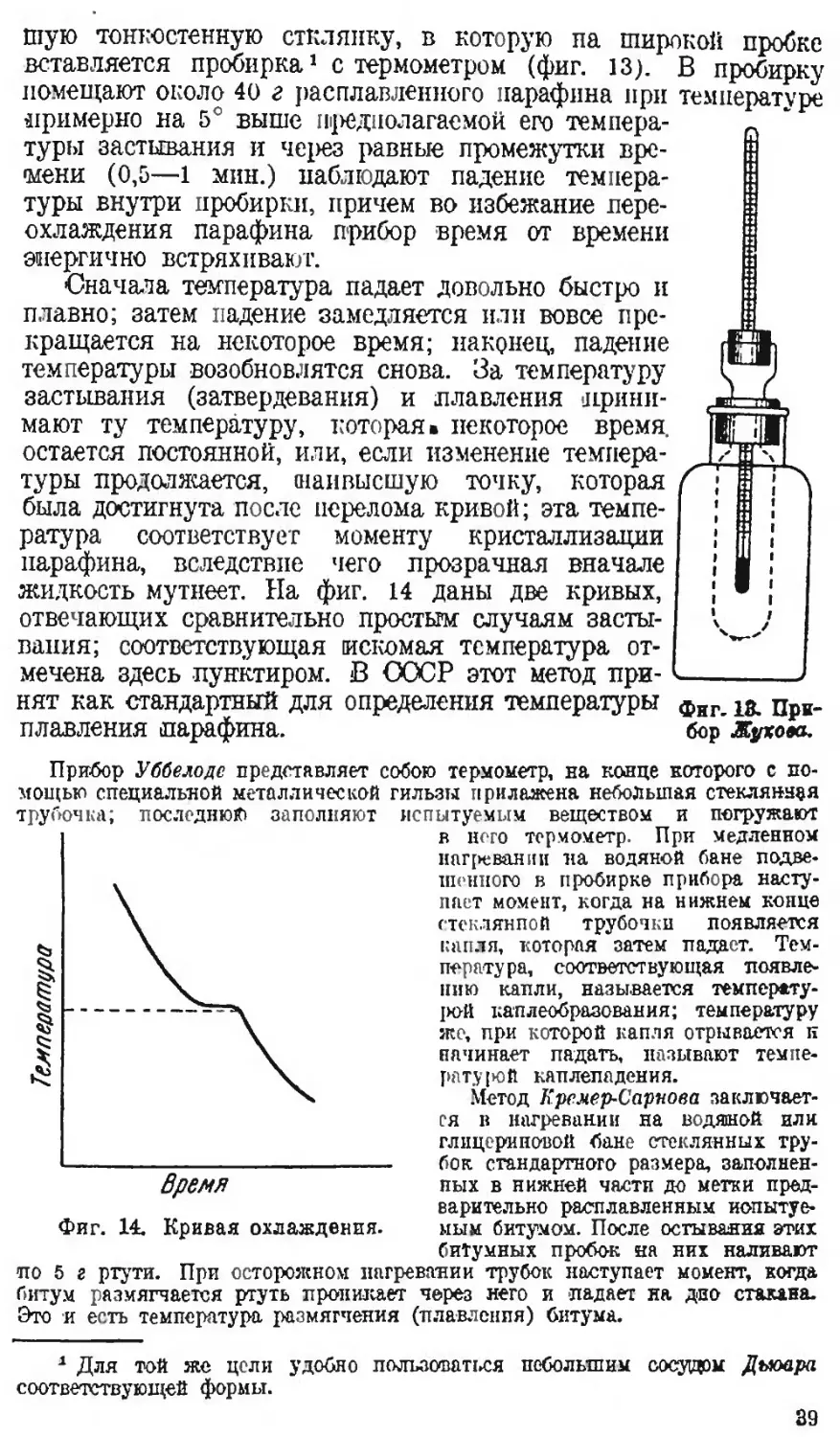




















































































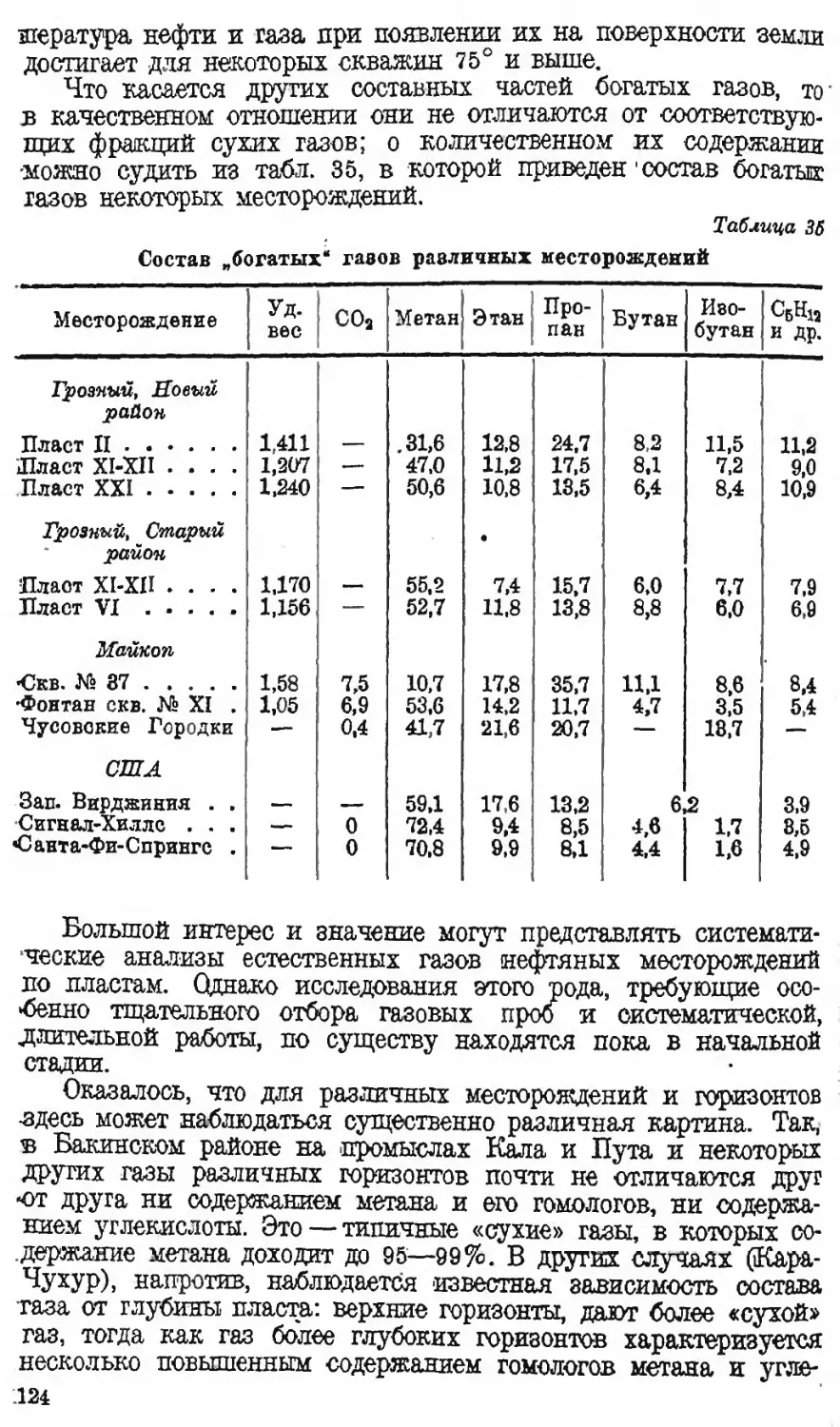
























































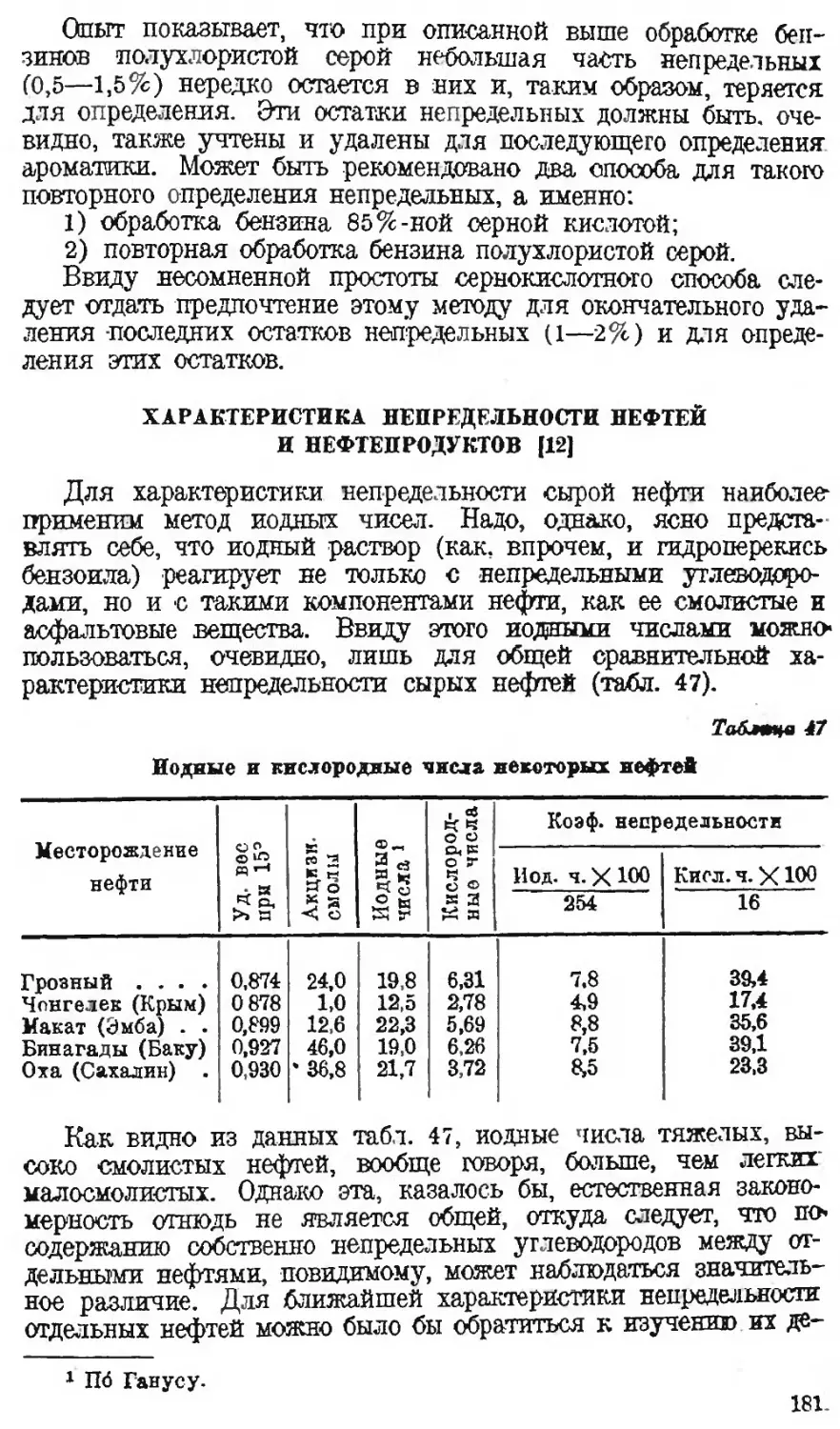









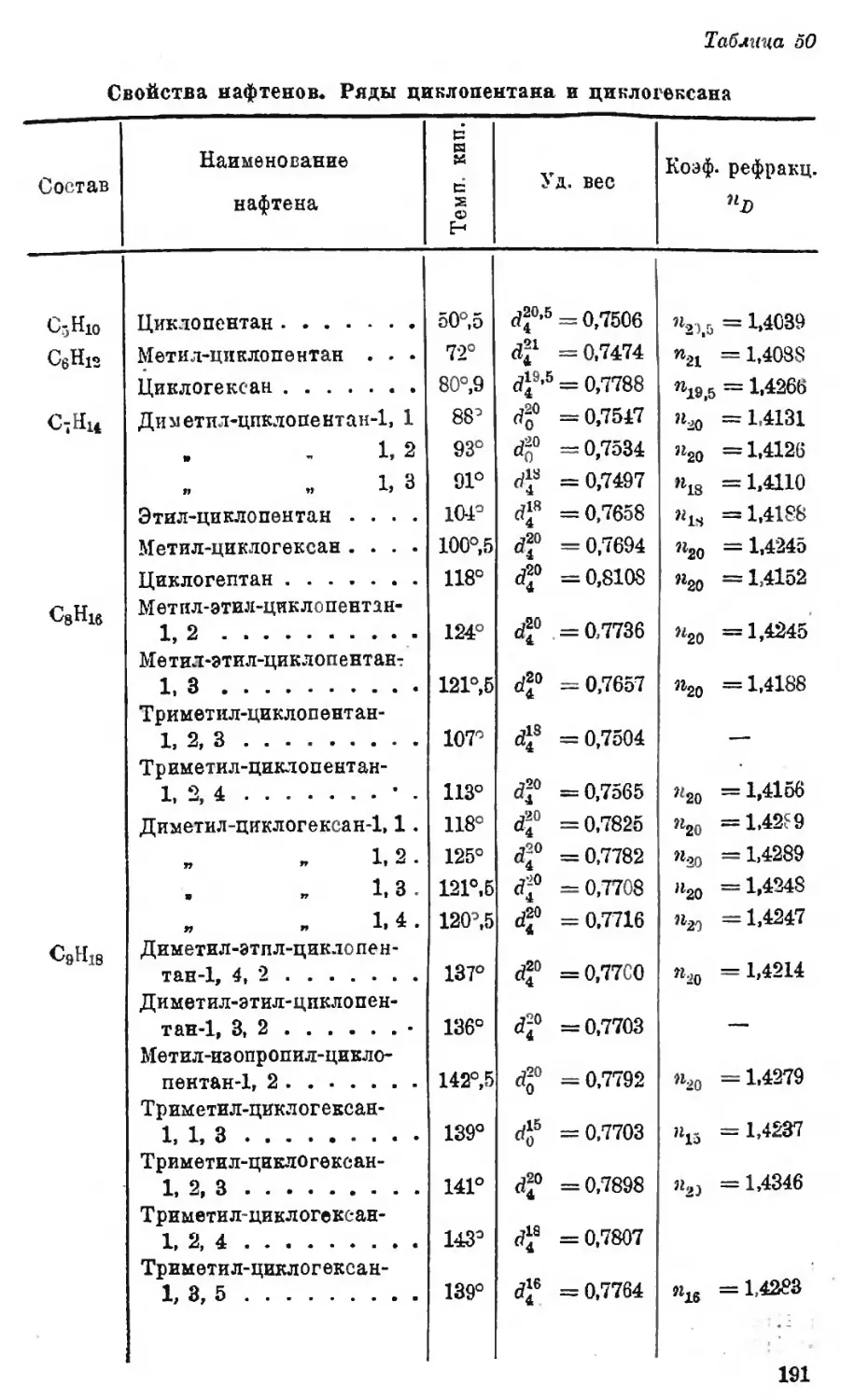




















































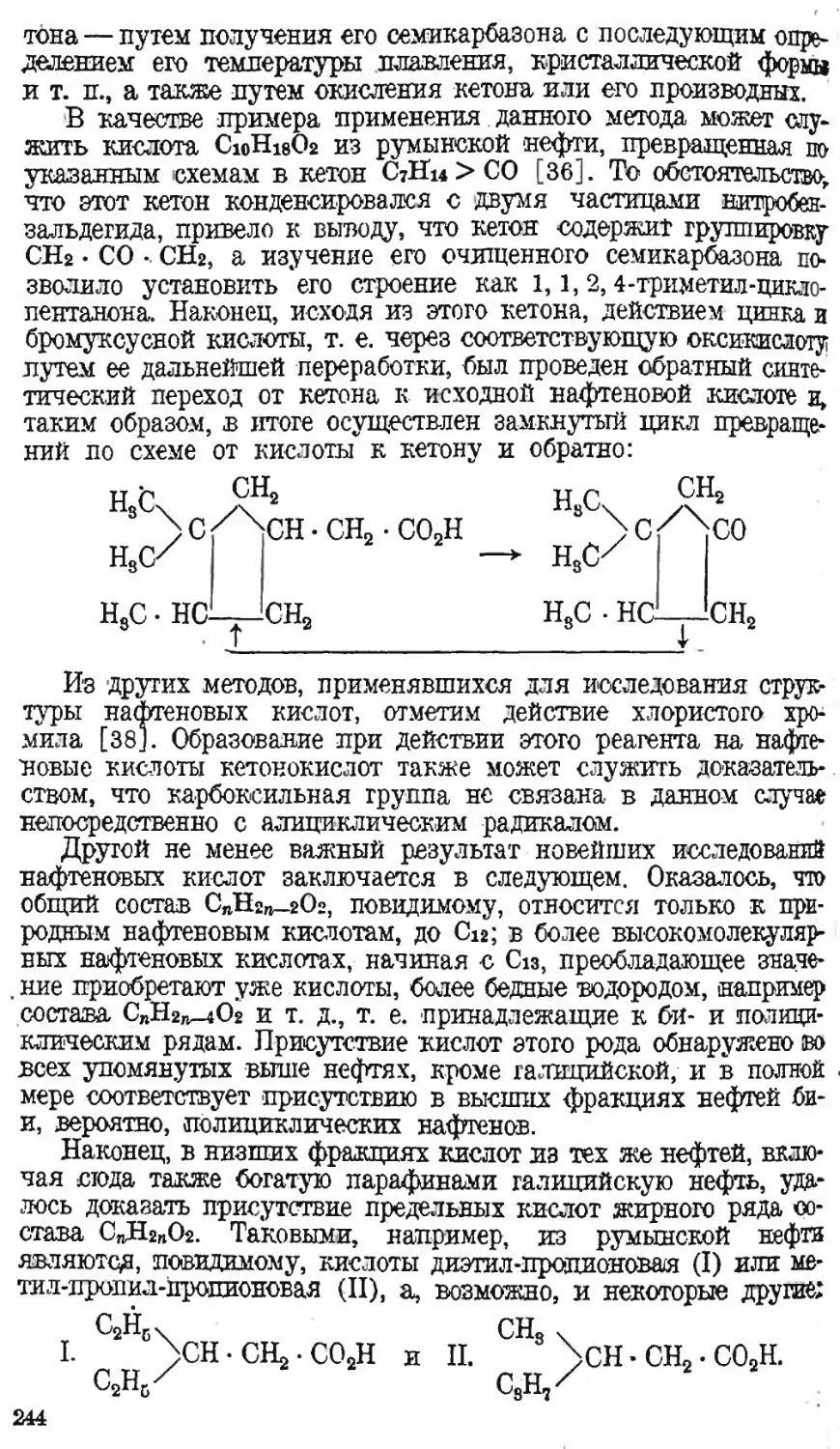

























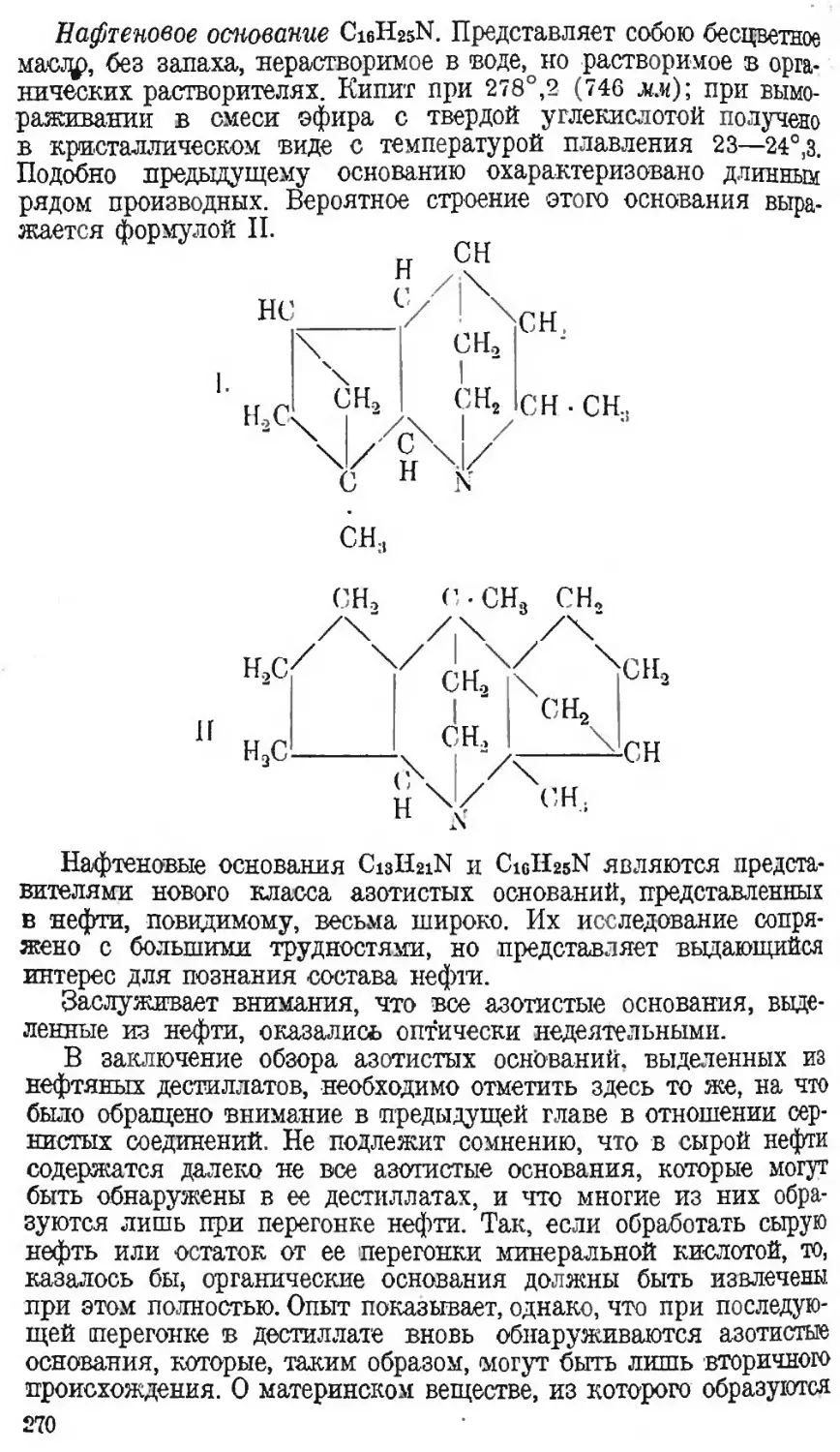


































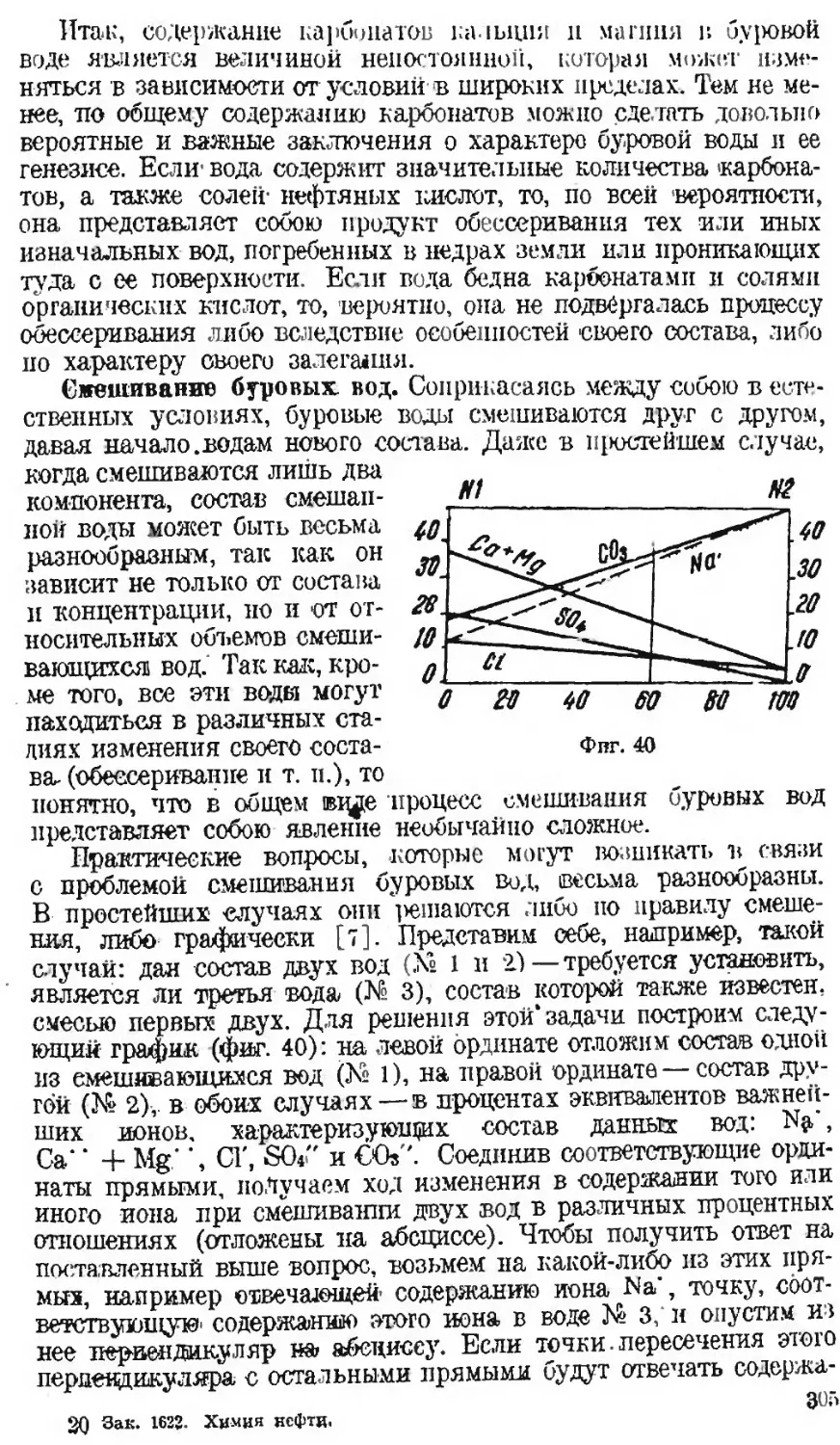






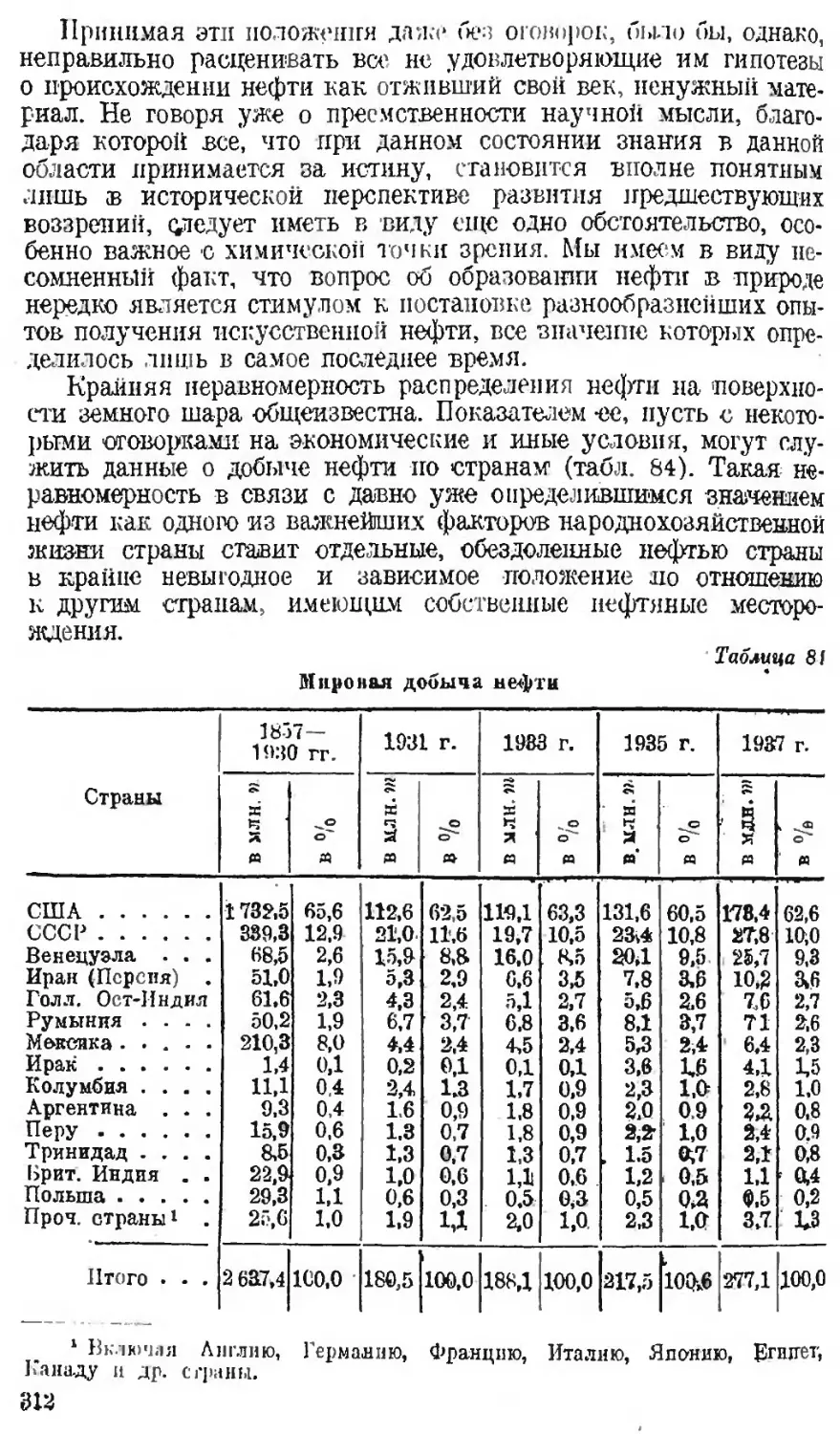




































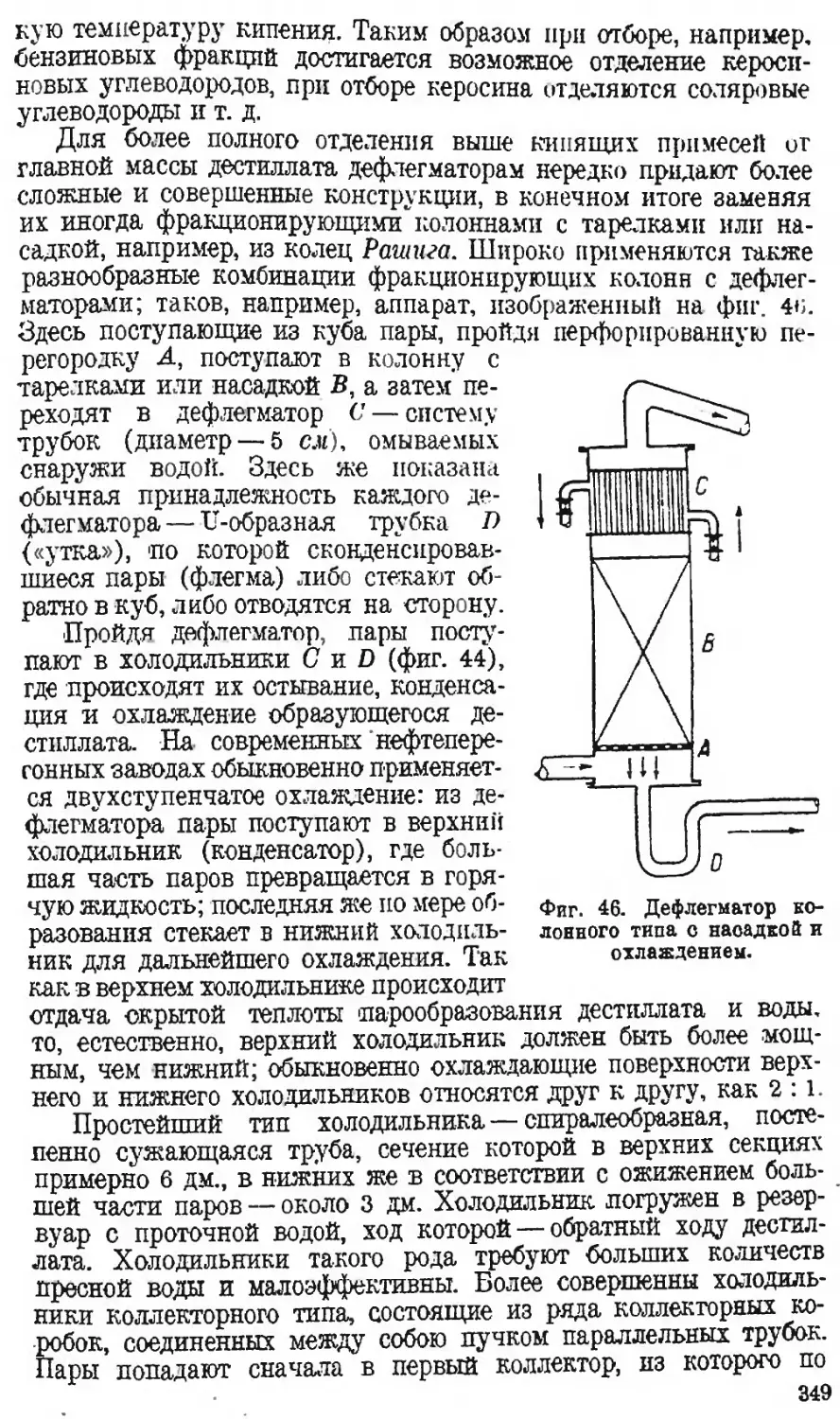


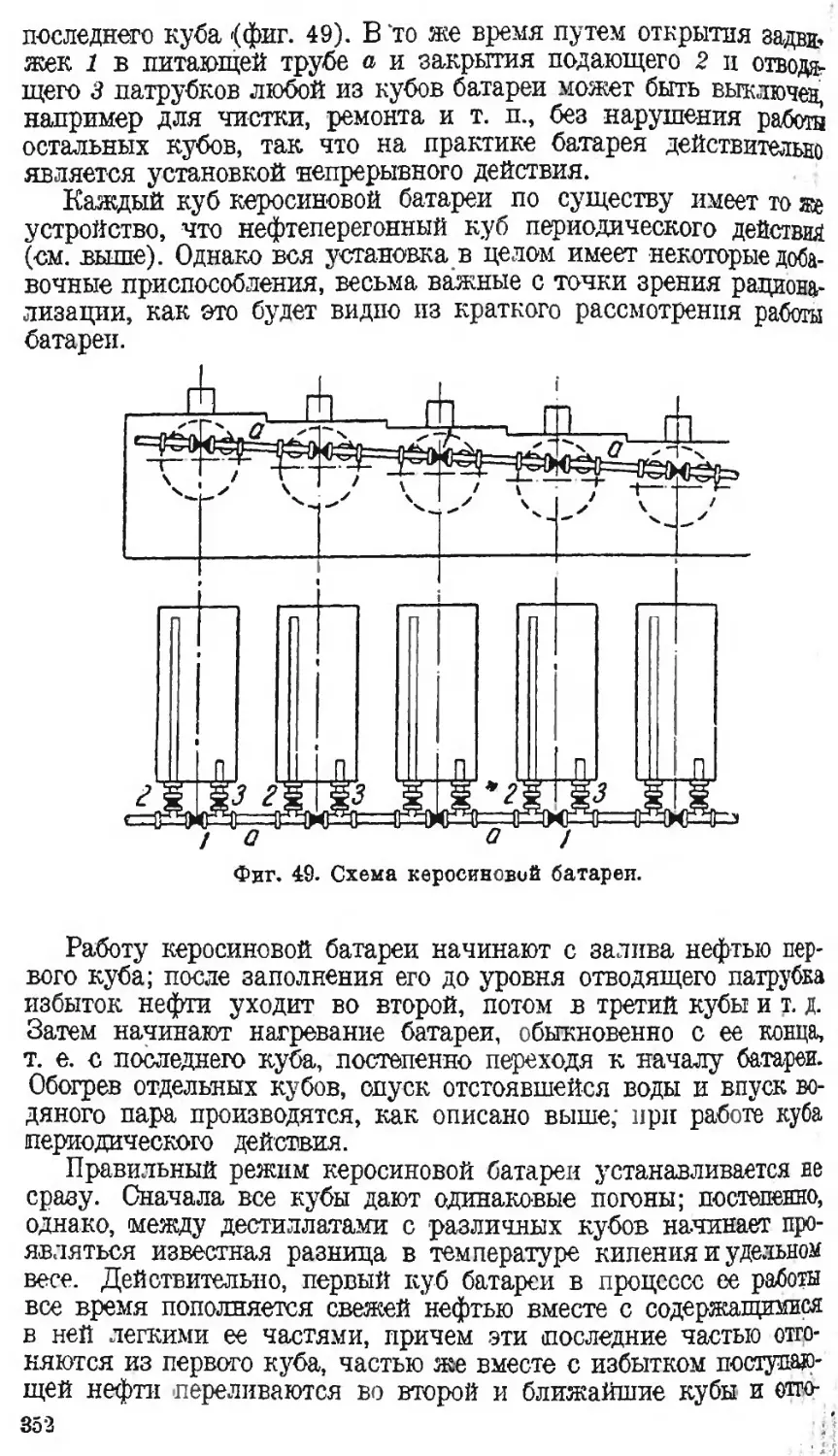




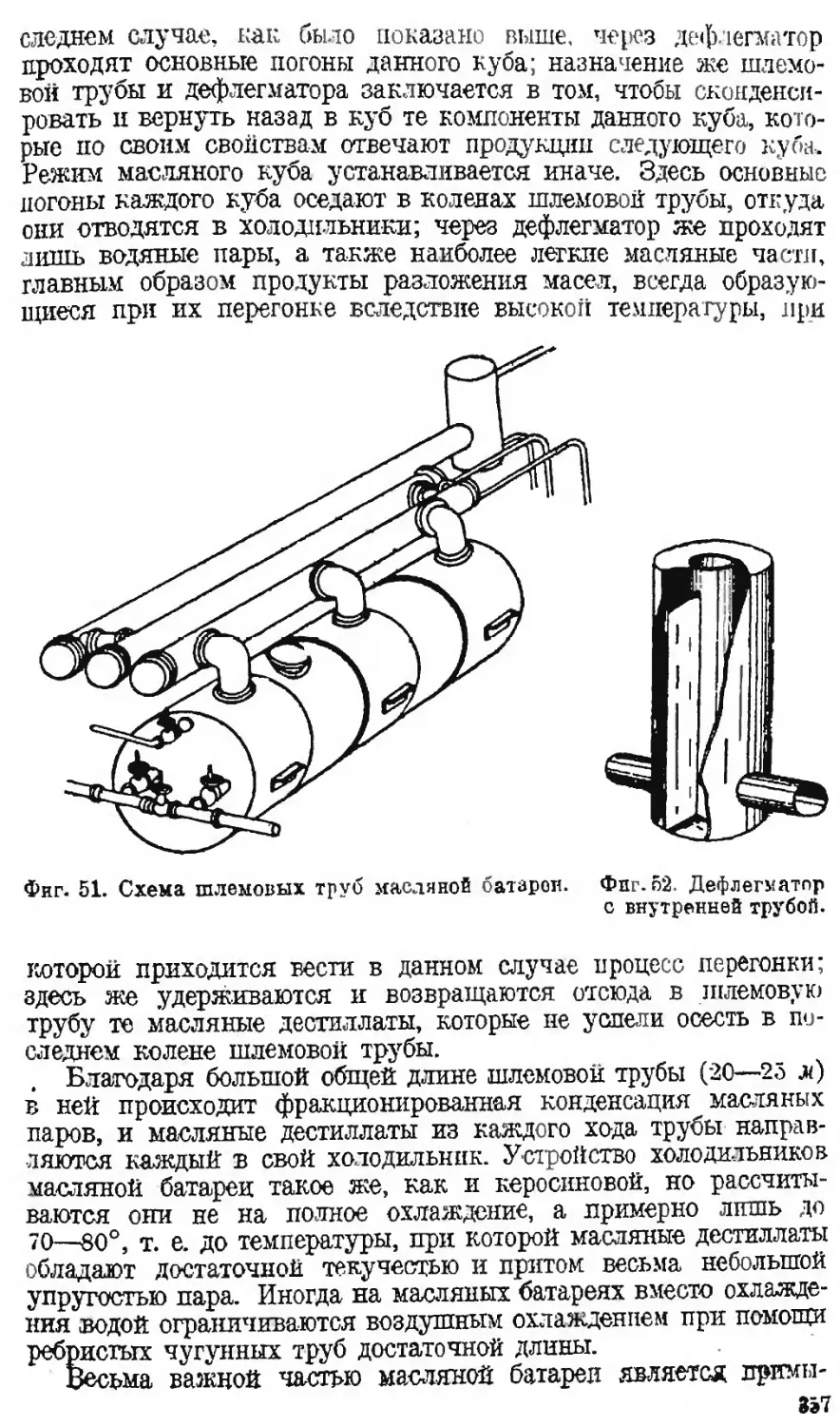











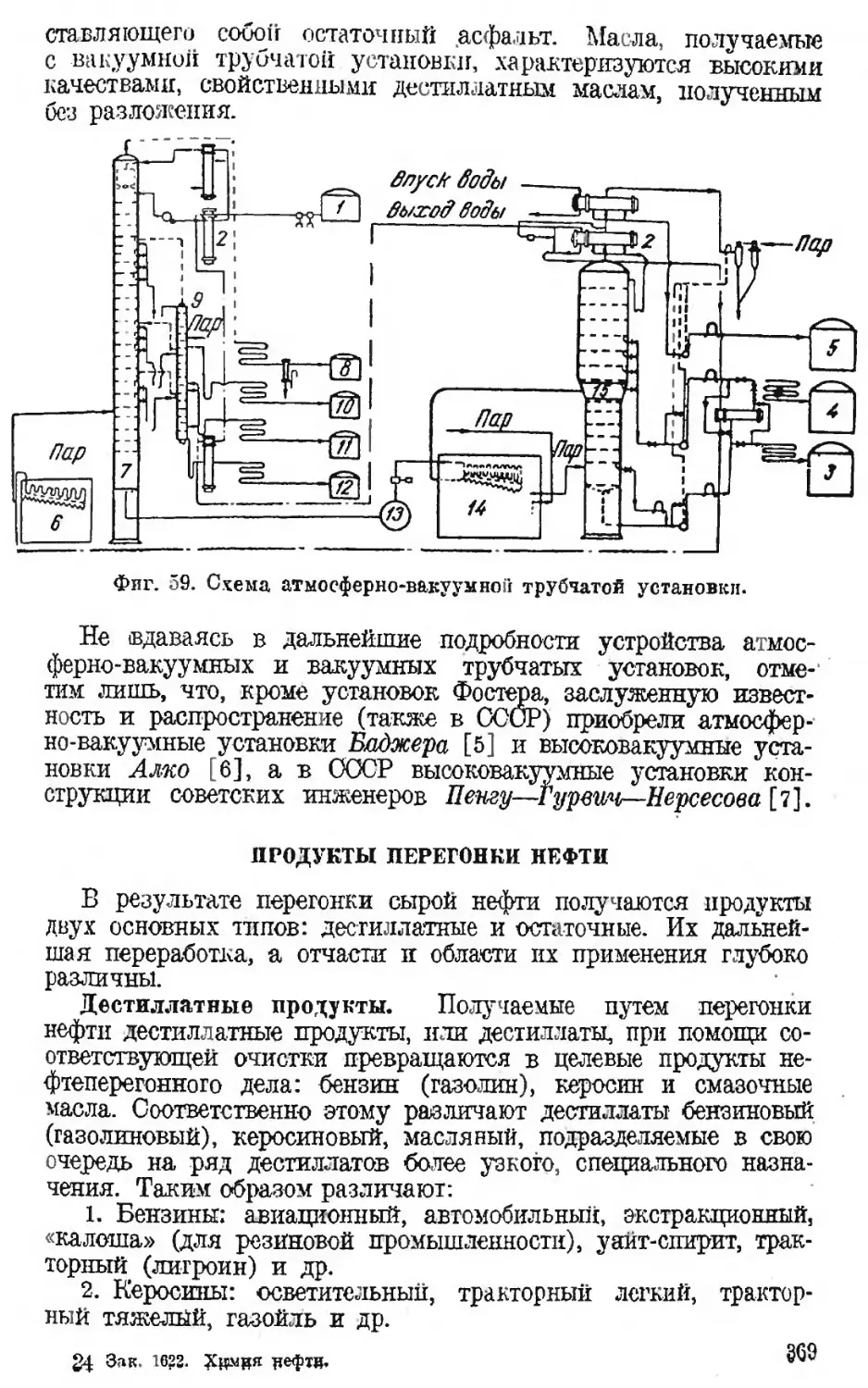












































































































































































































































































































































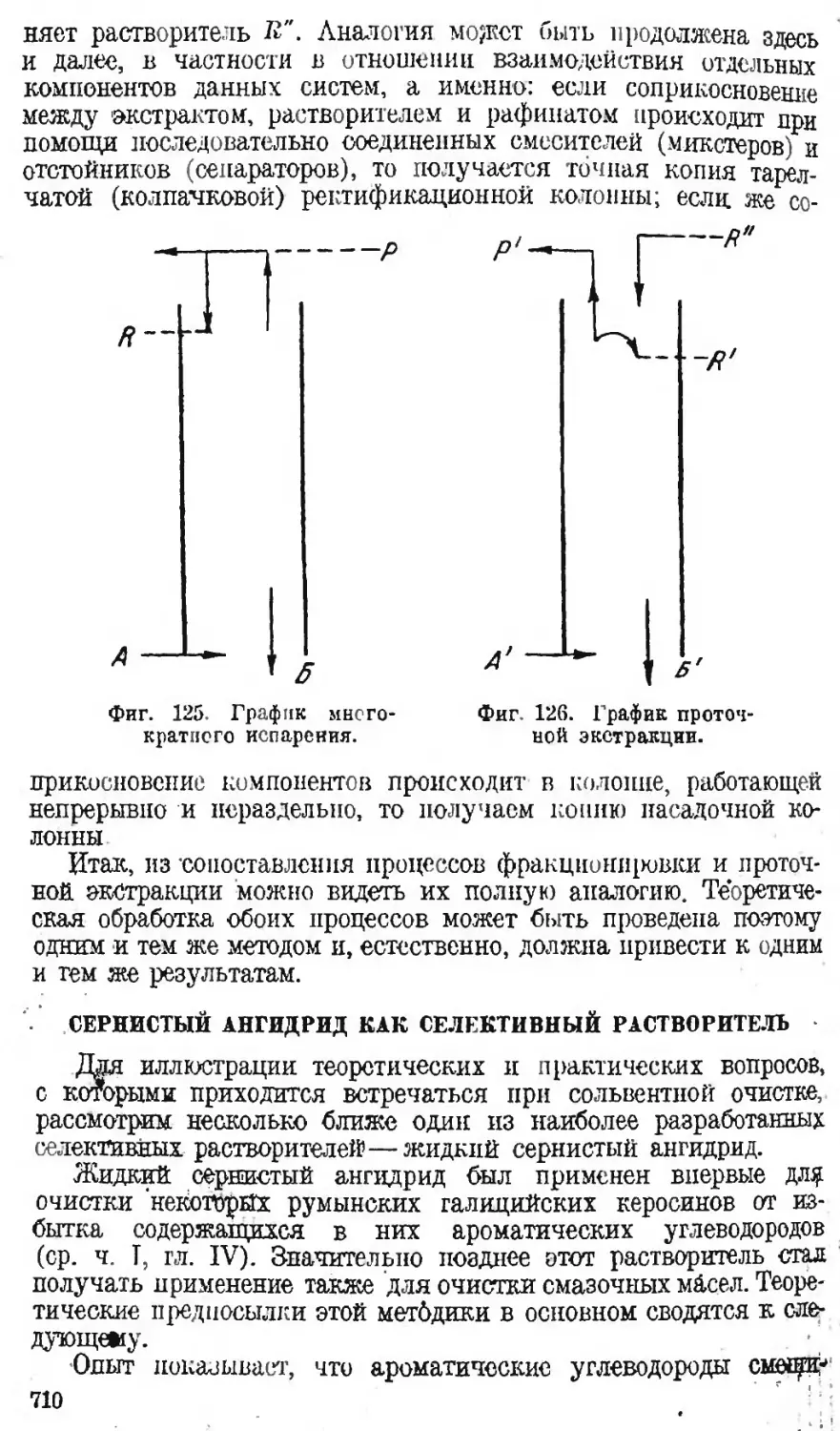



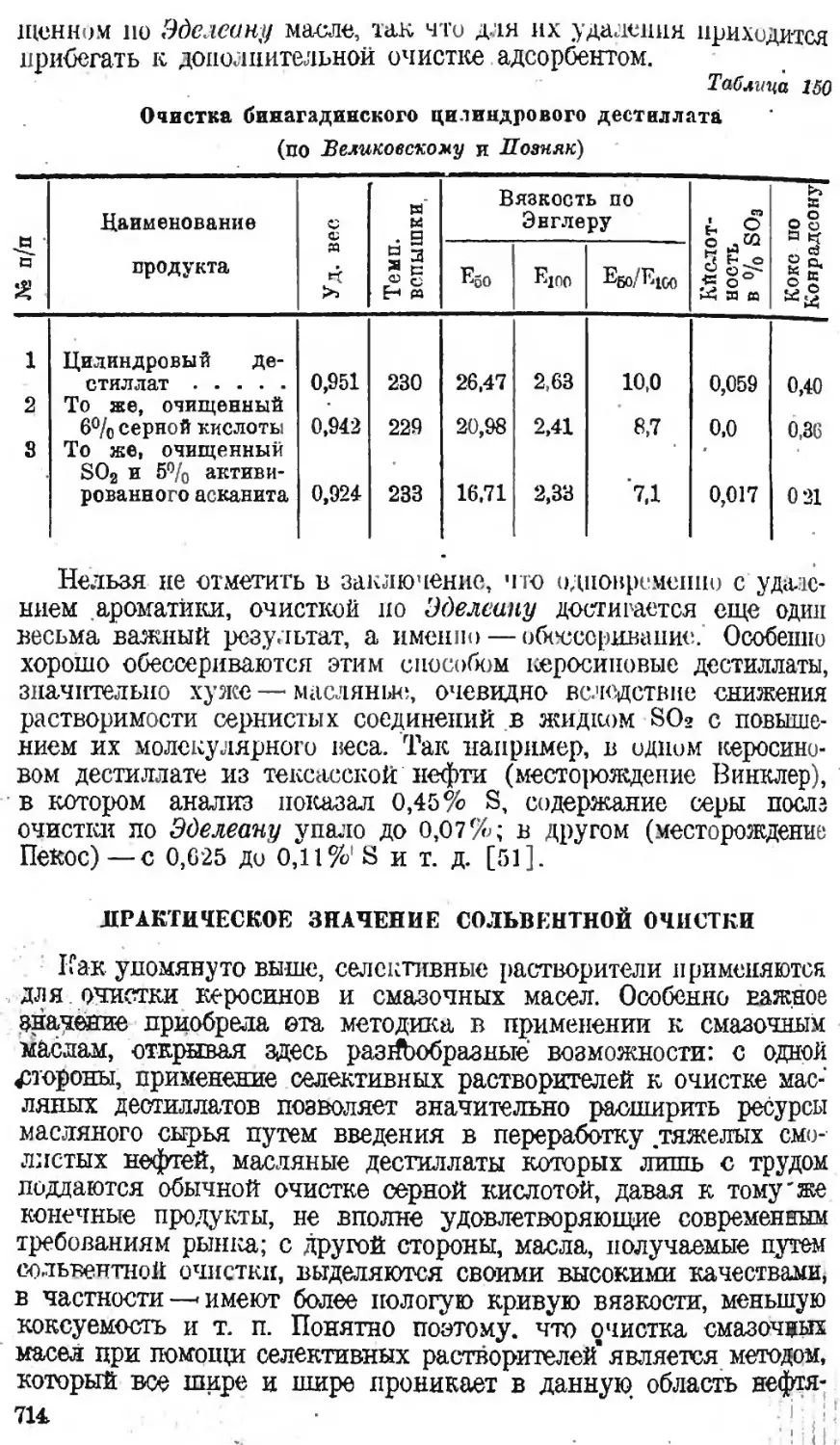














































































Автор: Наметкин С.С.
Теги: химия нефть органическая химия нефтяная промышленность нефтехимия
Год: 1939
Текст
ОПЕЧАТКИ
Стр. Строка Напечатано Следует читать По ВИНС
159 5 си. консистенция Консистентность автора
176 18 св. с реакциями наряду с реакциями тип.
178 25 п Одним и, пожалуй, Одним и еще недавно автора
единственным единственным
184 6 , прямой линии прямой гонки тип.
190 14 я Для замещения Для замещенных
197 1 сн. СбН8(НСз)2 С5Н8(СН3)2 9
292 19 св. солями слоями 99
307 11 л устойчивостью и еустойчивостью 99
325 16 , автором некоторыми авторами автора
338 1 сн. 1751° 175° я
346 17 св. заводской нефти заводской перегонки тип.
нефти
426 в гл. VI в ч. III гл. II Д автора
486 14 сн. циклогексена из бензола бензола из циклогексена 99
619 24 , ОДгвОдН C2H6OSO3H я
625 3 » сульфииированием сульфированием тип.
679 6 „ высокосортных высокосернистых автора
691 9 я чашки части тип.
693 13 св. пропана пропаном автора
762 6 сн. вязкости вязкостные тип.
>&s. 1622. Хлхяя кефтк.
Акад. С. С. НАМЕТКИН
ХИМИЯ НЕФТИ
ИЗДАНИЕ ВТОРОЕ
ИСПРАВЛЕННОЕ И ДОПОЛНЕННОЕ
Утверждено Всесоюзным Комитетом
по делам высшей шкош в качестве учебника
2/з%
ГОСУДАРСТВЕННОЕ
ОБЪЕДИНЕННОЕ НАУЧНО-ТЕХНИЧЕСКОЕ ИЗДАТЕЛЬСТВО
РЕДАКЦИЯ ГОРНО-ТОПЛИВНОЙ И ГЕОЛОГО-РАЗВЕДОЧНОЙ ЛИТЕРАТУРЫ
МОСКВА 1939 ЛЕНИНГРАД
АННОТАЦИЯ
Книга представляет ссбою второе, исправленное и рас-
ширенное издание Kjpca, читанного автором в течение ряда
лет в Московском нефтяном институте им. И. М. Губкина.
Ее задача — дать в сжатом изложении основные предста-
вления о составе, свойствах и происхождении нефти (I ч.),
химические основы переработки нефти (II ч.) и химические
основы очистки нефти и повышения качества нефтепродуктов
(III ч.). По сравнению с первым изданием, план курса остался
без изменения. Более или менее значительные дополнения
внесены во все главы. Кроме того введена новая обширная
глава .Повышение качества нефтепродуктов методом при-
садок “ (ч. III, гл. III).
Книга рассчитана на студентов нефтяных и химических
втузов, а также на аспирантов и работников научно-иссле-
довательских институтов.
Редактор Федотов И. /7. Технич. редактор Б. Модель
Сдано в набор 28 IV 1939 г. Тираж 6000 экз. ГГТ-30-5-2 Прот. Т. К. К. № 29 I 56,1 уч. авт. л. Объем { J 1 49у3 печ. л. Бум. листов 24% Заказ № 1622 Подписано к печати 28/VII 1939 г. Формат бумаги бОХ^’/тв Изд. № 170 Учетный № 1799. Леноблгорлит 3342 Тип. зн. в 1 бум. листе 103168
Бумага Вишерского бумкомбината
3-я тип. ГОНТИ им. Евг. Соколовой. Ленинград, пр. Красных Командиров, 29.
НРЕДИСЛОНИЕ W ВТОРОМУ ИЗДАНИЮ
Второе издание «Химии нефти» идет в печать, когда наша
страна Практически осуществляет третий пятилетии й план раз-
вития .народного хозяйства СССР. Задачи, выполнение которых
поставлено этим планом на очередь, поистине грандиозны по
всем отраслям нашего народного хозяйства. Но с особой силой про-
звучали слова, доклада В. М. Молотова и резолюций XVIII Съезда
ВКП(б) в части, касающейся химической промышленности. От-
ныне все живые химические силы страны должны проникнуться
положением, что «третья пятилетка— пятилетка химии» и что
одной из основных ее задач является задача: «превратить хими-
ческую промышленность в одну из ведущих отраслей промыш-
ленности полностью удовлетворяющих, потребное ги народного
хозяйства и обороны страны». • ' *'
Не менее ответственны задачи, поставленные планом третьей
пятилетки перед нефтяной промышленностью. Не касаясь здесь
всей совокупности мероприятий, намеченных планом в деле раз-
вития и рационализации нашей нефтяной промышленности, до-
статочно вспомнить, что одной из основных задач третьей пяти-
легки является проведение в жизнь Сталинской идеи создания
в районе Урата—Волги Второго Баку, — идеи, в которой кроется
обширнейшая и благодарная программа работ по всем разделам
науки о нефти и по всем отраслям нефтяной промышленности.
Важнейшими условиями успешного выполнения плана третьей
пятилетки, вне всякого сомнения, являются широко поставлен-
ная научно-исследовательская работа по всем очередным, в част-
ности, химико-технологическим задачам пашей промышленности
и надлежащая подготовка химико-технологических кадров для
освоения новых и рационализации старых технологических про-
цессов, намеченных к внедрению и расширению в течение третьей
пятилетки. Подходя с этой точки зрения к своей работе, позво-
ляю себе выразить надежду, что «Химия нефти» в ее новом
издании явится одним из посильных вкладов автора в великое
общее дело выполнения плана третьей пятилетки.
Потребность в новом издании «Химии нефти» обнаружилась
почти тотчас же после выхода II части книги. Уже одно это
обстоятельство свидетельствует о том, что в связи с бурным раз-
витием нашей нефтяной промышленности и ростом ее химиче-
ских кадров нужда в руководстве, которое освещало бы с хими-
ческой точки зрения 'основные вопросы состава и переработки
1* з
нефти, действительно ощущается. Потребовалось, однако, доста-
точно времени и труда, чтобы, внимательно пересмотрев обе
части старого издания, выправить его недочеты, снять все из-
лишнее, утратившее интерес и значение, и восполнить отдельные
главы результатами новейших исследований в данной, быстро
и широко развивающейся отрасли химической науки.
Более или менее обширные пополнения внесены во все главы
нового издания. Кроме того, автор счел необходимым выделить
в особую новую главу важный, имеющий большое теоретическое
и широкое практическое значение вопрос о повышении качества
нефтепродуктов методом специальных присадок (добавок), како-
выми являются ингибиторы, антидетонаторы и т. д. В основном
порядок и распределение материала в новом издании «Химии
нефти» остались без изменения. Лишь глава о происхождении
нефти перенесена из II части в I часть в соответствии с програм-
мой по «Химии нефти», утвержденной ГУУЗ НКТП 29 июля
1938 г. Вместе с тем в связи со значительным расширением
отдельных глав по переработке и очистке нефти признано целе-
сообразным в настоящем издании выделить вопросы очистки и
повышения качества нефтепродуктов в самостоятельную
(III) часть, сохранив во II части лишь вопросы переработки
нефти, т. е. обезвоживание ее и разные виды ее перегонки.
При подготовке к печати второго издания «Химии нефти»
были приняты во внимание полученные мною советы и указания
ряда товарищей. Пользуюсь отучаем выразить мою особую при-
знательность за это Б. П. Воинову, проф. А. К. Зайцеву, проф.
В. И. Исагулянц, проф. С. Н. Обрядиикову, П. В. Пучкову, Л. А.
Потоловскому, Е. А. Робинзон и А. В. Фросту. Позволяю себе
также вновь просить товарищей по работе в области химии и
переработки нефти не отказать мне в указаниях на замеченные
ими недочеты во втором издании этой книги.
Автор
ПРЕДИСЛОВИЕ к ПЕРВОМУ ИЗДАНИЮ
, «Химия нефти» представляет собою несколько расширенный
и дополненный курс, читанный мною в течение ряда лет студен-
там нефтяного факультета Московской горной академии, ныне
Московского нефтяного института им. акад И. М. Губкина. Курс
имеет своей задачей дать в сжатом виде химические основы тех-
нологии нефти, по возможности не затрагивая чисто технологи-
ческих вопросов ее переработки. Это ограничение должно иметь,
по мнению автора, весьма существенное значение для усвоения
химической стороны технологии нефти, т. е. той ее части, кото-
рой за сложностью новейшей аппаратуры и грандиозностью мас-
штабов нефтяной промышленности уделяется все еще недостаточ-
ное внимание. Такое положение дела является несомненно пере-
4
житном эпохи, когда на нефть смотрели почти исключительно
как на котельное топливо; оно — явно ненормально в настоящее
время, когда нефть постепенно приобретает характер сырья, под-
лежащего более или менее сложной химической переработке.
Действительно, не говоря уже о таких новейших методах пе-
реработки нефти, как крэкпнг и гидрогенизация, или о таких
направлениях технологии нефти, которые имеют задачей чисто
химическую переработку отдельных нефтяных фракций прямой
гонки или перегонки под давлением, достаточно вспомнить о том,
настолько усложнились требования, предъявляемые современной
техникой даже к таким нефтепродуктам, которые применяются
либо в качестве топлива для двигателей внутреннего сгорания,
либо в качестве смазочных материалов для машин разнообраз-
нейших конструкций и рабочих режимов. Чтобы удовлетворить
этим требованиям не только с качественной, но и с количествен-
ной стороны, старые, испытанные методы переработки нефти уже
недостаточны. На смену им является новая методика, в основу
которой приходится класть более глубокое знание состава нефти
и химизма способов ее переработки. Отсюда усиленная прора-
ботка разнообразнейпшх вопросов химии нефти в новейшее
время и громадная, быстро растущая литература по вопросам,
которыми еще недавно интересовались лишь отдельные предста-
вители «чистой науки».
Настоящий курс химии нефти распадается на две части. Его
первую часть составляет учение о составе и свойствах нефти; во
вторую часть курса вошли химия переработки и очистки нефти,
а также учение о происхождении нефти с химической точки зре-
ния. Как и в каждом специальном курсе, здесь представлялось
важным не только дать сжатое изложение важнейших результа-
тов исследования по отдельным вопросам и наметить проблемы
и очередные задачи дальнейшей работы, но по возможности кос-
нуться также самого процесса научного исследования нефти и
•его методологии. В связи с этим во всех главах, относящихся
к отдельным компонентам нефти, пришлось уделить внимание
методике анализа нефтей и нефтепродуктов й определения от-
дельных их составных частей, давая каждому методу критиче-
скую оценку с точки зрения его точности и достоверности полу-
чаемых результатов. Два шрифта, которыми отпечатана книга,
позволяют выделить материал первоочередного значения от того,
что имеет характер дальнейшего углубления вопроса.
Каждая глава снабжена- литературными ссылками общего и
специального характера. Первые из них либо дают указания на
источники, в которых материал соответствующей главы изложен
более подробно и которые в большей или меньшей степени были
использованы автором при составлении книги, либо специально
предназначены для первоначального ознакомления’ с предметом
и имеют своею целью подчеркнуть особую важность тщательного
изучения общей и -органической химии как базы для специаль-
ного изучения химии нефти. Ссылки второго рода отсылают чи-
тателя к первоисточникам. Само собою разумеется, что ссылки
5
эти отнюдь не исчерпывают журнальной литературы отдельных
вопросов полностью. В каждом случае автор стремился дать лишь
основные руководящие указания, воспользовавшись которыми,
читатель в порядке дальнейшей самостоятельной работы мог бы
охватить литературу вопроса с желаемой полнотой.
Не подлежит сомнению, что нефтяная промышленность уже
вступила в ту эпоху своего развития, когда не только нефть, но
и мазут будут целиком перерабатываться на полноценные про-
дукты самого разнообразного состава и характера. Мы живем
в начале этой -эпохи, но ее давно уже провидели наши научные
работники в области химического исследования нефти, своими
работами почти исключительно за свой страх и риск стремив-
шиеся приблизить наступление этой эпохи. Имена Менделеева.
Бейлъштейна и Курбатова, Марковникова и Оглоблина, Зелин-
ского, Конова wea, Гурвича и многих других исследователей кав-
казской нефти всегда будут памятны в истории развития хими-
ческого познания нефти вообще; но полагаться и в дальнейшем
на частную инициативу отдельных ученых-энтузиастов, очевидно,
невозможно. Наша нефтяная промышленность должна отчетливо
осознать, что ее дальнейшие успехи, особенно на новом этапе ее
развития, теснейшим образом связаны с успехами химии нефти,
что успехи эти всецело зависят от широкой и планомерной по-
становки научно-исследовательской работы в данной области п
что работы этого рода, несмотря на неотложные заботы сегодняш-
него дня, должны быть в полной мере обеспечены надлежащими:
с^дствами и кадрами. Внести посильную долю в <это большое
дело — главпая задача, котор, ю ставит себе автор настоящей
книги, надеясь, что труд его может послужить пособием не
только для студентов и аспирантов нефтяных факультетов и
вузов, но и ддя работников научно-исследовательских институ-
тов и промышленности.
ЧАСТЬ I
СОСТАВ, СВОЙСТВА. И ПРОИСХОЖДЕНИЕ НЕФТИ
ГЛАВА I
СОСТАВ И КЛАССИФИКАЦИЯ НЕФТЕЙ
Как известно, химическая природа вещества в первую оче-
редь определяется его составом. Ввиду этого изучение элемен-
тарного состава нефти и определение ближайших ее составных
частей являются первой естественной ступенью в химическом
познании нефтей. Эти же данные должны лечь в основу их хи-
мической классификации.
ЭЛЕМЕНТАРНЫЙ СОСТАВ НЕФТИ
Хотя нефть встречается -во всех странах света в весьма разно-
образных геологических условиях и на различных глубинах, тем
не менее элементарный состав ее колеблется в весьма узких пре-
делах (табл. 1). Основными образующими нефть элементами
являются углерод и водород. В громадном большинстве нефтей
содержание углерода колеблется в пределах от 84 до 85%, содер-
жание же водорода редко выходит за границы 12—147с. Отсту-
пления от ©тих норм наблюдаются лишь в тех случаях, когда,
кроме углерода и водорода, в составе нефти принимают заметное
участие другие элементы, а именно кислород, сера и азот. Часто
на долю этих трех элементов приходится в сумме меньше 1%;
-однако встречаются нефти, в которых содержание одной серы
может достигать 1—2% и более. Естественно, что в таких нефтях
содержание углерода и водорода не доходит до указанных выше
Йорм, однако во всяком случае не больше чём на 1% порознь
для углерода и водорода.
Другое свойство, тесно связанное с элементарным составом
нефти*'— ее удельный вес. Чем легче нефть, тем она при прочих
равных условиях меньше содержит углерода и больше водорода;
наоборот, чем тяжелее нефть, тем она при прочих равных усло-
виях больше содержит углерода и меньше водорода. Эта связь
между элементарным составом нефти и ее удельным весом ста-
новится совершенно понятной, если обратиться хотя бы к пред-
варительному рассмотрению ее химического состава.
Т
Определение элементарного состава нефти производится об-
щими методами анализа органических соединений, а именно:
углерод и водород определяются сожжением по Либиху, или в ка-
лориметрической бомбе; азот определяется по Дюма; сера — по
Кариусу, либо иными методами, которые будут рассмотрены
в гл. VIII; наконец, кислород определяется обыкновенно по
остатку, редко — методом непосредственного определения в виде
воды.
Таблица 1
Элементарный состав нефтей различных месторождений
№ п/п Месторождение УД. вес % с %н %s %N % О % золы
1 Сураханы (красная). - 0,793 85,34 14,14 0,03 0,49 ——.
2 Балаханы 0,930 87,01 12,15 0,40 0,44 —
3 Бинагады 0,911 87,01 12,30 0,15 0,54 —
4 Биби-Эйбат 0,901 86,72 12,72 0,20 0,36 —
5 Грозный 0,*50 85,95 13,00 0,14 0,07 0.74 ОДО
6 Грозный 0,906 86,41 18,00 0,10 0,07 0,40 0,12
7 Ухта 0,928 85,47 12,19 1,09 0,20 — 0,02
8 Ухта 0,897 85,30 12,46 0,88 0,14 — , 0,01
9 Галиция (Мразница). . 0,880 84,60 14,00 0,14 1,25 - —
10 Румыния (Буштенари) . 0,842 86,30 13,32 0,18 —
11 Германия (Ганновер; . 0,941 86,50 11,60 1,20 0,70 —
12 Эльзас (Пешрльбронн). 0,918 86,00 12,10 0,80 1,20 “ 1
13 США (Ойл Сити, Пен-
СИЛЬВ.) 0,810 85,80 14,00 —• 0.06 — •
14 США (Финдлей, Охайо) 0,836 84,57 13,62 0,72 0,11 — —
15 США (Канзас) — 84,20 13,00 1,90 0,45 0,45 —
16 США (Бомонт, Текс ас) 0,912 85,00 12,30 1,75 0,92 —
17 США (Вентура, Кали-
форния) 0,912 84,00 12,70 0,40 1,70 1.20 —
18 Канада (Ойл Спрингс) . 0,844 83,60 13,40 0,60 0,18 — —
19 Мексика 0,970 83,00 11,00 4,30 1,70 —
20 Саравак (о. Борнео) . . 0,902 86,47 12,37 0,35 0,13 0,68 —
21 Япония (Амацэ).... 0,829 84,66 13,22 0,22 0,35 1,32 0,23
22 Япония (Катцубо; . . - 0,881 84,52 13,12 0,83 0,97 0,77 0.27
23 Япония (Мийагава) . . 0,935 84,86 13,83 0,32 0,55 0.20 оде
24 Персия (Майдан-и-Наф-
тун) . . . . .... 0,837 85,40 12,80 1,(6 0,76 —
25 Египет 0,907 85,15 11,71 2,25* 0,89
ПРЕДВАРИТЕЛЬНЫЕ ДАННЫЕ О ХИМИЧЕСКОМ СОСТАВЕ НЕФТИ
Химический состав нефти в значительной степени опреде-
ляется уже ее элементарным анализом. Так как многие нефти но
своему элементарному составу более чем на 99% состоят из угле-
рода и водорода, то их главной составной частью являются, оче-
видно, углеводороды. Подавляющее большинство нефтяных угле-
водородов имеет предельный характер, чем резко ограничивается
выбор тех рядов, представители которых составляют главную
массу нефти: очевидно, это могут быть прежде всего углеводо-
роды ряда метана, или парафины, и полиметиленовые углеводо-
роды, или нафтены. Оба эти углеводородных ряда действительно
широко представлены в нефти, но далеко не одинаково в коли-
чественном отношении в одних и тех же погонах.
Углеводороды ряда метана общего состава CJ},H-2.W^2 пред-
ставлены в нефти наиболее полно и разнообразно, а именно, на-
чиная от газообразного метана, простейшего представителя ряда,
и кончая наиболее высокомолекулярными гомологами—твер-
дыми парафинами. Распределение этих углеводородов в различ-
ных нефтяных погонах и содержание их в различных нефтях
крайне неравномерны. Наиболее богаты парафинами газообраз-
ные части нефти, состоящие почти исключительно из низших,
газообразных гомологов метана; представители же других угле-
водородных рядов содержатся в этих газах лишь в качестве не-
большой примеси в виде паров. Жидкие нефтяные - погоны содер-
жат. более или менее зна зительнре. количество углеводородов
ряда метана..лщпь^й^пфеделах .до" 100—150°; -во фракциях же
с температурой кипения выше 150° содержание этих утлеводр-
дгиашо быстро -падает примерно „.до. q и ни,жег
Наконец, фракции пефтаг- начиная с самых легких
(соляровое, веретенное мшм ;т^..дХ .часто содержат"твердый
парафин, количество, которого, заставляет иногда 10—12 %л счи-
тая на сьщую..нефть (парафинистые нефти)? ОДНйко широко рас-
прбстранены также нефти, в которых содержание парафина либо
не превышает 1—2% (слабопарафинистые нефти), либо спу-
скается еще ниже — до таких количеств, точное определение ко-
торых с помощью обычно применяемых методов затруднительно
(беспарафиновые нефти). Наиболее яркие примеры парафи-
нистых нефтей OGCP дают грозненская, сураханская и челекен-
ская нефти; из парафинистых нефтей других европейских место-
рождений достаточно отметить румынские и галицийские нефти;
из американских — пенсильванскую нефть и некоторые мекси-
канские.
Даже в парафинистых нефтях уже значительно ниже 50°
к углеводородам ряда метана начинают присоединяться углево-
дороды других рядов и прежде всего простейшие нафтены, или
полиметилены. Эти углеводороды имеют непредельный состав, но
по своим химическим свойствам мало отличаются от гомологов
метана, т. е. имеют ясно выраженный предельный характер. Осо-
бенностью их строения является, как известно, кольчатое или
циклическое расположение углеродных атомов в частице угле-
водорода. Простейшие нафтены, состава СПН2И, содержат одно
такое углеродное кольцо- (цикл) и называются моноцикличе-
скими. Как показывает опыт, синтетически могут быть получены
моноциклические системы с весьма разнообразным числом угле-
родных атомов в цикле: от 3 до 15—17. а невидимому, и гораздо
больше (до 40 О). Замечательно, однако, что моноциклические
нафтены, обнаруженные до сих пор в нефти, принадлежат лишь
к двум нафтеновым рядам, а именно: к ряду циклопентана, т. е.
с 5 атомами углерода в цикле, и циклогексана, т. е. с 6 атомами
углерода в основном ядре; монощгелические же системы трех-
или четырехчленные, а также с 7, 8 и т. д. атомами углерода
9
в цикле до сих пор в нефти не обнаружены и, невидимому, в ней
не содержатся.
Кроме нафтенов рассмотренного типа, известно немало более
сложных кольчатых систем предельного характера, которые по
своей химической природе напоминают моноциклические наф-
тены. Их можно определить как би-, три- и вообще полицикли-
ческие нафтены. Простейшие из них генетически связаны с обще-
известными конденсированными системами ароматического ряда
(нафталин и т. п.). Все они соответственно своему циклическому
строению имеют различный непредельный состав, но вместе с тем
подобно моноциклическим нафтенам — ясно выраженный пре-
дельный характер. Примером бициклических нафтенов может
служить декагидронафталин или декалин, CioHie, обнаруженный
в различных нефтях. Судя по составу средних и высших нефтя-
ных погонов, весьма вероятно нахождение в нефти также многих
других би- и полициклических нафтенов; однако исследование
этих погонов представляет, как будет показано ниже, чрезвычай-
ные трудности и пока находится в начальной стадии.
Хотя содержание нафтенов в различных нефтях пока не
поддается точному учету, тем не менее не подлежит сомнению,,
что нафтеновые углеводороды .составляют -главную_ массу' .боль-
удаствал^фтейг--а.именно для средних и высших’погонов — до
60^70%^ jix состава и больше. Особенно это относится, конечно,
к нефтям слаГопарафинистым и беспарафиновым; но и парафи-
нистые нефти в своих погонах с температурой кипения выше 300°
.состоят преимущественно из углеводородов различных нафте-
новых рядов, поскольку об этом можно судить на основании их
предельного характера при непредельном составе. Наиболее бо-
гаты нафтенами из нефтей СССР — бакинские и эмбенские нефти,
из американских нефтей — калифорнские.
Кроме парафинов и нафтенов, одной из постоянных состав-
ных частей нефти являются бензольные или ароматические угле-
водороды. Низшие, наиболее изученные и интересные в практи-
ческом отношении углеводороды этого ряда, т. е. бензол, толуол
и ксилолы, содержатся в большинстве нефтей в весьма ограни-
ченном количестве. Выделенные из _них„лшрокие -бензольная,
толуольная и ксилольная фракции содержат обыкновенно от "1
до ,16%‘ ароматики на‘данную фракцию, и только для некоторых
нефтей эта величина поднимается до 12—17%. Наконец, хотя
и очень редко, встречаются нефти, в которых содержание бензола
в бензольной фракции (температура кипения 60—95°) достигает
20%, содержание же толуола и ксилолов в соответствующих по-
гонах (до 150°) поднимается до 30—60% и выше; в этих же при-
мерно границах удерживается содержание ароматики и в после-
дующих погонах. Таким образом для этих, правда, очень редких
нефтей ароматические углеводороды являются главной составной
частью,, т. е. играют в их составе такую же роль, какую для
большинства нефтей играют нафтены. Примерами особенно бо-
гатых ароматикой .нефтей могут служить из нефтей СССР ураль-
ская нефть (м. Чусовские Городки) и в значительна меньшей
Ю
степени — майкопская; из нефтей других месторождений — нефть
Зондских островов (Борнео. Ява).
Парафины, нафтены и ароматические углеводороды являются
главной составной частью нефти. Представители других углево-
дородных рядов, например непредельных, также обнаружены
в различных нефтях, но в крайне незначительных количествах.
Гораздо больший интерес для состава -нефти представляют не-
которые другие классы органических соединений, на (присутствие
которых указывает уже содержание в ней кислорода, серы и
азота (табл. 1). Правда, в количественном отношении, как об
этом можно судить по содержанию в различных нефтях соответ-
ствующих элементов (О, S и N), эти соединения играют в составе
большинства природных нефтей лишь второстепенную роль. Так
например, важнейшие из кислородных соединений нефти — неф-
тяные кислоты — содержатся в средних нефтяных погонах в ко-
личестве не более 1—2%, а в наиболее легких и самых тяжелых
дестиллатах (бензиновый, керосиновый, цилиндровый) их еще
меньше (0,0—0,5%). Что касается, наконец, сернистых и особенно
азотистых соединений нефти, то, как общее правило, они содер-
жатся в нефтях в количествах, значительно меньших, чем неф-
тяные кислоты.
Есть, однако, один тип кислород- и серу-содержащих соеди-
нений, которые занимают в составе некоторых нефтей видное
место; это— смолистые и--асфальтовые вещества, нефти Содер-
^.ание-этих.веществ в некоторых.тяжелых нефтях достигает Ш—
20%; а так как при 'этом нефти приобретают о<^бый-.ацецифиие-
скпй характер>..то. иногда их выделяют & -особую группу алв-
чистых, .нефтей Те из них, которые особенно богаты смолистыми
if асфальтовыми веществами, представляют собою уже переход-
ные формы от нефтей к природным асфальтам.
Этих предварительных данных о составе нефти вполне доста-
точно для объяснения отмеченной выше связи между удельным
весом нефти и ее элементарным составом. Чем тяжелее нефть,
тем больше содержится в ной высокомолекулярных углеводоро-
дов; в последних же по мере увеличения молекулярного веса
содержание углерода, естественно, возрастает, содержание же
водорода уменьшается, — правильность, которой подчиняются все
углеводородные ряды, за исключением нафтенов общей формулы
СнНг». Понятно поэтому, что чем тяжелее нефть, тем -в известных,
отмеченных выше пределах она богаче углеродом и беднее водо-
родом. Некоторую роль <в этоИ правильности может играть также
присутствие в" тяжелых нефтях других высокомолекулярных
соединений, например нефтяных смол и асфальтов.
ХИМИЧЕСКАЯ КЛАССИФИКАЦИЯ НЕФТЕЙ
В основу хпмшеекюй классификации нефтей должен быть
положен их*химический состав, -и в нефтяной литературе’ можно
найти несколько опытов в этом направлении [3, 7]. Так- какт
однако, данные о химическом составе нефтей крайне недоста-
11
точны и неполны, то в американской литературе обычно ограни-
чиваются характеристикой нефти, основанной на содержании,
в ней двух наиболее точно определяемых ее компонентов: пара-
фина и смолисто-асфальтовых веществ. Соответственно этому
нефти подразделяются на три следующих группы:
1) нефти парафинового основания;
2) нефти асфальтового основания;
3) нефти смешанного основания.
В настоящее время, в связи с несомненными успехами в деле
исследования химического состава нефтей, вопрос об их класси-
фикации вновь может быть поставлен на очередь. В основу ра-
циональной химической классификации нефтей должно- быть по-
ложено содержание в ннх какого-либо основного их компонента,
т. е. того или иного углеводородного ряда. Мы видели, что тако-
выми являются три ряда углеводородов: парафины, нафтены и
бензольные углеводороды (ароматика), соответственно чему в пер-
вую очередь намечаются следующие три основных типа нефтей:
I. Метановые нефти.
II. Нафтеновые нефти.
III. Ароматические или бензольные нефти.
Следует помнить, однако, что углеводороды тоге или иного
ряда никогда не являются единственным углеводородным ком-
понентом какой-либо нефти. Как явствует из вышеизложенного,
вместе, например, с нафтенами в нефти обыкновенно содержатся
в большем или меньшем количестве также парафины и арома-
тика. Таким образом при определении типа нефти надо иметь
в виду только преобладающее содержание в ней углеводородов
одного из вышеуказанных рядов, т. е. когда тот или иной выше-
указанный компонент нефти составляет больше ее половины
(50%). В подобного рода случаях вторая, меньшая, половина
нефти приходится на долю смолисто-асфальтовых и других вто-
ростепенных ее компонентов (нефтяные кислоты, сернистые со-
единения и т. д.), главным же образом — на долю других углево-
дородных рядов, сопутствующих основному. Если один из этих
дополнительных углеводородных рядов содержится в нефти в ко-
личестве не менее 25% ее состава, то вся нефть, естественно,
приобретает некоторые добавочные специфические особенности,,
свойственные данному ряду. Таким образом могут быть наме-
чены 6 смешанных типов нефтей, при обозначении которых
основной компонент должен занимать некоторое определенное,,
например последнее, место, а именно:
IV. Метано-нафтеновые нефти.
V. Нафтено-метановые нефти.
VI. Бензольно-нафтеновые нефти.
VII. Нафтено-бензолъные нефти.
VIII. Бензолъно-метановые нефти.
IX. Метано-бензолъные нефти.
Наконец, можно представить себе такие случаи, и они дей-
ствительно наблюдались, когда каждый из основных углеводо-
родных рядов, т. е. метановый, нафтеновый и бензольный, пред-
12
ставлены в нефти примерно >в равных количествах. Мы получаем
в таком случае последний смешанный тип:
X. Метано-бензолъно-нафтеновые нефти.
Раньше чем перейти к примерам, которые могут иллюстриро-
вать приведенную классификацию нефтей, необходимо отметить
следующее. Согласно применяемой в настоящее время и, как
будет показано йиже, пока довольно несовершенной методике
количественное определение углеводородов различных рядов про-
изводится не сразу во всей нефти, а в отдельных более или ме-
нее широких ее погонах. Так как наиболее точные результаты
получаются при этом для погонов, выкипающих до 300°, то при
определении типа нефти данные об этих погонах должны пре-
имущественно приниматься во внимание. Условие это особенно
важно в тех нередких случаях, когда тип нефти при переходе
от низших ее погонов к высшим несколько изменяется, что для
полной характеристики типа нефти должно быть, конечно, ого-
ворено. .
Приведем в заключение несколько примеров распределения
нефтей по вышеуказанным типам, воспользовавшись для этого
произведенными в ГрозНИИ анализами нефтей преимуще-
ственно СССР [7].
Как уже было указано, три основных типа нефтей встречаются
сравнительно редко. Наиболее распространенным из них
является нафтеновый тип, примерами которого могут служить
среди нефтей СССР прежде всего эмбенские нефти (Доссор, Ма-
нат); сюда же можно, невидимому, причислить некоторые наши
тяжелые нефти различных месторождений, например: тяжелую
майкопскую, калужскую, а также кирмакинейую нефть. Наконец,
довольно ярко выражен нафтеновый тип у некоторых бакинских
нефтей, например балаханской и сураханской, хотя отдельные
погоны этих нефтей, низшие п средние, вследствие значитель-
ного содержания парафинов, приобретают метано-нафтеновый
характер.
Нефти метанового типа в чистом виде не встречаются в СССР.
Так, даже грозненская парафинистая нефть в своих погонах до
300° имеет ясно выраженный нафтено-метановый характер; в по-
гонах же ее, кипящих выше 300°, нафтены по сравнению с пара-
финами выходят уже на первое место (метано-нафтеновый тип).
Таков же, повидимому, характер большинства других нефтей
Грозненского района (см. ниже).
Что касается третьего, основного типа нефтей, бензольного
или ароматического, то единственная нефть СССР, которая более
или менее' подходит к нему, это — уральская нефть (м. Чусов-
ские Городки). Однако значительное содержание в отдельных ее
погонах нафтенов заставляет отнести ее скорее к нафтено-бен-
зольному типу. .
Переходим к смешанным типам нефтей. Выше уже были при-
ведены примеры некоторых смешанных типов, а именно метШ>
нафтеновых, нафтено-метановых и нафгено-бензольных цефтей.
Примерами бенаольно-нафтенового типа могут служил» нефти
; 13
^иби-эйбатская и грозненская беспарафиновая (верхние пласты’
тяжелая); однако в своих легких погонах (до 150 ) обе эти нефти
носят иной характер, а именно являются метано-нафтеновыми.
Нефти метано-бензольного и бензольно-метанового типа, по-
водимому, в природе не встречаются.
Наконец, примером нефти, в которой все три основных угле-
водородных ряда представлены ботее или менее одинаково, мо-
жет служить майкопская нефть (метано-бензольно-нафгеновый
тип).
Приведенная выше химическая классификация нефтей может быть изо-
бражена графически, как показано на фиг. 1; дополнением к ней должна
служить табл. 2, в которой поименованы все 10 возможных типов нефтей
с их порядковыми литерами п числовыми индексами [8].
Таблица 2
Химическая классификация нефти
№ п/п Наименование классов Поряд- ковые литеры Число- вой индекс
I Метановые нефти А 1
II Нафтеновые „ В 2
III Бензольные „ ..... С 3
IV Метано-нафтендвые нефти м 12
V Нафтено-метановые , N 21
VI Бевзольно-нафтеновые „ 0 32
VII Нафтено-бензольные . р 23
VIII Бенвольно-метановые , R 31
IX Метано-бен зольные , Q 13
X Метано-нафтено-бензольные S 123
Фиг. 1. График химической клас-
сификации нефтей.
Согласно этому графику и табл. 2 смесь углеводородов какого-либо одного
ряда должна обозначаться однозначн*ым числом 1 (метайовая смесь), 2 (нефт-1-
новая смесь) или 3 (бензольная смесь),
и ее графическое изображение будет
представлять собою это число, ио мо-
щенное в центре круга (фиг. 2 или вну-
тренний круг фиг. 1). Двойные смеси,
к которым принадлежат некоторые при-
родные нефти, должны иметь индек-
сом двузначное число, причем цифра,
отвечающая преобладающей группе
углеводородов, должна занимать, со-
гласно сказанному выше, второе место.
Графически такие смеси углеводородов
могут быть изображены круговой стрел-
кой с двумя цифрами; стрелка должна
указывать на индекс преобладающей
в данной смеси группы углеводородов.
Кроме того, за каждой двойной смесью
подобного рода может быть закреплена
некоторая порядковая литера. Так на-
пример, нафтеяо-метановая нефть имеет
литеру N; ее индекс — 21 (график
см. фиг. з). Наконец, равномерная смесь углеводородов всех трех рядов
должна изображаться трехзначным числом 123; ее графиком является круг
с тремя разрывами (фиг 4), т. е. внешний круг фиг. 1
14
В заключение необходимо еще раз подчеркнуть условность
химической классификации нефтей, зависящую от нередко на-
блюдаемой изменчивости их состава при переходе от низших
погонов к высшим. Это обстоятельство, иллюстрированное выше
на примере грозненской парафинистой нефти, заставляет харак-
теризовать химический состав, нефти не в целом, а по отдельным
более или менее широким ее погонам. Естественно, что в такого
рода случаях можно говорить о принадлежности к тому или
иному типу не всей данной нефти, а лишь отдельных ее пого-
нов более или менее однородного состава.
Фиг. 3. График
смеси углеводоро-
дов двух рядов.
Фиг. 2. График
смеси углеводоро-
дов одного ряда.
Фиг. 4. График
смеси углеводоро-
дов трех рядов.
Несмотря на громадные трудности, встречаемые при рациональной
классификации нефтей, попытки в этом направлении продолжаются. Отме-
тим здесь новую классификацию американских нефтей, недавно разрабо-
танную и опубликованную Горным бюро США [9], в основе которой по-
ложены следующие операции и характеристики нефтей:
1. Разгонка нефти сначала при атмосферном давлении по стандартному
методу до 200° и далее на двадцатипятиградусные фракции, кончая
250—275° (показательная фракция легкой части нефти).
2. Разгонка нефти при 40 мм до 200° и дальше на двадцатипятигра-
дусные фракции, кончая 275—300° (показательная фракция тяжелой части
нефти).
3. Определение удельного веса обеих показательных фракций и отне-
сение легкой и тяжелой частей нефти к одному из трех классов соответ-
ственно границам удельных весов, установленным в табл. 3 для нефтей
парафинового основания, промежуточного основания и нафтенового осно-
вания- ‘ Таблица 3
Нормы к классификации нефтей по Горному бюро США.
Показательные фракции Удельны1? вес показательных фракций нефтей
Нефти пара- финовбго основания Нефти проме- жуточного основания Нефти нафте- нового осно- вания
Легкая часть нефти 0.8251 от 0,8255 0,8602
(фр’тция 250—275° при атмосферном давлении) и ниже до 0,8597 и выше
Тяжелая часть нефти 0,8762 от 0,8767 0,9340
(фракция 275—300° при 40 мм) и ниже до 0,9334 и выше
15
4. Наконец, на основе характера показательных фракций определяется
один из семи классов, к которому должна быть отнесена данная нефть
согласно табл. 4.
Таблица 4
Классификация нефтей по Горному бюро США
я 'и' Класс или основание нефти Основание легкой части нефти Основание тяжелой части нефти
1 Парафиновое Парафиновое Парафиновое
2 Парафино-промежуточное Промежуточное
3 Промежуточно-парафино- вое Промежуточное Парафиновое
4 Промежуточное W Промежуточное
5 Промежуточно-нафтеновое в Нафтеновое
6 Нафтено-промежуточное Нафтеновое Промежуточное
7 Нафтеновое и Нафтеновое
Новейшая классификация американских нефтей представляет несо-
мненный интерес прежде всего уже потому, что в ней находит отражение
характер как низших, так и высших нефтяных погонов. Приходится отме-
тить, однако, известную произвольность границ удельных весов показа-
тельных фракций, а также и то обстоятельство, что обозначения отдельных
классов здесь совершенно условны и отнюдь не отражают не только состав
нефти, но и те принципы, которые заложены в основу классификации.
Ввиду этого новейшая классификация американских нефтей имеет, оче-
видно, лишь временны^-л чисто служебный характер.
ЛИТЕРАТУРА
1. Гурвич Л. Г. Научные основы переработки нефти, 2-е пзд., М. — Л. (1925).
2. Исследование апшеронских нефтей, Центральная химическая лаборато-
рия Азнефти, Баку (1926).
3. Гефер Г. Нефть и ее ироизводные. пер с нем., М. (1907).
4. 'Engler С. und Hofer Н, Das Erdol, т- I (1913).
5. Redwood В., A. Treatise on Petroleum, т. I и HI, London (1922).
6. Hay H„ A. Handbook of the Petroleum Industry, т. I, New lork (1922)
7. Химический состав нефтей и нефтяных продуктов, «Труды ГрозНИИ4*
2-е ивд., М. —Л. (1935).
8. Частное сообщение Воинова Б. П. Баку.
9. U. S. Bureau of Mines. Rept. of Investigations, № 3279 (IX 1985k cm.
также Седых H., „Нефть* 20, 12 (1935).
ГЛАВА И
ФИЗИЧЕСКИЕ СВОЙСТВА НЕФТИ
Всякое химически чистое индивидуальное вещество характе-
ризуется совокупностью физических свойств, называемых его
константами. Таковы, например, удельный вес вещества, его тем-
пература кипения, температура плавления и т. д. Все эти свой-
ства или константы вещества всецело определяются его химиче-
ским составом и строением: изменяется состав или строение ве-
щества— изменяются и его константы.
Так как нефть представляет собою не химически индиви-
дуальное вещество, а чрезвычайно сложную смесь, к тому же
переменного состава, то говорить о константах нефти, как гово-
рят, например, о константах химически чистой воды, спирта или
какого-либо углеводорода, очевидно, невозздежно, тем более что
состав и свойства нефти, в зависимости от условий ее хранения,
улетучивания легких частей и т. п., могут весьма существенно
изменяться. И тем не менее, для характеристики нефти опреде-
ление ряда ее физических свойств имеет весьма важное значе-
ние. При всей простоте определения таких свойств, как удель-
ный -вес, температура кипения или застывания и т. п., они дают
первую, хотя и грубую характеристику нефти в отношении ее
состава и товарных качеств. Определение некоторых других фи-
зических свойств нефти является важным в ином отношении:
они дают основания для расчета и проектировки нефтепроводов,
аппаратуры для переработки данной нефти и т. п. Подробное
рассмотрение различных физических свойств нефти и методов
нх определения могло бы составить предмет специального курса
«ФЩ51Ёка.нефти»; в последующем изложении вопросы, относя-
:щйеся к этой обширной теме, будут освещены лишь в самых
общих чертах Ч
ОБЩАЯ ХАРАКТЕРИСТИКА
Нефть представляет собою жирную наощупь горючую жид-
кость, обыкновенно теынокоричневого или черного цвета. Кон-
систенция различных нефтей крайне разнообразна: от жидкой
1 Прекрасное начало курсу «Физика нефти» положил Кусаков М. М.
в своем фундаментальном труде: «Методы определения физико-химических
характеристик нефтяных продуктов», ОНТИ, М.—Л. (1S36).
2 Вак. 1622. Химия нефти.
17
маслянистой до густой смолообразной. Громадное большинство-
нефтей — легче воды. Все они обладают специфическим харак-
терным запахом, который от присутствия более или менее значи-
тельных количеств сернистых соединений может становиться’
крайне неприятным и даже трудно переносимым. Из нефтей
СССР ярким примером нефти такого рода может служить ураль-
ская нефть.
УДЕЛЬНЫЙ ВЕС
Для громадного большинства нефтей удельный вес их укла-
дывается в пределы от 0,75 до 1,00. Лишь как исключение встре-
чаются нефти с удельным весом меньше 0.75 и густые асфальто-
образные нефти с удельным весом больше 1,00.
Обыкновенно в каждом месторождении можно встретить как
легкие нефти с удельным -весом до 0,80—0,85, так и тяжелые
с удельным весом выше 0,90. Так например, удельный вес бакин-
ских нефтей колеблется к пределах от 0,78 до 0,93; для саха-
линских нефтей пределы этих колебаний от 0,83 до 0,94; для
калифорнских— от 0,77 до 1,01; для мексиканских — от 0,79
до 1,06; для тексасских — от 0,80 до 0,97 н т. д. Однако встре-
чаются месторождения и с более узкими пределами удельного
веса. Так например, для основных районов Грозненского место-
рождения колебания удельного веса нефтей ограничиваются' пре-
делами 0,84—о.-s7; для эмбенских нефтей — от 0,86 до 0,90 и
т. д.
Причину различия удельных весов у различных нефтей сле-
дует искать, конечно, в их составе и прежде всего в различии
химической природы их углеводородной части. Так как углево-
дороды различных рядов при одинаковой сложности их со-
става заметно различаются между собой удельным весом, то,,
естественно, преобладание в нефти того или иного углеводород-
ного ряда не может не отразиться на ее удельном весе: нефти
с преобладанием углеводородов парафинового ряда будут, вообще-
говоря, легче нефтей нафтенового типа; последние же — легче-
нефтей, богатых ароматическими углеводородами. Крайними при-
мерами, иллюстрирующими это положение, могут служить, с од-
ной стороны, легкие парафинистые пенсильванские нефти, с дру-
гой — тяжелая, богатая ароматикой уральская (пермская)-
нефть.
Наряду с химической природой образующих нефть углеводо-
родов большое влияние на величину ее удельного веса оказы-
вают и другие факторы. Так например, потеря нефтью легких
ее частей вследствие их испарения, естественно, должна сказы-
ваться на повышении ее. удельного веса. Если такое улетучива-
ние легких частей происходит в естественных условиях (выве-
тривание), то оно сопровождается обыкновенно побочными про-
цессами окислительного характера; в результате последних
нефть обогащается тяжелыми, смолистыми продуктами, еще бо-
лее повышающими ее удельный вес. Отсюда следурт, что влияние-
химической природы нефтяных углеводородов на удельный вес
18
л вообще физические свойства не4 та можно наблюдать лишь на
сравпимых объектах и лучше — не на сырых нефтях, а на со-
ответствующих, полученных из них дестиллатах и нефтепродук-
тах: бензине, керосине и т. п. с, одинаковыми пределами темпе-
ратуры кипения.
Большой интерес и значение представляют изменения удель-
ного веса нефтей отдельных месторождений в зависимости от глу-
бины их залегания. Характер этих -изменений может быть двоя-
кого рода.
В одних случаях удельный вес нефти уменьшается по мере
перехода от верхних нефтеносных пластов к нижним. Такого
рода изменение удельного веса нефтей с глубиной их залегания
наблюдается, например, у нас в Грозном и Майкопе, а также
в Галиции, Ганновере, Пенсильвании и некоторых других место-
рождениях. Оно легко объясняется большей сохранностью нефти
в нижележащих горизонтах, особенно если они защищены сверху
непроницаемыми, например глинистыми пластами, и сравни-
тельно большей склонностью ее к испарению зг окислению
в вышележащих, особенно в пористых слоях. Явления утяжеле-
ния нефти под влиянием указанных факторов наиболее ярко
наблюдаются при естественных выходах нефти, где зачастую она
является в виде густой вязкой массы, подобной горному дегтю.
В других случаях ход изменения удельного веса нефтей
с глубиной продуктивных горизонтов оказывается прямо проти-
воположным, т. е. по мере возрастания глубины залегания удель-
ный вес нефти не уменьшается, а увеличивается. Явления по-
добного рода, встречаемые в Бакинском районе, Эльзасе и дру-
гих месторождениях, объясняются следующим образом. Нередко
нефть встречается в залеганиях вторичного характера,, образо-
вавшихся в результате ее перехода с места образования в новые,
вышележащие горизонты. Эта миграция нефти может происхо-
дить по трещинам и расселинам; она может сопровождаться
также фильтрацией через пористые горные .породы, в ре-
зультате чего наиболее тяжелые смолистые части нефти, встре-
чая на своем пути некоторые породы, например типа флоридина,
могут ими задерживаться (адсорбироваться), облегчая, таким
образом, нефть, поступающую в верхние горизонты.
Что касается, наконец, изменения удельного веса для различ-
ных фракций какой-либо одной нефти, то, как общее правило,
удельный вес их возрастает по мере повышения температуры их
кипения: керосиновые погоны имеют более высокий удельный
вес, чем бензиновые, и т. д. Такая правильность наблюдается,
однако, лишь при грубой разгонке. Более тщательная фракцио-
нировка, когда влияние отдельных компонентов на физические
свойства смеси может сказываться более явственно, приводит
к гораздо более сложной зависимости, примеры которой будут
приведены в следующей главе при изложении результатов фра&-
ционировки бакинского бензина.
Удельный вес нефтей и нефтепродуктов находит широкое при-
менение для расчетов веса продукта по занимаемому им ббъему
’ 19
или, наоборот, объема продукта по его весу. Эти расчеты про-
изводятся по известной формуле:
P=VD,
где Р — вес тела, V — его объем и D — удельный вес.
Для ближайшей характеристики нефтей и нефтепродуктов и
их оценки удельный вес имеет довольно ограниченное значение.
Определение удельного ве^а нефтей п нефтепродуктов производится
либо ареометром, либо весами Вестфаля, реже—пикнометром. Относительно'
температурных условий, прп которых определяется удельный вес нефте-
продуктов, в разных странах полного единообразия не наблюдается. В СССР
вес нефтепродукта при 15° Ц относится к весу равного объема воды также
при 15° Ц, т. е. дается В США определение ©едется при 60° Ф или
16,66° Ц, т. е. определяется djjj- Такие же температурные нормы приняты
в Англии. Наконец в других европейских странах вес нефтепродукта при
15° Ц относится к весу воды при 4° Ц, т. е. определяется d™.
Отметим в заключение, что вместо обычных ареометров, которые сразу
дают удельный вес жидкости по отношению к воде, .в нефтяной промыш-
ленности США применяются обычно ареометры Бомэ (Ваитё), которые
дают плотность в градусах Боме (°Вб). Имеются две шкалы Боме, пока-
зания которых несколько отличаются между собою. Одна из них принята
Бюро стандартов США, другая, почти повсеместно употребляемая в неф-
тяной промышленности Штатов, недавно принята Американским нефтя-
ным институтом (API). Переход от этих шкал к обычным удельным весам
и обратно осуществляется при посредстве специальных таблиц.
ВЯЗКОСТЬ
'Вязкостью или внутренним трением жидкости называется
свойство, проявляющееся в сопротивлении, которое жидкость
оказывает перемещению ее частиц под влиянием действующей
на них силы
Различаются:
1. Абсолютная вязкость ?], представляющая собой силу сопро-
тивления взаимному перемещению двух слоев жидкости с по-
верхностью в 1 см2, находящихся друг от друга на расстоянии
1 см, если действующая сила вызывает взаимное перемещение их
со скоростью 1 см/сек. Абсолютная вязкость определяется по
скорости истечения жидкости из капиллярной трубки на основе
формулы Пуазеиля:
71 ~ SvL ’
где Р— давление, под которым происходит истечение; г—ра-
диус и L — длина капиллярной трубки; Т — время в секундах,
в течение -которого происходит вытекание объема жидкости v
(в си3).
Единица абсолютной вязкости называется пуазом’, для нее
указанная сила сопротивления равна 1 абсолютной единице
силы, т. е. 1 дине.
2G
2. Кинематическая вязкость представляющая собою отно-
шение абсолютной вязкости iq жидкости к ее удельному весу d
при той же температуре, т. е.
^~~d'
3. Удельная вязкость, JH, определяемая как отношение абсо-
лютной вязкости ц данной жидкости к абсолютной вязкости
?10 воды при той же температуре:
ff=i.
тю
4. Величина, обратная вязкости, 1
-честью (f).
—, называется теку-
М-
Вязкость является одним из важнейших физических свойств--
нефтей и нефтепродуктов. С ней приходится считаться при дви-
жении нефти по трубам (нефтепроводы), при работе освежитель-
ных и, особенно, смазочных масел и т. д. Определение абсолют-
ной вязкости — слишком сложно для подобного рода практиче-
ских задач, так как при этом пришлось бы предварительно опре-
делять такие величины, как длину капилляра, его радиус и т. д..
Эти определения совершенно отпадают для удельной вязкости,
как это ясно из следующего.
По формуле Пуазейля для абсолютной вязкости какого-
либо масла и абсолютной вязкости воды имеем:
т.Р^Т -p^tq
Г' = ~&Ъ И
Отсюда удельная вязкость масла получается, как отноше-
ние Tp'/j,/
п _ . РТ
тю РоЗУ.
где Р —давление, под которым происходит истечение масла из:
капилляра во время Т, а Ро и То — те же величины при том же
капилляре для воды.
Если истечение жидкости совершается под влиянием силы.’
тяжести, то, очевидно,
ветственным удельным
величины Р и Ро пропорциональны ооот-
весам d и <Zo; следовательно:
у] (IT
•/j0 rf02o
для определения удельной вязкости ка-
Как было указано,
кого-либо масла необходимо найти время истечения из капил-
ляра равных объемов масла и воды. Это соотношение положено
в основу техническою определения вязкости с помощью прибо-
ров, называемых вискозиметрами.
Число моделей вискозиметра предложенных в разное время,
весьма значительно. Они различаются между собой не только
.деталями своей конструкции, но отчасти также заложенными
в основу их принципами. Так, наряду с вискозиметрами, осно-
ванными на истечении жидкост I, имеются аппараты, в основу
которых положены другие явления, связанные с вязкостью,
.как-то: падение в исследуемой жидкости или вращение в ней
определенного тела. Наибольшее распространение получили при-
боры первого типа,
основанные на исте-
чении; таков, напри-
мер, вискозиметр
Энглера.
Вискозиметр Энглера
(фиг. б) состоит из
двух концентрических
латунных резервуаров А
п В, вставленных друг
в друга. Внутренний
из этих резервуаров
снабжен капиллярной
трубкой истечения, вы-
ложенной внутри пла-
тиной. палладием или
вообще нержавеющим
металлом. В резервуаре
находятся также указа-
тели уровня наливаемой
жидкости, термометр F
и специальная деревян-
ная палочка С, наглухо
закрывающая входной
отверстие в капилляр-
ную трубку. Наружный
резервуар служит тер-
мостатом для поддержа-
ния постоянной темпе-
ратуры внутреннего ре-
зервуара, последний во избежание, излишнего лучеиспускания -снабжен
также хорошо пригнанной крышкой Е. Вытекающая через капилляр жид-
кость собирается в специальную колбу D, калиброванную на 2ио c.w3 при 20°.
Размеры всех 'важнейших частей прибора, стандартизованы.
Первая задача при определении вязкости с аппаратом Энг-
лера заключается в нахождении времени истечения через капил-
ляр прибора 200 ел3 воды при 20°. Эта величина называется
водяной константой данного прибора. Точное определение ее
производится с помощью секундомера и время от времени повто-
ряется, так как ввиду накопляющихся в приборе .в процессе
работы изменений она с течением времени несколько изменяется.
У нормальных приборов Энглера истечение 200 см3 воды при 20е
должно происходить в течение 51—52 сек.
Когда определение или проверка водяной константы закон-
чено, переходят к определению времени истечения 200 см3 нефти
или исследуемого маета. Это определение производится или
22
также при 20р, или для вязких масел— при более высокой тем-
пературе, например при 50, 80 или 100°. В тех случаях, когда
надо проследить ход изменения кривой вязкости с температу-
рой, определения производятся последовательно при нескельких
температурах, например от 0° через каждые 10 или 20°. Обяза-
тельным условием точности всех этих определений является
неукоснительное выполнение ряда точно установленных приемов
•и правил, описание которых дается в специальных практических
руководствах.
Когда время истечения 200 см3 исследуемого масла и того же
объема воды определено, первую величину делят на вторую и
получают вязкость данного масла по Энглеру при температуре
истечения масла. Величину эту принято обозначать буквой Е
(или Э) с указанием соответствующей температуры, например
Его, Езо и т. д.
Вискозиметр Энглера применяется для определения вязкостей нефтей и
нефтепродуктов ® СССР и Германии, отчасти также в Румынии и Италии.
В других странах для этом цели пользуются вискозиметрами несколько
иной конструкции, но также основанными на истечении. Так, в США.
употребляют вискозиметр Сеиболта «универсальный», а для нефтетоплива
и особо вязких масел — «фурол», в Англии и ее колониях пользуются виско-
зиметром Редвуда, «обыкновенным» и «адмиралтейским»; во Франции —
иксометром Барбъе. Для приведения показаний этих вискозиметров к граду-
сам Энглера или обратно применяются специальные таблицы
Вискозиметром Энглера можно пользоваться также, хотя бы
масла для определения .вязкости имелось меньше 200 см3. В по-
добного рода случаях для вычисления вязкости по Энглеру
применяют либо специальные формулы, либо таблицы соответ-
ствующих поправок и множителей. Так например, при заливе
масла в вискозиметр 120 c..it3 и истечения 100 слг3 множитель
для перевода на истечение 200 сл3 равняется 1,65; при заливе
50 см3 и истечении -10 сл3 множитель равен 3,02 л т. д. Для той
же цели иногда находит применение специально сконструиро-
ванный по типу Энглера вискозиметр Уббелоде, требующий для
определения вязкости всего 30 с.м3 масла. Из вышеизложенного
ясно, что -вязкость, определенная по Энглеру или на любом из
других технических аппаратов, представляет собою чисто услов-
ную величину, пригодную лишь для сопоставления двух жидко-
стей, близких друг к другу .по своим физическим свойствам.
В тех случаях нефтяной практики, когда нельзя ограничиться
этими условными величинами, определяют абсолютную вязкость-
нефти "или нефтепродукта, наблюдая для этого продолжитель-
ность Пуазейлевского истечения с помощью капиллярного ви-
скозиметра.
Выше была выведена формула:
•г, _ dr
Подставляя в нее К — -—г, получаем:
•Л = KdT,
25
•где К представляет -собою константу данного прибора по отноше-
-кию к воде. Для расчета т)о, т. е. вязкость воды при различных
’температурах, может быть взята из таблиц; То определяется не-
посредственно, равным образом также d и Т. Зная абсолютную
вязкость жидкости или величины К и Г, не трудно рассчитать
и кинематическую вязкость щ. по формуле:
г1к — ~^ = К?-
а
Капиллярные вискозиметры, применяемые для определения
абсолютной вязкости, известны подобно техническим вискози-
метрам во многих модификациях. Простейшие из них—вискози-
метры Оствальда и капилляр Уббелоде— даны на фиг. 6 и 7.
•Оба они представляют собою U-образную трубку, в одно из колен
которой е впаян длинный капилляр к, заканчивающийся сверху
шарообразным расширением с метками с и d. Различие в устрой-
стве другого колена а видно на рисунках. Наблюдается время,
в течение которого жидкость в приборе, проходя капилляр, опу-
скается от с до d (у Оствальда) или поднимается в обратном на-
правлении от d до с (у Уббелоде). Последнее устройство более
правильно, так как при этом
устраняется необходимость введе-
лярный вискози-
метр Оствальда.
К
ния поправки на смачивание сте-
нок расширенной части левого
колена прибора cd, где измеряется
прошедший через капилляр объем
жидкости.
Таблица 5
Абсолютная вязкость воды
Фиг. 7. Капил-
лярный вискози-
метр Убеллоде.
Температура
0° 0,01797»)
103 0,013010
20е 0,010060
30= 0,007998
403 0,006563
50° 0,005500
60° 0,004735
70° 0,004075
80° 0,( 03570
90° 0,003143
Как было указано, для определения абсолютной вязкости ка-
кой-либо жидкости с помощью капиллярного вискозиметра не-
обходимо знать абсолютную вязкость воды. Эта константа не-
• однократно определялась различными.методами; значения ее для
различных температур от 0 до 90° даны в табл. 5. Как. видно,
абсолютная вязкость воды при 20° равна 0,01006. Отбрасывая
пятый десятичный знак, получаем, что при 20° для воды
'24
7)0 = 0,01 пуаза, что позволяет простым умножением на 100 пе-
рейти от абсолютной вязкости к удельной.
• Ввиду широкого распространения технических вискозиметров и сравни--
тельной простоты обращения с ними удобно иногда воспользоваться из по-
казаниями для расчета абсолютной, удельной или кинематической вязкости.
Для этого пользуются специальными эмпирическими формулами или таб-
лицами перевода одних величин в другие. Такова, например, формула Уб-
бело де, определяющая зависимость между удельной вязкостью R \ воде
при 0° и вязкостью в градусах по, Энглеру (£):
Я = = (4,(У12Е— d,
где d — у/]цельный .вес жидкости.
Если ввести в эту формулу значение = 0,01797 (при 0е), то после
соответствующих преобразований легко получаются формулы, связывающие
вязкость по Энглеру с абсолютной кинематической вязкостью:
Л ГГГО1 тп 0,0631'
'/] = I 0,07317?----—
X & <
и
т)Л = (0073137—
\ "
Чтобы составить представление о том, насколько по градусам
Энглера можно судить об истинной вязкости жидкости, сопоста-
вим в некоторых пределах градусы Энглера с кинематической-
вязкостью (табл. 6). Легко видеть, что для жидких масел вели-
чина щ. растет значительно быстрее, чем градусы Энглера; про-
порциональное же изменение обеих величин начинается при-
мерно только сЕ = 5. Отсюда можно судить о том, насколько-
условно определение вязкости по Энглеру и какие ошибки воз-
можны, если, например, при сопоставлении двух жидкостей вос-
пользоваться данными, полученными с помощью вискозиметра
Энглера без перевода их в абсолютные единицы Поэтому в на-
учно-исследовательской работе определение вязкости по Энглеру
не рекомендуется.
Таблица G
Перевод градусов Энглера в кинематическую вявкоет*
Градусы Энглера г1к Градусы Эмлера Ti*
1 0,0100 10 0,7247
2 0,1147 15 1,0926
3 0,1983 20 1,4588
4 0,2766 30 2,1900
5 0,3529 40 2,9224
7,5 0,5398 59 3,6637
Общеизвестно, что при охлаждении нефть и ее продукты
густеют, а при нагревании становятся более жидкими. Таким
образом вязкость зависит от температуры, и эта зависимость
ж.
.представляет большое значение и интерес для разрешения не-
которых практических вопросов, например при подборе смазоч-
ного масла для подшипников и других частей машины, работаю-
щих в различных температурных условиях в зависимости
от степени нагрузки и т. д.
Зависимость вязкости от температуры можно выразить следующей
эмпирической формулой:
'Go = (1 + at -+ ы'~ +•••).
•где и —вязкости данного масла при 0 и t°. Определив вязкость его
при трех разных температурах о, t и Л°, можно составить систему уравне-
ний первой степени, решением которых коэфпцпенты а и Ъ легко опреде-
ляются; зная же эти коэфпцпенты, можно на основе того же уравнения
составить таблиц) изменения вязкости масла с температурой.
Наиболее наглядное представление о том, как изменяется вяз-
кость масла с температурой, можно получить, если, определив
вязкость его при нескольких температурах, построить соответ-
ствующие кривые; при этом на оси абсцисс обыкновенно стала -
Фиг. 8. Температурные кривые кине-
матической вязкости по Блоху н До-
брянскому:
1— биби-8йбатская нефть; 2—бплахапская тяже-
лая нефть; з —балах&вская масляная нефть;
4—суахакская легкая нефть.
Фиг. 9. Температурные кривые
вязкости различных масел по
Блоху и Добрянскому:
I — спермацетовое масло; 2—сурепное
масли.
дываются температуры, на оси же ординат — вязкости, выражен-
ные в тех или иных единицах Примеры подобного рода кривых
можно видеть на фиг. 8 и 9. Фиг. 8 дает температурные кривые
кинематической вязкости нескольких бакинских нефтей. Как
видно, кривые эти имеют вполне плавный характер. Они на-
глядно показывают, что по мере повышения температуры вяз-
кость нефти сначала падает очень быстро, а затем — все более
26
и более медленно. У различных нефтей в ходе изменения этой
кривой наблюдается, однако, заметная разница. Вообще говоря,
у тяжелых нефтей температурная кривая вязкости имеет очень-
крутой вид, у более же легких нефтей — значительно более по-
логий. Не надо забывать однако, что, помимо влияния темпера-
туры, здесь могут иметь весьма существенное значение и другие
факторы, в первую очередь состав нефти, содержание в ней смо-
листых веществ и т. п. (см. ниже).
Ход изменения вязкости для различных масел (фиг. э) в об-
щем имеет такой же характер, как и для нефтей. В приведенных
примерах обращает на себя внимание чрезвычайно пологий вил
кривых вязкости для спермацетового и сурепного масел. Такой
ход изменения вязкости с температурой, характерный вообще
для растительных масел и жиров, составляет ценнейшее их свой-
ство как смазочного материала. Из минеральных масел очень
высоко стоят в этом отношении так называемые брайт-стоки.
Изменение вязкости смазочных масел с температурой часто
выражается в настоящее время при помощи специальных харак-
теристик, получивших наименование индекса вязкости. По Дину
и Дэвису [1] индекс вязкости масла представляет собою функ-
цию его вязкости У по Сейболту «универсальному» при темпе-
ратуре 100° Ф (т. е. 37,73° Ц) и вязкостей L п Н при той же
температуре масел двух серий, а именно:
1) сер-ии масел с наивысшим изменением вязкости от темпе-
ратуры; индекс вязкости этой серии масел условно принимается
равным О, самая же серия обозначается L;
2) серии масел с небольшим изменением вязкости от темпе-
ратуры; индекс вязкости этой серии масел условно принимается
равным 100, самая же серия обозначается Н.
Из каждой серии масел для сравнения с испытуемым берется
по образцу; вязкости этих образцов при 210 Ф (т. е. 9s,89 Ц) —
те же, что у испытуемого масла. В этих условиях индекс вяз-
кости вычисляется на основе следующего уравнения:
В. = 100^^- =
100(1 — V)
р
гдеВ = Ь —Я.
Здесь И. В. — индекс вязкости испытуемого масла; Лг—его
вязкость при 100° Ф; L и Н— вязкости при той же температуре
масел двух указанных выше серий, причем вязкости этих масел
при 210° Ф должны быть такие же, как у данного масла. Таким
образом индекс вязкости любого масла некоторой неизвестной
серии представляет собою выраженное в процентах отношение
разности между вязкостями при 100° Ф масла серии L и дан-
ного масла к разности О вязкостей при той же температуре ма-
сел серий L и Н при условии, что вязкости всех трех образцов-
этих масел при 210 Ф одинаковы.
Для вычисления индексов вязкости масел пользуются спе-
циальными таблицами или составленными на их основе номо-
граммами. Выдержка из такой таблицы приводился ниже
27
«(табл. 7). Ирл ее составлении использованы эмпирические зави-
симости для масел серий L и Н между 'Вязкостью при 100° Ф
ъ вязкостью при 210° Ф (по Сейболту). Зависимость эта выра-
жается следующими уравнениями:
Для К В. 100: ?/ = 0,0408л2 4-12,568^ — 475,4,
„ К В. 0: у = 0,2160л2 + 12,070л — 721,2,
тде у —вязкость при 100° Ф, а л— вязкость при 210е Ф.
Таблица 7
Выдержка из таблицы для вычисления индекса вязкости масел
по Дину и Дзвиеу
Вязкость при 310° Ф Вязкость L при 100° Ф (серия L) Вязкость Н при 100° Ф (серия Н) D = L — Н
40 138 107 31
60 . 781 426 355
100 2 646 1189 1457
120 3 838 1 620 2 218
140 5 202 2 084 3118
160 6740 3 580 4160.
Индекс вязкости для нефтепродуктов различного происхож-
дения колеблется в весьма широких пределах (от О до 100 и
больше). Колебания эти весьма значительны также для различ-
ных погонов одной и той же нефти в зависимости от их
вязкости. Так например, для различных погонов доссорской
нефти (Эмба) индекс вязкости меняется от 35 до 89; для оста-
точных же эмбенских продуктов здесь наблюдаются колебания
от 80 до 90. Очень большого размаха достигают эти -колебания
также для бакинских нефтей, причем у некоторых из них низ-
шие масляные погоны (от 400° и выше) нередко имеют отрица-
тельный индекс вязкости (табл. 8).
Весьма широкое распространение получила также другая ха-
рактеристика смазочных масел — вязкостно-весовая константа
(В.-в. К.).
Между вязкостью по Сейболту N и удельным весом D нефтяных фрак-
ции установлена следующая эмпирическая зависимость [2]:
7) = a-J-b log (Лг-т-с).
На основе этой зависимости были выведены следующие формулы для
.вычисления вязкостно-весовой константы масла:
10JD —1,0752 log (2Г — 38)
10 —log (X—38)
_ D — 0,24 — 0,022 log (N' — 35,5)
Л 0,755
Здесь A — вязкостно-весовая константа при 100° Ф (37,8° Ц); А’— вяз-
кость масла по Сейболту при той же температуре; D — его удельный вес
при 60° Ф (15.56° С); А' и N'— та же константа и вязкость масла при
210° Ф (98,9° Ц).
Для вычисления вязкостно-весовой константы на базе си-
28
стены определений и мер, принятых в Европе и в частности
в СССР, была предложена следующая формула [3]:
d — 0,24 — 0,038 logz
а =------!----’----__
0 755 — 0,01 log iq
Здесь а — вязкостно-весовая константа; d — удельный вес
нефтепродукта при 15° С и — кинематическая вязкость его
при 100° С, выраженная в сантистоксах.
Для нефтепродуктов из нефтей парафинового основания
вязкостно-весовая константа равна примерно 0,80—0,83; для
продуктов же пз нефтей нафтенового основания она выражается
величиной 0,87—0,90. Константы для различных нефтепродуктов
из одной и той же нефти отличаются между7 собою незначи-
тельно. В табл. 8 приведены характеристики некоторых совет-
ских масел с их индексом вязкости (И. В.) по Дину и Дэвису и
с их вязкостно-весовой константой (В.-в. К.) [3].
Таблица 8
Характеристики некоторых советских смазочных масел
Название продукта Уд. вес ^15 37,8°Ц Я 98,9Щ N' 100° ц Чй Я. В. В.-в. К.
Доссорская нефть
1 фракция (450—500°) . . . 2 „ (500—550°) ... . 3 » (550-600°) . . . Остаток 0,8948 0,9003 0,9009 09183 234,0 5254 1002,9 8 170,0 44.9 57.3 76,4 290,0 5.82 9,50 14,75 60.21 34,8 52,3 62,4 88,7 0,8366 0,8360 0,8285 0,8275
Легкая биби-эйбатская нефть •
1 фракция (400—450е) -. . 2 „ (450—500°) . . . 3 „ (500—510°) . . . Остаток 0,9078 0,91-95 0,9*293 0,9522 93,5 274,0 970.0 12 195,0 37,0 45,0 64,2 271.3 3.34 5.90 11,12 56,26 — 10,1 — 7.7 50,0 0Л64О 0,8690 0,8720 0,8750
Сураханский мазут •
1 фракция (408—425°) . . . 2 . (425—510°) . - . Остаток 0,8935 0,9159 09450 124,0 674,5 40,0 62,5 388,0 4.21 10,88 80,73 45,1 49,7 0,8410 0,8580 0,8590
Кроме температуры, вязкость зависит та<кже от давления. 0 характере
этой зависимости можно судитв по кривым фиг. 10, где на оси абсцисс
отложены давления в килограммах на 1 слг, а на оси ординат — отношения
вязкости при соответствующем давлении к -вязкости при атмосферном давле-
нии ( — для ряда минеральных и неминеральных масел. Как видно, с уве-
личением давления вязкость масел также увеличивается, причем характер
нарастания вязкости оказывается сначала довольно медленным, постепен-
ным; однако, начиная примерно с давления в 250—300 кг/см\ кривые дая
минеральных масел резко поворачивают вверх, указывая на чрезвычайно
быстрое увеличение вязкости при дальнейшем повышении давления: для
немпперальных масел характер кривых в общем сохраняется старый при
гораздо более высоких давлениях. В конечном итоге при очень высоких
давлениях (выше 1000 кг/см*) вязкость возрастает настолько, что масло
теряет характер жидкости и превращается в пластическую массу.
39
Помимо чисто физических, условий (температура, давление),
весьма существенное влияние на вязкость оказывают также
факторы химического характера.
Известно, -что при переходе от низших фракций какой-либо
нефти к высшим вязкость их повышается. Эта правильность
представляет собою проявление об-
Фиг. 10. Зависимость вязкости
от давления по Блоху и До-
брянскому.
Кривые т и 2 относятсяук органиче-
ским маслам, кривые з н_4 —к »ине-
ральным.
нов, п т. д. Иллюстрацией
собранный в табл. 9.
щей закономерности, согласно кото-
рой с повышением молекулярного
веса углеводородов в одном и том же
ряду вязкость их увеличивается.
Лишь для первых членов аромати-
ческого ряда наблюдается несколько
более сложная зависимость.
При сопоставлении соответствую-
щих угле водородов различных рядов
замечено, чго наибольшей вязкостью
обладают, повиД имому, нафтены,
далее следует ароматические угле-
водороды, затем парафины и, нако-
нец, непредельные углеводороды.
Существенное влияние оказывает
также строение углеводорода. Так
например, установлено, что у пара-
финов нормального строения вяз-
кость при прочих равных условиях
несколько выше, чем у нзопарафи-
сказанного может служить материал,
Таблица 9
Абсолютная вязкость углеводородов
Формула Название т] X Ю-5 Формула Назван пе 7) X IO”5
(*Б Н12 Пентан 287 (0°) Cfl Щн Октан 588 (20°)
С5 н10 Амилен 262 (0°) И io Этил-бензол 666 (20°)
Се н14 Гексан 320 (20°) л Ксилол-орто 807 (20°)
< 99 Гексан изо- 300 (20°) и Ксилол-мета 615 (20°) 1
Н6 Бензол 649 (20°) Ксилол-пара 648 (20°)
G; Н16 Гептан 410 (20°) ^8 Н16 Этнл-циклогексан 895 (20°)
Хотя данные по вопросу о влиянии химической природы
углеводородов на их вязкость пока далеко недостаточны и.к тому
же относятся почти исключительно к низшим представителям
различных рядов, тем не менее для химика-нефтяника они пред-;
ставляют выдающийся интерес. Действительно, мы подходим
здесь к вопросу об относительной важности в нефтепродукт
углеводородов различных рядов с точки зрения их вязкосиК
короче говоря, о так называемых «носителях вязкости», пред^
ставляющих особые интерес и значение для смазочных масел^
до
же на основании вышеприведенного материала следует ожи-
дать, что ни парафины, ни непредельные углеводороды с их
сравнительно малой вязкостью не могут быть носителями вяз-
кости смазочных масел. В самом деле, непосредственный опыт
показывает, что отделение парафина повышает вязкость сма-
зочного масла, и наоборог, добавлением Парафина вязкость сма-
зочного масла может быть понижена. С другой стороны, как
известно, в результате сернокислотной очистки содержание не-
предельных углеводородов в смазочных маслах становится
крайне незначительным; в некоторых же случаях оно делается
.даже равным нулю без существенного влияния на вязкость
масел.
Если, таким образом, ни парафины, ни непредельные угле-
водороды не могут быть носителями вязкости смазочных масел,
•то, очевидно, эта роль может принадлежать только одно- или
многоядерным представителям ароматического или нафтенового
рядов. Так как дальнейшее углубление этого вопроса
путем выделения отдельных компонентов смазочных масел
представляет, как будет показано ниже, чрезвычайные труд-
ности, то большой интерес представляет другой подход к его
изучению, а именно синтез высокомолекулярных ароматических
и нафтеновых углеводородов и выявление этим путем тех осо-
бенностей химического строения, которые оказывают то или
иное влияние на вязкость вещества (см. ч. Ш, гл. III).
Большой интерес для нефтяной практики имеет вопрос
о зависимости между вязкостью смеси и вязкостями образую-
щих ее компонентов.
Уже давно было показано, что вязкость смеси двух жидко-
стей, вообще говоря, не подчиняется закону аддитивности,
т. е. не является линейной функцией ре состава, и не может
быть вычислена по правилу смещения.
100—р . р
Т| ‘TJ’ —Ко Ь юо ’
где u Hi и — вязкости смеси и ее компонентов, а р— процент-
ное отношение между компонентами.
Если разница между вязкостями компонентов невелика, то
по только что приведенной формуле с достаточной точностью
может быть вычислена текучесть смеси, т. е. величина, обратная
вязкости; в общем же случае ошибки при такого рода расчете
как дтя вязкости, так и для текучести могут быть громадны
.(до 30%).
Теоретическое исследование вопроса о вязкости смеси двух
жидкостей приводит к довольно сложной формуле. Более на-
глядное представление по этому вопросу можно получить графи-
чески.'Если на оси абсцисс откладывать процентное содержание
в смеси одного из компонентов, а на оси ординат — вязкость
смеси, то ход изменения этого свойства выразится' не прямой
линией, а гиперболой, проходящей ниже прямой. Таким образом
вязкость смеси всегда оказывается меньше, чем этого требует
31
расчет по правилу смешения. На практике для определения
вязкости смеси из двух компонентов и для аналогичных расче-
тов удобно пользоваться специальной таблицей Молин-Гурвича*
составленной на основе чисто эмпирических данных.
ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ
Поверхностным натяжением называется сила, с которой жид-
кость сопротивляется увеличению своей поверхности. Отнесен-
ная к единице длины поверхностного слоя жидкости, сила эта„
выраженная в дпнах, принимается за единицу поверхностного
натяжения. С величиной поверхностного натяжения тесно свя-
зана другая физическая константа жидкости, ее капиллярная
постоянная, численно равная высоте подъема жидкости
в трубке, радиус канала которой равен 1 мм, а стенки которой
вполне смачиваются данной жидкостью. Соотношение между
поверхностным натяжением а и капиллярной постоянной а*
выражается формулой:
где д — удельный вес жидкости.
Существует целый ряд способов определения поверхностного-
натяжения: простейший из них — способ взвешивания капель —
заключается в следующем\ Если заставить жидкость медленно
вытекать из специальной бюретки с узким выхо/щпм отверстием
(сталагмометр), то на последнем образуются капли, которые, до-
стигнув определенного веса р, отпадают. Перед падением капля
поддерживается силой поверхностного натяжения по периметру
несколько суженной ее части. Отсюда
jo = 2пга,
где г — радиус суженной части капли, несколько меньший ра-
диуса выходного отверстия бюретки.
Величина у лепю определяется опытным путем, а именно
взвешиванием некоторого числа Ат капель. Определение вели*
чины г значительно труднее. Однако, зная из таблиц поверх-
ностное натяжение какой-нибудь жидкости (например для бен-
зола при 20° а = 28,8), можно прокалибровать ею данную бю-
ретку и вычислить для нее г из вышеприведенного уравнения.
Если требуется определить поверхностное натяжение жид|
гости, например нефти или нефтепродукта не по отношению
к воздуху, а на границе с какой-либо другой жидкостью, напри-
мер с водой, то капли первой жидкости выпускаются не на воз-
дух, а во вторую жидкость. В случае надобности капилляр дол-
жен быть загнут при этом кверху. Для определения г здесь
На этом принципе Рсбиндср построил удобный, получивший широко*
распространение прибор для определения поверхностного натяжения на*
ходгрхноегях раздела жидкость/газ и жидкость/жидкость.
3-
удобнее исходить из какой-либо изученной пары жидкостей (на-
пример поверхностное натяжение бензола к воде а = 32,б при
20е). Очевидно также, что в подобного рода случаях сила р вы-
шеприведенной формулы будет пропорциональна не удельному
весу жидкости, а лишь разности удельных весов обеих жидко-
стей.
В табл. 10 приведены величины поверхностного натяжения
некоторых нефтей и нефтепродуктов СССР по отношению к воз-
духу и воде по Гурвичу.
Таблица 10
Поверхностные натяжения нефтей п нефтепродуктов
(по X Гурвичу)
Нефти Уд. вес Поверхностное натяжение Нефте- продукты Уд. вес Поверхностное натяжение
в воз- духе 1 । в воде В В08«г духе в воде
Сураханская 0.797 25,8 27,8 Бензин 1 сорта 0714 20,4 47,9
Балаханская 0,875 28,9 27,1 Керосин очищ. 0,824 26,1 42,4
Биби-эйбатская 0,880 29,2 22,0 Веретен, масло 0,894 31,8 28,9
Бпнагадинская 0,932 31,0 19,0 Машинное , 0.9С9 35,7 34.0
б. Артема 0,918 30,7 17,3 Цилиндр. , 0,918 35,7 27,4
Как видно, при переходе от легких нефтей или нефтепродук-
тов к тяжелым поверхностное натяжение их в воздухе обнару-
живает явственную тенденцию к повышению; для поверхностного
же натяжения на границе с водой, наоборот, наблюдается тен-
денция к понижению
Поверхностное натяжение нефтей и нефтепродуктов имеет
первостепенное значение в ряде -вопросов нефтяной практики,
особенно в вопросе о нефтяных эмульсиях, природных и обра-
зующихся при очистке нефтяных дестиллатов, а также в вопро-
сах (применения некоторых нефтепродуктов, как-то: смазочных
масел, керосина и др.
ТЕПЛОВОЕ РАСШИРЕНИЕ
Расширение нефти и нефтепродуктов от нагревания подчи-
няется общей закономерности теплового расширения жидкостей,
определяемой формулой:
vt vQ (1 -f- at + bt2 + ciQ + • • • )•
Как общее правило, расширение жидкостей возрастает вместе
с температурой, иначе говоря, Ъ и с в приведенной формуле —
величины положительные. Нефть не является исключением из
этого правила. Однако абсолютное значение этих величин таково,
'вз
8 Зак. 1622. Химия нефти.’
Что Для практических целей можно пользоваться сокращенной
формулой, допуская, что Ь = с = О; следовательно:
== vo G + а)»
где vo и vt—объемы жидкости при 0° и tc; а — коэфициент
теплового расширения жидкости.
Подставляя в последнюю формулу вместо vo и vt обратные им
величины, т. е. удельные веса при соответствующих температу-,
рах1 do и df, получаем:
<?( = YqK. =Ч С -
или, отбрасывая в многочлене, стоящем в скобках, все члены,
содержащие величину а во второй и высших степенях, получаем
окончательно:
dt = d0(l — at).
Таким образом одним и тем же коэфициентом можно поль-
зоваться при пересчете от одной температуры к другой не только*
объема жидкости,' но и ее удельного веса.
По вопросу о тепловом расширении нефтей и нефтепродуктов
можно ограничиться следующими немногими данными.
Коофициентьг теплового расширения различных нефтей за?
метно различаются между собой, причем,- как общее правило,
чем легче нефть, тем больше ее жоэфициент расширения.
Для различных погонов одной и той же нефти коэфициент
расширения изменяется аналогичным образом: чем легче погон,
тем больше его коэфициент расширения.
Точное знание коэфициентов теплового расширения нефтей
и нефтепродуктов имеет большое практическое значение при
разного рода тепловых пересчетах их объемов.-До последнего
цремени (1935 г.) для этой цели пользовались температурными
поправками, которые были выведены много лет тому назад на
основании работ Д. И. Менделеева и Казанкина с бакинскими
нефтепродуктами и нефтями. Однако эти продут ты давно уже
утратили свое значение и даже перестали вырабатываться. Ввиду
этого большое значение и ценность приобрела большая работа^
Всесоюзного института метрологии и стандартизации по опреде-i
лению удельного веса 30 нефтепродуктов в интервале температур
от 0 до 50°, причем для трех из них удельный вес был определено
в еще более широком интервале температур, а именно от —20 да
100° [4]. Математическая обработка полученного таким образом
экспериментального материала дала температурные поправки для,
Наряду с градусами Цельсия (°Ц) в английской и особенно- америкай-
ской литературе до сих пор встречаются градусы Фаренгейта (°Ф). Перевей
одной пгколы в другую производится либо по формулам: г
Г Ц = (~ т+32)° Ф; Т,’® = Г| (Т, _ 82)1 °Ц,
либо по специалппым таблицам.
34
нефтей и нефтепродуктов СССР, принятые в настоящее время
как стандартные [5]. Эти поправки приведены в табл. п.
Таблица 11
Температурные поправки удельного веса нефтей
и нефтепродуктов СССР
Пределы уд. веса Поправки Пределы уд. веса d20 а 4 Поправки
0,7000—0,7100 0,000897 0,8500—0,8600 0,000699
0,7100—0,7200 0,000884 0,8600—0,8700 0,000686
0,7200—0,7300 0,000870 0,8700-ОЛ 800 0,000673
0,7300—0,7400 0,000857 0,8800—0,8900 0,000660
0,7400 -0,7500 0,000844 0,8900-0,9000 0,000647
0,7500-0,7600 0,000831 0,9000-0,9100 0,000633
0,7600—0,7700 0,000818 0,9100—0,9200 0,000620
0,7700—0,7800 0,000805 0,9200—0.9300 0,000607
0,7800—0,7900 0,000792 0,9300—0,9400 0,000594
0,7900—0,8000 0,000778 0 9400—0,9500 0,000581
0,8000—0,8100 0,000765 0,9500-0,9600 0,000567.
0,8100—0,8200 0,000752 0.9600—0,9700 0,000554
0,8200—0,8300 0,000738 0,9700-0,9800 0,000541
0,8300—0,8400 0,000725 0,9800—0^900 0,000528
0,8400—0,8500 0,000712 0.9900—ЦОООО 0,000515
Как показывает опыт, на величину коэфициента расширения
нефтей и нефтепродуктов оказывает влияние целый ряд факто-
ров. Большое значение, например, имеет содержание в нефти
парафина, который в известных температурных пределах может
находиться в ней в виде мельчайших взвешенных кристаллов;
заметное влияние имеют также степень очистки нефтепродукта;
•широта фракции, из которой он составлен, и т. д. Подробности,
сюда относящиеся, следует искать в специальных работах.
Для определения -коэфициента расширения нефтепродуктов
и других жидкостей пользуются либо дилатометром (фиг. 11),
либо пикнометром (фиг. 12) с капиллярной калиброванной шей-
кой. Для удобства наполнения дилатометр снабжается иногда
еще одной капиллярной трубочкой. Зная коэфициент расшире-
ния сосуда и производя измерение объема жидкости при раз-
ных температурах, можно вычислить ее коэфициент расшире-
ния. Устройство и применение пикнометра общеизвестны.-
Зависимость удельного веса от температуры для большинства
нафтепродуктов имеет линейный характер. В такого рода случаях
справедливо соотношение:
откуда
^ = <2, —Tf('a —<i).
8*
Зй
Так как здесь г — величина постоянная, то последняя фор-
мула позволяет вычислить удельный вес dz нефтепродукта при
температуре tz, если известен его удельный вес di при темпера-
туре ti. Очевидно, г представляет собою здесь изменение удель-
ного веса, отнесенное к 1°; иначе говоря, ото—температурная
поправка удельного веса для данного нефтепродукта. Значения
Y для нефтепродуктов различного удельного веса и приведены
в табл. 11.
Фиг. 11. Дилато-
метры.
Фиг. 12.
Пикнометр.
В тех случаях, когда изменение
удельного веса с температурой не
имеет линейного характера, обе по-
следние формулы оказываются уже
недостаточно точными. Пусть на-
пример, изменение удельного веса
какого-либо нефтепродукта выра-
жается приведенной выше формулой:
где а—средний термический коэ-
фициент расширения данного не-
фтепродукта в пределах от 0 до f.
Тогда
Отсюда
или приближенно:
^ = ^[1 — a(i2 — Q].
Сопоставляя эту формулу с при?
веденной выше:
d2 = dy О»
ВИДИМ, что
т. е. что в рассмотренном случае величина у равна произведению
среднего термического коэфициента расширения нефтепродукта
на его удельный вес di. Для парафинистых продуктов эти со-
отношения приводят к более точным результатам.
Во многих случаях большую практическую важность имеет,
знание коэфициента термического расширения нефтей и их про-
дуктов; таковы, например случаи расчета залива нефтехрани-
лищ, цистерн, баков и т. п. Средние коэфициенты термического ।
расширения для нефтепродуктов различного удельного веса (<№).
36
приведены в табл. 12. Они легко могут быть вычислены также
по формуле:
-у = ad20
где г берется из табл. 11.
Таблица 12
Средине коэфициенты термического расширения нефтепродуктов
Пределы уд. веса ai Средний коэфициент расширения Пределы уд. веса а30 Средний коэфициент расширения
0,700—0,720 0,001255 0,860—0,880 0,000782
0,720—0,740 0,001183 0.880—0,900 0,000734
0,740—0,760 0.001118 0,900—0,920 0,000688
0.760—0,780 . 0,001054 0,920-0,940 0,000645
0,780—0,800 0,000995 0,940—0,960 0,000604
0,800—0,820 0,000937 0,960- 0,980 0,000564
0,820—0,840 0,000882 0,980—1,000 0,000526
0,840—0,860 0,000831
Для практических расчетов изменения объема с температу-
рой имеются таблицы, позволяющие быстро определять процент-
ное изменение объема при повышении температуры для нефте-
продуктов данного удельного веса* а также объем, занимаемый
ими при разных температурах, по отношению к объему при 20°,
принятому за единицу.
ЗАСТЫВАНИЕ И ПЛАВЛЕНИЕ
Так как нефгь и ее продукты представляют собою вещества
не индивидуальные, то переход их из одного аггрегатного состоя-
ния в другое происходит постепенно. В частности, переходы их
из жидкого состояния в твердое, т. е. застывание, и обратно, из
твёрдого в жидкое, т. е. плавление, всегда сопровождаются не-
которой промежуточной стадией загустевания или размягчения,
вследствие чего соответствующие температуры (застывания или
плавления) всегда являются более или менее расплывчатыми и
условными, т. е. в большей или меньшей степени зависящими
от методики их определения. Очевидно также, что температуры
эти нельзя рассматривать как константы в обычном смысле этого
слова.
Обыкновенно нефть встречается в природе в жидком состоя-
нии. Однако в некоторых случаях она загустевает уже при не-
значительном охлаждении и становится трудно подвижной или
вовсе неподвижной. Явление это зависит от содержания
в цефти твердого парафина: чем больше содержание этою ком-
понента, тем легче застывает нефть, и обратно. Таким образом
температура застывания нефти или нефтепродукта может слу-
жить до известной степени мерой содержания в них парафина.
87
Кроме парафина, известное влияние на застывание нефти
оказывают, невидимому, также смолистые вещества, обладающие
свойством задерживать выделение, парафина. Таким образом
влияние смолистых веществ и парафина на застывание нефти
является, поводимому, прямо противоположным.
В отношении температуры застывания встречающиеся в при-
роде нефти .чрезвычайно разнообразны. Так например, грознен-
ская парафинистая нефть удельного веса 0,838 застывает уже
при 4-11°, тогда как грозненская же беспарафиновая нефть
удельного веса 0,863 застывает ниже — 20°; охинская нефть
(IV горизонт) остается текучей даже при самых сильных моро-
зах, которые бывают на о. 'Сахалине.
Еще больше разнообразия наблюдается в температурах за-
стывания различных нефтепродуктов. Так как и здесь высокая
температура застывания связана с нахождением в смеси тех
либо иных высокоплавких компонентов и в первую очередь твер-
дых парафинов, то наиболее низкую температуру застывания
(до—*80° и ниже), естественно, имеют бензины; за бензинами
следуют керосины, легкие масла и т. д. Если нефть — парафи-
нистая, то уже после отгонки бензина и керосина может полу-
читься мазут с температурой застывания до 30—35°. Такой ма-
зут неудобен в качестве топлива даже в летнее время, но он слу-
жит сырьем для заводского получения парафина. Получаемые
из парафинистого мазута масла также мало пригодны вследствие
высокой температуры застывания; они требуют обеспарафинива-
ния, благодаря которому температура их застывания может
быть резко понижена.
Температура застывания входит в технические нормы многих смазоч-
ных масел, а также мазута. Как уже было указано’, определение темпера-
туры -застывания нефтепродуктов крайне условно. Принятая в ССОР мето-
дика сводится к определению той температуры, яри которой охлажденное
в пробирке вещество при ее наклоне под углом 45° не перемещается в те-
чение 1 мин. Перед определением вещество предварительно подогревают до
60° ® течение 10 мин., а затем ему -дают остыть; такой «термической обра-
боткой» достигается полная однородность «вещества в связи с переходом в
раствор малейших кристалликов парафина, которые могли находиться в
парафинистом продукте, на вид вполне однородном, во взвешенном состоя-
нии.
Вместо температуры застывания для характеристики и нор-
мировки некоторых- нефтепродуктов пользуются их температурой
плавления. Определение последней, конечно, также крайне
условное и производится в специальной аппаратуре и строго
установленных условиях, Так например, температуру плавления
(застывания) парафина определяют в приборе Жукова', темпера-
туру плавления (каплепадения) вазелина и консистентных мазей
(солидол, тавот и т. п.) — в приборе Уббелоде; температуру
плавления (размягчения) асфальта — в приборе Кремер-Сар-
нова.
Прибор Жукова, применяемый для определения температуры
застывания и плавления парафина, представляет собой неболь-
38
Шую тонкостенную сткляпку, в которую па широкой пробке
вставляется пробирка1 с термометром (фиг. 13). В пробирку
помещают около 40 г рас плавленного парафина при температуре
примерно на 5° выше предполагаемой его темпера- п
туры застывания и через равные промежутки вре-
мени (0,5—1 мин.) наблюдают падение темпера-
туры внутри пробирки, причем во избежание пере-
охлаждения парафина прибор время от времени
энергично встряхивают.
Сначала температура падает довольно быстро и
плавно; затем падение замедляется или вовсе пре-
кращается на некоторое время; наконец, падение
температуры возобновлятся снова. За температуру
застывания (затвердевания) и плавления «прини-
мают ту температуру, которая» некоторое время,
остается постоянной, или, если изменение темпера-
туры продолжается, (наивысшую точку, которая
была достигнута после перелома кривой; эта темпе-
ратура соответствует моменту кристаллизации
парафина, вследствие чего прозрачная вначале
жидкость мутнеет. На фиг. 14 даны две кривых,
отвечающих сравнительно простым случаям засты-
вания; соответствующая искомая температура от-
мечена здесь пунктиром. В СССР этот метод при-
нят как стандартный для определения температуры фнг^ & прн.
плавления парафина. бор'Жукова.
Прибор Уббелоде представляет собою термометр, на конце которого с по-
мощью специальной металлической гильзы прилажена небольшая стеклянная
трубочка; последнюю заполняют
Время
испытуемым веществом и погружают
в него термометр. При медленном
нагревании на водяной бане подве-
шенного в пробирке прибора насту-
пает момент, когда на нижнем конце
стеклянной трубочки появляется
капля, которая затем падает. Тем-
пература, соответствующая появле-
нию капли, называется температу-
ря каплеобразования; температуру
же, при которой капля отрывается к
начинает падать, называют темпе-
ратурй каплепадения.
Метод Кремер-Сарнова заключает-
ся в нагревании на водямой или
глицериновой бане стеклянных тру-
бок стандартного размера, заполнен-
ных в нижней части до метки пред-
варительно расплавленным иопытуе-
Фиг. 14. Кривая охлаждения. мым битумом. После остывания этих
битумных пробок на них наливают
по 5 г ртути. При осторожном нагревании трубок наступает момент, когда
битум размягчается ртуть проникает через него и падает на дно стакана.
Это и есть температура размягчения (плавления) битума.
1 Для той же цели удобно полъзонат1ся небольшим сосудам Дьюара
соответствующей формы.
39
Детальное описание отмеченной здесь аппаратуры, а также многочис-
ленных других способов определения температуры плавления и застывания
нефтепродуктов следует искать в специальных методических руководствах.
"ИСПАРЕНИЕ И КИПЕНИЕ
Ъ о
Как известно, испарением называется такой процесс паро-
образования, который‘происходит только с поверхности жидко-
сти при самых разнообразных, непостоянных температурных
условиях. При испарении нефтей и Нефтепродуктов, как и
в случае других сложных смесей, в первую очередь улетучи-
ваются, конечно, наиболее легкие их части. При ©том, однако,
в зависимости от условий, в которых происходит испарение,
увлекаются также более или менее значительные количества
более тяжелых компонентов, хотя бы температура кипения их
намного отличалась от температурных условий испарения. Для
иллюстрации этого явления интересен следующий опыт
(Мэбери): нефть удельного веса 0,815, дававшая при перегонке
42% остатка выше 300°, была оставлена в плоской чашке при
сильной струе воздуха; через месяц ее удельный вес поднялся
до 0,862, остаток же составлял только 33,3%'; таким образом
в указанных условиях без всякого подогрева испарилось 8,7%
фракций, перегонявшихся выше 300°.
На скорость испарения жидкостей влияет целый ряд разно-
образнейших факторов, как-то: температура, величина поверх-
ности испарения, высота слоя жидкости в сосуде, скорость тока
воздуха, уносящего лары испаряющейся жидкости, и т. д.
В условиях хранения нефтей и нефтепродуктов большое зна-
чение имеют конструкции резервуаров, цвет, в который они окра-
шены, .и т. п. Ближайшее изучение и учет этих факторов состав-
ляет важную задачу проблемы борьбы с потерями при хранении
нефти и ее продуктов.
.При нагревании в открытом сосуде или колбе, соединенной
с холодильником, упругость паров жидкости, постепенно повы-
шаясь, достигает, наконец, внешнего, давления, и жидкость заки-
пает. Теперь парообразование происходит уже во всей массе
жидкости, и вещество начинает перегоняться. Если жидкость
представляет собою вещество индивидуальное, кипение ее проис-
ходит до конца при температуре, постоянной для данного ве-
щества и зависящей только от давления, т. е. при температуре
его кипения.
Если жидкость представляет собою смесь вроде, папример,
нефти или какого-либо нефтепродукта, явление значительно
осложняется. Очевидно, что при постепенном повышении тем-
пературы в первую очередь закипают и начинают перегоняться
наиболее легкие части данной смеси. Эти компоненты увлекают
в процессе своего парообразования бблыпие или меньшие коли-
чества паров более тяжелых частей (как при испарении), и, та-
ким образом, в каждый данный момент упругость паров кипя-
щей смеси определяется упругостью не одного какого-либо ве-
щества, а слагается из так называемых парциальных упругостей
40
отдельных находящихся в парах компонентов. По мере того, как
наиболее легкие части смеси выкипают, их место будут занимать
более тяжелые компоненты с более высокой температурой кипе-
ния и т. д. Таким образом температура кипения таких сложных
смесей, как нефть и ее продукты, не будет представлять собою
величины постоянной, «константы»; по мере перегонки она будет
непрерывно повышаться, так что здесь правильнее говорить не
о температуре кипения, а о температурных пределах, в которых
перегоняется данная смесь.
Определение температурных пределов кипения нефтей и неф-
тепродуктов, а- также определение процентного содержания
в них отдельных фракций, кипящих в известных пределах, про-
Фиг. 15. Аппарат для разгонки по Энглеру.
изводится путем разгонки и имеет большое значение особенно
для характеристики нефтей и легких нефтепродуктов (бензина и
керосина). Так как результаты такой разгонки зависят от усло-
вий, в которых она производится (аппаратура, скорость пере-
гонки и т. д.), то опыт ведется с соблюдением всех детально уста-
новленных условий. В настоящее время в СССР и в боль-
шинстве других стран для аналитических целей установлена
«разгонка по Энглеру» (фиг. 15); ее производят из небольшой
колбочки Вюрца со стеклянным холодильником \ причем раз-
1 При разгонке особенно легких нефтепродуктов, например легкого бен-
зина- с содержанием так называемого газового бензина, аппарат Энглера
обычного типа дает мало удовлетворительные результаты, так как (наиболее
легкие части анализируемого вещества не успевают одондансиррватъся
в стеклянном холодильнике и пропадают. Методы анализа нефтепродуктов,
принятые в США, предусматривают поэтому в приборе для разгонки ме-
таллический холодильник, охлаждаемый водой и льдом.
41
меры всех отдельных частей аппаратуры, залив, скорость пере-
гонки и т. д. строго стандартизованы. Обычно разгонка по
Энглеру производится лишь до 300°; поэтому ею пользуются,
дапример, для определения фракционного состава бензинов и
керосинов или соответствующих им дестиллатов. Разгонка более
высококипящих частей нефти производится уже в вакууме.
Нефть начинает кипеть тем ниже, чем богаче она легкими
частями, т. е. чем ниже ее удельный вес. Обычно, нефти
с удельным весом ниже 0,9 начинают кипеть ниже 100°, нефти
более тяжелые — выше 100°. Иногда на начале кипения нефти
отражается преобладание в ней того или иного Углеводородного
ряда с характерными особенностями его физических свойств.
Известно, например, ’ что нафтены — тяжелее парафинов, наи-
более же тяжелыми углеводородами являются ароматические.
Это значит, что нафтены и ароматические углеводороды кипят
ниже, чем парафины того же удельного веса. Отсюда следует,
что нефть, в которой преобладают нафтены или ароматика, мо-
жет иметь более низкое ’ начало кипения, чем парафинистая
нефть того же удельного веса.
Само собою разумеется, что разгонка по Энглеру даст лишь самые
общие и грубые представления о температурных пределах кипения данной
нефти или нефтепродукта и об отдельных содержащихся я них фракциях.
Более точные данные по этим вопросам можно получить при разгонке
с дефлегметором или колонкой, широко применяемыми нс только для чисто
научно-исследовательских целей, но за последнее в]>емя также для целей
технических, а именно для заводского получения узких нефтяных фрак-
ций. К этому вопросу мы еще вернемся ниже, в следующей главе, а более
подробно — в главе о перегонке нефти (ч. II).
ВСПЫШКА, ВОСПЛАМЕНЕНИЕ, САМОВОСПЛАМЕНЕНИЕ
Вспышкой, или температурой вспышки, называется та темпе-
ратура, при которой пары горючей жидкости, в нашем случае
нефти или нефтепродукта, в смеси с воздухом дают при прибли-
жении пламени короткое возгорание, вспышку. При более высо-
кой температуре в аналогичных условиях происходит возгора-
ние не только паров, но и самой жидкости. Температура, при
которой наступает такое явление, называется температурой вос-
пламенения. ! ।
Вспышка представляет собою одну из обычных характери-
стик нефтей и нефтепродуктов, по которой можно составить из-
вестное представление об огнеопасности вещества, о содержании
в нем легких частей и о некоторых других его качествах. Тем
не -менее, вспышка является едва ли ни самой условной констан-
той, на величине которой отражаются самые, казалось бы, мел-
кие детали аппаратуры и методики.
Большинство нефтей и нефтепродуктов имеют при обыкно-
венной температуре упругость паров, недостаточную для вспышки
с кислородом воздуха; для ее образования необходимо подо-
гревание горючей жидкости с периодическими пробами на
вспышку; для получения же при этом сходящихся результатов
42
Крайне важно, чтобы отдельные моменты определения были
тщательно отрегулированы. С этой целью предложен целый ряд
способов и аппаратов.
Аппараты для определения вспышки могут быть подразде-
лены на два основных типа: открытые и закрытые. Примером от-
крытых приборов для определения вспышки может служить при-
нятый в СССР прибор Бренкена, который' состоит из неболь-
шого фарфорового тигля, помещенного в песчаную баню, и тер-
мометра. В Америке вместо прибора Бренкена (Бр) употре-
бляется прибор К ливеланда— металлическая чашка на метал-
Фиг. 16- Аппарат Абель-
Ленского.
Фиг. 17. Аппарат Мартенс-
Пенского.
лической подставке. Все части обоих приборов, конечно, стан-
дартизованы. Из закрытых приборов наибольшее распростране-
ние получили: для низких вспышек (до 50°)—прибор Абелъ-
Пенского (А-П), а для более высоких вспышек — прибор Мар-
тенс-Пенского (М-П). Первый из этих приборов (А-П) употреб-
ляется для легких нефтей и керосина; второй (М-П)1—для тяже-
лых нефтей и смазочных масел. Их устройство в наиболее суще-
ственных чертах заключается в следующем (фиг. 1G и 17)..
Испытуемая нефть или нефтепродукт наливаются до опреде-
ленной метки в небольшой цилиндрический медный или латун-
ный сосуд, закрываемый .крышкой с небольшой заслонкой. По-
следняя прикрывает имеющееся в крышке небольшое окно, пе-
риодически открываемое при определении вспышки на короткий
промежуток -времени с помощью особого заводного механизма
(А-П), либо вручную с помощью пружинного рычага (М-П).
43
В момент отодвигания ййслонйи it оконцу автоматически
приближается пламя маленькой горелки. Оба прибора (А-П и
М-П) снабжены термометрами, опущенными в испытуемый неф-
тепродукт .через специальное отверстие крышки; у прибора
М-П, кроме того, имеется небольшая мешалочка, приводимая
в действие с помощью гибкого пружинного стержня. Цилиндри-
ческие сосуды, в которые наливается испытуемый нефтепродукт,
окружены воздушной баней; у прибора А-П последняя окружена
еще водяной баней. Нагревание производится извне с помощью
. показанных на рисунках горелок. Все части обоих приборов
стандартизованы. Определение вспышки производится в усло-
виях, также строго нормированных, описание которых можно
найти в специальных руководствах ио анализу нефтепродуктов;
здесь же приводится описание приборов для определения
вспышки других конструкций (приборы Люшер, Эллиот, Таг и
др.), применяемых в различных странах.
Чтобы иллюстрировать, насколько температура вспышки за-
висит от методики ее определения, обратимся к табл. 13, в кото-
рой сопоставлены вспышки различных нефтепродуктов, опреде-
ленные в открытом (Бр) и закрытом (М-П) аппаратах.
Таблица 13
Вспышки нефтепродуктов на равных аппаратах
по X. Гурвичу
Нефтепродукты Бр М-П Разница
Нефть 46 30 16
Мазут........ П9 £6 23
Пиронафт 115 106 9
Соляровое масло . . 160 148 12
Машинное „ . . 212 196 16
Цилиндровое„ . . . 236 215 21
Как видно из данных табл. 13, в открытом аппарате (Бр)
вспышка появляется всегда при более высокой температуре, чем
в закрытом (М-П). Это расхождение особенно значительно для
малооднородных продуктов (нефть, мазут); для более однородных
продуктов (различные масла) оно тем больше, чем тяжелее про-
дукт, т. е. чем выше его температура вспышки. Особенно велико
бывает такое расхождение при грубых загрязнениях тяжелых
нефтепродуктов легкими, как показывает следующий пример:
к маслу с температурой вспышки 200° по Бр и 180° по М-Н
было прибавлено 0,5 % бензина; после этого температура вспыш-
ки его оказалась 200° ио Бр (т. е. без изменения) и 60° по М-П.
Таким образом расхождение температур вспышки в открытом и
закрытом аппаратах достигло в данном случае 120°. Аналогич-
ные явления, хотя и в не столь резко выраженной форме, наблю-
даются при загрязнении масла продуктами его распада, например
44
от каких-либо неправильностей при перегонке и других причин.
Вообще же расхождение вспышки по Бр и М-П больше, чем на
25—30 , считается превышающим норму и может служить ука-
занием на загрязнение масла какими-либо легкими примесями.
Причина расхождения температур вспышки в открытом и
закрытом приборах понятна. В открытом приборе пары нефте-
продукта свободно диффундируют в воздух и рассеиваются; по-
этому необходимо значительное подогревание нефтепродукта для
достаточного повышения упругости его паров и получения смеси
их с воздухом, способной дать вспышку. В закрытом приборе,
наоборот, пары нефтепродукта накапливаются в сосуде, так что
необходимая для вспышки концентрация их достигается рань-
ше— при более низкой температуре. (Этими же соображениями
легко объясняется влияние на вспышку летучих примесей.
Из факторов, не связанных с конструкцией прибора, особое
влияние на температуру вспышки оказывают температура кипе-
ния вещества и давление, при котором производится 'определе-
ние вспышки.
Что между температурой кипения вещества и его температу-
рой вспышки должна существовать самая тесная связь, — само
собою понятно. Чем выше температура кипения вещества, тем
выше его температура вспышки, и наоборот. Для индивидуаль-
ных углеводородов зависимость эта, невидимому, может быть
выражена в форме простой пропорциальности £б1, если обе тем-
пературы выразить в градусах абсолютной шкалы, а именно:
Тг = К Т,
где Т' = 273 -М' представляет собою температуру вспышки t;
Т — 273 + t — температуру кипения t вещества по абсолют-
ной шкале;
К — коэфициент пропорциональности.
Так как нефтепродукты не имеют постоянной температуры
кипения, то к ним вышеприведенная формула, очевидно, не при-
ложима.
Заметное влияние на температуру вспышки оказывает также
давление, ввиду чего принято определение вспышки относить
к нормальному давлению (760 мм). Экспериментально устано-
влено, что понижение давления на 1 мм понижает температуру
вспышки на 0,033—0,036°. На основе этих данных легко соста-
вить таблицу поправок на барометрическое давление, которые
согласно сказанному для давлений ниже 760 мм будут иметь
положительное значение, для давлений же выше атмосферного —
будут отрицательны.
О влиянии легких примесей на температуру вспышки более
тяжелых нефтепродуктов уже было сказано выше. Более общим
вопросом того же порядка является вопрос о температуре
вспышки смеси двух нефтепродуктов. Последняя оказывается
всегда ниже той величины, которая может быть вычислена по
правилу смешения, и это отступление от аддитивности тем
45
больше, чем больше различие в температурах вспышки смеши-
ваемых компонентов.
По своей физико-химической сущности вспышка представляет
собою небольшой взрыв смеси паров горючей жидкости с возду-
хом. Как и всякий взрыв газовой смеси, вспышка может проис-
ходить лишь в сравнительно, узких пределах состава этой смеси:
при недостаточности паров горючего в смеси, равно как при
слишком большом их содержании взрыва вовсе не получается.
Отсюда — понятие о нижнем' и верхнем пределах взрывчатости
смеси, которые определяются наименьшим и наибольшим содер-
жанием горючего пара или газа в смеси, сохраняющей взрывча-
тые свойства.
При вспышке обыкновенно приходится иметь дело с нижним
пределом взрывчатости; таковы, например, случаи опреде-
ления вспышки керосина или смазочных масел, когда путем их
подогрева повышают содержание горючих паров в воздухе до
нижнего предела взрывчатости. Иначе обстоит дело с бензином,
который при комнатной температуре может вовсе не дать:
вспышки в закрытом аппарате (например А-П). Однако, пони-
жая упругость паров бензина путем охлаждения, можно подойти
к такой температуре, при которой вспышка произойдет. Оче-
видно, это уже верхний предел его взрывчатости.
Нижний и верхний пределы взрывчатости могут быть вычислены по
следующим простым формулам Торнтона:
Нижний предел..................Л Г 4- 2 (У—1)
Верхний • ..............• . Аг.
где У — число атомов кислорода, необходимого для полного сгорания,
М — молекул горючего вещества.
Так например, сгорание пентана, CsHis, происходит по уравнению:
СбИ12 +160 = 5СОо + 6Н2О;
здесь, следовательно, М — 1 и N = 16, так что для нижнего предела будем
иметь отношение объемов углеводорода и кислорода 1:15, а для верхнего
предела —1:4. Если взрывчатая смесь образована ие с кислородом, а с воз-
духом, то необходимо принять во •внимание, что 1 объем кисло|юдя содер-
жится в 4,85 или, округляя, в 5 объемах воздуха. Таким образом для смеси
пентана с воздухом нижний предел взрывчатости определится отношением
1:75, а верхний—отношением 1:20. Опыт довольно хорошо согласуется
с такого рода расчетами.
Определение температур вспышки имеет известное практиче-
ское значение главным образом для характеристики керосина и
смазочных масел.
Температура вспышки керосина может служить для характе-
ристики его огнеопасности. В прежнее время, когда бензин
являлся продуктом, не находившим достаточного сбыта, опас-
ность подмеси бензиновых фракций к керосиновым являлась
вполне реальной. Ввиду этого в целях обеспечения известного
минимума пожарной безопасности вспышка керосина нормирова-
лась во всех странах законодательным порядком с указанием
аппарата, с которым должно производиться определение. Так
46
например, в СССР нормальная температура вспышки керосина
должна быть не ниже 28° (А-П), в Германии и Австрии —21°
(А-П), во Франции — 35е (Гранъе, Люшер), в ОША —20—45°
(Сейболт, Таг) и т. д. Однако в настоящее время ввиду громад-
ного спроса на бензин опасность умышленного подмешивания
бензина к керосину, очевидно, исключается, а поэтому прежнее
номирование керосина по его вспышке в значительной мере
утратило свой практический смысл.
Что касается температуры вспышки смазочных масел, то как
уже было отчасти отмечено выше, эта характеристика может
служить здесь показателем однородности масла, степени его за-
грязнения (например бензином), а также соответствия его тем
образцам, которые были выбраны для той или иной цели. В за-
висимости от назначения смазочных масел температура вспышки
их может колебаться в очень широких пределах. Так например,
для наиболее легких типов масел (веретенные и т. п.) вспышка
колеблется от 120 до 170°; для масел средней вязкости (машин-
ные, турбинные и т. п.) — от 165 до 200° (Бр); масла для двига-
телей внутреннего сгорания (моторные, автолы), авиационные и
компрессорные имеют температуру вспышки от 185 до 240° (Бр);
наконец, для наиболее тяжелых масел, цилиндровых, вспышка
дается от 215 до 320° (Бр).
Если при определении вспышки в открытом приборе (Бр)
осторожно продолжить нагревание масла градусов на 30—50
выше температуры ево вспышки, поднося время от времени
зажигательное пламя, то, наконец, наступает возгорание масла
по всей его поверхности. Если такое возгорание продолжается
не менее 5 сек., соответствующую температуру называют темпе-
ратурой воспламенения нефтепродукта. Практическое значение
температуры воспламенения несравнимо меньше, чем темпера-
туры вспышки.
От воспламенения горючего вещества следует отличать его
самовоспламенение. Последнее должно происходить без сопри-
косновения смеси паров горючего и воздуха (или кислорода)
с пламенем, а лишь от соответствующего подогрева смеси извйе.
Температура самовоспламенения иногда на несколько сот граду-
сов выше температуры вспышки, причем методика определения
оказывает здесь еще большее влияние на его результаты, чем
в случае вспышки и воспламенения. Так например, для бензина,
температура вспышки которого около 15—20 ниже 0, различ-
ными авторами даются следующие температуры самовоспламене-
ния для смеси его с воздухом: 383 (Моор), 415° (Хольм) и в за-
висимости от материала трубки, в которой производился обогрев
смеси, —685° ((железо), 585° (кварц) и -545° (йенское стекло)
(Гвоздев). В последнее время методика определения температуры
самовоспламенения подвергалась значительному усовершен-
ствованию и нашла себе практическое применение, так как уста-
новлено, что существует несомненная связь между температурой
самовоспламенения горючего и склонностью его к детонации
в цилиндре двигателя внутреннего сгорания^
47
ТЕПЛОЕМКОСТЬ
Как известно, теплоемкостью называется количество тепла
(в калориях), которое необходимо затратить для нагревания 1 г
того или иного -вещества на один градус. Вместе с определением
ряда других тепловых констант нахождение теплоемкостей раз-
личных веществ составляет одну из задач специального отдела
физики, называемого калориметрией. Хотя данные различных
авторов по вопросу о теплоемкости нефтей и нефтепродуктов
значительно расходятся, тем не менее ввиду важности этой кон-
станты на некоторых наметившихся здесь правильностях необ-
ходимо остановиться.
Теплоемкости различных нефтей при температурах от 0 до
50° колеблются в довольно узких пределах, примерно от 0,4 до
0,5 кал; невидимому, как общее правило, с повышением удель-
ного веса теплоемкости нежней уменьшаются. Для иллюстрации
этой правильности может служить табл. 14; следует, однако,
иметь в виду, что данные некоторых авторов в нее не уклады-
ваются (ср. ниже). Табмца и
Теплоемкости различных нефтей между 0 и 50э
(по Мэбери и Гольдштейну)
Месторождение Уд. вес Теплоемкость
Пенсильвания .... 0,8095 0,5000
Уайоминг 0,8816 0,4323
Тексас • 0,9200 0,4315
Тексас 0,9466 0,4009
Калифорния 0,9600 0,3980
Ход изменения теплоемкости для различных погонов одной
и той же нефти, в зависимости от температурных пределов их
кипения или их удельного веса в отдельных случаях может ока-
заться довольно сложным. В первом приближении здесь наблю-
дается та же правильность, которая только что была отмечена
для нефтей: чем ниже удельный вес дестиллата, тем больше его
теплоемкость. По характеру изменения темплоемкости мы видим
здесь, таким образом, ту же 'закономерность, которая устано-
влена для теплоемкостей пормалыгых углеводородов ряда ме-
тана: как видно из данных табл. 15, по мере увеличения моле-
Таблица 15
Теплоемкости нормальных парафинов между 0 и 50°
Состав углеводорода Теплоемкость Состав углеводорода Теплоемкость
Св Н14 Cr П1я 0’10 И со 0,5272 0,5052 0,5021 CisHse СнН,0 СюН.ч ♦ 0,4997 0,4973 0,4957
48
кулярного веса, а следовательно, с повышением температуры
кипения и удельного веса темплоемкости парафинов правильно
уменьшаются.
Так как, однако, физические свойства веществ зависят не
только от их молекулярного веса, но также от их химической
природы, то понятно, что в отдельных случаях влияния отдель-
ных факторов на теплоемкость различных нефтяных фракций
могут перекрещиваться, тем более что зависимость теплоемкости
от молекулярного веса в других углеводородных рядах может
быть существенно иной, чем у парафинов; примером может слу-
жить ароматический ряд, в котором по мере увеличения молеку-
лярного веса теплоемкость углеводородов возрастает.
Для расчета теплового баланса нефтеперегонных установок
и в некоторых других случаях важно знать зависимость тепло-
емкости нефтепродуктов от температуры. Зависимость эту можно
выразить общей формулой:
Ct = а Ы 4- ct*...,
где Ct — теплоемкость нефтепродукта при температуре t.
Определив Ct данного нефтепродукта при трех различных
температурах, можно вычислить значения величин а, 6, с и по-
лучить искомую зависимость Ct от температуры.
Опыт показывает, что в пределах температур до 180—200°
зависимость теплоемкости нефтепродуктов от температуры часто
близка к линейной, причем с повышением температуры теплоем-
кость нефтепродуктов, как почти у всех жидкостей, равномерно
возрастает. При более высокой температуре в некоторых случаях
наблюдалось более интенсивное возрастание теплоемкости, и со-
ответствующая кривая начинала заметно менять свой вид. Для
иллюстрации может служить табл. 16, в которой приведены
теплоемкости некоторых бакинских нефтепродуктов [7].
Таблица 16
Теплоемкости бакинских нефтепродуктов
(по Тихомирову и Жузе)
Керосин дестиллат •Легкое соляр. масло <?1Б = 0,8845 Тяжелое соляр. масло d]6 = 0,8916
“15 =» 0,8112
t Ci t Ct t Ct
31,0 0,4418 28,6 0,4226 28,3 0,4307
101,2 0,5171 100,5 0,5315 100,2 0,5050
140,7 0,5936 179,0 0,6330 ' 181,6 0,5947
— 193 2 0,6367 201,4 0,6225
— — — — 219,0 0,6603
Для бакинского керосинового десгиллата изменение теплоемкости
с температурой может быть выражено формулой:
Ct = 0,424 + 0,000395# + 0.00000575#2.
4 Зак. 1822. Химия нефти.
49
Для легкого солярового масла эта зависимость принимает линейную
форму:
Ct = 0,387 + 0,00133/.
Наконец, для тяжелого солярового масла при температурах до 180°
получается также линейная зависимость:
Ct = 0,4004 4- 0,С01С7«;
при температурах же более высоких та же зависимость принимает для
тяжелого соляра более сложный вид, а именно:
Ct = 1,5856 — 0,0114414- 0,0300329/2.
СКРЫТАЯ ТЕПЛОТА ИСПАРЕНИЯ
Скрытой теплотой испарения называется количество теплоты
(в калориях), которое необходимо затратить для превращения
1 г жидкости в насыщающий пространство пар той же темпера-
• туры. Если скрытая теплота испарения отнесена к температуре
кипения жидкости, то ее называют иногда скрытой теплотой
кипения. Как показывают примеры табл. 17, в гомологических
рядах при переходе от низших членов ряда к высшим скрытая
теплота кипения уменьшается.
Таблица 17
Скрытая теплота кипения парафинов
Формула Наименование Темп, кипения Скрытая теплота кипения в кал
С5 Hjo Пентан 36° 85,8
Сс Ни Гексан (ИР 79,2
С7 Ню Гептан 98° 74,0
Cg Ни Октан 120° 71,1
СчоНи Декан 173° 60,5 •
Различают также полную теплоту испарения. Так называют суммарное
количество теплоты, необходимое для нагревания 1 г жидкости до некоторой
температуры Т с последующим превращением ее в насыщенный .нар той же
температуры, например температуры кипения данной жидкости. В гомоло-
гических рядах количество теплоты, необходимое для нагревания каждого
последующего гомолога от 0° до температуры его кипения, естественно,
возрастает, причем это возрастание отнюдь нс компенсируется постепен-
ным уменьшением скрытой теплоты кипения согласно, вышеизложенному.
Отсюда понятно, что полная теплота испарения в гомологических рядах
по мере возрастания молекулярного веса увеличивается.
Помимо общих экспериментальных трудностей, при опреде-
лении скрытой теплоты испарения нефтей и нефтепродуктов
встречаются еще специальные затруднения в отношении пол-
ного превращения их в парообразное состояние. В подобного
рода случаях особую важность приобретает закономерность, из-
вестная под именем «правила Трутона». Последнее заключается
в следующем:
Если Л/—молекулярный вес вещества, а Е— его скрытая
теплота кипения, то величина ME представляет собою молеку-
50
лярйую скрытую теплоту кипения. По Трутону молекулярная
скрытая теплота кипения различных веществ пропорциональна
их асболютной температуре кипения, т. е.
ME— КТ,
где Т—абсолютная температура кипения, а К —коэфициент
пропорциональности; для различных веществ, в том числе для
углеводородов, К близко к 20.
Чтобы воспользоваться правилом Трутона в применении
к нефтепродуктам, необходимо определить для них величину М.
Очевидно, здесь может быть речь только о средних молекуляр-
ных весах отдельных нефтяных фракций, т. е. о величинах, ко-
торые могут быть определены с достаточной точностью. Произве-
денные на основе ©тих определений подсчеты скрытых теплот
•испарения отдельных нефтяных фракций для различных неф-
тей, тяжелых и легких, парафинистых и беспарафинов.ых, дали
для соответствующих фракций хорошее совпадение и могут быть
приняты за основу при суждении о скрытой теплоте испарения
различных нефтяных фракций. Некоторый цифровой материал
ио этому вопросу собран в табл. 18.
Таблица 18
Скрытые теплоты испарения фракций бакинских и грозненских нефтей
(по Обрядчикову)
Наименование фракций Пределы темп, кип. Молекул, вес Скрытая теплота испарения в кал
Бензиновые . . 100—180° 100—137 66—74
Керосиновые . . . 235—285° 158—218 64-50
Соляровые .... 301—370° 209—260 54—51
Веретенные . . . 370—410° 260-290 51—47
Машинные .... 410—480° 300—380 47—40
Цилиндровые . . > 480° > 380 < 40
Прямые определенйя скрытой теплоты испарения, произво-
дившиеся различными авторами для различных нефтепродук-
тов, дали недостаточно совпадающие результаты.
ТЕПЛОТВОРНАЯ СПОСОБНОСТЬ
Теплотворной способностью называется количество теплоты
(в калориях), выделяемое 1 кг топлива при сгорании до конца,
т. е. до углекислоты и воды, короче говоря,—его теплота горения.
Определение теплоты горения производится с большой точностью
путем сожжения навески топлива в специальных бомбах при
повышенном давлении в атмосфере кислорода; выделяющаяся
теплота определяется калориметрически, причем непосредствен-
но получают так называемый верхний предел ее, т. е. теплоту
горения с образованием жидкой воды.
лф 51
От верхйего предела тсплотворной способности следует отличать Ниж-
ний ее предел, -или полезную теплотворную способность. Последняя соот-
ветствует тому случаю, когда образующаяся при сгорании вода не выде-
ляется в виде жидкости, а сохраняется или уносится в виде пара. Оче^
видно, нижний предел теплотворной способности равен верхнему ее пределу
без теплового эффекта сжижения воды.
Теплотворная способность нефтей колеблется в довольно
узких пределах, .примерно от 10 400 до 11 000 кал. При этом,
как видно из табл. 19, чем меньше удельный вес нефти, тем
больше ее теплотворная способность.
Таблица 19
Теплотворная способность некоторых нефтей СССР
(по данным АзНИИ)
Месторождение Уд. вес Теплотворная способность (верхний предел) в кал
Сураханы 0,7932 10916
Биби-Эйбат 0,8974 10610
Бинагады 0,9218 10430
Та же правильность наблюдается для теплотворной способно-
сти различных нефтепродуктов: как показывает табл. 20, при
переходе от легких нефтепродуктов. к тяжелым теплотворная
способность все время снижается, так что наибольшей тепло-
творной способностью обладают бензины, наименьшей — мазуты.
Таблица 20
Теплотворная способность бакинских нефтепродуктов
(по* Со к олову)
Наименование Теплотворная Наименование Теплотворная
нефтепродукта способность в кал нефтепродукта способность в КОЛ
Бензин 11230 Веретенное масло 10 894
Керосин 11059 Машинное , 10 899
Пиронафт 10 972 Цилиндровое , 10 883
Соляровое масло 10914 Мазут 10 843
Термохимия учит нас, что теплота горения всякого химиче-
ского соединения равна сумме теплот горения составляющих
его элементов за вычетом теплоты его образования из этих эле-
ментов. По сравнению с теплотой горения органических соедине-
ний теплоты их образования очень невелики. Отсюда возникает
возможность вычислять теплотворную способность топлива на
основе его элементарного состава, для чего различными авторами
были предложены разные формулы. Такова, например, формула
Менделеева:
Q = 810 + 300 Н— 28 (О — $),
где Q — теплотворная способность нефти; С, Я, О и S — про-
центное содержание в ней соответствующих элементов.
52
Некоторые из такого рода формул при расчете дают хорошее
совпадение с опытом. В основе их применения лежат теплоты
горения соответствующих элементов, например: углерода—
8 140 кал, водорода 34 500 кал, а за вычетом скрытой теплоты
его испарения — 30 000 кал и т. д.
Помимо формул, позволяющих вычислить теплотворную спо-
собность топлива по данным его элементарного состава, суще-
ствуют эмпирические формулы, в которых теплотворная способ-
ность выражается как функция удельного веса. Такова, напри-
мер, получившая широкое распространение, особенно в США,
формула и график Крэго:
Qv = 12 500 — 2100^;
Qp = Qv — 50,457/,
где Qv — высшая теплотворная способность при постоянном
объеме;
Qp — низшая теплотворная способность при постоянном
давлении;
Н—процентное содержание водорода в продукте, опре-
деляемое соотношением:
H = 2G —15d.
Применение формулы Крэго к бакинским продуктам дало
несколько пониженные результаты сравнительно с опытными
данными; однако расхождение не превышает 3—5%; для гроз-
ненских нефтепродуктов расхождение оказалось меньше чем
на 1%.
Особенно интересное обобщение в этой важной области получается,
если теплотворную способность (ее нижний предел) отнести не к единице
веса горючего вещества, а к единице веса кислорода (Д. П. Коновалов).
По уравнению горения данного вещества, составленному на основе элемен-
тарного состава горючего, не трудно вычислить кислородный коэфициент К
вещества, т. е. количество кислорода, необходимое для полного сгорания
одной его весовой единицы. Отношение нижнего предела теплотворной спо-
собности топлива Q1 к его кислородному коэфициенту можно назвать его
кислородным потенциалом. Опыт и вычисление показывают, что кислород-
ный потенциал твердого и жидкого топлива есть величина постоянная,
равная кислородному потенциалу аморфного угля, в среднем — 3,05, т. е.
Q1 = 3,057Г.
Этой закономерности подчиняется громадное количество органических
веществ. На ее основе, очевидно, может быть произведен расчет тепловорной
способности топлива [8].
ЦВЕТ НЕФТИ И НЕФТЕПРОДУКТОВ
В зависимости от содержания асфальтовых и смолистых ве-
ществ цвет нефти .колеблется в весьма широких пределах: от
коричневого, темнокоричневого и черного—до красноватого,
желтого и светложелтого. Как исключение, встречаются даже»
совершенно бесцветные нефти; такова, например, белая сура-
ханская нефть, в настоящее время, впрочем, почти исчезнув-
шая.
53
кроме той или иной окраски, большинство нефтей обладает1
также 'заметной флуоресценцией. Одни из них дат в отражен^
ном свете зеленоватый отлив (пенсильванская нефть), другие—
синеватый (бакинская нефть). Свойство это, несомненно, также
связано с присутствием в нефтях каких-то определенных соеди-
нений. Возможно, что это — различные многоядерные углеводо-
роды ароматического ряда типа хризена и ему подобных либо те
или иные родственные им системы.
Цвет сырых нефтей служит лишь для их общей характери-
стики. Напротив, ищет некоторых нефтепродуктов входит в их
технические нормы, так как по цвету можно судить о степени
их очистки и стабильности. По нормам, принятым СССР, цвет
предусматривается для крекинг-бензина, керосина, парафина и
масел различного назначения.
Определение цвета легких нефтепродуктов производится с помощью ко.
лориметра того или иного типа. Один из приборов этого рода, колориметр
Штаммера, установлен в СССР для определения цвета асфтопродучстов.
Сущность методики заключается в определении высоты столба нефтепро-
дукта, соответствующего по цвету одному из стандартных стекол, набор
которых является необходимой принадлежностью колориметра. Таких стекол
или марок у колориметра Штаммера шесть, а именно:
1) Water White W. W- 1 марка — бесцветная как вода.
2) Superfine White Su. W. 2 марка — со слабым желтоватым оттенком.
3) Prime White P. W. 3 Maptea — более интенсивное окра- шивание.
4) Standard White S. W. 4 марка — окрашивание еще более интенсивное.
5) Good Merchantable G- M. 5 марка — цвет чая средней крепости.
6) Not Good Merchantable N. G. M. 6 марка — цвет крепкого чая. • •
Каждая из этих марок — легко восстановляема: она соответствует цвету
определенного столба раствора хромовокислого калия (КаСгО«) в 50-процент-
пой серной кислоте определенной крепости. Для выражения степени окраски,
промежуточной между двумя марками, пользуются дробными величинами
(например 2,5 марки и т. п.).
Определение цвета цветных нефтепродуктов, например парафина, может
производиться либо в расплавленном состоянии, либо в растворе бензина.
Следует, однако, иметь в виду, что при этом обычно приходится иметь
дело с глубоки^ ^несоответствием оттенков исследуемого вещества и стекол
Штаммера. Ввиду этого удобнее определять цвет парафина в твердом со-
стоянии, для чего пользуются отраженным лучом света от’плитки парафина
толщиною в 1 е,м. Такая плитка кладется на соответствующую половину бе-
лой пластинки -колориметра (под пустую трубку), и цвет плитки сравни-
вается с переменным столбом бихромата калия или иного стандартного ра-
створа [91.
По техническим нормам СССР 1937 г. определение цвета по Штаммерл
предусматривается для крэкинг-бензинов, осветительного керосина, трактор-
ного керосина, парафина и некоторых видов масел специального назначения
(парфюмерное масло). Для ряда других -нефтепродуктов, как-то: дизельного
-топлива, некоторых видов индустриальных масел, масел для двигателей
внутреннего сгорания, в частности — авиационных, масел для паровах ма-
шин и компаундированных масел (судовые -масла), определение цвета
производится с другим колориметром, получившим широкое распростране-
ние в аналитических лабораториях, а именно с колориметром Дюбоска.
54
ЛУ ЧЕПРЕЛОМЛЕП ЦЕ
Фиг. 18. Рефрактометр Аббе
Коэфициенты лучепреломления или рефракции изменяются
для углеводородов различных рядов параллельно их удельным
весам. Наименьшими -коэфициептами рефракции обладают угле-
водороды парафинового ряда, наибольшими — ароматические
углеводороды: нафтены же зани-
мают промежуточное положение
между этими двумя крайними
рядами. Отсюда следует, что для
нефтей различных типов изме-
нение коэфициеитов рефракции
должно быть параллельно изме-
нению их удельных весов. Ход
изменения коэфициеитов рефрак-
ции для различных погонов
одной и той же нефти также
аналогичен ходу изменения их
удельных весов (ср. выше).
Кроме общей характеристики
нефтей и нефтепродуктов, этими
физическими свойствами их мож-
но пользоваться для аналитиче-
ских целей, например для опре-
деления парафина в полуфабри-
катах пли, как будет подробнее
показано ниже, для определения
в нефтях содержания ароматики.
Определение коэфициеитов ре-
фракции производится с помощью рефрактометров. Некоторые
приборы этого рода, например, цейсовский рефрактометр Аббе
(фиг. 18), ввиду его простоты и точности получили особенно
широкое распространение для характеристики химически одно-
родных веществ; применение его к нефти н нефтепродуктам
ввиду крайней сложности их состава сравнительно ограничено.
ОПТИЧЕСКАЯ ДЕЯТЕЛЬНОСТЬ
Способность нефтяных погонов вращать плоскость по 1яри-
зации света, иначе говоря, их оптическая деятельность, была
открыта более ста лет тому назад (Био, 1835 г.). В новейшее
время это свойство вновь привлекло к себе внимание различ-
ных исследователей и подвергнуто тщательному изучению па
громадном опытном материале.
Оптическую деятельность удобнее исследовать па отдельных
нефтяных фракциях, так как сырые нефти обыкновенно сильно
окрашены и плохо пропускают поляризованный свет. Оказалось,
что за отдельными немногими исключениями все нефти яв-
ляются оптически деятельными. Громадное большинство их имеет
слабое правое вращение, и только из некоторых нефтей (напрп-
55
мер с о. Борнео, о. Явы и др.) наряду с -правовращающими
фракциями были выделены также недеятельные и левовращаю-
щие.
Вращательная способность распределяется в нефтях по’ от-
дельным погонам крайне неравномерно. Низшие погоны, бензи-
новые и керосиновые, обычно вовсе недеятельны. Активность
начинает проявляться лишь с соляровых дести ллатов и по мере
повышения температуры кипения постепенно усиливается. Су-
ществует указание, что, достигнув далее некоторого максимума,
вращение для более высококипящих погонов начинает умень-
шаться. Однако последние исследования не подтвердили суще-
ствования такого максимума: изучение целого ряда мазутов по-
казало, *1то вращение их отдельных фракций возрастает до
конца перегонки без разложения, т. е. примерно до 320° при
6—10 мм давления.
Величины оптической деятельности у нефтей различных ти-
пов заметно разнятся между собой. Нефти метановые и нафте-
новые, а также нефти, занимающие промежуточное положение
между этими двумя основными типами, проявляют сравнительно
слабую активность. Из нефтей СССР сюда относятся нефти Ба-
лахано-Сабунчинского района, сураханская нефть, грозненские
парафинистые и слабопарафинистые, особенно же эмбенские
(Доссор, Макат). Значительно большую оптическую деятельность
проявляют нефти •бензольно-нафтенового типа, как-то: биби-
эйбатская, грозненская беспарафиновая и некоторые другие.
Следует, впрочем, отметить, что даже для этих последних неф-
тей угол вращения наиболее активных фракций при длине
трубки 100 мм обычно нс превышает одного градуса, для неф-
тей же первого тина этот угол колеблется всего от 0,1° до
нескольких десятых градуса.
Большой интерес в связи с проблемой происхождения нефти
представляет вопрос о носителях оптической деятельности,
иначе говоря, о тех веществах, которыми обусловливается ак-
тивность нефти.
Первоначально были высказаны предположения, что оптиче-
ская деятельность нефти связана с характером некоторых ее
второстепенных компонентов. Ближайшие исследования не под-
твердили, однако, этих предположений. Так например, после
того как было установлено, что естественные нафтеновые кис-
лоты являются оптически деятельными, естественно было по-
ставить вопрос, не ими ли обусловливается и оптическая дея-
тельность нефти. Оказалось, однако, что активность нефтепро-
дуктов сохраняется даже после тщательной обработки их ще-
лочью, т. е. после удаления нафтеновых кислот. Равным обра-
зом, очистка нефтепродукта от сернистых соединений, а также
полное отсутствие в некоторых из них азотистых соединений
нисколько не отражались на их оптической деятельности. От-
сюда можно сделать вывод, что носителями оптической деятель-
ности нефтей являются не второстепенные, а основные их ком-
поненты, т. с. углеводороды.
56
Как известно, оптическая деятельность органических соеди-
нений связана со вполне определенной особенностью их строе-
ния, а (именно с асимметричной структурой их молекул. В про-
стейших случаях эта особенность проявляется в том, что орга-
ническая молекула содержит но крайней мере один асимметри-
ческий атом углерода, т. е. углерод, связанный непосредственно
с четырьмя различными атомами или атомными группами.
Опыт и наблюдений показывают, что оптически деятельные ор-
ганические соединения могут принадлежать к самым разнооб-
разным классам органических веществ. Среди них имеются
также и углеводорода предельного характера, например опти-
чески деятельные нафтены, синтетически получаемые различ-
ными методами из оптически деятельного исходного материала.
Какова же химическая природа оптически деятельных угле-
водородов естественной нефти? Помимо только что приведенных
соображений общего характера, данные по этому интересному
вопросу пока далеко не достаточны.
Так как всякое оптически деятельное органическое соедине-
ние может быть получено либо из оптически деятельного исход-
ного материала, либо при участии оптически деятельного ве-
щества, последние же всегда в той или иной форме связаны
с оптически деятельными соединениями животного или расти-
тельного происхождения, то не без оснований неоднократно ука-
зывалось, что оптическая деятельность является одним из ре-
шающих соображений в пользу органического происхождения
нефти, точнее — ее оптически деятельных углеводородов. Энглер
высказал предположение, что источником образования этих
углеводородов нефти является холестерин.
Холестерин представляет собою бесцветное, жирное на-
ощупь, кристаллическое вещество состава О27Н46О с температу-
рой плавления 148°; оптически деятелен; его [а]л=—37,8°.
По своей химической природе это — вторичный полицикличе-
ский алкоголь; строение его полностью пока не установлено.
Холестерин широко распространен Bi животном царстве. Он
встречается, например, в мозговом веществе, желчи, белых кро-
вяных тельцах, животном жире, рыбьем печеночном жире, осо-
бенно у акул и т. д., причем в некоторых случаях содержание
его достигает нескольких процентов (до 4—0%). К холестерину
примыкают аналогичные вещества (стерины) растений, фито-
стерины, также широко распространенные в растительном
царстве.
Так как естественные нефти и ее дестиллаты дают некоторые
цветные реакции холестерина, например окрашивание в розовый
или фиолетовый цвет с трихлоруксусной кислотой (реакции Чу-
гаева), то прежде всего естественно возникает вопрос, не нахо-
дится ли холестерин в некоторых нефтях, как таковой, и не он
ли и обусловливает их оптическую деятельность. Против такого
предположения можно привести, однако, 'Следующие доводы:
1) как показало ближайшее исследование, реакция с трихлор-
уксусной кислотой вовсе не является специфической для холе-
67
Стер ина; 2) как было указано, холестерин вращает плоскость
поляризации света влево, тогда как большинство естественных
нефтей принадлежит к правовращающим соединениям; 3) нако-
нец, специальное исследование двух левых яванских нефтей на
содержание в них -холестерина с помощью очень чувствитель-
ной реакции (с дигитонином) привело к отрицательному ре-
зультату, т. е. показало полное отсутствие в нрх примеси холе-
стерина. Таким образом можно считать установленным, что
оптическая деятельность нефтей зависит отнюдь не от нахожде-
ния в них холестерина, как такового, и если связана с этим
веществом, то может зависеть только от продуктов его глубокого
распада.
Действительно, уже при медленной перегонке в вакууме ле-
вовращающий холестерин дает продукты с сильно правым вра-
щением. В присутствии инфузорной земли при этом получается
нефтеобразная смесь, содержащая, кроме газообразных и легких,
жидких углеводородов, также более высококипящие компоненты
типа смазочных масел. Особенно хорошо идет такое пирогене-
тическое разложение холестерина в присутствии безводного хло-
ристого алюминия: выход жидких углеводородов с температу-
рой кипения 60—400° достигает при этом 70%; по своему ха-
рактеру эта смесь углеводородов вполне аналогична природной
нефти и подобно последней имеет правое вращение. Последнее,
оДнако, почти полностью исчезает после обработки соответ-
ствующих погонов серной кислотой, хотя общая потеря от такой
очистки незначительна.
В противоположность неустойчивости оптической деятель-
ности холестериновых дестиллатов рядом специальных опытов
установлено, что оптическая деятельность погонов естественной
нефти сохраняется даже после обработки такими энергичными
реагентами, как дымящая серная кислота, азотная кислота и
хлористый алюминий. Отсюда следует, что оптически деятельные
углеводороды естественной нефти в отличие от носителей опти-
ческой деятельности холестериновых дестиллатов имеют пре-
дельный характер и что, если источником их образования в ес-
тественной нефти действительно является холестерин, продукты
распада последнего в процессе образования нефтяных углеводо-
родов претерпели превращение тидрогенизационного характера.
Невидимому, они должны принадлежать к пока еще мало изу-
ченному ’классу полициклических нафтенов; однако более опре-
деленных данных об их химической природе пока не имеется1.
Определение оптической деятельности нефтей и ее дестилла-
тов производится обычными поляризационными аппаратами.
ЭЛЕКТРИЧЕСКИЕ СВОЙСТВА.
Сухие нефти и нефтепродукты являются непроводниками или
диэлектриками. Диэлектрическая постоянная их колеблется
*- Вопрос о возможных источниках оптической деятелыюсти нефти* бу-
дет рясгмоцкпн также ниже, в главе о происхождении пефти. ,
58
в очень узких пределах (1,8—>2), мало отличается от диэлектри-
ческой постоянной воздуха (1,0006) и в 2—3 раза меньше ди-
электрической постоянной таких диэлектриков, как сургуч,
стекло и слюда.
Сопротивление, оказываемое сухими нефтями и нефтепродук-
тами электрическому току, чрезвычайно велико, и следова-
тельно, электропроводность их ничтожна. С размерах последней
можно судить из сопоставления электропроводности парафина
(около 10-1в) или чистого петролейного эфира (около 10“18)
с электропроводностью чистой воды (около 10~е при 0°).
Означенные свойства делают понятным применение некото-
рых нефтепродуктов в качестве изоляционных масел. Таковы
трансформаторные масла, применяемые для заливки трансфор-
маторов, .масляных реостатов и выключателей. Чтобы отвечать
своему назначению, эти масла должны выдерживать специаль-
ную пробу на пробиваемость при высоком напряжении электри-
ческого поля; так например, по нормам СССР при испытании
между двумя дисками с диаметром 25 мм на расстоянии 2,5 мм
при температуре 15—20' пробивное напряжение для сухого
масла должно быть не менее 25 кУ. Так как малейшие загряз-
нения могут вызвать резкое снижение пробиваемости, то испы-
туемое масло должно быть хорошо очищено и свободно от при-
меси воды и пыли. Интересно отметить, что пробиваемость
масла не находится в непосредственной связи с сопротивле-
нием, которое оно оказывает электрическому току, что явствует
уже из следующего: в то время как сопротивление -масла току
при повышении температуры уменьшается, сопротивление того
же масла пробиваемости с повышением температуры возрастает.
Нефть и нефтепродукты легко электризуются при трении и
как непроводники могут некоторое время сохранять электриче-
ские заряды на своей поверхности. Подобного рода явления на-
блюдаются, например, при погружении в бензин сухой шерсти
или шелка, при перекачке бензина по трубопроводам и т. д.
В результате такой электризации при разряде в подходящих
условиях может появиться искра, достаточно сильная для вос-
пламенения паров бензина со всеми вытекающими отсюда по-
следствиями (взрыв, пожар и т. п.).
Описание аппаратуры и методики определения таких элек-
трических свойств нефтей и нефтепродуктов, как диэлектриче-
ская постоянная, электропроводность и пробиваемость, можно
найти в специальных руководствах.
РАСТВОРИМОСТЬ
Взаимная растворимость воды и углеводородов очень неве-
лика и определение ее требует специальной методики.
Наиболее простой способ определения растворимости таких
трудно растворимых в воде жидкостей, как углеводороды, заклю-
чается в следующем. К определенной навеске вещества прили-
вают постепенно воду до тех пор, пока все вещество не перейдет
59
в раствор. Когда .раствор станет прозрачным или чуть-чуть
опалесцирующим, определяют количество прилитой воды и де-
лают пересчет на 100 г растворителя. При тщательной работе
расхождение двух повторных определений подобного рода не
превышает 2%. В табл. 21 приводятся растворимости в воде не-
которых нефтепродуктов и углеводородов жирного и ароматиче-
ского ряда, определенные указанным способом [10].
Таблица 21
Растворимость углеводородов в воде
(по Фюн еру, Успенскому и др.)
У глеводороды
и нефтепродукты
Растворимость в воде в % по весу
Пентан ................
Гексан ................
Гептан ................
Октан............. . .
Бензол.................
Толуол ................
Ксилол............ . . .
Пропил-бснзол..........
Бензин авио, грози.....
Бензин 1 сорта ........
Бензин II „ ...........
Бензин бакинский . . . .
0,0360 (16°)
0,0140 (15°.5)
0,0050 (15°,5)
0,0014 (16°)
0,1750 (10°)
0,0368 110°)
0,0076 (10°)
0,0030 (10°)
0,0011 (10э)
0,0009 (10°)
0,0017 (10°)
0,1856 (22°)
0,0492 (22°)
0,0180 (227)
0,0060 (15°)
0,0034 (22°)
0,0012 (22’)
0,0010 (22°)
0,0017 (22°)
Как видно, растворимость ароматических углеводородов
в воде значительно больше растворимости жирных. При этом
в обоих гомологических рядах растворимость быстро умень-
шается с увеличением молекулярного веса, и аналогичную пра-
вильность нетрудно проследить на примере различных собран-
ных в той же таблице бензинов. Что касается растворимости
в воде более тяжелых нефтепродуктов, особенно смазочных ма-
сел, то при обыкновенной температуре она является неопреде-
лимо малой.
Для определения растворимости воды в углеводородах в тех
случаях, когда «растворитель является легко летучим, удобно
пользоваться так называемым хлоркальциевым методом, кото-
рый заключается в следующем. Исследуемую жидкость взбал-
тывают с небольшим количеством воды и таким образом приго-
товляют сначала насыщенный водою раствор. Когда избыток
воды отстоится, раствор осторожно сливают и пропускают через
него сухой воздух. Последний, проходя через жидкость, увле-
кает с ее парами всю воду, которую и улавливают, направляя
воздух через взвешенную трубку с прокаленным хлористым
кальцием. Если затем вытеснить из этой трубки углеводородные
пары сухим воздухом и вторично взвесить ее, то ее привес дат
количество воды, содержавшейся во взятом объеме насыщенного
водою раствора. В табл. 22 собраны результаты, полученные по
60
этому методу для простейших ароматических углеводородов и
тех нефтепродуктов, что и в предшествующей таблице.
Растворимость воды в углеводородах
(по Успенскому)
Таблица 22
Углеводороды и нефтепродукты Уд. вес Растворимость воды в % по весу
при 10° нри 22°
Бензол 0,8887 0,0510 0,0662
Толуол 0,8743 0,0426 0,0526
Ксилол 0,8703 0,0185 0,0384
Бензин авио, грозн 0,7152 0,0072 0,0113
Бензин I сорта 0.7291 0,0059 0,0080
Бензин II сорта 0,7498 0,0062 0,9083
Бензин бакинский .... 0,7551 0,0049 0,0079
Мы видим из приведенных в табл. 22 примеров, что раство-
римость воды в ароматических углеводородах заметно меньше,
чем их растворимость в воде. Для бензилов наблюдается обрат-
ное явление, т. е. здесь растворимость воды в бензинах заметно
больше растворимости бензинов в воде; таковы же, невидимому,
эти соотношения и для чистых парафинов. Что касается изме-
нения растворимости воды в одном и том же гомологическом
ряду, то в этом отношении здесь сохраняется та же правиль-
ность, что и выше: с увеличением молекулярного веса углево-
дородов растворимость воды в них резко снижается. Для иллю-
страции этого положения, кроме материала табл. 17, интересно
отметить, что керосин при 18° растворяет только 0,005% воды,
парафиновое масло — 0,003%. Само собою разумеется, что
в этих последних и им подобных случаях хлоркальциевый ме-
тод определения растворимости воды уже не применим, и при-
ходится пользоваться иной, значительно менее точной методи-
кой, а именно: к определенному объему сухой жидкости (1 л)
прибавляют пр каплям воду и "взбалтывают, повторяя добавле-
ние воды до ясно видимого избытка.
Данные табл. 21 и 22 позволяют судить также о влиянии
температуры на взаимную растворимость воды и углеводородов.
Как видно, с повышением температуры растворимость воды
в углеводородах заметно увеличивается. В том же направлении
изменяется в зависимости от температуры и растворимость угле-
водородов в воде; однако в этом последнем случае повышение
температуры сказывается на растворимости гораздо слабее.
Вопрос о взаимной растворимости воды и нефтепродуктов,
главным образом легких, представляет значительный интерес
в связи с применением воды как защитной среды при хранении
горючих Жидкостей. Столь основные вопросы этой проблемы,
61
как вопрос о потерях горючих жидкостей вследствие растворе-
ния их в воде, а также возможном изменении свойств этих жид-
костей вследствие растворения в них воды, могут быть решены,
очевидно, только в результате тщательного изучения их взаим-
ной растворимости.
Большинство органических растворителей, как-то: бензол,
хлороформ, четыреххлористый углерод, сероуглерод, эфир и др.,
растворяют нефть и нефтепродукты (кроме парафина) во всех
отношениях. Спирты к этой обширной группе растворителей не
относятся. Хорошо растворяются в спиртах лишь наиболее лег-
кие составные части нефти; по мере же увеличения их молеку-
лярного веса, иначе говоря, по мере их утяжеления, раствори-
мость их быстро падает, чем можно пользоваться длй разделе-
ния на холоду исходного сырья на отдельные погоны (холодная
фракционировка).
Сами нефти и нефтепродукты служат хорошими раствори-
телями для целого ряда веществ, как-то: иод, сера, каучук,
многие другие смолы, а также большинство растительных и жи-
вотных масел.
Кроме истинных растворов, которые имелись в виду выше, из-
вестны также различные коллоидные растворы, в которых нефть
или продукты ее переработки принимают то или иное участие.
Так например, асфальтовые вещества (асфальтены), раство-.
ряясь в нефти и ее продуктах, образуют не истинные, а кол-
лоидные растворы. Сюда же должны быть отнесены разного рода
нефтяные эмульсии, при образовании которых в зависимости от
количественных взаимоотношений между компонентами нефть
может играть роль либо, дисперсионной среды (растворителя),
либо дисперсной фазы (растворенного вещества). Особый практи-
ческий интерес представляют водно-нефтяные эмульсии, с кото-
рыми нередко приходится иметь дело как при добыче пс({уги, так
и при очистке различных ее дестиллатов.
Совершенно иной характер носит растворяющее действие
углеводородов нефти на металлы. При обыкновенной темпера-
туре с хорошо очищенными нефтепродуктами действие это
крайне незначительно; оно усиливается, однако, при нагрева-
нии, особенно в присутствии воздуха, невидимому за счет пред-
варительного образования продуктов окисления нефти Действи-
тельно, нефти и их дестиЛлаты, содержащие значительные ко-
личества нафтеновых кислот (а также сернистых соединений),
нередко проявляют, особенно при повышенной температуре,
весьма энергичное разъедающее действие по отношению к раз-
личным металлам, и борьба с такого рода разъеданием или кор-'
розией металлов и металлической аппаратуры представляет со-
бою одну из серьезнейших задач технологии добычи и перера-
ботки нефти. Очевидно, однако, что мы имеем здесь уже не про-
стое растворение, а химическое взаимодействие металла с теми
либо иными компонентами, содержащимися либо образовавши-
мися в нефти в процессе ее изменения в природных ши искус-
ственных условиях.
62
ЛИТЕРАТУРА
Русаков М. Af., Методы определения физико-химически < характеристик
нефтяных продуктов, ОНТИ (1936).
ASTM, Американские стандартные методы испытания нефтепродуктов,
Стандартгиз (1936).
Седых Н. Ф., Таблицы для перевода английских и американских мер
в метрические по нефтепродуктам, М. — Л. (1932)
Гурвич Л. Г., Научные основы переработки нефти, 2-е изд., М. —Л. (1925).
Гефер Г., Нефть и ее производные, пер. с нем., М. (1907).
Добрянский А. Ф., Анализ нефтяных продуктов, М. — Л. (1925); изд. 2 е,
М. —Л. (1932).
Engler С. und Hofer Н., Das Erdol, т. I (1918) и т. IV (1930).
Голъде Д., Жиры и масла, т. I, Исследование нефтяных продуктов, пер.
под ред. Добрянского А. Ф., Л. (1931).
Библиотека ОСТ. Методы испытания нефтепродуктов.
jRedwood В. A., Treatise on Petroleum, т. I и III. London (1922).
Блох Л. С. ‘и Добрянский А. Ф., Вязкость нефтяных продуктов, М. — Л.
(1927).
Bakusin Af., Die Polarimetric der Erdole, Berlin (1910).
Химический состав нефтей и нефтяных продуктов, „Труды ГрозНИИ', 2-е
изд, ОНТИ (1935).
1. Davis, Lapeyrouse, Dean, .Oil and Gas J.* №46 (1932).
2. Hill, Coates, .Ind. Eng. Chem." № 6, 641 (1928).
3. Пинкечич 10. А. и Митрофанова H. А, Сборник .Технология топлив
и смазочных масел', ОНТИ (1936).
4. Худякова Л. Д. и Тистович И. С., „Труды ВИМС' в (22), 69, 93 (1934).
5. Кусаков М. М., „Нефт. хоз.' 10, 77 и 11, 67 (1935).
6. Ormandy, Craven, „J. Inst. Petr. Techn." 8, 145 (1922)
7. Тихомиров В. и Жузе, «Нефт. хоз." № 1, 74 (1929).
8. Коновалов Д. П., .Ж.Р.Х.О." 50, 81 (1918).
9. Равикович и Авакова, .Нефт. хоз." №*10 (1988).
10. Успенский С. П., „Нефт. хоз." № 11—12, 713 (1929).
ГЛАВА III
МЕТОДИКА ХИМИЧЕСКОГО ИССЛЕДОВАНИЯ
НЕФТЕЙ И НЕФТЕПРОДУКТОВ
Задачи химического исследования нефти, как и всякого дру-
гого, продукта, Встречающегося в природе в виде сложной смеси,
заключается в том, чтобы изолировать из нее отдельные инди-
видуальные вещества, по возможности их исследовать и наме-
тить их практическую ценность и пути их рационального ис-
пользования. В качестве исходного продукта при ©том часто
берут не сырую нефть, а разного рода технические продукты:
бензин, керосин и т. п. Общая методика исследования нефте-
продуктов прямой гонки, в применении к кавказской нефти и ее
продуктам разработанная главным образом Марковниковым и
его сотрудникам^, сводится к следующему [ 1 ].
ОБЩИЕ МЕТОДЫ
Исследуемый нефтепродукт прежде всего фракционирует 2—
3 раза с хорошим дефлегматором, собирая отдельные погоны
в пределах 10—15°.
Как известно, сырые погоны нефти еще па заводе подвер-
гаются обработке купоросным маслом для удаления смолистых
веществ И различных непредельных примесей, легко осмоляю-
щихся, а потому неблагоприятно влияющих на качество техниче-
ского продукта. Однако такой предварительной обработки да-
леко не достаточно для полного удаления непредельных соеди-
нений. Поэтому выделенные фракциопировкой нефтяные погоны
вновь должны быть обработаны крепкой серной кислотой. Для
полного отделения непредельных соединений обыкновенно до-
статочно взболтать углеводород с 10% по весу серной кислоты
удельного веса 1,84 в течение 15 минут. Кислотный слой, со-
держащий Продукты взаимодействия находящихся в нефти не-
предельных соединений с серной кислотой, до настоящего вре-
мени почти не исследован.
Дальнейшая задача заключается в отделении ароматических
углеводородов. В тех случаях, когда предполагается исследова-
ние ароматических углеводородов нефти, для изолирования их
нефтяные погоны обрабатывают дымящей серной кислотой и
64
изучают получающиеся ароматические сульфокислоты (см.
ниже); в противном случае для освобождения от ароматических
углеводородов можно ограничиться обработкой погонов поло-
винным объемом серно-азотной смеси сначала на холоду, затем
при комнатной температуре до тех пор, пока не прекратится
разогревание. Большая часть получающихся при последней
обработке ароматических нитросоединений растворяется в кис-
лотном слое, но часть их остается и в углеводороде. После про-
мывания щелочью углеводород отгоняют, а затем фракциони-
руют с хорошим дефлегматором, повторяя фракционировку не-
сколько раз и постепенно сокращая пределы температуры, при-
мерно до двух градусов. Более или менее постоянная темпера-
тура кипения отдельных фракций достигается обыкновенно
лишь после 8—9 тщательных фракционировок.
Фиг. 19. Графики разгонки бакинского бензина (по Марков ми-
ко ву).
Если для каждой полученной фракции определить теперь
количество и удельный вес, то результаты’ этой стадии работы
можно наглядно выразить графически. На оси абсцисс намеча-
ются температуры кипения отдельных фракций, а на оси орди-
нат откладываются соответствующие удельные веса; наконец,
отдельными вертикалями могут быть отмечены количества со-
ответствующих фракций. Примером могут служить приведенные
выше графики подобных перегонок бакинского бензина, произве-
денных Марковниковым (фиг. 19): одна из кривых (1) дает до-
вольно полную картину разгонки 32 кг бакинского бензина,
сравнительно богатого парафиновыми углеводородами; другая (2)
представляет собою частично разгонку’другого образца бакин-
ского бензина, более бедного парафинами. В обоих случаях раз.-
гонка велась сначала с небольшой колонкой типа Савалля дли-
ною 8/л м, затем — с дефлегматором Ле-Беля,. которого,
достигавшая 2 м для погонов с температурой кипения до 78°,
укорачивалась с повышением температуры кипения.
5 Зак. 1622. Химия нефти.
65
Как видно йЗ чертежа, Кривая изменений удельного Шй.
с температурой кипения имеет на протяжении от <30 до 13(г
три изгиба с (высшими точками около 50, 81 и 102°; это соот-
ветствует температурам кипения циклопентана, циклогексана к
метил-циклогексана. Количество этих фракций заметно превы-
шает количество соседних погонов, и указанные нафтены дей-
ствительно в нефти обнаружены. Низшие точки тех же изгибов,
вероятно, также должны соответствовать определенным инди-
видуальным составным частям. Первая из них (около СО0)
весьма близко подходит к константам одного из изомерных гек-
санов, диизо-пропила, также обнаруженного в бакинском бен-
зине; два других перегиба (около 91 и 113°) ближе пока не ис-
следованы. Для более высоких температур (кривая изменения
удельного веса с температурой кипения уже теряет свою на-
глядность. Так например, несомненное присутствие в нефти
октонафтена (температура кипения 118—120°) резко сказы-
вается на количестве соответствующей фракции, но совершенно
незаметно проходит для кривой изменения удельного веса с тем-
пературой кипения.
Чтобы в каждую стадию работы можно было охватить дости-
гнутые при фракционировке результаты и намечающиеся пер-'
спективы, удобно пользоваться графическим способом Юнга [2],
По предложению этого автора при вычерчивании кривых раз-'
гонки на оси ординат наносятся температуры, а на оси абс-
Ди/ v
цисс—величины отношения — , д,р — вес фракции, а д>—<
соответствующий ей температурный интервал. При таком по-
строении небольшим промежуточным фракциям будут соответ-
ствовать, очевидно, крутые подъемы кривой разгонки, тогда как
для Индивидуальных веществ, находящихся в данных пределах,
температуры,—горизонтальные или' близкие к горизонталям
части кривых.
Для иллюстрации приводим графики из произведенного Юн-
гом исследования фракции 27—41° пенсильванского бензина
для перегонок 1, 4, 7, 10 и 13. Как видно, при разгонке 1$
вполне отчетливо намечаются две составных части, на когорт
кривые разгонок 1 и 4 не давапи никаких указаний, а именно’
фракция с температурой кипения около 28° (изопентан) и фрак-
ция с температурой кипения около 36° (нормальный пентан).
Оба эти парафина действительно находятся в пенсильванской
нефти, но присутствие их, как показывают графики фиг. 2Q,
выявляются лишь постепенно, не ранее разгонки 7.
Фракционировка является той основной операцией, от ко-
торой зависит успех всей последующей работы по выделения
отдельных компонентов нефти из ее погонов. Отсюда понятно,
что для достижения возможной эффективности этой край®
важной, но длительной операции необходимо обеспечить
прежде всего надлежащей аппаратурой. Лабораторная технику
давно уже пользуется для подобных целей дефлегм я торя.ми $
колонками разного рода систем й конструкций, сравнительна^
ев
оценка которых неоднократно составляла предмет специальных
исследований и сочинений. В крупном масштабе такая аппа-
ратура широко применяется также в технике. Таковы, напри-
мер, ректификационные колонны на винокуренных и спирто-
очистительных заводах, дестилляционные аппараты для раз-
гонки легкого каменноугольного масла и т. д. В последнее
40°
33е
30°
25°
Фиг. 20. Графики разгонки фракции 27—41° бензина (по Юму).
время подобная аппаратура получила широкое распространение
также в нефтеперегонном деле, достигнув здесь невиданных ра-
нее масштабов и совершенства. Вое подробности по этому во-
просу отнесены к II 'я. (гл. II).
Кроме аппаратуры, большое значение имеет также Самая
методика фракционировки. В настоящее время для выделения
из нефти и ее погонов отдельных компонентов широко приме-
няются указанные ниже виды фракционировки [3, 4].
Фракционировка при постоянном давлении. Это — обычный
вид фракционировки, с которой начинают разделение нефти на
отдельные погоны, пользуясь, как было показано выше, сначала
грубой аппаратурой, а затем аффективными ректификацион-
ными колоннами современного типа. В конечном итоге при этом
получают либо тот или иной компонент нефти в чистом виде,
либо смесь двух и более компонентов, имеющих практически
одну и ту же температуру кипения, либо, наконец, нераздельно
кипящую, азеотропическую смесь двух компонентов. В двух
последних случаях дальнейшее разделение требует уже видоиз-
менения методики фракционировки путем изменения давления,
или введения нового компонента (см. ниже). Постоянные давле-
ния, при которых производится фракционировка, весьма разно-
образны. Если, например, бензиновые- погоны могут фракциони-
роваться при нормальном давлении, то при фракционировке мас-
ляных погонов во избежание разложения применяют высокий
вакуум (до 0,0001 мл ртутного столба и выше).
Фракционировка при переменной давлении. В тех случаях,
когда фракционировкой при определенном, постоянном. давле-
5*
67
нии не удается достичь полноты разделения нефтяных компо-
нентов, нередко бывает выгодно изменить давление, при котором
ведется перегонка, по следующим соображениям. Два компо-
нента, кипящие в условиях нормального давления практически
при одной и той же температуре и потому неразделимые, могут
иметь различные коэфициенты изменения упругости пара
с температурой; вследствие этого при пониженном давлении
здесь может оказаться заметное различие в относительных пар-
циальных давлениях паров отдельных компонентов и, следова-
тельно, дальнейшее разделение смеси может оказаться 'Возмож-
ным. Для некоторых нераздельно кипящих и особенно трудно
разделимых смесей углеводородов различных классов, например
смесей парафина с нафтеном, фракционировкой при изменен-
ном давлении действительно удалось достичь глубокого 'разде-
ления.
Фракциопировка с добавкой нового специального вещества.
Прибавление к нераздельно кипящей смеси углеводородов но-
вого вещества может дать при дальнейшей фракционировке осо-
бенно ценные результаты. Обычными добавками служат мети-
ловый или этиловый спирт, уксусная или пропионовая кислота-
и тому подобные вещества, образующие с одним из компонентов
исходной смеси азеотропическую смесь и легко удаляемые по
миновании надобности промывкой водой или щелочью. Образо-
вание новой азеотропической смеси резко нарушает установив-
шиеся в исходной смеси углеводородов соотношения между
парциальными упругостями их паров и делает возможным их
дальнейшее разделение путем фракционировки. Эта методика
дает в некоторых случаях исключительно четкие результаты.
Так например, добавка метилового или этилового спирта позво-
ляет разделить путем фракционировки постоянно кипящую
смесь норм, гексана с бензолом; прибавление уксусной кислоты
дало прекрасные результаты при разделении некоторых постоян-
но кипящих смесей нафтенов с парафинами, особенно же оказа-
лось эффективным при отделении некоторых ароматических
углеводородов от близко кипящих парафинов и нафтенов и т. д.
Фракционировка в различных се видоизменениях является
важнейшим общим методом выделения отдельных компонентов
из нефти и ее погонов. При оценке этого метода нельзя не от-
метить его чрезвычайной трудоемкости. Так например, некото-
рые исследователи американских нефтей (Ch. Iddbery, Sydney
Young, E. Fortcy и др.) производили в процессе своих работ до
30—40 и более перегонок отдельных нефтяных фракций и тем
не менее в большинстве случаев все-таки не могли изолировать
отдельных их компонентов в совершенно чистом виде. Хотя
аппаратура для точной фракционировки за последнее время
в высокой степени усовершенствована, тем не менее и теперь
при исследовании нефтей вслед за фракционировкой неизбежна
пользуются различными другими методами выделения углево-
дородов из их смесей. Широкое развитие и применение полу-
чили при этом некоторые методы физико-химического характера,
£8
которые вместе с фракциопировкой могут быть отнесены
к общим методам исследования нефтей. Таковыми являются
методы, описанные ниже.
Кристаллизация. Различают кристаллизацию простым вымо-
раживанием и кристаллизацию с добавлением подходящего рас-
творителя. Оба эти метода нашли применение при выделении
различных компонентов нефти.
В тех случаях, когда та или иная узкая фракция нефти со-
держит значительное количество отдельного компонента, кри-
сталлизующегося при охлаждении, можно ограничиться его
повторным вымораживанием. Для этого разделяемую смесь
охлаждают до достаточно низкой температуры и замораживают;
затем осторожно поднимают температуру смеси, перемешивают
образующуюся кашицу и центрифугируют жидкие примеси от
более или менее чистых кристаллов основного компонента. По-
вторяя эти операции, в отдельных случаях можно выделять из
нефтяных погонов углеводороды исключительной чистоты. При-
мером может служить получение из нефти чистого циклогек-
сана, после того как бензол из соответствующей фракции был
удален тем или иным путем (ср. ниже).
В тех случаях, когда данная смесь не содержит того или
иного углеводорода в преобладающем количестве и когда простое
вымораживание не дает ожидаемых результатов, смесь предвари-
тельно разбавляют подходящим растворителем и только после
этого ведут охлаждение и кристаллизацию. Растворитель дол-
жен быть подобран, конечно, таким образом, чтобы его после-
дующее удаление не вызывало трудностей и достигалось, напри-
мер, простым испарением без серьезных потерь разделяемых
углеводородов. В различных случаях в качестве растворителя
нашли применение метан, этан, пропан, этилен, дихлордифтор-
метан, простой диметиловый эфир и некоторые другие.
Адсорбция. Адсорбционные свойства некоторых веществ так-
же удобно использовать для разделения некоторых углеводород-
ных смесей. В качестве адсорбента наиболее подходящим ока-
зался силикагель, с помощью которого удается достичь, напри-
мер, полного отделения ароматических углеводородов от пара-
финов и нафтенов. Тот же адсорбент дает вполне заметный эф-
фект разделения некоторых смесей парафинов с нафтенами.
Обработка селективными растворителями. Метод селектив-
ного или избирательного растворения во многих случаях весьма
удобен для дальнейшего разделения компонентов нефтяной
фракции. В качестве растворителя нашли применение жидаий
сернистый ангидрид, уксусная кислота, ацетон, анилин и не-
которые другие. Их применение основано на том, что следствием
прибавления подходящего растворителя является распределение
компонентов данной нефтяной фракции между растворителем и
углеводородной смесью в отношениях, более или менее суще-
ственно отличающихся от тех, в которых разделяемые компоненты
образовали исходную смесь. Тем самым дальнейшее разделение
смеси становится практически возможным.
69
Таковы в самых сжатых чертах общие методы выделения из
нефти и ее погонов отдельных компонентов в чистом виде. Ха-
рактерной особенностью и достоинством этих методов является
возможность их применения без предварительной обработки
нефтяного погона какими-либо сильно действующими реагентами
(в том числе и крепкой серной кислотой), которые порою могут
весьма существенно изменить состав исследуемой смеси углево-
дородов. Их эффективность, вообще говоря, оставляет далеко за
собою старые методы выделения из нефти отдельных компонен-
тов; успехи же, достигнутые на этих путях, особенно за послед-
ние 10 лет, громадны как в отношении усовершенствования
самых методов и соответствующей аппаратуры, так и в отноше-
нии выявления ближайшего состава некоторых, в особенности
американских, нефтей.
СПЕЦИАЛЬНЫЕ МЕТОДЫ
Как видно из вышеизложенного, общие методы выделения из
нефти и ее погонов отдельных компонентов, как общее правило,
весьма сложны и длительны. Ввиду этого для выявления бли-
жайшего состава отдельных нефтяных фракций давно уже
нашли применение некоторые химические реакции, позволяю-
щие в отдельных случаях после сравнительно грубой фракцио-
нировки быстро решить вопрос о химической природе входящих
в данную фракцию углеводородов. Этим путем и шло вначале
исследование ближайшего состава различных нефтей и их пого-
нов. Так например, образование в надлежащих условиях из
нефтяной фракции хорошо кристаллизующихся ароматических
полинитросоединений может, очевидно, служить достаточным
доказательством присутствия соответствующей ароматики
в исходном погоне и т. д. Развивая эту чисто химическую мето-
дику, некоторые авторы шли дальше, а именно: с помощью тех
или иных реакций приготовляли из нефтяного углеводорода
ряд его производных и по возможности очищали их фракциони-
ровкой или иными методами; состав и свойства этих производ-
ных иногда позволяли полностью решить вопрос о химической
природе этих производных, а отсюда и о химической природе
исходного нефтяного углеводорода, особенно в тех случаях, когда
удавалось совершить обратный переход от этих производных
к исходному углеводороду и сравнить его с соответствующим
синтетическим продуктом. Именно этими путями старались
итги первые исследователи ближайшего состава нефтей, приме-
няя при этом специальные методы органического синтеза, слу-
жившие как для получения различных производных нефтяных
углеводородов, так и для синтеза последних. Исходными процес-
сами в этих более или менее сложных циклах превращений были
реакции, описанные ниже.
Хлорирование и разного рода превращения хлоридов (5).
Сухой хлор действует на нефтяные погоны тем легче, чем проще
их состав. Чтобы избежать по возможности образования поли-
то
хлоридов, следует вести реакцию в парах, пропуская хлор в пары
кипящего углеводорода. Для ускорения процесса, протекающего
весьма медленно в случае высших погонов (деканафтен), можно
пользоваться прямым солнечным светом; напротив, охлорение
низших погонов во избежание вспышки и обугливания следует
вести лишь на рассеянном свету.
Влажный хлор действует на нефтяные погоны более энергич-
но, чем сухой. Реакция идет здесь уже на холоду, лишь для на-
чала требуется небольшое подогревание (до 30—10°). И здесь
необходимо принять меры, чтобы предупредить образование по-
лихлоридов; для ©того хлор берется лишь в теоретическом коли-
честве из расчета на получение монохлорида.
В Обоих этих случаях реакция протекает по одному и тому
же уравнению; например, для какого-либо парафинового угле-
водорода, СлНеяч-2, она может быть выражена следующим образом:
+с12=спн^+1 ci-4-Hci.
Так как нафтены относятся к хлору совершенно так же, как
парафины, то понятно, что хлориды, полученные из нефтяного
погона одним из указанных методов, представляют собою не
индивидуальные вещества и всегда содержат примесь других
изомерных или близких по составу хлоридов. Тщательно фрак-
ционируя их, можно иногда значительно понизить процентное
содержание этих примесей по сравнению с тем, что было до
охлорения. Так1 йаятример, нефтяная'фракция 80—82р, состоя-
щая главным образом из циклогексана (температура кипения
81°) может содержать некоторую примесь нормального гексана
(температура кипения 69°) и метилциклопентана (температур
кипения 72°). В получающемся при ее охлорении хлор-цикло-
гексане (температура кипения 142°) может находиться поэтому
примесь нормального хлор-гексана (температура кипения
125—128°) и хлор-метил-циклопентана (температура кипения
125—127°). Отделение фракционировкой первого хлорида от
двух последних, очевидно, вполне осуществимо, тогда как отде-
лить тем же способом хлор-гексан от хлор-метил-циклоптентана
вследствие тождества их температур кипения, конечно, невоз-
можно. Подобные затруднения особенно возрастают для высших
нафтенов; здесь близость температур кипения как самых изо-
мерных или близких по составу углеводородов, так и их произ-
водных часто почти совершенно исключает возможность их раз-
деления путем фракционировки.
От хлоридов, настолько возможно очищенных, естественный
переход, с одной стороны, к непредельным углеводородам, с дру-
гой— к алкоголям.
Переход от нефтяных хлоридов к непредельным углеводоро-
дам легко осуществляется путем нагревания хлорида с раство-
ром едкого |кали или с органическим основанием <хинолин и
др.). Особенно часто применялась эта реакция для получения из
хлоридов нафт'енового ряда, CnHan-iCl, соответствующих не-
71
Предельных углеводородов, нафтиленов, состава СЛа» - 2, по
уравнению:
С1 + КОН = СМН2П_2 + КС1 + Н20.
Подвергнув затем тщательно расфракционированный наф-
тилен гидрогенизации, можно перейти от него обратно к. нафтену
и, сопоставив свойства последнего со свойствами исходного неф-
тяного погона, получить представление о степени индивидуаль-
ности ©того погона. В настоящее время наиболее простой
и надежный способ гидрогенизации непредельных углеводоро-
дов— каталитический метод Сабатъе и Сандерана (1905 г.).
Вместо него при гидрогенизации нафтиленов в 80—90-х годах
приходилось пользоваться иодисто-водородной кислотой при вы-
сокой температуре (до 256 ° и выше). Ввиду доказанного позднее
изомеризующего действия этого реагента (см. ниже) ценность
полученного этим путем материала, очевидно, значительно по-
нижается.
От нафтиленов можно осуществить еще один интересный пе-
реход, а именно: присоединением галоида (брома) получить
двугалоидозамещенное и отщеплением от последнего элементов
галоидоводородной кислоты перейти к следующему непредельному
ряду, к терпенам:
ОД,-, C„H2n_2Br2 СА-г
Такой переход открывал, казалось, возможность выяснения
связи между нефтью как источником нафтенов, с одной стороны,
и эфирными маслами растительного царства как источником
разнообразнейших терпенов —. с другой. В настоящее время
химическая сущность этой связи совершенно очевидна: большая
группа так называемых циклических терпенов (лимонен, тер-
пинен, сильвестрен и др.), как показало ближайшее исследова-
ние их химической структуры, содержит шестичленное яд|ю и,
таким образом, принадлежит к производным ряда циклогексана,
представители которого встречаются также и в нефти. Но в то
время, когда строение основного нафтенцого ядра представля-
лось еще загадочным (первая половина 80-х годов), казалось
крайне важным и интересным, что продукты гидрогенизации
терпенов состава Сп Н2п, т. е. тетрагидротерпены, получав-
шиеся из терпенов различными способами, по своим свойствам
.чрезвычайно напоминают нафтены. Понятны поэтому неодно-
кратные попытки осуществить обратный переход — от нафтенов
через нафтилены к терпенам. Однако в громадном большинстве
случаев попытки эти кончались неудачей: полное отщепление
галоида от дибромидов, получавшихся присоединением брома
к нафтиленам, удавалось осуществить лишь при повышенной
температуре, когда главными продуктами реакции оказываются
уже не терпены, а их полимеры. Лишь позднее, когда химиче-
ская природа нафтенов оказалась в значительной степени выяс-
ненной, переход от них к ряду терпенов удалось осуществить и
прежде всего на примере циклогексана, причем таким образом
72
были получены низшие простейшие терпены, СвН8, в природе не
встречающиеся.
Переход от нефтяных хлоридов к соответствующим алкого-
лям также встретил вначале затруднения. Сущность их сводится
к тому что почти независимо от условий реакция направляется
здесь не в сторону обмена, а главным образом в сторону отще-
пления галоидоводородной кислоты и образования нафтиленов.
Предварительное превращение хлоридов нагреванием с иодисто-
водородной кислотой в иодюры мало помогает делу. И с иодю-
рами, как при последующей обработке их влажной окисью се-
ребра, т. е. при непосредственном переходе к алкоголю, так и
при нагревании их с уксуснокислыми солями (Na, К, Ag), т. е.
при переходе к алкоголю через уксусный эфир, реакция идет
главным образом в сторону образования нафтиленов. В послед-
нем случае выходы на кислородные соединения значительно
лучше, однако процесс осложняется здесь новым весьма сущест-
венным обстоятельством: освобождающаяся уксусная кислота при
высокой температуре может присоединиться к нафтиленам с об-
разованием уксусного эфира, соответствующего уже не исход-
ному галоидозамещенному, а тому или иному его изомеру. В ре-
зультате такого рода осложнений получающиеся уксусные эфиры
кипят обыкновенно в пределах примерно 10°, так что выделение
продукта с более постоянной температурой кипения требует зна-
чительного количества этого трудно доступного вещества.
Итак, многочисленные попытки выработать метод перехода от
нефтяных хлоридов к алкоголям долго приводили к мало удо-
влетворительным результатам. Реакция Гриньяра позволила раз-
решить этот вопрос в новом направлении. Как показал Зелин-
ский [б], нефтяные хлориды в присутствии катализаторов (следы
J, А1С Js и т. д.) легко реагируют с магнием в присутствии эфира
с образованием гриньяровских комплексов. Последующим про-
пусканием кислорода и разложением продукта реакции подкис-
ленной водой автором был получен ряд нафтеновых алкоголей.
К сожалению, до настоящего времени работа эта опубликована
в виде лишь небольших протокольных заметок, не позволяющих
судить о значении метода для исследования нефтяных углеводо-
родов. Вся совокупность указанных превращений хлорида, на-
пример состава Cl, в соответствующий алкоголь может
быть выражена следующими уравнениями:
Сд,Н2П_1
СЛН2П_ j
_ &П-1
MgCl + 0==C„Hf
Cl + Mg^C^.^lgCl;
1V1&V1 ~Г V = V'nAlQn-l uMgCl;
OMgCl + H20 = С,Х-1 OH + MgCl (OH).
Нитрование по M. Коновалову [7]. Азотная кислота, как
показал М. Коновалов, может действовать нитрующим образом
одинаково как на нафтены, так и на парафины. Уравнение этой
важной реакции вполне аналогично уравнению реакции нитро-
вания ароматического углеводорода, например, для нитрования
какого-нибудь нафтена, СЛН2Л:
с„н2П+HNOS=ед»., NO,+Н30.
73
Опыт показывает, что после нитрования свойства нефтяного
углеводорода, не вошедшего в реакцию, почти не изменяются.
Таким образом сама по себе азотная кислота подобно хлору не
может служить для отделения нафтенов от парафинов. Однако
в противоположность галоидозамещенным нитросоединения, как
известно, существенно разнятся между собой в свойствах в за-
висимости от положения нитрогруппы, и в некоторых случаях
этим различием можно воспользоваться для разделения изомер-
ных углеводородов или их производных.
Существенный недостаток нитрования по М. Коновалову, как
метода исследования углеводородов предельного характера, за-
ключается в том, что реакцию эту приходится вести в запаян-
ных трубках, причем в каждую трубку берется обыкновенно не
более 5—10 см3 углеводорода и 25—30 см3 сильно разбавленной
азотной кислоты (удельного веса 1,075). Нагревание должно про-
должаться до 24 час. с перерывом для выпуска газов, которые
образуются в результате окислительных процессов, протекающих
одновременно с нитрованием. В этих условиях главными про-
дуктами реакции в зависимости от строения исходного углеводо-
рода являются третичное и вторичное нитросоединения предель-
ного характера; при этом, однако, значительная часть исходного
углеводорода все-таки не вступает в реакцию и получается об-
ратно. Отсюда ясно, что для получения более или менее значи-
тельных количеств нитросоединения приходится запаивать и на-
гревать десятки трубок. Температура нагревания в каждом от-
дельном случае определяется предварительным опытом: для раз-
ных углеводородов она колеблется между 115 и 140°; но в каж-
дом отдельном случае колебания температуры во избежание раз-
рыва трубок, не должны превышать примерно 5°.
Для упрощения методики нитрования углеводородов предельного ха-
рактера неоднократно ставились опыты в открытых сосудах. В этих усло-
виях слабая азотная кислота, которой ведется нитрование в напаянных
трубках (обыкновенно удельного веса 1,075, т. е. с. 12,5% HNOs), ужо не
действует. Зависит это, очевидно, от того, что температура реакционной
смеси в данном случае не может быть повышена в необходимой степени.
Чтобы компенсировать понижение температуры, приходится увеличивать
крепость азотной кислоты, что, однако, имеет свои неудобства. Главное
на них заключается в том, что с увеличением крепости азотной кислоты
возрастает разнообразие азот-содержащих продуктов реакции, а именно,
кроме ’ вторичных и третичных нитросоединений, появляются еще первич-
ные; полное же разделение всех трех видов нитросоединений предельного
характера пока не осуществимо. Таким образом, несмотря на крупные прак-
тические недостатки основного метода М. Коновалова—нитрования слабой
азотной кислотой в запаянных трубках, все же предпочтительное пользо-
ваться именно этим методом.
По окончании нитрования углеводород, не вошедший в реак-
цию, отгоняется, а смесь нитросоединений обрабатывается едкой
щелочью. Вторичные (и первичные) нитросоединения переходят
при этом в раствор в виде щелочных солей, третичное же —
остается без изменения и может быть нацело отделено от щелоч-
ного раствора; обработкой последнего углекислотой можно затем
выделить из него свободные нитросоединения обратно.
74
Первичные, вторичные и третичные нитросоединения подобно
спиртам различаются составом того радикала, в который входит
характерная в данном случае нитро-группа:
Нитросоединения
R—CH2N02 R^CHNOa R8^CN02
первичные вторичные третичные
Растворение первичных и вторичных нитросоединений в ще-
лочи совершается при небольшом подогревании очень легко и
сопровождается их превращением в таутомерные aci-формы,
обладающие кислотным характеров и называемые иногда нитро-
новыми кислотами. Сущность этого превращения заключается
в том, что один из водородов, стоящих при углероде, непосред-
ственно связанном с нитрогруппой, переходит (мигрирует) к этой
последней, образуя с ней радикал > NOOH; водород же группы
> NOOH подобно водороду карбоксила — СООН способен заме-
щаться на металлы с образованием солей. Так например:
R _ CHaNOs» —> R — CH = NOOH —► R — CH=NOOK.
В свободном состоянии aci-формы нитросоединений мало
устойчивы и превращаются обратно в обычные их модификации.
Так как в третичных нитросоединениях при углероде, связанном
с нитрогруппой, нет ни одного водородного атома, то растворение
в щелочи с образованием соли здесь, очевидно, невозможно.
Обыкновенно нефтяные нитросоединения, полученные описан-
ным выше путем, после разделения на вторичные и третичные
кипят в пределах нескольких градусов. Выделение более чистого
продукта достигается фракционировкой в вакууме и Не пред-
ставляет большого труда, если исходить из нефтяного углеводо-
рода, хорошо выфракционированного; в отдельных же случаях
некоторые нитросоединения получаются сразу почти в чистом
виде. Так например, при нитровании слабой кислотой нефтяного
метил-циклоиентана (темп, кипения 71—73°), содержащего зна-
чительную примесь нормального гексана (темп, кипения 69°), из
нафтена получается главным образом третичное нитро, из гек-
сана же, не содержащего третичного водорода, — вторичный ни-
трогексан. Обрабатывая смесь этих нитросоединений едкой ще-
лочью, можно сразу получить почти чистый третичный нитро-
метил-циклопентан.
От третичных нитросоединений можно легко перейти к ами-
нам, от вторичных (и первичных) в зависимости от условий вос-
становления, с одной стороны—<к аминам, с другой —к кетонам
(альдегидам). Амины и кетоны (альдегиды) в свою очередь мо-
гут быть легко превращены в алкоголи и далее — в галоидозаме-
щенные. Действием на галоидозамещенные водорода в момент
выделения можно, наконец, вернуться к исходному углеводороду.
Подобного рода замкнутый цикл превращений. полностью был
совершен Марковниковым с двумя сравнительно простийи по
составу нефтяными фракциями: 71—73° и 80—82°. Эти работы,
как будет показано ниже, дали богатый материал для характе-
76
ристики исследованных нефтяных погонов и вместе с тем исчер-
пывающее доказательство присутствия в первой из них метил-
циклопентана, а во второй — циклогексана.
Кетонизацня. Как известно, при действии на ароматические углеводо-
роды хлорангидридов кислот в присутствии безводных галоидных солей
алюминия. Aids или А1Вгз, образуются сложные комплексные соединения,
после разложения которых водой получаются соответствующие кетоны (реак-
ция Фриделя и Крафтса). Так например, из бензола СоНв и хлористого
ацетила CHs — СОС1 легко получается этим путем ацетофенон,
СвНв. 00 . СНз, я т. д. Зелинский показал [8], что с нафтенами эта реакция
идет в том же направлении, как с бензолом и его гомологами, т. е. приво-
дит к образованию кетонов с карбонильной группой в боковой цепи. Так
например, циклогексан с хлористым ацетилом в присутствии хлористого
или бромистого алюминия вступает 'в реакцию, которую, не касаясь ее слож-
ного и недостаточно еще разъясненного механизма, .можно выразить следую-
щим уравнением:
C6IIi2 + CHSCOC1 = CcIIu • СО СНа + ИС1.
Сопоставляя кетоны, полученные таким образом из разных нефтяных
фракций, с аналогичными продуктами из синтетических нафтенов, можно
получить более или менее определенное представление об извлеченном из
нефти цикле.
Оценивая работу, произведенную в далном направлении до настоящего
времени, необходимо прежде (всего иметь в виду значительные трудности,
с которыми сопряжена разработка этого вопроса, Известно, насколько
сильными реагентами являются безводные галоидные соли алюминия.
В ряду бензола реакция Фриделя и Крафтса сощювождается часто отще-
плением и перемещением боковых групп и тому подобными перегруппиров-
ками. Есть основания думать, что то отношению к нафтенам эта реакция
осложняется подобными же побочными процессами, к которым здесь может
присоединиться еще изомеризация основного циклического ядра. Кроме
того, как показали новейшие исследования [»], в реакцию Фриделя и
Крафтса могут быть введены также парафины. Хотя с парафинами эта
реакция идет гораздо труднее, чем с нафтенами и ароматическими углево-
дородами, тем не менее, эти новейшие данные делают реакцию Фриделя
и Крафтса, очевидно, уже явно неприменимой для той цели, которая ранее
имелась в виду в данном случае, т. е. для извлечения из нефтяных пого-
нов их циклических компонентов.
Каталитическая дегидрогенизация. Как показали Сабатье
и Сандеран [10 , ароматические углеводороды и разнообразные
их производные при пропускании в парах вместе с водородом
над специально приготовленным, мелко раздробленным метал-
лическим катализатором (лучше всего Ni) присоединяют б ато-
мов водорода, превращаясь в соответствующие алициклические
соединения. Эта гидрогенизация для ароматических углеводоро-
дов легко идет уже при 180—200°. При более высокой темпера-
туре протекает обратный процесс — дегидрогенизация, т. е. рас-
пад циклогексана и его гомологов на ароматические углеводо-
роды и водород; как показали позднейшие исследования, в под-
ходящих условиях, при температуре не выше 300°, эта реакция
может давать почти количественные выходы. Таким образом
совокупность обоих превращений, гидрогенизации и дегидроге-
низации, может быть выражена, например, для бензола следую-
щим уравнением:
CeHe + 6H^CeHI2.
76
Изучая явления гидрогенизации, и дегидрогенизации в раз-
личных направлениях, Зелинский показал [ц], что углеводо-
роды рядов циклопентана и циклогептана, а также парафины
(гексан) совершенно не подвергаются дегидрогенизационному ка-
тализу и остаются без изменения в тех условиях, при которых
циклогексан и его гомологи полностью превращаются в аромати-
ческие углеводороды. Это различие он предложил использовать
для исследования нефтяных углеводородов. Если ту или иную-
фракцию нефти пропустить через трубочку с палладиевой
чернью, нагретую до 300°, или с другим подходящим катализато-
ром (Ni и другие), то все углеводороды ряда циклогексана пре-
вращаются в ароматические; по количеству выделившегося при
этом водорода можно сделать заключение о содержании во взя-
той пробе углеводородов ряда циклогексана. Обрабатывая затем
полученный погон дымящей серной кислотой или оерно-азотной
смесью, можно по ароматическим продуктам реакции составить
представление об углеводородах ряда циклогексана, входящих
в состав исследуемой фракции. Очевидно также, что после уда-
ления всех углеводородов ряда циклогексана указанным путем
состав нефтяного погона сильно упрощается, а потому изолиро-
вание из него отдельных представителей тех родов, которые не
изменяются под влиянием дегидрогенизационного катализа, ста-
новится легче. Судя по результатам, полученным до настоящего
времени, систематическое приложение этого метода к исследова-
нию нефти обещает дать в высшей степени интересные резуль-
таты, тем более, что пока это—единственный способ разделения
столь близких по своим свойствам рядов, как ряд циклогексана,
с одной стороны, и ближайшие к нему нафтеновые ряды цикло-
иентана и циклогептана — с другой.
Действие серной кислоты. Крепкая серная кислота всегда
широко применялась и применяется к нефтяным углеводородам
не только в лаборатории для аналитических и иных целей, но
также в техническом масштабе для очистки дестиллатов. Оста-
вляя вопросы, связанные с очисткой, до одной из последующих
глав, рассмотрим здесь вкратце лишь вопрос о действии крепкой
серной кислоты на нефтяные углеводороды предельного ха-
рактера.
•При 0° не только серная кислота удельного веса 1,84, но даже
дымящая серная заметным образом не действуют на углеводо-
роды предельного характера. При комнатной температуре кис-
лота удельного веса 1,84 продолжает оставаться по отношению
к тем" же углеводородам инертной, тогда как с дымящей серной
наблюдается более или менее заметное взаимодействие. Если,
например, оставить какой-либо нафтен из ряда циклогексана,
хотя бы метил-циклогексан, с дымящей серной кислотой при
комнатной температуре, то углеводород постепенно переходит
в раствор, причем наблюдаются обильное выделение сернистого
газа и заметное самбразогревание. Так как в продукте реакции
обнаруживается ароматическая сульфокислота, то, очевидно,
нафтен претерпевает предварительно дегидрогенизацию за счет
77
окисляющего действия серной кислоты; при этом образуется со-
ответствующий гомолог бензола, который и сульфируется избыт-
ком серной кислоты:
С6Ни . CHS+ 3SO Д = С6Нб • сн3+ 3S02+вН2О;
При подогревании эта реакция может итти также и с менее
концентрированной серной кислотой, особенно если удалять из
сферы реакции образующуюся воду [12].
Что касается нафтенов ряда циклопентана, а также парафи-
нов, то хотя при соприкосновении с ними дымящей серной кис-
лоты углеводород частично и растворяется в кислоте,. которая
более или менее окрашивается, однако выделения сернистого
газа в данном случае не наблюдается. Имеется даже указание,
что при кипячении с дымящей серной кислотой парафины мо-
гут сульфироваться с образованием подобно ароматическим уг-
леводородам соответствующих сульфокислот [13]; однако при
исследовании нефти реакция эта пока не нашла применения.
Некоторые реакции углеводородов ряда циклогексана. В за-
ключение обзора специальных методов исследования углеводо-
родов нефти необходимо вкратце рассмотреть некоторые реакции,
с помощью которых могут быть обнаружены углеводороды ряда
циклогексана. Реакции эти, основанные на способности угле-
водородов ряда циклогексана давать в определенных условиях
производные ароматического ряда, сыграли важную роль в исто-
рии развития взглядов на углеводороды кавказской нефти. В не-
которых случаях они действительно могут служить для предва-
рительной ориентировки в составе тех или иных нефтяных пого-
нов. Сюда относятся:
1. Действие брома в присутствии бромистого алюминия. Реак-
ция эта, указанная Туставсоном для открытия ароматических
углеводородов, была затем с успехом применена М. Коноваловым
впервые к нононафтену [14]. Конечным продуктом ее в обоих
случаях являются пербромиды ароматического ряда, хорошо
кристаллизующиеся. Так например, из толуола и гептанафтена
с температурой кипения 160—102 (метил-циклогексана) полу-
чается пентабромтолуол, из ксилола и октонафтена с темпера-
турой кипения 118—120° (диметил-циклогексан) — тетрабром-
ксилол и т. д. Для толуола и метил-циклогексана эти превраще-
ния можно выразить следующими уравнениями:
СН3 - С§Н5 + lOBr = СН3СеВг5 4- бНВг;
СН3 • сени +1бВг = СВДВ^ -I- иНВг.
Позднее Зелинский показал, что с синтетическими гомоло-
гами циклогексана реакция эта протекает количественно, причем
для получения кристаллического пербромида достаточно 0,1 г
углеводорода. Так как другие нафтеновые ряды, а равно и пара-
фины не дают в тех же условиях ароматических пербромидов, то
получение последних из той или иной фракции нефти, предва-
рительно очищенной от бензола и его гомологов, должно указы-
78
вать на нахождение в нефтяном погоне соответствующего гомо-
лога циклогексана.
Известно, однако, что действие на ароматические углеводороды брома
в присутствии бромистого алюминия сопровождается иногда побочными про-
цессами отщепления или накопления боковых групп и т. п. По отношению
к синтетическим нафтенам при данной реакции также констатированы
процессы изомеризации. Так, например, при действии брома и бромистого
алюминия на циклогексан и метилциклопентан получается один и тот же
пербромид с температурой плавления 120—121° состава СоШВгв ялиШЬЕг?,
так что при одной из этих реакций происходит, очевидно, какая-то пере-
группировка. Возможно поэтому, что действие брома и бромистого алюми-
ния на нафтены, более сложные, чем это испытано до настоящего времени,
также будет сопровождаться изомеризационнымп процессами, подобно тому
как это наблюдается в ароматическом раду. Необходимо также иметь в
виду, что по отношению к нефтяным углеводородам реакция осложняется
обильным образованием малоустойчивых маслообразных продуктов, затруд-
няющих даже выделение ароматических пербромидов, не говоря уже о ко-
личественном их определении. Отсюда ясно, что действие брома и бромистого
алюминия может служить лишь для качественного открытия гомологов
циклогексана, да и то, невидимому, лишь низших, т. е. содержащихся в
бензине и низших погонах керосина.
2. Действие серы при высокой температуре. Ганн показал,
что дигидропиридиновые соединения при нагревании с серой
переходят обратно в тела пиридинового ряда. Так как, с другой
стороны, при нагревании о серой высококипящих частей нефти
наблюдается выделение сероводорода, то ясно, 'что и на нафтены
сера может действовать дегидрогенизирующим образом. Реакция
ведется в запаянных трубках при 210—220 , сопровождается
частичным обугливанием вещества и до конца не доходит. За
нагреванием следует поэтому обработка серно-азотной смесью
для извлечения ароматического углеводорода в виде нитросоеди-
нения. Этим способом из 70 г хорошо очищенного октонафтеиа
(темп, кипения 118-120°) после трех повторных обработок Мар-
ковникову и Шпади [15] удалось собрать до 5,3 г тринитро-изо-
ксилола, тогда как тот же углеводород без предварительного на-
гревания с серой давал лишь следы нитросоединений. Поздней^
шие исследования [16] показали, что конечным продуктом де-
гидрогенизации в данном случае оказываются не ароматические
углеводороды, а, невидимому, дигидробензолы. Так например,
после нагревания с серой циклогексана до 220-230° выделить
бензол не удалось: дегидрогенизация шла только до дигидробен-
зола, СбНв" который с нитрирующей смесью дал динитробензол.
Сколько-нибудь, широкого применения для исследования нефти
названная реакция до сих пор не получила, однако успешное
приложение ее при изучении структуры высших терпенов (сески-
терпенов) [17] позволяет ожидать что при исследовании масля-
ных фракций нефти реакция эта еще может принести пользу.
Существенным недостатком применения серы для целей дегидрогениза-
ции высокомолекулярных углеводородов является то обстоятельство, что
при этой реакции всегда образуется значительное .количество сернистых
соединений, осложняющих дальнейшую работу. В последнее время в каче-
стве вещества, способствующего дегидрогенизации, вместо серы предложен
70
селен, кристаллический или аморфный [18]. В (Присутствии селена реакция
дегидрогенизации протекает более гладко: не сопровождается побочными
процессами и дает несравнимо лучшие выхода на конечный продукт.
3. Действие дымящей азотной кислоты. Дымящая азотная
растворяет нафтены с саморазогреванием. Особенно энергично
протекает реакция с гомологами циклопентана: она сопрово-
ждается бурным вскипанием жидкости с обильным выделением
окислов азота и приводит к продуктам глубокого распада моле-
кулы исходного углеводорода. С гомологами циклогексана реак-
ция протекает несколько спокойнее: кроме продуктов глубокого
окисления углеводорода, здесь образуются также ароматические
полинитросоединения, например из 1,3-диметил-циклогекса-на—
тринитро-изоксилол и т. д. К сожалению, эта реакция изуча-
лась до настоящего времени главным образом на нефтяных, а не
на синтетических углеводородах. Применение се требует, ко-
нечно, предварительной очистки нефтяного погона от ароматиче-
ских углеводородов.
СИНТЕЗ НЕФТЯНЫХ УГЛЕВОДОРОДОВ
Химическое исследование всякого природного вещества всегда
идет сначала чисто аналитическим путем. После выделения ве-
щества в возможно чистом виде определяют его элементарный
состав и, применяя к нему разнообразнейшие методы- химиче-
ского исследования, пытаются установить его ближайшую хими-
ческую природу. Хотя этим путем зачастую удается установить
строение изучаемого вещества с полной несомненностью, тем не
менее задача может считаться окончательно решенной лишь в
том случае, если -выведенное аналитически сцюение вещест
удается подтвердить синтетическим путем. Таким образом хими-
ческое исследование нефти естественно выдвинуло вопрос о син-
тезе отдельных, выделенных из нефти углеводородов различных
урядов.
Изучение парафиновых и ароматических углеводородов про-
текало независимо от задач, выявлявшихся при исследовании
нефти. Лишь в отдельных случаях мог возникнуть вопрос о син-
тезе того или иного представителя этих рядов, выделенных из
нефти. Как общее же правило, исследование могло основываться
в этих случаях на обширном и надежном опытном материале,
собранном предшествующими исследованиями.
Иначе обстояло дело по отношению к нафтенам. Уже самый
термин «нафтены» возник при исследовании нефтяных углеводо-
родов. Химическая природа их некоторое время оставалась не
вполне ясной, и лишь синтетическое получение некоторых пред-
ставителей ©того класса углеводородов окончательно разрешило
этот основной вопрос химии нефти. Ввиду этого, говоря о мето-
дике исследования нефтей и нефтепродуктов, обойти вопрос
о синтезе нафтенов представляется невозможным.
Гидрогенизация ароматических углеводородов. При про-
должительном нагревании до 250° и выше самых разнообразных
органических соединений с избытком дымящей иодистоводород-
80
ной кислоты все они превращаются в конечном итоге в соответ-
ствующие углеводороды ряда метана (Бертло). Восстановление
(гидрогенизация)4 идет здесь, очевидно, за счет водорода, обра-
зующегося при диссоциации иодистоводородной кислоты при вы-
сокой температуре:
\ Ш^Н+1. • ' 1
Систематическое применение этой реакции к ароматическому
ряду (Вреден) показало [1р], что продуктами восстановления
(гидрогенизации) являются в данном случае действительно угле-
водороды предельного характера, но не ряда метана, а углеводо-
роды циклического строения, состава CnH2rt. Эти углеводороды
назывались обыкновенно гексагидробензолами; в них принимали,
как и в бензоле, шестичленное кольцо углеродных атомов. Не-
смотря на большие трудности, с которыми было сопряжено их
получение, они долгое время привлекали исключительное внима-
ние исследователей нефти, так как при большом сходстве их
с нафтенами естественно напрашивалась мысль о тождестве
этих двух углеводородных рядов (Бейлъштейн и Курбатов;
Шютценбергер и Ионин).
Основные трудности гидрогенизации ароматических соедине-
ний с помощью иодистоводородной кислоты заключаются в сле-
дующем:
а) для успешной гидрогенизации требуется очень высокая
температура (около 300°), вследствие чего приходится работать
в запаянных трубках с небольшими количествами вещества (на-
пример углеводорода);
б) работа сопряжена с затратой больших количеств ценного
продукта — иодистоводородной кислоты, применяемой в большом
избытке;
в) получаемый продукт реакции является крайне сложной
смесью углеводородов, из которой основное вещество, получаемый
гекса-гидробензол, может быть выделено лишь с большим трудом.
Но главное, что определило оценку как самой реакции гидро-
генизации ароматических углеводородов с помощью иодистоводо-
родной кислоты, так и полученных этим методом результатов,
заключалось в своеобразной ее особенности, которая долгое
время оставалась незамеченной. Оказалось [20], что реакция эта
протекает аномально, причем, например, бензол превращается
главным образом в углеводород не с шести-, а с пятичленным
циклом, а именно в метил-циклопентан (Кижнер, 1891 г.):
/СН = СНХ сн9 — СН2Х
НС< >СН + 6Н = | >сн- сн8.
хж-cir сщ—сн/
Позднейшие исследования показали, что аналогичные анома-
лии наблюдаются также при гидрогенизации гомологов бензола
с помощью иодистоводородной кислоты и что, таким образом,
реакция эта для получения нормальных продуктов гидрогениза-
81
6 Вак. 1622. Химия нефти.
ции ароматических углеводородов не пригодна. Эта последняя
задача была решена значительно позднее благодаря классиче-
ским исследованиям Сабатъе и Сандерана (1905 г.) в области
гидрогенизации при непосредственном действии водорода в при-
сутствии мелкораздробленных металлов (Ni и др., ср. выше).
К* этому времени вопрос о химической природе нафтенового ядра
уже получил окончательное разрешение; для этого был найден
путь синтеза нафтенов из двухосновных кислот.
Оинтев нафтенов из двухосновных кислот жирного ряда.
При сухой перегонке солей” двухосновных кислот жирного ряда,
начиная с адипиновой, НОгС . (СН2)4 СОгП, происходит образо-
вание циклических кетонов, содержащих на один атом углерода
меньше, чем исходная кислота [21]. Сначала эта реакция была
изучена на примере адипиновой кислоты (И. Вислиценус); глав-
ным продуктом реакции оказался в данном случае адипинкетон
или циклопентанон:
сн2—сна—соох сна—сщ.
I >Са = С0яСа+1 >С0.
СНа — СНа — СОСИ * СЩ —СП/
Почти одновременно та же реакция была изучена на приме-
рах пимелиновой (А. ф.-Байср) и пробковой кислот (Марковни-
ков)-, в обоих случаях также получились циклические кетоны;
из пимелиновой кислоты — циклогексанон:
,СН2 —СН2 —С00х
СН/ /Са =
ХСН,2 — сн2—соси
из пробковой—циклогептанон (суберон):
СНа—СЩ — СН2 — С00ч СНа — СНа—СПач
I >Са = СО„Са+1 )С0.
СН2-СН2-СН2- СОСИ СИ,—СН,—СП/
Хотя каждый из этих циклических кетонов был превращен;
обычным путем, т. е. через соответствующие алкоголь и галоидо-
производное, в углеводород состава С„Нгл, однако получение этих
нафтенов совершенно не отразилось на разъяснении вопроса,
о химической природе нефтяных углеводородов: внимание иссле-
дователей нефти занимали в это время главным образом другие,
более высокомолекулярные нафтены. Из нафтенов, присутствие
которых в нефти представлялось несомненным, первым был син-
тезирован 1,3-диметил-циклогексан (Зелинский, 1805 г.), по
своим свойствам оказавшийся весьма близким к нефтяному
октонафтену (темп, кипения 118—120°). Получение 1,3-диметил-
циклогексана следует рассматривать поэтому, как первый сиа-(
тез природного нафтена.
1,3-диметил-циклогексан был получен [22] из соответствую-
82
щего Кетона, приготовленного сухой перегонкой известковой ооли
а, а1-ди меги л-пи мели новой кислоты, по уравнению:
/СН2—СН(СН8) — С00ч
\с
ХСН2—СН(СНа) — cooz
/СЩ — СН(СНа)
= С08Са+СН2<
Вслед за тем в течение длинного ряда лет тем же автором были
синтезированы многочисленные другие представители класса
нафтенов из гомологов циклопентана и циклогексана, которые
и по настоящее время могут служить образцами для сравнения
с естественными нафтенами.
Сопоставление свойств синтетического и природного нафтенов
показывает, что при всей близости свойств этих углеводородов
между ними наблюдается все-таки некоторое различие. Так на-
пример, удельный вес синтетического нафтена заметно выше, чем
удельный вес нефтяного углеводорода; с бромом в присутствии
бромистого алюминия синтетический нафтен дает количествен-
ный выход тетрабромксилола, тогда как с нефтяным углеводоро-
дом та же реакция дает лишь несколько процентов пербромида,
и т. п. Это различие объяснялось сначала недостаточной чисто-
той нефтяного углеводорода.
Однако новей
е данные заста-
вляют искать истинную причину такого расхождения значи-
тельно глубже. Мы вернемся к этой теме ниже в связи с вопро-
сом о химической природе окто- и нононафтенов.
МЕТОДЫ ИДЕНТИФИКАЦИИ
Окончательное решение вопроса о химической природе той
или иной однородной фракции нефти достигается ее идентифи-
кацией, иначе говоря, установлением тождества ее с тем или
иным синтетическим продуктом, обычно углеводородом того или
иного ряда. Методы идентификации углеводородов весьма разно-
образны. Как общее правило, лишь тождество всех свойств при-
родного продукта с синтетическим достаточно для окончатель-
ного заключения об их тождестве; однако обыкновенно доволь-
ствуются совпадением лишь некоторых свойств, наиболее важ-
ных и доступных для их точного определения. На практике, ко-
нечно, нередки случаи, когда синтез выделенного из нефти ве-
щества пока еще не осуществлен, так что полная его идентифи-
кация невозможна. В таких случаях те же свойства природного
продукта используются для суждения о степени его чистоты,
или, как иногда говорят, об его индивидуальности. Наиболее
часто для этих целей пользуются определением состава и неко-
торых физических свойств.
Элементарный соетав и молекулярный вес. Эти характери-
стики являются, естественно, основными; из них легко вычис-
6* 83
ляется эмпирическая формула молекулы данного углеводорода.
Для их определения пользуются обычными методами элементар-
ного анализа органических соединений и определения их моле-
кулярного веса. Во многих случаях знание этих свойств дает
также указание *на принадлежность нефтяного углеводорода
к тому или иному основному классу углеводородов, например
к классу парафинов (СпНгп + г), нафтенов (СиНгп), к гомологам
бензола (СлН2п_ в) ит. п.
Удельный вес и коэфициент лучепреломления. Свойства эти,
как известно, являются обычной физико-химической характери-
стикой чистых веществ. О технике их определения уже было
сказано выше (гл. II). В ряду парафины — нафтены — арома-
тика обе характеристики растут в направлении от парафинов
к ароматике, что нередко позволяет ориентироваться в химиче-
ской природе углеводорода уже по одному его удельному весу
или коэфициенту лучепреломления. Так например, для угле-
водородов с С углеродами в частице (Се) в округленных цифрах
имеем следующие характеристики: для удельного веса — 0,67—
0,75 (для ряда циклогексана) — 0,88, для коэфициента лучепре-
ломления—1,38—1,40 (для того же ряда) — 1,50. Для углеводо-
родов с более высоким молекулярным весом «то глубокое разли-
чие несколько сглаживается, так кале .у парафинов и нафтенов
обе характеристики с повышением молекулярного веса растут;
у ароматики же, наоборот, понижаются. Равным образом появле-
ние в нафтене второго цикла [ср., например, декалии] нарушает
указанную правильность: наблюдается 1>езкое повышение как
удельного веса, так и коэфициента лучепреломления нафтена.
Широкое применение для идентификации органических соеди-
нений получили также удельное лучепреломление И и молеку-
лярное лучепреломление MR, определяемые по формулам Ло-
ренц-Лоренца:
п*—1 , м п-
т, и MR = -г • -т
где М— молекулярный вес вещества; d— его удельный вес [tff]
и п — коэфициент лучепреломления при 20°.
Обширные исследования, произведенные в этой области, поз-
волили рассчитать с высокой степенью точности атомные ко-
эфициенты лучепреломления как для углерода (С — 2,418) и
водорода (Н—1,100), так и для других элементов и элементар-
ных группировок, входящих в различные органические соедине-
ния, например для двойной связи (1 двойная связь— 1,733), для
гидроксила (ОН—2,625), карбоксила (С02Н—7,254) и т. д.
Тем самым получена возможность весьма ценного сопоставления
молекулярного лучепреломления, рассчитанного ио формуле на
основе найденных опытным путем величин 2И, d и я, и молеку-
лярного лучепреломления, вычисленного для данной молекулы^
исходя из атомных коэфициентов лучепреломления. Такие со-
поставления широко применяются особенно при исследований
жидких органических веществ и являются одним из основных
84
критериев, на основе которых нередко можно сделать безоши-
бочное заключение о степени чистоты органического вещества.
Следует, впрочем, иметь в виду, что примесь изомерного угле-
водорода того же класса, например парафина к изопарафину,
или наоборот, и т. п., не обнаруживается этим способом, и это
обстоятельство, естественно, резко ограничивает применение
данной методики для идентификации нефтяных углеводородов.
Температура кипения и температура плавления. Наряду
с удельным весом и коэфициентом лучепреломления темпера-
туры кипения и плавления (застывания) наиболее часто служат
для характеристики химических соединений. Для химически
чистого вещества при этих температурах мы имеем идеальное
равновесие между жидкой и паровой фазами вещества или со-
ответственно между его жидкой и твердой фазами. Равновесие
это, как известно, характеризуется тем, что во все время кипе-
ния или плавления (застывания) при постоянном давлении кри-
вая «температура — время» должна представлять собою горизон-
тальную прямую. Присутствие примесей резко сказывается на
ходе этой прямой, сообщая ей более или менее ясно выраженные
наклон и изломы. Обычные методы определения температур ки-
пения и плавления (застывания) описаны в гл. II; для более
точных определений, особенно при низких температурах, приме-
няется специальная аппаратура, причем вместо ртутного термо-
метра предпочтительно пользоваться хорошо выверенной плати-
новой термопарой.
Анилиновый метод. Критические температуры растворения
в анилине (а также в некоторых других растворителях, напри-
мер в нитробензоле и т. п.) настолько характерны для различ-
ных классов углеводородов, что свойством этим оказалось удобно
пользоваться как дня качественной характеристики отдельных
углеводородов, так и для количественного их определения в сме-
сях. Постепенно возрастая с увеличением молекулярного веса
углеводородов, температуры эти в ряду парафины — нафтены —
ароматика резко снижаются в направлении от парафинов к аро-
матике. Так например, для углеводородов с 8 углеродами в час-
тице (Се) в округленных цифрах имеем здесь следующие харак-
теристики: 70 (парафины), 45 (нафтены) и 20 (ароматика). Все
подробности, относящиеся к «анилиновому методу», изложены
в следующих главах (IV, V и VI) в связи с вопросами количе-
ственного определения ароматики, парафинов и нафтенов в угле-
водородных смесях; однако уже из приведенного примера ясно,
насколько яркой характеристикой углеводорода является его
«анилиновая точка».
Прочие методы.Вышеизложенным, конечно, далеко не исчер-
пываются все методы, которыми пользовались отдельные иссле-
дователи при изучении различных нефтяных погонов. Среди
физических и физико-химических характеристик, которые нашли
здесь применение, отметим, например: изменение вязкости с тем-
пературой, поверхностное натяжение, молекулярную дисперсию,
особенно спектры поглощения в области инфракрасных лучей и
85
Многие другие. Не менее разнообразны чисто химические методы,
нашедшие применение для установления химической природы
отдельных нефтяных углеводородов и их структуры. К некото-
рым из этих методов мы вернемся ниже при ближайшем рассмо-
трении состава отдельных нефтяных фракций.
ЛИТЕРАТУРА
1. Марковников и Оглоблин, „Ж.Р.Х.О/ 15, 237, 307 (1883); Морковников
„Ж.Р.Х.О." 80, 59 (1898).
2. Young 8., Destination Principles and Processes, London p. 113, (1922).
3. См. также Cooke M. B. and jRwe Я. P., Studies in the Fractional DistU-
lation of Crude Petroleum, Techn. Paper 431, Bureau of Mines (1928);
Лонгинов и Прянишников, Материалы к изучению дефлегматоров
и ректификационных колонн лабораторного типа, «Труды ИРЕА,*М.
(1929), Robinson С. 8. The Elements of Fraktional Di-tillation (1930)
4. Rossim F. D. „Refiner" 1в, № 11, 545 (1937); здесь же новейшая литера-
тура предмета.
5. Марковников, „Ж.Р.Х.О." 80, 84 (1898); см. также ряд статей и докладов
того же автора и его сотрудников в „Ж.Р.Х.О." (1884—1904).
6. Зелинский, .Ж.Р.Х.О." 85, 128*0 (1903); 88, 13, 767 (1905).
7. Коновалов, .Ж.Р.Х.О." 25, 889, 472, 500 (1893); он жз, Нитрующее дей-
ствие азотной кислоты на углеводороды предельного характера, М.
(1893); см. также Наметкин, К вопросу о действии азотной кислоты
на углеводороды предельного характера, М. (1911).
8. Зелинский, „Ж.Р.Х.О." 80, 310 (189о); 81, 60 (1899).
9. Ср. Nenilzescu и Jonescu, „Lieb. Ann." 491, 189 (1981); Hopff, „Вег.* <£
2739 (1931); 85, 482 (1932); Unger, „Вег." 85, 467 (1982); Zelinsky H. Д
и Tarassowa В. М. „Взг." 85, 1249 (1982) и т. д.
10. Sabatier et Seuderens, «Ann. Ch. et Phys." (8), t. IV (1905); см. также
Челинцев, Контактно-каталитические процессы в области органи-
ческих соединений, Л. (1927).
11. Зелинский, «Ж.Р.Х.О." 43, 1220 (1911); 45, 52 (1913); „Вег." 56. 1718 (1923);
12. Марковников и Шпади, „Ж.Р.Х.О.* 19, 519 (1887) и др.; Meyer Hans.
„Lieb. Ann." 488, 827 (1923).
13. Worstall, „Am. Chem. J." 20, 664 (1898)
14. Коновалов, „Ж.Р.Х.О." 22, 11 (1890).
15. Марковников и Шпади, «Ж Р.Х.О." 19, 171, 516 (1887).
16. Фридман, .Petroleum" 11, 978 (1915).
17. Ruzicka L. и Meyer J, „Helv. Chem Acta" 4, 507 (1921).
18. Diels О. и Karstens А., „Вег." 60 2323 (1927).
19. Вреден, „Ж.Р.Х.О." 8, 55 (V74); 8, 146 (1876); 9, 242 (1877); 10. 79 (18».
20. 2Г«лс«ер^„Ж.Р.Х.О." 28, 20 (1891); 24, 450 (1892); 28, 875 (1894); 29, 5&
21. Wislicenus J., „Lieb. Ann." 275, 309 (1898); v. Bayer A., „Lieb. Ащь‘
278, 111 (1893); Марковников, ,C. R" 110, 466 (1890); „Ж.Р.Х.О."»,
864 (1893).
22. Зелинский, ,Ber.“ 28, 780 (1895) и ряд статей того же автора с сотрудни-
ками в „Вег." н «Ж.Р.Х.О." за последующие годы.
ГЛАВА IV
АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ НЕФТИ
Основным источником получения ароматических углеводоро-
дов является, как известно, каменноугольный деготь [1]. Как
показала, однако, последняя война, при напряженном положении
с этим сырьем в качестве источника для получения ароматиче-
ских углеводородов можно пользоваться также нефтью и притом
не только в виде продуктов ее пиролиза, но также и непосред-
ственно в виде соответствующих нефтяных фракций прямой
гонки (например толуольной). Если прибавить к сказанному,
что присутствием ароматических углеводородов в значительной
степени определяются антидетонационные свойства топлива для
двигателей внутреннего сгорания, то станет понятным, что, не-
смотря на небольшое содержание ароматики в громадном боль-
шинстве исследованных в этом направлении нефтей, вопрос об
ароматических углеводородах нефти для химика-нефтяника
представляет не только теоретический, но и большой практиче-
ский интерес.
КРАТКАЯ ХАРАКТЕРИСТИКА АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ
Все ароматические углеводороды являются, как известно, про-
изводными бензола. Их можно разделить на следующие основ-
ные группы:
I. Бензол и его томологи (табл. 23).
11. Фенилированные бензолы: дифенил и его гомологи
(табл. 24).
III. Фенилированныс метан, этан и их гомологи с двумя и
более фенильными группами (табл. 24).
IV. Конденсированные ароматические системы: гомологиче-
ские ряды нафталина, антрацена и т. п. (табл. 25).
По богатству превращений ароматические углеводороды зани-
мают бесспорно первое место среди углеводородов различных
классов. Не вдаваясь в подробности [2J, укажем вкратце лишь
важнейшие их превращения, представляющие особый интерес
с точки зрения химии нефти.
1. Водород легко присоединяется к ароматическим углеводо-
родам в присутствии различных металлических катализаторов
87
Таблица 23
об
Свойства ароматических углеводородов. Бензол я его гомологи
Наименование Формула Темп, плавл. Темп. кип. Уд. вес. Коэф, рефр nD
df t’
Бензол С6Н6 4- 5,4 80,°4 0,879 20’ 1,5014
Толуол сн3 С6Н5 — 93’ 110,°6 0,866 20° 1,4955
Ксилол-орто ьгчснзЪСвЩ — 28° 144° 0,876 20’ 1,5024
Ксилол мета 1,ЗчСН3)2С6Н4 — 53’ 139° 0,874 8,°4 1,5032
Ксилол-пара 1.4-(СНз)5С6Н4 4-15’ 138° 0,868 14,°4 1,4991
Этпл-бензол С2НбСвН6 — 93° 136° 0,867 20° 1,4959
Гемэллитол 1, 2, 3-(СН3)зСвНз —- 175° 0,895 19,°6 1,5134
Псевдовумол 1, 2, 4-(СН3)зСвНз — 168’ 0,879 15/3 1,5067
Мезнтплен 1, 8, бЧСНзЬСвНз — 57’ 164° 0,865 17,°1 1.4980
Пропял-бензол С3Н7С6Н5 1 159° 0,868 12,°3 1,4955
Кумол нзо-С3Н7С6Н5 — 153’ 0,866 16/8 1,4944
Диэтил-бензол 1, — 182’ 0,868 16,°2 1,4990
Ивоамил-бенвол изо-С5Ни • С6Н- — 193’ 0,885 18’ '•
Дурол • 1, 2, 4, 5-(СН3)4С6Н2 4-80’ 192° — — 1 ——
Изодурол 1,3, 4, 5-(СН3)4С6Н2 196’ 0,896 0 ! —
Днмол-пара изо-СзН7СвН4СНз — тз/б 177,°3 0,862 13,°7 1,4926
Давтижчмтол 1, 3, Б-ССзНбИ-СвНз-СНз 200° 0,879 20° —
Таблица 24
Свойства ароматических углеводородов. Фенилировавные бензолы;
ди- и полифеиилированные парафины
Наименование Формула Темп, плавл. Темп, кип. • Уд. вес d
Бифенил С6НГ, • CcIIG 70,°5 264° 0,992 (73°/4°)
Битоли и-пара СП., • СвЩСеЩ СН3 121° — —
Дифенил-метан .... с6нБ-сн2.свнГ) 27’ 261° 1,001 (2G°/4°)
Трифенил-метан .... (С6НБ)3СН 92° ' ЗЕ9° —
Дибензил СеНс СН, • СН, • СсН6 52° ’ 284° —
(Ni, Pt, Pd и др.) с образованием гидроароматических углеводо-
родов (нафтенов). Количество присоединяющихся атомов водо-
рода находится при этом в полном соответствии с числом двой-
ных связей, содержащихся в данной ароматической системе
согласно бензольной формуле Кекуле, по месту которых происхо-
дит присоединение водорода (гидрогенизация). Так например:
СН СН2
СН,
нс^сн
2
НоС
сн2
бензол
СНГ СН
^\%сн
циклогексан
снс сн2
4Н
—>
6Н
--->
снссн
нафталин
С IL
С СН2
тет! алия
СН2нСН2
НС
>
НоС
сщ
сн2
2 \х \/
СН2Н сн
декалин
2. Крепкая азотная кислота, а равно серноазотная смесь
реагируют с Образованием сначала мононитросоединений арома-
тического ряда, а затем—ди- и полинитросоединений (нитрова-
ние)', например:
N02
CeHe + HN03 = H20 4- СвНБК02, а далее: CBHZ
2
2
89
Таблица 25
Свойства ароматических углеводородов. Конденсированные системы
Наименование Формула Темп, плавл. Темп, кип. Уд. вес
Нафталин Cjo^g 80° 218°
Метил-нафталин а . . . СН3-С;0Н7 —22° 241° 1,0005 (19°)
Метил-нафталин р . . . С Н3 • Охой? 32,°5 241° —
Этил-нафталин а .. . . ед-СюН, — 258° 1,018 (10°)
Этил-нафталин р . . . . С2НБ • С]оН7 —29° 251° 1,008 ( 0е)
Пропил-нафталин ₽ .. . U3H7 • С10Н7 — 265° 0,990 ( 0°)
Антрацеп Cj4,Hio 217° 351° —
3. С крепкой серной кислотой низшие ароматические углево-
дороды легко образуют сульфокислоты (сульфирование):
С6Н6+ H2S04 = C6H6SO3H+ Н20.
Гомологи бензола реагируют с серной кислотой аналогичным
образом; однако по мере увеличения боковой цепи энергия этой
реакции несколько падает.
4. Непредельные углеводороды легко конденсируются с бен-
золом и его гомологами в присутствии катализаторов (H2SO4,
безводный А1С1з или А1Вг3 и др.); образуются гомологи бензола
предельного характера, например с амиленом — амилбензол
и т. п.:
C6He+CcH10=C6H5.CGHn.
Другие реакции конденсации ароматических углеводородов
могут приводить к значительно более сложным продуктам. Так
например, при взаимодействии бензола и его гомологов с фор-
мальдегидом, СН2О, или его метиловым эфиром, СНг(ОСН3)2,
метилалем, в присутствии серной кислоты получаются разно-
образные в зависимости от условий реакции более или менее
сложные продукты конденсации, в конечном итоге — аморфные
порошки, «формолиты» неизвестного строения.
5. Ароматические углеводороды легко образуют многочислен-
ные комплексные соединения с разнообразными как неорганиче-
скими, так и органическими соединениями. Некоторые из этих
комплексов хорошо кристаллизуются и нашли применение при
выделении ароматических углеводородов, особенно высокомолеку-
лярных, из их смесей или растворов в других углеводородах. Та-
ковы, например, пикраты, более ити менее интенсивно окрашенные
комплексные соединения ароматических углеводородов с пикри-
новой кислотой (Р): пикрат бензола — Р- СвНв, пикрат нафта-
лина — Р • СюНв и многие другие. Сюда ж.е относится бесцвет-
ное комплексное соединение трифенилметана с бензолом,
(СвПд)з СН • СбНс, трудно растворимое в бензине, удобное для
открытия в бензиновых растворах небольших количеств бензола.
90
АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ БАКИНСКОЙ НЕФТИ
Впервые вопрос о нахождении в нефти ароматических угле-
водородов был систематически изучен Марко вныковым и Огло-
блиным [3] около 60 лет тому назад, и до сих пор данные этих
исследователей являются основными в этой области. Материа-
лом для работы послужили им нефти Бакинского района (Биби-
Эйбат, Балаханы). Для извлечения из них ароматических угле-
водородов авторы первоначально промывали заводские погоны
едкой щелочью, дробили в пределах 10°. и обрабатывали дымя-
щей серной кислотой или купоросным маслом; в иных случаях
промытые щелочью погоны предварительно фракционировались
3—4 раза, а затем уже обрабатывались кислотой. Впоследствии
для более тщательного отделения ароматических углеводородов
было признано необходимым обрабатывать нефтяные погоны
серно-азотной смесью. Однако в тех случаях, когда ароматиче-
ские углеводороды нефти не просто отделяются, а подлежат ис-
следованию, применение серной кислоты предпочтительно в ин-
тересах дальнейшей работы.
Обработкой нефтяных погонов серной кислотой содержащиеся
в них ароматические углеводороды превращаются в сульфоки-
слоты. Так как при продолжительном соприкосновении с дымя-
щей серной возможно -взаимодействие с этим реагентом также
некоторых нафтенов, то правильнее отнести на счет ароматиче-
ских углеводородов нефти, .по мнению Марковникова, лишь те
из получившихся сулгфокислот, которые содержатся в первой
порции серной кислоты (не больше 10% нефтяного погона).
Кислота эта, отделенная от углеводорода и разбавленная неболь-
шим количеством воды, иногда при стоянии на холоду превра-
щается в кристаллическую кашицу; в таком случае после не-
скольких перекристаллизаций сульфокислот^ может быть полу-
чена в чистом виде. Однако обыкновенно смесь серной кислоты
с сульфокислотами не кристаллизуется; тогда ее насыщают из-
вестью и по отделении сернокислого кальция разделяют извест-
ковые соли сульфокислот, пользуясь их различной раствори-
мостью в воде или алкоголе. Затем следуют выделение из извест-
ковой соли свободной сульфокислоты или превращение ее кипя-
чением с содой в натровую соль и, наконец, заключительный
переход к ароматическому углеводороду: ют свободной сульфо-
кислоты — сухой перегонкой ее с гашеной известью; от натровой
соли сульфокислоты—'Нагреванием с крепкой соляной
до 150—180°.
Разделение ароматических сульфокислот из низших нефтя-
ных погонов не представляет большого труда: сульфокислоты
эти — кристаллические вещества; и поскольку соответствующие
им ароматические углеводороды хорошо изучены, решение во-
проса о нахождении того или иного гомолога бензола в нефти
вполне осуществимо. Гораздо сложнее обстоит дело с высшими
нефтяными погонами. Получающиеся отсюда сульфокислоты—
уже густые, тягучие смолы, а соответствующие им ароматиче-
91
V
ские углеводороды — или вовсе не известны в чистом виде, или
изучены весьма мало. Понятно поэтому, что исследование выс-
ших нефтяных погонов на содержание в них ароматических
углеводородов встречает значительные- трудности. Главные ре-
зультаты, полученные в этом направлении, сводятся к следу-
ющему.
Погоны 105—115° и 115—125° бакинского керосина дали
сульфокислоты, из которых сухой перегонкой их известковых
солей с гашеной известью была получена смесь ароматически^
углеводородов с температурой кипения 75—141°. Фракциониро^
кой этой смеси и последующими реакциями в ней были обнару-
жены: бензол, СвНе, — получением кристаллического углеводо-
рода, уже по внешнему виду схожего с кристаллами бензола;
толуол, С7Н8, — при нитровании доказано образование динитро-
толуола; изоксилол, 1,3-СеН4(С1Тз)2, — при обработке серно-азот-
ной смесью получен триницю-изоксилол. Кроме того, было
установлено, что фракция этой смеси, отвечающая бензолу в
толуолу (температура кипения 75—1Ю°), при осторожном ни-
тровании дает смесь ароматических монопитросоединений, при
восстановлении которых действительно получилась смесь ани-
лина с толуидином.
Погон 156—165° керосина и нефтяной погон удельного веса
0,777 (при 17°) дали при обработке оорнютг кислотой совершенно
чистую сульфокислоту псевоокумола, 1, 2, 4-СеПз(СНз)з (темпе-
ратура кипения 168°), охарактеризованную несколькими про-
изводными (хлорапгидрид, амид). Полученный из нее углеводо-
род кипел главным образом при 109°. Отметим здесь же, что
в несколько более узкой фракции бакинской нефти (температуру
кипения 155—160°) доказано [4] также присутствие мезт-
лена получением некоторых производных этого гомолога бензола
(трихлор-мезитилен, тринигро-мезитилен).
Погон 180—190°. Получилась весьма сложная смесь сульфо?
кислот, из которой была приготовлена известковая соль. Часть
этой соли, в спирте легко растворимая, при медленном испаре:
нии ее водного раствора осталась в виде густого, сильно окра-
шенного сиропа. Соответствующая ей сульфокислота оказалась
также густой сиропообразной жидкостью. Нагреванием с креп-
кой соляной из этой сульфокислоты был получен углеводород
распавшийся при перегонке на несколько фракций. Из ни
фракция 178,7—182,7° по составу и по температуре • кипений
подходила к диэтил-бензолу, СбН^СгШ)^, (пара, температур!
кипения 182°); фракция же 187,с—192,6°— к изоамил-бензолу,
СвН5 • С5Нц (температура кипения 193°). Оба эти углеводорода
действительно дают легко расплывающиеся сульфокислоты; воз-
можно, что оба они действительно содержатся во фракция
180—190° исследованной нефти. ।
Погон 190—200° дал смесь известковых солей сульфокислот
которую по растворимости в алкоголе удалось разбить на две
.части: а) соль, труднорастворимая в алкоголе, была переведена
в натровую; последняя же — нагрета с крепкой соляной кисло-
92 '’'i
той; получился кристаллический углеводород (температура пла-
вления 78—80°), несомненно — дурол, 1, 2, 4, 5-СвН2(СНз)4
(температура плавления 80 , температура кипения 192°);
б) соль, легко растворимая в алкоголе, получилась в -виде сирсь
пообразней жидкости, из которой после разбавления водой выде-
лились бородавчатые кристаллы; кристаллы эти были также пе-
реведены сначала в натровую соль, а затем переработаны на
углеводород; последний кипел при 190—195°. Быть может, это
изо-дурол, 1, 3 ,4, б-СвН2(СН8)4 (температура кипения 196°).
Погон 200—210°. Натровые соли сульфокислот из этого по-
гона также оказались частью легко, частью трудно растворимы
в алкоголе. Углеводород, полученный из наиболее трудно раство-
римой соли, содержал дурол; углеводород же из соли, легко
растворимой в алкоголе, кипел главным образом при 200—208°.
В нем доказано присутствие диэтил -толуола, 1, 3, 5-СвН8-СН3-
• (СгН5)2 (температура кипения 200 ) как анализом углеводорода,
так и получением характерного для него кристаллического три-
бромида.
Число ароматических углеводородов, входящих в состав трех
последних нефтяных погонов, несомненно больше, чем указано
выше. Однако соответствующие им сульфокислоты не могли быть
выделены из смеси в чистом виде, и о строении их пока нельзя
сказать ничего определенного. То же относится к погонам, ки-
пящим выше 200°, в которых, кроме высших гомологов бензола,
начинают встречаться ароматические углеводороды конденсиро-
ванных рядов. Так например, из погона 240—250° керосина по- <
лучена сульфокислота, отвечающая, быть может, проъил-н&ф-
талину, С10Н7С3Н7. Есть указания на присутствие в бакинской
нефти и других углеводородов того же типа, а также иных аро-
матических рядов; данные эти носят, однако, лишь предвари-
тельный характер.
Итак, в бакинской нефти с несомненностью обнаружены: бен-
зол, толуол, ксилол -(мета), псевдокумол, мезитилен, дурол и ди-
этил-толуол; возможно также присутствие изодурола, диэтил-бен-
зола и изомил-бензола, а также некоторых гомологов нафталина.
АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ РАЗЛИЧНЫХ НЕФТЕЙ
Ио сравнению с бакинской нефтью ароматика других нефтей
СССР изучена значительно меньше.
Из нефтей кавказских месторождений отметим прежде всего
одну из первых исследованных наших нефтей, нефть Красных
Колодцев (близ Тифлиса), в которой давно уже были обнару-
жены бензол и толуол [б]. В грозненской нефти также обнару-
жено значительное количество бензола и толуола [6], а из по-
гона 160—165° получена сульфомезитиленовая кислота [7], чем
доказано присутствие мезитилена, 1, 3, 5-СвНз(СНз)з.
Особо должна быть отмечена богатая ароматикой пермская
нефть (Чусовские Городки), в которой путем получения из co-
os
ответствующих фракций полинитросоединений доказано присут-
ствие бензола, толуола и мета-ксилола [8].
В ряде других европейских нефтей также обнаружены от-
дельные представители углеводородов ароматического ряда. Так
например, в румынской нефти [9] путем нитрования обнару-
жены бензол, толуол, мета-ксилол, мезитилен и кумол. В гали-
цийской [10], кроме бензола, толуола, мета-ксилола и мезити-
лена, найден также параксилол. Имеются указания на присут-
ствие ароматических углеводородов также в ганноверской [11]
и некоторых других европейских нефтях (Тегернзее в Баварии,
Парма в Италии и др.).
Обращаясь к американским нефтям, мы и здесь должны от-
метить прежде всего недостаточность данных о химической при-
роде находящейся в них ароматики. Первые указания на присут-
ствие в пенсильванской нефти ароматических углеводородов
принадлежат Шорлеммеру [12], который еще в 1863 г. наблю-
дал, что при действии азотной кислоты на соответствующие
фракции этой нефти получаются нитробензол, нитротолуол и
динитротолуол. Позднее данные эти были подтверждены и рас-
ширены другими исследователями [13], причем, кроме бензола
и толуола, в пенсильванской нефти были обнаружены мета-кси-
лол, мезитилен, псевдокумол и цимол. В нефти Охайо (США),
кроме ароматических, найденных в пенсильванской нефти, обна-
ружены также изодурол и амил- или гексил-бешзол, все—обра-
боткой нитрующей смесью [14]. В калифорнской нефти [15]
найдены бензол, толуол, ксилолы (главным образом мета) и, воз-
можно, ютил-бензол, а в одном из образцов калифорнской нефти
(м. Пуэнте) оказалось такое количество нахвалила, что соответ-
ствующая фракция (температура кипения 220—221°) вся за-
кристаллизовалась. Наконец, в канадской нефти были обнару-
жены [16] в форме соответствующих нитросоединений лишь бен-
зол, толуол и два ксилола (пара и мета), высшая же ароматика
пока почти не изучена.
В последнее -время особенно тщательное исследование одного
из образцов мид-континентской нефти (Оклахома, Кей-Коунти,
South Ponca, США) обнаружило в его газолине следующие аро-
матические углеводороды [17]: бензол, толуол, этил-бензол, все
три ксилола (орто, пара и мета), изо-пропил-бензол (кумол), все
три триметил-бензола (гемеллитол, псовдокумол и мезитилен);
кроме того, вероятным оказалось присутствие: норм, пропил-бен-
зола и всех трех метил-этил-бензолов (орто, пара и мета). Выде-
ление этой ароматики производилось фракционировкой, обра-
боткой селективными растворителями (анилин, жидкий серни-
стый ангидрид), вымораживанием, кристаллизацией из димети-
лового эфира и других растворителей, адсорбцией силикагелем;
наконец, для контроля в отдельных случаях проводилось нитро-
вание. Некоторые из перечисленных ароматических углеводоро-
дов были -выделены со степенью чистоты 99,8—99,9%.
Укажем в заключение еще на один образец—на богатую
ароматикой нефть с о. Борнео; в ней найдены [18]: бензол.
'толуол, мета-ксилол, мезитилеи, а также нафталин, а и ₽-метил-
нафталияы и два изомерных диметил-нафталина; все нафтали-
новые углеводороды были выделены здесь в виде соединений
с пикриновой кислотой (пикратов), которые затем разлагались
водяным паром.
Как видно из всего вышеизложенного, выделенными или изу-
ченными из ароматических углеводородов нефти являются лишь
низшие гомологи бензола с температурой кипения ниже 200°.
Поразительно разнообразие этой ароматики: во фракциях с тем-
пературой кипения до 180° в различных нефтях, невидимому,
могут быть обнаружены все теоретически возможные ароматиче-
ские углеводороды, кипящие в указанных пределах. Что касается
ароматики с более высокой температурой кипения, то относи-
тельно нахождения ее в нефти имеются лишь немногие досто-
верные указания. Так например, указывается на присутствие
в некоторых нефтях нафталина и ближайших его гомологов.
Ароматика масляных фракций изучена еще меньше. Имеются
веские основания предполагать, что среди нее содержатся высо-
комолекулярные гомологи бензола, у которых бензольное ядро
связано с длинной цепью состава CHH2n + i нормального строе-
ния. Однако этот вопрос требует еще дальнейшей проработки.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ АРОМАТИКИ В. НЕФТЯХ
И НЕФТЕПРОДУКТАХ ПРЯМОЙ ГОНКИ
Для количественного определения ароматических углеводоро-
дов был предложен целый ряд специальных методов [19]. Одни
из них основаны на определении тех или иных физических кон-
стант углеводородных смесей, например их удельных весов, коэ-
ф'ициентов рефракции и т. д., которые, как известно, весьма
‘ существенно изменяются от большего или меньшего содержания
в подобных смесях бензола и его гомологов. Это — чисто физи-
ческие методы. Другие методы носят скорее химический харак-
тер. Они основаны на количественном выделении ароматических
углеводородов из анализируемой смеси в форме тех или иных
ароматических производных, как-то: сульфокислот, нитросоеди-
нений, формолитов и т. п. Не вдаваясь в подробную характери-
стику всех методов, остановимся лишь на наиболее употреби-
тельных из них.
Физические методы — по удельному весу или коэфициенту
рефракции. Как видно из примеров табл. 26, удельные веса и
Таблица 26
Формула и наименование Темп. кип. Уд. вес dj® Коэф. рефр.
С6Н34 гексан 69° 0,6603 1,3754
СбН12 циклогексан 81° • 0,7788 1,4296
Сейм метнл-циклопентан 72° 0,7474 J-W
С6Нв бензол 80,4° 0,8740 1,6014
95
коэфициенты рефракции парафинов, нафтенов и ароматических
углеводородов довольно резко различаются между собой по ве-
личине.
Хотя с повышением молекулярного веса углеводородов раз-
ница эта заметно сокращается, тем не менее ею с удобством
можно пользоваться для определения ароматики в смесях ее
с парафинами и нафтенами, например в различных нефтяных
фракциях. Для этого необходимо определить удельный вес или
коэфициент рефракции данной углеводородной смеси до и после
удаления из нее ароматики; удаление последней производится
обработкой отдельной пробы углеводорода крепкой серной кисло-
той (см. ниже). Зная повышение удельного веса а или коэфи-
циента рефракции Ъ данной фракции от прибавления к ней 1%
ароматики, для чего легко составить специальную таблицу, легко
рассчитать содержание ароматики А в исследуемой фракции по
формулам:
где d и п — удельный вес и коэфициент рефракции данной фрак-
ции до удаления ароматикщ a di и ni — те же константы после
удаления ароматики.
Простота методики делает ее, особенно при массовом опреде-
лении ароматики, чрезвычайно удобной. Однако но вопросу
о точности получаемых таким образом 'результатов мнения рас-
ходятся.
Следующие формулы позволяют вычислить содержание ароматики в
объемных и весовых процентах по правилу смешения:
У _ 100 (d — dt) 100D (d — dk)
D — di И (D — dt)d ’
где X— содержание ароматики в бензине в объемных процентах; У — со-
держание ароматики в весовых процентах; D—удельный нес чистой аро-
матики; d и di — удельные реса бензина до и после удаления ароматики.
Применение этих формул дает, однако, не вполне точные результаты,
так как при смешении ароматических углеводородов с парафинами и наф-
тенами происходит некоторое расширение смеси. Поправки к этим фор-
мулам, предложенные для получения более точных результатов, оказались
недостаточными, так как величина поправки зависит от природы неарома-
тической части бензина, а именно: при смешении ароматики с парафинами
отклонения от приведенных выше формул оказались наименьшими, при
смешении же ароматики с нафтенами эти отклонения значительно больше.
Ввиду этого здесь оставался один путь: приготовить искусственные смеси из
ароматики и соответствующих очищенных бензиновых фракций различного
удельного веса, т. е. с различным содержанием нафтенов, и определить
для этих смесей опытным путем величину коэфициента а в (зависимости,
от удельного веса неароматической части бензина. То же необходимо ока-
зать и относительно количественного определения ароматики рефрактометри-
ческим методом [20].
Сернокислотный метод. В основе этого метода лежит реак-
ция сульфирования ароматических углеводородов (см. выше).
Так как реакция эта с крепкой серной кислотой легко протекает
96
во многих случаях уже при комнатной температуре, причем
образующиеся сульфокислоты переходят в сернокислотный слой,
то все определение по существу сводится к обработке точно от-
меренного объема А углеводородной смеси серной кислотой и
объемному определению углеводородного остатка В, не вошед-
шего в реакцию. Разность между отмеренными объемами (А — В)
дает суммарное содержание ароматики во взятой пробе.
Практически определение ароматики сернокислотным спосо-
бом производится в сульфаторе (фиг. 21). Нижняя
расширенная часть этого прибора служит для сер-
ной кислоты; при определении она заполняется
в первую очередь до деления 0. Средняя часть
представляет собою бюретку на 25 см3, градуирован-
ную на 0,1 см3: она заполняется углеводородом и
служит для его отмеривания. Наконец, верхняя
расширенная часть сульфатора служит для пере-
мешивания обоих слоев.
Несмотря на кажущуюся простоту сернокислот-
ного метода, отдельные детали его далеко еще не
являются твердо установленными и продолжают
дискутироваться в нефтяной литературе до самого
последнего времени. Это относится к таким вопро-
сом как, например, необходимая и достаточная кре-
пость серной кислоты, температура и время встря-
хивания, пределы применимости метода в зависимо-
сти от состава анализируемой смеси и т. д.
Не подлежит сомнению, что первые три фак-
тора — крепость кислоты, температура и время
встряхивания — связаны между собою самым тес-
ным образом. Известно, например, что сульфирова-
ние некоторых ароматических углеводородов можно
провести даже с камерной серной кислотой при
условии надлежащего нагревания и’непрерывного
удаления воды из сферы реакции [21]. Однако
в этих условиях в реакцию вовлекаются также и
некоторые нафтены. О другой стороны, как показы-
вает опыт, при 0° те же нафтены заметно не реаги-
руют даже с дымящей серной кислотой (з—8%SOs),
которая в этих условиях нацело поглощает арома-
тику. Таким образом определяются различные более 7
Фпг. 21.
Сульфатор.
или менее удовлетворительные вариации сернокислотного метода.
Так например, при сравнительно небольшом содержании арома-
тики для ее удаления достаточно встряхивать углеводородную
смесь с 4 объемами 98 %-ной серной кислоты в течение 45 мин.
при 20° [22]. При более значительном содержании ароматики
(20% и более) предпочтительно пользоваться либо дымящей
серной (5—8% SO3) при 0°, либо 94—98%-ной серной с доба-
влением к ней фосфорного ангидрида [23] при комнатной тем-
пературе (20°). Химический смысл добавления к серной кислоте
ангидридов, серного или фосфорного, очевидно, один и тот же:
7 Эак. 1622. Химия нефти.
97
назначение их — связать образующуюся при сульфировании
воду и тем самым поддержать крепость серной кислоты до конца
на высоком уровне.
Так как 'непредельные углеводороды реагируют с крепкой
серной кислотой столь же легко, как ароматические, то одним
из непременных условий надежности сернокислотного метода
должно быть полное отсутствие в анализируемой -смеси непре-
дельных; в противном случае поглощение серной -кислотой даст,
очевидно, лишь суммарное содержание непредельных и арома-
тики вместе. Если содержание непредельных невелико, как это
имеет место в нефтях и нефтепродуктах прямой гонки, то ре-
комендуется предварительно очистить анализируемый углеводо-
род от непредельных обработкой его равным объемом 80—85%-ной
серной кислоты, не действующей на ароматику. В тех же слу-
чаях, когда содержание непредельных в нефтепродукте значи-
тельно (крэкинг-бензин), применение сернокислотного метода
в описанном выше виде явно недопустимо. К вопросу об ана-
лизе подобного рода нефтепродуктов мы вернемся ниже, в главе
о крекинге (ч. II).
Как уже было -отмечено выше, по мере удлинения боковой
цепи в ароматических углеводородах реакционная способность
их по отношению к серной кислоте несколько падает [24]; это
обстоятельство необходимо иметь в виду при определении аро-
матики сернокислотным методом.
Нитрационный метод. Реакция нитрования также иногда применяется
для количественного определения ароматики. Углеводородная смесь обра-
батывается для этого серно-азотной смесью или дымящей азотной кислотой,
образовавшееся ароматическое ннтрососдинсние собирается и взвешивается;
пересчет дает количество ароматического углеводорода, находившегося в ис-
ходной смеси.
Сопоставляя нитрационный метод с сернокислотным, необходимо отме-
тить- прежде ©сего, что реакция нитрования ароматических углеводородов
проходит, вообще говоря, полнее, чем реакция сульфирования. Особенно
это относится к бензольным смесям, что видно хотя бы из того, что по-
добная смесь, обработанная избытком дымящей сорной кислоты, продол-
жает содержать небольшое количество бензола, который можно открыть
серно-азотной смесью [25]. Зато учет продукта реакции проще при суль-
фировании (ср. выше), чем при нитровании, так как в этом последнем
случае:
1) остаток углеводородной смеси после нитрования всегда меньше, чем
можно ожидать по содержанию в ней ароматики, так как крепкая азотная
кислота всегда более или менее затрагивает также углеводороды парафи-
нового, особенно же нафтенового ряда: таким образом вести расчет по угле-
водороду, не вошедшему в реакцию, здесь невозможно;
2) наряду с мононитросоединениями ароматического ряда при нитро-
вании всегда может образоваться некоторое количество динитропродукта,
причем эти продукты реакции находятся здесь ие только в кислотном, но
и в углеводородном слое;
з) наконец, особенно осложняется расчет ароматики, когда анализируе-
мый продукт содержит не один, а, как обыкновенно, несколько аромати-
ческих углеводородов; в такого рода случаях учет, естественно, должен
быть проведен для каждого ароматического углеводорода отдельно, что воз-
можно лишь после их разделения путем фракционировки, всегда недоста-
точно совершенной.
Все вышеприведенные соображения ограничивают область возможного
98
применения нитрационного способа при анализе нефтепродуктов в лучшем
случае немногими специальными объектами; таков, например, случай бен-
зольно-бензиновых смесей, находящих применение в качестве топлива для
двигателей внутреннего сгорания, и т. п.
Метод анилиновых точек [26]. Этот метод, получивший за
последнее время особенно широкое применение при анализе
нефтепродуктов, основан на определении температуры взаимного
растворения анилина в углеводородной смеси до и после удале-
ния из нее ароматики.
В специальной пробирке смешивают 3 сл3 испытуемого нефте-
продукта, например бензина, с равным объемом чистого и су-
хого, свежеперегнанного анилина и осторожным нагреванием на
водяной бане переводят анилин в раствор. За ходом температуры
следят при этом по термометру с делениями на 0,1°, вставлен-
ному в пробирку, а для более равномерного нагревания окру-
жают пробирку другой пробиркой — муфтой. Когда содержимое
пробирки делается совершенно прозрачным и однородным, на-
гревание прекращают и, давая раствору медленно охлаждаться
при внутреннем перемешивании, наблюдают температуру, при
которой вследствие начавшегося выделения анилина в растворе
появляется белая муть. Эта температура Т1 называется анилино-
вой точкой исходного нефтепродукта. Далее берут вторую пробу
того же нефтепродукта (30—40 см3), обработкой серной кисло-
той удаляют из нее всю ароматику и в совершенно аналогич-
ных условиях определяют вторую анилиновую точку Т
нефтепродукта, освобожденного на этот раз от ароматических
углеводородов. Разность {Т—Т1) представляет собою депрессию
анилиновой точки, зависящую от содержания в первой пробе
ароматики; содержание последней в объемных процентах опре-
деляется затем по таблице коэфициентов, составленной на основе
определения величины депрессии для специально приготовлен-
ных бензольно-бензиновых смесей с различным содержанием
в них ароматического углеводорода.
Как показывает опыт, депрессия анилиновой точки зависит
от процентного содержания ароматических углеводородов и
в известной степени также от их молекулярного веса. Поэтому
при определении ароматики в бензине его приходится разбивать
путем фракционировки на три основных фракции: бензольную,
(температура кипения 60—95е), толуольную (температура кипе-
ния 95—122°), ксилольную (температура кипения 122—150°) и
фракцию выше 150°. Определение ароматики производится
в каждой из этих фракций отдельно. С другой стороны, природа
предельно-нафтеновой части бензина не оказывает заметного
влияния на результаты определения ароматики анилиновым, ме-
тодом. Все эти особенности данного метода необходимо учиты-
вать также при составлении таблицы коэфициентов, что, есте-
ственно, является особо ответственной частью методики.
В литературе вопроса можно найти’ несколько таблиц анили-
новых коэфициентов [26]. Приводимая ниже табл. 27 составлена
но методу, описанному выше, для различных фракций сурахан-
7* 99
ского и грозненского бензинов с различными добавками к этим
фракциям ароматических углеводородов, соответствующих им по
температуре кипения. Таблица составлена по формуле:
р = к(Т— Г1),
где (у—Т1) — депрессия анилиновой точки от добавки к бен-
зину р свесовых процентов ароматики; коэфициент же к пред-
сгавчяет собою, очевидно, процентное содержание ароматики, от-
вечающее депрессии анилиновой точки в 1° [27].
Таблица 27
Коэфициенты для определения ароматики анилиновым методом
(по К А. Робинвон)
Процентное содержа- Темп- кип. фракций бензина
ние ароматики во фракции 60—96° 95—122° 122—150° 150-200°
Коэфициенты
Около 1 1,00 1,00 1.00 1.00
3- б 1,17 1.17 1,21 1,26
5-10 1,16 1,15 1.21 1,28
10—20 1,12 1.12 1,19 1,29
20—30 1,09 1,09 1,15 1,27
80—40 1.С5 1,05 1,09 1.22
40—60 1,00 1.00 1,04 1.17
50-60 0,96 0,96 0,99 1,12
60—70 0.91 0.91 0,94 1,06
Существуют два видоизменения анилинового метода. Одни авторы опре-
деляют температуру взаимного растворения анилина и углеводорода, взятых
в ранных объемах, т. е. как ото описано выше. Очевидно, однако, что при-
нятое в этом случае объемное отношение двух смешивающихся жидкостей
(1:1) совершенно произвольно и получаемая таким образом анилиновая
точка отнюдь не должна рассматриваться как некоторая константа, харак-
теризующая данную пару жидкостей.
Действительно, при изменении отношений объемов смешиваемых жид-
костей можно наблюдать следующий ход изменения температуры взаимного
растворения: сначала, по мере прибавления компонента, взятого в меньшем
объеме, температура взаимного растворения повышается; при некотором
объемном отношении компонентов она становится максимальной, при даль-
нейшем же увеличении концентрации того же компонента температура
взаимного растворения вновь начинает постепенно понижаться. Таким обра-
зом для данной пары жидкостей, например анилин — углеводород, имеется
некоторое объемное отношение, при котором температура взаимного раство-
рения является наивысшей. Эту температуру называют критической темпера-
турой растворения, и некоторые авторы пользуются ею как константой
при определении ароматики в смеси с нафтенами и парафинами аналогично
анилиновым точкам, определяемым по вышеописанному способу. В табл. 28
приведены определенные различными способами анилиновые точки дева-
роматизированного бакинского бензина «Калоша» и того же бензина после
прибавления к нему различных количеств ароматического углеводорода.
100
Таблица 28
Анилиновые точки, определенные разными методами
Бенз, дезаро-
мат ....
Бенз.4-8% аро-
мат ....
Бенз. -|-13,9%
аромат . .
Бенз. 4- 46,2%
аромат . .
50,6 50,7 50,6 50,5 — 50,3 — — —
43,0 43,4 44,0 44,144,2 44,3 44,4 44,3 44,1
— — — 37,6 38,3 38,7 38,9 — 89,6
0 — - 4.7 — 7,9 — 9,0 —
— - — 50,7
— — — 44,4
— 39,6 89,4 89,6
9,8 9.9 9,7 9,9
50,5
44.1
37,6
4,7
Как видно из примеров табл. 28, разница между температурой взаим-
ного растворения анилина и нефтяных углеводородов, взятых в отношении
1:1, и их критической температурой растворения, небольшая при малом
содержании, ароматики, постепенно увеличивается по мере увеличения кон-
центрации последней и при содержании ее около 60% может достигать
нескольких градусов. Отсюда ясно, что должно быть соблюдаемо полное
соответствие' между методикой определения анилиновых точек при анализе
углеводородной смеси и тем методом, на основе которого была составлена
•таблица перевода депрессии анилиновой точки на процентное содержание
:ароматнки (т., е; табл. 27). В противном случае в результате получаются
•ошибки, которые; Например, при содержании ароматики около 80% могут
достигать 5% и более.
Ввиду того что метод анилиновых точек во многих странах
(в том числе в СССР) принят как стандартный, нелишне отме-
тить, что при определении значительных количеств ароматики
юн не дает существенных преимуществ перед сернокислотным
методом. Это ясно уже из того, что основная операция — коли-
чественное удаление ароматики с помощью крепкой серной ки-
слоты—одна и та же в обоих методах; учет же результатов
этой операции при сернокислотном методе несравнимо проще и
надежнее, чем при методе анилиновых точек. То же можно ска-
зать и о различных модификациях анилинового метода; таков,
например, нитробензольный метод, в котором вместо анилина
применяется нитробензол [28] и некоторые другие. В тех, од-
нако, случаях, когда содержание ароматики в анализируемом
.продукте невелико (например 1—2%), анилиновый метод вслед*
ствие его высокой чувствительности следует признать особенно
•подходящим- Таковы, например, случаи анализа бакинских бен-
зинов прямой гонки и др. Наконец, при полном групповом ана-
..лизе бензинов прямой гонки, когда вслед за ароматикой прихо-
.дится иметь дело с определением нафтенов и парафинов, анили-
новый метод, как будет показано ниже в соответствующих гла-
вах, также получил широкое заслуженное признание и приме-
нение.
101
Прл применении анилинового метода следует иметь в виду его особую
чувствительность к присутствию в анализируемом бензине разного рода
примесей. Так, присутствие в бензине серы и сернистых соединений должно
исключать применение анилинового и ему подобных методов (.например
нитробензольного); это обстоятельство приходится иметь в виду особенно
при работе с ъысокосернистыми бензинами, а также в случае применения
полухлористой серы для удаления непредельных из крэкинг-бензина.
Формолнтовый метод. Уже давно было сделано наблюдение [29], что
под влиянием серной кислоты ароматические углеводороды образуют с фор-
мальдегидом или его метиловым эфиром, метилалем, разнообразные про-
дукты конденсации. Воспользовавшись этой реакцией для характеристики
нефтей и нефтепродуктов [30], Наскоков предложил назвать порошкообраз-
ные аморфные вещества, получающиеся в этом случае, «формолитами»,
а выход формолита на 10(5 с.и8 (позднее —на 100 г) нефти или нефтепро-
дукта— их «формолитовым числом».
Так как ни парафины, ни нафтены не реагируют в аналогичных усло-
виях с формальдегидом, то образование формолптов, казалось бы, могло
служить для специальной характеристики нефти пли нефтепродукта на
содержание в них ароматических систем. Однако приходится констатиро-
вать, что количественная сторона формолитовой реакции до сих пор должна
считаться недостаточно разработанной и что в зависимости от условий
реакции, промывки и пр. выход на фюрмолит из одного и того же нефте-
продукта может колебаться в весьма широких пределах. Несколько лучшие
результаты получаются с метилалем; однако и в этом случае не только
выход формолита, но и его характер зависят от целого ряда факторов [31] г
концентрация и количество серной кислоты, концентрация ароматики, ко-
личество метилаля и т. д. Таким образом приходится придти к заключению,
что формолитовый метод может служить пока не для количественной,
а лишь для качественной характеристики нефтей и нефтепродуктов.
Химизм формолитовой реакции нельзя считать окончательно выяс-
ненным. Невидимому, первая фаза этого процесса конденсации состоит в
образовании углеводорода ряда дифенилметана [32]. В пользу такого тол-
кования говорит действительное нахождение дифснил-метана среди продук-
тов конденсации бензола с метилалем, особенно же — чрезвычайно легкое
образование димезитил-метана при конденсации симметричного триметил-
бензола (мезитилена) с формальдегидом в присутствии серной кислоты:
2C9Hi2 + СН2О = С9НП СН2 - С9НП + Н2О.
Надо думать, что последующий ход конденсации, приводящий в конеч-
ном итоге к аморфным' формолитам, идет по такой же схеме; однако даль-
нейших опытных данных в пользу подобного толкования этой реакции до
сих пор не получено.
КОЛИЧЕСТВЕННОЕ СОДЕРЖАНИЕ
АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ В РАЗЛИЧНЫХ НЕФТЯХ
Как видно их вышеизложенного, ароматические углеводороды
выделяются из нефти и ее погонов с помощью столь сильных
реагентов, как крепкая серная или азотная кислота. Так как
в аналогичных условиях некоторые нафтены способны давать
такие же ароматические производные (ср. выше), то естественно
возникает вопрос, содержится ли ароматика в нефти в готовом
виде и не образуется ли она в результате указанной переработки
нефтяных погонов или даже .просто при перегонке нефти [33]
как продукт пирогенетического разложения углеводородов дру-
гих рядов. Для освещения этого вопроса можно привести следу-
ющее соображения:
1. Образование сульфокислот наблюдается для погонов, полу-
ченных из сырой нефти при ее обработке легким паром или при
102
перегонке в вакууме с обогревом бани ниже 100° [34]. Это гово-
рит против предположения, что ароматические углеводороды
нефти получаются в результате как бы пирогенетического раз-
ложения нафтенов при перегонке нефти.
2. Количество сернистого газа, развивающегося при обра-
ботке нефтяных погонов дымящей серной кислотой, далеко не так
значительно, как должно было бы быть, если бы ароматические
углеводороды получались за счет окисляющего действия серной
кислоты на нафтены.
3. Ароматические углеводороды, полученные из сульфокислот,
по температуре кипения всегда соответствовали исходному по-
гону нефти, тогда как между температурами кипения нафтенов,
и соответствующих им гомологов бензола наблюдается довольно
значительная разница
4. Наконец, как показывает опыт, при обработке различных
погонов нефти дымящей серной кислотой достаточно переменить
кислоту три раза: третья порция уже почти не содержит суль-
фокислот; между тем, нефтяные погоны, даже трижды обрабо-
танные серной кислотой, продолжают содержать нафтены раз-
личных рядов и в том числе ряда циклогексана. Очевидно, та-
ким образом, что ароматические сульфокислоты получились из
этих погонов не за счет нафтенов.
Итак, ароматические углеводороды находятся в нефти в юто-
вом виде, а не образуются в процессе той или иной ее перера-
ботки-. С количественной стороны в этом! отношении между
нефтями различных месторождений наблюдается чрезвычайно
большое разнообразие. Это видно, например, из табл. 29, в кото-
рой приведены соответствующие данные для [важнейших нефтей
•СССР и некоторых американских по данным ГрозНИИ [35].
Определения производи тись для нефтяных погонов с темпера-
турой- кипения в пределах 30—50 , что позволяет судить о рас-
пределении ароматики в исследуемой нефти по различным ее
фракциям (1-я строка табл. 29 при каждом образце). Зная со-
держание отдельных фракций в данной нефти (2-я строка),
можно путем пересчета легко получить содержание тех пли
иных концентрирующихся в отдельных фракциях ароматических
углеводородов по отношению ко всей нефти. Метод определе-
ния — изменение удельного веса фракции до и после обработки
ее серной кислотой. В табл. 29 приводится материал по содер-
жанию ароматики для нефтяных фракций с температурой кипе-
ния только до 300° как наиболее надежный; однако и на него
следует смотреть как лишь на первый опыт сравнительной ха-
рактеристики различных нефтей на содержание ® них арома-
тики, что явствует уже из критической оценки современных ме-
тодов определения ароматики в нефтях и нефтепродуктах.
Как видно из табл 29, среди нефтей ССОР’ наиболее бедны
ароматикой бакинские нефти, особенно сураханские и Старого
района (Балаханы, Раманы, Сабунчи); Биби-Эйбат и особенно
Бинагады уже заметно богаче ароматикой. К апшеронским неф-
тям первого рода близко подходит по содержанию ароматики
Таблица 29
Содержание ароматических углеводородов в нефтях
Температура кипения фракций
в —- а й Месторождение Уд вес (15°) 60-95° сч см 7 о о о ю 7 см ooos-osi о О ю см 1 о со ooos—oss
1 Сураханы 0,848 1,5 1 4,5 6 5,5 17 7,9 18 10,1 16 10,6 I II
2 3 Балаханы Балаханы — Раманы — 0,876 1,7 1 2,8 6 2.8 12 4,9 15 9,9 19 8.0 1 II
Сабунчи 0,867 11 1 3,0 4 4.3 16 9,3 19 10.9 20 10,6 I 11
4 Бинагады 0 921 4 0,5 4 1.1 8 1.8 17 4,9 27 6,3 34 12,8 I II
5 Биби-Эйбат 0,865 3 2,6 3 4,8 7 6,9 12 12,0 22 13.3 27 7,6 I II
6 7 Грозный, параф, фонтан Грозный, слабо пара- 0,844 ык». со 6 3,5 9 4,4 14 8,7 18 8,7 17 11,0 I 11
8 фин. фонтан Грозный, беспарафин. 0,849 О 4,1 9 4,6 13 4.7 17 7,8 18 8,8 21 7,5 I II
легкая 0,860 5 4,2 9 5,4 13 6,0 18 9,3 24 8,3 27 5,5 I II
9 Вознесенская 0,900 3 0,2 2 0,8 3 19 11 4,0 22 8,1 25 10,2 I II
10 Майкоп 0,848 8 7,0 13 6,0 17 6,6 25 11,5 28 8,9 31 8.0 I II
11 Калужская 0,955 — — —- — 10 4,1 15 3,7 I II
12 Доссор (Эмба) 0,862 4 0,6 1 2,0 4 3.1 7 4,8 9 14,2 13 16,2 I II
13 Пермская 0 941 20 5,1 51 4,5 62 7.0 60 5.0 53 5,3 56 3,4 I II
14 Тонкава (Оклахома). . 0,821 6 4,6 9 6,5 12 5,9 20 М 22 13,4 24 9.0 I II
15 Девинпорт (Оклахома) 0,796 5 6,3 9 7,4 12 7,3 (16) (17) 18 8,0 I II
16 17 Мексиа (Тексас) . . . Хёнтингтон-Бич (Кали- 0,845 30 1,5 22 1,9 20 2,7 16 8,9 13 13,0 13 14,4 I II
форния) 0,897 4 2,4 6 4,4 10 8,5 17 6,6 25 6,6 28 6,2 I 11
I — процент ароматики во фракции.
II — процентное содержание фракции в нефти.
104
эмбенская нефть (Доссор). Значительно более богаты аромати-
ческими углеводородами из нефтей СССР — грозненские нефти,
особенно слабопарафинистые и беспарафиновые, но первое ме-
сто в этом отношении среди кавказских месторождений принад-
лежит Майкопу. Наконец, совершенно особое положение по со-
держанию ароматики среди наших нефтей занимает пермская
нефть (Чусовские Городки). Столь богатые ароматическими
углеводородами нефти, вообще говоря, чрезвычайно редки; в ка-
честве аналогичных примеров можно привести нефти Зондских
островов (Суматра, Борнео, Ява), некоторые нефти о. Сахалина
(Нутово), некоторые американские нефти, например месторожде-
ния Мексиа (Тексас) и другие. Что касается других американ-
ских нефтей, то для большинства из них содержание ароматики
определяется величинами примерно того же порядка, как для
наших апшеронских и грозненских нефтей. Из европейских
нефтей выделяются значительным содержанием ароматических
углеводородов некоторые галицийские и румынские нефти.
Та же табл. 29 показывает, что распределение ароматики по
различным фракциям одной и той же нефти крайне неравно-
мерно. В легких фракциях содержание ароматики обычно не ве-
лико; для более высоких фракций оно постепенно возрастает,
достигая для фракций 250—300° различных нефтей в среднем
20—25% и продолжая возрастать с дальнейшим повышением
температуры кипения фракций, в некоторых случаях до 40% и
выше. Наблюдались, однако, и прямо противоположные примеры,
когда содержание ароматики в различных фракциях одной и
той же нефти с повышением температуры их кипения не воз-
растало, а наоборот, уменьшалось. Таков, например, случай
тексасской нефти месторождения Мексиа (табл. 29, №16).
Большой практический интерес представляет характеристика
с точки зрения содержания ароматики некоторых нефтепродук-
тов и прежде всего бензинов и керосинов из различных нефтей.
Для советских бензинов и керосинов данные эти 'на основе
главным образом определений ГрозНИИ собраны в табл. 30;
дополнением к ней служит табл. 31 с данными содержания аро-
матики в пермском бензине [36].
Как показывают последние две таблицы, наши бензины, по
содержанию в них ароматики проявляют весьма большое разно-
образие. В полном соответствии с данными табл. 29, наиболее
бедными ароматикой (1,4—2%) являются эмбенский (Доссор)
и некоторые бакинские бензины (Сураханы, Балаханы). Следую-
щую группу (4—6% ароматики) составляют бензины из биби-
зйбатской, бинагадинской и грозненской парафинистой нефти.
Далее следуют бензины из грозненской слабопарафинистой и
беспарафиновой нефтей (9—13% ароматики) и особенно майкоп-
ский бензин (до 16% ароматики). Наконец, на первом месте по
содержанию ароматики должен быть поставлен бензин из перм-
ской нефти, содержащий только до 150°, т. е. суммарно бензола,
толуола и ксилола, свыше 31% ароматики.
105
Таблица 30
Содержание ароматических углеводородов в бензинах и керосинах
СССР
Бензины Керосины
Месторождения о ф л-1 О О fr- ee О ф ф Г S й а
с о © . % СО >» сз
***-* к «©’ нефти Уд. (15°) До 1 °/о И п ° РЙ И « а. Бен % Тол °/о • о Йе О о о **" о
1 2 Сураханы .... Балаханы .... 0,769 0,756 30 • 30 175 175 2 2 (0,00 0 0,6 0,4 0,824 0,834 30 30 17 15
3 Биби-Эйбат . . . 0,742 30 175 4 0,6 1,2 0,833 30 19
4 Бинагады .... 0,747 30 175 5 0,8 1,8 —•
5 Грозный, пара- фин. нефть . . — 65 130 4 — — — —
(авио)
6 Грозный, пара- фин. нефть . . 0,722 (легк.) 40 175 6 0,6 1,1 0,800 25 16
7 Грозный, пара- фин. нефть . . 0,741 20 200 8 0,810 0 18
(тяж.)
8 Грозный, слабо- параф. нефть . 0,730 40 175 9 1,2 2,0 0,807 30 19-
(легк.) «
9 Грозный, слабо- параф. нефть . 0,746 (тяж.) 20 200 11 — — 0,818 0 19-
10 Грозный, беспа-
рафпн. нефть (нижн. пласты) 0,730 (легк ) 40 175 8 1,0 2.1 0,ь20 30 •23-
11 Грозный, беспа-
рафии, нефть (нижн. пласты) 0,746 20 200 11 — —— - 0,838 0 £5-
(тяж.)
12 Грозный, беспа-
рафии, нефть (верхн. пласты) 0,742 (легк.) 40 175 11 0,9 2,0 0,830 30 26
13 Грозный, беспа- «ъ
рафин. нефть (верхи, пласты) 0,761 (тяж.) 20 200 13 — — 0,848 0
14 Майкоп — 65 130 10 -— — — —
(авио) 0,824 30 28.
15 Майкоп 0,751 20 200 16 1.5 2,3
• (тяж.) 20
16 Доссор (Эмба). . 0,778 25 200 1,4 — — 0,822 10
росинов колеблется сравнительно меньше. Керосины из бакин-
ских и грозненских парафинистой и слабопарафинистой нефтей
содержат от 15 до 19% ароматики, керосины же из грозненской
беспа рафиновой и майкопской нефтей — от 23 до 29%. Особо»
должны быть отмечены керосины из доссорокой и пермской неф-
106
Таблица 31
Содержание ароматики в пермском бензине
Наименование фракции Темп. кип. фракции Содержание ароматики в %
на фракцию' на бензин на нефть
Бейзольная 63— 93° 13,6 2,7 0,4
Толуольная 93—120° 35,2 12,6 2,0
Ксилольная 120—149° 59,7 16,0 2,5
тей: в доссорском керосине содержание ароматики оказывается
всего 10%, тогда как в керосине из пермской нефти оно, судя
по данным табл. 29, должно приближаться к 40%.
ПРИМЕНЕНИЕ И ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ АРОМАТИЧЕСКИХ
УГЛЕВОДОРОДОВ НЕФТИ
Ароматические углеводороды нефти могут быть использованы
в двух направлениях: 1) или после их выделения из нефти как
сырье для дальнейшей химической переработки на те или иные
соединения ароматического ряда; 2) или без выделения из •неф-
тяных погонов как составная часть последних, придающая им
известные специальные качества.
1. Как известно, каменноугольный деготь, являющийся
•основным сырьем для получения ароматических углеводородов,
содержит не более 1,5—2% бензола и его ближайших гомологов.
Хотя, как было показано, содержание этих основных углеводо-
.родов ароматического ряда во многих бензинах значительно
больше, тем не менее получение их из бензина в чистом виде
значительно труднее, чем из каменноугольного дегтя. Эти труд-
ности обусловливаются не только значительно более сложным со-
ставом соответствующих бензиновых погонов, но и специальным
характером тех "компонентов (парафины, нафтены), которые,
являясь в нефти спутниками ароматических углеводородов, не
поддаются удалению путем какой-либо несложной химической
обработки. Несмотря на это, в вопросе о получении ароматиче-
ских углеводородов из сырых нефтей в настоящее время имеется
-большой опытный материал и притом не только лабораторного,
но и заводского характера.
Еще до войны на одном из заводов в Германии стали выде-
лять из богатой ароматикой нефти с о. Борнео толуол путем
переработки ее толуольной фракции на нитротолуол. В 1914 г.
во Франции был построен завод для переработки толуола из той
же нефти на тринитротолуол (тротил), являющийся, как из-
вестно, одним из важнейших бризантных взрывчатых веществ,
а в 1915 г. было приступлено к постройке двух таких же заводов
у нас 'для аналогичной переработки майкопской нефти [37],.
107
Первая стадия получения тротила из нефти заключается
в выделении из нее толуольной фракции. Эта задача осуще-
ствлялась на наших заводах путем повторной перегонки газо-
лина или лигроина на колонной установке и дальнейшей кон-
центрации толуольной фракции путем дополнительной ее раз-
гонки. Таким образом получалась концентрированная толуоль-
ная фракция с содержанием от 20 до 32% толуола, поступавшая
далее в нитраторы. Здесь фракция подвергалась в течение
5—6 час. обработке серно-азотной смесью при энергичном пере-
мешивании и температуре не свыше 50° в начале и не свыше;
65° в конце реакции. После отделения кислотного слоя продукт
подвергался перегонке с ларом; не вошедший в реакцию бензиа
при этом отгонялся, а нитротолуол оставался в кубе, предста-
вляя собою вещество, вполне пригодное к выпуску с завода без.
какой бы то ни было дополнительной очистки в качестве полу-
продукта для дальнейшей переработки на тротил.
Для иллюстрации приводим в извлечении баланс нитротолуолового
завода по переработке 120 000 кг лигроина.
Взято
Лигроина............. 120000 кг
Нитрогруппа.......... 653 „
Получено
120653 кг
Нитротолуола ....... 1914 г
Легкого бензина .... 27 528
Тяжелого „ ...... 82902 „
Потери ................. 8309 „
120 653 кт
Итак, одно из направлений утилизации нефтяной ароматики
заключается в ее извлечении из соответствующих нефтяных по-
гонов путем их обработки подходящим реагентом с получением
соответствующего полупродукта ароматического ряда. Кроме
азотной кислоты, для аналогичной цели может быть применима,
очевидно, также серная кислота с последующей переработкой
образующихся сульфокислот на фенолы. В качестве сырья для
переработки нефтяной ароматики на нитросоединения или
сульфокислоты можно особенно рекомендовать соответствующие
погоны пермской нефти.
Значительно шире может’быть поставлено получение арома-
тических углеводородов из нефти после ее предварительного пи-
ролиза. Этот вопрос будет рассмотрен ниже (ч. II).
2. Как составная часть нефтепродуктов ароматические угле-
водороды нефти имеют особенно важное значение для повышения
антидетонационных качеств топлива для двигателей внутреннего
сгорания [38].
Горение смеси горючего с воздухом в цилиндрах двигателей
внутреннего сгорания, нормально протекающее сравнительно
спокойно и распространяющееся со скоростью нескольких метров
в секунду, приобретает в некоторых случаях характер взрыва,
распространяющегося с громадной скоростью (2000—3000 м/сек).
Это явление получило название детонации; оно ощущается при
работе мотора появлением особого рода стуков и влечет за собок>
108
не только резкое понижение мощности двигателя, но и усиленную
его изнашиваемость. Отсюда понятно, что изучение явления де-
тонации и борьбы с нею является одной из важнейших задач
технологии топлива двигателей внутреннего сгорания. Актуаль-
ность этой задачи особенно определилась за последнее время,
когда в конструировании всех двигателей внутреннего сгорания
легких типов, т. е. авиационных, автомобильных и тракторных,
отчетливо выявилась тенденция перехода к моторам с повышен-
ной степенью сжатия как более экономичным; между тем, с по-
вышением степени сжатия при данном топливе возрастает склон-
ность его к детонации.
Наряду со степенью сжатия горючей смеси важнейшим фак-
тором, определяющим появление стуков в моторе, является хими-
ческий состав применяемого топлива. Специальные исследования
показали, например, что углеводороды тяжелого бензина и керо-
сина более склонны к детонации, чем легкие углеводороды, обра-
зующие хотя бы газовый бензин. Сравнение в том же направле-
нии углеводородов разлшптых рядов привело к следующим важ-
ным результатам: наименее склонными к детонации оказались
углеводороды ароматического ряда и некоторые парафины
с сильно разветвленным углеродным скелетом (например триме-
тил-изобутил-метанц далее следуют нафтены и непредельные
углеводороды; па последнем же месте в этом отношении должны
быть поставлены парафины с нормальной цепью углеродных
атомов.
Громадное различие углеводородов различных рядов в отно-
шении их склонности к детонации указало на один из методов
борьбы с детонацией бензинов, богатых парафинами, вроде, на-
пример, нашего грозненского. Чтобы повысить антидетонацион-
ные качества такого рода бензинов, к ним добавляют достаточ-
ное количество ароматики в виде моторного бензола, т. е., в основ-
ном, смеси, бензола, толуола и небольшого количества ксилолов.
Такое добавление широко практикуется в настоящее время во
всех странах и, смотря по надобности, может составлять от 30
до 60% от бензина прямой гонки. Тем самым определяется роль
и значение ароматических углеводородов нефти как одной из
важнейших составных частей топливных бензинов, понижаю-
щих детонационные свойства последних. Такие богатые арома-
тикой нефти, как пермская и ей подобные, представляют в этом
отношении, естественно, особый интерес и значение как сырье
для получения антидетонирующего топлива.
Антидетонационные качества бензина характеризовались по
Рикардо следующим образом. Прежде всего на специальном
моторе с переменным сжатием определяли, при какой компрес-
сии данный бензин начинает детонировать. Далее, путем обра-
ботки крепкой серной кислотой освобождали пробу исследуе-
мого бензина от ароматики, а затем прибавляли к нему посте-
пенно столько толуола (бензола), чтобы получилась смесь, по
своим детонационным свойствам вполне соответствующая исход-
ному бензину. Процентное содержание толуола (бензола) в та-
109
кой смеси называется «толуолышм (бензольным) числом» ис-
следуемого бензина; оно может характеризовать склонность бен-
зина к детонации. Исследования последнего времени показали,
однако, что зависимость между содержанием толуола (бензола)
в бензине и склонностью последнего к детонации — чрезвычайно
сложная, ввиду чего для характеристики детонационных
свойств бензина стали пользоваться другими смесями перемен-
ного состава, например смесью нормального гептана с трпметил-
пзобутпл-метапом; к этому вопросу мы еще вернемся в главе
о парафиновых углеводородах, а также в специальном разделе
об антидетонаторах (ч. III, гл. III).
3. Следует отметить в заключение, что более или менее зна-
чительное содержание ароматики в нефтепродукте в некоторых
случаях может оказаться не только бесполезным, но даже вред-
ным и нежелательным для специальных целей его применения.
Один из таких -случаев — это некоторые специальные сорта
бензинов, применяемые в различных отраслях промышленности
в качестве растворителей. Несколько повышенная токсичность
ароматических углеводородов по, сравнению с углеводородами
других рядов заставляет во избежание случаев массового отра-
вления рабочих на производстве ограничивать содержание аро-
матики в такого рода бензинах весьма низкими пределами. Так
например, для бензина «Калоша», применяемого в резиновой
промышленности, содержание ароматики ограничено у нас нор-
мой «не более 1,5%»; бензины же с большим содержанием аро-
матических углеводородов в производство не допускаются.
Другой случай, когда повышенное содержание ароматики
нежелательно, это — случай осветительного керосина. Не под-
лежит сомнению, что до известных пределов содержание аро-
матики и вообще углеводородов, богатых углеродом, повышает
качество осветительного керосина, увеличивая силу света и по-
нижая -расход керосина на 1 свсчу/час. Однако содержание
такого рода углеводородов («карбюров») не должно превышать
известной нормы; в противном случае пламя керосиновой го-
релки приобретает неприятный красноватый' цвет, легко мигает
и коптит. Иногда эти нежелательные явления приобретают на-
столько резко выраженный характер, что приходится прини-
мать специальные меры для понижения содержания ароматики
в осветительном материале. Таков, например, известный случай
с керосином из некоторых румынских нефтей (Буштенари, Мо-
рели), которые смогли получить широкое применение в каче-
стве осветительного материала лишь после специальной очистки
от избытка ароматики жидким сернистым ангидридом по спо-
собу Эделеану (см. ч. III).
ЛИТЕРАТУРА
1. Веисгербер Р., Химическая технология каменноугольного дегтя, М.
(1929).; ТТоханский 1Г. П., Основы коксования и улавливания
побочных продуктов, Л. (1930).
2. См. курсы органической химии Реформатскою С. Н., Каррера IL,
Фаворского А. К., Шорыгина IT. П. и др.
110
3- Маркоеников и Оглоблин, „Ж.Р.Х-О.* 15, 237 (18Q3); Дорошенко, „ЖР.Х.О."
17, 285 (1885); Маркоеников, „Ж.Р.Х.О." 30, 73 и др. (1898).
4. Курбатое А. „Ж.Р.Х.О." 15, 129 (1883).
5. Бейлъштегт и Курбатов, „Ж.Р.Х.О." 15, 5 (1883).
6. Харичков. „Ж.Р.Х.О." 8, 655 (1899); Маркоеников, „Ж.Р.Х.О." 34, 635 (1902).
7. Коновалов М. и Плотникова, „Ж.Р.Х.О.* 33, 50 (1901).
8. Наметкин и Шахназарова, неопубликованное наблюдение.
9. Рот Р., „Ch. Zbl"., 1908, 1, 459; 1907, 11, 556.
10. Freund, „Lieb. Ann." 115, 19 (1860); Lachowicz, „Lieb. Ann." 220, 188
(1883); „Вег.* 18, 2663 (1883); Pawlewski В., „Вег.- 18, 1915 (1885);
Zaloziecki R, „Anzeig. Akad. d. Wissensch. Krakau", 1903, 22-8;
Zaloziecki R. и Frasch. „Вет.и 35, 886 (1902); Zaloziecki R. и Haus-
mann, „Вег." 85, 1584 (1902).
11. Kramer G. и Bottcher, „Вег.* 20, 595 (1887).
12. Schorlemmer, „Lieb. Ann.* 127, 311 (1863).
13. Бейлъштейн и Курбатов, „Ж.Р.Х.О." 15, 5 (1883); Fortey Е., ,J. Chem.
Soc." 73, 932 (1898); Mabery, „Am. Chem. J." 18, 43 (1896); Engler. C.,
„Вег." 12, 2187 (1879); 18, 2234 (1885).
14. Mabery Ch., „Am. Chem. J.' 17, 713 (1895); 18, 215 (1896).
15. Mabery Ch., „Am. Chem. J." 19, 797 (1897); Mabery a. Hudso.i E. „Am.
Chem. J.* 25, 283 (1901).
16. Mabery Ch, „Am. Chem. J.* 18, 43 (1896); 19, 419 (1897).
17. Литературу вопроса см. Rossini F. JD., „Refiner" 16, № 11, 545 (1937).
18. Jones H. a. Wootton H., „J. Chem. Soc.* 91, 1146 (1907).
19. Добрянский А. Ф., Анализ нефтяных продуктов, М.-Л. (1925); 2-е изд.
(1932).
20. Химический состав нефтей и нефтяных продуктов „Труды ГрозНИИ*,
2-е изд.. М.-Л. (1935). ’
21. Meyer Hans, „Lieb. Ann." 433, 327 (1928).
22. Inst, of Petroleum Technologists, Standard Methods of Testing Petroleum
and its Products, Sec. Ed., стр. 20, London (1929).
23. Kattwinkel, „Brennstoffchem.* 8, № 22, 353 (1927).
24. Тиличеев, „Нефт. хоз." XIX, 11, 586 (1930).
25. Маркоеников, „Ж.Р.Х.О.* 80, 67 (1898).
26. Tizard a. Marshall, „J. Inst. Petr. Techn.* 8, 578 (1922); Ormandy a. Craven,
Ibid. 12, 89 (1926); Тиличеев. Сборник „Химический состав нефтей
и нефтяных продуктов", ОНТИ (1935); Робинзон Е. А., „Нефт.
хоз." № 4 (193и); Робинзон Е. -4. и Покровская Е. С., Опреде-
ление группового состава бензинов, изд. Акад, наук СССР, М.-Л.
(1986).
27. Робинзон Е. А., Работа в печати.
28. Erskine, „Qil a. Gas J.* 144, 19 (1926); Добрянский и Хисин „Журн-
прикл. химии" 111, 143 (1930).
29. v. Ваеуег А.. ,Вет.а 5, 1094 (1872); 6, 220 (1873); 7, 1180 (1874) и др.
30- Настюков, „Ж.Р.Х.О." 36, 881 (1904) и другие работы того же автора.
31. Герр, „Chem. Ztg." 84, 893 (1910), Добрянский, „Нефт. хоз." XII, 57 (1927).
32. V. Ваеуег А., 1. с.
33. Aschan О., „Lieb. Ann.* 824, 1 (1902).
34. Pom Р., „Monit. petr. Roum." 569 (1907).
85. Химический состав нефтей и нефтяных продуктов, „Труды ГрозНИИ',
М.-Л. (1931).
36. Наметкин, „Нефт. хоз." XVII, Ха 7, 79 (1929).
37. Юрков, „Нефт. хоз, VI, №4, 661 (1924).
38. Engler С. Lnd Hofer И., Das Erdol, т. IV, 2-е изд. (1930); Памотин,
„Нефт. хоз." 20 № 1, 102 (1931); Клшерман, „Азерб. нефт. хоз."
И, № 2—3, 72 (1931).
ГЛАВА V
НЕФТЯНЫЕ УГЛЕВОДОРОДЫ ЖИРНОГО РЯДА
По богатству и разнообразию представителей, обнаруженных
в нефти, на первое место должны быть поставлены в настоя-
щее время углеводороды жирного ряда. Их можно разделить
на две основных группы:
I. Предельные углеводороды жирного ряда, т. е. метан и его
гомологи (парафины), представленные в нефти во всех трех
аггрегатных состояниях, а именно:
А. Низшие парафины, газообразные, образующие главную
массу естественного газа, почти всегда сопровождающие нефть
и находящиеся в ней в растворенном виде часто в весьма зна-
чительном количестве.
Б. Средние парафины, жидкие, образующие главную массу
некоторых жидких фракций нефти.
В. Высшие парафины, твердые, входящие в состав нефтя-
ного парафина.
II. Непредельные углеводороды жирного ряда, также содер-
жащиеся в нефти, хотя обыкновенно в крайне незначительном
количестве.
Так как методы исследования каждой из этих групп угле-
водородов жирного ряда имеют свои специфические особенности,
то каждая из них будет рассмотрена особо. •
ПРЕДЕЛЬНЫЕ УГЛЕВОДОРОДЫ ЖИРНОГО РЯДА
КРАТКАЯ ХАРАКТЕРИСТИКА УГЛЕВОДОРОДОВ РЯДА МЕТАНА
(ПАРАФИНОВ)
Несмотря на широкое распространение в природе, почти все
углеводороды ряда метана могут быть получены в чистом виде
лишь синтетически. Обычный путь для этого либо восстановле-
ние соответствующих галоидозамещенных (I), либо действие на
них металлического натрия {II), причем в этом последнем слу-
чае происходит сдваивание связанных с галоидом органических
радикалов. Так например:
I. СбН181-4-2Н == HI +CeHu (гексан);
II. 2C2Hrj I 2Na = 2NaI + C4H10 (бутан).
112
Немаловажное значение для получения углеводородов ряда
метана представляет также гидрогенизация непредельных угле-
водородов, легко протекающая в присутствии металлических
катализаторов, например:
СБН10 4- 2Н = СБН12 (пентан).
Лишь в отдельных случаях источником получения чистых
углеводородов ряда метана могут быть природные продукты. Та-
ков, например, естественный газ, представляющий собою иногда
почти чистый метан; таковы также некоторые фракции нефти,
представляющие собою после тщательной фракционировки и
•очистки в некоторых указанных ниже случаях тот или иной го-
молог метана почти в индивидуальном виде.
Состав углеводородов ряда метана определяется общей фор-
мулой: СпН2п+2. В отношении своих химических свойств они ха-
рактеризуются, как известно, необычайной устойчивостью при
обыкновенной температуре к самым разнообразным реагентам и
неспособностью к реакциям присоединения. Их превращения
можно отнести к двум основным типам:
1. Реакции замещения, а именно:
а) действие галоидов, из которых наиболее легко реагирует
о парафинами хлор (хлорирование, см. гл. III), наиболее труд-
но иод; Таблица 32
Свойства углеводородов ряда метана
Наименование Формула Темп, плавл. Темп, кип. Уд. вес Коэф. рефр. KD
d
Метан ‘ • СЩ -186 — 164 0.415 — 164 м—
Этан • Cj Не — 172 — 93 0,446 0 — —*
Пропан с3 н8 — — 44,5 0.536 0 —
Бутан норм С4 Ню — 135 4- 1 0,600 0 ——
Изобутан Ню — 145 — 11 — —• —
Пентан норм. . . . СБ Н12 — 131 4- 36,3 0,648 0 1,3649 (6°,5)
Изопентан СБ Й!2 — 158 4- 28 0,639 0 1,3549 (20°)
“Тетраметил-метан . СбНю — 20 4- 9,5 — — —
Гексан норм Св Hj4 — 94 4- 69 0,677 0 1,3769 (16^)
Гептан С? Ню — 97 4- 98.3 0,701 0 1,3867 (23°)
Октан Се Ню — 56 4-135,8 0,719 0 1,3988 (17° 6)
Нонан Сд Ноо — 51 4-150.5 0,733 0 1,4025 (25°)
Декан CioHjj — 31 4-173 0,745 0 1,4093
Ундекан Си Н-24 — 26 4-196 0,756 0 1,4182
Додекан С^Нзв — 12 4-215 0,766 0 1,4209
Тридекан С13Н28 — 6 4-234 0,772 0
Тетрадекан .... СцЩо + 5 4-252 0,772 10 —
Пентадекан .... С15Н32 4- ю 4-270 0,776 10 —
Гексадекан .... * С1бНз< + 19 4-287 0,771 25 ——
Гептадекан .... C17H3B + 22 4-зоз 0,777 22 1,4405
Октадекан СлвНзз 4- 28 4-318 0.777 28
Нонадекан С19 'Цо 4- 82 -4-330 0,777 32
Эйкоаан С20ЩЗ 4- 37 205 0,778 37
Дотриаконтан . . . С 32 Ще 4- .70 = * 310 0,775 79
Гексаконтан .... СеоНиг 4~102 -
8 Зак. 1622. Химия нефти.
113
б) действие разбавленной азотной кислоты яри нагревании,.
(нитрование по М, Коновалову, см. гл. III).
2. Реакции глубокого распада парафинов, куда относятся:
а) действие на парафины окислителей, из которых важней-
шим к практическом отношении является кислород воздуха;
в жестких условиях при этом получаются продукты полного
распада и окисления органических молекул, углекислота и вода;
в условиях же более умеренных получаются промежуточные
продукты — смесь органических кислот;
б) термический распад парафинов при температурах выше
400°, нашедший применение в новейших методах переработки
нефти — крекинг и пиролиз (ч. II).
Перечень важнейших представителей ряда метана с их фи-
зическими свойствами приведен в табл. 32.
А. Низшие (газообразные) парафины
Простейший углеводород парафинового ряда —метая— рас-
пространен в природе чрезвычайно широко. Он образуется при.
гниении растительных организмов без доступа воздуха на дне
стоячих -водоемов (болотный газ); выделяется в каменноуголь-
ных рудниках (рудничный газ); наконец, он образует обширные
подземные скопления, либо связанные с нефтяными месторожде-
ниями, либо независимые от них (естественный газ). В место-
рождениях последних двух типов метан вступается обыкновенно
в смеси со своими ближайшими гомологами. Неудко их сопро-
вождают также некоторые другие газы, чаше всего углекислота,
кислород, азот, иногда гелий и др.
ОБЩИЕ МЕТОДЫ ГАЗОВОГО АНАЛИЗА В ПРИЛОЖЕНИИ К АНАЛИЗУ
ЕСТЕСТВЕННОГО ГАЗА
Пробы естественного газа для исследования собираются и
хранятся обыкновенно ® опрокинутых бутылках над водой или
в специальных пинетках с кранами или без них. Ввиду различ-
ной растворимости отдельных компонентов газа ьода, применяе-
мая для предварительного наполнения этих бутылей или пипе-
ток, должна быть тщательно насыщена собираемым газом.
Измерение объема газа при его анализе пуизводптся в га-
зовых бюретках над водой; для получения же точных данных —
над ртутью. При каждом замеу отмечаются температура и да-
вление, функцией которых, как известно, являйся объем газа
согласно уравнению Мариотта — Гей-Люссака:
pv=povo(i + at).
Простейший тип газовой бюретки изображен на фиг. 22;
фит. 23 изображает другой, несколько более сложный, но чрез-
вычайно удобный тин газовой бюретки Гемпеля. Измерительная
трубка А бюретки, снабженная трехходовым краном К, окружена
здесь стеклянной муфтой с водой, благодаря чему при работе
114
достигается постоянство температуры. В той же муфте помещена
-запаянная с нижнего конца компенсационная трубка В, соеди-
няющаяся своим верхним концом с измерительной трубкой А
при посредстве сифонной манометрической трубки С с .ртутью
и крана К. Осторожно изменяя высоту уровня У, причем изме-
няются также объем и упругость газа в измер гге тьнон трубке А,
устанавливают ртуть в обоих коленах манометрической трубки
на одной высоте; в этот момент упругость газа в и мерительной
трубке А уравнивается, очевидно, с упругостью газа в компен-
Фиг. 22. Газо-
вая бюретка.
сационной трубке. Тогда за-
крывают кран.и производят от-
счет газа в измерительной труб-
ке А при постоянной темпера-
туре воды в муфте бюретки и
постоянном давлении газа
в компенсационной трубке В.
Фиг. 24. Прибор Орса.
Фиг. 23. Газовая
бюретка Гемпеля.
Для определения состава газовой смеси пользуются общими
методами газового анализа с поглощением отдельных составных
частей смеси соответствующими реактивами в определенной по-
следовательности. Так например, углекислоту поглощают креп-
ким раствором едкого кали, кислород — щелочным раствором
пирогаллола пли гидросульфита натрия и т. д. Самое поглоще-
ние ведется в специальных приборах, газовых пипетках, соеди-
няемых с прибором для измерения таза (бюреткой) толстостен-
ным каучуком встык. Удобны сблокированные системы таких
в* 115
пипеток в одном штативе; таковы, например, прибор Орса и мно-
гочисленные его видоизменения, получившие за последнее время
широкое распространение (фиг. 24).
Переходя к количественному определению содержащихся
в естественном газе углеводородов, приходится столкнуться
прежде всего с отсутствием таких реагентов, с помощью кото-
рых было бы возможно количественное поглощение метана и его
гомологов. Иниду этого при анализе такого рода газов их сме-
шивают с избытком воздуха и взрывают в особой пипетке при.
помощи электрической искры. Расчет производится далее двумя
способами: либо по количеству образовавшейся углекислоты^
определяемой обычным способом, т. е. поглощением крепким
раствором едкого кали, либо по сокращению объема, происходя-
щему вследствие образования при взрыве воды. Так например,,
полное сгорание метана происходит по следующему уравнению:
СН4 + 202 = 2Н20 + С03
1 объем 2 объема 1 объем
Как видно, объем углекислоты, образующийся при сгорании
метана, в точности равен объему сгоревшего газа; сжатие же,
одновременно происходящее вследствие образования воды, равно
удвоенному объему метана. Очевидно, каждая из этих экспери-
ментально определяемых величин может служить для суждения
как о составе горючего газа, так и о его количестве, взаимно
дополняя друг друга.
Для этана и других газообразных углеводородов уравнения
горения будут, конечно, несколько иными. Пользуясь ими, легко
рассчитать и здесь для каждого отдельного случая объем угле-
кислоты и сокращение объема, отвечающее воде, которые обра-
зовались при полном сгорании углеводорода; а этих данных до-
статочно не только для расчета содержания какого-либо одного
компонента, но и для определения состава несложной смеси,
нрлример смеси из двух углеводородов. При большем числе
компонентов этих данных для расчета уже недостаточно.
После определения углекислоты, кислорода и углеводородной
части анализируемой газовой смеси остаются так называемые
негорючие газы: азот, гелий и др. Суммарно их можно опреде-
лить как остаток после удаления всех вышеуказанных состав-
ных частей за вычетом того азота, который приходится на долю
воздуха, добавленного к газовой смеси для сожжения ее угле-
водородной части [1].
РАЗГОНКА ЕСТЕСТВЕННОГО ГАЗА В ВАКУУМЕ
В тех случаях, когда ввиду сложности состава углеводородной
части газовой смеси общие методы газового анализа оказыва-
ются недостаточными, приходится прибегать к комбинирован-
ному методу с предварительным упрощением состава газа путем
разгонки его в высоком вакууме при низких температурах. Схе-<
116
магическое изображение прибора для такой разгонки дано на
фиг. 25 1.
Прибор для разгонки естественного газа, монтированный на
специальной деревянной массивной подставке, состоит из двух
бюреток Е и F с подвижными уравнительными сосудами G и Я;
укороченного ртутного насоса TS; сосудов А, В, С, служащих
для сжижения и разделения углеводородов; соответствующих
каждому из этих сосудов манометров со ртутью М и прибора
для сожжения К; кроме того, к прибору присоединен сосуд 1>
с кокосовым углем и сосуды-промывалки с едким кали и хлори-
стым кальцием для поглощения содержащихся в анализируемом
газе паров воды и углекислоты. Отдельные части аппаратуры’
разделены рядом показанных на схеме герметических кранов.
Фиг. 25. Схема прибора для разгонки газа в вакууме.
Ход разгонки газа следующий. Прежде всего прибор эвакуи-
руется через кран 6 при помощи масляного или водоструйного
насоса; остатки же воздуха в сосудах А, В и С, в соединитель-
ных трубках и верхней части ртутного насоса Т поглощаются-
кокосовым углем, для чего баллон D с углем при открытом
кране 5 погружается в дьюаровский сосуд с жидким воздухом.
Спустя несколько минут после того как манометры 2И опустятся
до нуля, кран 5 закрывают; теперь можно считать, что в при-
боре установился достаточный вакуум. Чтобы при этой предва-
рительной операции ртуть из напорного резервуара насоса $ не*
заполнила сосуд Т и не разъединила его от насоса, необходимо^
1 Описанный ниже аппарат представляет собою значительно упрощен-
ное нидонзменениё установки Шеферд-Портера, описанный в книге Bur-
rell G., The Recovery of gasoline from natural Gas, New York (1926), стр-101-
Он был построен и с успехом применялся в ГИВИ при исследовании есте-
ственных газов СССР.
117
чтобы упругость воздуха в S была понижена так, чтобы ртуть
стояла ниже крана 7; для этого резервуар £ через особый кран
•9 соединяется с тем же действующим насосом. Если теперь пре-
кратить откачку воздуха из сосута 6’ и через кран 8 уравнять
давление в нем с атмосферным, то под действием последнего
ртуть устремляется в сосуд Т (кран 7 — закрыт) и заполняет
его: остатки воздуха в соединительной трубке ТЕ легко выво-
дятся в бюретку Ё, если при открытом кране бюретки несколько
опустить ее уравнительный сосуд. Остается удалить этот воз-
дух через кран 12, и аппарат готов для последующей работы.
Когда воздух из прибора удален, сосуды А, В и С погружают
в дьюары с жидким воздухом и приступают к загрузке аппа-
рата анализируемым газом. Для этого наполняют газом бю-
ретку Е, замеряют его, отмечая температуру и давление, и, открыв
кран 1, переводят газ в охлажденный сосуд А, где газ быстро
переходит в жидкое состояние б Эту операцию приходится по-
вторять несколько раз, чтобы обпщй объем взятого для анализа
газа был примерно 300—ООО см3. Предварительно анализируе-
мый газ пропускают через промывалки с едким кали и хлори-
стым кальцием, так как в противном случае углекислота и во-
дяные нары при температуре жидкого воздуха могут совер-
шенно закупорить соединительные трубки сосудов А, В и С.
Когда загрузка прибора анализируемым газом закончена,
начинают отгонку метана. Прежде всего с помощью водоструй-
ного насоса понижают давление в сосуде S так, чтобы ртуть
в левой части ртутного насоса опустилась ниже крана 7 и в .ре-
зервуаре Т образовался вакуум. Затем открывают кран 7;
тогда метан, пройдя последовательно сосуды В и С, поступает
в резервуар Т. Спустя некоторое время, кран 7 закрывают и
впускают в сосуд & воздух, так что ртуть перекачивается из
него обратно в резервуар Т и загоняет отогнанный метан
в верхнюю часть этого резервуара. Остается перевести этот газ
в бюретку Е (как это указано выше для остатков воздуха) и
здесь его измерить.
За первой откачкой тем же порядком производят вторую
и т. д., причем наблюдается, что спустя некоторое в|юмя коли-
чество откачиваемого каждый раз метана начинает постепенно
уменьшаться и соответственно упру гость газа, остающегося в со-
суде Л, начинает падать. Когда она доходит по манометру Л/ до
нуля, отгонку метана можно считать почти законченной. Не-
большое количество метана сохраняется еще в сосуде А в рас-
творе других оставшихся здесь сжиженных газов (этап, пропан
и др). Чтобы собрать и эти остатки метана, отнимают на некото-
рое время дьюары и дают упругости газов в сосудах Л. В и С
1 В позднейших моделях этого прибора, для разгчунгц газа в вакууме
вместо двух измерительных бюреток Е и F оставлена лишь одна F. Если
крап 12 сделать трехходовым и третий ход его соединить длинной трубкой
через кран 1 с сосудом А, то пробу анализируемого газа, взятую из бал.
лона, можно после замера в бюретке F перевести через трехходовой кран 12
в охлаждаемый сосуд Лит. д., как это описано в тексте.
118
повыситься до 100—150 мм. Затем вновь охлаждают эти сосуды
жидким воздухом и производят заключительные 2-3 откачки,
собирая последние (обычно несколько десятых 1 см3) метана.
В зависимости от состава анализируемого газа, а также от боль-
шего или меньшего содержания в охлаждающей смеси азота для
полной отгонки метана приходится производить 20—30 и более-
откачек, каждая из которых при налаженной работе занимает,
однако, не более 3-5 мин. Когда отгонка метана закончена, при-
водят отдельные измеренные объемы газа к 0° и 760 мм и их
суммируют.
За метаном следует отгонка из оставшегося в сосудах А, В
и С сжиженного газа других углеводородов и в первую очередь
этана. Для этого прежде всего отнимают дьюары из-под сосудов
В и С и весь газ переводят в сосуд А. Затем вместо дьюара
с жидким воздухом под него при закрытых кранах 2, 3 и 4 под-
водят другой дьюар с легким бензином (пентан), температура
которого при помощи пробирки с жидким воздухом предвари-
тельно понижена до 145—150° ниже 0°. Если теперь охладить
сосуд В жидким воздухом и открыть кран 2, то этан с неболь-
шой примесью пропана перегоняется из сосуда.А в сосуд В.
Затем погружают сосуд В в охлажденный до —150° бензин,
а сосуд С — в жидкий воздух. Открывая кран 3, позволяют
перейти этану в сосуд С, где он собирается уже в достаточно
чистом виде, и откуда при закрытых кранах 2 и 3 его откачи-
вают затем с помощью ртутного насоса в резервуар 2\и далее
в бюретку В вышеописанным способом. Затем весь газ, остав-
шийся в сосудах А, В и С, снова переводят в сосуд А и повто-
ряют отгонку этана до тех пор, пока упругость газа в этих сосу-
дах при —150° не сделается равной нулю.
После отгонки этана приступают последовательно к отгонке-
пропана и бутана. Ее производят аналогично отгонке этана, но
уже при температуре —120 для пропана и —90 для бутана.
В тех, однако, случаях, когда содержание этих газов невелико,
их показывают иногда совместно с остальными высшими угле-
водородами, содержащимися в образце газа (газовый бензин).
Для совместного определения всех этих углеводородов их соби-
рают сначала в сосуд А, после чего при закрытых кранах 1 и 2
дьюар с охлаждающей смесью убирают и дают сосуду принять
комнатную температуру. Зная объем сосуда А, давление содер-
жащегося в нем газа '(по манометру М) и температуру, имеем
все данные дня расчета общего содержания указанных компо-
нентов газа при обязательном, однако, условии, что содержание
это не велико; иными словами, если все эти компоненты при со-
ответствующей каждому из них парциальной упругости можно
рассматривать как постоянные тазы.
При разгонке естественного газа на его отдельные компо-
ненты в чистоте каждого из них убеждаются методом сожжения.
Такая проверка особенно необходима для метановой фракции,
так как вместе с метаном при разгонке отходят негорючие газы,
азот и гелий, а также кислород, если они содержатся в газе.
119'
•Сожжение отдельных фракций и последующее поглощение про-
изводят в обычных газовых пипетках, присоединенных непо-
средственно к аппарату для разгонки; определение же отдель-
лых компонентов смеси производят общими методами газового'
Анализа, как указано выше.
Описанный метод разделения естественного газа на компоненты основан
.на различной упругости пара различных углеводородов при разных тем-
пературах, и с этой точки зрения оп в полной мере аналогичен обыкновен-
:ной разгонке какой-либо жидкой смеси, например бензина. В табл, зз при-
ведены отношения упругостей первых членов ряда метана при низких тем-
пературах.
Таблица 33
Температура Формулы углеводородов Отношение упругостей
( — 180°) —( — 188°) СЩ :С2Нв 80:0,1
— 150° C2He:CsHs 4:0,4
— 120° С8Н8: С4НЗО 5:0,2
Как видно из этой таблицы, три первых члена ряда, метан — этан—
шропан, отличаются между собой по упругости пара при низких темпера-
турах весьма значительно; соответственно этому достигаемое при разгонке
их разделение при тщательной работе вполне удовлетворительно. Для сле-
дующих членов ряда, пропан — бутан — изобутан, различие в упругостях
пара уже гораздо меньше, и разделение их путем разгонки, естественно,
гораздо труднее. Чтобы облегчить эту задачу, можно увеличить число со-
судов-приемников А, В, С....играющих при разгонке роль секций гори-
зонтально расположенного дефлегматора.
Совершенно иначе построен -прибор для разгонки естествен-
ного таза, а также газового бензина, который описал и построил
ж ОША Подбильняк [2]; этот прибор благодаря своей простоте
и четкости работы получил ныне весьма широкое распростране-
ние. Устройство его в основном подобно обыкновенному аппа-
рату для фракционировки жидкостей. Сосуд, в котором нахо-
дится сжиженный газ, непосредственно соединен здесь с высо-
кой колонкой—дефлегматором, заключенной для •изоляции
в эвакуированную муфту. Верхняя, свободная от муфты часть
дефлегматора также охлаждается соответствующим образом.
Разгонка ведется при электрическом подогреве сжиженного газа
с регулировкой температуры при помощи термопары; переход
же от одного компонента смеси к другому определяется по ходу
кривой упругости газа, показываемой специальным манометром.
Несмотря на большие допускаемые загрузки (от 2 до 100 л газа
и больше), разгонка протекает быстро и даст хорошее разделе-
1 Детальное описание прибора Подбидъняка можно найти также в ру-
ководстве Соколова В. А., Методы иоследования природных газов, ОНТИ,
М. — Л. (1932).
Д20
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ БЕНЗИНА В ЕСТЕСТВЕННОМ ГАЗЕ
Содержание бензина в естественном газе является одним из-
основных условий, определяющих способ его утилизации. Ввиду
этого необходимо хотя бы вкратце остановиться на этом, испы-
тании газа, несмотря на то, что по существу оно имеет скорее-
технологический, чем химический характер.
Существует несколько методов для определения бензина,
в естественном газе. Одни из них, косвенные, основаны на опре-
делении того либо иного физического свойства газа, например-
его удельного веса, теплотворной способности и т. п., более или-,
менее резко изменяющихся в зависимости от содержания,
в газе 'бензина. Ввиду сложности и разнообразия состава есте-
ственного газа методы эти не могут дать точных результатов;,
они могут служить лишь для контроля
результатов, полученных иными спосо-
бами [3].
Более надежные данные получаются
прямым определением содержащегося
в газе бензина с применением либо мас-
ляных поглотителей, либо специальных
адсорберов с высоко-активированным дре-
весным углем. Последний метод, являю-
щийся наиболее простым и часто приме-
няемым, основан на способности свежего
активированного угля поглощать от 15
до ’20% по весу углеводородных паров и
газов.
Угольный адсорбер, употребляемый
для определения бензина в газе, предста-
вляет СОбОЮ ОЛОВЯННЫЙ ЦИЛИНДР еМКОСТЬЮ д.-.,
около 250 сдз с герметически привин- Фиг> 26' Со$ер
чивающимися крышками (фиг. 26); одна
из этих крышек снабжена медной труб-
кой, соединяющей адсорбер с прибором для измерения проходя-
щего через адсорбер газа (газовые часы и т. п.). Для ввода газа,
служит другая трубка, находящаяся в нижней части адсорбера,
а непосредственно над ней расположена сетка, поддерживаю-
щая активированный уголь. Последний берется в количестве
250 см3. Когда прибор собран, его соединяют с трубкой, подаю-
щей газ, и регулируют ток газа таким образом, чтобы закон-
чить испытание примерно в 30 мин. Скорость газа при этом мо-
жет колебаться от 0,4 до 0,8 м3/час, объем же пропускаемого-
газа устанавливают таким образом, чтобы в результате адсорб-
ции собрать примерно 25 см3 бензина (т. е. 10% от взятого-
угля). Этот предварительный подсчет газа, который следует'
взять для испытания, можно произвести на основании хотя бы
его удельного веса.
Когда пропускание газа через адсорбер закончено, пересы-
пают уголь в колбу с отводной трубкой и отгоняют адсорбиро-
121
ванный бензин перегретым паром, собирая его в тщательно ох-
лажденный градуированный приемник. Для более полной от-
гонки бензина некоторые авторы рекомендуют вместо пара
пользоваться глицерином при постепенном нагревании смеси до
260°; другие применяют для этой цели даже ртуть.
СОСТАВ ЕСТЕСТВЕННОГО ГАЗА
Естественным газом в нефтяном деле называют не только
газ, почти всегда сопровождающий нефть и находящийся в ней
в растворенном состоянии, но и горючий газ, источником кото-
рого являются так называемые газовые месторождения, видимо
не связанные с тем или иным месторождением нефти. О составе
этих газов имеется большая литература; однако среди опубли-
кованного материала имеется не мало данных, полученных не-
достаточно точными методами и поэтому неверных. Особенно
это относится к содержанию таких газов, как водород, окись
углерода и некоторые другие. Новейшие данные, полученные
с применением новой, более усовершенствованной методики и ап-
паратуры, в частности с разгонкой газов в высоком вакууме,
заслуживают большего доверия [4].
Главной составной частью горючих естественных газов, как
видно из табл. 34 и 35, является меган. В так называемых «су-
хих» газах содержание метана может доходить иногда до 93—
95%, а в отдельных случаях даже до 99% с лишним. Таким
образом в некоторых случаях естественный газ представляет со-
бою почти химически чистый метан.
Другой обычной составной частью естественных газов яв-
ляется углекислота. Содержание этого газа в скважинах раз-
личных месторождений колеблется в очень широких пределах —
•от нескольких десятых процента до 30—40% и больше. Инте-
ресно отметить, что подобного рода колебания наблюдаются
иногда даже для различных скважин одного и того же место-
рождения; таковы, например, Сураханы.
Соответственно колебаниям в содержании углекислоты, есте-
ственно колеблется в газе и содержание метана; в сумме же со-
держание обоих этих компонентов обыкновенно не менее 94—
95%, особенно если не принимать в расчет зачастую чисто слу-
чайной примеси к газу воздуха.
За вычетом метана и углекислоты на. долю всех остальных
компонентов сухого газа остается обыкновенно не более 4—5%.
В счет этого остатка входят этан (не более 2—3%), пропан (не
более 1,5—2%), бутан и изобутан (несколько десятых ггроцента)
и, наконец, более высокомолекулярные гомологи метана, обра-
зующие главную составную часть газового бензина. Примеры
«сухих» газов некоторых месторождений приведены в табл. 34.
«Сухой» газ является одним из основных типов естественного
газа; его называют иногда также «бедным» газом (считая на
выход из него бензина). Противоположностью сухого газа яв-
ляется «влажный» или «богатый» газ. Содержание метана в Со-
122
Таблица 3/
Состав «сухих* газов различных месторождений
Месторождение Уд. вес со2 Метан Этан Пропан Бутан и высшие
Бакинский район
Сураханы, пл. IV d, е
и V 0,778 19,5 76,8 2,0 1,2 С пропаном
Сураханы, пл. IV а . . 0,632 4,3 90.7 2,5 1,0
Раманы — 16,6 75 5 2,7 0,8 1,4
Сабунчи 0,667 8,8 84,6 0,9 0,2 0,5
Балаканы 0 691 11,5 83,7 0.6 0,2 С пропаном.
Нута — 13,2 81,3 1.6 2.9
Биби-Эйбат ...... — 6,3 90,7 2,1 0,4
Шубаны — 2,0 92.9 2,1 1.0 1.0
Кала 0,602 1.6 93,3 2,3 0,5 0,3
Дагестанский район >
Дагестанские Огни . . 0,660 7,8 88,2 1.9 1.1 С пропаном
Дувлак 0,712» 13,7 82,1 1.5 0.7
Берекей — 9,9 86,2 2,3 1,1
Мелитопольский район
Георгиевка, П-1П гориз. — 0,0 99,1 —— —
Покровка — 0,2 99,8 — —- —
Таманский район
Гора Нефтяная, южн. ч. — 5,2 84,0 8,3 2,5 € пропаном:
Гора Гнилая, сев.-вап. ч. — 0,4 99,6 — — —
Гряда Близнецы, зап. ч. — 0,7 99,3 — — —
США
Ventura, Калифорния . — — 89,3 2.0 4,3 4,4
Alien, Канзас 0,580 0,9 96,4 1.3 — —
Bartlesville, Оклахома . — 1.4 92,4 3.1 — —
Corsicanna, Тексас . . 0,7 98,0 0,0 — —
гатом газе уже значительно меньше, чем в сухом; оно спу-
скается здесь до 50—00% и ниже. Зато приобретают большее
значение гомологи метана, не только газообразные, но и жид-
кие, находящиеся в газе в парообразном состоянии.
Насколько разнообразно может быть содержание бензиновых
паров в естественном газе, показывает следующая справка:
тогда как выход бензина из сухих бакинских газов колеблется
обычно в пределах 30—50 г на 1 .и3 газа, в Грозном его богатые-
(влажные) газы дают бензина до 750 г на 1 м3 газа и больше.
Столь глубокое различие зависит вне всякого сомнения не
только от химической природы нефти и газа данного месторож-
дения, но и от температурных условий: чем выше температура,
при которой происходит выделение газа из недр, тем, очевидно,
более он должен быть насыщен, более богат бензиновыми па-
рами. Такого рода случай имеется как раз в Грозном, где тем-
123
шература нефти и газа при появлении их на поверхности земли
достигает для некоторых скважин 75° и выше.
Что касается других составных частей богатых газов, то1
в качественном отношении они не отличаются от соответствую-
щих фряк.пий сухих газов; о количественном их содержании
можно судить из табл. 35, в которой приведен 'состав богатых
газов некоторых месторождений.
Таблица 35
Состав „богатых- газов различных месторождений
Месторождение Уд- вес со2 Метан Этан Про- пан Бутан Иво- бут ан СбН12 и др.
Грозный, Новый район
Пласт II Пласт XI-XII .... Пласт XXI 1,411 1,207 1,240 1 11 .31,6 47,0 50,6 12,8 11,2 10,8 24,7 17,5 13,5 82 8,1 М 11,5 7.2 8,4 11,2 9,0 10,9
Грозный, Старый район •
Пласт Х1-ХП .... Пласт VI 1,170 1,156 — 55,2 52,7 7,4 11,8 15,7 13,8 6,0 8,8 7,7 6,0 7,9 69
Майкоп
•Скв. № 87 Фонтан скв. № XI . Чусовские Городки 1,58 1,05 7,5 6,9 0,4 10,7 53,6 41,7 17,8 14,2 21,6 35,7 11,7 20,7 11.1 4,7 8,6 3,5 18,7 8,4 5,4
США
Зап. Вирджиния . . •Сигнал-Хиллс . . . Оанта-Фи-Спрингс . — 0 0 59,1 72,4 70.8 17.6 9,4 9,9 13,2 8,5 8,1 6, 4,6 4,4 2 1.7 1,6 3,9 8,5 4,9
Большой интерес и значение могут представлять системати-
ческие анализы естественных газов нефтяных месторождений
по пластам. Однако исследования этого рода, требующие осо-
*бенно тщательного отбора газовых проб и систематической,
длительной работы, по существу находятся пока в начальной
стадии.
Оказалось, что для различных месторождений и горизонтов
-здесь может наблюдаться существенно различная картина. Так,
в Бакинском районе на 'Промыслах Кала и Нута и некоторых
других газы различных горизонтов почти не отличаются друг
ют друга ни содержанием метана и его гомологов, ни содержа-
нием углекислоты. Это — типичные «сухие» газы, в которых со-
держание метана доходит до 95—99%. В других случаях (Кара-
Чу хур), напротив, наблюдается 'Известная зависимость состава
таза от глубины пласта: верхние горизонты, дают более «сухой»
газ, тогда как газ более глубоких горизонтов характеризуется
несколько повышенным содержанием гомологов метана и угле-
.124
кислоты; содержание же самого метана падает в них до 87 и
даже до 76%.
В тех случаях, когда скважина нс истечении некоторого времени
оксплоатации начинает фонтанировать гаэом с нефтью, постепенно наблю-
дается некоторое обогащение газа гомологами метана и углекислотой по
сравнению с начальным,' чисто газовым фонтаном. Объясняется это сле-
дующим образом. При вскрытии нефтяного пласта с богатым запасом газа
в первую очередь выделяется метан; гомологи же метана и углекислота,
обладающие под давлением большей растворимостью в нефти, будут выде-
ляться лишь в незначительном количестве. По мере выхода газа и спа-
дания в пласте давления гомологи метана и углекислота начинают выде-
ляться в большем количестве, что нередко совпадает с появлением нефти
из буровой. В табл. 36 приведен пример того, как для одной из буровых
Йара-Чухура (VI горизонт) на протяжении 10 месяцев изменился состав
газа.
. Таблица 36
Состав пластового газа на Кара-Чухуре
Горизонт, глубина забоя Отбор пробы Состав газа Бензин в 1 *8 в г
СН4 С2Н6 с3н8 СО2
"VI горизонт, бур. 123, 2/VII 1933 г. 94,2 2,5 0,6 1,8 21,3
глубина 1075 м 18/1 1934 г. 86,4 4,6 2,0 3,5 133,0
Примеры, приведенные в .табл. 36, далеко не исчерпывают
всего разнообразия естественных газов, встречаемых в нефте-
носных и газоносных районах. Кроме метановых углеводородов
за углекислоты, в их состав нередко входит азот, содержание ко-
торого в них может колебаться в весьма широких пределах (от
О до 100%). В табл. 37 дано несколько примеров состава есте-
ственных газов различных месторождений Азербайджана по ра-
ботам АзНИИ. Как видно, здесь явственно намечаются три но-
вых типа естественных газов: азотисто-метановые газы (№ 1—3),
углекисло-азотисто-метановые газы (М 4 и 5) и азотистые газы
Состав газов некоторых месторождений Азербайджана
№ п/п I Месторождение газа Состав газа
Метановые углеводо- роды со2 Азот Воздух
1 •2 Набур, бур. 12 ...... . Хиналуг, Хачмас 55,2 89,3 5,4 39,4 7,8 2,9
3 Боз-Даг, Аджи-Булах . . . 84.4 — 15,6
4 Ахтинское, с. Ахты .... 55,5 13,6 23.3 7.6
5 Кусарское, Кутул 70,7 15,7 13,7 —
€ Пирсагат, скв. 81 — 1,2 98,8 —
7 Ленкорань 100,0
125
Особое место в составе некоторых естественных газов зани-
мает гелий Этот негорючий газ, лишь в два раза более тяже-
лый, чем водород, приобретает за последнее время все большее
и большее значение, главным образом в воздухоплавании.
В природе гелий встречается довольно часто (воздух, руднич-
ный и естественный газ, некоторые минералы и минеральные
источники), но обыкновенно в очень ограниченном количестве.
Содержание гелия в естественном газе также невелико; наибо-
лее богаты им некоторые месторождения естественного газа
в США, а именно: в Канзасе (до 1,28% гелия), в Тексасе (до
0,9%) и Оклахоме (до 0,63%). Недостаток содержания воспол-
няется здесь размерами дебита соответствующих скважин, и
промышленное получение гелия пошло именно по этому пути.
Постоянными спутниками гелия являются другие газы ну-
левой группы, а также азот, в количественном отношении зани-
мающий ' первое место среди негорючих газов. Отделение от ге-
лия всех сопутствующих газов и его количественное определе-
ние осуществляются путем последовательного поглощения газов
соответствующими реагентами: для горючих газов — после их
сожжения, для негорючих (азот и пр.) — обработкой кокосовым
углем при температуре жидкого воздуха. Лишь один гелий при
этом не поглощается и может быть определен по остатку. Тех-
ническое получение гелия из естественного таза достигается
с помощью специальных холодильных машин, основанных на
тех же принципах, как машины для жидкого воздуха. Так как
гелий является наиболее трудно сжижаемым газом (температура
кипения его —268° ниже 0°), то, превращая в жидкое состоя-
ние последовательно все находящиеся в смеси с гелием газы,
отделяя -их и постепенно обогащая, таким образом, остаток ге-
лием, можно получить последний желаемой чистоты, вплоть до
100%-ной1 2.
ПРИМЕНЕНИЕ ЕСТЕСТВЕННОГО ГАЗА
По сравнению с различными видами топлива естественный
газ при сгорании дает один из наиболее высоких коэфициеитов
полезного действия. Общеизвестно также, что при надлежащим
образом развитой газовой сети газом чрезвычайно удобно поль-
зоваться не только для лабораторных и домашних надобностей
(газовые горелки, плиты, ванны и т. п.), но также и для отопле-
ния. В Европе для этих целей используют преимущественно
искусственные нефтяной или светильный газы. В США с их
колоссально развитой газовой промышленностью3 для тех же
надобностей широко пользуются естественным газом. Как видно
1 Подробности о гелии на русском языке см. Лукашук А, Гелий, его
применение и добывание, Л. (1925).
2 О значении, которое придают в настоящее время запасам гелия, можно
судить уже из того, что ® США, которые в области промышленной добычи
гелия являются монополистами, экспорт гелия -воспрещен законом.
3 В 192S г. в США было добыто 44 млрд, л3 естественного газа.
126
из данных, .приведенных ниже, по своей теплотворной способно-
сти даже сухой, например, бакинский естественный газ почти
в два раза превышает светильный таз, приближаясь в атом от-
ношении -к лучшим видам твердого и жидкого топлива, но зна-
чительно превосходя последние в отношении удобства и эконо-
мичности использования.
Средняя теплотворная способность
различных видов топлива в кейфа
Дрова сухие............................. 4100
Торф (с 10% влаги).......................4750
Уголь каменный........................... 8 000
Нефть.................................. 10500
Газ светильный......................... 5 900
Газ естественный, сухой (Баку).......... 9000
Другой крайне важный вид использования естественного
таза — его отбензинивание, т. е. получение из него так называе-
мого газового бензина (natural gasoline) на специальных газоли-
новых заводах. Современная техника пользуется для этой цели
установками различных типов.
Компрессорные ааводы. На газолиновых заводах этого типа естествен-
ный газ подвергается при помощи компрессоров более или менее сильному
•сжатию с последующим охлаждением. Вследствие этого пары жидких угле-
водородов, находящиеся в естественном газе, конденсируются, растворяя
в себе частично также углеводороды, тазообразные при обыкновенных усло-
виях (бутан, йзобутан, пропан и др.)- Образуется летучая жидкость, га-
зовый бензин, легко отделимая от остальной, неконденсирующейся части
естественного газа.
Абсорбционные заводы. На этих заводах естественный газ, для извле-
чения из него бензина, пропускают через особые поглотительные колонны,
абсорберы, в которых сверху вниз стекает поглощающее бензин масло,
чаще всего соляровое1. Внутри для увеличения поверхности поглощения
.абсорберы оборудуются специальными тарелками или заполняются насадкой
{кольца Рашига и т. п.). Подача газа производится сниз} под небольшим
.давлением. Все бензиновые пары, а также часть постоянных газов задер-
живаются маслом; выходящий из абсорбера отработанный газ направляется
либо в топки, либо в газопроводы для перекачки в места потребления
(в Америке — на сотни километров), а масло, насыщенн е бензиновыми
парами, перекачивается из абсорбера через теплообменный аппарат в вы-
паритель. Здесь при помощи перегретого пара оно подогревается до 130—140°
и отдает поглощенные им в абсорбере бензиновые пары. Последние вместе
-с водяными парами направляются в холодильник и сепаратор, где проис-
ходят конденсация и отделение воды от бензина; остающееся же в выпа-
рителе абоорбцпнное масло направляется обратно в теплообменный аппарат.
Здесь оно частично охлаждается, отдавая теплоту свежему, направляюще-
муся в выпаритель маслу, а затем через орошаемый водою холодильник
подается в верхнюю часть абсорбера, начиная вновь свой кругооборот от
абсорбера к выпарителю, и обратно. *
Адсорбционные заводы. В отличие от абсорбционных заводов на уста-
новках адсорбционного типа извлечение бензина из естественного газа осу-
ществляется не жидким, а твердым адсорбентом — активированным углем
(char coal). Адсорбция производится в больших герметических резервуарах;
затем адсорбированный бензин отгоняется от угля перегретым паром, после
1 Прекрасным поглотителем для бензина является согласно патентам
Врежа технический тетралин (тетрагидро-нафталин).
127
чего остывший уголь вновь готов к употреблению. Как видно, в принципе
работа угольных («чаркольных») заводов идентична с описанным выше
способом количественного определения бензина в естественном газе; она.
характеризуется высокой производительностью и прекрасными качествами
получаемого бензина, но требует специальных, довольно дорогих сортов
угля высокой активности. Лучшими углями считаются ~ американский уголь
из скорлупы кокосовых орехов, немецкий уголь Байера и французский
Urbain\ оба последних изготовляются из торфа.
Наиболее распространенным типом газолиновых заводов является ком-
бинация компрессорного завода с абсорбционным. На такого рода заводах
естественный газ сначала компримируют при сравнительно небольшом
давлении; часть бензиновых паров при этом конденсируется, оставшийся
же полуотработанный газ направляют в абсорберы, где происходит оконча-
тельное извлечение бензина.' После национализации нефтяной промышлен-
ности в СССР построено несколько таких заводов (3 *в Баку и 8 в Гроз-
ном). За последнее- время довольно широкое распространение начинают по-
лучать также чаркольные заводы, особенно в Европе (Румыния, Польша).
В СССР имеется пока лишь один чаркольный завод (Сураханы).
Газовый бензин широко используется для подмешивания
к бензинам прямой гонки и крэкишЧ5ензинам в целях их облег-
чения. При этом его необходимо подвергнуть предварительной
стабилизации, т. е. освободить от растворенных в нем газообраз-
ных углеводородов, главным бразом пропана, бутана и изобу-
тана, присутствие которых слишком повышает упругость пара
бензина и его летучесть. Такая стабилизация достигается в же-
лезных, оборудованных тарелками колоннах-стабилизаторах.
В нижней части стабилизатора находятся паровые змеевики для
подогрева бензина. Последний подается в среднюю часть стаби-
лизатора; стекая по тарелкам вниз, где происходит подогрев до
требуемой температуры, он отдает свои наиболее легкие, не кон-
денсирующиеся части, которые уводятся через верхний конец
стабилизатора в топку или в специалыше компрессоры для
получения жидкого газа (см. ниже); более же тяжелые части
бензина из нижней части стабилизатора направляются в холо-
дильник и далее в резервуар для стабилизованного бензина.
Газовый бензин составляет около 9—10% всего бензина, вы-
рабатываемого на земном шаре, но его относительное значение
ввиду характера его утилизации несомненно значительно выше.
На первом месте в отношении добычи газового бензина должны
быть поставлены США. О росте газолиновой промышленности
в Америке можно судить по следующим данным,
1904 г.— 2 завода с производительностью
1911 г.— 176 .
1922 г. —1260 .
около ' 28 m бензина.
„ 21000 m ,
, 1800000 m
Газолиновая промышленность Европы находится пока еще
в начальной стадии.
Как уже было указано, при стабилизации газового бензина
освобождаются пропан и бутан. Несколько лет тому назад смесь
этих газов стали применять в США в качестве особого вида топ-
лива («жидкий газ») для домашних и промышленных целей.
Газовая смесь после стабилизации компримируется, нагнетается
в баллоны со специальными редукционными вентилями и доста-
128
вляется на места потребления. Постепенно жидкий газ получает
в Америке все более широкое распространение, особенно
в местах, удаленных от газовых районов и заводов. Кроме до-
машнего применения '(газовые плиты, печи и т. п.), жидкий газ
с успехом употребляется также при резке и сварке металлов
вместо ацетилена.
Потребление естественного газа в виде топлива в том или
ином виде представляет собою главный вид его утилизации.
В местах, удаленных от промышленных и населенных пунктов,
сухой и отработанный газы при большом газовом дебите уже
не находят сбыта, и на утилизации такого газа в США разви-
лась промышленность газовой сажи.
Для превращения в сажу естественный газ должен быть
разложен на элементы — углерод и водород. Такое разложение
требует, однако, очень высокой температуры (выше 1000°), и
соответствующие методы получения сажи (термический и эле-
ктрический) пока не получили сколько-нибудь широкого распро-
странения. Вся газовая сажа получается в настоящее время
почти исключительно путем сожжения газа при недостаточном
притоке воздуха в специальных аппаратах (желобчатые, ролико-
вые, дисковые) с многочисленными газовыми горелками-рож-
ками. Выделяющаяся вследствие неполного сгорания газа сажа
собирается, передается на сита для отделения от разного рода
посторонних примесей (окалина и т. п.) и подается в упаковоч-
ное отделение. Все эти операции, конечно^ механизированы.
При продолжительном воздействии высокой температуры физические
и химические свойства сащи заметно изменяются: отдельные частицы ее
плотно сцепляются, образуя твердые комки, напоминающие в некоторых от-
ношениях частицы графита. Ввиду этого для получения высококачествен-
ной сажи необходимо по возможности предохранять образовавшуюся сажу
от воздействия высокой температуры, для чего служат разного рода охлаж-
дающие приспособления (располагаемые над газовыми гирелками-рожками
желоба, вращающиеся диски и т. д.). Из других методов получения сажи
следует отметить:
1. Неполное сгорание без применения охлаждающих поверхностей. Ис-
ходным материалом служат при этом разного рода смеси жидких и твер-
дых углеводородов. Так получается ламповая сажа.
2. * Взрывные методы получения сажи. Подходящий для этого газ, на-
пример ацетилен, взрывают при недостаточном количестве воздуха. Один
из продуктов реакции — углерод—осаждается при этом в виде очень мел-
ких частиц.
3. Перегонка до кокса. Нефть и другие продукты перегоняют до конца,
в результате чего в перегонном кубе отлагается кокс. Для получения сажи
кокс подвергается затем измельчению в порошок.
4. Термическое разложение при отсутствии воздуха. Газ или .распы-
ленная нефть подвергаются воздействию высокой температуры без притока
воздуха. Метод имеет значение главным образом для получения водорода.
Все четыре последних метода получения сажи по сравнению с основ-
ным (желобчатым) имеют второстепенное значение, давая менее 10% от
общечч) количества получаемой в США сажи.
Выход сажи по методу неполного сгорания естественного газа
крайне незначительный—всего 1,5—2%; остальной газ сгорает
без остатка. Несмотря на столь ничтожные выходы, производство
129
9 Зак. 1622. Химия нефти. х и
газовой сажи, постепенно возрастая, достигло в 1931 г. в США
127 319 т. При этом: потребляются такие колоссальные количе-
ства газа, что в некоторых штатах США принимаются специаль-
ные меры к ограничению этого вида использования естественного
газа. Главная масса газовой сажи производится в штатах Тексас
и Луизиана. Около одной пятой ее вывозится из США в другие
страны.
Газовая сажа («микронекс» и др.) является лучшим и наи-
более ценным сортом сажи. Она находит широкое применение
в красочной, полиграфической и особенно резиновой промыш-
ленности, где она употребляется как лучший наполнитель при
изготовлении резиновых изделий (шины, галоши и т. и.). Ввиду
громадной потребности в газовой саже в СССР также присту-
пили к ее производству (Майкоп).
Переходим, наконец, к видам использования естественного
газа, в основе которых лежит его химическая переработка [5].
Такая переработка может быть осуществлена в трех различных
направлениях, а именно: 1) хлорирование, 2) окисление и 3) пи-
ролиз. Хотя все эти виды переработки естественного газа не
вышли из стадии лабораторных, в лучшем случае — иолузавод-
ских опытов, тем не менее они представляют выдающийся инте-
рес, так как именно в этих направлениях следует ncivaTb путей
рационального использования естественного газа. Соответствую-
щие реакции будут рассмотрены ниже по отношению к метану,
так как после отбензинивания метан является основной частью
всякого горючего естественного газа.
Хлорирование. При действии хлора на метан постепенно за-
мещаются все 4 его атома водорода на галоид, причем последо-
вательно образуются: 1) хлористый метил, 2) хлористый мети- «
лен. 3) хлороформ и 4) четыреххлористый углерод:
1) СН4 + С1о=НС1 + СНяС1; ‘
2) CHSC1 -Г CL = НС1 + СЩС12;
3) СН2С12 4- С12 = НС1 + СНС1,;
4) СНС1У+CL> = НС1 + СС1Ч. *
Хотя все эти реакции являются сильно экзотермическими,
тем не менее для начала хлорирования должна быть дана до-
вольно высокая температура (325—500'), которую необходимо
поддерживать и в дальнейшем. В связи с возникающими при
этом техническими трудностями для активизации процесса и
его урегулирования были заявлены многочисленные патенты.
В качестве катализаторов были предложены активированный
уголь и различные металлы: Си, Pb, Sn, Fe, Ni и др.; для облег-
чения же регулировки процесса рекомендуется разбавлять смесь
метана с хлором добавлением различных газов, как-то. воздуха,
азота, углекислоты, хлористого водорода, водяных п&1>ов и т. п.
При техническом хлорировании метана целевыми продуктами
процесса должны быть не только галоидопроизводные, но и хло-
ристый водород. При растворении в воде последний дает соля-
ную кислоту; что же касается галоидозамещенных метана, то,
130
смотря по степени охлорения, они находят весьма разнообразное
применение. Хлористый метил в технологии органических ве-
ществ применяется как хороший метилирующий реагент. Кроме
того, как легко сжижаемый газ (температура кипения —24°) он
может служить в качестве ценной по своей нейтральности охла-
ждающей жидкости для холодильных машин. Дешевый хлори-
стый метилен мог бы найти применение как легко летучий рас-
творитель (температура кипения 42°). Широкое применение
в медицине хлороформа как наркотического средства—общеиз-
вестно; хлороформ является также прекрасным негорючим рас-
творителем, а также исходным продуктом для многих органиче-
ских синтезов. Наконец, четыреххлористый углерод находит при-
менение не только в качестве дешевого негорючего растворителя1
(маслоэкстракционное и другие производства), но и в качестве
противопожарного средства. Получение всех этих продуктов
в'большом техническом масштабе является для газовой промыш-
ленности вопросом сегодняшнего дня; дешевый хлор для этой
цели обеспечен.
Окисление. Конечными продуктами окисления метана, как и
вообще всякого органического соединения, являются углекислота
и вода. Они образуются, например, в результате сгорания ме-
тана при избытке кислорода. Регулируя приток окислителя и
температурные условия, можно направить реакцию в сторону
образования различных промежуточных продуктов окисления
метана; важнейший из них, формальдегид, образуется из метана
по следующему уравнению:
СН4 + 20 == H2C0 + НоО.
По вопросу получения формальдегида из метана имеется об-
ширная патентная литература. Процесс окисления начинается
здесь при повышенной температуре (500—600°), которая под-
держивается далее теплотой самой реакции; для ее успешного
течения некоторые авторы рекомендуют применение давления,
а также катализаторов (Си, Fe, Ni, Со). Формальдегид находит
обширное применение в качестве дезинфецирующего вещества и
антисептика; в химической технологии он широко применяется
для изготовления органических красок (фуксин и др.), искус-
ственных смол (бакелит и т. п.) и т. д. Технически формальде-
гид получается пока окислением древесного спирта.
Вместо кислорода для окисления метана при высокой темпе-
ратуре можно пользоваться другими менее энергичными деше-
выми окислителями; таковы, например, водяной газ, углекислота
и окись углерода [6]. Так, с водяным паром окисление метана
может протекать в зависимости от температурных условий по
одному из следующих уравнений:
. СН4 + 2Н20 = С02 + 4Н2;
CH4-f- H2Q = CO Ч-ЗНг-
1 В настоящее время его получают действием хлора на сероуглерод.
5: ’ 131
Получающийся, таким образом, богатый водородом газ может
быть использован после удаления окислов углерода либо для
гидрогенизации, либо для синтетического получения аммиака и
некоторых органических веществ, например для известного син-
теза метилового спирта, осуществляемого ныне в большом тех-
ническом масштабе из окиси углерода и водорода:
СО + 4Н = СН30Н.
Сюда же, наконец, относится окисление метана за счет кисло-
рода некоторых окислов, например окиси бария; при этом обра-
зуются карбиды, а в присутствии азота — цианиды соответствую-
щих металлов; например:
ЗСН4 + ВаО = ВаС2 + СО + 6Н2.
Отметим в заключение, что реакции полного сгорания метана
в настоящее время также нашли применение в процессе утилиза-
ции естественного газа в 'ОША для производства жидкой и твер-
дой углекислоты [7].
Пиролиз. К этой группе можно отнести такие превращения
метана, при которых основную роль играет его термическая обра-
ботка. Простейшим из них является термический распад метана
на элементы, углерод и водород, представляющие значительно
большую техническую ценность по сравнению с естественным
газом: углерод — в виде сажи, в крайнем случае — в виде без-
зольного кокса, водород — как сырье для процессов гидрогениза-
ции, синтеза аммиака и т. п. Хотя распад метана да элементы
требует, как уже было отмечено, очень высокой температуры,
тем не менее некоторые новейшие опыты дают надежду, что про-
блема может быть успешно разрешена не только в лабораторном,
но и техническом масштабе [8]. ’
Не подлежит сомнению, что начальной стадией термического
распада метана является его диссоциация на водород и различ-
ные органические радикалы: СНз, СН2 и СН. В зависимости от
условий эти мимолетно образующиеся свободные радикалы либо
распадаются дальше на элементы, либо конденсируются между
собой с образованием высших углеводородов. Это последнее на-
правление реакций представляет, конечно, совершенно исключи-
тельный интерес, открывая перспективы превращения метана
в жидкое топливо для двигателей внутреннего сгорания или
в сырье для химической промышленности. Работы последнего,
времени, особенно Ф. Фишера и его сотрудников, установили
полную возможность такого превращения метана [9]. Его основ-
ными условиями являются: достаточно высокий нагрев метана
и быстрое выведение продуктов реакции из области высокой тем-
пературы. Давление, невидимому, также способствует конденса-
ции продуктов 'диссоциации. В получаемой таким образом жид-
кой смоле обнаружено присутствие жидких и твердых насыщен-
ных и ароматических углеводородов (бензол, толуол, ксилол,
нафталин и др.), в отходящих же газах найдены водород, этилен
и ацетилен. Аналогичные результаты получены также при дей-
Д32
ствии на метан электрических разрядов, и едва ли можно сомне-
ваться, что превращение метана в жидкие углеводороды займет
со временем видное место среди различных методов рациональной
утилизации естественного газа.
Большой интерес и значение представляет воздействие высо-
кой температуры на гомологи метана. Этот вопрос будет рассмо-
трен подробно в ч. II, в главе о крекинге (гл. III).
ЛИТЕРАТУРА
Стопневич А., Природные газы в России, изд. КЕПС’а, П. (1920).
Оберфелл и Алден, Газолин из природного газа, пер. под ред. Добрян-
ского А. Ф., М.— Л. (1926).
Шахназаров И. X., Естественный газ, его добыча и утилизация, 2-е изд.
М. — Л. (1932).
-Соколов В, А., Методы исследования природных газов М.—Л. (1932).
Burrell Gr., The Recovery of Gasoline from Natural Gas, New York (1925).
Титаренко И. И., Жидкие нефтяные газы, Азнефтиздат (1937).
Эллис В., Химия углеводородов нефти и их производные, пер. с англ.,
ОНТИ, М. (1936).
Исследование и химическая переработка природных и искусствен-
ных нефтяных газов, Сборник статей под ред. Потоловского Л. А.,
Гуллан Л. А. и Афиногенова П. 3. ОНТИ, Баку —Москва (1935).
1. Подробности следует искать в специальных руководствах и пособиях,
например: Лидов А. П., Анализ газов, 2-е из л. (1928); Францен Г.,
Газовый анализ, пер. с нем. Гвоздева С. Я.» М. — П. (1923);
Hempel W., Gasanalitische Methoden, и др.
2. Podbielniak Ж. Д „Oil a. Gas J.“ 27, № 85, 38; 27, № 52, 80 (1929); см.
также Fitch, „Nat. Petr. News" 28, № 25 (1981).
3. Описание ’ различных методов определения газолина в естественном
газе можно найти в цитированной выше книге Оберфелла
и Алдена и др.
4. Наметкин; Забродина, Карконас, Курсанов, Соколов, Успенский, „Журн.
прикл. химии* IV, № 2—3, 335 (1931): Доладугин и Лапкин,
Сборник „Химический состав нефтей и нефтяных продуктов*,
„Труды ГрозНИИ", М. —Л. (1931); Hill, „О1Г Gas J.“ 27, № 38,
140 ‘(1929) и др.
5. Черепенников А. А., „Нефт. хоз.’ 18, № 5, 796 (1930).
& См. особенно работы Фишера Ф. и его сотрудников, „Brennst.-Chem."
9. 39 (1928) и др.; также Клюквин и Клюквина, „Ж. хим. пром.*,
VII, № 11-12 и 13 (1930).
7. „Nat. Petr. News’. 23. № 31, 45 (1931).
8. Junker, „Oil a. Gas. J.“ 27, № 51, 111 (1929).
9. „Brennst.-Chem*. 9, 309 (1928); 10, 108 (1929).
Б. Средние (жидкие) парафины
Группа парафинов, жидких при обыкновенной температуре,
•охватывает обширный ряд гомологов метана — от пентана, CsHrs,
до гексадекана, CieHS4. Как было указано, они входят в состав
главной массы природной нефти, ее жидкой части; с этой точки
зрения они представляют для химии особый интерес. Ознакомле-
ние с ними удобно начать с парафинов бакинской нефти, кото-
рые благодаря работам ряда исследователей, особенно Марков-
никова и его учеников, изучены наиболее полно.
133
ПАРАФИНЫ БАКИНСКОЙ НЕФТИ
Бакинская нефть не богата парафинами. Как и в других
нефтях (см. ниже), они сосредоточены главным -образом во фрак-
циях до 100°, в более высоких погонах содержание их быстро
падает. Простейшими представителями этой группы парафинов
являются пентаны. состава С5Н12.
Пентаны СбН12. Все три пентана в бакинской нефти обнару-
жены: они составляют главную часть нефтяного погона до 40°.
Нахождение нормального пентана, СНз(СН2)з • СНз (температура
кипения 36°,3), указано впервые Менделеевым [1], а изопентана,
‘ С2Н5СН(СНз)2 (температура кипения 28'), —сначала Марковни-
новым [2], позднее — Ашаном [3]. В своих заключениях по дан-
ному вопросу эти исследователи руководствовались главным
образом составом и физическими свойствами соответствующих
погонов. Результаты фракционировки легкого петролейного
эфира, полученные Ашаном, приведены в табл. 3s.
Действительно, кроме состава и температуры кипения, удель-
ные веса соответствующих фракций также хорошо подходят
к удельным весам пентана и изопентана. Если же к соответствию
в составе и физических свойствах добавить, что и в отношении
к химическим реагентам (кислоты азотная, серная и др.) фрак-
ции эти оказались вполне подобны парафинам, то очевидно, что-
Таблица 38
Темп, кипения Колич. в t Уд. вес ^17,25
26-89 35 0.62443
30—32° 85 6,62527
32—35° 80 0,6-674
35-40° 28 0,63515
40—45° 17 0,65094
45—49° 58 0,66078
нахождение пентана и изопен-
тана в бакинской нефти мож-
но считать доказанным. Иа
табл. 38 видно также, что изо-
пентана в бакинской нефти
значительно больше, чем нор-
мального пентана.
В более легких частях бен-
зина Марковников (совместно ;
с Касаткиным) обнаружил [4]
присутствие еще одного угле-
водорода, вполне устойчивого
к крепкой азотной кислоте-
У глеводород кипел очень
9°. По всей вероятности, это—третий пен-
постоянно при 9 . По всей вероятности, это —третий пен-
тан, тетраметил-мет ан, С(СНз)4 (температура кипения. 9,5°).
Гексаны, СвНн. Из гексанов кавказской нефти раньше других
Менделеевым [5] был указан нормальный гексан, СНз • (СНг)! •
• СНз (температура кипения 69°).
Для получения этого углеводорода из нефти в чистом виде
Марковников [6] подверг соответствующую фракцию бензина
обработке дымящей азотной кислотой, которая, разрушая при-
месь циклического углеводорода -(главным образом метил-цикло-
пентана), не действует при обыкновенной температуре на геюсан.
После такой обработки получился углеводород со следующими
свойствами: температура кипения 68,5—69° (при 768 мм)»
d30°== 0,6681.
134
Очевидно, это — чистый гексан. Этим методом было найдено,
'что фракция 68—70° бакинского бензина содержит до 40% гек-
сана, фракция 70—72° — до 25% и фракция 72—74° —
-около 10%.
Для суждения о присутствии в кавказской нефти других
гексанов и вообще для предварительной ориентировки в составе
различных нефтяных фракций мы будем пользоваться при даль-
нейшем изложении результатами той разгонки бакинского бен-
зина, график которой приведен выше <фиг. 19); соответствующие
этому графику цифровые данные для фракций от 42—44° до 74—
76° приведены в табл. 39.
Таблица 39
Темп, кипения Колич. в г Уд. вес d15 "15 Темп. кипения Колич. в г Уд. вес d15 "16
42-44° 5 0,668 58—60° 144 0,6*8
44—46° 16 0,677 ' 60—62° 278 0,666
46—48° 3 0,704 62-64° 698 0,668
48-50° 24 0,718 64-66° 404 0,673
50—52е 51 0,716 66—68° 346 0,680
52-54° 19 C.T0 68—70° 547 0,692
54—56° 7 0.688 70—72= .. 971 0,712
56—58° 18 0,677 72-74° 1345 0,731
Из таблицы -видно, что вслед за небольшим повышением ко-'
личества погона около 44—46е, ближе пока не исследованного,
замечается значительное увеличение размеров фракций 48—50—
52° с максимальным подъемом удельного веса около 50°. Как
будет показано в гл. VI, это зависит от нахождения в данном
погоне одного из простейших нафтенов, циклопентана. Вместе
с этим нафтеном, как показал Маркоеников [7], в этих же фрак-
циях содержится один из гексанов, а именно триметил-этил-ме-
тан, (СНз)зС • С2Н5 (температура кипения 50°).
Открытие этого углеводорода в нефти и выделение его были
осуществлены следующим образом. При нитровании фракции
50—51° по Коновалову было замечено, что, кроме жидкого ни-
тросоединения, отвечающего циклопентану, получается какое-то
кристаллическое вещество с температурой плавления 40°. Ана-
лиз его привел к формуле нитрогексана, CeHisNOa, характер ни-
тро оказался вторичным. Нормальный гексан дает жидкое нитро-
соединение; изомеры его, содержащие третичный водород, должны
были бы дать преимущественно третичное нитро. Единственный
изомер нормального гексана, не .содержащий третичного водо-
рода,'— триметил-этил-метан. Если вышеупомянутое вторичное
кристаллическое нитро соответствует именно этому изомеру
тексана, то при восстановлении его должен получиться, оче-
видно, пинаколин:
(СН3)3 • С • СНХ02 • СН3 —> (СН3)3 С • СО • СН3. .
185
Соображения эти подтвердились полностью. При восстановле-
нии нитросоединения из указанной выше нефтяной фракции
действительно получился кетон, CeHisO, который не реагировал
с бисульфитом и дал семикарбазон с той же температурой плаь
вления, как семикарбазон синтетического иинаколина.
После трех последующих нитрований нефтяной фракции 50-*-
51° остался углеводород, который был прокипячен с азотной
кислотой удельного веса 1,4. Его физические свойства оказались
теперь следующие: температура кипения 48,5° при 739 мм,
удельный вес do0 = 0,6646.
Сравнение со свойствами синтетического триметил-этил-ме-
тана показывает почти полное совпадение. Выделение этого угле-
водорода из смеси с циклопентаном оказалось, таким образом,
возможным благодаря исключительной устойчивости его по от-
ношению даже к крепкой азотной кислоте.
Из других изомеров гексана с несомненностью доказано при-
сутствие в кавказской нефти диизопропила, (CIIsJaCH . СН.
• (СНз)2 (температура кипения 5ь°).
Для выделения этого углеводорода из бакинской нефти Мор-
ковников [8] нагревал фракцию 57—60° с серно-азотиой смесью
до 50°, взбалтывал ее затем с небольшим коли честном азотной
кислоты и кипятил над натрием. После такой очистки углеводо-
род перегонялся главным образом при 58—59} и имел удельный
вес di5 = 0,666; состав его отвечал формуле СвПк.
Ашан [9] подверг ту же фракцию бензина (температура ки-
пения 57—59°; d?7’25 = 0,(»б4ь5) охло^иию. Главная часть по-
лученного хлорида кипела 117—119J и имела состав CeHisCU
сравнение ее удельного вес с удельным весом третичного хлори-
стого диизопропила не оставляло сомнений в тождестве этих
препаратов.
Кроме рассмотренных гексанов, теоретически возможны и получена
еще два углеводорода состава СеНм:
(СНз)гСН • (СНг)2 • CII3 (температура кипения 60,2°) — 1,1-диметил-бутан.
СНз СН (СоНь)^ (температура кипения 63V-) — метил-диэтил-метан.
Обращаясь к табл. 39, мы замечаем прежде всего резкий скачок коли-
чества погона от фракции 56—5Ь° (1Ь г) к соседней 58—60° (144 г), отве-
чающей, очевидно, диизопропилу (см. выше). Затем происходит продолжи-
тельная задержка термометра на 62—04°, причем удельный вес, понижаю-
щийся неизменно от 50°, достигает при во—62° минимума, а далее начи-
нает постепенно повышаться, очевидно, от примеси более тяжелого нафтена
(метил-циклопентана). Сопоставляя удельные веса фракций 60—62—660,
с удельными весами только что упомянутых гексанов, мы замечаем весьма
близкое соответствие. По мнению Марков никоей [io] возможно ттовтфму,,
что один из этих гексанов, а быть может и оба, находятся в соответствую-
щих погонах нефти и притом в довольно значительном количестве. Не-
давно оба они действительно обнаружены в нефти Мид-Континента (см.
ниже).
Гептаны, С7Н16. Для суждения -о присутствии в бакинской
нефти более высококипящих парафинов необходимо продолжить
таблицу с результатами фракционировки бакинского бензина
(табл. 40).
136
Таблица 40
Темп, кипения Колич. в г УЙ- вес Я15 “15 Темп, кипения Колич. в г Уд. вес □15 “15
72—74° 1345 0,731 86— 88° 281 0,735
74—76° 831 0,745 88- 90° 474 0,729
76—78° 1124 0,742 90— 92° 11239 0,726
78—80° 1184 0,749 92— 94° 387 0,730
80—82° 1940 0,755 •94— 96° 298 0,738
82—84° 1С07 0,759 96— 98° 148 0,741
84-86° 400 0,754 98—100° 255 0,753
Судя по высокому удельному весу нефтяных фракций от 72°,
надо думать, что количество парафинов в бакинской нефти, на-
чиная с гептанов, не может быть велико. Естественно также,
что открытие их, не говоря уже о выделении в чистом виде
вследствие усложнения состава фракций становится все труд-
нее и труднее.
Для суждения о том. находится ли нормальный гептан (тем-
пература кипения 98,5°) в апшеронской нефти, можно восполь-
зоваться.-результатами, полученными Марновниковым [11] при
обработке дымящей азотной кислотой нефтяной фракции 100—
102°. Оказалось, что при частичном растворении углеводорода
в азотной кислоте удельный вес остатка <ючти не изменился,
при избытке же азотной кислоты весь углеводород растворился
без остатка. Так как по мнению самого автора трудно допустить,
чтобы фракция 100—102° не содержала примеси погона, .кипя-
щего всего на 2° ниже, то, учитывая полную устойчивость нор-
мальных парафинов к дымящей азотной кислоте, приходится
сделать вывод, что нормального гептана или вовсе нет в апше-
ронской нефти, или если он и содержится в ней, то в ничтож-
ном количестве.
Из гептанов ненормального строения в бакинской нефти обна-
ружены два: триметил-пропил-метан, (ОНз)зС • ОНг -ОНг • СНз
(температура кипения 78,5—79°), и симм. тетраметил-пропан,
(ОНз)гСН - ОНг СН(СНз)2 (температура кипения 83—84°).
Первый из этих углеводородов, постоянно сопутствующий
нефтяному циклогексану, был обнаружен в бакинской нефти
Марковниковым [12]. Чтобы изолировать его. фракция 80—82 е
бензина была подвергнута сначала вымораживанию с помощью
жидкого воздуха. Когда главная масса циклогексана была таким
образом удалена, углеводород был обработан дымящей серной,
а затем подвергнут нитрованию азотной кислотой удельного
веса 1,15 при 110—115°. Углеводород, не вошедший в эту реак-
цию, после кипячения с азотной кислотой удельного веса 1,4
показал состав, отвечающий формуле С7Н16, и свойства, близ-
кие к свойствам синтетического триметил-пропил-метана,
а именно: температура кипения 78-,5—79°; удельный вес =
= 0,7083; d20 =0,6921.
137
Строение ею было доказано превращением через вторичное
нитро в этил-бутил-пинаколин:
(СН8)8С • CHN02 • СН2СН8 — (СН3)8 с • со • С2Н6.
Второй из указанных выше изогептанов, тетраметил-пропан,
был получен синтетически и найден в нефти Хониным [13] при
исследовании третичных нитропродуктов, получающихся при ни-
тровании нефтяного циклогексана (фракция 80—82°). Амин,
приготовленный из такого третичного нитро, оказался неодно-
родным; тем не менее, при кристаллизации полученной из этого
амина тиомочевины удалось выделить вещество, вполне тожде-
ственное с тиомочевиной соответствующего амина из синтети-
ческого симм. тетраметил-пропана как по кристаллической
форме (ромбические пластинки), так и по точке плавления.
Хлороплатинаты обоих аминов оказались также тождественны'
по своим характерным многогранным кристаллам.
Октаны CeHi8 и другие парафины. Судя по анализам различ-
ных нафтенов, выделявшихся из фракций бакинского бензина
с температурой кипения выше 100d (см. гл. VI), присутствие
в них парафиновых углеводородов не подлежит никакому сомне-
нию. Сведения об этих парафинах, однако, крайне ограничены.
Так, Марковников [14] после обработки фракции 114—118° ды-
мящей азотной кислотой получил небольшой остаток, состав ко-
торого казался средним между С7Н14 и CbHJ8. Так как после ука-
занной обработки уже не могло остаться какого-либо углеводо-
рода с третичным водородом, то по мнению автора здесь вое-
можно присутствие трилетил-бутил-летана, (СН3)зС • СН2 • СН2,
СН2 • СНз.
Элементарный состав фракций бакинской нефти с температу-
рой кипения около 120° и выше также указывает на заметное
присутствие в .них углеводородов ряда метана, причем содержа-
ние -их заметно увеличивается к 124—126°. Это обстоятельство
может'служить косвенным указанием на присутствие здесь нор-
мального октана (температура кипения 125,8°).
Об отдельных, более высокомолекулярных углеводо!юдах ряда
метана бакинской нефти сведений пока не имеется, хотя нахо-
ждение их в различных ее погонах не подлежит сомнению.
ПАРАФИНЫ РАЗЛИЧНЫХ НЕФТЕЙ
По сравнению с парафинами бакинской нефти средние пара-
фины других нефтей СССР изучены мало. Так, для грозненской
нефти имеется указание на присутствие в ней пентана и изо-
пентана, гексана, гептана и изогептана [15]. Из этих парафин
нов только изогептан (температура кипения 90,5—91,5°) был
охарактеризован более или менее достаточно, да и то без вся-
ких данных об его строении.
Особо должно быть отмечено удачное применение к изучению
состава бензина хлорсульфоновой кислоты: при ее действии ваг
мещенные нафтены и изопарафины вступают с ней в реакцию,
188
тогда как нормальные парафины ею не затрагиваются. В резуль-
тате повторной обработки этим реагентом соответствующих фрак-
ций грозненского и майкопского бензинов удалось изолировать
из них следующие нормальные парафины: гексан, гептан, октан,
нонан и декан [16].
Переходя к нефтям других стран, остановимся прежде всего
на парафинах пенсильванской нефти — первой американской
нефти, химический состав которой был подвергнут детальному
исследованию и неоднократно изучался различными авторами
в последующем.
Первые химические исследования [17] пенсильванской нефти
относятся к 1862—63 г. (Пелуз и Кагур). Сначала нефть была
подвергнута фракционировке без предварительной очистки,
позднее—после предварительной очистки серной кислотой.
Фракционировкой из нее было выделено 11 углеводородов состава
СпНг» + 2, от СбН12 до С15Нз2: кроме того, получены указания на
присутствие в наиболее низкокипящих погонах также бутана,
С4Н10. Углеводороды были охарактеризованы анализами, плот-
ностью пара, устойчивостью к брому, серной и азотной кисло-
там, а также приготовлением соответствующих хлоридов. Полу-
ченные результаты дали авторам пород утверждать, что пенсиль-
ванская нефть состоит из углеводородов ряда метана, начиная
от самых низших его представителей.
Вскоре затем пенсильванская нефть была подвергнута более
тщательной разгонке и изучению [ 18], которые значительно по-
полнили результаты первых ее исследований (Шорлеммер, позд-
нее Морган; Уоррен я др.). Прежде всего было вновь показано,
что пенсильванская нефть содержит пентан, гексан, гептан и
'октан. Вместе с тем было обнаружено, что при нагревании соот-
ветствующих фракций с азотной кислотой всегда образуется не-
большое количество ароматических нитросоединений (нитробен-
зол, нитротолуол и др.); данные эти послужили доказательством
присутствия в пенсильванской нефти ароматических углеводо-
родов (гл. IV). Наконец, путем более тщательной фракциони-
ровки . было установлено, что парафины пенсильванской нефти
образуют два изомерных ряда: более высококипящие углеводо-
роды нормального строения и ниже кипящие, изопарафины. Это
последнее положение было подтверждено [19] впоследствии ря-
дом других исследователей (Мэбери и Гедеон, Сидней Юнг и др.)
на длинном ряде примеров, причем, однако, ближайшее строе-
ние выделенных изопарафинов в большинстве случаев остава-
лось невыясненным. Таким образом было установлено присут-
ствие в пенсильванской нефти следующих парафинов и изопа-
рафинов [20]:
С5Н12 — пентан (температура кипения 36,3°) и изопентан
(температура кипения 28°). После очистки соответствующих
фракций дымящей азотной кислотой углеводороды были полу-
чены в совершенно чистом виде, так что их свойства полностью
совпадали со свойствами синтетических углеводородов.
С6НИ — гексан (температура кипения 69°) и изогексан (тем-
139
лература кипения около 61°). Из этих углеводородов гексан был
выделен настолько чистым, что все его константы, включая упру-
гость пара при разных температурах, а также его критическую
температуру, вполне совпадали со свойствами синтетического
нормального гексана.
C7Hie — гептан (температура кипения 98,4 ) п изогептан
(температура кипения 90,3 ). После многократных перегонок
углеводороды эти были получены с небольшими примесями со-
ответствующих нафтенов. Лишь после бромирования с последую-
щей фракционировкой бромидов и их восстановлением удалось
очистить эти парафины рт. примесей, причем нормальный гептан
был получен в совершенно чистом виде.
С8Н1в — октаны. Нормальный октан (температура кипения
125,8°) был выделен в виде фракции 124—125 . которая по ана-
лизу, плотности пара и составу монохлорида вполне соответство-
вала синтетическому октану. Изомерный углеводород, изооктан,
был выделен в виде фракции 118,5—119,5', которая, однако, при
обработке серной кислотой и нитрующей смесью заметно изме-
няла свой удельный вес; повидимому, она содержала значитель-
ное количество октонафтенов.
С9Н20 — нонан. Имеются указания на присутствие в пенсиль-
ванской нефти только одного, невидимому, нормального нонана
(температура кипения 150,5°).
С10Н22 — декан. Сырые фракции, по температуре кипения со-
ответствующие нормальному декану (температура кипения 173°)
и его изомерам, имеют состав, отвечающий формуле СюН2о, а не
СюН22. Лишь после обработки серной кислотой и нитрующей
смесью и удаления ароматики состав их определился в соответ-
ствии с формулой С10Н22. Таким образом были выделены два де-’
кана: один с температурой кипения 173—174°, отвечающей
нормальному декану, и другой, изодекан, с температурой
162—163°.
С11Н24 — ундекан и (лгНм — додекан. Оба этих углеводорода
подобно деканам были обнаружены в соответствуюпщх фракциях
нефти после тщательной их обработки кислотами для удаления
ароматики; ундекан —во фракции 190—191°, додекан — во
фракции 214—216°.
Разгонка нефтяных фракций с температурой кипения выше
200’ обычно производилась в вакууме. Так, при исследовании
этих погонов, Мэбери [21] сначала перегонял сырую пенсиль-
ванскую нефть без доступа ‘воздуха при давлении 50 мм. После
8 перегонок каждая фракция подвергалась обработке серной кис-
лотой, а затем вновь фракционировалась при 50 мм до 30 раз
и более с отбором фракций в пределах 1° . Полученные таким
образом фракции анализировались и характеризовались различ-
ными константами (температура кипения при нормальном давле-
нии, удельный вес, коэфициент рефракции, плотность' пара);
изучались также продукты их хлорирования. Таким образом
в пенсильвинской нефти были обнаружены следующие пара-
фины:
140
С18Н28 — тридекан
С14Н80 — тетрадекан
С1бН32 — пентадекан
С16Н34 — гексадекан
СиНзд— гептадекан
С igHgg — октадекан
температура кипения 221—222'
я 236—238'
п » 256—257’
п п 275—276'
п 288—289'
п » 300—ЗОГ
и т. д. до СгвНбв, октоэйкозана, включительно.
Однако едва ли можно сомневаться, что все эти высптие па-
рафины уже не представляют собою индивидуальных веществ.
Так, при охлаждении полученных указанным путем углеводоро-
дов, начиная с гептадекана, С^Ндв, из жидкости выделялись
кристаллические парафины, по свойствам близкие к высшим
парафинам нормального строения; отфильтрованное же от кри-
сталлов масло обнаруживало явно непредельный состав.
Из других американских нефтей раньше других были изучены нефти
Охайс, Канады, Калифорнии и Тексаса.
В .низших фракциях юхайской нефти были обнаружены [22] те же.
парафины, что в пенсильванской нефти, начиная от пентана в. изопентана
и кончая дедеканом. Однако уже гептановая и октановая фракции охайской
нефти содержали настолько заметное количество углеводородов! непредель-
ного состава (нафтены, ароматика), что только после тщательной очистки
серной и азотной кислотами здесь удавалось получить углеводороды, по
своему составу отвечающие формулам гептана и октана. Фракционировка
высших фракций охайской нефти в общем дала тот же результат, что ж
для пенсильванской нефти [23], и здесь при охлаждении фракций выде-
лялся твердый парафин, отфильтрованное же масло имело непредельный
состав.
Исследование углеводородной части канадской нефти [24] в общем
дало такие же результаты, как нефтей пенсильванской и охайской. Однако
содержание углеводородов непредельного состава оказалось здесь больше
и соответственно содержание парафинов — меньше, чем в охайской нефти.
Что касается нефтей Калифорнии и Тексаса [25], то по своему составу
они удаляются от пенсильванской нефти несомненно еще дальше, чем
нефти Охайо и Канады. Тем не менее, в самых низших погонах кали-
форнской нефти присутствуют, невидимому, те же парафины, что и
в пенсильванской нефти, кончая гексаном.
Наконец, необходимо остановиться на отмеченной в предше-
ствующих главах нефти одного из месторождений Оклахомы
(Кой-Коунти, South Ponca, Мид-Континент, США); из ее бензина
в результате тщательной многолетней коллективной работы было
выделено свыше 20 парафинов и изопарафинов различного
строения и молекулярного веса, а именно [26]:
СеН14 — 2,3 диметил-бутан, температура кипения 58°,0
C6Hi4 — 2-метил-пентан „ „ 60е,3
СеН14 — 3-метил-пентан „ „ 63°,3
СеН14—норм, гекоан „ „ 68°,7
Выделение и разделение этих четырех гексанов производи-
лось фракционировкой, перегонкой с метиловым и этиловым
спиртом и вымораживанием. Чистота лучших образцов превы-
шала 95—98%. Для иллюстрации цолученных результатов при-
141
водится табл. 41, в которой сопоставлены свойства нефтяных и
синтетических гексанов.
Таблица 41
Гексаны нефти Ох лакомы
Гексаны Темп, кип. (760 мм) Темп. плав л. Уд. вес Коэф, рефр. flD W
Диизопропил (СН3)2 СН • СН (СН3)2 Нефтяной углеводород 58,0 -135 ' 0.668 1,378
Синтетический углеводород .... 58,1 —135 0,662 1,377
2-метил-пентан (СН3)2 СН(СН2)гСНз Нефтяной углеводород 60,4 -143 0.659 1,374 1,371
Синтетический углеводород .... 60,2 —' • 0,654
З-метил-пентан СН3 • СН, • СН(СНд) • СН£ • СН3 Нефтяной углеводород 63.3 -118 0,665 1,376
Синтетический углеводород . . . 63,2 0,665 1,876
Норм, чексан СНа(СН2\СН3 Нефтяной углеводород 68.8 —97,2 0,673 1,379
Синтетический углеводород .... 68,8 —95 0.659 1,Ь75
С7Н16 — 2,2-диметил-пентан (температура кипения 78.9°). Вы-
делен фракционировкой и вымораживанием в виде смеси 54%
2,2-диметил-пентана и 40% циклогексана.
C7Hie — 2-метил-гексан (температура кипения 9о,оВыде-
лен фракционировкой и кристаллизацией из смеси жидкого ме-
тана с пропаном. Степень чистоты 99,9%.
C7Hie — норм, гептан (температура кипения 98,4°). Выделен
фракционировкой, нитрованием для удаления следов толуола и
вымораживанием для отделения от метил-циклогексана и дру-
гих примесей. Степень чистоты 99,8%.
Callie — 2-метнл-гептан (температура кипения 117,2°). Выде-
лен фракционировкой и кристаллизацией из смеси жидкого ме-
тана с пропаном. Степень чистоты 97%.
CeHie — норм, октан (температура кипения 125,6’). Выделен
фракционировкой и вымораживанием. Степень чистоты лучшего
образца была 99,1%.
С9Н20 — 2,6-диметил-гептан (температура кипения 135,2°).
Выделен фракционировкой, извлечением жидким сернистым
ангидридом, адсорбцией селикагелем, перегонкой с уксусной кис-
лотой и кристаллизацией из жидкого дихлор-дифтор-метана.
Степень чистоты лучшего образца 99%.
CsHgo — изононан (температура кипения 140,9°). При фрак-
ционировке получается в смеси с нонанафтеном (температура
142
кипения 141,2е). Дальнейшее разделение производилось извле-
чением жидким сернистым ангидридом, адсорбцией селикаге-
лем, перегонкой с уксусной кислотой, перегонкой при понижен-
ном давлении, кристаллизацией из дихлор-дифтор-метана,
а также — из смеси жидкого метана с пропаном. Степень чи-
стоты лучшего .образца доходила до 85%.
С9Н20 *- 4-метил-октан, температура кипения 142°,4
С-9Н90— 2-метил-октан, ,, „ 143°, 8
С9Но0 — 3-метил- октан, „ „ 144°,2
Эти три изопарафина были выделены теми же методами,
какие были применены для получения предшествующего изо-
нонана. Степень чистоты отдельных углеводородов была со-
ответственно 80, 99,9 и 95%.
С9Н20 — норм, нонан (температура кипения 150,7°). Выде-
лен фракционировкой и вымораживанием. Степень чистоты
99,9%.
С9Н22 — норм, декан (температура кипения 174,0°). Выде-
лен как предыдущий. Степень чистоты лучшего образца выше
99,9%.
Кроме перечисленных парафинов и изопарафинов, в окла-
хомской нефти был обнаружен в процессе той же работы или
предполагается целый ряд других представителей ряда метана,
как-то: 3-метил-гексан, 2,2, 3,3-тетраметил-бутан 'л некоторые
другие изомеры октана и нонана. Что касается высших пред-
ставителей ряда метана, то ближайшее исследование вопроса
о нахождении отдельных их представителей в оклахомской
нефти находится еще в процессе работы.
Возвращаясь к европейским нефтям, остановимся в первую
очередь на парафинах галицийской и румынской нефтей.
Некоторые галицийские нефти содержат довольно значи-
тельное количество легких фракций, например до-30% и более
погона, выкипающего до 150°. Их ближайшее- исследование,
произведенное на нефти из Борислава [27], обнаружило содер-
жание следующих парафинов: пентан и изопентан, гексан и
изогексан, гептан, нонан и 2 декана. Все они были выделены
из соответствующих фракций путем тщательной фракцио-
нировки и очистки азотной кислотой. Свойства почти всех
этих парафинов довольно хорошо совпали со свойствами пара-
финов из американских нефтей.
В румынской нефти также обнаружен ряд парафиновых
углеводородов [28]. Так, из нефти месторождения .Колибааи
выделены: бутан и изобутан, пентан и изопентан, гексан и,
вероятно, изогексан, гептаны’, возможно также нахождение
в этой нефти октана и нонана>
Присутствие парафинов в других европейских нефтях' (гер-
манской, итальянской и др.), судя по 'Составу и свойствам от-
дельных их погонов, также не подлежит сомнению. Более точ-
ных данных по этому вопросу пока не имеется.
143
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ И СОДЕРЖАНИЕ ПАРАФИНОВ
В РАЗЛИЧНЫХ НЕФТЯХ
Непосредственное количественное определение парафинов
в углеводородных смесях пока невозможно, и содержание угле-
водородов этого класса в нефтяных дестиллатах производится
в настоящее время по остатку, т. е. путем вычета из общей
• массы углеводородов всего, что приходится на долю непредель-
ных, ароматических и нафтеновых углеводородов. •
Для суждения о содержании парафинов в различных нефтях
может служить табл. 42, составленная по данным ГрозНИИ [29]
аналогично табл. 29 для фракций различных нефтей с темпе-
ратурой кипения от 60 до 300°. Как видно из сопоставления
данных этих двух таблиц, различные нефти различаются между
собою по содержанию парафинов еще больше, чем по содержа-
нию ароматических углеводородов. Действительно, наряду
с нефтями, невидимому вовсе не содержащими парафинов (ка-
лужская нефть), мы находим в табл. 42 примеры нефтей, не-
которые фракции которых содержат парафинов во—70%
(до 86%).
Таблица 42
Содержание парафинов в различных нефтях
№ п/п I Месторождение Уд. вес (15°) % парафинов во фракциях 1 с тем- пературой кипения
60-95° • 95—122° 1 123-150° 150—200° • 1 с05<— 002 250—300°
1 Сураханы 0,848 24 18 43 17 38 45
2 Балаханы 0,876 44 31 28 13 11 19
3 Балаханы-Раманы-Са- бунчи 0,867 52 35 14 18 89 42
4 Бинагады ....... 0,931 48 41 36 13 8 6
5 Биби-Эйбат 0,865 47 37 36 19 27 31
6 Грозный, парафин., фон- тан 0,844 62 57 61 57 59 61
7 Грозный, слабопара- фин-, фонтан 0,849 61 52 56 48 52 51
8 Грозный, беспараф., легкая 0,860 60 ‘ 51 54 43 36 35
9 Вознесенская 0,900 86 68 48 18 17 27
10 Майкоп 0,848 53 42 43 36 35 31
11 Калужская 0,955 — .—- —
12 Доссор (Эмба) 0,862 56 36 39 24 24 26
13 Пермская 0,941 39 22 20 25 29 27
14 Тонкава, Оклахома . . 0,821 59 49 50 39 44 46
15 Девинпорт, Оклахома . 0,796 66 57 61 55 52 51
16 Мексиа, Тексас .... 0,845 48 52 61 63 68 59
17 Хентингтон-Бич, Кали- • форния 0,897 65 38 35 22 30 31
1 Содержание отдельных фракций в различных нефтях см. табл. 29
(строка II при каждом месторождении)-
144
Из нефтей СССР наиболее богаты парафинами нефти Гроз-
ненского района. Большинство бакинских нефтей содержат уже
значительно меньше парафинов, и это различие между гроз-
ненскими и бакинскими нефтями особенно увеличивается для
более высоких фракций. Эмбенская и пермская нефти по со-
держанию парафинов приближаются к бакинским, майкопская
же — к грозненским. Из американских нефтей на первом месте
по содержанию парафина должны быть поставлены пенсиль-
ванская и охайская нефти, а также некоторые нефти Мид-Кон-
тинента. Нефти Калифорнии и Тексаса более приближаются
в этом отношении к бакинским нефтям. Из европейских нефтей
богаты парафинами галицийские нефти; румынские, невиди-
мому, несколько уступают им в этом отношении.
Как общее правило, наибольшее содержание парафинов при-
ходится на фракции, выкипающие ниже 100—150°; для пого-
нов же с температурой кипения выше 150° содержание пара-
финов обыкновенно резко снижается. Значительно реже встре-
чаются такие нефти, в которых содержание парафинов в раз-
личных фракциях до 300 заметно не уменьшается; такое явле-
ние наблюдается, например, для грозненской парафинистой и
некоторых других нефтей. Наконец, еще реже встречаются
нефти, в которых содержание парафинов в более высококипя-
щих погонах не только не уменьшается, но даже возрастает
(Сураханы, Мексиа). Таким образом, как общее правило, рас-
пределение парафинов в различных нефтяных погонах ^изме-
няется в направлении, обратном тому, по которому изменяется
в тех же погонах содержание ароматических углеводородов. •
Никакой ярко выраженной связи между содержанием пара-
финов в нефтях и их удельным весом не наблюдается.
Как видно из табл. 42, высокое и низкое содержание
парафинов можно встретить как среди легких, так и среди тяже:
лых нефтей.
Вопрос о 'Содержании средних (жидких) парафинов в раз-
личных нефтепродуктах представляет особый интерес для бен-
зинов и значительно меныпий в отношении керосинов. Для
суждения по этим вопросам можно воспользоваться данными
той же табл. 42; дополнением к ней может служить табл. 43
Как видно, колебания в содержании парафинов в различных
бензинах значительно меньше, чем в сырых нефтях. Так на-
пример, содержание парафиновых углеводородов в большинстве
бензинов СССР колеблется между 40 и 60%. поднимаясь выше
60% только для бензинов из грозненской парафинистой нефти,
низшие фракции которой выделяются особенно ярко выражен-
ным метановым характером. С другой стороны, содержание тех
же углеводородов в бензинах из нефтей нафтенового типа (Ба-
лаханы, Сураханы) спускается до 30% и ниже.
Значительно бблыпие колебания в содержании парафино-
вых углеводородов наблюдаются для наших керосинов: если, на-
пример. с одной стороны, содержание их в керосине из гроз-
ненской парафинистой нефти достигает почти 60%. то, с дру-
145
Ю Зак. Г622 Химия нефти.
Таблица 4?
Содержание парафинов в бензинах и керосинах СССР 1
1 П/н .Месторождение нефти Содержание парафинов В /0 1 д/д Месторождение нефти Содержание парафинов в %
бенз, керос. бенз, кероок
1 2 3 4 5 6 7 8 9 10 Сураханы 26 34 Балаханы 82 13 Биби-Эйбат .... 40 24 Бинагады 41 — Грозный, парафин, нефть 67 — • авпо Грозный, парафин, нефть 64 59 легк. Грозный, парафин, нефть 63 59 тяж. Грозный, слабопа- рафин. нефть . . 57 50 легк. Грозный, слабопа- рафин. нефть . . .67 52 тяж. Грэзный, беспара- фин. нефть, нижн. пл 56 39 легк. 11 12 13 14 15 16 Грозный, беспара-. фин. нефть, нижн. пл 52 36 тяж. Грозный, беспара- фин. нефть, верхи, пл 44 • 16. легк. Грозный, беспара- фин. нефть, верхи, пл 40 10 тяж. Майкоп 48 — авпо Майкоп 44 30 тяж. Доссор (Эмба) 25
гой стороны, в керосинах из нефтей нафтенового типа (бала-
ханской, грозненской бесларафиновой и т. л.) содержание тех
же углеводородов составляет всего 10—15%.
ПРИМЕНЕНИЕ И ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ ПАРАФИНОВЫХ
УГЛЕВОДОРОДОВ НЕФТИ
Подобно тому, как это было сделано выше для ароматиче-
ских углеводородов нефти, мы рассмотрим сначала, какое прак-
тическое значение могут представлять парафины нефти в ка-
честве сырья для химической переработки или иных целей,,
а затем остановимся на тех специфических свойствах, которые
могут придавать парафины различным нефтепродуктам.
Нефтяные углеводороды, как известно, являются прекрас-
ными растворителями для самых разнообразных веществ, осо-
бенно органических, и широко применяются в этом направле-
нии в лабораториях и в промышленности в виде разлого рода
легких нефтяных погонов: петролейного эфира, лигроина и т. ш
Наряду с этими продуктами известное применение находят
1 Для характеристики этих нефтепродуктов см. также табл. 30.
146
также более узкие нефтяные фракции; таковы, например, неф-
тяной пентан или нефтяной гексан, которые можно найти
в прейскурантах всех крупных химических фирм. Эти нефтя-
ные углеводороды получаются -из соответствующих, более или
менее тщательно выделенных нефтяных фракций путем очистки
их кислотами — азотной и серной. Из них нефтяной пентан
получил за последнее время применение в качестве исходного
материала для приготовления амиловых спиртов и их уксус-
ных эфиров, которые широко применяются sb качестве раство-
рителей для нитролаков в лаковой промышленности.
Для получения из нефтяного пентана амиловых спиртов-
применяют нефтяную ‘ фракцию с температурой кипения 20—40',
которая в главной массе состоит из пентана и изопентана [30].
Фракция эта подвергается прежде всего хлорированию в паро-
вой фазе при температуре выше 100° и большом избытке угле-
водорода. Первыми продуктами реакции являются, первичные
и вторичные хлорпентаны по уравнению:
С5Н12 + С12 = С5Н11С1 + НС1.
В присутствии избытка хлора, хлорирование может пойти
дальше с образованием ди- и полихлоридов, например:
С6НПС1 + С12 == С6Н10С12 4- НС1.
Это последнее направление реакции является побочным и
ненужным; чтобы его избежать, необходимо, чтобы углеводород
был в избытке, и тогда ди- и полихлоридов образуется сравни-
тельно немного. Хлористый водород, образующийся при реак-
ции, поглощают водой и получают таким образом крепкую соля-
ную кислоту, легко очищаемую от возможных примесей органи-
ческих соединений (пентаны, хлорпентаны и т. п.).
При фракционировке продукта реакции в первую очередь
отходит не вошедший в реакцию углеводород, который идет за-
тем на повторное хлорирование. За углеводородной фракцией
следует смесь монохлоридов, которая почти нацело перего-
няется в пределах 85—107°; более тщательной фракциониров-
кой из этой смеси хлоридов удается выделить некоторые чистые
монохлориды, например первичный. хлористый амил и др. На-
конец, последним продуктом реакции является смесь дихлори-
дов пентана, не* представляющая особого интереса.
Смесь монохлоридов пентана и изопентана путем гидролиза
в присутствии слабых щелочей, соды или поташа превращается
далее в соответствующие спирты по уравнению:
СбНпС1 + Н20 = сБнпон+на.
Часть хлоридов расходуется при этом на побочную реак-
цию, не имеющую (производственного значения, а именно на
образование из хлоридов непредельных углеводородов, амиле-
нов, по уравнению:
СсНиС1 = СбН10 + НС1.
10*
147
Образование амиленов при гидролизе амилхлоридов наблю-
дается всегда; но применение слабых щелочей, особенно в при-
сутствии эмульгаторов, значительно ослабляет это побочное на*
правление реакции. •
Смесь амиловых спиртов, получаемых гидролизом нефтяных
амилхлоридов, либо превращается путем этерификации в смесь
соответствующих уксусных эфиров, либо подвергается тща-
тельной фракционировке для выделения отдельных компонент
тов смеси. Таким образом, исходя из смеси нефтяных пентанов,
в настоящее время из восьми изомерных амиловых спиртов
получаются в техническом масштабе следующие шесть:
1. Норм, амиловый спирт (первичн.) СН5- СН2 • СН2 СЩ • СН2 • ОН
2. Изоамиловый „ „ (СН8)2 • СН • СН9 СН2 .• ОН
3. Вторично-бутил-карбино л . . . СН8 • СН2 • СН(СН8) • СН2 • ОН
4. Метил-пропил-карбинол .... CHS • СН(ОН) • СН2 • СН2 • СН8
5. Диэтил-карбинол..............(СН8СН2)2 • СН • ОН
6. Амиловый спирт (третичный)' . (СН^2 С (ОН) • СН2 • СН8
Производство амиловых спиртов и их производных из неф-
тяных пентанов в настоящее время широко поставлено в США.
Так, фирма «Sharpies Solvent Corp.» (Филадельфия), кроме тех-
нической смеси амиловых спиртов (пентазол) и их уксусных
эфиров (пентацетат), выпускает на рынок следующие продукты
из смеси нефтяных пентанов
1) все шесть вышеуказанных амиловых спиртов;
2) нормальный хлористый амил;
3) смесь амилхлоридов, содержащую шесть изомерных хло-
ридов, соответствующих указанным выше спиртам:
4) смесь амиленов, состоящую главным образом из метил-
этил-этилена и триметил-этилена, а также в отдельности по-
следние два этиленовых углеводорода, выделенные из смеси пу-
тем тщательной фракционировки;
5) диамилен и диамиловый эфир, как продукты дальнейшей
переработки указанных амиленов и амиловых спиртов.
Наиболее широко поставлено, конечно, производство пента-
зола и пентацетата, которые отпускаются бочками и цистер-
нами; но и все другие перечисленные выше продукты можно
иметь в упаковках по 1, 5, ю, 50 и 1(Ю галлонов [31].
Значение парафинов в общей массе нефтяных углеводородов
весьма разнообразно, и его приходится учитывать при оценке
технических качеств самых разнообразных нефтепродуктов.
Наибольшее значение и интерес представляют парахрипы как
составная часть легких сортов горючего (бензин), ощюделяюща^
его антидетонационные качества, причем, как уже было ука-
зано выше1, качества эти зависят не только от общей массы
парафиновых углеводородов, входящих в состав данного горю-
чего, но также от химической структуры отдельных их предста-
вителей. Исследование детонационных свойств 27 отдельных
• ♦
1 См. гл. IV: подробнее» о детонации <?м. ч. Ш, гл. III.
148
парафиновых углеводородов, законченное в самое последнее
время [32], привело к следующим результатам в этом напра-
влении.
Наибольшей склонностью к детонации, как это отмечалось и
раньше, 'Обладают нормальные парафины. По мере увеличения
числа углеродных атомов в их цепи детонирующие свойства
этих углеводородов правильно возрастают, так что в ряду: нор-
мальные пентан, гексан, гептан, октан, нонан и декан наимень-
шей детонирующей способностью обладает пентан и наиболь-
шей— декан. Подобные же увеличения детонирующих свойств
с ростом длины углеродной цепи наблюдаются и в других ана-
логично построенных изомерных рядах, напримерtв серии:
2-метил-бутан, 2-метил-пентан, 2-метил-гексан, а также в дру-
гих сериях парафиновых углеводородов, сходственно построенных.
Совершенно иное влияние на склонность к детонации ока-
зывает увеличение в молекуле парафинового углеводорода
числа метильных групп. Так например, в серии 2-метил-бутан.
2,2-диметил-бутан, 2,2,3-триметил-бутан и 2,2,3,3-тетрамегил-бу-
тан, где число метильных групп постепенно увеличивается,
склонность углеводорода к детонации соответственно умень-
шается, несмотря на возрастание молекулярного веса; аналогич-
ная зависимость наблюдается также в других сходственно по-
строенных сериях углеводородов. .Особенно ярко сказывается
влияние строения молекулы на склонность ее к детонации при
сопоставлении изомерных углеводородов. Так например, из трех
изомерных пентанов наиболее склонным к детонации оказы-
вается норм, пентан, а наименее склонным — тетраметил-метан;
изопентан же помещается в этом отношении между своими изоме-
рами. Аналогичные соотношения наблюдаются также в изучен-
ных с этой точки зрения рядах гексана, гептана, октана и нонана.
Как ясно из вышеизложенного, детонационные свойства го-
рючего самым тесным образом связаны с химическим составом
и строением образующих его компонентов, и уже одно это об-
стоятельство объясняет то внимание, которое привлекает в на-
стоящее время изучение вопроса о ближайшем составе отдель-
ных, особенно наиболее легких нефтепродуктов.
Отметим в заключение некоторые случаи, когда парафино-
вые углеводороды являются нежелательными составными ча-
стями нефтепродукта. Таковы прежде всего смазочные масла.
Исключительная устойчивость парафиновых углеводородов
делала бы их, конечно, крайне желательным компонентом сма-
зочных масел, подвергающихся в процессе работы воздействию
таких факторов, как повышенная температура, окисляющее
действие кислорода воздуха и т. п. бднако высокая застывае-
мость парафинов нормального строения заставляет стремиться
при производстве смазочных масел к возможной их депарафи-
низации, т. е. к удалению из них тех компонентов, присутствие
которых делает масло при низкой температуре твердым или
полутвердым, вследствие чего пуск машины на холоду крайне
затрудняется.
ио-
Аналогичный пример представляют сорта керосина, предав
.значенные для горения при низких температурах (освещен^
улиц, маяков и т. п.). Присутствие в них парафиновых уш
.водородов, способных на холоду выкристаллизовываться, вызы-
вает помутнение керосина и засорение фитиля кристалликам^
парафина, вследствие чего сила света при горении данной
.керосина может значительно понизиться. Ввиду этого, при изцо*
товлении подобного рода керосина, особого внимания заслужи-
вают не только пределы его выкипаемости, но также и химичу
ский состав исходной нефти.
ЛИТЕРАТУРА
1. „Ж. Р. К. О.“ 15, 3, 189 (1883).
2. „Ж. Р. X. О.* 22, 23, (1890).
3. „Chem. Ztg." 21, 288 (1897).
4. „Вег." 82, 1449 (1899).
5. „Ж- Р. X. О.* 15, 191 (1883).
6. „Ж. Р. X. О/ 81, 222 (1899).
7. „Ж. Р. X. 0." 81, 523 (1899).
8. „Ж. Р. X. О. 22, 23 (1890).
9. „Вег". 81. 1801 (1898).
.10. „Ж. Р. X. О/ 80, 81 (1898).
Ч. „Ж. Р. X. О." 85, 1037 (1903).
12. „Вег." 88, 1905 (1900).
13. „Ж- Р- X. О." 87, 521 (1905); 40, 731 (1908); см. также Коновалов
„Ж. Р. X. О." 87, 910, 1122 (1905).
14. „Ж. Р. X. О.“ 84, 917 (1902).
15. Харичков, О составе и технических свойствах нефтей русских место-
рождений, Баку (1902); ом. также „Ж. Р. X. О." 81, 552 (1899).
16. „Нефт. хоз." 24, № 8, 158 (1933).
17. Pelouze et Cahours, „С. R.", 54, 1241 (1862); „Ann. Chim. et Phys.", (4)1
5 (1864).
18, Schorlemmer, „Lieb- Ann. 127, 311 (1863) и т. д.; Morgan Th., „Lieb. Ann.1
177, 304 (1875); Warren С. M., „Ztschr.f. Ch." 1885, 242, 666, „Jahresh
f. Ch.", 1888, 330 и др.
19. Mabery a. Hudson, „Am. Chem. J." 19, 243 (1897); Young S., ,J. Chem
Soc." 78, 905 *(1898) и др.
‘20. Библиографию по отдельным парафинам см. Engler С. u. Hofer, В.Д
230—248 (1913).
•21. Mabery, „Am. Chem. J." 28, 164 (1902).
22. Mabery, „Ат. Chem. J." 17, 718 (1895): 18, 215 (18945)-
23. Mabery, „Ат. Chem. J." 88, 251 (1905).
•24. Mabery, .Am. Chem. J." 18, 43 (1896); 19, 419 (1897); 88, 251 (1905); 85,
404 (1906).
25. Mabery с сотрудниками, „Ат. Chem. J.“, 19, 797 (1897); 25, 283 (1901k
88, 270 (1905); „J. Am. Chem. Soc." 22, 553 (1899); 28, 264 (1900);
• „Ind. Eng. Chem." 6, 101 (1914).
26. Rossini F. D., „Refiner" 16, № 11, 545 (1937), здесь же литература»
вопроса.
27. „Lieb. Ann." 220, 188 (1F83).
28. Poni P. .Chem. Zbl." 1900. II, 452:1902,11,1870; Costachescu, Ibid., 1911,1
682 и др.
29. Химический состав нефтей и нефтяных продуктов, „Труды ГрозНИИ*,
М, — Л. (1931); 2-е изд. (1935).
30. Добрянский и Гуревич. „Нефт. хоз." 15, № 9, 352 (1928); Koch а. Burrell,
„Ind. Eng. Chem." 19, № 4, 442 (1927); Ayres К, „Ind Eng. Chem-*
21, № 10, 899 (1929).
31. „Ж. прикл. химии" 2, № 2, 248 (1929).
32. Lovell, Campbell, Boyd, .Ind- Eng. Chem. 28, № 1 (1931); см. также
„Нефт. хоз.“ 21, № 8—9 (1931).
150
В. Высшие (твердые) парафины
К этой группе парафинов можно отнести все углеводороды
ряда метана, начиная с CieHs4 (гексадекан) и выше, которые
при обыкновенной температуре являются твердыми (ср. табл. 32).
Высшими их представителями являются гексаконтан, СеоНш,
и тетрагексаконтан, Св4Н13о, полученные реакцией Вюрца из
соответствующих галоидопроизводных [1], например сплавле-
нием мирицил-иодида, CsoHeil с натрием по уравнению:
2С30Н611 2Na = 2NaJ + С60Н122.
Искусственное получение многих других высших парафинов
было осуществлено обычными способами [2] через галоидо-
производные; последние же получались из соответствующих
кислородных соединений (кетоны, кислоты и т. д.), некоторые
из которых являются веществами легко доступными. Так на-
пример, при сухой перегонке известковой соли пальмитиновой
кислоты, CijHsiCOzH, может быть получен пальмитин-кетон,
(Ci5H31)2CO, который последовательным действием пятихлори-
стого фосфора и йодистого водорода был превращен в углеводо-
род С15Н31 • СН2. С15Н31:
cltHsl • соох
>Са=СО8Са+ (CI6HS1)2 • СО;
Cjeligi • coo
(С1Бн81)2 СО—>(С15Н81)2 CCl^C^CH^IV
Та же пальмитиновая кислота {восстановлением иодистым:
водородом была превращена в другой парафин, С1вНз4, и т. д.
С1БН31СО2Н —►С1БН81СН8.
Твердые парафины встречаются иногда в продуктах живот-
ного или растительного царства (пчелиный воск, некоторые
эфирные масла и т. п.). Они были обнаружены впервые в дре-
весном дегте (Рейхенбая, 1830 г.), и именно этот продукт полу-
чил наименование «парафин», которое укоренилось затем за
твердыми углеводородами ряда метана независимо от способа их.
добывания. В настоящее время парафин получают: 1) из про-
дуктов глубокой термической обработки битуминозных сланцев,
ббгхедов. бурого угля, 2) из природного озокерита (горный воск)
и 3) из парафинистых нефтей.
ПОЛУЧЕНИЕ ПАРАФИНА
В зависимости от исходного сырья техническое получение
парафина [3] характеризуется некоторыми заслуживающими
внимания особенностями.
Получение парафина из буроугольного и сланцевого дегти-
Еще недавно производство парафина было сосредоточено глав-
ным образом в Германии, где его получают из буроугольного*
дегтя, и в Шотландии, где сырьем служили продукты сухой пе-
151
регонки горючих сланцев. Получение парафина из буроуголь-
ного дегтя не сложно.
Когда при перегонке этого дегтя дестиллат начинает при охлаждении
кристаллизоваться, его собирают отдельно и оставляют на шк’колько дней,
<в течение которых кристаллизация парафина заканчивается. Масло, коте-.'
рым бывают пропитаны кристаллы парафина, отделяют сначала на
•фильтрпрессе, затем отжимают гидравлическим прессом, поело чего пара-
фин еще содержит от 5 до 15% масла и имеет желтый цвет. Для даль-
нейшей очистки сырец смешивают с 10—12% легкого буроугольного
бензина, нагревают открытым паром и расплавившуюся смесь выливают
в холодную воду; парафин застывает сплошным слоем на поверхности
воды. Застывший парафин разрезают на куски, завертывают в шерстяные
•салфетки и вновь прессуют на горизонтальных гидравлических прессах
при 200—250 ат давления. Если отделение масла не достаточно, к отпрес-
сованному парафину прибавляют 'Новую порцию бензина и повторяют всю
операцию еще раз; следы бензина, оставпгиеся после прессования, удаляют
продувкой паром. Полученный таким образом парафин не вполне бел,
а имеет зеленовато-желтый оттенок. Для обесцвечивания расплавленный
парафин перемешивают при 70—80° с отбеливающим порошком (костяной
или активированный уголь, отбеливающие глины и т. п.) при помощи ме-
шалки, после чего, дав смеси отстояться, фильтруют обесцвеченный па-
рафин через обыкновенную фильтровальную бумагу и разливают в формы.
•Отжатые маета и бензиновые растворы после отгонки растворителя идут
на изготовление мягкого парафина. Таковы в самых общих чертах основные
моменты получения парафина из буроугольного дегтя. Получение того же
продукта из сланцевого дегтя существенно отличается от описанного лишь
:в одном отношении: гвместо промывки (перекристаллизации) парафина-
сырца бензином в шотландской сланцевой промышленности применял!
почти исключительно способ потения парафина (швицевание). Об этом методе,
лежащем в основе получения нефтяного парафина, будет сказано ниже.
Переработка озокерита. Озокерит, или горный воск, пред-
•ставляет собою горючий минерал, относящийся к классу, из-
вестному под именем «минеральные носки». Ои состоит главны#
образом из особой разновидности парафина — церезина!
с большим или меньшим содержанием других веществ — мине-
ральных (песчаник) и органических <нефгеобраз!ше масла, смо-
листые вещества и т. п.). Наиболее богатое месторождение озо-
керита находится в Галиции (Борислав); вторым по запасам'
озокерита считаются залежи озокерита на о. Челекене (Каспий-
ское море). Озокерит встречается у нас также в Фергане, на
Апшеронском полуострове и в Забайкалье, но уже в значи-
тельно меньшем количестве, чем на о. Челекене.
Получение церезина из озокерита слагается из следующих
последовательных операций: а) отделение- озокерита от сопрово-
ждающих его минеральных пород и получение озокерита-сырца;
б) отгонка от сырца легких масел — получение озокерита-стан-
дарта и в) очистка озокерита-стандарта на желтый и белый
церезин.
Для отделения минеральных примесей чаще всего применяется горячая
выварка озокерита с водой. Выварка ведется в железных или чугунных
котлах, обогреваемых огнем, почти всегда недалеко от места добычи. Когда
..температура воды в котле достигает 60—70°, частицы озокерита начинают
плавиться и поднимаются наверх, на поверхность воды, минеральные же
примеси остаются на дне. Расплавленный озокерит счерпывается ковшом
152
и заливается в формы; по застывании он упаковывается в блоки и напра-
вляется па озокеритно-церезиновый завод дая дальнейшей переработки.
Поступающий на завод озокерит-сырец загружался в расплавителы.
плавится здесь, нагревается до 120° и посредством монжу подается в пере-
гонный куб для отгонки 20—35% содержащихся в нем масел. Отгонка
ведется при помощи перегретого пара в вакууме; под конец гонки темпе-
ратура внутри куба поднимается до 300°. Дестиллатные легкие масла
обыкновенно идут в топливо, более же тяжелые—утилизируются для по-
бочных производств. Главный продукт — остаток в перегонном кубе —
представляет собою озокерит - стандарт; его либо разливают в формы, либо
непосредствецно передают для очистки в кислотную мешалку.
Озокерит-стандарт представляет собою парафинообразную
массу черного цвета. Для очистки его подвергают обработке
смесью серной кислоты и олеума, взятых в отношении 2:1.
Очистка ведется в мешалке, снабженной двумя пропеллерами
для перемешивания, сначала при 130—150°, но постепенно тем-
пература повышается до 210°; при этом избыток кислоты уда-
ляется, остаток же ее нейтрализуется небольшим количеством,
извести. Затем без отстаивания кислого гудрона в мешалку за-
гружается адсорбент (животный уголь, глуховская тлина или
инфузорная земля), и перемешивание возобновляется еще на.
полчаса, после чего очищенную массу, охлажденную до 160—
170°, фильтруют на рамочном фильтре через специальные по-
лотна под давлением до 6—7 ат. Расход кислоты при такой
очистке для челекенского озокерита колеблется от 20 до 30%;
расход адсорбента — 5—10%. Весь процесс очистки длится 8—
10 час., причем теряется до 50% озокерита. Полученный таким
образом церезин—желтого цвета; для получения белого товара,
очистку приходится повторять два, а иногда и три раза с за-
тратой 5—10% серной кислоты и 10% адсорбента при каждой
новой очистке1.
Получение парафина из нефти. Парафинистые нефти яв-
ляются в настоящее время главным источником технического-
получения парафина. Производство его осуществляется на спе-
циальных парафиновых заводах и», в основном заключается
в следующем.
Парафинистый мазут подвергают разгонке на кубовой бата-
рее типа масляной либо на трубчатом кубе с ректификационной
колонной и получают так называемый парафинистый дестиллат.
Разгонка ведется обычно дважды, так как лишь после второй
перегонки парафин сполна переходит в кристаллическое состоя-
ние и легко фильтруется. В зависимости от исходного сырья
дрстиллат имеет удельный вес 0,848-М),875 и кипит в широких
пределах, захватывая главным образом соляровые и веретенные
фракции; содержание в нем парафина — от 12 до 20% и выше.
С батареи парафиновый дестиллат направляется в специальные
отстойники, снабженные змеевиками-подогревателями; здесь
происходит отделение грязи и воды, после чего дестиллат дере-
1 В СССР имеется озокеритно-церезивовый завод в с. Ростокино под
Москвой. Завод, оборудованный по последнему слову иностранной техники,
пущен в ход в 1927 г.
153-
качивают в громадные непрерывно работающие кристаллиза
торы-охладители с поверхностью охлаждения до 50 м2 и более.
•Обычно эти охладители представляют собою систему двойник
•труб; пр внутренним 6* (152,4 Мм) трубам перекачивается део-
тиллат, причем для облегчения хода жидкости вместе с ваде-'
лившимся парафином внутри труб имеется шнек, приводишь
в движение особым цепным колесом; снаружи эти трубы окру^
жены системой других 8” (203,2 мм) хорошо изолированны!
труб; охлаждающая жидкость (холодный соляной раствор
с холодильных установок) периодически перекачивается ад
межтрубному пространству. Температура охлаждаемого дестил-
лата поддерживается около 0°; при этом он вполне сохраняет
свою подвижность. С помощью плунжерных насосов его подают
далее на фильтрпрессы и отделяют здесь главную часть масла.
Фильтрация происходит через плотную хлопчатобумажную
ткань, пропускающую лишь масло, причем давление в хороши!
фильтрпрессах может быть доведено до 50 ат. Отжимное масло
спускается в приемники и может служить сырьем для перера-
ботки на смазочные масла; кристаллы же парафина, заполнив-
-шие камеры фильтрпресса, отгружаются по окончании фильтра-
ции в виде лепешек парафинового гача.
Отжатый гач подвергается прежде всего очистке серной кис-
лотой в мешалках обычного типа, но с обогревом до 70—75*;
так как гач при очистке должен быть в жидком состоянии. Раб-
ход кислоты достигает при этом 4—5%. За очисткой следует
промывка сначала едким натром, затем — водой в тех же тем-
пературных условиях. Задача очистки — освобождение гача от
смолистых веществ, присутствие которых нежелательно в после-
дующей стадии производства — процессе потения гача 1.
Потение гача в целях дальнейшего освобождения его от
масла производится в особых помещениях, называемых каме-
рами потения. Это — ряд неглубоких ящиков, устанавливаемых
по 8—10 штук один над другим на особых стойках в хороша
изолированном помещений. В новейших установках размеры
ящиков —15—18 м длины и 3 м ширины;" их днища ймеюФ
форму опрокинутых пологих пирамид; полезная емкость —от
5300 до 6300 л. Каждый ящик снабжен ^чпеткой из легкого’
углового или таврового железа, тщательно устанавливаемой по
уровню. Поверх решетки кладется сетка из оцинкованной про*
волоки с квадратными отверстиями в 6,25 см2: дал<ч!— вторая
сетка из • латунной проволоки с 50 отверстиями на каждые
2,5 см2 и, наконец, выше — змеевики для водяного охлаждения
гача.
Сначала ящики заполняют через особые трубы водой не-
много выше уровня сетки, затем в них накачивают расплавлен-
ный гач до образования слоя толщиной в 15 см. Если пропу-
1 Такой порядок очистки серной кислотой, практиковавшийся в первое
годы эксплоатации Грозненского парафинового завода, был .затем нзменей*
и в настоящее время очистка ведется после потения гача.
154
стать затем через змеевики холодную воду, гач охлаждается и
превращается в твердую массу. Тогда спускают воду из ящи-
ков, закрывают камеры и начинают пропускать в змеевики го-
рячую воду, поддерживая в камерах температуру немного ниже
температуры плавления гача. В этих условиях масло, остав-
шееся между кристаллами парафина, начинает выпотевать, сте-
кает через сетки и через спускные отверстия в центре .ящиков
отводится в резервуары для хранения и последующей обра-
ботки; конечно, вместе с маслом: отходит также некоторая часть
парафина. Осторожно поднимая температуру в камерах, можно
углублять процесс потения до почти полного удаления масла и-
получения парафина с желаемой температурой плавления.
Йо окончании процесса потения парафин расплавляют про-
пусканием через те же змеевики пара и спускают в сливной ре-
зервуар. Вся операция потения каждой загрузки продолжается-
40—48 час., но для некоторых сортов парафина это время может
быть несколько снижено. Загрузка каждой камеры потения со*
ставляет около 30 wi. Сырой парафин после первого потения
плавится 40—49° и находит разнообразное применение; если
требуется более высокоплавкий парафин, то его подвергают вто-
рому потению. Что касается, наконец, отпотевшего масла, то-
в зависимости от содержания в нем парафина его либо подвер-
гают вторичной перегонке, либо сразу пускают на кристаллиза-
цию и фильтрпрессы, а затем вновь в камеры на вторичное по-
тение.
Парафин после .потения имеет обыкновенно зеленый цвет и?
неприятный, керосиновый вкус и запах. Если необходимо изба-
виться от этих недостатков и получить очищенный парафин, то
его подвергают дополнительной очистке—отбеливанию — креп-
кой серной кислотой (олеум или моногидрат) с последующей
промывкой и обработкой флоридином, после чего парафин по-
лучается бесцветным, лишенным вкуса и запаха и стабильным
к свету.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ПАРАФИНА
'Содержание парафина в различных нефтях весьма разнооб-
разно. .Количественное определение его [4] представляет инте-
рес не только для общей характеристики нефти, но может
иметь также весьма существенное производственное и техниче-
ское значение. Основным методом определения парафина яв-
ляется способ Гольде-Энглера, сущность которого состоит в сле-
дующем.
Навеску нефти (100 г) подвергают перегонке из колбы*
Вюрца. Когда температура поднимется до 300Q, меняют прием-
ник и ведут перегонку до конца (до кокса); весь парафин со-
держится в этом втором дестиллате от 300и. ’ Для определения
в нем парафина к 10—15 г дестиллата прибавляют смесь эфира,
и абсолютного спирта (1 :1) до полного растворения взятой на-
вески дестидля та при комнатной температуре. Затем охлаждают
155-
этот раствор до —20°, благодаря чему весь парафин выделяется
из раствора в кристаллическом состоянии (хлопья); его быстро
на холоду отфильтровывают и промывают холодной спирте-
эфирной смесью. Затем растворяют парафин в горячем бензине
или бензоле, сливают раствор в тарированную чашечку и по уда-
лении растворителя взвешивают. Иногда выделенный таким спо-
собом парафин недостаточно чист — содержит, например, смоли-
стые вещества; в таких случаях его растворяют в бензине и
связывают смолистые вещества 1—2 каплями серной кислоты.
В методике Голъде-Энглера следует отметить два характер-
ных признака: применение определенного растворителя (спирте-
эфирная смесь) и предварительная перегонка нефти.
В отношении растворителя были сделаны предложения
вместо спирто-эфирной смеси применять метил-этил-кетон и не-
которые другие органические вещества. Эти предложения не
внесли в методику определения парафина и получаемые резуль-
таты чего-либо существенно нового.
Значительно более сложным оказался вопрос о предварителы
ной перегонке нефти при определении в ней парафина. Совер-
шенно очевидно, что перегонка эта сопровождается частичным
разложением парафина (крэкинг), в .результате чего получаемые
по Голъде-Энглеру результаты должны быть несколько понижен-
ными. Ввиду этого некоторые авторы предпочитают вести опре-
деление парафина без предварительной перегонки, хотя при этом
может возникнуть другая опасность: осаждение вместе с пара-
фином смол и асфальтенов. Одним из наиболее удачных виде-,
изменений методики определения парафина без предварительной
перегонки является следующий способ [5].
Навеска нефти около 10 г растворяется в легком бензине
(около 500 см3), и раствор зачищается 60—70 г флоридина при
комнатной температуре. Отработанный флоридин .подвергается
затем продолжительной экстракции бензином в аппарате Сок-
слета ддя. вымывания адсорбированного парафина, на что тре-
буется 2 дня. Далее весь бензиновый 'раствор очищенной нефти ;
упаривается до 15—20 см3, осаждается спирто-эфирной смесью,
и содержание парафина определяется по Голъде-Энглеру. Как
будет видно из приведенного ниже цифрового материала, опре-
деление парафина по только что описанному способу без пред-
варительной перегонки дает результаты в 2—з раза большие,
чем способ Голъде-Энглера с предварительной перегонкой.
СОДЕРЖАНИЕ ПАРАФИНА В НЕФТЯХ И ИХ ДЕСТИЛЛАТАХ [в}
Парафин содержится, повидимому, во всех нефтях. Однако
в количественном отношении здесь наблюдается весьма большое <
разнообразие. При рассмотрении имеющегося по этому вопросу
литературного материала необходимо принимать во внимание,
не только недостаточную точность методики, но также и то об-
стоятельство, что по отношению к различным, нефтях опублико-
ванные данные часто являются несравнимыми, так как они
156
были получены различными методами. Насколько могут расхо-
диться при этом количественные данные по отношению к одной
и той же нефти, показывает табл. 44, в которой по данным раз-
личных авторов приводится содержание парафина в важнейших
апшеронских нефтях, определенное двумя методами для каж-
дого образца: по Голъде-Энглеру с предварительной перегонкой
и без предварительной перегонки.
Таблица 44
Содержание парафина в некоторых апшеронских нефтях
Месторождение Уд. вес Содержание парафина в °/о
с предв. перегонкой без предв. перегонки
Сураханы 0,863 2,1 до 6
Балаханы-Сабунчи 0,868 0г5 1,95
Биби-Эйбат 0,867 0,7 1,26
Бинагады ... * 0,922 0,3 0,7—0,8
Кирмакинская нефть 0,951 0,1—0,2
Само собою разумеется, что количественные данные с рас-
хождением в зависимости от деталей методики на 200 и 300%
могут иметь лишь относительное значение; с этой оговоркой
можно перейти к рассмотрению материала по содержанию пара-
фина в различных -нефтях.
Большинство апшеронских нефтей относится к слабопарафи-
нистому типу. Однако в последнее время в бакинском районе
найдены нефти и другого типа с большим содержанием пара-
фина. Таковы, например, кара-чухурская нефть (9,9% пара-
фина), зыхская нефть (11,2% парафина) и др.
Наши -грозненские нефти делятся по содержанию в них пара-
фина на следующие четыре группы: грозненские парафинистые
нефти с содержанием парафина до 5,5%1; слабопарафини-
стые— 2,2—2,3% парафина; переходные — 0,8—0,9% и бес-
парафиновые, в которых содержание парафина колеблется от
0,1 до 0,7%.
В большинстве эмбенских нефтей содержание парафина не
превышает 0,4—0,6%, спускаясь для доссорской нефти до
0,1—0,2%. Лишь в отдельных месторождениях содержание пара-
фина поднимается здесь до 1,—1,2% (Шубар-Кудук, Сагиз, Но-
вобогатинское).
Выделяются из нефтей СССР по содержанию парафина нефть
с о. Челекена (Каспийское море)—до 6% парафина и некото-
рые ферганские нефти (Санто, Чимион), в которых содержание
парафина доходит до 8%.
1-По определениям без предварительной перегонки 9—10%. К парафи-
нистым нефтям относится также легкая малтобекская нефть, содержащая
от 6,2 до 8,9% парафина.
157
Заметное количество парафина (1,3%) содержится в перм-
ской нефти (Чусовские Городки), а также в Стерлитамакской
(1,5%) и ухтинской нефтях (2—2,5%).
Наконец, среди сахалинских нефтей встречаются как типич-
ные представители беспарафиновых нефтей (Оха), так и пара-
финистые нефти с содержанием до 3—4?о парафина (Нутово).
Из европейских нефтей выделяются содержанием парафина
нефти румынские (до 10% и выше) и галицийские (до 12%).
Из американских парафинистых нефтей прежде всего должна
быть упомянута знаменитая пенсильванская нефть.
Некоторые мексиканские нефти содержат от 7 до 10% пара-
фина.
Упомянем, наконец, нефть Зондских островов, в которой со-
держание парафина доходит до 7,57.
Что касается распределения парафина в нефти по ее пого-
нам, то в соответствии с температурой кипения твердых углево-
дородов ряда метана при перегонке нефтей на керосиновых ба-
тареях главная масса парафина остается в мазуте; лишь неболь-
шая часть парафина попадает в керосиновый дестиллат и мо-
жет быть обнаружена здесь вследствие кристаллизации парал
фина при сильном охлаждении. При разгонке мазута выделение
твердого парафина в дестиллатах, соляровом, веретенном и т. д.,
происходит, как это было отмечено выше, уже при обыкновен-
ной температуре, соответственно чему температура -застываний
парафинистых мазутов может быть довольно высокой (35° и
выше).
Хотя согласно вышеизложенному весь материал по количе-
ственному содержанию парафина в нефтях .может иметь липгь
ориентировочное значение, тем не менее заслуживают вниманий
попытки некоторых построенных на нем обобщений.
Мэбери [7] указал, что между содержанием парафина в
нефти и характером ее низших погонов существует известной
соответствие: чем богаче низшие погоны нефти углеводородами
ряда метана, тем больше парафина содержится в высших еб
фракциях, и наоборот. Эта закономерность действительно может
быть иллюстрирована целым рядом примеров из области нах
парафинистых, так и нафтеновых нефтей. Несомненно, однако,
здесь могут быть и исключения. Так например, парафинистая
сураханская нефть, как будет показано в гл. VI, в главной
массе своих низших погонов состоит из нафтенов, а не из угл^
водородов ряда метана и т. д.
Очевидно, для установления более точной зависимости между
содержанием в нефти отдельных компонентов требуются даль*
нейшее уточнение методики их определения и накопление болеет
надежного фактического материала в этой области. .
То же самое можно сказать по вопросу о зависимости содер-
жания парафина от глубины залегания нефти: по одним данным
с увеличением глубины залегания содержание парафина в неф-
ти возрастает (Сураханы, Галиция); однако в некоторых другие
случаях наблюдалась и обратная зависимость.
158
ФИЗИЧЕСКИЕ СВОЙСТВА ПАРАФИНА [8]
Очищенный (refined) парафин представляет собою бесцвет-
ную или белую, более или менее прозрачную, кристаллическую
массу, без запаха и вкуса, слегка жирную наощупь. Нераство-
рим в воде; мало растворим в абсолютном спирте; хорошо —
в эфире, хлороформе, бензоле, петролейном эфире, сероуглероде,
и минеральных маслах; при нагревании растворяется также во’
многих растительных маслах.
Удельный вес парафина в твердом состоянии определяется
общими методами определения удельного веса твердых тел с при-
менением вместо воды водно-спиртовых растворов, в которых па-
рафин тонет. Он существенно зависит от содержания в парафине
масла: очищенный парафин имеет в твердом состоянии удель-
ный вес 0,907—0,915 при 15°, для парафина же, не вполне очи-
щенного, после однократного потения, удельный вес колеблется
от 0,881 до 0,905, т. е. — значительно ниже. В жидком состоянии
все парафины имеют очень близкие удельные веса, например
при 60° ОКОЛО 0,776—0,781.
Температура плавления парафина ввиду неоднородности его
химического состава характеризуется недостаточной резкостью
и может простираться в пределах 10—12° и более. Так напри-
мер, очищенный грозненский парафин плавится при 49—60°,
а желтый — при 41—58°. Американские парафины представляют
собою более узкие фракции; их спецификации различают:
Парафин с температурой плавления 180—132° Ф, т. е. около 55° Ц
, . •„ . 124-127° Ф „ „ 52°Ц
, . „ , 117-120° Ф , . 48'Ц
Определяется температура плавления парафина лучше всего
в пробирке с погруженным термометром.
Более резкую характеристику, чем температура плавления,
дает температура застывания парафина, определяемая в приборе
Жукова-, она лежит в пределах между температурами начала и
конца плавления парафина и является главной константой, ха-
рактеризующей парафин. Следует, однако, иметь в виду, что
присутствие 3—4% масла почти не отражается на температуре
застывания парафина, понижая последнюю меньше, чем на 0,5°,
и может быть обнаружено только по характеру кривых засты-
вания.
Консистентностъ парафина, определяемая консистометром
Абрагама [9], является одной из важнейших его характеристик,
так как уже небольшие примеси масла оказывают на нее резкое
влияние. Так например, прибавление 0,5% масла понижает кон-
ей стентность парафина на 20%, прибавление 1% масла—на
30% и т. д. консистенция парафина имеет очень большое значе-
ние для некоторых видов его применения, в частности, напри-
мер, для производства свечей.
Поэтому весьма интересно отметить, что наш грозненский па-
рафин после перекристаллизации обладает значительно более
159
высокой консистентностью, чем перекристаллизованный же очи-
щенный американский парафин.
Консистометр Абрагама (фиг. 27) состоит из станины, несущей, наряду
с другими частями прибора, винт О. Движение, сообщаемое рукою этому
винту, передается поршеньку D, который бла-
годаря этому вдавливается в испытуемый про-
дукт, помещаемый под поршеньком в специаль-
ной чашечке J. Самое движение винта произ-
водится таким образом, чтобы стрелка цифер*
блата L двигалась с тою же скоростью, с какой
движется стрелка секундомера М. После одного
полного кругового движения стрелки испыта-
ние считается законченным, причем поршень
проходит в испытанном продукте всегда ровно
1 см. Потребовавшаяся для этого величина да-
вления в граммах и килограммах регистри-
руется на шкале Е от пружины В и может,
служить мерою консистентности испытуемого
вещества.
Фиг. 27. Консистометр
Абрагама.
Цвет парафина зависит от степени его
очистки и содержания масла. Хорошо
очищенные и свободные от масел пара-
фины— бесцветны и не изменяются на
свету. Недостаточно очищенный парафин
имеет светложелтый, желтйй и буровато-
желтый цвет, причем интенсивность его
окраски на свету увеличивается. Опре-
деление цвета парафина производится
в расплавленном или твердом состоянии
при помощи колориметра.
ХИМИЧЕСКИЙ СОСТАВ И СВОЙСТВА
ПАРАФИНА [10]
Элементарный состав парафина до не-
которой степени зависит от степени его
очистки. Сырой и недостаточно очищен-
ный парафин за счет примеси смолистых
и тому подобных веществ может содер-
жать некоторое количество кислородных
соединений, легко удаляемых дополни-
тельной очисткой серной кислотой или
нагреванием с металлическим натрием. Хорошо очищенный па-
рафин не содержит кислорода, и сумма процентного содержания
в нем углерода и водорода — близка к 100, независимо от того,
будет ли то парафин из нефти, озокерита или какого-либо дегтя-
Как видно из табл. 45, колебания в элементарном составе очи-
щенного парафина сравнительно не велики: около 0,8% для
углерода и 0,7% для водорода. Однако эти колебания значительно
превосходят различие в составе смежных гомологов ряда
из числа твердых парафинов; более того, данные некоторых авто-*
ров одинаково подходят как к составу высших представителей
160
Таблица 45
Элементарный состав парафина
Компоненты Найдею раз- ными авто- рами Вычислено
QjoHtf С24Щ0
% с 84,94—86,77 85,10 85,21 85,71
% н 14,21—14,87 14,90 14,79 14,29
ряда метана, тан и к ряду СЛН2Л, так что сделать какие-либо вы-
воды о ближайшей химической природе парафина на основе
одного его элементарного состава не представляется возможным.
Обычной составной частью всякого парафина является дестиллатноа
масло, содержание которого даже в хорошо очищенном парафине может
достигать 1,5—2% и более. Для некоторых видов применения парафина
примесь эта крайне нежелательна, соответственно чему количественное
определение масла в парафине имеет большое практическое значение. Про-
стейший способ такого определения заключается в следующем. Навеску
парафина (15—35 г) отжимают между несколькими кружками фильтро-
вальной бумаги и специальной ткани, помещенными в особом кольце; да-
вление доводится по 70 не/см2. По окончании отжима, механически при-
ставший парафин соскабливается, а общий привес бумаги и ткани прини-
мается за содержание масла в данной навеске. Несмотря на всю примитив-
ность этого метода для количественного определения, он широко применяется
в США, а также в Англии [11]. Кроме него существует еще несколько
методов определения масла в парафине, как то: рефрактометрический, ме-
тод селективного растворения, комбинированный метод и т. д. [12]. Окон-
чательное отделение масла от парафина достигается его перекристаллиза-
цией мз подходящих растворителей, каковы: спиртоэфирная смесь, ацетон
или метил-этит-кетон и особенно дихлор-этилен.
Хорошо очищенный парафин имеет ясно выраженный пре-
дельный характер, так что серная кислота при встряхивании
с ним либо вовсе не окрашивается, либо окрашивается в слабо-
желтый цвет. Неочищенные или недостаточно очищенные пара-
фины, наоборот, окрашивают серную кислоту более или менее
интенсивно и обнаруживают явное присутствие непредельных
соединений, образующихся в результате частичного разложения
при перегонке, т. е. при получении парафинового дестиллата.
Парафин весьма устойчив к самым разнообразным реаген-
там, как-то: кислотам (галоидоводородные, азотная и др.), щелоч-
ным металлам, щелочам и вообще основаниям (гидразин, амины
и др.). Хлор не реагирует с Твердым парафином даже после не-
скольких часов соприкосновения. При нагревании хлор, а также
бром действуют на парафин завещающим образом с выделением
галоидоводорода.
Окислители, как-то: хамелеон, хромовая кислота, азотная кис-
лота, на холоду не реагируют с парафином. При повышенной
температуре лучшим окислителем парафина является азотная
кислота; главными продуктами реакции являются в этом случае
лишь продукты глубокого распада, а именнокислоты уксус-
161
11 Зап. 1622. Химия нефти.
ная, масляная, валериановая, капроновая, эиантовая и др. Пз
высокомолекулярных кислот при этом была обнаружена цероти-
новая кислота, С26Н52О2 являющаяся 'главной составной частью
пчелиного воска. Реакция, которая за последние годы привле-
кает особое внимание в связи с проблемой получения высокомо-
лекулярных жирных кислот — окисление выоших парафиновых
углеводородов кислородом воздуха при нагревании — приводит
также лишь к продуктам более или менее глубокого распада
углеводородной молекулыК Перекись водорода, а также органи-
ческие перекиси и гидроперекиси (например гидроперекись бен-
зоила) с парафином не реагируют.
СТРОЕНИЕ ПАРАФИНА И ЕГО МОДИФИКАЦИИ
К вопросу о химическом строении парафина можно подходить
с точки зрения дальнейшего изучения отдельных, входящих в его
состав углеводородов, выделенных в индивидуальном состоянии.
Эта’ последняя задача представляет здесь громадные трудности
п, несмотря на большое число работ, пока не может считаться
решенной.
Первые опыты разделения парафина на ближайшие составные
части были проведены на образцах саксонского буроугольного
парафина Краффтом [13]. Парафин имел температуру плавле-
ния 30—35° и при 15 мм перегонялся 185—235'. После 12 фрак-
ционировок и перекристаллизации пз смеси спирта с эфиром
было выделено 7 предельных углеводородов, начиная с CnHse
(температура плавления 22°) и кончая С28Н48 (температура пла-
вления 45—48°), которые имели температуры кипения, темпера-
туры плавления и удельные веса одинаковые с константами по-
лученных тем же автором синтетических углеводородов того же
состава нормального строения1 2. Позднее более широкая фрак-
ция того же парафина была подвергнута фракционировке в ка-
тодном вакууме; при этом было выделено is высших гомологов
метана, от нонодекана, CisEko '(температура плавления 32°)
до гексатриаконтана, СзвНп (температура плавления около 76°).
Кроме этих 18 углеводородов, в головке и остатках после фрак-
ционировки наметилось присутствие ряда других гомологов
с меныпим и большим молекулярным весом, так что общее число
углеводородов ряда СиН2/1 + 2, входящих в состав исследованного
парафина, по мнению автора работы, не менее 35.
Кроме буроугольного парафина, в новейшее время аналогич-
но был изучен парафин из шотландского сланцевого дегтя. 'Раз-
гонка его в глубоком вакууме (0,3—0,5 мм) дала продукты, по
своим свойствам довольно близкие к синтетическим углеводоро-
дам Краффта нормального строения, а исследование этих про-
1 Подробнее об этом см. ч. II, гл. IVB.
2 Строение ю из этих высокомолекулярных углеводородов, вытекающее
из способов пх получения, подтвердилось в новейшее время исследованием
их по методу диффракцпи рентгеновых лучей [14].
162
= ОД + СН3 • CHgCCH,)» СН9 • CHg,
3
дуктов с помощью Х-лучей дало результаты, в общем подтвер-
дившие такое заключение о их строении [15].
Гораздо более сложным оказался вопрос о природе нефтяного
парафина. Давно уже было известно, что наряду с парафином,
выделяемым из парафинового дестиллата и обладающим ясно
выраженной кристаллической структурой, существует другой
парафин, более мягкий и по внешнему виду аморфный. Эта по-
следняя модификация парафина легко получается, если, не под-
вергая нефть перегонке, осадить из нее парафин, например
смесью амилового и винного спиртов; аналогичный характер
имеет церезин, получаемый из изокерита (см. выше) или из осадка
от парафинистых нефтей, который выделяется из них на буро-
вых инструментах, трубах и в резервуарах (rod wax). Различие-
этих парафинов полагали сначала не только в самом существо-
вании этих двух его модификаций, аморфной и кристаллической,
но и в том, что модификации эти имеют различное химическое
строение, а именно: находящемуся главным образом в сырой
нефти и не подвергавшемуся перегонке аморфному «протопара-
фину» придавали изо-строение (изопарафин)-, другую же моди-
фикацию, кристаллическую и более устойчивую считали про-
дуктом превращения первой под влиянием высокой температуры
перегонки («пиропарафин») приписывали ей нормальное строе-
ние [16]. Превращение протопярафипа в пиропарафин выража-
лось следующим уравнением:
СНЗХ /С Н3
>СН(СН2)ЯСН<
CHS
Эти представления, однако, не подтвердились при ближайшем
исследовании. Прежде всего было показано [17], что не только
сырые богатые парафином нефти (челекенская, чимионская, ру-
мынская, галицийская), но и аморфный на вид осадок, получае-
мый после осаждения парафина спиртом, обнаруживают под
микроскопом явное мелкокристаллическое строение. Далее было
установлено, что и в отношении некоторых других свойств, на-
пример растворимости в различных растворителях [18], между
обоими видами парафина нет никакой разницы. Наконец, осо-
бенно интересные и важные результаты были получены по этому
вопросу в самое последнее время [19]. Оказалось, что церезин
из сураханской нефти, этот типичный представитель «аморф-
ного» парафина, может перегоняться в катодном вакууме без
всякого разложения, совершенно не изменяя при этом своих
физических свойств. В менее глубоком вакууме действительно
происходит частичное разложение церезина с образованием па-
рафинов, но при 1—2 мм давления большая часть церезина пе-
регоняется, невидимому, без изменения.
Если, таким образом, под давлением нового эксперименталь-
ного материала старые представления о двух модификациях па-
рафина аморфной (протопарафин) и кристаллической (пиро-
парафин), и о генетической связи между ними должны быть
1Г 163
оставлены, то тот же материал (особено исследования ГрозНИИ)
не только не уничтожил, но еще более углубил различие между
двумя видами парафина. Сохраняя за дестиллатным продуктом
название «парафин» и обозначая другой его вид независимо от
способа его получения (переработка озокерита или rod wax’a,
осаждение из нефти) «церезином», проследим, в чем заклю-
чаются основные различия между ними, воспользовавшись для
этого главным образом обширным опытным материалом, опубли-
кованным в последнее время по парафинам и церезинам из
грозненской и сураханской нефтей [19].
Грозненская нефть является типичным примером парафини-
стых нефтей; она содержит преимущественно парафины и срав-
нительно небольшое количество церезинов, концентрирующихся
в гудроне. Сураханская нефть, наоборот, стоит в отношении со-
держащихся в ней высших гомологов метана особняком: наряду
с парафинами она содержит также значительное количество це-
резина, и ее гудрон может служить прекрасным материалом да
получения нефтяного церезина Сопоставление грозненских и
сураханских парафинов и церезинов приводит к следующим ре-
зультатам.
Нефтяные парафины (дестиллатные) имеют сравнительно не-
большой молекулярный вес (от СюЩ» до СзбН72) и соответ
ственно невысокие температуры кипения; ввиду этою при пере-
гонке нефти они переходят с соляром и другими масляными
дестиллатами. Нефтяные церезины имеют значительно более вы-
сокий молекулярный вес, например от С37Н76 до С53Н108 для су-
раханских церезинов; соответственно этому они концентри-
руются главным образом в гудроне и полугудроне и при пере-
гонке даже в глубоком вакууме (1,5—2,5 мм), как было указано
выше, частично разлагаются, переходя главным образом в преде-
лах 300—400°. .и
Температуры плавления выделенных грозненских парафинов
находятся в пределах примерно от 28 до 71°, для церезинов яр
заметно выше, а именно от 65 до 88° для грозненских и от 55 до
80° для сураханских.
При одной и той же температуре плавления церезины ши
сравению с парафинами имеют более высокий молекулярный и
удельный вес, более высокие коэфициенты рефракции, вязкость
(почти в 2 раза), а также нитробензольные точки.
Очень характерно различие кристаллической формы парафи-
нов и церезинов: парафины образуют пластинки или пластинча-
тые ленты, переплетающиеся между собою, причем парафины
легкоплавкие в общем дают пластинки и ленты большего раз-
мера, чем парафины тугоплавкие; от примеси масел и смол об-
щий характер этой кристаллической формы существенно не из-
меняется. Совершенно иное кристаллическое сложение имеют
церезины: они кристаллизуются в мелких иглах, плохо соединя-
ющихся между собой, и не дают таких прочных застывающих
систем, как парафины. В тесной связи с кристаллическим сложе-
нием находится различие между парафинами и церезинами,
164
имеющее большое производственное значение: в то время как
церезины прочно удерживают масла, трудно фильтрпрессуюгся
и плохо потеют, парафины проявляют противоположные каче-
ства, благодаря чему они сравнительно легко получаются в обез-
масленном состоянии.
Наконец, в химическом отношении между парафинами и це-
реЗИнаМй также наблюдается существенное различие/Так" на-
Дтример;‘"С*^мящёй серной кислотой (30% SO3)' парафины ни
на холоду, ни при подогревании до 100° практически не изме-
няются, тогда как церезины .после предварительного подогрева
с такой кислотой начинают с нею энергично и даже бурно реа-
гировать со вспениванием и саморазогреванием.
В отличие от парафинов церезины реагируют также с хлор-
сульфоновой кислотой, хамелеоном и азотной кислотой. Впро-
чем, в этом отношении в литературе вопроса имеются некоторые
противоречия, не вполне еще разъясненные [20].
Подводя итоги всему вышеизложенному, естественно притти
к представлениям, которые были высказаны уже давно (см.
выше), а именно, что парафиновые и церезиновые углеводороды
образуют два независимых гомологических ряда состава СпН^+2
и что, если парафины имеют ‘ нормальное строение, за церези-
нами должно быть закреплено изостроение, т. е. они предста-
вляют собою изопарафины. Мы подходим, таким образом, к дру-
гой стороне вопроса о химической природе парафина: к вопросу
о ближайшем химическом строении нефтяных парафинов и це-
резинов. . ’ .
Остановимся в первую очередь на нефтяных парафинах
Основной вопрос, подлежащий разрешению в этой области, за-
ключается в том, действительно ли нефтяные парафины имею!
нормальное строение и нет ли в их составе так называемых
изопарафинов.
Методика, довольно широко применяемая за последнее деся-
тилетие к исследованию вопроса о строении нефтяных парафи-
нов, заключается в следующем. Выделенный из той или иной
нефти парафин после тщательной перекристаллизации для осво-
бождения от масел фракционируют в глубоком вакууме на воз-
можно узкие фракции и снова перекристаллизовывают из раз-
личных растворителей. У полученных таким образом отдельных
нефтяных парафинов, правильнее — возможно суженных пара-
финовых фракций, определяют затем различные константы и,
нанося их на графики, изучают, насколько ход изменения этих
констант с температурой плавления или молекулярным весом
соответствует аналогичным кривым для ряда синтетических па-
рафинов нормального строения. Особенно интересные результаты
дают изучение рентгенограмм нефтяных парафинов и сопоставле-
ние их с рентгенограммами синтетических парафинов нормаль-
ного строения. Результаты, опубликованные до настоящего вре-
мени в указанных направлениях, сводятся к следующему.
Парафин из бирманской нефти [21] содержит, невидимому,
вое 14 нормальных углеводородов, от С21Щ4 до.СзДЪо; восемь из
165'
них были выделены в достаточно чистом виде. Однако некото-
рые из фракций имели аномальные температуры плавления; осо-
бенно аномальной оказалась фракция состава Сзз Нее с темпера-
турой плавления 66,5°, тогда как из синтетических парафинов
нормального строения такую температуру плавления должен был
бы иметь триаконтан, СзоН62.
Парафин из нефти Солт Крик, Уайоминг [22], был разогнан
на 'большое число узких фракций, которые показали лишь не-
большие отклонения от констант синтетических парафинов нор-
мального строения.
Грозненские парафины были также разогнаны [19] на ряд
фракций, оказавшихся довольно близкими по своим физическим
свойствам к соответствующим фракциям парафинов из других
нефтей и источников (буроугольные и сланцевые дегти). Так
как, однако, грозненские фракции перегонялись в пределах
30—50е, а высокоплавкие даже в пределах 60—90°, т. е. были
очень широкие, то едва ли было бы правильно делать какие-либо
заключения о их строении из вышеуказанного совпадения их
констант с константами других парафинов.
Особенно тщательному и разностороннему исследованию был
подвергнут-парафин из нефти Мид-Континента [231. Изучение
физических свойств ряда тщательно выделенных из этого пара-
фина фракций привело в основном к следующему заключению:
нефтяной парафин в главной массе состоит из углеводородов
нормального строения с приместью небольшого количества изо-
парафинов. Особый интерес представляет сделанное в самое по-
следнее время применение к тем же фракциям мид-континент-
ского парафина метода диффракции рентгеновых лучей. Оказа-
лось, что если построить кривую изменения периода идентич-
ности (identity period) в зависимости от молекулярного веса для
нормальных парафинов, то на этой кривой укладывается лишь
часть фракций, выделенных из нефтяного парафина. Такой ре-
зультат нельзя связывать с недостаточной индивидуальностью
некоторых исследованных фракций нефтяного парафина, так как
опыт показывает, что примесь углеводородов того же ряда и
строения почти не изменяет положения характерных линий фо-
тограммы чистых высокомолекулярных парафинов. Таким обра-
зом указанное несоответствие свидетельствует о примеси особого
рода, в данном случае, очевидно о примеси изомерных углеводо-
родов— изопарафинов. Окончательный вывод работы заклю-
чается в том, что содержание нормальных парафинов в исследо-
ванных фракциях не должно превышать 65%, содержание же
в них изопарафинов составляет по крайней мере 20^.
Итак, решение вопроса о химической природе бу^юугольного
п сланцевого парафина, нефтяного парафина и церезина наме-
тилось на основе изложенных выше опытных материалов в сле-
дующем виде: буроугольные и сланцевые парафины представляют
собою в основной своей массе смесь углеводородов ряда метана
нормального строения; нефтяные парафины состоят главным
образом также из-углеводородов ряда метана нормального строе-
166
ния, к которым, однако, примешано не менее 20% углеводородов
изостроения; наконец, церезины соответственно их некоторым
химическим реакциям должны быть отнесены к углеводородам
изостроения, т. е. представляют собою смесь изопарафинов.
•К совершенно аналогичным выводам привело применение
к тому же материалу реакции нитрования слабой азотной кисло-
той по М. Коновалову. Были исследованы тщательно очищенные
перекристаллизацией (обезмасленные) буроугольный парафин
(германский), нефтяной парафин (грозненский и американский)
и церезин (из сураханской пробки и челекенского озокерита).
Задача сводилась к выявлению прирохы образующихся нитро-
соединений; ясно, что в тех случаях, когда одним из продуктов
нитрования ’является третичное нитросоединение, в состав ис-
ходной смеси должны входить углеводороды изостроения (изопа-
рафины); наоборот, если среди продуктов данной реакции тре-
тичных нитросоединений не обнаруживается и образуются только
вторичные нитросоединения, то очевидно, исходные углеводороды
должны иметь нормальное строение, точнее — не имеют третич-
ного водорода.
Оказалось, что все три изученных типа углеводородных сме-
сей, т. е. буроугольный парафин, нефтяной- парафин и церезин,
дали при нитровании слабой азотной кислотой оба возможных
вида нитросоединений — вторичное и третичное1. Относитель-
ные количества и состав этих нитросоединений оказались,
однако, в каждом отдельном случае существенно различны.
Буроуголъный. парафин со средним молекулярным весом
образующих • его углеводородов дал при нитровании глав-
ным образом вторичное нитросоединение состава C^H-ssNO». Тре-
тичное нитро также образовалось при этой реакции, но лишь
в качестве побочного продукта (не свыше 5%); состав его отве-
чал формуле C22H45NO2.
Нефтяной парафин среднего молекулярного состава С24Н50
дал при нитровании в тех же условиях в 'Качестве главного про-
дукта также вторичное нитросоединение состава C24H4gNO2;
кроме того, в заметном количестве (25—35%) было получено'
третичное нитросоединение того же состава,
Наконец, церезин со средним молекулярным весом входящих
в его состав углеводородов С45Н92 дал при нитровании по
№. И. Коновалову главным образом третичный нитроцерезин со-
става C45H01NO2; побочными продуктами здесь оказались нитро-
парафины, вторичный и третичный, состав C22H4sNO2.
Причину столь глубокого различия в результатах нитрования
углеводородов исследованных парафинов и церезина следует
искать, конечно, в глубоком различии этих углеводородов между
собой и при этом не только в отношении их состава, но, оче-
видно, и их строения. Этот последний, весьма сложный вопрос
в* свете приведенных данных получает следующее разрешение.
1 Первичные нитросоединения при нитровании углеводородов слабой
азотной кислотой (удельного веса 1,075), как известно, не образуются.
167
Буроугольный парафин в главной своей массе состоит из
углеводородов ряда метана нормального строения. Однако даже
после тщательной перекристаллизации он содержит небольшую
примесь изопарафинов несколько более низкого молекулярного
веса, а именно: примерно C^Hie при среднем молекулярном весе
углеводородов нормального строения для германского буроуголь-
ного парафина CaeHsi.
Нефтяной парафин (грозненский) состоит преимущественно
из углеводородов ряда метана нормального строения, состава
С24Н50 со значительной примесью (25—35%) изопарафинов того
же состава.
Наконец, церезин, невидимому, состоит почти исключительно
из углеводородов ряда метана изостроения высокого молекуляр-
ного веса (в среднем около С45Н92), к которым примешаны более
ннзкоплавкие углеводороды того же ряда (т. е. изопарафины) со
средним молекулярным весом С22Н46.
Эти данные о строении углеводородов парафина и церезина,
по существу еще далеко не полные, интересно сопоставить между
собой с точки зрения термических условий получения соответ-
ствующих продуктов. Буроугольный парафин, получаемый к наи-
более жестких термических условиях (сухая перегонка), почти
нацело состоит из углеводородов нормального строения. Напро-
тив, церезин, техническое получение которото (очистка и т. п.)
осуществляется в наиболее мягких термических условиях, на-
цело состоит из углеводородов ряда метана изостроепия и, оче-
видно, сохранил без изменения строение углеводородов, содер-
жащихся в природном сырье. Наконец, нефтяной парафин (де-
сталлатный) находится как бы между этими двумя крайними
случаями. Таким образом знаменательно, что наиболее высоко-
молекулярные углеводороды, встречающиеся в природе (цере-
зин), имеют изостроение и что лишь путем термической перера-
ботки подходящего сырья получаются углеводородах ряда метана
нормального строения (парафины); интересно также, что чем
более жесткой является термическая обработка, тем богаче полу-
чаемый продукт углеводородами нормального строения, которые
являются, очевидно, продуктами вторичного происхождения. Мы
приходим, таким образом, к старому представлению о прото- и
пиропарафине, которое развивал еще Залозецкий [16], но кото-
рое ныне под влиянием новейших, кратко рассмотренных выше
исследований физико-химической природы церезина должно
быть освобождено от прежних неправильных представлений об
аморфной природе углеводородов церезина.
ПРИМЕНЕНИЕ ПАРАФИНА
Пи один из нефтепродуктов не находит столь разнообразного
применения, как парафин. Главная масса его идет на производи
ство свечей и спичек.
Для выделки свечей пригоден лишь твердый парафин с тем-
пературой плавления не ниже 52°; свечи из более легкоплав-
168
кого парафина мало удовлетворительны, так как гнутся в жар-
кур погоду и склеиваются в пачках друг с другом. Различают
«парафиновые» свечи, содержащие лишь 1,5—4% стеарина, и
«композиционные» свечи, в которых содержание стеарина дости-
гает 30% по весу и выше. При том же расходе вещества парафи-
новые свечи дают больше света, чем восковые, стеариновые и
спермацетовые. При сравнительной дешевизне парафин является
незаменимым материалом для свечного производства, так
как введение его освобождает громадные количества таких
значительно более ценных продуктов, как стеарин, воск
и т. п.
Не менее крупным потребителем парафина является спичеч-
ная промышленность. Здесь парафин применяется для пропитки
спичечной соломки с целью создать возможно легкий («резвый»)
переход пламени с головки зажженной спички на соломку. В от-
личие от свечного спичечный парафин принадлежит к типу
легкоплавких парафинов (температура плавления 36—17°) и не
подвергается глубокой очистке.
Из других областей применения парафина важнейшими
являются следующие.
В текстильной промышленности парафин применяется для
отделки (аппретуры) тканей; аналогичное применение находит
парафин в прачечном доле.
В бумажной промышленности парафин применяется для про-
питки особых сортов бумаги (вощеная бумага и т. п.).
Как изолятор парафин, широко применяется в электротех-
нике; как вещество, вполне устойчивое при комнатной темпера-
туре даже к крепким минеральным кислотам, парафин является
незаменимым материалом в граверном деле.
Парафин применяется далее в пчеловодстве для выделки
искусственной вощины и в промышленности естественных души-
стых для поглощения наиболее тонких ароматических веществ,
например из цветов, а также во многих других отраслях про-
мышленности и народного хозяйства.
Наконец, в химической промышленности и фармацевтическом
деле парафин находит применение при упаковке химических
веществ, разъедающих стекло, но главным образом: а) для изго-
товления разного рода мазей и композиций, важнейшей из ко-
торых является вазелин, и б) для переработки на жирные ки-
слоты.
Различают вазелины: естественный и искусственный, меди-
цинский и технический; во всех них парафин является важней-
шей составной частью.
Естественный вазелин был приготовлен и выпущен на рынок
впервые в 1871 г. американской фирмой Чисборо (R. А. Chesbo-
годк) из остатков парафинистой нефти. В настоящее время его
готовят главным образом, невидимому, путем холодного отстаи-
вания соответствующих дестиллатов парафинистых нефтей, ко-
торые идут далее на приготовление высокосортных смазочных
масел; осадок же, образующийся при отстаивании и состоящий
169
из твердых- и полутвердых парафинов с примесью масел, путем
соответствующей очистки перерабатывается на вазелин.
Искусственный вазелин приготовляется путем сплавления
парафина или церезина с подходящими нефтяными маслами.
Медицинские вазелины представляют собой вазелины наивыс-
шей степени очистки; они применяются для медицинских и кос-
метических целей (медицинские мази и т. п.). Технические вазе-
лины применяются для изготовления разного рода специальных
смазок, для предохранения металла от ржавчины и т. д.
Важнейшие требования, которым должен удовлетворять хо-
роший вазелин, заключаются в следующем: вазелин должен быть
однородным, прозрачным в тонком слое, должен легко тянуться
в нить или полотно, не должен расслаиваться и выделять масло
и вообще должен сохранять свою консистенцию при хранении.
В полной мере этими свойствами обладают лишь хорошие сорта
естественных вазелинов; они производятся в США. Из наших
нефтей прекрасным сырьем для получения вазелина являются
челекенская и особенно сураханская нефти. Однако это произ-
водство у нас еще не может считаться вполне налаженным.
В последнее время для парафина определилась новая об-
ширная область применения — как исходного сырья для получе-
ния жирных кислот [25]. Окислителем служит кислород воздуха,
иногда в присутствии подходящих катализаторов при надлежа-
щей температуре. Главным продуктом 'реакции являются высоко-
молекулярные жирные кислоты типа стеариновой, однако с при-
месью оксикислот жирного ряда. Продукт представляет собой
вполне удовлетворительный материал для мыловарения, и полу-
чение его, особенно при недостатке жиров, этого обычного сырья
мыловаренной промышленности, является одной из актуальных
задач химической переработки нефти и ее продуктов.
Нефтяной парафин производится главным образом в США,
где в 1934 г. было выработано свыше 200 000 т парафина. У нас,
в Грозном, имеется большой парафиновый завод, добывающий
парафин из грозненской парафинистой нефти. Завод оборудован
по последнему слову техники, находится в эксплоатации
с 1926 г. и удовлетворяет лишь внутреннюю потребность Союза,
в парафине. Около */* грозненского парафина расходуется у нас
на производства свечное и спичечное; остальное идет на другйе
отрасли промышленности народного хозяйства. Все ворзастаю-
щая потребность в парафине в связи с неуклонным развитием
производительных сил страны требует дальнейшего расширения
и развития у нас в Союзе парафинового производства.
ЛИТЕРАТУРА
1. Ле11 u.Hdgele, .Вег/ 22, 602 (18?9); Gaskard, .Ann. Chim.*(9) 1ft, 845 (1921).
2. Krafft, „Вег.- 15, 1687, 1711 (1882): 1в, 1714 (1883); 19, 2218 (1886); 29/
1323 (1896).
3. Шейгпхауер В., Буроугольные и сланцевые смолы, их получение и пере-
работка, пер. под ред. Валыис. В. X., П. (1921): В. В., Овокер?т:
и церезин, „Нефть* № 17, 15 (1931). Бедл А., Америкавскве:
170
методы переработки нефти, пер. с 1 англ, изд., М. —Л. (1925)*
Аблавацкий и Бражников, Парафиновый завод Грознефти Ааепб’
нефт. хоз/ № 6—7 (1928,; Bell, American Petroleum Refinin^’
2 изд. (1930); Прокофьев В. И.. Парафин, ОНТИ (1935). . °’
4. Добрянский А. Ф., Анализ нефтяных продуктов, М. — Л. (1925).
о. „Нефт. хоз.* XV, № 11—12, 639 (1928); ср. также Гухман и Гольдберг Д
„Азерб. Нефт. хоз/ № 10 (94), 74 (1929).
6. По различным источникам: см. также Гуревич Д. Г., Научные основы
переработки нефти, М. — Л. (1925): Engler С. u. Hofen, Н. Das
Erdol, т. I, Leipzig (1913): Жуков и Пантюхов, .Вести, жир. вещ/,
1900, X» 1 и др.; Беликовский А. С. и Павлова С. И, Нефти, би-
тумы и газы внекавказских месторождений СССР, ОНТИ (1934).
7. Mabery, „Proc. Amer. phil. Soc/ 86, 135 (1897).
К Наметкин, Беликовский, Нифонтова, .Нефт. хоз/ XVIII, № 10, 533 (1929).
9. Abraham Н., Asphalts and Allied Substances, стр. 498.
10. Любавин Н. Н, Техническая химия, т. V, ч. I, М. (1910), стр. 276 и т. д.
11. Standart Methods of Testing Petroleum and its Products, L. (1929).
12. Литературу вопроса см. в цитированной выше статье Наметкина,
Беликовского и Нифонтовой.
13. Krafft F., .Вег/ 20, 2259 (1888); 40, 4779 (1908).
14. Muller a. Saville, ,J. Chem. Soc/ 127, 599 (1925).
15. Francis, Watkins a. Wallington, ,J. Chem. Soc/ 121, 1529 (1922); Piper.
Brown, Dyment, Ibid. 127, 2194 (1925).
16. Engler u. Bohm, .Dingl. Polyt. J/ 262, 468 (1886); Engler C. u. Hofer H.
Das Erdol, т. I, стр. 159 (1913); Балозецкий (Zcdoziecki), .Ztschr-
angew. Chem/ 1888, 126.
17. Гурвич, .Научные основы"..., M.—Л (1922) стр. 12; ср. также Ракузин,
.Ж. Р. X. О/ 46, 154+ (1914).
18. .Нефт, хоз/ IX, № 9. 369 (1925).
19. Химический состав нефтей и нефтяных продуктов, М. — Л. (1931),
20. Ср. Бейлъштейн и Виганд, .Вег/ 16, 547 (1883); Маркуссен, .Chem. Zig/,
1914, 73 и 1915, 613.
21. Carpenter, .J. Inst. Petr/Techn/ 12, 288 (1926).
22. Buchter a. Graves, .Ind. Eng. Chem/ 19, 718 (1927).
23. Ferris, Cowles a. Henderson, Ibid. 21, 1090 (1929); Clark a. Smith, Ibid- 28,
697 (1931).
24. Наметкин и Нифонтова, .Известия Акад, наук СССР* (серия хими-
ческая) № 1 (1936).
25. Мошкин, „Нефт. и сланц. хоз/, т. 2, As 9—12 (1921) и т. 3, № 1—4
(1922).
НЕП НЕДЕЛЬНЫЕ УГЛЕВОДОРОДЫ ЖИРНОГО РЯДА
КРАТКАЯ ХАРАКТЕРИСТИКА НЕПРЕДЕЛЬНЫХ УГЛЕВОДОРОДОВ [1]
Непредельные углеводороды образуют несколько рядов, про-
стейшими из которых являются ряды этилена и ацетилена. Об-
щим способом их получения является отщепление элементов
галоидоводородной кислоты от соответствующих галоидозаме-
щенных при действии на них едких щелочей. Для получения
этиленовых углеводородов при этом исходят из одногалоидозаме-
щенных, для получения ацетиленовых — из двугалоидозамещен-
ных; например:
C2H6J 4- КОН = KJ + НоО + С2Н4 (этилен);
С2Н4Вг2 + 2К0Н = 2КВг + 2Н2О + C2HS (ацетилен).
Другим источником получения непредельных углеводородов
являются кислородные соединения. Так, из спиртов действием на
171
них водуотнимающих веществ при повышенной температуре'по-
лучаются этиленовые углеводороды, например:
С2Нь0Н — Н20 == С2Н4 (этилен).
В 'Качестве катализатора, способствующего этой реакции, осо-
бенно широкое применение приобрел в настоящее время глинозем.
Для получения ацетиленовых углеводородов в качестве
исходного материала могут служить альдегиды и кетоны, пре-
вращаемые сначала в двугалоидопроизводные с обоими галои-
дами при одном и том же атоме углерода.
Наконец в новейшее время источником получения различных
непредельных углеводородов могут служить специальные методы
переработки нефти, крэкинг и пиролиз, при которых наблюдается
массовое образование не только низших, газообразных, но и бо-
лее высокомолекулярных, жидких, главным образом этиленовых
углеводородов.
Состав непредельных углеводородов жирного ряда опреде-
ляется формулами: СяНг» — для ряда этилена и СЛал-® — для
ацетилена. Характерной особенностью их строения являются»
как известно, кратные связи — двойная и тройная.
Кроме этиленовых и ацетиленовых углеводородов, известно
немало других непредельных рядов. Одни из них, сохраняя от-
крытую группировку углеродных атомов, по своему составу и
свойствам характеризуются еще большей степенью ненредель-
ности, чем этилены и ацетилены, для объяснения которой в них
принимается наличие не одной, а двух или нескольких кратных
связей; в других допускается замкнутая группировка углеродных
атомов и вместе с тем также одна или несколько кратных связей
(нафтилены, терпены и т. п.). Для химии нефти наибольший
интерес представляют этилен и его гомологи, физические свой-
ства которых сведены в табл. 4G.
В отличие от углеводородов ряда метана все непредельные
углеводороды характеризуются большой реакционной способ-
ностью.
1. С особенной легкостью протекают здесь реакции присоеди-
нения, причем конечными продуктами всегда являются те или
иные соединения ряда метана 'предельного характера. Так напри-
мер, галоиды и галоидоводородные кислоты легко присоеди-
няются здесь уже при обыкновенной температуре:
ед+hi = ед1; с2н2+4Вг=едвг4.
Присоединение водорода происходит здесь под влиянием ка-
талитического воздействия мелкораздроблениых металлов либо
при обыкновенной температуре (в присутствии Pt, Pd), либо при
нагревании от 140° и выше в присутствии Ni).
ед+2Н=с2н
в*
ед+4Н=ед.
172
Таблица 46
Свойства углеводородов ряда этилена
Наименование Формула Темп, плав. Темп. кип. Уд. вес Коэф. рефр. nJD
d t
Этилен С2 В4 —169 -102.7 0,609 (яицк.)
Пропилен С3н6 — — 48.2 — —
Бутилен С*н8 *— —5 — ——
Диметил-этилен симы —J“1 —И ——
Изобутилен V —6 — —
Амилен сб Ню —— 39 — — —
Изопропил-этилен . . . 9 — 20 0,648 0 —
Метил-этил этилен
симм И —» 86 11 —
Метил этил-этилен не-
симм V — 31 0,667 0 1,378 (16)
Триметил-этилен . . . V — 36 0,678 0 —
Гексилен C<j Hjl2 — 69 0,683 15 —
Тетраметил-этилен . . V — 72 0,728 0 1.4128
Гептилен норм.. . С7 Ни 95 0,703 19,5 —
Октилен , Cg Hie — 122 0,722 17 —
Децилен „ . . С10Н20 —• 172 0,763 0 -
Унчецилен „ . . С11Н22 — 841 — — —
Додецилей , . . 12 -31 96 2 0.773 0 —
Тетрадецилен , . . -12 127 а 0,785 0 —
Гексадецилен . . . С16Щ2 + 4 155 2 0,784 15 1,442 (19)
Октадецилен , . . СюНзб +18 179 2 (,791 18
В подходящих условиях легко присоединяется к непредель-
ным углеводородам также вода. Так, этиленовые углеводороды
в присутствии серной кислоты, хлористого цинка и других ката-
лизаторов превращаются при этом в соответствующие спирты;
ацетиленовые же углевороды в присутствии солей окиси ртути,
присоединяя частицу воды, дают альдегиды или кетоны. На-
пример:
С2Н4 + Н20 = С2Нб0Н; С2Н2 + Н20 = СН8 • СНО.
2. К реакциям присоединения примыкают реакции окисле-
ния непредельных углеводородов, протекающие также очень
легко в самых разнообразных условиях. И здесь, по крайней
мере в первую фазу реакции, образуются различные продукты
присоединения. Так, этиленовые углеводороды с озоном (Оз)
дают взрывчатые озониды общей формулы СДЬпОз; кислородом
эти же углеводороды медленно окисляются с образованием
взрывчатых перекисей; органические гидроперекиси (надуксус-
ная кислота, гидроперекись бензоила) образуют с ними соответ-
ствующие окиси СЛЙ2ПО; со щелочным раствором марганцево-
кислого калия здесь сначала получаются гликолы общей фор-
мулы CftH2n(OH)2, окисляющиеся дальше уже с распадом орга-
1 При 18 мм.
2 При 16 мм.
178
нической молекулы. Вообще взаимодействие хамелеона в ней-
тральной или щелочной среде с непредельными углеводородами
протекает настолько легко, что хамелеоном обычно пользуются
как реактивом на непредельные углеводороды (или вообще на
кратные связи), так как при реакции вследствие раскисления
хамелеона здесь уже на холоду происходят резкое изменение
цвета реактива и выделение перекиси марганца.
3. Другой тип превращений, характерных для непредельных
углеводородов, это—реакции уплотнения и конденсации. Так,
под влиянием разного рода катализаторов (EbSCh, Z11CI2 и др.)
этиленовые ’углеводороды легко дают димерные, тримерные и
тому подобные формы. Например, из псевдобутилена, С4Н8,
в этих условиях легко образуются углеводороды Cellie, С12Н24
и т. д. Ацетиленовые углеводороды также легко полимеризуются,
причем в подходящих условиях здесь образуются углеводороды
ароматического ряда, например, из ацетилена, С2Н2— бензол.
СбНб. Наконец, непредельные углеводороды легко конденсируются
также с ароматическими углеводородами (см. гл. IV).
4. Нередко для характеристики и открытия непредельных
углеводородов применяются также различные их металлические
производные. Для этиленовых углеводородов характерны ком-
плексные ртутные производные, аморфные желтые осадки, полу-
чаемые при взаимодействии этиленов с сернокислой или уксус-
нокислой ртутью. Для ацетиленовых углеводородов более харак-
терны их взрывчатые медные или серебряные производные (кар-
биды), получаемые действием ацетилена или однозамещенных его
гомологов на аммиачные растворы закиси меди или серебра. И те
и другие металлические производные при действия на них ми-
неральных кислот легко разлагаются с обратным образованием
непредельных углеводородов.
НЕПРЕДЕЛЬНЫЕ УГЛЕВОДОРОДЫ РАЗЛИЧНЫХ НЕФТЕЙ
И НЕФТЕПРОДУКТОВ
В громадном большинстве нефтей непредельные углеводороды
либо обнаружены в крайне • незначительном количестве, либо
вовсе не были найдены. Таковы, например, нефти бакинская,
пенсильванская, галицийская и многие другие [2]. Легче обна-
руживаются непредельные углеводороды в нефтяных дестилла-
тах; однако обыкновенно они являются здесь продуктами вто-
ричного характера, образовавшимися в результате разложения
(крэкинга) основных углеводородов нефти при их перегонке. Для
иллюстрации можно привести следующие примеры.
В пешельбронском керосине (Эльзас) давно уже были обна-
ружены изомерные амилены и гексилены; надо думать, однако,
что они не содержатся в нефти в готовом виде, а образуются
в процессе ее перегонки [3].
Качественно непредельные углеводороды были обнаружены
также в некоторых дестиллатах пенсильванской нефти; однако
пробы с самой нефтью и ее дестиллатами, полученными перегон-
174
кой в вакууме, дали отрицательные результаты на содержание
непредельных [4].
Наиболее надежные указания на присутствие непредельных
углеводородов имеются для канадской нефти, из которой с по-
мощью серной кислоты удалось выделить небольшие количества
гексилена, гептилена, октилена и нонилена [5]. Эти углеводо-
роды, невидимому, не образуются в процессе переработки нефти,
а содержатся в ней в готовом виде, хотя <в крайне незначитель-
ном количестве.
Очень богаты непредельными нефтепродукты, получаемые
в результате крэкинга. Их состав будет рассмотрен ниже в связи
с вопросами переработки нефти.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ НЕПРЕДЕЛЬНЫХ В НЕФТЯХ
И НЕФТЕПРОДУКТАХ
Количественное определение непредельных в сложных смесях
представляет, вообще говоря, задачу весьма трудную. Сравни-
тельно проще решается она для легких нефтепродуктов; при
этом можно пользоваться одним из приводимых ниже мето-
дов [6].
Метод иодных чисел. Иодным числом какого-либо вещества
называется выраженное в граммах количество иода, которое мо-
жет быть связано 100 г вещества. Так как иод реагирует с не-
предельными углеводородами слишком медленно, то для харак-
теристики непредельности пользуются также бромом (бромные
числа), бромистым иодом (иодобромные числа) и хлористым
иодоМ; соответственно этому различают следующие важиейшие
видоизменения метода иодных чисел.
Способ Гюбля. В качестве реактива употребляют спиртовый
раствор иода с добавлением сулемы. В качестве растворителя
навески углеводорода применяют хлороформ. Навеска берется
с таким расчетом, чтобы для насыщения ее непредельных связей
хватило менее половины прибавляемого иодного раствора. После
смешения растворов навески и иода смесь оставляют на 24 часа
при комнатной температуре, после чего следует титрование гипо-
сульфитом с добавлением раствора йодистого калия.
Способ Гюбля-Баллера. Иодный раствор также готовится на
спирту, но с добавлением сулемы и соляной кислоты, благодаря
чему раствор делается более устойчивым. Прочие условия ана-
лиза и время — те же, что и по способу Гюбля.
Способ Гануса. Реактивом служит раствор однобромистого
иода в уксусной кислоте. Продолжительность реакции такого
раствора с непредельным углеводородом 15—30 мин. Титрование
гипосульфитом производится также с добавлением раствора
йодистого калия.
Способ Маргошеса. Применяется 0,2 N спиртовый раствор
иода. Навеску растворяют также в 96%-ном спирту к згрйба-
вляют 20 см3 иодного раствора; затем приливают 200 см9 воды,
встряхивают и образовавшуюся эмульсию оставляют на 3—5 мин.;
последующее титрование гипосульфитом ведется без добавления
йодистого калия.
Сравнительное исследование поименованных выше способов
на специально приготовленных растворах непредельных углево-
дородов в бензине приводит к следующим результатам [7].
При небольшом содержании непредельных (до 1—2%) способ
Гануса дает несколько повышенные числа. Способы Гюбля и
Гюбля-Баллера дают числа, очень близкие к теории, и обычно
рекомендуются как стандартные. Однако продолжительность
определения (свыше 24 час.) делает их нередко малопригодными
для практической работы. Вполне удовлетворительные резуль-
таты при небольшом содержании непредельных дает также способ
Маргошсса, что, принимая особенно во внимание быстроту опре-
деления, позволяет рекомендовать его для характеристики не-
предельности легких нефтепродуктов прямой гонки.
Главным источником ошибок при определении иодных чисел
является то обстоятельство, что при действии галоидов на угле-
водородные смеси с реакциями присоединения, на которых
базирует метод, могут иметь место также реакции замещения.
С легкими углеводородами и на холоду замещающее действие
иода и брома невелико; однако, по мере возрастания молекуляр-
ного веса углеводородов оно постепенно увеличивается, и прене-
брегать этим направлением реакции, очевидно, уже не прихо-
дится. Для его учета обыкновенно определяют количество галои-
доводородной кислоты, выделяющейся в процессе реакции заме-
щения.
Способ Мак-Илинея [8]. Реактивом служит здесь раствор
брома в четыреххлористом углероде, приливаемый к углеводороду
в том же растворителе. К этой смеси через несколько минут
прибавляется водный раствор йодистого калия. Бром, не вошед-
ший в реакцию, вытесняет из этого раствора иод, определяемый
титрованием раствором гипосульфита. Затем прибавляют йодно-
ватокислого калия (JOSK). Так как в растворе в случае реакции
замещения должен содержаться бромистый водород, то в резуль-
тате взаимодействия с йодноватой кислотой выделяется новое
количество иода, которое также определяется гипосульфитом.
Следует, однако, иметь в виду, что галоидоводородная кис-
лота, образующаяся при взаимодействии галоидов с органиче-
скими соединениями, может получаться не только в результате
типичных реакций замещения. Ход образования галоидоводо-
рода может быть, например, следующий: сначала бром присое-
диняется по месту непредельной связи, а затем ввиду малой
устойчивости того или иного из образовавшихся дибромидов
происходит частичное отщепление бромистого водорода, кото-
рый при его учете, например, по способу Мак-Илинея, будет
отнесен за счет реакции замещения, тогда, как в действитель-
ности он содержит часть галоида, пошедшего первоначально на
реакцию присоединения. Таким образом точный учет реакций
присоединения и замещения по этому способу, очевидно не воз-
можен.
176
Способ Кауфмана. Реактивом является раствор брома в чи-
стом метиловом спирте, насыщенном бромистым натрием.
В этих условиях в растворе образуется комплекс NaBr. пСНзОН,
избытком которого действуют на хлороформовый раствор тит-
руемого олефина или смеси олефинов; остаток брома оттитро-
вывают, как в предыдущем случае. Метод должен быть отнесен
к числу наиболее надежных способов характеристики непре-
дельности; однако общие, отмеченные выше недостатки мето-
дики в известной степени присущи и этому ее видоизмене-
нию [9].
Метод кислородных чисел. При действии гидроперекиси бен-
зоила или ацетила на непредельные углеводороды различных
рядов уже при обыкновенной температуре образуются окиси
этих углеводородов по следующему уравнению:
> С = С < + С6НбС0оН = > С — С < + CGH3C02H.
Y
В присутствии избытка гидроперекиси реакция эта проте-
кает количественно и может быть применена для точного опре-
деления непредельных углеводородов в смеси их с предельными
и ароматическими [10]. Сущность метода заключается в сле-
дующем.
К небольшой навеске углеводорода (0,1—0,3 г) прибавляют
избыток гидроперекиси бензоила в -растворе хлороформа. Смесь
оставляют при комнатной температуре на Р/г—2 дня, в течение
которых весь непредельный углеводород успевает окисляться.
Затем к смеси прибавляют водного раствора йодистого калия
и серной кислоты, а выделившейся от невошедшей в реакцию
гидроперекиси иод оттитровывают гипосульфитом. Зная коли-
чество активного кислорода гидроперекиси, взятой первона-
чально, по разности определяют количество кислорода, пошед-
шего на окисление взятой навески; пересчитывая далее полу-
ченный результат на 100 г анализируемого вещества, получают
его кислородное число, которое подобно бромному или иод-
ному числу может служить характеристикой непредельности
вещества.
Недостатками метода кислородных чисел являются продол-
жительность времени, необходимого для производства опреде-
ления, и некоторая сложность приготовления реагента (гидро-
перекиси). Эти недостатки искупаются, однако, надежностью
получаемых результатов, так как все углеводороды предельного
характера (парафины, нафтены, ароматика) совершенно не реа-
гируют с гидроперекисью бензоила и ацетила.
Иодные и кислородные числа представляют собою величины
несравнимые; однако путем простого пересчета легко подойти
к их сопоставлению. Для этого достаточно соответственно
основным реакциям, протекающим при определении иодных и
кислородных чисел, разделить: иодное число — на удвоенный
атомный вес иода (254), а кислородное число —на атомный вес
177
12 Зак. 1622. Химия нефти.
кислорода (16). Очевидно, мы получим, таким образом, те же
характеристики, выраженные, однако не просто в весовых (про-
центных) количествах, а для иодных чисел — в грамм-молеку-
лярных, для кислородных же чисел — в грамм-атомных соотно-
шениях. Помножая эти величины на 100, получаем величины,
которые можно назвать коэффициентом непредельности по иоду
или кислороду:
Иодное число><100 __ коэф. непредельности по поду;
= коэф, непредельности по кислороду.
Коэфициент непредельности выражает процентное содержа-
ние в анализируемом веществе некоторого условного непредель-.
него соединения, молекулярный вес которого равен 100. Чтобы
перейти от коэфициеитов непредельности к процентному содер-
жанию непредельных в различных нефтепродуктах или отдель-
ных их фракциях, необходимо в выражение их коэфициента не-
предельности ввести дополнительный множитель, например для
бензиновых погонов 0.7—1,4, для керосиновых — 1,5—2 и т. л,
соответственно среднему молекулярному весу углеводородов,
«образующих данный погон. Само собою разумеется, что чем
уже исследуемая фракция, тем ближе получаемый указанным
способом результат к истинному содержанию в ней непредель-
ных углеводородов.
Сернокислотный метод Для практических целей часто представляет
интерес суммарное определение непредельных в том или ином нефтепро-
дукте по объему. Одним и, пожалуй, единственным методом решения этой
задачи является сернокислотный метод в различных его видоизменениях,
при котором определяется уменьшение объема углеводородной смеси от
обработки ее серной кислотой. Метод основан на способности серной ки-
слоты растворять непредельные углеводороды с образованием в первую
очередь эфирно-серных кислот по следующему уравнению.
> С = С < -4- H2SO4 = > UH — С
О • SO3H
Следует, однако, иметь в виду, что вслед за этой первой фазой реак-
ции наступают последующие стадии ее, приводящие к образованию про-
дуктов полимеризацпи исходных непредельных углеводородов (димеров, •
тримеров и т. д.), а эти последние частично вновь переходят в углево-
дородный слой и могут быть удалены из него лишь путем последующей
перегонки. Еще сложнее протекает реакция непредельных углеводороде®
с серной кислотой в присутствии ароматики, когда, кроме полимеров, образу-
ются и остаются в углеводородном слое также продукты конденсации непре-
дельных углеводородов с ароматическими (см. гл. IV). Наконец, как будет
показано в гл. X, в реакцию с серной кислотой легко вступают также веще-
ства смолистого и асфальтового характера, которые всегда содержатся в неф-
тях и некоторых нефтепродуктах. Таким образом сернокислотный метод
даже в наиболее простых случаях даст педостаточпо точное содержание не-
предельных в нефтепродукте, и применение этого метода в работах еще не-
давнего прошлого оправдывается лишь отсутствием более точной методикя.
То же самое можно сказать о применении серной кислоты для удаления не1
предельных из углеводородных смесей — вопрос, который представляет
особую важность при анализе крэкинг-продуктов.
178 '
Метод полухлористой серы (11). Единственный метод, по-
зволяющий получить точное представление о содержании не-
предельных углеводородов в бензине, заключается в обработке
его на холоду полухлористой серой; по объему вошедшею в ре-
акцию углеводорода судят об объемном содержании в исходном
бензине непредельных.
Полухлористая сера (S»Ck) совершенно не реагирует на холоду с па-
рафинами, нафтенами и ароматикой; с непредельными асе углеводорода ми
она дает продукты присоединения типа охлоренных тиосульфидо-в. Так
например, реакция полухлористой серы с этиленом идет" по следующей
схеме:
SgCi^ 4“
СНо : С Но
С1Ь : СН2
= S:S
СП2-СИ4С1
СЩ.СН.СТ
Продукты присоединения полухлористой серы к
породам довольно устойчивы, ни кипят значительно
водородов. Поэтому при перегонке
реакционной смеси фракции крэкинг-
бензина с полухлористой серой не
вошедшие в реакцию парафины, на-
фтены и ароматика могут быть легко
отогнаны и, таким образом, нацело
освобождены от непредельных.
Определением непредельных
в бензине методом полухлори-
стой серы достигается одновре-
менное удаление их. необхо-
димое для последующего опре-
деления в бензине ароматики
Таким -образом метод особенно
удобен при так называемом
групповом анализе бензина,
когда опредетению подлежат
все четыре важнейших его ком-
понента: непредельные, арома-
тика, нафтены и парафины.
Определение производят либо
в цельном, неразогнанном бен-
зине, либо в тех его фрак-
неп редел ьным углево-
выше исходных угле-
Фпг. 28- Аппарат для определения
непредельных полухлористой серой-
О\ тг
циях, в которых обычно опре-
деляется ароматика, т. е. в бен-
зольной (температура кипе-
ния GO—95°), толуольной (температура кипения 95—120°) и
ксилольной фракциях (температура кипения 120—150°).
Аппаратура, которой удобно пользоваться для определения
непредельных методом полухлористой серы, состоит (фиг. 28)
из круглодонной колбы’А на 500 см3 q удлиненной шейной,
12*
179
в которую на шлифе вставлена пробка Ъ В эту пробку входит
капельная воронка с с оттянутым концом, соприкасающимся
с другой трубкой а, входящей в ту же пробку. Капельная во-
ронка служит для постепенного приливания в реакционную
колбу полухлористой серы, а в конце опыта через эту воронку
вводится вода и щелочь; трубка служит для последующего
введения пара. К шейке колбы припаяна отводная трубка Б
длиною в 10 см, разделяющаяся в конце на две части: одна
из них направляется вверх и заканчивается небольшим четырех-
или шестишариковым холодильником Г; другая часть напра-
влена вниз и заканчивается притертым проверенным сульфатором-
бюреткой В. Сульфатор этот в качестве приемника присоеди-
няется к прибору, когда реакция непредельных с полухлори-
стой серой закончена и предстоит перегонка непрореагировав-
шего бензина. До этой перегонки во избежание потерь летучих
частей бензина, следует нижний конец трубки Б закрывать
внутренней пробочкой м, жтграя улеживается на трубке при
помощи небольших каучуковых колец, надеваемых на припаян-
ные к пробочке и трубке Б крючочки. Этим же способом в свое
время прикрепляется к трубке Б бюретка В.
В случае, если по местным условиям изготовление подоб-
ного прибора затруднительно, можно пользоваться упрощенным
прибором, состоящим из тех же частей, но соединенных между
собою вместо шлифов простыми корковыми пробками.
В тех случаях, когда анализируемый бензин содержит не-
предельных не свыше 25%, определение последних производится
следующим образом. К 50 мл сухого бензина, охлажденного
до 0°, приливают по каплям при непрерывном перемешивании
и ледяном охлаждении 15 мл полухлористой серы и оставляют
опыт на 3—4 часа или на ночь. Затем добавляют к смеси сна-
чала холодной воды, а потом 25%-ной щелочи для разрушения
избытка полухлористой серы. Наконец, отгоняют невошедшую
в реакцию часть бензина водяным паром и собирают его в гра-
дуированный приемник-бюретку описанного выше прибора.
Разность между взятым и отогнанным объемами бензина пере-
считывают на 100 и получают содержание непредельных в объ-
емных процентах.
Если содержание непредельных в бензине по предваритель-
ному определению (превышает 25%, то применение метода полу-
хлористой серы требует разбавления бензина. В качестве рас-
творителя можно применять либо соответствующие по темпера-
туре кипения бензиновые фракции, освобожденные от непре-
дельных и ароматики, либо соответствующие ароматические
углеводороды. При последующем расчете такое разбавление
естественно, должно быть учтено.
1 Определение непредельных методом полухлористой серы можно про-
изводить также в приборах уменьшенного размера с доведением количества,
углеводорода до 20—5 и даже 2 лл. Точность определения при этом»
естественно, несколько снижается.
180
Опыт показывает, что при описанной выше обработке бен-
зинов полухлористой серой небольшая часть непредельных
СО,5—1,5%) нередко остается в них и, таким образом, теряется
для определения. Эти остатки непредельных должны быть, оче-
видно, также учтены и удалены для последующего определения
ароматики. Может быть рекомендовано два способа для такого
повторного определения непредельных, а именно:
1) обработка бензина 85 %-ной серной кислотой;
2) повторная обработка бензина полухлористой серой.
Ввиду несомненной простоты сернокислотного способа сле-
дует отдать предпочтение этому методу для окончательного уда-
ления -последних остатков непредельных (1—2%) и для опреде-
ления этих остатков.
ХАРАКТЕРИСТИКА НЕПРЕДЕЛЬНОСТИ НЕФТЕЙ
И НЕФТЕПРОДУКТОВ [12]
Для характеристики непредельности сырой нефти наиболее
применим метод иодных чисел. Надо, однако, ясно предста-
влять себе, что иодный раствор (как. впрочем, и гидроперекись
бензоила) реагирует не только с непредельными углеводоро-
дами, но и с такими компонентами нефти, как ее смолистые и
асфальтовые вещества. Ввиду этого иодными числами можно
пользоваться, очевидно, лишь для общей сравнительной ха-
рактеристики непредельности сырых нефтей (табл. 47).
Та&щя 47
Иодные и кислородные числа некоторых нефтей
Месторождение нефти о °_ © Ю И •“< _ и цизк. ОЛЫ * S слород- ;о числа Коэф, непредельности
Иод, я. X 100 Кисл. я. X100
5 >» и * я < о к 3 я 254 16
Грозный .... 0,874 24,0 19,8 6,31 7.8 39,4
Чонгелек (Крым) 0 878 1,0 12,5 2,78 4,9 17,4
Манат (Эмба) . . 0,899 12,6 22,3 5,69 8,8 35,6
Бинагады (Баку) 0,927 46,0 19,0 6,26 7,5 39,1
Оха (Сахалин) . 0,930 ' 36,8 21,7 3,72 8,5 23,3
Как видно из данных табл. 47, иодные числа тяжелых, вы-
соко смолистых нефтей, вообще говоря, больше, чем легких
малосмолистых. Однако эта, казалось бы, естественная законо-
мерность отнюдь не является общей, откуда следует, что по
содержанию собственно непредельных углеводородов между от-
дельными нефтями, невидимому, может наблюдаться значитель-
ное различие. Для ближайшей характеристики непредельности
отдельных нефтей можно было бы обратиться к изучению их де-
1 Пб Ганусу.
181.
стиллатов и нефтепродуктов. В табл. 48 приводятся некоторые
данные в этом направлении на основе определения иодных (по
Гану су) и кислородных чисел.
Таблица 48
Иодные и кислородные числа дестиллатов эмбенской нефти
Дестиллаты Уд. вес Акцизе. смолы Иодные числа Кислор. числа Коэф, непредель- ности
100 х И. ч. 100 X К. ".
254 16
Керосиновый . 0,823 5,3 0.61 2,1 3.8
Соляровый . . 0,880 2,0 10,8 1,63 42 10,3
Веретенный . 0,895 5.0 16,0 ‘\12 6.3 13,3
Машинный . . 0,899 6,0 16,9 2,39 6.7 14,9
Из табл. 48 видно прежде всего, что иодные и кислородные
числа дестиллатов и соответствующие коэфициенты непредель-
ности увеличиваются по мере утяжелёния дестиллатов, что отве-
чает постепенному возрастанию в них непредельных углеводо-
родов. Этот рост становится еще более явственным, если при-
нять во внимание, что множитель для перехода от коэфициен-
тов непредельностп к процентному содержанию непредельных
увеличивается по мере повышения молекулярного веса углеводо-
родов. Не подлежит сомнению, однако, что значительная часть
этих непредельных отнюдь не является естественной составной;
частью нефти, а образуется лишь при ее переработке в резуль-
тате частичного разложения отдельных ее компонентов (крэкинг).
Нельзя не отметить для всех приведенных выше примеров
значительного расхождения в характеристике непредельности.
одного и того же продукта на основе иодных и кислородных чи-
сел. Это расхождение особенно выделяется для тяжелых нефте-
продуктов, а также для сырых нефтей. Причина его заключается;:
невидимому, в присутствии среди компонентов, образующих эт£.
продукты, смолистых и асфальтовых веществ, которые значи-’
тельно энергичнее реагируют с гидросперекисью бензоила, чём;
с иодным раствором [12].
Переходим, наконец, к характеристике непредельности очи-
щенных нефтепродуктов. Как видно из данных табл. 49, в этом
направлении наблюдается чрезвычайно большое разнообразие,
зависящее для нефтепродуктов различных типов прежде всего
от степени их очистки. Нефтепродукты высокой степени очистки,
свободные от смолистых и непредельных веществ, имеют коэфй-
циент непредельности, выражаемый небольшим целым числом,
правильной дробью, или даже равный нулю; таковы, например;
•бензины прямой гонки, маловязкие масла наивысшей степени
очистки (веретенное, трансформаторное масла и др.), хорошо
182
Таблица 4&
Иодные и кислородные числа нефтепродуктов
Наименование нефтепродукта Уд. вес при 15° Цвет по Штаммеру Иодные числа Кислородные числа Коэф, непредельн.
Иодн. Ч-Х100 Кисл. ч.\100
254 16
Бензин автомобиль-
ный, II с., совет- ский, 1938 г. . . . 0,759 59,69 23,5
Бензин бак. II с.,
прямой гонки • • Motor gasoline „Те- 0,753 1,3 0,64 0,03 0,18 0,17
xas Со* 0,750 2,6 52,40 3,12 20,6 19,5
Керосин бакинский
(внутренний). • . Керосин .Standard 0,824 3,3 11,23 0,83 4.4 5Д
Oil Со* 0,807 2,7 27,04 1,77 10,6 ИД
Spindle Oil „Shell JI* Трансформаторное 0,869 0,882 — 4,51 0,28 0,63 1,8 3,4 1,8
масло эмбенское . -— 8,70 3,9
Transformator Oil
.Shell" 0,838 — 0,77 0,00 0,3 0Л
Турбинное масло эмбенское .... Машинное .2“ эм- 0,896 — 13,15 1,00 53 63
венское ..... Машинное масло ба- 0,698 16,0 15,04 1,28 5,9 83
кинское, эксп. . . 0,909 — 17,97 1,64 7,1 юз
Автол »М“ эмбен- 6,3
ский 0,898 — 15,96 132 83
Banop ,М* Цилиндровое ,М* 0,907 0,914 16,39 1,40 1,87 6.4 7,9 8,8 11,7
бакинское .... - - 20,29
Standard gas Engin „Standard Oil Co* Вазелин медицин- 0,892 12,0 19,43 2,94 7,6 183
ский белый, .Stan- dard Oil Со* . . • 0,828 180,5 1 9,19 0,90 3.6 5,6
- (60°)
Вазелин медпц. жел-
тый „Standard Oil Со* 0,841 7,0 1 14,45 1,23 5,7 7,7
(60°)
Парафин refined „Standard Oil Со* Парафин желтый 0,909 0,881 <1,5 3,1 — 0,00 — 0,0 1,6
0,26
.Standard Oil Со* — ——
Парафин белый спец, грозненский . - . 0,908 26 — 0,07 —• 0,44
Парафин желтый грозненский . . . 0,907 >5,0 1 — 0,12 — 0,75
1 С разбавлением бензином 1:10.
183-
очищенный белый парафин и др. Для нефтепродуктов, менее
тщательно очищенных, коэфициент непредельности выражается
несколько большим целым числом, причем для не слишком тя-
желых продуктов расхождение между коэфициентами непредель-
ности по иоду и кислороду сравнительно невелико; таковы, на-
пример, керосины прямой линии и наши маловязкие масла, не-
предельность которых зависит главным образом от нахождения
в них продуктов крэкинга. образовавшихся при перегонке и со-
хранившихся в масле вследствие его недостаточной очистки. На-
конец, для более тяжелых продуктов, например для большинства
смазочных масел и вазелинов, недостаточно очищенных, коэфи-
циент непредельности достигает 5—10 и более единиц, причем
расхождение между коэфициентами непредельности по иоду и
кислороду нередко достигает здесь 50% и более. Очевидно, мы
имеем здесь явление, аналогичное тому, которое было отмечено
выше для сырых нефтей, т. е. влияние примеси смолистых ве-
ществ. Особо следует отметить некоторые товарные образцы за-
граничных бензинов и керосинов, а в последнее время и наших.
Высокая непредельность их, вне всякого сомнения, зависит здесь
•от примеси к дестиллатам прямой гонки соответствующих кре-
кинговых фракций.
ЗНАЧЕНИЕ НЕПРЕДЕЛЬНЫХ КАК СОСТАВНОЙ ЧАСТИ
НЕФТЕПРОДУКТОВ
Будучи веществами малоустойчивыми и легко изменяющи-
мися под влиянием .различных факторов, непредельные углево-
дороды для большинства нефтепродуктов являются нежелатель-
ными компонентами. Особенно это относится к нефтепродуктам,
рассчитанным на длительную работу, каковы, например, смазоч-
ные масла. Основным условием доброкачественности смазочных,
масел является устойчивость их в работе, протекающей, вообще
говоря, при доступе воздуха и влаги в самых разнообразных тем-
пературных условиях. Естественно поэтому, что при изготовле-
нии смазочных масел малоустойчивые в этих условиях непре-
дельные углеводороды, как и вообще ненасыщенные соединения,
должны быть тщательно удаляемы из соответствующих дестил-
латов. То же относится и к осветительным маслам: подвергаясь
•осмолению в результате процессов окисления и полимеризации,
непредельные соединения способствуют здесь ускоренному засо-
рению пор фитиля и быстрому падению силы света. Таким
образом, и для осветительных масел непредельные соедине-
ния являются вредной примесью и должны быть из них уда-
ляемы.
Особое место в отношении содержания непредельных зани-
мают нефтепродукты, применяемые в качестве горючего для дви-
гателей внутреннего сгорания (автомобили, тракторы и т. п.).
Как мы видели, содержание непредельных в современных образ-
цах этих нефтепродуктов (бензины, керосины) может достигать
20% и более за счет добавки к нефтепродуктам прямой гонки
.1Р4
подходящих крэкинговых фракций. Соответствующим образом
стабилизованная примесь эта уже не является здесь вредной:
она не только значительно расширяет ресурсы горючего для дви-
гателей внутреннего сгорания, но и существенно повышает его-
качества в отношении детонирующих свойств.
ЛИТЕРАТУРА
1. См. также курсы органической химии Каррера, Л., Реформатского С.Н^.
Фаворского А. К., Шорыгина Л. П. « др.
2. Engler С. u. Hofer Н, Das Erdol, т. I, Leipzig (1913).
3. Le Bel, „С. 73, 499 (1871); 75, 267 (1872); 81, 967 (1875).
4. Balbiano u. Paolini, „Chem. Ztg.“. 25, 932 (1901); Tausz, „Ztschr. angew»
Chem.- 32, 233 (1919).
5. Mabery a. Quayle, „Am. Chem. J“. 35, 404(1906).
ft. Метод >i испытаний нефтепродуктов, Библиотека OCT № 5, M. (1930);
Добрянский, Анализ нефтяных продуктов, М. — Л. (1925); Marg-
osches, Hinner, Friedmann, „Ztschr. angew. Chem.* 37, 334 (1924);
Margosches, Krakowetz, Schnabel, „Petroleum* 25, № 35, 1179
(1929); Howes, „J. Inst. Petr. Techn*. 16, № 79,54 (1930); Робинзон E. A.
и Покровская E. С., Определение группового состава бензинов,.
изД. Акад, наук СССР.
7. Собственные наблюдения.
8. Mrllhiney, „J. Am. Chem. Soc.“ 21, 1084 (1899).
9. Галъперн Г. Д, Химия твердого топлива, 8, 384 (1937); 9, 175 (’938).
10. Прилежаев „Ж- Р- X. О.* 42, 1387 (1910); 43, 609 (1911); 44, 613 (1912У,.
Наметкин и Брюсова „Ж- Р. X. О*. 57, 372 (1926).
11. Наметкин С. С. и Робинзон Е. А., „Известия Акад-наук СССР", „Серия
химическая", № 4, 921 (1937); „Химия твердого топлива*. 9, № 1„
76 (1938). _
13. Наметкин в Абакумове кая, „Нефт- хоз.* 11, № 7, 58 (1926); Наметки»
и Пуцилло, „Нефт. хоз." 16, № 2, 231 (1929).
ГЛАВА VI
НАФТЕНЫ
При ближайшем исследовании химического состава нефтей,
эфирных масел и некоторых других природных веществ давно
уже определилась обширная группа органических соединений,
занимающих по своему составу и свойствам промежуточное по^
ложение между жирным и ароматическим рядами. По своим хи-
мических свойствам соединения эти очень близки к представи-
телям соответствующих классов жирного (алифатического) ряда;
в строении же их на основе их состава, методов получения и
некоторых превращений пришлось принять одну из характер-*
ных особенностей ароматического ряда — кольчатое или цикли-
ческое расположение атомов углерода. Соединения эти получили
название алициклических. Их простейшими представителями
являются нафтены — алициклические углеводороды предельного
характера, называемые иногда, также циклопарафинами.
КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА НАФТЕНОВ
Нафтены разделяются на несколько' основных групп, а именно:
1. Моноциклические нафтены состава CJH^, в свою очередь
разделяющиеся на подгруппы по числу углеродных атомов в их
цикле. В простейших нафтенах этого ряда кольчатые системы
(циклы) образованы из 3, 4, 5, 6, 7 и более углеродных атомов1;
из них в нефти обнаружены до настоящего времени лишь 5- к
6-членные циклы. Рациональное наименование таких цикличе-
ских систем составляется из приставки «цикло» и обозначе-
ния парафина, соответствующего по числу атомов углерода со-
ставу данного цикла. Обозначение их гомологов и производных»
производится обычными способами, принятыми в органической’
химии. Так, например:
СН2 СН8-СН —СН-СН3
ZCH2\ ВуУ^СН,
НаС СН* НаС^/СНг
СН,
циклопропан циклогексан
Н2С сн2
Сн2
1-2-диметнл-циклопентан
1 В новейшее время Ружичка [1] получил нафтены, содержащие 15 и
17 углеводородных атомов в цикле, а Н. Д. Зелинский [2j давно уже
синтезировал нафтены с 20 и 40 атомами в цикле: циклоэйкозан, CwH«, и.
никлотессараконтал, СюНэд.
186
В тех случаях, когда при обозначении нафтена не требуется
указания на состав его основного ядра или когда последнее не-
известно, удобно пользоваться более краткими и общими найме'
кованиями: таковы, например, пентанафтен для С5Ню, гекса-
нафтен для CeHig и т. п.
2. Бициклические нафтены. Принадлежащие к этой группе
нафтены имеют состав СпН2л_2, но сохраняют предельный ха-
рактер. Они относятся к моноциклическим нафтенам так же,
как, например, нафталин относится к бензолу. Простейшим при-
мером бициклических нафтенов может служить декагидронафта-
лин или декалин.
3. Три- и полициклические нафтены. Несмотря на еще ботее
непредельный состав (СпН2и_а и т. д.), представители этих наибо-
лее сложных групп нафтенов продолжают сохранять предель-
ный характер. Их отношение к простейшим нафтенам таково же,
каково, например, отношение антрацена и ему подобных конден-
сированных ароматических систем к ряду бензола.
КРАТКАЯ ХАРАКТЕРИСТИКА НАФТЕНОВ
Промежуточный характер, занимаемый нафтенами между па-
рафинами, с одной стороны, и ароматическими углеводородами—
с другой, не только выявляется на способах их получения и их
химических свойствах, но может быть подмечен также на их
физических константах и на ходе изменения этих констант с уве-
личением молекулярного веса углеводородов.
Основные методы получения нафтенов» По методике получе-
ния нафтены связаны более с жирным, чем с ароматическим ря-
дом. Переход к нафтенам </г соединений с открытой группиров-
кой .атомов связан с задачей замыкания соответствующего цикла;
переход от ароматического ряда требует коренного изменения так
называемого ароматического характера молекулы. Важнейшие
способы получения нафтенов — следующие.
1. Действие металлов, натрия или цинка, на двугалоидо-
замещенные ряда метана. Это — наиболее простой и удобный
способ получения простейшего нафтена, циклопропана, по урав-
нению:
СН2Вг СН2
Z) *
СН2 4~ 2Ха = 2ХаВг+ Н2
\н„Вг СН2
2. Восстановление галоидопроизводных или других соедине-
ний, например непредельных углеводородов, кетонов и др. с го-
товой алициклической системой. Принципиально эти способы не
отличаются от соответствующих способов получения парафинов
(ср. гл. V). Они требуют, однако, предварительного замыкания
соответствующей алициклической системы путем применения
187
того или иного специального метода. Таких методов известно
очень много. Простейший из них, сыгравший особенно важную
роль в истории химии нафтенов, заключается в сухой перегонке
двухосновных кислот жирного ряда или их солей (ср. гл. Ш).
Этим методом приготовлена главная часть нафтенов, поимено-
ванных в табл. 50.
3. Богатейшим источником алициклических соединений
являются также эфирные масла, из которых многие алицикли-
ческие соединения (терпены и т. д.) более или менее легко полу-
чаются в совершенно чистом виде; путем надлежащей обработки
•они могут быть легко превращены в соответствующие нафтены.
Так например, лимонен, один из наиболее простых и доступных
терпенов, путем каталитической гидрогенизации легко превра-
щается в метил-изопропил-циклогексан (ментан):
СНЧ — СН2 СН2
z II
CHSC СН—С + 4Н =
%Н си/, сн8
сн2—сн2 сн8
= СНуСН^ \ш-сн
\на—СН, СН.
Что касается выделения нафтенов из нефтяных погонов, то
:ввиду исключительной сложности состава исходного сырья такой
метод даже в применении к нефтя^, особенно богатым нафте-
нами, оказывается слишком сложным и мало надежным
(ср. ниже).
4. Получение нафтенов из ароматических углеводородов пу-
тем каталитической гидрогенизации уже было рассмотрено выше
(см. гл. IV). Применимость этого метода ограничивается, оче-
видно, одним рядом циклогексана.
Физические свойства. Известно, что удельные веса и коэфициенты
рефракции углеводородов ряда метана ниже, чем углеводородов ароматиче-
ского ряда. Нафтены занимают в этом отношении промежуточное положе-
ние: при одном и том же числе атомов углерода в частице удельный вес и
коэфициент рефракции нафтена всегда выше, чем у соответствующего па-
рафина, но ниже, чем у ароматического углеводорода. Такое различие
•особенно резко сказывается для низших представителей соответствующих
рядов и лишь постепенно, по мере увеличения молекулярного веса, сглажи-
вается для высших гомологов, на константах которых, естественно, должно
все сильнее и сильнее сказываться увеличение числа и ве?а боковых групп.
Очень характерен в различных рядах ход изменения тех же констант
•с увеличением молекулярного веса. Известно, что радикалы предельного
характера, Сп Н2п + 1, вступая в углеводород ряда метана, повышают его
удельный вес й коэфициент рефракции, которые, таким образом, по мере
увеличения молекулярного веса углеводорода возрастают. В ароматическом
ряду наблюдается обратное явление: вступление тех же самых радикалов
понижает удельный вес и коэфициент рефракции углеводорода, так что
•последние по мере увеличения молекулярного веса постепенно понижаются.
188
Тя-ким образом, если для изменения физических свойств с увеличением
молекулярного веса построить кривые, то в различных рядах углеводородов
кривые эти будут иметь существенно различный вид. В жирном ряду
в связи с увеличением удельного веса и коэфициента рефракции по мере
увеличения молекулярного веса кривая будет восходящая; наоборот, в аро-
матическом ряду та же кривая будет иметь нисходящий вид.
Изменения тех же физических свойств у нафтенов протекают иногда
в том же направлении, как у гомологов метана; однако в некоторых слу-
чаях оно принимает крайне своеобразный характер. Как видно из табл. 50,
при переходе от циклогексана к его гомологам удельный вес и коэфициент
рефракции сначала понижается (как у бензолов), а затем начинает повы-
шаться (как у парафинов). Столь же характерно изменение удельного веса
у гомологов циклопентана. Таким образом кривая изменения этих констант
будет иметь в данных случаях следующий вид: сначала в связи с паде-
нием удельного веса или коэфициента рефракции кривая будет пони-
жаться, а затем в связи с возрастанием тех же величин станет повышаться,
образуя, таким образом, некоторый изгиб. Подобный вид кривой наблю-
дается для удельного веса гомологов циклопентана, циклогексана и
циклогептана и для коэфициента рефракции гомологов циклогексана и цикло-
гептана. Следует отметить, однако, что даже в этих наиболее типичных
случаях нисходящая часть кривой сравнительно невелика, так что характер
изменения физических свойств у большинства нафтенов даже в этих рядах
таков же, как у парафинов.
Химические свойства. По своим химическим свойствам наф-
тены ближе всего подходят к парафинам. В некоторых отноше-
ниях они проявляют, однако, ряд своеобразных особенностей,
связанных с их циклическим строением, как видно из следую-
щего краткого обзора.
1. Реакции присоединения. Как общее правило, нафтены по-
добно парафинам не способны к реакциям присоединения.
Исключение, объясняемое стереохимическими соотношениями»
представляют лишь самые низшие и наименее прочные нафте-
новые ряды циклопропана и циклобутана, представители кото-
рых, присоединяя в подходящих условиях водород, галоидо-
водородные кислоты и галоиды, переходят в соединения с откры-
той цепью. Так например, для циклопропана СзН6 эти реакции
присоединения можно выразить следующим образом:
С3Н6 + Н2 = СНЧ • СН2 • СН8;
С8Н6 + Н1 = СН8 • СН9 • СЕЛ;
С3Н6+ 2Вг=ВгСН2 • СН2 • СЩВг.
Впрочем, исследования последних лет показали, что цикло-
пентан, циклогексан и их гомологи в соответствующих условиях
также способны присоединять водород, превращаясь в угле-
водороды с открытой цепью, т. е. в гомологи метана. Эта важная
реакция будет рассмотрена подробнее в гл. IV, ч. II в связи
с вопросом о гидрогенизации нафтенов.
2. Реакции замещения. Реакции этого типа наиболее ха-
рактерны для нафтенов. Важнейшими из них являются хлори-
рование и нитрование по М. Коновалову, т. е. те же реакции,
что для парафинов. Они были подробно рассмотрены выше,
в гл. III.
3. Окисление. По своей устойчивости к окислителям наф-
тены также вполне аналогичны парафинам: в обоих случаях
189
окисление наступает лишь при повышенной температуре и со-
провождается разрывом углеродной связи. Впрочем, конечны]!
результат реакции в обоих случаях существенно различен: в то
время как для парафинов реакция окисления приводит обычно
к продуктам глубокого распада исходного углеводорода, в ко-
нечном итоге нередко — к углекислоте и воде, нафтены, по край-
ней мере не замещенные, образуют в аналогичных условиях
двухосновные кислоты с тем 'же числом атомов углерода в ча-
стице. Так например, при окислении циклогексана получается
адипиновая кислота, циклопентан дает глутаровую кислоту и тд.:
СН., —сн2
СН./
\н2—С Но
СН, —СОоН
+ 50 = Н2(‘) Н-СН,
'с Но — С02Н
Для замещения нафтенов реакция окисления приводит обы-
кновенно к продуктам более глубокого распада. Лучшим окисли-
телем нафтенов является азотная кислота. Своеобразное дей-
ствие дымящей азотной и серной кислот на нафтены ряда цикло-
гексана рассмотрено выше, в гл. III.
4. Термический распад. Смотря по температуре, нагревание
вызывает либо частичный, либо глубокий распад нафтенов, при-
чем немаловажное влияние оказывает при этом наличие тех или
иных катализаторов. Так например, -простейший нафтен, цикло-
пропан, лишь при температурах выше 500° частью разрушается
частью изомеризуется в .пропилен; однако в присутствии влаги
п некоторых твердых катализаторов (Fe, Pt, А120з) разрыв этого
цикла может наступить уже при 100э. Циклогексан и его гомо-
логи при 300—350° в присутствии катализаторов (Pt, Ni) нацело
превращаются в ароматические углеводороды (дегидрогенизация,
см. гл. III). Более глубокий распад нафтенов наступает при тем-
пературах 500° и выше, когда наступает их крэкинг и пиролиз
(см. ч. II).
Перечень важнейших представителей некоторых, нафтеновых
рядов, представляющих особый интерес с точки зрения химии
нефти, приведен в табл. 50.
НАФТЕНЫ БАКИНСКОЙ НЕФТИ
Приступая к описанию естественных нафтенов, поставим пре-
жде всего вопрос, не находится ли в нефти простейший алици-
клический углеводород предельного характера — циклопропан?
Для выяснения этого вопроса Морковников исследовал [3] газо-
образные углеводороды нефти, содержавшиеся в специально при-
готовленном погоне бакинского бензина с температурой кипения
до 50°. Фракционировка этого погона велась в холодном поме-
щении; длинный холодильник был соединен с приемником,
охлаждавшимся холодной водой. Газообразные продукты, не сгу-
щавшиеся в приемнике, поступали из него последовательно
190
Таблица 50
Свойства нафтенов. Ряды циклопентана и циклогексана
Состав Наименование нафтена Темп. кип. Уд. вес Коэф, рефракц. nD
С5Н10 Циклопентан 50°, 5 d™* = 0,7506 = 1,4039
С6Н12 Метил-циклопентан . . • 72° =0,7474 w2i =1.4088
Циклогексан . 80°,9 t?i9’5 = 0,7788 W19,5 ~ 1.4266
Диметил-цпклопентан-1, 1 88° d“° =0,7547 n20 =1,4131
. - 1, 2 93° d*> =0,7534 n20 = 1,4126
„ ,, 1. 3 91° d1* =0,7497 4 9 h18 =1,4110
Этил-циклопентан .... 104° dj8 =0,7658 wis = 1.4188
Метил-циклогексан .... 100°,5 df =0,7694 «20 = 1.4245
CaHlR Циклогептан Метил-этил-циклопентан- 118° df =0,8108 «20 J— l,41o2
1,2 Метил-этил-циклопентан-: 124° d™ =0,7736 * «20 =1,4245
1, 3 Триметил-циклопентан- 121°,Б d*° =0,7657 «20 = 1.4188
1. 2, 3 Триметил-циклопентан- 107° d£ =0,7504 ——
1, 2, 4 ’ • 113° df =0,7565 «20 =1,4156
Диметил-циклогексан-1, 1. 118° df =0,7825 n20 = 1,42? 9
1. 2 • 125° dz° =0,7782 ?i20 =1,4289
. . 1.3. 121°,б d“° =0,7708 4 н20 =1,4248
C9H18 „ „ 1.4. Диметил-этпл-циклопен- 120°,5 df =0,7716 =1,4247
тан-1, 4, 2 Диметил-этил-цпклопен- 137° df =0,7700 n.1Q =1,4214
тан-1, 3, 2 • Метил-изопропил-цикло- 136° d;° =0,7703 —
пентан-1, 2 Триметил-циклогексан- 142°,Б df =0,7792 «20 =1,4279
1, 1, 3 Триметил-циклогексан- 139° dj5 =0,7703 n,- =1,4237 A J
1, 2, 3 Триметил-циклогексан- 1, 2, 4 Триметил-циклогексан- 141° 143° d“° =0,7898 dl8 =0,7807 4 =1,4346
1,3,5 139° dP =0,7764 4 =1,4283
191
Продолжение
Состав Наименование нафтена Темп. кип. Уд. вес Коэф, рефракц. nD
СюНао Тетраметил-циклогексан- 1, 1, 3, 5 148° d|4 = 0,7775 ^>4 = 4300
Диметил-этил- , 1, 3, 5 . 166° ^‘ = 0,7925 п22 — 1,4354
Диэтил-циклогексан-1, 3 . 170° df = 0,7957 п20 = 1,4388
Метил-ивопропнлцикло- гексан-1, 4 171° d‘20 = 0.7963 w2o = 14375
CioHig Декалин транс 185° df = 0,8695 n20 ~ 1|4695
Декалин цис 193° df = 0,8950 4 л н20 = 1,4805
в три промывные склянки, наполненные до половины олеонаф-
том, и в склянку с бромом. Непоглотивтниеся газы собирались
в газометр. Из нескольких сот литров получилось лишь весьма
незначительное количество бромюра: олеонафт также погло-
тил очень немного газа. Около 10 л газа пз газометра были оста-
влены на 4 суток с дымящей иодистоводородной кислотой при
комнатной температуре. Никакого поглощения не произошло,
и кислота по разбавлении водой не показала присутствия
иодюра. Следовательно, газ, заключавшийся в исследованном по-
гоне бакинской нефти, циклопропана не содержал; в противном
случае от взаимодействия циклопропана с иодистым водородом
должен был получиться иодистый пропил.
Другая (неопубликованная) попытка выделения из нефти низших наф-
тенов принадлежит Зелинскому. Действием хлористого ацетила в присут-
ствии хлористого алюминия на наиболее низкие погоны бензина был сде-
лан опыт кетонизации находящихся в них низших нафтенов. Однако по-
лученные этим путем продукты соответствовали преимущественно более
выеококипящим погонам, низших же нафтенов, т. е. простейших представи-
телей рядов циклопропана и циклобутана, в нефти обнаружено не было.
Последнее относится, впрочем, и к высшим гом< логам этих двух алицикли-
ческих рядов. Все более или менее изученные нафтены, выделенные до
настоящего времени из нефти, принадлежат к рядам циклопентана, цикло-
гексана и, быть мюжет, циклогептана; кроме того, в нефти несомненно при-
сутствуют углеводороды более сложных, вероятно конденсированных, али-
циклических систем. Что касается низших алициклических рядов — цикло-
пропана и циклобутана, то ни один представитель их до настоящего
времени в нефти не обнаружен.
После этих предварительных замечаний можно перейти к об-
зору нафтенов, выделенных из бакинской нефти.
Пентанафтен, С5Н10. Еще Менделеев указал на присутствие
в кавказской нефти какого-то углеводорода, кипящего около 5 5 °*
с бблывим удельным весом, чем гексан. Этому углеводороду
192
соответствует первый изгиб кривой изменения удельного веса
с температурой кипения нефтяных погонов, указанный впервые
этим автором. Существование такого изгиба видно также из
приведенных в гл. III кривой и таблицы с результатами пере-
гонки бакинского бензина: до 50—54° удельные веса потопов
постепенно возрастают; затем они начинают падать, так что
фракция 54—56° имеет’ такой же удельный вес, как фракция
42—44°. Количество фракции 50—52° в несколько раз превы-
шает количество смежных фракций, так что все говорит згх нахо-
ждение здесь какого-то индивидуального углеводорода. По тем-
пературе кипения фракция 50—52° ближе всего подходит к ци-
клопентану, С5Ыю, получаемому обычным путем, исходя из ци-
клопентанона. Правда, анализы фракции 50—52° (Морковников,
Аскан) дают числа, из которых еще нельзя заключить о нахо-
ждении пентанафтена, СэНю, в нефти: удельный вес этой фрак-
ции значительно ниже удельного веса циклопептана. Такое рас-
хождение может быть объяснено, однако, значительным содер-
жанием в этом нефтяном погоне какого-либо парафина.
Действительно, как мы видели выше, Морковников доказал
присутствие во фракции «50—52° триметил-этил-метана, (СНз)з-
• С СН2 • СНз, вторичное нитросоединение которого — кристал-
лическое. Из главного продукта нитрования фракции 50—51°
тем же автором было выделено еще одно нитросоединение также
вторичного характера, но жидкое [4]. Нитрбсоединение это
Т?ыло восстановлено в амия, охарактеризованный бензоильным
производным; температура плавления последнего оказалась
156—158°. Совершенно такое же бензоильное производное и по
внешнему виду кристаллов, и по температуре плавления дал
циклопентил-амин, полученный из оксима циклопентанона. Ока-
залось также, что в кислотном слое после нитрования фракции
50—51° содержится глутаровая кислота— характерный продукт
окисления незамещенного пятичленпого цикла.
Итак, присутствие циклопентана в бакинской нефти должно
считаться доказанным. Ито касается, однако, выделения этого
нафтена из нефти в сколько-нибудь чистом виде, то ввиду бли-
зости температур кипения циклопептана и триметил-этил-метана
такая задача должна быть сопряжена с чрезвычайными трудно-
стями, тем более что отношение обоих углеводородов к различ-
ным реагентам, в том числе и к азотной кислоте, крайне схоже.
Правда, вторичное нитросоединение триметил-этил-метана—
кристаллическое; однако полное его отделение от жидкого нитро-
соединения циклопентана едва ли осуществимо.
Гексанфтены, CeHiz. Из нафтенов состава CeHi2 в бакин-
ской нефти обнаружены два: метил-циклопентан и циклогексан.
а) Метил-цикл'опснтаи, С5Н9СН3. .В чистом виде этот бли-
жайший гомолог циклопептана был приготовлен впервые’ из со-
ответствующего кетона, ₽-метил-циклопептанона, полученного
сухой перегонкой кальциевой соли £ -метил-адипиновой кис-
лоты: кетон был восстановлен в алкоголь, последний же превра-
щен в углеводород нагреванием с иодистоводородпой кислотой
1! 3
13 Зак. 1П22. Химия нефти.
В запаянных трубках или восстайовлепией соответствующего
иодюра цинко-медной парой:
СН.,—СН-СН. Н2С— СН-СН3 Н2С —СН-СН3
1'1 II II
СН2 СН2 —*Н2С СН, —>н2с сн3
СОЛ со л со сн8
Фракция бакинской нефти, отвечающая метил-циклопеитану,
была выделена впервые Марковниковым и Шпади [5]. Осно-
вываясь на данных Вредеиа о гидрогенизации бензола и его про-
изводных (фенола) иодистоводородной кислотой, а также на
составе и плотности пара нефтяной фракции 69—70°, авторы
пришли к выводу, что в состав ее входит нормальный гексагидро-
бензол, т. е. циклогексан. Ошибочность такого заключения ста-
ла, однако, вполне очевидной, когда А. ф.- Байер показал (1894 г.),
что температура кипения истинного циклогексана лежит почти
на 10° выше. Действительно, нитрованием этой фракции Мар-
ковников и М. Коновалов получили вскоре третичный нитро-
продукт состава CeHnNOs, а из последнего амин; отсюда авто-
рами был сделан вывод о присутствии в нефти метил-циклолен-
тана [6], что и подтвердилось затем исследованием двухосновных
кислот, получающихся при окислении данной фракции азотной
кислотой [7].
Метил-циклопентан не может быть выделен из нефти в чи-
стом виде; близость его температуры кипения с температурой
кипения нормального гексана полагает разделению этих угле-
водородов непреодолимые препятствия. Несмотря на это, фрак-
ции 70—72° и 72—74° бакинской нефти с удобством могут слу-
жить для получения различных производных метил-циклопен-
тана, тем более, что количество их довольно значительно. Наи-
более удобной реакцией для этой цели является нитрование по
Коновалову.
Метил-циклопентан благодаря нахождению в нем третичного
водорода нитруется азотной кислотой удельного веса 1,075 уже
при 100°, тогда как нормальный гексан — лишь при 130° и
выше. Отсюда ясно, что если нитрование фракции 70—72° вести
при умеренной температуре, то вступать в реакцию будет преи-
мущественно метил-циклопентан; примесь нормального нитро-
гексана будет сравнительно невелика. Наилучшие результаты
по Марковникову получаются, если нитрование фракции 70—72°
вести при температуре не выше 115° и не повторять реакции
с одним и тем же углеводородом свыше двух раз. За отгонкой
левошедшего в реакцию углеводорода следует обработка смеси
нитросоединений едкой щелочью. Ввиду того что нормальный
гексан, представляющий собою главную примесь к нефтяному
метил-циклопептану, не содержит третичного водорода, а потому
не может образовать третичного нитро, после обработки щелочью
третичный нитро-метил-циклопентан сразу получается почти
194
в чистом виде и может быть использован для получения тре-
тичных производных метил-циклопентана: амина, алкоголя, про-
дукта дегидратации последнего— 1,1-метил-циклопентана и т. д.
Вторичный нитро-метил-циклопентан после выделения его из ще-
лочного раствора получается в гораздо менее чистом виде: к нему
всегда примешало некоторое количество вторичного нитрогек-
сана, хотя примесь эта при соблюдении вышеуказанных условий
нитрования сравнительно невелика.
Значительно легче, чем с азотной кислотой, реагирует мстил-циклопсн-
тан с хлором. Реакцию отриходится вести при сильном затемнении: тем
не менее, разогревание реакционной смеси достигает 70°. Однако разделение
полу чающихся при этом монохлоридов весьма затруднительно, так как
к изомерным хлоридам метил-циклопентана здесь примешано значительное
количество хлоридов жирного ряда, главным образом состава CeHuCL
б) Циклогексан, СбНг». Циклогексан (II) получен впервые
Л. ф.-Байерам [8 из циклогександиона, СбН80г (I) и цикло-
гексанона, СбНюО (III), обычным путем, т. е. через соответствую-
щие спирт СеНцОН и иодюр:
СО СНо СЩ
ilc^^cHo н,с/ХсНо нас/Чсн3
НГ^ -'ll! — | III |
Н2С СН2 НоС сн2 Н2С сщ
^СО ’ ^СНо ^со
Ввиду важности установления констант этого углеводорода
Зелинский повторил эту работу [9] и получил циклогексан в со-
вершенно чистом виде. Физические свойства его и важнейших
его производных приведены в табл. г>1; они вполне совпадают
с константами, найденными позднее Сабатье и Сандераном для
препаратов, полученных каталитическим методом, исходя из бен-
зола и фенола [10].
Первая попытка выделения циклогексана из нефти (Марков-
ников и Шпади, 1888 г), основанная на неправильных пред-
ставлениях о природе продукта гидрогенизации бензола иодисто-
водородной кислотой, как мы видели, была неудачна. Десять лет
спустя, когда свойства циклогексана быДи установлены, Марков-
ников посвятил этому вопросу новую большую работу 11.. На
этот раз, исходя из нефтяной фракции 80—82, автору удалось
не только доказать нахождение в бакинской нефти цикло-
гексана, но и получить как самый углеводород, так и целый ряд
его производных в достаточно чистом виде. По размерам фрак-
ция 80—82° бакинского бензина занимает первое место среди
других -погонов того же продукта. Этой же фракции, как пока-
зывает график, приведенный в гл. III, принадлежит высшая
точка второго изгиба кривой изменения удельного- веса нефтя-
ных погонов с температурой их кипения. Несмотря на это, обыч-
ная методика, т. е. обработка погона серно-азотной смесью и
13* 195
тщательная фракциоиировка, оказывается и здесь недостато той
для выделения углеводорода в чистом виде. Дальнейшая
очистка достигается обработкой фракции 80—82° дымящей азот-
ной кислотой, разрушающей главным образом примеси углсводо-
{юдов, содержащих третичный водород. Допуская при этом не-
которое разогревание, в лучшем 'Случае удавалось получить угле-
водород, отличающийся по своему удельному весу от чистою
циклогексана примерно на 0,01, т. е. все еще содержащий замет-
ное количество примесей. Тем не менее, как показал Марков-
ников, этим углеводородом можно с у спехом пользоваться для
получения различных производных циклогексана.
Основными реакциями для получения производных цикло-
гексана послужили хлорирование и нитрование по Коновалову.
Из полученных таким образом тщательно очищенных хлор-
циклогексана или нитро-циклогексана были приготовлены затем:
иодюр, циклогексен, циклогексил-амин, циклогексанол, цикло-
гексанон и т. д. Было констатировано таые, что в кислотном
слое после нитрования циклогексана содержится нормальный
продукт окисления незамещенного шестичленного ядра—адипи-
новая кислота. Наконец, исходя из иодюра, полученного нагрева-
нием хлор-циклогексана или циклогексанола (из амина) с иодисто-
водородной кислотой, быт получен обратно циклогексан и, та-
ким образом, был замкнут круг превращений этого важнейшего
природного нафтена по следующей схеме:
i— II о ф т я н о й
4-
хлорид, С6НИС1
1
иодюр, СвНп1
циклогексан, С6Н19
циклогексан
1
нитрососдинение, C6HUNO2
цикло гексил амин, CbHnNH2
цпклогексанол, С6Нн0Н
иодюр, СбНп1
!
циклогексан, C6Hj2
Интересно сопоставить физические свойства некоторых из этих препа-
ратов с синтетическими (тафл. 51).
Таблица^ 51
Формулы Синтетич. препараты Нефтяные препараты
Темп, кппения Уд. вес Темп, кипения Уд. вес
CcHhNOs 1095 ( 40 **) df =1,0680 109° ( 40 JWjk) = 1,6605
с6н]0о 151°,8 (748 мм) d± =0,9467 154°,5 (751 жл) d“° = 0,9473
Сб Н12 80°,1 (744 мм) d]9,5 = 0,7788 80°,6 (761 jw.h) = 0,7717
196
Эта таблица как нельзя лучше может иллюстрировать те трудности,
с которыми сопряжено получение из нефти чистого нафтена и его произ-
водных. Фракция 80—82°, несомненно, является одним из наиболее благо-
дарных объектов для этой цели, хотя бы уже потому, что все примеси,
содержащие третичный водород, при нитровании слабой азотной дадут
преимущественно третичное нитросоединение, легко отделимое с помощью
щелочи от главного продукта реакции, нитроциклогексана. Тем не менее,
как видно из табл. 51, очищенный нитропродукт из нефтяного углеводо-
рода заметно отличается (на 0,0075) по своему удельному весу от чистого
препарата. То же самое, хотя и в меньшей степени, наблюдается для угле-
водородов, СеН12: содержание какой-то примеси в нефтяном циклогексане,
даже переведенном через ряд производных, оказывается не только на его
более •низком' удельном весе, но и на значительной разнице (около 6°)
в температурах плавления; чистый циклогексан плавится 6,4°, тогда как
нефтяной препарат — около 0°. Такой примесью к хорошо очищенному неф-
тяному циклогексану является, как мы видели выше, триметил-пропил-ме-
тан, (СНз)зС - СНг • СН« -СНз. При нитровании этого парафина слабой
азотной кислотой может получиться лишь вторичное нитро, уже неотдели-
мое от вторичного нитронафтена. Таким образом примесь производных три-
метил-пропил-метапа должна сохраниться и-при последующих превращениях
нефтяного нитроциклогексана в амин, алкоголь, иодюр и, наконец, в угле-
водород. Исключение составляют лишь кетон СвНюО- Как видно из
табл. 51, здесь наблюдается полное совпадение физических свойств для
обоих препаратов, нефтяного и синтетического. Совпадение это, впрочем,
дВпЖце понятно: для очистки нефтяной кетон был переведен через бисуль-
фиттюе соединение; а так как кетон, соответствующий вторичному нитро
из тргиметил-ппбпил-метана, подобно простому пинаколину с бисульфитом
не реагирует, тр этим путем: и было достигнуто получение нефтяного цикло-
гексанона в совершенно ’щьртом виде.
Гептанафтепы, С7Ни, . Все шесть теоретически возможных
гептанафтенов рядов циклопентана, циклогексана и циклогеп-
тана известны. Физические свойства их сведены в табл. 50.
Большая часть этих углеводородов получена из алициклических
кетонов, приготовленных сухой перегонкой кальциевых солей со-
ответствующих двухосновных кислот. Наиболее доступным из
них является мстил-циклогексан, легко получаемый ныне гидро-
генизацией толуола по Сабатье:
СН = СН СН2—СН2
нс/ Sc • СН3+6Н=щс/ >сн • сна.
• СН = СН СН, — СН,
Чтобы разобраться в вопросе о нахождении гептанафтенов
в нефти, слова обращаемся к таблице, дополняющей график
фракционировки бакинского бензина, и продолжаем ее за 100°
л(табл. 52).
Как показывает кривая изменения удельного веса нефтяных
погонов с температурой их кипения (фиг. 11), в интересующих
нас пределах наблюдается минимум при 90—92°, за которым
следует подъем кривой с наивысшей точкой при 100—102°. Этим
двум перегибам кривой соответствует резкое повышение количе-
ства соответствующих фракций, что обусловливается, очевидно,
нахождением в данных пределах определенных химических инди-
видуумов. Как видно из табл. 50, таковыми могут быть для фрак-
ций 90—92° один из диметил-циклопентанов, СбНвЩСзХ для
197
Таблица 52
Темп. Количество Уд. вес Темп. Количество Уд. вес
кипения в г АЭ кипения В 1* л15 “15
100—102° 1041 0,758 120—132° 1046 0,756
102—104° 934 0,758 122—124° 410 0,762
104—106° 556 0,753 124—126° 367 0,765
106—108° 203 0,746 126-128° 304 0,766
108—110° 265 0,741 128—130° 182 0,765
110—112° 260 0,738 130—132° 251 0,765
112-114° 209 0,738 132—134° 137 0,765
114-116° 372 0,740 134—136° 272 0,761
116-118° - 4°0 0,743 136-138° 213 0,760
118—120° 1164 0,748
фракций же 100—102°—мзтил-циклогексан, СвИиСНз, или
этил-циклоггентан, С5Н9С2Н5. Рассмотрим порознь работы, отно-
сящиеся к исследованию этих двух нефтяных фракций.
Фракция 90—92° по своим размерам занимает третье ме-
сто среди различных погонов бакинского бензина. Несмотря на
это, исследование ее находится все еще в начальной стадии.
После того как Всйльштсйн и Курбатов [12] указали на присутствие
в кавказской нефти гексагидротолуола, СтНн (гептанафтена), впервые Миль-
ковский (в лаборатерип Морковникова) подверг погон 85—105° биби-эйбат-
ской и балаханской нефтей тщательной фракционировке [13]. Перегонка
велась сначала в пределах 5°, затем 2° и, наконец, 1°, причем каждая
порция, кипевшая в пределах одного градуса была перегнана в общем не
менее 30 раз. Количество отдельных фракций по весу оказалось различно:
для большинства из них оно не превышало 15—150 г и лишь для четырех
восходило до 500 г. Физические свойства и состав этих четырех фракций
приведены в табл. 53.
Таблица 53
Температура кипения 90-91° 93-94° 96—97’ 100—101°
Уд. вес dj7,° Состав: °/0 С %н 0,7320 84,97 14,86 0,7390 85,0 15,0 0,7515 85,52 14,90 0,7624 85,71 14,57
Теория для: С7Ни: % С —85,72; % Н —14,28.
Плотность пара всех этих фракций оказалась довольно близка к тео- •
ретической для СтНк, по составу же наиболее подходила к этому углево-
дороду фракция 100—101°. На нее то и было обращено затем главное вни-
мание (см. ниже); что касается фракции 90—91°, то изучение ее ограни-
чилось лишь получением из нее хлорида.
Позднейшие исследования восполняют этот пробел лишь в весьма ма-
лой степени. Как видно из табл. 53, элементарный состав фракции 90—91°
показывает, что, дсроме гептанафтена, СтНк, в нем должны содержаться
также парафины. Действительно, выше уже было указано, что Харичков
при исследовании соответствующей фракции (температура кипения 90—93°)
198
грозненской нефти, получил углеводород состава C7Hte (изогептан) с тем-
пературой кипения 90,5—91,5°. Этот же изогептан содержится, вероятно,
в бакинской нефти, но вместе с ним, судя по составу соответствующей
фракции, находится, несомненно, и гептанафтен. Чтобы изолировать этот
циклический углеводород, Зелинский (с Машковым) подверг фракцию
90—92° бакинского бензина обработке хлористым пропионилом в присут-
ствии хлористого алюминия [14]. Продуктом реакции действительно ока-
зался кетон состава СтНи. СО . СН*. CHs, отвечающий по мнению автора
одному из диметил-циклопентанов. Однако более точных данных о строении
этого гептанафтена до сих пор не имеется.
Метил-циклогексан, СвНпСНз. Начальные исследования
фракции 100—101 бакинской нефти ограничивались ее выде-
лением, характеристикой ее отношения к некоторым реагентам
(галоиды, азотная кислота) и приготовлением из нее хлорида.
Однако первые результаты по вопросу о ее химической природе
были получены иным путем, а именно при ее обработке бромом
в присутствии бромистого алюминия [15]. Оказалось что про-
дуктом ©той реакции является пентабромтолуол с выходом до
10% теоретического. Таким образом, впервые наметилось реше-
ние вопроса о природе главной составной части фракции 100—
102° бакинского бензина: очевидно, это нормальный гекса-гидро-
толу ол или метил-циклогексан.'
Вопрос получил окончательное разрешение, когда, исходя из
л-метил-циклогексанона, приготовленного сухой перегонкой каль-
циевой соли а-метил-лимелиновой кислоты, был синтезирован
метил-ци клогексан [16]:
СНа ’' ' СН2 СНз
Kcf'cH, НаС СН2
II 1 J I
Н2С СН. сн3—>Н2С сн.сн3 Н2С сн.сн3
но,с С02Н 'со СН,
Оказалось, что подобно другим гомологам циклогексана син-
тетический метил-циклогексан при обработке бромом в присут-
ствии бромистого алюминия дает ароматический пербромид. Та-
ковым в данном случае оказался пентабром-толуол, т. е. тот же
пербромид, который получается при аналогичной обработке неф-
тяного гептанафтена с температурой кипения 100—101'. Так как,
с другой стороны, физические свойства обоих углеводородов,
нефтяного и синтетического, оказались весьма близкими, то
вопрос о нахождении метил-циклогексана в бакинской нефти
можно было считать окончательно разрешенным в положитель-
ном смысле.
Из дальнейших исследований фракции 100—-102° бакинской
нефти [17], вполне подтвердивших нахождение в ней метил-
циклогексана, особый интерес представляет ее дегидрогениза-
ционный катализ.
Как уже было упомянуто (гл. III), Зелинский показал, что,
подвергая нефтяные погоны дегпдрогенизационному катализу,
199
Можно все нафтены ряда циклогексана перевести в ароматиче*
ские углеводороды, причем парафины, а также нафтены рядов
циклопентана и циклогептана остаются без изменения. Если
обработать затем продукт реакции дымящей серной или серно-
азотной смесью, то можно освободиться от ароматических угле-
водородов и, таким образом, значительно упростить состав неф-
тяного погона. Применив такую методику к фракциям 100—
100°,5 и 102—104° бакинской” нефти, Зелинский получил сле-
дующие результаты.
После катализа углеводород в обоих случаях содержал зна-
чительное количество толуола, получившегося, очевидно, за счет
дегидрогенизации метил-циклогексана. Обработанный серио-азот-
ной смесью углеводород показал прежний состав (C7Hi4); физи-
ческие свойства его до и после катализа сопоставлены в табл. 54.
Таблица 54
Темп, кипения Уд. вес Коэф. рефр.
До катализа После катализа 100—100°,5 100—101° d =0 766(18°) 4 = 0,7490 п 0 = 1,4210 н18 = 1,4142
До катализа Пос те катализа 102- 104° 101—102 ’,5 d = 0,7617 (18°) 4° = 0,7488 чк п13 = 1,4215 wi0 == 14101
Из табл. 54 видно, что исследованные фракции, кроме метил-
циклогексана, содержат еще какой-то гептанафтен, устойчивый
ио отношению к дегидрогенизационному катализу. Судя по фи-
зическим свойствам, невидимому, это — этил-циклопентан,
С5Н9 • С2Н5, константы которого приведены в табл. 50. Надо ду-
мать, однако, что к этому нафтену примешано некоторое коли-
чество какого-то парафина, чем и объясняются несколько более
низкие удельный вес и коэфициент рефракции нефтяного угле-
водорода. Правда, в анализах Зелинского мы находим лишь
слабое указание на возможность такой примеси к нефтяному
углеводороду; не следует забывать, однако, что примесь к гепта-
нафтену Ю—15% гептана или октана, как показывает расчет,
почти уже не может быть обнаружена элементарным анализом.
Фракция 114—118°. Нам остается рассмотреть вопрос о нахожде-
нии в бакинской нефти еще одного гептанафтена, приведенного в табл. 50,
а именно циклогептана. Для его освещения Марковников [18] исследовал
тщательно выделенную фракцию бензина с температурой кипения 114—118°.
Углеводород в количестве 795 ел3 был обработан сначала небольшим ко-
личеством азотной кислоты удельного веса 1,53 с разогреванием до 60°,
а под конец взбалтывался с 2—3 объемами кислоты при температуре не
выше 25—27°. После обычной очистки осталось всего 10 см3 углеводорода
с температурой кипения главным образом 11G—117°. Здесь-то и должен
был бы находиться циклогептап в виду его крайней устойчивости к дымя-
щей азотной кислоте. . . .
200
Действительно, анализ углеводорода после обработки его азотной кисло-
той показал несомненное присутствие в нем какого-то нафтена, но со зна-
чительной (до 50%) примесью парафинов. Так как все третичные углево-
дороды были уже разрушены азотной кислотой, то, судя по температуре
кипения остатка и ио его высокому удельному весу, из нафтенов в нем
мог содержаться лишь циклогептан, СЬНн. Таким образом, присутствие в
кавказской нефти циклогептана представляется вполне возможным.
Октонафтены, СвНж Известно не менее 12 различных синте-
тических октонафтенов, CeHie, принадлежащих к рядам цикло-
пентаиа, циклогексана и циклогептана. Так как соответствующие
нефтяные углеводороды изучены сравнительно мало и, неви-
димому, все группируются около 120°, то можно ограничиться
приведением лишь тех,синтетических нафтенов, температуры ки-
пения которых лежат также около 120°. Константы этих нафте-
нов сопоставлены в табл. 50. Наиболее доступными из них
являются три диметил-циклогексана (1,2-, 1,3- и 1,4-), легко
получаемые гидрогенизацией соответствующих ксилолов по Са-
батье [10].
Из бакинской нефти Марковниковым выделено два углеводо-
рода состава CeHie: октоиафтен и изооктонафтен. Рассмотрим
каждый из них в отдельности.
Октонафтен, CeHie. Как видно, из табл. 52, отвечающие окто-
иафгену фракции 118—120° и 120—122° занимают по своим
размерам одно из первых мест среди различных погонов бакин-
ского бензина. Впервые октонафген был выделен из бакинской
нефти Марковниковым и Оглоблиным [19]; позднее его неодно-
кратно выделяли и подвергали всестороннему изучению Яковхин
и другие исследователи [20]. Физические свойства двух препа-
ратов октонафтена сведены в табл. 53; судя по их анализам, оба
они содержат значительную примесь предельного углеводорода.
Таблица 55
Октонафтены Темп. кип. Уд. вес Анализ]
Марковнпкова и Оглоблина . . Гл. обр. 119° дй = 0,7714; <?п = 0,7582 С—85,39; Н—14,61
Яковкина .... 119° = 0,7649; dj8 = °-7503 С-85,3; Н—14,7
Теория для СпНэ» С—85,72; Н—-14,28
Основные данные о строении нефтяного октонафтена сводятся
к следующему:
1. При обработке октонафтена дымящей азотной кислотой по-
лучается немного тринитро-л0га-гссилол€!. Легче образуется это
нитросоединение, если октонафтен нагреть предварительно с се-
рой (ср. гл. III).
2. С бромом в присутствии бромистого алюминия октонафтен
дает некоторое количество тетрабром-пара-ксилола.
201
3. При обработке октонафтена избытком дымящей серной
кислоты до полного растворения углеводорода получается суль-
фокислота метаксилола.
4. Полученный Зелинским [21]. первый синтетический окто-
нафтен, 1,3-диметил-циклогексан, показал ту же температуру ки-
пения, что и нефтяной углеводород, но несколько больший удель-
ный вес, последнее обстоятельство зависит, надо думать, от со-
держания в нефтяном углеводороде какого-либо парафина, быть
может, нормального октана (температура кипения 125,8 ). В от-
ношении химических свойств синтетический октоиафтен обна-
ружил способность давать с дымящей азотной кислотой подобно
нафтену из нефти тринитрометаксилол, но с несравнимо луч-
шими выходами; с бромом же в присутствии бромистого алюми-
ния здесь получился тетрабром-лега-ксилол в количественных
выходах.
Хотя совокупность всех приведенных данных не оставляет
сомнения в том, что в состав фракции 118—120 бакинской
нефти входит 1,3-диметил-циклогексан (с примесью какого-то
октана), тем не менее-легко видеть, что в этом вопросе сущест-
вует некоторое противоречие, до сих пор не вполне разъясненное.
В самом деле, ближайшее исследование чистых синтетических
1,2, 1,3 и 1,4-диметил-циклогексанов показало, что каждый из
этих октонафтенов при действии брома в присутствии броми-
стого алюминия дает нормальный отвечающий ему ароматиче-
ский пербромид и притом с количественными выходами. Лишь
1,1-диметил-циклотексан представляет исключение, вполне по-
нятное впрочем, так как для этого октонафтена нет и не может
быть соответствующего гомолога бензола; ввиду этого при дей-
ствии брома и бромистого алюминия на 1,1-диметил-циклогексан
•происходит перегруппировка боковых групп: получается тетра-
бром-параксилол 22].
'Сопоставим эти данные с тем, что сказано выше относительно
нефтяного октонафтена. Как было указано, при действии на него
азотной и серной кислот получаются производные лета-ксилола;
с бромом же в присутствии бромистого алюминия образуется про-
изводное napa-ксияола. Как же объяснить такое противоречие?
Проще всего было бы допустить, что в состав данной нефтяной
фракции, кроме 1,3-диметил-циклогексана, входит также 1,4-ди-
метил-циклогексан. В таком случае, однако, производные пара-
ксилола должны были бы, очевидно, получаться и при других
сопровождающихся дегидрогенизацией процессах, например при
действии дымящей серной или азотной кислоты, чего на самом
деле не наблюдается. Возможно поэтому, что образование тетра-
бром-параксилола из октонафтена происходит в данном слу-
чае за счет не 1,4-, а 1,1-диметил-циклогексана, т. е. второго
октонафтена, дающего с бромом и бромистым алюминием тетра-
бром-параксилол. Этот гомолог циклогексана, стоящий особняком
в смысле его отношения к ароматическому ряду, при нагревании
с дымящей азотной или серно-азотной смесью (а вероятно, и
с дымящей серной) дает почти исключительно продукты глубо-
ко?
кого распада алициклической системы. Понятно поэтому, почему
в данных условиях природный октонафтен дает ароматические
нитропроизводные и сульфокислоты лишь за счет другого али-
циклического компонента фракции 118—120°, т. е. 1,3-диметил-
циклогексана.
В полном согласии с этим взглядом [23J на состав нефтяного октонаф-
тена находятся результаты, полученные при его каталитической дегидро-
генизации [24J. Как и в случае 1,1-диметил-циклогексана, пропускавие
нефтяного О'ктонафтена через трубочку с платиновой чернью при 300—310°
сопровождается лишь незначительным выделением водорода; свойства неф-
тяного углеводорода изменяются при этом также крайне незначительно,
очевидно за счет дегидрогенизации небольших количеств 1,3-диметил-цикло-
гексана, содержащихся .во фракции. Главная же масса углеводорода остается
без всякого изменения, т. е. ведет себя при дегидрогенизации так же, как
синтетический 1,1-диметил-циклогексан.
Итак в состав фракции 118—120° бакинской нефти входят 1,3-диметил-
циклогексан, какой-то ближе неисследованный октан и один из октонаф-
тенов, не изменяющийся при дегидрогенизационном катализе. Возможно, что
это — 1,1-диметил-циклогексан или один из гомологов циклопентана; однако
вопрос этот требует дальнейшего исследования.
Изооктонафтен, изо - С8Н16. При фракционировке промежу-
точных продуктов между окто- и нононафтеном замечается не-
которое постоянство температуры при 122—125°. Исследование
этого погона бензина, получившего наименование изооктонаф-
тсна, было начато [25] в лаборатории Марковникова Путохиным
и продолжено Паутынским [26]. Физические свойства изоокто
нафтена были следующие: температура кипения 122—124°;
удельный вес: do® = 0,7835, do20 = 0,7681. Его состав и плот-
ность пара приводили к формуле С8Н16, но, судя но анализу, и
к ©тому нафтену было примешано значительное количество
углеводорода ряда метана.
Паугынский получил из своего углеводорода целый ряд производаых:
хлорид, иодюр, нафтилен, уксусный эфир и спирт (весьма немного). Все
они имели несколько иные свойства, чем соответствующие производные
октонафтена. При получении модюра нагреванием хлорида изооктонафтена
с иодистоводородной кислотой в качестве побочного продукта образовалась
смесь углеводородов, которая, будучи снова нагрета с иодистоводородной
кислотой (до 2СЮ°), превратилась в углеводород предельного характера;
последний снова кипел при 122—124°, имел прежний состав и мало изме-
нившийся удельный вес, du° = 0,7807, т. е. был почти идентичен с исход-
ным продуктом, изо-CsHte. Все это, вместе взятое, считалось доказатель-
ством, что изооктонафтен представляет собою н-овое индивидуальное веще-
ство, новую составную часть бакинской нефти.
Рассмотрим, какие же данные можно привести для ближай-
шей характеристики химической природы фракции 122—124 .
1. Прежде всего совершенно очевидно, что химическую инди-
видуальность изооктонафтена можно принимать лишь с большой
оговоркой, так как трудно представить себе, чтобы фракция
122—124° не содержала значительной примеси соседнего по-
гона, т. е. фракции 118—120 . Надо думать поэтому, что изоокто-
нафтен в значительной степени должен состоять из октонафтена;
правильнее говоря, из углеводородов, образующих фракцию
118—120°» и наоборот.
203
2. В полном согласии с таким заключением о составе изоокто-
нафтена находятся следующие два наблюдения: а) подобно окто-
нафтену изооктонафтен дает с бромом в присутствии бромистого
алюминия тетрабром-тшра-ксилол (Маркоеников) и б) по отно-
шению к дегидрогенизационному катализу поведение изоокто-
нафтена оказалось совершенно подобно поведению октонафтена
(Зелинский); оба углеводорода почти не изменяли своих свойств
и выделяли лишь крайне незначительное количество водорода.
Естественно, что и выводы, какие можно сделать отсюда относи-
тельно химической их природы должны быть тождественны (ср.
выше).
3. Основываясь на близости физических свойств изооктонаф-
тена с продуктом гидрогенизации орто-ксилола иодистоводород-
ной кислотой, Маркоеников высказался 27] за тождество этих
углеводородов, приписав им обоим строение 1,2-диметил-цикло-
гексана. Однако в настоящее время эти соображения, очевидно,
отпадают, так как доказано, что иодистоводородная кислота при
высокой температуре обычно вызывает глубокую изомеризацию
циклической системы; кроме того, при этом всегда получается
слишком пестрая смесь углеводородов, так что выяснение ее
состава может составить лишь самостоятельную и притом крайне
сложную задачу.
4. Судя по аналитическим дапным, содержание парафинового
углеводорода во фракции 122—124° должно быть даже несколько
выше, чем во фракции 118—120°. Характерно при этом, что
переведение углеводорода через ряд производных не уменьшает
этой примеси, а скорее увеличивает. Это видно, например, из со-
поставления изооктонафтена с препаратом того же нафтена, по-
лученным путем переведения исходного углеводорода через хло-
рид, иодюр и нафтилен (ср. выше); удельный вес нафтена при
этом несколько понизился, очевидно вследствие обогащения пре-
парата углеводородом предельного ряда. Конечно, это—>тот же
парафин, возможно, нормальный октан (температура кипения
125,8°), о котором было упомянуто выше, при октонафгене.
Только здесь, во фракции 122—124°, он находится уже в значи-
тельно большем количестве.
Итак, изооктонафтен едва ли представляет собою индиви-
дуальное вещество; невидимому, он состоит из тех же углеводо-
родов, что октонафтен, с некоторым преобладанием, однако, угле-
водорода ряда метана, возможно нормального октана. Никаких
указаний на присутствие во фракции 122—124° еще каких-либо
углеводородов, например тех либо иных гомологов -циклогексана
или циклопеитана, пока не имеется.
Нононафтены, C9II18. Число синтетически полученных ноно-
нафтенов весьма значительно (не менее 15). Наиболее доступ-
ными из них являются представители ряда циклогексана, которые
могут быть получены гидрогенизацией соответствующих гомоло-
гов бензола.
Первое указание на присутствие в кавказской нефти ноно-
нафтена было сделано Марковниковым и Оглоблиным [19]. Вы-
204
деленный ими углеводород кипел при 135—136°, имел состав и
плотность пара, отвечающие формуле С9П18, и при слабом нагре-
вании с дымящей азотной кислотой дал небольшое количество
кристаллического нитросоединения, которое после кристаллиза-
ции плавилось, как тринитро-мезититен (230—-231°). Позднее
иононафтен послужил предметом обширного исследования М. Ко-
новалова [28].
Ноионафтен неоднократно выделялся Коноваловым как. из балахан-
ской, так и из биби-эйбатской нефти по общему методу, разработанному
н лаборатории Марковпикова. Как видно из табл. 56, различные препараты
нононафтена мало отличались друг от друга по своим физическим свой-
ствам. 1 । _____. ।
Таблица 66
Нононафтены Темп, кип. Уд. вес
1. Из биби-эйбатской нефти . 135—137° = 0,7Ш
2. Из балаханской „ 135—137° dff = 0,7694
Из производных нононафтена Коноваловым были получены: мопохлорид,
лодюр, уксусный эфир, алкоголь, СвНпОН, простой эфир этого алкоголя п
нафтилен; позднее сюда были добавлены днхлорид CoHieCh и углеводород
терпенного ряда СвНы, а затем нитропроизводные нононафтена, его амины
и кетон состава C»Hi«O. Анализы большинства этих соединений были
вполне удовлетворительны; лишь в некоторых случаях обнаружились недо-
чет углерода и избыток водорода, причем разница с теорией достигала ино-
гда 0,4—0,5% и даже 0,6%. Основываясь на известном постоянстве свойств
всех этих производных, М. Коновалов не только высказался за индиви-
дуальность изученного им нафтена, но и пытался разрешить вопрос о его
структуре.
Три обстоятельства считались решающими при обсуждении
вопроса о химической природе нононафтена, а именно:
I. При обработке нононафтена бромом в присутствии бро-
мистого алюминия получается трибром-нсевдокумол; выходы
7—8% теории.
2. С дымящей серной кислотой получается сульфокислота
псевдокумола.
3. По своим физическим свойствам иононафтен оказался
тождественным с гексагидро-псевдокумолом, который был полу-
чен Коноваловым гидрогенизацией псевдокумола иодистоводород-
ной кислотой.
Па основании этих данных Коновалов высказал уверенность,
что иононафтен в своей главной массе состоит из гексагидро-
псевдокумола, т. е. 1,2,4-триметил-циклогексана, и что в качестве
примесей в нем находятся лишь небольшие количества гекса-
гидромезитилена и одного или нескольких углеводородов ряда
метана (нонан, С9Н2о).
Достаточно, однако, одного сопоставления физических свойств
нононафтена со свойствами синтезированного |29] позднее 1,2,4-
триметил-циклогексана (см. табл. а0), чтобы убедиться, что
205
о тождестве этих углеводородов не может быть и речи. Мы ви-
дим разницу в температурах кипения до 7° ив удельных весах
больше чем на 0,010. Очевидно, столь глубокое различие в фи-
зических свойствах не может зависеть от одних примесей; при-
чина его должна лежать где-то глубже. Чтобы разобраться в этом
вопросе [30], подсчитаем примерное количество отдельных со-
ставных частей данной фракции и прежде всего содержание
в ней нонана, СаТЬо. Среднее арифметическое из трех анализов
нононафтена по Коновалову, С — 85,44% и Н —14,60%, должно
довольно точно выражать состав этого углеводорода. С другой
стороны, смесь состава 5СэН13 + C9II20, содержащая свыше 16%
нонана, имеет следующий элементарный состав: С— 85,49% и
Н —14,51%. Отсюда ясно, что содержание нонана, С9Н20, в ноно-
нафтене Коновалова может простираться до 16—20%. Возможно,
впрочем, что содержание этой примеси в действительности не-
сколько меньше и не превышает, например, 10%,
Переходим к рассмотрению других составных частей нефтя-
ной фракции 135—137°. Прежде всего в ней несомненно присут-
ствие 1,2,4 триметил-циклогексана (гексагидро-псевдокумола), до-
казательства чему были приведены выше. Однако, в настоящее
время уже нельзя, конечно, утверждать, что этот гомолог цикло-
гексана составляет главную часть данной фракции. Не говоря
уже о различии в физических свойствах, достаточно вспомнить,
что при действии брома и бромистого алюминия на нононафтен
Коновалов получил 7—8% ароматического пербромида, тогда
как синтетический 1,2,4-триметил-циклогексан дает приятой же
реакции количественные выходы на трибром-псевдокумол. Воз-
можно, конечно, что примеси, содержащиеся во фракции 135—
137°, понижают выходы на кристаллический бромид, но все же
трудно допустить, чтобы в нононафтене Коновалова содержалось
больше 10—15%, в крайнем случае 20% 1,2,4-триметил-цикло-
гексана. Главная часть 'Этого гомолога циклогексана в соответ-
ствии с его температурой кипения должна находиться во фрак-
ции 142—144°, и с этой точки зрения исследование ©той фрак-
ции должно представлять собою одну из очередных задач изуче-
ния бакинской нефти.
Наконец, как уже было указано, есть основание допустить
присутствие в нефти гексагидро-мезитилена, т. е. 1,3,5-триметил-
циклогексана. Нахождение этого нафтена во фракции 135—137°
тем более понятно, что но температуре кипения (139°) этот угле-
водород подходит к нононафтену Коновалова даже ближе, чем
1,2,4-триметил-циклогексан. Тем больше оснований утверждать,
что количество этой составной части значительно меньше содер-
жания в нефти 1,2,4-триметил-циклогексана. так как в против-
ном случае при соответствующей обработке получались бы про-
изводные мезитилена, а не псевдокумола. Распространяя эти
последние соображения также и на другие гомологи циклогек-
сана, приходим к выводу, что общее содержание их во фракции
133—137° должно быть весьма невелико, во всяком случае не
больше 5—10%.
203
Итак, на основании вышеприведенного расчета можно счи-
тать установленным состав 25—40% фракции 135—137°: 10%
парафинов 10—20% гексагидроисевдокумола и 5—10% других
гомологов циклогексана из ряда нононафтенов. Какова же хими-
ческая природа остальных 60—75% этой фракции? Так Как из
различных циклических систем с полной достоверностью обна-
ружены в нефти лишь системы циклогексана и циклопентана,
иг» которых первая уже рассмотрена, то остается допустить, что
главная часть нефтяной фракции 135—137° состоит из того или
иного гомолога циклопентана |3о]. Кроме указанных соображе-
ний, в пользу такого допущения можно привести еще следующие
доводы:
1. Как показал Коновалов, нононафтен по своим свойствам
чрезвычайно близко подходит к продукту гидрогенизации псевдо-
кумола иодистоводородной кислотой. В настоящее время, однако,
не может быть никакого сомнения, что гидрогенизация псевдо-
кумола иодистоводородной кислотой сопровождается глубокой
внутримолекулярной перегруппировкой. Судя по другим приме-
рам (бензол, толуол, ксилолы), надо думать, что и здесь сущ-
ность изомеризации сводится к превращению шести членного
ядра в пятичленное с соответствующим изменением в числе или
составе боковых групп. Отсюда следует, что природный нононаф-
тен в главной своей массе должен принадлежать к ряду цикло-
пентана.
2. Обращаясь к табл. 50, мы действительно встречаем два
гомолога циклопентана, и по температуре кипения, и по удельному
весу хорошо подходящие к .нононафтену Коновалова. Это—1,
3, 2- и 1,4,2-диметил-этил-циклопентаны, (СН»)г • С5Н7 • CiHs,
синтезированные Зелинским [31]. Возможно поэтому, что угле-
водороды эти, оба или один, составляют главную часть неф-
тяной фракции 135—137 . G этой точки зрения становится по-
нятным, почему при соответствующей обработке нононафтена
получается так мало ароматического пербромида, тогда как
с синтетическим 1.2,4-триметил-циклогексаиом та же реакция
протекает количественно.
3. Наконец, результаты недавних опытов показали (Зелин-
ский), что при дегидрогенизациомном катализе нононафтен
почти не изменяется, т. е. ведет себя как типичный представи-
тель ряда циклопентана [32]. Таким образом, и с этой точки
зрения высказанные выше соображения о составе главной
части фракции 135—137° бакинской нефти находят себе подтвер-
ждение.
Деканафтены, С10Н20. Число синтетически полученных дека-
нафтенов сравнительно невелико — около 5. Важнейшие из них
принадлежат к ряду циклогексана. Свойства их сведены
в табл. 50.
Из нефти выделено три углеводорода состава СюН2о: а-дека-
нафтен, Р-деканафтен и изодеканафтен. Физические свойства их
сопоставлены в табл. 57. Из этих трех природных нафтенов по-
следний (изодеканафтен) почти не изучен [33], и в дальнейшем
207
изложении будут рассмотрены лишь два первых а- и р-декаиаф-
тены.
Таблица 57
Деканафтены Темп, кипения Уд. вес
а-деканафтен 162—164° d® =0,7959
₽- я 168°,5—170° d® =0,8076 do° = 0,7929
Изодокаиафтен .... 150-153° dj =0,8043
В качестве исходного материала для получения декаиафте-
пов пользовались керосином. При его фракционировке наблю-
далось изменение удельного веса погонов с температурой их ки-
пения, как при фракционировке бензина. Это видно, например,
из табл. 58.
Таблица 58
Темп, кипения Уд. вес (15°) Темп, кппения Уд. вес (15°)
158—16'/° 0,7805 168—170° 0,7940
160—162° 0,7790 170—172° 0,7960
162-164° 0,7820 172-174° 0,7965
164—166° 0.7860 174—176° 0,7965
166-168° 0,7900 176—178° 0,7970
По размерам наиболее значительными оказываются в указан-
ных пределах фракции 10'2—164° и 168—170°; этим фракциям
соответствуют более или менее характерные изменения удельного
веса. Эти углеводороды описаны под именем а- и р-деканаф-
тенов.
Первый из этих нафтенов был выделен впервые Марковнико-
вым и Оглоблиным 119]; позднее он был вновь получен в той же
лаборатории Зубковым [34]. Исследование этого углеводорода
находится, однако, по настоящее время в самой начальной ста-
дии. Все, что можно считать установленным относительно а-де-
канафтена, это то, что по своим свойствам этот углеводород не
подходит ни к одному из известных в настоящее время синтети-
ческих деканафтенов. Никаких указаний на возможное строение
этого углеводорода не имеется, и даже индивидуальность его не
может считаться сколько-нибудь прочно установленной.
Значительно больше а-деканафгена изучен его изомер —
Р-деканафтен. Из табл. 50 и 57 видно, что физические свойства
Р-дек а нафтена особенно близко подходят к свойствам двух син-
тетических деканафтенов: 1,3-диэтил-циклогексана и 1,4-метил-
208
изопропил-циклогексаиа (ментана). Однако ближайшее сопоста-
вление химических свойств этих углеводородов не подтвердило
тождества ₽-деканафтена ни с одним пз этих двух синтетических
нафтенов.
Для получения производных 0-деканафтена, Рудевич пользовался как
охлорением, так и нитрованием [35]. Полученные при этом результата не
дали сколько-нибудь определенных указаний на с.Т{Юение исходного нафтена,
а лишь установили различие между' р-деканафтеном и ментаном. Так на-
пример, присоединением брома к ментену получат я дибромид, СщН^Вг»,
который при нагревании с хинолилом даст соответствующий терпен, СюНи;
те же превращения с ^-доканафтиленом привели, однако, пе к терпену,
а обратно к исходному нафтилену; так что хинолин отщепил в последнем
случае пе бромистый водород, 2НВг, а бром, Вг3. Точно также при обра-
ботке ментена безводным модным купоросом по Брюлю получается цимол,
СюНн, тогда как из ^-декапафтилена в аналогичных условиях Рудевич
получил углеводород, состав которого лучше всего подходил к CioHie.
Наиболее ценные и притом положительные указания на
строение Р-деканафтена были получены при действии на ©тот
нафтен иода. Деканафтен нагревался с иодом до температуры
кипения углеводорода в течение G чае.; при этом замечалось
обильное выделение йодистого водорода. Удельный вес оставше-
гося после реакции углеводорода несколько повысился. Бром
действовал на этот углеводород с обильным выделением броми-
стого водорода; после испарения избытка брома остались иголь-
чатые кристаллы с температурой плавления 218—220°. Анализ
и определение молекулярного веса этого вещества дали вели-
чины, приблизительно подходящие к формуле СюНцВг3. Неви-
димому, это трибромид симм. диметил-этилбензола, который по-
лучается по Якобсону приливанием ароматического углеводорода
к брому. Температура плавления пербромида по Якобсону 218°,
так что, если при указанных превращениях пе происходит ка-
кой-либо внутримолекулярной перегруппировки, присутствие
в бакинском К€|юсине 1, 3, 5-днметил-этил-циклогексана стано-
вится вероятным.
Обращаясь к таблице, видим почти полное совпадение удель-
ных весов |-деканафтена и 1, 3, 5-диметил-этил-циклогексана,
но заметную разницу в их температурах кипения. Было бы по-
этому крайне интересно, введя соответствующую поправку при
фракционировке, подвергнуть ближайшему исследованию ту
фракцию керосина, которая по температуре кипения ближе под-
ходит к указанному гомологу циклогексана.
Вышеизложенным исчерпываются все имеющиеся опытные
данные о моно-циклических нафтенах бакинской нефти. В самое
последнее время данные эти пополнились первыми достоверными
сведениями о бициклических нафтенах той же нефти; оказа-
лось 36], что в состав ее несомненно входит декалин.
Декагидронафталин (декалин), СюНщ. В целях ближайшего
изучения их состава ряд фракций бакинской нефти с темпера-
турой кипения от 173 до 196° был подвергнут каталитической
дегидрогенизации при 310° в присутствии платинированного
_ 209
14 Зак. 1622. Хядшя нефти.
угля. Уже сопоставление свойств этих фракций до и после деги-
дрогенизации (табл. 59) с несомненностью указывает на то, что
процесс дегидрогенизации действительно имел здесь место. Рез-
кому повышению удельного веса и коэфициента рефракции всех
погонов после катализа в полной мере соответствовало измене-
ние их отношения к крепкой серной кислоте: в то время как до
катализа при обработке моногидратом поглощалось 1—6,7% угле-
водорода (по объему), после катализа моногидрат удалял уже
27—48,5% углеводорода. Так как, наконец, почти все исследо-
ванные фракции (кроме первой) после катализа нс реагировали
ни с бромной водой, пи с хамелеоном, то очевидно, что все по-
именованные изменения их свойств могли произойти лишь за
счет образования новой ароматики путем дегидрогенизации ка-
ких-то гидроароматических углеводородов. Действительно, в от-
личие от исходных погонов, все фракции после катализа имели
явственный запах нафталина, а при дегидрогенизации одной из
фракций (четвертой) этот углеводород был получен даже в кри-
сталлическом состоянии. Тем самым присутствие в исходных
погонах бакинской нефти декагидронафталина (декалина) дол-
жно считаться доказанным.
Таблица 59
№ п/п До дегидрогенизации После дегидрогенизации
Темп. кип. Уд. вес и коэф, рефракции Темп. кип. Уд. вес и коэф, рефракции
1 173—180° d]° =0,7922 = 14432 160—191° J]9’5 = 0,8322 ng = 1,4687
2 178,5—182,5" dio,5 == 0р036 я#5 = ],4447 168—192° =0,8258 ng'5 = 1,4637
3 181—186° й®э =0,8023 — 1,4446 175—195° =0,8240 ng =1,4623
4 185—190° d}° =0,8081 «JJ*5 = 14457 172—194° df =0,8271 ng = 1,4633
5 190—196° d*° =0,8100 nl£ =1,4483 186- 204° 4° =0,8330 = 1,4665
Для характеристики состава бакинской нефти крайне важно
и интересно, что после удаления из продуктов катализа всей аро-
матики обработкой их моногидратом состав остатка продолжал
отвечать формуле СпНзп. Это показывает, что значительная,
а иногда и главная часть бакинских погонов с температурой
кипения от 173 до 196° подобно ряду других фракций с более
низкой температурой кипения состоит не из гидроароматических
углеводородов, а из нафтенов других рядов, не способных к ката-
210
литической дегидрогенизации, вероятнее всего— из высших
гомологов циклопентана.
Что касается более высокомолекулярных нафтенов бакинской
нефти, то все опытные данные об отдельных их представителях
ограничиваются пока тем, что содержится по этому вопросу
в работе Марковникова и Оглоблина [19]. Авторами были выде-
лены из бакинской нефти следующие высокомолекулярные наф-
тены (табл. 60).
Таблица 60
Название Формула Темп. кип. Уд. вес (17’)
Эндеканафтен СцН.^ 186-185° 0,7995
Додеканафтен 196—197° 0,8025
Тетрадеканафтен ^цН» 240—241° 0,8190
Пентадеканафтен 6'IG HlO 246-248° 0,8294
Молекулярные веса и элементарный состав этих нафтенов до-
вольно хорошо подходили к формулам от CuHst до Ci5HM;
с некоторыми из них были проделаны опыты хлорирования.
Однако, данных этих совершенно недостаточно не только для
определения ближайшей химической природы этих нафтенов,
но даже их индивидуальности. Во всяком случае, как ясно из
вышеизложенного, первые два нафтена табл. 60 должны содер-
жать значительную примесь декалина, CwHw.
Некоторые дополнительные данные для общей характери*
стики высших нафтенов будут приведены ниже в этой же главе.
НАФТЕНЫ РАЗЛИЧНЫХ НЕФТЕЙ
Нахождение нафтенов в различных наших нефтях, не отно-
сящихся к Бакинскому району, например в грозненских, эмбен-
ских и др., не подлежит сомнению. Однако по сравнению с наф-
тенами бакинской нефти эти нафтены почти не исследованы..
Имеются, например, краткие указания на присутствие в гроз-
ненской нефти циклогексана и метил-циклогексана [37]: что
же касается других нафтенов грозненской нефти, то о них можно
судить пока лишь на основе состава и общих свойств различных
нефтяных фракций; последнее относится также к содержанию
нафтенов в нефтях остальных наших месторождений.
Значительно полнее изучены нафтены некоторых заграничных
нефтей, причем оказалось, что нафтены являются и здесь столь
же постоянной составной частью нефти, как парафины и аро-
матика.
В пенсильванской нефти, а именно в погонах ее с темпера-
турой кипения выше 150°, углеводороды состава СДЬп были об-
наружены еще в самый первый период ее исследования (Уоррен),
однако ближе не были охарактеризованы [38]. Лишь после того
14* 211
как аналогичные углеводороды были выделены из бакинской
нефти и химическая природа их была более или менее установ-
лена, были предприняты поиски их в других нефтях и в тем
числе в пенсильванских. Низшие нафтены пенсильванской неф-
ти с большою тщательностью были изучены С. Юнгом и Форти
[39]. Путем тщательной фракционировки с последующей фи-
зико-химической характеристикой выделенных фракций ими
были обнаружены в пенсильванской нефти и выделены следую-
щие низшие нафтены:
Циклопентан, С5Н10, обнаружен во фракции с температурой
кипения около 50° на основе высокого удельного веса этой фрак-
ции, главное же — по образованию глутаровой кислоты при ее
окислении азотной кислотой.
Метил-циклопентан, CH3C5II9, был обнаружен в смеси с нор-
мальным гексаном во фракции 69,95—70,15е. Эта фракция
имела также повышенный удельный вес и в отличие от чистого
гексана реагировала, с дымящей азотной кислотой.
Циклогексан, CeHi2. Для выделения этого нафтена легкие по-
гоны пенсильванской нефти были сначала медленно разогнаны
9 раз; за этой предварительной разгонкой следовали химическая
очистка фракций, промывка и сушка; после 33 новых фракцио-
нировок углеводород перегонялся в пределах 80,55—80,65°, но
судя по несколько сниженному удельному весу (0.7722, вместо
0,7896), все еще содержал примесь парафинов. При его окисле-
нии получен характерный продукт окисления циклогексана,
адипиновая кислота. Путем вымораживания нефтяного углево-
дорода циклогексан был получен затем в почти чистом виде
(температура плавления 4,7° вместе 6,4°).
Диметил-циклопентан, (СНз)г С5Н8. Содержится, невиди-
мому, во фракции 91—93° пенсильванской нефти.
Метил-циклогексан, СНзСвНн. В небольшом количестве этот
нафтен также содержится в пенсильванской нефти, о чем можно
судить уже по слишком высокому удельному весу ее гептановой
фракции (температура кипения 98—99°).
По мере перехода к фракциям пенсильванской нефти с более
высокой температурой кипения содержание нафтенов, судя по
данным анализа, все увеличивается. Одновременно с этим, есте-
ственно, возрастают трудность их выделения в чистом виде и
даже простая их идентификация. В результате, сведения о выс-
ших нафтенах пенсильванской, как, впрочем, и других нефтей,
крайне недостаточны.
Как было указано в гл. V, Мэбери [40] путем выморажива-
ния фракций пенсильванской нефти с температурой кипения
выше 216°, тщательно разогнанных в вакууме, разделял их на
твердую часть, состоявшую из парафинов, и жидкую, состав
которой оказался непредельным. Такому разделению поддава-
лись, однако, лишь фракции с температурой кипения от 210 до
212' при 50 мм и выше; низшие же погоны либо вовсе не выде-
ляли кристаллов, либо, после отделения небольшого количества
легкоплавкого парафина (температура плавления около 20° и
212
ниже) сохраняли остальную часть парафиновых углеводородов
в своем составе, продолжая, таким образом, оставаться смесью
углеводородов различных рядов. В табл. 61 приведены некото-
рые наиболее интересные результаты разделения фракций пен-
сильванской нефти путем их вымораживания (депарафини-
зации).
Таблица 61
Высшие нафтены пенсильванской нефти
Темп. кип. фракций при 50 мм Твердая часть Жидкая часть
Формула Темп, плавя. Формула Уд. вес (20°)
199—200° СЛ 20° С1Д1з8—СщП g —
210-212° ^1оН4О 33-34° CjgHgg 0,8208
230—2323 С21Н44 40-41° C21Н42 0,8424
240 —242° 43° С«»Н411 0,8296
258—260° С23Н48 45° 1'23 Н46 0,6569
272—274° С24Н50 48° 6Л 0,8598
280—282 ’ Corll53 53—54° CajHjs 0,8580
292—291° 58° С27Н52 0,8688
310—312° CsyHps 60° С28Щ4 0,8694
Как видно из табл. 61, вымораживание фракции с темпера-
турой кипения 199—200° при 50 мм дало в остатке масло со-
става, среднего между СюНзз и СюНзв. Следующие фракции до
температуры кипения 280—282° после депарафинизации имели
состав СпН2и; последние же две фракции, а также углеводороды,
выделенные из продуктов естественного испарения нефти в неко-
торых пенсильванских скважинах, оказались еще более непре-
дельного состава, СпН2п_2. Все эти денарафинизованные фрак-
ции пенсильванской нефти по отношению к брому и дымящей
серной кислоте имели ясно выраженный предельный характер.
Так как, несмотря на тщательность разгонки (до 38 раз), трудно
ожидать, чтобы выделенные таким образом фракции даже после
вымораживания представляли собою индивидуальные вещества,
правильнее было бы охарактеризовать их как смесь соответ-
ствуюпщх моно- и бициклических нафтенов невыясненного строе-
ния.
Кроме пенсильванской нефти, наиболее изученными нефтями
северной Америки являются некоторые нефти Охайо, Канады,
Калифорнии и Мид-Континента.
Охайская нефть [41], как уже было отмечено в гл. V, по
своему составу очень близка к пенсильванской, особенно в низ-
ших ее погонах. Высшие фракции, начиная примерно от тем-
пературы кипения 210е при 760 мм, и здесь при выморажива-
нии, выделяют парафин, после отделения которого масло при-
обретает непредельный состав, а именно: начиная от Cis до Сп —
состав СпПзд, от Сю — состав Cw,H2ft_2 и, наконец, от Css — со-
став C/iiIIg» — 4. Все эти фракции охайской нефти имеют ясно
213
выраженный предельный характер, гак что образующие их угле-
водороды и здесь должны быть отнесены к нафтенам различных
рядов. Однако уже простое сопоставление соответствующих
фракций показывает, что в отношении непредельности состава
охайская нефть заметно превосходит пенсильванскую.
Следующей ступенью в направлении повышающейся непре-
дельности состава среди североамериканских нефтей являются
канадская и калифорнская нефти.
Низшие нафтены не выделены из канадской нефти, но об их
присутствии свидетельствуют анализы различных фракций, на-
чиная с октановой (температура кипения около 1*24—125°); во
фракциях же канадской нефти, кипящих примерно от 200° и
выше, нафтены отчетливо выдвигаются на первое место [42].
В калифорнских нефтях [43] нафтены обнаруживаются уже
в самых легких погонах. Так, в легкой нефти графства Фресно
(удельный вес &о —0,8423) уже фракция 68—70° представляет
собой смесь парафина с нафтеном, очевидно гексана с метил-
циклопентаном; в ближайших же ее фракциях наметились дру-
гие простейшие нафтены, невидимому те же, что в бакинской
нефти. Высшие нафтены калифорнской нефти изучены пока
также далеко недостаточно. Для их характеристики интересны
данные, полученные при исследовании тяжелой нефти из Санта-
Барбара {удельный вес dzo — 0,9845); фракционировкой ее при
60 мм был выделен ряд фракций в• пределах 5°, которые после
Таблица 62 Высшие нафтены кал ифор некой нефти очистки дымящей серной кислотой показали сле- дующие состав и свой- ства (табл. 62).
Темп. кип. • (60 мм) Формула Уд. вес • при 20° Несомненно, фракции эти не представляют со- бой. индивидуальных хи- мических соединений; тем не менее, (крайне интере- сен уже один их состав, выражаемый, как вид- но, формулами Сл,Н2п —2, СпДТ2л _ 4 -и C„H2ft_8. Так как характер всех этих углеводородов оказался авляют'собой нафтены раз-
150—155° 175—180° 190—195° 210—215° 250—255° 310—315° 340-345° предельным, № Ciebgo С^Взо С 18^1,2 ад» C27H4G С29Щ0 то, очевидне 0А621 С,Е8С8 0/919 0,8998 0,9299 0,9451 0,9778 ), они предст
личных рядов — би-, три-, и вообще полициклические.
Аналогичные результаты получены при исследовании ряда
нефтей Тексаса [44] и Луизианы [45]: нафтены различных
рядов пока не выясненного строения являются главной состав-
ной частью этих нефтей.
/v ^?™€ТИМ) наконец, нефть одного из месторождений Оклахомы
(Кей-Коунти, South Ройса.), являющуюся предметом подробного
исследования за последние 10—-12 лет в А. Р. J. (США). В
бензиновой части этой нефти обнаружены и выделены следую-
щие нафтены [46]:
214
Метил-циклопентан, СПз • CsHe. Выделен фракционировкой
с доследующим нитрованием для удаления примеси бензола, вы-
мораживанием и, наконец, перегонкой с метиловым спиртом.
Степень чистоты лучшего образца была 98,7%.
Цилкогексан, С6Н12. Выделен фракционировкой, нитрованием
для удаления бензола и вымораживанием. Степень чистоты
99,96%.
1,1-диметил-циклопентан (CHs^CsHg. Выделен фракциони-
ровкой с последующим нитрованием для удаления бензола.
Степень чистоты 95%.
Метил-циклогексан, СНз • СбНи. Выделен фракционировкой
с последующим нитрованием для удаления толуола и вымора-
живанием. Степень чистоты 99,8%.
1,3-диметил-циклогексан, (СНз^СвНю. Выделен фракциони-
ровкой и кристаллизацией из смеси жидкого метана с пропаном.
Степень чистоты 98%.
1,2-диметил-циклогексан, (СНз^СбНю. Способ Выделе-
ния— тот же, что и для предыдущего нафтена. Степень чи-
стоты 91%.
Этил-циклогексан, СгН5. СбНи. Выделен фракционировкой
с последующим извлечением жидким сернистым ангидридом,
адсорбцией силикагелем, перегонкой с уксусной кислотой и кри-
сталлизацией из смеси жидкого метана с пропаном. Степень
чистоты 95%.
Нононафтен, С&Н18 (температура кипения 136,7°). Выделен
фракционировкой с последующим извлечением жидким серни-
стым ангидридом, кристаллизацией из смеси жидкого здааж
с пропаном и кристаллизацией из дихлор-дифтормётанц.,?Сте-
пень чистоты лучшего образца выше 99%. По вопросу о строе-
нии этого нононафтена были сделаны выводы, аналогичные
тем, которые были даны выше для нононафтена из бакинской
нефти, т. е. что он представляет собою один из гомологов цикло-
пентана [47].
Нононафтен, С9Н18 (температура кипения 141,2°). Выделен
фракционировкой с последующим извлечением жидким серни-
стым ангидридом и адсорбцией силикагелем; затем следовали
перегонка с уксусной кислотой, фракционировка под уменьшен-
ным давлением и кристаллизация из дихлор-дифтор-метана,
а также—из смеси жидкого метана с пропаном.' Трудно отде-
ляется от изононана (температура кипения 140,8°); все же сте-
пень чистоты выделенного нафтена определяется в 95%. Пови-
димому, в основном, этот нононафтен представляет собою
1, 2, 4-триметилгциклогексан.
Кроме перечисленных нафтенов, выделенных из оклахомской
нефти в более или менее чистом виде, имеются основания пред-
полагать присутствие в ней следующих нафтенов, кипящих
в пределах 55—145°:
1,2-диметил-циклопентан (температура кипения 91°,8)
1,3-диметил-циклопентан ( „ „ 90°, 7)
215
Этил-циклопентан (температура кипения 103°,О)
1,2,4-триметил-циклопентан ( „ „ 112,5—113°о)
1,3,5-триметил-циклогексан
и некоторые другие гомологи циклопентана и циклогексана.
Выделение более выоококипящих нафтенов составляет задачу
дальнейших исследований.
Остановимся в заключение па нафтенах некоторых европейских нефтей.
Нафтены галицийской нефти пока почти не изучены, хотя присутствие
их в этой нефти и притом <в довольно значительном количестве не подле-
жит сомнению. Так, путем тщательной систематической фракционировки
погона галицийской нефти с температурой кипения 65—96° удалось выде-
лить циклогексан, совершенно * тождественный по своим константам с син-
тетическим углеводородом [48]. Повидимому, по содержанию нафтенов га-
лицийская нефть должна быть поставлена между нефтями пенсильванской
и бакинской [49].
Румынская нефть, видимо, несколько богаче нафтенами [50], чем гали-
цийская. В низших ее погонах обнаружены с различной степенью досто-
верности циклопентан, метил-циклопентан, циклогексан, метил-циклогексан
и этил-циклогексан. Высшие нафтены румынской нефти пока не изучены.
Несомненно также нахождение нафтенов в ряде других европейских
нефтей, например германской (Ганновер, Тегернзее) [51] и итальянской
(Парма) [52]. В последней присутствие низших нафтенов доказано мето-
дом окисления некоторых фракций; так например, при действии азотной
кислоты на ряд фракций с температурой кипения от 67 до 87° получены
нормальный продукт окисления циклогексана, адипиновая кислота и т. д.
Как видно из вышеизложенного, простейшие нафтены, обна-
руженные в различных нефтях, принадлежат лишь к двум рядам,
циклопентана и циклогексана. Известно, что эти алицикличе-
ские системы выделяются среди других простейших моноцикли-
ческих нафтенов наибольшей устойчивостью, и уже одно это
обстоятельство служит достаточным объяснением столь широ-
кого распространения их в природе.
Теоретические соображения, которые, казалось бы, могли разъяснить
различную устойчивость алициклических систем, лежат в области их сте-
реохимии [53].
Фиг. 29.
Фиг. 80.
Согласно тетраэдрическим представлениям классической стереохимии на-
правления линий, соединяющих центры тяжести двух углеводородных ато-
мов с центром тяжести третьего углерода при открытой их группировке,
например в пропане, образуют угол в 109°28'. Этим самым расположение
цепи из трех и более углеродных атомов в ^пространстве определяется яе
216
й-о Прямой, а по Некоторой Ломаной лйййи, которая для цепей йз 5 п G
углеродов почти естественно может замкнуться в циклическую систему
с минимальным уклоном от нормального расположения углеродных атомов.
Напротив, в цепях из 3, 4, 7, 8 и т. д. углеродов подобное замыкание коль-
чатой системы должно более или менее значительно отклонять углерод-
ные атомы от их нормального расположения,
вследствие чего вся система должна претер- Q-. qo qo
петь некоторое натяжение. Лг Лг Аг
Для различных кольчатых (алицикличе- >-
ских) систем такое натяжение может быть ЧР чг
легко вычислено, причем в простейших слу- JV ql
чаях получается хорошее соответствие между ООО
величиной натяжения и устойчивостью соот- -
ветствующей замкнутой системы: чем больше фиг‘ dl*
натяжение, тем меньше устойчивость системы,
и наоборот. Этим теоретическим представлениям («теория натяжения»
А. ф.-Байера, 1885 г.) противоречит, однако, ряд соображений, основанных
на более углубленном изучении изомерных превращений алициклических
соединений (изомеризация циклов), а равным образом на существовании и
выдающейся устойчивости многочисленных алициклических систем, синтези-
рованных в новейшее время1.
Впрочем, возможность существования такого рода систем со стереохи-
мической точки зрения можно представить себе, допуская расположение
углеродных атомов в них не по схемам, изображенным на фиг. 29 и 30,
а иначе, например по зигзагообразным кривым разного рода (фиг. 31).
Такого рода расположение углеродных атомов принимают в настоящее
время для некоторых высокомолекулярных соединений жирного ряда; воз-
можность подобных конфигураций для 'многочисленных алициклических
соединений также легко можно себе представить.,
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ НАФТЕНОВ В НЕФТЯХ
И НЕФТЕНРОДУЕТАХ
Определение нафтенов в сложных углеводородных смесях про-
изводится после предварительного удаления из (них непредель-
ных и ароматики, когда компонентами смеси являются только
парафины и нафтены1. Однако даже после такого упрощения
состава углеводородной смеси ввиду близости химических
свойств этих углеводородных рядов количественное определение
каждого из них продолжает оставаться задачей крайне трудной.
Таблица 63
ЭлементарныйГсостав некоторых углеводородов
Состав с6нм с,нч+ +10% свн12 СдЩ^з СтоНс2-{- + Ю'/о С10Н2о C-nHgn
% С 83.72 83,92 84,51 84 63 85,71
%Н 16 28 16,08 15,49 15,37 14,29
Казалось бы, что к ее решению можно подойти на основе различия
в элементарном составе парафинов, CnH^n-i-o и нафтенов СпВлп Однако,
как видно из табл. 63, 10%^ная примесь нафтена к парафину даже для
фракции с температурой кипения ниже 100°, например для гексана и его
1 См. примечание на стр. 186.
217
ближайших гомологов, вызывает лишь такие изменения в элементарном
состава, которые лежат в пределах ошибки анализа (0,2%); а так как
по мере повышения температуры кипения фракций влияние подобного рода
примеси сказывается на составе все меньше и меньше, то ясно, что эле-
ментарный состав нефтепродукта или его отдельных погонов может дать
лишь весьма приближенное представление об относительном содержании
в них парафинов и нафтенов.
Столь же мало применимы в данном случае и физические
методы, основанные на определении удельного веса.или коэфи-
циента рефракции смеси. Хотя нафтены и парафины заметно
различаются между собой по этим константам, однако влияние
изомерии на последние, особенно для низших представителей
различных рядов (ср. табл. 26), настолько значительно, что при-
менение этой методики в большинстве случае® могло бы при-
вести лишь к ошибочным результатам.
Метод анилиновых точек. Значительно меньше отражается
изомерия на критической температуре растворения различных
углеводородов ряда циклопентана и циклогексана -в анилине и
некоторых других растворителях. Ввиду этого описанный
в гл. ГУ в применении к определению ароматики метод анили-
новых точек (приложим также и к определению нафтенов в смеси
их с парафинами. Определение ведется здесь также на отдель-
ных, более или менее узких фракциях нефтепродукта, обыкно-
венно на тех же, которые применяются для определения арома-
тических углеводородов.
Как известно, для определения ароматики методом анилино-
вых точек необходимо найти две температуры взаимного раство-
рения анилина и углеводородной смеси: одну—до удаления из-
смеси ароматики, другую — после ее удаления. Так как метод
количественного отделения нафтенов от парафинов до сих пор
не известен, то при анализе на нафтены из соответствующих
температурных точек ((анилиновых) определяется только одна,
характеризующая данную смесь; что касается другой точки, не-
обходимой для вычисления соответствующей содержанию нафте-
нов депрессии, то .вместо нее приходится пользоваться некоторой
средней температурой, вычисленной из экспериментально найден-
ных критических температур растворения в анилине чистых па-
рафинов, кипящих в тех же пределах, как и изучаемая фракция.
Аналогично, пользуясь подходящими смесями чистых нафте-
нов и парафинов, можно определить величину депрессии, отве-
чающей 1% содержания нафтенов в растворе, а затем путем
пересчета составить таблицу коэфициеитов К, соответствующих
процентному содержанию нафтенов на 1° депрессии анилино-
вой точки. I
В табл. 61 приведены [52] средние критические температуры
растворения в анилине нафтенов и парафинов для ряда бензи-
новых фракций, а также коэфициеитов К, которыми можно поль-
зоваться для ^вычисления процентного содержания нафтенов
в бензинах. Как ясно из вышеизложенного, точность метода
анилиновых точек в данном случае значительно уступает точ-
ности его в применении к определению ароматики. Для более
218
высокобипящих нефтяных фракций ввиду недостаточной вы-
'ясненности природы образующих их углеводородов источники
ошибок при применении к ним анилинового метода должны быть,
естественно, еще значительнее, так что на весь полученный та-
ким образом цифровой материал следует смотреть пока как на
ориентировочный.
Таблица 64
Темп. кип. фракции Средняя крит. темп, растворения в анилине Коэфициенты AM
нафтенов парафинов
40— 60° 23 72 1,51
60— 95° 35 71 2,13
95—122° 43 71 2,76
122—150° 49 73 3,26
150-175° (сЗ) 76 3 45
175-200° 78 —-
Метод каталитической дегидрогенизации [53]. Метод основан
на дегидрогенизации, которую легко претерпевают нафтены ряда
циклогексана в присутствии катализаторов (Pd, Pt, Ni и др.)
при температуре около 300° (гл. III). Специальные опыты пока-
зали, что реакция эта протекает количественно, причем другие
компоненты нефтяных погонов (парафины, нафтены ряда цшо-
пентана, ароматика) остаются практически незатронутыми де-
гидрогенизационным процессом. Ввиду этого, если в хорошо очи-
щенной нефтяной фракции определить содержание ароматики,
затем подвергнуть ее дегидрогенизации и вновь определить в ней
содержание ароматических, то по количеству новообразовав-
шейся ароматики можно рассчитать содержание в исходной
фря-кции нафтенов ряда циклогексана. Метод дает весьма точ-
ные результаты, но, очевидно, может решить вопрос о количе-
ственном содержании нафтенов лишь частично, являясь, таким
образом, скорее методом дальнейшего уточнения и детализации
состава нафтеновой части нефти и ее продуктов.
КОЛИЧЕСТВЕННОЕ СОДЕРЖАНИЕ НАФТЕНОВ В РАЗЛИЧНЫХ
НЕФТЯХ
Характеристика различных нефтей в отношении содержания
в них нафтенов уже была дана несколько выше. Остается допол-
нить ее данными количественного характера, для чего может
служить табл. 65, составленная подобно ранее приведенным та-
блицам 29 и 42 на основе определения ГрозНИИ [56].
1 Величина коэфициента пр? определении нафтенов не зависит от ме-
тодики определения анилиновой точки, т. е. от того, определяется ли ани-
линовая точка методом равных объемов или методом максимальной анили-
новой точки (ср. определение ароматики анилиновым методом).
219
Таблица 65
Содержание нафтенов в различных нефтях
№ п/п I Месторождение нефти Уд- вес (15°) % нафтенов во фракциях 1 с темп, кипения
ю СТз О о п см см I СП o02I~SSI О о ю 200—250° § со о U3 см
1 Сураханы 0,848 76 81 51 66 44 39
2 Балаханы 0,876 56 68 66 75 74 62
3 Балаханы-Раманы-Са- бунчи . . 0/67 48 64 82 66 43 38
4 Бинагады 0,921 48 55 56 70 65 58
5 Биби-Эйбат 0,865 50 60 57 69 51 42
6 Грозный, парафин, фон- тан 0,844 24 37 30 29 23 22
7 Грозный, слабопара- фин. фонтан 0,849 34 39 31 35 30 28
8 Грозный, беспараф. лег- кая 0,860 35 40 33 39 40 -37
9 Вознесенская 0,900 11 30 49 71 61 48
10 Майкоп 0,848 40 45 40 39 36 36
11 Калужская 0,955 — — — 90 85
12 Доссор (Эмба) 0,862 40 63 57 69 67 61
13 Пермская 0,941 41 27 18 15 18 17
14 Тонкава, Оклахома . . 0,821 85 42 88 41 34 30
15 Девинпорт 0,796 2) 34 27 29 31 31
16 Мексиа, Тексас .... 0,845 22 26 19 21 19 28
17 Хентингтон-Бич, Кали- форния 0,897 40 56 55 61 45 41
Как видно, по содержанию нафтенов между нефтями наблю-
дается столь же большое разнообразие, как по содержанию пара-
финов (гл. V). Очевидно также, что при сравнительно неболь-
шом содержании ароматики в громадном большинстве нефтей
численные данные для содержания нафтенов и парафинов
должны находиться в обратной зависимости друг к Другу: чем
больше в нефти нафтенов, тем меньше в ней парафинов, и на-
оборот.
Из нефтей СССР наиболее богаты нафтенами бакинские и эм-
бенские нефт, а также некоторые нефти Грозненского района
(грозненская бесларафиновая, Вознесенская). Обращает на себя
внимание высоким содержанием нафтенов тяжелая калужская
нефть. Менее богаты нафтенами нефти грозненские парафини-
стая и слабопарафинлстая, майкопская и особенно пермская.
В отношении распределения по различным фракциям здесь
наблюдаются те же три основных типа нефтей, которые были
намечены в гл. V для фракционного распределения парафинов.
1 Содержание отдельных фракций в различных нефтях см- табл. 29
(строка II при каждом месторождении'.
220
У .одних нефтей содержание нафтенов по мере повышения
температуры кипения погонов постепенно повышается; таковы,
например, большинство бакинских нефтей, эмбенская и Возне-
сенская нефти.
Ко второму типу относятся нефти, у которых содержание
нафтенов в различных фракциях (до 300°) сохраняется более или
менее постоянным; сюда относятся нефти: грозненские беспара-
финовая и слабонаряфинистая, майкопская, калужская и др..
Наконец, третий тип нефтей характеризуется постепенным по-
нижением содержания нафтенов во фракциях по мере повыше-
ния температуры кипения последних. Таковы, например, нефти
сураханская, грозненская парафинистая и особенно пермская.
Значительный интерес представляет содержание нафтенов
в различных нефтепродуктах. Наиболее изученными в ©том
отношении, естественно, являются бензины и керосины.
Таблица 66
Содержание нафтенов в беззинах и керосинах СССР 1
1 п/ц «х 1 Месторождение нефти Содержание нафтенов в % № п/п Месторождение нефти Содержание нафтенов в %
бенз. керос. бенз керос.
1 Сураханы 72 49 10 Грозный, беспарафин. 36 99
2 Балаханы 66 72 йефть, нижн. ил. . легк-
3 Биби-Эйбат .... 56 57 •
4 Бинагады 54 —• 11 Грозный, беспарафин. 87 ' Ш
5 6 Грозный, парафин, нефть ’. Грозный, парафин. 29 авпо 30 25 12 нефть, нижн. пл. . Грозный, беспарафин. тяж. 45 58
7 нефть Грозный, парафин. легк. 29 23 13 нефть, верхи, пл.. Грозный, беспарафин. легк. 47 61
8 нефть Грозный, слабопа- тяж. 34 31 14 нефть, верхи, пл. . Майкоп тяж. 42 «мм
9 рафии, нефть . . Грозный, стабопа- рафин. нефть . . легк. 34 тяж. 29 15 16 Майкоп Доссор (Эмба) . . . авио 40 тяж. 37 65
Как видно из табл. 66, содержание нафтене в в различных
бензинах СССР колеблется в довольно широких пределах: от
70% и выше до 30% и ниже. В бакинских бензинах содержание
нафтенов превышает 50%; особенно выделяется в этом отноше-
нии сураханский бензин (72% нафтенов). Противоположной
крайностью являются бензины из грозненской парафинистой
нефти, содержащие лишь 29—30% нафтенов. В остальных бен-
зинах СССР содержание нафтенов колеблется примерно между
35—15%.
J Характеристику этих нефтепродуктов см. табл. 50.
221
Не менее широкие колебания в содержании нафтенов можно
наблюдать на керосинах СССР. Наиболее богаты нафтенами ке-
росины из нефтей б ал аданской (72% нафтенов), эмбенской
(65%) и грозненской беспарафиновой верхних пластов (свыше
60%> нафтенов). В большинстве других наших керосинов содер-
жание нафтенов ниже 40%, для керосинов же из грозненской
парафинистой нефти оно спускается до 25% и ниже.
Кроме общего содержания нафтенов, большой интерес для
характеристики состава нефтей представляет получекный не-
давно путем каталитической дегидрогенизации материал по со-
держанию отдельных нафтеновых рядов, циклонентана и цикло-
гексана, в бензинах СССР. Благодаря этому материалу предста-
вляется возможным охарактеризовать низшие погоны важней-
ших наших нефтей с точки зрения количественного содержания
в них 4 основных углеводородных рядов: ароматика, нафтены
ряда циклогексана, нафтены ряда циклопептана и парафины
(табл. 67).
Таблица 67
Состав различных бензинов СССР (фракции 60—150е)
А» I /гг Месторождение нефти Темп. кип. фракций Состав фракций в % по ве< у
Арома- тика Нафтены Пара- фины
р. цпкло-!р. цикло- гексана j пентана
60— 95е 37 Зэ 27
1 Сураханы | 95—122° 1 47 33 19
122—150° 6 38 30 26
60— 95° — 33 29 38
2 Балаханы—Сабунчи . | 95—122° 1,5 49 27 22.5
122—150° 4 40 32 24
3 Биби-Эйбат | 95—122° 3 33 27 37
122—150° 7 20 41 32
4 Грозный, фонтан, па- 60— 95° 3 11 26 60
рафпн. нефть . . . . • 95—122° 7 14 22 57
122—150° 9 8 22 61
5 Грозный, беспарафи- ( 60— 95° 4 17 25 54
новая нефть . . . . { 95—122° 8 29 16 47
122—159° 12 24 23 41
6 Доссор—Эмба .... | 95—122° 2 23 37 38
122—150° 4 33 26 37
Из табл. 67 видно, что в бензинах, богатых нафтенами, на-
блюдается либо преобладание ряда циклогексана над рядом ци-
клопентана (нефти сураханская, балахано-сабунчинская, гроз-
ненская беспарафиновая), либо оба нафтеновых рада предста-
влены [примерно одинаково (доссорская нефть). Напротив, в неф-
тях, сравнительно бедных нафтенами, преобладающее значение,
невидимому, приобретает ряд циклопентана (грозненская пара-
финовая нефть).
ПРИМЕНЕНИЕ И ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ НАФТЕНОВ
КАК СОСТАВНОЙ ЧАСТИ НЕФТИ
Несмотря на. богатейшее содержание нафтенов во многих
нефтях, выделение и утилизация отдельных их представителей
в техническом масштабе до сих пор являются вопросом буду-
щего. Тем не менее, ввиду выдающегося интереса, который пред-
ставляют некоторые из возможных путей утилизации нафтенов
этого рода, мы остановимся сначала на них, после чего рассмо-
трим вкратце те специфические особенности, которые придают
нафтены различным нефтепродуктам.
Важнейшим методом утилизации нафугенов является превра-
щение их в ароматические углеводороды. Как известно, переход
к углеводородам этого ряда от нефти можно осуществить сле-
дующими способами:
а) способом непосредственного выделения ароматических
углеводородов из нефти и ее дестиллатов (гл. IV);
б) способом лирогенетического разложения нефти, который
будет рассмотрен ниже, в главе о переработке нефти (ч. II);
в) способом каталитической дегидрогенизации, уже неодно-
кратно упоминавшимся выше. Так как сырьем при образовании
ароматики путем дегидрогенизационного катализа являются со-
держащиеся в нефти нафтены ряда циклогексана, то на этом
способе здесь необходимо остановиться.
В настоящее время, невидимому, едва ли можно сомневаться,
что все основные предпосылки для проведения в технику полу-
чения ароматических углеводородов способом каталитической
дегидрогенизации нафтенов имеются налицо. Самый способ пред-
ставляется в следующем виде. Путем фракционировки выделяют
из нефти подходящую фракцию, подвергают ее дегидрогенизации
и выделяют из катализггга ароматический углеводород путем но-
вой фракционировки. Более или менее значительная примесь
нафтенов ряда циклопентапа и парафинов, которые могут со-
держаться в выделенном ароматическом углеводороде, не ме-
шает в большинстве случаев его дальнейшей переработке; непре-
дельных же соединений в катализате почти не содержится, так
что никакой предварительной очистки полученного ароматиче-
ского углеводорода обыкновенно не требуется. Простота аппа-
ратуры и прекрасные выходы являются лучшей рекомендацией
метода, и едва ли не единственным вопросом, требующим спе-
циального внимания в этой области, является вопрос о сохран-
ности в процессе работы катализатора. Этот вопрос решается со-
блюдением двух основных условий данной реакции: 1) тщатель-
нейшей очисткой нефтяной фракции, так как малейшие следы
сернистых соединений нефти отравляют катализатор, и 2) под-
223
бором надлежащей температуры дегидрогенизации, так. как
слишком высокая температура (выше зио—31о°) вызывает ча-
стичное обугливание и быстрое снижение активности катализа-
тора.
Ценной особенностью метода дегидрогенизации в применении
его к техническому получению ароматических углеводородов
является его гибкость. ’Подбирая соответствующую фракцию
нефти, можно' варинровать процесс в зависимости от большего
или меньшего спроса на тот или иной ароматический углеводо-
род и, используя одну и ту же аппаратуру и метод, направить
работу в сторону удовлетворения наибольшей потребности дан-
ного момента. Крайне важно также, что, как это видно из дан-
ных табл. 60, в большинстве нефтей наивысшее содержание на-
фтенов ряда циклогексана приходится на долю фракции 95—
122°, которая в главной массе должна состоять из метил-цикло-
гексана; последний же при каталитической дегидрогенизации
дает толуол.
О тех возможностях, которые таит в себе метод каталитиче-
ской дегидрогенизации для получения ароматических углево-
дородов, можно судить, например, на основании следующих не-
давно опубликованных данных [57!: из пермского бензина (Чу-
совские Городки), кроме толуола, который содержится в его
фракции ПО—112° (гл. IV), путем дегидрогенизации фракции
100—102° оказалось возможным получить еще 21,5% толуола,
считая на исходный бензин; из фракции же 100—102% сура-
хаиского 'бензина, почти не содержащего ароматики, после
дегидрогенизации было получено ароматики (главным образом
толуола) до 68%.
Кроме превращения в ароматические углеводороды, можно
упомянуть следующие возможные пути химической переработки
нафтенов ряда циклогексана.
Нефтяной циклогексан, C6Hi2, действием хлора может быть
легко превращен в хлорциклогексан, СбНцС1; от последнего же
легко представить себе два перехода: либо к соответствующему
непредельному углеводороду, циклогексену, СбНю, либо к алко-
голю, циклогексанолу, СсНиОН. Оба эти соединения, предста-
вляя выдающийся интерес с чисто химической точки зрения, мо-
гут найти и отчасти уже находят практическое применение.
Циклогексанол, в чистом виде получаемый гидрогенизацией фе-
нола, находит применение в мыловаренной промышленности, так
как добавка его сильно увеличивает пенообразующую 'способ-
ность мыла.
Циклогексен, а также метил-циклогексен при пропускании
через железную трубку, нагретую до 6005, разлагаются на эти-
лен и бутадиен или метил-бутадиен (изопрен). Как известно, бу-
тадиен и его гомологи легко полимеризуются с образованием ка-
учукоподобной массы, которая по своим основным техническим
качествам вполне соответствует естественному каучуку. Таким
образом, исходя из нефтяного циклогексана, путем несложных
превращении можно получить искусственный каучук. Основная
224
реакция этого метода получения бутадиена1 * * * * ‘ может быть выра-
жена следующим уравнением:
сн2—сн2
н.с'7 \н,=
“ А
сн==сн
СН2 = СН2 (этилен)
СН2 = СН— СН = СН2 (бутадиен)
Наконец, нельзя не отметить, что оба указанных производ-
ных циклогексана—циклогексен и циклогексанол — ври окис-
лении почти количественно превращаются в адипиновую кис-
лоту; последняя же является важнейшим исходным материалом
для синтетического получения соединений ряда циклопентана.
Кроме того, эфиры адипиновой кислоты находят применение
в качестве растворителя нитроцеллулозы в процессе ее желати-
нирования.
Не подлежит сомнению, что не только в количественном, но
и в качественном, отношении нафтены являются важнейшим ком-
понентом всех трех основных видов нефтепродуктов: бензинов,
керосинов и смазочных масел.
Как составная часть бензина нафтены имеют исключительное
значение, сообщая этому нефтепродукту весьма ценные антидето'-
национные качества. Мы уже видели, (гл. IV), что. по своим
антидетонирующим свойствам нафтены идут, тотчас же за аро-
матическими углеводородами; в этом отношении они превосхо-
дят даже непредельные углеводороды. Вполне понятно поэтому,
что бензины, богатые нафтенами, например наш бакински#,
должны представлять с этой точки зрения особую .ценность, и
опытное испытание действительно вполне подтвердило их высо-
кие антидетонационные качества [58].
Для осветительного керосина нафтены являются столь же'
нормальной составной частью, как парафины. Однако, по сравне-
нию с последними нафтены имеют важное преимущество-более
низкой застываемости, так что керосины, более богатые нафте-
нами. характеризуются более. низкой температурой помутнения
(гл. V). На качество керосина, (применяемого как топливо для
двигателей внутреннего сгорания (трактора и т. и.), нафтены
должны оказывать такое же влияние, как на качество бензина.
Что касается, наконец, смазочных масел, то, как ясно из
вышеизложенного, после удаления парафинов замещенные на-
фтены различных родов являются их главной составной частью.
Особенно важное значение среди них имеют, повидимому, на-
фтены с длинной боковой цепью, и высокие качества (Минераль-
ных смазочных масел связаны, повидимому, именно с их при-
родой. '
1 Бутадиен получается также пирогенетическим разложением т^ефтй (
или ее дестиллатов, и на этом методе’ основан один из способов получений
синтетического каучука (способ Бызова). Однако выходы (ий. бутадиен-з?е
превышают при этом 10% от веса нефти.
15 Зак. 1622. Химия нефти-
225
ЛИТЕРАТУРА
Эллис К., Химия углеводородов нефти и их производных, т. I и II, пев.
с англ., ОНТИ, Москва (1936—1938).
Brooks В. Т., The Chemistry of the non-benzenoid Hydrocarbons, New-York
(1922).
Aschan, 0. Chemie der alicyklischen Verbindungen, Braunschweig (1905).
1. Ruzicka, Brugger, Pfeiffer. ScJiinz, Stoll, „Helv. chim. Acta“ 9, 499 (1926У
,Ch. Zbl.‘, 1926, II, 186.
2. Зелинский, „Ж. P. X. O.“ 41, 720 (1909).
3. .Ж. P. X. 0." 80, 69 (1898).
4. wBer.“ 80, 974 (1897).
5. „Ж. P. X. ОЛ 20, 118 (1888)
6. .Ж. P. X. O.“ 27 (2), 179 (1895); 28 (2), 125 (1896).
7. Маркоеников, „Ж. P. X. О." 80, 186 (1898); 81, 215 (1899); Наметкин,
.Ж. P. X. ОЛ 48. 1603 (1911).
8. .Lieb. Ann.- 278, 111 (1893).
9. ,Ber." 28, 780, 1022 (1895); 84, 2799 (1901).
10. .Ann. Ch. et Phys." 4 (8). 319 (1905).
11. „Ж. P. X. 0." 80, 151 (1898).
12. „Ж. P. X. ОЛ 15, 21 (1883).
13. „Ж.-Р. X. 0." 17, (2), 37 (1885).
14. „Ж. P. X 0." 81, 405 (1899).
15. „Ж. P. X. 0.Л 28, 40 (1891).
16. Зелинский и Генерозов. „ВегЛ.29, .729 (1896); 30, 1537 (1897).
17. Маркоеников, .Ж. Р. X. 0." 82,302 (1900): 85, 1023 (1903): Наметкин,
„Ж. Р. X. О." 42. 691 (1910); Зелинский, .Ж. Р. X. О.* 45, 54 (1913).
18. .Ж. Р. X. 0." 84, 917 (1902).
19. .Ж. Р. X. ОЛ 15, 237, 807 (1883).
20 Яковкин, .Ж. Р. X. ОЛ 16 (2), 293 (1884); Ижевский, Ibid. 20 (2), 116
(1888): Жуковский, Ibid. 24, 202 (1892); Маркоеников и Коновалов,
Ibid. 27 (2), 2 (1895).
21. „В er Л 28, 780 (1895).
22. Зелинский. „Ж. Р. X. ОЛ 45, 613 (1913).
23. Наметкин, ,Нефт. и сланц. хозЛ 2, № 5—8, 35 (1921)
24. Зелинский, „Вег." 56, 1718 (1923).
25. „Ж. Р. X. ОЛ 16, (2), 293 (1884).
26. „Ж. Р. X. ОЛ 18, (2), 256 (1886).
27. „Ж. Р. X. ОЛ 27. (2), 180 (1895).
28. „Ж. Р. X. О Л 19, 255 (1887); 22, 4, 118 (1890); 28, 446 (1891); 25, 389
(1893).
29. Зелинский и Реформатский, „ВегЛ 29, 214 (1896).
30. Наметкин, „Нефт. и еланц. хоз." 2, № 5—8, 35 (1921).
31. Зелинский и Глинка, „Ж. Р. X. О Л 39, 1170 (1907); Зелинский и Паппе,
Ibid. 87, 627 (1905).
32. .Вег." 56, 1720 (1923).
S3. Стародубский, „Ж. Р. X. ОЛ 22, 64 (1890).
34. „Ж. Р. X. О." 25. 382 (1893).
35. „Ж. Р. X. ОЛ 25, 385 (1898); 26, (2), 68 (1894); 80, 586 (1898).
36. Зелинский и Еазанскяй, „ВегЛ 64, 2265 (1931). ,
37. Маркоеников, „Ж. Р. X. ОЛ 84, 635 (1902): Харичков, „О составе и тех-
нических свойствах нефтей русских месторождений", стр. 189,’
Баку (1902).
38. Warren С. М„ „Ztschr. f. СЬЛ 1865, 666.
39. Young Sydney, „Chem. Soc." 78, 905 (1898); Fortey Emily., Ibid. 73, 93^
(1898); Young S. a. Fortfy E., Ibid. 75, 880 (1899).
40. Mabery Ch„ „Am. Chem. J." 28, 164 (1902); 83, 270 (1905).
41. Mabery, „Am. Chem. J." 17, 713 (1895); 18, 215 (1896); 88, 251 (1905).
42. Mabery, Am. Chem. J.„ 18. 43 (1896): 19, 419 (1897).
43. Mabery, „Am. Chem. J.“ 19, 797 (1897); Mabery a. Hudson, Ibid. 25,288-
(1901); Mabery a. Zoule., Ibid. 88, 270 (1905).
226
44. Mdbery a. Buck, „J. Am. Ch. Soc.", 22, 553 (1900); Mdbery, Ibid. 23, 264
(1901). »Ind. Eng. Chem.", 6, 101,
45. Coates a. Best. „J. Am. Ch. Soc." 25, 1153 (1903.; 27, 1317 (1905).
46. Rossini F. D., „Refiner* 16, № 11, 545 (1937).
47. White a. Rose, „Journ. of the Bureau of Standards* 18, № 6, 799 (1934)
48. Fertey, E. .J. Chem. Soc.* 78. 932 (189S).
49. Lachowicz, „Lieb. Ann." 220, 188 (1883).
50. Poni P., „Chem. Zbl.“ 1901, 1, 6Э. •
51. Kramer u. Bottcher, „Ber.“ 80, 595 (1897). Ahrens u. Riemer, „Ztschr.
angew. Chem." 20, 1557 (1907); Sachse, „Chem. Zbl.* 1903, 1, 368.
52. Cecchi-Mengarini, .Gazz. chim. Ital.“ 29, 1. 460 (1899;; Balbiano. Ibid-, 82,
1, 437 (1902); Balbiano, Zeppa, Ibid. 83, II. 42 (1903).
53. Varit-Hoff, J. К., Расположение атомов в пространстве, рус. пер. Бер-
кенгейма Б. под ред. Зелинского, Н. Д., И. (1911); Aschan О., Chemie
der alicyklischen Verbindungen (1905); Wittig, G. Stereochemie,
Leipzig (1930).
54. По Тиличееву, см. сборник „Химический состав нефтей и нефтяных про-
дуктов", гл. Ill, М. — Л. (1931); здесь же — основная литература
вопроса.
55. Литература вопроса см. гл. III. 9; также Таиьз и Putnoky, »Вег.“ 52,
1573 (1919); Danaila u. Stoenescu, .Petroleum* 28, № 4, 107 (1927);
Доладугин я Егорова, тот же сборник, что и в 54), гл. VI.
56. Химический состав нефтей и нефтяных продуктов, III, IV и VI. .Труды
Н.-И. Ин-та Грознефти," М. — Л. (1931).
57. Зелинский и Юрьев, „Изв. Акад, наук СССР," стр. 851 (1980).
58. Клигерман, „Азерб. нефт. хоз.* II, № 2—3, 72 (1931); Еланский, Ibid., П,
№ 2—3,78 (1931); Калаюпао Н., Ibid. II,. № 2—3, 85 (1931).
ГЛАВА VII
КИСЛОРОДНЫЕ СОЕДИНЕНИЯ НЕФТИ
Среди кислород-содержащих соединений нефти на первом ме-
сте должны быть поставлены органические кислоты состава
СяНа^Ог предельного характера. Это — так называемые нефтя-
ные кислоты, отделяемые от нейтральных частей нефти обработ-
кой щелочью. И по составу и по свойствам нефтяные кислоты
вполне соответствуют нафтеновым кислотам, т. е. синтетическим
кислотам, являющимся производными нафтенов. Ввиду этого
описанию нефтяных кислот должна быть предпослана краткая
характеристика кислот нафтеновых.
МЕТОДЫ ПОЛУЧЕНИЯ НАФТЕНОВЫХ КИСЛОТ
По характеру исходных материалов методы получения нафте-
новых кислот можно разделить на следующие основные группы:
1. Из соединений парафинового ряда. Эти методы получения
нафтеновых кислот чрезвычайно разнообразны. Наиболее инте-
ресными из них для химии нефти являются следующие:
а) Действие дибромидов парафинового ряда на натрий-мало-
новый (также — натрий-ацетоуксусный) зфир [ 1 ]; образующийся
при этом в первую очередь эфир дикарбоновой кислоты при его
гидролизе дает соответствующую дикарбоновую кислоту, которая,
выделяя углекислоту, превращается в одноосновную нафтеновую
кислоту. Этим методом был синтезирован целый ряд нафтеновых
кислот; так например, взаимодействием натрий-малонового эфира
с 1,4-дибром-пентаном получена метил-циклопентан-карбоновая
кислота и т. д.:
СН2— СНВг-СЩ
СНо —СН —СН3
сн2 ->
CH2—C = (C02R)9
co2r сщ—ch-ch3
CNa» =2NaBr+CH2
\ \ zC02R
C02R СН2—С<
xC0oR
СН2—СН—СН2 сн2—сн—сн3
CH2 —-СЩ
СН2—С = (СОаН), сн2—сн—со2н
228
б) Конденсация эфиров двухосновных кислот под влиянием
алкоголята натрия [2]. Образующиеся при этом эфиры цикличе-
ских ₽-кетонокислот могут быть превращены далее путем гидро-
лиза и восстановления карбонильной группы в нафтеновые ки-
слоты. Этим путем, например, эфир адипиновой кислоты может
быть превращен в циклопентанон-карбоновую кислоту и т. д.:
СНа — СЩ — COOR СН2—СН * COOR
—> | \со
СЩ—СН2—COOR сн2—сн2
2. Из соединений алициклического ряда. Из соединений с го-
товой алициклической системой нафтеновые кислоты могут быть
получены- различными методами, например через нитрилы путем
их гидролиза и т. д. Важнейшим из методов этого рода является
синтез нафтеновых кислот с помощью магний-органических сое-
динений [3]; так например, при действии углекислоты на маг-
ний-хлор-циклогексан с последующим разложением образующе-
гося комплекса водой получается циклогексан-карбоновая ки-
слота и т. д.:
ч- COs Ч- що
C6HuMgBr —> C6HuC02MgBr —> СбНпС02Н
Этим способом было приготовлено большое количество нафте-
новых кислот рядов циклопентана, циклогексана и циклогептана.
3. Из кислот ароматического ряда. Нафтеновые кислоты ряда
циклогексана давно уже,были получены гидрированием аромати-
ческих кислот. В зависимости от условий реакция здесь либо
останавливается на промежуточной стадии образования непре-
дельной циклической кислоты, либо доходит до конца, т. е. др,
стадии циклогексан-карбоновой кислоты. Так, при действии на
бензойную кислоту амальгамы натрия получается тетрагидробен-
зойная кислота, СбН9СО2Н [4], тогда как восстановление на-
трием в растворе амилового спирта приводит к гексагидро-бен-
зойной (циклогексан-карбоновой) кислоте, СвНцСОгН [5|.
Гидрогенизация бензойной кислоты и ее гомологов водородом
легко протекает даже при комнатной температуре в присутствии
таких катализаторов, как коллоидная платина или платиновая
чернь [6]. В присутствии никеля гидрогенизация ароматиче-
ского ядра протекает, как известно, лишь при 180—200° С.
В этих условиях одновременно с гидрогенизацией происходит
также отщепление углекислоты, так что, например, из бензой-
ной кислоты получается циклогексан [7]:
Ч-бн
С6НбС02Н —> сбн12 + со2
Со сложными эфирами ароматических кислот та же реакция
проходит без всяких осложнений и притом в зависимости от тем-
пературных условий обратимо, т. е. вполне аналогично гидроге-
низации ароматических углеводородов [-8]:
C6H5C00R+6Н C6HUCOOR
229
4. Из нефти и ее дестиллатов. Богатейшим источником нафте-
новых кислот являются нефть и различные ее дестиллаты. Мето-
дика выделения их из этого сырья будет рассмотрена ниже.
л
КРАТКАЯ ХАРАКТЕРИСТИКА НАФТЕНОВЫХ КИСЛОТ
Нафтеновые кислоты представляют собой труднолетучие, ча-
стью жидкие, частью кристаллические вещества, мало раствори-
мые в воде, с запахом, напоминающим соответствующие по моле-
кулярному весу кислоты жирного ряда. Низшие представители
нафтеновых кислот летучи с водяными парами. Несмотря на
свой непредельный состав, СпН2^_2 02, они подобно нафтенам
имеют ясно выраженный предельный характер и не обесцвечи-
вают хамелеона.
По своим химическим свойствам нафтеновые кислоты пред-
ставляют собой типичные карбоновые кислоты. При нейтрали-
зации они легко образуют разнообразные соли, из которых
щелочные и частью щелочно-земельные довольно хорошо раство-
римы в воде. Со спиртами нафтеновые кислоты легко этерифи-
цируются в обычных условиях с образованием сложных эфиров;
последние представляют собою жидкости с довольно неприятным
привязчивым запахом прогорклого масла. Подобно другим кар-
боновым кислотам, нафтеновые кислоты легко образуют также
соответствующие им хлорангидриды (I), ангидриды (II) и амиды
(III). Эти последние прекрасно кристаллизуются и особенно
удобны для характеристики нафтеновых кислот.
свнпсо
I. СбНпСОС1
хлорангидрид
цик л or ею аа-к&р боновой
II.
свн„со
ангидрид
ill. C«HnCON'H,
амид
кислоты
Нафтеновые кислоты, не замещенные в a-положении по от-
ношению к карбоксилу, подобно соответствующим кислотам
жирного ряда легко бронируются с образованием бром-замещен-
ных кислот:
При сухой перегонке нафтеновые кислоты разлагаются с вы-
делением углекислоты и образоваписм углеводородов и других
нейтральных соединений.
В табл. 68 приведен список простейших нафтеновых кислот
рядов циклопентана и циклогексана с указанием их констант и
температур плавления их амидов.
280
Таблица 68
Некоторые синтетические нафтеновые кислоты
Формула кислоты Наименование кислоты Тема, илжвл. Темп. киг. Уд. вес Темп, ллавл. амида
Ряд цикло Cg Hg.COg Н сн3.с5н8..со2н сн3.с5н8.согн СЩ.ед.СОгН <WCH2.CO2H Ряд ЦИКЛО свни со,н СН3. CflH10. СО2Н . <СН3.СвНЛ.СО2Н CH3.ceHlfl.c02H СН3. С6Н10. СО2Н С Н3. СаНю. СО2Н СвНц.СНо.СОзН ' Св Нп.СНа.СН2.СО3Н СН3.СвН10.СНа.СО2Н пентана Циклопентан-карбо- новая Метил-ц.-г.-карбо- новая—1, 2 Метил-ц.-п.-карбо- новая—1, 3 Метил-ц-п.-карбо- новая—1, 1 Циклопентил-уксус- ная гексана Циклогексан-карбо- новая Метил-ц.-г.-карб. (cie)—1. 2 Метил-ц.-г.-карб. (trans)—1, 2 Метил-ц.-г.-карбе- новая—1, 3 Метил-ц.-г.-карбо- новая—1, 4 Метил-ц.-г.-карбо- новая—If 1 Цпклогекспл-уксус- ная Циклогексил-пропио- новая Метил-ц.-г.-уксус- ная—1, 4 7 14 31 51 Ш 88 32 74 216 217 115 15 мм 219 228 232 237 242 245 245 234 246 277 1 — 1,051 (15°) °’957 < О') 1,006 (у) 1.сэт(^) 1,027 (т) /20\ 1,009 (-т) \ 4 / 1.007 С.996 (^) 179 123,5 150 124 144 184 153' 181 155 22) 69 172 120 162
231
ПРИРОДНЫЕ НАФТЕНОВЫЕ (НЕФТЯНЫЕ) КИСЛОТЫ
Природные нафтеновые кислоты были обнаружены в сурахан-
ской нефти Эйхлером (1874 г.) л им же впервые охарактеризо-
ваны [9]. Почти одновременно их выделили из румынской нефти
и подвергли изучению Геллъ и Мейдингер [10]. Этими же авто-
рами был установлен правильный химический состав, СПН2Л - 2 О2,
природных нафтеновых кислот и дана их химическая характе-
ристика: слабая кислотность, способность к этерификации, на-
сыщенный характер при непредельном составе и т. д.
Природные нафтеновые кислоты можно было бы обозначать
так же, как синтетические, т. е. по имени основного ядра, с ко-
торым связана карбоксильная группа. Так как, однако, у боль-
шинства природных нафтеновых кислот строение основного ядра
еще недостаточно выяснено, то при их обозначении приходится
довольствоваться ссылкой на эмпирический состав кислоты,,
иначе -говоря, указанием того нафтена, естественным продуктом
окисления которого является данная кислота или же из кото-
рого она может быть выведена замещением водорода на карбо-
ксильную группу. Необходимо ясно представлять себе различие
этих способов обозначения нафтеновых кислот; так например,,
кислота СНз • СзНеСОгН по первому способу должна быть на-
звана метил-циклопентан-карбоновой кислотой, по второму спо-
собу— гептанафтеновой кислотой, по третьему — гексанафтен-
карбоновой кислотой.
Получение и очистка. Получение вв сырой нефти или
нефтепродуктов смеси нафтеновых кислот сводится к следующим,
операциям. Исходный материал обрабатывают слабым раствором
щелочи при энергичном перемешивании и дают слоям разде-
литься. Если отстаивание происходит медленно и образуются
более или менее стойкие эмульсии, то смесь подогревают до 60—
70°. Когда нижний щелочной слой,, куда в виде солей (мыла}
переходят нафтеновые кислоты,' просветлеет, его отделяют от
углеводородного слоя и подкисляют разбавленной серной кисло-
той. Выделяется маслянистый слой темного цвета -и крайне не-
приятного запаха; это — сырые нафтеновые кислоты.
Даже в тех случаях, когда щелочной раствор нафтеновых
солей при отделении от углеводородного слоя совершенно прозра-
чен, он содержит более или менее значительное количество ней-
тральных масел. Количество последних особенно зависит от при-
роды исходного материала. Так например, нафтеновые кислоты,
выделенные из различных дестиллатов обработкой однопроцент-
ным раствором едкого натра с последующим подкислением ще-
лочного раствора минеральной кислотой, показали при анализе
[11] следующее содержание нейтральных масел (табл. 69).
Количество нейтральных масел, которые переходят из щелочного раст-
вора в сырые нафтеновые кислоты, м-ожет быть иногда значительно выше,
чем показано в табл. 69 (до 80%), особенно если выщелачивание' велось
недостаточно разбавленной щелочью. Чтобы понизить содержание этих при-
месей, рекомендуется еще раз повторить растворение смеси нафтеновых
232
жислот в слабой щелочи с последующим подкислением щелочного pacrtopa.
Хорошие результаты дает также следующий способ: щелочный раствор
.нафтеновых кислот разбавляют равным объемом спирта и. полученный
таким образом водно-
спиртовый раствор встря-
хивают несколько раз
с легким бензином; по-
следний извлекает при
этом нейтральные масла,
находившиеся в щелоч-
ном растворе. В лите-
ратуре вопроса можно
найти также ряд дру-
гих методов очистки сы-
рых нафтеновых кислот,
рекомендуемых для при-
менения в техническом
масштабе. Таковы, на-
пример, методы очистки
Таблица 69
Содержание масел в нафтеновых кислотах
Наименование исходного дестиллата Содержа- ние в °/0 нейтральн. масел
Соляровое масло легкое 9,9
• „ тяжелое 16,6
Веретенное масло 29,0
Машинное масло 48,0
с применением аналина, парафина, плавиковой кислоты (смесь плавикового
шпата с серной кислотой) и др.
Количественное определение [12]. Нафтеновые кислоты,
очищенные одним из вышеуказанных способов, содержат значи-
тельное количество примесей, среди которых преобладают веще-
ства нейтрального характера, «необмыливаемые». Количественное
определение этих примесей позволяет судить о степени чистоты
нафтеновых кислот и составляет поэтому одну из обычных за-
дач при анализе нафтеновых кислот («асидол») или их щелоч-
ных солей («мылонафт»). Определение производится следующим
образом.
Сначала определяют для анализируемой смеси нафтеновых
кислот ее кислотное число Ki, т. е. число милиграммов едкого
кали, необходимого для нейтрализации 1 г анализируемого ве-
щества1 *. Для этого точную навеску вещества (около 1.г) тит-
руют полунормальным раствором едкого калц в присутствии
фенолфталеина как индикатора. Если на нейтрализацию взятой
навески Ai пошто Bi см3 полунормальной щелочи, то Kt ана-
лизируемого вещества вычисляется по формуле:
~ 0,028 1000. Bi 28 В,
Kl~ At ~ Aj •
Затем тем же порядком определяют кислотное число чистых
нафтеновых кислот анализируемой смеси. Для этого 10 г сырых
кислот нейтрализуют в присутствии спирта и фенолфталеина
нормальным раствором едкого кали и, добавив затем спирта или
воды с таким расчетом, чтобы получился 50%-ный водно-спир-
товый раствор, трижды извлекают отделившееся, нерастворимое
в щелочи масло легким петролейным эфиром. Водно-спиртовый
раствор нагревают далее для удаления спирта на водяной бане,
после чего остаток, водный раствор калийных солей нафтеновых
1 В некоторых старых работах кислотность выражают эквивалентом
серного ангидрида (SOa) на 100 г масла. Для пересчета следует отмечать,
что 1% 80з эквивалентен 14 мг едкого кали на 1 г навески.
233
кислот, обрабатывают для выделения свободных нафтеновых
кислот слабой серной кислотой в присутствии метил-оранжа как
индикатора. Выделившиеся нафтеновые кислоты извлекают
петролейным эфиром и, дав слоям хорошо отстояться, промы-
вают эфирный раствор насыщенным раствором поваренной соли.
.Остается профильтровать эфирный раствор, отогнать петролей-
ный эфир, тщательно удалить последние следы его вместе со
следами воды (5 мин. в шкафу при 103—105°) и определить
кислотное число #2 полученных таким образом чистых нафте-
новых кислот, как это описано выше:
_ 0,028-1000-К _ 28
2 А% А%
Отношение кислотного числа сырых нафтеновых кислот Ki
к кислотному числу чистых нафтеновых кислот К% может слу-
жить мерой процентного содержания Р чистых нафтеновых кис-
лот в исследуемой смеси:
Р, = 100 S
* Л2
Если содержание нафтеновых кислот определяется в продукте, в кото-
ром кислоты находятся в виде солей, то необходимо их выделить в сво-
бодном состоянии. Для этого 100 г средней пробы вещества, например мыло-
нафта, растворяют в воде и, (профильтровав, доводят объем раствора до 1 л.
Далее берут 100 см3 этого раствора ( = 10 г мылонафта) и выделяют из
них свободные нафтеновые кислоты разбавленной серной кислотой в присут-
ствии метил-оранжа. Затем извлекают выделившиеся нафтеновые кислоты
легким петролейным эфиром, причем в раствор переходят также нейтраль-
ные масла, и ведут дальнейшую обработку эфирного раствора, как показано
выше. После высушивания взвешивают полученные сырые нафтеновые
кислоты и, умножая их вес на 10, получают процентное содержание (7
сырых нафтеновых кислот в мылонафте. Определив затем, как описано
выше, кислотные числа Ki и К? для сырых и чистых нафтеновых кислот
данного мылонафта, получают содержание чистых нафтеновых кислот -в
мылонафте по формуле:
А = С^.
Титрование с индикаторами при анализе большинства сма-
зочных масел и топлив, имеющих темную окраску, нередко дает
лишь приближенное значение кислотности при значительном
расхождении между собой параллельных опытов. Кроме того,
Описанная выше методика обладает еще одним весьма суще-
ственным недостатком: применение экстракции требует для по-
следующего отстаивания растворителя часто довольно продол-
жительного времени (сутки и более). Эти недочеты совершенно
отпадают при электрометрическом методе определения кислот-
ности. Сущность этого метода заключается в следующем: между
двумя платиноводородными электродами, опущенными в про-
дукты разной кислотности (различные pH), при замыкании цепи
возникает электрический ток вследствие разности потенциалов.
Один из продуктов является при этом контрольным и может
вовсе не содержать кислоты. В другом продукте при титрований
щелочью концентрация водородных ионов будет уменьшаться.
234
При уравнении pH в> обоих продуктах, например в момент полной
нейтрализации анализируемого продукта, разность потенциалов
становится равной нулю, и ток в цепи прекращается, что легко
регистрируется капиллярным электрометром или чувствитель-
ным нулевым гальванометром. Зная кислотность контрольного
продукта и количество щелочи, пошедшей на титрование анали-
зируемого продукта, кислотность последнего легко можно опре-
делить. Метод особенно удобен для быстрого определения кислот-
ности темнцх и мутных продуктов [13].
Распределение в нефтях и дестнллатах. Содержание нафте-
новых кислот в нефтях, вообще говоря, невелико; тем не менее,
для различных нефтей здесь наблюдаются заметные колебания.
Как общее правило, нефти нафтенового типа значительно бо-
гаче нафтеновыми кислотами, чем нефти парафиновые [13]. Так
например, богатые нафтенами нефти бакинские, грозненская бес-
парафиновая, доссорская (Эмба) содержат нафтеновых кислот
значительно больше, чем такие богатые парафинами нефти, как
челекенская, грозненская парафинистая, пенсильванская и др.
Для нефтей одного илп близких типов содержание нафтено-
вых кислот, вообще говоря, повышается с увеличением удельного
веса нефтей. Однако в связи с различием в составе углеводород-
ной части различных нефтей в отдельных случаях, естественно,
могут наблюдаться более или менее значительные уклонения от
этой правильности (табл. 70).
Таблица 70
Содержание нафтеновых кислот в бакинских нефтях
(по Гуреичу [14])
№Ks н/п Наименование нефти Уд. вес нефти Кпслотн. число в mi КОН
1 Сураканская белая . . . 0,789 0,08
2 „ красная . . 0,803 0,56
3 „ тяжелая • . 0,858 0,43
4 Раманинская 0,878 0,84
б Сабунчинская 0,880 1,46
6 Б1лаканская 0.882 1,40
7 Биби-эйбатская 0.888 1,01
8 Бинагадинская ..... 0,924 1,90
Что касается, наконец, распределения нафтеновых кислот по
различным дестиллатам одной и той же нефти, то таковое крайне
неравномерно. Это видно из данных табл. 71, в которой приво-
дится содержание чистых нафтеновых кислот в различных
погонах балаханской нефти (по Гурвичу): наиболее богаты наф-
теновыми кислотами средние ее. достиллаты — соляровый и вере-
тенный, остальные же дестиллаты, не только более легкие—бен-
зиновый и керосиновый, но и более тяжелые — машинный и ци-
линдровый, — содержат нафтеновых кислот тем меньше, чем
дальше они отстоят от солярового дестиллата.
283
Таблица 71
Содержание нафтеновых кислот в дестиллатах
№№ п/п Наименование дестиллатов Кислотное чис- ло в хг КОН Содержание нафтеновых кислот в %
1 Бензиновый . • • • Следы Следы
2 Керосиновый .... 1,26 0,5
3 Соляровый 3,92 2,2
4 Веретенный .... 2,94 1,9
5 Машинный 1,68 1,4
6 Цилиндровый . . . 0,42 0,4
Отдельные представители, их выделение и свойства. Очи-
щенные одним из вышеописанных способов нафтеновые кислоты
могут служить исходным материалом для получения химически
чистой смеси нафтеновых кислот и выделения из этой смеси от-
дельных представителей. * Задача этой высшей степени их очи-
стки заключается прежде всего в отделении от кислот веществ
фенольного и смолистого характера, которые упорно удержи-
ваются нафтеновыми кислотами при предыдущей их очистке-.
Для достижения этой цели предлагалось [15] разлагать щелоч-
ные соли нафтеновых кислот углекислотой; при этом, однако;
наряду с фенолами частично выделяются также нафтеновые
кислоты, так что метод не достигает поставленной цели. Для от-
деления смолистых веществ удобно перевести нафтеновые кис-
лоты в известковые соли и промыть последние бензином или
бензолом; выделенные из промытых таким образом солей нефтя-
ные кислоты почти не содержат смолистых веществ [16]. Но
лучшим методом получения-чистых нафтеновых кислот является
переведение очищенных кислот в сложные, метиловый или эти-
ловый, эфиры с последующим их гидролизом после тщательной
фракционировки. лучше в вакууме [17].
Получение сложного, например метилового, эфира нафтеновых кислот
производится действием на них избытка абсолютного метилового спирта в
присутствии крепкой серной кислоты или при пропускании в смесь спирта
с кислотой сухого хлористоводородного газа. По окончании реакции смесь
выливают в холодную воду, отделяют выделившийся слой сложного эфира н
тщательно промывают его на холоду сначала водой, затем—слабой щелочью-
При этом отделяются не только кислотные примеси, но, что особенно важно
в данном случае, также вещества фенольного характера. Просушенную в
перегнанную смесь сложных эфиров либо омыляют далее кипячением со
спиртовым раствором едкого кали, либо предварительно подвергают ее тща-
тельной фракционировке в вакууме для выделения возможно более узких
фракций эфиров. Эта последняя операция подобно выделению отдельных
нефтяных углеводородов из тсго или иного нефтепродукта, требует крайне,
много времени и труда: до 30 последующих фракционировок смеси эфиров
и более.
Вместо глубокой фракционировки смеси сложных эфиров
нафтеновых кислот для разделения последних можно пользо-
226
Природные нафтеновые кислоты Таблица 72
Формула кислоты Наименование кислоты Происхожд. нефти Кие лота Метил, эфир
Темп. кип. 1 Уд. вес Темп. кип. Уд. вес
Св Нц СОа П Гексанафтен- Баку 216—217° d0 = 0.971 165,5-167°,5 dte = 0,905
карбоновая Румыния 216-220° d4® = 0,951 165—166° d.*8 = 0.9С6 Ojr — 0,936
с7 н13согн Гептанафтен- Баку 287—289° d416 == 0.986 189-190°
карбоновая Румыния Галиция 284— 288° 236—288° d4® = 0,976 188—192° 185—193° а» = одюв a,.=Олп
С8Н1БСОЯН Октонафтен- Баку 251—258* do = 0,980 dfi « 0,984 211—218° mo сч co а с&сь ООО IIII11 I SS Е
карбоновая Румыния Галиция 248—252° 250—258° 210-215° 210—214°
СВНП СО2П Нононафтея- Баку 255-260° — 220—225° —
карбоновая Румыния Галпдпя 257-261° 260—26401 = 0,985 223-226° 219-225е» d4*> = 0,941 djg 0,935
СюНмСОД1 Деканафтен- Баку 267—269° dn =» 0,982 d4® = 0.988 — —
карбоновая Румыния Галиция 268—266° 255—25801 225—288° 205—21001 85 8 И II II ppp > "со'со'са > liUccos
CiiHaCOjH Эндеканафтен- Румыния 272—275° d4«o = 988 288—241°
карбоновая Галиция 268—272° — 226—230° tfjg — u,wo
все выделенные до сих пор природные нафтеновые кислоты ни
в коем случае нельзя считать за индивидуальные соединения.
За немногими исключениями, все они при дальнейшей, более
тщательной фракционировке могут быть разделены на 2—з
фракции и, таким образом, представляют собой более иди ме-
нее сложную смесь изомерных нафтеновых кислот [25 . Бли-
жайшее рассмотрение данных табл. 7? показывает, что при
хорошем соответствии температур кипения как самих кислот
различных месторождений, так и их метиловых эфиров, для
их удельных весов наблюдается известное различие’, прости-
рающееся в отдельных случаях даже на второй десятичный
знак. При имеющемся материале трудно сказать зависит ли это
от различной тщательности выделения кислот, или от различия
их химической природы в зависимости от месторождения.
Для характеристики нафтеновых кислот из различных де-
сти ллатов одной и той же нефти может служить табл. 73, в ко-
торой сведены свойства кислот, выделенных из бакинской
нефти [26].
Таблица 73-
Нафтеновые кислоты из бакинской нефти
Наименование Свойства исходи, дестиллата Свойства нафтеновых кислот
исходного - Кислотн.'
с дестиллата Уд. вес Вязкость Уд. вес Вязкость Иодн.
£ при 15° Е (30°) число при 15° Е (30°) число
1 Керосиновый . . 0,825 1,06 230 0,950 —— 2,62
2 Пиронафт . . . 0,864 1,45 186,5 0,952 8,35 —
3 Соляровый легк. 0,882 2,10 170 0,951 15,0 2 42
4 Вазелинов, масло 0,889 3,20 152 0 944 —
5 Соляров. тязгел. 0,892 3,40 136 0,942 18,90 2,50
6 Веретенный . . 0,902 7,12 108 0,936 34,76 6,17
7 Машинный . . . 0,910 16,12 87,5 0,935 47,71 7,18
8 Цилиндровый . 0,921 — 32,6 0,929 92,92 1136
Как видно из данных табл. 73, кислотные числа природных
нафтеновых кислот колеблются в весьма широких пределах, по-
степенно понижаясь с утяжелением исходного дестиллата. А так
как кислотность S находится в обратной зависимости от моле-
кулярного веса М кислоты, по формуле для одноосновной кис-
лоты М = 56100 :8, то те же данные указывают, что нафтеновые
кислоты, выделенные из различных дестиллатов, глубоко разли-
чаются между собой своим молекулярным вес’ом. Уже средний
молекулярный вес кислот, выделенных из керосинового дестил-
лата, как показывает их кислотность, равен 244, т. е. примерно
молекулярному весу пальмитиновой кислоты, С15Н31СО2Н
(М = 254). С утяжелением исходного дестиллата молекулярный
вес кислот постепенно повышается, для наиболее тяжелого де-
стиллата (цилиндрового) далеко превышая М = 1000, если
только на резкое падение кислотности здесь не оказала влияния
некоторая примесь нейтральных масел.
238
Удельный вес нафтеновых кислот, ио мере утяжеления исходного де-
стиллата заметно падает (табл. 73), т. в. подчиняется той же закономерно-
сти, как удельный вес одноосновных кислот жирного ряда. Впрочем, для
низших представителей ряда, выделенных из керосинового и легкого соля-
рового дестиллатов, это изменение мало заметно; оно начинает ясно проя-
вляться, начиная только со средних и тяжелых соляровых дестиллатов;
для более же летних кислот, выделенных с большей тщательностью, можно
наблюдать даже обратный ход изменения удельного веса (ср. табл. 73).
Аналогичные закономерности наблюдаются также для природных нафтено-
вых кислот других месторождений (Япония).
Вязкость нафтеновых кислот по- мере утяжеления исходного дестиллата
постепенно повышается, все время значительно превышая вязкость послед-
него.
Из других физических свойств природных нафтеновых кислот заслужи-
вает внимания их крайне низкая температура застывания. Тогда как неко-
торые синтетические нафтеновые кислоты ряда циклогексана имеют темпе-
ратуру плавления выше 30° (ср. табл. 66), природные нефтеновые кислоты
остаются жидкими даже при 60° ниже нуля.
Химическая характеристика. Как уже было указано, прп-
, родные нафтеновые кислоты при непредельности их состава,
СпНг^-аОг, характеризуются насыщ иными свойствами. Свой'
ства эти проявляются как в устойчивости этих кислот к хамеле-
ону, так и в неспособности их соединяться с бромом и галоидо-
водородными кислотами. Правда, как видно из табл. 73, иодное
число природных нафтеновых кислот, крайне незначительное
для низших представителей; заметно повышается с увеличением
молекулярного веса кислот. Надо думать, однако, что здесь ска-
зывается частичное разложение этих высокомолекулярных сое-
динений в той или иной стадии их получения.
Из солей нафтеновых кислот наибольший интерес представ-
ляют щелочные, особенно натронные, широко применяемые у нас
в качестве суррогата натрового мыла (см. ниже). Соли эти
легко получаются нейтрализацией нафтеновых кислот щелочью,
содой или поташом. Они представляют собою мягкие 1Жйла, легко
растворимые в воде и спирте, трудно — в легких нефтяных
дестиллатах. Однако в присутствии кислот те же соли (кислые
мыла) растворяются во всех нефтяных дестиллатах в значитель-
ном количестве.
Другие соли нафтеновых кислот легко получаются из щелочных реак-
цией обменного разложения. Наиболее характерны из них щелочно-земель-
ные соли нафтеновых кислот, представляющие собою аморфные или неясно
кристаллические осадки, трудно растворимые в воде, спирте и бензине. Ив
индивидуальных кислот некоторые из этих солей получены в хорошо обра-
зованных кристаллах.
Остальные соли нафтеновых кислот не получены в кристаллическом
состоянии. Все они — нерастворимы в воде и алкоголе, но более или менее
растворимы в эфире, бензине и других органических растворителях. Из
этих растворов они легко выделяются в виде желатинообразной массы,
частью пластической каучукоподобной (соли А), Мн), частью—-вязкой и.
клейкой (соли Сг). Некоторые из солей нафтеновых кислот, растворяясь в
органических растворителях, окрашивают их в различные характерные
цвета. Так например, соли кобальта растворяются в бензине с фиоле-
товым окрашиванием; медаые соли дают голубовато-зеленое окрашивание.
Интересно отметить, что аналогичную качественную реакцию дают также
некоторые синтетические нафтеновые кислоты, а именно кислоты ряда
циклопентана, но не циклогексана [27].
239
Из ближайших производных нафтеновых кислот, кроме солей/-
хорошо изучены их сложные эфиры, хлорангидриды и амиды. 1
Кроме метиловых эфиров, рассмотренных -выше в связи с во-
просами выделения чистых нафтеновых кислот, известны их эти-
ловые, бензиловые, глицериновые и многие другие эфиры. Осо-
бенно интересны из них глицериновые эфиры, нафтеновые ана-
логи естественных жиров. Они получаются нагреванием нафте-
новых кислот с глицерином до 250° и представляют собою более
или менее густые, вязкие масла [28].
При восстановлении метиловых эфиров нафтеновых кислот
натрием в среде абсолютного спирта (метод Буво и Блан) были
получены соответствующие первичные нафтеновые спирты [29].
Так например, эфир гептанафтен-карбоновой кислоты был пре-
вращен этим путем в гептанафтен-карбинол и т. п.:
С7Н18С00СН8 —> С7Н18СН2ОН.
Хлоргидриды нафтеновых кислот,- легко получаемые дей-.
ствием на нафтеновые кислоты трех- или пятихлористого фос-
фора, представляют собою жидкости с резким запахом, довольно
устойчивые по отношению к воде.
Амиды нафтеновых кислот легко получаются действием ам-
миака на их сложные эфиры или хлорангидриды. Они -прекрасно
кристаллизуются из воды, бензола и ацетона и применимы для
выделения чистых нафтеновых кислот из их смесей (ср. выше).
Нерастворимы в бензине. Подобно другим амидам карбоновых
кислот—устойчивы к щелочам, но легко гидролизуются под
влиянием кислот. При действии фосфорного ангидрида амиды
нафтеновых кислот превращаются по общей схеме в -соответ-
ствующие нитрилы, которые в свою очередь кипячением с раз-
бавленными минеральными кислотами могут быть обратно пре-
вращены I? исходные кислоты:
Н,0 • 4- 2HtO
—> CJE^CN —> ОД^СООН.
Большой интерес для характеристики амидов нафтеновых
кислот представляет также реакция Гоффмана — действие на
них брома в присутствии едкого натра. По общей схеме этой ре-
акции здесь получаются первичные амины с промежуточным
образованием соответствующих изоцианатов (I) или уретанов,
если вести реакцию в среде метилового спирта (II):
I- CnH2n_1C0NH2—>CnH2n_x • NCO—>
П. CnH9n_1C0NH2—>CX-1NH • C02CH3—>
Из остальных превращений нафтеновых кислот должны быть отмечены
их сульфирование, окисление и сухая перегонка.
Хотя нафтеновые кислоты легко растворяются в крепкой серной
кислоте (удельного веса 1,84) с саморааогреванием, однако сульфирование
их протекает веюьма медленно. Подогревание ускоряет реакцию; тем не ме-
нее и в этих условиях получить индивидуальные сульфонафтеновые кислоты
до сих пор не удалось [30].
Столь же устойчивыми являются нафтеновые кислоты по отношению
к окислителям. Даже такие окислители, как щело.чный раствор марганцево-
240
кислого калия и бихромат, на холоду вовсе не действуют на нафтеновые
-кислоты, при нагревании же происходит образование лишь продуктов более
ели менее глубокого их распада.
Как известно, при сухой перегонке известковых солей жирных кислот
получаются соответствующие кетоны, например из уксусной кислоты —
.ацетон и т. д. С нафтеновыми кислотами эта реакция протекает гораздо
сложнее: наряду с веществами кетонного характера здесь получается много
таза, а также жидкие углеводороды непредельного характера. Несравнимо
более интересные результаты получены при пропускании смеси нафтеновых
кислот с избытком уксусной кислоты над специально приготовленным ка-
тализатором (закись марганца) при 410—420° [31]. В этих условиях нафте-
новые кислоты удалось превратить в соответствующие метил-кетоны, на-
пример, для гептанафтен-карбоновой кислоты — по уравнению:
С7Н18 • СООН + СН3 • СООН = СО2 + ЩО + СтНи - СО • СНа.
Строение. Первые исследователи нафтеновых кислот, осно-
вываясь на некоторых их свойствах, в частности —на сравни-
тельно слабой их кислотности, полагали, что по своему строению
нафтеновые кислоты представляют собой внутренние ангидриды,
а именно лактоно-опиргы [32]. Эти представления оказались
ошибочными. Вся совокупность свойств нафтеновых кислот
с полной несомненностью свидетельствует, что соединения эти
представляют собою типичные карбоновые кислоты; слабая же
кислотность их по аналогии с высшими гомологами ряда уксус-
ной кислоты легко объясняется влиянием сравнительно высо-
кого их молекулярного веса. Впрочем, ближайшее исследование
показало, что нафтеновые кислоты не только легко разлагают
углекислые и азотистокислые соли, но в подхода
«ш:».
: условиях
даже выделяют хлористый водород из хлористого кальция.
Значительно сложнее оказался вопрос о строении радикалов,
-связанных в нафтеновых кислотах с карбокислом. Введу того
что как нафтеновые кислоты, так и нафтены при непредельности
их состава проявляют вполне насыщенный характер, само собою
напрашивалось положение об общности строения их основного
ядра. Положение это действительно подтвердилось при его про-
верке опытным 'путем: оказалось, что при восстановлении гепта-
нафтен-карбоновой (октонафтеновой) кислоты иодистым водоро-
дом в присутствии красного фосфора получается тот* самый
'октонафтен, который может быть выделен из нефти (0. Ашан
[33]). Тем самым вопрос о строении основного ядра нафтеновых
кислот сводился, очевидно, к вопросу о строении нафтенов.
В гл. VI было показано, что при всей сложности состава от-
дельных, выделенных из нефти, даже простейших моноцикличе-
ских нафтенов их основное ядро должно быть отнесено либо
к ряду циклопентана, либо к ряду циклогексана. В этом же при-
мерно направлении наметилось решение вопроса о строении ос-
новного ядра нафтеновых кислот. Важнейшими моментами
в этой, пока еще не вполне' законченной проблеме являются сле-
дующие.
Маркоеников показал [34], что амид гексанафтен-карбоновой
кислоты превращается с помощью реакции Гоффмана (ср. вьапе)
в ямин состава СвНиКНа, оказавшийся тождественным с амином
16 Зак. 1622. Химия нефти.
241
(II) из вторичного нитросоединения (I), полученного нитрова-
нием метил-циклопентана. Тем самым не только было устано-
влено, что я,мия гексанафтен-карбоновой кислоты представляет
собою производное кислоты 1,2-метил-циклопентан-карбоновой
(III), но вместе с тем на указанном примере было доказано и
более общее положение, что в состав основного ядра нафтеновых
кислот может входить пятичленный цикл:
I CHN02 II. chnh2 III. CHCONH» а
н2сх рнещ Н2С снсн3 Н2С снсн. _ 1 1 8
Н2С—СН2 -> 1 1 н26—сн» Н2С—сн2
Зелинский [35] подверг некоторые тщательно выделенные из
бакинской нефти нафтеновые кислоты, а именно гепта-, окто-;
ноно- и деканафтен-карбоновые, следующему ряду превращений:
метиловые эфиры этих кислот были восстановлены по Буво и
Блану (ср. выше) в соответствующие карбинолы; последние же
через галоидопроизводные (иодюры) были превращены в угле-
водороды (нафтены) по следующей общей схеме:
Для выяснения химической природы основного ядра этих
углеводородов, очевидно тождественного с ядром исходных нафл
теновых кислот, углеводороды были подвергнуты далее дегидро
генизационному катализу '(см. гл. III). При этом оказалось, что
константы их почти не изменяются и водорода при катализе
почти не выделяется. Отсюда следует, что основное ядро главной
части нафтеновых кислот во всяком случае не является гидро-
ароматическим.
Результаты, полученные путем дегидрогенизационного катализа, нахо-
дятся в полном соответствии с более ранними данными других исследовав
телей (0. Ашан, Марковников), которые пытались установить’ природу
основного ядра нафтеновых кислот путем сопоставления свойств некоторых
из них* со свойствами соответствующих по составу гидроароматичесиц
кислот; полученных гидрогенизацией одноосновных кислот ароматического
ряда (бензойной, толуиловой и др.). Сопоставление это не дало положи-
тельных результатов, так что вопрос о- нахождении в нефти кислот гидро-
ароматического ряда должен был остаться открытым.
В последнее время [36] в вопросе о строении природных на:
фтеновых кислот сделан новый важный шаг вперед
(Ю. ф-Браун).
На ряде жирных и нафтеновых кислот известного строения
было показано, что их монозамещенные ямищ относятся к ня*
тихлористому фосфору различно в зависимости от строения
кислоты, а именно: амиды кислот общей формулу
R' • СН2 • СОгН могут быть превращены этим путем в дигало*
вдозамещенные амиды состава R' • О(С12) • CONHR; амтпты кие*
лот общей формулы (R')s: СН • СОзН превращаются тем же ено*
242
собой в моногалоидозамещенные амиды состава (R')*:C-C1-
• CONHR; наконец амиды кислот обшей формулы (R')i.:C.
- СО2Н при действии пятихлористого фосфора вовсе не дают
галоидозамещегпщх амидов. Схематическое действие пятихлори-
стого фосфора на монозамещенные кислотные амиды (иначе на-
зываемые «имидами») с различным, т. е. первичным (I), вторич-
ным (II) или третичным (III) положением карбоксильных групп
можно выразить следующим образом:
4-pcis
I. R' • СН, • СО • NHR —> R' • ССЦ СО • NHR;
4- РС1В
IL R' 2:CH-C0-NHR —-> ! R'|2: СС1 • СО • NHR;
III. R' 3: С • СО • NHR не реагирует с РС1Б.
Применение этой методики (получившей наименование имид-
хлоридного метода) к различным нафтеновым кислотам, выде-
ленным из калифорнийской, румынской, галицийской и северо-
германской нефтей, привело к одному и тому же результату: ока-
залось, что в основном природные нафтеновые кислоты до Си
построены по типу > СН • СН2. СОгН, т. е. карбоксиль-
ная группа в них отделена от алициклического радикала одной,
а в некоторых случаях, возможно, и несколькими группами
СН2 Ч Напротив, кислоты типа СпН2п > СН • СО±Н, в которых
карбоксильная группа связана непосредственно с алицикличе-
ским радикалом, подобно, например, рассмотренной выше 1,2-ме-
тил-циклопентан-карбоновой кислоте, встречаются среди ’ при-
родных нафтеновых кислот несравнимо реже.
Выявление природы алициклического радикала нафтеновых
кислот удобнее всего производить путем их превращения в со-
ответствующие кетоны с последующей характеристикой послед-
них. Так например, для кислот с первичным положением кар-
боксильной группы переход от кислоты к кетону может быть со-
вершен по следующим схемам:
+ NH,
I. R > СН • СН, • СОоН —* R > СН • СН, • CONH2;
+ ВгОК
II. R > СН • СН, • CONHg —> R>CH-CH2-NH2;
Щ. R > СН • СЩ • N (CH9)S ОН —> R > С = СНа+ ЗДНДгЬЩО;
IV. R > С = СН,—-*R >СО.
Переход от кислоты к амину можно осуществить не только
методом Гофмана, т. е. окислением амида кислоты бромновати-
стой щелочью (I—II), но и действием на нафтеновую кислоту
азотистоводородной кислоты по Шмидту. Следующий переход от
амина к непредельному углеводороду надежнее всего осуще-
ствляется методом исчерпывающего метилирования (III). Нако-
нец, превращение углеводорода в кетон (IV) достигается озони-
рованием непредельного углеводорода; характеристика же ке-
1 Ср., например, в табл. 6S ’кислоты: циклопентан- или циклогексан-
уксусная, циклогексан-пропионовая и т. и.
16* 24»
тона — путем получения его семикарбазона с последующим опре-
делением его температуры плавления, кристаллической форма
и т. п., а также путем окисления кетона или его производных.
В качестве примера применения. данного метода может слу-
жить кислота С10Н18О2 из румынской нефти, превращенная до
указанным схемам в кетон С?Н14>С0 [36]. То обстоятельство-,
что этот кетон конденсировался с двумя частицами нитробен-
зальдегида, привело к выводу, что кетон содержит группировку
СН2 • СО •. СНг, а изучение его очищенного семикарбазона по-
зволило установить его строение как 1,1, 2,4-триметил-цикло-
пентанона. Наконец, исходя из этого кетона, действием цинка и
бромуксусной кислоты, т. е. через соответствующую оксикислоту
путем ее дальнейшей переработки, был проведен обратный синте-
тический переход от кетона к исходной нафтеновой кислоте и,
таким образом, в итоге осуществлен замкнутый цикл превраще-
ний по схеме от кислоты к кетону и обратно:
Н8С • НС—СН2
Из других методов, применявшихся для исследования струк-
туры нафтеновых кислот, отметим действие хлористого хро-
мила [38J. Образование при действии этого реагента на нафте-
новые кислоты кетонокислот также может служить доказатель-
ством, что карбоксильная группа не связана в данном случае
непосредственно с алициклическим радикалом.
Другой не менее важный результат новейших исследований
нафтеновых кислот заключается в следующем. Оказалось, что
общий состав СяН2п-2О2, поводимому, относится только к при-
родным нафтеновым кислотам, до С12; в более высокомолекуляр-
ных нафтеновых кислотах, начиная с С13, преобладающее значе-
ние приобретают уже кислоты, более бедные водородом, например
состава СпНгп^Ог и т. д., т. е. принадлежащие к би- и полици-
клическим рядам. Присутствие кислот этого рода обнаружено во
всех упомянутых выше нефтях, кроме галицийской, и в полной
мере соответствует 'Присутствию в высших фракциях нефтей би-
и, вероятно, полициклических нафтенов.
Наконец, в низших фракциях кислот из тех же нефтей, вклю-
чая сюда также богатую парафинами галицийскую нефть, уда-
лось доказать присутствие предельных кислот жирного ряда со-
става СяНгпОг. Таковыми, например, из румынской нефти
являютця, невидимому, кислоты диэтил-пропионовая (I) или ме-
тил-лролил-црепионовая (II), а, возможно, и некоторые другие:
С2НбХ СН8 ч
I. ^СН-СЩ-СОаН и II. >СН • СН2 • С02Н.
ОД/ ед/
244
Имеются указания на присутствие жирных кислот'также и;
в нашей кавказской нефти [37].
Интересные данные были получены при сопоставлении нафтеновых кис-
лот из сырой нефти и из нефтяных дестиллатов. Оказалось, что все разли-
чие между нефтяными кислотами этих двух различных способов получения»
заключается в отсутствии среди кислот, выделенных непосредственно из
сырой нефти, наиболее низкокипящих представителей этих кислот, во всем
же остальном наблюдалось полное тождество. Таким образом можно думать,
что эти низшие нефтяные кислоты появляются в дестиллатах в результате
какого-то более глубокого окислительного процесса, происходящего при пе-
регонке нефти.
Подведем итоги всему, что известно о строении природных,
нафтеновых кислот:
1. Природные нафтеновые кислоты являются типичными кар-
боновыми кислотами; основное ядро их принадлежит к одному
или нескольким нафтеновым рядам.
2. Кислоты гидроароматического ряда во всяком случае не
составляют главной массы природных нафтеновых кислот, и если
входят в состав последних, то лишь в незначительном коли-
честве.
3. Несомненно установленной является принадлежность не-
которых природных нафтеновых кислот к кислотам ряда цикло-
пентана, которые, повидимому, и составляют главную массу на-
фтеновых кислот. !
4. Присутствие в нефти кислот других нафтеновых рядов,
как-то кислот .ряда циклобутана и циклогептана, а также ряда,
циклогексана, замещенных в положении 1,1, до настоящего вре-
мени является не доказанным.
5. По своему строению некоторые из природных нафтеновых
кислот несомненно относятся к типу СпНгп>СН (СН2)ГСО«Н,
где х равно или больше единицы.
6. Среди наиболее низкокипящих нафтеновых кислот, вы-
деленных из нефтяных дестиллатов (но не из сырой нефти), при-
сутствуют кислоты Ж1грного ряда состава СпН2лО?.
7. Высокомолекулярные нафтеновые кислоты, начиная с Cis,
принадлежат, повидимому, преимущественно к соединениям^
у которых их основное ядро является би- или полициклическим;
более определенных данных о их химической природе пока не
имеется.
ДРУГИЕ КИСЛОРОДНЫЕ СОЕДИНЕНИЯ НЕФТИ
Кроме нафтеновых кислот, в состав нефти входят некоторые-
другие кислородные соединения, из которых наиболее изучен-
ными являются фенолы.
При обработке сырых дестиллатов или нефтей щелочью со-
держащиеся в них фенолы переходят в щелочной раствор вместе*
с нафтеновыми кислотами и выделяются вместе с последними
при подкислении щелочного раствора. Присутствие в сырых на-
фтеновых кислотах фенолов может быть легко обнаружено при
245-
помощи продиазотированяой сульфаниловой кислоты: полу-
чается интенсивное красное окрашивание вследствие ооразования
азокраски (оксиазоооединения).
Для отделения фенолов от нафтеновых кислот проще всего
после перегонки для освобождения от смолистых вещ ств с пе-
регретым паром подвергнуть перегон обработке 5—6%-ным рас-
твором соды; кислоты переходят при этом в раствор в виде со-
лей, тогда как фенолы с примесью углеводородов, всегда содер-
жащихся в сырых нафтеновых кислотах, выделяются в виде
маслянистого слоя. Так как часть фенолов остается в содовом
растворе, то для полного отделения фенолов необходимо экстра-
гирование раствора эфиром. Получив, таким образом, фенолы
•с примесью углеводородов, обрабатывают эту смесь щелочью, от-
деляют углеводородную часть и выделяют фенолы из щелочного
раствора при помощи минеральной кислоты. При всей простоте
этого метода он. невидимому, страдает одним весьма, существен-
ным недостатком: надо думать, что при помощи содового рас-
твора количественйое отделение нафтеновых кислот едва ли воз-
можно, так как часть их благодаря гидролизу всегда будет оста-
ваться в смеси с фенолами.
Другой метод отделения фенолов от нафтеновых кислот за-
ключается в следующем. К смеси кислот и фенолов прибавляют
•метиловый или винный спирт и в присутствии хлористого водо-
рода или серной кислоты превращают кислоты в «сложные эфиры.
Затем выливают смесь в холодную воду и, отделив водный слой,
извлекают фенолы слабой щелочью на холоду, чтобы предупре-
дить гидролитическое расщепление эфиров.
Сведения о содержании фенолов в нефти крайне разноречивы.
Возможно, что до известной степени это зависит от недостаточно
точной методики их определения, но несомненно также, что боль-
шую роль здесь играют индивидуальные особенности различных
нефтей. Так например, два исследованных в последнее время
образца сырых нафтеновых кислот из галицийских нефтей пока-
зали: один (месторождение Борислав) содержание фенолов около
30%, тогда как другой (месторождение Битков) — только 3.1%.
Оба определения произведены одним и тем же (содовым) мето-
дом [39]. Содержание фенолов в бакинской нефти, невидимому,
ничтожно: так, сырые нафтеновые кислоты из бакинского керо-
синового дестиллата содержали фенолов менее 0,2% [30].
Химическая природа фенолов нефти начинает получать в по-
следнее время разъяснение.- Так например, после тщательной
•фракционировки фенолов из богатой' ими бориславской нефти
(см. выше) в ней обнаружены и охарактеризованы следующие
•отдельные представители этого ряда [39]: все три крезола (орто-,
мета-, и пара-), два ксиленола (1,3,5-, и 1,3,4-) и ₽-нафтол; веро-
ятно также присутствие третьего ксиленола (1,2,4-) и фенола
с тремя метильными группами. Ни простейшего фенола (карбо-
ловой кислоты), ни фенолов с длинными боковыми цепямивдан-
ной нефти не было найдено. Интересно также, что в ней не уда-
лось обнаружить соединений с метоксильными группами типа
246-
гваякола, которые являются обычной составной частью древес-
ного, например букового, а также буроугольного дегтя.
Кроме кислот и фенолов, в нефти находятся также какие-то эфирообраз-
ные соединения, способные обмыливаться при нагревании со спиртовой
щелочью. Их присутствие было обнаружено на основе заметного превыше-
ния коэфициента обмыливания некоторых нефтей по сравнению с их кислот-»
ностью, причем разность (эфирное число) для тяжелых нефтей может до-
стигать здесь нескольких единиц (в миллиграммах КОН на 1 а нефти).
Химическая природа ц состав этих эфирообразных соединений ближе пока
не изучены. По мнению некоторых авторов [40] они напоминают собою те
воскообразные соединения, которые встречаются в буром угле и по своему
происхождению могут быть связаны с воскообразными веществами некото-
рых водорослей.
В заключение следует отметить, что, кроме вышепойменовалт-
ных кислородных соединений нефти, в продуктах ее перегонки
могут быть обнаружены и другие, образующиеся в результате
вторичных процессов распада и окисления различных ее компо-
нентов. Так например, вода после взбалтывания с легкими пого-
нами пенсильванской нефти показала заметное содержание
уксусного альдегида [41 j и т. п.
ПРИМЕНЕНИЕ КИСЛОРОДНЫХ СОЕДИНЕНИЙ НЕФТИ
Из кислородных соединений нефти, рассмотренных в настоя-
щей главе, до настоящего времени нашли специальное практиче-
ское применение только нефтяные кислоты. -Область практиче-
ского их применения довольно разнообразна. Главнейшим видом
использования 'нафтеновых кислот является применение их
в мыловарении, где они с успехом частично замещают жирные-
кислоты — пальмитиновую и стеариновую. Применение нафтено-
вых кислот для мыловарения началось еще до последней войны,
но особенно расширилось ввиду недостатка жиров в послевоен-
ное время. Для получения нафтенового мыла щелочные отбросы
после очистки соответствующих- дестиллатов, обычно керосино-
вых, упариваются и для выделения мыла подвергаются искус-
. ственному охлаждению или обычному высаливанию поваренной
солью.
Натровое мыло из нафтеновых кислот, выпускаемое в настоя-
щее время под наименованием «мылонафт» (до войны—«мы-
лоин», «суррогат» и др.), обладает хорошей моющей способно-
стью и дезинфекционными свойствами. Оно имеет, однако, недо-
статочно твердую консистенцию и неприятный запах, особенно
препятствующий его широкому применению. Для устранения
этого основного недостатка нафтенового мыла было предложено
много способов, как-то: обработка нафтеновых кислот перегре-
тым паром, сукновальной глиной, формалином, различными
окислителями и т. д.; тем не менее, проблема дезодорации нафте-
новых кислот и получаемого из них мыла пока еще не может
считаться решенной полностью.
Так как мылонафт содержит значительные количества воды,
то в целях облегчения транспорта наши нефтеперегонные заводы
частично перешли к выработке свободных нафтеновых кислот,
247
«асидола», получаемого подкислением щелочного отброса. Аси-
дол, являясь сырьем для мыловаренного производства, имеет и
самостоятельные области применения, например при валянии
шерсти. Кроме того, свободные нафтеновые кислоты были реко-
мендованы в качестве дезинфицирующего и антисептического
средства, в частности как материал для пропитки железнодо-
рожных шпал, предохраняющий их от гниения, как растворитель
для некоторых смол и для регенерации каучука из старых вул-
канизированных его изделий и т. д.
Кроме щелочных солей нафтеновых кислот, практический интерес пред-
ставляют и некоторые другие их соли, по разнообразному применению ко-
торых имеется обширная, главным образом патентная литература. Так на-
лример, большое значение имеют известковые соли нафтеновых кислот, на-
.шедшие широкое применение как прекрасный материал для получения кон-
еистентных мазей. Эти же соли рекомендуется добавлять к некоторым расти-
тельным маслам (кунжутное и др.) для повышения вязкости их в смеси
с минеральными маслами. Многочисленные патенты заявлены также по
.вопросам применения некоторых солей нафтеновых кислот, главным обра-
зом алюминия и хрома, для получения сиккативов, пластических и изоля-
ционных масс, линолеума и т. д.
Производство мылонафта и- асидола сосредоточено главным
•образом в СССР. Главным производителем их являются Азнеф-
тезаводы. Вся наша продукция мылонафта потребляется исклю-
чительно внутри страны.
литература
Aschan О., ^Chemie der alicyklischen Verbindungen (1905).
ЛТершевский H., Нефтяные кислоты и их применение, пер. о франц, пол пел
Сурабекова Г. С. (1915).
JBudowski j.. Die Naphtehsauren (1922).
JPaphtali M„ Chemie, Technologie und Analyse der Naphten-SSuren (1927).
JTолъдберг Д. О., Нафтеновые кислоты, их получение и применение, ОНТИ(1932).
1. Perkin jun., „Вег/ 17, 54 (1884); 2L 739 (1888) и др.
2. Dieckmann, „Lieb. Ann.* 817, 27 (ПЮ1).
3. Зелинский, „Вег/ 85, 2687. 4415 (1902); 88, 208 (1903); 41, 2627 (1908) и др.
4. Aschan О., .Lieb- Ann/ 271. 231 (1892).
5. Морковников, ,Вег/ 25, 370, 3355 (1892) и др.
6. Wills tatter u. Mayer, „Вег/ 41, 1475 (1908).
7. Sabatier et Murat, „С. R/ 154, 923 (1912).
g. Sabatier, Senderens et Murat, „С. R.“ 154, 924 (1912); 158, 424, 751 (1913);
Зелинский и Уклонскал „Вег/ 45, 3677 (1912).
9. .Bull. Soc. Natur. Moscou", 1874, 46, 274.
10. „Вег/ 7, 1216 (1874); 10, 850 (1877).
11. Schmitz. „Mat. grasses", 1912, 250.
12. Cm. OCT № 5- Методы испытаний нефтепродуктов, 2-е изд. М. (1930);
Добрянский А. Ф., Анализ нефтяных продуктов, М.-Л. (1925); Гольде Д,
Жиры и масла, т. I, Исследование нефтяных продуктов, пер. под
ред. Добрянскою А. Ф., Л. (1931).
13. Павловский М. Т., „Нефт- хоз/ 28, № 3, 75 (1935).
14. Гурвич „Научные основы", 2-е изд., стр. 111 и 112 (1925).
15. Марковников и Оглоблин, „Ж. Р. X. О/ 15, 345 (1883).
16. Weiwart, „Seifensieder-Ztg." 1918, 367.
17. Эйхлер (1. с.) и последующие исследователи.
18. Годаскин и Завершинская, „Ж- Р. X. О/ 45, 377 (1913).
19. Харичков, Холодная фракционировка нефти (1903).
•20. Гурвич, Научные основы, 2-е изд. стр. 117 (1925).
248
21. См. гл. образом работы Ascharia О., Марковникова, Зелинского и Пюхаля.
22. Frangopol, Диссертация, Мюнхен (1910).
23. Kozicki und Pilot, .Petroleum" 11, 310 (1915).
24. Tanaka a. Hagai, ,J. Fakulty Eng.“, Токио (1923—1925),
25. Ср. Зелинский, „Вег.* 67, 46 (1924).
26. По данным Пюхаля, .Chem. Revue* 21, 128 (1914); .Chem. Zbl.“ 1914.
И, 273.
27. О солях нафтеновых кислот, см. Харичков, .Ж. Р. X. О.“ 29, 691 (1897);
.Chem. Ztg." 84, 479 (1910); Пюхаля, „Chem. Ztg * 38, 869 (1912);
Тютюнников, Seifeneileder-Ztg." 1923, 591 и др.
28. Шершевский, „Seifensieder-Ztg." 1911, 852; Харичков, .Petroleum*,
102 (1907); Wischin, 8, 1066 (1907).
29. Зелинский, .Вег." 58, 47 (1925).
30. Гурвич, Научные основы, 2-е изд. (1925).
31. Зелинский и Ряхина, „Вег." 57, 1932 (1924).
32. Hell и Meidinger, 1. с.; те же воззрения развивались некоторыми nos-,
днейшини исследователями, как-то Залозецким и Харичковим.
33. „Вег." 28. 867 (1890); 24, 2710 (1891).
34. .Ж. Р. X. О.“ 81. 214 (1899).
35. „Вег." 57, 42. 51 (1924).
36. „Lett) Ann." 490. 100 (1931); „Вег.“ 86, 1499 (1933); 87, 218 (1934).
37. Доклады Академии наук СССР, Серия А, 1930, 382; ср. также более
ранние указания Марковникова и Оглоблина и других исследова-
телей кавказской нефти.
38. Тютюнников, „Нефт. хоз “ 10, № 6, 804 (1926).
39. Holzmann и Pilot, „Btennst-Ch." 11; 409 (1930).
40. Kramer, „Chem, Ztg.“ 1907, 676.
41. Robinson, „Ch. Zbl." 1899, 1,1065.
ГЛАВА VIII
СЕРНИСТЫЕ СОЕДИНЕНИЯ НЕФТИ
Как было указано в гл. I, сера является одной из постоян-
ных составных частей нефти. Иногда она находится в ней в сво-
бодном состоянии или в виде сероводорода, но обыкновенно—1
в виде сернистых органических соединений, различающихся по
своему составу и свойствам, но характеризующихся одним общим
для всех них качеством — крайне неприятным запахом. Впро-
чем, характер этого запаха — довольно разнообразен в зависи-
мости от химической природы сернистых соединений.
КРАТКИЙ ОЧЕРК СЕРНИСТЫХ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ [1]
Как известно, сера — ближайший аналог кислорода, а серо-
водород— аналог воды. В -связи с этим существует ряд серни-
стых органических соединений, которые являются полными ана-
логами соответствующих кислородных соединений. Таковы, на-
пример, важнейшие из сернистых соединений, представляющие
особый интерес для химии нефти: R • SH, меркаптаны, аналоги
спиртов, В • ОН; К • S • R, сульфиды, аналоги простых эфиров,
В . О - R и т. д.
Меркаптаны, В * SH. Эти -простейшие сернистые органиче-
ские соединения легко получаются действием кислых сернистых
щелочей (сульфгидратов) на органические галоидопроизводные;
так например:
CHSI + KSH = KI + СН3 - SH.
В ароматическом ряду, кроме меркаптанов, известны также
тиофенолы с группой SH в бензольном ядре.
Меркаптаны — легко летучие жидкости с чрезвычайно силь-
ным, отвратительным запахом. Так например, метил-меркаптан
кипит уже при 20е и может быть обнаружен по запаху в коли-
честве в 250 раз меньшем, чем натрий при помощи спектраль-
ного анализа. Не растворимы в воде, но легко растворяются
в органических растворителях. Подобно сероводороду обнаружи-
вают слабокислотные свойства и, растворяясь в щелочах, дают
металлические производные, меркаптиды, например СНз • SNa
и т. -п. Легко образуются также меркаптиды тяжелых металлов,
например свинца и особенно ртути. Минеральные кислоты разру-
шают меркаптиды и выделяют обратно свободные меркаптаны.
250
Сульфиды {тиоэфиры), R S • К. Сульфиды получаются дей-
ствием галоидных соединений на меркаптиды (I) или средние*
сернистые щелочи (II):
I. CH8I-CH3SK= KI + CHS - S • CHg;
II. 2CHSI -h K2S = 2KI + CH3 • S • CH*
Это — нейтральные, нерастворимые в воде вещества, кипя-
щие выше, чем меркаптаны. При действии окислителей (азотная
кислота, хамелеон) превращаются в соединения, в которых сера
проявляет свою высшую валентность, а именно:
сульфиды сульфоксиды сульфоны
Сульфоксиды — бесцветные вещества, без запаха; проявляют
свойства слабых оснований. Сульфоны — кристаллические веще-
ства, также без запаха, отличающиеся чрезвычайно большой
устойчивостью не только к окислителям, но и к восстановителям.
Дисульфиды, R • S • S - R. Отличаются от сульфидов на один'
атом серы. Образуются действием иода на меркаптиды (I) или
окислением меркаптанов (II):
I. 2CHsSNa -4-21-4- 2NaI + CHS • S., S CH8;
IL 2CHsSH + 0 = H20 + CHS • S • S CH8.
Дисульфиды представляют собою тяжелые жидкости непри-
ятного запаха. При восстановлении цинковой пылью в кислой
среде превращаются в меркаптаны, а действием окислителей —
в сульфокислоты. <
Сульфокислоты, R SOsH. Представляют собою высшую степень окисле-
ния сернистых органических соединений. Особое значение сульфокислоты,
имеют* в ароматическом ряду, где они получаются действием серной кис-
лоты на ароматические углеводороды (см. гл. IVy Сульфокислоты жирного
ряда проще всего могут быть получены действием галоидных соединений
на соли сернистой кислоты:
CHgl —|- SOgKao = Nal f- CHgSOgNaj
CH3SOsNa 4- HCI = NaCI + CH8SOSH.
Сульфокислоты — кристаллические вещества, легко растворимые в воде.
Представляют собою сильные кислоты, аналогичные карбоновым кислотам и,
как таковые, образуют соли, хлор-ангидриды, амиды и другие типичные
производные кислот.
Тиофены и тиофаны. Эти сернистые соединения принадлежат
к группе циклических соединений; в них атом серы принимает;
участие в образовании пятичленного цикла:
тиофен
н2с—сн2
тиофаи
25h
Тиофен, представляющий .собою легко летучую, бесцветную
жидкость (температура кипения 84°), является одной из состав-
ных частей каменноугольного дегтя и в количестве до 0,5 %- со-
держится в неочищенном каменноугольном бензоле. Входит
также в состав сланцевого дегтя, а в небольшом количестве со-
держится в буроугольном дегте и некоторых нефтях. Синтетиче-
ски может быть получен различными способами, например дей-
ствием серы или сероводорода на ацетилен и т. д. По своим
свойствами тиофен очень напоминают бензол: способен нитро-
ваться, сульфурироваться и т. д. На легкости взаимодействия
'тиофена с крепкой серной кислотой основано отделение его из
растворов в ароматических и других углеводородах. Присутствие
►серы в тиофене выражается способностью его к реакции с ртут-
ными солями (меркурирование); так например, с уксуснокислой
ртутью получается смешанное металло-органическое соединение,
из которого кислоты легко выделяют обратно тиофен. Этой реак-
цией удобно пользоваться для количественного отделения тио-
/фена:
C4H4S + (CHsC00)2Hg = СЩСООН+С4 H4S • Hg - 00С • СН3;
C4H8SHgOOC - CH8+2HCI = C4H4S + HgCI24-CH8C00H.
Тиофен способен давать ряд цветных реакций; особенно ха-
'рактсрна реакция тиофена с изатином (производное индиго) и
* серной кислотой: получается интенсивное синее окрашивание
(индофениновая реакция).
Подобно тому как к бензолу примыкает ряд его гомологов
(толуол, ксилол и т. д.), так же точно тиофен является лить
первым членом своего гомологического ряда. Гомологи тиофена
.(тиотолен и т. д.) встречаются там же, где тиофен, и по своим
-свойствам вполне аналогичны первому члену ряда.
Тиофаны представляют собою тетрагидротиофцны, т. е. отно-
сятся к тиофенам так же, как, например, циклогексан к бензолу.
* Они встречаются в сланцевом дегте и несомненно входят в состав
некоторых нефтей. Синтетическое получение тиофанов сводится
к действию сернистых щелочей на дибромиды с открытой цепью
и атомами брома в положении 1,4. Так например, 1,4-дибром-бу-
тан при действии сернистого калия дает простейший тиофан
И Т‘ д‘ СН2—СН.,Вг СН, —СН.Х
| +К28 = 2КВг4- | ‘ >S.
СН2—СЩВг СЩ —«Н/
Тиофаны- — бесцветные жидкости с крайне неприятным запа-
хом, перегоняющиеся без разложения; нерастворимы в воде,
легко растворяются в обычных органических растворителях. По
своим химическим свойствам -напоминают сульфиды жирного
ряда (см. выше). Подобно последним они при действии окисли-
телей превращаются в соответствующие сульфокиси и сульфоны.
Отличаются чрезвычайно большой устойчивостью, особенно по
отношению к щелочам и щелочным металлам (могут пере-
гоняться над натрием).
'252
КАЧЕСТВЕННОЕ И КОЛИЧЕСТВЕННОЕ О ПРЕДЕЛЕНИЕ СЕРЫ
В НЕФТЯХ И НЕФТЕПРОДУКТАХ {>]
Фпг. 32. Ламповый при-
бор.
Качественное открытие серы может представлять практиче-
ский интерес лищь в тех случаях, когда содержание ее в нефти
настолько невелико, что обычный признак сернистых соедине-
ний — их неприятный запах чувствуется слабо. В таких случаях
высушенную нефть или нефтепродукт нагревают некоторое время
с металлическим калием1. Затем по охлаждении осторожно до-
бавляют воду и открывают в водном слое сероводород при по-
мощи нитропруссидного натрия (красно-фиолетовое окрашива-
ние). Другое испытание (на активную серу) производится сле-
дующим образом. Пробу масла (100 е) нагревают 12 час. при 85°
в присутствии медной, отполированной пластинки. Если на по-
следней образуется черный осадок, его
растворяют в азотной кислоте и и раство-
ром проводят обычным путем испытание
на серную кислоту. Реактив — хлористый
барий.
Количественное определение серы в
нефтях и нефтепродуктах можно произ-
водить различными способами. Наиболее
общим методом является сожжение ана-
лизируемого вещества в калориметриче-
ской бомбе. Сера получается при этом
в виде частью серной кислоты, частью
сернистого газа, который в случае надоб-
ности может быть дополнительно пре-
вращен в серную кислоту путем окисле-
ния, например, бромноватистой щелочью
для последующего весового определения
в форме сернокислого бария. Неудобство
этого метода, помимо необходимости при-
менения специальной, дорогой аппара-
туры, заключается в крайней ограничен-
ности размеров навески (не более 0,7 г);
при небольшом же содержании серы малые навески могут дать
в процентном отношении значительные ошибки.
Прекрасные результаты, особенно по отношению к легким
нефтям и нефтепродуктам (бензин, керосин, легкие масла), дает
так называемый, «ламповый метод». Сущность этого метода за-
ключается в следующем. Навеска анализируемого нефтепродукта
сжигается в небольшой лампочке а под ламповым стеклом б,
соединенным с поглотительным прибором в, который в свою
очередь соединен с водоструйным насосом (фиг. 32). Поглоти-
тельный прибор заполнен стеклянными бусами, которые смачи-
ваются точно отмеренным объемом титрованного раствора соды.
При надлежащей скорости просасывания через поглотительный
* Вместо калия можно применять также натрий, магний (стружки) и
цинковую пыль.
253
прибор поглощение сернистого газа происходит полностью, и
лишь небольшая часть е о задерживается в отпотевшем лампо-
вом стекле; по окончании сожжения она должна быть присоеди-
нена к содовому раствору.
Когда сожжение законченох, отпггровывают невошедшую'
в реакцию избыточную соду и по количеству израсходованной
соды судят о содержании серы в анализированном нефтепро-
дукте. Ввиду необходимости вести работу в очень разбавленных
растворах (промывные воды) применение обычного объемно-ин-
дикаторного метода дает в данном случае иногда недостаточно
отчетливые результаты; предпочтительнее поэтому пользоваться
здесь методами электрометрического объемного анализа. Эту же
методику удобно комбинировать с методом калориметрической
бомбы, пропуская газообразные продукты сгорания через по-
глотительный прибор в и не изменяя последующих операций.
Менее надежные, но во многих случаях вполне приемлемые результаты
получаются при определении серы методами прокаливания навески нефте-
продукта с веществами, связывающими серу в той или иной форме. Таков,
например, широко применяемый метод Эшка, заключающийся в следующем.
Точную навеску нефтепродукта (около 1 г) тщательно перемешивают в.
тигле со смесью Эпика (1 часть К’аэСОз + 2 части MgO) и нагревают’ от-
крытый тигель, постепенно повышая температуру до красного каления дна
тигля. Когда весь углерод сгорит, тигель охлаждают, выщелачивают его
содержимое водой п, окислив бромной водой, определяют серу в виде сер-
нистого бария. Очень хорошие результаты дает также метод Тютюнникова.
навеску вещества нагревают с металлическим магнием; при этом углеводо-
роды разлагаются на уголь и водород, сера же связывается в виде сер-
нистого магния. Выделяя пз последнего сероводород и определяя его тем
или другим способом, находят содержание серы во взятой навеске. Область
применения обоих этих методов, а равно и многочисленных других, им
подобных, ограничивается, очевидно, лишь тяжелыми нефтями и нефтепро-
дуктами, для которых один из наиболее опасных источников ошибки —
частичное улетучивание анализируемого вещества при прокаливании его
в открытом сосуде — сводится до минимума.
СОДЕРЖАНИЕ СЕРЫ В РАЗЛИЧНЫХ НЕФТЯХ
И НЕФТЕПРОДУКТАХ (4]
В большинстве нефтей общее содержание серы меньше 1%;
лишь как, исключение встречаются нефти, в которых сера содер-
жится в количестве, превышающем 1%. Однако и в этих узких
пределах для нефтей различных месторождений наблюдаются,
как показывает табл. 74, значительные колебания.
Содержание серы в важнейших нефтях СССР (бакинских,
грозненских и эмбенских) не достигает 0,5%; в большинстве
этих нефтей оно колеблется около 0,2%, спускаясь для некото-
рых (сураханские, некоторые эмбенские и др.) до 0,1% и ниже.
Из нефтей СССР, в которых содержание серы превышает 0,5%,
Для устранения влияния примесей, имеющихся в воздухе лабора-
тории, и более полного сгорания навески удобно пользоваться прибором
с подводкой воздуха, предварительно очищенного или обогащенного кисло-
родом L3J.
*254
Таблица 74
Содержание серы в различных нефтях
Нефти СССР Разные не ф т и
Месторождение %s Месторождение °/oS
Сураханы 0,04—0,07 Гялиппя . . . . 0,03—0,30
Балаханы 0,07—0,32 Румыния . 0,'об—ОДО
Сабунчи 0,06—0,12 Эльзас . 0,60—0,80
Раманы 0,07-0,10 Ганновер 1.2»
Биби-Эйбат 0,10—0,18 Пенсильвания (США) . 0,08—0,10
Бинагады . 0,08-0,22 Канзас . 0,14-035
О. Челекен 0,25 Оклахома , 0,13—0,73
Грозный 0,20—0,25 Уайоминг . 0,08-2.62
Доссор (Эмба)..... 0,11—0,20 Калифорния , 0,28—3,58
Макат „ 0,22—0,23 Тексас „ 0,13—2,54
Ухта (Чибью) 1,12 Мексика 5,34
Чусовские Городки . . 5,40 Венецуэла 0,38—2,65
Ишимбаево 3,00 Персия 1,06
Фергана (Шорсу) . . . 2,07 Египет 2,25
О. Сахалин 0,10—0,4 Индия 0.10-0,15
должны быть отмечены нефти ухтинские, некоторые ферганские
(Чимион, Шорсу), особенно же уральская нефть (Чусовские Го-
родки), в которой содержание серы достигает 5,4%.
Переходя к месторождениям, лежащим за пределами СССР,
отметим прежде всего, что нефти важнейших после Кавказа евро-
пейских месторождений — Галиции и Румынии—по содержа-
нию серы напоминают наши бакинские и грозненские нефти; за-
метно богаче серой нефти некоторых других европейских место-
рождений (Ганновер, Эльзас, Италия).
Что касается нефтей внеевропейских месторождений, то и
здесь можно наблюдать довольно большое разнообразие. Так,
среди американских нефтей встречаются такие, в которых со-
держание серы не превышает 0,1—0,2% (Пенсильвания, Нью-
Йорк, Колорадо и некоторые другие), но большинство месторо-
ждений Северной Америки дают нефть смешанного типа, т, е.
такую, среди которой можно встретить как образцы, небогатые
серой (0,2—0,3%), так и образцы, в которых содержание серы
превышает 0,5%. Особенно богаты серой некоторые нефти Аркан-
заса (до 2,0%), Калифорнии (свыше 3,0%), Тексаса (свыше
2,5%) и Мексики (свыше 5,0%). Отметим здесь же несколько
нефтей внеамериканских месторождений, богатых серой, как-то:
нефти персидская (до 1,0%) и египетские (свыше 2,0%).
Распределение серы по различным погонам одной и той же
нефти зависит не только ст природы сернистых соединений, но
и от других причин. Если нефть перегоняется без разложения, то
’содержание серы, как общее правило, возрастает от низших по-
гонов к высшим: меньше всего ее содержится в бензине,
больше — в керосине, еще больше, обычно свыше 50,0 % всей
серы,—в мазуте. В тех случаях, когда перегонка нефти сопро-
255
вождается разложением, указанная правильность может нару-
шаться: часть серы может перейти при этом из высших фракций
в низшие, которые в силу этого могут оказаться несколько бо-
гаче серой последующих погонов. Впрочем, несмотря на это, при
перегонке с разложением до кокса остаток (кокс) обыкновенно
сильно обогащается серой, содержание которой в нем может под-
ниматься до нескольких процентов.
ТЕХНИЧЕСКИЕ НОРМЫ НЕФТЕПРОДУКТОВ НА СЕРУ
Как видно из данных табл. 74, наши основные нефти не бо-
гаты серой. Ввиду этого в СССР содержание серы до последнего’
времени не нормировалось даже для таких нефтепродуктов, как
бензин и керосин; норма на серу (0,5%) была установлена у нас
лгишь для одного из видов нефтетоплива: «мазут морской». Тех-
нические нормы нефтепродуктов СССР последнего времени
(1937 г.) ввиду ввода у нас в эксплоатацию ряда новых место-
рождений, дающих высокосернистую нефть, значительно расши-
ряют область нормирования серы. Здесь, например, находим
нормы на серу для авиационных бензинов (не больше 0,05% S),
для крэкинг-бензинов (не больше 0,1% S), для дизельного и
моторного топлива (не больше 0,2% S) и т. д.
В Америке, где в переработку идут нефти с самым разно-
образным содержанием серы, нормы на серу введены для различ-
ных нефтепродуктов. Так, например, согласно правительствен-
ной спецификации США содержание серы в моторном бензине
не должно превышать 0,10%; для разного рода керосинов оно
может колебаться от 0,10 до 0,135%? и т. д.
Необходимость нормирования серы вытекает из того обстоя-
тельства, что как сами сернистые соединения, особенно серово-
дород и меркаптаны, так и сернистый газ, образующейся при
сгорании различных сернистых соединений, оказывают на раз-
личные части двигателей внутреннего сгорания - (трубопроводы,
клапаны, цилиндры) разъедающее действие, которое, естественно,
особенно усиливается при повышенном 'Содержании серы и при
высокой температуре. Не менее вредное влияние некоторые виды
сернистых соединений оказывают на стенки металлических ре-
зервуаров для хранения нефтепродуктов; особенно сильную кор-
розию вызывают элементарная сера, сероводород и меркаптаны.
Ввиду значительных трудностей, с которыми сопряжено полное
удаление некоторых видов сернистых соединений (см. ч. III,
гл. II), весьма важным представляется вопрос о рациональных
нормах на серу в нефтепродуктах, так как практические тенден-.
ции здесь очевидны: во избежание излишних расходов на
очистку стремятся сохранить повышенное содержание серы
в нефтепродуктах, выпускаемых на рынок. Таким образом воз-
никает вопрос о максимально допустимом содержании серы’
в различных нефтепродуктах; особенно важное значение этот
вопрос имеет для моторного топлива, и для его освещения про-
деланы большие и важные работы.
256
Специальные опыты показали. что особенно важное влияние
на коррозию двигателя при работе на сернистом топливе оказы-
вает жидкая вода, очевидно ввиду образования при ее взаимо-
действии с сернистым газом сернистой кислоты и окисления по-
следней под влиянием кислорода до серной кислоты. Отсюда по-
нятны те меры, которые рекомендуются для предотвращения кор-
розии под влиянием указанной причины: моторы снабжаются
специальными приспособлениями, вентиляторами, устраняю-
пцгми возможность конденсации воды в картере (особенно важно
в зймнее время), а также термостатами, регулирующими темпе-
ратуру водяной рубашки, и фильтрами для масла. При нали-
чии этих приспособлений содержание серы в моторном топливе
по мнению некоторых специалистов может быть повышено без
всякого вреда для двигателя до 0,3—0,5% и выше, для дизель-
ных же топлив — до 1.5—2%, причем, так как дизеля обычно
работают в закрытом помещении, в данном случае конденсация
воды исключается даже в отсутствие указанных выше специаль-
ных приспособлений. В целом, этот вопрос представляет собою
одну из важнейших проблем народного хозяйства, которая тре-
бует самого внимательного изучения.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ СЕРЫ И РАЗЛИЧНЫХ ТИПОВ
СЕРНИСТЫХ СОЕДИНЕНИЙ
Новейшие исследования не ограничиваются суммарным опре-
делением содержания серы в нефти. Не говоря уже о глубоком
научном интересе, который представляет ближайшее изучение
вопроса о тех формах (соединениях), в которых сера содержится
в нефти и ее дестиллатах, исследование этого вопроса предста-
вляет также серьезный практический интерес. В самом деле,
, не все сернистые соединения одинаково активны к различным
металлам; не все они одинаково относятся к различным реаген-
там. Отсюда понятно, что ближайшее определение химической
природы сернистых соединений данной нефти может оказаться
важным при выборе оборудования для ее переработки (коррозия),
при подборе надлежащих методов ее очистки и т. п. Отсюда
практическое значение, которое может представлять анализ
нефти и ее дестиллатов на содержание в ней различных типов
сернистых соединений.
Систематический ход анализа сернистых соединений нефти
заключается *в следующем [5]. Прежде всего определяют и уда-
ляют из образца сероводород, H’S; затем последовательно ве-
дутся определение и удаление элементарной серы, меркаптанов,
дисульфидов и сульфидов, после удаления которых производится
• определение остаточной серы — тиофенов (ост. S). Во всех слу-
чаях, кроме сероводорода, количественное определение произво-
дится ламповым способом по разности двух определений: до и
после удаления соответствующего типа сернистых соединений.
Более подробное описание методики анализа дается ниже. При
ее оценке необходимо иметь в виду, что надежность отдельных
2',7
17 Зак. 1622. Химия нефти.
испытаний нередко является здесь недостаточной; применимость
же ее к случаям, когда анализируемый продукт содержит зна-
чительное количество непредельных, совершенно исключается.
Сероводород, H2S. Прежде чем приступать к определению
других сернистых соединений, должен быть удален сероводород,
так как он мешает определению элементарной серы и меркапта-
нов. Для этого пользуются слабо подкисленным раствором хло-
ристого кадмия (10 г хлористого кадмия в 100 см3 воды, к кото-
рой прибавлен 1 см3 крепкой соляной кислоты). После встряхи-
вания исследуемого образца с небольшим избытком этого рас-
твора весь сероводород удаляется в виде сернистого ‘кадмия.
Собрав и взвесив последний, можно рассчитать процентное со-
держание сероводорода в исследуемом образце.
Элементарная сера. Определение и удаление серы произво-
дятся встряхиванием исследуемого образца с металлической
ртутью. Выделяется сернистая ртуть, которую отфильтровывают.
Определяя ламповым способом содержание 'Серы до и после обра-
ботки ртутью, по разности получают процентное содержание эле-
ментарной серы в анализируемом образце.
После удаления элементарной серы образец делится на две
неравные части- меньшая идет на определение меркаптанов,
большая — на определение дисульфидов и для последующего
анализа.
Меркаптаны, R - SH. Определение меркаптанов может быть
произведено следующим методом. Небольшая часть образца
встряхивается с половинным объемом спиртовою раствора плум-
бита (1 объем 95%-ного спирта + 1 объем водного раствора плум-
бита натрия). Встряхивание продолжается до тех пор, пока от
прибавления небольшого количества 'серы к пробе смеси уже не
будет получаться окрашивания. После двух таких последова-
тельных обработок все меркаптиды свинца переходят в свинцо-
вый раствор; затем обработанный образец промывают водою и
фильтруют его. Содержание серы, которая приходится на долю
меркаптанов, равно разности между процентным содержанием
серы в образце до и после обработки плумбитом. Определе-
ние— ламповым способом.
Другой метод определения меркаптанов заключается в следующем. Осво-
божденное от сероводорода и серы масло растворяется в бензоле и обраба-
тывается суспензией основного уксуснокислого свинца. Образуются мерка-
птиды, легко растворимые в бензоле. Если теперь отделить водный слой от
бензольного и прибавить к последнему точный объем дединормальной сер-
ной кислоты, то при встряхивании меркаптиды разрушаются, и весь входив-
ший в их состав свинец переходит в осадок в виде сернокислого свинца,
который может быть определен весовым способом; отсюда легко рассчитать
количество меркаптанов, которое было связано этим свинцом. Можно также
вместо весового определения свинца оттитровать не вошедшую в реакцию
серную кислоту, положив в основу расчета то количество этой кислоты,
которое связывается при разрушении меркаптидов и осаждении сернокис-
лого свинца. г
•
Дисульфиды, R • S - S • R. Определение дисульфидов произво-
дится в большей части образца, оставшегося после удаления
258
сероводорода и элементарной серы; меньшая часть его, как было
указано, пошла на определение меркаптанов. Анализируемое
масло смешивают с равным объемом ледяной уксусной кислоты
и нагревают 3 часа с цинковой пылью. Все дисульфиды превра-
щаются при этом в меркаптаны. Отфильтровав избыток цинка и
отмыв уксусную кислоту водой, обрабатывают масло спиртовым
раствором плумбига натрия, как описано выше, и определяют
общее количество меркаптанов, находящихся в исследуемом
масле и образовавшихся при восстановлении дисульфидов. Про-
центное содержание серы, которая приходится на долю дисуль-
фидов, выразится разностью между содержанием серы до и по-
сле их восстановления и удаления.
Сульфиды, В • S - В. Масло, освобожденное от меркаптанов и
дисульфидов, подвергают обработке порошкообразной азотнокис-
лой закисью ртути. Образуется нерастворимое комплексное со-
единение, и ’сульфиды переходят в осадок. Процентное содержа-
ние сульфидной серы равно разности между содержанием серы
до и после обработки азотнокислой закисью ртути. И здесь оба
определения серы производятся ламповым способом.
Остаточная сера. Содержание серы в масле после обработки
азотнокислой закисью ртути дает остаточную серу, которая при-
ходится на долю тиофенов и тиофанов.
Схематически ход анализа на содержание серы и различных
типов сернистых соединений представлен на приводимой ниже
диаграмме. Такой анализ удобнее производить не с сырой
нефтью, а с ее дестиллатом, причем, однако, надо учитывать
следующее важное обстоятельство. Не подлежит сомнению, что
целый ряд простейших сернистых соединений, обнаруженных
в нефтяных дестиллатах, отнюдь не предсуществуют в самой
исходной нефти, а образуются лишь при ее перегонке. Это
явствует уже из того обстоятельства, что запах неочищенного
нефтяного дестиллата обыкновенно бывает сильнее, неприятнее,
чем запах исходной нефти. Вместе с тем нередко в дестиллате
можно обнаружить такие сернистые соединения (сероводород
и др.), которые не открываются в исходной нефти. Образование
подобного рода сернистых соединений при перегонке нефти мо-
жет быть связано либо с распадом каких-то более сложных серу-
содержащих соединений нефти, либо, наоборот, с их синтезом
в результате взаимодействия элементарной серы с нефтяными
углеводородами. С этой важной оговоркой можно перейти к об-
зору сернистых соединений, обнаруженных в различных нефтях
и их дестиллатах.
СЕРНИСТЫЕ СОЕДИНЕНИЯ РАЗЛИЧНЫХ НЕФТЕЙ
Сероводород встречается в сырых нефтях, невидимому, до-
• вольно редко. Присутствие его обнаружено, например, в галиций-
ской [6], канадской [7] и особенно в некоторых тексасских [8]
нефтях; однако, даже столь богатая серой нефть, как наша перм-
ская (Чусовские Городки), сероводорода не содержит. С другой
17* 259
Схема определения серы и сернистых соединений
Образец содержит:
H,S, S, RSH, R,S», R,9 и остаточную S
Обработка CdCl,
I часть. Опредепенне R-SH
Разлепить па
2 части.
Метол I. Спирт, плумбит
Метод II. Титрование
II часть — восстановление
Масло содержит
R»S„ Ki3 и остаточную S.
Ламповое определение JA 3
Масло содержит
RjSjy R,9 я остаточную 9
Ламповое определение № 3
Масло содержит
RSH, R,S и остаточную S
Обработка спиртовым плумбитом
Сульфиды'—в осадке
Масло содержит
оотаточную S.
Ламповое определение № б
стороны, выделение сероводорода при перегонке нефти — обычное
явление; однако в этих случаях сероводород представляет собою
уже вторичный продукт, образующийся либо в результате раз-
ложения более сложных сернистых соединений, либо как про-
дукт взаимодействия углеводородов известных типов, например
нафтенов, с находящейся в нефти элементарной серой при по-
вышенной температуре.
Элементарная сера встречается в сырых нефтях значительно
чаще сероводорода; при хранении таких нефтей она может кон-
центрироваться в иле, собирающемся на дне нефтехранилищ.
Сера обнаружена, например, в ряде нефтей Румынии [9], Те-
ксаса [10] и др.
Меркаптаны также встречаются в нефти довольно редко.
Вполне достоверно доказано их присутствие в иранской нефти
(месторождение Майдан-и-Нафтун) при исследовании ее бензино-
керосинового погона, составляющего 36% по объему от
нефти [11]. Меркаптаны были изолированы от других сернистых
соединений обработкой 50%-ным раствором едкого кали с по-
следующим подкислением воднощелочной жидкости. Полученное
таким образом масло было перегнано с паром, а затем тщательно
расфракционировано с дефлегматором Гемпеля. Таким образом
были выделены изопропил-меркаптан (температура кипения 56—
58°) и изобутил-меркаптан (температура кипения 86—88°). Пер-
вый из них был получен в количестве 55—60% от смеси мер-
каптанов, второй — в количестве 18%. Оба меркаптана были
охарактеризованы рядом производных Кроме указанных фрак-
ций, разгонка сырой смеси меркаптанов дала около 9% нижеки-
пяших фракций и вышекипящий остаток. В первых вероятно
присутствие этил-меркаптана (температура кипения 37°), во
втором — изоамил-меркаптана (температура кипения 120°).
Меркаптаны содержатся, невидимому, также в уральской,
а возможно, и некоторых других нефтях, но ближе этот вопрос
пока не исследован.
Кроме меркаптанов, при исследовании того же погона иран-
ской нефти были обнаружены диэтил-, диизопропил- и диизобу-
тил-дисулъфиды; было доказано, однако, что они образовались
за 'счет окисления соответствующих им меркаптанов.
Значительно чаще, чем в форме каких-либо иных соединений,
нефтяная сера бывает представлена, как показали исследования
Мэбери и его сотрудников, в виде сульфидов.. Для их выделения
одна из нефтей Охайо была подвергнута следующей обра-
ботке [12]. Наиболее богатые серой фракции этой нефти (темпе-
ратура кипения 200—300°) были обработаны крепкой серной
кислотой; после разбавления водой кислый раствор нейтрали-
зовался углекислым свинцом или известью и подвергался пере-
гонке с паром. Таким образом получено светложелтое масло, со-
державшее почти 15% серы. Масло было разогнано в вакууме на
пятиградусные фракции, которые очищались далее получением
и кристаллизацией двойных соединений с сулемой. Наконец, пу- .
тем разложения этих двойных соединений сероводородом серни-
261
стые 'Соединения нефти выделялись обратно и вновь перегонялись
в вакууме. Таким образом были выделены в пределах несколь-
ких градусов и охарактеризованы следующие сульфиды общей
формулы (CnH2n+i)S: диметил-, диэтил-, вероятно этил-пропил-,
дипропил-, этил-пентил-, диизобутил-, дибутил-, бутил-пентил-,.
. дипентил- и дигексил-сулъфиды. Ни меркаптаны, ни тиофены не
могли быть обнаружены ни в одной из фракций этой нефти.
Другой тип сульфидов, а именно циклические сульфиды или
тиофаны, были выделены Мэбери из канадской нефти [13 .
Нефть была обработана серной кислотой, и полученный кислый
гудрон после разбавления водой был нейтрализован. Выделив-
шееся богатое серой масло было разогнано в вакууме и дважды
очищалось переведением через двойные соединения с сулемой;
последние разлагались затем сероводородом, а полученное масло
тщательно расфракционировано в вакууме. Таким образом был
выделен длинный ряд сульфидов различной степени чистоты,
общей формулы (CJb^sS, начиная от CeHisS (температура ки-
пения 125—130°) и кончая С1вНзб8 (температура кипения 290—
295°). По своим свойствам эти сульфиды несколько напоминали
сульфиды охайской нефти; например, при действии хамелеона
подобно последним они превращались в соответствующие суль-
фоны. Глубокое различие между ними сказывалось, однако", не
только в их составе, но и в отношении к брому; тогда как суль-
фиды предельного ряда (тиоэфиры) при действии брома давани
кристаллические продукты присоединения общей формулы
(СпН2м-1)28Вг2, сернистые соединения канадской нефти реагиро-
вали с бромом чрезвычайно энергично, с обильным выделением
бромистого водорода и образованием малохарактерных продуктов
замещения. Ввиду всех этих опытных данных Мэбери принял
сернистые соединения канадской нефти за тетрагидротиофены
(тиофаны), и позднейший синтез этих сернистых соединений и
исследование их свойств [14] в полной мере подтвердили такое
заключение.
Тиофаны принадлежат к наиболее трудно удаляемым серни-
стым соединениям нефти. Несомненно, они встречаются, кроме
канадской нефти, также и в некоторых других богатых серой
нефтях. Повидимому, их содержит, например, наша уральская
нефть; однако более точных данных по этому вопросу пока не
имеется.
Встречается ряд указаний, что в некоторых нефтях (румын-
ской [15], грозненской [16] и др.) содержатся также тиофен и
его гомологи. Эти указания имеют чисто качественный характер
(индофениновая реакция). Во всяком случае, содержание этих
сернистых соединений в нефтях чрезвычайно незначительно;
для грозненского бензина оно определяется, например, как одна
часть тиофена на 10 000 000 частей бензина.
В отличие от других составных частей нефти, находящих то
или иное применение, сернистые соединения являются тем ее
262
компонентом, который вследствие корродирующего действия на
металлы имеет лишь отрицательное значение. Поэтому одна из
задач очистки нефти и ее дестиллатов заключается в том. чтобы
по возможности освободиться от сернистых соединений. Соответ-
ствующая методика — способы обессеривания нефтепродуктов —
будет рассмотрена ниже, в ч. II.
ЛИТЕРАТУРА
1. Броун А. С. я Сиверцев А. П„ Химия сернистых соединений жидкого
топлива, ОНТИ, Л. (1937).
2. Добрянский А. Ф., Анализ нефтяных продуктов, 2-е изд-, ОНТИ., М.-Л.
(1932); Engler С. u Hofer Н., Das Erdol, т. IV, 2-е изд. (1930);
Голъде Д., Жиры и масла, т. I, Л. (1931); Корсаков М. П.,
„Нефт. хоз/ 14, № 1, 68 (1928).
3. Чернов Г. и Соболев Б., ,Нефт. хоз/ 25, № 9, 55 (1933).
4. По разным справочникам и журнальным источникам.
5. Faragher, Morrel a. Monroe, „Ind. Eng. Chem/ 19, 1281 (1927).
6. Nawratil, „Dingl. pol. J.u 246, 423 (18b2).
7. Mabery, „Proc. Amer. Acad/ 31, 17 и 43 (1894).
8. Richardson a. Wallace, .J. Soc. Chem. Ind/ 20, 690 (1901); Richardson,
„J. Frankl. Inst/ 162, 115 (19C6).
9 Bourquoi, „Chem. Ztg/ 1902, 492 (Kiselings Bericht).
10. Richardson a. Wallace, „J. Soc. Chem. Ind/ 21, 316 (1902).
11. Birch a. Harris, „J. Chem. Soc/ 127, 898 (1925).
12. Mabery a. Smith, .Вег/ 22, 3303 (1889); 24, Ref. 456 (1891); cp. Kost u.
Lagai, „Dingi. pol. J/ 284, 69 (1892).
13. Mabery a Quayle, „Am. Chem. J/ 85, 404 (1906).
14. v. Braun I. u. Trwnpler, „Ъет.и 48, 545 (1910); Гришкевич-Трохимовскнй
.Ж. P. X. О/ 48, 880, 901, 928 (1916).
15. Edeleanu a. Filiti, .Bull/ [3], 23, 884 (1900) и др.
16. Харичков, „Ж. Р. X. О/ 31, 655, (1899).
ГЛАВА IX
АЗОТИСТЫЕ СОЕДИНЕНИЯ НЕФТИ
Азотистые соединения, являющиеся наряду с кислородными
и сернистыми соединениями одной из постоянных составных ча-
стей нефти [1], принадлежат к органическим основаниям. Про-
стейшие представители этого обширного и разнообразного класса
органических соединений имеют открытую группировку атомов,
т. е. принадлежат к жирному ряду; наиболее сложные из них
относятся к так называемым гетероциклическим соединениям.
С точки зрения химии нефти представляют интерес, как будет
показано ниже, и те и другие.
КРАТКАЯ ХАРАКТЕРИСТИКА АЗОТИСТЫХ ОРГАНИЧЕСКИХ СОЕДИ-
НЕНИИ ОСНОВНОГО ХАРАКТЕРА
Простейшие органические основания пли амины являются,
как известно, производными аммиака; они могут быть получены
действием на аммиак галоидных соединений жирного ряда. Так
например, действием йодистого метила могут быть замещены на
метильную группу все три водорода аммиака, причем последо-
вательно образуются в виде солей амины трех типов: первичный
(метил-амин), вторичный (диметил-амин) и третичный (триме-
тил-амин):
СН8 • NHg • Hl; (CH3)2NH • НI; (СН3)3Х Hl.
Последней фазой взаимодействия галоидного соединения
с аммиаком является образование солей четырехзамещенных
оснований, являющихся, очевидно, полными аналогами аммо-
нийных солей, например хлористого или йодистого аммония.
Если для взаимодействия с аммиаком взять не одно-, а двух-
замещенное галоидопроизводное жирного ряда, то аналогичным
путем получается» не амин, а диамин жирного ряда, точнее —
его галоидоводородная соль. Так например, из бромистого пен-
таметилена, ВгСН2(СНг)з СНгВг, и аммиака образуется бромисто-
водородная соль пентамети лен-диамина:
/СН,— СН,Вг XI-L ,СН2— СН,- NH, • НВг
н.2с< - + =н.с<
ХСН2 — CH2Br NH:l “ ЧСН2 —СН.2—NH2-HBr .
Амины и диамины жирного ряда представляют собою силь-
ные основания. С кислотами они образуют прочные соли, из
264
которых щелочи выделяют свободные основания. Низшие пред-
ставители этого класса — газообразные вещества; более высоко-
молекулярныежидкости с характерным запахом.
При нагревании галоидоводородных солей -некоторых диами-
нов, например тетраметилен- и пентаметилен-диамина, происхо-
дит характерная реакция: отщепляется частица аммонийной
соли, например хлористого аммония, и образуется новое органи-
ческое основание уже вторичного характера, в котором - азот
в виде группы NH замыкает пяти- или шестичленную гетероци-
клическую систему. Так например, из хлористоводородной соли
пентаметилен-диамина этим способом. получается пиперидин:
/СЩ—CH.,NH,HC1 /СЕ» — СНо\
Н.С< - - ==\ТН4С1-РН.ЭС/ “ >NE-HC1.
ХЖ.-»-СН2ХН»НС1 “ ХСЕ» —СЕ/
Пиперидин является типичным и сильным вторичным осно-
ванием; его характерной особенностью является способность
в разнообразных условиях терять 6 атомов водорода с превра-
щением в новое гетероциклическое основание — пиридин:
СН2 СН. СН СИ
Н.зс/ \.ХН—* нс/
сн, сн/ сн“ сн
пиперидин пиридин
Пиридин имеет такое же отношение к пиперидину, тик бен-
зол к циклогексану. Подобно бензолу пиридин получается из
каменноугольного дегтя, но он встречается также в продуктах
сухой перегонки торфа, дерева и животных остатков. Пиридин
является простейшим представителем пиридиновых оснований,
образующих особый гомологический ряд. Сам пиридин и его
гомологи — слабые основания; тем не менее, в некоторых отно-
шениях он напоминает бензол, например: подобно бензолу пири-
дин способен присоединять С атомов водорода (образуется пипе-
ридин), может бромироваться, нитроваться и сульфироваться,
хотя и несколько труднее, чем бензол. Аналогия между пириди-
ном и бензолом проявляется еще в одном отношении: подобно
тому как бензольное ядро может конденсироваться с другим
бензольным ядром, причем образуется нафталин, точно так же
пиридиновое кольцо может конденсироваться с бензольным; при
этом образуются две новых гетероциклических системы — хино-
лин и изохинолин:
нафталин хинолиа изохваолик
265
Подобно пиридину хинолин и изохинолин являются слабыми
основаниями; они образуют два самостоятельных гомологических
ряда гетероциклических соединений, которые обычно назы-
ваются хинолиновыми и изохинолиновыми основаниями.
КАЧЕСТВЕННОЕ И КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ АЗОТА
В НЕФТЯХ
И то и’ другое определение производится по общим методам
анализа органических соединений [2].
Для качественного открытия азота пробу высушенной нефти (1—2 г)
нагревают с кусочком металлического калия, постепенно повышая нагрева-
ние до красного каления. Органическое вещество при этом частью улетит,
но весь азот окажется связанным в виде цианистого калия. Если теперь
горячую еще пробирку опустить в стакан с небольшим количеством водыг
пробирка лопается (обычно с воспламенением избытка металлического ка-
лия), и цианистый реалий переходит в раствор. Отфильтровав его, прибав-
ляют к фильтрату по нескольку капель растворов солей закиси и окиси
железа и нагревают. Затем по охлаждении смеси прибавляют к ней соля-
ную кислоту до растворения выделившихся гидратов закиси и окиси же-
леза. Если испытуемая проба содержала азот, то раствор оказывается окра-
шенным в зеленовато-голубой цвет берлинской лазури; при более значи-
тельном содержании азота получается синий осадок этого трудно
растворимого вещества.
Количественное определение азота’® нефтях производится ме-
тодом сожжения по Дюма. Азот выделяется при этом в свобод-
ном состоянии и определяется по объему.
СОДЕРЖАНИЕ АЗОТА В РАЗЛИЧНЫХ НЕФТЯХ И НЕФТЕПРОДУКТАХ
Содержание азота в различных нефтях обычно выражается,
как видно из данных табл. 75, десятыми и даже сотыми долями
процента [3]. Лишь как исключение встречаются нефти, в ко-
торых содержание азота достигает 1,5—2% или немного более.
Особенно выделяются в этом отношении некоторые нефти Кали-
форнии, Тексаса и Японии, а также одна нефть Алжира. Со-
держание азотистых соединений в главнейших наших нефтях
(Баку, Грозный) — ничтожно.
Таблица 75
Содержание азота в различных нефтях
Месторождение % N Месторождение %N
Баку Грозный Нефтяная гора .... Ухта . . Пешельбронн (Эльзас) . Сиди-Брагим (Алжир) . Саравак (о. Борнео) . . Амаце (Япония).... Кусодсу „ .... Ойл-Спрпнгс (Канада) . 0,05 0,07 0,14 0,14—0,20 0,25—1,10 2,17 0,18 0,35 1,33 0,18 • Петролиа (Канада) . . Зап. Вирджиния (США) Бельмонт Юж. Охайо (США) Сев. Охайо Джпфферсон, Тексас . Санта Барбара, Кали- форния Лос-Анжелес, Калифор- нпя Вентура, Калифорния . ___ С,16 0,54 0,26 0,02—0,35 >1,00 1,25 1,20 0,56-1,46
266
Распределение азотистых соединений по различным погонам
одной и той же нефти очень неравномерно; соответственно
своей температуре кипения они сосредоточены главным образом
в высококипящих частях нефти. Интересно, что даже нефтяной
кокс содержит заметное количество азота (0,3—0,4%) [4].
Вопрос о зависимости содержания азота от глубины залегания нефти
исследован пока недостаточно. Как показало изучение этого вопроса на при-
мере месторождений южного Охайо [5], при прочих равных услоЛях, со-
держание азота возрастает с увеличением глубины залегания. Однако явле-
ние это слишком сложно, и для окончательных выводов необходимо даль-
нейшее накопление фактического материала.
АЗОТИСТЫЕ СОЕДИНЕНИЯ РАЗЛИЧНЫХ НЕФТЕЙ
Выделение простейшего азотистого основания, аммиака, на-
блюдалось при перегонке многих нефтей [6]. Имеются также
указания на присутствие в нефти некоторых простых аминов,
например: метил-амина в грозненской нефти [7], триметил-
амина— в галицийской [8] и т. д. Но главная масса азотистых
соединений нефти принадлежит к более высокомолекулярным
органическим основаниям. Их выделение производится промыв-
кой нефти или ее дестиллатов серной кислотой, с которой они
образуют соли; разлагая последние щелочью, получают смесь
свободных оснований, которую для ближайшего исследования и
разделения подвергают прежде всего фракционировке в вакууме.
Далее следуют получение солей и хлороплатинатов, их кристал-
лизация и ближайшая характеристика отдельных выделенных
оснований, точнее — более или менее узких их фракций. Таким
образом с различной степенью полноты были исследованы орга-
нические основания кавказских, галицийских, румынских и не-
которых американских нефтей; при этом, в общих чертах были
получены следующие результаты.
При исследовании одной румынской нефти было выделено ма-
слянистое 'вещество с запахом пиридина и температурой кипе-
ния около 117°; элементарный анализ дал числа, близкие к со-
ставу пиридина; при действии водорода в момент выделения ве-
щество превратилось в пиперидин. Таким образом выделенное
основание состояло, повидимому, главным образом из пири-
дина [9].
Значительно более сложные по составу и бедные азотом осно-
вания были выделены, из керосина и мазута кавказской нефти
[10]. Эти основания также имели пиридиновый запах, но пере-
гонялись они при 260—370° без разложения; давали кристалли-
ческие двойные соли с хлорной платиной. Содержание в них
азота было только 5,6—6,6% (против 17,7% N для пиридина).
Таким образом в состав кавказской нефти, повидимому, наряду
с пиридиновыми основаниями входят также основания более
бедных азотом рядов, например ряда хинолина и ему подобных.
Наиболее полно были исследованы [11] органические основа-
ния калифорнских нефтей. Путем фракционирован из смеси
267
этих оснований, полученной обычным способом, был выделен и
охарактеризован ряд отдельных оснований от CiaTIijX до C17H21N
включительно. Вещества эти представляли собою маслянистые
жидкости с резким запахом, напоминающим никотин. Их соли
мало характерны; с хлорной платиной они давали хло^кшлати-
наты; с иодистым метилом — продукты присоединения подобно
типичным аминам; при действии хромовой кислоты были полу-
чены только продукты глубокого распада в отличие от хиноли-
новых оснований, которые в аналогичных условиях окисления
дают пиридин-карбоновые кислоты. На основании совокупности
всех этих данных было высказано предположение, что основания
калифорнской нефти следует отнести к гидрированным пириди-
новым и хинолиновым основаниям, находящимся в связи со сла-
быми органическими кислотами [12].
В последнее время азотистые основания 1;алифорнской
нефти были вновь подвергнуты исследованию [13], причем хи-
мическую природу некоторых из этих оснований удалось, нако-
нец, установить.
Интересны масштаб и методика этого исследования, проводи-'
мого (с конца 20-х *гг.) в одном из американских университетов
(Ajastin, Texas). Исходным продуктом был керосиновый дестиллат
фирмы «Union Oil Со of California» с содержанием 0,055 %, азота,
очищавшийся жидкой сернистой кислотой. 150 т остатков от
такой очистки были обработаны разбавленной серной кислотой,
откуда затем было выделено 50 л нерастворимых в воде основа-,
ний. Они были разогнаны примерно на 50 фракций с темпера-
турами кипения от 180 до 335°; однако путем дальнейшей фрак--
ционировки не удалось выделить ни одного соединения в чистом
виде. Лишь путем фракционированной нейтрализации и после-
дующего выделения свободных оснований удалось, наконец, по-
лучить кристаллизующиеся соли и пикраты, которые и были
применены далее для получения чистых оснований. В некото-
рых случаях весьма ценные услуги оказала также обработка
смеси жидким сернистым ангидридом, который образует с осно-
ваниями нефти продукты присоединения различной степени
устойчивости: одни из них диссоциируют уже при обыкновен-
ной температуре, другие, напротив, являются (весьма устойчи-
выми; это различие в отдельных случаях также приложимо
к частичному разделению смеси оснований.
Как показало ближайшее исследование, основания калифор-
нийской нефти принадлежат к двум основным типам: 1) негнд-.
рированные гетероциклические основания ряда хинолина и
2) гидрированные гетероциклические основания более сложного
строения. Для их разделения удобно иногда пользоваться мето-
дом «постепенного извлечения», который заключается в еле--
дующем: основания растворяют в избытке разбавленной соляной
кислоты и экстрагируют раствор хлороформом или хлористым
этиленом. Разделение достигается вследствие большей раствори2
мости в этих растворителях хлоргидратов гидрированных основа- .
ний по сравнению с хлоргидратами оснований негидрированныг.
268
Применение указанных методов позволило изолировать из
калифорнийского керосинового дестиллата следующие основания.
2,4-диметил-хинолин ChHuN и
2,3-диметил-хинолин.
Эти основания были выделены из фракции смеси оснований
с температурой кипения 263—270°. Методика разделения —
экстракция жидким сернистым ангидридом и последующее пере-
ведение оснований через пикраты с кристаллизацией последних
из ледяной уксусной кислоты. Легче растворимый пикрат ока-
зался тождественным с пикратом синтетического 2,4-димегил-
хинолина; пикрат же, труднее растворимый в ледяной уксусной
щгслоте, дал свободное основание, тождественное с 2,з.-диметил-
хинолином. •
2,8-диметил-хинолин. Этот третий изомерный диметил-хино-.
лин был выделен из фракции керосиновых оснований с темпера-
турой кипения 253—256° методом «постепенного извлечения».
.Основание охарактеризовано рядом производных, как-то: кислый
сульфат, перхлорат, двойная соль с хлорной ртутью и т. д.
2, 4, 8-триметил-хинолин, CisHisX. Основание было выделено
из фракции с температурой кипения 273° методом «постепенного
извлечения» и также охарактеризовано рядом производных
(хлоргидрат, сульфат, пикрат и др.).
2, 3, 8-триметил-хинолин. Это — первое хинолиновое основа-
. ние, выделенное из смеси керосиновых оснований в чистом виде.
Оно составляет около 18% фракции оснований, кипящей при
276—277° при нормальном давлении. В чистом виде основание
получается либо в виде пикрата осаждением из уксуснокислого
раствора, либо в виде кислого сульфата Свободное основание,
выделяемое из пикрата или сульфата нагреванием с аммиаком,
представляет собой кристаллическое вещество с температурой
плавления 55—56°: при 747 лл оно кипит с частичным разло-
жением при 280°. Целый ряд полученных из него производных
оказался тождественным с соответствующими производными син-
тетического 2,3,8-триметил-хинолина.
Природа гидрированных оснований калифорнийского керосина
оказалась гораздо сложнее. Имеются веские основания предпола-
гать, что основания эти (их ‘называют «нафтеновыми основа-
ниями») представляют собой гидрированные производные пирин-
дена, т. е. конденсированной системы пиридинового ядра с пятй-
членным циклом, и пириндацина, т. е. конденсированной симме-
трической системы пиридинового кольца с двумя пятичленными
циклами. Из этих оснований следующие два выделены в чистом
виде.
Нафтеновое основание C13H21N. Основание выделено из фрак-
ции керосиновых оснований, кипевшей в пределах 215—216°,
дробной перекристаллизацией пикратов. Представляет собою бес-
цветное масло, нерастворимое в воде и смешивающееся с обыч-
ными растворителями во всех отношениях. Его температура ки-
пения 225°,6 при 750 мм; температура плавления 24°,5. Вероят-
ное строение этого основания выражается формулой I.
269
Нафтеновое основание C16H25N. Представляет собою бесцветное
масл£>, без запаха, нерастворимое в воде, но растворимое в орга-
нических растворителях. Кипит при 278°,2 (746 лл); при вымо-
раживании в смеси эфира с твердой углекислотой получено
в кристаллическом виде с температурой плавления 23—24°,з.
Подобно предыдущему основанию охарактеризовано длинным
рядом производных. Вероятное строение этого основания выра-
жается формулой II.
Нафтеновые основания C13H21N и C1GH25N являются предста-
вителями нового класса азотистых оснований, представленных
в нефти, повидимому, весьма широко. Их исследование сопря-
жено с большими трудностями, но представляет выдающийся
интерес для познания состава нефти.
Заслуживает внимания, что все азотистые основания, выде-
ленные из нефти, оказались оптически недеятельными.
В заключение обзора азотистых оснований, выделенных из
нефтяных дестиллатов, необходимо отметить здесь то же, на что
было обращено внимание в предыдущей главе в отношении сер-
нистых соединений. Не подлежит сомнению, что в сырой нефти
содержатся далеко не все азотистые основания, которые могут
быть обнаружены в ее дестиллатах, и что многие из них обра-
зуются лишь при перегонке нефти. Так, если обработать сырую
нефть или остаток от ее перегонки минеральной кислотой, то,
казалось бы, органические основания должны быть извлечены
при этом полностью. Опыт показывает, однако, что при последую-
щей перегонке в дестиллате вновь обнаруживаются азотистые
основания, которые, таким образом, могут быть лишь вторичного
происхождения. О материнском веществе, из которого образуются
270
эти основания при перегонке нефти, пока ничего не из-
вестно [14].
Не исключена возможность практического применения азо-
тистых оснований нефти. Уже в настоящее время имеется ряд
патентов для их использования в качестве инсектисидов, а также
антиокислителей (ингибиторов) для стабилизации крекинг-де-
стиллатов нефти и улучшения качества смазочных масел, и не-
сомненно, что в дальнейшем, по мере изучения состава и свойств
азотистых оснований нефти, возможности в данном направлении
будут все более и более расширяться.
ЛИТЕРАТУРА
1. Эллис Ё., Химия углеводородов нефти и их производных*, т. II, пер.
с англ., ОНТИ, Москва (1938).
2. Гаттерзлан-Виланд, Практические работы по органической химии. 3-е
русск. изд., Л. (1930).
3- По разным старым и новейшим справочникам и журнальным источникам.
4. МаЪегу, ,Proc. Am. Acad." 81, 10, 50 (1894).
5. МаЪегу a. JDunn, „Ат. Chem. J." 18, 215 (It 96).
6. Zaloziecki, -„Chem. Ztg." 1891, 1204 и др.
7. Харичков, »Ж. Р. X. О." 88, 1275 (19С6).
8. Grabowski, ср- Zaloztecki, „Monatshefte" 18, 498 (1692).
9. Griffiths et Blwnann. „Bull." (ЗЦ 25, 725 (1901).
10. Тищенко В. В., „Ж Р. X. О-“ 25, 51 (1893); Шестаков, „Ж. Р. X. О." 80
874 (1898); Xлопин, „Вег." 38, 2837 (1900).
11. МаЪегу, ,J. Soc. Chem. Ind.* 19, 504 (1900) и др.
12. Pekham, „Jahresber. Ch.* 1894, 748.
13. Poth, Schulze, King, Thomphson, Slagle, Floyd a. Bailey. „J. Am. Ch. Soc.*
52, 1239 (1930); см. такяе ряд позднейших работ Bailey с сотруд-
никами; ibid. 55, 4136, 4141, 4145 (1938); Ьв, 2741 (1934) и др.
14. По вопросу об азотистых соединениях нефти, в частности нефтей СССР»
см. только что появившиеся работы Яихушина К. Д. в „Сборнике
по вопросам нефтепереработки", .Труды АзНИИ" (1938).
ГЛАВА X
СМОЛИСТЫЕ И АСФАЛЬТОВЫЕ ВЕЩЕСТВА НЕФТИ
К этой группе -относятся наиболее сложные и наименее изу-
ченные компоненты нефти, являющиеся в то же время важней-
шими составными частями природных и искусственных асфаль-
тов. При выделении из нефти или асфальтов они образуют слож-
ную смесь соединений различной химической природы, и задача
исследования заключается прежде всего в том, чтобы разделить
эту смесь па более или менее резко разграниченные группы ве-
ществ. характеризующихся общими свойствами.
КЛАССИФИКАЦИЯ И РАЗДЕЛЕНИЕ СМОЛИСТЫХ
И АСФАЛЬТОВЫХ ВЕЩЕСТВ НЕФТИ
После отгонки легких и масляных фракций нефти остаток
(гудрон) представляет собою смолистую массу разнообразной
консистенции, содержащую смолистые и асфальтовые вещества,
частью находившиеся в нефти в готовом виде, частью образовав-
шиеся во время ее перегонки вследствие разложения и после-
дующей полимеризации и конденсации других составных частей
нефти. Получаемый таким .образом продукт по своему составу
и свойствам весьма близок -к некоторым природным асфальтам.
Главнейшими составными частями его являются:
Масла — остатки высокомолекулярных углеводородов, не ото-
гнавшиеся при перегонке нефти.
Нефтяные смолы и асфальтены — нейтральные вещества,
являющиеся главнейшими представителями смолистых и асфаль-
товых веществ нефти.
Асфалътогеновые кислоты и %ix ангидриды — смолистые ве-
щества кислотного характера.
Разделение этих основных групп смолистых и асфальтовых
веществ и количественное определение их в различных битумах,
естественных и искусственных, производится по Маркуссону [1].
1. Прежде всего определяют и отделяют свободные асфалъто-
геновые кислоты. 5 г битума, т. е. смеси смолистых и асфальто-
вых веществ, растворяют в 25 см* бензола и обрабатывают
200 см* спирта. АсфаДьтогеновые кислоты остаются в растворе,
тогда как нейтральные компоненты смолистого и асфальтового
характера выпадают в осадок. Когда раствор отстоится, его отде-
272
ляыг и титруют в 1грисутствии фенолфталеина спиновым рас-
твором едкого натра, (О,IN) до слаГм>|хмовой окраски. Затем при- •
башляют к раствору равны» объем воды и экстрагируют бензолом
неомыленные вещества, после чего водно-спиртовый раствор
упаривается на водяной бане, и из него действием минеральной
кислоты получают асфальтогеновые кислоты в своботном состоя-
нии в виде бурой смолистой массы.
2. Наряду со свободными кислотами Наличные битумы, осо-
бенно естественные, содержат также ангидршУы асфальтогеновых
кислот. Для их определения соединяют выделенный спиртом оса-
док из начальною бензольного раствора битума и неомыленные
компоненты, извлеченные бензолом из водно-спиртовою раствора
при предыдущем определении. Эту смесь подвергают обработке
нормальным спиртовым раствором едкого кали: при этом анги-
дриды асфальтогеновых кислот превращаются в калийные соли,
из которых свободные кислоты выделяются действием минераль-
ных кислот после предварительного извлечения неомыляемых
бензолом, как это описано выше.
3. После отделения асфальтогеновых кислот и их ангидридов
приступают к выделению асфальтенов. Для этою все смолистые
и асфальтовые вещества от предыдущего определения, т. е. не
реагирующие со щелочью, растворяют в 10 см* бензола и приба-
вляют к раствору 200 см* бензина, выкипающею до 50°. Аофать-
тены осаждаются при этом в виде чернобурого аморфною по-
рошка, который отфильтровывают, промывают таким же бензи-
ном и взвешивают.
4. Бензиновый раствор, отфильтрованный от асфальтенов, со-
держит нейтральные смолы и масла. Для их разделения раствор
упаривается до 25 см* и обрабатывается 25 г твердого адсор-
бента (сукновальная глина, флоридин, силикагель). Подвергая
эту смесь экстракции легким бензином в приборе Сокслета, из-
влекают сначала масла, получаемые после отгонки бензина
в виде светлоокрашенной густой жидкости, тогда как нейтраль-
ные сметы остаются в адсорбенте. Наконец, возобновив после
удаления масел экстракцию адсо1)бента новым растворителем,
а именно хлороформом, извлекают последний компонент смеси —
нейтральные смолы.
Анализ по Маркуссону дает наиболее полное представление
о битуме; однако метод этот довольно сложен и требует много
времени. Его можно, впрочем, значительно упростить, если отка-
заться от определения асфальтогеновых кислот и их ангидри-
дов, содержащихся в нефтяных битумах в сравнительно неболь-
ших количествах. Такой упрощенный анализ битумов произво-
дится по Саханову [2] следующим образом.
Навеска битума растворяется в небольшом количестве бен-
зола или хлороформа и осаждается легким бензином. Выпадают
асфальтены, вместе с которыми выделяется также большая часть
асфальтогеновых кислот и их ангидридов. Обработкой осадка
спиртом на холоду кислоты и ангидриды переводятся в раствор
и после испарения растворителя получаются в свободном виде;
273
18 Зак- 1622. Химия нефти
в остатке — чистые асфальтены. Бензиновый раствор после оса*
ждения асфальтенов и кислот содержит масла и смолы; их раз-
деление производится по Маркуссону.
ХАРАКТЕРИСТИКА ОТДЕЛЬНЫХ ГРУПП СМОЛИСТЫХ
И АСФАЛЬТОВЫХ ВЕЩЕСТВ НЕФТИ
Сопоставление результатов исследования смолистых веществ
различного происхождения, например нефтяных, каменноуголь-
ных, древесных и др., убеждает в глубоком различии между
ними. Напротив, смолистые и асфальтовые вещества нефтяного
происхождения, хотя бы из различных нефтей, оказались
в основном одинаковыми. Таким образом смолистые и асфальто-
вые вещества нефти являются столь же характерными ее компо-
нентами, как и остальные, например углеводороды различных
рядов, нафтеновые кислоты и т. д. Чтобы составить полное пред-
ставление о химической природе нефти, необходимо поэтому
ближе охарактеризовать различные виды ее 'смолистых и асфаль-
товых веществ и выявить состояние, в котором они находятся
в нефти.
Асфальтены |2]. С внешней стороны асфальтены предста-
вляют собой темнобурые или черные аморфные порошки. Они
не плавятся при нагревании, но при температурах выше 300°
разлагаются с образованием газов и трудно сгорающего кокса.
Удельный вес их выше единицы. При прокаливании в пробирке
они вовсе не выделяют дестиллатов. Нерастворимы в спирту и
легком бензине (петролбйном эфире); легко растворяются в бен-
золе, хлороформе и сероуглероде. В табл. 76 сопоставлен элемен-
тарный состав нескольких образцов асфальтенов, выделенных
из .различных исходных материалов.
Таблица 76
Элементарный состав асфальтенов
— - ------ - _ _
Происхождение асфальтенов Уд. вес % С % н % 8 °/оО
Грозненский мазут, беспарафин. . . . . 85,2 7,4 0,7 6,7
Грозненский гудрон , ... 1,14 86,7 6,8 0,6 5,9
Грозненский гудрон, окисленный . . . 1,14 85,3 6,4 0,6 7,7
Из табл. 76 видно, что нефтяные асфальтены независимо от
исходного материала, взятого для их получения, имеют весьма
близкий элементарный состав. Интересно, что даже процесс окис-
ления гудрона, проводимый в весьма жестких условиях, практи-
чески не изменяет ни свойств, ни состава асфальтена; откло-
нения же от полного тождества в составе, возможно, объясняются
трудностью получения вполне однородных асфальтенов, как тел
коллоидных.
Крайне характерны для асфальтенов явления растворимости.
Оказалось, что растворимость асфальтенов в таких растворите-
274
лях, как бензол, сероуглерод, хлороформ, четыреххлористый
углерод и др., тесно связана с предварительным набуханием их
в этих жидкостях. В других растворителях, как легкий бензин,
винный спирт, этиловый эфир и др., асфальтены не набухают
и практически не растворяются.
Другая характерная особенность.растворов асфальтенов в не-
которых растворителях — их беспредельная растворимость в них
без образования насыщенных растворов. Так например, можно
приготовить 30%-ный раствор асфальтена в бензоле или в хлоро-
форме и еще более сконцентрировать его путем испарения рас-
творителя без нарушения его однородности. Такие растворы
почти тверды и совершенно лишены подвижности.
Приведенные особенности асфальтеновых растворов являются
характерными для коллоидальных растворов. Таким образом
асфальтены—типичные коллоиды, «лиофильные» по отношению
к одной группе растворителей, в которых они набухают и раство-
ряются .с образованием коллоидальных растворов, и «лиофоб-
ные» по отношению к другим растворителям, в которых они не
набухают и не растворяются. С этой точки зрения понятно оса-
ждение асфальтенов-из их растворов, применяемое для аяатати-
ческих целей (ср. выше) й основанное на замене одного раство-
рителя, лиофильного (например бензола), другим — лиофобным
(например петролейным эфиром); вследствие такой замены, на-
ходящийся в виде коллоидального раствора асфальтен под-
вергается коагуляции и выпадает в виде аморфного осадка, ко-
торый может быть отфильтрован и взвешен1.
Все вышеизложенное позволяет выявить то состояние, в ко-
тором асфальтены находятся в нефти и ее остатках. Нефть
представляет собой по отношению к асфальтенам смесь раство-
рителей, лиофобных (метановые углеводороды и. вероятно, на-
фтены) и лиофильных (ароматика и, вероятно, тяжелые масла
и особенно смолы). Поэтому легкие нефти, бедные ароматиче-
скими углеводородами, и их мазуты содержат в растворенном со-
стоянии лишь ничтожное количество асфальтенов; небольшое
количество их находится иногда еще во взвешенном, грубо ди-
сперсном состоянии. Напротив, тяжелые, богатые смолами нефти,
особенно же их гудроны, могут содержать значительное коли-
чество’асфальтенов в виде устойчивого коллоидального раствора.
Химическая природа асфальтенов изучена пока далеко не-
достаточно. Для их характеристики интересны следующие дан-
ные.
Асфальтены имеют довольно высокие бромные и иодные
числа, обнаруживая, таким образом, непредельный характер.
Возможно, впрочем, что с галоидами здесь образуются соедине-
ния юкоониевого или сульфониевого типов с присоединением га-
лоидов к атомам кислорода или серы.
1 Кроме петролейного эфира, для осаждения асфальтенов было предло-
жено пользоваться также другими растворителями, как-то: метил-этпл-ке-
тоном (бутанолом) в сме<н1 с водой, уксусным эфиром и др.; по отношению
к ним коллоидальные растворы асфальтенов также, очевидно, лиофобны.
18* 375
При обработке хлороформового раствора асфальтенов дымящей азотной
кислотой при —10° получаются, повидимому, нитросоединения, коричне-
вые порошки, растворимые в щелочи; при нагревании их щелочного рас-
твора происходит отщепление элементов азотистой кислоты. При нагревании
с крепкой или дымящей серной кислотой асфальтены образуют продукты
с 6—8% серы. При нагревании этих продуктов с соляной кислотой серная
кислота отщепляется, по щелочи на них не действуют.
Хамелеоном асфальтены окисляются в кислоты. С формали-
ном и 'Серной кислотой образуют формолиты. Асфальтены не реа-
гируют с пячихлористым фосфором; не сочетаются с диазосо-
единениями; почти не подвергаются омылению и обладают
весьма малыми ацетильными числами. С сулемой и хлорным же-
лезом образуют комплексные соединения. Опыты определения
молекулярного веса асфальтенов (в камфоре, до методу Раста)
дали величины порядка нескольких тысяч.
Подводя итоги, можно сказать, что асфальтены представляют
собой высокомолекулярные органические соединения, содержа-
щие, .кроме углерода и водорода, также серу и кислород. Сово-
купность всех приведенных выше реакций показывает, что
асфальтены не представляют собой ни спиртов или фенолов, ни
кислот или их ближайших производных (сложные эфиры, лак-
тоны), ни альдегидов или кетонов. Возможно, что это—гетеро
циклические соединения с серой или кислородом в ядре, причем
эти два элемента (О и S) способны взаимно замещать друг
друга. В пользу этого последнего положения юворит, между
прочим^ состав асфальтенов из природного асфальта. Повторяя
в общем основные свойства нефтяных асфальтенов, асфальтен,
например, из тринидадского асфальта вовсе не содержит кисло-
рода, но зато имеет 10,9% серы; остальное приходится на долю
углерода (82,0%) и водорода (7,8%).
Нефтяные смолы [4. 5] Нейтральные нефтяные смолы пред-
ставляют собою полужидкие, иногда почти твердые, тягучие,
темножелтые или коричневые вещества удельного веса несколько
выше единицы. Они вполне растворимы во всех нефтяных мас-
лах и легком бензине, а также в бензоле, хлороформе, эфире.
Труднее растворяются в винном спирте и ацетоне. В отличие от
асфальтенов они образуют при этом истинные, а не коллоидаль-
ные растворы. Элементарный состав, молекулярный вес и другие
свойства смол находятся в зависимости от того продукта, из
которого они выделены, а также от методики их выделения.
Как было указано, методика выделения смол, применяемая
в настоящее время, сводится к адсорбированию их тем или иным
твердым адсорбентом с последующим извлечением из адсорбента
подходящим растворителем1. В качестве адсорбентов наиболее
часто применяются при этом силикагель и флоридин, причем
результаты, получаемые в этих случаях, существенно разнятся
друг от друга. Как общее правило, флоридин извлекает значи-
1 Другие методы, например непосредственное применение к минераль-
ным маслам различных растворителей (винны^ спирт, ацетон), гораздо
менее совершенны и ныне оставлены. •
276
тельно больше смол, чем силикагель, причем разница может до-
ходить до 50—60% и выше. Эта разница зависит, невидимому,
от конденсирующего действия флоридина па вещества промежу-
точного характера и масла, тал: что цифры, получаемые с фло-
ридином («флоридиновые смолы»), являются слишком высокими;
силикагель дает величины («силикагелевые смолы»), более отве-
чающие истинному содержанию смол, хотя, ловидимому, не-
сколько ниже действительности. Ввиду этого в дальнейшем мы
будем касаться только силикагелевых смол.
Нейтральные смолы перегоняются в катодном вакууме без
разложения [6]; при перегонке же в менее глубоком вакууме
они разлагаются. Частично нейтральные смолы перегоняются
также с парами в обычных условиях перегонки нефти и могут
быть обнаружены в различных дестиллатах, начиная с кероси-
нового.
Образование смол легко наблюдается при подогреве дестил-
латов, особенно если пропускать при этом в нагретую жидкость
кислород. В зависимости от исходного материала, послужившего,
для выделения смол, интересно проследить изменение их состава
и молекулярного веса
Таблица 77
Молекулярный вес и элементарный состав нейтральных смол
Исходный материал Молек. вес смол Элементарный состав смол •
% С % н % s |%О
Нефть беспараф. грози- уд. вес 0,876 589 84,1 9,8 0,8 5,3
Нефть балахано-сабунч., , . 0,878 738 83Д 10,4 0,8 5,3
Дестиллат керосин, грозненский . . . 290 77,9 10,0 1Д 103
. , бакинский. . . 265 79.3 9,8 1.7 9,2
Дестил чат соляровый грозненский - . 298 89,9 9,9 1.6 7,6
„ , бакинский . . . 345 80,6 9,7 3,6 7,1
Дестиллат машинный грозненский . . 466 82,3 10,2 1,3 6.2
, , бакинский . . . 433 82.2 9,7 1,0 7,1
Дестиллат цилиндров, грозненский . . 471 82.6 10,1 1,2 6,1
ж „ бакинский. . . 501 83,6 10,4 1Д 4,9
Гудрон грозненский 757 84,8 9,8 0,5 4,9
. бакинский 915 , 85,4 10,4 0,4 3.R
Как-видно из табл. 77, элементарный состав смол, получен-
ных из соответственных нефтепродуктов, одинаков, независимо
от того, из какой нефти они получены. Для молекулярных ве-
сов соответственных смол также получаются величины одного
по-рядка. Таким браэом молекулярные формулы смол, вычислен-
ные на основе этих данных должны быть близки между собой,
независимо от того, из какой нефти получен исходный дестиллат.
К иным выводам приводит сопоставление молекулярного веса
и состава нейтральных смол, полученных из различных дестил-
латов. Легко видеть, что молекулярные веса смол оказываются
тем больше, чем тяжелее дестиллат, иначе говоря, чем выше сред-
штй молекулярный вес образующих его углеводородов. Так как
смолы представляют собою, повидимому, первую переходную сту-
пень от нефтяных углеводородов к асфальтенам (ср. ниже), то
такое соотношение вполне понятно. С другой стороны, нельзя
не заметить также изменения в составе при переходе от низших
смол к высокомолекулярным: наблюдается заметное увеличение
процентного содержания углерода и уменьшение содержания
серы, ^ргда как процент водорода заметно не изменяется, коле-
блясь в пределах 9,7—10,4%.
Нейтральные смолы обладают интенсивной окраской и весьма
сильной красящей способностью. Особенно это касается тяже-
лых смол, выделенных из гудронов и сырых нефтей: их красящая
способность в 10—20 раз выше, чем смол из солярового или керо-
синового дестиллатов, и достаточно, например, 0,005% тяжелой
нейтральной смолы, чтобы окрасить бесцветный бензин в соло-
менно-желтый цвет. Окраска дестиллатов и не содержащих
асфальтенов сырых нефтей обусловливается наличием именно
нейтральных смол.
По своим химическим свойствам нейтральные смолы очень
напоминают асфальтены. К азотной и серной кислотам, пяти-
хлористому фосфору, формалину и другим реагентам они отно-
сятся подобно асфальтенам. Их иодные числа невелики, так
что только для самых тяжелых смол оказывается возможным
допустить наличие одной двойной связи.
Очень характерна для нейтральных смол способность превра-
щаться в асфальтены. Такое превращение особенно легко про-
исходит при доступе воздуха и при нагревании; но оно может'
происходить и без доступа воздуха, очевидно, за счет процессов
дальнейшей конденсации, которые могут протекать уже при
обыкновенной температуре. Химизм превращения смол в асфаль-
тены пока еще недостаточно ясен, поскольку химическая при-
рода как смол, так и асфальтенов требует дальнейших иссле-
дований.
Асфа.тьтогеновые кислоты и их ангидриды (кислые смолы). Этот вид
смолистых веществ изучен меньше рассмотренных выше. Они представляют
собою смолообразные вещества удельного веса выше единицы. Растворимы
в спирту и хлороформе, но мало в бензине. Сера содержится в них в пере-
менном количестве. При продолжительном нагревании до 120° сначала пе-
реходят в ангидриды, затем в вещества, уже потерявшие способность обмы-
ливаться.
Молекулярный вес цсфалътогеновых кислот показывает, что они
образовались в результате соединения двух молекул высококипящего углево-
дорода. Их можно назвать поэтому полинафтеновыми кислотами. От обыкно-
венных нафтеновых кислот они отличаются содержанием серы, более высо-
ким молекулярным весом, трудной растворимостью в воде натровых солей
и нерастворимостью в бензине медных солей. Как показывает табл. 78,
асфальтогеновые кислоты, выделенные из нефтей (тексасской), по ряду
своих свойств довольно близко стоят к кислотам из природного асфальта
(тринидадского).
Карбены и карбоиды. С внешней стороны карбены и кар*
-бонды похожи на асфальтены, отличаясь от последних лишь бо-
лее темной окраской и несколько ‘ повышенным ^содержанием
278
Таблица 7&
Исходный материал Свойства асфальтоген оных кислот
Растворим, в бензине Кислотн. число Число омылив. Иодное число % S
Нефть (Тексас) .... Почти не 89.3 147,0 17,3 1.2
Асфальт (Тринидад) . растворимы 98,5 120,4 22,4 3,1
кислорода. Сходство это распространяется также на их химиче-
ские свойства: все три рода веществ дают аналогичные реакции
с азотной и 'Серной кислотами, хлорным железом, сулемой и т. д.
Характерно различие их в растворимости: карбены не раство-
римы в четыреххлористом углероде, но легко растворимы в серо-
углероде; карбиды подобно углям не растворимы ни в каких
растворителях. От каменных углей они отличаются тем, что при
перегонке вовсе не образуют фенолов.
Карбены и карбоиды не встречаются в сырых нефтях и
редко — в природных асфальтах, но они составляют обычную
часть так называемых асфальтитов (см. ниже). Довольно много
карбенов и карбоидов образуется также при продувке сквозь-
нефть воздуха или при действии на нее серы при 120°. При
более низкой температуре в тех же условиях образуются глав-
ным образом нефтяные смолы и асфальтены.
Акцизные смолы При техническом анализе нефтей и некоторых нефте-
продуктов одним из обычных испытаний является определение смолистых
веществ по акцизному способу или, короче говоря, «акцизных смол». Сущ-
ность испытания заключается в обработке сухого нефтепродукта (50 сл*>
в бензиновом растворе серной кислотой удельного веса 1,84 (10 см9) с по-
следующим определением увеличения сернокислотного слоя (кислого гуд-
рона). Это увеличение нижнего слоя, будучи помножено на 2, дает процент-
ное содержание по объему акцизных смол в испытуемом нефтепродукте.
Акцизные смолы отнюдь не представляют собою какого-либо особого
вида смолистых веществ При обработке нефти или нефтепродукта серной
кислотой в указанных условиях в кислотный слой переходят, кроме-
рассмотренных выше смолистых и асфальтовых веществ нефти, также ее-
непредельные углеводороды, а частично, несомненно, также и некоторые вы-
сокомолекулярные углеводороды предельного характера. Таким образом опре-
деление акцизных смол, представляя собою совокупность ряда разнообраз-
ных процессов физико-химического характера, отнюдь не может служить
для характеристики содержания в нефтепродукте одних смолистых и асфаль-
товых веществ. Научная ценность таких определений весьма ограничена и
условна, и такое заключение вполне оправдывается существованием описан-
ных в литературе явно парадоксальных случаев, когда акцизная проба имеет
отрицательное значение, очевидно за счет растворимости кислого гудрона
в бензиновом растворе нефтепродукта, или когда содержание акцизных смол
получается явно преувеличенным, например больше 100%, очевидно за счет*
перехода в кислый гудрон части бензинового раствора.
ВЗАИМНЫЕ СООТНОШЕНИЯ МЕЖДУ РАЗЛИЧНЫМИ ВИДАМИ
СМОЛИСТЫХ И АСФАЛЬТОВЫХ ВЕЩЕСТВ
Как уже было отмечено, (первыми продуктами превращения
нефтяных углеводородов в ‘ смолистые и асфальтовые вещества
279»
являются,' невидимому, нейтральные и кислые смолы. Образо-
вание смол происходит уже при стоянии масла на воздухе,,
быстрее — при нагревании, о чем можно судить на основания
его постепенного потемнения Очевидно, при этом происходит
процесс окисления масла; однако схема этого процесса до сит
пор не может считаться вполне выясненной. На основе соло--
ставления средних молекулярных весов нефтяных фракций и
образующихся из них смол можно предполагать, что в громад-
ном большинстве случаев реакция окисления ограничивается
здесь действием на частицу углеводорода двух атомов кислорода
без глубокого распада органической молекулы. Однако механизм
этой реакции остается пока еще совершенно не ясным.
Далеко не все углеводороды дают при окисляющем действии
кислорода (также серы) одинаково легко нейтральные и кислые
смолы. Тяжелые углеводороды окисляются легче, чем легкие; не-
предельные— легче предельных, особенно парафинов. Отсюда
понятно, что каждая фракция нефти, начиная с керосиновой,
дает* свои смолы, для нее характерные.
Пре(вращение нейтральных смол в асфальтены уже было рас-
смотрено выше. Хотя механизм этого превращения также
•остается еще не выясненным, несомненно, однако, одно обстоя-
тельство: так как молекулярный вес у асфальтенов по крайней
мере в 2—з раза больше чем у смол, то, очевидно, в образовании
частицы асфальтена принимает участие несколько частиц ней-
тральных смол, иначе говоря, мы имеем здесь какую-то реакцию
глубокого уплотнения (конденсации) более простых молекул
в частицы более сложные.
Отношение асфальтенов к образующимся из них карбенам и
карбоидам является наименее ясным; естественно, что о меха-
низме превращения первых во вторые пока ничего не известно.
Все вышеизложенное о взаимных соотношениях между раз-
личными видами смолистых и асфальтовых веществ и об их
образовании может быть выражено следующей схемой:
нефтяные углеводороды
Ф
нейтральные смолы
Ф
асфальтены
карбены и карбоиды
В этой схеме не достаточно ясно лишь положение кислых
смол. Что они образуются наряду с нейтральными смолами из
углеводородов нефти, едва ли подлежит сомнению. Но какова
их дальнейшая судьба в процессе постепенного изменения смо-
листых и асфальтовых веществ нефти, остается невыясненным.
СОДЕРЖАНИЕ В РАЗЛИЧНЫХ НЕФТЯХ И НЕФТЕПРОДУКТАХ
Вопрос о содержании смолистых и асфальтовых веществ
в различных нефтях представляет большой интерес с точки зре-
ния их сравнительной характеристики. Основным и необходимым*
280
условием для такого сопоставления различных нефтей должно
быть, очевидно, сравнимость имеющегося цифрового материала
и, следовательно, известная общность методики, применявшейся
для его получения. В этом направлении, по отношению к нефтям
СССР в ГрозНИИ проделана большая исследовательская ра-
бота [3], позволяющая охарактеризовать наши нефти в отно-
шении содержания в них смолистых и асфальтовых веществ,
вполне научно хотя, быть может, и недостаточно полно *.
Таблица 79
Содержание смол я асфальтенов в нефтях СССР
№ п/п Месторождение нефти Уд. вес нефти при 15° Содержание
ас фальтенов В °/о смол сила- кагел. в %
1 Сураханы (Баку) 0.848 0,0 • 4,0
2 Балаганы „ 0,876 следы 4,8
3 Сабунчи , 0,872 следы 4,9
4 Бннагады ...... 0,918 0,6 11,9
5 Биби-Эйбат „ 0,865 0,3 9,0
6 Шубаны » 0,919 1,2 13,7
7 О. Артема , 0,924 0,5 15,8
8 Грозный, парафин, нефть . . 0,841 0,9 4,3
9 „ беспараф. . . . ода 1,8 7,8
10 „ ел.-параф. „ . . 0,845 . хз 5Л
11 Майкоп, легкая ' „ . . 0,843 0,3 6,5
12 „ тяжелая „ . . ода 03 10,7
13 Калужская . . 0,955 0,5 20,0
14 Доссор (Эмба) 0,873 0,0 1,6
15 Макат 0.905 0,0 5,0
Как видно из данных табл. 79. по содержанию смолистых и
асфальтовых веществ большинство нефтей СССР можно разде-
лить на две группы нефти 1егкие и тяжелые. Первая группа
нефтей с удельным весом до O,SSO—0,ь90 характеризуется отсут-
ствием или незначительным содержанием асфальтенов и срав-
нительно небольшим содержанием смол (до Ъ%). Примерами
могут служить легкие бакинские и особенно эмбенские нефти
(Доссор). Тяжелые нефти, относящиеся ко второй группе, имеют
удельный вес около 0,9 и выше, в них наблюдается уже заметное
содержание асфальтенов, содержание же смол поднимается в них
до 10—15% и выше. Примерами этого рода являются тяжелые
бакинские, майкопские и калужская нефти. Полным отсут-
ствием асфальтенов и небольшой смолистостью и здесь выде-
ляется эмбенская нефть (Макал). Между легкими и тяжелыми
нефтями намечается еще промежуточный тип, не укладываю-
щийся в очерченные выше границы; сюда относятся, например,
биби-эйбатская нефть и легкая майкопская.
1 Содержание смол определялось суммарно без разделения их на кис
лые и нейтральные.
281
Совершенно особое место по содержанию смолистых и асфаль-
товых веществ занимают грозненские нефти (№№ 8—10). Прд
-своих малых удельных весах (0,841—0.855) грозненские нефти
всех трех типов, т. е. парафинистая, слабопарафинистая и бес-
парафиновая, содержав до 1% и выше асфальтенов, тогда как
по содержанию смол они довольно близки к легким нефтям дру-
гих месторождений. Таким образом грозненские нефти выде-
ляются из ряда других нефтей ССОР своим резко выраженным
асфальтеновым характером.
Переходя к другим европейским и американским нефтям,
прежде всего можно встретить среди них в отношении содер-
жания смолистых веществ те же два основных типа, что были
намечены выше для нефтей СССР. Легкие румынские, галиций-
ские, а также чехословацкие нефти либо вовсе не содержат
асфальтенов, либо содержат их не свыше 0,15%; примерно то же
относится к легким нефтям Пенсильвании, Оклахомы и друш
американских -месторождений. Напротив, тяжелые нефти Гер-
мании, Эльзаса, Калифорнии, Тексаса и т. д. содержат до 1%
и больше асфальтенов и повышенные количества смол. Нефти
асфальтенового характера, аналогичные грозненским, также
можно встретить среди американских нефтей. Такова, например,
легкая нефть месторождения Мексиа (Тексас) с удельным весом
0,845, в которой содержание асфальтенов равно 1,3%, а смол
силикагелевых — 5 % [7 ] •
Как вещества нелетучие или перегоняющиеся лишь с боль-
шим трудом, асфальтены и смолы при отгоне светлых продуктов
концентрируются в остатках, т. е. в мазуте и гудроне. Как уже
было указано, одновременно при ©том происходят также ново-
образование смол за счет конденсации масел и дальнейшее пре-
вращение смол в асфальтены. Понятно поэтому, что содержание
смол и асфальтенов, возрастая по мере углубления отбора, до-
стигает наибольших величин для данной нефти в масляном гуд-
роне. При дальнейшей концентрации процесс обогащения
остатка смолами и асфальтенами идет все дальше, так что в ре-
зультате получаются гудроны, твердые при обыкновенной тем-
пературе, т. е. нефтяные остаточные асфальты. Некоторые ана-
литические данные для характеристики этих асфальтов будут
приведены ниже.
ЗНАЧЕНИЕ И ПРИМЕНЕНИЕ СМОЛИСТЫХ
И АСФАЛЬТОВЫХ ВЕЩЕСТВ НЕФТИ
Присутствие смолистых веществ в дестиллатных нефтепро-
дуктах влияет на их качество лишь в отрицательном смысле:
ухудшает цвет, увеличивает нагаро- или коксообразование. Осо-
бенно вредное влияние оказывают -смолистые вещества на каче-
ство смазочных масел, так как при хранении масел, особенно
же в работе, их смолы легко превращаются в асфальтены; по-
следние же оказывают еще более отрицательное влияние на ка-
чество масел — понижают их смазывающую способность и еще
282
более повышают коксообразование. Именно с этими обстоятель-
ствами связана одна из важнейших задач очистки нгфъшротук-
тов: по возможности освободиться от содержащихся в них смо-
листых и асфальтовых веществ.
Есть однако, особый вид нефтепродукта, в котором смоли-
стые и асфальтовые вещества являются главной составной ча-
стью. Это — природные асфальты, получившие настолько обшир-
ное и разнообразное применение, что для восполнения их ре-
сурсов в настоящее время широко развилось производство искус-
ственного асфальта. Так как и природные и искусственные
асфальты своим происхождением теснейшим образом связаны
с нефтью, то ниже в связи ^вопросом об утилизации смолистых
и асфальтовых веществ нефти они подлежат краткому рассмо-
трению.
Природные асфальты. Природные асфальты делятся на не-
сколько групп:
1. Собственно асфальты или горные смолы. Это — более или
менее обширные скопления естественного асфальта на поверх-
ности земли, образующие иногда целые озера. Таковы, напри-
мер, асфальтовые озера на о. Тринидаде (Малые Антильские
острова), в провинции Бермудец (Венецуэла), в Мексике, на
о. Сахалине (Охинское асфальтовое озеро) и др. Добыча асфальта
может производиться здесь лопатами, в некоторых местах — кир-
ками. Продукт содержит обыкновенно довольно значительную
примесь минеральных (веществ; применяется главным образом
в строительном: деле.
2. Асфальтиты. Отличаются от собственно асфальтов до-
вольно значительной твердостью и блеском; почти не содержат
минеральных примесей, являясь, таким образом, почти чистыми
битумами. Таков, например, сирийский асфальтит и ряд других,
известных под различными названиями: гельсонит (Юта и Ко-
лорадо), грэемит (Зап. Вирджиния), альбертит (Куба, Мексика)
и-др. Асфальтиты добываются обыкновенно горными разработ-
ками, так как не встречаются па поверхности земли, а заполняют
ее трещины; сирийский асфальтит выносится в Мертвое море
соседними горячими источниками и по охлаждении застывает
кусками. Применяется главным образом в лаковом (производстве.
3. Асфальтовые породы. Характерной особенностью-является
здесь преобладание нал битумом минеральных частей извест-
няков и песчаников Встречается в разных странах, в частности
в СССР (около Сызрани и др.). Добыча в зависимости от мест-
ных условий ведется или открытыми разработками, или в шах-
тах и штольнях. Применяются в больших количествах для по-
крытия дорог в форме асфальтового порошка, получаемого путем
измельчения асфальтовой породы, или в виде гудрона после ее-
вываривания.
Едва ли можно сомневаться, что асфальты образовались из
нефтей. Самый процесс асфальтизации нефти представляет со-
бою результат испарения легких частей нефти с одновременным
обогащением остатка смолами и асфальтенами, образующимися
283.
путем одновременных процессов окисления и конденсации высока
молекулярных нефтяных углеводородов. Наиболее подхо-
дящим материалом для такого рода превращения являются
нефти нафтенового типа, особенно тяжелые, богатые смолами д
сернистыми соединениями. О том, что некоторую роль в образо-
вании природных асфальтов играют сернистые соединения,
можно заключить из того, что содержание серы в ‘асфалътц
довольно высокое (2—12%). По Гсферу 8 —участие сервь
стых 'соединении в процессе асфальтообразования на о. Трини-
даде сводится к следующему: из скрытых трещин смоляного
озера выступают сульфатные воды, содержащие главным образом
гипс, который восстаиовляется битумом до сероводорода анало-
гично следующему уравнению:
CaSO4<+ СН4 = CaS + С02 + 2Н20.
Образующаяся при этой реакции углекислота вытесняет даже
из сернистого кальция свободный сероводород, который частью
выделяется в виде газа или уносится водой, частью, действуя
на битум, замещает в нем кислород серой. В пользу подобного
участия 'Сернистых соединений в процессе образования асфальт!
говорит не только значительное содержание углекислоты и серо-
водорода в газах, вырывающихся из смоляного озера на о. Три-
нидаде, но также нередко наблюдаемая близость мес^рожденф
серы, сероводородных вод и асфапьта в других странах (Сици-
лия, Ганновер и т. д.). Следует, впрочем, иметь в виду, что про-
цесс обогащения серой отнюдь не является обязательным спут-
ником асфальтообразования, так как встречаются асфальты, пра-
вильнее асфальтиты, в которых содержание серы сравнительное
велико (меньше 2%).
Таблица 80
Состав природных и искусственных асфальтов
(по И/аркуссону)
Материал Кислоты асфаль- тогено- вые °/о Ангид- риды асфаль- товых кислот °/о Асфаль- тены % Смолы °/о Масла °/о
Яриродн«е асфальты
Тринидадский сырой . 6,4 3.5 37,0 23,0 31,0
Бермудецский очищ. . Искусственные асфальты 3,9 2,0 35,8 14,4 39,6
Канзасский (медиум) . 0 3,0 24,0 11,0 62.0
Германский (мягкий) . 0 4,0 4,4 8,6 83,0
Русский 0 2,0 15,5 16,1 66,0
Что касается состава природных асфальтов, то основные •ком-
поненты самого битума уже были подробно рассмотрены выш^
284
1ак например, важнейшие природные шфаняы — тринидадский
и бермудецский1 — состоят, кроме минеральной части, исклю-
чительно из смол, нейтральных и кислых, асфальтенов и масел
(табл. 80). В составе асфальтитов заметное участие принимают
также карбены и карбоиды.
Искусственные (нефтяные) асфальты. Различают два типа
нефтяных асфальтов* остаточные и окисленные. Первые полу-
чаются в остатке после отгонки масляной части нефти в резуль-
тате частью концентрации смолистых и асфальтовых веществ
нефти, частью новообразования их при отгонке масел. как это
уже было отмечено выше. При получении вторых процесс со-
провождается продувкой воздуха при не слишком высокой тем-
пературе (около 200° и выше), благодаря чему новообразование
смолистых и асфальтовых веществ ускоряется и протекает более
энергично.
Искусственные асфа шты представляют собой вещества едва
текучие, иногда твердые при обыкновенной температуре, темно-
бурого цвета. Удельный вес их около единицы. При сухой
перегонке дают масляные лестиллаты и кокс. Горят коптящим
пламенем, оставляя коксообразный остаток. От природных
асфальтов отличаются низким содержанием серы (обычно
ниже 1%), что, впрочем, зависит от содержания серы в исход-
ной нефти.
Подобно природным искусственные асфальты, как было уже
указано, состоят из смол, асфальтенов и масел. Однако в коли-
чественном отношении здесь наблюдается значительная разница.
Как видно из Данных табл. 80, искусственные асфальты отли-
чаются от естественных, во-первых, полным отсутствием асфаль-
тогеновых кию тот и. во-вторых, значительно большим содержа-
нием масел. Кроме того, природные асфальты (всегда содержат
большее или меньшее количество минеральных примесей, тогда
как в искусственных асфальтах содержание золы редко превы-
шает 1%, так что они представляют собою почти чистый битум.
Наконец, должно быть отмечено еще одно различие природных и
искусственных асфальтов: в то время как природные асфальты
либо вовсе не содержат парафина, либо содержат его крайне не-
значительное количество, в искусственных асфальтах содер-
жание парафина может достигать нескольких процентов.
Техническое получение искусственных асфальтов, которые по своим
качествам не уступали бы естественным или по крайней мере к ним при-
ближались, составляет важную и нелегкую технологическую задачу [10].
Успех процесса естественно связан как с выбором сырья, -так н с надлежа-
щими условиями его переработки. Вопрос о сырье более или менее ясен:
наиболее подходящим материалом для искусственной асфальтизации, как
уже было указано, являются тяжелые, богатые смолами нефти нафтенового
типа. Гораздо сложнее вопрос о надлежащей переработке этого сырья: тем-
пературные условия и глубина отбора масла, особенно же условия после-
дующей продувки воздухом, — все эти факторы, вместе взятые, а частью
и каждый из них в отдельности, могут оказать решающее влияние на оо-
1 Близким по составу к бермудецскому асфальту оказался природный
асфальт с большого асфальтового Охинского озера на о. Сахалине [9].
285
став конечного продукта, а именно на соотношение между тремя важней-
шими компонентами асфальта, определяющими его технические качества:
масла — смолы — асфальтены. Успехи, достигнутые в этой области, в ча!
стности за последние годы в СССР, громадны. Они позволяют широко ис-
пользовать дешевый искусственный асфальт там, где раньше применялся
исключительно природный асфальт, которым европейские страны, в том
числе и СССР, слишком бедны, чтобы покрыть все возрастающую потреб-
ность в этом материале.
Для полноты обзора искусственных (нефтяных) асфальтов следует упо-
мянуть в заключение об асфальте из кислого гудрона. При разбавлении этого
гудрона водой на поверхности разбавленного сернокислотного слоя виде,
ляется слой кислой смолы в виде более или менее густой массы, после
надлежащей обработки которой (промывка, отгонка легких частей и про-
дувка воздухом) получается темнобурая смолистая масса, хрупкая на хо-
лоду, но размягчающаяся при нагревании. Кроме смол, асфальтенов и масел,
асфальт из кислого гудрона содержит еще продукты взаимодействия асфаль
тов с серной кислотой, устойчивые по отношению к щелочи. По своим
качествам кислые асфальты значительно уступают другим видам искусствен-
ного асфальта.
ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ АСФАЛЬТА
Важнейшими областями применения асфальтов являются до-
рожное и строительное дела, потребляющие глазную массу при-
родного и искусственного асфальта для таких назначений, как
мощение улиц и тротуаров, заливка полов на заводах и в лабо-
раториям, а также под паркет, изоляция от сыро ли, покрытие
канализационных труб и т. п. Далее, асфальт применяется для
приготовления кровельного толя; в электротехнике как изолятор
для изготовления оболочки кабелей; для приготовления разного
рода специальных замазок и клея и т. д. Наконец, значительные
количества асфальта потребляет лаковая промышленность для
получения асфальтовых лаков. Почти во всех этих случаях
с одинаковым успехом могут применяться как природные
асфальты и асфальтиты, так и подходящие по качеству сорта
искусственных асфальтов. Природный сирийский асфальт нахо-
дит, кроме того, интересное применение в репродукционной тех-
нике, основанное на его замечательном свойстве превращаться
в нерастворимую модификацию после воздействия на него в тон- .
ком слое солнечного света.
О размерах и росте продукции нефтяных асфальтов в Аме-
риго могут дать представление следующие данные [11]:
Добыча асфальта в тоннах
В 1914 г....... 674 470
, 1924 .......3078 000
„ 1927 ....... 3 528 080
. 1930 „..... 5139000
В СССР лучшим сырьем для получения нефтяного асфальта
считаются калужская нефть, тяжелая бинагадинская и грознен-
ская беспарафино-вая. В связи с грандиозным размахом дорож-
ного строительства в СССР производство нефтяного асфальта
должно быть отнесено к категории наиболее актуальных и ответ-
ственных отраслей нефтяной промышленности Союза, продукция
которой быстро возрастает с каждым годом.
286
ЛИТЕРАТУРА
Маркуссон И., Асфальт, пер. с нем. Кузнецова С. И., М. — Л. (1926).
Абрахам Г., Асфальты и другие битумы, пер. с англ., ОНТИ (1934).
1. Marcusson, „Chem. Ztg.* 37, 822 (1914); .Ztschr. angew’ Chem.“ 29, 346
(1916); 31, 113 (1918); „Petroleum* 12, 1149 (1917).
2. „Нефт. и сланц. хоз.“ VII, № 11—12, 933 (1924).
3. „Нефт. хоз." IX, № 8, 222 (1925).
4. .Нефт. хоз.* XIII, № 9, 334 (1927).
5. Васильев Н. А. и Жирнова Л. В., „Нефт. хоз.* XVIII, А» 11—12, 707 (1S29).
6. Но опытам Коровкиной В. А. в Ш И Акад, наук (частное сообщение
Левенсон В. Э.).
7. „Химический состав и классификация нефтей", сборник .Химический
состав нефтей и нефтяных продуктов*, „Труды ГровНИИ", М. — Л.
(1931). f
8. Engler-H6fer, Das ErdoL т. П, 52, 1909.
9. Наметкин С. С. и Павлова С. Я., .Нефт. хоз.* XX. № 1, 86 (1931).
10. Ср. Воронов А. и Логвинова Н. II., „Нефт. хоз.* XVIII, № 3, 449 (1930).
11. Кети С., .Oil a. Gas J.*, 192S, 112.
ГЛАВА XI
МИНЕРАЛЬНЫЕ ВЕЩЕСТВА НЕФТИ
Под минеральными веществами нефти разумеются:
1) зола, получающаяся .при сгорании нефти, представляющая
собою остатки уех веществ минерального состава, которые всегда,
правда в крайне незначительном количестве, содержался
в нефти;
2) вода, почти всегда сопровождающая нефть и в небольшом
количестве находящаяся в ней в растворенном состоянии, а ино-
гда образующая с ней трудно разделимую эмульсию.
Таким образом зольные вещества и вода являются кошю-
нентами нефти, подлежащими рассмотрению в настоящей главе.
А. Зола
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ И СОДЕРЖАНИЕ ЗОЛЫ В НЕФТИ
Количественное определение золы в нефти и ее продуктах
производится осторожным нагреванием навески вещества (200—
300 г) в платиновой чашке. Сначала при этом удаляются лету-
чие ‘составные части нефти; затем остаток обугливается, и
в чашке, вместе с зольными веществами, остается кокс, который
при последующем прокаливании сгорает. Привес чашки после
прокаливания определяет количество золы во взятой навеске.
Сведения о количественном содержании золы в различных
нефтях далеко недостаточны и на первый взгляд даже несколько
противоречивы. Так, согласно старым определениям Марковни-
кова и Оглоблина [1] балаханский профильтрованный мазут
содержит 0,14% золы, что при пересчете на нефть дает в сред-
нем 0,09%. С другой стороны, более поздние определения Тур-
вина показали для важнейших бакинских нефтей следующее
содержание золы (табл. 81).
Таблица 81
Содержание золы в бакинских нефтях
Нефть Уд. вес Акцизные СМОЛЫ в % Золы в %
Сураханская 0,852 2,8 0,0(22'
Балаханская 0,8^2 10,0 0,0026
Биби-эйбатская 0,88У 13.1 0,0032
Бинагадинская 0,924 19,6 0,0138
288
Как видно, по Турвйчу содержание зольных веществ в балй-
ханской нефти в 36 раз меньше, чем по определению Маркович-
нова и Оглоблина; такое противоречие т{>ебует разъяснения.
Известно, что при перегонке нефть всегда более или менее
значительно разъедает стенки перегонного куба, причем иод
влиянием кислотных компонентов нефти железо частично может
переходить в раствор. Естественно поэтому, что определение
зольного остатка в мазуте (как у Марковникова и. Оглоблина)
всегда должно давать преувеличенное содержание золы по срав-
нению с тем, что получается при определении золы непосред-
ственно в самой нефти. Известное обогащение железом, хотя,
конечно, в значительно меньшей степени, чем при перегонке,
возможно также при подъеме нефти по обсадным трубам. Так
как коррозия обсадных труб, естественно, будет тем сильнее, чем
богаче нефть кислотными компонентами, то понятно, что нефти
тяжелые, смолистые являются обыкновенно также и наиболее
зольными (ср. табл. 79 и 81).
С зольными компонентами нефти не следует смешивать ее
механические примеси, каковыми могут быть песок, мельчайшие
частицы.железа обсадных труб, стираемых переносимым нефтью,
песком, и т. п. .Большая часть этих примесей остается в отстой-
никах, но наиболее мелкие частицы могут удерживаться нефтью
во взвешенном состоянии и уноситься дальше, особенно если
нефть принадлежит к типу высокосмолистых нефтей. Ввиду
этого яеред определением золы нефть обязательно должна быть
.профильтрована, причем в случае надобности она может быть
разбавлена;каким-либо растворителем (бензином и т. п.); послед-
ний после фильтрования раствора отгоняют, а затем уже при-
ступают к определению золы. К сожалению, не все описанные
в литературе определения золы нефти сопровождаются оговор-
кой относительно фильтрования; соответствующий опытный
материал не представляет, очевидно, никакой научной цен-
ности.
Возвращаясь с вышеприведенными оговорками к вопросу
•о количественном содержании в нефтях зольных веществ, огра-
ничимся указанием, что в этом, отношении между различными
нефтями наблюдается довольно большое разнообразие. Если
в одних нефтях, как мы видели (табл. 81), содержание золы не
превышает тысячных долей процента, то, с другой стороны,
имеются нефти, в которых содержание зольных веществ дости-
гает сотых и даже десятых долей процента. Как уже было ука-
зано, особенно выделяются в этом отношении тяжелые нефти
с высоким содержанием смолистых веществ.
Отметим в заключение, что зола обнаруживается не только
в нефти, но также в нефтяных дестиллатах и продуктах.
Однако в этих последних случаях она уже-не является состав-
ной частью исходной нефти: соответствующие вещества попа-
дают здесь либо в результате разъедания аппаратуры (холодиль-
ника), либо вследствие неполноты промывки при очистке де-
стиллатов.
19 Зак. 1622. Химия нефти.
2^9
ХИ^ИЧЁСКИЙ СОСТАВ НЕФТЯНОЙ 3(кШ
Обычными компонентами нефтяной золы являются соедине-
ния Са и Mg, Fe, Al и «SdOa. Значительная часть этих зольных
веществ попадает в нефть либо из воды и пород, с которыми
нефть приходит в соприкосновение, либо в результате разъеда-
ния рабочей аппаратуры (ср. выше). Хотя на долю этих компо-
нентов обыкновенно приходится подавляющая часть нефтяной
золы, тем не менее с точки зрения химии нефти они, очевидно,
не представляют большого интереса.
Новейшие систематические исследования состава нефтяной
золы позволяют расположить ее элементы ио степени их важ-
ности (встречаемости) в следующий ряд [2]:
SO N V Р К Ni J Si Са Fe Mg
Ха Al Мп Pb Ag Au Cu Ti U Sn As
Хотя этот ряд пока еще нельзя считать окончательно уста-
новленным, тем не менее рассмотрение его представляет выдаю-
щийся интерес. Мы видим прежде всего, что перечисленные выше
металлы (Са, Mg, Fe, Al) и даже S.iO2 занимают в ряду с дру-
гими элементами нефтяной золы далеко не первое место; они
отодвинуты ближе к середине ряда; на первое же место вслед
за такими элементами как S, О, N, соединения которых были
рассмотрены выше в отдельных главах, выступают ванадий (V),
фосфор (Р), калий (К), никель (Xi) и иод (J). Эти элементы
нефтяной золы щюдставляют, очевидно, особый интерес.
Никель. Открытие этого элемента .в нефтяной золе было сделано недавно
(Рамзай, 1923 г.). Систематическое исследование большого числа нефтей
в этом направлении «приводит к заключению, что никель, невидимому,
является постоянной составной частью нефтяной золы. Он обнаружен также
в золе некоторых торфов, бурых углей и неоднократно в растениях. Содер-
жание никеля в нефтяной золе может достигать 2% и выше, но часто оно
выражается лишь долями процента. Так, анализ золы бакинской нефти по-
казал в ней содержание лишь 0,01% NiO [з]. Ввиду того что никель как
катализатор обладает высокими гидрирующими свойствами, некоторые
авторы рассматривают его присутствие в нефтяной золе как указание на
то, что в процессе нефтеобразовапия весьма существенную роль, играли
реакции гидрирования.
Ванадий. Ванадий довольно широко распространен в природе. Он
является составной частью ряда специальных минералов (ванадинит, кар-
нотит и др.), не образуя, однако, столь богатых скоплений, как, например,
железные, медные и другие руды, в которых ванадий нередко встречается
в небольших количествах. Далее, ванадий обнаружен в золе углей, сланцев
и торфа, в золе естественного асфальта, в золе некоторых морских растений
и животных (голотурии асцидии и др.); наконец ванадий часто является
составной частью нефтяной золы в количествах, которые колеблются
в весьма широких пределах (от 0 до 40% и выше); в частности, например,
в золе бакинской нефти ванадий обнаружен не был [3].
Вопрос о форме, в которой ванадий содержится в нефти, а также’
r указанных выше животных и растительных организмах пока • еще не
достаточно ясен; однако уже самый факт нахождения «ванадия в золе
столь разнообразных видов топлива, как нефть, уголь, сланцы и т. д., ука-
зывает на известную общность процессов их образования и с точки зрения
их геохимии представляет исключительный интерес.
2fl0
О практическом значении ванадия ясфиший зилы можно «удить из
того обстоятельства, что на утилизацию ванадия из нефти заявлены спе-
циальные патенты.
Фосфор, найденный впервые в золе канадской нефти, неоднократно
обнаруживался затем в различных нефтях; его соединения пользуются,
повидимому, столь же широким распространением средн нефтей, как соеди-
нения ванадия. Содержание фосфора в нефтяной золе достигает, считая на
фосфорный ангидрид, 5% и выше, а самое нахождение его рассматривается
некоторыми авторами как подтверждение теории происхождения нефти из
животных остатков.
Не останавливаясь подробнее на различных компонентах неф-
тяной золы, отметим в заключение, что присутствие многих из
них, особенно тяжелых и драгоценных металлов, не является по-
стоянным и носит, надо думать, случайный характер. В качестве
типичного примера привода! анализ золы иранской нефти
(Майдан-и-Нафтун):
Fc2O8, А]20з .......... 26,96 NiO ......................2,70
СаО......................2,31 МпО . . . •...............4,39 '
MgO......................1,18 Р905 .................... 5,53
V,O5.....................5,03 SO3......................35,29
SiO,.....................2,29
Б. Вода
Нефть, свободная от воды, получается лишь в сравнительно
редких случаях при добыче; обыкновенно же ее сопровождает
так называемая «буровая вода», количество которой иногда в не-
сколько раз превышает количество добываемой нефти. При хра-
нении свежедобытой нефти большая часть буровой воды обыкно-
венно легко отстаивается и может быть механически отделена.
В таких случаях в нефти остается лишь небольшое количество
воды, которая находится частью в растворенном, частью во взве-
шенном состоянии. Нередко, однако, отделение воды от нефти
задерживается: нефть образует с водой на вид совершенно одно-
родную эмульсию, для разделения которой требуется подогрева-
ние или даже особые специальные приемы (см. ч. II). Таким
образом вода является не только спутником нефти, но с полным
правом может рассматриваться как одна из ее составных частей.
Буровые воды всегда содержат большее пли меньшее количе-
ство минеральных солей, состав которых не может, очевидно, нс
отражаться на составе зольных составных частей нефти, и об-
ратно; уже одно это обстоятельство оправдывает рассмотрение
золы и воды нефти в одной, настоящей главе.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ И СОДЕРЖАНИЕ ВОДЫ В НЕФТИ
Из разнообразных -способов определения воды в нефти и неф-
тепродуктах наибольшее распространение получили способ от-
стаивания и способ перегонки.
Способ отстаивания. По этому способу одновременно произ-
водится определение воды и механических примесей. Для этого
291
пользуются специальным сосудом— отстойником на особой де-
ревянной подставке; для обогрева в процессе работы его перено-
сят в стакан с горячей водой (фиг. 33}.
Испытуемую нефть или нефтепродукт взбалтывают в течение
10 мин. для получения однородной смеси с осевшей водой и на-
ливают в сухой отстойник до метки 50 сл3. Затем добавляют
к смеси сухой бензин до метки 500 см3, хорошо взбалтывают
течение 3—5 мин. и, поместив отстойник в деревянную под-
ставку, дают смеси отстояться в течение примерно 20 час. Если
по истечении этого времени получилось полное разделение жид-
Фиг. 33. Отстойник для опреде-
ления воды.
костей, то вся вода и грязь соби-
раются в нижнюю, суженную
часть отстойника. Тогда произво-
дят отсчет и, помножив отмерен-
ное количество отстоявшейся
воды на два, получают содержа-
ние се в объемных процентах.
Если отстаивание задерживается и
между солями воды и масла наблю-
дается слой эмульсии, то для разруше-
ния последней отстойник переносят
в стакан с теплой водой (70°) и оста-
вляют е>ч> ид1!г.ь на 20—40 мин. В боль-
шинстве случаев после этого наблюдает-
ся полное разделение водного и масля-
ного слоев, и вода частью оседает
в нижней части отстойника, частью же
в виде капелек прилипает к его стен-
кам. С помощью специальной медной
палочки а все эти капельки (росу)
собирают в нижнюю часть отстойника,
после чего производят отсчет, дав
предварительно прибору с его содер-
жимым принять комнатную темпера-
туру.
Наконец, в тех случаях, когда не-
смотря на нагревание, полного раз-
рушения эмульсии все-таки не наблю-
дается, к исследуемой смеси приба-
вляют 1—2 c.w-1 нафтеновых кислот или в крайнем случае 1—2 см2 со-
ляной кислоты удельного веса 1,12, вновь перебалтывают смесь и ведут
определение, как описано выше. В случае прибавления соляной кислоты
при отсчете, естественно, делают соответствующую поправку.
Способ иерогрнкп. Характерной особенностью этого метода
является определение воды после ее отгонки от нефти или нефте-
продукта. Особенно широкое распространение получила та моди-
фикация этого способа определения воды, которую предложили
Дин и Старк.
'Прибор ьДина и Старка (фиг. 34) состоит из круглодонной
медной колбы Л на 500 см3 и специального градуированного
приемника В, соединяющего колбу с обратно поставленным холо-
дильником С. Для определения воды отмеривают 100 см3 хорошо
взболтанной нефти (или нефтепродукта) в колбу с помощью
пипетки и смывают пипетку 100 см3 бензина с температурой ки-
292
в
Фиг. 34. Прибор
/Тина и Старка.
пения выше 93°, сливая бензин в ту же колбу. Затем, соединив
кол оу с холодильником через градуированный приемник, нагре-
вают ее, регулируя кипение жидкости так, чтобы из холодиль-
ника в приемник падало 2—4 капли в секунду.
Градуированная часть приемника быстро запол-
няется жидкостью: внизу собирается вода, свер-
ху же—бензин, избыток.которого стекает обратно
в колбу. Когда количество воды в приемнике
перестанет увеличиваться, нагревание колбы
прекращают, дают приемнику принять комнат-
ную температуру и, отсчитав объем собравшейся
в нем воды, получают се содержание в объемных
процентах.
Если бы нефть или нефтепродукт совершенно
не образовали с водою эмульсий, содержание
в них воды определялось бы, очевидно, ее раство-
римостью, которая, как было указано в гл. II,
выражается примерно лишь тысячными долями
процента. Непосредственный опыт показывает,
однако, что воды содержится даже в отстоявшей-
ся нефти нередко в количестве 1—2% и более, и
уже одно это обстоятельство евпдете: ьствует, что
главная часть (воды находится здесь нс в раство-
ренном, а во взвешенном состоянии, в виде
громадного количества микроскопических капелек, т. е. в форме
эмульсии. Физико-химическая природа нефтяных эмульсий бу-
дет рассмотрена во второй частп настоящего курса в связи
с вопросами обезвоживания нефти.
ХИМИЧЕСКИЙ СОСТАВ БУРОВЫХ ВОД [4 —8|
По своему составу буровые воды подобно другим видам есте-
ственных вод характеризуются содержанием более или менее
значительных количеств минеральных и отчасти органических
солей. Общее содержание солей в различных буровых водах («су-
хой остаток») — чрезвычайно разнообразно. Так например, в бу-
ровых водах Грозненского района оно не превышает 6,3%. спу-
скаясь во многих случаях до десятых, а изредка даже до сотых
долей процента. В других случаях минерализация буровых вод
значительно больше. Так, содержание солей в буровых водах
Балаханской площади (Баку) достигает 17%, в некоторых ^же
пенсильванских буровых водах оно превышает 26%. Таким обра-
зом по своей минерализации многие буровые воды значительно
превышают воды открытых морей и океанов1 и нередко прибли-
жаются к типу так называемых рассолов.
1 Содержание солей в «водах Атлантического и Великого океанов ко-
леблется в пределах 3,2—3,7%. В Средиземном море высший предел —
также около 3,7%. .В закрытых и полузакрытых ‘морях -содержание солей
‘ 298
Переходя к ближайшей характеристике солей, входящих в.со-
став буровых вод, остановимся прежде всего на способах выраже-
ния результатов их химического анализа.
Еще недавно результаты химического анализа естественных
вод выражали в процентах содержащихся в них окислов (NagO,
СаО и т. д.) или солей (NaCl и т. д.). Такой способ в настоящее
время совершенно оставлен. Так как естественные воды содержат
обыкновенно несколько солей, обычно сильно диссоциированных
и образующих в растворе крайне сложную систему равновесия,
то более правильно выражать химический состав естественных
вод в ионной форме, например в миллиграмм-ионах на 1 л воды.
Такой способ пользуется в настоящее время широким распро-
странением и вполне отвечает существу данного вопроса.
Катионами солей, входящих в состав буровых вод, являются
чаще всего Na’, Са' * и Mg", значительно реже Ге" и К’;
обычные анионы буровых вод — СГ и НСОз', реже — SO/' и
СОз". Кроме того, в состав буровых вод нередко входят недиссо-
циированные окислы, например АПОз, Гс-зОз и особенно S1O2,
находящиеся в растворе в коллоидном состоянии. Из перечислен-
ных ионов с количественной стороны на первом месте должны
быть поставлены ионы Na’ и СГ. В некоторых буровых водах
относительные количества этих двух ионов таковы, что в пе-
ресчете па соль не менее 90% от сухого остатка должно быть
отнесено за счет хлористого натрия. По сравнению с ионом Na’
другие катионы, далее Са’ ' и Mg", представлены'в буровых во-
дах несравненно слабее.
Что касается анионов буровых вод, то первенствующее зна-
чение вместе с ионом СГ здесь имеет ион ПС Оз', в количествен-
ном отношении нередко выдвигающийся даже на первое место.
Ион серной кислоты, SO/', представлен в буровых водах на-
столько слабо, что еще недавно одним из характерных признаков
буровых вод считалось полное отсутствие в них сернокислых со-
лей. Еще слабее представлен в буровых водах ион СОз". Кремне-
кислота является одной из' постоянных составных частей буро-
вых вод, однако в сравнительно небольших количествах (14—
50 мг/л). Сероводород встречается лишь изредка. Большой инте-
рес представляет присутствие в буровых водах небольших коли-
честв брома и особенно иода (см. ниже). Наконец, имеются ука-
зания на присутствие в некоторых буровых водах радия, причем
радиоактивность некоторых вод по своей величине приближается
к радиоактивности некоторых классических источников [9].
Одной из’ обычных составных частей буровых вод являются
органические кислоты, содержащиеся в них в форме солей. При
подкислении соляной кислотой такие воды дают характерный бе-
лый осадок или муть от этих кислот. При определении щелоч-
ности буровых вод соли органических кислот ведут себя подобно
обыкновенно оначитсльно меньше— влияние пресноводных рек. Так, в во-
дах Черного моря сухой остаток составляет 1,S%, а в водах Каспийского
моря — только 0,6%.
294
углекислым солям и определяются вместе с ними титрованием
соляной кислотой в присутствии метилоранжа в качестве инди-
катора. Таким образом найденная этим способом щелочность
буровых вод заключает в себе все слабые кислоты, в том число
и органические, соли которых тидролизованы. Если кроме об-
щей щелочности определить в буровой воде содержание ионов
СОз", НСОз' и S", то по разности можно рассчитать в ней со-
держание органических кислот, например в эквивалентах угле-
кислоты, в предположении, что все органические кислоты —
одной основности, например, одноосновны. По своей величине со-
держание органических солей в буровых водах обыкновенно
меньше содержания углекислых солей, редко равно ему и еще
реже превышает его.
Ближайшее исследование органических кислот, содержащихся
в буровых водах, давно уже [10] обнаружило в них жирные кис-
лоты, начиная с муравьиной (НСОгН) и кончая капроновой
(C5H11CO2II). Позднейшими работами было доказано присутствие
в них также нафтеновых кислот.
Приведенная характеристика состава буровых вод дана по наиболее
типическим образцам. В какой мере здесь возможны отклонения от ука-
занного типа, показывает [11] анализ воды, сопровождающей уральскую
нефть (месторождения Чусовские Городки). Из анионов на первом место
здесь оказался ион SO/7; содержание пажа СГ здесь в два раза меньше,
чем иона SO/7; углекислых солей в воде, сопровождавшей первые тонны
уральской нефти, вовсе не было обнаружено. Из катионов первое место
по содержанию в этой воде занял ион NH* * \ второе место — ион Са''
(в два раза меньше, чем иона NH**), далее — ион К', и только четвертое
место оказалось за ионом Na' (в два раза меньше, чем Са‘').
ХИМИЧЕСКАЯ КЛАССИФИКАЦИЯ БУРОВЫХ ВОД
Несмотря на кажущееся разнообразие, буровые воды могут
быть сведены по своему составу к очень немногим основным
группам. Чтобы определить положение их (как и всякого дру-
гого типа естественной воды) в химической системе естественных
вод, прежде всего необходимо выразить результаты их анализа
в более наглядной и сравнимой форме, а именно не в миллиграм-
мах ионов на 1 л воды, как это делается обыкновенно, а в осо-
бых единицах, которые могут быть названы «реакционной ем-
костью» ионов (Стеблер, Палъмер). Для перехода, от обычного
способа выражения химического состава какой-либо воды к этому
новому способу, достаточно найденное содержание каждого ион£,
выраженное в миллиграммах на 1 л воды, разделить на величину
эквивалента данного иона, например для Na' —на 23,0; для
СГ —на 35,40; для Mg" — на 12,6; для СОз —на 30 и т д.
Таким образом получают содержание каждого иона в особых
1 Азотистые основания еще раньше были обнаружены в некоторых
грозненских буровых водах [12]. Отметим, что когда примерно через полгода
после первого анализа чусовской воды мы получили и проанализировали
второй образец этой воды, то аммонийных оснований в нем почти не ока-
залось.
295
единицах, которые названы выше «реакционной емкостью» и ко-
торые представляют собой в сущности миллиграмм-эквиваленты
данного тнша. 'Выражая содержание того или иного иона в мил-
лиграмм-эквивалентах на 1 л, ставят- обыкновенно перед соот-
ветственной числовой величиной индекс г.
Химические свойства воды как раствора почти полностью
определяются содержащимися в Ней анионам it и катионами,
в частности их относительной концентрацией. Выражая послед-
нюю в миллиграмм-эквивалентах, можно дать числовое значение
целому ряду основных химических свойств воды. Очевидно так-
же, что.для любой воды сумма эквивалентов всех катионов дол-
жна быть равна сумме эквивалентов всех анионов, в ней нахо-
дящихся. Это соотношение позволяет проверять результаты ана-
лиза воды путем простого суммирования реакционных емкостей
отдельных анионов и катионов. Если в результате сопоставления
суммы эквивалентов всех анионов с суммой эквивалентов всех
катионов наблюдается небольшое несоответствие (1—-2%), его
можно отнести за счет ошибки анализа. Если несхождение более
значительно, это указывает на нахождение в воде какого-либо
иона, который при анализе не определялся и • может быть опре-
делен количественно путем простого расчета, т. е. вычитания из
суммы реакционных емкостей всех анионов суммы реакционных
емкостей всех катионов (или паоборо'А. В некоторых случаях,
в частности при анализе суровых вод, таким приемом пользуются
иногда для определения реакционной емкости некоторых ионов,
например щелочных (ср. табл. 82 и 83), Особенно наглядно и
просто получаются все эти соотношения, если результаты ана-
лиза воды выразить в процентах реакционных емкостей отдель-
ных ионов.
Основными свойствами естественных вод по Пальмеру явля-
ются их соленость и щелочность.
Соленость воды обусловливается солями, которые водой не
гидролизуются; она может быть связана с любым из катионов,
но зависит лишь от ионЛ сильных кислот, а именно: соленость
воды равна удвоенному числу миллиграмм-эквивалентов ионов
сильных кислот (СГ, SO/).
Различают первичную и вторичную соленость. Первичная со-
леность 'Si определяет это свойство воды лишь в отношении ще-
лочных ионов, т. е. главным образом ионов Na; и К*; вторичная
соленость 82 характеризует его в отношении щелочно-земельных
ионов, т. е. Са’ ’ и Mg‘ *. Вторичная соленость совпадает с поня-
тием «постоянная жесткость» воды.
Щелочность воды обусловливается-гидролизом солей и зави-
сит от присутствия в воде солей слабых кислот, главным образом
углекислых, а в буровых водах — также органических (см. выше).
Щелочность воды равна удвоенному числу миллиграмм-эквива-
лентов ионов слабых кислот.
Различают: первичную щелочность 41, отвечающую содержа-
нию в воде ’щелочных солей слабых кислот, и вторичную щелоч-
ность Л 2. соответствующую содержанию щелочно-земельных со-
296
Таблица сР2
Состав буровых вод
I К J а с е. по П а л ь м е р у '
• 1 г 2 1 2 * 3 1 4 1 5 г 6 з
Jft-ноны на 1 л Ха, К’ 9 593,0 1995,0 1 714,7 1683,4 а) 1007,0.
Са" 29,8 36,0 5,6 2,9 71,5 10,0
Mg" 84,4 57,1 4,7 3,6 36,2 6,3,0
СГ 4210.0 2 076,8 1126,7 352,1 23 9000 147,0
so/ 613,2 — — —. 312,0 111,0
со/ 372,0 1110,0 1308,0 1917,0 4 554,4 1148,0
ЛА-эквпваленты на 1 л г. Na, ГС ... . 417,45 90,68 75,35 73.38 825,0 43,7
г. Са" 1,49 0,18 0,27 0.15 4,0 0,5
г. Mg" 6,92 4,70 0,76 0,30 3,0 0,5
г. СГ • 400,70 58,56 31,78 9,93 674,0 4,1
г. SO/ 12.77 6,0 2,3
г. СО/ 12.39 37,00 43,60 63,90 152,0 38,3
Минерализация . ед,72 191,12 150,75 147,66 1664,0 89,4
Реакцион. емкость в % Na, К’ 49.01 47,45 49,32 49.70 49,6 49,0
Са" 0,17 0,09 0,18 0,10 1 0.2 0,5
Mg" . 0,82 2,46 0,50 0,20 0,2 ал
СГ 47,05 30,64 21,08 6,72 40,5 4,6
SO/ 1,49 .—. — 0,4 2,6
СО/ 1,46 19,36 28,92 43,28 9,1 42,8
Первичная соле- ность 97,08 61,28 43,16 13,44 81,8 14,4
Вторичная соле- ность 0 0 0 0 0 0
Первичная ще- лочность . . . 0,94 33,62 56,48 85,96 17,4 83.6f
Вторичная ще- лочность . . . 1,98 5,10 1,36 0,60 0,8 2,0
лей тех же кислот4 Вторичная щелочность совпадает с поня-
тием «временная жесткость» воды.
Обозначим реакционные емкости в процентах для щело-
чей — а, для щелочных земель — Ъ, для других катионов — с, для
сильных кислот — т, для слабых кислот — я. Между этими ве-
личинами могут существовать различные соотношения, опреде-
ляющие пядь основных классов естественных вод, по Палъмеру:
а) Непосредственно не определялось.
* № 1—4—грозненские буровые воды по данным ГрозНПИ [о] с пе-
ресчетом Малярова К. Л. [8].
2 № б — буровая врдд Бакинского района. (Хорос ан ы 13]).
« № 6 —буровая вода месторождения Коалинга (Калифорния [ь]).
* Третичные соленость и щелочность, отвечающие содержанию таких
ионов, как F^‘‘, АГ” и т. и., могут быть оставлены без рассмотрения
ввиду крайне незначительной пх величины для буровых вод.
Таблица 83
Состав буровых вод
• 11 класс III класс IV класс
7 1 8 2 9 2 10 3 И 3 12 2
ЛГг-ионы на 1 л
Na, К‘ 4137,0 а) а) 10476,0 а) а)
Са” 81.0 400,2 6 217,9 319,4 380,9 1143,5
Mg" . 40,0 223,2 2 708,4 242.5 53,1 1007,3
Cl' 6 367,0 27900,0 78 300,0 14154,0 14 510,0 13 900,0
S04" а) 528,0 — 4 285,7 — 2 820,0
С03" 218,0 927,3 1363,6 92,1 — —
лИг-эквп валенты
на 1 л
г. Na’, IC . . . . 179,9 790,0 1719,0 455,55 385,80 311,0
г. Са” 4,0 20,0 311,0 15.94 19,00 57,0
г. Mg ’ 3,3 18,0 223,0 19,94 4,40 83,0
г. Cl' 179,6 786,0 2 208,0 399,14 409,20 392,0
г. S04" 0,3 11,0 — 89.21 —— 59,0
г. С03" 7.3 31,0 45,0 3 07 — -—
374,4 1 656,0 4 506,0 982.86 818,40 902,0
Реакцион. емкость
в %
Na*, К’ 481 47,7 38,2 4635 4 719.00 34,5
Са” 1.0 1,2 6,9 1,62 2,37 6,3
Mg” 0,9 1.1 4,9 2,03 0,44 9,2
Cl' 48,0 47,4 49,0 40,61 50,00 43,5
S04" 0,1 0,7 — 9,08 — 6,5
С03" 1.9 19 1.0 0.31 . -— —
Первичная соле-
нооть 96,2 95,4 76,4 92,70 94,38 69,0
Вторичная соле-
ность 0 0.8 21,6 6,68 5,62 31,0
Цервичная ще-
~-ТОЧНОСТЬ . . . 0 0 0 0 0 0
Вторичная ще-
лочность . . . 3,8 3,8 2,0 0,62 0 0
I. а > т, т. с. реакционная емкость щелочей (в процентах)
больше реакционной емкости сильных кислот, так что в воде
имеется избыток щелочи, связанной со слабыми кислотами, на-
пример с угольной кислотой; этот избыток щелочи определяет
первичную щелочность воды.
II. а==щ, т. е. реакционная емкость щелочей (в процентах)
равна реакционной емкости сильных кислот, так что щелочи и
я) Непосредственно не определялось.
1 '—буровая вода месторождения Коалинга (Калифорния) [б].
2 Лг 8, 9 и 12—воды Бакинского района: Illy-баны, Сабунчи и Шихов
мыс, [13J.
3№ 1о и 11 — грозненские буровые воды по данным 4ГрозН1Ш ’6])
с пересчетом Малярова К. Л. [18],
29$
сильные кислоты полностью нейтрализуют друг друга; первичная
щелочность и вторичная соленость у вод II класса отсутствуют.
III. а < m < а 4- Ь, т. е. реакционная емкость щелочей, (в про-
центах) меньше реакционной емкости сильных кислот, которая
в свою очередь меньше суммы реакционных емкостей щелочей
и щелочных земель. Таким образом воды III класса содержат
избыток сильных кислот, которые дают со щелочными зем ними
вторичную соленость.
IV. а +: Ъ = т, т. е. сумма реакционных емкостей щелочей и
щелочных земель (в процентах) равна реакционной емкости
сильных кислот; щелочи и щелочные земли полностью нейтра-
лизованы здесь сильными кислотами. Таким образом первичная
и вторичная щелочность в водах IV класса отсутствуют.
V. а + Ъ < т, т. е. сумма реакционных емкостей щелочей и
щелочных земель (в процентах) меньше реакционен емкости
сильных кислот. Таким образом воды V класса характеризуются
присутствием в них свободных сильных кислот.
Соотношения эти наглядно и просто выражаются графически следую-
щим образом [8]. Разделим линию на 50 равных часп-й и допустим,
что вдоль нее скользят три перпендикуляра: а, 0 л т. Установим перпен-
дикуляры аир так, чтобы площади прямоугольников а, b и с соответ-
ствовали процентам реакционных ёмкостей щелочей а, щелочных земель h
и других металлов с, а перпендикуляр Y—так, чтобы площади т и п
отвечало реакционным емкостям сильных и слабых кислот. Перемещая
перпендикуляры а, 0 и Y вдоль линии АВ, можно выразить состав любой
води в процентах реакционных емкостей. Фиг. 35, 36, 37, 38 и 39 дают
графические изображения всех пяти классов естественных вод по Палъмеру;
здесь же приведены характеристики их солености и щелочности, вытекаю-
щие из самого определения этих понятий.
Наблюдение и опыт показывают, что громадное большинство
буровых вод относится к I классу Пальжра * *. Наибольшее зна-
чение для характеристики вод I класса имеют первичная соле-
ность Si и первичная щелочность Ai. Как ясно из их определе-
ний, величины Si и Ai должны изменяться в обратном напра-
влении: чем больше Si, тем меньше Ai, и наоборот. Пределы ко-
лебаний этих величин для буровых вод чрезвычайно велики:
так, для грозненских буровых вод I класса первичная соленость
изменяется в пределах от 8,5 до 97, а первичная щелочность —
от 0,12 до 87. Тем самым, в зависимости от величины отноше-
ния Si: Ai буровые воды I класса Палълера могут быть разоиты
на следующие три основных группы [8]:
1. Соленые воды, в которых первичная соленость значительно
превосходит первичную щелочность, так что отношение Si : не
меньше 2.
2. Щелочные воды, в которых первичная соленость значи-
тельно меньше первичной щелочности, так что отношение Si:
меньше 0,5.
1 Кроме буровых вод, к I классу Палъмера принадлежат многие воды,
связанные с изверженными породами. Сюда относятся воды таких озер,
как Байкал, Онежское и др.
*2‘.9
3. Промежуточные типы вид, а именно щелочно-соленые
(2>jS'i ; Ai>i), т. е. воды с преобладающей соленостью и солено-
щелочные (1 > Sj : Al > 0,5), т. е. воды с преобладающей щепол-
ностью.
Приведенные в табл. 82 примеры принадлежат исключительно
к 1 классу Палъмера (№ 1—о).
Фиг. 36. Класс II: ш = о; *91=5«=5т;
^1л = 2b.
Фиг. 85. Kiacc I: ♦«<«; *9; = 2м;
= 2{а — w); А о = 2b.
Фпг 37. Kiacc III: a<^m<^a-j-b
S. = 2a; & = 2 (»i — ar, J2 = 2 {a I-
hb —ш).
Фиг. 38. Класс IV: m == a -|- b;
= 2a; 8* = 2b.
Фиг 39. Класс V: m > a + b,
81 = 2a; = 2b.
Среди суровых вод различных месторождений можно встре-
тить немало примеров, не удовлетворяющих основному условию
для вод I класса (a>w); эти воды относятся главным образом
к III классу \ для ното-рого а < т < а Ъ. -Первичная щелоч-
ность вод III класса равна нулю: такова же. очевидно, пер-
вичная щелочность вод II, IV и V классов по Палъмеру. Таким
образом отношение £i: Ai, позволившее разделить буровые воды
1 К III классу Палъмера относятся -воды, связанные с осадочйыми по-
родами: атс воды большинства рек и многих тгеточнико-в.
300
i класса на несколько характерных групп, для вод других палЬ-
меровских классов теряет свое знамени**.
Характерной особенностью вод III класса является их вторич-
ная соленость л*2. Величина вторичной солености колеблется для
различных вод в очень широких пределах. Для вод III класса
она больше нуля и достигает для буровых вод 60%, а дтя вод
некоторых источников 70% и выше. Все воды III класса
являются более пли менее жесткими, причем жесткость их обу-
словливается двумя факторами: хлоридами и сульфатами щелоч-
ных земель («постоянная жесткость») и бикарбонатами щелоч-
ных земель («временная жесткость»).
111 класс представлен среди буровых вод значительно беднее,
чем 1 класс. Тем не менее, образцы вод III класса можно встре-
тить среди буровых вод различиях месторождений; несколько
примеров этого рода приведено в табл. 83 (№ 9—10).
Промежуточное положение между водами I и III классов за-
нимают воды II класса: для них а — т, т е. реакционная емкость
щелочных ионов в точности равна реакционной емкости анионов
сильных кис тот. Очевидно, это — .тишь особый случаи, для ко-
торого и первичная щелочность и вторичная соленость равны
нулю. Воды II класса можно представить себе в результате сме-
шения вод I и Ш классов. Среди буровых вод они встречаются
очень редко, но некоторые из них чрезвычайно близки к этому
классу (табл. S3 № 7—8).
При постепенном возрастании реакционной емкости сильных
кислот эта величина может оказаться, наконец, равной сумме
реакционных емкостей щелочных и щелочно-земельных ионов.
Этим условием (w = а 4- Ь) определяется IV класс Палъмера;
к нему принадлежат воды морей и соляных озер. Среди буровых
вод воды IV класса встречаются почти столь же редко, как воды
II класса (табл. 83, Ла 11—12).
Воды V класса вовсе не встречаются среди буровых вод.
Приведенная классификация по существу лишь определяет
положение буровых вод в общей химической системе естествен-
ных вод без всякого учета не только их геологических признаков,
но и таких основных факторов, как процессы изменения их хи-
мического состава во времени под влиянием тех или иных спе-
циальных условий отдельных месторождений. Такие процессы
могут в корне изменить состав изнача1ьной пластовой воды; они
также сообщают буровым водам ряд специфических особенностей,
частью уже отмеченных. Эти процессы представляют поэтому
в выдающийся интерес.
В заключений следует отметить, что выражение состава вод в ионной
форме, а следовательно, и в форме реакционной емкости не может отра-
зить содержания в воде таких компонентов, как растворенные газы, кол-
лоиды и т. и. Некоторые авторы «полагают поэтому, что лишь выражением
состава воды в весовых или атомных процентах элементов могут быть
достигнуты полная обднюсть способа количественного выражения состава
ноды в отношении всех растворенных в ней веществ, а. также независимость
выражения аналитических данных от меняющихся теоретических пред-
ставлений [14].
. 3ul
Химические процессы в буровых водах
Важнейшие из процессов изменения состава буровых вод,
наблюдаемые в естественных условиях, следующие.
Обессеривание буровых вод. Как уже было указано, ион сер-
ной кислоты (SO/')» как общее правило, или совершенно отсут-
ствует в буровых водах, или находится в них в крайне незначи-
тельных количествах. Объяснение такой особенности состава бу-
ровых вод наметилось в двух основных направлениях: либо при-
нимают, что обессеривание буровой воды произошло в результате
изменения состава изначальной. («первообразной») воды под
влиянием восстановляющего действия на ее сульфаты нефтяных
углеводородов, либо рассматривают процесс обессеривания буро-
вой воды как процесс биохимический, протекающий при ближай-
шем участии в нем особых микроорганизмов.
Восстановление сульфатов под влиянием углеводородов нефти
может происходить по уравнению (Энглер и Г сфер):
• CaS04 + CH4 = CaS + G02 + 2H20. (1)
Следующей фазой реакции является вытеснение серово-
до|юда углекислотой:
CaS + С02 + Н20 = СаСОя + H2S. (2)
Наконец, под влиянием окисляющего действия кислорода
воздуха или других окислителей сероводород может превра-
щаться в элементарную серу:
H.2S 4- 0 = S + Н20. (3)
Термодинамический расчет показывает [г>], что восстановле-
ние различных сульфатов нефтяными углеводородами должно
происходить далеко не с одинаковой легкостью. Наиболее легко
должен восстанавливаться сернокислый кальций; в этом случае
восстановителем могут быть самые разнообразные углеводороды
нефти, начиная с метана. Наиболее трудно должны восстанавли-
ваться сульфаты щелочных металлов; здесь реакция восстано-
вления может итти лишь при участии непредельных углеводоро-
дов, например этилена или его гомологов, которые находятся
в нефти в очень небольших количествах. Экспериментальная
проработка этого вопроса находится пока в начальной стадии.
Весьма вероятно, что неодинаковая легкость восстановления
различных сульфатов является одной из причин существования
сульфатных буровых вод. Характерно, что большинство этих вод’
(табл. 82 и S3, № 1—7) не имеет постоянной жесткости (^2 = 0),
либо она является у них крайне незначительной (N® 8). Следо-
вательно, ион SO/' связан в этих водах лишь со щелочными
ионами, главным образом с ионом Na’, а эта соль, как было
отмечено, восстанавливается углеводородами нефти с особым тру-
дом. Правда, изредка встречаются сульфатные воды .(№ 10 и 12),
ооладающш» некоторой постоянной жесткостью ($2 > 0). В этих
302
случаях можно допустить, что процесс обессеривания боды еще
•не успел дойти до конца, и ближайшее рассмотрение состава
этих вод вполне подтверждает такое предположение.
Действительно, обращаясь -к уравнению (2), видим, что вслед
за восстановлением сульфатов должны происходить вытеснение
сероводорода углекислотой и образование карбонатов. Накопле-
ние последаих в обессеренной буровой воде представляет обычное
явление. Наоборот, в тех случаях, когда процесс обессеривания
буровой воды еще не закончился содержание в ней карбонатов
будет невелико и во всяком случае не достигает возможного
максимума для данной воды.
Что касается второго продукта реакции (2) — сероводорода,
то как в свободном состоянии, так и в виде солей он действи-
тельно нередко встречается в буровых водах нефтяных место-
рождений. Правда, содержание сероводорода в буровых водах
обыкновенно очень невелико, но уже самый факт его нахождения
может служить признаком, что данная вода находится в стадии
обессеривания согласно вышеприведенным уравнениям. Извест-
ная неустойчивость сероводорода, особенно к окислителям
[уравнение (3)], вполне разъясняет путь его дальнейшего пре-
вращения, и нахождение во многих нефтяных месторождениях
более или менее значительных залежей серы (Луизиана, Тексас,
Италия, в СССР — район Эмбы) становится с этой точки зрения
вполне понятным.
Как видно из вышеизложенного, основной реакцией процесса
обессеривания буровых вод является восстановление сульфатов
до сероводорода;. Вопреки мнению, что такое восстановление мо-
жет [происходить под влиянием разного рода органических ве-
ществ, в частности углеводородов, биохимическое объяснение
-того же процесса требует, чтобы обязательным условием этого
восстановления в природе являлось участие в процессе микро-
организмов. Факты восстановления сульфатов под влиянием
жизнедеятельности специальных микроорганизмов [Microspira de-
sulf uricans, М. aestuarii, Vibrio termodesulfuricans известны уже
давно. Доказано также чрезвычайно широкое распространение
этих микроорганизмов в почвенных водах, реках, озерах, морях
и океанах, в глубоких артезианских колодцах, а также, пови-
димому, в буровых водах. Участие их в процессе обессеривания
буровых вод является поэтому весьма вероятным; однако пол-
ного единства во взглядах по этому вопросу пока еще не до-
стигнуто. Одни авторы представляют себе, что процесс обессе-
ривания морской воды может протекать под влиянием микро-
организмов на морском дне, после- чего обессеренная вода должна
оказаться погребенной в осадочных породах. Некоторые новейшие
авторы, напротив, считают, что нахождение микроорганйзмов
определенного типа в недрах нефтяных месторождений доказано
и что обессеривание буровых вод необходимо связывать с жизне-
деятельностью именно этих микроорганизмов. Какой из этих
взглядов отвечает действительности, должны показать дальней-
шие исследования [15].
303
Следует отметить в заключение, что в некоторых случаях сульфаты,
обрануя нерастворимые осадки (например SO«Ri, SthSr), также должны
исчезать из буровых вод. Образование этих трудно растворимых сульфа*
тов может произойти, очевидно, в результате встречи и смешения вод,
несущих растворимые сульфаты, с водами, содержащими растворимые
соль бария1 или стронция. Очевидно, однако, что такие случаи- имеют да*
лево не столь общий характер, как рассмотренные выше.
Обогащение буровых вод карбонатами. Как мы видели,
сущность процесса обессеривания буровой воды заключается
в снижении содержания в ней серной кислоты (сульфатов) за
счет одновременного увеличения содержания угольной кислоты
(карбонатов). Очевидно также, что общее содержание сильных и
слабых кислот, выраженное в эквивалентах, должно оставаться
при этом неизменным, так что процесс обессеривания буровой
воды можно выразить следующей схемой:
т • SO/' —> г • СО/.
Эта простая схема процесса обессеривания сильно ослож-
няется вследствие присутствия в буровых видах ионов щелочно-
земельных металлов.
Действительно, если растворимость в воде сульфатов н карбо-
натов щелочных металлов значительно * превышает встречаю-
щиеся в буровых водах концентрации щелочей, связанных
с угольной кислотой, то но отношению к щелочным землям тот
же вопрос решается совершенно иначе. Опыт показывает, что
растворимость сульфатов кальция и магния во много раз пре-
вышает растворимость соответствующих карбонатов. Таким обра-
зом при наличии достаточной концентрации в Дуровой воде
ионов Са'' и Mg'' процесс ее обессеривания, естественно, дол-
жен сопровождаться выпадением карбонатов щелочных земель
и, следовательно, глубоким изменением ее минерализации не
только, в качественном, но и в количественном отношениях.
Следует, впрочем-, иметь в виду, что выпадению карбонатов
может оказать серьезное препятствие содержащаяся в ’воде угле-
кислота, образование которой можно представить себе, например,
как результат взаимодействия карбонатов с органическими ки-
слотами, находящимися в нефти. Известно, что в присутствии
углекислоты растворимость щелочно-земельных карбонатов в воде
значительно возрастает за счет образования соответствующих
бика!>бонатов по уравнениям:
СаС034- СО.>+ Н.,0 = Са(НС0,.)2;
MgC08 + С02 -J- Н20 = Mg(HC03)2;
Несомненно, растворимость карбонатов но этим уравнениям
должна зависеть -прежде всего от парциальной упругости углеки-
слоты. Таким образом получается довольно сложное равновесие
между содержащимися в растворе карбонатами, бикарбонатами
и углекислотой, зависящее от таких разнообразных факторов,
как давление, температура, присутствие в растворе других со*-
лей и г. д.
Итак, содержание карбонатов кальция и магния в буквой
воде является величиной непостоянной, которая может изме-
няться в зависимости от условий в широких пределах. Тем не ме-
нее, по общему содержанию карбонатов можно сделать довольно
вероятные и важные заключения о характере буровой воды и ее
генезисе. Если- вода содержит значительные количества карбона-
тов, а также солей- нефтяных кислот, то, по всей вероятности,
она представляет собою продукт обессеривания тех или иных
изначальных вод, погребенных в недрах земли или проникающих
туда с ее поверхности Если вода бедна карбонатами и солями
органических кислот, то, вероятно, она не подвергалась процессу
обессеривания либо вследствие особенностей своего состава, либо
по характеру своего залегания.
Слгешиванив буровых вод. Соприкасаясь между собою в есте-
ственных условиях, буровые воды смешиваются друг с другом,
давая начало.водам нового состава. Даже в простейшем случае,
когда смешиваются лишь два
компонента, состав смешан-
ной воды может быть весьма
разнообразным, так как он
зависит не только от состава
и концентрации, но и от от-
носительных объемов смеши-
вающихся вод. Так как, кро-
ме того, все эти воды могут
находиться в различных ста-
Фпг. 40
диях изменения своего соста-
ва- (обессеривание и т. п.), то
понятно, что в общем процесс смешивания буровых вод
представляет собою явление необычайно сложное.
Практические вопросы, -которые могут возникать в связи
с проблемой смешивания буровых вод, весьма разнообразны.
В простейших случаях они знаются либо по правилу смеше-
ния, либо графически [7]. Представим себе, например, такой
случай: дан состав двух вод (Х« 1 и 2)—требуется установить,
является ли третья вода (№ 3), состав которой также известен,
смесью первых двух. Для решения этой* задачи построим следу-
ющий график (фиг. 40): на левой ординате отложим состав одной
из смешивающихся вод (М 1), на правой ординате — состав дру-
гой (№ 2),. в обоих случаях — в процентах эквивалентов важней-
ших ионов, характеризующих состав данных вод: N$>*,
Са* * 4- Mg“, СГ, SO4" и СОз". Соединив соответствующие орди-
наты прямыми, получаем ход изменения в содержании того или
иного иона при смешивании двух вод в различных процентных
отношениях (отложены на абсциссе). Чтобы получить ответ на
поставленный выше вопрос, возьмем на какой-либо из этих пря-
мых, например отвечающей содержанию иона Па*, точку, соот-
ветствующую* содержанию этого иона в воде № 3,‘м опустим ю
нее перпендикуляр на абсциссу. Если точки. пересечения этого
перпендикуляра с остальными прямыми будут отвечать содержа-
303
2Q Зак. 1623. Химия нефти.
нию соответствующих ионов в виде № 3, то. следи вате л ыю, эта
последняя действительно образована в результате смешивания
первых двух вод; отношение, в котором произошло это смешива-
ние, отмечается тем же перпендикуляром на абсциссе. Подобным
образом может -быть решен ряд аналогичных задач.
В тех случаях, когда одновременно со смешиванием воды пре-
терпевают обессеривание, состав их, как мы видели, может чрез-
вычайно глубоко изменяться. При этом, однако, у каждой данной
воды должна оставаться неизменной о;ща ее характеристика,
а именно отношение г - tfa* : г • СГ. Если это отношение возросло,
то, очевидно, к данной воде примешалась другая вода с большим
содержанием иона Na’ и меньшим содержанием иона СК. Если
то же отношение понизилось, то ясно,» что произошло смешивание
с водой, в которой содержание иона Na* —меньше, содержание
же иона СГ — больше, чем в данной воде.
В простейших случаях задачи на идентификацию буровых
вод, претерпевших одновременно и смешивание и обессеривание,
могут быть решены количественно методами, аналогичными
вышеуказанным [7].
Изменение минерализации. Изменение общей минерализации
буровых вод может итти в двух направлениях: в сторону ее по-
нижения или в сторону повышения.
Некоторые случаи понижения общей минерализации буровых
вод уже отмечены выше. Таков прежде всего случай выпадения
карбонатов; сюда же относятся случаи смешивания данной воды
с другой, слабее минерализованной, например с какой-либо по-
верхностной водой.
Повышение минерализации буровой воды может произойти
либо в результате смешения данной вс^ы с водой другого пласта,
сильнее минерализованной, либо как следствие выщелачивания
водой тех гарных пород и отложений, с которыми она прихода
в соприкосновение. Весьма важным фактором в процессе повы-
шения общей минерализации буровых вод может явиться также
испарение воды под влиянием быстрого движения нефтяных га-
зов, происходящего к тому же нередко при сравнительно высоких
температурах глубин земли [16]. Иногда концентрация солей
в буровой воде достигает таких пределов, что происходит засоре-
ние скважины вследствие кристаллизации в ней значительных
количеств поваренной соли. Такие случаи нередки, например,
в Пенсильвании. Так как растворимость поваренной соли мало
зависит от температуры, то причина ее массового выпадения
заключается, очевидно, в удалении части растворителя вслед-
ствие его испарения.
ГЕНЕЗИС БУРОВЫХ ВОД
Как видно из вышеизложенного, процессы изменения состава
буровых вод могут быть весьма глубоки и разнообразны. Есте-
ственно, что эти же процессы в применении к водам, послужив-
шим основой для образования того или иного образца буровой
?06
йоды, -могут также в корне изменить их состав, так что вопрос
о генезисе буровых вод, вообще говоря, является весьма слож-
ным. Для его постановки с химической точки зрения рассмотрим
вкратце основные типы естественных вод по их происхождению.
Таких типов — два: воды атмосферного происхождения и воды
первичного происхождения.
1. Воды атмосферного происхождения. Это — дождевые воды,
просочившиеся в почву и растворившие некоторое количество
наиболее растворимых составных частей горных пород; они ха-
рактеризуются более или менее значительным содержанием
сульфатов и относительной устойчивостью своего состава.
2. Воды первичного происхождения. Это — зю преимуществу
морская вода, удержанная осадочными породами при их отло-
жении и в дальнейшем сохранившая основные черты своего со-
става. Характеризуется небольшим содержанием сульфатов и
относительной устойчивостью своего состава.
Смешением этих двух основных типов вод можно получить
различные производные воды, а л вменение в их составе под вли-
янием соприкосновения с нефтью и газом, горными породами и
отложениями может дать начало разнообразнейшим видам есте-
ственных, в частности буровых, вод. В какой мере отдельные из
указанных основных типов вод принимали действительное уча-
стие в образовании буровых вод каждого данного месторождения,
этот -вопрос может быть выяснен лишь на основе совокупности
не только химических, но и геологических данных.
ЗНАЧЕНИЕ БУРОВЫХ ВОД И ИХ УТИЛИЗАЦИЯ
Систематическое изучение состава буровых вод имеет важное
значение в различных стадиях нефтяного хозяйства. Так напри-
мер, если при разведочном бурении обнаружено, что на некото-
рой Глубине в скважине появилась вода, не содержащая сульфи-
дов, то близость нефтеносного или газоносного горизонта во мно-
гих случаях становится весьма вероятной. С другой стороны,
если, например, вода, встреченная скважиной на глубине 1 000 —
1 500 м, по своему составу оказалась аналогичной воде вблизи
поверхности земли и если дальнейшее углубление скважины
сопряжено с известными затруднениями, то может быть поднят
вопрос о прекращении дальнейшего бурения.
Так как нефтеносные горизонты месторождения обыкновенно
чередуются с водоносными, то при' эксплоатационном бурении
крайне важно знать, какой состав имеет вода различных пла-
стов; это имеет большое практическое значение при решении
таких вопросов, как правильный выбор горизонта для закрытия
воды, является ли вода в данной скважине пластовой или ояя
попала в нее из других горизонтов и т. д.
Только имея ясное представление о составе и возможных из-
менениях состава вод различных пластов (ср. выше), можно на-
метить и. принять правильные меры к тому, чтобы остановить
20* 307
Происходящее обводнейие нефтеносных пластов и восстановить
правильный режим месторождения.
Хотя сами по себе буровые воды доставляют нефтяному хо-
зяйству немало хлопот и нередко требуют больших расходов для
их отвода за пределы месторождения, тем не менее в некоторых
случаях воды эти можно частично утилизировать, извлекая из
них некоторые более или менее ценные продукты. На сегодняш-
ний день таковыми продуктами для некоторых месторождений
являются поваренная- соль и иод.
Для получения хорошей и дешевой поваренной соли из бу-
ровых вод необходимо наличие нескольких условий, как-то: вы-
сокая концентрация хлористого натрия в буровой воде, относи-
тельно небольшое содержание в ней других солей, дешевизна
дальнейшей концентрации рассола и т. д. Все эти условия
имеются налицо, конечно, далеко не всегда. Одним из удачных
примеров возможности широкой утилизации буровых вод для
получения поваренной соли является оз. Беюк-шор (Балахан-
ское), ,в 10 км к северо-востоку от г. Баку [17—18].
Оз. Беюк-Шор представляет собою продолговатый бассейн длиною
в’ 6,7 к.к при ширине от 1 до 2 км. Площадь его водного зеркала опреде-
ляется в S31 ?а, глубина же в зависимости от времени года — от 0,2 до 2 л.
Общий фонд ловарелной соли озера исчисляется в 841 тыс. т; сверх того
он постоянно пополняется буровыми водами, которые при желании могут
быть целиком направлены в озе[х> с промыслов Романы— Сабунчк —«Вала-
халы. При. крайне благоприятных климатических условиях для дешевой
концентрации рассола и прп условии надлежащей постановки дела (бас-
еейнизация) оз. Беюк-Шор могло бы с избытком обеспечить все Закав-
казье п Азербайджан* поваренной солью, которая по своим качествам не
уступает лучшим образцам этого продукта.
Большой и также несомненно промышленный интерес пред-
ставляет нахождение в некоторых буровых водах мода, содержа-
щегося в них частью в виде иона J' (иодистьгй натрий), частью
в виде иона <Юз' (йодноватокислый натрий). Интересны, напри-
мер, в этом отношении буровые воды азербайджанских месторо-
ждений. Подсчитано, что две-основных канавы, отводящие тгр\
мысловые воды в Бакинском районе, Сурахано-Рамаиинская и
Кишлинская, уносят ежегодно в море свыше 500 000 кг иода при
его содержании 12—24 мг на 1 л воды. Буровые воды Сальян-
ского района (Нефте-Чала) значительно богаче иодом: они со-
держат от 30 до 50 мг иода на 1 л воды.
Способы выделения иода из его солей весьма разнообразны;
для этой цели пользуются, например, действием на ник перекиси
марганца с серной кислотой при нагревании, действием аэоти-
стой кислоты (нитрит + серная кислота), хлора, гипохлорита
и т. д. Главные трудности, с которыми приходится иметь депо
при получении иода из буровых вод, следующие: 1) небольшое
содержание иода в буровых водях при высокой их обтиай мине-
рализации и невозможности дальнейшего концентрирования рас-
творов, так как при этом происходит значительная потеря иода;
2) нещ'дко высокая щелочность буровых вод и содержание в -ник
солей нафтеновых кислот, что вызывает высокий расход серной
3>.8
кислоты и загрязнение иода нафтеновыми кислотами. Дтя прео-
доления этих трудностей и разработки промышленных методов
получения иода из буровых вод за последние 10—15 лет в СССР
была проделана большая -исследовательская работа [19], в ре-
зультате которой иодная проблема может считаться ныне решен-
ной у нас полностью. Лабораторные исследования испытывались
на полузаводских установках сначала па оз. Беюк-Шор, а затем
в HecJiTe-Чала (Сальяны), где буровые воды значительно богаче
иодом, имеют незначительную щелочность и почти не содержат
нафтеновых кислот. Техническое получение иода из буровых
вод может быть осуществлено следующим образом [19].
Буровая вода, подается в большой бассейн и подкисляется
здесь серной кислотой. Выделяющиеся при этом нафтеновые
кислоты всплывают наверх, отстаиваются и спускаются. В целях
снижения расхода на серную кислоту предпочтительно пользо-
ваться не чистой кислотой, а кислотой с нефтеперегонных заво-
дов, регенерированной из масляного гудрона. Подкисленные и
освобожденные примерно на 95% от нафтеновых кислот воды
смешиваются далее с нитритом. Выделяется свободный иод. Опыт
показал, что для улавливания иода при очень слабых его кон-
центрациях наиболее удобным является метод адсорбции. В ка-
честве адсорбентов можно пользоваться при этом активирован-
ным углем, крахмалом, клетчаткой и т. л. О крахмалом, напри-
мер, иод дает довольно прочное соединение, но оно легко разру-
шается при действии восстановите, ши, например сернистого газа,
причем иод снова переходит в раствор в виде иона J'. Две по-
следних операции могут производиться прямо на фильтре типа
обыкновенного нутча, на котором равномерным слоем расположен
слой крахмала толщиною в 2—3 см. Когда крахмал насытится
иодом (проба фильтрата на иод), прекращают доступ иодной
воды и, переключив спуск, направляют на фильтр 2%-ный рас-
твор сульфита, слабо подкисленный, заставляя его медленно
фильтроваться через крахмал под слабым вакуумом. Крахмал
при этом снова делается светлым и готов для нового поглощения
иода; сульфитный же раствор поступает на выделение иода, для
каковой цели теперь ввиду большей концентрации иона J можно
пользоваться любым из -вышеуказанных способов.
В настоящее время переработка буровых вод на под и бром
производится в СССР также на о. Челекене Температура буро-
вой воды на выходе из скважины здесь GO—«5 . Челекенская
вода к тому же совершенно не содержит нафтеновых кислот. Вы-
деление брома и иода производится здесь хлором после предва-
рительного подкисления буровой воды. Первое поглощение иода
ведется железными стружками; новое выделение иода произво-
дится также хлором с почти теоретическими выходами
(свыше 99%).
ЛИТЕРАТУРА
Сулин В. А., Воды, нефтяных месторождений СССР, ОНТИ (1935).
ТДуэвич X. Г., Научные основы переработки нефти, М.-Л. (1925).
Добрянский А. Ф., Анализ нефтяных продуктов, 2-е изд., М.-Л. (1931).
309
Добрянский А. Ф-, Itvpc технологии неф^и, £-е изд., М.-Л. (1983).
Engler С. u. u Hofer H.j Das Erdol, В. 1 (1913); В. II (1909)
1. .Ж. Р. X. О." 15, 253 (1883).
2. Hackford, „Oil a. Gas J." 1924. 18 Сент., 108.
3. „Нефт. хоз.* 12, № 6, 843 (1927).
4. Голубятников Д. В., Детальная геологическая карта Апшеронского
полуострова. Ьиби-Эйбат, ч. II (1916).
5. Роджерс, Химическое соотношение вод нефтяных месторождений, пер.
под ред. Калинкою (1924).
6. „Нефт. и сланц. хоз." 6, № 2, 239 и 7, № 8, 323 (1924).
7. „Нефт. и сланц. хоз." 8, № 6 (1925).
8. Маляров. К. JL, Химический состав буровых вод Грозненского района,
М. (1929).
9. Тверцгт В. С. и Милин В. Б. „Нефт. хоз." 18, № 11—12, 656 (1929).
10. Потылицин, „Ж. Р. X. О." 14, 800 (1882).
11. Наметкин С. С. и Успенский С. Й, „Нефт. хоз.“ 17, № 7, 79 (1929).
12. Коричное К. В., „Ж. Р. X. О.“ 38, 881 (1906).
13. „Азерб. нефт. хоз," 6, № 2, 39 (1926).
14. Вернадский В. Й, История минералов земной коры, т. II. История при-
родных вод,, ч. I. вып. 1. Госхимтехиздат, Л. (1933); см. также
„Природа* № 9 (1929).
15. Гинзбург-Карагин ееа Т. и Лавренев В., „Аз. нефт. хоз," № 3, 23 (19271;
Гинзбург-Карагичева Т., Микробиологические очерки, 2-е изд., М.-Л.
(1936).
16. Mills nd Wells, The Evaporation and Concentration of Waters asscociated
with Petroleum, Washington (1919).
17. Чингиз-Илъдръ1м, „Ж- хим. пром.* VI, № 2, 110 (1929).
18. Уразов Г. Г., Бакинские иодные озера, Материалы КЕПС (1919).
19. Магидсон О., „Ж. хяьн пром." IV, № 1, 3 (1927).
ГЛАВА XII
ПРОИСХОЖДЕНИЕ НЕФТИ
К вопросу об образовании нефти в природе можно подходить
с двух точек зрения — химической и геологической. Изучив фи-
зико-химические свойства нефти и возможности ее образования
в лабораторных условиях из того либо иного исходного мате-
риала, химик может дать первые руководящие указания по во-
просу о томлив чего и каким образом могла образоваться нефть
в природе. Как показывает опыт, возможности эти чрезвычайно
разнообразны: с химической точки зрения материнскИхМ веще-
ством нефти могли быть вещества минерального характера (угле-
родистое железо), углеродистые газообразные вещества и разного
}юда органические остатки животного и растительного происхо-
ждения. Однако эти чисто химические возможности далеко не
равноценны с геологической точки зрения. Изучая условия за-
легания нефтей в различных месторождениях земного шага, гео-
логия давно уже установила ряд основных положений, которым
должна удовлетворять всякая теория происхождения нефти, при-
емлемая с геологической точки зрения. Вкратце положения эти
сводятся к следующему:
1. Все нефтяные месторождения приурочены к осадочным
породам, образовавшимся в морских бассейнах.
2. Образование нефти происходило во все геологические пе-
риоды, за исключением архейского, не содержащего также расти-
тельных или животных остатков.
3. Образование нефти происходило при высоких давлениях,
по не слишком-высокой температуре.
4. В большинстве случаев нефть встречается в местах пер-
вичного залегания; нефтеносные свиты образуются чередованием
непроницаемых глинистых или мергелистых пород с пропитан-
ными нефтью песками и песчаниками, реже — известняками.
5. Нетронутая нефтяная залежь занимает замкнутое со всех
сторон пространство, в котором должны находиться все конеч-
ные продукты процесса нефтеобразования.
6. Места вторичного залегания, куда нефть могла переме-
ститься из первичного месторождения по трещинам земной коры,
не должны иметь никакого значения в вопросе об образовании
нефти в природе.'
311
Принимая эти положения даже без оговорок, било бы, однако,
неправильно расценивать все не удовлетворяющие им гипотезы
о происхождении нефти как отживший свой век, ненужный мате-
риал. Не говоря уже о преемственности научной мысли, благо-
даря которой все, что при данном состоянии знания в данной
области принимается за истину, становится вполне понятным
лишь ав исторической перспективе развития предшествующих
воззрений, следует иметь в виду еще одно обстоятельство, осо-
бенно важное с химической точки зрения. Мы имеем в виду не-
сомненный факт, что вопрос об образовании нефти в природе
нередко является стимулом к постановке разнообразнейших опы-
тов получения искусственной нефти, все значение которых опре-
делилось лишь в самое последнее время.
Крайняя неравномерность распределения нефти на поверхно-
сти земного шара общеизвестна. Показателем -ее, пусть с некото-
рыми -оговорками на экономические и иные условия, могут слу-
жить данные о добыче нефти по странам (табл. 84). Такая не-
равномерность в связи с давно уже определившимся значением
нефти как одною из важнейших факторов народнохозяйственной
жизни страны ставит отдельные, обездоленные нефтью страны
в крайне невыгодное и зависимое положение ио отношению
к другим странам, имеющим собственные нефтяные месторо-
ждения.
Таблица 81
Миро нал добыча нефти
18-37— 1 ИЗО гг. 1931 г. 1983 г. 1935 г. 1937 г.
Страны о- S а я (И ’HffK
я О 3 о" Я О сГ~ г? я С о ‘ <5 о
« я » » И « я
США 1732,5 65,6 112,6 62,5 119,1 63,3 131,6 60,5 178,4 62,6
СССР 339,3 12,9 21,0. 11,6 19,7 10,5 2&4 10,8 27,8 10,0
Венецуэла . . . 68,5 2,6 15,9 8,В 16.0 8,5 20,1 9,5 25,7 9,3
Иран (Персия) . 51,0 1,9 5,3 2,9 6,6 3,5 7,8 3,6 ю,2 36
Гол л. Ост-Индия 61,6 2,3 4,3 2,4 5,1 2,7 5,6 2.6 7,6 2,7
Румыния .... 50,2 1,9 6,7 3,7 6.8 3,6 8,1 3,7 71 2,6
Мексика ..... 210,3 8,0 4,4 2.4 4,5 2,4 5,3 2,4 ' 6,4 2,3
Ирак 1,4 0,1 0,2 0,1 0,1 0,1 3,6 1,6 4,1 W
Колумбия .... 11,1 0,4 2,4 1.3 1,7 0,9 2,3 1.0 2,8 1.0
Аргентина . . . 9.3 0.4 1,6 0,9 1.8 0,9 2.0 0.9 0,8
Перу 15,9 0,6 1.3 0.7 1.8 0,9 2,2* 1,0 2,4 0.9
Тринидад .... 8,5 0,3 1,3 0,7 1.3 0,7 , 1.5 0,7 2,1 0,8
Крит. Индия . . 22,9 0,9 1,0 0,6 1,1 0.6 1,2 । 0,5 1,1 - 0,4
Польша 29,3 1,1 0,6 0,3 . 0,5 0,3 0,5 ОД Ф.5 0,2
Проч, страны1 . 25,6 1.0 1.9 1Д 2,0 1,0. 2,3 1.0Г 3,7. W
Итого . - . 2 637,4 юо.о • 180,5 100,0 188,1 100,0 217,5 100кб 277,1 100,0
.
1 Включая Англию, Германию, Францию, Италию, Японию, Египет,
Канаду и др. страны.
312
/(ругой не мепее несомненный факт, это — постепенное исто-
щение старых нефтяных месторождений. Подсчеты, произведен-
ные некоторыми американскими исследователями, свидетель-
ствуют, что через 40—50 лет нефтяные запасы США будут исто-
.щеяы. Возможно, что расчеты эти слишком пессимистичны и что
на смену старых, истощенных месторождений будут открыты но-
вые, мощные залежи нефти. Такие открытия могут, однако, лишь
отсрочить полное истощение мировых запасов нефти, самый же
факт их постепенного оскудения остается несомненным. Приме-
ром тому могут служить знаменитые месторождения Пенсильва-
нии, на которых создалась нефтяная промышленность США; как
видно ггз табл. 85, эти когда-то богатейшие месторождения по ко-
личеству добываемой нефти занимают ныне среди других место-
рождений США весьма скромное место вследствие "непрерывно
продолжающей падать добычи.
Таблица 85
Дневная добыча* нефти в США в баррелях
(январь 1928 г.)
Оклахома 736 200 1ойоминг 52 300
Тексас 673050 Иллинойс 30000
Калифорния 627400 Охайо 29000
Мвкснка 180000 3-ап. Вирджиния . . . 22300
Канзас 106050 Пенсильвания 23000
Арканзас 96 100 Кентукки ’ . 21 ОСО
Луизиана 62250 Монтана . 13 050
Неравномерярсть распределения нефти на земной поверхности
в связи с исключительной .важностью, которую приобрела нефть
как в развитии промышленности и транспорта (воздушного, вод-
ного и сухопутного), так и в вопросах обороны страны, а равным
образом грозный призрак грядущего истощения мировых запа-
сов нефти в условиях все возрастающей -их эксплоатации вполне
оправдывают'то исключительное внимание, которое за последнее
время повсеместно уделяется проблеме получения искусственной
нефти. Несмотря на ряд трудностей, которые еще недавно стояли
на пути полного разрешения этой проблемы в большом техниче-
ском масштабе, едва ли можно сомневаться здесь в конечном
успехе и в близости времени, когда нефтяникам и потребителям
придется считаться в одинаковой степени как с естественной
нефтью, доминирующей в настоящее время, так и с искусствен-
ной нефтью, которой принадлежит будущее. По существу говоря,
это время, как было показано в гл. IV, уже наступило, и при рас-
смотрении вопроса об образовании нефти в "рироде необходимо
все время иметь в виду те пути искусственного получения нефти
и ее продуктов, которые после проработки в лаборатории ча-
стично шии полностью уже освоены также и промышленностью.
Теории происхождения природной нефти, чрезвычайно много-
численные и разнообразные, можно разбить на следующие основ-
ные группы:
BI3
1) 'теории минерального происхождения нефти;
2) теории происхождения нефти пз животных остатков;
3) теории происхождения нефти из растительных остатков;
4) теории смешанного типа.
Ниже будут изложены лишь наиболее типические и важные,
теоретические представления по каждой группе в отдельности.
Минеральная теория происхождения нефти. Эта теория про-
исхождения нефти связывается обыкновенно с именем Менде--
леева, хотя и раньше высказывались предположения, что нефть
образовалась из минеральных веществ (Абих, Вертело и др.).
Представления эти получили известное обоснование в опытах
Клоэца [1], который показал, что при обработке зеркального
чугуна минеральными кислотами или при действии воды на бо-
гатый углеродом ферромантан при 100—300° образуется смесь
жидких углеводородов, напоминающая естественную нефть. На
основе этих наблюдений и была развита минеральная теория
происхождения нефти, принятая тотчас же некоторыми геологами
(Абих).
Материнским веществом нефти по Менделееву [2] является
углеродистое железо, значительные количества которого должны
быть сосредоточены в глубинах земли. Такое допущение вытекает
уже из сопоставления средней плотности земли (5) с относи-
тельно малой плотностью (2,5) 'большинства минеральных ве-
ществ, встречающихся на поверхности земли; отсюда следует,
что внутри земли должны преобладать вещества более тяжелые,
например широко распространенные в природе железо и другие
металлы. Со столь же широко распространенным в природе угле-
родом эти металлы должны образовать в недра$ ’земли соответ-
ствующие карбиды, например углеродистое железо и т. п. Если
теперь представить себе, что к этим размягченным от высокой
температуры, а быть может, и жидким металлическим массам, со-
держащим карбиды, по трещинам, образующимся в процессах
горообразования, проникает вода, то в результате взаимодей-
ствия ее с карбидами, естественно, образуется газообразная смесь
углеводородов, которые, перемещаясь с места своего образования,
конденсируются в подходящих местах земной коры (пустоты,
пористые осадочные породы и т. и.) и, постепенно изменяясь со-
ответственно условиям своего залегания, превращаются в то со-
стояние, в котором мы встречаем их в виде нефти.
С химической точки зрения минеральная теория не вызывает
в настоящее время никаких сомнений.
Правда, ближайшие исследования состава углеводородной
смеси, получаемойв условиях опытов Клоэца, показали, что
предельные углеводороды содержатся здесь лишь в газообразной
части (вместе с водородом); жидкие же продукты реакции состоят
лишь из этиленовых углеводородов с ничтожной примесью в выс-
щих фракциях, вероятно, нафтенов [з]. Так как, однако, этиле-
новые углеводороды под влиянием температуры и давления,
а также подходящих катализаторов (флоридин) способны поли-
меризоваться с образованием не только соединений с открытой
314.
группировкой атомов, но также и циклических систем, причем
в присутствии водорода одновременно здесь легко могут происхо-
дить также явления гидрогенизации, то не подлежит сомнению,
что образование всех основных типов углеводородов нефти легко
объяснимо с точки зрения минеральной теории происхождения
нефти.
Образование кислородных, сернистых и азотистых компонен-
тов нефти также не встречает в настоящее время каких-либо за-
труднений с точки зрения минеральной теории. Так например,
сернистые соединения могли образоваться при взаимодействии
перегретого водяного пара с сернистыми и серусодержащими
углеродистыми металлами. Исходным материалом для образова-
ния азотистых соединений нефти могли оказаться нитриды ме-
таллов и продукты взаимодействия их с водой и т. д. Наконец,
в образовании кислородных соединений могла принять участие
окись углерода, присутствие которой вместе с водородом действи-
тельно обнаруживается, например, в вулканических газах. До-
пуская образование смеси водорода и окиси углерода (водяной
газ) как результат действия перегретою водяного пара на угле-
род, свободный или карбидный, легко представить себе, что от
взаимодействия этих газов в присутствии подходящих катализа-
торов в природе может образоваться не только пестрая смесь
кислородных соединений, но и углеводородов различных рядов,
как это показал Ф. Фишер сначала в лабораторных, а затем
в полу заводских и заводских условиях.
Основные возражения против теории минерального происхо-
ждения нефти лежат в области геологии [4]. Не входя в подроб-
ное их рассмотрение, укажем лишь, что минеральная теория про-
тиворечит требованию первичности нефтяных месторождений и
вместе с тем’оставляет необъяснимым отмеченное выше нахожде-
ние нефти лишь в осадочных породах, содержащих остатки жив-
ших когда-то организмов. Из соображений того же порядка, ле-
жащих в области геологии, заслуживает внимания указание на
недоказанность существования карбидов в недрах земли и невоз-
можность проникновения к ним воды, если бы они там даже на-
ходились. Это последнее обстоятельство обусловливается суще-
ствованием так называемого срединного пояса земли — оливгпю-
вого или базальтового, который вследствие высокой температуры
должен находиться в пластическом (вискозном) состоянии, что
исключает возможность образования в ©том поясе каких-либо
трещин или пор для проникновения сквозь них воды. Наконец,
указывают на очевидное родство между нефтью и ископаемыми
углями, что, в свою очередь, заставляет ожидать известной общ-
ности в составе материнского вещества, давшего начало нефти,
с одной стороны, и ископаемым углям — с другой; для углей же
органическое происхождение является несомненным.
Из доводов, приводившихся против теории минерального про-
исхождения нефти и лежащих вне соображений геологического
характера, необходимо остановиться на вопросе об оптической
деятельности нефти.
315
Как известно, получение оптически деятельных соединении
в современной химии связано в той или иной форме с веще-
ствами растительного или животного происхождения. Так как,
с другой стороны, все попытки синтезировать оптически деятель-
ное соединение, исходя из материала оптически недеятельного,
долгое время приводили либо к оптически недеятельному веще-
ству, либо к рацемическому соединению, то, казалось, оптическая
деятельность нефти является одним из неоспоримых доводов про-
тив минеральной теории щюисхождения нефти. Однако в настоя-
щее время, когда показано, что оптически деятельное соединение
может образоваться из оптически недеятельного под влиянием
оптически деятельного катализатора [б , неопровержимость рас-
сматриваемого довода должна считаться сильно поколебленной:
всегда можно представить себе, что образовавшаяся в процессе
рассмотренных выше реакций начальная смесь веществ (первич-
ная нефть), претерпевая в период длительного своего Х1>анения
в недрах различные превращения (гидрогенизация, конденсация
и т. д.), всегда -могла встретить те или иные- оптически деятель-
ные остатки каких-либо организмов, которые или сыграли по от-
ношению к первичной нефти роль оптически деятельного катали-
затора, или, растворившись в первичной нефти, приняли непо-
средственное участие в тех или иных ее превращениях, войдя, та-
ким образом, в состав нефти. Впрочем, последнее допущение ча-
стично приводит нас уже к теориям происхождения нефти из
органических остатков.
Теория происхождения нефти из животных остатков. Мате-
ринским веществом нефти согласно этой теории послужили
остатки морских животных (рыб, рептилий, а также низших
организмов), живших в отдаленные геологические эпохи и погре-
бенных под мощными слоями осадочных пород. Накопление этих
остатков происходило в результате не только постепенного, есте-
ственного вымирания фауны отдельных бассейнов в различные
эпохи, но могло являться также следствием катастрофических
причин, например внезапного изменения солености вследствие
прорыва могучего источника пресной воды в солено-водный бас-
сейн, или наоборот \ и т. п. Известным основанием для такого
представления об образовании нефти являете^ уже то отмечен-
ное выше обстоятельство, что нефть залегает в осадочных поро-
дах морского прохождения. Так например, пояс нефтяных зале-
ганий, начинающийся у подножия Карпат и проходящий через
Молдавию и Валахию, Крым, Тамань, северные и южные пред-
горья Кавказского хребта, Апшеронский полуостров и Каспий-
ское море по направлению к Средней Азии (Фергана),---весь
этот пояс находится в области осадочных пород Третичного моря
и т. д.
Как известно, важнейшими углеродистыми соединениями жи-
вотных организмов являются белковые вещества и жиры. Первые
1 Примеры подобного рода массовой гибели водной фауны неоднократно
описывались в новейшую эпоху.
316
из них крайне нестойки. Опыт и наб подгпие показывают, что все
белковые вещества животных трупов, погребенных под землей,
быстро и без остатка разрушаются, превращаясь в газообразные
или легко растворимые в воде и вымываемые вещества. Иначе
ведут себя в тех же условиях жиры. Сначала они гидролизуются
на глицерин и жирные кислоты. Первый из этих продуктов в за-
висимости от условий легко разлагается дальше на простейшие
вещества либо вымывается водой; что же касается кислот, вхо-
дящих в состав жиров, то в условиях пребывания под землей они
могут сохраняться без заметного изменения годами. Естественно
возникает вопрос, не им ли и обязана нефть своим образованием
в природе.
Для выяснения вопроса о возможной роли жиров и жирных
кислот в процессе нефтеобразования Энглер произвел обширную
экспериментальную работу [б]. По его поручению на одном из
заводов была произведена сухая перегонка большого количества
(429 кг) рыбьего жира (ворвань).
Условия перегонки: температура 420°; давление —10 ат.
Получены:
1. Маслообразный продукт........339 кч (69%)
2. Кокс, пек....................65 „ (13%)
3. Потери (вода и газ)....... 88 „ (18%)
Маслообразный продукт был подвергнут затем новой пере-
гонке; при этом было получено 91% дестиллата, который и был
подвергнут ближайшему исследованию, а именно — разгонке,
очистке кислотами и т. д.
Разгонка дала:
до 150°............25,9% (уд. вес 0,712)
150—300®...........58 % (уд. вес 0,817)
выше 300"...........16Д%
Первый погон (до 150 ) при обработке серной кислотой поте-
рял 37% по объему, откуда следует, что он содержал весьма зна-
чительное количество непредельных. Действительно, при более
детальном исследовании из дестиллата были выделены в виде
дибромидов гексилен, гептилен, а также другие низшие и высшие
гомологи этилена; кроме того, в форме соответствующих нитро-
соединений были обнаружены бензол, толуол и метГаксилол; нако-
нец, из предельных углеводородов были найдены в том же де-
стиллате нормальный пентан, нормальный гексан, диизопропил,
нормальный и вторичный гептаны (этилизоамил), нормальный и
вторичный октаны (диизобутил) и нормальный нонан. Из тяже-
лых фракций того же дестиллата был выделен твердый парафин,
хотя и в не вполне чистом виде.
Что касается, наконец, нафтенов, то ио мнению Энглера среди
продуктов перегонки жиров имеются также углеводороды и эгого
ряда; однако анализы отдельных, выделенных Энглером фракций
заставляют сомневаться в таком заключении, и для объяснения
присутствия в некоторых природных нефтях значительных коли-
317
честв нафтенов приходилось допускать образование их как ре-
зультат последующего уплотнения непредельных углеводородов,
Лишь значительно позднее было показано, что при сухой пере-
гонке рыбьего жира в присутствии алюмосиликата получается
дестиллат, состоящий преимущественно из нафтенов и, что осо-
бенно интересно, содержащий также нафтеновые кислоты [7].
Итак, животные жиры могут служить источником образова-
ния всех основных типов нефтяных углеводородов. Каков же мо-
жет быть последовательный путь указанного превращения их
в природе?
Первая, стадия превращения жиров в процессе нефтеобразова-
пия, — несомненно, их гидролиз с образованием свободных жир-
ных кислот. Надо думать, что весьма существенная роль при
этом выпадает на долю соответствующих бактерий или гидроли-
зующих энзимов. Дальнейшее превращение жирных кислот
в природе могло происходить в различных условиях давления и
температуры, соответственно чему процесс нефтеобразования мог
протекать в различные периоды времени. В зависимости от этого
начальный продукт, образующийся из жирных кислот в природ-
ных условиях, * «протопетролеум» (первичная нефть), может
весьма существенно, отличаться от природной нефти, более или
менее приближаясь по своему составу к тому веществу, которое
было получено Энглером сухой перегонкой жиров в лаборатор-
ных условиях. Лишь постепенно, претерпев дальнейшие превра-
щения (полимеризация и т. п.), протопетролеум переходит в на-
стоящую нефть, которую мы встречаем в природе.
Таковы в общих чертах основные положения теории происхо-
ждения нефти из животных жиров. Разработанная главным обра-
зом Энглером с химической стороны и Г сферам с точки зрения
геологии нефти теория эта пользуется в настоящее время широ-
ким признанием со стороны как химиков, так и геологов. Тем
большего внимания заслуживают некоторые ее стороны, которые,
могут вызвать те или иные вопросы и возражения. Как и раньше,
мы остановимся, конечно, и здесь преимущественно на химиче-
ской стороне темы.
1. Как было отмечено выше, одним из продуктов сухой пере-
гонки животных жиров в описанных выше условиях является,
довольно значительное количество кокса. (13%). А между тем,
в местах залегания нефти никогда не обнаруживается сколько-
нибудь значительных скоплений углистых веществ. Это кажу-
щееся противоречие было блестяще [разрешено Энглером. Как по-
казали поставленные им специальные опыты в изогнутых запаян-
ных трубках, сухая перегонка жирных кислот под высоким да-
влением протекает нацело, без образования какого бы то ни было
j глистого остатка.
2. Как могли образоваться азотистые и сернистые вещества
нефти? Представить себе образование их из жирных кислот, оче-
видно, химически невозможно. С точки зрения теории животного
происхождения нефти остается допустить, что источником их
образования явились 'белковые тела. Однако такое допущение на-
318
Ходится в противоречии с той легкостью, с которой белки в при-
родных условиях подвергаются полному разрушению (ср. выше).
Таким образом, вопрос об образована азотистых и сернистых
соединений нефти с точки зрения теории животного ее проис-
хождения требует да.ТЬнейшей проработки.
3. Высокая оптическая деятельность некоторых нафтеновых
нефтей также непонятна, если принять во внимание, что жиры
сами по себе оптически недеятельны.
4. Как было отмечено выше, получение из животных жиров
нефтеобразных продуктов в лабораторных условиях происходит
при значительных давлениях и температуре, превышающей 40о .
Возможны ли такие условия в природе? Вопрос о давлении не
вызывает, конечно, никаких сомнений. Совершенно очевидно,
что в природных условиях животные остатки могут оказаться на
больших глубинах под давлениями даже гораздо большими,
чем те, с которыми имел дело Энглер в своих опытах. Иначе
обстоит тот же вопрос с температурой. Даже принимая во вни-
мание совокупное действие всех тепловых факторов, способных
вызвать повышение температуры на глубинах залегания нефти
(геотермический коэфициент, бактериальные процессы, внутрен-
нее трение деформирующихся напластований и др.), нельзя пред-
ставить себе, чтобы температура достигла здесь 400°. Однако не-
достаток температуры в природных условиях может быть воспол-
нен, с одной стороны, повышенным давлением, с другой — вре-
менем. Хотя значение этого последнего фактора в масштабе гео-
логических эпох не поддается учету, тем не менее влияние его
в процессе нефтеобраэования не подлежит никакому сомнению.
Для объяснения скоплений таких количеств жиров, которые обеспе-
чивали бы образование нефти в пророде, некоторыми авторами делалось
допущение, что под водой или во влажной почве при отсутствии кисло-
рода воздуха трупы животных могут претерпевать особого рода жировое
перерождение,* каковое приводит к превращению белков в высокомолеку-
лярные жирные кислоты, т. е. к образованию трупного воска (адипоцира).
В пользу такого допущения приводились случаи образования трупного
воска на кладбищах с влажной почвой, особенно же те изменения, которые
претерпевают трупы людей и животных при пх продолжительном пребыва-
нии под вбдой. Ближайшее исследование этого явления было проведено
на таких случаях, которые позволили произвести количественный учет
образовавшегося жировоска, а также на специальных опытах, поставленных
в целях установления характера химических изменений органического ве-
щества морских животных с момента их смерти до окончательного' погре-
бения их в .минеральных отложениях. Эти исследования показали, что
пакопление в природе больших количеств жиров за счет жирового пере-
рождения белков невозможно; что при анаэробном разложении трупов
животных жиры сохраняются сначала в химически неизмененном виде, при-
чем, однако, структура жировой ткани совершенно исчезает вследствие
полного разрушения белковой ткани; что, наконец, образование жировоска
(трупного жира) происходит вследствие постепенного накопления первона-
чально находившихся в трупе жировых веществ, причем другие вещества
трупа, в частности белки, нацело разрушаются.
Теорйя происхождения нефти из растительных остатков.
Органические соединения, встречающиеся в природе как остатки
растительных организмов, чрезвычайно ‘разнообразны. Совокуп-
на
И ость новейших данных из этой области показывает, что одйй-йЗ
этих веществ, а именно все углеводороды .и их производные, на-
пример так называемые пектиновые вещества *, а равным обра-
зом растительные белки, подвергаются в естественных условиях
глубокому распаду до газообразных и лейке вымываемых
соединений; другие растительные вещества, а именно—углеводо-
роды, смолы, воска, жирные кислоты и лигнин, не разрушаются,
а накапливаются св местах отложения растительных остатков х
путем целого ряда сложных реакций, постепенно обогащаясь
углеродом, превращаются последовательно св торф, бурые.и камен-
ные угли. Эти погребенные под землей, нередко чрезвычайно
мощные скопления остатков растительных организмов неодао-
кратно рассматривались как материнское вещество нефти, кото-
рая якобы могла образоваться из них либо путем сухой пере-
гонки, либо при действии на них перегретого водяного пара.
Однако представление о происхождении нефти из раститель-
ных остатков встречает весьма серьезные возражения с различ-
ных сторон. Прежде всего приходится считаться с тем твердо
установленным фактом, что всякая перегонка- растительных остат-
ков, сухая или с паром, сопровождается образованием угли-
стой массы, что, как $то уже было отмечено выше, совершенно
не согласуется с характером нефтяных залеганий. С другой сто-
роны, деготь, получаемый из растительных остатков при их пе-
регонке, особенно сухой, по своей химической природе весьма
существенно отличается от нефтей: он содержит, например, много
фенолов и непредельных соединений, много низкокипящих кето-
нов, смол и асфальтенов, т. е. характеризуется присутствием -та-
ких веществ, которые либо вовсе не встречаются в естественных
нефтях, либо если и встречаются в них, то обычно в количествах
весьма незначительных.
Нельзя не отметить, однако, что, несмотря на указанные не-
дочеты, теория. растительного происхождения нефти* имеет и не-
которые положительные стороны. С нею, например, согласуются
бедность нефтей азотистыми соединениями, а также высокая
оптическая деятельность некоторых нефтей, которую в данном
случае можно связать с оптической деятельностью таких веществ,
как растительные смолы, воска и эфирные-масла [8]. Чтобы оп-
тическая активность этих веществ сохранилась в.процессе.нефте-
образования, необходимо, чтобы во избежание рацемизации про-
цесс протекал при сравнительно невысокой температуре и чтобы,
таким образом, главная роль принадлежала при этом давлению.
1 Пектиновые вещества — соединения класса. углеводов, широко рас-
пространенные в природе, например в плодах (яблоки), -ягодам
(земляника, крыжовник), корневищах (сахарная свекла) и т. ,д. .Лри .их
гидролизе получаются, с одной стороны, полиозы, распадающиеся :затвм да
различные мо.нозы (арабиноза, галактоза и .др.), с другой—продукты
с лен ия мон оз, кислоты глюкуроновая, галактуроновая и т. п.; кроме того,
при этом образуются метиловый спирт и уксусная кислота. Таким образом,
пектиновые вещества представляют собою, невидимому, сложные -проэт$-
рифицированные продукты конденсации полиоз с некоторыми- кислотамз
из класса углеводов.
320
А в этих условиях, ио мнению некоторых авторов, возможно су-
щественное изменение в составе конечных продуктов превратив-
ния растительных остатков и, в частности, отсутствие в них
продуктов обугливания.
В последнее время теория растительного происхождения
нефти получила новое, в высшей степени ценное подтвержде-
ние [9]. При исследовании вытяжек, полученных экстракцией
сланца с верхнего Изара, месторождение которого расположено
в доломитах верхнего альпийского триаса, установлено нахожде-
ние в этом горючем ископаемом порфирина—одного из ближай-
ших производных характернейшего растительного пигмента, хло-
рофилла, с нахождением которого в зеленых частях растений,
как известно, неразрывно связан основной процесс жизнедеятель-
ности растительного организма — процесс усвоения углекислоты,
иначе говоря, ассимиляция углерода. Очевидно, тем самым преж-
де всего получено неопровержимое доказательство ближайшего
участия растительных организмов в образовании сланцевых зале-
жей. Но это наблюдение, как было установлено далее, имеет го-
раздо более общее значение. Оказалось, что те же самые произ-
водные хлорофилла обнаруживаются в нефтях, асфальтах и озо-
керитах. Таким образом образование и этих горючих ископаемых
должно быть связано с какими-то глубокими превращениями
в недрах земли остатков растительных организмов.
Хлорофилл представляет собою сложное гетероциклическое соедине-
ние, основным ядром которого является ядро пиррола (CtH*N). Хлорофилл
состоит пз смеси двух близких по составу и строению веществ: хлоро-
филла a (CeeHraOsNiMg) и хлорофилла b (CwHzoOeNiMg). Атом магния
соединен в хлорофилле с атомом пиррольного кольца. По своей химиче-
ской природе хлорофилл должен быть отнесен к воскообразным веществам
типа сложных эфиров. При действии на него щелочей происходит его
омылпвание с выделением метилового спирта, а затем и более глубокие
превращения, которые в итоге приводят к продуктам, еще содержащим
магний и называемым фил тинами. При действии кислот филлины теряют
магний и дают вещества, называемые порфиринами. Хлорофилл и его
ближайшие производные имеют характерные спектры поглощения, позво-
ляющие судить об их присутствии даже в тех случаях, когда они нахо-
дятся в недостаточно чистом виде и в крайне незначительных количе-
ствах.
Чтобы оценить значение и общность этих новейших данных
о нахождении в различных горючих ископаемых порфиринов
(А Трейбс), важно отметить, что из 53 нефтей, исследованных
в этом отношении, лишь 6 нефтей (некоторые месторождения
Галиции, Пенсильвании и зап. Вирджинии) не показали при-
сутствия в них порфиринов. Все эти 6 нефтей были совершенно
бесцветны или почти бесцветны, очевидно, вследствие адсорбции
смолистых красящих веществ нефти теми или иными твердыми
адсорбентами, через которые данные нефти подвергались филь-
трации в недрах земли. Так как порфирины принадлежат
к числу веществ, которые также легко задерживаются твердыми
адсорбентами, в частности, например, глинами, то естественно
лритти к заключению, что ненахождение порфиринов в указан-
ных шеста образцах нефтей вызвано факторами вторичного ха-
• 821
21 Зак. 1622. Химия нефти.
рактера, т. с. адсорбцией порфиринов теми же адсорбентами,
которые обусловили обесцвечивание данных нефтей.
Кроме нефтей, на содержание порфиринов были изучены
9 образцов асфальтов различных месторождений и 3 образца озо-
керита. Во всех них было также показано содержание порфи-
ринов в более или менее значительных количествах.
Само собою .разумеется, что в процентном отношении содер-
жание порфиринов >в отдельных изученных объектах было не-
велико и поддавалось определению только с помощью методов
спектрометрии. В некоторых случаях содержание порфиринов
было ниже 0,0004%; в других — оно поднималось до 0,004 и
даже до 0,04% (тринлдадская нефть). Это последнее содержание
порфирина в нефти должно быть признано весьма высоким,
особенно если 'принять в расчет, что содержание его при пере-
счете на сухое вещество зеленого листа составляет около 0,8%
и что листья отнюдь не составляют главной массы отмершего
растения.
Теория смешанного типа.Как было показано выше, основные
направления научной мысли в вопросе о происхождении нефти
не дают исчерпывающего разрешения этой обширной проблемы.
Не возвращаясь к рассмотренным выше отдельным их недоче-
там, отметим лишь, что теория минерального происхождения
нефти, имея явно выраженный эманационный характер, логиче-
ски приводит к заключению, что запасы нефти, непрерывно
возобновляясь в недрах земли, являются неисчерпаемыми. Мы
видели, однако, в начале этой главы, что факты противоречат
такому заключению. Теории, допускающие образование нефти
в природе из остатков животных или растительных организмов,
имеют другой общий недочет: все они являются дестиллацион-
ними, т. е. предполагают массовую перегонку материнского ве-
щества нефти и, следовательно, вторичный характер ее залега-
ния, что противоречит данным геологии. Последнее относится,
очевидно, также к теориям смешанного типа, которые допу-
скают, что в образовании нефти принимали одновременное ущ-
елие как животные, так и растительные остатки. Хотя по суще-
ству такие представления (Шталь, Потонъе и др.), конечно, оо-
лее правильны, потому что там, где имеются большие скопления
животных остатков, естественно должны находиться и остаМ
растительного 'Происхождения, однако, поскольку такого рюйа
теории допускают массовую дестиллацию, к ним приложимы те
же возражения, что и к другим рассмотренным выше органиче-
ским теориям.
В проблеме происхождения не^гги с химической точки зре-
ния необходимо различать две стороны вопроса: вопрос о воз-
можном материнском .веществе нефти и вопрос о тех химических
реакциях, которые претерпевает это вещество в процессе его
превращения в природную нефть. Первый из этих вопросов под-
дается экспериментальному изучению: благодаря работам Зе-
линского с сотрудниками здесь можно считать установленным,
что при надлежащей обработке нефтеобразные продукты полу-
822
чаются из самого разнообразного исходного материала. Так, при
перегонке с хлористым алюминием такие результаты показалп
следующие продукты животного и растительного происхождения:
холестерин, пальмиппювая, стеариновая и олеиновая кислоты,
пчелиный воск, абиетиновая кислота, бетулпя, каучук.
Холестерин, весьма устойчивый алициклический" спирт, со-
става СгтШбО, чрезвычайно широко распространенный r живот-
ных организмах, в частности входящий в состав мозга, желчных
камней и т. п., при нагревании с хлористым алюминием дал.
кроме газообразных углеводородов, нефтеподобное вещество
с обильным содержанием легких углеводородов бензинового типа,
в составе которых преобладали циклические формы рядов цикло-
гексана и циклопентана. Таким образом бензин пз холестерина
по своему составу приближался к бензину из бакинской нефти.
Высшие погоны того же вещества (с температурой кипения
до 400°) имели масляный характер; они перегонялись без разло-
жения даже при обыкновенном давлении, слабо реагировали
с хамелеоном и серной кислотой и, таким образом, состояли по
преимуществу также из углеводородов предельного характера.
Интересно, что фракции этого масла с температурой кипения
выше 250° обладали таоким же вращением (т. е. правым), как
большинство естественных нефтей [Ю].
Пальмитиновая и стеариновая кислоты. Эти наиболее устой-
чивые компоненты естественных жиров при перегонке с хлори-
стым алюминием, кроме углекислоты и небольшого количества
газообразных продуктов, обесцвечивающих хамелеон, дали глав-
ным образом твердый парафин. Таким образом вполне вероятно,
что именно эти кислоты являются в природе источником про-
исхождения нефтяного парафина или вообще парафинистых
нефтей [11].
Олеиновая кислота при перегонке с хлористым алюминием
дала более сложную картину [12]. Оказалось, что твердых пара-
финов при этом вовсе не получается; образуется сложная смесь
углеводородов предельного и непредельного характера, среди ко-
торых анализ обнаружил: небольшое количество ароматических
углеводородов, предельные и главным образом непредельные
углеводороды рядов циклогексана и циклопентана и углеводороды
жирного ряда, предельные и непредельные, примерно в равных
количествах.
Пчелиный воск дал при аналогичной переработке, кроме угле-
кислоты и горючих газов, главным образом углеводороды ряда
метана, жидкие и твердые, с выходом до S0% по весу от исход-
ного материала [13].
Из веществ растительного происхождения в этом направлении
было также изучено несколько характерных образцов, а именно:
абиетиновая кислота как пример смоляных кислот [14]; бету-
лин как пример фитостеринов [15]; наконец, как образец расти-
тельных смол каучук — естественный и искусственный [16]. Во
всех этих случаях продуктом взаимодействия с хлористым алю-
минием при нагревании оказалось нефтеобразное вещество, для
91* 323
каждого отдельного случая — своего определенного состава. Так,
абиетиновая кислота дала смесь углеводородов с преобладанием
ароматики; из бетулина получилось вещество, в низших пого-
нах которого преобладали углеводороды ряда циклогексана,
а в высших — ароматика; наконец, естественный каучук, так же
как и искусственный, при перегонке с хлористым алюминием
дал смесь углеводородов предельного характера., богатую угле-
водородами ряда циклогексана.
Итак, целый ряд веществ как животного, так и растительного
происхождения в определенных условиях может являться источ-
ником образования нефтеобразных продуктов, причем в зависи-
мости от природы исходного («материнского») вещества получае-
мая искусственная нефть характеризуется той или иной осо-
бенностью состава, встречаемой также среди естественных неф-
тей различных типов и месторождений. Эти опытные данные
могут служить, очевидно, лишь иллюстрацией теории органиче-
ского происхождения нефти, так как условия лабораторного
образования нефтеподобных продуктов из перечисленных выше
веществ глубоко отличны от тех условий, в которых, надо ду-
мать, процессы нефтеобразования протекали в 'Природе. Правда,
экспериментально доказало, что аналогичные результаты могут
быть получены также и в других условиях под влиянием мине-
ральных веществ, встречающихся в недрах земли, в частности,
например, под влиянием кизельгура [17]. Тем не менее, основ-
ные возражения, которые были приведены выше против «лестил-
лационных» теорий происхождения нефти, вполне применимы,
очевидно, также п к поименованным опытам, «г, таким образом,
второй основной вопрос проблемы происхождения нефти, вопрос
о сущности химических реакций, лежащих в основе процесса
нефтеобразования, попрежнему остается открытым.
Если по вопросу о химизме процесса нефтеобразования в при-
роде теории происхождения нефти из сырья смешанного типа,
т. е. из животных и растительных остатков, оказались бессильны
дать такую картину процесса, которая была бы приемлема как
с геологической, так и с химической точек зрения, то по вопросу
об изначальном, материнском веществе нефти полученные в по-
следнее время новые, крайне важные факты позволяют считать
данную проблему, повидимому, окончательно разрешенной.
Как было указано выше, в целом ряде нефтей, асфальтов и
озокеритов в настоящее время твердо установлено нахождение
продуктов распада хлорофилла, так называемых порфиринов,
точнее — растительных порфиринов (филлопорфиринов). Из-
вестно, однако, что аналогичные продукты распада (порфирины)
образуются также при соответственной обработке пигмента крас-
ных -кровяных шариков, гематина, CsaBssNaOiFeOH, и его
соли, гемина, C'siHraXaOiFeCl, т. е. продуктов явно живот-
ного происхождения. Эти два основные типа порфиринов су-
щественно отличаются друг от друга по составу, особенно же
по своим характерным спектрам поглощения, позволяющим де-
лать безошибочное заключение не только о природе исследуе-
324
мого порфирина, но и о количественном содержании его в рас-
творе. В высшей степени знаменательно, что точные методы со-
временной спектрометрии позволили с полной несомненностью
установить {присутствие в различных нефтях, асфальтах и биту-
минозных породах порфиринов обоих указанных типов, т. е. пор-
фиринов, являющихся производными не только хлорофилла, но
и гемина; в некоторых случаях порфирины последнего типа ока-
зались даже в преобладающем количестве. При всей осторож-
ности в выводах из этих фактов, очевидно, необходимо иритти
к заключению, что материнское вещество нефти имело смешан-
ный характер, т. е. представляло собою остатки не только расти-
тельных, но и животных организмов прежних геологических эпох.
Новейшие воззрения на происхождение нефти. В самое по-
следнее время сделана новая интересная попытка найти пути
для разрешения проблемы происхождения нефти, причем в ка-
честве основной химической реакции этого процесса автором
выдвинута деструктивная гидрогенизация.
Успехи гидрогенизации в различных областях химии и техно *
логии, и в частности, за самое последнее время также в области
технологии нефти давали и ранее повод к привлечению этой ре-
акции для объяснения массового образования в природе углево-
дородов предельного характера. Так, еще Сабатье и Сендеран,
с именами -которых связано самое начало последнего, блестящего
периода в истории гидрогенизации, выдвинули следующие пред-
ставления по вопросу о происхождении нефти: в глубинах земли
наряду с такими металлами, как железо, никель, кобальт и т. л.,
в некотором количестве находятся также щелочные и щелочно-
земельные металлы и притом частью в свободном состоянии,
частью в виде карбидов Реагируя с водой, проникающей к ним
с поверхности земли или из водных бассейнов, первые из них
дают водород, карбиды же — ацетилен. Эти газообразные веще-
ства под влиянием каталитического воздействия железа, никеля
и других металлов вступают во взаимодействие друг с другом и
в результате реакций уплотнения и гидрогенизации дают смесь
более высокомолекулярных углеводородов уже предельного ха-
рактера — нефть.
Совершенно очевидно, что представления эти, являясь одним
из видоизменений теории минерального происхождения нефти,
вызывают вое те же возражения, которые были рассмотрены выше
в связи с теорией Менделеева. Очевидно также, что они значи-
тельно уступают последней в отношении (вероятности исходного
минерального материала при образовании нефти (щелочные и
щелочно-земельные металлы вместо углеродистого железа), но
зато успешно разрешают крайне важный вопрос о предельном
характере углеводородов нефти1.
1 Само собою разумеется, что, дополняя явлениями гидрогенизации те
процессы, в результате которых по Менделееву образовалась нефть в при-
роде, можно легко устранить все возражения против минеральной теории
происхождения нефти, основанные на составе углеводородней смеси, по-
лучаемой при взаимодействии воды с углеродистым железом (ср. выше).
325
Совершенно иную точку зрения выдвигают некоторые новей-
шие авторы. Вместе с Потонъе [18] они допускают, что мате-
ринским веществом нефти являются богатые жирами сапропели-
товые отложения, мощные скопления которых являются резуль-
татом постепенного отмирания богатого жирами планктона,
т. е. микрофлоры и макрофауны бассейнов прежних геологиче-
ских эпох. К этому основному материнскому веществу нефти
в некоторых случаях примешивался материал гумусового ха-
рактера (гуминовые кислоты и пр.) как остатки прежней макро-
флоры, т. е. высокоорганизованных растений до древесных пород
включительно * 1. Эта смесь сапропеля с гуминовыми веществами,
скопляясь на дне соленоводного бассейна, претерпевала здесь
ряд превращений химико-биологического характера и постепенно,
с течением времени, становилась все более и более однородной.
После гидролиза жиров оставшиеся в сапропелитовых отло-
жениях жирные кислоты под влиянием анаэробных микроорга-
низмов частично превращаются в углеводороды и кетоны, не-
предельные же кислоты, кроме того, медленно полимеризуются.
Образующиеся таким образом вторичные -продукты превращения
жиров, растворяясь в общей массе жирных кислот, образуют
среду, в которой диспергируются гуминовые вещества; все же
это, вместе взятое, представляет собою гомогенную смолообраз-
ную массу большой вязкости и высокого удельного веса, которая
в смеси с минеральными веществами (песок, глина) продолжает
оставаться на дне водного бассейна и (постепенно покрываться
мощными минеральными отложениями. Эта смолообразная масса
может быть названа «первичной нефтью».
Дальнейшая судьба первичной нефти геологически связана
с газообразующими процессами, которые поднимают ее со дна
бассейна, химически же—с процессом гидрогенизации.
Выше уже было отмечено, что одна термическая обработка
материала растительного происхождения дает продукт, по своему
составу резко отличающийся от природной нефти. Напротив,
как. будет показано в ч. II, современная техника в процессе
гидрогенизации приобрела метод, с помощью которого в соответ-
ствующих условиях температуры и давления оказывается воз-
можным превратить в нефтеобразную смесь углеводородов не
только тяжелые нефтяные остатки й смолы, но даже каменный
уголь. Аналогичные превращения могла претерпеть в природе
первичная нефть, причем, однако, так как процесс этот протекал
здесь в течение геологических периодов, условия его были не
столь жестки, как в современной технике гидрогенизации,
Образование необходимого для гидрогенизации водорода можно предста-
вить себе в результате взаимодействия воды с железом или углеродом при
высокой температуре.
1 Гумусовыми веществами называются продукты гниения органических
веществ, главным образом растительного происхождения. Это — бурые или
черные, мало изученные вещества, образующие вместе с выветрившимися
и наносными породами верхний, покрытый растительностью слой земли
(гумус).
326
я именно: при высоких давлениях температура процесса могла
•быть по мнению некоторых авторов примерно всего около 200°,
что, повидимому. допустимо в природных условиях, если при-
нять во внимание, что нередко у забоя нефть имеет температург
около 100'.
Итак, образование нефти в природе можно представить как
результат гидрогенизации первичной нефти, образовавшейся ил
•смешанного гумусо-сапропелитового материала путем постепен-
ного изменения его в природных областях соленоводного бас-
сейна в условиях анаэробного разложения. В зависимости от
•ближайшего химического состава сапропелитового материала и
•большего или меньшего содержания в нем гумусовых веществ
-состав первичной нефти может колебаться в довольно широких
пределах, соответственно чему продуктом ее гидрогенизации мо-
гут оказаться нефти различных типов. Так например, если
в первичной нефти преобладали жирные кислоты предельного
ряда и продукты их превращения, то конечным продуктом ги-
дрогенизации окажется нефть метанового типа: если при обра-
зовании первичной нефти первенствующее место занимали не-
предельные кислоты жирного ряда и продукты их уплотнения
циклического строения, то в конечной нефти будут преобладать
нафтены; наконец, в тех случаях, когда материнское вещество
нефти содержало значительные количества гумусовых веществ,
производных ароматического ряда, в составе конечной нефти
видное место займет ароматика. В соответствии со всем выше-
изложенным легко видеть также, что образование кислородных,
азотистых и сернистых соединений, а равным образом такие
•свойства нефти, как ее оптическая деятельность, объясняются без
особых затруднений.
Новейшие воззрения на происхождение нефти представляют
собою стройную теорию в которой в выошей степени удачно
•объединены, взаимно пополняя друг друга, основные начала тео-
рий минерального и ор1анического происхождения нефти. Как
мы видели, теория эта дает законченное объяснение не только
образованию всех основных компонентов нефти, но и таких ее
свойств, как оптическая деятельность. Вполне справедливо 'По-
этому мнение некоторых авторитетов в этой области (Губкин),
что в настоящее время данная теория является наиболее обосно-
ванной не только с химической, но и с геологической точки зре-
ния. Представляет поэтому интерес вкратце остановиться на та-
ких ее сторонах, которые могут вызвать известные сомнения, и.
•таким образом, определить некоторые направления дальнейшего
развития научной мысли в этом сложном вопросе.
Основным элементом новейших взглядов на происхождение
нефти, как ясно из вышеуказанного, является допущение, что
•одним из главнейших процессов нефтеобразования является про-
цесс гидрогенизации, июнимаемый в том же смысле, как говорят
в настоящее время о деструктивной гидрогенизации топлива.
*0 химической точки зрения можно с уверенностью утверждать,
’что такое допущение вполне правильно и что процесс деструк-
327
тивной гидрогенизации должен войти как один из основных мо-
ментов во всякую новейшую теорию происхождения нефти.
Однако две стороны этого вопроса еще требуют более углублен*
кого изучения, а именно: о происхождении водорода, необходи-
мого для гидрогенизации, и об условиях, при которых протекает
эта стадия процесса нефтеобразования.
Образование водорода, необходимого для гидрогенизации пер-
вичной нефти, можно представить себе как результат взаимо-
действия воды с раскаленным раствором карбидов в ферроман-
гане, т. е. по существу тем же путем, -как представлял себе обра-
зование углеводородов нефти Менделеев. Очевидно поэтому, что
к этой части теории применимы все те возражения, которые
были приведены выше против массового газообразования в нед-
рах земли по Менделееву. Однако эти возражения нисколько не
подрывают возможности процесса гидрогенизации в природе, так
как для осуществления этого процесса недры располагают дру-
гим источником водорода, а именно — -некоторыми реакциями
биохимического характера, протекающими в присутствии микро-
организмов.
Микробиологическими исследованиями последнего времени
[19] установлено нахождение в водах и -породах целого ряда
нефтяных месторождений, притом- на громадных глубинах
(до 1000 м), анаэробной нефтяной микрофлоры, которая, возбу-
ждая некоторые специальные виды брожения, вызывает распад
продуктов животного и растительного происхождения с выделе-
нием горючих газов: метана, водорода и др. Среди этих биохи-
мических процессов особый интерес представляют: метановое к
водородное брожение белков, метановое брожение клетчатки и др.
Таким образом не исключена возможность, что источником водо-
рода для процессов гидрогенизации, протекающих в природе,
являются не пирогенетические реакции разложения воды, а про-
цессы бактериального характера:
Условия, в которых протекал процесс гидрогенизации в при-
роде, также требуют дальнейшего уточнения и разработки. Ка-
ково бы ни было ближайшее материнское вещество нефти, на-
пример «первичная нефть» или керогеновое вещество сланцев,
как представляют себе американские геологи, с химической
точки зрения совершенно очевидно, что, исходя из этих слож-
ных смесей -высокомолекулярных, богатых углеродом веществ,
легкие и средние углеводороды нефти могли образоваться лишь
путем разложения материнского вещества с последующим обо-
гащением продуктов этого разложения водородом, короче говоря,
путем деструктивной гидрогенизации. По сравнению с промыш-
ленной гидрогенизацией топлива этот процесс в природе дол-
жен характеризоваться значительно более высокими давлениями
(до 500—600 ат и выше), а также участием другого важного,
хотя и не поддающегося учету фактора — времени. Но даж
принимая во внимание эти факторы, в настоящее время трудно
было бы ответить на вопрос, достаточна ли для осуществлен
гидрогенизации материнского вещества нефти та температура
328
(около 200 ), которая мыслима в данном случае с геологической
-точки зрения. Судя по тому, что процессу гидрирования в дан-
ном случае, как и вообще при гидрогенизации жидкого топлива,
должен предшествовать процесс распада (крэкинга) высоко-
молекулярных соединений, надо думать, что температура около
200° здесь окажется недостаточной, а в таком случае естественно
возникает вопрос, не имеет ли и эта стадия процесса нефгеобра-
зования биохимического характера. Однако за недостатком опыт-
ных данных этот вопрос должен быть оставлен пока открытым.
Наконец, нельзя не отметить в заключение еще раз, что но-
вейшие исследования нефтей, асфальтов и битуминозных по^юд,
как было указано выше, с полной несомненностью доказали при-
сутствие в них продуктов распада не только растительного, но
и животного происхождения, а именно по»рфиринов, образовав-
шихся в результате распада не только хлорофилла растений, но
и гемина животных. Новейшие теории происхождения нефти
отныне не могут оставлять этого факта без внимания, и со-
ответственно этому вопрос о материнском веществе нефти дол-
жен быть подвергнут в них переработке и дополнению.
В предыдущем изложении вопрос о происхождении нефти
был освещен почти исключительно с химической точки зрения.
Другая, не менее важная сторона этого вопроса — геологиче-
ская— была затронута выше лишь в самых общих чертах; более
детальное освещение ее 'следует искать в специальных геологи-
ческих трудах и руководствах.
Как ясно из вышеизложенного, процесс образования нефти
в природе пока еще не может считаться полностью разъяснен-
ным; лишь отдельные стадии его в настоящее время являются
более или менее определившимися и притом, как мы видели, не
только с химической, но и с геологической точки зрения. Зало-
гом и основным условием успеха в дальнейшем развитии наших
представлений о происхождении нефти должно быть не взаимное
исключение основных, изложенных выше воззрений в этой слож-
ной области, а их взаимная координация и пополнение в со-
ответствии с данными и требованиями современной химии и
геологии.
ЛИТЕРАТУРА
Гефер Г. Нефть и ее производные, пер. со 2-го нем. изд. Крейцера Г., Спб.
и М. (1907).
Губкин II. М. Учение о нефти, М.-Л. (1932).
Engler С. и Hofer Н., Das Erdol., В. II., Leipzig (1L09).
Вернадский В. II., Очерки геохимии, 4-е изд. (1934).
1. .Jahresber. d. Ch.“ 367 и 1196 (1878).
2. Менделеев Д., Нефтяная промышленность в североамериканском штате
Пенсильвания и на Кавказе, гл. IV. Спб. (1877): см. такав «Основы
химии*, гл. VIII.
5. «Ж- Р. X. О.» 43, 1437 (1911).
4. Ср., например. Балицкий К., Геология нефти, гл. XIII и т. д. И. (1921).
5. Ср. работы Бредила, «Вег.* 41, 752 (1908); «Biochem. Ztschr.» 46, 7 (1912).
329
6. «Вег.» 21, 1816 (1888); 22. 529 (1889); 26, 1449 (1893); 28. 2501 (1895)- 8ft
2865 (1897). ’
7. Kobayashi., «J. Soc. Chem. Ind.» A, 250 (1921).
8. Walden P., <Chem. Ztg.» 391 (1906).
9. Treibs A., «Lieb. Ann.» 509,103 (1934); 517, 172 (1935); См. также Stutzer 0.k
Erdolmuttersubstanz (1935).
10. Зелинский H. Д. ъ Лавровский К. П., «Вег.» 60,1798 (19i7); 61,1291 (1928).
11. Зелинский И. Д. и Лавровский К. И. «Вег.» 61; 1054 (1928).
12. Ibid.; см. также Зелинский Н. Д. и Левина Р. Я., «Ж. првкл. хим.» в
20 (1933).
18. Зелинский Н. Д., и Лавровский Л. И. «Вег.» 62, 1264 (1929).
14. Зелинский Н. Д., Семигановский Н. Н., Козлов. Н. С., «Вег » 62, 2202 (1929)'
64, 2130 (1931).
15. «Вег.> 64, 2130 (1931).
16. «Ж. прпкл. хим.» 6, 86 (1933).
17. Steinkopf, «J. рг. Ch.» [2], 100. 65 (1920).
18. См. «Материалы по вопросу о происхождении нефти из сапропелит»
со вступительными статьями Горбова А. И. и Калиикого К.г на-
печатанные в «Нефт. и сланц, хоз.» № 9—12 (1921).
19. См. Гинзбург-Карашчева. Микробиологические очерки, 1-е пзд. М. (1932);
2-е ивд.» М.-Л. (1936).
ЧАСТЬ Ц
ПЕРЕРАБОТКА НЕФТИ
ГЛАВА I
ОБЕЗВОЖИВАНИЕ НЕФТИ
В I части (гл. XI) было указано, что вода является почти
постоянным спутником нефти, попадая в нее либо из того же
продуктивного пласта, либо из слоев, лежащих выше или ниже
продуктивного. В некоторых случаях разделение смеси воды
t нефтью на две фазы достигается самопроизвольно уже при
отстаивании. Часто, однако, вода образует с нефтью совершенно
однородную на вид эмульсию, т. е. чрезвычайно стойкую смесь,
состоящую из мельчайших капелек воды, равномерно распреде-
тенных в нефти. Разделение подобного рода эмульсин нередко
требует применения специальных методов и аппаратуры. Так
тсак при основном технологическом процессе переработки нефти,
при ее перегонке, во избежание разного рода осложнений (пере-
бросы, образование накипи и т. п.) необходимо, чтобы нефть не
содержала воды, то первой, нередко достаточно сложной задачей
переработки нефти является ее обезвоживание.
СПОСОБЫ ОБЕЗВОЖИВАНИЯ НЕФТИ
В тех случаях, когда вода легко отстаивается от нефти, их
разделение осуществляется в технике в специальных отстойниках
различных конструкций. Нефть направляется в эти отстойники
непосредственно из буровой. Здесь же происходит осаждение
механических примесей (буровая грязь, песок), выносимых
нефтью из скважины иногда в значительных количествах.
Простейший тип отстойника для отделения воды от нефти
изображен на фиг. 41. Нефть поступает через трубы а и Ъ сна-
чала в первый резервуар, в слой соленой воды, нагреваемый
паровым змеевиком. Здесь происходит расслаивание, и нефть,
постепенно собираясь, заполняет первый резервуар и через
трубу с переливается во второй резервуар, где происходят окон-
чательные отстаивание и разделение: нефть отводится через
верхнюю трубу т; вода же и грязь, не отделившиеся в первом
резервуаре, удаляются через сифон d и спуск и. Более оовер-
331
шейные отстойники во избежание потери легких фракций нефп
устраиваются герметическими.
Гораздо более сложную задачу представляет собою отделен^
воды от нефти в случае нефтяных эмульсий пли, как обыкно-
венно их называют, эмульсионной нефти. Различные сносей
разрушения подобного рода эмульсий можно разделить на
две группы: способы физико-механические и способы физико-
химические.
Физико-механические способы разрушения эмульсий. Спо-
собы эти весьма разнообразны. Простейшие из них основаны на
подогреве эмульсионной нефти или ее фильтрации; более слож-
ные — на применении центробежной силы или электричества.
Применение обогрева. Нагревание — один из давно употре-
бляемых и весьма распространенных способов разрушения неф-
Фпг. 41. Схема отстойника для нефти.
тяных эмульсий, в простейшей форме применяемый уже в обы-
кновенных отстойниках (ср. выше): при повышении температуры
понижается вязкость эмульсионной нефти, появляются 'Конвек-
ционные токи, и в результате соединения мелких капелек воды
в более крупные эмульсия распадается.
В наиболее совершенных современных установках [1] нагре-
вание эмульсионной нефти производится в особых подогревате-
лях и трубчатых печах с доведением температуры до 115—140°
и выше при 5—14 ат давления. В этих условиях, естественно,
происходит частичное отбензинивание нефти, причем вместе
с парами бензина частично отгоняется также и вода. У становка
этого рода снабжены ректификационной и стабилизационной ко-
лоннами, а также аппаратурой для промывки отогнанного бен-
зина; в результате обезвоживания на таких установках прямо
с промыслов можно получать, кроме чистой, обезвоженной нефти,
несколько процентов легкого стабилизированного бензина. Более
упрощенные установки для обезвоживания, основанные на подо
греве. эмульсионной нефти, не всегда оправдывают себя как по
причине значительного расхода топлива для нагревания, так.
вследствие потерь легких частей нефти от испарения.
Фильтрация. В этих способах обезвоживания подогретую
эмульсионную нефть под давлением заставляют проходить чере?
382
ту или иную, предварительно смоченную соленой водой среду:
песок, диатомовую землю, подходящую ткань и т. п. Предпола-
гается, что при этом может происходить настолько глубокое
изменение формы частиц эмульсии, что устойчивость последней
нарушается, и наступает ее разделение. Повидимому. в некото-
рых случаях путем фильтрации удается достичь удовлетвори-
тельного обезвоживания нефти; однако потери легких фракций
могут достигать здесь весьма значительных размеров, особенно
ввиду частого засорения фильтров.
Центрифугирование [2]. Центрифуга — аппарат, давно и
широко применяемый в некоторых видах лабораторной и про-
мышленной техники. Работа ее основана на действии центро-
бежной силы, которая, как учит механика, изменяется прямо
пропорционально массе вращающегося тела ад, радиусу круга
вращения г и квадрату числа оборотов и, т. е.:
f — кпггп-,
где к— коэфициент пропорциональности.
Непременным условием конструкции центрифуг, применяе-
мых в технике, является непрерывность их действия. Разделяе-
мая эму 1ьсия, например эмульсионная не(}пь. подается в осе-
вую часть вращающейся центрифуги и, попадая в сферу дей-
ствия центробежной силы, распадается: более тяжелые части
(вода, грязь) отбрасываются к периферии, более же легкие—
отодвигаются по направлению к центральной части аппарата,
его валу. Таким образом разделившиеся жидкости могут быть
выведены из аппарата каждая через свою особую отводную трубу.
Так как величина центробежной силы возрастает пропорцио-
нально квадрату числа оборотов в единицу времени, то понятно,
что для увеличения эффективности действия центрифуги стре-
мятся по возможности повысить скорость ее вращения. В со-
временных центрифугах эта скорость громадна. Так, центрифуга
Лаваля дает 6 оои об/мин; у сверхцентрпфуги Шарплесса ра-
бочая скорость достигает 17 000 об/мин и выше.
Центробежная сила при центрифугировании играет ту же
роль, какую играет сила тяжести при обыкновенном отстаивании
эмульсии. Понятно поэтому, что ряд факторов, содействующих
разрушению эмульсии при отстаивании, оказывает положитель-
ное влияние и при центрифугировании. Так, например, разде-
ление эмульсии наступает тем быстрее, чем больше разница
удельных весов ее компонентов: в тех же случаях, когда раз-
ница эта невелика, рационально увеличить ее искусственным
путем, например добавлением какой-либо дешевой минеральной
соли (глауберовой или поваренной); такого рода методика оди-
наково применима как при обыкновенном отстаивании, так и
при центрифугировании. То же можно сказать и о подогреве,
нередко применяемом для снижения вязкости эмульсий не
только при отстаивании, но и при центрифугировании.
Центрифугирование — один из наиболее совершенных спосо-
бов борьбы с эмульсиями. Разделение последних происходит при
ззз
этом значительно быстрее, чем при упрощенных способах, вслед-
ствие чего потери от испарения легких частей нефти значительно
снижаются.
Электрические методы [3]. Переменный электрический ток
высокого напряжения широко применяется как для осаждения
мельчайших частиц твердых тел (пыль, дым), взвешенных в га-
зовой фазе или увлекаемых газовым потоком (аэрозоли), так и
для разрушения эмульсий, в частности нефтяных (гидрозолей),
что по существу — одно и то же. В типичном аппарате этого
рода, дегидраторе Коттрелла, нефтяная эмульсия поступает сверху
в рабочий цилиндрический резервуар (3 м высоты и 1 .о диа-
метре), по вертикальной оси которого расположен медленно вра-
щающийся вал с несколькими дисками. Этот вал вместе с ди-
сками представляет собою рабочий электрод установки, тща-
тельно изолированный от других ее частей; он соединен с одним
из полюсов трансформатора, напряжение которого достигает
11 000 V. тогда как другим, заземленным электродом установки
служит наружная стенка рабочего резервуара. В процессе ра-
боты эмульсия проходит кольцевое пространство между дисками
и стенками рабочего цилиндра и здесь распадается: разделив-
шиеся же вода и нефть через выводную трубу, расположенную
в нижней части рабочего цилиндра, отводятся в отстойник, где и
производится их отделение. Установка снабжена змеевиком для
регулирования температуры эмульсии, а также специальными
автоматическими противопожарными приспособлениями.
Механизм действия дегидратора Коттрелла в общих чертах
заключается в следующем [3]. Мельчайшие частицы воды, вхо-
дящие в состав нефтяной эмульсии, приобретают в переменном
электрическом поле высокого напряжения соответствующие за-
ряды, располагаются по силовым линиям поля и сближаются
между собой, будучи отделены друг от друга лишь тончайшим
слоем диэлектрика — нефти. Однако при достаточно высоком
напряжении поля эти тончайшие нефтяные пленки могут разо-
рваться, в результате чего мельчайшие частицы воды начинают
соединяться в более крупные, способные отделяться и оседать
уже под влиянием одной силы тяжести.
Совершенно иначе построен электрический аппарат для раз-
бивки эмульсий Сейберта п Бреди, работающий на постоянном
токе, в среднем при напряжении около 500 V. Действие этою
аппарата «становится понятным, если принять во внимание, что
в эмульсиях взвешенные частицы и окружающая их среда несут
противоположные заряды. В нефтяных эмульсиях, как правило,
вода заряжена положительно, тогда, как нефть — отрицательно.
Вследствие этого водяные капельки эмульсии начинают пере-
двигаться в постоянном электрическом поле по направлению
к катоду и здесь выделяются. Естественно, что подобного рода
перенос (катафорез) должен сопровождаться частичным электро-
лизом растворенных в воде солей; однако это побочное явление
в условиях быстрого продвижения эмульсии через электрическое
поле не достигает сколько-нибудь значительных размеров. До-
334
стечь этим способом полного разрушения эмульсии удается срав-
нительно редко.
Физико-химические способы разрушения эмульсий. К этой
обширной группе относятся такие способы борьбы с нефтяными
эмульсиями, которые основаны на прибавлении к последним
различных химических веществ, разрушающих эмульсии, — тан
называемых деэмульгаторов. Действие эгпх веществ по существу
сводится к нарушению устойчивости тех пленок, которые разъ-
единяют капельки воды от окружающей их нефти, и, таким обра-
зом, имеет физико-химический характер. По своей природ- и
механизму действия на нефтяные эмульсии эти вещества могут
быть подразделены на несколько основных подгрупп, приводимых
ниже.
Электролиты. Различные кислоты, щелочи и соли с успехам
могут применяться для разрушения некоторых эмульсий. Из
кислот хорошее действие оказы-вают серная, соляная, а также
уксусная и нафтеновые кислоты; из щелочей — едкий натр,
а также известь; из солей — длинный ряд веществ, среди кото-
рых могут быть отмечены: поваренная соль, глауберова соль, сода
двууглекислая, хлористый кальций, железный купорос, хлорное
железо, азотнокислое железо и многие другие. Механизм дей-
ствия этих веществ на нефтяные эмульсии довольно-разнообра-
зен. Одни из них при достаточных концентрациях вызывают
коагуляцию веществ, из которых состоит пограничная между
водой и нефтью «защитная пленка» (см. ниже), и, таким обра-
зом, разрушают эмульсию; другие, притягивая воду и соединяясь
с нею, тем самым нарушают стабильность эмульсии и разби-
вают ее; третьи вступают во взаимодействие с некоторыми: со-
лями, входящими в состав эмульсии, образуя с ними нераство-
римые осадки, и т. д. В своем простейшем виде применение
электролитов для борьбы с эмульсиями заключается в том, что
эмульсию заставляют проходить сквозь слой подогреваемой соле-
ной воды, как это описано выше.
Неэлектролиты. К этой группе веществ, разрушающих неф-
тяные эмульсии, относится целый ряд органических соединений
различного состава и строения; таковы, например, газолин, бен-
зол, сероуглерод, четыреххлористый углерод, спирт, фенол, эфир,
ацетон и многие другие. Одни пз них, являясь хорошими раство-
рителями для соединений, образующих «защитную пленку.;
эмульсии (смолы, нафтеновые мыла и т. п.), растворяют эти
соединения, следствием чего и является разрушение эмульсии.
Другие, .смешиваясь с нефтью, понижают ее вязкость и тем са-
мым способствуют ее отстаиванию. Эффективность действия не-
которых из подобного рода веществ поразительна. Так например,
добавкой 0,1—0,3% технического фенола удавалось разбить неф-
тяную эмульсию, которая сохранялась при нагревании даже под
давлением; при 100’ та же эмульсия разбивалась уже от при-
бавления всего лишь 0,01% фенола. [4].
Коллоидные вещества. К этой группе веществ, рекомендо-
ванных для борьбы с нефтяными эмульсиями, относятся много-
335
численные и разнообразнейшие коллоиды, как-то: мелко раз-
дробленные кремнезем и глины; натровые соли высокомолекуляр-
ных кислот, жирных, смоляных и сульфокислот; крахмал, клей
и альбумин; наконец, некоторые более или менее сложные со-
ставы (композиции) из подобного рода веществ, специально при-
готовляемые либо являющиеся отходами некоторых видов про-
мышленности, в частности нефтеперерабатывающей.
Деэмульгаторы этого рода получили за последнее время ши-
рокое распространение, особенно в Америке. Примером их могут
служить различные композиции, выпускаемые под торговой мар-
кой Tret-o-Lite и прекрасно зарекомендовавшие себя во многих
случаях борьбы с эмульсиями. Состав этих композиций со-
образно с природой нефти и местными условиями довольно раз-
нообразен; одни из них — твердые, другие — жидкие; одни рас-
творимы только в воде, другие — только в нефти, третьи —и
в воде и в нефти. Приводим состав [5] одной из композиций
Tret-o-Lite (в процентах):
Олеиновый натрий.........83,0
Смоляное мыло (натровое) . 5,5
Кремневокислый натрий . . 5,0
Фенол....................4,0
Парафин..................1,5
Вода.....................1.0
Не менее действительными деэмульгаторами во многих слу-
чаях являются сложные смеси коллоидных веществ, гораздо
более доступные в условиях нефтяной промышленности, осо-
бенно там, где добыча и основная переработка нефти, как
в СССР, связаны и объединены хозяйственно и территориально
(Азнефть, Грознефть). Таким сложным деэмульгатором может
служить кислый гудрон от очистки керосина или, что значи-
тельно лучше, от обработки «солярового (вазелинового) дестил-
лата дымящей серной кислотой на предмет «получения так назы-
ваемого «контакта» [б]. Активным началом в данном случае
являются главным образом заключающиеся в гудроне сульфо-
кислоты, деэмульгирующее действие которых весьма значи-
тельно. Так например, стойкая биби-эйбатская эмульсия, содер-
жащая до 50% воды и почти не изменяющаяся после нагрева-
ния на водяной бане в течение нескольких часов, легко расслаи-
вается от прибавления 0,3—0,4% по весу гудрона в водном (рас-
творе, причем в нефти остается не больше 1% воды. Еще более
активным: и удобным деэмульгатором является кислый гудрон
после сульфирования керосина или газойля, если этот гудрон
(так называемый «черный контакт») подвергнуть предваритель-
ной нейтрализации содой. Этим деэмульгатором с успехом поль-
зуются ныне в Баку для разбивки калинской эмульсии
(до 5—7 тыс. т/сутки), являющейся одной из наиболее стойких
нефтяных эмульсий в СССР.
Физико-химические способы разрушения нефтяных эмульсий
характеризуются простотой и дешевизной применяемой аппара-
туры. Все оборудование состоит здесь из системы резервуаров н
836
нефтепроводов, подающего насоса и отопительного устройства.
Деэмульгирующее вещество смешивается с эмульсионной
нефтью при требуемой температуре, после чего смесь оставляется
в покое для разделения воды и нефти.
ПРИРОДА НЕФТЯНЫХ ЭМУЛЬСИЙ
Вода может образовать с нефтью, как ясно из вышеизложен-
ного, чрезвычайно тесную омесь различных степеней устойчи-
вости. В промысловой практике такие смеси называют обыкно-
венно эмульсионной нефтью. Вода может находиться в эмуль-
сионной нефти прежде своего в виде более или менее крупных
капель, которые пребывают в ней во взвешенном (суспендиро-
ванном) состоянии. Такое состояние является, однако, мало
устойчивым: даже при достаточной вязкости нефти, задерживаю-
щей расслаивание подобного рода смесей, все же мелкие капли
воды собираются постепенно в более крупные, которые оседают
на дно, тогда как более легкая нефть всплывает наверх.
Полное расслаивание эмульсионной нефти на два резко от-
граниченных друг от друга слоя наступает, однако, далеко не
всегда. Нередко между водой и нефтью упорно сохраняется про-
межуточный слой, также состоящий из воды и нефти, но по
внешнему виду совершенно однородный. Обыкновенно это — бо-
лее или менее густая мазеобразная либо пузырчатая рыжеватая
масса, характеризующаяся громадной устойчивостью: известны
примеры, когда такая масса сохраняла свою структуру в тече-
ние нескольких лет без заметного изменения. Это особое устой-
чивое состояние смеси воды и нефти представляет собою нефтя-
ную эмульсию в более узком смысле этого термина.
Внешний вид нефтяных эмульсий весьма разнообразен. Одни
из них имеют ячеистое, ноздреватое строение, другие — сметано-
образны или имеют консистенцию сбитого масла.
Цвет эмульсий, обыкновенно рыжеватый, иногда становится
яркорыжим, напоминающим глинистый раствор. Однако в слу-
чае значительного преобладания нефти цвет эмульсии стано-
вится темнее, приобретая бурый или черно-бурый оттенок.
Удельный вес эмульсии — важное свойство, позволяющее на
основе удельных весов частично отделившихся воды и нефти
определить, пользуясь специальными таблицами и номограм-
мами, содержание в эмульсии воды и нефти, т. е. состав
эмульсии.
Вязкость нефтяных эмульсий по сравнению с вязкостью
нефти и воды громадна. Для ее определения обыкновенно поль-
зуются вискозиметром Энглера, причем для придания эмульсии
подвижности иногда приходится поднимать температуру до 90°.
Для иллюстряпии приводим свойства и состав двух грозненских
эмульсий из скважин Старого (Xs 1) и Нового (№ 2) районов
(табл. 86). к
Опыт показывает, что при встряхивании с водой какого-либо
индивидуального углеводорода или тщательно очищенного неф-
337
22 Зак. 1622. Химия нефти.
Таблица 8$
Состав и свойства двух образцов грозненских эмульсий
(по Вышетраескому)
№ п/п Свойства эмульсии Состав эмульсии . Свойства нефти
уд. вес вязкость Ego % нефти % воды уд. вес вязкость Ей
1 0,969 42,49 14%1 86%i ОЛ'43 1,65
2 0,959 21,19 6%2 94W 0,844 0,77
тяного дестиллата сколько-нибудь устойчивой нефтяной эмуль-
сии не образуется. Если, однако, к смеси тех же веществ доба-
вить небольшое количество готовой нефтяной эмульсии, напри-
мер после отслаивания эмульсионной нефти на центрифуге,
либо того или иного коллоида, растворимого в воде или угле-
водороде, например натрового или известкового мыла, то та
самая смесь при встряхивании легко образует устойчивую
эмульсию. Интересен также обратный опыт. Если взять сьгрук>
смолистую нефть, легко образующую с водой нефтяную эмуль-
сию, и предварительно обработать ее подходящим адсорбентом,
например сукновальной глиной, то вследствие потери асфаль-
товых и смолистых веществ нефть потеряет свою характерную
темную окраску, а вместе с тем и способность давать с водой
устойчивую эмульсию. Этот небольшой опытный материал от-
четливо свидетельствует, что для образования устойчивой эмуль-
сии недостаточно двух веществ, не растворимых друг в друге,
например воды и углеводорода; для этого необходимо участие
некоторого третьего компонента, играющею при образования
эмульсии какую-то, очевидно весьма существенную роль. Для
уяснения этой роли необходимо ближе ознакомиться с приро-
дой эмульсии.
При рассмотрении под микроскопом или на микрофотогра-
фии эмульсионная нефть представляется в виде темного поля
нефти, на котором рассеяны более или менее крупные каши
воды. В одних случаях эти капли настолько велики (фиг. 42), ,
что при малой вязкости нефти легко представить себе их осе-
дание под влиянием силы тяжести; устойчивой эмульсии здесь,
очевидно, не образуется. В других случаях размеры капелек
воды могут оказаться настолько малы (фиг. 43), что не только
самопроизвольное разделение эмульсии оказывается невозмож-
ным, но даже искусственная ее переработка нередко мало
успешна. Такого рода устойчивые эмульсии представляют собою
особое состояние образующих эти эмульсии веществ, которое
1 После нагревания в автоклаве при 6 ат и 192°.
2 После нагревания в автоклаве о нафтеновой кислотой яри 6 ат и1751°-
338 *
должно рассматриваться как коллоидальный раствор одной жид-
кости е другой.
Во всяком коллоидальном растворе необходимо различать:
растворитель или внешнюю (дисперсионную) фазу и распреде-
ленное в этой фазе другое вещество, называемое внутренней или
дисперсной фазой. В нефтяных эмульсиях внешней фазой
обыкновенно является нефть, внутренней же или дисперсной
фазой — вода; эмульсию такого рода можно кратко обозначить
«вода в нефти». Однако легко получаются нефтяные эмульсии
также и обратного типа. т. е. «нефть в воде», в которых внеш-
ней фазой служит вода, дисперсной же фазой — нефть.
Устойчивость коллоидального раствора зависит от многих
факторов, в частности от степени дисперсности его внутренней
Фиг. 42. Нефтяные эмульсии под Фиг. 43. Нефтяные эмульсии под
микроскопом. микроскопом.
фазы: чем больше распыление, тем устойчивее коллоидальный
раствор. В такого рода системах размеры частиц определяются
в среднем от 0,1 у до 1 у у. Такие частицы, как показывает не-
посредственное микроскопическое и ультрамикроскопическое на-
блюдение, не остаются в коллоидальном растворе в покое, а со-
вершают быстрые зигзагообразные движения (Броуново движе-
ние), зависящие, как полагают в настоящее время, от постоя н-
ных столкновений -коллоидальных частиц с молекулами раство-
рителя. На определенной ipannne дисперсности эти столкно-
вения оказываются вполне достаточными, чтобы компенсировать
действие на частицы дисперсной фазы силы тяжести: в таком
случае эти частицы остаются во внешней фазе во взвешенном
состоянии до тех лор, пока вследствие взаимного слияния и
соответственного увеличения их размеров не нарушится устой-
чивость коллоидального раствора и не начнется в'ыпадение рас-
творенного вещества — его коагуляция. Все эти соображения
полностью применимы к коллоидальным растворам одной жид-
кости в другой, в частности — к нефтяным эмульсиям.
22* 33»
Другое важное условие устойчивости коллоидальных раство-
ров и эмульсий заключается в присутствии некоторых посторон-
них веществ, обволакивающих частицы дисперсной фазы и не
позволяющих им укрупняться путем взаимного слияния. В при-
менении к нефтяным эмульсиям необходимость присутствия по-
добного рода веществ уже была выявлена выше; их называют
обыкновенно эмульгаторами. В отношении нефтяных эмульсий
эмульгаторами могут являться, например, зольные, асфальтовые
и иные вещества, и накопление этих веществ в нефтяной эмуль-
сии доказано непосредственным опытом [7]. В свете современ-
ных представлений роль эмульгаторов при образовании нефтя-
ных эмульсий представляется следующим образом.
По мере дробления на мелкие части данного объема жидкости
или вообще какого-либо тела поверхность его, слагающаяся из
суммы поверхностей его отдельных частей, естественно, будет все
более и более возрастать; то же самое, конечно, должно наблю-
даться для коллоидальных растворов, или эмульсий, по мере
увеличения степени их дисперсности. Очевидно также, что при
этом все большее и большее значение будут приобретать свой-
ства пограничных слоев между двумя соприкасающимися фа-
зами, т. е. поверхностное натяжение на границе этих двух фаз—
внешней (растворитель) и внутренней, или дисперсной. Пред-
ставим теперь себе, что одна из этих фаз служит растворителей
для некоторого третьего компонента смеси, присутствие которого
понижает поверхностное натяжение его растворителя на границе
со второй фазой. Оказывается, что это третье вещество не рас-
пределяется в первой фазе равномерно, а накопляется (адсорби-
руется) на границе обеих фаз и притом тем в большей степени,
чем сильнее понижается поверхностное натяжение от его [при-
сутствия. Это важное положение коллоидальной химии, выве-
денное впервые математическим путем (Джиббс), может быть
иллюстрировано многочисленными опытными примерами. Вместе
с тем оно полностью разъясняет ту весьма существенную роль,
которую играют эмульгаторы при образовании разного род
устойчивых эмульсий [8].
Действительно, представим себе, что на границе двух несме-
шивающихся жидкостей, из которых одна распылена в другой
в виде мельчайших капель, накопляется третье вещество, эмуль-
гатор. В таком случае поверхности этих капель будут предста-
влять -собою как бы особые пленки из вещества эмульгатора»
которые, естественно, придадут дисперсной фазе особую стабиль-
ность: даже при соприкосновении друг с другом капли талого
рода, заключенные как бы в особые оболочки, не будут сливаться,
и коллоидальный раствор»сохранит свое состояние устойчивости
до тех лор, пока под действием тех или иных внешних факторов
указанные пленки не будут разрушены. В тех случаях, когда
между коэфициентами рефракции поверхностной пленки и окру;
жающей ее жидкости имеется достаточная разность, поверхност-
ная пленка может оказаться видимой под микроскопом или дая®
невооруженным глазом; хорошо видны эти пленки также в тег
340
случаях, когда эмульгаторами смеси минеральное масло — вода
служат такие коллоидальные вещества, как сажа, основные
сернокислые соли меди, железа или никеля и т. и. В других
случаях поверхностные пленки могут быть обнаружены иными
способами. Так, например, если к бензину, в котором растворено
1—2% нефтяных смол, прибавить воду и переболтать эту смесь
в закупоренном цилиндре, то посте отстаивания между‘слоями
воды и бензина окажется пузырчатый слой эмульсии; поверх-
ностная пленка этой эмульсии настолько прочна, что небольшой
кусочек стекла, опущенный в бензиновый слой, не тонет, а за-
держивается на поверхности пленок, не разрывая их.
Новейшие исследования показывают, что стабилизация эмульсии зави-
сит не только от способности эмульгатора понижать поверхностное натяже-
ние на поверхности раздела данных жидкостей. Так например, мыла
маслено-нафтеновых кие тот (кислотное число 145) при концентрации 0,01У
понижают поверхностное натяжение на границе бензол — вода примерно
в два раза и образуют весьма стойкую эмульсию. Напротив, мыла керо-
спно-нафтеновых кислот (кислотное число 315) при концентрации 0,5(W
хотя и снижают поверхностное натяжение на той же границе (бензол — вода)
более чем в 5 раз, тем не менее не образуют сколько-нибудь прочной
эмульсии. Очевидно, устойчивость эмульсии 'зависит не только от сниже-
ния поверхностного натяжения на границе двух жидкостей; решающую
роль здесь играет, повидимому, прочность образующей защитной пленки [9].
ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ БОРЬБЫ С ЭМУЛЬСИЯМИ
Концентрируясь в пограничных пленках, отделяющих одну
фазу эмульсии от другой, эмульгаторы обусловливают не только
устойчивость, но и самый тип эмульсии. Действительно, допу-
стим, например, что тот или иной эмульгатор на-границе мине-
ральное масло — вода понижает поверхностное натяжение больше
со стороны воды, чем со стороны масла. В таком случае погра-
ничная пленка, естественно, будет стремиться сделаться выпук-
лой в сторону воды и вогнутой в сторону масла: другими сло-
вами, образуется эмульсия «масло с водой». Такого 1>ода соотно-
шения наблюдаются для данной пары жидкостей в присутствии
натрового мыла, основных сульфатов меди, железа и никеля,
мелко измельченного кремнезема и некоторых других эмульга-
торов.
Пусть теперь какой-либо другой эмульгатор, концентрируясь
на границе той же пары жидкостей (минеральное масло — вода),
понижает поверхностное натяжение пленки больше со стороны
масла, чем со стороны воды. В этом случае пограничная пленка
будет стремиться стать выпуклой со стороны масла и вогнутой
со стороны воды, следствием чего должно получиться образова-
ние эмульсии «вода в масле». Примерами подобного рода эмуль-
гаторов для системы минеральное масло — вода могут служить
известковое мыло, сажа и т. п.
Обобщая все только что сказанное, приходим к следующему
важному выводу: для каждой пары жидкостей, не растворимых
одна в другой, можно подыскать такие эмульгаторы, которые бу-
дут давать эмульсии двух видов: либо первого компонента во
341
втором, либо второго в первом. Но этот же вывод может быть
.выражен в несколько иной форме.
Эмульгаторы как типичные коллоиды «лиофильны» к одним
жидкостям, в которых они набухают и растворяются, и «лио-
•фобны» к другим жидкостям, в которых они не набухают и не
растворяются. Естественно поэтому, что лиофильный коллоид,
концентрируясь у границы раздела двух жидкостей, будет по-
нижать поверхностное натяжение со стороны именно той жид-
кости, по отношению к которой он является лиофильным; иначе
говоря, в соответствии со сказанным выше, коллоиды, лиофиль-
ные к той или иной жидкости, способствуют образованию эмуль-
сий, в которых эта жидкость является внешней фазой; коллоиды
же. ’ лиофобные к определенной жидкости, способствуют образо-
ванию эмульсий, в которых эта жидкость будет внутренней
(дисперсной) фазой. У нефтяных эмульсий одна из фаз—вода,
по отношению к которой коллоиды могут быть либо гидрофильны'
либо гидрофобии. Ввиду этого по отношению к нефтяным эмуль-
сиям только что сформулированный принцип может быть выра-
жен следующим образом: гидрофильные коллоиды-эмульгаторы
содействуют образованию эмульсий, в которых вода является
внешней фазой; гидрофобные же коллоиды способствуют обра-
зованию таких эмульсий, в которых вода служит внутренней
фазой (Банкрофт).
Все вышеизложенное представляет исключительно важное
значение для борьбы с нефтяными эмульсиями. Действительно,
чтобы разбить' эмульсию, необходимо, очевидно, разрушить по-
граничные пленки, в которых заключена внутренняя фаза
эмульсии; как мы видели, эта задача может быть решена раз-
личными способами.
Так, например, пограничные пленки могут быть разрушены на-
греванием, так как заключенная в них жидкость при повыше-
нии температуры, естественно, должна расширяться, а частично,
быть может, даже превращаться в пар; при этом на известной
стадии нагрева упругость пленок может быть превзойдена, и
пленки разрываются, а заключенные в них капельки жидкости
-освобождаются от своих оболочек и соединяются в более круя-
ные капли.
Другой путь разрушения эмульсий — путь применения кол-
лоидов, прямо противоположных по своему характеру тому
эмульгатору, с присутствием которого связано образование дан-
ной эмульсии. Пусть, например, мы имеем систему масло —
вода, в которой содержится небольшое количество какого-либо
гидрофильного коллоида, например натрового мыла. Такая си-
стема стремился, как сказано выше, образовать эмульсию «масло
в воде». Добавим теперь к этой системе немного гидрофобного
коллоида, например известкового мыла, которое стремится эмуль-
гировать ту же смесь в обратном направлении, т. е. с образованием
эмульсии «вода в масле». Легко представить себе, что одновре-
менное действие на данную систему обоих этих эмульгаторов,
гидрофильного и гидрофобного, может взаимно компенсиро-
342
ваться, так что никакой эмульсии не образуется. Это и наблю-
дается в действительности, если оба эмульгатора взяты в соот-
ветствующих количествах. Отсюда следует также, что водные
эмульсии, образованные при участии гидрофильного эмульга-
тора, могут быть разрушены добавкой гидрофобного эмульгатора,
и наоборот, и это простое положение [10] должно быть положено
в основу таких физико-химических методов борьбы с эмуль-
сиями, которые заключаются в добавке к стойкой эмулы. шт пот-
хедящего коллоида (ср. выше).
Как было отмечено выше, разрушение эмульсий иногда до-
стигается также действием на них тех ити иных электролитов:
кислот, солей, щелочей; на этом основано, между прочим, при-
менение соленой воды в нефтяных отстойниках разных типов.
В этих случаях дело сводится по существу к коагуляции тех
эмульгаторов, с присутствием которых связано образование по-
граничных пленок данной эмульсии: под влиянием подходящих
электролитов эмульгаторы как типичные коллоиды могуу’ коагу-
лировать, т. е. выделяться из раствора в виде крупных хлопьев,
что в свою очередь делает дальнейшее существование погранич-
ных пленок, а «вместе с темщ самой эмульсии невозможным.
Коллоидная химия учит нас, что явления коагуляции сопро-
вождаются потерей частицами дисперсной фазы тех электри-
ческих зарядов, которые появляются на них, правильнее — на их
пограничных пленках, в результате адсорбции ионов соприка-
сающимися поверхностями двух несмешивающихся жидкостей.
В случае нефтяных эмульсий дисперсная фаза, т. е. мельчай-
шие капельки воды, почти всегда заряжены положительно,
адсорбируя на своей поверхности положительные ионы; внешняя
же фаза, т. е. нефть, соответственно приобретает отрицательный
заряд. Будучи заряжены одноименно, частицы дисперсной фазы
взаимно отталкиваются, что не позволяет им сливаться в более
крупные агрегаты и, таким образом, иоддерживать устойчивость
эмульсии. Прибавка к такой эмульсии электролита вносит в си-
стему существенные изменения: начинается адсорбция части-
цами эмульсии ионов электролита, противоположных но знаку
тому заряду, который несут сами эти частицы. Так. положи-
тельно заряженная дисперсная фаза, например частицы воды
в нефтяной эмульсии, адсорбируют анионы электролита: послед-
ние же, естественно, будут нейтрализовать у этих частиц их соб-
ственные электрические заряды. В результате, утратив вслед-
ствие нейтрализации свои заряды, а вместе с ними и свию устой-
чивость, частицы эмульсии начинают сливаться в более круп-
ные капли, и эмульсия расслаивается.
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ ОБЕЗВОЖИВАНИЯ НЕФТИ
Обезвоживание нефти как самостоятельная проблема нефтя-
ного дела определилось не сразу. Пока нефть была дешева, ее
особо легкие продукты (бензин) не представляли особой цен-
ности и относительный процент устойчивых нефтяных эмульсий
343
при добыче был не велик, ограничивались одним непродолжи-
тельным отстаиванием эмульсионной нефти; устойчивые же неф-
тяные эмульсии вместе с водой спускали в отводные канавы,
либо собирали эмульсию в земляные ямы (амбары, пруды) и
время от времени сжигали отстоявшуюся нефть. Однако со вре-
менем, в связи с переходом к экоплоатации пластов, содержа-
щих более тяжелую нефть, и постепенным их обводнением,
а также в связи с применением некоторых специальных спосо-
бов экоплоатации нефтяных скважин (особенно эрлифта), отно-
сительное количество нефти, получаемой в виде более или ме-
нее стойкой эмульсии, стало все больше и .больше возрастать.
Так например, по американским данным, в 1920—1921 гг. в районах
Мидконтинента и побережья Мексиканского залива почти одна пятая часть
общей добйчи приходилась на эмульсионную нефть; в Калифорнии за
1922 г. свыше одной десятой всей добычи добытой нефти было подвергнуто
обезвоживанию одними лишь электрическими методами [10]. Насколько
широко распространение нефтяных эмульсий в наших добывающих райо-
нах, видйо уже из следующего: при микроскопическом обследовании гроз-
ненских нефтей, взятых непосредственно из 80 действующих скважин,
почти »/ю образцов содержали эмульсии, а около 10% всей грозненской
нефти требуют применения тех или иных способов обезвоживания [11].
Столь же широко распространение нефтян&х эмульсий в Бакинском районе,
причем некоторые их виды характеризуются большой устойчивостью; таковы,
например, некоторые биби-эйбатские, сураханские и другие эмульсии.
Борьба с нефтяными эмульсиями диктуется их свойствами.
Не только перегонка их вследствие перебросов и образования
накипи невозможна, но даже перекачка их по трубопроводу
вследствие большой их вязкости нередко крайне затрудни-
тельна. Свободное отстаивание нефтяных эмульсий требует не-
редко нескольких десятков дней и, следовательно, громадной
запасной емкости; кроме того, естественно, такое отстаивание
должно сопровождаться большими потерями ныне наиболее цен-
ных, легких (газолиновых) частей эмульсионной нефти. Тем
самым определяется большая важность искусственного разруше-
ния нефтяных эмульсий теми или иными способами, вкратце
очерченными выше. К сожалению, природа нефтяных эмульсий
столь разнообразна, что, несмотря на большую проделанную ра-
боту, универсальных методов борьбы с нефтяными эмульсиями
пока не найдено. Ввиду этого каждая природная эмульсия
должна быть предварительно изучена с точки зрения ее физико-
химических свойств в лаборатории, и лишь на основе получен-
ных данных могут быть предприняты опыты ее разрушения
в производственных условиях. Впрочем, такое предварительное
исследование нефтяной эмульсии отнюдь еще не гарантирует
успеха в поисках практически применимого метода ее разру-
шения. Бывают случаи, когда, несмотря на техническую воз-
можность разрушения данной эмульсии тем или иным спосо-
бом, сама эта операция оказывается неприемлемой с экономиче-
ской точки зрения. Таким образом, в области борьбы с нефтя-
ными эмульсиями впереди еще большая исследовательская ра-
бота.
344
ЛИТЕРАТУРА
Гурвич J. Г., Научные основы переработки нефти, М.-Л. *1925).
Дау Д. К, Нефтяные эмульсии, Нефт. изд.-во (1928).
Манчо А., Разрушение нефтяных эмульсий, .Нефт, хоз.“. 8, № 9—12 (1922)-
Вишетравский С., Американские, бакинские и грозненские нефтяные
эмульсии, „Аз. нефт. хоз." 10, № 8—9 и 10 (1929).
1. Ср, Бат. В. М., ,Аз. нефт. хоз." 13, № 11—12, 146 (1933).
2. Ayers Б'. Е, „Nat. petrol. News" 15 (10/X 1923).
3. Eddy W. G. and Eddy H. C., ,lnd- Eng. Chem." 13, № 11, 1016 (1921)-
4. Kuczynsky, „Petroleum" 19, 420 (1923).
5. Matthews R. К a. Crosby, „Ind. Eng. Chem.“ 18, 1015 (1921)
6. Шнейдер Ф., .Аз. нефт. хоз." 10, № 12, 138 (1930); Бух. Д. и сотрудники^
„Аз. нефт. хоз." 13, № 10, 48 (1933); 15, № 12, 49 (1935); 16, № 11,
51 и № 12, 54 (1936).
7. Дау. Д. Б., Нефтяные эмульсии, стр. 106—109.
8. Briggs Т. Р., .Ind. Eng. Chem.“ 18,1008 (1921).
9. Тихомиров В. И. и Пипик 0. Г., „Труды АзНИИ", вып. 3 (1931),
10. Ср. Дау. Д. Б., Нефтяные эмульсии, стр. 7.
И. Сельский В. А., „Грози, нефт. хоз." № 3—4, 57, 1923; Сельский Л. А-
Итоги исследования грозненских нефтей, стр. 154, М.-Л. (1927)-
ГЛАВА II
ПЕРЕГОНКА НЕФТИ
Основным технологическим моментом переработки нефтз
является ее перегонка, благодаря которой из сырой обезвожен-
ной нефти получаются ее дестиллаты, перерабатываемые затем
путем очистки на различные нефтепродукты: бензин, керосин
и масла. В зависимости от условий процесс заводской перегонки
нефти почти всегда сопровождается более или менее сильным ее
разложением, причем нередко такое разложение (крэкинг) ста-
вится специальной задачей данного технологического процесса.
Этим последним, весьма важным случаям перегонки нефти будет
посвящена ниже особая глава; в настоящей же главе будут рас-
смотрены лишь такие случаи перегонки нефти, которые проте-
кают либо вовсе без разложения, либо с минимальным и неиз-
бежным ее разложением, т. е. случаи перегонки, обычно назы-
ваемые «прямой гонкой».
ТЕХНИКА ЗАВОДСКОЙ НЕФТИ
В процессе развития нефтеперерабатывающей промышленно-
сти заводская перегонка нефти («прямая гонка») последова-
тельно (прошла три основных этапа, характеризующихся приме-
нением различной, постепенно совершенствовавшейся аппара-
туры: от маломощных кубов периодического действия, через не-
прерывно действующие кубовые батареи, до современных трубча-
тых кубов с их мощным погоноразделительным устройством
(ректификационные колонны). Хотя в основе каждого из этих
способов перегонки нефти лежат одни и те же физические явле-
ния нагрева, кипения и конденсации, тем не менее, для уясне-
ния ряда теоретических вопросов перегонки нефти в техниче-
ском масштабе важно хотя бы в самых общих чертах рассмо-
треть аппаратуру и технику каждого из этих способов заводской
перегонки нефти.
Перегонка сухой нефти с целью получения из нее светлых продуктов
известна давно. Первыми организаторами ее в промышленном масштабе
являются, повидимому, крестьяне, бр. Дубинины в районе Моздока (Сев.
Кавказ, 1823 г.). Около того же времени Рейхенбах (1830 г.) получил иг
нефти керосин лабораторным путем, но лишь в конце 50-х годов прошлого
столетня перегонка нефти на керосин прочно утвердилась в промышленном
346
масштабе в Америке. Как осветительный материал керосин быстро завоевал*
всеобщее признание и стал служить предметом вывоза из Америки в jpv-
гие страны.
В 60-х годах, когда инициатива бр. Дубининых была уже забыта,
нефтепер 'онные з воды стали вновь появляться в различных местностях
старой России: в Сураханах (Баку), Шираках (Грузия), на Тамани и т. л
Тем не менее, американский керосин ввозился к нам вплоть до конца
70-х годов.
Перегонка нефти в кубах переодического действия. •Этит
первоначальный, простейший вид перегонки нефти в промышлен-
ном масштабе напоминает перегонку на обычных лабораторных
аппаратах (колба, кубик); различие заключается лишь в разме-
рах и некоторых деталях аппаратуры. Перегонка велась сначала
в кубах, которым придавали форму вертикально поставленного
цилиндра, высота которого примерно равна диаметру его попе-
речного сечения. Позднее перешли к цилиндрическим кубам ле-
жачего типа, который сохранился до настоящего времени
(фиг. 44).
Фиг. 44. Схема перегонного куба.
Перегонный куб представляет собою цилиндр из листового
железа толщиною 0,25—0,38 дм. В зависимости от емкости раз-
меры его бывают весьма разнообразны: нефтеперегонный куб на
25 т (1500 пуд.) имеет в длину около 6,5 м при диаметре попе-
уючного сечения 2,5 л. Наполнение куба производится с помощью
подающей трубы о, уровень же нефти в кубе определяется отво-
дящей трубой, по которой избыток нефти может вытекать JI3
куба. Когда куб наполнен нефтью до уровня отводящей трубы
вентили на обеих трубах, подающей и отводящей, закрываются,
и начинается обогрев куба.
Нагревание перегонного куба обыкновенно производится с по-
мощью нефтяной форсунки f; для этой цели можно пользоваться,
кроме нефти, также и другими видами топлива, в частности ка-
менным углем, особенно пылевидным топливом (США). ^Чтобы
увеличить поверхность обогрева, нефтеперегонные кубы обыкно-
венно имеют одну или две жаровых трубы А (фиг. 45).
Когда температура внутри куба поднимается примерно до
100°, на дне куба отделяются остатки воды, содержавшейся в сы-
347
рой нефти. Чтобы обеспечить в дальнейшем спокойную перегонку,
без перебросов, эту воду спускают через особый кран е и продол-
жают нагревание: когда же в холодильнике появляются первые
фракции дестиллата, для облегчения перегонки начинают вво-
дить в куб перегретый водяной пар (с температурой 225—250°).
Водяной пар вводится в куб через специальный коллектор
(парораспределитель), представляющий собою расположенную на
дне куба трубу Ь, изогнутую по кривизне днища; диаметр кол-
лектора— 2,5—3 дм. От коллектора по направлению к обоим
концам куба отходят восемь более узких разводящих труб (диа-
метр— 1 дм.) с отверстиями для выхода пара. Расход последнего
зависит от природы дестиллата: чем тяжелее дестиллат, тем, как
общее правило, больше расходуется пара при перегонке.
Пары, образующиеся при пере-
г4ц гонке, поступают из куба прежде
•Фиг. 45. Поперечное сечение
перегонного куба.
всего в шлем с и шлемовую трубу d.
Назначение шлема заключается
в том, чтобы задерживать капельки
нефти, увлекаемые потоком пара, и
возвращать их обратно в куб. В це-
лях наиболее успешного разреше-
ния этой задачи кубовые шлемы
оборудуются разного рода приспо-
соблениями для механического за-
держивания капель (перегородки,
щетки и т. д.). Из шлема через шле-
мовую трубу пары направляются да-
лее в дефлегматоры В. Для куба
емкостью 25 т высота шлема на
практике колеблется в пределах
80—100 см, а его диаметр — от 60 до 75 см\ диаметр шлемовой
трубы обыкновенно бывает 35—40 см. В различных конструк-
циях нефтеперегонных (кубов шлемовые трубы бывают либо го-
ризонтальные, либо имеют небольшой наклон в сторону куба или
в сторону дефлегматора.
В дефлегматоре, который представляет собою одну из наибо-
лее ответственных частей нефтеперегонного куба, происходит кон-
денсация паров высококипящих компонентов, увлекаемых из
куба главной массой паров дестиллата. Такая частичная конден-
сация паров в дефлегматоре происходит за счет развития поверх-
ности охлаждения в дефлегматоре, которому придают для этой
цели разнообразнейшие конструкции. В простейшем случае это -г
цилиндр высотою около 1,8 м при диаметре в 1 м, разделенный
двумя, не доходящими до конца вертикальными перегородками,
из которых одна укреплена на нижней, а другая на верхней
конечности дефлегматора (фиг. 44). Благодаря такому устрой-
ству парам дестиллата приходится, проходя через дефлегматор,
совершать удлиненный путь, во время которого, естественно/бу-
дут конденсироваться по преимуществу такие компоненты, • ко-
торые имеют меньшую упругость пара, иначе говоря, более вда
348
кую температуру кипения. Таким образом при отборе, например,
бензиновых фракций достигается возможное отделение кероси-
новых углеводородов, при отборе керосина отделяются соляровые
углеводороды и т. д.
Для более полного отделения выше кипящих примесей ог
главной массы дестиллата дефлегматорам нередко придают более
сложные и совершенные конструкции, в конечном итоге заменяя
их иногда фракционирующими колоннами с тарелками или на-
садкой, например, из колец Раги ига. Широко применяются также
разнообразные комбинации фракционирующих колонн с дефлег-
маторами; таков, например, аппарат, изображенный на фиг. 4G.
Здесь поступающие из куба пары, пройдя перфорированную пе-
регородку А, поступают в колонну с
тарелками или насадкой В, а затем пе-
реходят в дефлегматор С— систему
трубок (диаметр — 5 сл), омываемых
снаружи водой. Здесь же показана
обычная принадлежность каждого де-
флегматора — U-образная трубка D
(«утка»), по которой сконденсировав-
шиеся пары (флегма) либо стекают об-
ратно в куб, либо отводятся на сторону.
Пройдя дефлегматор, пары посту-
пают в холодильники С и D (фиг. 44),
где происходят их остывание, конденса-
ция и охлаждение образующегося де-
стиллата. На современных нефтепере-
гонных заводах обыкновенно применяет-
ся двухступенчатое охлаждение: из де-
флегматора пары поступают в верхний
холодильник (конденсатор), где боль-
шая часть паров превращается в горя-
Фиг. 46. Дефлегматор ко-
лонного типа с насадкой и
охлаждением.
чую жидкость; последняя же по мере об-
разования стекает в нижний холодиль-
ник для дальнейшего охлаждения. Так
как в верхнем холодильнике происходит
отдача скрытой теплоты 'парообразования дестиллата и воды,
то, естественно, верхний холодильник должен быть более мощ-
ным, чем нижний; обыкновенно охлаждающие поверхности верх-
него и нижнего холодильников относятся друг к другу, как 2:1.
Простейший тип холодильника — спиралеобразная, посте-
пенно сужающаяся труба, сечение которой в верхних секциях
примерно 6 дм., в нижних же в соответствии с ожижением боль-
шей части паров — около 3 дм. Холодильник погружен в резер-
вуар с проточной водой, ход которой — обратный ходу дестил-
лата. Холодильники такого рода требуют больших количеств
пресной воды и малоэффективны. Более совершенны холодиль-
ники коллекторного типа, состоящие из ряда коллекторных ко-
робок, соединенных между собою пучком параллельных трубок.
Пары попадают сначала в первый коллектор, из которого по
349
узким параллельным трубкам направляются во второй коллек-
тор; далее они переходят в новый пучок параллельных трубок и
третий коллектор и т. д. Коллекторные холодильники также по-
гружены в один или два резервуара с проточной водой.
Кроме холодильников, погруженных в воду, в нефтеперегон-
ном деле применяются также холодильники оросительного типа.
Они предстал яют собой систему расположенных друг над другом
и соединенных между собой труб, на которые с помощью спе-
циального распределительного устройства подается сверху оро-
шающая их вода, свободно стекающая вниз и при этом частично
испаряющаяся. Благодаря использованию скрытой теплоты испа-
рения орошающей воды расход последней на конденсацию десгил-
латных паров и на охлаждение дестилла-
Фиг. 47. Скруббер.
та оказывается значительно меньше, чем
в системе погруженных холодильников.
Фиг. 48. Сепаратор с фонарем.
Хотя верхний холодильник, как было указано, служит для
конденсации попадающих в него паров, тем не менее, часть наи-
более легких углеводородов успевает пройти через него в нескон-
денсированном состоянии. Чтобы собрать эти весьма ценные бен-
зиновые части перегоняемой нефти, пары этих углеводородов на-
правляют в особое приспособление, скруббер, куда попадают
также и газы, всегда выделяющиеся при перегонке нефти (воздух^
СО, СОг, и др.).
В простейшем виде скруббер при нефтеперегонном кубе пред-
ставляет собою железный цилиндр высотою 4—4,5 м при диа-
метре поперечного сечения около 75 см, с насадкой, орошаемой
сверху через особое устройство водой или тяжелым бензином
(фиг. 47). Газы и несконденсировавшиеся лары поступают
в скруббер снизу по трубе, проходят через решетку, поддержи-
вающую насадку скруббера (битый кирпич, кокс, кольца Ра-
шига и т. п.), и на своем дальнейшем пути встречают орошаю-
щую жидкость. При достаточно низкой ее температуре лары лег-
ких углеводородов, прошедшие через первый холодильник, кон-,
денсируются теперь, превращаясь в легкий бензин, который
350
вместе со скрубберной жидкостью стекает в нижнюю часть скруб-
бера и отводится далее через трубу А; газы же свободно выходят
.наружу. Дальнейшая задача заключается: при орошении во-
дой— в механическом отделении воды от бензина, что легко
достигается с помощью сифона В; при орошении же тяжелым
бензином — в перегонке полученного продукта для выделения
легкого бензина либо в ином его использовании соответственно
качеству продукта.
Из второго холодильника дестиллат вместе с паровой водой
направляется в приемно-сортировочное отделение и прежде
всего попадает здесь в сепаратор для отделения воды от масла.
По своей конструкции сепараторы бывают, весьма разнообразны.
Обычно они основаны на принципе сообщающихся сосудов и
снабжены фонарем для наблюдения за цветом дестиллата
(фиг. 48). Пройдя сепаратор, дестиллат поступает в распредели-
тель, представляющий собою систему узких ящиков, соединен-
ных каждый со своим резервуаром. Руководствуясь удельным
весом дестпллата, приемщик направляет его в соответствующий
ящик, пользуясь для этого подвижной трубой или желобом, со-
единенными с питающей трубой.
Таковы в самых общих чертах устройство нефтеперегонного
куба и основные моменты работы на нем. Несмотря на ряд су-
щественных недостатков кубов периодического действия и слабую
их эффективность, они находят применение в некоторых процес-
сах нефтеперегонного дела вплоть до настоящего времени; та-
ковы, например, вторичная перетЬнка бензина, перегонка нефти
до кокса и т. и.
Перегонка нефти на кубовых батареях. Основными недо-
статками кубов периодического действия являются сравнительно
небольшая эффективность установки и невозможность рацио-
нальной утилизации тепла горячих остатков, получаемых при
перегонке (мазут, гудрон). Эти недостатки хорошо устраняются
в специально сконструированных системах нескольких кубов или
батареях непрерывного действия. Различают керосиновые'и мас-
ляные батареи, существенно отличающиеся друг от друга по
своему устройству и условиям работы.
Батареи непрерывного действия предлагались для перегонки
нефти в Америке еще в 60-х годах прошлого столетия, но при-
вивались крайне медленно. В Баку непрерывная перегонка была
установлена впервые в 1883 г. на заводе Нобеля, откуда соответ-
ствующие установки получили наименование нобелевских ба-
тарей.
Керосиновая батарея. Еще недавно керосиновая батарея
была основной установкой нефтеперегонного завода, на которой
получались главные продукты нефтеперегонного дела: газолин
керосин и мазут. Она состоит из нескольких нефтеперегонных
кубов (от 10 до 15), расположенных параллельно друг другу
очень пологой лестницей и соединенных друг с другом после-
довательно таким образом, что нефть самотеком может перехо-
дить из первого куба во второй, из второго — в третий и т д. до
851
последнего куба (фиг. 49). В ‘то же время путем открытия задви?
жек 1 в питающей трубе а и закрытия подающего 2 и отводя-
щего 3 патрубков любой из кубов батареи может быть выключен,
например для чистки, ремонта и т. п., без нарушения работа
остальных кубов, так что на практике батарея действительно
является установкой непрерывного действия.
Каждый куб керосиновой батареи по существу имеет то же
устройство, что нефтеперегонный куб периодического действия
(см. выше). Однако вся установка в целом имеет некоторые доба-
вочные приспособления, весьма важные с точки зрения рациона-
лизации, как это будет видно из краткого рассмотрения работы
батареи.
Фиг. 49. Схема керосиновой батареи.
Работу керосиновой батареи начинают с залива нефтью пер-
вого куба; после заполнения его до уровня отводящего патрубка
избыток нефти уходит во второй, потом в третий кубы и т. д.
Затем начинают нагревание батареи, обыкновенно с ее конца,
т. е. с последнего куба, постепенно переходя к началу батареи.
Обогрев отдельных кубов, спуск отстоявшейся воды и впуск во-
дяного пара производятся, как описано выше; ври работе куба
периодического действия.
Правильный режим керосиновой батареи устанавливается не
сразу. Сначала все кубы дают одинаковые погоны; постепенно,
однако, между дестиллатами с различных кубов начинает про-
являться известная разница в температуре кипения и удельном
весе. Действительно, первый куб батареи в процессе ее работы
все время пополняется свежей нефтью вместе с содержащимися
в ней легкими ее частями, причем эти последние частью отго-
няются из первого куба, частью же вместе с избытком поступаю-
щей нефти переливаются во второй и ближайшие кубы и отго-
352
йяются take из них. Однако в последующие кубы эти наиболеё
легкие компоненты уже не доходят, нацело отгоняясь раньше,
так что, например, дестиллаты из средних кубов батареи при
установившемся режиме ее работы содержат уже только кероси-
новые фракции, а дестиллаты из последних кубов—лишь соля-
ровые погоны; наконец, самый последний куб керосиновой ба-
тареи дает тяжелый соляровый дестиллат; остаток же, вытекаю-
щий из этого куба, представляет собою мазут. Для иллюстра-
ции изменения физических
свойств дестиллатов, полу-
чаемых с керосиновой бата-
реи, может служить табл. 87,
в которой приводятся темпе-
ратура паров и удельные веса
цогонов, отобранных с раз-
личных кубов 12-кубовой
батареи.
Пропускная способность
керосиновой батареи в сутки
составляет примерно 4—5
объемов ее залива. Так как
каждый куб батареи дает,
как было указало, продукт
со своими особыми свой-
ствами, то каждый из де-
стиллатов либо собирается
отдельно, либо комбини-
руется в приемно-сортиро-
вочном отделении по удель-
ному весу с соседними дестиллатами
Таблица 87
Свойства дестиллатов с 12-кубовой
батареи
(по Добрянскому)
№ куба Темпера- тура паров Уд. вес .DO- ГОНОВ dJJ
1 120°
О *4 130° 0,758
3 151° 0,761
4 172° 0,769
5 184° 0,776
6 194° 0,787
7 211° 0,800
8 221° 0,810
9 236° 0,819
10 254° 0,880
11 258° 0,844
12 262э 0,856
для получения товарных
продуктов.
Большое значение при перегонке на керосиновой батарее
(так же как при перегонке на кубах периодического действия)
имеет впуск водяного лара, который в высшей степени способ-
ствует перемешиванию нефти, облегчая, таким образом, тепло-
передачу и предупреждая пригорание и образование продуктов
разложения. Расход водяного пара для разных кубов керосино-
вой батареи различен: в первых кубах при отгонке бензина рас-
ходуется не больше 3—5% пара, в последнем же кубе батареи
расход пара возрастает до 35—40%; в, среднем отгонка всего
керосинового дестиллата требует примерно 12% водяного пара,
считая* на нефть.
Расход топлива на керосиновой батарее нобелевского типа
составляет обыкновенно 3—3,5% от перегоняемой нефти, из ко-
торых 2—2.5% идут на огневой обогрев, остальное — на получе-
ние и перегрев водяного пара, потребляемого при перегонке
нефти; для нефтей, очень богатых бензиновыми фракциями, рас-
ход топлива может быть даже несколько ниже. Столь малое по-
требление топлива керосиновой батареей возможно, однако, лишь
при соблюдении двух условий: самой тщательной теплоизоляции
353
23 Зак. 1622. Химия нефти.
отдельных ее элементов и утилизации тепла горячих остатков
(мазута) из последнего куба батареи. Для этой последней цели
при керосиновой батарее имеется особая установка—подогрева*
тель нефти, в которой за счет горячего мазута из последнего
куба происходит первоначальное подогревание нефти, поступаю*
щей далее в первый куб батареи для перегонки.
По своей конструкции подогреватели для нефти весьма разно*
Образны. Простейшие из них представляют собою обширные
резервуары вместимостью в 1,5—2 полезной емкости батареи и
больше, снабженные расположенными внутри их (горизонтально
или вертикально) змеевиками, в которые поступает горячий
мазут. Снаружи эти змеевики омываются сырой нефтью, которая
и подогревается, таким образом, за счет тепла мазута, при этом,
естественно, охлаждающегося. Нередко устраивают двухступен-
чатое подогревание нефти в двух последовательных подогревате-
лях. Так например, на одном из грозненских нефтеперегонных
непосредственно из последнего куба с температурой 276°, а вы-
заводов в ближайший к батарее подогреватель поступает мазут
ходит из подогревателя с температурой 226°, подогревая при
этом нефть от 84 до 126°; в другой подогреватель поступает ча-
стично уже остывший мазут, охлаждаясь здесь далее (от 226 до»
146°) и нагревая сырую поступающую в него нефть (от 38 до 84°)..
Как ясно из вышеизложенного, подогреватели для нефти при нефте-
перегонной батарее представляют собою типичные теплообменные аппараты,
пли теплообменники. Так как эффективность их действия, естественно,
зависит от величины поверхности теплообмена и скорости прохождения
жидкостей, то в целях объединения обоих этих принципов в одной уста-
новке теплообменники обыкновенно содержаа* сильно развитую трубчатую
поверхность, заключенную в обширный резервуар с веществом, подлежащим
нагреванию. Теплообменники другого типа строятся по принципу противотока,
а именно: горячая жидкость идет внутри трубы в одном направлении, хо-
лодная же протекает в другой, наружной трубе, концентрической с первой,
в противоположном направленпп (труба в трубе)-
Кроме утилизации тепла горячего мазута, подогреватели при
нефтеперегонной батарее выполняют ряд других, весьма важных
функций, а именно: " . - ’ j
1. Подогревание нефти способствует быстрому и полному от-
делению содержащейся в ней воды, которая, таким образом, от-
слаивается в подогревателе и не попадает в кубы.
2. Введение в батарею подогретой нефти заметно повышает
производительность батареи, тем более что отгонка наиболее
легких бензиновых частей начинается уже в подогревателе;
образующиеся при этом пары бензина направляются в холодиль-
ники и скруббера первых головных кубов батареи.
3. Одновременно с подогревом нефти происходит также охла-
ждение мазута, которое совершенно необходимо во избежание
воспламенения горячего мазута при выпуске его из куба и хра-
нении в открытых амбарах.
Наиболее узким местом керосиновой батареи обычного типа
является ее слабое погонорааделителъное устройство. Каждый
куб батареи в процессе его работы дает лишь небольшую часть
354
(20—25%) характерного для него погона; остальное, как пока-
зывает опыт,, должно быть отнесено к погонам, характерным для
соседних кубов. Таким образом, например, широкая керосиновая
фракция, получаемая на керосиновой батарее обычного тина,
всегда содержит более или менее значительную головку, т. е.
тяжелый бензин, и хвосты, т. е. соляровые фракции; вместе с тем
некоторая часть керосиновых фракций, с одной стороны, всегда
попадает в гцзолин, с другой — задерживается в мазуте или
попадает в соляровое масло и т. п.
С целью повышения фракционирующих свойств кубовых ба-
тарей их стали снабжать за последнее время колонными аппара-
тами ио нескольку на .батарею, так что каждый' аппарат одно-
временно обслуживает несколько кубов [1]. Хотя путем такого
усовершенствования достигается более совершенное погоноразде-
ление, тем не менее, его следует рассматривать лишь как пере-
ходное мероприятие, направленное к возможно более рациональ-
ному использованию имеющейся аппаратуры. Более совершен-
ное решение данного вопроса достигается в настоящее время
наиболее совершенной перегонной аппаратурой современной тех-
ники— трубчатыми кубами (см. ниже).
Другая важная задача, к решению которой направлено усовершенство-
вание нефтеперегонной кубовой батареи, заключается в дальнейшем, воз-
можно большем снижении расхода топлива на ее эксплоатацию. Кроме
тщательной теплоизоляции всей довольно громоздкой аппаратуры кероси-
новой батареи и утилизации теплоты горячего мазута из последнего куба,
такое снижение достигается возможно широким использованием тепл»; ко-
торое содержится в горячих парах воды и дестиллатов, получаемых с ба-
тареи. Особенно важное значение -в нефтеперегонном деле приобрели так
называемые -пародесгиллатные теплообменники, которые улавливают и пе-
редают подлежащей перегонке нефти не только теплоту охлаждения воды
и дестиллата, но и скрытую теплоту их парообразования. Заводы, на ко-
торых производится утилизация тепла этого рода, получили наименование
регенеративных. В СССР примером регенеративных установок является
Константиновский нефтеперегонный завод (на Волге, под Ярославлем), ана-
логичный завод в Баку и др. Значение их явствует уже не того обсто-
ятельства, что, например, на Константиновском заводе расход топлива
почти в два раза меньше, чем на керосиновой батарее обычлого типа
той же мощности [2]. Использование тепла дестиллатов на кубовых ба-
тареях значительно облегчается при постановке на них ректификационных
колонн, так как при этом, как указано выше, в каждой колонне соби-
раются пары из нескольких кубов.
Масляная батарея. Назначение масляной батареи заклю-
чается в том, чтобы путем перегонки выделить из тяжелых ча-
стей нефти отдельные ее погоны, которые после соответствую-
щей обработки находят применение в виде смазочных и иных
масел. Разгонке подвергается при этом уже не сырая нефть,
а мазут из последнего куба керосиновой батареи либо непосред-
ственно, либо после прохождения через теплообменный аппарат
керосиновой батареи.
По своему устройству масляная батарея в общем напоминает
керосиновую; все же здесь имеются некоторые существенные
особенности, связанные со свойствами исходного материала и его
компонентов (фиг. 50). Основными элементами масляной батареи
23* 355
являются: масляный куб А, шлем и шлемовая труба В, дефлег-
матор С, эксгаустер D, холодильник Е и водонапорная башня Е:,
в тех случаях, когда по местным условиям для масляной бата-
реи может быть предоставлен лишь холодным мазут, батарея
снабжается еще подогревателем для предварительного подогрева
мазута аналогично тому, как это было указано выше при опи-
сании керосиновой батареи.
Масляная батарея состоит обыкновенно из 12—15 кубов, но
встречаются и более мощные батареи (до 20 кубов). Масляный
куб подобно керосиновому имеет чаще всего форму лежачего ци-
линдра, по своим же размерам обыкновенно он меньше кероси-
нового куба; обычная его емкость 10—15 т, реже 20 т и больше.
Фиг. 50. Схема масляной батареи.
Куб не содержит жаровой трубы, и дно его должно быть изоли-
ровано от непосредственного действия топочных газов. Нередко
внутри масляного куба имеются специальные распорки во из-
бежание его деформации в процессе работы.
На своей верхней поверхности параллельно своей оси куб не-
сет 2—3 низких шлема, соединенных с общей шлемовой трубой.
Последняя представляет собою либо одну длинную трубу, на
протяжении которой расположены несколько (3—4) дефлегмато-
ров, либо чаще состоит из нескольких последовательно соеди-
ненных между собою колен, заканчивающихся одним дефлегма-
тором (фиг. 51). Устройство дефлегматора у масляных кубов
столь же разнообразно, как у кубов керосиновой батареи. Часто
применяются дефлегматоры с перегородкой; нередко они устраи-
ваются по типу трубы в цилиндре (фиг. 52), причем пары про-
ходят по кольцевому пространству между трубой и цилиндром,
охлаждаясь благодаря свободному течению воздуха в трубе и
частично здесь конденсируясь.
Функции шлемовой трубы и дефлегматора у масляного куба,
существенно иные, чем у куба керосиновой батареи, В ЭТОМ ио-
356
следам случае, как было показано выше, через дефлегматор
проходят основные погоны данного куба; назначение же шЧехо-
вой трубы и дефлегматора заключается в том, чтобы сконденси-
ровать и вернуть назад в куб те компоненты данного куба, кото-
рые по своим свойствам отвечают продукции следующего куба..
Режим масляного куба устанавливается иначе. Здесь основные
погоны каждого куба оседают в коленах шлемовой трубы, откуда
они отводятся в холодильники; через дефлегматор же проходят
лишь водяные пары, а также наиболее легкие масляные части,
главным образом продукты разложения масел, всегда образую-
щиеся при их перегонке вследствие высокой температуры, при
Фиг. 51. Схема шлемовых труб масляной батареи. Фиг. 52 Дефлегматор
с внутренней трубой.
которой приходится вести в данном случае процесс перегонки;
здесь же удерживаются и возвращаются отсюда в шлемовую
трубу те масляные дестиллаты, которые не успели осесть в по-
следнем колене шлемовой трубы.
, Благодаря большой общей длине шлемовой трубы (20—25 л)
в ней происходит фракционированная конденсация масляных
паров, и масляные дестиллаты из каждого хода трубы направ-
ляются каждый в свой холодильник. Устройство холодильников
масляной батареи такое же, как и керосиновой, но рассчиты-
ваются они не на полное охлаждение, а примерно лишь до
70—80°, т. е. до температуры, при которой масляные дестиллаты
обладают достаточной текучестью и притом весьма небольшой
упругостью пара. Иногда на масляных батареях вместо охлажде-
ния водой ограничиваются воздушным охлаждением при помощи
ребристых чугунных труб достаточной длины.
Весьма важной частью масляной батарея является примы-
Зэ7
кающий к дефлегматору водяной эксгаустер (фиг. 53). Эксгау-
стер представляет собою многоэтажный инжектор той или иной
конструкции, в сопло которого под давлением поступает холод-
ная вода (из водонапорной башни), а в боковое отверстие — газы
и несконденсировавшиеся в дефлегматоре пары — водяные и
масляные. Благодаря энергичному охлаждению пары, попадаю-
щие в эксгаустер, быстро конденсируются в нем и вместе с водой
выносятся через гидравлический затвор в особые отстойники;
газы же, не конденсирующиеся в эсгаустере и образующиеся
в результате частичного разложения масел при перегонке, отво-
дятся через особый патрубок в общую трубу, служащую для вы-
равнивания давления во всей батарее, а из трубы с помощью
специального насоса выкачиваются наружу. Что касается, на-
конец, масел, конденсирующихся в эксгаустере (эксгаусгерные
масла), то, являясь продуктами ча-
стичного разложения (крэкинга)
исходного сырья, они не поддаются
очистке и могут применяться лишь
в качестве топлива.
Как видно из вышеизложенного,
эксгаустер выполняет на масляной
батарее целый ряд функций, важ-
нейшие из которых следующие:
1. Благодаря непрерывному от-
сасыванию легких паров и газов
в кубе и шлемовоп трубе создается
поток в сторону дефлегматора, вслед-
ствие чего пары масла быстро уда-
ляются из куба, т. е. из области
высокой температуры.
2. В процессе перегонки происходит разделение неразложив-
шихся масел от продуктов их разложения, которые вытягиваются
эксгаустером.
3.,Быстро конденсируя водяные пары, эксгаустер сокращает
объем их, т. е. создает некоторое разряжение (до полуатмосферы
остаточного давления и больше), благодаря чему до известной
степени облегчается перегонка масел.
Работа масляной батареи начинается с наполнения ее обез-
воженным мазутом, после чего начинают подогрев батареи с по-,
следнего ее куба, осторожно впуская в него также водяной пар.’
Когда появляются первые дестиллата, количество водяного пара
усиливают и постепенно вводят в работу остальные кубы ба-
тареи.
Расход водяного пара при перегонке мазута весьма значите-
лен: для легких масляных дестиллатов он составляет 30—40%
от веса масла, для тяжелых — поднимается до 100%.
Расход топлива на масляных батареях составляет 5—9%,
причем большая часть его вдет на получение и перегрев водя-
ного пара.
Ввиду высоких температур кипения углеводородов, образую-
358
Щйх масляные дестиллаты, их перегонка, естественно, требуем
довольно жестких температурных условий. Применение водяного
пара, а также работа эксгаустера заметно улучшают условия
перегонки. Их дальнейшее смягчение в целях предупреждения
разложения масел достигается применением более высокого ва-
куума, для чего пользуются специальными мощными насосами;
таковы, например, установки ТПу&ъце, работающие под давле-
нием 3—5 мм, и др. [3].
В настоящее время масляные кубовые батареи доживают свой век.
Новых таких батарей не строится, потому что недостатки, лтмече-яныр.
выше для кубовых батарей, 'усугубляются при перегонке мазутов на масла:
выхода масел значительно ниже потенциальных возможностей сырья, ка-
чество дестиллатов — не высокое, остаток же из-за длительного пребывания
в кубах при высокой температуре претерпевает частичное разложение и
осмоляется, что резко снижает его качества как сырья для получения
остаточных масел.
Перегонка нефти на трубчатых установках. За последние
10—15 лет, несмотря на громадные успехи, достигнутые введе-
нием перегонки нефти на кубовых батареях, нефтяная промыш-
ленность пережила новую реконструкцию в этой основной обла- 1
сти переработки нефти; в результате этой реконструкции кубо-
вые батареи постепенно сходят со сцены и заменяются перегон-
ными установками нового типа — трубчатыми установками (труб-
чатками). Причины, заставившие взамен кубовой батареи искать
новой, более совершенной аппаратуры, заключаются не только
в ряде отчасти уже отмеченных выше недостатков кубовой бата-
реи, но также и в том, что в связи в успехами машиностроения
промышленность и потребитель стали предъявлять к качеству
нефтепродуктов такие требования, которые уже не могли быть
выполнены на старой аппаратуре.
Основные недостатки нефтеперегонных кубовых батарей заключаются
в следующем:
1. Громоздкость аппаратуры; кубовые батареи требуют на себя больших
количеств металла, занимают много места и представляют значительные
трудности при регулировке, особенно в моменты пуска батареи и пеобходи- '
мости быстрой ее остановки.
2. Большие размеры батареи затрудняют ее теплоизоляцию, вследствие,
чего значительные количества тепла во время работы батареи пропадают
непроизводительно.
3. Необходимость медленного обогрева батареи и принцип перетока
нефти из первого куба к последнему заставляют большие массы нефти
находиться долгое время и совершенно непроизводительно в зоне высоких
температур; помимо пожарной опасности, здесь кроется другое неудобство,
а именно неизбежность частичного разложения дестиллатов под влиянием
длительного высокотемпературного нагревания и снижение их качества.
Крайне слабая погоноразделительная способность кубовой батареи, на
что было указано выше; очевидно также, что добавочное оборудование
батареи фракционирующими колоннами сводит отдельные кубы до роли
простых парообразователей.
Трубчатая установка для перегонки нефти на бензин, керо-
син и соляровые масла представляет собой сложный агрегат, ос-
новными элементами, которого являются: трубчатый куб и печь,
в которых происходят нагревание и частичное испарение нефти;
359
иногда выделяемый в самостоятельную часть установка экйайо-
ратор (сепаратор), где паровая фаза отделяется от жидкой; мощ-
иая ректификационная колонна (одна или несколько) с дефлег-
матором; теплообменники и холодильники; насосы для пере-
качки продуктов и особый куб для получения водяного пара.
Фиг. 54 дает схематическое изображение трубчатой установки
для перегонки нефти с одной колонной, а именно установки
Фостер-Уилера [4J, являющейся одной из простейших и в то
же время весьма совершенных установок этого рода.
Пройдя теплообменные аппараты 5, 4, где происходит предва-
рительный подогрев сырья примерно до 175°, подлежащая пере-
Фиг. с 4. Схема трубчатой установки Фост-ер- Уилера.
гонке нефть подается из резервуара насосом 2 в трубчатую
печь, где она нагревается до 400 . Трубчатая лечь Фостера
представляет собою особую самостоятельную часть установки
(внутренние размеры оо 5,5 X 5,9 X 6,8 л), обогреваемую нефтью
или газом. Внутреннее ее пространство можно подразделить на
три части, а именно: 1) камера сгорания 6, где происходит сго-
рание топлива; 2) разделенная на несколько секций узкая кон-
векционная камера 1, отделенная от камеры сгорания переваль-
ной стенкой 5, и 3) радиантная камера, занимающая верхнюю
потолочную часть камеры, сгорания. В конвекционной и радиант-
ной камерах заложены трубы, образующие при помощи особых
соединений («ретурбенты») непрерывный змеевик, по которому
протекает подлежащая обогреву нефть, направляясь из нижней
секции конвекционной камеры в ее верхние секции и далее в ра-
диантную камеру. Направления, в которых заложены трубы
в конвекционной и радиантной камерах, взаимно перпендику-
лярны. В конвекционной камере находится также специальная
секция (4 ряда до 5 труб в каждом) для перегрева водяного пара.
Обогрев труб радиантной и конвекционной камер протекает
неодинаково. Трубы, подвешенные под потолком камеры сгора-
ния, образуют почти сплошной экран (2 ряда труб по 28 труб
в каждом), воспринимающий тепло, главным образом за счет
радиации тепловых лучей факела форсунки и раскаленных пиж-
Ш1Х стенок камеры сгорания; лишь в малой степени в обогреве,
экранных труб принимает участие непосредственное соприкосно-
вение их с горячими топочными газами. Отсюда название пото-
лочных или экранных труб — радиантная секция (камера). Обо-
грев труб конвекционной камеры, образующих три секции (всего
52 трубы), напротив, происходит за счет непосредственного со-
прикосновения их с горячими топочными газами, прошедшими
через перевальную стенку и направляющимися в боров в ниж-
ней части конвекционной камеры. Таким образом нефть дви-
жется в печи навстречу топочным газам, т. е. по принципу про-
тивотока, благодаря чему достигается не только постепенный обо-
грев нефти, но и более полное .экономическое использование
топлива.
По выходе из радиантной камеры нагретая нефть поступает
в колонный аппарат (колонну), которая вместе с трубчатой
псчыо представляет собою центральную часть всякой трубчатой
установки.
Колонна Фостер-Уилера (фиг. 55), несколько видоизменяв-
шаяся в процессе разработки и усовершенствования отдельных
частей установки, представляет собою громадный цилиндр высо-
тою 22,6 jf и диаметром около 2 м. К нижнему перфорирован-
ному днищу этого цилиндра приклепан другой цилиндр мень-
ших размеров, высотою около 2 м и диаметром 0,84 м. Последний
цилиндр представляет собою вместилище для стекающего сверху
жидкого остатка от перегонки (мазут, bottom), подвергаемого
здесь для удаления последних следов легких дестиллатов обра-
ботке открытым перегретым водяным паром, поступающим через
проложенные маточники. Уровень мазута в этой нижней секции
колонны поддерживается на высоте 1 м с помощью специального
автоматического контроллера, пройдя который мазут отводится
в мазутный теплообменник 4 (фит. 54); здесь мазут частично
охлаждается, отдавая тепло направляющейся в трубчатую печь
нефти, а далее через холодильник 8, с помощью особого насоса
9 перекачивается в мазутный резервуар 10 (фиг. 54 .
Главная часть колонны Фостер-Уилера на всем своем протя-
жении (кроме самой верхней части) оборудована 34 тарелкам]!
специального устройства. Нижние 6 тарелок отделены от 28 верх-
них свободным пространством; здесь находится ввод в колонну
поступающей из трубчатой печи нагретой нефти.
Ввод горячей нефти в колонну представляет собою шестидюй-
мовую трубу тангенциально присоединенную к корпусу колонны
(фиг. 56). Внутри колонны, являясь как бы продолжением ввода,
установлена спирально изогнутая пластина, одним своим концом
зы
Разрез по а-а
Разрез по D-D
Разрез по Е-Е
Фиг. 55. Ректификацион-
ная колонна Фостер-
Уилера.
вид сверху
Разрез по в-в
Разрез по С-С
примыкающая к стенке ко-
лонны, другим же концом
отходящая от стенки на
35,6 см. Примерно одна
треть (1) этой пластины,
примыкающая к вводу,
является утолщенной н
сплошной; остальные две
трети С?) ее усеяны много-
численными отверстиями.
Благодаря такому устрой-
ству горячая нефть, по-
ступающая вместе с па-
рами в колонну, отбрасы-
вается по спирали к цен-
тру, а через отверстия пла-
стины — к стенкам колон-
ны, одновременно разбрыз-
гиваясь в виде тончайшей
пленки; в результате до-
стигаются сильное распы-
ление нефти и высокая
эффективность ее испа-
рения.
Отделившись от жид-
кой фазы, пары нефти
поднимаются вверх но ко-
лонне и, постепенно рек-
тифицируясь на ее тарел-
ках, дают начало основ-
ным, получаемым с уста-
новки дестиллатам, кото-
рые отводятся каждый
в свой холодильник 11 и
далее—-в свой резервуар
17—20. Лишь самый лег-
кий дестиллат, газолин,
покидает колонну в паро-
образном состоянии; прой-
дя теплообменник 3
(фиг. 54), два последова-
тельно соединенных холо-
дильника 13 и водоотде-
литель 14, газолин отво-
дится на промывку в
скруббера 15 и в соответ-
ствующий резервуар .16;
часть его с помощью на-
соса 7 подается в . ко-
лонну обратно, на ороше-
ние (12).
362
Иижяяя секция колонны ФостерУ и.i с ра, служащая для разделеййя
жидкой и паровой фаз, очевидно, играет здесь роль эвапоратора, т. е. при-
способления, специально предназначенного для отделения жидкой и паро-
вой фаз. Такое отделение должно производиться по возможности тщательно,
так, чтобы брызги нефти ле увлекались в среднюю часть колонны и не
перемешивались здесь с дестиллатом. Ввиду этого на некоторых установ-
ках эвапораторы представляют собою обособленные части со специальными
приспособлениями для задерживания капелек жидкости (решетки, сетки
и т. и.), через которые лишь паровая фаза проходит свободно. В нижнюю
часть эвапоратора обыкновенно подается водяной пар для отдувания по-
следних следов легких дестиллатов.
Фиг. 56. Ввод нефти в ко-
лонну.
Наиболее существенной частью каждой колонны является
расположение и устройство ее тарелок (фиг. 55). Как было ука-
зано выше, первые 6 тарелок в колон-
не Фостер-Уилера расположены ниже
ввода нагретой .нефти 1. Следующие
4 тарелки расположены непосред-
ственно над ее вводом. Далее, следуют
3 тарелки DE особого устройства,
образующие’ так называемую первую
«испарительную секцию» (stripping
section); с нее при перегонке парафи-
нистой нефти отводится некристалли*
зующийся парафинистый дестиллат
(slop-wax). Выше расположены 10 таре-
лок обычного типа; с седьмой из них
отводится обычный парафинистый де-
стиллат 5. Следующие три тарелки
и образуют вторую «испарительную
секцию», аналогичную первой; с нее отводится газойль 4. Нако-
нец, следуют последние 8 тарелок, из которых ла последнюю,
самую верхнюю, поступает орошение 2, с четвертой же (сверху)
отводится керосиновый дестиллат 3. Все тарелки должны быть
расположены строго горизонтально.
В описываемой колонне лишь два продукта отводятся пз специальных
устройств, остальные отводятся с обыкновенных тарелок. В новейших ко-
лоннах того же типа все продукты отводятся только с «испарительных
секций».
Кроме поименованных выше тарелок, в колонне, над вводом
нефти из трубчатой печи, расположены два ряда пластин (жа-
люзи), перекрывающих друг друга в шахматном порядке. Это
устройство играет роль отбойников, препятствующих увлече-
нию брызг жидкости. Такие же приспособления имеются в верх-
ней части колонны, над вводом орошения, а также под каждой
из семи верхних тарелок; .впрочем, последние жалюзи перекры-
вают собою не всю площадь сечения колонны, а только часть ее,
расположенную над отверстиями для паров.
Каждая из основных 28 тарелок (т. е. все тарелки колонны,
за исключением шести тарелок испарительных секций) имеет
следующее устройство. На тарелке имеется 8 продольных щеле-
видных отверстий (91,5 X 5,08 ел) с закругленными краями и
363
бортиками Высотою < ,62 см. Каждое из отверстий покрыто кол-
пачком той же формы, но несколько большего размера (99,1 у
X 11,4'X 10,16 см), с краями, зазубренными в виде пилы. От-
верстия на тарелке расположены параллельно друг другу и за-
нимают только ее среднюю часть, оставляя крайние сегменты
свободными. Один пз таких сегментов каждой тарелки отгоро-
жен от остальной ее части бортом (высота 17,75 см), образуя
собою как бы чашку, наполненную во время работы колонны
жидкостью. В эту жидкость погружены концы двух: сливных
трубок, опускающихся с расположенной выше тарелки, так что
создается гидравлический затвор, препятствующий прорыву
ларов вверх через сливные трубки. У противоположного сегмента
борт значительно ниже (5.0S с.к); он определяет толщину слоя
падкости в тон части тарелки, которая занята колпачками.
Со дна этого сегмента вниз отходят две сливные трубки в пер-
вым сегмент следующей нижней тарелки. Таким образом
флегма, поступающая через сливные трубки в первый сегмент,
переливается через его борт, проходит по тарелке вдоль ее диа-
метра по каналам, образуемым стенками колпачков, барботи-
руется здесь парами, проходящими через щели между стенками
отверстий тарелки и колпачками, теряя при этом cbqh легкие
части и обогащаясь тяжелыми, попадает, наконец, на противо-
лежащий сегмент и через сливные трубки стекает на нижеле-
жащую тарелку, чтобы проделать здесь такой же путь.
Испарительные секции колонны имеют иное устройство.
Нижняя тарелка каждой из них не имеет ни покрытых колпач-
ками отверстий, ни сливных трубок; только посредине ее
имеется большое прямоугольное отверстие, продолжающееся
в виде канала вверх сквозь следующие две тарелки. Образо-
ванные таким образом на каждой из двух последних тарелок
две пары сегментов несут на себе: одна пара — по два щеле-
видных отверстия на каждом сегменте, прикрытые колпачками,
другая пара — сливные трубки для ввода и вывода флегмы..
Таким образом нижняя тарелка каждой испарительной секции
представляет собою как бы основание (дно) стоящей на ней
части колонны, на котором собирается и задерживается соот-
ветствующая флегма. На второй тарелке проложены маточники
для открытого пара, и с нее по трехдюймовой линии отводится
в виде жидкости освобожденный от легких фракций «отпарен-
ный» продукт, направляемый в холодильники.
Сливные трубки ближайшей, расположенной над испаритель-
ной секцией тарелки соединяются со специальной двухходовой
задвижкой, одна часть которой расположена внутри колонны,
другая — снаружи. Регулируя эту задвижку от руки, напра-
вляют флегму, стекающую с этой тарелки, в два потока: один из
них идет по сливным трубкам на верхнюю тарелку испаритель-
ной секции, другой же с помощью шестидюймовой трубы, нахо-
дящейся снаружи колонны и снабженной гидравлическим за-
твором, направляется на ближайшую тарелку, расположенную
иод испарительной секцией, и служит орошением для этой
Ь64
части колонны. Таким образов, та £тасть флегмы, которая пб-
надает в испарительную секцию, постепенно заполняет ее, под-
вергается здесь обработке водяным паром и т. д., кате это опи-
сано выше; другая же часть флегмы, перемешиваясь с флегмой
части колонны, расположенной ниже испарительной секции,
отдает здесь своп легкие части в виде паров, которые проходят
в верхнюю часть колонны через прямоугольный канал, прони-
зывающий тарелки испарительной секции. Путем регулировки
двухходовой задвижки, вплоть до закрытия отдельных потоков,
можно изменять количества и качества отдельных дестиллатов
в желаемой степени и, таким образом, направлять перегонку
сообразно потребности производства в зависимости от качества,
сырья.
Сливные трубки трех тарелок, с которых продукты отводятся
непосредственно, без испарительных секций, также снабжены
двухходовыми задвижками, которые одной части конденсата
дают возможность стекать на следующую, нижележащую та-
релку, другую же часть его отводят в холодильник и приемник.
Регулирование этих задвижек также определяет количество и
' качество отдельных дестиллатов с установки.
Детали устройства тарелок колонных аппаратов весьма
разнообразны. Так, отверстия в тарелках делаются не только
щелевидные, но также овальные и круглые. Соответственно
этому разнообразную форму имеют и колпачки; кроме продол-
говатых, в большом ходу колпачки трехгранные (установка
Кросса), шестигранные (установка Винклер-Коха) и т. п. Раз-
меры колпачков н число их в колонном аппарате также весьма
разнообразны. Имеются колонны, в которых количество кол-
пачков определяется несколькими тысячами штук.
Весьма существенную часть работы колонны составляет оро-
шение, достигаемое вводом одноименных фракций в соответ-
ствующую секщпо колонны. Такое орошение производится, как
было указано выше, -перетоком флегмы в местах нахождения
испарительных секций; подобное же орошение имеется в самой
верхней секции колонны. Газолин, получаемый из этой секции,
после конденсации и промывки частью направляется в резер-
вуар, частью же отдельным центробежным насосом непрерывно
подается обратно в колонну, на верхнюю ее тарелку, как оро-
шение (reflux). Температура вверху колонны поддерживается
постоянной с помощью регулирующего пирометра, автоматиче-
ски изменяющего количества подаваемого орошения, которое
является, таким образом, основным регулятором работы колонны.
Описанная трубчатая установка Фостер-Уилера перераба-
тывает в среднем около 450 т нефти в сутки; имеются также
установки этой системы, перерабатывающие 1000—2000 т
в сутки и больше. Так например, в Баку и Грозном в настоя-
щее время построено несколько трубчатых установок с произ-
водительностью 3500 т в сутки (до 1 млн. г нефти в год). Диа-
метр колонны такой трубчатой установки достигает 4 м, а ее
высота — 30 х
366
Расход тол ша на трубчатой установке Фостер-Уилера в за-
висимости от сырья и глубины отбора составляет 1,5—2,5% от
перерабатываемой нефти, расход водяного пара — 4—6%. Уста-
новка работает под небольшим избыточным давлением, а именно
0,2 ат наверху колонны и 0,8 ат внизу. Для характеристики
трубчатой установки большое значение имеет высокий коэфи-
циент использования тепла, особенно ио сравнению с кубовой
установкой. В то время как на кубовых батареях этот коэфи-
циент обычно не превышает 35% и лишь в редких случаях до-
стигает 50%, в трубчатой печи Фостера полезная утилизация
тепла достигает 75%.
Трубчатая установка Фостер-Уилера, как. было показано
выше, имеет только одну ректификационную колонну, которая
является нс только погоноразделителем,, ио в нижней своей
части также эвапоратором. В свою очередь погоноразделптеть-
ную часть этой колонны можно рассматривать состоящей из не-
скольких поставленных друг на друга ректификационных ко-
лонн соответственно числу получаемых с установки конденса-
тов (флегм). В ряде других трубчатых установок проведено бо-
лее глубокое разделение этих элементов установки па не-
сколько отдельных колонн меньшего размера, каждая из кото-
рых выполняет свою специальную функцию: одни из них яв-
ляются эвапораторами или сепараторами, другие дают бензино-
вый или керосиновый дестиллат и т. д. Таковы, например, труб-
чатые установки Вилъке, Борланна, Пиниа, Баджера и др.
Большинство современных трубчатых установок снабжается
ректификационными колоннами колпачкового типа, устройство
которых, как это видно на примере колонны Фостер-Уилера,
довольно сложно. Значительно проще и дешевле так называе-
мые насадочные колонны, получившие благодаря этому весьма
широкое применение в нефтяной промышленности, но пригод-
ные только при малых производительностях и диаметрах ко-
лонны.
Насадочная колонна представляет собою высокий цилиндр,
заполненный для увеличения поверхности соприкосновения
паров с флегмой тем или иным материалом. Отношение диа-
метра колонны к ее высоте определяется в пределах от 1 :6 до
1:8. Материалом для насадки являются: битый кирпич или
стекло, кокс и специальные насадки различной формы
(фиг. 57), приготовляемые из огнеупорных и кислотоупорных
материалов; таковы, например, кольца Рашига (открытые ци-
линдры различных размеров с отношением диаметра к высоте
1:1), насадки типа «Ргуш» (трехгранные призмы, полые или
с продольными перегородками и наружными выступами, пре-
пятствующими плотному прилеганию отдельных призм друг
к другу) и т. д. Во всех этих случаях, как показал опыт, важно
лишь возможное развитие поверхности; материал же насадки и
его свойства, в частности теплоемкость и теплопроводность, по-
видимому, пе имеют значения. Насадка засыпается в колонну
или в беспорядке, или каким-либо ощюделенным образом
36Ъ
<с щелью воспрепятствовать образованию струйного потока паров.
,Дця поддержания насадки колонны снабжаются тарелками, ко-
торые здесь представляют собою решетки, способные задерживать
1на ‘себе тонкий слой флегмы (около 1 сл высоты). Кроме того,
iHасадочные колонны снабжаются нередко перегородками с кол-
пачками с целью отвода части флегмы на сторону в качестве
щелевого продукта.
Сопоставление колпачковых и насадочных колонн приводит
к следующим результатам. При средней мощности колпачковые
колонны можно считать равноценными с колоннами насадоч-
ного типа. Напротив, для колонн, рассчитанных на большие
мощности, преимущества оказываются на стороне колпачковых
колонн. С другой стороны, насадочные колонны являются весьма
Фиг. 57. Различные типы насадок.
удобными при работе на сильно разъедающем сырье, так как
в данном случае возможно применение насадок из тех или иных
кислотоупорных материалов.
Описание колонного аппарата было бы не по; но, если не
остановиться еще на одной ответственной его части — дефлег-
маторе (конденсаторе). Назначение дефлегматора в колонном
аппарате заключается в том. чтобы сконденсировать часть про-
ходящих через него паров в жидкость и вернуть последнюю на
орошение котонных тарелок. В простейшем случае дефлегматор
представляет собою помещенный в верхней части колонны
змеевик, по которому протекает охлаждающая жидкость (вода,
нефть или ее дестиллаты). Более совершенные дефлегматоры
имеют следующее устройство: это — цилиндр с двумя попереч-
ными перегородками А и В, сквозь которые проходит большое
количество трубок с кососрезанными нижними концами
(фиг. 58). По трубкам проходят пары из колонны, поступающие
через патрубок В и выходящие через С; между трубками —
охлаждающая жидкость. Регулируя температуру этой жидкости
таким образом, чтобы она была немного выше температуры про-
ходящих паров, достигают частичной конденсации и возвра-
щают их в колонну на одну из верхних ее тарелок. Дефлегма-
торы ставятся или наверху колонны, или сбоку от нее в виде
отдельного аппарата.
Дефлегматоры описанных выше типов применяются только
при колоннах малой
г
В
бодо
мощности. Для больших колонн орошение
подается специальным насосом из дестил-
латного приемника головного продукта.
Что касается, наконец, таких частей
трубчатой установки, как теплообменные
аппараты, холодильники и т. и., то на
них можно не останавливаться, так как
здесь не наблюдается каких-либо прин-
ципиальных особенностей по сравнению
с аналогичными устройствами кубовых
батарей.
Вакуумная: перегонка на трубчатых
установках. Трубчатые установки приме-
няются в- настоящее время для получе-
ния не только легких нефтепродуктов
(бензин, керосин), но также и масляных
дестиллатов. В последнем случае уста-
новка должна работать, естественно, под
вакуумом во избежание разложения де-
стиллатов. Таким образом переработка
нефти на светлые продукты и масла и
здесь ведется в две ступени на двух труб-
чатых установках: атмосферной и вакуумной, которые нередко
блокируются в одну атмосферно-вакуумную установку. Лишь
в отдельных случаях возможно получение легкого масляного
дестиллата, например — парафинистого, при атмосферном да-
влении.
Soda I
X.
А
Фиг. 58. Дефлегматор.
Фиг. 59 дает общую схему атмосферно-вакуумной трубчатой
установки. Нефть из резервуара 1 нагревается здесь сначала за
счет теплообмена с парами бензина 2 наверху атмосферной ко-
лонны, затем за счет теплоты паров солярового масла 2 на-
верху (Вакуумной колонны,’ далее за счет теплообмена с горя-
чими дестиллатами вакуумной колонны и, наконец, поступает
в трубчатую печь 6 атмосферной установки. Отсюда нефть вы-
ходит о температурой около 275° и направляется в колонну 7,
работающую при атмосферном давлении, где одна часть нефти
перерабатывается на легкие дестиллаты (8—12), другая же по-
кидает колонну в виде горячего мазута, направляемого горячим
насосом 13 в печь вакуумной установки 14. В этой печи мазут
нагревается до 425° и по выходе из нее направляется (в вакуум-
ную колонну 15, давя я здесь начало образованию различных
масляных дестиллатов 3—5 солярового, веретенного, машинного
и цилиндрового. В зависимости от сырья на подобного рода ат-
мосферно-вакуумной установке удается отгонять до 88—92%
от сырой нефти с получением р остатке 8—12% гудрона, ПреД-
^8
ставляющего собой остаточный асфальт. Масла, получаемые
с вакуумной трубчатой установки, характеризуются высокими
качествами, свойственными дестиллатным маслам, полученным
без разложения.
Фиг. 59. Схема атмосферно-вакуумной трубчатой установки.
Не вдаваясь в дальнейшие подробности устройства атмос-
ферно-вакуумных и вакуумных трубчатых установок, отме-
тим лишь, что, кроме установок Фостера, заслуженную извест-
ность и распространение (также в СССР) приобрели атмосфер-
но -вакуумные установки Баджера [5] и высоковакуумные уста-
новки Алко 6], а в СССР высоковакуумные установки кон-
струкции советских инженеров Пенгу—Гурвич—Нерсесова [7].
ПРОДУКТЫ ПЕРЕГОНКИ НЕФТИ
В результате перегонки сырой нефти получаются продукты
двух основных типов: десгиллатные и остаточные. Их дальней-
шая переработка, а отчасти и области пх применения глубоко
различны.
Дестиллатные продукты. Получаемые путем перегонки
нефти дестиллатные продукты, или дестиллаты, при помощи со-
ответствующей очистки превращаются в целевые продукты не-
фтеперегонного дела: бензин (газолин), керосин и смазочные
масла. Соответственно этому различают дестиллаты бензиновый
(газолиновый), керосиновый, масляный, подразделяемые в свою
очередь на ряд дестиллатов более узкого, специального назна-
чения. Таким образом различают:
1. Бензины: авиационный, автомобильный, экстракционный,
«калоша» (для резиновой промышленности), уаит-спирит, трак-
торный (лигроин) и др.
2. Керосины: осветительный, тракторный легкий, трактор-
ный тяжелый, газойль и др.
3G9
24 Зак. 1622. ХДОВЯ рефти.
3. Масла: соляровое, трансформаторное. веретенное, Турбин-
ное, машинное, моторное, автомобильное, авиационное, цилин-
дровое и др.
На заводе соответствующие дестиллаты отбираются но удель-
ному весу; для получаемых же из них нефтепродуктов устана-
вливаются определенные технические нормы, включающие,
кроме удельного веса, ряд других физико-химических -и техно-
химических свойств данного нефтепродукта. Лишь в редких
случаях дестиллаты употребляются -в сыром виде без очистки.
Соответственно качеству сырья, которым располагает нефтяная
промышленность той или иной страны в данное время, а также.
в связи с успехами нефтеперегонного дела и прогрессом маши-
построения технические нормы на различные нефтепродукты не
остаются постоянными, а изменяются, варьируя нередко в до-
вольно широких пределах.
Следует, впрочем, отметить, что в настоящее время при пе-
регонке нефти получаются не товарные продукты, а так назы-
ваемые компоненты; товарные же продукты готовятся смеше-
нием этих компонентов. Так например, керосиновый компонент
получают после полного отбора лигроина с началом кипения
выше 200, а затем добавкой небольшого количества лигроина
доводят содержание в компоненте фракций до 200 сообразно
требованиям стандарта.
Остаточные продукты. По сравнению с дестиллатными про-
дуктами остаточные продукты, получаемые при перегонке
нефти, значительно менее разнообразны. Сюда относятся пре-
жде всего мазуты, которые идут главным образом на крэкинг и
частично как специальные топливные мазуты и смазочные ма-
зуты, полугудроны и гудроны, т. е. остатки, получаемые после
отгонки от сырой нефти, либо только бензиновых и керосиновых
фракций (мазуты), либо в большей или меньшей степени также
и масленых фракций (гудроны и полу гудроны). Качество этих
продуктов в высокой степени зависит от химического состава
исходной нефти, в частности от содержания в ней парафина и
смолистых продуктов.
Другой тип остаточных нефтепродуктов — нефтяные битумы
или асфальтовые гудроны; одни из них являются типично
остаточными продуктами, остатками после перегонки нефтей,
по преимуществу нафтенового типа; другие получаются путем
продувки в остаточные продукты воздуха при повышенной тем-
пературе и, таким образом, являются окисленными асфальтами.
II те и другие находят широкое применение в дорожном строи-
тельстве. К остаточным продуктам относятся также некоторые
наиболее тяжелые сорта смазочных масел, как-то: вискозины,
вапора и брайт-стоки.
Кроме дестиллатных и остаточных нефтепродуктов, нефте-
перегонные заводы и связанные с ними масловарки производят
значительный ассортимент составных, компаундированных
продуктов. Сюда относятся разного рода консистентные мази и
смазки, некоторые компаундированные масла и т. д.
370
ТЕОРИЯ ЙЁРЕГОЙКИ. ИНДИВИДУАЛЬНЫЕ ЖИДКОСТИ
Со свободной поверхности .жидкости всегда происходит ис-
парение, т. е. отделение паров во внешнее п]к>странство. Эти
пары называются ненасыщенными: их упругость меньше внеш-
него давления, но она постепенно увеличивается по мере повы-
шения температуры жидкости. В момент, когда упругость паров
подогреваемой жидкости сравняется с внешним давлением, паро-
образование начинается во всей массе жидкости: начинается ее
кипение, и ее пары приобретают наибольшую при данных усло-
виях упругость. Такие пары называются насыщенными. Отводя
образующиеся при кипении нары из сферы их образования и
понижая их температуру путем искусственного охлаждения
в холодильнике, можно понизить их упругость; часть паров при
этом должна, очевидно, вновь превратиться в жидкость, т. е.
сконденсироваться, и может быть собрана в приемник. Такова
в самых общих чертах физико-химическая сущность процесса
перегонки.
Как ясно из вышеизложенного, процесс перегонки слагается
из двух прямо противоположных стадий: кипения, т. е. парооб-
разования во всей массе жидкости, и конденсации, т. е. пере-
хода насыщенных паров в жидкое состояние с образованием
«конденсата». Наиболее просто протекает. весь. этот процесс
в случае индивидуальных жидкостей.
Как известно, характерная особенность кипения индивиду-
ального вещества заключается в постоянстве его температуры
кипения, т. е. той температуры, при которой упругость его ла-
ров достигает при данном давлении наибольшей величины.
Между упругостью ненасыщенных паров Р и температурой ки-
пения t данного вещества существует сложная функциональ-
ная зависимость, различная для разных веществ:
Р = f (t).
Определение этой зависимости опытным путем является
одной из задач экспериментальной физики, требующей специ-
альной методики и точной аппаратуры.
Опытное определение упругости насыщенных паров при
разных температурах. Для определения сопряженных значе-
ний упругости насыщенного пара Р при разных температурах t
можно пользоваться одним из следующих методов [9]:
1. Статический метод. Сущность метода заключается в опре-
делении понижения высоты ртутного столба барометра при вве-
дении в баро-метрическую пустоту изучаемого вещества в коли-
честве, достаточном для ее заполнения насыщенным паром.
Упругость Р измеряется разностью высот ртутных столбов до
и после введения жидкости при условии, если температура t
в течение опыта не изменилась.
2. Динамический метод основан на непосредственном опре-
делении температуры кипения t вещества под определенным да-
влением, устанавливаемым произвольно • и точно измеряемым.
24* 371
Еелйчййа этого Дайлёййя и нрсдстйвляет собою, как была ука-
зано, упругость Р насыщенного пара данного вещества при
температуре t.
3. Метод изучения изотерм основан на исследовании зави-
симости между упругостью Р и объемом V ненасыщенного пара
при постоянной температуре. Принимая V и Р за оси коорди-
нат, выражают зависимость между этими величинами в виде
ряда кривых (изотерм), каждая из которых соответствует опре-
деленной температуре. При малых упругостях и больших объ-
емах ненасыщенные пары по своим свойствам близки к идеаль-
ным газам, так что можно принята PV = const; кривая мало
отличается от гиперболы. По мерс уменьшения объема V и уве-
личения упругости Р кривая все более и более отходит от ги-
перболы; в некоторой точке наступает перелом, и кривая пре-
вращается в прямую, параллельную оси К, уравнение которой
Р = const. С этого момента упругость уже не зависит от объема,
так как пар становится насыщенным; его упругость при данной
температуре определяется по точке перелома.
Одним из обязательных условий при определении упругости
насыщенного пара является совершенная чистота объекта ис-
следования.
Зависимость между упругостью насыщенных паров и тем-
пературой. Второй закон термодинамики позволяет вывести
следующую зависимость между упругостью насыщенного пара Р
и температурой в градусах абсолютной шкалы Т\
(IP Л
7/г ~‘7’04
где L — скрытая теплота парообразования па 1 граммомоле-
кулу жидкости, a vi и тв — удельные объемы насыщенного пара
и жидкости, взятых также в количестве 1 граммолекулы. Это
уравнение (Клапейроне-Клаузиуса) (вполне точно, однако при-
менение его встречает ряд практических трудностей и ограни-
чивается поэтому лишь областью теоретических выводов термо-
динамики. Вместо него в практической работе пользуются упро-
щенными эмпирическими формулами и диаграммами, позво-
ляющими с большей или меныпей точностью получить требуе-
мую зависимость.
Практические задачи, требующие знания зависимости упру-
гости пара от температуры или, что то же, температуры кипе-
ния от давления, заключаются обыкновенно в расчете темпе-
ратуры кипения жидкости при заданном давлении на основе
температуры кипения ее при некотором другом, например нор-
мальном давлении. Подобного рода задачи могут быть решены
путем сопоставления данной жидкости с некоторой другой, для
которой зависимость температуры кипения от давления хорошо
изучена. Таковы, например, вода, нормальные гексан или октан
и некоторые другие вещества. Для сравнения лучше выбирать
жидкости, имеющие сходные кривые упругости, т. е. по возмож-
ности близкие друг к другу по своей химической природе.
872
При этоМ: Mojito йолЬзойатйсй следующими формулами!
1. Формула Дальтона. Эта наименее точная формула имеет
следующий вид
'о~ ' = V —
где to и t'o — температуры кипения двух жидкостей при нор-
мальном давлении, a t и Г— их же температуры кипения при
некотором другом, но одном и том же давлении. Физиче-
ский смысл формулы Дальтона заключается, очевидно, в сле-
дующем: при температурах, равноотстоящих от температуры ки-
пения двух жидкостей при нормальном давлении, упругости их
насыщенных паров одинаковы. Эта правильность оправды-
вается, однако, лишь в отдельных случаях.
2. Формула Дюринга. Зависимость между теми же величи-
нами, что и в предшествующей формуле, выражается по Дю-
рингу следующим образом:
где К — некоторая постоянная, изменяющаяся для различных
жидкостей при сопоставлении их с водой от 0,52 до 2,29; для
углеводородов эта величина колеблется в пределах примерно от
1,1 до 1,5, чем и определяется точность результатов, которые мо-
гут быть получены с помощью этой формулы.
3. Формула Рамсай-Юнга. Эта формула имеет следующий
вид:
где То и Т — абсолютные температуры кипения одной жидкости
при разных давлениях, а Т'о и Т’ — такие Же температуры дру-
гой жидкости при тех же давлениях; С — постоянная для дан-
ной пары жидкостей, очень малая величина, для углеводоро-
дов порядка 0,00007—0,00003.
На практике особенно часто приходится прибегать к подоб-
ного рода формулам для расчета температуры кипения в ва-
кууме того или иного вещества, например какого-либо углево-
дорода нефтяной фракции и т. п. Для сравнения можно поль-
зоваться при этом температурой кипения при различных давле-
ниях нормального октана (табл. 88).
Таблица 88
Температуры кипения нормального октана
(по Томасу и Юнгу)
Давление в мм [1g 760 100 50 30 20 10 5
Температура кипения . . 125°,8 65°,6 50°,2 39°,3 31°,3 19°,1 7Э,7
Получаемые при подобного рода пересчетах данные часто
хорошо согласуются с опытом. Однако нередки случаи, когда
3
расхождения Достигают при этом нескольких градусов, а ййогда
могут превышать даже 10°.
При С —о формула Ражаи-Юнга может быть написана
в следующем виде:
То- Т_ ТУ~Г
т0 - ту ’
т. е. отношение изменения температуры кипения различных
веществ, в зависимости от давления, к температуре кипения
при нормальном давлении — величина постоянная.
Этой закономерностью, хорошо оправдывающейся для род-
ственных веществ при небольших изменениях давления, можно
пользоваться для приведения температуры кипения к нормаль-
ному давлению. На ней основано правило Крафтса:. если при
давлении Я температура кипения жидкости — Z, то при нор-
мальном давлении эта температура Тй может быть найдена по
формуле:
7’0==7’_|_а(760— Н).
Величина а определена для многих 'веществ; около нормаль-
ного давления (760 .о) для многих веществ изменение Т на 1 мм
давления равно 0,04°.
Формулы Дюринга и Рамсай-Юнга находят широкое применение для
пересчета температуры кипения при некотором, например нормальном,
давлении на температуру кипения при каком-либо ином заданном давле-
нии, и наоборот.' Недостатком их является необходимость знать темпера-
туру кипения некоторой стандартной жидкости при двух различных
давлениях, что иногда представляет известное неудобство. Последнего,
однако, можно избежать, если в качестве стандарта принять не одну,
а две жидкости, например два углеводорода. В таком случае формула
Дюринга для трек жидкостей а, b и с примет следующий вид:
<0°-<n ь- „ *0Ь-'Ь. ,,
if—Iе 1 4ОС_ (с «•
Если преобразовать эти выражения, вычтя из обеих частей их по
единице и разделив одно из полученных таким образом выражений на
другое, то получаем соотношение:
у —у-(^_<С) _ —1 _
— tV — К2 — 1 3’
Легко видеть, что это соотношение является следствием пропорции;
*og —*oc_Jg-*c_ к
t^ — ty 3'
Вычитая из обеих частей этой пропорции по единице, получаем окон-
чательно:
tb 1 _ Кх-К2
tb—te 1-Л4- а-2_х ,
где К4 от давления не зависит [8].
Графическое изображение зависимости между упругостью
насыщенных паров и температурой. Проследить, как изменяется
упругость паров с изменением температуры, удобнее всего гра-
374
фически, построив на основе опытных данных кривые упруго-
сти пара на координатных осях р, Г. На фиг. со приведены та-
кие кривые для бензола 1 и толуола 2. Как видно, кривые эти
имеют вид парабол, которые при низких температурах касаются
оси абсцисс Т, при высоких же температурах делают резкий по-
ворот в сторону оси ординат р Допустим, что уравнение какой-
либо из этих кривых может
быть действительно выра-
жено уравнением параболы:
р == а7«,
где а и п — постоянные
числа.
Если через р0 и То обоз-
начить некоторую пару со-
пряженных значений коор-
динат данной параболы, то,
очевидно:
Д’о = «V»
откуда
Фиг. 60. Кривые упругости пара:
1—для беязол»; х,—для толуола.
где 7’0 и Т—температуры кипения данной жидкости по абсо-
лютной шкале температур при давлениях р$ и р.
Логарифмируя последнее уравнение, получаем:
1g Р — 1g Ро = п (ig Т — 1g /’о)-
Таким образом в координатных осях 1g р и 1g Т зависимость
между упругостью насыщенных паров и нх температурой должна
бы изображаться прямой линией. В действительности, вместо
прямых при этом получаются несколько изогнутые линии, как
это видно на фиг. 61, так что истинный вид кривых изменения
упругости насыщенных паров от температуры в координатных
осях р, Т может быть выражен параболой лишь с известной до-
лей приближения.
Кроме вышеприведенных формул, для выражения зависимо-
сти между температурой и упругостью насыщенных паров раз-
личных жидкостей был предложен ряд эмпирических формул н
диаграмм, некоторые из которых очень удобны для расчета и
дают хорошие результаты. Таковы, например, диаграммы Аш-
ворта, Вилъс&на и др. Их рассмотрение выходит из рамок на-
стоящего курса [10].
Если проследить изменение температуры кипения какого-
либо вещества с давлением, то легко заметить следующую кар-
тину: при больших давлениях, например близких к атмосфер-
ному, температура кипения снижается с понижением давления
крайне медленно-; постепенно, однако, по мере падения давле-
ния снижение температуры кипения усиливается и, наконец,
при низких давлениях становится весьма значительным. Приме-
ром может служить ход изменения температуры кипения гепта-
декана (СпНзо) по Краффту (табл. 89).
Таблица 89
Температура кипения гептадекана
(по Краффту)
Дав тонне в л/.н Hg 760 1С0 50 . 30 20 10 Катодный вакуум
Температура кипения . 303° 223° 20V,5 187°,5 177° 161° 81э
Нанося температуры кипения гептадекана на оси ординат,
а .давление на осн абсцисс, можно построить кривую, наглядно
изображающую ход изменения температуры кипения с давлением
для данного примера (фиг. 62). Как видно, снижение темпера-
туры кипения, достигаемое благодаря вакууму, может быть гро-
мадно: для выоококипящих
Фиг. 61. Кривые упругости паров
в координатных осях (lg Т, lg Р):
1 — бенволи; 2 — толуола.
Фиг. 62. Кривая изменения темпе-
ратуры кипения гептадекана в за-
висимости от давления.
Перегонка индивидуальных жидкостей. Как уже было отме-
чено, перегонка индивидуальных жидкостей представляет собою
процесс, протекающий при условии постоянства давления изо-
термически: температура должна оставаться здесь постоянной
во все время процессов перегонки и конденсации, и это обстоя-
тельство позволяет широко пользоваться перегонкой для харак-
теристики степени чистоты подвергаемого перегонке вещества.
376
Прибор устраиваемся таким образов, чтобы
термометр находился в парах жидкости, для
чего пользуются либо обыкновенной кол-
бой Вюрца (фиг. 03), либо круглодонной
колбой с разного рода специальными отвод-
ными насадками для пара (фиг. 64). Кипе-
ние должно происходить равномерно, без
толчков, во избежание последних в перегон-
ный сосуд вносят небольшие кусочки пори-
стых тел (пемзу, кусочки неглазурованного
фарфора и т. п.), либо пропускают в ки-
пящую жидкость мельчайшие пузырьки
воздуха через тончайший капилляр. При
ОТСУТСТВИИ ЭТИХ приспособлений кипение Фиг. 63. Колба Вюрца.
легко приостанавливается, и тогда может
наступить перегрев жидкости и паров, который сказывается на
показании термометра. При соблюдении надлежащих условий
перегонки точность определения температуры кипения индиви-
дуальных веществ может достигать 0,01°.
Фиг. 64. Отвогные насадки для пара.
ТЕОРИЯ ПЕРЕГОНКИ. НЕСМЕШИВАЮЩИЕСЯ ЖИДКОСТИ
Среди разнообразных случаев жидких смесей простейшим
является случай двух жидкостей, нерастворимых друг в друге.
Правда, жидкостей, абсолютно нерастворимых друг в друге, не
существует; однако известно немало случаев, когда взаимная
растворимость двух жидкостей настолько незначительна, что
практически их можно считать нерастворимыми. Таковы, напри-
мер, смеси: вода и различные углеводороды, например бензол
и др. При сливании вместе такие две жидкости образуют два
резко отграниченных друг от друга слоя: более тяжелый— внизу,
более легкий — наверху. При нагревании такой смеси упругость
паров каждого компонента будет оставаться такой же, как и при
его нагревании в отдельности до той же температуры; общая же
упругость паров смеси.Р по закону Дальтона будет равна сумме
парциальных упругостей отдельных компонентов Pi и Рг:
Р==?1+Р2.
в
377
к например, из таблиц упругостей .па.|юв различных }к>
щрств при разных температурах можно найти, чти для бензола
упругость паров, постепенно возрастая с температурой, при
г»9°,2 достигает 535 мм; при той же температуре упругость во-
дяного пара оказывается равной 225 мм. Таким образом упру-
гость паров смеси этих двух веществ при 69 \2 равна 760 ж
Отсюда следует, что при 69°,2 и нормальном давлении смесь
бензола с водой должна закипать и перегоняться; это и наблю-
дается в действительности, хотя каждый из компонентов дан-
ной смеси в отдельности кипит значительно вьппе: бензол при
80° ,2, вода при 100°.
Рассмотренные простые соотношения лежат в основе важного
технологического и лабораторного процесса перетопки с водяным
паром и поэтому подлежат ближайшему изучению.
Основные газовые законы. В последующем изложении при-
дется иметь дело с рядом основных газовых законов, подробно
.излагаемых в курсах физики и химии. Законы эти — следующие:
1. Закон Бойля-Мариотта. При постоянной температуре упру-
гость газа Р изменяется обратно пропорционально его объему V;
иначе, говоря:
РГ=С,
где С—величина постоянная.
2. Закон Гей-Люссака. При постоянном давлении коэфп-
циент объемного расширения газов <х — величина для идеальных
газов постоянная и равная отсюда:
Г0(14-аО,
где Jo — объем газа при о, а V—объем газа при темпера-
туре t.
3. Закон Авогадро- Ампера-Жерара. В равных объемах паров
или газов при одинаковых температурах и давлениях содер-
жится одно и то же число молекул. Таким образом если
масса газа Q при некоторой температуре и давлении зани-
мает объем К, то, обозначая молекулярный вес этого газа
через М, получаем для любой единицы объема:
Q: Vjf = const.
Это последнее равенство можно преобразовать в другое:
т/. Q.
' V = const,
т. е. объем одной граммолекулы (моля) для всех паров
и газов — величина постоянная. При 0° и 760 мм давления ,
эта величина равна 22,414 л.
4. Уравнение Клапейрона представляет собою следствие двух
первых газовых законов:
•378
или иначе:
РГ=7ГД
где 7’==/4-273— абсолютная температура, а // — газовая по-
стоянная, зависящая лишь от объема взятого газа (или его •
массы) и от выбора единиц измерения объема и давления.
Так, если взять одну граммолекулу газа и выразить давле-
ние в атмосферах, а объем в литрах, то газовая постоянная
получает следующее значение:
й=^==Ч^£=0>0821-
5- Закон Дальтона. Давление (упругость) смеси несколь-
ких газов В равно сумме их парциальных давлений, т. е.
тех давлений (Рр Р2, и т. д.), которые показал бы каждый из
газов смеси, если бы он один занимал весь объем, зани-
маемый данной смесью, т. е.
Р = Р1 + Р2 + 1,з+.--
Все приведенные законы приложимы только к идеальным
газам. Реальные газы и пары отступают от них в большей или
меньшей степени, вообще говоря, тем значительнее, чем газ или
пар ближе к состоянию насыщения.
Правило фаз. Наряду с основными газовыми законами боль-
шое значение при изучении различных случаев равновесия по-
лучил один из основных законов термодинамики, известный пол
именем «правило фаз» (Гиббс, 1876 г.). В самых общих чертах
сущность этого закона заключается в мелующем.
Во всякой равновесной системе следует различать фазы си-
стемы и ее компоненты.
Фазой называется каждая составная часть системы, которую
можно отделить от системы каким-либо чисти механическим
способом. Так например, система «лед — вода — нар» состоит из
трех фаз; система «вода — пар» содержит две фазы и т. д.
Компонентами (веществами) называются такие химически
разнородные составные части системы, которые могут перехо-
дить из одной фазы в другую путем обратимого процесса. Так
например, во всех приведенных выше примерах— один компо-
нент (вода); система «вода — бензол» содержит два компонента;
система «вода—(жидкость)—бензол (жидкость) — их пары»
состоит также из двух ‘компонентов, но содержит три фазы и т. д.
Если в системе, состоящей из одной или нескольких фаз, не
происходит каких-либо изменений, сопровождающихся перехо-
дом вещества из одной фазы в другую, говорят, что система на-
ходится -в равновесии. Равновесие системы определяется темпе-
ратурой t, давлением В и составом фаз; от абсолютной величины
массы отдельных фаз равновесие не зависит. С другой стороны,
изменение температуры, давления или состава фаз, естественно,
влечет за собою нарушение равновесия данной системы.
379
Различают три вида равновесия, для каждого из которых су-
ществует свое особое соотношение между числом фаз У и чис-
лом компонентов п:
1. Равновесие абсолютное характеризуется тем признаком,
• что самое существование данной равновесной системы возможно
лишь при одной определенной температуре t и одном определен-
ном давлении Р. Таг. например, система «лед — вода — пар» мо-
жет находиться в равновесии лишь при t = 0°,0074 и Р = 4,62
{путного столба.. Здесь У — п 4- 2.
2. Равновесие полное характеризуется тем, что одна из ве-
личин, определяющих равновесие, например температура или
давление, могут .быть выбраны произвольно; в таком случае,
однако, другая величина определяется однозначно, по уравне-
нию P = f (t). Примерами могут служить системы «вода—пар»;
«лед — пар» и т. п. В этом случае У = п4- 1.
3. Равновесие неполное характеризуется (возможностью про-
извольного в известных пределах выбора температуры и давле-
ния. Так например, система, состоящая из одного пара, или
одного льда, или из одной жидкой воды, находится в неполном
равновесии, так как во всех этих случаях температура и давле-
ние могут изменяться независимо друг от друга в широких пре-
делах без изменения физического состояния системы. Для всех
с гучаев этого рода У — н.
Объединяя все три случаи равновесия одной формулой, по-
лучаем общее выражение правила фаз:
У £ п 4- 2,
т. с. в условиях равновесия число фаз У пе может превышать
число компонентов системы н больше чем на два.
Другое выражение правила фаз связано с понятием о сте-
пени свободы равновесной системы, т. е. о возможности произ-
вольного выбора температуры, давления и состава фаз, характе-
ризующих данную систему. Число степеней L для абсолютного
равновесия, очевидно, равно нулю, для полного равновесия
L = 1, а для .неполного равновесия L = 2. Соединяя эти равен-*
ства с приведенным выше выражением правила фаз, получаем
второе общее выражение этого закона:
Л = и4-2—У.
Примеры. 1) Две несмешивающиеся жидкости и их пары, например
бензол и вода; число компонентов п = 2; число фаз N = 3; число степеней
свободы L = 1, т. е. равновесие полное; следовательно, здесь можно вы-
брать произвольно лишь одну переменную: пли температуру, или давление,
или состааз паровой фазы; тогда две другие переменные определяются
однозначно.
2) Две смешивающихся жидкости к их пары, например бензол и то-
луол; число компонентов п = 2; число фаз У = 2; число степеней свободы
L = 2, т. е. равновесие неполное; следовательно, пе выходя ва известные
пределы, в данном случае можно произвольно выбрать две переменные,
иащ/пмер температуру и давление; тогда третья переменная, т. е. состав
жидкой или паровой фазы, определится однозначно»
380
С( став пара. 13 случае двух пе смешипающихся жидкостей
состав пара может быть легко определен в общем виде путем при-
менения основных газовых законов.
Пусть Ql и Q-2 — массы паров двух не смешивающихся друг
с другом жидкостей; п и v2 — занимаемые ими объемы; pi и р*
их упругости; З/i и 1Z2— их молекулярные веса.
Смешаем эти пары в объеме V, не изменяя температуры.
Пусть теперь общая упругость паров смеси равна Р, парциаль-
ные упругости отдельных компонентов смеси — Pi и Ра.
По закону Бойля-Мариотта имеем:
Fl 1 — И ^2 ~ Г2/’2»
откуда
/> fl Pt ,r /> e‘>Pi
1 1 — jr J 2 — "p" •
Но ПО закону Дальтона’.
I‘=l\+A,;
следовательно:
n___ rlPl 1 ^>P2
]' -r >
откуда
VP^^p^v.^.
Далее, на основе уравнения Клапейрона для каждого из
взятых паров должно существовать соответствующее соотно-
шение:
^1?! = ^ и %р9 — г2Т,
где rt и г2 — постоянные, зависящие от количества взятых
паров Qi и Qit а также от выбора единиц измерения объемов
и упругостей. Если от масс Qj и Q2 каждого вещества пе-
рейти к их граммолекулам и условиться об измерении объ-
емов в литрах, а упругостей — в атмосферах» то последние
соотношения принимают такой вид:
»
’^iPl •З/] _ 'Гдрл Л/о _ 7>7’
~0Г------
где газовая постоянная 7? = 0,0821.
Отсюда:
91 — EiZikVl
Qi
или, наконец:
& Р2^2‘
Это последнее, общее выражение зависимости состава пара
двух не смешивающихся жидкостей от парциального давле-
ния этих паров и их молекулярного веса удобно может быть
выражено следующими двумя способами;
831
а) Допустим, что Q, -Н Q.2 = 1; тогда ,
_Qt____Pjit,
1—Qi
откуда легко рассчитать:
о _ AJA- _ п 0 _ _ AJA
“ PpVi + А ™ А^А-ралл*
б) Пусть теперь & -1- (А = 100; тогда аналогично получаем
соотношение между компонентами, выраженное в процента!:
_ Ю0ЛЛ4 о/ п о __ _ МВД о/
г 1 ~ PPVi + А*1 А /о t2 J>i-’А + А-1А '°‘
Пример, Для смеси бензола (J/i = 78) с водой (3/2 = 18) при темпе,
ратуре кипения смеси <59°,2 имеем Pi = 535 мм и Рз = 225 мм. Подставляя
эти величины в последние выражения для (Л и 0_> и произведя вычло,
ленпя, получаем: 01 = 91,1% и Оз = 8,9%, т. е. при температуре мяпеим
смеси бензола с водяным паром при нормальном давлении Р = 760 жж
пары должны содержать 91,1% бензола и 8,9% воды, что находится в под.
ном соответствии с опытом.
Температура кипения. Температура кипения смеси двух не-
растворимых друг в друге жидкостей легко определяется по упру-
гости пара компонентов* смеси при разных температурах. Так
например, для .бензола и воды в соответствующих таблицах на-
ходим следующие данные (табл. 90).
Таблица 90
Температура 66° 68° 70° 72’
Упругость паров бензола в мм ........ Упругость паров воды „ „ ........ 472 196 505 214 544 234 580 255
Упругость паров смеси в мм 668 719 778 835
Для определения температуры кипения смеси бензола и воды
при заданном, например нормальном, давлении можно восполь-
зоваться этими данными различными способами.
Построим кривую (фиг. G5) изменения температуры кипения
смеси бензола и воды в зависимости от давления, откладывая
для этого на оси абсцисс температуры, а на оси ординат — со-
ответствующие упругости паров смеси. Проведем затем прямую,
параллельную оси абсцисс на высоте, отвечающей 760 мм. Точка
пересечения этой прямой с кривой изменения температуры ки-
пения смеси в зависимости от давления будет соответствовать
температуре кипения этой смеси при нормальном давлении.
К тому же результату легко притти путем. следуй щего простого рас-
чета. H-j табл. 90 видно, что при G68 мм давления ьмесь бензола с водой’
«82
Дол/Kitii кипсТь при 66°, я при .v.v — при 72 . iinrpptKunpvH в npciP'
лах ятих температур, находим точку, при которой эта гм<чч, -ш.т.кна ки-
петь в условиях нормального давления (7.йо .и.и):
п-с 1 г 760 - 668
Г,Ь + ° 835~668
6JP.3.
Фиг. 65. Кривая температуры
кипения смеси бензол — вода
в зависимости от давления.
Небольшие отклонения от опыта, которые получаются иногда при по-
добного рода расчетах, объясняются тем обстоятельством, что при теорети-
ческом исследовании вопросов перегонки
пары рассматриваются в первом прибли-
жении как идеальные газы с применением
к ним соответствующих законов газового
состояния, что заведомо неверно п, есте-
ственно, приводит к известному расхожде-
нию между теоретическим расчетом и дан-
ными опыта.
Перегонка двух не смешиваю-
щихся жидкостей. Если паровая фаза
состоит только из паров, выделяемых
жидкой фазой, то, очевидно, в усло-
виях равновесия упругость этих па-
ров будет равна внешнему давлению,
т. е. будет удовлетворять условию,
характерному для температуры
кипения. Отсюда следует, что
в условиях равновесия со своей па-
ровой фазой жидкость должна нахо-
диться при точке кипения. Это усло-
вие применимо также для случая
двух не смешивающихся жидкостей.
С точки зрения правила фаз си-
стема из двух не смешивающихся
жидкостей и их паров в состоянии
равновессия имеет одну степень
свободы и, как только что сказано,
должна находиться при температур кипения; давление и
состав паровой фазы здесь однозначно определяются темпе-
ратурой.
Так как упругость насыщенных паров двух не смешиваю-
щихся жидкостей равна сумме упругостей паров отдельных ком-
понентов, то понятно, что температуры кипения подобного родя
систем при данном давлении лежат ниже, чем у отдельных ком-
понентов системы. Как уже было отмечено, температура эта за-
висит от химической природы компонентов, но не зависит от их
относительного количества.
Если путем подогрева сообщить находящейся в равновесий
системе из двух не смешивающихся жидкостей и их паров не-
которое количество тепла, то равновесие ее. естественно, должно
нарушиться. Допуская, однако, что подогрев системы происхо-
дит бесконечно медленно, можно считать, что происходящие при
этом процессы будут протекать также крайне медленно, так что
-система будет оставаться в состоянии, бесконечно близком к со-
383
стоянию равйовесйя. Такое допущеййе, Позволяет рассмотри
.явления испарения жидкостей и конденсации их паров с точка
зрения учения о равновесии, изложенного выше.
Если испарение жидкостей производить в замкнутом про-
стран-стве, то упругость паровой фазы, естественно, будет возра-
стать; одновременно согласно условию полного равновесия L=i
будет повышаться температура системы, и наступит изменение
состава ее паровой фазы. Если, однако, образующиеся пары бу-
дут отводиться из системы на сторону, так что упругость их бу-
дет оставаться постоянной, то согласно тому же условию темпе-
ратура системы и состав ее паровой фазы также останутся без
изменений. Таким образом процесс выкипания обеих не смеши-
вающихся жидкостей -под влиянием притока к системе теплоты
будет происходить при постоянном давлении, постоянной темпе-
ратуре и постоянном составе паровой фазы, который опреде-
ляется выведенным выше уравнением:
М2
Если не смешивающиеся жидкости взяты в количествах,
определяемых последним отношением, то вместе они выкзпшот
без остатка; в противном случае в остатке окажется компонент,
взятый в избытке, и дальнейшее его испарение потребует повы-
шения температуры до точки кипения этого компонента.
Если вместо нагревания производить бесконечно медленное
охлаждение равновесной системы двух не смешивающихся жид-
костей, то общая картина по существу не изменится, и равнове-
сие также не нарушится. Все различие будет заключаться лишь
в том, что под влиянием охлаждения часть паровой фазы си-
сТемы сконденсируется при постоянной температуре, которую
.можно назвать в данном случае температурой конденсации.
Если такая конденсация произойдет в замкнутом пространстве;
то создаваемое парами давление, естественно, понизится; одновре-
менно согласно правилу фаз должны будут понизиться темпе-
ратура системы и измениться состав паровой фазы. Если же
конденсация будет происходить при постоянном давлении, на-
пример при постоянном подводе все новых и новых паров, то ш
температура системы, так и состав паровой фазы останутся не-
изменными. Таким образом при изобарическом процессе все
действие охлаждения сведется к процессу конденсации паров,
причем состав получаемого дестиллата (конденсата) должен
быть, очевидно,- тот же самый, что у паровой фазы, т. е. опреде-
ляться тем же уравнением:
~Q.> ' •J
Как ясно из вышеизложенного, перегонка с водяным паром,
понижая температуру кипения жидкости, дает тот же эффект,
что и перегонка под уменьшенным давлением. В тех случая^
когда требуется еще большее снижение температуры кипеЫя,
384 *
можно прибегнуть к перегонке с паром под уменьшенным Да-
влением. При этом, как показывают примеры табл. 91, могут
встретиться два случая: с понижением давления количество по-
требляемого водяного пара либо увеличивается, либо умень-
шается.
Таблица 91
Расход пара при перегонке несмешивающнхся веществ
(по К. Эллиот)
Вещества Темпера- тура кипения Парциаль- ное давте- ние паров вещества, в jwjk Парциаль- ное давле- ние паров воды, в мм Содержание вещества в парах, в % по весу Расход пара в кг на 1 кг вещества
Бензол Г 16°,0 62,50 13,50 95,3 0,06
и 1 69°,2 535,00 226,00 91,0 0,10
вода 1 112°, 6 1867,00 1173,00 86,9 •0,15
Нафталин f 46°,1 0,57 75,40 5,0 19,00
и вода 1 99°, 3 18,10 741,80 14,4 5,90
Как видно из табл. 91, при перегонке смеси бензол — вода
понижение общего давления и соответственно снижение темпера-
туры кипения смеси сопровождаются уменьшением расхода пара.
Очевидно, что в подобного рода случаях перегонка с паром в ва-
кууме должна бы представлять особые выгоды; однако эти пре-
имущества резко снижаются вследствие тех затруднений, с кото-
рыми приходится встречаться при конденсации водяных паров
в условиях глубокой вакуумной перегонки.
В других случаях, как нафталин — вода, снижение общего
давления и соответственно температуры кипения смеси сопро-
вождается резким повышением расхода пара; то же наблюдается
и при получении средних и тяжелых нефтяных дестиллатов. Не-
смотря на это, перегонка с паром в вакууме широко применяется
в нефтяной промышленности, так как достигаемое при этом сни-
жение температуры кипения имеет в ряде случаев, например
при перегонке мазута «а масла, чрезвычайно важное значение.
Расход пара достигает при этом 120% и больше, но он вполне
оправдывается снижением температуры перегонки и рядом дру-
гих практически важных результатов, каковы: энергичное пере-
мешивание обогреваемого масла проходящим водяным паром,
благодаря чему устраняется пригорание; ускорение хода тяжелых
масляных паров в дефлегматорах и холодильниках и т. д.
ТЕОРИЯ ПЕРЕГОНКИ. ДВЕ СМЕШИВАЮЩИЕСЯ ЖИДКОСТИ
Кроме только что рассмотренного случая двух жидкостей, не
смешивающихся, т. е. нерастворимых друг в друге, возможны
ЗЬЕ
25 Зак. 1622. Химия нефти.
разнообразнейшие смеси Двух жидкостей, либо частично раэд.
римых друг в друге, либо растворимых полностью, т. е. во вд
отношениях. Для всех смесей (растворов) подобного рода упру-
гость насыщенных паров 'зависит не только от температуры, но и
от взаимной растворимости компонентов смеси. Сущность ад
последней зависимости заключается в том, что взаимная' раство-
римость жидкостей понижает упругость их паров, так что пар-
циальное давление насыщенных ларов каждого компонента дан-
ной смеси ниже упругости паров тех же компонентов, взятых от-
дельно друг от друга при той же температуре. Эти соотношения
для различных возможных здесь случаев удобно выразить гра-
фически.
Изотермы упругости паров смеси двух жидкостей. Отложим
на оси абсцисс молекулярные концентрации низкокишшщо
компонента данной смеси, а на двух ординатах — упругости на-
сыщенных паров этих компонентов. Тогда в зависимости от их
упругость насыщенных паров различ-
ных смесей может оказаться выражен-
ной одной из следующих кривых, изо-
терм (фиг. 66).
Кривая 1 соответствует случаю,
когда компоненты смеси — взаимно не-
растворимы. Для этого случая, как
было показано выше на примере смеси
бензола с водой, упругость паров сме-
си Р равна сумме упругостей Pi + Pt
отдельных компонентов, независимо
от состава смеси. Поэтому кривая
упругости пара такой смеси должна
представлять собою прямую, парал-
лельную оси абсцисс. Так как, однако,
полная нерастворимость двух жидко-
стей существует только в теории, то
для очень слабых растворов здесь бу-
дет наблюдаться лишь постепенный пе-
реход к предельному значению упругости пара, и соответствую-
щая кривая упругости пара будет иметь закругленные концы.
Кривые 2, 3 и 4 изображают ход изменения упругости пара
смеси двух компонентов, взаимно растворимых друт в Друге.
При постепенно возрастающей взаимной растворимости компо-
нентов упругость пара их смесей будет снижаться все больше и
больше, оставаясь, однако, выше упругости пара высококипя-
щего компонента. Примерами смесей подобного рода могут слу-
жить смеси: фенол и вода, винный или пропиловый спирт и’вода
и др. Некоторые из такого рода смесей имеют для определенных
концентраций максимальные упругости пара (кривая 3).
Кривая 5 отвечает изменению упругости пара раствора осо-
бого типа: две соответствующие жидкости обладают полной взаим-
ной растворимостью, причем кривая упругости пара изменяется
здесь постепенно по прямой без каких-либо максимальных точек.
386
взаимной растворимости
Фиг. 66. Изотермы упруго-
сти паров смеси двух жид-
костей.
Такие растворы называются идеальными; примером их может
служить смесь бензола с толуолом или нормального гексана и
нормального октана.
Известно немало случаев, когда растворитель и растворенное
в нем вещество, взаимодействуя друг с другом, образуют проч-
ное соединение, имеющее по сравнению с его компонентами зна-
чительно более высокую температуру кипения и, следовательно,
меныпую упругость пара. Примером такого рода соединений
может служить соляная кислота с содержанием 20,3% хлоро-
водорода; такой водный раствор имеет постоянную точку кипе-
ния (110°), причем концентрация его при перегонке не изме-
няется. Аналогичные постоянно кипящие водные растворы из-
вестны для бромоводорода крепостью 48,2% НВг (температура
кипения 126е) и иодоводорода крепостью 57% (температура ки-
пения 127°), а также для кислот азотной и серной. Из органи-
ческих соединений подобное же отношение к воде обнаруживает
муравьиная кислота: ее водный раствор с содержанием 77,5%
кислоты кипит при нормальном давлении постоянно 107°, 1, то-
гда как безводная муравьиная кислота имеет температуру ки-
пения 101°.
В идеальном случае при перегонке подобного рода растворов
различных концентраций должно наблюдаться следующее: сна-
чала должен отогнаться тот компонент, который находится
в данной смеси в избытке; затем наступает перегонка постоянно
кипящей при данном давлении смеси, причем состав ее жидкой
и паровой фаз, равно как упругость паровой фазы во все время
перегонки сохраняются постоянными. Такого рода растворы на-
зываются азеотропическими. Графически (фиг. бб) перегонка
азеотропической смеси может быть выражена кривой 9, предста-
вляющей как бы прямую противоположность -кривой 1. Для ре-
альных смесей этого рода кривая и здесь имеет закругленные
концы, так как в случае слабых растворов общая упругость па-
ров приобретает свое предельное значение не сразу, а лишь по-
степенно.
Наконец, кривые 6, 1 и 8 представляют собою как бы проме-
жуточные случаи между идеальными и азеотропическими рас-
творами. Некоторые из них имеют выступающие вниз точки, ко-
торые показывают, что при определенных концентрациях соот-
ветствующие смеси имеют минимальную упругость пара и, сле-
довательно, максимальную температуру «кипения.
Аналогичные кривые получаются, если для смеси двух жид-
костей рассматривать. зависимость от их взаимной растворимо-
сти не для упругости пара, а для температуры кипения. Кривые,
построенные для этого последнего случая (изобары), будут иметь
обращенный вид по сравнению с изотермами.
г* Упругость пара идеального раствора. Как ясно из вышеизло-
женного, идеальным раствором следует называть смесь двух
весьма ‘близких по своей химической природе жидкостей, рас-
творимых друг в друге во всех отношениях. По закону Долгтона
общая упургостъ паров данной смеси' Р должна быть равна
25* 887
сумме парциальных упругостей pi и р-2 паров, находящихся над
этой смесью. Однако эти парциальные упругости pi и Рг должны
быть здесь меньше упругостей Pi и Pz паров тех же веществ,
взятых в отдельности при той же температуре, так как упругость
паров каждого из компонентов смеси от их взаимной раствори-
мости, естественно, должна понижаться.
Обозначим через х молекулярную концентрацию одной из
смешивающихся жидкостей. Тогда содержание в смеси другого
компонента выразится величиной 1 — х.
Пусть, например, смесь содержит пс весу 47,1% бензола (М = 78)
и 52,9% толуола (Л/ = 92). Разделив процентное содержание каждого жом.
лонента на соответствующий молекулярный вес, получаем число, опреде.
ляющее отношение, в котором молекулы бензола и толуола образуют дан-
ную смесь, а именно: для бензола (47,1 :78) = 0,6038 и для толуола
(52,9 : 92) = 0,5750.
Вычисляя отсюда молекулярную концентрацию, получаем:
для бензола: 0603S 0,>038 + 0,5750 0,5750
для толуола: '— —01 R 0,6088-|-0,5750 ’
или в процентах 51,2% бензола и 48,8% толуола.
Совершенно очевидно, что упругость пара смеси двух жид-
костей зависит от ее состава:
/*(*).
где х — молекулярная концентрация одной из жидкостей данной
смеси.
В общем случае эта зависимость должна быть весьма слож-
ной. Однако в тех случаях, когда компоненты смеси по своим
свойствами и химической природе весьма близки друг к другу,
к ним приложима, как показывает опыт, следующая простая за-
кономерность: парциальная упругость pi пара летучего веще-
ства над раствором его в каком-либо растворителе пропорцио-
нальна молекулярной концентрации х вещества в данном рас-
творе, т. е.:
Pi = С.х,
В случае идеальных растворов двух жидкостей, очевидно»
любая из них может быть принята за растворитель, другая-
за растворенное вещество.
Поэтому, если
А =
то
_??2 ~ ^2 (1 а')’
Только что приведенная закономерность представляет собою,
очевидно, не что иное, как специальное выражение закона
Генри, согласно которому растворимость паров и газов про-
порциональна их парциальному давлению над раствором.
Представим себе, что в предпоследнем равенстве
368
т. е. что мы имеем одну из жидкостей данной смеси в чистом
виде; тогда, очевидно:
т. е. постоянная С представляет собою упругость пара 1\
чистого растворенного вещества.
Таким образом окончательно имеем:
Pi = Р&
т. е. парциальная упругость паров растворенного вещества
равна произведению упругости его паров в чистом виде на
его молекулярную концентрацию в растворе (закон Рауля).
В случае идеального раствора двух жидкостей имеем
также:
^2 = Д(1 — *)•
где Р2 — упругость паров чистого растворителя.
Наконец, для упругости паров раствора по закону Даль-
тона имеем:
Фиг. 67. Вывод уравнения равно-
весия фаз.
упругостей обоих компонентов
Применимость закона Рауля, ямба® говоря, ограничивается
довольно узкими пределами, примерно растворами, содержащими
не свыше 5—10% растворенного вещества. Закон вовсе не при-
меним к йзеотропическим смесям и к растворам электролитов.
Напротив, в применении к идеальным растворам и, в частности,
к некоторым углеводородным смесям закон Рауля оправдывается
в широких пределах [11]. Графически он выражается, очевидно,
прямой линией.
Состав пара и жидкой фазы идеального раствора. Для тех
случаев, когда общее давление паров смеси двух жидаостей вы-
ражается прямой (идеальные ра-
створы), нетрудно вывести фор-
мулу, связывающую состав пара
с составом жидкой фазы.
Отложим на оси абсцисс
(фиг. 67) молекулярные концен-
трации идеальной смеси двух жид-
костей; пусть также ординаты BD
и АС выражают собою упругости
пара Pi и Рг этих веществ в чи-
стом состоянии при некоторой
постоянной температуре. Оче-
видно, что прямые AD и СВ вы-
ражают изменения парциальных
смеси, линия же CD представляет собою изотерму упругости
пара смеси в зависимости от ее состава, уравнение которой дано
выше:
Р=Р1а;+Р#(1-®).
№9
На абсциссе возьмем точку Я, отвечающую некоторому
определенному составу смеси, и восстановим из нее перпен-
дикуляр. Теперь имеем две пары подобных треугольников:
ACBcv HBG и ADBcvAFH, из которых получаем:
FH_AH GH__BH
BD~~ АБ 11 AC АВ'
Отсюда:
FH 1W-AH
GH~~ AC • БН '
Ho FH и GH— парциальные давления компонентов данной
смеси Pt и т?2, которые для идеальных растворов, как было
указано, пропорциональны молекулярной концентрации этих
компонентов в парах у1 и у.2\ АН и НВ — молекулярные кон-
центрации тех же компонентов в жидкой смеси xt и я2; на-
конец, ВВ = Р1 и АС — Ръ Таким образом, подставляя эти
значения в последнее равенство, получаем:
3/1 Xl'P1
3/2 х2-Р2’
Отношение Рг: Р2 при Данной температуре — величина по-
стоянная; введем для него обозначение К. Пусть далее
+ — 1 и ai+#2 = l; тогда последнее уравнение приобре-
тает такой вид:
У1 у1
1 — У1 1 — *1
или иначе
У1 + (К—1) х1у1 — Кх1 = О,
где 2/1 и Xi — молекулярные концентрации одного из компонен-
тов идеального раствора в паровой и жидкой фазе.
Это уравнение называется уравнением равновесия фаз, Tait
как оно выражает зависимость между молекулярными концен-
трациями одного из компонентов идеального раствора в жидкой
и паровой фазе (яч и у г).
Как видно из уравнения равновесия фаз, состав последних находится
в зависимости от величины К, определяемой отношением упругостей пара
компонентов при данной температуре. Так как, однако, упругость пара за-
висит не только от состава компонента, но также и от температуры, то
уравнение равновесия фаз содержит в скрытом виде также зависимость от
температуры. Таким образом, состав фаз смеси двух растворимых друг
в друге жидкостей определяется двумя переменными, температурой и да-
влением, как рто и должно быть с точки зрения правила фаз для случая
неполного равновесия: L = 2.
На координатных осях xi, yi, уравнению равновесия фаз со-
ответствует кривая равновесия, имеющая вид гиперболы (фиг. 68),
которая проходит через начало координат ял ==2/1 = О и через
точку Xi = t/i=l; необходимые для построения кривой равно-
весия другие сопряженные значения yi для различных концен-
траций Xi жидкой фазы могут быть получены либо путем рае*
чета на основе последнего уравнения, либо графически,
390
Необходимым условием, чтобы кривая равновесия имела вид
гиперболы, является постоянство коэфициента К, т. е.
Такое условие -соблюдается, например, ври изотермическом
испарении. В условиях обычного испарения, т. е. при постоян-
ном давлении и непрерывно повышающейся температуре, коэфи-
циент К уже не является величиной постоянной, и кривая рав-
новесия отступает от вида гиперболы.
Если Pi— упругость паров нижекилящего компонента рас-
твора, то коэфициент
АГ всегда больше еди-
ницы; отсюда легко
притти к выводу,
ЧТО У1 должно быть
больше «1, т. е. что
содержание нижеки-
пящего компонента
в паровой фазе боль-
ше, чем в жидкой.
Ввиду этого отноше-
ние Pi: Р2 называют
коэфициентом обо-
гащения.
J5O0M
Ш-
700
500
* 0,2
me
toco;
900
-JL
ъ
&
10.6
са
§•
(OJ
aB'AB
00
760лл
I „ 4>’ " * 1 . . . А-.|Д .1. -
0“ 20®40 60®100 V
300
100
Фиг. 68. Кривая равно-
весия.
б Ц2 б/ £б цз ТЛ
Ландеятрацця Жидкой фазы
Фиг. 69. Построение кривой равновесия для
смеси бензол—толуол
Графическое построение кривой равновесия на координатных
осях и ух может быть осуществлено для случая испарения при
постоянном давлении какого-либо идеального раствора, напри-
мер смеси бензол—толуол, следующим образом (фиг. 69). Пре-
жде всего на координатных осях Р, строим несколько изотерм
упругости пара данной смеси -в зависимости от ее состава, на-
пример для температур 85, 90, 95, 100 и 105°. Каждая из этих,
изотерм будет представлять собою, как ясно из вышеизложен-
ного, наклонную прямую, крайние точки которой будут иметь
своими ординатами Рг (для чистого толуола) и Pi (для чистого
бензола). Так как компоненты смеси кипят при нормальном да-
влении 80°,5 (бензол} и 110°,6 (толуол), то при построении кри-
391
вой равновесия для атмосферного давления нет Надобности вы-
ходить за пределы указанных выше температур.
Точки пересечения построенных изотерм с линией атмосфер,
кого давления определяют пять значений концентраций xi зщ.
кой фазы, для которых концентрации в паровой фазе yi могут
быть найдены графически следующим построением.
На основании законов Дальтона и Рауля имеем:
Отсюда следует:
= и у, = -^-1,
где pi — парциальное давление компонента, содержание кото-
рого в паровой фазе равно у г, Pi — упругость паров того же ком-
понента в чистом состоянии, и Р — общая упругость паровой
фазы.
Для построения yi (фиг. 70) на осях Р, х проводим изотерму
упругости пара смеси бензола и толуола CD и изотерму упруго-
сти пара одного из компонентов смеси, а именно бензола AD.
Фиг. 70. Построение ординат для
кривой равновесия идеального
раствора.
Фиг. 71. График равновесия па-
ров с жидкостью.
Из точки Е, отвечающей условию АН = xi, проводим линию, па-
раллельную оси абсцисс до пересечения с осью Pi, а из точки
F—параллельную той же оси до пересечения с линией, соеди-
няющей точки А и К. Наконец, из точки L опускаем перпенди-
куляр до пересечения с осью абцисс в точке N. Легко показать,
что AN = yi.
В самом деле, из подобных треугольников АКБ и ALN
имеем: AN : АВ — NL : КВ, откуда, так как LN — FH, получаем:
AN = FH. АВ : КВ. Далее из подобных треугольников AFH и
ADB имеем: FH: DB — АН: АВ, откуда FH-AB = DB-AK
Теперь подставляя в выражение для AN вместо FH • АВ равную
величину DB -.АН и принимая во внимание, что DB = Pi, АН=
= xi и КВ = ЕН = Р, получаем окончательно: AN-——,
892
т. е. линия AN действительно равна yi. Откладывая теперь на
осях гл по линии FH от точки Я отрезок ANполучаем
одну из точек искомой гиперболы (кривой равновесия). Другие
сопряженные значения yi, xi находятся аналогичным путем; они
дают ряд новых точек гиперболы, по которым последняя и может
быть построена.
Как ясно из вышеизложенного, коэфициент К в уравнении
равновесия фаз является функцией температур кипения компо-
нентов данной смеси Для выражения этой функции предложены
различные формулы [10], которыми можно пользоваться при по-
строении кривых равновесия.
Опытное определение состава жидкой и паровой фаз^Так как
реальные жидкости более или менее отступают от основных га-
зовых законов, то во многих случаях для построения кривых
равновесия приходится прибегать к опытному определению со-
става жидкой и паровой фаз. Для этой цели можно пользо-
ваться различными методами, из которых остановимся на сле-
дующих двух:
1. В специальном приборе производят разгонку данной смеси
двух жидкостей определенного состава, собирая ряд небольших
последовательных фракций с тщательным учетом их веса и по-
следующим определением состава каждой из них. Затем путем
простого пересчета определяют состав ряда последующих %е-
стиллатов и получают, таким образом, веса и составы первого де-
стиллата (I фракция), второго дестиллата (I + II фракции),
третьего дестиллата (I +' II + Ш фракции) и т. д. Откладывая
затем веса этих дестиллатов на оси абциос, а состав в молеку-
лярных процентах одного из компонентов—на оси ординат,
строят кривую А (фиг. 71) изменения состава ряда последую-
щих дестиллатов и, продолжив эту кривую до пересечения
с осью ординат, получают состав начального дестиллата беско-
нечно малых размеров. Очевидно, что состав этого дестиллата,
определяемый лишь путем экстраполяции, должен соответство-
вать составу паров, находящихся в равновесии с исследуемой
смесью при ее начальном составе; зная же состав паров, можно
легко определить согласно вышесказанному парциальные упру-
гости 'Каждого компонента смеси при данном ее составе.
Тот же опытный материал может быть использован и в ином
направлении. Допустим, например, что при перегонке исследуе-
мая смесь была разбита на 6 фракций, веса и состав которых
были определены опытным путем так же, как вес и состав
остатка от перегонки. Сделаем теперь пересчет состава и веса
отдельных дестиллатов не в прежнем, а в обратном порядке, т. е.
переходя от дестиллата VI (VI фракция) к дестиллату V (VI4-
4- V фракции), дестиллату IV (VI4- V 4- IV фракции) и т. д.
Если построить теперь кривую В (фиг. 71) изменения состава
для ряда последующих дестиллатов и продолжить эту новую
кривую до пересечения ее с осью ординат, то по аналогии с пре-
дыдущим построением получим, очевидно, состав бесконечно
малого последнего дестиллата, соответствующий составу паров,
398
находящихся в равновесии с остатком от разгонки исследуемой
смеси. Отсюда легко рассчитать парциальные упругости каждого
компонента при составе жидкой смеси, определяемой анализом
остатка от разгонки.
, Для иллюстрации описанной методики может служить разгонка смеси
хлороформа и толуола, результаты которой сведены ,в табл. 92 и нанесены
на график (фиг. 71). Начальное содержание хлороформа в смеси было
65,33%, содержание его по анализу в остатке—51,30% (молекулярных).
Путем экстраполяции получается, что пары, находящиеся в равновесна
с начальной смесью, содержат 89,53% (молекулярных) хлороформа; пары
же, находящиеся в равновесии с остатком, — 82,55% (молекулярных) хло.
реформа.
Таблица 92
Разгонка смеси хлороформа и толуола
(по К. Эллиот)
№ соединенных фракций Вес соедин. фракций, в » Содержание CHClg в соедин. фракциях в мол. °/о № соединенных фракций Вес соедин. фракций, в 4 Содержа- ние CRCI3 в соедин. фракциях в мол. %
I 13,24 89,36. VI 20,05 88,58
I, II# 29,10 89,28 VI, V 37,11 84,37
1,11, III . . . . 44,90 88,87 VI,V,IV . . . 57,99 85,24
1,11, III, IV . . 65,78 88,21 VI, V, IV, III . 73,79 85,88
ЦП,III,IV,V . 82,84 87,67 VI, V, IV, III, II, 89,65 86,45
1,11, III, IV, V, VI 103.89 86.82 VI, V, IV, III, II, I 102,89 86,82
Проведя подобного рода опыты и расчеты со смесями данных жид.
костей различного состава, можно получить исчерпывающее представление
о зависимости состава насыщенных паров, а следовательно, и упругости
образующих компонентов от состава данной жидкой смеси.
Как видно из вышеизложенного, одним из основных условий
применимости описанной методики к исследованию смеси даух
жидкостей является возможность быстро и достаточно точно
анализировать состав различных ее дестиллатов. В зависимости
от природы смешанных жидкостей такой анализ может быть
произведен различными методами. Чрезвычайно удобным для
анализа двойных смесей является иногда рефрактометрический
метод, основанный на определении коэфициеитов лучепреломле-
ния (рефракции) отдельных погонов с помощью получивших
столь широкое распространение рефрактометров Пульфриха й
Аббе. Само собою разумеется, что для определения состава смеси
двух жидкостей по ее рефракции предварительно должны быть
составлена таблица и построен график, показывающие связь
между рефракцией смеси и содержанием в ней того либо иного
компонента данной смеси.
Простой и очень удобный аппарат для определения состава
паров в зависимости от состава жидкой смеси представлен на
фиг. 72. Он состоит из грушеобразного сосуда с длинной шей-
394
кой, снабженной в верхней части четырьмя отверстиями для вы-
хода ларов. Сосуд заключен в стеклянную муфту, припаянную
к краю шейки сосуда; нижняя, суженная часть этой муфты со-
единяется с хорошим холодильником, не
•показанным на фиг. 72. К холодильнику
присоединяются оптированные прием-
ники. Шейка грушеобразного сосуда, за-
крыта непроницаемой для пара пробкой,
через которую проходят узкая трубочка
с воронкой для внесения и выливания
жидкости и электрический нагреватель,
при помощи которого жидкость внутри
сосуда доводится до кипения. Весь при-
бор погружается в баню, температура ко-
торой несколько выше температуры ки-
пения исследуемой жидкости. Таким
образом во время опыта пары кипящей
жидкости быстро, без образования стекаю-
щей обратно флегмы проходят груше-
образный сосуд и муфту, попадают в хо-
лодильник, конденсируются здесь и соби-
раются .в отдельные приемники. Даль-
нейшее исследование отдельных фракций
и остатка, расчет состава дестиллата и
построение графика производятся, как
Фиг. 72. Аппарат для
определения состава па-
ров в зависимости от
состава хидкой^смесн.
описано выше.
Описанный метод [12] характеризуется простотой и удоб-
ством, особенно при работе с несложными смесями.
Фиг. 73. Схема прибора для анализа жидкой и паровой фаз
смеси.
2. Другой метод построения кривых равновесия основан на
разделении жидкой и паровой фаз в условиях адиабатического
процесса, а именно при частичном однократном испарении иссле-
дуемого сырья при постоянной температуре [13].
Прибор, в котором производятся образование и разделение
жидкой и паровой фаз в указанных условиях, состоит (фиг. 73)
из следующих частей: воронка с краном 1, из которой иссле-
дуемое сырье поступает в змеевиковый подогреватель 2, а затем
в адиабатический эвапоратор 3, где и происходит разделение
жилкой и паровой фаз, направляющихся далее каждая в свой
холодильник и приемник 4. Подогрев сырья до требуемой тем-
пературы производится в змеевике простой горелкой. Для со-
хранения адиабатичности процесса эвапоратор имеет наружный
кожух, в 'который в первую очередь поступает нагретое сырье,
чтобы затем перейти во внутрен-
нюю часть эвапоратора, где и
происходит разделение фаз. По
окончании опыта определяется
состав обеих фаз, паровой (де-
стиллата) и жидкой (остатка), и
на основе полученных результа-
тов строится кривая равновесия.
Опыт показывает, что для по-
фиг. 74. Кривые разгонов равно-
весных фаз.
строения кривой равновесия со-
вершенно безразлично, на какие
фракции разбивать при пере-
гонке в адиабатическом эвапора-
торе исследуемое сырье, если
только оно выкипает равномерно
и в не слишком широких преде-
лах, хотя для сложной смеси
кривая равновесия вообще долж-
на изменяться с изменением
величины отбора. Для неши-
роких погонов такую перегонку
достаточно осуществить 1—3 раза при различных заданных
температурах а затем, для определения состава полученных
продуктов (фаз) нужно произвести их разгонку по Энглеру
или на более совершенном приборе и построить график этих
разгонок.
Для иллюстрации метода могут служить условия и результаты разгонок
на адиабатическом эвапораторе погона с третьего куба 15-кубовой батареи
Грознефти (табл. 93), которые легли в основу построения графика (фиг. 74).
Таблица 93
Разгонка на адиабатическом эвапораторе
(по Обрядчикову и Хохрякову)
№ разгонок 1 2 8
Продолжительность в мин. . . . 15 12 9.5
Температура входящего сырья . 77°,9 92°, 4 97°,2
. пара дестиллата . 76°,4 88°,5 96°,4
• остатка 77°,5 89°,5 96°,9
Количество отгона в г 78,5 120,0 130,5
• остатка 659,0 166,0 80,0
Если теперь в точках, соответствующих температурам h /•
h и т. д., построить перпендикуляры до пересечения с кривыми
разгонок дестиллата / и остатка //, то соответствующие ординаты
будут представлять собою про-
центы «легкокипящих» в парах
(2/1, 2/2 и т. д.) и «легкокипящих»
в жидкости (®i, яг и т. д.). При-
нимая на новых осях координат
за абциссы si, xz и т. д., за орди-
наты— 2/1, 2/2 и т. д., получаем
ряд точек, по которым может
быть вычерчена кривая равнове-
сия для исследуемого случая.
Фиг. 75 дает несколько таких
кривых для различных фракций
нефти.
Беря для построения кривой
равновесия данные разгонок по
Энглеру, тем самым строят кривую
равновесия в объемных процентах.
Можно показать, однако, что ход
фиг. 75. Кривые равновесия
нефтяных погонов:
1 — авиобекзин; 2—легхяй бевзин;
3 —тяжелый бензин; 4 — авиобензив
4- тяжелый соляр.
кривой равновесия для смеси из
цвух компонентов (но не для слож-
ной смеси) не зависит от того, в каких процентах выразить состав
паровой и жидкой фаз: в молекулярных, весовых или объемных.
Однократное испарение двух смешивающихся жидкостей.
Как и в других рассмотренных выше случаях равновесие жидкой
фазы двух смешивающихся жидкостей с их насыщенными пара-
ми возможно лишь при температуре кипения данной смеси. Так
как, однако, получающееся в данном случае равновесие является
неполным и имеет две степени свободы (Б=2), то, как мы ви-
дели, состав фаз определяется здесь двумя переменными—тем-
пературой и давлением, так что для каждой данной температуры
при заданном давлении имеются вполне опрделенный состав
жидкой фазы данной смеси, а также вполне определенный, хотя,
вообще говоря, и неодинаковый с первым, состав паровой фазы.
С другой стороны, например при заданном давлении, могут су-
ществовать довольно широкие температурные пределы, при кото-
рых возможно равновесие жидкой и паровой фаз данной смеси
при строго определенном, однако, составе фаз для каждой тем-
пературы.
В последующем изложении будут рассмотрены лишь такие
случаи, когда две смешивающиеся жидкости вполне растворимы
друг в друге и не образуют азеотропических смесей, т. е. случаи,
к которым относятся, например, смеси двух углеводородов (бен-
зол и толуол, гексан и октан и т. п), иначе говоря, упрощенные
случаи угловодородных смесей, с которыми приходится иметь
дело при перегонке нефти и ее дестиллатов. Кипение смесей
подобного рода характеризуется той особенностью, что при по-
стоянном давлении в определенном температурном интервале
897
сначала .испаряется преимущественно нижекипящий компонент
смеси, так что содержание другого, вышекипящего компонента
в жидкой фазе (в остатке) постепенно возрастает. Отсюда
явствует, что по мере испарения температура кипения смесей
подобного рода должна постепенно повышаться, что, как из-
вестно, и наблюдается в действительности. Понятно также, что
при конденсации паров подобного рода смесей должно наблю-
даться обратное явление: сначала конденсируется преимуще-
ственно второй, вышекипящий компонент, содержание же пер-
вого компонента (нижекипящего) в парах постепенно возрастает
и, следовательно, температура системы должна понижаться.
Испарение смеси двух жидкостей можно производить в раз-
личных условиях. Простейшим случаем испарения двух смеши-
вающихся жидкостей является такой случай, когда переход си-
стемы из жидкого состояния в газообразное совершается одно-
кратно-, причем паровая jr жидкая фазы нагреваются совместно,
как это происходит, например, при испарении в трубчатых кубах.
Для такого однократного испарения легко определить отношение
массы образовавшихся паров к массе оставшейся жидкости для
одного из компонентов смеси и построить кривые (изобары) из-
менения паровой и жидкой фаз с температурой.
Допустим, что Qo— масса смеси двух жидкостей до испаре-
ния и Q— масса той же смеси после испарения из нее части,
масса которой, очевидно, равна Qo— Q; пусть далее жо и да-
концентрации в данной смеси какого-либо одного, например ни-
жекипящего, компонента до и после испарения, а у — концентра-
ция того же компонента в образовавшихся парах. Легко видеть,
что масса этого компонента определяется следующим равенством:
(Qo ~ Q) У = Qoxo —
откуда
Qo __ у—ж
Q у — *0*
Определяя отсюда отношение массы образовавшихся паров
(Qo—Q) к массе оставшейся жидкости Q, получаем:
Q)— Q xq — x
Q у — ?o'
Зависимость молекулярного состава жидкой и паровой фаз
(т.- е. величин да и у) от температуры для случая однократного
испарения можно вывести из уравнения равновесия фаз
(см. выше), если в этом уравнении выразить коэфициент К как
функцию температуры. Однако ввиду того что функция ©та—
весьма сложная, удобнее и проще проследить зависимость тем-
пературы кипения от величин да и у графически путем построе-
ния соответствующих кривых, изобар, в -координатных осях
да, t и у, t.
Построение кривой температуры кипения для смеси двух вполне рас-
творимых друг в друге жидкостей, например бензола и толуола, в зави-
симости от состава смеси при постоянном давлении, например при 760 мм,
производится на тех же основаниях, которые были положены выше для
398
построения кривой равновесия фаз. Действительно, как мы видели, при
построении этой -кривой сначала находят для некоторого постоянного,
например нормального, давления и ряда температур концентрации одного
из компонентов в жидкой фазе т, а затем путем применения законов
Дальтона и Рауля графически находят для тех же температур концентра-
ции того же компонента и в паровой фазе у. Таким образом имеется пол-
ная возможность на осях х, t и у, t построить искомые изобары, которые
покажут изменение состава жидкой и паровой фаз системы в зависимости
от температуры.
На фиг. 7G представлены изобары системы бензол — толуол.
Одна из кривых, кривая жидкого состояния CAD, изображает
ход изменения температуры кипения для различных смесей, со-
став которых в молекулярных концентрациях отложен на оси
абсцисс. Другая кривая CBD относится к паровой фазе той же
смеси (кривая парового состояния), показывая изменение со-
става пара, находяще-
гося в равновесии
с жидкой фазой данной
системы при том же
давлении. Температуры
отложены на оси орди-
нат. Начальные точки
обеих кривых общие;
одна из них (80°,б)
отвечает температуре
кипения чистого бен-
зола, другая (11О°»6)
— температуре кипе-
ния чистого толуола.
Каждая промежуточ-
ная точка кривой CAD
Фиг. 76. Схема изобар для смеси бензол —
толуол.
жидкого состояния от-
вечает температуре кипения при 7G0 мм, соответствующей
ординате этой точки для смеси бензола с толуолом, состав кото-
рой определяется абсциссой той же точки.
Пусть имеется некоторое начальное состояние системы бен-
зол— толуол -40 при температуре 6 и составе жидкой фазы хе.
Состав паровой фазы для той же температуры to определится,
очевидно, точкой Во на кривой CBD с концентрацией того же
компонента уо. После того как система потеряет некоторое коли-
чество обоих компонентов вследствие их однократного испаре-
ния, пусть температура ее поднимется до t с концентрациями
хну для жидкой и паровой фаз. Продолжим ординату точки
Ао до пересечения с прямой АВ в точке Е. Йз построения
видно, что АЕ = хо — х и ВЕ = у— хо\ таким образом найден-
ное выше отношение массы образовавшихся паров Qo — Q к массе
оставшейся жидкости Q после однократного испарения опреде-
ляется графически отношением отрезков АЕ и BE.
Представим себе, что система нагребается дальше и дости-
гает некоторой температуры ti, определяемой точками. Ai и Bi,
которым соответствуют концентрации жидкой и паровой фаз
' 399
£1 и 2/i. Если при этом оказывается, что yi *=яо, то последняя
система, полученная путем однократного испарения, будет со-
стоять, очевидно, из одних лишь паров, так как состав пара
может оказаться тождественным с начальным составом жидкой
фазы лишь при условии полного испарения взятого раствора
Очевидно также, что наименьшая концентрация, которой может
достичь в жидкой фазе ниэкокипящий компонент, определяется
величиной $i, т. е. той коцентрацией, которую должна иметь по-
следняя капля раствора, находящаяся в равновесии с насыщен-
ными парами этого раствора.
Для однократного испарения .можно легко представить себе
также обратный процесс — однократную конденсацию. Действи-
тельно, пусть точкам Bi и Ai на тех же кривых парового и жид-
кого состояния (фиг. 76) отвечает некоторое состояние равно-
весия, характеризуемое температурой ti и концентрациями па-
ровой и жидкой фазы 3/1 и si. В результате однократной конден-
сации пусть новое состояние системы определится точками В и
А при температуре t л концентрациях у и х. Опуская из точки
Bi перпендикуляр на линию АВ, легко находим, что АЕх
и BE = у — yi. С другой стороны, полагая, что начальная
масса паров при концентрации yi была gi, что после однократ-
ной конденсации масса их стала q при концентрации у и что
в результате конденсации образовалась жидкая фаза в количе-
стве qi — q с концентрацией ж, находим по предыдущему:
(?i —д)ж = д12/1 — д?/,
откуда
— 7 у — yi
7 У1 — X
Так как, однако, у — yi = BE и yi — х== АЕ, то, как видим,
после однократной конденсации отношение массы образовав-
шейся жидкости к массе оставшихся паров определяется графи-
чески отношением тех же отрезков АЕ и BE; однако в данном
случае отношение это обратно тому, которое как было показано
выше, определяет при однократном испарении отношение массы
образовавшегося пара к . массе оставшейся. жидкости.
Если продолжить охлаждение системы в условиях однократ-
ной конденсации, может' быть достигнута некоторая температура
to, при которой концентрация низкокипящего компонента в жид-
кий Фазе х0 окажется равной первоначальной концентрации того
же компонента в паровой фазе yi. Такое состояние .системы Л
будет характеризоваться полным ожижением ее паров, так как
при однократной конденсации состав жидкой фазы может ока-
заться тождественным с начальным составом паровой фазы,
очевидно, лишь при соблюдении этого условия. Очевидно так-
же, что последний остаток паров при однократной конденсации
будет иметь концентрацию з/о, определяющую предельное наи-
большее значение, которого может достичь концентрация низко-
кипящего вещества в паровой фазе данной системы при данных
условиях;'
400
Многократное испарение двух спешивающихся жидкостей.
Многократное испарение заключается в повторении процесса
однократного изменения состояния системы, т. е. в повторении
однократного испарения с удалением из системы образующихся
каждый раз паров. Для уяснения этого процесса можно вос-
пользоваться тем же графиком, как при однократном испарении
(фиг. 76).
Сохраняя все прежние обозначения для перехода путем одно-
кратного испарения из состояния Ао с координатами io, jo в со-
стояние А с координатами i, х, получаем следующее отношение:
Q:Q0 = BE: АВ,
откуда
е=^с<>.
где Qo — масса начальной смеси и Q — масса жидкого остатка
после образования паров.
Если теперь удалить из системы образовавшиеся пары и под-
вергнуть ее вторичному однократному испарению путем подогрева
до температуры ti, то система перейдет в новое состояние, жид-
кая фаза которого определится точкой Al с координатами ti, л;
для массы этой жидкости Qi аналогично только что сказанному
получаем:
_B^L п_В& BE
^~AvBl 'ав'™
После нового удаления паров из системы жидкий остаток мо-
жет быть подвергнут однократному испарению в третий раз
с аналогичными результатами и т. д. Уменьшая температурные
интервалы и увеличивая число последовательных однократных
испарений системы с удалением образующихся паров, получаем
в пределе картину постеленного изменения состояния системы
в процессе ее многократного испарения; иначе говоря, картину
обыкновенной перегонки данной смеси без дефлегмации.
Легко видеть, что выделение из смеси низкокипящего компо-
нента путем многократного испарения достигается полнее, чем
при однократном испарении, так как при этом удается поднять
температуру несколько выше с сохранением жидкой фазы.. Оче-
видно также, что пределом повышения температуры здесь будет
точка С, т. е. температура кипения вышекипящего компонента,
для которой концентрация х равна нулю, так что последняя
капля выкипающего раствора должна состоять здесь из одного
высококипящего компонента. Понятно также, что в точке С
должно произойти полное -выкипание смеси, так как выведенное
выше для массы жидкого остатка выражение представляет собою
в пределе произведение бесконечного числа правильных дробей,
стремящееся к нулю.
К аналогичным выводам легко притти и для случая много-
кратной конденсации паров, которую можно рассматривать как
повторение процесса однократного изменения состояния системы
26 8ак. 1622. Химия нефти.
с одновременным удалением из нее образующейся жидкости (вод;,
денсата). Сохраняя здесь обозначения, которые были введены
выше для однократной конденсации, и представляя многократ-
ную конденсацию как совокупность весьма большого числа
однократных конденсаций с удалением каждый раз 'образовавше-
гося конденсата, легко находим для каждой однократной кон-
денсации следующие сотношения:
АЕ Л) Jin ЛЕ
9о = л^-3в«> пт-
где gi, q и до — массы смеси паров для кая той отдельной одно-
кратной конденсации, определяемой точками Bi, В, Во и т. д
с постепенно понижающимися температурами fi, £, /о и т. д. и
постепенно повышающимися концентрациями yi, у, у о и т. д.
Уменьшая температурные интервалы и увеличивая число
последовательных однократных конденсаций системы с удале-*
нием образующихся конденсатов, получаем в пределе картину
многократной конденсации, иначе говоря, — обыкновенную кон-
денсацию паров без повторного испарения конденсата. Легко
видеть, что этот процесс идет полнее, чем при однократной кон-
денсации. Его пределом является, очевидно, точка В, т. е. тем-
пература кипения нижекипящего компонента, при которой
должна произойти полная конденсация паров вышекипящего
компонента, так как в этой точке концентрация низкокипящего
компонента в последнем остатке паров достигает своей предель-
ной величины (2/~ = 1). Наконец, в этой же точке D происхо-
дит окончательная конденсация паров нижекипящего компо-
нента, так как выведенное выше соотношение для массы паров
(g, до и т. д.) в пределе, как произведение бесконечно большого
числа правильных дробей, превращается в нуль.
•Как ясно из вышеизложенного, концентрации х и у жидкой
и паровой фаз для системы из двух вполне смешивающихся
компонентов определяются при постоянном давлении одной лишь
температурой; таким образом величина их для данной равновес-
ной системы не зависит от того, каким способом достигнуто рав-
новесие: способом ли однократного испарения или каким-либо
иным. Различие будет однако, в массе фаз: так как полное
выкипание жидкости при однократном испарении наступает при
более низкой температуре, чем при постепенном выкипании, то
очевидно, при заданной температуре остаток не испарившейся
жидкости в случае однократного испарения должен быть мень-
ше, а общий процент отгона больше, чем при многократном
испарении. Равным образом, так как полная конденсация паров
в случае постепенной конденсации наступает при более низкой
температуре, чем в случае однократного процесса, то при одной
и той же температуре и однократной конденсации получается
больше конденсата и меньше ларов в остатке, чем при -конденса-
ции многократной.
Определение массы жидких фаз при постепенном испарении
или конденсации представляет большое значение для решения
402
йекоторых практических вопросов. Для решения этой заДаЧй,
для случая постепенного шпарения, допустим, что масса Q дан-
ной смеси двух жидкостей с концентрацией х в жидкой фазе
низкокипящего компонента претерпевает бесконечно малое испа-
рение, получая при этом отрицательные приращения массы и
концентрации dQ и dx; пусть, далее, выделившиеся пары при-
обретут при этом концентрацию у, сохраняя равновесие с жид-
кой фазой. Легко видеть, что начальная масса Qx низкокипя-
щего компонента, распределившаяся благодаря испарению между
жидкой и паровой фазами, может быть выражена следующим
образом:
Qx = (Q — dQ) (z — dx)-{-ydQ.
Раскрыв скобки и преобразовав это выражение, получаем
дпференцпальное уравнение:
Qdx = (у — х) dQ
или
dQ__ dx
Q “ у—х '
Проинтегрировав это выражение в пределах от Qo, z0 до
Q, х, получаем:
пли
dx
у — я *
Значение интеграла, нахо-
дящегося в правой части по-
следнего равенства, может
быть найдено различными спо-
собами. Простейший из них—
графический, легко осуще-
ствимый, если известна кри-
вая равновесия фаз. Для
этого на ординатах —и
у —
Фиг. 77. Кривая
бензол — толуол.
для смеси
абсциссах я строим кривую
(фиг. 77); тогда площадь, ограниченная этой кривой и осью
абсцисс в пределах от х до z0, даст искомую величину инте-
грала.
Для случая постепенной конденсации задача о массе па-
ровых фаз решается аналогичным образом. Если в системе
из двух компонентов общую массу пара обозначим через д,
а концентрации одного из компонентов—через у в паровой
фазе и через х в конденсате, то для массы этого компонента
до и после бесконечно малой конденсации, получаем:
= — dq) (У — dy) + xdq,
26*
403
и далее, после преобразования:
dq_____________________________d,y
V У — « *
Интегрируя это уравнение в пределах от начального со-'
стояния системы (д0, У о) Д° данного (q, х, у), будем иметь
соотношение, вполне аналогичное тому, которое было выведено t
•выше для случая постепенного испарения, а именно:
Пользуясь только что выделенными формулами и графиками
входящих в них интегралов, можно решать различные задачи;
можно, например, решить вопрос о том, каковы будут масса и
состав дестиллата, полученного обыкновенной перегонкой дан-
ной смеси из двух компонентов до остатка заданной концен-
трации, и другие задачи. Само собой разумеется, что для ре-
шения подобного рода задач необходимым условием является на-
личие данных о зависимости между основными переменными, ха-
рактеризующими состояние данной системы, причем данные эти
могут быть основаны либо на опытных определениях (общий
случай), либо на соотношениях, выведенных из основных газо-
вых законов (идеальные растворы).
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ФРАКЦИОНИРОВКИ И РЕКТИФИКАЦИИ
Простая перегонка смешивающихся жидкостей, не слишком
различающихся температурами кипения, не приводит к полному
их разделению. Процесс перегонки такой смеси характеризуется
постепенным повышением температуры, начиная примерно от
температуры кипения низкокипящего компонента до точки ки-
пения высококипящего компонента данной смеси, причем по
мере повышения температуры дестиллаты будут все более и
более обогащаться вышекипящим компонентом и содержать все
меньше и меньше нижекипящего компонента. Применение вся-
кого рода специальных приспособлений (дефлегматоры, фракци-
онирующие и ректификационные колонны) значительно улуч-
шает погоноразделение и поэтому находит широкое применение
как в лабораторной, так и в заводской практике.
Основные принципы погоноразделения. Для разделения
’ фракций путем перегонки применяются два процесса: последо-
вательное испарение и последовательная ректификация. Оба
эти процесса могут совмещаться в одном и том же аппарате.
Так, например, в разного рода перегонных сосудах (колба, куб),
снабженных дефлегматором того или иного типа или колонкой
с насадкой либо тарелками процесс последовательного испа-
рения осуществляется непосредственно в перегонном сосуде;
в дефлегматоре же происходит последовательная конден-
сация. Перегонка на этих аппаратах, называемая фракциониро-
. 404
ванной перегонкой или фракционировкой, протекает всегда при
постепенно повышающейся температуре, благодаря чему переход
сырья в парообразное состояние совершается здесь не сразу,
а начиная с наиболее легких частей и с постепенным вовлече-
нием в процесс перегонки все более и более тяжелых фракций.
Повышение температуры происходит при этом за счет тепла, не-
прерывно поступающего в дефлегматор или колонну из куба
вместе с вышекипящими частями, которые частично конденси-
руются при этом и возвращаются в перегонный сосуд. Отсюда
ясно, что погоноразделительные устройства, в основе которых
лежит принцип последовательного испарения, работают периоди-
чески даже в тех случаях, когда предусмотрена подача сырья
во время перегонки, так как работа такого рода перегонных аппа-
ратов всегда лимитируется вместимостью их перегонного сосуда
(куба).
Примером погоноразделительных устройств, работающих по
принципу только последовательной конденсации, могут служить
шлемовые трубы кубовых батарей, особенно масляных. В насто-
ящее время такие погоноразделительные устройства почти не
применяются, так как осуществляемое ими погоноразделение —
крайне несовершенно. Вместо процесса последовательной кон-
денсации в настоящее время при перегонке нефти применяется
процесс ректификации, представляющий собою ряд чередую-
щихся процессов испарения и конденсации, осуществляемых
в ректификационной колонне. Примером могут служить опи-
санная в начале настоящей главы перегонная установка
Фостер-Уилера и другие аналогичные аппараты. Характер-
ная особенность их работы заключается прежде всего
в том, что подогрев сырья производится здесь отдельно,
в трубчатой печи, и поступающее в колонну сырье, нагретое до
соответствующей температуры, сразу, всей своей массой, пре-
вращается в парообразное состояние. Последующая затем ректи-
фикация паров совершается постепенно: наиболее тяжелые ком-
поненты, сконденсировавшись, падают в нижнюю часть колонны
и отводятся наружу, лары же более легких компонентов подни-
маются по колонне вверх, постепенно конденсируются здесь на
разной высоте в порядке понижения их температур кипения и
также отводятся с разных высот колонны наружу в виде разного
рода дестиллатов. Таким образом в колонных аппаратах обеспе-
-чивается прежде всего непрерывность работы, крайне важная
• с практической точки зрения. Вместе с тем в процессе • ректифи-
кации отдельные зоны ректификационной колонны сохраняют
в течение всего процесса перегонки известное постоянство как
материального, так и теплового режима: состав и температура
в каждой точке ректификационной колонны должны оставаться
во все время перегонки без изменения. Благодаря этому метод
ректификя пии легче поддается теоретическому обоснованию и
подлежит рассмотрению в первую очередь.
Погоноразделение в лабораторных условиях. Если погоно-
разделительные аппараты, применяемые в промышленности,
405
Должны строиться, как было показано выше, по принципу после-
довательной конденсации, то большинство погоноразделительных
приспособлений лабораторного типа, наоборот, основано на прян,
цине последовательного испарения. Сюда должны быть отнесены
прежде всего дефлегматоры и насадки различных типов, приспо-
собляемые к перегонным сосудам {колбы, кубики и т. п.) в целях
более совершенного погоноразделения. Приспособления этого рода
чрезвычайно разнообразны: от простых трубчатых насадок
(фиг. 64) до дефлегматоров различных типов, например Глин-
ского (фиг. 78), Ле-Беля Геннингера (фиг. 79), Нортон-Оттен
(фиг. 80) и т. д. Сюда же относятся колонны лабораторного типа
с различным наполнением, например с бусами (по Гемпелю),
короткими стеклянными трубками (по Роберу) или обрезками
алюминиевой проволоки (по Гадаскину), а также барботирую-
щие дефлегматоры различных конструкций (например по Арбу-
зову, фиг. 81) и колонны новейших типов с разного рода спе-
циальной изоляцией, насадкой и орошением. Сравнительному
исследованию разнообразнейших приборов этого рода и опреде-
лению их относительной эффективности посвящен целый рад
работ [14]. Не задерживаясь на подробностях, отметим лишь
Фик. 78. Деф-
легматор Глин-
ского.
Фиг. 79. Деф-
легматор Ле-
Белъ-Геннингера.
Фиг. 80. Деф-
легматор Нор-
тон-Оттен.
Фиг. 81. Дефлег-
матор Арбузова.
Весьма важным фактором, определяющим мощность действия
колонны, является ее длина: чем длиннее колонна, тем сильнее
ее погоноразделительная способность, особенно в 'Случае изолиро-
ванных колонн с наполнением и без конденсатора. Напротив,
увеличение диаметра колонны оказывает сравнительно слабое
действие на ее мощность, особенно при небольших скоростях
гонки. г
Очень большое влияние на работу колонны оказывает скорость
406
гонки; чем медленнее вести перегонку, тем яснее проявляется
погоноразделительная способность колонны. Большое влияние
оказывает также активность наполнения колонны, особенно изо-
лированной.
Наконец, максимальное влияние на эффективность колонны
оказывает орошение, особенно при больших скоростях гонки.
Орошение поступает в колонну обычно за счет действия особого
дефлегматора (конденсатора), работа которого регулируется либо
скоростью просасываемого воздуха, либо проточной водой со
специально подобранной температурой; широкое применение по-
лучили также наливные конденсаторы различных типов (фиг. 82),
наполняемые различными жидкостями в зависимости от темпе-
ратуры кипения компонентов разгоняемой смеси.
Среди разнообразнейших колонных аппаратов лабораторного
типа все большее и большее применение для разгонки нефтей и
нефтепродуктов приобретает за последнее время стандартный
аппарат с колонной, сконструированный в США [13]. Колонна
этого аппарата обнаруживает прекрасные погоно-
разделительные свойства, и получаемые на ней
результаты лежат в основе построения графиков,
которые получили условное наименование кри-
вых ИТК (истинных точек кипения).
Стандартный аппарат (фиг. 83) для определе-
ния ИТК состоит из круглодонной колбы в 5 л
с тубусами для термометра и капилляра (на слу-
чай перегонки в вакууме) и припаянной к колбе
фракционирующей колонны с дефлегматором и
муфтой для изоляции; все — из стекла Рутех,
хорошо противостоящего температурным колеба-
ниям. Колонна заполнена жестяными кольцами
стандартного размера (7X7 мм), а снаружи
окружена кожухом с обогревом при помощи элек-
трообмотки и тока горячего воздуха, темпера-
тура которого регулируется в особых подо-
гревателях. Таким образом снаружи колонны
поддерживается та же температура, что и
внутри ее, так что перегонка осуществляется
в адиабатических условиях. Наверху колонны
имеется дефлегматор; поддерживая с помощью
струи холодного воздуха, снаружи от дефлегма-
тора, ту или иную температуру, можно регули-
ровать " количество орошения. Все * управление
процессом перегонки сосредоточено на особом
Фит. 82. Налив-
ной конденса-
тор.
щите, на котором смонтирован ряд контрольных и регулирую-
щих приборов. Все размеры аппарата стандартизованы.
Перегонка на стандартной установке с колонной производится
в строго определенных условиях с постоянной скоростью
(б см3/мин) и отбором приблизительно трехпроцентных фракций
по объему. "Разгонка нефти начинается при атмосферном давле-
нии и продолжается до тех пор пока температура в колбе не до-
407
стигает 340—345°; затем. продолжают разгонку при 1—1,5 мм
остаточного давления до того же предела температуры в колбе.
В этих условиях в зависимости от природы нефти удается ото
брать в общем 70—80% от сырой нефти без сколько-нибудь за-
метного разложения.
Полученные в результате разгонки нефтяные фракции взве-
шиваются и пересчитываются на процентное содержание их
в исходной нефти. Затем, пользуясь тем или иным методом, на-
пример графиками Кокса, Вильсона или Максвелла, приводят
наблюдавшиеся при разгонке температуры кипения к нормаль-
ному давлению и, наконец, для построения кривой ИТК на оси
абсцисс откладывают процентное содержание отдельных фракций
в нефти, а на оси ординат — соответствующие им температуры
кипения.
408
Опытное определение четкости фракционировки с помощью
описанного стандартного аппарата может быть произведено пу-
тем вторичной фракционировки полученных погонов. Такие
определение показывает [16], что четкость разделения здесь не
хуже той, которая достигается на атмосферных ректификацион-
ных колоннах, и значительно лучше той, которую дают вакуум-
ные масляные колонны. Ввиду этого описанный аппарат может
считаться достаточно точным для определения потенциального
содержания в нефти отдельных товарных фракций. Заслужи-
вает внимания, что кривые, чрезвычайно близкие с кривыми
ИТК, получаются при разгонке нефти из кубика со стандарт-
ной колонкой Гадаскина [16].
Зависимость между различными кривыми разгонок. Кривые ИТК
показывают более или менее точно истинный фракционный состав смеси;
разгонка же по Энглеру по существу представляет собою процесс посте-
пенного испарения, когда отходящие в каждый данный момент пары на-
ходятся в равновесии с остатком в перегонном сосуде. Поэтому начало
кипения по Энглеру почти совпадает с началом однократного испарения;
точно так же и конец кипения по Энглеру часто почти совпадает с концом
кипения по ИТК. Расхождения <в конце иереи нки между данными по .ИТК
и по Энглеру обусловливаются случайными причинами, например перегревом
в конце перегонки по Энглеру и т. п. ___
Ввиду легкости, с которой могут быть получены кривые ИТК, и срав-
нительной простоты необходимой для этого аппаратуры в высшей степени
интересной и важной является задача построения по этим кривым кривых
однократного испарения (ОИ), нахождение которых экспериментальным пу-
тем значительно более сложно. В самых общих чертах эти задача решается
следующим образом.
Экспериментально установлено, что для большинства нефтяных фрак-
ций в пределах 10—70% отгона кривые ИТК и ОИ выражаются прямыми
линиями. Таким образом, имея экспериментально найденную прямую ИТК,
для построения кривой ОИ достаточно по каким-либо параметрам по-
строить в дополнение к первой прямой вторую прямую. В качестве тако-
вых параметров удобно взять в данном случае два, для которых на основе
обширного экспериментального материала установлена известная зависи-
мость, а именно:
1) угол наклона кривой ИТК к кривой ОИ, причем наклоны обеих
кривых выражаются в градусах Цельсия, приходящихся на 1% отгона;
2) точка пересечения кривой ИТК с кривой ОЙ.
По этим двум параметрам, углу и точке, кривая ОИ определяется
однозначно. Более подробное изложение этого важного вопроса выходит
из рамок настоящего курса 117].
Схема процесса ректификации. Для уяснения работы рек-
тификационной колонны представим себе, что пары, поднимаю-
щиеся от ее тарелок, находятся в равновесии с жидкостью, сте-
кающей на следующую тарелку. Такое условие может быть вы-
полнено только в теории; построенная на подобных основаниях
колонна должна быть названа идеальной. Аналогично, тарелки
колонны, удовлетворяющие указанному условию, называются
теоретическими или идеальными.
Легко представить себе, что на каждой из тарелок идеаль-
ной колонны в процессе ее работы имеются четыре потока ве-
щества: считая тарелки сверху, на n-ую тарелку поступают
жидкость (флегма) с ближайшей верхней, т. е. (эд—1)-й та-
релки, и пар с ближайшей нижней, т. е. (м4-1)-й тарелки;
с другой стороны, уходит с той же я>й тарелки жидкость на
409
л-1
Фиг. 84. Схема баланса веще-
ства для теоретических та-
релок.
(л 4- 1)-уЮ тарелку, т. е. вйиз, а пары — на (•»— 1)-ую тарему,
т. е. вверх. На фиг. 84 эти движения жидкости через- теоретик
скую тарелку изображены графически, причем, как выше,
масса 'жидкости обозначена здесь через Q, а масса пара—че-
рез д; индексы при величинах Q и q обозначают номер тарелки,
с которой берется данный продукт, а х и у обозначают конден,
трации низкокипящего компонента в жидкой и паровой фазах
Из четырех потоков вещества, поступающих на тарелку, два»
а именно Q„ и qn, находятся согласно определению идеальной
тарелки в равновесии. Два других потока, а именно поток жид-
кости с («та — 1)-й тарелки, т. е. и поток пара с (и+ 1)-й
тарелки, т. е. qn +1, напротив не могут находиться в равнове-
сии, но при соприкосновении стре-
мятся к этому состоянию путем из-
менения своего состава в следую-
щих направлениях: из жидкого по-
тока Qn.-i некоторое количество
низкокипящего компонента превра-.
щается в пар, благодаря чему кон-
центрация его понижается c^_i
до х-п', с другой стороны,’из паро-
вого потока qn+л часть высококи-
пящего компонента конденсируется
в жидкость, благодаря чему кон-
центрация в парах другого компо-
нента, нижекипящего, должна будет
повыситься от Уп+л до уп. Таким
образом материальное равновесие на
тарелке восстановливается, причем
более летучий компонент перехо-
дит из жидкого состояния в паро-
образное и поднимается вверх,
тогда как менее летучий компонент, ’наоборот, переходит из паро-
образного состояния в жидкое и стекает вниз. Что касается, на-
конец, теплоты1 парообразования, необходимой для превращения
более летучего компонента в пар, то таковая, очевидно, компен-
сируется *за счет теплоты, выделяющейся при конденсации паров
более тяжелого компонента, так что в этом отношении тарелка
работает как теплообменный аппарат.
Материальный и тепловой балансы для теоретической та-
релки. Материальный баланс для теоретической тарелки
легко вычисляется на основе схемы ее -работы, представленной
на фиг. 84. Сохраняя все прежние обозначения и принимая,
что масса поступающих на тарелку (веществ равна массе ве-
ществ, уходящих с нее, получаем следующее равенство:
+ “ Qn + ffn-
Для более летучего компонента, принимая в расчет его кон-
центрации в жидкой и паровой фазе (х и у) с соответствую-
щими индексами, аналогично получаем:
Qn-l “Ь ^п+ 1 Уп4 1 = Qn^n “Ь О.пУп'
410
Для составления теплового баланса в качестве исходной
температуры возьмем температуру t°n жидкости на n-й тарелке.
Примем теперь для краткости следующие обозначения: а —
скрытая теплота парообразования (кипения) для пара q»+1;
Ъ—теплота тех же паров при их поступлении с Н- 1)-й та-
релки на “я-ую тарелку; с — теплота жидкости в d — те-
плота смеси; е — скрытая теплота парообразования для пара
q», f — потеря теплоты от охлаждения. При учете этих теплот
необходимо иметь в виду, что пары д» +1 образуются из кипя-
щей жидкости, содержащей нижекипящего компонента не-
сколько меньше, чем жидкость на w-й тарелке, вследствие чего
температура этих паров должна быть несколько выше и что,
наоборот, температура жидкости должна быть несколько
ниже, чем t п, так как жидкость на (а— 1)-й тарелке содержит
нижекипящего компонента несколько больше, чем n-я тарелка,
и поэтому должна кипеть при более низкой температуре. Прини-
мая во внимание знаки величины a, d, с, d, ей/, получаем:
а-|-Ь4_^ = е~Ьс4~Л
В этом равенстве величины Ъ и с должны быть крайне незна-
чительны, так как разность температур двух смежных тарелок
очень невелика; величины d и / — также ничтожны. Кроме того,
попарно величины Ъ 4- d и с + / взаимно уравновешивают друг
друга в последнем равенстве, так что -всеми ими, очевидно,
можно пренебречь. Таким образом из этого равенства оконча-
тельно получаем:
а = е,
т. е., при составлении теплового’ баланса можно принимать в рас-
чет лишь скрытую теплоту кипения.
Крайне важные выводы получаются из последнего соотноше-
ния, если присоединить к нему «правило Трутона» (ч. I, стр. 37),
согласно которому молекулярная скрытая теплота кипения ME
различных веществ пропорциональна их абсолютной темпера-
туре кипения Т:
ME = КТ,
где К, коэфициент пропорциональности, для различных веществ,
в том числе и для углеводородов, близок к 20.
Если теперь массы паров, поступающих с (w 1)-й тарелки
на n-ую, т. е. д,н-л, и паров, уходящих с «-й тарелки на
(»— 1)-ую, т. е. д», выразить в граммолекулах, то для скрытых
теплот а и е получаем следующие выражения:
а == МЕдп^ и е = М'Е'дп.
Поделив обе части этих равенств на Т, и Т' и принимая во
внимание, что а = е, если пренебречь разностью температур на
смежных тарелках, по сокращении получаем:
Qn+l ===
и далее, на основе выведенного выше материального .баланса
теоретической тарелки;
Два последних равенства могут быть применимы, очевидно
к любой тарелке идеальной колонны, за исключением самой
верхней или первой тарелки, а также за исключением той, на
которую в колонну подается (вещество. Эти равенства показы-
вают, что работа идеальной колонны характеризуется постоян-
ством граммолекулярного парообразования и перелива, иначе
говоря, количество граммолекул пара, поступающего на каждую
тарелку, равно количеству граммолекул пара, оставляющего эт\'
тарелку, а также—количество граммолекул жидкости, притека-
ющей на тарелку, равно количеству жидкости, стекающей с этой
тарелки. Наконец, применяя два последних равенства к выве-
денному выше уравнению материального баланса для более ле-
тучего компонента, получаем:
Q^n-i "Н 1= Qxn + 0Уп»
а после преобразования:
Уп -1 __
an-l хп Ч
В этих последних равенствах индексы при величинах Q и q
опущены, так как ввиду постоянства граммолекулярного пе-
релива и парообразования они утратили для аиих величин вся-
кое значение.
Расчет числа теоретических тарелок колонны. Вычисление
количества теоретических (идеальных) тарелок ректификацион-
ной колонны, которое необходимо для получения дестиллата не-
которой определенной концентрации относительно того илн
иного, например более летучего, компонента, наиболее просто
производится следующим графическим способом [18]. •
Пусть дана кривая равновесия данной смеси из двух компо-
нентов (фиг. 85). На этой кривой возьмем точку с координатам
fan, уп), которые соответствуют равновесным концентрациям бо-
лее легкого компонента в жидкой и паровой фазе на n-й та-
релке. Нанесем далее еще две точки с координатами, отвечаю-
щими составу потоков жидкости и пара для той же тареж
а именно (хп, уп^) и уп). На отмеченных трех точках
строим треугольник, две стороны которого по построению парал-
лельны осям координат; следовательно, это прямоугольный тре-
угольник, у которого вершина прямого угла лежит в точке
уп) на кривой равновесия.
Легко видеть, что точка уп+х) отвечает составу жид-
кости, стекающей с ^й тарелки, и составу пара, поступающего
на ту же тарелку, т. е. условиям, которые имеются непосред-
ственно под я>й тарелкой. Ясно также, что наблюдаемое здесь
изменение состава пара определяется разностью уп—Уянл, т. е-
размерами вертикального катета построенного прямоугольного
треугольника.
Равным образом точка fan—i, уп) отвечает составу жидкости,
поступающей на -n-ую тарелку, и составу пара, покидающего
эту же тарелку, т. е. условиям, которые имеют место непосред
412
ственно над тарелкой. Очевидно, что наблюдаемое в данном
случае изменение состава жидкой фазы определяется разностью
•2'п—-1 я», т. е. длиной горизонтального катета того же треуголь-
ника.
Выше была найдена пропорция:
(Уп УпЛ 1) • ^И) — Q • 7»
в которой, как было только что показано, величины ул — ул+1
и яп-1—хп определяются катетами прямоугольного треуголь-
ника. Так как, однако, отношение вертикального катета к гори-
зонтальному, иначе говоря, тангенс угла, противолежащего вер-
тикальному катету, определяет наклон гипотенузы к оси абс-
цисс, то ясно, что для данного случая этот наклон определяется
отношением Q : q.
Совершенно такое же построение можно сделать для (п — 1)-й
тарелки идеальной колонны. Вершинами углов нового треуюль-
Фиг. 86. Вывод числа теоретических
тарелок колонны.
Фиг. 86. Баланс вещества для
верхнего конца колонны.
ника на этот раз будут точки с координатами: im),
(хл _ 1, уп _ i) и (жп_2, i/n-i). Как видно, первая из этих точек
совпадает с Одной из вершин треугольника, построенного для
™-й тарелки; вторая точка лежит на кривой равновесия; нако-
нец, третья точка вместе с первой дает направление гипотенузы
треугольника, наклон которой к оси абсцисс определяется тем
же соотношением, что и для w-й тарелки, т. е. Q: q. Отсюда
ясно, что гипотенузы обоих рассмотренных треугольников лежат
на одной прямой, образующей с осью абсцисс угол, тангенс ко-
торого равен Q : q. Распространяя этот вывод на другие тарелки
колонны (кроме первой), приходим к выводу, что гипотенузы
всех аналогично построенных треугольников должны предста-
влять собою отрезки одной и той же прямой, наклон которой
к оси абсцисс определяется отношением Q: q.
Самая верхняя, т. е. первая тарелка идеальной колонны под-
лежит особому рассмотрению. На эту тарелку (фиг. 86) посту-
пают пары со второй тарелки колонны и жидкое орошение
(reflux) из конденсатора; уходят с первой тарелки пары в кон-
413
денсатор и жидкая флегма на вторую тарелку. При атом, оче-
видно, составы орошения и окончательного продукта должны
быть одинаковы (яо); кроме того, состав паровой фазы с 'первой
тарелки yi также должен быть равен яо. Таким -образом, выве-
денное выше соотношение принимает для первой таре тки еле-
дующий вид: .
'Построим теперь на диаграмме (фиг. 85) прямоугольный
треугольник для первой тарелки, приняв за вершины
углов точки с координатами («i, yi = яо), (ал, 2/2) и
(жо, yi — хо). Легко видеть, что и в этом -случае наклон гипоте-
нузы треугольника определяется той же самой величиной Q:q.
Итак, процесс ректификации в целом можно изобразить при
помощи совокупности прямой, проходящей через точку .(яо, ж0)
с наклоном Q : q к оси абсцисс, и ряда прямоугольных треуголь-
ников, гипотенузы которых расположены на этой прямой, а вер-
шины прямых углов — на кривой равновесия. Указанную пря-
мую называют прямой концентрации или рабочей линией. Что
касается прямоугольных треугольников, то, начиная от точки
fa?o, xq), они образуют между кривой равновесия и рабочей ли-
нией ряд ступеней (фиг. 85); каждая из этих ступеней соответ-
ствует отдельной тарелке идеальной колонны; число же ступе-
ней должно соответствовать числу идеальных тарелок, которые
необходимы для осуществления заданного разделения данной
смеси.
Уравнение рабочей линии, проходящей через точку (жо, яо)
и имеющей наклон Q: q к оси абсцисс, должно иметь следую-
щий вид: п
Для промежутка времени, в течение которого 1 граммо-
лекула вещества покидает аппарат в виде продукта, баланс
вещества будет равен:
0. — Q +-1,
откуда
(2/ —а?о) (Ф+1) = <2(я — -*0),
или, определяя у:
и = ® х 4- —°—
У Q-|-1 ж * Q-j-1 *
В этом последнем выражении для у величина пред-
“Г *
ставляет собою отрезок, отсекаемый рабочей линией на оси
ординат, благодаря чему крайне облегчается построение ра-
бочей линии по двум точкам: первая из них лежит на оси у
и имеет координаты (о, j); вторая точка определяется пе-
ресечением диагонали с перпендикуляром к оси абсцисс х = х0,
т. е. имеет координаты (я0,‘я0).
Расчет числа теоретических тарелок для колонны непрерыв-
ного действия, оборудованной тарелками выше и ниже места
414
ввода -сырья, производится отдельно для верхней и нижней части
колонны. Верхняя часть -колонны рассчитывается, как это опи-
сано выше для простой ректификационной колонны, т. е._ графи-
чески, путем построения рабочей линии колонны до точки пе-
ресечения ее с линией х = хп, где хп определяет состав жидко-
сти, питающей колонну. Эта же точка ‘является одной из точек
рабочей линии нижней части колонны. Другую точку, необхо-
димую для построения этой второй рабочей линии, получают
от пересечения диагонали графика с линией х = хт, где хт
определяет 'состав остатка, покидающего колонну. Система пря-
моугольных треугольников, построенных на обеих рабочих ли-
ниях, определяет число теоретических тарелок обеих частей 1ю-
лонны (фиг. 87).
Фиг. 87. Полная диаграмма ректи-
фикационной колонны.
Фиг. 88. Графическое определение
коэфициента полезного действия
тарелки.
Все предыдущие выводы сделаны для колонны тарелочного
типа с колпачками. Для колонн другого типа с разного рода
насадками задача расчета заключается в определении высоты
колонны, которая необходима для достижения того или иного
эффекта погоноразделения. Для решения этой задачи можно
пользоваться следующим методом. Подразделяют колонну па ряд
частей, каждая из которых работает как теоретическая тарелка,
т. е. имеет -высоту, достаточную для создания в колонне равно-
весия между жидкой и паровой фазами данной смеси. Такая
высота получила наименование эквивалентной высоты одной те-
оретической тарелки'1. Если для данной смеси жидкостей и для
определенной -конструкции колонны эта величина найдена, то
общая высота колонны получается путем простого перемноже-
ния этой величины на рассчитанное число теоретических та-
релок.
Коэфициент полезного действия тарелки. Все вышеприведен-
ные расчеты- числа тарелок были сделаны при допущение, что
1 Сокращенно— НЕТР— Hight Equivalent of Theoretical Plate.
415
колонна оборудована теоретическими (идеальными) тарелкш
удовлетворяющими условию равновесия между парами, подни-
мающимися с данной тарелки, и жидкостью (флегмой), стекш
щей на следующую тарелку. Тарелки, которыми оборудованы ре-
альные колонны, не удовлетворяют этому условию: в них кон-
центрация легкого компонента в паровой фазе не достигает тех
величин, при которых устанавливается равновесие с флегмой;
соответственно этому работа реальной тарелки представляет со-
бою лишь часть работы тарелки теоретической. Для характери-
стики работы реальной, например n-й, тарелки пользуются вели-
чиной тр которая называется коэфициентом полезного действия
тарелки [19]:
4=1па ^*-100,
‘ Уп° — Уп+1 ’
где ул — состав пара, поднимающегося с и-й тарелки, ум-i—со-
став пара, поступающего на ту же тарелку снизу, а 2/»°—состав
пара, который находится в равновесии с флегмой, стекающей
с n-й тарелки.
Для графического определения коэфициента полезного дей-
ствия тарелки может служить диаграмма (фиг. 88). Здесь тре-
угольник acd соответствует теоретической тарелке, треугольник
же abe — действительной. При прохождении пара через теорети-
ческую тарелку происходит изменение его концентрации от
2/n+i до г/п°; из графика видно, что уп°—yn+i = ac. При прохо-
ждении лара через действительную тарелку происходит измене-
ние его концентрации от г/»+1 до уп, т. е. на величину уп—
— уп + 1 — аЪ. Коэфициент полезного действия • определяется
поэтому отношением отрезка аЪ к отрезку ас.
Коэфициент полезного действия тарелки определяется опыт-
ным путем для данной колонны и данного состава перегоняемой
жидкости, причем допускают, что для всех тарелок колонны этот
коэфициент примерно один и тот же. Если, например, этот сред-
ний коэфициент полезного действия тарелки данной колонны
равен 60%, то для перехода от числа теоретических тарелок
к числу тарелок, которое необходимо в действительности, первая
величина должна быть разделена на 0,6.
На величину коэфициента полезного действия оказывают
влияние различные факторы. Важнейшим из них является лег-
кость диффузии при парообразовании, с одной стороны, и кон-
денсации — с другой: чем меньше, например, пузырьки, на ко-
торые разбивается пар, покидая жидкую фазу, тем больше по-
верхность соприкосновения жидкой и паровой фаз и тем выше
коэфициент полезного действия тарелок. Поэтому устройство и
размеры колпачков, форма и размеры прорезов в тарелке, глу-
бина погружения колпачков в жидкость (флегму) крайне важны
для повышения коэфициента полезного действия тарелки. Боль-
шое влияние оказывает также скорость движения паров в ко-
лонне,* определяемая их плотностью. При атмосферном давлении
скорость паров в колонне может доходить до 0,6—0,8 м/сек; в ва-
416
куумных колоннах скорость паров значительно выше
(2—3 м/сек); в колоннах, работающих иод давлением, наоборот,
она может снижаться до 0,1—0,3 м/сек. В прорезах колпачков
скорость ларов в 5—10 раз больше, чем в свободном сечении
колонны. Если скорость движения паров при данной их плот-
ности становится слишком большой, то вместе с парами может
оказаться увлеченной также и жидкость, так что нормальная
работа колонны нарушается.
Орошение колонны. Для нормальной работы ректификацион-
ной колонны необходимо, чтобы часть вещества, покидающего ко-
лонну в виде окончательного и наиболее легкого продукта ректи-
фикации, возвращалась обратно в колонну на первую тарелку
в виде так называемого орошения. При этом, как было показано
выше, наклон рабочей линии к оси ординат может быть выражен
следующи м образом:
tga== QTI’
где v — масса орошения, которое должно поступать на первую
тарелку в течение времени, необходимого для получения 1 грам-
молекулы окончательного продукта.
Орошение представляет собою один из важнейших факторов,
определяющих режим работы колонны. Действительно, пусть ас,
ad, ае и af представляют собою
рабочие линии, определяю-
щие различные режимы работы
некоторой колонны (фиг. 89).
Крайними случаями здесь
являются два режима, опреде-
ляемые рабочими линиями ас
и af. Оба эти случая практи-
чески неосуществимы по сле-
дующим соображениям. В слу-
чае рабочей линии ас произве-
сти нормальное построение для
расчета числа тарелок колонны
с доведением системы прямо-
угольных треугольников до ли-
нии х = х„, очевидно, не пред-
ставляется возможным. Таким
образом данный пример, как и
всякий другой, где рабочая линия пересекается с кривой равно-
весия в некоторой точке д, не лежащей на линии х = хп, пред-
ставляет собою случай, явно практически невозможный. Для
второго случая,' характеризуемого рабочей линией af, угол a
наклона рабочей линии к оси абсцисс равен 45°, и следова-
тельно, tg а — 1; отсюда Q : (Q + 1) = 1, -что возможно лишь при
условии Q — co, т. е. орошение — величина бесконечно большая.
Очевидно, при этом условии производительность колонны равна
нулю, так что и этот случай практического значения не имеет.
417
27 Зак. 1622. Химия нефти.
Особого внимания Заслуживает случай, характеризуемый р&.
бочей линией adf пересекающей кривую равновесия в точке, ко-
торая лежит на линии х—хп. Теоретически для этого случая
можно произвести графическое построение для расчета числа
тарелок; очевидно, что для обоих отрезков рабочей линии, т. е.
выше и ниже точки d, число это — бесконечно большое. Легко
показать, что для этого случая орошение получается минималь-
ное. Действительно, обозначая для этого случая орошение че-
рез Qo, получаем из уравнения рабочей линии, проходящей через
точки (so, уо) и (sn, уп):
Qo__ go Уп
Qo + 1 а’о — л п ’
откуда
™ Уп—Хп
По мере уменьшения угла наклона рабочей линии вели-
чина уп, как видно из фиг. 89, увеличивается; отсюда ясно, что
величина Qo по мере уменьшения того же угла должна умень-
шаться, достигая в точке d своего минимума.
Наконец, рабочая линия ае соответствует режиму, возможному
не только теоретически, но и практически. В этом случае вели-
чина орошения больше минимальной, причем количество теоре-
тических тарелок по мере увеличения наклона рабочей линии,
как видно из фиг. 89, уменьшается. Наиболее выгодное орошение
определяется конструкцией и размерами колонны; при этом при-
ходится принимать в расчет расход топлива, так как тепло, от-
данное орошением охлаждающей воде в дефлегматоре (конден-
саторе), обычно теряется. Практика показывает, что орошение
для ректификационных колонн в различных случаях может из-
меняться в (пределах от 1,4 до 4—5-кратной величины его мини-
мума.
ТЕОРИЯ ПЕРЕГОНКИ. СЛОЖНЫЕ СМЕСИ
Системы, составленные более чем из двух компонентов, могут
быть весьма разнообразны. Сюда относятся, между прочим, си-
стемы, состоящие более чем из двух вполне смешивающихся
жидкостей, в частности нефть и ее дестиллаты. Такие системы
можно объединить в одну большую группу под названием «слож-
ные смеси».
По правилу фаз система, состоящая из п компонентов, вполне
растворимых друг в друге, так что число (N) фаз ее равно двум
(жидкость и пар), имеет п степеней свободы согласно равенству:
X == п + 2 — N — п.
Таким образом и здесь мы имеем неполное равновесие. При-
мерами подобного рода систем могут служить нефть и ее дестил-
латы. Как явствует из правила фаз, подобные системы опреде-
ляются п переменными, которые могут быть выбраны произ-
вольно; например, можно задаться температурой системы и кон-
418
центрациями (н— 1) компонентов в жидкой фазе (концентрация
последнего, n-го компонента становится при атом, очевидно, из-
вестной); тогда остальные характеристики системы, как-то: со-
держание всех компонентов в паровой фазе и in упругость, опре-
деляются однозначно.
Состав пара и жидкой фазы сложной смеси.. В основу
общей теории равновесия сложной смеси может быть положено
допущение, что число п компонентов данной смеси бесконечно
велико, так что молекулярная концентрация одного из компонен-
тов может быть выражена бесконечно малой величиной dr.
Допуская, далее, непрерывное изменение молекулярных весов
в данной системе и температуры, кипения, получаем сложную
смесь бесконечно большого числа компонентов, .вполне раство-
римых друг в друге, для которой парциальное давление dp паров
компонента, имеющего в жидкой фазе молекулярную концентра-
цию dr, выражается согласно закону Рауля следующим равен-
ством:
dp — Pdr.
Здесь Р— упругость насыщенных паров данного компонента
смеси над этим же компонентом в жидкой фазе при той же тем-
пературе.
Так как общая упругость р паров всякой системы равна
сумме парциальных упругостей ее компонентов, то для нашей
сложной смеси жидкостей будем иметь:
J Fdx.
о
Эта формула позволяет определить то давление р, под кото-
рым при избранной температуре данная смесь начинает кипеть;
для этого необходимо, однако, иметь таблицы упругостей Р на-
сыщенных паров всех компонентов и знать их концентрации
в жидкой фазе данной смеси.
Парциальное давление р паров какого-либо компонента, вхо-
дящего в состав сложной смеси, может быть выражено не только
вышеприведенной формулой, вытекающей из закона Рауля; дру-
гое выражение давления р того же компонента в зависимости от
его молекулярной концентрации dy в паровой фазе получается
на основании законов Дальтона и АвогшУро величиной pdy.
Таким образом имеем:
Р
Fdx —pdy пли dy — - dr.
Интегрируя это уравнение, получаем для молекулярной кон-
центрации и в паровой фазе следующее выражение:
419
27*
Это уравнение характеризует состав у паровой фазы, отве-
чающей составу жидкой фазы равновесной системы при данной
температуре. Оно называется поэтому уравнением равновесии
фаз сложной смеси и применяется как для построения кривой
равновесия, так и для определения температур точек росы (на.
чала конденсации) и точек закипания. Расчет ведут, разбивая
кривую разгонки сложной смеси на ряд участков, причем все
компоненты, кипящие в пределах некоторого участка, приравни-
ваются к условному компоненту, имеющему средние свойства.
Температура системы Т определяется такими значениями упру,
гости паров отдельных компонентов, при которых общая упру-
гость паров смеси, определяемая по уравнению равновесия, равна
внешнему давлению.
Уравнение равновесия фаз сложной смеси удобно изобразить
графически в координатных осях х и у\ для этого необходимо
знать упругость насыщенных паров Р отдельных ее компонентов
при температуре Т и молекулярный состав смеси. Соответствую-
щая кривая называется кривой равновесия фаз сложной смеси.
По своему виду эта кривая похожа на кривую равновесия фаз
смеси двух жидкостей (фиг. 68), но отличается от нее тем, что
все точки ее отвечают одной и той же температуре состояния
равновесия паров и жидкости, в то время как для бинарной
смеси каждая точка кривой равновесия отвечает своей темпера-
туре состояния равновесия.
Таковы в самых общих чертах теоретические основы опреде-
ления состава пара и жидкой фазы с южной смеси жидкостей,
вполне растворимых друг в друге. Как видно, они в полной мере
соответствуют тому, что подробно было рассмотрено выше для
«идеального» раствора двух жидкостей. Опытное определение со-
става жидкой и паровой фаз для сложных смесей также рассмо-
трено выше. Само собою разумеется, что такое определение по
отдельным компонентам смеси — задача, чрезвычайно сложная
и при современном уровне наших знаний даже невыполнимая.
Поэтому в данном случае ограничиваются определением в см си
содержания отдельных, более или менее значительных групп
компонентов, искусственно выделяемых из общей их массы; та-
ковы, например, «легкокипящие» и «тяжелокипящие» нефтяные
дестиллаты и т. п. Так как практика требует выделения из нефтй
не индивидуальных компонентов, а лишь отдельных погонов, ки-
пящих в более или менее широких пределах, то такое упрощение
задачи является вполне приемлемым. За молекулярный вес таких
групп компонентов принимается их средний молекулярный вес,
определяемый опытным путем. Выше (фиг. 75) даны примеры
кривых равновесия для нескольких сложных смесей, а именно
для некоторых нефтяных дестиллатов и их смесй.
Испарение сложной смеси. Сложная смесь может испа-.
ряться в условиях либо однократного, либо многократного-испа-
рения. Оба эти вида процессов испарения были подробно рас-
смотрены выше для случая двух вполне смешивающихся жид-
костей; там же описаны 'методика и аппаратура для опытного
420
их проведения, применимая также для случая сложной смеси.
Естественно, однако, что соотношения, характеризующие про-
цесс испарения сложной смеси, будут значительно сложнее.
Обозначим начальную массу некоторой сложной смеси че-
рез Q. Если концентрация в ней некоторой группы компонентов
равна j-o, то их масса в начальной смеси равна Qxo. Пусть в про-
цессе однократного испарения некоторая часть е этой смеси пре-
вратилась в пар; масса испарившейся части будет тогда eQ,
а масса остатка Если концентрацию той же группы
компонентов в жидком остатке обозначить через х, а в парах —
через у, то получаем:
ф:0 = (1 — с) Qx-1-eQy,
откуда
(1 — с) х + су = т0
пли
У = | [*о—О— «)*]•
В координатных осях х, у, е это уравнение представляет собою
некоторую поверхность, являющуюся геометрическим местом рав-
новесных кривых для различных степеней е отгона. Для случая
полного испарения всей .системы (е == 1) последнее уравнение
принимает вид:
У = я0.
Это обозначает, что при полном: испарении смеси паровая
фаза приобретает тот состав в отношении определенной группы
компонентов, который имела начальная смесь, как это и тре-
буется по смыслу процесса однократного испарения.
Для многократного испарения эти соотношения значительно
сложнее. Пусть начальная масса сложной смеси равна Q; тогда
после испарения части е этой смеси в остатке будем иметь массу
(1 — e)Q с некоторым средним молекулярным весом М. Если кон-
центрация некоторой группы компонентов в остатке равна х,
то их общая масса в этом остатке составит (1 —e)Qx.
Пусть, далее, произошло незначительное испарение остатка
с выделением паров, масса которых составит бесконечно малую
величину Qde- если концентрацию в этих парах той же группы
компонентов обозначить через у, то масса их в парах окажется
равной yQde.
Теперь после указанного дополнительного испарения, будем
иметь новый остаток, масса которого равна ф(1 — е — de); кон-
центрация в этом остатке все той же группы компонентов теперь
изменится и станет (х + dx); масса же их в этом остатке ока-
жется равной (1 — е — de)Q(x dx). Таким образом, для общей
массы избранной группы компонентов получается следующее
равенство:
(1 —е) Qx = yQde (1 — е — de) Q (х -f- dx).
431
Сокращая обе части равенства на Q, раскрыв скобки, отбро-
сив бесконечно малую величину высшего порядка и преобрази
вав полученное равенство, окончательно, после приведения, по-
лучаем:
Этим диференциальным уравнением устанавливается связь
между концентрациями некоторой группы компонентов в жидкой
и паровой фазах х и у после многократного (постепенного) ис-
парения некоторой части е данной сложной смеси. В системе
координатных осей ж, у, е это — также уравнение некоторой по-
верхности, которая представляет собою геометрическое место
кривых равновесия, отвечающих различным степеням отгона е.
Под конец испарения, когда с — 1, последнее уравнение превра-
щается в уравнение прямой у = х', это означает, что в системе
к этому моменту остается только один высококипящий компо-
нент, как и должно быть по смыслу данного процесса.
Ректификация сложной смеси. Основное уравнение мате-
риального баланса для случая ректификации сложной смеси по-
лучается из положения, что масса q паров, поднимающихся по
колонне через определенное ее сечение, равна сумме двух вели-
чин: массы Q флегмы (орошения), проходящей через то же се-
чение вниз по колонне, и массы D дестиллата, покидающего
колонну, т. е.
Q = Q +
Обозначим концентрации некоторой группы компонентов, на-
пример «легкокипящих», в жидкой и паровой фазах соответ-
ственно через х и 2/, а концентрацию той же группы компонен-
тов в дестиллате через z. Тогда будем иметь:
qy = Qx-]-Dz.
Исключая из последнего уравнения величину д, после соот-
ветствующих преобразований получаем:
г—у = ^(у—X).
В координатных осях х, у, z это — уравнение некоторой плос-
кости, которая проходит через начало координат (ж = у = 2=0)
и точку, координаты которой х = у = z = 1; кроме того, в этой
плоскости лежат еще две точки, расположенные в координатной
плоскости з/= о и обладающие координатами x — D\ y=Q't
z — l и ж=1; = z = D.
Пусть из колонны выходит дестиллат, представляющий собою
данную группу компонентов в чистом виде; в этом случае я=1
и вышеприведенное уравнение приобретает более простой ВД»
а именно: < '
1zz_2/_ _2_
у — аг D "
422
В координатных осях ж, у это—• уравнение прямой линии,
направление которой определяется значениями величины Q : D.
Обозначим эту последнюю величину через К, т. е. допустим, что
Q-D = Kt и сделаем графическое построение уравнения:
1 — у == К {у — х).
Пусть х= 1; тогда при любом значении К величина у также
равна единице. Следовательно, точка с координатами х — у = i
является общей для всех прямых, выражаемых данным j равне-
нием. Вторая точка, характеризующая эти прямые, получается,
если допустим, что я = 0; тогда для у определяется следующее
общее выражение:
где К — отношение Q, т. е. массы флегмы (орошения), к D, т. е.
к массе дестиллата. Таким образом, для разных значений К по-
лучается пучок прямых, исходящих из точки (х—у = 1) и пе-
ресекающих ось у
в различных точках,
011 ределяемых послед-
ним равенством. На
фиг. 9о дано изображе-
ние этого иучкапрямых,
называемых иногда пря-
мыми орошения; здесь
же даны кривые равно-
весия для ректифика-
ции сложной смеси.
Все приведенные
выводы и построения
(фиг. 90) относятся
к верхней части колон-
ны (концентрацион-
ной); здесь поднимаю-
щиеся снизу пары,
встречая на своем пути
жидкость, отдают ей
увлеченные ими «тя-
Фиг. 90. Прямые орошения и кривые равно
весия для верхней части колонны.
желокипящие» и взамен получают содержавшиеся в жидкости
«легкокипящие». Выведенные уравнения позволяют вычислить
величину орошения Q при заданном составе паровой фазы.
В нижней части колонны процесс протекает несколько иначе?
поднимающиеся снизу пары, богатые тяжелокипящими, встреча-
ются с жидкостью, содержащей легкокипящие. В результате об-
мена пары и здесь отдают жидкости тяжелокипящие, сами же
получают от жидкости легкокипящие, так что в нижней части
колонны собирается жидкость, не содержащая легких фракций
либо содержащая их в очень ограниченном количестве. Для
498
общего случая материальный баланс нижней части колонны
получается на основе соотношения:
Q =.«+’.
где Q —- масса некоторой группы компонентов, например «легко-
кипящих», стекающих через данное сечение колонны в единицу
времени; q—масса паров тех же компонентов, проходящих че-
рез то же сечение; г— их масса, сохраняющаяся в жидком
остатке.
Обозначая, как и выше, соответствующие концентрации че-
рез х, у' и z\ получаем:
Исключаем из этого уравнения д; после соответствующих
преобразований, получаем:
у' —г'= у-(/ — «')
или, вводя обозначение Q : г = К':
у'-г'=К'(у'-х').
Это и есть уравнение материального баланса для нижней
части колонны в общем виде.
Допустим теперь, что в нижней части колонны «легкокипя-
щих» вовсе не остается, т. е. я' = 0; тогда последнее уравнение
принимает более простой вид:
у’=ь" (.у'—И-
Очевидно, это — уравнение ряда прямых, направление кото-
рых зависит от величины К'. Для их построения предположим,
что ж' = 0; тогда независимо от величины К' ордината также бу-
дет равна нулю: у' = 0. Это значит, что все прямые проходят че-
’рез начало координат. Вторая точка для каждой из этих линяй
получается, если допустить, что у' = 1, тогда для величины s
получается следующее выражение:
или, подставляя вместо К его значение:
Таким образом, для нижней части колонны получается пучок
йрямых, исходящих из начала координат, в направлении, обрат-
ном тому, которое было найдено для верхней части колоннн
(фиг. 90).
Что касается расчета числа тарелок колонны для ректифи-
кации сложной смеси, исчисления необходимого и достаточного
орошения, которое обеспечило бы нормальную работу -колонны,
424
и других (Вопросов, связанных с задачей расчета и конструкций
ректификационных колонн, то ближайшее их рассмотрение уже
выходит из рамок настоящего курса. Однако с принципиальной
стороны все эти задачи могут быть разрешены теми же мето-
дами, основания которых изложены выше в связи с ректифика-
цией смеси двух вполне растворимых друг в друге жидкостей;
необходимо лишь за чистые компоненты смеси условно принять
некоторые отдельные группы компонентов, например «легкокипя-
щие» и «труднокипящие», -как это и было сделано в пред-
шествующем изложении.
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ ПЕРЕГОНКИ КАК МЕТОДА
ПЕРЕРАБОТКИ НЕФТИ
Перегонка нефти является тем основным методом ее перера-
ботки, который, открыв в свое время возможность получения
из нефти осветительных, а позднее и других масел, положил на-
чало современной нефтяной промышленности. Представляя со-
бою обычно первую после обезвоживания глубокую техническую
операцию, которой подвергается сырая нефть, перегонка до сих
пор занимает первенствующее положение среди всех прочих ме-
тодов переработки нефти. Ее задача — отделение от углеводоро-
дов нефти смолистых продуктов, которые вместе с остатками
наиболее тяжелых масел либо находят применение в качестве
котельного топлива, либо перерабатываются на асфальт, тогда
как углеводородная часть, подвергаясь в процессе перегонки
разделению, дает начало разного рода дестиллатным продуктам,
начиная с наиболее легких (бензины, керосины) и кончая тя-
желыми смазочными маслами.
Так как при перегонке на кубовых батареях нефть вследст-
вие длительного нагревания подвергается частичному разложе-
нию, то еще недавно техническая мысль искала таких путей
переработки нефти, которые совершенно исключали бы перегонку
(«холодная фракционировка» и т. п.). В настоящее время по-
требность в такого рода методике может считаться отпавшей:
в современных трубчатых установках нефтяная промышленность
приобрела мощнутб и высокосовершенную перегонную аппара-
туру, которая позволяет осуществлять перегонку нефти с мини-
. мальной затратой топлива, практически без разложения, с отбо-
* ром в некоторых случаях свыше 90% от сырой нефти. Таким
образом надо думать, что перегонка нефти и впредь останется
основой заводской переработки нефти.
Как. правило, перегонка нефти даже на аппаратах, снабжен-
ных мртпньгми ректификационными колоннами, не может произ-
вести разделение нефтяных компонентов по их составу, что не-
редко требуется существом дела. Ввиду этого за последнее время
нефтяная промышленность наряду с перегонкой на трубчатых
батареях стала пользоваться некоторыми новейшими методами,
имеющими по существу то же назначение, что и перегонка, т. е.
425
разделение различных компонентов нефти. Такова селективная
очистка с помощью избирательных растворителей, которая будет
рассмотрена ниже в гл. VI.
ЛИТЕРАТУРА
Трегубов А. М., Основные начала перегонки и ректификации. 2-е
Баку —Москва (1988). ИЗД''
Эллиот JT., Принципы перегонки, пер. под ред. Тищенко И, Л., Л. (lm
Эллиот Е., Перегонка на практике, пер. под ред. Тищенко И. А., Л. (1m
Робинзон Е. ПС, Основные начала дпобиой перегонки, пер. под
Ерасуского И. А., Харьков (1930). m
Обрядчиков С. Н. и Хохряков П. А., Расчет и конструкция ректификацион-
ных ко тонн для нефтеперегонных установок, ОНТИ (1933).
Бэджер В. и Мак-Кэб В., Основные процессы и аппараты химических про-
изводств, ОНТИ (1933).
Добрянский А. Ф., Курс технологии нефти, М. Л. (1931).
Белл, Американские методы переработки нефти, ОНТИ (1933).
Sidney Young, Distillation Principles and Processes, London (1922).
1. Perdew, „Нефт. хоз." 18, № 2, 249 (1930).
2. Пархоменко, „Нефт. хоз." 16, № 3, 365, (1919).
3. Схемы новейших способов переработки нефти в США, пер. под. вел
Седых Н. Ф-, ОНТИ (1933).
4. Еострин, „Нефт. хоз." 18, № 1, 62 (1930).
5. Американские нефтеперегонные заводы, „Нефт. изд.' (1929).
6. Вакуумные трубчатые установки Алко, Баку (1933).
7. Еацнелъсон, Вакуумные трубчатые установки Пенгу—Гурвич—Нерсесова,
Баку — Москва (193о).
8. Трегубов А. М., „Нефт. хоз." 16, № 1, 68 (1935).
9. Хволъсон, Курс физики, т. III.
10. См., например, Трегубов, Основные начала перегонки и ректификации,
Баку -г Москва (1938).
11. Ср. Rodgers М. С. a. Brown G. G. „Ind Eng. Chem." 22, № 3 (1930).
12. Rosanoff, Bacon, White, „Journ. Am. Chem. Soc." 86, 2, 1803 (1914).
13. Обрядчиков и Хохряков. Расчет и конструкция, стр. 12 и сл.
14. См. Лонгинов В. и Прянишников А., Материалы к изучению дефлег-
маторов и ректификационных колонн лабораторного типа, „Труды
Ин-та хим. чистых реактивов", М. (1929); здесь же литература
вопроса.
15. Peters W. A. a. Baker, „Ind. Eng. Chem.' 18, 69 (1926).
16. Гурвич В. Л., Еаминер, Луценко, Кривые разгонок нефтей, 2-е изд.,
Баку — Москва (1933).
17. Pyroomow a. Beiswenger, „дз. нефт. хоз." 12, N 2, 43 (1932).
18. Me. Cabe a. Thiele, ,lnd. Eng. Chem." 17, 605, 1925.
19. Murphree, „Ind. Eng. Chem.' 17, 747 (1925).
ГЛАВА III
ПЕРЕГОНКА С РАЗЛОЖЕНИЕМ (КРЭКИНГ И ПИРОЛИЗ)
Общеизвестно, что перегонка многих органических веществ
сопровождается их частичным или полным разложением. Угле-
водороды нефти в этом отношении не являются исключением, и
в тех случаях, когда перегонка нефти протекает при температу-
рах выше 300и, частичное разложение ее наблюдается вполне
явственно. Так как с понижением давления температура кипе-
ния жидкости понижается, то, как мы видели в предыдущей
главе, получение высококипящих нефтяных дестиллатов (масел)
производится при пониженном давлении. С другой стороны, со-
вершенно очевидно, что, повышая давление в перегонном аппа-
рате, легко достичь как раз обратной цели: температура кипения
жидкости повышается, а вместе с тем увеличивается возмож-
ность ее разложения при перегонке. Продуктами разложения
при такого рода перегонке нефти являются нардду с жидкими
дестиллатами также газообразные углеводороды ’ и твердый, не
перегоняющийся остаток (кокс); самому же процессу перегонки
нефти с разложением обыкновенно дают наименование крэкинг-
лроцесс или просто -крэкинг (англ, распад, расщепление). Ввиду
исключительно важного значения, -какое приобрела перегонка
с разложением в нефтяной промышленности за последние 15—
20 лет, рассмотрению ее химизма посвящена эта специальная
глава.
ОСНОВНЫЕ ВИДЫ ПЕРЕГОНКИ НЕФТИ С РАЗЛОЖЕНИЕМ
Различают несколько видов перегонки с разложением, полу-
чивших многообразное оформление в нефтяной промышленно-
сти, а именно:
1. Перегонка до кокса.
2. Крэкинг в жидкой фазе.
3. Крэкинг в паровой фазе.
4. Пирогенетичеокое разложение (пиролиз).
Не вдаваясь в их детальное 'рассмотрение с технологической
стороны, ограничимся краткой характеристикой, необходимой
для уяснения их химизма.
Перегонка до кокса. Этот простейший вид перегонки нефти
с разложением производится в кубах периодического действия
427
Фиг. 91. Схема куба для перегонки до
кокоа.
с утолщенным днищем (до 0,5 дм. толщины). Кубы — подвесного
типа для предоставления им большей свободы расширения и
сжатия при обогреве и остывании (фиг. 91). Кубы А снабжены
фракционирующей колонной В; однако пары по мере их утяже-
ления могут направляться по желанию также прямо в холо-
дильник С, минуя колонну. Рабочий цикл каждого куба — 48 час.
Сырьем для перегонки до кокса обычно служат мазут или
сырая нефть парафинового типа. Перегонка начинается обыч-
ным порядком с отбором
газолиновых, керосиновых
и соляровых фракций.
В этой последней фазе ха-
рактер перегонки Посте-
пенно начинает сущест-
венно изменяться: начи-
нается разложение (крэ-
кинг), которое постепенно
усиливается. Образую-
щиеся при этом пары
легких дестиллатов и газы,
пройдя колонну, напра-
вляются в холодильник;
дестиллаты сортируются
далее обычным порядком, а газы для улавливания наиболее
легких паров направляются в газолиновую установку. Посте-
пенно усиливая обогрев куба, доводят температуру жидкости'
в нем до 450° или несколько выше и заканчивают процесс.
Конец перегонки определяется появлением тяжелых желтых
паров, состоящих, невидимому, главным образом из высокомоле-
кулярных конденсированных систем типа антрацена, хризена
и др.
Крэкинг в жидкой фазе. Этот важнейший вид перегонки
с разложением получил свое промышленное оформление впер-
вые в 1915 г. (Бёртон в США). Основная идея его заключается
в том, что перегонка нефти или нефтепродукта ведется под по-
вышенным давлением, благодаря чему удается достичь значи-
тельно более высокого нагрева сырья и, как следствие, более глу-
бокого его крэкинга, чем при перегонке под обыкновенным давле-
нием. Выдающийся успех, выпавший на долю первых же уста-
новок жидкофазного крэкинга, вызвал появление громадного
числа систем и патентов, многие из которых получили практи-
ческое осуществление. Они .различаются между собой не только
деталями общей схемы и механических приспособлений, разра-
ботка которых потребовала в процессе развития крэкинга гро-
мадной изобретательности и труда; различие между ними за-
ключается нередко также в относительных величинах основных
факторов крэкинг-процесса, температуры (от 400 до 480°) и дав-
ления (от 5—6 до во ат и выше). Не вдаваясь в подробности,
рассмотрим вкратце важнейшие схемы промышленных установок
жидкофазного -крэкинга.
428
Авторами первых патентов на установки крекинга в жидкой
фазе были Бентон (1886 г.), Дъюар и Редвуд (1890 г.), Шутов и
Гаврилов (1891 г.). Практического осуществления их патенты не
получили.
Система Бёргошь Первая промышленная установка жидко-
состояла из толстостенного куба 1,
фазного крэкинга (фиг. 9*2)
в котором сырье, обычно. со-
ляровое масло, нагревалось
до 400—410° при давлении
4—5 ат. Образовавшиеся
пары из куба направлялись
в дефлегматор (на фиг. 92
не показан), откуда газы и
легкие пары уходили в хо-
лодильник 2, тяжелые же
части стекали
в куб. Вследствие
кой температуры
крэкинг протекал
медленно. Кроме
обратно
невысо-
обогрева
крайне
того, на
дне куба В зоне непосред- фиг, 92. Схема крэкинг-установки Бер-
ственного обогрева быстро тона.
оседал кокс, создавая опас-
ность местных перегревов. Для устранения этих недостатков
были предложены различные усовершенствования. Их сущность
сводилась к разрешению следующих первоочередных задач:
а) обособить .область непосредственного обогрева от той части
аппаратуры, где происходит собственно крэкинг, всегда сопут-
ствуемый коксообразованием;
Фиг. 93. Схема крэкинг-установки Елэрка.
б) усилить циркуляцию жидкости, особенно в местах непо-
средственного обогрева, и тем по возможности воспрепятствовать
скоплению в этих местах кокса.
Первая йе этих задач была решена обособлением и вынесе-
нием в топочное пространство обогреваемой части куба в виде
системы наклонных труб (установка Клэрка, фиг. 93).
429
Для .решений второй задачи куб одной из новейших устано-
вок этого типа (Дженкинс) снабжен пропеллером, работа кого-
рого обеспечивает усиленную циркуляцию крэкируемого сырья
Система Дженкинса. Куб этой системы (фиг. 94),
сколько установок которой имеются в СССР [1], снабжен
двумя цилиндрическими коллекторами А, А, соединенными
между собой как у Клэрка, системой непосредственно обогревае-
мых труб числом 120. В более длинном коллекторе находится
пропеллер, приводимый во вращение особым электромотором-
работа этого пропеллера обеспечивает движение сырья по обогре-
ваемым трубам установки со скоростью 3,5 м/сек. Из куба 2, ра-
ботающего в данном случае при 410—420° и 9—12 ат давления,
пары поступают в эвапоратор 5* Здесь происходит первое разде-
ление: тяжелая флегма стекает обратно в куб, более же легкие
Фиг. 94. Схема крэкинг-установки Джэюсипса.
пары направляются дальше и "через верхнюю часть эвапоратора
при давлении, пониженном до 2 ат, поступают во фракционирую-
щую колонну 7, где наступает второе разделение: газолиновые
пары через верхнюю часть колонны направляются в очистные
башни и далее в холодильники 8, Р; более же тяжелая флегма
стекает во второй испаритель 4. Здесь флегцд несколько обогре-
вается путем теплообмена и через дополнительную колонну
отдает свои легкие части, направляющиеся ио пути газолиновых
паров, тогда как более тяжелые части отводятся в особый холо-
дильник 8, 10.
Что касается тяжелых остатков, скопляющихся в кубе, то
через особый трубопровод они направляются из куба прежде
всего в расширительную камеру или первый испаритель 5.
Здесь вследствие понижения давления до 2 ат также происхо-
дит разделение: тяжелые части уходят в коксоотделитель 6 и
приемник 12, более же легкие части направляются в упомяну-
тый выше второй испаритель 4 и далее в особый холодиль-
ник 8, и, предварительно отдав во втором испарителе через
430
теплообменник часть своей теплоты для -нагревания флегмы
из фракционирующей колонны 1.
Сырьем для установки Дженкинса служат газойль или со-
ля|ювое масло. К ним щюдварительно прибавляют небольшое
количество гашеной извести (0,15—0,30%), назначение которой
двоякого рода: во-первых, нейтрализация кислых продуктов га-
зойля п соляра, необходимая для снижения их корродирующего
действия; во-вторых, содействие образованию кокса в более
рыхлом состоянии, благодаря чему он не столь плотно пристает
к стенкам аппаратуры
Рабочий цикл установки Дженкинса — не менее 30 дней,
после чего .в течение примерно 24 час. производится чистка ап-
паратуры от кокса. Выходы на газолин — не менее, чем в дру-
гих наиболее совершенных системах, т. е. около 50%.
Система Даббса. Эта система является одной из самых рас-
пространенных в США, а ее различные модификации позво-
Флг. 95. Схема крэкинг-установки Даббса.
ляют сделать выбор установки соответственно самым разнооб-
разным условиям и заданиям. Характерной особенностью си-
стемы Даббса (фиг. 95) является прежде всего полное обособле-
ние места обогрева крокируемого сырья от реакционной камеры.
Обогрев производится в трубчатой печи 1, откуда подаваемое на-
сосами 7. и 8 сырье, нагретое до 450—480°, направляется в хо-
рошо изолированную реакционную камеру 2— толстостенный
стальной цилиндр в несколько метров высоты. Здесь происходит
крэкинг: пары и газы отходят через верхнюю часть камеры У
и направляются во фракционирующую колонну 3, а кокс осе-
дает в камере и постепенно заполняет ее. Специальная система
труб 10 служит для контроля и регулировки уровня в камере 2.
Из колонны пары и газы последовательно поступают в холо-
дильник 4, газоотделитель 5, 11 и приемник б, 12; лишь в при-
емнике начальное давление (12—15 ат) снижается; таким об-
разом, реакционная камера этой установки, колонна и холо-
дильник работают под одним и тем же давлением, и в этом ее
вторая характерная особенность.
431
В описанной модификации системы Даббса происходит «крэ-
кинг до кокса»: жидкого крэкинг-остатка почти не получается
В таких условиях рабочий цикл установки невелик; при р&.
боте на мазуте и тяжелых нефтях он не превышает 6 дней, щ,
как по истечении этого времени реакционная камера омы-
вается заполненной коксом. Столь небольшой рабочий цикл
установки до известной степени искупается высокими выходами
на газолин, которые в зависимости от сырья достигают 60—70%
Для получения товарного продукта собираемый с установки
крэкинг-дестиллат должен быть подвергнут вторичной перегонке
и очистке.
Другие видоизменения системы Даббса работают с получе-
нием жидкого остатка. Последний выходит из реакционной ка-
меры и либо отводится сразу в холодильник, либо поступает
в испарительную камеру с пониженным давлением. Здесь на-
ступает его разделение: более легкие части его отходят и вместе
со свежим сырьем поступают на повторный крэкинг, остаток же
направляется .в резервуары и находит применение в качестве
котельного топлива. Само собой разумеется, что выходы на
газолин при подобного рода вариациях процесса значительно
ниже, чем при «крэкинге до кокса».
Система Кросса. Характерными признаками этой системы
являются:
а) По гное обособление процесса нагрева сырья в трубчатой
печи (как у Даббса), куда сырье нагнетается насосом 1
(фиг. 96) и откуда оно поступает в реакционную камеру 2—
горизонтально расположенный цилиндр из цельного куска
стали длиною 12 м при внутреннем диаметре 1 м и толщине
стенок 7,5 см.
б) Введение особой испарительной камеры (эвапоратора 3),
куда нагретое сырье поступает непосредственно из реакционной
камеры и где вследствие резкого снижения давления (до 2,5 ат)
происходит энергичное парообразование с разделением легких
частей от тяжелого остатка.
в) Наконец, благодаря снабжению установки мощной ректи-
фикационной колонной 4 (15,5 м высоты) установка дает гото-
вый продукт, не требующий вторичной перегонки.
Наряду с Даббсом установки Кросса (фиг. 96) -принадле-
жат к числу наиболее распространенных в США. Они работают
при высоком давлении (до 60 ат и выше); обогрев сырья—
в трубчатой печи достигает 450—470°, при выходе же из реакци-
онной камеры эта температура снижается до 410—420°. Рабо-
чий цикл при работе на газойле в среднем равен 9—12 дням;
однако в некоторых новейших установках этой системы продол-
жительность рабочего цикла удалась увеличить до 30—40 дней.
Выхода на галозин столь же высоки, как у Даббса; коксообра-
зование не превышает 1—2%.
К системе Кросса очень близка по своей конструкции си-
стема Виккерса. Две установки этой фирмы имеются в СООР
[2]. Это были первые промышленные установки крэкиша
432
в ССОР, сооруженные в 1927 г., одна —в Баку, другая —
в Грозном. В 1935 г. они были исключены из эксплоатации и
в настоящее время используются частью для вторичной пере-
гонки (Баку), частью же в качестве опытной (Грозный).
Характерной особенностью ряда описанных выше систем
является наличие в них реакционной камеры, в которой проис-
ходят задержка крэкируемого продукта при высокой темпера-
туре и крекинг его с выделением кокса. Накопление последнего
в реакционной камере и определяет одну из важнейших сторон
работы установки—продолжительность ее рабочего цикла. Вне-
Фиг. 96. Схема крэкинг-установки Кросса.
которых новейших жидкофазных установках различных систем
сделана поэтому удачная попытка обойтись вовсе без реакцион-
ной камеры. Удлинив дтя этого соответствующим образом труб-
чатую печь высокого давления, направляют из нее нагретое
сырье сразу в эвапоратор, несколько снижая при этом его тем-
пературу за счет разбавления его другим, более холодным
сырьем и тем приостанавливая дальнейшее развитие крэкинга
и слишком энергичное коксообразование. Таковы предпосылки
системы Винклер-Коха (фиг. 97), установки которой имеются
в СССР [3].
В самых общих чертах работа установки Винклер-Коха за-
ключается в следующем. Насос 1 подает мазут в теплообменник
2, расположенный при фракционирующей колонне 5. Здесь про-
исходит подогревание сырья до 280°, и с этой температурой оно
поступает в трубчатую печь низкого давления 3. Здесь оно на-
гревается до 405—110°, подвергаясь при этом лишь легкому
крэкингу, и переходит в эвапоратор 4, смешиваясь по пути
с нефтепродуктом из второй трубчатки 7, нагретым до 480° при
428
28 бак. 1622. Хигмип нефти.
28 ат давления. В эвапораторе температура смеси поддержи
вается около 410°, так что дальнейшее уг дубление крэкикга
здесь прекращаете^. Зато вследствие резкого снижения далле-
ния (до 2 ат) здесь происходит усиленное парообразование
Тяжелый остаток удаляется при этом в холодильник 8, тогда
как легкие фракции через верхнюю часть эвапоратора перехо-
дят в ректификационную колонну 5 и здесь подвергаются но-
вому разделению: пары пресс-дестиллата через верхнюю часть
Фиг. 97. Схема крэкинг-установки Винклер-Коха.
колонны уходят в холодильник ,9 и сепаратор 10, откуда часть
пресс-дестиллата возвращается в виде орошения 6; керосиновые
же и газойлевые части через нижнюю часть колонны возвра-
щаются в печь высокого давления для повторного крэкиро-
вания.
Отложения кокса в установке Винкмр-Коха ничтожны, бла-
годаря чему рабочий цикл достигает 40 дней и больше. Выходы
пресс-дестиллата — до 44%.
Согласно проекту установки Винклер-Кота должны были давать
175 т/сутки пресс-дестиллата с глубиною крэкинга за однократный пропуск
17%. При этом температура на выходе из трубчатой печи должна была
держаться порядка 480—490°. Стахановцы наших крэкинг-заводов не-
сколько изменили режим установки и добились крупных успехов. Да-
вление в насосе было повышено с 40 до 45—60 ат, давление на выходе
из печи было увеличено до 35 ат, температура же на выходе из большой
печи была повышена до 505°. В этих условиях глубина крэкинга за один
пропуск повысилась с 17 примерно до 25%, т. е. почти в полтора раза.
Тем самым удалось существенно снизить количество сырья для повтор-
ного крекирования («рисайкл») и, следовательно, увеличить производитель-
ность установки как по сырью (с 500 до 750 т/сутки и выше), так и сто
пресс-дестиллату (с 175 до 300 т/сутки).
Современные крэкинг-установки представляют собою весьма
сложные сооружения, в которых обыкновенно производят сна-
чала отбензинивание сырой нефти, а затем крекинг остатка. Их
пропускная способность достигает 1500 т/сутки сырой нефти и
больше. Основными элементами их является система теплооб-
менников и трубчатые печи (не меньше 2), реакционная камера
и эвапоратор, 2 ректификационных колонны (одна для сырья,
другая для продуктов крэкинга), холодильники и насосы. Тем-
434
иература в трубчатых печах достигает 495—510° и выше, в ре-
акционной камере — несколько ниже (480—485 ). Давление —
более или менее высокое, постепенно снижающееся от печей
к ректификационной колонне и приемнику.
Основные тенденции в развитии современной крокинг-аппа-
ратуры сводятся к следующему [4]:
1. Раздельное крэкирование различных фракций сырья дтя
повторного крэкинга (рисайклов) с целью добиться возмож-
ности довести глубину распада каждой фракции до возможного
предела.
2. Возможное углубление крэкинга тяжелых частей исходного
сырья и соответственное увеличение выходов бензина путем
разбавления тяжелого сырья и перенесения части процесса
в реакционную камеру, что уменьшает рисайкл, разгружает печи
и увеличивает производительность установки.
3. Применение повышенных давлений в погоноразделитель-
ных устройствах крэкинг-установки; это позволяет снизить со-
держание бензиновых паров в отходящих газах, а также при не-
котором оптимуме давления добиться максималы^й темпера-
туры для поступающего в печь рисайкла, что в свою очередь
ведет к снижению тепловой загрузки печи.
4. Возможное углубление отбора рисайкла и соответственно
возможное утяжеление остатка, что также позволяет несколько
поднять выход бензина на сырье.
5. Возможное углубление крэкинга за цикл, что должно
привести к снижению кбэфициента рисайкла, в основном опре-
деляющего пропускную способность установки.
Ниже дается схема одной из новейших установок Даббса
с производительностью до 1700 т калифорнийской нефти
в сутки (фиг. 98); выходы крэкинг-бензина в зависимости от
сырья’ колеблются от 49 до 549', считая на отбензиненную
нефть [б].
Крэкинг в паровой фазе. Важнейшими недостатками крэ-
кинга в жидкой фазе являются:
а) необходимость работать под давлением и, как следствие,
применение особо прочной, дорогой аппаратуры;
б) сравнительная медленность ]>еакции и вытекающая от-
сюда необходимость повторного крэкирования одного и того же
материала.
Если представить себе, однако, что сырье, подлежащее крэ-
кингу (керосин, газойль), превращено сначала в парообразное
состояние, а затем эти пары нагреты до температуры всю—650
при которой их крэкинг должен заканчиваться практически
мгновенно, то в этих условиях, естественно, всякая нужда
в аппаратуре высокого давления уже отпадает. Эти соображе-
ния лежат в основе третьего вида перегонки с разложением —
крэкинга в паровой фазе [6].
Преимущества парофазного крэкинга перед жидкофазным,
особенно возможность работать при атмосферном или немного
лишь повышенном давлении, выявляются в настоящее время
2Ь* 1 435
все с большей и большей определенностью. Однако, несмотря
на эти преимущества, развитие и продвижение в промышлен.
иость крекинга в паровой фазе происходит крайне медленно: до
сих пор число установок парофазного крэкинга исчисляется
единицами, тогда как число установок жидкофазного крэ-
кинга — многими сотнями. Основная причина такого несоответ-
ствия заключается прежде всего в тех трудностях, с которыми
сопряжено экономическое нагревание до столь высоких темпе-
ратур парообразных веществ при их крайне малой теплопровод-
ности. Отсюда громадный перерасход топлива при крэкинге
в паровой фазе: в то время как новейшие системы жидкофаз-
Фиг. 98. Схема новейшей крэкинг-установки Даббса:
1 —сырьевой насос; 9 и з—теплообменники; 4 — отстойник; 6 —теплообменник для крэкинг-
остатка; б—пародестиллатный теплообменник; 7 —колонна для отбензинивания нефти; в—ков-
деноатор бензина прямой гонки; S — неконденсирующийся газ; ю—бачки для бензина прямо!
голки; 11—бензин прямой гонки; 19 — стриааинг для лигроина прямой гонки; 13 и 14—холо-
дильник для лигроина и лигроин прямой гонки; is—отбензиненная нефть; 16—ректяфям-
ционная колонна для крекинг-продуктов; 17 — стриппинг для крэкинг-лигроииов я керосинов;
18—горячий насос; 19—печь для глубокого крекинга (температура 510°. давление 22,4 от);
«о —тяжелая флегма; 91 и 99 —наооо и печь для утежеленного сырья (температура 496°; дав-
ление 18 ат); 93 — общая реакционная камера; 94 и as — вентили; £6 —эвапоратор; 97—испари-
тель для крэкинг-остатка под сниженным давлением; 98—конденсатор; 99 — гае; 80—приемник;
31 — васоо для крэкинг*остатка; 39— холодильник; 33 — крэкинг-остаток; 34—пары крэкивг-бон-
аина; Эб —холодильник для крэкияг-5ензияа;-3в—газосепаратор; 37 — крэкннг-бензин на отабв-
лизацию; 38 — газ на абсорбцию.
кого крэкинга расходуют на топливо хге больше 3—5% от пере-
рабатываемого сырья, при парофазпом крэкинге по этой статье
расходуется нередко свыше 20% сырья. Отсюда ясно также, что
одним из решающих моментов в проблеме парофазного крэкинга
является момент теплотехнический, а именно — вопрос о радио-.
калькой конструкции печи для нагревания крэкируемого сырья
в парообразном состоянии.
Гораздо меньшее значение имеет в настоящее время ряд
других особенностей парофазного крэкинга, которые совсем еще
недавно расценивались как крупные его недостатки. Указывалось,
например, на чрезмерно большое газообразование при парофаз-
ном крэкинге (до 25% газа). Однако в настоящее время, как
будет показано в конце этой главы, газы от крэкинга в паровой
436
производится предварительный подогрев
Фиг. 99. Схема крэкинг-установки Джайро.
фазе приобретают самостоятельное значение как ценнейшее
сырье для их последующей химической переработки. Обраща-
лось также внимание на значительные трудности и потери,
с которыми сопряжена очистка парофазного бензина. Однако
трудности эти с избытком покрываются высокими антидетона-
ционными качествами парофазниго крекинг-бензина, потери же
при условии применения новейших методов очистки бензинов
крэкинга и пиролиза (гидроочистка) совершенно отпадают.
Важнейшими системами парофазного крэкинга в настоящее
время являются системы Джайро (Gyro), Нокс (Кпох) и
де-Флорез (de Florez).
Система Джайро. В парофазной установке Джайро (фиг. 99)
крокируемый продукт проходит последовательно две печи: эко-
номайзер 6, в котором
до 370°, и конвертер
5, где температура
паров доводится уже
до 590°. По выходе
из этой последней
печи перегретые па-
ры направляются
в особую камеру, ар-
рестер 3, где они
встречаются со све-
жим сырьем из бака
1, которое подается
насосом 2. Здесь
температура смеси
сразу падает до 315°
и всякий дальней-
ший крэкинг, есте-
ственно, приостанав-
ливается. Пз арре-
стера смесь жидко-
сти и паров переходит в ректификационную колонну 4
для разгонки: дестиллатные пары с пределами кипения по зада-
нию уходят через верхнюю часть колонны по направлению к хо-
лодильнику 7, газоотделителю 8 и сборнику 9; конденсат же
забирается насосом и перекачивается в экономайзер 6, причем
избыток его автоматически может ‘быть возвращен в сырьевой
бак 1. Регулирование работы колонны 4 производится с по-
мощью парциального конденсатора 13, который может дагь весь
необходимый в производстве ^водяной нар. Разделение воды и
пара производится в особом сепараторе 14; вода подается из во-
дяного резервуара 15 с помощью насоса 16.
Как уже было отмечено, продукт выходит из экономайзера
с температурой около 370 ; теперь он направляется в эвапора-
тор 10, где происходит второе разделение: остаток со дна эва-
поратора через теплообменный аппарат и холодильник 11 от-
водится в мазутный резервуар 12; пары же, подлежащие крэки-
437
рованию, направляются в конвертер, причем в эвапоратбде
к ним подмешивается около 3% перегретого водяного пара.
В конвертере’ смесь углеводородных и водяных паров рас-
пределяется сначала по трубам, работающим параллельно, за-
тем переходит в секции, соединенные и работающие в последо-
вательном порядке. Здесь’и протекает процесс парофазного кре-
кинга, причем в трубах, работающих параллельно, пары прихо-
дят в соприкосновение с контактной массой из смеси окиси и
закиси железа. Повидимому, роль этого контакта заключается
в следующем: за счет кокса, оседающего в трубах конвертера,
окись железа восстановляется в закись, последняя же оки-
сляется кислородом диссоциировавшей воды обратно до окиси;
при этом в процессе восстановления окиси железа углерод
(кокс) превращается в окись углерода:
Fe208-pC = 2Fe0 + C0;
Н20^Н2 + 0;
2FeO + 0 = Fe203.
В пользу этой схемы говорит то обстоятельство, что в газах,
получаемых в процессе Джайро, действительно содержатся во-
дород и окись углерода, а также продукт дальнейшего окисле-
ния последней, углекислота.
Установка Джайро дает около 55—00% газолина, 15% остат-
ков (мазута) и 25—30% газа, содержащего до 40% непредель-
ных углеводородов. Обычным сырьем служит газойль.
Система Нокса. Эта система парофазного крэкинга инте-
ресна по применяемому методу обогрева паров крэкируемого
сырья: обогрев до требуемой температуры (около 560°) произво-
дится смешением паров нефтепродукта (обычно газойля) с горя-
чим инертным газом, сильно нагретым в специальной печи.
В качестве переносчика тепла от печи к парам крэкируемого
продукта можно применять различные газы, в частности есте-
ственный газ (метан), азот, дымовые газы, предварительно осво-
божденные от остатков кислорода и водяных паров и т. п. По-
дробное описание системы Нокса см. [?].
Как ясно из вышеизложенного, характерная особенность
парофазного крэкинга заключается в том, что процесс проте-
кает в данном случае при высокой температуре и низком да-
влении. В настоящее время, однако, почти повсеместно пришли
к выводу, что проведение крэкипг-тгроцесса в этих условиях не
рационально, и основная тенденция в дальнейшем развитии
крэкинг-процесса сводится к работе при повышенных давле-
ниях. Эта тенденция определяется следующими соображениями:
1. При крэкинге под низким давлением выход бензина
меньше, а выход газа больше, чем при крэкинге под повышен-
ным давлением. Одной из причин того различия является поли-
меризация простейших олефинов, которая под давлением,
естественно, протекает легче. С другой стороны, повидимому,
давление, способствует преобразованию осколков молекул (ра-
дикалов) в молекулы, тогда как при низком давлении они пре-
438
ления удельный объем паров и газов
ик. Вследствие этого производитель-
ВмдоВ Zffjo В fm ёаЗойля
Фиг. 100. График выхода бензина — выхода
газа при различных давлениях.
имущественно распадаются дальше до метана и этана. Фиг 100
может иллюстрировать это положение; на ней даны кривые за-
висимости выхода бензина (в процентах по объему) при одно-
кратном крэкинге от выхода газа при различных давлениях I8 ].
2. Бензин крэкинга под низким давлением значительно
богаче непредельными, чем бензин крэкинга под повышенным
давлением.
3. В силу низкого ;
оказывается слишком г
ность установки не мо-
жет быть достаточно
большой, так как для
этого потребовалась бы
аппаратура (колонны,
эвапораторы) чрезмер-
ной величины.
4. Вследствие низ-
кого давления в колон
пах загрузка поступает
в печь с низкой тем-
пературой; в резуль-
тате в печи сырью при-
ходится сообщать много
тепла и расход топли-
ва значительно выше,
чем при крэкинге под
давлением. Так напри-
мер, при высоком давлении в колоннах сырье поступает в печь
при температуре порядка 380° и нагревается в лечи до
510—520°, т. е. всего на 130—140°; в установках же парофаз-
ного крэкинга сырье подается в печь с температур не выше
200—260° и должно нагреться в печи др 530—540°, т. е. на
300—340°.
Пирогеиетическое разложение нефти (пиролиз). Перегонку
нефти с разложением можно вести при еще более высоких тем-
пературах, чем те, которые характерны для парофазного крэ-
кинга. В этом случае как самый процесс, так и его техническое
оформление приобретают ряд специальных особенностей, кото-
рые дают основание выделить новый тип перегонки с разложе-
нием— пирогенетическое разложение или пиролиз нефти [9].
При очень высоких температурах (около 1000°) нефть на-
цело разлагается на углерод и водород, но эта форма пиролиза
нефти не находит пока практического применения. При более
низких температурах наряду с газом и твердыми продуктами
пиролиза нефти (сажа, кокс) начинают получаться также жид-
кие продукты, образуя так называемую нефтяную смолу или
деготь.
По своему химическому составу нефтяной деготь -резко отли-
чается от исходного сырья, нефти. Полученный в надлежащих
условиях температуры (около 700°), он в главной своей части
439
состоит из ароматических углеводородов и может служить ис-
точником их промышленного получения. Таким образом опре-
деляется специальный вид термического разложения нефти,
пирогенетическое разложение или пиролиз, который может быть
назван по его целевому продукту ароматизацией нефти.
Пирогенстичсское разложение нефти нашло практическое применение
прежде всего как способ получения горючего газа. Этот нефтяной газ еще
в 70-х годах получил довольно широкое распространение, и у нас, напри-
мер, им освещались такие города, как Киев, Казань и др. А. Летний
(1878 г.) впервые показал [10], что дегтеобразный продукт, образующийся
при получении нефтяного газа, богат ароматикой, и, таким образом, поло-
жил основание переработке нефти па ароматические углеводороды. В 1885 г.
на кустарно-промышленной и сельскохозяйственной выставке в Нижнем-
Новгороде уже были выставлены образцы бензола, нафталина и антрацена,
полученные из нефтяного дегтя на заводе Рагозина. Позднее вопросом об
ароматизации нефти много занимался Никифоров, работа которого привела
к организации завода, где пз нефтяного бензола получали нитробензол, ани-
лин и другие продукты. Наконец, в последнюю войну недостаток ароматиче-
ских углеводородов для изготовления .взрывчатых веществ заставил обратить
на пирогенетическое разложение нефти особое внимание, и в Баку на раз-
личных заводах было поставлено несколько крупных установок для пере-
работки нефти н^ нефтяной деготь и далее на бензол и толуол.
Новейшая практика пиролиза нефти с целью получения’ аро-
матических углеводородов была разработана главным образом
в Баку во время войны (1916—1917 гг.). Аппаратура, применяе-
мая в этом процессе, принадлежит к двум основным типам: ре-
торты и генераторы. Реторты, применяемые для пиролиза нефти,
бывают двух (Видов: чугунные или стальные горизонтальные ре-
торты типа Пинча и вертикальные шамотовые реторты типа
Пиккеринга.
Фиг. 101. Схема установки для пиролиза нефти. Реторты Пинча.
Металлические реторты имеют круглое или овальное сечение
либо они—сводчатого характера. Обычно реторта состоит из
3 соединенных последовательно, расположенных друг над другом
колен, длиной 2 м каждое при. диаметре 30 см. На концах ре-
торты приспособлены герметические крышки, из которых верхняя
имеет вводную трубу для подачи сырья, нижняя же — выводное
отверстие, соединенное с гидравликой 2, для продуктов пиролиза.
Реторты 1, сблокированные по две — три* в одной печи (фиг. 101),
обогреваются топочными газами от сжигания газообразного или
440
жидкого топлива. Температура различных колен реторты не оди-
накова. Верхнее колено, где происходит главйым образом испа-
рение сырья, редко нагревается выше 4'20"; в нижнем же колене,
где протекает главный процесс пиролиза, температура достигает
670—680температура среднего колена обычно около 500—550°.
В гидравлике, куда продукты пиролиза попатают из нижнего
колена реторты, оседает гидравлическая смола, которую посте-
пенно отводят в особый сборник; газообразные же и парообразные
продукты пиролиза направ-
ляются последовательно в холо-
дильники, воздушные 3 и водя-
ные 4, и скруббера 5 для улавли-
вания наиболее летучих арома-
тических углеводородов. По вы-
ходе из последнего скруббера
г.аз практически уже не содер-
жит паров бензола; такой газ на-
правляют в газгольдеры и топки.
Пропускная способность од-
ной трехколенчатой реторты —
до 16 кг/чая. Продолжитель-
ность ее работы в среднем около
48 час. По истечении этого вре-
мени реторта очищается от кокса,
после чего она снова вступает
в работу. Расход топлива — 25%
от сырья.
Шамотовые реторты типа
Пиккеринга (фиг. 102) имеют
круглое сечение диаметром 30 сл;
высота их 3 л. В количестве
6—8 штук эти реторты А объе-
диняются в одну башню — печь,
обогреваемую форсункой Б. По-
дача сырья производится сверху,
выход же продуктов писолиза —
внизу — в шамотовый горшок
с отводом в гидравлику К, откуда
Фиг. 102. Печь Пиккериню.
нары и газы направляются в хо-
лодильники и скруббера. Выделяющийся кокс довольно быстро
засоряет реторты и примерно один раз в сутки должен выжи-
гаться; механическое удаление его в данном случае невозможно
ввиду хрупкости реторты.
Пропускная способность реторты описанного выше типа — •
14—16 кг нефти в час при средней пиро генез ирующей поверх-
ности 22 000—30 000 см2; расход топлива—19—21% по отно-
шению к сырью.
Генераторы. Генераторные установки, широко применяемые
для газификации разных видов топлива, твердого (уголь, торф
и т. п.) и жидкого (нефть и др.), весьма разнообразны. Нефтяной
441
Фиг. 103. Генератор.
генератор (фиг. 103) в главной своей части состоит из двух ци-
линдрических башен 1 и 2 (высота 6 м, диаметр около 2 .it), вы-
ложенных внутри шамотовым кирпичом с такой же насадкой
В своей нижней части обе башни соединяются между собой w
рокой трубой. Первая башня (испаритель) в верхней своей части
имеет штуцер с форсункой, подающей нефть; зажигая послед-
нюю, нагревают продуктами ее сго-
рания обе башни, причем топочные
газы выходят в трубу через особый
лаз 3, находящийся в верхней части
второй башни (перегреватель).
Когда температура внутри системы
поднимается до 700°, прекращают
действие форсунки и через особый
вентиль 4 начинают подавать в ис-
паритель нефть, предварительно
закрыв лаз 3 для выхода топочных
газов. Попадая на раскаленную
насадку, нефть испаряется и в пе-
регревателе подвергается оконча-
тельному пиролизу, продукты кото-
рого, пройдя гидравлику, поступают
в холодильник и т. д.
Примерно через полчаса после
начала -пиролиза температура вну-
три генератора настолько падает
(примерно до 650°), что дальней-
-----------------------[ прянем направлении становится неце-
лесообразным/ Тогда прекращают подачу нефти через вентиль
4 и вновь зажигают форсунку для подогрева. генератора и вы-
жигания осевшего кокса; на это требуется также около полу-
часа, после чего, прекратив действие форсунки, можно вновь
возобновить подачу нефти для пиролиза и т. д. Таким образом
работа генератора протекает периодически.
Пропускная способность генератора указанных выше разме-
ров превышает 30 кг нефти в час при расходе топлива около
10%, считая па сырье. Однако выхода на ароматические углево-
дороды на генераторных установках значительно меньше, чем на
ретортных (см. ниже).
шее. ведение процесса -в
ПРОДУКТЫ ПЕРЕГОНКИ НЕФТИ С РАЗЛОЖЕНИЕМ
Как видно из вышеизложенного, продуктами перегонки нефти
с разложением являются газообразные вещества, жидкие масла
и кокс. Выхода, состав и относительное значение каждого из
этих продуктов при различных видах перегонки* с разложением
далеко не одинаковы и . должны быть рассмотрены поэтому от-
дельно.
Газообразные продукты. Количество и состав газообразных
412
продуктов, образующихся при различных видах перегонки с раз-
ложением, зависят от условий процесса.
При перегонке до кокса, т. е. в наиболее мягких условиях
крэкинга, выхода на газ не превьфгают, считая на сырье, 1,5—2%.
Естественно, что газы эти не могут представлять самостоя-
тельного значения и после извлечения газолина направляются
в топки. То же относится и к газам жидкофазного крэкинга.
хотя газообразование в этом случае несколько выше, чем при
перегонке до кокса, и в некоторых случаях достигает 5—6%,
считая на сырье.
В отношении своего состава газы жидкофазного крэкинга
также не представляют особого интереса. В главной своей массе
(75—80% по объему и выше) они состоят из углеводородов ряда
метана; содержание в них наиболее реакционно способных и
поэтому практически наиболее интересных компонентов — непре-
дельных углеводородов — редко превышает 18—20% по объему.
Несравнимо большее значение имеют газообразные продукты
парофазного крэкинга и пиролиза. Содержание непредельных
углеводородов в этих газах поднимается до 30—35% (по объему)
от их состава и выше, общие же выхода на газ от исходного
сырья для некоторых видов парофазного крэкинга (Джайро) до-
стигают 25—30%; при пирогенетическом разложении нефти вы-
хода на газ — еще больше, поднимаясь для ретортных систем
до 45—50% на сырье. Столь высоцре газообразование при ра-
боте на жидкие продукты крэкинга и пиролиза ложилось бы
тяжелым балластом на эти технические процессы в целом, если
бы за последнее время для газов, богатых непредельными угле-
водородами, не была найдена новая специальная область их
применения, а именно: химическая переработка их на такие
ценные органические соединения, как спирты, гликолы и их про-
изводные. Этот важный вопрос будет подробно освещен ниже,
в конце настоящей главы.
Жидкие продукты. Целевым продуктом крекинга является
прежде всего крэкинг-бензин, представляющий собою после со-
ответствующей очистки (ч, III) прекрасное топливо для двига-
телей внутреннего сгорания. Не только выхода на этот продукт,
но и его состав в высокой степени зависят от оформления крэ-
кинг-процесса и от таких факторов, как температура, давление,
сырье и т. и.
При перегонке до кокса общие условия процесса и прежде
всего температура недостаточны для углубленного крэкинга, и
главными продуктами процесса являются в данном случае не
крэкинг-бензин, а более тяжелее погоны — керосиновые и мас-
ляные. Эти последние могут служить в качестве моторного топ-
лива или же применяются в качестве материала для последую-
щего жидкофазного крэкинга. Следующие, еще более тяжелые
дестиллаты, парафинистый и масляные, либо поступают на
фильтр-ттрессы для переработки на парафин и смазочные масла,
либо также идут на крэкинг. Наконец, отмеченный выше (стр. 428)
последний погон, получаемый в количестве всего 1—1,5%
443
сырья, имеет характер смолы или пека (gum, still wax, wax
tailings); он находит применение для пропитки дерева и для
изоляционных целей.
Для жидкофазного и лароф^зного крэкинга бензин является
уже фактически целевым продуктом процесса. В количественном
отношении важнейшие современные виды жидкофазного крэкинг.
процесса дают примерно одни и те же выхода на крэкинг-
бензин, достигающие в лучших современных системах, считая на
сырье, 50% и выше (до.70%). В качестве сырья в подавляю-
щем большинстве современных систем крэкинг-процесса служит
газойль и другие дешевые дестиллаты. Одной из основных задач
дальнейшего развития крэкинг-процесса является возможное
расширение его сырьевой базы в ту сторону, чтобы сырьем для
него могли служить также тяжелые нефти и мазут. В этом на*
правлении в настоящее время уже достигнуты существенные
успехи (системы Даббса, Винклер-Коха), но в основном непо-
средственный жидкофазный и парофазный крэкинг мазута и
тяжелых нефтей пока еще продолжает оставаться одной из оче-
редных проблем крэкинга будущего.
В отношении своего состава жидкофазный и парофазный бен-
зины различаются между собой довольно значительно. Бензины
жидкофазного крэкинга примерно на 30—35% состоят из.не-
предельных углеводородов; ароматики в них не больше 15—20%;
остальное приходится на додю углеводородов предельного харак-
тера, т. е. парафинов и нафтенов. Парофазпые крэкинг-бензины
содержат непредельных углеводородов значительно больше, чем
жидкофазные, а именно — иногда до 40—45%; содержание в них
ароматики также поднимается до 40-—45% (в отдельных слу-
чаях до 60%) при соответствующем снижении нафтенов и па-
рафиновых углеводородов. Понятно поэтому, ^гго по своим анти-
детонационным свойствам парофазный бензин значительно пре-
восходит бензин жидкофазный. Его «октановое число» в зависи-
мости от исходного сырья и системы крэкинга колеблется от
71—76 (де-Флорез) до 84—88 (Джайро). Этот бензин предста-
вляет собою поэтому весьма ценный продукт для подмешива-
ния к бензину прямой гонки в целях повышения его антидето-
национных качеств.
Переходя к крэкинговым погонам с температурой кипения
выше 200°, следует отметить, что использование их по анало-
гии с соответствующими фракциями прямой гонки встречает
ряд серьезных трудностей. Так например, крэкинг-керосины (тем-
пература кипения 200—300°) в отличие от керосина прямой
гонки мало пригодны в качестве осветительного материала, так
как-дают слишком большое нагарообразование. Применение со-
ответствующих погонов (крэкинг-керосин и крэкинг-газойль)
в качестве тракторного и моторного топлива также встречает
известные трудности ввиду содержания в этих погонах весьма
склонных к осмолению непредельных ' углеводородов; все же,
однако, показано [11], что крэкинг-керосины с концом кипения
300° (до 200е — ю%) после очистки 1% технической серной
444
кислоты и вторичной перегонки могут применяться в качестве
тракторного топлива как в неразбавленном виде, так и в смеси
с бакинским керосином прямой гонки. Как смазочный материал
крэкинг-масла, очевидно, мало пригодны уже вследствие своей
легкой окисляемости. Ввиду всего изложенного керосиновые и
соляровые крэкинг-фракции обычно идут в качестве материала
на повторный крэкинг; остатки же применяются как котельное
топливо, причем, однако, содержание в них мелкораздроблен-
ного 'взвешенного оседающего кокса может вызывать иногда и
здесь ряд затруднений при хранении крэкинг-остатков, особенно
же при их перекачке.
Особое место среди жидких продуктов перегонки с разложе-
нием занимают продукты пиролиза нефти. В этом случае глав-
ным целевым продуктом являются ароматические углеводороды,
из которых в настоящее время этим путем получают в промыш-
ленном масштабе бензол, толуол и нафталин.
Для .выделения бензола и толуола нефтяной деготь из гидравлики и
холодильников, а также скрубберное масло, абсорбировавшее наиболее ле-
тучие продукты пиролиза, не сконденсировавшиеся в холодильниках, под-
вергают отдельно разгонке из куба периодического действия. Продуктами
этой разгонки являются:
Темп. кпп. Уд. вес
Легкое масло.................... до 170°
Среднее (нафталиновое) масло . . . 170—240°
Скрубберное масло............... 240—300°
0,845-0,875
0,900
0,950
Вбсь бензол и толуол содержатся, конечно, в легком масле, которое,
для выделения этих ароматических углеводородов, подвергается следую-
щей переработке. Прежде всего подвергают легкое масло ректификации
с целью выделения более или- менее узких бензольной и толуольной фрак-
ций (первая ректификация). Ректификацию ведут на колонном аппарате
либо с водяным паром, руководствуясь при отборе фракций их удельными
весами, либо без пара после предварительной вторичной перегонки легкого
масла на обыкновенном кубе для отсечения тяжелого хвоста. В этом по-
следнем случае легкое масло- разбивается в результате ректификации по
температуре кипения на следующий ряд фракций:
Головная часть................до 73°
Легкий бензол............... 73— 79'* .
Бензол сырой..................79— 83°
Бензол-толуол................ 83—108°
Толуол сырой.................108—113°
Тотуол-ксилол................113—122°
Ксилол сырой................ 122—150°
После ректификации главная масса бензола оказывается сосредото-
ченной во фракции 79—83° («бензол сырой»), толуол же — во фракции
108—113° (толуол сырой»). Так как фракции эти содержат еще значи-
тельное количество непредельных углеводородов, то для получения чистых
бензола и толуола их подвергают прежде всего очистке 98%-ной серной ки-
слотой с последующей промывкой водой и щелочью. Общий расход серной
кислоты достигает при этом 10—15%, потери же составляют примерно 14—
15% от подвергаемого очистке продукта.
Очищенные и промытые бензол и толуол подвергают, наконец, новой
ректификации па мощном колонном аппарате высотою до 6 л (чистая
ректификация). Задача этой последней операции заключается в возможно
полном разделении бензола и толуола друг от друта и в освобождении их
445
от продуктов полимеризации и конденсации, всегда образующихся при
сернокислотной очистке продуктов крэкинга и пиролиза. В результате по-
лучают бензол и толуол со следующими свойствами:
Бензол
Толуол
Темп. кип.
. . 79,4— 80,4°
. . 109,7-110,5°
Уд. вес
0 8806
0,8682
Промежуточные фракции по мере их накопления подвергаются повтор-
ной ректификации.
Хорошо очищенные нефтяные бензол и толуол по своим ка-
чествам должны быть поставлены выше соответствующих угле-
водородов из каменноугольного дегтя. В отличие от каменно-
угольных бензола и толуола они не 'Содержат тиофена и его го-
мологов: реакция с изатином дает отрицательный результат; на
99—юо% они растворяются в серной кислоте: бензол —в ды-
мящей (10% 80з), толуол — в 98%-ной; выдерживают хамелео-
новую пробу, т. е. не обесцвечивают водно-спиртового раствора
марганцевокислого калия, бензол — в течение 1 мин., толуол—
в течение 70 сек. Весьма существенным их недостатком является,
однако, их высокая себестоимость, зависящая как от затраты
больших количеств серной кислоты при их очистке, так и от
громадных отходов, получаемых в процессе самого пиролиза.
Лишь за последнее время утилизации этих отходов начинает
делать заметные успехи, значительно снижая себестоимость
нефтяных бензола и толуола.
Из жидких масел, получаемых при пиролизе нефти, кроме
бензола и толуола, в настоящее время выделяются и находят
применение следующие продукты:
Головная фракция (амилены). Эти наиболее легкие фракции
продукта пиролиза нефти (температура кипения до 70е) содер-
жат около 60% непредельных углеводородов, главным образом,
изомерных амиленов (С^Ню) и гексиленов (C«Hi2), которые
могли бы быть использованы для получения путем гидратации
амиловых и гексиловых спиртов. Кроме того, те же погоны со-
держат заметное количество изопрена, т. о. углеводорода, кото-
рый мог бы служить исходным материалом для получения синте-
тического каучука. Однако утилизация головной фракции в этих
интересных направлениях—дело будущего, хотя в этой области
рте проделана большая работа частью в лабораторном, частью
r полузаводеком масштабе [12]; в настоящее же время пере-
работка -головной фракции идет по несколько иному пути,
а именно — по пути приготовления из нее так называемой лак-
олифы (лаколь).
Приготовление этого суррогата олифы заключается в следую-
щем. Сначала обрабатывают головную фракцию отработанной
серной кислотой после очистки бензола и толуола; затем после
отделения кислоты промывают углеводородный слой водой и от-
работанной щелочью, В результате получается сложная смесь
446
непредельных полимеров, обладающая способностью к аутоксй-
дации, т. е. к медленному присоединению кислорода воздуха
с образованием лакообразных продуктов. Лак-олифа применяется
для покрытия железа и в других случаях вместо настоящей
олифы.
Другой, более совершенный способ получения суррогата
олифы заключается в обработке бензольной готовки 1—2% хло-
ристого алюминия [13]. Реакцию проводят в течение 2—4 час.
при температуре 10—20° в железной аппаратуре и получают
вязкое, высокомолекулярное высыхающее масло, которое в тон-
ком слое образует пленку и может служить суррогатом олифы.
Выход высыхающих полимеров из бензольной головки достигает
45—50%; остальные 50—55% составляет бензол, который в ука-
занных условиях практически не участвует в реакции и может
быть выделен из ее продуктов путем ректификации с последую-
щей доочисткой серной кислотой.
Следует отметить, впрочем, что получаемый этим путем смолистый
продукт по своим свойствам оказался более подходящим в качестве весьма
ценного сырья для лакокрасочной промышленности, чем в качестве заме-
нителя олифы, так как здесь прежде всего имеется глубокое различие в хи-
мическом составе: олифа представляет собою, как известно, специально
обработанное растительное масло (льняное, конопляное и т. и.), тогда как
указанные выше заменители олифы являются смесью высокомолекулярных
полимеров, образовавшихся в результате процесса полимеризации непре-
дельных углеводородов бензольной головки. Не подлежит сомнению, что
возможность получения из бензольной головки значительных количеств
полимерных смол, оказавшихся применимыми для изготовления масляных
лаков, смолок, сургучей, а также в электротехнической промышленности,
в частности для изготовления кабельной массы, открывают для процесса
пиролиза нефти совершенно новые техно-экономические перспективы.
Химический состав бензольной головки весьма сложен и пока еще
недостаточно выяснен [13]. Кроме нахождения в ней бензола (до 60%) п
низших олефинов, различными авторами доказано несомненное присут-
ствие здесь значительного количества диеновых углеводородов, а именно:
бутадиена (I), 0-метилбутадиена (изопрена, И) и особенно циклопентя-
диена (Ш):
I. СН2 = СН—СН = СН2;
И. СН„ = СН-С = СН2;
Сп3
СН —СН
Hl. II
сн
СИ
Бензин пиролиза. При выделении бензольной и толуольной
фракций '(первая ректификация) всегда получается значитель-
ное количество богатых ароматикой побочных промежуточных
фракций. После их повторной ректификации в отходах остаются
погоны, которые, будучи очищены и смешаны в надлежащих
пропорциях для получения плавной кривой выкипания, дают
бензил высоких антидетонационных качеств. Его применяют
в качестве добавки для повышения антидетонационных качеств
бензинов прямой гонки.
447
Изменяя режим работы реторты, металлической или шамот-
ной, можно сделать .бензин пиролиза уже не побочным, а целе-
вым продуктом пирогенетического разложения нефти. В качестве
сырья при этом применимы сырая нефть или любой нефтепро-
дукт; температурные условия пиролиза — обычные (около 700°),
но подача сырья должна быть увеличена с 14—16 кг на. реторту
в час до 70 кг. В этих условиях удается получать до 20—35%
бензина, выкипающего до 175°, с содержанием ароматики около
25% [14].
Очистка продуктов пиролиза, в частности пиробензина, обыч-
ным путём, т. е. серной кислотой, сопровождается большими по-
терями за счет полимеризации и осмоления непредельных угле-
водородов. Кроме того, сернокислотная очистка пиробензина, уда-
ляя из него легкие олефины (например, амилены), резко снижает
его пусковые качества. Столь нежелательные последствия могут
быть устранены заменой сернокислотной очистки пиробензина
легким его гидрированием («стабилизационная гидрогенизация»)
в присутствии сероустойчивых катализаторов, например гидри-
рованием при температуре* около 350° и давлении 60—70 от
в присутствии двухсернистого молибдена [15].
Сольвент. Широкая фракция с началом кипения от 135° и
выше. При разгонке ее до 250° должно отгоняться не менее 98%.
Содержит ксилолы, выделение которых оказалось, однако, не вы-
годным. Является удобным материалом для получения раствори-
теля типа сольвент-нафта, применяемого в лакокрасочной про-
мышленности; употребляется также при изготовлении составов
для борьбы с насекомыми.
Нафталин. Этот углеводород является важнейшим и наибо-
лее легко выделяемым компонентом среднего масла из нефтяного
дегтя. Нафталин получают из этого масла вымораживанием (при
10—14° ниже 0°) с последующим центрифугированием выде-
ляющихся кристаллов. В заключение следует возгонка в спе-
циальных камерах, в результате чего получается очень чистый
нафталин. Выхода на него не превышают 5%, считая на сырье
Выделение из нефтяного дегтя такой высокомолекулярной,
находящейся в нем' ароматики, как антрацен, фенантрен и хри-
зен, пока еще недостаточно разработано.
Зеленое масло. Так называется широкая тяжелая фракция
продуктов пиролиза нефти с началом кипения не ниже 150°; до
350° должно отгоняться от него не меньше 95%. Содержание
нафталина в зеленом масле не больше 2%. Зеленое масло упо-
требляется главным образом как сырье для выработки сажи;
оно может служить также материалом для получения путем де-
структивной гидрогенизации высокооктанового бензина.
Пек. Жидкий или твердый пек получается в остатке после от-
гонки от нефтяного дегтя зеленого масла. Употребляется для
приготовления кровельного толя, а также для покрытия жетез-
ных и деревянных частей, устанавливаемых в грунте. Является
основным сырьем для производства нефтяного кокса.’
Кокс. Твердый остаток в кубе, получаемый при перегонке
448
нефти до кокса; представляет собою почти чистый углерод, бо-
лее или менее пропитанный маслом. Он образует на Дне куба
слой толщиною 20—24 см в зависимости от характера исходного
сырья. Его удаляют из куб<1 вручную через особые лазы после
предварительной продувки паром и остывания. Иногда для облег-
чения этой операции в кубы перед их загрузкой закладывают
особые цепи; после перегонки их вытягивают воротом, разрых-
ляя, таким образом, кокс и облегчая его удаление.
Нефтяной кокс на 96% состоит из углерода. Он содержит не-
редко не больше 0,1—о,2 % золы, т. е. практически является без-
зольным; лишь в тех его частях, которые непосредственно при-
легали к стейкам куба, содержание золы вследствие растворения
железа повышается до о,6—0,8 7с. В этом отношении нефтяной
кокс значительно выше, чем кокс с установок жидкофазного кре-
кинга, в котором содержав^ золы достигает 1,5% и выше. Со-
держание серы в нефтяном коксе за висит от исходного сырья,
но даже при малосернистом сырье достигает 0,5—0,6%. Его
теплотворная способность равна 7 500—8 000 кал.
Нефтяной кокс благодаря своей малой зольности находит
применение главным образом для выделки электродов, особенно
в алюминиевой промышленности.’ Меньшее значение имеет неф-
тяной кокс как топливо и как сырье для металлургической про-
мышленности. Ограниченность применения нефтяного кокса на-
ряду с периодическим характером перегонки до кокса является,
невидимому, главной причиной сравнительно слабого распро-
странения этой системы переработки нефти [16].
При крэкинге в жидкой фазе коксообразование, как уже
было отмечено, также зависит* прежде всего от характера сырья
и при работе на мазуте может достигать 8—10% от сырья и
выше. При средней пропускной способности крэкинговой уста-
новки 120—150 т сырья в сутки кокс занимает, ‘таким образом,
видное место в ее продукции. На сегодняшний день приходится
констатировать, однако, что единственное место его применения
это — топки. Однако и в виде топлива крекинговый кокс требует,
либо специальной обработки (бр!гкеты), либо специальных при-
способлений (топки для пылевидного топлива). По своим каче-
ствам (содержание масел, зольность) крэкинговый кокс значи-
тельно уступает продукту, получаемому при перегонке нефти до
кокса, и должен расцениваться как балласт крэкинг-процесса.
При парофазпом крэкинге, сырьем для которого, как было
указано выше, обычно служат сравнительно легкие дестиллаты
(керосиновый, газойль и т. п.), коксообразование крайне незна-
чительно и во всяком случае не превышает 1% от сырья. Часть
кокса в виде мелкой пыли или дыма уносится движущимися па-
рами и газами, другая часть уходит вместе с тяжелыми остат-
ками и оседает в мазутном резервуаре. Ввиду небольших выхо-
дов вопрос о коксе как о продукте парофазного крэкинга, оче-
видно, не может иметь серьезного значения и должен рассма-
триваться лишь с точки зрения возможности засорения аппа-
ратуры.
449
29 Зак. 1622. Химия нефти. ш
Гораздо большее значение имеет .кокс, получаемый при intpo.
лизе нефти, когда выхода на него достигают по крайней мере
5% от сырья. Этот кокс является плотным материалом, почти не
содержащим золы и серы, весьма ценным для некоторых спе-
циальных целей (производство электродов и т. и., см. выше).
Однако из всего получаемого таким образом кокса при работе
в металлических ретортах удается утилизировать не больше по-
ловины; остальная же часть вследствие чрезвычайной плотности
не поддается механическому откалыванию и устраняется выжи-
ганием. При работе на шамотных ретортах кокс получается рых-
лый и загрязненный, в генераторах же он вовсе пропадает, сго-
рая- при продувке.
В качестве сырья для получения беззольного кокса из нефти
удобно пользоваться пеком, побочным продуктом пиролиза
нефти, остающимся после отгона нефтяного дегтя всех дру-
гих продуктов вплоть до антраценовой его-части [17]. Пиролиз
пека производят в‘ муфельной печи специальной конструкции
с приспособлением для загрузки жидкого (подогретого) пека ме-
ханическим путем и с гидравликом общепринятого устройства
для изоляции муфеля от внешнего воздуха. Достигаемая в печи
температура—1100—’1200 , благодаря чему вырабатываемый
кокс совершенно свободен от летучих углеводородов, содержит
до 99% углерода и в среднем о,1—о,2 золы. Выгрузка кокса не
представляет затруднений, так как вследствие значительной
разницы коэфициентов расширения шамота и кокса последний
после снятия ретортных крышек разрыхляется сам и легко вы-
гребается.
ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ ПЕРЕГОНКИ С РАЗЛОЖЕНИЕМ
Перегонка с разложением представляет собою крайне сложный
процесс, в котором чисто физические явления тесно перепле-
таются с химическими реакциями разложения, конденсации- и
изомеризации. Изучение всей совокупности этих явлений пред-
ставляет собою крайне трудную задачу, исключительное значе-
ние которой совершенно очевидно: только этим путем можно
представить себе постепенный переход; в технике крэкинга и пи-
ролиза от грубого эмпиризма к полному овладению процессом
с точным учетом значения каждого отдельного его фактора и
радикальным использованием всех его продуктов. Особенно цен-
ные результаты дало исследование термического распада угле-
водородов не на таких сложных смесях, как нефть и ее про-
дукты, а на отдельных индивидуальных углеводородах различ-
ных классов. * 9
Авторами первых исследований термического распада углеводородов
были еще Бертло [18], Тирпе и Юнг [19], позднее Габер [20]. В новейшее
время изучением физико-химических основ процесса занимались Лесли и
Иетгоф [21], особенно же Сахапов и Тмличсев, исследования которых
должны быть положены в основу наших современных представлений о хи-
мизме крэкинглпроцесса в жидкой фазе. Теоретические основы других форм
крэкинга изучены пока совершенно недостаточно, так что в последующем из-
ложении мы будем останавливаться по преимуществу на крэкинге в жидкой
фазе.
450
Методика исследования. Опы ное исследование крэкинг-нро-
цеос-а удобно производить в небольшом аппарате — автоклаве—
на разные загрузки, испытанном на давлении от 40 до 150 ат.
Образец такого прибора, выработанный в практике работ
ГрозНИИ, представлен на фиг. 104.
Прибор состоит из автоклава для нагрева крэкируемого про-
дукта 1, небольшой колонки-дефлегматора з для возвращения
в автоклав более тяжелых частей, ’змеевикового холодильника 6
и приемника 7, в котором собираются крэкинг-дестиллаты. Для
измерения давления в автоклаве и приемнике служат маноме-
тры’4, 8; температура измеряется термометром, опускаемым
в особый карман 2, заканчивающийся в реакционной жидкости.
Три вентиля служат: один 5 — для разъединения автоклава от
Фиг. 104. Лабораторная установка для крэкинга в жидкой фазе.
холодильника в начале опыта и для последующего уравнивания
давления в автоклаве и пржмнике; другой О — для выпуска из-
лишних газов и поддержания давления в аппарате на требуе-
мой высоте; третий 10— для спуска крэкинг-дестиллата* Авто-
клав и приемник снабжены крышками на солидных болтах.
Методика опытного исследования крэкинга в жидкой фазе
в основном заключается в следующем.
Навеска крэкируемого вещества (нефть, нефтепродукт или
индивидуальный углеводород) помещается в автоклаве примерно
до половины его емкости. Крышки автоклава и приемника гер-
метически закрывают и завинчивают болтами; все три вентиля
также закрывают и начинают нагревание автоклава. Нагревание
можно производить непосредственно сильными горелками (Теплу
и т.п.). Когда давление в автоклаве достигает требуемой вели-
чины, излишние газы постепенно перепускают с помощью вен-
тиля 5 в приемыш; 7 так, чтобы во всем приборе установилось
• 29* 461
одинаковое давление. Избыток, газов выводят далее через'вен-
тиль 9; для учета их пропускают через газовые часы, для ис-
следования же их состава собирают в газометре над насыщенным
раствором поваренной соли.
Когда температура крэкируемого продукта достигает примерно
375°, усиливают нагревание, чтобы но возможности быстрое
пройти промежуточный этап до тии температуры, при которой
предполагают вести крэкирование; далее поддерживают темпе-
ратуру в пределах 1—2° простым регулированием пламени го-
релок. Во все время опыта через каждые 3—5 мин. ведут запись
температуры внутри автоклава и давления в автоклаве и‘при-
емнике.
Когда время, намеченное для крэкинга, истекло, убирают
горелки для скорейшего охлаждения, снимают с автоклава изо-
лирующий асбест и дают автоклаву остыть. Затем выпускают из
прибора избыток газа через вентиль .9, сливают- крэкинг-дестил-
лат через другой вентиль 10 и направляют его на исследование;
наконец, вскрывают автоклав извлекают и взвешивают непере-
гнавшийся остаток и подводят баланс опыта.
Систематические исследования жидкофазного крэкинга как
индивидуальных углеводородов, так и разного рода нефтепро-
дуктов в автоклаве привели к ряду весьма ценных результатов,
которые в значительной степени разъяснили физико-химическую
сущность этого процесса. Однако условия копирования в авто-
клаве слишком далеки от тех условий, в которых ведется про-
цесс в современных крэкинговых установках, и на основе,дан-
ных автоклавного крэкирования невозможно дать исчерпываю-
щий ответ на обычный и основной запрос промышленности: ка-
ковы должны быть оптимальные условия крэкинга данною
сырья на данной установке для получения крэкинг-бензина, по
своим качествам Отвечающего требованиям потребителя в дан-
ное время. Ответ на этот* вопрос может быть получен лишь на
специальной экспериментальной установке, построенной с со-
хранением всех основных принципов современных промышлен-
ных крэкинг-установок и на которой можно воспроизвести и
вариировать условия крэкинг-пропясса в достаточно широких
пределах. Примером экспериментальной установки такого рода
может ^служить установка Кед лога, получившая в США до-
вольно широкое распространение [22].
Опытная крэкинг-установка Келлога (фиг.’ 105) состоит из следующих
основных частей. Резервуары 1, 4, 5. служат для различного сырья: све-
жего сырья (I), смешанного сырья (4), тяжелого лигроина (5) и т. д.
Насосы 6 подают это сырье в стальные змеевики различных зон. Эти
змеевики сделаны .из тяжелых труб, погруженных в цилиндрические
ванны, заполненные свинцом, с .электрообогревом; температура изме-
ряется двумя термопарами, из которых одна помещена в ванну, другая —
внутрь змеевика. Змеевики отдельных зин делятся на самостоятельные
секции, так что с помощью центрального распределительного клапана
сырье может быть направлено в ту либо иную нагревательную секцию.
Из змеевиков сырье поступает в реакционную камеру 8, а затем через
редукционные клапаны .9 — в испарительную* камеру 10, работающую при
атмосферном давлении. Здесь происходит разделение; легкие части цосту*
452 л
иают далее в ректификационную кояоп-ну, и, откуда легкие погоны -(крэ-
кипг-бснзин), пройдя холодильник, направляются на стабилизацию, более
же тяжелые погоны возвращаются назад для подмешивания к сырью; тяже-
лые части пз атмосферной испарительной камеры 10 переходят в вакуумную
.испарительную камеру 12 и здесь также разделяются на более тяжелую
часть (гудрон) и более легкую часть, которая возвращается частью для
подмешивания к сырью, частью же идет назад в ректификационную ко-
лонку.
Опытные крекинговые установки непрерывного действия
строятся в США с пропускной способностью на 50—100 кг
сырья в час *(1;5—2,5 т/сутки) и более. Очно из основных усло-
!6
Фиг. 105. Опытная крэкинг-установка -Ее-мога:
1—резервуары для исходного сырья; 5 резервуары для рециркуляционного ма-
териала; з—центральный раопределительн А коллектор для переключения
трубопроводов; 4 и б—змеевики I и II зон; 6 и 7 —змеевики и реакционные
камеры III и IV зон; 8—свинцовые нагреватели для подогрева сырья перед
вступлением его в змеевнкв; 9 — реакционные камеры; 10 — свинцовые ванны
реляционных камер; 11 — колонна испарительная; 12 — колонна испарительная
вакуумная; 13 и 14 — колонна I и И насадочные; is — колонка вакуумная ректи-
фикационная: 16 — приемник; 17— тяжелый дестиллат из колонны 1г>; 18—про-
дукт* со дна колонны Ь5; 19 — гудрон со два колонны 12; 20 — бензиновый
конденсатор; 31 — колонна для ректификации бензина; 32 — резервуар для оро-
шения; 23—приемник для бензина; 24— бензпкопровод; 3s— спускная линия
для бензина со дна стабилизационной колонны; 26—стабилизационная колон-
на; 27 — газовый счетчик.
вий, которому должна удовлетворять опытная установка, заклю-
чается в том, чтобы скорость прохождения сырья была на ней
такая же, как и на установке промышленного типа. При соблю-
дении этого условия и надлежащем технологическом оформле-
нии ее основных частей опытная установка может служить сле-
дующим назначениям: *
1. Контроль производства, т. е. нахождение режима доя дан-
ной заводской у-становкй, обеспечивающего наибольшие выхода
крэкинг-бензина заданных качеств из данного сырья.
2. исследовательская работа в области изыскания новых и
усовершенствовании старых способов крэкирования,
453
• 3. Получение опытных данных для проектирования новых
крэкинговых установок.
Показательно, что результаты работы опытной крэкинговой
установки Келлога расходятся с результатами работы промыт-,
ленной установки при соответствующих режимах -не более чем
на 2—3%.
Несравнимо меньше изучен химизм других видов перегони
с разложением и, в частности, пиролиза. Экспериментальные
работы в этой области 23] носят пока чисто технологический
характер, имея своим назначением определение условий для по-
лучения оптимальных выходов на ароматические углеводороды.
Для экспериментального исследования пиролиза служит
(фиг. 106) железная реторта 1 длиною в 1 л с внутренним диа-
Фиг. 106. Лабораторная установка для пиролиза нефти.
метром 5 см. Реторта обогревается электрическим током, прохо-
дящим через хром-никелевую проволоку, намотанную по всей
длине реторты. Температура нагрева регулируется реостатом 5
и измеряется пирометром, состоящим из термопары 2 и милли-
вольтметра 4. Во время опыта температура поддерживается по.-
стоянной с колебаниями ± 5° О. Исходное сырье поступает
в реторту-из градуированной капельной воронки 7 с заданной
скоростью, обычно 2,5 г!мин. Полученная в результате пиролиза
смола конденсируется в гидравлике 8 и во втором приемнике Р,
соединенном через трубку Юс холодильником 3; охлажденный
газ с целью отделения от него паров жидких углеводородов про-
пускается через промывные склянки 11 с соляровым маслом.
Количество полученного газа замеряется газовыми часами 12;
для анализа пробы газа время от времени собираются в газо-
метр, 13 с насыщенным раствором поваренной соли. Давление
в реторте поддерживается немного выше атмосферного, около
4-5 —10 мм ртутного столба открытого манометра 15, и регу-
лируется водоструйным насосом 14.
454
Общий выход смолы определяется iio привесу всех прйемнй-
ков — 8. Р, 11, взвешенных до опыта. В оптимальных условиях,
пиролиза (температура 700—710°) выход смолы, например, из
суря,ханского мазута составляет 45—50% по весу от исходного
сырья.
Для разделения на фракции ’ смола из гидравлика и второго
приемника (гидравлическая и холодильничная смола) соеди-
няются вместе и перегоняются из железного кубика с отбором-
фракций: до 175° 175—225°, 225—250° И 250—325°; остаток —
пек/ Скрубберное масло из промывных склянок также соеди-
няется вместе и подвергается разгонке с отбором легкого- масла
до 175°. Легкое масло из смолы и скрубберного масла соеди-
няется вместе (выход 17—19% от сырья) и подвергается фрак-
ционированной перегонке с отбором фракций, отвечающих сы-
рому бензолу, толуолу и ксилолу.
Общий выход газа определяется на основе показания газо-
вых часов и удельного веса газа и составляет 40—50% по весу
от исходного сырья. Анализ газа производится по Темпелю
с определением:
1) высших непредельных углеводородов, начиная с пропи-
лена,— поглощением 85%-ным раствором серной кислоты без
встряхивания;
2) углекислого газа—поглощением раствором едкого кали;
3) кислорода — поглощением щелочным раствором пирогал-
лола»
4) этилена — поглощением бромной водой.
Кислород и углекислый газ обычно содержатся в газах пи-
ролиза в незначительном количестве. Содержание этилена в газе
от пиролиза сурахаяского мазута составляет около 22%, высших
непредельных углеводородов—15—20%.
Для определения кокса последний выгружается из реторты
и взвешивается. Выход его при пиролизе сураханского мазута
составляет 1—3% от исходного сырья.
Химизм крэкинг-процесса. Химические реакции, протекаю-
щие при крэкинг-процеосе, приводят прежде всего к продуктам
разложения крокируемого вещества; таковы, например, газы
крэкинга и вообще легкие углеводороды, образовавшиеся в про-
цессе расщепления исходных, более сложных молекул. Реакции
разложения являются основным типом химических превраще-
ний, наблюдаемых при крэкинг-процессе.
Наряду с разложением при крэкинге наблюдаются также ре.-
акции прямо противоположного типа, которые приводят к про-
дуктам более сложного, состава но сравнению с исходным сырьем.
Образование таких продуктов хорошо наблюдается, например,
. ври крэкинге узких нефтяных фракций, когда путем последую-
щей фракционировки крэкипг-дестиллата легко могут быть вы-
делены новообразовавшиеся более тяжелые фракции крэкинг-
масел. Образование продуктов этого рода происходит несомненно
за счет реакций уплотнения первичных продуктов разложения
крэкируомого вещества и, следовательно, имеет вторичный ха-
455
рактер. Таким образом наряду с процессами расщепления, раб-
лада при крэкинг-процессе имеют место также реакции, уплот-
нения. I
Реакции разложения и уплотнения являются важнейшими
превращениями, наблюдаемыми при крэкипг-нроцессе. Не под-
лежит, однако, сомнению, что наряду с ними здесь могут про-
исходить также разного рода внутримолекулярные перегруппи-
ровки, сопровождающиеся изменением углеродного скелета
исходной молекулы, перемещением боковых цепей и т. п. Реак-
ции этого рода могут1 .быть объединены в одну большую группу—
? роцессы изомеризации. Эти процессы чрезвычайно разно-
образны, но при исследовании крэкинга. они вследствие их ма-
лой изученности должны быть поставлены на второе место.
Ввиду этого при последующем изложении мы сосредоточим
главное внимание на наблюдаемых при крэкинге реакциях раз-
ложения и уплотнения.
Реакции разложения. Типичные примеры реакций разложе-
ния, представленных при крэкинг-процессе наиболее резко, дает
крэкинг парафиновых углеводородов.
Легко представить себе, что распад молекулы какого-либо
углеводорода может происходить в самых разнообразных напра-
влениях. Так например’, распад одного из простейших парафи-
нов, бутана, теоретически мож т произойти по следующим важ-
нейшим схемам:
СИ8 - СЩ • СНо = СН3 = СН.. • СН., - СН : СН., + Н.,;
сн8 • с и.; • .сн.; • сн3=сн3 - сн:+сн4+с,- *
СНу. сн; . сн; • СН3= СН3 • СН : СНо4-СН4;
СН3-СН;.СНа-СН8 = СН3.СН34~СН2 : СН, ит. д.
Для более сложных парафинов число возможных направле-
ний распада, естественно, должно быть еще больше, и едва, ли
можно сомневаться, хгто многие из этих направлений в усло-
виях высокотемпературного крэкинга действительно имеют место.
При более умеренных температурах, свойственных жидкофаз-
ному крекингу, число фактических направлений распада пара-
финового углеводорода значительно сокращается. Действительно,
изучение крэкинга парафина, главная масса которого состоит из
углеводородов ряда метана состава от С24Н50 до СгбН34, привело
к следующим важным результатам.
Независимо от давления (от 10 до 40 ат) и времени (от 1
до 3 час.) при 425° среди разнообразных продуктов крэкинга
ш рафина кокса вовсе не наблюдается; содержание же водорода
в газах крэкинга не превышает 0,5—1%, считая на газ. Отсюда
ясно, что направления распада парафина, аналогичные дам
первым из вышеприведенных реакций, при 425° либо совершенно ,
либо почти нацело отпадают.
Изучение жидких продуктов- низкотемпературного крэкинга
парафинов также крайне важно для суждения о механизме
этого процесса. Уже одно разнообразие этих продуктов, не по-
звол яющее выделить каких-либо определенных веществ при крэ-
4М)
кингс даже индивидуальных углеводородов, заставляет притти
к выводу, что распад высокомолекулярного углеводорода проис-
ходит не в каком-либо одном, а в нескольких направлениях.
Заметное преобладание среди них при температурах ниже 500е
имеет, повидимому, распад молекулы тяжелого углеводорога на.
две равные части, например для додекана —по следующей схеме:
ОН, (СН.,)4. СН,СН2(СН2\ • СН3 = СН3(СН.,)3 СН': СИ, 4-
додекан (норм.) гексилен
СН:1(СИ,)4СН,,.
гексан
. Однако эго направление реакции разложения парафинового
углеводорода далеко не единственное.
Прежде всего, совершенно очевидно, что образовавшаяся при
'крэкинге молекула парафинового углеводорода в свою очередь-
будет подвергаться разложению по аналогичной схеме с обра-
зованием еще более простых по своему составу углеводородов.
Газообразование, всегда наблюдаемое при крэкинге. можно рас-
сматривать, ’ очевидно, как результат ряда подобного рода реак-
ций разложения вторичного, третичного и т. д. порядков. На-
глядной иллюстрацией сказанного может служить следующая
схема крэкинга парафинового углеводорода нормально]’© строе-
ния, состава С24Н50, с распадом его по средине молекулы:
С-24Н50 = СгДб + С12Н24
ад4+с6и12
С3нД-С3Н6 и т. д.
Еще большее разнообразие в состав крэкпнг-продуктов
должны вносить такие направления реакций разложения, кото-
рые не укладываются в только что приведенную схему. Сюда
относится прежде всего, очевидно, крэкинг нормальных пара-
финов с нечетным чис. ом- углеродных атомов, особенно же раз-
ложение изопарафинов, чрезвычайно разнообразных по своему
строению. Но и для нормальных парафинов с четным числом
углеродных атомов линию распада сложной молекулы можно
♦ представить себе, очевидно, не только в том направлении, как
это изображено на последней или какой-либо аналогичной иной
схеме, но и в любом другом месте молекулы, например ближе
к ее середине или дальше от нее. Тем самым легко объясняется
отмеченная выше сложность состава . крэкинг-нродуктов не
только такого сырья, как нефть или нефтепродукт, по и чистых
индивидуальных парафиновых углеводородов нормального строе-
ния [18].
Ещё более сложную картину могут дать реакции разложе- ,
ния этиленовых углеводородов, нафтенов и ароматики с боко-
выми цепями -различного состава и строения. Этот вопрбс будет
рассмотрен несколько" ниже в связи с крекингом углеводородов
различных рядов.
457
Реакции уплотнения. Реакции уплотнения проявляются при
крэкинг-процёссе прежде всего в увеличении удельного веса
крэкинг-оиатков. Таким образом одновременно с образованием
при крэкинге масел таких легких продуктов, как крэкинг-бензин
и крэкинг-керосин, происходит новообразование каких-то тяже-
лых продуктов, собирающихся в остатках с температурой кипе-
ния выше 300°. Среди этих продуктов в первую очередь необхо-
димо отметить асфальтены, содержание которых в остатках по
мере удлинения продолжительности крэкинга сначала довольно
быстро возрастает. Но уже через 1—часа после начала крэ-
кинга и достижения некоторого предела содержание асфальтенов
начинает резко падать; одновременно наблюдается и постепенное
образование кокса.
Крекинговый кокс необходимо рассматривать как один из ко-
нечных продуктов реа щий уплотнения, имеющих место при кре-
кинге. По своему составу он представляет собою вещество до-
вольно сложное; как будет показано ниже, он содержит от 13
до 20% летучих (до 500°) и заметное количество асфальтенов,
в главной же своей части он состоит из карбоидов. Опыт показы-
вает, что образование кокса в значительной степени зависит от
химической природы крокируемого сырья. Так например, пара-
финистые нефтепродукты, бедные циклическими углеводородами
*и особенно ароматикой, менее склонны к коксообразованию,
тогда как тяжелые смолистые нефти ароматического характера
при прочих равных условиях проявляют склонность к коксо-
образованию в гораздо более резкой степени. Прекрасной иллю-
страцией этого положения могут служить данные табл. 94, в ко-
торых сведены результаты крэкинга (450°, 10 ат, 36 мин.) гроз-
ненского парафинистого дестиллата до и после удаления из него
ароматики - путем обработки дымящей серной кислотой
(с 7.5% SOs). ’ .
Таблица 94
Крэкннг парафинистого дестиллата до и после очистки
(по ГрозНИИ)
Наименование исходного продукта Уд. вес остатка (выше 300°) Выход на исходный продукт в %
асфальтены карбоиды
Грозненский парафинистый дестил- 1,029 1,99 3,49
лат до удаления ароматики . . . Тот же продукт после удаления
ароматики 0,930 0,08 0,11
Как видно из табл. 94, до удаления ароматики при крэкииге
наблюдаётся резко выраженная картина глубоких процессов
уплотнения, выражающихся как в повышении удельного веса
остатка, так и в образовании значительных количеств асфалъ-
456
Тейов и карбоидов; после удаления ароматики те же явлений
оказываются 'выраженными несравненно слабее. Отсюда можно
сделать следующий важный вывод по вопросу о механизме реак-
ций уплотнения при крэкинге: парафиновые углеводороды в усло-
виях низкотемпературного крэкинга мало склонны к реакциям
уплотнения и образованию кокса; эти процессы, как ясно из
вышеизложенного, тесно связаны прежде всего с превращениями
углеводородов ароматического ряда.
Другим источником образования продуктов уплотнения при
крэкинге являются этиленовые углеводороды. Образуясь в про-
цессе разложения парафинов, этиленовые углеводороды в усло-
виях низкотемпературного крэкинга легко претерпевают уплотне-
ние, образуя как высокомолекулярные продукты полимеризации
непредельного характера, так и продукты конденсации с арома-
тикой. Наряду с этим ио мере течения крэкинга в его продук-
тах наблюдается накопление все больших и больших количеств
нафтенов и, повидимому, ароматики, которые в свою очередь мо-
гут давать в процессе крэкинга не только продукты разложения,
но и продукты еще более глубокого уплотнения. Совокупность
всех этих реакций создает, естественно, весьма сложную картину,
столь характерную для крэкинг-процесса; к некоторым ее де-
талям мы еще вернемся несколько ниже.
Процессы изомеризации. Как будет ясно из последующего,
углеводороды обладают различной степенью устойчивости к по-
вышенной температуре: одни из них поддаются воздействию
этого фактора легче, другие — труднее. Кроме рассмотренных
выше реакций разложения и уплотнения, углеводороды нередко
претерпевают менее глубокие изменения, при которых размеры
молекулы не меняются и процесс ограничивается внутримолеку-
лярной перегруппировкой исходной .системы в новую систему,
более устойчивую в данных условиях; короче говоря, — изомери-
зацией или изомерным превращением исходной системы.
Изомерные превращения углеводородов нередко в высокой
степени облегчаются присутствием (контактом) некоторых сто-
ронних веществ: металлов, окислов, щелочей, солей и т. и.
Примеры подобного рода превращений чрезвычайно многочис-
ленны и разнообразны. В одних случаях процесс ограничивается
одним перемещением кратной связи. Так например, давно было
показано, что при пропускании над кремнеземом мри 500—505°
иди над окисью алюминия при 525—535° изопродилэтилен на
80% изомеризуется в триметилэтилен:
сн8Ч сн8Х
>СН - СН : СН2 -► )С : СН - СН3.
СН/ Сн/
Равным образом доказано, что при медленном пропускании
над глиноземом и ортофосфорной кислотой бутен-1 может быть
количественно превращен в бутен-2:
СН, • СНо • рн : СН3 -> СН3 • СН : СН • СН3.
459
В более сложных случаях Изомерное превращение углеводо-
рода сопровождается изменением углеродного скелета исходной
системы. Так например, установлено, что нормальный гептан и
нормальный октан при нагревании в присутствии некоторых
катализаторов частично изомеризуются в соответствую^^ изо-
парафины, изогептан и изооктан. Равным образом циклогексан
при нагревании над окисью алюминия при 510° частично изо-
меризуется в метилциклопентан:
СЩ .
ПР .'сН.,
(Я I,
п./.'zx ci г,
И.,С— ГЦ
I
СИ..
По подлежит сомнейию, что многие изомерные превращения
подобного рода, имеют место также в процессе крэкинга нефтя-
ных углеводородов; совершенно ясно, что их изучение предста-
вляет поэтому не. только теоретический интерес, например в це-
лях выявления всего разнообразия и сущности 'превращения
углеводородов при крэкинг-процессе.. Такие превращения, как
изомеризация нормальных парафинов в изопарафины, с точки
зрения проблемы получения высокооктанового .моторного топ-
лива, несомненно, привлекают в настоящее время самое серьезное
внимание последователей; однако не только детали этих иссле-
дований, но даже достигнутые в данном направлении несомнен-
ные успехи по соображениям военного характера не опублико-
вываются. Ниже, говоря о «реформинге», а также о крэкинге
с хлористым алюминием, мы еще встретимся с этим вопросом.
Кинетика крэкинг-процесса.Окорость всякой химической ре-
акции есть функция целого ряда переменных, определяемая на
основе учета того или иного ее продукта. Определяя скорость
крэкинг-процесса по количеству образующихся в единицу вре-
мени крэкинг-бензина или вообще продуктов разложения, инте-
р<ч но проследить ее зависимость от таких факторов, как продол-
жительность процесса, природа крэкируемого сырья, темпера-
тура н давление.
Продолжительность процесса. При постоянной температуре
скорость ’ крэкинг-нроцесса вначале не зависит от времени. Со-
ответственно этому, как видно из данных табл. н.‘>, количество
Таблица 95 .
Крэкинг парафинистого мазута яри 400°
(по ГрозНИИ)
[|ридолжцтсльность опыта в час. 1.6 3 6
Выход крукппг-бензпна в % . . 5,й 11.2 20.4 «
4G0
крэкинг-бензина, образующегося, например, при медленном кре-
кинге парафинистого мазута при 400 , изменяется вначале про-
порционально времени.
С измененной условии крэкинга эта простая зависимость
легко нарушается, что является прежде всего прямым следствием
уменьшения концентрации исходного вещества. Соответственно
этому и скорость крэкинг-процесса с течением времени умень-
шается, как это видно из данных табл. 96.
• Таблица 96
Крэкннг фракции газойля при 425°
(по ГрозНИИ)
Продолжительность опыта в мин 84 64 184
Выход крэкппг-бензина в % . . . 9.4 • 15,8 34,6
В тесной связи с зависимостью скорости крэкипг-процесса от
его продолжительности находится влияние состава крэкируемого
материала на скорость его крэкинга.
Состав крэкируемого материала. Для выявления зависимо-
сти между составом сырья и скоростью его крэкинга могут слу-
жить следующие опыты (табл. 97). Ряд узких фракций прямо»
гонки был прокрэкирован (425°, 1 час) с последующей разгонкой
и учетом выходов на различные продукты крэкинга. Если сум-
мировать затем в отдельных опытах выхода на газообразные и
жидкие продукты крэкинга, то по полученным результатам
можно судить о суммарных выходах на продукты разложения и,
следовательно, о скорости крэкинг-процесса. для разных исход-
ных материалов при одинаковых условиях крэкинга.
Таблица 97
Крэкииг различных дестиллатов (по ГрозНИИ)
Наименование и темп, кип. исходного материала Выхода в °/о на прод. разложения Выход на крэ- кинг-бензин в %
Лигроиновая фракция 180—220° Керосиновая фракция 220—270° Газойль 270—300° . . . Соляровая фракция 300-325° Машинная фракция 250—280° (6 мм) . . . 17,0 25,0 33,0 46,0 £5,5 12,1 14,9 15,8 17,6 22,4 4KL
Данные табл. 97 показывают, что скорость к-рэкпнг-проЦесей.
для легких дестиллатов наименьшая, но она постепенно возра-
стает по мере утяжеления крэкируемого материала. Особенно
резню сказывается такое возрастание при учете всех продуктов
разложения, так что, например, при переходе от лигроиновой
фракции к машинной скорость крэкинга увеличивается в 5 раз.
Образование одних крэкинг-бензипов из разного исходного
сырья без учета других продуктов разложения зависит от исход-
ного материала не в такой сильной (’тепеии. Так, наиболее лег-
кая, лигроиновая, фракция дала при крэкинге в вышеуказанных
условиях 12% крэкинг-бенз-ина. тогда как наиболее тяжелая,
машинная, — 22% крэкинг-бензина.
Кроме фракционного состава, 'большое влияние на скорость
крэкинга имеет также химический состав крэкируемого сырья
Наиболее резко сказывается такое влияние при повторном крэ-
кинге (recycling), когда, как. это будет показано ниже, вместе
с постепенным изменением состава сырья скорость его крэкн-
рования при прочих равных условиях резко снижается.
Температура крэкинга. Как известно, скорость химических
реакций с повышением температуры быстро возрастает. Эмпири-
ческое правило Вант-Тоффа (1884 г.) гласит, что повышение тем-
пературы на 10е вызывает увеличение скорости реакции при-
мерно в 2 раза. Эта закономерность оказалась применимой
также и к крэкинг-процессу, по крайней мере в пределах 400—
450°. В табл. 98 приведены средние значения времени, потреб-
ного при разных температурах для получения 10, 20 и 30%
крэкинг-бензина при крэкинге обычно применяемого сырья (со-
ляровое масло, мазут) без учета газообразных продуктов раз-
ложения.
Таблица 08
Зависимость скорости крэкинга от температуры
(по ГрозНИИ)
Температура опыта Время (в минутах) образования крэкинг-бензина
Ю% 20% ‘ 30%
400° 180 360 720
о Ю Я Т 30 60 120
450° 5 10 20
475° 1 1.7 3
500° 0,1 0.25 05
Данные табл. 93 показывают, что температура 450—475° обес-
печивает практически достаточно высокую скорость для про-
ведения крэкинг-процесса. В этих примерно условиях и рабо-
тают. код; мы видели в начале этой главы, новейшие трубчатые
системы жидкофазного крэкинга. Дальнейшее повышение тем-
462
исратуры в с 1учас жидкофазного крэкинга бы ю бы перацио--
нально, тал; как оно создавали бы лишь условия для усиленного
образования кокса, а вместе с том чрезмерно увеличило бы да-
вление внутри аппаратуры.
Следует иметь в виду, что данные табл. 98 должно рассма-
тривать лишь как первое приближение к характеристике ско-
рости крэкинг-процесса. Действительно, так- как они взяты без
учета газообразных продуктов крэкинга, то, очевидно, уже по
одному этому они не могут служить мерой скорости крэкинг-
процесса в целом. К тому же правило Вант-Гоффа весьма при-
близительно: как показали ближайшие исследования этого во-
проса, границы возрастания скорости реакций для разных реак-
ции при повышении температуры на 10° должны быть значи-
тельно расширены — от 1,4 до 7; с другой стороны, термиче-
ский коэфициент скорости зависит также от температуры. Так.
.некоторые новейшие данные показывают, что удвоение скорости
реакции крэкинга происходит около 430° при повышении тем-
пературы не на 10°, а на каждые 14е и при 600° — на каждые
215 [24].
Ближайшее изучение -крэкинга чистых углеводородов предельного ха-
рактера показало (см. ниже), что, невидимому, это — реакция моломолеку-
лярная, так что константа скорости се К может быть вычислена по уравне-
нию: f
„ 1 . а
А — — • In-------,
t а — х
Где а — исходное количество углеводорода и а — х — количество угле-
водорода, не успевшего подвергнуться крекингу за время t. В случае неф-
тяных фракций задача определения константы К, естественно, осложняется,
однако в первом приближении и здесь оказывается применимой только что
приведенная формула [24].
Давление. В противоположность температуре давление не
оказывает заметного действия на скорость жидкофазного крэ-
кинга. Это важное положение проверено на примере различных.
нефтепродуктов (как-то: парафин, соляровый и парафинистый
дестиллаты, мазут) и для давлений от 10 до 100 ат должно счи-
таться экспериментально • установленным [21. 25]. Его иллю-
страцией может служить табл. 99 с результатами крэкинга па-
рафина при 450° и разных давлениях.
Хотя, таким образом, давление при крэкицге в жидкой фазе
не оказывает заметного влияния на выхода легких крэкинг-де-
сгиллатов, особенно бензина, тем не менее не подлежит сомне-
нию, что для жидкофазного крэкинга в целом оно все же имеет
важное служебное значение. Обеспечивая сохранение больших
количеств сырья в жидком состоянии при более высоких тем-
пературах,. давление улучшает теплопередачу, уменьшает воз-
можность местных перегревов и, таким образом, в высокой сте-
пени содействует эффективности работы установки; в этом и со-
стоит основная роль давления при крэкинге в жидкой фазе.
Очень серьезное влияние оказывает давление на состав про-
дуктов крэкинга, Как видно из последнего столбца табл. 99,
ф>3
Таблица О')
Крэкинг парафина при разных давления^
(по ГрозНИИ)
Условия крекинга Выхода продуктов крэки (по весу) нга, % Иодные числа
темпе- ратура давле- ние в ат время в мин. газы, потери кр.-бенз. до 200° кр.-керос. 200—300° остаток выше 300° крекинг- бензинов
450° 10 64 9,9 41,9 31,8 11,7 129,(1
450° 20 64 11,5 42,8 19,7 21,3 110,9
450° 30 65 14,6 42,7 22 7 17,6
450° 30 65 12,9 46,9 ‘ 17,4 20,5 93,1
450° 40 63 13,8 38,4 22,9 23,2 79 7
450° 100 40 7,2 33,2 36 6 21.3 - 63.G
450°, 100 81 26,7 40,3 18,9 13.3 0
ио мере повышения давления при крэкинге в жилкой фазе
иодное число образующеюся крэкинг-бенэина резко снижается,
так что, например, для крэкипг-бепзина, полученного при 100 ат
давления, иодное число оказалось в два раза меньше, чем для
бензина, полученного при 10 ат давления. Очевидно, таким
образом, что с повышением давления при жидкофазном крэ-
кинге количество непредельных, содержащихся в крэкпиг-беп-
зине, резко снижается; ближайшее изучение этого вопроса за-
ставляет думать, что причиной такого снижения является пре-
вращение непредельных углеводородов частью в углеводороды
нафтенового ряда, частью в высококипящие продукты полиме-
ризации.
*
Менее ярко сказывается влияние давления в тех с 1учаях, когда изме-
нение этого фактора достигается нагнетанием в аппаратуру (автоклав)
какого-либо нейтрального газа, например азота, что по существу не должно
оказывать влияния на парциальные давления отдельных компонентов
системы в процессе крэкинга. Тем не меиес, и в подобного рода случаях
повышение суммарного давления в системе вызывает заметное снижение
непредельных и увеличение ароматики в бензине. Примером может слу-
жить (табл. 100) бензин до 150°, полученный крекированием в автоклаве
грозненского парафинистого дестиллата в течение 1 часа при 450°, причем
давление создавалось нагнетанием в автоклав азота и суммарно вариировало
от 70 до 200 ат [2б].
Влияние давления на ход крэкинг-процесса легко объясняется с точки
прения закона Ле-Шатедъе, который может быть выражен в общем виде сле-
дующим образом: если в системе, находящейся в устойчивом равновесии,
изменить одно из условий последнего (например давление, температуру и
т. п.), то система претерпевает такое изменение, которое стремится ослабить
влияние этого изменения пли оказать ему противодействие. Этот закон
является одним из основных законов природы; ® применении к химическим
реакциям, протекающим под давлением, он принимает такую форму: если
реакция протекает с уменьшением объема, то увеличение давления ускоряет
эту реакцию;, наоборот, если реакция сопровождается увеличением объема,
то повышение давления может лишь замедлить реакцию.
Применяя закон Ле-Шателье к различным реакциям, протекающим при
крэкинге, получаем следующие результаты.
464
Таблица 100
Содержание непредельных и ароматики в куэкннг-бензинз
в зависимости от давления
(по ИГИ Акад, н ijk)
№ п/п Темп. опыта Время в час. Сум. давл. в ат Содержание в °/0 по объему
Непредельн. Аромат.
1 450° 1 70 17,3 6 7
•> -J 450° 1 120 16.2 8.5
3 450? 1 150 15,4 9,1
4 45(Р 1 20 14,8 9,9
Реакции изложения, например: ।
С4И10 = С2ПЙ 4- С2Щ или C4HW СЩ + С$Нв
могут замедляться лишь с увеличенном давления.
Наоборот, реакции полимеризации или гидрогенизации с повышением
давления будут ускоряться:
С3Нв 4“ Н2 = СЙН8 или "C^Hg 4~ CgH^.
Наконец, при крэкинге могут иметь место и такие реакции, На которые
изменение давления не должно оказывать никакого влияния. Таково, напри-
мер, образование дифенила из бензола и другие реакции, протекающие без
изменения объема:
2С«Нв = Н-. + С«Нг, • СвНз.
Газообразование при крэкинг-процессе [27]. Наряду с неко-
торыми чисто физическими показателями, как-то: изменения
цвета, вязкости и т. и., газообразование является одним из
Основных -признаков разложения, наступающего в известных
условиях при перегонке. В частности, газообразные вещества,
гсак было отмечено выше, являются постоянными продуктами
перегонки с разложением нефти и ее продуктов независимо от
того, ведется ли такая перегонка в вакууме, при повышенном
или при обыкновенном давлении. В главной своей массе эти
газы состоят из низших, газообразных углеводородов различных
рядов с примесью водорода и небольших количеств окиси угле-
рода.
Температура, при которой в процессе термического разложе-
ния начинается газообразование, зависит прежде всего от при-
роды нефти или нефтепродукта и колеблется в достаточно широ-
ких пределах. Частичное разложение (крэкинг) некоторых неф-
тей, сопровождавшееся выделением газообразных продуктов, на-
блюдалось уже при температурах около 250° и ниже. При 300—
3509 выделяющиеся газы обыкновенно содержат водород. Про-
должительное назревание, особенно же присутствие катализа Го-
465
30 Зак. 16?2. Хцмия нефти.
ров, может значительно снизить температуру начала газообра-
зования.
Для правильной ориентировки в этим вопросе необходимо иметь
в виду, что, кроме газов, образующихся в результате разложения (крэ-
книга), при перегонке нефти и се продуктов в первую очередь должны
выделяться газы, которые находились в исходном продукте в растворен-
ном состоянии. Таковы, например, компоненты естественного газа, являю-
щцеся главной составной частью вбкондснснрующихся газов, получаемых
при перегонке сырой нефти. Выделение этих газов, естественно, значи-
тельно осложняет вопрос о начале газообразования, наступающего в ре.
зультате деструкции. Одним из признаков начавшегося крэкинга, казалось
бы, может служить появление в газах водорода, который, как известно,
в естественном газе не содержится. Однако частичный крэкинг нефтепро-
дукта начинается, повидпмому, несколько раньше появления в газах во-
дорода, так что и этот признак можно принимать как указание па начало
крэкинга лишь с оговоркой.
Температура является важнейшим фактором газообразования
при перегонке нефти .с разложением. В основном, влияние этого
фактора сводится к следующему: чем более жестки температур-
ные условия перегонки, короче говоря, чем выше ёе температура,
тем сильнее газообразование.
Действительно, как было отмечено выше, при наиболее мяг-
ких условиях крэкинга, при перегонке нефти до кокса, выхода
газа не превышают 1,5—2% по весу на сырье. В условиях жид-
кофазного крекинга-, т. е. в среднем при температурах 450—ISO0,
газообразование достигает 5—С % по весу.’ Наконец, в условиях
парофазного крэкинга, т. е. при температурах 550—600°, выхода
на газ поднимаются для некоторых систем уже до 25—30% по
весу на сырье. Во всех этих -случаях газ, очевидно, является
лишь побочным продуктом процесса термического разложения
нефти.
Известно, однако, что уже в 70-х годах прошлого века «неф-
тяной газ», т. е. газ, получаемый термическим разложением
нефти, находил применение для освещения целых городов.
'У нас, например, нефтяным газом освещались Киев, Казань и
некоторые другие города. Позднее, когда для получения нефтя-
ного газа были сконструированы небольшие, портативные уста-
новки, этот газ стал находить широкое применение, для освеще-
ния поездов, а также для технических предприятий: заводов,
лабораторий й т. и. В настоящее время нефтяной газ выраба-
тывается -в громадных количествах в различных городах СССР
(заводы- «Нефтегаз») и за границей, являясь, очевидно, уже це-
левым продуктом, при производстве которого стремятся к воз-
можно высоким его выходам. Путь к этой цели был найден
давно: необходимо, чтобы термическое разложение нефти произ-
водилось при достаточно высокой температуре.
Действительно, как видно из табл. 101, по мере повышения
температуры выше 550° газообразование для всех нефтепродуктов
неизменно -возрастает. Лишь в области 650—750°.в отношении
суммарных выходов на газ наступает известная стабилизация.
При этом, однако, уже начиная с 700° (и даже ниже), некоторые
тяжелые углеводородные газы становятся недостаточно устой-
466.
чйвыми и начинают разлагаться на более простые компоненты?
при 800 из углеводородов практически устойчивым остается
только метан.
Зависимое ть скорости газообразования от температуры
крайне характерна для некоторых химически однородных угле-
водородов. Известно, например, что циклогексан при 600° дает
лишь 1,4% газа, а при 650е —32,5%, так что в пределах 600—
650° скорость газообразования для циклогексана-делает громад-
ный скачок Аналогичный характер изменения скорости газо-
образования с температурой наблюдается для некоторых других
углеводородов, а также, естественно, для некоторых узких неф-
тяных фракций. Примером может служить газообразование при
крэкинге и пиролизе парафина, цилиндрового масла, отчасти
бензина (табл. 101). Наоборот, для широких нефтяных иогонов
(керосин, мазут) наблюдается ботее постепенное нарастание ско-
рости газообразования с повышением температуры
Таблица 301
Выхода газа иа»нефтепродуктов
(в % по весу)
(но Добрянскому)
• Исходное сырье Температура
550° 600° 650° 700° 750°
Бензин - .. — 210 56,0 62,0
Керосин 24,2 34,2 51,2 52,0 ' 52,0
Газойль 15,4 36,5 49,0 48,5 54,0
Парафин ==^= — 65,0 64,8 —-
Цилиндровое масло . . - — 15,6 410 44,0 42,0
Мазут ,Г", парафинистым 27,0 47,0 44,0 56,0 —
Зеленое масло —— 21,2 26,6 30,7
Из сказанного • следует, что, наряду с температурой на газо-
образование при крэкинге и пиролизе нефтяных продуктов дол-
жен оказывать большое влияние характер исходного сырья. Как
правило, чем тяжелее сырье, тем легче (т. е. при более низкой
температуре) наступает газообразование. Сопоставление мазута
с бензином (табл. 101) дает вполне убедительную иллюстрацию
этого положений. Большое влияние оказывают в данном отно-
шении также химический состав сырья и степень его однород-
ности. Из той же таблицы видно, цапример, что при 600 выход
газа из цилиндрового масла составляет 15,6%, тогда как нро:
дукт с более широкой температурой кипения, мазут, при той
же температуре дает 47%.
Что касается, наконец, конечных выходов на газ при опти-
мальных для данного сырья условиях, то, очевидно, здесь надо
искать связь прежде всего с химическим составом сырья. Лег-
кое, т. е. более богатое водородом сырье, например парафин, ха-
рактеризуется высокими выходами на. газ. Наоборот, тяжелое,
30* ' 467
Ф. е. бедное водородом, сырье, богатое ароматикой, дает понижен-
ные выхода.на газ; таково, например, зеленое масло (табл. 101).
Влияние* давления на газообразование при крэкинге ti пиро-
лизе нефти и ее продуктов изучено пока недостаточно, но не
подлежит сомнению. Установлено, например, что при 5ио° и
повышении давления от 1 до 12 ат выхода газа (в процентах по
объему) постепенно и равномерно возрастают. При 600° повыше-
ние давления почти не отражается на выходах газа, а при 750°,
когда значительные количества непредельных претерпевают ре-
'акции «полимеризации и уплотнения, выхода газа при повыше-
нии давления от 1 до 12 аг заметно падают. О влиянии давления
ла состав газов крэкинга и пиролиза будет сказано ниже.
Состав газов при термическом разложении нефти весьма су-
щественно меняется прежде всего в зависимости от температура
реакции. Из табл. 102 и 103 можно составить .представление
о характере этого изменения: в табл. К 2 дано -сопоставление со-
става тазов, получаемых при жидкофазном крэкинге, парофаз-
ном крэкинге и пиролизе нефтепродуктов; по табл, юз можно
детально проследить изменение «состава газов пиролиза газойля
в железной трубке при емкости реакционного пространства
100 см*, скорости подачи сырья 3 см3/мин и в пределах темпе-
ратур от ООО до 850°.
я
В табл. 102 приведены типичные анализы газов крэкинга и пиролиза
бакинских заводов [2S], а именно: газ жидкофазного крэкинга, с завода
Винклер-Koza при работе на сураханском парафинистом ’мазуте и режиме
495° и 35 ат давления; газ парофазнига крэкинга с быв. завода «Советский
крэкинг» системы инж. Шулова-Ь'апслюшникова при работе ла тяжелом
бензине или керосине прямой гонки (температура при выходе из печи
020°); газ пиролиза при температурном- режиме 660—670° и работе либо
на ];рэ1$и)1Г-керосине (реторты), либо на легкой солярке (газогенераторы).
Таблица 10.2
Состав бакинских газов крэкинга н пиролиза
(по АзНИП)
Компоненты Жидкофазн. крэкинг Парофазн. крэкинг Пиролиз
Водород 6,0 •8,0 15,0
Метан 30,5 32.0 <5,0
Этилен 4,5 12,5 17,0
Этан 18.0 14,0 7,0
Пропилен * 7,5 15,0 8,0
Пропан 15,U 6,5 1,0
Бутилены 6,0 6,0 2,8
Бутаны 6.0 2,0 0,2
Бутадиен . . . 1 — — 1,5
Высшие углеводороды 6.0 5,0 2,0
Окись углерода 0,5 — 0,5
Итого. . . а 100,0 100,0 100,0
468
Ближайшее 'рассмотрение табл. 102 приводит к следующим
выводам:
1. Наивысшее содержание водорода наблюдается в газе пи-
ролиза; при переходе от жидкофазного крэкинга к пиролизу
содержание .водорода возрастает с G до 1~>%, т. е. в 2,5 раза.
2. Содержание метана при переходе от жидкофазного крэ-
кинга к пиролизу возрастает в полтора раза (от 30,5 до 45%).
3. Содержание этана, пропана и бутана, особенно двух по-
следних, при переходе от жидкофазного крэкинга к пиролизу
резко снижается. Для жидкофазного крэкинга характерно до-
вольно высокое содержание пропана и бутана (в сумме'^о 21%)
4. Газы жидкофазного ырэкинга содержат сравнительно
мало непредельных углеводородов (в сумме около 18%). В га-
зах парофазного крэкинга и пиролиза''содержание этих углево-
дородов значительно выше (32—35%).
5. Наименьшее содержание этилена, наблюдается в газах жид-
кофазного крэкинга (4—5%), наибольшее — в газах пиролиза
(17% и выше); в газах парофазиого крэкинга содержание эти-
лена достигает 12—13%?.
6. Содержание пропилена в газах жидкофазного .крэкинга и
пиролиза примерно одинаково (7—8%); в газах парофазного
крэкинга содержание пропилена почти в 2 раза больше
(до 15%).
7. Содержание бутиленов в газах жидкофазного и парофаз-
ного крэкинга одинаково (6%); в газах пиролиза содержание
бутиленов значительно меньше (около 3%).
Интересно проследить теперь, как изменяется 'состав газов
пиролиза при постепенном изменении температурного режима
процесса от 600 и выше (табл. 1 оз).
Таблица 103
Состав газа при пиролизе газойля
(в процентах по объему)
(но Гроллу)
Газы —т-* Температуры
• 600° 650° 700’ 750° too0 850°
Водород 8,3 9,1 10,1 10,6 13.2 23,4
Метан 26,9 26.4 31,0 89,0 38,4 40,9
Ацетилен .... — ' — Сл еды 0,8
Этилен 29,4 29.1 28,6 29.6 32,6 27,7
Этан . - 10,2 7,9 7,0 6.7 5,2 3,1)
Пропилен 17,0 16.8 15,7 10.0 10,0 3,4
Пропан 1,8 2.4 2 1 1,4 06 0,3
Бутилен .... 5,7 7,2 4,6 2,0 0,0 0,0
Бутан Сумма нспре- .. 0,7 1,0 0.4 0,2 0,0 0,0
дельных . \ . 52,1 53,1 48,9 42,1 42,6 31,9
469
Как видно из Данных табл. 103, объемное содержание про-
стейших компонентов газов пиролиза — водорода и метана--
с повышением температуры процесса все время возрастает. Осо-
бенно обращает на себя внимание скачок в содержании водо-
рода при переходе от 800 до 850е, что нельзя не поставить
в связь с падением содержания в газе других основных ком-
понентов. Одни из этих компонентов характеризуются, особой
неустойчивостью к высоким температурам, вследствие чего со-
держание их с повышением температуры пиролиза быстро сни-
жается. Таковы, например, пропан, СзН8, бутан, С4Н10, в менвг
шей стейени — этан. СгН6, пропилен, СзН8 и бутилены, С4Н8.
Наконец, этилен, наиболее устойчивый из олефинов к высоким
температурам, обнаруживает тенденцию к снижению содержания
в газах пиролиза’лишь при температурах выше 800° [29].
Вполне отчетливое влияние на состав газов пиролиза оказы-
вает давление [27]. Уже повышение давления до 3 ат при 750°
заметно .снижает в газе пиролиза содержание тяжелых газов,
особенно непредельных, которые под влиянием давления легче
полимеризуются и уплотняются, а следовательно, уходят из га-
зообразной фазы. Естественным следствием удаления непредель-
ных является другое изменение состава газов пиролиза, а имен-
но— повышение содержания в них водорода, а также отчасти
метана и этана. Понижение давления оказывает противоположное
влияние: общее содержание непредельных в газе пиролиза,,
в частности содержание этилена и бутиленов, возрастает, со-
держание же метала и этана понижается. Для иллюстрации
этих последних может служить табл. 104.
Таблица 104
Состав газов пиролиза керосина при 750° и
равных давлениях
(по Добрянскому)
Газы « Давления
40 мм • 760 мм
Водород . • 15,6 14,8
Метан . 23,3 , 34,3
Ацетилен . 0.8 —
Этилен . 29,2 21,6
Этан . . 2,5 8,4
Пропилен 10,1 10.9
Пропан . 0,5 2,0
Бутилены 3,5 1.5
Бутан и высшие 9.2 5,0
Бутадиен 4.6 1,5
470
Заслуживает внимания, что аналогичное изменение состава газа наблю-
далось также при более низких температурах, например при крэкинге
в автоклаве грозненского керосинового дестиллата при 450° в условиях,
когда давление. создавалось нагнетанием инертного газа (азота). Как по-
называют данные табл? 105, и-здесь, аналогично тому, что наблюдается
при изменении состава бензина в подобных же условиях (табл. 100), со-
держание в газе непредельных СпНзп, но мере повышения суммарного
давления .в системе постепенно снижается; одновременно повышается со-
держание в газе углеводородов предельного характера С,гН2п+2; содержа-
ние же водорода, напротив, снижается, очевидно • за счет • протекающих
реакций гидрирования непредельных газов и их полимеров [26].
Таблица 105
Состав газов крэкинга грозненского керосинового дестиллата
(по ИГИ Академии наук)
Газы в % по объему Общее давление в ат
70 95 145 180 240 308 380 425
Водород 5,5 3,2 5,0 4,5 3,0 2,9 1.3 1,1
* 7.4 6,2 4,8 2,9 1,8 0,9 0,5 04
C/Jbjj+a • 86,2 87,7 89,9 91,6 94,4 i 96,6 97,9 97,9
До сих пор речь шла лишь о газообразных компонентах га-
зов крэкийга и пиролиза. Газы эти по выходе из холодильников
содержат также значительную примесь более высоко кипящих
углеводородоаз, т. е. паров легкого крэкинг-бензина. Содержание
этого компонента зависит преимущественно от температуры про-
цесса. В наиболее благоприятных условиях оно может достигать
400 г бензина на 1 .и3 газа. Для улавливания этого бензина за-
водские установки крэкинга и пиролиза снабжаются специаль-
ными скрубберами.
Термическое разложение нефти и ее продуктов при 430—550°
дает, как было показано, мало газа, который, однако, весьма бо-
гат бензином. По мере повышения температуры (до ~ 650°) при
нарастающем газообразовании содержание бензина в газе посте-
пенно падает вследствие параллельно нарастающего крэкинга
легких, т. е. бензиновых компонентов. При еще более высоких
температурах (~ 6'50—750°), когда основная масса парафинов
и нафтенов уже претерпела распад, газ начинает обогащаться
ароматикой, в первую очередь бензолом. Наконец, при темпера-
турах выше 750° наблюдается новое снижение в содержании
парообразных компонентов вследствие все возрастающего про-
цесса распада ароматики с образованием нелетучих полицикли-
ческих углеводородов и кокса.
Коксообразование. Как. было указано выше, коксообразование
при крэкинге представляет собою сложную реакцию уплотнения,
сопровождающую основные реакции крекинг-процесса — реак-
цми разложения. Коксообразование теснейшим образом связано
с теми же основными факторами . крэкинг-процесса, как обра-
зование крэкинг-бензина.' Однако зависимость эта при коксо-
образовании носит в некоторых отношения^ существенно иной-
характер.
Весьма важное значение при коксообразования имеет пре-
жде всего характер крокируемого материала. При жидкофазном
крэкинге легких дестилатов и при не слишком высокой тем-
пературе процесса (425—450°), коксообразование вначале вовсе
не наблюдается: происходит лишь накопление тяжелых продую
тов уплотнения (см. выше), и только после их образования на-
чинается выделение кокса. Естественно поэтому, что чем тяже-
лее нефть или нефтепродукт, тем легче происходит коксообра-
зование при крэкинге, так как подобного рода сырье содержит
уже в готовом виде материал для образования кокса, в виде тя-
желых масел, смол и асфальтенов. Такое влияние тяжелых со:
ставных частей нефти на коксообразование хорошо иллюстри-
руется данными табл. Ю6: чем тяжелее исходный материал, тем
легче идет коксообразование при его крэкинге.
Таблица 106
Коксоойрйзов^ние при крэкинге (423°, 1 час)
(по ГрозППИ) ’
Крэкирусыыи продукт Уд. вес ' продукта Выхода на кокс в %
Парафинистый дестиллат . . 0,883- 0,00
Сураханскпй мазут ........ 0,890 0,24 ’
Грозненский парафинистый мазут . 0,906 0,95
Вознесенская нефть ’ . 0,929 1,05
Бнби-эйбатский мазут ....... 0,931 1.51 ‘
Бинагадинская нефть 0,922 99
Калужская нефть •. . . . 0,957 492
Очень характерно изменение скорости коксообразования со
временем: если скорость образования крэкинг-бензина, как было
показано выше, по мере хода процесса постепенно уменьшается,
то для коксообразования наблюдается как раз обратное явле-
ние. Причина понятна: если количество и концентрация масел,
при разложении которых получается крэкинг-бензин, по мере
течения процесса постепенно уменьшается, то в то же время ко-
личество и концентрация продуктов уплотнения,’ из которых
образуется кокс, постепенно возрастают, а вместе с тем, есте-
ственно, возрастает и скорость коксообразования. В табл. 107
приведены результаты крэкинга парафинистого дестиллата (425°,
5—ю аг), которые могут служить иллюстрацией сказанного.
473
Таблица id?
ОЙряйование бензина н к-окса во вр$иенн
(по ГрозНИИ)
Продолжительность крэкинга в мин. Выхода продуктов крэ- кинга в %
фракция до 200° фракция до 300° кокс
34 14,0 31.0 0,00
63 21,5 46,9 0,00
94 34.2 63,2 0,62
300 38,0 63,0. 8,59
Фиг. Ю7 дает наглядное изображение данных, приведенных
в табл. 107.
Практическое значение только что сказгтнного громадно: оче-
видно, слишком большая продолжительность крэкинг-лроцеоса и
стремление получить ма-
ксимальный выход бен-
зина в одном процессе не-
выгодны из-за усиленного
коксообразования; техни-
чески выгоднее система
повторного крэкинга (см.
ниже).
Что касается, наконец,
влияния температуры и
давления на процесс ко-
ксообразования, то в этом
отношении здесь имеется
полная аналогия с теми
закономерностями, КОТО- Фиг. 107. График к табл. 107.
рые наблюдаются при
образовании крэкинг-бензина, а именно: скорость коксообра-
зования увеличивается вдвое при повышении температуры па
каждые 10° (или в б раз на каждые 25э); от давления в преде-
лах Ю—40 ат коксообразование не зависит. Приведенный
в табл. 108 материал по крекингу грозненского -парафинистого
мазута может служить иллюстрацией этих положений.
Как уже было указало, крэклиговый кокс представляет соиою вещество,
крайне сложное и к тому же далеко не однородное. Анализ типичного
образца крэкингового кокса дает следующие результаты (табл. 109).
Состав крэкингового кокса в зависимости от условий крэкинга может
существенно изменяться. Как показывает табл. 110, крэкинговый к кс
состоит из масел, смол, асфальтенов и карбоиЛов. По море углубления
крэкинга содержание в коксе к’арбоидов быстро возрастает, содержание
же всех остальных компонентов резко снижается. В конечном итоге по пи
вся органическая масса кокса может превратиться в карбон ды, т- е. про-
дукты наиболее глубокой конденсации углеводородов, ‘ олизкие к каменно-
угольному кокбу.
473
ibfijuua 1Ьё
КоксообраЛ ванЙе при крэкинге грозненского мазута
(по ГрозНИИ)
Условия крэкинга Выхода на коке, в %
темпера- тура давление в ат время в мин.
425° 62 0,95
425° 10 1,05
425 > 10 32 0,59
425° 20 32 0,57
425° 30 32 0,83
425° 40 32 0,84
Таблица ЮН
Анализ крэкингового кокса
(по Кроссу)
Наименование составных частей
Летучие вещества (до 500°)........
Углерод .............. ...........
Зола..............................
Содержание в %
13,08
80,42
6,50
Сера.............................
Железо ..........................
100,00
1,83
2,76
Таблица 1Щ
Состав крэкингового кокса
(по ГрозНИИ)
Крэкинговый материал Условия крэкинга Состав кокса в %
темпера- тура давле- ние в ат время в мин. смол и масел асфаль- тенов карбои-! дов ,
Масляный гудрон 450° 20 33 32,0 10.5 57,5
Калужская нефть 450’ 20 63 19,4 3,2 77,4’
Вопрос об индивидуальных компонентах крэкингового кокиа-
и их ближайшей химической природе тесно связан с вопросом
о химической природе основных источников его образования и
будет рассмотрен- ниже в связи с крэкингом ароматических
углеводородов.
474
Крэкинг углеводородов различных рядов.
В предыдущем изложении поведение углеводородов различ-
ных рядов при крэкинге неоднократно подвергалось обсужде-
нию. Этот материал должен быть теперь систематизирован и по-
полнен новыми данными [зо].
Парафиновые углеводороды. Как было указано выше, пара-
финовые углеводороды нормального строения при крэкинге
в умеренных температурных условиях (около 425°) претерпе-
вают разложение, которое происходит главным образом как бы
в середине или вблизи середины молекулы. Продуктами '/того
разложения является смесь предельных и непредельных угле-
водородов, усложненная продуктами реакций вторичного по-
рядка, а именно: продуктами уплотнения непредельных углево-
дородов первичного распада, а также продуктами распада от
вторичного и последующих разложений парафинов. Такой после-
довательный крэкинг парафинового углеводорода можно выра-
зить следующей примерной схемой:
с82н6в=с16н31+с1вн;и —>
10
полимеры
8
Углубление процесса путем повышения температуры до 600°
и выше смещает место разрыва нормального парафина ближе
к концу цепи, так что газообразование усиливается: образуются
низшие парафины и высокомолекулярные этиленовые углеводо-
роды [20, 30], например:
СН3 • (СН^ • СН2 • CHj = СН3 • (СН2)Ь • СН : СН2 + СН4 и т. п.
Одновременно увеличивается количество продуктов уплотне-
ния. Табл. 111 может служить иллюстрацией этих положений
на примере крэкинга додекана (С12Н26).
Таблица 111
Крэкинг додекана
(по Тиличееву и Фешину)
Продукты крэкинга 425°, 1 час 425°, 3 часа. 450°, 1 час
Газы и потерн Крэкинг-бензин до 200° Продукты уплотнения. 2,8% 15.5% П.7% 10,4% 29,7% 20,4% 16,6% 42,3% 17,5%
Сунна продуктов крэкинга .... 30,0% 60,5% 76,4%
475
Для характеристики крэкинга нормальных парафинов очень
важно также, что в начальной стадии этого процесса водород
в газообразных продуктах крэкинга и. кокс в остатках совер-
шенно отсутствуют.
Скорость разложения парафиновых углеводородов нормаль-
ного строения находится в несомненной зависимости от их мо-
лекулярного веса. Так, при крэкинге в одинаковых условиях
(425°, 1 час) декан,- С10Н22. дал 27,3% продуктов разложения,
тогда как дотриаконтан, СзгНбб, —84,5%. С другой стороны, вы-
хода бензина при крэкинге в тех же условиях додекана, Ci2H2e,
гексадекана, СюПзг, и дотриаконтана СзгИов, практически ока-
зались почти одинаковыми (15.5; 16,7 й 16,4%), т. е. не обна-
ружили какой-либо зависимости от молекулярного веса исход-
ного материала.
Крэкинг изопарафиновых углеводородов изучен пока -недо-
статочно. Те немногие примеры, которые были исследованы в этом
направлении, например диизоамил; C10II22, и один из диметил-
тетрадеканов, С16Н34, заставляют думать,.что скорость крэкинга
изопарафинов и соответственно выхода- на продукты разложения
при 425° здесь несколько больше, чем в случае парафинов нор-
мального строения.1 К аналогичным выводам приводит исследо-
вание изомерных октанов: нормальный октан оказался в усло-
виях крэкинга значительно устойчивое, Чем его изомеры. Отме-
чено также, что в газах крэкинга изопа-рафинов содержится за-
метное количество водорода. Однако для более полных заклю-
чений по ^сем этим вопросам требуются дополнительные иссле-
дования.
Большого внимания кап: с теоретической, так и с практиче-
ской точек зрения заслуживают крэкинг и пиролиз низших па-
рафинов. Очевидно прежде всего, что низшие парафины предста-
вляют собою наиболее простые и благодарные объекты для изу-
чения кинетики термического распада углеводородов; понятно
поэтому, что именно с этой стороны следует ожидать оконча-
тельного разъяснения вопроса о механизме процессов этого рода.
Вместе с тем изучение термического распада низших парафинов
представляет интерес с точки зрения проблемы рациональной
утилизации естественного газа и вообще наиболее простых ком-
понентов нефти. Естественно поэтому, что новейшая литература
по вопросам крэкинга и пиролиза метана и его ближайших го-
мологов громадна.
Из всех парафиновых углеводородов метан наиболее устой-
чив к высокой температуре. При наг.ревапии до 480° в течение
в дней он совершенно не изменяется, и даже ..при .700° в отсут-
ствии специальных катализаторов разложение его протекает
крайне медленно; только при температурах выше 900° распад
метана приобретает заметную скорость, причем реакция про-
текает но уравнению:
СН4 = С-|-2Н2
1 Ииирогив, при температурах порядка 600—650° изопарафины, пони-
д if миму, более устойчивы, чем нормальные парафины.
476
Иллюстрацией может служить таил. 112, в которой приве-
дены результаты опытов разложения метана в кварцевой трубке
при 600—1000° и атмосферном давлении; о ходе распада метана
можно судить здесь ио количеству водорода, образующегося
в данных условиях в течение определенного времени, например
в течение 10 мин [31]. •
Таблица 112
, Термическое разложение метана
Температура 600’ 7СС° 800° 850° 900° 950° 1000°
Содержание водорода в газе 0,2 0,9 2,1 3.4 14,7 22,1 37,5
В присутствии металлических катализаторов термическое раз-
ложение метана значительно облегчается; например, над палла-
дием реакция начинается уже при 230°, над никелем — при
320°, над железом — при 330° и т. д. .
Разложение метана па углерод (уголь, графит) и водород со-
гласно вышеприведенному уравнению является обратимой реак- •
цией; доказано, что при достаточно высоких температурах можно
не только разложить метан на составляющие его элементы, но и
обратно — синтезировать метан из этих элементов. И та и дру- ,
гая реакции должны приводить, очевидно, к равновесной си-
стеме:
СН4 zi С + 2Щ.
Это равновесие неоднократно подвергалось как эксперимен-
тальному изучению, так и теоретическому исследованию на базе
термодинамики (см. ниже). В тех случаях, когда данное равно-
весие не осложнялось реакциями вторичного характера, для
его константы по тину мономолекулярной реакции неоднократно
но ручались хорошие совпадения между' теорией и опытом.
Термическое разложение метана нередко сопровождаете я
образованием из промежуточных продуктов его распада (радика-
лов) более высокомолекулярных углеводородов, каковы: этап,
олефины, бутадиен, ацетилен, ароматика. Синтез этих продук-
тов можно выразить следующей схемой [32]:
2СН4 -> Н,4-2СН3' -> 2Н, + 2СН/ -> ЗЩ + 2С11'" -> 4HS4-2C
И И II
с.,н6 с3н4 с.,щ
Образование из метана олефинов наблюдается преимуще-
ственно при сравнительно низких температурах (до 1000°). и
пониженном давлении. Суммарные выхода олефинов могут дости-
гать при этом 8—9% от количества разложившегося метана,
однако в патентной литературе можно встретить и более высо-
кие выхода. Образование в данных условиях высших олефинов
477
(пропилена и т. д.) можно 'представить себе по обобщенной схеме
синтеза этилена из метана:
w(CIV) -> C„H2,t.
Ацетилен, напротив, образуется из метана, преимущественно
при высоких температурах, порядка 2000° и выше, хотя в не-
бе 1ыпих количествах ето образование наблюдается почти при
всех температурах, когда происходит термический распад ме-
тана.
Что касается, наконец, ароматики — этой наиболее интерес-
ной части продуктов пиролиза метана, то основные условия для
ее получения заключаются, невидимому, в особо тщательно по-
добранной для реакции температуре (до 1200°), возможно крат-
ком времени пребывания газа в зоне реакции и специальных ка-
тализаторах. Главным компонентом получаемой при этом смолы
является бензол (до 50% и больше) Г По вопросу о механизме
образования ароматики при пиролизе метана взгляды до сих
пор расходятся: по мнению более ранних исследователей арома-
тика образуется здесь в результате уплотнения ацетилена, тогда
как новейшие авторы считают источником ее образования эти-
лен и бутадиен (см. ниже).
Этан. По сравнению С метаном этан значительно менее устой-
чив -к высокой температуре. При отсутствии катализаторов он
начинает разлагаться около 483°, но под влиянием некоторых
металлов эта температура значительно снижается; так напри-
мер, в присутствии никеля этан разлагается уже при 325°.
Начальными продуктами распада (диссоциации) этана при
температурах порядка 500° являются этилен и водород. Несо-
мненно, что эта реакция также проходит через промежуточную
стадию мимолетного образования радикалов (СН3 и т. п.). Но
так как, с другой стороны, наряду с диссоциацией этана на эти-
лен и водород -в данных условиях несомненно имеет место и
обратная реакция, т. е. гидрогенизация этилена с образованием
этана, в то в совокупности весь этот процесс может быть выра-
жен следующей равновесной схемой:
Н3С —СН3 2СН/ -> 2СН/ + И2-+С2Н4 + и2.
Кроме этого основного направления реакции, при пиролизе
этана|ыожет наблюдаться ряд побочных превращений.
Прежде всего за счет водорода, накопляющегося iiq мерс дис-
социации этана, может происходить гидрирование промежуточных
радикалов (СНз' и СН-г") с образованием метана. Очевидно, для
1 Пиролиз метана и ого ближайших гомологов представляет большой
интерес также с точки зрения увеличения ресурсов моторного топлива (ср.
гл. V). Исходным материалом для этой цели могут служить естественный
газ, не конденсирующиеся газы нефтеперегонных установок, отходящие
газы от стабилизации газового бензина и т. п. Выхода бензина (до 200°)
из чистого метана — не велики: около 5 л бензина на 100 .и3 газа. Однако
эти выхода сильно возрастают по мере обогащения метана его ближайшими
гомологами: так например, газ от стабилизации газового бензина дал уже
49,3 л бензина ца каждые 100 л3 газа [33].
478
этого необходимо известное накопление водорода, т. е. возмож-
ное углубление диссоциации этапа., что в свою очередь требует
надлежащего 'повышения температуры. 11 действительно, замет-
ное образование мотана при пиролизе этапа наблюдается лишь
при температурах выше 700°.
Второе побочное .направление реакции при пиролизе этана —
это распад метана. На основании вышеизложенного это напра-
вление реакции требует температур порядка 900° и выше. Про-
дукты этого направления реакции рассмотрены выше.
Наконец, третье побочное направление реакции при пиролизе
этана заключается в образовании жидких продуктов, в частно-
сти богатого ароматикой бензина. Химическая сущность этого
направления сводится, очевидно, к процессам полимеризации,
в которых может принимать участие не только основной про-
дукт диссоциации этана, этилен, но и промежуточно образую-
щиеся радикалы (СН/' и СН "). joo м3 чистого этана могут дать
при Пиролизе в надлежащих условиях до 24 л бензина, состоя-
щего примерно на 75% из ароматических углеводородов. Примесь
к этану высших гомологов метана существенно повышает выхода
бензина.
Иллюстрацией термического распада этана может служить
табл. 113 [34].
• Таблица ИЗ
Термический распад этана
(по Хм-Унлеру)
Темпе- ратура Выход в % от взятого этана 4 Состав газа в % по объему
жидкие про- дукты уголь ацети- лен этилен водород • метан этан
700° 0 0 2,8 21,3 21,9 2,7 49,6
750° 2,1 Следы 4,3 24,3 32,3 13,3 21Д
800° 9,7 3.0 21,1 38,4 21,1 12/7
850° 17 9 2,3 14,7 41,4 ’ 32,4 7,5
900° 21,9 3,1 1,8 5,0 44 3 38,9 8,4
950° 12.8 13,9 10 3,8 52,6 40.8 1,4
1000° 6,5 16,2 0,8 2,4 58,5 33,9 4,1
Как видно из таблицы, при 700( в основном этан действи-
тельно распадается на этилен и водород. Жидкие продукты по-
являются в продуктах реакции лишь *при 750° и выше,
а уголь — при 900°. Содержание водорода в газе непрерывно
возрастает по мере повышения температуры процесса; содержа-
ние же этилена достигает максимума при 750 и при дальней-
шем повышении температуры быстро падает, надо думать, за счет
частью образования жидких полимеров, частью дальнейшего рас-
пада этилена (см. ниже).
Пропан и бутаны. Термическая устойчивость этих углево-
дородов заметно меньше, чем у этана: пропан начинает разла-
479
ГггГЬсй прй йагреВаиий ойоЛо 460й, а йорм‘а.пьйый бутай и изо-
бутан— около 435°. Металлические катализаторы (никель, же-
лезо и т. п.) и здесь более или менее сильно снижают темпера-
туру начала разложения. Тате например, в присутствии никеля
иропан диссоциирует уже при 200° и быстро распадается при
350—400°.
Продуктами пиролиза пропана и бутанов являются низшие
парафины и олефины, бутадиен, ацетилен, некоторые аромати-
ческие углеводороды, уголь и водород. Можно представить себе
целый ряд путей образования этих продуктов из данного сырья,
однако действительное значение, естественно, имеют лишь немно'
тле из теоретически возможных схем и прежде всего те, кото-
рые разъясняют начальные стадии пиролиза пропана и бутанов.
Основными реакциями начальных стадий пиролиза простей-
ших гомологов метана являются их Дегидрогенизация, т. е. рас-
падение на водород и соответствующей олефин, и расщепление
на два низших углеводорода — предельный и непредельный.'
Для случая пропана эти две реакции приводят к четырем про-
дуктам: первая — к водороду и пропилену, вторая — к метану и
этилену, по следующим схемам:
С3Н8 = Но-р С3Н6:
’ ' с3н8 = сн4+С2н4-
Примерно до 575° пиролиз пропана идет одновременно и оди-
наково по обоим этим направлениям. При более высоких темпе-
ратурах процесс дегидрогенизации отходит на второй план, усту-
пая место второй реакции с отщеплением метана. Все другие
продукты пиролиза пропана несомненно являются продуктами
вторичного происхождения.
Пиролиз изобутана происходит в основном аналогично пи-
ролизу пропана и может быть выражен • следующими уравне-
ниями:
(СН,)2СН СН, = Но + (СН,)2С : СНО;
(CHJ2CH • СН3 = СН4 + СА3 СН : СН2.
Эти реакции при 600° и даже при 700° являются, повиди-
мому, преобладающими при’ пиролизе изобутана [35]; однако
уже при 750° на первое место среди продуктов разложения
(больше 50%), выступает метан, очевидно в значительной мере
за счет реакций вторичного характера.
Что касается, наконец, пиролиза норм, бутана, то хотя и здесь
начальными реакциями являются дегидрогенизация и расщепле-
ние углеводорода, однако легко видеть, что в- соответствии со
строением норм, бутана каждая из этих реакций может иметь
два различных направления, так что в итоге мы имеем четыре
начальных реакции термического распада норм, бутана;
СН, • СНо • СН2 - СН3 = Но + СН, • СН2 • СН : СН>;
(Я1, • сн; • СН2 • СН3 = Н2“Ч- СЩ • СН : СН • СН3Г
СН’ • сн; - СН2 - СН3 = СН4 + СН, • СН : СН2;
OIL. сн; - СНо • СН3 = СН3 - СН8+ СНо: СНо.
480
При температурах порядка 400°, по крайней мере в присут-
ствии некоторых катализаторов (СггОз), пиролиз бутана идет
главным образом в направлении его дегидрогенизации, причем
образуются, повидимому, оба бутилена нормального строения
(а и Р). Однако уже при 600—700 на первое место выступают
здесь реакции распада бутана по обеим приведенным выше схе-
мам, т. е. с образованием либо этана и этилена (распад по сере-
дине молекулы), либо метана и пропилена (распад с краю); ре-
акции же дегидрирования отходят на второй план. Характерно,
что чем выше температура пиролиза бутана, тем, повидимому,
больше отодвигается место его распада к краю молекулы; на
это указывает, между прочим, непрерывное возрастание содержа-
ния метана в газообразных продуктах- реакции вплоть до 900°.
Образование остальных продуктов пиролиза бутана можно рас-
сматривать как результат вторичных процессов.
Большой практический интерес представляет пиролиз смеси естествен-
ных газов, получаемой при стабилизации газового бензина и состоящей
в основном из этана, пропана л бутана (пропан-бутановая фракция). Пи-
ролиз этой газовой смеси имеет своей задачей получение из нее по преиму-
ществу жидкого топлива. Реакцию приходится вести при довольно высокой
температуре (800—900°). Некоторые авторы рекомендуют применение ка-
тализаторов, особенно меди. Выхода дегтя достигают по некоторым ука-
заниям до 350 см3 дегтя на 1 м3 газовой смеси; в состав его наряду с про-
стейшими непредельными и ароматическими углеводородами входят также
конденсированные ароматические системы, как-то: нафталин, фенантрен,
аценафтен, пирен и хризен [36].
Пентаны и высшие парафины. Пиролиз пентана и ближай-
ших гомологов (до декана включительно) изучены гораздо
меньше, чем пиролиз низших парафинов. Точное установление I
температуры начала распада для большинства этих углеводоро-
дов отсутствует; повидимому, температуры эти ниже 600°. При;
сравнительно низких температурах (порядка 650—750 ) здесь
наблюдаются явления дегидрогенизации с образованием высших
олефинов и водорода. Однако с повышением температуры реак-
ции, а также с увеличением молекулярного веса исходного угле-
водорода реакции дегидрирования отходят на второй план; на
первое же место и здесь выступают типичные реакции распада.
На это указывают как исчезновение в продуктах пиротиза выс-
ших олефинов, так и постепенное накопление в этих продуктах
по мере повышения температуры реакции водорода, метана и эти-
лена; очевидно и здесь с повышением температуры распад исход-
ного углеводорода смещается преимущественно к краю моле-
кулы. Само собой разумеется, что термический распад всех этих
углеводородов сопровождается также вторичными процессами
полимеризации и уплотнения и реакциями распада первичных
продуктов реакции.
Из ботее высокомолекулярных парафинов лучше других были
изучены додекан и гексадекан. Картины крэкинга и пиролиза
этих углеводородов в общем 'вполне аналогичны только что рас-
смотренным и частично разобраны выше.
31 Зак. 1622. Химия нефти.
481
Подводя итоги всему, что известно по данному вопросу,
можно оказать, что крэкинг парафиновых углеводородов -предста-
вляет собою сложный процесс, начальные стадии которого состоят
из двух основных реакций: дегидрирования молекулы - углевода,
рода и ее распада. Первая из этих реакций легко наблюдается
для низших, особенно 'газообразных, парафинов при умеренных
температурах процесса. Для более высокомолекулярных парафи-
нов, начиная примерно с CeHu, эта -реакция отходит на второй
I план, и, начиная с 450—500°, единственной первичной реакцией
| крэкинга остается здесь распад парафина на два углеводорода,
из которых один принадлежит к ряду парафинов, другой—
к ряду олефинов.
Большое влияние на термический распад парафина имеют
температура и давление. При умеренных температурах (до 500°)
и высоких давлениях распад парафина происходит симметрично,
т. е. ближе к середине молекулы; соответственно этому газообра-
зование .может быть в данных условиях невелико. Наоборот, вы-
сокие температуры (выше 500°) и низкие давления способствуют
сдвигу распада молекулы к ее краю и вызывают усиленное газо-
образование.
Остается отметить, что изучение -кинетики крэкинга пара-
финов привело к тому же заключению,’ которое было отмечено
для термического распада метана: неглубокие начальные стада
крэкинга парафинов в полном соответствии с приведенными
выше схемами должны рассматриваться в первом приближении
как реакции мономолекулярные (т. ё. первого порядка).
Этиленовые углеводороды. Как известно, нефти и обычно
применяемые для крэкинга нефтепродукты почти или вовсе не
содержат непредельных углеводородов. Эти углеводороды, однако,
образуются при крэкинге сырья предельного характера и, оста-
ваясь в сфере действия основных факторов крэкинг-процесса,
вследствие своей высокой реакционной способности вступают
в разнообразнейшие реакции распада и уплотнения как друг
с другом, так и с другими молекулами (ароматика). Таким обра-
зом этиленовые углеводороды принимают самое деятельное уча-
стие в образовании крэкинг-лродуктов. Ближайшее изучение
их поведения в условиях крэкинга на индивидуальных углево-
дородах этого ряда должно представлять поэтому выдающийся
интерес с точки зрения уяснения химизма крэкинг-процесса
Общее представление о поведении этиленовых углеводородов
в условиях крэкинг-процесса- можно составить себе из ближай-
шего рассмотрения результатов жидкофазного крэкинга капри-
лена, CeHie, и гексадецилена, СхвНэг, результаты которого [37]
сведены в табл. 114.
Как это видно на примере каприлена, в умеренных темпера-
турных условиях (425°) реакция разложения этиленового угле-
водорода протекает сначала крайне медленно и дает лишь не-
большое количество продуктов распада. Процесс идет сначала
главным образом в другом направлении, а именно—в сторону
образования продуктов уплотнения. Лишь постепенно коляде-
482
Таблица 114
Крэкинг каприлена и гексадецнленж
(по Тиличееву и Фейгину)
Продукты крэкинга Каприлен Гексадецилен
425°, 14 мин. 425°, 60 мин. 425°, 14 мин. 425°, 60 мнн.
Газы и потери .... Жидкие продукты раз- ложения Продукты уплотнения . 3,2% 1.8% 23,8% юз% 10.9% 38,8% 3,2% 12.4% 36,9% 5.2% 37,9% 27.2%
Сумма продуктов крэ- книга 28,5% 6О,О0/о 52,5% 70,3%
ство продуктов разложения начинает ’накопляться, и такое на-
копление связано, очевидно, с разложением не только исходного
углеводорода, но и продуктов его уплотнения, образовавшихся
в первые фазы процесса.
Механизм реакции уплотнения этиленовых углеводородов под
влиянием высокой температуры и давления’ остается' пока не-
выясненным. Можно лишь условно принять, что реакция проте-
кает здесь в первую очередь по той же схеме, ок и уплотнение
тех же углеводородов под влиянием серной кислоты, хлористого
цинка и других подобных реагентов, т. е. приводит к образова-
нию ряда непредельных полимеров исходного углеводорода,
а именно: димера, тримера и т. п. [38]. В полном согласии с та-
ким допущением находится высокая непредельность продуктов
кратковременного крэкинга этиленовых углеводородов, которую
можно видеть на примерах табл. 115.
Таблица 115
Свойства крэкннг-фракцнй каприлена
(по Тиличеееу и Фейгину)
Температура кипения фракций 425°, 14 мин. 425°, 60 мин.
’<*г содержание непредель- ных jis d4. содержание непредель- ных
50 — 80° 0.7С2 74,5 0,701 37,2
80 — 110° 0,707 84,7 0,718 —
110 — 135° 0,725 93,2 0,741 50,5
135 — 160° 0,737 94,2 0.776 48,8
160 — 180° 0,761 84.6 0,796 54.9
180 — 203° 0,782 81,2 0,807 52,7
Вслед за реакциями уплотнения ближайшей стадией крэ-
кннга этиленовых Углеводородов являются реакции разложения,
31* 483'
которые в первую очередь должны привести, очевидно, тщ»
к непредельным углеводородам.
Действительно, как видно из данных табл? 115. низшие
фракции продуктов кратковременного крэкинга каприлена (тем-
пература кипения 122,5—123,5°) по крайней мере на 75—85%
состоят из непредельных углеводородов с температурой кипения
какнцже, так и выше температуры кцпения каприлена. Оче-
видно, это — продукты разложения (крэкинга) как самого ка-
прилена, так и его полимеров. На примере нормального бути-
лена '(I) и его димера (II) эти первые этапы последовательного
хода крэкинга этиленовых углеводородов могут быть выражены-
следующей приблизительно схемой:
I. 2СН3 • СН2 • СН : СН2
U И. СН3 • СН2 • СН<СН8) • СН : СН • СН2 • СН8-
LIII. СН3 • СН2 • CH(CHS) • СН: СН2+ОД4
Хотя последняя (III) стадия этой схемы является, лишь при-
мерной, тем не менее, совершенно ясно, Что одним из конечных
результатов рассмотренного этапа крэкинг-процесса этиленовых
углеводородов должно быть появление новых высокомолекуляр-
ных углеводородов этиленового ряда изостроения.
Данные’ той же табл. 115 позволяют сделать дальнейшие
важные заключения о последующем ходе крэкинг-процесса эти-
леновых углеводородов. Действительно, сопоставив содержание
непредельных в соответствующих фракциях крэкинг-продукта
после 14-минутного и часового крэкинга, мы видим, что по мере
хода процесса непредельность соответствующих фракций резко
снижается; так например, для фракции ПО—135°, в которой
должна быть сосредоточена главная масса исходного каприлена,
после часового крэкинга содержание непредельных снижается
почти вдвое и т. д. Отсюда ясно, что в процессе крэкинга эти-
леновых углеводородов наряду с процессами уплотнения и .раз-
ложения происходит также новообразование углеводородов пре-
дельного характера.
Вопрос о ближайшей природе углеводородов предельного ха-
рактера, образующихся при крэкинге этиленовых углеводородов,
изучен пока недостаточно. Исследование этого вопроса на раз-
личных примерах показало, что эта часть продуктов крэкинга
этиленовых углеводородов состоит из смеси парафинов, нафте-
нов и ароматических углеводородов. Чтобы проследить механизм
и условия их образования, необходимо рассмотреть поведение
в условиях крэкинга низших олефинов и в первую очередь,
этилена.
Этилен при отсутствии катализаторов под влиянием уме-
ренных температур (порядка 350—450°) претерпевает сначала
лишь полимеризацию и уплотнение с образованием главным об-
разом высших олефинов. При более высоких температурах; пр®-
484
мерно от 550е, среди продуктов превращения этилена появля-
ются также метан, водород и, наконец, уголь, т. е. продукты раз-
ложения этилена, а также этан как продукт его гидрирования.
В зависимости от условий, т. е. температуры, давления и вре-
мени пребывания газа в зоне высокой температуры, выхода от-
дельных продуктов термического превращения этилена колеб-
лются в широких пределах. Иллюстрацией может служить
табл. 116, в которой сведены результаты опытов термического
распада этилена при пропускании его через кварцевую трубку,
причем время пребывания газа при соответствующих темпера-
турах изменялось от 46 до 58 сек. [39].
Термическое разложение этилена
(по Уилер-Вуду)
Таблица 116
Темпе- ратура Выхода в % Состав газа в % по объему
жидкие про- дукты уголь н2 СН4 С2Н6 с2щ СзЩ и С4Нв
650° 1.6 Нет 0,7 0,5 2,0 89,9 3,8
700° 12,2 п 3,2 4.9 4.9 66,2 5,0
750° 28,2 Следы 7,2 16,7 8,6 47,6 3,2
800° 36,1 1,4 17,3 33,7 6,9 29,0 1,1
850° 31,4 11,9 35,8 49,7 3,3 12.2 0,5
900° 13,4 13.4 51,0 55,2 2,2 4,6 Нет
Как видно из табл. 116, в указанных выше условиях измене-
ния, претерпеваемые этиленом даже при 650°, не велики и
в основном сводятся к образованию продуктов полимеризации и
уплотнения (пропилен, бутилен, жидкие продукты). Начиная
с 700°, наблюдается постепенное возрастание в газах водорода и
метана;.однако уголь появляется лишь при 760—800°. Количе-
ство этих продуктов распада с повышением температуры непре-
рывно возрастает, а содержание этилена в газе соответственно
падает. Характерно нарастание выходов на жидкие продукты
до 800° (maximum), тогда как содержание пропилена й бути-
лена возрастает лишь до 7«00°. Состав жидких продуктов разъ-
ясняет это кажущееся несоответствие: в основной своей массе
они состоят из ароматических углеводородов; так например,
жидкие продукты, полученные пиролизом этилена при 700°,
примерно на 50% состояли из бензола.
•Одним из важнейших продуктов уплотнения этилена является
бутадиен (в табл. 116 не показан). Так как полимеризация эти-
лена в наиболее умеренных условиях в первую очередь должна
приводить к бутилену, то, повидимому, бутадиен представляет
собою не что иное, как продукт дегидрирования бутилена, так
485
что образование этого диена можно представить следующей
ехемой: 2С„ . = сщ. сн . GH. CHj.
СН3 • СН2 • СН : СН2 = Н2+СН2: СН • СН : СЩ.
Ввиду общеизвестной лабильности диеновых систем темпе-
ратурные пределы, в которых при пиролизе этилена можно на-
блюдать и улавливать образование бутадиена, оказываются до-
вольно ограниченными. Имеются указания, что при 600° бута-
диен еще не наблюдается, что оптимальная температура его
образования — около 750° и что при 900° он снова исчезает
из продуктов пиролиза этилена, претерпевая более глубокое
уплотнение и в конечном итоге превращаясь в ароматику.
Роль бутадиена и его гомологов как промежуточных продук-
тов при образовании ароматических углеводородов в процессе
крэкинга и пиролиза олефинов в настоящее время получила ши-
рокое признание. Тем не менее, детали химизма этого процесса
до сих пор не могут считаться полностью установленными. Наи-
более вероятным представляется следующий путь этого слоав-
кого процесса’ [40].
Поскольку твердо установлено, что диеновые углеводороды
легко конденсируются с непредельными соединениями с образо-
ванием циклических систем («диеновый синтез»), то следует
ожидать, что первой стадией превращения бутадиена в бензол
является образование циклогексена' по схеме:
/СН, /\2
НС сн2 НС сн2
.1 + II = II I
НС сн2 НС сн2
х
Второй стадией процесса должно быть образование циклогек-
сена из бензола, что можно представить себе как результат
одновременно протекающих процессов гидро- и дегидрогенизации
циклогексена в циклогексан и бензол по схеме:
СН
нс^Чзн
II • I *-
HC//CH
сн
бензол
сн,
н2с/\сн2
I I
НзС^/СЩ
СН, •
циклогексан
В пользу приведенных схем образования ароматики при пи-
ролизе этиленовых углеводородов можно привести целый ряд
доводов и соображений, как-то:
1. Экспериментально установлено, что циклогексен и его го-
мологи действительно содержатся среди продуктов нироавдл
олефинов при умеренных температурах.
486
2. Установлено также, что циклогексен и другие нафтилены
при нагревании, правда, в присутствии некоторых катализаторов,
действительно легко претерпевают реакции одновременной гидро-
и дегидрогенизации с образованием нафтена и ароматического
углеводорода [41]. Для циклогексена ©тот процесс протекает по
следующему уравнению:
3.. Наконец, присутствие циклогексана и вообще нафтенов
в продуктах пиролиза олефинов также является давно устано-
вленным фактом; дальнейшая же судьба их в данных условиях
понятна: путем дегидрогенизации при температурах" порядка
Ь00° циклогексан и его гомологи превращаются в соответствую-
щие ароматические углеводороды.
Переход в ароматические углеводороды представляет собою
лишь одно из возможных превращений этилена и его гомологов
под влиянием высокой температуры. В более умеренных темпе-
ратурных условиях превращения эти на первом этапе могут
ограничиться одной полимеризацией по типу образования не-
предельных димеров, тримеров и вообще полимеров. Для эта-
лена эти превращения, особенно легко осуществляемые под
влиянием катализаторов и под давлением, выражаются следую-
щей схемой:
-> С4Н8 -> С8Н16 и т. д.
»тилен бутилен дябутжлея
Дальнейшие превращения образующихся таким образом поли-
меров могут происходить в нескольких направлениях. Путем
гидрогенизации за счет водорода, всегда содержащегося в крэ-
кинг-продуктах этиленовых углеводородов, непредельные поли-
меры могут легко превратиться в соответствующие им углеводо-
роды предельного характера с открытой группировкой углерод-
ных атомов. С другой стороны, те же полимеры или изомерные
им этиленовые углеводороды в результате частичного крэкинга
с отщеплением конечных групп (в виде метана и т. п.) могут
превратиться в двуэтиленовые углеводороды; последние же пу-
тем циклизации с последующей гидрогенизацией и дегидрогени-
зацией должны дать сначала нафтилены, а затем нафтены и
ароматику по схемам, рассмотренным выше. Таким образом ста-
новится понятным нахождение в продуктах крэкинга этиленовых
углеводородов всех трех основных типов углеводородов предель-
ного характера: парафинов, нафтенов и ароматики. Большее или
меныпее преобладание одного из этих типов над другими зави-
сит от условий процесса. Так например, в процессе крэкинга
этиленовых углеводородов при атмосферном давлении нафтены
образуются в ничтожном количестве, тогда как под давлением
они составляют, повидимому, главный продукт крэкинга [42]
и т. д.
Весьма вероятно, что одним ив основных превращений' олефийов
в условиях крэкинга является реакция, получившая наименование «гидро-
487
полимеризации» (см. стр. 622). Реакция эта заключается в полимервзапд
этиленового углеводорода с одновременным гидрированием образующий*
полимеров, подобно тому как это описано выше для гидро- и дагюпл
генизации циклогексена. Особенно легко происходит гидрополшериа&щц
олефинов над влиянием катализаторов (серная кислота, хлористый &л
миний и т. д.); однако и в отсутствие их, например при нагревании о»,
финов под -давлением, гидрополимеризация также иногда наблюдается.
Смотря по типу непредельного углеводорода, в результате гидрополипе»
зации образуются парафины, нафтены, ароматика и глубоко непредельна
легко осмоляющиеся системы.
Пропилен и другие олефины. Термический распад пропилена
сходен с пиролизом этилена. Продуктами его являются: метц
пропан, этилен, бутилен и жидкие продукты, содержащие бен-
зол и толуол. В меньших количествах наблюдаются этан, ацеш-
лен, бутадиен и уголь. Выхода этих продуктов зависят от усло-
вий (процесса, а химизм их образования аналогичен химизму
образования тех же и аналогичных продуктов из этилена. То ж
относится и к воздействию высокой температуры на различные
бутилены.
Как было показано выше, с увеличением молекулярного веса
крэкинг парафиновых углеводородов значительно ускоряется.
Та же самая правильность наблюдается и для этиленовых угле-
водородов. Примеры табл. 114 показывают, что при 14-минугном
крэкинге гексадецилен, С1вНз2, дает в з раза большее количеств
продуктов разложения и в полтора раза больше продуктов по-
лимеризации, чем каприлен, CeHie, за то же время и в тех se
условиях. Интересно также, что содержание полимеров при крэ-
кинге высокомолекулярных этиленовых углеводородов довольно
быстро достигает своего максимума и в дальнейшем начинает
падать за счет усиленного образования продуктов разложения.
Как среднее можно принять, что при переходе от каприлена
к гексадецилену общая скорость при прочих равных условнят,
увеличивается в начале процесса почти в два раза. [37, 43].
Интересно в .заключение сопоставить отношение к термиче-
скому воздействию соответственных углеводородов двух рассмо-
тренных классов—парафинов и олефинов.
При умеренных температурах (400—500° и ниже) олефинн
легко подвергаются полимеризации, являясь, в общем, менее
устойчивыми в данных условиях, чем парафины. Существенное
влияние на этот равновесный процесс оказывают уже небольшие
давления (до 20 аг), смещая его в сторону образования новш
количеств полимеров. С кинетической точки зрения этот , про-
цесс должен быть отнесен к реакциям второго порядка.
При температурах порядка 600° и выше низшие олефинн
оказываются уже более устойчивыми, чем соответствующие
парафины, поскольку первые образуются в этих условиях. $
последних путем их дегидрирования (реакция первого порядка).
Что касается высших олефинов, то как при высоких, так я .ф
умеренных температурах они являются в термическом отноше-
нии менее устойчивыми, чем соответствующие парафины.
Ароматические углеводороды. Характер реакций, имекнщах
488
место при крэкинге ароматических углеводородов, существенно
зависит от сложности их состава и строения.
Простейшие ароматические углеводороды — бензол и его бли-
жайшие гомологи — чрезвычайно устойчивы в условиях жидко-
фазного крэкинга; особенно большую устойчивость к высокой
температуре обнаруживает простейший представитель конденси-
рованных ароматических систем — нафталин.
При пропускании паров бензола через раскаленную докрасна фарфо-
ровую трубку наблюдается, как это показал еще Бертло [44], разложение
этого углеводорода: образуются газообразные продукты, состоящие из
почти чистого водорода, и смолистые вещества, из которых были выделены
некоторые многоядерные ароматические углеводороды.
Новейшие исследования показали, что разложение бензола
под влиянием высокой температуры начинается ниже 500°, однако
до 600° эта реакция протекает крайне медленно. В присутствии
катализаторов процесс заметно ускоряется; особенно энергично
действует в этом направлении никель, значительно слабее —
железо, в еще меньшей степени — медь [45].
Продуктами термического распада и уплотнения бензола
являются:
а) Газы, в составе которых, кроме водорода (88—91%), обна-
ружены непредельные углеводороды (7—8%); однако ацетилен
среди этих газов согласно новейшим Данным не находится [45].
б) Смолистые продукты, образующиеся в результате уплот-
нения бензола при высокой температуре. Простейшими продук-
тами уплотнения бензола являются дифенил и дифенилбензолы
(пара и мета), образование которых можно представить следую-
щей схемой:
2СвНв = На + Свн5 • СвН6 (дифенил);
СвН6 • CeH3+С6Н„ = Н, + СвН6 С6Н< • С6Н6 (дифенилбензол).
Кроме этих двух многоядерных углеводородов ароматического
ряда, среди продуктов уплотнения бензола обнаружены также
некоторые другие—'Жидкие и кристаллические. Интересно,
однако, что среди продуктов пиролиза чистого бензола вовсе не
встречается нафталин.
в) Кокс, охарактеризованный выше как конечный продукт
конденсации в процессе крэкинга и пиролиза нефтепродуктов.
Образование кокса при пиролизе бензола происходит до-
вольно медленно; зато в некоторых конденсированных ароматиче-
ских системах склонность к коксообразованию при высоких тем-
пературах проявляется необычайно ярко. Так например, антра-
цен, СиНю, после нагревания при 475° в течение З1/^ час. (при-
чем’давление доходило до 50 ат) превратился в сплошную коксо-
образную массу без признаков жидкости, содержавшую
59,0% карбоидов [46].
Переходя от бензола к его гомологам [47], необходимо отме-
тить прежде всего, что низшие представители ряда, в которых
боковыми группами являются метильные радикалы, в условиях
4S9
жидкофазного крэкинга оказываются почти столь же усгой^м-
выми, как бензол. Ближе всех к бензолу, в этом отношен
стоит толуол. Метильная труппа удерживается в толуоле весь®
прочно, так что бензола при пиролизе толуола получается д.
много; 'Преобладают продукты уплотнения толуола и продув®
их дегидрирования, например дибензил (I) и стильбен (П)
а также конденсированные системы типа нафталина,
цена и т. и.:
2СЩ • СвНБ - Н2 + СвНб СН2 • СЩ • СвНБ; m
С6НЙ СНа - СН2 • СйНб -> Н2 + С6Н5 •СН:СН• ОД. $
Этилбензол значительно менее устойчив по сравнению с тс-
луолом, но ведет себя при пиролизе вполне аналогично толуолу,
образуя как продукты распада (бензол, толуол), так и продукта
уплотнения.
Ксилолы. Продуктами термического распада ксилолов
являются, с одной стороны, продукты их деметилирования,
т. е. толуол и бензол, с другой — продукты их уплотнения раз-
личных типов. Наиболее устойчивым из ксилолов является мета,
ксилол, чем, повидимому, и объясняется преобладание его в тех-
нических продуктах пиролиза (каменноугольный деготь, нефтя-
ной деготь и т. п.).
У гомологов бензола с более длинными боковыми цепями тер-
мическая устойчивость постепенно снижается по мере удлине-
ния боковой цепи. Поскольку эти углеводороды можно рассма-
тривать как высокомолекулярные парафины, у которых один во-
дород замещен на фенильный или ;вообще ароматический оста-
ток, их крэкинг, естественно, развивается в двух основных на-
правлениях: • боковые цепи отпадают, расщепляясь при крэкинге
подобно парафинам на более мелкие части, ароматическое же
ядро подвергается реакциям уплотнения, как описано выше.
Из более сложных ароматических углеводородов наибольший
интерес представляют конденсированные системы, в первую оче-
редь— нафталин и антрацен.
Нафталин, СюН8, исключительная устойчивость которого
к высокой температуре уже была отмечена выше, подобно бен-
золу образуется при самых разнообразных процессах пиролиза
и поддается термическому воздействию лишь при температурах
выше 750°. Первым продуктом превращения нафталина в дан-
ных условиях является динафтил, образующийся из нафталина
по той же схеме, как из бензола образуется дифенил, но зна-
чительно легче последнего: согласно данным некоторых авто-
ров [47] при пиролизе нафталина удается получать до 40% ди-
нафтила: ; ;
2С10Н8 = н2 + с10н7. с10н7.
нафталин динафтвл
С другой стороны, характерно, что нафталин (а также антра-
цен) не образуют при пиролизе бензола, так что основные пре-
вращения, претерпеваемые нафталином при пиролизе,* заклю-
чаются в реакциях уплотнения, а не разложения.
490
Механизм образования нафталина при пиролизе предста-
вляют себе различно. Наиболее вероятным является допущение,
что нафталин (III) образуется при пиролизе аналогично той
схеме, которая приведена выше для образования бензола, т. е.
при участии бутадиена, а именно путем уплотнения цикло-
гексена с бутадиеном (I) с последующей дегидрогенизацией про-
межуточного углеводорода (П):
Антрацен, С14Н12, подобно нафталину при пиролизе образует
в первую очередь простейший продукт уплотнения, диантрил:
Сам антрацен является, невидимому, продуктом уплотнения
толуола через дибензил (ор. выше):
/СН2
Сен/ I .
сн
;вн6 - 2Н9+с,н1^|\с.и4
сн
дибекзнл автрацеи
Особо ярко выраженной склонностью к образованию высоко-
молекулярных многоядерных продуктов уплотнения в процессе
термического воздействия характеризуются некоторые полици-
клические углеводороды, в которых бензольное или нафталино-
вое ядро сконденсировано с пятичленным циклом, предельным
или непредельным. Таковы, например, инден, CbHs, аценафтен,
СиНю, аценафтилен, С^Нв, и т. п., уплотнение которых в высоко-
конденсированные системы может наблюдаться уже при низких
температурах:
инден
аценафтен
аценафтилен
Как ясно из вышеизложенного, одной из характерных особен-
ностей крэкинга ароматических углеводородов является реакция
коксообразования. Вспомним, что в противоположность арома-
тике парафины не обнаруживают склонности к коксообразова-
нию: можно, • например, часами крекировать, парафин (418°,
491
10 ат, 2 час.) без образования кокса [48]. Отсюда сам собою
напрашивается вывод, что именно ароматика является основным
-источником образования кокса при крэкинг-процессе [49] j
этот 'вывод вполне оправдывается на опыте тяжелых, богатых
•ароматикой, нефтей и нефтепродуктов, действительно дающщ
при крэкировании обильное образование кокса.
Существуют две основные точки зрения на механизм обрат
зования кокса в процессах крэкинга и пиролиза.
Согласно взглядам одних авторов кокс представляет собою
свободный углерод, образующийся в результате распада того иля
иного углеводорода на элементы, например:
С6Н6-6С + ЗН2.
О этой точки зрения естественно ожидать, что аналогичный
распад могут претерпевать не только углеводороды ароматиче-
ского ряда, ио и углеводороды других рядов, например пара-
фины. Мы видели, однако, что опыт не согласуется с такого рода
допущением и что источником коксообразования в условиях
крэкинга являются в первую очередь ароматические углеводо-
роды. К тому же, как было показано выше (стр. 474), по своему
составу кокс представляет собою отнюдь не простое тело, а ве-
щество очень сложного состава. Отсюда ясно, что последнее
и ему подобные уравнения во всяком случае не выражают со-
бою химической сущности процесса коксообразования.
Другой взгляд на механизм образования кокса, неоднократно
отмеченный в предшествующем изложении, рассматривает этот
процесс как ряд последовательных реакций уплотнения, претер-
певаемых при высокой температуре ароматическими углеводоро-
дами. Первыми .продуктами реакций этого рода для бензола
являются, как мы видели, дифенил и дифенилбензол; промежу-
точным продуктом при этом, повидимому, служит дигидродифе-
нил по уравнению:
2СбНв = С12Н12.
Конечными продуктами дальнейших реакций уплотнения
с этой точки зрения являются вещества, близкие к каменно-
угольному коксу, т. е. карбоиды. Последние при достаточно вы-
сокой температуре являются единственным твердым остатком
крэкинга или пиролиза углеводородов, и тем не менее,-это все
же — не элементарное вещество.. При сухой перегонке карбоиды
дают около 3% летучих, элементарный же анализ их показы-
вает, что они содержат только 92% углерода:
Элементарный состав карбоидов (по ГрозНИИ):
С - 92,07%; Н - 4,15%; О - 3,78%.
Итак, карбоиды представляют собою сложное вещество,.вер-
нее— смесь веществ, образующихся при крэкинге и пиролизе
492
ароматических углеводородов как конечный продукт ряда После-
дующих стадий уплотнения. Для уяснения механизма этих ре-
акций важное значение имеет нахождение в продуктах пиролиза
бензола углеводородов типа дифенила. Некоторые авторы пола-
гают поэтому, что процесс коксообразования следует предста-
влять себе по следующей схеме:
Бензол Дифенил ->
Высшие продукты
уплотнения бензола
Кокс
Надо думать, однако, что эта схема отвечает лишь одному
из возможных путей, по которым в действительности идет про-
цесс образования кокса при пиролизе ароматических углеводо-
родов.
В самом деле, наиболее типичным случаем коксообразования
является, как известно, процесс сухой перегонки каменных и
бурых углей. При этом, однако, кроме одноядерных и много-
ядерных ароматических углеводородов типа дифенила, обра-
зуются многочисленные конденсированные системы, как-то: наф-
талин, СюН8, антрацен, СиНю, и его изомер, фенантрен, а также
углеводороды еще более сложного состава и строения, например
пирен, CieHio, хризен, CieHi2, пицен, С22Н14, и, наконец, неизвест-
ного строения крэкен, C^His; сюда же должны быть отнесены
конденсированные предельные и непредельные системы, образо-
ванные при участии пятичленного цикла, каковы, например,
инден, аценафтен и т. п. Некоторые из этих конденсированных
систем, как было показано выше, являются продуктами уплотне-
ния более простой ароматики и чрезвычайно склонны к коксо-
образованию при повышенной температуре, так что не исключена
возможность, что процесс образования кокса при крэкинге и
пиролизе ароматики проходит через стадию промежуточного
образования некоторых из этих конденсированных систем.
Что касается, наконец, образования кокса при крэкинге и
пиролизе неароматического сырья, то во всех подобного рода
случаях легко представить себе в качестве некоторой промежу-
точной стадии образование той или иной ароматики, которая и
является ближайшим материалом для последующего процесса
коксообразования.
Нафтены.'Как было показано в ч. I, нафтены, встречающиеся
в нефти, принадлежат к рядам циклопентана и циклогексана.
Высшие нафтены также являются либо гомологами циклопентана
и циклогексана, либо представляют собою соответствующие кон-
денсированные системы. Крэкинг этих углеводородов изучен до
сих пор совершенно недостаточно.
Циклопентан и его гомологи. Циклопентан значительно бо-
лее устойчив по отношению к высокой температуре, чем соответ-
ствующий парафин нормального строения (т. е. норм, пентан).
Установлено, что главными продуктами высокотемпературного
крэкинга циклопентана (при 574—650 ) как при атмосферном,
так и при пониженном давлении являются этилен и пропилен,
493
представляющие
пентана:
собою, очевидно, продукты распада цикло-
Н2С —СН2 Н2С = СН2
Н2С СН2 Н2С СН3
\/ X/
сн, сн
4
Наряду с реакцией распада циклопентан (I) претерпевает при
высокой температуре также реакцию дегидрирования, продуктами
которого являются водород, циклопеитен {11) и циклопента-
•диез (П1):
Н2С — СН2
Н2С СН2
СН2
Таким образом основные реакции, претерпеваемые циклопен-
таном при температурах порядка 575—650°,—те же, что и для
простейших парафинов: расщепление и дегидрогенизация; сюда
присоединяются, конечно, вторичные реакции гидрирования и
уплотнения, продукты которых (этан, пропан, -высшие угле-
водороды) тоже обнаруживаются в продуктах крэкинга цикло-
пентана.
Ближайший гомолог циклопентана, метилциклопентан, отно-
сится к высокой температуре примерно так же, как циклопентан.
Здесь, однако, изучены пока лишь продукты распада, каковыми
оказались этилен, пропилен и изобутилен. Очевидно, распад
метилциклопентана происходит в двух основных направлениях:
Н2С---------СН • СН3 , 2СН3 • СН: СН2
"‘"х"........... / сн3
Н2^ ХХХ X сн2: С + Н2С: СН2
X '
Циклогексан и его гомологи. Циклогексан по своей устойчи-
вости к высокой температуре занимает среднее место между бен-
золом и норм, гексаном. При 500° он значительно устойчивее
гексана (а также циклопентана), но менее устойчив, чем бензол.
Характерно, что в отсутствие катализаторов основной реакцией
является не дегидрирование (т. е. не образование бензола), а рас-
пад с образованием непредельных углеводородов. При более вы-
494
сокой температуре, а именно при 650—700°, наряду с продуктами
распада циклогексана появляется также продукт его дегидро-
генизации— бензол. При еще более высокой температуре (750°)
количество жидких продуктов резко снижается, а ’ состав их
осложняется присутствием продуктов конденсации, нафталина
и антрацена. Кроме ароматики, жидкие продукты пиролиза
циклогексана содержат также высокомолекулярные олефины.
Газообразные продукты пиролиза циклогексана состоят* из
водорода, низших парафинов (главным образом метана), низших
олефинов (этилен и преимущественно пропилен) и бутадиена.
Вполне вероятно, .что бутадиен и является источником образо-
вания бензола в данном с ту чае (ср. выше). Так как бутадиен
особенно гладко образуется при пиролизе циклогексана, то воз-
можно, что распаду циклогексана с образованием бутадиена пред-
шествует предварительное дегидрирование циклогексана до ци-
клогексена по схеме:
СН2
Н2С СН2
Н2С сн2
сн.
сн2
НС ' сн2
Воздействие на циклогексан и его гомологи высоко! темпера-
туры в присутствии некоторых катализаторов будет рассмотрено
в следующей главе.
Особенно сложно протекает крэкинг конденсированных нафте-
новых систем. Так например, при крэкинге декалина происхо-
дят реакции образования предельных и непредельных углеводо-
родов жирного ряда, непредельных циклических углеводородов
(циклогексен и циклогексадиен) и ароматики (тетралин и наф-
талин). Естественно, что взаимные связь и соотношения между
реакциями образования отдельных углеводородов этой сложной
смеси остаются пока далеко невыясненными.
Что касается, наконец, нафтенов с длинными боковыми це-
пями, то при их крэкинге, надо думать, будут происходить в пер-
вую очередь те же самые реакции, которые происходят при крэ-
кинге аналогично построенных гомологов бензола, т. е. отще-
пление боковых цепей последующим распадом циклической
системы соответственно ее строению.
термодинамика’ крэкинг-процесса
В предшествующем изложении было показано, что крэкинг
даже индивидуальных углеводородов, не говоря уже о таких
сложных смесях, как нефтяные дестиллаты, представляет собою
комплекс разнообразнейших реакций разложения, уплотнения
495
и изомеризации. Полное овладение этим-, как и всяким другим
химическим .процессом, требует точного выявления ряда физико-*
химических его характеристик, которые были рассмотрены выше
лишь в самых общих чертах, а именно: какова кинетика реак-
ций крэкинга и его механизм, т. е. тот путь, по которому данное
вещество или система стремятся в заданных условиях к состоя-
нию равновесия; какие продукты могут образоваться в резуль-
тате данного процесса и в каких относительных количествах*
каковы условия равновесия данной системы при данном про-
цессе и т. д. Хотя сложность химических превращений, сово-
купность которых образует крэкинг-процесс, не дает возможно-
сти получить большинство его физико-химических характери-
стик опытным путем, тем не менее, с помощью термодинамики
ответ на некоторые из поставленных выше вопросов, правда
лишь качественного характера, может быть получен [51].
I. Один из методов приложения термодинамических расчетов
к крэкинг-процессу и вообще к пирогенетическим реакциям осно-
ван на уравнении изменения свободной энергии А при химиче-
ской реакции:
Л = — RTIvlK,
где R — газовая постоянная, Т—абсолютная температура и К—
константа равновесия данного процесса; зпак минус показывает,
что реакция протекает в сторону уменьшения свободной энергии.
Это уравнение по существу применимо лишь к обратимым
химическим реакциям; однако для случаев, когда скорости даль-
нейших -превращений продуктов реакции очень малы, а в пре-
деле равны нулю, некоторые авторы расширяют область его при-
менения на реакции, по существу необратимые, в частности—,
на реакции образования и распада углеводородов. Таким обрат'
зом могут быть произведены, например, вычисления свободной
энергии образования углеводородов различных типов из элемен-
тов и тому подобные расчеты. Хотя все вычисления этого .рода
за недостатком необходимых опытных данных неизбежно
являются весьма приближенными, тем не менее, ими можно
пользоваться для приблизительного расчета изменения свобод-
ной энергии при отдельных реакциях крэкинга и пиролиза; зная
же величину А, можно на основании вышеприведенного урав-
нения вычислить константу равновесия К для данного процесса,
которая, указывая на количественные соотношения концентра-
ций веществ, принимающих участие в данном равновесии, по
существу определяет направление реакции: чем больше кон-
станта равновесия, тем глубже протекает реакция в данном на-
1гравлении, и наоборот’. Таким образом с помотаю расчета ока-
зывается, по крайней мере, возможным предугадать возможность
или невозможность реакции в данном направлении.
По Аррениусу * медленное течение многих химических реакций
объясняется тем обстоятельством, что непосредственное участие
в них принимают не все молекулы данной системы, а лишь те'
из них, которые содержат некоторый избыток энергии по срав-
496
нению с остальными. Такие молекулы называются активиро-
ванными; теплоту же Е, необходимую для превращения обыкно-
венной молекулы в активированную, называют энергией акти-
вации.
Пусть между концентрациями активированных и обыкно-
венных (не активированных) молекул имеется равновесие, кон-
станта которого равна КА. Применяя к этому равновесию урав-
нение изохоры, получаем зависимость изменения константы рав-
новесия К а от температуры:
d In КА Е
от = rt1’
где Е— энергия активации дацных молекул, независимая от Т
в малых интервалах температур.
Интегрирование этого уравнения дает:
In КА — — + Kq,
где Ь'о—постоянная интегрирования, которая не может быть
определена термодинамически.
Для вычисления энергии активации Е можно воспользо-
ваться эмпирической формулой Аррениуса, выражающей зави-
симость коэфициента скорости реакции от температуры в сле-
дующей форме:
d In fc _ Е
dT ~~ ВП ’
Допуская, что Е от температуры не зависит и что коэфи-
циенты скорости реакции \ и Л2 известны при двух разных
температурах и 72> легко получаем:
Отсюда
In
ш k3 В к
т2)’
Е—В1п^~
Т1-Т2
Ъ-тг
И. Уравнение изохоры (Вант-Гофф) многократно и успешно
применялось за последние годы при изучении различных
обратимых реакций, наблюдаемых при крэкинге углеводоро-
дов, каковы, например:
1. Реакции образования простейших углеводородов из эле-
ментов и обратные реакции, т. е. распад этих углеводородов
на углерод (графит, аморфный уголь) и водород, например:
С + 2Н9 Z2 СН4.
2. Реакции гидрирования олефинов и обратные реакции
дегидрирования соответствующих парафинов, например:
C2H4 + H9ziC2H6.
32 Зак. 1622. Хииия нефти.
497
3. Реакции гидрирования ароматических углеводородов и
обратные реакции дегидрирования соответствующих нафтенов,
например:
зн2 с6н12.
4. Реакции уплотнения ароматических углеводородов и
обратные реакции распада этих продуктов уплотнения, на.
пример:
2СбНв^С6Нб.С6НБ + Н2.
Ниже рассматривается один из простейших примеров этого
рода, а именно гидрирование бензола и обратный процесс
дегидрирования циклогексана.
Для некоторой обратимой реакции:
константа равновесия при постоянном давлении Кр согласно
закону действующих масс выражается следующим образом:
к ~рп' ’Рп'-
где р — соответствующие парциальные давления реагирующих
веществ, пропорциональные их концентрациям.
Изменение этой константы равновесия с температурой
согласно уравнению изохоры выражается следующим образом:
dlnK^_ Q
ат “ ят*'
В этом уравнении Q — тепловой эффект реакции, зависи-
мость которого от температуры может быть выражена следую-
щим соотношением:
• -^=2 и'с/—2ис,=Дс1>-
Смысл этого последнего соотношения заключается в том,
что изменение теплового эффекта реакции при изменении
температуры равно изменению разности теплосодержаний
реагирующих веществ при том же изменении температуры.
Интегрируя последнее выражение в пределах от нуля до Т,
находим:
г
о
Через Qo здесь обозначен условный тепловой еффект ре-
акции' при абсолютном нуле; для определения которого необ--
ходимо экспериментальное определение теплового эффекта
реакции Q при какой-либо одной температуре; величина же
ДСр находится из эмпирических формул, определяющих зави-
498 . , .
симость от температуры теплоемкостей веществ, реагирующих
и получающихся в результате реакций.
Подставляя теперь в уравнение изохоры значение Q из
последнего уравнения, находим:
т
О
Интегрируй это уравнение, получаем:
In Кр = - А.+-L J (± f Лср4т) dT+J.
о о
Здесь постоянная интегрирования J находится из эмпи-
рических данных для Кр, причем берут среднее для несколь-
ких значений Кр, соответствующих различным температурам.
Рассмотрим теперь применение последнего уравнения
к протекающей в газообразной фазе реакции:
*“ СеН6 + ЗН2.
Теплоемкости циклогексана, бензола и водорода в зависи-
мости от температуры выражаются следующими эмпириче-
скими формулами:
Для С6Н12 (пар) ср = в,8 + 0,07.5 Т;
„ СеНв „ ^ = 6,55 + 0,052 Т;
„ Н2 „ ср = 6,65 + 0,0007 Т.
Так как-
Ч = СР (для C6Hg) + Зср (для Н2) — ср (для СеН12),
то
= 19,7 — 0,021 Т.
Таким образом для теплового эффекта рассматриваемой
реакции в зависимости от температуры мы получаем следую-
щее соотношение:
Q = QO+19,7 Т—0,010 Т2.
Опытное определение теплового эффекта реакции при ка-
кой-либо температуре производится термохимическим путем
из теплот сгорания веществ, принимающих участие в данной
реакции. Для циклогексана, бензола и водорода наиболее
надежными для температуры 25° С (т. е. Т =298,1) являются
следующие теплоты сгорания:
ДЛЯ С6Н12 (ЖИДК.) 938 900 КС1Л[м0ЛЬ
„ СвН6 „ 782 800
, Н2 „ 63 310
32*
499
Чтобы перейти к газообразному состоянию, необходимо
к указанным теплотам сгорания циклогексана и бензола при.
бавить их теплоты парообразования, а именно:
Для С6Нб 8040 кал)моль
„ С6Н12 • 8100 „
Из этих данных легко рассчитать тепловые эффект^ рас-
сматриваемой реакции, а именно:
j = 48 800 кал и Qo = 43 800 кал}моль.
Внося теперь выражения найденные для Qo и &ср в урав-
нение, определяющее 1пКр, выполняя интегрирование и пере-
ходя к десятичным логарифмам, находим:
438С0 , 19,7 1 r 0.010Т
— ]да. 2.303Т “ 1,986 ь 1,986 • 2,803 '
где JR принято равным 1,986.
Произведя соответствующие вычисления, окончательно
находим:
lgKp= --^+9,81g Т—0,0022 Т-1-с.
. е
В этом окончательном уравнении константа С может быть
найдена лишь опытным путем. Для этого должен быть произве-
ден хотя бы один опыт дегидрирования циклогексана при неко-
торой постоянной температуре, например при 275° (1’ = 548°),
и определен точный состав полученных продуктов,’ который на
основе закона действующих масс позволит вычислить опытную
константу Кр. Анализ жидких продуктов реакции (бензол + ци-
клогексан) удобно производить в данном случае определением
коэфициента рефракции смеси углеводородов и сопоставлением
найденной реакции со специальным трафиком изменения ре-
фракции смеси бензола с циклогексаном в зависимости от ее
состава. Кроме того, в газах, выходящих из прибора после ре-
акции, определяется содержание водорода.
Эти опытные данные позволяют’ высчитать, что при 275°
константа равновесия данной реакции Кр =— 0,2; откуда
С == — 6,56.
Наконец, подставляя найденное значение С в последнее урав-
нение для 1g Кр, можно рассчитать значение этой константы для
разных произвольно выбранных температур и сравнить вычис-
ленные значения Кр с опытными. Как показывают приводимые
ниже примеры, при этом получается вполне удовлетворительное
совпадение:
t Т 1g Кр (вычисл.) 1g Кр (найден)
230° 503 - 2,0 — 2,13
250° 523 —1.2 —1,10
275’ 548 — 0,2 — 0,20
500
III. Основанием второго метода термодинамических, также
весьма приближенных расчетов в области крэкинга и пиролиза
может служить тепловая теорема Нернста, которая приближенно
выражается следующим уравнением:
lg — 2 «1.75 1g Т—
В этом уравнении К — копстанта равновесия реакции, зави-
сящая только от абсолютной температуры Т; Q—тепловой эф-
фект реакции крэкинга, определяемый ла основе термохимиче-
ского соотношения:
Q = 2 ?о 2 9»
где 2 до— сумма теплот сгорания исходных продуктов и Уд—
сумма теплот сгорания конечных щюдуктов реакции.
2п представляет собою алгебраическую сумму молекул, уча-
ствующих в реакции, причем исчезающее молекулы берутся со
знаком (+), появляющиеся же молекулы—со знаком ми-
нус (—).
Наконец, 2пС является алгебраической суммой произведений
числа молекул, исчезающих (+) и появляющихся (—), на хи-
мические постоянные веществ, участвующих в реакции; для во-
дорода химическая постоянная по Нернсту равна 1,6; для угле-
водородов ориентировочно она может быть принята равной 3,0;
для метана — 2,5.
•
Из величин, входящих в уравнение, выражающее теорему Верного,
наиболее сложной является химическая постоянная С. Так называют ве-
личину, имеющую для -каждого вещества определенное численное значение,
не зависящее от состояния или модификации, в котором находится ве-
щество. Химическая постоянная определяется в термодинамике из уравне-
ния, выражающего общую зависимрсть упругости пара вещества от темпера-
туры; она может быть вычислена также по формулам кинетической теории.
В шервом приближении химическая постоянная может быть вычислена
из правила Трутона, константа которого находится в простой зависимости
от химической постоянной, а именно:
С = 0,14^',
где ME — молекулярная скрытая теплота испарения жидкости, а Т — ее
абсолютная температура кипения при атмосферном давлении. Принимая
во внимание, что для углеводородов близко к 20 (ч. I, стр. 51), полу-
чаем значение химической постоянной для всех углеводородов близким
к 3 [42].
в
Для иллюстрации применения тепловой теоремы Нернста
к изучению крэкинг-процесса рассмотрим крэкинг какого-либо
индивидуального углеводорода, например нормального гексана.
Основное направление этой «реакции должно быть выражено
следующим уравнением:
СеН14 Z2 С3Н8 + С8НЬ( — 32 000 кал).
501
&та реакция приводит к некоторому состоянию равйовесйя,
подчиняющемуся закону действия масс, а именно:
[С8н8] • [С3н6]
[С6н14]
где [СвНн], [СзН8] и [С3Нв]—концентрации гексана, пропана
и пропилена в состоянии равновесия, а К — константа равно-
весия, зависящая только от температуры.
Вычислим эту константу для данного случая на основе /те-
пловой теоремы Нернста, для чего в приведенное выше уравнение
этой теоремы подставим:
Q — тепловой эффект реакции распада гексана на пропан и
пропилен, т. е. — 32 000 кал;
Ей — 1 — 1 — 1= — 1, согласно приведенным выше опреде-
лениям и правилу знаков;
ЕпС = 3-1 — 3 •> 1 — 3 • 1 = — 3, по тому же правилу.
Таким образом общее уравнение «принимает для данного слу-
чая следующий вид:
Вг+1’751^7+3-
Пусть теперь для примера Т = 750° абс. или, что то же,
477° С, что близко отвечает температурным условиям жидао-
фазного крэкинга. Подставляя в последнее уравнение Т = 760
и произведя расчет, получаем, что 1g# =—1,2 откуда К=0,06.
Таким образом при 477° С гексан распадается лишь в очень
слабой степени.
Иная картина получается, если повысить температуру реак-
ции крэкинга гексана. Так например, при Т = 873°" абс. или
600° С по тому же уравнению для 1g К вычисляется 4- 0,2, от-
куда К = 1,58. Таким образом при 600° С большая часть гек-
сана должна быть уже разложена *на пропан и пропилен.
Развивая далее термодинамический анализ разных направле-
ний реакции крэкинга для различных парафинов и других неф-
тяных углеводородов при разных температурах, можно аналогич-
ным путем притти к следующим выводам:
1. Среди различных возможных 'способов разложения пара-
финов при крэкинге распад их посредине молекулы является
преобладающей реакцией, особенно для высокомолекулярных па-
рафинов. Так например, для углеводородов С20Н42 при 477° С
константа равновесия К оказывается равной 40 000, т. е. при
указанной температуре разложение этого парафина практически
идет необратимо и нацело. Напротив, если разложение этого
парафина на водород и олефин с тем же числом атомов углерода
в частице и может иметь место в указанных выше температур-
ных условиях, то липть в крайне ограниченных количествах.
2. Реакции разложения этиленовых углеводородов должны
итти значительно труднее, чем парафинов. Теоретически здесь
более вероятны реакции полимеризации, по крайней мере для
низших олефинов.
502
3. Превращение этиленовых углеводородов в соответствующие
нафтены с точки зрения термодинамики вполне возможно; однако
скорость этой реакции, как показывает опыт, крайне незначи-
тельна.
4. Из ароматических углеводородов гомологи бензола с корот-
кими цепями '(метил-, отчасти этил-бензол) являются наиболее
прочными и практически не должны разлагаться в условиях
жидкофазного крэкинта. . Напротив, гомологи бензола с длин-
ными боковыми цепями сравнительно легко разлагаются с отще-
плением боковых цепей либо в виде олефинов, либо в виде низ-
ших парафинов.
5. Нафтены с боковыми цепями должны разлагаться с обра-
зованием этиленовых углеводородов приблизительно в такой же
степени, как и ароматические углеводороды.
6. Нафтены ряда циклогексана должны легко дегидрогенизи-
роваться в условиях крэкинга с образованием соответствующих
ароматических углеводородов, причем водород, образующийся
при ©той реакции, может пойти на гидрогенизацию этиленовых
углеводородов.
7. Образующиеся при крэкинге ароматические углеводороды
способны вступать в реакции уплотнения как между собой, так
и с непредельными углеводородами; в первом случае должны
получаться производные дифенила, динафтила, а также полици-
клические углеводороды; во втором — производные антрацена,
фенантрена и других полициклических углеводородов.
Таковы в самых общих чертах выводы, которые можно сделать
относительно поведения углеводородов различных радов тгря
жидкофазном крэкинге на основе термодинамических соображе-
ний. Легко видеть, что выводы эти полностью согласуются с теми
опытными данными, которые были приведены выше в связи с во-
просом о крэкинге индивидуальных углеводородов различных
рядов и отдельных нефтяных фракций. Надо помнить, однако,
что выводы эти имеют лишь качественный характер; о количе-
ственной же стороне реакций, имеющих место при крэкинг-про-
цесое, можно судить лишь в отдельных случаях на основании
опытных данных, к тому же крайне недостаточных и неполных
ввиду чрезвычайной сложности всего процесса в целом.
ТЕОРИИ КРЭКИНГ-ПРОЦЕССА
Как ясно из вышеизложенного, крекинг-процесс предста-
вляет собою сложный комплекс одновременно протекающих реак-
ций дегидрогенизации и разложения исходного сырья, полимери-
зации и уплотнения входящих в его состав непредельных и
ароматических углеводородов и нередко более или менее глубо-
кой изомеризации начальных систем в системы нового строе-
ния. Уже одно ©то обилие и (разнообразие реакций, совокупность
которых образует крэкингнпроцеос, делает понятным и неизбеж-
ным то положение, что нет и не может быть единой, универ-
сальной теории, которая •разъясняла бы весь данный комплекс
503
в целом. Речь может итти. очевидно, лишь о механизме отдель-
ных реакций, наблюдаемых при крэкинг-процеосе и в нерву©
очередь о механизме реакций, наиболее характерных для кре-
кинг-процесса. — реакций разложения.
Теории реакций разложения. Разложение 'представляет собою
основную реакцию, претерпеваемую при крэкинг-процессе пара-
финами. Именно поэтому естественно начать рассмотрение тео-
рий крэкинг-процесса с обзора теоретических представлений
о механизме термического разложения парафинов.
Наиболее ранней теорией механизма разложения углеводо-
родов является положение Бертло (1866 г.), согласно которому
основным продуктом распада каждого углеводорода должен быть
ацетилен, во многих случаях являющийся, однако, лишь проме-
жуточным продуктом для образования более сложных веществ,
например ароматики, смолистых продуктов и т. п. Так напри-
мер, распад метана, этана и этилена по Бертло можно предста-
вить следующими схемами:
и
и
и
Хотя некоторыми новейшими авторами [52] при высоко-
температурном пиролизе метана (1120°) действительно удавалось
получать значительные выхода на ацетилен, тем не менее, поло-
жение Бертло давно оставлено; в частности, образование аро-
матики при пиролизе углеводородов различных рядов, как было
отмечено выше, разъясняется в настоящее время совершенно
иначе.
Согласно другому представлению (Торпе и Юнг и др.) сущ-
ность термического распада углеводородов заключается в том,
что связь между углеродными атомами углеводорода в определен-
ном месте разрывается, причем одновременно происходит переме-
щение водородного атома с одного осколка молекулы на другой
В результате, как было показано выше, происходит образование
двух новых молекул — предельной и непредельной. Место раз-
рыва исходной молекулы зависит от температурных условий.
При умеренных температурах (жидкофазный крэкинг) разрыв
происходит по середине или вблизи середины молекулы; при бо-
лее высоких температурах (парофазный крэкинг, пиролиз) моле-
кула распадается ближе к одному из своих концов, в резуль-
тате чего наблюдается повышенное образование газообразных
продуктов ее распада, т. е. низших углеводородов и водорода.
Хотя эти представления довольно правильно рисуют картину
результатов термического распада углеводородов, по крайней
мере в условиях недостаточной глубины процесса, все же они
совершенно недостаточны для разъяснения самого механизма
процесса. В частности, остается неясным, когда и почему насту-
пает один из основных этапов всего процесса — перемещение во-'
дородного атома с одного осколка молекулы на другой—икав
5041
надо представить себе эти осколки. Ответ на эти вопросы пы-
тается дать современная теория радикалов.
Первые попытки изолирования свободных органических ради-
калов делались еще на заре органической химии, но лишь
в 1900 г. эта задача -получила свое разрешение в синтезе дрифе-
нилметила как продукта диссоциации гексафенилэтана:
(СвН6)8С - С(СвН5)з Z2 2(СвН-)3С'.
С тех пор число подобного рода тяжелых радикалов весьма
возросло, и существование их в настоящее время уже не вызы-
вает никаких сомнений.
Несравнимо сложнее оказался вопрос о существовании про-
стейших алифатических радикалов, например метила, СН'з,
этила, С2Н5', и т. п. Лишь в 1929 г. Панет успешно разрешил
эту задачу, доказав образование некоторых из -этих радикалов
(правда, лишь косвенным путем) при термическом распаде тяже-
лых металлоорганических соединений, как-то: тетраметилсвинца,
(СН3)4РЪ, тетраэтилсвинца, (СаНз^РЬ, и др.
Сущность первоначального метода Панета, раз [вшившего вопрос о
существовании простейших свободных радикалов, заключается в следую-
щем. Пары чистого тетраметилсвинца пропускают в токе чпстого водорода
через узкую кварцевую трубку (диаметр трубки 2 мм, длина ее 60 см)
под давлением 1—2 лл со скоростью 10—20 м/сек. Если на некотором рас-
стоянии от ввода газа нагреть такую трубку газовой горелкой, то вследствие
разложения металлоорганического соединения внутри трубки появляется
свинцовое зеркало; остальные продукты! разложения уносятся насосом.
Остудив после этого кварцевую трубку, -переносят газовую горелку не-
сколько ближе к вводу газа. Теперь вследствие возобновившегося разло-
жения тетраметилсвинца на новом месте обогрева появляется новое
свинцовое -зеркало, старое же зеркало постепенно исчезает вследствие
взаимодействия его со свободными радикалами. Этот опыт можно повто-
рять неограниченное число раз, причем, изменяя расстояние между зер-
калами, скорость прохождения газа и другие условия опыта, можно по-
лучить ряд весьма ценных данных относительно устойчивости отдельных
радикалов, продолжительности их жизни и других их свойств.
После того -как было установлено, что при термическом рас-
паде тяжелых металлоорганических соединений действительно
образуются простейшие свободные радикалы, и радикалы эти
были охарактеризованы, Райс высказал положение, что анало-
гичные радикалы должны образовываться также при термиче-
ском распаде других органических соединений и что радикалы
эти могут взаимодействовать не только между собой, но и
с цельными, не разложившимися или новообразовавшимися мо-
лекулами. Это последнее направление реакции, естественно,
должно быть особенно важным, так как ввиду незначительной
концентрации свободных радикалов взаимодействие их друг
с другом не может иметь сколько-нибудь серьезного значения
в общем балансе процесса термического распада. С другой сто-
роны, сталкиваясь и реагируя с цельными молекулами, сво-
бодные радикалы могут отнимать у них водород; соединяясь
с ним, они превращаются при этом в соответствующие моле-
кулы, тогда как из молекул, потерявших атом водорода, обра-
<"05
зуются новые радикалы, которые в свою очередь сами реагируют
с новыми молекулами, и т. д. Таким образом в итоге реакция
приобретает цепной характер.
Качественные подтверждения основных положений этой кон-
цепции были получены сначала самим Райсом с сотрудникам^
а затем и другими авторами. Было показано, что образование
свободных радикалов действительно наблюдается при термиче-
ском распаде этана, пропана, бутана, пентана, гептана, некото-
рых кислородных соединений (ацетона) и т. д. Правда, доказать
существование радикалов тяжелее этила пока не удалось, что
очевидно, находится в связи с их крайней неустойчивостью’
Последнее обстоятельство не являлось, однако, препятствием
в обобщению приведенного выше опытного материала и к укре-
плению теоретических представлений Раиса, которые под именем
теории радикалов получили вскоре широкую известность и при-
менение для разъяснения механизма термического распада угле-
водородов [53].
Итак, по Райсу первая стадия термического распада углево-
дородов парафинового ряда заключается в расщеплении их на
два одинаковых или различных радикала. На примерах этаяа
и норм, бутана реакции образования соответствующих радика-
лов могут быть выражены следующими уравнениями:
СН8.СН8 -> СН/4-СН/;
СН3 - СН2 - СН2 СН3 -> СН3 - СН2 - СН/ + СН/;
СН, • СН2 • СН2 • СН, -> СНд • СН/ 4- 'СН2 СНо.
Реакционная способность свободных радикалов должна быть
громадна. Интересно проследить поэтому их превращения и ре-
акции, так как ими, очевидно, определяется ближайший состав
продуктов крэкинга в первые, еще недостаточно глубокие и ме-
нее сложные фазы процесса. Превращения эти следующие:
1. Взаимодействие радикалов друг с другом, в результате
которого из них могут образоваться старые либо новые углево-
дороды. Так например, радикалы метил, этил и- пропит, образо-
вавшиеся из норм, бутана, соединяясь с такими же радикалами,
дадут этан, бутан, гексан и другие парафины:
СН/ + СН/ = СН8 - СН8;
СН8 - СН/ 4- 'СН2 • СН8 == СН3 - СН2 • СН2 - СН*
СНд - СН2 - СН/ + 'СН, - сн2 - сн8 =
= СНд • сн2 • сн2 • сн2 - сн2 - СНд
И т. Д.
Как. было отмечено выше, превращения радикалов по этой
схеме вследствие небольшой их концентрации должны иметь
второстепенное значение.
2. Разложение радикалов. Теоретически разложение свобод-
ных радикалов можно представить себе в различных направле-
ниях. Однако лишь некоторые из этих направлений можно счи-
тать реально существующими при термическом распаде парат*
финов на основе изучения состава продуктов, получаемых в пер-
вые стадии данной реакции. К этому вопросу можно подходить
506
также с энергетической точки зрения, исходя из положения,
что наиболее (Вероятными должны быть такие направления рас-
пада радикалов, которые требуют наименьшей энергии актива-
ции. На этих основах можно представить себе в каждом отдель-
ном случае наиболее и наименее вероятные направления распада
свободного радикала; подвергая, далее, этот вопрос сравнитель-
ному изучению на различных объектах, можно сделать отсюда
обобщающие выводы и сопоставить их с новыми фактами из
данной области. В 'Качестве примеров приводим схемы некото-
рых типичных случаев разложения радикалов:
♦ СН3 СН2 • СН2' -► СН2 : СН2 + СН8'
СН8 • СН2 • СН2 СН2' -> ‘ СН3 • СН2' 4- СН2: СНд
и т. д.
Наиболее типичные случаи разложения радикалов сводятся
к разрыву простой связи между двумя углеродами с образова-
нием нового, более простого радикала и углеводорода ряда эти-
лена. Более детальное рассмотрение этого вопроса с энергетиче-
ской точки зрения позволяет установить также наиболее вероят-
ное место разрыва открытой цепи углеродных атомов, что осо-
бенно важно для случая распада радикалов большого веса,
а именно: распад радикалов наиболее легко происходит по месту
второй связи, считая от углерода со свободной валентностью.
Примерами могут служить приведенные выше случаи распада
радикалов пропила и норм, бутила.
Напротив, отрыв атома водорода от радикала является
с энергетической точки зрения менее вероятным, чем разрыв
связи между двумя углеродами. Поэтому распад радикала с от-
щеплением атома водорода, вообще говоря, имеет второстепенное
значение и может наблюдаться лишь в отдельных случаях. Та-
ков, например, распад этила на этилен и водород (атом) и не-
которые другие:
СН8.СН2' ЩСгСЩЧ-Н'.
3. Взаимодействие радикалов с молекулами (исходного угле-
водорода. Радикалы Н', СН/, СгН5', в образовании которых
при термическом распаде парафинов не может быть никаких
сомнений, реагируя с молекулами -исходного углеводорода, дают
начало новым радикалам, а сами превращаются в соответствую-
щие молекулы. Так например:
с4н10+с2н/ = с2нв+с4нй'-
Само серою разумеется, что новый радикал, бутил, разлагаясь,
тотчас же превращается в более устойчивые формы согласно
сказанному выше.
Для суждения о химической природе продуктов, образую-
щихся при взаимодействии радикалов с молекулами, большое
значение имеет вопрос, какой водород легче отрывается от моле-
кулы под влиянием свободного радикала: первичный (т. е. один
из водородов группы НзС-), вторичный (т. е. водород группы
507
П2С:) или третичный (т. е. водород группы НС;). Термохимиче-
ский расчет, а равным образом и чисто химическая характе-
ристика приводят к согласному заключению, что наиболее под.
вижным является третичный водород, который, очевидно, и дод.
жен наиболее легко отпадать при воздействии на молекулу ра-
дикала. Вторичный водород связан с углеродом прочнее третич-
ного, а первичный — наиболее прочно. В этом именно порядке
и следует ожидать отщепления водородных атомов от молекулы
при действии на нее свободного радикала.
Таковы в самых общих чертах основания теории радикалов
Райса в применении к термическому распаду парафинов. Если
теперь на основе вышеизложенного произвести расчет наиболее
вероятного состава продуктов термического распада того или
иного парафина, то оказывается, что в первые стадии процесса,
когда последний еще не осложнен реакциями вторичного по-
рядка, для ряда объектов получается хорошее совпадение с тем
что дает непосредственный анализ продуктов реакции. Такие
сопоставления здесь, конечно, чрезвычайно интересны и важны:
с одной стороны, в тех случаях, когда между теорией и опытом
наблюдается соответствие, нельзя не видеть в этом вескою под-
тверждения теории; с другой стороны, в тех случаях, когда
между теорией и опытом имеются расхождения, последние могут
дать ценные указания на те поправки, в которых нуждается тео-
рия. Для теории радикалов, которая находится ныне в стадии
усиленного развития и проверки, подобные указания являются,
конечно, особенно ценными.
В качестве иллюстрации ниже приводятся полные схемы и расчет по
Райсу образования продуктов термического распада пропана, изобутана я
тетраметилметана. При этом на основе изменения энергии активации
принимается, что вероятности реакций первичных, вторичных и третич-
ных водородов при ЬСОЭ находятся между собою в отношении 1:2:10.
Вводя обозначения Р2... для первичных реакций. Sz...—для вто-
ричных реакций и Ai, А2... —- для цепных реакций, получаем следующие
отношения:
Пропан С3Н8. Механизм термического распада пропана в указанных
выше условиях по Райсу можно выразить следующим образом:
Pt СзНй —> СНз' + СН3 • СН/
£l CHg • СН2' + С3НЧ —> СЙН6 + СН3 • СН2 СН/
&> СН3 • СН/ + С3Н8 —> С2Нв + (СН3)2 : СН'
АГ С3Н8 4- R —> RH + СН3 • СН2 - СН/—> RH + С2Н4 4- СН/
А2 CsH84-R—> RH + (CH3)2 : СН' —> RH-j-CHg- СН:СН24-Н'
где R = СН/ пли Н',
Конечные продукты распада, определяемые цепными реакциями
(Ai и -As), состоят здесь из смесей в равных количествах а) метана 4-эти-
лена и б) водорода 4~ пропилена. Принимая во внимание общее число пер-
вичных и вторичных водородов в пропане и их относительные реакцион-
ные способности, конечный результат крэкинга пропана можно предста-
• вить следующими уравнениями:
А 6С3Н8= 6СН44-6С2Щ
А 4С3Н8 = 4С3Н6 4* 4Н2
At 4- А2 10С3Н8 = 6СН4 4- 6С2Н4 4- 4С8Н6 4- 4Н2
508
Отсюда для состава смеси продуктов распада пропана в молекуляр-
ных процентах получаем:
СН4 —30%; С2Н4-30%; С3Н6 —20%; Н2-20%.
Как видно из фиг. 108,1, общий характер теоретического графика,
построенного на этих данных, хорошо совпадает с экспериментальной
кривой.
Изобутан, C4Hi0. Распад этого изопарафина можно выразить по
Райсу — следующим образом:
р С4Н1П —> СН3' + (сад СИ'
61 (СН3)2 СН' —> С3Н6 + Н'
С4Ню + R —> ЕН 4- (СНз)2 СН • СН2' —> RH 4- С3Нв4- СНз'
А. С4Ню 4- R ~> ЕН 4- (СН3)3 С' —> ЕН 4" (СНз)2 С = СН2 4- И',
где Е = СН3', Н'.
Конечный результат крэкинга изобугана представляется следующим
образом:
Л1 9C4HW = 9СН4 4-9С3Нв
А% 10С4Ню — IOCjHr (изо) 4~ ЮН2
Л4-А3 19С4Н10 = 9СЩ 4-9С3Нв 4-1СС4Н8 (изо)4-1ОН2
В молекулярных процентах это составит:
СН4 —23,7%; CdHe —28,7%; С4НЙ (изо) - 26,3%; Н2 — 26,3%.
И здесь вычисленные данные хорошо совпадают с данными опыта
(фиг. 108, II).
Тетраметилметан, СБН12. Единственной цепной реакцией, определяю-
щей состав продуктов крэкинга этого изопарафина, является:
А СбН12 4- R —> ЕН 4- (СН3)з • С • СН/ —> ЕН 4- С4Н8 -J- СН/,
(взо-J
где R = СНз'-
Следовательно, продуктами термического распада тетраметилметана
должна быть по теории Рейса смесь метана и изобутилена в экви-
молекулярных количествах, что вполне со-
гласуется с данными опыта (фиг. 108, III).
В предыдущем изложении вопрос
о механизме реакций разложения рас-
смотрен па примерах углеводородов
ряда метана, для которых этот тип
реакций является при крэкинге наи-
более характерным. Что касается меха-
низма разложения углеводородов дру-
гих рядов, то следует отметить прежде
всего, что этот вопрос разработан пока
совершенно недостаточно. Можно огра-
ничиться поэтому лишь самым крат-
ким изложением тех попыток, которые
были сделаны в данном направлении
в применении к углеводородам ряда
этилена.
Энергетический расчет показывает,
Фиг. 108. Графики продук-
тов термического распада
парафинов:
1 — опытный гуафих; э — рассчя-
--- --- ___________________ и___________ тайный.
связи (> С = С <) требуется 145 кал,
тогда как на разрыв простой углеродной связи (-* С — С <—)
должно быть затрачено лишь 82,5 кал, на разрыв же связи угле-
509
рода с водородам (-* С — Н) —100 кал [ 54 ]. Отсюда следует, что
при воздействии высокой температуры на этиленовые углеводе-
роды нельзя ожидать в первую очередь разрыва двойной связи, что
в полной мере соответствует относительной термической ycrog.
чивости таких простейших олефинов, как этилен и его ближай-
шие гомологи. Термического распада этиленовых углеводородов
следует ожидать поэтому не по месту двойной связи, а в других
возможных направлениях: можно представить себе, что под
влиянием (высокой температуры происходит активация некото-
рых молекул непредельного углеводорода, сущность которой за-
ключается в том, что двойная связь этих молекул превращается
•в простую. В результате образуется двухвалентный радикал, ко-
торый может претерпевать дальше распад и разнообразные пре-
вращения по образцу рассмотренных выше одновалентных ради-
калов [55]. На примерах этилена и отчасти пропилена эти пре-
вращения представляются следующим образом.
Этилен, С2Н4. Вслед за- активацией молекулы этилена (I) мо-
жет происходить взаимодействие активированной молекулы с не-
активированной (II); при этом в результате отнятия атома во-
дорода от’ молекулы этилена двухвалентный радикал превра-
щается в предельный одновалетный радикал этил, а молекула
этилена — в непредельный одновалентный радикал (винил):
Н2С : СН2 -> Н2С' • СН2'; (I)
Н2С' - СН2' + Н2С : СН2 -> Н3С • СН/+ Н2С : СН'. (II)
Дальнейшие превращения образовавшихся одновалентных
радикалов, этила и винила, сводятся к следующему: радикал
этил отнимает атом водорода от молекулы этилена, образуя, та-
ким образом, Частицу этана и радикал винил (III), а радикал
винил, отдавая атом водорода, превращается в ацетилен (IV):
Н8С СН/ + НОС : СН2 -> Н3С • СН3 + Н2С : СН'; (III)
Н2С : СН' -> HC:CH-j-H'. (IV)
Образование этана и ацетилена действительно наблюдается
при крэкинге этилена.
Пропилен, СзНе. Активация пропилена происходит во воем
согласно с активацией этилена; дальнейшие же превращения
активированной молекулы, т. е. соответствующего двухвалент-
ного радикала, в данном случае значительно сложнее. Здесь
можно ожидать также непосредственного распада частицы npoi
пилена на два радикала—метил и винил .(I), которые, отнимая
от частицы пропилена водород, будут превращаться далее в ме-
тан (II) и этилен (III). Судьба образующегося одновременно не-
предельною радикала, пропенила, СзН'5, представляется пока
недостаточно ясной.
СН3 • СН : СН2 -> СН/ + СН2 : СН'; (I)
СН/ + СН3 • СН: СН2 -> СН4+СН2 : СН • СН/; ' (Ш
СН2: СН' + СН8 • СН : СН2 -> С2Н4 + СН2 : СН • СН/ (HI)
Метан и этилен действительно являются основными газо-
образными продуктами крэкинга пропилена.
510
Итак, допуская существование двухвалентных и непредель-
ных радикалов, можно объединить механизм термического рас-
пада углеводородов ряда метана и ряда этилена общей идеей
образования на первых этапах этих реакций свободных ради-
калов.
Весьма важно отметить, что существование двухвалентных
радикалов в настоящее время уже не является гипотетическим:
экспериментально доказано, например, что метан при соприкос-
новении с платиновой проволочкой, нагретой до 1000°, распа-
дается на водород и двухвалентный радикал, метилен СНУ*, оха-
рактеризованный некоторыми весьма характерными свойствами,
в частности, например, способностью при воздействии на зер-
кало иода давать иодистый метилен, CH2J2, и т. п.
Механизм реакций разложения был рассмотрен выше главным образом
с точки зрения теории радикалов. Из других, новейших теорий механизма
этих реакций особого внимания заслуживают теоретические представления
Бэрка (1931 г.) с дополнениями Касселя (1933 г.), а также теория Шмидта
(1932—34 гг.). Изложение этих теорий выходит из рамок настоящего ру-
ководства.
Теории реакций уплотнения. Как известно, реакции уплот-
нения особенно характерны для начальных стадий термического
воздействия на непредельные углеводороды. Естественно поэтому
поставить вопрос, в какой степени применимы к этим реакциям
развитые выше представления о механизме первых этапов крэ-
кинга олефинов. Допуская попрежнему, что первым этапом
этого процесса является активация олефина с образованием
двухвалентного радикала; легко представить себе, что одним из
возможных ближайших этапов здесь может быть присоединение
данного радикала к ближайшей частице этилена с образованием
нового двухвалентного радикала, димерного по отношению к ис-
ходному; образовавшийся таким образом димерный радикал пу-
тем перегруппировки водорода превращается далее в димер ис-
ходного олефина. На примере этилена эта реакция уплотнения
может быть выражена следующей схемой:
НоС: СН2 -> НоС' • СН/;
Н2С : СНо + НоС' • СН/ Н2С' • СЩ • СН2 • СН/;
Н2С' • СН2 • СН2 • СН/ Н3С • СН : СН • СЩ.
Ход дальнейшего уплотнения этилена с образованием соот-
ветствующих тримеров, тетрамеров и вообще полимеров рисуется
в следующем виде: промежуточный димерный радикал, С4Н8",
реагируя с новой молекулой этилена, образует тримерный ради-
кал СвН-м", в свою очередь реагирующий со следующей части-
цей этилена и т. д. Реакция приобретает, таким образом, цепной
характер, причем активными центрами ее все время остаются
промежуточные продукты, двухвалентные радикалы различного
типа; димерные, тримернью и т. д. Одновременно с дальнейшим
уплотнением под влиянием различных причин вое время дол-
жна происходить также дезактивация некоторых йз образовав-
щихся радикалов, в результате которой образуются соответ-
511
ствующие полимеры исходного^ олефина, т. е. его димеры, трз-
меры и вообще полимеры. На примере этилена эта схема меха-
низма полимеризации олефинов может быть выражена следуя,
щим .образом:
2Н47
4н8" + ->
Здесь jp0=jp+ q; p=Pi~\~Qi и т. д.
дт ->^ос2н
^оС4Н8"
j?ceHl2"
ьС8н16"
".
V
4 "
±12
№
ьС8н1в''+д2сХ*
Pq^
рС
pfi
Несмотря на всю наглядность и вероятность этой схемы, она
еще нуждается в дальнейшей проработке и проверке [55].
ТЕПЛОТА РЕАКЦИИ КРЭКИПГ-ПРОЦЕОЗА
Одной из характеристик крэкинг-процесса, весьма важных
особенно для тепловых расчетов крэкинг-аппаратуры, является
теплота реакций крэкинг-процесса. Эту величину можно опреде-
лить различными способами [57].
Экспериментально теплота реакции крэкинга была опреде-
лена впервые в специальном приборе при крэкинге парафина
в присутствии хлористого алюминия. Найденная величина коле-
балась в пределах —(550—1000) кал, считая на 1 кг образо-
вавшегося крэкинг-бензина; однако, принимая во внимание спе-
цифические особенности методики, принято считать эту вели-
чину равной приблизительно —500 кал на 1 кг образовавшегося
•крэкинг-бензина [21].
Теплота реакции крэкинг-процесса может быть вычислена
также путём тептовых подсчетов. В основу последних должно
быть положено уже приведенное выше термохимическое соотно-
шение:
Q = 2 9о 2 Q»
где Q—теплота реакции; — сумма теплот сгорания началь-
ных продуктов и — сумхма теплот сгорания конечных про-
дуктов того же процесса.
При выборе величины Ego можно пользоваться либо экспе-
риментально найденной теплотой горения крэкируемого сырья,
либо одной из тех формул, которые позволяют вычислить эту
теплоту на основе элементарного состава вещества (ер. ч. £
стр. 52). Что касается суммы теплот сгорания продуктов крэ-
кинга Eg, то при ее вычислении необходимо принимать во bhs- ‘
мание не. только жидкие крэкинг-продукты разного рода с их
выходами и соответствующими теплотами горения, но также
побочные продукты, газы и кокс. В качестве примера в табл. Ш
приводится подсчет теплот реакции крэкинга для двух грознен-
ских продуктов — парафинистого дестиллата и парафинистого
мазута с исправленными выходами бензина и газа, причем, те-
плоты сгорания были рассчитаны по уравнениям и графику-
Крэго (см. ч. I, стр. 53). ,
512
Таблица 117
Подсчет теплоты реакции крэкинга
(по Обрядчикову)
Исходное сырье Грозненский парафин, дестиллат Грозненский парафин, мазут
Уд. вес при 15° ... . 0,833 0,906
Весовой выход про-
дуктов:
Гае и потери 2,8 3.9
Фракция до 200° . . . 14,2 19,5
„ 200—300° . 17,0 17,7
Остаток 64,6 57,1
Карбоиды — 0,9
Сумма продуктов . . . 98,6 99.1
Уд. вес фракции до 200° 0.744 0,733
, „ „ 200—»
300° 0,829 ода
Уд. вес фракции остатка 0,901 0,933
Теплоты сгорания:
Газ 11400-0.028 = 319 11400-0,039= 445
Фракция до 200° . . . 10475-0.142 = 1487 10500-0,195 = 2048
„ 200-300° . . 10265-0,17 =1745 10275-0,177 = 1819
Остаток 10070-0,646 = 6505 9970-0,571 = 6693
Карбоиды — 7870-0,009 = 71
Сумма теплот сгорания 10056 10076
Теплота сгорания сырья 10 125 - 0,986 = 9983 10055-0,991 = 9965
Теплота реакции: 1
кал/к» сырья — 73 • —111
кал/кг бензина .... —510 -570
Теплоты реакций крэкинга могут быть вычислены также пу-
тем анализа теплового баланса хорошо изученной крэкинг-уста-
новки. Произведенные в этом направлении у нас в Союзе под-
счеты дали .для теплоты этой реакции величину порядка
—(300—350) кал на 1 кг образовавшегося бензина [58]. При-
мерно того же порядка, а именно —(200—300) кал на 1 кг бен-
зина, найдена теплота крэкинг-процесса опытным путем. С углу-
блением крэкинга теплота процесса существенно изменяется и
при глубоких формах крэкинга может сделаться даже положи-
тельной.
ПОВТОРНЫЙ КРЭКИНГ
Однократный крэкинг того или иного легкого нефтепродукта
(газойль, соляр) обычно дает не больше 25—30% крэкинг-бен-
зина. Чтобы повысить выход на этот основной продукт крэкинг-
процесса .без'излишнего газо- и коксообразования, применяют
повторный крэкинг (recycling), сущность которого заключается
33 Зак. 1в22. Химия нефти.
в следующем: легкие части отгоняют, разделяя продукт одно-
кратного крэкинга на крэкинг-бензин и более тяжелые десгнл-
латы; последние подкачиваются затем к сырью и снова посту-
пают на крэкинг, для них уже вторичный. Ввиду громадного
значения, какое имеет повторный крэкинг в практике крэклнь
процесса, его кинетика и конечные результаты заслуживают
специального рассмотрения [52].
Для изучения повторного крэкинга можно применять сле-
дующий метод. То или иное сырье подвергается в определенных
условиях однократному крэкингу с последующей разгонкой в
три части: крэкинг-бензин (до 200°), более тяжелый нож
с температурой кипения 200—350° и остаток выше. 350°; фрак-
ция 200—350° подвергается затем новому крэкингу, и получен-
ный таким образом продукт двухкратного крэкинга вновь разго-
няется на такие же фракции, как после однократного крэкини
Новая фракция 200—350° опять подвергается крэкингу и т. д.
В табл. 118 приведены результаты шести проведенных таким
образом последовательных крекингов эмбенского солярового
масла, которые могут служить для характеристики повторного
крэкинга.
Таблица 118
Повторный крэкинг эмбенского соляра при 450° и 40 атм
(по ГрозНИИ)
Наименование повторных крэкингов Время крэкинга в мин. Выход продуктов в весовых процентах *
газы и потери ДО 200° 200—300° • 300—350° выше 350° варбо- ПДЫ
Однократный . - 42 8,3 28,7 33,1 15,6 10,5 0
Двухкратный . . 52 8,6 21,3 44,5 151 8,6 0
Трехкратный . . 75 7,0 16,3 51,3 15,7 7,9 0
Четырехкратный 101 7,9 12, L 51,2 15,6 10,3 0
Пятикратный . . 101 7,7 3,9 65,8 9,2 12,4 0
Шестикратный . 160 13,8 8,0 54,3 4,2 19.0 0
Данные табл. 118 показывают прежде всего, что скорость раз-
ложения крэкингового соляра (фракция 200-^350°) по мере
углубления крэкинга постепенно падает, так что, например, по
сравнению со скоростью однократного крэкинга скорость п№
кратного -крэкинга примерно в 14 раз меньше. Таким образок
кинетика повторных крэкингов резко отличается от кинетики
крэкинга продуктов прямой гонки: последние крэкир-уются зна-
чительно быстрее.
Относительное количество образующихся газов крэкинга уве-
личивается по мере углубления крэкинга: каждый последующи®
крэкинг дает относительно больше газов, чем предыдущий. Эта
особенность повторного крэкинга прекрасно /наблюдается О2
пересчете газообразования на 1 кг образующегося при крэдак
Г.14
бензина. Так, если при первичном крэкинге, как видно из
табл. 118, количество образующегося газа составляет 182 л
(а в других случаях оно может падать даже до 40—50 л), то
при шестикратном крэкинге оно поднимается до 1500 л на 1 кг
крэкинг-бензина.
Кокоообразование при повторном крэкинге всецело зависит
от характера сырья. В одних случаях (табл. 118) даже после
шестикратного крэкинга заметного образования кокса не наблю-
дается; на другом сырье, например на газойле из грозненской
парафинистой нефти, наблюдается сильное коксообразование
уже после четырехкратного крэкирования, несмотря на полную
аналогию других условий крэкинга. *
Очень характерны изменения состава и свойств крэкинг-бен-
зинов, образующихся при повторных крекингах.
Удельный вес эмбенского соляра после крэкинга сначала
уменьшается, очевидно ввиду образования более легких продук-
тов распада молекул исходного сырья. Однако уже после трех-
кратного крэкинга удельный вес нефтепродукта начинает систе-
матически возрастать, очевидно за счет образующихся продук-
тов уплотнения и конденсации. Удельные веса как крэкинг-бен-
зинов, так и других фракций, а равно и остатка также увели-
чиваются по мере углубления крэкинга. Так, 'фракция с темпе-
ратурой кипения 300—350° уже после четырехкратного крэкинга
имеет удельный вес больше единицы, хотя у той же фракции
исходного соляра удельный вес равен 0,871.
Для суждения о химическом составе продуктов повторного
крэкинга интересно проследить изменение не только их удель-
ного веса, но и таких характеристик, как иодные числа и ани-
линовые точки. Для бензинов повторного крэкинга из эмбен-
ского соляра эти данные приведены в табл. 119.
Таблица 119
Свойства бензинов повторного крэкинга из эмбенспого соляра
(по ГрозНИИ)
Наименование повторного крэкинга Удельный вес (15°) Иодное число Анилиновая точка
Однократный 0,746 118,5 + 41^
Двухкратный 0,7б9 — 4-34.0
Трехкратный 0,797 — + 24,2
Четырехкратный 0,833 548 + 8,4
Пятикратный 0,^39 47,8 + 0,8
Шестикратный 0,873 41,2 <—18,0
Как видно из данных этой таблицы, по мере углубления крэ-
кинга иодные числа крэкинг-бензинов уменьшаются, и, следова-
тельно, содержание в них непредельных постепенно падает. Так
как, с другой стороны,» анилиновые точки тех же бензинов по-
степенно снижаются, то очевидно, содержание в них ароматиче-
зз* • 515
бензин после шести собою практ1гаеска ЧИС1ув
л®®^? таЛ® ^зо^если с точки зрения выхода крекинг,
ароматику. Такта ооразо , ОдЯ<жра,тный, двухкратный и, »-
бен8^_( ?оет>атшй крекинги являются практически цежоо-
жет быть, тРехи₽ат^^_„ *1ие повторные крэкинги могут найти
образными, то метода получения либо бензинов
cn^b^„S" Девиационными свойствами, либо аро
матаЙ Хймйет найти то или иное техническое приме-
пение. которые можно сделать в результате
Практически^вывода, ^орые * ^p^ и в
исследования ш®™!® .,„№KKOg практики. Так как скорость ш>
сойпадают с данными Углубления процесса постепенно па-
вторного п°ет ^точных оснований возвращать од-
дает, то, очевидно, .н * й материал для повторного кра
нажды уже на свежем сырье. Правильнее
книга в Установку,.^™^ледовательном порядке (in to
вести повторный кре _ полученный после однократного кре-
dem, гуськом) а ^Хазойль «Дастся в стд£
книга и отгонки бензи крэкинга передается на сдель-
на Т ко?5 темвда поддерживается н.
Газойль
4<
I крэкирование
(1000 г)
разгонка__________________
------------ Т
4- . У „ остаток
газ бе?зин (d'»)
(fl't) (b г)
промеж,
фракция
(сЧ)
+ (a'4-b' + d')1
у
[I крэкирование
(1000 1)
, разгонка_________________
—------------~L ——- 4,
4 * остаток
гав
а" г) Ф" г) v
у
промеж,
фракция
(c"t)
। + d")t
III крэкирование
И Г. д.
516
сколько выше, чем на установке для однократного крекинга; для
последующего крэкинга крекинг-газойль после двукратного кре-
кинга вновь передается на отдельную установку, работающую
на несколько ином режиме, и т. д. Практика повторного крэ-
кинга в таком порядке отмечена в Америке на заводах некото-
рых крупных фирм.
Методика повторного крэкинга, описанная выше, весьма существенно
отличается от того, с чем приходится иметь дело в производственных усло-
виях, когда на повторное крэкирование идет не одна промежуточная фрак-
ция между бензином и остатком (рисайкл), а смесь этой фракции со
свежим сырьем. Значительно ближе к производственным условиям
другая методика повторного крекирования, смысл которой, как ясно из
прилагаемой схемы, заключается в следующем.
Пусть при первичном крекировании некоторого количества газойля
(например, 1000 г) получено а г газа, Ь' г бензина (до 200°), с' г проме-
жуточной фракции (200—350°) и d' г остатка (выше 350°). Тогда для
вторичного крэкирования к полученной промежуточной фракции (с' г) до-
бавляют столько свежего газойля, чтобы общее количество сырья для пов-
торного крэкинга вновь составило 1000 г (т. е. а' + д' 4- d' г). Пусть, да-
лее, при новой разгонке получено а" г газа, Ъ” г бензина, с" г промежу-
точной фракции и d" г остатка. Для третичного крекирования в качестве
сырья берут с" г фракции с добавкой к ней {(Г 4- b" 4- d")z
свежего газойля, т. е. вновь 1000 г сырья, и т. д. Опыт показывает, что
после двух-трех повторных крэкингов лодбного рода рисайкл становится
постоянным как по составу, так и по относительной массе, и, как видно,
весь процесс в целом в значительной мере приближается к тому, что имеет
место при крэкинге на непрерывно действующей установке [60].
РЕФОРМИНГ
В связи с повышением требований на антцдетовационные
качества бензина и перепроизводством в США бензина прямой
гонки все большее и (к)лыпее значение за последние годы при-
обретает особый' вид крэкинг-процесса, получивший наименова-
ние «реформинг» (reforming). Не представляя по сравнению
с ранее рассмотренными жидкофазным и парофазным крекингом
ничего принципиально нового, .реформинг имеет одну характер-
ную особенность: сырьем для него служат наиболее легкие де-
стиллаты прямой гонки, тяжелый бензин и лигроин [б1].
Сущность реформинг-процесса с технологической точки зре-
ния заключается в следующем. Сырье (бензин, лигроин или
смесь этих продуктов) при помощи подающего насоса загру-
жается в трубчатую печь, откуда оно выходит при температуре
около 560° под давлением 13—20 ат. Из печи продукт напра-
вляется в эвапоратор, причем путем подкачки холодного сырья
достигается прекращение реакции крэкинга. Наконец, из эвапо-
ратора через теплообменники реформированный продукт посту-
пает в ректификационную колонну, где происходит разделение:
реформированный бензин направляется в сборник, флегма же
из нижней части ректификационной колонны идет на подмеши-
вание к сырью перед поступлением его в печь. На реформинг
работают некоторые жидкофазные установки, особенно Даббса,
а также парофазные установки де-Флореза, Джайро и некоторые
другие
517
Принимая во внимание режим процесса, следует ожидать,
что при реформинге происходит глубокое изменение химического,
состава исходного сырья. Действительно, на газы (и потери) лрк"
реформинге бензина приходится 10—12%; одновременно происхо-
дит образование тяжелых масел крэкинг-мазута (до 10% к
больше). Таким образом при реформинге можно наблюдать пол-
ный ассортимент тех продуктов, которые образуются при глубо-
ком крекинг-процессе. Так как, кроме того, при этом происходит
резкое повышение антидетонационных свойств бензина, то есте-
ственно притги к выводу, что реформинг сопровождается обиль-
ным новообразованием тех углеводородов, которые столь харак-
терны для продуктов крэкинга, т. е. .непредельных и ароматики.
По своим температурным условиям реформинг соответствует
крэкингу в паровой фазе; однако, принимая во внимание боль-
шую термическую устойчивость бензиновых углеводородов по
сравнению с углеводородами обычного сырья крэкинг-процесса,
следует думать, что реформированный бензин по своему составу
занимает промежуточное место между жидкофазным и парофаз-
ным крэкинг-бензином.
Больших перспектив следует ожидать от каталитического
реформинга,'т. е. от такой переработки бензина и особенно ли-
гроина, когда под влиянием надлежащим образом подобранного
катализатора парафины могли бы, например, превращаться
в изопарафины и т. п. В этом направлении, повидимому, воз-
можно искать и найти один из путей получения высокооктано-
вого топлива.
АНАЛИЗ И СОСТАВ КРЭКИНГ-БЕНЗИНА
Несмотря на широкое распространение, которое получил в на-
стоящее время крэкинг-процесс, вопрос о составе даже основ-
ного крэкинг-продукта — бензина — не может считаться пока
окончательно разрешенным. Отсутствие стандартной методику
при необычайном разнообразии предложенных способов анализа
крэкинг-бензина, громадная, все продолжающая возрастать ли-
тература этого вопрЪса с полной очевидностью свидетельствуют,,
что здесь мы встречаемся с одним из наиболее трудных вопро-
сов нефтяной химии.
Анализ крэкинг-бензина [62J. Задача анализа крэкинг-бен-
зина, как и бензина прямой гонки, заключается в определении
его группового состава. Эта задача чрезвычайно осложняется,
здесь значительным содержанием в крэкинг-бензине непредель-
ных углеводородов, которые, как известно, содержатся в бензи-
нах прямой гонки в совершенно ничтожных количествах. В связи,
с этой особенностью состава крэкинг-бензина первым этапом его*
анализа является определение в нем непредельных. Эта задача
легко решается методом иодных чисел с последующим пересче-
том полученных результатов на процентное содержание непре-
дельных в бензине; при этом условно притшмялот, что средний*
молекулярный вес непредельных углеводородов, содержащихся.
618
в анализируемом нефтепродукте, совпадает со средним молеку-
лярным весом всего нефтепродукта. Наиболее простым и точ-
ным методом определения иодных чисел в крэ кинг-бензинах
.является метод Маргошеса (см. ч. I).
Определение непредельных различными галоидными методами приво-
дит нередко к противоречивым результатам, так как последние зависят
от целого ряда факторов, не всегда принимаемых во внимание.
Важнейшими из них являются:
1) химическая природа непредельных;
2) величина избытка галоидцрго раствора и его концентрация;
3) физико-химические свойства растворителя?
4) пррдолжительность опыта.
Перечисленные факторы оказывают влияние также на результаты, по-
.яучаемые по методу Маргошеса, и для получения надежных результатов,
как показало специальное исследование, необходимо соблюдение следую-
щих основных условий:
1. Иодный раствор должен быть взят о таким расчетом, чтобы избыток
его составлял примерно 90%, считая на раствор, взятый для определения.
2. Концентрация иодного раствора рекомендуется 7e N.
3. Продолжительность взаимодействия иодного раствора с углеводо-
родом 5 мин.
4. В качестве растворителя для иодного раствора может применяться
либо спирт, либо смесь эфир — ацетон.
Другой метод определения непредельных, особенно удобный
для анализа крэкинг-бензина, это — объемный метод полухлори-
стой серы. Весьма существенные преимущества этого метода,
подробно описанного в ч. I, по сравнению с галоидными мето-
дами заключаются в следующем:
1. Результаты анализа (в процентах но объему) по методу
полухлористой серы получаются непосредственно и не требуют
всегда несущих в себе источник ошибки пересчетов на основе
среднего молекулярного веса анализируемого продукта.
2. Метод полухлористой серы позволяет производить опреде-
ление непредельных не только в отдельных фракциях крэкинг-
бензина, но и в цельном, неразогнанном бензине, что невоз-
можно при пользовании галоидными методами.
3. Определение непредельных методом полухлористой серы
включает в себя последующую, крайне ответственную операцию
анализа крэкинг-бензина— удаление непредельных; после опре-
деления их методом полухлористой серы исследуемый образец
.вполне готов для последующего определения ароматики.
4. Особо должна быть отмечена высокая надежность резуль-
татов, получаемых методом полухлористой серы, зависящая от
полной индиферентности полухлористой серы на холоду ко всем
другим компонентам крэкинг-бензина, а именно: к парафинам,
нафтенам и ароматике.
Вторым этапом при анализе крэкинг-бензина является уда-
.ление непредельных, совершенно необходимое для последующего
определения ароматики. Эта операция при анализе крэкинг-бен-
зина производится либо с цельным, неразогнанным бензином,
либо с теми его фракциями, которые должны пойти затем на
определение ароматики; фракции эти—следующие:
519
Бензольная фракция, температура кипения 60— 95°
Толуольная * • . 95—120°
Ксилольная „ . • 120—150°
этим трем фракциям крэкинг-бензина иногда присоеди-
няют четвертую, высшую фракцию с температурой кипения
150—200°.
Как было указано выше, удаление непредельных при н
определении методом полухлористой серы производится одновре-
менно и притом почти количественно. В бензине остается всего
0,4—0,8% непредельных, которые легко удаляются либо повтор-
ной обработкой бензина полухлористой серой, либо обработкой
его 85 %-ной серной кислотой. Само собою разумеется, что эти
последние следы непредельных должны быть учтены и добавлены
к основному их количеству, которое было получено в результате
первой обработки бензина полухлористой серой.
Кроме описанного метода, для освобождения от непредельных был
•предложен ряд других менее надежных и удобных. Важнейшие из них еле-
дующие:
1. Сернокислотный способ. Известны многочисленные вариации метода
удаления непредельных с помощью серной кислоты различной концентра,
ции (от 80- до 95%-ной). Все они основаны на способности непредельных
углеводородов давать в присутствии серной кислоты продукты полимери-
зации, для отделения от которых фракция анализируемого бензина после
обработки ее серной кислотой должна быть тщательно перегнана. Основ-
ной недостаток сернокислотного способа заключается в том, что при обра-
ботке серной кислотой углеводородных смесей, содержащих, кроме непре-
дельных, также ароматику, всегда имеет место образование продуктов
конденсации ароматических углеводородов с непредельными (ср. ч. I,
стр. 90). Так например, есть указания, что даже 80%-ная серная в при-
сутствии непредельных, удаляет до 30% ароматики, а 88%-ная кислота—
до 50%. Вся эта ароматика образует с непредельными высокомолекулярные
продукты конденсации, которые при последующей отгонке бензина остаются
вместе с продуктами полимеризации непредельных и поэтому теряются для
определения в отгоне ароматики.
2. Способ бромирования при нивкой температуре [вз]. Метод заклю-
чается в действии на фракцию крокинг-бензипа брома при охлаждении
до —20° с последующей отгонкой не вошедшего в реакцию углеводорода
от образовавшихся дибромидов сначала’ в вакууме, затем 2—з раза при
обыкновенном давлении. Основным] недостатком метода являются побоч-
ные процессы, всегда возможные при непосредственном действии брома на
углеводородные смеси, несмотря на хорошее охлаждение (реакции замеще-
ния и т. п.).
3. Способ полимеризации при участии хлористого алюминия [63]. Под
ваавнием хлористого алюминия непредельные углеводороды столь же легко
образуют продукты полимеризации, как и в присутствии серной кислоты.
Оказалось, однако, что применение этой методики для анализа крекинг-
бензина всегда дает сильно преуменьшенные результаты при последую-
щем определении ароматики, которая также вовлекается в реакцию с мо-
ристым алюминием иногда на 10—20%, а иногда на 50—75%. Тем самым
устанавливается, очевидно, непригодность этого метода для анализа крэ-
кинг-бензина.
4. Способ каталитической гидрогенизации. В литературе по катализу
имеются указания, что могут быть приготовлены такие катализаторы
(Ni, Си), которые, не будучи способны гидрировать ароматику, сохраняют
эту способность по отношению к непредельным углеводородам. Применив
такого рода катализатор к смеси ароматических и непредельных углеводо-
родов в присутствии водорода, можно, очевидно, подобрать такие условия,
при которых непредельные прогидрировались бы полностью, ароматика же
520
осталась бы незатронутой и могла бы быть определена далее одним из
методов, применяемых к ее определению в бензинах прямой гонки. Не-
смотря на кажущуюся простоту этого метода, практика его до сих пор не
может считаться окончательно разработанной.
После удаления непредельных дальнейший анализ крэкинг-
бензина сводится, очевидно, -к случаю анализа бензина прямой
гонки. Не вдаваясь в подробное рассмотрение этого вопроса
(см. ч. I, стр. 95, 144, 217), отметим здесь лишь основные его
моменты.
Определение ароматики в крэкинг-бензинах и его фракциях
после удаления непредельных производится проще всего серно-
кислотным методом: углеводородную смесь обрабатывают в сулъ-
фаторе либо тремя объемами раствора 30 г фосфорного ангид-
рида в 100 см3 серной кислоты удельного веса 1,84 (по Каттвин-
келю), либо двумя объемами дымящей серной кислоты с содер-
жанием 3—8% серного ангидрида при охлаждении и учитывают
изменение объема углеводорода вследствие поглощения арома-
тики серной кислотой. Лишь в тех случаях, когда содержание
ароматики в бензине не превышает 2—з%, вместо сернокислот-
ного способа рекомендуется применять метод анилиновых точек
[64], причем для удаления ар’оматики можно пользоваться теми
же реагентами, как для ее определения, т. е. либо раствором
Каттвинкеля, либо дымящей серной. Применение для той же
цели более слабой серной .кислоты, например моногидрата, есте-
ственно требует значительно больших количеств реагента; так
английские стандартные нормы рекомендуют для полного удале-
ния ароматики применять в общей сложности 4 объема моно-
гидрата [65]. Несомненно, однако, что при столь значительных
количествах серной кислоты на сцену должен выступить один
трудно поддающийся учету источник ошибки, а именно—про-
стая растворимость различных углеводородов бензина в серной
кислоте.
Последним этапом анализа крэкинг-бензинов является опре-
деление в них нафтенов и парафинов. Определение произво-
дится методом анилиновых точек, предварительное же удаление
ароматики — как рассмотрено выше.
Как ясно из вышеизложенного, основной задачей анализа крэкинг-
бензинов является определение в них содержания непредельных и арома-
тики. В процессе разработки этого вопроса неоднократно предлагалась
значительно более простая методика по сравнению с рекомендованной,
а именно: после определенйя непредельных, как это было описано выше,
можно, казалось бы, путем обработки бензина избытком серной кислоты
удалить -из него непредельные и ароматику вместе; производя, таким обра-
зом, суммарное определение обоих этих компонентов и зная уже содержа-
ние в бензине непредельных, по разности можно вычислить содержание
в нем также и ароматики. Ближайшее исследование показало, однако, не-
достаточную надежность этой методики, так как образующиеся при’ дей-
ствии серной кислоты на смесь ароматики с непредельными продукты
полимеризации и конденсации чрезвычайно трудно поддаются в дальней-
шем удалению из бензина. Ошибки, мсорые получаются при анализе
крэкинг-бензина по этому способу, могут*юстигать 30% и выше от общего
содержания непредельных и ароматики [62].
5Й1
Состав крэкинг-бензина. Ввиду трудностей, с которыми со-
пряжен анализ крэкинг-бензина, и отсутствия стандартной ме-
тодики к литературным данным о составе крэкинг-бензина сле-
дует относиться с большою осторожностью. Очевидно, например,,
что сопоставление состава различных образцов крэкинг-бензина^
проанализированных различными авторам без точного описа-
ния методики анализа, строго говоря, совершенно невозможно,
и только отсутствие более точных данных может оправдать такое
сопоставление. Приводим прежде всего данные о составе совет-
ских товарных бензинов прямой гонки и крэкинга (жидкофаз-
ного), а также о крэкинг-бензине, полученном крекированием
бакинского газойля на установке Кросса (табл. 120).
Таблица
Состав советских бензинов1
(по Елигерману)
Наименование бензина Уд. вес Нача- ло ки- пения в °* Конец кипе- ния в ° Группов. состав в %
непре- дельн. аро- мат. наф- тены пара- фины
Бакинский прямой гонки . . 0,753 50 188 0 3,7 52,7 43,6
Грозненский легкий 0,723 40 190 0 3,0 32,3 64,7
Грозненский тяжелый .... 0,740 49 207 6,0 6,7 27,0 66,3
Бакинский крэкинг-бензин . 0,731 2:9 187 25,6 14,7 9,4 53,3
Грозненский . 0,718 26 188,5 27,6 16,4 6,2 49,8
Крэкинг-бензин с уетановкч Еросса 0,752 32 191 34.2 2,1 11,9 51,8
Сравнивая состав -бензинов прямой гонки и крэкинг-бензина,
мы видим прежде всего глубокое различие между ними в содер-
жании непредельных: в то время как в бензинах прямой гонки
непредельных вовсе не содержится или содержится всего не-
сколько процентов, содержание их в бензинах жидкофазного
крэкинга повышается до 25% и выше. Резко повышается в бен-
зинах жидкофазного крэкинга по сравнению с обычными бен-
зинами прямой гонки также содержание ароматики; зато по со-
держанию нафтенов бензины крэкинга значительно уступают
бензинам прямой гонки. Что касается, наконец, углеводородов
парафинового ряда, то содержание их в обоих типах бензинрв.
примерно одного и того же порядка.
Большой интерес представляют данные о групповом составе
крэкинг-бензинов различных систем парофазного крэкинга
(табл. 121). Само собою разумеется, что сделанные выше ого-
ворки должны иметь в данном случае особое значение.
1 Приведенное в табл. 120 содержание ароматики в бензинах' жидко-
фазного крэкинга несколько ниже действительного. Причина—недостаточ-
ность примененной автором [661 методики ее определения по разнести
между двумя величинами: суммарным количеством непредельных и аро-
матики, определенным путем удаления их серной кислотой, и количеством
непредельных, найденным методом иодных чисел.
5?)
Таблица 121
Состав парофявных крэкинг-бенвннов
(по разным источникам)
Наименование установки Групповой состав в %
непредельн. аро матич. нафтены парафины
Джайро 45—50 40—45 остальное
Воке 36.0 64,0 —
де-Флорез 36.2 12.0 11,9 32.9
^Моторин" Герра {14] . 31,0 59.0 10,0
,Химгаз“ 69.0 13,0 17.8
В табл. 120 и 121 видно, что во всех бензинах парофазного
крэкинга суммарное содержание непредельных и ароматики за-
метно выше, чем в бензинах жидкофазного крэкинга. Характерно
также, что именно в бензинах парофазного крэкинга содержа-
ние непредельных достигает своей наибольшей величины; как
«бензины жидкофазного крэкинга, так и бензины пиролиза со-
держат непредельных меньше, чем бензины парофазного крэ-
кинга: первые за счет повышенного содержания в них парафи-
нов, вторые — за счет ароматики.
Что касается, наконец, продуктов пиролиза, то, как уже было
указано в начале этой главы, низшие, бензиновые погоны, полу-
чаемые в результате ректификации жидких продуктов этого про-
цесса при нормальном его режиме, состоят после надлежащей
очистки почти нацело из ароматических углеводородов.
ХИМИЧЕСКАЯ ПЕРЕРАБОТКА ПРОДУКТОВ КРЭКИНГА
Значение крэкинг-процесса далеко не ограничивается тем,
что на базе этого способа переработки нефти нефтяная промыш-
ленность приобрела новый грандиозный источник получения
главного на сегодняший день целевого продукта своего производ-
ства— бензина. Наряду с разрешением этой чрезвычайно важ-
ной проблемы переработки нефти крэкинг-процесс стимулировал
невиданный еще размах исследовательских работ в области освое-
ния продуктов крэкинга путем их чисто химической перера-
ботки. Особенно крупных успехов достигла в настоящее время
химическая переработка газообразных продуктов'крэкинга, ко-
торые первоначально рассматривались как нежелательный, хотя
и неизбежный отход крэкинг-процесса и либо выпускались на
воздух, либо направлялись в топки.
Мы видели, что в отличие от естественных газов газы крэ-
кинга и пиролиза характеризуются значительным содержанием
непредельных углеводородов. Высокая реакционная способность
этих последних, естественно, выдвинула вопрос об их более ра-
циональном использовании путем превращения их с помощью
той или иной химической реакции в галоидопроизводные, спирты
523
я другие вещества, на которые предъявляется спрос различными
отраслями промышленности. Так создается на наших глазах но-
вая отрасль нефтяного дела, теснейшим образом связанная
с крэкинг-процессом: синтез разнообразнейших органических ве-
ществ на базе сырья нефтяного происхождения.
Подробные данные о составе газов крэкинга и пиролиза раз-
личных нефтепродуктов были даны в табл. 102, юз и 104. По
содержанию непредельных углеводородов лучшим сырьем да
химической переработки, вообще говоря, являются газы паро-
фазного крэкинга и пиролиза. Как и во всякой другой отрасли
химической промышленности, ближайший выбор и подготовка
сырья являются и здесь первой и основной задачей.
Выбор и подготовка сырья. Выбор сырья для химической
переработки производится в зависимости от местных условий,
а также от целевой установки. Для получения, например, про-
изводных этилена выгодно применять газы пиролиза; перера-
ботку пропилена и бутиленов целесообразнее базировать на га-
зах парофазного крэкинга. Газы жидкофазного крекинга
являются наименее ценным сырьем для химической переработки.
Выбранный для переработки технический газ должен быть
подвергнут предварительно специальной подготовке, сущность
которой -заключается в концентрировании наиболее ценных со-
ставных частей газа и отделении баластных его частей (метан,
водород).
Предложено несколько методов для разделения газовых сме-
сей углеводородов на образующие их компоненты [67]:
1. Метод адсорбции. Сущность метода заключается в насы-
щении газом того или иного адсорбента, обыкновенно активиро-
ванного угля, и в последующей регенерации адсорбированного
газа перегретым паром. Уже после одной такой операции содер-
жание этилена в регенерированном газе пиролиза повышается
до 2-5%, а пропилена — до 12%. При многократном повторении
этой операции содержание этилена в этиленовой фракции газа
может быть доведено до 85%, а пропилена в пропиленовой
фракции — до 50—60%. Несмотря на такие результаты, метод
концентрирования газов путем адсорбции активированным углем
едва ли найдет применение в промышленности, так как он имеет
ряд весьма существенных недостатков: дороговизна адсорбента,
высокий расход пара и воды, потребность в больших емкостях
(газгольдеры, угольные адсорберы), наконец длительность про-
цесса И T. Д'
2. Метод жидкостной абсорбции. Газы парафазного крэкинга
или пиролиза растворяются в подходящем растворителе, напри-
мер в крэкинг-бензине («Химгаз»), под давлением (до 15 от)
в специальной поглотительной койонне с тарелками, причем
растворитель подается сверху, газ же — снизу колонны. Затем
за счет разности давлений растворитель вместе с растворенным
в нем газом подается последовательно на ряд ректификацио]тных
колонн, где в порядке снижения давление происходит последо-
вательное выделение (ректификация) растворенных газов: в ко-
524
лонне первой ректификации 'выделяется главным образом пропи-
лен с примесью этилена; в следующей колонне происходит вы-
деление бутиленов, к которым всегда подмешаны амилены; нако-
нец, в последней колонне выделяются амилены с примесью выс-
ших олефинов. Что касается этилена, то значительная часть
IIISKl
его проходит через поглотительную колонну свободно и уходит
вместе с метаном и водородом через верхнюю часть этой колонны
в газгольдер. Само собою разумеется, что вместе с олефинами
переходят в раствор, а затем выделяются из раствора также низ-
шие гомологи метана, главным образом пропан, бутан, изобутан
и пентаны. Самое выделение газа в отдельных колоннах регули-
руется не только снижением давления, но и надлежащим подня-
тием температуры.
С технической точки зрения процесс жидкостной абсорбции
осуществляется значительно проще процесса адсорбции. К со-
жалению, однако, и в этом случае получаемые газовые концен-
траты крайне неоднородны; для выделения же более однородных
газов необходимо повторение всего цикла жидкостной .абсорб-
ции, что крайне осложняет процесс.
3. Метод фракционировки. Фракционировка газов при низ-
ких температурах и высоких давлениях на сегодняшний день —
наиболее совершенный способ выделения из технических газов
отдельных компонентов, хотя и здесь главными продуктами раз-
гонки являются углеводородные смеси. Обыкновенно получают
следующие фракции: амилен-пентановую, бутилен-бутановую,
пропилен-пропановую и этилен-этановую; последние два компо-
нента— этилен и этан —иногда получаются раздельно, причем
некоторые системы доводят концентрацию этилена до 98%. Не
останавливаясь на аппаратурной стороне метода фракционировки
газов крэкинга и пиролиза, ограничимся здесь указанием, что,
повидимому, этот метод является наиболее надежным как с тех-
нологической, так и с экономической ’ стороны особенно в тех
случаях, когда дальнейшая переработка требует возможно более
однородного сырья.
4. Селективная химическая переработка. При химической пе-
реработке газов крэкинга и пиролиза по самому существу при-
меняемых реакций нередко с успехом можно.пользоваться газо-
выми смесями. Так например" примесь к этиленовым углеводо-
родам гомологов метана, как правило, не оказывает влияния на
чистоту получаемого конечного продукта переработки, так как
парафины значительно устойчивее к обычно применяемым
в данном случае реагентам либо вовсе с ними не реагируют.
Аналогичным примером является действие на различные этиле-
новые углеводороды серной кислоты. Как будет показано ниже,
ввиду существенного различия в скоростях взаимодействия раз-
личных этиленов с серной кислотой одной и той же концентра-
ции представляется возможным проводить поглощение различ-
ных этиленов последовательно, пропуская их смесь через, скруб-
беры, орошаемые серной кислотой с различной, постепжно по-
вышающейся концентрацией.
525
Важнейшие, определившиеся в настоящее время пути химв-
ческой переработки газов крэкинга и пиролиза следуют^
[67, 68]. ’
Действие хлористого водорода. Присоединяясь к этилену к
его гомологам, хлористый водород образует с ними соответствую-
щие монохлориды. Так например, с этиленом при этом обра-
зуется хлористый этил (температура кипения 136); с пропиле-
ном— почти исключительно хлористый изопропил (температура
кипения 36°) с небольшой лишь примесью хлористого пропила.
Е Т’ Д" 1) СН2: СН2 4- НС1 = СНУ - СН2С1;
2) СН3 • СН : СН2 + НС1 = СН3 • СНС1 • СНЧ;
3) СН3 • СН: СН2 + НС1 = СН8 - СН2 - СН„С1.
Взаимодействие низших этиленовых углеводородов с хлори-
стым водородом протекает лишь при повышенной температуре и
в присутствии катализаторов. Лучшим катализатором эад
процесса является, повидимому, хлористый висмут. Реакцию
ведут, быстро пропуская, например, смесь этилена с хлороводо-
родом (1) при 150—200° над катализатором, нанесенным на ту
или иную насадку. Улавливание хлористого этилена может
быть почти нацело произведено применением высокой компрес-
сии (до 100 аг) или с помощью активированного угля. Получе-
ние хлористого изопропила (2) протекает в аналогичных усло-
виях, но может быть осуществлено при несколько более низкой
температуре. Разделение этих хлоридов ввиду значительной раз-
ницы в температурах кипения труда не представляет.
Еще легче присоединяется хлористый водород к бутиленам, притек
частично эти реакции идут уже при комнатной температуре. Однако он
осложняются здесь образованием изомерных хлоридов, а также некоторыми
вторичными процессами: отщеплением хлороводорода, уплотнением двух
частиц хлористого бутила в- октилхлорид и т. п.:
C4HSC1 -J- С4НвС1 = НС14- С8НПС1.
Низшие монохлориды находят широкое применение в хими-
ческом синтезе в качестве алкилирующих веществ. Хлористый
этил применяется также для местной анестезии.
Действие хлора [69]. Хлор присоединяется к низшим оле-
финам довольно медленно, особенно вначале. Ускорителями этой
реакции могут служить различные вещества, например древес-
ный уголь; в присутствии конечного продукта реакции процесс
значительно ускоряется. Большое влияние на ход реакции ока-
зывают также температура, скорость пропускания смеси газов
через катализатор и тщательность перемешивания газообразной
смеси.
Начальным продуктом взаимодействия этилена с хлором
является хлористый этилен (температура кипения 85°), по
уравнению:
СН2: СН2 4- С12 = СН2С1 • СН2С1. ,...; J
526
Кроме хлористого этилена, при этой реакции образуются
также высшие хлориды этана, и в первую очередь 1,2,2-три-
хлорэтан (температура кипения 114е):
СЩС! • СН2С1+ С12 = НС1 + CHoCl • СНСЦ.
Хлористый этилен и трихлорэтан являются -прекрасными
растворителями и широко применяются в лакокрасочной про-
мышленности и промышленности пластических масс. Кроме
того, хлористый этилен находит применение как сырье для по-
лучения различных производных гликола. Так например, дей-
ствием на хлористый этилен сухого уксуснокислого натрия по-
лучают один из наиболее ценных растворителей лакокрасочной
промышленности — уксусный эфир гликола: .
СН2С1 СН2—OOC-CHj
| + 2Na00C - СН3 = 2NaCl + |
СН2С1 СН2—00С-СН8
При нагреваний хлористого этилена с раствором соды под
давлением получается свободный гликол, а действием на него
аммиака получают этилендиамин, применяемый в качестве
ускорителя при вулканизации каучука, и т. д..
СН2С1 СЩ - NIL
| +4NH = 2NH4C1+ I
СН2С1 СЩ - NIL,
Действие серной кислоты. Взаимодействие серной кислоты
с олефинами, в зависимости от условий’ может протекать в раз-
личных направлениях. Для целей химической переработки эти-
лена и его гомологов, находящихся в газах крэкинга и пиро-
лиза, особый интерес представляет реакция присоединения (
с образованием алкилсерных кислот, которые при последующем
действии воды гидролдзуются с образованием сответствующего
спирта; серная кислота .при этом получается обратно и после
концентрации может быть вновь направлена в реакцию. Так
например, с этиленом эти.реакции могут быть выражены сле-
дующей схемой:
СЩ: СН2 + ILS04 = СН8 • СЩСЗО^Н;
СН8 • СЩ 0S03H + Н20 == HgSO, + СНдСЕЛН.
Скорость поглощения этиленовых углеводородов серной кис-
лотой с образованием алкилсерных кислот зависит от различных
факторов, важнейшими из которых являются природа углеводо-
рода и крепость серной кислоты. Так например, 90%-ная сер-
ная кислота практически вовсе не поглощает этилена, тогда как
изобутилен легко поглощается уже 54 %-ной кислотой. На этой
разнице в отношении к серной кислоте различных этиленовых
углеводородов основано промышленное получение спиртов из га-
зов. крэкинга без предварительной их ректификации. Смесь га-
зов пропускают сначала в скруббер, орошаемый 55—58%-ной
серной кислотой. Здесь поглощаются высшие гомологи этилена,
527
содержащиеся в смеси: амилены .и все бутилены, за исключе-
нием нормального бутилена, для связывания которого газы на-
правляют в особый скруббер с 65 %-ной серной кислотой. Дяд.
логичным образом для поглощения пропилена применяют
85 % -ну ю кислоту, для «этилена же — 98%-ную.
, Взаимодействие этиленовых углеводородов с серной кисло
той — реакция экзотермическая и в зависимости от темпера-
турнъгх условий может привести к образованию разного рода
полимерных веществ. Ввиду этого во избежание побочных про-
цессов «приходится вести ее при охлаждении. Так например,
при поглощении пропилена температура не должна превышать
5—10°. С другой стороны, для возможного ускорения прощеоса,
что особенно важно в случае этилена и пропилена, предложены
•различные катализаторы, например сернокислое серебро и др.
Что касается, наконец, последующего гидролиза алкилоер.
ных кислот, то и здесь в каждом отдельном случае пользуются
своей специальной методикой. Так например, для получения
этилового спирта можно обрабатывать этилсерную кислоту не-
посредственно водяным паром, тогда как при гидролизе изопро-
пилсерной кислоты во избежание образования полимерных ве-
ществ требуется 'Предварительное разбавление водой и т. п.
[12, 70].
Химическая сторона проблемы превращения этиленовых углеводоро-
дов в соответствующие спирты выяснена давно. Основные задачи, от раз-
решения которых зависит внедрение этих процессов в промышленность,
заключаются в следующем:
1. Обеспечение промышленности надлежащими запасами дешевого
сырья, этиленовых углеводородов.
2. Сведение до минимума расхода серной кислоты, которая при совре-
менном уровне наших знаний является необходимым участником превра-
щения этиленовых углеводородов в спирты.
3- Проведение процесса гидролиза промежуточных продуктов реакции
алкилосерных кислот в таких условиях, чтобы целевой продукт реакции—
спирт — получался с наилу чшими выходами без излишнего разбавления
кислоты, которая должна возвращаться в производство.
Первый из указанных вопросов решается в настоящее время просто;
крэкинг и пиролиз дают неисчерпаемый источник этиленовых углево-
дородов— этого основного сырья для получения спиртов. Два других
вопроса потребовали большой исследовательской работы; весьма удачное
разрешение для случая превращения этилена в винный спирт они полу-
чили в работах АзНИИ.
По методу АзНИИ [71] взаимодействие этилена с серной кислотой про-
водится при температуре около 70° и повышенном давлении (ю—15 от).
Компоненты тщательно перемешиваются в аппарате специальной конструк-
ции. В этих условиях расходный коэфициент серной кислоты удалось
снизить почти до теоретической величины. Последующий гидролиз этмо-
оерной кислоты осуществляется в АзНИИ на непрерывно действующей ко-
лонне с отгонкой образующегося спирта; отработанная серная кислота по-
лучается с концентрацией до 60—85% и выводится через нижнюю чаоть
колонны.
Вопрос об утилизации отработанной серной кислоты при получении
винного спирта из этилена наиболее полно решается параллельной Орга-
низацией производства других спиртов: изопропилового, изобутиловых
и т. д.; как было указано выше, при этом требуется серная кислота’по-
степенно падающей концентрации. •'
528
Получение спиртов из газов крэкинга производится в ОША
на нескольких предприятиях (Standard Oil Со of New Jeraey,
Union Carbide Co и др.). Это производство обогатило различные
отрасли промышленности рядом новых, ранее не доступных
в большом масштабе спиртов и их производных. Таковы осо-
бенно изопропиловый спирт (1) и дня, бутиловых: третичный
(2), получаемый из изобутилена, и вторичный (3) —из нор-
мальных бутиленов. Они получаются по следующим схемам:
СНЗХ СНЗЧ СНЗХ
>СН-> >CH0S03H-> >СН • ОН;
С-Н/ СН/ СН/
снзх
/С = СНз -► (CH8)3C0S03H -> (СН3)3С - ОН;
СН/
сн3. СН2. СН=CHL С0НбХ с2нбЧ
> “ >CH0S03H-> >сн-он.
сн3• сн=сн • СН/ СН/ СН/
(1)
(2)
(3)
Эти три спирта широко применяются в качестве растворите-
лей в промышленности пластических масс и лако-красочной,
а изопропиловый — также в качестве заменителя винного спирта,
в парфюмерном деле и других отраслях промышленности. Полу-
чение их в промышленном масштабе открыло также возможность
широкого использования их для промышленного синтеза*
в частности, например, для каталитического получения соответ-
ствующих кетонов: из изопропилового спирта — ацетона, из вто-
ричного бутилового спирта — метилэтилкетона. Находят приме-
нение также некоторые ближайшие производные этих спиртов,
например’ уксусный эфир изопропилового спирта, который
можно получать непосредственно взаимодействием изопропил-
серной кислоты с уксуснокислым кальцием:
2C3H7OSO3H -И (СН3С00)2Са = 2СН3С00С3Н7 + SO4Ca + SOA-
Особенно важное значение приобретает простой изопропило-
вый эфир, получаемый в больших количествах дегидратацией
изопропилового спирта с помощью серной кислоты при нагре-
вании:
2С3Н70Н = С3Н7 • 0 • С3Н7 4- ЩО.
Изопропиловый эфир — хороший растворитель для живот-
ных, растительных и минеральных масел, а также для некото-
рых восков и смол. Он представляет несомненный интерес и зна-
чение и как добавка к моторному топливу, повышающая его ан-
тидетонационные свойства (октановое число 100).
Получение спиртов из газов крэкинга через алкилоерные
кислоты представляет большие неудобства, связанные с затра-
той «больших количеств серной кислоты и последующей ее. кон-
центрацией. .Большой интерес представляют поэтому указания
на возможность получения этилового спирта непосредственным
529
34 Зак. 1622. Химия нефти.
действием на этилен воды при высоких температурах (400—
500°) и давлениях (до 200 ат) в присутствии подходящих ката-
лизаторов (уголь, покрытый фосфорной кислотой и др.) Однако
эта методика еще не вышла, повидимому, из стадии предвари-
тельной разработки.
Действие хлорноватистой кислоты. [72]. Хлорноватистая ки-
слота легко присоединяется к этилену и его гомологам с обра-
зованием соответствующих хлоргидринов; так, например, обра-
зование хлоргидрина этилена может быть выражено следующим
уравнением:
СН2 : СН2 + СЮН = СН2С1 • СН2ОН.
Необходимую для этой реакции хлорноватистую кислоту при-
готовляют пропусканием хлора в дестиллированную воду на хо-
лоду; вслед за этим тем или иным способом осуществляют воз-
можно более тесное соприкосновение хлорноватистой кислоты
с этиленом при температуре не свыше 10 , регулируя процесс
таким образом, чтобы концентрация образовавшегося хлорги-
дрина была не выше 8—10%; в противном случае параду
с хлоргидрином образуется заметное количество побочною про-
дукта реакции—хлористого этилена.
Выделение хлоргидрина (температура кипения 129°) из про-
дукта взаимодействия этилена с хлорноватистой кислотой про-
изводится путем ректификации и высаливания. В ряде случаев
для дальнейшей переработки хлоргидрина этилена можно поль-
зоваться непосредственно его водный раствором. Важнейшими
продуктами такой переработки являются:
1. Этиленгликол. Этот простейший гликол получают в боль-
шом масштабе, нагревая хлоргидрин этилена со слабым раство-
ром бикарбоната или щелочи в автоклаве при небольшом дав-
лении. Реакция протекает по уравнению:
СН2С1 • СН20Н + COsHNa = NaCl + С02 + СН20Н СН20Н.
Этиленгликол (температура кипения 197°; температура пла-
вления— 17,4°)—бесцветная, сиропообразная жидкость слад-
кого вкуса; может применяться прежде всего как суррогат гли-
церина в пищевой промышленности.
Чрезвычайно важно и широко применение гликола в каче-
стве радиаторной жидкости для автомобилей и самолетов. Бла-
годаря высокой температуре кипения и низкой температуре за-
стывания этиленгликол обладает ценнейшими преимуществами
перед обычной радиаторной жидкостью (водой), а также легко*
изменяющими свой состав водно-спиртовыми и соляными рас-
творами; последние, кроме того, в противовес нейтральному гли-
ко лу действуют разъедающим образом на металлические части
радиатора. • •
Из ближайших производных гликола, получивших залго-
следнее время промышленное значение, особенно важным,
является «нитрогликол», т. е. азотнокислый эфир этиленгли-
кола. Получаемый аналогично нитроглицерину, этот низший
530
«го аналог находит широкое применение в технике взрывчатых
веществ, особенно в качестве добавки к нитроглицерину для по-
нижения его температуры застывания.
Глико л является также полупродуктом при изготовлении
«ряда ценных растворителей, широко применяемых в некоторых
видах химической промышленности.
2. Окисъ этилена. Легко получается из хлоргидрина дей-
ствием щелочи:
сн2 • он сн2
СН Cl + Na0H = ХаС1 + **2° + /°
Окись этилена (температура кипения 12,5°), один из важ-
нейших продуктов переработки этилена и его хлоргидрина, при-
обретает за последнее время все большее значение в лаборатор-
ном и промышленном синтезе органических веществ. Так, на-
пример, при взаимодействии окиси этилена с водой под неболь-
шим давлением получается гликол; при этом в зависимости от
условий реакция может пойти также по линии взаимодействия
избытка окиси и гликола с образованием диэтилен- и триэтилен-
тликола; например:
СНЗХ Н УСН2 • СН20Н
>0+1 =о/
сн/ о сн2 • СН20Н хсн2. СНйОН
Диэтиленгликол находит применений в промышленности пла-
стических масс в качестве растворителя.
С этиловым спиртом окись этилена дает моноэтиловый эфир
гликола — один из наиболее ценных растворителей для той же
промышленности («Целлосольв») :
СЕ/ сн, — он
)ОН-СоНБОН= •
СН/ " СЩ —О-С2Н6
•Наконец, с крепким раствором аммиака окись этилена дает
-смесь моно-, ди- и триэтаноламинов:
СН2Х
NEL + 3 I )0 -+ КН/ЩСЩОН.. .N (СЩСЩОН^.
сн/
Смесь этаноламинов находит в настоящее время применение
зв различных отраслях промышленности (текстильная, кожевен-
ная и др.); в нефтяной промышленности смесь этаноламинов
предложена в качестве добавки (в сотых долях процента) к сма-
зочным маслам из нефтей парафинового основания для пониже-
ния температуры их застывания.
Кроме поименованных случаев, хлоргидрин этилена находит
. применение в большом числе лабораторных и промышленных
‘синтезов различных органических веществ, имеющих более или
*менее широкое применение. Таковы, например: новокаин, широко
Е31
известное средство для местной анестезии; иприт, важнейшее на
боевых отравляющих веществ, и многие другие.
Отметим в заключение, что, кроме хлоргидрина и окиси эти-
лена, немаловажное значение успели приобрести за последнее
время также соответствующие производные пропилена.
Полимерные бензины. Давно известно, что под влиянием
минеральных кислот, некоторых солей (ZnCl2, А1СЪ и т. и.)- и
других соединений (фтористый бор) этиленовые углеводороды
претерпевают полимеризацию с образованием полимеров различ-
ной степени сложности (димеры, тримеры, тетрамеры и т. д.). За
последние годы эта давно известная реакция была применена
к газам крэкинга и дала начало новому, многообещающему на-
правлению промышленности переработки нефтяных газов—по-
лучению полимерных бензинов.
Переработка газов крэкинга на полимерные бензины произ-
водится в присутствии катализатора; в качестве последнего ока-
зались наиболее подходящими фосфорная кислота на таких но-
сителях, как шамот, кизельгур, активированный уголь и т. п.
[73], а также разбавленная серная кислота [74]. Действие этих
катализаторов на непредельный углеводород сводится.в первую
очередь к образованию кислого эфира, который разлагается за-
тем с образованием полимера и выделением свободной кислоты;
па примере пропилена и фосфорной кислоты эта реакция может
быть выражена следующим образом:
/ОН /ОН УСН3
О : Pz0H-l~CH2 : СН • СН8= 0 : PZO —СН<
\ОН \0Н ХСН3
Н3СЧ /ОН Н0\ /СН8
>сн-о-р^о +о = р-о-сн =
H8CZ хон но/ \сн8
Процесс полимеризации газообразных олефинов под влия-
нием фосфорной кислоты протекает наиболее легко с бутиленами,
особенно с изобутиленом; пропилен полимеризуется значительно
труднее, наиболее же трудно протекает полимеризация этилена.
Со смесью олефинов, находящихся в газах крэкинга и пиролиза,
реакцию полимеризации удобно проводить, пуская газ через
стальную трубку с катализатором на носителе при температуре
230—250° и давлении 7—12 ат; полезная длина трубки. 60—
65 см; ее диаметр 2,5—4 см. Получаемые этим путем полимерные
бензины—-весьма высокого качества. До 150° они выкипают
в количестве 60—70%, до 200°—в количестве 80—90%. Их.
октановое число — 78—82. По составу они почти целиком со-
стоят из непредельных углеводородов, а стабилизация их лучше*
всего достигается путем легкого гидрирования (гидроочистка).
Полимеризация олефинов изучена настолько хорошо, что ее
532
применяют в настоящее время в промышленном масштабе не
только для получения полимерного бензина, но и для пригото-
вления из бутиленовых газов крекинга диизобутилена, который
путем гидрирования превращается далее в изооктал; последний
же, как будет показано ниже (ч. III, гл. III), представляет собою
наиболее ценную добавку к бензину для получения высоко-
октанового топлива. Ресурсы сырья для получения полимерных
бензинов громадны, и уже в настоящее время продукция поли-
мерных бензинов в США исчисляется сотнями тысяч тонн.
Термическая обработка газов крэкинга. Как - видно из
табл. 102, значительную часп» газов крэкинга составляют угле-
водороды ряда метана, не принимающие участия в образовании
полимерных бензинов. Для использования этих газов в данном
направлении они должны быть подвергнуты воздействию высокой
температуры, короче говоря, пиролизу, в процессе которого они
превращаются сначала в низшие непредельные углеводороды,
а затем — в жидкие продукты, богатые ароматикой. Так полу-
чают особый вид высокооктановых бензинов, называемых пиро-
бензинами.
Сырьем для получения пиробензина могут служить газы
жидкофазного и ларофазного крэкинга, а также естественный
газ подходящего состава,’особенно пропано-бутановая фракция
от стабилизации бензинов прямой гонки. Процесс осуществляется
при температурах от 700 до 900° и давлении от 1 до 7 ат. Уста-
новка состоит из трубчатого подогревателя, в котором газ подо-
гревается до 500°, и реакционного змеевика, в котором при тем-
пературах 700—900° происходит пиролиз сырья; далее следуют
холодильник, смолоотделитель, приемник и стабилизатор, т. е.
обычная аппаратура установки для пиролиза. Подогревателем
установки служит обычная крекинговая трубчатка: реакцион-
ная трубчатка изготовляется из высокосортной, нержавеющей
хромоникелевой стали. Для предотвращения изгиба труб реак-
ционной трубчатки их располагают вертикально и укрепляют
только сверху.
В зависимости от условий получения и сырья ппробензины полу-
чаются со следующими свойствами:
• Удельный вес.................... 0,796—-0,880
Начало кипения............. 39—70°
Конец кипения •............ 187—211°
Темп, плавления............от — 38 до — 70°
Анилиновая точка...........ниже 0°
Октановое число (в смесях) .84—104.
Процесс успешно развивается в США в заводском масштабе и в 1936 г.
дал свыше 300 000 т высокооктанового бензина.
Другое не менее важное направление пиролиза нефтяных га-
зов, а равно и жидких нефтепродуктов приводит к получению
бутадиена. (дивинилу) — исходного материала для получения
синтетического "каучука.
Бутя,диен (температура кипения —9°) образуется в процессе
разложения различных органических веществ, в частности —
538
нефти. Основные задачи при его получении заключаются в w
чтобы задержать процесс уплотнения образовавшегося бутадиена
.а затем выделить его возможно более полно из продукта реакдв£
Первая из этих задач решается быстрым выделением образовав-
шегося бутадиена в смеси с другими продуктами из сферы вы-
сокой температуры; для решения второй отделяют сначала ям.
кие, смолистые продукты пиролиза, газообразные же направляй
в абсорберы с подходящим поглотителем (маслом). Дальнейшая
задача сводится главным образом к проблеме отделения бута-
диена от других близких по свойствам продуктов реакции, ооо-
'бенно от бутиленов. Для этой цели можно пользоваться избира-
тельной растворимостью различных углеводородов в разных рас-
творителях и другими методами, детали которых, естественно,
составляют секрет производства. Совокупностью этих методов
удается довести выход бутадиена при пиролизе -нефти до ю%
(Вызов), при пиролизе же спирта — до 30% (Лебедев). Новейшие
исследования [75] показывают, что термической обработкой эти-
лена можно, невидимому, получить бутадиен с еще большймн
выходами (до 70%, считая, вероятно, на этилен, вошедший в ре-
.акцию). -Эта последняя реакция протекает по следующему урав-
нению: j л I
СН,: СЩ + СНо : СН2 = Н2 + СН2 : СН • СН : СНа.
Если результаты этих исследований подтвердятся и будут
освоены промышленностью, проблема технического получения
искусственного каучука на базе нефтяного сырья окажется окон-
чательно разрешенной.
Синтетические масла. В предшествующем изложении речь
шла о химической переработке почти исключительно газообраз-
ных продуктов крэкинга. В самое последнее время обращено вни-
мание и на другую часть этой обширной проблемы — химическую
переработку жидких углеводородов крэкинга.
Давно уже было известно [76], что при действии на непре-
дельные углеводороды жирного ряда хлористого или бромистого
.алюминия, а также таких катализаторов, как фтористый бор,
флоридин, фосфорная и серная кислоты и т. д., образуются вяз-
кие продукты уплотнения, имеющие характер минеральных сма-
зочных масел. Из всех этих катализаторов хлористый алюминий
дает, как оказалось, особенно благоприятные результаты, а лег-
кая доступность как исходного сырья в виде разного рода крэ-
'кинг-дестиллатов, так и катализатора (см. гл. IV) особенно стн-
мулироЬала исследования в этой многообещающей области [77].
Их основные задачи заключаются не только в том, чтобы прора-
ботать технологическую сторону процесса получения синтети-
ческих масел, что в основном оказалось весьма несложно, но
гтакже и в том-, чтобы путем систематических опытов'сопоставить
химический характер исходного сырья р физико-химическими
свойствами получающихся масел и, таким образом, наметить
наиболее благоприятные условия процесса.
<584
Опыты получения синтетических смазочных масел можно
производить в автоклаве с мешалкой, снабженной гильзой для
термометра, манометром и двумя отверстиями с пробками, из ко-
торых одно, в крышке, служит для загрузки, другое, в днище, —
для разгрузки автоклава. Для обогрева автоклав оборудован либо
рубашкой, либо внутренним паровым змеевиком. Самый процесс-
получения синтетических масел заключается в следующем.
В автоклав загружают ту или иную фракцию крэкинг-дестил-
лата, или раствор какого-либо индивидуального непредельного-
углеводорода в очищенном хлористым алюминием лигроине, до-
бавляют туда же 2—3% безводного хлористого алюминия и при-
ступают к нагреванию смеси при постоянном ее перемешивании..
Обогрев ведется до 60—70° и продолжается от 8 до 30 час., после
чего дают смеси отстояться. Затем нижний слой, содержащий
комплексное соединение хлористого алюминия с углеводородами,
отделяют, а верхний, маслянистый слой промывают раствором,
едкого натра, затем — водой и подвергают перегонке с перегретым
паром либо в вакууме, чтобы довести вязкость масла до тре-
буемых пределов.
Из нижнего слоя путем аналогичной обработки также может
быть получено немного синтетического масла; однако по своим
качествам оно всегда оказывается значительно ниже, чем масло,,
полученное из верхнего слоя.
Систематическое исследование процесса образования синтети-
ческих масел пока еще далеко не закончено. На сегодняшний
день, в основном, оно привело к следующим интересным и важ-
ным результатам.
Оказалось, что синтетические масла, изученные в отдельных-
случаях, обладают особо высокими качествами. Так, масла типа
автолов из продуктов крэкинга различного грозненского пара-
финистого сырья характеризуются прежде всего весьма низким
удельным весом (0,862—0,383), совершенно не обычным для ана-
логичных естественных масел. Они обладают низкой застывае-
мостью (ниже —20°) и весьма пологой кривой вязкости. Масла,
имеют хороший цвет, к тому же улучшающийся при стояниш
масла, и очень низкие коксовые числа {по Конрадсону). Окисле-
ние масла продувкой воздуха (150°, 24 часа) с Последующей
разбавлением петролейным эфиром не вызывает выпадения смо-
листого осадка,- так что синтетические масла ведут себя в этом
отношении как лучшие минеральные смазочные масла, например*
масла из пенсильванской нефти.
Большой интерес представляют данные, правда пока недоста-
точные, по получению синтетических масел из некоторых инди-
видуальных непредельных углеводородов. Данные эти показы-
вают, что под влиянием хлористого алюминия этилен и его гомо-
логи образуют синтетические масла, имеющие наиболее низкие
температурные индексы вязкости и по своим свойствам прибли-
жающиеся к маслам из парафинистого сырья. Напротив, масла,,
полученные аналогичным путем из нафтиленов и терпенов,,
имеют более высокие индексы вязкости и по своим качествам при-
535»
ближаются к маслам из нафтеновых нефтей. Химизм превращу,
ния того и другого сырья в синтетические масла сводится, надо
думать [78], не только к реакциям полимеризации под влиянием
хлористого алюминия, но также к процессам гидро- и дегидро
лимеризации, которые наблюдаются, например, при действии на
.этиленовые углеводороды крепкой серной кислоты, а именно:
часть непредельного углеводорода превращается в продукты
уплотнения, претерпевая при этом восстановление до угле-
водорода предельного характера за счет другой части исход-
ного сырья, которое подвергается превращению обратною
рода с образованием тех или иных продуктов конденсация
окисленного типа, в частности—смолистых и асфальтообразных
веществ.
Приводим для иллюстрации баланс переработки крэкинг-дестиллата от
крекинга отека с парафинового завода (Грозный), Содержавшего 33,2%
•масла. Температура кипения крэкинг-дестиллата 161—281,5°; его перера-
ботка на синтетические масла дала:
Моторное масло (-Е\оо = 2,8)........... 27,7%
Маловязкое масло = 1.75)................ 9,7%
Фракция 162— 301° ^лигроин — керосин) . . 50,0%
Масло из осадка........................ 5,7%
Потери.................................. 6,9%
100,0%
В приведенном опыте выход на моторное масло составил всего 27,7%,
•считая на сырье; однако в других условиях в связи с более удачным под-
бором сырья и условий опыта выхода на синтетическое масло достигали
40—50% и выше. Если принять, таким образом, во внимание простоту
аппаратуры и процесса получения синтетических масел, особенно отсут-
ствие необходимости в сернокислотной очистке масла и в его • обеспарафв-
нивании; если учесть, наконец, высокие качества получаемых масел, то
становится понятным, что процесс получения синтетических масел из кре-
кинг-дестиллатов так быстро вышел из стадии лабораторных опытов и вне-
дрился в промышленность, хотя целый ряд вопросов, с ним связанных,
еще’ продолжает оставаться не достаточно выясненным, как это было
отмечено выше.
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ КРЭКИНГА И ПИРОЛИЗА НЕФТИ
1. Исключительное значение крэкинга как системы перера-
ботки нефти определяется прежде всего получением новых гро-
мадных ресурсов бензина высоких качеств при все возрастающих
требованиях на этот продукт.
Чтобы оценить значение крэкинг-процесса с точки зрения
количественной стороны бензиновой проблемы, достаточно ука-
зать, что в настоящее время свыше половины всего количества
бензина, добываемого в США, получается путем крэкинга. Рас-
четы, произведенные за 1935 г., показывают, что благодаря пере-
работке на бензин более тяжелых масел путем их крэкинга
только за один этот год удалось сберечь свыше 153 000 000 т сырой
нефти, которую при отсутствии крэкинг-процесса пришлось бы
для удовлетворения потребности в легком топливе перерабаты-
536
вать на бензин прямой гонки. За все же время своего существо-
вания крэкинг-процесс сэкономил для США на 1 января 1938 г.
свыше 1,5 млрд, т нефти. Принимая во внимание выдающиеся
успехи крэкинг-процесса не только в отношении увеличения вы-
ходов на бензин и расширения ресурсов сырья, пригодного для
крэкирования, но и в смысле возрастания возможностей исполь-
зования различных продуктов крэкинга, можно смело утвер-
ждать, что удельное значение крэкинг-процесса в общей системе
переработки нефти продолжает неуклонно возрастать. Наиболее
убедительным доказательством этого положения являются но-
вейшие успехи в области получения полимеризационных бен-
зинов на базе крэкинг-процесса.
В табл. 122 приведены данные о производстве крэкинг-бензина-
и бензина прямой гонки за 1922—1935 гг.
Таблица 122
Производство бензина в США
fno Г. Эглофу)
Годы Получено бензина прямой гонки в млн. m Продукция крэкинг-бензина в
млн. m % ко всей бензиновой продукции % к бензину прямой гонки
1922 14,40 8,60 20,0 25,0
1925 20,10 8,25 29,1 41J0
1927 28,15 14,46 38,9 513
1929 34,98 20,53 86,8 58,7
1931 30,14 24,86 45,3 Я2,5
1935 24,40 29,79 54,9 121,1
Как видно из этой таблицы, производство крэкинг-бензина.
неуклонно растет как в абсолютных цифрах, так и по отношению
к бензину прямой гонки. Производство бензина прямой гонки,
напротив, за последние годы заметно падает и уже значительно
отстает от производства крэкинг-бензина.
2. Не менее важное значение имеет крэкинг-процесс с точки
зрения повышения качества бензина как топлива для двигателей
внутреннего сгорания. Мы уже видели (ч. I, стр. 109), что
в связи с постепенным переходом на двигатели с повышенной
степенью сжатия современная техника предъявляет к бензину
требования, которым многие бензины прямой гонки совершенно-
не удовлетворяют в отношении их антидетонационных свойств
и что свойства эти в высокой степени улучшаются путем разба-
вления бензинов прямой гонки бензинами крэкинга. Содержание
последних в современных товарных бензинах достигает 40—50%
и более, в корне изменив состав автомобильного топлива по-
сравнению с топливом ’недавнего прошлого, главным образом
в отношении содержания в нем непредельных и ароматики.
Вспомним далее, что с улучшением антидетонационных свойств
топлива при прочих равных условиях увеличивается сохран-
БЗТ
ность двигателя и одновременно значительно повышается его
мощность. Лишь сопоставив все эти обстоятельства, можно полу-
чить представление о том по-истине громадном технико-эконо-
мическом значении, которое приобрел в настоящее время кт.
кинг-процесс не только в нефтяном деле, но и в общей системе
народного хозяйства.
3. Большое значение в современной системе переработку
нефти занимает высокотемпературный крэкинг, т. е. пиролнз
Целевым продуктом пиролиза нефти, как мы видели, являются
ароматические углеводороды, особенно бензол, толуол, ксилол,
.значение которых как сырья для получения органических кра-
сок, фармацевтических препаратов, синтетических душистых и
взрывчатых веществ общеизвестно. Полученные путем пиролиза
нефти ароматические углеводороды выгодно отличаются от аро-
матики из каменноугольного дегтя полным отсутствием оерни-
стых соединений (тиофены); однако полная очистка их от непре-
дельных соединений, всегда сопровождающих ароматику пиро-
лиза, требует слишком больших количеств серной кислоты и по
этому мало выгодна. Значительно легче достигается стабилиза-
ция бензина пиролиза, основанная, как мы видели, на удаления
.лишь наиболее легко осмоляющихся компонентов. После такой
стабилизации бензин пиролиза становится прекрасным топливом
’для двигателей внутреннего сгорания, по своим антидетонациои-
ным качествам дайте превосходящим бензин крэкинга, что глав-
ным образом и определяет практическую ценность этого метода.
4. Выше уже было очерчено современное состояние вопроса
о химической переработке газов крэкинга. Чтобы судить о поло-
жении этого вопроса в промышленности, достаточно отметить, что
уже в настоящее время прейскурант одной из американских
•фирм, работающих в этой области (Carbide and Carbon Chemicals
Corp.), содержит свыше 25 названий разного рода органических
веществ, производящихся из газов крэкинга в крупном завод-
ском масштабе. Главное место среди них принадлежит таким
еще недавно мало доступным веществам, как спирты изопропило-
вый и нормальный бутиловый, этиленгликол и окись, этилена,
лропиленгликол и окись пропилена, ди- и триэтиленгликолы и
многие другие. Можно считать, таким образом, что нефтяная
промышленность имеет здесь твердую базу для дальнейшего раз-
вития проблемы получения из нефти высоких химических цен-
'ностей, и неудивительно, если даже среди нефтяников высказы-
вается мнение, что в ближайшем будущем газы крэкинга ока-
жутся в положении целевого продукта нефтяной промышлен-
ности как сырья для его дальнейшей химической переработка
Представляется также несомненным, что одним из ведущих на-
правлений этой переработки будет синтез высококачественного
моторного топлива путем полимеризации газов крэкинга и пиро-
лиза. , , ।
ЛИТЕРАТУРА
Добрянский А. Ф., Курс технологии нефти, М.-Л. (1931).
Стрижов И. Н., Американские нефтеперегонные заводы (1929).
538
Сахаме А. Н. и Тиличеев М. Д., Крэкинг в жидкой фазе, М.-Л. (1928).
Белл Г. С., Американские методы перереботки нефти. 2-е изд.. М.-Л. (1933).
Еострин К. В., Производство крэкинг-бензина, Азнефтиздат (1933).
Sedlaczek Е., Die Krackverfahren unter Anwfcndung von Druck, Berlin (1929).
Труды конференции по крэкингу и гидрогенизации в Грозном",
апрель (1931).
Ямгй Ch., The Pyrolysis of the Organic Compounds, New-York (1929).
Ewec В., О крэкинге пер. под ред. Тычинина, М.-Л. (1924).
Сабатье П. Катализ в органической химии, гл. XVII (1932).
Эьлофф Г., IHaad Р., Лоури ?., Болльман Г., Левинсон Б., Разложение и поли-
меризация углеводородов, пер. под ред. Зелинского Н. Д, ОНТИ
(1935).
Добрянский А. Ф., Научные основы крэкинга нефти, ОНТИ (1935).
Эллис Е., Химия углеводородов нефти и их производных, ОНТИ, т. I (1936),
т. II (1938).
1. Чиркин Б., „Аз. нефт. хоз." № 10, 67 (1930).
2. Вальгис В., „Аз. нефт. хоз.* № 10, 53 (1927); Богословский Ю. Е. и
Маргулов А. П., „Труды Н. Т. С. Нефт пром", вып. 1, Крэкинг (1929).
3. Еострин Е.. „Нефт. хоз.* 18, №10, 520 (1929); „Аз. нефт. хоз." № 2, 72
(1930).
4. Обрядчиков С- Н., „Нефт. хоз." 17, № 12, 14 (1936).
б. Morrell J. С., Nelson Е F., Egloff G., „Nat. Petr. News* 28, N. 45, 78.
(1936).
6. Седых H. Ф.» „Нефт. хоз.* 1», № 11—12, 677 (1928); № 5, 767 (1929).
7. Стрижов В. Н-, Американские нефтеперегонные заводы, Нефт. изд.-
стр. 9-320, (19-9).
8. Keith Р. С., Ward I. F., ВиЫп L. С., „Am. Petr. Ints. Proc.* 14, М. Ш
49 (май 1933).
9. Покровский А. Л., „Нефт. хоз.* 2, № 1—4, 63 и № 5—6,. 49 (1921);
Добрянский А. Ф., Пирогенетическое разложение нефти, П. (1922);
Еванов Г. Е., „Аз. нефт- хоз." № 12, 86 (1923); Добрянский А. Ф.
Научные основы крэкинга нефти, ОНТИ (198£); Бутков Н. А., Вве-
дение в технологию пиролиза, ОНТИ (1937).
10. „Ж. Р. X. О." 9, 269 (11-77); „Dinglers polyth. Journ.* 229, 653 (1878).
11. „Нефт. хоз.“ .28, № 8, 37 (1935).
12. См. ниже [67—72]; литература вопроса у Эллиса Е., Химия углеводо-
родов нефти и их производных, т. I и 11.
13. Потоловский Л., Атальян А., Буйницкая В., „Аз. нефт. хоз.* 14, № 10,.
91 (1934): 16, № 4, 73 (1936).
14. Герр В. Ф., „Нефт. хоз.* 9, № 9 343 (1928).
16. См. ниж. в гл. VI „Очистка гидрогенизацией или гидроочистка".
16. „Нефт. хоз." 10, № 9, 862 (1929).
17. Герр В. Ф. и Ульянов Г. П„ „нефт. хоз.* 8, № 6, 840 (1927).
18. Berthelot М., „Ann. Chim. phys.“ [4] 9, 445 (1866).
19. Thorpe u. Young, „Lieb. Ann." 165, 1 (1873).
20. Haber, „Вег." 29, 2691 (1896).
21. Leslie a. Potlhof, „Ind. Eng. Chem.* 18» 776 (1926).
22. Седых H. Ф., „Нефт. хоэ." 15, № 7, 67 (1984).
23. Бутков H. А., Рабинович, Еолпенская, „Нефт. хоз." 14, № 6, 29 (19 33).
24. Geniesse, Beuter, „Ind. Eng. Chem." 24, № 2, 219 (1982).
25. „Нефт. хоз * 10, № .2, 223 (1929).
26. Наметкин и Герасимов, „Ж. прикл. химии" И, 413 (1938).
27. Добрянский А. Ф., Научнее основы..., ОНТИ (1935).
28. Потоловский Л. А., Гутыря В. С., „Аз. нефт. хоз." 14, № 5 (149), W •
(1934). „ v
29. Groll, „Ind. Eng. Chem." 22, № 10, 1081 (1930).
30. Литературу вопроса см. Эглофф Г. и др., Разложение и полимеризация
углеьодородов, ОНТИ (1985).
80а. Gault, Hessel, „Ann. de Chimie" [10] 2, 319 (1924).
629 •
£1. Jamaguti, «Bull. Chem. Soc. Japan* 2, 289 (1927).
32. Stanley, Nash, ,011 and Gas J.“ 28, № 86, 125 (1930).
33. Dunstan, Hague, Wheeler, ,Ind. Eng. Chem." 26, № 3, 307 (1934).
34. Hague, Wheeler, ,J. Chem. Soc.*, 19*29, 378.
35. Pease, „J. Am. Chem, Soc." 50, 1779 (1928); Hurd, Spence, Ibid 51 йага
(1929). '
36. Zanetti, „Ind Eng. Chem.* 8, 674 (1916); Zanettz, Egloff, Leslie Ibid s
777 (1916); 9, 474 (1917); Dawidson, Ibid. 10, 901 (1918). ’ ’
37. Тиличеев, Фейгин, «Труды конференции по крэкингу..(1931).
38. Ср. Бутлеров А. Й., «Ж. Р. X, О.“ 5, 187 и 302 (1873); 8,279 и 351Л firm-
9, 38, (1877); 11, 197 (1879); 14, 199 (1882); Лебедев С. В., fox»?
ский, «Ж- Р. X. О.* 61, 2175 (1929) и многочисленные работы дру-
гих авторов.
39. Wheeler, Wood, .J. Chem. Soc.*, 1980, 1819.
40. Ср. Davidson, .Ind. Eng. Chem.* 10, 901 (1918); ср. также Staudin&r
Endle, Herold, .Bet.* 46, 2467 (1913).
41. Зелинский H. Д, .Ber.* 57, 2055 (1924); ср. также ’Петров 4 л
„Ж. P. X. 0.* 60, 1441 (1928). ‘
42. Ипатьев и Петров, ,Ber.“ 59, 2035 (1926); 60, 753 (1927).
43. Gault et Altchidj an, «Ann. de chim.* [10], 2, 209 (1924).
44. Bertelot, „Ann. de Chim.“ [4] 9, 445 (1866); см. также Schmidt u. Schults
„Lieb Ann.* 174» 230 (1874).
45. Zanetti and Egloff, ,Ind. Eng. Chem.* 9, 350 (1917).
46. Соханов A. H. и Тиличеев M. Д., Крэкинг в жидкой фазе, М.-Л. (19281
стр. 143.
47. Литература вопроса см. Добрянский А. Ф., Научные основы крэкинга
нефти.
48. Carpenter, «Journ. Inst. Petr Techn.* 14, 68, 460 (1928).
49. Brooks, ,Ind. Eng. Chem.* 18, 521 (1916).
50. Pyl, .Ber.* 60, 1138 (1927).
51. Еолосовский H. А., Химическая термодинамика, Л. (1932); Партшмпм
Дж, Р, Раковский А, В., Курс химической термодинамики, М.-Л
(1932); Francis A. W., ,Ind. Eng. Chem.* 20, 277 (1928).
52. Frolich, White и др., ,Ind. Eng. Chem." 22, 13 (1930).
53. Райс Ф. О. и Райс E. £., Свободные алифатические радикалы, пер.
под ред. Фроста А. В., ОНТИ (1937).
.54. Pauling, ,J. Am. Chem. Soc.“ 54, 8570 (1932).
55. Гапон, „Ж. P. X. О.* 62, 1388 (1930); Hurd, «Ind. Eng. Chem." 26, 50
(1934).
56. Belchetz L., .Trans. Far. Soc.“ 30, 170 (1934).
57. Обрядчиков, Тепловые вопросы крэкинга, «Труды конференции по крэ-
кингу и гидрогенизации,* Грозный (1931); его же, Физико-химиче-
ские основы расчета крэкинг-аппаратуры, ОНТИ (1934).
58. Обрядчиков и Великанов, „Нефт. хоз.“ 10, № 9, 370 (1929).
59. .Нефт. хоз.* 10, №2, 225 (1929); ср. также работы Тиличеева ъ Селеджн-
ева, Тиличеева п Александрова в „Трудах конференции по крэкингу
и гидрогенизации*, Грозный (1931).
'60. Обрядчиков С. Н. и Еарасик, ,Нефт. хоз.* 16, As 7, 17(1935).
61. Фейгин и Бушкова, .Нефт. хоз.* 14, As 10, 45 (1933); 15, № 1, 40 (1934);
Egloff G., „Oil and Gas. J.“ 80, № 3, 56 (1933).
62. Наметкин и Робинзон, .Нефт. хоз.* 14, № 3, 184; № 4, 230; № 5, 292
(1933).
63. Тиличеев и Масина, Сборник „Химический состав нефтей и нефтяных
продуктов*, М.-Л. (1981).
64. Ср. Лихушин Е„ «Азерб. нефт. хоз.* 6, № 10 (58), 45 (1926).
65. Stand. Met. of Testing Petroleum and its Products, 2 Ed. (1929).
66. Елигерман, «Аз. нефт. хоз.* 12, № 8—9, 59 (1932).
67. Потоловский Л. А. и Гутыря В. С-, «Аз. нефт. хоз.* 14, № 5 (149), 62
(1934).
68. «Труды заводской сессии Н. Т. С.“, вып. 1 (1930); Пипик О. Г, Искус-
ственный нефтяной газ, Азнефтеиздат (1932); Рамайя, Береж,
540
Файнгар, Пархоменко, Нефть как сырье для промышленности пла-
стических масс, ОНТИ (1933); Эллис К., Химия углеводородов...,
т. I и II.
<59. См. новейшую патентную литературу; также Brooks and Irwin Hum*
phrey, .Ind. Eng. Chem." 9, 750 (1917).
70. Пигулевский В. В. и Назаров С. А., .Нефт. ▼оз/ 12, № 3, 292 (1931):
Межебовская Е. .Аз. нефт. хоз.“ 18, № 6—7, 53 (1933); Пипик О. и
Межебовская Е., .As- нефт. хоз." 18, № 6—7, 64 (1933).
71- Гутыря В. и Далин М., Пипик О., Межебовская Е. и Полякова 3. и
др., Сборник, Исследование и химическая переработка природных
и искусственных нефтяных газов". Баку—Москва (1935).
72. Brooks, ,Chem. Met. Eng." 22, 631 (1920); Curine and Joung, .Chem. Met.
Eng." 25, 1091 (1921).
73. ,Ж. общей химии" 5, 321, 327, 335 (1936) и др.
74. McAllister, .Refiner" 16, N. 11, 493 (1937).
75. Schneider, V, Frolich K., „Ind. Eng. Chem." 28, 1405 (1931).
76. Engler C-t Routala O., .Ber." 48, 388 (1910); Braunly, .Nat. Petr. News"
9, 35 (1919) u др.
77. Hash A., Stenley H., Bonten A., ,J. Inst. Petr. TechnoL" 88, 30 (1930);
Sullivan, Voorhees, Hecley, Shankland, .Ind. Eng. Chem." 28, 604
(1931); Жердева Л. Г. .Нефт. хоз." 18, № 10, 236 (1932) и 15, № 4,
53 (1934); Петров Л. Д., Анцус, Пожилъцова, „Ж. прикл. химии*
5, 790 (1932); Гухман и Андреева, ,Аз. нефт. хоз." 14, № 7—8, 117
(1934).
78. Наметкин и Абакумовская, ,Ж. общей химии" 2 (64). 608’ (1932); Намет-
кин и Руденко, .Ж. общей химии" 7, 763 (1937).
ГЛАВА IV
СПЕЦИАЛЬНЫЕ ВИДЫ КРЭКИНГА
Кроме видов крэкинг-процесса, описанных в предшествующей
главе, известны и уже нашли промышленное применение некото-
рые виды перегонки нефти и ее дестиллатов с разложением, про.
текающие в особых специальных условиях. Их назначение—
либо возможное смягчение обычных условий крэкинг-процесса
и,' как следствие, упрощение соответствующей аппаратуры, либо
то или иное специальное видоизменение режима процесса с целью
получения особых продуктов, отличных от обычных продуктов
крэкинга. Таким образом намечаются три категории специаль-
ных видов крэкинга:
А. Крэкинг в присутствии катализаторов и в первую оче-
редь— в присутствии хлористого алюминия.
Б. Восстановительный крэкинг (гидрогенизация), иначе го-
воря, крэкинг в атмосфере водорода.
Б. Окислительный крэкинг, т. е. крэкинг в атмосфере кисло-
рода (воздуха).
Эти три специальных вида крэкинг-процесса подлежат рассмо-
трению в настоящей главе.
А. Крэкинг в присутствии катализаторов
Как известно, катализаторами называются вещества, кото-
рые, будучи введены в сферу той или иной реакции, либо уско-
ряют, либо замедляют ее течение. Катализаторы первого рода
называются положительными, катализаторы второго рода — отри-
цательными. В реакции деструктивного крэкинга катализаторы
до сих дор не получили широкого применения, хотя и было бы
крайне заманчиво несколько смягчить те жесткие условия тем-
пературы и давления, в которых обычно проводится этот процесс.
Исключение составляет, пожалуй, лишь одно вещество, приме-
нение которого в целях получения бензина из тяжелых'нефте-
продуктов уже проложило себе дорогу в промышленность. Речь
идет о безводном хлористом алюминии.
ХЛОРИСТЫЙ АЛЮМИНИЙ
Безводный хлористый алюминий представляет -собою бесцвет-
ное кристаллическое вещество состава А1С1з. Будучи нагрет
542
в запаянном сосуде, хлористый алюминий плавится при 194°,
в открытом же сосуде он уже при 183°, не плавясь, переходит
в парообразное состояние и возгоняется. Чрезвычайно гигроско-
пичен; водою разлагается на глинозем и хлористый водород.
При осторожном испарении его водных растворов (в вакууме)
получается кристаллический гидрат состава А1С1з • 6HtO, кото-
рый при нагревании распадается на глинозем, хлористый водо-
род и ’воду. Безводный хлористый алюминий образует много-
- численные комплексные соединения и широко применяется
.в органическом синтезе, особенно при многочисленных реакциях
конденсации. Получается в лабораторных условиях нагреванием
алюминиевых стружек в струе сухого хлороводорода, в техниче-
-ском же масштабе — действием хлора на смесь боксита и кокса
при 900°:
А1Д + ЗС -f- ЗС12 = ЗСО + 2А1С13.
Технический хлористый алюминий всегда более или менее
-окрашен в желтый цвет от примеси хлорного железа. Благодаря
многочисленным патентам за последние 10—15 лет его производ-
ство было весьма усовершенствовано [1], а резкое снижение его
стоимости открыло для него широкую возможность применения,
в частности в нефтяной промышленности.
КРЭКИНГ В ПРИСУТСТВИИ ХЛОРИСТОГО АЛЮМИНИЯ
Давно уже были сделаны наблюдения, что при нагревании
с хлористым алюминием нефть и ее тяжелые части дают газо-
образные и легкие углеводороды бензинового типа. Техника такой
«бензинизации» нефти весьма проста: обезвоженную нефть или
ее погоны нагревают с 5—10% безводного хлористого алюминия
до 2601—280°. Сначала при этом образуется тяжелый маслени-
стый слой, нерастворимый в нефтепродукте; затем начинается'
' отгонка легкого дестиллата, который после промывки щелочью
получается совершенно прозрачным, бесцветным и стабильным.
В зависимости от сырья выхода на такой бензин бывают раз-
личны; в лучших случаях они достигают 70% и выше.
Первые опыты [2] разложения тяжелых углеводородов нефти под
влиянием хлористого алюминия были сделаны русским химиком Густавсе-
ном (1881 г.). Много позднее этим вопросом занимался Зелинский [з].
исследования которого привели к его проработке у нас в полузаводском
масштабе. В США особенно много работал над этим вопросом Мак-Аффи
(с 1915 г.), которому удалось осуществить способ получения бензина из
тяжёлых нефтей и нефтепродуктов с помощью хлористого алюминия
н крупном промышленном масштабе [4].
Химизм процесса. Образование легких углеводородов из тя-
желых под влиянием хлористого алюминия представляет собою
типичный кракинг-процесс. Для суждения о его химизме боль-
шое. значение должен иметь прежде всего вопрос о составе полу-
чающегося таким образом крэкинг-бензина. Характерная особен-
ность состава этого бензина заключается в том, что в отличие
543
от обыкновенного крэкинг-бензина бензин, получаемый, реакцией
хлористого алюминия на нефть и ее продукты, не реагирует ни
с бромом, ни с раствором марганцевокислого калия и, таким
образом, совершенно не содержит непредельных углеводородов-
насколько можно судить по имеющимся пока далеко неполным
данным, составными частями этого бензина являются низшие
парафины,- нафтены и ароматика.
Образование низших представителей ‘различных углеводо-
родных рядов при действии хлористого алюминия на более тя-
желые нефтяные масла можно рассматривать как результат дро-
бящего действия этого реагента на более тяжелые углеводород-
ные молекулы. Насколько глубоко может итти такое расщепле-
ние показывают опыты с нефтяным гептанафтеном: оказалось
что главным продуктом крэкинга является в данном случае
изобутан С4Н10. Таким образом действие хлористого алюминия
приводит здесь к полному расщеплению исходного нафтена с об-
разованием одного из простейших представителей ряда метана.
Как уже было отмечено выше, при взаимодействии галоидных
солей алюминия с нефтяными продуктами всегда происходит
также образование какого-то тяжелого масла. Выделение этого
масла значительно облегчается, если в смесь углеводорода с хло-
ристым или бромистым алюминием пропускать ток сухого га-
лоидоводорода. Ближайшее исследование показало [2, 5], что
этот продукт представляет собою комплексное соединение галоид-
ного алюминия с углеводородным остатком. Так например, по-
видимому, все предельные углеводороды, за исключением метана
и его ближайших гомологов, при взаимодействии с бромистым
алюминием в указанных условиях образуют комплекс состава
AlBra. С4Нв. При разложении этого комплекса водой образуются
масла с ясно выраженным непредельным характером; судя до
их температуре кипения, они представляют собою продукты
уплотнения каких-то более простых, но мало устойчивых в дан-
ных условиях углеводородных молекул или остатков.
Простые стехиометрические соотношения требуют, чтобы при
крэкинге всякого предельного углеводорода происходило образо-
вание по крайней мере двух более простых молекул: одна из них
может представлять собою предельный углеводород, другая же
обязательно должна быть непредельного состава. Казалось бы,
что при крэкинге с хлористым алюминием распад высокомоле-
кулярных углеводородов идет как раз по этой простой схеме:
продукты крэкинга предельного характера выделяются в свобод-
ном состоянии в виде крэкинг-бензина, непредельные же угле-
водороды образуют с галоидным алюминием комплексное соеди-
нение отмеченного выше состава А1Вгз • С4Н8. Нетрудно показать,
однако, что в действительности крэкинг в присутствии хло-
ристого'алюминия протекает гораздо сложнее.
Действительно, как показывает опыт, процесс бензинизации
тяжелых нефтяных продуктов успешно протекает уже в присут-
ствии сравнительно небольших количеств хлористого алюминия
(5—10%). Совершенно очевидно, что такого количества соли не
544 *
Может Хватить йа связывание всей массы непредельных угле-
водородов, которые должны образоваться в процессе крэкинга
с хлористым алюминием согласно вышеизложенному, особенно
если принять во внимание высокие выхода на бензин при этом
процессе. Таким образом естественно притти к выводу, что, кроме
образования комплекса с галоидными солями алюминия, а быть
может, именно через этот комплекс, непредельные углеводороды
крэкинга претерпевают в данной случае какие-то дополнитель-
ные превращения. О характере этих превращений можно
строить пока лишь предположения. Возможно, например, что
превращения эти представляют собою процессы того же порядка,
как при высокотемпературном крэкинге этиленовых углеводоро-
дов (гл. III). Нахождение водорода в газах, выделяющихся при
крэкинге с хлористым алюминием [5], а также новообразование
ароматических углеводородов при ‘этом процессе, наблюдавшееся
некоторыми авторами, говорит как будто в пользу такого пред-
положения; однако вопрос требует еще дальнейшей эксперимен-
тальной проработки.
Не менее сложную картину рисует ближайшее исследование
продуктов, получаемых в результате крэкинга с хлористым алю-
минием чистых углеводородов различных рядов. Характер этих
продуктов свидетельствует, что реакции, протекающие при крэ-
кинге этого рода, весьма разнообразны, и здесь, кроме реакций
расщепления, вне всякого сомнения имеют место .также про-
цессы и реакции других типов, как-то: процессы изомеризации,
циклизации, уплотнения (конденсации) и др. [5]. Так например:
нормальный гексан, СвНн, при кипячении с хлористым алюми-
нием превратился в нормальный пентан, С5Н12, изогексан, СвНн,
(2- или 3-метилпентан) и циклогексан, CeHi2; нормальный нонан,
С9Н20, при нагревании с хлористым алюминием до 100—120°
дал нормальный бутан, СЛю, и амилен, СбНю, изомеризовав-
шийся в циклопентан, С5Н10.
Из высших парафинов в том же направлении изучены их
смеси, содержащиеся в грозненском парафине и сураханском це-
резине (ч. I, гл. V В). Опыты были проведены с продуктами хо-
рошо очищенными и с продуктами, специально обезмасленными
путем четырехкратной перекристаллизации из дихлорэтана.
Бензины, полученные из этих продуктов путем их крэкинга
с хлористым алюминием, выкипали до 210—-240° и обладали
предельным характером (содержание непредельных равно нулю);
их групповой состав по фракциям показан в табл. 123 [6].
Данные эти показывают, что основными продуктами крэкинга
с хлористым алюминием нефтяного ‘ парафина и церезина
являются углеводороды ряда метана (парафины), составляющие
более 99% некоторых бензиновых фракций продуктов этой реак-
ции. Ни непредельные, ни нафтены практически не образуются
из хорошо очищенных и обезмасленных парафина и церезина
при их крэкинге -с хлористым алюминием. Что касается арома-
тики, то лишь небольшие количества 'углеводородов этого ряда
содержатся в низших фракциях бензина этого рода (темпера-
545
35 Зак. 1622. Химия нефти.
Таблица
Состав бензинов крэкинга с А1С13 из парафина и церезина
Исходное сырье । Темп. кип. фракций Фр. состав бензина из сырья очищенного Фр. состав бензина из сырья обезмасленного
аромат. нафт. параф. аромат. нафт. параф
Парафин 60— 95° 0,5 0 99,5 0,4 0 99,6
95—122° 2.9 0 97,1 2,2 0 97,8
122—150° 7,8 0 92 2 7,0 0 93,0
150—200° 26,3 0 73,7 — —
Церезин 60- 95° 2,1 0 97,9 0,7 0 99,3
95-122° 4,3 1,5 94,2 2,4 1,2 96,4
122—150° 10,4 4,1 85,5 8,5 0 91,5
150-200° 20,9 0,7 78,4 16,3 0 83,7
тура кипения 65—95° и 95—122°); однако в высших погонах
того же бензина содержание ароматики резко возрастает, доходя
во-фракциях с температурой кипения 150—200° до 16—2-6%.
Характерно, что содержание ароматпки особенно значительно для тех
случаев, когда крэкируемое сырье было очищаю, но не обезмаслено, и
что бензины, полученные путем крэкинга церезина, содержат значительно
больше ароматики, чем бензинЪт, полученные путем крэкинга парафина
Вполне возможно поэтому, что ароматика образуется здесь не за счет крэ-
кинга углеводородов ряда метана, составляющих главную массу
нефтяного парафина и церезина, а за счет той маслянистой примеси,
полное освобождение от которой нефтяного парафина и особенно церезина
представляет значительные трудности. /Такое заключение становится
особенно ясным, если содержание ароматики во фракциях, приведенное
в табл. 123, перечислить на цельный, неразогнанный, бензин; как показы-
вают данные табл. 124, содержание ароматики выражается в таком слу-
чае для большинства фракций бензина, полученного путем крэкинга с хло-
ристым алюминием нефтяного парафина и церезина, всего лишь десятыми
долями процента.
Таблица Ш
Содержание ароматики и фракций в бензине крэкинга парафина и
церезина с А1С13
Сырье Температура кипения фракции Содержание в бензине
60-95° 95—122° 122-150° 150-200°
Парафин ( ОД 0.3 0,6 1,6 ароматики
очищенный 1 20,0 11,5 7,1 6,1 фракций
Парафин | 0,1 0,2 0,3 — ароматики
обезмасленный 1 15,6 10,4 4.8 фракций
Церезин J 0,5 0,6 1.4 3,8 ароматики
очищенный | 25,2 13,7 13,1 18,2 фракций
Церезин f 0,2 ’ 0,3 0,9 1,6 ароматики
обезмасленный [ 32,8 10,6 10,0 9,7 фракций
546
Углеводороды этиленового ряда претерпевают под влиянием
хлористого алюминия, полимеризацию и конденсацию с образо-
ванием более сложных углеводородных систем. (Эти реакции
легли в самое последнее время в основу нового производства,
получения* синтетических масел (см. гл. III), химизм которого
сводится в основном, как было указано выше, к реакциям по-
лимеризации, гидрополимеризации и дегидрополимеризации не-
предельных углеводородов исходного сырья. Однако при этом
всегда наблюдается также образование легких продуктов с тем-
пературой кипения более низкой, чем температура кипения
исходных углеводородов; это обстоятельство свидетельствует,
что одновременно с реакциями уплотнения (полимеризации) здесь
имеют место также процессы обратного типа, т. е. реакции распада
или деструкции, иначе говоря, крэкинга исходных угле-
водородов. А так как эти процессы уплотнения, с одной стороны,
и крэкинга (деструкции) —с другой — протекают одновременно,
то при действии хлористого алюминия на углеводороды этилено-
вого ряда можно наблюдать образование продуктов реакций
совершенно особого рода, которые могут быть названы реакциями
деструктивной полимеризации и гидрополимеризации. По
своему составу продукты деструктивной полимеризации и гидро-
полимеризации характеризуются той особенностью по сравнению
с продуктами полимеризации и гидронолимеризации обычного
типа, что число углеродных атомов в частицах, образующихся
при этих своеобразных реакциях, не является кратным, числу
углеродов в молекуле исходного олефина. Так например, при дей-
ствии хлористого алюминия на амилен, С5Н10, наблюдается обра-
зование не только таких полимеров как СюН2о (димер), С10Н22
(гидродимер), С15Н32 (гидротример) и т. д., но и таких продуктов
деструктивной гидрополимеризации как С9Н20, СмНгв и т. д. [67].
Ароматические углеводороды при длительном нагревании
с хлористым алюминием изменяются в двух основных направле-
ниях: происходят перескок коротких боковых цепей с одной
молекулы на другую и образование высокомолекулярных- про-
дуктов уплотнения. Так например, толуол при действии хлори-
стого алюминия дал бензол, ксилол, триметилбензол и смесь,
продуктов конденсации; ксилол частично перешел в аналогичных
условиях в толуол и триметилбензол и т. д.
Метилированные нафтены ряда циклогексана при нагревании
с хлористым алюминием частью давали продукты глубокого рас-
пада исходной молекулы — газообразные парафины (ср. выше),
частью изомеризовались, например орто- и пара- диметилцикло-
гексаны переходили в мета-соединение. Гомологи циклогексана
с более длинными цепями (С2Н5, С3Н7 и С5Н11) претерпевали
превращения более сложного типа: за счет отщепления боковых
групп здесь частью происходило образование низших парафинов
и циклогексана, который превращался под влиянием хлористрго
алюминия в высокомолекулярные продукты полимеризации;
частью же наблюдалось образование метилированных гомоло-
гов циклогексана, очевидно за счет отщепления метильных групп
55* 547
от исходных углеводородов с последующим их присоединением
в новом положении. Так например, диэтилциклогексан превра-
щался в этих условиях главным образом в тетраметилциклогек-
сан, бутан и продукты уплотнения циклогексана и т. п.
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ И ПЕРСПЕКТИВЫ ПРОЦЕССА
Несмотря на несложность аппаратуры, прекрасные выхода на
бензин и простоту его очистки, крэкинг-процесс в присутствии
хлористого алюминия до сих пор не получил широкого промыш-
ленного применения. Причина этого обстоятельства, помимо
сравнительно высокой стоимости хлористого алюминия, заклю-
чается в ряде его отрицательных сторон, которые выявляются
лишь при ближайшем знакомстве с техникой этого процесса.
Важнейшие из них следующие:
1. Процесс протекает сравнительно медленно, и проведение
его в большом масштабе требует непрерывного механического пе-
ремешивания.
2. До сих пор при крэкинге с хлористым алюминием не уда-
лось наладить аппаратуры непрерывного действия, так что про-
цесс приходится проводить в аппаратуре, действующей периоди-
чески.
3. Во время хода процесса наблюдается возгонка хлористого
алюминия, что засоряет аппаратуру.
4. При процессе получаются слишком большие выхода кокса
(до 20%)-и малоценных газов (до 10% низших гомологов метана).
5. В отношении антидетонационных свойств получаемый бен-
зин также оставляет желать многого: его октановое число—
всего около 60.
6. Наконец, техническое оформление процесса требует гро-
моздкого и емкого транспорта для перевозки на заводе сухих про-
дуктов в общей сумме до 40% от загружаемого сырья (кокс,
хлористый алюминий и пр.).
Некоторые из перечисленных недостатков крэкинга в присут-
ствии хлористого алюминия в процессе дальнейшей его прора-
ботки, (возможно, будут устранены. Имеются, например, указа-
ния, что при надлежащем подборе сырья удается путем крэкинга
с хлористым алюминием получить бензин с хорошим октановым
числом. Надо думать, что такой же результат может быть до-
стигнут другим способом, а именно путем реформинга (гл. III)
низкооктанового бензина, который обычно получается при крэ-
кинге с хлористым алюминием.
Для полноты освещения вопроса о крэкинге в присутствии
катализаторов необходимо отметить, что влияние, аналогичное
хлористому алюминию, оказывает на крэкинг-процесс также ряд
других веществ, как-то: хлорное железо, хлористый цинк и т. д.
Однако практического применения эти вещества до сих пор не
получили.
Б. Восстановительный крекинг (гидрогенизация)
Как было показано в гл. III, одним из постоянных спутников
крэкинг-процесса является коксообразование. Появление кокса
548
представляет собою следствие того обстоятельства, что газы и
другие легкие продукты крэкинга '(крэкинг-бензин) в процентном
отношении значительно богаче водородом, чем другие более
тяжелые продукты этого процесса. Ввиду этого, чем дальше идет
крэкинг, т. е. чем больше образуется газов и крэкинг-бензина,
тем меньше водорода будет сохраняться на долю остатка, который
при достаточной глубине процесса может нацело превратиться
в кокс. Чтобы избавиться от этого наименее желательного про-
дукта крэкинга, естественно было восполнить указанный недо-
статок водорода путем его искусственного ввода в среду реакции
крэкинга. В этом и заключается сущность процесса гидрогени-
зации в применении к проблеме получения легкого топлива из
тяжелых нефтей, дегтей и даже угля путем их гидрирования.
Гидрирование органических соединений действием молекулярного водо-
рода в присутствии катализаторов известно еще со времени Фарадея, полу-
чившего этан действием на этилен водорода в присутствии губчатой платины
(1844). Однако лишь в XX столетии, благодаря работам Сабатье
с сотрудниками (Саидеренс, Мэйл, Мюра и др.), Фокина, Зелинского, Вилъ-
штеттера, Пааля, Скита и многочисленных других исследователей, гидриро-
вание как метод получило широкое применение не только в лабораторной
практике, но и в промышленности [8].
ТЕХНИКА ГИДРИРОВАНИЯ
Молекулярный водород представляет собою вещество мало
активное. Самые разнообразные соединения не только парафи-
нового или ароматического ряда, но даже вещества с ясно выра-
женным непредельным характером могут оставаться -в соприкос-
новении с водородом неограниченное время без заметного взаи-
модействия. Картина резко меняется в присутствии катализа-
тора: в одних случаях при надлежащем подогреве, в других —
даже при обыкновенной температуре легко наступает гидрирова-
ние, чаще всего простое присоединение водорода, протекающее
нередко количественно. Методика гидрирования органических
соединенней чрезвычайно разнообразна.
По Сабатъе и Сандеренсу [8, 9] пары гидрируемого вещества
в смеси с чистым водородом пропускают через трубку, заполнен-
ную металлом-катализатором. В качестве катализаторов приме-
няются платиновая чернь или восстановленные из соответствую-
щих окислов никеть, кобальт, железо и медь, причем восстал ов-
.ление их производится в самой реакционной трубке; наибольшее
применение из этих металлов получил никель. Для каждого
рода гидрирования существует некоторый оптимальный интервал
температуры. Нижняя граница этого интервала определяется
температурой, при которой как исходный материал, так и про-
дукт реакции находятся в парообразном состоянии; впрочем, это
последнее условие, как показывает опыт, отнюдь не является
обязательным. Верхняя граница температурного интервала
связана с обратимостью реакции гидрирования, которая при вы-
сокой температуре может пойти в обратном направлении, т. е.
в сторону дегидрирования (дегидрогенизации). Типичными при-
549
мерами гидрирования по Сабатье может служить гидрирование
непредельных и ароматических углеводородов и их производных,
легко протекающее при 150—200° в присутствии порошкообраз-
ного никеля-катализатора .
По Фокину [101 и Вимитеттеру [111 гидрирование орга-
нических соединений можно производить в присутствии 1—^2%
платиновой черни даже при комнатной температуре. Реакцию
ведут в спиртовом или уксусном растворе при энергичном встря-
хивании реакционного сосуда и постоянном подводе водорода
по мере его поглощения.
Как показали Паалъ [12], а позднее Скита [13], гидрирова-
ние успешно протекает также в присутствии коллоидных пла-
jl тпны и палладия. В качестве защитных кол-
«-tiO--..4=uxZ7\ лондов, повышающих стойкость коллоидных
, растворов этих металлов, были предложены
1 у различные вещества: наиболее доступным из
I них является гуммиарабик. И в этом случае
Ц реакцию ведут в подходящем растворе при
S ''Iff энергичном встряхивании с непрерывной по-
дачей водорода.
Роль давления при реакции гидрирования
оценена давно [4]. Реакцию вели сначала
в стальных бомбах (фиг. 109), поднимая дав-
ление до 200 ат, а температуру до 400°.
В качестве катализатора при гидрировании
обыкновенно пользуются продажной закисью
или окисью никеля. Водород также может’
J применяться продажный, непосредственно из
бомбы, без предварительной очистки. В этих
Фиг. юэ. Бомба условиях гидрирование протекает, очевидно,
для гидрирования. в фазе, притом нередко с боль-
шой скоростью, и часто дает количественные выхода на конеч-
ный продукт.
В своем первоначальном виде метод гидрирования мог найти
применение лишь в лабораторных исследованиях. Позднее,
в связи с успехами химического машиностроения в вопросах кон-
струкции и постройки крупных аппаратов высокого давления,
метод гидрирования при высоких давлениях и температурах на-
шел применение в различных видах химической промышленно-
сти. Чтобы оценить его значение, достаточно вспомнить промыш-
ленный синтез аммиака из азота воздуха и синтез метилового
спирта (метанола) из (водяного газа. Громадное значение при-
обрело также промышленное гидрирование некоторых органиче-
ских веществ, которое, как показал опыт, можно успешно про-
изводить при сравнительно невысоком давлении (около 2 ат);
таковы, например, гидрирование («отверждение») жиров, гидри-
рование нафталина и т. д.
В новейшее время техника работы под высокими давлениями
сделала громадные успехи, благодаря которым оказалось возмож-
ным осуществить синтез аммиака при давлении до 4500 ат.
550
В этих условиях образование аммиака из смеси азота с водоро-
дом протекало количественно без всякого катализатора; при
2000 ат и прочих равных условиях выхода при этой реакции па-
дали до 40%. В какой степени освоение такого рода сверхвысо-
ких давлений отразится на методике гидрирования органических
соединений, покажут дальнейшие исследования.
ТЕОРИЯ ГИДРИРОВАНИЯ
Гидрирование в присутствии мелко раздробленных металлов
представляет собою частный случай каталитических реакций,
протекающих в гетерогенной среде («гетерогенный катализ»).
Хотя некоторые примеры подобного рода реакций известны уже
много десятков лет, тем не менее, их механизм до сих нор не
может считаться вполне выясненным. Основной, крайне слож-
ный вопрос заключается здесь в той роли, которую играют в этих
реакциях металлические катализаторы.
По мнению одних авторов каталитическое действие металлов
при реакциях гидрирования зависит от адсорбции водорода, а
также гидрогенизируемого вещества на поверхности катализа-
тора. Происходящие при этом местное разогревание и активиро-
вание водЪрода являются основной причиной возникновения
реакции, которая в отсутствии катализатора могла бы пойти
лишь при значительно более высокой температуре. То обстоя-
тельство, что для приготовления активного катализатора прихо-
дится прибегать к специальным приемам повышения его адсорб-
ционных свойств, свидетельствует, как будто, в пользу подобного
рода представлений. Хотя с этой точки зрения некоторые сто-
роны вопроса (например, специфический характер действия раз-
личных катализаторов на одно и то же вещество и т. и.) остаются
непонятными, тем не менее, не подлежит сомнению, что явлениям
адсорбции должно быть отведено весьма важное место в реакциях
гетерогенного катализа.
Другое объяснение каталитических реакций заключается
в допущении образования некоторых промежуточных, мало-
устойчивых и чрезвычайно реакционно-способных соединений
катализатора с одним из взаимодействующих веществ. В приме-
нении к реакциям гидрирования этими промежуточными веще-
ствами являются образующиеся на поверхности катализатора
продукты присоединения водорода к металлу, т. е. так называе-
мые гидриды. Примерами таких гидридов могут служить давно
известный водородистый палладий, а также водородистые пла-
тина и никель. Все эти гидриды — крайне малоустойчивы;
благодаря этому они чрезвычайно легко отдают свой водород
другим соединениям, т. е. энергично гидрируют их, являясь,
таким образом, передатчиками водорода гидрируемому веществу.
С другой стороны, способность соединяться с водородом может
проявляться у некоторых металлов настолько сильно, что они
оказываются способными отнимать водород даже от некоторых
сложных веществ, т. е. действовать на них дегидрирующим обра-
.551
Зом. Так, с точки зрения теорий промежуточных соединений
объясняются явления гидрирования и дегидрирования в присут-
ствии мелко раздробленных металлических катализаторов.
Как ясно из вышеизложенного, первые стадии гидрирования
в присутствии металлических катализаторов рассматриваются
одними авторами как процесс чисто физический, другими — как
явление химического ' порядка. Между этими крайними точ-
ками зрения за последнее время наметилось как бы некоторое
примирение. Новые представления, кроме общей или физической
адсорбции, представляющей собою чисто физическое явление
сгущения адсорбируемого вещества на поверхности адсорбера,
допускают также существование особой химической адсорбции,
действующей лишь в пограничной области фаз и являющейся
основой гетерогенного катализа. Несомненно, что основную
роль при этом играет соприкосновение реагирующих атомов
с катализатором, как на это указывают твердо установленные
соотношения между формой реагирующих молекул и строением
кристаллической решетки катализатора. Таким образом можно
принять, что границы между физической и химической теориями
адсорбции согласно новейшим представлениям постепенно сгла-
живаются [15].
ГИДРИРОВАНИЕ ТОПЛИВА
Гидрирование топлива, или, обозначая этот процесс услов-
ным термином «гидрогенизация», точнее — «деструктивная гид-
рогенизация», как было указано выше, имеет своей конечной
целью получение легкого топлива из тяжелых • нефтей, разного
рода дегтей, а также из угля и продуктов его переработки *. Тех-
ника этого процесса напоминает гидрирование в бомбе, отли-
чаясь от этого метода лишь более рационально построенной
аппаратурой [16].
Техника гидрогенизации. В лабораторной обстановке гидроге-
низация ведется в специальных автоклавах высокого давления
различной конструкции, а именно: в автоклавах с мешалкой
(фиг. 110) или вращающихся около горизонтальной оси
(фиг. 111). Оба аппарата приспособлены к проведению реакции
при температурах до 450—500° и давлениях до 300 ат. Подача
в них водорода осуществляется либо непосредственно из бомбы,
либо с помощью компрессора. Подогрев производится или газом,
или при помощи электричества.
Техника гидрирования жидкого топлива в автоклаве заклю-
чается в общих чертах в следующем. В тщательно вычищенный
от предыдущих опытов автоклав загружаются требуемое количе-
ство сырья и, если нужно, тот или иной катализатор. Загрузка
берется с таким расчетом, чтобы другого компонента реакции,
водорода, не только хватило на полную гидрогенизацию сырья,
1 Некоторые авторы называют этот процесс «бергинизацией» по имени
Бергиуса, главного инициатора работ в этой области.
552
НО чтобы вместе с тем его парциальное давление изменялось
во время опыта по возможности в узких пределах. После сборки
автоклава и заполнения его водородом до требуемого давления
аппарат выдерживается в течение 20—30 мин, для проверки
герметичности; затем начинается его обогрев, причем за темпе-
ратурой следят по особому термометру, опущенному в специаль-
ный карман. Примерно за 50 до заданной температуры вклю-
чается мотор для вращения либо мешалки, либо всего автоклава:
одновременно через каждые 3 мин.
начинают запись давления (по
манометру)- и температуры, регули-
руя нагревание таким образом, что-
бы температура держалась в преде-
лах 2—Зь около заданной. По исте-
чении назначенного на обогрев
времени нагревание прекращают и
по возможности быстро снижают
температуру автоклава, а на сле-
дующий день отмечают остаточное
давление и, выпустив избыток газа,
вскрывают автоклав. Остается со-
брать его содержимое, отделить его
от катализатора и подвергнуть ис-
следованию на состав.
Работа на автоклавах обычного
типа носит периодический харак-
тер. Одним из основных вопросов
проведения гидрогенизации в про-
мышленность явилось построение
непрерывно действующей аппара-
туры высокого давления. В настоя-
щее время такая аппаратура уже
получила известное распростране-
ние в работах по гидрированию как
в лабораторном, так и в промышлен-
ном масштабе. Ее основные элементы
были разработаны в таких отраслях
промышленности, как отверждение
жиров, промышленный синтез ам-
миака и др. Примером может слу- ,
жить аппарат для непрерывного гидрирования ИГИ Академии
паук С-ССР, описанный недавно в специальной литературе 17 ;
схема его изображена на фиг. 112.
Исходный продукт из градуированного резервуара 1 жидкостным на-
сосом 2 закачивается в подогреватель 3 емкостью около 5 л, являющийся
одновременно испарителем продукта. Сюда же поступает циркулирующий
водород из циркуляционного насоса 9 и маслоотделителя 10. Подогреватель
снабжен внутри длинным карманом, позволяющим измерять т мпературу
по всей его длине (для выпуска неиспарившегося продукта внизу подо-
гревателя имеется спускной вентиль).
Испарившийся продукт вместе с водородом из подогревателя через
558
Флг. ПО. Автоклав высокого
давления:
а —• выпускной вентиль; Ь — холо-
дильник высокого давления; с — вен-
тиль длж впуска или выпуска rasa;
d — монометр; в — масленка; f— шкив;
д — карман для термометра; А —ме-
шалка; » — газовая горелка; к — ста-
кан а'токлава.
верхний штуцер поступает в нижнюю часть реакционной камеры 4
емкостью 4,8 л, наполненной до верху (4,7 л) катализатором. Обогрев
реактора производится электропечью. Температура в реакторе измеряется
в двух точках: 1) снизу на расстоянии 15 см от днища и сверху на рас-
Фиг. 111. Вращающийся автоклав.
стоянии 12 см от верха- Реактор имеет спускное отверстие для выпуска
жидких продуктов в приемник 13.
Из реакционной камеры газообразные продукты реакции направляются
в змеевиковый холодильник высокого давления 5. Охлажденные продукты
поступают в приемник высокого давления б>, из которого жидкие продукты
Фиг. 112. Схема опытной установки для непрерывного
гидрирования.
перепускаются при помощи редукционного вентиля в стеклянный градуи-
рованный приемник 11, находящийся под. атмосферным давлением. Выде-
ляющийся при этом газ (прессдестиллатный) измеряется газовыми ча-
сами 12. Несконденсировавшийся в приемнике 6 газ направляется из него
в сепаратор 7, служащий для улавливания могущих увлечься легких бен-
эийовых фракций. Из сепаратора 7 газ поступает в очиститель высокого
давления 8, наполненный известковым тестом, где освобождается от серо-
водорода и поступает в циркуляционный газовый насос 9, а отсюда го-
нится через маслоотделитель 10 обратно в подогреватель 3.
Таким образом осуществляется замкнутый цикл без вывода всего
газа. Как будет ясно из дальнейшего, циркуляционный газ состоит почти
из одного водорода лишь с небольшим содержанием метана. Почти весь
углеводородный газ растворяется в прессдестиллате и выделяется из по-
следнего при выпуске продукта.
Для возмещения водорода, пошедшего на реакцию и отходящего с га-
зами прессдестплтата, в установку из буфера. 14 при помощи вентиля
непрерывно подается свежий водород.
” Гидрирование угля [16]. Превращение угля в жидкое топ-
ливо, т. е. в углеводороды, может быть выражено следующими
уравнениями:
иС + 2nH = СПН2П или пС-}- (2% 2) Н = CnH2,i+o.
Практическое осуществление подобного рода реакций встре-
чает большие трудности.
Впервые гидрирование угля осуществил уже более 60 лет
тому назад Бертло, воспользовавшись в качестве восстановителя
дымящей иодистоводородной кислотой в большом избытке [18].
Он получил при этом сложную смесь углеводородов, в которой
могли быть констатированы лишь немногие отдельные компо-
ненты (гексан, бензол). Хотя в новейшее время возможность
превращения этим способом угля в жидкое топливо в общем
подтвердилась, однако, практического значения данная методика
иметь не может. Столь же безрезультатными с практической
точки зрения оказались опыты гидрирования угля с примене-
нием таких восстановителей, как муравьинокислые слои, окись
углерода и вода, двууглекислая сода, водород и т. д. [19]. Лишь
в новейшее время удалось разрешить эту трудную задачу путем
гидрирования угля непосредственно водородом при высоких дав-
лениях и температурах.
Бергиуе, которому принадлежит главная заслуга широкой и
успешной опытной разработки вопроса гидрирования угля непо-
средственно водородом, еще в 1913 г. заявил патент, основное со-
держание которого сводится к следующему [20].
Каменный уголь или другие подобные материалы (бурый
уголь, торф и т. л.) подвергаются в течение нескольких часов
действию водорода в автоклавах высокого давления тори темпера-
турах примерно 300—400° и давлении около 200 ат. Примене-
ния каких-либо катализаторов при этом не требуется; однако
для ускорения процесса необходимо присутствие вещества,
например бензина, растворяющего образующиеся из угля орга-
нические соединения. В этих условиях большая часть обрабаты-
ваемого угля (до 85%) переходит в растворимое или жидкое
вещество, содержащее, кроме углерода, главным образом водород
и отчасти кислород. По своей химической природе вещества, об-
разующиеся при гидрировании угля, являются по преимуществу
1 См. ниже продукты гидрогенизации.
Е55
углеводородами, подобными нефтяным, с различными температу-
рами кипения. Кислородные соединения имеют характер фено-
лов. Несжиженный остаток представляет собою окрашенное
в темный цвет вещество, состоящее по преимуществу из углерода
и водорода с примесью золы. Наконец, содержащиеся в угле азо-
тистые соединения выделяются в процессе гвдрирования в виде
аммиака и аммиачных соединений и могут быть в этом виде
использованы.
Техническое оформление процесса гидрирования угля встре-
тило сначала значительные трудности, и лишь в 1917 г., после
громадных материальных затрат на исследовательскую работу
(до 12 млн. германских марок), в Маннгейме-Рейнау была по-
строена первая опытная установка «бергинизации угля». Сле-
дующим этапом была постройка близ Мерзебурга (Германия)
громадных принадлежащих I. G-. Farbenjndustrie заводов Leuna-
Werke. Процесс гидрирования угля осуществляется здесь сле-
дующим образом [21].
Сырьем производства служит бурый уголь, залегающий
в окрестностях Биттерфельда на глубине 40 м пластами мощ-
ностью до 100 м. Сначала его подсушивают и -измельчают, за-
тем затирают с тяжелым минеральным маслом в пасту. Эщ
паста с помощью специальных прессов вдавливается в верти-
кальные печи 6—7 м высотой при диаметре в 1—2 м и подвер-
гается здесь действию водорода при 200 ат давления и темпера-
туре около 450°. Пройдя через лечь, угольная паста на 80%
превращается в жидкие продукты, которые разделяют на среднее
масло, легко подвижную жидкость, и на густое, полутвердое
при обыкновенной температуре масло. Твердый остаток подвер-
гают обработке растворителем; полученный экстракт вместе
с упомянутым густым маслом применяют на ‘получение новых
порций угольной пасты, остаток же после экстракции — полу-
кокс— употребляют в качестве топлива под котлами. Наконец,
главный полупродукт — среднее масло — подвергают новой гидро-
генизации при 200 ат давления, причем масло целиком превра-
щается в искусственный бензин.
Превращение угля путем гидрирования в жидкое топливо яв-
ляется, бесспорно, одним из наиболее блестящих достижений со-
временной нам науки и техники. Несмотря на это, дальнейшее
развитие проблемы гидрирования топлива несколько уклонилось
от своего первоначального направления и пошло -по пути визи-
рования тяжелых масел.
Гидрогенизация тяжелых смол, нефтей и т. п. Установки,
разработанные на заводах Leuna-Werke для гидрирования уголь-
ной пасты, оказались вполне применимыми также для деструк-
тивной -гидрогенизации тяжелых масел, как-то: буроутольной
смолы, тяжелых нефтей, мазута и т. п. Схематически такая уста-
норзка изображена на фиг. 113.
Из резервуара 1 с помощью насосов высокого давления 2 под-
лежащее гидрированию масло (с температурой кипения
250—100°) подается в трубчатую печь .9, захватывая по пути
надлежащий катализатор. Здесь‘сырье нагревается до 425°;
556
в той же печи имеется другая трубчатка 4 для нагревания до
той же температуры водорода. Из печи нагретое масло и водо-
род поступают в реакционную камеру—вертикальный толсто-
стенный цилиндр высотою 12 м с внутренним диаметром 0,9 м.
Здесь при 200 ат давления происходит реакция, за счет теплоты
которой температура поднимается до 450°. Из реакционной ка-
меры продукты направляются в холодильник 7 и сепаратор-при-
емник 8. Здесь происходит разделение; газы, в том числе избы-
точный водород, отводятся в резервуар для водорода 11; жидкие
же продукты поступают в перегонный куб 10, работающий при
обыкновенном давлении. Разгонка из этого куба дает следующие
продукты:
1) сырой бензин (температура кипения до 155°), поступаю-
щий (13) на очистку 15 и далее на колонную установку 5 для
получения товарного бензина 3;
Фиг. 113. Схема установки для гидрогенизации тяжелых
смол, нефтей и т. п.
2) среднее масло (температура кипения 155—300°), идущее
(12) на повторную гидрогенизацию через новую трубчатую
печь 16;
3) остаток (температура кипения 300—410°), направляемый
на подмешивание к сырью для новой гидрогенизации 14.
Как видно из схемы, среднее масло и водород, подогретые во
второй трубчатой печи 16 и 17 до 480 \ поступают во вторую
реакционную камеру 18. Здесь при давлении 200 ат благодаря
теплоте реакции температура смеси вновь поднимается до
500—'510°; продукт реакции направляется отсюда в свой особый
холодильник 19 и сепаратор-приемник 20; дальше, газы отходят
в резервуар для водорода 11, жидкие же продукты поступают для
разгонки в тот самый перегонный куб 10, в котором ведется раз-
гонка жидких продуктов из первой реакционной камеры.
Смесь, поступающая в реакционные камеры для гидрировал
ния, содержит обыкновенно 7,5% водорода и 92,5% жидкого
сырья (буроугольная или сланцевая смола, мазут, гудрон, тяже-
лая нефть и т. п.). Выход готовых продуктов по объему соста-
вляет нередко 1О0—105%, причем по желанию реакцию можно
направлять в сторону получения либо бензина, либо керосина.
Так например, буроугольная смола путем гидрогенизации может
быть превращена в бензин с выходом 85%; остальные 15% при-
557
ходЯтсЯ на газы, кокса же вовсе не получается. Соляровое масло
с температурой кипения 245—325° дало 81% бензина, 19%
газа и т. д.
Крайне важно отметить, что одновременно с превращением
тяжелых масел в легкие при гидрогенизации в присутствии спе-
циальных катализаторов легко достигается обессеривание конеч-
ных продуктов. Так как в надлежащих условиях гидрогениза-
ции самые устойчивые сернистые соединения, тиофены и тио-
фаны, распадаются, отдавая свою серу в виде сероводорода
то в продуктах гидрогенизации после промывки щелочью содер-
жание серы определяется обыкновенно лишь сотыми долями
процента.
Гидрирование горючих газов [19]. Особое место среди спо-
собов получения из угля жидкого топлива путем гидрогенизации
занимают такие методы, когда непосредственным сырьем для
гидрирования является не самый уголь, а легко получаемая из
него техническая смесь горючих газов, так называемый «водя-
ной газ». Этот газ образуется из угля действием на него водя-
ного пара при температуре около 1000° по реакции:
С + Н20 = СО + Н2 — 26 990 кал.
Синтезы различных органических соединений из угля, про-
ходящие через стадию водяного газа, получают за последнее
время все более и более широкое развитие. Чтобы оценить зна-
чение этой методики, достаточно вспомнить широко поставлен-
ное в настоящее время производство синтетического метилового
спирта из обогащенного водородом водяного газа. Образование
этого спирта в присутствии сложных катализаторов (а именно
ZnO с примесью Сг20з. V-2O5 и другие окислы) происходит чрез-
вычайно гладко при температуре около 400э и давлении
150—200 ат по уравнению:
С0 + 2Н2=СНа0Н.
В Противоположность этой задаче Ф. Фишер и Троши поста-
вили себе целью получить из водяного газа не индивидуальное
вещество, а смесь органических веществ, пригодную в качестве
жидкого топлива. Эта задача была ими успешно разрешена пу-
тем каталитического «гидрирования окиси углерода в автоклаве
в аналогичных условиях температуры и давления и в присут-
ствии в качестве катализатора железа и поташа. Полученное
таким образом синтетическое жидкое топливо было названо авто-
рами «синтол» *.
Состав синтола оказался весьма сложным. Главными его ком-
понентами являются различные спирты, альдегиды и кетоны;
углеводороды же входят в него лишь в ничтожном количестве
(не более 1,5—2%). Соответственно такому составу теплотвор-
ная способность синтола значительно ниже, чем у бензина
(только — 7,540 кал)\ тем не менее, испытание синтола на мото-
цикле дало вполне удовлетворительные результаты.
1 Сокращенное synthetisches Oel.
558
Путем дополнительного нагревания в автоклаве до 400—450°
синтол может быть превращен б смесь, состоящую главным обра-
зом из углеводородов, — «синтин» *. Углеводороды эти лишь
частью растворяются в серной кислоте; они образовались, оче-
видно, за счет отщепления воды от компонентов синтола с после-
дующим уплотнением их в углеводороды предельного характера;
надо думать, — циклического строения.
Получение синтола производилось сначала в специальной
стальной аппаратуре при давлениях до 1-30 ат и температуре
б зависимости от природы катализатора — 400—450°. Дальней-
шие исследования показали [-22], что в присутствии надлежа-
щих катализаторов превращение,смеси окиси углерода с водоро-
дом в нефтеподобные углеводороды можно воспроизвести в один
прием и притом, что особенно важно, при обыкновенном давле-
нии. Лучшими катализаторами этой реакции являются окислы
железа, никеля и кобальта в присутствии окислов некоторых
других металлов (Zin, Al, Мп, Сг и др.), а также следов щелочей
[23]. Техническими продуктами гидрогенизации окиси углерода
в этих условиях оказались: газообразные углеводороды (этан,
пропан и частично бутан); Синтетический бензин с температурой
кипения 20—180°, напоминающий пенсильванский; синтетиче-
ский керосин с температурой кипения 180—330° и синтетиче-
ский твердый парафин, плавившийся после перекристаллизации
при 61° и выше.
Получение синтетического бензина из окиси углерода и водо-
рода давно уже прошло стадию лабораторных и полузаводских
опытов [*24]. Имеются веские основания полагать, что значи-
тельная часть бензина, потребляемого ныне в Германии, изгото-
вляется именно этим синтетическим путем; однако все детали
этого вопроса, естественно, держатся в строжайшем секрете, что
вызывается, надо думать, соображениями не только патентного
характера. Тем больший интерес представляет ряд работ, про-
деланных в этой области и частично опубликованных в других
странах, как-то: в Англии, Америке, Японии, Испании, а также
в ССОР [25].
Важнейшими моментами синтеза жидких углеводородов из
окиси углерода и водорода являются подбор и приготовление ка-
тализаторов и подготовка газовой смеси для синтеза.
Первые успехи по синтезу жидких углеводородов из окиси
углерода были получены с железными катализаторами. Одно
пирофорическое железо, получаемое восстановлением окиси же-
леза водородом, превращает окись углерода при 230—240°
лишь в газообразные гомологи метана (тазоль). Однако добав-
ка к железному катализатору окисей некоторых металлов (Со,
Zn, Си и др.) позволила получить в аналогичных условиях не
только газообразные, но также жидкие и твердые углеводороды.
Лучшей добавкой оказалась медь. Однако в дальнейшем желез-
1 Сокращенное synthetisehes Bensin.
559
ные катализаторы ввиду сравнительно малой их активности
были оставлены и заменены никелевыми и кобальтовыми. Так
например, катализатор состава СоО . O2O3 давал жидкие про-
дукты уже при 70°. В результате большой работы был найден
целый ряд сложных катализаторов весьма высокой активности
как, например: катализатор состава Ni 4- 20% Мп 4- io%A12o3’
(нормальный никелевый контакт), который за один проход при
190—200° давал до 130 см3 масла на 1 м3 газа; катализатор со-
става Со4*15%Ж дал в аналогичных условиях до 144,5 сл3
масла и т. д.
Так как синтез углеводородов из окиси углерода — реакция
сильно экзотермическая, то во избежание перегрева необходима
обеспечить удаление тепла из сферы реакции. С другой сто-
роны, для придания катализатору большей поверхности его обык-
новенно наносят (осаждают) на то или иное пористое вещество
(кизельгур и т. п.); этим способом готовят так называемые оса-
жденные катализаторы. Оба эти условия хорошей работы ката-
лизатора успешно разрешаются в так называемых сплавных или
скелетных катализаторах, открытие которых является важным
этапом в развитии каталитического гидрирования.
Сплавные катализаторы приготовляются сплавлением двух
или нескольких специально подобранных металлов. Чаще
всего применяют сплавы Ni или Со с А1 или Si. При обработке
таких сплавов после их раздробления едкой щелочью А1 и Si
переходят в раствор, a Ni и Со остаются в виде пористого скелета.
Катализатор тщательно промывают водой и во влажном состоя-
нии загружают в трубку или в аппарат для гидрирования.
После просушки в токе водорода катализатор готов к работе;
иногда его подвергают дополнительной активации нагреванием
в токе водорода при 300—350 \ -Сплавные катализаторы характе-
ризуются выдающейся активностью; так, например, катализатор
состава Ni—Со — Sf дает выхода до 161 сл3 масла на 1 л3 газа
Весьма важное значение для синтеза углеводородов из окиси
углерода имеет также подготовка газовой смеси. Окись углерода
и водород должны быть тщательно освобождены от сернистых
примесей (ядов) как-то: CS2 и H2S; для этой цели газовая смесь
очищается в лабораторных условиях пропусканием через раска-
ленную медную сетку, щелочный раствор пирогаллола и активи-
рованный уголь; в технике для той же цели пользуются из-
вестью, болотной рудой и другими очистителями. Большое зна-
чение имеет также освобождение газовой смеси от инертных
газов (СО2, СН4, N2), так как присутствие их снижает скорость
реакции; столь же вредное влияние оказывает присутствие в га-
зовой смеси водяных паров, так что поступающий на синтез газ
должен быть тщательно высушен сплавленным хлористым каль-
цием, фосфорным ангидридом и т. п. Наконец, большое значе-
ние имеет относительное содержание в газе окиси углерода и
водорода. Наиболее благоприятно соотношение: СО : Н2 = 1:2;
при таком составе газа удавалось получать до 190 см3 масла из
1 л3 газа. При более высоком содержании окиси углерода увели-
560
чивается содержание непредельных в конечном продукте син-
теза, избыток же водорода явно способствует образованию ме-
тана.
Так как гидрирование окиси углерода приводит к сложной
смеси углеводородов, то, естественно, какое-либо определенное
химическое уравнение может выразить здесь лишь одно из на-
правлений реакции, имеющих место при данном процессе. Так
например, при относительном составе газовой смеси СО: Н2 —
= 1:2 реакция идет в среднем с образованием нонана по сле-
дующему уравнению:
9С0 + 19Н2 = 9Н2О + С9Н20.
Кроме нонана, при этом происходит образование также це-
лого ряда других углеводородов—жидких, газообразных и твер-
дых. Сложность состава этой смеси делает затруднительным рас-
чет теоретического выхода при данной реакции; обыкновенно его
принимают равным 250 е.и3 масла на 1 м3 газа.
Аппаратура для получения жидкого топлива из окиси угле-
рода и водорода в основном состоит из следующих частей:
1) аппараты для очистки и контроля газа;
2) реакционные камеры '(трубки) с катализатором;
в) приемники и уловители для продуктов реакции.
Самый процесс не сложен, но требует активного катализа-
тора и высокой чистоты газа; контроль реакции может осуще-
ствляться измерением сжатия газа (контракция), которое со-
ставляет в среднем 50—60% начального объема газа.
Как было указано выше, при гидрировании окиси углерода без давле-
ния получаются газообразные, жидкие и твердые углеводороды. Кислород-
ные соединения синтольного типа при работе без давления почти не
образуются. По терминологии Ф. Фишера образующиеся при реакции про-
дукты обозначаются следующим образом:
1. Газолъ (нефтяной эфир) — смесь наиболее легких углеводородов
ряда метана (СгНв, СзН8, G«Hio и отчасти СзНи), с примесью низших эти-
ленов (от 16 до 35%). Газоль обладает высокой теплотворной способностью
(до 23 000 кал/м3); он может применяться как газообразное топливо, а его
этиленовые углеводороды — как сырье для химической переработки.
2. Когазин 1 — легкокипящее масло (бензин) с концом кипения 220°,
является целевым и наиболее ценным, продуктом синтеза. Путем разгонки
из него легко получаются бензины с различной выкипаемостью и в зави-
симости от режима процесса с различным содержанием непредельных.
Главными компонентами его являются нормальные парафины: пентан, гек-
сан, гептан, октан и нонан; из олефинов обнаружены пентен-1, пентен-2,
гексен-1, гексен-2, гептен-2, октен-2 и др. Нафтены в когазине I, повиди-
мому, отсутствуют; ароматика содержится в крайне незначительном коли-
честве.
Когазин является хорошим горючим для двигателей внутреннего сго-
рания; однако его октановое число не велико (около 58 и даже ниже), и
для получения топлива- с октановым числом 76—77 необходимо подмеши-
вание к нему значительных количеств бензола или спирта. Зато когазин I
не содержит сернистых и кислородных соединений и, таким образом,
является некорродирующим топливом.
3. Когазин II.— высококипящее масло с началом кипения выше 220°,
является прекрасным дизельным топливом Содержание непредельных
в нем незначительно.
36 Зак. 1622. Химия нефти.
561
4. Парафин, образующийся в процессе гидрирования окиси углерода,
представляет собою продукт высокой ценности. Его накопление в масле
может доходить до 15% и выше.
Большой интерес представляет вопрос о механизме синтеза
углеводородов из окиси углерода. Здесь следует различать про-
цессы, 1Протекающие при обыкновенном и при повышенном дав-
лениях.
При обыкновенном давлении промежуточными продуктами
•при гидрировании окиси углерода в присутствии металлических
катализаторов являются, невидимому, их карбиды; последние
при действии водорода или соответствующего металлического
гидрида дают свободный металл и те или иные углеводородные
радикалы (СП, СН2 или СП3), которые полимеризуются затем
в соответствующие углеводороды. Так например, с никелевым
катализатором реакция может быть выражена следующей схе-
мой:
2Ni-|-СО = NiC J-NiO;
NiO “Б Н2 == Ni “t- R2R;
(Ni + R2==NiH2);
NiC + Ha = Ni+^>CH2;
(NiC + NiH2 = 2Ni + \CH2);
»CH2 = C„H2„;
CrtR2H + R2 — CrtH.2u ; 2-
Суммарно это направление реакции можно выразить сле-
дующим уравнением:
иСО -J- (2w -J-1) Н2 = С^Нод |-2 *4~ яН20.
Одновременно, особенно в присутствии железного катализа-
тора и при избытке окиси углерода, реакция идет в несколько
ином направлении, а именно вместо воды побочным продуктом
при образовании углеводородов является углекислота:
2пС0 -f- (п -f-1) Н2 = СпН^ 12 ~Ь %С02.
В качестве предшествующей фазы обоих этих процессов не-
редко допускают адсорбцию окиси углерода и водорода на по-
верхности контакта, сопровождающуюся разрыхлением в актив-
ных точках связи С — О в С... О. Вслед за этим под влиянием
реакционно-способного водорода происходит отрыв кислорода
с образованием воды; углерод же, как сказано выше, образует
с металлом карбид и т. д. Образующиеся углеводороды сначала
адсорбированы катализатором.' Скорость их десорбции, есте-
ственно, тем больше, чем меньше их молекулярный вес; поэтому
понятно, что в низших погонах продукта реакции (газоль) со-
держится значительное количество непредельных, тогда как бо-
лее высокомолекулярные продукты успевают почти полностью
прогидрироваться. Чрезмерному 1>осту цепей препятствует их
крэкинг, и с этой точки зрения понятно, что чем выше темпера-
562
тура реакции, тем больше в ее продуктах газообразных углеводо-
родов.
Совершенно иначе протекает синтез углеводородов из окиси
углерода под давлением. Первой фазой процесса здесь является,
повидимому, присоединение водорода к окиси углерода с обра-
зованием формальдегида:
СО + Н2 = СЩО.
В отсутствии щелочи формальдегид гидрируется далее до
метана, в присутствии же щелочи формальдегид претерпевает
превращение с образованием метанола и окиси углерода:
2СН20 = СН30НН-С0.
Механизм дальнейшего усложнения продуктов реакции, ко-
торая приводит к образованию «синтола», весьма сложен.
Можно полагать, что в первую очередь здесь происходит при-
соединение к метанолу окиси углерода с образованием уксус-
ной кисло гы, которая восстановляется далее до уксусного
альдегида и випното спирта:
СН30Н + СО = СН3 • СООН;
СН» • СООН сн» сно сн» • сн. • он.
О V О «
Винный спирт в свою очередь присоединяет окись углерода
с образованием пропионовой кислоты, которая при восстановле-
нии дает пропионовые альдегид и спирт и т. д.
Основанием другой схемы образования синтола являются
реакции конденсации альдегидов по типу альдоля и кротоно-
вого альдегида с последующим гидрированием. Исходным про-
дуктом этих превращений и здесь является муравьиный альде-
гид. Дальнейшие превращения его могут быть выражены сле-
дующей схемой:
СН20 4- СН20 + СНоОН • СНО;
СНоОН • СНО 4 Н2 = СН3 • СНО 4 ЩО;
СН.. СНО 4- СН3 • СНО = СН3. СНОН • СН2 • СНО;
СН. • СНОН • СН, СНО — Н20 = СН, СН = СН • СНО;
С Н3 • СН = СН • СНО + 2Н2 = СН3 • СН2 • СН, • СН2 • он
и т. д.
Имеются и другие попытки истолкования механизма гидри-
рования окиси углерода под давлением; однако вопрос этот еще
не может считаться окончательно выясненным. Несомненно
лишь одно: при обыкновенном и повышенном давлениях эта ре-
акция идет по совершенно различным схемам, что явственно
сказывается уже на составе конечных продуктов этих процес-
сов; как мы видели, гидрирование окиси углерода при обыкно-
венном давлении приводит к смеси углеводородов, тогда как
при повышенном давлении здесь получается сложная смесь
кислородных соединений («синтол»). Таким образом давление
играет в процессе гидрирования окиси углерода решающую
з/ 563
роль. Впрочем, внутреннее содержание этой роли остается не-
достаточно понятным: надо думать, оно находится в тесной
связи с общим механизмом реакции гидрирования окиси угле-
рода, остающимся в отдельных своих стадиях, как было ука-
зано выше, пока еще недостаточно ясным.
Синтез жидкого горючего, в частности — бензина, из окиси
углерода представляет собою, бесспорно, одно из величайших до-
стижений современной химии. Знаменательно, что с экономиче-
ской точки зрения этот синтетический бензин уже в настоящее
время является вполне приемлемым, особенно для стран, бед-
ных нефтью; в дальнейшем же вследствие роста цен на нефть
и дальнейших несомненных успехов в деле получения синтети-
ческого бензина, особенно в отношении его выходов, общая конъ-
юнктура будет становиться, естественно, все более и более благо-
приятной для синтетического продукта.
Для иллюстрации современного положения и роста продукции искус-
ственного жидкого топлива может служить табл. 125, в которой имеют я
данные по этому вопросу за 1935—1937 гг. для Германии, Великобритании,
Франции и Италии с подразделением по способу получения топлива на
две группы: а) гидрогенизация угля, ® частности бурого' утля, по Бергиусу
и б) гидрирование окиси углерода по Фишеру-Тропш [26].
Таблица 125
Продукция искусственного жидкого топлива за 1935—1937 гг.
Страны и способы получения 1935 г в тыс. m 19с6 г. в тыс. m 1937 г. в тыс. тп
Число установок Продукция Число установок Продукция Число установок Продукция
проект. дей- ств. проект. дей- ств. проект. дей- ств.
Германия: Гидрогенизация . 1 350 250 4 800 320 5 840 750
Фишер-Тропш . . 0 0 0 3 200 20 4 250 200
Великобритания: Гидрогенизация . 1 150 35 1 150 123 1 150 140
Фишер-Тропш . . 0 0 0 0 0 0 0 0 0
Франция: Гидрогенизация . 0 0 0 2 36 0 2 36 9
Фишер-Тропш . . 0 0 0 0 0 О 1 20 3
Италия: Гидрогенизация . 0 0 0 0 0 0 2 ЗСО 0
Фишер-Тропш . . 0 0 0 0 0 0 0 0 0
Отметим в заключение другой интересный случай гидриро-
вания горючих газов с образованием жидких продуктов,
а именно—гидрирование ацетилена. Если вести эту реакцию
564
в присутствии подходящих катализаторов, то, кроме газообраз-
ных продуктов (этан, этилен), получается до 66% жидких угле-
водородов, в частности—углеводородов бензинового типа [27].
Техническое значение этой методики остается пока недостаточно
выясненным.
ПРОБЛЕМА ВОДОРОДА ПРИ ГИДРОГЕНИЗАЦИИ
Так как водород является обязательным участником всякого
восстановительного каталитического процесса, то понятно, что
получение достаточно чистого дешевого водорода должно рас-
цениваться как одна из основных проблем технического гидри-
рования, особенно когда этот процесс осуществляется в крупном
масштабе.
Наиболее сложной оказывается водородная проблема в слу-
чае каталитического гидрирования при обыкновенном давлении,
так как обычные катализаторы подобного рода реакций1 (Ni, Pt,
Pd и др.) чрезвычайно чувствительны к таким постоянным за-
грязнениям технического водорода, как сернистые и мышьяко-
вистые соединения. Во избежание довольно сложной очистки
технического водорода при работе с подобного рода катализато-
рами под обыкновенным давлением удобнее пользоваться элек-
тролитическим водородом, легко получаемым путем электролиза
подкисленной пли щелочной воды, причем катодом служит же-
лезная пластинка, анодом — такая же, но никелированная пла-
стинка. Электролитический водород не требует очистки и прямо
из электролизера может быть направлен в осушительный аппа-
рат и далее в гидрогенизационную печь.
При гидрировании топлива под давлением слишком тща-
тельной очистки водорода не требуется; можно пользоваться не-
посредственно техническим водородом либо прямо из водород-
ного баллона, либо через -компрессор.
Методы получения водорода в технике весьма разнообразны.
В качестве сырья при этом пользуются почти исключительно
водой [28].
Ближайшим материалом для получения водорода в технике
часто служит «водяной газ», легко получаемый пропусканием
водяного пара через раскаленный до 1000—1100° кокс по урав-
нению:
С Н20 — СО Н2.
После очистки «водяного газа» от таких примесей, как водя-
ной пар, углекислота и сероводород, остаток подвергают глубо-
кому охлаждению, почти нацело освобождаются от окиси угле-
рода и азота (из угля) и получают водород со степенью чис-
тоты 97—97,5%.
«Водяной газ» всегда содержит некоторое количество угле-
кислоты, образующейся по уравнению:
со + н3о = со2+Щ.
665
Эта реакция лежит в основе другого важного способа полу-
чения водорода в технике: смешав водяной газ с избытком во
дяного пара, пропускают эту смесь при 500° для ускорения ре-
акции через катализатор (Ре20з с Сг20з и другими окислами
после чего удаляют углекислоту *и сероводород, промывая газы'
водой под давлением 15—40 ат. В итоге получают газ, содер-
жащий 95% водорода и .тишь 3% окиси углерода.
Водород получают в технике также взаимодействием водяною
пара с железом «при 550—800° по уравнению:
зре + 4Н20 F3(\ + 4Н2.
Реакция, выражаемая этим уравнением, обратима: при более
высокой температуре процесс идет преимущественно в сторону
восстановления окисла железа, при пониженной же темпера-
туре —в обратную сторону, т. е. в сторону образования водо-
рода. Последнее направление реакции осуществляется в тех-
нике пропусканием водяного лара через восстановленную ад-
тактную массу (углекислая окись железа и т. п.); образую-
щаяся же в результате этого процесса закись-окись железа
(FeaCh) последующей операцией восстановляется. В качестве
восстановителя употребляют обыкновенно «водяной газ», оба
компонента которого — водород и окись углерода—одинаково
применимы для этой цели; водород — по только что приведен-
ной схеме, окись же углерода—но уравнению:
Fe304 + 4С0 == 400, 4- 3Fe.
Крайне интересное видоизменение последнего метода получе-
ния водорода заключается в том, что осуществляют реатЬда
железа с жидкой водой, работая под давлением (процесс Бер-
гнуса). В присутствии некоторых катализаторов при 300—340°
реакция протекает с большой скоростью, давая водород высокой
степени чистоты (до 99,95% Н2). Так как реакция эта—-сильно
экзотермическая, то подогрев требуется только в начале про-
цесса; в дальнейшем же температура поддерживается теплотой
реакции. Необходимое давление также создается за счет самого
процесса. Регенерация образующейся закись-окиси железа осу-
ществляется нагреванием ее с углем при 1000°.
Что касается, наконец, электролитического получения водо-
рода, то, несмотря на чистоту этого водорода, а также на эф-
фективность процесса и другие его достоинства, большие рас-
ходы на оборудование завода, а также на электроэнергию
являются серьезными препятствиями к внедрению этого способа
получения водорода в промышленности.
Гораздо меньшее значение имеют методы получения водо-
рода, в которых сырьем служит не вода, а другие водородсодер-
жащие вещества, например углеводороды, в частности газооб-
разные (метан, ацетилен) или* жидкие (нефтяные масла, смолы.
Хотя в надлежащих условиях и этими «путями можно получить
водород высокой степени чистоты, однако термическое разложе-
ние этого сырья требует слишком высоких температур (выше
£66
1000°) и в промышленном отношении уступает вышеописанным
методам. Впрочем, в присутствии^ водяного пара температура
разложения углеводородов до свободного водорода может быть
значительно снижена, и на этом пути можно ожидать серьез-
ных успехов в деле промышленного получения чистого водорода.
ПРОДУКТЫ ГИДРОГЕНИЗАЦИИ
При гидрировании топлива получаются тс же основные виды
продуктов, как при крэкинге, т. е. газы, жидкие продукты и
твердые вещества. Однако относительные количества и значение
их, а также их состав весьма существенно отличаются от соот-
ветствующих продуктов обыкновенного крэкинга.
Газы. Получающиеся при -гидрогенизации различных нефте-
продуктов газы обыкновенно в главной массе состоят из водо-
рода. В зависимости от начального давления и глубины гидро-
генизации содержание водорода в этих газах составляет обыкно-
венно 70—80%, доходя иногда до 90% и выше. Другим основ-
ным компонентом газов гидрогенизации являются низшие гомо-
логи метана, т. е. метан, этан, пропан и бутан. Их суммарное
содержание в газах гидрогенизации составляет дополнение до
100% к содержанию водорода. Третьим компонентом газов гидро-
генизации являются непредельные углеводороды; однако содер-
жание их в противоположность газам крэкинга в общем не пре-
вышает нескольких десятых процента, спускаясь нередко до
нуля. Наконец, в случае гидрогенизации нефтей и нефтепродук-
тов, содержащих сернистые соединения, газы, гидрогенизации
всегда содержат сероводород [29].
Жидкие продукты. Жидкие продукты гидрогенизации столь
же разнообразны, как продукты крэкинга или прямой гонки.
Среди них наибольший интерес представляют бензины, по
своему составу резко отличающиеся от обычною крэкинга-бен-
зина. G точки зрения фракционного состава бензины, получен-
ные гидрогенизацией, хотя бы, например, грозненских нефте-
продуктов (парафинистый дестиллат, парафинистый полугуд-
рон), характеризуются обилием легких' фракций. Так, бензины
с концом кипения 200—215° содержат в среднем фракций до
80° —15%, фракций до 100°—35% и фракций до 140° —70%.
Крэкинг-бензины обычного типа не содержат в таком количестве
легких фракций. Причина такого различия заключается, неви-
димому, в следующем: в то время как в условиях крэкинга об-
разующиеся под влиянием термического - распада исходного
сырья легкие непредельные углеводороды полимеризуются и
дают углеводороды с более высокой температурой кипения, в ус-
ловиях гидрогенизации те же легкие непредельные углеводороды
гидрируются, переходя в углеводороды предельного характера
с примерно той же температурой кипения [30].
Для суждения о химическом составе бензинов гидрогениза-
ции может служить табл. 126, в которой приведены как суммар-
ное содержание углеводородов различных типов в бензине (из
567
грозненского парафинистого полугудрона (3 час. при 425°
200 ат начального давления; 10% окиси никеля), так и солен
жание тех же углеводородов в отдельных 'фракциях. А '
Таблица 126
Химический состав бензин) гидрогенизации
(по ГрозНИИ)
Пределы кипения фракций Содержание в весовых процентах
ароматич. непред. нафтены парафины
До 60° 0,0 10,0 0,0 90,0
60—95° 6,1 8,9 27,0 58,0
95—122° 3,0 11.3 34,0 51,7
122—150° 5,5 11,3 36,0 47,2
150—200° 10.3 93 30,0 50,4
Нач. кип.—200° .... 5,0 10,0 26 0 59,0
Сопоставляя данные табл. 120 и 120, приходим к выводу,
что по сравнению с крэкинг-бензинами бензин гидрогенизации
содержит значительно меньше непредельных и отчасти арома-
тики, но заметно больше нафтенов. Содержание парафинов
в обоих типах бензинов примерно одинаковое. Весьма ценным
качеством бензинов гидрогенизации являются их высокие анти-
детонационные свойства [31]. Так, из парафинистого сырья
путем деструктивной гидрогенизации могут быть получены
бензины с октановым числом 72,6—75,3, а из сырья, богатого
ароматикой, — с октановым числом 85—86. Столь высокие ан-
тидетонационные свойства бензинов гидрогенизации при указан-
ном выше их составе можно объяснить лишь допущением, что
парафины, являющиеся их главной составной частью, имеют
здесь изостроение.
Необходимо отметить еще одно важное качество бензинов ги-
дрогенизации—простоту их очистки, сводящейся обыкновенно
к одной промывке щелочью. Так как в условиях гидрогениза-
ции даже такие устойчивые сернистые соединения, как тиофены
и тиофаны, разрушаются, отдавая свою серу в виде сероводорода,
то даже из высокосернистого сырья получаются бензины гидро-
генизации, содержащие после промывки щелочью нередко лишь
сотые доли процента серы. Естественно, что такие бензины вы-
держивают пробу на коррозию и докторскую пробу; вместе
с тем они оказываются стабильными также в отношении смоло-
образования. Лишь в некоторых случаях в зависимости от ре-
жима процесса они требуют небольшой дополнительной очистки.
Принимая во -внимание все вышеизложенное, необходимо
568
притти к выводу, что в отношении качества бензины гидроге-
низации должны быть отнесены к лучшим образцам нефтепро-
дуктов этого рода. Однако необходимо иметь в виду, что в неко-
торых случаях современная техника предъявляет к бензинам
еще более высокие требования, особенно в отношении антидето-
национных качеств, удовлетворить которым бензины гидрогени-
зации уже не в состоянии.
Керосины гидрогенизации также могут представлять выдаю-
щийся интерес в общей системе переработки некоторых нефтей,
а именно в тех случаях, когда керосиновые и газойлевые де-
стиллаты прямой гонки получаются неудовлетворительного ка-
чества (высокое содержание серы, плохой цвет и т. п.). В ре-
зультате гидрогенизации таких дестиллатов, в подходящих усло-
виях температуры и давления от сырья получается 65—85%
высококачественного керосина, удовлетворяющего самым высо-
ким требованиям в отношении стабильности, содержания серы,
цвета и нагарообразования. Остальные 20—40% от сырья при-
ходятся на долю более легких погонов ('бензин); общие же вы-
хода продуктов гидрогенизации по отношению к сырью соста-
вляют и здесь 100—105%' (по объему).
Масла. Еще больший интерес представляет гидрогенизация
масляных дестиллатов низкого качества [32]. Оказалось, что и
в этом случае при работе в надлежащих условиях получается
60—85% (по объему) смазочного масла с несколько пониженной
вязкостью, но значительно более резко выраженным парафини-
стым характером, чем у загруженного сырья. Сопоставление
с маслами прямой гонки приводит к заключению, что масла
гидрогенизации по своим качествам не только не уступают, но
нередко превосходят некоторые, даже наиболее высокосортные
масла: при данной вязкости они имеют высокие удельные весц;
более высокие, чем у других масел, температуры вспышки и вос-
пламенения; небольшую коксуемость (по Конрадсону); низкую
вастываемость при охлаждении; наконец, пологую кривую из-
менения вязкости с температурой. В отношении химического
состава масел гидрогенизации нельзя не отметить также, что
процесс гидрогенизации почти нацело разрушает сернистые и
кислородные соединения, находившиеся в исходном материале,
обеспечивая, таким образом, для масел гидрогенизации ничтож-
ную кислотность и резкое снижение в содержании серы (на
80—90% по сравнению с начальным).
Итак, по своим физико-химическим свойствам масла гидро-
генизации удовлетворяют совокупности всех требований,..предъ-
являемых современной техникой смазочного дела к наиболее
высокосортным маслам для авто- и авиамоторов. Испытание их
на двигателях внутреннего сгорания полностью подтвердило их
высокие качества как смазочного материала.
Кокс. Являющийся постоянным продуктом обычного крэ-
кинга, кокс, как это будет показано ниже, может иногда полу-
чаться также и в процессе гидрогенизации. Однако как техни-
ческий продукт он не имеет в данном случае самостоятельного
569
значения, так как, изменяя режим процесса в сторону повыше-
ния давления водорода или введения катализаторов, легко
можно свести коксообразование до нуля при гидрогенизации
даже тяжелых нефтепродуктов.
чФИЗИКО-ХИМИЧЕСКЙЕ ОСНОВЫ ГИДРОГЕНИЗАЦИИ
Как было показано выше, деструктивная гидрогенизация
т. е. процесс гидрирования, жидкого'топлива (тяжелые нефти-
гудроны и т. п.)? протекает при такой температуре (около 4ЭД°\
при которой эти продукты должны подвергаться частичном}’
разложению, т. е. крэкингу. Таким образом деструктивная гидро-
генизация представляет собою не что иное, как крэкинг в атмо-
сфере водорода. Интересно проследить, в какой степени сохра-
няют в данном случае свою силу те закономерности, которые
были установлены в гл. III для обычного жидкофазного крэ-
кинга, и в какой мере сказывается присутствие водорода на ходе
отдельных, имеющих в данном случае место реакций.
Методика исследования. Опытное исследование процесса
гидрогенизации производится в автоклавах лабораторного типа,
в которых перемешивание' достигается различными способами:
либо при помощи мешалки или вращения автоклава, как опи-
сано выше, либо с помощью качания автоклава на специальной
качалке. Во всех случаях температура опыта должна заме-
ряться внутри автоклава, для чего в крышке автоклава имеется
карман для термометра. Так как температура является одним
из важнейших факторов гидрогенизации и крэкинга, то на пра-
вильность определения температуры должно быть обращено осо-
бое внимание. Специальные опыты показали [30], что в авто-
клавах с мешалками правильный замер температуры является
затруднительным и для выяснения основных факторов гидро-
генизации предпочтительнее пользоваться качающимися авто-
клавами. Одним из способов выяснения правильности измерения
температуры на данной гидрогенизационной установке может
служить сопоставление результатов проведенных на ней опы-
тов крэкинга того или иного нефтепродукта с результатами, по-
лученными в аналогичных условиях на стандартной установке,
описанной в гл. III.
Химизм гидрогенизации. Так как температурные условия
гидрирования топлива примерно те же, что и при крэкинге, то
основные реакции, имеющие место при обоих этих процессах—
одни и те же. Это — реакции разложения сложных органиче-
ских молекул на более простые, реакции уплотнения продуктов
разложения друг с другом, приводящие к более сложным до со-
ставу системам,' и процессы изомеризации -(ср. ч. II, гл Ш).
Впрочем, последние имеют для основной характеристики гидро-
генизации топлива второстепенное значение; поэтому они будут
рассмотрены ниже в связи с гидрогенизацией углеводородов
отдельных классов. Наконец, должен быть отмечен еще один
тип реакций, в значительной степени определяющий природу
570
продуктов деструктивной гидрогенизации топлива; это — реак-
ции присоединения водорода, энергично протекающие в усло-
виях гидрирования топлива. Для правильного понимания хи-
мизма гидрогенизации каждый из основных типов реакций,
протекающих при гидрогенизации, должен быть рассмотрен здесь
отдельно. .
Реакции разложения. Как было показано выше, при гидро-
генизации тяжелых нефтепродуктов всегда образуются газы,
бензиновые фракции, керосины и т. д. Массовое образование
низших углеводородов из высших является признаком того,
что и. в данном случае имеют место реакции разложения. Чтобы
составить себе представление о ходе этих реакций при гидроге-
низации, интересно проследить, какое влияние оказывают на них
основные факторы процесса: время, температура и давление, а
также характер сырья, взятого для гидрогенизации.
Прежде всего, как видно из данных табл. 127, при постоянной
температуре и давлении количество продуктов разложения при
гидрогенизации (как и при крэкинге) с течением времени по-
степенно возрастает, но при достаточной продолжительности
процесса достигает некоторого предела. Этот предел для того
пли иного вида продуктов разложения, например для бензина,
зависит от характера сырья и от температуры процесса; так на-
пример, -гидрогенизация парафинистого дестиллата при 423°
дает предельный выход бензина примерно в 43%.
Таблица 127
Кинетика гидрогенизации парафинистого дестиллата
(по ГрозНИИ)
№ Продолжит. Газы и Бензин Керосин
процесса потери до 2С0° 200—300°
п/п в час. ' в % в % в %
Температура 425°; начальное давление
водорода 100 am
1 0,5 2,0 7,9 14.4
2 1.5 5,8 22,2 22,3
3 3,0 9,7 36,0 24,0
4 6,0 13,2 42,7 21,3
5 9,0 34.0 41,4 11,6
6 12,0 25,4 42,9* 15,4
Дальнейшее накопление бензина оказывается уже невозмож-
ным, так как таковое сначала компенсируется разложением уже
образовавшегося бензина на газообразные углеводороды; при
более же жестком углублении процесса выхода на бензин
в связи с дальнейшим увеличением газообразования начинают
понижаться. Табл. Г28, в которой приведены данные гидрогени-
зации парафина при 450° и 100 ат начального давления, может
служить иллюстрацией этого положения,
Гидрогенизация парафина
(по ГрозНИИ)
Таблица 128
Ко п/п Продолжитель- ность процесса Газы и потери В 7о Бензин до 200° в % Керосин 200- 300° в % Остаток выше 30QP в %
Температура 450°, начальное давление 100 ап
1 17 мин. 7,9 26,0 19,5 48,9
2 34 „ 28.0 54,6 13,7 6,0
3 1 ч. 01 „ 41,5 49,9 7,4 3,5
4 2 „ 00 „ 40.3 50,9 8,6 2,5
5 з „00 . 43,9 48,1 6,8 3,4
6 6 „ 00 „ 58,7 33,8 5,9 8,9
7 11 ч. 68 мин. 65,3 15,5 3,9 17,6
Данные следующей табл. 129 позволяют проследить влияние
на ход реакции разложения при гидрогенизации другого важ-
ного фактора — давления водорода, а также сопоставить гидро-
генизацию с крэкингом (давление водорода О ат). Как видно,
в начальных стадиях процесса скорость разложения при гидро-
генизации несколько замедлена ио сравнению со скоростью
крэкинга. Так например, при 425° в течение 30 мин. из парафи-
нистого дестиллата образуется в условиях обычного крэкинга
около 16% бензина, тогда как в условиях гидрогенизации при
100 ат начального давления водорода получается бензина
только 8%. Однако по мере углубления процесса скорости крэ-
кинга и гидрогенизации постепенно выравниваются, так что,
например, трехчасовые процессы при 425° дают примерно оди-
наковые выхода на бензин (36—38%) как в случае крэкинга,
так и в случае гидрогенизации.
Те же опыты показывают, что величина давления водорода
при достаточной глубине процесса, не влияя существенно на его
развитие (ср. опыты № 6—11), оказывает Заметное влияние на
его конечный результат. Так например, при 6-часовых опытах
выхода бензина, которые, очевидно, можно считать уже пре-
дельными, для крэкинга составили 38%, для гидрогенизации же
три 100 ат давления — 43%, а при 200 ат — 47%. Таким обра-
зом предельный выход бензина в случае гидрогенизации пара-
финистого дестиллата заметно выше, чем в случае крэкинта
при прочих равных условиях. Повидимому, это зависит от не-
сколько большей термической устойчивости углеводородов пре-
дельного характера по сравнению с непредельными углеводо-
родами, составляющими, как известно, весьма значительную
часть продуктов крэкинга.
Для суждения о влиянии температуры на скорость реакции
разложения при гидрогенизации могут служить данные
табл. 130.
Б72
Таблица 129
Гидрогенизация парафинистого дестиллата
(по ГрозНИИ)
№ п/п Начальное давление водорода в ат Газы и потери • в % Бензин до 200° в % Керосин 200—300° в %
Температура 425°, продолжительность 0,5 чао.
1 0 2,4 15,8 18,8
2 100 2,0 7,9 *14,4
Температура 425°, продолжительность 1,5 час.
3 0 16,6 36,5 20,4
4 70 4,6 27,6 22,5
5 100 5,8 22,2 22,2
Температура 425°, продолжительность 3 час.
6 0 9,0 37,0 27,0
7 25 13,4 39,3 19,5
8 50 16,0 40,3 18,1
9 100 9.7 36,0 24.0
10 150 11,4 S6.2 24,6
11 200 9,1 35,5 23,6
Температура 425°, продолжительность 6 час.
12 0 14,0 38,0 25,0
13 100 13,2 42,7. 21,3
14 150 17,0 44,2 18,9
15 200 13,0 46,8 24,5
Таблица 130
Влияние температуры на скорость гидрогенизации
(по ГрозНИИ)
Темп. в СС Продолжит. процесса Газы и потери в % Бензин до 200° в % Керосин 200-300° в %
Начальное давление 100 ат
375 18 час. 3.2 6.6 14,5
400 3 „ 5,0 6.6 12,5
425 0.5 „ 2,0 7,9 14,4
425 6 час. 13,2 42,7 21,3
450 1 „ 22,2 41,0 18,4
475 11 мин. 21.0 39,5 17,5
578
Как видно из двух серий опытов, приведенных в этой
таблице, с повышением температуры опыта на 25° при одновре-
менном сокращении его продолжительности в 6 раз выхода на
бензины гидрогенизации остаются в пределах ошибки опыта не-
изменными, а это значит, что при повышении температуры на
каждые 10° скорость гидрогенизации увеличивается вдвое. Той
же закономерности, как было показано в гл. III, подчиняется
крэкинг-процесс, хотя естественно, что в связи с громадной
сложностью обоих этих процессов—гидрогенизации и крэкинга—
приводимые опыты и выведенные пз них закономерности дол-
жны рассматриваться лишь как первое приближение.
Что касается, наконец, влияния характера сырья на ход ре-
акций разложения при- гидрогенизации, то пока по этому во-
просу опытных данных еще не достаточно. Так как крэкинг
более высокомолекулярных углеводородов -протекает, вообще го-
воря, сравнительно легче, то следует ожидать, что более тяжелые
нефтепродукты будут гидрироваться несколько успешнее, чем
более легкие. Основные закономерности, рассмотренные выше,
должны, таким образом, сохраниться, очевидно, и в этих слу-
чаях. К вопросу о гидрогенизации тяжелых нефтепродуктов мы
еще вернемся несколько ниже.
Реакции уплотнения, Как было показано в гл. III, реакции
уплотнения проявляются при крэкинге в новообразовании бо-
лее высокомолекулярных, тяжелых продуктов, в1 конечном
итоге— карбондов (кокса). Эти характерные признаки реакций
уплотнения, протекающих при высокой температуре, наблюда-
ются также при гидрогенизации, хотя далек® не в таких разме-
рах, как при простом крэкинге. Опыт показывает, что весьма
существенное значение при этом оказывает давление водорода
и температура (табл. 131).
Таблица 131
Реакции уплотнения при крэкинге и гидрогенизации парафинистого
дестиллата
(по ГрозНИИ)
Продолжитель- ность опыта в час. Крэкинг (425°) Гидрогенизация (425°)
УД. вес остатка от 300° колич. карбопд. В % Нач. давление 50 ат Нач- давление 100 аиг
уд. вес остатка от 300° количество карбоидов в % уд. вес остатка от 300° количество карбоидов В %
0,5 0,991 0,01 0.896 0,875 0,01
1,0 — — 0,900 0,00
1,5 1,04 0,62 — —— 0,891 0,01
3.0 1,00 — 0,991 3,00 0,940 0,10
6,0 —— 8,60 — — 0966 0,02
12.0 — — — — 1,041 0,07
574
Как видно из данных табл. 131, при недостаточном Давлении
Водорода (ниже 100 ат) общая картина процессов уплотнения
при крэкинге и гидрогенизации примерно одна и та же: по мере
увеличения длительности процесса наблюдается быстрое возраста-
ние удельного веса продукта, особенно остатка с температурой
кипения выше 300 , в котором концентрируется главная масса
продуктов уплотнения; в то же время среди продуктов реакции
появляются более или менее значительные количества карбои-
дов (кокса) — этих наиболее высокомолекулярных продуктов
уплотнения. Но уже при начальном давлении водорода в 100 ат
(а также при более высоггих давлениях) возрастание удельного
веса по мере углубления процесса заметно снижается, коксо-
образование же при температуре 425° и выше резко падает и
даже совершенно прекращается. Таким образом, чтобы при
гидрогенизации парафинистого дестиллата (грозненского) при
425° избежать коксообразования, необходимо работать при на-
чальном давлении пе ниже 100 ат, что соответствует оператив-
ному давлению при 425°—235 ат.
Не меньшее влияние на процессы конденсации и коксообра-
зования оказывает температура, при которой ведется процесс
гидрогенизации (табл. 132).
Таблица 132
Гидрогенизация парафинистого дестиллата
(по ГрозНИИ)
№ п/п | Темп. процесса Продолжи- тельность процесса Бензиновые фр. Керосиновая фр. Уд. вес остатка 3 мс я е Й да
уд. вес иод. число уд. вес иод. число
Начальное давление 100 «w; выход бензина 40%
1 400 18 час. 0,723 0.847 —• 0,968 0,01
2 425 3 „ 0,728 52 0.840 31 0,940 0,1')
3 475 7 мин. 0,735 41 0,564 25 1,039 —
Начальное давление 150 ат; выход бёнёАна 40%
4 425 3 час. 0,724 46 0,824 я— 0,918 0.С8
5 450 0,5 . 1 0,721 46 0840 27 0,932 о,со
• Начальное давление 200 ат; выход бензина 40 Уо
с 425 3 час. 0.725 46 0,831 28 0,900 —
7 450 0.5 „ 0,721 39 0 846 27 0940 0,01
Из табл. 132 видно, что с повышением температуры процесса
и несмотря на соответствующее сокращение времени для полу-
чения одних и тех же выходов на бензин, удельные веса остатка
при достаточной глубине процесса всегда возрастают. Это влия-
ние температуры может, однако, в значительной мере, а иногда
' 575
и полностью компенсироваться соответствующим повышением
давления, так что даже при сравнительно жестких условиях
гидрогенизации (450—475°), но при соответственно высокой на-
чальном давлении (150—200 ат) коксообразование при проле
может оказаться практически равным нулю.*
Данные таблиц 131 и 132 показывают, что для грсзненскбго
парафинистого дестиллата и, надо думать, для аналоговых
нефтепродуктов типа тяжелых газойлей температура 425____450°
и начальное давление 100—150 ат являются наиболее благо-
приятными для гидрогенизации, особенно, если не задаваясь
слишком высокими выходами, ограничиваться в одном процессе
(«первичная гидрогенизация») выходами на бензин в 30—35%
и тем самым снизить продолжительность процесса. Для более
легкого сырья, например для легких газойлей (температура ки-
пения 200—350°), оптимальная температура гидрогенизации
может оказаться несколько выше указанной нормы; наоборот,
для более тяжелого сырья, например для тяжелых остатков, эта
температура должна быть относительно ниже.
Реакции присоединения водорода. Эти реакции, чрезвычайно
характерные для гидрогенизации топлива, заключаются в при-
соединении водорода либо непосредственно к радикалам, обра-
зующимся в результате отщепления от более или менее слож-
ных молекул исходного материала, либо к. получающимся в про-
цессе крэкинга непредельным углеводородам по месту их двой-
ных связей. Количественное представление о реакциях присое-
динения при гидрогенизации топлива можно составить путем
учета израсходованного водорода по разности между количе-
ством водорода, введенного в автоклав, и количеством водорода,
оставшегося после гидрогенизации. Для грозненского парафини-
стого дестиллата данные о расходе водорода в различных усло-
виях гидрогенизации приведены в табл 133.
Таблица 133
Расход водорода при гидрогенизация
Грозненского парафинистого дестиллата
(по ГроэНИИ)
п/п к Условия гидрогенизации Выход, в °/о по весу Количество присоедив. водорода в °/| по весу
темпера- тура нач. давл. водорода в ат время’ в мин. бензин и потери карбоиды
1 425 25 144 39,3 4,0 0,3
2 425 50 182 42,6 3,4 06
3 425 100 182 860 01 1.1 1,5 ’
4 450 2С0 31 42,0 * 0.0
Как видно из данных табл. 133, на количество водорода, при-
соединившегося в процессе гидрогенизации, особенно си&ное
влияние оказывает давление. Так, для грозненского парафинй-
576
стого дестиллата при 25 ат начального давления расход Водо-
рода составил лишь 0,3% по весу при весьма значительном
коксообразовании (4%), тогда как при 200 ат за время, почти
в 5 раз меньшее, было израсходовано водорода 1,5% по весу
при полном отсутствии коксообразования. Данные эти особенно
.знаменательны, если при их оценке учесть, что температурные
условия этих опытов были примерно одни и те же (425—450°),
равно как и выхода на бензин в пределах допустимых отклоне-
ний для отдельных опытов были одинаковы.
Присоединение водорода в условиях гидрогенизации никогда
не бывает полным. Даже если взять водород в заведомо недоста-
точном количестве, то он все же целиком не используется и
при достаточной продолжительности опыта. Так например,
в опыте гидрогенизации грозненского парафинистого дестиллата
при 425° и 25 ат начального давления (табл. 133) водород был
загружен в количестве 0,6% по весу, тогда как расход его соста-
вил лишь 0,3%, т. е. всего 50% от его загрузки, хотя выход на
бензин, полученный в этом опыте, свидетельствует о значитель-
ной глубине процессов разложения. При более высоких давле-
ниях абсолютное количество присоединяющегося водорода при
прочих равных условиях, конечно, увеличивается, но относи-
тельное его количество уменьшается, так что при начальных
давлениях 100—200 ат и достаточно глубоком процессе гидроге-
низации на присоединение идет не более 30—35% взятого
водорода; остальная же часть его может быть обнаружена в сво-
бодном состоянии в газах гидрогенизации.
Эти данные приводят к выводу, что присоединение водорода
в условиях гидрогенизации топлива — процесс обратимый, и,
таким образом, высокие давления, применяемые при гидрогени-
зации топлива, необходимы здесь не для чего иного, как для уве-
личения концентрации водорода и соответствующего смещения
равновесия в сторону продуктов его присоединения. К такому
же заключению, как это подробнее будет показано в дальней-
шем изложении, приводит исследование реакций гидрирования
индивидуальных углеводородов при высоких температурах.
Совокупность теоретических и опытных данных по вопросу
о гидрировании этиленовых и ароматических углеводородов при-
водит к выводу, что в температурных условиях крекинга и
гидрогенизации жидкого топлива этилен и его ближайшие гомо-
логи, присоединяя водород, могут нацело или почти нацело пре-
вращаться в соответствующие парафины. Напротив, более вы-
сокомолекулярные гомологи этилена, а также ароматика в тех
же температурных условиях присоединяют водород лишь ча-
стично, образуя со своими продуктами гидрирования равновесие,
которое при небольших давлениях резко сдвинуто в сторону
почти полной диссоциации этих продуктов. Положения эти на-
ходятся в полном соответствии с химическим составом различ-
ных продуктов гидрогенизации топлива. Действительно, анализ
газов гидрогенизации показывает [30], что непредельных угле-
водородов в них почти не содержится; очевидно таким образом,
37 Зак. 1622. Химия нефти.
присоединение водорода к газообразным непредельным углеводо-
родам, образующимся в процессе крэкинга исходного сырья, про-
исходит в условиях гидрогенизации нацело и практически не-
обратимо. Напротив, как видно из данных табл. 132, бензины и
керосины гидрогенизации (всегда имеют довольно значительные
иодные числа и, следовательно, всегда содержат более или ме-
нее заметные количества непредельных, несмотря на присутствие
в условиях процесса избытка водорода.
Температурными границами реакций присоединения водорода
в значительной мере определяются условия процесса гидрогени-
зации топлива в технике. В самом деле, как ясно из вышеизло-
женного, при температурах, близких к 500°, процессы присоеди-
нения водорода даже при высоких давлениях резко сокращаются,
особенно для тяжелых этиленовых углеводородов, появляющихся
уже среди начальных продуктов крэкинга тяжелых газойлей я
нефтяных остатков. Ввиду этого, как общее правило, первичная
гидрогенизация тяжелых нефтепродуктов ведется обыкновенно
при температуре не выше 425—450°, т. е. в таких условиях тем-
пературы, когда скорости реакций, протекающих при гидрогени-
зации топлива, в том числе и реакций присоединения водорода,
должны сильно понизиться. Так как увеличение давления водо-
рода, как уже было указано выше, отражается лишь на смеще-
нии равновесия этого последнего процесса, то для повышения
его скорости при недостаточной температуре остается лишь один
путь, широко используемый в настоящее время техникой: путь
применения катализаторов, ускоряющих процесс присоединения
водорода к продуктам термического распада углеводородов
исходного сырья.
Гидрогенизация некоторых нефтепродуктов может протекать
вполне удовлетворительно в отсутствии каких-либо специальных
катализаторов; таковы, например, неоднократно упоминавшийся
ранее грозненский парафинистый дестиллат и другие аналогич-
ные нефтепродукты, содержащие сравнительно немного веществ,
легко конденсирующихся и склонных к коксообразованию. Если,
однако, нефтепродукт содержит значительное количество высшей
ароматики, а также смолистых или асфальтовых веществ, то
процессы уплотнения и коксообразования начинаются уже с са-
мого начала их термического распада, и для предупреждения
этих нежелательных направлений реакции необходимо примене-
ние катализаторов, ускоряющих присоединение водорода. При-
мером нефтепродуктов, гидрогенизация которых в отсутствие ка-
тализаторов не дает удовлетворительных результатов, могут слу-
жить мазут или полугудрон из той же грозненской парафини-
стой нефти. Гидрогенизация этих нефтепродуктов без катализа-
торов неизменно дает значительное кокоообразование. Напротив,
в присутствии окиси никеля, взятого в качестве катализатора
в количестве 5—10%, гидрогенизация этих тяжелых остатков
протекает весьма гладко, без образования сколько-нибудь значи-
тельного количества кокса [30, 32]. ' 'У.
Катализаторы, применяемые при гидрогенизации топлива,
578 ; г 1.
весьма разнообразны [32, 33], причем приготовление многих из
них защищено многочисленными патентами. Наиболее часто при-
меняется, пожалуй, все-таки обычный гидрирующий катализа-
тор— никель, употребляемый в данном случае в виде его про-
дажной окиси. Кроме никеля, при деструктивной гидрогенизации
находят применение также некоторые другие металлы, как-то:
железо, кобальт, медь и т. д., или осаждаемые в виде их окислов
на тот или иной субстрат (пемза, силикагель, уголь и другие
пористые материалы), или применяемые непосредственно в вида
их солей. Широкое признание получают за последнее время
смешанные катализаторы, представляющие собою смеси окислов
различных металлов (NiO 4- AI2O3, NiO + Сг#Оз, Co±Os 4- Сг±Оз
и т. п.), или тот либо иной катализатор с «присадкой», повы-
шающей его активность. В качестве активирующей присадки ре-
комендуются некоторые кислородные соединения первой группы
металлов — щеточные земли, редкие земли, кислородные соеди-
нения магния, бора и т. д. Особенно важное значение при гидро-
генизации топлива имеют также катализаторы, не отравляемые
присутствием сернистых соединений. В качестве такого серо-
устойчивого катализатора особое внимание привлекает двухсер-
нистый молибден (MoS2).
Следует отметить в заключение, что, помимо разнообразней-
ших катализаторов, рекомендуемых и применяемых при гидроге-
низации топлива, бесспорное каталитическое действие при ©том
процессе оказывают также стенки автоклава, в котором происхо-
дит гидрогенизация. На каталитическое действие стенок реакци-
онного сосуда имеются многочисленные указания в литературе,
так что реакция присоединения при гидрогенизации топлива -по-
добно многочисленным другим реакциям гидрирования, протека-
ющим в присутствии сторонних металлов или их окислов (ср.
выше), должны рассматриваться как типично каталитические
даже в тех случаях, когда они проходят без применения специ-
альных катализаторов.
ГИДРОГЕНИЗАЦИЯ УГЛЕВОДОРОДОВ РАЗЛИЧНЫХ РЯДОВ
Для уяснения химизма процесса гидрогенизации топлива,
так же как при крэкинге, представлялось бы особенно целе-
сообразным изучение в этом направлении индивидуальных угле-
водородов различных рядов. К сожалению, опытный материал
в этой области пока крайне ограничен.
Парафиновые углеводороды. Углеводороды ряда метана (па-
рафины) представляют собою насыщенные системы, не способ-
ные к реакциям присоединения. Ввиду этого их гидрогенизация
может происходить лишь по одной основной схеме: сначала—
крэкинг, затем — гидрирование. Такая схема находит подтвер-
ждение в исследовании гидрогенизации нефтяного парафина.
Особенно показательно сопоставление гидрогенизации парафина
с обыкновенным его крэкинтом [34].
Оказалось, что в условиях одной и той же лродолжитель-
07* 579
ности опыта (1 час) и температуры (около 450°), независимо <п
того, вести ли обычный крэкинг парафина или его гидрогениза-
цию под высоким или малым давлением, выхода на бензин, ке-
росин и тяжелый* остаток (температура кипения выше зоо°)
а также на газ получаются практически одни и те же. Однако
в отношении физико-химических ’свойств и состава между про-
дуктами крэкинга и гидрогенизации парафина оказалось боль-
шое различие: удельные веса продуктов гидрогенизации были
ниже, чем у продуктов крэкинга, и, соответственно цвет—
лучше.. Одновременно бромные числа бензинов и керосинов гидро-
генизации оказались значительно ниже, чем: у соответствую-
щих продуктов крэкинга, что ясно указывает на реакции гидри-
рования (присоединения водорода), имеющие место в процессе
гидрогенизации парафина.
Ближайший состав продуктов крэкинга и гидрогенизации
парафина изучен пока еще недостаточно. Однако на основе тща-
тельной фракционировки этих продуктов и определения физи-
ческих свойств выделенных фракций можно сделать заключение,
что наиболее легкие из них в главной своей части состоят из
низших парафинов (пентан, гексан и др.) и этиленовых угле-
водородов нормального строения, причем количественно в про-
дуктах гидрогенизации шире представлены парафины, в продук-
тах же обычного крэкинга — этиленовые углеводороды, а также
повидимому, нафтены. Такого рода соотношения сохраняются
также для более тяжелых продуктов крэкинга и гидрогенизации
вплоть до тяжелых остатков, что явствует уже из их элемен-
тарного состава: в то время как содержание водорода в тяжелом
остатке от крэкинга парафина составляет 8,2%, в соответствую-
щем продукте гидрогенизации анализ показал 13,8% во-
дорода.
Присоединение водорода при гидрогенизации парафина по
мнению некоторых авторов [34] происходит лишь по месту обра-
зующихся в процессе крэкинга свободных осколков молекул
(радикалов), а не по месту кратных связей более стабильных
продуктов крэкинга, непредельных углеводородов, так как вы-
сокая температура процесса должна препятствовать этому по-
следнему направлению реакции. Однако ближайшее исследова-
ние гидрогенизации непредельных углеводородов не вполне со-
ответствует такому представлению.
В самое последнее время результаты гидрогенизации пара-
фина нашли полное подтверждение в опытах деструктивной
гидрогенизации норм, октадекана, CieHss, в присутствии в каче-
стве катализатора двухсернистого молибдена, M0S2 (29). Резуль-
таты этих опытов сведены в табл. 134.
Как видно из табл. 134, распад норм, октадекана при де-
структивной гидрогенизации проходит чрезвычайно легко: уже
через 1 час образуется здесь до 37,2% бензиновой фракции
(до 200°), считая на полученный тидрогенизат (или 33,2% на
исходный углеводород); примерно такое же количество бы^о по-
лучено при этом и керосиновой фракции, содержавшей» пссом-
580
Таблица 134
Гидрогенизация норм, октадекана
(440°, 90 ат, 1,3% MoSa)
(по Пучкову)
1 № п/п 1 Продолжи- тельность в чао. Выход гидрогени- зата в % Выхода в % на гидрогенизат Иодные числа
фракция 28—200° фракция 200—300° остаток выше 300° фракция до 200° фракция 200-300°
I 1 89.2 37.2 39.6 15,5 18,9 6,9
2 4 68,6 63,4 19,6* — 6,3 3,9
* . 8 52,6 71,52 — 4,8 —
ненно, также неизмененный октадекан. Общий выход гвдрогени-
зата в том же опыте (№ 1) составлял 89,2%; газ и потери — 8%.
С увеличением продолжительности опыта за счет возраста-
ющего газообразования постепенно снижаются, как это видно
из табл. 134, выхода на гидрогенизат. Одновременно возрастают
выхода легких фракций; выхода же на керосиновые погоны и
остаток постепенно снижаются до нуля. Следует отметить, что
полученная в четырехчасовом опыте керосиновая фракция цели-
ком выкипала до 255°, а это показывает, что за 4 часа октаде-
кан в данных условиях «распадается целиком на более легкие
продукты. Необходимо добавить еще, что по мере увеличения
продолжительности опыта наблюдается постепенное уменьшение
иодных чисел обеих фракций гидрогенизата, т. е. снижение
в них содержания непредельных.
Что касается, наконец, химической природы продуктов де-
структивной гидрогенизации октадекана, то в основном, как и
следовало ожидать, они оказались углеводородами ряда метана
с большей'или меньшей примесью олефинов. После удаления
последних из продукта одночасовото опыта [№ 1 ] была получена
смесь углеводородов, высокая анилиновая точка которой (72,1)
в полной мере отвечала анилиновой точке соответствующей
смеси парафинов. Однако заметное снижение анилиновой точки
(с 72,1 до 67,0) у соответствующего продукта восьмичасового
опыта (№ 3) заставляет принять, что гидрогенизат, полученный
в данных условиях, содержит небольшую примесь нафтенов.
Образование ароматики во всех опытах, собранных в табл. 134,
оказалось совершенно ничтожным.
Интересно отметить в заключение, что при деструктивной
гидрогенизации парафинов недавно доказано несомненное нали-
чие процессов изомеризации исходной системы (29). Опыты, про-
водившиеся с норм, гексаном, CeHu, показали, что в присут-
* Конец кипения 255°.
8 Кенец ктщеивя 17Р.
581
ствии двухсернистого молибдена продуктами гидрогенизации
(2 часа при 400—440° и 90 ат начального давления) является
смесь углеводородов, главной составной частью которой ока-
зался, естественно, исходный гексан, не вошедший в реакцию
Кроме гексана, в продукте реакции обнаружен углеводород с бо-
лее низкой точкой кипения, который при нитровании слабой
азотной 'Кислотой по М. Коновалову' дал некоторое количество
третичного нитросоединения. Тем самым присутствие третичного
водорода в одном из продуктов деструктивной гидрогенизации
норм, гексана, а следовательно, наличие изомеризации нормаль-
ной цепи исходного углеводорода можно считать доказанным. По-
добного рода случаи изомеризации в процессе деструкции исход-
ного вещества могут быть названы деструктивной изомериза-
цией. •
Этиленовые углеводороды. В отличие от углеводородов ряда
метана этилен и его гомологи в подходящих условиях могут не-
посредственно соединяться с водородом, превращаясь в соответ-
ствующие парафины; обратно, отщепляя при высокой темпера-
туре водород, парафины превращаются в этиленовые углеводо-
роды. Между этими двумя прямо противоположными реакциями
можно представить себе равновесие, которое в простейшем слу-
чае для этилена выражается следующим равенством:
СаН4Н-Н2^С2Нб.
Опытное исследование этого равновесия было произведено [35]
при температурах G00, Ь50 и 700°; оно привело к следующему
эмпирическому уравнению для константы равновесия К этой
реакции:
— ИТ In К= 31,244 — 28,88 Г,
где R — газовая постоянная, а Т — абсолютная тепература.
Если экстраполировать это уравнение за пределы указанных
выше температур, а именно рассчитать константу равновесия:
r_JCsHJ[H2]
[С2Н61
для обычных температурных условий гидрогенизации (400—500°),
то легко притти к выводу, что при этих температурах гидрирова-
ние этилена идет до конца даже при атмосферном давлении; при
давлении же водорода в 50 ат то же самое наблюдается даже
при 600° [36].
В вопросе о гидрировании высших гомологов этилена по
уравнению:
СпНзп+2Н СпН2пх3
за отсутствием опытных данных пока приходится ограничиваться
лишь приблизительными расчетами на основе тех же положений
термодинамики, которыми можно пользоваться при аналогичных
подсчетах для крэкинг-процесса (гл. III). Эти расчеты приводят
к выводу, что при увеличении молекулярного веса этиленовых
582
углеводородов равновесие постепенно сдвигается в сторону мейее
полного их гидрирования. Однако для этиленовых углеводородов
с небольшим молекулярным весом гидрирование практически
дойдет до конца уже при невысоких давлениях 'Водорода, по-
рядка 50—100 ат [36, 37 J.
Качественной иллюстрацией к сказанному может служить
деструктивная гидрогенизация гексилена, CeHi2, в присутствии
двухсернистого молибдена [29]. Оказалось, что при 400° и на-
чальном давлении 90 ат гексилен, судя по падению давления,
нацело гидрируется уже в течение первых 5—10 мин. Иодное
число гидрогенизата оказалось равным нулю. Главным продук-
том реакции оказался норм, гексан; кроме того, получена ниже-
кипящая фракция, давшая при нитровании слабой азотной ки-
слотой по М. Коновалову заметное количество нерастворимого
в щелочи третичного нитросоединения. Очевидно, и здесь, как
при деструктивной гидрогенизации норм, гексана, частично про-
исходит деструктивная изомеризация исходной системы с нор-
мальной цепью углеродных атомов в углеводород ряда метана
изостроения.
Ароматические углеводороды. Как было показано в гл. III,
бензол и его ближайшие гомологи в условиях жидкофазного
крэкинга весьма устойчивы. Свойство это они сохраняют также
при нагревании в атмосфере водорода. Так например, бензол при
нагревании в атмосфере водорода до 485° почти не изменяется;
наблюдается лишь небольшое образование дифенила по реакции:
свнв+свнв=свнб.с6нб+н2.
Аналогично ведет себя при нагревании в атмосфере водорода
толуол; однако, кроме образования дитолила (ближайшего гомо-
лога дифенила), здесь наблюдается также частичный распад мо-
лекулы с образованием бензола и метана.
В присутствии катализатора, например восстановленного ни-
келя, палладия и др., отношение бензола и его гомологов к на-
греванию в атмосфере водорода до 400—500° существенно изме-
няется в сторону гидрирования ароматического ядра. Образую-
щиеся при этом равновесия в случае бензола (1) и толуола (2):
— СН3СвНп,
в настоящее время хорошо изучены [38, 39]. Для обеих реакций
опытным путем были найдены константы соответствующих рав-
новесий при нескольких температурах; затем были выведены за-
коны изменения этих констант с температурой; наконец, при
помощи термодинамических соотношений были рассчитаны кон-
станты равновесий, а также степени диссоциации циклогексана
и метилциклогексана для широких пределов температуры и да-
вления. В табл. 135 приводятся некоторые данные для характе-
ристики диссоциации циклогексана -или, что то же, реакции
гидрирования бензола.
5S8
Таблица 13S
Диссоциация циклогексана
(по Фросту)
Температура Процент бензола в системе бензол-циклогексан при давлении водорода
1 ат 10 ат 50 ат 100 ат 200 ап
200° 0,04 4-10”Б 3 • 10-’ 4 • 10-8 6-10-*
250 ‘ 6,94 7-Ю-» 6 • 10-5 710-6 9-Ю-ч
300° 85,00 0,56 4,6 • IO”3 6•10-* 7-10-5
350° 99,54 18,00 0,18 0,02 3-10-»
400° 99,98 83,80 3,86 0,50 0,06
450° 99,99 98,70 37,80 7,07 0,95
500° 100,00 99,90 86,70 44,80 9,20
Мы видим, таким образом, что в обычных условиях гидроге-
низации (400—600°) лишь при достаточно высоких давлениях во-
дорода (200 аг и выше) гидрирование является полным; .при
нормальном же давлении водорода, наоборот, практически весь
циклогексан является диссоциированным на бензол и водород
уже при 350°.
Аналогичную картину диссоциации в соответствующих усло-
виях температуры и давления дает метилциклогексан, лишь
с несколько большим сдвигом равновесия в сторону ароматиче-
ского углеводорода. Вероятно, для высших гомологов бензола
такого рода сдвиги будут- выражаться еще более явственно
[39, 40].
Как было разобрано в предшествующей главе, бензол уже
при температурах ниже 500° начинает уплотняться с образова-
нием дифенила. С другой стороны, дифенил при нагревании
в атмосфере водорода под давлением уже при 450—480° легко
превращается обратно в бензол [41]. Таким образом и в данном
случае мы имеем, очевидно, равновесную систему:
2С6Нвщ±
н2+ свн5 • С6Н
с-
Это равновесие в настоящее время также хорошо изучено не
только с качественной, но и с количественной стороны [39, 42]:
были непосредственно измерены константы равновесия для тем-
пературы от 840 до 920°; далее те же константы были вычи-
слены для интервала температур 45—320° на основе третьего на-
чала термодинамики. Построенная на этих данных кривая по-
зволила вычислить зависимость константы равновесия от тешк-
ратуры и рассчитать процент’конденсации бензола в различных
условиях опыта (табл. 136). ,
584
5 I
Таблица 136
Конденсация бензола в дифенил
(по Фросту)
Темпера- тура в °C Конден- сация бензола В п/о Р дифенила в % к Р бензола
Р бензола —0,1 1 ат 10 ат
Р водорода —0,1 1,0 10 100 1,0 10 103
300° 32 5»6 5,6 0,56 0,06 56 5.6 0,56
400° 34 6.3 6,3 0,63 0,06 63 6Д 0,63
500® 35 7.6 7.6 0,76 0,08 76 7,8 0,76
600° 37 8.9 8.9 0,89 0,09 89 8,9 0,89
800° 43 14,0 14,0 1,40 0,14 138 14,0 140
1000’ 50 25,0 25.0 2,51 0,25 251 25,0 2,50
Данные табл. 136 показывают, что в условиях отсутствия
отвода водорода и дифенила (столбец 2) процесс конденсации
бензола имеет значительную величину и медленно -растет с по-
вышением температуры. Избыток водорода препятствует конден-
сации, так что при увеличении парциального давления Р водо-
рода до 100-кратного давления паров бензола конденсация по-
следнего в дифенил сходит до десятых долей процента. Вообще
же, процент конденсации при данной температуре обратно про-
порционален отношению давления водорода к давлению бензола
Реакция конденсации бензола идет крайне медленно. Однако
в процессах, протекающих продолжительное время, особенно же
в присутствии веществ, ускоряющих конденсацию, а также в от-
сутствии водорода 'реакции конденсации бензола и его гомологов
могут пойти очень глубоко. Если же принять во внимание, что,
как показывает расчет, дальнейшая конденсация дифенила в бо-
лее сложные продукты (дифенилбензол и т. д.) термодинами-
чески вполне оправдывается и обладает константами равновесия,
близкими к константе конденсации бензола в дифенил, то ста-
новится понятным, что в благоприятных условиях (крэкинг)
конденсация ароматики может привести к продуктам очень глу-
бокого уплотнения, вплоть до карбоидов. Наоборот, под сильным
давлением водорода реакции рассмотренного типа практически
могут совсем прекратиться, что, как мы видим, также находится
в полном соответствии с опытом (гидрогенизация).
Значительно сложнее протекает деструктивная гидрогениза-
ция бензола и его гомологов в присутствии двухсернистого мо-
либдена под высоким давлением водорода. Как показали иссле-
дования последнего времени [29], в этих условиях простейшая
ароматика претерпевает одновременно, кроме нормальной гидро-
генизации до циклогексана и соответствующих его гомологов,
следующие превращения:
585
1) изомеризацию бензольного -ядра в пятичленный цикл
в результате чего из бензола получается метил-циклопентан’
а из гомологов бензола — другие, высшие гомологи циклопен-
тана;
2) дециклизацию бензольного ядра, иначе говоря, его раскры.
тие; в итоге продукты гидрогенизации бензола и его гомологов
полученные в указанных выше условиях, содержат более или
менее значительные количества парафинов;
з) отщепление боковых групп, например от толуола--ме-
тильной группы, от этилбензола — этильной и т. д.; в итоге
происходит обогащение гидрогенизата бензолом и продуктами
его деструктивной гидрогенизации.
Оптимальные условия для изомеризации и раскрытия бен-
зольного ядра (пп. 1 и 2) — примерно 400° при начальном да-
влении водорода около 140 ат. При более высокой температуре
(около 470°) большое значение приобретает отщепление боковых
групп (п. з). В табл. 137 сведены 'результаты деструктивной
гидрогенизации бензола, толуола и этилбензола в присутствии
двухсернистого молибдена, в отношении группового состава
основных фракций полученных гидрогенизатов после удаления
из них не вошедшей в реакцию ароматики.
Таблица 137
Гидрогенизация бензола, толуола и этилбензола
(400°, 140 ат начального давления, 1—2 час., 10% MoS2)
(по Пучкову)
Исходные углеводороды Состав гидрогенизата в %
шестичленные нафтены пятичленные нафтены парафины
Бензол 37,5 57,1 5,4
Толуол 34,9 51.4 18,6
Этилбензол . . . 89,0 46,0 15,0
Для установления полной картины процессов, имеющих ме-
сто при гидрогенизации различных нефтепродуктов, существенно
знать поведение в данных условиях не только простейшей аро-
матики, но и более сложных ароматических углеводородов, в ча-
стности ди-, три-- и полифенилированных, а также конденсиро-
ванных ароматических систем. Однако до сих лор в этих напра-
влениях имеются данные лишь качественного характера [43].
Гидрогенизация большинства полифенилированных аромати-
ческих углеводородов в условиях высоких температур протекает
сравнительно легко, особенно в присутствии катализаторов. Про-
дуктами этих реакций являются продукты крэкинга исходах
систем — низшая ароматика. Так например, трифенилметан при
нагревании до 500° при начальном давлении водорода 60 аг
распадается на дифенилметан и бензол:
(СеНе)ьСН + Щ = (СвН:)2СН, + С6Нв.
В аналогичных условиях гидрогенизация дифенилметана
приводит также -к образованию бензола; равным образом, про-
дуктами гидрогенизации дифенилпропана и дифенилбутана
является низшая ароматика.
Сложнее протекает гидрогенизация конденсированных арома-
тических систем в условиях высоких температур и давлений.
Как было упомянуто в предыдущей главе, системы эти, вообще
говоря, весьма устойчивые, при нагревании под давлением соб-
ственных паров до 500° и даже ниже легко превращаются в кок-
оообразную массу. Таково, например, поведение в означенных
условиях нафталина и антрацена, причем в первую очередь ана-
логично образованию дифенила из бензола и здесь происходит
сдваивание исходной молекулы с выделением водорода: нафта-
лин, СюН8, превращается в динафтил, СгоНи; антрацен, CuHw,—
в диантрил, С28Н18. В условиях гидрогенизации конденсирован-
ные системы уже не образуют кокса, и процесс протекает по
следующей схеме: сначала одно из крайних бензольных колец
системы гидрируется, например нафталин превращается в тетра-
лин (тетрагидронафталин) и т. п.; затем прогидрированное
кольцо разрывается, и с отщеплением боковых групп может обра-
зоваться низшая конденсированная система; последняя снова
гидрируется по месту одного из своих крайних бензольных ко-
лец и т. д., пока в результате последнего гидрирования я после-
дующего распада прогидрированного кольца не образуется про-
стейшая ароматическая система — бензол. Так например, для
фенантрена эта схема может быть выражена следующим обра-
зом:
Само собой разумеется, что полное отщепление боковых групп,
образующихся в. результате распада прогидрированных колец,
в действительности не наблюдается, вследствие чего основными
продуктами этого распада являются ближайшие -гомологи новой
образовавшейся конденсированной системы или бензола.
Подводя итоги, мы видим, что в процессе гидрогенизации выс-
шая. ароматика стремится к возможно большему упрощению.
587
своего состава, в конечном итоге — к превращению в бензол или
его низшие гомологи. В этом заключается глубокое отличие
гидрогенизации от крэкинга, ib процессе которого, наоборот, низ-
шая ароматика путем реакций уплотнения стремится к превра-
щению в более сложные ароматические системы, в конечном
итоге — в карбоиды (кокс).
Нафтены. Данные о поведении нафтенов в условиях гидроге-
низации крайне недостаточны и в основном сводятся к следую-
щему.
Циклопентан при гидрировании в присутствии катализатора
Pt превращается в нормальный пентан. Аналогично ведут себя
гомологи циклопентана, превращаясь при гидрогенизации в го-
мологи пентана изостроения [44]. Тем самым получают вероят-
ное объяснение высокие антидетонационные свойства бензинов
гидрогенизации при несомненном парафинистом их составе
(ср. табл. 126). При более высоких температурах (460° и выше)
гомологи циклопентана довольно легко отщепляют метильные и
иные группы, причем такой распад бывает тем легче, чем больше
относительный вес боковой парафиновой труппы данного угле-
водорода.
Из шестичленных нафтенов циклогексан, CeHi2, довольно
устойчив при 460°; но уже при 490° он частью изомеризуется
в метилциклопентан (примерно на 20%), частью же (па 15%),
претерпевает глубокий распад с образованием метана. Гомологи
циклогексана менее устойчивы при повышенной температуре,
особенно в присутствии различных катализаторов (NiO, MoSj и
др.). В итоге термические превращения циклогексана и его го-
мологов могут итти по следующим основным направлениям:
1. Дегидрогенизация шестичленных нафтенов до соответ-
ствующих ароматических углеводородов. Для циклогексана и его
гомблогов в присутствии активных катализаторов (Pt, Ni) такая
ароматизация, как известно, происходит легко уже при 300°.
Однако в условиях деструктивной гидрогенизации, т. е. при тем-
пературах порядка 400—450Q и под давлением водорода, это на-
правление реакции приобретает подчиненное значение и зача-
стую практически 'сводится на-нет.
2: Отщепление боковых групп. При деструктивной гидрогени-
зации особенно легко происходит отщепление длинных боковых
цепей, которые распадаются затем по типу термического распада
углеводородов жирного ряда. Несколько труднее отпадают более
простые боковые группы; они дают начало. соответствующим
простейшим углеводородам жирного ряда (метан, этан и т. л.).
В итоге одновременно происходит обогащение продукта деструк-
тивной гидрогенизации замещенных нафтенов продуктами де-
струкции циклогексана.
з. Глубокий распад циклогексана и его гомологов. Такой
распад может наступить для гомологов циклогексана вслед за
отщеплением у них боковых групп, особенно под влиянием
«дробящего» действия некоторых металлических катализаторов
(активный Ni). В условиях обычной деструктивной гидрогбяиЗа-
588
дии (температура порядка 450°) глубокий распад тпестичленных
нафтенов является незначительным, приобретая заметные раз-
меры лишь лри более высоких температурах.
4. Изомеризация шестичленных нафтенов в пятичленные. Это
направление реакции имеет при деструктивной гидрогенизации
циклогексана и его гомологов, повидимому, первостепенное зна-
чение, особенно в присутствии некоторых катализаторов (M0S2),
как показали это исследования последнего времени.
5. Дециклизация шестичленных нафтенов, т. е. раскрытие их
шестичленного кольца с образованием соответствующих гомоло-
гов ряда метана.
Весьма тщательно была изучена за последнее время деструк-
тивная гидрогенизация циклогексана и некоторых простейших
его гомологов в присутствии двухсервмстого молибдена (MoS«).
Оказалось, например, что циклогексан при длительном (26 час.)
нагревании в присутствии 10% MoS2 при 400° и начальном да-
влении водорода 140 ат (оперативное давление 365—325 ат) дал
гидрогенизат, который при анализе показал следующий со-
став [29]:
Метилциклопентан...............62% (по объему)
Норм, гексан.................19 , „
Изогексаны...................15 , „
Циклогексан....................4, „
Ароматических углеводородов в этом гвдрогенизате практи-
чески не содержалось.
Аналогичные результаты были получены.в соответствующих
условиях при деструктивной гидрогенизации метилциклогексана
и симм. триметилциклогексана. Таким образом в указанных
выше условиях деструктивная гидрогенизация циклогексана и
его ближайших гомологов в основном идет по направлениям,
указанным в пп. 4 и 5, т. е. по линиям изомеризации шести-
членного цикла в пятичленный и дециклизации, т. е. раскрытия
цикла. Интересно отметить, что дециклизация шестичленных
нафтенов идет, повидимому, по обоим возможным здесь напра-
влениям, как это видно из состава парафиновой части продукта
гидрогенизации циклогексана, а именно: по линии раскрытия
шестичленного цикла данного нафтена с образованием норм,
гексана и по линии раскрытия пятичленного цикла метилцикю-
пентана с образованием изогексанов.
Пять основных .разобранных выше направлений термического
распада циклогексана и его ближайших гомологов под высоким
давлением водорода сохраняют свое руководящее значение также
и для более сложно построенных нафтеновых систем. Так, де-
структивная гидрогенизация полинафтенов типаСбНп(СН2)лСвНц,
где п = о, 1, 2 и т. д.г в присутствии катализатора (А120з + FeiOs)
приводит к образованию простейших парафинов, в конечном
итоге метана, СН4, и простейших нафтенов, например циклогек-
сана, С6Н12, по следующей схеме [45]:
С6Н, ,(С ну,, • С0Н,,+(п+1) Н,=иСН. + 2СвНи.
589
Естественно, что вслед за этой начальной фазой деструктив-
ной гидрогенизации полинафтена наступают последующие ее
этапы, «когда распаду подвергаются уже молекулы образовавше-
гося простейшего нафтена, направления же их распада опреде-
ляются всей совокупностью условий, в которых протекает про-
цесс (температура, давление, катализатор и т. д.).
Что касается, наконец, конденсированных нафтеновых систем,
то о поведении их в процессе деструктивной гидрогенизации
можно судить по декалину — простейшему примеру углеводоро-
дов этого рода. Декалин при 450° и 100 ат начального давления
водорода расщепляется с образованием циклогексана и его про-
стейших гомологов (16%) и метана (3%). При дальнейшем углу-
блении процесса сюда присоединяются, очевидно, продукты де-
структивной гидрогенизации циклогексана и его ближайших го-
мологов в зависимости от условий реакции. Более сложные кон-
денсированные нафтены распадаются по аналогичным схемам,
напоминающим, очевидно, схемы распада при деструктивной
гидрогенизации ароматических конденсированных систем (ср.
выше).
ПОВТОРНАЯ ГИДРОГЕНИЗАЦИЯ
Как было отмечено выше, однократная гидрогенизация того
или иного нефтепродукта приводит к некоторому предельному
выходу бензина гидрогенизации (40—50%); при более глубоком
процессе этот выход начинает уменьшаться за счет разложения
бензина на газы (см. табл. 128). Однако практически достижение
такого выхода представляется нерациональным, так как при
этом заметная часть образовавшегося бензина гидрогенизации
уже успевает подвергнуться распаду и, таким образом, теряется.
Чтобы полнее использовать сырье и в конечном итоге получить
максимальные выхода на бензин, необходимо, как и при крэ-
кинге, прибегать к отгонке образовавшегося бензина, остаток же
подвергать новой переработке в прежнем направлении, т. е.
в данном случае — повторной гидрогенизации [30]. В табл. 138
приведены два примера такого рода повторной гидрогенизации,
а именно: грозненского парафинистого дестиллата без катализа-
тора и такого же полутудрона с катализатором (10% окиси ни-
келя). В обоих случаях после первичной гидрогенизации (I) про-
изводилась отгонка бензина до 200°, остаток же с температурой
кипения выше 200° подвергался вторичной гидрогенизации (II),
а затем, после новой отгонки бензина, — третичной гидрогени-
зации (III).
Как видно из таблицы, суммарный выход бензина после трех-
кратной гидрогенизации при практически полном отсутствии
коксообразования, в обоих случаях составляет 63—65% на
сырье, что значительно выше предельного выхода на тот же про-
дукт при однократной гидрогенизации. Однако эти 63—65 % от-
нюдь не исчерпывают всех возможностей метода гидрогенизации
в отношении данного сырья. Действительно, табл. 138 показы-
590
Таблице 138
Повторная гидрогенизация
(по ГрозНИИ)
№ повт. гид- рогенизация Время опыта, в час. Выход в % по весу на гидр, продукт Выход в % по весу на вех. продукт
газы п потери бензин до L00° 1 остаток выше 200° 1 карбо- иды газы и потеря бензин до 200° £8 см остаток выше 300°
Грозненский парафинистый дестиллат; темп. 425°, нач. давл. 150 ат
I 8 8,5 31,8 59,7 0.0 8,5 31,8 — — "1 »
11 6 14,8 41,8 43,4 0.0 8,8 24,9 — —
Ш 6 18,6 •33,1 52,9 0,4 3.5 8,6 9,8 4,5
Итог о . . . 20,8 65,8 9,3 4Л
Грозненский парафинистый полугудрон; темп. 425°, нач. давл. 200 ат
I 8 4,8 27,4 66,9 — 4,8 27,4 — —
И 6 13,8 37,8 48,4 —— 9 Я 25,3
III 6 24,5 32,3 43,2 — 7.» 10,5 8,7 ы
Итог О . . . 21,9 63,2 8Д м
вает, что после трехкратной гидрогенизации, например, парад-
кистого дестиллата получается около 15% остатка с температу-
рой кипения выше 200°, вполне пригодного для новой гидрогени-
зации. Некоторое дальнейшее увеличение выходов на бензин гидро-
генизации можно представить себе также за счет части потерь
(21%), всегда слишком больших при работе на -лабораторной
установке, т. е. в условиях получения цифрового материала
табл. 138. Если хотя бы ориентировочно ввести вое эти по-
правки, то получаем, что предельный выход бензина из парафи-
нистого дестиллата при ряде повторных его гидрогенизаций со-
ставляет примерно 77% по весу на исходное сырье, что хорошо
согласуется с литературными данными о выходах на бензин
гидрогенизации при работе на газойле (80% по весу на сырье).
Нельзя не обратить внимания на относительное увеличение
газообразования с каждой новой гидрогенизацией (табл. 138).
В этом отношении повторная гидрогенизация ничем не отли-
чается от процесса повторного крэкинга, где также наблюдается
постепенное увеличение газообразования.
Скорость образования бензина при повторных процессах гид-
рогенизации, как видно из данных той же таблицы, постепенно
падает. Однако падение скорости бензинообразования здесь да-
леко не так стремительно, как при повторном крэкинге (гл. III),
591
и это обстоятельство должно, конечно, находиться в связи с со
ставом продуктов повторной гидрогенизации.
Табл. 139, являющаяся дополнением к предшествующей, может дат
некоторое представление по этому вопросу в отношении грозненского пл
рафинового дестиллата. . ца>
Таблица 13S
Свойства продуктов повторных гидрогенизаций
(по ГрозНИИ)
№ пов- тори. гидроге- низации Продол- жительн. опыта в час- Уч- веса дестиллатов, d]5 Иодные числа
фракции до 200° фракции 200—800° остаток выше 300° фракции до 200° Фракции 200—300'*
Грозненский парафинистый дестиллат; темп. 425°, нач. давл. 150 ат
I 3 0,722 0,822 0,900 48 — - -
11 6 0,78» 0.860 1,01 19
III б 0,7 i 8 0,915 1,06 15 9
Как видно из данных таблицы 130, удельные веса отдельных фракций
продуктов повторной гидрогенизации систематически возрастают. Особенно
обращает на себя внимание повышение удельного веса остатка в темпера-
турой кипения выше 300°. Однако одновременно с этим иодные числа
продуктов повторной гидрогенизации, по крайней мере легких,. постепенно
снижаются, что указывает на постепенное исчезновение в них предельных.
Интересные результаты дает таюке сравнительная разгонка (при з лл)
продукта повторной гидрогенизации и исходного материала парафинистого
дестиллата (табл. 140).
Таблица 140
Сравнительная разгонка парафинистого дестиллата и остатка от его
вторичной гидрогенизации
(по ГрозНИИ)
Температурные пределы фракций при 3 мм Парафинистый дестиллат Остаток вторичной гидрогенизации
выход в % уд- вес (15°) выход в % уд. вес (16°)
До 200° 7,0 0,850 • 28,6 1,00
200 — 250° 42,5 0,868 42,9 1.06
250 — 300° 84,7 0,892 13,8 1,11
300 — 350° 4,6 0,907 11.2 —
Выше 350° ... ..... 10,7 0,929 2.0 ——
Как видно, фракционный состав остатков после гидрогенизации зна-
чительно легче состава исходного парафинистого дестиллата, откуда
явствует, что образования тяжелых, высокомолекулярных продуктов при
этом не наблюдается. Таким образом повышение удельного веса продуктрв
повторной гидрогенизации вызывается какой-то ино.й причиной, и таковой,
повидимому, может быть только постепенная ароматизация этих продуктов.
С этой точки зрения становится понятным отмеченное выше падение скр-
592
ростп бензинообразования при повторных гидрогенизациях. Очевидно,
накопление в продуктах повторной гидрогенизации более устойчивой аро-
матики не может не отразиться в данном направлении на скорости этого
процесса. Однако падение скорости процесса при гидрогенизации не может
быть столь резким, как при крэкинге, когда наряду с ароматизацией
наблюдается также образование тяжелых продуктов уплотнения, в конеч-
ном итоге карбоидов, а в этом и заключается одно из наиболее характерных
.отличий между крекингом, с одной стороны, и гидрогенизацией — с другой.
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ И ПРИМЕНЕНИЕ ГИДРОГЕНИЗАЦИИ
Разработка промышленных методов деструктивной гидроге-
низации топлива была проведена сначала в Германии, в тресте
I. G. (Interessengememschaft d. Deutschen Far benindus trie), кото-
рый владеет также патентами Б&ргиуса. Учитывая громадное
значение, которое должна иметь гидрогенизация для нефтяной
промышленности, ощн из крупнейших нефтяных трестов США,
Standard Oal О of New Jersey, вошел с трестом I. G. в соглаше-
ние и после дополнительной лабораторной и полузаводской про-
работки вопроса построил и пустил в ход три гидротенизацион-
ных завода с суточной производительностью около 700 т каж-
дый (1929 г.). Организованное одновременно акционерное обще-
ство Hydro-Patents С° еще в 1930 г. объединило около 80 % всей
нефтяной промышленности ОША, и уже одно это обстоятель-
ство показывает, каких исключительных результатов ожидало
промышленность от гидрогенизации как нового метода перера-
ботки нефти.
Как ясно из вышеизложенного, на сегодняшний день можно
наметить следующие шесть видов применения процесса гидро-
генизации в нефтяной промышленности:
1. Превращение тяжелых высокосернистых смолистых неф-
тей, а также нефтяных остатков в бензин и другие дестиллаты
с низким содержанием серы, не содержащие смолистых веществ.
2. Превращение низкосортных керосиновых и газойлевых де-
стиллатов в высококачественный керосин с одновременным обра-
зованием небольших количеств бензина.
3. Улучшение качества смазочных масел в отношении их
удельного веса, температуры вспышки, коксообразования и кри-
вой изменения вязкости с температурой.
4. Обессеривание различных дестиллатов, особенно бензинов,
с одновременным повышением их качества путем придания им
стабильности и повышения их антидетонационных свойств.
5. Превращение легких газойлей из парафинистых нефтей
в хорошие, стабильные бензины с низким содержанием серы и
хороших антидетонационных качеств.
6. Наконец, гидрогенизация предложена как метод стабили-
зации крэкинг-бензинов, так как путем легкого гидрирования
последних легко удается превратить их наименее стабильные со-
ставные части (диены и т. п.) в устойчивые углеводороды пре-
дельного характера.
Как метод переработки нефти гидрогенизация обладает рядом
выдающихся достоинств. Важнейшие из них следующие:
38 Зак. 1622. Химия нефти.
593
а) Гибкость процесса как в отношении сырья, так и в отно-
шении получаемой продукции. По существу ‘все перечисленные
выше применения гидрогенизации можно осуществлять на од-
ной и той же установке с самым разнообразным сырьем.
б) Качество сырья для гидрогенизации может быть весьма
низкое: тяжелые смолистые нефти, гудроны и т. п.; в результате
процесса это сырье превращается в бензин, керосин и масла.
в) Высокие выхода на конечный продукт, как, это явствует
из следующего примера: для получения 100 г бензина (путем
гидрогенизации достаточно 110 т сырья, тогда как при крэкинге
для той же цели требуется в среднем примерно 220 т.
т) Полное отсутствие коксообразования при надлежащим
образом отрегулированном процессе, благодаря чему рабочий
цикл установки может быть удлинен до 6—у мес. и больше.
д) Исключительно высокое качество получаемой продукции,
что особенно должно быть отмечено в отношении бензинов и
смазочных масел, конечно при надлежащем подборе сырья для
гидрогенизации. Полученные в этом отношении результаты от-
крывают инженеру авто- и авиапромышленности возможность
приступить к конструированию моторов, работающих с более
высокой компрессией, при более высоких температурах и с боль-
шими скоростями, достигая этим путем дальнейшего повыше-
ния коэфициента полезного действия.
Отметив высокие достоинства гидрогенизации как нового ме-
тода переработки нефти, укажем в заключение его едва ли не
единственный недостаток, препятствующий, однако, быстрому и
широкому его распространению. Недостаток этот заключается
в том, что гидрогенизацию топлива на сегодняшний день мы
умеем проводить лишь в очень жестких условиях температуры
и давления. Осуществление этих условий требует дорогой и до-
вольно сложной аппаратуры из специальных сортов стали, про-
тивостоящих не только указанным факторам, но также воздей-
ствию таких веществ, как образующийся в процессе гидрирова-
ния топлива сероводород. Состав и способы изготовления этих
сортов стали составляют секрет немногих фирм, которым удалось
разрешить задачу их приготовления; совершенно несомненно,
что даже американской нефтяной промышленности удалось
овладеть проблемой гидрогенизации в сравнительно короткий
срок лишь благодаря упомянутому выше соглашению с трестом
I. G., в руках которого скопился исключительной ценности опыт
по работе с высокими давлениями и температурами благодаря
его 'Предшествовавшим работам по синтезу аммиака и метанола.
В. Окислительный крэкинг
Третий специальный вид крекинга, подлежащий рассмотре-
нию в настоящей главе — окислительный крэкинг—имеет своей
задачей получение продуктов окисления нефтяных углеводоро-
дов за счет окисляющего действия на них кислорода воцдуха.
Таким образом окислительный крэкинг, или крэкинг в амо
594
сфере воздуха (кислорода), есть один из способов чисто химиче-
ской переработки нефти и ее дестиллатов.
Первые указания на возможность получения органических кислот пз неф-
тяных углеводородов находим у Энглера [4G]. В 1ка4 г. па получение кислот
окислением нефтяных углеводородов кислородом воздуха был заявлен па-
тент (Шааля), не нашедший, однако, практического применения [47]. Во
время мировой войны и в последующие годы, в связи с жировым кризи-
сом, этим вопросом занимался целый ряд выдающихся исследователей
Германии и Австрии, как-то: Бергманн [48], Ф. Фишер [49], Грюн [50],
в последнее время Цернер [51] и многие другие. Эти исследователи поль-
зовались в качестве сырья для окисления главным образом парафином —
нефтяным и буроугольным. Из русских исследователей вопрос окисления
нефтяных углеводородов кислородом -воздуха изучали Харичков [52],
а после 1917 г. — Мошкин, Тютюнников [53], Тычинин и Иванов [54],
Г. С. Петров, Данилович и Рабинович [55]. Благодаря работам ©тих иссле-
дователей лабораторный и полузаводекой этапы проблемы получения орга-
нических кислот путем окисления нефтяных углеводородов могут счи-
таться в СССР пройденными.
ТЕХНИКА ОКИСЛИТЕЛЬНОГО КРЭКИНГА
Окисление нефтяных углеводородов с целью получения орга-
нических кислот производится продувкой через подогретый неф-
тепродукт воздуха. Условия ©того процесса, рекомендуемые для
разных нефтепродуктов, различны.
Окисление парафина при обыкновенном давлении в лабора-
торных условиях удобно производить в стеклянном цилиндре
длиною 60 см и шириною 5 см. В цилиндр загружают 200 г па-
рафина и при температуре 160° энергично продувают через масло
воздух, со скоростью до 50б л/час. Летучие продукты окисления
улавливают охлаждаемыми ловушками, из которых одна- может
быть пустой, а две следующих наполнены водой или водным
раствором щелочи. Нелетучие продукты окисления остаются
в цилиндре; через 6 час. после начала продувки воздуха они
получаются в количестве 80—95% от взятого парафина в за-
висимости от условий опыта и, кроме одноосновных кислот жир-
ного ряда, содержат более или менее значительное количество
оксикислот.
Для опытов в более крупном масштабе удобно пользоваться
аппаратами из алюминия. Эти аппараты, построенные по тому
же принципу, как стеклянные, оборудуются наружным водяным
охлаждением для поддержания во время процесса постоянной
температуры; это — необходимо, так как окисление парафина
сопровождается значительным выделением тепла. Аппарат дол-
жен быть снабжен также достаточным числом -промывателей для
улавливания летучих продуктов окисления и хорошей воздухо-
дувкой, энергично подающей воздух, пропускаемый предвари-
тельно через счетчик, а после этого в реакционную камеру [56].
Другим удобным материалом для получения кислот может
служить вазелиновое масло [55]. Предварительная очистка со-
ответствующего дестиллата представляется крайне желательной,
так как удаление непредельных и смолистых веществ содей-
в8* 5^5
ствует получению более чистых продуктов окисления. Очистка
дестиллата можно производить обычными способами: серной
кислотой, адсорбентами и т. п. Последующее окисление вазели-
нового масла производится в общем так же, как парафина, но
в несколько иных температурных условиях, а именно: масло
доводится в реакционном сосуде до 90°, после чего начинают
продувание воздуха. Во все время процесса температуру поддер-
живают в пределах 95—98°; ввиду экзотер^ичности реакции и
здесь приходится применять охлаждение. Для обогрева масла
перед началом продувки воздуха и для его охлаждения во время
процесса удобно пользоваться одним и тем же глухим змееви-
ком, расположенным внутри реактора. Процесс окисления ре-
комендуется производить в данном случае в присутствии неко-
торых катализаторов, например нафтеновокислого марганца;
однако и в этих условиях окисление идет крайне медленно и
продолжается много часов. Так например, при окислении в ука-
занных условиях вазелинового масла, очищенного дымящей сер-
ной кислотой, анализ про-
Таблица 141 дукта окисления пока-
зал следующие результаты
(табл. 141).
Окисление вазелинового масла
воздухом
(по Петрову, Даниловичу и Рабиновичу)
Время окисления, Получено кислот
в час. в % по весу
15 2,09
20 7,33
3) 12,42
40 17,88
48 20,79
Так как при продолжи-
тельной продувке воздуха
в процессе окисления могут
принимать участие наряду
с углеводородами также об-
разовавшиеся кислоты (см.
ниже), то следует, прерывая
время от времени процесс
окисления масла, удалять
образовавшиеся кислоты
путем их выщелачивания,
неоскислившееся же масло подвергать дальнейшему окислению,
постепенно используя путем повторных переработок все подле-
жащее окислению сырье.
Значительно быстрее протекает окисление нефтяных угле-
водородов под влиянием кислорода воздуха при повышенном
давлении; ввиду этого • некоторые авторы рекомендуют прово-
дить этот процесс в автоклаве [49]. Однако усложнения аппа-
ратуры, связанные с применением довольно значительных да-
влений (до 30 ат), повидимому, не оправдываются достигаемыми
результатами, и способ получения органических кислот путем
окисления высокомолекулярных нефтяных и иных углеводородов
при повышенном давлении не получил практического приме-
нения.
Особый вид окислительного крэкинга представляет собою
крэкинг нефти, ее дестиллатов и разного рода смол при высокой
температуре и недостатке кислорода. Если вести такой процесс
в аппаратуре генераторного типа (ср. гл. III), то можно осуще-
ствить непрерывное получение крэкинг-бензина с образованием
598
лишь небольших количеств кислородных соединений в конечном
продукте и без заметного коксообразования. Таков, например»
разработанный в СССР крэкинг Дубровая. Эта система работает
при обыкновенном давлении и характеризуется поэтому сравни-
тельной простотой аппаратуры. Однако состав получаемого по
атому способу прессдестиллата, естественно, в высокой степени
зависит от условий процесса (температура, подача воздуха
и т. п.), соответственно чему очистка сырого дестиллата здесь
значительно осложняется.
АНАЛИЗ И РАЗДЕЛЕНИЕ ПРОДУКТОВ ОКИСЛИТЕЛЬНОГО
КРЭКИНГА
Главными продуктами окислительного крекинга нефтяных
углеводородов являются органические кислоты — одноосновные
и оксикислоты. И те и другие представляют собою продукты глу-
бокого распада исходного материала. Лишь наиболее легкие и
летучие из них собираются в ловушках, основная же масса
одноосновных кислот и все оксикислоты остаются в реакторе;
здесь же находится главная часть «неомыляемых» продуктов,
состоящих по преимуществу из не вошедших в реакцию угле-
водородов исходного нефтепродукта.
Отделение «неомыляемых» составляет первую задачу анализа
и переработки продуктов окислительного крэкинга. Такое отде-
ление достигается обработкой этих продуктов водной или спир-
товой щелочью, после чего «неомыляемые» частью удаляются
механически, как всплывший наверх слой, частью извлекаются
из щелочного раствора бензином.
Для аналитических целей б г окисленного продукта кипятят в течение
получаса в колбе с обратно поставленным холодильником с 25 сл* спирта,
и раствором 3 г едкого кали в возможно меньшем количестве воды. После
охлаждения щелочной раствор смешивают с 25 см* воды и 3—4 раза
взбалтывают е легким бензином (температура кипения-до 60°). Затем отгон-
кой растворителя сгущают бензиновые вытяжки, к остатку вновь приба-
вляют 25 см* спиртового раствора едкого кали и после разбавления водой
снова извлекают «неомыляемые» тем же растворителем. В заключение,
отгоняя растворитель, соединяют все бензиновые вытяжки во взвешенной
колбочке и, доведя остаток до постоянного веса, определяют «неомыляемые»
весовым путем.
Отделение неомыляемых для производственных целей, естественно,
должно быть по возможности упрощено; в общих чертах оно сводится
к следующему: окисленный продукт обрабатывают в течение нескольких
часов крепким раствором едкого натра; после отстаивания часть «неомыляе-
мых» всплывает наверх и отделяется, остальная же часть их извлекается
бензином при нагревании. После отгонки остатков бензина, абсорбирован-
ного щелочным раствором, последний разлагают серной кислотой, в ре-
зультате чего получается смесь органических кислот с содержанием около
5% «неомыляемых».
Следующим этапом анализа и переработки продуктов окис-
лительного крэкинга являются определение и отделение окси-
кислот, представляющих собою один из постоянных продуктов
данного процесса. Для суждения о содержании связанных окси-
кислот (см. ниже) может служить эфирное число анализируемого
597
продукта, т. е. число миллиграммов едкого кали, которое не-
обходимо затратить для нейтрализации связанных органических
кислот, содержащихся в 1 г вещества.
«Эфирное число» обыкновенно определяют простым вычитанием кислот-
ного числа (см. ч. I, гл. VII) из числа омыления. Последнее представляет
собою число миллиграммов едкого кали, которое идет на нейтрализацию
всех содержащихся в 1 г вещества кислот — свободных и связанных. Для
его определения 1,5—2 г испытуемого вещества нагревают с водно-спитгго!
вой щелочью в течение У2—1 часа в колбочке с притертым холодильником
(трубкой). Параллельно ставится слепой опыт в совершенно таких же усло-
виях. Затем охлаждают растворы и титруют пх полунормальной соляной
кислотой в присутствии алкали-бляу как индикатора. Число омыления
получается из этих данных по формуле:
(от —п)»28
А
где А — навеска анализируемого вещества, а т и п — число куб. санти-
метров полуформальной соляной кислоты, которые пошли на титрование
в слепом опыте и в опыте с анализируемым веществом.
Оксикислоты представляют собою продукты дальнейшего, бо-
лее глубокого окисления исходного материала. Так как все по-
пытки задержать процесс до стадии их образования остаются
пока безуспешными, то на сегодняшний день одной из основных
задач переработки сырых кислот, получаемых окислением неф-
тяных углеводородов, является возможно полное удаление из
них оксикислот. В производственных условиях эта задача ре-
шается проще всего обработкой сырых кислот 15—25 объемами
бензина. Жирные кислоты переходят при этом в раствор, из ко-
торого они выделяются затем отгонкой растворителя. Оксикис-
лоты не растворимы в бензине и получаются при обработке
сырых кислот бензином в осадке. Количество бензина, необходи-
мого для отделения оксикислот, может быть значительно сни-
жено (до 4—5 объёмов), если смесь сырых кислот с ‘растворите-
лем насытить хлористым водородом; и в этом случае жирные
кислоты остаются в растворе, оксикислоты же выпадают в виде
осадка.
До сих пор речь шла об анализе и разделении наиболее тя-
желых продуктов окислительного крэкинга, остающихся в реак-
торе. Вместе с не вошедшими в реакцию углеводородами эта
часть в зависимости от продолжительности процесса окисления
составляет от 40—50 до 75—80%, считая на общую массу про-
дуктов реакции. Остальная, более летучая часть продуктов окис-
ления перегоняется в ловушки. Здесь собирается небольшое ко-
личество отогнавшихся углеводородов, низшие кислоты жирного
ряда, а также недоокислившиеся продукты, альдегиды и спирт.
Разделение этих продуктов легко достигается обычными мето-
дами: кислоты отделяются промывкой слабой щелочью, альде-
гиды— переведением через бисульфитное соединение, наконец
спирты—обработкой фталевым ангидридом или борной кисло-
той через соответствующие сложные эфиры.
598
ПРОДУКТЫ ОКИСЛИТЕЛЬНОГО КРЭКИНГА
Итак, продуктами окислительного крэкинга являются одно-
основные кислоты, оксикислоты, альдегиды и спирты. Необхо-
димо несколько ближе рассмотреть их химическую природу и
их значимость как продуктов данного технологического про-
цесса.
Кислоты. Химическая природа кислот, получаемых при
окислительном крекинге, всецело зависит от природы исходного
сырья.
При окислительном крэкинге парафина несомненно полу-
чаются одноосновные кислоты жирного ряда. Так например,
среди продуктов окисления парафина кислородом воздуха под
давлением оказался ряд твердых кислот, которые после тщатель-
ной очистки по своему составу и температуре плавления хо-
рошо подошли [49] к следующим кислотам жирного ряда нор-
мального строения:
Формула Название кислоты Темп, плавл.
CtfHpgOs Тридекановая................... 40°, 5
СюНэдОз Пентадекановая.................. 5F
С17Н34О2 Маргариновая................... 60°
С19Н33О2 Нонадекановая.................. 66°, 5
Эти данные показывают, что высшие кислоты жирного ряда,
получающиеся при окислении парафина под давлением, содер-
жат нечетное число углеродных атомов в частице. Было бы,
однако, неправильно делать отсюда какие-либо выводы и обобще-
ния, так как высшие продукты окисления парафина кислородом
при обыкновенном давлении, как показало их исследование [57],
состоят, как раз наоборот, по преимуществу из кислот с четным
числом углеродных атомов. Среди них были обнаружены сле-
дующие кислоты:
Формула Название кислоты Темп, плавл.
СюНэдОг Каприновая *.................. 81°,4
^14^28^2 Миристиновая.................. 54°
С1вНйО2 Пальмитиновая................. 62°,6
С18НзбО2 Стеариновая................... 69°,3
СэдЩоОа Арахниновая................... 77°
и друг.
Совершенно иной оказалась химическая природа однооснов-
ных кислот, получающихся при окислении кислородом воздуха
эмбенского вазелинового масла 58 . В отличие от кислот, полу-
чаемых при окислении парафина, одноосновные кислоты,
являющиеся продуктами окисления вазелинового масла, оказа-
лись жидкими. Для установления природы их основного ядра
широкая фракция этих кислот была проэтерифицирована, смесь
же полученных метиловых эфиров восстановлена металлическим
натрием по Бу во и Блану (ср. ч. I, гл. VII). Полученная та-
ким образом смесь первичных спиртов действием йодистого фос-
фора была переведена в смесь соответствующих иодюров; послед-
ние же восстановлены цинковой пылью в водно-спиртовой среде.
£99
Образовавшаяся смесь углеводородов не растворялась в серной
кислоте, не обесцвечивала брома и по своему составу соответ-
ствовала общей формуле моноциклических нафтенов, CJI.
Отсюда явствует, что одноосновные кислоты из эмбенского
липового масла представляют собою смесь нафтеновых кислот
что в полной мере отвечает химическому характеру исходного
сырья. Ближайшее строение этих кислот остается пока неиссле-
дованным.
Итак, одноосновные кислоты, получающиеся при окислитель-
ном крэкинге нефтяных углеводородов, в зависимости от хими-
ческой природы исходного сырья принадлежат либо к одно-
основным кислотам жирнбго ряда, либо к нафтеновым кислотам
Как уже было отмечено, одновременно образуются также про-
дукты более глубокого распада исходных молекул, а именно-
му равьиная, уксусная, пропионовая, масляная и другие низшие
кислоты жирного ряда, собирающиеся главным образом в ло-
вушках.
Оксикислоты. Как уже было указано, отделение оксикислот
от одноосновных кислот производится при помощи бензина.
Однако нерастворимостью в бензине характеризуются лишь сво
бодные оксикислоты; что же касается их внутренних ангидри-
дов (лактоны, лактиды), то при обработке бензином они перехо-
дят в раствор вместе с одноосновными кислотами, и содержание
их в этих последних может быть снижено лишь путем их -гидра-
тации, например путем нагревания с водой [58] с последующей
новой обработкой смеси бензином.
Ни одна из оксикислот, получающихся при окислении нефте-
продуктов кислородом воздуха, в индивидуальном виде пока не
выделена. По своей химической природе они представляют со-
бою, повидимому, смесь свободных оксикислот с их внутренними
ангидридами. Некоторые авторы [52] называют их также
«асфальтогеновыми» или «полинафтеновыми» кислотами (см.
ниже).
Оксикислоты представляют собою густые сиропообразные
жидкости с удельным весом больше единицы. Они легко раство-
римы в спирте, эфире, бензоле, хлороформе и сероуглероде: мало
растворимы в бензине. С уксусным ангидридом оксикислоты
образуют сложные эфиры; не реагируют с гидроксиламином,
-фенилгидразином и семикарбазидом, но восстановляют аммиач-
'ный раствор серебра, а также феллингов раствор. Крепкими
щелочами разлагаются уже на холоду с образованием асфаль-
товых веществ (карбоидов).
Молекулярный вес оксикислот в известной степени зависит,
естественно, от среднего молекулярного веса углеводородов
исходного нефтепродукта. Так как, однако, при окислении кис-
лородом воздуха происходит более или менее глубокий распад
молекулы исходного углеводорода, причем в зависимости от
строения окисляющихся углеводородов глубина этого распада
может быть существенно различной, то естественно, что одного
молекулярного веса оксикислот, получаемых при окислении того
• воо
или иного нефтепродукта, далеко недостаточно для суждения
о ближайшем соотношении между исходным материалом и про-
дуктами его окисления. Так например [52], при окислении
фракции бакинского керосина с температурой кипения 1G4—1бби
воздухом при 150° была получена смесь оксикислот, по своему
составу и молекулярному весу приблизительно подходившая
к формуле С24Н34О4; отсюда можно сделать вывод, что процесс
окисления сопровождался в данном случае уплотнением моле-
кулы исходного углеводорода. С другой стороны, при окислении
тем же методом бакинского машинного масла были получены
оксикислоты приблизительно с тем же молекулярным весом
(М ?^404); отсюда явствует, что либо окисление данного масла
протекает без уплотнения молекулы, либо, что более вероятно,
окисление сопровождается в данном случае глубоким распадом
исходных углеводородов, причем продукты окисления уплот-
няются далее с образованием соединений типа лактидов, т. е.
с удвоением системы.
Для характеристики оксикислот, получающихся при окисле-
нии эмбенского вазелинового масла, в табл. 142 приведены не-
которые их свойства после I и II очисток бензином.
Таблица 142
Свойства оксикислот
(по А. Д. Петрову и Иванову)
Свойства оксикислот После I очистки После II очистки
Кислотное число 88,3 96
Число омыления 143 169
Эфирное число 64,7 73
Иодное число 14 —
Ацетильное число 123 —
Процент лактонов 27 37
(по стеаролактону)
В отличие от одноосновных кислот оксикислоты при пере-
гонке даже в вакууме разлагаются. Путем повторной вакуумной
перегонки они нацело превращаются с отщеплением воды и
частично углекислоты частью в непредельные кислоты, частью
в непредельные углеводороды.
Альдегиды и спирты. Кроме кислот, среди продуктов окис-
ления нефтяных углеводородов кислородом воздуха легко обна-
руживаются также промежуточные продукты этой реакции —
альдегиды и спирты. Эти нейтральные, летучие продукты сосре-
доточиваются в так называемой «альдегидной фракции», т. е.
в продукте, получаемом из ловушек после обработки его для
отделения летучих кислот раствором соды или слабой щелочью.
При окислении, например, различных сортов парафина «альде-
гидная фракция» получается в количестве до 9%, считая на
601
окислившийся парафин; содержание же ее в Лову Шейном про-
дукте (дестиллате) нередко превышает 20%. • рц-
Техническая «альдегидная фракция» все еще содержит не-
которое количество свободных кислот, так что для ее дальнейшей
переработки необходимо повторное взбалтывание ее с раствором
соды до полного удаления кислых примесей. После этого при-
ступают к выделению альдегидов, для чего нейтрализованное
масло обрабатывают крепким раствором бисульфита. Образую-
щиеся бисульфитные соединения переводят в водный раствор
если нужно при подогревании, а не вошедшее в реакцию масло
(углеводороды, спирты) отделяют, извлекая остатки масла из
бисульфитного раствора взбалтыванием с подходящим раствори-
телем (бензол, толуол и т. п.). Наконец, разлагая бисульфитные
соединения содой, получают альдегиды в свободном состоя-
нии.
'Спирты после обработки «альдегидной фракции» бисульфи-
том остаются в смеси вместе с углеводородами. Их разделения
можно достичь различными методами. Наиболее простой и на-
дежный из них, так называемый «боратный метод», заключается
в следующем [59].
К бензольному раствору смеси спиртов с углеводородами при-
бавляют борную кислоту в количестве, несколько большем, чем
это требуется по предварительному анализу для превращения
всех спиртов в полные эфиры борной кислоты — B(OR)s. Смесь
нагревают на масляной бане в колбе с дефлегматором, медленно
отгоняя бензол, с парами которого, естественно, отгоняется также
вода, образующаяся в процессе этерификации спирта. Когда тем-
пература начнет подниматься выше 80°, перегонку переносят
в вакуум и продолжают отгонку более легких продуктов до тех
пор, пока, судя по температуре кипения, не прекратится отход
углеводородной части смеси. В остатке получается смесь борных
эфиров спиртов, находившихся в исходной смеси. После омыле-
ния щелочью эти спирты получаются в свободном виде в слож-
ной смеси; для выделения из этой смеси отдельных представи-
телей ее подвергают тщательной фракционировке, а их иденти-
фикация производится обычными методами, например окисле-
нием в соответствующие кислоты и т. д.
Ближайшее изучение показало, что состав «альдегидных
фракций», получаемых при окислении различного сырья, весьма
существенно зависит от состава этого сырья.
«Альдегидная фракция» от окисления парафина и парафи-
нового гача, как оказалось [60], содержит ряд альдегидов жир-
ного ряда, повидимому нормального строения. Среди них не-
сомненно присутствуют, например, ценный этантол, С7НмО,
а также высшие его гомологи с 8, 9 и т. д. до 17 атомов углерода
в частице. После определения этих альдегидов из остатка был
выделен весь ряд нормальных спиртов жирного ряда, начиная
с пропилового, СзН7ОН, и кончая октадециловым, СквНэтОН.
Выхода на эти нейтральные продукты окисления парафина ока-
зались следующие: на спирты — до 12%, на альдегиды же, очё-
602
видно в связи с их меньшей устойчивостью к окисляющему дей-
ствию кислорода, — несколько меньше, а именно около 8%?.
Совершенно иная картина получилась при исследовании
«альдегидной фракции» от окисления вазелинового масла. Ока-
залось, что фракция эта почти исключительно состоит из угле-
водородов; спиртов в ней содержалось только 1%, альдегидов
же лишь 0,6%. Таким образом как по качеству кислот, полу-
чаемых при окислении кислородом воздуха, так и по выходам
на побочные продукты этого процесса — альдегиды и спирты,—
парафин (гач) в качестве сырья для окислительного крэкинга
имеет несомненные преимущества перед вазелиновым маслом;
различие же в результатах, получаемых при их окислении кис-
лородом воздуха, легко объясняется различием в составе исход-
ного сырья: в то время как парафин состоит преимущественно
из углеводородов ряда метана нормального строения, главной
составной частью вазелинового масла, например эмбенского,
являются высокомолекулярные нафтены.
ХИМИЗМ ОКИСЛИТЕЛЬНОГО КРЭКИНГА
Окисление нефтепродуктов кислородом воздуха при повы-
шенной температуре представляет собою дальнейшее углубление
реакции, давно известной под именем аутоксидации. Для раз-
личных углеводородов этот процесс самопроизвольного окисле-
ния под влиянием кислорода воздуха протекает существенно
различно.
Давно уже экспериментально устновлено [61], что первыми
продуктами аутоксидации непредельных соединений, в частности
непредельных углеводородов, являются перекиси (Бах, Энглер).
Эти перекиси («молокиси»), образующиеся уже при обыкновен-
ной температуре в результате присоединения молекулы кисло-
рода по месту кратной связи, представляют собою либо густые
сиропообразные жидкости обыкновенно красновато-желтого
цвета, либо порошкообразные продукты. Это—чрезвычайно ре-
акционно-способные, нередко взрывчатые соединения. С водою
они образуют перекись водорода; из раствора йодистого калия
выделяют иод; обесцвечивают синий раствор индиго; окисляют
мышьяковистую кислоту в мышьяковую и т. п. При обыкно-
венной температуре образование их идет обыкновенно крайне
медленно и в отдельных случаях продолжается месяцами. Так
например, при аутоксидации 20 г циклогексена по уравнению:
СН,—СИ,—СН сн,—сн3—сн—о
I I +02 = 1 I I
СН,-СН,-СН сн,—сн,—сн—о
поглощение кислорода продолжалось 151 день, причем окисли-
лась примерно только половина взятого циклогексена [62].
Аналогично аутоксидации непредельных может происходить
окисление кислородом воздуха ароматических углеводородов по
месту ароматического ядра. Очевидно, п в этих случаях можно
60S
представить себе в первую очередь присоединение кислород»
с образованием соединения перекисного типа, которое р?
гается затем с распадом бензольного кольца. Таковы, напримеп
нашедшие в последнее время практическое применение реакт
окисления бензола в малеиновую кислоту (I) и нафталина»)
фталевую кислоту (II), легко протекающие в присутствии окис-
лов ванадия при действии кислорода:
,СО2Н
нс/
НС
Значительно более сложным является вопрос о механизме
окисления элементарным кислородом углеводородов и боковых
цепей предельного характера. Приходится констатировать, что
полного единства во взглядах здесь на сегодняшний день еще не
достигнуто, и в последующем изложении мы вкратце рассмотрим
две основных точки зрения на этот сложный химический про-
цесс.
Не подлежит сомнению, что предельные углеводороды подобно
непредельным поддаются воздействию элементарного кислорода
даже при обыкновенной температуре. Реакция эта, естественно,
протекает в данном случае еще более медленно, и среди продук-
тов ее не удается обнаружить начальных перекисей (молоки-
сей), а лишь продукты окисления вторичного и последующи
порядков. Тем не менее, учитывая несомненную неустойчивость
начальных продуктов взаимодействия углеводорода с Кислоро-
дом и обобщая развитые выше представления о процессе аутокси-
дации, можно представить себе, что мимолетное образование ко-
локиси происходит и в данном случае. Очевидно далее, что
в случае взаимодействия кислорода с углеводородом предель-
ного характера уже невозможно представить себе на основе
обычных представлений о валентности молокись как некоторую
молекулу; это — лишь некоторое состояние молекул углеводо-
рода и кислорода в момент их активного столкновения, вслед
за которым тотчас же должна наступать перегруппировка с об-
разованием новых, более устойчивых форм. В качестве таковых
можно принять, например, алкильные гидроперекиси, предста-
вляющие собою перекись водорода, в которой один из водородов
замещен на органический радикал. Так например, для случая
окисления элементарным кислородом метана эта первая фаза
реакции может быть выражена следующей схемой:
СН4 09 — [СН4О2] СН3ООН. :
604
• Гидроперекиси алкилов, изученные пока далеко недостаточно,
представляют собою мало устойчивые соединения, взрывающиеся
при самых разнообразных механических, термических и ката-
литических воздействиях. Лишь в совершенно чистом виде и
слабых концентрациях они могут выдерживать в нейтральной
среде довольно значительное нагревание. Основными продуктами
распада гидроперекисей алкилов являются карбонильные соеди-
нения, т. е. альдегиды и кетоны [63]. Так например, распад
гидроперекиси метила приводит к образованию муравьиного
альдегида и воды, по схеме:
СНзООН = СНгО + Н2О.
Следующей фазой процесса должна быть аутоксидация аль-
дегида, протекающая уже значительно быстрее. На примере бен-
зальдегида этот процесс был изучен особенно тщательно, причем
образование гидроперекиси бензоила, CeH5COOOII, доказано
в данном случае с полной несомненностью. Обобщая эту реак-
цию, можно представить себе, например, окисление муравьиного
альдегида с образованием в первую фазу также гидроперекиси,
распадающейся далее на муравьиную кислоту и кислород:
СНаО 4 02 -> [СН,002] -> НСОООН;
нсооон=нсоон+о.
Применяя эти схемы к высшим представителям углеводоро-
дов предельного характера, можно, казалось бы, составить пол-
ное представление о механизме их окисления, которое, как мы
видели, действительно приводит к образованию альдегидов и
кислот. Тем не менее, легко показать, что с точки зрения разви-
тых выше представлений некоторые явления, наблюдаемые при
окислении углеводородов предельного характера элементарным
кислородом, остаются совершенно непонятными и даже им про-
тиворечат. Их основной недостаток заключается в том, что в них
нет места для спиртов, которые, как было показано выше, явля-
ются одним из основных продуктов данного окислительного про-
цесса.
Другое представление о механизме окисления углеводородов
предельного характера элементарным кислородом, не отрицая
образования при этом процессе перекисей, рассматривает их как
продукты вторичной реакции, заключающейся в аутоксидации
образующихся в условиях реакции непредельных углеводородов
или углеводородных радикалов. Образование этих первичных
продуктов можно рассматривать как результат своеобразного
крэкинга исходных углеводородов, весь же процесс в целом —
как крэкинг окислительный. Рассмотрим прежде всего, в какой
степени возможен процесс распада (крэкинга) высокомолеку-
лярных углеводородов в условиях окисляющего действия на них
элементарного кислорода.
Как известно, процесс окисления углеводородов элементар-
ным кислородом — реакция сильно экзотермическая. Если, не-
смотря на крайнюю медленность течения этого процесса, все же
605
во избежание слишком большого поднятия температуры прихо-
дится вести эту реакцию при охлаждении извне, естественно
притти к выводу, что в местных центрах, где сосредоточен про-
цесс окисления, температура может подниматься до тех прем
лов (375—400°), при которых уже начинается крэкинг в жидкое
фазе. Правда, окисление парафина обыкновенно проводится при
температуре не свыше 150°, для некоторых же нафтенов, напри-
мер для 1,4-диметилциклогексана, окисляющее действие элемен-
тарного кислорода наблюдается даже при 100°; следует помнить
однако, что это лишь средняя температура продукта, поддержи-
ваемая за счет небольшого числа активных центров, в которых
протекает окислительный процесс. Естественно также, что
участки, прилежащие к этим центрам, могут приобретать темпе-
ратуру, значительно более высокую, чем средняя температура
всего продукта, и приближающуюся к температуре жидкофаз-
ного крэкинга.
Как было показано в предшествующей главе, крэкинг угле-
водородов предельного характера приводит к образованию не-
предельных углеводородов и, вероятно, свободных радикалов.
И в том, и в другом случаях дальнейшее образование перекисей
в атмосфере кислорода совершенно понятно, равно как и после-
дующее образование из этих перекисей спиртов, альдегидов и
кислот.
Действительно, известно, например, что перекись этила раз-
лагается с образованием уксусного альдегида и этилового
спирта [64] по уравнению:
СН3СН2 • О • О СН2СН8= СН3СН0 + СН3СН20Н.
Образование же подобного рода перекисей можно представить
себе как результат окисления соответствующих радикалов, воз-
никших в результате крэкинга соответствующего углеводорода,
например:
СНВ(СН2)„ СНа - CH2(CH2)W СН3 -> I
I - СН3(СНг),.СН/ + СН2(СН,)„СН8 I
I -» CHsCCH^CHaO-O-CHsCCH^CHs -» |
| -> CHe(CH,)n CHjOH+0НС(СНД» сн3
Изучение взаимодействия различных парафинов (от метана
до гептана включительно) с воздухом при постоянном объеме;
в кварцевом сосуде, соединенном капиллярной трубкой с ртрт
ным манометром, дало следующие важные результаты [65].
Оказалось, что давление смеси углеводорода с воздухом изме-
няется сначала до определенной температуры нагрева согласно
закону Гей-Люссака; затем происходит внезапный скачок давле-
ния, очевидно соответствующий моменту взаимодействия угле-
водорода с кислородом воздуха; далее, в известном интервале
температуры давление вновь изменяется согласно закону Гей-
Люссака, потом новый скачок давления и т. д. В общем, прй
постепенно повышающейся температуре таких скачков давления
606
наблюдается столько, сколько углеродных атомов содержится
в данном углеводороде, причем каждый скачок давления соответ-
ствует, очевидно, определенной ступени реакции. Аналогично
протекает окисление первичных спиртов жирного ряда, так что
на основании всего вышеизложенного приходится притти к вы-
воду, что процесс окислительного крекинга происходит не по
середине молекулы, как это представлено на предыдущих схемах,
а с краю, с отгоранием последнего углерода. Для нормального
пентана такое ступенчатое окисление может быть выражено сле-
дующей схемой:
СН8 • СНо • СНо • СНо СН3 -> СН3 СН2 • СНо • СН/ + СН1;
' СН8 • СН2 • сн2. СНо. о • о. сн*
СНо. CH2. CH2• СНоОН-4- СНоО
пли СН3 • СН9. СЩ. СНО H-CEjOH и т. д.
Как видно, и в том и в другом случаях перекиси являются
лишь вторичными продуктами данного окислительного процесса,
и в этом, как уже было указано, заключается характерная осо-
бенность представлений о механизме этого процесса как об
«окислительном крэкинге».
Хотя до окончательного решения вопроса о механизме окисления угле-
водородов предельного характера элементарным кислородом еще нехватает
опытных данных, тем не менее, в пользу представления об ©том процессе
как об окислительном крэкинге можно привести некоторые весьма убеди-
тельные соображения косвенного порядка, а именно:
1. Известно, что окисление элементарным кислородом хорошо очищен-
ных углеводородов предельного характера протекает крайне трудно и ме-
дленно. Путем предварительного нагрева или добавки продукта, уже нахо-
дившегося в данной реакции, окисление тех же углеводородов может быть
значительно ускорено. Таким образом ясно, что для начала нормального
течения реакции окисления элементарным кислородом необходимо созда-
ние в среде углеводорода активных центров, в которых процесс мог бы
полупить, свое начальное развитие.
2. При пропускании смеси воздуха с парами углеводорода ряда метана
(пентан, гексан) через нагретую трубку с платиной наблюдается образова-
ние наряду с такими продуктами окисления, как окись углерода, угле-
кислота, формальдегид и др., также непредельных углеводородов (амилен и
гексилен). Совершенно очевидно, что уже один факт образования этих
углеводородов в данных условиях представляет выдающееся значение для
суждения о механизме этого окислительного процесса.
Кроме спиртов, альдегидов и кислот, продуктами окисления
нефтяных углеводородов элементарным кислородом являются,
как мы видели, также оксикислоты. Поскольку ближайшее
строение этих соединений пока не известно, говорить о химизме
их образования можно, очевидно, лишь предположительно. По-
видимому, эти оксикислоты являются нормальными продуктами
дальнейшего окисления одноосновных кислот и, надо думать,
относятся к л-ряду. В таком случае их образование можно пред-
ставить. по следующей схеме: кислота (I), частично превратив-
шись в свою непредельную форму (II), окисляется по месту двой-
ной связи с образованием соответствующей молокиси (III); по-
следняя изомеризуется затем в кислоту перекисного типа (пер-
607
кислота, IV), которая, наконец, с выделением свободного кисло-
рода превращается в а-оксикислоту (V):
II
R. СЩСООН
III
/ОН
IV V
R • СН(0Н)С000Н R • СН(0Н)С00Н4-0
Наконец, их внутренние ангидриды должны образоваться
в таком случае по лактидной схеме:
НО • СН • СООН
носо-сн-он
R
О —СН—СО
= 2На0 + I I
со-сн—о
В какой степени эти последние схемы отвечают действитель-
ности, может показать лишь более детальное изучение химиче-
ской природы этих соединений.
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ И ПЕРСПЕКТИВЫ ОКИСЛИТЕЛЬНОГО
КРЭКИНГА
Как уже было указано, окислительный крэкинг прошел пока
лишь полузаводскую стадию своего развития. Таким образом,
говоря об его практическом значении, приходится расценивать
лишь те перспективы, которые сулит эта новая отрасль перера-
ботки нефти. Рассмотрим с этой точки зрения, какую практи-
ческую ценность могут представлять все три основных вида
продукции окислительного крэкинга: кислоты, нейтральные
продукту (альдегиды и спирты) и оксикислоты.
1. Главный, целевой продукт окислительного крэкинга—
одноосновные кислоты, твердые и жидкие, в зависимости от
исходного материала для их получения, может служить прежде
всего сырьем для мыловаренной промышленности. Однако мыла,
сваренные на этих кислотах, даже после предварительного отде-
ления большей части оксикислот, по своим качествам значи-
тельно ниже, чем мыла из обычного сырья — Ж1гвотных жиров.
По своей моющей способности они уступают так называемым
ядровым мылам, получаемым омылением жиров. Кроме того, они
имеют темный цвет и неприятный запах, обесцвечивание же и
дезодорация кислот, получаемых окислением нефтяных угле-
водородов, оказалось задачей весьма сложной и технически пока
не разрешенной (ср. ч. I, гл. VII). Ввиду всего вышеизложен-
ного заграничная промышленность отказалась от употребления
608
этих кислот как сырья для мыловарения [51]. Низшие рас'
римые в воде кислоты, получаемые из ловушек, могут найти
применение в текстильной промышленности для замены уксус-
ной кислоты.
2. Альдегиды и спирты, получаемые при окислении нефтяных
углеводородов в качестве побочных продуктов, должны предста-
влять выдающийся интерес и значение для экономической сто-
роны нового производства. Действительно, некоторые из этих
веществ находят широкое применение в современной парфюмер-
ной промышленности и ценятся в нем весьма высоко. Таковы,
например, нониловый и дециловый альдегиды, являющиеся со-
ставной частью целого ряда цветочных .композиций, в част-
ности — искусственного розового масла; таков же далее энанто-
вый альдегид или энашол, С7НиО, являющийся одним из исход-
ных материалов для получения так называемого жасмин-альде-
гида (запах жасмина).
Можно наметить и другие пути утилизации некоторых ком-
понентов «альдегидной фракции». Так например, содержащийся
в ней нормальный гептиловый спирт мог бы служить исходным
материалом для получения нормального гептана, являющегося
составной частью стандартной смеси, которой пользуются для
определения детонационных свойств бензина по так называемой
октановой шкале [66]. Наконец, грубо выделенная из альдегид-
ной фракции смесь спиртов могла бы служить хорошим стаби-
лизатором для спирто-бензиновых смесей, представляющих со-
бою весьма ценное антидетонационное топливо, для двигателей
внутреннего сгорания с высокой степенью сжатия.
Сказанного достаточно, чтобы утверждать, что хотя «альде-
гидная фракция» представляет собою лишь побочный продукт
при окислении нефтяных углеводородов элементарным кислоро-
дом, тем не менее, широкая и планомерная утилизация ее мо:
жет оказать решающее влияние на себестоимость целевого про-
дукта этого производства и, следовательно, на всю его эконо-
мику.
3. Утилизация оксикислот является наиболее трудной и
сложной задачей в проблеме использования продукции окисли-
тельного крэкинга. Некоторые перспективы здесь уже намечены
в направлении получения из оксикислот искусственной олифы,
лаков и пластических масс [53, 55]. Тем не менее, в целом раз-
решение этого вопроса — задача будущего.
Подводя итоги, мы видим, что окислительный крэкинг как
специальный метод химической переработки нефти имеет все
данные для широкого развития в ООСР. Вопрос о наилучшем,
сырье для этого производства с химико-технологической точки
зрения совершенно ясен: таковым должен быть парафин (гач),
и лишь недостаток этого продукта может оправдать применение
для этой цели какого-либо иного сырья1.
1 Ориентировка на вазелиновое масло, принятая у нас для получения
синтетических кислот, диктуется соображениями комплексной постановки
этого нового производства: имеется в виду получать одаовремеино синте-
609
39 Эаи. 1622. Химия нефти.
ЛИТЕРАТУРА
Добрянский А. Ф., Крэкинг с хлористым алюминием, ОНТИ (1938).
Эллис К., Гидрогенизация органических соединений, пер. с англ »» i
II и Ш, ОНТИ (1934-35). Выц*
.Деструктивная гидрогенизация топлив*, Сборник статей, НИС, ТЯХттплп
и ГЛАВГАЗ НКТП (1934).
Долгов Б. Н., Методы химического использования окислов углерода, ОНТИ
Дубровой К. и Шейнман А. Окислительный крэкинг, ОНТИ (1936).
Марек Л. Ф. и Ган Д. А., Каталитическое окисление < рганических оплгж
нений, пер. с англ., ОНТИ (1936).
1. Ральсон О. С., Безводный хлористый алюминий, пер. под ред. Тычинии
М.—Л. (1924).
2. Густавсон, .Ж. Р.Х. О." 18, 149 (1881); 16, 95, 235 (1884).
8. Зелинский Н. Д, .Неф’’. хоз.“ 1, № 9—12. 8 (1920); 2, № 5 —8, 44 (1921>
2, № 9—12, 1 (1921); 9, № 9, 872 (1925); см. также Гольдштейн и
еЭнллин, „Нефт. хоз." 17, № 8 (1936).
4. McAfe А. И., „Ind. Eng. Chem." 7, 737 (1915); также многочисленные патен-
ты этого автора; ср. Gross R.t A Handbook of Petroleum (1928) стр. 318,
5. Nenitzescu G. u.Dragann А., „Вег." 66, 1892 (1933); Cox, „Bui. soc. chim.’
87, 1549 (1925); Hunter a. Jone, „J. Am. Chem. Soc.“ 55, 1248(1933);
Тронов Б. и Григорьев, „Труды Сиб. физ.-техн. ин-та", т. I, вып. £
1 (1934); Grihnald et Stratford, „С. R.“ 178, 2149, 1924; Зелин-
ский Н. Д. и Смирнов В. А., „Нефт. хоз". 9, № 9, 372 (1925).
6. По опытам Баталовой Е. Г. и Голенова П. М. и МНИ им. И. М.
кина, Москва (1935).
7. Наметкин С. С- и Гуденко М. Г., „Ж. общей химии” 7, 768 (1937).
8. См. особенно: Сабатье II., Катализ в органической химии, Л. (1932).
9. Sabatier et Senderens, „Ann. ch." [8] 4, 319 (1905).
10. „Ж. P. X, О." 89, 607 (1907).
11. „Ber." 43, 1176 (1910); 44, 3423 (1911) и др.
12. „Вег." 88, 1406, 2414 (1905) и др.
13. „Вег." 42, 1627 (1909); 48. 3393 (1910) и др.
14. „Ж. Р. X, О." 88, 75 (1902); „Вег." 40, 1270, 1282 (1907) и др.
15. Новые идеи в области катализа. Сборник переводных статей под. ред.
Рогинского С. 3. и Фроста (1932); см. также Баландин А. А., „Ж. Р.
X. О." 61, 909 (1929); 62, 703 (1930).
16. Фишер Ф., Превращение углей в жидкое топливо, пер. о нем. Новосиль-
цева, М.—Л. (1926); Велъблинг X, Гидрирование топлива, Л. (1932);
Ellis G., Hydrogenation of organic, Substances, Нью-Йорк (1930).
17. Пучков П. В., „Petroleum" 82, № 15, 1 (1936) или „Химия твердого тол-
лива", 6,852 (1985); см. также Елюквин Н., Воронов Ф. и Прейс М.,
„Химия твердого топлива" 3, № 2—3, 135 (1932) и,4, 241 (1933);
Немцов М. С. и Фрост А. В., „Химия твердого топлива* 3, №7—8,
552 (1932).
18. „Bull." (2), 11, 275 (1869).
19. Фишер Ф., Превращение углей в жидкое топливо, М.—Л. (1926).
20. DRP, 301231 (1919); „Zeitschr. f. angew. Chem." 42, 400 (1924).
21. Велъблинг X., Гидрирование топлива и продуктов его перегонки, пер.
Лебедева А. Д, ОНТИ (1932), стр. 133 и сл.
22. Fischer F. u. Tropsch Н, „Вег," 59, 830, 832, 923 (1926); 60, 1330 (1927);
23. „Brennstoffchemie" 12, 225 (1931).
24. „Brennstoffchemie" 11, 489 (1930).
25. Елюквин Н. А. и Вольнов Ю. Н., „Химия твердого топлива" 4, № 4,355
(1933); Раппопорт Н.,Блюдов А., Шевякова Л., Француз Е., „Химия
твердого топлива" 5, № 3, 221 (1935).
тические кислоты как продукт окисления очищенного вазелинового’ масла
и сульфонафтеновые кислоты («контакт»)’ как побочный продукт от пред-
варительной очистки вазелинового масла крепкой серной киыкхкА
(см. ч. III, гл. I).
610
26. „Oil a. Gas Journ.* в, 1 (1938).
27. Sabatier et Sender ens, „С. R.“ 128,1173 (1899); Fischer F. u. Peters. Brenn-
stoffchemie* 12, 286 (1931).
38. Тейлор Гп Производство водорода, пер. под ред. Шульц В. Н., Л. (1980).
29. Пучков Н. В., О гавах гидрогенизации, .Химия твердого топлива" 7
172 (1936); 8,502 (1937); ею же, диссертация: Деструктивная гидро^
генивация углеводородов и тяжелых нефтяных i родуктов, М.
(1938); см. также „Ж. общей химии* 8, 11с8 и 1159 (1938) и другие*
работы того ясе автора.
Й). Соханов А. Н. и Тиличеев М. Д., Гидрогенизация нефтяных продуктов,
„Труды конференции по крэкингу и гидрогенизации", Грозный (1931).
31. Haslam R. Т. a. Bussell В. Р., „Nat. Petr. News,* 22, № 40, 58 (1930),
Haslam R. T. a. Bauer U7., .Refiner* 10, N. 2, 110(1981); Beall, .Re-
finer* 9, N. 8, 77; N, 9, 66; N. 10, 147 (1930); см. также .Ind. Eng.
Chem.* 24, 1129 (1932). 6
32. .Труды конференции по крэкингу н гидрогенизации,* Грозный (1931).
33. См. Ellis С., Hydrogenation of organic substances (1930); Сабатье П.,
Катализ в органической химии, гл. XX, Л. (1932).
34. Waterman Н. J. a. Perquins Н. J., „J. Inst. Petr. Pechn." 10, 670 (1924):
11, 42 (1925); 18, 413 (1927).
35. Pease R. H, a. Burgau E. 8., „J. Am. Chem. Soc.* 50, 2715 (1928).
36- Немцов M. С. и Фрост А. В., „Химия твердого топлива", 8, 588 (1932),
87. Francis „Ind. Eng. Chem." 20, 277 (1928).
38. Berrouss Ысаггпг, .J. Am. Chem. Soc." 49,1157 (1927); Жаркова и Фрост*
.Ж. общей химии* 2, 712 (1982).
39. Фрост А. В., .Химия твердого топлива" 4, 171 (1933).
40. Введенский А. А., Винникова, Жаркова и Фундилер, „Ж. общей химии"
3 (65), 718 (1933).
41. „Вег.* 60, 1955 (1927).
42. Введенский А. А. и Фрост А. В., „Ж. общей химии" 2, 542 (1932).
43. „Ж. Р. X. О." 60, 1448 (1928); Лихачев, „Ж. Р. X. О." в1, 1840 (1929); см.
также „Вег." во, 1950(1927); в2, 719 (1929); 68. 2179 (1930).
44. Зелинский, Казанский, Плате, „Вег." 6в, 1419 (1933).
45. .Ж. Р. X. О." 61, 1339 (1929).
46. Engler С, „Вег.* 12, 2186 (1879).
47. Schaal Е., DRP. 82705 (1884).
48. Bergmann, „Ztschr. angew. Chem.* 31, 69, 115, 148, 252 (1918).
49. Fischer F. u. Schneider, Gesammelte Abhand. zur Kenntnis der Kohle
(1920); подробный реферат Мошкина Л., „Нефт. хоз.* 2, № 9—12,
25 (1921) и 8, № 1-4,165 (1922).
50. Grun Ad., „Вег.* 58, 987 (1920).
51. Zemer, „Chem. Zt." 54, N. 27 (1980).
52. Харичков, „Ж. P. X. О.* 40, 1757 (1908); 41, 345 (1909).
53. Тютюнников, „Нефт. хоз.* в, 471 (1924).
54. Тычинин и Иванов, „Маслобойно-жировое дело" № 3—5 (1930).
55. Петров Г. Данилович и Рабинович, Развитие методов окисления
нефтяных и минеральных масел и техническое использование
получаемых продуктов, Госхимтехиздат (1933).
56. По отчетам проф. Мошкина', см. также Варламов, „Маслобойно-жировое
дело* № 2—3 (1931), стр. 38.
57. Keiber С., „Ъег.и 58, 66, 1567 (1920).
58. По данным Петрова А. Д. и Иванова, см. [55].
59. Шорыгин и Макаров-Землянский, “Ж. Р. X. О.* 62, 2047 (1930).
60. Наметкин и Звор-ыкина, „Ж. общей химии" 4, 906 (1934);
fl. Бах Л. Н., „Ж. Р. X. О-* 29, 378 (1897); Engler С., „Вег". 80, 1669 (1897);
Engler С. u Weisiberg J., Kritische Studien iiber die Vorgange der
Autoxidation (1904); Бах. А. И., „Успехи химии" 8, № 2, 177 (1934).
62. Зелинский и Борисов, „Ж. Р. X- О." 62, 2051 (1980).
63. Медведев и Алексеева, „Вег.и 65, 138 (1982).
64. Wieland И.. „Вег.* 68, 1028 (1930). _ _ /т
65. Minoku Akita, „Journ. Soc. Chem. Ind. Japan* (Supp.) 86,486, B. (Juli 1933).
66. Петров А. Д., Андреев и Чаплыгин, „Журн. прикл. химии." 5,578(1982).
39*
ЧАСТЬ III
ОЧИСТКА И ПОВЫШЕНИЕ КАЧЕСТВА
НЕФТЕПРОДУКТОВ
ГЛАВА I
ОЧИСТКА НЕФТИ И ЕЕ ДЕСТИЛЛАТОВ
ОБЩИЕ МЕТОДЫ
Третьей и последней стадией переработки нефти является
очистка, которая обыкновенно следует за перегонкой нефти,
редко ей предшествует. В первом случае очистке подвергаются
нефтяные дестиллаты с целью удаления из нит .разного рода
составных частей, понижающих качество нефтепродуктов: та-
ковы легко осмоляющиеся непредельные соединения, кислород-
ные и сернистые соединения нефти и т. п. Во втором случае
предварительной очистке подвергаются некоторые сырые нефт
в целях облегчения последующей их переработки, например на
высокосортные смазочные масла, а также для предохранения
нефтеперегонной аппаратуры от разъедания. В дальнейшем из-
ложении мы будем различать:
А. Общие методы очистки, которым посвящена настоящая
глава; сюда относятся наиболее широко применяемые способы
очистки нефтей и нефтепродуктов, а именно:
1) кислотная очистка и
2) щелочная очистка и промывка.
В. Специальные методы очистки, которые будут рассмотрены
в следующей главе.
С каждым из способов очистки связаны свои особые химиче-
ские вопросы. Поэтому каждый из них подлежит особому рас-
смотрению.
ТЕХНИКА ПРОЦЕССА ОЧИСТКИ
Кислотная очистка заключается в обработке нефти или ее
дестиллатов крепкой серной кислотой. С технологической точка
зрения различают периодическую и непрерывную очистку.
632
Периодическая очистка легких дестиллатов производится
в кислотных мешалках (фиг. 114), в которых перемешивание
осуществляется либо специальным механическим приспособле-
нием (бензин), либо энергичной продувкой воздуха (керосин,
масла). После отстаивания отделяют кислотный слой (кислый
гудрон) и повторяют очистку свежей порцией кислоты. Общий
расход серной кислоты (удельный вес 1,84) при очистке бензина
может достигать 1,5% \ при очистке керосина—0,5—1,0% по
весу от дестиллата. Кислота при очистке бензина и керосина
подается в два
приема: сначала
дают от Vs до Vs
кислоты и переме-
шивают смесь в те-
чение нескольких
минут; затем дают
кислоте отстояться,
спускают кислый
гудрон и подают
остальную кислоту,
после чего следует
новое перемешива-
ние, на этот раз бо-
лее длительное, а
именно для бензина
до 30 мин., для ке-
росина 1—2 час.
После тщательного
отстаивания, кото-
рое для бензина на-
ступает быстро, для
керосина же требует
несколько часов
(4—8 час.), отде-
ляют кислый гуд-
рон, чем в сущно-
сти и заканчивается
Очистка масляных дестиллатов или остатков (мазут, полу-
гудрон) производится также периодически в мешалках
•(фиг. 115). Однако по сравнению с очисткой легких дестиллатов
кислотная очистка на масла имеет некоторые существенные осо-
-бенности. Во-первых, очистка на масла требует значительно
большего расхода серной кислоты: уже для очистки веретенного
дистиллята ее расходуется 2—2,5%, для цилиндрового же де-
стиллата этот расход повышается до 4 и даже до 6%. Еще
-больше серной кислоты идет на изготовление некоторых спе-
циальных сортов масел (трансформаторное масло, paraffinum
Фиг. 114. Периодическая очистка легких дестил-
латов: А — кислотная мешалка; В — щелочная
мешалка:
а — ввод дестиллата;^—ввод воздуха с—ыонжу; d —спусж
гудрона.
кислотная очистка бензина и керосина.
1 Некоторые бензины прямой гонки, применяемые в качестве топлива
для двигателей внутреннего сгорания, вовсе не подвергаются очистке.
613
liquidum и др.), причем наряду с кислотой удельного веса 1,84
здесь находит применение также дымящая серная с содержанием
серного ангидрида до 20%. Вторая особенность сернокислотной
очистки масел заключается в применении обогрева, для чего
мешалки маслоочистных отделений снабжаются паровыми змее-
виками; так например, очистка цилиндрового дестиллата про-
изводится при подогреве до 60° и т. п. Эти же змеевики служат
для предварительной подсушки масляных дестиллатов, с чего
обычно начинается их очистка. Наконец, следует отметить, что
по сравнению с очисткой бензина и керосина очистка масляных
дестиллатов требует значительно больше времени: уже одно пе-
ремешивание дестиллата с кислотой требует здесь 2,5—з час.,
Фиг. 115. Периодическая очистка масляных дестиллатов
а — кислотная мешалка; Ъ — натровые чаны; с — промывочный чан; d - сушильный ящик
е — монжу.
отстаивание же продолжается до 12 час. и заканчивается лишь
в специальных отстойниках, снабженных змеевиками для обо-
грева.
•После кислотной очистки дестиллат всегда имеет кислую
реакцию, которая зависит частью от сохранившихся в нем сле-
дов серной кислоты, частью от органических кислот. Чтобы сде-
лать дестиллат нейтральным, как общее правило, его подвергают
после кислотной очистки промывке водой, затем обработке вод-
ным раствором щелочи, т. е. щелочной очистке, и, наконец, но-
вой промывке водой. Первая промывка водой освобождает де-
стиллат от минеральной кислоты, снижая тем самым последую-
щий расход щелочи; щелочная очистка удаляет из дестиллата
органические кислоты и делает его нейтральным, в чем убе-
ждаются обыкновенно реакцией на фенолфталеиновую бумажку;
наконец, заключительная промывка имеет своей задачей вымы-
614 '
ванне из масла зольных частей (мыла), которые нередко упорно
удерживаются очищенным маслом и, как будет показано ниже,
относятся к вредным примесям, снижающим его качество.
Промывка и щелочная очистка производятся в щелочных ме-
шалках, которые всегда ближайшим образом связаны с кислот-
ными мешалками (фиг. 115). Кислый дестиллат переводится из
кислотной мешалки в щелочную с помощью особой трубы, для
чего щелочные мешалки располагаются всегда несколько ниже
кислотных. Перемешивание и здесь производится либо продув-
кой воздуха, либо специальными механическими приспособле-
ниями. Отстаивание после промывки и щелочной очистки иногда
крайне осложняется вследствие образования эмульсий, для раз-
рушения которых в случае масляных дестиллатов приходится
прибегать к специальным приемам (нагревание и т. и.). Расход
едкого натра, обычно применяемого для щелочной очистки неф-
тяных дестиллатов, для бензина прямой гонки и крэкинг-бен-
зина в Баку не превышает 0,04—0,07%, для керосинового де-
стиллата составляет 0,1—0,2%, для масел же повышается до
0,5% по весу, считая на взятый в очистку дестиллат.
Наряду с периодической очисткой легких дестиллатов в на-
стоящее время все большее распространение приобретают спо-
собы их непрерывной очистки. Не вдаваясь в подробности тех-
нической стороны этого важного метода, ограничимся указанием
на его основные принципы: дестиллат и серная кислота подаются
в очистную аппаратуру непрерывным потоком; здесь благодаря
тем или иным приспособлениям (инжектор, система диафрагм
и т. п.) они тщательно перемешиваются, вступают во взаимодей-
ствие и уносятся далее, в последующие части аппаратуры, где
происходят отстаивание и отделение отработанной кислоты.
В более простых конструкциях дестиллат через перфорирован-
ные трубы поступает в нижнюю часть цилиндрического, вер-
тикального резервуара и выходит через его верхнюю часть,
пройдя в виде мельчайших пузырьков через серную кислоту,
подаваемую в тот же резервуар снизу, и направляясь далее
в последующие резервуары, где он претерпевает дальнейшие
фазы очистки (отстаивание, промывка и пр.). Весь процесс не-
прерывной очистки ведется в герметически закрытой аппаратуре,
благодаря чему потери на улетучивание наиболее легких частей
дестиллата сведены здесь, Очевидно, до минимума. Кроме того,
благодаря более полному использованию серной кислоты расход
ее при непрерывной очистке резко снижается (до 50%).
Фиг. по дает схему непрерывной очистки бензина, в кото-
рой объединены все последующие фазы процесса, а именно:
1) предварительное отстаивание дестиллата;
2) очистка дестиллата серной кислотой;
3) отстаивание кислоты, увлеченной потоком бензина;
4) промывка дестиллата водой;
5) отстаивание воды, увлеченной бензином;
6) очистка едким натром;
7) отстаивание едкого натра, увлеченного бензином;
615
8) промывка водой;
9) отстаивание воды и выход готового продукта.
Как показано на схеме, бензин поступает в нижнюю часть ка-
ждого резервуара и оставляет его в верхней части. Реагенты—
серная кислота и едкий натр—подаются также в нижние часта
соответствующих резервуаров и по мере использования заме-
няются свежими. В тех случаях, когда желательно более тесное
соприкосновение дестиллата с реагентом, таковое достигается
циркуляцией реагента внутри резервуара от нижней его часта
Фиг. 116. Схема непрерывной очистки бензина.
к верхней путем применения небольшого вспомогательного на-
соса. Что касается, наконец, промывки, то обыкновенно вода
непрерывно вбрызгивается в верхние части промывных резер-
вуаров и выводится снизу. На некоторых установках допускается
иное устройство: водой заполняют около одной трети промыв-
ного резервуара и периодически сменяют ее свежей.
По своей емкости очистные мешалки бывают от 25 до 800 л»
для легких дестиллатов и от 80 до 300 .и3 для смазочных масел.
Пропускная способность небольшой установки для непрерывной
очистки бензина составляет 3—4 м3/час. Естественно, она зави-
сит от многих факторов, в частности — от качества дестиллата.
химизм кислотной очистки
Обращаясь к вопросу о химизме сернокислотной очистки, мы
не будем отделять в дальнейшем изложении периодическую
очистку от непрерывной, так как в том и в другом случаях
химические реакции, лежащие в основе технологического про-
цесса, совершенно одинаковы. Некоторые, более или менее су-
щественные особенности, связанные частью с составом дестил-
лата, частью с условиями очистки, могут быть отмечены, однако,
при действии серной кислоты на различные дестиллаты — лег-
кие, масляные, особенно же крэкинговые, а также сырую нефть
и ее остатки (мазут). Ввиду этого при изучении химизма серно-
кислотной очистки естественно рассмотреть каждый из этих важ-
нейших случаев нефтяной практики отдельно.
Кислотная очистка легких дестиллатов пряной гонки. Как
было отмечено в ч. I (гл. V и VI), бензины и керосины прямой
гонки в главной массе обыкновенно состоят из парафиновых
углеводородов и нафтенов; ароматические углеводороды играют
в их составе гораздо более скромную роль. Так как потери при
очистке для легких дестиллатов прямой гонки обычно не пре-
616
вышают 2—4%, то, очевидно, в составе этих дестиллатов глав-
ное место принадлежит углеводородам тех же самых трех видов.
Таким образом при суждении о химизме сернокислотной очистки
легких дестиллатов прямой гонки необходимо прежде всего рас-
смотреть, каким изменениям могут подвергнуться в условиях
данного процесса эти главные составные части бензинового и
керосинового дестиллатов.
Устойчивость парафинов и нафтенов к серной кислоте обще-
известна. Не возвращаясь вновь к этому, уже ранее рассмотрен-
ному вопросу (ч. I, гл. V и VI), мы можем здесь лишь констати-
ровать, что с кислотой удельного веса 1,84 при обыкновенной
температуре ни парафины, ни нафтены заметно не реагируют.
Таким образом эти главные компоненты бензиновых и керосино-
вых дестиллатов в результате сернокислотной очистки остаются
без изменения — обстоятельство, которое, очевидно, и лежит
в основе этой общепринятой методики их очистки.
Несколько сложнее вопрос о поведении в условиях сернокис-
лотной очистки ароматических углеводородов, входящих в си-
став легких дестиллатов. Мы видели (ч. I), что серная кислота
удельного веса 1,84 не только реагирует с ароматическими угле-
водородами в направлении их сульфирования при комнатной
температуре, но даже применяется для их количественного опре-
деления. Для этого, однако, требуется громадный избыток серной
кислоты (например, при определении ароматики в бензине —
3 объема), так как в противном случае образующаяся при суль-
фировании вода быстро снижает концентрацию серной кислоты
до такого предела, при котором дальнейшее сульфирование аро-
матики уже не идет. Таким образом очевидно, что при серно-
кислотной очистке легких дестиллатов, когда количество серной
кислоты по отношению к массе дестиллата крайне незначительно
(0,5—1,5%), серная кислота, если и затрагивает ароматику, то
в совершенно ничтожной степени.
Только что высказанные соображения определяют следую-
щие основные условия сернокислотной очистки легких дестил-
латов прямой гонки:
1. Количество серной кислоты, применяемой для очистки, не
должно быть велико.
2. Применение слишком крепкой серной кислоты (дымящей)
исключается, если не считать таких случаев, когда (как, напри-
мер, при изготовлении некоторых специальных сортов бензина)
требуется несколько снизить в них содержание ароматики.
3. Температура, при которой ведется очистка, должна быть
умеренной, во всяком случае не требующей обогрева.
Условия эти диктуются не только соображениями экономиче-
ского характера, яо и самым существом дела, так как уклонение
от них может иметь своим последствием снижение в нефте-
продукте ароматики, которая должна расцениваться для бензи-
нов, а также для керосинов в известных пределах как положи-
тельный компонент. В тех случаях, когда снижение ароматики
'требовалось для повышения качества нефтепродукта (румынские
Л17
керосины), эта задача успешнее решалась без применения серной
кислоты, а именно—сольвентным методом (см. ниже гл. II).
Итак, при сернокислотной очистке легких дестиллатов щ
главные компоненты — парафины, нафтены и ароматика—прак-
тически не затрагиваются; действие реагента направляется, та-
ким образом, на другие составные части этих дестиллатов,
а именно на такие примеси, как некоторые непредельные,
а также кислородные, сернистые и азотистые соединения. Эта
последние ведут себя при кислотной очистке наиболее просто:
обладая основным характером, они в виде солей нацело вымы-
ваются из дестиллата (например, керосинового и др.). Действие
серной кислоты на другие примеси мы рассмотрим на более
ярких примерах, чем обычно дают легкие дестиллаты прямой
гонки, а также при специальных методах очистки (обессери-
вание).
Кислотная очистка крэкинг-бензин». По своему составу бен-
зины крэкинга — жидкофазного и парофазного—отличаются от
бензинов прямой гонки высоким содержанием непредельных и
отчасти ароматических; эта особенность их состава не может,
очевидно, не отразиться на характере тех химических процессов,
которые имеют место при кислотной очистке крэкинг-бензинов.
Задача очистки — получить -стабильный бензин стандартных
качеств — в данном случае, вообще говоря, чрезвычайно ослож-
няется. Очевидно, и здесь эту задачу надо -понимать как уда-
ление из бензина наиболее изменчивых его компонентов, с при-
сутствием которых связаны образование и выделение смол, изме-
нение цвета бензина и вообще понижение его стабильности.
К таковым компонентам крэкинг-бензинов в первую очередь
должны быть отнесены присутствующие в них так называемые
диеновые углеводороды, т. е. углеводороды с двумя двойными
связями типа бутадиена и ему подобные, особенно легко осмо-
ляющиеся; возможно, что сюда же следует отнести некоторые
наименее устойчивые непредельные углеводороды с одной двой-
ной связью. Очевидно, однако, что очистка крэкинг-бензинов ни
в коем случае не должна быть направлена к полному удалению
из них непредельных углеводородов, так как такая очистка со-
провождалась бы слишком большими потерями не только в ко-
личественном, но и в качественном отношении вследствие сни-
жения антидетонационных свойств бензина. Таким образом при
очистке крэкинг-бензинов приходится иметь дело с задачей уда-
ления из них легко подвергающихся осмолению, наименее устой-
чивых углеводородов при условии возможно меньших потерь
более устойчивых систем предельного и непредельного характера.
Надлежащее решение этой 'сложной задачи при помощи серно-
кислотной очистки представляет значительные трудности, кото-
рые лишь отчасти разрешены обширной практикой крэкинговых
заводов.
Итак, в основу химических процессов, протекающих при
очистке крэкинг-бензинов серной кислотой, должны быть поло-
жены реакции взаимодействия серной кислоты с непредельными
618
углеводородами [1]. Как показывает изучение этих реакций на
индивидуальных углеводородах, их можно свести к следующим
основным типам:
Реакции присоединения. Серная кислота может образовать
с углеводородами этиленового ряда продукты присоединения
двоякого рода: кислые эфиры, иначе называемые алкилсерными
кислотами, и средние эфиры. Так например, с этиленом эти
реакции приводят к образованию либо кислых (I), либо сред-
них (II) эфиров этилового спирта:
I. СН2 : СН2 + НО • SOsH = СН3 • СН.2 • OSCLH;
II. 2СН2 : СНп + НО • S02 • ОН =
==c2h5.o.so2.o.c2h6
С гомологами этилена образуются обыкновенно соответствую-
щие производные либо вторичных, либо третичных спиртов. *
Кислые эфиры серной кислоты представляют собою вещества кислот-
ного характера, легко растворимые в воде. При нейтрализации они обра-
зуют соответствующие соли. Водой, а еще легче водной щелочью легко гидро-
лизуются. При осторожном нагревании образуют с выделением серной
кислоты ее средние эфиры, по уравнению
2C2H6OSO8H = SO4H2 + SO2(OC2H5)2.
Средние эфиры серной кислоты — бесцветные маслянистые жидкости,
не растворимые в воде. Растворяются в органических растворителях, в част-
ности в бензине. Холодная вода медленно гидролизует их с образованием
сначала спирта и алкилсерной кислоты, в конечном же итоге—спирта и
серной кислоты. При нагревании со спиртами дают простые эфиры:
SO2(OC2H6)a + С2П5ОН = С2Нб • S08H 4- (С^НвЬО.
При кислотной очистке крэкинг-дестиллатов наблюдается
образование и кислых и средних эфиров серной кислоты.
Не растворимые в дестиллате кислые эфиры остаются в кислом
гудроне, а последние следы их могут быть легко удалены из
дестиллата последующей промывкой. Присутствие кислых эфи-
ров серной кислоты в гудроне легко обнаруживается путем раз-
бавления гудрона водой и последующей перегонки с водяным
паром: образовавшиеся при этом вследствие гидролиза спирты
собираются в приемнике и могут быть легко обнаружены здесь
обычными реакциями на алкоголи.
Средние эфиры серной кислоты, не растворимые в воде, но
легко растворяющиеся в бензине, будут оставаться, очевидно,
в дестиллате и тем самым могут заметно повысить в нем содер-
жание серы. При нагревании такого дестиллата с анилином
происходит взаимодействие последнего с эфирами серной кис-
лоты, в результате чего образуется не растворимый в дестиллате
сернокислый анилин. Собрав и взвесив эту соль, можно полу-
чить представление о количестве средних эфиров серной кис-
лоты, содержащихся в данном дестиллате [2]. Очевидно, обра-
зование средних эфиров серной кислоты следует рассматривать
как наименее желательное направление кислотной очистки.
Уравнения взаимодействия углеводородов этиленового ряда
с серной кислотой, приведенные выше, показывают, что на одно
619
li то же количество углеводородов при образовании кислого
эфира требуется вдвое больше кислоты, чем- при образованна
среднего эфира. Таким образом, чтобы избежать образованна
средних эфиров, следовало бы, казалось, вести очистку неко-
торым избытком крепкой серной кислоты, но при этом, есте-
ственно, пришлось бы примириться с повышенными потерт
непредельной части крэкинг-бензина, что, как было указало
выше, совершенно нерационально. Мы встречаемся здесь с одно
из тех противоречивых требований, которые крайне характерна
для процесса кислотной очистки крэкинг-бензина. Эти противо-
речия стараются разрешить в каждом отдельном случае чисто
эмпирическим путем. Как общее правило, при очистке крэкищ-
бензина стараются применять небольшие количества (0,4—о,7%
по весу) крепкой серной кислоты с последующей тщательной
промывкой (см. ниже). При этом приходится иметь в виду, что
количество средних эфиров серной кислоты, содержащихся
в очищенном дестиллате до его промывки, может достигать
вполне заметных величин.
Известно, что кислые и средние эфиры серной кислоты легко
гидролизуются с образованием соответствующих спиртов ио
уравнениям:
СоН50 • SO8H + Н2О = SO4H2 + С2Н6ОН;
(C2H60)2S02 + 2Н,0 = S04H2 + 26ДОН.
В тех случаях, когда для кислотной очистки была взята кхс-
лота недостаточной концентрации шли когда концентрация ее
понизилась вследствие тех или иных причин во время очисти,
реакции гидролиза могут начаться уже в процессе очистки де-
стиллата. Образующиеся при этом спирты растворяются часть»
в дестиллате, частью же в кислоте, почему может создаться впе-
чатление, что этиленовые углеводороды принимают непосред-
ственное участие в образовании кислого гудрона. При длитель-
ном соприкосновении с кислотой количество спиртов в гудроне
уменьшается; очевидно, постепенно они вовлекаются во взаимо-
действие с серной кислотой с промежуточным образованием
эфиросерных кислот и новых этиленовых углеводородов, которые,
реагируя по нижеприведенным схемам, дают начало разнообраз-
нейшим продуктам полимеризации и конденсации.
Для спиртов, .образующихся в результате гидролиза эфиров
серной кислоты, при очистке нефтепродуктов можно представать
себе еще одно направление дальнейших превращений—их окис-
ление. Реакции этого рода возможны, однако, лишь в условиях,
когда серная кислота начинает проявлять свои окислительные
свойства, т. е. при несколько повышенной температуре. Вяя&
в связи с очисткой масляных дестиллатов мы еще вернемся
к этому направлению процесса очистки.
Полимеризация. Под влиянием серной кислоты углеводорода
этиленового ряда легко полимеризуются с образованием непре-
дельных полимеров различной сложности. Так например, изо-
бутилен (СлНе) при действии разбавленной серной кисжяй
620 -
(& частей крепкой серной 4- 1 часть воды) дает преимуще-
ственно триизобутилен (CisHs*); амилен (С5Н10) под влиянием
серной кислоты (4 частей крепкой серной + 1 часть воды) пре-
вращается главным образом в диизоамилен (СюНвд), и т. д. Пер-
вой фазой этих процессов полимеризации несомненно является
образование соответствующих алкилсерных кислот, при взаимо-
действии которых с новой молекулой непредельного углеводорода
образуются соответствующие димеры, тримеры и вообще поли-
меры, серная же кислота выделяется в свободном виде [з]. Так,
для изобутилена эти реакции могут быть выражены следующими
уравнениями:
(СН3)аС : СНз + НОЗОзН = (СН^С 0S08H;
(CHg^C: СНз + (СН3)3С • OSOsH = На804+(СНд)2С : СН • С(СНз)8;
C(CHS);
8
и т. д.
Значительно легче, глубже и сложнее, чем этиленовые угле-
водороды, полимеризуются под влиянием серной кислоты разного
рода «диены». Продуктами реакции являются в данном случае
густые смолообразные вещества неизвестного строения, концен-
трирующиеся, повидимому, главным образом в кислом гудроне.
Процессы полимеризации непредельных углеводородов —
обычное явление при кислотной очистке крокингчбензина. Смо-
лообразные продукты при этом остаются преимущественно в кис-
лом гудроне, продукты же менее глубокой полимеризации (ди-
меры, тримеры и т. п.) почти целиком сохраняются в очищен-
ном дестиллате, так как они мало растворимы в серной кислоте
и, несмотря на свой непредельный характер, крайне слабо с ней
реагируют. Оставаясь в очищенном дестиллате, полимеры зна-
чительно понижают его технические качества: увеличивают его
удельный вес, особенно же повышают его выкипаемость. Вос-
становление нормальной выкипаемости дестиллата легко дости-
гается повторной его перегонкой, так как температуры кипения
полимеров, образующихся при кислотной очистке крэкинг-бен-
зина, лежат значительно выше нормальных пределов темпера-
туры кипения товарных бензинов.
Важнейшими факторами, оказывающими влияние на обра-
зование полимеров при кислотной очистке, являются продолжи-
тельность контакта дестиллата с серной кислотой и температура,
при которой осуществляется этот процесс.
Опыт показывает, что чем продолжительнее соприкосновение
дестиллата с серной кислотой, тем больше образуется полимеров
и, следовательно, тем больше при кислотной очистке непроизво-
дительных потерь углеводородов этиленового ряда. Для возмож-
ного снижения этих потерь кислотную очистку крокинг-бензина
ведут по возможности короткое время, стараясь при этом обеспе-
чить наилучший контакт между кислотой и дестиллатом с по-
G21
мощью тех или иных специальных приспособлений (ср. выше)
Так, в некоторых современных очистных установках непрерыв-
ного действия дестиллат остается в соприкосновении с серной
кислотой не больше 1—1,5 мин., и этого короткого времени ока-
зывается достаточно, чтобы диеновые углеводороды дестилла*га
успели нацело прореагировать с серной кислотой.
Второй важный фактор—температура, при которой ведется
кислотная очистка. Как общее правило, можно считать, неви-
димому, что при пониженной температуре, например при 10°
ниже 0, процесс полимеризации этиленовых углеводородов вдет
более умеренно [4]. Имеются, однако, указания, что в неко-
торых других отношениях, например для возможно полного отде-
ления смолистых веществ, для получения бензина хорошего
цвета и т. п. выгоднее вести очистку при обыкновенной или даже
несколько повышенной температуре. Таким образом оптималь-
ную температуру кислотной очистки крэкинг-дестиллата обык-
новенно приходится устанавливать чисто эмпирическим путем.
Гидрополимеризация. Кроме только что рассмотренной поли-
меризации обычного типа, протекающей под влиянием более или
менее разбавленной серной кислоты, этиленовые углеводороды,
как показали исследования последних лет [5], легко претерпе-
вают в присутствии избытка крепкой серной кислоты (удельный
вес 1,84) полимеризацию более сложного тшгя получившую наи-
менование «гидрополимеризации». Сущность этого процесса за-
ключается в том, что в указанных условиях этиленовые угле-
водороды превращаются в соответствующие димеры, тримеры и
прочие полимеры с одновременным их гидрированием, т. е. в по-
лимеры предельного характера, коротко говоря, в гидрополи-
меры. Так например, для циклогексена (СеНю) этот процесс
может быть выражен следующей схемой:
+Щ +Н2
С6Ню * С 12^22 *
И т. д.
Аналогичные превращения претерпевают в подобных же усло-
виях этиленовые углеводороды с открытой группировкой угле-
родных атомов, например изобутилен, амилен и др.
При изучении гидрополимеризации углеводородов этилено-
вого ряда прежде всего естественно возникает вопрос, откуда
берется водород, благодаря которому вместо полимеров обычного
типа здесь получаются гидрополимеры предельного характера.
Ответ на этот вопрос дает исследование кислотного слоя данной
реакции: при его разбавлении водой выделяется маслянистое
вещество резко непредельного характера, сначала бесцветное,
затем быстро превращающееся в окрашенное — сначала в зеле-
новато-желтый, а под конец в бурый цвет; при стоянии это
масло постепенно переходит в темнокоричневую асфальтообраз-
ную массу. Так как вещества эти надо рассматривать как про-
дукты одновременного дегидрирования и полимеризации, ко-
роче— дегидрополимеризации все того же исходного углеводо-
4522
рода этиленового ряда, из которого по приведенной выше схеме
образуются его гидрополимеры, то, очевидно, мы имеем здесь
своеобразный случай протекающей под влиянием серной кислоты
сопряженной реакции восстановления — окисления.
Доказано экспериментально [6], что гидрополимеризация
углеводородов этиленового ряда с образованием полимеров пре-
дельного характера несомненно имеет место при обработке креп-
кой серной кислотой крэкинг-дестиллатов, особенно же дестил-
латов пиролиза. Но те же опытные данные показывают, что
в условиях обычной кислотной очистки крэкинг-дестиллатов
образование гидрополимеров предельного характера по сравне-
нию с продуктами полимеризации и конденсации других видов
незначительно. С возможностью образования гидрополимеров
необходимо считаться лишь в некоторых специальных случаях,
например при обработке крэкинг-дестиллата большим избытком
крепкой серной кислоты в аналитической практике и тому подоб-
ных случаях.
Конденсация. Как было указано выше, в процессе полимери-
зации углеводородов этиленового ряда в первую фазу образуются
эфиры серной кислоты, реагирующие затем со второй частицей
того же углеводорода. Если в данной смеси, кроме непредельных
углеводородов, имеется также ароматика, то действие кислого
эфира направляется преимущественно в сторону ароматики; при
этом образуются продукты конденсации ароматических угле-
водородов с этиленовыми, представляющие собою соответствую-
щие высшие гомологи бензола предельного характера. Так на-
пример, из смеси бензола с гексиленом в присутствии 10%-ной
крепкой серной кислоты был получен соответствующий иэо-
гексилбензол [7] и т. д.:
/СН3
С6Н6 + СН2: СН • (СН2)8. СН8 = СбН5. сн/
ХС4Н9
Конденсация непредельных углеводородов с ароматикой
является одной из важнейших реакций, которые имеют место
при кислотной очистке крэкинг-бензина. Как ясно из выше-
сказанного, эта реакция приводит к новообразованию высоко-
молекулярной ароматики, которая трудно реагирует с крепкой
серной кислотой, почти нерастворима в ней и концентрируется
в очищенном дестиллате, а после вторичной перегонки послед-
него—в остатке от перегонки. В технике продукты полимериза-
ции и конденсации, образующиеся при кислотной очистке крэ-
кинт-дестиллатов, нередко объединяют под общим наименованием
«полимеры».
Окисление. Серная кислота, раскисляясь до сернистбго газа,
может действовать на органические вещества и, в частности, на
углеводороды окисляющим образом. В условиях кислотной
очистки такое действие серной кислоты особенно наблюдается
в тех случаях, когда процесс ведется при повышенной темпе-
ратуре (см. очистка масляных дестиллатов). Но и в отсутствии
623
нагревания, например при очистке крэкинг-бензина, некоторое
выделение сернистого газа иногда наблюдается. Повидимому,
явление связано с особой реакционной способностью диеновых
углеводородов, всегда присутствующих в неочищенном крэкинг-
дестиллате и подвергающихся под влиянием серной кислоты не
только полимеризации, но одновременно и частичному окисле-
нию. Насколько глубоко могут итти подобного рода процессы
при кислотной очистке, показывает иногда наблюдаемое выде-
ление углистого осадка из некоторых фракций очищенного
дестиллата. Выпадение этого осадка несомненно связано с усло-
виями кислотной очистки: чем больше взять серной кислоты для
очистки, тем больше получается фракций, в которых образуется
углистый осадок; уже одно это обстоятельство показывает, что
здесь приходится иметь дело с процессом одновременного окис-
ления и полимеризации или конденсации каких-то компонентов
исходного дестиллата, особенно легко и глубоко претерпевающих
взаимодействие с серной кислотой [8].
Итак, кислотная очистка крэкинг-бензина представляет собою
сложнейший комплекс реакций серной кислоты с различными
компонентами дестиллата, в первую очередь — с углеводородами
этиленового ряда и ароматикой. Реакции этого последнего рода,
только что подробно рассмотренные, имеют типовой характер.
При очистке крэкинг-бензина в связи с его составом они выра-
жены особенно ярко и показательно, но в той или иной мере эти
реакции могут, конечно, иметь место при кислотной очистке
также и других дестиллатов, поскольку в составе последних
находятся одновременно непредельные углеводороды и арома-
тика. Что касается, наконец, парафинов и нафтенов, входящих
в состав крэкинг-бензина, то по вопросу об их поведении в усло-
виях кислотной очистки соответствующих дестиллатов здесь при-
шлось бы. очевидно, повторить лишь то, что уже было сказано
в связи с кислотной очисткой легких дестиллатов прямой гонки,
так как присутствие углеводородов этиленового ряда нисколько
не отражается на их общеизвестной устойчивости по отношению
к серной кислоте, по крайней мере в условиях кислотной
очистки легких дестиллатов.
Кислотная очистка масляных дестиллатов. Хотя состав мас-
ляных дестиллатов изучен пока далеко недостаточно, тем'не ме-
нее можно считать установленным, что их главными компонен-
тами, являются углеводороды тех же трех рядов, которые обра-
зуют’легкие дестиллаты прямой гонки, т. е. парафины, нафтены
и ароматика; притом, как общее правило, по мере • утяжеления
дестиллатов содержание в них парафинов уменьшается, содер-
жание же ароматики возрастает. Важно отметить здесь также
повышенное содержание в масляных дестиллатах второстепенных
составных частей нефти, которые и в данном случае должны
рассматриваться как вредные примеси; таковы непредельные,
кислородные и сернистые соединения различных типов. Этим
последним обстоятельством в первую очередь и объясняется одна
из особенностей кислотной очистки масляных дестиллатов—
624
значительно больший расход серной кислоты по сравнению
с тем. что имеет место при очистке легких дестиллатов.
Другая важная особенность кислотной очистки масляных де-
стиллатов заключается в применении подогрева. Причина, за-
ставляющая вести в данном случае очистку при повышенной
температуре,— более или менее значительная вязкость масля-
ных дестиллатов. Повышение температуры снижает вязкость,
благодаря чему смешение дестиллата с кислотой и последующе^
отстаивание кислого гудрона значительно облегчаются, хотя во
же требуют, как было отмечено выше, весьма продолжитель-
ного времени. Чтобы по возможности сократить это время, прак-
тика кислотной очистки выработала целый ряд практических
приемов, которые сводятся к добавке к отстаивающемуся маслу
разного рода сторонних ингредиентов, как-то: паровой воды,
крепкого раствора едкого натра, растворимого стекла и т. п
Если после добавления этих веществ продолжить еще некоторое
время перемешивание, мельчайшие частички, в виде которых
кислый гудрон был распределен в масле, довольно быстро соби-
раются в крупные хлопья, оседающие затем на .дно мешалки.
Проследим теперь, как могут отразиться условия очистки
масляных дестиллатов. а равно и состав последних на химизме
этого процесса.
Увеличение количества серной кислоты и повышенная темпе-
ратура, при которой ведется очистка масляного дестиллата,
должны, казалось бы, прежде всего содействовать полноте уда-
тения из него ароматики. Принимая, однако, во внимание, что
по мере усложнения состава ароматических углеводородов ре-
акционная способность их с крепкой серной кислотой быстро
снижается и что количество серной кислоты, применяемой при
очистке масляных дестиллатов, по отношению к общей массе де-
стиллата остается обыкновенно все же крайне недостаточным
(2—6%), легко притги к выводу, что в громадном большинстве
случаев и здесь сколько-нибудь значительного снижения аро-
матики -после кислотной очистки ожидать не приходится. Исклю-
чение могут -составлять лишь те дестиллаты. при очистке кото-
рых применяются более или менее значительные количества ды-
мящей серной кислоты; таковы, например, вазе типовое масло,
при получении которого расходуется до 20% дымящей серной
КИСЛОТЫ С содержанием ДО 20% 80з. И paraffinum liquidum. при
очистке которого расход дымящей серной кислоты достигает 50%.
Эти случаи будут рассмотрены ниже несколько подробнее
Переходя к другим важнейшим компонентам масляных де-
стиллатов— парафинам и нафтенам, — приходится отметить
прежде всего, что высшие парафины столь же устойчивы к креп-
кой серной кислоте, кал; и низшие. Что касается нафтенов, то.
как известно (ч. I, гл. VI). при взаимодействии с избытком
крепкой серной кислоты они более или менее легко могут под-
вергаться дегидрогенизации с последующим сульфинированием
образующегося ароматического углеводорода. То обстоятельство,
что при очистке масляных дестиллатов выделяется значительное
40 Зак 1622. Хкашя вефтп.
количество -сернистого газа, является косвенным подтверждением
подобного рода реакций при этом процессе, особенно при боль-
шом избытке крепкой или дымящей серной кислоты, т. е. в слу-
чаях, только что отмеченных. Как общее правило, следует ду-
мать, что действие главной массы серной кислоты и здесь напра-
вляется на другие компоненты дестиллатов, которые занимают
в количественном отношении в их составе второстепенное зна-
чение, и прежде всего на непредельные углеводороду.
Непредельные углеводороды, образуясь при перегонке нефти
и мазута в результате частичного крэкинга, являются постоян-
ными" составными частями различных нефтяных дестиллатов,
особенно масляных. Об этом можно судить, как было показано
в ч. I (гл. V, 11), уже по иодным или кислородным числам раз-
личных дестиллатов, возрастающим по мере утяжеления послед-
них. Правда, главная часть продуктов разложения масел, как
мы видели выше (ч. II, тл. II), не попадает в масляные дестил-
латы, а отходит в так называемые эксгаустерные масла; тем не
менее, наиболее высокомолекулярные из продуктов разложения
все же остаются в масляных дестил латах, и представляется инте-
ресным проследить их вероятные превращения в условиях кис-
лотной очистки.
Как было показано выше, в связи с кис« отной очисткой крэ-
кинг-бензинов реакции этиленовых углеводородов с серной кис-
лотой сводятся либо к образованию кислых или средних эфиров
серной кислоты с последующим переходом к соответствующим
спиртам, либо к реакциям полимеризации и конденсации, либо,
наконец, к реакциям окисления. В надлежащих условиях можно
наблюдать все эти виды взаимодействия серной кислоты с не-
предельными углеводородами; однако сравнительное исследова-
ние в этом направлении этиленов различного молекулярного веса,
показало [9], что склонность их к различным реакциям указан-
ных типов неодинакова. Так, например, если ближайшие гомо-
логи этилена сравнительно легко, а иногда и очень легко при-
соединяют серную кислоту с образованием кислых и средах
эфиров, которые могут превращаться далее в соответствующие
спирты, то у высших этиленов склонность к образованию сред-
них эфиров и спиртов сильно падает; вслед за первой фазой —
образованием кислого эфира — реакция направляется здесь глав-
ным образом в сторону процессов полимеризации и конденсации,
продукты которых почти целиком удерживаются маслом. Таким
образом непосредственное участие высших этиленовых угле-
водородов масляных фракций в образовании кислого гудрона
должно быть признано незначительным.
Как было уже отмечено при кислотной очистке масляных де-
стиллатов, в связи с повышенной температурой, при которой
ведется процесс, наблюдается обитьное выделение сернистого
газа, что указывает на наличие здесь каких-то реакций окисле-
ния. Эти окислительные процессы должны достигать особой
интенсивности в упомянутых выше случаях применения дымя-
щей серной кислоты. Такая кислота естественно должна дей-
626
стэовать прежде всего на ароматику данного дестиллата, превра-
щая ее в соответствующие сульфокислоты, поскольку эта аро-
матика еще сохранила способность сульфироваться (ср. ч. I,
гл. IV). С другой стороны, действуя на нафтены гидроаромати-
ческого ряда, крепкая серная кислота может сначала дегидри-
ровать их до соответствующей ароматики, а затем просульфи-
ровать эту последнюю. Такой путь образования ароматических
сульфокислот можно наблюдать уже при взаимодействии с сер-
ной кислотой простейших гомологов циклогексана в условиях
повышенной температуры.
Аналогичные реакции дегидрирования с последующим обра-
зованием сульфокислот имеют место при действии крепкой, осо-
бенно же дымящей серной кислоты на масляные дестиллаты.
Наиболее характерные примеры такого рода реакций дает очистка
некоторых соляровых дестиллатов, которая приводит, с одной
стороны, к вазелиновым маслам высокой степени очистки, с дру-
гой— к смеси высокомолекулярных «нефтяных сульфокислот»,
частью остающихся в кислом гудроне, частью растворяющихся
в очищенном дестиллате. Эти сульфокислоты представляют
большую практическую ценность, и выделение их из кислого
дестиллата является, как будет показано ниже, важной отраслью
утилизации побочных продуктов очистки нефтяных дестиллатов
дымящей серной кислотой.
Состав и природа нефтяных сульфокислот в известной сте-
пени зависят как от природы исходного дестиллата, так и от
методики их получения и выделения.
Сульфокислоты, образующиеся в результате обработки при
нагревании до 70—80 е соляровых дестиллатов (удельный вес
0,880—0,882) дымящей серной кислотой с содержанием 20—30%
SOs и выделяемые йз кислого масла обработкой небольшим ко-
личеством водного спирта, представляют собою густую, бурую,
сильно флюоресцирующую жидкость, которая во всех отношениях
смешивается с водой, образуя коллоидальные растворы; при
взбалтывании в водных .растворах сильно пенится подобно мыль-
ным растворам и образует с минеральными маслами стойкие
эмульсии. Они хорошо растворяются также в маслах и при ще-
лочной промывке кислых.масел являются главными потребите-
лями щелочи. Очищенные от примеси масел и смолистых ве-
ществ, нефтяные сульфокислоты имеют светложелтую окраску
и твердую, вязкую консистенцию. Их средний состав отвечает
формуле C20H27SO3H, что соответствует углеводороду ряда
CftH^_12 [10].
Путем нагревания 50 %-него водного раствора нафтеновых
сульфокислот в автоклаве при 25 ат давления они были превра-
щены в соответствующие углеводороды, кипевшие, естественно,
в широких пределах. По своему среднему составу эти углево-
дороды отвечали общей формуле СпЙ2л -12 и при действии креп-
кой серной кис юты образовали смесь сульфокислот, по своим
свойствам напоминавших исходные нефтяные сульфокис-
лоты [101.
40* 627
При очистке крепкой серной кислотой более тяжелых дестил-
латов, чем только что указано, образуются сульфокислоты,
имеющие несколько больший молекугярный вес. Так, при ис-
следовании щелочных отбросов от очистки смазочных масел
моногидратом из этих отбросов действием слабой соляной кислоты
были выделены сульфокислоты, по своим свойствам напоминав-
шие вышеописанные, но. судя по анализу их бариевой соли,
уже иного среднего состава, а именно — С-2сН37 • SO3H, что отве-
чает общей формуле углеводородного ряда СА-к [11].
Еще больший молекулярный, вес имеют сульфокислоты и
сульфоны от очистки высококипящих фракций нефти дымящей
серной кислотой, выделенные из кислого гудрона. Соединениям
этим, получившим наименование туменолсульфокислоты и
туменолсульфоны, приписываются следующие формулы [12]:
СЛО^ОзН
ттменолсульфокиолоты туменолсульфоны
Хотя индивидуальность этих соединений, равно как и при-
веденный их состав не могут не возбуждать известных сомнений,
тем не менее, высокий молекулярный вес их и еще большая сте-
пень непредельности их состава по сравнению с нефтяными
сульфокислотами, рассмотренными выше, совершенно очевидны.
Серная кислотами второстепенные компоненты нефти. Выще
было рассмотрено отношение к серной кислоте в условиях кис-
лотной отгистки различных углеводородов. Кроме последних
в состав тяжелых нефтяных дестиллатов и сырых нефтей вхо-
дят кислородные соединения (нафтеновые кислоты и фенолы),
а также вещества смолистого и асфальтового характера. Ка-
ково же их отношение к серной кислоте и каково их участие
в образовании кислого гудрона? •
Фенолы и отчасти нафтеновые кислоты могут реагировать
с крепкой серной кислотой, образуя соответствующие сульфо-
производные, переходящие в кислый гудрон. Кроме того, нафте-
новые кислоты, особенно высокомолекулярные, способны перехо-
дить в сернокислотный слой в растворенном виде и могут быть
выделены из гудрона в неизмененном состоянии. Таким обра-
зом фенолы и нафтеновые кислоты должны быть отнесены к со-
ставным частям нефти и ее дестиллатов, принимающим ближай-
шее участие в образовании кислого гудрона. В количественном
отношении это участие не может быть значительно, так как со-
держание фенолов и нафтеновых кислот в нефтях и нефтяных
дестиллатах обыкновенно невелико.
Более существенная роль в образовании кислого гудрона
должна принадлежать смолистым и асфальтовым веществам
нефти, которые в сырых нефтях содержатся нередко в весьма
значительном количестве, в тяжелых же дестиллатах более или
менее легко образуются за счет процессов полимеризации и кон-
денсации продуктов прямой гонки.
По всем признакам действие крепкой серной кислоты на
смолы и асфальтены носит смешанный, чисто химический и фи*
(528
зико-химический характер [13]. Несомненно, здесь происходят
прежде всего какие-то химические реакции, которые приводят'
к продуктам, более богатым серой по сравнению с исходным ма-
териалом, как свидетельствуют об этом результаты анализа орга-
нического вещества, выделяемого из кислого гудрона Отсюда
естественно притти также к выводу, что главная масса серы,
содержащейся в этом веществе, имеет своим источником пе серу
первоначального дестиллата, а серную кислоту, реагирующую
со смолами и асфальтенами и входящую в состав образующихся
таким образом более сложных продуктов.
С другой стороны, как показывает подсчет, значительное ко-
.тичество серной кислоты, взятой для очистки дестиллата, исче-
зает в этом процессе и не может быть выделено обратно пз кис-
лого гудрона. Так, при очистке бакинского керосинового дестил-
лата (0,35% кислоты) исчезает около 28% взятой для очистки
серной кислоты; при очистке машинного дестиллата (3% сер-
ной кислоты) исчезает уже около 45% кислоты и т. д. Эта
исчезающая кислота идет, очевидно, на те или иные химиче-
ские реакции. Особенно характерен здесь параллелизм между
природой дестиллата и количеством исчезающей серной кис-
лоты: чем тяжелее очищаемый дестиллат, тем больше расхо-
дуется и связывается серной кислоты при его очистке. Часть
этой кислоты, как мы видели, может расходоваться на окисли-
тельные и иные процессы и выделяться в виде сернистого газа;
но несомненно также, что другая часть серной кислоты, вступая
во взаимодействие с характерными для тяжелых нефтей и де-
стиллатов компонентами, смолами и особенно асфальтенами, пе-
реходит в кислый гудрон в связанном состоянии.
Какова же природа соединений, образующихся при действии
крепкой серной кислоты на смолы и асфальтены в условиях
кислотной очистки? Едва ли можно сомневаться, что в этих
условиях смолы, содержащиеся в дестиллате, быстро превра-
щаются в асфальтены, так что в поставленном вопросе можно
ограничиться рассмотрением лишь одних асфальтенов. Но и
при таком ограничении в нем остается на сегодняшний день еще
много неясного.
Если некоторое количество асфальтена растворить в бензиле
или парафиновом масле и подействовать на такой раствор креп-
кой серной кислотой, то из раствора выпадает осадок, по внеш-
нему виду похожий на исходный асфальтен, но в отличие от
последнего не растворимый даже в кипящем бензоле. Содержа-
ние серы в этом осадке много больше, чем в исходном асфаль-
тене (7% вместо 4,2%). При нагревании его с соляной кислотой
даже при обыкновенном давлении наблюдается отщепление сер-
ной кислоты, так что, повидимому, здесь действительно прихо-
дится иметь дело с каким-то продуктом взаимодействия асфаль-
тена с серной кислотой. Однако сказать что либо более опреде-
ленное о химической природе этого продукта пока было бы
преждевременно [14].
Кроме рассмотренных выше реакций, при взаимодействии
62'J
крепкой серной кислоты с нефтяными смолами и асфальтенами
имеют, повидимому, место также процессы физико-химического
характера, а именно явления двухсторонней абсорбции. Эти
явления хорошо наблюдаются на примере тяжелых остаточных
масел типа вискозинов, всегда содержащих смолистые веще-
ства. При действии крепкой серной кислоты на эти масла иди
на растворы их в бензине в одних случаях можно наблюдать
переход некоторой части в сернокислотный слой, в других —
•обратно — серной кислоты в слой углеводородов. Лишь неболь-
шая часть этой абсорбированной кислоты находится в такого
•рода растворе в свободном виде, в состоянии мельчайшего рае
пыления, и может быть выделена из него центрифугированием;
главная же масса серной кислоты, перешедшей в углеводород-
ный слой, образует здесь какие-то малоустойчивые соединения,
легко разлагающиеся не только при промывке холодной водой,
но отдающие серную кислоту даже самопроизвольно при хране-
нии подобного рода растворов. Ближайшая природа этих мапо-
устойчивых соединений пока неизвестна, но по всем признакам
они содержатся также в кислом гудроне [13].
Действительно, кислый гудрон почти не растворим в бензине.
Если, однако, отмыть содержащуюся в гудроне серную кислоту,
то получается органический остаток, который, будучи высушен,
почти целиком растворяется в бензине; такие же растворители,
как ксилол, извлекают органическую массу гудрона даже без
предварительной промывки его водой. Таким образом связь ме-
жду серной кислотой и органическими компонентами кислого
тудрона смолистого характера настолько слаба, что ее можно
нарушить действием не только воды, но и некоторых раствори-
телей [13].
С факторами физико-химического характера при кислотной
очистке масел приходится встречаться особенно в связи с вопро-
сом об отделении кислого гудрона. Осаждение последнего на дно
мешалки при очистке некоторых масел происходит крайне ме-
дленно, так как гудрон находится в масле в виде мельчайших,
равномерно распределенных в масле частичек, т. е. в виде кол-
лоидального раствора, достаточно устойчивого при повышенной
вязкости масла несмотря на сравнительно слабую степень дис-
персности. Если, однако, по окончании собственно очистки, не
прекращая перемешивания, прибавить в мешалку немного креп-
кого раствора едкого натра или паровой воды, то наступает бы-
страя коагуляция: частицы кислого гудрона собираются в круп-
ные хлопья, которые быстро оседают на дне мешалки.
В некоторых случаях, например при очистке масляного полу-
гудрона, т. е. мазута со средних кубов масляной батареи, под-
вергаемого у нас переработке на наиболее тяжелые цилиндро-
вые масла типа вапоров и вискозинов, последние следы кислого
гудрона со следами серной кислоты все-таки остаются в масле.
Для ее удаления приходится прибегать к крайней мере: путем
пропускания перегретого водяного пара раскисляют серную кис-
лоту до сернистого газа, который отходит вместе с паром, после
4530
чего начинают перегонку мазута, точнее — его концентрацию, до
получения такого остатка в кубе (концентрата), который имел
бы нужную температуру вспышки.
Прочие случаи кислотной очистки. В предыдущем изложении
на примерах легких и тяжелых дестиллатов прямой гонки (бен-
зины, керосины, масла), а также крэкинг-бензина были рассмо-
трены основные химические процессы, имеющие место при кис-
лотной очистке этих дестиллатов. Само собой разумеется, что
все сказанное может быть распространено и на другие случаи
кислотной очистки, встречающиеся в практике нефтеперераба-
тывающей промышленности. Таковы, например, случаи парафи-
нового гача и озокерита, подвергаемых очистке для последующей
переработки на парафин и церезин (ч. I, гл. V В), или очистка
некоторых масляных нефтей, обрабатываемых крепкой сер-
ной кислотой для удаления смолистых веществ, благодаря
чему при последующей перегонке удается получить дестиллаты
более высокого качества, и т. д. Можно не останавливаться ближе
на этих новых случаях кислотной очистки, так как в зависимо-
сти от ее условий и состава исходного сырья здесь должны на-
блюдаться по существу те же самые химические реакции и фи-
физико-химические процессы, которые были подробно рассмо-
трены нами на вышепоименованных примерах.
ХИМИЗМ ЩЕЛОЧНОЙ ОЧИСТКИ И ПРОМЫВКИ
Щелочная очистка нефтяных дестиллатов обыкновенно сле-
дует за кислотной очисткой, реже ей предшествует. Рассмотрим
сначала реакции, которые имеют место при щелочной очистке
обычного типа.
Реакции при щелочной очистке. Как было указано в начале-
настоящей главы, задача щелочной очистки заключается в ней-
трализации нефтяных дестиллатов, т. е. в связывании и вымыва-
нии тех кислот, которые в той или иной форме содержатся в де-
стиллате. Таковыми являются:
1) нафтеновые кислоты (а также фенолы), содержащиеся
в сыром дестиллате;
2) кислоты, появившиеся в дестиллате после его кислотной
очистки, а именно:
а) серная кислота, свободная, взвешенная в дестиллате, или.
связанная, в виде средних эфиров и, быть может, в виде эфиро-
серных кислот;
б) сульфокислоты ароматического ряда, или сульфонафтено-
вые кислоты.
При действии едкого натра все свободные кислоты, содержа-
щиеся в дестиллате, независимо от их природы нейтрализуются
и переходят в виде солей в щелочный раствор; связанные же
кислоты, например эфиры серной кислоты, обмыливаются ще-
лочью; в .результате и эти кислоты превращаются в соответ-
ствующие соли натрия (мыла), переходящие в водно-щелочный
слой. Так для среднего эфира серной кислоты, например для
С31
днэтил сульфата, эта реакция может быть выражена следующим
.уравнением:
S02(0C2H5)2 + 2NaOH = 2С2Н50Н+SO.Na,.
Водные растворы едкого натра, применяемые для щелочной
очистки, бывают различной крепости. Для очистки бензинов
употребляют щелочь крепостью 16—20° Вё, т. е. с содержанием
11—14,5% едкого натра; для более тяжелых дестиллатов во из-
бежание образования слишком устойчивых эмульсий приходится
брать более слабую щелочь, а именно: для керосина 5—7° Вё,
т. е. с содержанием 3,5—4,5% едкого натра, для масляных же
.дестиллатов — еще более слабую, а именно 2,5—4,5° Вё, т. е.
с содержанием 1,2—3% NaOH.
Применение слишком слабой щелочи в процессе очистки не-
которых дестиллатов имеет своим естественным следствием явле-
ние, которое значительно осложняет щелочную очистку, а имен-
но— усиленное гидролитическое расщепление образовавшихся
в результате нейтрализации солей нафтеновых и сульфонафте-
новых кислот. Для простейших нафтеновых солей эта 'реакция
может .быть выражена следующим уравнением:
СпН2п_1 • СООЬ’аЧ- Н20 NaOH-j- CrtHSn-i СООН:
Очевидно, что равновесие, выражаемое этим равенством, на
основании закона действующих масс должно сдвигаться слева
направо, т. е. в сторону нарастания гидролиза, по мере увели-
чения действующей массы воды, иначе говоря, по мере умень-
шения крепости щелочного раствора. Легко видеть также, что
в условиях щелочной очистки нефтяных дестиллатов имеется
еще один фактор, способствующий гидролизу солей нафтеновых
и сульфонафтеновых кислот. Фактор этот — присутствие подвер-
гаемого очистке дестиллата.
Действительно, представим себе, что образовавшиеся в про-
цессе гидролиза кислоты растворимы в дестиллате, как это имеет
место -в действительности при щелочной очистке нефтепродук-
тов. В таком случае переход этих кислот в дестиллатный слой,
естественно, нарушит равновесие, установившееся в водно-ще-
-лочном слое, и для восстановления этого равновесия по закону
действующих масс остается лишь один путь: гидролиз некото-
рого нового количества находящихся в водно-щелочном растворе
солей. Факты находятся в полном согласии с этими требова-
ниями теории. Для их иллюстрации может служить табл. 143,
в которой приведены результаты следующих опытов: разные
объемы керосина встряхивались с 50 с.и8 раствора среднего на-
•фтенового мыла с содержанием 12% нафтеновых кислот (моле-
кулярный вес 180), после чего определялось количество кислот,
перешедших в керосин [15]. Как видно из табл. 143, с возра-
станием объема керосина, действующего здесь как растворитель,
количество солей, подвергшихся гидролизу, правильно возра-
стает.
R32
Таблица 143
Гидролиз нафтенового ныла
(по Юркоеу)
Объем керосина, в мл % нафтеновых кислот в керо- сине "/о нафтеновых кислот, перешед- ших в керосин, от всего их количества
50 0,144 1.2
100 0,090 1.5
200 0,072 2.4
400 0,036 24
800 0,027 3,6
Из других факторов, кроме концентрации щелочи (см. выше),
известное влияние на гидролиз имеет также температура. Инте-
ресно, что в крепких растворах нафтеновых солей повышение
температуры, как оказалось, заметно усиливает гидролиз, тогда
как для слабых растворов наблюдается обратное явление: с по-
вышением температуры гидролиз не усиливается, а ослабе-
вает [151. Причина такой аномалии не вполне понятна.
Итак, щелочная очистка не удаляет из дестиллата всех на-
фтеновых кислот" некоторое количество их остается в дестиллате
вследствие гидролизующего действия воды; в силу этого в де-
стиллатный слой может перейти также известное количество
средних нафтеновых солей. Эти нежелательные осложнения
очистки, казалось бы, проще всего устранить, ведя очистку
крепкой щелочью при низкой температуре. Однако в этих усло-
виях нередко приходится иметь дело с еще большим осложне-
нием процесса очистки — с образованием эмульсий.
Эмульсии при щелочной очистке. Образование эмульсий при
щелочной очистке нефтяных масел — явление обычное и, можно
сказать, неизбежное. С внешней стороны такие эмульсии пред-
ставляют собою непрозрачные жидкости молочного цвета или
густые, сметанообразные смеси. Невооруженному глазу они
представляются вполне однородными, но под микроскопом обна-
руживается типичная картина эмульсии либо масла в водной
щелочи, либо водной щелочи в масле. Степень устойчивости
этих эмульсий может быть почти столь же разнообразна, как
у естественных нефтяных эмульсий, рассмотренных в ч. II, гл. I.
Эмульсия «кислое масло—водная щелочь» может образо-
ваться самопроизвольно уже при простом соприкосновении этих
двух жидкостей. Энергичное встряхивание или перемешивание
этих жидкостей, естественно, еще более содействует эмульгирова-
нию: жидкости разбиваются на отдельные более или менее мел-
кие капли, которые после прекращения механического воздей-
ствия на смесь частью собираются в более крупные агрегаты и
постепенно отслаиваются, частью же сохраняют свое состояние
633
дисперсности, образуя эмульсию. В результате после отстаива-
ния обыкновенно получают три слоя: верхний масляный слой,
<более или менее мутный вследствие содержания в нем мельчай-
ших капелек водной щелочи; нижний водно-щелочный слой, со-
держащий также соли нафтеновых и сульфонафтеновых кислот и
•более или менее опалесцирующий вследствие содержания в нем
растворенного масла, и средний эмульсионный слой более или
менее тустой консистенции. Легко убедиться, что причины,
обусловливающие устойчивость эмульсий подобного рода, и здесь
должны быть те же, что были подробно рассмотрены в ч. II,
гл. I в связи с вопросом о нефтяных эмульсиях вообще.
Действительно, мы видели, что образование эмульсий каких-
либо двух жидкостей всегда теснейшим образом связано с нали-
чием в их системе некоторого третьего компонента (эмульга-
тора), который накопляется на границе раздела двух соприка-
сающихся жидких фаз. понижает поверхностное натяжение того
из компонентов, по отношению к которому он является лио-
фильным, и образует на поверхности отдельных капелек эмуль-
сии те оболочки (пленки), которые придают эмульсии большую
или меныпую устойчивость. В случае системы «кислое масло—
водная щелочь» такими эмульгаторами, очевидно, будут на-
тгриевые соли нафтеновых и сульфонафтеновых кислот, т. е. на-
фтеновые и сульфонафтеновые мыла, образующиеся при щелоч-
ной очистке в результате взаимодействия щелочи с соответ-
ствующими кислотами Поскольку натровые масла гидрофильны,
присутствие их в данной системе неизбежно вызывает образова-
ние эмульсии, в которой вода является внешней фазой.
В процессе щелочной очистки имеет значение не столько са-
мый факт образования эмульсии, как степень ее устойчивости,
зависящей от прочности тех мыльных оболочек (пленок), в ко-
торые заключены отдельные элементы дисперсной фазы эмуль-
сии, т. е. капельки масла. В свою очередь, прочность этих обо-
лочек должна обусловливаться накоплением на границе двух
жидких фаз эмульгатора, в данном случае — мыла. Так как, на-
конец, по мере увеличения концентрации этого эмульгатора по-
верхностное натяжение воды на границе ее с маслом будет все
более и более понижаться, то для эмульсий типа «масло в воде»
естественно ожидать, что чем больше это снижение поверхност-
ного натяжения, тем прочнее эмульсия, и наоборот. Эти инте-
ресные и важные выводы полностью подтверждаются при сопо-
ставлении снижения поверхностного натяжения под влиянием
накопления эмульгатора с прочностью эмульсий, образуемых
мыльной водой с различными нефтяными дестиллатами
<табл. 144).
Действительно, данные табл. 144 показывают, что снижение
поверхностного натяжения воды на границе ее с тем или иным
нефтепродуктом прежде всего возрастет по мере накопления на
водном слое эмульгатора — нафтенового мыла. Одновременно из
той же таблицы видно, что при одной и той же концентрации
мыльного раствора снижение для системы «вода —- бензин»
634
Таблица 14 £
Поверхностное натяжение мыльной воды на границе
с нефтепродуктами
(по Гурвичу)
Поверхностное натяжение в системе Вода + 0,26% мыла Снижение поверхн. натяжения + 1% мыла Сниже- ние по- верхн. натяже- ния
Бензин 48,1 6$ 41,8 3,8 25
Керосин 42,4 3,6 38,8 2,8 0,8
Веретенное масло .... 28,9 3,0 25,9 2,7 0.3
является наибольшим, для следующей системы «вода — керосин»-
оно несколько меньше и, наконец, для системы «вода—веретен-
ное масло» — еще меньше. Опыт показывает, что устойчивость-
соответствующих эмульсий находится в полном соответствии,
с этими данными. Так, при встряхивании в пробирке бензина
с однопроцентным раствором натрового мыла получается на-
столько гглотная и устойчивая эмульсия, что пробирку можно
опрокинуть и никакой жидкости не прольется; с веретенным,
маслом при прочих равных условиях также образуется эмуль-
сия, но уже гораздо менее плотная сметанообразная [16].
Аналогичные соотношения можно проследить для тех же де-
стиллатов при замене нафтенового мыла щелочными солями
нефтяных сульфокислот или даже свободными сульфокислотами,
образующимися при очистке масел крепкой, особенно же дымя-
щей серной кислотой. Будучи сильными эмульгаторами* даже-
в свободном состоянии, эти сульфокислоты несомненно являются
тем основным фактором благодаря которому образование эмуль-
сий наблюдается при промывке кислого масла не только ще-
лочью. но даже чистой водой.
Сказанного достаточно, чтобы понять, почему в производ-
ственных условиях образование эмульсий редко наблюдается при
щелочной очистке легких дестиллатов — бензина и керосина,—
31 наоборот, как обычное явление встречается при очистке ма-
сел. Все объясняется тем, что содержание нафтеновых кислот,
соли которых, как мы видели, являются сильнейшими эмульга-
торами, в бензиновых дестиллатах ничтожно, в керосиновых
обыкновенно также сравнительно не велико, ,но оно резко повы-
шается для соляровых, веретенных и машинных дестиллатов-
(ср. ч. I, гл. VII). С другой стороны, количество серной кис-
лоты, применяемой при очистке легких дестиллатов, и темпера-
турные условия этой очистки таковы, что сколько-нибудь замет-
ного образования сульфонафтеновых кислот здесь ожидать не
приходится. Таким образом, как общее правило, ввиду отсут-
ствия или недостатка эмульгаторов при очистке бензиновых и
керосиновых дестиллатов эмульсии либо вовсе не образуются,
635-
либо если и образуются, то в малоустойчивой, легко расслаи-
вающейся форме. Встречаются, правда, хотя и очень редко, ис-
ключения. Так например, имеются указания на образование
иногда при щелочной очистке керосиновых эмульсий, настолько
плотных, что их можно резать ножом [16]. Причину образова-
ния и устойчивости таких эмульсий следует искать, очевидно,
в повышенном содержании в соответствующих дестиллатах либо
нафтеновых кислот, либо каких-либо иных эмульгаторов, напри-
мер сернистых соединений; эти же обстоятельства, как ясно из
вышеизложенного, являются причиной образования устойчивых
эмульсий при очистке масляных дестиллатов.
До сих пор речь шла об эмульсиях, образующихся при уча-
стии гидрофильных эмульгаторов, т. е. об эмульсиях типа
«масло в воде». В процессе очистки нефтяных дестиллатов ино-
гда образуются эмульсии и противоположного типа, т. е. типа
«вода в масле», когда вода является дисперсной фазой, а масло —
внешней. Как было показано в ч. II, гл. I, образование таких
эмульсий требует участия гидрофобных эмульгаторов, не раство-
римых и не .набухающих в воде, но растворимых и набухающих
в дестиллатах; таковы, например, щелочно-земельные мыла,
асфальтены, смолы и т. п. Такого рода эмульгаторы могут обра-
зоваться в производственных условиях от различных причин. Так
например, вещества смолисто-асфальтового характера появляются
в дестиллате при более или менее продолжительном хранении
его в неочищенном виде; щелочно-земельные мыла образуются
при промывке, если вместо паровой (дестиллированной) воды
взять обыкновенную, особенно жесткую, воду и т. п. В нормаль-
ных условиях очистки, а также при употреблении чистою
едкого натра и паровой воды подобного рода эмульгаторы не мо-
гут образоваться, так что эмульсии типа «вода в масле» встре-
чаются при очистке масляных дестиллатов сравнительно редко.
Напротив, если выщелачивание дестиллата. или нефти ведется
до их кислотной очистки, образование эмульсий этого типа
вполне естественно.
Новейшие последования показывают, что специфичность действия эмуль-
гаторов прп образованип эмульсией того или иного типа зазпсит от целого
ряда условий, как-то: температуры, концентрации щелочи и т. д. Измене-
ние этик условий не только существенно изменяет устойчивость эмульсии, но
может изменить даже самый тип эмульсии, т. е. “вызвать ее обращение (ин-
версию). Так например, натриевые соли жирных и нафтеновых кислот,
как оказалось, могут образовать не только эмульсии типа «масло в воде»,
но также эмульсии обратного типа, т. е. «вода в масле». Как общее правило,
масляные эмульсии с увеличением в известных пределах концентрации
едкого натра (а также сульфата натрия) и с понижением до известного
•предела температуры претерпевают обращение по правилу: эмульсия типа
«масло в воде» превращается в эмульсию типа «вода в масле». Причина
образования под влиянием одного и того же мыла эмульсий различного
типа заключается, повидимому, в том, что с изменением указанных усло-
вий существенно меняется распределение мыла между фазами эмульсии,
а следовательно, изменяется характер основного фактора, определяющего
не только устойчивость, но и самый тип эмульсии [17].
Разрушение масляных эмульсий. В тех случаях, когда
эмульсии, образующиеся при очистке масляных дестиллатов,
63G
являются слишком устойчивыми и расслаиваются медленно,
приходится прибегать к тем или иным методам искусственного
их разрушения. В основе — это те же приемы, которыми поль-
зуются для разрушения естественных нефтяных эмульсий
(ч. II, гл. I); однако разнообразие этих методов, фактически при-
меняемых при очистке, здесь значительно меньше.
Из физико-механических методов разрушения эмульсий при
очистке масляных дестиллатов пользуются лишь одним нагре-
ванием. Для этой цели маслоочистная аппаратура всегда снаб-
жается паровыми змеевиками, и для скорейшего отстаивания
как щелочного раствора, так и воды после промывки подогрев
доводится до 70—80 . Несмотря на это, отстаивание щелочного
раствора после очистки различных масляных дестиллатов про-
должается 5—8 час., для отстаивания же воды посте промывки,
требуется 2 суток и более.
Механизм действия обогрева при разрушении нефтяных
эмульсий уже был рассмотрен нами в ч. II, гл. I. В случае мас-
ляной эмульсии разрушение ее при обогреве, очевидно, должно
произойти по тем же причинам, т. е. в результате разрушения
оболочек, в которые заключены капельки масла. Такое объясне-
ние действия обогрева особенно применимо к тем случаям, когда
нагревание эмульсии или даже самая обработка щелочью про-
изводятся при температуре выше 100° в автоклаве, как это ре-
комендуется некоторыми авторами. Надо думать, однако, что
при этом еще более важное значение имеет изменение, которое
претерпевает при нагревании поверхностное натяжение между
водно-щелочным раствором и маслом.
Действительно, прямой опыт показывает, что если поверх-
ностное натяжение масла на границе с чистой водой при повы-
шении температуры постепенно снижается, то для системы «ми-
неральное масло — раствор щелочи» может наблюдаться как раз
обратное явление: при очень слабых растворах щелочи (тысяч-
ные доли нормального) поверхностное натяжение между дестил-
латом и раствором щелочи при повышении температуры сильно
возрастает. А так как с повышением поверхностного натяжения
увеличивается сопротивляемость жидкости к раздроблению на
мельчайшие частицы, т. е. к ее эмульгированию, то становится
понятным, почему щелочная очистка масляных дестиллатов
должна вестись при повышенной температуре с применением
лишь очень слабых растворов щелочи.
Физико-химические методы разрушения нефтяных эмульсий,
как было показано в ч. II, гл. I, заключаются в прибавлении
различных деэмульгаторов, нарушающих устойчивость оболочек
(пленок) данной эмульсии. Деэмульгаторы, применяемые для
разрушения эмульсий, образующихся в процессе очистки ма-
сляных дестиллатов, менее разнообразны, так как применение
некоторых веществ, употребляемых для разрушения естествен-
ных нефтяных эмульсий, могло бы отразиться на качестве, ма-
сляного дестиллата. Важнейшими из деэмульгаторов для масля-
ных эмульсий являются следующие вещества:
R37
Спирты. Прибавление спирта, винного или метилового, к рас-
твору едкого натра является одним из давно известных спосо-
бов борьбы с образованием масляных эмульсий. Механизм дей-
ствия этого деэмульгатора заключается в следующем. Как уже
было отмечено, натровые мыла, образуя с водой коллоидальные
растворы, являются теми эмульгаторами, которые, накопляясь
на границе двух данных фаз, снижают поверхностное натяжение
своего растворителя (воды) на границе с другой фазой (маслом}
и тем самым вызывают образование эмульсии. Известно, однако,
что те же самые мыла, которые с водой дают коллоидальные
растворы, в водном спирте образуют не коллоидальные, а истин-
ные растворы, в которых мыла находятся в виде отдельных рас-
пределенных в растворителе молекул [18]. Ввиду этого есте-
ственно ожидать, что с прибавкой спирта способность мыля,
резко снижать поверхностное натяжение своего растворителя на
границе с маслом должна сильно понизиться, и непосредствен-
ные измерения поверхностного натяжения на границе подобного
рода двух фаз полностью подтверждают эти соображения [16].
С другой стороны, прямым следствием такого изменения при-
роды мыльного раствора в присутствии спирта должно явиться
исчезновение возможности появления прочных пленок и, следо-
вательно,— вообще образования эмульсий.
Препятствием к широкому употреблению винного или мети-
лового спиртов для борьбы с образованием масляных эмульсий
является их сравнительно высокая стоимость, и применение
спиртов при щелочной очистке масляных дестиллатов ограничи-
вается случаями получения особо высокосортных масел, например
paraffin u m liquidum и некоторых других.
Кислоты и мыла. Гораздо более широкое применение для
предупреждения и разрушения нефтяных эмульсий получило
прибавление небольших количеств таких веществ, как олеиновая
или керосинонафтеновые кислоты, а также нафтеновые мыла и
соли нефтяных сульфокислот. Повидимому, действие этих де-
эмульгаторов основано на том, что при известных концентра-
циях эти вещества начинают понижать поверхностное натяже-
ние на границе дестиллат — водная щелочь со стороны дестил-
лата., вследствие чего компенсируется то снижение поверхно-
стного натяжения со стороны водно-щелочного раствора, благо-
даря которому только и возможно образование эмульсии «масло
в воде». Таким образом здесь имеется, повидимому, лишь част-
ный случай (взаимной компенсации двух эмульгаторов, ко-
торая при известных количественных соотношениях между
последними может либо вовсе задержать образование эмульсии,
либо, привести к полному разрушению эмульсии, уже образо-
вавшейся.
Электролиты. Для разрушения масляных эмульсий иногда
применяются также поваренная соль, глауберова соль, серная
кислота и другие электролиты. Механизм действия этих -веществ
должен быть здесь, очевидно, тот же, что и при разрушении
естественных нефтяных эмульсий.
ез?
Некоторые видоизменения методики щелочной очистки.
Как было указало, щелочная очистка обыкновенно следует за
кислотной и заключается в обработке кислого масла слабым рас-
твором едкого натра. В этот обычный способ щелочной очистки
вносятся иногда изменения, а именно: либо изменяют порядок
очистки, либо заменяют едкий натр другими реагентами, напри-
мер: содой, гашеной известью. Рассмотрим эти видоизменения
щелочной очистки несколько ближе.
Если сырую нефть или неочищенный дестиллат подвергнуть
обработке водным* раствором едкого натра, то нафтеновые ки-
слоты, фенолы, а также некоторые сернистые соединения пере-
ходят в водно-щелочной раствор; эту методику начинают при-
менять у нас в настоящее время для возможно полного извлече-
ния и использования содержащихся в сырой нефти или ее де-
стиллатах нафтеновых кислот [19]. После такого выщелачива-
ния дестиллат должен быть подвергнут обычной кислотной,
а затем и щелочной очистке, нефть же подвергается предвари-
тельно обычной перегонке. Заводские опыты очистки, проведен-
ные по этому способу, показали, что не только расход серной
кислоты, но и суммарное потребление едкого натра при этом
значительно меньше, чем при обычном способе очистки. Оказы-
вается также, что после предварительной очистки щелочью, ве-
роятно. вследствие удаления легко разлагающихся при перегонке
нафтеновых и полинафтеновых кислот, получающиеся дестил-
латы обладают гораздо более высокими качествами по сравне-
нию с теми, которые получаются из того же сырья без предва-
рительной его очистки щелочью. Однако, несмотря на все ска-
занное, очистка дестиллатов с предварительным их выщелачи-
ванием пока не получила достаточно широкого применения, так
как проведение ее требует лишней операции и дополнительного
оборудования [16].
Попытки замены довольно дорогого едкого натра другими ре-
агентами делались неоднократно. Наиболее естественно, казалось
бы, заменить едкий натр более дешевой гашеной известью. Опыт
показывает, что такая замена может быть произведена лишь ча-
стично, а именно — для нейтрализации находящейся в дестил-
лате свободной серной кислоты, что составляет, например, для
машинного дестиллата обыкновенно около 50% общей кислот-
ности [20]. Напротив, попытки полной замены едкого натра из-
вестью дали малоудовлетворительные результаты вследствие
специфических свойств известковых мыл: эти мыла трудно рас-
творимы в воде и медленно оседают, растворимы в масле и не
вымываются из него водой, так что работа с ними вносит ряд
практических осложнений в задачу очистки дестиллата и за-
трудняет последующую утилизацию нафтеновых кислот [15].
Большее, повидимому, значение может иметь замена едкого
натра содой [21]. Так,’ при очистке керосинового дестиллата
10—15%-ным раствором соды удается нейтрализовать около
90% всех -кислот, содержащихся в дестиллате, и только послед-
ние 10% кислот требуют для своего удаления едкого натра.
639
Обязательным условием успешного применения соды при очи-
стке является достаточная температура (25—30°), при которой:
выделяющаяся углекислота удалялась бы из сферы реакции воз-
можно полнее. В противном случае между компонентами реак-
ции устанавливается равновесие:
2CnHa«-i • C02H + C0sNa2^2CnH&„_1 • СО2Ха+СО2+Н.2О,
и дальнейшая нейтрализация приостанавливается.
В заключение— несколько слов о перемешивании при очи-
стке нефтепродуктов. При периодической очпбтке, которая, не-
смотря на громоздкость аппаратуры и сравнительно малую ее
производительность, до сих пор еще широко распространена
в нефтяной промышленности, перемешивание обыкновенно про-
изводится продувкой воздуха. Если с технологической точки
зрения такой способ перемешивания для всех не слишком ле-
тучих продуктов является вполне рациональным и удобным, то-
с точки зрения химической он должен быть признан малоудо-
влетворительным. Действительно, как было показано в ч. II,
гл. IV, воздух (кислород) при несколько повышенной темпера-
туре, особенно же в присутствии щелочи, действует на нефтя-
ные углеводороды окисляющим образом с образованием спиртов,
альдегидов и кислот. Отсюда естественно притти к выводу, что
механическое перемешивание при щелочной очистке с химиче-
ской точки зрения должно быть признано более рациональным,
так как в этих условиях окисляющее действие кислорода воз-
духа должно быть, очевидно, значительно слабее. К аналогич-
ному заключению приводит опытное исследование данного во-
проса в отношении кислотной очистки [22].
ВТОРИЧНАЯ ПЕРЕГОНКА В ПРОЦЕССЕ ОЧИСТКИ
В тех случаях, когда исключается необходимость приме-
нения тех или иных специальных методов очистки, подлежащих
рассмотрению в следующей главе, обработка дестиллата серной
кислотой и щелочью с последующей промывкой водой вполне
достаточна для получения из дестиллата конечного очищенного
нефтепродукта. Этот обычный порядок очистки существенно из-
меняется для крэкинг-дестиллата. После обработки серной -кис-
лотой вследствие взаимодействия последней с непредельными
углеводородами и сернистыми соединениями и образования
рассмотренных выше продуктов полимеризации и конденсации
крэкинг-дестиллат получается окрашенным в желто-коричневый
цвет с повышенными удельным весом и концом кипения. Для
освобождения от этих примесей, снижающих качество крэкинг-
дестиллата, его после кислотной очистки подвергают новой пере-
гонке, которую обычно называют вторичной, считая первичной
ту перегонку, которая была проведена на крэкинг-установке и
дала сырой крэкинг-дестиллат [23].
Количество продуктов полимеризации и конденсации («поли-
меров»), появляющихся в крэкинг-дестиллате после его кислот-
но
йой очистки, йе велико (3—5%). Их отделение могло бы,
однако, повести к значительным потерям крэкинг-бензина, так
как по условиям заводской перегонки необходимо, чтобы остаток
от перегонки составлял в среднем 15—20% от загрузки перегон-
ной аппаратуры. Таким образом, если отбор крэкинг-дестиллата
для последующей его очистки производить с нормальным кон-
цом кипения, 'весьма существенные потери крэкинг-бензина при
вторичной перегонке неизбежны. Чтобы избежать этих потерь,
отбор дестиллата при перегонке на крэкинг-установке (первич-
ная перегонка) производится в несколько более широких грани-
цах, а именно — из расчета, чтобы дестиллат, подлежащий очи-
стке, содержал примерно лишь 60—85% крэкинг-бензина, удо-
влетворяющего норме. Благодаря этому при вторичной перегонке
дестиллата после очистки его серной кислотой можно получить
от 15 до 40% остатка, состоящего из крэкинга-керосина и крэ-
кинг-газойля, вместе с которыми удерживаются также и поли-
меры.
Во избежание перегрева и возможного разложения полимеров
при перегонке, что привело бы к снижению качества дестиллата,
вторичная перегонка ведется в возможно более мягких' усло-
виях, а именно — с водяным паром, расход которого в различ-
ных случаях колеблется от 15 до 40%; нагревание доводится до
160—180°. В этих условиях полимеры сохраняют полную устой-
чивость, и разложение претерпевают лишь некоторые сернистые
соединения очищенного дестиллата; выделяющийся при этом
сернистый газ легко отмывается водой, образующейся при кон-
денсации водяного пара.
Аппаратура, применяемая для вторичной перегонки, весьма
разнообразна. На одних заводах для этой цели пользуются ку-
бовыми батареями, специально дооборудованными для этой цели
достаточной мощности колоннами. Другие заводы предпочитают
пользоваться для той же цели трубчатками, причем иногда
встречается соединение двух перегонных установок: одна из них
работает под обыкновенным давлением и служит для получения
собственно крэкинг-бензина, другая работает под вакуумом и t
служит для перегонки более тяжелых (хвостовых) фракций очи-
щенного дестиллата.
Применение, которое в настоящее время находят полимеры,
и их ценность как нефтепродуктов весьма ограничены. Первона-
чально их пробовали подмешивать к крекинговому сырью и на-
правлять на повторный крэкинг. Однако за последнеее время от
такого использования полимеров стали отказываться, так как по
сравнению с исходным сырьем они заметно богаче сернистыми
соединениями, которые при крэкинге разъедают аппаратуру и
затрудняют последующую очистку крэкинг-дестиллата. Ввиду
этого в настоящее время предпочитают подмешивать полимеры
после их перегонки к различным сортам легкого топлива, на-
пример к тракторному, дизельному или так называемому печ-
ному топливу, обслуживающему небольшие домашние отопитель-
ные устройства.
41 Вак. 1622. Химия нефти.
641
ОТХОДЫ ОЧИСТКИ И ИХ УТИЛИЗАЦИЯ
Общие методы очистки нефтепродуктов дают громадное ко-
личество отходов, утилизация которых составляет важную за-
дачу нефтеперерабатывающей промышленности: после кислот-
ной очистки получаются кислые гудроны, после обработки ще-
лочью— щелочные отбросы. Состав этих отходов и химические
основы их утилизации должны составить предмет особого рас-
смотрения.
Кислые гудроны. В зависимости от характера подвергаемого
очистке нефтепродукта и условий очистки, в частности крепости
применяемой серной кислоты, кислые гудроны можно разделить
на несколько разновидностей, заметно отличающихся друг от
друга своим составом и свойствами, а также способами их ути-
лизации.
Важнейшими из этих разновидностей являются следующие
[24, 25]:
1. Кислые гудроны от очистки легких дестиллатов прямой
гонки. Получаемые от очистки бензиновых и керосиновых де-
стиллатов кислые гудроны представляют собою легко подвиж-
ную смолистую жидкость черного цвета, содержащую 70—80%
серной кислоты и от 10 до 20% органической массы; последняя
легко выделяется разбавлением гудрона небольшим количеством
воды (5—10%) с последующим нагреванием до 60° и сохраняет
свою подвижность продолжительное время. Ближайший состав
органической массы изучен пока недостаточно; в нее входят
продукты глубокой полимеризации и конденсации непредельной
части исходных дестиллатов, сульфо- и эфирокислоты, а также
продукты гидролиза эфирокислот, т. е. спирты и продукты их
окисления.
2. Кислый гудрон от очистки крэкинг-бензина и продуктов
пиролиза нефти '(бензола, толуола и т. д.). Эта разновидность
кислого гудрона также содержит много свободной серной кис-
лоты (от 50 до 75%); содержание органической массы в ней ко-
леблется от 20 до 40%. Текуч, но при хранении более или менее
быстро густеет.
3. Кислые гудроны от очистки смазочных масел. Подвижные
или малоподвижные смолистые вещества черного цвета, до-
вольно разнообразные по составу и свойствам в зависимости от
природы масляного дестиллата и условий его очистки.
Гудроны, получаемые при очистке легких масляных дестил-
латов (соляровых и веретенных) крепкой серной кислотой, со-
держат 40—45% свободной кислоты и 45—50% органических
веществ. ^Текучи; при разбавлении водой разделяются на 'Кис-
лотный слой и органическую массу; последняя сначала под-
вижна, но при стоянии постепенно твердеет.
Гудроны от очистки машинного и цилиндрового дестиллатов
содержат .25—40% серной кислоты и 45—во % органической
массы. Текучи, но при хранении густеют и постепенно делаются
твердыми. Легкость разделения на кислоту и органическую
642
массу довольно разнообразна н зависит, повидимому, также от
природы исходной нефти. Органическая масса обыкновенно
быстро твердеет.
Наконец, Гудроны от очистки наиболее тяжелых масел типа
брайтстоков и тяжелых автолов, а также масляных полугудро-
нов содержат уже только 1Л—30% свободной серной кислоты;
органическая масса —твердая; содержание ее поднимается до
60—80%. Эти гудроны представляют собою малоподвижные смо-
листые вещества, быстро твердеющие.
4. Кислые гудроны от очистки нефтепродуктов дымящей сер-
ной кислотой. Эта разновидность кислых гудронов занимает
среди них особое место. Это — густая черная или черно-корич-
невая жидкость, густеющая при хранении. Содержание серной
кислоты достигает здесь 60—70%, содержание органической
массы не превышает 30—40%. Органическая масса легко отде-
ляется при разбавлении кислого гудрона 3—5% воды с нагре-
ванием до 80 '; она состоит главным образом из сульфокислот,
легко растворима в воде и является основной частью так на-
зываемого «контакта».
Кислые гудроны в главной своей массе представляют собою
типичные отходы, балласт нефтеперерабатывающей промышлен-
ности. За исключением некоторых их разновидностей (см.
ниже), подвергаемых специальной переработке, кислые гудроны
из года в год свозятся и спускаются в особые пруды (амбары),
загромождающие заводскую территорию и отравляющие завод-
ской район вредными газами (SO2 и т. п.). О количестве этих
отходов можно судить уже по следующей справке: в заводском
районе Азнефти имеется 5 прудов дня кислого гудрона, в кото-
рых находится примерно около 300 тыс. т кислого гудрона;
ежегодное поступление кислых гудронов но Азнефти исчис-
ляется примерно в 50 тыс. т.
Гудроны, находящиеся в прудах, обыкновенно выделяются
в особую группу — прудовые гудроны. По существу они не пред-
ставляют собою особого вида, так как являются смесью дру-
гих разновидностей кислых гудронов. Однако условия и длитель-
ность их хранения налагают на их состав и свойства особый
отпечаток.
Содержание серной кислоты в прудовых гудронах колеблется
от 3 до 15%, органической же массы — от 85 до 95%. В раз-
личных слоях содержание кислоты различно; так, верхние
слои вследствие разбавляющего действия атмосферных осадков
содержат кислоты меньше, чем средние стой. Органическая
масса прудовых гудронов представляет собою шламообразное
вещество с примесью увлеченного гудроном масла; это последнее
отстаивается в теплую погоду и собирается в виде верхнего мас-
ляного слоя.
Утилизация кислого гудрона. Использование кислого гудрона
можно представить себе по двум направлениям: утилизация со-
держащейся в гудроне серной кислоты и утилизация органиче-
ской массы гудрона.
ил» 643
Для утилизации серной 'кислоты, содержащейся в кислом
гудроне, необходимо отделить от неё органическую массу гуд.
рона. Для этого были предложены различные способы. Можно,
например, действием подходящего растворителя, а именно—
так называемого сольвента, т. е. фракции нефтяного или ка-
менноугольного дегтя с температурой кипения 120—180°, рас-
творить органическую массу гудрона и, отделив раствор, под-
вергнуть кислоту сгущению до возможной концентрации (см.
ниже). Отгоняя, далее сольвент от раствора органической массьд
получают последнюю в виде более или менее твердого асфаль-
та, растворитель же применяют к повой загрузке гудрона [20].
Метод обладает большими достоинствами, каковы: применимость
его даже к прудовым (амбарным) гудронам, полнота отделения
серной кислоты, высокий выход асфальта и т. д. Его широкому
применению препятствует лишь трудная доступность больших
количеств сольвента, который находит в настоящее время более
важное применение, а именно — как материал для пиролиза.
Другой способ выделения кислоты из кислого гудрона заклю-
чается в разбавлении гудрона водой при нагревании. Повиди-
мому, наиболее рациональна следующая методика [24]. Смотря
по разновидности гудрона и его подвижности, загружают в ме-
шалку от 5 до 15% паровой воды и, пропуская через змеевик
водяной пар, прибавляют постепенно кислый гудрон, перемеши-
вая смесь продувкой воздуха. Содержапщеся в гудроне эфиро-
серные кислоты подвергаются при этом обмыливанию. Затем,
прекратив перемешивание, дают смеси отстояться и спускают
кислоту, органическую же массу подвергают вторично такой же
обработке. Обогрев смеси, как правило, производится не свыше
60—90°. В этих условиях удается получать, считая на гудрон:
из кислого гудрона от очистки керосина — 40—45% серной
кислоты светложелтого цвета, крепостью 75—80%; из кислого
гудрона от очистки машинного масла — 25—30% серной кис-
лоты темного цвета, крепостью бо—65%; наконец, из кислого
гудрона от очистки цилиндровых масел —10—12% серной кис-
лоты крепостью 55—60%. Вместо обработки водой при нагре-
вании удобно также действовать па гудрон открытым паром: за
счет воды, обазующейся при его конденсации, разделение кис-
лотного слоя и органической массы происходит без излишнего
разведения серной кислоты.
Регенерированная из кислого гудрона серная кислота всегда
более или -менее окрашена в темный цвет за счет растворенных
в ней смолистых веществ. Тем нс менее, она с успехом может
применяться непосредственно, без дальнейшей очистки и кон-
центрации, для разных назначений, например для выделения
нафтеновых кислот из щелочных отходов (см. ниже), для выде-
ления иода из буровых вод (см. ч. I, гл. XI Б), для получения
железного купороса и других сернокислых солей и т. д. В тех
случаях, когда требуется иметь более концентрированную кис-
лоту, чем она получается в процессе регенерации из кислого
гудрона, повышение крепости ее достигается либо добавкой не-
644
обходимого количества олеума, либо упариванием регенериро-
ванной кислоты сначала под обыкновенным давлением, в свин-
цовых чренах, а затем либо при обыкновенном давлении в спе-
циальных чанах с продувкой горячего воздуха (система Che-
mlco), либо в вакууме. Затраты на получение концентрирован-
ной серной кислоты из кислого гудрона настолько значительны,
что регенерированная крепкая серная кислота обходится лишь
немногим дешевле чистой. Однако при оценке этого процесса в
целом необходимо иметь в виду, что регенерация серной кислоты
из кислого гудрона одновременно освобождает нефтеперегонный
завод от тягостного отброса производства.
Что касается органической массы кислого гудрона, получае-
мой после отделения серной кислоты, то использование ее воз-
можно «в нескольких направлениях. Можно, например, перера-
батывать органическую массу на суррогат асфальта [24], для
чего после дополнительной промывки органической массы во-
дой и удаления из нее остатков серной кислоты продувают через
массу в течение нескольких часов воздух для отгонки воды при
температуре ПО—120°, а затем продолжают эту операцию еще
3—4 часа при более высокой температуре (180—220°). Подучае-
мый таким образом асфальт оказывается, однако, более низкого
качества, чем нефтяные асфальты из смолистых нефтей (бина-
гадипской и др.). Имеются и другие предложения утилизации
органической массы кислого гудрона как-то: изготовление из
нее в смеси с древесными опилками, угольной пылью и нефтя-
ным коксом топливных брикетов, переработка ее на консистент-
ные мази и т. п. Повидимому, однако, все эти способы утилиза-
ции органической массы кислого гудрона требуют еще дополни-
тельной проработки.
Совершенно особое место среди различных способов утилиза-
ции кислого гудрона занимает переработка кислых продуктов от
очистки некоторых видов соляровых масел дымящей серной кис-
лотой. Процессы этого рода служат источником получения
весьма важного технического продукта, известного под наимено-
ванием «контакт» [27].
«Контакт» представляет собою вещество сложного состава,
получаемое в процессе обработки дымящей серной кислотой не-
которых соляровых, вазелиновых и других дестиллатов и нашед-
шее применение в различных видах промышленности. Впервые
«контакт» был применен как эмульгатор в процессе расщепле-
ния жиров еще в 1914 г. (патенты Г. С. Петрова). В настоящее
время положение «контакта» как расщепительного реагента
в жировой технике является вполне утвердившимся Ч Действие
его как расщепителя основано на его высокой эмульгирующей
способности как в кислой, так и в щелочной средах. Другое
важное применение «контакта» основано на его высокой мою-
* Подобного рода расщепители имеют в жировой промышленности боль-
шое значение. Достаточно .вспомнить, «например, широко применяемый
за границей реактив Твичвля, получаемый обработкой смеси нафталина
я олеинороЦ нцедота концентрированной серной кислотой при нагревании.
645
щей и смачивающей способности, благодаря которой «контакт»
применяется ® текстильной промышленности для замывания
пятен на ткани, удаления грязи и т. п., при изготовлении
шлихты (проклеивание и шлихтование основы) и расшлих-
товке товара, в процессах подготовки его к отбелке и крашению
и т. д.; равным образом «контакт» рекомендован для промывки
шерсти и шерстяной ткани. В кожевенной промышленности
«контакт» как прекрасный эмульгатор находит применение не
только в процессе обезжиривания голья, но также при изготов-
лении дубильных масел и жировальных смесей. Наконец, благо-
даря способности участвовать в образовании продуктов конден-
сации фенолов с формальдегидом «контакт» находит применение
при изготовлении пластических масс («карболит»), обладающих
ценными качествами.
Уже простое перечисление различных видов применения
«контакта» показывает, что получение его могло бы представлять
собою один из специальных видов переработки известных неф-
тяных дестиллатов на этот ценный продукт. Однако высокий
расход серной кислоты, потребляемой при этом, заставляет
объединять процесс его приготовления с очисткой некоторых
видов масел, а именно: с очисткой вазелинового дестиллата на
вазелиновое масло, а в самое последнее время с очисткой соля-
рового масла как сырья для получения синтетических кислот
(см. ч. П, гл. IV В).
Как было указано выше, при обработке легких масляных де-
стиллатов дымящей -серной кислотой получается сложная
смесь сульфокислот, распределяющихся частью в сернокислом,
частью в масляном слое. Ввиду того что ближайшая химиче-
ская природа этих сульфокислот пока еще не .может считаться
установленной, рационально обозначать их как «нефтяные суль-
фокислоты», указывая, таким образом, лишь на сырье, служа-
щее для их получения, без указания на природу нефтяных
углеводородов, из "которых они" образуются. Нефтяные сульфо-
кислоты являются главной составной частью «контакта; содер-
жание их в «контакте» различных марок не меньше .40%.
Остальные 60% приходятся на долю минерального масла («не-
омыляемые»), серной кислоты (от 1 до 3%) и воды; содержание
неомыляемых в зависимости от марки «контакта» может коле-
баться от 5% {«контакт» для текстильной промышленности) до
20% («контакт» для расщепления жиров).
Основными моментами технического получения «контакта»
из соляровых дестиллатов (удельного веса 0,878—0,882)
являются следующие операции:
1) Просушка дестиллата в специальных железных резер-
вуарах при 90—95°.
2) Обработка просушенного дестиллата серной кислотой
(с 20% серного ангидрида) в освинцованных агитаторах при
перемешивании подсушенным воздухом.
3) Отстаивание и отделение кислого гудрона.
4) Перевод кислого масла в новый освинцованный резервуар
646
подходящей емкости и продувка масла воздухом для удаления
сернистого и серного ангидрида, которые могут оказаться
в масле.
5) Экстракция в этом же резервуаре кислого масла для из-
влечения из него сульфокислот; последнее производилось сна-
чала небольшими количествами водного спирта, но затем оказа-
лось, что удобнее производить извлечение сульфокислот одной
дестиллированной водой в количестве 1—2%, считая на исход-
ный дестиллат.
6) Отделение экстракционной вытяжки от отстоявшегося
масла и осушение последнего продувкой воздухом.
7) Подача подсушенного масла в агитатор для повторной
обработки серной кислотой той же крепости и для дальней тих
описанных выше операций процесса, повторяемого 4 раза, при
общей затрате дымящей серной кислоты до 18—20%, считая
на исходный дестиллат.
8) Заключительная обработка сырых экстракционных вытя-
жек с целью снижения в них содержания минерального масла.
Такое обезмасливание водных вытяжек может производиться
различными способами, как-то: обработкой вытяжек небольшими
количествами спирта, разбавлением их водой с последующим на-
званием, извлечением масла из водных вытяжек бензином
и т. п. О целью наилучшего обезмасливания и получения наи-
более концентрированного «контакта» рекомендуется также
упаривание водных вытяжек «контакта» или отсаливание их
серной кислотой, поскольку взаимная растворимость «нефтяных
сульфокислот» и серной кислоты незначительна; повышенное со-
держание серной кислоты в «контакте», полученном путем от-
саливания, может быть снижено путем диализа.
Получение «контакта» связано с утилизацией тех продуктов
сернокислотной очистки масел, которые остаются главным обра-
зом в масляном слое. Вне всякого сомнения, что частично суль-
фонафтеновые кислоты содержатся также в кислом гудроне; бу-
дучи выделены из этого гудрона в свободном виде или в форме
солей, эта наиболее ценная часть кислого гудрона находит себе
применение особенно как расщепитель жиров. Другое важное
применение кислого гудрона связано с его деэмульгирующими
свойствами, благодаря которым он находит применение при
борьбе с эмульсиями в процессах обезвоживания нефти (см.
ч. II, гл. I), при щелочной очистке масел (ср. выше) и т. и.
Наконец, благодаря высокому содержанию серной кислоты кис-
лые гудроны от очистки масел дымящей серной кислотой нахо-
дят успешное применение в сернокислотной очистке тяжелых
остаточных масел, заменяя собою свежий моногидрат [28].
Наиболее рациональный способ утилизации кислого гудрона
заключается повидимому, в следующем. Гудрон прокачивают че-
рез специальный конвертор, где в надлежащих условиях орга-
ническая часть гудрона превращается в пористый, хрупкий
кокс, а сера — в сернистый газ (SO2); последний после необхо-
димой очистки смешивается с требуемым объемом воздуха и пре-
647
вращается контактным методом в серный ангидрид и дымящую
серную кислоту [29].
Необходимо отметить в заключение, что на некоторых заво-
дах США кислый гудрон утилизируется как топливо. Тепло-
творная способность кислого гудрона от очистки смазочных ма-
сел в среднем равна 7250 кал; содержание в нем серы—около
6,9%, золы — около 1,4%. Кислый гудрон перекачивают из ме-
шалки в смесительный чан и разбавляют мазутом в отношении
7:3; чтобы гудрон оставался во взвешенном состоянии и не
давал твердых комков, к смеси добавляют немного извести, на-
гревают эту смесь до 38° с энергичным перемешиванием и на-
правляют в топки. Вся сера этого топлива превращается в про-
цессе сгорания в сернистый газ и сероводород; тем не менее,
пока вода, образуемая при горении, не конденсируется, котлы
и другая аппаратура заметно не разъедаются. Коррозия начи-
нается лишь после того, как топка тушится, и со сконденсиро-
вавшейся водой начинается образование серной и сернистой
кислот, действующих сильно корродирующим образом. Для пре-
дупреждения коррозии в данном случае рекомендуется: а) про-
изводить растапливание топок обычным топливом и вводить
в них -гудрон лишь после того, -как температура топки подни-
мется достаточно высоко, и б) после того как топка потушена,
следует обрызгать ее раствором щелочи.
Щелочные отбросы и их утилизация. Щелочные отбросы,
получаемые после щелочной очистки нефтяных дестиллатов,
представляют собой разбавленные водные растворы солей серной
кислоты и нафтеновых кислот; количество свободной, не исполь-
зованной щелочи редко достигает в них 1%. Нафтеновые кис-
лоты, содержащиеся в щелочных отбросах от чистки некоторых
дестиллатов, например керосинового, либо самостоятельно вымы-
ваемые путем обработки щелочью, например газойля, предста-
вляют значительную ценность и утилизируются у нас для по-
лучения мылонафта и асидола.
Для получения мылонафта щелочные отбросы сначала упа-
риваются в котлах или колоннах [30], после чего нафтеновые
мыла высаливаются поваренной солью. Они собираются при
этом на поверхности в виде рыхлой или полужидкой массы,
легко отделяемой от соляного раствора, куда уходит также боль-
шая часть сернокислых солей, содержавшихся в сыром мыло-
нафте. Ввиду недостаточно твердой консистенции нафтеновые
мыла применяются в жировой технике не как таковые, а лишь
в виде подмеси к твердым мылам.
Асидол представляет собою смесь свободных нафтеновых кис-
лот, содержащихся главным образом, в керосиновом и соляро-
вом дестиллатах. Получение его производится обработкой соот-
ветствующих щелочных растворов серной кислотой. Сырые наф-
теновые кислоты всегда содержат бдлыпую или меньшую примесь
минерального масла (см. ч. I, гл. VII), полное удаление кото-
рого представляет собою довольно сложную задачу.
В последнее время у нас обращено внимание на то обстоя-
649
тельство, что так как при перегонке нефти часть нафтеновых
кислот несомненно разрушается, то для повышения выходов на
эти кислоты следует выщелачивание их производить до пере-
гонки нефти 19]. Такой порядок представляет несомненный
интерес и значение еще в одном отношении: удаление нафтено-
вых кислот из сырой нефти в некоторых случаях несомненно
снижает коррозию нефтеперегонной аппаратуры; таков, напри-
мер, случай легкой майкопской нефти и т. п. [31].
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ ОБЩИХ МЕТОДОВ ОЧИСТКИ
Несмотря на отмеченные выше недостатки очистки нефтяных
дестиллатов серной кислотой и щелочью, методы эти до сих пор
являются широко применяемыми и по справедливости могут
быть названы общими методами очистки. Отдельные задачи, вы-
двигаемые техникой применения различных нефтепродуктов, не
всегда успешно разрешаются этими методами, либо разрешаются
ими частично; таковы, например, задача обессеривания нефтя-
ных дестиллатов и некоторые другие. Для решения этих задач
приходится прибегать к другим, более совершенным методам
очистки, пополняющим то, что может быть достигнуто лишь
отчасти с помощью общих методов. Рассмотрению этих спе-
циальных методов очистки посвящена следующая глава.
ЛИТЕРАТУРА
Гурвич Л. Г., Научные основы переработки нефти, М.—Л. (1926).
Белл Г. С., Американские методы переработки нефти, изд. 11, ч. II, ОНТИ,
(1933).
Добрянский А. Ф., Курс технологии нефти, М.—Л. (1931).
Ру X. П. и Эспейч Р. Г., Очистка легких нефтепродуктов, ОНТИ (1982).
Черножуков Н. И., Теория очистки нефтепродуктов, ОНТИ (1932).
Лострин Л., Производство крэкинг-бензина, Азнефтенздат (1983).
Каличевский В. А. и Стагнер Б. А., Химическая очистка нефтепродуктов,
пер- с англ., ОНТИ (1936).
Гретц А., Химпя нефти и искусственного жидкого топливт пер. под род.
Петрова А. Д, ОНТИ (1936).
1. Brooks В. Т. a. Irwin Humphrey, „J. Am. Chem. Soc.’40, 822(1 18); Mor-
rel J. C., „Ind. Eng. Chem.“ 19, N 4, 794 (1927).
2. Heusler u, Bennstedt, „Ztschr. angew. Chem.’ 17, 264 (1904).
3. Бутлеров A. M., „Ж. P. X. 0.’ 5, 187 и 302 (1878); 8, 279 и 351 (1876);
9, 88 (1877); 11, 197 (1879); 14, 199 (1882) и многочисленные работы
позднейших авторов: см. также Лебедев С. В. и Коблянский, „Ж.
Р. X, 0." 61, 2175 (1929); Norris J. F. a. Joubert, „J. Am. Chem.
Soc.“ 49, 873 (1927) и др.
4. Klemgard Е. N., .Refiner’ 6, № 5, 51 (1927).
5. Ormandy a. Graven, .J. Inst Petr. Techn.* 18, № 65, 844 (1927), Намет-
кин и Абакумовская, „Ж. общей химии’ 2, 608 (1932); 7, 759 (1937).
6. Тарасов и Попова, „Нефт. тоз.“ 18, № 2, 260; № 6, 991 (1930).
7. Brocket, Bull. [3], 9, 137 (1898).
8. Ру X. П. и Эспейч Р. Гч Очистка легких нефтепродуктов, стр. 62 и сл.
ОНТИ (1933).
9. Brooks a. Humphrey, ,J. Am. Soc.’ 40, 622 (1918); Brooks a. Dunstan,
„Ind. Eng. Chem.’ 14, 1112 (1922).
10. Петров Г. С. и Рабинович А. Ю., Нефтяные сульфокислоты и их техни-
ческое применение, Госхимтехиздат (1932).
649
11. Pilat u. Dawidson E., „Petroleum" 24, 1559 (1928).
12. Spiegel, „Dingier's polytech. J.“ 287, 41 (1893).
13- Гурвич Л. Г., Научные основы-.., ото. 420—427.
14. Ср. Mar cusson, „Petroleum" 12, 1149 (1916).
15. Юрков, „кг. нефт. хоз." 3, № 10, 85 (1923).
16. - Гурвич Л. Г.. Научные основы..., стр. 447—471.
17. Абрамович-Дворецкая Р, М., „Аз. нефт. хоз." 15, № 2, 100 (1935); здесь ае
новейшая литература предмета.
18, Krafft, „Вег." 82, 1595 (1899).
19. Беликовский А. С, и Дружинина А. В., „Нефт. хоз." 25, № 6, 48 (193Я1
20. Черножуков И. И., „Аз. нефт- X' з." 8, К» 11, 89 (1923). ’
21. Гурвич В. Л., „Аз. нефт- х^з." 8, № 11, 90 (1923).
22. С . Гурвич Л. Г., Научные основы..., стр. 439 и сл.
23. Еострин К. В., Производство крэкинг-бензина, Азьефтеиздат (1938).
24. Власенко Б. Е., „къ нефт. хоз." 8, № 3, 95 (1926).
25. Рейснер В., „Аз. нефт. хоз." 12, № 4, 58 (1932).
26. Seidenschnur, „Petroleum" 7, 1175 (1911); Воинов В. П., Гинсбург И. Аз
нефт. хоз." 9, № 12, 51 (1929).
27. Петров Г. С. и Рабинович А. Ю., Нефтяные сульфокислоты и вх техни-
ческое применен”е, Госхимтехиздат (1932).
28. „Нефт. хоз." 15, № 8, 217 (1928).
29. Патенты J. Hechenbleikner u. „Chemical Construction Corporation" на
способ „Hechenbleikner-Sludge-Contaktprocese".
30. „Труды заводской сессии НТС“, локлад Гуреича В. Л., вып. III (1930)’
31. Выборова А., „Аз. нефт. хоз." 10, № 9, 117 (1930); 12 № 7, 79 (1982).
ГЛАВА II
ОЧИСТКА НЕФТИ И ЕЕ ДЕСТИЛЛАТОВ
СПЕЦИАЛЬНЫЕ МЕТОДЫ
В этой главе рассмотрены такие методы очистки нефти и
повышения качества ее продуктов, которые, не имея столь об-
щего значения, как методы, рассмотренные в предыдущей главе,
вое же находят широкое применение в практике нефтеперегон-
ного дела. Сюда должны быть отнесены:
А. Адсорбционные методы, основанные на применении
к очистке нефтепродуктов различного рода адсорбентов.
Б. Методы обессеривания при помощи разного рода реаген-
тов (плумбит, гипохлорит и т. п.) и катализом.
В. Методы очистки с помощью некоторых солеи, например
хлористого цинка, хлористого алюминия и т. п.
Г. Метод очистки гидрогенизацией или гидроочистка.
Д. Методы очистки с помощью селективных растворителей,
короче солъвентная очистка.
Задачей этих методов является восполнение тех или иных
недочетов общих методов очистки. Каждый из них имеет свою
собственную теоретическую базу и свои специфические особен-
ности, и каждый из этих методов подлежит поэтому особому
рассмотрению.
А. Адсорбционные методы очистки
Из всех специальных методов очистки нефтепродуктов адсорб-
ционные методы имеют наиболее общее значение. Достаточно
отметить, что эта методика применяется для очистки крэкинг-
бензина, для очистки разного рода специальных (парфюмерных,
трансформаторных, турбинных и т. и.) и автомобильных масел
(брайтстоки), для очиски керосина и т. д., при общей тенден-
ции к дальнейшему расширению областей ее применения. Сущ-
ность адсорбционных методов заключается в обработке дестил-
лата-тем или иным адсорбентом, чаще всего — так называемой
отбеливающей землей.
АДСОРБЕНТЫ В НЕФТЯНОЙ ПРОМЫШЛЕННОСТИ
Давно известно, что некоторые виды естественных земель
(глин) обладают моющей и отбеливающей способностью. Это их
6С1
свойство давно уже обеспечило им применение как в домашнем
обиходе, так и в промышленности. Широкое промышленное зна-
чение эти отбеливающие земли нашли себе первоначально
в Англии для обезжиривания тканей (fuller’s earth — фуллерова
земля, сукновальная глина *), позднее — для очистки раститель-
ных, животных и минеральных масел, жиров и восков. В конце
XIX в. громадные залежи отбеливающих земель были открыты
в Америке, во Флориде (флоридин), а в начале XX в. — в Бава-
рии. В пределах СООР отбеливающие глины также давно из-
вестны; таковы, например, гжельская глина (мыловка) подмо-
сковного района, крымская глина «кил», шемахинское «горное
мыло» и др.; за последние 20 лет в СООР был найден целый ряд
месторождений отбеливающих глин, среди которых наибольшую
известность приобрели глуховский каолин (с. Полошки Глухов-
ского у. Черниговской обл.) и гумбрин (с. Гумбри около Ку-
тайся).
Кроме отбеливающих земель, высокими отбеливающими свой-
ствами обладают такие вещества, как силикагель, т. е. искус-
ственно приготовленная аморфная водная кремнекислота; не-
которые породы, богатые естественным силикагелем (трепела,
опоки); животный и активированный растительный уголь и не-
которые другие пористые материалы [1].
В нефтяной промышленности адсорбционная очистка приме-
няется не только для обесцвечивания, но также и для других
целей, каковы, например, нейтрализация масел и стабилизация
крэкинг-бензина. Соответственно этому не только методика при-
менения, но и самый род применяемого адсорбента бывают раз-
личны. Широкое применение здесь получили, однако, лишь
глины двух основных типов:
1. Естественнее глины, обладающие высокой способностью
поглощать кислоты, но очень незначительными обесцвечиваю-
щими свойствами. Типичным представителем такого рода глин
является флоридин.
2. Бентониты, в сыром виде совершенно не обладающие обес-
цвечивающими свойствами; однако после обработки минераль-
ной (серной) кислотой из них получается продукт весьма высо-
ких качеств как в отношении нейтрализации масел, так и в от-
ношении их обесцвечивания; таковы, например, американская
глина palex, немецкие глины различных фирм, советский крым-
силь и др.
Глины представляют собою мягкие, кристаллические или
аморфные породы, в состав которых входят главным образом
кремнекислота, глинозем и вода; кроме того, в них всегда содер-
жатся также железо (окисное и закисное), кальций, магний и
щелочные металлы. В качестве адсорбентов применимы лишь
глины, обладающие характером геля общей формулы:
А1(0Н)3 • wSiO2 • wH20.
1 Fuller — сукновал, валяльщик сукон.
652
Таким образом отбеливающие глины должны быть отнесены
по своему составу к гидросиликатам алюминия. Большая часть
их малоактивна в качестве адсорбентов; путем обработки серной
кислотой часть связанной кремнекислоты данного алюмосили-
ката выделяется в свободном состоянии, и адсорбционная спо-
собность глины значительно повышается. Такие отбеливающие
глины называются активированными или искусственными.
ТЕХНИКА АДСОРБЦИОННОЙ ОЧИСТКИ
Очистка адсорбентами может производиться либо в паровой,
либо в жидкой фазе.
Парофазная очистка при помощи адсорбентов применяется
для очистки бензинов крэкинга; особенное распространение по-
лучил здесь способ Грея, сущность которого заключается в сле-
дующем (фиг. 117).
204°Ц
Фиг. 117. Схема очистной установки ио Грею:
1 — колонна крэкинг-установки; 2 — кондевсаторы; 3 — газос₽паратор;
4 — насосы для орошения; г> — вода; 6 — газ; 7 —камеры Грея; 8 — сбор-
ник для полимеров; 9 — насос; ю — холодильник; 11 — выход полимеров;
13 — дополнительная колонка; 13 — газооепаратор; 14 — бензин.
Пары крэкинг-бензина прямо из ректификационной колонны
крэкинг-установки пропускают через специальные башни 7, точ-
нее— камеры, наполненные флоридином. Камеры Грея новей-
ших конструкций представляют собою громадные цилиндры вы-
сотою до 5,2 м при диаметре до 5,5 м, с двойным перфорирован-
ным дном, на котором покоится сплошной слой адсорбента.
В цилиндрической части камеры имеются три горизонтальных
перегородки также из перфорированного железа, прикрытые, как
и дно, медной сеткой для поддерживания зерен флоридина и
свободного прохода паров бензина.
653
Ввод паров — сверху через специальный патрубок; выход—
снизу после нижней перегородки. На нижнем конусе имеются
также особый патрубок для спуска образующихся полимеров и
змеевик с закрытым паром для подогрева. Для загрузки и раз-
грузки адсорбента камеры снабжены обыкновенно четырьмя лю-
ками: один наверху, два на боковой поверхности и один внизу.
Камеры устанавливаются на эстакаде и снаружи хорошо изо-
лированы. На фиг. 73 представлена схема очистной установки,
непосредственно соединенной с ректификационной колонной
крэкинг-установки 1. Обыкновенно такая установка имеет две
камеры 7: пока работает одна, другая разгружается от исполь-
зованного адсорбента и загружается свежим. В других случаях
на две ректификационные колонны ограничиваются тремя ад-
сорбционными камерами. Для заполнения камер применяется
зернений флоридин в виде крупки размером 30 X 60 меш
Сущность очистного действия флоридина ври повышенной
температуре на некоторые углеводороды крэкинг-бензина,
а именно на его диолефины, заключается в их полимеризации,
тогда как на ароматику, нефтены, парафины и олефины крэкинг-
бензина флоридин не оказывает заметного действия даже при
повышенной температуре. Образующиеся полимеры, имея более
высокую температуру кипения, чем основная масса углеводоро-
дов крэкинг-бензина, сгущаются в жидкость, и таким образом
происходит легкое удаление наименее устойчивых частей крэ-
кинг-бензина от главной его массы, находящейся в парообразном
состоянии. Сконденсировавшиеся полимеры направляются в спе-
циальные сборники 8, откуда их перекачивают насосом 9 в рек-
тификационную колонну крэкинг-установки 1 для выделения
из них увлеченного ими бензина. Окончательно отбензиненные
полимеры вместе с флегмой колонны могут быть направлены
в лечь на новое крэкирование; что же касается освобожденного
от полимеров бензина, то для получения товарного продукта его
направляют в специальную колонну очистной установки 12, на
которой и производится окончательное отделение полимеров от
основной массы бензина.
Очистка крэкинг-бензина флоридином в паровой фазе полу-
чила широкое распространение. Тем не менее, приходится отме-
тить, что очищенный этим способом бензин имеет меньшую ста-
бильность, чем крэкинг-бензин, очищенный серной кислотой.
Поэтому очистка 'флоридином в паровой фазе применяется
только в таких случаях, когда очищенный бензин сразу же по-
ступает на рынок, без длительного хранения. Экономически па-
рофазная очистка крэкинг-бензина флоридином в несколько раз
выгоднее сернокислотной очистки. Кроме того, при очистке фло-
* Mash (англ.)— петля, ячейка сети, сита; также мера числа отверстий
в сите на 1 дюйм или величина размола сыпучих тел. Наиболее распро-
страненные сита имеют 16, 30, 60, 50, 200 отверстий на 1 линейный дюйм.
Крупка размола 30 X 60 меш обозначает такую величину зерен, которые
целиком проходят через сито с 30 отверстиями на 1 дюйм и нацело за-
держиваются ситом с 60 отверстиями на 1 дюйм.
654
ридином не происходит потери антидетонационных свойств бен-
зина, что частично имеет место при очистке его серной кисло-
той за счет увлечения серной кислотой части олефинов.
кофазная очистка адсорбентами применяется в двух ви-
доизменениях, а именно: в форме перколяции1 или в виде кон-
тактного фильтрования.
Перколяция при помощи глины заключается в том, что вер-
тикальный цилиндрический сосуд загружают сплошным слоем
зерненой глины размола 16X 30, 30 X 60 или 60X 90 меш и
через ©тот слой, как через фильтр, пропускают тот или иной
нефтепродукт. В процессе этого фильтрования глина адсорби-
рует смолы и кислые продукты, и нефтепродукт выходит из
фильтра освобожденным от большей части содержавшихся в нем
смол со значительно улучшенным цветом. Этим способом удобно
производить очистку керосина и смазочных масел, особенно
легких; тяжелые масла, например цилиндровые, приходится
фильтровать в целях снижения вязкости в растворе лигроина.
Перколяционное фильтрование имеет ряд весьма существен-
ных недостатков, важнейшие из которых следующие. Перколя-
ция не дает прежде всего равномерной очистки нефтепродукта.
Первые его порции, проходя через свежий адсорбент, очищаются
прекрасно, даже чрезмерно, но затем по мере загрязнения адсор-
бента степень очистки постепенно снижается, и вскоре насту-
пает недоочистка. От смешения продуктов различных степеней
очистки можно, конечно, получить продукт, удовлетворяющий
тем или иным техническим нормам, однако продукт этот—явно
неоднородный, что и сказывается как при его хранении, так
в процессе его работы. Естественным следствием сказанного
являются необходимость смены адсорбента и его регенерация.
Такую регенерацию можно производить многократно, до 30 раз;
однако расходы по регенерации адсорбента ложатся на весь про-
цесс очистки этим способом крайне обременительно, тем более
что применение глины мелкого размола и более эффективной
в данном случае исключается: при такой глине фильтр быстро
засоряется, фильтрование же можно осуществлять лишь при по-
вышенном давлении, что еще более увеличивает расход как на
оборудование, так и на эксплоатацию (мощные насосы).
Второй вид очистки нефтепродуктов в жидкой фазе при по-
мощи адсорбента — контактное фильтрование — состоит в сле-
дующем: нефтепродукт смешивается с пылевидной глиной такой
степени размола, что 85—90% ее проходит через сито в 200 меш,
смесь нагревается затем определенное время до более или менее
высокой температуры, а затем освобождатся от глины на фильтр-
прессе. Благодаря мелкому размолу глины и повышенной тем-
пературе, снижающей вязкость масла, между глиной и маслом
осуществляется весьма близкое соприкосновение («контакт»), и,
таким образом, достигается весьма высокая и притом равномер-
ная степень очистки.
1 Percolation — фильтрование, просачивание.
665
Для контактного фильтрования в США широко применяются
различные сорта глин, как природных (из Флориды, Аттапульгус
и Тексаса), так и активированных (Палекс, Фильтрол и др.).
Основными условиями успеха при контактном фильтровании,
кроме выбора подходящего для данного случая адсорбента, яв-
ляется соблюдение ряда основных условий работы, а именно:
для получения равномерно очищенного продукта необходимо,
чтобы глина была возможно более равномерно распределена
в масле, что достигается энергичным механическим перемешива-
нием смеси вплоть до поступления смеси на нагревание. Послед-
нее производится в трубчатой печи обычного типа; смотря по
нефтепродукту д природе адсорбента, температура смеси, выхо-
дящей из печи, может колебаться в различных случаях от 175
до 315°. Так как глина, применяемая для контактного фильтро-
вания, содержит значительное количество влаги, то при нагре-
вании образуется значительное количество водяного пара, и по
выходе из печи нагретая смесь должна быть направлена в сепа-
ратор. Если смесь не особенно вязкая, то по охлаждении -(до
95—150°) она прямо направляется на фильтр-пресс; смеси высо-
кой вязкости предварительно разбавляются лигроином, и только
после этого их фильтруют.
Контактное фильтрование имеет существенные преимущества
перед перколяцией, из которых главное — однородность получае-
мого готового продукта при сравнительно небольших первона-
чальных затратах. Если добавить сюда, что очистка с помощью
глины освобождает от необходимости очистки масла щелочью
(процесса, сравнительно дорогого и сопровождающегося иногда
известными осложнениями), то становится понятным, что лишь
высокие цены на хорошую глину служат препятствием к все-
общему распространению контактного фильтрования.
Парожидкофазная очистка при помощи адсорбентов, иначе
говоря, очистка в смешанной фазе, также находит применение
для некоторых нефтепродуктов, особенно парофазного крэкинг-
бензина. Основания этого метода заключаются в том, что с по-
вышением давления и температуры эффективность действия
глины на крэкинг-бензин сильно повышается. По способу Остер-
стрема взаимодействие глины и бензина проводят в трубчатой
печи при давлении 15—35 ат и температуре 315—345°; из
печи продукт поступает в эвапоратор, где происходит разделе-
ние: полимеры и глина оседают на дно эвапоратора, откуда их
отводят в холодильник и на фильтр-прессы; очищенный же бен-
зин через верхнюю часть эвапоратора направляется в ректифи-
кационную колонну для окончательного освобождения от послед-
них следов полимеров. Таким образом данный способ представ-
ляет собою довольно удачную комбинацию контактного фильтро-
вания и парофазной очистки со вторичной перегонкой крэкияг-
десгиллата.
ОБЩАЯ ТЕОРИЯ АДСОРБЦИИ
Выше было показано, что в процессах, в которых участвуют
две соприкасающиеся фазы, чрезвычайно важное значение
6Б6
имеют свойства поверхности раздела, т. е. пограничного слоя»
отделяющего одну фазу от другой. Особенно важную роль начи-
нают приобретать эти пограничные слои в тех случаях, когда
повышается степень дисперсности данной системы, т. е. когда
поверхность, занимаемая данной массой тела, сильно возрастает
по сравнению с объемом (ср. ч. II, гл. I).
Если размеры тела, например (для простоты) куба или шара,
уменьшаются, то объем его V уменьшается быстрее, чем поверх-
ность $. Действительно, для куба, ребро которого—а, имеем
V = а3 и Ь' = ба2, откуда удельная поверхность, т. е. поверх-
ность, отнесенная к единице объема:
8: Г=6а®:а®=—.
а
Аналогично для шара радиуса г имеем:
Таким образом удельная поверхность тела изменяется обрат-
но пропорционально его линейным размерам, т. е. стороне куба
или радиусу шара. Отсюда понятно, что при делении тела на
все меньшие и меньшие части при его диспергировании объем
тела остается, естественно, постоянным; общая же поверхность
всех его частей непрерывно возрастает. Понятно также, что
у пористых тел, каковы силикагель, уголь (особенно активиро-
ванный), некоторые виды глины и т. п., общая поверхность всех
частиц их в данном объеме чрезвычайно велика. Мерой степени
их дисперсности может служить отношение:
где г —средний линейный размер частиц данного тела и а — по,-
стоянная, зависящая от формы частиц. В табл. 145 для иллю-
страции сказанного показан рост удельной поверхности (размер-
ность— слй-1) при возрастающем дроблении 1 ел8 какого-либо
тела на кубики меньших размеров.
Таблица 145
Возрастание удельной поверхности
(по В. Оствальду)
Длина стороны куба Число кубов Суммарная поверхность Удельная поверхность в см—1
1 см 1 6 см2 6
1 мм 10« 60 • 6-10
0,1 , 106 600 ff 6-103
0,01 . 109 6000 tl 6-10»
1р. 1012 6 м? 6-10*
1pp. 1021 6000 б-Ю1
42 Зак. 1622. Химия нефти.
657
Известно, что поверхности, отделяющие одну фазу системы
от другой, короче говоря, поверхности раздела, характеризуются
некоторым избытком энергии, называемой поверхностной энер-
гией. В тех случаях, когда одна из фаз — жидкая, эта энергия
проявляется в поверхностном натяжении, т. е. в особой между-
молекулярной силе, которая, будучи направлена перпендикурно
к поверхности внутрь жидкости, стремится уменьшить эту по-
верхность до минимума, как бы втягивая находящиеся на по-
верхности молекулы внутрь жидкости. Благодаря поверхно-
стному натяжению капля жидкости, освобожденная от действия
силы тяжести, как известно, самопроизвольно принимает форму
шара (опыт Плато). Представим теперь себе, что некоторое
число частиц жидкости должно быть переведено изнутри на ее
поверхность, которая, таким образом, претерпевает некоторое
увеличение. Очевидно, при 'этом должна быть затрачена неко-
торая работа против сил поверхностного натяжения, и эта ра-
бота, отнесенная к созданию 1 смг новой поверхности1, может
служить мерой поверхностного натяжения а для данной погра-
ничной среды при данных условиях:
а = Н : откуда В = aS,
где В— работа, которую требуется затратить для образования
новой поверхности жидкости 8.
Соотношение между поверхностным натяжением а и капил-
лярной постоятгной а2, а равно методика определения поверх-
ностного натяжения вкратце даны в ч. 1, гл. II.
Если увеличение поверхности жидкости производится в изо-
термическом и обратном процессе, то вся работа, производимая
пар'-фаза 2
Фиг. 118. Схема молекулярных сил на границе
двух фаз.
при этом против мо-
лекулярных сил,
превращается в' за-
пас свободной энер-
гии F на границе
раздела соответст-
вующих фаз; при
этом очевидно:
F — 7? = aS.
Указанные выше
соотношения могут
быть иллюстрирова-
ны фиг. 118. Пусть
Р—радиус сферы действия молекул некоторой жидкой среды,
для которой имеется поверхность раздела £ с другой фазой, на-
пример с паром. Тогда действие молекулярных сил на любую
из молекул жидкости, лежащих в се глубине на расстоянии р
от поверхности раздела или еще глубже, очевидно, взаимно
1 Или, что то ж.с, сила, отнесенная к единице длины поверхностного
слоя.
658
уничтожается. Напротив, если молекула Находится на расстоя-
нии от поверхности раздела, меньшем, чем р, то часть между-
молекулярных сил, действующих на эту молекулу, сохранится
от в аимн ii ком и ации и даст начало некоторой слагающей
поверхностного натяжения, направленной внутрь жидкости.
Наибольшего значения эти слагающие достигают, очевидно, для
молекул, расположенных непосредственно на поверхности’ раз-
дела, так как в этих случаях они получаются от полных мо-
лекулярных полусфер радиуса р.
Поверхностное натяжение жидкости зависит от многих при-
чин и прежде всего от характера пограничной фазы. Так напри-
мер, поверхностное натяжение для пары вода—бензол а = 3бд
для пары же вода — эфир а = 9,7 (при 20°). С другой стороны,
от прибавления различных веществ к данной жидкости поверх-
ностное натяжение ее может либо понижаться, либо возрастать.
Примером первого рода может служить добавка к воде пропило-
вого или иного спирта, резко снижающая поверхностное натя-
жение воды; подобного рода вещества называются поверхностно-
или капиллярно-активными; кроме спиртов, к ним относятся
жирные кислоты и их эфиры. Напротив, большинство электроли-
тов, а также белковые вещества изменяют поверхностное натя-
жение лишь в малой степени, обычно лишь слегка повышая его;
вещества этого рода называются поверхностно- или капиллярно-
неактивными.
Крайне важно, наконец, что величина поверхностного натя-
жения связана, с концентрацией молекул, растворенных на по-
верхности. Действительно, так как система всегда, стремится по-
строить свою поверхность с минимальной затратой энергии, то
иа этой поверхности будут сосредоточиваться по преимуществу,
очевидно, те молекулы, с присутствием которых связана мини-
мальная поверхностная энергия. Отсюда следует ожидать, что
вещества, понижающие поверхностное натяжение, т. е. поверх-
ностно-активные, будут скопляться на поверхности раздела, и
наоборот, вещества поверхностно-неактивные не будут нако-
пляться на поверхности, а распределятся преимущественно
внутри жидкости. Применив к этим соображениям качествен-
ного характера второй закон термодинамики, Гиббс, придал им
следующую количественную формулировку:
й__ с da
— ЯГ ‘ de ’
где 8 — разность концентраций вещества на поверхности и во
всем объеме жидкости, выраженная в граммолекулах; с — кон-
центрация вещества в растворе.
Последнее уравнение полностью приложимо к явлениям
адсорбции, под которыми в наиболее общем смысле следует под-
разумевать всякое изменение концентрации раствора у его по-
верхности раздела с какой-либо иной твердой, жидкой или газо-
образной фазой. Вещества, которые с возрастанием концентра-
ции понижают поверхностное натяжение, т. е. капиллярно-актив-
ные .<0), адсорбируются положительно (8>0); напро-
тив, вещества, с повышением концентрации которых в растворе
поверхностное натяжение возрастает, т. е. капиллярно-неактив-
ные >0), адсорбируются отрицательно (8<0), т. е. вы-
тесняются с поверхности раздела. Ряд примеров подобного рода,
имеющих ближайшее отношение к химии нефти и ее переработке,*
был дан в ч. II, гл. I в связи с вопросом об обезвоживании
нефти и в ч. III, гл. 1 в связи с вопросом о разрушении эмуль-
сий, образующихся при щелочной очистке.
Еще более наглядную картину явлений адсорбции дают те
случаи, когда адсорбированное вещество практически нераство-
римо в адсорбирующем (адсорбенте). 'Примерами явлений этого
рода может служить, с одной стороны, адсорбция углем, особенно
активированным, силикагелем, глинами и другими подобными
адсорбентами разного рода газов и паров, например в угольных
противогазах, в адсорбционных установках для поглощения па-
ров растворителей из воздуха и газового бензина из естествен-
ного газа и т. и.; с другой стороны, адсорбция теми же или
иными адсорбентами разного рода жидких и твердых веществ из
их растворов, например обесцвечивание разного рода растворов,
окрашенных ют присутствия смолистых и иных веществ, про-
цессы крашения и т. д. Сюда же должны быть отнесены описан-
ные выше случаи адсорбции, нашедшие применение в процессе
очистки разного рода нефтепродуктов.
Действительно, допустим, что в тот или иной раствор вно-
сится твердое тело с достаточно развитой поверхностью. Если
поверхностное натяжение раствора на границе с этим телом
с повышением концентрации раствора уменьшается, то' концен-
трация раствора на этой границе в действительности стремится
к возрастанию, снижаясь тем самым в остальной массе раствора;
в таких случаях говорят, что твердое тело адсорбировало на
своей поверхности часть растворенного вещества. Наоборот,, если
поверхностное натяжение на границе раствора с твердой фазой
с повышением концентрации раствора возрастает, то концентра-
ция раствора на этой границе в действительности стремится
к снижению, возрастая вследствие этого в остальной массе рас-
твора; подобного рода случаи известны под наименованием от-
рицательной адсорбции.
Количественные соотношения для зависимости степени
адсорбции от концентрации раствора и количества адсорбента
во многих случаях хорошо выражаются следующей эмпириче-
ской формулой изотермы адсорбции (Фрейндлих):
где А — количество адсорбированного вещества на единицу
массы (1 г) адсорбента; с — концентрация раствора в равнове-
сии с адсорбентом; К и п — постоянные для данных веществ:
660
адсорбента, растворителя и растворенного вещества. Для
адсорбции пз раствора показатель -~- колеблется для разных
веществ от 0,1 до 0,5.
Недостаток ©той формулы состоит в том, что она дает непре-
рывное возрастание количества адсорбированного вещества А
с увеличением концентрации с без указания на существование
некоторого предела, связанного с насыщением адсорбционного
слоя.
Между тем, в действительности изотерма адсорбции, как по-
казывает опыт, должна иметь совершенно иной вид, а именно:
первые порции поверх-
ностно-активного веще-
ства дают резкое сни-
жение поверхностного
натяжения а с повыше-
нием концентрации, и
адсорбция растет пер-
воначально пропор-
ционально концентра-
ции с. Затем постепен-
но наступает насыще-
ние адсорбционного
слоя, адсорбция начи-
нает отставать от роста
концентрации с, и, на-
конец, всякая зависи-
мость между адсорб-
цией и дальнейшим
Фиг. 119. График адсорбции А п поверх-
ностного натяжения а в зависимости от кон-
центрации.
повышением концентрации с практически исчезает: адсорбция
достигает своего предельного значения и практически перестает
изменяться от увеличения концентрации. Фиг. 119 может слу-
жить иллюстрацией сказанного; здесь же представлено измене-
ние поверхностного напряжения а с концентрацией для дан-
ного случая (изоамиловый спирт на границе системы вода —
воздух).
Такой изотерме адсорбции хорошо удовлетворят следующее
уравнение (Лангмюир):
с
с -j- о ’
А —
где го —предел адсорбции, к которому последняя стремится
при безграничном увеличении концентрации (с—>счэ). Очевид-
но, это—'уравнение гиперболы.
При ближайшем ознакомлении с явлениями адсорбции
прежде всего бросаются в глаза необычайное разнообразие этих
явлений, а затем насомненная их специфичность (см. ниже): не
всякое вещество может быть адсорбировано любым адсорбентом.
Здесь наблюдаются соотношения, до известной степени напоми-
нающие явления химического сродства, и, естественно, возни-
661
кает вопрос, какими же' силами обусловливаются явления
адсорбции. По ©тому, вопросу имеются различные взгляды. По
мнению одних исследователей в основу явлений адсорбции
должны быть положены электрические силы, поскольку согласно
современным представлениям междумолекулярные силы должны
иметь электрическую природу. Другие авторы пытались разъя-
снить явления адсорбции чисто механическими представле-
ниями, допуская в данном случае проявление физико-химиче-
ской силы притяжения. Во всяком случае, с количественной
точки зрения ни те, ни другие представления пока еще не могут
считаться окончательно оформившимися [2].
ТЕОРИЯ-АДСОРБЦИОННОЙ ОЧИСТКИ
Очистка с помощью отбеливающих глин и порошков осно-
вана, как ясно из вышеизложенного, прежде всего на специфи-
ческой способности применяемых адсорбентов удерживать на
своей поверхности некоторые компоненты {примеси) нефтяных
дестиллатов, которые снижают качества данного продукта, а так-
же на способности адсорбентов содействовать явлениям полиме-
ризации и конденсации других компонентов тех же дестиллатов.
На первом месте среди подобного рода примесей, удаление кото-
рых при помощи адсорбентов производится особенно легко и
удобно, должны быть поставлены смолистые вещества и продукты
их дальнейших превращений — асфальтены.
Смолистые вещества придают нефтепродуктам более или ме-
нее интенсивную окраску, увеличивают коксуемость масел и
являются для нефтепродуктов не только нежелательной, но и
вредной примесью. Они представляют собой наиболее тяжелые
части каждого нефтепродукта, и молекулярный вес их возра-
стает по мере утяжеления (повышения температуры кипения)
нефтепродукта (ср. ч. I, гл. X). Они легко адсорбируются фло-
ридином, углем и другими подходящими адсорбентами и тем
прочнее удерживаются ими, чем больше их молекулярный вес.
Табл. 146 может служить иллюстрацией сказанного.
Таблица 146
Адсорбция смол флоридином
(по Гурвичу)
Обозначение смол До промыв- ки в °/о После про- мывки в %
Смолы из цилиндрового масла . 11,8 8,0
„ , машинного „ 10,1 7,1
„ . веретенного 8,2 4.8
В первом столбце этой таблицы показаны проценты смол (от
веса флоридина), адсорбированных при действии 1 г флоридина
662
на 100 г однопроцентного раствора смол в бензине; во втором
столбце — количество смол, удержанное адсорбентом после про-
мывки большим избытком бензина. Соотношения эти вполне
понятны, если рассматривать адсорбцию как проявление между -
молекулярных сил между адсорбентом и адсорбируемым веще-
ством; при этом, очевидно, при прочих равных условиях весовое
количество адсорбированного вещества должно быть тем больше,
чем выше его молекулярный вес.
Аналогичные явления наблюдаются при адсорбции нафтено-
вых кислот из высших углеводородов, например парафинов. II
здесь высокомолекулярные соединения адсорбируются флориди-
ном сильнее и удерживаются прочнее, чем низкомолекулярные.
Специфический характер явлений адсорбции при ‘ очистке
нефтепродуктов проявляется в высшей степени многообразно.
Так например, американский флоридин обладает высокой ней-
трализнрующей способностью по отношению к кислому маслу,
тогда как у гумбрина эта способность выражена настолько слабо,
что при очистке гумбрином бакинских масляных дестиллатов
требуется последовательная комбинированная обработка дестил-
латов щелочью, кислотой и отбеливающей глиной.
Но особенно ярко выступает специфический характер явле-
ний адсорбции в условиях применения одного и того же адсор-
бента, папример флоридина, к углеводородам различных рядов.
Опыт показывает, что наиболее легко поддаются адсорбции угле-
водороды непредельного характера, причем вслед за адсорбцией
их на поверхности адсорбента они нередко претерпевают более
или менее глубокую полимеризацию. Так например, под влия-
нием флоридина изобутилен превращается в триизобутилен,
амилен — в диамилен и т. д.
Вслед за непредельными углеводородами по способности ад-
сорбироваться флоридином должны быть поставлены углеводо-
роды ароматического ряда; значительно слабее последних адсор-
бируются нафтены, на последнем же месте в этом отношении
стоят углеводороды ряда метана.
Если сравнить, однако, твердые парафины с полинафтенами,
то как оказывается, последние адсорбируются труднее первых,
и в этом случае специфический характер сил, обусловливающих
адсорбцию и напоминающих силу химического сродства, вы-
является особенно отчетливо.
До сих пор речь шла о влиянии на адсорбцию растворенного
вещества; при этом, однако, известную роль не может не играть
также природа растворителя: чем легче адсорбируется данным
адсорбентом растворитель, тем меньше адсорбируется при про-
чих равных условиях растворенное в нем вещество. Поэтому,
например, адсорбция смол легче осуществляется из бензинового
раствора, чем из 'бензольного и т. п. В тех случаях, когда
адсорбция уже произошла, обратное извлечение смол из адсор-
бента может быть осуществлено различными растворителями в
порядке постепенно возрастающей склонности их к данному
адсорбенту. Так например, смолы, поглощенные флоридином,
663
извлекаются различными растворителями в следующем порядке:
после того как флоридин был промыт избытком бензина, бензол
извлекает из него некоторое количество смол с более высоким
удельным весом и иодным числом, чем смолы, извлекаемые
бензином; когда извлечение бензолом закончено, переход к но-
вому растворителю, хлороформу, приводит к смолам нового типа
с еще более высоким удельным весом и иодным числом и т.-д.
Очевидно, и здесь приходится иметь дело со специфическим ха-
рактером тех междумолекулярных сил (в данном случае между
адсорбентом и растворителем), с проявлением которых связаны
явления адсорбции.
В тех случаях, когда количество адсорбента недостаточно для
поглощения всех веществ, содержащихся в нефтепродукте и спо-
собных адсорбироваться в данных условиях, между этими веще-
ствами может возникнуть своего рода соревнование за возможно
полное поглощение адсорбентом. При этом в случае таких
сложных смесей, как тот или иной дестиллат, могут происходить
разнообразнейшие процессы взаимного вытеснения одних ве-
ществ другими и установление в конце концов некоторого рав-
новесия. Естественно, что при этом наиболее прочно удержи-
ваются те вещества, которые в отдельности наиболее энергично
адсорбируются данным адсорбентом, а именно: из нефтепродук-
тов— смолистые и асфальтовые вещества, содержащиеся в дан-
ном дестиллате. Так как, однако, некоторая часть адсорбента
может расходоваться при этом также и на другие компоненты,
например на нафтеновые кислоты, азотистые и сернистые соеди-
нения и т. п., то следует по возможности удалять эти вещества
предварительно путем обработки дестиллата подходящим реаген-
том: нафтеновые кислоты—щелочью, азотистые соединения —
слабой серной кислотой и т. п.
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ АДСОРБЦИОННЫХ МЕТОДОВ ОЧИСТКИ
Как было указано в начале этой главы, адсорбционным ме-
тодам по общности их применения и значимости должно быть
отведено первое место среди специальных методов очистки. Не
говоря уже о том, что адсорбенты применяются для очистки са-
мых разнообразных видов нефтепродуктов, начиная с наиболее
легких (бензины, керосины) и кончая самыми тяжелыми (цилин-
дровые масла), достаточно отметить здесь, что очистка путем
адсорбции обходится значительно дешевле сернокислотной; ре-
зультаты же ее весьма эффективны и обыкновенно значительно
выше результатов, получаемых путем применения общих мето-
дов очистки. По отношению к важнейшим видам нефтепродук-
тов эти результаты в самых общих чертах сводятся к следую-
щему: крэкинг-бензины приобретают известную стабильность
без потери антидетонационных свойств; керосины улучшают
цвет и запах; у масел резко снижаются кислотность и коксуе-
мость с одновременным улучшением цвета и т. д. Впрочем, лри-
664
менение адсорбционных методов очистки следует рассматривать
не с точки зрения возможности замены ими общих методов, опи-
санных в предыдущей главе, а лишь с точки зрения исправления
и дополнения этих методов, как мы видели, далеко несовершен-
ных и недостаточно эффективных. В этом разрезе введение ад-
сорбционной очистки следует расценивать едва ли не как наи-
более важное усовершенствование методики очистки нефтепро-
дуктов, особенно после того, как на смену животного угля, давно*
уже применявшегося для очистки некоторых избранных нефтя-
ных продуктов (парафиновое масло, вазелин и др.), были введены
отбеливающие земли, получившие столь широкое применение
в современной нефтеперерабатывающей промышленности.
Б. Методы обессеривания
Под методами обессеривания подразумеваются некоторые спе-
циальные методы очистки, задача которых заключается в воз-
можном снижении содержания серы в том или ином нефтепро-
дукте. Методы эти довольно разнообразны; однако применение
их, естественно, ограничивается теми случаями, когда содержа-
ние серы в нефтепродукте превышает известные нормы или
когда сера находится в нефти в особенно активной форме, так
что во избежание коррозии нефтеперегонной аппаратуры или
резервуаров для хранения необходимо прибегнуть к возможному
снижению ее содержания. Достигается это путем воздействия
на нефтепродукт того или иного реагента или путем высокотем-
пературной обработки нефтепродукта в определенных условиях.
Среди реагентов, воздействием которых может быть достигнуто
«обессеривание», в первую очередь должны быть упомянуты
щелочи, щелочной раствор окиси свинца (плумбит), соли хлор-
новатистой кислоты (гипохлориты), некоторые другие соли,
а также окислители '(азотная кислота, озон); из специальных
методов обработки нефтепродуктов в целях обессеривания отме-
тим здесь также пропускание паров нефтепродукта через неко-
торые металлы и металлические окислы, особенно же гидрогени-
зацию.
ТЕХНИКА ОБЕССЕРИВАНИЯ
Аппаратура и технические приемы, применяемые при обессе-
ривании нефтепродуктов, зависят от избранной методики.
Для обессеривания щелочью, раствором плумбита и гипохло-
ритом можно применять обычную очистную аппаратуру. При
обессеривании бензинов часто пользуются мешалками с цирку-
ляционным насосом (фиг. 120), который обеспечивает циркуля-
цию очищаемого продукта, забирая его с поверхности жидкости
в мешалке и перекачивая с разбрызгиванием в нижние слои
•реагента. Удобно пользоваться здесь также непрерывно дей-
ствующей аппаратурой различных конструкций. При очистке
665
более тяжелых продуктов, например керосина, можно пользо-
ваться мешалками обычного типа либо также аппаратурой не-
прерывного действия.
Обессеривание при помощи металлических окислов в некото-
рых случаях дает весьма интересные результаты. Так, по Фрашу
[3], обработкой легких дестиллатов охайской^ нефти смесью
окислов меди (75% СиО), железа
р
4
Фиг. 120. Мешалка с циркуляцией:
1 — поступление бензина; 2 — поступление реагентов и
воды; з — выход очищенного бензина; 4 — циркуляцион-
ный насос; б — спускные линии; С — разбрызгиватель
реагента.
(15% ГегОз) и свинца
(10%-РЪО) при нагре-
вании удавалось сни-
зить содержание серы
с 0,75 до 0,08 7с. Одна-
ко в других случаях,
в частности с дестил-
латами, в которых сера
содержится главным
образом в виде тиофа-
нов, например для
дестиллатов нашей
уральской нефти, ме-
тод Фраша оказался
недействительным [4].
Среди всех методов
обессеривания наи-
большее общее значе-
ние имеет бесспорно
метод гидрогенизации.
Как будет показано
ниже, сера всех видов
сернистых соединений,
встречаемых в нефтях
и нефтепродуктах, пе-
реходит при гидроге-
низации в сероводород,
который, будучи слабо
растворим в углеводо-
родах, частью выде-
ляется в свободном ви-
де, частью, легко отмы-
вается при последую-
щей обработке дестил-
лата щелочью. Обес-
серивание при помощи
гидрогенизации требует обычной гидрогенизапионной аппара-
туры (ч. II, гл. IV); условия температуры и давления будут рас-
смотрены ниже.
Что касается, наконец, методов обессеривания с помощью
окислителей, то. несмотря на выдающийся интерес, которой
представляют эти методы с теоретической стороны (см. ниже),
они до сих пор еще не вышли из стадии лабораторной прора-
ботки.
666
ХИМИЗМ ВАЖНЕЙШИХ ПРОЦЕССОВ ОБЕССЕРИВАНИЯ
Химические реакции, имеющие место при различных процес-
сах обессеривания, весьма многочисленны; они будут 'рассмо-
трены ниже применительно лишь к наиболее важным методам
Обессеривание серной ки-лотой. Действие серной кислоты на
различные соединения весьма разнообразно. В одних случаях
здесь происходят чисто химические реакции (окисление, суль-
фирование); в других—наблюдаются лишь фишко-химические
процессы растворения; наконец, такие сернистые соединения, как
тиофаны, а также молекулярная сера вовсе не затрагиваются
крепкой серной кислотой. Отсюда понятна практическая важ-
ность знания как той формы, в виде которой сера находится
в нефтепродукте, так и тех превращений, которые сернистые со-
единения, находящиеся в нефтепродукте, могут претерпеть под
влиянием серной кислоты.
Как окислитель серная кислота может действовать на серо-
водород и «а меркаптаны. В первом случае продуктом реакции
является молекулярная сера, во втором — дисульфиды. Реакции
эти протекают по следующим уравнениям:
H2S H2S04 = S —j— 2Н0О -4- S02;
2RSH+H2S04 = R S • S‘. K+ S02+2НД
Практические выводы, которые можно сделать из этих урав-
нений, весьма важны. г
Так как молекулярная сера пе растворяется в серной кис-
лоте и сравнительно хорошо растворима в бензине, то, оче-
видно, в результате обработки серной кислотой нефтепродукта,
содержащего сероводород, последний лишь превратится в моле-
кулярную серу, которая целиком останется в нефтепродукте.
Таким образом ясно, что нефтепродукты, содержащие сероводо-
род, до их обработки серной кислотой должны быть освобождены
от сероводорода каким-нибудь иным способом, например обра-
боткой щелочью.
В отношении очистки от меркаптанной серы на первый
взгляд кажется, что и здесь нельзя ожидать сколько-нибудь ре-
альных результатов: происходит лишь превращение сравни-
тельно летучих меркаптанов в менее летучие дисульфиды. Но
так как дисульфиды довольно легко растворяются в крепкой
серной кислоте, то процесс обессеривания фактически все же
имеет здесь место и тем в большей степени, чем больше берется
серной кислоты.
Легко растворяются в крепкой серной кислоте и ею уда-
ляются также алкйлсульфиды, а равным образом некоторые сер-
нистые продукты вторичного происхождения, как-то: сульфо-
кислоты, средние и кислые эфиры серной кислоты и т. п. Наи-
более трудно поддаются действию серной кислоты, как уже было
отмечено выше, тиофаны. Как общее правило, более гжококипя-
щие погоны обессериваются труднее, чем легкие,
667
Как ясно из вышеизложенного, серная кислота в некоторых
случаях может дать вполне ощутимый эффект обессеривания,
причем, особенно благоприятствует этому работа при понижен’
ной температуре [5]. Так например, двукратная обработка бен-
зина из чусовской нефти 1% купоросного масла снизила в нем
содержание серы с 0,25 до 0,1%, т. е. больше чем на половину.
С другой стороны, после очистки керосина из той же нефти 1%
крепкой серной кислоты содержание серы в нем упало с 1,72
лишь до 1,27%; даже после повторной очистки 5% крепкой сер-
ной кислоты содержание серы снизилось здесь лишь до 0,84%.
Общий вывод, который можно сделать из всего вышеизложен-
ного, очевидно, следующий: как реагент для обессеривания креп-
кая серная кислота может дать иногда несомненный положи-
тельный результат; в других случаях эффект ее применения
явно недостаточный.
Обессеривание щелочью. Простейшим методом обессеривания
нефти или нефтяного дестиллата является обработка их водной
щелочью, короче говоря, щелочная очистка. В этих условиях
в водно-щелочной раствор переходят сероводород и меркаптаны
по следующим уравнениям:
NaOH + H.2S т=± NaSH + H90;
NaOH + NaSH Na2S + H20;
NaOH + RSH RSNa+H20.
Все эти реакции обратимы. Тем не менее, в случае серово-
дорода благодаря сравнительно небольшой растворимости его
в нефтяных маслах и дестиллатах повторной обработкой ще-
лочью легко достигается полное его удаление. С меркаптанами,
которые, наоборот, хорошо растворимы в нефтепродуктах, дело
обстоит гораздо сложнее: образующиеся меркаптиды легко гидро-
лизуются водой; получающиеся же таким образом свободные
меркаптаны извлекаются нефтяным маслом. Таким образом здесь
должно происходить распределение меркаптана между двумя
растворителями (вода и масло), так что полное удаление мер-
каптана невыполнимо.
Имеются указания, что меркаптаны могут реагировать со щелочью в раз-
личных направлениях, например:
RSH + 2NaOH = ROH + Na2S + Н2О;
2RSH + 2NaOH = RaS -j- NaoS + 2ЩО;
R • CH2 CH2 • SH 4- 2NaOH = R - CH : CH2 + Na2S + 2H2O.
Характер и полнота реакции зависит здесь, однако, от разнообразных
причин: от молекулярного веса меркаптана, его строения, концентрации
щелочи, температуры и т. д. Таким образом, даже принимая во внимание
всю совокупность реакций меркаптанов со щелочью, приходится признать,
что полное удаление меркаптановой серы щелочью, одк правило, — задача
неосуществимая.
Как показывают приведенные выше уравнения, сероводород
может реагировать со щелочью с образованием- не только кислых
солей '(сульфидов), например NaSH, но и средних солей (бисуль-
668
фидов) — Na2S. Последние могут получаться в сколько-нибудь
значительном количестве, естественно, лишь в присутствии из-
бытка концентрированной щелочи, но уже самая возможность
их образования представляет выдающийся интерес с точки зре-
ния проблемы обессеривания. Дело в том, что водные растворы
сернистых металлов легко растворяют серу с образованием мно-
госернистых металлов. Так, для натрия известен целый ряд та-
ких соединений: Na2S2 • 6Н2О, Na^Ss • 8Н2О, Na2S< - 8Н«О и
NasSs • 6Н2О. Все эти соединения окрашены от темножелтого до
оранжевого щюта; все они хорошо растворимы в воде и легко
гидролизуются ею, однако без выделения свободной серы. Ввиду
легкости их образования этой реакцией можно пользоваться для
извлечения молекулярной серы из ее растворов в нефтяных де-
стиллатах [б]; для этой цели дестиллаты достаточно подвергнуть
промывке концентрированным раствором сернистого натрия
(Na2S).
Что касается других сернистых соединений, встречающихся
в нефтепродуктах, как-то: сульфиды, дисульфиды, тиофены и
тиофапы, то обработка щелочью не оказывает на них никакого
влияния.
Итак, водная щелочь представляет собою реагент, явно недо-
статочный для обессеривания нефтепродуктов. В лабораторных
условиях для освобождения углеводорода от меркаптана удобно
пользоваться спиртовой щелочью; понятно, однако, что в произ-
водственных условиях такая методика явно неприемлема ввиду
ее высокой стоимости. Более подходящим реагентом дня этой
цели является так называемый плумбит, т. е. щелочный раствор
окиси свинца (глета); в смеси с серным цветом этот реагент
известен в США под наименованием «докторского раствора».
Обессеривание „докторский раствором". Растворение глета
в едком натре при энергичном перемешивании смеси, например
продувкой воздухом, происходит довольно легко по уравнению:
РЬО + 2NaOH = Pb(0Na)2+Н20.
После отстаивания получается раствор плумбггга натрия
в избытке щелочи, который в смеси с тонко измельченной серой
применяется при очистке, называемой обычно плумбитной. Ме-
ханизм действия этого раствора в основном сводится к следую-
щему.
При взаимодействии меркаптана со щелочным раствором
окиси свинца прежде всего происходит образование соответ-
ствующего меркаптида свинца по уравнению:
2RSH + Pb(0Na)2 R. S • Pb • S R + 2NaOH.
Меркаптиды свинца, особенно высокомолекулярные, хорошо
растворимы в нефтяных маслах; поэтому, образуясь при очистке
плумбитом, они не выпадают, а переходят в масляный слой,
окрашивая его сначала в желто-зеленый, а затем и темнозеле-
ный цвет. Известно, что подобные растворы меркаптидов свинца
легко реагируют с элементарной серой, и этой реакцией поль-
669
зуются иногда для качественной пробы на серу (докторская
проба, doctor test): нефтепродукт взбалтывают с 'раствором плум-
бита, затем прибавляют1 к смеси немного серного цвета, снова
взбатгывают и наблюдают цвет серы, располагающейся на гра-
нице раствора плумбита и нефтепродукта; если масло содер-
жало 'сернистые соединения, которые прореагировали с плумби-
том, сера темнеет и даже чернеет вследствие выделения серии-
стого свинца. Для случая взаимодействия серы с меркаптидом
свинца реакция протекает по уравнению 7]:
R • S • РЬ • S • R + S = PbS4-R • S • S • R.
Вместо серы для разрушения меркаптидов свинца можно применять
и другие реагенты, например серную кислоту, сернокислый магний и т. д.
С серной кислотой и ее солями реакция приводит к превращению меркап-
тидов также в дисульфиды, свинец, же выделяется в виде сернокислого
свинца:
RS • Pb - SR + 2H2S04 = RS • SR + SO3 + ЩО •{- PbSO4.
Как видно из приведенного выше уравнения, кроме серни-
стого свинца, выделяющегося в ввде осадка, при «докторской
пробе» в случае нахождения в растворе меркаптидов образуются
дисульфиды (R • S . S - R), тяжелые жидкости, легко раствори-
мые в масле, что крайне важно для установления баланса серы
для случая очистки нефтепродукта «докторским раствором».
Действительно, пусть нефтяной дестиллат, содержащий мер-
каптаны, подвергнут очистке «докторским раствором»; количе-
ство добавленной серы пусть строго соответствует последнему
уравнению, для чего предварительно пусть было сделано спе-
циальное определение процентного содержания в дестиллате
меркаптанов (ср. ч. I, гл. VIII). Принимая в расчет оба по-
следних уравнения, легко иритти к важному выводу, что общее
содержание серы в нефтепродукте после обработки его «доктор-
ским раствором» не уменьшается, так как в виде сернистого
свинца при этом оседает лишь та сера, которая была добавлена
вместе с плумбитом. Таким образом смысл и значение очистки
«докторским раствором», довольно широко применяемой в на-
стоящее время в процессе так называемого «подслащивания»
(sweetening) дестиллата, заключается не в снижении -содержания
серы, а в изменении ее характера: вместо сильно корродирующих
меркаптанов после очистки «докторским раствором» в дестил-
лате остаются гораздо менее активные дисульфиды.
Так как дисульфиды, образующиеся из меркаптанов в про-
цессе очистки «докторским раствором», кипят значительно выше,
то при вторичной перегонке очищенного дести плата можно до-
биться некоторого снижения содержания серы; последнее, впро-
чем, не превышает обычно десятых долей процента.
Кроме меркаптанов, плумбитные растворы, естественно, легко
реагируют с сероводором; другие сернистые соединения, встре-
чающиеся в нефтяных дестиллатах, как-то: сульфиды, дисуль-
фиды и таофаны, а также элементарная сера, сероуглерод и тщс
670
фены при комнатной температуре не реагируют ни с плумби-
том, ни с «докторским раствором» [8].
Вышеприведенные уравнения выражают лишь основные про-
цессы, протекающее при взаимодействии меркаптанов с «доктор-
ским раствором». Ближайшее изучение этой реакции [9] обна-
руживает, что одновременно здесь может иметь место ряд побоч-
ных «реакций, нередко значительно осложняющих процесс
очистки. Важнейшие из них следующие.
Так как в водных растворах плумбит легко подвергается
гидролизу на едкий натр и гидрат окиси свинца, то прежде
всего легко 'представить себе, что взаимодействие последнего
с меркаптаном (может привести к образованию не только ука-
занного выше среднего, но также и основного меркаптида; по-
следний же при нейтрализации едким натром может образовать
соответствующую соль:
Pb(OH)o + RSH R • S • РЬ • ОН 4- Н20;
R • S • РЬ OH-j-NaOH R S • Pb - 0Na + ЩО.
Основные меркаптиды свинца, как показывает опыт, хорошо
растворимы в масле; их образованию особенно содействует при-
сутствие избытка концентрированного раствора илу мбита. На-
против, под влиянием избытка воды, как видно из последних
двух уравнений, здесь должен наступить гидролиз с обратным
образованием свободных меркаптанов.
Еще более разнообразные побочные процессы имеют места во
второй фазе очистки «докторским раствором», т. е. в результате
взаимодействия образовавшихся меркаптидов с серой. Исследо-
вание показывает, что при этом процессе происходит образова-
ние ряда промежуточных частью мало устойчивых продуктов
характера полисульфидов, которые уже при хранении постепенно
изменяются с образованием сернистого свинца и органических
дисульфидов. В некоторых отдельных случаях такие промежу-
точные полисульфиды удавалось изолировать, так что с извест-
ной долей вероятности* здесь можно представить себе несколько
последующих реакций. Так например:
.SR /SR SR
Pb< +S->Pb< ->PbS+'
\SR XS —SR SR
/SR RS4
Pb< + )Pb = RS4 /SR SR
Ns—SR RS/ \pb-S-Pb/ +1
SR
И T. Д.
Аналогичные полисульфиды образуются при взаимодействии
серы с основными меркаптидами. Эти полисульфиды, однако,
еще менее устойчивы; они легко превращаются в соединения,
уже не содержащие органических радикалов, типа основных
сульфидов свинца, например:
Pb.2S3(0H)2, Pb2S4(0H).2 и т. д.
671
Обессеривание «Докторским раствором» — Метод, требующий
весьма тщательной работы, согласованной с данными лаборато-
рии. Главная опасность заключается здесь в избытке специально
добавляемой элементарной серы; поэтому количество последней
должно быть строго дозировано путем специальных лаборатор-
ных опытов в соответствии с количеством образующихся меркап-
тидов. Наилучшие результаты, в частности — снижение общего
содержания серы, обработка докторским раствором дает в тех
случаях, когда нефтепродукт содержит меркаптаны и элемен-
тарную серу, т. е. когда специальной добавки серы не требуется.
Но и в тех случаях, когда такая добавка необходима, при надле-
жащей рецептуре и дозировке, вырабатываемых для каждого
частного случая отдельно, положительный эффект очистки несо-
мненен. В основном, он определяется, как указано выше, пере-
водом сильно корродирующей серы меркаптанов в неактивную
дисульфидную серу.
Плумбитная очистка нефтепродуктов нашла некоторое при-
менение также в СССР. В Батуми этот способ применялся, на-
пример, к очистке экспортного керосина, а в последнее время —
к очистке крэкинг-бензина. С помощью центробежного насоса
бензин подается сначала в сульфаторы, загруженные комовой
серой. Здесь он растворяет необходимое количество серы, а за-
тем подается в следующую часть установки на обработку плум-
битом. Промывка водой завершает очистку. Пропускная способ-
ность установки — 600 т/сутки.
Очистка «докторским раствором» требует расхода значитель-
ных количеств свинцового глета, который для снижения стоимо-
сти очистки подлежит регенерации. Такую регенерацию отрабо-
танных остатков плумбитной очистки можно производить раз-
личными способами, важнейшие из которых следующие.
Плумбитные остатки смешивают со свежим раствором едкого
натра и при температуре около 100° продувают через такую
смесь воздух. Происходит окисление сернистого свинца до серно-
кислого с образованием свежего раствора плумбита; суммарно
эти две реакции можно выразить следующим уравнением:
PbS + 4NaOH+202 == Pb(0Na)2 + S04Na2 + 2Н20.
Рекомендуется также расчленять эти реакции на два отдель-
ных процесса: сначала действием серной кислоты (регенериро-
ванной из кислого гудрона) переработать сернистый свинец
на сернокислый, последний же обработкой едким натром превра-
тить по предыдущему в плумбит:
PbS + SO4Ha = SO4Pb + H2S;
PbS04 + 4NaOH = Pb(0Na)2 + S04Na2+2Ha0.
Наконец, некоторыми авторами для регенерации плумбитных
остатков рекомендуется хлорная вода, которая одновременно
действует растворяющим образом на сернистый свинец и окис-
ляющим образом на выделяющийся сероводород, так что в ре-
672
зультате сернистый свинец превращается в смесь. состоящую из
окиси свинца, серы и хлористого свинца:
С12-рНоО = СЮН + НС1;
PbS + СК) Н = Pb<) + S -р Н< ’1;
РЬО 4- 2НС1 = РЬС12 4- Н20.
При дальнейшем действии щелочи окись свинца и \ юрислыГг
свинец превращаются в плумбит, который содержит уже доста-
точное количество серы и может идти прямо в дело’как ('док-
торский раствор».
Обессеривание гипохлоритом. Значительно более совершен-
ным методом обессеривания в применении к бензинам и кероси-
нам прямой гонки по сравнению с очистной их щелочью и нлум-
бптом является обработка соответствующих дестиллатов гипо-
хлоритом [10]. В качестве такового обычно применяются ще-
лочные растворы солей натрия или кальция хлорноватистой
кислоты.
Хлорноватистые соли действуют на сернистые соединения
окисляющим образом, превращая их частью в сернистые соеди-
нения, растворимые в воде и поэтому удаляемые из дестиллата
в результате очистки, частично же — в сернистые соединения,
остающиеся в дестиллате, подобно тому как это имеет место при
плумбитной очистке. Во всех случаях хлорноватистые соли, те-
ряя свой кислород, превращаются в соли хлористоводородной
кислоты:
СЮХа = ХаС1 + 0.
Реакции окисления различных сернистых соединений под
влиянием гипохлорита протекают весьма разнообразно и нередко
довольно сложно, приводя в условиях очистки не к одному,
а к нескольким продуктам. Отдельные направления этих реакций
в некоторых случаях пока еще недостаточно изучены; основные
же процессы, имеющие место при действии гипохлорита на сер-
нистые соединения нефти, сводятся к следующим реакциям [11].
Сероводород при действии гипохлорита образует главным
образом серу, а также небольшие количества серной кислоты;
для главной реакции имеем уравнение:
H2S + NaOCl = NaCl + Н20 + S.
Так как элементарная сера, образующаяся в результате та-
кой очистки, растворяясь в дестиллате, сообщает ему сильные
корродирующие свойства, то необходимо, чтобы сероводород был
удален из дестиллата до очистки его гипохлоритом, т. е. отмыт
водным раствором едкого натра.
Глубина окисления меркаптанов гипохлортпом зависит от
условий реакции (см. ниже). При недостаточной ее эффектив-
ности главными продуктами ее оказываются дисульфиды по
схеме:
2RSH 4- СЮХа = NaCl4~ Н2О 4- R • S • S • R
43 Зак. 1622. Химия нефти.
673
Одновременно образуются небольшие количества соответствую-
щих сульфокислот. Напротив, при высокой эффективности реак-
ции в качестве главных продуктов окисления меркаптанов полу-
чаются сульфокислоты, в качестве же побочных — небольшие
количества дисульфидов и серной кислоты. Образование суль-
фокислот при действии гипохлорита на меркаптаны можно вы-
разить следующим уравнением:
RSH + 3C10Na = ЗХаС1 + RSO3H.
Так как органические сульфокислоты хорошо растворимы
в воде, то. очевидно, в результате последнего направления ре-
акции содержание серы в дестиллатах должно резко снижаться.
Столь же легко окисляются гипохлоритом сульфиды, пре-
вращаясь при этом в сульфоны:
К R\ /О
>S4-2C10Na = 2XaCl+ >S<
Rz Rz ^0
Низшие сульфоны хорошо растворимы в воде и ею вымы-
. ваются; высшие сульфоны легче .растворяются в дестиллате и
могут быть отделены от последнего лишь с помощью вторичной
перегонки, концентрируясь благодаря своей высокой темпера-
туре кипения в остатке от перегонки. В том же направлении,
как сульфиды жирного ряда, хотя значительно слабее, реаги-
руют с гипохлоритом циклические сульфиды, или тиофаны, то-
гда как тиофен, напротив, с гипохлоритам вовсе не реагирует.
Эффективность очистки гипохлоритом в надлежащих усло-
виях для некоторых нефтей громадна. Имеются указания, что,
пользуясь этим методом обессеривания, можно снизить содер-
жание серы на 90% и даже на 98% от начального ее содержа-
ния в дестиллате. Кроме надлежащего состава сернистых соеди-
нений подлежащих удалению, для этого необходимо соблюдение
следующих условий, вытекающих из сущности химических ре-
акций, протекающих при очистке гипохлоритом.
Как известно, хлорноватистая кислота принадлежит к весьма
слабым кислотам, вследствие чего щелочные соли ее в разба-
вленных водных растворах почти полностью гидролизованы:
ClONa + Н20 NaOH + СЮН.
Таким образом окисляющее действие гипохлорита следует
правильнее приписывать не солям, а самой хлорноватистой кис-
лоте, и общие оптимальные условия очистки гипохлоритом,
установленные опытом, находятся в полном соответствии с та-
кими представлениями об этом процессе.
Действительно, как показывает опыт, очистка протекает зна-
чительно эффективнее, если применять не крепкие, а разбавлен-
ные растворы гипохлорита: увеличивается не только скорость
реакции, но и глубина процесса окисления сернистых соедине-
ний, которое протекает в этих условиях преимущественно до
сульфокислот и сульфонов. На практике обычно применяют рас-
674:
творы с содержанием от 1,5 до 2,5% гипохлорита, и более в за-
висимости от содержания серы в очищаемом дестиллате. Боль-
шая эффективность разбавленных растворов гипохлорита
с точки зрения последнего уравнения понятна: очевидно, здесь
сказывается действие массы воды, благодаря которой гидролиз
гипохлорита и образование активной хлорноватистой кислоты
становятся полнее.
Другое условие успешности процесса очистки гипохлоритом
заключается в том, что применяемый раствор гипохлорита дол-
жен содержать некоторое избыточное количество свободной ще-
лочи (от 0,05 до 0,13% едкого натра). Присутствие свободного
едкого натра должно согласно последнему уравнению понижать
диссоциацию хлорноватисто-натриевой соли и делает поэтому
раствор гипохлорита более стабильным. С другой стороны неко-
торое количество свободной щелочи необходимо здесь для свое-
временной нейтрализации продуктов окисления сернистых со-
единений кислотного характера (сульфокислоты, серная кис-
лота); в противном случае в процессе очистки может образо-
ваться кислая среда, и наступит взаимодействие образующихся
одновременно из гипохлорита хлорноватистой и соляной кислот
с выделением свободного хлора:
ClONa-4- HoS04 = NaHSO, + СЮН; NaCI-J-HaS04 ==
= NaHS04J-HCl; C10H -f-HC1 = H20 %- Cl2.
Как видно, два рассмотренных условия успешности очистки
нефтепродукта гипохлоритом, можно сказать, взаимно противо-
речат друг другу. Ввиду этого оптимальные условия такой
очистки для каждого отдельного случая могут быть установлены,
лишь эмпирически, путем тщательной лабораторной проработки
вопроса. В частности, очевидно, крайне важно установить ми-
нимальное количество свободного едкого натра, необходимого
в каждом данном случае, так как избыток едкого натра в пол-
ном соответствии с уравнением гидролитической диссоциации
гипохлорита и с данными опыта лишь замедляет скорость
очистки и направляет реакцию в сторону неполного окисления
сернистых соединений, например меркаптанов—в дисульфиды,
и т. п.
Одним из неблагоприятных моментов процесса очистки гипо-
хлоритом является отмеченная выше возможность образования
в данном случае свободного хлора; последний же, действуя на
углеводороды дестиллата хлорирующим образом, может оста-
ваться в очищенном продукте в виде хлоридов, что, конечно,
нежелательно. Однако присутствие в растворе гипохлорита не-
большого избытка свободной щелочи почти полностью устраняет
это побочное направление процесса очистки, оставляя в очищен-
ном продукте лишь следы галоида (0,01% хлора и меньше). На-
против, недостаток щелочи, а также длительная обработка гипо-
хлоритом могут привести к повышенному содержанию хлора
в продукте, которое может подняться до нескольких десятых до-
лей процента и выше.
43* 675
Как было указано, гипохлорит применяется обыкновенно для
очистки бензинов и керосина прямой гонки или широкой бен-
зино-керосиновой фракции. После промывки щелочью для уда-
ления сероводорода (ср. выше) дестиллат подвергают последо-
вательно дважды обработке гипохлоритом. Далее следует новая
промывка едким натром и вторичная перегонка, в процессе ко-
торой происходит отделение дисульфидов и растворимых в де-
стиллате сульфонов. Одновременно при этом происходит разде-
ление бензиновых фракций от керосиновых. Первые после про-
мывки щелочью и водой дают готовый продукт, вторые же < тя
дальнейшей очистки подвергают фильтрации через адсорбент
(ср. выше).
Кроме дести ктатов прямой гонки, гипохлорит применяют
иногда, также для очистки крэки ir-бензина. В этом случае,
однако, процесс очистки осложняется иногда присоединением
элементов хлорноватистой кислоты по месту двойной связи
с образованием соответствующих хлоргидринов по уравнению:
СН, С1 СН,С1
сн2 + он СЩОН
Таким образом в результате очистки крэкинг-бензина гипо-
хлоритом в очищенном продукте может оказаться хлор. Так кал;,
однако, для образования хлоргидринов -в данном случае тре-
буется свободная хлорноватистая кислота, то, очевидно, это на-
правление реакции может быть ослаблено тем же самым спосо-
бом, как реакция хлорирования углеводородов предельного ха-
рактера в присутствии хлорноватистой кислоты (ср. выше), т.е.
введением свободной щелочи, снижающей степень гидролиза
гипохлорита [12].
Очистка крэкинг-бензина гипохлоритом производится обык-
новенно в следующем порядке: дестиллат промывают щелочью,
затем обрабатывают щелочным раствором гипохлорита; далее
следуют сернокислотная очистка, промывка водой, щелочью,
наконец вторичная перегонка и заключительная промывка
5 %-ной щелочью. Как видно, очистка гипохлоритам отнюдь не
может заменить очистки крэкинг-бензина серной кислотой с по-
следующей перегонкой (вторичной) очищенного бензина.
Обессеривание азотной кислотой и другими окислителями.
Как видно из вышеизложенного, обессеривание гипохлоритом
сводится к окисляющему действию данного реагента на серни-
стые соединения нефти. Однако по отношению к некоторым сер-
нистым соединениям, например, к тиофанам, гипохлорит хгак
окислитель является недостаточно энергичным, и применение его
в такого рода случаях не дает надлежащего эффекта. Так, на-
пример, в опытах обессеривания дестиллатов уральской нефти
получены следующие результаты: в легких дестиллатах с содер-
жанием от 0.2S до 0,42% S после обработки гипохлоритов содер-
жание серы упало до 0,03—0,04%, тогда как с более тяжелыми
676
и богатыми серой дестиллатами та же методика уже яр тала удо-
влетворительных результатов [i3j.
Известно, однако, что тиофаны, лишь с трудом поддающиеся
действию гипохлорита, легко окисляются при действии более
энергичных окислителей, например азотной кислоты и хаме-
леона; в первом случае продуктами окисления оказываются
сулъфоокисп, во втором — сульфоны, и те и другие — раствори-
мые в воде соединения:
W -> cnH2nso сдл\
тилфаны сульфоокиси сульфоны
Тем самым намечается новый путь освобождения нефтяных
дестиллатов от наиболее трудно удаляемых сернистых соедине-
ний, от тиофанов.
Лабораторные опыты [14] показали полную применимость
данной методики к керосиновым дестиллатам уральской нефти.
Так например, в керосиновом дестиллате удельного веса 0.SS3.
кипевшем в пределах 140—315°, с содержанием 2.77% S после
обработки серной кислотой и гипохлоритом содержание серы
снизилось только до 2,1S% S. Зато последующей обработкой
этого дестиллата разбавленной азотной кислотой (удельного веса
1,2 с содержанием 32% HNOs) при комнатной температуре и энер-
гичном перемешивании удалось спустить содержание серы до
0,24% S. Несмотря на выдающийся интерес с точки зрения вы-
яснения природы сернистых соединений отдельных нефтей,
практического применения данная методика не полущила.
Большего практического значения следует ожидать от приме-
нения для обессеривания нефтепродуктов озонированного воз-
духа [15]. Химическая сущность процесса заключается здесь
в окисляющем действии реагента на сернистые соединения де-
стиллата, в том числе на наиболее устойчивые из них (тио-
фаны) с превращением их в соединения (сульфокислоты, сер-
ная кислота и т. д.), отмываемые далее водой и водной ще-
лочью, После перегонки с водяным паром обработанный таким
образом чусовский бензин (содержание серы 0,24% S) показал
резкое снижение в содержании серы; последнее при достаточной
длительности процесса озонирования (1,5—2 часа) могло быть
доведено до нуля. Если обработку озоном производить при по-
ниженной температуре (—15, —20°), то процесс протекает осо-
бенно гладко: не происходит осмоления бензина, которое имеет
место, если проводить озонирование при комнатной темпера-
туре [16].
Обессеривание гидрогенизацией. Как было указано выше,
гидрогенизация представляет собой наиболее общий метод обес-
серивания: в атмосфере водорода и надлежащих условиях тем-
пературы и давления все виды сернистых органических соедине-
ний, даже наиболее устойчивые из них (тиофены и тиофаны),
могут быть разрушены с образованием соответствующего угле-
водорода предельного характера и сероводорода. Так например.
677
для диметилтиофена эту реакцию можно выразить следующим
уравнением:
(CH3)2C4H2S + 4Но == H2S + (\.Н14.
Условия, в которых могут быть достигнуты подобные резуль-
таты, значительно более умеренны, чем рассмотренные в ч/п
гл. IV условия гидрогенизации жидкого топлива; в то же время,’
чтобы достичь полного обессеривания, необходимо одно спе-
циальное обстоятельство, а именно — присутствие подходящего
сероустойчивого катализатора. В этих условиях вся сера при-
сутствующих в нефтепродукте сернистых соединений уходит
в виде сероводорода, который в главной своей массе выделяется
в свободном состоянии; остатки же его, растворившиеся в неф-
тепродукте, легко отмываются щелочью.
Особенно наглядные результаты обессеривания получаются
при работе с высокосернистыми продуктами, например с буро-
угольными и сланцевыми бензинами [17]. Так например, 'бен-
зин, выделяемый путем разгонки кашшгрской сланцевой смолы,
содержит 10,6% S, главным образом в виде гомологов тиофена;
его удельный вес — 0,891 (>15°/15°); его фракционный состав но
Энглеру: начало кипения—145°; от 150 до 175'—85%; конец
кипения — 200°. После повторной гидрогенизации при 350° ц
начальном давлении водорода 50 ат (оперативное давление около
100 ат) получается продукт, полностью или почти полностью
обессеренный (содержание серы от 0,0 до 0,2%); его удельный
вес 0,750—0,760; его фракционный состав: до 150э — от 50 до
65%; от 160 до 175° — 20—30%, конец кипения — прежний.
В качестве катализатора для получения подобных результатов
наиболее подходящим оказался даух1сернистый молибден (MoS2).
В этих результатах заслуживает внимания не только полное
или почти полное обессеривание высокосернистого исходного
продукта; крайне важно также резкое изменение фракционного
состава бензина -в сторону увеличения легких его частей; ново-
образование последних происходит, очевидно, за счет тидрогени-
зационного распада тиофенов исходного сланцевого бензина. Ме-
ханизм этого распада пока еще не может считаться окончательно
установленным; вполне возможно, что гидрогенизация тиофенов
протекает через промежуточное образование соответствующих
тиофанов и меркаптанов по следующей схеме:
НС —СН
НО СН
Аналогичные результаты легко получаются при гидрогениза-
ции содержащих серу нефтепродуктов. Полнота обессеривания
легко достигается и здесь в условиях, значительно более мяг-
ких, чем те, которые необходимы для деструктивной гидрогени-
678
зации тяжелых нефтей и остатков. Так например, при гидро-
генизации бензино-керосиновой фракции ишимбаевской нефти
с начальным содержанием серы 1—1,3% 'содержание серы в за-
висимости от условий -процесса, легко удавалось снижать до со-
тых долей .процента. В качестве катализатора и здесь приме-
нялся обыкновенно двухсернистый молибден при начальном да-
влении водорода 40 ат (оперативное давление около 90 ат) и тем-
пературе 400 18а]. Прекрасные результаты были получены
с теми же нефтепродуктами в аналогичных условиях темпера-
туры (400—420 ) при невысоких давлениях (начальное давле-
ние ют 5 до 20 ат) в присутствии другого катализатора — окиси
хрома [19].
Гидрогенизация под давлением как метод обессеривания тре-
бует специальной, довольно сложной и дорогой аппаратуры;
в этом — основной недостаток этого метода. Отсюда понятны по-
пытки различных авторов подыскать такой катализатор, кото-
рый позволил бы осуществить обессеривание путем гидрогени-
зации при обыкновенном давлении. Опыт показывает, что в при-
сутствии подходящих катализаторов (Pt, Ni на окиси алюминия)
при значительном избытке водорода обессеривание высокосерни-
стых бензинов (нефтяных и других) действительно может быть
достигнуто. При этом, однако, катализатор довольно быстро от-
равляется серой, и дальнейшее обессеривание приостанавли-
вается; для его возобновления катализатор должен быть регене-
рирован, что связано со значительными расходами и наруше-
нием непрерывности процесса. В этом — основные трудности
обессеривания при обыкновенном давлении, и, повидимому, даже
лучшие из опубликованных в этой области работ [20] пока еще
не решают полностью данной проблемы.
Кроме только что рассмотренных методов обессеривания,
известны и частью находят практическое применение некоторые
другие способы, как-то: обессеривание некоторыми солями
(Aids ZnCl2) и с помощью селективных растворителей; методы
эти будут рассмотрены ниже; относительно обессеривания путем
адсорбции было сказано выше.
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ МЕТОДОВ ОБЕССЕРИВАНИЯ
Так как -сера и сернистые соединения являются одними из
наименее желательных составных частей нефти и ее продуктов,
то значение обессеривания как метода повышения качества неф-
тепродуктов следует расценивать чрезвычайно высоко. Действи-
тельно, если вспомнить, что уже простая перегонка некоторых
высокосортных нефтей нередко связана с чрезвычайно быстрым
изнашиванием нефтеперегонной аппаратуры и что присутствие
сернистых соединений всегда вызывает более или менее значи-
. тельную коррозию не только в разнообразнейших случаях ри
менения нефтепродуктов (топливо, смазка), но даже при их хра-
нении, то становится ясным, почему содержание серы является
679
одним из важнейших признаков, определяющих качество нефти
и вырабатываемых из нее продуктов. Понятно также, что лишь
путем обессеривания возможно введение в круг переработки и
применения высокосернистых нефтей, примеры которых были
приведены в I ч., гл. VIII.
Как ясно из изложенного, в некоторых простейших случаях
обессеривание нефтепродуктов достигается сравнительно легко
в других случаях обессеривание требует применения наиболее
эффективных методов вплоть до гидрогенизации. Не возвра-
щаясь к сравнительной оценке этих методов, отметим лишь, что
до сих пор нефтяная промышленность не располагает простым
и достаточно мощным методом обессеривания, который был бы
применим ко все-м видам сернистых соединений. В этом отно-
шении особые надежды следует возлагать, как было указано
выше, на методы каталитической гидрогенизации при обыкновен-
ном давлении.
В. Методы очистки с о ляни
Давно известно, что некоторые соли хлористоводородной
кислоты могут применяться для очистки нефтяных дестиллатов.
В порядке убывающей способности к такой очистке важнейшие
из этих солей могут' быть расположены в следующий ряд: хло-
ристое олово (SnCh), хлористый цинк (ZnCl2), хлористый алю-
миний (А1С1з) и хлорное железо (FeCle); другие хлориды, как-то:
хлористый свинец (РЬС12), сулема (HgCl2), хлорная медь (СиС12),
хлористый магний (MgCIs) и т. д., гораздо менее пригодны для
этой цели, оказывая на дестиллаты лишь слабое воздействие.
Особого внимания среди всех этих солей заслуживает хлористый
цинк [21].
Безводный хлористый цинк готовится прокаливанием смеси
сернокислого цинка с хлористым натрием:
SOJZn + 2NaCl ZnCl2 + Na2S04.
Хлористый цинк возгоняется при этом и уходит из сферы
реакции, так что процесс превращения сульфата в хлорид про-
ходит в данном случае до конца. Сплавленный безводный хло-
ристый цинк (температура плавления 250°) во влажном воздухе
быстро расплывается, образуя с водой концентрированные рас-
творы. Водные растворы хлористого цинка готовятся растворе-
нием в соляной кислоте металлического цинка, окиси цинка,
сернистого цинка и других его ‘Соединений. На некоторых произ-
водствах, например при регенерации олова, хлористый цинк
является побочным продуктом и даже отбросом. Если не считать
употребления хлористого цинка в лабораторной и заводской
практике в качестве водуотнимающего н конденсирующего ре-
агента, то едва ли нс единственное его применение в крупном
масштабе — это пропитка дерева, особенно шпал, дтя предохра-
нения от гниения. Вообще можно считать, что как материал
для очистки нефтяных дестиллатов хлористый цинк предста-
вляет собою вещество, легко доступное н недорогое.
680
ТЕХНИКА ОЧИСТКИ ХЛОРИСТЫМ цинком
Очистка нефтяных дестиллатов как прямой гонки, так и крэ-
кин а при помощи хлористого цинка может производиться
в жидкой или паровой фазе.
Для жидкофазной очистки хлористый цинк предварительно
наносится пз водного раствора или расплавленного состояния на
пемзу, асбест или глину, и дестиллат -медленно перегоняется
с этим реагентом, взятым примерно в количестве 5% к весу де-
стиллата. Очистка завершается промывкой дестиллата 10%-ным
раствором соды и -водой.
Несмотря на свою простоту, жидкофазная очистка хлористым
цинком заключает в себе ряд технологических недостатков, важ-
нейшие из которых следующие. Так как процесс очистки проис-
ходит в гетерогенной среде, то скорость реакции должна зависеть
здесь от интенсивности перемешивания; вместе с тем зависи-
мость этой скорости от количества реагента оказывается весьма
сложной, в частности — различной для разных дестиллатов.
И наконец, жидкофазная очистка хлористым цинком не может
разрешить весьма важного вопроса о беспрерывности этого про-
цесса.
Парофазная очистка хлористым цинком предложена в двух
видоизменениях.
По американскому способу [22] неочищенные пары пропу-
скаются непрерывным потоком в очистной резервуар или ко-
лонну, где они промываются горячим раствором хлористого
цинка. Очистные аппараты могут быть помещены между фрак-
ционирующей колонной и ее конденсатором; можно также скон-
денсировавшийся бензин снова превратить в парообразное со-
стояние и направить пары в очистную установку. Окончатель-
ный выбор того или иного способа зависит от местных условий.
Ближайшие условия очистки бензина хлористым цинком по
американскому способу следующие.
Средняя температура, при которой успешно протекает очистка
крэкинг-бензина хлористым цинком, лежит в пределах 150—175“.
Концентрация раствора хлористого цинка в зависимости от
материала, подвергаемого очистке, меняется от 70 до 85% по
весу. В процессе работы она должна поддерживаться постоянной.
Время соприкосновения между ларами и раствором хлори-
стого цинка, достаточное для производства очистки, измеряется
секундами: от 2 до 12 сек.
Крайне важное значение для успешности ^очистки имеет хо-
рошее перемешивание, которое может быть обеспечено примене-
нием башен с разного рода насадкой или ректификационной ко-
лонной с колпачками, или, наконец, инжекторами и механиче-
скими мешалками.
По другому способу (советскому) очистка бензина, и керосина
в паровой фазе производится твердым хлористым цинком [21].
Сущность способа заключается в том, что пары очищаемого де-
стиллата, образовавшиеся в кубе или трубчатой печи, пропу-
скаются через колонну с хлористым цинком на насадке; затем
6*1
эти пары проходят пере 5 небольшую ректификационную колонну
и конденсируются в холодильнике. Наиболее существенными мо-
ментами парофазной очистки хлористым цинком по этому ело*
собу являются следующие.
Набивка, на которую наносится хлористый цинк, должна
иметь большую поверхность и обладать слабо основным или ней-
тральным характером. Лучшей набивкой является пемза помо-
лом 3—8 мм (диаметр зерна), которую погружают на 10—15 мин.
либо в расплавленный хлористый цинк, либо в горячий водный
раствор этой соли, после чего его подсушивают при температур?
150—200°. Количество хлористого цинка, которое удерживается
пемзой при указанной обработке, может достигать 50% веса
набивки.
Верхний температурный -предел иарофазной очистки хлори-
стым цинком лежит при 250°. так как при этой температуре
безводный хлористый цинк плавится. Таким образом керосины
практически можно чистить хлористым цинком лишь в жидкой
фазе. Нижний температурный предел очистки обозначается ме-
нее резко; во всяком случае, хлористый цинк действует на пары
некоторых углеводородов крэкинга (диолефины) уже при 20е.
Оптимальная температура для очистки крэкинг-бензина хлори-
стым цинком лежит в интервале 150—205°.
Эффект очистки получается только при определенных скоро-
стях паров нефтепродукта в отгистной колонне, обеспечивающих
•минимум времени для соприкосновения их с хлористым цин-
ком. Скорости эти зависят от различных факторов, как-то: ка-
чество набивки, характер очищаемого дестиллата и т. д., и мо-
гут .быть определены лишь опытным путем.
При расходе хлористого цинка до 0,1—0,2% по весу к очи-
щаемому сырью тяжелые бензины и лигроины выходят вполне
удовлетворительных качеств, причем содержание смол в этих
продуктах, определяемое выпариванием в фарфоровой чашке,
обычно бывает равно нулю; для бензинов внутреннего рынка
расход реагента‘может быть сокращен вдвое.
Потери при очистке хлористым цинком крэкинг-бензина
в паровой фазе сводятся главным образом к потерям на испаре-
ние легких частей и колеблются от 3 до 7%; они должны зави-
сеть особенно от качества аппаратуры.
Качество бензина, очищенного хлористым цинком в паровой
фазе, весьма высокое: бензин приобретает -высокую стабильность,
общее же содержание в нем серы уменьшается в среднем на.
40%. Даже парофазный крэкинг-бензин, очищенный хлористым
цинком и доочищенный 1% серной кислоты (вместо 10—12%
кислоты, которая требуется без хлористого цинка), получается
вполне стабильным, не изменяющимся при хранении в течение
нескольких месяцев и больше.
ХИМИЗМ ОЧИСТКИ ХЛОРИСТЫМ цинком
Химические процессы, протекающие при очистке хлористым
цинком, пока еще изучены не достаточно; судить о них можно
682
лишь на основе взаимодействия этого реагента, с отдельными
классами органических соединений, которые встречаются в очи-
щаемых дестиллатах.
Парафины, нафтены и ароматика не реагируют с хлористым
цинком, тогда как этиленовые углеводороды полимеризуются под
влиянием этого реагента так же, как под влиянием серной кис-
лоты. Эта реакция протекает, однако, крайней медленно, и прак-
тически реакции полимеризации и конденсации в процессе
очистки хлористым цинком претерпевают лишь такие компо-
ненты крэкинг-бензина, как диеновые углеводороды, а также кис-
лородные соединения, например альдегиды, если они присут-
ствуют в дестиллате. Продуктами этих реакций являются высо-
комолекулярные вещества смолистого характера неизвестного
строения.
Другое важное направление очистного действия хлористого
цинка заключается в связывании или переработке сернистых со-
единений дестиллата. Так, сероводород, почти всегда присут-
ствующий в крэкинг-дестиллате, реагирует с хлористым цинком
с образованием сернистого цинка:
ZnCl2 4- HaS = ZnS 4- 2 на
Меркаптаны под влиянием хлористого цинка, превращаются
в сульфиды:
2RSH-f-ZnCI2 = 2НС1 4- ZnS 4- R S • R.
Тем самым количество хлористого цинка в насадке должно
постепенно уменьшаться.
Другая реакция, приводящая к более или менее значитель-
ным потерям цинка, заключается в гидролизе хлористого цинка
за счет 'воды, содержащейся в дестиллате или образующейся
в процессе очистки при тех или иных процессах конденсации
кислородных соединений дестиллата:
ZnCL 4- Н20 = Zn(OH)Ul 4- НС1.
Необходимо, впрочем, иметь в виду, что при наличии неболь-
ших количеств и даже в крепких водных растворах хлористый
цинк дает с водою комплексные соединения типа аквокислот,
способных диссоциировать с выделением иона водорода, напри-
мер:
ZnCl2 + Н20 = [ZnCL, ОН] Н;
ZnCl24-2H.2O = [ZnCU(0H)2] Н.2.
Эти аквокислоты имеют сильные кислотные свойства, но при
разбавлении водой разрушаются с образованием продуктов ги-
дролиза. Зато в -концентрированных водных растворах, а также
в твердом виде с содержанием небольших количеств влаги эти
аквокислоты реагируют подобно минеральной кислоте, например
серной, обладая к тому же перед последней ценными преиму-
ществами: тогда как очистку серной кислотой во избежание
глубокого разрушения отдельных ценных частей дестиллата прИ-
Ь.43
ходится вести в умеренных температурных условиях и даже на
холоду, очистку хлористым цинком, правильнее его аквокисло-
тами, можно, как было выше указано, производить при повы-
шенной температуре, усиливая тем самым ее эффективность.
Когда очистное действие хлористого цинка снижается, он мо-
жет быть подвергнут регенерации. С химической точки зрения
задача регенерации заключается в данном случае не только
в удалении смолистых веществ, образовавшихся в процессе
очистки, но также и в том, чтобы превратить обратно в хлори-
стый цинк различные продукты его изменения, как-то сернистый
цинк, хлорокись и гидроокись цинка и т. п. Если хлористый
цинк был на насадке, его регенерация начинается с промывки
насадки горячей водой, подкисленной соляной кислотой: при
этом содержавшаяся вместе с хлористым цинком смола всплы-
вает наверх и может быть легко отделена от водного раствора.
Последний выпаривают затем досуха и получают хлористый
цинк черного цвета вследствие примеси к нему углистых частиц.
Таг: а я соль может быть применяема для очистки дестиллатов
с таким же успехом, как и свежий хлористый цинк. В этих
условиях, применяя для выщелачивания насадки разбавленную
соляную кислоту, можно регенерировать до 70—75% хлористого
цинка; применяя же крепкую серную кислоту — до 90%. При
сильном загрязнении соли после ее повторных регенераций
можно осторожным обжигом перевести ее в окись цинка, которая
действием соляной кислоты легко превращается в хлористый
цинк.
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ ОЧИСТКИ СОЛЯМИ
Практическое применение очистки солями ограничивается
пока несколькими установками в США. работающими на хло-
ристом цинке по американскому способу. Повидимому, однако,
этой методике предстоит ’большая будущность, так как на основе
литературных данных [22] очистка крэкинг-бензина хлористым
цинком обходится значительно дешевле, чем очистка серной
кислотой.
Г. Очистка гидрогенизацией или гидроочистка
Как было показано выше, гидрогенизация является наиболее
общим и надежным методом обессеривания жидкого топлива,
позволяющим в сравнительно мягких условиях температуры и
давления освободиться от всех видов сернистых соединений, ко-
торые встречаются не только в природных нефтях, ио и вообще
в жидком топливе различных видов. Таким образом, кроме того
специального назначения, которое гидрогенизация имеет как спе-
циальный вид крэкинга (деструктивная гидрогенизация), эта
методика может применяться также как специальный метод
очистки. Помимо обессеривания, гидроочистка была предложена
в последнее время еще для одной весьма важной специальной
очистной операции — для стабилизации моторного топлива.
684
Независимо от источника и способа получения, все виды мо-
торного топлива нуждаются в тщательной очистке, более или
менее специальной • для каждого частного случая. Особенно
осложняется очистка для таких видов моторного топлива, кото-
рые в настоящее время заняли доминирующее положение в то-
пливном балансе: для бензинов крэкинга, особенно парофазного,
для моторного бензола и других продуктов пиролиза, например
пиробензина, и наконец, для бензинов из сланцевых и первич-
ных угольных смол. Все эти виды моторного топлива в неочи-
щенном виде характеризуются .большим или меньшим содержа-
нием веществ, весьма склонных к смолообразовании: присут-
ствие этих смолообразователей делает, таким образом, эти виды
топлива неустойчивыми, нестабильными при хранении вслед-
ствие выделения смолистых осадков, которые загрязняют резер-
вуары для хранения, трубопроводы и карбюраторы моторов, за-
бивают топливную систему и нередко снижают антидетонацион-
ные свойства бензина. Таким образом удаление смолообразова-
телей может рассматриваться как одно пз условий получения
стабильного высококачественного моторного топлива.
ХИМИЗМ СМОЛООБРАЗОВАНИЯ
Химическая природа смолообразователей, равно как и сущ-
ность тех химических превращений, которые приводят к выделе-
нию смол, не могут считаться вполне выясненными. Обыкно-
венно различают смолообразование в продуктах крэкинга,
а именно — в моторном топливе, и смолообразование в смазоч-
ных маслах; последнее будет рассмотрено ниже, в гл. IIГ.
Смолообразование в сырых продуктах крэкинга часто отно-
сят за счет присутствия в них высоконелредельных углеводоро-
дов— диеновых и триеновых. Как известно, эти углеводороды
в присутствии кислорода воздуха особенно легко претерпевав >т
так называемую аутоксидацию (самоокисление), сопровождаемую
процессами полимеризации и конденсации, продуктами которых
являются смолообразные вещества, собирающиеся главным обра-
зом на дне и стенках сосуда. Повышение температуры, прямой
солнечный свет и присутствие некоторых сторонних примесей
(сернистые соединения, некоторые металлы, мыла) ускоряют эти
процессы смолообразования; напротив, содержание высоконепр<-
дельных углеводородов в темноте, в отсутствии кислорода и ве-
ществ, действующих каталитически на процесс смолообразова-
ния, замедляют этот процесс. Впрочем, процесс смолообразова-
ния, правда, обычно гораздо более медленный, наблюдается
также при хранении более простых углеводородов непредельного
характера (моноолефинов) и даже в углеводородных смесях,
вовсе не содержащих непредельных, например в бензинах пря-
мой гонки.
Как было показано при рассмотрении окислительного крэ-
кинга (ч. II, гл. IV), начальными продуктами взаимодействия
углеводородов с кислородом являются особого рода перекиси
(молокиси), легко претерпевающие далее превращения с образо-
ванием разнообразных органических соединений, как-то: альте
гидов, кислот и т. д. Можно считать установленным, что обоь
зование этих перекисей является первой фазой также ’в процесс'
смолообразования в крэкинг-бензине. Табл. 147 дает -картпву
течения этого процесса в различных условиях; определение
перекисей производилось здесь иодным методом и выражено
дВ эквивалентах иода на 100 гл3 бензина [23].
Таблица 14т
Образование перекисей в крэкинг-бензине
Время действия Солнечный свет и кислород Солнечный свет и воздух Солнечный свет без кислорода Кислород в темноте
часы Мил и граммы пода на 100 еж3
0 О 0 0 0
2 22,0 12,0 4.0 Следы
4 44,0 24,0 4.0
10 102,0 36,0 4,0
14 232,0 60.0 8,0
36 548,0 • 300,0 i 8.0 !»
Образование перекиси является лишь первой фазой про-
цесса смолообразования. Как соединения, мало устойчивые, осо-
бенно при нагревании, и к тому же обладающие высокой реак-
ционной способностью, перекиси, образующиеся при хранении
крэкинг-бензина, частью затем разлагаются, а частью действуют
окисляющим образом на другие компоненты данной смеси. Так
например, разложение перекиси углеводорода, имеющего груп-
пировку > С = СН2, можно представить следующей схемой,
предусматривающей образование альдегида и кетона [23]:
Вещества альдегидного и кетонного характера., а также про-
дукты их окисления (органические -кислоты) действительно по-
являются -в крэкинг-бензине при его хранении; таким образом,
эти вещества можно рассматривать как продукты ближайших
превращений образующихся в первую очередь перекисей непре-
дельных углеводородов крэкинг-бензина.
Для суждения о характере дальнейших реакций в процессе
смолообразования существенный интерес представляют данные
табл. 148. в которой содержатся результаты анализа и элемен-
тарный состав сухих смол из неочищенного крэкинг-бензина,
полученных в медной чашке [23]:
686
Состав и свойства енол
Таблица 14#
Смолы Неомыляе- мые Кислоты нераство- римые Кислоты раствори- мые
•/0 100,00 13,00 55,00 30,50
% • , °/0 н .... Анализ 0/° о % золы . . . 70,73 6,95 64,20 6,32 73,22 6,81 66,70 7,20
19,51 2,00 13,81 15,38 18,58 0,98 24,23 0.80
Иодное число 72 — 89 84
Молекулярный ве<- . . Температура плав ле- 200 — 119 ’—
ния в °C 98—105 192--195 —
Как видно, смолы из крэкинг-бензина в главной своей массе
имеют кислотный характер («кислые смолы»), и, следовательно,
основной реакцией процесса смолообразования должна быть при-
знана реакция окисления. Судя ио среднему молекулярному
весу смол, известную роль при этом должны играть также реак-
ции уплотнения (полимеризация, конденсация); однако для бли-
жайшей их характеристики пока не хватает опытного материала.
Итак, смолообразование в крэкинг-бензине происходит за
счет его особо активных компонентов, которые, естественно,
должны быть сосредоточены в тех или иных отдельных его фрак-
циях. Между различными фракциями крэкинг-бензина может
наблюдаться громадное различие в склонности к смолообразо-
ванию [24]. Так например, парофазный крэкинг-бензин, разо-
гнанный на 10 равных частей, после хранения в течение 2 ме-
сяцев при 35° в одинаковых условиях показал следующее со-
держание смол по фракциям (табл. 149).
Таблица 1 №
Распределение смол по фракциям крэкинг-бензина
через 2 месяца
Температура
кипения фракций
в СС
Смолы в медной
чашке в мг
в 100 гм3
Смолы в
условных
единицах
До 64
64 — 70
70 — ЬО
80 — 87
87 — 100
100 — 108
108 — 115
115 — 122
122 —135
135 — 150
4 200 210,0
130 6,5
7Э 3,5
87 4,4
135 6.8
25 1,2
20 1.0
6800 340,0
150 7,5
3 650 182,5
687
ОПРЕДЕЛЕНИЕ И СОДЕРЖАНИЕ СМОЛ В КРЭКИНГ-БЕНЗИНЕ
Различают истинные смолы, действительно содержащиеся
jb крэкинг-бензпне, и потенциальные смолы; последние предста-
вляют собой смолы, образовавшиеся из соответствующих мало-
устойчивых компонентов бензина в процессе определения смол
например при выпаривал ш бензина, при продувке воздуха
:и т. п. Несмотря на принципиальное различие между этимн
двумя видами смол, до сих пор не существует метода раздель-
ного определения истинных и потенциальных смол. Более того
.даже совместное определение смол различными методами редко
.дает сходящиеся результаты и представляет собою поэтому одну
из наиболее условных характеристик крэкинг-бензина.
Обычно определение смол в бензине производится выпарива-
нием юи см3 бензина во взвешенной чашке па водяной бане,
после чего чашку с остатком высушивают и взвешивают. Перво-
начально при этом применяли медную чашку стандартных раз-
меров; позднее, когда выяснилось каталитическое влияние меди
на процесс образования смол, стали пользоваться чашками фар-
форовыми или стеклянными (из стёкла « Pyrex»). Чтобы избе-
жать добавочного смолообразования в процессе испарения бен-
зина при нагревании, рекомендовано производить испарение при
обыкновенной температуре с отдувкой образующихся паров бен-
зина струей воздуха. Аппаратура и здесь, конечно, строго стан-
дартизована.
Определение смол в чашке не дает содержания истинных
смол, так как в процессе испарения бензина часть малоустой-
чивых его компонентов успевает осмолиться. Более точное со-
держание истинных смол дает метод однократного испарения
25 см3 бензина в специальном приборе: спирально согнутой
трубке, предварительно взвешенной и нагреваемой на масляной
бане до 220°. Бензин подается в прибор по каплям, пары же
его уносятся струей воздуха. Определение смол и здесь произво-
дится вторичным взвешиванием прибора.
Другие методы определения смол основаны па предваритель-
ном поглощении бензином кислорода для возможно полного пре-
вращения потенциальных смол в истинные, после чего следует
весовое определение смол в чашке. Поглощение кислорода осу-
ществляется либо в стеклянной колбе на водяной бане при нор-
мальном давлении, либо в бомбе при 100° и начальном давле-
нии 7 ат. И в том, и в другом с гучаях аппарат соединяется
с манометром, показания которого позволяют судить о ходе ад-
сорбции кислорода. Б обоих случаях в начале опыта наблю-
дается некоторый начальный период индукции, в течение кото-
рого поглощение кислорода протекает крайне медленно. Счи-
тают, что для стабильного бензина такой индукционный период
должен быть не менее 4 час. Повидимому, в течение этого про-
межутка времени в бензине происходит накопление веществ,
действующих автокаталитически на процесс окисления бензина,
после "чего этот процесс приобретает более значительную скорость.
688
Содержание смол в крэкинг-бензине колеблется в весьма ши-
роких пределах в зависимости от его происхождения, продол-
жительности хранения, главное же — от степени его очистки.
Ниже приводятся некоторые данные по этому вопросу на основе
определения смол методом выпаривания в чашке.
В неочищенном крэкинг-бензппе содержание смол доходит
до 250—300 мг и выше на 100 см3 для жидкофазного и до
2000—4000 мг на 100 с.и3 для парофазного крэкинг-бензина.
В продукте, очищенном серной кислотой и глиной с последую-
щей промывкой, содержание смол уже значительно меньше, но
все еще высоко: от 25 до 100 л/з на 100 о3, и только после
перегонки очищенного бензина содержание смол в нем сни-
жается до 3—б мг на 100 см3. Технические нормы нефтепродук-
тов ССОР требуют, чтобы содержание смол в крэкинг-бензине
(очищенном) не превышало 5 мг на 100 см3. Многие американ-
ские авторы полагают, однако, что даже при содеря^нии 10—
15 мг смол на 100 см3 крэкинг-бензин остается пригодным для
применения в двигателях внутреннего сгорания.
ГИДР00ЧИС1КА
Очистка бензинов крэкинга и пиролиза имеет в основном
своей задачей освободиться от таких веществ, которые, являясь
причиной смолообразования, делают бензин в большей или мень-
шей степени нестабильным. Эта задача, как было показано
выше, может быть решена обычными методами очистки, напри-
мер обработкой бензина серной кислотой и адсорбентом с после-
дующей перегонкой очищенного бензина. При этом, однако, мо-
гут получаться настолько значительные потери вследствие явле-
ний полимеризации непредельных и конденсации их с аромати-
кой, что возникает вопрос о рентабельности всего данного про-
цесса в целом. Особенно это относится к некоторым видам паро-
фазных бензинов и легких продуктов пиролиза. В такого рода
случаях весьма важную, если не решающую роль должна
сыграть очистка гидрогенизацией, или гидроочистка [25].
Сущность гидроочистки заключается в легком гидрировании
данного продукта крэкинга или пиролиза в сравнительно мягких
условиях температуры п давления в присутствии подходящего
сероустойчивого катализатора. Так например, для получения
стабильных бензинов парофазното крэкинга достаточно подвер-
гнуть неочищенный бензин гидрированию в следующих усло-
виях:
Температура процесса.......................320—400
Давление . • • ...................*4ат
Время контакта.................•...........0,1—0..» мин.
Концентрация Но на 1 объем продукта........Зл> о ьема
Расход водорода от веса сырья . ...........0.5—1\ о
Наиболее подходящим катализатором для этого процесса ока-
зался смешанный катализатор: двухсернистый молибден на
окиси цинка (MoSs + ZnO). Потери, присущие другим методам
она
44 Зак. 1622. Химия нефти.
очистки, при работе методом гидроочистки практически отсут-
ствуют. J
Основная задача, которую приходится иметь в -виду вд
гидроочистке, заключается в том, чтобы, подвергая бензин очи-
стке, не снизить его антидетонационных свойств. Такое сниже-
ние, естественно, должно произойти, если бензин будет подвер-
гнут глубокому гидрированию, когда непредельные углеводо-
роды бензина полностью превратятся в углеводороды предель-
ного характера. Опыт показывает, однако, что для получения
стабильного бензина вовсе нет надобности в столь глубоком гид-
рировании. Очевидно, в процессе гидроочистки происходит селе-
ктивное гидрирование, причем в первую очередь происходит
гидрирование наиболее неустойчивых компонентов бензина.
В -результате может получиться продукт, достаточно стабильный^
хотя и с высоким содержанием непредельных углеводородов.
Еще более важные результаты получаются от применения
гидроочистки к продуктам пиролиза [26]. При очистке легкого
масла пиролиза серной кислотой, как это делается на нефтега-
зовых заводах, выход очищенного масла не превышает обычно
56% от сырья. При этом из легкого масла вычищаются все не-
предельные углеводорода; очищенное масло имеет начало кипе-
ния около 85° и лишено так называемых пусковых фракций,
вследствие чего к нему приходится прибавлять до 20% бензина
прямой гонки, значительно понижающего октановое число ис-
ходного продукта. Применение гидроочистки к легкому маслу
резко изменяет эти малоудовлетворительные результаты.
Легкое масло пиролиза легко гидрируется при температуре
около 350° и рабочем давлении от 2—3 ат и выше. В качестве
катализатора- и здесь с успехом может служить двухсернистый
молибден (M0S2). Расход водорода составляет 0,5—о,7%. Выход
готового продукта—около 93^ при образовании полимеров до
7%. Получаемые, таким образом, пиробензины имеют вполне
удовлетворительный фракционный состав и высокие антидето-
национные свойства: октановые числа их ’ в зависимости от
конца кипения колеблются от 84 до 93, а с добавкой этиловой
жидкости могут быть подняты до 100. Аналогичные -результаты
дает гидроочистка в применении к бензольной головке пиролиза,
с тем лишь отличием, что получаемый продукт выделяется зна-
чительным содержанием пусковых фракций (начало кипения
35—40°).
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ ГИДРООЧИСТКИ
Гидроочистка представляет собою один из новейших спе-
циальных методов очистки продуктов крэкинга и пиролиза.
Она требует специальной гидрогенизационной аппаратуры и того
или иного источника водорода. Впрочем, аппаратура для гидро-
очистки характеризуется значительно большей простотой, чем
для деструктивной гидрогенизации, так как рабочие давления,
как было указано выше, здесь не велики. Водород для гидро-
690
очистки требуется средней чистоты. Принимая во внимание, что
гидроочистка дает высококачественные продукты при самых не-
значительных потерях, следует ожидать, что этот метод очистки
продуктов крэкинга и пиролиза займет в нефтеперерабатываю-
щей промышленности выдающееся место.
Д. Очистка с помощью растворителей
За последние 10 лет явно обозначилось новое направление
нефтеперерабатывающей промышленности, получающее наряду
с адсорбционными методами очистки все более и более широкое
и плодотворное применение, особенно в области производства
смазочных масел. Сущность этой методики заключается в замене
химической очистки нефтяных дестиллатов более совершенными
методами, а именно: обработкой специальными растворителями,
обладающими избирательной (селективной) растворимостью
к отдельным компонентам нефтяных продуктов. Преимущества
такой методики перед рассмотренными выше методами чисто хи-
мической очистки очевидны: здесь удается избежать воздей-
ствия на углеводороды нефти разного рода сильно действующих
реагентов (серная кислота и т. п.), которые могут весьма" суще-
ственно изменить химическую природу этих углеводородов. Не
менее интересно и плодотворно должно быть применение такой
методики к лабораторному исследованию состава нефтяных де-
стиллатов, хотя громадные трудности, встречаемые на этом пути,
очевидны, поскольку вопрос должен стоять в данном случае не
только о качественном, но и о количественном разделении угле-
водородов различных рядов [27].
РАСТВОРИТЕЛИ ПРИ ПЕРЕРАБОТКЕ НЕФТИ
Избирательное (селективное) растворение в применении
к нефтяным продуктам может иметь двоякого рода цель: либо
отделение тяжелых нефтяных фракций от более легких («холод-
ная фракционировки»), либо отделение тех или иных составных
частей неявных продуктов, снижающих их качество. Для до-
стижения той или другой цели были предложены различные
растворители. , ,
В качестве растворителей для разделения нефтяных фрак-
ций были предложены спирты: этиловый и изоамиловый.
Этиловый спирт (96 ) действительно оказался способным раз-
делитТ мазут с вязкостью Э50 = 7,7 на две чашки [28]; бсктее
легкую (950 = 2.7), растворившуюся в спирте, и более тяжелый
остаток (050 = 14,6). Такое разделение требует, однако, громад-
ного количества спирта (10 объемов) и к тому же крайн
вершенно: как показало ближайшее исследование, полученные
продукты гораздо менее однородны, чем продукты, получаемые
обыкновенной перегонкой.
Аналогичные результаты были получены с изоамиловым
спиртом [29]: мазут растворялся в изоамиловом спирте и иод-
691
44*
вергался дробному осаждению винным спиртом. Таким образом
из грозненского мазута удалось выделить кристаллический
углеводород с температурой плавления 87 \ т. е. углеводород
типа церезина, в количестве до 10,7%. Эти результаты не напщ
практического применения.
Несравнимо большую важность представляет второе напра-
вление работ в данной области, т. е. применение селективных
растворителей для отделения от нефтяных продуктов отдельных
составных частей, снижающих их качество. В качестве раство-
рителей этого рода на первом месте должен быть отмечен жвд-
кий сернистый ангидрид (SO?), давно уже предложенный для
этой цели Эделеану [30].
Жидкий сернистый ангидрид (температура кипения—ю°) хо-
рошо растворяет ароматику и непредельные углеводороды,
а также смолистые вещества нефти; так например, бензол и то-
луол смешиваются с жидким сернистым ангидридом даже при
—20° во всех отношениях. Напротив, нафтены и парафины рас-
творяются в жидком сернистом ангидриде гораздо хуже. С дру-
гой стороны, сам сернистый ангидрид довольно хорошо раство-
ряется в некоторых углеводородах, например, в ароматике, так
что лишь в избытке сернистого ангидрида наступает расслаива-
ние: на верхний слой, представляющий собою раствор серни-
стого ангидрида в нафтенах и парафинах данного нефтепро-
дукта, и нижний слой — раствор ароматики и непредельных,
а также смолистых веществ в сернистом ангидриде; в этом же
нижнем слое концентрируется большая часть сернистых соеди-
нений, которые хорошо растворимы в жидком сернистом анги-
дриде. Разделяя слои и повторяя извлечение верхнего слоя 2—
3 раза, удается резко снизить содержание в очищаемом продукте
не только ароматики, но и сернистых соединений.
Другой растворитель, также давно предложенный для разде-
ления ароматики от углеводородов с открытой цепью, это—ди-
метилсулъфат SO^CIIsh [31]. Как показало, однако, ближай-
шее исследование, диметилсульфат (температура кипения 187°)
растворяет значительные количества легко кипящих углеводо-
родов парафинового ряда и только для высококипящих масел
дает хорошее разделение. Тем не менее, вследствие своих силь-
ных ядовитых свойств дпметплсу льфат как селективный раство-
ритель не получил сколько-нибудь широкого распространения.
В новейшее" время число- селективных растворителей, пред-
ложенных для отделения ароматики от углеводородов парафи-
нового ряда, чрезвычайно возросло, причем часть их нашла
применение не только в лабораторной работе, но n в промышлен-
ности. Важнейшие из них указаны ниже.
Пропан, СзНе. Чистый пропан кишит при —12,2°; упругость
его паров при 21° равна 8.5 ат. Его критическая температура
—100,1°; критическое давление — 43,8 ат. Удельный вес жид-
кого пропана при 21° — около 0,5, а при 100° — 0,25. По мере
приближения к критической температуре вязкость и поверхно-
стное натяжение пропана снижаются до весьма малой величины.
692
Равным образом резко падает вблизи критической температуры
удельный вес жидкого пропана, так что газообразный пропан
под давлением 70 пт при 104 обладает гораздо большим удель-
ным весом, чем жидкий пропан при 100° и соответствующем
давлении.
Как селективный растворитель пропан стал применяться
сначала для депарафинизации смазочных масел. В настоящее-
время пропан считается одним из лучших и наиболее разно-
сторонних растворителей, применяемых для различных целей*
как-то: 1) для депарафинизации смазочных масел пропан весьма,
удобен вследствие его малой вязкости, ничтожной растворяю-
щей силы в отношении парафина и свойства самоохлаждаться;
наиболее благоприятные температуры для депарафинизации про-
пана лежат в пределах от —42° до 4-21°; 2) для деасфальтиза-
ции масел пропан удобно применять при температурах от 38 до
60°, когда асфальтовые вещества весьма мало растворяются
в пропане и осаждаются, тогда как масло и парафин полностью
растворимы в нем; 3) при температурах от 60 до 100° раствори-
мость в пропане масел и парафина постепенно уменьшается*
причем начинают осаждаться сначала наиболее тяжелые соеди-
нения; около критической температуры пропана из раствора
выделяется все масло, так что разделение возможно здесь путем
простой декантации.
Пропан не ядовит, чрезвычайно стабилен, не вызывает кор-
розии аппаратуры и одновременно может выполнять роль рас-
творителя и охладителя. Запасы пропана громадны; он полу-
чается на нефтеперегонных заводах при стабилизации газовых
бензинов и на промыслах как один из компонентов естествен-
ного таза; уже под небольшим давлением пропан может быть пе-
реведен в жидкое состояние. Таким образом как растворитель
пропан очень дешев и легко регенерируется Существенным не-
достатком его является необходимость для работы с ним распо-
лагать спецальной герметичной аппаратурой, выдерживающей
довольно высокие давления [32].
Хлорекс, или ₽'-дихлордиэтиловый эфир (СЮНгСН^гО.
Хлорекс представляет собою жидкость с температурой кипения
178°; упругость паров его при 38 равна 10 мм. Приготовление
и методика применения хлорекса составляют собственность од-
ной из американских фирм (Carbide and Carbon Chemical Corp.).
Хлорекс является одним из лучших селективных растворите-
лей, применяемых для очистки смазочных масел [33]. При вы-
дающейся селективной силе хлорекс выделяется крайней просто-
той аппаратуры, которая необходима в процессе его применения:
экстракцию хлорексом можно проводить в обычных мешалках,
а регенерацию растворителя — перегонкой в обычных периоди-
чески действующих кубах. Само собою разумеется, что такая
упрощенная аппаратура имеет ряд недостатков, которые заста-
вили впоследствии перейти к аппаратам более совершенной кон-
струкции (экстракторы противоточной системы и т. д.).
Основные достоинства хлорекса заключаются в следующем.
693.
Хлорекс тяжелее воды; его удельный вес 1,22. Так как раство-
римость его в воде невелика, то хлорекс может храниться под
слоем воды, благодаря чему потери его от испарения могут быть
сведены до минимума. Впрочем, потери эти и без того крайне
незначительны благодаря низкой упругости его паров при обык-
новенном давлении. Растворяющая сила хлорекса такова, что
для получения хороших результатов возможно применение его
в весьма умеренных отношениях к маслу, например в отноше-
нии 1,5 :1. После экстракции разделение слоев происходит бы-
стро и резко. Так как хлорекс не вступает ни в какие реакции
с компонентами сырья в условиях процесса, то его регенерация
как из рафината, так и из экстракта легко производится путем
отгонки. Потери хлорекса при очистке крайне незначительны.
Температура замерзания хлорекса низка (—51,7°), что дает воз-
можность работать с этим растворителем при низкой темпера-
туре. Наконец, теплоемкость и скрытая теплота испарения хло-
рекса малы, что весьма важно для снижения расхода топлива
при регенерации растворителя. Впрочем, большинство этих по-
ложительных качеств хлорекса принадлежит также и другим
селективным растворителям, нашедшим применение в промыш-
ленности, а именно нитробензолу, фенолу и фурфуролу.
Из числа недостатков хлорекса была отмечена склонность его
к разложению при высоких температурах. Так как при этом вы-
деляется хлористым водород, который может действовать на ап-
паратуру корродирующим образом, то полная регенерация рас-
творителя требует в данном случае введения водяного пара, что
сопряжено с дополнительными расходами. С другой стороны,
имеются указания, что применение хлорекса нередко требует
предварительной подготовки сырья путем обработки его серной
кислотой или деасфальтизации с помощью пропана; это отно-
сится в особенности к получению остаточных масел, когда ука-
занная подготовка сырья необходима даже при умеренном со-
держании смолистых веществ (5—15%).
Нитробензол, С6Н5 • NO*. Нитробензол представляет собою тя-
желую жидкость (удельный вес 1,204) с температурой кипе-
ния 209е; температура плавления нитробензола 5,7°. Это—деше-
вый, легко доступный растворитель, изготовляемый в громадных
количествах как полупродукт анилокрасочной промышленности.
Хотя для селективной очистки масел нитробензол был рекомен-
дован одним из первых, тем не менее .применение его в заводской
практике довольно органичено. Повидимому, это связано с силь-
ными ядовитыми свойствами, присущими нитробензолу (кровя-
ной яд).
Нитробензол обладает как растворитель выдающимися каче-
ствами: при высокой растворяющей силе он выделяется своими
селективными свойствами. Он обладает высокой стабильностью
в рабочих условиях и не вызывает коррозии аппаратуры; имеет
низкую упругость паров (0,5 мм при 32°), так что. потери его
от испарения незначительны, содержание же его ларов в атмо-
сфере рабочего помещения невелико, что чрезвычайно важно
ввиду его ядовитых свойств. Расходный коэфициент для нитро-
бензола при селективной очистке масел исключительно мал: от
0,5 до 2,0 объемов растворителя на один объем сырья; благодаря
этому стоимость регенерации растворителя в данном случае не-
велика. Применение нитробензола в качестве селективного рас-
творителя не требует предварительной подготовки сырья путем
перегонки или кислотной очистки. Тем не менее, обработка
нитробензолом в итоге дает наивысшие выхода смазочных масел
весьма высокого качества; в частности, эти масла выделяются
своим цветом, так как нитробензол обладает выдающейся спо-
собностью извлекать из масел красящие вещества.
Рафинат, полученный после очистки нитробензолом, еще не
представляет собою готового смазочною масла: он требует депа-
рафинизации и доочистки глиной; кислотная очистка не тре-
буется. Депарафинизация может производиться в большинстве
случаев либо до, либо после нитробензольного процесса; лишь
сырье, очень богатое парафином, требует предварительной, хотя
бы частичной депарафинизации [34].
Фенол, СеНб 'ОН. При обыкновенной температуре фенол — кри-
сталлическое вещество (температура плавления 42,5°); его тем-
пература кипения 182°. Фенол представляет собою селективный
растворитель, нашедший широкое промышленное применение для
очистки не только авиационных и моторных масел, но и таких
специальных нефтепродуктов, как масла трансформаторное, тур-
бинное и т. д.
Обработка фенолом производится при температурах от 38 до
82°; в отдельных случаях для получения рафината особо высо-
кого качества температуру повышают до 121°, причем, однако,,
выход рафината, естественно, уменьшается, Расход растворителя
по отношению к маслу не превышает 1,75 :1, т. е. весьма ограни-
чен; несмотря на это, очистка фенолом дает масла превосход-
ного качества. Индекс вязкости полученных масел зависит, есте-
ственно, от глубины очистки фенолом, т. е. от числа последова-
тельных ступеней очистки; при заданном же индексе вязкости
готового масла очистка фенолом дает масла с лучшим цветом и
меньшим процентом кокса. Фенольный процесс не требует ка-
кой-либо предварительной обработки сырья. Широкая масляная
фракция и даже цилиндерсток прямо поступают на обработку
фенолом, после чего рафинат подвергается вторичной перегонке.
Таким образом при фенольном процессе кислотная очистка- и
даже обработка глиной оказываются излишними. Особенно хоро-
шие результаты получаются, если при повторной обработке фе-
нолом добавлять к нему небольшие количества воды. Безводный
фенол является прекрасным растворителем для нафтеновых.ком»
понентов масла, полностью удаляя их из рафината. Водный Фе-
нол, будучи плохим растворителем для парафиновых компонен-
тов масла, предупреждает переход их в экстракт. Таким образом
последовательно разбавляя экстракты небольшими количествами
воды (около 5%) и подвергая получаемые при этом отстой но-
вой обработке фенолом, можно получить до 75% рафината пре-
695
несходного качества (индекс вязкости до 96). Применением боль,
шинства других растворителей не удается получать столь высо-
ких выходов на масла столь высокого качества [35].
Фурфурол, В свежеперегнанпом виде фурфурол представляет
собою бесцветную жидкость с температурой кипения 162°. Теу-
СН — СН
сн с-сно
о
пература застывания его —37°. Удельный вес фурфурола 1,164
(при *15°), упругость его паров при 38 равна 7 xw. Раствори-
мость фурфурола Б в°Де Довольно значительная — 8,96 %
при 38°. ”
Фурфурол получается кипячением с разбавленными кисло-
тами различного дешевого сельскохозяйственного сырья и отбро-
сов, содержащих пентозы и пентозаны; таковы, например, со-
лома, початки кукурузы, шелуха подсолнуха и т. д. В настоящее
время фурфурол является одним из весьма доступных раствори-
телей, нашедших применение в 'Промышленной очистке смазоч-
ных масел.
Фурфурол обладает рядом выдающихся качеств, которые ста-
вят его в один ряд с самыми лучшими селективными раствори-
телями. Он обладает высокой селективностью и растворяющей
силой: имеет сравнительно низкую температуру кипения, при
которой он может отгоняться без разложения; процесс экстрак-
ты фурфуролом ведется при довольно высокой температуре
(от 80 до 120°), благодаря чему облегчаются смешение раствори-
теля с маслом и последующее отстаивание. Недостатком фурфу-
рола является некоторая неустойчивость его при хранении, вслед-
ствие чего фурфурол темнеет и даже несколько осмоляется.
Впрочем при хорошей герметизации потери фурфурола от его
нестабильности, растворимости в воде и других причин весьма
незначительны (от 0,025 до 0,0757с).
Расходный коэфициент фурфурола по отношению к маслу не
больше 3 по объему. Дестиллатное сырье подвергается обработке
Фурфуролом непосредственно, без предварительной подготовки.
В зависимости от характера сырья получают рафинат в количе-
стве от 66 до 84% по объему с индексом вязкости до 100 и выше.
При работе с тяжелыми остатками (резидиумы), с более или ме-
нее значительным содержанием смол, необходима предваритель-
ная деасфальтизация сырья; как и в других случаях, такая под-
готовка наиболее успешно осуществляется при помощи пропана.
Лишь в отдельных случаях, например с пенсильванскими остат-
ками, почти не содержащими смол, предварительная деасфаль-
тизация сырья может не применяться 36 I.
Кроме рассмотренных, известен ряд растворителей, также ха-
рактеризующихся высокой селективностью по отношению к раз-
личным компонентам смазочных масел и большой растворяющей
<596
силой; таковы, например, анилин, фенил гидразин, левулиновая
кислота и др. Большинство из них может применяться лишь,
в лабораторных условиях по различным причинам, как-то: вы-
сокая токсичность, дорогая стоимость и т. д.
Анилин, CttHjXH® (температура кипения 183^; температура плавления
—6°), как известно, является прекрасным растворителем для ароматических
углеводородов, тогда как парафины и нафтены растворяются в нем лишь
в малой степени. Чтобы несколько снизить способность анилина растворять
масла, рекомендуется применять влажный анилин; количество его по от-
ношению к маслу в зависимости от состава последнего может келебаться
от 0,5 до 5 частей по весу. Применение анилина в качестве селективного
растворителя особенно удобно ввиду возможности количественного удаления
его из масла, для чего достаточно промывки слабой солянай кислотой; ани-
лин переходит при этом в водный раствор, в виде анилиновой соли [37]..
Фенилгидразин, CefbNHNHs (температура кипенпя 243,5°; тем-
пература плавления 19,6°) как основание подобно анилину может быть,
удален из масла также количественно путем промывки соляной кислотой-
Обладает высокой растворяющей способностью по отношению к ароматическим
углеводородам и может служить для глубокого отделения ароматики от пара-
финов в маслах, причем все операции можно производить здесь при одной и
той же температуре (комнатной). С легко кппящими смесями, содержа-
щими свыше 25% ароматики, получаются ошибки, которые могут дости-
гать 10%. Недостатки фенилгидразина как селективного растворителя
являются его сравнительно высокая стоимость, а также легкая окисляемость
на воздухе [зь].
' Левулиновая кислота, СНз • СО - СНг СНг • СО?Н (температура ки-
пения 250°; температура плавления 37°), получается кипячением вино-
жадного сахара с крепкой соляной кислотой. Вследствие своей высокой
стоимости едва ли может найти промышленное применение, но удобна
для лабораторных исследований, причем, как и в случае фенилгидразина,.
все операции можно производить при комнатной температуре. Рекомендуется
применять трехкратное количество левулиновой кислоты на одну часть,
масла. В смеси с ароматическими углеводородами (до 90%) все виды лег-
ких и машинных масел обнаружили почти теоретическое количество аро-
матики. Будучи легко растворима в воде, левулиновая кислота легко от-
мывается от масла [38].
Особо должны быть отмечены получающие все более и бо-
лее широкое признание процессы с парными растворителями, на-
пример с растворами сернистого ангидрида в бензоле, пропана —
в орто-крезоле и т. п.
Жидкий сернистый ангидрид представляет собою раствори-
тель с большой селективностью, но слабой растворяющей силы.
Это последнее обстоятельство заставляет при пользовании дан-
ным растворителем применять его в значительно больших коли-
чествах по сравнению с другими растворителями, что при обще-
известных свойствах сернистого ангпдрнда представляет весьма
существенные неудобства. Вместе с тем сернистый ангидрид,
давший при очистке богатых ароматикой румынских керосинов,
столь выдающиеся результаты (ч. I, гл. IV), в применении
к смолистому масляному сырью оказался явно недостаточным, -
способным при условии рентабельности давать смазочные масла,
лишь средних качеств, правда в .достаточной мере стабиль-
ные, но с недостаточным индексом вязкости. Значительно луч-
шие результаты были получены при замене сернистого ангидрида
раствором его в бензоле (7 : 3), причем экстракция производи-
лась не один, а два раза [39]. При этом, естественно, полу-
чаются рафинаты двух сортов, общий выход на очищенные
масла повышается, и производство смазочных масел даже из вы-
сокосмолистого сырья становится рентабельным.
Особенное распространение за последнее время получает так-
же так называемый Duo-sol-процесс, в котором растворителями
являются пропан и о-крезол. В этой паре растворителей пропан
является растворителем для рафината, крезол же —для
экстракта. Для очистки в этом процессе применяются масляные
остатки Президиумы). В простейших случаях очистка заклю-
чается здесь в экстракции сырья смесью пропана с крезолом,
после чего следует либо доочистка рафината обычным способом1,
либо перколяция в лигроиновом растворе. Процесс заканчи-
вается депарафинизацией рафината- тем или иным способом [40].
ТЕХНИКА СОЛЬВЕНТНОЙ ОЧИСТКИ
О технике применения селективных растворителей, жидких
при обыкновенной температуре, можно судить на основании
следующей схемы [зз] очистки хлорексом :(фиг. 121).
Фиг. 121. Схема очистки селективными растворителями.
Подлежащее очистке масло и растворитель проходят ряд по-
следовательно расположенных смесителей D-l, D-2, D-3 и отстой-
пи ко н-сепараторов В-1, К-2, K-:i, причем масло поступает из ре-
зервуара А, растворитель же — из бака В. Предварительно
смесь проходит через обогреватель С, где смесь принимает тре-
буемую температуру. Свежее масло смешивается с растворите-
лем, уже дважды использованным (из £-2); отстоявшиеся в Е-1
(>98
слои направляются: нижний слой растворителя с а|юматикой
непредельными и смолами в специальный резервуар / верхний
же слой, частично очищенное масло, во второй смеситель Z)-9
захватывая по пути растворитель из третьего отстойника E-:i
Пройдя далее второй отстойник, масло переходит в третий сме-
ситель, захватывая при этом свежий растворитель Из послед-
него отстойника E-:i очищенное масло поступает в другой резер-
вуар G.
При обоих резервуарах F
и G имеются свои перегон-
ные кубы Н и ./ для отгонки
растворителя Отгонка про-
изводится водяным паром,
и регенерированный раство-
ритель направляется в свой
бак В; вода же, сконденси-
ровавшаяся из водяного па-
ра. собирается в особый ре-
зервуар М и для полноты
регенерации растворителя
перегоняется еще раз из
отдельного котла N В этих
условиях потери раствори-
теля оказываются мини-
мальными.
После отгонки раствори-
теля в котле Н остается
экстракт, в котле же J—
очищенное масло; и то и
другое направляются в со-
ответствующие резервуары
О и Р.
Приведенная схема очи-
стки вполне применима
также к таким случаям,
когда растворитель имеет
температуру застывания вы-
ше комнатной. Единственное дополнение в подобного рода слу-
чаях заключается в добавочном подогревателе для чистого рас-
творителя.
Более сложную конструкцию имеет аппаратура для раство-
рителей. газообразных при обыкновенной температуре. Приме-
ром может служить процесс Эделеану. Не останавливаясь на
промышленных установках Эделеану, ограничимся здесь описа-
нием крупно табораторной установки этого рода 41].
В мешалку А (фиг. 122), снабженную воронкой, рубашкой,
термометром и смотровым окошком, загружается подлежащий
очистке дестиллат. Растворитель, жидкий сернистый ангидрид,
вводится в мешалку из резервуара В, снабженного мерным стек-
лом, при помощи вентилей 4 и 5, причем для уравнения давле-
699
ний в обоих сосудах J п В открываются также вентили 6. 7 и 8
Источником для получения новых порций растворителя служит
баллон с сернистым ангидридом Z. Перемешивание масла с рас-
творителем производится короткое время (10 мин.) лопастной
мешалкой, приводимой в действие мотором, при температурах
ниже 0°, после чего дают смеси отстояться. Нижний слой
(экстракт) спускают далее в приемник С с помощью вентилей 9
и 10, а также 8, 11 и 12, причем последние три вентцля служат
для уравнения давления. Через смотровое окошко можно на-
блюдать момент прохождения поверхности, разделяющей жид-
кость на два слоя. Тогда вентили О и 10 закрывают и присту-
пают к обработке масла свежей порцией растворителя в преж-
нем порядке; только экстракты, получаемые в результате каж-
дой новой обработкп масла растворителем (например, второй и
третий), собираются каждый раз в особые приемники D и Е. На-
полнение и опорожнение отдельных частей аппаратуры произ-
водится, как видно из схемы, с помощью системы соединяющих
труб и вентилей (/—26).
Очищенное масло после удаления пз него небольшого коли-
чества оставшегося сернистого ангидрида спускается из ме-
шалки через вентиль 14; растворитель же (SO2j, составляющий
главную массу экстрактов, подвергается регенерации. Для этого
приемники С? D и Е подогреваются; находящийся в них серни-
стый ангидрид отводится че.|юз трубку t в змеевиковый холо-
дильник F, снабженный охлаждающей рубашкой, и здесь кон-
денсируется. стекая в резервуар В.
В обеих вышеприведенных схемах смешение растворителя
с маслом и последующее отстаивание производятся периодически
в различных сосудах смесителях (микстерах) и отстойниках
(сепараторах). В проточных системах смесители и отстойники
-соединены в одном сосуде, обычно представляющем собою длин-
ный, горизонтально расположенный цилиндр, разделенный вер-
тикальными перегородками на 8—-9 отделений. Эти отделения
одновременно являются смесителями и отстойниками, причем
более тяжелый экстракт перекачивается из отделения в отделе-
ние насосами в одном направлепип, например от VIII к I отде-
лению; более же легкий рафинат с помощью специальных ин-
жекторов переходит из отделения в отделение в обратном направ-
лении. Если экстракция ведется парными растворителями, то
один из них подается на одном конце цилиндра, другой—на
противоположном конце, причем движение растворителей произ-
водится также в противоположных направлениях: растворитель
для экстракта движется по пути экстракта п выходит в ниж-
ней части I отделения, тогда как растворитель для рафината
движется в направлении хода рафината и выходит в верхней
части последнего отделения.
ХИМИЗМ И БЛИЖАЙШИЕ ЗАДАЧИ СОЛЬВЕНТНОЙ ОЧИСТКИ
Опыт показывает, что очистка подходящими селективными
растворителями повышает индекс вязкости смазочных масел,
*700
Как 'было показано при рассмотрении нефтяных углеводоро-
. различных рядов [ч. I], наши познания ближайшего со-
уменьшает их склонность к нагарообразованию в процессе ра-
боты на моторе, снижает их окисляемость. Так как в основе
данного метода очистки лежат исключительно процессы физико-
химиче к г хара те )а без каких-либо химических реакций
между растворителем и компонентами масла, то характеристика
химизма процесса сотьвен ной очистки сводится по существу
’ к решению вопроса, какие составные части масляного сырья
удаляются (экстрагируются) при его обработке тем или иным
растворителем и какие сохраняются в рафинате. В свою очередь
этот вопрос упирается в крайне сложную проблему состава выс-
ших погонов нефти и ее остатков (резидиумов).
Как было показано при рассмотрении нефтяных углеводоро-
дов различных рядов [ч. I], наши познания ближайшего со-
става нефти ограничиваются погонами до 160—200°; изучение
же вопроса о составе более высококипящих погонов, особенно
масляных, находится еще в самой начальной стадии и по суще-
ству сводится к изысканиям надежных методов для характери-
стики и определения отдельных групп соединений, содержа-
щихся в масляном сырье. Однако и эта задача является чрез-
вычайно сложной в методическом отношении, и многочисленные
исследования, произведенные в данном направлении, позволили
пока наметить лишь следующие основные типы компонентов
сырья, служащего для получения смазочных масел:
1. Парафины и изопарафины. Сюда относятся высокомолеку-
лярные углеводороды ряда метана как с нормальной цепью угле-
родных атомов, так и пзостроения (ч. I, гл. V В). Они имеют
различную консистенцию—от мягкой, мазеобразной до твердой
и хрупкой, низкую вязкость, неудовлетворительную кривую из-
менения ее с температурой, высокую температуру плавления.
Совокупность этих свойств заставляет считать парафины и изо-
парафины за нежелательные компоненты смазочного масла и
стремиться к их удалению, т. е. к депарафинизации масла.
2. Бензолъно-парафиновые и нафтено-парафиновые углеводо-
роды. Это — гомологи бензола, дифенила, нафталина и, быть мо-
жет, некоторых других простейших ароматических систем,
а также соответствующих нафтенов с длинными открытыми бо-
ковыми цепями предельного характера. Эти углеводороды харак-
теризуются высоким индексом вязкости (100—200 ) и дают не-
большое нагарообразование. По своему отношению к кислороду
бензольно-парафиновые углеводороды являются более устойчи-
выми, чем нафтено-парафиновые. Поэтому некоторые авторы
[38] считают бензольно-ароматические углеводороды главной и
наиболее важной частью хорошо очищенного масл . По мнен
других авторов [39] таковой являются нафтено-парафиновые
углеводороды, которым, кроме вышеуказанных качеств, свой-
ственны также хорошая маслянистость и высокая вспышка.
3. Многоядерная ароматика и продукты ее неполного гидри-
рования, представляющие собою системы, склонные к нагаро-
образованию; под влиянием кислорода воздуха при повышенной
температуре и давлении они также легко окисляются с образов^
пием осадков. Поэтому они должны расцениваться как нежела-
тельные компоненты смазочного масла, подлежащие удалению
4. Полициклические нафтены — также малоценные масляные
компоненты: они характеризуются сравнительно крутой кривой
вязкости и низкой вспышкой, а также склонностью к образова-
нию нагаров и осадков.
5. Асфальтены — твердые высокоплавкие вещества, являю-
щиеся наименее желательными компонентами смазочного масла
То же можно сказать о смолах, которые, как известно, легко
превращаются в асфальтены.
6. Наконец, масляное сырье может содержать небольшие ко-
личества таких компонентов нефти, как кислородные соедине-
ния [нафтеновые кислоты, фенолы] и сернистые соединения, ко-
торые также подлежат удалению из смазочного масла, как ве-
щества корродирующие.
В отличие от бензиновых и отчасти керосиновых погонов, химическая
природа которых может быть охарактеризована в первую очередь принадлеж-
ностью к тому или иному ряду углеводородов (парафины, нафтены, аро-
матика), для компонентов более тяжелых погонов такая характеристика
нередко явно недостаточна. Так например, гомологи бензола или нафта-
лина с длинной боковой цепью либо соответствующие им нафтены с длин-
ной боковой цепью п подобные им сложные углеводороды отражают в
своих свойствах принадлежность по i райней мере к двум различным типам
углеводородов, а- именно, с одной стороны, к углеводородам кольчатого
строения (ароматика, нафтены), с другой — к углеводородам с открытой
группировкой атомов углерода (парафины). Начальной характеристикой
такого рода сложных систем может служить < тносительяая значимость
числа углеродных атомов, образующих в них кольцевые группировки и по
разности до 10и открытые цепи. Выраженная в процентах к общему числу
углеродных атомов данной системы первая из этих величин может быть
названа «кольцевой характеристикой» системы, а нахождение такого рода
характеристик в применении к отдельным углеводородам или к их смесям
называется «кольцевым анализом» (44).
С принципиальной стороны кольцевой анализ как метод начальной ха-
рактеристики химического состава смесей высокомолекулярных углеводо-
родов (масел) не встречает возражений. Однако при современном состоянии
наших знаний свойств углеводородов масляных фракций нефти нахождение
достаточно точных кольцевых характеристик масла встречает большие
трудности и во всяком случае требует значительных уточнений ло сравне-
нию с литературными данными по этому вопросу.
Как ясно из изложенного, единственно ценными компонен-
тами смазочных масел являются бензоль но-парафиновые или
нафтено-парафиновые углеводороды, являющиеся носителями та-
ких ценных качеств хорошего смазочного масла, как масля-
нистость, пологая кривая изменения вязкости с температурой
(высокий индекс вязкости), высокая вспышка при данной вяз-
кости, малая склонность к нагарообразованию (небольшие числа
Конрадсона) и к образованию осадков при нагревании масла
в атмосфере кислорода по Сляю, Вуткову или иными методами..
Поэтому основная задача при получении хорошею смазочного
масла заключается, очевидно, в том, чтобы, сохранив в надле-
жаще выбранном сырье наиболее ценные компоненты без изме-
ненная, удалить из него все то, что, являясь для масла либо-
702
ненужным балластом, либо вредной примесью, снижает его ка-
чество. Обраоотка масла селективным растворителем является
для ©той цели наилучшим средством.
Действительно, представим себе случай, когда иодлежапщй
очистке продукт представляет собою масляный мазут (резидиум,
остатки). Его очистку можно провести по следующей схеме.
Прежде всего разгонкой в вакууме полу 1ают широкую масля-
ную фракцию. Эта фракция подвергается затем деасфальтизации
при помощи того или иного селективного растворителя. Одно-
временно со смолами и асфальтенами при этом удаляются боль-
шая часть сернистых соединений и некоторые другие примеси,
снижающие качество масла, например тяжелая ароматика.’
В рафинате, кроме бонзбльно-парафиновых или нафтено-парафи-
новых углеводородов, содержатся также парафины, нередко выпа-
дающие из рафината после отгонки растворителя. Поэтому сле-
дующая задача очистки — депарафинизация рафината, которая
осуществляется различными способами. Простейшими из них
являются «холодное отстаивание» и центрифугирование.
Процесс «холодного отстаивания» осуществляется в самых
общих чертах следующим образом. Профильтрованное через
глину масло (30—40%) растворяется в бензине (70—60%) и
заливается в отстойный резервуар, постепенно охлаждаемый до
—12, —13°. Здесь масло отстаивается при этой температуре
в продолжение суток. При этом происходит высаживание пара-
фина (петролатум), который собирается в нижней части резер-
вуара; масло же в бензиновом растворе занимает его остальную
часть и может быть удалено отсюда с помощью подвижной
трубы, устанавливаемой над уровнем петролатума После уда-
ления масла из резервуара выкачивается отстоявшийся петро-
латум; в некоторых случаях предварительно удаляют еще полу-
отстоявшееся масло, которое идет на вторичное отстаивание.
Растворитель удаляется как из масла, так и из петролатума
путем отгонки водяным паром, после чего масло готово к изго-
товлению разного рода товарных продуктов, а именно цилин-
дровых и моторных масел, петролатум же после обесцвечивания
и очистки идет на изготовление вазелинов и мазей. С помощью
холодного отстаивания удается снизить температуру застывания
масла примерно до —2°.
Центрифугирование, как было указано в ч. П, гл I, во много
раз превосходит действие силы тяжести этого основного фактора
при всяком процессе отстаивания. Масло растворяется в бен-
зине, охлаждается до температуры, определяемой в зависимости
от требуемой температуры застывания готового масла, и про-
пускается через центрифугу Шарп леса в непрерывном процессе;
выходят из центрифуги два продукта: депарафинизированное
масло (брайт-сток) й петролатум. По сравнению со способом
холодного отстаивания центрифугирование дает более однообраз-
ный продукт, лучше депарафинизированное масло (температура
застывания до —15—20°) и более высокого качества петролатум.
Как было указано выше, депарафинизация смазочных масел
70S
удобно осуществляется в настоящее время также в растворе
пропана. Для этого деасфальтизированный рафинат перекачи-
вают в колонну — сепаратор, куда в необходимом количестве
добавляется также пропан. Путем испарения части этого рас-
творителя температуру раствора понижают до —43°, ускоряя
охлаждение отсасыванием пропана с помощью компрессора. За-
тем отстоявшуюся смесь перекачивают на громадные фильтры,
где и заканчивается процесс депарафинизации. Если требуется
осветление масла, его подвергают обработке глиной, после чего
следует концентрация масла, т. е. отгонка от него легких ча-
стей до получения остаточного масла с требуемой температурой
вспышки.
В тех случаях, когда исходное масляное сырье мало удовле-
творительно для получения высокосортных масел, например ха-
рактеризуется высоким содержанием смол, богато полицикличе-
скими нафтенами и многоядерной ароматикой, процессов де-
асфальтизации и депарафинизации иногда бывает недостаточно:
полученное масло вследствие содержания в нем малоценных
компонентов может оказаться неудовлетворительным по своим
вязкостным свойствам, по низкой температуре вспышки и дру-
гим качествам. В таких случаях для получения высокосортною
смазочного масла приходится прибегать к повторной доочистке
рафината тем же или новым селективным растворителем, в ре-
зультате чего получают два продукта: рафинат высшего качества
с индексом вязкости 92—95 и экстракт, используемый далее
как сырье для получения индустриальных масел. Как и в пред-
шествующем случае, сольвентная очистка завершается ио мере
надобности обработкой масла глиной и его концентрацией.
На основании изложенного не трудно прптти к выводу, что далеко
не всякий органический растворитель применим в процессе очистки нефте-
продуктов. Назначение селективного растворителя заключается в том, чтобы
отделить наиболее ценные «парафинистые» составные части масла, которые
обычно остаются нерастворимыми, от других компонентов масла, извлекае-
мых растворителем. Эти последние компоненты, снижающие качество масла,
для краткости будут называться далее «ароматикой», подразумевая иод
нею не только ароматические углеводороды различных типов, но также
непредельные углеводороды и разного рода производные углеводородов,
содержащие кислород, серу и а;гот. Важнейшие требования, которым дол-
жен удовлетворять хороший селективный растворитель, следующие [45]:
1. Коэфициент распределения «ароматики» между растворителем и па-
рафиновыми углеводородами масла должен быть достаточно высок, иначе
говоря, сродство «ароматики» к растворителю должно быть гораздо больше,
чем к парафиновым углеводородам масла. Тем самым, естественно, дости-
гается не только снижение расхода растворителя, нс и уменьшение числа
этапов, на которых должна происходить обработка масла растворителем.
2. Растворитель должен об падать возможно более низкой раствориющен
способностью по отношению к парафиновым углеводородам; тем самым
повышаются выхода на очищенное масло.
з. Растворитель должен быть по возможности нерастворим в парафи-
новых углеводородах. Чем меньше растворимость растворителя в масле,
тем меньше расход растворителя, а также неизбежные затраты на его ре-
генерацию из маелйноуо слоя.
4. В условиях ('••лективной очистки растворитель должен быть вполне
устойчив во изПежанне неизбежных потерь и загрязнения конечных про-
дуктов ичисткп.
704
5. .Растворитель должен иметь относительно низкую температуру кипе-
ния, чтобы можно было легко произвести полную его отгонку как от
экстракта, т. к и от -очищенкого масла. В противном случае затрудняется
^генерация растворителя, тем солее что значительное повышение темпе»
ратури при отгонке растворителя от масла может снизить качество послед-
него.
G. Процесс обработки дестиллата селективным растворителем должен
протекать по возможности в условиях комнатной температуры и нормаль-
ного давления; отступления от этого условия приводят к повышенным
эксплоатационным расходам. Оптимальная температура гипения раствори-
теля определяется в пределах 30—110°.
<• Растворитель не .должен обладать резко выраженными токсическими
свойствами.
я. Растворитель должен быть по возможности легко доступным и де-
шевым, так как, принимая во внимание невозможность полной его реге-
нерации и неизбежность потерь в процессе очистки, чем дороже раствори-
тель, тем больше, будут сказываться его потери на стоимости очистки.
9. Растворитель не должен вызывать коррозии очистной аппаратуры,
так как явления подобного рода также отражаются на стоимости очистки.
Само собой разумеется, что перечисленные требования для хорошего
селективного растворителя в совокупности должны рассматриваться лишь
как желательные, но отнюдь не обязательные. Для иллюстрации этого по-
ложения достаточно сопоставить свойства рассмотренных выше селектив-
ных растворителей, уже получивших применение в промышленности.
ОБЩИЕ ОСНОВАНИЯ СОЛЬВЕНТНОЙ ОЧИСТКИ
В основу очистки селективными растворитетями должно
быть положено учение о распределении растворенного вещества
между двумя растворителями. Независимо от деталей методики
сольвентной очистки, всегда можно представить себе здесь сле-
дующую картину: те или иные компоненты данного нефтепро-
дукта, например ароматика, непредельные смолы, находятся
в растворе смеси других углеводородов (парафины, нафтены);
прибавление нового селективного растворителя, в котором озна-
ченные компоненты растворяются лучше, чем в основной массе
углеводородов нефтепродукта, имеет своей задачей извлечение
этих компонентов из остальной массы нефтепродукта, которую
можно рассматривать как начальный растворитель тех же ком-
понентов. Такое извлечение регулируется основным законом рас-
пределения растворенного вещества между двумя несмешиваю-
щимися растворителями (фазами): если в обоих растворителях
растворенное вещество обладает одним и тем же молекулярным
весом, то отношение его объемных концентраций в этих двух
соприкасающихся между собой растворителях (фазах) при дан-
ной температуре, оказывается величиной постоянной и независи-
мой от количества растворенного вещества. Обозначая объемные
концентрации растворенного вещества в верхней (разе через i.
а в нижней фазе через (\ получаем:
где X — постоянная величина, называемая коэффициентом рас-
пределения. л ,
Допустим, что растворенное вещество взято в количестве, до-
статочном для насыщения обоих растворителей. 1о1да вели
705
45 Зак. 1022. Химия нефти.
Чины Ci й С 2 будут представлять собой растворимости ДапйогО
вещества в данных растворителях при данных температурах
а коэфициент распределения А' определится как отношение этих
растворимостей. * •
Исходя из закона распределения и допуская, что один рас-
творитель совершенно не смешивается с другим, можно вывести
общие формулы, определяющие степень извлечения вещества из
раствора.
Действительно, пусть р — первоначальное содержание раство-
ренного вещества в растворе; m = V2:Vi — отношение объема
второго растворителя к объему первого растворителя, К — коэфи-
циент распределения (Сз: Ci) на — количество вещества, пере-
ходящего во второй растворитель. Тогда имеем:
где F2C2 = а.
Отсюда легко определяется:
mfi
а = Рт—,--F-
J 1 -j-
Извлечение растворенного вещества можно производить также
другим способом, а именно: поделив второй растворитель на не-
которое число н равных частей, произодить извлечение каждой
порцией растворителя последовательно, отделяя использованную
часть растворителя перед введением новой. Очевидно, отношение
у2: Vi — т придется заменить в данном случае новым отноше-
нием: V?: nVi == пь: п. Вводя это обозначение, получаем для
количества щ — вещества, извлекаемого первой порцией раство-
рителя:
к.
п. — Р —;—Г ’
1 J н -u tn К
а для остатка после первого извлечения:
м К п
г. = }> — р —---- = })-------.
1 J * и -j-жд. J н -j- шК
Отсюда следует также, что величина «j может быть выра^
жена иначе, а именно:
Р п wK (1 л-f-mli) ’
Прилагая те же рассуждения ко. второму извлечению, ана-
логично получаем: для остатка г2 после извлечения второй
порцией растворителя: ‘
/ 11 \2
? о-Р I ’ F" } >
- . J \j! 4“ К /
а для количества а.2 извлеченного вещества после двух извле-
чений:
/ н . / и у
°--’ =? L1 ~ \7^к) J •
706 ' ' .
Наконец, для остатка г„ после извлечения л-й порцией
растворителя получим аналогично: ‘
71
,п 4- тК
для суммарного же количества а„ вещества/ извлеченного зал
извлечений, будем иметь:
/ // '.»»
и.
Гн =р
Н- ’ »
ait=p—p
Легко показать, что вл всегда больше а, увеличиваясь с вели-
чиной п. Гак например, допуская что А<=3, м = 1 и я = 5,
получаем для однократного извлечения «=5/в р, а для пяти-
кратного извлечения суммарно тем же количествам раствори-
теля а = 31/з2 р, т. е. во втором случае извлечение оказывается
слишком в 5 раз полнее, чем в первом. Отсюда следует, что
одно и то же количество растворителя используется ’ гораздо
более рационально, если применять его не один раз, а предвари-
тельно поделить его на несколько частей и применять для из-
влечения каждую порцию растворителя отдельно. Понятно
также, что наиболее совершенное извлечение достигается путем
применения разного рода специальных экстракционных аппара-
тов, в которых одно и то же количество растворителя произво-
дит извлечение (экстракцию) систематически и непрерывно, бу-
дучи регенерируемо в процессе работы путем непрерывной от-
гонки от экстракта.
Все предшествующие выводы относятся к случаям, когда два
растворителя, между которыми происходит распределение рас-
творенного вещества, совершенно нерастворимы друг в друге.
Такие случаи, естественно, можно представить себе только в тео-
рии. В действительности пе существует двух жидкостей, совер-
шенно не растворимых друг в друге, вследствие чего распределе-
ние растворенного вещества происходит не между чистыми рас-
творителями, а между насыщенными растворами одного раство-
рителя в другом. Кроме того, совершенно ясно, что при сольвент-
ной очистке нефтепродуктов растворенное вещество, подлежащее
извлечению, является весьма сложной смесью различных соеди-
нений (ароматика, смолы, сернистые соединения и т. д.), и не
менее сложную смесь представляет собою тот растворитель (па-
рафины, нафтены и т. д.), из которого подлежит удалению в дан-
ном случае указанная смесь растворенных веществ. Само собою
разумеется, что в силу этих обстоятельств предшествующие вы-
воды должны значительно, усложниться, хотя теоретические
основы их, естественно, останутся без изменения.
ТЕОРИЯ СОЛЬВЕНТНОЙ очистки
Как было указано выше, в основу теории сольвентной очистки
Должно быть положено учение о распределении вещества *м *жду
двумя растворителями. Однако применение этого учения к драк-
707
тически наиболее важному случаю — к очистке смазочных ма-
сел — ввиду крайней сложности состава, подлежащего очистке
продукта, приводит к слишком сложным уравнениям и диаграм-
мам [27, 46].
Гораздо проще поддается теоретической обработке данный во-
прос на основе аналогии, которую можно провести между основ-
ным процессом сольвентной очистки, т. е. извлечением или
экстракцией, и процессами перегонки и ректификации. Действи-
тельно, и тот и другой процессы но существу преследуют цель
одного и того же порядка: разделение составляющих нефть или
нефтепродукт компонентов. При перегонке и [юктификации кри-
терием для такого разделения служит температура кипения,
иначе говоря, молекулярный вес компонентов; при экстракции
же критерием является растворимость компонентов в том или
ином растворителе или, по существу дела, их химическая при-
рода. Аналогия между этими, на первый взгляд столь различ-
ными, процессами оказалась в высокой степени плодотворной
для теоретического освещения метода сольвентной очистки. От-
сылая читателя к оригинальным работам в этой важной области
очистки нефтепродуктов 27, 47 1, ограничимся ниже .лишь са-
мым общим освещением данного вопроса.
Однократное испарение и однократная экстракция. Как
было показано выше, простейшим случаем перегонки двух сме-
шивающихся жидкостей является однократное испарение (ч. II,
гл. II), когда переход данной системы из жидкого состояния ’
в парообразное с последующим разделением жидкой и паровой
Фиг. 123. Изотерма одно-
кратного иопарения.
Фиг. 124. Изотерма одно-
кратной экстракции.
708
фаз совершается однократно. Там же дана кривая «темпера-
тура-концентрация» (изобара) для однократного испарения
(фиг. 76); другая диаграмма для случая «давление — концентра-
ция» (изотерма) может быть построена аналогичным образом
(фиг. 123) с тою, однако, разницей, что верхняя часть кривой
(О АР) изображает здесь изменение упругости компонентов
в жидкой фазе, нижняя же часть кривой (ОВР) служит анало-
гичной характеристикой для паровой фазы. Для каждого давле-
ния состав жидкой и паровой фаз (ди и $2), на которые рше-
1 ' •
I > .
ляется данная’ смесь, определяется при однократном испаншии
однозначно либо но изобаре, либо по изотерме данного процесса.
„ 1усть теперь смесь двух взаимно растворимых жидкостей
оорабатывается селективным растворителем при некоторой по-
стоянной температуре, причем растворимость данных компонен-
тов в растворителе пусть будет различная. После взбалтывания
происходит разделение слоев, подобно тому как. прй однократном
испарении пар отделяется от жидкости; в данном случае экс-
тракт, очевидно, аналогичен паровой фазе, а рафинат (остаток) —
жидкой. Тате как, однако, экстракт и рафинат представляют со-
бою растьоры. то, очевидно, ту роль, которую, при однократном
испарении играет внешнее давление, в случае экстракции должно
играть осмотическое давление. Таким образом для случая одно-
кратной экстракции получаем кривую «осмотическое давление —
концентрация» (фиг. 124), вполне аналогичную кртгвой «давле-
ние— концентрация» при однократном испарении. Легко видеть
из этого построения. 1) что осмотическое давление двух находя-
щихся в равновесии фаз — одно и то же i(jti = j2) и 2) что те
простые отношения между массой образовавшихся паров и мас-
сой оставшейся жидкости, которые были введены для случая
однократного испарения, могут быть распространены также на
случай однократной экстракции для количественного соотноше-
ния между массой экстракта и массой рафината.
Многократное испарение и проточная экстракция; Много-
кратное испарение, как было показано выше (ч. II, гл. II), пред-
ставляет собою ряд последовательных процессов испарения сме-
шивающихся жидкостей с удалением из'системы: образующихся
каждый раз паров. Фракционпровка и ректификация являются
в практическом отношении наиболее важными видами многократ-
ного испарения; они обслуживаются специальной аппаратурой
с колоннами насадочного или тарелчатого (колпачкового) типа,
которые «могут обеспечить непрерывный ход процесса перегонки.
Там же приведены основания графического метода расчета важ-
нейших показателей данных процессов. Поэтому здесь можно
ограншшться лишь графическим изображением одного из этих
процессов, а именно процесса фракционировки, представляю-
щего, как видно из последующего, глубокую аналогию с проточ-
ной экстракцией.
Действительно, уже одно сопоставление диаграммы фрад що-
нкровки (фиг. 125) с диаграммой проточной экстракции
(фиг. 126) ясно обнаруживает эту аналогию. И в том, и в дру-
гом случаях ввод сырья производится в одном конце аппарата
(на диаграммах А и А'); здесь же происходит вывод одного из
конечных продуктов процесса, а именно нижнего продукта (жид-
кости) при фракционировке Б и экстракта при сольвентной
очистке Б'. На другом конце тех же диаграмм показан вывод
других продуктов: верхнего продукта (пара) в случае фракцио-
нировки Р и рафината при экстракции Р\ На этом же (верхнем)
конце диаграммы фракционировки показан ход рефлюкса >. роль
которого в процессе проточной экстракции Р часшчио вы пол-
тин
няет растворитель Л". Аналогия мо?кст быть продолжена здесь
и далее, в частности в отношении взаимодействия отдельных
компонентов данных систем, а именно: если соприкосновение
между экстрактом, растворителем и рафинатом происходит при
помощи последовательно соединенных смесителей (микстеров) и
отстойников (сепараторов), то получается точная копия тарел-
чатой (колпачковой) ректификационной колонны; если же со-
Фиг. 125. График много-
кратного испарения.
Фиг. 126. График проточ-
ной экстракции.
прикосновение компонентов происходит в колонне, работающей
непрерывно и нераздельно, то получаем копию насадочной ко-
лонны
Итак, пз сопоставления процессов фракционщювки и проточ-
ной экстракции можно видеть их полную аналогию. Теоретиче-
ская обработка обоих процессов может быть проведена поэтому
одним и тем же методом и, естественно, должна привести к одним
и тем же результатам.
СЕРНИСТЫЙ АНГИДРИД КАК СЕЛЕКТИВНЫЙ РАСТВОРИТЕЛЬ
Для иллюстрации теоретических и практических вопросов,
с которыми приходится встречаться при сольвентной очистке,
рассмотрим несколько ближе один из наиболее разработанных
селективных растворителей—жидкий сернистый ангидрид.
Жидкий сернистый ангидрид был применен впервые дл$
очистки некоторых румынских галицийских керосинов от из-
бытка содержащихся в них ароматических углеводородов
(ср. ч. Т, гл. IV). Значительно позднее этот растворитель стал
получать применение также для очистки смазочных мйсел. Теоре-
тические предпосылки этой методики в основном сводятся к сле-
дующему.
-Опыт показывает, что ароматические углеводороды смелей*
710
взаимной растворимости
ьаются с жидким сернистым ангидридом в практических усло-
виях температуры во всех отношениях. Столь же легко раство-
ряются в SO2 олефины [48]. Напротив, углеводороды парафино-
вого ряда образуют ; с сернистым ангидридом систему
С ограниченной растворимостью, зависящей от температуры.
Эта зависимость для каждого гомолога метана может быть вы-
ражена некоторой кривой, причем для углеводородов нормаль-
ного строения общий характер кривых, как видно из фиг. 127,
сохраняется примерно одинаковым [49].
Чтобы уяснить себе соотношения, наблюдаемые для систем
подобного рода, • проследим изменение
при разных температурах жидкого сер-
нистого ангидрида и хотя бы, напри-
мер, тетрадекана (СыНзо) (фиг. 1'27).
Левая половина соответствующей кри-
вой 6 может служить иллюстрацией
растворимости сернистого ангидрида в
углеводороде (содержание 80s в смеси
меньше 50%), правая половина, наобо-
рот, — растворимости углеводорода в
сернистом ангидриде (содержание
80г — больше 50%). Та же кривая
изображает ход температуры смешения,
т. е. исчезновения или появления мути
в растворе при различных количе-
ственных соотношениях между обоими
компонентами,, что соответствует со-
стоянию насыщения при данной тем-
пературе.
Как видно из левой половины кри-
вой, растворимость сернистого ангид-
рида, точнее — содержание SOs в насы-
щенном растворе его в тетрадекане,
при 0° сравнительно невелико (около
8%), но уже при 26° оно поднимается
до 19,3%. Иначе говоря, для смеси, со-
держащей 8% SO* и 92% СнНзо, тем-
пература смешения лежит около 0°,
для смеси же 19,3% SO2 4- 80,7»% СыНзо — при 26°. При даль-
нейшем повышении содержания SO* в смеси, температура смеше-
ния будет повышаться вплоть до 55.5°. что соответствует составу
смеси 66,1% SO2 и 33,9% СнНзо. т. е. уже раствору тетрадекана
в жидком’ сернистом ангидриде.
•Переходя к правой половине кривой 6, мы видим, что при
низких температурах содержание тетрадекана (СцНзе) в насы-
щенном растворе его в жидком сернистом ангидриде ничтожно.
Так, даже при 11,5° такой насыщенный раствор содержит всего
1,2% СнНзо и 98,8% SO2. С повышением температуры раствори-
мость углеводородов в сернистом ангидриде сначала медленно
возрастает. Так, при содержании 2,0%> СиНзо и 98,0% SOs рас-
711
Фиг. 127. Кривая взаимной
растворимости парафинов
и жидкого сернистого анги-
дрида:
1 — бутхи и SO,; 2~ гексан и S08;
з—октан и 308; 4 — декан и SO»;
л — додекан и SOs; в — тетраде-
кан и SO,; 7 —дот] иаконтан и SO,.
твор мутится при 21.0°, но уже при содержании 23,7% С14ТТ30
и 7б.з% S(-’>2 температура смешения поднимается до 53,1 °. На-
конец, как было только что указано, при составе смеси 33,9%
СиПзо и бб.1% SO* температура смешения достигает своей мак-
симальной величины, 55,5°.
Максимальная температура смешения, к которой можно по-
дойти с обеих сторон кривой, в данном случае растворяя сер-
нисты it ангидрид в углеводороде или углеводород в сернистом
ангидриде, называется критической температурой растворения.
При этоН температуре и выше ее данные жидкости смешиваются
во всех отношениях; иными словами, при этих температурах две
данные жидкости всегда образуют одну фазу. Напротив, при
более низких температурах те же жидкости вполне или частично
расслаиваются, т. е. образуют две фазы, и полное смешение воз-
можно здесь лишь при строго определенных для каждой данной
температуры количественных соотношениях между данными ком-
понентами смеси.
Фиг. 128. График критической тем пера-
туры растворения парафинов jt серни-
стом ангидриде.
Кривые фиг. 127 показы-
вают, что критическая тем-
пература растворения нор-
мальных парафинов в жид-
ком сернистом ангидриде за-
висит от их молекулярного
веса. Так, для бутана эта
температура лежит пример-
но при —4/, для гекёана—
при для декана —при
37.3 , для додекана — при
47.3м, для тетра декана—
при 55.5°. для дотриаконта-
на.— при 110°. Таким обра-
зом критическая те^ператур<1 растворения нормальных парафи-
нов в жидком сернистом* ангидриде постепенно повышается с уве-
личением молекулярного веса углеводородов; графически это
повышение можно выразить кривой, изображенной на фиг. 128.
Что касается, наконец, взаимной растворимости жидкого сер-
нистого ангидрида и нафтенов, то хотя этот вопрос изучен пока
недостаточно полно, тем не менее, есть основания полагать, что
критические температуры растворения для систем этого рода
лишь в малой степени отличаются от соответствующих темпе-
ратур для смесей сернистого ангидрида л парафинов с тем же
числом атомов углерода в частице. Непосредственные определе-
ния показали [50], например, что критическая температура рас-
творения для смеси циклогексана и сернистого ангидрида лежит
при 13,5°,*т. е. примерно всего лишь на 3° выше критической
температуры растворения для смеси нормального гексана и сер-
нистого ангидрида. Кроме того, обнаружено, что с повышением
молекулярного веса растворимость нафтенов в сернистом анги-
. дриде уменьшается, т. е. изменяется в том же направлении, как
для нормальных парафинов.
Па основе данных о взаимной растворимости углеводородов
различных рядов и о растворимости их в жидком сернистом ан-
гидриде м жно составить представление о тех отношениях, кото-
рые имеют место при обработке данным .растворителем нефтя-
ных дестиллатов. Дестиллаты прямой гонки, как известно, со-
держат парафины, нафтены, ароматику и в небольшом количе-
стве также олефины: между собою углеводороды этих рядов обла-
дают полной взаимной растворимостью. Напротив, в отношении
к жидкому сернистому ангидриду полной растворимостью, как
мы видели, обладаю! только ароматика и олефины, тогда как.
парафины и нафтены, особенно высшие, и при пониженной тем-
пературе обладают по отношению к этому растворителю лишь
ограниченной и небольшой растворимостью. Отсюда следует, что
при обработке жидким сернистым ангидридом ароматика и оле-
фины должны распределяться между двумя растворителями:
жидким сернистым ангидридом и не растворимой в последнем
парафиновой и нафтеновой частью дестиллата.
Прежде всего очевидно, что для извлечения ароматики из
масла необходимо, чтобы наступило расслаивание, т. е. чтобы
слой сернистого ангидрида отделился от углеводородного слоя.
Температура такого расслаивания зависит, естественно, от со-
держания ароматики: чем больше в масле ароматики, тем. ниже
температура, при которой в данном случае происходит расслаи-
вание. Так например, при обработке 66 процентами SO2 одного
мексиканского керосинового дестиллата с 17% ароматики рас-
слаивание наступило уже при 10°, тогда как керосиновый де-
стиллат из нефти с о. Борнео, содержавший 40% ароматики, при
обработке таким же количеством‘сернистого ангидрида показал
расслаивание только при— Ю°.
Другой важный вопрос заключается в полноте извлечения
ароматики из данного дестиллата. Так как коэфициент распреде-
ления не зависит от абсолютного количества растворенного ве-
щества (т. е. ароматики) и должен сохраняться постоянным при
повторных операциях извлечения, то, очевидно, полное извлече-
ние ароматики в данном случае не может быть достигнуто. По-
следнее при значительном содержании ароматики идет сначала
интенсивно, но постепенно ослабевает и, наконец, практически
вовсе останавливается. Тем не менее, нескольких обработок
жидким сернистым ангидридом вполне достаточно, чтобы иолу-*
чить вполне отчетливый эффект очистки, выражающийся, на-
пример, для масляных дестиллатов в снижении удельного веса
при некотором повышении температуры вспышки, снижении
коксообразования и, что особенно важно, в заметном улучшении
индекса вязкости масла. Для иллюстрации приводим сравни-
тельные результаты 41 очистки цилиндрового дестиллата из
бинагадинской нефти в одном случае серной кислотой, в ЛРУ-
ром — жидким SO2 и 5% активированной глины (асканита). ш-
менение глины диктуется в данном случае тем обстоятельством,
что некоторые виды смол (а также нафте новые кислоты) плохо
извлекаются жидким сернистым ангидридом и остите я в < и-
щенком no Эделеину масле, так что для их удаления приходится
прибегать к дополнительной очистке адсорбентом.
Таблица 150
Очистка бинагадинского цилиндрового дестиллата
(по Беликовскому и Позняк)
№ п/п I Наименование продукта Уд. вес Темп, вспышки, Вязкость по Энглеру Кйслот- ность В % SO3 । Кокс по Конрадсону
Е5о E1Q0 Ебо/Екю
1 Цилиндровый де- стиллат 0,951 230 26,47 2,63 10,0 0,059 0,40
2 То же, очищенный 6% серной кислоты 0,942 229 20,98 2,41 8,7 0,0 0,36
3 То же, очищенный SO2 и 5% активи- рованного асканита 0,924 233 16,71 2,33 7,1 0,017 0 21
Нельзя не отметить в заключение, что одновременно с удале-
нием .ароматики, очисткой но Эделеину достигается еще один
весьма важный результат, а именно — обессеривание/ Особенно
хорошо обессериваются этим способом керосиновые дестиллаты,
значительно хуже — масляные, очевидно вследствие снижения
растворимости сернистых соединений в жидком SO* с повыше-
нием их молекулярного веса. Так например, в одном керосино-
вом дестиллате из тексасской нефти (месторождение Винклер),
в котором анализ показал 0,45% S, содержание серы послэ
очистки по Эделеану упало до 0,07%; в другом (месторождение
Пекос)—с 0,025 до 0,11 %' S и т. д. [51].
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ СОЛЬВЕНТНОЙ ОЧИСТКИ
пак упомянуто выше, селективные растворители применяются
для. очистки керосинов и смазочных масел. Особенно важное
значение приобрела ©та методика в применении к смазочным
маслам, открывая здесь разйЬобразныё возможности: с одной
^тороны, применение селективных растворителей к очистке мае-’
ляных дестиллатов позволяет значительно расширить ресурсы
масляного сырья путем введения в переработку тяжелых смо-
листых нефтей, масляные дестиллаты которых лишь о трудом
поддаются обычной очистке серной кислотой, давая к тому'же
конечные продукты, не вполне удовлетворяющие современным
требованиям рышга; с другой стороны, масла, получаемые путем
сольвентной очистки, выделяются своими высокими качествами,
в частности—-имеют более пологую кривую вязкости, меньшую
коксуемость и т. п. Понятно поэтому, что очистка смазочных
масел при помощи селективных растворителей является методом,
который все шире и шире проникает в данную область нефтя-
714
Иого Дела. Жидкий сернистый ангидрид, нитробензол, хлорекс,
фено.I, фурфурол, пропан, а, также парные растворители
соперничают и конкурируют между собой, и следует лишь по-
желать скорейшего и возможно более широкого внедрения этой
уже зарекомендовавшей себя методики в советскую нефтяную
промышленность. Чтобы составить представление о значении
сольвентной очистки за границей, достаточно отметить, что
в 1937 г. свыше 90% масляной продукции США очищались пу-
тем обработки селективными растворителями.
ЛИТЕРАТУРА
Гурвич Л. Г., Научные основы переработки нефти, М.—Л. (1925).
Бе.м Г. С., Американские методы переработки нефти, изд. П. ч. П, ОНТИ.
М.-Л. (1933).
Добрянский А.-Ф., Курс технологии нефти, М.—Л. (1931).
Лострин К. В., Производство крэкинг-бензина, Баку—Москва, (1933).
Баррель Ж. А., Теория и практика переработки нефти, М.—Л. (1932).
Трети А.> Химия нефти п искусственного жидкого топлива, ОНТИ, М.—Л.
(1936).
Каличевский В. А. и Сттнер Б. А., Химическая очистка нефтепродуктов,
ОНТИ, М.-Л. (1936).
Броун А. С. и Сиверцев А. П., Химия сернистых соединений жидкого топ-
лива, ОНТИ, Л. (1937).
5< Залозецкий, „Chem. Ztg. ...
6. Riqfy, „J. Inst. Petr. Techn.* 15, 241 (1929).
7. Wood A. B., „Ind. Eng. Chem.“, 16, 1116 (1924).
S. Wood A. E, Sheely C., Trusty, „Ind. Eng. Chem/ 18, Д69 (1926).
9. Ott E. a. Reid E., „Ind. Eng. Chem." 22, 884 (1930).
10. Кострин К. В., Очистка гипоху ори том? Баку (1932).
11. Dunstan,
1. Лауш, Отбеливающие глины, М.—Л. (1929); февножукое Я. JST, Петти к
Хохрякова В. А., „Труды масляной комиссии ВСНХ СССР*, т. I, М.—Л.
(1930); Блаюдаров М., Отбеливающие земли, их свойства н приме-
нение для очистки нефтепродуктов, ОНТИ — Азнефтеиздат (1934).
2. Наумов В., Химия коллоидов, глава „Поверхностные явления и адсорб-
ция", составленная Ребиндером П. Л., Л. (1932).
3. Frash. „Ind- Eng. Chem." 4, 136 (1912).
4. Постовский и Плюснин, „Ж. хим. прод.“ в, № 11, 778 (1929); „Нефт. хоз.
11, № 11, 561 (1930).
19, 78 (1895)- Robinson, Амер, патент 910584.
Wood А. В., „Ind. Eng. Chem.", 16, 1116 (1924). ...__
Ott E. a. Reid K, „Ind. Eng." Chem." 227,884 (1930).
~ 'w., ,J. Inst Petr. Techn.", стр.Т112 '(1922) и стр. 201 (1924):
Ayers G. W., „Oil a. Gas J.", стр. 201 (3/X 1929); Блаюдаров M. Л.,
„Ла. нефт. хоз." 9, № 2 (1929).
12. Birch 3. F. a. Norris W. 3., „Refiner", 90 (июль 1928).
13. Виробьян P. А., „Нефт. хоз." 11, № 2, 198 (1930).
14. Наметкин С. С. и Соснина А. С., „Ж. прикл. химии’, 7, № 1—2,123 (1934).
15. Ма песку М., „Braunkohlen Arch.* 40, .53 (1930).
16. Кирсанов А. и Суслина В., „Ж. прикл. химии 8, 2 <7 (193э).
17. Наметкин С. С, Санин П. И., Маковер С. В. и Цыба А. Н., „Ж. прикл.
ХИМИИ* в, 494(1933). HQQnV nv
18. Лавровский К., Арбузов, Пучков, „Нефт. хоз. 16, № 8, 69> (193d), ом.
также Пучков П., „Химия твердого топлива.9, 852 (193г).
19. Молдавский Б. и Пекорский Е., -Нефт. хоз. 17 (1УЗЬ). н
20. Тиц И. Н., „Ученые записки МГУ" 3 ,165 £934); Тиц И., Шуикин В.,
Епифанский П., „Нефт. хоз.* 16, № Й^Зэ). Е
21. Мусатов К. А., „Нефт. хоз." 15, №11» (1934), р. „ д д'
• „Нефт. хоз." 15, № 11, 37 (1984); Задолин С. А. и Бутков В. а.,
22. Rachman А^, ^Nat/petn News (ноябрь 1931); рус. пер. см. -Нефт. хоз.*
18, 55 (1932).
715
23. Le Hoy, Sion/ G., Provine В. TF., Bennet H. T., „Ind. En^ Chem • 01
1079 (1929).
24. Martiles a. Jfos*-, „J. Inst. Tetr. Techn.' 15, 657 (1929).
25. Молдавский, Вокорский, Андреевский, „Нефт. хоз.* 16, 52 (1935).
26. Бутков В. А., Введение в технологию пиролиза, ОНТИ (1937).*
27. Литературу и многие детали вопроса см. сборники: «Избирательные
растворители в нефтепереработке' и * Селективные растворите in"
Аонефтеиздат (1934 и ]936); см. также Auld, ,,J. Inst- Petr ТнЛт »’
22, 57(1935). an‘
29. Aisinman, „Ding. Politech- J.“ 297, 14.
29. Харичков E. В., Холодная фракционировка нефти (1903)
<Ю. Edekanu; „Petroleum* 9, 862 (1913—1914).
31. Valenta, „Chem. Ztg.“ 30, 266 (1906); Oriife, „Chem. Umscbau' 14 11t>
’ (1907). ’ "
32. Rahlke W. H., Giles R, JET., Adams С . E., .Refiner' 12, № 6, 229 (1933У
Брей, Свифт, Kapp, „Oil a. Gas J.“ 82, XI (1933). ’ h
33. Balrike IF. H., Brown A- B.,D<woky F. F.. .Refiner' 12, N. 11, 445 (1933)-
Page J. M., Buehler С. C., Biggs S. H., .Oil a. Gis J.“ 81, N. 4S u
(1933). ’
34. Ferris S. TF. a. Houghton W. F., .Reimer' 11, N. 11, 560 (1932); 12, X. p
328 (1933); Ferris S. IF., Meyers TIC A. a. Pcterkin Я. G„ Ibid 12
X 11, 435 (1933). ’ ’
35. Stratford R. K., Pokorny 0. S., Raggett J. L., .Refiner' 12, X. Ц 45$
(1933); Stratford R. K., Moora, Pokorny, .Oil a. Gas J.“ 31, N.’45 8
(1933).
36. Manley R. E.,Mc Carty В. У., Gross H. R,t .Refiner' 12, N. 11, 420 (1933),
37. Holde, „Petroleum" 18, 853 (1922); „Brennstoff chemie 4, 177 (1923).
38. Go-lire 0., „Petroleum" 23, 73 (1927); Ferris S. IT.. Birkhimer JS. R„ Ren-
derson L. M., „Ind. Eng. Chem.' 23, 753 (1931).
89. Cottrell О. P., „Refiner' 12, *N. 11, 432 (1933); Cain IF., Ibid. 11 N 11
553 (1932). ’ ' ’
49. Tuttle M. a. Max. B. Miller, „Refiner" 12, N. 11, 453 (1933).
41. Беликовский А. С. и Возняк И. В., „Нефт. хоз.' 20, № 6, 475 (1931).
42. Церножуков И. И. и Крейн С. Э., Окисляемость минеральных масел,
ОНТИ (1936).
43. Сп. Mikeska, „Ind. Eng. Chem." 28, N. 8 (1938).
44. Vlugter, Waterman, van Westen, „J. Inst. Petr. Techn. , 18, N. 107, 735
(1932); 21; N. 141, 666 (1935).
45. Pook J. W! a. Wadsworth 1. M., „Refiner' 12. N- 11, 412 (1933).
46. См; доклад Гунтер T. и Нвш А. на Всемирном нефтяном конгрессе
в Лондоне 1933 г., т. II,. стр. 340.
47. См доклад Саалъ Р. и Ван-Дик, Ibid. (46); т И, стр, 352.
48. Moore, Morrell, Egloff G., „Met. a. Chem. Eng.“ 18; N. 8, 396 (1918).
49. Seyer W. F. a. Todd. E., „Ind- Eng. Chem.' 23, N. 3, 325 (1931).
50. Seyer IF. F. a. Dunbar, „Trans. Roy. Soc. Can." 16, 307 (1922).
51. BifiiuH, „Ing. End. Chem.' 22, N. 6 (1930).
ГЛАВА Ш
ПОВЫШЕНИЕ КАЧЕСТВА НЕФТЕПРОДУКТОВ
МЕТОДОМ ПРИСАДОК
Нередко ни общие, пи специальные методы очистки не могут
обеспечить надлежащего [качества таких нефтепродуктов, как
моторное топливо или смазочные Масла. В связи с этим боль-
шое; все растущее значение получают методы повышения каче-
ства нефтепродуктов путем добавления к ним разного рода спе-
циальных веществ, присадок, наличие которых в нефтепродукте
сообщает ему то или иное недостающее качество. Обзору важ-
нейших из этих методов посвящена настоящая глава, распадаю-
щаяся соответствен1но целевому назначению различных присадок
па следующие разделы:
А. Присадки, снижающие склонность нефтепродуктов к само-
окислению и смолообразованию т. е. ингибиторы.
Б. Присадки, повышающие антидетонационные качества мо-
торного топлива, т. е. антидетонаторы.
В. Присадки, понижающие температуру застывания смазоч-
ных масел, короче говоря, депарафинизаторы.
Г. Присадки, улучшающие смазывающие качества масел;
разнообразные присадки этого рода могут быть объединены
в одну группу иод общим наименованием мобриканторы Г
«
А. Ингибиторы
Как было показано в гл. II (см. Гидроочистка), самые разно-
образные нефтепродукты нередко содержат особого рода смоли-
стые вещества, короче — смолы, в большей или меньшей степени
снижающие качества этих продуктов. Свежеперетнанные нефте-
продукты обыкновенно содержат немного смолистых веществ;
однако при продолжительном стоянии содержание смол в нефте-
продукте, как общее правило, возрастает. Таким образом разли-
чают истинные смолы, действительно содержащиеся в нефтепро-
дукте и потенциальные смолы, образующиеся в нем за счет
реакций аутоксидации и конденсации малоустойчивых, прейду- ,
щественно высоконепределъных компонентов нефтепродукта. Одни
1 Не смешивать с люб/шкаторсмш— йехапичеекими приспоеоблкниями
разного рода для подачи смязктг к трущимся поверхностям.
717
йз способов борьбы с такого рола смолообразованием заключаются
в коренном изменении состава нефтепродукта либо путем уда-
лснпя наиболее неустойчивых смолообразователсй (очистка сер-
ной кислотой и т. п.), либо путем иного изменения их хими-
ческой природы (гидрирование в умеренных условиях и т. и.).
Во многих случаях, однако, та же цель может быть достигнута,
гораздо проще, а именно добавкой небольшого количества (иногда,
о,1—0,2% и меньше) некоторого вещества, ингибитора или
антиокислителя, одно присутствие которого в нефтепродукте де-
лает его стабильным в течение того или иного времени, вполне
достаточного для практических целей. ] [игибиторы могут приме-
няться для стабилизации не только таких легких нефтепродук-
тов. как моторное топливо, но и для смазочных масел.
I
ИНГИБИТОРЫ МОТОРНОГО ТОПЛИВА
Смолообразование, как было показано выше, вызывает ослож-
нения как при хранении моторного топлива, так и при его при-
менении в двигателе внутреннего сгорания и заметно снижает
аитидетонациопные качества бензина. Прибавка ингибитора
практически ликвидирует эти нежелательные явления и пред-
л -w «-г А Л л - * <• п п
Фиг. 129. Смолообразование в моторном
бензоле:
I—моторный бензол. 2—то же с присадкой 0,005° 0
пм>акр*зола; з— то ясе с присадкой 0,01°/о гтара-
крёзола; 4—-то же с присадкой 0,ОБо,0 паракре-
аола.
етавляет сооою простейшим
способ стабилизации мотор-
ного топлива. Фиг. 129 дает
примеры смолообразования
в моторном бензоле в при-
сутствии и отсутствии инги-
битора (паракрезола).
Число веществ, которые
были исследованы в каче-
стве ингибиторов для мотор-
ного топлива, чрезвычайно
велико. Испытание их актив-
ности можно производить
путем сопоставления перио-
дов индукции одного и того
же бензина при его нагре-
вании в металлической бом-
бе в атмосфере кислорода
при стандартных условиях в присутствии или отсутствии тех
или иных ингибиторов. .Наблюдающееся при этом падение давле-
ния в бомбе наносится’ на график; за меру активности ингиби-
тора принимают удлинение периода индукции бензина под влия-
нием данного ингибитора.
Сравнительные исследования подобного рода показали, что
простейшие соединения жирного ряда не обладают свойствами
ингибиторов для крэкинг-бензина. Те же отрицательные каче-
ства показало и большинство классов соединений ароматиче-
ского ряда, как-то: углеводороды, спирты, галоидопроизводные,
эфиры, кетоны, кислоты, нитросоединения и т. д. Напротив, мно-
гис ароматические ямины и фенолы. особенно же яминофенолы,
показали себя сильными ингибиторами. В табл. 151 приведен
ряд бопее или менее активных ингибиторов при концентрации
0,01% с соответствующими им периодами индукции для пен-
сильванского крэкинг-бензина согласно приведенным выше ука-
заниям. В последнем столбце этой таблицы обозначены раство-
рители, в которых производилась добавка ингибиторов, трудно-
растворимых в крэкинг-'бензпне [1].
Таблица / 5/
Ингибиторы моторного топлива
Наименование ингибитора Период индукции в мин. - Раствори- тель
Анилин 45
Орто-толуидин 60 Ацетон
Нафтиламин-а 105 —
Нафтиламин-р Дифениламин 75 Ацетон
105 *—
Фенил-нафтиламив-а 720 Метанот
Орто-фенилендиамин 450 Ацетон
Мета-фенилендиамин 165 Метанол
Нара-феви лен диамин 960 Этанол
Диамино-дифениламин-/;, р' ... . 495 Ацетон
Фенол, мета-крезол 75 — —•
Орто-крезол • . 135 ♦ —
Пара-крезол 210 —
Нафтол-а- ...... 2250
Нафтол-? 330
Пирокатехин 2 400 Бензол
Резорцин 150 Ацетон
Гидрохинон 85 Ацетон
Пирогаллол • . . . . . 2'185 Этанол
Орто-аминов енол .... 1320 Ацетон
Мета-аминофенол 330 Метанол
Пара-аминофенол 2840 Метанол
Амино-гпдрокситолуол-2,5 .... 1785
Как, видно из данных табл. 151, простейшие первичные амины
ароматического ряда (анилин, толуидины) мало эффективны
как ингибиторы. Несколько более эффективны нафтиламины, при-
чем а-снафгиламин действует заметно сильнее, чем ^-нафтиламин.
Значительно возрастает эффективность действия ароматш еских
аминов при переходе от первичных аминов ко вторичным (дифе-
ниламин, особенно же фенил-а-нафгиламин), а также от вв ц-
иия второй аминогруппы (фенилендиамины). I последнем слу-
чае особенно заслуживает внимания зависимость эффективности
изомерных ингибиторов от их -строения: из трех феиилендиами-
нов наиболее сильным ингибитором оказался нара-фениленди-
амин и наиболее слабым — мета-фенилендкамин; ортб-соедииение
заняло в этом отношении промежуточное место.
718
Простейший фенол (карболовая кислота) является слабым
ингибитором. Несколько сильнее его оказались изомерные кре-
золы, причем и здесь на первом месте должен быть поставлен
пара-изомер, а на последнем — мета-соединение. Значительно
более сильными ингибиторами оказались фенолы ряда нафта-
лина, т. е. нафтолы; из них особенно должен быть отмечен
а-нафтол, являющийся одним из наиболее сильных ингибиторов.
Вступление второй гидроксильной группы в ароматическое ядро
сильно повышало эффективность ингибитора лишь в том случае,
если оба гидроксила оказывались в орто-положении (пирокате-
хин) ; два других изомерных двухатомных фенола, т. е. резорцин
и гидрохинон, показали слабую активность. Наконец, трехатом-
ный фенол с рядовым положением гидроксилов, т. е. 1,2,3-три-
оксибензол (пирогаллол), должен быть отнесен к числу наиболее
мощных ингибиторов.
Наиболее сильными ингибиторами являются, невидимому,
аминофенолы. Интересно, что в трех изомерных простейпшх
аминофенолах порядок изменения эффективности оказался тот
же,’что и в фенилендиаминах: наиболее сильное действие про-
являл цара-аминофенол и наименее сильное — мета-аминофецол.
К этому же ряду соединений относится один из сильнейших при-
меняемых в настоящее время ингибиторов — бензил-аминофенол
(«Баф»)..
Испытания ингибиторов только что рассмотренным методом,
когда определяется изменение периода индукции при окислении
крэкинг-бензина в бомбе, -дают лишь первые ориентировочные
данные для суждения об эффективности ингибитора. Чтобы
составить представление о пригодности данного ингибитора для
практических целей, необходимо провести его испытание в усло-
виях продолжительного хранения бензина при контакте с воз-
духом. Несколько примеров подобного рода приведено в табл. 152
для характеристики стабилизации неочищенного крэкинг-беп-
зина при его хранении в присутствии ингибитора в стеклянной
посуде из бурого стекла в продолжение б месяцев.
Данные табл. 152 показывают, что ингибиторы чрезвычайно
задерживают смолообразование, так что во многих случаях коли-
чество смол, образовавшееся в бензине при хранении его в тече-
ние 6 месяцев в присутствии ингибитора, лишь в малой степени
отличается от начального содержания смол в том же бензине.
.Интересно отметить также случай глубокого несоответствия в ха-
рактеристике ингибитора, получаемой при его испытании в бомбе
и методом хранения: гидрохинон, показавший сравнительно ко-
роткий период индукции (табл. 151), оказался прекрасным инги-
битором в условиях хранения бензина при обыкновенной темпе-
ратуре. Возможно, что в условиях испытания в бомбе, т. е. при
100° и под давлением кислорода, сам гидрохинон претерпевает
глубокое изменение, чем и объясняется отмеченное несоответ-
ствие. ’ ’ d
Ингибиторы находят применение для стабилизации как очи-
щенных, так и неочищенных крэкицг-бензинов. Предваритель-
720
Таблица 152
Испытание бензина с ингибитором
Цвет Смолы В Ж1 R ЮГ) C..V f Октано-
Наименование продукта по Сей- вое
болту медн. чашка етекл. чашка число
Неочищенный бензин:
начальные качества 39 85 4 77
после 6 мес 11 8+2 »38 65
Бензин с ингибитором (0,01%)
Пирокатехин:
начальные качества 20 14 о U 77
после 6 мес. 22 7 12 75
Гидрохинон:
начальные качества 21 2 О 77
после 6 мес . 21 5 6 75
Нафтол: •
начальные качества 19 14 6 77
после 6 мес 16 12 2 74
Пара-аминофенол:
начальные качества 19 6 0 77
после 6 мес ' 7 109 6 75
пая очистка т|>ебуется. например, в таких случаях, когда не-
обходимо снизить содержание серы в бензине или достичь пол-
ного обесцвечивания нефтепродукта. Последующая добавка инги-
битора часто позволяет значительно сократить расход реагентов
при очистке и снизить потери, так как в этом случае в задачу
очистки не входит стабилизация бензина; эту задачу успешно
выполнит ингибитор. Но еще более важным является примене-
ние ингибиторов к неочищенным крэкинг-бензинам, когда для
получения товарного продукта и его стабилизации оказывается
возможным полностью исключить очистку бензина. Экономиче-
ское значение такого упрощения процесса получения моторного
топлива само собою понятно.
Ингибиторы, применяемые для стабилизации товарных бензинов,
должны удовлетворять некоторых! специальным требованиям. Они должны
быстро и хорошо растворяться в бензине и но должны ухудшать его цвет.
В воде они не должны растворяться, так как в противном случае, прн
соприкосновении бензина с водой, например в специальных условиях хра-
нения и т. п., ингибитор может быть полностью извлечен водою и оставит
бензин незащищенным. Наконец, ингибитор должен быть достаточно ста-
бильным и по возможности жидким и летучим, так как нелетучие кристал-
лические вещества, отлагаясь в виде осадка, могут вызывать осложнения
з работе мотора. Сами собою разумеется, что удовлетворение всем перечне
л 721
Зак. П»22. Химця нефти.
лепным условиям — задача, практически весьма трудно разрешимая,’и ра-
бота в области изыскания новых ингибиторов является одной из наиболее
интересных и актуальных задач химии нефти. Иностранная патентная ли-
тература последних лет содержит по этому вопросу обширный материал.
ИНГИБИТОРЫ ДЛЯ ИЗОЛЯЦИОННЫХ И СМАЗОЧНЫХ МАСЕЛ
При соприкосновении с воздухом минеральные масла, недо-
статочно очищенные, часто выделяют смолистые осадки В про-
цессе работы масла, особенно при повышенной температуре и со-
прикосновении с воздухом, при каталитическом воздействии*
металлических поверхностей, образование подобного рода смоли-
стых осадков значительно ускоряется; как говорят, -масло ста-
реет и требует регенерации, иначе говоря, повой, дополнительной
очистки.
Состав смолистого осадка (шлама, sludge), образующегося
в отработанном масле, весьма сложен. В него входят:
1) кислые смолы, образовавшиеся в результате окислишя и
конденсации наиболее неустойчивых углеводородов масла;
2) тяжелые смолы и даже асфальтены, образовавпшеся в ре-
зультате дальнейших превращений смолистых веществ, которые
находились в исходном масле;
3) медные и железные соли высокомолекулярных кислот,
образовавшихся при окислении тех или иных компонентов масла
и прореагировавших затем с металлической поверхностью аппа-
рата или мотора;
4) наконец, разного рода, механические примеси, как-то: пыль,
песок, частички металла и т. и.
Таким образом, оставляя в стороне составные части шлама,
имеющие либо случайный, либо вторичный характер, т. е. меха-
нические примеси и соли, необходимо притти к выводу, что
в основном шлам представ, яет собою смесь продуктов реакций
аутоксидации и уплотнения углеводородов смазочного масла.
Большая или меньшая окисляемость минерального масла,
т. е. склонность образующих его углеводородов к реакциям ауто-
ксидации и уплотнения, зависит от целого ряда факторов; среди
них на первом месте следует поставить, конечно, химическую
природу углеводородов данного масла.
Как известно, наиболее легко подвергаются реакциям аутокси-
дации и уплотнения углеводороды непредельного характера.
Присутствие непредельных углеводородов в минеральных маслах’
прямой гонки не подлежит сомнению; они появляются здесь
главным образом в результате частичного крэкинга исходного
сырья в процессе его перегонки. Что касается очищенного масла,
то, поскольку цри очистке непредельные соединения удаляются
в первую очередь, присутствие в хороню очищенном масле не-
предельных углеводородов в* сколько-нибудь значительном коли-
честве 1гредставляется маловероятным. Правда, в литературе
имеются указания, что, например, минеральные масла, подобно
терпенам озонируют воздух; или что при окислении трансфор-
маторного масла получаются смоляные кислоты, близкие по со-
7#
сШу К смоляным кислотам канифоли; однако подобного рОДА
указания еще недостаточны для утверждения, что в хороню
очищенном масле содержатся подобные терпенам непредельные
углеводороды. Во всяком случае следует считать установлен-
ным, что удаление непредельных углеводородов достаточно
плодотворно влияет на стабильность масла.
Парафиновые углеводороды несравнимо более устойчивы, чем
непредельные углеводороды. Как было показано в главе об
окислительном крэкинге (я. И, гл. IV), требуется длительное
взаимодействие при повышенной температуре, чтобы ввести в ре-
акцию с кислородом высокомолекулярные углеводород ряда
метана. Продуктами этой реакция являются в конечном итоге
спирты, альдегиды, кислоты и оксикислоты жирного ряда; все
они хорошо растворимы в исходном углеводороде, так что при
аутоксидации парафиновых углеводородов, как общее правило,
не наблюдается выделения каких-либо осадков.
Большой интерес представляет аутоксидации ароматических
углеводородов и нафтенов. Систематические исследования этого
вопроса [2 привели к следующим результатам.
Ароматические углеводороды без боковых цепей, особенно
простейшие (бензол, нафталин), чрезвычайно устойчивы к кисло-
роду даже при повышенных температуре и давлении (150',
15 ат). Менее стойки в аналогичных условиях более сложные
конденсированные системы антрацена и фенантрена, особенно
при более высокой температуре (250°): получается сложная
смесь продуктов аутоксидации, среди которых можно обнаружить
вещества фенольного и кислотного характера^ а также смолы
и, невидимому,' асфальтены.
Наличие боковых цепей в ароматическом углеводороде сильно
увеличивает склонность его к аутоксидации. Большое влияние
на результат реакции оказывают число боковых цепей, их взаим-
ное расположение и длина. Так, пз трех изомерных ксилолов
наиболее легко поддается аутоксидации пара-ксилол; более
устойчивым по отношению к кислороду является орто-ксилол,
наиболее же стабилен мета-ксилол. С увеличением длины боко-
вой цепи в ароматическом углеводороде стабильность его посте-
пенно снижается: чем длиннее -боковая цепь в ароматическом
углеводороде, тем легче он поддается аутоксидации. Продуктами
этих превращений в основном являются фонолы, кислоты (частью
летучие) и смолы; кроме того, в отдельных случаях здесь наблю-
дается образование соответствующих спиртов, альдегидов, кето-
нов и оксикислот.
Ароматические системы, содержащие в своем составе поли-
метиленовые циклы, иначе говоря, не полностью гидрированная
ароматика, например тетралин и т. п. системы, характеризуются
еще большей склонностью к реакциям аутоксидации и уплотне-
ния под влиянием кислорода. Основными продуктами этих пре-
вращений и здесь являются кислоты и оксикислоты, фенолы и
смолы; характерно также образование нередко обильного тем-
ного осадка (шлама), не растворимого в петролейном эфире и
46* 733
лишь частично растворимого в спирте, простом эфире и бен-
золе.
Шестичленные нафтены различных типов поддаются действию
кислорода значительно легче ароматики, причем более сложные
системы и здесь менее стабильны. Аналогично также влияние
боковых цепей: чем больше в нафтене боковых цепей (алкилов)
и чем они длиннее, тем легче поддается он воздействию кисло-
рода. Основными продуктами аутоксидации нафтенов являются
кислоты и оксикислоты, образование которых обыкновенно со-
провождается разрывом полиметиленового кольца по месту при-
соединения к нему боковой цепи. Дальнейшими продуктами
аутоксидации являются и в данном случае смолы и в незначи-
тельном количестве асфальтены. Количество этих вторичных
продуктов аутоксидации нафтенов по сравнению с кислой частью
невелико.
Совершенно особый интерес представляет аутоксидация сме-
сей нафтенов с ароматикой, т. е. углеводородных смесей,- по
своему составу в большей или меныпей степени приближаю-
щихся к естественным нефтяным маслам [2]. Специальными
опытами установлено, что присутствие в углеводородной смеси
1—10% либо ароматики без боковых групп, либо не полностью
гидрированной ароматики, либо, наконец, углеводородов типа
дифенил- и трифенилметана задерживает аутокспдацию нафте-
нов. Ароматические углеводороды с боковыми цепями действуют
на окисление нафтенов кислородом воздуха аналогичным обра-
зом; при этом, однако, тщчбуется значительно более высокая кон-
центрация ароматики в данной смеси, а именно не менее
20—30%. Характерно, что обильное выделение шлама, столь
обычное при аутоксидации не полностью гидрированной арома-
тики, наблюдается также при окислении углеводородных смесей,
содержащих такую ароматику.
В свете только что рассмотренных фактов становятся прежде
всего понятными старые наблюдения различных авторов, что
высокоочищенные трансформаторные масла обладают большей
склонностью к аутоксидации и выделению шлама в процессе их
работы, чем аналогичные масла меньшей степени очистки [3].
Подобного рода перечищенные масла легко получаются, напри-
мер, при обработке масляного дестиллата дымящей серной кис-
лотой. С другой стороны, показано, что добавка к перечищен-
ному маслу неочищенного или слабо очищенного масла повы-
шает стабильность перечищенного масла [4]. Очевидно, что
недоочшценное масло содержит какие-то вещества— ингибиторы,
которые, будучи способны стабилизировать малоустойчивые
компоненты масла, удаляются при достаточно глубокой ег<
очистке; такими ингибиторами могут быть согласно вышеизло-
женному ароматические углеводороды недоочищенного масла,
точнее — продукты окисления этой ароматики, например фенолц
смолы ароматического происхождения и т. п. Действительно,
добавлением к хорошо очищенному («перечищенному») вазели-
новому маслу 3—ю% смол из масляного дестиллата, богатого
724
тяжелом ароматикой (например, из тяжелой балаханской йефти),
удавалось значительно повысить стабильность вазелинового
масла в отношении его к. окисляющему действию кислорода
Еще более наглядную картину стабилизации масел дает
добавка к ним некоторых специальных ингибиторов. В этом на-
правлении в настоящее время исследовано весьма большое ко-
личество веществ; некоторые пз них проявляют громадную
эффективность и находят применение в качестве ингибиторов
для защиты как изоляционных, так и смазочных масел.
Выдающимися качествами ингибитора для изоляционных ма-
сел обладает элементарная сера: в присутствии 0,01% этого ве-
щества образец трансформаторного масла нагревался при 120°
под давлением кислорода в продолжение 100 час. и не показал
никакого окисления, тогда как контрольный образец масла
в аналогичных условиях оказался'сильно окисленным. Чрезвы-
чайно сильными антиокислительными свойствами обладает также
лгзоамитсульфид, (СяНн^З [5].
Из кислородсодержащих органических веществ наиболее
активными ингибиторами окисления являются фенолы. В отно-
шении аутокепдании вазелинового масла наибольшими анти-
окислительными свойствами обладают, невидимому, ^-нафтол и
пирогаллол, а также гидрохинон и пирокатехин. Менее активны
]юзорцин и флороглюцин. Простейший фенол (карболовая кис-
лота) вовсе не активен. Действие фенолов в первую очередь
сказывается на снижении образования кислых продуктов и
(в несколько меньшей степени) продуктов уплотнения. Характер
последних резко изменяется. Вместо смолистых веществ, раство-
римых в петролейном эфире, в присутствии фенолов образуются
темные осадки, по внешнему виду и нерастворимости в летро-
лейном эфире напоминающие асфальтены [2].
•Из других кислородных органических соединений свойствами
ингибитора окисления обладают хиноны. Напротив, альдегиды,
кетоны, кислоты, оксикпслоты, фенолокислоты и в меньшей „сте-
пени спирты понижают стабильность минеральных масел [2].
Своеобразное влияние оказывают соли нафтеновых кислот; следы
последних могут находиться в минеральном масле: нафтеновые
соли одних металлов (Na, Fe, Мп, Znf Pb, Си, Ag) снижают ста-
бильность масла, тогда как. аналогичные соли других металлов
(Са. Ba, Hg, особенно Sn), наоборот, повышают ее [6].
Среди азотсодержащих органических соединении многие обла-
дают ясно выраженными свойствами ингибиторов окисления.
Таковы, например, анилин и особенно ₽-нафтиламин. Следует,
впрочем, отметить, что в присутствии этих ингибиторов, как и
в случае применения фенолов, существенно изменяется состав
продуктов уплотнения; среди последних появляются асфальтены,
которые вовсе не образуются дри окислении кислородом чистого
вазелинового масла. Табл. 153 может служить для иллюстра-
ции действия некоторых ингибиторов на ау гоксидацию вазели-
нового масла в течение 3 час. при 150 и 1о ат давления ки .го-
рода [2].
72q
Таблица 1Г)3
ОкиёлЯеыость ваВелйновогй шасла
(по Черно Жукову и KpeicH)
f »> * % инги- битора Кислот- ность в ж КОН °/о про- дуктов уплотне- ния % всех продук- тов окис- ления
Вазелиновое масло 0 39,5 20,5 45,0
То же 4-пирокатехин .’ 0,1 Следы 1,6 1,8
я И 1,0 1.5 1,6 1.6
Э» я » 3,0 1.4 2,1 2,4
То же + р-нафтол 0,1 0,5 1.5 1.8
Я 99 • 1.0 0,9 1,4 2.7
1» 3* 33 3,0 1,9 3,2 5,6
То же 4- р-наф тилам пн . , 0,1 8,3 10,6 19,0
99 » » 1,0 0,6 3,7 6,2
п " и ....... 3,0 Следы 1,6 3.2
Кроме приведенных ингибиторов, в патентной литературе
можно встретить немало иных органических веществ, предло-.
женных в качестве антиокислителей для минеральных масел.'
Среди них следует отметить: гексаметилен-тетрамин, мочевину,
дифенилгуанидин, диэтиланилин, аминофенол, фенилендиамин,
4-оксибифепил и многие другие. Как видно, некоторые из них
рекомендованы также как стабилизаторы для крэкинг-бензина.
Кроме того, имеется ряд ингибиторов неизвестного состава и
строения, распространяемых под тем или иным запатентованным
наименованием.
ХИМИЗМ ДЕЙСТВИЯ ИНГИБИТОРОВ
Замедляющее (тормозящее) действие ингибиторов на процессы
окисления .и уплотнения легких и тяжелых минеральных масел
под влиянием кислорода представляет собою частный • случай
обширной группы явлений, известных под общим наименованием
явлений отрицательного катализа. Отдельные случаи подобного
рода известны давно. Таковы, например, стабилизация раствора
сернистокислого натрия под влиянием маннита, бензилового
спирта и бензальдегида; торможение аутоксидации бензальдегида
под влиянием добавки гидрохинона, иодистоводородното метил-
амина и других антиокислителей; стабилизация под влиянием тех
же ингибиторов крайне неустойчивого акролеина; действие раз-
личных ингибиторов на процесс высыхания непредельных расти-
тельных жиров, например льняного масла, и т. д. Знаменательно,
что действие ингибиторов зачастую проявляется уже при крайне
незначительных концентрациях. Так например, тормозящее
действие «гидрохинона на процесс аутоксидации бензальдегида
вполне отчетливо наблюдается. уже при концентрации ингиби-
тора 1 : 1 000 000; при концентрации же 1 :1000 гидрохинон
726
практически полностью приостанавливает аутокспдацию бенз-
альдегида.
Чтобы уяснить механизм тормозящею действия ингибиторов,
необходимо прежде всею обратить внимание на то обстоятель-
ство, что все ингибиторы окисления более или менее легко окис-
ляются сами в тех условиях, в которых они проявляют свое
тормозящее действие в отношении реакций окисления других
веществ. С этой точки зрения понятно, ггто ингибитор, приме-
няемый для стабилизации крэкинг-бензина или масла, не
остается неприкосновенным; он постепенно расходуется, так что
при хранении бензина в присутствии ингибитора наступает мо-
мент, когда торможение основной реакции прекращается и на-
ступает усиленный процесс окисления крэкинг-бензина или
масла, протекающий с большой энергией и скоростью.
Во время хранения крэкинг-бензина с ингибитором количество смол
возрастает сначала очень медленно. Через некоторое время образование
смол1 ’ в фарфоровой чашке начинает быстро ускоряться. Обыкновенно
считается, что содержание ю мг омол на юо г бензина при его испарении
в воздушной струе указывает па окончание периода индукции во время
хранения; содержание же 15 мг смол служит признаком начала быстрой
аутоксидации бензина. Повышение периода индукции бензина путем до-
бавки ингибитора является весьма надежным признаком стабильности бен-
зина при хранении. Так, увеличение периода индукции на 150—200 мин.
может служить довольно надежным признаком стабильности бензина на
протяжении 10—12 месяцев, что практически вполне достаточно.
Большое значение для эффективности действия ингибитора
имеет содержание в крэкинг-бензине перекисей. Ингибитор про-
являет максимальную эффективность лишь в тех случаях, когда
он добавляется к свежеперегнанному бензину. В тех же слу-
чаях, когда крэкинг-бензин стоял некоторое время на воздухе
и успел накопить некоторое количество перекисей, добавка инги-
битора не дает полного эффекта, и период индукции бензина со-
кращается. Разрушая в бензине пероксиды, можно добиться по-
вышения эффективности ингибитора.
Механизм действия ингибиторов окисления пока не может
считаться окончательно установленным. Этот сложный вопрос
должен быть увязал прежде всего с другим основным вопросом
данной области — с вопросом о механизме самопроизвольного
окисления органических и иных соединений под 'влиянием кис-
лорода. воздуха (аугоксидация). Как было показано в ч. II, гл. IV
(«Химизм окислительного крэкинга»), начальная стадия этого
широко распространенного процесса, согласно воззрений, разви-
тых Бахом и Энглером свыше 40 лет тому назад, представляет
собою присоединение к окисляемому веществу молекулы кисло-
рода с образованием особого малоустойчивого соединения пере-
кисного типа, так называемой «молокиси».
Воззрения Баха —Энглера на механизм процесса аутоксидации
являются далеко не общепринятыми. 1 настоящее время наряду с этими
воззрениями, дающими наиболее простую и наглядную картин ки ления
кислородом, разрабатываются ц дискутируются две основных рочкц зрения.
727
По Виланду [7] процессы окисления приставляют обою частный слу-
чай реакции дегидрирования, в основе которого лежит предварительная
активизация водорода окисляющегося вещества. Так, при окислении r при-
сутствии металлического катализатора, например палладия (Pd), металл
образует с окисляемым телом МН) промежуточный комплекс (JPdH), рас-
па дающийся затем на окисленное тело (Л) и водородистый палладий (PdH).
Роль кислорода при таком процессе сводится к отнятию водорода от водо-
родистого металла, т. е. j; рспнерацни катализатора:
Л1 + Pd —> -IPdH —> Л + PdH.
Очевидно, в этой интерпретации окисление представляет собою процесс
обратный реакции восстановления:
Л |-Р<1Н —> ДР<Ш —> JL11-4- Pd‘.
В тех случаях, когда действие кислорода протекает в отсутствие ме-
таллических катализаторов, реакция, по Вилано, должна быть" тесно свя-
зана о непредельным характером кислорода, а именно: непредельные сое-
динения присоединяются к молекуле кисло]юла непосредственно, предель-
ные ate — после предварительной активации водородного атома окисляю-
щейся частицы вещества.
Согласно другой концепции ' (Боне) в основу аутоксикации должен
быть положен процесс образования гидроксильных групп (ОН), короче го-
воря, процесс гидроксилирования окисляющегося соединения [8]. С этой
точки зрения начальным продуктом окисления в атмосфере кислорода, оче-
видно, должна быть не перекись (как у Баха — Энглера), а. спирт. В на-
стоящее время нахождение спиртов среди продуктов окисления углеводо-
]>одов предельного характера, как было отмочено выше (ч. II, гл. IV), до.
казано с полной несомненностью. Повидимому, однако, они являются лишь
вторичными продуктами данной реакции. Одним пз наиболее слабых мест
гидроксильной теории явтяется также требование предварительной дис-
социации молекулы кислорода, что явно необходимо для образования
гидроксила, но маловероятно с энергетической точки зрения.
На базе воззрений Баха— Энглера построена получившая
• широкое признание теория действия ингибиторов, разработанная
французскими химиками Мурё и Дюфресс [9]. Согласно этой
теории окистяющееся вещество (Aj и прибавленный к нему
ингибитор (В) при взаимодействии с кислородом образуют неста-
бильные перекиси, Л (О*) и 7? (Оз), которые при взаимодействии
друг с другом (разлагаются с обратным выделением молекуляр-
ного кислорода и неактивных частиц А и В (схема I); или, если
ингибитор реагирует не со свободным кислородом, а с перекисью
4(Ог), то в качестве промеягуточных продуктов можно пред-
ставить себе образование также малоустойчивых промежуточных
окислов 4(0) и В(0). которые и здесь при взаимодействии друг
’с другом приводят к начальным частицам, 4L и В, и молекуляр-
ному кислороду (схема II):
Схема Т. Л 02 = А (02);
В -И 02 = В (02);
А (0.) + В (02) = Л + В + 20,.
Схема II. Л4-0о=А(02);
J (Оа) + Б = Л(0) + Ь‘(0);
А (О) -f- В (0) = А + В + Ou.
1 Ср. ч. И, гл. IV «Теории гидрирования».
738
При всей простоте приведенных схем они удачно разъясняют
целый ряд харл мерных особен постой действия ингибиторов,
1. Все ингибиторы, как было указано выше, более или менее
легко окисляются и в то же время легко регенерируются в про-
цессе своей работ, благодаря чему небольшие количества инги-
битора (В) способны вызвать стабилизацию значительного коли-
чества вещества. (А).
2. Ингибиторы постепенно расходуются в процессе своей ра-
боты. Для объяснения этого факта следует лишь допустить, что
малоустойчивые продукты окисления ингибитора, например В (fl),
не регенерируют* я, а превращаются в продукты дальнейшего
окисления, которые выводятся из реакции по схеме:
В (0) -> ВО и т. д.
3. Одно и то же вещество в одних случаях может1 замедлять
реакцию, т. е. действовать как ингибитор, в других случаях —
ускорять процесс как типичный катализатор. Так например, иод
является ингибитором при аутоксидации бензальдегида и поло-
жительным катализатором при окислении стирола и т. д. Со-
гласно представлений Муре подобные случаи можно объяснить
следующим образом: представим себе что в последней фазе
схемы II малоустойчивый окисел В (fl) реагирует нс с -4(0),
а с молекулой Я по схеме:
Л+Б(0) = + В-
«
в таком случае ясно, что вещество В является уже не ингиби-
тором, а передатчиком кислорода, т. е. положительным катали-
затором реакции окисления вещества А.
Одно из главных возражений против схем Мурё и Дюфресса
заключается в том, что вопреки требованию теории перекиси,
образующиеся в результате аутоксидации, отнюдь не склонны
к выделению молекулярного кислорода при взаимодействии
С другими веществами; скорее, они проявляют склонность
к образованию более устойчивых продуктов окисления исходив
вещества., причем нередко наблюдается выделение атомарного
кислорода. Отсюда понятны попытки найти иные истолкования
механизма действия ингибиторов, которые не находились бы
в столь явном противоречии с фактами; весьма важным шагом
в этом направлении явился опыт подведения под теорию Баха —
Энглера кинетических основ на базе теории цепных реакций [ 10].
Сущность цепного механизма химической реакции в общем
виде заключается в следующем. Под. влиянием термического
движения или поглощения света отдельные молекулы веществ»
активируются, становясь активными центрами и, быть может,
иескольтсо изменяясь в своем строении (1); три этом они при-
обретают способность соединяться с молекулой кислорода (-)•
Образующаяся таким образом перекись реагирует со второй
молекулой вещества (3); в результате образуется более устойчи-
вый продукт окисления и второй активный центр цепи. 1 го-
729
апеДйий в свою очередь соодййяеш с Кислородом, образуя
новую молекулу перекиси и т. д. Таким образом получается цепь,
которая, теоретически, может продолжаться без конца. Так на-
пример, для аутоксидации ацетилена была предложена следую-
щая цепная схема:
НС = СН-> НС = СН; m
НС = СН.+ О2-> НС = СН;
НС = СН НС —СН НС = СН. (з)
I | +нс^сн-> II II + I I
0 — 0 0 0
И т. д.
Может, однако, случиться, что на определенном звене прои-
зойдет разрыв цепи, так как реакция уклоняется в сторону от
нормального своего течения. Для случая аутоксидации ацети-
лена можно представить себе, например, следующие побочные
течения реакции (4) и (5):
p[£j: * Qрг
I I + 02 НСООН + С0о; (4)
0 — 0
НС = СП
| I ->Н0СО + СО. (5)
0 — 0
Экспериментально установлено [11], что при аутоксидации
ацетилена действительно получаются: глиоксаль (3), углекислота
и муравьиная кислота (4), формальдегид и окись углерода (5).
Аналогичные цепные схемы можно представить себе для ауто-
ксидации этиленовых углеводородов, альдегидов и других соеди-
нений.
Цепные реакции, как учит новейшая химическая кинетика^
характеризуются некоторыми особенностями, важнейшими из
которых являются: высокий квантовый выход в соединении
с сильно экзотермическим характером реакции и весьма силь-
ное влияние на ход процесса ничтожных следов сторонних при-
месей, Прекрасным объектом для изучения цепных реакций
являются фотохимические процессы, в частности фотохимиче-
ское окисление некоторых альдегидов, например бензальдегида
и энантола.
Второй закон фотохимии (Эйнштейн) гласит, что фотохими-
ческие процессы являются квантовыми реакциями: их цервой
стадией является поглощение реагирующей молекулой одного
кванта энергии, hv, где v — частота колебаний, характерная для
поглощающей молекулы. Между тем, установлено [12], что при
аутоксидации бензальдегида на квант световой энергии, погло-
щенной альдегидом, окисляется до 10 000 его молекул. Столь
ненормально высокий квантовый выход при реакциях подобного
730
рода объясняется с точки зрения теории псиных реакции тем об-
стоятельством, что'энергия, развивающаяся при цепной реакции,
не рассеивается, а- передается соседним молекулам вещества,
активирует их и вводит в цепь соответствующих превращений.
С этой точки зрения фотохимическую аутокспдацию альдегидов,
а также непредельных углеводородов следует рассматривать как
типичную цепную реакцию.
Другое характерное свойство цепных реакций—их чувстви-
тельность к сторонним примесям — объясняется тем обстоятель-
ством, что стороннее вещество, ингибитор, может либо принять
энергию активации на себя, либо способствовать ее рассеянию.
И в том. и в другом случаях происходит обрыв данной цепи про-
цесса. А так как в цепных реакциях цепь может состоять из
нескольких тысяч звеньев, то понятно, что обрыв цепи па каком-
либо звене имеет своим следствием дезактивацию громадного
количества молекул, которые могли бы возглавить последующие
звенья цепи, если бы последняя не была оборвана. Тем самым
становится попятным тот громадный тормозящий эффект. кото-
рый может оказать небольшое количество подходящего ингиби-
тора на процесс аутоксидации того или иного вещества, легко
окисляющегося под влиянием кислорода. Опытное исследование
этого процесса было проведено па примере фотохимической
аутоксидации бензальдегида и энангола; оно показало, что тормо-
зящее действие различных ингибиторов на эти реакции проте-
кает почти тождественно как на свету, так и в темноте; это
обстоятельство в свою очередь позволило сделать вывод, что
аутоксидация бензальдегида и энантола протекает по цепной
схеме не только на свепу, но и в темноте и что схему действия
ингибиторов в обоих этих случаях необходимо строить на базе
механизма цепных реакции [1'2]. Все основные характерные
особенности действия ингибиторов с этой точки зрения действи-
тельно получают весьма простое объяснение.
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ ИНГИБИТОРОВ В НЕФТЯНОМ ДЕЛЕ
Несмотря на новизну этого вопроса, метод повышения каче-
ства нефтепродуктов с помощью ингибиторов успел получить
в нефтяной промышленности широкое применение,
в производстве моторного топлива. Так, по сведениям из США
здесь в 1937 г. свыше 40% крэкинг-бензина стабилизировались
различными ингибиторами и выпускались на рынок без серно-
кислотной очистки. Применение ингибиторов к стабилизации
минеральных масел, особенно трансформаторных и турбинных,
также приобретает все большее признание, таким образом не
подлежит никакому, сомнению, что ингибиторам принадлежит
в нефтяном деле самое широкое будущее.
Б. Антидетонаторы
В ч. I при-рассмотрении основных компонентов нефти, т. е.
ароматических, парафиновых и нафтеновых углеводородов, уже
было отмечено, что в известных случаях спокойная работа дви-
гателя внутреннего сгорания нередко нарушается детонацией,
проявляющейся особыми стуками в моторе («мотор стучит»)’
Там же было указано, что склонность данного топлива к дето-
нации растет со степенью сжатия горючей смеси в цилиндре
двигателя и, кроме того, в высокой -тепепи зависит от химиче-
ского состава топлива. Эта последняя зависимость связана
с глубоким различием углеводородов различного строения в их
склонности к детонации при прочих равных условиях; в основ-
ном это различие может быть выражено следующим образом:
1. Наименьшей склонностью к детонации характеризуются
ароматические углеводороды; отдельные представители этого
класса различаются между собой в отношении детонационных
свойств сравнительно немного.
2. Наиболее склонны к детонации парафины нормального
строения, причем склонность эта у них повышается с увеличе-
нием молекулярного веса. Изопарафины, напротив, проявляют
значительно меньшую склонность к. детонации, к тому же сни-
жающейся по мере разветвления цепи углеродных атомов, иначе
говоря, но мере увеличения в частице углеводорода числа ме-
тильных групп (ср. ч. 1, гл. V. 1 Б).
3. Гомологи этилена менее склонны к детонаций, чем пара-
фины, и на этом их свойстве основано антидетонациоиное прей-,
мущество крэкинг-бензина по сравнению с бензином прямой
гонки. Большое влияние на детонационные свойства отдельных
углеводородов ряда этилена оказывает их строение.
4. Нафтены по своим антидетонациопным свойствам стоят
тотчас вслед за ароматикой, нередко превосходя в этом отно-
шении даже непредельные углеводороды.
Приготовление’ высококачественного (не детонирующего) то-
плива, которого т]юбуют современные скоростные моторы, пред-
ставляет собою сложную задачу, к разрешению которой можно
подойти различными путями. В одних случаях задача успешно
разрешается смешением надлежащим образом подобранных ком-
понентов. В других — для снижения детонационных свойств то-
плива прибегают к помощи специальных присадок, антидетона-
торов. Поскольку иотучение ие детонирующего моторного то-
плива является в настоящее время одной из наиболее важных
задач нефтяной промышленности, в настоящем разделе должны
быть освещены с химической точки зрения все основные, отно-
сящиеся к этой проблеме вопросы, как-то: природа детонации
топлива и ее характеристика, состав современного не детонирую-
щего моторного топлива. антидетонаторы моторного топлива и
механизм их действия [13, 14].
НОРМАЛЬНОЕ ГОРЕНИЕ И ДЕТОНАЦИЯ
Во многих случаях горение газовой смеси распространяется
от горящего слоя к соседнему благодаря подогреву этого .слоя
путем теплопроводности. Такой тип горения обыкновенно назы-
вают нормал ъным.
732
Нормальное горение может проявляться в различных фор-
мах (видах). Простейшим из них является равномерное распро-
странение^ пламени с постоянной небольшой скоростью- его
можно наб подать, например, при зажигании газовой смеси у от-
крытого конца горизонтальной трубы, противоположный конец
которой закрыт. Другой аналогичный случай —это сгорание
у выхода из трубки, через которую подается горючая смесь под
атмосферным давлением: если скорость распространения пла-
мени окажется больше скорости истечения смеси, пламя .переме-
стится внутрь трубки: наоборот, при скорости распространения
пламени, меньшей скорости истечения горючей смеси, пламя ото-
двинется от отверстия трубки и потухнет. Эти явления удобно
наблюдаются, например, с газовой горелкой Бунзена. При непо-
движном фронте пламени, определяемом грающей его внутрен-
него конуса, нормальные к поверхности горения слагающие ско-
рости газового потока и распространения пламени, очевидно,
равны, откуда получается основное равенство:
Q = rS,
где Q— расход газа в секунду, v— линейная скорость распро-
странения пламени и 6' — площадь поверхности горения, послед-
няя может быть измерена путем фотографирования стационар-
ного пламени. Зная же Q и S, определяют линейную скорость
распространения пламени, и. Опытные исследования показали,
что скорость эта при нормальном горении быстро возрастает при
повышении начальной температуры газовой смеси.
Более сложный случай нормального горения представляет
собою горение с отбросом пламени. Такого рода отбросы наблю-
даются, если находящаяся в трубе горючая смесь воспламеняется
от искры. В этом случае в месте воспламенения вследствие рас-
ширения газа вознш ают элементарные волны сжатия, которые
направляются к обоим концам трубы впереди фронта пламени,
образующегося при воспламенении последующих слоев газовой
смеси. Каждая следующая волна сжатия, распространяясь в бо-
лее -нагретой среде’, догоняет предыдущую волну и налагается на
нее; в ‘результате образуется крутой перепад давления, харак-
терный для ударной волны, которая, отразившись от конца трубы,
отбрасывает фронт пламени в направлении, обратном его перво-
начальному распространению.
В некоторых случаях нормальное сгорание горючей смеси
в закрытой трубе завершается чрезвычайно быстрым продвиже-
нием фронта пламени, резким скачком давления и сильным уда-
ром (стуком), происходящим вследствие тотчка во. ны сжатия
о заглушку трубы. Явление это получило название детонацион-
ного сгорания или детонации. Опыт показывает, что возникно-
вение детонации зависит от целого ряда причин, среди которых,
как было указано выше, весьма важное значение имеет состав
горючей смеси. При прочих равных условиях наступлению дето-
национного сгорания способствуют повышенная начальная тем-
пература горючей смеси п повышенное давление, а также уве-
738
личение длины трубы. Так напри.ш р, горение эквимолекулярной
смеси метана и кислорода (СГЦ 4- 0-2) происходит с детонацией,
если его производить в трубе длиною в 1.3 л при дав, ениях
больше 500 мм; в той же трубе, но при более низких давлениях
или при тех же давлениях, но в более короткой трубе горение
эквимолекулярной смеси метана и кислорода протекает нор-
мально (с отбросами). Эти примеры показывают, что между нор-
мальным горением и . етонапией нет резкой границы.
Переход от нормального горения к детонации можно пред-
ставить себе следующим образом [13]. Сначала горение тазовой
смеси раенро траняется обычным путем, т. е. на основе подо-
грева последовательных слоев смеси путем теплопроводности.
При этом, однако, воспламенение каждого последующего слоя
постепенно облегчается вследствие непрерывного подогрева све-
жей смеси волнами сжатия. Одновременно волны сжатия, нс
принимая непосредственного участия в передаче горения, обусло-
вливают вследствие прогрессирующего подъема температуры не-
прерывный саморазгон скорости реакции и соответственно по-
следней саморазгон в распространении пламени. В результате
при наличии подходящих условий (состав смеси, длина тр\бы,
начальное давление смеси)'горение переходит в детонацию, при-
чем фронт пламени (волна горения) совпадает с ударной вол-
ной, образуя взрывную или детонационную волну.
Экспериментальное изучение процессов горения и взрывов произво-
дится фотографическим методом: распространяющееся по горизонтальной
трубе пламя фиксируется па фотографической пленке, навернутой па вра-
щающийся барабан. Получается наклонный след пламени, дающий воз-
можность измерить скорость для любого1 момента времени и для любого
положения фронта пламени в трубе. Современные высокоскоростные фо-
тографические камеры позволяют закреплять па пленке движение пла-
мени далее для таких громадных скоростей, какими характеризуется рас-
пространение детонационной волны.
ОСНОВНЫЕ СВОЙСТВА ДЕТОНАЦИОННОЙ ВОЛНЫ
Опыт и применение фотографической съемки позволяют уста-
новить ряд характерных особенностей детонационного сгорания,
отличающих его от других видов распространения пламени.
Важнейшие из этих особенностей следующие:
1. Возникновение детонации сопровождается образованием де-
тонационной волны, распространяющейся в среде догорающего
газа с весьма большой скоростью.
2. Скорость детонационной. волны представляет собою кон-
станту, которая характеризует тазовую смесь и почти не зависит
от физических условий распространения волны.
3. При образовании детонационной волны происходит само-
воспламенение газовой смеси впереди распространяющегося
фронта юрепия, например при нормальном давлении и обыкно-
венной температуре — на расстоянии примерно 10 см впереди
фронта.
4 Весьма характерной для фотоснимков йолиь! детонаций
является своеобразная долосатпсть. наблюдаемая для самых
разнообразных горючих смесей. Эта полосатость является след-
ствием того, что поступательное распространение волны дето-
нации происходит не по прямой, а ио спирали. Спиральный след
детонационной волны можно воспроизвести, если стеклянную
трубку покрыть изнутри порошком мела, который хорошо при-
стает к стеклу; если затем произвести в этой трубке воспламе-
нение теоретической смеси окиси углерода и кислорода с не-
большой примесью водорода (0,7%), то после прохождения волны
детонации на стенках трубки остается отчетливый спиральный
след (фиг. 130).
Фиг. 130. Спиральный след волны детонации (по Бону и Фрезер),
5. Применение теории распространения ударных волн в упру-
гих жидкостях к яв гениям детонации позволило построить
гидродинамическую теорию распространения детонацпопных*волн
и вычислить для различных частных случаев величины основ-
ных параметров детонационной волны, ее давление и скорость
распространения. Сравнение вычисленных таким образом пара-
метров с найденными экспериментально показало хорошее совпа-
дение теории с опытом.
6. Опытное определение давлений, развиваемых детонацион-
ной волной, производилось неоднократно. Новейшие измере-
ния [15] .были произведены в стеклянной трубе с медными диа-
фрагмами различной толщины, прокалибрированньгми на разрыв
по давлению сжатого воздуха. Точность опыта была такой, что
ври уменьшении толщины диафрагмы на 0,02 :мл она разрыва-
лась при взрыве данной горючей смеси. Одновр< менно произво-
дилось фотографирование пламени, что позволяло измерить ско-
рость детонационной волны. В табл. 154 приводятся опытные и
вычисленные давления, развиваемые детонационной волной при
взрывах некоторых горючих смесей
7. В табл. 155 приведены вычисленные и опытные скорости
волны при взрывах смеси водорода с кислородом, в большинстве
случаев в присутствии инертного газа (азот, гелий). Как видно
из"данных этой таблицы, наблюдается хорошее приближение рас-
чета к опыту, если принять во внимание степень диссоциации
конечного продукта процесса (воды), которая при температурах
детонации должна быть весьма значительной [16].
8. Наиболее трудной задачей для экспериментальной про-
верки является определение температурных изменений на фронте
пламени, происходящих здесь в связи с быстро следующими
735
Таблица 154
Давления детонационных воЛй
* Состав смеси Давление в ат
вычислено В найдено
Водород: 2Но 4-0-2 Метан: ОЩ-р-О’ " , СЩ4-203 , СЩ + 802 Зтилен: С*ТЦ 4- 2СЬ „• ед+зо. Ацетилен: С?>Н<> 4- О-> „ c.;hs 4- ioo2 17,5 36,3 29,2 25,9 21.2 41,1 31,6 22,0 20,4 34,0 31,0 26,0 22,4 41,5 34,0 41,5 22,0
Таблица 155
Скорость детонационной волны для смесей водорода и кислорода
Состав смеси Вычислено Найдено и'м/сек
без учета диссоциации с учетом диссоциа- ции
2И24-О2 2907 2806 2819
2Н2 4“ Оо 4~ N2 2712 2378 2407
zh24-O24-8N, 2194 2033 2055
2H24~024-5No 1927 1850 1832
2Вз 4“ О2 4~ 1,5 Н е 3722 3200 ЗОЮ
2Н34- Оо4-ЗПе 3990 3432 3130
2Н24-О24-г>Не 4С83 3613 3160
друг за другом взрывными процессами. Невидимому, эти затруд-
нения мотут разрешиться лишь при переходе от одиночных дето-
наций к взрывам, которые периодически возобновляются, как
это имеет место в двигателе внутреннего сгорания.
л;- ^.’Око-рость волны детонации в трубе не зависит от материала
трзМй, ее диаметра, если он не слишком мал, начальной темпе-
ратуры изначального давления смеси (в пределах, например,
1—3 ат и 10—100°), характера зажигания и места воспламене-
ния (у открытого' или закрытого конца трубы и т. и.).
10. Наиболее активными смесями являются смеси с недостат-
ком кислорода (ср. табл. 154). Примесь инертных газов оказы-
вает различное влияние в зависимости ’от характера изменения
плотности газовой смеси от прибавления инертного газа
(ср. табл. 155): если плотность горючей смеси от прибавления
736
инертного газа (например, азота) увеличивается, скорость дето-
национной волны падает; наоборот, если плотность смеси от
прибавления инертного газа (например, гелия) снижается, ско-
рость волны может существенно возрасти. Очевидно, что ско-
рость детонационной волны зависит не только от химического
состава горючей смеси, но также от среднего молекулярного
веса или ее плотности.
ДЕТОНАЦИЯ В ДВИГАТЕЛЕ ВНУТРЕННЕГО СГОРАНИЯ
В предыдущем изложении приведены основные свойства де-
тонационной волны для случая ее возникновения в длинной, го-
ризонтально расположенной трубе. Уменьшая длину трубы и не-
сколько увеличивая ее диаметр, можно постепенно цритти
к цилиндрической бомбе, т. е. к аппарату, который по своим раз-
мерам и форме является подобием цилиндра двигателя внутрен-
него сгорания, а вводя новые параметры — периодическую подачу
газовой смеси и постоянное изменение ее плотности в связи
с движением поршня в цилиндре, — можно подойти, наконец,
к явлениям воспламенения горючей смеси и детонации в дви-
гателе внутреннего сгорания.
Опыт и фотоснимки фронта пламени свидетельствуют, что его
распространение при воспламенении неподвижной горючей смеси
в бомбе имеет сначала сферический характер; лишь после со-
прикосновения со стенкой бомбы это первоначальное направле-
ние изменяется, а именно: под влиянием охлаждающего действия
стенки скорость перемещения пламени уменьшается, и сфери-
ческая форма его поверхности нарушается, так как в местах со-
прикосновения фронт пламени начинает перемещаться вдоль
стенки бомбы. Такой же характер распространения пламени, как
показывает опыт, сохраняется и в цилиндрах работающего дви-
гателя в широких пределах от 600 до 1200 об/мин; при этом,
поскольку сохраняется нормальный тип сгорания, повышение
степени сжатия, изменение химического состава горючей смеси,
а также изменение местоположения свечи, от которой происхо-
дит зажигание смеси в цилиндре, заметно не отражаются на
форме распространяющегося фронта пламени.
Нормальная работа двигателя тесно связана со своевремен-
ным, нормальным ходом процесса сгорания горючей смеси,
а именно — сгорание должно заканчиваться, когда поршень ци-
линдра находится вблизи верхнего, мертвого положения. В неко-
торых случаях, однако, сгорание в двигателе может перейти
в иную, более бурную форму — в детонацию, которая характери-
зуется следующими основными признаками [13]:
1. Появление детонации в двигателе сопровождается образова-
нием детонационной волны, распространяющейся в цилиндре
двигателя с весьма большой скоростью, подобно тому как это
наблюдается для случая детонаций в трубе. Основными факто-
рами образования детонационной волны и здесь являются доста-
точно высокая скорость сгорания топлива в цилиндре двигателя
787
47 Зак. 1622. Хммиа нефтж.
и соответственно этому быстрое и резкое поднятие температуры
горючей смеси.
2. Образовавшаяся детонационная волна подобно молоту уда-
ряет о стенки цилиндра и вызывает характерный металлический
стук («knock»). Ближайшая связь между явлениями стука в мо-
торе и образованием детонационной волны подтверждается пол-
ным совпадением физических условий, определяющих возникно-
вение обоих ©тих явлений.
3. Появление детонации сопровождается резким падением
мощности и экономичности двигателя. Отсюда — известное, вы-
зываемое детонацией ограничение в отношении допустимой сте-
Фиг. 131. Изменение
мощности двигателя при
разных степенях сжа-
тия:
— без детпжации; Ъ — при
детонации.
пени сжатия в моторе. В самых общих
чертах ограничение это сводится к сле:
дующему. Без детонации повышение сте-
пени сжатия вызывает увеличение сред-
него эффективного давления, а следова-
тельно, и мощности двигателя, одновре-
менно понижается удельный расход то-
плива и следовательно, повышается ко-
эфициент полезного действия мотора
(фиг. 131, кривая а). С появлением дето-
нации при определенной, часто неболь-
шой степени сжатия наступает резкое па-
дение величины среднего эффективного
давления, а следовательно и мощности
мотора (фиг. 131, кривая Ь).
4. Вследствие полного расстройства
работы мотора при детонации на выхлопе
временами появляется черный дым (не-
сгоревшие частицы углерода), соответ-
ственно чему температура выхлопных газов заметно снижается.
5. При детонации увеличивается теплоотдача тазов стенкам
цилиндров, следствием чего могут явиться механические повре-
ждения отдельных частей двигателя, например прогар поршней
л т. п.
Следует иметь в виду, что появление стуков в моторе, правда, не-
сколько иного характера, может наблюдаться и без детонации, по другой
причине, а именно — вследствие самовоспламенения горючей смеси в ци-
линдре. Такое самовоспламенение может появиться до детонации либо как
следствие относительно высокой степени сжатия и низкой температуры
самовоспламенения топлива, либо как результат длительной детонации
вследствие сильного разогревания стенок камеры сгорания. Самовоспламене-
ние происходит в цилиндре до появления искры от свечи и характеризуется
преждевременным сильным повышением давления еще в течение хода
сжатия. И здесь одновременно наблюдаются глухой стук или шум в мо-
торе, неравномерность его работы, потеря мощности и ухудшение экономич-
ности работы мотора. Самовоспламенение и детонация отнюдь не исклю-
чают друг друга, а наоборот, нередко наблюдаются совместно.
МЕТОДЫ ОЦЕНКИ ДЕТОНАЦИОННЫХ СВОЙСТВ ТОПЛИВА
Хотя появление детонации при работе мотора обнаруживается
сравнительно легко (наслух), тем не менее количественная
738
оценка этого явления представляет значительные трудности и до
сих пор является условной. Основная задача, с которой прихо-
дится иметь здесь дело экспериментатору-практику, заключается
в сравнительной характеристике детонационных свойств различ-
ных топлив для двигателей внутреннего сгорания.
Приборы, применяемые для характеристики детонации, весьма
разнообразны. Их главная часть представляет собою двигатель
внутреннего сгорания, на котором и производится оценка дето-
национных свойств топлива. Для этого камера сгорания двига-
теля снабжена в верхней своей части отверстием, в которое ввин-
чивается специальная наружная втулка. В этой втулке свободно
перемещается тонкий стержень (игла), нижний конец которого
опирается на чувствительную диафрагму; через верхний конец
стержня при появлении детонации вследствие резких скачков да-
вления, передаваемого через диафрагму, и энергичного подпры-
гивания стержня происходит периодическое замыкание особого
электрического тока. В цепь этого тока включается специальный
электролизёр; по объему гремучего газа, выделяющегося при ра-
боте двигателя в единицу времени, можно судить о силе детона-
ции, которая имеет место в камере сгорания двигателя в данных
условиях. На изложенных основаниях построены получившие
широкое признание различные приборы для определения дето-
нации (Midgley, Ethyl Gasoline Corporation и т. д.); ® других
аппаратах вместо измерения гремучего газа замеряют силу тока
в цепи в миллиамперах или другие показатели, характеризую-
щие работу двигателя.
Выше (ч. I, гл. IV) уже был приведен один из способов коли-
чественного выражения относительной детонационной способно-
сти топлива, а именно — по толуольному, или бензольному числу
(эквиваленту). Числа эти представляют собою проценты толуола
или бензола, которые необходимо добавить к исследуемому, пред-
варительно дезароматизированному бензину, чтобы получить то-
пливо, по своим детонационным свойствам равноценное исход-
ному бензину. Основной недостаток этого метода заключается
в крайне сложной зависимости между количественным содержа-
нием ароматики в бензине и величиной основных показателей,
характеризующих детонацию бензина, что в высокой степени
осложняет применение данного метода для 'Практических целей.
Таким же недостатком обладают варианты этого метода, осно-
ванные на добавке к дезароматизированному бензину некоторых
иных веществ, понижающих детонационные свойства бензина,
например анилина и тетраэтилсвинца. Поэтому все подобные вы-
ражения детонационных свойств топлива ныне оставлены, усту-
пив место так называемой октановой (точнее — изооктановой)
характеристике бензина.
Октановая характеристика получается в результате сопоста-
вления детонационных свойств бензина со свойствами искус-
ственного топлива, составленного из смеси сильно детонирующего
нормального гептана (I) и не детонирующего изооктана (2, 2, 4-
триметил-пентана, II).
’ 739
I. CH8 (CH.2)0 • сн8
темп. кип. 98°,3
II. (СН8)8С . СН2. СЩСН^
темп. кип. 106 —107°
Детонационные свойства смеси норм, гептана и изооктана
изменяются -в зависимости от процентного содержания компонен-
тов по прямой, так что, изменяя состав такой смеси, можно по-
лучить с точки зрения детонационных свойств почти любой бен-
зин. Октановое число бензина определяется процентным содер-
жанием изооктана в такой смеси его с норм, гептаном, которая
по своим детонационным свойствам равноценна испытуемому
бензину. Так например, 100-октановый бензин имеет те же дето-
национные свойства, как чистый 100%-ный изооктан и т. д. Со-
поставление детонационных свойств исследуемого бензина и ис-
кусственной смеси производится на специальных приборах с дви-
гателем, кратко отмеченных выше.
Получение нормального гептана и изооктана для оценки дето-
национных свойств бензинов является важной задачей обслужи-
вания нефтяной промышленности и потребителей легкого мотор-
ного топлива.
Нормальный гептан в ОША выделяют из эфирного масла ка-
лифорнийской сосны (Pinus Jeffreyi Murray), в котором этот угле-
водород содержится в количестве до 96%. В других странах,
в частности в ССОР, где нет источника готового гептана, для по-
лучения этого углеводорода может служить гептиловый альдегид
(энантол), легко получаемый пиролизом эфиров рицинолевой
кислоты (касторовое масло). Переход от энантола к гептану осу-
ществляется через стадию гаптилового спирта по следующей
схеме [17]:
С6Н13 • СНО С6Н18 • СН20Н -> С7Н,6.
Исходным продуктом для получения изооктана является бу-
тиленовая фракция газов крэкинга. Обработкой этой фракции
разбавленной серной кислотой можно связать лишь один из ком-
понентов этой смеси, изобутилен (ср. ч. II, гл. III), который че-
рез изобутилсерную кислоту может быть превращен в третичный
изобутиловый спирт и далее в чистый изобутилен; последний же
путем полимеризации в присутствии подходящего катализатора
(серная кислота, фосфорная кислота и т. п.) перерабатывается,
наконец, в диизобутилен. В подходящих условиях температуры
и крепости серной кислоты все эти отдельные операции могут быть
соединены вместе: образовавшаяся изобутилсерная кислота (I)
тотчас же реагирует со второй частицей изобутилена с образова-
нием диизобутилена. (II), который выделяется из продуктов реак-
ции (высшие полимеры) фракционировкой. а затем путем отдель-
ного гидрирования превращается в изооктап (III) по следующим
схемам:
СН8Х
I. >С : СН2 + H.,S04 = (СН3)8С • 0S( J’;
СП,/
<
740
СН8К
II. (CH^C • 0S08H + )C : CH. =
СН/
(CHS)8 • C • CH :
III. (CHS)8C • CH :
+ H2S04;
+Щ=
,CH3
о
'Ч’н/
= (CH3)8C -CH2-CH
Получение изооктана представляет собою в настоящее время
важную побочную отрасль нефтяной промышленности, так как
этот углеводород, как будет показано ниже, является одним из
компонентов наиболее высокосортных видов моторного топлива
[17, 18].
АНТИДЕТОНАЦИОННЫЕ ДОБАВКИ И АНТИДЕТОНАТОРЫ
С полным основанием можно утверждать, что склонность
к детонации является важнейшим признаком, определяющим ра-
бочие качества топлива для двигателя внутреннего сгорания.
Действительно, опыт показывает, что при недетонирующем
топливе повышение степени сжатия с 5 до 8 увеличивает мощность
двигателя легкого типа примерно на 25% и снижает удельный
расход топлива примерно на 23%. Понятно поэтому, что основ-
ное направление в развитии современного моторостроения опре-
делилось в сторону конструирования легких двигателей с повы-
шенной степенью сжатия. Серьезным препятствием на этом пути
является, однако, детонация топлива. Так например, при работе
на многих выдающихся по своим качествам бензинах прямой
гонки, в частности на грозненском и краснодарском авиабензи-
нах, применение двигателей со степенью сжатия выше 5,4—5,8
оказывается уже невозможным из-за наступления детонации
последняя же, как было показано выше (ср. фиг. 131), вызывает
резкое падение мощности и экономичности двигателя. Есте-
ственно, таким образом, что снижение детонационных свойств
моторного топлива является одной из актуальнейших задач то-
пливной проблемы.
Повышение качества моторного топлива может быть достиг-
нуто либо существенным изменением его химического состава пу-
тем разбавления его каким-либо компонентом с высоким октано-
вым числом (антидетонационные добавки), либо с помощью при-
садки (антидетонатора), коренным образом изменяющей процесс
горения данного топлива. Оба эти способа повышения качества
топлива должны быть рассмотрены отдельно.
Антидетонационные добавки. Горючие вещества, которые мо-
гут применяться в качестве добавок для улучшения детонацион-
741
пых качеств моторного топлива, должны удовлетворять трем
основным условиям. Во-первых, они должны обладать высоким
октановым числом; во-вторых, они должны быть легко доступны
и дешевы; наконец, они не должны снижать основных качеств
моторного топлива, например, путем неблагоприятного изменения
его физико-химических свойств, а также не должны препятство-
вать дальнейшему повышению октановых чисел топлива при по-
мощи антидетонаторов, обычно применяемых для этой цели
в топливной промышленности.
Чтобы ориентироваться в перечисленных условиях, рассмо-
трим несколько примеров, воспользовавшись для этого данными
табл. 156, 157 и 158.
Таблица 156
Октановые числа парафинов
Наименование углеводородов Температура кипения Октановое число
Изобутан — 12 2 100
Бутан норм 0,6 92
Изопентан 28,0 91
Пентан норм 36,2 65
Гексан норм 69,0 45
Гептан норм 98,4 0
Изооктан 106,5 100
Октан норм 124,6 — 32
Нонан норм 150,6 — 44
Декан норм 174,0 — 53
Таблица 157
Октановые числа нафтенов
Наименование углеводородов Температура кипения 0 ктановое число
Циклопентан 49,4 122
Метилциклопентан 71,9 89
Этилциклопентан 103,0 57
Диметилциклопентан-1,3 90,5 85
Пропилциклопентан 130,6 27
Бутилциклопентан 157,2 0
Циклогексан 80,9 97,5
Метилциклогексан 101,0 75
Диметилциклогексан-1,2 127,9 81
Диметилциклогексан-1,3 121,6 68,5
Днметилциклогексан-1,4 121,7 73,5.
Этилциклогексан . 131,6 41
Пропилциклогексан . 155,7 14
Бутилциклогексан 180,5 -5-
742
Таблица 158
Октановые числа ароматических углеводородов
Наименование углеводородов Температура кипения Октановое число
Бензол 80,4 96
Толуол 110,4 106,5
Этилбензол 136,1 123
Ксилол-1,2 144,0 114
Ксилол-1,8 139,0 125,5
Ксилол-1,4 137,0 128
Ксилолы, смесь изомеров 139,0 112,5
Изопропилбензол (кумол) 153,4 104,5
Пропилбензол норм 158,6 120
Мезитилен 164,0 131
Метил-изоп ропил-бензол-1,4 (цимол). 177,3 128
Бутилбензол вторичный 175,0 91
Бутилбенвол третичный 189,3 114
Бутилбензол норм 182,1 110,5
Амилбензол третичный — 114
Среди парафинов выделяются своими высокими октановыми
числами изобутан, бутан, изопентан и изооктан. Совершенно оче-
видно, что ни изобутан, ни бутан не годятся в качестве антиде-
тонационных добавок вследствие их слишком низкой темпера-
туры кипения: начало кипения для наиболее легких бензинов
(авиабензины) по техническим нормам СССР — около 40°. Бо-
лее подходит для этой цели изопентан; однако, чистый, синтети-
ческий изопентан слишком дорог, нефтяной же изопентан всегда
содержит примесь нормального пентана, октановое число кото-
рого значительно ‘ ниже. Поэтому пентановую фракцию, всегда
содержащуюся в легком бензине, следует расценивать не с точки
зрения ее антидетонационных свойств, а как фракцию «пуско-
вую», облегчающую пуск мотора в холодное время года.
Из всех парафинов наибольшее значение в качестве антиде-
тонационной добавки приобрел в настоящее время изооктан.
0 техническом получении его полимеризацией изобутилена ска-
зано выше. В США изооктан выделяют также путем тщательной
фракционировки некоторых бензинов, и себестоимость получае-
мого этим способом продукта является, невидимому, наименьшей
(около 1 цент, -за 1 галлон). Изооктан входит в состав авиабен-
зина ОША в количестве от 30 до 50%; роль пусковой фракции
в этом бензине выполняет изопентановая фракция (10—15%),
получаемая из газового бензина; остальное -приходится на долю
отборного нафтенового бензина прямой гонки. Такое авиатопливо
имеет высокое октановое число, которое добавкой антидетонатора
может быть поднято до ТОО.
. Переходя к нафтеновым компонентам нефти (табл. 157), мы
видим прежде всего, что высокими октановыми числами характе-
ризуются циклогексан и особенно циклопентан. Как известно,
743
в чистом виде циклопентан чрезвычайно трудно доступен; к тому
же содержание его в нефтях крайне невелико. Таким образом
значение циклопентана с точки зрения антидетонационных
свойств бензина должно быть признано ничтожным. Из других
пятичленных нафтенов метилциклопентан и диметилциклопен-
таны несомненно имеют положительное значение как компо-
ненты с относительно высокими октановыми числами; в извест-
ной мере они нейтрализуют влияние соседних нормальных пара-
финов, гексана и гептана. Из шестичленных нафтенов выде-
ляется своим высоким октановым числом циклогексан. У гомоло-
гов циклогексана октановые
Фиг.
кого
132. Зависимость допусти-
сжатия от содержания бен-
зола.
числа уже значительно ниже; осо-
бенно резко снижаются они для
гомологов циклогексана, кипящих
выше 150°. Понятно поэтому,
что температуру выкипания наф-’
теневых бензинов, применяемых
в качестве компонентов для вы-
сокооктанового топлива, прихо-
дится ограничивать 140—150° и
даже ниже. Вследствие сравни-
тельно дорогой стоимости нафтены
пока не находят применения в
качестве антидетонационных до-
бавок.
Что касается, наконец, арома-
тических углеводородов, то, как
видно из данных табл. 158, мы
имеем здесь наиболее ровный ряд
в отношении октановых характе-
ристик: для ароматики с темпера-
турой кипения до 200° октано-
вые числа колеблются всего лишь
в пределах от 91 до 96 (бутил-
бензол, бензол) до 128—130 (па-
ра-ксилол, цимол, мезитилен). Ка-
залось бы, что ароматический
может дать большой выбор добавок с высоким октано-
числом. Однако большинство ароматических углеводородов
ряд
вым
либо забронированы для других ответственных целей (толуол),
либо слишком дороги и трудно доступны; последнее относится •
в известной степени даже к такой ароматике, как этилбензол и
кумол, получаемой действием этилена и пропилена на бензол
в присутствии хлористого алюминия. В итоге из всей ароматики
в качестве компонента с высоким октановым числом остается
почти один бензол, действительно нашедший широкое применение
при изготовлении моторного топлива в смеси с бензином прямой
гонки.
Добавка бензола существенно повышает антидетонационные
свойства моторного топлива. Как видно из графика фиг. 132, до-
бавка 50% бензола повышает допустимую степень сжатия на
744
25%, а добавка 60% бензола — на 3S%. Таким образом присут-
ствие бензола в моторном топливе позволяет существенно увели-
чивать эффективность и мощность мотора. Однаки, с другой сто-
роны, добавка бензола имеет некоторые отрицательные послед-
ствия. Прежде всего наличие бензола (температура плавле-
ния 5,°4) должно более или менее существенно повысить темпе-
ратуру застывания топлива, что крайне нежелательно, особенно
для авиатоплива при •высокоскоростных и зимних полетах. Впро-
чем, данное влияние бензола в значительной мере ослабляется
тем обстоятельством, что в качестве добавки к топливу приме-
няют не чистый бензол, а бензол моторный, который, как видно
из табл. 159, содержит лишь 55% бензола и применение кото- *
рого, очевидно, экономически более выгодно.
Гораздо более важное зна- Таблица г>э
чение имеет другое отрицатель-
ное следствие присутствия в
Состав моторного авнабенвола в %
моторном топливе значитель-
ного количества ароматики и в
частности .бензола, а именно —
резкое снижение приемистости
Бензол ..................
Толуол ..................
Ксилолы..................
Высшие гомологи..........
55
35
I
3
такого топлива к тетраэтил-
свинцу. Ввиду этого в настоящее время при получении высших
сортов моторного топлива стремятся не к повышению, а к по-
нйжению в них ароматики.
Из других антидетонационных добавок большое внимание при-
влекает за последнее время изопропиловый эфир, СзН7 • О • СзНт
(октановое число 100).
Изучение детонационных свойств различных углеводородов и их смесей
ведется в двух направлениях. С одной стороны, исследуется связь между
детонацией, составом и строением различных углеводородов. В результате
больших работ, проделанных в этом направлении, удалось установить ряд
эмпирических правильностей, которые уже были освещены в ч. I (гл. IV.
V Б и VI). Второе направление данных работ имеет своей задачей иссле-
дование детонационных свойств отдельных технических продуктов и в пер-
вую очередь бензинов по возможно более узким фракциям. Ближайшей
задачей такого детализированного исследования детонационных свойств
является установление точек максимальной и минимальной детонации
(т. е. пик и провалов) для данного продукта, который получает, таким обра-
зом, полную характеристику его детонационных свойств. В этом направле-
нии в ГрозНИИ была проделана большая работа для характеристики трех
важнейших видов нашего бензина, а именно — бакинского, грозненского и
майкопского по их трехградусным фракциям. Относящиеся сюда подроб-
ности следует искать в оригинальных работах [19].
Практическое значение детализированного исследования де-
тонационных свойств моторного топлива по узким фракциям мо-
жет быть весьма велико. Действительно, зная «пики» детонации,
т. е. те фракции, которые являются преимущественными носи-
телями детонационных свойств данного топлива, можно поставить
вопрос об удалении этих фракций путем углубленной фракцио-
нировки или о соответствующем снижении конца кипения то-
плива в целях улучшения его детонационных свойств. Так на-
пример, совершенно ясно, что удаление из бензина путем тща-
745
тельной его ректификации нормального гептана (температура ки-
пения 98,3°) или снижение конца кипения бензина ниже темпе-
ратуры кипения нормального октана (125,8°) должно улучшить
детонационные свойства бензина. С другой стороны, знание «про-
валов» детонации, т. е. тех фракций бензина, которые характе-
ризуются наиболее высокими антидетонационными свойствами,
дает возможность поставить вопрос о специальном выделении не-
которых из этих фракций как ценнейших компонентов высоко-
октанового топлива. По имеющимся сведениям в США уже всту-
пили на ©тот путь для выделения из некоторых нефтей изооктана
(температура .кипения 106—107°). Тщательно выделенная и очи-
щенная соответствующая фракция является, невидимому, наи-
более дешевым изооктаном для авиатоплива.
Антидетонаторы. Как ясно из вышеизложенного, антидетона-
пионные добавки представляют собою высокооктановые горючие
продукты, которые, будучи прибавлены к моторному топливу
в более или менее значительном количестве (например, несколько
десятков процентов), коренным образом изменяют его химический
состав и тем самым исправляют его детонационные свойства.
Совершенно иной характер имеют антидетонаторы. По своей хи-
мической природе это — нередко довольно труднодоступные орга-
нические или металлоорганические вещества, добавляемые к то-
пливу в небольших количествах (например, десятые доли про-
цента) и, очевидно, неспособные существенно изменить при этих
условиях химический состав топлива. Действие антидетонато-
ров направляется, таким образом, в другую сторону: подобно ка-
тализаторам они оказывают влияние на самую химическую ре-
акцию, протекающую в их присутствии, в данном случае на
процесс горения, и лишь этим путем улучшают детонационные
свойства моторного топлива.
Количество веществ, испытанных в качестве антидетонаторов
моторного топлива, чрезвычайно велико, однако практическое
значение из них получили только отдельные единицы. В табл. 160
приведено несколько наиболее интересных антидетонаторов, эф-
фективность которых выражена в условных единицах, а именно:
за единицу эффективности принят антидетонационный эффект,
вызываемый прибавлением к дезароматизированному бензину
30% бензола (по объему), что соответствует увеличению наивыс-
шей полезной степени сжатия в моторе примерно на 10%; при-
веденные в последнем столбце величины указывают, во сколько
раз может быть уменьшена добавка соответствующего антидето-
натора по сравнению с бензолом для прлучения того же эффекта.
Данные табл. 160 показывают, что ароматические амины по
своей антидетонационной эффективности в 10—15 раз превосхо-
дят бензол. Отдельные простейшие первичные и вторичные амины
ароматического ряда мало различаются между собой; зато они
значительно превосходят в этом отношении третичные амины
(например, диметиланилин и т. п.), а также амины жирного
ряда (и те и другие в табл. 160 не значатся). Но особенно
выделяются как антидетонаторы некоторые металлоорганические
746
Таблица 160
Относительная эффективность различных антидетонаторов
Название антидетонатора
Химическая
формула
Эффектив-
ность
Бензол 1,0
Этиловый спирт с2н5он 2,U
Толуидин СвЩСНзКНо 10,0
Метиланилип CeHsNHCHs 12.0
Анилин ... С«НбЫН, 13,5
Ксилидин CsHsCCHsliNH, 15,0
Дифениламин (CeHsJsNH 16,0
Тетраэтил-олово Sn (СзЩ)* 25,U
Диэтил-селен Se(C2H5), 60,0
Диэтил-теллур Те (СзЦоЬ 200,0
Днхлор-диэтил-свинец РЬ(С2Нб)2 С12 300,0
Тетракарбонил-никель Xi (СО), 300,0
Хлор-триэтил-свинец РЬ«М1ЛС1 4500
Пентакарбонил-железо Fe (COh 500,0
Тетраэтил-свинец РЬ (С.,Н5)4 600,0
соединения; среди них отметим здесь тетракарбонил-никель и
пентакарбонил-железо, особенно же тетраэтил-свинец, превосхо-
дящий по своей эффективности бензол примерно в 600 раз и по-
лучивший благодаря этому широкое применение для улучшения
детонационных качеств моторного топлива.
Тетраэтил-свинец, (СгН^РЪ, в лабораторных условиях легко
получается действием магний-бром-этила, CsHsMgBr. на сухой
хлористый свинец с последующим разложением продукта реак-
ции водой и перегонкой выделившегося маслянистого слоя сна-
чала при обыкновенном давлении (с паром), а затем в вакууме.
В технике тетраэтил-свинец получают действием хлористого
этила на сплав металлического натрия со свинцом.
Тетраэтил-свинец представляет собою тяжелую бесцветную
жидкость с довольно приятным эфирным запахом. Его темпера-
тура кипения 81,5° при 10 мм; удельный вес d184 = i,66. При
стоянии на воздухе, особенно на солнечном свету, тетраэтил-
свинец частично разлагается: сначала выделяется белая муть и
осадок; под конец — даже металлический свинец. Присутствие
аминов жирного ряда, некоторых их производных и ароматиче-
ских вторичных аминов, особенно в присутствии галоидопроиз-
водных, действует на тетраэтил-свинец стабилизирующим обра-
зом.
•Тетраэтил-свинец — сильный яд; ввиду этого получение и
применение его должны быть обставлены крайними мерами пре-
досторожности (сильные тяги, спецодежда, перчатки и даже
маски). Длительная и неосторожная работа с тетраэтил-свинцом
тем более опасна, что действие его на организм носит комулятив-
ный характер (т. е. накопляется) и может привести к самым
тяжелым нервным расстройствам и смерти.
747
Открытие органически связанного свинца в моторном топливе является
ъ настоящее время немаловажной задачей. Для решения этой задачи пред-
ложено несколько методов. Качественно вопрос решается проще всего
экспозицией исследуемого бензина на прямом солнечном свету или на
свету ртутной лампы. Если бензин содержал тетраэтил-свинец, в только
что указанных условиях происходит выделение окиси свинца, которая
легко обнаруживается в форме йодистого или сернистого свинца. Количе-
ственное определение свинца в моторном топливе сводится к разрушению
металлоорганического соединения действием крепкой соляной кислоты или
сильных окислителей с последующим определением свинца в виде, напри-
мер, сульфата и т. п.
Открытие высоких антидетонационных свойств тетраэтил-
свинца создало новую эпоху в деле получения высокооктанового
моторного топлива. Тетраэтил-свинец прибавляется к бензину
в виде так называемой «этиловой жидкости»; последняя содер-
жит около 55% (по объему) тетраэтил-свинца, остальные же 45%
приходятся в этой жидкости на долю некоторых галоидопроиз-
водных (бромистый этил, бромистый этилен, а-хлорнафталин и
др.). Эти галоидопроизводные, сгорая в цилиндре двигателя вме-
сте с тетраэтил-свинцом, создают условия, при которых свинец
не отлагается в виде окиси в цилиндрах, а уносится в виде га-
лоидоводородной соли вместе с выхлопными газами. Кроме того,
присутствие галоидопроизводных, повидимому, действует на те-
траэтил-свинец стабилизирующим образом.
Этиловая жидкость, прибавленная к бензину в количестве
1—3 см3 на 1 л, вызывает повышение октанового числа бензина
иногда на 15—20 пунктов; в других случаях это повышение зна-
чительно меньше (5—7 пунктов и меньше). Столь различная
приемистость к тетраэтил-свинцу связана с составом бензина.
Наивысшую приемистость к тетраэтил-свинцу показывают угле-
водороды предельного характера, в частности бензины, состоящие
из парафинов, изопарафинов и нафтенов. Напротив, бензины, бо-
гатые непредельными и ароматическими углеводородами, сравни-
тельно мало отзываются на добавление этиловой жидкости.
Именно ввиду этого высшие сорта моторного топлива в настоящее
время составляются без участия ароматических и непредельных
углеводородов; отсутствие последних дает возможность поднимать
с помощью тетраэтил-свинца октановое число некоторых видов
моторного топлива до 100 и выше.
Из других антидетонаторов, отмеченных в табл. 160, бесспорно заслу-
живает внимания лентакарбонил-железо, Fe(CO)s, получаемое обработкой
порошка железа окисью углерода при 100—200° и давлении 150—200 ат.
В чистом виде это — бесцветная жидкость с температурой кипения 104°,6;
ее температура г плавления около—20°. По своей эффективности занимает
место тотчас вслед за тетраэтил-свинцом.
ХИМИЗМ ДЕЙСТВИЯ АНТИДЕТОНАТОРОВ
Вопрос о механизме действия антидетонаторов при сгорании
топлива в моторе в настоящее время еще нельзя считать полно-
стью разрешенным, тем более что химизм процесса сгорания и
детонации углеводородов и их смесей, как это было указано
748
выше, также остается пока еще недостаточно ясным. Многочи-
сленные попытки дать теорию, или, по крайней мере, схему ан-
тидетонирующего действия некоторых веществ можно разбить на
две основных группы: одни пытаются разъяснить данный про-
цесс с чисто физической точки зрения, не касаясь его химизма;
другие, напротив, делают попытки разобраться в химической сто-
роне явления, истолковывая его физико-химическую сущность.
Ниже дается краткий очерк обоих этих направлений научной
мысли в данном вопросе. Как будет показано, различные теории
действия антидетонаторов теснейшим образом переплетаются
с различными -взглядами на сущность явления детонации то-
плива..
С чисто физической точки зрения детонацию топлива в мо-
торе можно представить [20] следующим образом (Рикардо),
Пусть смесь топлива с воздухом, сжатая в цилиндре двигателя
и нагретая до температуры, близкой к температуре ее самовос-
пламенения, возгорается, например, от свечи. Вначале образую-
щийся фронт пламени распространяется нормально; при этом
газообразные продукты сгорания, естественно, должны увеличи-
вать общую упругость газовой смеси и ее температуру. Когда
температура песгоревшей части газовой смеси достигает темпе-
ратуры ее самовоспламенения, происходит мгновенное самовозго-
рание, сопровождаемое резким скачком давления, которое и пере-
дается стенками цилиндра в виде характерного стука. Опыт по-
казывает, что между температурой воспламенения топлива и
склонностью его к детонации, характеризуемой наивысшей полез-
ной степенью сжатия (Н. П. С. С.) для данного топлива, дей-
ствительно наблюдается прямая зависимость (табл. 161).
Таблица 161
Температура самовоспламенения и склонность топлива
к детонации
Топливо Температура самовоспламе- нения в °C Н.П.С. С.
Гептан • 430 3,7-4,7
Гексан 470 4,8—5,1
Пентан 515 5,7
Циклогексан 535 6,0
Бензол 675—710 6.9
Толуол >760 >7,0
Спирт 515 7,0
Данные табл. 161 свидетельствуют, что для ряда углеводоро-
дов температура самовоспламенения й Н. П. С. С. изменяются
в одну сторону: чем -выше температура самовоспламенения, тем
выше Н. П. С. С., т. е. тем меньше склонность данного топлива
к детонации. Однако имеются и обратные случаи: таков, напри-
744
мер, спирт, у которого температура самовоспламенения ниже, чем
у циклогексана и бензола, а Н. П. О. С. выше. Особенно должен
быть отмечен в этом отношении сероуглерод, обладающий крайне
низкой температурой самовоспламенения (около 140°) и в то же
время чрезвычайно трудно детонирующий, что находится в яв-
ном противоречии с рассматриваемой концепцией. Не менее важ-
ным в этом отношении является твердо установленный факт [21],
что в зависимости от состава бензина добавка тетраэтил-свинца
может либо повышать, либо понижать температуру самовоспла-*
менения этого топлива, тогда как склонность его к детонации от
этой добавки понижается. Все эти факты с полной очевидностью
показывают, что самовоспламенение и детонация отнюдь не по-
крывают друг друга, а могут наблюдаться, как это уже было
отмечено выше, совместно и независимо друг от друга и что, сво-
дить, таким образом, действие антидетонаторов к повышению тем-
пературы самовоспламенения топлива неправильно.
Другая физическая теория действия антидетонаторов, теория актив-
ных ядер (Каллендер), подходит к явлениям детонации несколько иначе.
При распылении топлива в (карбюраторе под влиянием струи воздуха испа-
рение капелек топлива должно происходить, очевидно, постепенно: сначала
испаряются наиболее легкие части бензина, затем — более тяжелые; под
конец остаются наиболее тяжелые и наименее летучие микроскопические
капельки парафиновых углеводородов (ядра). Опыт показывает, что тем-
пература самовоспламенения капель всегда ниже, чем температура само-
воспламенения паров того же вещества. С другой стороны, известно, что
более тяжелые парафины самовоспламеняются легче, чем более легкие
(табл. 161). Отсюда ясно, что указанные ядра, т. е. микроскопические ка-
нелькп тяжелых парафинов, при сжатии и надлежащем повышении темпе-
ратуры горючей смеси могут оказаться активными центрами: самовоспла-
меняясь, они зажигают смесь сразу в громадном числе точек, другими
словами, вызывают ее детонацию [22].
Действие антидетонационных добавок теория активных ядер рисует
следующим образом. Добавки б высокой температурой самовоспламенения,
например ароматические углеводороды, разжижают бензин, так что относи-
тельное количество активных ядер уменьшается. Одновременно парциаль-
ное давление паров бензина вследствие его разбавления уменьшается, что
способствует лучшему испарению капель. Все это вместе взятое, есте-
ственно, должно уменьшать склонность к детонации бензина, к которому
добавлено достаточное количество ароматики.
Действие металлоорганических антидетонаторов согласно той же теории
заключается в том, что при распаде их в цилиндре двигателя поверхность
активных капель покрывается тончайшим слоем металла, который должен
предохранять ядра от самовоспламенения; тем самым предотвращается и
детонация топлива.
Основные возражения против теории активных ядер сводятся к еле-
дующему. Прежде всего установлено, что явления детонации наблюдаются
не только с жидкими, но и с газообразными горючими, т. е. в таких слу-
чаях, когда образование активных капельножидких ядер невозможно себе
представить. Что касается металлоорганических антидетонаторов, то допу-
скаемое теорией металлическое покрытие активных ядер не только не мо-
жет предохранить их от самовозгорания, а скорее наоборот: в условиях:
сгорания топлива в моторе металлические частицы, образующиеся в ре-
зультате распада антидетонатора, должны быстро окисляться с выделением
большого количества тепла, что в свою очередь должно не предохранять
активные ядра, а содействовать скорейшему их самовоспламенению. Оче-
видно, таким образом, теория активных ядер также не в состоянии разъ-
яснить механизм детонации и действия антидетонаторов.
750
Все, что характеризует явление детонации, свидетельствует
прежде всего о том, что в основе этого сложного явления лежит
резкое увеличение скорости основной реакции, протекающей
в цилиндре двигателя, т. е. реакции сгорания топлива, и что,
с другой стороны, действие антидетонатора или антидетонацион-
ной добавки по существу сводится к отрицательному катализу,
т. е. к задержке саморазгона реакции, иначе говоря, к удержа-
нию скорости реакции сгорания топлива в тех пределах, которые
соответствуют нормальному сгоранию. Отсюда ясно, что механизм
действия антидетонаторов должен быть увязан с механизмом
реакции сгорания топлива в моторе, т. е. в основном с механиз-
мом окисления углеводородных смесей под влиянием кислорода
воздуха. Мы подходим, таким образом, к рассмотрению меха-
низма действия антидетонаторов с химической точки зрения.
Выше, в связи с учением об окислительном крэкинге (ч. II,
гл. IV), и позднее, в связи с учением об ингибиторах (ч. Ill,
гл. III), было показано, что наиболее простое л полное разъясне-
ние химизма реакций окисления под влиянием кислорода воз-
духа дает теория, предусматривающая промежуточное образова-
ние при этих реакциях перекисей (молокисей), причем процесс
образования этих промежуточных веществ и их дальнейшие пре-
вращения протекают, невидимому, по схеме цепной реакции.
Если теперь представить себе, что вследствие быстро протекаю-
щего образования перекисей в данной горючей смеси к моменту
воспламенения смеси в цилиндре мотора произойдет чрезмерное
накопление этих в высокой степени активных веществ, то, оче-
видно, их дальнейший распад в надлежащих условиях может
принять чрезвычайно бурный характер, т. е. вызвать детонацию.
С другой стороны, перекиси (молокиси) могут и не накапли-
ваться в горючей смеси, если они претерпевают дезактивацию по
мере своего образования вследствие превращения в вещества, не-
способные к продолжению цепи. Таким образом, если во многих
точках системы будет происходить обрыв цепей, процесс окисле-
ния горючей смеси будет итти спокойно, без всякой детонации.
Отсюда отчетливо вырисовываются роль и значение антидетона-
торов моторного топлива: очевидно, антидетонаторы должны пре-
пятствовать накоплению перекисей в горючей смеси, реагируя
с ними, содействуя превращению их в малоактивные соединения
и, таким образом, своевременно обрывая образовавшиеся реакци-
онные цепи [23].
Таковы в самых общих чертах основы пероксидной теории
детонации и действия антидетонаторов. Ближайший анализ во-
проса показывает, что в пользу этой теории можно привести ряд
весьма веских соображений.
Действительно, непосредственные опыты показывают, что уже
небольшая добавка различных органических перекисей явственно
повышает детонационные свойства топлива; перекиси же, осо-
бенно нестойкие, могут снижать Н. П. С. 0. в той же или боль-
шей степени как наиболее мощные детонаторы топлива подобно,
например, нитритам, т. е. эфирам азотистой кислоты 24 . В пол-
751
ном согласии с этими данными находится наблюдение [25 , что
такие малосклонные к детонации углеводороды, как амилен и
циклогексан, после продолжительного соприкосновения с возду-
хом теряют свои антидетонационные свойства, очевидно за счет
образовавшихся в них перекисей. С другой стороны, доказано,
что даже такие наиболее реакционно-способные продукты превра-
щения перекисей, как альдегиды, не только не увеличивают
склонности топлива к детонации, но скорее даже, наоборот, не-
сколько повышают И. П. С. С., т. е. снижают детонационные
свойства топлива [24]. Таким образом ясно, что причину дето-
нации топлива следует искать прежде всего в возможности обра-
зования в процессе сгорания топлива веществ перекисного ха-
рактера.
В какой степени возможно образование перекисей в условиях,
предшествующих моменту зажигания горючей смеси в цилиндре
двигателя? Этот вопрос был также подвергнут эксперименталь-
ному освещению. Особенно убедительные результаты были полу-
чены при изучении состава выхлопных газов в условиях работы
горячего двигателя от электромотора ’24]. Как показали опыты,
при степени сжатия 5 перекиси в выхлопных газах не обнару-
живались, по мере же увеличения степени сжатия (до 8) проба
на перекиси становилась все более интенсивной.
Опыты производились на двигателе с 1200 об/мин; температура ру.
башки была 80°; топливо начальное — смесь бензина с бензолом. Вначале
двигатель работал на газе нормального составу. После установления тепло-
вого режима двигатель переключался на другое топливо — ундекан, СиНм,
зажигание выключалось, число же оборотов поддерживалось электромото-
ром. Выходившая из двигателя смесь, газов подвергалась анализу.
Итак, можно считать установленным, что органические пере-
киси действительно образуются в процессе сжатия горючей смеси
в цилиндре двигателя и что перекиси эти являются основной
причиной детонации. Остается рассмотреть несколько ближе,
в чем заключается с точки зрения перекисной теории химизм
действия антидетонационных добавок и антидетонаторов.
Как мы видели выше, антидетонационные добавки предста-
вляют собою горючие вещества (изооктан, бензол, изопропиловый
эфир и др.), прибавляемые к топливу в более или менее значи-
тельном количестве и тем самым существенно изменяющие его
состав. Снижение детонационных свойств топлива связано в дан-
ном случае прежде всего с его разбавлением, вследствие чего
интенсивность образования перекисей, естественно, должна
уменьшиться. Характерным свойством антидетонационных доба-
вок является их высокое октановое число, что, надо думать, на-
ходится в непосредственной связи с относительно малым темпе-
ратурным коэфициентом образования перекисей у этих соедине-
ний: сравнительное изучение в этом направлении углеводородов
различных рядов показало, что наибольшей склонностью к оки-
слению кислородом воздуха при повышенной температуре обла-
дают нормальные парафины, ароматика же значительно уступает
им в этом отношении.
752
Помимо разбавляющего действия, антидетонационные * до-
бавки, повидимому, могут вступать также во взаимодействие
с перекисями, обрывая, таким образом, цепную реакцию подобно
типичным антидетонаторам. Так например, показано [26 , что
лри окислении кислородом смеси гексана с бензолом наблюдается
образование фенола даже при таких температурах, которые не-
достаточны для окисления одного бензола. Повидимому, реакция
протекает в данном случае по следующей схеме: сначала обра-
зуется высокоактивная перекись гексана, которая реагирует да-
лее с бензолом, так что в итоге образуются гексиловый спирт и
-фенол:
CeHu + 0s = С6Н14О2;
С6Н14О, + с6н6 = С6Н130Н -4- СеН-0Н.
Как видно, бензол играет в данном случае роль типичного
антидетонатора вроде, например, какого-либо ароматического
амина.
Как было доказано выше, многие ароматические амины
являются не только антидетонаторами, но и хорошими ингиби-
торами; взятые уже в небольшом количестве, они энергично за-
держивают процессы самоокисления углеводородов, протекающие
при обыкновенной температуре. Как показывает опыт, это свое
свойство ароматические амины сохраняют также при повышенной
температуре, например при 230° [27], а. также при сгорацйи
углеводородов. Таким образом здесь наблюдается несомненная
общность действия присадок, которые на первый взгляд принад-
лежат к двум различным типам: ингибиторов и антидетонаторов
моторного топлива. С точки.зрения пероксидной теории такая
общность понятна: и в том, и в другом случая .роль амина' сво-
дится к тому, что он принимает на себя действие высокоактив-
ных первичных продуктов окисления (перекисей) и тем самым
обрывает цепную реакцию окисления.
Что касается, наконец, металлоорганических антидетонаторов,
то необходимо отметить прежде всего, что применение к ним пе-
роксидной теории требует некоторых дополнительных пояснений.
Дело заключается в том, что, будучи весьма сильными антиде-
тонаторами, малоорганические соединения, вообще говоря, зна-
чительно уступают ароматическим аминам, как ингибиторы. Так
например, установлено, что тетраэтил-свинец примерно в 100 раз
-слабее динафтиламина в качестве ингибитора и примерно во
столько же раз сильнее этого амина в качестве антидетонатора
[28]. Это кажущееся противоречие легко разъясняется однако,
если допустить, что такие металлоорганические антидетонаторь,
как тетраэтил-свинец, пентакарбонил-железо и т. п., действуют
не сами по себе, а своим металлом, когда он выделяется в мелко-
раздробленном виде при разложении этих соединений под влия-
нием повышенной температуры 291. Так например, тетраэтил-
свинец начинает разлагаться лишь при температурах около 200 ,
понятно поэтому, что он может оказывать лишь небольшое
влияние на процессы окисления, протекающие при более низких
753
48 Зак. 1622. Химия нефти.
температурах, но он оказывается весьма эффективным при про-
цессах, идущих при температурах порядка 200° и выше.
Действительно, прямыми опытами показано, что введение
в горючую смесь вместо паров тетраэтил-свинца продуктов его
распада от предварительного нагревания до 300° сразу же вы-
зывает подавление детонации. Показано также, что целый ряд
металлов в мелко распыленном состоянии производит аналогич-
ное действие, причем максимальный эффект обнаруживают: тал-
лий, калий, свинец, железо, никель и силен. Таким образом в ос-
новном, можно считать установленным, что непосредственное
участие в реакции сгорания моторного топлива в присутствии
металлоорганического антидетонатора принимает не сам антиде-
тонатор, а входящий в его состав металл; роль же этого металла»
очевидно, вполне аналогична роли типичного антидетонатора:
принимая на себя действие перекисей (молокисей), образовав-
шихся при непосредственном окислении отдельных компонентов
топлива, частицы металла обрывают цепную реакцию окисления
топлива и тем предотвращают его детонацию.
Пероксидная теория, наиболее просто и полно разъясняющая явления
детонации топлива и действия антидетонаторов, получила в настоящее
время широкое признание. Тем не менее, некоторые вопросы в этой слож-
ной области и поныне остаются не вполне понятными и требуют дальней-
шего исследования. Так например, известно, что такие углеводороды, как
олефины, тетралин и некоторые другие, обладая ярко выраженной склон-
ностью к образованию перекисей, тем не менее значительно уступают пара-
финам в скл нности к детонации; известен ряд наблюдений, когда приба-
вление тетраэтил-свинца, этого наиболее изученного антидетонатора, вызы-
вало не положительный а нулевой или даже отрицательный эффект
в смысле снижения детонации; непонятен также ярко выраженный избира-
тельный характер действия тетраэтил-свинца на топлива различного состава,
например слабая приемистость к этому антидетонатору олефинов и особенно
ароматики. Эти и многие другие факты показывают, что наука еще далеко
не достигла исчерпывающего познания природы детонации топлива и ее
предупреждения.
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ АНТИДЕТОНАТОРОВ
Антидетонационные добавки и антидетонаторы приобрели
в современной топливной промышленности исключительно важ-
ное значение, уже освещенное в предшествующем изложении.
И те, и другие соединения получаются на специальных установ-
ках и заводах, и обеспечение ими авио- и автотранспорта имеет
не только громадное народнохозяйственное, но и оборонное зна-
чение. Несомненно, этим именно обстоятельством объясняется
в первую очередь, что, несмотря на крайне ядовитые свойства
тетраэтил-свинца и весьма существенные возражения, которые
вызывает его применение в моторах с точки зрения интересов
общественного здравоохранения, этот антидетонатор, как наибо-
лее эффективный, получил широкое распространение во всем
мире (Ethyl-Gasolin Corp.). Производство и применение других
антидетонаторов, например пентакарбонил-железа (Германия)
вызывается, повидимому, патентными соображениями. Что ка-
сается антидетонационных добавок, то наибольшее значение среди
754
них ныне приобрели изооктан и изопропиловый эфир; их про-
изводство в США суммарно уже перешло за 1 млн. г в год,
строительство же соответствующих заводов приобретает все бо-
лее и более широкое распространение.
В. Депарафинизаторы
Низкая температура застывания является одним из важней-
ших свойств смазочных масел. Так например, низкая темпера-
тура застывания автола обеспечивает легкий пуск машины в хо-
лодную погоду, облегчает непрерывный приток масла к подшип-
никам и тем самым уменьшает износ двигателя. А между тем,
смазочные масла из нефтей с парафиновым основанием, т. е.
масла, обладающие наилучшими качествами, имеют слишком вы-
сокую температуру застывания. Так например, цилиндрсток из
грозненской парафинистой нефти с вязкостью Еюо = 2,б засты-
вает при 50°, а соответствующая фракция сураханской нефти
имеет температуру застывания около 35°. С помощью различ-
ных методов, путем частичного выделения парафина можно зна-
чительно снизить температуру застывания масла: однако сниже-
ние это часто бывает совершенно недостаточно. Так например,
сураханский брайтсток, обеспарафиненный методом выморажива-
ния и центрифугирования на установке Макс Миллера (Баку),
имеет тем не менее, температуру застывания 4-13°. Дальнейшая
депарафинизация масел с температурой застывания 10—15°, как
показывает опыт, не экономична и к тому же вредно отражается
на качестве масел, а именно: снижает индекс вязкости масла,
понижает смазочные свойства масла (его «масленистость») и
стабильность в отношении окисляемости и, наконец, повышает
склонность масла к коксообразованию и увеличивает летучесть
масла. Для такого рода случаев большое значение приобретают
в настоящее время специальные масленые присадки, депара-
финизаторы, небольшая добавка которых к маслу резко снижает
его застывание, не изменяя существенно других его качеств.
ДЕПАРАФИНИЗАТОРЫ СМАЗОЧНЫХ МАСЕЛ
Среди веществ, добавка которых вызывает стабильное сниже-
ние температуры застывания масел, наибольшее значение при-
обрели следующие:
1) алюминиевая соль стеариновой кислоты (стеарат алюми-
ния);
2) триэтанолшшн—продукт взаимодействия аммиака с окисью
этилена (ср. ч. П, гл. III), в смеси со стеаратом алюминия;
3) синтетическое масло («парафлоу»), получаемое конденса-
цией нафталина с хлорированным парафином в присутствии
хлористого алюминия (по реакции Фриделя-Краффтса).
Применение перечисленных добавок требует предваритель-
ных лабораторных опытов, которые должны определить, при
какой качественной и количественной добавке к данному маслу
получается наибольшее снижение температуры его застывания,
которая к тому же должна оставаться стабильной независимо от
температурной обработки масла в процессе его работы, т. е.
охлаждения его или нагревания. В табл. 162 приведены при-
меры обработки одного из образцов балаханского масла смесью
стеарата алюминия с триэтаноламином и синтетическим маслом
«парафлоу « [30].
Таблица 16%
Влияние добавок на свойства масла
(по Рамайа ТСаладжала)
До обработки масла (бала- После^обработки масла
ханского) 0,25% стеарата А1 и 0,25% триэтаноламина 0,5% парафлоу
Застывание 4-2° .... Е^о = 10,7; Eiqo = 2,05 . . Золы нет Застывание — 20° — 10,9; Е%о = 2,33 Золы 0,035 Застывание — 20° Ь’б') в П,7; ®юо= 2,02 Золы 0,004
Как видно из таблицы, добавка к балаханскому маслу равных
количеств стеарата алюминия и триэтаноламина (по 0,25%)'или
прибавление к маслу 0,5% «парафлоу» почти не отражаются на
вязкости масла; зольность масла, особенно от первой добавки,
естественно, несколько возрастает. Зато наблюдается резкое
снижение температуры застывания масла: с 4-2° до обработки
масла температура застывания снижается до—20° после со-
ответствующей добавки.
Давно известно, что присутствие некоторых веществ, напри-
мер асфальтовых и смолистых, заметно понижает температуру
застывания масел, что полное удаление этих веществ из масла,,
наоборот, повышает температуру его застывания и что, таким
образом, добавкой асфальтовых *и смолистых веществ к очищен-
ному маслу может быть достигнуто снижение температуры его
застывания (ср. ч. I, гл. II). Действие перечисленых выше спе-
циальных добавок, снижающих температуру застывания масла,
представляет собою, очевидно, явление того же порядка: во всех
этих случаях под влиянием тех или иных веществ в данных
условиях происходит как бы задержка выделения из раствора
парафина, с выпадением которого, как известно, и связано явле-
ние застывания масла. Практически задача заключается, таким
образом, лишь в подборе таких веществ, добавка которых, сни-
жая температуру выпадения парафина из масла, по возможно-
сти меньше отражалась бы на других его свойствах.
756
„ПАРАФЛОУ*, ЕГО ПОЛУЧЕНИЕ И СВОЙСТВА
Из всех предложенных депарафинизаторов наибольшее рас-
пространение и значение приобрело в настоящее время синтети-
ческое масло «парафлоу» f31]. Ныне на основе ряда опублико-
ванных исследований методика получения, состав и свойства
«парафлоу» могут быть освещены в достаточной мере '32j.
Получение «парафлоу» слагается из нескольких последую-
щих операций; первой из них является хлорирование парафина.
Хлорирование производят, пропуская хлор в продажный пара-
фин при температуре около 95° до тех пор, пока содержание
хлора в продукте реакции не поднимется до 10—12%. По охла-
ждении отфильтровывают не вошедший в реакцию парафин,
а маслообразное вещество, представляющее собой смесь хлорза-
мещенных продуктов с небольшой примесью растворенного па-
рафина, приводят во взаимодействие с нафталином в присут-
ствии безводного хлористого алюминия. По существу эта реак-
ция представляет собою, очевидно, один из частных случаев
синтеза гомологов в ароматическом ряду по известному методу
Фриделя-Краффтса, когда в первую очередь образуется простей-
ший, однозамещенный (одноалкильный) гомолог, а затем —
двух- и полиалкильные гомологи нафталина. Допуская, что
средний состав хлорзамещенного грозненского парафина выра-
жается формулой С24Н4еС1, получаем для образования одно- и
двухалкильного нафталина следующие уравнения:
С10Н8+С.24Н4вС1 = С2.Н4й С1ПН, 4- НС1;
ад9С1 — C24n46 • с10н6 • с24н4Э-|- hcl
Вполне возможно, что кроме одно- и двухалкильных нафта-
линов при этой реакции образуются также продукты более глу-
бокого замещения. Они находятся, вероятно, в смоле, всегда об-
разующейся при реакции Фриделя-Краффтса: об этой смоле
будет сказано несколько ниже.
«Парафлоу» представляет собою двухалкильное производное
нафталина. Для получения его в очищенном виде прежде всего-
необходимо отделить от него соответствующее одноалкильное
производное и ту небольшую примесь парафина (7—8%), кото-
рая содержалась в хлориде, применявшемся в реакции Фриделя-
Краффтса. Такое отделение достигается либо перегонкой в ва-
кууме, либо методом селективного растворения.
Наиболее удобным способом очистки «парафлоу» является, повидимому»
метод селективного растворения [32]. В качестве растворителя наиболее
подходящим оказался ацетон при 45—47°. В этих условиях парафин и
одноалкильное производное нафталина переходят в раствор, а двухалкилъ-
ный нафталин «парафлоу») остается в нижнем слое и отделяется от ра-
створа декантацией.
Другой способ очистки сырого «парафлоу» — перегонка в глубокое
вакууме. При этом отгоняются почти исключительно парафин и одно-
алкильное производное нафталина, «парафлоу» же остается в перегонном
сосуде. Этот способ очистки «парафлоу» менее удобен, так как перегонка-
столь высокомолекулярных веществ всегда может сопровождаться их ча-
стичным разложением.
75-
При обоих способах очистки «парафлоу» всегда получается
< некоторым содержанием смол (около 7%), количественное опре-
деление которых можно произвести обычным путем с помощью си-
ликагеля. «Парафлоу», очищенный отгонкой более легких частей,
естественно, содержит больше смолистых частей, чем «парафлоу»1,
•очищенный обработкой ацетоном. Однако практического значе-
ния это обстоятельство почти не имеет: непосредственный опыт
показывает, что смолы «парафлоу» вызывают примерно такое
же снижение температуры застывания масла, как очищенное
«парафлоу». Вполне возможно поэтому, что смолы эти в значи-
тельной мере состоят также из алкилнафталинов, но более глу-
•бокой степени замещения, чем «парафлоу», например из три-,
тетра- и вообще из полиалкилнафталинов.
«Парафлоу» представляет собою густое масло зеленого цвета
в отраженном свете. Физические свойства «парафлоу» в опреде-
. лении различных исследователей несколько колеблются, ве-
роятно в зависимости от степени очистки его от указанных выше
примесей. • Ниже (табл. 163) приводятся константы «парафлоу»
согласно данным основной работы по этому вопросу [31] после
соответствующих переводов и пересчетов американских показа-
телей на принятые в нашей нефтяной литературе.
Таблица 163
Физические свойства .парафлоу"
(по Дэвису и Блэквуду)
Удельный вес (15°б6/15°, 56) ...................
Вязкость по Энглеру, Ез7°, 8 ..............♦ . .
* „ „ Еэ8°, 9 ...................
Индекс вязкости ................................
Застывание......................................
Вспышка.............................•...........
'Кокс по Конрадсону .....................
0,9111
108,5
6,46
104
— 3°,9
290°,6
1.99
Вследствие высокого молекулярного веса (в среднем — около
800) «парафлоу» не перегоняется без разложения даже в ва-
жууме.
Снижение температуры застывания от добавки «парафлоу»
и стабильность этого снижения зависят от ряда условий, кото-
рые будут рассмотрены ниже.
«Парафлоу» — углеводород предельного характера; его иод-
ное число равно нулю.
Химическое строение «парафлоу» не может считаться пол-
ностью установленным: не ясны положение боковых цепей и их
•строение, а равно и способ соединения их с нафталиновым ядром.
Действительно, пусть первая боковая группа С24Н49 по аналогии с за-
местителями более простого состава (например с СгШ) вступает в нафта-
линовое ядро в /3-положение. Если вторая группа того же состава согласно
вышеприведенным уравнениям для получения «парафлоу» поместится
также в 0-положение, то легко представить себе образование трех различ-
ных 0,0-изомеров «парафлоу». Если же вторая группа частично буд-ет ста-
новиться также в а-положение, то в общей сложности по теории должно
'758
предвидеть 10 двухзамещенных изомеров. Вполне возможно, что если не-
все, то некоторые из этих изомеров действительно образуются при полу-
чении технического «парафлоу», так что формулу (I), предложенную для
«парафлоу» как для 0-даалкил-нафталина [3*1], следует относить во-
всяком случае лишь к одному из углеводородов, образующих эту сложную*
смесь («парафлоу»):
Образование системы (I) и ей подобных требует допущения, что реак-
ция происходила здесь при участии монохлорпарафина. Другая концепция
исходит из предположения, что при получении «парафлоу» в реакции уча-
ствует дихлорпарафин и притом также в количестве двух молекул на одну
молекулу нафталина и что при этом происходит образование двух новых
жирноароматических колец по схеме (П) или какой-либо иной, ей подоб-
ной [30]. Возможность образования дихлоридов при хлорировании пара-
фина, конечно, не исключается; кроме того, в настоящее время едва ли
можно привести в пользу формулы (II) какие-либо иные, более веские
соображения.
Из побочных продуктов, образующихся при взаимодействии
хлорпарафина с нафталином, наибольший интерес предста-
вляет, конечно, моноалкильное производное нафталина. Оно со-
ставляет около 25% и выше от общего количества продуктов
данной реакции и несомненно является промежуточным продук-
том при получении «парафлоу». Оно представляет собою жидкое
масло предельного характера (иодное число равно нулю) с не-
большим содержанием хлора (примесь хлорпарафина), кипящее
в широких пределах: основная масса его кипит при 200—250°
л 1—2 мм давления; при повторном взаимодействии с хлор-
парафином в присутствии хлористого алюминия дает продукт,
по своим свойствам не отличимый от «парафлоу».
ТЕОРИЯ ДЕЙСТВИЯ ДОБАВОК ТИПА .ПАРАФЛОУ'
Теоретическая сторона действия «парафлоу» и ему подобных
добавок заключает в себе два самостоятельных вопроса».
769
а именно: во-первых, вопрос о том, в какой мере снижающее
, действие этих препаратов на температуру застывания масла свя-
зано с составом и строением добавки и каковы здесь другие
основные факторы; во-вторых, в чем заключается механизм сни-
жающего действия этих добавок.
Опытный материал по вопросу о зависимости действия до-
бавки на температуру застывания масла от состава и строения
добавки пока крайне ограничен. Все же здесь можно считать
экспериментально установленными некоторые основные руково-
дящие положения [32].
Прежде всего установлено, что снижение температуры засты-
вания масла вызывается добавкой лишь двухалкильного произ-
водного нафталина типа «парафлоу» и что, напротив, одноал-
кильные производные нафталина, как оказалось, таким свой-
ством не обладают. Это последнее положение было подтверждено
затем на таких специально приготовленных, правда неоднород-
ных, гомологах нафталина, как однозамещенные продукты кон-
.денсации нафталина с цетеном, CieH82, и*октадециленом, С18Н86,
т. е. на цетил-нафгалине (I) и октадецил-нафталине (II):
I. С10Н7 • С16Н88; II. С10Н7 • C1SH87.
Второе положение, также установленное опытным путем, за-
ключается в несомненном значении для синтеза добавок типа
«парафлоу» характера ароматического ядра, с которым связаны
указанные боковые цепи. Так например, опыт синтеза аналога
«парафлоу» на базе 1,4-метил-изопропил-бензола (цимола) и
хлорпарафина, т. е. углеводорода типа «парафлоу» с бензольным
.ядром вместо нафталинового, дал отрицательный резулвтат: по-
. лученный препарат не обладал свойством снижать температуру
.застывания масла. С другой стороны, однако, синтез аналога
«парафлоу» на базе дифенила, СбН5 ..CeHs, и хлорпарафина дал
положительный результат: полученный препарат по своему дей-
ствию на застывание минерального масла почти не отличался
-от «парафлоу» из нафталина [32].
Таковы немногие данные по вопросу о связи состава и строе-
ния добавок типа «парафлоу» с их способностью снижать темпе-
ратуру застывания минерального масла. Вкратце выводы по
этому интересному вопросу можно формулировать следующим
образом: препараты типа «парафлоу», вызывающие снижение
температуры застывания- минеральных маоел, представляют со-
бою высокомолекулярные жирноароматические углеводороды,
в которых ароматическое ядро состоит из двух бензольных ядер,
либо конденсированных по типу нафталина, либо связанных
между собою по типу дифенила. С этим ароматическим ядром
должны быть связаны, кроме того, по крайней мере два больших
радикала жирного .ряда. Остается невыясненным, какие арома-
тические системы, кроме нафталина и дифенила, способны да-
вать замещенные типа «парафлоу», а равным образом, каковы
строение и минимальный вес боковых групп, введение которых
.способно придать получаемой таким образом жирноароматиче-
760
ской системе способность вызывать стабильное снижение темпе-
ратуры застывания минерального масла.
Кроме состава и строения добавки, на снижение температуры
застывания масла оказывают влияние некоторые другие факторы
и прежде всего характер или тип испытуемого масла, тип и со-
держание в нем парафина, способ обработки масла и т. д.
Влияние этих факторов сказывается в том, что некоторые масла,
в частности, например, масла из грозненской парафинистой
нефти, или мало, или вовсе не восприимчивы к -парафлоу»,
тогда как бакинские масла (а именно: балаханские, сураханские,
биби-эйбатские и др.) показывают хорошую восприимчивость
к той же добавке: 0,5—1,0% «парафлоу» дают стабильное сни-
жение температуры их застывания на 14—is и выше. Повиди-
мому, малая восприимчивость масла к «парафлоу» в первую
очередь зависит от количественного содержания в” масле пара-
фина, так как разбавление слабовосприимчивого грозненского
масла бакинским дает смесь, способную удовлетворительно ре-
агировать на добавку «парафлоу» [33].
Для одного и того же масла снижение температуры застыва-
ния зависит от количества добавки: чем больше взято добавки,
тем больше снижение температуры застывания: это снижение,
впрочем, сильно отстает от роста количества добавки (в про-
центах). Так например, у одного американского машинною масла
с температурой застывания —1°,1 снижение этой температуры
от различных добавок «парафлоу» выразилось следующим обра-
зом:
От добавки 0,5% >п1рафлоу“.......на 1ГС
1-2% „ ..........17=С
„ . 4г-6% ...............„ 25ЭС
На практике ввиду соображений экономического порядка раз-
меры добавки «парафлоу» обыкновенно не превышают 1% от
веса масла.
Таковы основные факторы, оказывающие влияние на сниже-
ние температуры застывания масла под влиянием добавок типа
«парафлоу» и ему подобных. Вопрос о механизме этого явления
чрезвычайно сложен. Попытки подойти к его изучению с по-
мощью микрофотографии показали, что в некоторых случаях
явление может быть связано с изменением размера ‘кристаллов
парафина, выпадающих из масляного раствора. Так, небольшие
примеси стеарата алюминия или «парафлоу» уменьшают размеры
кристаллов, одновременно изменяя их форму, и температура за-
стывания масла понижается. Наоборот, в присутствии значитель-
ных количеств стеарата алюминия и триэтаноламина получаются
крупные кристаллы парафина, и температура застывания масла
повышается. Однако эти простые отношения чрезвычайно ослож-
няются явлениями адсорбции, которые несомненно имеют место
в условиях выпадения парафина из масла, и на сегодняшний
день вопрос о механизме действия добавок на температуру засты-
вания масла пока еще не может считаться окончательно выяс-
ненным.
761
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ ДЕПАРАФИНИЗАТОРОВ
Глубокая депарафинизация масел, достигаемая применением
холода и растворителей, хотя и позволяет резко снизить темпе-
ратуру застывания масла, тем не менее экономически мало вы-
годна и, кроме того, нередко сопровождается падением ряда
□весьма важных качеств масла, а именно: уменьшается индекс
вязкости масла, возрастает склонность его к коксообразованию,
увеличивается летучесть масла (следовательно, и его расход),
’снижаются масленистость масла и стабильность его к окисле-
нию. В связи с этим применение добавок — депарафинизаторов
(«парафлоу» и т. п.), небольшие количества которых способны
вызвать стабильное снижение температуры застывания масла
без ухудшения других его качеств, — приобретает все большее
и большее значение. Перед другими способами депарафинизации
масел метод добавок—депарафинизаторов — выделяется доступ-
ностью и дешевизной, позволяющими ставить вопрос о примени-
мости его не только к смазочным маслам, но даже к таким деше-
вым и массовым продуктам, как, например, дизельное топливо.
Г. Любриканторы
В этом разделе будут рассмотрены присадки, назначение ко-
•торых — повышение смазывающих свойств масел, иначе говоря,
присадки, которые мы объединяем под общим наименованием
любриканторы (от lubricants—смазочные масла).
Качество смазочного материала («смазки»), как известно,
определяется совокупностью целого ряда свойств, большая
часть которых уже была рассмотрена выше в различных главах.
Таковы, например, вязкость масла и ее изменение с температу-
рой («индекс вязкости»), температура вспышки, устойчивость
смазки к кислороду воздуха («окисляемость») и т. д. Кроме
этих общеизвестных характеристик, весьма существенных для
определения степени пригодности смазочного масла в работа
при данных условиях, всякая смазка должна обладать еще
'Одним весьма важным свойством, природа которого долгое время
оставалась в высшей степени загадочной. Свойство это получило
наименование масленистости или липкости (oiliness); оно харак-
теризует прилипаемость смазки к смазываемой поверхности, т. е.
по существу полностью определяет смазывающее действие дан-
ного смазочного материала. Присадки, повышающие масле-
нистость масла, будут объединены ниже под общим наименова-
нием собственно любриканторы.
Особое положение среди присадок к смазочным маслам за-
нимают некоторые вещества, обладающие способностью улуч-
шать вязкости свойства масла, а именно—повышать его ин-
декс вязкости. Такие присадки будут рассмотрены ниже отдельно
под наименованием велоситоры.
Описанию различных присадок к смазочным маслам должен
«быть предпослан краткий очерк смазочных материалов, их ка-
честв и применения.
'762
СМАЗОЧНЫЕ МАТЕРИАЛЫ И МАСЛА
По своему происхождению смазочные материалы разделяются"
на растительные, животные и минеральные.
Растительные и животные масла-и жиры, как известно, пред-
ставляют собою глицериновые эфиры (глицериды) высших кис-
лот жирного ряда, предельных (пальмитиновая, стеариновая),
непредельных (олеиновая) и более сложных по своему составуг
какова, например, рицинолевая кислота. Все эти масла характе-
ризуются способностью при гидролизе, протекающем особенно
легко в присутствии минеральных кислот и щелочей (обмыли-
вание), давать глицерин и свободную органическую кислоту. Так
например:
С8Нб(С18Н8Б02)з+ЗЩО = С8НБ(0Н)д + ЗС18Н86О2,
три стеарин вода глицерин стеариновая «-та
До половины прошлого столетия растительные и животные-
жиры и масла были единственными смазочными материалами.
Они применялись сначала даже для смазки подвижного состава,
железных дорог; так например, в Германии для этой цели упо-
требляли сурепное масло, во Франции, Бельгии и Англии —
кокосовое масло. Лишь в 60-х годах стали появляться в практике
смазочного дела минеральные масла, а именно — сначала камен-
ноугольные и сланцевые, а затем и нефтяные.
Растительные масла содержатся в семенах и плодах различ-
ных растений и получаются из них либо прессованием, либо-
экстракцией. Таковы, например, масла: касторовое—из семян
клещевины, рапсовое или сурепное — из семян сурепицы, льня-
ное— из льняного семени, хлопковое — из семян хлопчатника,
горчичное — из семян горчицы, оливковое—из плодов маслич-
ного дерева (оливки), пальмовое—из сердцевины кокосового ореха
и многие другие. Некоторые из этих масел являются прекрасным
смазочным материалом; таковы, например, масла касторовое, гор-
чичное, оливковое, находящие известное применение и в настоя-
щее время, главным образом в виде смесей с минеральными мас-
лами (компаундированные масла). Другие растительные масла
не могут применяться в качестве смазочных в виду их склон-
ности более или менее легко подвергаться аутоксидации, т. е.
поглощать кислород воздуха, с превращением в твердые эластич-
ные лаки: таково, например, льняное масло, принадлежащее
к типу высыхающих растительных масел, а также лолувысы-
хающие масла вроде хлопкового и др.
Животные масла и жиры получаются вытапливанием из туш
животных, наземных и морских (киты, тюлени, дельфины и т. п.).
Они отличаются от растительных масел более высокой темпера-
турой плавления и также часто представляют собою прекрасный
смазочный материал, который находит широкое применение-
главным образом при изготовлении компаундированных и кон-
систентных смазок. Таковы, например, говяжье, баранье и сви-
ное сало; говяжье и свиное масло — жидкий отжим от соответ-
ствующих сортов сала; спермацетовое масло — отжим от
768-
китового спермацета; тюленье и дельфинье масло — из соответ-
ствующей ворвани и др. Помимо дефицитности, большим не-
достатком этих смазочных материалов является их сравнительно
малая устойчивость в рабочих условиях, в результате чего на-
ступает их разложение (прогоркание) и даже осмоление.
Минеральные масла при современном масштабе применения
^смазочных материалов в различных видах промышленности пред-
ставляют собою основной материал этого рода. Получаются они
почти исключительно соответствующей переработкой масляных
мазутов и нефтей, а их сортимент в соответствии с разнообразием
их применения чрезвычайно велик. Ниже приводятся некоторые
.данные относительно наиболее важных видов минеральных сма-
зочных масел соответственно техническим нормам, принятым
в СССР, с указанием важнейших областей их применения [34].
Индустриальные масла, особо легкие и легкие машинные.
К этой группе относятся наиболее легкие смазочные масла
,с удельным весом от 0,860 до 0,912, низкими температурами за-
стывания (от —25 до —15°) и вспышки (120—175°), небольшой
'.вязкостью (Ебо = 1,3'-н 3,3). В прежнее время масла этого типа
«объединялись в одну группу веретенных масел, так как основ-
ным их назначением была смазка веретен в текстильной промыш-
ленности. Это свое назначение они сохранили и в настоящее
время; однако в овязи с широким развитием машиностроения
область их применения чрезвычайно расширилась. Так напри-
мер, масла типа велосит 0«Л» и «Т»), кроме смазки веретен,
ватеров и крутильных машин в хлопчатобумажном и шерстяном
производствах, нашли себе применение во многих других слу-
чаях для смазки трущихся частей легких механизмов с боль-
шим числом оборотов и легкой нагрузкой (небольшие шпиндель-
ные станки, контрольно-измерительные приборы и счетчики, лег-
кие трикотажные и швейные машины и т. п.); сепараторные
масла («Л» и «Т») в основном применяются для смазки под-
шипников легких и тяжелых сепараторов; швейное масло упо-
требляется для смазки швейных, вязальных и трикотажных ма-
шин; сюда же относятся веретенные масла («2», «3». «ЗВ»),
а также масло вольта «Л», применяемое главным образом для
.подшипников малой и средней мощности динамомашин и эле-
ктромоторов.
Индустриальные масла, средние и тяжелые машинные. Сюда
относятся более тяжелые смазочные масла (удельный вес
0,886—0,931) с температурой вспышки от 180 до 215° (Бр), зна-
чительной вязкостью (jE?5o = 4 -н 10 и £100 до 2,2) и с темпера-
турой застывания не выше 5°. Область применения масел этой
группы громадна. Таково, например, наиболее распространенное
масло машинное «Л», широко применяемое для смазки большин-
ства станков металлообрабатывающей промышленности, для
смазки подшипников и движения паровых машин мощностью до
500 л. с., для смазки цилиндров и движения маломощных нефте-
двигателей (не дизелей), наконец, в банкоброшах, чесальных
машинах и ватерах (текстильная промышленность). К машин-
7К4
ному «Л» примыкают машинное «ЛВ ,, машинное <:2», машин-
ное «2В»1 и машинное «Т», применяемые в аналогичных, н<?
более тяжелых условиях работы, например для сма-зки тяжелых
станков с небольшой скоростью движения и т. п.
Кроме машинных масел, к этой же группе относится наиболее
легкое масло группы — вольта «Т», применяемое для сма-зки
подшипников динамомашин и электромоторов средней и большой
мощности, делающих до 1 тыс. об/мин. а также наиболее тяже-
лые (вязкие) масла группы: морское масло, употребляемое глав-
ным образом для смазки подшипников паровых машин речных
и морских судов, и цилиндровое «2». применяемое как тяжелое
машинное масло для смазки механизмов, работающих с большой
нагрузкой и малой скоростью, во всех тех случаях, когда про-
исходит нагревание трущихся частей, а также "для смазки горя-
чих частей некоторых двигателей внутреннего сгорания, вклю-
чая и дизеля.
Специальные смазочные масла. Таковы турбинные масла
(«Л», «ЯМ» и «И»), компрессорные масла («Л», «М» и «Т») и
фригус.
Турбинные масла служат для смазки подшипников всевоз-
можных турбин. Это—тяжелые масла (удедьный вес
0,901—0.906) со средней вязкостью (Е50 = 2.9 ч- 4.5). средней
температурой вспышки (-180—185е) — и достаточно низкой тем-
пературой застывания (от —10 до —15°). Так как в условиях
работы турбинные масла находятся в постоянном соприкоснове-
нии с металлом и кислородом воздуха, т. е. в условиях способ-
ствующих окислению масла и образованию в нем осадка, то при
изготовлении турбинных масел обращается особое внимание на
их стабильность и возможно низкую начальную кислотность;
вместе с тем хорошее турбинное масло не должно давать устой-
чивых эмульсий с водой, так как образование таких эмульсий
нередко приводит к авариям турбин.
Компрессорные масла служат для смазки воздушных цилин-
дров компрессоров п воздуходувных машин, а масло фригус—
для цилиндров холодильных установок, работающих на аммиаке
и углекислоте. От турбинных масел компрессорные масла отли-
чаются широкими нормами удельного веса (0,891—0,926), боль-
шей вязкостью (Eso до 6.5—7.0; Еюо до 2,3—2,8). более высокой
температурой вспышки (до 240°) и, так как масло работает на
сжатие, отсутствием нормы на температуру застывания. Напро-
тив, фригус принадлежит к типу легких, маловязких масел
(удельный вес 0,876—0,896; Е5о = 2.0-~2,3) с низкой темпера-
турой вспышки (160°) и низкой застываемостью (—25 ).
Масла для двигателей внутреннего сгорания. К этой группе
масел относятся моторные масла («М» и «Т»), автотракторные
масла или автолы разных марок («6», «8». «10», «10В» и «18»)
и нигрол тракторный. Все это — масла с высоким удельным
1 Буква «В» в маркировке масла обозначает, что масло — выщелочен
ное.
765
весом (0,911—0,926), с высокой вспышкой (185—215° Бр.) и
вязкостью (для моторных масел и автолов Eso==5,5 — 18, для
нигрола еще выше, Е«о = 4,0 4,5), с достаточно низкой’тем-
пературой застывания (от 0 до —8°). Моторные масла приме-
няются главным образом для смазки цилиндров и движения ди-
зелей, автолы — для смазки автомобилей и тракторов, нигрол__
исключительно для залива коробки скоростей и диференциала
тракторов.
Авиационные масла. Авиамасла употребляются для смазки
двигателей авиационного типа. Для изготовления их приме-
няют специальные сорта сырья и наиболее совершенные способы
очистки, в частности сольвентные методы. Несколько уступая
автолам в отношении удельного веса (0,886—0,906), авиамасла
превосходят автолы своею вязкостью (Еюо = 2,6 -т- 3,7) и
вспышкой (225—230° МП, а для «экспортного брайтстока даже
275° Бр); они характеризуются также низкой застываемостыо
(от —10 до —35г). Весьма важное значение для характери-
стики авиамасла имеет его индекс вязкости. Согласно нашим
техническим нормам индекс вязкости различных авиамасел (по
Дину и Дэвису) колеблется от 92 до 98.
Масла для паровых машин. Различают цилиндровые масла
для машин, работающих насыщенным паром, и для мятпин,
работающих перегретым паром. Основные требования, предъя-
вляемые к маслам этой группы, сводятся к следующему: масло
должно обладать хорошей смазывающей способностью и легко
распыляться при подаче в цилиндры; вместе с тем оно должно
давать минимальное нагарообразование и не оказывать корро-
дирующего влияния на металлические поверхности.
Для машин, работающих насыщенным паром, наша нефтяная
промышленность рекомендует в зависимости от давления пара
(5—12 ат) следующие масла: отмеченное выше цилиндровое «2»,
вискозин «3» и нигрол «Л». Все они—тяжелые вязкие масла,
лишенные каких-либо механических примесей, а также воды и
минеральных кислот. Температура вспышки их выше 200°, т. е_
выше температуры насыщенного пара среднего давления. Наи-
более важной характеристикой этих масел является их вязкость
при повышенных температурах (Еюо от 1,8—2,2 до 5,о—7,0).
Для машин, работающих перегретым паром, рекомендуется
в зависимости от температуры перегрева (до и выше 350°) также
три масла: вапор «М», вапор «Т» и цилиндровое «6» — дестил-
лат. Это наиболее тяжелые и вязкие масла (Еюо = 4,5 — 6,0),
характеризующиеся соответственно условиям их работы наивыс-
шей температурой вспышки <290—320° Бр).
Кроме смазочных масел, широкое применение в качестве
смазочных материалов находят также смазочные мазуты, полу-
гудроны и гудроны.. Они применяются в железнодорожном транс-
порте для смазки вагонных букс, деревянных осей телег, повозок
и для смазки грубых частей многих сельскохозяйственных ма-
шин, как-то: сеялок, косилок, жаток, веялок и т. п.
Меньшее значение в качестве смазочного материала имеют
763
iio сравнению co смазочными Маслами вазелины естественный и
искусственный (ч. I, гл. V, IB), а также церезин. Они употре-
бляются 1греимуществено для изготовления разного рода спе-
циальных смазок, особенно предохранительных, каковы, напри-
мер, смазки ружейная, пушечная, аммуничная, снарядная
И ДР-
Консистентные мази. В тех случаях, когда вследствие конструктивных
особенностей машины смазочное масло не может долго удерживаться на
трущихся поверхностях, давно уже стали применять мазеообразные, полу-
твердые смазки, которые плавятся и превращаются в жидкость лишь при
повышенной температуре, в частности когда такое повышение происходит
в результате трения при движении машины. С остановкой машины, когда
температура трущихся поверхностей снижается, смазка вновь густеет и бла-
годаря этому удерживается на месте.
Консистентные мази изготовляются нагреванием смеси минерального
к. растительного масел с едким натром или гашеной известью, реже —
с едким кали. Образующиеся при этом мыла (загустители) вызывают загу-
стение смазки; степень же загустения (консистенция) смазки и ее качества
могут изменяться в весьма широких пределах в зависимости от выбора
сырья и способа его переработки, особенно от относительных количеств
минерального масла >и мыла, а также от природы разного рода наполни-
телей (мел, гипс, тальк, графит и т. п.), прибавляемых к смазке частью
для увеличения ее веса, частью для повышения некоторых ее технических
качеств.
Различают мази, растворимые в воде, и мази водоупорные. Первые
готовятся на натровых и калиевых мылах, вторые — на кальциевых, реже
ни магниевых, свинцовых и алюминиевых мылах. Примерами консистент-
ных маэей могут служить солидолы (тавот, мадия), паровозные смазки, бри-
кеты для прокатки, мази волковая, шариковая, канатная, колесная, гра-
нитная и др.
Масла специального назначения. Кроме смазочных масел,
нефтяная промышленность вырабатывает ряд масел специаль-
ного назначения. К таковым, например, принадлежат
трансформаторные масла, применяемые главным образом для
охлаждения сердечников трансформаторов и масляных реоста-
тов, нагревающихся при прохождении электрического тока.
Трансформаторные масла являются, таким образом, маслами
охлаждающими и изоляционными. Они изготовляются из лег-
кого масляного сырья с особо тщательной очисткой. Удельный
вес масла не больше 0,896; вязкость Eso не больше 1,8: вспышка
не ниже 140° МП. Особое внимание обращается на отсутствие
в масле влаги, а также на минимальную кислотность масла,
так как присутствие в масле воды и кислот резко снижает
диэлектрические свойства масла. От хорошего трансформатор-
ного масла требуется также, чтобы при продолжительном нагре-
вании (до 100 час. при 100—200°) оно не выделяло большого
осадка, так как последний, собираясь на дне трансформаторного
бака, может явиться причиной аварии’ (короткое замыкание).
К маслам специального назначения относятся также масла
парфюмерное и медицинское вазелиновое. Это — тоже легкие
масла высокой степени очистки, без запаха и вкуса. Удельный
вес их 0,875—0,890; вязкость Ебо = 2,5 н- 4,5. Применяются
соответственно для парфюмерных и медицинских целей.
7G7
Особое место среди смазочных материалов занимают масла, применяе-
мые при обработке металла; назначение, их — служить не только
смазочным, но и охлаждающим средством. Сюда относятся: а) режущие-
эмульгирующие масла, представляющие собою либо прозрачное масло
(эмулъсол), легко образующее с водой стойкие эмульсии, либо пасты сме-
танообразной консистенции (паста «Л» и паста «Т»), также легко эмуль-
гирующие с водой, и б) сульфофрезолы, представляющие собою осерненные
нефтяные масла. Эмульгирующие масла и пасты применяются в виде
эмульсии с водой для поливки инструмента при обработке металла реза-
нием, а также при обдирочных и шлифовальных работах. Сульфофрезолы
употребляются как таковые для поливки фрез и резцов фрезерных, ре-
вольверных, токарных, автоматических и тому подобных станков, когда
необходима особая тщательность обработки металла (работа по калибрам)
и когда желательно увеличить производительность станка путем увеличе-
ния скоростей.
ТРЕНИЕ И ЕГО ОСНОВНЫЕ ФАКТОРЫ
Общеизвестно, что всякое перемещение Друг относительно
друга соприкасающихся поверхностей каких-либо двух твердых
тел вызывает появление силы трения, касательной к поверхности
соприкосновения тел и препятствующей их взаимному переме-
щению. Различают следующие виды трения:
1. Сухое трение. Так называют трение между двумя соприка-
сающимися твердыми поверхностями, движущимися одна по
отношению к другой, если между ними совершенно отсутствует
какое-либо смазочное вещество. Сухое трение пропорционально
давлению между трущимися поверхностями и пе зависит ни от
велггчины этих поверхностей ни от скорости движения их друг
относительно друга (законы Кулона). При нормальном ходе ма-
шины сухое трение ввиду большого износа недопустимо за
исключением некоторых специальных случаев (тормоз и т. п.).
2. Жидкостное трение. Этот вид трения развивается в тех
случаях, когда между поверхностями двух тел, движущихся друг
относительно друга, находится слой смазывающей жидкости,
которая не допускает их непосредственного соприкосновения.
С практической точки зрения жидкостное трение представляет
собою наиболее совершенный вид трения: при нем износ тру-
щихся поверхностей отсутствует, расход энергии на трение резко
снижается по сравнению с другими видами трения, особенно
сухим трением, и наконец, температура трущихся поверхностей
повышается лишь незначительно. Естественно поэтому, что при
конструировании всякой машины одной из основных задач
является создание для трущихся поверхностей машины таких
условий, которые полностью обеспечили бы для них жидкостное
трение.
Между отмеченными двумя крайними видами трения суще-
ствуют промежуточные формы, а именно — трение полусухое и
полужидкостнов. При полусухом трении трущиеся поверхности
лишь в отдельных местах разделяются смазкой, в остальных ме-
стах они непосредственно соприкасаются между собой и поэтому
сильно изнашиваются. Полусухое трение допустимо лишь в ис-
ключительных случаях, например в моменты пуска ма-
768
шины и т. п. Полужидкостное трение более совершенно. Оно
характеризуется хорошей смазкой большей части трущихся по-
верхностей, так что лишь в отдельных местах наблюдается непо-
средственное их соприкосновение, т. е. сухое трение. Очевидно
поэтому, что и в данном случае имеется такой вид трения, с ко-
торым приходится лишь мириться ввиду тех или иных специаль-
ных условий (конструкция машины, малая скорость и т. п.).
Жидкостное трение подчиняется закону, который может быть
выражен следующей формулой (Я. Я. Петров, 1883 г.):
вдесь н— коэфициент трения — представляет собою отношение
силы трения к нагрузке; — абсолютная вязкость смазывающей
жидкости, иначе говоря, ее коэфициент внутреннего трения; v —
скорость трущихся поверхностей друг относительно друга; А —
толщина масляного слоя между трущимися поверхностями;
Р — нагрузка на единицу трущейся поверхности; Ai и А« —
коэфициенты трения между смазывающей жидкостью и поверх-
ностями трущихся тел (коэфициенты внешнего трения).
По отношению к поверхности данного твердого тела необхо-
димо различать жидкости, смачивающие эту поверхность и не
смачивающие ее. Первые быстро и равномерно растекаются по
поверхности твердого тела, образуя на ней сплошной, обыкно-
венно весьма тонкий слой, т. е. как бы смачивают ее; жидкости
второго рода собираются на поверхности твердого тела отдель-
ными каплями, т. е. не смачивают ее. Лишь жидкости первого
рода, да и то далеко не все, применяются в качестве смазочного
материала, так как, оказавшись между двумя поверхностями
твердого тела, они растекаются между ними тончайшим слоем.
Напротив, не смачивающие жидкости не сохраняются между
двумя сближенными поверхностями твердого тела, а легко выжи-
маются из пространства между ними нацело. По отношению
к железной поверхности смачивающими жидкостями являются
минеральные, растительные и животные масла, а также вода,
спирт и некоторые другие: типичным примером жидкости, не
смачивающей железную поверхность, является ртуть.
Опытные измерения показывают, что внешнее трение А», А«.
во много раз больше внутреннего тления смазывающей жид-
кости т]. Ввиду этого в приведенном выше выражении для коэ-
фициента трения у- сумма ?]/Xi -f- *п/А 2 представляет собою вели-
чину, настолько малую по сравнению с величиной h, что ею во
многих случаях можно пренебречь. Таким образом формула для
коэфициента трения р получает упрощенный вид:
Итак, в условиях жидкостного трения коэфициент у, во-пер-
вых, прямо пропорционален абсолютной вязкости смазывающего
* 469
49 Важ. 1622. Хижня нефти.
масла п и относительной скорости трущихся поверхностей v
во-вторых, обратно пропорционален нагрузке на единицу тру’
щейся поверхности р и толщине масляного слоя h. Из этих
переменных при жидкостном трении особенно важное значение
имеет абсолютная вязкость масла, в частности ее изменение
с температурой; к этому вопросу мы еще вернемся ниже в раз-
деле о велоситорах. Из других переменных величина скорости г
и нагрузки р определяется рабочими и конструктивными усло-
виями, заранее заданными для данного механизма; при устано-
вившейся его работе оии являются постоянными. Наконец, тол-
щина слоя масла h — величина наиболее сложная; она зависит
не только от конструктивных особенностей данного механизма.
иг. 133. Влияние скорости вращения вала и на-
грузки на коэфициент трения.
Опытное исследование зависимости р от вышеуказанных фак-
торов приводит к весьма показательным результатам. Если на
оси ординат отложить величины коэфициента н а на оси
абсцисс — одну из независимых переменных (*n, v или р). то при
условии постоянства двух остальных изменение у приобретает
вид крайне характерной кривой. Так например, для изменения р
в зависимости от скорости вращения вала в кольцевом подшип-
нике при трех различных нагрузках [р = 1} р = 2 и
Р = 8 кг/см2] были получены три кривых, изображенных на
фиг. 133 [35].
Ближайшее рассмотрение кривых фиг. 133 показывает, что
с уменьшением скорости вращения коэфициент трения и посте-
пенно убывает, достигает некоторого минимума и начинает далее
быстро возрастать. Очевидно, что изменение коэфициента тре-
ния у в соответствии с законом жидкостного трения наблюдается
в данном случае лишь для скоростей, не ниже определенного
минимума, иначе говоря, лишь для правых половин кривых
770
фиг. 133; незадолго до перегиба этих кривых коэфициент трения
Начинает изменяться по новому закону, после же их перегиба
он начинает быстро возрастать с уменьшением скорости враще-
ния вала. Физическая сущность этого явления заключается
в следующем.
При некоторых небольших скоростях вращения вала масля-
ная пленка смазки начинает разрываться, вследствие чего жид-
костное трение постепенно начинает переходить в полужидкост-
ное. Наступает момент, соответствующий минимальному значе-
нию р для данного механизма. При дальнейшем уменьшении
скорости вращения вала полужидкостное трение скоро превра-
щается в полусухое; естественным же следствием такого превра-
щения должно явиться быстрое -возрастание коэфициента р.
При жидкостном трении громадное значение на величину и
оказывает вязкость смазочного масла; в процессе полужидкост-
ного трения роль вязкости отступает на задний план, главное же
влияние приобретает особое свойство смазки — ее масленистость,
от которой зависит прочность масляной пленки между трущи-
мися поверхностями.
Кривые фиг. 133 иллюстрируют также зависимость коэфициента трения
от давления на единицу трущихся поверхностей, а именно — снижение
коэфициента р< по мере возрастания давления при одних п тех же ско-
ростях вращения. Что касается зависимости от вязкости смазки, то со-
ответствующие кривые имеют примерно такой же вид, как кривые фиг. 133.
Что же касается зависимости коэфициента н от толщины масляного слоя, то
ета зависимость оказывается значительно более сложной, чем ©того тре-
бует вышеприведенная формула, так как величина слоя является также
функцией скорости вращения и других переменных (ср. выше).
МАСЛЕНИСТОСТЬ И ЕЕ ФИЗИКО-ХИМИЧЕСКАЯ ПРИРОДА
Как было отмечено выше, смазывающим действием обладают
лишь такие жидкости, которые смачивают данные трущиеся по-
верхности. С другой стороны, давно известно, что смачивание
находится в тесной связи с явлениями поверхностного натяже-
ния и капиллярности. Отсюда понятно, что причину смазываю-
щего действия некоторых жидкостей давно уже пытались увя-
зать прежде всего с явлениями поверхностного натяжения [зп].
Не подлежит сомнению, что явления капиллярности, в част-
ности форма мениска, образуемого данной жидкостью, являются
одним из основных факторов смазывающего эффекта: ведь
именно капиллярные силы заставляют смазку проникать
в самые узкие зазоры между соприкасающимися поверхностями
и смачивать их. С другой стороны, как показывает опыт, луч-
шими смазывающими свойствами обладают масла с наименьшим
поверхностным натяжением, и наоборот, жидкости, хотя бы и
вязкие, но обладающие большим поверхностным натяжением,
мало или вовсе не пригодны в качестве смазочного материала;
таковы, например, антраценовые масла, меласса, сульфитные ще-
лока и т. п. Естественно таким образом, что, определяя поверх-
ностное натяжение данного масла на границе с определенной
49*
металлической поверхностью, можно составить представление
о степени пригодности этого масла в качестве смазочного мате-
риала для данной поверхности.
Существует немало методов для косвенного определения по-
верхностного натяжения на границе твердое тело — жидкость;
описание этих методов можно найти в соответствующих главах
того или иного курса физики. Весьма ценные и нередко вполне
достаточные указания для оценки различных смазочных масел
можно получить также более простым путем, а именно — сопо-
ставлением поверхностного натяжения масел на границе масло —
вода с помощью весьма простого и удобного способа взвешива-
ния капель (ч. I, гл. II). Так как поверхностное натяжение
масла по отношению к воде обратно пропорционально числу ка-
пель, подсчитываемых по этому способу, то, принимая поверх-
ностное натяжение одного из масел за 100, легко получить ряд
чисел, характеризующих поверхностное натяжение различных
масел на границе с водой. В табл. 164 приведены подобные
характеристики для некоторых минеральных и растительных
масел.
Таблица 164
Поверхностное аатяженпе на границе масло — вода
(по Трилъя)
Масла Число капель Поверх- ностное
Минеральные масла
Парафиновое . . . ...............
Соляровое.........................
Маловязкое нейтральное ..........
Растительные масла
Оливковое .......................
Сурепное ........................
Кокосовое .......................
95
102
99
100
93
95
132
138
161
72
68
59
Как видно из данных табл. 164, на границе с водой поверх-
ностное натяжение растительных масел значительно ниже по-
верхностного натяжения минеральных масел; эта особенность
растительных масел не может не быть сопоставлена с другим
давно известным их качеством — более высокой их смазывающей
способностью.
Другое важное наблюдение, позволившее ближе подойти
к уяснению природы смазывающей способности различных ма-
сел, заключается в следующем. Было замечено, что свежие, хо-
рошо очищенные растительные и животные масла обладают
меньшей смазывающей способностью, чем масла менее свежие,
772
например, прогоркшие. Как известно, прогоркание этих масел
(жиров) вызывается частичным их гидролизом и сопровождается
появлением в них свободных жирных кислот. С другой стороны,
непосредственный опыт показывает, что примесь к жирам свобод-
ных кислот вызывает снижение их поверхностного натяжения на
границе с водой. Таким образом естественно было притти к прак-
тически весьма важному выводу, что для повышения качества
смазочных масел полезно прибавлять к ним небольшие количе-
ства свободных жирных кислот. В табл. 1G5 приводятся некото-
рые данные, иллюстрирующие эффект, производимый неболь-
шими добавками высокомолекулярных жирных кислот на сниже-
ние поверхностного натяжения по границе масло —вода, что
в свою очередь, как было отмечено выше, неизменно сказывается
на повышении смазывающей способности масла.
Таблица 166
Влияние жирных кислот на поверхностное натяжение по границе
масло— вода
(ео Трилъя)
Масла Примесь1 жирных кислот в % Число капель Поверх- ностное натяжение
Минеральное (0,903) 0 101 100
1.9 125 80
28 130 78
Оливковое 0.1 110 92
2.2 125 80
4,5 140 73
Сурепное 015 108 93
н 2,5 132 76
Естественно возникает вопрос, в чем же кроется причина
столь характерного влияния примеси жирных кислот на смазы-
вающие качества масел? Чтобы ответить на этот вопрос, потре-
бовалось углубленное исследование тех соотношений, которые,
как оказалось, возникают между молекулами смазки, с одной
стороны, и смазываемой поверхностью — с другой. В основе этих
новейших представлений о физико-химической сущности дей-
ствия смазочных веществ лежит химическая теория поверхност-
ного натяжения и адсорбции.
Явления поверхностного натяжения и адсорбции и теперь
еще нередко расцениваются как явления чисто физические. Тем
интереснее, что именно со стороны физики раздался авторитет-
ный призыв к рассмотрению явлений этого рода с химической
точки зрения (Лангмюир). Согласно этих представлений все
междуатомные и междумолекулярные соотношения тел должны
рассматриваться с точки зрения проявления одних и тех же сил
1 Выражено в процентах олеиновой кислоты
778
химического сродства, которые обозначаются обыкновенно тер-
минами -основная и дополнительная валентность. С особой отчет-
ливостью и плодотворностью эта идея была применена к явле-
ниям смачивания, адсорбции и смазки трущихся поверхностей.
Представим себе, что небольшое количество растительного
масла, например оливкового, или высокомолекулярной органиче-
ской кислоты, например олеиновой, помещено на поверхности
воды. Масло быстро распространяется по этой поверхности и
покрывает ее тончайшим слоем, пленкой. Измерения показывают,
что наименьшая толщина такой пленки равна длине молекулы
вещества, образующего данную пленку. Для триолепа это соста-
вляет около 1,3 10~7 см, для олеиновой кислоты — около
1,1 • 10~7 см. Таким образом в данных условиях образуются мо-
помолекулярные поверхностные пленки; естественно, возникает
вопрос, каким же образом располагаются в этих пленках обра-
зующие их молекулы как по отношению друг к другу, так и по
отношению к поверхности воды.
Известно, что в противоположность углеводородам низшие
спирты и кислоты легко растворяются в воде. Так как взаим-
ной растворимостью обладают лишь вещества, родственные друг
другу по своей химической природе, то, очевидно, причину рас-
творимости в воде простейших спиртов и кислот следует видеть
во взаимодействии молекул воды с гидроксильными группами
спирта или карбоксилами кислоты. Правда, по мере повышения
молекулярного веса растворимость спиртов и кислот постепенно
снижается; очевидно, однако, такое снижение вызывается встреч-
ным возрастающим влиянием углеводородных остатков, которые
соответственно крайне незначительной растворимости в воде
углеводородов не обладают сколько-нибудь заметно выраженным
сродством к воде. Распространяя эти представления с целых
молекул на их части, легко получить картину построения моле-
кулярной пленки на поверхности воды, например из олеиновой
кислоты: молекулы кислоты должны быть здесь строго ориенти-
рованы, а именно — активные группы, в данном случае карбо-
ксилы, обращены внутрь и даже погружены в воду вследствие
ярко выраженного их притяжения молекулами воды, тогда как
углеводородные остатки держатся над поверхностью 'воды, обра-
зуя на ней нечто вроде ворса. Такая структура в настоящее
время хорошо изучена на нерастворимых в воде веществах, на-
несенных на ее поверхность, и может считаться совершенно не-
сомненной. Применение этих представлений к явлениям смачи-
вания твердых поверхностей и смазки дало вполне отчетливую
картину этих явлений и составило эпоху в их разъяснении.
Если небольшое количество олеиновой или расплавленной
стеариновой кислоты поместить на полированную металлическую
пластинку, покрыть эту пластинку другой такой же стеклян-
ной и, нажимая на них, заставить их перемещаться относи-
тельно друг друга взад и вперед, а затем разъединить их путем
скольжения, то на каждой пластинке останется тонкий слой
кислоты. Рентгеновский анализ показывает, что молекулы кис-
774
io гы в таком слое уже ориентированы согласно фиг. 134, если
представить каждый такой слой мопомолекулярным. Здесь моле-
кула кислоты представлена кружком с прямой чертой; кружок
условно обозначает карбоксил, черта — связанный с карбокси-
лом радикал. Таким образом и здесь на каждой поверхности
твердого тела получается, как и выше на поверхности воды,
своеобразный молекулярный ворс (частокол Лангмюира)
Как показывает фиг. 131, молекулы кислоты располагаются
своими продольными осями перпендикулярно к трущимся по-
верхностям. Активные группы, в данном случае карбоксилы,
располагаются в непосредственном соприкосновении с металли-
ческой поверхностью и прочно удерживаются (адсорбируются)
последней, очевидно за счет дополни-
тельного сродства карбоксильных
групп; связанные же с этими группами
радикалы, например СпНзэ для стеари-
новой кислоты и СпНзз — для олеино-
вой, образуют между металлическими
поверхностями нечто вроде своеобраз-
ного молекулярного ворса, который
разделяет металлические поверхности
и не допускает их непосредственного
.^прикосновения. Таким образом сколь-
жение происходит не между металли-
ческими поверхностями, а между око-
пшпшп
1ШШШШ
жиж
«I иг. 134. Ориентация ак-
тивных молекул между
трущимися поверхностями.
т. е. между отдельными
нечностями ориентированных молекул,
ворсинками, которые, зацепляясь друг за друга, и являются
причиной трения при перемещении одной металлической поверх-
ности относительно другой.
В свете этих представлений становится понятным повышение
смазывающих качеств масла от прибавления к нему небольшого
количества жирных кислот. Действительно, молекулы нейтралы
ного масла, например масла, состоящего из смеси углеводородов,
не ориентируются на металлической поверхности, так как такое
масло не содержит активных (полярных) групп; вследствие этого
в данном случае, очевидно, нет оснований к образованию той
прочной пограничной пленки, которая препятствует непосред-
ственному соприкосновению двух металлических поверхностей
даже при высоком давлении на них. Напротив, если к ней-
тральному маслу добавить немного высокомолекулярной жирной
кислоты, то молекулы последней быстро ориентируются между
металлическими поверхностями и создают на них те погранич-
ные слои, по которым и происходит скольжение. Отсюда выте-
кает важный вывод, что смазывающая способность, иначе го-
воря, масленистость, определяется степенью легкости, с которой
молекулы смазочного материала ориентируются на соприкасаю-
щихся поверхностях.
Иллюстрацию этих отношений дает фиг. 135. Здесь на оси
абсцисс нанесены проценты активного вещества и растворителя,
например олеиновой кислоты, и нейтрального масла; на оси
776
ординат—коэфициент трения. Характер кривой ясно показы-
вает, что уже при небольшой концентрации активного вещества
коэфициент трения резко снижается; дальнейшее же увеличение
этой концентрации не вызывает существенного изменения коэ-
фициента трения. Таким
образом роль нейтраль-
ного масла сводится здесь
к роли растворителя для
активного вещества.
Большой интерес пред-
ставляет зависимость ко-
эфицнента трения от хи-
мической природы актив,
ных молекул смазки. Хо-
тя большинство органиче-
ских веществ (кислоты,
спирты, в известной мере
даже углеводороды) явля-
ются в данном отношении
активными, однако степень той активности, естественно, может
быть глубоко различна в зависимости как от химического ха-
рактера активной группы, так и от величины ее молекулы. За-
висимость смазываюшего действия от величины активной моле-
кулы в одном гомологическом ряду выражается простой законо-
мерностью, которая
фиг. 136
[Активы,
малеоилы
Фиг. 135. Загисимость коэфициента тре-
ния от концентрации активного вещества.
может быть иллюстрирована графиком
50 ЮО !50 200 250 300 350 400 450 500550 600
Молекулярный бес
Фяг. 136. Зависимость коэфициента трения от вели-
чины активной молекулы.
Здесь на оси абсцисс отложены молекулярные веса активных
молекул, на оси ординат — коэфициенты трения у, так что ка-
ждая точка на графиках выражает коэфициент трения для со-
ответствующего представителя того или иного гомологического
ряда: жирных кислот (I), спиртов (II) и парафинов (III). Полу-
ченные точки, как видно, располагаются для каждого ряда на
одной прямой и притом таким образом, что при переходе к ка*
776
ждому ближайшему члену ряда, т. е. при увеличении веса мо-
лекулы на группу СН?, коэфициент трения снижается на одну
и ту же величину.
Интересно также проследить наклон трех прямых фиг. 136.
Для жирных кислот имеем прямую, круто спускающуюся вниз,
что соответствует быстрому повышению их активности с увеличе-
нием молекулярного веса в этом ряду. Для спиртов получается
кривая, значительно более пологая, что соответствует значи-
тельно меньшей активности высших представителей этого ряда,
сравнительно с кислотами. Наконец, для парафинов, наименее
активного ряда из трех данных гомологических рядов, полу
чается наиболее пологая кривая.
Наглядное объяснение благоприятного влияния величины
активных молекул на их смазывающее действие можно получить,
если две трущиеся поверхности, отделенные молекулярными
слоями ориентированных молекул смазки, уподобить двум щет-
кам, соприкасающимся одна с другой своей щетиной. Чем длин-
нее щетина, тем легче осуществить смещение таких щеток отно-
сительно друг друга, так как с увеличением длины волоса воз-
растает его способность к деформации и наклону. Аналогичной
гибкостью несомненно обладают также молекулярные цепи, при-
чем гибкость эта, очевидно, должна возрастать с увеличением
длины цепи, а вместе с тем должна увеличиваться и легкость
перемещения относительно друг друга тех поверхностей, на ко-
торых данные цепи прочно ориентированы.
Новейшие исследования показывают, что коэфициент трения
определяется аддитивно из следующих переменных: величины Ь,
вависящей от природы трущихся поверхностей, величины d, за-
висящей от природы активной группы смазки, и величины пс.
определяемой природой гомологического ряда, к которому при-
надлежит активное вещество смазки с числом п углеродных ато-
мов в цепи молекулы этого вещества. В итоге получается про-
стая формула, которая позволяет вычислить коэфициент трения
для данных трущихся поверхностей при различных смазках:
р. = Ь — d — пс,
где у — коэфициент трения (Гарди).
Слой смазки между двумя трущимися поверхностями рассма-
тривался выше преимущественно как слой молекулярный: один
слой ориентированных молекул прочно удерживается (адсорби-
руется) одной трущейся поверхностью, другой слой — другой
поверхностью. Практически после образования таких молеку-
лярных слоев при достаточном количестве активного вещества
в смазке остается еще достаточно активных молекул, которые,
естественно, также стремятся ориентироваться под влиянием
окружающего поля сил. В итоге происходит образование двух но-
вых слоев ориентированных молекул, плотно прилегающих к пер*
вым слоям, причем вследствие взаимного притяжения углеводо-
родных остатков молекулы второго слоя должны быть ориенги-.
рованы не так, как ориентированы молекулы первого слоя*
тп
а в противоположную сторону. На вторые слои могут налагаться
третьи слои молекул с той же ориентацией, как первые слои,
и т. д. Таким образом смазка приобретает между двумя трущи-
мися поверхностями сложную структуру с периодической ориен-
тацией молекул активного вещества в направлении, перпендику-
лярном к поверхности твердого тела. Активные группы обоих
первых слоев прочно адсорбируются твердыми поверхностями;
активные группы второго слоя прочно удерживаются активными
группами третьего слоя и при скольжении остаются относительно
друг друга неподвижными. Таким образом плоскостями сколь-
жения являются слои, соприкасающиеся друг с другом наименее
активными группами, например группами СНз.
В тех случаях, когда активное вещество растворено в мало-
активном, также происходят ориентация активных молекул и их
адсорбция на поверхности трущихся тел. Между образовавши-
мися таким образом двумя адсорбционными слоями (ворс или
частокол) могут разместиться прежде всего последующие слои
активного вещества, а затем слой слабо или вовсе не ориентиро-
ванных молекул масла (сольватный слой). При невысоких да-
влениях такой слой может достигать значительной толщины в
играть большую роль, уменьшая износ и расход энергии на тре-
ние. При высоких давлениях, когда ббльшая часть смазки вы-
давливается из зазора, исключительное значение приобретают
ориентированные активные молекулы, прочно адсорбированные
трущимися поверхностями и составляющие с ними как бы одно
целое.
СОБСТВЕННО ЛЮБРИКАНТОРЫ И ИХ ПРИМЕНЕНИЕ
Как ясно из вышеизложенного, добавка некоторых активных
веществ к смазочным маслам усиливает их смазочное действие
(масленистость) за счет строгой ориентации активных молекул
с адсорбцией их трущимися поверхностями и с образованием
ряда параллельных слоев таких молекул, препятствующих не-
посредственному соприкосновению твердых поверхностей и обра-.-
зеванию сухого трения. Такие активные вещества были названы
выше любриканторамщ точнее — собственно любриканторами.
Некоторые из любрикйнторов, например олеиновая кислота, из-
вестны и находили применение уже давно, задолго до того, как
был разъяснен механизм их действия число их особенно воз-
росло и продолжает возрастать за последнее время в связи
с прогрессом машиностроения и ростом мощностей, рабочих на-
грузок, скоростей и температур в машинах самых разнообраз-
ных назначений. В связи с этим все большее и большее значе-
1 Вот рассказ, который мне пришлось слышать от одного старого ра-
ботника водного транспорта. На Волге, под Казанью, находятся перекаты,
которые нередко бывали не под силу буксирам, перегруженным большим
караваном баржей. Выход из такого положения был известен давно: ста-
новились на якорь и посылали нарочного в Казань на местный мыловарен-
ный завод за олеиновой кислотой. Надлежащая добавка этой кислоты
к смазочному маслу резко повышала эффективность работы машины, я
буксир легко преодолевал встреченное им на пути препятствие.
*78
вне приобретают специальные смазки для сверхвысоких давле-
ний или Е. Р. смазки (Extreme Pressure Lubrikants).
Условия работы некоторых частей современных машин чрез-
вычайно жестки. Известно, например, что давления, которые
приходится выдерживать зубьям гипоидной шестерни автомо-
биля, достигают 25 000—29 000 кг/см2 и выше. В этих условиях
всякое чисто минеральное масло должно нацело выжиматься
из пространства между трущимися поверхностями, в результате
чего может появиться сухое трение со всеми его последствиями:
чрезмерным повышением температуры, свариванием отдельных
неровностей (пик) трущихся поверхностей, образованием между
ними «мостиков», заеданием и усиленным износом тех же поверх-
ностей. Во избежание всех этих крайне губительных для машины
последствий к минеральному смазочному маслу прибавляются
те или иные любрикапторы, подбор которых до сих пор произво-
дится в значительной степени чисто эмпирическим путем.
В качестве любриканторов на сегодняшний день изучено
громадное количество различных веществ как неорганических,
так и органических, причем изучению подвергались как проч-
ность образующихся пограничных пленок и их эффективность,
гак и влияние их на износ машины.
Из неорганических веществ в качестве любрикантора осо-
бенно широкое признание получила элементарная сера. Изуча-
лись также минеральные кислоты в различных концентрациях
и разные газы, мало растворимые в маслах и способные созда-
вать между трущимися поверхностями газовое давление; таковы
углекислота, аммиак, сернистый газ, сероводород и др. Очень
эффективным оказалось введение небольших количеств сероводо-
рода (до 0,005%), показавшего себя примерно в 100 раз более
активным, чем элементарная сера.
Подробно изучались в качестве возможных любриканторов
также различные классы органических соединений, как-то:
спирты, кислоты, их соли и сложные эфиры, простые эфиры, ке-
тоны, нитросоединения и амины, галоидопроизводлые, а также
производные серы и фосфора.
Из галоидопроизводных поразительный эффект дает добавка
к маслу уже 1% четыреххлористого углерода; однако лету-
честь этого продукта, повидимому, исключает возможность его
широкого применения. Большое значение приобрел хлорирован-
ный парафин, получаемый непосредственным действием на па-
рафин хлора при 70° с последующей промывкой полученного
продукта водой и едким натром.
широкое применение в качестве любрикантора приобрел
метилдихлорстеарат (метиловый эфир дихлорстеариновой ^кис-
Я0ТЫ)' СН, • (СН2)15 • СС12 • C00CHs.
Автомобильные масла с этой присадкой в количестве 1,5% от
веса масла имелись в продаже В США уже в 1935 г. и в резуль-
тате широко поставленных испытаний (4,8 млн. км пробега, из
т
которых организованного и изученного около 1,6 млн. км), заре-
комендовали себя с лучшей стороны.
Осерненные масла получают, вводя в минеральное, расти-
тельное и животное масла элементарную серу в количестве от
0,5 до 1,5—2,0%. Получаемые таким образом масла весьма
активны, но сильно корродируют аппаратуру. Химически свя-
занная сера менее активна, но зато не корродирует в широких
температурных пределах (до 100° и выше). Для получения такого
рода масел можно пользоваться нефтями, богатыми серой (до
2% S). Наибольшей эффективности достигают, добавляя к осер-
ненным маслам свинцовое мыло. Присадка серы, иногда вместе
со свинцовым мылом, получила наиболее широкое распростране-
ние на практике.
Из многочисленных новейших любриканторов одним из наи-
более удачных является трикрезил фосфат. Особенно хорошие
результаты дает смесь 5% осерненного жирного масла с 3%
трикрезил фосфата.
Итак, действие собственно любрикантора сводится к преду-
преждению ряда крайне тяжелых последствий, которые несет
за собой появление на некоторых участках трущихся по-
верхностей сухого трения. Одной из таких функций любрикан-
тора является предупреждение сваривания трущихся поверхно-
стей на их отдельных участках, так что смазка играет в данном
случае роль «антифлюса». Очевидно, что действие любрикантора
проявляется в условиях контакта, определяемого сферой моле-
кулярного взаимодействия, и что, как всякое взаимодействие
молекул, т. е. всякая реакция между ними, действие любрикан-
тора должно носить явственно специфический характер. Отсюда
прежде всего следует, что нет и не может быть универсального
любрикантора и что присадка, вполне пригодная и полезная
в одних случаях, может оказаться в других случаях не только
непригодной или бесполезной, но даже вредной. Необходимо
также иметь в виду, что применение любриканторов, определяе-
мое вышеуказанными условиями, ограничено областью высоких
давлений и нагрузок, когда вследствие крайнего сближения тру-
щихся поверхностей масляной пленке все время угрожает опас-
ность разрыва. Только в этих условиях и проявляется замеча-
тельное свойство любриканторов: укреплять масляную пленку и
почти мгновенно (в тысячные доли секунды) восстанавливать ее
в случае ее нарушения. С другой стороны, следует помнить, что
в условиях жидкостного трения, когда трущиеся поверхности
отстоят друг от друга довольно далеко, а давление между ними
не достаточно велико для проявления молекулярных сил и
ориентации активных молекул смазки, применение любрикан-
торов совершенно бесполезно; мало того, при значительной хи
мической активности любриканторов они могут оказаться не-
редко даже вредными, так как, разъедая металл, они загрязняют
масло продуктами коррозии, которые шлифуют металлические
поверхности и повышают степень их износа. Все указанные осо-
бенности действия собственно любриканторов являются для них
760
в высокой степени характерными и определяют собой ту их
специфичность, которая весьма существенно отличает их от
других масляных присадок.
ВЕЛОСИТОРЫ И ИХ ПРИМЕНЕНИЕ
Получение высоковязких смазочных масел с пологой кривой
вязкости в зависимости от температуры, т. е. с высоким инде-
ксом вязкости (90—100), представляет собою одну пз важнейших
задач современного производства минеральных смазочных ма-
сел, связанную с быстрым прогрессом моторостроения и широким
использованием авиа- и автомоторов в условиях низких темпе-
ратур. Между тем, производство такого рода масел сопряжено
со значительными трудностями и нередко приводит к резкому
снижению выходов на смазочное масло. За последние годы,
однако, в разрешении этой задачи достигнуты выдающиеся
успехи принципиально совершенно новым путем, а именно по-
казано, что повышение вязкости масла и улучшение ее индекса
легко достигаются путем прибавления к маслу небольших коли-
честв специальной высокомолекулярной присадки.
Углеводороды, обладающие чрезвычайно высокой вязкостью,
известны давно. Таков, например, углеводород, получаемый взаи-
модействием псевдокумола (2 мол) с аллиловым спиртом (1 мол)
в присутствии серной кислоты по уравнению [37]:
СН8
2CJUCIU+сщ=сн • сщон (СНД&Щ - с - сданд,
СН3
Несмотря на свой сравнительно небольшой молекулярный
вес (280)-, этот углеводород обладает громадной вязкостью (775
при 15° по отношению к воде).
С другой стороны, как было отмечено в ч. I (гл. II), вязкость
смеси двух масел всегда может быть вычислена из вязко-
стей компонентов либо с помощью таблиц, составленных, правда,
на основе чисто эмпирических данных, либо с помощью спе-
циальных графиков. При этом, как оказывается, хотя вязкость
смеси двух масел различной вязкости всегда меньше, чем того
требует расчет по правилу смешения, тем не менее добавка ком-
понента с большей вязкостью всегда вызывает повышение вяз-
кости другого менее вязкого компонента. Таким образом вопрос
об улучшении вязкостных свойств масла методом присадок сво-
дится к получению такой присадки, велоситора, который был
бы приемлем для данного назначения не только с технической,
но и с экономической точки зрения. В высокой степени удачно
разрешила эту задачу американская фирма Standard Oil Com-
pany of New Jersey выпуском на рынок особого высоковязкого
масла «паратона».
«Паратон» представляет собою прозрачное желтоватое масло
весьма вязкой консистенции; состав его приближается к фор-
муле CwH2m. В табл. 166 приведены важнейшие константы этого
масла [38].
781
Свойства „паратона*
Таблица 7б<г
Уд. вес <f420 Рефракция при 20° Вязкость Вспышка по Бр. Застыва- ние Иодное число Зольность Кокс по Конрад- сону Индекс вязкости по Д. и Д. Молеку- лярный вес
Esc
0,8842 1,4892 Е65 74 234 — 12 6.0 0,0009 0,107 122 518
Изучение влияния, которое оказывает добавка «паратона»,
было проведено на самых разнообразных маслах и привело
в основном к следующим результатам. Не оказывая заметного
влияния на большинство свойств масла, добавка «паратона» по-
вышает вязкость масла и увеличивает его индекс вязкости; при
этом, чем ниже индекс вязкости исходного масла, тем эффектив-
нее сказывается влияние «паратона».' Смешение масел с «пара-
тоном» ‘подчиняется законам смешения минеральных масел;
в частности, увеличение индекса вязкости масла зависит исклю-
чительно от первоначального индекса вязкости масла и вели-
чины добавки к нему «паратона». Так например, добавка 10%
«паратона» к автолу повышает его индекс вязкости на 40—50
единиц и обеспечивает автолу индекс вязкости выше СО; та же
добавка «паратона» к авиационному маслу повышает его индекс
вязкости на 20—25 единиц, обеспечивая авиамаслу индекс вяз-
кости выше 100.
Интересные результаты получены при разгонке «паратона»
в глубоком вакууме [39]. Оказалось, что около 80% «паратона»
могут быть от него /гегко отогнаны в виде масла типа машинного
с умеренной вязкостью (Eso — 5,3). В остатке от такой разгонки
получается прозрачный, смолообразный продукт темнокрасного
цвета; после промывки эфиром это вещество светлеет и приобре-
тает янтарный цвет. Оно может быть выделено из «паратона»
также без перегонки путем разбавления «паратона» эфиром.
Этому веществу присвоено наименование эксанол.
Эксанол, как показало его изучение, обладает колоссальной
вязкостью: при пересчете с кинематической вязкости на вяз-
кость по Энглеру ориентировочно получена величина Еюо по-
рядка 9600. Очевидно, таким образом, эксанол является носите-
лем вязкости «паратона». Ближайшее изучение его химической
природы представляет, естественно, выдающийся интерес.
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ ЛЮБРИКАНТОРОВ
Практическое значение любриканторов и их применение для
улучшения качества смазочных масел уже были освещены
в предшествующем изложении. Повышение масленистости и
улучшение вязкостных свойств масел — все это задачи первосте-
пенной важности, и разрешение этих задач методом присадок
782
является одним из наиболее блестящих достижений новейшей
химии и технологии нефти. Практическое значение этих дости
жений громадно, так как в данном случае удается весьма зна-
чительно увеличить ресурсы высококачественных масел и одно
временно значительно упростить методику их получения
ЛИТЕРАТУРА
Наш А. и Хоуэс Д., Принципы производств! и П1имененпя моторных топ
лив, т. II, пер. с англ, под ред. Седых Я. Ф., ГОНТИ, 1938.
Марек А.Ф. н Ган Д. А., Каталит-ческое окисление органических соеди-
нений, пер. с англ., ОНТИ, 1936.
Маслен ников М. М., Сгорание и детонация, ОНТИ, 1933.
Еланский В_. И., Детонация топлив и способы се измерения, ОНТИ, 1933.
Каличевский В. А. и Стайнер Б. А., Химическая очистка нефтег родуктов
пер. о англ., ОНТИ, 1936.
Арчбктт А. и Диллеи Р. М., Трение, смазка и смазочные материалы, п-р-
с англ, под ред. Мананеева Ф. Я., ОНТИ, 1934.
Klemgard Е. N., Lubricating Greases, their Manufacture and Use, New York
1937.
The Science of Petroleum, v. II, London — New-York — Toronto 1938.
1. Egloff, Morrel, Lowry, Dryer, „Ind. Fng. Chem.1* 24, 1375 (1932); 26, 497
886 (1984) и т. д.; Hoffert W. H. a. Claxton G., „J. Soc.Cht-m. Ind.*
52, 25 (1433).
2. Черножуков Я. И. и Крейн С. Э., Окистяемость минеральных масел.
ОНТИ, 1936; .Ж. прикл. химии' 8, 2F1 (1936); 10, 1485 (1987).
8. Тычинин и Бутков, «Нефт. и сланп.хоз." № 8, 341 (1924); № 1. 38 (1925):
Бутков 2Z. А., „Нефт. тоз." № 8, 189 (1927).
4. Бутков Я. А., „Нефт. хоз." 10 № 3, 3SS (1926): Гои der ffevden м. Турке.
„Erdol п Теег' 8, 839 (1928) и др.
5. Mizushima u. Tamada, „Petr. limes'. 9/IX. (CO (1929).
6. Тычинин и Иванов, вНс1фт. хоз." 18, № 6, 879 (1930); lamada, „J. Soc
Chem. Ind. (Japan)- 33, 318 1930).
7. Wieland H., ,Ber.“ 45, 484 (1912); 46, 3327 (1913) и т. д.
8. Bone с сотрудниками „J. Chem. Soc.“ 87, 1232 (1935); 89, 660 (1906) и т. д
9. Moureu et Duffraisse, „Cfinpt. rend." 174, 258; 175, 127 (1922) и т. д.
10. Семенов Я Я., Цепные реакции, ОНТИ, 1934.
Ц. Кистяковский, Ленер, Спенс, „J- Am. Chem. Soc. “52, 3785, 4837 (1930)
12. Backstrom, „J. Am. Chem. Soc." 49, 1460 (1927).
13. Соколик А., Горение и детонация в газах, IТТИ, Л. — М. (1934).
14. Марек Л. Ф. и Ган Д. А., Каталитическое окисление органических си
единений, пер. с англ., ОНТИ, М. (1936); Сгорание и детонация
сборник статей, ОНТИ, М. (1932); Масленников М. М., Сгорание
и детонация, ОНТИ, М. — Л. (1933).
15. Campbell, Littler. Whitworth, „Proc. Roy- Soc." 187, 380 <1932).
16. Lewie, Fnanf, B«J. Am. Chem. Soc.“ 52, 8905 (1930).
17 Петров А. Д. Андреев Д. Я., Чаплыгин Б. Л.. „Ж. прикл. химии". 5
578 (1932). л .
18- McAllister 8. Я., .Refiner" 16, N. 11, 492 (1937).
19. Химический состав нефтей и нефтяных продуктов, .Труды I розНИИ
изд. 2, гл. VII, М. — Л. (1935).
20. Рикардо, Быстроходные двигатели внутреннего сгорания, OIИ8
21. Masson, Hamilton, „Ind. Eng. Chem." 21, 544 (1919).
22. .ARC", Report N. 1013 (1926). о n ,
23 Moureu, Dufraisse, Chaux, ,C. R." 184, 29 (1937): Callend at. „ARC , Re-
port N. 1062 (1927) и др-
24. CalleMar, „ARC", Report N. 1062 (1927)
783
26. Hardies. Moss, ,J. Inst. Petr. Techn.“ 16, 77 (1929)
26. Hardies, „J. Chem. Soc.“, 1928. 226.
27. Бутков, .Известия B.T. И.’ №1,9 (1?29).
28. Layng, Jouker, .Ind. Eng. Cbem.- 20, 1048 (1923).
29. Eger ion, .Trans Farad. Soc.“ 24, 697 (1928).
80. Рамана, Халаджала, .Нефт. хоз.“ 26, № 4, 40 (1934); Рамана, Халадмсала
и Хайман С., .Нефт. хпз.* 26. № 12, 37 (1934).
31. U. S. Р. 1816 022 (14 июля 1931, 3 мая 1930); Dawis, .Nat. Petr. News*
48 (14 окт. 1931), 32—84 (28 дек. 1932); Dewis, Blackwood, ,Ind. Eng.
Chem.- 28, N. 12. 14c2 (1931).
82. Исагулянц В. И., частное сообщение го работам тт. Богуш, Русского,
Михневской (МНИ им. акад. Губкина).
38. Доладугин А. И., Солодовник М. С., Энглин Б. А., „Нефт. хоз." 27, № 4
68 (1%Б).
84. Таблицы технических норм нефтепродуктов, ежегодное издание Орга«
нэ<1 ти.
ЗБ. По опытам Стрибека-, цитируется по книге: Попич А. Г. и Лютер X. А.,
Смазочные материалы, ГИЗ, 1919.
38. Арчбютт Л. и Дилей Р. М., Трение, смазка и смазочные материалы,
ОНТИ, 1934; Trirat J. J., „Science et Industrie" № 1с8 (1927); Деря-
гин Б. В. Трение и смазочное действие, Москва 1938.
87. Кгаекег u. Spilker, „Вэг.“ 24, 2786 (1691).
38. Пало X. X. и Саранчук Л. И., „Нейт. хоз.“ 19, № 3, 61 (1938).
89. По неопубликованным данным Медведева Г. В. (работа бригады Ру-
денко М. Г., ИГИ Акад, наук СССР).
ОГЛАВЛЕНИЕ
Предисловие ко второму изданию.............................. 3
Предисловие к первому изданию...................... ’ ‘ ’ * 4
ЧАСТЬ ПЕРВАЯ
СОСТАВ, СВОЙСТВА И ПРОИСХОЖДЕНИЕ НЕФТИ
Глава I. Состав и классификация нефтей...................... 7
Элементарный состав нефти................................. 7
Предварительные данные о химическом составе нефти....... н
Химическая классификация нефтей............................. ц
Литература.........................................’ . ig
Глава II. Физические свойства нефти.......................... 17
Общая характеристика...................................... 17
Удельный вес . •........................................... 18
Вязкость................................................... 20
Поверхностное натяжение. ........................ . . . 32
Тепловое расширение..............................* . . . 33
Застывание и плавление................................... 37
Испарение и кипение...................................... 40
Вспышка, воспламенение, самовоспламенение................ 42
Теплоемкость............................................. 48
Скрытая теплота испарения................................ 50
Теплотворная способность................................. 51
Цвет нефти и нефтепродуктов.............................. 53
Лучепреломлэние.......................................... 55
Оптическая деятельность ................................. 55
Электрические свойства................................... 58
Растворимость . ......................................... 59
Литература............................................... 03
Глава III. Методика химического исследования нефтей и нефте-
продуктов ................................................ 64
Общие методы............................................. 64
Фракционировка при постоянном давлении (67). Фракцпонп-
ровка прп переменном давлении (67). Фракционировка с до-
бавкой нового специального вещества (68). Кристаллизация (69).
Адсорбция (69). Обработка селективными растворителями (69).
Специальные методы....................................... 70
Хлорирование и разного рода превращения хлоридов (70). Ни-
трование по ЛГ. Коновалову (73). Котонизация (76). Каталити-
ческая дегидрогенизация (76). Действие серной кислоты (77).
Некоторые реакции углеводородов ряда циклогексана (80).
Синтез нефтяных углеводородов........................... 80
Гидрогенизация ароматических углеводородов (80). Синтез на-
фтенов пз двухосновных кислот жирного ряда (82).
785
50 Зак. 1622. Химия нефти.
Методы идентификации..................................... 83
Элементарный состав и молекулярный вес (83). Удельный вес
и коэфициент лучепреломления (84). Температура кипения и
температура плавления (85). Анилиновый метол (85). Прочие
методы (85).
Литература............................................... 86
Глава IV. Ароматические углеводороды нефти................. 87
Краткая характеристика ароматических углеводсродов..... 87
Ароматические углеводороды бакинской нефти.............. 91
Ароматические углеводороды различных нефтей.............. 93
Количественное определение ароматики в нефтях и нефтепро-
дуктах прямой гонки .............-.................... 95
Физические методы (95). Сернокислотный метод (96). Нитра-
ционный метод (98). Метод анилиновых точек (99). Формол п-
товый метод (102).
Количественное содержание ароматических углеводородов в раз-
личных нефтях..............•........................ 102
Применение и практическое значение ароматических углеводоро-
дов нефти......................................... 107
Литература............................................. 110
Глава V. Нефтяные углеводороды жирного ряда.............. 112
I. Предельные углеводороды жирного ряда.................. 112
Краткая характеристика углеводородов ряда метана (парафинов) 112
А. Низшие (газообразные) парафины...................... 114
Общие методы газового анализа в приложении к анализу есте-
ственного газа....................................... 114
Разгонка естественного газа в вакууме .................. Ц6
Определение содержания бензина в естественном газе 121
Состав естественного газа.............................. 122
Применение естественного газа.......................... 126
Компрессионные заводы (127). Абсорбционные заводы (127). Ад-
сорбционные заводы (127).
Литература........................................... 133
Б. Средние (жидкие) парафины .................. ...... 133
Парафины бакинской нефти............................... 134
Парафины различных нефтей.............................. 138
Количественное определение и содержание парафинив в различ-
ных нефтях........................................... 144
Применение и практическое значение парафиновых углеводоро-
дов нефти........................................... 146
Литература............................................. 150
JB. Высшие (твердые) парафины.......................... 151
Получение парафина..................................... 151
Получение парафина из буроугольного и сланцевого догтя
(151). Переработка озокерита (162). Получение парафина из
нефти (158).
Количественное определение парафина.................... 155
Содержание парафина в нефтях и их дестиллатах.......... 156
Физические свойства парафина........................... 159
Химический состав и свойства парафина.................. 160
Строение парафина и его модификации.................... 162
Применение парафина . . . ............................. 168
Литература.............................* е ’....... . 170
786
Стр.
II. Непредельные углеводороды жирного ряда.................. 171
Краткая характеристика непредельных углеводородов . . , . ’ 171
Непредельные углеводороды различных нефтей и нефтепродук-
тов ........................................... • Л . 174
Количественное определение непредельных в нефти и нефтепро-
дуктах .............................................. 175
Метод иодных чисел (175). Метод кислородных насел (177).
Сернокислотный метод (178). М- тод полухлористой серы (179).
Характеристика непредельности нефтей и нефтепродуктов . . . . 181
Значение непредельных как составной части нефтепродуктов . . 184
Литература......................................* . . . . 185
Глава VI. Нафтены ч........................................ IfO
Классификация и номенклатура нафтенов................... 188
Краткая характеристика нафтенов......................... 187
Основные методы получения нафтенов (187). Физические свой-
ства (188). Химические свойства (189).
Нафтены бакинской нефти . .............................. 190
Нафтены различных нефтей . *............................ 211
Количественное определение нафтенов в нефтях и нефтепро-
дуктах ................................................ 217
Метод анилиновых точек (218). Метод каталитической дегидро-
генизации (219).
Количественное содержание нафтенов в различных нефтях ... 219
Примен'ние п практическое значение нафтенов как составной
части нефти............................................ 223
Литература..................*.......................... 228
Глава VII. Кислородные соединении нефти.................... 228
Методы получения нафтеновых кислот...................... 228
Краткая характеристика нафтеновых кислот................ 230
Природные нафтеновые (нефтяные) кислоты............. . 232
Получение и очистка (232). Количественное определение (238).
Распределение в нефтях и дестиллатах (235). Отдельные пред-
ставители, их выделение и свойства (236). Химическая харак-
теристика (239). Строение (241).
Другие кислородные соединения нефти..................... 245
Применение кислородных соединений нефти................. 247
Литература............................................ 248
Глава VIII. Сернистые соединения нефти......................... 250
Краткий очерк сернистых органических соединений............. 260
Качественное и количественное определение серы в нефтях и
нефтепродуктах........................................... 263
Содержание серы в различных нефтях и нефтепродуктах .... 254
Технические нормы нефтепродуктов на серу.................... 256
Количественное определение серы и различных типов сернистых
соединений ..........................................
Сернистые соединения различных нефтей...............
Литература...........................................
Глава IX. Азотистые соединения нефти......................
Краткая характеристика азотпстых органических соединений
основного характера ..........................• • • •
Качественное и количественное определение азога в нефтях - .
Содержание азота в различных нефтях и нефтепродуктах . . . .
Азотистые соединения различных нефтей..................
Литература..............»..............................
50*
264
264
266
266
267
271
787
Стр.
Глава X. Смолистые и асфальтовые вещества нефти............. 272
Классификация и разделение смолистых и асфальтовых веществ
нефти................................................. 272
Характеристика отдельных групп смолистых и асфальтовых
веществ нефтп......................................... 274
Асфальтены (274). Нефтяные- смолы (276). Лсфальтогеновые
кислоты и их ангидриды (276). Карбены п карбоиды (278).
Акцизные смолы (279).
Взаимные соотношения между различными видами смолистых и
асфальтовых веществ................................... 279
Содержание в различных нефтях и нефтепродуктах........... 280
Значение и применение смолистых и асфальтовых веществ нефти 282
Природные асфальты (288). Искусственные (нефтяные) ас-
фальты (285). Практическое применение асфальта......... 28b
Литература............................................... 287
Глава XI. Минеральные вещества нефти........................ 288
А. Зола..................................................... 288
Количественное определение и содержание золы в нефти.... 288
Химический состав нефтяной золы.......................... 290
К. Бода..................................................... 291
Количественное определение и содержание воды в нефти .... 291
Способ отстаивания (291). Способ перегонки (292).
Химический состав буровых вод............................ 293
Химическая классификация буровых вод..................... 295
Химические процессы в буровых водах...................... 302
Обессеривание буровых вод (302). Обогащение буровых вод
карбонатами (304). Смешивание буровых вод (805). Изменение
минерализации (80b).
Генезис буровых вод...................................... ЗОВ
Значение буровых вод и их утилизация..................... 307
Литература............................................... 309
Глава XII. Происхождение нефти.............................. 331
Минеральная теория гроисхождения нефти (314). Теория про-
исхождения нефти из животных остатков (316). Теория проис-
хождения нефти ив растительных остатков (319). Теории
смешанного типа (322). Новейшие воззрения на происхожде-
ние нефти (325).
Литература. . . ,........................................ 329
ЧАСТБ ВТОРАЯ
ПЕРЕРАБОТКА НЕФТИ
Глава I. Обезвоживание нефти............................ 381
Способы обезвоживания нефти........................... 331
Физико-механические способы разрушения эмульсий (335).
Природа нефтяных эмульсий............................. 337
Физико-химические основы борьбы с эмульсиями.......... 341
Практическое значение обезвоживания нефти............. 348
Литература. . т . т . . , , . , . т . . . т . т , . . . , ,
788
Стр.
Глава II. Перегонка нефти................................... 346
Техника заводской перегонки нефти..............'......... 346
Перегонка нефти в кубах периодического действии (347). Пе-
регонка нефти на кубовых батареях (351). Керосиновая бата-
рея (361). Масляная батарея 6355). Перегонка нефти на труб-
чатых установках (359). Вакуумная перегонка на трубчатых
установках (868).
Продукты перегонки нефти............................... 36'.)
Дестиллатные продукты (369). Остаточные продукты (870>.
Теория перегонки. Индивидуальные жидкости................ 371
Опытное определение упругости насыщенный 'паров при раз-
ных температурах (371). Зависимость между упругостью на-
сыщенных паров и температурой (372). Графическое изобра-
жение зависимости между упругостью насыщенных паров н
температурой (374). Перегонка индивидуальных жидкостей (376).
Теория перегонки. Несмешивающиеся жидкости............... 377
Основные газовые законы (878). Правило фаз (379). Состав
пара (380). Температура кипения (382). Перегонка двух не-
смешивающихоя жидкостей (383).
Теория перегонки. Две смешивающиеся жидкости............. 385
Изотермы упругости паров смеси двух жидкостей (386). Упру-
гость пара идеального раствора (387). Состав пара и жидкой
фазы идеального раствора (889). Опытное определение состава
жидкой и паровой фаз (395). Однократное испарение двух
смешивающихся жидкостей (397). Многократное испарение
двух смешивающихся жидкостей (401).
Теоретические основы фракционировки и ректификации...... 4<Ч
Основные принципы погоноразделения (404). Погоноравделе-
нив в лабораторных условиях (405). Зависимость между раз-
личными кривыми разгонки (409). Схема процесса ректифика-
ции (409). Материальный и тепловой балансы для теорети-
ческой тарелки (410). Расчет числа теоретических тарелок
колонны (412). Коэфициент полезного действия тарелки (415).
Орошение колонны (417).
г1еория перегонки. Сложные смеси......................... 41«
Состав пара и жидкой фазы сложной смеси (419). Испарение
сложной смеси (420). Ректификация сложной смеси (422).
Практическое значение перегонки как метода переработки нефти 425
Литература............................................... 426
Глава III. Перегонка с разложением (крэкинг и пиролиз). . . . 427
Основные виды перегонки нефти с разложением................ 427
Перегонка до кокса (427). Крэкинг в жидкой фазе (428). Крэ-
кинг в паровой фазе (435). Пирогенетичеокое разложение
нефти (пиролиз) (489).
Продукты перегонки нефти о разложением...................... 442
Газообразные продукты (442). Жидкие продукты (443). Кокс
(448).
Физико-химические основы перегонки с разложением ........... 450
Методика исслеповаипя (451). Химизм крэкинг-процесса (455).
Кинетика крэкинг-гроцесса (460). Газообразование при крэкинг-
процессе (465). Крэкинг углеводородов различных рядов
Термодинамика крэкинг-процесса.............................
Теории крэкинг-процесса....................................
Теории реакций уплотнения (511).
Теплота реакции крэкинг-процесса............................ §12
Повторный крэкинг..........................................
Стр.
Реформинг.................................................... 517
Анализ и состав крэкинг-бензина.............................. 518
Анализ крэкинг-бензпна(518). Состав крэкинг-бензина (522).
Химическая переработка продуктов крэкинга.................... 523
Выбор и подготовка сырья (524). Действие хлористого водо-
рода (526). Действие хлора (526). Действие серией кислоты
(527). Действие хлорноватистой кислоты (530). Полимерные
бензины (582). Термическая обработка газов крэкинга (533).
Синтетические масла (584).
Практическое значение крэкинга п пиролиза нефти.............. 536
Литература................................................... 539
Г л а в а IV. Специальные виды крэкинга......................... 542
Л. Крекинг в присутствии катализаторов....................... 542
Хлористый алюминий........................................... 542
Крэкинг в присутствии хлористого алюминия.................... 543
Химизм процесса (548).
Практическое значение и перспективы процесса................. 548
Б. Восстановительный крекинг (гидрогенизагсия)............... 548
Техника гидрирования......................................... 549
Теории гидрирования.......................................... 551
Гидрирование топлива......................................... 552
Техника гидрогенизации (5Ь2). Гидрирование угля (555). Гидро-
генизация тяжелых смол, нефтей и т. п. (556). Гидрирование
горючих газов (558).
Проблема водорода при гидрогенизации......................... 565
Продукты гидрогенизации...................................... 567
Физико-химические основы гидрогенизации...................... 570
Методика исследования (570). Химизм гидрогенизации (570).
Гидрогенизация углеводородов различных рядов (579).
Повторная гидрогенизация................................... 590
Практическое значение п применение гидрогенизации.......... 598
В. Окислительный крекинг................................. 594
Техника окислительного крэкинга.......................... 595
Анализ и разделение продуктов окислительного крэкинга . . . 597
Продукты окислительного крэкинга......................... 599
Кислоты (599). Оксикислогы (600). Альдегиды и спирты (601).
Химизм окислительного крэкинга........................... 603
Практическое значение и перспективы окислительного крэкинга . 608
Литература............................................... 610
ЧАСТЬ ТРЕТЬЯ
ОЧИСТКА И ПОВЫШЕНИЕ КАЧЕСТВА НЕФТЕПРОДУКТОВ
Глава I. Очистка нефти и ее дестиллатов. Общие методы . . . 612
Техника процесса очистки . . ........................... 612
Химизм кислотной очистки................................ 616
Кислотная очистка легких дестиллатов прямой гонки (616).
Кислотная очистка крэкинг-бензина (618). Кислотная очистка
масляных дестиллатов (624). Серная кислота и второстепен-
ные компоненты нефти (628). Прочие случаи кислотной
очистки (631). г
799
Стр.
Химизм щелочной ОЧИСТКИ И промывки..................* , 631
Реакции при щелочной очистке (631). Эмульсин при щелочной
очистке (663). Разрушение масляных эмульсий (636). Некото-
рые видоизменения методики щелочной очисткп (639).
Вторичная перегонка в процессе очисткп.................. 640
Отходы очистки я их утилизация.......................... 642
Кислые гудроны (642). Утилизация кислого гудронт (643). Ще-
лочные отбросы и их утилизация (648).
Практическое значение общих методов очистки............. 649
Литература.............................................. 649
Глава II. Очистка нефти и ее дестиллатов. Специальные ме-
тоды ........................._........................... 651
А. Адсорбционные методы очистки......................... 651
Адсорбенты в нефтяной промышленности.................... 651
Техника адсорбционной очистки л -....................... 653
Общая теория адсорбции.................................. 656
Теория адсорбционной очисткп............................ 662
Практическое значение адсорбционных методов очистки..... 664
Б. Методы обессеривания................................. 665
Техника обессеривания . ................................ 665
Хпмизм важнейших процессов обессеривашы................. 667
Обессеривание серной кислотой (667). Обессеривание щелочью
(668). Обессеривание „докторским раствором*4 (669). Обессери-
вание гппохлоритом (673). Обоссорнванис азотной кислотой и дру-
гими окислителями (676). Обессеривание гидрогенизацией (679).
Практическое значение методов обессеривания............. 679
Б. Методы очистки солями ................... 680
Техника очистки хлористым цинком........................ 681
Химпзм очистки хлористым цинком........................ 682
Практическое значение очистки солями.................... 684
Г. Очистка гидрогенизацией или гидроочштка.............. 684
Хпмизм смолообразования................................. 985
Определение и содержание смол в крэкпнг-бензпне......... 688
Гидроочистка............................................ 689
Практическое значение гидроочистки...................... 690
Д. Очистка с помощью растворителей .............. 691
Растворители при переработке нефти...................... 691
Техника сольвентной очистки............................. 698
Химпзм и ближайшие задачи сольвентной очистки........... 700
Общие основания сольвентной очистки..................... 705
Теория сольвентной очистки..................._.......... 707
Однократное испарение и однократная экстракция (708). Много-
кратное испарение и проточная экстракция (709).
Сернистый ангидрид как селективный растворитель......... 710
Практическое значение сольвентной очисткп .............. 714
Литература..............................................
Глава III. Повышение качества нефтепродуктов методом при-
садок ................................................. Ш
А. Ингибиторы . . ........................................ Ш
Ингибиторы моторного топлива........................... 718
Ингибиторы для изоляционных п смазочных масел........... 722
Стр.
Химизм действия ингибиторов..................; . . . . 726
Практическое значение ингибиторов в нефтяном деле.... 731
Б. Антидетонаторы ....................... 731
Нормальное горение и детонация....................... 730
Основные свойства детонационной волны................ 784
Детонация в двигателе внутреннего сгорания........... 787
Методы оценки детонационных свойств топлива.......... 738
Антидетонационные добавки и антидетонаторы............. 741
Антидетонационные добавки (741). Антидетонаторы (746).
Химизм действия антидетонаторов...................... 748
Практическое значение антидетонаторов................ 764
Б. Депарафинизаторы t .................... . 755
Депарафинизаторы смазочных масел..................... 755
„Парафлоу", его получение и свойства................. 767
Теория действия добавок типа „парафлоу".............. 758
Практическое значение депарафинизаторов.............. 762
Г. Лкбриканторы...................................... 762
Смазочные материалы и масла.......................... 763
Трение и его основные факторы........................ 768
Масленистооть и ее физико-химическая природа........... 771
Собственно любриканторы и их применение.............. 778 *
Велооиторы и их применение........................... 781
Практическое значение лгобрнканторов.................. 782
Литература........................................... 783
11 — 1*
ОПЕЧАТКА
Стр. Строка Напечатано Следует читать По вине
159 5 сн. консистенция Ковгнстентноеть автора
176 18 св. с реакциями наряду с реакциями тип.
17S 25 „ Одним и, пожалуй, Одним я еще недавно автора
единственным единственным
184 6 „ прямой линии прямой гонки тип.
190 14 „ Для замещения Для замещенных 99
197 1 си. С6НЙ(НС3)2 Ct,H8(CH8)2
292 19 св- солями слоями 99
307 П „ устойчивостью неустойчивостью 99
325 16 , автором некоторыми авторами автора
338 1 СБ. 1751° 175°
346 17 св. заводской нефти заводской перегонки тип.
нефти
426 в гл. VI в ч. Ш гл. II Д автора
486 14 сн. циклогексена из бензола бензола вз циклогексена 99
619 24 „ C2H5-SO8H C2H6OSOsH 9г
625 3 „ сульфинированием сульфированием ТИП.
679 6 - . высокосортных высокосернистых автора
691 9 . чашки чаети тип.
693 13 св. пропана пропаном автора
762 6 сн. вязкости вязкостные тип.
Sax. 1622. Химия нефти.