



































































































































































































































































































































































































































































Автор: Черных В.П.
Теги: органическая химия химия органические вещества учебное пособие органические реакции химическая наука реакции и реагенты физические и химические свойства
ISBN: 966-615-155-3
Год: 2003




МИНИСТЕРСТВО ОХРАНЫ ЗДОРОВЬЯ УКРАИНЫ НАЦИОНАЛЬНЫЙ ФАРМАЦЕВТИЧЕСКИЙ УНИВЕРСИТЕТ
К ЮО-летию НФаУ
ЛЕКЦИИ
ПО ОРГАНИЧЕСКОЙ ХИМИИ
В.П. ЧЕРНЫХ
Учебное пособие для студентов высших учебных заведений
Харьков Издательство НФаУ «Золотые страницы» 2003
УДК 547
ББК 24.2р73
Л 43
Рекомендовано Министерством образования и науки Украины (письмо № 14/18.2-690 от 14.04.2003).
Рецензенты:
В.Д. Орлов, доктор химических наук, профессор, зав. кафедрой органической химии Харьковского национального университета им. В.Н. Каразина; П.А. Безуглый, доктор фармацевтических наук, профессор, зав. кафедрой фармацевтической химии Национального фармацевтического университета.
Лекции по органической химии В.П. Черных: Учеб, пособие Л 43 для студ. высш. учеб, заведений. — Харьков: Изд-во НФаУ: Золотые страницы, 2003. — 456 с.
ISBN 966-615-155-3
ISBN 966-8032-69-1
В лекциях по органической химии, которые на протяжении многих лет читаются студентам фармацевтического университета, в простой и доступной форме изложены общие подходы в рассмотрении теоретических вопросов современной органической химии. Даны представления о реакциях и реагентах, механизмах органических реакций и их направлении, физических и химических свойств органических веществ. Подчеркнута роль и значимость органической химии как науки, показана генетическая связь между классами органических соединений.
Для студентов фармацевтических вузов и факультетов.
УДК 547
ББК 24.2р73
ISBN 966-615-155-3
ISBN 966-8032-69-1
© Черных В.П , 2003
© НФаУ, 2003
Посвящается моим дочерям
Юле и Владе
ПРЕДИСЛОВИЕ
Вот уже четыре десятилетия я каждый раз с волнением вхожу в лекционную аудиторию, где на меня устремлены более двухсот пар пытливых и внимательных глаз студентов.
Моя задача — донести до каждого из них все богатство содержания и красоту структурных формул королевы химических наук — Органической химии. Именно она является тем фундаментом, на котором возводится величественное и значимое здание фармацевтических специальностей. Без знания органической химии невозможно успешно освоить фармацевтическую, токсикологическую, биологическую, аналитическую химию, фармакогнозию и другие дисциплины учебного плана.
Мой опыт чтения лекций и наблюдения за восприятием их студентами убедили меня в необходимости издания курса органической химии именно в предлагаемом виде.
Кафедра органической химии Национального фармацевтического университета имеет большой опыт создания и издания учебной литературы. Написание и издание учебников и учебных пособий по дисциплине преподавательский коллектив считает своей первейшей задачей и святым долгом перед студентами. Исключительно богатый многолетний опыт преподавания, творческий подход позволили создать учебно-методический комплекс по органической химии в виде учебника, практического руководства и методических указаний.
Учебник «Органическая химия» в 3-х книгах, изданный на русском и украинской языках, удостоен Государственной премии Украины, широко используется в преподавании курса в фармацевтических вузах и классических университетах Украины, России, Беларуси и других стран СНГ. Практическое руководство к лабораторным и семинарским занятиям на русском и украинском языках выдержало три издания. Новый подход к органическому синтезу, а именно комбинаторный синтез, воплошен в учебнике, который органично дополняет фундаментальный учебник по органической химии.
Нужно сказать, что курс лекций по органической химии претерпевал качественные изменения в зависимости от количества часов, выделенных на дисциплину в типовых учебных планах, с неизменной тенденцией к их уменьшению. Все это требовало творческого подхода к чтению лекций со стороны преподавателей. И следует отдать должное, преподавательский корпус кафедры органической химии всегда
4
Лекции по органической химии
отличался высокой требовательностью к лекторскому мастерству, высокой культурой, эмоциональностью, интеллигентностью, свободным владением материала.
Профессор П.А. Петюнин, доцент Н.Н. Валяшко и я читали курс в полном объеме, постоянно совершенствуя его и дополняя новой информацией. Сегодня из-за уменьшения количества лекционных часов преподавателям приходится читать урезанный курс, что очень сложно. Но сложно и студентам разобраться в большом объеме материала, изложенного в учебнике или вынесенного на самостоятельную работу, возникает масса вопросов. Поэтому я счел возможным предоставить студентам тексты своих лекций, которые не заменяют учебник, а скорее дают курс в сжатом изложении, акцентируют внимание на наиболее трудноусваиваемых студентами понятиях, разделах, механизмах реакций.
Достоинством издания, с моей точки зрения, является и то, что органическая химия здесь представлена в системе фармобразования, подчеркнута ее роль в освоении специальных дисциплин.
Основные понятия изложены не сухим, академичным стилем, а живым, понятным языком, приближенным к разговорной речи.
Буду рад, если мой труд поможет вам в освоении курса органической химии, в самостоятельной работе, при подготовке к практическим занятиям, коллоквиумам и семинарам.
Всю жизнь (сведения об авторе лекций вы можете прочесть в конце этой книги) я посвятил любимому делу — органической химии, науке, которая верой и правдой служит человеку. Ее роль и значение в нашей жизни как нельзя лучше характеризуют слова великого М.В. Ломоносова: «Широко распростирает химия руки свои в дела человеческие. Куда ни посмотрим, куда ни оглянемся, везде обращаются пред очами нашими успехи ея прилежания».
Я надеюсь, что познав и поняв органическую химию, вы полюбите ее так же, как и я, и перед вами откроются широкие горизонты ее практического применения в вашей жизни, в вашей профессии.
1. КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
КЛАССИФИКАЦИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Каждый период развития органической химии отмечен попытками ученых каким-то образом привести разнообразие химических соединений в единую систему.
Важнейшими признаками, которые положены в основу современной классификации органических соединений, являются строение углеродной цепи и природа функциональных групп.
Классификация по строению углеродной цепи
В зависимости от расположения углеродных атомов в молекуле органические соединения делят на несколько больших групп.
Различают два типа органических соединений: ациклические и циклические. Ациклические или алифатические (от древнегреч. алифар — жир) — вешества с открытой (незамкнутой) цепью, другое их название — соединения жирного ряда. По строению углеводородной цепи среди ациклических соединений различают: насыщенные (предельные) вешества, содержащие только простые углерод-углеродные связи и ненасыщенные (непредельные) алифатические — структуры с кратными (двойными, тройными) углерод-углеродными связями.
6
Лекции по органической химии
сн3— сн3
сн3—сн2—соон
предельные алифатические
соединения
сн2= сн2 сн2=сн—соон
непредельные алифатические соединения
К циклическим относятся соединения, содержащие в своей структуре замкнутые цепи атомов — циклы (от греч. циклос — круг). Природа атомов, входящих в цикл, лежит в основе деления всех циклических соединений на две большие группы: карбоциклические и гетероциклические. В молекулах карбоциклических соединений цикл состоит только из атомов углерода. Гетероциклические соединения имеют в своей структуре циклы, содержащие наряду с атомами углерода атомы других элементов, чаще всего О, S, N.
н2с—сн2
н2с—сн2
алициклическое соединение
гетероциклическое соединение
карбоциклические соединения
Карбоциклические соединения в свою очередь делятся на алициклические и ароматические.
Алициклические структуры подобно алифатическим соединениям по степени насыщенности подразделяются на насыщенные и ненасыщенные:
насыщенное алициклическое соединение
ненасыщенное алициклическое соединение
Среди гетероциклических соединений различают насыщенные,
ненасыщенные и ароматические структуры:
1. Классификация и номенклатура органических соединений
7
насыщенный гетероцикл
ароматический гетероцикл
Соединения, молекулы которых состоят только из атомов углерода и водорода, называются углеводородами. Замещение одного или нескольких атомов водорода на функциональные группы ведет к образованию других классов органических соединений.
Классификация по природе функциональной группы
Функциональная группа — структурный фрагмент молекулы, характеризующий свойства соединений данного класса. Например, свойства карбоновых кислот характеризуются наличием карбоксильной
группы С ; в спиртах функциональная группа — спирто-ОН
вый гидроксил —ОН; к аминам относятся соединения, содержащие группу -NH2ht. д.
По количеству и однородности функциональных групп органические соединениия делят на моно-, поли- и гетерофункциональные.
Вещества с одной функциональной группой называют монофункциональными, с несколькими одинаковыми функциональными группами — полифункциональными. Соединения, содержащие несколько различных функциональных групп, — гетерофункциональные.
Соединения одного класса объединены в гомологические ряды. Гомологический ряд — это ряд органических соединений с одинаковыми функциональными группами и однотипным строением, каждый представитель гомологического ряда отличается от предыдущего на постоянную единицу (—СН2—), которую называют гомологической разностью. Члены гомологического ряда называются гомологами.
Н3СОН — метанол
Н5С2ОН — этанол ►
Н7С3ОН — пропанол и т.д. -
гомологический ряд одноатомных спиртов (алканолов) СлН2п+|ОН
НОМЕНКЛАТУРА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Химическая номенклатура — совокупность названий индивидуальных химических веществ, их групп и классов, а также правила составления их названий.
8
Лекции по органической химии
Соблюдать соответствия между существующей классификацией веществ и их наименованиями позволяют номенклатурные системы.
Номенклатура органических соединений складывалась на протяжении всего периода возникновения и становления органической химии как науки. Для названий органических соединений применяют несколько номенклатурных систем: тривиальную, рациональную, международную (ИЮПАК).
Тривиальная номенклатура
На первых этапах развития органической химии соединения назывались случайно. Это было связано с их нахождением в природе: щавелевая кислота, яблочная кислота и другие, или с источником их получения: древесный спирт, муравьиная кислота и др. Многие тривиальные названия прочно укоренились и до сих пор широко применяются.
Рациональная номенклатура
В основе рациональной номенклатуры используется принцип деления органических соединений на гомологические ряды. Вещества рассматриваются как производные простейшего представителя данного ряда: для алканов — метана, алкенов — этилена, алкинов — ацетилена и т. д., например:
।-----------1 I------- 1
НС=СН нзс—LCZ=£“Hj н3с—с=с—сн3
ацетилен метилацетилен лиметилацетилен
В настоящее время применение рациональной номенклатуры ограничено. Основные ее принципы нашли свое отображение в радикало-функциональной номенклатуре.
Международная номенклатура (ИЮПАК)
Первая попытка создать номенклатурную систему, которая позволяла бы дать однозначное название любому органическому соединению, была предпринята химиками в 1892 году на международном конгрессе в Женеве (женевская номенклатура). Правила современной номенклатуры были разработаны на XIX конгрессе Международного союза теоретической и прикладной химии в 1957 году. Эти правила известны под названием номенклатуры ИЮПАК.
Номенклатурные правила ИЮПАК предусматривают несколько способов образования названий органических соединений. Наиболее широко применяются заместительная и радикало-функциональная номенклатуры.
Заместительная номенклатура
Прежде чем перейти к рассмотрению заместительной номенклатуры, дадим определение основным понятиям.
1. Классификация и номенклатура органических соединений
9
Родоначальная структура — структурный фрагмент молекулы (молекулярный остов), лежащий в основе названия соединения: главная углеродная цепь атомов для ациклических соединений, для карбо-
и гетероциклических — цикл:
Родоначальное название может быть систематическим, тривиальным или полусистематическим.
В органической химии для лр’-гибридизованного углерода существует такое понятие, как первичный, вторичный, третичный.
Атом углерода, связанный a-связью только с одним атомом углерода, называется первичным, с двумя — вторичным, с тремя — третичным.
Радикал — остаток углеводорода, образующийся в результате удаления одного или нескольких атомов водорода. Свободную валентность в радикалах обозначают черточкой.
По количеству свободных валентностей различают одно-, двух-,
трехвалентные радикалы:
н,с—сн—сн=
—сн—сн—сн2—
радикал двухвалентные
радикалы
н3с—сн—сн—
одновалентный
н3с—с=
трех валентный радикал
В зависимости от того, у какого атома углерода находится свобод
ная валентность, различают первичные, вторичные и третичные радикалы:
10
Лекции по органической химии
Н,С-СН-СНг-СНг- н3с-сн—сн—сн3 н,с-с—снгсн,
J| 2 2 л । । j j | 2 J
сн3 сн3 сн3
первичный радикал
вторичный радикал
третичный радикал
Заместителем называют любой атом или группу атомов, включая радикал и функциональную группу, которые не входят в родоначальную структуру.
Положение заместителей в молекуле указывают с помощью цифр или букв, которые называют локантами. Для обозначения нескольких одинаковых заместителей или кратных связей в данной молекуле применяют множительные (умножающие) приставки: ди- (два), три- (три), тетра- (четыре), пента- (пять) и т. д.
Согласно заместительной номенклатуре органические соединения рассматривают как производные углеводородов, в молекулах которых один или несколько атомов водорода замешены на другие атомы или атомные группы.
Составление названий проводят в определенном порядке:
1. Среди всех функциональных групп, имеющихся в соединении, выбирают старшую. Следующие группы перечисляют в порядке уменьшения их старшинства:
-СООН, -SO3H, -COOR, -CONH2, —C=N
,0
—С^ , /С=О , -ОН, -SH, -NH2 н
В названии органического вещества лишь старшая функциональная группа обозначается в суффиксе, все остальные — в префиксе, но некоторые функциональные группы всегда находят свое отражение в префиксе:
-F, -Cl, -Вг, -I, -OR, -SR, — N=O,-
4 О
Их не рассматривают по старшинству.
2. Устанавливают родоначальную структуру. Если соединение содержит кратные связи, то они должны войти в родоначальную структуру.
3. Проводят нумерацию атомов родоначальной структуры таким образом, чтобы старшая функциональная группа получила по возможности меньший номер.
4. Составляют название соединения в целом: первым указывают в алфавитном порядке функциональные группы (кроме старшей) и уг
1. Классификация и номенклатура органических соединений ' ’
леводородные радикалы в префиксе, затем — название родоначальной структуры в корне и в конце названия — старшую функциональную группу в суффиксе.
Степень насыщенности обозначается специальными суффиксами: -ан — для насыщенных, -ен — для двойной, -ин — для тройной связи.
Локанты, буквенные или цифровые, и множительные приставки располагают перед названием заместителей или кратных связей.
Пример составления названий:
заместители
З-амино-2,4-диметилпентановая кислота
старшая функциональная группа
сн3
5 4 3 2 I I
б ) сн3—с=сн—с—сн2—он
Вг снз
4-бром-2,2-диметил-3-пентен-1 -ол
Ра дикало-функциональная номенклатура
В основе радикало-функциональной номенклатуры лежит название класса (спирт, кетон и др.), перед которым перечисляют названия радикалов и функциональных групп (кроме старшей), например:
<5 Г <Р *
н3с—сн—сн—сн2он
nh2
|^аминобутиловый спирт
н3с—о—сн3
диметиловый эфир
н2с=сн—С! винилхлорид, хлористый винил
н,с—с—С7Н
3 II 2 о
метил этилкетон
Родоначальную структуру чаще обозначают с помощью тривиального названия, а положение радикалов — с помощью буквенных локантов: а. р. у. 8 (греческий алфавит). Буквой а обозначают ближайший к старшей функциональной группе атом углерода.
В дальнейшем при изучении различных классов органических соединений мы расширим приведенные краткие пояснения на многочисленных примерах.
2. ХИМИЧЕСКАЯ СВЯЗЬ. ВЗАИМНОЕ ВЛИЯНИЕ
АТОМОВ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Остановимся на одном из важнейших вопросов химии, как осуществляется связь атомов в молекулах? Используя знания, полученные в курсе неорганической химии, рассмотрим вопросы природы химической связи между атомами или типы химических связей.
Современная теория химической связи базируется на квантовомеханических представлениях о строении молекулы. Впервые электронную теорию химической связи, предложили в 1916 г. немецкий ученый В. Коссель и американский ученый Дж. Н. Льюис. Авторы электронной теории выдвинули идею о том, что химическая связь — результат взаимодействия внешних электронных оболочек атомов. Образуя химическую связь, каждый атом стремится заполнить внешнюю электронную оболочку до конфигурации, присущей инертным газам. При этом он принимает участие в образовании обшей электронной пары, отдает или принимает электроны. Принцип заполнения валентных оболочек до конфигурации инертных газов получил название ок-тетное правило.
ТИПЫ ХИМИЧЕСКИХ СВЯЗЕЙ
Согласно принятой в настоящее время классификации существует несколько типов химической связи: ионная, ковалентная, водородная, металлическая и др. По способу образования химической связи различают две основные: ионную и ковалентную.
ИОННАЯ СВЯЗЬ
Ионная связь — характерна для атомов значительно отличающихся по электроотрицательности. Ионный тип связи характерен для неорганических соединений.
Рассмотрим, как осуществляется связь в натрие хлориде:
Na- -г С!: ------ Na+Cl'
ионная связь
Хлор по сравнению с натрием обладает большей электроотрицательностью, то есть большим сродством к электрону. Вследствие этого электрон переходит с внешней оболочки натрия на внешнюю оболочку хлора, при этом образуются положительный и отрицательный заряды, между которыми действуют силы электростатического притя
2. Химическая связь. Взаимное вп/яние атомов в органиче' ких соединениях
13
жения. Ион натрия приобретает электронную конфигурацию неона, а ион хлора — конфигурацию аргона.
Соединения с ионной связью характеризуются относительно большим дипольных' моментом, хорошей электропроводимостью, высокими температурами плавления.
КОВА ЛЕНТН \Я СВЯЗЬ
Ковалентная связь — основной тип связи в органических соединениях, характеризующийся увеличением электронной плотности меж ду химически связанными атомами в молекуле по сравнению с распределением электронной плотности в свободных атомах.
Ковалентная связь возникает между атомами, имеющими одинаковую или близкую по значению электроотрицательность. Для образования ковалентной связи каждый из атомов предоставляет по одному электрону, при этом образуется общая пара электронов, которая в равной степени принадлежит обоим атомам. На письме ковалентная связь изображается черточкой между атомами.
Классическим примером ковалентной связи является молекула метана:
Атом углерода содержит на внешнем электронном уровне четыре валентных электрона и, чтобы доукомплектовать этот слой до октет-ной конфигурации, он образует четыре обшие электронные пары с четырьмя атомами водорода.
В зависимости от электроотрицательности атомов между которыми образовалась ковалентая связь, она может быть полярной или неполярной.
Если электроотрицательность атомов одинакова, то обшая электронная пара находится на одинаковом расстоянии от ядра каждого из атомов. Такая связь называется ковалентной неполярной:
ковалентная неполярная связь
14
Лекции по органической химии
При возникновении ковалентной связи между атомами с различной электроотрицательностью общая электронная пара смещается к более электроотрицательному атому. В этом случае образуется кова-1ентная полярная связь.
ковалентная полярная связь
Стрелка в формуле указывает на полярность ковалентной связи. С помощью греческой буквы 8 («дельта») обозначают частичные заряды на атомах: 8+ — пониженную, 8~ — повышенную электронную плотность.
По числу электронных пар, образующих ковалентную связь, различают связи простые — с одной парой электронов и кратные — с двумя или тремя парами.
кратная ковалентная связь
Н । /Н ^c=c-Z н /Хн
простая ковалентная связь
Основные характеристики ковалентной связи: длина связи (расстояние между центрами атомов в молекуле); энергия связи (энергия, которую необходимо затратить на разрыв связи); полярность связи (неравномерное распределение электронной плотности между атомами, обусловленное различной электроотрицательностью); поляризуемость (легкость, с которой смещается электронная плотность связи к одному из атомов под влиянием внешних факторов); направленность (ковалентная связь, направленная до линии, соединяющей центры атомов).
Атомы некоторых элементов (кислорода, азота, серы, галогенов и др.) расходуют не все внешние электроны для формирования октет-ной оболочки. Такие электроны называют неподеленные, необобществленные или л-электроны.
Н
• N : + ЗН --------Н : N : неподеленная
• . пара электронов
н
Донорно-акцепторная связь или координационная — разновидность ковалентной связи, которая отличается способом образования.
2. Химическая связь. Взаимное влияние атомов в органических соединениях 15
Донор — атом, который должен иметь пару неподеленных электронов, а акпепторэч может быть любой атом (в том числе и протон), у которого не хватает до образования октета двух электронов.
Например, образование иона аммония протекает по донорно-акцепторному механизму. Ковалентная связь образуется за счет того, что атом азота (донор электронов) отдает неподеленную пару электронов на образование ковалентной связи протону (акцептору электронов):
Вновь образовавшаяся связь Н—N ничем не отличается от остальных связей Н—N, которые имелись у аммиака. Такая связь называется еще координационной.
Семиполярная связь — частный случай донорно-акцепторной связи. Она образуется между атомами, один из которых имеет неподеленную пару электронов, а другой содержит на внешнем энергетическом уровне шесть электронов (секстет), то есть имеет вакантную атомную орбиталь.
Рассмотрим образование семиполярной связи в молекуле оксида триметиламина:
Н.С
акцептор
н3с
донор
Н3С\ +
Нзс-^N-О' == (CH3)3N —О
Н3С
В результате образования связи атом кислорода (акцептор) приобретает отрицательный заряд, а атом азота (донор) — положительный. Такой вид связи обозначают следующим образом:
—N—О
ИЛИ
—N—*0
Соединения с семиполярной связью не проводят электрический ток, несмотря на наличие наряду с ковалентной связью и ионного взаимодействия.
ВОДОРОДНАЯ СВЯЗЬ
Водородная связь образуется в результате электростатического взаимодействия между атомом водорода, несущим дробный положительный заряд, и неподеленной парой электронов другого атома. Обычно такие атомы водорода называют активными.
Водородную связь графически изображают тремя точками:
16
Лекции по органической химии
водородная связь
Водородная связь бывает внутримолекулярная и межмолекулярная.
Примером соединений с внутримолекулярной водородной связью могут быть салициловый альдегид и о-хлорфенол.
салициловый альдегид о-хлорфенол
Межмолекулярные водородные связи (МВС) возникают между двумя или большим числом молекул с образованием димеров или ассоциатов:
- Н-СК нх=сн-с< _^с-сн=сн2
о—н - О
димер акриловом кислоты
(мвс
I сл н5с—о—н- о—н— о—н
С2Н5 ассоциат этанола
Водородная связь тем сильнее, чем более электроотрицателен элемент, с которым связан атом водорода. По сравнению с обычной ковалентной связью (Е = 340 — 360 кДж/моль) энергия водородной связи невелика (10 — 40 кДж/моль), но наличие этого вида связи существенно отражается на физико-химических свойствах веществ. За счет межмолекулярной водородной связи повышается температура плавления «-нитрофенола (114 °C), по сравнению с о-нитрофенолом (45 °C). Образование ассоциатов этиловым спиртом сказывается на его температуре кипения (78 °C), которая значительно выше, чем у неспособного образовывать водородную связь диметилового эфира (—24 °C).
Прежде чем говорить о строении молекулы и о свойствах различных классов соединений, остановимся на строении атома с точки зрения квантово-механических представлений.
Химическая связь. Взаимное влияние атомов в органических соединениях
17
Как уже отмечалось, современная теория химической связи основана на квантово-механическом рассмотрении молекулы как системы из электронов и атомных ядер.
Из курса неорганической химии и физики известно, что электроны представляют собой вид материи, обладающий одновременно свойствами частицы и электромагнитной волны.
Согласно квантовой теории состояние электронов в атоме описывается с помощью четырех квантовых чисел: и — главное квантовое число, / — азимхгальное квантовое число, т — магнитное квантовое число и л — спиновое квантовое число.
Электрон в атоме находится на определенной атомной орбитали. Атомная орбиталь — это область пространства внутри которой наиболее вероятно нахождение электрона.
Состояние электрона определяется расстоянием электронного облака от ядра, его формой, ориентацией в пространстве и вращением электрона вокрс г собственной оси.
В зависимости от расстояния электрона от ядра атома изменяется траектория его движения, то есть форма атомной орбитали. Существуют s.p. «/./-атомные орбитали, которые отличаются друг от друга запасом энергии.
з-орбпталь р-орбитнль «/-орбиталь
Рис. 2.1. Геометрическая форма s-,p- и «/-атомных орбиталей
Для атомных орбиталей .s-типа характерна сферическая симметрия, для электронов p-типа существуют три одинаковых по энергии гантелеобразной формы орбитали, которые отличаются друг от друга лишь ориентацией в пространстве: ре рг, p.-атомные орбитали. В каждой из них существует узловая область р-орбитали. где вероятность нахождения электрона равна нулю. Для «/-атомных орбиталей существуют пять более сложных геометрических форм
Электроны 5-орбитати ближе находятся к атомному ядру и с большей силой притягиваются к нему, чем р-электроны, которые более удалены и имеют большую подвижность. Энергия электрона падает в следующем ряду:
f>d>p>s
Атохшая орбиталь, не занятая электронами, называется вакантной и условно обозначается как □.
_________________________________Лекции по органической химии
ГИБРИДИЗАЦИЯ АТОМНЫХ ОРБИТАЛЕЙ
Согласно квантово-механическим представлениям о химической связи число образуемых атомом ковалентных связей определяется количеством одноэлектронных атомных орбиталей, то есть количеством неспаренных электронов. Однако в действительности атомы элементов образуют большее число ковалентных связей, чем содержат неспаренных электронов на внешнем энергетическом уровне. Например, атом углерода в основном состоянии имеет два неспаренных электрона (1л2 Is2 Ip2), а образует четыре ковалентных связи. Это можно объяс-
Таким образом, на внешнем энергетическом уровне атома углерода находятся четыре неспаренных электрона: один-s и три-р. Поскольку химические связи образуются валентными электронами, то связи, например, в молекуле метана СН4 должны были бы быть неравноценными: одна связь С—Н образована^-электроном, а три остальные — р. В действительности в молекуле метана все связи совершенно равноценны. Для объяснения этого факта в квантовой механике вводится понятие о гибридизации атомных орбиталей (АО). Слово гибридизация означает взаимодействие, перекрывание, перемешивание. При взаимодействии одного s-электронного облака с тремя p-электронными облаками образуются четыре качественно новых гибридизированных электронных облака или атомные орбитали:
1s + Зр = 4SP3
Таким образом, из нескольких различных по форме и близких по энергии АО путем комбинирования (смешивания, сочетания) образуется такое же количество одинаковых по форме и равных по энергии гибридизованных атомных орбиталей:
Гибридизованные орбитали по сравнению с негибридизованны-ми более выгодны геометрически, т. к. позволяют увеличить плошадь перекрывания с орбиталями других атомов, что ведет к образованию более прочных связей. Результатом перекрывания большей доли гибридной орбитали с орбиталями других атомов является ковалентная связь.
2. Химическая связь. Взаимное влияние атомов в органических соединениях ’ ”
Атом углерода характеризуется тремя видами гибридизации с участием 5- ир-орбиталей, каждому из которых соответствует определенное валентное состояние атома.
Первое валентное состояние углерода (sp3-гибридизация).
Образование О-связи.
Состояние sp’-гибридизации — результат взаимодействия одной s- и трехр-атомных орбитатей.
25-орбиталь
2д-орбиталь
2ру-орбиталь
2р.-орбиталь
Рис. 2.2. Схема образования и расположение в пространстве гибридных 5р3-орбиталей
Четыре равноценные орбитали между собой образуют угол 109°28' и ориентированы в пространстве от центра правильного тетраэдра к его вершинам. Такое размещение связано со стремлением АО к максимальному удалению друг от друга за счет взаимного электростатического отталкивания. Расположение атомных орбиталей определяет название состояния sp3 гибридизации как тетраэдрическое.
Доля 5-облака в каждой из четырех гибридных 5р3-орбиталей равна '/4. В результате перекрывания таких орбиталей с другими орбиталями (5, р, d и гибридными sp3, sp1, sp) вдоль линии, соединяющей центры атомов, образуются только простые ковалентные связи — о (греч. «сигма») связи. Перекрывание атомных орбиталей вдоль линии, соединяющей центры атомов, называют о-перекрыванием или осевым, так как максимальная электронная плотность при этом находится на оси, соединяющей два ядра.
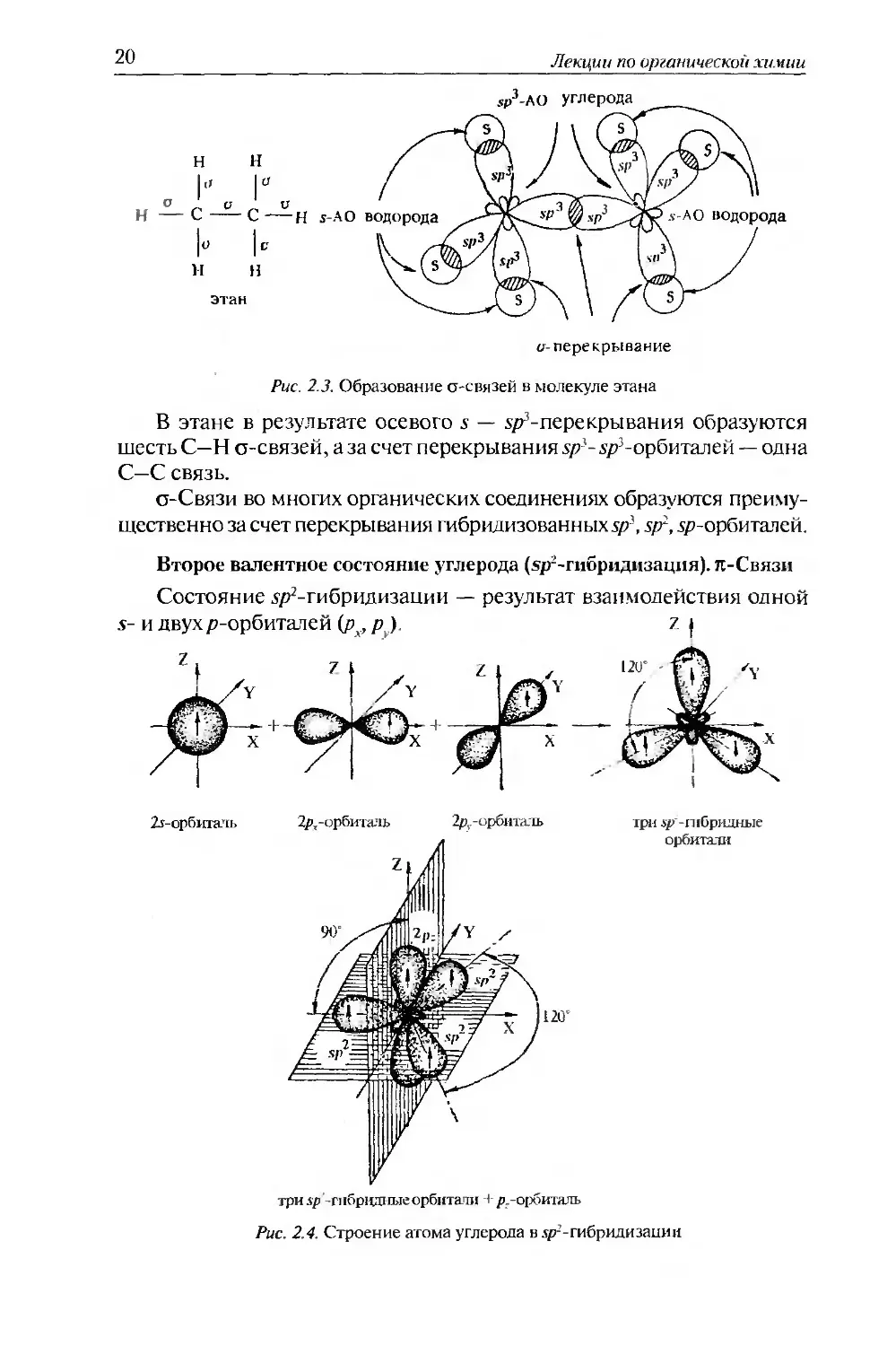
Состояние sp3-гибридизации характерно для алканов. Рассмотрим образование о-связей на примере этана.
20
Лекции по ор/анической химии
Рис. 2.3. Образование о-связей в молекуле этана
В этане в результате осевого .s — х/?-перекрывания образуются шесть С—Н о-связей, аза счет перекрывания .у/-уР-орбиталей — одна С—С связь.
о-Связи во многих органических соединениях образуются преимущественно за счет перекрывания гибрилизованных уД sp~, гр-орбиталей.
Второе валентное состояние углерода (вр2-гибридизация). тг-Связи
Состояние лр’-гибридизаиии — результат взаимодействия одной 5- и двухр-орбиталей (pv, р)
2у-орбиталь
три sp’-гибридные орбитали
Рис. 2.4. Строение атома углерода в sp2-гибридизации
2. Химическая связь. Взаимное влияние атомов в органических соединениях
21
Образованные три эквивалентные з/л-гибридные орбитали находятся в одной плоскости подугто.м 120°, поэтому .^-гибридизация называется тригональной. Негибридизованная р_-орбиталь расположена в плоскости, которая перпендикулярна плоскости расположения гибридных орбиталей. Условно доля s-облака в каж юй из трех .^-гибридных орбиталей равна '/3. Такая гибридизация характерна для соединений с двойными связями, например, для этилена.
Рис. 2.5. Образование л-связи в молекуле этилена
Атомы углерода в этилене находятся влр2-гибридизации. Перекрывание трех гибридных орбиталей каждого из углеродов дает о-связи (четыре С-Н и одну С-С). Кроме того, перекрывание двух негибридизо-ванныхр-орбиталей в плоскости, перпендикулярной плоскости о-связи. называют л-перекрываннем, а образующаяся в результате связь — л-связью. Ее максимальная электронная плотность сконцентрирована в двух областях — выше и ниже оси, соединяющей центры атомов. л-Связь менее прочна, чем о. л-Связь образуется только между атомами. которые находятся vsp1- или sp-гибридизации.
Третье валентное состояние (sp-гибридизация)
Состояние sp-гибридизации — результат взаимодействия одной s- и одной р-орбитали (д).
зр-Гибридизацию называют еше линейной потому, что две sp-гиб-ридные орбитали расположены под углом 180°. Остальные две негиб-ридизованные р- и д-орбитали находятся в двух взаимно перпендикулярных плоскостях и расположены под прямым углом к sp-гибрид-ным орбиталям. Долял-облака в каждой из двух гибридных sp-орбита-лей равна '/ Такой тип гибридизации характерен для соединений с тройной связью, например, для ацетилена.
В молекуле ацетилена sp-гибридизованные атомы образуют две простые С—Н о-связи и одну тройную связь между двумя атомами углерода, которая состоит из одной о- и двух л-связей, расположенных во взаимно перпендикулярных плоскостях.
22
Лекции по арктической химии
две лр-гибридные орбитали
б
Рис. 2.6. Строение атома углерода в .sp-гибридизации: а — схема образования гибридных sp-орбиталей; б — взаимное размещение орбиталей при зр-гибридизации
Рис. 2.7. Образование л-связи в молекуле ацетилена
Для описания химической связи с позиций квантовой механики пользуются двумя основными методами: валентных связей (ВС) и молекулярных орбиталей (МО).
Метод валентных связей был предложен в 1927 году В. Гайтлером и Ф Лондоном. Основные положения метода заключаются в следующем. Химическая связь представлена в виде пары электронов с про-’И’К'положными спинами. Она образуется в результате перекрывания атомных орбиталей.
2. Химическая связь. Взаимное влияние атомов в органических соединениях
23
При образовании молекулы атомные орбитали остаются без изменений, а пара связывающих электронов локализована между двумя атомами.
В отличие от метода валентных связей метод молекулярных орбиталей рассматривает молекулу не как совокупность атомов, сохраняющих свою индивидуальность, а как единое целое. Предполагается, что каждый электрон в молекуле движется в суммарном поле, создаваемом остальными электронами и всеми ядрами атомов. Иначе говоря, в молекуле различные АО взаимодействуют между собой с образованием нового типа орбиталей, называемых молекулярными орбиталями (МО).
Перекрывание двух атомных орбиталей приводит к образованию двух молекулярных орбиталей.
Рис. 2.8. Схема образования ковалентной связи Н—Н
Одна из них имеет более низкую энергию, чем исходные АО, и называется связывающей орбиталью, другая обладает более высокой энергией, чем образующая ее АО, и называется разрыхляющей или ан-тисвязывающей орбиталью. Заполнение молекулярных орбиталей электронами происходит аналогично заполнению атомных, то есть по принципу Паули и в соответствии с правилом Гунда. Молекулярная разрыхляющая орбиталь в основном состоянии остается вакантной. Ее заполнение электронами происходит при возбуждении молекулы, что ведет к разрыхлению связи и распаду молекулы на атомы.
ВЗАИМНОЕ ВЛИЯНИЕ АТОМОВ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Согласно современным представлениям, природа и механизм взаимного влияния атомов определяется характером распределения электронной плотности в молекуле и поляризуемостью ковалентных связей.
24
Лекции по органической химии
Электронные смешения в органических соединениях подразделяют на два вида: индуктивный эффект — смешение электронной плотности по цепи о-связей и мезомерный эффект — смешение по системе л-связей.
Индуктивный эффект. Рассматривая типы химических связей, мы отмечали, что между атомами с одинаковой электроотрицательностью пара электронов связи в равной мере принадлежит обоим участникам связи (ковалентная неполярная связь). Например, связи в молекулах метана, бутана — неполярные, электронная п лотность в них распределена симметрично и молекула не имеет дипольного момента. Если же в молекуле бутана один атом водорода заместить на галоген — хлор, то электронная плотность ковалентной связи С—С1 смешается к более электроотрицательному атому хлора (ковалентная полярная связь)
8'"' 8"+ 8'+ 8* 8
4 3 2 1
НС - СН, - СН, - СН, Н,С -э СН, -э СН, -> СН, -> С1
л 2 2 3 3 222
«-бутан, ц=0 1-хлорбутан. ц*0
Пара электронов о-связи принадлежит как углероду, так и хлору, но несколько смешена к хлору, поэтому хлор приобретает частично отрицательный заряд (8~), а атом углерода связи С—О — равный по величине частично положительный заряд (8‘).
Уменьшение электронной плотности на С’ приводит к тому, что последний, проявляя акцепторные свойства, смещает на себя электроны 8-связи от соседнего атома углерода. Происходит поляризация связи С2—С1 и частичный положительный заряд возникает также на С2, что в свою очередь ведет к поляризации связи С2—С3 и возникновению частичного положительного заряда на С3 и т. д. При этом дробный положительный заряд на атомах углерода в цепи от С до С4 уменьшается: 8+ > 8'+ > 8"+ > 8"'+.
Поляризация одной связи углерод-галоген вызывает поляризацию молекулы в целом и следовательно появление дипольного момента.
Индуктивный (индукционный) эффект — передача электронного влияния заместителя вдоль цепи о-связей, которая возникает в силу различной электроотрицательности атомов.
Индуктивный эффект обозначается буквой /, а смешение электронной плотности изображают с помощью стрелки вдоль простой о-связи. острие которой указывает на направление смещения.
По направлению электронного влияния заместителей различают положительный +/и отрицательный -/индуктивный эффект.
Отрицательный индуктивный эффект проявляют заместители, притягивающие электроны о-связи, например: —МО2 —CsN, —СООН, -Hal, -ОН.
2. Химическая связь. Взаимное влияние атомов в органических соединениях
25
Отрицательный индуктивный эффект, как правило, увеличивается с ростом электроотрицательности атомов. Он сильнее выражен для заместителя с тройной связью, т. к. в его составе находится более электроотрицательный зр-гибридизованный атом углерода. В свою очередь атом углерода в зр3-гибридизации, как менее электроотрицательный, в составе заместителя проявляет +/ по отношению к атомам углерода в зр и зр2-гибридизации:
Положительный индуктивный эффект проявляют заместители, отталкивающие от себя электроны о-сьязи. чаше всего это алкильные группы (Aik). Электронодонорные свойства у алкильных заместителей возрастают с ростом длины углеводородной цепи (-С4Н9 > -СН3) и увеличиваются в ряду от первичных до третичных радикалов ((СН3),С— > (СН,),СН- > СН3СН2— > СН3—). Последнее объясняется тем, что индуктивный эффект затухает по цепи.
Суммируя вышеизложенное, кратко остановимся на основных свойствах индуктивного эффекта:
1. Индуктивный эффект проявляется лишь при наличии в молекуле атомов с различной электроотрицательностью.
2. Индуктивный эффект распространяется только через о-связи в одном направлении.
3. Индуктивный эффект быстро затухает по цепи. Максимум его действия — четыре о-связи.
4. Индуктивное смещение определяется наличием дипольного момента: рД).
Мезомерный эффект (эффект сопряжения). Прежде чем рассматривать передачу электронного влияния заместителей по системе тг-свя-зей, определим понятия сопряженная система и сопряжение.
Сопряженной называется система, в которой имеет место чередование простых и кратных связей, либо соседство атома, имеющего вакантную р-орбиталь или неподеленную пару р-электронов. Сопряженные системы бывают с открытой и замкнутой цепью:
Н2С=СН-СН=СН2 СН2=СН-С1 сн2=сн-сн2+
нафталин
бутадиен-1.3
хлорэтен аллильный катион
Каждая из приведенных цепей сопряженных связей называется еще цепью конъюгации (от латин. — перекрывание, наложение). В них
26
Лекции по органической химии
имеет место сопряжение — дополнительное перекрывание л- ир-ор-биталей, имеющих параллельные оси симметрии (компланарные). За счет сопряжения происходит перераспределение (делокализация) л-электронной плотности и образование единой л-электронной системы.
От вида перекрываемых орбиталей различают несколько видов сопряжения: л,л-сопряжение (прекрывание двух л-орбиталей), /?,л-со-пряжение (перекрывание р- и л-орбиталей):
Рис. 2.9. Сопряженные системы бутадиена-1,3. хлористого винила и аллильного катиона
Сопряжение — энергетически выгодный процесс, происходящий с выделением энергии. Сопряженные системы характеризуются повышенной термодинамической устойчивостью.
Дав определение сопряжению и сопряженным системам, рассмотрим электронные эффекты, которые наблюдаются при введении в такие системы различного рода заместителей.
2. Химическая связь Взаимное влияние атомов в органических соединениях
Эффект сопряжения или мезомерный эффект (М) — процесс передачи электронного влияния заместителя по сопряженной системе п-связей Смешение электронной плотности в сопряженных системах возможно лишь при включении в систему электронодонорных или электроноакцепторных заместителей.
Например, в молекуле бензола имеется сопряжение, но нет заместителей. поэтому мезомерный эффект отсутствует. Гидроксигруппа в молекуле фенола входит в сопряженную систему и проявляет мезо-мерный эффект, а в молекуле бензилового спирта —ОН группа изолирована от сопряженной системы дву мя о-связями и не проявляет ме-зомерного эффекта.
бензол
фенол
бензиловый спирт
Мезомерный эффект обозначают буквой М, а смещение электронной плотности в сопряженной системе — изогнутой стрелкой. По направляющему действию заместителя мезомерный эффект делится на положительный (+Л/) и отрицательный (-М).
Положительный мезомерный эффект проявляют заместители (электронодонорные атомы или атомные группы), предоставляющих электроны в сопряженную систему, т. е. имеющие неподеленные пары электронов или отрицательный заряд:
...СН, .. .. .. .. .........
-О ;-N —NHCH —NH,;—осн,;-ОН;—F;—ci; —Вг
СН, ’
Максимальный +Му атомов с отрицательным зарядом. Заместители, содержащие неподеленные пары электронов, имеют тем больше +М, чем меньше в пределах периода электроотрицательность атомов, содержащих неподеленные пары электронов.
Отрицательный мезомерный эффект проявляют заместители, смещающие на себя электронную плотность сопряженной системы:
+ + х. ^>0
—OR2;-NR,;—C'=N;/С=0; —С;* ;
О OR’
^О ^0
он nh2
28
Лекции по органической химии
Максимальный — Мпроявляют заместители, несущие положительный заряд. В ненасыщенных группировках — Л/-эффект возрастает с увеличением разности электроотрицательности атомов кратной связи.
Рассмотрим несколько примеров проявления мезомерного эффект;
фенол
хлорэтен
л-аминобензолсульфо кислота
Мезомерный эффект по сравнению с индуктивным вызывает более сильное смещение электронной плотности и практически не затухает.
Совместное проявление индуктивного и мезомерного эффектов заместителя
Мезомерный и индуктивный эффекты одного заместителя могут совпадать и не совпадать по направлению. Например, в молекуле ак-
ролеина альдегидная группа — С ' проявляет — I и — М, а гидро-
ксильная группа в молекуле фенола обладает —I, но +М-эффектом.
2. Химическая связь Взаимно" зияние атомов в органических соединениях
29
акролеин
Как видно из приведенного примера, в молекуле фенола противоположное электронное смешение приводит к тому, что эти два эффекта как бы «гасят» друг друга. А в молекуле акролеина индуктивный и мезомерный эффекты усиливают друг друга. Мезомерный эффект заместителя обычно больше, чем индуктивный, так как л-связи поляризуются легче, чем о-связи.
Поляризация, обусловленная мезомерным эффектом, имеет альтернирующий характер: под влиянием заместителя смешаются не только л-электронные облака, но и облака о-связей. Это явление наблюдается в системах с открытой и замкнутой цепью сопряжения:
анилин
Хотя аминогруппа проявляет -/-эффект, вызывает понижение электронной плотности на всех атомах углерода ароматического цикла, но за счет -гЛ/-эффекта пары электронов атома азота, который больше —1. в целом наблюдается повышение электронной плотности на атомах углерода бензольного кольца, особенно в положениях 2. 4. 6. Происходит альтернирующая поляризация.
В молекулах с открытой цепью сопряжения обычно указывают частичные заряды, которые сосредоточены на концах сопряженной системы:
8- ЛА
Н2С=СН-^С—N
акрилонитрил
Эффект сверхсопряжения (гиперконъюгация). Наряду с л,л- и р,п-сопряжением имеет место особый вид сопряжения — гиперконъюгация (сверхсопряжение) или о.л-сопряжение.
Эффект сверхсопряжения — взаимодействие, которое возникает при перекрывании электронного облака о-орбиталей связи С—Н
30 Лекции по органической химии
с л-орбиталями кратной связи. Такой вид перекрывания электронных облаков представляет собой о,л-сопряжение. которое присутствует как в алифатическом, так и в ароматическом ряду соединений. Смешение электронов при этом изображают с помощью изогнутой стре ikm. Любая из о-связей метильной группы пропена может участвовать в о.л-со-пряжении.
О, п-сопряжение
Рис. 2.10. Схема перекрывания а-орбиталей связи С—Н с п-орбиталью кратной связи в молекуле пропена
Величина эффекта гиперконъюгации тем выше, чем больше атомов водорода при углероде, связанном с ненасыщенной системой. Понятие о сверхсопряжении объясняет повышенную реакционную способность, подвижность а-водородных атомов в молекулах альдегидов, кетонов, кислот и их производных. Иногда сверхсопряжение называют по имени открывших его ученых эффектом Натана — Бекера.
3. ИЗОМЕРИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ. ПРОСТРАНСТВЕННОЕ СТРОЕНИЕ МОЛЕКУЛ
Термин изомерия (от греч. isos — одинаковый, meros — часть) впервые был введен в 1830 г., когда стали известны вещества, имеющие одинаковый качественный и количественный состав, но обладающие различными физическими и химическими свойствами.
Изомерия — это явление, заключающееся в существовании соединений, имеющих одинаковую молекулярную формулу, но различающихся порядком связывания атомов в молекуле или расположением атомов в пространстве, и вследствие этого различающихся по физическим и химическим свойствам.
Такие соединения называются изомерами. Различают два основных вида изомерии — структурную (изомерия строения) и пространственную (стереоизомерия).
СТРУКТУРНАЯ ИЗОМЕРИЯ
Структурные изомеры — это соединения, имеющие одинаковую молекулярную формулу, но отличающиеся друг от друга порядком связывания атомов в молекуле.
32
Ле,Щ1‘и по органической химии
Структурная изомерия подразделяется на изомерию углеродной цепи, изомерию положения и изомерию функциональных групп.
Изомерия углеродной цепи. Обусловлена различной последовательностью связывания атомов, образующих углеродный скелет молекулы. Например, для алкана состава С4Н|0 можно написать два изомера:
С4Н10 СН — СН—СН—СН3 СН —СН —СН
и-бутан J,
изобутан
Для органических соединений циклического строения изомерия цепи может быть вызвана величиной цикла.
с6н12
циклогексан
метилциклопентан
Изомерия положения обусловлена разным положением функциональных групп, заместителей или кратных связей в молекуле.
с,н,„о сн-сн-сн-сн,-он сн—сн—сн—сн,
бутанол-1 ОН
бутанол 2
с4н8 нс2=сн-сн2-сн3 сн3-сн=сн-сн3
бутен-1 бутен-2
Изомерия функциональных групп обусловлена присутствием в изомерах одинакового состава различных по природе функциональных групп.
С,Н,0 сн,—сн.-он
z О 3 2
этанол
сн—о—сн3 диметиловый эфир
ПРОСТРАНСТВЕННАЯ ИЗОМЕРИЯ (СТЕРЕОПЗО ЧЕРНЯ)
Пространственные изомеры — это соединения, имеющие одинаковую молекулярную формулу, одинаковый порядок связывания атомов в молекуле, но отличающиеся друг от друга расположением атомов в пространстве.
Пространственные изомеры называют также стереоизомерами (от греч. stereos — пространственный).
3. Изомерия органических соединений. Пространственное строение '-олекул 33
Пространственная изомерия подразделяется на конфигурационную и конформационную.
Но прежде, чем перейти к рассмотрению этих видов стереоизомерии, остановимся на способах изображения пространственного строения молекул органических соединений.
Для изображения пространственного строения молекул, их конфигурации или конформации используют молекулярные модели и специальные стереоформулы.
Молекулярные модели — наглядное изображение молекул органических и неорганических соединений, позволяющее судить о взаимном расположении атомов, входящих в состав молекулы.
Наиболее часто используют три основных типа моделей: шаростержневые (модели Кекуле — Вант — Гоффа), скелетные (модели Драйдин-га) и полусферические (модели Стюарта — Бриглеба). Модели позволяют судить не только о взаимном расположении атомов в молекуле, но они удобны и для рассмотрения валентных углов и возможности вращения вокруг простых связей. Модели Драйдинга учитывают и межатомные расстояния, а модели Стюарта — Бриглеба отражают и объемы атомов. Ниже на рисунке приведены модели молекул этана и этилена.
сн—сн3
этан
н,с=сн2
этилен
Рис. 3.1. Модели молекул этана (слева) и этилена (справа): а — шаростержневые; б — Драйдинга; в — полусферические (Стюарта — Бриглеба)
34 Лекции по органической химии
Стереоформулы. Для изображения пространственного строения молекулы на плоскости чаще всего используют стереохимические и перспективные формулы, а также проекционные формулы Ньюмена.
В стереохимических формулах химические связи, расположенные в плоскости чертежа, изображают обычной чертой; связи, находящиеся над плоскостью — жирным клином или жирной чертой, а расположенные под плоскостью — штриховым клином или пунктирной линией:
Н
С
этан
Перспективные формулы описывают пространственное строение на плоскости с учетом рассмотрения молекулы вдоль одной из углерод-углеродных связей. По внешнему виду они напоминают лесопильные козлы:
При построении проекционных формул Ньюмена молекулу рассматривают в направлении одной С—С-связи таким образом, чтобы атомы, образующие данную связь, заслоняли друг друга. Из выбранной пары ближний к наблюдателю атом углерода изображают точкой, а дальний — окружностью. Химические связи ближнего атома углерода с другими атомами представляют линиями, берущими начало отточки в центре круга, а дальнего — от окружности:
сн-сн3
3. Изомерия органических соединений. Пространственное строение молекул ^5
Существуют проекционные формулы Фишера, которые применяют обычно для изображения на плоскости пространственного строения оптических изомеров.
Рассмотрим получение проекционной формулы Фишера для бутанола-2 (СН3—СН(ОН)—СН2—СН3). Для этого модель молекулы располагают таким образом, чтобы атом углерода, связанный с гидроксильной группой находился в плоскости чертежа, а заместители, расположенные горизонтально, были над плоскостью, расположенные вертикально — за плоскостью чертежа. При проецировании такой модели на плоскость получают проекционную формулу Фишера, в которой связи, находящиеся над плоскостью, изображают горизонтальной линией, а связи, находящиеся за плоскостью, — вертикальной линией. В точке пересечения этих линий находится атом углерода, который обычно не обозначается символом:
сн3
сн—сн—сн—сн3 но—с—н
он
СН
НО
с2н5
проекция Фишера
бутанол-2
с2н5
КОНФИГУРАЦИОННАЯ ИЗОМЕРИЯ
К конфигурационной относится оптическая и геометрическая изомерия.
ОПТИЧЕСКАЯ ИЗОМЕРИЯ
В1815 г. Ж. Био открыл существование оптической активности для органических соединений. Было установлено, что некоторые органические соединения имеют способность вращать плоскость поляризации поляризованного света. Вещества, которые обладают такой способностью, называются оптически активными.
Если луч обычного света, в котором, как известно, электромагнитные колебания распостраняются в разных плоскостях, перпендикулярных к направлению его распространения, пропустить через призму Николя, то выходящий свет будет плоскополяризованным. В таком луче электромагнитные колебания совершаются только в одной плоскости. Эту плоскость называют плоскостью поляризации (рис. 3.2).
При прохождении поляризованного луча света через оптически активное вещество плоскость поляризации поворачивается на определенный угол а вправо или влево. Если вещество отклоняет плоскость
36
Лекции по органический химии
поляризации вправо (при наблюдении навстречу лучу), его называют правовращающим, если влево — левовращающим. Правое вращение обозначают знаком (+), левое — знаком (—).
Рис. 3.2. Схема образования поляризованного света и вращения плоскости поляризации оптически активным веществом
Оптическую активность измеряют с помощью приборов, называемых поляриметрами.
Явление оптической активности распространено среди органических веществ, особенно среди природных (гидрокси- и аминокислот, белков, углеводов, алкалоидов).
Оптическая активность большинства органических соединений обусловлена их строением.
Одной из причин возникновения оптической активности органических молекул является наличие в их структуре з/Р-гибридизирован-ного атома углерода, связанного с четырьмя разными заместителями. Такой атом углерода называется хиральным или асимметрическим. Часто для него применяют более общее название — хиральный центр. В структурных формулах асимметрический атом углерода принято обозначать звездочкой — С*:
Н
I сн,—с*—сн,—СП.
С1
2-хлорбутан
н
I сн,—с*—соон
3 I он
молочная кистота
Соединения, содержащие один асимметрический атом углерода, существуют в виде двух изомеров, относящихся друг к другу как пред-
* Призма Николя — призма, изготовленная из двух кристаллов исландского шпата (СаСО3), склеенных канадским бальзамом
3. Изомерия органических соеоинений. Пр<л транств^ иное строение .ио. е^у.ч
37
мет к своему зеркальному отображению. Такие изомеры называются энантиимерами
а б в
Рис. 3.3. Модели энантиомерных молекул бромиодхлорметана
Для изображения пространственного строения оптических изомеров на плоскости могут быть использованы стереохимические формулы. Например, энантиомеры бутанола-2, изображенные с помощью стереохимических формул, имеют следующий вид:
Однако стереохимические формулы не всегда удобны для описания пространственного строения молекул. Поэтому чаше всего оптические изомеры изображают на плоскости с помощью проекционных формул Фишера. Например, так выглядят энантиомеры 2-бромбута-на. изображаемые с помощью проекции Фишера.
СН3
Н-------Вг
С2Н5
СН
с2н5
Энантиомеры очень похожи друг на друга, но тем не менее не тождественны. Они имеют одинаковый состав и последовательность связывания атомов в молекуле, но отличаются друг от друга относительным расположением их в пространстве, т. е. конфигурацией. В том, что эти молекулы разные, легко убедиться при попытке наложения их моделей друг на друга.
Свойство молекул не совмещаться со своим зеркальным изображением называется хиральностью (от греч. cheir— рука), а также молекулы называют хиральными. Наглядным примером могут служить левая
38
Лекции по органической химии
и правая руки, которые являются зеркальным отражением друг друга, но вместе с тем их нельзя совместить. Молекулы, которые совместимы со своим зеркальным изображением называют ахиральными.
Хиральность молекул является обязательным условием для проявления веществом оптической активности.
Как установить является ли молекула хиральной? Хиральность молекулы можно легко обнаружить путем построения модели молекулы и модели ее зеркального изображения с последующим их совмещением. Если модели не совмещаются — молекула хиральна, если совмещаются — ахиральна. Такой же вывод можно сделать и на основе стереохимических формул молекул по наличию или отсутствию элементов симметрии, так как причиной оптической активности органических соединений является их асимметрическое строение. Поскольку молекула представляет собой трехмерное образование, ее строение можно рассматривать с точки зрения симметрии геометрических фигур. Основным элементами симметрии являются плоскость, центр и ось симметрии. Если в молекуле отсутствует плоскость симметрии, то такая молекула хиральна.
Энантиомеры обладают одинаковыми физическими и химическими свойствами (температура кипения, температура плавления, растворимость, электропроводность и другие константы будут одни и те же), вращают плоскость поляризации поляризованного луча на один и тот же по величине угол, но имеются и различия.
Энантиомеры отличаются знаком вращения, один вращает плоскость поляризации поляризованного луча влево, другой — вправо; они с различной скоростью реагируют с другими хирал ьными соединениями, а также имеется различие в физиологическом действии. Например, лекарственный препарат левомицин — антибиотик широкого спектра действия. Если его эффективность принять за 100, то правовращающая форма составит только 2 % от эффективности левовращающей формы.
Значение величины угла поворота плоскости поляризации поляризованного света зависит от природы активного вещества, толщины слоя оптически активной среды, через которую проходит поляризованный свет, и длины его волны. Для растворов угол а зависит также от природы растворителя и концентрации оптически активного вещества. В меньшей степени оптическое вращение зависит от температуры.
Для сравнительной оценки оптической активности различных соединений используют значение удельного вращения [а]. Удельное вращение является константой оптически активного вешества. Оно характеризует оптическую активность раствора с концентрацией оптически активного вещества 1г/мл при толщине слоя 1 дм.
3. Изомерия органических соединений. Пространственное строение молекул
Удельное вращение вычисляют по одной из приведенных формул:
а
а 100
для веществ в растворе
1с
для жидких веществ а
а
/р ’
где а— измеренный угол вращения, град.; / — толщина слоя, дм; с — концентрация оптически активного вещества, г/100 мл раствора; р — плотность жидкого вещества.
Если молекула имеет один асимметрический атом, то она существует в виде двух изомеров, если же молекула имеет несколько асимметрических атомов углерода, то число возможных изомеров у вели-чивается. Число оптических изомеров определяют по формуле:
N = 2", где N — число изомеров; п — число асимметрических атомов углерода. Так при наличии в молекуле двух асимметрических атомов углерода число изомеров равно 21 = 4, трех — 23 = 8, четырех — 24 = 16 и т. д.
Например, бромяблочная кислота, содержащая два асимметрических атома углерода, существует в виде четырех стереоизомеров (I—IV).
СООН 1 соон соон соон соон
*
Vxrlvjrl L1 ии но 11 11 _О11 11О 11
Rr
СНВг н ВГ В1 11 ВГ 11 н иг
СООН соон соон соон соон
I п III IV
Стереомеры I и II, атакже III и IV относятся друг к другу как предмет и его зеркальное изображение и являются энантиомерами.
Стереоизомеры I и III, I и IV, а также II и III, II и IV не являются зеркальными отображениями друг друга, они отличаются конфигурацией при одном из асимметрических атомов углерода. Такие стереоизомеры называют диастереомерами. В отличие от энантиомеров диастереомеры имеют различные физические и химические свойства.
Для соединений, содержащих два хиральных атома углерода, связанных с одинаковыми заместителями, общее число стереоизомеров уменьшается до трех. Например, винная кислота должна существовать в виде четырех стереоизомеров (22 = 4), а известно лишь три. Это обусловлено появлением у одного из стереоизомеров такого элемента, как плоскость симметрии.
40
Лекции п(. органической химии
СООН с зоон - гли ( зоон ( ЗООН
* ‘ 11
СМОп Н ОН но н н он
игл 14 гл 1 I
*снон НО Н 11 он н П ПЛОСКОСТЬ
1 симметрии
СООН СООН СООН СООН
I III
Стереомеры I и II являются энантиомерами. Стереоизомер III (.мезо-форма) является оптически неактивным Молекула мезовинной кислоты ахиральна. Каждый энантиомер винной кислоты по отношению к .мезо-форме является диастереомером.
Номенклатура оптических изомеров
В номенклатуре наряду с названием соединения указывают также конфигурацию и направление вращения плоскости поляризованного света. Последнее обозначают знаком (+) для правовращающего изомера или знаком (—) для левовращающего изомера.
Для обозначения конфигурации оптических изомеров существуют D,L- и /?,S-стереохимические системы.
D,/.-система обозначения конфигурации. Установить абсолютную конфигурацию молекул оказалось для химиков довольно сложной задачей. Впервые это удалось лишь в 1951 г методом рентгеноструктурного анализа. До этого времени конфигурацию оптических изомеров устанавливали методом сравнения со специально выбранным стандартным веществом. Такая конфигурация получила название относительной. В 1906 г. русским ученым М.А. Розановым в качестве стандарта для установления относительной конфигурации был предложен глицериновый альдегид.
/О
с< н
н------он
сн2он
£>- (+)-глицериновый
альдегид
с< н
но-------н
сн2он
L- (-)-глицериновый
альдегид
Для правовращающего изомера выбрали формулу Фишера, в которой гидроксильная группа у хирального атома углерода находится справа, а для левоврашаюшего — слева. Конфигурация правовращающего изомера обозначается буквой D, а левоврашаюшего — L.
3. Изомерия органических соединена ' Пространственное строение можкул 4 1
С использованием в качестве эталона сравнения глицеринового альдегида была разработана D./.-система стереохимической классификации хиральных соединений, т. е. отнесения соединений соответственно к D- или /.-стереохимическому ряду.
D.A-система главным образом применяется в ряду многоатомных спиртов, гидрокси-, аминокислот и углеводов:
( 2НО соон ( 2ООН
Г И и
L Н_ ОН 11 ОН гт2ГМ'
( :н2он сн, 1 2 ( зн2
СООН СООН
Р-(+)-глицериновый альдегид
D- яблочная кислота
/.-аспарагиновая кислота
Для соединений с несколькими асимметрическими атомами углерода. таких как а-гидроксикислоты, а-аминокислоты, винные кислоты. конфигурацию условно определяют по верхнему асимметрическому атому углерода (по гидроксикислотному ключу), в то время как в молекуле углеводов конфигурацию устанавливают (условно) по нижнему асимметрическому атому углерода.
сно
( :но
11 ОН
1
сн2он
D- (-^(-глицериновый альдегид
( 2ООН
н— —ОН
но— — Н
( 200Н
Р-винная кислота
соон
гЦГ\ 11
н- он
сн3
/-треонин
н— —он
но— —н
н— —он
н— —онп
1
сн2он
D- глюкоза
/?,5-снстема обозначения конфигурации. D,L-система оказалась практически неприемлемой для соединений мало похожих на глицериновый альдегид. Поэтому Р. Каном, К. Ингольдом и В. Прелогом была предложена /?,.S'-система обозначения абсолютной конфигурации оптических изомеров. R,5-система построена на определении старшинства заместителей у хирального центра.
Старшинство заместителей определяется величинами атомных номеров элементов. Чем больше атомный номер, тем старше замести-
4^Лекции по органической хилшг тель. Например, в молекуле бромйодхлорметана старшинство заместителей уменьшается в ряду:
53 > 35®Г > 17^1 > .Н
Н*
I—С— Вг35
С117
После установления старшинства заместителей модель молекулы ориентируют так, чтобы заместитель с наименьшим порядковым номером был направлен в сторону, противоположную глазу наблюдателя. Если старшинство трех остальных заместителей убывает по направлению часовой стрелки, то молекула имеет конфигурацию, обозначаемую буквой R (от лат. rectus — правый), а если старшинство заместителей убывает против часовой стрелки, конфигурацию обозначают буквой S (от лат. sinister—левый). Например, для молекулы бромйодхлорметана:
5-конфигурация
/^-конфигурация
Рис. 3.4. Определение конфигурации по /?,5-системе для молекулы бромйодхлорметана
Рассмотрим определение старшинства заместителей и конфигурации для более сложных молекул на примере молочной кислоты (рис. 3.4). Уже по первому слою (80,6С, ,Н, 6С) становится понятно, что старшим заместителем является группа ОН, а младшим — водород. Для выяснения старшинства двух других заместителей СН3 и СООН с одинаковым атомным номером (6С) по первому слою, необходимо рассмотреть второй слой. Сумма атомных номеров второго слоя СН3-группы = 1 + 1 + 1 = 3, а группы СООН = 8 + 8-2 = 24. Значит -СООН-группа старше группы —СН3. Старшинство заместителей вокруг асимметрического атома углерода в молекуле молочной кислоты уменьшается в ряду: ОН > СООН > СН3 > Н
СН3—СН--СООН
ОН
молочная кислота
3. Изомерия органических соединений. Пространственное строение молекул
43
Я-конфигураиия 5-конфигурация
Рис. 3.5. Определение конфигурации по Я.Л'-систе.ме для молочной кислоты
Рацематы. Смесь равных количеств энантиомеров оптически неактивна, ее называют рацемической смесью (рацематом). Рацематы отличаются от индивидуальных энантиомеров физическими свойствами, они могут иметь различную температуру плавления, растворимость; отличаются спектральными характеристиками.
На практике чаще приходится сталкиваться не с индивидуальными энантиомерами, а рацематами, которые образуются в результате химических реакций, протекающих с образованием хиральных молекул.
Для разделения рацематов на энантиомеры пользуются тремя методами:
1. Механический метод. В результате кристаллизации некоторых оптически активных соединений могут образовываться две формы кристаллов, похожих друг на друга как предмет и его зеркальное отображение. Их можно отделить под микроскопом препаративной иглой (механически)
2. Биохимический метод основан на том, что определенные виды микроорганизмов предпочитают одну из энантиомерных форм и поедают ее, вторая остается и может быть легко выделена.
3. Химический метод. В основе химического метода лежит перевод энантиомеров при помощи оптически активных реагентов в диастереомеры, которые уже отличаются друг от друга по физическим свойствам. Диастереомеры гораздо легче разделить.
Например, следует разделить рацемическую смесь двух кислот (А + В). Для этого к смеси добавляют оптически активное основание (С). Между рацемической формой и оптически активным основанием протекает реакция
(А + В) + С —> АС + ВС.
АС и ВС — это диастереомеры. Они обладают различной растворимостью и методом последовательной кристализации можно выделить два диастереомера отдельно.
44
Лекции по органической химии
Но так как АС и ВС образованы слабой органической кислотой и основанием, то используют для их разложения минеральные кислоты,
АС + НС1 —>А + СНС1
ВС + НО —> В + С • НО
Таким образом получают чистые энантиомеры А и В.
ГЕОМЕТРИЧЕСКАЯ ИЗОМЕРИЯ
Причиной возникновения геометрической изомерии является отсутствие свободного вращения вокруг с-связи. Этот вид изомерии характерен для соединений, содержащих двойную связь, и для соединений алициклического ряда.
Геометрические изомеры это вещества, имеющие одинаковую молекулярную формулу, одинаковую последовательность связывания атомов в молекулах, но отличающиеся друг от друга различным расположением атомов или атомных групп в пространстве относительно плоскости двойной связи или плоскости цикла.
Причиной возникновения данного вида изомерии является невозможность свободного вращения вокруг двойной связи или <7-связей, образующих цикл.
Например, бутен-2 СН3—СН=СН—СН3 может существовать в виде 2-х изомеров, которые различаются расположениями метильных групп в пространстве относительно плоскости двойной связи.
Чис-бутен-2
транс-бутен-2
или 1,2-диметилциклопропан существует в виде двух изомеров, которые различаются расположением метильных групп в пространстве относительно плоскости цикла:
дис-димстил циклопропан троис-диметилциклопропан
Для обозначения конфигурации геометрических изомеров используют цис-, транс-систему. Если одинаковые заместители расположе-
3. Изомерия органических соединений. Пространственное строение чолекул ны по одну сторону от плоскости двойной связи или цикла — конфигурацию обозначают цис-, если по разные стороны — транс-.
Для соединений, у которых при атомах углерода с двойной связью находятся различные заместители, применяют Z.E-систему обозначений.
Z.E-система является более обшей. Она применима к геометрическим изомерам с любым набором заместителей. В основе этой системы лежит старшинство заместителей, которое определяют у каждого атома углерота отдельно. Если старшие заместители из каждой пары расположены по одну сторону от двойной связи, конфигурация обозначается буквой Z(от нем. zusammen — вместе), если по разные стороны — буквой Е(от нем. entgegen — напротив).
Так для 1-бром-1-хлорпропена возможно два изомера:
1 17 I 35
Н С1 Н\ Вт
с=с с=с
6 / \35 6 / \17
Н,С Вт и Н.С С1
3 11 3
1 II
Z-1 -бром-1 -хлорпропен Е-1 -бром -1 -хлорпропен
Старшим заместителем у одного атома углерода является метильная группа (заместители 'Н и (’СН.), а у другого — атом брома (заместители 'CI и 3?Вг). В изомере I старшие заместители расположены по одну сторону от плоскости двойной связи, ему приписывают Z-koh-фигурацию, а изомеру II Е-конфигурацию (старшие заместители расположены по разные стороны п юскости двойной связи).
Гео* 1етрические изомеры имеют разные физические свойства (температуру плавления и кипения, растворимость и т. д.), спектральные характеристики и химические свойства. Такое различие в свойствах позволяет довольно легко установить их конфигурацию с помошью физических и химических методов.
КОНФОРМАЦИОННАЯ ИЗОМЕРИЯ
Конформационная (поворотная) изомерия обусловлена вращением атомов или атомных групп вокруг одной или нескольких простых a-связей. В результате вращения вокруг С—С-связей молекулы могут иметь различные пространственные формы, которые называют конформациями.
Например, молекула этана вследствие вращения вокруг углеродуглеродной связи может принимать бесконечное множество конформаций, каждая из которых характеризуется определенным значением
46
Лекции по органической химии
потенциальной энергии. Две крайние конформации называют заслоненной и заторможенной.
заслоненная конформация этана
заторможенная конформация этана
Для представления конформаций на плоскости используют формулы Ньюмена и перспективные формулы.
В заслоненной конформации этана атомы водорода метильных групп, если смотреть вдоль связи углерод-углерод, расположены друг за другом. В заторможенной — атомы водорода одной метильной группы максимально удалены от атомов водорода другой. Между заслоненной и заторможенной конформацией молекула в процессе вращения принимает множество скошенных конформаций.
Каждая из конформаций молекулы этана характеризуется различной потенциальной энергией. Заслоненная конформация имеет максимальную энергию, а заторможенная — минимальную.
Энергетическая равноценность различных конформаций объясняется существованием в молекуле так называемого торсионного напряжения (напряжения Питцера), которое обусловлено взаимодействием (отталкиванием) электронных облаков противостоящих связей. В заслоненной конформапии противостоящие связи максимально сближены, поэтому взаимодействие между ними наибольшее. Разность энергий заслоненной и заторможенной конформаций называется энергетическим барьером вращения. Для этана энергетический барьер невелик, он составляет около 12 кДж/моль и легко преодолевается молекулой при обычных температурах за счет энергии теплового движения.
При вращении вокруг С2—С3 связи в н-бутане возможны четыре крайние конформации, из которых две заторможенные и две заслоненные:
сн-сн -сн -сн3
анти-
гош-
заторможенные конформации
заслоненные конформации
3. Изомерия органических соединении Пространственное строение молекул
Заторможенная конформация, в которой метильные группы (объемные заместители) максимально удалены друг отдруга, получила название ондаи-конформации. Другая заторможенная конформация называется гош-конформапией.
Заторможенная гош-конформация обладает несколько большей потенциальной энергией (за счет метил-метильного взаимодействия), чем онлпи-конформация (в ней взаимодействие между метилами вообще отсутствует).
Хотя в молекуле н-бутана существует свободное вращение вокруг центральной С—С связи, однако в каждый момент времени большая часть молекул представлена наиболее энергетически выгодной конформацией.
Конформации с наименьшим запасом энергии называют конформерами или конформационными (поворотными) изомерами.
Так, н-бутан при 25 °C существует примерно на 70 % в форме анти-конформера и на 30 % го!«-конформера.
В отличие от конфигурационных изомеров, конформеры превращаются друг вдруга без разрыва химических связей и не поддаются разделению. Они обнаруживаются только физико-химическими методами.
4. КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ РЕАКЦИЙ И РЕАГЕНТОВ
Наиболее широко в органической химии используют классификацию химических реакций, основанную на представлениях об их механизме. Учитывается способ разрыва химической связи, природа интермедиатов, природа реагента и другие факторы.
В соответствии со способом разрыва связи реакции делятся на гомолитические (симметричный разрыв двухэлектронной ковалентной связи с образованием двух свободных радикалов А- [ -В -э А- + В), гетеролитические (несимметричный разрыв связи, приводящий к появлению двух частиц с противоположными зарядами А: । В —> А- + В*) и перициклические (молекулярные).
По природе интермедиатов реакции могут быть ионными, радикальными и др.
Ионные реакции осуществляются при участии заряженных частиц. К этому же типу относятся многие органические реакции, интермедиатами в которых являются карбкатионы или карбанионы, образующиеся в результате гетеролитического распада исходных веществ.
Карбкатионами называют органические катионы, содержащие положительно заряженный атом углерода. Атом углерода в карбкатионе, несущий положительный заряд, находится в ^/-гибридном состоянии: три его о-связи расположены в одной плоскости, а р-атомная орбиталь является вакантной.
Вакантная
4. Классификация химических реакций и реагентов
Карбанионами называют органические анионы, содержащие отрицательно заряженный атом углерода, т. е. трехвалентный атом углерода с неподеленной парой электронов.
Неподеленная пара электронов находится на лр’-гибридизованной атомной орбитали в случае карбанионов ряда алкинов, а карбанионы, в которых неподеленная пара электронов сопряжена с кратной связью, содержат эту пару электронов нар-атомной орбитали.
Радикальные реакции протекают с участием свободных радикалов.
Свободными радикалами называют незаряженные частицы, содержащие неспаренный электрон (одноэлектронную атомную орбиталь).
Неспаренный электрон может находиться на ^-гибридизованной атомной орбитали или нар-атомной орбитали. В зависимости от этого свободный радикал может иметь тетраэдрическое или плоское строение.
Плоское строение имеют простые алкильные радикалы (неспаренный электрон находится на р-АО).
Третичные алкильные радикалы имеют тетраэдрическое строение (неспаренный электрон занимает лр’-гибридную орбиталь).
мстил mpem-бутил
Перициклические реакции протекают без образования промежуточных активных частиц, в таких реакциях происходит синхронный разрыв старых и образование новых химических связей.
Например:
50
Лекции по органической химии
Реакции, протекающие по ионному механизму, делят на нуклеофильные и электрофильные в зависимости от природы атакующего реагента.
Нуклеофильными называют реагенты, которые предоставляют электронную пару для образования химической связи с субстратом. К нуклеофильным реагентам относятся: молекулы, содержащие хотя бы одну неподеленную пару электронов,
NH3, R-NH2, R-OH и др.; ионы, несущие отрицательный заряд (анионы).
О Н, CN. RO. Cf, Вг’ и др.; молекулы, имеющие центры с повышенной электронной плотностью,
С=С
алкены.
арены и др.
Нуклеофилы способны образовывать ковалентную связь с субстратом, атакуя в его молекуле центры с пониженной электронной плотностью.
Электрофильными называют реагенты, принимающие электронную пару от субстрата при образовании с ним химической связи. К электрофильным реагентам относят катионы (Н+, NO2+,C1+ и др.), нейтральные молекулы, имеющие вакантную орбиталь (А1СЦ, ЕеВг3и др.) или центры с пониженной электронной плотностью,
S+5/O
R— Н
6+^0
R—
ОН
галогены С12, Вг2,12 (в присутствии кислот Льюиса).
Электрофильные реагенты способны образовывать ковалентную связь с субстратом, атакуя в его молекуле центры с повышенной электронной плотностью.
Заслуживает внимания широко используемая в органической химии классификация химических реакций по формальным признакам:
1. Реакции присоединения обозначаются символом «А» (от англ addition — присоединение).
А=В+Х —У
А----В
I I
X У
Эти реакции характерны для соединений, имеющих кратные связи. Реакции присоединения могут протекать по следующим возможным механизмам:
4. Классификация химических реакций и реагентов
а) электрофильное присоединение (АЕ);
б) нуклеофильное присоединение (Ач);
в) свободнорадикальное присоединение (AR);
г) молекулярное (синхронное) присоединение.
2. Реакции замещения обозначаются символом «S» (от англ. substitution — замещение).
А—В + X—У ------------*- А—X + В—У
Они характерны для всех классов органических соединений и могут протекать по следующим механизмам:
а) электрофильное замещение (SE):
б) нуклеофильное замещение (SN);
в) свободнорадикальное замещение (SR).
3. Реакции отщепления (элиминирования) обозначаются символом «Е» (от англ, elimination — отщепление)
А----В -----»- А=В + Х —У
X У
Реакции отщепления характерны для галогенопроизводных углеводородов, спиртов, гетерофункциональных карбоновых кислот (галогене-, гидрокси-, аминокислот).
4. Перегруппировки.
А—В ----------*- 'А---В
I • I
X X
Происходит переход (миграция) отдельных атомов или групп атомов от одного фрагмента молекулы к другому.
5. Реакции окисления и восстановления. Сопровождаются изменением степени окисления ато на углерода, являющегося реакционным центром. Механизм реакции окисления органических веществ достаточно сложен.
По количеству молекул, принимающих участие в лимитирующей стадии, различаютмономолекулярные и биомолекулярные реакции (обозначают цифрами 1 и 2 соответственно). В лимитирующей (самой медленной) стадии мономолекулярной реакции принимают участие молекулы одного реагента, в бимолекулярной — молекулы двух реагентов.
Обширную группу химических реакций составляют каталитические реакции, осуществление которых возможно только в присутствии катализатора.
Большое значение имеют реакции полимеризации, заключающиеся в образовании высокомолекулярного вещества (полимера) из низкомолекулярных веществ (мономеров).
5. КИСЛОТНОСТЬ и основность ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
В настоящее времядля оценки кислотности и основности соединений, в том числе и органических, применяют две теории: теория Брен-стеда и теория Льюиса. Теория Бренстеда (1923 г.) является протолитической или протонной теорией кислот и оснований, а теория Льюиса (1925 г.) — апротонной или электронной теорией кислот и оснований.
КИСЛОТНОСТЬ И ОСНОВНОСТЬ ПО БРЕНСТЕДУ
Согласно теории Бренстеда, кислотой называют соединение, способное отдавать протон, т. е. кислота — донор протона, а основание — соединение, способное присоединять протон, т. е. основание является акцептором протона.
Теория Бренстеда называется протонной или протолитической теорией кислот и оснований, так как определения кислота и основание взаимосвязаны между собой процессом присоединения и отщепления протона.
Кислотно-основный процесс, согласно теории Бренстеда, может быть представлен следующей схемой:
А—Н
+ В
А' + В-Н
кислота основание
сопряженное сопряженная
основание
кислота
Кислота А—Н, теряя протон, превращается при этом в основание А-, которое называется сопряженным основанием данной кислоты. Основание же В, присоединяя протон за счет неподеленной пары электронов, превращается в кислоту ВН+, которая является сопряженной данному основанию.
Таким образом, кислота А—Н и сопряженное основание А-, основание В и сопряженная кислота ВН ’ образуют две сопряженные кислотно-основные пары.
Кислотность и основность соединений — понятия относительные. Эти два свойства неразрывно связаны между собой и проявляются одновременно, т. е. если одно соединение в данной реакции выступает в роли кислоты, то другое обязательно — в роли основания. Определенное соединение может быть кислотой по отношению к одному веществу и основанием по отношению к другому. Относительность
5. Кислотность и основность органических соединений
этих понятий становится очевидной при рассмотрении конкретного примера.
СН3СООН + НОН < - - —СН3СОО + Н(О+
кислота основание сопряженное сопряженная
основание кислота
zp: ,р-н
Нзс-с' + Н5ОЛ^=^= Нзс-с' + HSO4
ОН " он
основание
кислота
сопряженное сопряженная
основание
кислота
Уксусная кислота, которая в воде проявляет кислотные свойства, в безводной серной кислоте ведет себя как основание.
Количественно силу кислот и оснований можно выразить с помощью константы равновесия данной реакции. Величину константы равновесия чаше всего определяют в водной среде. Для реакции:
RCOOH + НОН
кислота основание
Кравн.
RCOO- + Н3О+
сопряженное сопряженная основание кислота
[RCOO | [Н3О ] [RCOOH] [Н2О]
Учитывая то, что вода в данной реакции выступает в роли растворителя, т. е. концентрация ее практически не изменяется (55,6 моль*л'’), концентрацией воды можно пренебречь. В данном случае константу равновесия называют константой кислотности или константой диссоциации Кя и выражают формулой:
[RCOO ] [Н3О+ ] к =---------------
[RCOOH]
Любая протолитическая реакция обратима и характеризуется константой диссоциации.
Большинство органических соединений, проявляющих кислотные свойства, в водной среде являются слабыми кислотами, константы которых выражаются малыми величинами. Например, Ко уксусной кислоты при 25°С равна 1,7610'5. Оперировать такими малыми величинами в практической работе не удобно, поэтому используют зна
Лекции по органической химии
чения рКо — показатель константы — отрицательный логарифм константы диссоциации: рКо = — lgKo (например: рКи для СН3СООН равняется 4,75).
Рассматривая зависимость между значениями величин Кя, рК, и силой кислоты, следует отметить одну закономерность: чем больше величина Кя (константа кислотности), тем сильнее кислота, чем больше величина рКо, тем кислота слабее.
Подобно кислотам силу оснований иногда выражают величиной К4 (константа основности), характеризующей легкость, с которой основание отрывает протон от воды:
к,
R-NH-, + НОН -—.........* R-NH3 + ОН
_ [RNH3+ ] [ОН ]
^7»
[RNHJ
Для удобства пользуются величиной рКЛ
pK»=-igKft
При этом, чем меньше рКЛ, тем сильнее соответствующее основание. Однако намного удобнее выражать степень ионизации кислот и оснований в одной шкале (в шкале рКо) подобно тому, как значение pH одинаково хорошо характеризует и кислотность, и основность. Для основания величина рКо обычно означает кислотность сопряженной
кислоты — протонной формы основания рКвн. Сопряженная кисло-
+
та в виде R— NH3 отдавая протон, превращается в основание R—NH,.
Типы органических кислот
В зависимости от природы элемента, который связан с атомом водорода, кислоты разделяют на четыре основные группы:
1. ОН-кислоты: карбоновые кислоты, спирты, фенолы и др.
2. SII-кислоты: тиолы, тиоловые кислоты и др.
3. NH-кислоты: амины, амиды, имиды и др.
4. СН-кислоты: углеводороды и их производные.
Наряду с указанными типами кислот известны кислоты: Si-H, Р—Н, As—Н, т. е. кислотой может быть любое соединение, содержащее связь А—Н и способное к отщеплению протона. Теория Бренстеда применима к любым типам кислот, способным к диссоциации с разрывом связи элемент—водород.
5 Кислотность и основность органических соединений___________22
Кислотность соединений в основном определяется устойчивостью образующегося аниона, которая обусловлена делокализацией (распределением) отрицательного заряда.
Чем устойчивее сопряженное основание, тем сильнее кислота. Устойчивость аниона определяется следующими факторами: природой атома, связанного с атомом водорода (реакционный и кислотный центр); электронодонорными или электроноакцепторными свойствами заместителя, связанногос реакционным центром, и природой растворителя.
При равных других факторах устойчивость анионов, а следовательно. и кислотность возрастают с увеличением электроотрицательности и поляризуемости атомов кислотного центра. С увеличением электроотрицательности увеличивается сила, с которой атом может удержать пару электронов после отщепления водорода. Поскольку в пределах периода периодической системы электроотрицательность атомов возрастает слева направо (поляризуемость не меняется), то ОН-кислоты сильнее соответствующих NH-кислот, а те, в свою очередь, сильнее СН-кислот, например:
О
НзС-С....... ! о-н: L_______।
уксусная кислота рКй4.7
zP н3с-с
ИЧН-'Н!
I_______I
ацетамид
рК„ 45,4.
'г
Н3С-С
Гсн'^'н:
L---А-__1
ацетон
ркя ао,о
В пределах группы периодической системы электроотрицательность атомов уменьшается сверху вниз, но увеличивается их объем, а следовательно, возрастает поляризуемость, т. е. возможность делокализации внешнего электронного облака. Это способствует повышению стабильности аниона и приводит к возрастанию кислотности. Поэтому SH-кислоты обладают большей кислотностью, чем ОН-кислоты, например:
СН—СН—ОН СН—СН—SH
этанол (рК„ 18 0) этантиол (рК„ 10,5)
Наличие электронодонорных заместителей (+7, +М) при кислотном центре приводит к уменьшению кислотности, а присутствие электроноакцепторных атомов или атомных групп (-7, -М) повышает кислотные свойства.
О
С1-*-сн2—с
ОН
о Н3С^С он
хлоруксусная кислота (рК„^5)
уксусная кислота (рКо 4,7)
56
Лекции по органической химии
Наряду с природой кислотного центра и строением радикала, значительное влияние на проявление кислотных свойств оказывает растворитель. Наиболее эффективным растворителем является вода, она имеет высокую диэлектрическую постоянную и способна связывать катионы и анионы за счет сольватации. Эффект сольватации приводит к устойчивости образовавшегося аниона, а следовательно, кислотность соединений возрастает.
Типы органических оснований
Согласно теории Бренстеда, любое соединение, способное присоединять протон, может выступать в роли основания; это могут быть как нейтральные молекулы, так и заряженные частицы. В нейтральных молекулах для проявления основных свойств необходимо наличие атома с неподеленной парой электронов или наличие кратной связи. Основаниями могут быть анионы: алкоксид-ион RO ; алкилмер-каптид-ион RS-, карбанион R3C ~ (триалкилметанид-ион) и другие.
Различают два типа оснований Бренстеда: «-основания или оние-вые (наличие атомов с неподеленной электронной парой) и л-основа-ния (наличие л-связи).
В «-основаниях центром основности, или центром протонирования (местом присоединения протона), чаше всего выступают такие элементы, как азот, кислород, сера. В зависимости от центра основности ониевые основания делятся на три основные группы:
1. Аммониевые, к которым относятся первичные (RNH2), вторичные (R2NH) и третичные (R3N) амины, азометины (RCH=NR), нитрилы (R~C=N) и азотсодержащие гетероциклы;
2. Оксониевые, к которым относятся спирты (ROH), простые эфиры (R— О—R), альдегиды (RCHO), кетоны (RCO) и функциональные производные кислот (сложные эфиры, хлорангидриды, амиды и др.);
3. Сульфониевые, к которым относятся тиоспирты (R—S—Н) и тиоэфиры (R—S—R).
Основность органических соединений в основном определяется устойчивостью образовавшегося катиона, которая обусловлена делокализацией положительного заряда.
На основность значительное влияние оказывает электроотрицательность и поляризуемость элементов, составляющих центр основности, электронодонорное или электроноакцепторное влияние радикала, связанного с центром основности, и природа растворителя.
5. Кислотность и основность органических соединений
57
Большая электроотрицательность атомов основного центра способствует бо iee прочному удержанию им неподеленной электронной пары, что характеризует меньшую основность соединения, иначе говоря, чем больше электроотрицательность атома, тем меньше основность. Таким образом, аммониевые основания проявляют более сильные основные свойства по сравнению с оксониевыми. Увеличение же поляризуемости атома приводит к уменьшению основности сопряженного основания и увеличению кис тотности сопряженной кислоты. Поэтому сульфониевые основания слабее оксониевых.
Существенную роль в проявлении основных свойств играет растворитель. Эффект сольватации способствует устойчивости образовавшегося катиона, а следовательно, приводит к повышению основности.
Центром основности л-оснований, к которым относятся алкены, алкадиены и арены, является л-связь. В процессе взаимодействия протона с соединениями, содержащими кратную связь, происходит частичное перекрывание свободной 5-орбитали протона со связующей л-МО кратной связи, в результате чего образуется короткоживущая частица — л-комплекс.
По сравнению с ониевыми л-основания являются слабыми основаниями.
КИСЛОТЫ И ОСНОВАНИЯ ЛЬЮИСА
Теория кислот и оснований Льюиса, в отличие от теории Бренсте-да, является более обшей. Согласно этой теории основанием считается любая частица (атом, молекула или анион), способная отдавать электронную пару для образования ковалентной связи, а кислотой — любая частица (атом, молекула, катион), способная принимать пару электронов с образованием ковалентной связи.
То есть, по Льюису, основание является донором, а кислота — акцептором пары электронов. Из приведенного определения видно, что основания Льюиса тождественны основаниям Бренстеда. Однако кислоты Льюиса охватывают более широкий круг соединений.
Кислотой Льюиса считается любая частица, имеющая вакантную орбиталь.
Согласно теории Льюиса, к кислотам относятся не только соединения, отшепляюшие протон (протонные кислоты), но и другие вещества, имеющие вакантную орбиталь и способные принимать па
58
Лекции по органической химии
ру электронов (апротонные кислоты). Кислотами Льюиса, например, являются такие соединения, как BF4, AtCl3, FeCl3, SbCl3, ZnClr HgCl3 и др.
Кислотно-основный процесс по Льюису состоит в образовании ковалентной связи между основанием и кислотой за счет электронной пары основания и вакантной орбитали кислоты. Так, основания Льюиса, имеющие неподеленные пары электронов, образуют с кислотами Льюиса «-комплексы:
8+ 8-
Н3С—*С1: + А1С13^=^
хлорметан хлорид
(основание алюминия
Льюиса) (кислота
Льюиса)
СН—CI—А1С1, —► СН3[А1С14]
«-комплекс
А основания, имеющие кратную связь, образуют с-комплекс.
+ Вг+
Легкость протекания кислотно-основного взаимодействия по Льюису определяется силой кислоты и основания.
УГЛЕВОДОРОДЫ
6. АЛКАНЫ
Алканами называют углеводороды алифатического ряда, в молекулах которых атомы углерода связаны между собой только простыми о-связями.
Обшая формула алканов — СлН,п+2. Алканы образуют гомологический ряд, в котором каждый представитель отличается от последующего на группу —СН,— (гомологическая разность):
сн4 метан с2н6 этан с3н8 пропан
с4н|0 бутан с5н12 пентан с6н|4 гексан
с7н16 гептан CgH|g октан Цн20 нонан
с,он22 декан
Номенклатура алканов
Первые четыре члена гомологического ряда алканов — метан, этан, пропан и бутан — имеют тривиальные названия. Названия последующих углеводородов с нормальной углеродной цепью образуют от названия греческих числительных, указывающих количество атомов углерода в молекуле, с добавлением суффикса -ан.
Названия алканов с разветвленной углеродной цепью образуются согласно заместительной номенклатуре И ЮПАК следующим образом:
1. Для построения названия выбирают наиболее длинную нераз-ветвленную углеродную цепь (главную углеродную цепь); если имеется несколько цепей одинаковой длины, выбирают ту, которая имеет наибольшее число разветвлений:
jCH3-CH2-CH2-CH—СН2-СН3:
....—-.....—1..........'
СН3
[СНууСН2—СН2—CH-j-CH2—сн3 :сн4сн3
:СН3:
2. Нумерацию атомов углерода главной цепи проводят с того конца, к которому ближе заместитель:
12 3 4
СН3—CH—CHj-CH3
сн3
2-метилбутан
60
Лекции по органической химии
Если одинаковые заместители равноудалены от обоих концов молекулы, начало нумерации определяется алфавитным порядком:
6 5 4 3 2 1
сн3—сн—сн—сн—сн—сн3
|н2 сн3
СН3
З-метил-4-этил гексан
Если одинаковые заместители равноудалены от обоих концов молекулы, нумерацию начинают с того конца, где число заместителей больше:
СН3
1 2 з1 4 5 6
СН3—сн-с— СНз—сн-сн3
сн3 сн3 сн3
2,3,3.5 -тетраметил гексан
3. Составляют название соединения, учитывая следующие правила:
• названия заместителей перечисляют в алфавитном порядке, указывая цифровой локант, соответствующий положению каждого заместителя в главной углеродной цепи;
• число одинаковых заместителей! обозначают множительными приставками дн-, три-, тетра- и т. д.;
• затем называют углеводород, в основе которого выделена главная углеродная цепь.
Если в молекуле алкана две метильные группы расположены на одном конце углеродной цепи и другие ответвления отсутствуют, используют приставку изо-; если три метильные группы — приставку нео-: сн3 сн3
сн3—сн—сн2— сн3 сн—с—сн2—сн3
сн3
изопентан J
неогексан
Алкил — остаток углеводорода, образующийся в результате отщепления атома водорода.
Необходимо обратить внимание на то, что не следует путать понятия «углеводородный радикал» как остаток алкана (R—) со «свободным радикалом» (R-), который имеет неспаренный электрон.
Важно запомнить, что при переходе от алкана к алкилу следует заменить суффикс -ан на -ил. Для двухвалентных радикалов с двумя свободными валентностями возле одного углеродного атома суффикс -ан изменяется на -илиден, для трехвалентных радикалов с тремя свободными валентностями возле одного углеродного атома соответственно суффикс -ан изменяют на -илидин.
6. Алканы
61
Наиболее часто встречающиеся радикалы:
СН3-сн3—сн2— СНз СН3/СН —
метил
этил
изопропил
метилен
этилидеи
сн3-сн2-сн2-сн2— н-бутил
сн3— сн2-сн пропилидеи
СНз\
JXh—сн,— СН< СН3-СН2-СН— сн3
СНз 3 сн3-с—
изобутил
втор-бутил
mpem-бутил
—сн2—СН2—СН2—триметилен
метип, метилидин
этилидин
Алканы по рациональной номенклатуре рассматривают как производные метана, в молекуле которого один или несколько атомов водорода замещены на углеводородные радикалы. Заместители вокруг «метанового» углерода перечисляют в порядке увеличения сложности их строения:
СН3
СНз—СН2-Г-С7- СН2—СН2-------СНз
'*-К
СНз
диметил этилпропилметан
Изомерия
Структурная изомерия алканов обусловлена различной последовательностью связывания атомов углерода в молекуле (изомерия пели
СН3
СН3—СН2—сн2—СН3 СН3—СН—сн3
бутан п изобутан,
2-метилпропан
В ряду алканов, начиная с гептана С7Н|6, возможно проявление оптической изомерии:
сн3—сн2-сн—сн2 —сн2- сн3
<^н3
62
Лекции по органической химии
сн3
I,>н
/сч
сн3—СН2 сн2_СН2—сн3
R-3-метилгексан
СНз
Н4, I "с
СНз—сн2—Н2с н2с—сн3
S-3-метилгексан
Способы получения алканов
Природными источниками алканов являются нефть, газ, уголь, торф, сланцы и др. Основная цель промышленного получения алканов — выделение различных фракций или индивидуальных соединений.
Синтетические способы получения
Оксосинтез алканов (метод Фишера — Тропша). Каталитическое гидрирование оксида углерода (11) позволяет получить алканы нормального строения (состав С6— С|0). Смесь оксида углерода (II) и водорода пропускают над катализатором Fe — Со при температуре 180— 300 °C.
Fe t лСО+ (2л + 2)Н2 ——► С„Н2и+2 + лН2О
Каталитическое гидрирование алкенов и алкинов. Присоединение водорода по месту разрыва кратной связи алкенов и алкинов осуществляют в присутствии катализаторов (тонко измельченных Pt, Pd или Ni).
IH1 [Н)
нс=с— сн3---------► н2с=сн—сн3 —► сн3—сн2—сн3
пропин пропен пропан
Взаимодействие галогеналканов с металлическим натрием (реакция Вюрца). Используется для синтеза симметричных алканов и протекает через стадию образования металлорганических соединений.
СН3-1 +2Na + I-CH3---► СН3—СН3 + 2NaI
метилйодид этан
Реакция Вюрца протекает в две стадии:
СН31 + 2Na ----► CH3Na + Nal
метилнатрий
CH3Na + CH3I------► СН3 — снз + Nal
этан
Если в качестве исходных веществ используют два разных галоген-алкана, образуется смесь алканов, которую трудно разделить.
6. Алканы
63
СНз-1 + CH3CH2-I +2Na
----► СН3-СН3 + СН3-СН2СН3 + СН3СН2- СН2СН3 + 2Na! этан пропан бутан
Сплавление солей карбоновых кислот со щелочами. Соли щелочных металлов карбоновых кислот при сплавлении со щелочью образуют алканы.
СН3—COONa-1- NaOH ——►CH4f + Na2CO3 ацетат натрия метан
Гидролиз металлорганических соединений. Действием воды на карбид алюминия получают метан в лабораторных условиях.
AI4C3 + 12Н2О----* 3CH4f + ЗА1(ОН)3
карбид алюминия
Электролиз водных растворов солей карбоновых кислот (реакция Кольбе). Чаще электролизу подвергают калиевые или натриевые соли карбоновых кислот:
X) сн,-сн9-с^ ONa
X) СНГСНГС^ +Na
О’
натрия пропионат
.О
СНГСН7~С^ СНГСНГС^ —*
3 2 О’ -е О' -с°1
сн3-сн2+сн2-сн, —— сн3-сн2-снгсн3
бутан
На аноде анион карбоновой кислоты окисляется, образуя свободный радикал, который легко декарбоксилируется. Алкильные радикалы вследствие рекомбинации превращаются в алкан. На катоде образуется водород и соответствующий гидроксид щелочного металла.
Физические свойства
Первых четыре представителя гомологического ряда алканов — газы. Алканы нормального строения состава С5—С17 — жидкости, последующие представители — твердые вещества. Физические константы (Гкиг1, ( ) в гомологическом ряду алканов возрастают с увеличением молекулярной массы.
64
Лекции по органической химии
Большое влияние на физические свойства алканов оказывает строение углеродной цепи: температура кипения алканов разветвленного строения ниже, чем алканов нормального строения.
Алканы газообразные и твердые — без запаха, жидкие — обладают характерным «бензиновым» запахом. Не растворимы в воде, хорошо растворимы в неполярных растворителях. Увеличение молекулярной массы алканов приводит к уменьшению их растворимости.
Химические свойства
Предельные углеводороды называют парафинами (от лат. рагит affinitas — мало свойств), что указывает на их сравнительно низкую реакционную способность. Алканы — инертные, малоактивные соединения, устойчивы к действию кислот, щелочей и окислителей.
Чем же обусловлена их химическая инертность? Следует отметить прочность о-связей С—С и С—Н. Различие значений электроотрицательности лр’-гибридизованного атома углерода (2,5) и атома водорода (2,1) незначительно. Поэтому о-связи С—С и С—Н в алканах практически неполярны и не склонны к гетеролитическому разрыву.
Химические превращения алканов сопровождаются гомологическим расщеплением связей. Характерны реакции свободнорадикального замещения (SR). При высоких температурах возможен гомолитический разрыв углерод-углеродных связей.
I. Реакции радикального замещения (SR).
1. Галогенирование. Алканы реагируют с галогенами с образованием моно- и полигалогеналканов. Реакция протекает по цепному свободнорадикальному механизму.
При взаимодействии метана с хлором, протекающем при освещении или нагревании, происходит постепенное замещение атомов водорода атомами хлора:
СН4+ С12—~—►СН3С1 + НС1
хлорметан
СН3С1 + С12 —-----► СН2С12+ но
дихлорметан
СН2С12 + а2—СНС!3 + на трихлорметан, хлороформ
СНС13 + Clj—СС14 + НС!
тетрахлорметан, четыреххлористый углерод
6. Алканы 65
Механизм данной реакции был изучен академиком, лауреатом Нобелевской и Государственной премии СССР Н.Н. Семеновым.
В цепной реакции выделяют три последовательные стадии: инициирование, рост цепи и обрыв цепи.
Инициирование. Молекула галогена под действием квантов света (hv) или нагревании претерпевает гомолитический разрыв связи с образованием свободных радикалов:
ci-i-ci hv >2СГ
Рост цепи. Образовавшиеся свободные радикалы хлора атакуют связь С—Н в молекуле СН4, отрывают атом водорода с образованием НС1 и свободного метильного радикала СН3.
С Н4 + СГ-----*- СН3 + НС1
Метильный радикал атакует молекулу хлора, отрывает атом галогена и образует хлорметан СН3С1 и свободный радикал хлора:
Cl- -Cl + CHj----*- СН3С1 + Cl-
Образовавшийся радикал С1 повторяет цикл указанных превращений, т. е. происходит цепная реакция, в которой атом хлора, прореагировавший на предыдущей стадии роста цепи, способствует высвобождению нового радикала хлора на последующей стадии. Образуется смесь моно-, ди-, три- и тетрахлорпроизводных метана.
Цепная реакция завершается только после исчезновения всех образовавшихся в ходе превращений свободных радикалов.
Обрыв цепи происходит в результате рекомбинации (димеризации) свободных радикалов.
СН3 + СН3-----► СН3—СН3
СН3+ CI------►СН3С1
Следует обратить внимание на региоселективность (избирательность) галогенирования (особенно бромирования) алканов. В первую очередь замещается атом водорода при третичном углеродном атоме, затем — при вторичном и в последнюю очередь — при первичном.
СН3 СН3
СНз— СН-СН2- СН3 + Вг2 ——►СН3—С—СН2—СН3 + НВг
2-метилбутан Вг
2-бром-2-метилбутан
66
Лекции по органической химии
Такую последовательность замещения определяет устойчивость образующихся свободных радикалов. Региоселекгивность галогенирования алканов возрастает при понижении температуры реакции. Чем активнее галогенирующий реагент, тем ниже избирательность процесса.
2. Нитрование алканов проводят разбавленной азотной кислотой при нагревании и повышенном давлении (реакция Коновалова):
i,p
СН—снз + HNO, R--------- снз—СН2—NO, + Н,0
j j j разб л z z /
нитроэтан
Механизм SR:
I t>p
НО- -NO,----- НО- + NO,-
СН—СН3+ НО—~СН3—СН2 + Н2О
сн—сн2+ ho-no2—- сн—сн—no2 + НО
Следует отметить, что алканы в обычных условиях не взаимодействуют с концентрированной азотной кислотой.
3. Сумфохлорирование. Алканы при совместном действии оксида серы (IV) и хлора в условиях УФ-облучения образуют алкансульфо-нилхлориды R-SO2C1.
СН4+ SO2 + C12-^—CH3SO2C1 + HCI
метансульфохлорид
Реакция протекает по свободнорадикальному механизму (SR):
he
Cl--Cl -----* 2C1-
CH4 + Cl-------* HC1 +CH3
//Q______
ch3 + ns ---------* CH3SO2
о
CH3sb2 + ci2 -----ch3so2 + 'ci
Сульфохлорирование алканов позволяет получить смесь первичных и вторичных алкансульфохлоридов. Третичные алкансульфохлориды в силу пространственных препятствий не образуются.
II. Окисление. Алканы в избытке кислорода или на воздухе горят с выделением большого количества теплоты:
6. Алканы
67
СН4 + 2О2---► С02 + 2Н2О + Q
Окисление алканов в присутствии катализатора (соли марганца, хрома и др.) и при нагревании (~ 150—200 °C) протекает по радикальному механизму и сопровождается разрывом С—С связей.
о, сн,—сн,—сн,
СН-СНгСН—*СН,СН-СН,—►СН-СН-СН,--------:---
3 2 3 3 3 I * 3 —сн,—сн—СН.
0-0 3
СН—СН—СН, о—ОН
сн,—с—сн,
3 II
о
ацетон
.О
-сн,он + сн—с<
II
метанол о п
уксусный альдегид
нсоон сн—СООН
муравьиная кислота уксусная кислота
III. Крекинг алканов — это процесс термического расщепления алканов.
Термический крекинг проводят при t~ 800 °C и выше, каталитический — при t = 450...550 °C в присутствии алюмосиликатных катализаторов.
Высшие алканы в условиях термического крекинга образуют смесь низших алканов и алкенов.
1500°с
2СН4-------- НС=СН + ЗН2
сн—СН3 б0°-800<,с» н2 + СН=СН2
сн—сн—сн3 —-—- СН4 + СН=СН2
Крекинг-процесс имеет важное промышленное значение: получение высокооктановых бензинов, непредельных и ароматических углеводородов.
7. ЦИКЛОАЛКАНЫ
Циклоалканами называют алициклические углеводороды, в которых углеродные атомы, образующие цикл, находятся вsp3-гибридизации.
Классификация циклоалканов
Циклоалканы классифицируют по величине цикла, числу циклов и способу соединения циклов в молекуле.
По величине цикла различают:
• циклоалканы с малыми циклами (трех- и четырехчленные);
• циклоалканы с обычными циклами (пяти-, шести- и семичленные);
• циклоалканы со средними циклами (от восьми- до одиннадцатичленных);
• макроциклы (двенадцатичленные и более).
В зависимости от числа циклов в молекуле выделяют:
• моноциклические;
• бициклические;
• полициклические.
Бициклические циклоалканы по способу соединения циклов подразделяют на 3 группы:
спираны (два кольца имеют один общий атом углерода);
конденсированные (два кольца имеют два общих соседних атома углерода);
мостиковые (два кольца имеют три и более общих атомов углерода).
Номенклатура циклоалканов
Названия моноциклических циклоалканов образуют в соответствии с правилами ИЮПАК путем добавления к названию алкана с соответствующим количеством атомов углерода префикса цикло-.
циклогексан
циклопропан циклобутан циклопентан
Названия одновалентных радикалов образуют путем замены в названии соответствующего циклоалкана суффикса -ан на -ил.
7. Циклоалканы
69
циклопропил
Бициклические циклоалканы называют в зависимости от числа углеродных атомов циклической системы и способа соединения циклов.
Спираны называют путем прибавления префикса спиро- к названию алкана с соответствующим числом атомов углерода. Между префиксом и корнем (названием алкана) в квадратных скобках указывают в порядке возрастания число атомов углерода в каждом из циклов, исключая общий. Нумерацию атомов углерода спирановой системы начинают с меньшего цикла, с углеродного атома, расположенного рядом с общим.
Конденсированные и мостиковые бициклические циклоалканы называют путем прибавления префикса бицикло- к названию алкана с соответствующим числом углеродных атомов. В квадратных скобках между префиксом и названием алкана в порядке убывания указывают число углеродных атомов в каждой из трех цепей, соединенных общими узловыми (третичными) атомами углерода. Нумерацию атомов углерода следует начинать с одного из узловых атомов. В первую очередь нумеруют самую длинную углеродную цепь, затем — более короткую. В мостиковых системах в заключение нумеруют самую короткую углеродную цепь — мостик:
бицикло[4.2.0]октан
бицикло[2.1.0]пентан
бицикло[2.2.1]гептан
Наряду с систематическими названиями многие бициклические системы имеют тривиальные названия.
70
Лекции по органической химии
4
камфан.
1,7,7-триметилбицикло[2.2.1]гептан
пинан.
2,6,6-триметилбицикло[3.1.1 ] гептан
Полициклические соединения содержат более двух циклов, соединенных мостиками. Названия каркасных полициклических углеводородов происходят от эмпирических названий соответствующих геометрических фигур, напоминающих их строение.
призман
кубан
ХЛЗ^СН/СН
Н2С^^ СН2^^СН2
адамантан
Изомерия циклоалканов
Для циклоалканов характерны все виды изомерии.
Структурная изомерия циклоалканов обусловлена величиной цикла, природой и взаимным расположением заместителей:
а) изомерия размера цикла;
метилциклопропан
циклобутан
б) изомерия положения заместителей;
^СН3
'СН3
1,3-диметил циклогексан
1,2-диметил циклогексан
7. Цикяоалканы
71
в) изомерия боковых цепей.
пропилциклопропан изопропилциклопропан
Геометрическая изомерия циклоалканов обусловлена различным расположением заместителей относительно плоскости цикла.
транс-} .2-диметил-циклобутан (Е- изомер)
цпс-1,2-диметил-циклобутан (Z изомер)
Следует учитывать, что циклоалканы, как правило, имеют не плоское (кроме циклопропана), а пространственное строение в виде определенных конформаций.
При рассмотрении геометрических изомеров условно считают строение циклоалканов плоским.
Оптическая изомерия характерна для циклоалканов, молекулы которых не имеют плоскости симметрии.
Способы получения
Циклопропан, циклогексан и их гомологи могут быть выделены в чистом виде из некоторых видов нефти.
72
Лекции по органической химии
Синтетические способы получения циклоалканов
1. Взаимодействие а,(я-дигалогеналканов с металлическим натрием и цинком. Данный метод — внутримолекулярный вариант реакции Вюрца, позволяющий получить трех-, четырех- и пятичленные циклоалканы.
^СН2Вг
СН2 +2Na-------------/СН2 + 2NaBr
^^CH2Br Н2С^
циклопропан
2. Пиролиз кальциевых, бариевых или ториевых солей дикарбоновых кислот. При пиролизе (сухой перегонке) солей дикарбоновых кислот образуются циклические кетоны, которые затем восстанавливают до соответствующих циклоалканов.
X)
CH2-CH2-cf сн2-сн2 сн2-сн2
I >н2
СН2-СН2-С^ сн2-сн2 сн2-сн2
о
кальциевая соль циклопентанон циклопентан
адипиновой кислоты, адипинат кальция
Позволяет получить пяти- и шестичленные циклоалканы.
3. Циклоприсоединение. Это процесс соединения двух или более ненасыщенных молекул с образованием продукта циклического строения. В зависимости от числа атомов, принимающих участие в образовании цикла, различают [2+1], [2+2] и [4+2] циклоприсоединение. Например, димеризация алкенов ([2+2] циклоприсоединение) позволяет получить циклобутан и его гомологи.
сн2=сн2 hv н2с—сн2
сн2=сн2 н2с—сн2
этен циклобутан
Реакцию Дильса — Альдера (диеновый синтез) относят к реакциям [4+2] циклоприсоединения.
^сн2 ™ + Г— СН СНт ^сн2 бутадиен-1,3 этен циклогексен циклогексан
73
7. Циклоалканы
Реакция широко используется для получения циклогексана и его производных.
4. Электроциклические реакции. Электроциклической реакцией замыкания цикла называют реакцию, в которой происходит образование о-связи между концами сопряженной системы молекулы.
Н
СН^
сн2 нс^ сн2
i hv I I
H Сч хСН2 нс^ х н2 сн^
н
гексатриен-1,3,5 циклогексен циклогексан
Данный способ позволяет получить ненасыщенные алициклические соединения, которые могут быть восстановлены до циклоалканов.
Физические свойства
В обычных условиях циклопропан и циклобутан — газы; циклоалканы, содержащие от 5 до 11 атомов углерода в цикле, — жидкости; последующие представители — твердые вещества.
Температуры кипения и плавления циклоалканов выше по сравнению с константами соответствующих алканов. Практически не растворимы в воде.
Строение циклоалканов
Циклоалканы представляют собой в определенной степени жесткие структуры.
Для циклоалканов характерны следующие виды напряжений:
1) торсионное (Питцеровское) напряжение — связано со взаимодействием химических связей в заслоненной или частично заслоненной конформациях;
2) напряжение Ван-дер-Ваальса — обусловлено взаимным отталкиванием заместителей при сближении на расстояние, близкое сумме их ван-дер-ваальсовых радиусов;
3) угловое (Байеровское) напряжение — присуще отдельным циклоалканам и связано с отклонением валентных углов между углеродуглеродными связями в цикле от нормального (тетраэдрического) значения.
Теория напряжения циклов была предложена немецким химиком-органиком А. Байером в 1885 г. Согласно этой теории циклоалканы представляют собой плоские многоугольники. Единственным фактором, определяющим прочность цикла, считалось наряжение, вызван
74
Лекции по оритиче^кой химии
ное отклонением внутренних валентных углов цикла по сравнению с тетраэдрическим углом.
Если рассматривать форму циклоалканов в виде простых геометрических фигур, имеющих плоское строение, то можно отметить, что при переходе от одного цикла к другому происходит изменение валентного угла.
Чем значительнее отклонение, тем выше угловое напряжение и менее устойчив цикл.
В соответствии с вышеприведенным, трехчленный цикл менее устойчив, чем четырехчленный, а последний — менее устойчив, чем пятичленный. Эти представления подтверждались накопленным к тому времени экспериментальным материалом. Однако для шестичленного цикла экспериментальные данные вступали в противоречия с теорией. Шестичленные циклы (внутренний угол 120°), имеющие значительное отклонение валентных углов от тетраэдрического, оказались устойчивее пятичленных, в которых внутренние углы наиболее близки к тетраэдрическим.
Причиной несоответствия теории Байера с экспериментальным материалом явилось ошибочное представление о плоском строении циклов. В действительности же циклоалканы (исключая циклопропан) не имеют плоского строения.
Пространственное строение циклоалканов определяется разной конформационной подвижностью углеродных атомов, зависящей от числа звеньев в цикле. Молекула любого циклоалкана стремится принять в пространстве такую форму (конформацию), в которой сумма углового, торсионного и ван-дер-ваальсового напряжений была бы минимальной.
Наиболее жесткую структуру имеет циклопропан.
/ В соответствии с правилами геометрии три точки /сх\СН2 всегда лежат в одной плоскости. Именно поэтому и Z-cSA молекула циклопропана имеет плоское строение.
П2С \-JoU .
VSL / Атомы водорода в данной молекуле находятся в зас-С Н 2 лоненной конформации, что и обуславливает силь-' ное торсионное напряжение. Свободное вращение
относительно углерод-углеродных связей невозможно. Внутренние валентные углы в молекуле циклопропана сильно отклонены от тетраэдрического значения, в результате чего и возникает большое угловое напряжение. Искажение валентных углов при .^-гибридизации орбиталей углеродных атомов должно составлять а = (109°28’ - 60°):2 = 24’44'.
7 Циклоалканы
75
В действительности в молекуле циклопропана валентный угол равен 106°. Согласно квантовохимическим представлениям изменение угла является результатом изменения гибридизации атомов углерода. Гибридизация орбиталей в молекуле циклопропана ближе кхр2-гибри-дизации алкенов, чем к ^'-гибридизации алканов (промежуточное положение).
Вследствие взаимного отталкивания электронных облаков углерод-углеродных связей максималь
Рис. 7.1. Образование «банановых» связей в молекуле циклопропана
ная электронная плотность перекрывающихся орбиталей атомов углерода в молекуле циклопропана расположена не по прямой, соеди
няющей центры связываемых атомов, а за пределами треугольника
молекулы.
Образующиеся связи называют «банановыми» или т-(греч. «тау») связями. Они занимают промежуточное положение между о и л-свя-зями. Образование т-связей выгодно, так как валентные углы увеличиваются до 106° (вместо теоретически рассчитанных 60°) и снижается угловое напряжение молекулы.
Четырехчленный цикл, в отличие от трехчленного, все же обладает незначительной гибкостью. Валентные углы искажены меньше, чем в трехчленном цикле, несколько ниже и угловое напряжение. Один из углеродных атомов циклобутана выходит из плоскости трех атомов на угол 25—30°, что приводит к уменьшению торсионного напряжения цикла.
В пятичленном цикле практически отсутствует угловое напряжение (отклонение внутренних валентных углов от тетраэдрического составляет менее Г). Однако в плоском пятичленном цикле связи С—Н находятся в заслонен ной конформации, что обуславливает значительное торсионное напряжение. Каждый из пяти углеродных атомов циклопентана, стремясь уменьшить торсионное напряжение в цикле, поочередно выходит из плоскости, в которой расположены четыре оставшихся атома углерода. Кольцо находится в непрерывном волнообразном движении — псевдовращении. Эта неплоская осциллирующая структура называется конформацией «конверта». В конформации «конверта» угловое напряжение увеличивается, однако это в полной мере компенсируется снижением торсионного напряжения молекулы.
Если представить шестичленный цикл плоским, то его внутренние валентные углы должны быть равными 120°. Это обусловило бы значительное угловое напряжение. Следует отметить, что в плоской
76
Лекции по органической химии
структуре появляются взаимодействия, связанные с заслонением С—Н -связей, то есть торсионное напряжение.
Однако же циклогексан не является плоской структурой и существует без углового напряжения, так как все валентные углы в нем тетраэдрические (109°28').
До недавнего времени считали, что наиболее устойчивыми структурами циклогексана являются две изомерные конформации, взаимопрев-ращающиеся друг в друга за счет поворота вокруг о-связей без их разрыва: «кресла» С (отангл, chair— кресло) и «ванны» В (от англ, boat—лодка).
Более устойчивой является конформация «кресла», так как лишена торсионного напряжения (все атомы углерода и водорода находятся в заторможенной конформации). В конформации «ванны» происходит заслонение связей, расположенных вдоль двух параллельных боковых сторон молекулы, что обуславливает торсионное напряжение.
В настоящее время с помощью физико-химических методов исследования установлено, что циклогексан может существовать в различных конформациях: «кресло», /ивдсти-форма (искаженная ванна), «полукресло», «ванна». Второй по устойчивости является дгвмс/и-форма.
Она образуется в результате «скручивания» конформации «ванны» в продольном направлении, что уменьшает напряжение в цикле.
В обычных условиях преобладающая часть молекул циклогексана (99,9 %) существует в конформации «кресла». В результате вращения вокруг углерод-углеродных связей одна конформация «кресла» переходит в другую, энергетически равноценную, форму кресла. Такой процесс называют инверсией цикла.
конформация «кресла»
конформация
«ванны»
твист -конформация
конформация «кресла»
7. Циклоалканы
Две конформации «кресла» могут взаимно превращаться как с промежуточным образованием конформации «ванны», wewcw-конформа-ции, так и без прохождения через конформацию «ванны».
Изучая пространственное строение циклогексана, было установлено, что шесть С—Н связей расположены вдоль оси симметрии молекулы, а другие шесть — под углом 109°28’. Связи, параллельные оси симметрии, называют аксиальными (обозначают символом «а»), а расположенные радиально — называют экваториальными (обозначают символом «е»). Каждый атом углерода имеет одну аксиальную, а другую экваториальную связь С—Н. При инверсии цикла (-100 000 раз в секунду при 25 °C) все экваториально связанные атомы водорода становятся аксиальными, а все аксиально связанные — экваториальными. Взаимопревращения в циклогексане протекают настолько быстро, что все атомы водорода циклогексана становятся эквивалентными.
Две конформации «кресла» монозамещенного циклогексана энергетически не равноценны. Более стабильной является конформация с экваториальным положением заместителя. Аксиальное положение заместителя для циклогексана менее выгодно, так как возникает сте-рическое отталкивание, обусловленное взаимодействием заместителя с аксиально расположенными атомами водорода в положении 3 и 5.
78
Лекции по органической химии
Химические свойства циклоалканов
Для циклоалканов характерны реакции свободнорадикального замещения (SR).
циклогексан
бромциклогексан
Циклоалканы с малыми циклами проявляют своеобразные химические свойства, связанные с особенностями их строения. Из-за значительного углового и торсионного напряжения трехчленный и, в меньшей степени, четырехчленный циклы являются неустойчивыми. Именно поэтому для циклопропана и циклобутана наряду с реакциями замещения характерны реакции присоединения, сопровождающиеся раскрытием цикла.
7. Гидрирование
Н2; Ni, 50 °C
сн3-сн2-сн3
пропан
циклопропан
циклобутан
H2;Ni, 200 "С
—---------► СН3-СН2-СН2-СН3
бутан
2. Галогенирование
циклопропан
+ Вг2
Br-CH2-CH2-CH2-Br
1,3-дибромпропан
Следует отметить, что с бромом циклобутан вступает в реакцию замещения SR.
7. Циклоалканы
79
3. Гидрогалогенирование
+ НВг —
циклопропан
СН3-СН2-СН2-Вг
1-бромпропан
Представляет интерес реакция присоединения галогеноводородов к алкилзамещенным циклопропана, протекающая в соответствии с правилом Марковникова:
СН3
сн
Вт
+ НВг н2с—сн2
СН3-СН-СН^-СНз
2-бромбутан
метилциклопропан
Однако циклобутан не реагирует с галогеноводородами.
Для циклоалканов и их производных характерны реакции суже ния и расширения циклов. Данные реакции являются каталитическими и протекают в присутствии кислот Льюиса:
этил ци клобутан
сн2-сн3
AlCh, t
метилциклопентан
В связи с низкой реакционной способностью и отсутствием функциональных групп идентификация алканов с помощью каких-либо аналитических реакций невозможна. Прежде всего используют физико-химические константы (температуры кипения, плавтения, коэффициент преломления, удельное вращение — для оптически активных жидких циклоалканов и др.); определяют процентное содержание углерода и водорода путем сжигания, молекулярную массу и др.; данные физических (инструментальных) методов исследования (ИК-, ПМР-спектроскопии и масс-спектрометрии).
8. АЛКЕНЫ
(ЭТИЛЕНОВЫЕ УГЛЕВОДОРОДЫ, ОЛЕФИНЫ)
Углеводороды алифатического ряда, содержащие одну двойную связь, называют алкенами.
\ п /
Двойная связь С=С состоит из о-связи, которая образо-
вана ур2-гибридизованными атомными орбиталями (АО) атома углерода и п-связи, образованной негибридизованными р-орбиталями. Строение двойной связи более подробно было рассмотрено ранее.
Общая формула алкенов С Н .
Номенклатура
Простейшим представителем углеводородов ряда этилена является сам этилен, или этен: С,Н„, Н,С=СН,. Названия алкенов по замес-тительной номенклатуре ИЮПАК образуются путем замещения суффикса -ан в названии предельного углеводорода на -ен, положение двойной связи указывается цифрой, при этом нумерация начинается с того конца цепи, к которому ближе расположена двойная связь:
Н2С=СН—СН3 пропен, или пропилен
Н2С=СН—СН2—СН, бутен-1
Н,С—СН=СН—СН, бутен-2
1 2 3 4 5
Н3С — СН — СН — СН — снз 4-метилпентен-2
сн3
Для низших членов гомологического ряда алкенов более употре-бимы тривиальные названия — этилен, пропилен, для остальных членов гомологического ряда применяют заместительную номенклатуру.
Иногда используют рациональную номенклатуру. По этой номенклатуре углеводород рассматривают как производное этилена, в котором атомы водорода замещены на радикалы.
Например:
CH,---СН----CH=CH2 СН:—СН=СН,
J | 2 J 2
сн3
изопропилэтилен метилэтилен
8. Алкены (этиленовые углеводороды, олефины)
Если в молекуле этилена два атома водорода замещены на радикалы, то алкены рассматривают как симметричные или несимметричные.
СН
СН^
С=СН2
несимметричный диметилэтилен
Название одновалентных радикалов этого ряда углеводородов получают добавлением к названию алкена суффикса -ил, атом углерода со свободной валентностью должен иметь наименьший номер, например:
3 2 I
н3с—сн=сн—
пропен-1-ил
I 2 н3с—с=сн2 1-метилэтенил
Для некоторых радикалов используются тривиальные названия:
СН2—СН— винил (этенил)
СН==СН—CH-алл ил (пропен-2-ил)
Изомерия
Для алкенов характерна изомерия углеродного скелета, изомерия положения двойной связи и геометрическая (цис-, транс-) изомерия:
изомерия положения
сн—сн—сн=сн2 сн—сн=сн-сн3
бутен-1 бутен-2
изомерия цепи
сн—с—сн2
сн—сн-сн=сн2 I
/г j сзн □
бутен-1 з
2-метилпропен
Бутен-2 существует в виде двух пространственных изомеров:
цис-бутен-2
транс-бутен-2
82
Лекции по органической химии
Если у атомов углерода, связанных двойной связью, имеется три или четыре разных заместителя, используют Е, Z-систему обозначений конфигурации геометрических изомеров.
Способы получения
1. Природные источники (нефть, природный газ).
2. Крекинг высших алканов, то есть термическое разложение предельных углеводородов с образованием низкомолекулярных предельных и непредельных соединений.
< 3. Дегидратация предельных одноатомных спиртов. Алкены легко могут быть получены дегидратацией спиртов. В лабораторных условиях в качестве водоотнимающего средства обычно используют серную или фосфорную кислоту.
H2so4
н3с—сн—ОН -----------* H2C=CH2f + Н2О
этанол г этилен
В промышленности применяют оксид алюминия. Пары пропилового спирта пропускают над катализатором при температуре 300—400 °C
А12о3
сн—сн—сн—он —-—* сн3—сн=сн2 + Н2О
пропанол-1 пропилен
Реакция протекает на границе раздела между твердой фазой и газообразной (гетерогенный катализ).
Отщепление воды от спирта с разветвленной структурой подчиняется правилу А. Зайцева (1875 г.): при образовании молекулы воды водород отщепляется от наименее гидрогенизированного атома углерода, находящегося в P-положении по отношению к ОН-группе.
Н2$О
сн,—сн—сн—сн,-------------сн,—сн=с—СН, + Н,0
3 I I 3 ' 3 I
он сн3 сн3
З-метилбутанол-2 З-метилбутен-2
4 . Отщепление галогеноводородов от галогеналканов. При действии спиртового раствора щелочи на моногалогенопроизводные отщепляется галогеноводород и образуются алкены.
NaOH
Н3С-СН—Вг ------------* Н2С=СН2 + NaBr + Н2О
спирт, р-р
5 .Дегалогенированиедигалогеналканов. Вицинальные дигалогенал-каны (с атомами галогена у соседних атомов углерода) при нагревании с Zn ил и Mg в водноспиртовом растворе отщепляют два атома галогена, образуя алкены:
8. Ллкены (этиленовые уггевоОороды. олефины)
83
Н,С —СН—СН- + Zn
I I
Вг Вг
Н3С—СН—СН, + ZnBr2
6 . Дегидрирование алканов. Это промышленный метод получения алкенов. Реакцию проводят при температуре не выше 600 °C во избежание разрыва С—С связей (крекинг). В качестве катализатора используют мелкораздробленный никель, оксид хрома (III) Сг2О3 и др.
н3с—сн—сн—сн3
------- сн,=сн—сн,—сн,
- Н,-2 2 3
Сг,О, бутен-1
300-400 °с
—3—СН,—сн=сн-сн, - л, J J
бутен-2
Физические свойства
Первые представители гомологического ряда алкенов (С2—С4) — это газы, дальше — (С5—С|7) жидкости и твердые вещества. С увеличением молекулярной массы увеличивается их температура плавления, температура кипения, удельный вес*. Алкены практически нерастворимы в воде, сами являются хорошими растворителями. Они горят более коптящим пламенем, чем алканы, что объясняется большим процентом содержания углерода.
Химические свойства
Химические свойства алкенов обусловлены наличием двойной С=С-связи (углерод-углеродной).
\ 71 Z
Двойная связь С=С значительно короче (0,134 нм) про-
стой о-связи и прочнее. Энергия этой связи 611 кДж/моль, на разрыв о-связи приходится 339,6 кДж/моль, тогда энергия разрыва л-связи будет равна: 611 — 339 = 272 кДж/моль. то есть энергия л-связи невелика. Этим объясняется легкость, с которой непредельные углеводороды вступают в реакции присоединения.
В свою очередь двойная связь влияет на реакционную способность связей С—Н у соседнего с ней ур3-гибридизированного атома углерода
Алкены с нормальным строением кипят при более высокой температуре, чем их разветвленные изомеры. Температура кипения «г/с-изомеров выше, чем транс-томе-ров, а температура плавления — наоборот
84
Лекции по органической химии
(а-атом) (благодаря о,тг-сопряжению). Атомы водорода приобретают подвижность и способность вступать в реакции замещения (SR).
Для алкенов характерны также реакции окисления, восстановления и полимеризации.
» 7 Гидрогенизация (гидрирование) — присоединение водорода протекает в присутствии катализаторов (тонкоизмельченных Pt, Pd или Ni).
сн2=сн2 + Н2 м > сн3-сн3
2. Присоединение галогенов (галогенирование). Алкены легко присоединяют хлор и бром, труднее — йод, в результате образуются вицинальные дигалогенопроизводные алканов.
СН=СН2 + Вг2-------------* сн2—СН2
Вг Вг
1,2-дибромэтан
Присоединение происходит по электрофильному механизму — А£ (присоединение электрофильное). Под влиянием п-электронной плотности двойной связи молекула брома поляризуется, при этом образуется тг-комплекс:
Н2С=СН2 + Вг2
СН2 5+
J—-----► Вг—-Вг
сн2
л-комплекс
Электронодефицитный атом брома забирает пару электронов тг-связи на образование о-связи с атомом углерода, в результате образуется о-комплекс:
СН2 8+ §_
Ц---- Вг—-Вг
СН2
ион карбония
бромоний катион
(о-комплекс)
о-Комплекс представляет собой равновесную систему, состоящую из карбкатиона и бромоний-иона.
Взаимодействие о-комплекса с бромид-ионом происходит стереоселективно, то есть бромид-ион атакует о-комплекс с противоположной стороны от имеющегося галогена. Такое присоединение называется /иранс-присоединением.
8. Алкены (этиленовые углеводороды, олефины)
85
+ Вг
Вг
Н,С~СН, I
Вг
Эта реакция имеет аналитическое значение для качественного и количественного определения соединений, содержащих двойную углерод-углеродную связь.
Направление реакции галогенирования может изменяться в зависимости от условий реакции. Так, алкены с галогенирующими реагентами (хлором, N-бромсукцинимидом) в присутствии инициаторов процесса образования свободных радикалов (пероксиды, температура, УФ-свет) вступают в реакцию свободнорадикального замещения. При этом атом водорода у атома углерода в «-положении к двойной связи замещается на галоген. Такое замещение получило название аллильного.
500-600”С
н2с=сн—сн3 + С12-------------* н2с=сн—CH^—Cl + НС1
аллил хлорид
Реакция идет через образование аллильных радикалов
Ч2с=сн—сн. -—* сн—сн=сн2.
которые являются более устойчивыми по сравнению с обычными алкильными.
3. Присоединениегалогеноводородов (niapoi алогенирование). Алкены вступают в реакцию присоединения сф всеми галогеноводорода-ми. В случае этилена или симметричных алкенов образуется один продукт реакции независимо от того, к какому атому углерода присоединяется водород или галоген:
СН=СН2 + НВг --------* СН3—СН2Вг
бромэтан
Н,С—СН=СН—СН, + НВг -------------► н,с—сн,—сн—сн,
3 3 J 2 । J
Вг
2-бромбутан
Если галогеноводород присоединяется к несимметричному алкену, то во время реакции могут образоваться два продукта, например:
86
Лекции по органической п- i;
----->► СН,—CH—CH,
I
H c—CH=CH2 HBr Br
J 2 2-бромпропан
------* CH3—CH—CH2Br
1-бромпропан
В 1869 г. русский химик В. В. Марковников на основании экспериментальных данных установил закономерность в присоединении га-логеноводородов и родственных им соединений к несимметричным алкенам, известную под названием правила Марковникова. Его сущность в следующем:
при взаимодействии галогеноводоров и родственных им соединений с несимметричными алкенами атом водорода присоединяется к более гидрогенизированному атому углерода, т. е. к атому углерода, содержащему большее число атомов водорода.
Это эмпирическое правило, а теперь, используя электронные представления, дадим пояснения к нему.
Направленность присоединения определяется поляризацией алкена в нереагирующем состоянии. Вследствие +/-эффекта и о,л-со-пряжения со стороны алкильных групп п-электронная плотность двойной связи смещена к более гидрогенизированному атому углерода, что определяет наиболее вероятное место присоединения протона.
5+<~Х5- 5+ 5-
СН3----С = СН2 + Н-------Вг ------- Н3С—С\—сн3
1н, н’с Вг
Кроме того, из двух возможных вариантов присоединения протона преимущественно реализуется тот, при котором в качестве промежуточного продукта присоединения образуется более устойчивый карбкатион:
—1 н3с /С—сн2 — и3с -—н3с—с— сн3 сн3 третичный карбкатион (более устойчив) . н,с 1 X + —- нс—сн2 Н3С^ первичный карбкатион
8. Алкены (этиленовые углеводороды, олефины)°'
4. Присоединение воды (гидратация). Гидратация алкенов идет в присутствии минеральных кислот, чаще всего серной, и приводит к образованию спиртов:
5+
сн,—►СН===СН + нон
3 К /
сн-—( н—сн3
он
проппнпл-2
Механизм реакции следующий:
+ медленно
н3с—сн=сн2 + н ------------
н3с—сн—сн3
изопропил катион
быстро
н3с—сн—сн3 + ОС
44
изопропилоксониевы й катион
---► н3с—сн—сн3 + н+
он
5. Присоединение гипогалогенных кислот (гипогалогенирование). Гипогалогенные кислоты (НОС1, HOBr, HOI) присоединяются к алкенам подвойной связи с образованием галогенгидринов. Механизм реакции электрофильный:
Н2С=СН2 + НОС1
Н2С---СН2
ОН С1
этиленхлоргидрин
6. Озонирование алкенов. При взаимодействии алкенов с озоном образуются перекисные соединения, которые называются озонидами. При обработке цинком в уксусной кислоте они разрушаются с образованием альдегидов или кетонов в зависимости от строения алкена.
88
Лекции по органической химии
сн—сн=сн—сн3+о3 бутен-2
O-ft-0
/ ; \ zn
сн,—сн/ \ сн—сн,----------»
сн3соон
2СН3—С ^Н
этаналь
Сопоставляя продукты окисления, можно судить о наличии и положении двойной связи в этиленовом углеводороде.
7. Окисление раствором калия перманганата (реакция Вагнера, 1888г.) — гидроксилирование. Большое значение имеет реакция окисления алкенов КМпО4, которая известна как реакция Е. Вагнера. При взаимодействии алкенов с разбавленным водным раствором КМпО4 в слабощелочной или нейтральной среде образуются двухатомные спирты (гликоли):
СН, сн—Оч О’ _ _ СН,—ОН
II 2 KMnO. I 2 \ / Н2О; ОН | 2 I
II но * | Mrk ----------------*| + МпО2|+КОН
СН2 2 СН—О О СН2—ОН
этиленгликоль
Эта реакция является качественной на кратные связи, по обесцвечиванию раствора перманганата калия можно судить о непредельнос-ти соединения.
8. Полимеризация алкенов. Полимеризация является важным свойством этиленовых углеводородов. Полимеризация — это образование высокомолекулярных соединений (полимеров) из низкомолекулярных (мономеров) в результате соединения молекул мономера.
полиэтилен (полимер)
п СН2 = СН2
этилен (мономер)
Группа СН2 СН2—Ь — это элементарное звено полимера, то есть элементарный состав полимера не отличается от состава мономера.
9. АЛКАДИЕНЫ
(ДИЕНОВЫЕ УГЛЕВОДОРОДЫ, ДИОЛЕФИНЫ)
Диеновые углеводороды — это углеводороды, содержащие в своей структуре две двойные связи. Общая формула — СН 2.
Простейшим представителем алкадиенов является аллен, или пропадиен Н2С=С=СН2.
Названия алкадиенов по заместительной номенклатуре ИЮПАК образуют путем прибавления к корню предельного углеводорода суффикса -диен с указанием положения каждой из двойной связи. Например:
12 3 4
н2с=сн—сн =сн2
бутадиен-1,3, дивинил
1 2 3 4 5
н2с=с—сн2—сн=сн2
сн3
2-метилпентадиен-1,4
Для некоторых алкадиенов сохранились тривиальные названия.
н,с=с—сн=сн
2
сн3
изопрен
В зависимости от взаимного расположения двойных связей алкадиены делятся на три группы.
\ /1. Алкадиены с кумулированными двойными связя-
^С=С=С ми — алленовые углеводороды. В алленовых угле-водородах двойные связи находятся в положении 1,2.
Представителем является аллен.
н2с=с=сн2
аллен
с=с—(СН,)—с—с z п
2. Алкадиены с изолированными двойными связями. Двойные связи отделены друг от друга двумя и боль-шим числом простых связей.
1 2 3 4 5 6
Н2С=СН—СН—СН—СН—сн2
гексадиен-1,5
90
Лекции по органичен кои химии
хс=сн—НС=С^
3. Алкадиены с сопряженными, конъюгированными двойными связями. Двойные связи разделяются одной простой связью. Пер-
вым представителем этого ряда является дивинил, или бутадиен-1,3.
н2с=сн-нс=сн2
дивинил
Строение алкадиенов
Строение алкадиенов с кумулированными двойными связями рассмотрим на примере аллена. В его молекуле центральный атом углерода находится в ^^-гибридизации, а атомы углерода метиленовых групп — в ^-гибридизации. Все три атома углерода расположены в пространстве линейно, а л-связи находятся в двух взаимно перпендикулярных плоскостях.
Рис. 9.1. Строение молекулы аллена
В молекулах алкадиенов с сопряженными двойными связями происходит дополнительное перекрываниер-электронных облаков соседних л-связей (л,л-сопряжение) и образуется единая л-электронная система. Ниже (рис. 9.2) показано образование единого л-электрон-ного облака в молекуле бутадиена-1,3.
лл-сопряжение
Рис. 9.2. Строение молекулы аллена
делокализованная
гс-система
9. Алкадиены (диеновые углеводороды, диолефины)
91
В результате тгл-сопряжения происходит укорачивание о-связей.
Так, в молекуле бутадиена-1,3 длина связи С2—С3 составляет 0,148 нм, тогда как длина связи С—С в этане — 0,154 нм. Энергия сопряжения бутадиена-1,3 составляет примерно 15 кДж/моль.
Алкадиены с изолированными двойными связями построены аналогично алкенам.
Особенности строения алкадиенов различных типов сказываются на их реакционной способности. Диены с кумулированными и изолированными связями напоминают по химическим свойствам алкены, вступая в реакции присоединения с участием двух двойных связей самостоятельно.
Для сопряженных диенов характерны некоторые особенности в химических свойствах.
АЛКАДИЕНЫ С СОПРЯЖЕННЫМИ СВЯЗЯМИ
Способы получения
1. Каталитическое дегидрирование алканов и алкенов. Это промышленный метод получения дивинила (бутадиена-1,3) и изопрена (2-ме-тилбутадиена-1,3). Катализаторами реакции являются Сг2О3/А12О, (смешанный оксидный алюмохромовый катализатор).
н3с—СН—СН—сн3
н2с=сн—сн—сн3
650цс - н7с=сн—сн=сн, бутадиен-1,3
н3с—сн—сн—сн3 сн3
н3с—сн—сн=сн2 сн3
550 °C
н2с=с—сн=сн2
сн3
2-метилбутадиен-1,3
92
Лекции по органической химии
2. Способ С.В. Лебедева. Поэтому методу бутадиен-1,3 получают из этилового спирта путем одновременного каталитического дегидрирования и дегидратации на смешанном цинкалюминиевом катализаторе:
2С2Н5ОН -Z—Н2С=СН—НС=СН2+ 2Н2О + Н2
Химические свойства
Сопряженные диены проявляют ненасыщенный характер. Наличие сопряженной системы в их молекуле приводит к тому, что они присоединяют различные вешества не только по месту одной двойной связи (1,2-присоединения), но и к крайним атомам сопряженной системы с перемещением двойной связи (1,4-присоединение). Соотношение этих продуктов зависит от условий проведения реакции и природы электрофильного реагента.
1. Гидрирование. Водород в момент выделения образует с алкадиенами-1,3 обычно продукты 1.4-присоединения, например:
н,с=сн—сн=сн,------------
2 2 2СН.ОН + 2Na
н3с—сн=сн—сн3 бутен-2
В присутствии катализаторов (Ni, Pt) диены-1,3 присоединяют водород в 1,2- и 1,4-положения с образованием соответствующих алкенов, которые подвергаются дальнейшему гидрированию до алканов:
Н3С-СН2-СН=СН2-
1LC==CH~CH=CH,-^- бу™'1
2 2 Kt
Н2
Kt
н3с-сн=сн-сн3-'
бутен-2
н3с-сн2-сн2-сн3
бутан
2. Присоединение галогенов. Присоединение галогенов приводит к образованию смеси продуктов 1,2- и 1,4-присоединения. Как правило, при повышении температуры и переходе от хлора к йоду возрастает выход продукта 1,4-присоединения. Например, в процессе бромирования бутадиена-1,3 при температуре —80 °C образуется преимущественно продукт 1,2-присоединения, а при 40 °C — продукт 1,4-присоединения:
9. Алкадиены (диеновые углеводороды, диояефцны)
93
1,2-присоединение
н2с=сн—сн=сн2
Вг Вг
I I
н2с—сн—сн=сн,
3,4-дибромбутен-1
1,4-присоединение
н2с—сн=сн—СН
Вг
Вг
1,4-дибромбутен-2
Присоединение галогенов к сопряженным алкадиенам происходит по электрофильному механизму. Особенность механизма состоит в том, что электрофильная частица атакует концевой атом углерода сопряженной системы, поскольку при этом образуется мезомерно стабилизированный карбкатион, строение которого можно представить граничными структурами I и II:
Вг2
Н2С=СН—СН=СН2-----
Н2С=гСН—СН—СН2
| 5+ &-
Вг---»-Вг
л-комплекс
—» н,с—сн,—сн=сн
-Вг- 2| 2
Вг I
+
н2с—сн=сн—сн2
Вг ц
Последующая атака карбкатиона бромид-ионом приводит к образованию продуктов 1,2-(1П) и 1,4-присоединения (IV):
IV
94
Лекции по органической химии
3. Присоединениегалогеноводородов. Присоединение галогеноводородов также происходит с образованием продуктов 1,2- и 1,^присоединения:
1,2-присоединение
НВг
Н2С=СН—СН=СН2---!
1,4-присоединение
Вг
н3с—сн—сн=сн2
З-бромбутен-1
н,с—сн=сн—сн, I 1-бромбутен-2 „
4. Реакция Дильса — Альдера (диеновый синтез). Реакция основана на взаимодействии сопряженных диенов с веществами, имеющими в своем составе двойную или тройную углерод-углеродную связь (так называемыми диенофилами). В процессе реакции диеновые углеводороды присоединяют диенофилы в положение 1,4 с образованием циклических структур:
бутадиен-1,3 малеиновый
ангидрид
ангидрид 1,2,3,6-тетра-гидрофталевой кислоты
Эта реакция является качественной на соединения, содержащие сопряженные двойные связи. Протекает по молекулярному механизму, который характеризуется синхронным процессом разрыва и образования связей в реагентах ([4+2]-циклоприсоединение).
5. Полимеризация. Важным свойством сопряженных диенов является их склонность к полимеризации, причем полимеризация происходит предпочтительно по положениям 1 и 4 как по ионному, так и по свободнорадикальному механизму:
лН,С=СН—нс=сн,—*- -есн,— нс=сн—сн,-э-
2 2 2 2 п
бутадиен-1,3
1,4-полибутадиен
9. Алкадиены (диеновые углеводороды диолефины)
При полимеризации замещенных диенов 1,4-присоединение осуществляется по принципу «голова к хвосту»
п н,с=с—нс=сн,-^* н-сн,-с=сн -н2с-н,с-С=СН-СН,-К
I I I z I * z I ll
сн3 сн3 сн3
изопропен полиизопрен
Реакция полимеризации широко используется в производстве синтетического каучука.
Ачтожетакое натуральный каучук? Его получают излатекса (млечный сок) тропического растения гевеи, произрастающей главным образом в Бразилии. Выделяющийся при подсочке деревьев млечный сок содержит 20—60 % каучука, который осаждают добавлением муравьиной или уксусной кислоты.
По химическому строению натуральный каучук представляет собой линейный стереорегулярный полимер изопрена (полиизопрен), имеющий 1<мс-конфигурацию изопреновых звеньев:
Молекулярная масса натурального каучука составляет в среднем 100 000-150 000.
10. АЛКИНЫ
(АЦЕТИЛЕНОВЫЕ УГЛЕВОДОРОДЫ)
Углеводороды алифатического ряда, содержащие одну тройную связь, называют алкинами.
Алкины образуют гомологический ряд общей формулы СяН , родоначальником ряда является ацетилен НС=СН. Как видно из общей формулы, алкины являются изомерами алкадиенов
Номенклатура и изомерия
По номенклатуре ИЮ ПАК названия алкинов образуют от названий соответствующих алканов, заменяя суффикс -ан на -ин с указанием положения тройной связи в цепи углеродных атомов. Нумерацию углеродной цепи начинают с того конца, к которому ближе расположена тройная связь.
Например, 5 4 3 2 1
н3с—сн—с = с—сн3
сн3
4-метилпентин-2
Согласно рациональной номенклатуре ацетиленовые углеводороды рассматривают как производные ацетилена, в молекуле которого атомы водорода замещены на радикалы:
Н3С—С^=СН метилацетилен
Н С---CHj—С^=СН этилацетилен
Н3С---С^=С-----СН3 диметилацетилен
Названия радикалов образуются путем добавления окончания -ил к названию углеводорода:
Р((2 = С этинил
н3с-с=с-нс=с—сн— 1-пропинил 2-пропинил (пропаргил)
Изомерия ацетиленовых углеводородов начинается с третьего члена гомологического ряда.
Для алкинов характерны структурная изомерия, которая обусловлена различной структурой углеродной цепи, и изомерия положения тройной связи.
10. Алкины (ацетиленовыеуглеводороды)
изомерия цепи
н3с—сн—с=сн
I ice—сн—сн—с=сн
пентин-1
З-метилбутин-1
изомерия положения
н3с—сн—с=сн н3с—с=с—сн3
бутин-1 бутин-2
Способы получения
В промышленном синтезе широко используется ацетилен, для получения которого существует ряд специфических способов. Гомологи ацетилена чаще всего получают дегидрогалогенированием дига-логеналканов и алкилированием ацетиленидов.
Промышленные способы получения
1. Карбидный метод. При взаимодействии карбида кальция с водой образуется ацетилен:
СаС2 + 2Н2О -------- НС=СН + Са(ОН)2
2. Пиролиз метана или нефтяных фракций. Метан подвергают действию высокой температуры в течение короткого времени и затем быстро охлаждают с целью избежать дальнейшего расщепления ацетилена на углерод и водород:
2СН4 —00°С- НС=СН + ЗН2
Лабораторные способы получения
1. Дегидрогалогенирование дигалогеналканов и галоген алкенов. При нагревании вицинальных или геминальных дигалогеналканов со спиртовым раствором щелочи образуются алкины:
С,н5он
Н,С—СН, + 2NaOH ——--------* НС=СН + 2NaBr + 2Н2О
II
I I ацетилен
Вг Вг
1,2-дибромэтан
С7Н.ОН
Нзс—СН—CHBr2 + 2NaOH —5 > Н3С—С=СН+2NaBr + 2Н2О
1,1-дибромпропан пропин
В аналогичных условиях алкины образуют также галогеналкены, содержащие атом галогена у атома углерода с двойной связью.
98
Лекции по органической химии
2. Реакции алкилирования ацетиленов. При взаимодействии металлических производных ацетиленовых углеводородов с алкилгалогени-дами образуются алкины с большим числом атомов углерода в молекуле:
с,н,- Вг
HC=CNa —~~—* НС=С~С2Н5
- NaBr z
ацетиленид бутин-1
натрия
Нзс—C=CMgl — н—> н3с—с=с—сн3
пропинилмагний- ' бутин-2
йодод
Физические свойства
Три первых представителя гомологического ряда представляют собой газы, далее следуют жидкости (С5—С15), а начиная с углеводорода С16Н30, — твердые вещества. Изменения температур плавления и кипения в ряду алкинов подчиняются основным закономерностям, характерным для алканов и алкенов.
Химические свойства
Химические свойства ацетиленовых углеводородов обусловлены наличием в их структуре тройной связи. Атомы углерода, связанные тройной связью, находятся в состоянии sp-гибридизации. Вы знаете, что тройная связь — это сочетание одной о-связи и двух л-связей, расположенных во взаимно перпендикулярных плоскостях, ее длина около 0,120 нм, энергия связи 812 кДж/моль. Для алкинов, как и для алкенов, характерны реакции электрофильного присоединения. Но по сравнению с алкенами, алкины менее активны, реакции протекают, как правило, в две стадии. Это объясняется большей электроотрицательностью хр-гибридизованного атома углерода по сравнению с sp3-гибридизованным атомом, что приводит к уменьшению длины связи —С=С—. Этим объясняется и слабый —СН-кислотный характер алкинов: R—С=С8<Н8+.
Таким образом, наряду с реакциями присоединения для ацетиде-новых углеводородов характерны и реакции замещения атома водорода концевой С—Н-группы. Наконец, аналогично алкенам алкины вступают также в реакции окисления, восстановления и полимеризации.
Реакции присоединения
1. Гидрирование. Присоединение водорода проходит ступенчато по месту тройной связи в присутствии катализаторов. Такой процесс называется стереоселективным гидрированием. Конечным продуктом будет алкан.
10. Алкины (ацетиленовые углеводороды)
99
[Н| Нзс\ /И
н3с-с=с-сн— бутин-2 Li(NH3) ХСН3 /ира«с-бутен-2 [Н1 Нх /Н — с=с [Н],
и<с> н,с'с=с>
<д<с-бутен-2
н3с-н2с-сн2-сн3
бутан
2. Галогенирование. Алкины довольно легко присоединяют по месту тройной связи одну молекулу хлора или брома, в результате образуется преимущественно/пронс-дигалогеналкен. Присоединение второй молекулы галогена идет труднее. Механизм реакции аналогичен галогенированию алкенов (АЕ).
н3с—с=сн
Вг,
Нзс /Вг
с=с
BrZ ЧН
Вг2
Вг Вг
I I
н3с—с—СН
I I
Вг Вг
транс-Х ,2-дибромпропен
1,1,2,2-тетрабромпропан
Обесцвечивание бромной воды — качественная реакция на тройную связь.
3. Гидрогалогенирование. Присоединение галогеноводородов (НО, НВг) происходит также в две стадии с образованием соответственно моногалогензамещенных или геминальных дигалогеналканов.
Реакция может проходить по механизму электрофильного или радикального присоединения. В первом случае соблюдается правило Марковникова. При радикальном механизме наблюдается противоположное направление присоединения.
Вг
а) Н,С—С=СН • НВг» Н,С—С=СН, -Br ~ Н3С~С—СН,
J J | Z J । " J
Вг Вг
2-бромпропен 2,2-дибромпропан
б) н,с—с=сн ,1,1Вг> Н,С-СН—СНВГ-^* Н3С-СН,-СНВг,
Н2О2 1 Н2и2 J z z
1 -бромпропен- 1 1,1 -дибромпропан
4. Гидратация (реакция Кучерова). Ацетилены очень легко присоединяют воду в кислой среде в присутствии солей ртути (II) в качестве катализатора. Присоединение происходит в соответствии с правилом Марковникова. При этом из ацетилена образуется уксусный альдегид, а из других алкинов — кетоны:
100
Лекции по органической химии
H,SO4 нс^сн + нон —:—
HgSO4
8+
Н2С=СН—ОН' L_________
виниловый спирт
► н3с-сх н
уксусный альдегид
8+^8- h,so. сн,-^с=сн + нон—— 3 HgSO,
н,с—с—сн
о н'
" ' 8+
н,с—с—сн
о
ацетон
Гидратация алкинов протекает через стадию образования непредельных спиртов, содержащих гидроксильную группу у атома углерода с двойной связью. Такие спирты неустойчивы. В процессе образования они изомеризуются в карбонильные соединения. Эту закономерность установил в 1877 г. русский химик А.П. Эльтеков, и она получила название правила Эльтекова.
Реакция Кучерова лежит в основе производства уксусного альдегида, уксусной кислоты и уксусного ангидрида.
5. Присоединение спиртов и кислот. Эти и способом получают эфиры не существующего в свободном состоянии винилового спирта. Реакция протекает по механизму нуклеофильного присоединения (AN):
RONa
НС=СН + С2Н5ОН----------- н2с=сн—ОС2Н5
винплэтиловый эфир
Катализаторами нижеприведенной реакции являются соли ртути (II), меди (I), кислоты Льюиса.
нс=сн + н3с —сх
н2с=сн—о—с—сн3 виниловый эфир уксусной кисюты
Виниловый эфир уксусной кислоты используется для производства полимера поливинилацетата (ПВА).
Реакции замещения
1. Образование ацетиленидов. Способность ацетиленов с концевой тройной связью отдавать протон позволяет говорить о них как о соединениях, проявляющих кислотные свойства. И хотя их кислотность очень мала, тем не менее алкины способны к реакциям замещения и реагируют с сильными основаниями, образуя при этом соли. Такие соли называют ацетиленидами.
10 Линины (ацетиленовые углеводороды)
101
Если пропускать через аммиачный раствор солей одновалентной меди или серебра ацетилен, из раствора выпадает соответственно красно-коричневый осадок ацетиленида меди или белый осадок ацетиленида серебра:
НС=СН + 2[Ag(NH3)JOH-----Ag—С=С—Ag + 4NH3 + 2Н2О
ацетиленид серебра
НС=СН + [Cu(NH3)2]OH-----* Си—с=с—Си + 2NH3 + Н2О
ацетиленид меди
Ацетилениды тяжелых металлов в сухом виде чувствительны к трению, удару и легко взрываются.
Ацетилениды натрия представляют особый интерес как промежуточные соединения для синтеза других алкинов. Они легко вступают в реакцию нуклеофильного замещения с галогеналканами:
НС=СН + 2NaNH2 --------*- Na—С=С~Na + 2NH3
ацетиленид натрия
Na—С=С—Na + 2CH3I ----------* Н3С—С=С—СН3 + 2NaI
Аналогично протекает реакция с реактивами Гриньяра (магний-органическими соединениями). Русскими учеными Чугаевым и Це-ревитиновым было предложено определение подвижности атомов водорода с помощью магнийорганических соединений в органических соединениях:
НС=СН + 2CH3MgI----------- IMgC=CMgI +2CH4f
По количеству атомов водорода выделяется эквивалентное количество СН4. Метан собирают, определяют объем и рассчитывают число подвижных атомов водорода. Этот метод имеет аналитическое значение.
2. Замещение водорода при Csp на галоген. Атомы водорода при углероде с тройной связью замещаются на атомы галогенов (хлор, бром, йод) при действии галогенов в присутствии щелочей.
нс=сн -^aQC1 НС=С—Cl N-aQCI » Cl—с=с—Cl
хлорэтин дихлорэтан
Реакции окисления
Алкины, подобно алкенам, подвергаются окислению по месту тройной связи. Продуктами реакции являются соответствующие кар
102 Лекции по органической химии
боновые кислоты. В качестве окислителей используют перманганат калия в нейтральной или щелочной среде, озон и др.
fol ,0 О
нс=сн п .» с—С -----------------------*- СОТ + н—с
КМпО., Н,О z \ 21 \
ИО он он
щавелевая кислота муравьиная кислота
При окислении ацетилена в нейтральной среде образуется щавелевая кислота, которая в этих условиях декарбоксилируется и конечным продуктом будет муравьиная кислота. Гомологи ацетилена окисляют в щелочной среде с разрывом по месту тройной связи.
[О] н,с-сн-с=с-сн,------------- н,с-сн,-с + н,с-с
32 КМпО4;ОН- ОН 3 ЧОН
пентин-2
пропионовая кислота уксусная кислота
Реакция окисления алкинов используется для определения их строения.
Полимеризация алкинов (димеризация, циклотримеризация)
Алкины способны полимеризоваться в нескольких направлениях. В присутствии хлорида меди (I) и хлорида аммония ацетилен димеризуется с образованием винилацетилена:
НС=СН + НС=СН
CuCl, NH„C1
н2с=сн—с=сн
винилацетилен
Винилацетилен является исходным продуктом в производстве хлоропреновых каучуков.
При нагревании в присутствии активированного угля или комплексных никельорганических катализаторов ацетилен циклотриме-ризуется с образованием бензола:
знс=сн
Это один из синтетических способов получения ароматических углеводородов.
АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ (АРЕНЫ)
Ароматическими первоначально называли органические соединения. которые или сами имели приятный запах, или же выделялись из природных вешеств, обладающих приятным запахом. В последствии среди них были обнаружены соединения с приятным и неприятным запахом, а также соединения без запаха. Однако название за большой группой органических соединений, проявляющих сходные с бензолом свойства, сохранилось.
Кароматическим углеводородам или аренам относятся соединения, молекулы которых содержат одно или несколько бензольных колец.
В зависимости от числа бензольных циклов, входящих в состав молекулы, различают одноядерные (моноциклические) и многоядерные (полициклические) арены.
11. МОНОЯДЕРНЫЕ АРЕНЫ
Простейшим представителем одноядерных ароматических соединений является бензол (С6Н6).
Впервые бензол был получен английским ученым М. Фарадеем в 1825 г. из светильного газа, образующегося в процессе переработки каменного угля. Однако строение его молекулы в течение многих лет оставалось загадкой для химиков. Несмотря на то, что формула С6Н6 предполагает достаточно выраженный ненасыщенный характер, бензол, в отличие от непредельных соединений, оказался относительно инертным веществом. Он сравнительно устойчив к нагреванию и действию окислителей, практически не вступает в характерные для ненасыщенных соединений реакции присоединения. Наоборот, для бензола более характерными оказались не свойственные непредельным соединениям реакции замещения.
Составу С6Н6 приписывались разные структурные формулы, но все они не объясняли в полной мере химических свойств бензола.
В 1865 г. немецкий химик Кекуле предложил формулу бензола, представляющую собой цикл из шести атомов углерода с чередующимися простыми и двойными связями:
.СН
НС ^сн
II I
НС' ^сн
сн
104
Лекиии по органической химии
Формула Кекуле предполагает равноценность всех атомов углерода и водорода в молекуле.
В соответствии с формулой Кекуле бензол должен иметь два 1,2-дизамещенных изомера:
Экспериментально же было установлено, что 1,2-дизамегценные производные бензола не имеют изомеров положения, т. е. они существуют в виде одного соединения.
Для объяснения этого противоречия в 1872 г. Кекуле выдвинул ос-цилляционную гипотезу, согласно которой двойные связи в молекуле не фиксированы, а непрерывно перемещаются (осциллируют) между двумя возможными положениями:
Правильно отображая некоторые свойства бензола, формула Кекуле тем не менее не согласовывалась с рядом установленных фактов. Все это возвращало химиков к пересмотру структуры бензола.
В соответствии с современными представлениями, основанными на данных квантовой химии и физико-химических исследований, молекула бензола представляет собой правильный плоский шестиугольник. Все углеродные атомы находятся в состоянии .sp2-гибридизации. За счет^-гибридизованных атомных орбиталей каждый атом углерода образует три о-связи (одну с атомом водорода и две с соседними атомами углерода). Негибридизованная р-атомная орбиталь участвует в образовании ароматического секстета. Облака р-электронов имеют форму объемной восьмерки и расположены перпендикулярно плоскости цикла, которая разделяет их пополам.
11. .Миноядерные арены‘ J
Если посмотреть на проекцию л-электронной плотности сверху, то мы можем увидеть такую картину:
Образование замкнутой сопряженной системы (ароматического секстета) является для молекулы бензола энергетически выгодным. Экспериметально установлено, что сопряжение в цикле приводит к уменьшению энергии на 150.7 кДж/моль, по сравнению с расчитанной для циклогексатриена. Эта разность составляет энергию сопряжения.
В бензольном кольце нет простых и двойных связей в прямом понимании этого слова. Такую связь называют ароматической. Если длина простой связи С—С в алканах составляет 0,154 нм, длина двойной связи в алкенах — 0,134 нм, то длина С—С связи в молекуле бензола равна 0,140 нм, т. е. является промежуточной между длиной одинарной и двойной связи.
Совокупность специфических свойств бензола — высокая стабильность, инертность в реакциях присоединения, склонность к реакциям замещения, получила общее название «ароматичность», или «ароматические свойства».
Что же требуется для того, чтобы мы имели право отнести соединение к ароматическому ряду?
а) для проявления ароматического характера молекула должна прежде всего иметь плоское строение;
б) молекула должна иметь замкнутую сопряженную систему;
в) количество л-электронов должно соответствовать формуле 4/7 + 2, где п = 0, 1, 2, 3 и т. д. (данная закономерность была сформулирована в 1931 г. немецким ученым Э. Хюккелем).
Номенклатура и изомерия
По заместительной номенклатуре ИЮПАК одноядерные арены рассматривают как производные бензола.
метилбензол, толуол
106
Лекции по органической химии
При наличии в кольце двух и более заместителей их положение указывают цифрами. Нумерацию атомов углерода бензольного кольца осуществляют таким образом, чтобы заместители имели возможно меньшие номера.
В дизамещенных производных бензола наряду с цифровым обозначением положений заместителей применяют приставки: орто-(о-) положение — 1,2; мета-(м-) положение — 1,3 и пара-(п-) положение — 1,4.
1,3-диметил бе нзол, м-ксилол
1 -изопропил-4-метил бензол Л-ЦИМОЛ
Кроме названий по заместительной номенклатуре сохранились и тривиальные названия: толуол, ксилол, кумол и др.
Одновалентные радикалы аренов имеют общее название—арилы (Аг). Двухвалентные радикалы бензола называют фениленами (о-, м-, п-)'.
фенил
Изомерия гомологов бензола обусловлена разными структурами, числом и положением заместителей в бензольном кольце.
Для однозамещенных гомологов бензола характерна изомерия, связанная с разной структурой заместителя.
Дизамещенные производные бензола существуют в трех изомерных формах, в зависимости от взаимного расположения в бензольном кольце (изомеры положения).
11. Моноядерные арены
107
1.2-диметилбензол, 1,3-диметилбензол. 1,4-диметилбензол,
о-ксилол
л<-ксилол
и-ксилол
Для тризамешенных бензолов с одинаковыми заместите зми в бензольном кольце существуют также три изомера:
1,2,3-триметилбензол рядовой триметилбензол вицинальный триметил бензол
1,2,4-асимметричный тринитробензол
1,3,5-триметилбензол симметричный триметилбензол
Способы получения аренов
1. Циклотримеризация аминов
Ацетилен пропускают над активированным углем при повышенной температуре. Реакция была открыта Зелинским.
>t
знс=сн-----
2. Обработка ацетона конц. H2SO4
!б[ _ СНз
сн —с 1 ~\-сн
\ г- .-// 3
СНз; : О;
:о< хсн':
‘^с 1
I сн3
конц. H2SO4
мезитилен
108
Лекции по органической химии
3. Дегидрогенизация алициклических соединений
СИ2 *'СН2
сн2 хсн2
'СН2
циклогексан
Pd, >t
-3H2f
Эта реакция показывает взаимосвязь между ароматическими и алициклическими соединениями.
4. Реакция Вюрца —- Фиттига
C6H5Br + CH3Br +2Na —* С6Н5СН3 + 2NaBr
бромбензол метилбензол
толуол
Эта реакция чаще всего используется для получения гомологов бензола.
5. Алкилирование ароматических углеводородов по Фриделю — Крафтсу.
FeBr.
-НВг
Физические свойства
Бензол и его низшие гомологи представляют собой жидкости, обладающие специфическим запахом. Ароматические углеводороды не растворимы в воде и хорошо растворяются в органических растворителях. Многие из них сами являются хорошими растворителями для других органических веществ. Из-за высокого содержания углерода горят коптящим пламенем.
Химические свойства
Реакционная способность бензола и его гомологов определяется главным образом наличием в структуре замкнутой п-электронной системы, которая является областью повышенной электронной плотности. Ароматические углеводороды, как и алкены, обладают нуклеофильным характером. Однако, в отличие от ненасыщенных соединений, при взаимодействии с электрофильными реагентами арены более склонны к реакциям замещения, а не присоединения, поскольку при этом сохраняется их ароматический характер. Эти реакции носят название реакций электрофильного замещения SE.
11. Мономерные арены
Реакции присоединения для аренов менее харакгерны, так как они приводят к нарушению ароматичности, с трудом вступают ароматические углеводороды и в реакции оксиления.
1. Реакции электрофильного замещения (SE)
При атаке электрофильной частицей л-электронной системы бензольного кольца в результате электростатического взаимодействия образуется неустойчивый л-комплекс:
л-комплекс
Е +
Далее электрофил «вырывает» пару электронов из ароматического секстета бензольного ядра и между ним и одним из атомов углерода образуется о-связь. Таким образом нарушается ароматичность бензольного ядра, образуется карбкатион — о-комплекс.
Делокализацию положительного заряда в о-комплексе можно представить с помощью резонансных структур (I—111):
Образование о-комплекса является наиболее высокоэнергетичной стадией реакции, определяющей ее скорость. о-Комплекс не устойчив, он отщепляет протон от атома углерода, связанного с электрофилом, благодаря чему восстанавливается ароматичность бензольного кольца.
-Н+
110
Лекции по органической химии
К наиболее важным реакциям SEотносятся реакции нитрования, сульфирования, галогенирования, алкилирования и ацилирования.
1. Нитрование. В качестве нитрующих агентов чаше используют концентрированную азотную кислоту или смесь концентрированной азотной и серной кислот (нитрующая смесь):
+ HNO3
конц.
H2SO4 конц.
60 °C
нитробензол
+ Н2О
Атакующей электрофильной частицей в реакции является ион нитрония NO2+, который образуется в результате кислотно-основного взаимодействия между азотной и серной кислотами, где азотная кислота играет роль основания:
HONOj + H2SO4
Н—О—NO2 + HSO4
Н
протонированная азотная кислота
+
н-о-NO2 н2о + no2
J-| ион
нитрония
Ион нитрония атакует п-электронную систему бензольного ядра. В результате реакции образуется нитробезол.
+ Not
к-комплекс
NO+2
о-комплекс
2. Сульфирование — это процесс замещения атома водорода в бензольном ядре на сульфогруппу — SO3H. Для сульфирования бензола и
11. Моноядерные арены
111
его гомологов применяют концентрированную серную кислоту или олеум (раствор триоксида серы SO3 в серной кислоте):
+ H2SO4 конц.
бензол сульфокислота
Особенности механизма сульфирования аренов из> чены недостаточно. Однако экспериментальные данные свидетельствую о том, что атакующей электрофильной частицей служит триоксид огры SO3:
2h2so4 so3 + н3о+ + hso;
3. Галогенирование. Бензол и его гомологи хлорируются, бромиру-ются и йодируются. Замещение атома водорода в бензольном ядре на атом хлора или брома осуществляют в присутствии катализаторов — кислот Льюиса (AlCl3, FeBr3, ZnCl2 и др.):
AlCl3
+ HCl
хлорбензол
Под действием катализатора молекула галогена поляризуется. Атакующей электрофильной частицей служит либо комплекс поляризованной молекулы галогена с кислотой Льюиса, либо катион галогена, образующийся в процессе ионизации данного комплекса:
5+ 5+8- + -
Cl—С1 + А1С13 — [ С1-*С1 • • • А1С13 ] ' СГ[А1С14]
4. Алкилирование по Фриделю — Крафтсу. Для введения алкильной группы в молекулу ароматического соединения в качестве электрофильных реагентов чаше всего используют галогеналканы. Взаимодействие происходит в присутствии катализаторов — кислот Льюиса:
хлорэтан
> AIC1,
| + С2Н5С1 --------
НС1
этилбензол
112
Лекции по органической химии
Атакующей электрофильной частицей является карбкатион, который образуется при взаимодействии алкилирующего агента и катализатора:
S+ 8—
С2Н5------* С1 + А1С13 С2Н5 + AIC1’
карбкатион
Для алкилирования аренов также могут быть использованы спирты (реакции протекают в присутствии кислот Льюиса или минеральных кислот — Н3РО4, H2SO4) или алкены (в этом случае алкилирование требует присутствия кислот Льюиса и минеральной кислоты как источника протонов)
По своему механизму реакция алкилирования аналогична реакциям нитрования, сульфирования и галогенирования.
5. Ацилирование по Фриделю — Крафтсу. Ацилированием называют процесс введения в молекулу органического соединения ацильной
группы — R—С
Ацилирование бензола и его гомологов обычно осуществляют га-логенан гидридами карбоновых кислот в присутствии кислот Льюиса:
ацетофенон, метилфенилкетон
Электрофилом, атакующим бензольное кольцо, является либо аци-+
лиевый ион СН—С=О , либо комплекс ацилгалогенида с катализа-
тором СН3СОА1С14:
RCOC1 + А1С13 ~ RCOA1C14 ~ У R—С—О + А1С14
Для введения ацильной группы могут быть использованы и ангидриды карбоновых кислот.
II. Реакции присоединения
Реакции присоединения не характерны для аренов, они протекают в жестких условиях.
11. Моииядерные арены*
1. Гидрирование. При повышенных температуре и давлении, в присутствии катализаторов (мелкопористый никель — никель Ренея) бензол и его гомологи присоединяют три молекулы водорода:
+ ЗН2
Ni
циклогексан
Остановить реакцию на стадии образования продуктов частичного гидрирования невозможно, поскольку они гидрируются значительно легче, чем сам бензол.
2. Хлорирование. При интенсивном солнечном освещении или под действием ультрафиолетового излучения бензол присоединяет хлор. Реакция протекает по радикальному механизму с образованием гексахлорциклогексана:
III. Реакции окисления
1. Окисление бензольного цикла. Бензольное кольцо устойчиво к действию окислителей. В обычных условиях ни перманганат калия, ни азотная кислота, ни оксид хрома (VI), ни другие сильные окислители не окисляют бензол. В жестких же условиях, например, при действии кислорода воздуха в присутствии оксида ванадия (V2O5), при температуре 400-500 °C бензольное ядро окисляется, образуя малеиновый ангидрид:
^0
сн—
II 2^0 + 2СО2
сн—С\
+ 2Н2О
малеиновый ангидрид
2. Окисление гомологов бензола. Алкилбензолы, в отличие от незамещенного бензола, окисляются значительно легче. В этом случае при действии сильных окислителей (КМпО4, К2Сг2О? идр.) подвергаются окислению боковые цепи:
114
Лекции по органической химии
пропилбензол
бензойная кислота
изофталевая кислота
Продуктами реакции являются ароматические карбоновые кислоты. Каждый алкильный радикал в бензольном кольце, независимо от длины углеродной цепи, окисляется до карбоксильной группы.
3. Озонирование. Подобно алкенам, бензол и его гомологи реагируют с озоном, образуя продукты присоединения, — триозониды:
Триозониды взрывоопасны. Это маслянистые жидкости, они нестойкие и под действием влаги разрушаются с образованием дикарбо-нильных соединений и продуктов их дальнейшего окисления — дикарбоновых кислот.
11. Моноядерные арены‘
Правила ориентации в бензольном ядре
В молекуле незамещенного бензола электронная плотность распределена равномерно, поэтому электрофильный реагент может атаковать в равной степени любой из шести атомов углерода.
Если же в бензольном кольце содержится какой-либо заместитель, то под его влиянием происходит перераспределение л-электронной плотности и новый заместитель вступает в определенные положения по отношению к имеющемуся.
По влиянию на направление реакций электрофильного замещения и реакционную способность бензольного кольца заместители можно разделить на две группы — заместители I рода (орто-, пара-ориен-танты) и заместители II рода (л/елга-ориентанты).
Заместители I рода — атомы и атомные группы, проявляющие положительный индуктивный (+/) или положительный мезомерный (+М) эффекты (доноры электронов):
-М<2, -NHR, -NH2, -ОН, OR, -NHCOR, -OCOR, -SH, -Aik. -Hal
Заместители I рода (за исключением галогенов) увеличивают электронную плотность в бензольном кольце, тем самым активируют его в реакциях St и направляют следующие заместители в орто- и пара-положения.
Заместители II рода — группы, проявляющие отрицательный индуктивный (-1) или отрицательный мезомерный (-М) эффекты (элек-троноакцепторы):
-NR3, -NO2, -SO3H, -CN, -CHO, -COR, -COOH, -COOR, -CCI3
116
Лекции по органической химии
Заместители II рода уменьшают электронную плотность в бензольном ядре и снижают скорость реакций SE по сравнению с незамещенным бензолом. Вновь входящий заместитель направляется преимущественно Bjwe/по-положение.
При введении третьего заместителя необходимо учитывать природу двух уже имеющихся в бензольном ядре.
Ориентация в дизамещенных производных бензола
В зависимости от электронной природы заместителей и их взаимного расположения различают согласованную и несогласованную ориентацию.
При согласованной ориентации оба заместителя направляют новый заместитель в одни и те же положения бензольного кольца. Согласованная ориентация характерна для дизамещенных производных бензола, в которых соблюдаются условия:
1) ориентанты одного и того же рода находятся в люотй-положс-нии относительно друг друга:
м толуидин
2) заместители находятся в орто- или zzapa-потожении по отношению друг к другу, но один из них является ориентантом I рода, а другой — II рода:
11 Моноядерныс арены
117
ОН(-)
* о-нитрофенол
При несогласованной (несовпадающей) ориентации один из заместителей направляет новую группу в одни, а другой — в иные положения бензольного кольца. Предпочтительное направление замещения можно определить, используя следующие правила:
1) если один из заместителей является ориентантом 1 рода, он определяет направление замещения:
ОН(-)
л-нитрофенол
2) если оба заместителя являются ориентантами I рода, направление замещения определяется более сильным ориентантом:
о-толуидин
3) если оба заместителя являются ориентантами II рода, замещение осуществляется с трудом, а место вхождения третьего заместителя определяется более сильным ориентантом:
о-нитробензойная кислота
Наряду с электронной природой заместителей на соотношение продуктов замещения оказывают влияние пространственные факторы.
МНОГОЯДЕРНЫЕ АРЕНЫ
Ароматические углеводороды, содержащие два и более бензольных ядер, относятся к многоядерным аренам.
В зависимости оттого, каким образом связаны бензольные кольца, многоядерные арены делятся на две группы:
• арены с конденсированными (анелированными) бензольными циклами;
• арены с неконденсированными (изолированными) бензольными циклами.
12. МНОГОЯДЕРНЫЕ АРЕНЫ
С КОНДЕНСИРОВАННЫМИ БЕНЗОЛЬНЫМИ ЦИКЛАМИ
Конденсированные многоядерные арены содержат в своем составе два или более бензольных ядра, имеющих общие атомы углерода.
Наиболее важными представителями конденсированных аренов являются нафталин, антрацен и фенантрен.
нафталин
антрацен
фенантрен
НАФТАЛИН
Нафталин состоит из двух конденсированных бензольных колец. Два атома углерода (9 и 10) общие для двух колец.
В отличие от бензола атомы углерода в молекуле нафталина неравноценны. Положения 1, 4, 5, 8 равнозначны, их принято обозначать буквой а и называть «-положениями. Положения 2, 3, 6
12. Многоядерные арены с конденсированными бензольными циклами* 1“ и 7 также равнозначны и их обозначают буквой Р и называют р-по-ложениями.
Для монозамешенного нафталина возможно 2 изомера (а- и Р~), а при наличии двух одинаковых заместителей — 10 изомеров.
ГН
а-метилнафталин р-метилнафталин
В номенклатуре дизамещенных нафталинов наряду с цифровым обозначением положений заместителей применяют также и приставки: орто-положение —1,2, мета —1,3, пара —1,4, пери —1,8, ам-фи — 2, 6. Например:
1,2-диметилнафталин, орто -диметил нафтали н
2,6-диметилнафталин, амфи - д и мети л нафтал и н
Радикалы нафталина называют нафтилами:
а-нафтил
р-нафтил
Способы получения
/. Природные источники. В промышленности нафталин, главным образом, получают из каменноугольной смолы, где его содержание составляет около 10 %. Наряду с нафталином из каменноугольной смолы выделяют также некоторые его моно- и диметильные производные.
2. Получение нафталина из ацетилена. При пропускании ацетилена через нагретые до 700—800 °C трубки наряду с бензолом образуется также и нафталин.
5СН=СН
700-800 "С
+ Н2
120
Лекции по органической химии
Физические свойства
Нафталин — бесцветное кристаллическое вещество с характерным запахом, сублимируется при 81 °C. Не растворяется в воде, растворяется в органических растворителях. Применяется в быту для борьбы с молью, в химии красителей, для получения лекарств, пластмасс, глиф-талевых смол.
Строение нафталина
Электронное строение нафталина сходно со строением бензола. Его молекула плоская. Дипольный момент равен нулю, но электронная плотность распределена не так равномерно, как в молекуле бензола. Более высокая электронная плотность «-положений нафталина делает их более реакционноспособными по сравнению с Р-положениями.
Длина связей в молекуле нафталина разная.
С—С (Балканах) 1,54А
С — С (в бензоле) 1,40 А
Cj—С2 (в нафталине) 1,36 А
С3 — С4 (в нафталине) 1,43 А
Химические свойства нафталина
Нафталин, как и бензол, проявляет свойства ароматических соединений и для него характерны, прежде всего, реакции электрофильного замещения, но он также легко вступает в реакции присоединения и окисления.
1. Реакции электрофильного замещения. В реакции электрофильного замещения (нитрование, сульфирование, галогенирование) нафталин вступает значительно легче бензола. При этом образуются в основном продукты «-замещения. Это обусловлено тем, что в «-положении нафталинового ядра выше электронная плотность и при атаке в «-положение образуется более стабильный о-комплекс, чем в Р-положении:
12. МногояОерные арены с компенсированными бензольными циклами
121
Как видно, при атаке a-положения делокализация положительного заряда в о-комплексе происходит с сохранением ароматичности одного из бензольных ядер в возможных резонансных структурах.
В случае атаки электрофилом p-положения, лишь в одном случае возможно сохранение ароматичности бензольного ядра. Следовательно, замещение по a-положению энергетически более выгодно.
Нитрование. Нафталин довольно зегко нитруется нитрующей смесью с образованием в основном а-изомера:
hno3
H,SO4:60 °C
а-нитронафталин
Сульфирование. Для сульфирования нафталина используют концентрированную H2SO4, причем в зависимости от температуры, при которой проводится реакция, получают а- или P-продукты замещения. При температуре 80 °C реакционной среды образуется а-на-фталинсульфокислота, а при 160 °C — Р-нафталинсульфокислота:
+ H,SO4
Р-нафталинсульфокислота
При нагревании а-изомера до температуры 160 °C он полностью превращается в Р-нафталинсульфокислоту.
122
Лекции по органической хи.чип
Галогенирование. При температуре 90—110 °C в присутствии катализатора — FeCl3, нафталин хлорируется с образованием преимущественно а-хлорнафталина. Реакция протекает по механизму SE:
С12
FeCl,
Галогенирование нафталина возможно и без катализатора. Тогда вна
чале протекает реакция присоединения галогена, а затем отщепления галоген водорода. Например, при взаимодействии брома с нафталином, бром присоединяется в положения 1 и 4, а затем продукт присоединения отщепляет молекулу бромводорода, образуя а-бромнафталин:
Вг
1,4-дибром-
1,4-дигидронафтапин
Правила ориентации в нафталиновом ядре
Правила ориентации в нафталиновом ядре имеют свои особенности.
Направление электрофильного замещения в монозамещенных производных нафталина определяется электронной природой уже имеющегося заместителя и большей реакционной способностью а-положения.
При наличии в нафталиновом ядре электронодонорного заместителя электронная плотность повышается прежде всего в том кольце, с которым связан заместитель, и поэтому реакция электрофильного замещения происходит именно в этом кольце. Замещение, прежде всего, идет по 4 положению (а-положение).
Например, нитрование 1 -метилнафталина приводит к образованию 1 -метил-4-нитронафталина:
12. Миогояоерные арены с коноежированными б^нзо.; ьными циклами
123
Электроноакиепторный заместитель понижает электронную плотность в нафталиновом ядре и прежде всего в том кольце, с которым он связан. Поэтому реакции идут с участием атомов углерода второго бензольного ядра.
Так как a-положения более реакционноспособны, то замещение происходит преимущественно в положения 5 и 8.
Например, нитрирование 1-нитронафталина нитрующей смесью приводит к образованию смеси изомеров:
1,8-динитро-нафталин
2. Реакции присоединения
Реакция восстановления. Присоединение водорода идет вначале по a-положению. Затем образуется 1.2,3,4-тетрагидронафталин, который при 200 °C гидрируется с образованием декагидронафталина—декалина:
1,4-дигидро-нафталин
1,2,3.4-тетрагидро- декагидронафта-нафталин, тетралин дин, декалин
124
Лекции по органической химии
Тетралин используется как топливо и растворитель для жиров и смол Декалин используется как растворитель для лаков, является заменителем скипидара.
3. Реакции окисления
В отличие от бензольного, нафталиновое ядро легко окисляется и при окислении гомологов нафталина окисляется само ядро.
Например:
метилнафталин
СгО,
сн,соон
2-метил-1,4-нафтохинон
Окисление нафталина в присутствии V2O5 приводит к образованию фталевого ангидрида:
АНТРАЦЕН
Это многоядерное соединение, состоящее из трех линейно конденсированных бензольных колец.
В молекуле антрацена нумеруются лишь атомы углерода, несущие водород. Положения 1,4, 5, 8 — называют «-положениями; 2, 3, 6, 7 — Р-положениями; 9, 10 — у-положениями или ц- (мезо — средний) положениями.
При одном заместителе в ядре возможно 3 изомера — а, р, у.
Способы получения
Антрацен находится в каменноугольном дегте, а именно в антраценовом масле, в количестве 0,5 %, откуда его и добывают в промышленности.
В лаборатории можно получить антрацен по реакции Фриделя — Крафтса из бензола и 1,1,2,2-тетрабромэтана в присутствии А1Вг3:
12. МногоМерные арены с конденсированными бен юльными циклами
125
ахн вг-сн-Дг Л|В'<
1 b:+Z'd 1--------*
JL _Вг-сн-Ьг_
-4HBr
Физические свойства
Антрацен — это бесцветное кристаллическое вещество, г равна 217 °C, перегоняющееся с водяным паром, растворимо в бензоле, эфире, нерастворимо в Н.О. Растворы обладают голубой флюоресценцией.
Химические свойства
Антрацен обладает меньшей ароматичностью, чем бензол и нафталин. Он в большей мере «ненасыщенное» соединение и для него более характерны реакции присоединения, чем электрофильного замещения. Наиболее реакционноспособными в молекуле антрацена являются л/езо-по южения (положение 9 и 10).
1. Реакция восстановления. Антрацен легко присоединяет водород с образованием 9,10-дигидроантрацена. Дальнейшее его гидрирование приводит к пергидроантрацену.
пергидроантрацен
2. Реакции замещения. Галогенирование антрацена происходит по механизму присоединения-отщепления. Например:
Н С1
9, Ю-дигидро-9,10-дихлор-антрацен
126
Лекции пи ормнической химии
С1
9-хлорантрацен
Нитрование антрацена азотной кислотой в среде уксусной кислоты при комнатной температуре ведет к образованию 9-нитроантраце-на. В результате сульфирования антрацена серной кислотой при нагревании образуются а- и Р-изомеры, которые являются более устойчивыми, чем у-.
а-антрацен- Р-антрацен-
сульфокислота сульфокислота
3. Реакция окисления. Антрацен очень легко окисляется концентрированной азотной кислотой или хромовой смесью до антрахинона.
О 9,10-антрахинон
Антрахинон — это желтое кристаллическое вещество, обладающее свойствами хинонов.
ФЕНАНТРЕН
Структурным изомером антрацена является фенантрен.
В антрацене три бензольных кольца соединены линейно, тогда как в фенантрене они соединяются ангулярно, т. е. под углом.
антрацен
фенантрен
12. МногояОерные арены с конденсированными бензольными циклами Ш
С увеличением числа ароматических колец в многоядерных соединениях растет число монозамешенных изомеров: так для нафталина возможны два продукта (а. Р). для антрацена — три. для фенантрена — 5.
Цифрами обозначены положения, в которых могут находиться за местители.
Получение
Получают фенантрен в основном из каменноугольной смолы.
Физические свойства фенантрена
Фенантрен — это твердое кристаллическое вещество с гп1 101 °C, г 340,2 °C. Растворяется в эфире, бензоле трудно — в спирте. Растворы имеют голубую флуоресценцию.
Химические свойства
Фенантрен, как и антрацен, обладает более слабым ароматическим характером, чем нафталин, и тем более, чем бензол. Электронная плотность в его молекуле распределяется неравномерно, ароматичность среднего бензольного кольца здесь настолько нарушена, что связь между 9 и 10 атомами углерода приобретает характер двойной связи. Реакции замещения протекают по механизму присоединения-отщепления.
Так, вначале присоединяется бром в 9 и 10 положениях с последующим отщеплением НВги образованием 9-бромфенантрена:
9,10-дибром-9.10-дигид ро-фенантрен
>t
-нв7
9-бромфенантрен
9,10-Дибром-9,10-дигидрофенантрен может быть выделен в свободном состоянии.
128
Лекции по органической химии
При окислении фенантрена образуется фенанренхинон. При дальнейшем окислении идет разрыв связи С9—С|0 и образуется дифеновая кислота:
[о]
При восстановлении фенантрена образуется продукт, который носит название пергидрофенантрен.
Конденсированная система пергидрофенантрена и циклопентана называется циклопентанпергидрофенантрен, или стеран.
стеран.
циклопентан перпирофенантрен
Данная структура лежит в основе стероидов.
13. МНОГОЯДЕРНЫЕ АРЕНЫ С ИЗОЛИРОВАННЫМИ БЕНЗОЛЬНЫМИ ЦИКЛАМИ
Наиболее важными представителями этой группы соединений являются бифенил, дифенилметан и трифенилметан.
трифенилметан
БИФЕНИЛ
Молекула бифенила содержит два бензольных кольца, соединенных <7-связью. Нумерацию каждого бензольного ядра проводят отдельно.
3 2 2' з'
Способы получения
1. Бифенил выделяют из каменноугольной смолы, где он содержится в небольшом количестве.
2. Дегидрогенизация бензола. Пары бензола пропускают через раскаленные трубки. Выход бифенила достигает 70 %.
3. Реакция Ульмана. Нагревание йодбензола в присутствии порошка меди:
Си 200-250 "С • + 2Си1
130
Лекции по органической химии
Физические свойства
Бифенил — твердое вещество с гпд 70 °C, легко растворим в спирте, эфире.
Строение. Химические свойства
Два бензольных кольца в молекуле бифенила соединены между собой о-связью, длина которой (0,148 нм) несколько меньше длины углерод-углеродной связи в алканах (0,154 нм).
Вокруг о-связи в молекуле бифенила возможно свободное вращение, при этом барьер вращения невелик. Но если вор.до-положениях обоих бензольных колец содержатся объемные заместители, то из-за пространственных препятствий вращение вокруг о-связи, соединяющей два цикла, сильно затрудняется или становится невозможным. Бензольные кольца располагаются под углом.
Если в o/wio-положениях каждого бензольного ядра молекулы бифенила находятся разные заместители, то такие соединения проявляют оптическую активность. Обусловлено это тем. что эти соединения хотя и не содержат асимметрического атома углерода (центра хиральности), не имеют также и плоскости симметрии. Их молекула асимметрична в целом.
Так, 6,6 -динитродифеновая кислота существует в виде двух энантиомеров (зеркальных изомеров):
СООН
Вид пространственной изомерии, обусловленный ограничением свободного вращения вокруг простой связи, называется атропизоме-рией (от греч. атроло — нет поворота)
Для проявления атропизомерии необязательно наличие четырех заместителей в ор/ло-положениях. иногда достаточно трех или даже двух объемных заместителей.
Химические свойства
Химические свойства бифенила аналогичны свойствам моноядер-ных аренов. Для него характерны реакции электрофильного замещения (нитрование, галогенирование, сульфирование и т. д.). В эти реакции бифенил вступает легче, чем бензол, при этом образуются преимущественно продукты пара- и opwo-замешения. Связано это с тем,
13. Многоядерные арены с изолированными бензольными циклами
что фенильные группы оказывают по отношению друг к другу слабые электронодонорные свойства, т. е. одно из колец при этом выполняет роль заместителя I рода.
4-нитробифенил
4,4' -д и н итробифен ил
2,4'-динитробифенил
Нитрогруппа в молекуле 4-нитробифенила уменьшает электронную плотность, прежде всего в бензольном кольце, в котором она находится, поэтому вторая нитрогруппа вступает в незамещенное ядро.
ДИФЕНИЛМЕТАН
В молекуле дифенилметана два бензольных цикла связаны через метиленовую группу. Нумерацию углеродных атомов в каждом цикле проводят отдельно:
Способы получения
Дифенилметан получают по реакции Фриделя — Крафтса алкилированием бензола бензилхлоридом или дихлорметаном:
бензил хлорид
132
Лекции по органической хи мии
Физические свойства
Дифенилметан — твердое кристаллическое вещество с г 26— 27 °C и запахом герани, он легко растворим в спирте, диэтиловом эфире, хлороформе.
Химические свойства
Для дифенилметана характерны реакции с участием бензольных циклов и активной метиленовой группы.
Как ароматический углеводород дифенилметан вступает в реакции электрофильного замещения, образуя 4,4 -дизамешенные и 2,4,2 ,4 -тетразамещенные продукты:
4,4'-динитродифенилметан
2,4,2’,4'-тетранитродифенилметан
Атомы водорода метиленовой группы в молекуле дифенилметана подвижны вследствие электроноакцепторного действия фенильных групп. В результате дифенилметан легко окисляется:
Бензгидрол является исходным продуктом при получении димедрола— (C6H5)2CHOCH2CH2N(CH3)2 HC1 — антигистаминного средства.
Водородные атомы метиленовой группы также легко замещаются
на галоген:
13. Мныояоерныеаренъ’ с изояироеанньми йен юльными циклами
133
+ НВг
дифенилбромметан
Дифенилметан используется в парфюмерной промышленности для отдушки мыл; некоторые его производные являются пестицидами.
ТРИФЕНИЛМЕТАН
В молекуле трифенилметана три бензольных цикла связаны через мет лновую группу (СН):
Способы получения
Трифенилметан получают по реакции Фриделя—Крафтса из хлороформа и бензола:
Физические свойства
Трифенилметан — кристаллическое вещес!во с 76 °C, трудно растворимое в воде, легко — в органических растворителях.
Химические свойства
По химическим свойствам трифенилметан во многом напоминает дифенилметан. Для него также характерны реакции электрофильного замещения с участием бензольных циклов и реакции с участием атома водорода мети новой группы.
Фенильные радикалы проявляя -/-эффект смещают на себя электронную плотность с атома углерода СН-группы. вследствие чего атом водорода этой группы приобретает подвижность и легко замещается на металлы и галогены. За счет метиновой группы трифенилметан легко окисляется.
Так, при действии амида натрия в жидком аммиаке трифенилметан образует трифенилметилнатрий:
134
Лекции пс орг '.ическои хц мии
Трифенилметилнатрий окрашен в желтый цвет, он имеет ионное строение, его эфирные растворы проводят электрический ток.
Стабильность трифенилметил-аниона и наличие окраски объясняется тем, что в делокализации отрицательного заряда принимают участие все три бензольных ядра:
трифенилметил-анион
При взаимодействии трифенилметана с хлором атом водорода СН-группы замешается на хлор, продуктом реакции является трифе-нилхлорметан. Атом хлора в его молекуле очень подвижен. При растворении трифенилхлорметана в жидком SO2 (растворитель с высокой диэлектрической постоянной) в присутствии А1С13 происходит его ионизация с образованием трифенилметил-катиона:
жидкий SO,, AIC1,
трифенилкарбкатион
При окислении трифенилметана образуется трифечил карбинол который подвергается ионизации в концентрированной серной кислоте:
13. Многоядерные арены с изолированными бензильными циклами
135
трифенилкарбинол
H2SO4
Устойчивость трифенилметил-катиона также обусловлена участием бензольных колец в делокализации положительного заряда за счет сопряжения:
трифенилметил-катион
При обработке трифенилхлорметана цинком в бензольном растворе образуется свободный трифенилметильный радикал, находящийся в равновесии со своим димером:
производное циклогексадиена
136
Лекции по органической химии
Трифенилметильный радикал — это первый радикал, полученный в свободном виде. Он был получен в 1900 г. американским химиком М. Гомбергом. Столь высокая устойчивость трифенил метила по сравнению с алкильными радикалами объясняется значительной делокализацией неспаренного электрона по всем бензольным кольцам.
Свободный радикал трифенилметила окрашен в желтый цвет.
Красители трифенилметанового ряда
К красителям трифенилметанового ряда относятся бриллиантовый зеленый и фенолфталеин.
Бриллиантовый зеленый — представительаминопроизводныхтри-фенилметана. Его получают конденсацией бензальдегида с N,N -ди-этиланилином в присутствии кислотного катализатора (хлороводородной кислоты) согласно схеме:
N(C2H5)2
4,4'-бис-(диэтила.мино)трифгнилмета:< лейкооснование бриллиантинового зеленого (бесцветное вещество)
13. Многоядерные арены с изолированными бензольными циклами
137
4.4'-бис-(диэтиламино)трифенилкарбинол карбонильное основание бриллиантинового зеленого (бесцветное вещество)
бриллиантиновый зеленый
Бриллиантиновый зеленый применяется в медицине в виде I—2 % водных или спиртовых растворов как антисептическое средство, а также при изготовлении бактерицидных пластырей.
_ Фенолфталеин является представителем гидроксипроизводныхтрифенилметана. Получают фенолфталеи н конденсацией фенола с фталевым ангидридом в присутствии концентрированной кислоты:
138
Лекции по органической химии
Фенолфталеин представляет собой белое кристаллическое вещество с Гп1 259—263 °C, практически нерастворимое в воде, хорошо растворимое в этаноле. Фенолфталеин применяется в аналитической практике как кислотно-основный индикатор. В кислой и нейтральной среде он находится в бесцветной лактонной форме, в щелочной среде (при pH 8,2—10,0) приобретает малиновую окраску вследствие образования хиноидной структуры.
фенолфталеин фенолфталеин
(бесцветная форма) (форма с малиново-красной окраской)
pH 8,2-10.0
бесцветная форма pH > 12
Фенолфталеин применяется в медицине как слабительное средство при хронических запорах.
14. ГАЛОГЕНОПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ
К галогенопроизводным углеводородов относят соединения, у которых один или несколько атомов водорода замешены на атомы галогенов.
Галогенопроизводные углеводородов классифицируют в зависимости от природы углеводородного радикала (алифатические, алициклические и ароматические), числа атомов галогена в молекуле (моно-, ди-, три- и полигалогенопроизводные), характера галогена (фтор-, хлор-, бром-, йодпроизводные). характера атома углерода, с которым связаны атомы галогена (первичные, вторичные и третичные галогенопроизводные).
Номенклатура
По заместительной номенклатуре ИЮПАК названия галогенопроизводных углеводородов составляют аналогично названиям соответствующих углеводородов. Вначале цифрой указывают положение замещения (если это необходимо) затем называют галоген (если нужно — перед ним количество атомов) и прибавляют название родоначальной структуры (в алифатических галогенопроизводных это главная углеродная цепь, в алициклических и ароматических — цикл).
Нумерацию начинают с ближнего к галогену конца углеродной цепи.
Н3С—СН—СН2 Вг
1 -бромпропан
хлорбензол
Простейшие галогенопроизводные называют и по радикало-функциональной номенклатуре. Тогда названия составляют из названия углеводородного радикала, связанного с галогеном, и суффикса -фторид, -хлорид, -бромид или -йодид или прибавлением слов: фтористый, хлористый, бромистый, йодистый.
qH—С1
Н2С=СН—СН—Вг
сн,—СН—С1 о з 2
этилхлорид, хлористый ЭТИЛ
алл и л бром ид, бромистый аллил бензилхлорид, хлористый бензил
140
Лекции по орган! че< кой химии
За некоторыми галогенопроизводными сохранились тривиальные названия:
СНС13 хлороформ
HC=C—СН=СН, хлоропрен 2 I 2
С1
ГАЛОГЕНАЛКАНЫ
Общая формула: СпН2(|+)На1.
Способы получения
Существует много разнообразных методов введения галогена в молекулу соединения. Рассмотрим наиболее общие.
1. Галогенирование алканов. Взаимодействие алканов с галогенами протекает по свободнорадикальному механизму SR при УФ-облучении.
С12,/>у С12,Ау СЬ.Ау
СН4 -НС1 НзС с -НС1 СН2С12 НС1 СНС13 НС1 СС14
метан хлорметан дихлорметан трихлорметан тетрахлорметан
Недостатком этого способа является образование смеси моно-, ди-и полигалогеналканов.
2. Присоединение галогеноводородов к алкенам. Реакция идет обычно на холоду, способ особенно удобен для получения моногалогенопроизводных, так как реакция не сопровождается образованием побочных продуктов полигалогенопроизводных.
Н,С-НС=СН, + НВг ----------- НС—СН—СН,
3 I 3 I J
пропен gr
2-бромпропан
Механизм реакции АЕ — электрофильное присоединение.
3. Взаимодействие спиртов с галогенирующими реагентами. Часто галогенопроизводные получают замещением гидроксильной группы спиртов на галоген. Для этого используют галогеноводороды (НО, НВг, HI), галогениды фосфора (РС13, РС15, РВг3, РВг5), тионилхлорид (SOC12).
Взаимодействие спиртов с галогеноводородами протекает по схеме:
С2Н5ОН + НС1 С2Н5С1 + Н2О
хлорэтан
Легче всего замещается на галоген гидроксильная группа у третичного атома углерода. Проводить реакцию с первичными спиртами не
14. Галогенопроизводн ые углеводороде»
141
обходимо в присутствии катализаторов: минеральных кислот, галогенидов пинка и т. д.
С более высокими выходами галогеналканы образу ются при взаимодействии спиртов с галогенидами фосфора (111) ити фосфора (V), а также тионилхлоридом:
ЗС2Н5ОН + РС13 -----»ЗС2Н5С1 + Н3РО3
С2Н5ОН +РС15
С2Н5С1 + РОС13 + НС1
C2H5OH + SOC12
С2Н5С1 +SO2+Het
4. Взаимодействие галогеналканов с солями галогеноводородных кислот (реакция Финкельштейна). Реакцию используют для получения фтор- или йодалканов из более доступных хлор- или бромпроизвол-ных:
C2H5Br + Nal _ацетон^ C2H5I + NaBr
бромэтан йодэтан
Физические свойства
Низшие галогеналканы — газы или жидкости со своеобразным сладковатым запахом, средние — жидкости, высшие — твердые вещества. Большинство из них практически нерастворимы в воде, нелегко растворяются в органических растворителях.
Химические свойства
Галогеналканы обладают высокой реакционной способностью и для них характерны реакции нуклеофильного замещения (SN), элиминирования (отщепления) (Е). Они также вступают в реакции восстановления и взаимодействуют с некоторыми металлами.
Реакции нуклеофильного замещения
Способность галогеналканов вступать в реакции SN обусловлено полярностью связи углерод-галоген. Атом галогена, имея большую электроотрицательность, чем атом углерода, смешает на себя электронную плотность связи С-Hal. В результате атом галогена приобретает частичный отрицательный заряд (5—), а атом углерода — частичный положительный заряд (5+). Галогеналканы вступают в реакции с нуклеофильными реагентами, и при этом происходит замещение галогена на нуклеофил.
142
Лекции по органической химии
элек рофильный центр
— С-Hal + Nu
—C-Nu
+ Hal
галогеналкан нуклеофил
продукт уходящая
замещения группа
В зависимости от строения галогеналкана, природы нуклеофила и растворителя реакции S4 протекают по двум основным направлениям: SN1 и Sn2.
Механизм SN2 (бимолекулярное нуклеофильное замещение)
По механизму SN2 реагируют первичные и несколько труднее вторичные галогеналканы. Реакция протекает в одну стадию через образование переходного состояния. Вначале нуклеофил атакует атом углерода, связанный с галогеном (электрофильный центр), со стороны, противоположной связи С-Hal , т. е. атака идет с тыла. В результате происходит постепенное вытеснение нуклеофилом галогенид-иона (уходящей группы). Этот процесс включает переходное состояние, т. е. момент, когда связь С-Hal еще не разорвалась, а связь C-Nu еще не полностью образовалась.
Z_ \Ч 5+ Nu +—;С-Hal
Nu—С— + Hal
переходное состояние
Образование переходного состояния сопровождается изменением гибридного состояния атома углерода с sp3 Hasp1. Одна доля негиб-ридизованной р-атомной орбитали атома углерода в переходном состоянии частично перекрывается с орбиталью атакующего нуклеофила, а вторая — с орбиталью атома галогена.
Возвращение атома углерода в ^-гибридное состояние после отщепления галогенид-иона происходите обращением конфигурации.
14. Гамгенопроимодные углеводородов
143
Протеканию реакции по механизму Ss2 способствуют активные нуклеофильные реагенты — они легче образуют переходное состояние — и апротонные растворители, поскольку протонные полярные растворители сольватируют нуклеофил, тем самым снижая его реакционную способность.
По предложению английского химика К. Ингольда описанный механизм получил обозначение SN2. Буква S указывает на замещение, N — на нуклеофильный тип реакции, а цифра 2 обозначает, что реакция является бимолекулярной, т. е. в стадии, определяющей скорость реакции в целом (в данном случае образование переходного состояния), участвует два реагента (галогеналкан и нуклеофил). Скорость реакций, протекающих по механизму S42. зависит от концентрации обоих реагентов.
Механизм SN1 (мономолекулярное нуклеофильное замещение)
По этому механизму происходит нуклеофильное замещение в третичных и, в определенных условиях, во вторичных галогеналканах. В молекуле третичных галогеналканов объемные заместители при атоме углерода, связанном с галогеном, создают пространственные препятствия для подхода нуклеофила к электрофильному центру, и его атака с тыла становится невозможной. Вместе с тем третичные галогеналканы способны в сильнополярных средах к ионизации. По механизму SN1 реакция протекает в две стадии:
Стадия 1
I
—С—Hal
I
медленно
+ Hal
карбкатион
Стадия 11
быстро
Nu ---------
—C-Nu
На первой стадии происходит диссоциация молекулы галогенал-кана при участии молекул протонного полярного растворителя. В результате образуются карбкатион и галогенид-ион. Поскольку процесс ионизации протекает медленно, то 1 стадия определеят скорость всей реакции. На второй стадии образовавшийся карбкатион быстро реагирует с нуклеофилом.
Протеканию реакции по механизму SN1 способствуют высокая ионизирующая и сольватирующая способность растворителя, а также стабильность образующегося карбкатиона. Устойчивость алкильных карбкатионов обусловлена делокализацией положительного заряда за счет +/-эффекта алкильных групп и возрастает в ряду:
144
Лекции по органической химии
сн, сн,
+ + . чх+ ч.+
СН3 < СНу*-СН2 < ^сн < сн3
сн3 сн3
Поэтому третичные галогенопроизводные легче всего подвергаются ионизации.
Механизм нуклеофильного замещения, протекающий по рассмотренной схеме, называется мономолекулярным, т. к. на стадии, определяющей скорость всего процесса (стадия 1), принимает участие молекула только одного реагента — галогеналкана. Такой механизм обозначают SN1.
Таким образом, на основании вышеизложенного можно сделать вывод, что первичные галогеналканы обычно реагируют по механизму Sn2, третичные — по механизму S^l. Вторичные галогеналканы, в зависимости от природы нуклеофила и растворителя, могут реагировать как по механизму SN2, так и по механизму SN1.
1. Гидролизгалогеналканов. Галогеналканы гидролизуются до спиртов. Реакцию обычно проводят в присутствии водных растворов щелочей, т. к. с водой она протекает медленно.
Н,0
Н3С-С1 + NaOH -----------*- Н3С-ОН + NaCl
2. Реакция Вильямсона. Эта реакция является одним из лучших способов получения простых эфиров. Она заключается во взаимодействии галогеналканов с алкоголятами или фенолятами.
Н3С-Вг + NaOC2H5-----*- Н3С-О~С2Н5 + NaBr
Н3С-Вг + NaO
метилэтиловый эфир
метил фениловый эфир
+ NaBr
3. Взаимодействие ссолями карбоновых кислот (ацетолиз). При действии солей карбоновых кислот на галогеналканы образуются сложные эфиры. Реакцию проводят в среде апротонного полярного растворителя.
14 Галогенопроизводные углеводородов
145
СН,СН — Вг + NaO-C-CH3 ---* СНДСН,О-С~СН, + NaBr
11 J 3 2 п 3
о о
этил ацетат
4. Взаимодействие с аммиаком, алкил- и ариламинами (аммонолиз и аминолиз). При взаимодействии с аммиаком и аминами галогеналка-ны алкилируют их с образованием смеси первичных, вторичных и третичных аминов, а также солей — четвертичных аммониевых оснований. Например, первичный амин образуется по схеме:
I- -i+_ NH3
н3с—i + nh3-----*- [ch3nhJ I------► h3c-nh2 + NH4I
5. Взаимодействие с солями циановодородной кислоты. Первичные и вторичные галогеналканы с цианидом калия или натрия в среде апротонного полярного растворителя образуют нитрилы (SN2):
СН3СН—Вг + NaCN ---------*- СН3СН—C=N + NaBr
пропанонитрил
6. Взаимодействие с солями азотистой кислоты. Продукты, образующиеся в результате этой реакции, зависят от условий ее проведения. строения галогеналкана и соли.
Н5С—Вг +NaNO2 -----------*- HSC—NO2 + NaBr
нитроэтан
НА Н3С
СН-Вг + AgNO2-----------► СН-О-N=O + AgBr
Н3С н3с
изопропил-
нитрит
Реакции элиминирования
Реакции элиминирования сопровождаются отщеплением галоге-новодорода от галогеналкана и приводят к образованию алкенов.
nV I ____________\ /
/С-с— -----------* /С=С\ + Н + На1
Hal
Поскольку отщепление водорода происходит от Р-атома углерода, то такие реакции называются Р- или 1,2-элиминированием.
146
Лекции no органической химии
Реакции элиминирования (Е) и нуклеофильного замещения (SN) конкурируют друг с другом, но в определенных условиях каждая из ни> может стать доминирующей. Отщепление галогеноводорода от галогеналкана становится основным процессом в присутствии нуклеофильных реагентов, обладающих высокой основностью. К ним относятся спиртовые растворы гидроксидов щелочных металлов или алкоголяты щелочных металлов. Элиминированию способствуют также повышение температуры реакционной смеси и концентрации реагентов. Так, при взаимодействии йодэтана с водным раствором щелочи основным направлением реакции является нуклеофильное замещение, а продуктом реакции — этиловый спирт. При использовании спиртового раствора щелочи доминирующим процессом становится реакция элиминирования, продуктом реакции — этилен.
Н3С-СН2-1
NaOH (Н2О) нуклеофильное замещение
NaOH (С2Н5ОН)~ Р-элиминирование
Н3С—СН-ОН + Nal этанол
Н2С=СН2 + Nal + Н2О этен
Реакции элиминирования галогеналканов могут протекать по мо-номолекулярному (Е1) и бимолекулярному (Е2) механизмам.
Механизм Е2 (бимолекулярное элиминирование).
В реакции отщепления по механизму Е2 наиболее легко вступают первичные галогеналканы.
&+I pl а
В + Н-С-С-На1
переходное состояние
--------- в-н + /С=С + На1
Реакция отщепления, протекающая по бимолекулярному механизму, требует присутствия основания, идет в одну стадию с образованием переходного состояния, в формировании которого принимают участие молекулы двух реагентов. И поэтому скорость такой реакции зависит от концентрации обоих реагентов. Процессы разрыва и образования связей в переходном состоянии происходят синхронно.
В отличие от механизма SN2 в механизме Е2 частица с неподелен-ной парой электронов или несущая отрицательный заряд действует не как нуклеофил, а как основание, атакуя атом водорода при 0-углерод-
ном атоме.
14 Галогенопрои вводные угле- ооорооов
147
Механизм Е1 (л.ономолекулярное элиминирование).
Наиболеелегко происходит элиминирование по данному механизму у третичных галогеналканов. Реакция не требует основания как реагента, но для ее протекания необходим ионизирующий растворитель. Процесс является двустадийным.
медленно
Стадия I -----С —С-Hal « *
I
Н
галогеналкан
Стадия II
быстро
карбкатион
— с-с+ + Hal Н ।
карбкатион
алкен
Стадией, определяющей скорость реакции, является образование карбкатиона. Вторая стадия включает в себя стабилизацию карбкатиона путем отщепления протона.
Если в молекуле галогеналкана имеется несколько альтернативных путей отщепления галогеноводорода, то реализуется тот из них, при котором двойная связь образуется у наиболее замещенного атома углерода, то есть вместе с галогеном уходит водород от наименее гидрогенизированного соседнего атома углерода.
Эта закономерность получила название правила Зайцева:
СН, н3с-сн-сн-сн3 Вг 2-бром-З-метилбутан
СНз
—*н3с-с=сн-сн3 2-метилбутен-2 (основной NaOH(C,HsOH) продукт
-NaBr;—Н2О реакиии)
СН,
—- н3с-сн-сн=сн.
3-метилбутен-1 (побочный продукт реакции)
Взаимодействие с металлами
При взаимодействии галогеналканов с такими металлами, как магний, образуются магнийорганические соединения — реактивы Гриньяра:
148
Лекции по органической химии
эфир
C2H5Br + Mg ---------- C2H5MgBr
этилмагний-бромид
Магнийорганические соединения являются сильными нуклеофильными реагентами и сильными основаниями. Их активность обусловлена полярностью связи углерод-магний:
6— 8+
H3c-CH2—MgBr
Восстановление галогеналканов
При каталитическом гидрировании галогеналканов или действии на них водорода в момент выделения, а также йодоводородной кислотой происходит замещение атома галогена водородом:
Н5с—С1
н2
Kt
С2Н6 + НС1
Н5с—С1 + HI ——* С2н6 + НС1 + 12
ДИГАЛОГЕНАЛКАНЫ
Дигалогеналканы — это углеводороды, содержащие в своем составе два атома галогена. В зависимости от их положения дигалогеналканы делятся на 3 группы:
1) геминальные дигалогеналканы, атомы галогена находятся у одного и того же атома углерода:
С1
Н3С-СН
С1
1,1-дихлорэтан, хлористый этилиден
Нередко в этом случае применяют приставку -гем
2) вицинальные дигалогеналканы. атомы галогена находятся у соседних атомов углерода:
н2с—СН—СН3
С1 С1
1,2-дихлорпропан
14. Галогегопроизвоёвые углеводородов
3) атомы галогена разделены несколькими углерод-углеродными связями:
Н,С—сн,—сн—СН,
2| 2 2 I 2
Cl Cl
1,4-ди**юрбутан.
хлористый тетр;1 стилен
Способы получения
Геминальные галогенопроизводные получают при взаимодействии альдегидов и кетонов с пентагалогенилами фосфора (PCL, РВг5):
,0
Н,С-С + PCI, ------------- H,C-CHCI, + POU3
1,1- дихлорэтан уксусный альдегид
н,с— С—сн, ,,
3 II 3 + РС15-----* Н3С-С-СН3 + Р0С13
0 С1 С1
ацетон 2,2-дихлорпропан
Путем присоединения двух молекул галогеноводородов к алкинам:
НС1 НС1
нс=сн ---------* Н2С=СН-С1 --------*- Н3С-СНС12
Вицинальные дигалогеналканы образуются в результате присоединения галогенов к алкенам:
Н3С—НС=СН2 + Вг2 -------------- н3с—сн—сн2
Вг Вг
1,2-дибромпропан
Дигалогенопроизводные с атомами галогенов у различных углеродных атомов получают при действии галогеноводородов, галогенидов фосфора, тионилхлорида на гликоли или другие многоатомные спирты:
н2с-сн— сн2 + РВг3--------► н,с-сн2-сн2 + Н3РО3
I I I J J
ОН ОН Вг Вг
пропандиол-1,3
1,3-дибромпропан
150
Лекции по органической химии
Физические и химические свойства
Дигалогеналканы — тяжелые маслянистые жидкости или кристаллические соединения, нерастворимые в воде. С введением второго атома галогена в молекулу возрастают температуры кипения (кроме фтор-производных). Дигалогеналканы являются хорошими растворителями.
Химические свойства дигалогеналканов во многом сходны со свойствами моногалогеналканов. Но есть и различия. При нагревании геминальных и вицинальных дигалогеналканов со спиртовым раствором щелочи образуются алкины:
Н3С—СН2-СНС12 + NaOH C2Hs°H > Н3С—С=СН + NaCl+ Н2О
1,1 -дихлорпропан пропин
С н он
Нзс-сн-СН2 + NaOH 2 5 > Н3С-С=СН+ NaCl + Н2О
| | пропин
С1 С1
1,2-дихлорпропан
Щелочной гидролиз вицинальных дигалогеналканов ведет к образованию гликолей, а геминальных — альдегидов и кетонов:
С1 ОН о
/ NaOH ' „ *
н3с-сн -- ,нп. * н3с-сн ----------------- н3с-с
3 \ (Н2О) J X и о J х
Cl он 2 н
гаи-диол уксусный альдегид
н3с-сн-сн
Cl C1
NaOH (Н2О)
1,2-дихлорпропан
H3C-CH-CH2+NaCl+ Н2О
ОН ОН
пропиленгликоль
ГАЛОГЕНАЛКЕНЫ
Галогеналкенами называют производные алкенов, в которых один или несколько атомов водорода замещены атомами галогенов.
По взаимному расположению двойной связи и атома галогена га-логеналкены можно условно разделить на 3 группы:
1) винилгалогениды — соединения, содержащие атом галогена у углерода с двойной связью:
Н2С=СН-С1
хлористый винил
Н3С-НС=СН-Вг
1-бромпропен-1
С1 I н;с=с-сн3 2-хлорпропен
14 Галогенопро’/зводнне углеводородов
151
2) аллилгалогениды — соединения, в которых атом галогена находится в a-положении к углероду, образующему двойную связь:
а а
н2с=сн-сн-а Н.С—НС—НС=СН2
хлористый аллил С1
3-хлорбутен-1
3) соединения, в которых атом галогена и атом углерода, образующий двойную связь, разделены двумя и более простыми углерод-углеродными связями:
н2с=сн—сн—сн-ci н3с—нс=сн—сн—сн—С1
4-хлорбутен-1 5-хлорпентен-2
Способы получения
Галогеналкены получают:
— гидрогалогенированием алкинов:
НС1
НС=СН --------- Н2С=СН-С1
винилхлорид
— галогенированием алкенов в аллильное положение:
500 °C
Н2С=СН-СН3+С12 --------- Н2С=СН-СН—С1 + НС1
аллилхлорид
— действием галогенирующих реагентов на непредельные спирты:
н2с=сн-сн—ОН + SOC12—* Н2С=СН-СН—Cl + so2+ НС1
аллилхлорид
Физические и химические свойства
Физические свойства в гомологическом ряду галогеналкенов подчиняются обычным закономерностям.
Реакционная способность галогеналкенов зависит от взаимного расположения двойной связи и атома галогена в молекуле. Так, ви-нилгалогениды характеризуются низкой реакционной способностью связи С-Hal и двойной связи. Причиной малой реакционной способности связи С-Hal является сопряжение неподеленной пары электронов атома галогена с л-электронами двойной связи.
Н2С^СН-^ Hal
-I, +М
152
Лекции по органической химии
В результате сопряжения (+М-эффект) связь С-Hal укорачивается и становится значительно прочнее, чем в галогеналканах. Реакции нуклеофильного замещения для винилгалогенидов в большинстве случаев осуществить не удается.
Винилгалогениды под действием концентрированных растворов щелочей превращаются в алкины; присоединяют галогены, галогено-водороды (присоединение идет по правилу Марковникова); в присутствии катализатора они легко полимеризуются:
NaOH
НС^СН + NaCl + Н2О
ацетилен
Н2С=СН-С1
НС1
Н3С-СНС12
1,1-дихлорэтан
Kt
4н2с-нсХ
С1« пол ив ин илхлорил
В молекулах аллилгалогенидов атом галогена обладает повышенной подвижностью. Аллил галогениды вступают в реакции нуклеофильного замещения легче, чем галогеналканы.
Замещение, как правило, происходит по механизму S41, что объясняется склонностью аллилгалогенидов к ионизации. При этом образуется аллильный катион, повышенная стабильность которого обусловлена делокализацией положительного заряда по сопряженной системе.
-Hal +
Н2С=СН—СН—Hal ^==ГН2С=СН-СН2-—* СН2~НС=СН2
аллильный катион
Присоединение галогеноводородов в аллилгалогенидах также протекает по правилу Марковникова:
Н2С=СН—*'СН2—* С1 - -1 - н3с-нс—сн2
хлористый аллил
С1 С1
1,2-дихлорпропан
В соединениях, где атом галогена и двойная связь разделены двумя или большим числом простых С—С-связей, каждая из этих групп ведет себя независимо друг от друга.
14 Галогенопроизвсюные углевооиродчи
АРОМАТИЧЕСКИЕ ГАЛОГЕНОПРОИЗВОДНЫЕ
153
Ароматическими галогенопроизводными называют производные ароматических угтеводородов. у которых один или несколько атомов водорода замещены атомами галогенов.
В зависимости от положения атомов галогенов различают две группы галогенопроизводных ароматического ряда:
• галогенарены — соединения, у которых атомы галогена непосредственно связаны с бензольным кольцом:
о-хлортолуол
бромбензол
• арилалкилгалогениды — соединения, содержащие атомы галогена в боковой цепи (алкильном радикале):
Способы получения
Для введения галогена в бензольное ядро используют:
1. Реакцию галогенирования аренов. Реакция протекает по ионному механизму в присутствии катализаторов хлоридов металлов А1С13, FeBr3, SbCl3:
Cl
хлорбензол
Катализатор способствует образованию положительного иона галогена, который осуществляет замещение в бензольном ядре.
2. Реакцию солей арилдиазония с галогенидами меди:
Си2С12
хлорбензол
N2f
154
Лекции по органической химии
Для введения галогена в боковую углеродную пень используют:
1. Галогенирование алкиларенов. Реакция идет при нагревании до высоких температур или при облучении УФ-светом. Механизм радикальный:
бензилхлорид
Галогенированию подвергается в основном a-положение относительно бензольного ядра, поскольку в этом случае образуется устойчивый свободный радикал бензильного типа.
С12 —------* 2СГ
С6Н5СН3 + С1*----------- С6Н5СН2 HU
С6Н5СН2 + С12 ---------С6Н5СН2С1 + СГ
2. Реакцию хлорметилирования. При действии на ароматические углеводороды формальдегида и хлороводорода в присутствии катализатора (А1С13, ZnCl2) атом водорода в кольце замещается на хлорметильную группу:
СН2С1
(1^1 11 z + .„CL + НС1 »С12 , —- + Н,0
н н Механизм этой реакции: О II . t + НС1 н н Г J + СН2ОН —7* ( „^\_CH2C1 - СН2ОН + С1 гидроксиметил-катион . ,сн,он * Н2О; ZnCl3 J —Zn(OH)Cl
14. Галогенопроизво^ные углевосюрооов
155
Физические свойства
Галогенопроизводные ароматического ряда — это жидкие или кристаллические вещества. Температуры кипения галогенаренов возрастают в ряду: фтор- , хлор-, бром-, йодпроизводные. Соединения этого ряда не растворимы в воде, но легко растворимы в органических растворителях.
Химические свойства
Реакционная способность ароматических галогенопроизводных зависит от положения галогенов. Для соединений, содержащих галоген в боковой цепи, характерны все реакции галогеналканов.
Соединения, в которых галоген непосредственно связан с бензольным ядром, характеризуются низкой реакционной способностью связи С-Hal. Это обусловлено сопряжением неподеленной пары электронов атома галогена с л-электронной системой бензольного ядра:
Галогенарены похожи в этом отношении на винилгалогениды. В результате сопряжения происходит укорочение и уменьшение полярности связи С-Hal. Поэтому реакции нуклеофильного замещения идут лишь в очень жестких условиях:
NaOH
300 "С
NH3
200 °C
анилин
Подвижность атома галогена возрастает с введением в орто- или дара-положения по отношению к галогену электроноакцепторного заместителя в бензольное ядро, который увеличивает дробный положительный заряд на атоме углерода связи С-Hal. Реакция замещения галогена протекает в более мягких условиях и идет по механизму SN2:
156
Лекции по органической химии
Образование о-комплекса является лимитирующей стадией реакции.
По ароматическому ядру галогенарены вступают в обычные реакции электрофильного замещения. Атом галогена, связанный с бензольным ядром, проявляет — I и +М-эффекты.
Поскольку в статическом состоянии —/-эффект > +М-эффекта, то атом галогена проявляет элктроноакцепторные свойства и снижает реакционную способность бензольного ядра в реакциях SE . В динамическом состоянии +Л/-эффект > -/-эффекта, поэтому атомы галогена выступают как ориентанты I рода, направляя заместители в орто- и /шро-положения. Например:
В результате реакции образуются два изомера: о-нитрохлорбензол и и-нитрохлорбензол.
15. НИТРОСОЕДИНЕНИЯ
Нитросоединениями называют производные углеводородов, содержащие в своем составе одну или несколько нитрогрупп — NO,.
Нитрогруппа имеет плоское строение, атомы азота и кислорода находятся в состоянии^ -гибридизации. Электронное строение нитрогруппы можно представить с помощью граничных (резонансных) структур:
Олин из атомов кислорода соединен с атомом азота двойной связью, а другой — семиполярной связью (от англ, semi — наполовину), т. е. связь нитрогруппы является полуполярной.
В действительности оба атома кислорода в нитрогруппе равноценны, поэтому ее строение можно представить следующим образом:
zp-'A
R-NA ю - 'А
Вещества с семиполярными связями имеют большой дипольный момент, они обычно кипят при более высоких температурах, чем изомерные им вещества без семиполярных связей.
Классификация и номенклатура нитросоединений
В зависимости от природы углеводородного радикала, с которым связана нитрогруппа, различают:
Алифатические нитросоединения
• насыщенные (нитроалканы)
СН3—NO2 нитрометан
СН3—СН2—NO2 нитроэтан
• ненасыщенные (нитроалкены, нитроалкины)
н3с—сн=с—СН
no2 нс=с—сн—сн—сн.
2-нитробутен-2
NO2
3-нитропентин-1
158
Лекции по оргсчической химии
Ароматические нитросоединения
Ароматические нитросоединения делятся на две группы: соединения, содержащие нитрогруппу, связанную с атомом углерода ароматического ядра, и соединения, содержащие нитрогруппу в боковой цепи:
нитробензол фенилнитрометан
В зависимости оттого, у какого (первичного, вторичного, третичного) атома углерода находится нитрогруппа, нитросоелинения бывают первичные, вторичные или третичные.
сн—сн—сн3
СН,— СНт-NO, I
322 NO,
нитроэтан, 2
первичное нитросоединение 2-нитропропан,
СН вторичное нитросоединение
I сн3—с—сн3 no2 2-метил-2-нитропропан, третичное нитросоединение
Названия нитросоединений образуют путем добавления префикса нитро- к названию соответствующего углеводорода с указанием по-
ложения нитрогруппы:
сн,—сн—сн—СН,
3 । 2 3
no2
2-нитробутан
Нитроарены, содержащие нитрогруппу в боковой цепи, рассматривают как производные предельных углеводородов, содержащих в качестве заместителей ароматический радикал и нитрогруппу:
1-нитро-2-фенилэтан
СН—СН—СН
NO2
2-нитро-1 -фенилпропан
159
15. Hump '''оединения
НИТРОСОЕДИНЕНИЯ АЛИФАТИЧЕСКОГО РЯДА
Способы получения
1. Нитрование алканов (реакция Коновалова). На предельный углеводород действуют разбавленной азотной кислотой (10—25%) при повышенной температуре и давлении.
Н3С—СН—СН3+HNO3 (разб.) —НДС—СН~СН3 + Н2О пропан J
NO2 2-нитропропаи
2. Действие солей азотистой кислоты на галогенопроизводные алканов:
Н.С—CH2I + NaNO2
йодэтан
Н3С—сн—NO2+ Nal
нитроэтан
Данную реакцию целесообразно проводить в среде апротонного растворителя для уменьшения образования побочных продуктов — эфиров азотистой кислоты.
3. Окисление трет-алкиламинов. Данный способ используют только для получения третичных нитросоединений:
н,с—с—СН I nh2
сн3
н3с—с—сн3 + no2
Н2О
Нитроалканы по физическим свойствам — бесцветные жидкости или вещества желтого цвета. Причиной окрашивания является наличие хромофора — группы -NO2 Нитроалканы имеют приятный запах, ядовиты. Мало растворимы в воде, растворимы в большинстве органических растворителей.
Химические свойства
Для нитросоединений характерны два ряда реакций: реакции с участием нитрогруппы и реакции с участием подвижных атомов водорода при а-углеродном атоме.
а
Н,С—СН—-
&+Н
О
160
Лекции по органической химии
1. Таутомерия и образование солей. Благодаря наличию подвижных атомов водорода при а-углеродном атоме первичные и вторичные нитросоединения являются таутомерными веществами.
нитроформа
ОН
R—СН—N
О а««-нитрофор.ма
В растворе между этими формами устанавливается динамическое равновесие. Такой вид таутомерии получил название — аци-нитро-гзугоме-рии. В нейтральной среде равновесие почти полностью смещено в сторону нитроформы. В щелочной среде равновесие смещается в сторону аци-нитроформы. Так, первичные и вторичные нитроалканы растворяются в водном растворе щелочи, образуя соли нитроновых кислот.
нзс\
/СН—N
Н.С \
3 О
2-нитропропан
NaOH
НС1
Н3С\ О Na"
натриевая соль диметилнитроновой кислоты
Соли нитроновых кислот легко разрушаются минеральными кислотами с образованием исходных нитроалканов.
Третичные нитросоединения в виду отсутствия подвижных атомов водорода при а-углеродном атоме не способны к таутомерии, а следовательно не взаимодействуют со щелочами.
2. Реакция с азотистой кислотой. Первичные, вторичные и третичные нитросоединения по разному относятся к действию азотистой кислоты. В реакцию с HNO2 вступают только те нитросоединения, у которых есть подвижные атомы водорода при а-углеродном атоме.
Первичные нитропроизводные образуют алкилнитроловые кислоты:
СН-CH-NO,+O=N-OH —*Н,С—C-NO,—*CH-C-NO2
3 I 2 -Н,О 3 ' ' 2 3 и 2
н &+н V n-oh
О метилнитроловая
кислота
нитрозонитросоединение
Нитроловые кислоты растворяются в щелочах, образуя соли, окрашенные в красный цвет.
15. Нитрос'>ег1инения lt>3
Вторичные нитросоединения с азотистой кислотой образуют псев-юшпролы(нитрозо-нитоосоединения):
С - NO,
KC-C-fH +HOrN=O-----------------H,C-C-N=O
3 1 ----------J - H,0 3 |
CH3 2 CH3
псевдонитрол
2-нитрозо-2-нитропропан
Псевдонитролы — это бесцветные вещества, которые в кристаллическом состоянии являются ассоциированными соединениями, но в растворе или в расплаве ассоциаты разрушаются и появляется синее окрашивание.
Третичные нитросоединения не реагируют с азотистой кислотой.
Реакцию с азотистой кислотой используют для отличия первичных. вторичных и третичных нитросоединений друг от друга.
3. Реакция конденсации с альдегидами и кетонами. За счет подвижных атомов водорода в a-положении нитросоединения способны в слабощелочной среде вступать в реакции конденсации с альдегидом с образованием нитроспиртов (нитроалканолов):
снз— сх h + Н2С —NO,—►H.C-CH-CH-NO, -------------------*-
н \ | 2 3 г-1——1-; 2 -Н2о
этаналь [ОН___Н;
&+ 1-нитропропанол-2
----- НС—СН—СН—NO, -н,о 3 2
1 -нитропропен-1
Нитроспирты легко дегидратируются с образованием непредельных нитросоединений.
4. Реакция восстановления. При восстановлении нитроалканов образуются алкиламины:
ЗН2
CH3-NO2 -------► сн—NH2 + 2Н2О
метиламин
Нитросоединения алифатического ряда обладают сильным местным раздражающим действием и являются относительно токсичными веществами. Они относятся к клеточным ядам общего действия, особенно опасны для печени. Очень токсичны хлорсодержащие нитропарафины и непредельные нитросоединения.
162
Лекции по органической химии
НИТРОСОЕДИНЕНИЯ АРОМАТИЧЕСКОГО РЯДА
Способы получения
1. Нитрование аренов. Нитросоединения, содержащие нитрогруппу, связанную с ароматическим радикалом, получают нитрованием аренов смесью концентрированных азотной и серной кислот, называемой «нитрующей смесью». Реакция протекает по механизму электрофильного замещения (S,).
hono2
(H2so4), f
-Н2О
бензол
hono2 (H,SO4).z
-H2O
нитробензол
HONO, (H2SO4)~, t
-Н,О
jw-динитробензол
Максимально в бензольное ядро можно ввести три нитрогруппы. Нитрогруппа настолько дезактивирует бензольное ядро, что для введения второй нитрогруппы требуются более жесткие условия, а третья вводится с большим трудом.
2. Введение нитрогруппы в боковую цепь. Введение нитрогруппы в боковую цепь осуществляется по реакции Коновалова разбавленной азотной кислотой при повышенной температуре и давлении. Реакция протекает по радикальному механизму (SR).
толуол
HNO3 (разб.)
и с—NO
фенилнитрометан
Другим способом введения нитрогруппы в боковую цепь является реакция взаимодействия алкилгалогенидов с солями азотистой кислоты:
15. Нитросоединения
163
бензилхлорид
фенилнитрометан
КО
По физическим свойствам нитросоединения ароматического ряда — это жидкие или кристаллические бесцветные или окрашенные в желтый цвет вещества с приятным запахом горького миндаля, не растворимые в воде, имеют высокие температуры кипения и плавления.
Химические свойства
Обладая электроноакцепторными свойствами (-1, -М), нитрогруппа в целом понижает электронную плотность бензольного ядра, тем самым уменьшает его реакционную способность в реакциях электрофильного замещения (S,) и создает возможность для протекания реакций, идущих по механизму нуклеофильного замещения (SN).
Для нитроаренов возможны реакции по нитрогруппе и по ароматическому ядру.
1. Реакции с участием нитрогруппы
Реакция восстановления (реакция Зинина). При восстановлении ароматических нитросоединений образуются ароматические амины. В зависимости от pH реакционной среды процесс восстановления может идти по двум направлениям, отличающимся образованием разных промежуточных продуктов.
В нейтральной и кислой среде (pH < 7) в качестве промежуточных соединений образуются ароматические нитрозосоединения и арилгидроксиламины:
нитробензол
- 2Н2О
нитрозобензол
фенилгидроксиламин
- Н2О
анилин
В щелочной среде (pH >7) происходит конденсация образующихся в процессе реакции нитрозосоединений с арилгидроксиламином и образуются азоксисоединения. Последние присоединяют водород и превращаются в гидразосоединения, которые, в свою очередь, легко переходят в ариламины:
164
Лекции по органической химии
нитробензол
нитрозобензол
азобензол
гидразобензол
Реакцию восстановления нитроаренов в щелочной среде (рН>7) можно остановить на любой из приведенных стадий. Она служит основным способом получения азо- и гидразосоединений. Реакция открыта в 1842 году русским ученым Н.Н. Зининым.
2. Реакции по ароматическому ядру
Реакции электрофильного замещения (SE). Под влиянием нитрогруппы (-/; -М) понижается реакционная способность бензольного ядра в реакциях электрофильного замещения. Так, нитробензол не алкилируется в условиях реакции Фриделя — Крафтса, но может вступать в реакции нитрования, галогенирования, сульфирования с образованием л/е/ла-замещенных нитроаренов:
зи-хлорнитробензол
15. Нитросоединения
165
Реакции нуклеофильного замещения (SN). Пониженная электронная плотность в ароматическом ядре нитроаренов создает возможность для протекания реакций нуклеофильного замещения в аренах. В реакциях SN нитрогруппа направляет заместитель в орто- и ияра-положения:
мп
+ КОН
Соединения, содержащие две и более нитрогруппы, придают повышенную подвижность атомам водорода, находящимся в орто- и иара-положениях бензольного ядра. Так, сам нитробензол не окисляется, а динитробензол окисляется легко.
По этой же причине легко замещается атом хлора в молекуле о-нитрохлорбензола. Это позволяет получать различные нитропроизводные ароматического ряда:
о-нитроанизол
1РР Лекции no органической хи чип
При действии на тринитробензол гидроксиламина атом водорода легко замещается на аминогруппу:
2,4,6-тринитроанилин
В молекуле динитрозамешенного толуола две нитрогруппы активируют атомы водорода метильной группы. Такие соединения могут вступать в реакции с ароматическими альдегидами:
2,4-динитротолуол
2,4-дин итростил ьбе н
Нчтросоединения ароматического ряда имеют большое значение. Это исходные вещества в производстве синтетических красителей, полимеров, моющих средств и ингибиторов коррозии. Ряд нитросоединений находят применение в качестве биологически активных веществ. Так, эфиры фосфорной кислоты, содержащие нитроарильные фрагменты, являются инсектицидами, производные 2,4-динитрофенола — гербициды; на основе нитропроизводных, обладающих антибактериальным действием, созданы лекарственные препараты (левомицетин, синтомицин, фуразолидон и др.). Некоторые ароматические нитросоединения применяются как душистые вещества.
Ароматические нитросоединения угнетают нервную и особенно кровеносную системы, нарушая снабжение организма кислородом.
Метаболизм нитросоединений связан с окислительно-восстановительными реакциями и, в частности, с окислительным фосфорилированием. Например, 2,4-динитрофенол — один из наиболее мощных реагентов, разобщающих процессы окисления и фосфорилирования в организме, что препятствует образованию АТФ в клетке.
16. АМИНЫ
Амины — производные аммиака, в молекуле которого один, два или три атома водорода замещены углеводородными радикалами. По числу замешенных атомов водорода они делятся на первичные, вторичные и третичные амины.
NH, аммиак
h,c-nh2
первичный амин, метиламин
н3с NH н3с-сн2 вторичный амин, метил этиламин
Н3С H3C-N н3с-сн2 третичный амин, диметилэтиламин
Существуют и четвертичные аммониевые соли, например хлорид тетраметиламмония [(CH3)4N’]Cr, соответствующее ему основание — гидроксид тетраметиламмония [(CH3)4N+]OH“, который представляет собой сильное основание, аналогичное гидроксидам щелочных металлов, так как связь с гидроксильной группой здесь ионная.
В зависимости от природы углеводородных радикалов амины подразделяются на алифатические, алициклические, ароматические и смешанные (имеющие алифатический и ароматический радикалы):
h3c-nh2
алифатический алициклический амин, ароматический амин, смешанный амин, амин, метиламин циклогексиламин анилин Ы,Ы-диметиламин
Для аминов характерна метамерия:
h3c-ch2-ch2-nh2
Н3СХ
н5с2/
н3с H3C-N Н3С
Эти соединения имеют общую формулу C3HgN, но отличаются друг от друга строением углеводородных радикалов.
Названия простых по строению аминов образуют от названий соответствующих углеводородных радикалов, связанных с атомом азота, добавляя в конце корень -амин. Кроме того, многие ароматические амины имеют тривиальные названия.
168
Лекции по органической химии
н3с-сн-сн3 nh2
изопропиламин
Н3С NH / н3с-сн2-сн2
метилпропиламин
фениламин.
аминобензол, анилин
дифениламин
Производные толуола, содержащие аминогруппу в бензольном
о-толуидин
.м-толуидин
л-толуидин
АМИНЫ АЛИФАТИЧЕСКОГО РЯДА (АЛКИЛАМИНЫ)
Способы получения
1. Действием аммиака на галогенпроизводные (реакция Гофмана).
Нзс—Cl +NH3
+ _ NH,
Нзс-NH, С1 ---»
н3с—nh2+ nh4ci
Первоначально аммиак с хлористым метилом образует соль, которая под действием избытка аммиака превращается в первичный амин.
Метилирование может протекать и дальше. Образовавшийся первичный амин реагирует со следующей молекулой галогеналкана и т. д. В результате образуется вторичный амин, затем третичный и соль четвертичного аммониевого основания:
Н3С—NH2 + Н3С—Cl
н3сх +
NH
Н3С
Cl NH3 Н3СХ ------* NH + NH4C1
Н3С
хлорид
диметиламмония
диметиламия
16. Амины
169
НзСх
NH +Н3С—Cl—*
Н3С
Н3С
H3C-N +
Н3С
С1“ NH, НзС
—* Нзс-N +NH4C1
H3CZ
хлорид
триметиламин
триметиламмония
Н3С
3 \
H3C-N + Н3С—Cl
Н3С
Н3С
3 \ +
Н3С— N—СН3
H3CZ
хлорид
тетраметиламмония
Преимущество этого метода — это простота, недостаток — трудно разделить образующиеся амины, однако для промышленного производства этот метод очень удобен.
2. Перегруппировка Гофмана. При обработке незамещенных амидов карбоновых кислот гипобромитом натрия (смесь брома и гидроксида натрия) образуются первичные амины, при этом углеродная цепь укорачивается на один атом:
О
Н3С—CZ + г48вг
I 2 ।
//
н,с—с —-
\ - NaBr
N—Na
амид
уксусной кислоты
метилизоцианат
//
н3с—с
3 \ №
бирадикал
н3с—nh2+ со2|
Этот способ имеет большое значение и дает возможность получать чистые амины.
3. Способ Габриэля. Конденсацией фталимида калия с алкилгалоге-нидами с последующим гидролизом получают только первичные алифатические амины:
170
Лекции по op viuuet кои химии
фталимид калия
N-метил фталимид
фталевая кислота
4. При восстановлении нитроалканов и нитрилов образуются первичные амины:
h3c-ch2-no2 нитроэтан
H.C-CEN
ацетонитрил
h3c-ch2-nh2
этанам ин
h3c-ch2-nh2
этанамин
Физические свойства
Низшие амины (метиламин и этиламин) — газы, с увеличением количества атомов углерода — жидкости, высшие амины — твердые вещества. Они обладают аммиачным запахом, а также запахом соленой сельди.
Низшие амины хорошо растворимы в воде, с увеличением молекулярной массы растворимость снижается, а высшие амины не растворимы в воде.
Химические свойства
Реакционная способность алифатических аминов определяется наличием у атома азота неподеленной пары электронов, которая обуславливает то, что амины активно выступают в роли нуклеофилов, а также являются сильными органическими основаниями.
Основность. Являясь производными аммиака, алкиламины подобно аммиаку проявляют ярко выраженные основные свойства, кото
16. Амины
171
рые обусловлены способностью атома азота с неподеленной электронной парой к присоединению протона с образованием иона замещенного аммония. При этом амины в 10 раз сильнее, чем аммиак. Это связано с изменением электронной плотности на атоме азота.
NHj CHy*NH2
+/
Алкильные радикалы увеличивают электронную плотность на атоме азота за счет проявления +/-эффекта, что приводит к заметному усилению основности. Поэтому можно предположить, что третичные алкиламины, имеющие три донорных заместителя, будут более сильными основаниями, чем вторичные, а вторичные, в свою очередь, более сильными, чем первичные и аммиак:
СН, CH^*N сн3
СН3 )NH сн3
> СНу—NH2 > NH3
Такая закономерность изменения основности алкиламинов наблюдается в газовой фазе и в неводных растворах. В водных растворах наряду с электронными эффектами заместителей влияет и сольватационный эффект растворителя.
В результате совместного проявления этих двух факторов основность третичных аминов в водных растворах ниже основности первичных и вторичных:
сн,
'SjH > сн3—nh2 > ch3-*n
сн3 СН3
1. Водные растворы алкиламинов имеют щелочную среду:
R-NH2 + HOH R-NH3 + ОН’
2. С кислотами алкиламины образуют соли алкиламмония:
c2h-nh2 + НС1
C2H5-NH3 Cl-
хлорид этиламмония
3. Взаимодействие с галогеналканами (реакция алкилирования)*.
См. способы получения (реакция Гофмана).
172
Лекции по органической химии
4. Взаимодействие аминов с ацилирующими реагентами (реакция ацилирования). Первичные и вторичные алкиламины вступают в реакцию с функциональными производными карбоновых кислот — гало-генангидридами, ангидридами и сложными эфирами, образуя N-за-мещенные амиды карбоновых кислот:
н3с-с +
С1
хлорангидрид уксусной кислоты
h2n-ch3 —► Н3С-С + НС1
N-CH3 метиламин Н
N-метиламид уксусной кислоты
В процессе реакции атом водорода при азоте в молекуле амина замещается на остаток карбоновой кислоты. Третичные амины не содержат при атоме азота водорода и поэтому в реакцию ацилирования не вступают.
5. Взаимодействие с азотистой кислотой. Реакция с HNO2 применяется для идентификации алифатических аминов.
Первичные алкиламины под действием азотистой кислоты превращаются в спирты с выделением азота и воды:
на Г 1 н,о
H3C-NH,+HO-N=O—► H3C~N=N Cl —* H3C-OH+HCi +N2|
-2H2O L
метилдиазоний хлорид (неустойчивое соединение)
Вторичные алкиламины в реакции с азотистой кислотой образуют N-нитрозоамины:
НДС
3 \
NH + HO-N=O
Н3С
НДС
3 \ N-N=O + НО
/ НС
N-ннтрозодиметиламин (тяжелая маслянистая жидкость)
Третичные амины при обычной температуре с азотистой кислотой не реагируют:
Н,С
3 \
Н3С—N +H0-N-0
3 /
Н3С
16. Амины
173
6. Изонитрильнаяреакция. Специфическая реакция обнаружения первичных аминов. При нагревании первичных алкиламинов с хлороформом в присутствии шелочи в спиртовой среде образуются изонитрилы (изоцианиды):
С2Н5ОН .
н3с—NH2 +СНСЦ+ ЗКОН------------- H3C-N=C + ЗКС1 + ЗН2О
метиламин метил изоциан ид
Изонитрилы имеют очень неприятный запах. Эта реакция очень чувствительна.
7. Реакция окисления. Первичные алкиламины при окислении озоном образуют нитроалканы:
о3
н3с—nh2 —- h3c-no2 + н2о
метиламин нитрометан
Под действием сильных окислителей первичные алифатические амины образуют смесь вешеств, в которой преобладают альдегиды, а вторичные — тетразамешенные гидразины.
При окислении третичных аминов действием Н2О, или пероксикислотами RCOOOH образуются N-оксиды аминов:
Н3С
3 \
Нзс-N + Н2О2 н3с
н,с
3 \
Нзс—N—о + Н,0
/ 1
н3с
N-оксил триметиламина
N-Оксиды аминов являются биологически активными соединениями.
АМИНЫ АРОМАТИЧЕСКОГО РЯДА (АРИЛАМИНЫ)
Способы получения
1. Восстановление нитросоединений (реакция Зинина).
Н2О
2. Взаимодействие галогенпроизводных с аммиаком и аминами.
аС1
+ 2NH3 ',Р »
3 Си
NH4C1
анилин
хлорбензол
174
Лекции по органической химии
При взаимодействии галогенаренов с ариламинами образуются вторичные и третичные ариламины:
хлорбензол
анилин
t,p
Си
>,Р Си
+ НС1
дифениламин
3. Алкилирование первичных ариламинов.
N-метиланилин
Физические свойства
Ариламины представляют собой бесцветные высококипящие жидкости или твердые кристаллические вещества со слабым неприятным запахом. Мало растворимы в воде, сильно токсичны, окисляются кислородом воздуха, из-за чего при хранении приобретают желтоватую окраску.
Химические свойства
Для ариламинов характерны реакции с участием атома азота и реакции с участием атомов углерода ароматического ядра.
Основность. Ароматические амины обладают основным характером. Однако они слабее, чем амины жирного ряда и даже слабее аммиака.
Снижение основности обусловлено сопряжением неподеленной пары электронов атома азота с л-электронной системой ароматического ядра:
16. Амины
175
+М- эффект
Основные свойства падают в ряду:
Являясь слабыми основаниями, ариламины образуют соли только с сильными минеральными кислотами:
анилиния хлорид, хлористый фениламмоний, солянокислый анилин
Анилин не образует соли с Н2СОГ
На основность ариламинов существенное влияние оказывают заместители в бензольном кольце. Электронодонорные заместители увеличивают основность, а электроноакцепторные — уменьшают ее.
При переходе от первичных к третичным основность ароматических аминов снижается.
1. Реакция алкилирования. Первичные и вторичные ариламины реагируют с галогеналканами, образуя N-алкил- и М,1Ч-диалкиларил-амины. Реакция протекает труднее в виду снижения нуклеофильных свойств атома азота.
с6н—nh2+ Нзс—I -------- C6H-NH-CH3 + hJ
N-метиланилин
сн3
С6Н—NH-CH3 + Нзс-1 —*с6н—Nx
сн3
N.N-диметиланилин
176
Лекции по органической химии
2. Реакция ацилирования. Первичные и вторичные амины при взаимодействии с хлорангидридами или ангидридами карбоновых кислот образуют N-замещенные амиды карбоновых кислот. N-Ацильные производные анилина и его гомологов называют анилидами.
N-фениламид уксусной кислоты.
ацетанилид, анилид уксусной кислоты
Амиды карбоновых кислот легко гидролизуются в кислой или щелочной среде с образованием исходного амина и карбоновой кислоты:
Способность ацильных производных легко подвергаться гидролизу позволяет применять эту реакцию в органическом синтезе для временной защиты аминогруппы от окисления и протекания по ней нежелательных реакций.
Ацильные производные аминов имеют большое значение как фармацевтические препараты. Например, ацетанилид — это жаропонижающий препарат, который долгое время применялся в медицинской практике, а в настоящее время — в ветеринарии.
3. Реакция образования изоцианидов (качественная реакция) Аналогичноалифатическим аминам первичные ароматические амины при нагревании с хлороформом и щелочью в спиртовой среде образуют изоцианиды — вещества с неприятным тошнотворным запахом:
NH2
|| ~ с,н,он
I. + СНС13 + ЗКОН —5 >
+ ЗКС1 + ЗН2О
фенилизоцианид
16. Амины
177
4. Реакция взаимодействия с азотистой кислотой. Алифатические и ароматические амины по-разному реагируют с азотистой кислотой, эту реакцию можно использовать для отличия первичных, вторичных и третичных аминов алифатического и ароматического рядов.
Первичные ароматические амины при действии азотистой кислоты в присутствии сильной минеральной кислоты образуют соли диа-зония. Эта реакция получила название реакции диазотирования.
бензолдиазоний хлорид
Вторичные ариламины и N-алкилариламины в этих условиях превращаются в N-нитрозоамины:
N - метил - N - н итрозоан ил ин
Третичные ароматические амины при взаимодействии с азотистой кислотой нитрозируются по бензольному кольцу обычно в пара-положение, если лара-положение занято, то нитрозогруппа занимает ор/ло-положение:
Ы,1Ч-диметиланилин
л-нитрозо-Ы,К-диметиланилин
5. Реакция взаимодействия с ароматическими альдегидами. Первичные ароматические амины реагируют при нагревании с ароматическими альдегидами, образуя азометины (основания Шиффа):
178
Лекции по органической химии
сйн>н2+ q=c-c6h5—* c6h5n=ch-c6h5+ н2о
N-бензилиленанилин, основание Шиффа
Реакции с участием атомов углерода ароматического ядра
Для ариламинов характерны реакции электрофильного замещения по ароматическому ядру.
Аминогруппа проявляет +Л/-эффект и выступает в качестве сильного электронодонора по отношению к бензольному кольцу, тем самым активируя его в реакциях электрофильного замещения.
6. Реакция галогенирования.
Анилин взаимодействует с бромной водой в отсутствие катализатора, образуя сразу 2,4,6-триброманилин:
Вг
5,4,fr-Tpn6poMaHiuinH
Для введения хлора в ароматическое ядро требуются особые условия (присутствие НО, СС14, С2Н.ОН, 16 °C).
7. Реакция нитрования. Поскольку аминогруппа чувствительна к действию окислителей, а ее влияние на ароматическое ядро обуславливает сравнительно легкую окисляемость аренаминов до хиноними-на, нитрование ароматических аминов проводят предварительно их проацилировав. Это необходимо для защиты аминогруппы, а также для понижения реакционной способности ароматического ядра.
16. Амины
179
Образовавшееся ацильное производное подвергают гидролизу и выделяют свободные нитроанилины.
8. Реакции сульфирования. При взаимодействии анилина с концентрированной серной кислотой образуется иара-аминобензолсульфо-кислота. которую чаще называют сульфаниловой кислотой. Реакция протекает через стадию образования N-фенилсульфаминовой кислоты, которая перегруппировывается в ла/ю-аминобензолсульфокислоту:
анилиний гидросульфат
>t
-н,о
N-фенилсул ьфам иновая кислота
сульфаниловая кислота
В сульфаниловой кислоте имеются две функциональные группы: -NH2 (основная) и -SO3H (кислотная). Это приводит к тому, что сульфаниловая кислота существует в виде биполярного иона (внутримолекулярной соли), называемого иногда цвиттер-ионом. Именно этим объясняется его высокая температура плавления.
Сульфаниловая кислота довольно сильная, с основаниями оналег-ко образует соли:
h2n
SO,H+NaOH
h2n
SO3Na+ H2O
я-аминобензолсульфонат натрия
Из-за биполярной структуры сульфаниловая кислота не способна образовывать соли с минеральными кислотами, т. е. несмотря на наличие аминогруппы, она не обладает основными свойствами.
Сульфаниловая кислота широко применяется в производстве красителей и лекарсвенных средств. Наибольший интерес представляет амид сульфаниловой кислоты (сульфаниламид, стрептоцид), структура
180
Лекции по органической химии
которого лежит в основе сульфаниламидных препаратов, которые составили целую эпоху в лечении инфекционных заболеваний.
стрептоцид, амид сульфаниловой кислоты
В качестве исходного вещества для получения стрептоцида используют анилин. Синтез осуществляют в четыре стадии:
NHj
л-ацетамино-
бензол сульфамид
л-ацетаминобензол-
сульфаниламид, амид сульфаниловой кислоты
В молекуле стрептоцида содержится аминогруппа и сульфамидная группа. За счет 1ЧН2-группы стрептоцид растворяется в кислотах, а за счет SO2NН2 — растворяется в щелочах, т. е. обладает амфотерными свойствами:
Стрептоцид был получен немецким студентом Гельмо. Но потребовалось 30 лет, чтобы его внедрить в медицинскую практику.
Стрептоцид и другие сульфаниламидные препараты действуют на стрептококковые микроорганизмы, отсюда и произошло название «стрептоцид» Однако сульфаниламиды действуют не бактерицидно, а бактериостатически, т. е. задерживают рост микроорганизмов.
16. Амины
181
В основе бактериостатического действия сульфаниламидов лежит их сходство с и-аминобензойной кислотой (ПАБК). которая участвует в биосинтезе фолиевой кислоты, необходимой для жизнедеятельности микроорганизмов.
Подобие этих соединений как в структурном, так и в химическом отношении приводит к тому, что сульфаниламиды способны вступать в конкурентное замещение л-аминобензойной кислоты, препятствуя синтезу фолиевой кислоты, что и подавляет рост и размножение микроорганизмов.
Производные стрептоцида:
альбуцид, сульфацил
уросульфан
норсульфазол
букарбан
Конечно, нам не предоставляется возможным рассмотреть все сульфаниламидные препараты. Более длительно вы будете знакомиться с ними в курсе фармацевтической химии, только помните одно: что есть сульфамидные препараты и есть сульфаниламидные. Сульфамиды — это общее понятие, а сульфаниламидные обязательно содержат Ч- NH-С6Н-SO2-NHR.
Амины — промежуточные продукты в производстве красителей, пестицидов, полимеров, ингибиторов коррозии, ПАВ, адсорбентов, лекарственных препаратов.
Алифатические амины поражают нервную систему, вызывают нарушения проницаемости стенок кровеносных сосудов и клеточных мембран, функций печени и развитие дистрофии.
Ароматические амины вызывают образование метгемоглобина, угнетающего центральную нервную систему. Некоторые ароматические амины — канцерогены.
17. ДИАЗО- И АЗОСОЕДИНЕНИЯ
ДИАЗОСОЕДИНЕНИЯ
Диазосоединениями называют органические вещества, содержащие в своей структуре группировку из двух атомов азота, связанную с углеводородным радикалом и остатком минеральной кислоты.
По природе углеводородного радикала различают алифатические и ароматические диазосоединения.
Ароматические диазосоединения имеют общую формулу ArN2X, где Аг — ароматический радикал, X — кислотный остаток.
Если X — остаток сильной минеральной кислоты (СГ. Вг-, HSO4 , NO3~), диазосоединения имеют ионное строение и называются соли диазония (Ar-N =NX‘), а если X — остаток слабой минеральной кислоты (CN-, HSOj-, ОН-, SH-), то они имеют ковалентное строение — Ar—N=N—X. Диазосоединения общей формулы Ar—N=N—О-М+, где М — металл, называютдиазотаты.
В зависимости от pH среды эти формы способны взаимопревра-щаться.
В кислой среде диазосоединения существуют в виде солей диазония, в щелочной — в виде диазотатов, близкой к нейтральной — в виде изомерных диазогидратов:
кислая среда нейтральная среда
+ - NaOH +
Аг—N=NC1 -------------► Ar—N=N — ОН
арилдиазогидроксид
*• Ar—N=N—ОН
NaOH
щелочная среда
Аг—N=N—О Na+
арилдиазотат натрия
Номенклатура. Изомерия
По номенклатуре ИЮПАК названия ароматических диазосоединений образуются путем прибавления к названию углеводорода суффикса -диазо, а названия солей диазония — добавлением окончания -диазоний с последующим указанием аниона.
17. Диазо- и аэосоединения
183
C6H5-N= N-OH
бен золдиазогидгхжсид
C6H5-N = N-O-Na+
бензолдиазотат натрия
4-метилбензолдиазоний бромид
Поскольку ковалентно построенные диазосоединения в своем составе содержат двойную связь, то для них характерна геометрическая изомерия, или так называемая син-(иис-) и анти-(транс-)изомерия. например:
аити-арилдиазогилроксид
сии-арилдиазогидроксил
Способы получения солей арендиазония
1. Реакция диазотирования. В основе реакции лежит взаимодействие первичных ароматических аминов с азотистой кислотой в среде сильной минеральной кислоты. Так как азотистая кислота не устойчива, то в реакции используют ее соль. В реакции диазотирования на один моль ароматического амина и один моль соли азотистой кислоты используется не менее 2,5 моль минеральной кислоты: один моль расходуется для вытеснения азотистой кислоты из ее соли, второй моль — для образования соли диазония, а избыток кислоты необходим для создания кислой среды. Кислота также способствует растворению ароматического амина. Необходимо отметить, что в реакцию диазотирования вступает не соль амина, а свободный амин, находящийся в равновесии с этой солью:
C6H5NH3C1" C6H5NH2+ НС1
Реакцию также проводят при охлаждении до 0—5 °C. Это связано с тем, что диазотирование протекает с выделением тепла, а соли диазония и азотистая кислота разлагаются уже при комнатной температуре.
184
Лекции по органической химии
NaNO2, НС1 изб.
NaCl + Н2О
бензолдиазоний хлорид
Механизм реакции
Кинетические исследования Ингольда показали, что реакция протекает по ионному механизму. Диазотирующим агентом является не азотистая кислота, а образующийся из нее, в зависимости от условий, реагент:
Н
+_____ I + _____ +
НО—N==O + Н H-O-N=O N=O + Н2О
протонированная нитрозоний-катион азотистая кислота
NO+ + Н—О—N=O -—O=N—О—N=O---------N,O,
-н
I оксид
Н азота (Ill)
Наиболее активным является нитрозоний-катион.
Вначале нитрозоний-катион образует с ароматическим амином N-нитрозоамин, который в кислой среде переходит в таутомерную форму — диазогидроксид. На завершающей стадии диазогидроксид превращается в соль арилдиазония:
Н Н
+ 1+ -н+ 1 5
C6H5-NH2 + NO C6H5-N-N=O C6H5-N-N=O
|_| N-нитроза.мин
C6H5-N=N-OH C6Hj-N«NCl + H2O диазо гидроксид
Полученные водные растворы диазония, не выделяя, используют в дальнейших синтезах.
2. Взаимодействие ароматических аминов с алкилнитритами. В среде этанола в присутствии минеральных кислот взаимодействие первичных ароматических аминов с эфирами азотистой кислоты приводит к образованию солей арилдиазония в кристаллическом виде: C.H,NH,+ C,H-ONO + НС1—*С,Н5-^М сГ+ C,HSOH + H2O о j Z Z J о J Z J Z
17Диазо- и азосоединения
185
В 96% спирте содержится 4 % воды, которая идет на гидролиз эфира. Выделившаяся при этом азотистая кислота диазотирует ароматический амин, а образовавшаяся соль диазония как плохо растворимая в спирте выпадает в осадок.
Химические свойства солей диазония
Соли диазония являются очень реакционноспособными соединениями. Их активность в химических превращениях связана с наличием диазокатиона. Атомы азота в диазокатионе находятся в состоянии хр-гибридизации. Один из них имеет неподеленную пару электронов, а второй содержит положительный заряд, который распределен в основном между двумя атомами азота и частично по л-электронной системе бензольного кольца. За счет делокализации каждый атом азота приобретает частичный положительный заряд, что можно представить в виде резонансных структур:
-*----- <^~^-N=N
Все реакции солей диазония можно разделить на две группы:
1. Реакции с выделением азота;
2. Реакции без выделения азота.
Реакции с выделением азота
Эти реакции сопровождаются разрывом связи С—N и замещением диазогруппы на другие атомные группы.
От способа разрыва связи С—N замещение может происходить по механизму Ss1 или SR.
По механизму S41 на первой медленной стадии связь С—N в диазокатионе разрывается гетеролитически. При этом выделяется молекула азота и образуется неустойчивый диазокатион, который на второй стадии быстро взаимодействует с нуклеофилом:
+___ _ медленно _i_
Ar—N=N X ----------------*- Ar + N., + X
арилкатион + -
Ar + Nu ------* Ar—Nu
По радикальному механизму (SR) замещение диазогруппы происходит в присутствии солей одновалентной или порошкообразной меди, которые являются донорами электронов. На первой стадии Си* отдает диазон иевому катиону один электрон, превращаясь в Си2* (ион меди (II)). При этом образуется нестойкий диазорадикал, который превращается в более стойкий арильный радикал и молекулу азота. На второй стадии он отрывает атом галогена от галогенида меди (II), регени-рируя галогенид меди (1):
186
Лекции по органической химии
C6H5-N^NCr+ CuCl —* C6H5-N=N--------------*- N2 + С6Н5-
u 2 диазорадикал арильный
t радикал
C6H<f + Cl^-Cu-Cl---► С6Н5С1 + CuCl
Замещение диазогруппы на гидроксигруппу. При нагревании подкисленных водных растворов солей диазония образуются фенолы:
c6h5-n = NHSO4 + н2о С6Н5ОН + N2 + H2SO4
бензолдиазоний гидросульфат
Замещение диазогруппы на атом йода. При нагревании соли диазония с йодидом натрия или калия образуются арилйодиды:
C6H5-N = NC1' + KI С6Н51 + N2 + КС1
Это наиболее удобный метод введения йода в бензольное кольцо.
Замещение диазогруппы, катализируемое солями одновалентной меди (реакция Зандмейера). В присутствии саней одновалентной мели диазогруппа замещается на атом хлора, брома, нитро-, циано-, хлорсульфонильную и другие группы:
C6H5SO2C1 + N2 so2. CuCl, t
C6H5CI + n2 CuCl.
C6H5-№NC1 бензолдиазоний Cu. t хлорид
КВг, Си. t
C6H5NO2 + N2 + NaCl CuCN t C6H5Br + N2 + KC1
C6H5CN + N2 + KC1
Замещение диазогруппы на атом водорода. При взаимодействии солей диазония с восстанавливающими агентами (фосфорноватистая кислота, формальдегид, спирты и др.) происходит замещение диазогруппы на атом водорода:
С6Н5Й=МС1 + Н3РО2 - H-'f- С6Н6 + N2 + НС1 + Н3РО3
При использовании спирта реакция протекает в двух направле
ниях:
17. Диазо- и ^соединения
187
C6H5f!i=NCl + С2Н5ОН
-С6Н6 +N2 + НС1 + СН3СОН
- С6Н5-О-С2Н5 + N2 + НС1
фенилэтиловый эфир
Реакция замещения на атом водорода применяется для удаления аминогруппы из ароматического ядра после ее использования для необходимой ориентации замещения, например:
Реакции без выделения азота
Образование диазопроизводных. Являясь электрофильными реагентами, соли диазония вступают в реакции с нуклеофильными реагентами, образуя диазопроизводные, например:
2NaOH
CzHc-l\I—N Cl -NaCl;-H2O * N N 0Na натрия диазотат ch3Nh2 CfiH<;-N=N-NH СНз
1 J IN ——— |\ -HCt , . 3-метил-1 -фенилтриазен NaCN NI CM
-NaCl " 7°"J бензолдиазоцианид Восстановление солей диазония приводит к арилгидразинам: + |Н] C6H5-N = NC1~— — +4НС[ “ C6H5NHNH2- НО + 2SnC14
В качестве восстановителей используют хлорид олова в НО или цинковую пыль в CHjCOOH.
Реакция азосочетания. Соли арилдиазония вступают во взаимодействие с фенолами и ароматическими аминами, образуя азосоединения общей формулы Ar—N=N—Аг:
188
Лекции по органической химии
Н*
-HCI
\.ЮН
-NaCI; -Н2О
Эти реакции называются реакциями азосочетания и протекают по механизму электрофильного замещения Sr
Поскольку диазокатион является слабым электрофилом, то он реагирует только с теми ароматическими соединениями, которые имеют сильные электронодонорные заместители (—ОН; —NH2; —NHR; —NR2 и дп) Замещение идет преимущественно в ипрп-положении.
Соль диазония в реакциях азосочетания называют диазосоставляющей (диазокомпонента), а фенол или ароматический амин — азосоставляющей (азокомпонента). Влияние заместителей в азосоставляющей противоположно влиянию заместителей в диазосоставляющей.
Азосочетание с фенолами проводят в слабощелочной среде, где они превращаются в реакционноспособные феноляты:
С ароматическими аминами реакцию проводят в слабокислой среде.
Сильнокислая среда недопустима, так как аминогруппа превращается в аммонийную, которая дезактивирует кольцо и уменьшает нуклеофильность азосоставляюшей.
Механизм реакции азосочетания.
Являясь электрофильным реагентом, катион арилдиазония образует с ароматическим амином или фенолом о-комплекс, который отщепляет протон и превращается в азосоединение:
о-комплекс
N=N
N(CH3)2
17. Диазо- и азосоединения
189
Следует помнить, что азосочетание с первичными или вторичными аминами может привести к образованию диазоаминосоединений (триазенам). Нагревание последних в кислой среде приводит к азосоединениям:
н+, г
пере групп ировка
4-а м и н оазобе н зол
АЗОСОЕДИНЕНИЯ
Названия азосоединений с одинаковыми радикалами составляют из префикса азо- и названия углеводорода. Положение заместителей обозначается цифрами или локантами:
азобензол 4-амино-2-гилроксиазобензол
В случае различных радикалов азосоединения рассматривают как производные более сложного по структуре углеводорода, который содержит в качестве заместителя ареназогруппу (Ar—N=N—)
2-бензолазонафталин
Азосоединения существуют в виде двух геометрических изомеров — син-(цис-) и анти-(транс-)'.
хс6н5
N=N
N—N
сил-арилдиазогидроксид
c,h.z
с6н5
анти- арил д и азо гид роке ид
Химические свойства
Обусловлены наличием в структуре азогруппы —N=N—. За счет наличия неподеленных пар электронов на атомах азота, азосоедиенения проявляют слабые основные свойства:
190
Лекции по органической химии
C6H5-N=N-C6H5 + НС1 C6H5-NH=N-C6H5 + Cl
Образующиеся соли хорошо растворимы в соответствующих кислотах.
С участием азогруппы азосоединения вступают в реакции окисления и восстановления. Так, при действии пероксикислот происходит окисление с образованием азоксисоединений:
о
II
~ и м-м г и 1О1’ СНГС-О-ОН +
C6H5-N-N-C6H5-----------------► QHj-N-N-^ ч
О"
азоксибензол
Восстановление в мягких условиях приводит к гидразосоединени-ям, а действием хлорида олова (П) в среде хлороводородной кислоты получают ариламины:
c6h5-n=n-c6h5 -J”1 » c6h5-nh-nh-c6h5
(Zn + NaOH) гидразобензол
Азосоединения представляют собой окрашенные кристаллические вещества, применяются как красители и лекарственные препараты.
салазопиридазии
салазодиметоксин
Химиотерапевтические средства салазопиридазии и салазодиметоксин используют для лечения больных неспецифическим язвенным
колитом.
ГИДРОКСИЛЬНЫЕ ПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ. СПИРТЫ. ПРОСТЫЕ ЭФИРЫ. ТИОЛЫ. ТИОЭФИРЫ. ФЕНОЛЫ
Гидроксильными производными называют производные углеводо родов, в которых один или несколько атомов водорода замещены на гидроксильную группу.
Гидроксипроизводные углеводородов классифицируют в зависимости от типа гибридизации углеродного атома, связанного с гидроксильной группой:
• спирты (гидроксигруппа расположена при 5р3-гибридизован-ном углеродном атоме);
• фенолы (фенольный гидроксил связан с5р2-гибридизованным углеродным атомом ароматического кольца);
' • енолы (гидроксигруппа находится прил/г-гибридизованном углеродном атоме двойной связи);
Следует отметить, что неизвестны гидроксипроизводные углеводородов, содержащие ОН -группу при 5р-гибридизованном углеродном атоме.
18. СПИРТЫ
Гидроксипроизводные углеводородов, содержащие одну или несколько гидроксильных групп при лр’-гибридизованных углеродных атомах, называют спиртами.
В зависимости от числа гидроксильных групп в молекуле различают одно-, двух-, трех- и полиатомные спирты.
сн3—он НэС—СНэ -1 | ~ н2с—сн—сн2 II 1 н,с—сн—сн—сн2 ~ 1 1 1 1 z
ОН он он он он он он он он
одноатомный двухатомный трехатомный четырехатомный
спирт спирт спирт спирт
По природе углеводородного радикала спирты делят на алифатические, алициклические и ароматические:
алициклический спирт
СН2ОН
ароматический спирт
СН3—ОН н2с=сн—сн2—ОН
алифатические спирты
192
Лекции по органической химии
В зависимости от расположения гидроксигруппы в углеводородной цепи молекулы выделяют первичные, вторичные и третичные спирты. Первичные спирты содержат гидроксигруппу при первичном атоме углерода, вторичные и третичные — соответственно при вторичном и третичном углеродных атомах.
R
R—СН2—ОН
.СН—ОН
R—С—ОН
первичный спирт
вторичный спирт
R" третичный спирт
ОДНОАТОМНЫЕ СПИРТЫ
Одноатомными спиртами называют производные углеводородов, содержащие одну гидроксигруппу, связанную с $р3-гибридизованным углеродным атомом.
Одноатомные спирты классифицируют в зависимости от природы углеводородного радикала:
• насыщенные (гидроксипроизводные алканов и циклоалканов);
• ненасыщенные (гидроксипроизводные ненасыщенных углеводородов, в которых гидроксигруппа не связана с кратной связью):
• ароматические (гидроксипроизводные ароматических углеводородов с группой —ОН в боковой цепи)
Номенклатура
По заместительной номенклатуре названия одноатомных спиртов образуют из названия родоначального углеводорода, суффикса -ол и цифрового локанта, указывающего положение гидрокейгруппы.
4 3 2 14 3 2 1
сн3—сн2—он сн3—сн2~сн—сн3 сн3—сн=сн—сн2—он
ОН , , .
бутанол-2 бутен-2-ол-1
этанол
>—он
циклогексанол 2-фенилэтанол
Для названия спиртов широко используют радикально-функциональную номенклатуру, в соответствии с которой к названию углеводородного радикала добавляют слово спирт.
Н2С=СН—СН2—ОН аллиловый спирт
сн3 изопропиловый
спирт
бензиловый спирт
18. Спирты
193
Рациональная (карбинольная) номенклатура спиртов используется в отдельных случаях для упрощения образования названий. Спирты рассматривают как производные метилового спирта СРЦОН, получившего название карбинол. Достаточно распространенными остаются тривиальные названия отдельных спиртов- СН3ОН — древесный спирт, С,Н5ОН — винный спирт и др.
Изомерия
Структурная изомерия спиртов обусловлена различным строением углеводородного скелета и положением гидроксигруппы в углеродной цепи:
• изомерия углеродного скелета
СН3—СН2—СН2—СН2—ОН
бутанол-1
• изомерия положения
СИ3\ СНу"""
СН—сн2—ОН
2-метилпропанол-1
СН3-СН2-СН2-СН2-ОН СН3-СН-СН2-СН3 он бутанол-2
бутанол-1
Н2С=СН СН2 СН2-ОН СН3-СН=СН-СН2-ОН
бутен-З-ол-1 бутен-2-ол-1
Геометрическая изомерия спиртов обусловлена наличием кратной связи в углеводородном радикале.
Н3С\С=С/СН2-ОН н^
н3С\С(^н
1К ""СН2-ОН
чис-бутен-2-ол-1
тралс-бутен-2-ол-1
Оптическая изомерия характерна для спиртов, содержащих в своем составе асимметрический атом углерода.
* СН2-СН2-СН3
СН3-СН-СН2-СН2-СН3 I чХн
ОН НО'^Ч
сн3
И-пентанол-2
СН2-СН2-СН3 H/,1
H3CZ он
5-пентанол-2
пентанол-2
Способы получения
Спирты получают различными синтетическими способами.
1. Гидратация алкенов. При гидратации этилена получают первичный спирт, его гомологи образуют вторичные и третичные спирты:
194
Лекции по оршническои химии
н2с=сн2 + н2о —> сн3-сн2-он
этилен этанол
Н,С=СН-СН3 + Н2О -~2S°4’f » СНгСН-СН, * J Л I J
пропен ОН
пропанол -2
2. Гидрирование карбонильных соединений. При восстановлении альдегидов, карбоновых кислот и сложных эфиров образуются первичные, а при восстановлении кетонов — вторичные спирты.
сн3-сн2-сС
Н пропаналь
сн3-сн2-с-сн3 о бутанон
-Н|;-1 сн3-сн2- сн2-он
пропанол-1
|Н|; Pt
-----► СН3-СН2-СН-СН3 он бутанол-2
Для получения ненасыщенных спиртов из соответствующих альдегидов и кетонов в качестве мягкого селективного восстановителя используют борогидрид натрия NaBH4 (не затрагивает кратные углерод-углеродные связи).
н2с=сн-снгс^ ---------► Н2С=СН-СН2-ОН
н
бугсн-3-аль пропен-2-ол-1
3. Гидролиз галогенопроизводных углеводородов. В присутствии водных растворов щелочей при нагревании галогенопроизводные углеводородов подвергаются гидролизу с образованием спиртов (S41 или Sn2).
R—Hal Н2°;ОН ♦ R-OH + Hal-
4. Взаимодействие карбонильных соединений с магнийорганически-ми соединениями (реактивами Гриньяра). Магнийорганическое соединение присоединяется к молекуле карбонильного соединения (альдегиды, кетоны, сложные эфиры) по месту разрыва л-связи карбонильной группы. Образовавшийся алкоголят гидролизуется до спирта.
195
18. Спирты
формальдегид мстилмагнийбромид
н I
CH3-C-OMgBr+ н2о--------
н
СН3
----- Н--C-OMgBr
н
броммагнийалкоголят
СН3-СН2-ОН + Mg(OH)Br
этанол
Первичные спирты получают при действии магнийорганических соединений на формальдегид, с другими альдегидами образуются вторичные спирты, с кетонами и сложными эфирами — третичные спирты
о 'тг|3 СП3
С н CH^MgRr^ С2н5—C-OMgBr ——----►С2Н5—6-он
2 5 I -Mg(OH)Br 2 э 1
НН Н
лропаналь бром магний бутанол-2
алкоголят
сн3 сн3
с2н5-с-сн3 с2н5-с-OMgBr_Mg(OH)Br - С2Н5-С-ОН
о сн3 сн3
бутанон 2-метилбутанол-2
5. Спиртовое брожение углеводов (глюкозы, фруктозы, крахмала или целлюлозы) позволяет получить этиловый спирт:
с6н12о6
дрожжи*
глюкоза
2С2Н5ОН + 2СО21
этиловый спирт
Физические свойства
Спирты — это жидкости или твердые вещества, растворимы во многих органических растворителях. Низшие спирты, в отличие от высших, хорошо растворимы в воде. Спирты состава Cj — С3 обладают характерным алкогольным запахом, С4 — С5 — неприятным «сивушным», высшие алканолы — с приятным фруктовым запахом.
Температура кипения спиртов значительно выше температуры кипения соответствующих углеводородов, их галогенопроизводных. Это объясняется наличием водородных связей, которые обуславливают образование ассоциатов:
5+ 5“ 5+ 5“ 5+ 8“
• • • Н—*-О: • • • Н—►О: • • • H-^Q: •
R R R
196
Лекции по of. анической химии
Прочность водородных связей невелика, но для их разрыва при переходе молекулы из жидкого в газообразное состояние требуется дополнительная энергия.
Химические свойства
Химические свойства спиртов обусловлены наличием полярных связей О—Н и С—О, а также не поделенных электронных пар атома кислорода. Для спиртов характерны реакции, протекающие с разрывом связи О—Н (образование алкоголятов, этерификация, окисление, дегидрирование, взаимодействие с реактивами Гриньяра), с разрывом связи С—О (нуклеофильное замещение ОН-группы. образование простых эфиров, дегидратация) и протонирование.
1. Кислотно-основные свойства. Спирты являются амфотерными соединениями.
Кислотность спиртов. Полярность связи О—Н в спиртах определяет ее склонность к гетеролитическому разрыву.
Спирты как ОН-кислоты реагируют со щелочными металлами с образованием алкоголятов.
2С2Н5ОН + 2Na —2C2H5ONa + Hjf
этилат натрия этоксил натрия
Алкоголяты легко разлагаются водой до исходных спиртов, что подтверждает более низкую кислотность спиртов по сравнению с водой:
C2H5O~Na+ + НОН
С2Н5ОН + NaOH
Из-за низкой кислотности спирты почти не вступают в реакцию со щелочами.
Кислотные свойства спиртов уменьшаются при переходе от первичных спиртов к третичным:
5+СН3х сн3—н > р 8+>8'+>8"+ СНз
5'+ СНК.. д’
н > сн3-^с—►о~«—н°
СН3
Такая зависимость объясняется проявлением -(-/-эффекта со стороны углеводородного радикала.
Основные свойства. Обусловлены способностью неподеленных электронных пар атома кислорода гидрокси группы спиртов реагировать с протоном с образованием оксониевых солей.
5+5-
R—О—И + ННа!
R—О:
Hal"
18. Спирты
197
Алкилоксониевые соли во многих реакциях образуются в качестве промежуточных продуктов.
Основные свойства спиртов возрастают при переходе от первичных спиртов к третичным.
5-
снз\
СН/
5- СН3\ 5”-
0-*—Н <СН3~^С—н сн3
2. Образование сложных эфиров. Спирты реагируют с органическими и кислородсодержащими минеральными кислотами с образованием сложных эфиров.
+ Н2О
сн3-сн2-он_+ HO-NO2
О /О
конц. H.SO.: t '/
сн3-сч + НО-СН2-СН3 „ - - сн3—с
он о-сн-сн
уксусная кислота этиловый спирт этилацстат 2 3
CH3-CH2-O-NO2 + Н2О
этил нитрат
3. Дегидратация спиртов. Для спиртов характерна межмолекулярная и внутримолекулярная дегидратация.
В результате межмолекулярной дегидратации образуются простые эфиры.
------, н+, t
СН3-СН2-0Н + НОФСН2-СН3----------- СН3-СН2-О-СН2-СН3 + н2о
этанол диэтиловый эфир
Межмолекулярная дегидратация становится доминирующей при нагревании избытка спирта в присутствии каталитических количеств минеральной кислоты при температуре 140—160°C и протекает по механизму SN1 или Sn2
Молекула спирта под действием минеральной кислоты протони-руется с образованием оксониевого катиона, а затем происходит замещение группы —ОН:
Н I .. сн3—с—он +н
н этанол
Н н
I +/
СИ3-С-С) л н этилоксоний катион
Механизм SN2 включает образование переходного состояния, которое формируется в процессе нуклеофильной атаки электрофильного атома углерода оксониевого катиона второй молекулой спирта:
198
Лекции по органической химии
нуклеофильный центр
электрофильный центр
Н \ н / j_[
I •• I Х+ / 1
СН3-С-ОН+СН3-С-О\ = н н + н
этанол этилоксоний
катион
Н + н
«—сн3-с—о—с—сн3-
н н н
? .. <FH’ zH СН3-С-О-”С”-ОЧ
н нН ЧН н
переходное состояние
-Н2О
н н
СН3-С—О-С-СНз н н
диэтиловый эфир
Механизм SN1 протекает через сталию образования карбкатиона:
СН3
СНз-t + Н2О сн3
сн3-с-оч
СН Н
СН]
СНз сн3
1+ ..I
СН3~С +:о—С-СН3
СН3 н СН3
сн3
сн3 + сн3
сн3-с—6—с-сн3 —г сн3 н сн3
сн3
-77Г СН3-С-О-С-СН3
-Я I | J
сн3 сн3
Первичные спирты вступают в реакцию межмолекулярной дегидратации по механизму SN2, третичные — по механизму SN1, а вторичные — как по Sn2, так и по SN1.
Продуктами внутримолекулярной дегидратации являются алкены.
Н
I н-с-сн, Г k -I- ? н он L ___ _ _ J
Следует помнить, что спирты, в которых с атомом углерода, несущим гидроксильную группу, связаны неравноценные атомы углерода, отщепление молекулы воды происходит по правилу Зайцева, т. е. водород уходит от соседнего, менее гидрогенизированного атома углерода:
сн3-сн2-сн-сн-сн3-т^сн3-сн2-сн=с-сн3
ОНСН3 . СН3
2-метилпентен-2
Н\ t
------ н2с=сн2 +Н2о
этилен
18. Спирты 199
Вн> гримолекулярная дегидратация становится доминирующей реакцией при нагревании спиртов с избытком минеральной кислоты при температуре выше 170 °C.
Следует отметить, что для спиртов более характерны реакции отщепления, протекающие по механизму £1. Это связано с кислотностью реакционной среды, в которой сильное основание — алкоксид-анион RO” не существует, так как быстро взаимодействует с протоном.
Дегидратация третичного спирта протекает по механизму £1.
СН3 СН3
I .. н+ I ±-^Н
сн3-г-он CHj-i-c^ —
СН3 СНз
2-метилпропанол-2 трет-бутилоксонии катион
2-.метилпропен
СН3 СНз-<£+ сн2*-Н карбкатион
4. Взаимодействие с галогеноводородными кислотами. При взаимодействии спиртов с галогеноводородными кислотами (НС1, НВг, HI) гидроксильная группа замешается на атом галогена и образуются га-логеноалканы. С первичными спиртами реакция протекает по механизму Sn2, с третичными — по SNI. Вторичные спирты реагируют как по механизму SN2, так и по механизму SN1.
Механизм S„ 2:
N
R-CHj-OH —
первичный спирт алкилоксоний катион
---► R-CHi-Br
Н2О 2
б- S+zH
- Вг..-Ь--О
. н и н
переходное состояние
галогеналкан
Механизм SN1:
R-^C-OH -Ы*
R\ + /И R\ + вг- R\
R-^C — С< R ->C R-^C-Br
R ^H “H2° R^ R<
третичный спирт
алкилоксоний катион
карбкатион
галогеналкан
200
Лекции по органической химии
С йодоводородной и бромоводородной кислотами реакция протекает легко, с хлороводородной — значительно труднее. Первичные и вторичные спирты реагируют с хлороводородной кислотой только в присутствии хлорида цинка (кислоты Льюиса).
Первичные, вторичные и третичные спирты при взаимодействии с реактивом Лукаса (эквимолярными количествами концентрированной хлороводородной кислоты и цинка хлорида) с различной скоростью образуют галогенопроизводные углеводородов. Проба Лукаса используется для распознавания первичных, вторичных и третичных спиртов.
5. Взаимодействие с галогенангидридами неорганических кислот. Спирты взаимодействуют с галогенидами фосфора (РС13, PCL и др.) и тионилхлоридом (SOC12) с образованием галогеналканов:
ЗСН3-ОН + РС13 —► ЗСН3-С1 + Н3РО3
СН3-ОН + РС15 —-СНз-С! + РОС13 + НС1
СН3-ОН + SOC12---► СН3-С1 + SO2 + НС1
Реакция с тионилхлоридом SOC1, протекает по механизму внутримолекулярного нуклеофильного замещения SN.i:
8+
СН3-СН2-ОН + SOC12---►СПз—ClI3 44/S=O—СН3-СН2-С1
-на “ лк ~s°2
этанол хлорэтан
этиловый эфир хлорангидрида сернистой кислоты
6. Окисление. Спирты окисляются под действием кислорода воздуха в присутствии катализаторов (Си, СиО) и различных неорганических окислителей — оксида хрома (VI), хромовой смеси (дихромата калия в серной кислоте), перманганата калия в серной кислоте и др.
Первичные спирты образуют альдегиды (при дальнейшем окислении — карбоновые кислоты с тем же числом углеродных атомов).
Н
I
R—С—ОН
I н
Ю1
альдегид
карбоновая
кислота
Вторичные спирты окисляются до кетонов:
R—СН—R' -»-R— С—R'
-И2О ||
ОН О
кетон
18. Спирты
201
Третичные спирты более устойчивы к окислению. При действии сильных окислителей происходит расщепление углеродного скелета молекулы третичного спирта с образованием смеси карбоновых кислот и кетонов.
В промышленности спирты превращают в альдегиды или кетоны путем кагалитического дегидрирования. При пропускании паров спирта над катализатором (Zn. Си. Ag) при температуре 300 °C происходит отщепление водорода и образуются альдегиды или кетоны: R-CH2-OH Cu~' R-Cf ° R— R’ -н2 * R~‘—R
2 он OH °
альдегид кетон
7. Взаимодействие с магнийорганическими соединениями. Спирты реагируют с реактивами Гриньяра. Атом водорода гидроксигруппы замешается на магнийгалоген и выделяется газообразный углеводород.
R-OH + CHjMgBr-------► R-OMgBr + CH4f
бромалкоголят магния
Измерив объем выделившегося газа, определяют количество гидроксильных групп в молекуле спирта (реакция Чугаева — Церевитинова).
ДВУХ-. ТРЕХ- И ПОЛИАТОМНЫЕ СПИРТЫ
Спирты, содержащие две гидроксильные группы, называются двухатомными спиртами, диолами или гликолями.
Классифицируют гликоли в зависимости от положения гидроксильных групп:
R-CH-CH-R’
I I
ОН он
а-гликоль
R— СН-СН,-СН-R’
I 2 I
ОН он
R—СН-СН2-СН-СН-R’
ОН ОН
р-ГЛИКОЛЬ
у-гл и коль
Двухатомные спирты, содержащие две гидроксильные группы при одном углеродном атоме, называются геминальными диолами. Эти спирты неустойчивы, в момент образования они отщепляют воду, превращаясь в альдегид или кетон:
R ОН
R ОН
-н2о
R
,С=О
гем-дмол
По систематической номенклатуре названия гликолей образуют от названия соответствующего углеводорода, добавляя суффикс -диол и цифровые локанты, указывающие положение гидроксильных групп. По радикально-функциональной номенклатуре двухатомные спирты
202
Лекции по органической химии
называют путем добавления к названию соответствующего двухатомного радикала суффикса -гликоль.
н2с—сн—сн3
он он
н2с—сн2
он он этандиол-1,2, этиленгликоль
пропандиол-1,2, пропиленгликоль
Спирты, содержащие три гидроксильные группы, называются трехатомными спиртами, триолами или глицеринами. Названия трехатомных спиртов образуют путем добавления к названию соответствующего углеводорода суффикса -триол и цифровых локантов, указывающих положение ОН-групп:
Н2С— СН—СН2
ОН ОН ОН пропантриол-L 2, 3, глицерин
Спирты, содержащие более трех гидроксильных групп, называют многоатомными спиртами, или полиолами. Четырехатомные спирты называют эригритами, пятичленные — пеититами, шестичленные — гек-ситами и т. д.
Способы получения
Двух- и трехатомные спирты получают теми же способами, что и одноатомные, используя в качестве исходных веществ соответствующие полифункциональные производные. Существует ряд специфических способов получения.
Гидроксилирование алкенов (реакция Вагнера) позволяет получить а-гликоли:
ЗСН3-СН=СН2+ 2КМпО4 + 4Н2О ---------
пропен
---* ЗСН3-СН-СН2+ 2КОН + 2МпО2
ОН ОН
пропандиол-1,2
Гидратация оксирана и его производных проводится в кислой среде и сопровождается раскрытием цикла:
Н2С—СН2 + Н2О н2с-сн2
О ОН ОН
оксиран этиленгликоль
Данная реакция лежит в основе промышленного способа получения этиленгликоля.
18. Спирты
Гидролиз жиров. Жиры, представляющие собой сложные эфиры трехатомного спирта глицерина и высших карбоновых кислот, в кислой и шелочной средах гидролизуются до глицерина.
h2c-o-co-r н2с-он
HC-O-CO-R ЗН?—- НС-OH +3R-COOH
Н+ или ОН- I
Н2с-о-CO-R Н2С-ОН
триглицерид глицерин
В промышленности глицерин получают синтетически на основе нефтегазового сырья. Синтез глицерина окислением пропилена считается наиболее перспективным:
н2с=сн-сн3 °2’^°С > н2с=сн-с(-^н2с=сн-сн2-он-^
пропилен акролеин Н аллиловый спирт
...Н°С| »н2С—CH~CH2 -^-н2с-сн-сн2 С! он он он он он
З-хлорпропандиол-1,2 глицерин
Физические свойства
Двухатомные и трехатомные спирты представляют собой жидкости или кристаллические вещества с высокими температурами кипения и плавления (наличие водородных связей). Отличительной особенностью спиртов с несколькими гидроксильными группами является их сладковатый вкус, усиливающийся с увеличением числа ОН-групп.
Химические свойства
Двух- и трехатомные спирты вступают в аналогичные одноатомным спиртам реакции. Однако различное количество гидроксильных групп в спиртах определяет и некоторые их особенности.
Кислотные свойства. С увеличением числа гидроксильных групп в молекуле кислотные свойства спиртов усиливаются. Это связано с электроноакцепторным влиянием одной гидроксильной группы на другую (-/-эффект).
Гликоли являются более сильными ОН-кислотами, чем одноатомные спирты. Они образуют алкоголяты (гликоляты) не только со щелочными металлами, но и другими активными металлами (Al, Mg и т. д.), а также со щелочами и гидроксидами тяжелых металлов.
204
Лекции по органической химии
Н2р-ОН NaOH
Н2с—он -н2о
^Н2С—ONa NaOH
н2с—ОН моногликолят натрия (неполный гликолят)
-Н2О
Н2С—ONa ^Н2С—ONa дигликолят натрия (полный гликолят)
а-Гликоли способны реагировать с гидроксидом меди (II) с образованием комплексных соединений. Данная реакция является качественной на спирты, содержащие а-гликольный фрагмент.
н.с-он
-1
Н2С-ОН
+ |Си(ОН)2
2
н I
Н2С—Оч ХО-СН2 | | + 2Н,О
н2с-о о-сн2
Н гликолят меди (II)
но- сн2
сн
pi глицерат меди (11)
н2с-он Н2С-
2 НС-ОН +1Си(ОН)2—---------- I
I —4Н2О НС
н2с-он
глицерин
Образование простых и сложных эфиров. Гликоли образуют неполные и полные производные при взаимодействии со спиртами (простые эфиры), кислородсодержащими минеральными или карбоновыми кислотами (сложные эфиры).
Н2С-ОН R-ОН; Н+ Н,с-OR R-ОН; н+ H,C-OR
H2c-OH -н2о н2с-ОН - Н2° h2c-or
простой эфир
н2с-он H2SO4 н2р- -ono2
2 НС-ОН + 3HNO3- 2 НС- -ONO2 +3н2о
н2с-он н2с- -ono2
нитроглицерин
Н2( :-о-с^°
н,( е-он Г пНз
2HC-OH +3HO-CC ^0 н‘ СНт -* н L (уС^() + ЗН2О г ^сн3
тт 1пн уксусная п2С UH кислота н2< ^>о
глицерин три ацетат
18. Спирты
Окисление. Окисление гликолей в зависимости от природы окислителя может протекать по-разному:
/Р
Н->С-ОН(О1 С-о
-| —► I н -
Н2С-ОН Hit-ОН
Ю1
гликолевый альдегид
| он сн2-он
гликолевая
кислота
[О]
соон
|О1
"н ^о хн
глиоксиловая кислота
СООН
- I соон
щавелевая кислота
глиоксаль
Для идентификации а-гликолей используют реакцию окисления йодной кислотой Н1О4.
Н2С-ОН О О
I // //
нс-он +ню4---------- н-сч +сн3-сч +ню3 + н2о
сн3 н н
пропилен гл и коль метаналь этаналь
При окислении происходит разрывхимической связи между углеродными атомами а-гликольного фрагмента и образуются соответствующие карбонильные соединения. По продуктам окисления устанавливают положение диольного фрагмента в молекуле.
Дегидратация. Гликоли под действием водоотнимающих реагентов подвергаются внутри-и межмолекулярной дегидратации. Направление реакций определяется условиями их проведения.
н2с—сн2 он он
Н+;г
-Н2О
н2с=сн он виниловый спирт
zo
------*н3с-с'
1_1
уксусный 11 альдегид
ZOH НО' о
ИХ 9Н2 H2S0<;' н?с' "сн?
н2с^ СН2 —н2о Нэс\ ^сн,
он но 2 о 2
диоксан
Глицерин подвергается внутримолекулярной дегидратации с образованием непредельного альдегида акролеина:
206
Лекции по органическойхшши
KHSO4;r „ О— I ЛЛ. 5+ н2с—сн—сн2 н0 - н2с—сн=сн±~о—н
он он он он
' //° — н£-сн-с!; —ттг* н2с=сн-с
•но Н. Н н
акролеин
Поликонденсация. Этиленгликоль в результате реакции поликонденсации способен образовывать полиэфир — полиэтиленгликоль но-[-сн2-сн2-о-]л-н.
Полиэтиленгликоль с молекулярной массой до 400 используют в фармации в качестве растворителя лекарственных веществ, основы для мазей, связывающего вещества в производстве таблеток.
аминоспирты
Аминоспиртами называют производные углеводородов, содержащие аминогруппу (N-алкил- или №,М-диалкиламиногруппу) и спиртовый гидроксил.
Н,С—СН,—ОН
2-аминоэтанол, р-аминоэтиловый спирт
н2с—сн—он N(C2H5)2
2-М,М-диэтмламиноэтанол
Получают аминоспирты путем аммонолиза и аминолиза оксирана и его производных, галогеноспиртов, а также восстановлением соответствующих нитроспиртов.
СН2~СН2 NH3 сн2-сн2
О —Н2° NH2 ОН
2-аминоэтанол
СН2-СН2—ОН
С1
2-хлорэтанол
(CH3)2NH
---------► СНэ-СН2-ОН
на | *•
N(CH3)2
2-N.N диметиламиноэтанол
|Н|
сн2-сн2-он---------- сн2-сн2-он
I -Н2О |
no2 nh2
2-аминоэтанол
Простейшие аминоспирты — густые жидкости с аммиачным запахом, растворимые в воде.
18. Спирты
Важное биологическое значение имеют холин и ацетилхолин. Холин — [2-(гидроксиэтил)-триметиламмония гидроксид]
[HO-CH2-CH2-N+(CH3).]OH-играет важную роль в обмене веществ, входит в состав лецитинов, содержится в нервных тканях, мозге, печени, почках, мышне сердца, участвует в построении клеточных мембран.
+
[Сн3-С-о-CH2-CH2-NH2 (СН3)3]ОН
о
Ацетилхолин является медиатором и участвует в передаче нервных импульсов.
ПРОСТЫЕ ЭФИРЫ
Простыми эфирами называют органические соединения, в которых два углеводородных радикала соединены атомом кислорода.
Общая формула простых эфиров R—О—R', где R, R' — углеводородные радикалы.
Различают симметричные простые эфиры (содержат одинаковые углеводородные радикалы) и несимметрические или смешанные (углеводородные радикалы различны).
По радикало-функциональной номенклатуре названия простых эфиров образуют путем добавления к названию углеводородных радикалов суффикса -овый и слова эфир. В смешанных простых эфирах углеводородные радикалы перечисляют в алфавитном порядке.
сн3-о-сн3 сн3-о-с2н5
диметиловый эфир метилэтиловый эфир
По заместительной номенклатуре ИЮПАК простые эфиры рассматривают как производные углеводородов, в которых атом водорода замещен ал кокс и группой RO-. Более сложный по структуре радикал принимают за родоначальную структуру.
1 2 з
СН3-СН2-О-СН2-СН2-СН3
1-этоксипропан
Простые эфиры — бесцветные легколетучие жидкости с характерным запахом, плохо растворимы в воде, хорошо растворимы в органических растворителях.
Способы получения
1. Межмолекулярная дегидратация спиртов. При нагревании спиртов в присутствии концентрированной серной кислоты образуются простые симметричные эфиры.
208
Лекции по органической химии
СН3-СНгоХ+Н<5-СН2-СН3^7-^^СН3-СН2-О-СН2-СН3+Н2О
диэтиловый эфир
2. Синтез Вильямсона. Алкилирование алкоголятов и фенолятов алкилгалогенидами позволяет получить несимметричные простые эфиры.
СН3—1 + С2Н5—ONa
- СН3-О-C2HS + Nal метилэтиловый эфир
СН3-1 +
^2^-ONa'
Oo-CHl+ Nal
метоксибензол
Химические свойства
Простые эфиры являются достаточно инертными веществами.
Образование оксониевых солей. Наличие в молекуле простого эфира атома кислорода со свободной электронной парой определяет их основные свойства. При взаимодействии простых эфиров с концентрированными минеральными кислотами образуются оксониевые соли.
С2Н5-б-С2Н5 + НС1^— диэтиловый эфир
с2н5-о-с2н5 н диэтилоксоний
Cl-
хлорид
Расщепление простых эфиров.
а) Расщепление под действием щелочных металов. Простые эфиры под действием щелочных металлов (Na, К, Li) при нагревании способны расщепляется с образованием алкоголятов и натрпйорганичес-кихсоединений.
С2Н5-О-С2Н5 +2Na—-—- C2H5-ONa + C2H5Na
этилат этилнатрий
натрия
б) Расщепление под действием кислот (ацидолиз). Простые эфиры разлагаются концентрированной йодоводородной кпслотоп уже на холоду с образованием алкилйодида и спирта.
С2Н—О-С2Н5 +Н1 диэтиловый эфир
С2Н5-ОН +С2Н5—1 этиловый этилиодид спирт
Реакция проходит по механизму Ss2.
Если реакцию проводят при нагревании, то образуется вторая молекула этилйодида.
18. Спирты
209
СН3-СН2-ОН
н* 5+
—сн3-сн
СН3-СН2-1 + Н2О
Окисление простых эфиров. Простые эфиры окисляются на воздухе при нормальной температуре. Легче всего окисление протекает по а-углеродному атому.
СН3-СН,-О-СН2-СНз + О2------~СН3-СН2-О-СН-СН3
О-О-Н
пероксид
Образующиеся пероксиды неустойчивы, легко взрываются. Работать с эфирами необходимо с большой осторожностью, предварительно убедившись в отсутствии пероксидных соединений.
ТИОЛЫ. ТИОЭФИРЫ
Тиолами (тиоспиртами, меркаптанами) называют соединения общей формулы R—SH (аналоги спиртов). Их можно рассматривать как производные сероводорода H2S, в котором один атом водорода замещен углеводородным радикалом.
По систематической номенклатуре названия тиолов образуют путем добавления к названию родоначального углеводорода суффикса -тиол. Префикс меркапто- используют, если в молекуле SH-группа не является старшей
По рациональной номенклатуре тиолы называют меркаптанами.
СН3—СН2—SH — этилмеркаптан, этантиол
Тиолы представляют собой жидкие или твердые вещества (за исключением метантиола), с неприятным запахом, ядовиты.
Получают тиолы из сероводорода и его кислых солей реакцией алкилирования галогеналканами или спиртами.
H2S + C2H5OH C2H5SH + H2O
NaHS + C2H5C1 -----► C2H5SH + NaCI
Химические свойства
Тиолы во многом сходны со спиртами, обладают более выраженными кислотными свойствами. Реакции обусловлены поляризацией связи S—Н и нуклеофильными свойствами атома серы.
Образование тиолятов (меркаптидов). Тиолы образуют соли не только со щелочными металлами, но и их гидроксидами, а также ионами тяжелых металлов.
210
Лекции по органической химии
C2H5SH + NaOH-------► C2H5ONa + Н2О
этантиолят натрия
Ацилирование. Реакция аналогична этерификации карбоновых кислот.
н-
СН3’СО HS-C2H5-------------- СНз-С^ + НОН
LJil о С2п5
этиловый эфир
тиоуксус ной кислоты
Окисление. В отличие от спиртов тиолы окисляются не по углеродному атому, а по атому серы. При окислении тиолов в мягких условиях (Н2О2, СиС12 и др.) образуются диалкилдисульфиды, в жестких (KMnO4, HNO3 или НО1) — сульфокислоты.
C2H5-$H + O + HS-C2H5 I___________J
C2H5-S-S-C2H5+H2O
диэтилдисул ьфид
о
[о] II
C2H5SH С2н5—s—он
о
этансульфокислота
Тиоэфирами (сульфидами) называют производные сероводорода H2S, в которых два атома водорода умещены углеводородными радикалами. Их можно рассматривать как тиоаналоги простых эфиров (R-S-R').
СН3—S—С?Н5 — метилтиоэтан, метилэтилсульфид
Получают сульфиды в результате алкилирования алкилгалогени-дами тиолятов и сульфидов щелочных металлов.
C2H5SNa + C2HjI
2C2H5I + Na2S
----►C2H5-S-C2H5*f4<t'3
этилтиоэтан.
диэтил сульфид
-----C2HrS-C2H5 ♦ 1I\U
Сульфиды — бесцветные вещества с неприятным запахом. Практически нерастворимы в воде. При взаимодействии с кислотами об-
разуют сульфониевые соли
CH3-S-CH3 + H2SO4-
CH3-S-CH3
Н
HSO4
диметилсульфония гидросульфат
18. Спирты ___________________________________________2__
Окисление сульфидов в зависимости от природы окислителя и условий реакции протекает с образованием сульфоксидов R2SO или сульфонов R2SO2.
Н2О2 9
CH3-S-CH3-----► CH3-S-CH3
диметилсульфоксид
CH3-S-C2H5-^^CH3-S-CH2-CH3
о
метилэтил сульфон
Сульфоксиды менее стабильны, чем сульфоны. Сульфоксиды относительно легко окисляются до сульфонов и восстанавливаются до сульфидов.
19. ФЕНОЛЫ
Фенолами называют производные ароматических углеводородов, у которых один или несколько атомов водорода в ароматическом ядре замещены на гидроксильную группу.
По числу гидроксильных групп фенолы делят на: одноатомные, двухатомные, трех- и полиатомные.
ОДНОАТОМНЫЕ ФЕНОЛЫ
К одноатомным фенолам относятся гидроксилсодержащие ароматические соединения, имеющие в своем составе одну гидроксильную группу:
1 -гидроксинафталин, а-нафтол
Для названия фенолов применяют заместительную номенклатуру, в этом случае перед названием арена ставится префикс -гидрокси.
При наличии заместителей в ароматическом ядре за родоначальную структуру берут фенол.
Наряду с заместительной номенклатурой в ряду фенолов широко применяются тривиальные названия.
Метильные производные фенола называют крезолами, а 2-изо-пропил-5-метилфенол называют тимолом.
Для некоторых фенолов чаще используют тривиальные названия (ксилол, тимол и т. д.). Для названия гомологов фенола в качестве основы используют слово фенол.
о-крезол, 2-метил фенол
л/-крезол, 3-метил фенол
4-метил фенол
тимол
19 Ф^и.'.ы
213
Способы получения
1. Природные источники. Фенол получают при сухой перегонке каменноугольной смолы. Вторая фракция с /кип 150—230 °C содержит фенол. Впервые фенол был получен таким способом Ф. Рунге в 1834 г.
2. При нагревании галогенопроизводных бензола со щелочами в присутствии катализатора:
8% NaOH. Си
300 °C. 200 атм
хлорбензол
3. Сплавление натриевых солей сульфокислот со щелочами:
NaOH сплавл.
бензолсульфо-кислота
NaOH сплавл.
натриевая соль фенолят натрия,
бензолсульфокислоты феноксид натрия
4. Разложение солей диазония:
бензолдиазония
хлорид
НОН. г
5. Окисление кумола (кумольный способ). Это основной промышленный способ получения фенола:
фенол
ацетон
214
Лекции по органической химии
Способ является экономически выгодным, так как позволяет кроме фенола получить и дру1 ой важный продукт — ацетон.
Химические свойства
Для фенолов характерны два направления реакций: реакции по фенольному гидроксилу и по ароматическому ядру.
В молекуле фенола имеет место сопряжение неподеленной пары электронов атома кислорода гидроксильной группы с л-электронами бензольного ядра. Это приводит к довольно сильной поляризации связи О<— Н, что объясняет более выраженный кислотный характер фенолов в сравнении со спиртами. Атомы водорода гидроксильной группы приобретают подвижность и довольно легко замещаются на другие атомы или атомные группы.
С другой стороны, наличие положительного мезомерного эффекта приводит к укорачиванию связи С—ОН, что делает практически невозможными реакции замещения фенольного гидроксила.
Реакции с участием группы ОН
1. Кислотные свойства. Фенолы обладают кислотным характером, они способны образовывать соли уже при действии щелочей:
ОН + NaOH
Н2О +
фенолят натрия
В водном растворе феноляты подвергаются частичному гидролизу, т. к. образованы слабой кислотой и сильным основанием. По этой причине их растворы имеют щелочную реакцию. Минеральные кислоты, в том числе и угольная, вытесняют фенолы из их солей.
На кислотность фенола оказывают влияние заместители в ароматическом ядре. При наличии в «-положении электроноакцепторных заместителей (-NO2, -Вг, -Cl, -CN и др.) кислотные свойства усиливаются. Если в «-положение ввести электронодонорные заместители (-NH2, -ОСН3 и др.), то происходит снижение кислотных свойств, поскольку уменьшается смещение электронов связи О—Н к атому кислорода, что затрудняет отрыв протона:
19. Фенолы
215
2. Образование простых и сложных эфиров. Простые эфиры фенола непосредственно из фенола получить не удается. Для их получения используют реакцию Вильямсона: взаимодействие феноксида натрия с галоидными алкилами или арилами.
о—сн-сн3
фенилэтиловый эфир (фенетол)
При взаимодействии фенолятов с галогенаренами образуются ароматические простые эфиры:
дифениловый эфир
Дифениловый эфир имеет запах герани.
Ароматические простые эфиры имеют схожие химические свойства с простыми эфирами алифатического ряда. Они не взаимодействуют со щелочами, разложить их можно только йодоводородной кислотой.
Взаимодействие фенолятов с галогенангидридами карбоновых кислот приводит к образованию сложных эфиров:
/О сн—
С1
—NaCl
фенилацетат
ацетилхлорид
Аналогичные продукты также получают по реакции ацилирования самих фенолов с ангидридами карбоновых кислот:
-сн,соон
уксусный ангидрид
Такая реакция называется О-ацилированием.
В отличие от спиртов фенолы не этерифицируются непосредственно карбоновыми кислотами из-за недостаточной электронной плотности на атоме кислорода.
216
Лекции по органической химии
J. Взаимодействие фенолята натрия с хлоруксусной кислотой. Примером реакции О-алкилирования является реакция получения фенок-сиуксусной кислоты:
ONa+ClCH2COOH
О—СН—COOH+NaCl
феноксиуксусная кислота
Феноксикислоты являются твердыми полупродуктами с четкой температурой плавления и служат для идентификации фенолов. Феноксиуксусная кислота имеет большое значение в синтезе фармацевтического препарата феноксипенициллина. Пенициллин — это природный антибиотик, который образуется в результате размножения плесени. В настоящее время пенициллин получают в медицинской промышленности в больших чанах, в которых произрастает плесень. Однако природу пенициллина можно изменять. Если в питательную среду плесени добавить феноксиуксусную кислоту, получают феноксиметил-пенициллин, который более устойчив, чем обычный пенициллин.
Аналогично получают 2,4-дихлорфеноксиуксусную кислоту — стимулятор роста растений.
Реакции по ароматическому ядру
Гидроксильная группа является одним из самых сильных доноров электронов (+М), она значительно повышает электронную плотность в бензольном ядре и тем самым увеличивает его реакционную способность в реакциях SE.
1. Бромирование. При взаимодействии фенола с бромной водой в отсутствии катализатора в молекулу вводится три атома брома и образуется 2,4,6-трибромфенол:
ОН
ЗВг,
2,4,6-трибромфенол
+ ЗНВг
Эта реакция используется для качественного и количественного определения фенола.
19 Фенолы
217
Для введения одного или двух атомов брома в бензольное кольцо реакцию бромирования проводят в низкополярных растворителях (СС14, СНС13):
ОН
2Вг2, СС14
-2НВг
л-бромфенол (преимущественный продукт)
2. Нитрование. Если бензол нитруется только нитрующей смесью, то фенол можно нитровать разбавленной азотной кислотой при комнатной температуре:
ОН
2HNO3 разб., 25 °C
—2Н2О
л-нитрофенол
В этих условиях в молекулу фенола вводится одна нитрогруппа. Полученную смесь нитрофенолов можно разделить перегонкой с водяным паром. В связи с тем, что в молекуле о-нитрофенола гидроксильная и нитрогруппа находятся рядом, между атомом водорода ОН-груп-пы и атомом кислорода МО2-группы образуется внутримолекулярная водородная связь (ВВС):
неассоциированный о-нитрофенол
Подобные соединения легколетучи. Как видно, образуется 6-членный цикл за счет хелатной водородной связи (внутримолекулярная водородная связь называется хелатной).
Молекулы лоро-изомера образуют межмолекулярные водородные связи (МВС):
218
Лекции по органической химии
ассоциированный «-нитрофенол
Для п- и о-нитрофенолов характерна бензоидно-хиноидная таутомерия. А/е/ио-нитрофенолы не являются таутомерными вешествами, так как в них нет сопряжения между ОН- и NOj-группами.
бензоидная форма «-нитрофенола
хиноидная форма «-нитрофенола
Свободные нитрофенолы — бесцветные вещества или имеют желтую окраску. При добавлении щелочей образуются соли нитрофенолов, окрашенные в ярко-желтый цвет.
При действии концентрированной азотной кислоты на фенол образуется осадок 2,4 6-тринитрофенола:
ОН
3HNO3 конц.
-ЗН2О
2,4,6-тринитрофенол, пикриновая кислота
2,4,6-Тринитрофенол называется пикриновой кислотой, по кислотности он приближается к серной кислоте.
3. Сульфирование. Сульфирование фенола проходит очень легко и в зависимости от температуры приводит к о- или и-изомерам:
19. Фенолы
219
о-гидроксибензол-сульфоки слота
л-гидроксибензол-сул ьфоки слота
SO3H
4. Алкилирование и ацилирование. Реакции алкилирования и ацилирования по бензольному кольцу (С-алкилирование и С-ацилиро-вание фенолов) проводят в условиях реакции Фриделя — Крафтса:
о-метилфенол л-метилфенол
4-гидроксиаиетофенон
5. Азосочетание. Фенолы в качестве азосоставляющей в слабощелочной среде вступают в реакцию азосочетания с солями диазония с образованием азосоединений:
4'гидроксиазобензол
220
Лекции по органической химии
6. Синтез фенолкарбоновых кислот (реакция Кольбе — Шмитта). При пропускании углекислого газа через раствор фенолята натрия образуется салицилат натрия, который при действии минеральных кислот образует салициловую кислоту.
салицилат натрия
салициловая кислота
7. Гидроксиметилирование. При обработке фенолов формальдегидом в кислой или щелочной среде образуется смесь о- и и-гидрокси-метилфенолов. Реакция протекает по механизму SE, электрофильной частицей в условиях кис лотного катализа является гидроксиметил-катион СН2ОН:
формальдегид
* СН2ОН
гидроксиметилкатион
2-гидрокси-метилфенол
сн2он 4-гидрокси-метил фенол
В присутствии минеральных кислот гидроксиметилфенол вступает в реакцию конденсации с молекулой фенола:
19. Фенолы
221
2.2’-ди гидроксиди фенилметан
В более жестких условиях образуются высокомолекулярные продукты поликонденсации — фенолоформальдегидные смолы.
CH2-jOH + Hi • «
он
СН2ОН
-н,о
В результате такой реакции поликонденсации могут образовываться смолы линейного или сетчатого строения.
Реакции восстановления и окисления
При каталитическом гидрировании (восстановлении) фенола образуется циклогексанол:
/Сн2
Н,С 'СН-ОН
2| I
Н7с. /СН?
СН2
ииклогексанол
Циклогексанол является исходным продуктом в синтезе капрона и нейлона. Цинковая пыль может восстановить фенол даже до циклогексана:
С6Н5ОН + Zn ------- ZnO + Сбн12
Фенолы легко окисляются даже кислородом воздуха. Наличие примесей приводит к ускорению окисления. Окисленный фенол краснорозового цвета, очищенный — бесцветный.
ОН
СгО3, Н
222
Лекции т органической химии
Фенолы, в зависимости от природы окислителя, могут образовывать разные продукты.
В щелочной среде фенолы окисляются в большей степени, чем в нейтральной, это связано с образованием феноксид-иона. В результате сопряжения увеличивается электронная плотность на бензольном ядре, и оно становится еще более чувствительным к действию окислителей.
При окислении фенола персульфатом калия K2S2O8 в щелочной среде образуется гидрохинон (реакция Эльбса):
ОН
K2S2O8, ОМ-
ОН
ОН
гидрохинон
ДВУХ- И ТРЕХАТОМНЫЕ ФЕНОЛЫ
Двухатомные фенолы содержат в своей структуре две группы ОН,
трехатомные — три группы ОН.
ОН
гидрохинон, 1,4-ди гидрокс ибе н зол
пирокатехин, 1,2-ди гидроксибензол
ОН
пирогаллол, 1,2,3-тригидроксибензол
флороглюцин, 1,3,5-тригидроксибензол
оксигидрохинон, 1,2,4-тригидроксибензол
Способы получения
Двухатомные фенолы получают теми же способами, что и одноатомные, но используют для этой цели дизамещенные производные бензола.
1. Сплавление натриевых солей арендисульфокислот со щелочами:
л-бензолдисульфо- резорцин
кислота
19. Фенолы
223
Этим способом чаше всего получают резорцин.
2. Взаимодействие галогенозамещенных фенолов или дигалогенпро-изводных бензола со щелочами:
.и-бензолдисульфо- пирокагехин
кислота
Эта реакция применяется для получения пирокатехина.
3. Восстановление хинонов:
хинон
гидрохинон
Химические свойства
Для многоатомных фенолов характерны те же реакции, что и для одноатомных.
Они могут галогенироваться, нитроваться, сульфироваться.
С увеличением числа ОН-групп в молекуле усиливаются кислотные свойства по сравнению с фенолом. Но несмотря на это, кислотные свойства двух- и трехатомных фенолов остаются слабее по сравнению с угольной кислотой.
Двухатомные фенолы образуют соли не только со щелочами, но и с тяжелыми металлами:
Q ОН
ОН
(CHJtOO)2Pb
—2СН3СООН
При окислении пирокатехина и гидрохинона образуются соответствующие о- и л-хиноны.
Важнейшие представители многоатомных фенолов
ГИДРОХИНОН (л-дигидроксибензол). Является сильным восстановителем, очень легко окисляется, в том числе и раствором FeClr
гидрохинон
л-бензохинон
224
Лекции по органической химии
В результате окисления образуется и-хинон.
Гидрохинон применяется как проявитель в фотографии. Встречается в природе в виде гликозида арбутина, который содержится в растении толокнянка.
ПИРОКАТЕХИН (о-дигидроксибензол) легко окисляется, являясь хорошим восстановителем. Восстанавливает аммиачный раствор Ag2O, при нагревании восстанавливает раствор Фелинга:
о-бензохинон
Монометиловый эфир пирокатехина — гваякол применяется в синтезе ванилина (4-гидрокси-З-метоксибензальдегида), ароматических и лекарственных веществ.
РЕЗОРЦИН (jw-дигидроксибензол). Кристаллическое вещество, хорошо растворяется в воде, Гп1110 °C.
Применяется в аналитической практике в растворе НО как реактив на кетозы (проба Селиванова). Применяется в производстве красителей и резорцинформальдегидных смол. Является медицинским препара-
том, используется в мазях как антисептическое средство.
ОН
НО
ПИРОГАЛЛОЛ (1,2,3-тригидроксибензол) (от слова «пирос» — огонь). Белое кристаллическое вещество с rnj] 134 °C, растворяется в воде и спиртах.
Получают при декарбоксилировании галловой кислоты:
ОН
Пирогаллол является сильным восстановителем Серебряные соли пирогаллола легко восстанавливаются до металлического серебра, поэтому он используется в фотографии. Щелочные растворы пиро-
19. Фенолы
225
галлола хорошо поглотают кислород, что используется для количественного определения его содержания в газовой смеси.
ФЛОРОГЛЮЦИН (1,3,5-тригидроксибензол). Кристаллическое вещество с г 223 °C, возгоняется, растворимое в спиртах и плохо — в воде.
Получают при нагревании водных растворов 1,3,5-триаминобен-зола в кислой среде.
Для флороглюцина возможна кето-енольная таутомерия:
никлогексантрион-1,3,5
Наличие гидроксильных групп доказывают реакцией с FeCl3 (образуется темно-фиолетовое окрашивание). Наличие кетогрупп подтверждают реакцией с NH,OH с образованием триоксима.
Флороглюцин используется в аналитической практике для количественного определения пентоз. При нагревании пентоз образуется фурфурол, который сфлороглюциномдаетокрашенный продукт конденсации.
АМИНОФЕНОЛЫ
Аминофенолами называют производные ароматических углеводородов, содержащие в своем составе фенольный гидроксил и аминогруппу.
По взаимному расположению —ОН и — NH2-rpynn в аминофенолах возможны три изомера:
226
Лекции по органической химии
Аминофенолы можно получить из соответствующих нитрофенолов путем восстановления, при взаимодействии двухатомных фенолов с аммиаком или при электролитическом восстановлении нитробензола в присутствии кони. H2SO4 (промышленный способ).
Химические свойства аминофенолов обусловлены наличием двух функциональных групп — фенольного гидроксила и ароматической аминогруппы.
Аминофенолы проявляют как основные свойства (за счет NH,-группы), так и кислотные свойства (за счет ОН-группы). и в целом являются амфотерными соединениями.
л-гидроксианилиний хлорид
л-аминофенол
л-аминофенол ят натрия
Аминофенолы вступают в реакцию ацилирования, причем реакция происходит по NH,-группе как более нуклеофильному центру.
+ (СН3СО)2О _СНзСООН
NHCOCH
ОН
л-аминофенол
л-аиетиламинофенол, парацетамол
и-Ацетиламинофенол, известный как лекарственный препарат парацетамол, применяется как жаропонижающее и болеутоляющее средство.
Метиловый эфир (анизидин) и этиловый эфир л-аминофенола (фенетидин) используются в синтезе лекарственных препаратов и красителей.
метиловый эфир л-аминофенола, л-аниэидин
H2N
ОС2Н5
этиловый эфир л-ами нофенола, л-фенетидин
19 Фенолы
227
Лекарственный препарат фенацетин является ацетильным производным и-фенетидина и находит применение как жаропонижающее и антиневрологическое средство.
С2Н5О
NH-С—СН3
О фенацетин
п- и о-Аминофенолы легко окисляются с образованием хинони-минов.
и-аминофенол
л-хинонимин о-аминофенол
о-хинонимин
Восстанавливающая способность аминофенолов используется в фотографии.
АЛЬДЕГИДЫ И КЕТОНЫ
Альдегидами и кетонами называют производные углеводородов.
содержащие в своем составе карбонильную группу С—О. Поэтому их еще называют карбонильными соединениями.
В альдегидах карбонильная группа связана с углеводородным ра-дикалом и атомом водорода. Общая формула альдегидов R—Сх , Н
группировка —Сх получила название альдегидная группа. Н
В кетонах карбонильная группа связана с двумя углеводородными радикалами.
Общая формула кетонов
. Карбонильную группу в кето
нах часто называют кетогруппой.
В зависимости от строения углеводородного радикала альдегиды и кетоны подразделяют на алифатические, алициклические и ароматические. Среди алифатических альдегидов и кетонов различают насыщенные и ненасыщенные (предельные и непредельные).
20. АЛЬДЕГИДЫ И КЕТОНЫ АЛИФАТИЧЕСКОГО РЯДА
НАСЫЩЕННЫЕ АЛЬДЕГИДЫ II КЕТОНЫ
Номенклатура альдегидов и кетонов весьма многообразна. Используют как тривиальные названия, так и номенклатуру ИЮПАК. Поэтому многие карбонильные соединения имеют несколько названий.
Тривиальные названия альдегидов происходя г от названия кислот, в которые они превращаются при окислении. По заместительной номенклатуре ИЮПАК названия альдегидов образуют от названия углеводородов с тем же числом атомов углерода (включая углерод альдегидной группы), прибавляя суффикс -аль Нумерацию главной углеродной цепи начинают с атома углерода альдегидной группы.
20. АлъОегиоы и кетоны алифатическо. > ряда
229
о
н-с
н
муравьиный альдегид, формальдегид,
метаналь
О н3с-с н уксусный альдегид, ацетальдегид, этаналь
н3с—сн—сн—с
3 2 2 \
н
масляный альдегид, бутаналь
4 3 2
снгсн-сн,-с
сн3 'н
изовалериановый альдегид, 3-метилбутанал ь
О
Нередко в названиях альдегидов положения заместителей указывают греческими буквами а, р, у и т. д. При этом буквой а обозначают атом углерода, соединенный с альдегидной группой:
О
у Р « ,р сн,-сн,-сн-с
I 4 сн3 н а-метилмасляный альдегид
8 Y Р а
н,с-сн-сн,-сн,-с 3 I z z \
ОН Н
у-гидроксивалериановый альдегид
Кетонную группу по заместительной номенклатуре обозначают суффиксом -он и цифрой обозначают атом углерода, входящий в кетогруппу. Нумерацию проводят таким образом, чтобы атом углерода карбонильной группы получил меньший номер:
5 4 3 2 1
Н.С—CH-CH—С—СН, 3 I 2 II 3 сн3 о
1 2 3 4 5 6 7
сн,-сн,-с-сн,-сн,-сн,-сн3
_* L 11 Z L L J
О
4-метилпентанон-2
гептанон-З
По радикало-функциональной номенклатуре названия кетонов состоят из названия углеводородных остатков в порядке алфавита и суффикса -кетон.
СН'-С-СН'-СН, 3 II 23
о
метилфенилкетон
метилэтилкетон
Способы получения
/. Реакция окисления спиртов. Первичные спирты окисляются до альдегидов, а вторичные — до кетонов.
230
Лекции по органической химии
° ''
н3с—сн-он Ti^*H3C-C
этанол этаналь Н
н3с
сн-он
н3с
пропанол-2
[°]. н3с\ хсн3
-н2о V
О
пропанон
2. Реакция гидратации алкинов (реакция Кучерова). В условиях реакции Кучерова из ацетилена образуется уксусный альдегид, все гомологи ацетилена дают кетоны.
О
НС=СН -нон' Hg2\ н3с-с
3 \
ацетилен Н
этаналь
НОН, Hg2+
н3с-сн2-сесн------------► н3с-сн7с-сн3
о
бутин-1 бутанон
3. Реакция гидролиза дигалогеналканов. При гидролизе геминальных дигалогеналканов с атомами галогена у первичного атома углерода образуются альдегиды, а у вторичного — кетоны:
/ НОН, ОН- '/
Н3С-СН,-СН ---------------* Н3С-СН,-С + 2НС1
3 2 \ 3 2 \
С1 Н
1,1-дихлорпропан пропаналь
НОН ПН'
н,с-с-сн3 -----------сн3-с-сн3 + 2НС1
з z \ з J и J
Cl Cl О
2,2-дихлорпропан пропанон
4. Реакция термического разложения солей карбоновых кислот. Из смешанной соли муравьиной и другой карбоновой кислоты при термическом разложении (пиролизе) получают альдегиды, а в остальных случаях образуются кетоны:
/6 "'х О о
v 11 \ II >t 'г
zCa * СН-С + СаСО,
Н3С '„О Н хн
кальциевая соль уксусной и муравьиной этаналь
кислот
20. А.и оегиды и кетоны алифатического ряоа
231
X zCa \сч нс'С'сн
Нзс ^0 „О СНз ' '
СаСО3
кальциевая соль уксусной кислоты
пропанон
5. Реакция прямого карбонилирования (оксосинтез). В промышленности альдегиды получают взаимодействием алкенов с оксидом углерода (II) и водородом при повышенной температуре и давлении в присутствии катализатора.
zO нзС ,0
2Н3С-НС=СН2 +2СО+ 2Н2----------- HjC-CHjCHjC' + 3 СН-с'
бутаналь Н Н,С Н
2-метилпропаналь
Физические свойства
Насыщенные альдегиды и кетоны являются бесцветными жидкостями со своеобразным запахом (формальдегид — газ с острым запахом). Карбонильные соединения имеют более низкие температуры кипения, чем соответствующие спирты, т. к. не способны образовывать водородные связи.
Кетоны являются хорошими растворителями. Высшие альдегиды обладают цветочным запахом и широко применяются в парфюмерии.
Альдегиды раздражают слизистые оболочки глаз и верхних дыхательных путей, пагубно влияют на нервную систему. С увеличением числа атомов углерода в молекуле раздражающее действие ослабевает. Ненасыщенные альдегиды обладают более сильным раздражающим действием, чем насыщенные.
Химические свойства
Химические свойства альдегидов и кетонов определяются наличием в их молекуле карбонильной группы, которая является сильно полярной группой и имеет значительную поляризуемость.
Карбонильная группа образована из двух атомов с сильно различающимися электроотрицательностями. Атом углерода карбонильной группы находится в состоянии ^-гибридизации и связан с тремя окружающими его атомами о-связями, расположенными в одной плоскости под углом 120°. Негибридизованная р-орбиталь атома углерода перекрывается с р-орбиталью атома кислорода, образуя л-связь.
Атом кислорода, как более электроотрицательный элемент, притягивает к себе о- и л-электроны. В результате этого двойная связь карбонильной группы сильно поляризована, на атоме кислорода возни
232
Лекции по органической химии
кает частичный отрицательный заряд, а на атоме углерода — частичный положительный:
—с
Благодаря такой поляризации альдегиды и кетоны способны вступать в реакцию с нуклеофильными реагентами, которые атакуют атом углерода карбонильной группы. Альдегиды, как правило, более реакционноспособны, чем кетоны. Поскольку алкильные радикалы за счет +/-эффекта уменьшают положительный заряд на атоме углерода карбонильной группы, наличие в молекуле кетона двух алкильных групп приводит к большему понижению положительного заряда, чем в молекуле альдегида.
Все реакции альдегидов и кетонов условно можно разделить на следующие группы:
• реакции нуклеофильного присоединения (AN);
• реакции присоединения-отщепления;
• реакция конденсации;
• реакция с участием а-углеродного атома;
• реакции полимеризации;
• реакции окисления и восстановления.
Реакции нуклеофильного присоединения
1. Присоединение синильной кислоты. Синильная (циановодородная) кислота присоединяется к карбонильных- соединениям, образуя циангидрины или а-гидроксинитрилы:
°5" Х+ я 5+ // &-
сн-с + Н-(=\ 3 \
н
он I
> H3C-CH-C=N
н итрил а- гидрокс и -пропионовой кислоты
Кетоны также образуют а-гидроксинитрилы:
СН.-С + H~C=N 3 \
сн3
пропанон
ОН I
—H3C-C-C=N
СН3
нитрил а-мети.та-гидрокси-пропионовой киелоть'
Образующиеся нитрилы можно легко гидролизовать до соответственных а-гидроксикислот, что используется в синтезе этих кислот.
20. Альдегиды и кетоны алифатического ряда 4м
2. Присоединение гидросульфита натрия. Альдегиды и кетоны (содержащие группировку Н,С—СО—R) реагируют с гидросульфитом (бисульфитом) натрия, образуя бисульфитные соединения.
л
СНгС + :S-ОН
3 \ I
Н О Na+
ОН I
- Н3С-С—SO3Na+
Н
бисульфитное соединение уксусного альдегида
Бисульфитные соединения — твердые вещества, трудно растворимые в воде, легко образуются, поэтому применяются для качественного обнаружения альдегидов, а также для выделения альдегидов из реакционной среды.
3. Присоединение воды. Растворение альдегидов в воде сопровождается образованием гидратов. Реакция представляет собой обратимый процесс. Продукты реакции гидратации обычно неустойчивы и выделить их практически невозможно из-за разложения на исходные компоненты:
/? ?Н
сн3-с + нон И с-с-он
3 \ 3 I
Н н
гидрат уксусного альдегида (гем-диол)
4. Присоединение спиртов. В спиртовом растворе альдегиды образуют полуацетали, а в присутствии следов минеральных кислот — ацетали:
// C2HSOH, Н+ ^С2Н5
СН3С +С2Н5ОН^:Н3С-С-ОС2Н5 < -- Н3С-С-ОС2Н5+Н2О
НН н
полуацеталь ацеталь
Кетоны из-за низкой реакционной способности и пространственных препятствий со спиртами не взаимодействуют.
Реакции присоединения-отщепления
Альдегиды и кетоны взаимодействуют с азотистыми основаниями с образованием неустойчивых продуктов нуклеофильного присоединения, которые стабилизируются благодаря отщеплению молекулы
234
Лекции по органической химии
воды. Эта группа реакций получила название реакций присоединения-отщепления.
1. Взаимодействие с аммиаком. Альдегиды, присоединяя молекулу аммиака, образуют альдимины. В процессе реакции в начале образуется неустойчивый аминоспирт, от которого затем отщепляется вода.
Ж----.
снз-с^ + :NH3^=^ н
он
I h3c-c-nh2^=^h3c-ch=nh + Н2О |4 этанимин,
ацетальдимин
Кетоны также взаимодействуют с аммиаком, но при этом образуются продукты более сложного строения.
2. Взаимодействие с аминами. Первичные амины, реагируя с альдегидами или кетонами дают имины. Если реакция протекает с ароматическими аминами, то образующиеся имины называют также основаниями Шиффа.
О //
сн3-с + H2N-C2H5^=^H3C-CH=N-C2H5+Hi_O н
3. Взаимодействие с гидроксиламином. Продукты конденсации альдегидов и кетонов с гидроксиламином называют альдоксимами и ке-токсимами.
О // снз-с + H,N—ОН
3 \ 2 н
H3C-CH-N—ОН + К0 оксим ацетальдегида
н3с-с—сн3 + h2n-oh н3с—с—сн3 *
о N-OH
оксим ацетона
Реакцию образования оксимов используют для выделения и идентификации альдегидов и кетонов.
4. Взаимодействие с гидразином. Подобно взаимодействию с первичными аминами протекают реакции оксосоединений с гидразином, фенилгидразином, семикарбазидом, тиосемикарбазидом.
20 Алъоегиоы и кетоны алифатического ряда
235
R
4c=n-nh;
,/ 2
гидразон
H,N—NH2 HjN—NH—C—NH2
гидразин семикарбазид
R \
C=N-NH-C-NH, / И
R' О
семи карбазон
R
'c=N-NH-C6Hs
R фенилгидразон
H,N-NH-CtH, фенил гидразин
-Н20
h2n-nh-c—nh2 тиосемикарбазид
-Н,0
R
\
C=N-NH-C-NH,
/ У
R’ S
тиосем и карбазон
Образующиеся производные оксосоединений обычно представляют собой кристаллические вещества с четкими температурами плавления. Эти реакции используются для идентификации исходных оксосоединений.
Реакции конденсации
1. Альдольная конденсация. Альдегиды и кетоны, содержащие атомы водорода у а-углеродного атома, в присутствии каталитических количеств основания способны вступать в реакцию самоконденсации, называемую альдольной конденсацией. При этом молекулы альдегидов соединяются друг с другом посредством связей С—С и образуют альдоль — р-гидроксиальдегид:
-Р ОН- /Р
2Н С-С ------------ ILC-CH-CH-C
J \ J I L \
Н ОН н
3-гидроксибутаналь,
Р-оксимасляный альдегид,
альдоль
Рассмотрим более подробно механизм реакции в условиях основного катализа. В слабощелочной среде гидроксид-ион (катализатор) отщепляет протон от cc-углеродного атома альдегида с образованием енолят-аниона:
Н О
\ /
енолят-анион
236
Лекции по органической химии
На следующей стадии сильный нуклеофил — енолят-анион атакует электронодефицитный атом углерода карбонильной группы второй молекулы альдегида с образованием альдоля:
&-я Со о
сн,-с + сн,—с
и н
он <
I //
=₽=*= н.с—с-сн,-с
I 2 \
+ ОН’
альдоль
При нагревании Р-оксиальдегиды легко теряют воду, превращаясь в ос, Р-ненасышенные альдегиды.
°н ° 0
Н3с—с-сн,-с * н,С-НС=СН—Q + Н2о
I ы 4
н н н
кротоновый альдегид,
бутен-2-аль
Легкость, с которой происходит отщепление воды, объясняется подвижностью а-водоролного атома и образованием сопряженной системы двойных связей. Реакция образования ненасыщенного альдегида из альдоля известна в органической химии под названием кротоновая конденсация.
2. Сложноэфирная конденсация (реакция Тищенко). При нагревании альдегидов в присутствии этилата алюминия образуются сложные эфиры карбоновых кислот:
/Я (С,Н,О),А1
2Н3С-С ---------
Н
Н3С~С
Ь-сн,-сн3
этилацетат
В этой реакции одна молекула альдегида восстанавливается до спирта, а вторая — окисляется до кислоты Такая реакция самоокисления-самовосстановления, получила название диспропорционирования или дисмутации.
20 Алъсегиоы и кетоны алифатического ряда 2-5 z
Реакции с участием а-углеродного атома
Электроноакцепторное влияние карбонильной группы приводит к повышению подвижности атомов водорода, находящихся при o'-углеродном атоме (СН-кислоты):
К числу реакций, протекающих с участием а-углеродного атома, относится реакция галогенирования и рассмотренная ранее альдольная конденсация.
Реакция галогенирования. Альдегиды и кетоны легко вступают в реакции с галогенами с образованием а-галогенпроизводных:
н.с-сн,—С ------
\т -НВг
пропионовый альдегид
н.с-сн—с
L и
а-бромпропио-
новый альдегид
а-Галогенопроизводные альдегидов и кетонов проявляют слезоточивое действие и называются лакриматорами (от лат. lacrima — слеза).
При галогенировании соединений типа (СН,—СО-) в щелочной среде образуются тригалогенкарбонильные соединения, которые затем расщепляются с образованием карбоксильного аниона и тригало-геналкана (CHHal,). Например, в случае йодирования в щелочной среде происходит выделение желтых кристаллов йодоформа:
1„ он-
R-C-CH, -------- R-C-CL
II 3 II 3
О О
R-C + СН1,| О йодоформ
Реакция образования йодоформа (йодоформная проба) используется в аналитической практике.
Реакции восстановления и окисления
1. Реакции восстановления. Реакцию восстановления альдегидов и кетонов широко используют для получения спиртов. Карбонильные соединения восстанавливаются как при каталитическом гидрировании, так при действии таких восстановителей, как алюмогидрид лития LiAlH4. Альдегиды восстанавливаются до первичных спиртов:
238
Лекции по органической химии
О
// Н2
н3с-сн-с - - н3с-сн—сн2—он н
Кетоны восстанавливаются до вторичных спиртов:
Н.С—С—СН,
3 II
О пропанон, ацетон
LiAlH ---- н.с-сн-сн, I он пропанол-2
2. Реакции окисления. Альдегиды и кетоны по разному относятся к действию окислителей.
Альдегиды очень легко окисляются, даже при действии слабых окислителей (ионы Ag4. Cu:*).
2.1. Реакция «серебряного зеркала». При действии аммиачного раствора нитрата серебра (реактив Толлеса) на альдегид происходит окислительно-восстановительная реакция. Альдегид окисляется в соответствующую кислоту, а катион серебра восстанавливается в металлическое серебро, которое дает блестящий налет на стенках пробирки.
О
//
CH3-C +2[Ag(NH,)2]OH
Н
О
I Ч
2Ag| + СН—С + Н2О +3NH3
О" NH4+
Эта реакция является качественной для обнаружения альдегидной группы.
2.2. Реакции взаимодействия альдегидов с медно-виннокислым комплексом. Альдегиды также восстанавливаются реактивом Фелинга (смесь раствора сульфата меди со щелочным раствором натриикалие-вой соли виннокаменной кислоты):
CuSO4 + 2NaOH
Cu(OH)2 + Na2SO4
COOK I
2 HC-OH Cu(OH)2
HC-OH I
COONa
COO\ NaOOC I X ।
HC-OH X HQ-CH I *Z----Cu--:: I
HC-OH \HO-CH I \ I
COOK ooc
сегнетова соль
медно-виннокиелый комплекс
20 Хъдегиды и купоны а.-ифатическчси ряда
239
COQ NaOOC
I X, 1
нс-онХ но—сн
2 I _----Cu- = :. I нс-он \HO-CH
I X I
COOK ООС
/?
CFL-C
3 \
Н
он
COOK
он- // НС-ОН
СНГ С + 2СиОН + 2 Н2О + 2 I
---► 3 \ nC_Un
° Na+ COONa
2CuOH —'—* Cu2O| + Н2О
красно-коричневый
Кетоны к действию этих окислителей инертны. Не окисляются они и кислородом воздуха. Только при действии более сильных окислителей кетоны удается окислить. При этом происходит разрыв углеродуглеродной связи между атомом углерода карбонильной группы и углеводородного радикала и образуется смесь кислот.
Реакции полимеризации
Альдегиды, в отличие от кетонов, способны полимеризоваться. Реакция полимеризации протекает в обычных условиях и ускоряется в присутствии минеральных кислот. При хранении 40 % водного раствора формальдегида (формалин), особенно при температуре ниже 9 °C, наблюдается выпадение белого осадка продукта линейной полимеризации (параформа): О
п Н-С + Н2О Н
но-сн2-1 осн
параформ
Уксусный альдегид в присутствии следов серной кислоты приводит к образованию, в зависимости от условий, двух циклических продуктов — паральдегида и метальдегида:
Н,С^ /°х JZH,
3 СН НС 3
л=3
г-20 "С
о nH3C-CZ н
л=4
г=0“С
С
I паральдегид
Н,С СНз сн-о о I хн н3с -
нс/СНз
Н/° о-с метальдегид "сн3
240
Лекции по органической химии
Паральдегид — жидкость, метальдегид — твердое вещество, используется в быту как сухое горючее под названием «сухой спирт».
НЕПРЕДЕЛЬНЫЕ АЛЬДЕГИДЫ
В непредельных альдегидах карбонильная группа связана с углеводородным радикалом, содержащим кратную связь. Положение двойной связи в углеводородном радикале обозначают греческими буквами. В соответствии с этим различают а, р- или р, у- и т. д. непредельные альдегиды:
7 ₽ а
н,с—сн=сн- erf н
а,р- непредельный альдегид
ура
Н3С-СН=СН—СН—dj
Р.у- непредельный альдегид
Наибольший интерес представляют а, p-непредельные альдегиды, у которых двойная связь сопряжена с карбонильной группой:
Л) н,с=сн—erf н акролеин, пропен-2-аль
Л) н,с—сн=сн—с' н кротоновый альдегид, бутен-2-аль
Для непредельных альдегидов характерны реакции карбонильных соединений, а также ненасыщенных углеводородов.
Альдегиды, содержащие в своей структуре сопряженную систему, имеют ряд особенностей, отличающих их от остальных альдегидов. Присоединение галогеноводородов и воды протекает против правила Марковникова.
Л) н,с=сн—-н акролеин, пропен-2-аль
НВг
сн—сн=с
Вг
^он
енол
-О сн,—сн,—с^ | 2 2 и
Вг
3-бромпропаналь
Непредельные альдегиды используются в синтезе ряда соединений, в том числе и лекарственных препаратов.
21. АЛЬДЕГИДЫ И КЕТОНЫ АРОМАТИЧЕСКОГО РЯДА
АРОМАТИЧЕСКИЕ АЛЬДЕГИДЫ
Ароматические альдегиды — это производные ароматических углеводородов. которые содержат в своей структуре альдегидную группу:
Различают две группы этих соединений: альдегиды, содержащие альдегидную группу в бензольном ядре и альдегиды, у которых альдегидная группа содержится в боковой цепи.
Простейшие представители:
Альдегиды, содержащие альдегидную группу в боковой цепи, называют как производные альдегидов жирного ряда:
фенилуксусный альдегид
Способы получения ароматических альдегидов
Ароматические альдегиды можно получить теми же способами, что и жирные.
1. Окисление спиртов. Как все первичные спирты, бензиловый спирт легко окисляется с образованием бензальдегида:
242
Лекции по органической химии
[°]
С.Н,СН,ОН * 6 3 - -н,о
бензиловый
спирт
В качестве окислителей чаще всего используют оксид хрома (VI), хромовую смесь и др.
2. Перегонка кальциевых солей (соль ароматической кислоты и муравьиной). При сухой перегонке смешанных кальциевых солей ароматических кислот и муравьиной кислоты легко образуются ароматические альдегиды:
/О сн,-с< 6 5 сх /Са /О Н-(Х о
/°
СаСО3 + Н5С—
Н
3. Издигалоидозамещенных — п) тем омыления. Гидролиз геминальных галогенаренов, содержащих атомы галогена у первичных атомов углерода (в боковой цепи), приводит к получению ароматических альдегидов:
/С1 м-
СН—СН
5 ХС1 Н>°
хлористый бензилиден
Но есть и специфические способы получения ароматических альдегидов, не характерные для жирного ряда.
Окисление ароматических углеводородов.
При окислении толуола и других соединений, содержащих метильную группу, связанную с бензольным ядром, довольно легко образуются ароматические альдегиды. При использовании в качестве окислителя оксида хрома (VI) реакцию проводят в среде уксусного ангидрида:
СН
СТО, (СН3СО)2О
о 11 хо-с-сн3 нс^о-с-сн
II
О ___
3
Н2О; Н+
-СН3СООН
гем-диацетат
21 Альдегиды и кетоны ароматического ряда 446
Образовавшееся диацетатное производное толуола не способно к окислению, а при его гидролизе легко образуется бензальдегид.
Способ Гаттермана — Коха (реакция карбонилирования).
Прямое введение в молекулу альдегидной группы позволяет получать замешенные ароматические альдегиды:
СН3
о н
и-толуиловый альдегид
Физические свойства
Ароматические альдегиды — это жидкие или кристаллические вещества нейтрального характера, трудно растворимые в воде, легко в органических растворителях. Они очень легко окисляются кислородом воздуха.
Бензойный альдегид имеет запах горького миндаля.
Химические свойства ароматических альдегидов
Ароматические альдегиды во многом повторяют свойства альдегидов жирного ряда.
I. Дают реакцию «серебряного зеркала», так как окисляются.
2. Восстанавливаются до спиртов.
3. Присоединяют бисульфит натрия с образованием бисульфитного производного.
4. Присоединяют НСN с образованием циангидринов.
5. С фенилгидразином образуют гидразоны.
6. С гидроксиламином образуют альдоксимы.
Однако ароматические альдегиды проявляют ряд специфических свойств:
1. Ароматические альдегиды не способны вступать в альдольную конденсацию. Как вы помните, для того чтобы осуществилась эта реакция, необходимо наличие подвижного атома водорода при а-углеродном атоме. В ароматическом альдегиде такого атома водорода нет.
Н
244
Лекции по органической химии
2. Взаимодействие с аммиаком. Реакция ароматических альдегидов с аммиаком — одна из отличительных реакций альдегидов этого ряда.
С6Н5~СЧ + H2NH —- с„нгс
Н -Н2О 6 3 Xх
н 2 n4h
альдимин 1
z° С6Н5~<
н
- хЧ
,с-с6н5 н
c6h5ch=n
C6H;CH=N
'хн-с.н.
Ь 5
H,NH—С6НГС ; !
-Н2° n-h :
гидробензамид
н ;
адьдимин
В этой реакции три молекулы бензальдегида взаимодействуют с двумя молекулами аммиака. Реакция протекает через стадию образования альдимина, который вступает в реакцию с альдегидом с образованием гидробензамида.
3. Реакция Канниццаро — Тищенко. В присутствии сильных оснований или концентрированных шелочей ароматические альдегиды вступают в реакцию диспропорционирования (самоокисления-самовосстановления).
<2° NaOH - +
2С6Н—Сх --------- C6H$COONa + С6Н5СН2ОН
Н
Механизм реакции:
он“ '>5+
н5с—\ ==* Н5С—С--н; + НС-С6Н5 =5=^
Н он' <0
5“
б-
слн,соон + с,н,—с—н----------
од о 5 ।
н
-----► с,н,соо + с,н,сн,он О Д о 3 2
Реакция протекает с переносом гидрид-иона (Н“).
Одна молекула альдегида восстанавливается, а другая окисляется и образуются ароматический спирт и кислота.
21 Альдегиды и кетоны ароматического ряда гАЗ
4. Действие галогена на альдегид. Реакция галогенирования. В отличие от альдегидов жирного ряда, которые образуют в этих условиях а-галогенсодержашие альдегиды, ароматические альдегиды образуют галогенангидриды ароматических кислот.
с,н.—с' + С1 н
/° с6н—с' + НС1
С1
хлорангидрид бензойной кислоты
5. Бензоиновая конденсация. Альдегиды, которые содержат альдегидную группу в бензольном ядре, способны под действием солей циановодородной кислоты вступать в реакцию бензоиновой конденсации с образованием бензоина.
2С,Н—С' о J X
он о
бензоин (кетоноспирт)
6. Реакция конденсации ароматических альдегидов с альдегидами жирного ряда. В присутствии оснований ароматические альдегиды вступают в реакцию конденсации с альдегидами или кетонами жирного ряда, содержащими подвижные атомы водорода при o'-углеродном атоме.
Эта реакция подобна альдольной конденсации и называется перекрестной альдольной конденсацией, она протекает по тому же механизму.
он” хР
сбн5-сх + СНз-С\ ----------- С,Н-СН-СН2-С\ -------
Н н 6 3 I н
ОН п
З-гидрокси-5-фенилпропаналь (альдоль)
-777Г С,Н5-СН=СН—С<
-н2о 6 э
коричный альдегид
Образовавшийся в этой реакции альдегидоспирт (альдоль) легко теряет воду, превращаясь в непредельный ароматический альдегид 3-фенилпропеналь (коричный альдегид).
246
Лекции по органической химии
7. Конденсация альдегидов ароматического ряда с ангидридами карбоновых кислот жирного ряда в присутствии ацетата натрия — реакция Перкина.
.О
ZO Н3С-< CH,COONa
C,H,-Cz +
о J X /
н н3с-с\ чо
,0 /О с6н5-сн=сн-сч + н3с-с^ он он
фенилакриловая кислота, коричная кислота
Реакция протекает при длительном нагревании, в результате реакции образуются а,0-непредельные кислоты.
Механизм реакции:
CHj—С СН—С
СН3СОО- + о -- о + сн3соон сн,-с. сн—с
J Х^ х^.
о о
с,н,-с
6 5 \
н
СН3СООН
-СН3СОО~
с,н,—сн-сн,—с—о—с—сн, —>
6 5 I 2 II II 3 -н2о он о о
н,о
CJLCH-CH- с—о—с—сн,——
65 II II 3
о о
с6н5сн=снсоон+сн3соон
8. Реакции по бензольному ядру. В реакции электрофильного замещения (SE) альдегиды вступают в соответствии с правилами ориентации. Альдегидная группа — электроноакцепторная группа, она проявляет -Л/-эффект и относится к.мето-ориентантам.
21 Альдегиды и кетоны ар^матичес^оги ряда
247
Например:
кони. HNO3 кони. H2SO4
кони. H,SO,; t
.м-формилбензолсульфокислота
.и-нитробензальлегид
АРОМАТИЧЕСКИЕ КЕТОНЫ
Соединения, содержащие карбонильную группу, связанную с двумя углеводородными радикалами, называют кетонами. У нас речь будет идти об ароматических кетонах.
Ароматические кетоны делятся на две группы:
Жирно-ароматические — это кетоны, у которых карбонильная группа связана с ароматическим и жирным радикалами.
метилфенилкетон или ацетофенон
Чисто ароматические — это кетоны, у которых карбонильная группа связана с двумя ароматическими радикалами.
дифенилкетон или бензофенон
В названии ароматических кетонов часто наряду с систематическим названием используют тривиальные.
Получение
Ароматические кетоны можно получить теми же методами, что и жирные. Например:
I. Окисление вторичных спиртов.
2. Перегонка кальциевых солей соответствующих кислот и др.
Чаще всего для получения ароматических кетонов применяют реакцию ацилирования по Фриделю — Крафтсу.
248
Лекции по органической химии
3. Реакция Фриделя — Крафтса (реакция ацилирования).
На ароматический углеводороддействуютхлорангидридом кислоты в присутствии хлорида алюминия (III), это обычная реакция аци-
лирования в ароматическом ряду.
Физические и химические свойства кетонов
Ароматические кетоны — это твердые или жидкие вещества, растворяющиеся в органических растворителях.
В химическом отношении обладают всеми свойствами кетонов.
Из отдельных представителей нас будет интересовать ацетофенон.
Это жидкость с запахом черемухи, обладает снотворным действием. Используется в парфюмерии.
Атомы водорода в группе СН1 подвижны и легко замещаются на атомы хлора.
+ НС1
со-хлорацетофенон
® Обозначает, что хлор находится в крайнем положении боковой цепи.
Аюм хлора обладает значительной подвижностью и легко замещается на другие атомы или атомные группы.
Производным ацетофенона является и-нитроаиетофенон, который используют как полупродукт для синтеза антибиотиков синтомицина и левомицетина.
NO,
Т 2
[I \ и-нитроаиетофенон
ох сн3
21 Альдееидь1 и кетоны ароматического ряда
249
ФЕНОЛОАЛЬДЕГИДЫ
Соединения, содержащие в своей структуре фенольный гидроксил и альдегидную группу, связанную с бензольным ядром, называются фенолоальдегидами. Их называют как производные альдегидов. Кроме химических названий, существуют и тривиальные.
Например:
альдегид
альдегид
Способы получения фенолоальдегидов
1. Способ Реймера — Тимана. Это один из способов получения ароматических о-оксиальдегидов. Примером является получение салицилового альдегида.
Реакция проводится при нагревании фенола и хлороформа в присутствии щелочи.
ОН
СНС13, NaOH. t
+ NaCl + Н2О
Механизм реакции:
При взаимодействии хлороформа со щелочью образуется дихлор-карбен, представляющий собой бирадикал, имеющий вакантную атомную орбиталь.
СНС13 + NaOH---------► ССС12 + NaCl + Н2О
дихлоркарбен
Дихлоркарбен — сильный электрофильный реагент, он легко взаимодействует с феноксид-ионом, который образовался при взаимодействии фенола со щелочью.
+ 2НС1
Дихлоркарбен легко присоединяется в о-положении бензольного ядра. Образовавшийся карбанион стабилизируется за счет отщепления протона от ядра и присоединения его к атому углерода с отрица
250
Лекции по органической химии
тельным зарядом. о-Дихлорметилфеноксид-ион под действием воды в условиях реакции превращается в салициловый альдегид.
2. Способ получения п-оксибензальдегида. 4-Гидроксибензальдегид получают действием на фенол циановодородной и хлороводородной кислот.
хлорангидрид имино-муравьиной кислоты
Предполагают, что реакция протекает через стадию образования хлорангидрида иминомуравьиной кислоты. Образующийся имин л-оксибензальдегида при действии воды гидролизуется с образованием л-оксибензальдегида.
Химические свойства фенолоальдегидов
Фенолоальдегиды обладают свойствами как фенолов, так и альдегидов. Введение альдегидной группы в молекулу фенола повышает кислотные свойства последнего, так как альдегидная группа проявляет электроноакцепторные свойства.
-М
Наличие гидроксильной группы, проявляющей +Л/-эффект, в молекуле и-оксибензальдегида приводит к уменьшению степени электрофильности альдегидной группы, т. к. электрофильность определяется величиной 5+ на атоме углерода карбонильной группы.
Интересно отметить, что электрофильные свойства о-оксибенз-альдегида значительно выше, чем у и-изомера.
Это связано с тем, чтоуо-изомера имеет место внутримолекулярная водородная связь, которая увеличивает 5+ на углеродном атоме карбонильной группы.
КАРБОНОВЫЕ КИСЛОТЫ
Карбоновыми кислотами называют производные углеводородов, которые содержат в своем составе карбоксильную группу.
По природе углеводородного радикала различают алифатические, алициклические, ароматические, гетероциклические карбоновые кислоты и др. По количеству карбоксильных групп они делятся на моно-, ди-, три- и поликарбоновые. Алифатические карбоновые кислоты по мере насыщенности углеводородного радикала разделяют на насыщенные и ненасыщенные.
22. МОНОКАРБОНОВЫЕ КИСЛОТЫ
Монокарбоновыми кислотами называют производные углеводородов, содержащие в своем составе одну карбоксильную группу
/О
—с\
ОН
НАСЫЩЕННЫЕ МОНОКАРБОНОВЫЕ КИСЛОТЫ
Номенклатура и изомерия
Для кислот более употребимы тривиальные названия, которые связаны с нахождением их в природе или источником получения. Например, муравьиная кислота (впервые выделена из муравьев), а уксусная кислота получается в результате уксусно-кислого брожения сахаристых веществ.
Положение заместителей относительно карбоксильной группы в тривиальных названиях обозначают греческими буквами а, р, у и т. д.:
Т|3а Ра
сн3-сн2-сн-соон СН2-СН2-СООН
снз Вг
а-метилмасляная кислота p-бромпропионовая кислота
По заместительной номенклатуре ИЮПАК названия кислот образуют из названий углеводородов с таким же числом атомов углерода, включая и атом углерода карбоксильной группы, прибавлением суффикса -овая и слова кислота.
252
Лекции по органической химии
нсоон
муравьиная кислота, метановая кислота
3 2 I
сн3-сн2-соон пропионовая кислота, пропановая кислота
3 2 I
сн3-сн-соон
сн3
изомасляная кислота, 2-метилпропановая
кислота
Остаток карбоновой кислоты, образующийся при отщеплении протона карбоксильной группы, называется ацилоксигруппой
R—С . Название ее образуют от тривиального названия кис-
О—
лоты и суффикса -ат:
>° /О
н— сн3 C/f СН.—СН,—
формиат о— хо— Хо ацетат пропионат
При отнятии атома водорода от карбоксильной группы образовы-
вается ацильная группа R—
Ее название образуют от триви-
ального названия кислоты и суффикса -ил. По заместительной номенклатуре названия ацильных групп образуют от названия кислоты и заменяя -овая кислота на суффикс -оил:
формил, метаноил
СН3-С^ ацетил, эта но ил
сн3-ср-с^ снз изобутирил 2-метилпропаноил
Изомерия насыщенных монокарбоновых кислот обусловлена строением углеводородного радикала (структурная изомерия), начинается с 4 представителя гомологического ряда:
сн3-сн2-сн2-соон
масляная кислота
СН3-СН-СООН сн3 изомасляная кислота
Число изомеров увеличивается по мере возрастания длины углеводородного радикала.
22. Монокарбоновые кислоты
253
Способы получения
1. Окисление первичных спиртов и альдегидов.
R----СН2----ОН
первичный спирт
^О
R----
хн
альдегид
карбоновая кислота
О
В качестве окислителей используют К2Сг2О7, КМ пО4 в кислой срс де и др.
2. Гидролиз геминальных тригалогенопроизводных углеводородов. Ре акция протекает в кислой или щелочной среде с образованием промежуточной ортокислоты, которая в свободном состоянии не существует. Она легко теряет молекулу воды, превращаясь в карбоновую кислоту.
^С1
R-C —С1
"С1
н,о
он,' н+
тригалогено-производное
>>н />
R-C-OH ——-* R-С—ОН
"он ’Н2°
ортокислота
3. Гидролиз нитрилов. Нитрилы при нагревании с водным раствором кислоты или щелочи гидролизуются до карбоновых кислот:
н,о R—C=N —Г— ОН или Н *
нитрил
R—Сч
nh2 амид карбоновой кислоты
НОН ------►
ОН или Н’
R—С—ОН
карбоновая кислота
Реакция протекает через стадию образования амидов.
4. Взаимодействие магнийорганических соединений с оксидом углерода (IV). При взаимодействии магнийорганических соединений с оксидом углерода (IV) получают соли карбоновых кислот, при подкислении которых выделяют соответствующие кислоты:
R—MgCl + СО2
net.
К ----5
OMgCl
R—+ MgCl2
ОН
соль карбоновой кислоты
карбоновая кислота
254
Лекции пс органической химии
5. Гидрокарбоксилирование алкенов. Карбоновые кислоты образуются также при нагревании алкенов с оксидом углерода (II) в присутствии кислотного катализатора и повышенном давлении:
н* СН,=СН, + СО + Н,О -------------
2 2 2 t, р
этен
сн3-сн2-соон
пропионовая кислота
Полученные кислоты содержат на один углеродный атом больше, чем исходный алкен.
Физические свойства
Кислоты с числом атомов углерода не более трех представляют собой жидкости с острым запахом. Кислоты с С4—С9 — маслянистые жидкости с неприятным запахом. С числом атомов углерода 10 и выше — твердые вещества. Низшие карбоновые кислоты смешиваются с водой в любых соотношениях.
С увеличением углеводородного радикала растворимость в воде уменьшается.
Температура кипения кислот выше температуры кипения спиртов с тем же числом атомов углерода, что свидетельствует о большей степени их ассоциации.
В результате образования межмолекулярных водородных связей карбоновые кислоты образуют как линейные, так и циклические ассоциаты, существующие в виде димеров.
” н—О
R—^с—R о—н ••• О'''
Химические свойства
Реакционная способность карбоновых кислот характеризуется наличием карбоксильной группы, внутри которой осуществляется сопряжение между неподеленной парой электронов атома кислорода с л-электронами карбонильной группы (р, л-сопряжение).
Вследствие этого связь Ос-Н становится полярной и водород способен отщепляться в виде протона, что обуславливает кислотный ха
22. Монокарбоновые кислоты
рактер карбоновых кислот. Смешение электронной плотности к ато-
му кислорода группы приводит к возникновению 5+ на
атоме углерода карбоксильной группы, чем больше величина 8+, тем более реакционноспособны карбоновые кислоты.
Основные реакции карбоновых кислот условно можно разделить на группы:
• с участием связи ОН (кислотные свойства);
• нуклеофильное замещение с участием атома углерода карбок сильной группы;
• замещение атома водорода при а-углеродном атоме;
• окисление и восстановление.
Кислотные свойства
В водных растворах карбоновые кислоты диссоциируют с образованием карбоксилат-иона:
z°
R—С + Н2О
ОН
'Лэн карбоксилатной
+ н3о+ ион гидроксония
Строение карбоксилат-иона можно представить с помощью двух граничных структур:
В карбоксилат-ионе оба атома кислорода равноценны, отрицательный заряд равномерно распределен между ними.
Сила карбоновых кислот зависит от стабильности образовавшегося карбоксилат-иона и степени делокализации отрицательного заряда в нем.
На силу карбоновых кислот также оказывает влияние структура углеводородного радикала и природа заместителей в нем. Электронодонорные заместители дестабилизируют карбоксилат-ион и ослабляют кислотные свойства. Электроноакцепторные заместители за счет -/-эффекта способствуют делокализации отрицательного заряда в карбоксилат-ионе, повышая его устойчивость, что приводит к повышению кислотных свойств.
256
Лекции по органической химии
Образование солей. При взаимодействии с активными металлами, основными оксидами, гидроксидами и карбонатами щелочных мета-лов карбоновые кислоты образуют соли:
СН3-СООН + NaOH
СН3-СООН + NaHCO3
---*- CH3-COONa + Н2О
------* CH3-COONa + СО2| + Н2О
Взаимодействие с гидрокарбонатом натрия является простей шей качественной реакцией на карбоксильную группу.
Названия солей образуются от названий анионов карбоновых кислот:
н—
ONa формиат натрия
СНз-----С\
ок
ацетат калия
Соли пропионовой кислоты — пропионаты, .масляной — бутираты и т. д.
Реакции нуклеофильного замещения. Благодаря наличию дробного положительного заряда на атоме углерода карбоксильной группы карбоновые кислоты способны вступать в реакции нуклеофильного замещения (SN).
Взаимодействие со спиртами (реакция этерификации). Карбоновые кислоты реагируют со спиртами при нагревании в кислой среде. Реакция протекает с образованием сложных эфиров.
н3с—С + Н3С-ОН
\QH метанол уксусная кислота
HI: г
.« н3с—С + н2°
ОСН3
метилацетат
Реакция этерификации обратима, и чтобы сместить равновесие в сторону образования сложного эфира, необходим избыток спирта или кислоты или же удаление воды.
Механизм реакции этерификации:
z-S- — ж ОН
S.P Н* >/ОН нй-СН. I .
сн—с' СН,—С * н, с-с—о—сн,^
'он Хон I L
карбкатион ОН Н
22. Монокарбоновые кислоты
1 /°
н3с—с—о—СН3 м '** Н3С—+ Н2о + н+
Cl ОСН3
О метилацетат
н н
Карбоновая кислота на первой стадии протонируется по атому кислорода карбонильной группы с образованием карбкатиона, электрофильные свойства которого значительно выше, чем у исходной кислоты.
Карбкатион присоединяет молекулу спирта за счет неподеленной пары электронов атома кислорода гидроксильной группы с образованием оксониевого иона, который стабилизируется путем отщепления молекулы воды и протона. В результате образуется сложный эфир.
Взаимодействие с галогенирующими реагентами. При действии на карбоновые кислоты хлоридов фосфора (III) и (V), бромида фосфора (III) или тионилхлорида образуются галогенан гидрилы карбоновых кислот:
Н3С-Н2С-СООН + РС13--------Н3С-Н2С-С + Н3РО3
пропионовая кислота Q
хлорангидрид пропионовой кислоты, пропионил хлор ид
/О
н3с-соон + soci2 ---------► H3c-cz+ so} + HClt
4 Cl
хлорангидрид уксусной кислоты, ацетил хлорид
Галогенангидриды кислот используются как ацилирующие реагенты.
Взаимодействие с аммиаком и аминами. При взаимодействии карбоновых кислот с аммиаком, первичными или вторичными аминами образуются аммониевые соли, которые при пирролизе превращаются в амиды:
# 200 °C
Н3С-СООН + NH3^—Н3С-С —J77*H3C-C
J J \ _ + Г1?и J К
onh4 nh2
ацетат аммония ацетамид
Образование ангидридов кислот. При действии водоотнимающих средств, таких, как пентаоксид фосфора (Р2О5) или трифторуксусный ангидрид (CF3CO)2O, образуются ангидриды карбоновых кислот:
25r
Лекции no органической химии
2Н3С-Сх
ОН
Р2О5
-Н3РО4
ангидрид
уксусной кислоты
Ангидриды карбоновых кислот являются активными ацилирующими средствами.
Замещение водорода при а-углеродном атоме (реакция Геля — Фольгарда — Зелинского). Под влиянием карбоксильной группы, которая проявляет отрицательный индуктивный эффект (—Г), увеличивается подвижность атомов водорода при а-углеродном атоме.
При взаимодействии карбоновых кислот с галогенами в присутствии тригалогенидов фосфора атом водорода при а-углеродном атоме замешается на галоген.
СН3-СН-С + Вг,
I он н
пропионовая кислота
РВг3
----► СН.-СН-СООН + НВг 31
Вт
а-бромпропионовая кислота
В результате реакции образуются галогенкарбоновые кислоты. Реакция протекает через стадию образования галогенангидридов, которые галогенируются легче, чем кислоты:
C1L-C1L-C.
ОН
РВг3 -н3ро33
^О СН.-СН.-С.
Вг
Вг2
-НВг
вг2
-НВг
нон
СН3-СН-С —НВг *
Вг Вг
бромангидрид а-бромпропионовой
кислоты
СН.-СН-С
I \
Вг ОН
ct-бром п роп ионовая кислота
Окисление Монокарбоновые кислоты устойчи-
вы к действию окислителей, кроме муравьиной кислоты, которая легко окисляется КМпО4 и другими окислителями с образованием угольной кислоты:
22. Монокарбоновые кислоты
259
[О]
Н-СООН -----------
муравьиная кислота
со2 + н2о
В связи с особенностью строения муравьиная кислота обладает восстанавливающей способностью, она подобно альдегидам дает реакцию «серебряного зеркала»:
Н—:+2[Ag(NH3)2]OH —*- НО-С-ОН+2 Ag|+4NH3 + Н2О ой / О \
со2 Н2О
Отличительной реакцией муравьиной кислоты является также реакция ее разложения при нагревании с конц. H2SO4.
н—
он
конц. H2SO4; t
cof + н2о
В результате выделяется оксид углерода (II) и вода.
НЕНАСЫЩЕННЫЕ МОНОКАРБОНОВЫЕ КИСЛОТЫ
К ненасыщенным карбоновым кислотам относятся кислоты, которые содержат в углеводородном радикале кратную связь.
Номенклатура и изомерия
В номенклатуре ненасыщенных карбоновых кислот широко применяются тривиальные названия:
СН2=СН-СООН СН2=С-СООН
акриловая кислота 1*3
метакриловая
кислота
СН2=СН-СН2-СООН НС=С-СООН
винилуксусная пропиоловая
кислота кислота
По номенклатуре ИЮПАК названия образуются аналогично насыщенным кислотам, используя суффикс -ен или -ин для обозначения соответственно двойной или тройной связи с цифровым указанием положения этой связи в углеводородном радикале:
4 3 2 1
СН2=СН-СН2-СООН
бутен-3-овая кислота
В зависимости от положения кратной связи относительно карбоксильной группы различают а, [3—; 0, у- и др., например:
260
Лекции по органической хи мии
Р а сн2=сн-соон
а,Р-ненасышенная кислота
Y Ра
СН2=СН-СН2-СООН
Р,у-ненасыщенная кислота
Для ненасыщенных карбоновых кислот характерна структурная изомерия, изомерия положения кратной связи и геометрическая изомерия:
сн—сн=сн—
бутен-2-овая кислота О Н
/О сн==сн—сн—
бутен-3-овая кислота ОН
изомеры положения
Чме-бутен-2-овая кислота
транс-бутен-2-овая кислота
Способы получения
В синтезе ненасыщенных карбоновых кислот используют ненасыщенные соединения, например, окисление в мягких условиях ненасыщенных спиртов и альдегидов, гидролиз нитрилов и др. Кроме этих используют также и специфические методы получения.
Гидрокарбоксилирование алкинов (Реакция Реппе). В присутствии карбонатов алкины с оксидом углерода (11) и водой образуют сс.р-не-насыщенные монокарбоновые кислоты:
СП = СН + СО + Н2О
сн2=сн-соон акриловая кислота
Элиминирование ft-галогено- и ft-гидроксикарбоновых кислот. При нагревании с водой Р-галогенокарбоновые кислоты отщепляют галогеноводород и превращаются в а,р-ненасыщенные кислоты:
СН,-СН7-СООН —----------*- СН,=СН-СООН + НС1
I 2 2 2
d акриловая кислота
Р*Гидроксикарбоновые кислоты при нагревании отщепляют воду и также образуют а,Р-ненасыщенные кислоты:
22. Монокарбоновые кислоты
261
СН3-СН-СН2-СООН
ОН
СН3-СН=СН-СООН + Н2О кротоновая кислота
Физические свойства
Ненасыщенные монокарбоновые кислоты представляют собой бесцветные жидкости или кристаллические вещества. Низшие представители хорошо растворяются в воде. А с увеличением молекулярной массы растворимость уменьшается. Высшие ненасыщенные кислоты в воде не растворяются, но хорошо растворимы в органических растворителях.
Химические свойства
Реакционная способность ненасыщенных карбоновых кислот обусловлена наличием карбоксильной группы и кратной связи.
За счет карбоксильной группы они образуют соли, галогенангид-риды. ангидриды, сложные эфиры, амиды, т. е. вступают в реакции характерные для карбоновых кислот.
По кратной связи ненасыщенные карбоновые кислоты вступают в реакции присоединения, окисления, полимеризации. Так. они обесцвечивают раствор КМпО4, раствор бромной воды. Необходимо отметить, что присоединение галогеноводородов к а.р-ненасышенным кислотам проходит против правила Марковникова. что объясняется отрицательным мезомерным (-М) и отрицательным индуктивным (-/) эффектами карбоксильной группы:
8^018-936 8'8' /°
Н =СН-С + НВг --------------»- СН,-СН,-С
I 2 2 \
ОН Вг ОН
0-бромпропионовая кислота
В сравнении с соответствующими насыщенными кислотами а,р-ненасышенные кислоты проявляют более сильную кислотность, что объясняется повышением устойчивости образующегося аниона за счет делокализации заряда по сопряженной системе.
Полимеризацией метилового эфира метакриловой кислоты получают пол и метилакрилат — органическое стекло или плексиглас.
О
сн3
«СН2=С-С
СН, ОСН3
сн,-с
2 I
о осн3
полиметилметакрилат
262
Лекции по органической химии
Плексиглас — небьющееся, обладающее большой прочностью стекло.
Примером взаимного превращения геометрических изомеров в ряду ненасыщенных карбоновых кислот служит олеиновая кислота, которая при действии азотистой кислоты или УФ-облучения изомеризуется в элаидиновую кислоту, которая является ее тронс-изомером:
СН,-(СН,)7-СН II
НООС-(СН2)7-СН олеиновая кислота цгл?-изомер
СН.-(СН7)7-СН
II
НС-(СН2)7-СООН
элаидиновая кислота тра«с-изомер
Олеиновая кислота — это жидкость, а элаидиновая — кристаллическое вещество. Смесь этиловых эфиров олеиновой (около 15 %), линолевой (около 15 %) и линоленовой кислоты (около 57 %) входит в состав препарата «линетол», который применяется для профилактики и лечения гипертонии и атеросклероза.
АРОМАТИЧЕСКИЕ МОНОКАРБОНОВЫЕ КИСЛОТЫ
Ароматическими карбоновыми кислотами называют соединения, в молекулах которых карбоксильная группа связана с бензольным кольцом.
СООН
бензойная кислота
кислота
бензойная кислота
Метилбензойные кислоты называются толуиловыми кислотами:
о-толуиловая кислота
Физические свойства
Ароматические карбоновые кислоты представляют собой бесцветные кристаллические вещества, растворимые в этаноле и эфире.
22. Монокарбоновые кислоты
263
Химические свойства
Химические свойства ароматических карбоновых кислот обусловлены наличием карбоксильной группы и бензольного ядра.
По карбоксильной группе ароматические карбоновые кислоты образуют соли. галогенангидриды. ангидриды, эфиры и т. д„ например:
бензоилхлорид, хлорангидрид бензойной кислоты
Кислотность ароматических кислот превышает кислотность алифатических (кроме муравьиной кислоты) и ненасыщенных карбоновых кислот, это объясняется делокализацией отрицательного заряда бензоат-иона по сопряженной системе бензольного кольца.
За счет бензольного кольца они вступают в реакции электрофильного замещения SE (нитрование, сульфирование, галогенирование). Карбоксильная группа, являясь ориентантом второго рода, дезактивирует бензольное кольцо в реакциях SE и направляет заместители
При нагревании выше 200 °C ароматические карбоновые кислоты в присутствии катализатора декарбоксилируются:
бензойная кислота бензол
Бензиловый эфир бензойной кислоты входит в состав перуанского бальзама. Натриевая соль бензойной кислоты (бензоат натрия) применяется как отхаркивающее средство при бронхитах.
23. ДИКАРБОНОВЫЕ КИСЛОТЫ
Дикарбоновыми кислотами называют производные углеводородов, содержащие в своем составе две карбоксильные группы.
НАСЫЩЕННЫЕ ДИКАРБОНОВЫЕ КИСЛОТЫ
По заместительной номенклатуре название дикарбоновых кислот строят исходя из соответствующих углеводородов с добавлением множительной приставки ди- и суффикса -овая кислота. Наряду с заместительной номенклатурой широко применяются тривиальные названия.
НООС-СООН ноос-сн2-соон ноос-сн2-сн2-соон
щавелевая кислота. малоновая кислота, этандиовая кислота пропандиовая кислота
янтарная кислота, бутандиовая кислота
Для них характерна структурная изомерия.
Физические свойства
Дикарбоновые кислоты представляют собой кристаллические вещества. Низшие гомологи хорошо растворимы в воде. С увеличением молекулярной массы кислоты ее растворимость уменьшается.
Способы получения
Дикарбоновые кислоты получают теми же методами, что и монокарбоновые кислоты, используя в качестве исходных веществ соответствующие бифункциональные соединения.
Окисление двупервичных гликолей, диальдегидов и гидроксикислот:
[°! % //° [°]
но-сн2-сн2-сн2-он -----* ^с-сн—с -------------
пропандиол-1,3 J-] jq
[°]
—НООС-СН,-СООН
малоновая кислота
НО-СН2-СН2-СООН
НООС-СН2-СООН
малоновая кислота
23. Дикарбоновые кислоты
265
Одной из важных реакций получения дикарбоновых кислот является реакция омыления динитрилов.
N=C—СН—СН—С=\
динитрил янтарной кислоты
нин » НООС-СН,-СН,-СООН
Н’ 2
янтарная кислота
Химические свойства
Имея в своем составе две карбоксильные группы, дикарбоновые кислоты диссоциируют ступенчато, образуя анион (рКо,) и дианион (рКп2).
Высокая кислотность по первой ступени объясняется взаимным влиянием второй карбоксильной группы, которая способствует делокализации образующегося отрицательного заряда карбоксилат-иона и тем самым повышает его устойчивость. По мере удаления карбоксильных групп ослабевает их взаимное влияние и кислотность по первой ступени падает. Отрыв протона от второй карбоксильной группы происходит труднее вследствие низкой стабильности дианиона, поэтому кислотность дикарбоновых кислот по второй ступени значительно ниже, чем по первой.
При максимальном удалении карбоксильных групп последние не оказывают взаимного влияния и через 5—6 связей каждая из них ведет себя независимо.
По химическим свойствам дикарбоновые кислоты, так же как и монокарбоновые, способны образовывать одни и те же функциональные производные. Только в зависимости оттого, одна или две карбоксильные группы участвуют в реакции, получают кислые или средние соли, полные и неполные эфиры, галогенангидриды, амиды и др. Например:
2С,Н50Н
I ОС2Н5 * _2Н 0
ос2н5
диэтиловый эфир щавелевой кислоты (полный эфир)
лз
он
I с.н,он
———
^ОС2Н5 - Н2° моноэтил овый эфир щавелевой кислоты (неполный эфир)
NaOH
-Н,О
HOOC-COONa
монон атриевая соль щавелевой кислоты, кислый оксалат натрия
ноос-соон
2NaOH
—2H2O
NaOOC-COONa
оксалат натрия
266
Лекции по органической химии
Вместе с тем следует отметить, что дикарбоновые кислоты проявляют и ряд специфических свойств. В частности, они по-разному относятся к нагреванию.
1. Отношение дикарбоновых кислот к нагреванию. Щавелевая и малоновая кислоты при нагревании свыше температуры плавления отщепляют оксид углерода (IV) и превращаются в монокарбоновые кислоты:
НООС-СООН -200-С* НСООН + СО,1 муравьиная z I кислота
ноос-сн2-соон » сн3-соон + co2f
уксусная кислота
При нагревании янтарной и глутаровой кислот, взаимное влияние карбоксильных групп которых слабее, декарбоксилирования не происходит, а осуществляется процесс внутримолекулярной дегидратации с образованием циклических ангидридов:
н2с-соон
I —
н2с—СООН ’Н-°
янтарная кислота
А) н2с—с^
I /° Н1С-<0 янтарный ангидрид
СН — СООН сн2 ^сн—СООН
глутаровая кислота
хсн2— с^
Н2С^ \ сн2-с/
о
глутаровый ангидрид
Адипиновая кислота в этих условиях подвергается декарбоксилированию и дегидратации с образованием циклического кетона — циклопентанона:
Н2С---СН
СООН
Н2 С-----сн2-
адипиновая кислота
СООН
-Н2О, -СО2
циклопентанои
2. Образование циклических амидов. При нагревании янтарной и глутаровой кислот или их ангидридов с аммиаком образуются циклические имиды:
23. Дикарбоновые кислоты
267
% z°
C-CH2-CH2-C + NH3 но он
янтарная кис юта
-2Н2О
Важное значение в органическом синтезе имеет диэтиловый эфир малоновой кислоты или малоновый эфир:
В результате электроноакцепторного влияния двух сложноэфир-ных групп за счет -/-эффекта возрастает подвижность атомов водорода метиленовой группы, что обуславливает наличие СН—кислотного центра. При действии на малоновый эфир алкоголята натрия образуется натриймалоновый эфир, который взаимодействуете электрофильными реагентами. Так. при алкилировании малонового эфира образуется алкилмалоновый эфир, который, гидролизуясь, дает алкилмалоновую кислоту. Последняя легко декарбоксилируется и образует монокарбоновую кислоту:
С2Н5О Na
-С2Н5ОН
О
ОС2Н5
натриймалоновый эфир
СН31
-Nal
СН,1
-Nal
Л)
I ос2н5 нс—сн3
Н2О: н"
-2С2Н5ОН
^ос2н5
метилмалоновый эфир
268
Лекции по органической химии
метилмалоновая кислота
сн3—сн2-----
он
пропионовая кислота
Алкилированию может подвергаться и второй атом водорода метиленовой группы, что дает возможность осуществлять синтез разнообразных монокарбоновых кислот.
При ацилировании натриймалонового эфира ос-галогенкарбоно-выми кислотами образуются различные дикарбоновые кислоты.
НЕНАСЫЩЕННЫЕ ДИКАРБОНОВЫЕ КИСЛОТЫ
Простейшими представителями этого класса являются малеиновая и фумаровая кислоты, которые являются геометрическими изомерами 2-бутенлиовой кислоты:
ноос
^соон с = с
Нх ^соон с = с ноосх
малеиновая кислота, чис-бутендиовая кислота
фумаровая кислота znpowc-бутендиовая кислота
Способы получения
Малеиновую кислоту можно получить дегидратацией яблочной кислоты с последующей гидратацией образующегося малеинового ангидрида:
НООС-СН,-СН-СООН------
2 I —2Н2О
он
малеиновый ангидрид
яблочная кислота, гидроксиянтарная кислота
сн —соон
II
сн—соон
матеиновая кислота
Фумаровая кислота получается конденсацией глиоксалевой кислоты или изомеризацией малеиновой кислоты:
23. Дикарбоновые кислоты
269
хсоон он- хсоон t
НООС-С + Н,С —~*НООС-СН=С ---*
хн ' соон ’H2° соон со
НООС\ соон с = с
НХ ЧН
150 “С
---------с=с
изомеризация
ноос н
/СООН
малеиновая кислота
фумаровая кислота
Химические свойства
По сравнению с насыщенными дикарбоновыми кислотами ненасыщенные дикарбоновые кислоты являются более сильными кислотами. Это объясняется взаимным влиянием двух карбоксильных групп.
Реакционная способность ненасыщенных дикарбоновых кислот обусловлена наличием двух карбоксильных групп и кратной связи По кратной связи протекают реакции электрофильного присоединения и реакции окисления. Например:
НООС-СН,-СН2-СООН-^-
янтарная кислота
НОН.Н- НООС-СН,-СН-СООН
сн
НООС-СН-СН-СООН I I Вг Вг дибромянтарная кислота
НО
ноос-сн3-сн-соон - I
С1 хлорянтарная кислота
сн
СООН
соон
яблочная кислота
ОН
НООС-СН-СН-СООН
ОН он
виноградная кислота
По карбоксильной группе образуются кислые и средние соли, неполные и полные сложные эфиры, амиды и т. д.
Особенностью малеиновой кислоты является ее способность при нагревании образовывать циклический ангидрид. Фумаровая кислота в аналогичных условиях ангидрида не образует по причине пространственной удаленности карбоксильных групп.
нс—соон нс—с-°
II -Йо * II /О
нс—соон 2 НС----------
малеиновая кислота малеиновый ангидрид ®
270
Лекции по органической химии
АРОМАТИЧЕСКИЕ ДИКАРБОНОВЫЕ КИСЛОТЫ
Ароматическими дикарбоновыми кислотами называют производные ароматических углеводородов, содержащие две карбоксильные группы, непосредственно связанные с ароматическим ядром.
Важными представителями этого класса являются фталевая, изо-фталевая и терефталевая кислоты:
СООН
фталевая кислота, изофталевая кислота, терефталевая кислота, 1,2-бензоддикарбоновая 1,3-бензолдикарбоновая 1,4-бензолдикарбоновая
кислота кислота кислота
Они имеют общее название — фталевые кислоты.
Способы получения
Основным способом получения ароматических дикарбоновых кислот является каталитическое окисление ксилолов кислородом воздуха.
Так, при окислении л-ксилола получают терефталевую кислоту:
Фталевую кислоту получают из о-ксилола или нафталина:
23. Дикарбоновые кислоты 271
Физические и химические свойства
Фталевые кислоты представляют собой кристаллические вещества с высокими температурами плавления. Фталевая и терефталевая кислоты мало растворимы в воде, в то время как изофталевая кислота в воде легко растворима.
По степени кислотности они превышают бензойную кислоту.
Арендикарбоновые кислоты образуют кислые и средние соли, полные и неполные сложные эфиры и амиды и т. д. Фталевая кислота в отличие от своих изомеров при нагревании легко теряет молекулу воды с образованием ангидрида, который при взаимодействии с аммиаком образует фталимид:
Фталимид широко используется в органическом синтезе.
24. ФУНКЦИОНАЛЬНЫЕ ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ
К важнейшим функциональным производным карбоновых кислот относятся: галогенангидриды, ангидриды, сложные эфиры, амиды, гидразиды, гидроксамовые кислоты, нитрилы и др.
ГАЛОГЕНАНГИДРИДЫ КАРБОНОВЫХ КИСЛОТ (АЦИЛГАЛОГЕНИДЫ)
Ацилгалогениды — это такие производные карбоновых кислот, в которых гидроксильная группа, входящая в состав карбоксильной, замешена на атом галогена:
Номенклатура
Названия ацилгалогенидов образуют из названий соответствующих кислот или ацильных групп и названий галогенов:
хлорангидрид бензойной кислоты, бензоилхлорид
хлорангилрид уксусной кислоты, анетилхлорид
Способы получения
Хлор- и бромангидриды могут быть получены при действии гало-
тонирующего реагент .о а на карбоновые кислоты: РС1? Л) :—*R—С< + Н.РО. X л » С1 PCI, /О
он к + РОС13+НС1 С1 soci, /О —*R—С<^ + HC1 + SO2|
С1
24. Функциональные производные карбоновых мклот 416
Йодпроизводные карбоновых кислот получают следующим образом
0 /°
н.с—с^ + р + 12 —- н,с—с^ + Н3РО3
ОН 1
Физические свойства
Низшие галогенангидриды карбоновых кислот — жидкости с рез-к<1 м запахом, раздражающие слизистые оболочки.
Химические свойства
Галогенангидриды — сильные электрофильные реагенты, сильнее карбоновых кислот. Галоген в этих соединениях обладает исключительно бо 1ьшой подвижностью:
Электрофильные свойства подобных соединений зависят от величины дробного положительного заряда (8') на углероде карбонильной группы. Со стороны галогена проявляется выраженный —I, который в статическом состоянии больше +М, поэтому на атоме углерода возникает выраженный 8’, что и обуславливает сильные электрофильные свойства галогенангидридов. Они легко вступают в реакции нуклеофильного замещения:
О
R—
XNH2
амид кислоты
2NH,
-NH4C1
нон
HC1
о
►R—C^
OH
карбоновая к-та
О
R—07 XNHOH
nh2oh
-НС1
/О R-C<
Cl
R'OH
-HC1
гидроксамовая к-та
__o'/ -
xnhnh2
2NH2NH2
-nh2nh2-hci
гидразид кислоты
/О
- R—C^
OR'
сложный эфир
ZO R'-C^
ON a
-NaCl
R'-C^ О
О
R— C^
О
ангидрид кислоты
274
Лекции по органической химии
В подобных реакциях в молекулу нуклеофила вводится ацильная группа, поэтому такие реакции называют реакциями ацилирования, а галогенангидриды карбоновых кислот — ацилирующими агентами.
Галогенангидриды в силу своей высокой активности нашли чрезвычайно большое применение в органическом синтезе.
АНГИДРИДЫ КАРБОНОВЫХ КИСЛОТ
Ангидридами называют производные карбоновых кислот, в молекулах которых атом водорода карбоксильной группы замешен на ацильную группу:
О
R—
О
R-< о линейный ангидрид
циклический ангидрид
Номенклатура
Названия ангидридов образуют из тривиальных названий соответствующих кислот:
уксусный ангидрид.
ангидрид уксусной кислоты
.малеиновый ангидрид, ангидрид малеиновой кислоты
Способы получения
1. Дегидратация карбоновых кислот. При пропускании паров кислоты над соответствующим катализатором (пентаоксид фосфора, трифторуксусный ангидрид) происходит выделение воды — реакция дегидратации:
О
нзС—С\
Г.9-Й; Р2О5
Н3С—С
+ 2НРО3
о
уксусный ангидрид
24. Функциональные проимобные карбоновых кислот
275
2. В промышленном масштабе ангидриды получают взаимодействием галогенангидридов с безводными солями карбоновых кислот:
О н3с— < С1\ анетилхлорид * \
v \
О'INat' н3с-с^ о
—NaCl
/О н3с—
о н3с-с/
о
уксусный ангидрид
натрия ацетат
3. Взаимодействие карбоновых кислот с кетенами (промышленный метод получения уксусного ангидрида):
;-----, г~7~1
О i 8+*8- н2с=с^ н3с-с^
Н3С-С^ ! СН = С=О --- О -->- О
9'н t н3с-с^ н3с-с^
! ! о о
Физические свойства
Ангидриды — кристаллические вещества или жидкости, малорастворимые в воде, обладают резким запахом.
Химические свойства
Ангидриды карбоновых кислот имеют менее выраженный электрофильный характер, чем галогенангидриды, но большую электрофильность, чем соответствующие карбоновые кислоты, поскольку +М-эффект атома кислорода приходится на две ацильные группы:
8+
8+' R—С
276
Лекции по органической хи чии
Ангидриды карбоновых кислот легко вступают в реакции нуклеофильного замещения, используются как ацилирующие реагенты:
НОН, г
/О
R—
2RCOOH
О
R-C/
О
с,н.он. t
nh2ch.
/° R—СГ
ОС2Н5
/° R—СГ
NHCH3
RCOOH
RCOOH
С,Н,ОН
хо н,с—
I "о
с—сн-сн2-с
но ос2н5
неполный эфир (моноэфир)
О
янтарный ангидрид
RNH,
°\ /°
X—СН2-СН2-С\
но
неполный а.мид (моноамид)
NHR
О
О
Уксусный ангидрид используется в синтезе синтетических волокон, фармацевтических препаратов (ацетилсалициловая кислота).
СЛОЖНЫЕ ЭФИРЫ
Сложные эфиры — производные карбоновых кислот, в молекулах которых гидроксильная группа, входящая в состав карбоксильной группы, замещена на остаток спирта или фенола —OR':
О
R—С
OR'
Номенклатура
Сложные эфиры называют по исходным кислоте и спирту или фенолу:
О
н3с—С
ос2н5
этиловый эфир уксусной кислоты, уксусно-этиловый эфир, этилаиетат, этил этаноат
24. Функциональные производные карбоновых кислот'
Способы получения
1. Взаимодействие галогенангидридов и ангидридов карбоновых кислот со спиртами и феноксидами щелочных металлов'.
с2н5он
-НС1
/О
НзС-С( ~
С1
/О Н3С—с\ ос2н,
этила цетат
С,Н.ОН
С6Н5 О Na
-NaCl
О С6Н5 О Na
Н3С~ С\
ОС6Н5
Н3С-С^ о
фенилацетат
2. Взаимодействие карбоновых кислот со спиртами, катализируемое протоном кислоты (реакция этерификации):
//° Г’"! H?s°.
R-c
Х!он ;
+ Н2О
Равновесие наступает, когда прореагирует примерно 2/3 моля исходного вещества. Для смещения равновесия в сторону образования конечных продуктов отгоняют полученный эфир или берут какое-либо из исходных веществ в избытке.
Физические свойства
Сложные эфиры — чаще жидкости с приятным запахом с более низкими температурами кипения, чем соответствующие кислоты и спирты, что связано с отсутствием ассоциации.
Химические свойства
Сложные эфиры — типичные электрофилы. Из-за +Л/-эффекта атома кислорода, связанного с углеводородным радикалом, они проявляют менее выраженный электрофильный характер по сравнению с галогенангидридами и ангидридами кислот:
R' = алкильный радикал
278
Лекции по органической хи.чии
Электрофильность эфиров увеличивается, если углеводородный радикал образует с атомом кислорода сопряженную систему, т. н. активированные эфиры:
ариловый эфир
виниловый эфир
Сложные эфиры вступают в реакции нуклеофильного замещения.
1. Гидролиз сложных эфиров проходит как в кислой, так и в щелочной среде.
Кислотный гидролиз сложных эфиров — последовательность обратимых превращений, противоположных реакции этерификации:
О
R— Q7/ + Н2О
О—R'
сложный эфир
+ R'OH
карбоновая кислота
Механизм этой реакции включает протонирование атома кислорода карбонильной группы с образованием карбкатиона, который реагирует с молекулой воды:
ОН
I .
с—о-н
перегруппировка
R—С—ОН
Cl
/O-R'
н +
< • R-C +R'OH
-Н+ Чо-н
разложение
Щелочной гидролиз. Гидролиз в присутствии водных растворов щелочей проходит легче, чем кислотный потому, что гидроксид-анион более активный и менее объемный нуклеофил, чем вола. В отличие от кислотного, щелочной гидролиз необратим:
24. Функциональные производные карбоновых кислот
279
Н2О //*
R— CZ + NaOH --------*• R-C' + R'OH
О—R' ONa
Щелочь выступает не в роли катализатора, а в роли реагента. Гидролиз начинается с нуклеофильной атаки гидроксид-ионом атома углерода карбонильной группы. Образуется промежуточный анион, который отщепляет алкоксид-ион и превращается в молекулу карбоновой кислоты. Алкоксид-ион, как более сильное основание, отрывает протон от молекулы кислоты и превращается в молекулу спирта:
8Х°6" . 7°'
R—CZ + ОН R—С —ОН —*
\)-R' C^O-R’
о хо
—►R-CT + R'O— R-C\ + R'OH
OH o'
Щелочной гидролиз необратим потому, что карбоксилат-анион имеет высокую делокализацию отрицательного заряда и не восприимчив к атаке спиртового гидроксила.
Часто щелочной гидролиз сложных эфиров называют омылением. Термин произошел от названия продуктов щелочного гидролиза жи
ров — мыла.
2. Взаимодействие с аммиаком (аммонолиз) и его производными протекает по механизму, аналогичному щелочному гидролизу:
+ R'OH
+ R'OH
+ R'OH
+ R'OH
NH—ОН
гидроксиамид
280
Лекции по органической химии
3. Реакция переэтерификации (алкоголиз сложных эфиров) катализируется как минеральными кислотами, так и щелочами:
/° /°
н3с-с( + с2н5он — - н3с-с^ + сн3он
осн3 ос2н5
Для смещения равновесия вправо отгоняют более летучий спирт.
4. Сложноэфирная конденсация Кляйзена характерна для эфиров карбоновых кислот, содержащих атомы водорода в a-положении. Реакция п; отекает в присутствии сильных оснований:
СЛ°“Na °
2Н3С—С' Н3С-С-СН2-С\ +С2Н5ОН
ОС2Н5 ОС2Н5
ацетоуксусный эфир
(Р-кетоэфир)
Алкоксид-ион отщепляет протон от а-углеродного атома молекулы эфира. Образуется мезомерно стабилизированный карбанион (I), который, выступая в роли нуклеофила, атакует атом углерода карбонильной группы второй молекулы эфира. Образуется продукт присоединения (II). Он отщепляет алкоксид-ион и превращается в конечный продукт (III). Таким образом, всю схему механизма реакции можно разделить на три стадии:
1. н3с—с' + с2нр ос2н5 2. Нзс—С^-"’ СН-С' ос2н5 Q л з. н3с—с—сн—— ос2н5 ос2н5 // - г сн— С\ + с2н5он ос2н5 (I) -—- н,с—с-сн—с' ч I 2 X ОС2Н5 ос2н5 ОС2Н5 (И) ° О =: Н3С-С-СН-С\ + С2нр ОС2Н5 (III)
24. Функциональные производные карбоновых кислот
281
Если в реакцию вступают два сложных эфира, содержащие ос-ато-мы водорода, то образуется смесь четырех возможных продуктов. Реакция используется для промышленного получения ацетоуксусного эфира.
5. Восстановление сложных эфиров:
V + Н2 осн3
Ni
—- Н3С—СН—ОН + СН3ОН
Первичные спирты образуются при действии газообразного водорода в присутствии скелетного никелевого катализатора (никель Ренея).
6. Действие магнийорганических соединений с последующим гидролизом приводит к образованию третичных спиртов:
8- —
5+^,°
Н3С-С <___ СН3 Mgl ос2н'”’
OMgl
I CH3MgI н,с—с—сн,-----2—-
3 | 3 -C2H5OMgI
ос2н5
з
CH3MgI
-C2H5OMgl
OMgl н3с-с-сн3
нон
-Mg(OH)l
он
н3с—с—сн сн3
Сложные эфиры имеют большое значение как ацилирующие реагенты, растворители, используются для синтеза альдегидов, кетонов, полимеров («органическое стекло» — плексиглас), лекарственных веществ: этилформиат — для производства витамина В,. Бензилбензоат используют для лечения чесотки. Эти вещества известны как отдушки в парфюмерии (этилформиат, этилацетат) и компоненты пищевых эссенций: грушевой — изоамилацетат, яблочной — изоамилвалерат, ромовой — этилформиат, ананасовой — этилбутират.
АМИДЫ КАРБОНОВЫХ КИСЛОТ
Амидами называют производные карбоновых кислот, в молекулах которых гидроксильная группа карбоксила замещена на аминогруппу:
/О
R—
NHR'
Эти соединения можно рассматривать как ацильные производные аммиака, первичных и вторичных аминов.
282
Лекции по органической химии
Номенклатура
Названия амидов образуют от названий соответствующих кислот
и аминов. В тривиальных названиях ацилов заменяют суффикс -ил на -амид. Согласно заместительной номенклатуре ИЮПАК в названиях соответствующих кислот часть -овая кислота заменяется на суффикс
-амид. Символом N обозначают положение заместителей у атома азо
та амидной группы:
О
з
NHCH3 метила,мид уксусной кислоты. N - метилацетам ид.
N-.метилэтанамид
Способы получения
Амиды получают в результате взаимодействия галогенангидридов, ангидридов или сложных эфиров карбоновых кислот с аммиаком, первичными или вторичными аминами; при нагревании аммонийных солей карбоновых кислот, при гидролизе нитрилов:
н3с-с
С1
NH3
-НС1
анетилхлорид
(Н3ССО)2О уксусный ангидрид
NH,
-СН.СООН
.° н,с-с -nh2
ацетамид
-Н2О
О н3с-с
3 4 - +
о NH,
аммония ацетат
о
Н,С-С
з \
NH,
H2O(HjSO4) 8<ч8
-------- H3C-CEN
ацетонитрил
метилацетат
о-снз -СНз°Н
Физические свойства
Амиды — кристаллические вещества или жидкости, растворимые в воде и органических растворителях. Это ассоциированные соединения, в которых я-электроны карбонильной группы смешаются к наиболее электроотрицательному атому кислорода, пара электронов азота смещена в сторону кислорода. За счет этого водород, находящийся при атоме азота, обладает способностью к образованию водородной связи с другой молекулой амида:
24. Функциональные производные карбоновых кислот
283
_ R' _ _ R' О R'
8+ | 8- 8+ | 8- 8+ । 8-
H-*N~C=O ••• H--N-C-0 - H-*N-C=O
j I d 1 ЗА I
R R R
Поскольку амилы являются ассоциированными, то они имеют более высокие, по сравнению с соответствующими карбоновыми кислотами, температуры плавления и кипения.
Химические свойства
Амиды — очень слабые электрофилы.
Как же распределена электронная плотность в амидах?
За счет сильного смещения неподеленной пары электронов атома азота (+Л/-эффект) к карбонильной группе частичный положительный заряд на атоме углерода С=О группы в амидах меньше, чем у га-логенангидридов, ангидридов и сложных эфиров:
SXP5-" S+-CP5”"
R—С. С—О—С R—С R—<\
6’ * 'r (ун,
Вследствие такого электронного строения амиды практически не вступают в реакции с нуклеофильными реагентами.
1. Амфотерность амидов. Амиды — нейтральные вещества. Они проявляют амфотерный характер.
Основные свойства. Амиды можно рассматривать как производные аммиака, у которого атом водорода замещен на ацильный остаток. Но ацильный остаток содержит карбонильную группу, находящуюся в сопряжении с неподеленной парой электронов атома азота, поэтому основные свойства NH2 группы значительно понижены. Амиды образуют соли лишь с сильными минеральными кислотами, вырывая пару электронов из р,я-сопряжения:
©
он ОН он
- л -- «« * R-с -«—r_c©^—► R-Q@
NH2 NH2 NH2
р,Я-сопряжение
Солеобразование наступает в отсутствие влаги. Эти соли быстро разлагаются водой, так как они образуются слабым основанием и сильной кислотой.
Итак, по сравнению с аммиаком основные свойства амидов понижены вследствие сильного смещения пары электронов в сторону карбонильной группы:
2X4
Лекции по органической химии
..,Н *чР
H-N R-C
Н ^Н2
Кислотные свойства. Однако по сравнению с аммиаком амиды обладают большей кислотностью. В молекулах незамещенных и N-замещенных амидов атомы водорода связи N—Н приобретают подвижность за счет сопряжения неподеленной пары электронов атома азота с л-электронами карбонильной группы.
Такие амиды проявляют свойства NH-кислот:
2. Гидролиз амидов. В нейтральной среде амиды гидролизуются значительно труднее, чем другие функциональные производные карбоновых кислот. Катализируют этот процесс кислоты или щелочи:
Р
R—С + NaOH
NH2
о Na r-сАДн Cnh2
R-C + NH3
ONa
8+Го
R-C' 4- H
/nh2
HOH
R—C -
NH, © протонированная форма амида
?н н
R-C-OJ®
NH2 Н
Со- н
R-C-OH
Cnh3
'! +
R-C + NH4
ОН
3. Дегидратация. При нагревании незамещенных амидов с сильными водоотнимаюшими средствами (Р2О5 или РОС1}) образуются нитрилы:
24. Функниона чьные производные карбоновых кислит
285
^°\ R-О \
NH,’
р2о5. t
-Н,0
R—C=N
4. Расщепление незамещенных амидов до первичных иминов. Реакция открыта в 1881 г. немецким химиком А.В. Гофманом, потучила название «перегруппировки Гофмана»:
ц NaOBr .
Н3С-Н2С-С - Н ;C-H2C-NH2 + СО2* + NaBr
NH, 2
этиламин
5. Восстановление амидов под действием алюмогидрида лития LiAlH4 идет до образования аминов:
/,° [Н]
н3с-н2с-с -- > h3c-h2c-h2c-nh2+н2о
NH, ’
I пропиламян
N-замешенные амиды дают вторичные или третичные амины.
6. Замещение атома водорода в группе NH? на галоген. Реакция обычно проходит в присутствии оснований:
9
н3с-с + С12 nh2
-----* Н3С-с' + НС1
NHC1
Полученные N-галогенамиды — нестабильные соединения со свойствами окислителя. Они используются в качестве галогенирующих реагентов.
Амиды карбоновых кислот находят широкое применение как растворители (формамид, диметилформамид и др.), в производстве синтетических волокон, лакокрасочных материалов, биологически активных веществ. Они довольно часто используются для идентификации кислот. Для подтверждения того, что получена та или иная кислота различными способами, можно ее идентифицировать по ее производным, в том числе амидам.
Например, четкая температура плавления амида масляной кислоты часто служит для однозначного решения вопроса о получении соответствующей кислоты, которая при нормальных условиях представляет собой жидкость.
286
Лекции по органической химии
ГИДРАЗИДЫ КАРБОНОВЫХ КИСЛОТ
Гидразиды — производные карбоновых кислот, в которых гидроксильная группа карбоксила замешена на остаток гидразина, алкил-или арилгидразина:
О
R-c\ Л'
NH-NZ
\
R
Номенклатура
Названия гидразидов образуют от названий соответствующих карбоновых кислот и гидразинов. Согласно заместительной номенклатуре ИЮПАК заменяют часть названия карбоновой кислоты -овая кислота на суффикс -гидразид:
HX-HjC-c'
NH—NH2
гидразид пропионовой кислоты, пропаногидразид
N'-фенилгидразил уксусной кислоты
Способы получения
Гидразиды получают при действии гидразина на хлорангидриды, ангидриды, эфиры карбоновых кислот:
Н3С-С
Cl
О NH,NH, //
-HCI
ацетилхлорид
(Н3ССО)2О
NH2NH
-сн,соон
уксусный ангидрид
л
н3с-с
nh-nh2
гидразид уксусной кислоты
О н3с-с
Ь-сн
NH,NH
-СН3ОН 3
метилацетат
24. Функциональные производные карбоновых кислот
Физические свойства
287
Гидразиды — кристаллические вещества с четкой температурой плавления, что часто используется для идентификации органических соединений.
Химические свойства
По химическим свойствам гидразиды во многом напоминаютами-ды. Они имеют два атома азота: у первого (а-атома азота) амидного атома азота пара электронов смещена в сторону карбонильной группы. Вместе с тем неподеленная пара электронов [3-атома азота не участвует в сопряжении с карбонильной группой, поэтому гидразиды об ладают более выраженными основными и нуклеофильными свойствами, чем амиды. Уже с разбавленными кислотами они образуют устойчивые соли, ацилируются, реагируют с карбонильными соединениями, азотистой кислотой:
на
н3с—с +
4HN-NH3 а
N-ацетил гидразина гидрохлорид
(Ч)6-8+Чи
Н3С-С>) ..
xnh-nh2 а р
СН31
CHjCOCl
-НС1
(СН3СО)2О
-СН3СООН
го н,с—с' з ч
________н
-Н2О
hno2
—2Н2О
н3с-с
NH~NH-CH3
О О
—• н3с-с с-сн
NH-NH
N.N'-диацетилгидразин
з
н3с-с
3 \
HN-N=CH-CH3 этилиденгидразид уксусной кислоты
О н3с-с
N-N=N
азид уксусной кислоты
288
Лекции по органической хи чии
Гидразиды находят широкое применение в синтезе лекарственных веществ. Среди них найдены противотуберкулезные препараты (фти-вазид).
НИТРИЛЫ (ЦИАНИДЫ)
Нитрилы — органические соединения, содержащие одну или несколько цианогрупп —ChN, связанных с углеводородным радикалом:
R-CsN
Номенклатура
Названия нитрилов образуют из тривиальных названий ацильных остатков соответствующих кислот или систематических названий карбоновых кислот, имеющих то же количество атомов углерода, с последующим добавлением суффикса -нитрил:
H,C-C=N ацетонитрил, этанонитрил, нитрил уксусной кислоты
С Н —СН —С=1\ о э 2 фенилацетонитрил, фенил этанонитр ил, нитрил фенилуксусной кислоты
Способы получения
1. Дегидратация амидов сильными водоотнимаюшими средствами:
А р,о5,г
Нзс-С . -------* H3c-C=N + Н2О
2. Замещение других групп цианогруппой — взаимодействие галогеналканов с солями циановодородной кислоты (цианидами):
Н3С—I +KCEN
H3C-C=N +К1
3. Дегидратация альдоксимов в присутствии кислот:
О N-ОН -
// // н _
R-C + NH,OH----------* R-C ------------- R-C=N
Н -н2о 'н -н,о
альдоксим
Физические свойства
Нитрилы — жидкие или твердые вещества, не растворяющиеся в воде, но растворимые в органических растворителях нейтрального характера.
24. Функциональные производные карбоновых кислот
Химические свойства
289
Цианогруппа, проявляя отрицательный индуктивный эффект, увеличивает подвижность атомов водорода при а-углеродном атоме и за счет этого возможны реакции конденсации. Атомы углерода и азота образуют полярную связь, по месту разрыва которой нитрилы вступают в реакции нуклеофильного присоединения.
а
R-CH
8+<х8--*C=N
f
Н
1. Присоединение нуклеофильных реагентов. Гидролиз нитрилов проходит при нагревании с водными растворами кислот или щелочей с образованием амидов, которые гидролизуются до кислот:
нон
Н3С-CEN —--------
Н или ОН
3
Н,С—C=N-Н
Г1 '•он
/Р нон /Р
Н3С-С —---------г Н3С-С + NH3
NH2 Н или он ОН
Реакции со слабыми нуклеофилами обычно катализируются кислотами:
8+ 8- 8- 8+
H3C — CEN +Н3С-О-*Н
HC1
H3C-C=NH
ОСН3 .
НС1
на
h3c-c=nh2
ОСН3
С1
иминоэфира гидрохлорид
2. Реакция с участием а.-углеродных атомов. Конденсация нитрилов (реакция Торпа) проходит в присутствии оснований: амидов, алкоксидов металлов. Реакция аналогична альдольной конденсации:
290
Лекции по органической химии
' 3+<\5-
r-ch2-cen
“ C,H.ONa
R-CH-C=N —:-------
н
C,f-U0xa ₽ а
—;------«► R-H,C-C—CH-C=N
2 II I
NH R
р-иминонитрил
3. Восстановление нитрилов алюмогидрилом лития или водородом протекает до соответствующих аминов:
[Н]
Н3С-CEN кат > H3C-H2C-NH2
Нитрилы широко используются в органическом синтезе. С помощью нитрила муравьиной кислоты в структуре соединения увеличивают углеродную цепь. Ацетонитрил — хороший растворитель для жирных кислот, используется в производстве витамина В Кристаллическое вещество — малонодинитрил легко вступает в реакции конденсации, широко используется для получения гетероциклических соединений: витаминов Вг В6, пестицидов, красителей.
ЖИРЫ
Жиры (триацилглицерины, триапилглипериды) — сложные эфиры трехатомного спирта глицерина и высших алифатических кислот. Их общая формула:
О
II
Н2С-о-с-R
О
II
HC-O-C-R’
О
II H2C-O-C-R”
В состав жиров преимущественно входят одноосновные кислоты с неразветвленной цепью, которые содержат четное число углеродных атомов от 4 до 26.
По кислотному составу триацилглицериды подразделяют на простые, содержащие остатки одинаковых кислот (R=R' =R") и смешанные, в составе которых встречаются разные кислотные остатки.
24. Функциональные производные карбоновых кислот
291
Природные жиры — чаше смешанные триацилглицерины. Кислотный состав жиров человеческого организма главным образом представлен стеариновой, пальмитиновой кислотами, которые поступают как с пишей, так и образуются путем биосинтеза. Ненасыщенные жирные кислоты: олеиновая, линолевая, арахидоновая не образуются в организме человека, а лишь поступают с пишей. Они получили название незаменимые.
Номенклатура
По систематической номенклатуре ИЮПАК для жиров в начале перечисляют в алфавитном порядке названия жирных кислот, а затем указывают родоначальную структуру — глицерин. Согласно тривиальной номенклатуре в жирах часть названия жирных кислот -иновая заменяют суффиксом -ин:
0 Н
н2с-о-с-с15н31 н2с-о-с-с„н23
0 °
нс-о-с-с15н31 нс-о-с-с15н31
0 п
н с-о—с-с н н,с-о—с—С17Н„
трипальмитоилглицерин, I -лауроил-2-пальмитоил-З-стеароил-
трипальмитин глицерин,
1 -лауриноил-2-пальмитостеарин
Получение
Для синтеза триацилглицеринов можно использовать реакции о-ацилирования глицерина (этерификация, взаимодействие глицератов натрия с хлорангидридами кислот):
II II
Н2С-ONa С1-С—(СН2)|4-СН3 Н2С-О-С—(СН2)^-СН3
I О О
I II II
НС—ONa +С1-С-(СН2)14-СН3-^ НС-О-С—(СН2)^-СН3+3NaCl
I 0 I О
I II I II
Н2С-ONa С1-С-(СН2)|4-СН3 H2C-O-C-(CH2)-j7CH3
трипальмитоилглицерин
Синтетические способы получения жиров из глицерина не имеют промышленного значения. Чаще триацилглицерины выделяют из измельченных растительных и животных тканей экстракцией, прессованием, вытапливанием.
292
Лекции по органической химии
Физические свойства
Жиры — твердые или жидкие, но не газообразные вещества. Консистенция жиров зависит от их кислотного состава. Твердые трианил-глицерины, как правило, содержат остатки насыщенных жирных кислот, это чаше жиры животного происхождения. В состав жидких жиров, которые называют маслами, входят в основном остатки ненасыщенных кислот. Растительные жиры, как правило, — жидкие. Исключение составляет масло какао, которое при нормальных условиях твердое вещество, и рыбий жир — жидкость.
Природные жиры являются смесями триацилглицеринов, поэтому не имеют четких температур плавления. Жиры нерастворимы в воде, но хорошо растворяются в органических растворителях: углеводородах, эфире, хлороформе.
Химические свойства
Как сложные эфиры жиры способны к гидролизу, а при наличии в их структуре ненасыщенных кислот триапилглицериды проявляют свойства алкенов.
1. Гидролиз жиров катализируют разбавленные растворы кислот или щелочей:
н2с-о-со-(СН2)14- СН3 НС-О-СО-(СН2),-СН3 Н2С-О-СО-(СН2)14-СН3
трипальмитоил глицерина
НОН
Н или ОН
нон н,с-он
н + или ОН- НС-ОН + ЗНООС—(СН2)14—сн3 Н2С—ОН пальмитиновая кислота глицерин
В промышленности гидролиз ведут при нагревании с водой в присутствии сульфокислот или нагреванием паром Нередко для гидролиза используют фермент липазу.
При взаимодействии жиров с водными растворами щелочей образуется смесь глицерина и натриевых (калиевых) солей жирных кислот, которые называют мылами. Сам процесс щелочного гидролиза триацилглицеридов, ведущий к получению мыла, имеет название омыление. Этим же термином часто обозначают реакции щелочного гидролиза других соединений.
24. Функциональные производные карбоновых кис ют
293
Полученную в результате гидролиза смесь пальмитиновой и стеариновой кислот используют для изготовления свечей.
Остановимся на качестве жиров. В аналитической практике реак-пия омыления жиров используется для установления их качества. Определяют так называемое число омыления — количество мг КОН, расходуемых на гидролиз 1 г жира. Избыток калия гидроксида нагревают с триацилглицеридом и методом обратного титрования определяют количество щелочи, которая ушла на нейтрализацию кислот. Таким образом определяют общее содержание как свободных, так и связанных в триглицериды кислот.
2. Гидрогенизация жиров — присоединение водорода по месту разрыва двойных связей в остатках ненасыщенных кислот. Процесс проходит в присутствии никелевого или платинового катализатора при повышенной температуре и давлении:
Н2С-О-СО-(СН2)7-НС=СН-(СН2)7-СН3
НС-О-СО-(СН2)7 -НС=СН-(СН2)7-СН3
Н2С-О-СО-(СН2)7-НС=СН-(СН2)7-СН3
гн1
Ni. г,Р
триолеоил гл и церин, жидкий жир, масло
[Н] Н2С-О-СО-(СН2)16-СН3
' Ni, г,р * нс-о-со-(СН2)16-снз н2с-о-со-(СН2)16- СН3 тристеароил гл и церин
Гидрогенизации подвергаются в основном растительные жиры и жиры морских животных. Этот процесс лежит в основе производства маргарина, мыла. Жидкие жиры омыляют, получают глицерин и непредельные кислоты, которые восстанавливают, а из них получают мыло.
Маргарин — пищевой жир с добавлением вкусовых веществ и отдушек, например, диацетила со вкусом и цветом сливочного масла. Промышленные жиры, полученные в результате гидрогенизации, называют саломасами.
3. Присоединение галогенов к жирам имеет большое аналитическое значение. Остатки ненасыщенных кислот в структуре жира обнаруживают по обесцвечиванию бромной воды.
294
Лекции по органической химии
Н2С-О-СО-(СН2)7-НС=СН-(СН2)7-СН,
ЗВг,
НС-О-СО-(СН2)7 - НС=СН-(СН2)7 -сн3 ---------*
Н2С-О-СО-(СН2)7-НС=СН-(СН2)7-СН3
триолеин
Н2с-о-СО-(СН2)7-СНВг —СНВг -(СН2)7-СН3 ЗВг, I -----НС-О-СО-(СН2)7-СНВг -СНВг -(СН2)7-сн3
Н2С-О-СО-(СН2)7-сНВг-СНВг-(СН2)7-СН3
три(9,10-дибромстеароил)-глииерин
4. Окисление жиров. Наличие двойных связей в молекулах жиров способствует легкой окисляемости, что ведет к «прогорканию». Различают два типа «прогоркания»: гидролитическое — расщепление до свободных кислоте короткой цепью, которое происходит под действием ферментов либо микроорганизмов, и окислительное. Окисление жиров ведет к образованию альдегидов и кетонов с короткой цепью, имеющих неприятный запах и вкус:
Н2С-О-СО-(СН2)7-НС=СН-(СН2)7-СН3
О2
HC-O-CO-R -----*
I
H2C-O-CO-R' о-о
о? Н2С-О-СО-(СН2)7-НС-СН-(СН2)7 - сн3
* HC-O-CO-R
I
Н2с-о-со-R’
нон
* ,<
Н2С-О-СО-(СН2)7—с
I г
HC-O-CO-R
I
HT-O-CO-R'
остаток азелаинового
полуальдегида
+ Н3С—(СН2)7-С
н
пелларгоновый альдегид
Жиры, образованные насыщенными жирными кислотами, при окислении образуют кетоны.
25. ГЕТЕРОФУНКЦИОНАЛЬНЫЕ КАРБОНОВЫЕ КИСЛОТЫ
К гетерофункциональным карбоновым кислотам относятся производные карбоновых кислот, в углеводородном радикале которых один или несколько атомов водорода замещены на другие атомы или атомные группы: галоген, гидроксигруппу, альдегидную группу, кетон-ную группу, аминогруппу. Гетерофункциональные карбоновые кислоты — это бифункциональные соединения, они обладают свойствами карбоновых кислот и свойствами того класса органических соединений, который определяет заместитель в углеводородном радикале.
ГАЛОГЕНОКАРБОНОВЫЕ КИСЛОТЫ
Галогенокарбоновыми кислотами называют производные карбоновых кислот, которые в углеводородном радикале содержат один или несколько атомов галогена.
По природе углеводородного радикала различают алифатические, алициклические и ароматические галогенокарбоновые кислоты.
Алифатические галогенокарбоновые кислоты по взаимному расположению атома галогена и карбоксильной группы делятся на: а, р, у, А и т. д.
Например:
а
Н3С—СН—СООН
С1
а-хлорпропионовая кислота, 2-хлорпропановая кислота
Р а н2с—сн2-соон
С1
[3-хлорпропионовая кислота, 3-хлорпропановая кислота
ура н2с—СН2-СН2-СООН
а
у-хлорпропионовая кислота, 4-хлорпропановая кислота
Способы получения
1. Действие галогенов на органические кислоты.
8+ 8+/>°
Н3С-СН*С\ М он
н
С12
Н3С-СН-СООН +НС1
С1
В этом случае, как правило, получают а-галогеносодержащие карбоновые кислоты.
296
Лекции по органической химии
2. Реакция Геля — Фольгарда — Зелинского. Этот способ имеет большое значение. Он заключается в действии галогена на предельные карбоновые кислоты в присутствии фосфора.
уО
Нзс—с' + РВг3 хон
(Br2 + Р)
-н3РО, 3
бромангидрид уксусной кислоты
НОН
Н2^~С\ -НВг
Вг ВГ
а н,с-сс
X ОН
бро.ман гидрид бро.муксусной кислоты
бромуксусная
кислота
Реакция Геля — Фольгарда — Зелинского осуществляется значительно легче, чем в предыдущем случае. На первой стадии образуется бромангидрид уксусной кислоты. Бром в значительной степени усиливает смещение электронной плотности от а-углеродного атома. Водороды при а-углеродном атоме становятся более подвижны и легко замешаются на галоген. Образовавшийся бромангидрид бромуксусной кислоты при взаимодействии с водой образует бромуксусную кислоту.
3. Действие галогеноводородов на непредельные карбоновые кислоты.
н2с=с-с
он
НВг
<-0
H,CZHC=C — 2| \
Вг он
//° Н2С-Н2С-С\ Вг ОН
акриловая кислота
Р-бромпропионовая кислота
Реакция присоединения протекает против правила Марковникова.
Физические свойства
Галогенокарбоновые кислоты — это бесцветные жидкости или кристаллические вешества, хорошо растворимые в воде; с увеличением молекулярной массы растворимость кислот в воде уменьшается.
Химические свойства
Галогенокарбоновые кислоты более сильные кислоты, чем карбоновые. Введение галогенов в молекулу увеличивает силу кислот.
25. Гетерофункциональные карбоновые кислоты
297
уксусная монохлоруксусная дихлоруксусная
кислота кислота кислота
трихло руксусная кислота
Сила кислот зависит от величины дробного положительного заряда на атоме углерода карбоксильной группы. А величина 8+ увеличивается с введением галогенов, которые обладают отрицательным индуктивным эффектом. Например, трихлоруксусная кислота по силе приравнивается к серной кислоте. Значит, сила кислоты зависит от количества атомов галогена в углеродном радикале. Кроме того, сила кислоты зависит от положения галогена в цепи. Поскольку индуктивное смешение эффективно на протяжении четырех — пяти о-связей, то ос-галогенокарбоновые кислоты более сильные, чем [3-, а р— сильнее, чем у-галогенокарбоновые кислоты.
Для галогенокарбоновых кислот характерны реакции по карбоксильной группе и реакции с участием атома галогена.
По карбоксильной группе они образуют все функциональные производные карбоновых кислот: соли, сложные эфиры, ангидриды, галогенангидриды, амиды и др.
Например:
/О н,с-сн-с' -
I, он
а-хлорпропионовая кислота
NaOH
---—* H.C-CH-COONa
—н2о 3 I
Cl
натриевая соль а-хлорпропионовой кислоты
SOC12
-SO2; -НС1
„О
н.с-сн-с
'°
хлорангидрид а-хлорпропионовой кислоты
С2Н5ОН; Н+, t
-Н2О
О этиловый эфир
Н3С—СН-С" а-хлорпропионовой (Ц ОС2Н5 кислоты
Алифатические галогенокарбоновые кислоты, подобно галогенал-канам, вступают в реакции замещения галогена по механизму нуклеофильного замещения (Sx). Причем, подвижность галогена зависит от взаимного расположения галогена и карбоксильной группы. Карбо-
298
Лекции по органической мии
ксильная группа, как электроакцепторная группа (-/ ), увеличи-
вает подвижность атома галогена в молекуле:
ГО8-а 8+ S
С1
ОН
Больше всего галоген будет подвижен при а-углеродном атоме. Подвижность атома галогена можно подтвердить целым рядом реакций:
О
сн—сн—с
3 I ^он он
молочная кислота
+ NaCl
сн3—сн—СООН
C^N
- + сн —сн -coonh4
nh2
а-иианопропионовая кислота
аммонийная соль а-аминопропионовой кислоты
Галоген в p-положении менее подвижен, а в у-положении влияние карбоксильной группы практически не сказывается.
Галогенокарбоновые кислоты находят широкое применение для получения окси- и аминокислот, широко используются в органическом синтезе лекарственных препаратов, например: уреид а-бромизо-валериановой кислоты — бромизовал — это снотворный препарат.
ГИДРОКСИКИСЛОТЫ (СПИРТОКИСЛОТЫ, ФЕНОЛОКИСЛОТЫ)
Гидроксикислоты — это производные карбоновых кислот в молекуле которых помимо карбоксильной группы содержится одна или несколько гидроксильных групп. Для них вводятся понятия основность и атомность. Основность определяется числом карбоксильных групп в молекуле, а атомность — общим числом гидроксильных групп, включая гидроксильную группу карбоксила.
25 Гетерофункционалъные карбоновые кислоты
299
Например:
Л)
сн2—С-^он
он
гидроксиуксусная кислота (одноосновная, двухатомная)
Для названия гидроксикислот наряду с заместительной номенклатурой широко применяются тривиальные названия. По заместительной номенклатуре ИЮПАК за родоначальную структуру принимается кислота, а положение гидроксильной группы в молекуле обозначают буквами греческого алфавита или цифрами.
По положению гидроксильной группы относительно карбоксильной гидрокислоты делятся на а. £. у и т. д.
3 2 ° 1 ^0
сн-сн-сСон
он
а-гидроксипропионовая кислота, 2-гидроксипропановая кислота, молочная кислота
3₽ 2“ 1
СН—сн—с
он
о
он
[3-гидроксипропионовая кислота, 3-гидроксипропановая кислота
Способы получения
1. Окисление гликолей. Для проведения этой реакции берется такое количество окислителя, чтобы не затронуть вторую гидроксильную группу.
сн2он [0]
сн2он
сн2он [О|
I —*
с С
^о сд I он сн2он
гликолевый альдегид
гидроксиуксусная кислота
2. Гидролиз галогенкарбоновых кислот.
сн2-соон
С1
НОН
NaOH
хо
сн—
I он + NaCI
он
гликолевая кислота
Эти способы дают возможность получать а, р, у, 8-оксикислоты.
300
Лекции по органической хрчии
3. Омыление циангидринов альдегидов и кетонов.
" ОН
8+ '8+ 8- |
СН,—С^ + HCN-----------* Н,С~С—C=N
3 Н / 3 |
\ г Н
— циангидрин
уксусного альдегида
нон
[ОН]
НОН
[ОН]
X) сн—сн—с< 3 | ^он он
+ NH3
молочная кислота
Обычно этим способом получают а-оксикислоты.
4. Восстановление кетонокислот.
сн,—сн—CZ
3 I х>н он
пировиноградная кислота
молочная кислота
Физические свойства
Это вязкие бесцветные жидкости или кристаллические вещества, хорошо растворимые в воде.
Химические свойства
Гидроксикислоты обладают более сильными кислотными свойствами, по сравнению с незамещенными карбоновыми кислотами.
Сила кислоты зависит от положения гидроксильной группы в молекуле и уменьшается в следующем порядке: а > Р > у > 8, то есть а-гидроксикарбоновые кислоты более сильные, чем Р-, а Р— сильнее, чем у и т. д.
Х0" но— сн2— с^
-1 ОН
Со стороны гидроксильной группы проявляется -/-эффект. За счет этого увеличивается 8+ заряд на углероде карбоксильной группы. По
25. Гетерофункционалъные карбоновые кислоты
мере удаления гидроксильной группы от карбоксильной снижается ее влияние и сила кислоты уменьшается.
Оксикислоты образуют все производные по карбоксильной группе и вступают в реакции с участием спиртового гидроксила.
Кто хорошо знает свойства спиртов и кислот, тот легко усвоит свойства оксикислот. К примеру, по СООН-группе гидроксикислоты об разуют хлорангидриды, сложные эфиры, соли, ангидриды и др.:
NaOH
Д)
Н2^-Сх + Н О
ОН ONa
н,с-сС _
21 он он
натриевая соль гликолевой кислоты
CH,OH.H+,r н г-С<О СН..ОН, Нт, f <0
---------** । С
он осн3
-Н,0
- Н,0
метиловый эфир гликолевой кислоты
I о-сн, о-сн3 3 диметиловый эфир гликолевой кислоты, метиловый эфир метоксиуксусной кислоты
В реакцию со спиртами, прежде всего, вступает карбоксильная группа и образуется сложный эфир, затем может вступить и ОН-груп-па с образованием диэфира.
С участием гидроксильной группы гидроксикислоты алифатического ряда вступают в реакции, характерные для спиртов:
<0
н2с----Сх + HCI
ососнзон
аиетоксиуксусная кислота
н2с-сх + н2о
С1 ОН
хлоруксусная кислота
°. *0
,C-Cs
н он
глиоксалевая кислота
В результате взаимного влияния гидроксильной и карбоксильной групп появляются и новые свойства у оксикислот.
302
Лекции по органической химии
Специфические свойства оксикислот
1. Отношение оксикислот к нагреванию. Оксикислоты в зависимости от положения ОН-группы по разному себя ведут при нагревании. Это позволяет отличить а, р, у и другие оксикислоты друг от друга.
а) а-гидроксикислоты:
гликолевая кислота
-2Н2О
с----СН
//
о
лактид гликолевой кислоты
В случае а-оксикислот происходит взаимодействие между двумя молекулами кислоты, при этом выделяются две молекулы воды, и образуется циклический сложный эфир, который называется лактидом
б) Р-гидроксикислоты:
О
Р а
сн =сн —с ""он
акриловая кислота
На а-углеродном атоме электронная плотность уменьшается,
вследствие чего водород становится подвижен — отщепляется молекула воды и образуется непредельная кислота. Таким образом Р-окси-кислоты образуют а, Р-непредельные кислоты.
в) у-гидроксикислоты.
Y Р
/СН2—сн,
НО \н2
но—с II о
-н2о
у-гидроксимасляная кислота у-лактон
При нагревании у-гидроксикислоты претерпевают внутримолекулярную циклизацию с образованием циклических сложных эфиров, которые называются лактонами
25. Гетерофункциональные карбоновые кислоты
Расщепление а-гидроксикислот. Для а-оксикислот характерна еще одна специфическая реакция. При действии концентрированной серной кислоты а-оксикислоты разлагаются с разрывом С—С связи:
,0 и m Л)
сн—сп—сС конц 2 > Н—С'^ +СН—
3 | ^ОН >' ОН ^Н
ОН I H2SO4
со +н2о
В этих условиях, как правило, образуется муравьиная кислота и соответствующий альдегид или кетон. Образовавшаяся муравьиная кислота при действии концентрированной серной кислоты и нагревании разлагается с выделением оксида углерода (II) и воды. Это реакция используется для определения а-гидроксикислот.
Несколько слов о молочной кислоте, так как из а-гидроксикислоз она имеет наиболее важное значение.
МОЛОЧНАЯ КИСЛОТА (а-оксипропионовая)
* Она содержится в кислом молоке, откуда и по-
СНз СН СО Н ЛуЧила свое название.
QPI В промышленности молочную кислоту получа-
ют молочнокислым брожением углеводов:
СН,А ------* 2СН,—сн—соон
о 12 б J ।
ОН
Получают молочную кислоту и при восстановлении пировиноградной кислоты:
[н]
сн,—с—соон -—-сн —сн—соон
II I
о он
В молочной кислоте содержится один ассиметрический атом углерода. Поэтому она существует в виде двух оптических изомеров:
С я ХЮН он с 2ООН
( Д(->-мол< 2Н3 зчная кислота ни ( L( +)-молс ... |_| 2Н3 )чная кислота
304
Лекции но органической химии
Представителями двухосновных гидроксикислот являются яблочная (оксиянтарная) кислота и винная (диоксиянтарная) кислота.
ЯБЛОЧНАЯ КИСЛОТА
СООН Яблочная кислота — это двухосновная трехатомная кис-I лота. Молекула яблочной кислоты содержит один асим-
* СНОН метрический атом углерода, поэтому существует в виде двух
СН оптических изомеров и оптически неактивной рацемической форме:
СООН
( 2ООН ( 2ООН
11 1 1 11
( ЗН.СООН ( ЗН2СООН
D(+)-яблочная кислота £<->-яблочная кислота
В природе встречается L(—^-яблочная кислота.
СООН
*СНОН
*СНОН
I
СООН
ВИННАЯ КИСЛОТА
Винная кислота —двухосновная, четырехатомная. Молекула виннои кислоты содержит два асиметрических атома углерода и существует в виде трех стереоизоизомеров: £>-винная кислота, /.-винная кислота и мезовинная кислота.
( ХЮН
Н" он
но— —Н
СООН
£)-винная кислота
( 1’ООН
11О I1
н— —он
( ХЮН
/.-винная кислота
( :оон Ol_J
11 О11
II
11 О11
СООН .мезовинная кислота
При хранении вина она выделяется в виде нерастворимой кислой калиевой соли (калий виннокислый).
25. Гетерофункщюналъные карбоновые кислоты
305
ЛИМОННАЯ КИСЛОТА
сн2соон
аС
/ОН ""COOH
СН2СООН
Это четырехатомная, трехосновная кислота. Относится к ос-гидроксикислотам.
Как и все ос-оксикислоты, лимонная кислота при нагревании с концентрированной H2SO4 разлагается с выделением муравьиной кислоты и ацетона дикарбоновой кислоты:
сн2соон /ОН
С'""СООН
сн2соон
конц. о СН2СООН сн3
H2SO4 | I
-----*н—С +С=о —* 2СО, + С—О он | |
конц. сн,соон сн,
H,SO4 ацетондикарбоновая ацетон
СО + Н2О кислота
Ацетондикарбоновая кислота при нагревании декарбоксилируется с образованием ацетона. Соли лимонной кислоты называют цитратами. Лимонная кислота, также как и винная, относится к пищевым кислотам. Цитрат натрия используется при консервировании крови.
ФЕНОЛОКИСЛОТЫ
Фенолокислотами называют производные аренкарбоновых кислот, у которых один или несколько атомов водорода в ароматическом ядре замещены на гидроксильную группу, то есть это соединения, которые содержат фенольную и карбоксильную группы.
Например:
о-гидроксибензойная
кислота; льгидроксибензойная
салициловая кислота кислота и-гидроксибензойная
3,4,5-тригидроксибензойная 2- гидроксикоричная
кислота кислота
306
Лекции по органической химии
Даже если карбоксильная группа находится в боковой цепи, но в молекуле содержится фенольный гидроксил, — это тоже фенолокислоты.
Способы получения
1. Сплавлением сульфобензойных кислот со щелочами:
2. Карбоксилирование фенолов оксидом углерода (IV) (Реакция Кольбе — Шмитта). Нагревание фенолов в щелочной среде с оксидом углерода (IV) приводит к образованию салициловой кислоты:
Для двухатомных фенолов присоединение СО2 идет более легко:
Физические свойства
Фенолоксикислоты — кристаллические вещества, труднорастворимые в воде, перегоняются с водяным паром. Они более сильные кислоты, чем бензойные, с FeQ3 дают фиолетовое окрашивание.
Химические свойства
В химическом отношении это бифункциональные соединения. Они дают все производные по функциональны и группам:
25. Гетерофункциональные карбоновые кислоты
307
+СО21 + Н2О
+ 2Н2О
В реакцию с гидрокарбонатом натрия вступает только карбоксильная группа. При этом образуется мононатриевая соль салициловой кислоты, а при действии щелочей образуется ди натриевая соль салициловой кислоты.
Благодаря близости двух функциональных групп в молекуле салициловая кислота, в отличие от своих изомеров, взаимодействует с гидроксидом меди (II):
Си(ОН)2
С участием карбоксильной группы протекают реакции со спиртами, галогенирующими реагентами:
с2н5он, н
SOC12
хлорангидрид салициловой кислоты
При взаимодействии салициловой кислоты с уксусным ангидридом образуется ацетилсалициловая кислота:
308
Лекции по органической линии
Салициловая кислота по кислотным свойствам превосходит бензойную кислоту, а также о- и л-гидроксибензойные кислоты. Это можно объяснить тем, что происходит дополнительная стабилизация салици-лат-иона за счет образования внутримолекулярной водородной связи:
При нагревании салициловая кислота легко декарбоксилируется:
Применение
Производные салициловой кислоты используются в качестве анальгетических, жаропонижающих и противовоспалительных средств, например:
^^.COONa
натрия салицилат
ацетилсалициловая кислота (аспирин)
о-ОКСИКОРИЧНАЯ КИСЛОТА
Содержит в своей структуре непредельную связь, карбоксильную группу в боковой цепи и фенольный гидроксил.
Получают по реакции Перкина:
__
ОС 'н ''"ОН салициловый альдегид
(СН3СО)2О; t CH3COONa
1^^Т-СИ=СН-СООН ^Х)Н
о-оксикоричная кислота
25. Гетерофункциональные карбоновые кислоты 2
Для о-гидроксикоричной кислоты известны два изомера цис- и транс-'.
транс-томер
opzno-кумаровая кислота
цис- изомер кумариновая кислота
Из полигидроксикарбоновых кислот отметим протокатеховую и галловую кислоты.
ПРОТОКАТЕХОВАЯ КИСЛОТА
СООН (3.4-дигидроксибензойная кислота)
2 Протокатеховая кислота — производное пирокатехина. L Дз С РеС13дает сине-зеленую окраску, входит в состав дубиль-
41 ОН ных веществ.
ОН
ГАЛЛОВАЯ КИСЛОТА
(3,4,5,-тригидроксибензойная кислота)
СООН
Л
НО 5>4 он
он
Получают при гидролизе танина, это промышленный способ получения ее из дубильных веществ. С FeCl3 дает сине-черное окрашивание. Щелочные растворы галловой кислоты являются сильными восстановителями, восстанавливают серебро из
AgNO,, поэтому применяют как проявитель в фотографии. Галловая кислота легко декарбоксилируется при нагревании:
СООН
ОН
ОН
пирогаллол
ОКСОКИСЛОТЫ
К оксокислотам относятся альдегидо- и кетонокислоты. Уже само название указывает на то, что в структуру соединений входит карбоксильная группа и альдегидная или кетонная группы. В зависимости от взаимного расположения функциональных групп различают а, р, у и т. д. оксокислоты.
310
Лекции по органической химии
Наиболее важными представителями альдегидо- и кетонокислот являются:
н—с—соон сн,—с—СООН
сн,—с—сн,—соон
II о
глиоксиловая кислота, пировиноградная кислота, ацетоуксусная кислота, глиоксалевая кислота 2-оксопропановая кислота 3-оксобутановая кислота
Оксокислоты получают окислением гидрокси кислот. При окислении гидроксикислот, содержащих первичный спиртовой гидроксил, образуются альдегидокислоты, а при окислении гидроксикислот со вторичным спиртовым гидроксилом образуются кетонокислоты:
сн3—сн— —
Ан он
молочная кислота
/° сн3—с—с
6 он
пировиноградная кислота
Пировиноградную кислоту также получают из виноградной, то есть из рацемической смеси винных кислот, при нагревании (пирос — нагревание).
СООН
СН2ОН
снон Анон
со2+снон
соон
глицериновая кислота
СООН
сЧ^-Н^
О
сн3—с—с
II ""ОН
О
Вначале происходит декарбоксилирование и образуется глицериновая кислота, которая теряет воду с образованием непредельной оксикарбоновой кислоты как промежуточного соединения. В свободном состоянии такая кислота существовать не может. Согласно правилу Эльтекова, гидроксильная группа не может находиться при углероде с двойной связью в открытой цепи, так как имеет место сопряжение неподеленной пары электронов атома кислорода с р-электронами двойной связи. Атом водорода становится протонированным и переходит к атому углерода СН2-группы, при этом образуется пировиноградная кислота.
а-Оксикислоты легко декарбоксилируются в присутствии концентрированной серной кислоты, превращаясь при этом в альдегиды:
25. Гетерофункциональные карбоновые кислоты
311
О
Z кони. H,SO4 ^_r,o4
СН,—с—С ----------------►СНз с +<-О2|
|| ^он н
о
пировиноградная
кислота
Пировиноградная кислота применяется в синтезе гетероциклов и целого ряда фармацевтических препаратов (атофан). Это жидкость не очень приятного запаха.
Ацетоуксусная кислота относится к Р-оксокислотам, обладает свойствами кетонов и свойствами карбоновых кислот.
(3-кетомасляная кислота
Если сравнивать рКа пировиноградной кислоты и ацетоуксусной, то следует отметить, что рКа пировиноградной кислоты выше, чем у ацетоуксусной. Это объясняется удаленностью С=О группы от СООН в ацетоуксусной кислоте.
Второе свойство этой кислоты — легкое декарбоксилирование:
сн—с—сн—с
ОН
О
2
о
+
н
со, +сн,—с—сн, -----*- сн,—с—сн
3 II 2 3 II
о о
Декарбоксилирование происходит уже при комнатной температуре или при незначительном нагревании. В отличие от кислот, сложные эфиры Р-кетонокислот являются устойчивыми соединениями. Этиловый эфир р-кетомасляной кислоты называют ацетоуксусным эфиром:
.0
СНз—с—сн2—с
ОС2Н5
312
Лекции по органической химии
Ацетоуксусный эфир получают по реакции Кляйзена. Исходным продуктом в этой реакции является этиловый эфир уксусной кислоты. Катализирует этот процесс алкоголят натрия. Эта реакция походит на альдольную конденсацию. Этилат натрия реагирует с этиловым эфиром уксусной кислоты:
C2H5ONa
Na+ + С2Н56
(Л)6- /°
8+ 8+ // - - //
сн2-^ с + С2н5о--------- с2н5он + сн—С\
I ХОС2Н5 ос2н
Н2 5 J
карбанион
Образовавшийся карбанион реагируете молекулой сложного эфира по карбэтоксилъной группе:
'"ОН - С2Н5О'
о
сн3—с—сн,—с
анион, который стабилизируется
за счет отщепления этоксид-иона
о
ос,н.
Ацетоуксусный эфир — тахтомерное соединение для него характерна кето-енольная таутомерия, которая обусловлена наличием подвижных атомов водорода метиленовой группы:
кетонная форма (93%)
енольная форма (7%)
Между этими двумя формами устанавливается динамическое (подвижное) равновесие. Кетонная форма более выгодна в энергетическом отношении и поэтому ее значительно больше.
25. Гетерофункциональные карбоновые кислоты
313
Наличие кетонной и енольной форм доказывается с помощью химических реакций. Кетоформа ацетоуксусного эфира взаимодействует с гидроксиламином с образованием оксима; восстанавливается водородом в момент выделения и дает другие реакции кетонов:
NH;OH
сн,-с-сн,-с
II Х
О
ос2н5
сн—с—сн—С
N—ОН ОС2Н5
оксим ацетоуксусного эфира
сн,—с—сн,—С
I \
ОН ОС2Н
этиловый эфир
Р-гидроксимасляной кислоты
Существование енольной формы доказывается образованием красного окрашивания с FeCl3, обесцвечиванием бромной воды:
И
СН—С—СН-
ОН
СН,—С—СН—С
II I 4
о Вг 0СЛ
Для ацетоуксусного эфира характерно два типа расщеплений: ке-тонное и кислотное.
Кетонное расщепление протекает под влиянием разбавленных кислот или щелочей. Называется кетонным, так как образуются при этом кетоны:
сн3—с—сн—сС' н+или0Н ’
/'ОС2Н5 _ с2н5ОН
О
—► сн3—с—сн2-<содн.'—сн3—с—СН3
|| -СО2 Ц
о о
В этих условиях происходит гидролиз по сложноэфирной группе, образуется ацетоуксусная кислота, которая декарбоксилируется с образованием кетона.
314
Лекции по органической химии
Кислотное расщепление протекает пол влиянием концентрированных растворов щелочей.
8+ сн—С -
сн
ос2н5
ОН
сн—с—сн—с
ОСгНг
8"
СН,—с КП
¥н
сн2—с
ОС2Н5
о
з
О
О
О
НОН
СН3—С +СН3—С
О'
он-ОС2Н5
2СН3СОО + С2Н5ОН
Ацетоуксусный эфир широко применяется для получения различных кетонов или кислот, например:
„Т1 н C,HsO Na+ СНу— С = СН — СООС2Н5 СН31
СНу— С—сн2—С С -----
ОС2Н5
он
снг с--сн-сС° ass II | ОС2Н5
О'Ма+ натрийацетоуксусный эфир h (гНз
- СНу— С = С — СООС2Н5 сн31
О СН3 метилаиетоуксусный эфир
O’Na+ метил натрийацетоуксусный эфир
кислотное расщепление
СНз
CHj—С—с—с
--------►СНз-С
NaOH. конц.
о
-ONa+CHr|H_C^ONa+^H5OH
Н3С
ос2н5
о сн3
лиметилацетоуксусный эфир
кетон ное расщепление
СНу-С—сн —сн3+со2 + с2н5он НОН, Н+или ОН- I I о сн3
Таким образом, из ацетоуксусного эфира мы получили натриевую соль изомасляной кислоты и метилизопропилкетон.
Ацетоуксусный эфир широко применяется в синтезе многих лекарственных препаратов.
25. Гетерофункциональные карбоновые кислоты
АМИНОКИСЛОТЫ
315
Это производные кислот, в молекуле которых один или несколько атомов водорода в радикале замешены на группу NHr
По положению группы NH, аминокислоты делятся на а, р, уит. д.
р а Р а у Р а
СН'-СООН 1 2 СИ Ь-СН-СООН СН2-СН2-СООН J 1 . Z Z СНХ-СН2-СН2-СООН
NH, nh2 nh2 nh2
аминоуксусная а-а.мино- Р-а.мино- у-амино-
кислота, пропионовая пропионовая масляная
глицин кислота, кислота. кислота
а-аланин Р-аланин
Ароматические аминокислоты бензольного ряда рассматривают как производные бензойной кислоты:
СООН
и-аминобензойная кислота, антраниловая кислота, 4-аминобензойная кислота, о-а.минобензойная кислота
2-аминобензойная кислота
Способы получения
1. Гидролиз белков. Таким способом получают 31 аминокислоту, из них 30 ос-аминокислот и одну Р-аминопропионовую кислоту.
2. Действием NH3 на галогенокарбоновые кислоты. При этом получают различные (а, р, у) аминокислоты в зависимости от положения галогена.
3. Метод Зелинского — Стадникова. В процессе реакции образуется 2-аминопропанонитрил, который гидролизуется до кислоты:
Т) СНз—с С +nh4cn-3 н ацетальдегид НОН [Н+],[ОН] ►Н2О + CH3-CH-C=N Н0Н -Анг 2-аминопропанонитрил СН3-СН-СООН + NH3 nh2 а-аминопропионовая кислота
Этим способом получают только а-аминокислоты.
316
Лекции по органической химии
4. Присоединение NHt к непредельным карбоновым кислотам. Реакция присоединения протекает против правила Марковникова.
8+Хр>8- 4
СН2—СН—СООН + NH3
акриловая кислота
сн—сн—СООН
nh2
(3-аминопропионовая кислота
5. Восстановление нитробензойных кислот.
СООН
СООН
NO2
nh2
л-нитробензойная л-аминобензойная
кислота
кислота
В молекуле аминокислоты содержится кислотный центр — ^О
~C-~-Qp_|-группа и основный центр — -NH2-rpynna, поэтому аминокислоты кристаллизуются из водных растворов в виде внутримолекулярных солеи.
X)
R-СН—С<_ . I 0 +NH3
В химическом отношении аминокислоты обладают свойствами карбоновых кислот и первичных аминов. Они взаимодействуют и с кислотами, и со щелочами, т. е. проявляют амфотерные свойства:
N,OH > сн,—СН,—С?’0 + Н,0
ан; x°Na
СН. СН —с -
4 он
nh2
натриевая соль а-аланина
НС1
^О сн—СН2— С' I хон +NH С1
хлороводородная соль а-аланина
25- Гетерофинкциональные карбоновые кш ,оты
317
Специфические свойства аминокислот
1. Отношение к нагреванию. В этом они напоминают окси кислоты.
а-Аминокистоты образуют дикетопиперазин (межмолекулярный циклический диамид):
сн2-с^^ /СН2_сх
nh^ 4£н2о
lhq' ' 'j ^с-сн2
О дикетопиперазин
P-Аминокислоты при нагревании отщепляют аммиак и образуют непредельные кислоты:
сн2—СН2-С
NH2
'^он
СН2=СН-СООН + NH3 акриловая кислота
у-Аминокислоты претерпевают внутримолекулярную дегидратацию с образованием циклических амидов — лактамов:
ЛЭ сн,-сн,-сн,-сС ---------- СН2—СН2-СН2-С=О
| ОН I I
NH2 1----NH------1
у-л актам
2. Для а-аминокислот характерно образование комплексных соединений с ионами меди (II).
СН,—СООН 12 .
+ Си
NH2
При этом образуется растворимый в воде устойчивый комплекс интенсивно синего цвета. Только а-аминокислоты дают подобную синюю окраску.
Значение аминокислот заключается в том, что они являются исходными веществами при построении белковых тел.
В состав белка входит 31 аминокислота, из них 30 а-аминокислот и одна р-аминокислота.
Отметим некоторые из аминокислот.
318
Лекции по органической химии
гликокол
(глицин, аминоуксусная кислота)
сн2-соон nh2
Относится к ос-аминокислотам.
АЛАНИН (oc-аминопропиововая кислота)
Различают а- и р-аланины, которые входят в состав белка.
сн^н-сСон nh2
сн2-сН7-сС°н
nh2
а-агчинопропионовая кислота, а-аланин
Р-аминопропионовая кислота, Р-аланин
Молекула а-аланина содержит асимметричный атом углерода и существует в виде пары энантиомеров.
( :оон ( 2ООН
Г1 IN Г12 н2м Г1
сн3 гн3
£)-аланин £-аланин
В построении молекулы белка принимает участие только Л-ала-нин.
у-АМИНОМАСЛЯНАЯ КИСЛОТА (ГАМК)
сн2—сн2—сн2—
nh2
,о с С
ОН
ГАМК содержится в клетках мозга и участвует в процессах торможения.
ПОЛИПЕПТИДЫ
Полипептиды это продукты конденсации аминокислот. По количеству остатков аминокислот различают ди-, три- и полипептиды.
25. Гетерофункционапные карбоновые кислоты
319
х°
снг-с^.он+ hnhch;cooh —
nh2
СН2—С— nh-ch2-cooh nh2
ГЛИЦИЛ-ГЛИЦИН
дипептид
Полипептиды — соединения нейтрального характера. Хара?л еп-ной реакцией на полипептиды является реакция гидролиза, в результате чего образуются аминокислоты.
Когда говорят о полипептидах, то имеют в виду высокомолекулярные соединения, содержащие большое количество пептидных связей.
26. ПРОИЗВОДНЫЕ УГОЛЬНОЙ кислоты
^о
Угольная кислота Н—О—ССГ в свободном состоянии не суще-
он
ствует, разлагается на СО2 и Н2О. Как двухосновная кислота угольная кислота может образовывать два ряда функциональных производных: неполные и полные галогенангидриды, сложные эфиры, амиды и др.
ХЛОРАНГИДРИДЫ
НО-С—С1 II о
.монохлорангидрид угольной кислоты, хлоругольная кислота
С1—с—С1
II
о
дихлорангидрид угольной кислоты.
фосген
Фосген — полный хлорангидрид угольной кислоты, в переводе означает — рожденный светом. Получают при смешивании газов оксида углерода (II) и хлора. Реакция идет только при облучении УФ-све-том:
С1\
СО + С12 /с=о
СГ
Фосген — это газ удушающего действия с Гкип 7.8 °C. Обладает запахом свежепрелого сена. Его пары тяжелее воздуха. Раздражающе действует на легкие, вызывая отек.
Химические свойства
1. Взаимодействие с Н2О. Как хлорангидрид легко разлагается водой с образованием угольной и хлорводородной кислот.
Ск
/С=О + нон с г
С=О + 2НС1 нох |
н2о + со2
26. Производные угольной кислоты
321
2. Взаимодействие с аммиаком.
сц H2N )с=0 + 2NH3 *^2=0 + 2НС1
ciz h2n
.мочевина — полный амид угольной кислоты
3. Взаимодействие со спиртами.
С1 с2н5ох
/С=О + 2С,Н5ОН ----«- £=О + 2НС1
СГ С2Н5С)
диэтиловый эфир угольной кислоты, диэтилкарбонат
АМИДЫ УГОЛЬНОЙ кислоты
Неполный амид угольной кислоты называется карбаминовой кислотой:
h2n-c—ОН
Карбаминовая кислота нестойкая и не встречается в свободном состоянии, т. к. легко разлагается при комнатной температуре:
H2N—С\ --------- co2t + nhJ
ОН
Производные карбаминовой кислоты легко разлагаются.
Нагревание карбаминовокислого аммония ведет к разложению его до мочевины и Н20:
H2N-C— ONH4 H2N-C—NH, +Н20
о о
Эфиры карбаминовой кислоты называются уретанами. Общая формула уретанов:
H,N—С—OR 2 II
О
Получают их из фосгена и соответствующего спирта с последующим действием аммиака:
322
Лекции пс органический химии
Cl
С1.
NH3 H2N.
Cl
-НС1
С2н5о'
или из диэтилового эфира карбаминовой кислоты:
-на /
С2Н5О
h2n^
С=О +С2Н5ОН
С2Н5О\
С=о +NH3 C2H5OZ С2Н5б
Уретаны — вещества с четкими температурами плавления и служат для идентификации спиртов. Находят применение как снотворные препараты.
Мочевина — это диамид угольной кислоты:
H2N—С—NH2
О
Мочевина является конечным продуктом распада белков. Она имеет большое биохимическое значение.
Впервые мочевина была получена Велером в 1828 г. из аммонийной соли циановой кислоты:
NH4O—C=N
h2n—с—nh2
о
В промышленности ее получают из СО2 и NH3:
СО2 + 3NH3
H2N—С—NH., + Н2О
О
Мочевина — это бесцветное кристаллическое вещество, хорошо растворимое в воде, нейтрального характера.
Химические свойства
1. Взаимодействие мочевины с кислотами. Протонирование мочевины происходит по атому кислорода, поскольку основность группы
—NH2 значительно понижена в результате сопряжения:
h2n-c—nh2 + HNO3
H2N~d—NH2 NO3'
*^S-
OH
нитрат мочевины
26. Производные игольной кислоты
323
2. Гидролиз мочевины. Мочевина при нагревании легко гидролизуется водой или водными растворами кислот и щелочей.
H2N\ | А
С=О + 2Н2О---------- 2NH3J + coj
h2n
3. Взаимодействие с азотистой кислотой. При взаимодействии с азотистой кислотой мочевина разлагается с выделением азота, оксида углерода (IV) и воды:
н’\ .
2HO-N=O + С=О——НО—N=N—С—N=N—ОН—- НО-С-ОН+ 2 N-J н/ I а
СО2 + Н2О
4. Отношение мочевины к нагреванию.
При нагревании мочевины образуется биурет:
h2n-c—NH2+ h,n—c-nh2^— h2n—c—nh—c—nh2+ nhJ
I I II I 1
О 0 0 0
биурет
Биурет хорошо растворяется в воде.
Дальнейшее нагревание приводит к образованию циануровой кислоты:
H,N —С—NH, ~т—* Н—N=C=O 2 II 2 NH3
II J изоциановая
кислота
н—о—C=N циановая кислота
О
ОН
циануровая кислота
Циануровая кислота не растворима в воде, с раствором CuSO4 в присутствии NH3 образуется комплексное соединение, окрашенное в сиреневый цвет.
324
Лекции по органической химии
5. Мочевина взаимодействует с ацилирующими реагентами с образованием ацилмочевины. N-ацильные производные мочевины называются уреидами.
\
сн—+ С=О------------ СН—
Cl h2nz "hc1 nh-c-nh2
о
уреид уксусной кислоты
Мочевина и ее производные широко используются в синтезе лекарственных препаратов.
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Классификация и номенклатура
Гетероциклическими соединениями называют большую группу органических веществ, циклы которых содержат в своем составе, кроме атомов углерода, атомы других элементов. Эти атомы называют гетероатомами (от древнегреческого «гетерос» — иной, другой).
Гетероатомами могут быть многие элементы, но особый интерес представляют гетероциклы с атомами кислорода, азота, серы. Гетероциклические соединения имеют чрезвычайно важное значение: они входят в состав витаминов, хлорофилла, гемоглобина, нуклеиновых кислот, пигментов, алкалоидов и др. Более половины лекарственных препаратов как растительного происхождения, так и синтетических, содержат в своей структуре гетероциклические фрагменты.
Классификация гетероциклов
В основе классификации гетероциклов лежат различные признаки:
• величина цикла;
• природа гетероатома;
• количество гетероатомов;
• степень насыщенности гетероцикла.
В особую группу выделяют конденсированные гетероциклы.
Первичным в классификации гетероциклов является размер цикла, который определяется количеством атомов, входящих в цикл.
По величине цикла различают трех-, четырех-, пяти-, шести-, семичленные гетероциклы.
азетидин
По природе гетероатомов выделяют кислород-, азот-, серусодержа-
щие гетероциклы:
О фуран
тиофен
326
Лекции по органической химии
По количеству гетероатомов различают гетероциклы, содержащие один, два, три и более гетероатомов.
оксазол
N-N
4 У
оксадиазол
По степени насыщенности гетероциклы делятся на насыщенные, ненасыщенные и ароматические:
пирролин ненасыщенный гетероцикл
пиридин ароматический гетеропикл
тетрагидрофуран насыщенный гетероцикл
Примерами конденсированных гетероциклических соединений являются хинолин, пурин и др.
хинолин
Номенклатура гетероциклов
Для гетероциклических соединений используют тривиальные и систематические названия. Тривиальные названия допускаются номенклатурой ИЮПАК и являются наиболее употребительными.
В систематических названиях гетероциклов отражают природу, количество и положение гетероатомов, размер и степень насыщенности цикла.
В названии выделяют префикс, корень и суффикс. В префиксе отражают природу и количество гетероатомов, в корне — размер цикла, а степень насыщенности — в суффиксе. Дтя обозначения гетероатомов используют префиксы окса- (кислород), тиа- (сера) и аза- (азот).
Размер цикла обозначается корнями -ир- (трех-), -ет- (четырех-), -ол- (пяти-), -ин- (шести-), -еп- (семичленный), а степень насыщенности — суффиксами -идин (насыщенный цикл с атомом азота), -ан (насыщенный гетероцикл без атома азота), -ин (ненасыщенный цикл).
27. Трех-, четырех- и пятичленные гетероциклические соединения...427
Для частично восстановленных соединений используют приставку дигидро-, тетрагидро- с указанием номеров атомов, к которым присоединен водород. Если атом водорода присоединен только к одному атому цикла, то в названии указывается номер гидрированного атома и буква Н. В шести- и семичленных азотсодержащих гетероциклах полная насыщенность цикла обозначается приставкой пергидро-. Количество гетероатомов одного вида указывается в названии с помощью множительных приставок ди-, три-, тетра- и т. д. При наличии в молекуле гетероцикла различных гетероатомов их нумерация определяется старшинством: 0>S>NH>N. При составлении названий делается ряд упрощений. Например:
оксиран
окса + ир + ан
префикс корень суффикс
27. ТРЕХ-, ЧЕТЫРЕХ- И ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ С ОДНИМ ГЕТЕРОАТОМОМ
Большой практический интерес для фармации представляют трех -и четырехчленные кислород- и азотсодержащие гетероциклы:
азиридин
NH
оксетан
азетидин
ОКСИРАН и ОКСЕТАН
оксиран, этиленоксид, элжсиэтан
оксетан
Оксиран — бесцветный, очень ядовитый газ с эфирным запахом, /кип *0’7 °C- Хорошо растворим в воде и органических растворителях.
Оксетан — жидкость с /кип 47,8 °C. Хорошо растворяется в воде, этаноле и диэтиловом эфире.
Способы получения
1. Циклизация $-галогеноспиртов под действием концентрированных растворов щелочей (реакция Габриэля).
328
Лекции по органической химии
а р
СН,----СН,
| г___.Ц + NaOH
ОШ СН ।________।
2-хлорэтанол.
Р-хлорэтиловый спирт
н2с—сн2
-----*- \ / + NaCl + Н2о
оксиран
Использование у-галогеноспиртов в условиях реакции Габриэля позволяет получить оксетан и его производные:
а р у
СН,— СН,—СН ,
I ‘
ан си
I_________1
+ NaOH —*-
З-хлорпропанол-г у-хлорпропиловый спирт
Н2С-СН2 н2с—о
+ NaCl + Н2О
оксетан
Образование четырехчленного гетероцикла, в отличие от трехчленного, протекает труднее из-за удаленности реакционных центров.
2. Окисление этилена кислородом воздуха при температуре 300— 400 С над серебряным катализатором (промышленный способ получения).
Н2С=СН. + О2
300-400 °C
Н2С—сн
о
2
Химические свойства
Оксиран и оксетан являются реакционноспособными соединениями. Их рассматривают как производные циклопропана и циклобутана, в которых одна группировка —СН2— замещена на гетероатом. Высокая реакционная способность оксирана и оксетана обусловлена наличием углового п торсионного напряжения (подобно малым циклоалканам) и полярных связей С—О. Большинство реакций сопровождается раскрытием цикла по месту разрыва связи «углерод — кислород» и протекает в условиях кислотного катализа.
Реакции, катализируемые кислотами. Оксиран легко протониру-ется за счет неподеленной пары электронов гетероатома:
8+5+ 8'+-5'+
V-—V
О О протонированный
оксиран
н
Образовавшийся протонированный оксиран гораздо легче подвергается действию нуклеофильных реагентов: 8Т/ > 8+.
27. Трех-, четырех- и пятичленные гетероциклические "О’динения...
329
Н2О н2с— —сн2 этандиол-1,2,
-н+ - 1 он 1 он этиленгликоль
с2н5он н,с— —сн, 2-этоксиэтанол,
~н* HC1 ” 1 ОН дне— 1 ос2н5 —сн2 этил целлозольв 2-хлорэтанол, тиленхлоргидрин
-н+ 1 ОН 1 э C1
Реакции взаимодействия оксирана с сильными нуклеофилами (аммиак, амины, металлорганические соединения).
Оксиран с аммиаком в зависимости от соотношения реагентов образует моно-, ди- и триэтаноламины:
Н2С—СН2
NH3
Н,С—СН, 2 \ / 2
о
НО—СН2-СН2\
NH но—сн2—сн/
но—сн2—сн2—nh2
этанола.мин, 2-аминоэтанол
НО—СН2-СН2\ но—ch2-ch2-n но—сн2—сн/
диэтаноламин
триэтаноламин
Оксиран реагирует с алифатическими аминами с образованием N-алкиламиноэтанолов:
Н2С\ /Н2 + R_Nh2 ---------► НО-СН2— СН2—NH— R
о
N -а л кила м и ноэта нол
Взаимодействие оксирана с реактивами Гриньяра. С магнийорга-ническими соединениями оксиран образует продукты присоединения, легко гидролизующиеся до спиртов.
8+
Н,С----СН
CH3-MgBr^ Н2^ ^Н2 нон, н+х
BrMgO СН3 -Mg(OH)Br пропанолятбромидмагний
сн3-сн2-сн2-он
пропанол-1
330
Лекции пи органической химии
Полимеризация оксирана. Возможна в присутствии сильных оснований с образованием полиэтиленоксида (полиэтиленгликоля):
Н,С—СН « \ /
О
основание
но--сн2—сн2—о—н
п
полиэтиленоксид
Полиэтиленоксид (ПЭО) — содержит различное число мономеров и может иметь различное агрегатное состояние. ПЭО находит широкое применение в качестве вспомогательных веществ в приготовлении лекарственных форм (основы мазей и суппозиториев, связывающие вещества в производстве таблеток).
Оксетан по химическим свойствам сходен с оксираном. Меньшая степень напряжения в четырехчленном цикле оксетана способствует тому, что реакции присоединения с раскрытием цикла протекают гораздо медленее. Многие реакции оксетана приводят к образованию 1,3-дизамещенных пропана.
Оксиран, оксетан и их производные имеют широкое практическое применение. Оксиран обладает дезинфицирующим действием, широко используется в промышленном органическом синтезе (производство этиленгликоля, этаноламинов, эпоксидных смол, растворителей, пластификаторов, эмульгаторов). Оксиран и оксетан входят в состав антибиотиков.
АЗИРИДИН и АЗЕТИДИН
Н |С—сн2
Н2С--NH
NH
азиридин, этиленихмин
азетидин, три метил е н и м и н
Азиридин — бесцветная жидкость с (кип 55 °C. Хорошо растворяется в воде и органических растворителях.
Азетидин — бесцветная жидкость с аммиачным запахом, /кип 63 °C. Хорошо растворяется в воде и спиртах.
Способы получения
Циклизация галогенаминов в присутствии щелочи (общий способ получения азиридина и азетидина).
а р
СН,-СН, NaOH Н2С — СН,
.2.2 NaOH 2 х / 2 + +
nh2 а NH
p-хлорэтиламин
азиридин
27. Трех-, четырех- и пятичленные гетероциклические соединения...
331
а Р У сн2-сн2-сн2
NH2 L
у-бромпропиламин
NaOH Н2С—СН2
Н2С—NH
азетидин
+ NaBr + Н2О
Взаимодействие 1,2-дихлорэтана с аммиаком в присутствии оксида кальция СаО (промышленный способ получения).
СН, — СН, ГйП Н,С—СН, /''„г! л- и о
I 2 I 2 + NH3 —° > 2 \ / 2 +СаС12 + Н2О
d Cl NH
Химические свойства
Азиридин и азетидин по химическим свойствам сходны с кислородсодержащими гетероциклами — оксираном и оксетаном.
Реакции, сопровождающиеся раскрытием цикла. Азиридин и азетидин вступают в реакции присоединения, протекающие по месту разрыва связи «углерод — азот».
NH.
h2n—ch2-ch2-nh2 этандиамин-1,2
сн2-сн2 RNH, —► R— NH—CH2-CH2-NH2
N I НОН диамин но—ch2-ch2-nh2 2-аминоэтанол Cl—ch2-ch2-nh2 2 -хлорэта на м ин
Н НС1 —►
Реакции, протекающие с сохранением цикла. Азиридин и азетидин представляют собой вторичные циклические амины и вступают в реакции алкилирования, ацилирования и нитрозирования. Наличие неподеленной пары электронов на атоме азота азиридина и азетидина обуславливает их основные свойства.
О
N—СН3
N-метилазиридин
N-ацетилазиридин
N-нитрозоазиридин
з
332
Лекции пи органической химии
Применение производных азиридина и азетидина
Производные азиридина применяются в промышленности при производстве пестицидов, инсектицидов, полимерных материалов, компонентов ракетного топлива. В медицинской практике производные азиридина нашли применение как эффективные противоопухолевые препараты (тиофосфамид и бензотэф).
Из производных азетидина важное значение имеет азетидинон-2 ф-лактам), входящий в состав антибиотиков группы пенициллина:
Н2С--СН2
HN---С%
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ С ОДНИМ ГЕТЕРОАТОМОМ
Важнейшими представителями этой группы соединений являются пиррол, фуран и тиофен:
пиррол, азол
Фуран, оксол
тиофен, тиол
Названия одновалентных радикалов приведенных гетероциклов образуют с помощью суффикса -ил, указывая цифрой или буквой греческого алфавита положение свободной валентности:
а-пиррил; p-пиррил; а-фурил; 0-фурил; а-тиенил; р-тиенил;
2-пиррил 3-пиррил 2-фурил 3-фурил 2-тиенил 3-тиенил
Способы получения
Общие способы получения пиррола, фурана и тиофена
1. Циклизация 1,4-дикарбонильных соединений (синтез Паале — Кнорра). При действии на 1,4-дикарбонильные соединения водоотнимающих реагентов (конц. H2SO4, Р2О5) получают фуран и его производные, при использовании аммиака — пиррол и его гомологи, пентасульфида фосфора P2S5 — тиофен и его производные.
27. Трех-, четырех- и пятичченные гетероциклические соеоинения...
2. Цикл реакций Юрьева. Позволяет осуществить взаимные превращения фурана, пиррола и тиофена (г ~ 450 °C, катализатор — А12О3).
Специфические способы получения
1. Получение пиррола:
а) из природных источников путем перегонки каменноугольной смолы, а также богатых белками материалов (кости, костный мозг и т. д.);
б) синтетическим путем при нагревании диаммонийной соли слизевой кислоты:
ho^j ^он
Н^с/С ‘
H4N+ -OOC/C\ /C^COO +NH4 -2СО2;-4Н2О;-NH3 X /
<Jrl HU 4 [X н
диаммонийная соль слизевой кислоты пиррол
в) при перегонке сукцинимида в присутствии цинковой пыли'.
Н,С —СН, / \
2Zn
пиррол
2ZnO
сукцинимид
334
Лекции по органической химии
2. Получение фурана:
а) в лабораторных условиях фуран получают сухой перегонкой слизевой кислоты'.
>ОН\
j \СООН-зн2о НООС^х(-)/''СООН -со2
/НО?
НООС^ 'сРц
слизевая кислота дегидрослизевая кислота
пирослизевая кислота фуран
б) в промышленности фуран получают из альдопентоз, которые при нагревании с водоотнимающими средствами подвергаются внутримолекулярной дегидратации.
Образовавшийся фурфурол окисляют до пирослизевой кислоты, термическое декарбоксилирование которой приводит к фурану:
н+<
-зн2о
3. Получение тиофена. В промышленности тиофен получают двумя методами:
а) при парофазной циклизации бутана с серой;
б) по реакции Чичибабина при пропускании смеси ацетилена с сероводородом над катализатором А1203:
27. Трех-, четырех- и пятичленные гетероциклические соединения...
Физические свойства
335
Пиррол — бесцветная жидкость с запахом, напоминающим запах хлороформа; гкип 130 °C; мало растворим в воде, хорошо растворим в этаноле и бензоле. На воздухе темнеет и осмоляется.
Фуран — бесцветная жидкость со своеобразным запахом, напоминающим запах хлороформа: гкип32 °C: нерастворим в воде, хорошо растворяется в этаноле и диэтиловом эфире.
Тиофен — бесцветная жидкость со слабым запахом сернистых соединений; г.иг] 84 °C; нерастворим в воде, хорошо растворим в этаноле, эфире и бензоле. Устойчив к высокой температуре. Окисляется на свету.
Химические свойства
Ароматичность пятичленных гетероциклов с двумя л-связями обусловлена тем, что в сопряжение с л-электронами двойных связей вступает неподеленная пара электронов гетероатомов О, У или S. Образуется за ткнутая, сопряженная система, в которой число обобщенных электронов отвечас" правилу Хюккеля (4« + 2).
В молекуле пиррола атомы углерода и атом азота находятся в состоянии хр2-гибридизации. За счет хр2-гибридизованных орбиталей
каждый атом, входящий в состав цикла, образует три о-связи, расположенные в плоскости кольца. При этом у атомов углерода и атомов
азота остается по одной негибрицизованной р-атомной орбитали, ко
торые расположены параллельно друг другу в плоскости, перпендикулярной плоскости кольца. Каждая изр-атомных орбиталей атомов углерода имеет один электрон, а на р-орбитали атома азота находится неподеленная пара электронов. При перекрывании р-орбиталей образуется единый ароматический секстет, охватывающий все атомы
цикла.
Атом азота в хр2-гибридизации, имеющий электронную конфигурацию, в которой неподеленная пара электронов занимает негибридизован-ную р-атомную орбиталь, называется пиррольным.
По аналогии с пирролом гетероатом, вносящий в л-электронную систему два электрона, занимающих p-АО, и образующий с другими атомами только о-связи, называется гетероатомом пиррольного типа.
Реакционная способность пиррола, фурана и тиофена определяется наличием в их структуре цикла с л-электроноизбыточной ароматической системой (шесть л-электронов приходится на пять атомов цикла). Однако степень ароматичности указанных гетероциклов ниже, чем у бензола, и зависит от природы гетероатома. Тиофен по своему химическому поведению в наибольшей степени напоминает бензол.
336
Лекции по органической химии
Фуран имеет наименее выраженный ароматический характер. В некоторых реакциях фуран ведет себя как ненасыщенное (диеновое) соединение. Пиррол занимает промежуточное положение.
Вследствие различной электроотрицательности гетероатома в молекулах пиррола, фурана и тиофена, в отличие от бензола, электронная плотность распределена неравномерно. Следует отметить, что на атомах углерода в «-положении электронная плотность выше, чем в P-положении, что и определяет направленность протекания реакции электрофильного замещения.
А. Общие химические свойства пиррола, фурана и тиофена
1. Взаимодействие с минеральными кислотами. Фуран и пиррол проявляют высокую реакционную способность в реакциях даже с таким слабым электрофилом, как протон. В присутствии минеральных кислот гетероциклы подвергаются протонированию, которое может осуществляться преимущественно по «-положению. Процесс протонирования сопровождается потерей ароматичности.
Х = О, NH
Образовавшиеся структуры 1 и II лишены ароматичности, и в дальнейшем происходит либо разрыв цикла с образованием полимера (наиболее вероятный процесс для фурана), либо полимеризация с сохранением цикла.
Присоединение протона может происходить и по гетероатому с образованием фуроксониевых и пирролиевых солей с последующей их полимеризацией:
пирролиевый
катион
полимер
Таким образом, фуран и пиррол в присутствии минеральных кислот осмоляются, образуя полимерные продукты темного цвета. Такое отношение к кислотам называется ацидофобностью, от латинского acidum — кислота и греческого «фобос» — страх.
"27. Трех-, четырех- и пятичленные гетероциклические соединения...
337
Введение в фурановое и пиррольное кольцо электроноакцепторных заместителей (-NO,, -СООН. -СОН и др.) приводит к снижению ацидофобности.
Тиофен из-за своей жесткой ароматической структуры не обладает ацидофобностью. Это связано со способностью атома серы в данных условиях изменять свою гибридизацию и неспособностью образовывать сульфониевые соли.
2. Реакции электрофильного замещения (SE). Как отмечалось выше, электронная плотность в молекулах фурана, пиррола и тиофена распределена неравномерно, что связано с наличием и природой гетероатома. Пара электронов гетероатома сопряжена с л-электронами кольца, электронная плотность на гетероатоме понижается, а в «-положениях — повышена (аналогично влиянию донорных заместителей в opwo-положениях аренов).
В первую очередь замещается атом водорода при а-углеродном атоме; если это положение занято, то замещение протекает по Р-положе-нию. Такая направленность замещения обусловлена тем, что при участии а-углеродных атомов образуется более устойчивый о-комплекс, благодаря большей возможности для делокализации положительного заряда.
Сульфирование. При проведении реакций SE необходимо помнить об ацидофобности и высокой реакционной способности фурана и пиррола.
Для сульфирования фурана и пиррола используют мягкий сульфирующий реагент — пиридинсульфотриоксид:
пиридин-сул ьфотриокс ид
пиридин
фуран-2-сульфокислота (X = О);
пиррол-2-сульфокислота (X - NH)
338
Лекции по органической химии
Пиридинсульфотриоксид не обладает кислотными свойствами, но сохраняет сульфирующее действие.
Тиофен легко сульфируется концентрированной серной кислотой на холоду:
тиофен
конц. H2SO4
тиофен-2-сульфокислота
Нитрование. Нитрование фурана, пиррола и тиофена проводят аце-тилнитратом — продуктом взаимодействия азотной кислоты с уксусным ангидридом:
СН — С\
/О + hono2 сн3—с_
3 "о
сн—с* + СН3СООН
'o-no2
О +сн—с*
X ~"o-no2
Х=О, S, NH
ацетил нитрат
2-нитрофуран (X = О);
2-нитропиррол (X = NH);
2-нитротиофен (X = S)
+ сн3соон
Нитрование тиофена можно проводить концентрированной азотной кислотой, однако предпочтение отдают ацетилнитрату.
Ацилирование. При ацилировании ацидофобных гетероциклов в качестве ацилирующих реагентов используют ангидриды карбоновых кислот в присутствии катализаторов — кислот Льюиса (тетрахлорид олова, хлорид цинка и др.). Тиофен ацилируют хлорангидридами карбоновых кислот в присутствии катализатора хлорида алюминия А1СЦ:
нс-сн
НСХ ХСН + (СН3СО)2О х
Х= О, NH
нс-сн
SnCl4
сн3соон
2-анетилфуран (X = О);
2-ацетилпнррол (X = NH)
2-ацетилтиофен
27. Трех-, четырех- и пятичленные гетероциклические соединения...
339
Галогенирование. Прямое галогенирование фурана и пиррола протекает очень бурно, реакция поддается контролю с трудом и может привести к разрушению гетероциклических ядер. Галогенирование осуществляют с помощью сульфурилхлорида SO2C12. Происходит постепенное замещение атомов водорода на галогены.
2-хлорпиррол (X = NH) 2.5-хлорпиррол (X = NH)
2-хлорфуран (X = О) 2,5-хлорфуран (X = О)
тетрахлорпиррол (X = NH) тетрахлорфуран (X = О)
Тиофен галогенируется на холоду непосредственным действием галогена (хлора или брома). Образуются моно-, ди-, три- и тетразаме-щенные производные тиофена:
тиофен 2-хлортиофен
2,5-дихлортиофен
тетрахлортиофен
Реакция с йодом протекает медленно в присутствии катализатора HgO.
3. Реакции присоединения. Для пятичленных гетероциклов реакции присоединения менее характерны, чем реакции электрофильного замещения. Рассмотрим реакции восстановления, окисления и диеновый синтез.
1) Реакции восстановления. Восстановление фурана происходит при высокой температуре (140 °C) при давлении 100—150 атм в присутствии катализатора (никель Ренея, палладий).
фуран
тетрагидрофуран (оксолан)
340
Лекции по органической химии
+ 2Н2
тетра гидротиофен
Восстановление тиофена происходит легко при комнатной температуре и давлении в присутствии катализатора — палладия.
Частичное восстановление пиррола с образованием 2,5-дигидро-пиррола (пирролина) происходит при действии цинка в уксусной кислоте (водородом в момент выделения). Полное восстановление пиррольного цикла до тетрагидропиррола (пирролидина) осуществляют в условиях каталитического гидрирования над платиновым или палладиевым катализатором.
пиррол ИН
[ Н]
Zn + СН3СООН
2Н2
(Pt)
пирролидин
2) Реакции окисления. Фуран и пиррол чрезвычайно чувствительны к действию окислителей. Легко окисляются даже кислородом воздуха. Окисление проходит с разрывом гетероциклического ядра и образованием пиррольных и фурановых полимерных смол. Пропускание смеси фурана с воздухом над катализатором V2O. и температуре 320 °C приводит к образованию малеинового ангидрида:
фуран
[О| v2o5
малеиновый ангидрид
При окислении пиррола хромовой кислотой образуется имид малеиновой кислоты.
[О]
н2Сг2°4
малеинимид
Тиофен окисляется с большим трудом.
3) Диеновый синтез. Занимая промежуточное положение между ароматическими соединениями и 1,3-диенами. фуран вступает в характерную для сопряженных диенов реакцию Дильса — Альдера:
27. Трех-, четырех- и пятич тнные гетероциклические соединения
341
4. Реакции, идущие с расширением цикла, а) При нагревании фури-лалкилкетонов с аммиаком получают замещенные 3-гидроксипири-дины:
метил фурил кетон
NH3 ---►
б) При взаимодействии пиррол-калия с хлороформом в присутствии этилата натрия пиррольное ядро превращается в пиридиновое. Реакция происходит через сталию образования дихлоркарбена:
пиррол-калий
C2H5ONa
+ СНС13
-КС1; -NaCl; -C2H5ONa
5. Взаимные превращения пятичленных гетероциклов с одним гетероатомом. Советский химик-органик Ю.К. Юрьев в 1936 г., учитывая высокую реакционную способность гетероциклов, показал возможность взаимных превращений фурана, пиррола и тиофена (см. «Общие способы получения фурана, пиррола и тиофена», с. 333).
Методы идентификации пятичленных гетероциклов с одним гетероатомом
Для обнаружения фурана и пиррола применяют очень простой и доступный метод — окрашивание сосновой лучины: сосновая лучинка, смоченная хлороводородной кислотой и фураном, окрашивается в интенсивно-зеленый цвет; в парах пиррола она приобретает ярко-красную окраску.
342
Лекции по органической химии
Тиофен открывают индофениновой пробой: смесь изатина с концентрированной серной кислотой в присутствии даже следов тиофена окрашивается в синий цвет.
Б. Специфические химические свойства пиррола
Пиррол относится к природным соединениям. Это бесцветная жидкость с температурой кипения 130 °C, малорастворимая в воде.
Отметим свойства, характерные для пиррола и не отмеченные в общей характеристике реакционной способности пятичленных гетероциклов.
1. Кислотно-основные свойства пиррола и продуктов его восстановления, которые не свойственны фурану и тиофену.
I
Н
пиррол проявляет слабокислые свойства
пиррол ин проявляет свойства непредельных соединений и аминов
пирролидин проявляет свойства вторичных циклических аминов
Пиррол — слабая NH -кислота (рК*= 17,5), пара электронов атома азота вступает в сопряжение с л-электронами, что приводит к поляризации NH-связи и увеличению подвижности атома водорода.
NaNH2
N
Н8+
пирролид натрия
CH3Mgl
I “СН4
Mgl н
пирро.1
N'^Na’1’ -NH3
пиррилмагний иодил
2. Соли пиррола являются реакционноспособными веществами и широко применяются в органическом синтезе для введения в молекулу пиррола алкильных и арильных заместителей. Направление реакции зависит от температурного режима:
27. Трех-, четырех- и пятичленные гетероциклические соединения...
343
Наряду с хлорангидридами карбоновых кислот в реакциях ацилирования используют сложные эфиры.
N-форм ил пиррол
3. В условиях реакции Кольбе — Шмитта пирролид натрия карбок-силируется оксидом углерода (IV):
СЗ +С°2 N Na+ пирролид натрия
натриевая соль пиррол-2-карбоновой кислоты
пиррол-2-карбоновая кислота
4. Пирролид натрия формилируется в условиях реакции Раймера — Тимана:
+ СНС13 + NaOH Na+
2-формилп иррол
пирролид натрия
344
Лекции по органической химии
5. И наконец, еще одна особенность — пиррол вступает в реакцию азосочетания.
4-
+ [C6II5-N~N]C1-
q/- бензолазопиррол
+ на
пиррол бензолдиазоний хлорид
Важнейшие производные пятичленных гетероциклов с одним гетероатомом
Производные фурана
ТЕТРАГИДРОФУРАН (ТГФ, оксолан)
Жидкость с гкип 60 °C, хорошо растворяется в воде, имеет приятный запах. По химическим свойствам ТГФ напоминает простые эфиры.
Широко применяется как растворитель в магнийоргани-
ческом синтезе.
ТГФ является основой фуранозных форм углеводов.
ФУРФУРОЛ (фуран-2-карбальдегид)
Бесцветная жидкость с /кип 162°С. На воздухе быстро темнеет в результате окисления.
Фурфурол впервые был выделен из отрубей, название происходит от латинского furfur — отруби.
В промышленности фурфурол получают путем кислотного гидро
лиза полисахаридов, в частности, пентозанов, содержащихся в соломе, кукурузных початках, шелухе подсолнечника, хлопковых коробочках и др.
Н Н
/ НОЛс—с-^ОН \ / Н// \ \н у
С C'V' / \ г— н qh он;Сч -------J н
H+,t° ---------►
-ЗН2О
фурфурол
альдопентоза
Качественная реакция на фурфурол: образование красного окрашивания с анилином в среде уксусной кислоты.
27. Трех-, четырех- и пятичленные гетероциклические соединения...
Химические свойства
а) Реакции по альдегидной группе:
345
КОН
реакция Канниццаро
NaHSOj
nh2oh
-Н2О
KCN
}—СН2ОН + \ />—с—ОН
О фурфуриловый спирт О пирослизевая
Шн К1,слота
----SOjNa гидросульфитное
U j_j производное
2NH3
-ЗН2О
оксим фурфурола
б) Реакции по фурановому кольцу. Фурфурол легко вступает в реакции SE (преимущественно по положению 5). Поскольку нельзя провести нитрование непосредственным действием концентрированной азотной кислоты, используют концентрированную азотную кислоту в среде уксусного ангидрида:
5 -нитрофурфуролдиацетат
5- н итрофу рфу рол
о
Введение двух ацетильных остатков, обладающих электроакцеп-торными свойствами, увеличивает устойчивость фурфурола в кислой среде.
346
Лекции по органической химии
Фурфурол применяется как исходный продукт в производстве ряда медицинских препаратов: фурацилина, фурадонина, фуразолидона и др.
фурадонин, 14N45-HHTpcHt)yp0ypitnKaeH)-ам и ио] гилантои н
фуразолидон,
3- [ N-(5-н итрофурфу рил иден)-амино]оксазолидон-2
Препараты нитрофуранового ряда обладают высокой антибактериальной активностью. Особенно ценным их свойством является способность проявлять эффект против устойчивых к сульфаниламидам и антибиотикам форм возбудителей.
Производные пиррола
Соединения, содержащие пиррольные ядра, синтезируются в процессе жизнедеятельности животных и растений. Важное значение имеют относящиеся к «-аминокислотам гетероциклического ряда пролии и оксипролин. Они содержат в своей структуре пирролидиновое кольцо.
пролин оксипролин
Производные пиррола входят в состав полимеров, используемых в медицинской и фармацевтической промышленности (пирролидон-2, поливинилпирролидон):
н2с—сн2 h2cXq^\^o
у-бутиролактон
NH3
-н2о
н2с—СН.
о NH
пирролидон-2 у-аминомасляная кислота
27. Трех-, четырех и пятичленные гетероциклические соединения...
М-винилпирролидон-2 поливинилпирролилон
Низкомолекулярный ПВП, молекулярная масса полимера 12 GOO-13 ООО, образует коллоидные растворы в воде и применяется для изготовления кровезаменителя «Гемодез»: среднемолекулярный ПВП (35 000—40 000) используют в фармации как связывающее средство в производстве пилюль и таблеток.
Сополимеризацией акриламида, винилпиролидона и этилакрилата получают биорастворимый полимер для глазных пленок, обеспечива
ющих продолжительное действие лекарственных веществ.
Продолжая рассматривать большую значимость производных пиррола, отметим структуру порфина, состоящую из 4-х пиррольных колец, соединенных между собой метиновыми группами. Структура порфина входит в состав таких производных соединений, как гемоглобин и хлорофилл. В свободном состоянии в природе порфин не встречается, но широко представлены его производные, которые называют порфиринами.
Производные тиофена
Находят широкое практическое применение в производстве красителей, пластмасс, инсектицидов.
Витамин Н (биотин) содержит в своей структуре гидрированный тиофен.
КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СИСТЕМЫ С ОДНИМ ГЕТЕРОАТОМОМ
Кумарон (бензо[Л]фуран) — система, состоящая из бензола и фу-
кумарон, бензо|Л|фуран
348
Лекиии по органической химии
Кумарон — жидкость с /кип 174 °C, встречается в каменноугольной смоле. Кумарон легко полимеризуется, применяется для приготовления лаков.
Индол (бензо[Л|пиррол) — конденсированная гетероциклическая система, состоящая из бензольного и пиррольного колец.
индол, бензо[Л]пиррол
Индол — кристаллическое вещество с / 52 °C, обладает неприятным запахом. Содержится в выделениях человека как продукт белкового обмена. В малых концентрациях имеет приятный запах и применяется в парфюмерии (масло жасмина).
Способы получения
1. Индол может быть выделен в чистом виде из каменноугольной смолы (до 3—5 %).
2. Циклизация N-формил-о-толуидина. Относится к реакциям конденсации кротонового типа, осуществляется в присутствии сильного основания:
N-формил-о-толуидин
NaNH2
-Н2О
индол
3. Реакция Чичибабина (конденсация анилина с ацетиленом):
4. Перегруппировка фенилгидразонов альдегидов или кетонов в присутствии кислотного катализатора (метод Фишера). Используют для получения гомологов индола с алкильными заместителями в пирроль-
ном ядре.
фенилгидразон ацетона
h2so4; д -NH3
2-метилиндол
27. Трех-, четырех- и пятичленные гетероциклические соединения...
Химические свойства
349
Индол является ароматическим соединением. По химическим свойствам оно напоминает пиррол. Апидофобен под действием минеральных кислот осмоляется. Подобно пирролу индол является слабой NH-кислотой, реагирует с сильными основаниями (щелочными металлами, щелочами, алкоксидами металлов, магнийорганическими соединениями):
индолкалий
индол
индолмагний йодид
Индол вступает в реакции электрофильного замещения SE.
В отличие от пиррола реакции SE для индола протекают по Р-по-ложению. Это объясняется преимущественным образованием более устойчивого о-комплекса, чем в случае «-замещения. Образовавшийся о-комплекс по P-положению энергетически более выгоден, т. к. сохраняется ароматичность бензольного ядра. В другом случае ароматический характер бензольного кольца нарушен, следовательно, энергетический запас молекулы существенно возрастает, что и обуславливает нестойкость такой частицы.
350
Лекции по органической хи чип
Индол восстанавливают водородом в присутствии катализатора платины:
Важнейшие производные индола
Наибольший интерес представляют производные индола, образующиеся в живом организме или применяющиеся в качестве лекарственных препаратов.
ИНДОКСИЛ (3-гидроксииндол; 3-оксоиндолин)
Желтое кристаллическое вещество с сильным фенольным запахом, rnji 85 °C. Существует в двух таутомерных формах: кетонной и енольной (кето-енольная таутомерия):
енольная форма 3-гидроксииндол
кетофор.ма
3-оксодигидроиндол
В промышленности индоксил получают при взаимодействии анилина с натриевой солью хлоруксусной кислоты. Последующее нагревание промежуточного продукта — натриевой соли N-фенилуксусной кислоты — с амидом натрия приводит к индоксилу.
анилин
NaNHj
180-200°С
индоксил
натриевая соль
N-фениламиноуксусной кислоты
Индоксил вступает в реакции, характерные для карбонильных соединений. О наличии енольной формы (фенольного гидроксила) свидетельствуют растворимость индоксила в щелочах, образование кристаллических солей со щелочными металлами и окрашенных комлек-сов с FeClr
27. Трех-, четырех- и пятичлениые гетероциклические соединения...^э!
Производные индоксила широко распространены в природе в качестве метаболитов растительных и животных организмов.
Индоксил в щелочной среде легко окисляется кислородом воздуха до красителя индиго:
индоксил
101
он-
индиго
ИНДИГО
Индиго — темно-синее с медным отливом кристаллическое вещество с гпл 390—392 °C (с разложением).
Это один из самых древних органических красителей. Характеризуется яркой окраской, высокой светоустойчивостью. Был известен еще древним египтянам и народам Индии (от португальского in digo — индийский), выделен из растений-индигоносов.
В настоящее время широкое распространение получил способ, основанный на взаимодействии анилина с натриевой солью хлоруксусной кислоты с последующим окислением образовавшегося индоксила кислородом воздуха (см. «Индоксил»),
Индиго синее легко восстанавливается глюкозой с образованием бесцветного лейкооснования — индиго белого, которое в отличие от синего хорошо растворимо в воде. На воздухе очень легко идет обратный процесс — индиго белое окисляется до синего.
индиго синее
Na2S2O„
352
Лекции по органической химии
Индиго синее имеет транс-строение, при таком расположении колец относительно двойной связи образуются ВВС между карбонильными и NH-группами.
Сульфирование индиго приводит к образованию индигокармина (динатриевая соль индиго-5,5 -дисульфокислоты).
Индигокармин используется в пищевой промышленности как краситель, в аналитической практике — как индикатор.
При окислении индиго азотной или хромовой кислотами образуется изатин:
О Н
Н О
индиго
[О]
ньо3
изатин
ИЗАТИН
Ярко-красное кристаллическое вещество (/ 203 °C). По строению представляет собой внутримолекулярный циклический амид (лактам) изатиновой кислоты. Об этом свидетельствует реакция изатина со
щелочью:
изатин
NaOH
НС1
натриевая соль изатиновой кислоты
Нагревание натриевой соли изатиновой кислоты с хлороводородной кислотой приводит к образованию изатина.
Для изатина характерна лактам-лактимная таутомерия с преобладанием лактимной формы.
27. Трех-, четырех- и пятичленные гетероциклические соединения...
353
лактамная форма лактимная форма
В изатине имеются две карбонильные группы. Более реакционноспособной является С=О группа в P-положении (кетонный карбонил), а-Амидный карбонил сопряжен с неподеленной парой электронов атома азота. Сопряжение приводит к повышению электронной плотности на а-атоме углерода, а следовательно, и к уменьшению ее реакционной способности в реакциях с нуклеофильными реагентами.
Атом водорода группы NH в молекуле изатина подвижен и может замещаться на щелочной металл.
Изатин широко применяется в органическом синтезе, а также как аналитический реагент для фотометрического определения примеси тиофена в бензоле (индофениновая реакция).
[2-амино-
ТРИПТОФАН
|-3-(^-индолил)пропионовая кислота]
Относится к незамещенным «-аминокислотам, входящим в состав белка.
Триптофан содержит асимметрический атом углерода, вследствие чего проявляет оптическую активность. Природный триптофан относится к левовращающим соединениям.
354
Лекции по органической химии
СЕРОТОНИН [ 5 -гидрокси - 3 -ф -аминоэтил )индол]
Является биогенным амином. Это гормоноподобное вещество, способствует передаче нервных импульсов, вызывает сокращение гладкой мускулатуры внутренних органов и сужение сосудов, участвует в регуляции многих жизненно важных функций организма. Нарушение обмена серотонина в организме приводит к такому заболеванию, как шизофрения.
Р-ИНДОЛИЛУКСУСНАЯ КИСЛОТА
(гетероауксин)
Это природный гормон роста (стимулятор роста) растений. Применяется в сельском хозяйстве для ускорения роста растений.
На основе индолил-3-уксусной кислоты создан противовоспалительный нестероидный лекарственный препарат индометацин:
2-метил-5-мегокси-1 -(и-хлорбензоил)инлолил-З-уксусная кислота
28. ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ С ДВУМЯ ГЕТЕРОАТОМАМИ
В данной лекции будут рассмотрены пятичленные ароматические гетероциклические соединения, содержащие два гетероатома, один из которых обязательно атом азота. Поэтому такие гетероциклические соединения объединены под общим названием — азолы. Другим гетероатомом может быть азот, кислород, сера (N, О, S).
Какие нас будут интересовать азолы? Это прежде всего:
I I
Н Н
пиразол, имидазол,
1,2-диазол 1,3-диазол
тиазол, 1,3-тиазол
оксазол, изоксазол,
1,3-оксазол 1,2-оксазол
Все приведенные гетероциклы обладают ароматическим характером. Постоянным гетерофрагментом азолов является так называемый «пиридиновый» атом азота, который для образования ароматического секстета предоставляет лишь один р-электрон. Неподеленная электронная пара «пиридинового» атома азота расположена на лд2-гибридизированной атомной орбитали и находится в плоскости гетероциклического ядра. Второй гетероатом на образование ароматического секстета предоставляет пару электронов, которая расположена нар-атомной орбитали (рис. 28.1).
Риг- 28.1. Электронное строение молекулы 1,2-азола (X = NH, О, S)
356
Лекции по органической хилши
Таким образом, вклад каждого гетероатома в образование единой л-электронной системы различен.
Азолы — соединения с высокими температурами плавления и кипения, хорошо растворимые в полярных растворителях и плохо — в неполярных.
Рассмотрим некоторые особенности азолов.
1. Кислотность и основность
Все азолы обладают слабыми основными свойствами за счет неподеленной пары электронов атома азота пиридинового типа. При взаимодействии с минеральными кислотами они образуют соли.
Например:
Н
тиазол
тиазолий хлорид
При присоединении протона к молекуле азола образуется катион, в котором сохранена ароматическая структура, а положительный заряд равномерно распределен по всей молекуле — этим можно объяснить то обстоятельство, что азолы не ацидофобны и не окисляются в присутствии сильных минеральных кислот.
В ряду азолов пиразол и имидазол, помимо основных свойств, проявляют и слабокислотные свойства за счет атома азота пиррольного типа. Следовательно, пиразол и имидазол являются амфотерными соединениями и способны вступать в реакции как с минеральными кислотами, так и со щелочами, образуя при этом соли, например:
Н
Г
N
I
Н
имидазолин хлорил
КОН
-Н2О
имидазол
имидазол калий
I н
+ к
Аналогично протекают реакции и для пиразола.
2. Азольная таутомерия
В ряду пятичленных гетероциклов с двумя гетероатомами для пиразола и имидазола характерна азольная таутомерия. Азольная тауто-
28 Пятичленные гетероциклы с двумя гетероатомами
мерия обусловлена перемещением атома водорода NH-группы к атому азота пиридинового типа:
.4. I
таутомерные формы пиразола, положения 3,5 равноценны
таутомерные формы имидазола, положения 4,5 равноценны
В молекулах диазолов происходит постоянная миграция водорода и определить, у какого из атомов азота находится водород, не представляется возможным.
В результате таутомерных превращений положения 3 и 5 в молекуле пиразола становятся равноценными, 3-метилпиразол и 5-метилпира-зол являются таутомерными формами одного и того же соединения:
11 н
3(5)-метилпиразол
А в случае имидазола равноценными являются положения 4 и 5:
н3с
н
4(5)-метилимидазол
Миграция водорода происходит настолько быстро, что выделить индивидуальные таутомеры не представляется возможным. Поэтому эти соединения так и называют 3(5)-метилпиразол и 4(5)-метил-имидазол.
358
Лекции по органической химии
ПИРАЗОЛ
пиразол, 1,2-диазол
Кристаллическое вещество с температурой плавления 70 °C, температурой кипения 187 °C, легко растворимое в воде, растворимое в спирте, эфире, бензоле; обладающее запахом пиридина.
Способы получения
1. Присоединение диазоалканов к алкинам. Реакцию используют для получения пиразола и его производных.
Пиразол получают в результате реакции присоединения диазометана к ацетилену:
ацетилен
диазометан
пиразол
2. Взаимодействие гидразина, алкиларилгидразинов с 1,3-ди-карбонильными соединениями. Этот способ чаще применяют для получения гомологов пиразола. Например, при взаимодействии гидразина с ацетилацетоном образуется 3,5-диметилпиразол:
/СНз
Н7С—С^ + H,N—NH,-
2| % 2 2
с °
II
о
3,5-диметилпиразол
ацетилацетон, пентандион-2,4
Химические свойства
1. Кислотность и основность. Пиразол проявляет амфотерные свойства, благодаря наличию в его структуре атомов азота пиридинового и пиррольного типов.
Кислотный центр
Основный центр
28. Пятичленные гетероциклы с двумя гетероатомами
359
За счет атома азота пиридинового типа пиразол проявляет основные свойства, а за счет атома азота пиррольного типа — слабые кислотные свойства:
2. Реакции с электрофильными реагентами. Направление реакции с электрофильными реагентами существенным образом зависит от природы атакующего реагента и условий их проведения.
Алкилирование, ацилирование пиразола протекает обычно с образованием продуктов N-замещения. Так, при взаимодействии пиразола с йодистым метилом в нейтральной или щелочной среде образуется N-метилпиразол:
пиразол N-метилпиразолий N-метилпиразол
йодид
Реакции электрофильного замещения с участием атомов углерода пиразольного цикла возможны только с сильными электрофильными реагентами (нитрование, сульфирование, галогенирование).
Это объясняется наличием в молекуле атома азота пиридинового типа, обладающего электроноакцепторными свойствами, что приводит к понижению реакционной способности азолов в реакциях SE по сравнению с фураном, пирролом и тиофеном. Замещение происходит преимущественно по положению 4 (наиболее удаленному от атомов азота положению).
Поскольку пиразол не обладает ацидофобными свойствами, его нитрование и сульфирование проводят концентрированными азотной и серной кислотами соответственно. Обе реакции протекают через стадию образования неактивного катиона пиразолия, что значительно затрудняет протекание реакции:
360
Лекции по органической химии
(олеум) МН
пиразол-4-сульфокислота
Галогентрование пиразола происходит сравнительно легко:
пиразол 4-хлорпиразол
3. Реакции восстановления. В зависимости от условий восстановления пиразола образуется частично гидрированный продукт — пира-золин или полностью восстановленное производное — пиразолидин:
пиразолидин
2Нэ
ТГ
пиразолин-2
Производные пиразола
Из производных пиразола нас будет интересовать пиразолон-5, структура которого лежит в основе многих лекарственных препаратов. Пиразолон-5 — таутомерное вешество и может существовать в СН2-, ОН- и NH-формах.
Ссн-сн
•ЛС. N но N"
(СН2-форма) пиразолон-5
(ОН-форма)
5-гидроксипиразол
(NH-форма) пиразолон-3
28. Пятичленние гетероциклы с двумя гетероатомами
361
В приведенном равновесии существенно преобладает СН2-форма, поэтому предпочтение отдают названию пиразолон-5.
Важнейшим производным пиразолона-5 является 3-метил-1-фе-нилпиразолон-5, который является полупродуктом для получения ряда лекарственных препаратов жаропонижающего и болеутоляющего действия — антипирина, амидопирина, анальгина.
3-Метил-1-фенилпиразолон-5 впервые был синтезирован в 1883 г. немецким химиком Людвигом Кнорром из ацетоуксусного эфира и фенилгидразина:
СН3
9^0'" h2Min-c6h5 -_-Н20 > фенилгидразин
О ОС2Н5
ацетоуксусный эфир
О HIN
сн3
сн3
100°C . „J.. N
-С2Н5ОН
С6Н5
QH5
этиловый эфир 3-фенилгидразономасляной кислоты
3-метил-1 -фенил-пиразолон-5
QH5
З-Метил-1 -фенипиразолон-5 существует также в трех таутомерных
формах:
СН2 -форма
сн3 НС—С
с6н5 ОН-форма
„СН, нс=с
Л\
ю N Н
с6н5
NH-форма
АНТИПИРИН
Получают метилированием 3-метил-1 -фенилпиразолона-5 (в NH-форме):
с6н5
3 -метил-1 -фенилпиразолон-5
снл
-HI
с6н5
антипирин, 2,3-диметил-1 -фенилпиразолон-5
Антипирин — препарат жаропонижающего действия.
362
Лекции по оргаиическс л химии
АМИДОПИРИН
Атом водорода СН-группы в молекуле антипирина обладает повышенной подвижностью, что обусловлено близостью и акцепторным действием соседней СО-группы, и легко замещается на другие атомы и атомные группы.
Амидопирин получают по следующей схеме: антипирин нитрози-руется нитритом натрия в присутствии НО, затем восстанавливают 4-нитрозоантипирин до 4-аминоантипирина, последний алкилируют йодистым метилом.
НО NO
4-нитрозоантипирин
-Н2О
2СН3] ------*-
-2 HI
амидопирин, 2,3-диметил-4-диметиламино-1 -фенилпиразолон-5, 4-(Ы,М-диметиламино)антипирин
Образование 4-нитрозоантипирина, вещества изумрудно-зеленого цвета, является качественной реакцией на антипирин.
АНАЛЬГИН
В химическом отношении анальгин представляет собой производное антипирина.
анальгин,
2,3-диметил-4-метиламино-1-фенил-гшразолон-5-Ы-метансульфонат натрия
Качественной реакцией, позволяющей отличить антипирин, амидопирин и анальгин друг от друга, является их взаимодействие с хлоридом железа (III).
28. Пятичленные гетероциклы с двумя гетероатомами
363
образуется комплекс, окрашенный в красный цвет
образует фиолетовое образует фиолетовое
окрашивание, исчезающее окрашивание, не исчезающее
при добавлении при добавлении
минеральной кислоты минеральной кислоты
Для лекарственных препаратов этого ряда характерно сочетание жаропонижающего и болеутоляющего действия.
ИМИДАЗОЛ
Бесцветное кристаллическое вещество с температурой плавления 91 °C и температурой кипения 256 °C, растворимое в воде, спирте и эфире.
Н имидазол 1,3-диазол
Способы получения
Получают имидазол из глиоксаля, аммиака и формальдегида. Поэтому имидазол иногда еще называют глиоксамином.
глиоксаль формальдегид имидазол
Химические свойства
Для имидазола характерны такие же реакции, как для пиразола. Имидазол проявляет амфотерные свойства:
имидазол калий
N I
Н
ба
N
1
Н имидазол ий хлорид
364
Лекции ио органической химии
Алкилирование и ацилирование протекает по пиридиновому атому азота:
N
-НВг
N-метилимидазол
I Н
имидазол
I
н
N-метилимидазолий бромид
Нитрование и сульфирование протекают преимущественно по 4-му положению. Реакции протекают в жестких условиях, вследствие образования в кислой среде малоактивного катиона имидазолия.
O2N
HNO,
N
I Н
Н ✓
Н
4-нитроимидазол
H,SO.
-ЗНВг ЗВг2
2,4,5-трибромимидазол
катион имидазолия
ho3s
п
имидазол-4-сульфокислота
I н
С бромом в воде или хлороформе имидазол легко образует три-бромпроизводное.
Имидазольный цикл устойчив к действию окислителей и восстановителей. Однако под действием Н2О2 происходит разрушение цикла с образованием оксамида:
Су + 2н2о2 N
I Н
О NH,
/ 2
С I
о nh2
+ СО2+ 2Н7О
оксамид,
диамид щавелевой кислоты
28. Пятичленные гетероциклы с двумя гетеро'’,помами
365
БЕНЗИМИДАЗОЛ
Бензимидазол представляет собой конденсирован ную гетероциклическую систему, которая сос~ои'т " s имидазольного и бензольного ядер.
Получают бензимидазол и его производные кондеи сацией о-фенилендиамина с кисло гам и:
о-фенилендиамин
н
По химическим свойствам бензимидазол подобен имидазолу. Он проявляет амфотерный характер, для него характерна азольная таутомерия, реакции алкилирования протекают по «пиридиновому» атому азота. А вот реакции нитрования и сульфирования бензимидазола идут с участием бензольного ядра (в положение 5).
Производным бензимидазола является такой лекарственный пре парат, как дибазол. Его получают взаимодействием о-фенилендиамина с фенилуксусной кислотой:
Х[_[ фенилуксусная кислота о-фенилендиамин
2-бензилбензим идазол
дибазол.
2-бензилбензимидазола гидрохлорид
Дибазол обладает сосудорасширяющим, спазмолитическим и гипотензивным действием.
ТИАЗОЛ
“N Бесцветная жидкость с неприятным запахом , 117°С).
Д 2 v Тиазол в своей структуре содержит два гетероатома —- азот и \1X серу.
тиазол, 1,3-тиазол
366
Лекции по органической химии
Способы получения
Основным методом синтеза тиазола и его замещенных является взаимодействие а-галогенокарбонильных соединений с тиамидами (синтез Ганча).
Взаимодействие амида тиомуравьиной кислоты с хлоруксусным альдегидом ведет к образованию тиазола:
h2n
/Я-н S
хлоруксусный альдегид
тиоформамид
-Н-,0;
-НС1
ДбН.НТ1^ + С-1
ч..../
енольная тиольная форма форма
НС—N
4 \\
НС СН
S
1,3-тиазол
Химические свойства
Наличие атома азота пиридинового типа сообщает тиазолу основные свойства (взаимодействие с НС1), по этому атому происходит алкилирование тиазольного ядра; реакции электрофильного замещения протекают по 5-му положению, а реакции нуклефильного замещения протекают по положению 2. Тиазол трудно окисляется и восстанавливается.
2-аминотиазол N-метилтиазолий
йодид
Производные тиазола
Важное значение имеет 2-аминотиазол как полупродукт в синтезе лекарственных препаратов.
НС<0 H2Nx н2к + 2 Cl s хлоруксусный тиомочевина НС-Р.Н. _HN4 rN ”"нс , - -+-, -с'КНг <Лнн, 'ci HS s 2 4 - - - ' 2-аминотиазол
альдегид
28. Пятичленные гетероциклы с двумя гетероатомами
367
2-Аминотиазол проявляет более выраженные основные свойства по сравнению с тиазолом. Центром основности по-прежнему остается циклический атом азота, так как пара электронов Ь1Н2-группы сопряжена с ароматической системой.
Алкилирование протекает по циклическому атому азота, а реакции ацилирования — по аминогруппе. 2-Аминотиазол вступает в реакцию диазотирования:
2-Аминотиазол широко используется в производстве лекарственных препаратов. Производными 2-аминотиазола являются сульфаниламидные препараты норсульфазол и фталазол, которые обладают антимикробным действием:
норсульфазол
фталазол
Особую значимость имеет также продукт полного восстановления тиазола — тиазолидин, который входит в состав антибиотиков пенициллинового ряда общей формулы:
ноос
тиазол идиновое ядро
Н3С
.0 NH-C' R
Р-лактамный цикл
368
Лекции по органической химии
ОКСАЗОЛ И ИЗООКСАЗОЛ
о о
оксазол, изооксазол,
1,3-оксазол 1,2-оксазол
Оксазол и изооксазол — бесцветные жидкости (г оксазола 69 °C, изооксазола 95 °C).
Оксазол и изооксазол проявляют ароматические свойства. Реакции электрофильного замещения протекают преимущественно по 4-му положению. Оба гетероцикла проявляют слабые основные свойства.
Ядро изооксазола входит в состав противотуберкулезного препарата — циклосерина:
Среди производных оксанола известны вещества, обладающие антибактериальным, жаропонижающим, анальгетическим и снотворным действием.
29. ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ С ОДНИМ ГЕТЕРО АТОМОМ
К наиболее распространенным представителям этой группы соединений относятся гетероциклы, содержащие в качестве гетероатома атом азота (азины):
пиридин хинолин
ИЗОХИНОЛИН
акридин
и неароматические кислородсодержащие гетероциклические соединения, такие, как:
а-пиран у-пиран
1
ПИРИДИН
Пиридин — бесцветная жидкость с характерным запахом, смешивается с водой и эфиром во всех соотношениях. Образует азеотропную смесь состава c5h5n • ЗН2О.
Положения 2,6 в молекуле пиридина называют сх,сх'-положсния-ми, положения 3,5 обозначают — Р,Р', а положение 4 — у-положение.
а-этилпиридин, 2-этилпиридин
Р-этилпиридин, 3-этилпиридин
а-этилпиридин, 4-этилпиридин
Способы получения
Пиридин и его монометильные производные — а, р, у-пиколины получают из каменноугольной смолы.
Кроме того, существуют синтетические методы получения:
370
Лекции по органической химии
1. Нагревание до 400 °C уксусного альдегида с аммиаком в присутствии Alflj приводит к смеси метильных производных пиридина (пико-гинов):
а-пиколин у-пиколин
2. При нагревании акролеина с аммиаком получается ft-пиколин:
//
н2с=сн-сч
Н
+ NH3
3. Взаимодействие ацетилена с синильной кислотой'.
сн=сн
+ №СН
СН=СН
4. Конденсация ацетальдегида и формальдегида с аммиаком:
2СН3-С-Н +Н-С-Н +NH3
+ зн2о + н|
Используя другие альдегиды и их смеси, получают различные ал-килпиридины.
Строение и химические свойства
Пиридин относится к ароматическим соединениям. Его молекула имеет плоское строение. Атомы углерода и азота находятся в состоянии .$р2-гибридизации. В образовании ароматического секстета принимают участие пять />-атомных орбиталей атомов углерода и одна />-атомная орбиталь атома азота. Неподеленная пара электронов атома азота занимает хр2-гибридную орбиталь и не принимает участия в образовании ароматического секстета, что сообщает пиридину основные свойства. Атом азота такой конфигурации получил название пиридинового. Атом азота, обладая большей электроотрицательностью, понижает электронную плотность на атомах углерода в молекуле пиридина. В меньшей степени затрагиваются положения 3 и 5 ф-положения), и максимальная электронная плотность сосредоточена на атоме азота.
29. Шестичленные гетероциклические соединения с одним гетероатомом
371
Влияние атома азота на электронную плотность пиридинового ядра сравнимо с влиянием нитрогруппы на бензольное кольцо в молекуле нитробензола, но более сильное.
Гетероциклы, в которых гетероатом понижает электронную плотность на атомах углерода ароматического кольца, называют л-дефи-цитными. То есть пиридин является л-дефицитной ароматической системой.
Характерные для пиридина реакции можно разделить на три группы:
I. Реакции, протекающие с участием гетероатома.
II. Реакции замещения атомов водорода пиридинового ядра.
III. Реакции восстановления и окисления.
I. Реакции, протекающие с участием гетероатома
1. Взаимодействие с кислотами. За счет неподеленной пары электронов атома азота пиридин проявляет слабые основные свойства (рКвн+ 5,25).
При взаимодействии с сильными минеральными и органическими кислотами он образует хорошо растворимые пиридиниевые соли:
+НВг
Вг
пиридиний бромид
2. Реакция с оксидом серы (VI). С участием неподеленной пары электронов атома азота пиридин легко реагирует с триоксидом серы, образуя донорно-акцепторный комплекс:
+ SO3
пиридинсульфотриоксид
372
Лекции по органической химии
3. Взаимодействие с алкил- и ацилгалогенидами. В этих реакциях пиридин образует четвертичные соли N-алкил- и N-ацилпиридиния соответственно.
I
СН3
N-метилпиридиний йодид
CH.I
СН,СОС1
N-ацетилпиридиний хлорид
Реакции взаимодействия пиридина с алкил- и ацилгалогенидами указывают на нуклеофильные свойства пиридинового атома азота. Соли N-ацилпиридиния используются как ацилирующие реагенты.
II. Реакции замещения атомов водорода пиридинового ядра
Для пиридина характерны как реакции электрофильного замещения (SE), так и нуклеофильного замещения (S.J.
1. Реакции электрофильного замещения (SE). Электроноакцепторное влияние атома азота в молекуле пиридина приводит к тому, что реакции электрофильного замещения для пиридина протекают с трудом.
Реакции нитрования, сульфирования и галогенирования проходят медленно в жестких условиях и с низкими выходами. При этом электрофильный реагент направляется в Р-положение:
3-нитропиридин
3-бромпиридин
H2SO4
Hg2-, 220 °C
3-пиридинсульфоки слота
2. Реакции нуклеофильного замещения В реакции нуклеофильного замещения пиридин вступает легко. Замещение протекает по положениям 2,4,6, легче всего нуклеофильный реагент вводится в положения 2,6 («-положения).
29. Шестичленные гетероциклические соединения с одним гетероато.шш
373
Ярким примером реакции этого типа является аминирование пиридина амидом натрия по Чичибабину.
Реакция протекает по механизму SN2:
NaNH-,
о-комплекс
2-аминопиридин, а-аминопиридин
Аналогично протекает гидроксилирование:
КОН, 300 X
-Н2
2-гидроксипиридинолят 2-гидроксипиридин, калия а-гидроксипиридин
IIL Реакции восстановления и окисления
1. Восстановление. Пиридин восстанавливается легче, чем бензол. В зависимости от природы восстановителя и условий проведения реакции образуются различные продукты.
6Н
Na + C2HSOH ЗН2
Pt, Pd, Rh 4H2
Ni, 180 "С
н
пиперидин
CH3-CH2-CH2-CH2-CH2-NH2
пентанамин-1
Реакция с раскрытием пиридинового цикла протекает при высокой температуре в присутствии никелевого катализатора.
2. Окисление. Пиридиновое кольцо устойчиво к действию окислителей. Алкилпиридины, подобно алкилбензолам, окисляются легко с образованием соответствующих пиридинкарбоновых кислот:
никотиновая кислота
СООН
+ Н2О
Лекции по еретической химии
374
При действии пероксикислот пиридин окисляется по атому азота с образованием N-оксида.
сн3сооон
(Н2О2 + CHjCOOH)
О’
N-оксид пиридина
N-оксид пиридина и его производные легко восстанавливаются:
РС13
+ РОС13
Следует отметить, что N-оксид пиридина в отличие от пиридина довольно легко вступает в реакции электрофильного замещения. Это обусловлено электронодонорным влиянием атома кислорода. В результате смещения электронной плотности от атома кислорода в кольцо на атомах углерода в а и у-положениях электронная плотность повышается.
Замещение идет легче по у-положению, так как оно пространственно не затруднено. К тому же рядом стоящий атом азота понижает электронную плотность в а-положениях.
При нитровании N-оксида пиридина азотной кислотой с высоким выходом образуется ^оксид-4-нитропиридин:
пиридин
N-Оксид пиридина используют для получения у-замещенных производных пиридина:
29. Шестичленные гетероциклические соединения с одним гетероатомом
375
Алкилирование и ацилирование N-оксида проходит по атому кислорода с образованием солей N-алкокси- и N-ацилоксипиридиния соответственно:
N-метоксипиридиний йодид
СН3СОС1
N-ацетоксипиридиний хлорид
Для N-оксида пиридина характерны также реакции нуклеофильного замещения, протекающие по положениям 2,4,6 (а и у-положе-ниям).
Важнейшие производные пиридина
ПИКОЛИНЫ
Пиколинами называют монометильные производные пиридина:
а-пиколин, 2-метилпиридин
По химическим свойствам пиколины сходны с пиридином. Они образуют соли с сильными кислотами и алкилгалогенидами, окисляются до N-оксидов, восстанавливаются до производных пиперидина, аналогично метилпроизводным бензола окисляются до пиколиновой, никотиновой и изоникотиновой кислоты.
376
Лекции по органической химии
Например, при окислении Р-пиколина образуется никотиновая кислота:
Р-ПИКОЛИН
[О]
В молекулах а- и у-пиколинов водородные атомы в метильных группах подвижны (о,л-сопряжение), поэтому они в отличие от Р-пиколинов вступают в реакции конденсации с альдегидами ароматического ряда.
С6Н—, ZnCl н
180 °C,-Н2О
>CH=CH-C,HS
О J
а-стирил пиридин, 1-(2-пиридил)-2-фенилэтен
ГИДРОКСИПИРИДИНЫ
В зависимости от положения гидроксильной группы различают три изомера:
а-гидроксипиридин р-гцдроксипиридин у-гидроксипиридин
Гидроксипиридины представляют собой кристаллические веще-
ства, хорошо растворимые в этаноле, ацетоне, малорастворимые в диэтиловом эфире и бензоле.
Особенностью а- и у-изомеров является то, что они существуют в двух таутомерных формах:
2-гид роксипиридин пиридинол-2
(гидроксиформа)
пиридон-2 4-гидроксипиридин
(оксоформа) пиридинол-4
(гидроксиформа)
пиридон-4
(оксоформа)
29. Шестичленные гетероциклические соединения с одним гетероатомом
377
В молекуле Р-гидроксипиридина отсутствуют сопряжение неподеленной пары электронов атома кислорода гидроксильной группы с гетероатомом, что делает невозможным таутомерию.
Р-Гидроксипиридины в водных растворах существуют в нейтральной и биполярной формах в соотношении 1:1.
Н нейтральная биполярная
форма форма
Гидроксипиридины являются бифункциональными соединениями, они проявляют свойства пиридина и фенола. Так, с водным раствором FeCl3 все они образуют пурпурное окрашивание различной интенсивности. Свойства фенола в большей степени проявляет 3-гидроксипиридин, гидроксильная группа которого не сопряжена с гетероатомом.
С алкилгалогенидами 2- и 4-гидроксипиридины образуют N-ал-килпиридоны, а 3-гидроксипиридин дает соли N-алкилпиридиния:
пиридинол-2
пиридинол-3 N-метил-З-гидрокси-пиридиний йодид
Из производных гидроксипиридинов отметим пиридоксин (витамин В6).
Применяется пиридоксин в виде соли с хлороводородной кислотой при В(-гиповитами-нозе, токсикозе, анемиях, лейкопениях и заболеваниях нервной системы.
АМИНОПИРИДИНЫ
В зависимости от положения аминогруппы различают:
а-аминопиридин, 2-аминопиридин пиридиламин-2
Р-аминопиридин, 3-аминопиридин пиридиламин-3
У -аминопиридин. 4-аминопиридин пиридиламин-4
378
Лекции по органической химии
Аминопиридины — белые кристаллические вещества, легко растворимые в воде, этаноле и других органических растворителях.
Аминопроизводные пиридина обладают более сильными основными свойствами, чем пиридин и анилин. Они содержат в своем составе два ониевых основных центра — атом азота пиридинового типа и атом азота аминогруппы.
В молекулах а- и у-аминопиридинов аминогруппа находится в сопряжении с гетероатомом, что приводит к понижению основных свойств аминогруппы. Поэтому 2 и 4-аминопиридины образуют соли только с одним эквивалентом кислоты:
+ НС1 ~ -
Н
4-аминопиридиний
хлорид
В молекуле Р-аминопиридина аминогруппа не сопряжена с гетероатомом, поэтому он образует соли с двумя эквивалентами кислоты:
дихлорид
а- и у-Аминопиридины являются таутомерными веществами и существуют в аминной и иминной формах:
Химические свойства аминопиридинов зависят от положения аминогруппы. Так, 3-аминопиридин проявляет свойства ароматических
29. Шестичленные гетероциклические соединения с ооним гетероатомом
379
аминов, легко вступая в реакции алкилирования, ацилирования, диазотирования по аминогруппе с образованием устойчивых солей диазония.
2 и 4-Аминопиридины алкилируются по циклическому атому азота, а ацилируются по атому азота аминогруппы:
4-амино- 1-метил-пиридин йодид
ПИРИДИНКАРБОНОВЫЕ КИСЛОТЫ
В зависимости от положения карбоксильной группы в молекуле различают а-, Р- и у-пиридинкарбоновые кислоты:
СООН
пиколиновая кислота, 2-пиридинкарбоновая кислота
никотиновая кислота, 3-пиридинкарбоновая кислота
изоникотиновая кислота, 4-пиридинкарбоновая
кислота
Пиридинкарбоновые кислоты представляют собой белые кристаллические вещества, способные возгоняться, растворимые в горячей воде. Их получают окислением соответствующих пиколинов.
По карбоксильной группе они образуют соли, сложные эфиры, галогенангидриды, амиды, гидразиды и другие функциональные производные. Для них характерны реакции декарбоксилирования, причем а-кислоты декарбоксилируются легко, а у— только при нагревании со щелочью. Вследствие наличия кислотного и основного центров пиридинкарбоновые кислоты проявляют амфотерные свойства и реагируют с кислотами и щелочами:
3-карбоксипиридиний хлорид
380
Лекции по органической химии
В кристаллическом состоянии и частично в растворах они существуют в виде внутримолекулярных солей (ивиттер-ионы).
I Н
пвиттер-ион никотиновой кислоты
Пиридинкарбоновые кислоты в результате электроноакцепторного влияния атома азота являются более сильными кислотами, чем бензойная.
Кислотность а- и у-пиридинкарбоновых кислот выше, чем Р-кис-лоты, так как в а- и у-пиридинкарбоновых кислотах карбоксильная группа находится в сопряжении с атомом азота, что приводит к усилению ее депротонизации.
Никотиновая кислота и ее амид (никотинамид) известны в медицине как провитамин витамина РР. При недостатке витамина РР развивается заболевание кожи — пеллагра, поэтому его называют анти-пелларгическим.
М,М-Диэтиламид никотиновой кислоты под названием кордиамин (25% — водный раствор) применяется как средство, стимулирующее центральную нервную систему.
Схема синтеза амида и М,М-диэтиламида никотиновой кислоты:
Производные изоникотиновой кислоты применяются в медицине как противотуберкулезные средства:
29. Шестичленные гетероциклические соединения с одним гетере томом
381
гидразид изоникотиновой кислоты, изониазид
фтивазид
Изониазид и фтивазид применяют в медицине для лечения различных форм туберкулеза.
ПИПЕРИДИН
Пиперидин (гексагидропиридин) — бесцветная жид-| । кость с резким аммиачным запахом, смешивается с водой
и большинством органических растворителей. Обладает N свойствами вторичных аминов и является более сильным основанием, чем пиридин (рКвн+ 11,22).
Образует соли с кислотами, с HNO2 — N-нитрозопроизводное, bctv пает в реакции алкилирования и ацилирования по атому азота и др.:
СН3СОС1
V
сосн3
Ы-ацетилпиперидин
КИСЛОРОДСОДЕРЖАЩИЕ ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ С ОДНИМ ГЕТЕРОАТОМОМ
а-пиран, 2Н-пиран
4
у-пиран, 4Н-пиран
а- и у-Пираны представляют собой неустойчивые соединения.
В отличие от а- и у-пирана, их оксопроизводные являются довольно устойчивыми ароматическими соединениями.
382
Лекции по органической химии
а-Пирон — бесцветная жидкость с запахом свежего сена, у-пирон — бесцветное кристаллическое вещество.
В молекулах а- и у-пиронов неподеленная пара электронов циклического атома кислорода находится в сопряжении с л-электронами двойной связи оксогруппы.
Делокализацию электронной плотности в их молекулах можно представить в виде резонансных структур:
у-Пирон проявляет слабые основные свойства и при взаимодействии с минеральными кислотами и алкилгалогенидами образует соли
пирилия:
4-гидроксипирилий хлорид
4-метоксипирилий йодид
Пирилиевый катион содержит замкнутую л-систему из шести электронов и обладает ароматическим характером. у-Пирон не вступает в характерные для альдегидов и кетонов реакции (образования оксимов, гидразонов).
кумарин (бензо[/>]пирон-2)
КУМАРИН
Кумарин представляет собой конденсированную систему, состоящую из бензольного и a-пиронового ядра. По строению он является лактоном дис-о-гидроксикоричной кислоты (кумариновой кислоты).
29. Шестичленные гетероциклические соединения с одним гетероатомом
Синтетический кумарин получают по реакции Перкина из салицилового альдегида:
салициловый альдегид
CH.CCCNa
+ (СН,СО),О---------•
3 2 180 "С
Химические свойства кумарина обусловлены наличием лактонного и бензольного колец. При нагревании со щелочами образуется соль о-гидроксикоричной кислоты, при подкислении которой происходит быстрая рециклизация в кумарин:
Ц /Н
динатриевая соль кумариновой кислоты
По бензольному кольцу кумарин вступает в реакции электрофилы ного замещения (нитрование, сульфирование) по положению 6:
На основе производных кумарина созданы лекарственные препараты, проявляющие антикоагулянтную активность. — неодикумарин. фепромарон, синкумар и др.
О
ХРОМОН
Хромон представляет собой конденсированную гетероциклическую систему, которая состоит из бензольного и у-пиронового циклов. По химическим свойствам хромон сходен с у-пироном, при обработке сухим НО он образует соль бензопирилия:
384
Лекции по органической химии
хромо н
Реакции электрофильного замещения по бензольному кольцу для хромона затруднены и протекают лишь при наличии в молекуле электронодонорных заместителей.
Широко распространенными в природе производными хромона являются хромон и изофлавон, называемые флавоноидами:
изофлавон, 3-фенилхромон
В растениях они находятся в виде гликозидов, проявляют биологическую активность.
30. КОНДЕНСИРОВАННЫЕ ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ С ОДНИМ ГЕТЕРОАТОМОМ
Важнейшими пиридинсодержашими конденсированными системами являются хинолин, изохинолин, акридин.
хинолин
изохинолин
акридин
хинолин, бензо[Л]пиридин
ХИНОЛИН
Хинолин представляет собой бициклическую систему, содержащую конденсированные пиридиновое и бензольное кольца. Его также можно рассматривать как гетероциклический аналог нафталина (1-азанафталин).
В пиридиновом кольце положения обозначаются греческими буквами а(2), Р (3), у (4). Хинолин — бесцветная жидкость с неприятным запахом (гкип 237 °C), хорошо смешивается с водой,
этанолом, диэтиловым эфиром, перегоняется с водяным паром.
Способы получения
Хинолин и его метильные производные получают из продуктов перегонки каменноугольной смолы.
Среди синтетических способов получения хинолина и его производных важнейшими являются синтез Скраупа и синтез Дебнера — Миллера.
7. Синтез Скраупа. Для получения хинолина по методу Скраупа нагревают анилин с глицерином и конц. H2SO4 в присутствии окислителя — нитробензола:
CHL-OH I 2
+ сн-он I сн2-он
C6H5NO2, t
kohu.H2SO4
Механизм реакции включает три последовательные стадии.
386
Лекции по органической химии
На первой стадии глицерин под действием конц. H2SO4 подвергается дегидратации с образованием акролеина:
СН?-ОН I сн-он
снгон
H2SO4, t II 2
---------* сн
-2Н2° 1^0
с<н
акролеин
На второй стадии образовавшийся акролеин вступает в реакцию конденсации с анилином:
н\ Ян’; сс-н N-CH2
I н
конц. H2SO4
-Н20
1,2-дигидрохинолин
На третьей стадии реакции 1,2-дигидрохинолин окисляется нитробензолом в хинолин:
c6h5no2
- c6h5nh2>
-н2о
Для получения производных хинолина, содержащих заместители в бензольном кольце, вместо анилина используют его соответствующие производные со свободным рр/ио-положением.
30. Конденсированные шестичленные гетероциклические соединения...
387
Реакция была открыта в 1881 г. австрийским химиком-органиком З.Х. Скраупом. В литературе эту реакцию иногда называют скраупиро-ванием.
2. Синтез Дебнера — Миллера (реакция открыта в 1881 году). Является модификацией синтеза Скраупа.
Его применяют для получения производных хинолина, содержащих алкильный заместитель в пиридиновом кольце.
В качестве исходных веществ используют ароматический амин и альдегид, который может вступать в реакцию кротоновой конденсации. Синтез проводят в присутствии хлорида цинка (II), хлороводородной или другой минеральной кислоты.
Например, для получения 2-метилхинолина (хинальдина) используют анилин и ацетальдегид.
На первой стадии происходит кротоновая конденсация двух молекул альдегида с образованием а,Р-ненасыщенного альдегида:
/О но, t
2сн—с' _но> сн3—СН=СН—
н 2 ХН
кротоновый альдегид,
бутен-2-аль
Далее происходит взаимодействие кротонового альдегида с анилином. Окислителями выступают азометины С6Н5—N=CH—R, которые образуются в процессе реакции:
s£os‘ ^6' сн, .
Г fl / СН-СН-С^ ---------------- I Др. I н ,
^NH2 1 Н H2so4
СН3 СН3
3-анилинобутаналь
2-метил- 2-метил-
1,2-дигидро- хинолин,
хинолин хинальдин
Химические свойства
Хинолин является ароматическим соединением: его молекула имеет плоское строение и единую сопряженную 10 л-электронную систему.
По химическим свойствам хинолин схож с пиридином. Для него характерны реакции:
388
Лекции по органической химии
• с участием гетероатома;
• электрофильного и нуклеофильного замещения;
• окисления;
• восстановления.
7. Реакции по гетероатому. Наличие в молекуле хинолина атома азота пиридинового типа обуславливает основные свойства. Как основание хинолин немного слабее пиридина (рКвн+ хинолина в Н2О = = 4,94, рКвн+пиридина — 5,25).
Хинолин образует соли с сильными кислотами, алкил- и ацилга-логенидами:
хинолиний хлорид
N-метилхиноли ний йодид
N-ацетилхинолиний хлорид
2. Реакции электрофильного и нуклеофильного замещения. В результате электроноакцепторного влияния гетероатома в молекуле хинолина электронная плотность распределена неравномерно: в пиридиновом кольце она ниже, чем в бензольном. Поэтому при действии электрофильных реагентов замещение проходит по бензольному кольцу, а нуклеофильное — по пиридиновому.
кают преимущественно по положению 5 и 8.
При обработке нитрующей смесью образуются 5- и 8-нитрохи-нолины. Сульфирование концентрированной серной кислотой при 220 °C
30 Конденсированные шестичлениые гетероциклические соединения...
389
приводит к образованию 8-хинолинсульфокислоты, а при 300 °C — 6-хинолинсульфокислоты (в этих условиях происходит перегруппировка 8-изомера в более термодинамически выгодный 6-изомер):
В реакции нуклеофильного замещения хинолин вступает значительно легче, чем пиридин. При этом реакции протекают по положению 2.
2-аминохинолин
NaNH, (NH,)
КОН, 300 °C
2-гидроксихинолин
он
3. Реакции восстановления и окисления. При восстановлении хинолина в первую очередь восстанавливается пиридиновое ядро. Образование продуктов реакции зависит от катализатора и условий проведе-
ния.
1,2,3,4-тетрагидрохинолин
390
Лекции по органической химии
Окисление хинолина перманганатом калия в щелочной среде приводит к расщеплению бензольного кольца и образованию хинолиновой (2,3-пиридиндикарбоновой) кислоты. В присутствии пероксикислот хинолин образует N-оксид:
КМпО4, он
КМпО4, он
НООС^. .
J J + 2СО2| + Н2О НООС N
хинолиновая кислота,
2,3-пиридиндикарбоновая кислота
Важнейшие производные хинолина
Хинолиновое ядро является структурным фрагментом многих алкалоидов и лекарственных препаратов.
8-ГИДРОКСИХИНОЛИН
он
Бесцветное кристаллическое вещество с гпл 75—76 °C и характерным запахом, мало растворимо в воде, диэтиловом эфире, бензоле. Его получают нагреванием орто-аминофенола с глицерином в присутствии концентри-
рованной серной кислоты (синтез Скраупа) или сплавлением 8-хинолинсульфокислоты со щелочью:
SO3H
3NaOH (сплавл.)
—Na2SO3
НС1
ОН
С катионами многих металлов (Mg2\ AI3+, Zn2+ ит. д.) 8-гидрокси-хинолин способен образовывать нерастворимые координационные комплексы — хелаты, что используется в качественном анализе:
ОН
MgCl2
2NaOH
30. Конденсированные шестичленные гетероциклические соединения...
391
Это свойство 8-гидроксихинолина также лежит в основе его при
менения в медицине.
Множество производных этого соединения (хинозол, нитроксо-лин (5-НОК), энтеросептол) обладают антибактериальной и противогрибковой активностью.
энтеросептол, 8-гидрокси-7-йод-5-хлорхинолин
хинозол.
8-гидроксихинолиния сульфат
нитроксолин (5 НОК), 8-гидрокси-5-нитро-хинолин
ХИНОЛИН-4-КАРБОНОВАЯ КИСЛОТА (ЦИНХОНИНОВАЯ)
СООН
Из ее производных следует отметить лекарственный препарат совкаин, который является местным анестетиком.
С н
CONH(CH,)2bC 2 5 22 С2Н5-НС1
ОСН2СН2СН2СН3
гидрохлорид диэтилам и ноэтил ам ид а 2-и-бутоксицинхониновой кислоты
изохинолин
8 I
ИЗОХИНОЛИН, бензо[с] пиридин
Изохинолин является изомером хинолина, но в отличие от него, циклы соединены вдоль связи С3—С4 пиридинового кольца и поэтому его называют бензо[с]пиридин. Нумерацию атомов в молекуле изохинолина проводят как в конденсированных аренах с учетом положения гетероатома.
Изохинолин — это бесцветное кристалли
ческое вещество с гпл 24,6 °C и запахом, напоминающим запах хинолина, растворяется в воде и большинстве органических растворителей.
Способы получения
Изохинолин содержится в хинолиновой фракции каменноугольной смолы (1%).
392
Лекции по органической химии
В лаборатории синтез изохинолина и его производных осуществляют по реакции Бишлера—Наперальского.
При взаимодействии Р-фенилэтанамина с хлорангидридами карбоновых кислот образуется N-ацил-Р-фенилэтанамин, который при действии водоотнимающих средств (РОС13, Р2О5 или полифосфорная кислота) подвергается циклодегидратации с образованием замещенных 3,4-дигидрохинолина.
Последующее дегидрирование приводит к образованию изохинолина.
Р-фенилэтиламин
N-ацил-Р-фенилэтиламин
l-R-замещенный
3,4-дигидроизохинолин
l-R-замещенный изохинолин
Химические свойства
По химическим свойствам изохинолин во многом напоминает хинолин. За счет гетероатома он проявляет слабоосновные свойства, взаимодействует с сильными кислотами. Как основание изохинолин немного сильнее хинолина (рКвн+ изохинолина 5.14; рКвн+ хинолина 4,94). Алкилирование и ацилирование протекает по гетероатому с образованием солеобразных продуктов присоединения.
Изохинолин вступает в реакции электрофильного замещения, как и хинолин, в положениях 5 и 8 бензольного кольца.
Нуклеофильное замещение протекает в положении 1.
30. Конденсированные шестичленные гетероциклические соединения...
393
Изохинолин труднее восстанавливается, чем хинолин.
H2(Ni)
1,2,3,4-тетрагидроизохинолин
При окислении изохинолина щелочным раствором КМпО4 окислению подвергаются оба ядра и в результате образуется смесь кислот: фталевой и 3,4-пиридиндикарбоновой кислот.
КМпО4, он
СООН
ноос
ноос
СООН
фталевая кислота 3,4-пиридин-дикарбоновая кислота
394 Лекции по органической химии
Подобно хинолину при действии пероксикислот изохинолин образует N-оксид изохинолина.
Производными изохинолина являются многие алкалоиды изохинолинового ряда — морфин, папаверин, наркотин и др.
АКРИДИН
акридин
дибензо[Ь,е]пиридин
Акридин представляет собой конденсированную систему из двух бензольных и одного пиридинового ядер. Акридин (дибензо[Ь,е]пиридин) подобен антрацену, в котором группа =СН- замещена на атом азота (10-азаантрацен). Нумерацию атомов проводят таким образом, чтобы азот получил наибольший номер — 10. Положения 9 и 10 называют л/езо-поло-жениями.
Акридин — светло-желтое кристаллическое вещество с гпл 111 °C и характерным запахом, легко возгоняется. Хорошо растворяется в органических растворителях, мало растворим в воде. Разбавленные растворы имеют синюю флуоресценцию. Растворы солей обладают зеленой флуоресценцией, при разбавлении водой происходит гидролиз и зеленая флуоресценция переходит в синюю, характерную для свободного акридина. Акридин оказывает на слизистые оболочки и на кожу раздражающее действие, вызывает чувство жжения, откуда и произошло его название «асег» — едкий, акридин.
Акридин выделяют из антраценовой фракции каменноугольной смолы.
Чаще акридин получают синтетическим путем, используя следующие методы:
1. Конденсация дифениламина с карбоновыми кислотами. Акридин получают по способу Бернтсена при нагревании дифениламина с одноосновными карбоновыми кислотами в присутствии /пСЦкак водоотнимающего средства.
дифениламин
ZnCl2
акридин
+ 2Н2О
30. Конденсированные шестичленные гетероциклические соединения...^95
2. Циклизация N-фенилантраниловой кислоты по реакции Дроздова—Магидсона—Григоровского:
9,10-ди гидроакридин,
акрилан
В молекуле 9-хлоракридина атом хлора проявляет большую подвижность, поэтому его используют для получения производных по положению 9:
акридан
396
Лекции по органической химии
Химические свойства
Реакции с участием гетероатома. Акридин является слабым основанием. Он обладает более слабыми основными свойствами, чем пиридин и хинолин.
Акридин, подобно пиридину, взаимодействуя с кислотами, образует соли, которые называются акридиниевыми. С надкислотами образует N-оксид акридина:
акридиний хлорид
С1
йодистый N-метилакридиний
С электрофильными реагентами акридин взаимодействует с боль
шим трудом и неоднозначно.
Реакции нуклеофильного замещения для акридина идут достаточно легко по положению 9. Например, при действии на акридин амида натрия образуется 9-аминоакрид ин:
NH3 (жидкий)
NaNH,
9-аминоакридин
Акридин достаточно устойчив к действию окислителей.
При действии дихромата калия в уксусной кислоте акридин окисляется в акридон-9, который является таутомерным веществом:
30. Конденсированные шестичленные гетероциклические соединения...
Хотя в акридоне-9 и содержится группа С=О, однако она не обладает кетонным характером. Это объясняется тем, что необобщенная электронная пара атома азота сопряжена с карбонилом, что снижает ее электрофильные свойства.
Молекулы акридона-9 благодаря межмолекулярным водородным связям образуют димеры. Акридон-9 в свободном состоянии представляет собой димер с высокой температурой плавления:
При окислении акридина в жестких условиях (КМпО4 в щелочной среде) затрагивается одно из бензольных ядер с образованием хинолин-2,3-дикарбоновой кислоты:
КМпО4 (ОН Г'
СООН
СООН
+ 2СО-,
хинолин-2,3-дикарбоновая кислота, акридиновая кислота
Восстановление акридина происходит подобно антрацену по наиболее активным л/езо-положениям. Так, при действии натрия в спиртовом растворе или при каталитическом гидрировании акридин легко восстанавливается в 9,10-дигидроакридин (акридан):
Акридан обладает очень слабыми основными свойствами. Так же, каки у дифенилами на, неподеленная электронная пара атома азота уча-
398
Лекции по органической химии
ствует в сопряжении с л-электронами бензольных колен. Только с сильными кислотами он образует соли, которые легко гидролизуются.
Важнейшие производные акридина
9-АМИНОАКРИДИН
9-Аминоакридин — желтое кристаллическое вещество с t 236—237 °C, ра-створяется в этаноле, ацетоне. 9-Аминоакридин является более сильным основанием, чем акридин. Несмотря на то, что в нем имеется два основных центра, соль образуется только по кольцевому атому азота. Пара электронов аминогруппы вступает в сопряжение с л-электронами пиридинового кольца, смещаясь к кольцевому атому азота и повышая на нем электронную плотность. В результате чего аминогруппа утрачивает способность к образованию солей.
Ацилирование протекает по аминогруппе:
9-ацетамидоакридин, 9-Ы-ацетиламиноакридин
-на
НС1
хлористоводородная соль 9-аминоакридиния, 9-а.миноакридиний хлорид
Из производных акридина отмстим лекарственные препараты акрихин и риванол:
30. Конденсированные шестичленные гетероциклические соединения...
399
акрихин
9(4'-диэтиламино-Г-метилбутиламино)-2-метокси-6-хлоракридина дигидрохлорид
(лактат 6,9-диам и но-2-этоксиакридина, иди этакридина лактат)
Солеобразование с кислотами идет по кольцевому и диэтиламинному атому азота.
Акрихин нашел применение при лечении малярии. Риванол — бактерицидный препарат.
31. ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ С ДВУМЯ ГЕТЕРОАТОМАМИ. ПУРИН И ЕГО ПРОИЗВОДНЫЕ. АЛКАЛОИДЫ
Ароматические шестичленные гетероциклические соединения, содержащие в своей структуре в качестве гетероатомов два атома азота, называются диазинами.
4
4 4
1,2-диазин, 1,3-диазин, 1,4-диазин,
пиридазин пиримидин пиразин
Для этих соединений чаще применяют тривиальные названия: пиридазин, пиримидин, пиразин. Как видно из приведенных формул, эти три диазина являются изомерами.
Существуют также шестичленные гетероциклы, содержащие в своей структуре в качестве гетероатомов атомы азота и кислорода или азота и серы,например:
оксазин
Оксазин и тиазин не обладают ароматическим характером и по своим свойствам напоминают ациклические соединения, содержащие аналогичные функциональные группы.
Пиридазин, пиримидин и пиразин представляют собой ароматические соединения, во многом напоминающие пиридин. Наличие в молекулах двух атомов азота придинового типа, которые дезактивируют друг друга, приводит к тому, что эти гетероциклические соединения являются более слабыми основаниями, чем пиридин. Несмотря
31. Шестичленные гетероциклические соединения с двумя гетероатомами... 401
на наличие двух основных центров в молекуле диазины образуют соли с одним эквивалентом минеральной кислоты:
пиримидин
В реакции электрофильного замещения эти гетероциклы вступают с трудом. Реакции SL возможны только при наличии в молекуле сильных электронодонорных заместителей, таких, как-МН2-группа, -ОН-группа и других, способных проявлять +М-эффект.
Реакции же нуклеофильного замещения (SN) протекают легко.
ПИРИДАЗИН (1.2-диазин)
4
Пиридазин — бесцветная жидкость с /кип 207 °C. Пиридазин и его производные получают реакцией конденсации гидразина с 1,4-дикарбонильными соединениями (предельными или непредельными):
H,N—NH2
гидразин
малеинди альдегид
С минеральными кислотами и алкилгалогенидами пиридазин образует соли по одному атому азота:
СН3
пиридазиний хлорид
N-метилпиридазиний йодид
402
Лекции по органической химии
Подобно пиридину при действии пероксикислот образует N-ok-сид:
сн—сооон
При восстановлении или каталитическом гидрировании происходит раскрытие цикла и образуется тетраметилендиамин:
H2N СН2—СН2—СН2—СН2—NH2
Производные пиридазина применяются в качестве лекарственных препаратов и гербицидов.
ПИРАЗИН
(1,4-диазин)
Пиразин — бесцветное кристаллическое вещество с Гп157 °C. Пиразин и его производные получают реакцией конденсации 1,2-диаминов с 1,2-дикар-бонильными соединениями:
.NH2
Н2С
н2с
^nh2
2,3-Дигидропиразин
пиразин
этилендиамин глиоксаль
На первой стадии образуется 2,3-дигидропиразин, который легко окисляется до пиразина.
Как и пиридазин, пиразин является слабым основанием и образует соли с кислотами по одному атому азота, при взаимодействии с пероксикислотами окисляется до N-оксида:
31. Шестичленные гетероциклические соединения с двумя гетероатомами...
403
СН3СОООН
N-оксид пиразина
При восстановлении пиразина водородом в момент выделения образуется циклический диамин — пиперазин:
Н I
H2f fH2
H2CxNXH2 I H пиперазин
Пиперазин — сильное основание, обладающее свойствами вторичных алифатических аминов, образует соли с двумя эквивалентами кислоты. Пиперазина адипинат применяют как глистогонное средство.
Из производных пиперазина отметим циклические амиды «-аминокислот, которые называют дикетопиперазинами.
Н
Н
сн2
nh2.
>ОН";
но
। -_:
NH2 I -2Н2О
/N.
н2с с
н2
/N
н2с сн2
сн2
/СП, N
Н,С. /СН, N
а-аминоуксусная кислота
н
ди кетопиперазин
о
О
н
Восстановлением дикетопиперазина можно получить пиперазин.
4
пиримидин
ПИРИМИДИН
Пиримидин (1,3-диазин) — кристаллическое вещество с Z 225 °C, легко растворимое в воде, этаноле. Образует соли с одним эквивалентом сильных кислот:
404
Лекции по органической хи мии
Реакции электрофильного замещения протекают с большим трудом, только при наличии в молекуле электронодонорных заместителей, при этом реакция SE протекает по положению 5:
ОН ОН
2-амино-4-гидроксипиримидин 2-амино-4-пшрокси-5-нитропири иидин
Атомы азота пиридинового типа, вследствие электроноакцепторного действия, обуславливают общее уменьшение электронной плотности и прежде всего в положениях 2, 4. 6, что способствует протеканию реакций нуклеофильного замещения (SN):
Пиримидин получают реакцией взаимодействия мочевины с малоновым эфиром по следующей схеме:
сн2
//с\
о ос2н5
малоновый эфир
H2N\ C2H,ONa'
С=О ----------
/ - 2С,НЮН
h2n мочевина
барбитуровая кислота (оксо-форма)
но
с—N
// SS
нс с—он
\ /
C=N
/
НО
гидроксиформа
2,4,6-трихлор-пиримидин
31. Шестичленные гетероциклические соединения с двумя гетероатомами... 40э
Барбитуровая кислота (2,4,6-тригидроксипиримидин) является производным пиримидина и из нее можно его получить, действуя трихлороксидом фосфора с последующим восстановлением.
БАРБИТУРОВАЯ КИСЛОТА
О Важнейшее производное пиримидина. Это
С Н циклический уреид малоновой кислоты, малонил-Н2С N мочевина. Представляет собой бесцветное кристал-
q q лическое вещество, легко растворяется в теплой
''Ч) воде, трудно в холодной. В результате щелочного I гидролиза барбитуровая кислота разлагается, обра-
Н зуя малоновую кислоту, NH3 и СО2. Эта реакция
свидетельствует о неароматическом характере соединения.
CO2f +NH3t+ НООС-СН2-СООН
Группа -СН2 в положении 5 обладает высокой реакционной способностью. Метиленовая группа в барбитуровой кислоте фланкирована с обеих сторон кетогруппами, которые повышают подвижность атомов водорода.
Барбитуровая кислота — это таутомерное вещество. Для нее характерны два вида таутомерии: кето-енольная и лактам-лактимная.
кетонная форма
енольная форма
лактамная форма
лактимная форма
406
Лекции по органической химии
Таким образом, барбитуровая кислота существует в двух таутомерных формах: триоксо-форме и тригидрокси-форме:
I Н
Говоря о барбитуровой кислоте, следует отметить, что она в 5—6 раз сильнее, чем уксусная. Кислотность барбитуровой кислоты обусловлена существованием подвижных атомов водорода в 5 положении. Если один из атомов водорода замешен на радикал, то сила кислоты несколько уменьшается, при замене же двух атомов водорода сила кислоты резко снижается.
Используя в качестве исходных продуктов производные малоновой кислоты, можно получить реакцией конденсации с мочевиной целый ряд фармацевтических препаратов.
К примеру:
в. 0
К, II о R=R'=C H5 —барбитал(5,5-диэтилбарбитуроваякис-r-C N лота)
R= R'= C6H5 — люминал или фенобарбитал • (5-этил-5-фенилбарбитуровая кислота).
К производным пиримидина следует отнести аллоксан, который получается при конденсации мезоксалевой кислоты и мочевины.
О СООН с=о + I соон мезоксалевая кислота
аллоксан
Важными производными пиримидина являются пиримидиновые основания, которые входят в состав нуклеиновых кислот:
О ОН
I
Н урацил
2,4-дигидроксипиримидин
31. Шести членные гетероциклические соединения с двумя гетероатсма ни.
407
ЦИТОЗИН, 4-амино-2-гидроксипиримидин
Пурин и его производные
Группа пурина включает природные продукты, не менее важные для физиологии животных и растений, чем моносахариды или аминокислоты.
ПУРИН
имидазо[4,5-с1]пиримидин
Пурин — бициклическая система, состоящая из пиримидинового и имидазольного колен. Пурин представляет собой бесцветное кристаллическое вещество нейтрального характера, легко растворимое в воде. Об
разует соли как с кислотами, так и со щелочами, обладает ароматическим характером.
Амфотерность пурина связана с наличием в его структуре имида-
зольного ядра.
НС1
Н
СГ
пуриний хлорид
натриевая соль пурина
408
Лекции по органической химии
Слабые кислотные свойства пурин проявляет за счет атома азота пиррольного типа, а основные — за счет атома азота пиридинового типа в имидазольном фрагменте молекулы.
Как и для имидазола, для пурина характерна азольная таутомерия:
Важнейшими производными пурина являются оксо- и аминопроизводные. К оксопуринам относятся мочевая кислота, ксантин и гипоксантин.
мочевая кислота,
2,6,8-триоксопурин
ксантин
МОЧЕВАЯ КИСЛОТА
В нормальной моче человека и животных содержится незначительное количество мочевой кислоты. Основным продуктом азотистого обмена является мочевина. При некоторых заболеваниях, в частности при подагре (откладывание мочевой кислоты в виде Na солей с суставах), увеличивается количество мочевой кислоты. У птиц и змей мочевая кислота является главной составной частью экскрементов (у змей до 90%).
Мочевая кислота — бесцветное кристаллическое вещество, труднорастворимое в воде, не растворимое в С,Н.ОН и эфире. Мочевая кислота очень слабая кислота; ее нельзя обнаружить при помощи обычных индикаторов, так как она не изменяет их цвет. Она образует соли с одним или двумя эквивалентами одновалентных металлов.
Для мочевой кислоты характерна лактам-лактимная таутомерия.
оксо-форма, лактамная форма
гидрокси-форма, лактимная форма
31. Шестичленные гетероциклические соединения с двумя гетероатомаж
409
В кристаллическом состоянии мочевая кислота находится в лактамной (оксо-) форме, а в растворе между лактамной и лактимной формами устанавливается динамическое равновесие, в котором преобладает лактамная форма.
Мочевая кислота является двухосновной кислотой. Кислотные свойства мочевой кислоты определяются лактимной формой. Солеобразующими являются гидроксильные группы, находящиеся в положении 2 и 8. Как двухосновная мочевая кислота образует два ряда солей — кислые и средние.
NaOH
-н2о
моно натриевая соль мочевой кислоты
динатриевая
соль мочевой кислоты
Соли мочевой кислоты называются уратами. Средние соли хорошо растворимы в воде, из кислых наиболее растворим урат лития. Поэтому для лечения подагры применяют соли лития.
Из мочевой кислоты получают пурин.
пурин
410
Лекции по органической химии
1. Строение мочевой кислоты установлено при помощи реакций окисления:
а) окисление азотной кислотой:
HNO3
+ H,N-C-NH.
2 II
О
мочевина
аллоксан
Таким образом, при окислении азотной кислотой сохраняется пиримидиновое ядро и раскрывается имидазольный цикл.
б) окисление перманганатом калия в нейтральной или щелочной среде:
H,N—С—NH
2 II
+ СО2
аллантоин
Эта реакция доказывает наличие в мочевой кислоте пятичленного имидазольного цикла.
2. Мурексидная проба — качественная реакция на мочевую кислоту. Мочевую кислоту нагревают с конц. HNO3 и добавляют концентрированный раствор аммиака — образуется пурпурно-фиолетовое окрашивание. При этом образуется аммонийная соль пурпурной кислоты следующего строения:
пурпурная кислота
мурексид,
аммонийная соль пурпурной кислоты
При частичном восстановлении мочевой кислоты образуется ксантин.
31. Шестичленные гетероциклические соединения с двумя гетероатомами...
411
2,6-диоксопурин, ксантин
+ Н20
Ксантин проявляет амфотерные свойства. Для него характерна лактам-лактимная и азольная таутомерия.
гидрокси-форма, лактимная форма
лактам-лактимная таутомерия ксантина
азольная таутомерия ксантина
Из аминопроизводных пурина отметим аденин или 6-аминопурин, который входит в состав нуклеиновых кислот, содержится в печени. Окисляется азотистой кислотой до 6-оксипурина (гипоксантина):
6-аминопурин, аденин
ОН
HONO
6-гид роксипурин,
гипоксантин
В природе встречаются N-метильные производные ксантина.
412
Лекции по органической химии
Теофиллин, теобромин и кофеин относятся к алкалоидам.
ТЕОФИЛЛИН
1,3-диметилксантин
Этот алкалоид содержится в листьях чая. Для него возможна азольная таутомерия. Он проявляет основные свойства за счет атома азота пиридинового типа имидазольного цикла. Применяется как диуретическое средство, стимулятор ЦНС.
ТЕОБРОМИН
3,7 - диметилксантин
Раньше для медицинских целей теобромин получали из плодов дерева какао, в настоящее время применяется синтетический теобромин. Это соединение обладает амфотерными свойствами; основные свойства обусловлены атомом азота пиридинового типа имидазольного цикла, а лактимная форма теобромина проявляет кислотный характер.
КОФЕИН
1,3,7-триметилксантин
Кофеин получают из отходов при производстве чая. Он содержится в листях чая и зернах кофе. В промышленных масштабах кофеин получают при метилировании мочевой кислоты диметилсутьфатом. Кофеин возбуждает ЦНС, повышает кровяное давление, стимулирует сердечную деятельность, обладает мочегонным действием.
АЛКАЛОИДЫ
Что же такое алкалоиды?
Алкалоиды — это группа азотсодержащих органических соединений, преимущественно растительного происхождения, обладающих основными свойствами и высокой биологической активностью.
По строению большинство алкалоидов относится к гетероциклическим соединениям.
Алкалоиды широко распространены в растительном мире.
Алкалоидов в алкалоидоносных растениях может содержаться до 2 и более процентов. Они могут содержаться в различных частях рас-
31. Шестичлеиные гетероциклические соединения с двумя гетероапюмами... тений — в коре и корневищах, листьях и цветах, а также в стеблях и плодах.
Некоторые семейства являются наиболее богатыми по содержанию алкалоидов: такие как кутровые (Аросупасеае), маковые (Papaveraceae), бобовые (Fabaceae), сложноцветные (Asteraceae) и др.
Интересно, что некоторые семейства не содержат алкалоидов, такие как розовые (Rosaceae), губоцветные (Lamiaceae) и др. Содержание алкалоидов в растениях может изменяться в процессе роста.
Алкалоиды в растениях содержатся в большинстве случаев в виде солей органических (реже минеральных) кислот. Чаше алкалоиды образуют соли со следующими кислотами: яблочной, винной, лимонной, молочной, янтарной, роданистоводородной, фосфорной, серной или с танином.
Азкалоиды — это кристаллические вещества, не растворимые в воде, растворимые в органических растворителях. Они обладают основными свойствами, с минеральными кислотами образуют соли. Соли хорошо растворяются в воде и плохо в органических растворителях. Природные алкалоиды — оптически активные соединения, вращают плоскость поляризованного луча влево. Обладают горьким вкусом, некоторые алкалоиды применяются как лекарственные средства.
В настоящее время из растений выделено свыше 5000 различных алкалоидов.
Для алкалоидов принята химическая классификация, в основу которой положена природа гетероцикла, входящего в состав алкалоидов.
Существуют следующие основные группы алкалоидов: производные пиридина и пиперидина, хинолина.изохинолина, индола, пурина и др. Таким образом, теофиллин, теобромин и кофеин относятся к алкалоидам группы пурина.
УГЛЕВОДЫ (САХАРА)
Понятие об углеводах впервые было введено в 1844 г. русским химиком К. Г. Шмидтом. Углеводы — это соединения, которые наиболее широко представлены в природе. Они образуются в зеленых частях растений в результате фотосинтеза — сложного биохимического процесса, который сопровождается усвоением солнечной энергии, воды и углекислого газа в присутствии хлорофилла.
СО2 + 2Н2О
(Н-С—ОН) + Н2О + О2
Молекулы углеводов состоят из атомов углерода, водорода и кислорода, причем соотношение атомов водорода и кислорода такое, как в молекуле воды [Сх(Н2О)у]. Например, в молекуле глюкозы С6Н12О6 [С6(Н2О)6].
Однако дальнейшее изучение строения этих соединений и открытие веществ с составом, который не соответствует эмпирической формуле, показали, что отнесение их к гидратам углерода является только формальным, но принятое название углеводы сохранилось.
С 1927 г. к углеводам относят многочисленную группу природных и синтетических веществ, которые являются по химическому строению полигидроксильными соединениями, содержат альдегидную или кетонную группы, или же образуют их при гидролизе.
Углеводы делятся на моносахариды — монозы (простые сахара), которые не гидролизуются водой, и полисахариды (сложные сахара), соединения, способные гидролизоваться до моносахаридов.
Сложные углеводы, в свою очередь, классифицируются на олигосахариды, которые образуют при гидролизе от двух до десяти молекул моносахаридов, и полисахариды (полиозы), которые при гидролизе образуют более 10 молекул моносахаридов.
32. МОНОСАХАРИДЫ
Моносахариды представляют собой полигидроксильные соединения, которые содержат альдегидную или кетонную группы. Их еще называют монозами, или простыми углеводами (сахарами).
Классификация и номенклатура
Моносахариды классифицируют с учетом двух признаков — характера оксогрупп (альдегидная или кетонная) и длины углеродной цепи.
32. Моносахариды
В зависимости от наличия в структуре моносахаридов альдегидной или кетонной групп их делят на альдозы и кетозы.
В зависимости от числа атомов углерода в молекуле моносахариды классифицируют на:
триозы (С3) пентозы (Ср
тетрозы (Ср гексозы (С6) и т. д.
Большинство природных моносахаридов являются пентозами и гексозами.
С учетом обоих классификационных признаков существуют: альдопенозы альдогексозы
кетопентозы кетогексозы
В названиях моносахаридов, как правило, используют тривиальную номенклатуру, в которой все сахара имеют окончания -оза (глюкоза, фруктоза, галактоза, рибоза и т. д.).
Номенклатура ИЮ ПАК для названия углеводов не используется.
Стереоизомерия
Моносахариды — оптически активные соединения, для их классификации применяют Р-,А-стереохимическую систему названий. Принадлежность к D- или Z-стереохимическому ряду определяют по конфигурации асимметричного атома углерода, максимально удаленного от альдегидной или кетонной группы (для пентоз — С4, для гексоз — С5).
Сахара, которые имеют сходное строение с D-глицериновым альдегидом, относят к D-ряду, если со строением Z-глицеринового альдегида, то к Z-ряду.
СН2ОН
н.с..о
но£.Щ сн2он
L-гл инериновый
альдегид
D-глицериновый альдегид
н, ( ( :н,он
н- -он ( 2=0
но- —н но- —н
н— -он н— -он
:н- -он; ;н— -он;
( зн2он сн2он
D-глюкоза D-фруктоза
нс"° н—I—он
но—н
[HO--HJ
СН2ОН
СН2ОН с=о но—|-н н—I—ОН [но--’н: " сн’бн
/.-арабиноза
Д-сорбоза
416
Лекции по оргаиическог/ xu.mii
Пространственные D- и £-изомеры являются энантиомерами (оптическими антиподами). Большинство природных моносахаридов относится к D-ряду.
Рассмотрим важнейшие — пентозы и гексозы.
ПЕНТОЗЫ
Среди пентоз наиболее распространенными в природе являются альдопентозы. Общая формула альдопентоз:
.О се I н *снон
*снон
Как видно из формулы, пентозы имеют 3 асимметрических атома углерода. Следовательно, количество оптических изомеров будет: 23 = 8 или они должны иметь 4 пары антиподов.
*снон I сн2он
Важнейшие представители альдопентоз:
сГ
и н
1 i ОН
ПО' И
игл и
ПО 11
( :н2он
L-арабиноза
с j "т. чо
п С£11
игл и
ПО 11
11 ОН
( :н2он
D-ксилоза
о ; и
н
IT гли
11 ОН
тт гли
11 OII
1Д глы
ОН
( :н2он
D-рибоза
/О
сс
н
II
11 н
ГТ
11 он
и гли
L1 О11
( зн,он
2 - дезо кс и - D-рибоза
ГЕКСОЗЫ
Общая формула гексоз:
.О сб I н *снон
Гексозы содержат в структуре 4 асимметрических атома углерода, то есть имеют 24 = 16 оптических изомеров или 8 пар энантиомеров.
*снон
*снон хСНОН *СН2ОН
417
32. Моносахариды
Важнейшие представители гексоз:
( .0 / - X о v J / \\ 2 х о ?н2он ( =0 ( —н но— —он н— он но ЗН2ОН ( фруктоза L-ci :н он 2 3=0 —н —он —н гн2он грбоза
н он но I 1 п н но
но н но Н НО Г1 н н—
н он н ОН но г! он н—
п ( D-m он н зн2он ( юкоза D- м ОН н зн2он ( анноза D- га Un :н,он ( лактоза 7)-<
альдогексозы кетогексозы
Пространственные изомеры моносахаридов, которые отличаются конфигурацией одного или нескольких асимметрических атомов углерода, называются диастереомерами.
Диастереомеры, которые отличаются конфигурацией только одного асимметрического атома углерода, называются эпимерами.
Сравним строение Д-глюкозы и D-маннозы. Эти моносахариды отличаются конфигурацией только одного — верхнего асимметричного атома углерода, то есть являются эпимерами.
Эпимерами также являются: Л-арабиноза и D-ксилоза, D-ксилоза и D-рибоза, D-глюкоза и D-галактоза.
О I \\/ Г 'I о х \\ / г \
:н— ! ! 2 —ОН; ;Н0— — Н;
но— 4 н— 5 н— 6 ( —н но— —он н— —он н— 6 :н2он ( —Н —он —он :н2он
Строение моносахаридов
Длительное время в науке существовало представление, что моносахариды являются соединениями только с открытой углеродной цепью.
Однако при более глубоком изучении их строения было установлено, что цепные формулы не объясняют некоторых химических свойств и не объясняют таких фактов:
1. Почему моносахариды не присоединяют бисульфит натрия?
418
Лекции по органической химии
2. Не дают окрашивания с фуксинсернистой кислотой.
3. Чем обусловлены превращения маннозы, глюкозы и фруктозы друг в друга под действием щелочей?
4. Почему у моносахаридов появляется мутаротация, которая связана с изменением угла вращения при стоянии свежеприготовленных растворов? Если приготовить раствор глюкозы и поместить в поляриметр, то угол вращения равен 11JJ°. Глюкоза отклоняет плоскость поляризованного луча света на 112°, через 2 часа на 90°, через 3 часа — на 60° и окончательно на 52,5°.
5. Почему при рассмотрении формулы глюкозы в ней можно насчитать 5 гидроксильных групп и только одна отличается своей реакционной способностью?
Чтобы ответить на эти вопросы, надо познакомиться с двумя видами таутомерии: карбонильно-ендиольной и цикло-оксо-таутомерией.
Углеродный скелет молекулы моносахаридов может изгибаться, приобретая форму «ухвата» (клешни). При этом альдегидная группа и гидроксильные группы при С4 или С5 находятся на расстоянии, равном примерно одной ковалентной связи, и между ними происходит химическое взаимодействие, подобно реакции между альдегидами и спиртами:
ZO он
R—CZ + R—ОН R—С—OR
н хн
Продуктами реакции являются полуацетали.
В соответствии с теорией напряжения циклов наиболее выгодно взаимодействие, приводящее к образованию пяти- или шестичленных циклов.
Шестичленный цикл образуется при взаимодействии альдегидной группы с гидроксильной группой при С5 альдогексоз или С6 кетогексоз. Такой цикл называется пиранозным.
P-D-глюкопираноза, Д-глюкоза, а-Д-глюкопираноза,
циклическая форма оксо-форма циклическая форма
32. Моносахариды
419
При взаимодействии альдегидной группы с гидроксильной группой при С4 альдогексоз или С, кетогексоз образуется пятичленный цикл, который называется фуранозным.
Так образуются полуацетальные циклические формы глюкозы. Приведенные циклические формы моносахаридов называются формулами Колли — Толленса.
Внутримолекулярное образование полуацеталя приводит к тому, что атом углерода карбонильной группы превращается в асимметрический. Этот новый хиральный центр называется аномерным. При этом образуется два новых стереоизомера, которые получили названия а- и [3-аномеров.
Гидроксильная группа при аномерном центре называется полуацетальной, или гликозидной.
Поскольку формулы Колли — Толленса неудобны для изображения циклических структур, более удобны для написания и понимания проекционные формулы Хеуорса. В этом случае циклические формы моносахаридов изображаются в виде плоских многоугольников с атомом кислорода в цикле.
Для того, чтобы перейти от формул Колли — Толленса к формулам Хеуорса, необходимо учитывать такие правила:
1. Заместители, расположенные в формуле Колли — Толленса слева, в формуле Хеуорса изображаются над плоскостью, а заместители, расположенные справа, — под плоскостью цикла.
2. У альдоз £)-ряда в пиранозной форме группа —СН2ОН, а в фуранозной — группа —СН(ОН)СН2ОН всегда расположены вверху, над плоскостью цикла.
420
Лекции по органической химии
а-С-глюкопираноза
Р-/)-глюкофураноза
Аналогично изображаются с помощью формул Хеуорса пиранозные и фуранозные формы кетогексоз.
а-Р-фруктопираноза Р-Р-фруктофураноза
Для изображения смеси а- и p-аномеров в формуле Хеуорса расположение полуацетального гидроксила показывают волнистой линией.
Р-глюкопираноза
Таутомерия
Цикло-оксо-таутомерия. Моносахариды являются таутомерными веществами. В кристаллическом состоянии они имеют циклическое строение. В водном растворе циклическая форма через открытую оксоформу превращается в другие циклические формы — пиранозные и фуранозные. Цикло-оксо-таутомерия объясняет отсутствие некоторых реакций на альдегиды.
Таким образом, в водном растворе моносахариды существуют в виде пяти таутомерных форм: открытой, а- и p-пиранозных и а- и р-фуранозных.
32. Моносахариду
421
Такой вид таутомерии называется цикло-оксо-таутомерией, или кольчато-цепной.
Переход одной формы в другую происходит непрерывно. В определенный момент наступает динамическое равновесие, при котором количество всех форм становится постоянным.
Р-О-глюкофураноза
Р-О-глюкопираноза
В равновесной смеси преобладают пиранозные формы: -63% P-D-глюкопиранозы и 36% а-Л-глюкопиранозы. Фуранозные формы и открытая форма присутствуют в очень маленьких количествах.
Способность моносахаридов к цикло-оксо-таутомерии объясняет явление мутаротации.
Мутаротация — это изменение величины угла удельного вращения свежеприготовленных растворов оптически активных соединений.
Вначале в свежеприготовленном растворе глюкозы наблюдается величина угла удельного вращения 112° (a-D-глюкопираноза), через некоторое время величина угла становится равной +52,5° и остается неизменной, что есть результатом установившегося динамического равновесия между пятью таутомерными формами моносахаридов.
Карбонильно-ендиольная таутомерия. Как известно, альдегидная группа проявляет электроноакцепторные свойства '
), это вызывает поляризацию связи С,—С2, атом водорода при С2 становится подвижным и мигрирует к атому кислорода карбонильной группы. При этом образуется ендиольная форма.
422
Лекции по органической химии
сн2он с=о
ендиольная НО
D-глюкоза, форма
карбонильная форма Н------ОН
н—он сн2он D-манноза
Такие превращения протекают в слабощелочной среде. В ендиоль-ной форме при двойной связи имеются две равноценные гидроксильные группы. Согласно правилу Эльтекова, гидроксильная группа не может находиться при атоме углерода с двойной связью, вследствие р. п-сопряжения. В зависимости от того, какой гидроксил в данный момент времени будет вступать в сопряжение, образуются либо альдегидная, либо кетонная формы моносахаридов. Ендиольная форма является общей для эпимеров. Она едина для эпимерных моносахаридов D-глюкозы и D-маннозы и изомерной им D-фруктозы.
Физические свойства
Моносахариды — это бесцветные кристаллические вещества, растворимые в воде, спирте, не растворяются в эфире. Большинство из них сладкие на вкус, но встречаются и горького вкуса.
Способы получения
Кислотный гидролиз полисахаридов является важнейшим способом получения моносахаридов:
~ \ иН2О, н+
I^6^10^5 ) ------------иС6Н12О6
\ In
крахмал
глюкоза
32. Моносахариды
423
Окисление многоатомных спиртов. При окислении пятиатомных спиртов образуются пентозы, а шестиатомных — гексозы.
сн2он
сн2он
сн2он
гексит
альдогексоза
Химические свойства моносахаридов
Являясь полигидроксикарбонильными соединениями, моносахариды проявляют свойства карбонильных соединений, многоатомных спиртов и циклических полуацеталей.
Химические превращения в ряду моносахаридов можно условно разделить на две группы:
• реакции при участии открытых форм моносахаридов;
• реакции при участии циклических форм.
I. Реакции открытых форм моносахаридов
1. Общая реакция на углеводы (проба Молита). При действии на углеводы конц. H,SO4 и а-нафтола образуется фиолетовое окрашивание. В химическом отношении вначале образуется фурфурол или 5-гидроксиметилфурфурол, которые конденсируются с сс-нафтолом и образуют окрашенные продукты.
2. Отношение к действию минеральных кислот (внутримолекулярная дегидратация). Альдопентозы при нагревании с минеральными кислотами (НС1, H,SO4) образуют фурфурол, который с анилином дает красное окрашивание, а с флороглюцином — кристаллический осадок.
'НО' ’ЮН]
/сн-нс \ н:„ Н / \ z- -С .ТСН С-г'
ОН HOJ н
НС1, t
-зн2о
фурфурол
альдопентоза
При взаимодействии альдогексоз с минеральными кислотами выделяется 3 молекулы Н2О и образуется нестойкий 5-гидроксиметил-фурфурол, который разлагается с образованием муравьиной и левулиновой кислот:
424
Лекции по органической химии
/но/ <он\
/ /сн-нс'\нЛ
'-Н-?с c-cz
НОН2С 0-H’HOj н
альдогексоза
5-пшроксиметилфурфурол
нсоон + сн—с—сн—сн—СООН
муравьиная
кислота О
левулиновая кислота
5-Гидроксиметилфурфурол дает красное окрашивание с резорцином. Данная реакция имеет аналитическое значение и позволяет отличить пентозы от гексоз.
3. Специфическая реакция на кетозы (реакция Селиванова). Кетозы легче, чем альдозы, превращаются в 5-гидроксиметилфурфурол, который является нестойким соединением. Он взаимодействует с резорцином в среде хлороводородной кислоты в момент выделения (реакция in statum nascendi) с образованием вишневокрасного окрашивания.
Альдозы эту реакцию дают, но в течение длительного времени.
4. Окисление. Моносахариды легко окисляются, но, в зависимости от природы окислителя и условий окисления, образуются разные продукты.
• Окисление в кислой и нейтральной среде:
При действии слабых окислителей (бромной воды или разбавленной азотной кислоты) альдозы окисляются до одноосновных альдоновых кислот.
н ( ^0
Н он
но— —н
н— —он
н— —он
сн2он
D-глюкоза
Вг7 + н,о (НВгО)
нох ( ^0
н— —он
но— —н
н— —он
н— —он
СН2ОН
D- глюконовая кислота
Сильные окислители, такие, как концентрированная азотная кислота, окисляют в молекуле альдоз альдегидную и первичную спирто-
32. Моносахариды 423
вую группы с образованием дикарбоновых гидроксикислот, которые называются альдаровые, или сахарные, кислоты:
нх ( х>0 соон
н— —он н— —он
но— — н кони. HNO, НО —н
н— —он н— —он
н— —он н— —он
( :н2он соон
/J-глюкоза ZJ-глюкаровая кислота
D- сахарная кислота
При избирательном окислении в молекуле альдозы первичной спиртовой группы без участия очень склонной к окислению альдегидной группы образуются уроновые кислоты.
Окисление в этом случае проводят, предварительно защитив альдегидную группу.
• Окисление в щелочной среде.
Подобно альдегидам, моносахариды окисляются аммиачным раствором оксида серебра (реактив Толленса), гидроксидом меди (II) в щелочной среде (реактив Троммера) или же реактивом Фелинга. При окислении в щелочной среде моносахариды расщепляются до смеси продуктов окисления.
альдоза
+ [Ag(NH3)2]OH
реактив Толленса
। продукты окисления
ХО R-C^
Н
альдоза
+ Си (комплекс)
реактив Фелинга
। продукты окисления
Эти реакции являются качественными на альдозы и кетозы.
5. Восстановление. При восстановлении моносахаридов водородом в присутствии катализатора (Ni, Pd) образуются многоатомные спирты.
Из £)-глюкозы образуется Д-сорбит, из Д-маннозы — Д-маннит, из Д-ксилозы — Д-ксилит и т. д.
426
Лекции по органической химии
Н ( /О ГЛ Г-Д
11 OI1
но— —н
н— —он
н— —он
( зн2он
Д-глюкоза
( :н2он
н О1 1
но— —н
н— —он
н— —он
( зн2он
D- сорбит
Фруктоза при восстановлении образует смесь двух шестиатомных спиртов D-сорбита и D-маннита.
с :н2он ( :н,он с :н2он
( "° н г! "ОН ио н
НО "11 но + н
11О_ II
11 TJ "О11 11 Г31Д ГД он
11' Un
11 ( D-c ОН 11 :н2он орбит Д-с ОН :н2он аннит
II ( ОН 2Н2ОН
6. Реакции с участием оксогруппы. Альдозы, подобно альдегидам, легко вступают в реакцию с циановодородной кислотой с образованием циангидринов. При взаимодействии с гидроксиламином образуются оксимы.
нс : = N—ОН
н— —он
но— —н NH,OH
н— —он -н2О
н— —он
СН2ОН
ОКСИМ глюкозы
^О
С хн
н— —он
но— H нсх
н— —он
н— —он
СН2ОН
D- глюкоза
ОН I
н—с—C=N
н— —он
но— —н
н— —он
н— —он
СН2ОН
циангидрин (гидроксинитрил) глюкозы
32. Моносахариды
427
Взаимодействие с фенилгидразином (образование озазона). При нагревании моносахаридов с фенилгидразином в соотношении 1 : 3 образуются бмс-фенилгидразоны. которые получили название озазонов:
н----ОН
но----н
н-----он
н-----он
сн2он
D- глюкоза
3C6H.NHNH,
-C6KNH;. -NH.
-2Н,О
HC=N—NH—C,H,
| о J
C=N—NH —C,H.
o 5
но----H
H----OH
н----OH
CH2OH
но
,О
С"Н
он
Н С„н—NHNH,
ОН -Н,о
он
сн,он
озазон глюкозы
н
1 Н.
он
но
н
но
н
он
н
н
он
н
он
сн2он фенил гидразон глюкозы
XNH-c6h5
н _________а
()[[ -C6H5NH;
сн2он
D- глюкоза
н
н
н
HC=NH
с=о
но---н
н---он
HC=N—NH-C6H5
C=N—NH-C6H5
2C6H —NHNH,^ но-н
-h2o,-nh, н-----он
н----он н----он
сн2он
сн2он
моноимин озазон
Эпимеры D-глюкоза и D-манноза и изомерная им фруктоза образуют один и тот же озазон.
Образование озазона используется для определения подлинности моносахаридов.
428
Лекции по органической химии
II. Реакции при участии циклической формы
1. Образование гликозидов. Моносахариды, являясь циклическими полуацеталями, реагируют в присутствии кислотного катализатора со спиртами и фенолами с образованием циклических ацеталей — гликозидов.
a—D- глюкопираноза
метил-a-D-
глюкопиранозид
СН ОН
Н ОН
метил- 0-D-
глюкопиранозид
Называют гликозиды по названию циклических форм моносахаридов с изменением окончания -оза на -озид. Например, глюкопиранозид. глюкофуранозид.
Гликозиды построены по типу простых эфиров. Молекулы гликозидов состоят из сахарной части и агликона (несахарная часть). Гликозиды широко представлены в растительном мире, являются важными биологически активными соединениями. Примером могут быть сердечные гликозиды, флавоноиды и др.
В названии гликозида должно быть отражено все: исходный моносахарид, величина цикла, конфигурация, радикал (несахарная часть), его расположение.
Например:
метил-а-1)-глюкопиранозид
метил-0-.О-глюкопиранозии
Окончание -ид указывает на отсутствие в молекуле свободного полуацетального гидроксила.
Гликозиды фуранозного строения приблизительно в 100 раз менее стойки, чем гликозиды пиранозного строения.
32. Моносахариды
Гликозиды устойчивы к действию щелочей, но в водных растворах кислот они гидролизуются.
При гидролизе происходит расщепление гликозида на агликон и сахарный компонент и образуется смесь а- и p-аномеров глюкопиранозы.
Гликозиды не способны мутаротировать.
В природе встречаются самые различные гликозиды. В качестве агликонов могут быть кислородсодержащие соединения, стероиды и моносахариды. Связь агликона с аномерным атомом углерода осуществляется через атом кислорода, поэтому такие гликозиды называются О-гликозидами. N-гликозиды содержат в качестве агликона алифатические, ароматические амины или N-содержащие гетероциклические амины. S-гликозиды — производные тиоспиртов и тиофенолов.
2. Алкилирование. При взаимодействии моносахаридов с галогенал-канами или диметилсульфатом (CH3)2SO4 алкилированию подвергаются все гидроксильные группы, включая и полуацетальный гидроксил. В кислой среде такие соединения гидролизуются только по гликозидной связи:
D- глюкопиранозид
2,3,4.6-тетраметил-
Р-глюкопираноза
430
Лекции по органиче! кои химии
3. Ацилирование. При ацилировании моносахаридов ангидридами карбоновых кислот легко образуются сложные эфиры:
сн.он
о н
СН2ОАс
Н 7-----О
5(СН,СО),О 7/н
ОН н он a-D-глюкопираноза
CH3COONa
Н ОАс
'°^ОАс + 5АсОН
где Ас = СНЧСО пентаацетил гл юкоп ираноза
Отдельные представители
К производным моносахаридов относится аскорбиновая кислота (витамин С) и глюкозамин — представитель аминосахаров.
ВИТАМИН С (аскорбиновая кислота)
В промышленности аскорбиновую кислоту получают из £)-глюкозы.
н ( ^0 ( зн2он ( 2Н2ОН
Н О11 И он 1° 11 ОН
1 1О" ио- н но н
Н J ~ П 1 п он —
н— —он н— —он с=о
( ?н2он ( зн2он СН2ОН
D-глюкоза D-сорбит
/.-сорбоза
сн2он с=о
но----н
- н------он
СООН
но——н сн2он /.-сорбоза
н-----он: ----->
НО——н" ~Н2° сн,он
2-кето-£-гулоновая
аС-ОН
II о р c-ОН I у СН--1
НО—I—н
сн2он
аскорбиновая кислота
кислота
32. Моносахариды
431
Для удобства формулу аскорбиновой кислоты принято изображать таким образом:
Аскорбиновая кислота представляет собой у-лактон-2,3-дигидро-А-гулоновой кислоты. Соединение проявляет кислотый характер за счет одной из ОН-групп.
Витамин С содержится в шиповнике, лимонах, капусте, апельсинах и других продуктах, играет большую роль в жизнедеятельности организма.
Потребность в витамине С для человека — 50—70 мг в сутки. Его недостаток в организме снижает сопротивляемость к инфекционным заболеваниям.
ГЛЮКОЗАМИН (2-дезокси-2-аминоглюкопираноза)
Глюкозамин — это основная часть полисахарида хитина. Из этого полисахарида построен панцирь черепахи, раков и др. Он входит в состав антибиотика стрептомицина.
6 ГН пн
Это сильное основание, которое с минеральными кислотами образует соли.
33. ДИ- И ПОЛИСАХАРИДЫ
ДИСАХАРИДЫ
Дисахаридами называют углеводы, молекулы которых состоят из двух остатков моносахаридов одинаковой или разной природы, соединенных между собой гликозидной связью.
Дисахариды легко гидролизуются в кислой среде с образованием двух молекул моносахаридов. В зависимости от способа образования гликозидной связи, дисахариды разделяют на две группы — восстанавливающие и невосстанавливающие.
В восстанавливающих дисахаридах гликозидная связь образуется из гликозидной (полуацетальной) гидроксильной группы одного и любой, но не полуацетальной гидроксильной группы второго моносахарида. В большинстве случаев гликозидную связь образует гидроксильная группа у 4-го углеродного атома. Таким образом, дисахарид имеет свободную гликозидную гидрокильную группу, и следовательно, обладает восстанавливающими свойствами. В свежеприготовленных растворах таких дисахаридов наблюдается явление мутаротации. Представителями восстанавливающих дисахаридов является мальтоза, целлобиоза, лактоза.
Номенклатура дисахаридов является сложной. Название образуется по типу О-замещенных производных моносахаридов, исходя из названия восстанавливающего звена с указанием всех имеющихся заместителей. Наиболее употребительны тривиальные названия дисахаридов, обычно связанные с источником получения вещества.
МАЛЬТОЗА
(солодовый сахар)
Молекула мальтозы состоит из двух остатков Л-глюкопиранозы, связанных 1,4-гликозидной связью.
сн2он СН2ОН hJ—он н>—он tW1—-НО№-?ЮН НОуП-Y он Н ОН" ' н он a- D- глюкопираноза СН7ОН сн.он - н0^°>н H0N?HjfL0JVl#0H н он н он O'-мальтоза, 4-0-(а-/>глюкопиоанозило) a-D- глюкопираноза
33. Ди- и полисахариды
433
В растворе мальтоза существует в нескольких таутомерных формах — а- и р-циклнческой и альдегидной По свойствам мальтоза напоминает свойства глюкозы. Она мутаротирует, дает положительную реакцию с реактивом Толленса и реактивом Фелинга. С участием альдегидной формы вступает в реакции с фенилгидразином. гидроксила мином, циановодородной кислотой. При окислении бромной водой превращается в мальтобионовую кислоту.
мальтоза мальтобионовая кислота
За счет полуацетального гидроксила мальтоза образует гликозиды.
мальтоза метилмальтозид
С участием циклических форм мальтоза, аналогично моносахаридам, образует простые и сложные эфиры по всем гидроксильным группам.
ЦЕЛЛОБИОЗА
Это изомер мальтозы, состоит из двух остатков D-глюкопирано-зы, связанных гликозидной связью в положении 1,4. В отличие от мальтозы глюкоза, полуацетальный гидроксил которой участвует в образовании гликозидной связи, имеет Р-конфигурацию.
434
Лекции по органической химии
Р-/)-глюкопираноза Р-Р-глюкопираноза Р-целлобиоза,
4-О-(р-£>-г1юкопиранозидо) p-ZJ-глюкопираноза
В растворе целлобиоза существует в нескольких таутомерных формах — а- и p-циклической и альдегидной. Целлобиоза — восстанавливающий дисахарид (дает реакцию с раствором Толленса и Фелинга). Для нее характерны реакции с фенилгидразином, гидроксиламином, циановодородной кислотой. При окислении в мягких условиях превращается в целлобионовую кислоту. За счет полуацетального гидроксила образует гликозиды. По всем гидроксильным группам образует простые и сложные эфиры.
Целлобиоза и мальтоза имеют разное пространственное строение. В молекуле целлобиозы один остаток глюкозы по сравнению с мальтозой повернут на 180 °C.
Целлобиоза легко растворяется в воде, но не расщепляется в организме человека и поэтому не может быть использована в качестве продукта питания.
ЛАКТОЗА (молочный сахар)
Молекла лактозы состоит из остатков Р-Д-галактопиранозы и Д-глюкопиранозы, связанных 1,4-гликозидной связью.
Р-О-галактопираноза р-О-глюкопираноза Р-лактоза,
4-О-ф- D- гапактопиранозидо), р-/)-глюкопираноза
В растворе существует в нескольких таутомерных формах — а- и p-циклической и альдегидной. Лактоза восстанавливает раствор Фелинга и реактив Толленса. Ряд реакций обусловлен также наличием потенциальной альдегидной группы: образование фенилгидразона и других производных. При окислении концевой альдегидной группы лактозы образуется лактобионовая кислота. Лактоза гидролизуется под влиянием кислот и ферментов — Р-галактозидаз (лактаз), содержащих
33-Ди- и полисахариды
435
ся в некоторых видах бактерий и грибов, а также в кишечнике млекопитающих. Спиртовому брожению лактоза не подвергается, она сбраживается молочнокислыми бактериями, под влиянием которых она вначале гидролизуется, а затем превращается в молочную кислоту. Лактоза является важнейшим углеводным компонентом молока млекопи тающих. В коровьем молоке содержится до 4,5% лактозы, в женском молоке — до 7,5 %. Производят лактоз} из молочной сыворотки, которая является отходом при изготовлении масла и сыра. Применяют лактозу в технологии лекарств как наполнитель в порошках, таблетках и экстрактах. Сладость в 4—5 раз меньше, чем у сахарозы, и как заменитель сахара ее не используют.
В невосстанавливающих дисахаридах гликозидная связь образуется за счет полуацетальных (гликозидных) групп обоих моносахаридов.
Эти дисахариды не имеют свободного полуацетального гидроксила. Поэтому в растворах они существуют только в циклической форме, их растворы не мутаротируют и не обладают восстанавливающими свойствами (не дают реакцию «серебряного зеркала»).
Такие дисахариды не дают реакций по альдегидной группе и гликозидному гидроксилу. Они способны лишь к образованию простых и сложных эфиров. Представителем невостанавливающих дисахаридов является сахароза.
САХАРОЗА (тростниковый или свекловичный сахар)
Молекула сахарозы состоит из остатков а-Р-глюкопиранозы и P-D-фруктофуранозы. Гликозидная связь в сахарозе соединяет аномерные атомы углерода обоих сахаров, причем гликозидный гидроксил у
глюкозы находится в «-положении, а у фоуктозы в p-положении. Сахароза не образует анамеров.
Исходя из химической структу-
сахароза
ры, сахарозу можно назвать как а-Р-глюкопиранозидо-р-Р-фруктофуранозид.
436
Лекции по органической химии
Сахароза содержится в сахарной свекле, сахарном тростнике, в соках различных плодов, откуда ее и получают. В растениях сахароза служит растворимым резервным сахаридом, а также той транспортной формой, которая легко переносится по растению.
Растворы сахарозы оптически активны [ар20=+66,5и]. мутарота-ции не наблюдается, и они не проявляют восстанавливающих свойств.
Сахароза при кипячении водных растворов в присутствии кислот или при действии ферментов гидролизуется с образованием смеси глюкозы и фруктозы. При этом происходит изменение знака удельного вращения, то есть характерное для сахарозы вращение плоскости поляризации вправо [сх20 =3-66,5°] изменяется на левое вращение [а2о=—39,5°]. Процесс изменения знака удельного (оптического) вращения по величине и знаку в процессе гидролиза называется инверсией. Образующаяся в процессе гидролиза смесь равных количеств D-глюкозы и D-фруктозы называется инвертным сахаром. Инвертный сахар является основной частью пчелиного меда.
Сахароза легко расщепляется ферментативно в организмах человека и животных под действием фермента сахаразы.
Сахароза является ценным пищевым продуктом и исходным сырьем для получения этилового спирта. В виде стеарата сахароза нашла применение как эмульгатор и стабилизатор лекарственных препаратов.
ПОЛИСАХАРИДЫ
К полисахаридам относятся соединения, молекулы которых содержат более десяти моносахаридных звеньев, связанных О-гликозидной связью.
Чаще всего полисахариды состоят из нескольких сотен и даже тысяч моносахаридных остатков, образующих линейные или разветвленные полимерные цепи.
Полисахариды, построенные из моносахаридных звеньев одного типа, называются гомополисахариды (гомогликаны), а построенные из различных моносахаридных звеньев — гетерополисахариды (гетерогли-каны). Оба полимера могут быть линейными или развлетвленными
Гомополисахариды (гомогликаны) постороенные из остатков пентоз, называются пентозанами, из остатков гексоз — гексозанами. Подавляющее большинство природных полисахаридов — гексозаны. К ним относятся крахмал, целлюлоза, гликоген, декстраны и др.
КРАХМАЛ
Крахмал является неоднородным полисахаридом и состоит из 20% растворимой в воде фракции, называсмой амилозой, и около 80% нерастворимой фракции, называемой амилопектином. Они отличаются друг от друга химическим строением
33. Ди- и полисахариды
Амилоза — линейный полимер, в котором D-глюкопиранозные остатки связаны а-1,4-гликозидной связью состоит из 200—350 мономерных звеньев:
Амилопектин — полимер разветвленной структуры, который может содержать 1000 и более остатков D-глюкозы в молекуле.
В цепи полисахарида остатки глюкозы соединены а-1,4-гликозидными связями, а боковые отвлетвления связаны с основной цепью а-1,6-гликозидными связями.
Амилоза при действии I образует комплексное соединение, окрашенное в синий цвет. На этом свойстве крахмала основано использование его в качестве индикатора.
Амилопектин связывает 12 в незначительных количествах с образованием красного окрашивания.
Крахмал в воде набухает, но не растворяется. При нагревании крахмала с разбавленной H,SO4 образуется a-D-глюкопираноза.
(С6Н10О5)л -----(С6Н10О5)х-----*С,2Н22О,,------- С6Н|2О6
декстрин мальтоза а-О-глюкопираноза
л = 6000, м.в. 1 000 000
Декстрин — это растворимый крахмал, он восстанавливает реактив Фелинга.
Крахмал служит основным источником резервной энергии в растениях; встречается главным образом в семенах, клубнях, корнях.
В природе встречается животный крахмал, который называется гликогеном. Этот полисахарид снабжает организм глюкозой при повышенных физических нагрузках и в промежутках между приемами пищи. Гликоген построен также как и крахмал растительный, но он
438
Лекции по органической химии
состоит из 100% амилопектина, но представляет собой еще более разветвленную структуру. Гликоген — важнейший резервный полисахарид животного мира. Он содержится в печени и мышцах.
ЦЕЛЛЮЛОЗА (клетчатка)
Молекула целлюлозы представляет собой линейную цепь, состоящую из остатков p-P-глюкопиранозы, связанных между собой {3-1,4-гликозидной связью:
Целлюлоза — широко распространенный в природе полисахарид, являющийся составной частью оболочек растительных клеток.
Крахмал — это энергетический материал, клетчатка — опорный материал. Чистая целлюлоза — это молодые клетки. В процессе старения клеток в них появляются пектиновые вещества, лигнин и др.
В клеточных стенках растений целлюлоза составляет 40—50%, а в таком важнейшем сырье, как хлопковое волокно — 98%.
Природная целлюлоза обладает высокой механической прочностью, устойчива к химическому и ферментативному гидролизу.
Клетчатку получают из древесины, обрабатывая ее бисульфитом кальция или щелочью (Ca(SO3H)2). Все сопутствующие вещества растворяются в щелочах, а клетчатка как гликозид, стойкий к щелочам, остается в неизменном виде, ее промывают водой и сушат.
Клетчатка не растворяется ни в воде, ни в органических растворителях, но растворяется в аммиачном растворе гидроксида меди (реактив Швейцера (Cu(NH. )4(ОН)2).
Клетчатка с 12 не дает окрашивания. Растворяется в растворах сильных кислотах, таких как H2SO4 и Н3РО4. Если сернокислый раствор
33. Ди и полисахариды
439
вылить в воду, то образовавшаяся масса, называемая амилоидом, уже будет давать с 1, окрашивание. На этом свойстве основан способ получения пергамента. Обыкновенную бумагу опускают в 70%-ный раствор H,SO , а затем в Н,О. Образовавшийся амилоид затягивает поры бумаги и образуется водонепроницаемая бумага.
При гидролизе целлюлозы водным раствором серной кислоты получают раствор глюкозы, который после связывания серной кислоты используют для получения этилового спирта (гидролизный спирт).
(CfcH10O>
нон с-
-----целлобиоза-------»-
[н+]
0-7)-глюкопираноза
В качестве пищевого продукта целлюлоза не может быть использована, так как в организме человека отсутствуют ферменты, расщепляющие целлюлозу до глюкозы. В этот состоит принципиальное отличие целлюлозы от крахмала.
Бактерии, живущие в пищеварительном тракте жвачных животных (коров, овец и др.), вырабатывают фермент, расщепляющий целлюлозу до глюкозы. Поэтому жвачные животные могут питаться продуктами, содержащими целлюлозу.
Целлюлозу используют для изготовления различных сортов бумаги (в том числе фотографической) и картона, химической переработки на искусственные волокна (ацетатные, вискозные, медноаммиач-ные), пластмассы (этролы), пленки полимерные, кино-, фотопленки, лаки и эмали, бездымный порох, моющие средства.
К гомополисахаридам относится декстран — полисахарид бактериального происхождения, построенный из остатков а-Ц-глюкопи-ранозы. Получают их из сахарозы при участии бактерий Leoconostoc mesenteroides. Частично гидролизованные декстраны используют в фармации в производстве плазмозаменителей полиглюкин и реополи-глюкин.
ИНУЛИН
Инулин — полигликан глюкозы, содержится в клубнях сложноцветных и других растений. Молекула инулина имеет линейное строение и состоит из остатков 0-Ц-фруктофуранозы. Используется как заменитель крахмала в питании диабетиков.
34. ТЕРПЕНЫ
Терпены — группа липидов или жироподобных веществ природного происхождения, которая включает в себя отерпет/овь/еуглеводороды и их кислородсодержащие производные (спирты, альдегиды, кетоны), называемые терпеноидами.
Терпеновые углеводороды построены из фрагментов изопрена:
н2с=с—сн=сн2, (С5нк)„ сн3
где п — число изопреновых фрагментов, которых обычно бывает от 2 до 8.
Связывание молекул изопрена происходит по принципу «голова к хвосту» (изопреновое правило, сформулированное в 1921 г. Л. Ружич-кой), которое позволило выяснить строение многих терпенов и родственных им соединений.
f ’
н,с=с—сн=сн, + н-с=с—сн=сн, I >< Ч^\ I
СН3 н СН3
изопрен
н2с=с—СН,-СН,-СН=С-СН=СН,
2 I 2 I сн3 сн3
оцимен
Натуральный каучук также можно считать терпеном. Однако изопрен, который является родоначальником класса терпенов (п = 1), не принято относить к терпенам.
Классификация терпенов
I. По числу изопреновых звеньев терпены подразделяют на следующие ряды:
• монотерпены, или собственно терпены (л = 2, соединения, содержащие 2 изопреновых фрагмента);
• сесквитерпены (л = 3, название произошло от сескви — полтора);
34. Терпены
44:
• тритерпены (л = 6);
• тетратерпены (л = 8);
• политерпены (л > 8).
П. По наличию или отсутствию циклов, по количеству имеющихся циклов каждый из вышеприведенных рядов подразделяется на следующие группы:
Природные источники терпенов
В природе терпены встречаются в растениях и входят в состав эфирных масел. Кроме терпенов в эфирных маслах находятся вещества самой разнообразной структуры: спирты, альдегиды, сложные эфиры и др.
По консистенции эфирные масла походят на масла, хотя по химическому строению таковыми не являются. В отличие от жирных масел, эфирные масла — летучие жидкости. Они полностью испаряются, не оставляя жирных пятен. Свое название эфирные масла получили благодаря их свойству обладать приятным запахом.
АЦИКЛИЧЕСКИЕ ТЕРПЕНЫ
В основе углеродного скелета ацилкических терпенов лежат структуры изомерных димеров изопрена — оцимена и мирцена:
9 7 6 5 4 3 2 1
н,с=с—сн,-сн,-сн=с-сн=сн, I I
сн, сн,
8 оцимен. 3,7-диметилоктатриен-1,3,7
8 7 6 5 4 3 2 1
СН,—С=с сн,—СНт-С-СН=СН,
3 I н II
сн, сн,
9 3 2
мирцен, 7-метил-3-метиленоктадиен-1,6
442
Лекции по органической химии
При написании терпеноидов широко применяются общепринятые формулы, в которых показан только углеродный скелет без отражения алкильных заместителей.
Мирцен содержится в эфирном масле лавра благородного и хмеля обыкновенного, а оцимен — в эфирном масле базилика обыкновенного, васильков.
Производные этих монотерпенов — гераниол и нерол содержатся в эфирном масле розы и герани и являются геометрическими изомерами. При их окислении получают альдегиды — цитраль А и цитраль В — природные компоненты лимонного масла. В кислой среде нерол легко циклизуется в сс-терпинеол:
- н2о
цитраль А. гераниаль, цис-3,7-диметил-2,6-октадиеналь
цитраль В, нераль, транс-3,7 -риметл-2,6-октадиеналь
Смесь цис- и ц/ранс-изомеров цитраля — желтая маслянистая жидкость с антисептическим, болеутоляющим, противовоспалительным действием, используется в офтальмологии; наряду с гераниолом и неролом применяется как отдушка в парфюмерно-косметической промышленности.
Цитраль отпугивает некоторые виды муравьев, используется как репеллент (лат. repello — отпугиваю).
34. Терпены
443
МОНОЦИКЛИЧЕСКИЕ ТЕРПЕНЫ
Лимонен и ментан — наиболее важные представители моноцик-лических терпенов.
, МЕНТАН
Ментан — родоначальник группы моноциклических терпеноидов, в природе в свободном виде не встречается. Он может быть получен гидрированием лимонена или «-цимола. Ментан — жидкость со свойствами предельных циклических углеводородов.
ЛИМОНЕН
СН, Лимонен — жидкость, обладающая
свойствами непредельных углеводоро-/Чк дов. (-)-Лимонен и его рацемат входят или в состав скипидара, эфирных масел
хвойных деревьев, (+)-лимонен — в эфирное масло цитрусовых (до 90 %), \ /%\ бергамота, тмина, сельдерея. Лимонен
Н2С СН3 обуславливает запах лимонного масла,
из которого был впервые получен.
Лимонен имеет один асимметрический атом углерода и существует в виде двух зеркальных изомеров. Рацемическую форму — (+)-ли-монен (дипентен) — получают изомеризацией изопрена (С.В. Лебедев, 1908—1913 гг.) или при сухой перегонке каучука. При 500—700 °C происходит обратный процесс — крекинг дипентена.
Получение
500-700 °C
150 °C, Р
лимонен, ментадиен-1,8
изопрен
Химические свойства
Как ненасыщенное соединение лимонен способен вступать в реакции гидратации, гидрирования. При дегидратации лимонена образуется и-цимол.
444
Лекции по органической химии
гексагидроцимол
При нагревании терпина в присутствии кислоты образуются изомерные а- и Р-терпинеолы:
терпин, моногидрат ментадиола-1,8
Р-терпинеол (ментен-8-ол-1)
Ментадиол — двухатомный спирт, его моногидрат под названием терпингидрат проявляет антисептическое, слабое мочегонное действие, используется как отхаркивающее средство.
В парфюмерно-косметической промышленности используются а-терпинеол с запахом сирени и p-терпинеол с запахом ландыша.
34. Терпены
445
МЕНТОЛ
шее значение из изомеров имеет (—)
Из кислородсодержащих производных ментана большое значение имеет ментол (л-мен-танол-3) — основной компонент эфирного масла мяты перечной. Соединение имеет 3 асимметрических центра и может существовать в виде 8 оптических изомеров. Наиболь-ментол, у которого гидроксиль-
ная, метильная и изопропильная группы размещены экваториально. Такой изомер обладает специфическим запахом, антисептическим действием и вызывает характерное чувство холода при действии на
кожу.
Получение
Ментол можно получить вымораживанием мятного масла или восстановлением (—)-ментона, содержащегося до 20 % в эфирном масле мяты:
ментол
(+)-Ментол в промышленности получают алкилированием л/-кре-зола по реакции Фриделя — Крафтса с последующим каталитическим гидрированием образующегося тимола:
ТИМОЛ
(+ )-ментол
446
Лекции по органической химии
Химические свойства
Ментол — вторичный спирт, при взаимодействии с изовалериа-новой кислотой образует сложный эфир:
ментилизовалерианат
Ментол — твердое кристаллическое вещество с запахом мяты, используется как слабое антисептическое, успокаивающее, обезболивающее средство в составе лекарственных веществ (бороментол, пекту-син и др.), 30% раствор ментола в ментилизовалерианате под названием валидол используют при стенокардии. Ментол применяют в кондитерской и парфюмерно-косметической промышленности.
БИЦИКЛИЧЕСКИЕ ТЕРПЕНЫ
Чаще всего в основе строения терпеноидов этой группы лежат 4 структуры:
4-метил-
бицикло[3.1.0]гексан
Производныетуйана (туйон) и карана (карены) содержатся в эфирном масле шалфея. сс-Пинен — ненасыщенное производное пинана — наиболее распространенный в природе представитель этой группы.
сс-ПИНЕН
или
а-Пинен находится в различных эфирных маслах, но основная масса — до 75 % в скипидаре (терпентинном масле) Это жидкость, которая не смешивается с водой.
34. Терпены
447
Молекула сс-пинена содержит 2 асимметрических центра и обладает оптической активностью. Левовращающий сс-пинен преобладает в сосне приморской, а правовращающий — в сосне лесной.
Химические свойства
сс-Пинен — типичное непредельное соединение с присущими этому классу свойствами.
1. Взаимодействие с разбавленными кислотами. При нагревании с разбавленными кислотами (азотной, серной) сс-пинен превращается в сс-терпинеол и терпин.
2. Окисление. Пинен и скипидар окисляются кислородом, превращаясь в сс-пероксид.
а-пинен
пероксид
вербеной
Полученный а-пероксид можно обнаружить по окислительным свойствам. Если к крахмалу добавить KI, а затем скипидар, то при взбалтывании образовавшийся пероксид сс-пинена вытесняет 12 из KI и окрашивает крахмал в синий цвет.
448
Лекции по органической химии
Воздух вокруг хвойных деревьев озонирован. Это связано с тем, что в составе их эфирных масел в большом количестве содержится образующий пероксиды а-пинен. Пероксиды разлагаются с образованием атомарного кислорода, который, соединяясь с кислородом воздуха, образует озон.
3. Взаимодействие с хлороводородом. а-Пинен с НС1 образует бор-нилхлорид — твердое вещество с запахом камфоры, которое используется для синтеза камфоры:
4. Взаимодействие с органическими кислотами. Органические кислоты, присоединяясь к а-пинену, с небольшим выходом дают сложные эфиры борнеола:
борнилацетат
а-Пинен и скипидар используют как растворители, ускорители затвердевания (высыхания) красок за счет пероксидов. Скипидар применяют в медицине как местнораздражающее, обезболивающее и антисептическое средство. а-Пинен и борнеол используют для синтеза камфоры.
БОРНЕОЛ
(борниловый спирт, камфанол-2)
Это спирт камфанового ряда, имеющий за счет трех асимметрических атомов углерода оптические изомеры: энантиомеры, диастереомеры борнеола и изоборнеола.
Свое название борнеол получил от борнейского лавра, из эфирного масла которого он был впервые по
лучен на острове Борнео.
Это кристаллическое вещество с температурой плавления 204—208,5 °C, температурой кипения 212 °C, растворим в спирте и не растворим в воде.
34. Терпены
449
Химические свойства
Борнеол обладает свойствами вторичных спиртов, вступает в реакции окисления с образованием кетона — камфоры, в реакции замещения ОН-группы на галоген, образует сложные эфиры с органическими кислотами.
камфан
При кипячении с металлическим натрием в ксилоле происходит взаимное превращение друг в друга изоборнеола и борнеола.
Борнеол и его эфиры используют как душистые вещества в мыловарении, парфюмерной, кондитерской промышленности. Они понижают кровяное давление.
КАМФОРА
(камфара; 2-борнанон; 1,7,7-триметилбицикло[2.2.1]гептан-2-он)
или
Камфора является бициклическим кетоном на основе камфана (борнана). В структуре молекулы содержится 2 асимметрических атома, но вместо четырех ожидае
мых изомеров камфора существует только в виде двух изомеров. Отсутствие диастереомеров объясняется тем, что асимметрические ато
450
Лекции по органической химии
мы жестко связаны между собой тремя углеродными цепями, что исключает возможность иной конфигурации.
Получение камфоры
1. Из природных источников. D-камфору получают из 60-100-летних деревьев камфорного дерева, которое произрастает во Вьетнаме, Китае, Японии, Тайване, а также культивируется в тропиках. Камфора содержится в эфирных маслах камфорного лавра, сибирской пихты, базилика, полыни, камфорного шалфея.
Природные источники не удовлетворяют потребности в камфоре, поэтому используются полусинтетические и синтетические способы получения.
2. Получение из борнилацетата (метод Н.В. Вершинина). Этот метод получения камфоры из пихтового масла, содержащего 30—40% борнилацетата, имеет промышленное значение. Борнилацетат отделяют, подвергают омылению, и полученный (—)-борнеол окисляют хромовой смесью в (—)-камфору.
3. Получение рацемической камфоры из а-пинена (синтез В.Е. Ти-
а-пинен камфен борнилацетат
борнеол
Камфора — бесцветное кристаллическое вещество (Гпл 178 °C) с резким характерным запахом, флуоресцирует в УФ-свете, растворима в спирте, малорастворима в воде. Вращение и хаотическое движение кристаллов по поверхности воды связано с тем, что камфора летуча (легко сублимируется). Упругость ее паров на разных поверхностях кристалла неодинакова, вследствие этого кристалл с разными гранями с различной силой отталкивается от поверхности пленки воды, что и вызывает его вращение.
Химические свойства камфоры
Камфора — типичный кетон. С участием оксогруппы она образует оксимы, семикарбазоны, фенилгидразоны.
34. Терпены
1. Взаимодействие с гидроксиламина гидрохлоридом:
камфора
+ NH2OHHC1
+ НС1 + н2о
оксим камфоры
Реакция применяется для количественного определения камфоры по выделяемому эквивалентному количеству HCI.
2. Окисление и восстановление камфоры:
борнеол
HNO3
I^^COOH kX-COOH
камфорная кислота
3. Взаимодействие с бромом. Карбонильная группа активирует атомы водорода в «-положении, метиленовая группа проявляет С—Н-кис-лотный характер, в результате чего возможно замещение на галоген:
3-бромкамфора, а-бромкамфора
НВг
В медицине камфору используют как кардиотоническое, аналеп-тическое, местнораздражающее и антисептическое средство, в промышленности — как пластификатор, компонент бездымного пороха, репеллент. Бромкамфору применяют при заболеваниях центральной нервной системы как успокаивающее средство.
452
В.П. ЧЕРНЫХ
Валентин Петрович Черных родился 5 января 1940 г. в с. Речина Орловской области.
В 1959 г. с отличием закончил Харьковское медицинское училище, в 1964 г. — Харьковский фармацевтический институт.
После окончания аспирантуры ХФИ служил в армии.
В 1968 г. защитил кандидатскую диссертацию, в 1977 г. — диссертацию на ученую степень доктора фармацевтических наук, в 1990 г. стал доктором химических наук, в 1997 г. — членом-корреспондентом НАН Украины, в 2001 г. — лауреатом Государственной премии Украины.
С 1980 г. заведует кафедрой органической химии.
Прошел школу административного управления: 1971—1974 — декан, 1976—1980 —проректор по учебной работе, с 1980 г. — ректор Национального фармацевтического университета.
Автор 544 научных статей, 15 монографий, 8 учебных пособий, практикума и учебника по органической химии в 3-х книгах.
За 40 лет научно-педагогической деятельности им подготовлены тысячи высококвалифицированных специалистов для фармацевтической отрасли, высшего образования. Научной школой В.П. Черных подготовлено 13 докторов и 39 кандидатов наук.
Автор 16 оригинальных лекарственных препаратов. На 34 лекарственных препарата-генерика разработана научно-техническая документация и освоен их промышленный выпуск. Получено 57 патентов и 306 авторских свидетельств.
Основное научное направление: целенаправленный синтез новых биологически активных веществ — производных дикарбоновых кислот и гетероциклических структур, изучение их реакционной способности, механизмов химического взаимодействия; разработка оптимальных препаративных методов получения лекарственных субстанций, установление закономерностей связи между химической структурой, физико-химическими свойствами и биологической активностью; создание экологически чистых технологий получения лекарственных субстанций: исследование путей циклизации многофункциональных реагентов.
Удостоен почетных званий — заслуженный изобретатель Украины, заслуженный деятель науки и техники Украины, лауреат Государственной премии Украины 2001 года. Награжден орденами Украины «За заслуги» II и III степени, орденами Трудового Красного Знамени и «Знак Почета», медалями, почетными грамотами.
453
СОДЕРЖАНИЕ
Предисловие............................................... 3
1. Классификация и номенклатура органических соединений .5
2. Химическая связь. Взаимное влияние атомов в органических соединениях........................................... 12
3. Изомерия органических соединений. Пространственное строение молекул............................................. ..31
4. Классификация химических реакций и реагентов. 4й
5. Кислотность и основность органических соединений. .. 52
Углеводороды
6. Алканы................................................. 59
7. Циклоалканы..........................................68
8. Алкены (этиленовые углеводороды, олефины) ...........80
9. Алкадиены (диеновые углеводороды, диолефины).........89
10. Алкины (ацетиленовые углеводороды)..................96
Ароматические углеводороды (арены)
11. Моноядерные арены......................................103
Многоядерные арены
12. Многоядерные арены с конденсированными бензольными циклами..................................................118
13. Многоядерные арены с изолированными бензольными циклами......................................... 129
14. Галогенопроизводные углеводородов..................139
15. Нитросоединения .................................. 157
16. Амины..............................................167
17. Диазо- и азосоединения.............................182
Гидроксильные производные углеводородов. Спирты.
Простые эфиры. Тиолы. Тиоэфиры. Фенолы
18. Спирты................................................191
19. Фенолы.............................................212
Альдегиды и кетоны
20. Альдегиды и кетоны алифатического ряда............... 228
21. Альдегиды и кетоны ароматического ряда ............241
Карбоновые кислоты
22. Монокарбоновые кислоты................................251
23. Дикарбоновые кислоты.............................. 264
24. Функциональные производные карбоновых кислот.......272
25. Гетерофункциональные карбоновые кислоты............295
26. Производные угольной кислоты.......................320
454
Гетероциклические соединения
27. Трех- и четырехчленные гетероциклические соединения с одним гетероатомом.......................................327
28. Пятичленные гетероциклы с двумя гетероатомами..........355
29. Шестичленные гетероциклические соединения с одним гетероатомом...............................................369
30. Конденсированные шестичленные гетероциклические соединения с одним гетероатомом............................385
31. Шестичленные гетероциклические соединения с двумя гетероатомами. Пурин и его производные. Алкалоиды..........400
Углеводы (сахара)
32. Моносахариды..............................................414
33. Ди- и полисахариды.....................................432
34. Терпены................................................440
В лекшях зоргашчно!'xiMi'i. яю протягом багатьох роюв читаються студентам фар-мацевтичного утверситету. в проспи i доступшй форм! викладет загальн! пшходи до розгляду теоретичних питань сучасно! оргашчо! xiMi'i. Даються уявлення про рсакци та реагента, мехашзми оргашчних реакщй та 1'х напрями. фвичш i xiMiHHi властивосп оргашчних речовин. Пшкреслена роль i значимость оргашчно!' xiMi'i як науки, показаний генетичний зв’язок м!ж квасами оргашчних сполук.
Для студенпв фармацевтичних вуз!в i факультепв.
Навчалъне видання
ЛЕКЦП
3 ОРГАН1ЧНО1 XIMII В.П. ЧЕРНИХА
Навчальний псхЯбник для студенпв вищих навчальних заклал в
Росшською мовою
Редактор А. М. Миколюк
Коректор М.М. Поточняк Комп’ютерна верстка О. Б. IcaeeoT Оформления обкладинки К.М. Коротко!
Пшписано до друку 30.04.2003. Формат 60х90'/|6. Патр офсетний.
Гарштура Times. Друк офсетний. Ум. друк. арк. 28,5-Тираж 500 прим. Зам. 212.
Видавнинтво Нашонального фармацевтичного ушверситету. Укра!на, 61002, м. Хармв, вул. Пушкшська, 53.
Свщоцтво cepi'i ДК № 33 вш 04.04.2000.
ТОВ «Золоп стор1нки».
Укра'ша, 61145, м. Харив, вул. Косм1чна, 26.
Тел./факс: (0572) 30-32-10, 19-56-65.
Свщоцтво cepi'i ДК № 276 вщ 12.12.2000.
ОПЕЧ \ТКИ
Стр. Напечатано Стелет чгтатъ
1 3 4
45 /.А-система Е, Z-система
73 Н ^С-НСГ CHj НС- ^СН2 н циклогексен Н НС^ сн, • 1 1 НС- /СН2 ^с н циклогексадиен-1,3
76, 77 «ванна» ванна
136, 137 бриллиантиновый бриллиантовый
189 ,с6н5 xN=N c6h5z син-арилдиазогидроксид N=N с6н/ \н5 анти-арилдиазогидроксид с6н5 n=n C6HZ анти- азобензол N=N СбН5 C6HS син-азобензол
295 Ура Н2С—СН—CHj—СООН а у-хлорпропионовая кислота; 4-хлорпропановая кислота ура H2<j:—сн—сн—соон а у-хлормасляная кислота; 4-хлорбутановая кислота
353 незамещенным незаменимым
368 изооксазол изоксазол