
















































































































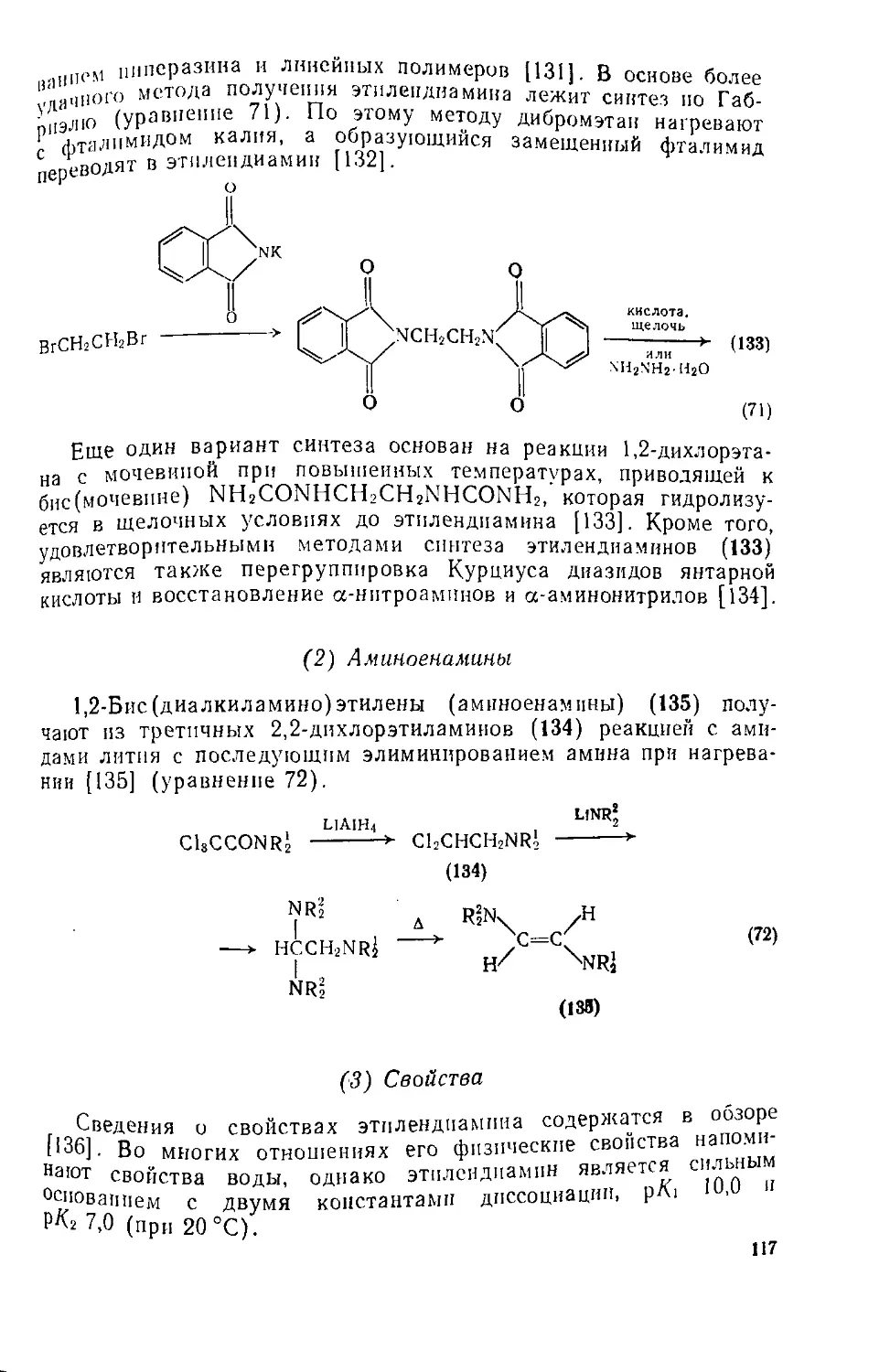























































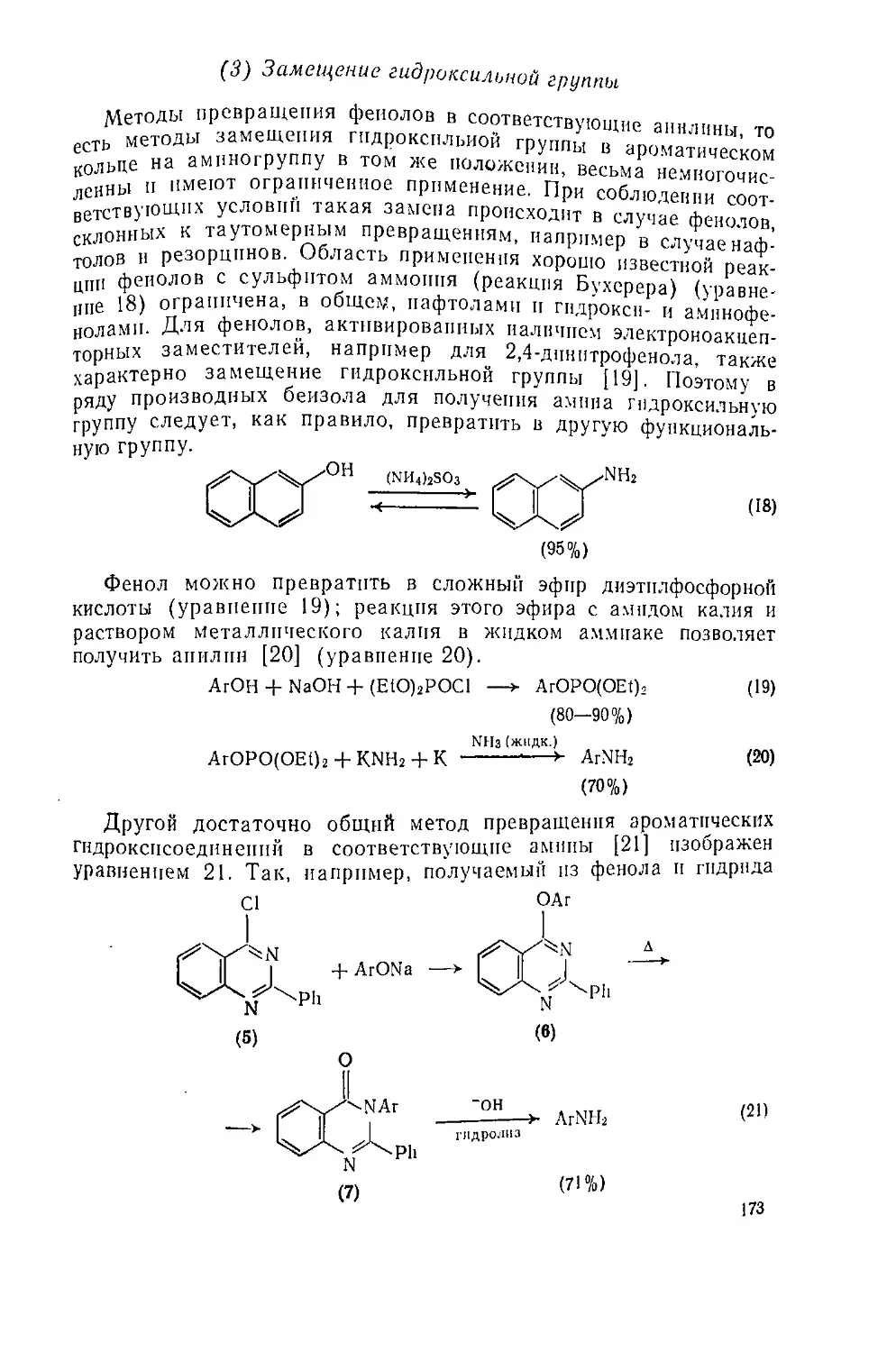





























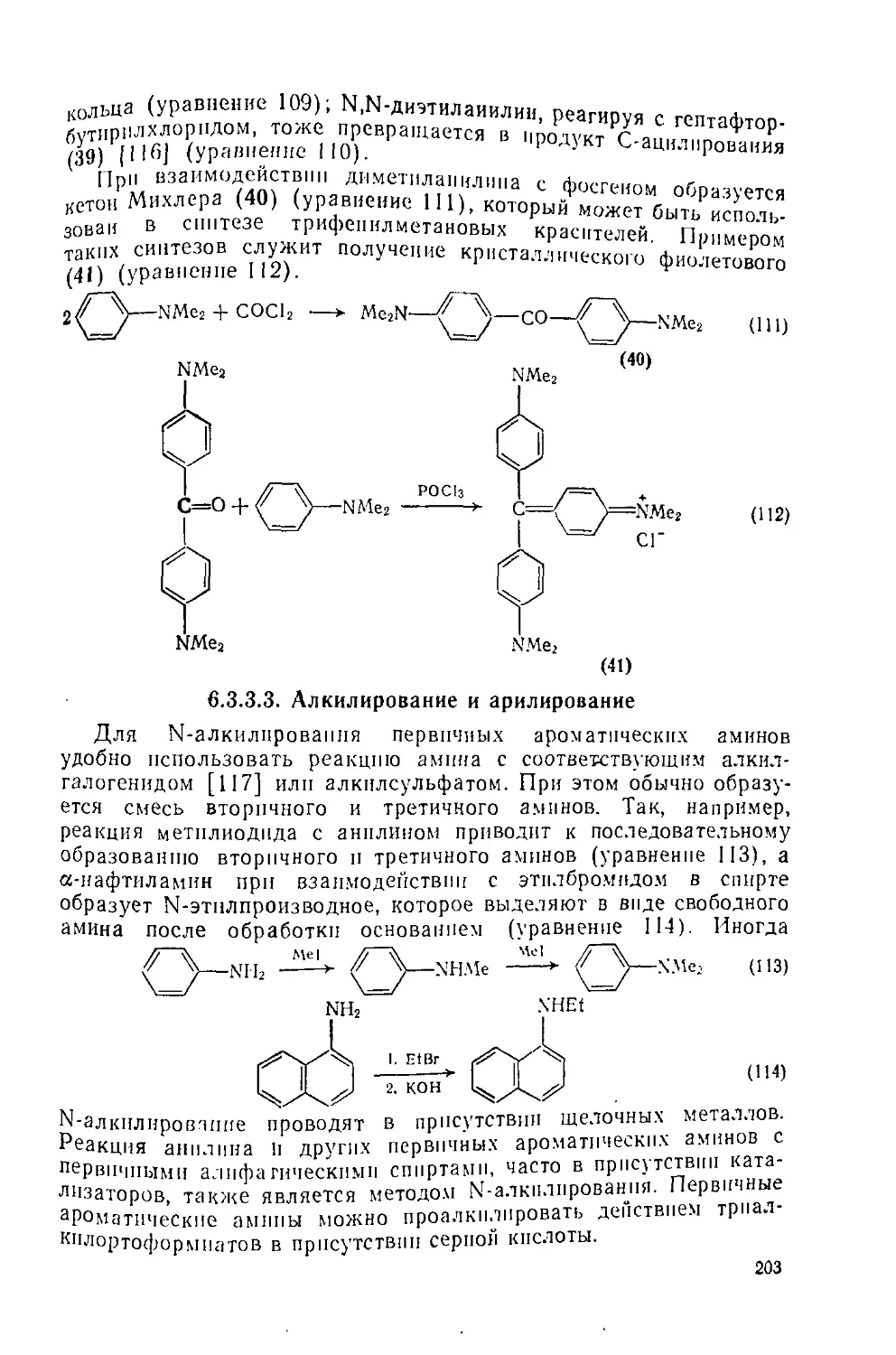














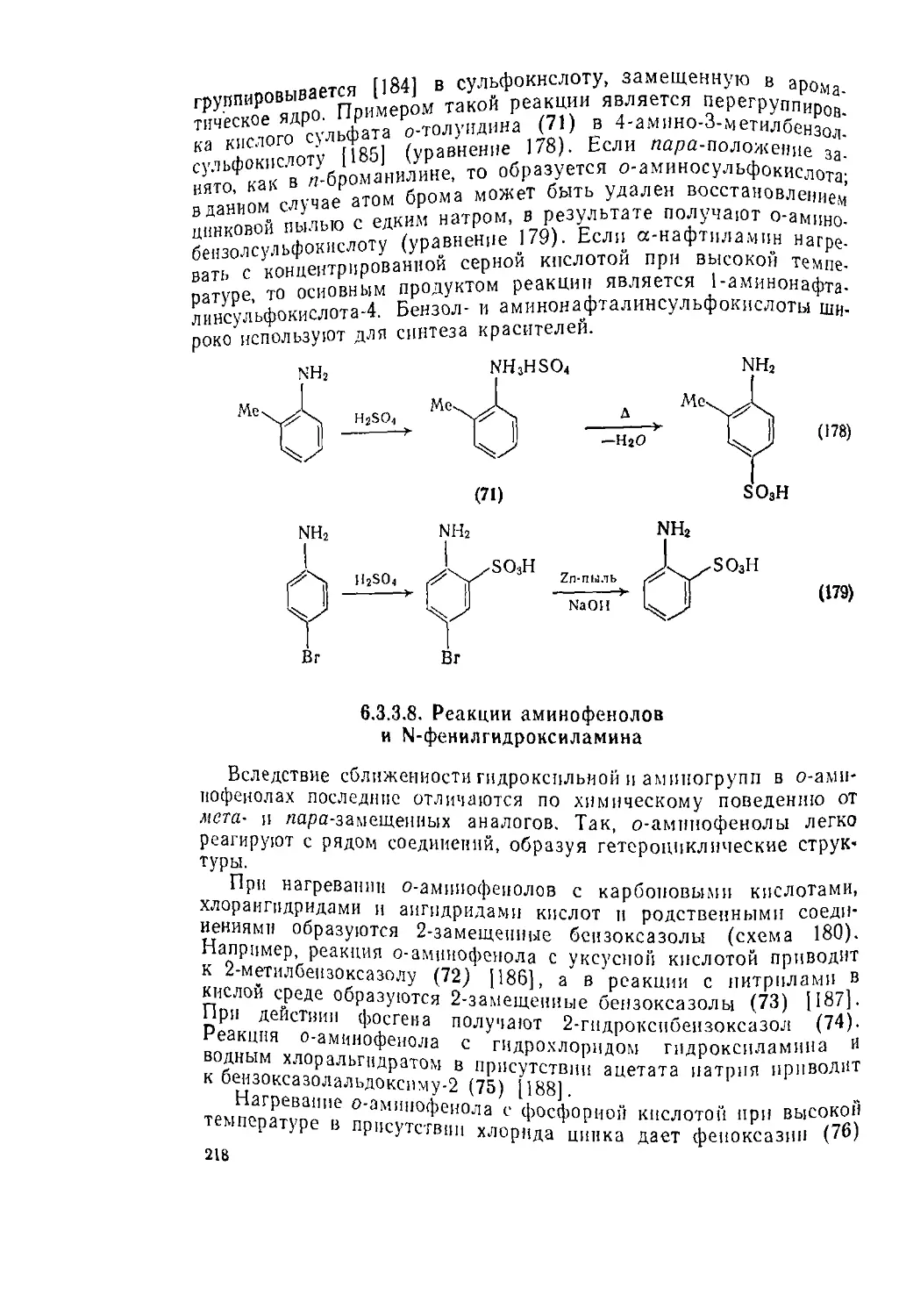





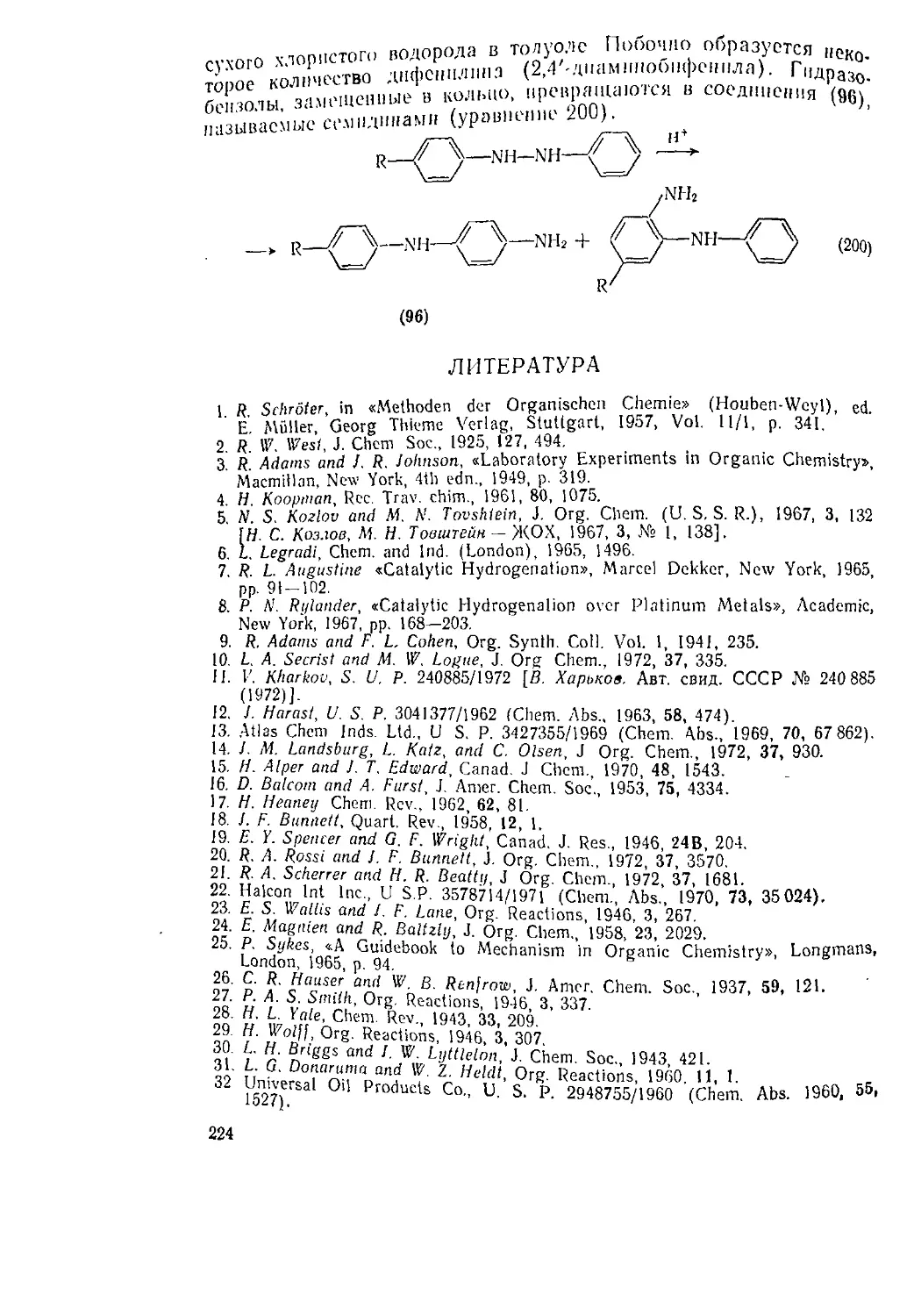













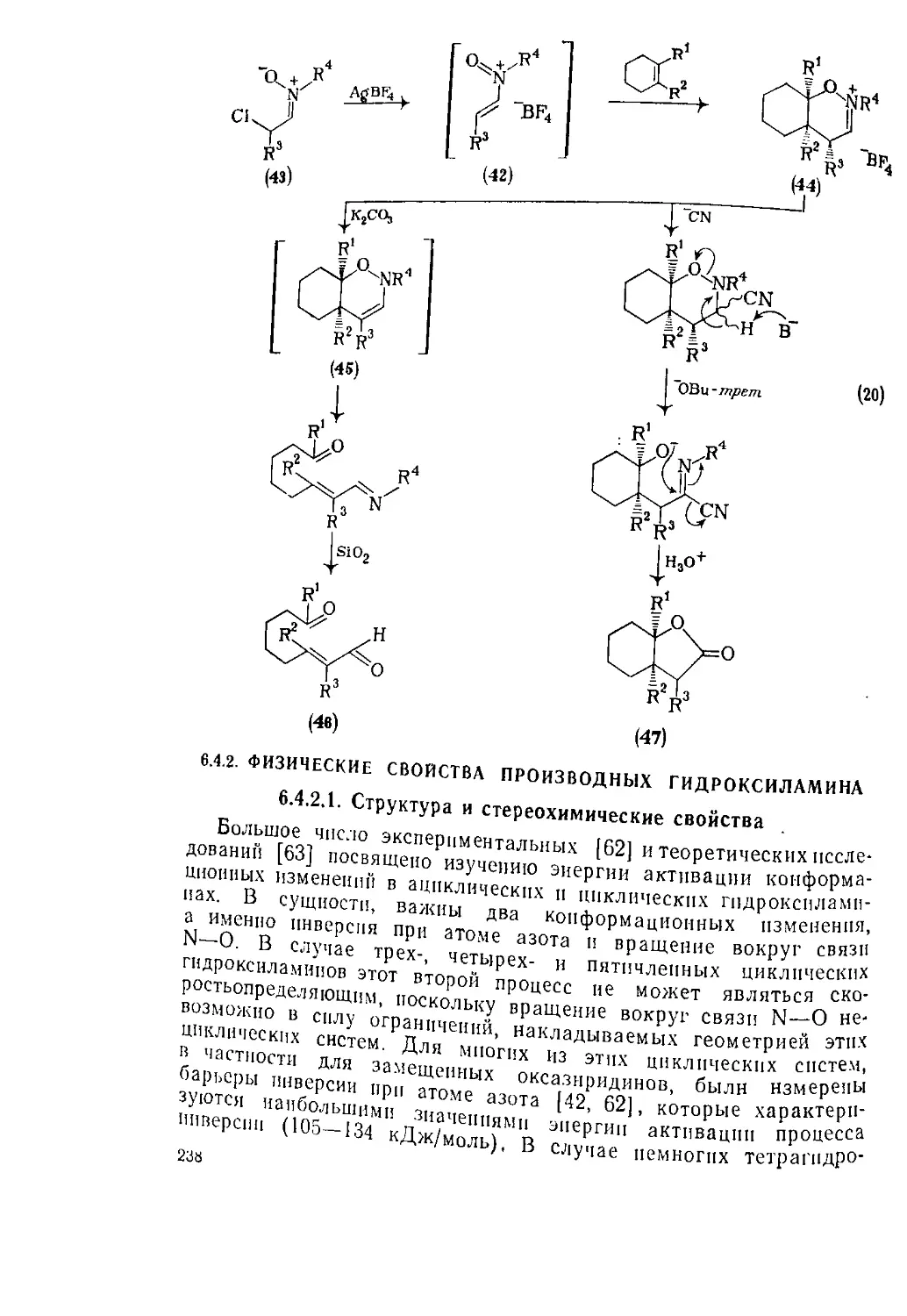











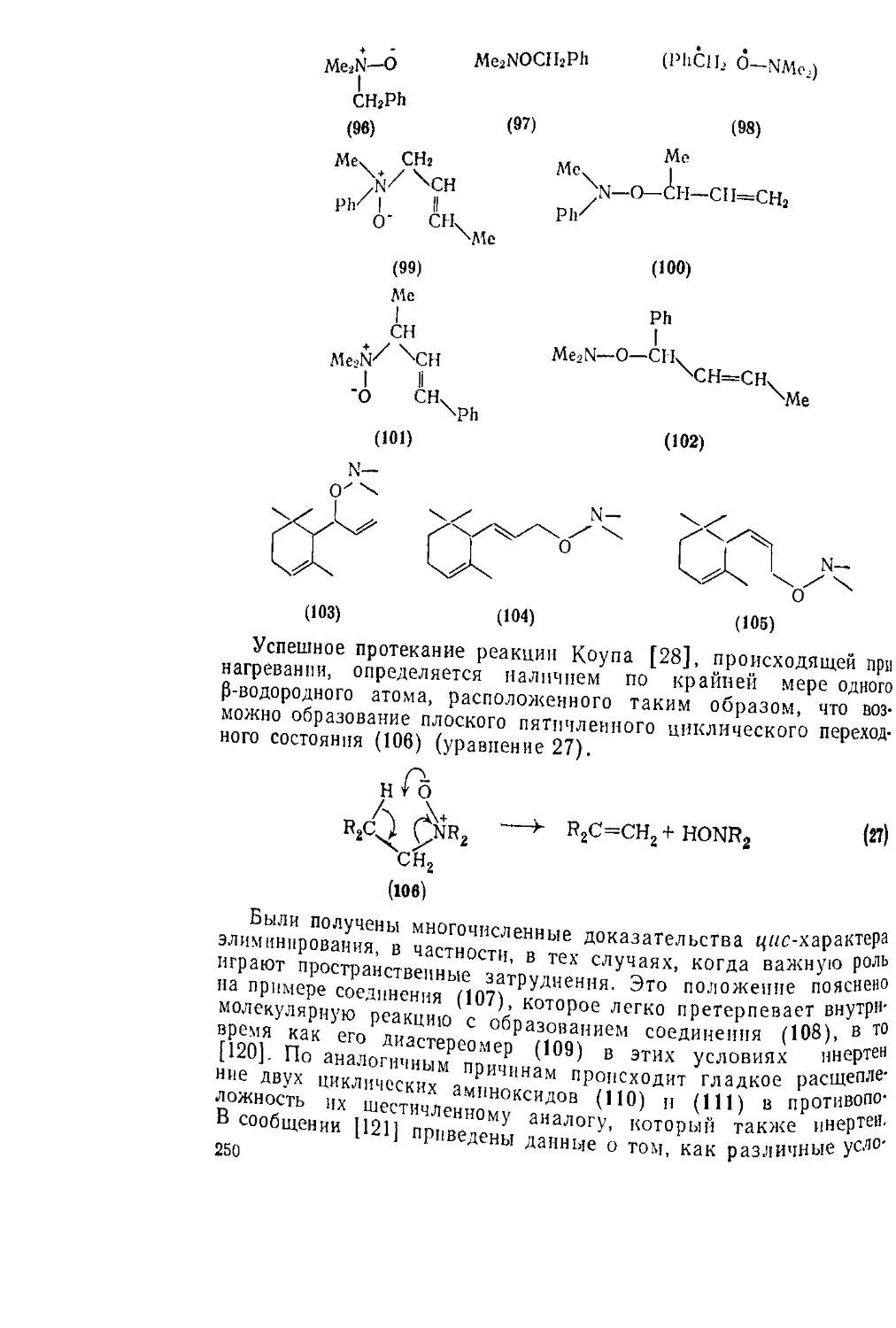
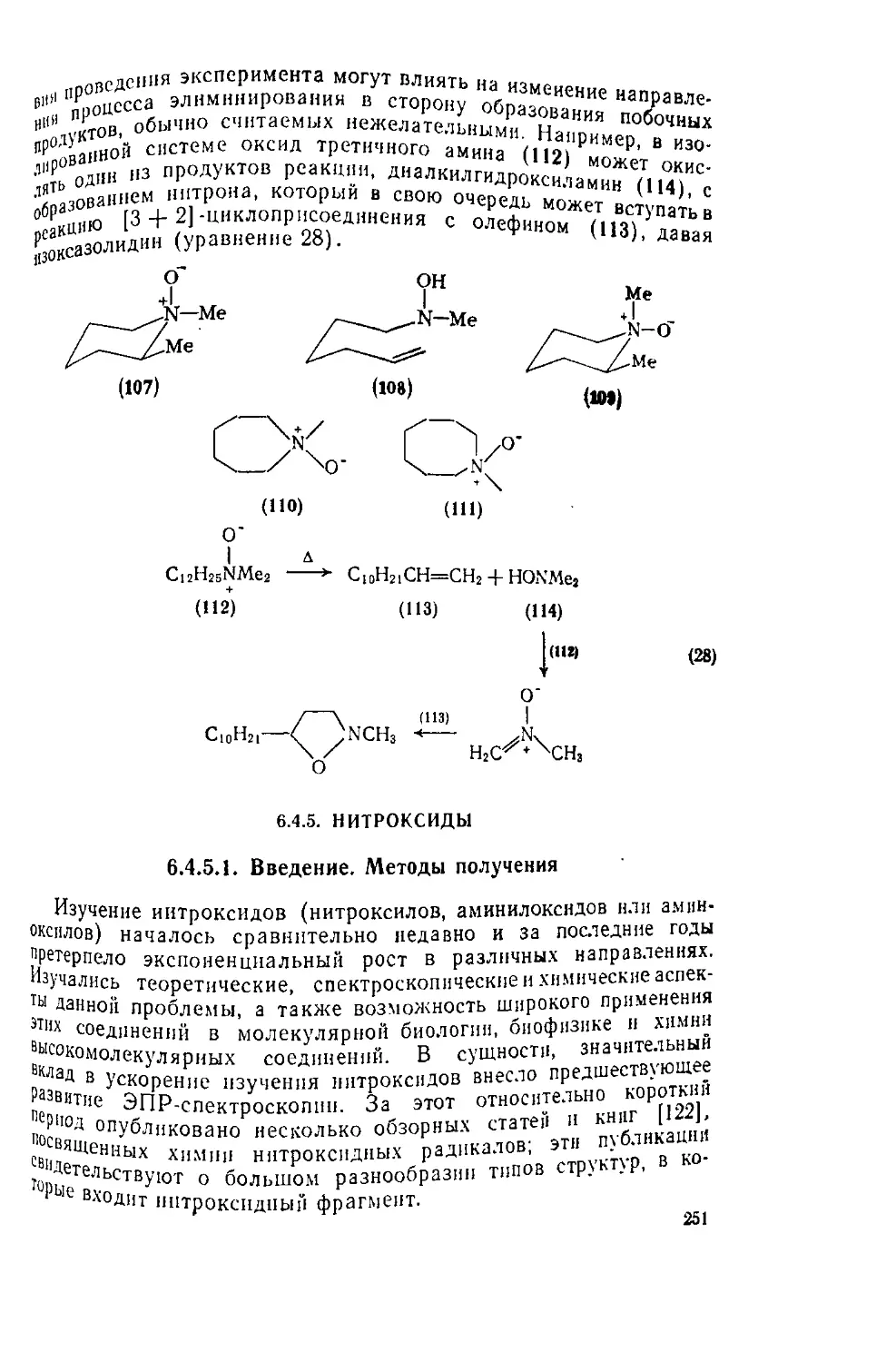










































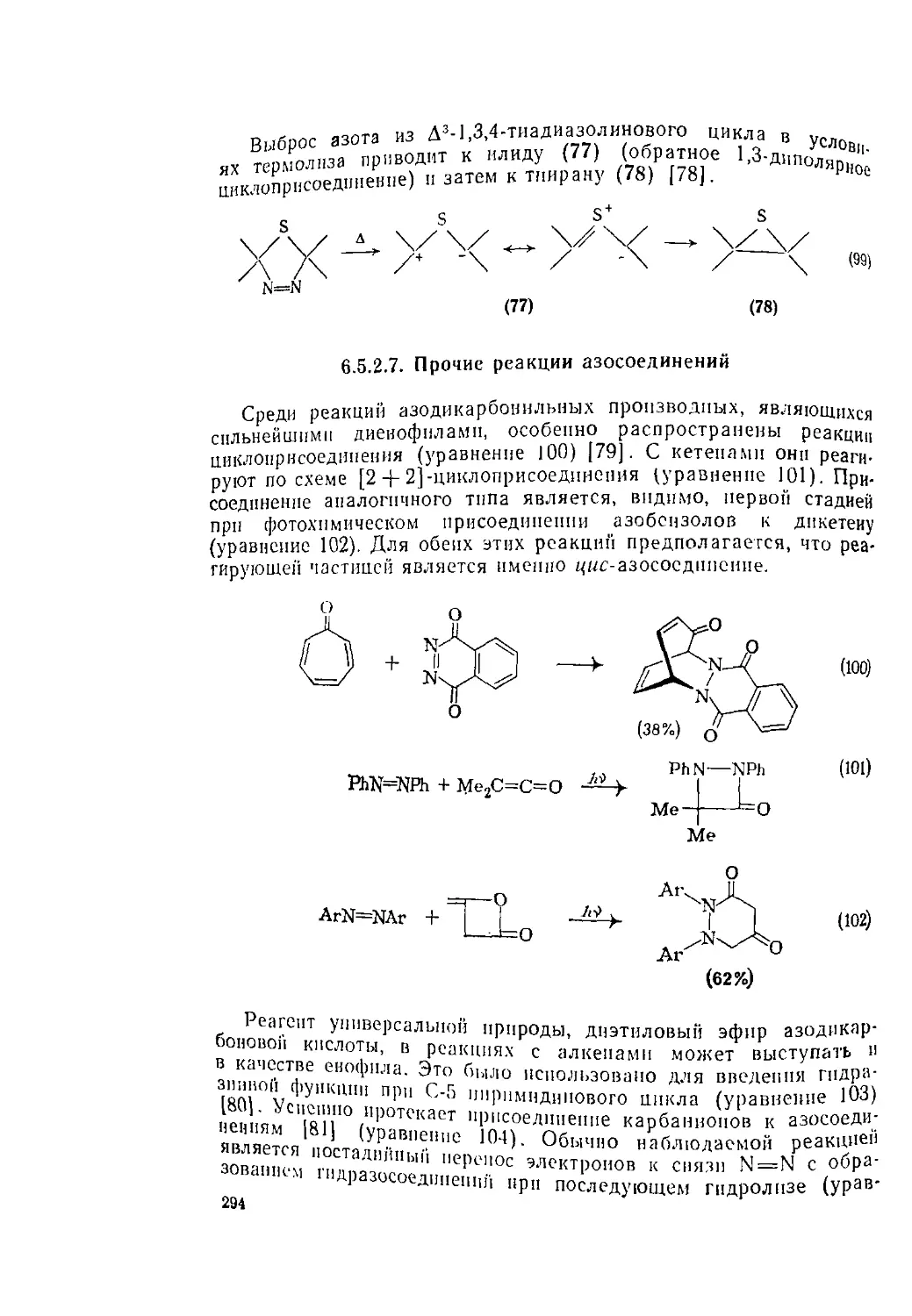















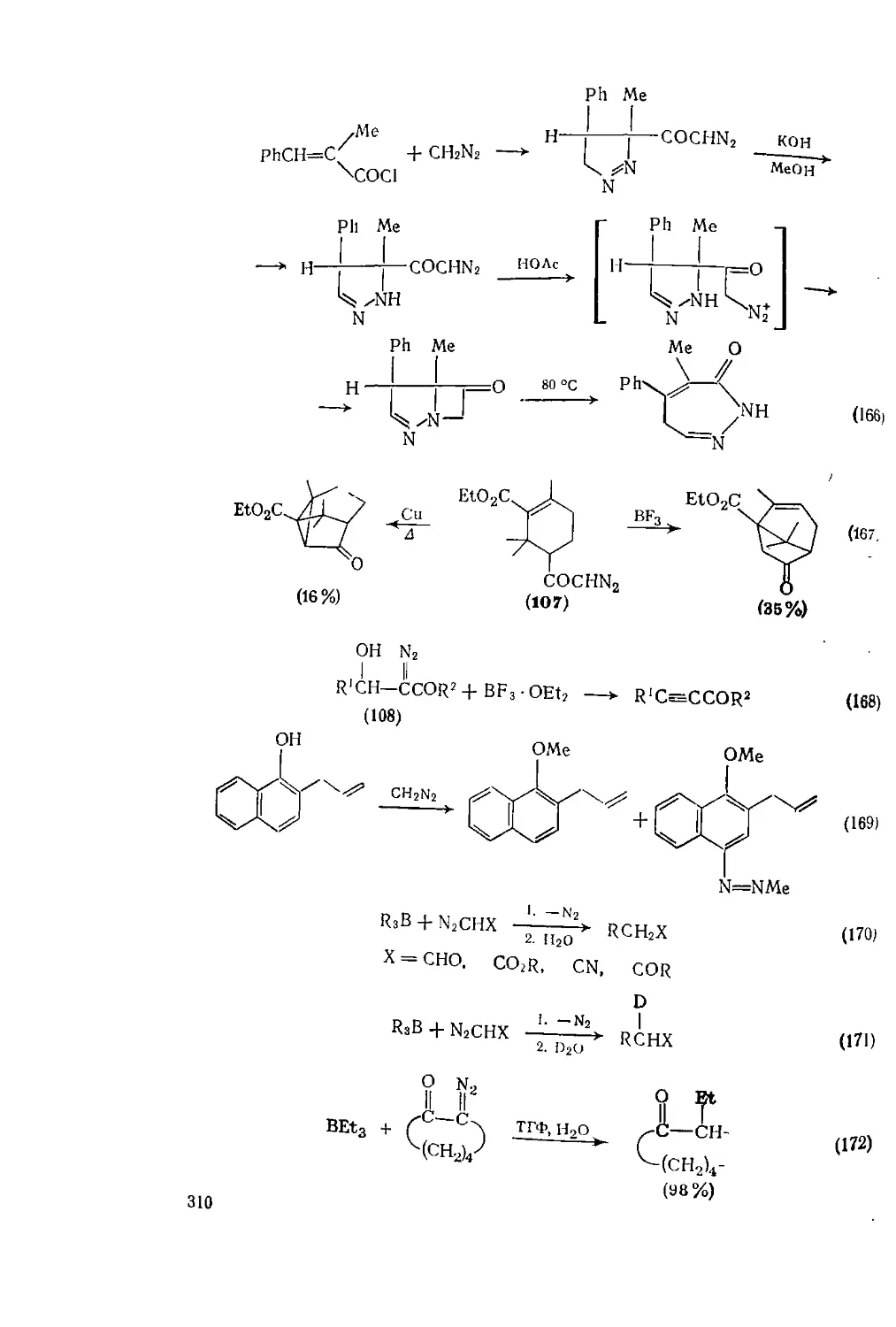

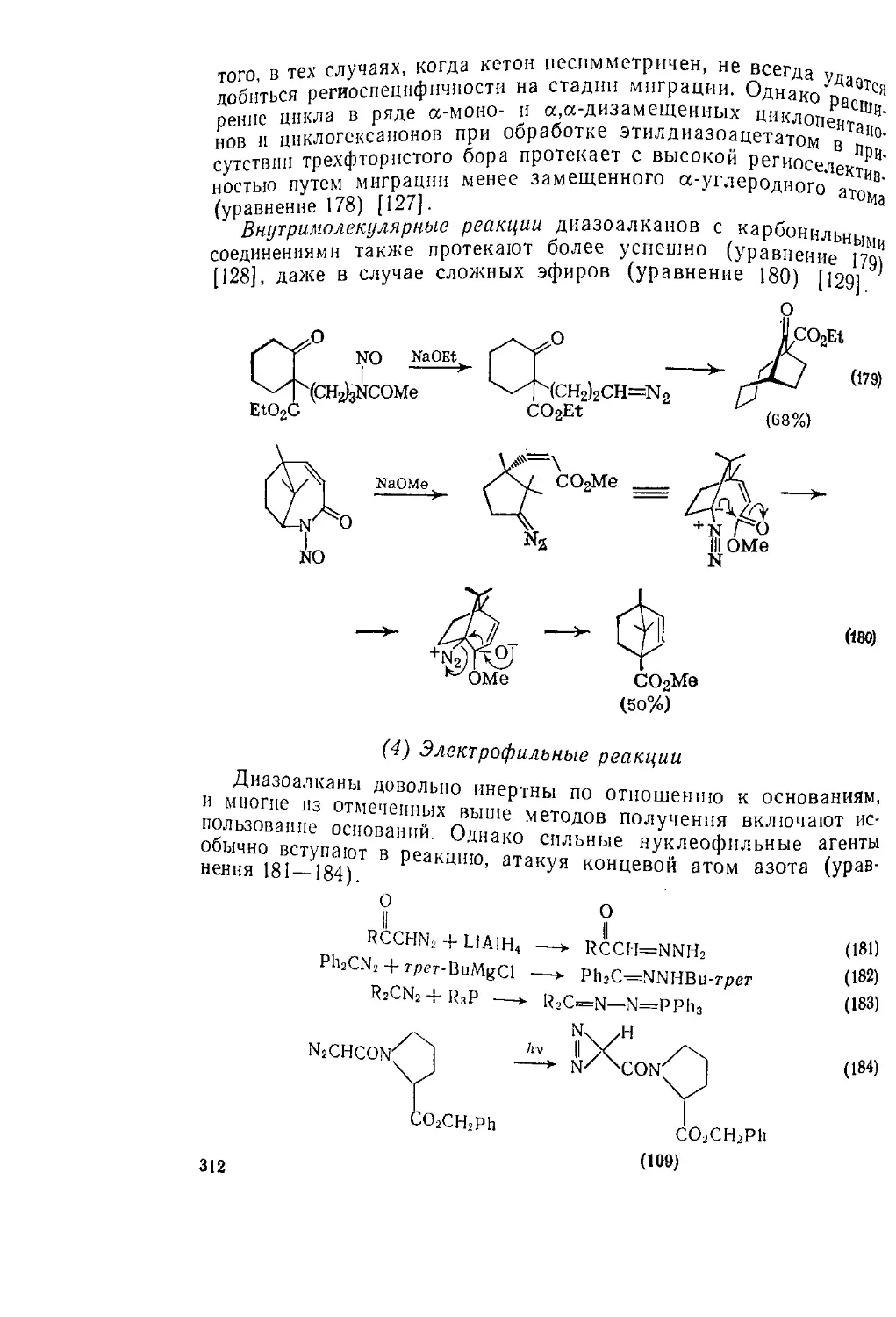







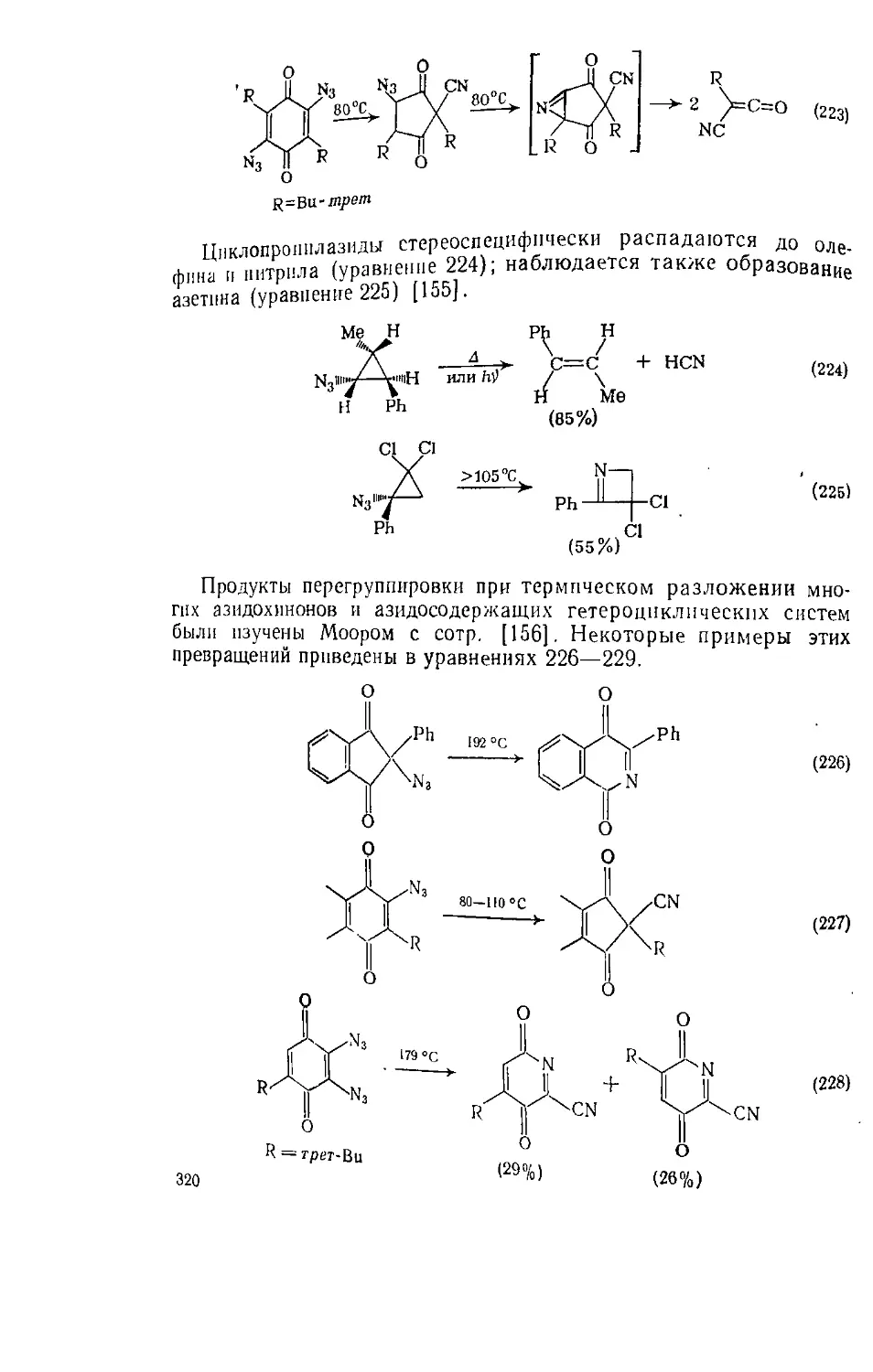




















































































































































































































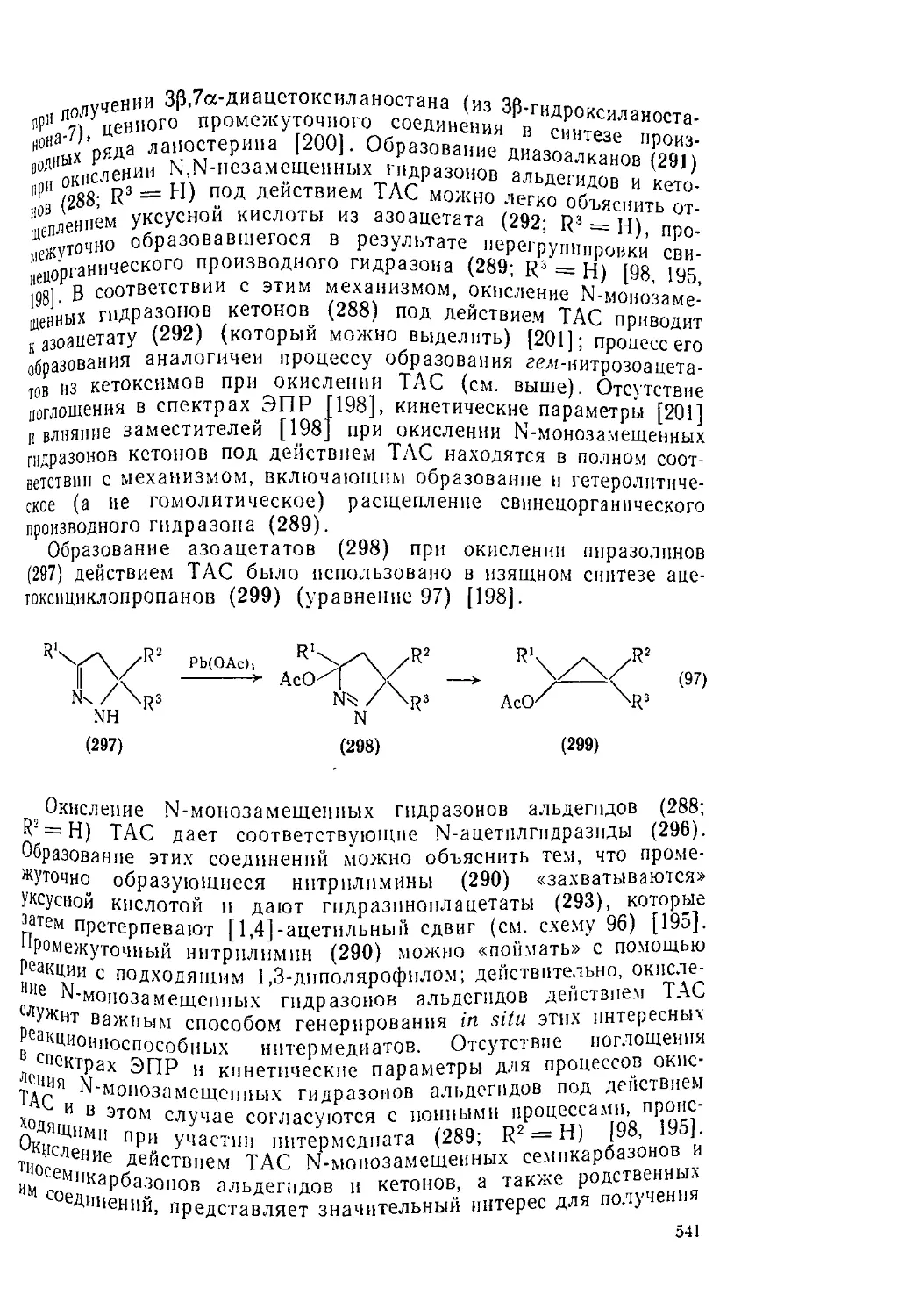








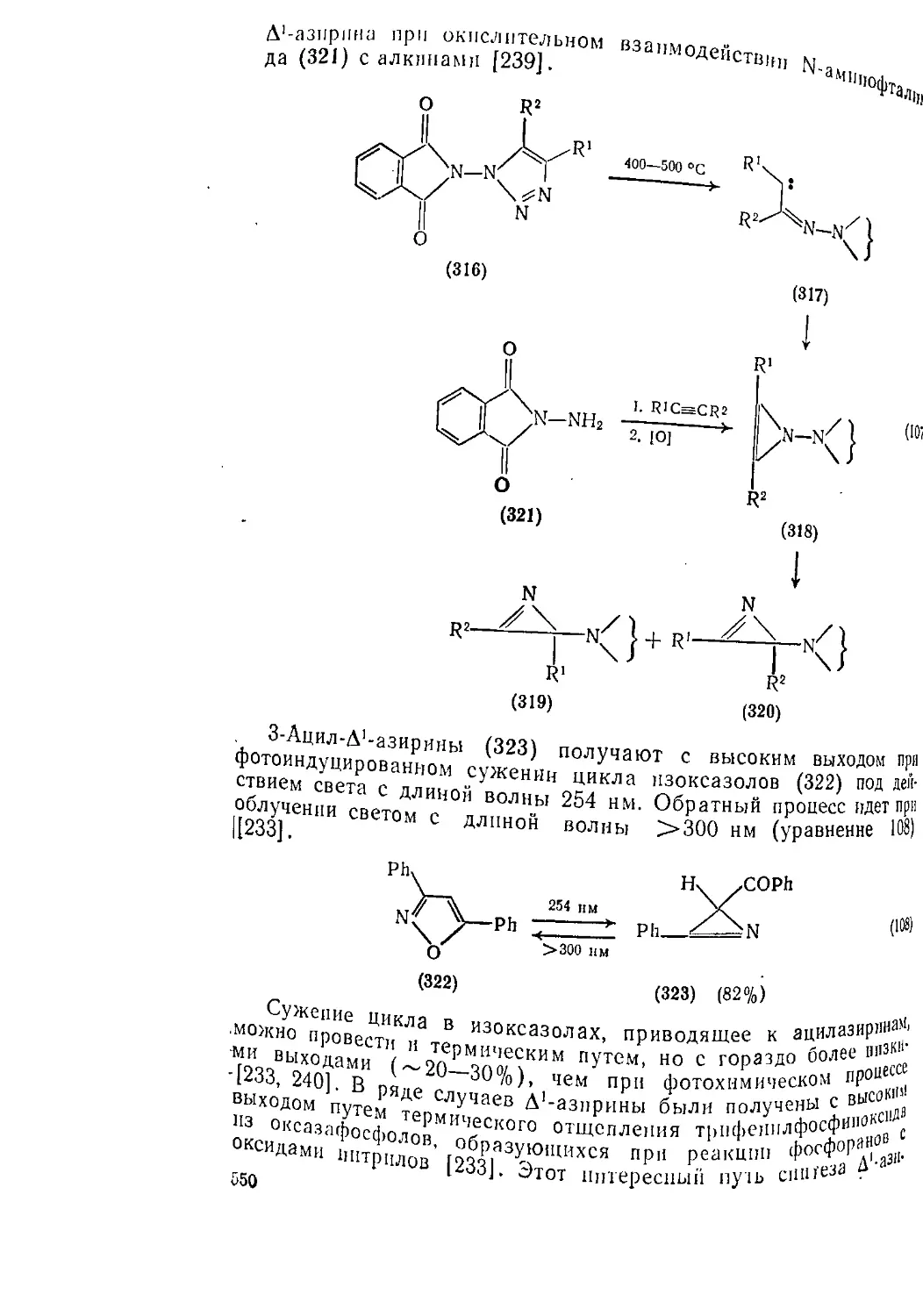






























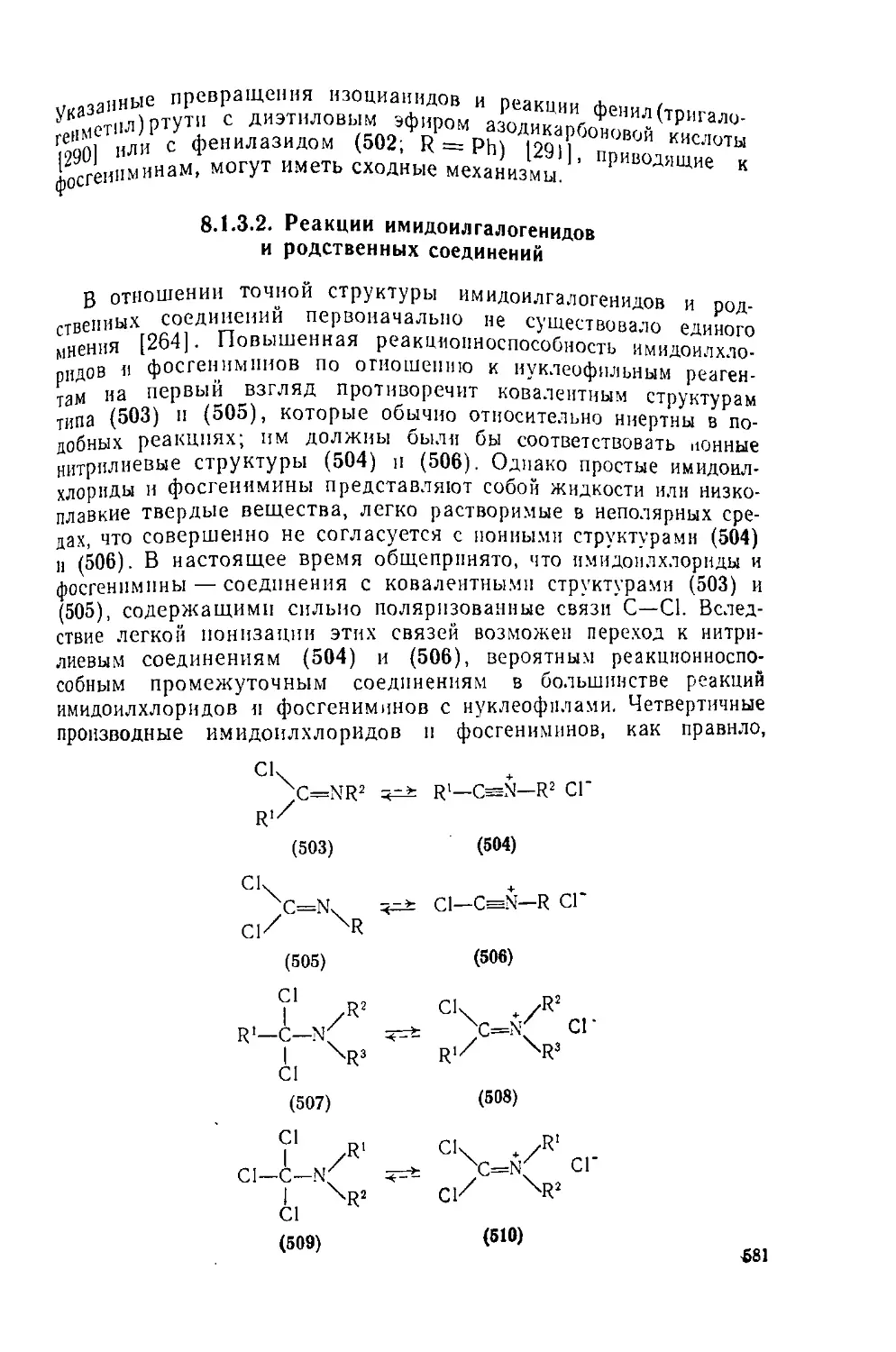









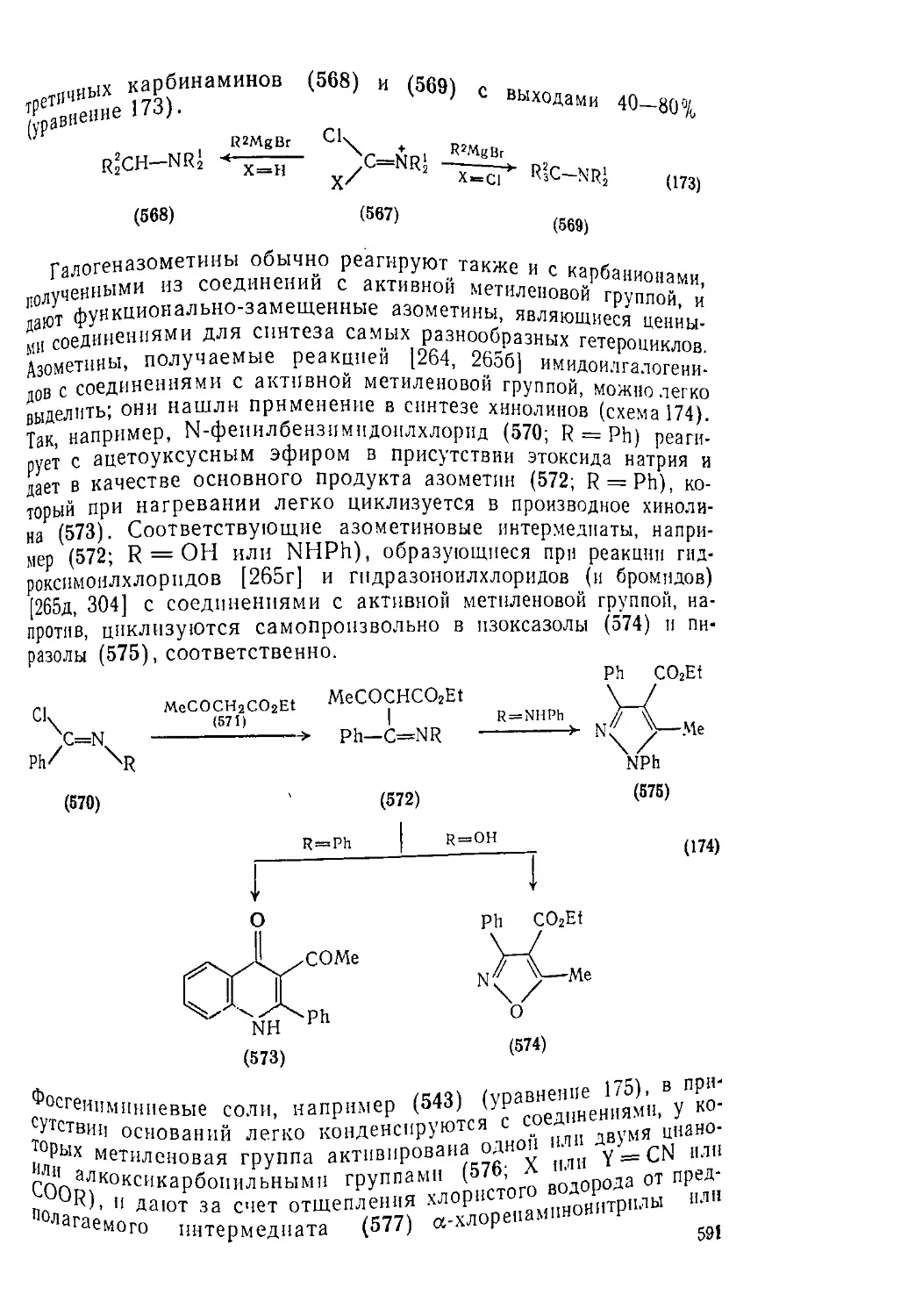



















































































































































Текст
Больше химической литературы и
прочих полезных материалов для
химиков на https://vk.com/chemzone
More chemistry books and other
useful resources for chemists are
available on https://vk.com/chemzone
СНВйПИЕ
vk.com/chemzone
COMPREHENSIVE
ORGANIC CHEMISTRY
The Synthesis and Reactions of Organic
Compounds
CHAIRMAN AND DEPUTY CHAIRMAN OF THE EDITORIAL BOARD
SIR DEREK BARTON, F.R.S.
AND
W. DAVID OLLIS, F.R.S,
Volume 2 Nitrogen Compounds
Edited by I. O. SUTHERLAND
Universily of Liverpool
e
PERGAMON PRESS
D NEW YORK • TORONTO . SYDNEY PARIS FRANKFURT
ОБЩАЯ
ОРГАНИЧЕСКАЯ
ХИМИЯ
том 3
АЗОТСОДЕРЖАЩИЕ СОЕДИНЕНИЯ
Перевод с английского
канд. хим. паук
А. Я. ЧЕРНЯКА
Под ред. акад.
Н. К. КОЧЕТКОВА
и канд. хим. паук
Л. В. БАКИНОВСКОГО
МОСКВА,
«ХИМИЯ», 1982
УДК 547
Общая органическая химия./Под род. Д. Бартона
п У. Д. Оллпса. Т. 3. Азотсодержащие соединения./
Под ред. И. О. Сазерленда.— Пер. с англ./Под
ред. Н. К. Кочеткова н Л. В. Бакпновского. — М.:
Химия, 1982. — 736 с., нл.
Третий том перевода настоящего многотомного издания по*
священ азотсодержащим органическим соединениям — аминам,
производным гвдроксплампна и гидразина, азоторгаппческим
нонам к радикалам, нитренам, нитро- и пнтрозосоедкнениям,
иминам, нитронам, нитрилам и изоцианидам; описаны методы
получения этих соединении, структура, свойства, реакции, при-
менение.
Издание предназначено для научных работников, инже-
неров-химиков, работающих на предприятиях химической, неф-
техимической п других отраслей промышленности, преподава-
телей и аспирантов химических н хвмвко-техпологическнх ву-
зов, биохимиков и биологов.
736 с., 12 табл., 2210 литературных ссылок.
л 1803000000-108
° "050(01J-82 -Подписное
© 1979 Pergamon Press Ltd.
© Перевод на русский язык. Издательство «Химия», 1982 г.
СОДЕРЖАНИЕ
Предисловие к тому 2 апглнГп hoiq издания 9
ЧАСТЬ 6. АМИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ 11
6.1. Алифатические и циклические амины. Дж. Р, Малпасс 11
6.1.I. Методы получения аминов И
6.1.1.1. Алкилирование и деалкилирование аминов 11
6.1.1.2. Методы восстановления 16
6.LI.3. Синтез аминов с помощью реакций присоединения 21
6.1.1.4. Применение металлоргаинческих реагентов в синтезе аминов 24
6.1.1.5. Гидролитические методы получения аминов 27
6.1.1.6. Синтез аминов с помощью внутримолекулярных перегруппиро-
вок 28
6.1.1.7. Получение циклических аминов 31
6.1.1.8. Разделение хвральных аминов 37
6.1.2. Свойства аминов 39
6.I.2.I. Структура и стереохимия простейших аминов 39
6.1.2.2. Спектроскопические свойства аминов 45
6.1.2.3. Основность аминов 53
6.I.2.4. Физиологическая активность аминов 58
6.1.3. Реакции аминов 61
6.1.3.1. Нуклеофильные свойства аминов 61
6.1.3.2. Основные свойства, соли четвертичных аммониевых оснований
н катализ 67
6.I.3.3. Окисление аминов 72
6.1.3.4. Кислотные свойства аминов 74
6.1.3.5. Введение защитных групп в амины и их удаление 78
6.1.3.6. Реакции азиридинов 81
Литература 86
6.2. Полнфункциональные амины. Дж. М. 3. Гладых, Д. Хартли 91
6.2.1. Ненасыщенные амниы 91
6.2.1.I. Енамины 91
6.2.1.2. Аллиламниы 161
6.2.1.3. Амины алленового ряда 104
6.2.1.4. Инамниы 105
6.2.1.5. Амины ацетиленового ряда НО
6.2.2. Диамнны и полиамины 113
6.2.2.I. М-Диамнны 113
6.2.2.2. 1,2-Диамнны 116
6.2.2.3. 1,3-Диамины 121
6.2.2.4 Полиамииы 123
6.2.3. Амииоспнрты 126
6.2.3.I. 1,1-Амнноспирты 126
6.2.3.2. 1,2-Ампиоспнрты 128
6.2.3.3. 1,3-Амнноспирты 138
6.2.4. Простые амнноэфнры 139
6.2.4.1. 1,1 -Амнноэфнры 139
6.2.4.2. 1,2-Аминоэфкры 143
6.2.4.3. 1,3-Аминоэфиры 145
6.2.4.4. Макроциклические аминоэфиры 146
6.2.5. Галоген амины (С-галогсизамещеиные) 148
6.2.5.1. а-Галогенамииы 148
6.2.52. P-Гало! енамины 155
6.2.5.3. у-Галогеиамины 159
Литература 161
s
S/иолрешДожеских аминов
Вечные ,?Рт7е™"нь""ро«"™ческ|1е амины
Полнфупкциоиальные ароматические амины
Свойства ароматических аминов
в 3. Ароматические амины.
6.3.1-1-
6.3.1.2.
6.3.1.3.
6.3.1.4-
6.3.2.
й Ч 2 1 Основность
6 3 2 2 Спектроскопические свойства
6 3 2 3 Физиологические свойства
633 Реакции ароматических аминов
6 3 3.1. Реакции с азотистой кислотой
Б.з'з'2. Ацилирование
63 3 3 Алкилирование и арилнроваиие
6 3 ЗЛ Образование изоцианатов и производных мочевины
6.3.3.5. Окисление
6 3 3.6. Реакции с. альдегидами и кетонами
6.3.3.7. Галогенирование, нитрование и сульфирование
6 3.3.8. Реакции а.минофеиолов и N-феиилгндрокснламина
6.3.3.9. Реакции ароматических диаминов и феинлгпдразнна
Литература
6.4 . Производные гидроксиламииа. Дж. С. Робертс
6.4.1. Методы получения производных гидроксиламииа
6.4.1.1. Номенклатура и вводные замечания
6.4.1.2. N-Моиозамещенные ациклические гпдрокенламины
6.4.1.3. О-За.мещснные ациклические гпдрокенламины
6.4.1.4. N.N-Дизамещенные ациклические гидроксиламниы
674.1.5. ЦО-Дизамещенные ациклические гидроксиламниы
6.4.1.6. К.М.О-Тризамещештые ациклические гидроксиламниы
6.4.1.7. N.N-Днзамещештые циклические гидроксиламниы
6.4.1.8. N.N.O-Тризамещеииые циклические гидроксиламниы (7)
6.4.1.9. N.O-Ди- и М,М,О-триза.мещениые циклические гидроксиламниы
(8) и (9)
6.4.2. Физические свойства производных гидроксиламииа
6.4.2. 1, Структура и стереохимические свойства
64.2.2. Спектроскопические и кислотно-основные свойства
6.4.3. Реакции производных гидроксиламина
6.4.3.1. Нуклеофильные свойства
6.4.3.2. Окисление и восстановление
6.4.3.3. Перегруппировки
6.4.4. Оксиды аминов
6.4.4.1. Методы получения
6.4.4.2. Физические свойства
6.4.4.3. Реакции
6.4.5. Нитроксиды
кНп' ^ведение- Методы получения
влЙ P«S?a стереохимия и спектроскопические свойства
Лтература СПИН™аЯ Л°ВуШКа " СПШ10вая «етео
6.5 Производные гидразина и родственные с------- -
-3-1. Алифатические, ацпк'шческнр п ,7„ ‘ ‘
гидразины и полифуикцио. аИ" щ,кличе«ие
6-01.1. Методы получения 1ЩИОиалЬ||1« системы
Реакции'13 " СтеРеох"мая
I-ндразил-радикалы
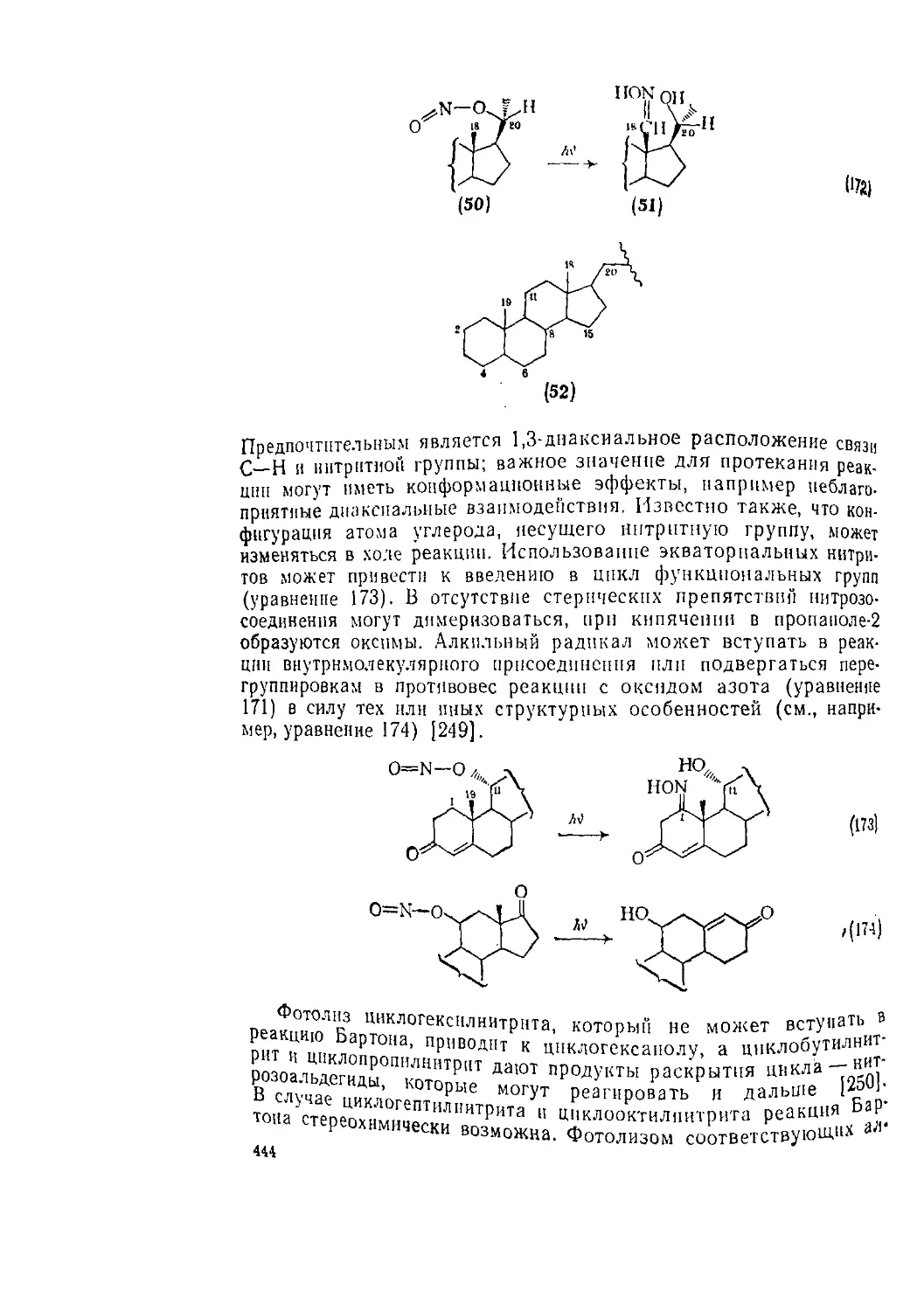
168
168
168
176
181
182
191
191
193
195
196
196
201
203
206
207
211
214
218
220
224
228
228
228
229
231
231
232
232
233
234
234
238
238
240
240
240
243
245
247
247
247
248
251
251
255
259
262
263
268
268
268
275
278
284
285
285
гидразины.
арнл-
6.5.1.2.
6.5.1.3.
6.5.1.4.
6.5.2.
6.5.2.1.
6
в.5.2.2. Циклические азосоедииения 287
6.5.23. Ароматические азосоедииения 288
6.5.2Л. Мопозамсщепиые диазены и дипмииы 289
6.5.25. цис-транс-Изомерия 289
6.5.2.6. Термическое и фотохимическое разложение 290
65.2.7. Прочие реакции азосоединеиий 294
6.5.2.8. Азоксисоедииення 296
6.5.2.Э. Азодиоксисоединения 299
6.5.3. Диазосоедииеиия 300
6.5.3.I. Методы получения 300
6.5.3.2. Реакции 305
6.5.4. Азиды 313
6.5.4.1. Методы получения 313
6,5.4.2. Структура и устойчивость 315
6.5.4.3. Реакции 316
6.5.5. Системы, содержащие полиазафрагмеиты 326
Литература 329
6.6. Азоторганические ионы и радикалы, нитрены и родственные частицы.
Т. Л. Джилкрист 334
6.6.1. И-Галогепамины и катионы нитрения 335
6.6.1.1. Методы получения N-галогенаминов 335
6.6.1.2. Реакции с олефинами 336
6.6.1.3. Реакции с ароматическими соединениями 339
6.6.1.4. Хлорирование связей С—Н. Реакция Гофмана — Лефлера 340
6.6.1.5. Перегруппировки N-хлорамииов. Промежуточное образование
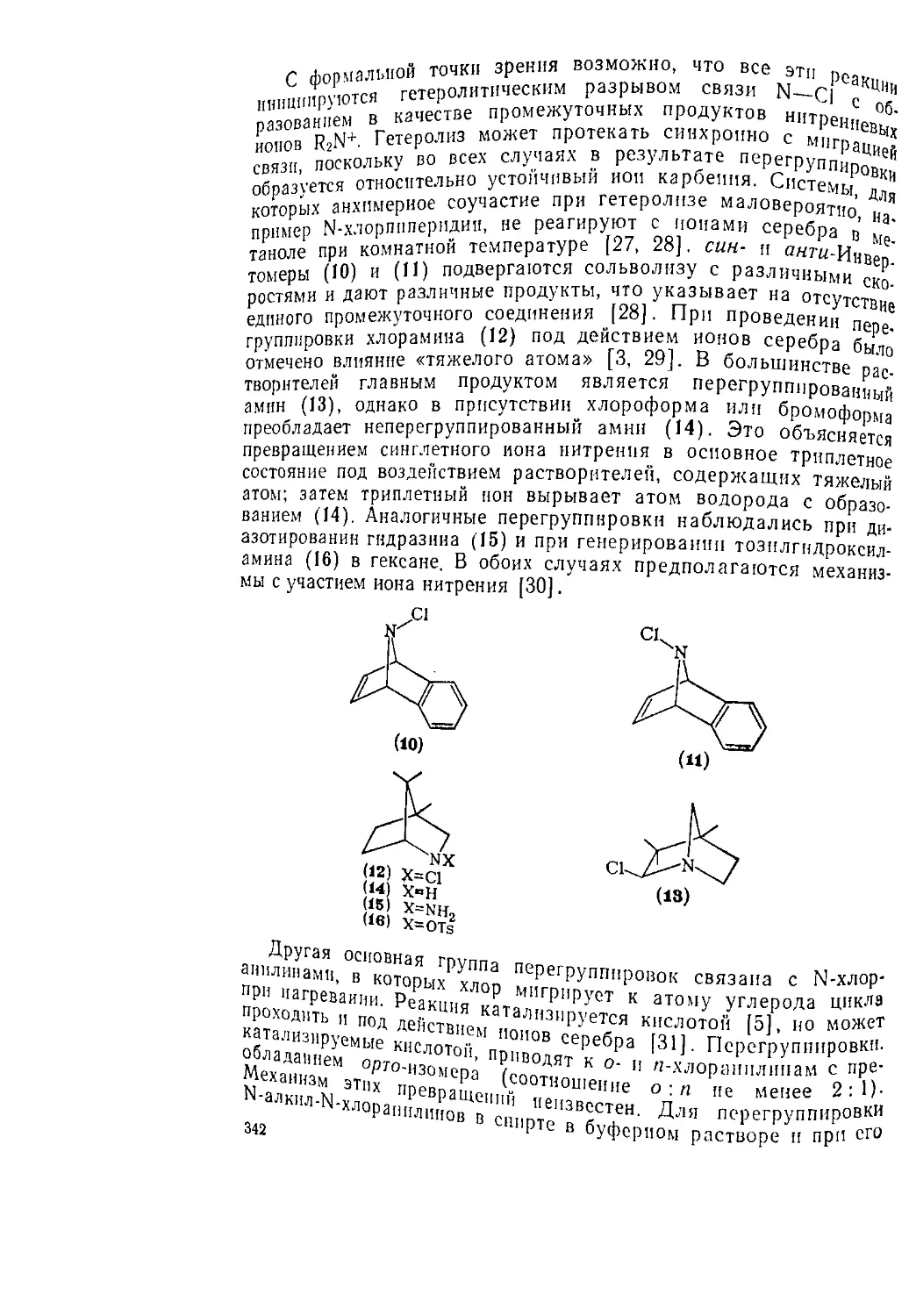
ионов питрепня 341
6.6.1.6. Прочие реакции N-хлораминов 343
6.6.2. Азоторганические анионы 344
6.6.3. Азоторганические радикалы 348
6.6.3.I. Аминильиые радикалы 348
6.6.3.2. Радикалы амннпя 351
6.6.4. Нитрены 353
6.6.4.1. Методы генерирования 354
66.4.2. Структура 359
6.6.4.3. Реакции 359
Литература 367
ЧАСТЬ 7. НИТРО- и НИТРОЗОСОЕДИНЕНИЯ. Р. Дж. Кумбс 372
7.1. Нитрозосоединения 372
7.1.1. Методы получения алифатических и ароматических иитрозосо-
сдииеинй 372
7.1.2. Свойства иитрозосоединений 382
7.1.3. Реакции иитрозосоединений 387
7.1.4. Иитрозоловые кислоты 399
7.2. Нитросоединения 399
7.2.1. Методы получения ароматических и алифатических иитросоеди-
пеппй 399
7.2.2. Свойства питросоедпнеиий 411
7.2.3. Реакции питросоедпнеиий 419
7.3. Нитриты и нитраты 440
7.3.1. Эфиры азотпегой кислоты 440
7.3.2. Эфиры азотной кислоты 445
7.4. Нитрозамины 449
7.4.1. Методы получения N-питрозосоединений 449
7.4.2. Свойства М-пптрозосоедш1еш|й 451
7.4.3. Реакции N-иитрозосоединений 454
7.5. Нитрамины 459
7.5.1. Методы получения нитраминов 459
7
7 5.2. Свойства нитраминов 461
7'5.3. Реакции нитраминов 463
Литература ' 466
ЧАСТЬ 8. ИМИНЫ, НИТРОНЫ. НИТРИЛЫ II изоцилииды.
Дж. Тэннант '476
8.1. Азометины 476
8.1.1. Введение 476
8 111. Общие методы создания С = М-свяЗн 476
8 112. Геометрическая (син-анти) изомерия, прототропная таутомерия
н молекулярные перегруппировки азометпнов 488
8.1.2. Алифатические, алициклические и содержащие ароматические
заместители азометппы 494
8.1.2.1. Специфические методы синтеза простых азометпнов 494
8.1.2.2. Реакции простых азометпнов 499
8.1.2.3. Циклические азометнцы с малым циклом (азирины, азетипы и
родственные соединения) 547
8.1.2.4. Сопряженные полиазаены 559
8.1.3. Имидоплгало1енпды и родственные соединения 576
8.1.З.1. Методы получения имндоилгалогенидов и родственных соедине-
ний 577
8.1.3.2. Реакции импдонлгалогенндов и родственных соединений 581
8.1.4. Имидаты и родственные соединения _ 598
8.1.4.1. Методы получения имндатов, тнонмидатов и родственных со-
единений 598
8.14.2. Реакции имидатов, тнонмидатов и родственных соединений 601
8.1.5. Амидины и родственные соединения 607
8.1.5.1. Методы получения амидинов и родственных соединений 607
8.1.5.2. Реакции амидинов и родственных соединений 609
8.1.6. Днполярные системы 613
8.1.6.1. Методы получения N-оксндов азометинов (нитронов) 613
8.1.6.2. Реакции N-окспдов азометпнов (нитронов) 618
8.1.6.3. Азометипимнпы и азометин-илиды 626
8.1.7. Гетерокумулены 629
8.1.7.1. Методы получения изоцианатов, изотиоцианатов, карбодиимидов
и кетенимпнов 630
8.1.7.2. Реакции изоцианатов, изотиоцианатов, карбодцимидов и кетен-
иминов 639
8.2. Нитрилы н иэоцианиды
8.2.1. Введение
8.2.2. Нитрилы
8.2.2.1. Общие методы получения алифатических, алициклических и аро-
матических нитрилов
8.2.2.2. Реакции алифатических, алициклических и ароматических нит-
рилов
8.2.3. Полнциаиосоедннеиня
o n o V Методы получения полицианосоедпиеинн
о.2.3.2. Реакции полициацосоединепий
8.2.4. Днполярные системы
8.2.4.1. Методы синтеза оксидов нитрилов, нитрплимицов и ннтрил-илп-
8 82.5. Имщшипды”108 НИТРИЛОВ' читрилцминов п питрил-плидов
ЯОЧ О Методы получения изоциаиидов
O.2.O.2. Реакции изоциаиидов
Литература
Предметный указатель
647
647
647
647
661
677
677
683
692
693
695
700
700
702
711
728
в
ПРЕДИСЛОВИЕ К ТОМУ 2
АНГЛИЙСКОГО ИЗДАНИЯ
Классификация материала органической химии, в основе которой лежат
химические особенности функциональных групп, стала уже традиционной. Этот
принцип классификации не потерял своего значения, несмотря на возникновение
других подходов к систематизации, например, таких, в основе которых лежат
механизмы реакции. Традиционный подход, который был принят при написании
«Общей органической химии», позволяет уделить особое внимание реакциям
органических соединений и современным методам синтеза. Химики-органики, за-
нимающиеся научно-исследовательской пли преподавательной деятельностью,
ощущают наибольшую потребность в информации подобного рода. Можно на-
деяться, что принятый в даиио*м томе принцип систематизации даст возможность
специалистам и тем, для кого органическая химия является менее знакомой дис-
циплиной, достаточно легко отыскать необходимую информацию.
Основные руководства по органической химии, такие, как Губен-Вейль, со-
держат полную сводку методов синтеза и реакций. Имеется также большое
число монографий, которые столь же подробно охватывают специальные разделы
органической химии. «Общая органическая химия» является попыткой найти
компромиссное решение между специализацией н огромным объемом материала.
Поэтому внимание было сконцентрировано на критическом отборе включаемого
в книгу материала, и это накладывало особую ответственность на составителей.
Сколь успешно справились они с этой задачей, ясно покажет то, как будет вос-
принята эта книга химиками-органиками.
Характерными чертами научных исследований в прошедшие два десятилетня
были развитие новых синтетических методов и увеличение числа функциональных
групп, используемых в органической химии. Эти успехи стали возможны благо-
даря мастерству н энтузиазму талантливых исследователей, а также применению
физических методов при установлении структур, использованию эффективных
методов разделения и признанию общих закономерностей, сформулированных
специалистами по физической органической химии. Побудительным мотивом этих
исследований, приведших к успехам, был поиск путей к полному синтезу при-
родных соединений сложной структуры. Вехами на этом пути являются синтезы
стрихнина, резерпина, витамина Bi2> макролидных антибиотиков, антибиотиков
р-лактамной природы и простагландинов. Потребность в синтетических соеди-
нениях, которые должны отвечать специальным требованиям, например требова-
ниям, предъявляемым фармацевтической промышленностью, также создает по-
стоянный стимул для разработки эффективных методов синтеза.
Отмечая эти достижения, некоторые ученые (средн них редко встречаются
химикн-органпкн) считают органическую хнмнв наукой, достигшей полного раз-
вития н даже отживающей свой век, и обращаются к биологической науке,
переживающей, по их мнению, больший подъем и дающей ббльшие стимулы
к исследованиям. Ограниченность этой точки зрения становится очевидной при
знакомстве с материалом, изложенным в «Общей органической химии». Не-
смотря на все возрастающее количество утонченных приемов н методов, пока
9
,ся при проведении синтезов в лаборатории аб-
далеко не всегда УДа"цЯнф„ЧВОСТн, которая характеризует биохимические
содютной стерео- в РеП‘°^Х не сформулированы понятия субстратной сие-
„„оиессы. К тому же, в cjM сформулированы на функциональной
ццфнчвости, отлнч1ше °ТЧТ0 даа последних десятилетия этого столетия станут
основе. Можно ожидал , хИМИИ> н эффективность проводимых в
свидетелями пР0ГРесса 1 станет сравнимой с эффективностью про-
лабораториях хпмиче пожелатЬ1 чтобЬ1 «Общая органическая
ХТя/в"вов скромный вклад как источник информации, а может быть,
лжи X источник вдохновения для нынешнего поколения химиков-органнков.
Том 2 охватывает соединения, характеризующиеся наличием азот- » фос-
форсодержащих функциональных групп, а также карбоновые кислоты и их про-
взводные. (В том. 3 перевода вошли части 6—8 английского издания, посвящен-
ные азотсодержащим соединениям. —Прим, ред.) Читатель обнаружит, что в
последние годы некоторые разделы претерпели существенно большее развитие,
чем другие, что и нашло отражение в характере цитируемой литературы. Так,
большая доля ссылок, относящихся к последним годам, указывает на прогресс
синтетической органической химии. Однако в некоторых более традиционных
областях исследований достижения в последние годы менее значительны и доля
ссылок, относящихся к более раиинм годам, существенно
преследовалась цель дать читателю представление о
органической химии.
Любая книга подобного типа является результатом
и я хотел бы выразить мою искреннюю благодарность
выше. В обоих случаях
современном состоянии
труда ее составителей,
.. ..... .. каждому из них. План
издания был рассчитан на сжатые сроки, которые очень трудно было выдер-
жать, но авторы сделали все, чтобы решить эту задачу. Заслуживают благодар-
ности издательство Пергамои Пресс н д-р Колин Дрэйтон за энергичную и эф-
тозданХнигн б°ТКУ 3аХЛестыва10щег0 их потока рукописных материалов. Идея
“ю □ СЭРУ Дер№У БарТ°ИУ И "Р°феСС^ Оллнсу, Ла-
ввичат.,,» Р б° Ы М °° М11ОГОМ обязаны их усилиям и энтузиазму Все
хетт, которая тЗеТивоТ'"' МИ™ В“раЖа,0Т также благодарность Джинн Бо-
связанными с и3даинем ХГ 3а‘,ИМаЛаСЬ °р1 ^«^‘Онньши вопросами,
ме1)е одного из редакторов том’а.И ,,СПР°СТЬ,е Треб°Ва™я "° хР-пе«
ЛИВЕРПУЛЬ
О. САЗЕРЛЕНД
ЧАСТЬ 6.
АМИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ
6.1. АЛИФАТИЧЕСКИЕ И ЦИКЛИЧЕСКИЕ АМИНЫ
ДЖ- Р. МАЛПАСС (University oj Leicester)
6.1.1. МЕТОДЫ ПОЛУЧЕНИЯ АМИНОВ
Существует большое число препаративных методов получения
аминов. Многие из них не новы, но выдержали испытание време-
нем; упоминание о других, хотя сами реакции и не нашли широ-
кого применения, часто встречается в литературе. Границы при-
менимости недавно разработанных методов еще предстоит опреде-
лить. Из этого множества методов выбраны и кратко изложены
ниже принципиально важные методы. Поскольку в синтезе аминов
сложной структуры неизбежно приходится использовать более
простые амины, распределение материала между этим разделом п
разделом 6.1.3 (реакции аминов) является достаточно условным и
субъективным. Более детально многие реакции получения аминов
рассмотрены в фундаментальных обзорах [1—8]. Из литературы
последних лет по возможности были выбраны ссылки обобщаю-
щего характера, несмотря на то что при этом в ряде случаев, не-
видимому, менее очевидными становятся достижения отдельных
авторов.
6.1.1.1. Алкилирование и деалкилирование аминов [1,9,10]
В принципе наиболее очевидный путь синтеза аминов заключа-
ется в алкилировании аммиака пли другого амина действием ал-
кнлгалогеппда. Однако при проведении этой реакции возможно
дальнейшее алкилирование (уравнения 1—4).
RX + NH3 —> RNH3 X" —> RNH2 (первичный) (1)
RX + RNH2 —> R2NH2 X’ —> R2NH (вторичный) (2)
RX + R2NH —> R3NH X' —> R3N (третичный) (3)
RX + R3N —> R<N X" (четвертичная аммониевая соль) (4)
Несмотря на свойственные аминам различия в нуклеофильности,
вряд ли можно ожидать, что эти реакции удастся остановить на
11
какой-либо определенной стадии, поэтому значение данного мето-
да для лабораторной практики, в общем, ограничено. Однако пер-
вичные или вторичные амины во многих случаях довольно успеш-
но могут быть получены при использовании достаточного избытка
аммиака или первичного амина, соответственно (конечно, если это
оправдано экономически и не вызывает трудностей при последую-
щем выделении продукта реакции). Используя 1 экв алкилирую-
щего агента в реакции со вторичными аминами, можно получить
третичные амины с высоким выходом. Наибольшее значение среди
реакций данного типа имеет внутримолекулярное аминирование
замещенных алканов, в результате которого получают цикличе-
ские амины (см. с. 31).
Реакционная способность алкилгалогенндов уменьшается в ря-
ду; RI > RBr С?> RC1 3> RF. Добавление в реакционную смесь
иоднд-иона инициирует обмен галогена и облегчает реакцию с дру-
гими галогенидами. Реакционная способность понижается при уве-
личении степени замещения в амине или RX; это обстоятельство
может быть с успехом использовано, чтобы остановить приведен-
ные выше реакции на более ранней стадии. Возрастание степени
разветвленности алкильного радикала в RX приводит к увеличе-
нию вероятности протекания конкурентного процесса — вызывае-
мого аминами Р-элимннирования в соединениях, содержащих под-
ходящим образом расположенные атомы водорода. С потенциаль-
ной возможностью элиминирования следует считаться также и
тогда, когда в качестве нуклеофильного агента используется поп
R2N_ для того, чтобы увеличить реакционную способность соот-
ветствующего амина. Получение первичных аминов из первичных
алкилгалогенндов при действии амида натрия является, как пра-
вило, удовлетворительным методом синтеза (незначительные коли-
чества алкена удаляются легко), а превращение вторичных ами-
нов in situ в их литиевые соли может оказаться полезным при по-
лучении третичных аминов (уравнение 5) [11].
[O~lNH+K-BuBr [O”]2nbu'h (5)
Избежать опасности элиминирования НХ помогает использование
ненасыщенных галогенидов в сочетании с гидрированием на ко-
нечной стадии синтеза. Этот вариант позволяет, кроме того, син-
тезировать амины, получение которых прямым алкилированием
невозможно из-за пространственных затруднений (уравнение 6)
[[12].
Et Et Et
। „ трет-BuNH-i ] Hj.NI I
Me—C—C=CH —---------->- Me—C—CsCH --------> Me— C—C2H6 (6)
C1 трет-BuNH rper-BuNH
^Уравнения 1—4 не ограничены только теми случаями, когда
л — галоген. В промышленных масштабах с помощью подходящих
12
катализаторов может быть осуществлено прямое превращение
спиртов в амины, а в лаборатории для получения аминов тра-
диционно используют сложные эфиры. На практике можно встре-
титься со множеством примеров подобных превращений, но осо-
бенно широко применяют эфиры сульфоновых кислот (например
X = п-толуолсульфоиилокси), в частности, для синтеза ампносаха-
ров и аминостероидов, причем в соответствии с 5^2-характером
реакции наблюдается стереоспецифичность замещения. Получае-
мые из спиртов фосфониевые соли гладко реагируют с первичны-
ми и вторичными аминами, приводя с высокими выходами со-
ответственно к вторичным и третичным аминам (после выделения
с помощью основания) (уравнение 7) [13].
+ ДМФ или бензол
R'R2NH+ R—О—PPh3 ---------------> R'R2NR + O=PPh3 (7)
Однако направленное получение конкретных первичных, вто-
ричных или третичных аминов методом алкилирования требует,
как правило, приемов, позволяющих в большей степени контро-
лировать ход реакции. Удачные способы синтеза аминов R3-xNHx,
имеющих при азоте атомы водорода (х = 0, 1, 2), можно разде-
лить на две группы.
В случае синтеза первичных п вторичных аминов несколько
связен N—Н (их число соответствует значению х) предваритель-
но могут быть блокированы заместителями, из которых по край-
ней мере один понижает, кроме того, реакционную способность
амина. Остающийся при азоте протон может быть затем удален
с помощью основания, и последующее моноалкплнрованпе не ос-
ложняется более глубоким алкилированием или конкурентным
элиминированием (в случае первичного или вторичного RX). Син-
тез завершает удаление защитной группы (или групп). Ниже при-
ведены хорошо известные синтезы Габриеля [14] (уравнение 8)
и Гипзберга [1, 2] (уравнение 10) наряду с более новыми реак-
циями (уравнение 9 [15], 11 [16] и 12 [17]). Особым преимуще-
ством последних является использование мягких условии для уда-
ления защитных групп па заключительной стадии синтеза.
О О
BuLI ТГФ RX НС1
HN(SPh)2 -----_'2о'о~с LiN(SPh)2 -—> RN(SP1i)2 -----------* Wft
К конц. кислота
. или щелочь
R2N— SO2Ar -------> R'R2NSO2Ar ------------->" R‘R2NH
(8)
О)
(Ю)
13
(cr,S02)20.CH2c±> r!NHSOiCFs
Li/МИ)
R2NHj EtjN _78 оС
—► R'R2NSO2CF3
основание
R*R2NH
(И)
(ЕЮ)2РОП. СС1,
r2nh2 —---------------
R2NHP(OEt)2
Rix
основание
о
О
HCI
---> R’R2NH
(12)
—> R'R2NP(OEt)2
При алкилировании субстрата, в котором блокированы (х + 1)
связей N—Н, используют образование четвертичной соли; после-
дующее удаление защитных групп приводит к целевому амину
(уравнение 13—16). Традиционными примерами являются реак-
ции Делепина (уравнение 13) н Риттера (уравнение 14) [1, 2].
Реакция Риттера применима также и к карбениевым ионам,
образующимся из алкилгалогенндов при действии кислот Льюиса
и из алкенов под влиянием протонных кислот, но при этом следу-
ет учитывать возможность перегруппировок карбениевого нона.
Образующиеся из аминов и карбонильных соединений имины (азо-
метины, основания Шиффа) важны как исходные соединения в
синтезе многих аминов. Алкилирование подобных соединений (1)
является ключевой стадией в уравнении 15; в уравнение Гб
дит стадия алкилирования фосфинпмпнов (2) [18].
вхо-
,N.
•N
RX
R1—N=-CR2
rnh2
EtOH 2
R
(13)
H2SO4
R*OH --------
« r2c=n
R' --------
гидролиз
-------* R‘NH2
(14)
o--.c
r2nh2
R2N=C:
r'x
R'R2N=C;
гидролиз
-------R’R2NH
(15)
(1)
X’
R2N=PPh3
(2)
R‘R2N—PPIlj •-дрол“> RIR2NH
(16)
На первый взгляд этот
тивпым, когда речь вдет о
подход может показаться бесперспек-
ои может оказчткго с,,итезе третичных аминов (х = 0). Но
четвертичных амипшо» езНым ? тех случаях, когда расщепление
бирательным поонесгп» ЫХ солей (Декватерннзация) является из-
мер уравнение°П ив ПР" Восста1,овле”1'ч ™ Эмде (нанри-
11 1е 17), или в примере, приведенном в уравнении 18,
где используется циклическая трпазасистема
СН2СН=СН2 СНзСН—СН2 г'и prj И-Prl l+ Na, Ilg -I6CH=CH2
СНз—N—СНгРЬ > CH3—N—CH2Ph K'Pr—N—CH2Ph н-Pr Г (17) R2N'XX'NR2 1. Rix R1^ LJ 2-h2°’ D2/n-CH3 <18> NR2 R
Деалкилирование аминов, в частности деметилирование, сыгра-
ло важную роль в синтезе и при установлении строения, особенно
в химии алкалоидов. Успешное применение деалкилирования
третичных аминов с помощью бромциана [реакция Брауна (урав-
нение 19)] имеет давнюю историю [1—3]; по легкости удаления
отдельные группы описываются следующим рядом: аллил, бен-
зил > метил > этил и т. д. Для удаления алкильных групп мож-
но использовать фосген (С12С=О) или эфиры хлормуравьнной ки-
слоты (уравнение 20). Было показано, что фенилхлорформиат
(R1 = Ph) [20] является более эффективным реагентом, чем
алкиловые сложные эфиры, и дает результаты, сравнимые с ре-
зультатами реакции Брауна. По-вндимому, аллилхлорформиат на-
столько эффективен как реагент для удаления алкильных групп,
что ряд авторов серьезно обсуждает использование простых ал-
кильных групп для защиты вторичных аминов во многостадийных
синтезах [21]. Деметилирование тропннола (3) действием этого
реагента (уравнение 21), протекающее с выходом 77%, выгодно
отличается от реакции с этилхлорформиатом (выход 16%) и де-
метилирования в условиях реакции Брауна (еще более низкий вы-
ход). Закономерности, определяющие избирательность расщепле-
ния, еще не выяснены, но, по-видимому, предпочтительными явля-
ются разрывы при бензильном и трег-алкильном центрах; так, в
случае М.М-диметил-тргт'-бутиламина образуется диметиламин.
R3N + BrCN —>- RBr-|-R2NCN ► R2NH (19)
(3)
Особый интерес для синтеза нередко представляет модифика-
ция аминов за счет С-алкплироваипя в а- пли 0-положенпе к атому
азота. Однако алкилирование в такое положение (при насыщенном
15
„гярпояном атоме) прямыми методами невозможно, посколь-
ку обоазованпе карбаниона рядом с атомом азота крайне новы-
голно- гораздо легче генерируется электрофильным «-углеродный
атом (со с реакцией Манниха, с; 23). Зеебах описал получение
замаскированных вторичных а-ампнокарбанпонов путем питрози-
пования вторичных аминов с последующим металлированием в
а положение к азоту при низких температурах [22]. Алкилирова-
ние можно осуществлять различными алкилгалогенндамн (реакции
с карбонильными соединениями, нитроалкепами, «^-непредель-
ными кетонами и подобными соединениями здесь не рассмат-
риваются), в результате чего получают модифицированные вторич-
ные амины. В качестве примера приведена проводимая в одном
реакционном сосуде последовательность реакций, завершающаяся
удалением нитрозогруппы с помощью НС1 или никеля Ренея
(уравнение 22). Другие методы алкилирования при атоме азота
и'углерода рассмотрены в последующих разделах.
/СНз
изО’Рг—
C2H5ONO
— ElOH
/СНз
uso-Pr—N
'NO
тгф
-------------->
LiX'fPr- 430)2
/CH2Li
изс-Рг—Nz
\no
PhCHH -CH2CH2Ph
-----* изо-Рг—л
,СН2СН2Р11
uso-Pr—tsK
'H
HC1 или
Ni Ренея
(22)
Прямое введение аминогруппы в циклические и ациклические
алканы, не содержащие функциональных групп, можно осущест-
вить действием трихлорида азота в присутствии кислоты Льюиса
в качестве катализатора [23]. Таким способом удается превратить
адамантан в 1-амнноадамантан с выходом 95%.
6.1.1.2. Методы восстановления
Многие азотсодержащие соединения различных классов пре-
вращаются в амины при восстановлении. Ниже рассмотрены осо-
бенности наиболее важных реакций, а также приведен ряд при-
меров, отобранных из большого разнообразия полезных, но не
очень широко используемых реакций. В полном объеме такие ме-
тоды получения аминов изложены в других руководствах [1, 3,
9, 24а].
(1) Восстановление амидов, гидразидов,
уретанов, изоцианатов
Амиды легко доступны, п их восстановление до соответствую-
щих первичных, вторичных или третичных аминов с помощью
алюмогцдрнда лития L1AIH4 (уравнение 23) протекает легко п
с высокими выходами. При добавлении амида к избытку восста-
16
навливающего реагента реакция обычно протекает без осложпр
кий: при недостатке восстановителя из некоторых N N-дизамр
шейных амидов можно получить альдегиды [246]. Применение
гидрида алюминия дает хорошие выходы аминов в мягких vcio-
виях, даже при получении пространственно затрудненных аминов
и при использовании ограниченного количества восстановителя.
Применение днборапа позволяет провести восстановление более
селективно [25] (уравнение 24). Борогидрид натрия можно ис-
пользовать только в том случае, когда амид предварительно ак-
тивирован путем превращения в четвертичную иминиевую соль
(уравнение 25 [26, 27]), однако ацилоксиборогидрид натрия не
требует такоГг активации [28].
о
II ЫА1Н4
R'CNR2R3 ~Et2O „ЛИ ТГФ* R,CH2W (23)
/СО2Ме ,СО2Ме
W о
NCH2PI1 NCH2Ph
О X
|| Et3o+ “BE., | + NaBHj
R'CNR2 pci5R‘C=NR*--------------” r'ch2nri (25)
(X = OEt ИЛИ CI)
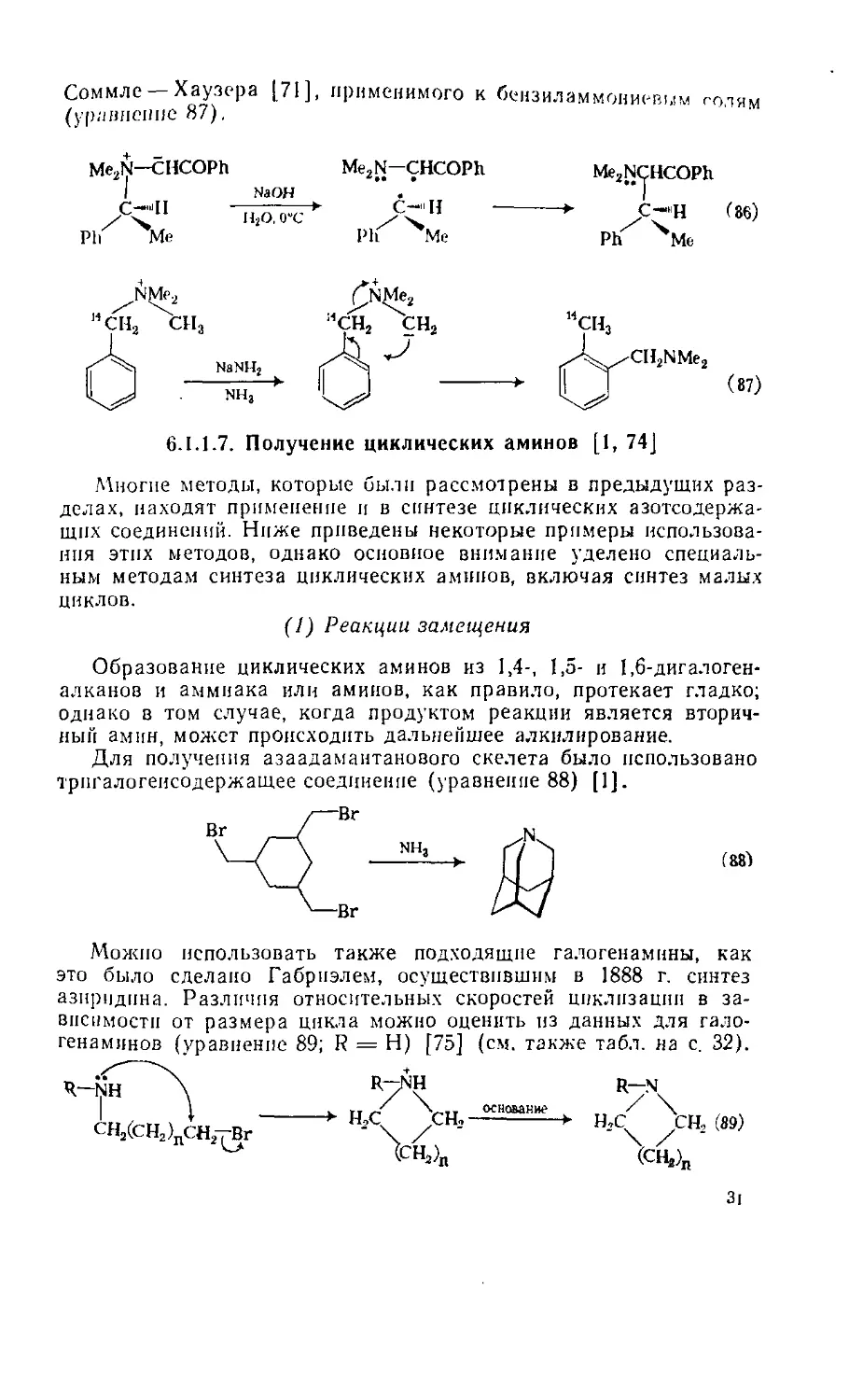
Менее распространено восстановление ацилгндразинов (гидра-
зидов), однако оно протекает столь же эффективно, приводя к
аминам (уравнение 26). Уретаны (4) и изоцианаты (5) nptf вос-
становлении LIA1H4 дают N-метиламины (уравнение 27), в то
время как каталитическое гидрирование или обработка натрием
в жидком аммиаке сопровождаются потерей карбонильного атома
углерода (уравнение 28).
|| шлиц
rcnhnh2 --------*- rch2nh2
о
II ЫЛПЦ Ы'АНН
RNHCOR ---------► RNHCHs *--------- RN=C==O
(4) <5)
О
И Pd. И2
RNHCOR ------v --Д»- RNH2
или Na, NHa
(4)
(26)
(27)
(28)
(2) Восстановление азидов и нитросоединений
Легкость синтеза азидов в условиях стереоспецифической
8п2-реакции из галогенидов или сульфонатов пли путем присо-
единения к алкенам (см. разд. 6.1.1,3) позволяет использовать их
Ц.
первичных аминов. L1AIH.1 или диборан обычно
для получения перв сохра11С1111см конфигурации, а каталитц.
восстанавливают азид CVTCTB1Hl палладиевого или илатиново-
ческое ™ДРХГХаРе? ™" несмотря на разбавление а°т.
го катализатора пр азотом. В последнем случае
м0СферЫ водорода вьд ₽ 'не измеНяетСя „ поэтому дл/н/
суммарный ооъем газа ир.^
должны быть использованы другие
блюдення за х А иРк.спектр0СК0Ш1я). Иногда в случае катали-
тпческого 'гидрирования выходы свободных аминов после выделе-
е Z. данно оказывались заниженными. Это происходило, 8
частного! когда в реакционной среде присутствовали следы хло-
рнрованных углеводородов (например, растворителей для снятия
спектоов ЯМР), поскольку при их гидрогенолизе выделяется НС1,
образующий с амином соль. Это было удачно использовано в слу-
чае реакций гидрирования, приводящих к неустойчивым аминам:
добавление небольшого количества хлороформа приводит к обра-
зованию соли выделяющегося амина; это более мягкий метод за-
щиты, чем добавление каких-либо кислот [29]. Применение ката-
лизатора Линдлара позволяет избирательно восстанавливать не-
насыщенные азиды (уравнение 29) [30].
/=\ ________________
'К // \ катализатор
'-' "---у Линдлара
(29)
'N3
NH2
Ограниченная доступности алифатических нитросоединений
(по сравнению с ароматическими) снижает их значение для син-
теза аминов, несмотря на легкость восстановления. В литературе
отмечаются преимущества использования комплексов рутения
[например, RuC12 (PPh3) 2I для восстановления нптрогруппы в
условиях гомогенного катализа [31].
(3) Восстановление нитрилов, оксимов, гидразонов
Реакции восстановления кратных связей углерод — азот за-
нимают особое место в арсенале синтетических методов: введение
циано!руппы и ее восстановление позволяют нарастить углерод-
впинпуПЬ»(УРаВНеНПе 30)’ Каталитическое гидрирование до пер-
~вого ,?В осуществляют в присутствии никеля Ренея, пал-
будет свяДи«пла™™в-о катализатора при условии, что амин
аХ (с помп„пНпе,В0Л“ (СМ- "Радь-ДУШпй раздел) или в виде
акционной смеси/ уксУсного ангидрида, присутствующего в ре-
среду Если не ппйипмя ПР” добавлеШш аммиака в’реакционную
реагировать1/ / //ХР/,1)СЛОсТОР°«™' амин может
вторичные или третичные амиш,Р^у10ЩИМпс« иминами (6), давая
ное Направление реакции nn „n' Следует отметить, что созпатель-
оправдано [32]. Эффективным аУ ПУТ" В не1<оторых случаях было
ется алюмогндрид лития ппЛ осста11авл1ШаЮЩ11М агентом явлЯ-
дрид лития, применение гидрида алюминия и в этом
10
случае позволяет осуществить избирательное восстановление в
присутствии нитрогрупп и сложноэфирпых группировок и сохпа-
НПТЬ незатронутыми сопряженные двойные связи (уравнение 31)
[ЭЭ] • „2
RCs.N [RCH=NH] RCH2N'H2 (30)
(6)
/=Х _Л||^ /=\
г м V(31)
'— 44—C==N \—'J W—CH2NH2
Последовательное проведение алкилирования и восстановле-
ния позволяет получать вторичные амины из нитрилов (уравнение
32) [34]. Восстановительное удаление циапогруппы было исполь-
зовано в синтезе оптически активных аминов из оптически актив-
ных аминокислот [35].
+ BF; ОМе
(EtO)2CH вг; + МеОН | NaBH,
RCsN -------------> RC=NEt ------> RC=NEt------—RCH2NHEt (32)
Восстановление оксимов (7) и гидразонов (8) в различных ус-
ловиях протекает достаточно гладко (уравнение 33) и открывает
возможности превращения альдегидов и кетонов в амины. Ката-
литическое гидрирование оксимов обычно протекает стереоселек-
тивно, с присоединением водорода к менее пространственно
затрудненной стороне молекулы (уравнение 35). Реакция с нат-
рием в спирте (или сходные условия, для которых характерен
термодинамический контроль реакции) может приводить к другой
стереохимии продукта восстановления (уравнение 34) [36].
r2C=NOH —> R2CHNH2 <— R2C=NNHR (33)
(7) (8)
?н
НССНз
На. пропанол .
11 3 ----------11- о(- NH2 (экваториальный) (34)
| [^ -—у- Ц-6 -NIL (аксиальный) (35)
1 J НОАс 1
ПО'
Для восстановления широко используют гидриды металлов,
особенно L1A1H.!, который избирательно атакует пространственно
затрудненные оксимы с наиболее доступной стороны. Однако в
ходе восстановления этим реагентом могут происходить перегруп-
пировки (см. с. 33).
(4) Восстановительное алкилирование иминов
Реакция альдегидов и кетонов с аммиаком или первичными
аминами и присутствии восстанавливающего агента (уравнение оь)
Называют обычно восстановительным алкилированием амии в,
19
восстановительным аминоалкилнрованием
аминированием карбонильных соединений.
или восстановительным
C=O + H2NR
-н2о
C=NR
восстановление
н
\|
J)C—NHR
(36)
Амин может быть заменен нитросоедннением пли другим азот-
содержащим соединением, превращающимся в первичный амин
при восстановлении. Можно предусмотреть меры, сводящие к ми-
нимуму дальнейшее алкилирование; так, в синтезе первичных
аминов обычно используют избыток аммиака. Избыток первично-
го амина в реакционной смеси обеспечивается медленным добав-
лением карбонильного соединения; одновременно уменьшается
возможность конкурирующей реакции — альдольной конденсации.
Наиболее эффективно реакция проходит при использовании ти-
пичного катализатора гидрирования (никель Ренея или платина)
или при использовании гидридов металлов, например цианоборо-
гидрида натрия или лития. Довольно часто реакция сопровожда-
ется конкурентно протекающим процессом восстановления. Про-
межуточно образующиеся имины могут быть, конечно, получены
и другими методами (см. гл. 8.1) п гладко восстановлены с по-
мощью L1AIH4 или NaBH4. Для восстановления иминов в оптиче-
ски активные амины применяют хиральные борогидрпды [37].
Получение третичных аминов описанным методом было огра-
ничено в основном метилированием с использованием метаналя
(формальдегида). Восстановительное метилирование с использо-
ванием борогидридов (уравнение 37 [38] и 38 [39]), как пра-
вило, обладает преимуществами по сравнению с традиционным ме-
тодом Эшвайлера — Кларка (уравнение 39). В качестве восста-
навливающего агента в последнем методе используют муравп-
ную кислоту; его применение иногда осложняется протеканием по-
бочных реакций [40]. Фактически эта реакция является частным
случаем общей реакции Лёйкарта [41] (уравнение 40); ей можно
отдать предпочтение перед другими вариантами восстановитель-
ного аминирования, особенно в тех случаях, когда имеются функ-
циональные группы, чувствительные к воздействию других восста-
навливающих агентов. Амин вводят в реакцию в виде формиата
или в виде формильного производного HCONR2.
/н
PhCH2N^
xEt
НСНО, NaBH3CN
H2O, pH 6-8
(37)
1. НСНО, MeOH
2. NaBH, *
> PhCIKN
(38)
RNHS + Н2СО (изб.) + НСООН —> —> RNMe2 (39)
;nh + o=c' +hcooh
\ I
,N—С—H
/ I
(40)
20
(5) Восстановительное деалкилирование
Восстановительное деалкилирование четпон
с0Леи можно осуществить действием L1A1H гг"НЬ1х аммоииевых
явственным выходом проходит селективп™1ракт,1Чески с Ко-
с помощью триэтплборогндрнда лития- этот по лемет,'лирование
точником попов Н , обладающих зпачйтел .JaX'" ЯВляется ис-
и регенерирующих третичный амин из четгХ укл.еоФилыюстыо
соли [42] (уравнение 41). Бензильная групп? » "1? аммоНиевой
ЛЯА- легко удаляется каталитическим гидр0Ге((0?(,п ВерТ1,чнь,х С0‘
что эта реакция происходит и в случае свобод??’ тот факт-
ляет использовать бензильную группу в качеств dM“H0B. позво-
удаленпя алкильных групп использовали и тиоляты Для
ненне 42); проведено сравнение этого метода с размбпт (ураВ’
ранее [43]. ДЛ С РазРаоотанными
+ LiEt3BH
CH’NMe’ r -^^77
CH2NMe2
(41)
RI
EtNMe2 +
------> R—NMc:,
I
Et
PrSLl
ГМФТА
(42)
6.1.1.3. Синтез аминов с помощью
реакций присоединения
Нуклеофильное присоединение аммиака и аминов к низшим
алкенам протекает с трудом; эта реакция осуществима при повы-
шенных температуре и давлении в присутствии катализаторов.
Стирол реагирует с аминами (уравнение 43) в присутствии ката-
литического количества бутнллптпя (т. е. с литиевой солью ами-
на); 1,4-присоедпненпе к диенам в аналогичных условиях приво-
дит к аллиламинам [44] (уравнение 44).
PhCH=CH2 + Et2NH
BuLi, циклогексан
PhCH2CH2NEt2 (43)
Et2NH
BuLl, циклогексан
___________________
NEt2
(44)
Неактивированная тройная углерод-углер Д чем Р в01-1Ная
ет в условиях нуклеофильного присоединен присутствии ка-
связь. Реакция первичного аминаi с а1*е™ е 45) а не к проме-
тализатора приводит к имину [45] UPdB ..,,ем 126). Ацети-
Жуточно образующемуся енамину (ср. с ур 11ЧНых аминов
ленид медн(1) катализирует присоедине е молекулы ацети-
(Уравненне 46), причем в реакцию вступаю дв дает еиам11.
лена. Присоединение к активированной р
Ны [46] (см. разд. 6.2.1). ___
.—. zn(OActe / \_м=СНМе (45)
\ )--МНг с<1(ОЛс)2 \_J
\----/ 21
НСэСН +
CuCl /Н HCsCH H^I
HCsCH + RsNH --► H2C=C CuCI L—с=сн (46)
XNR2 R2Nz
Подходы к синтезу аминов, использующие способность алкенов к
электрофильному присоединению, являются, в общем, более пер-
спективными, хотя для превращения первичного аддукта в амин
может потребоваться вторая стадия. Разработаны многочисленные
методы синтеза азидов и других предшественников аминов. Азид
иода присоединяется к алкенам стерео- и региоспецпфично по
ионному механизму. Эта реакция лежит в основе схем синтеза
азиридинов (см. разд. 6.1.1.7) и азиринов (см. гл. 17.7), однако
при восстановлении азидов дибораном и восстановительном удале-
нии иода из гидрохлорида амина получают амины (уравнение47)
[47].
1. НС1
2. LiAlH4
LiAlH4
Присоединение изоцианата иода протекает аналогичным обра-
зом, приводя к р-иоди.зоцианату и затем к 0-иодуретану (после
реакции со спиртом). При каталитическом действии оснований
Р-иодуретаны превращаются в азиридины; при действии цинка
обычно удаляется иод и образуется уретан (уравнение 48) [48].
1. INCO
2. МеОН
PhCH=CHCH3 - -> PhCHCH2CH3 (48)
3. Zn» ЛсОН, EtjO J
hnco2ch3
Непосредственное присоединение азотистоводородной кислоты
к напряженным алкенам или алкенам с концевой двойной связью
может не сопровождаться перегруппировкой, катализируемой кис-
лотами (см. разд. 6.1.1.6), если реакция проводится в присутствии
ацетата ртути с последующим восстановительным удалением рту-
ти (уравнение 49) [49].
1. HN3, Hg(oAc)2, ТГФ, Н20
2.К0Н, NaBH.,
(49)
22
Алкены можно вводить и в реакцию Риттера (см с
,/n R недавно разработанном варианте этой nLv. , од‘
Мнения к алкену применяют нитрат ртути и аиХХ^Тв
этом случае за стадиен присоединения также следует восстанете
Не промежуточно образующегося ртутьорганического со“ния
действием борогидрида. Тем самым избегают использования сил,
„ой кислоты и связанной с этим возможности перегруппировки
промежуточно образующихся соединений [50]. Други? примеры
синтезов, в основе которых лежит электрофильное присоединение
к алкенам, встретятся в разделах 6.1.1.4 и 6.1.1.7. Возможно
же аминоалкилнрование
(уравнение 50) [51].
:с=С/ + со + HN^ + Н2О
п ....~ так-
алкенов в присутствии катализатора
ИЬгОз. нагревание
Fe(CO)s, давление
•> H-C-C-CH2-n( (50)
—с—н+ ;с=о + hn'
—С—C—N;
(51)
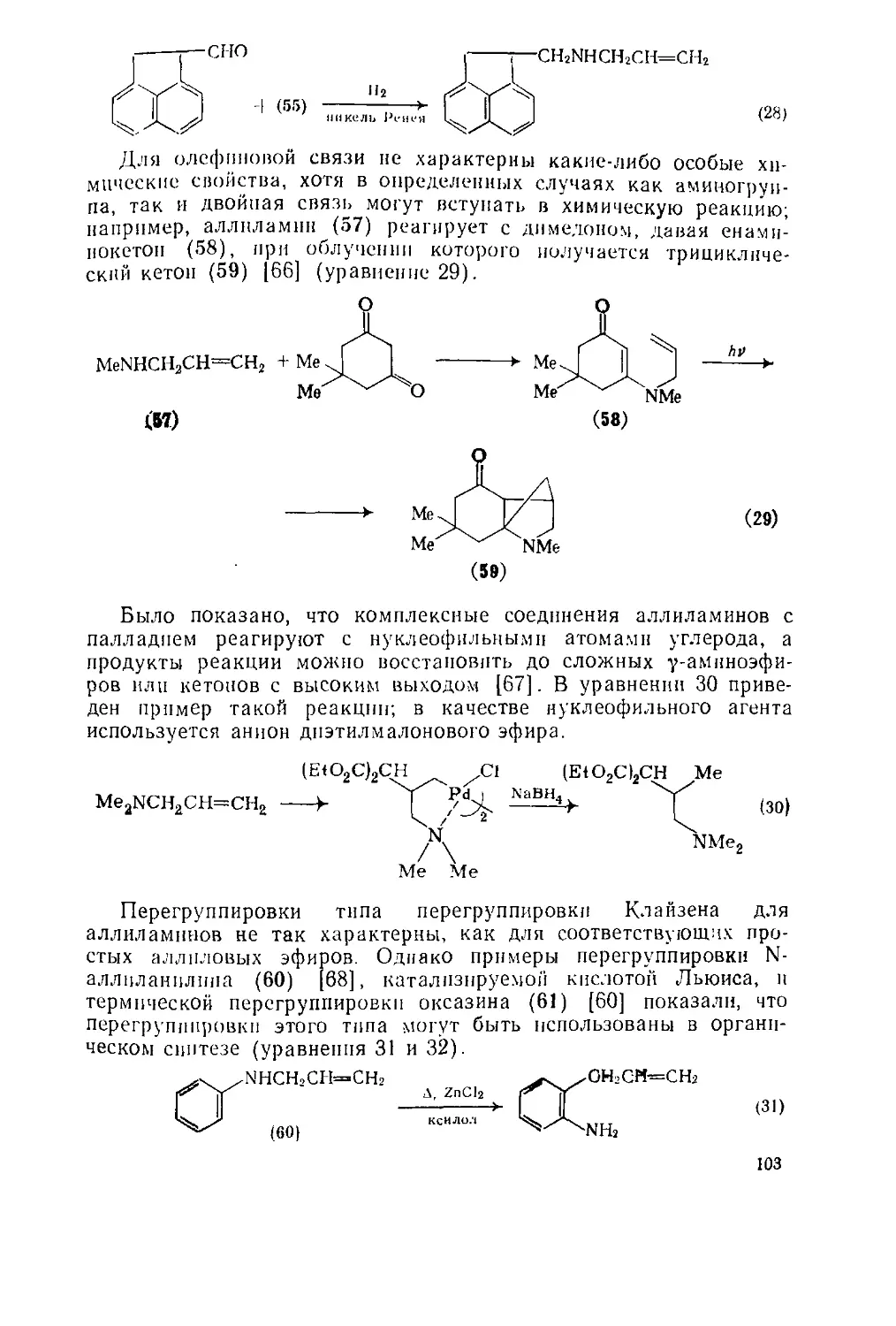
Реакция Манниха дает возможность получить разнообразные
а-аминоалкильные производные соединений с активным водоро-
дом, используя альдегид (обычно формальдегид) и амин [24г]
(уравнение 51). Обычно амин используют в виде гидрохлорида;
реакцию проводят в слабокислой среде. Уравнение 52 иллюстри-
рует аминоалкилнрование циклогексанона, другие примеры реак-
ции Манниха приведены в последующих разделах.
МеСОС!
Для модификации реакции Манниха были предложены новые
Удобные источники основания Манниха (часто оии могут быть син-
тезированы заранее и в больших количествах). Примеры синтезов
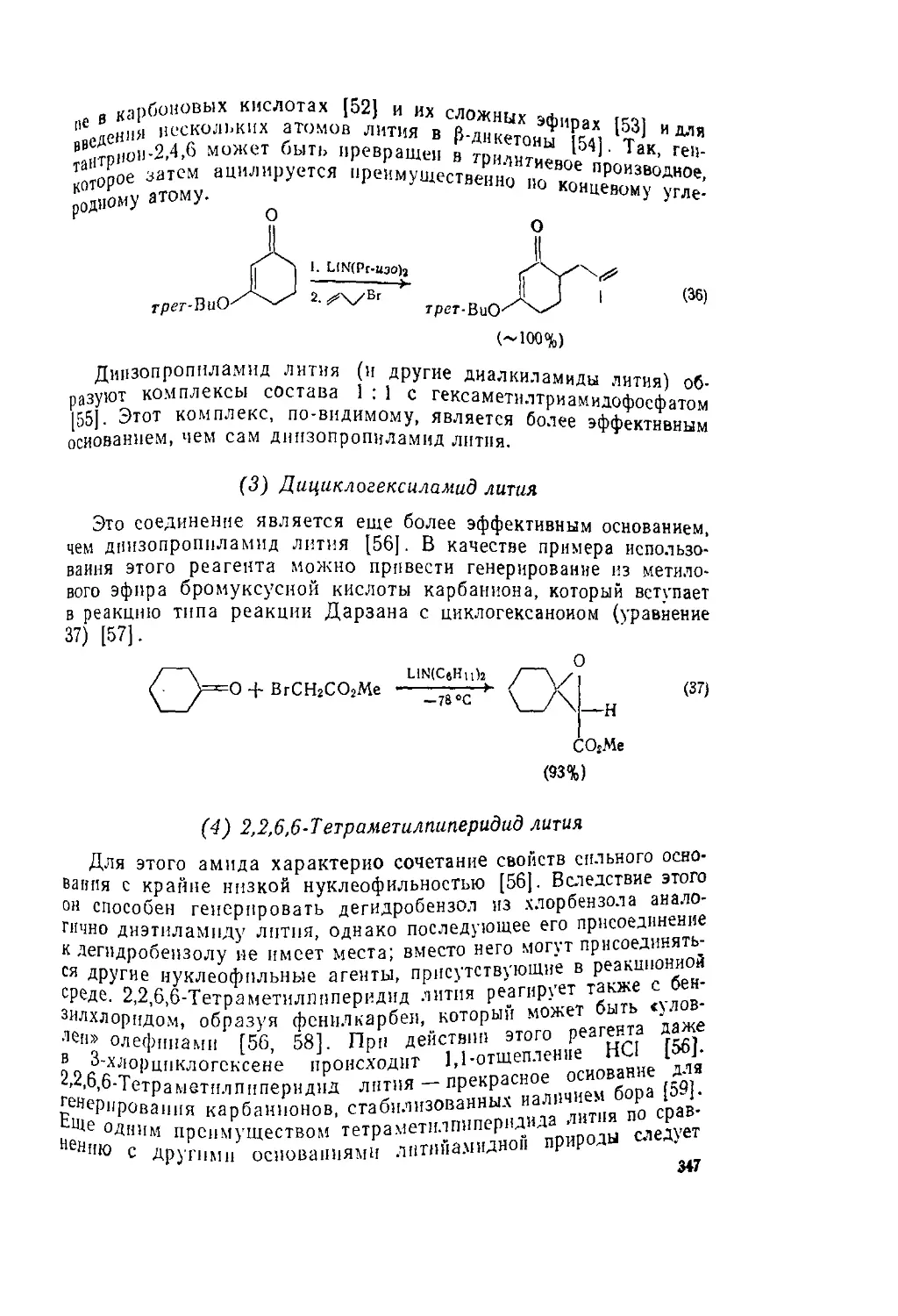
таких соединений приведены в уравнениях 53 [5-J и 1°
r2nch2nr2 R2N=c№ '0Ас (53)
Me2NCH2NMc2 ——Me2N=CH2 Cl (5?1)
Кроме того, возможность получения силиловых ^"Р0®
”ых Ф°рм не только кетонов, но и альдегидов, к р „ к_
лот и сложных эфиров расширяет границы применен’’ J£ в 0Рбыч.
Ции и позволяет использовать многие соединен,, , Р gblJI0 п0.
“х условиях нереакциоииосиособиы [54]. Так™ А прОдукт в
^ено соединение (9)-ключевой "ромежуто ньш прод
с«нтезе а-метилен-у-бутпролактоиа (Ю), который в свою 1
собой структурный элемент, присущий многим
ХЛ»»«™етр»«11Я.
о
при.
OSiMe3
MC3SICI
-° Et3N; ДМФ
О
'O
I. MeLi
2. Me2N=CH2 CFjCOJ
o
Me2N-
о
I. Mel
2. Води. NaHCO3
o
(9) (W)
Дегидрирование аминов над палладием приводит к иминам,
которые могут реагировать как с исходным амином, так и с вво-
димым в реакционную среду первичным или вторичным амином,
давая соответственно вторичные или третичные амины (например,
уравнение 56, выход 90%) [55].
HN
Pd
PhCH2NHMe ——t [PhCH=NMe] ------------->- PhCH2N? I (56)
1 QU 1—< J
6.1.1.4. Применение металлорганических реагентов
в синтезе аминов
Борорганические соединения, получаемые из алкенов (в том
числе и из пространственно затрудненных алкенов) реагируют с
хлорамином (11), гидрокснламин-О-сульфокислотой (12) [56]
или О-мезитиленсульфонилгидроксилампном (13) [57] с высокой
степенью стереоспецифичности, давая первичные амины (уравне-
ние 57). 1
(И)Х=с1; (12)x=oso3h
(•») x=oso2
24
Браун разработал метод синтеза вторичных аминов на основе
реакции борорганических соединений с алкилазидами. Триалкнл
бораны реагируют при нагревании в ксилоле; повышенная реак-
ционная способность дналкплхлорборанов позволяет получать
„.алкилампны при обычной температуре, а в более жестких усло-
виях - и пространственно затрудненные амины. Применение ал-
цилднхлорборанов [58] (уравнение 58) еще больше увеличивает
скорость реакции и способствует полному использованию алкиль-
ных групп при атоме бора. В каждой реакции происходит переме-
щение алкильного радикала и отщепление молекулы азота, по-
следующий гидролиз во всех случаях приводит к вторичным 'ами-
нам с хорошим выходом. Схема синтеза и его стереоспецифичность
иллюстрируются уравнением 59.
R'BCh
R2N—ВС12
+ 1 -
+N'2
—> R'R2N—BCI2
на
н2о
R'R2NH
(58)
R2Na
бензол
3.Гидролиз
1.BHCl2-Et2O, BCl3,'пентан
2. < У-N», бензол
(59)
Для получения третичных аминов борорганические соединения
использовали не очень широко, но отдельные примеры подобных
синтезов описаны (уравнение 60). Характер последней реакции
(как и реакции 57) определяется особенностями структуры произ-
водного амина R2NX. Получено аминопронзводное, содержащее
две «уходящие» группы, 0-2,4-дпнитрофспил-М-хлоргидроксиламин
(И), которое дает возможность переносить сразу две алкильные
группы из борорганического соединения на один и тот же атом
азота; это позволяет использовать борорганические соединения и
Для синтеза циклических аминов. В примере, приведенном в урав-
нении 61, образование третьей связи углерод—азот осуществляет-
ся обычным способом [59].
R3B + Me2NCI —►—► Me2NR <60)
11,,Рбш'ая схема превращения хлор- и бром производных в ПВРВ11
То„рам1,ны Реакцией реактивов Гриньяра с x^°PaN"1H0M ("йртиие
реа„ИамИ1Юм) иллюстрируется уравнением 62. Взапьюд
Щ.егоУИВа Гр|||1ьяра с нитрилом н последующий гидро оп0щ0 из-
' Устный Пр°лукта присоединения представляетсо” ное восста-
|ю ныи метод С11птсза кеТ0110В1 ОдНако непосредственно
Роцц1м"е аДДукта (15) позволяет получить перво' а реактива
63). Пр» “ведения
Ра и последующем гидролизе можно д
образуется также
двух алкильных заместителей, однако
(уравнение 64) [60].
Mg, ei2o
RC1 -------> RMgCI -
С1МН2
RNH2
и кетон
(62)
NMgX
I! ,
R'MgX+R2CN —+ R’CR2
LiAll-h
nh2
R'CHR2 (63)
(15)
R'MgX (изб.) + R2CN
L Нагревание в толуоле
2, Гидролиз
о nh2
R’CR2+R2C—R2 (64)
Присоединение алкильной пли арильной группы к углерод-
ному атому в a-положении к атому азота происходит при реакции
соответствующего литиевого производного с гидразоном (уравне-
ние 65) [1, 9]; как известно, при восстановлении образующихся
гидразинов водородом в присутствии никелевого или палладиево-
го катализатора получаются первичные амины [61]. Исходя из
алкилиденарепсульфеиамида (16) —замаскированного амина,
можно также получить первичные амины, поскольку связь S—N
,в продукте присоединения легко гидролизуется (уравнение 66)
R R
--->- NH—^С—N!!2 (65)
/ \ 2, 1НДРОЛЯЗ / \ Р<1/С /
R Li R
RL1 \| | гидролиз \| -V
/C=NSAr --► /С—NSAr-------и—NH2 (66>
(16)
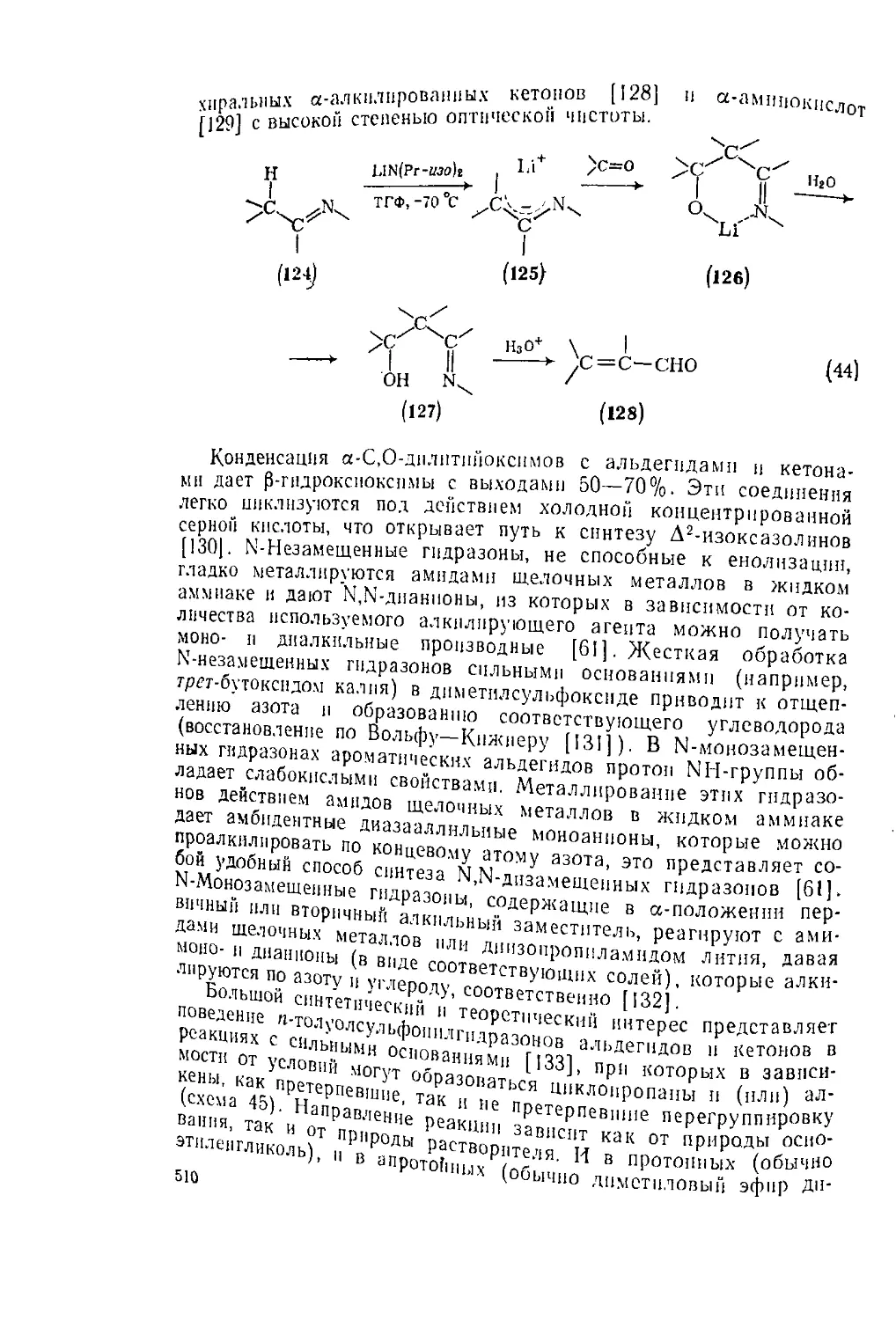
Вторичные амины можно получать а-алкилироваиием
(уравнение 67). Для метплирс----
фопнйметнлпды (уравнение 68)
26
ПМИИОИ
ирования удобно использовать сули-
RQ), причем можно видеть, что хираль-
НЫЙ реагент позволяет получить оптически чистый
р-хиральпыи центр) {63]. 1НСГЫ“ амии из имина
R2
\ R2Mgx \ | /MgX R2
V—NR' ---------> хс—5Г г"дроли,
Z 7 ” /С-nhr" (67)
о
II-
ArSCII-j
phCH=NPh ----------
О
II
CH2SAr СН3
, I Ni Ренея I
PhCHNHPh ---------»- PhCHNHPh
* •
(68)
Общий принцип «а-алкилнровання» может быт> „
на синтез третичных аминов. Возможность введения₽тпеу°СТРаИеН
ных заместителей (уравнение 69) демонстрирует значение мета тт'
органических соединении для синтеза пространственно затруднен'
ных третичных аминов. Это же подтверждает превращение NN
диалкиламндов в амины при действий большого избытка магний
органического соединения (уравнение 70); эту реакцию часто нщ
зывают реакцией Буво [3]. d
3BuMgBr
Et2NCCl3 --------► EhNCBus
(69)
(70)
6.1.1.5. Гидролитические методы получения
аминов [1—3]
Как было показано выше, восстановительные “e^11Helilll| в
ют превращать амиды, изоцианаты и Родс между азотом и
амины; при этом характерно сохранение с углёродноп ча-
карбоинльным углеродом (или, в общем у п’аСщепления связи
стыо молекулы). При гидролизе в РезУдь 'ется весь дпапа-
N—СО образуются амины. Ниже кратк 1ак0 не рассмат-
зон гидролитических методов синтеза ами > спосОбЫ введения
рнваются подробности механизмов реаки• тся в других ру-
Функциональных групп; эти проблемы о ) ' .,клеофильно1”1 атаке
ководствах. Амиды менее чувствительны достижениядо-
молекулой воды, чем сложные эфиры, поэ у _ 11агревание и
ваточной скорости гидролиза обычно Р тве катализатора
применение кислоты или основания в и11Ч11Ваются толь-
, М- Реакции сольволиза амидной связ! 1ЬдОвание кислотно
° случаями гидролиза (уравнение 71), /реаКцпя ,,PnB0;l!'LIO
к^ализатора и соответствующего ^"^шествить избирательную
. ,1НУ и сложному эфиру) позволяет У с,юж11оэфнр>1011 г
Otofi11"0 По амид"01"1 группе в присутс ‘ амиды, подвер
Uto6<> Устойчивые амиды, в частности N ари 27
мртанониу в присутствии трехфтористого бора [1]. Предваритель-
ное превращение амида в четвертичную нмпиисвую соль (как по-
казано в уравнении 25)_позволяет осуществлять превращение 8
амии под действием одной лишь воды.
о ®
II RSOH. катализатор II „
R'NHCR2 ——-------------R'NH2 + R3OCR3
(71)
R5 = H, Aik
Аналогичные методы применяют в случае имидов, а аминолиз
с помощью гидразина особенно успению протекает в случае
N-замещенных фталимидов в синтезе Габриэля (см. уравнение 8),
причем в довольно мягких условиях. Гидролиз сульфонамидов
(см. уравнение 10) требует, как правило, жестких условий реак-
ции; компонентом наиболее эффективных реагентов является
48%-ная бромистоводородная кислота (в присутствии фенола,
связывающего выделяющийся бром). Альтернативные методы —
это восстановление натрием в нафталине или днметоксиэтане
[1,2].
В соответствии с теорией при гидролизе изоцианатов, изоти-
оцианатов, мочевин п уретанов в присутствии кислоты пли осно-
вания образуются амины. Удобным методом удаления защитных
уретановых групп оказалось действие хлористого водорода в нит-
рометане [65]. Образующиеся в ходе деградации по Брауну
(см. уравнение 19) цианамиды гидролизуются и декарбоксилиру-
ются с помощью разбавленной кислоты. N-Нитрозампны легко
гидролизуются до аминов (см. уравнение 22); расщепление п-нит-
розо-Ы.М-дпалкилаиилинов является общепризнанным способом
удаления защитных групп в синтезе вторичных аминов (уравне-
ние 72).
он. н2о
NR1R2 --------з- R>R2NH + O=N
6.1.1.6. Синтез аминов с помощью внутримолекулярных
перегруппировок [1, 66]
Перегруппировки могут быть разбиты на две группы: пере-
группировки, которые включают миграцию алкильной или ариль-
ной группы к электроподефицитному атому азота, и перегруппи-
ровки, для которых характерна миграция от азота к углероду.
Большинство перегруппировок относится к первой группе, и их
принято подразделять на типы А — Г в соответствии с обобщен-
ными механизмами, приведенными в уравнениях 73—76. Отдель-
ные перегруппировки рассматриваются в разделах, касающихся
соответствующих функциональных групп,'Для ознакомления с дан-
ными о механизме, стереохимии и степени согласованности актов
выброса заместителя X и миграции R читателю следует обратить-
ся к этим разделам. г
28
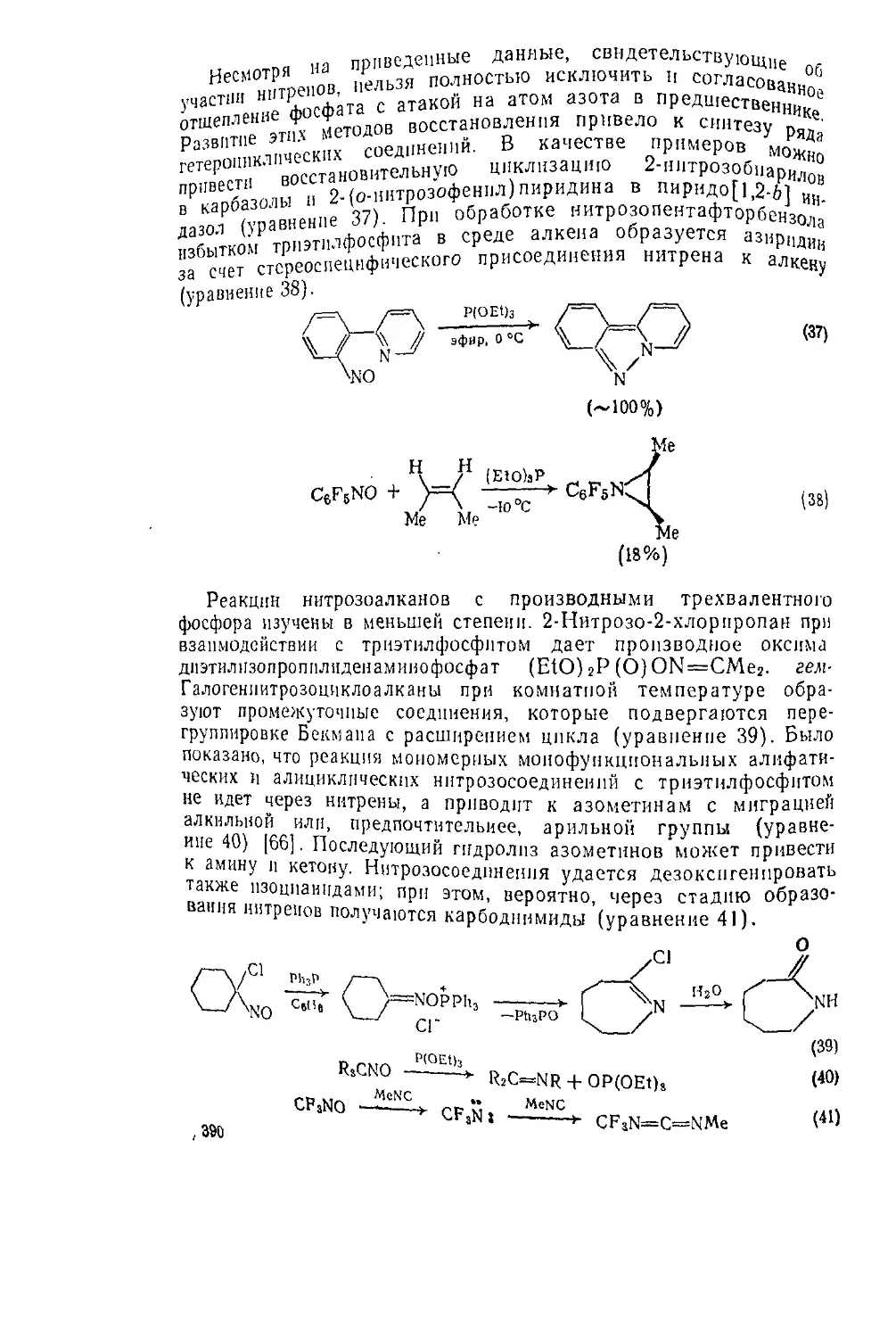
A. O=C=N—R'----
Б O=C=NII-R’-1
B. R‘<=Nrx —R2-C=N~R’ —
—> r’nh2
о
J1
> r2cnhr’
(73)
(74)
(75)
(76)
Ниже поиведены отдельные примеры перегруппировки Гофма-
к галогеяашдаи [67) (уравнение 77) а паре-
группировки ацилазндов по Курциусу [68] (уравнение 78) Пе-
регруппировка ацнлгидроксиламинов (перегруппир ' _,,ппи.
отвечает типу А для случая X = ОН (см. выше), _ Р Ру _
ровка Шмидта для карбоновых кислот (уравнение / ) У •
(77)
/L- ' Ас2О, .ЧаОАс
/-\ 2. Гидролиз
) СО2Н з Восстановление
^nh2
Ph (А)
•СОЫз
СН3(СН2)1бСООН
conh2
.Ph
(78)
(79)
\—' \\Нг
С11з(СН2)16МНз
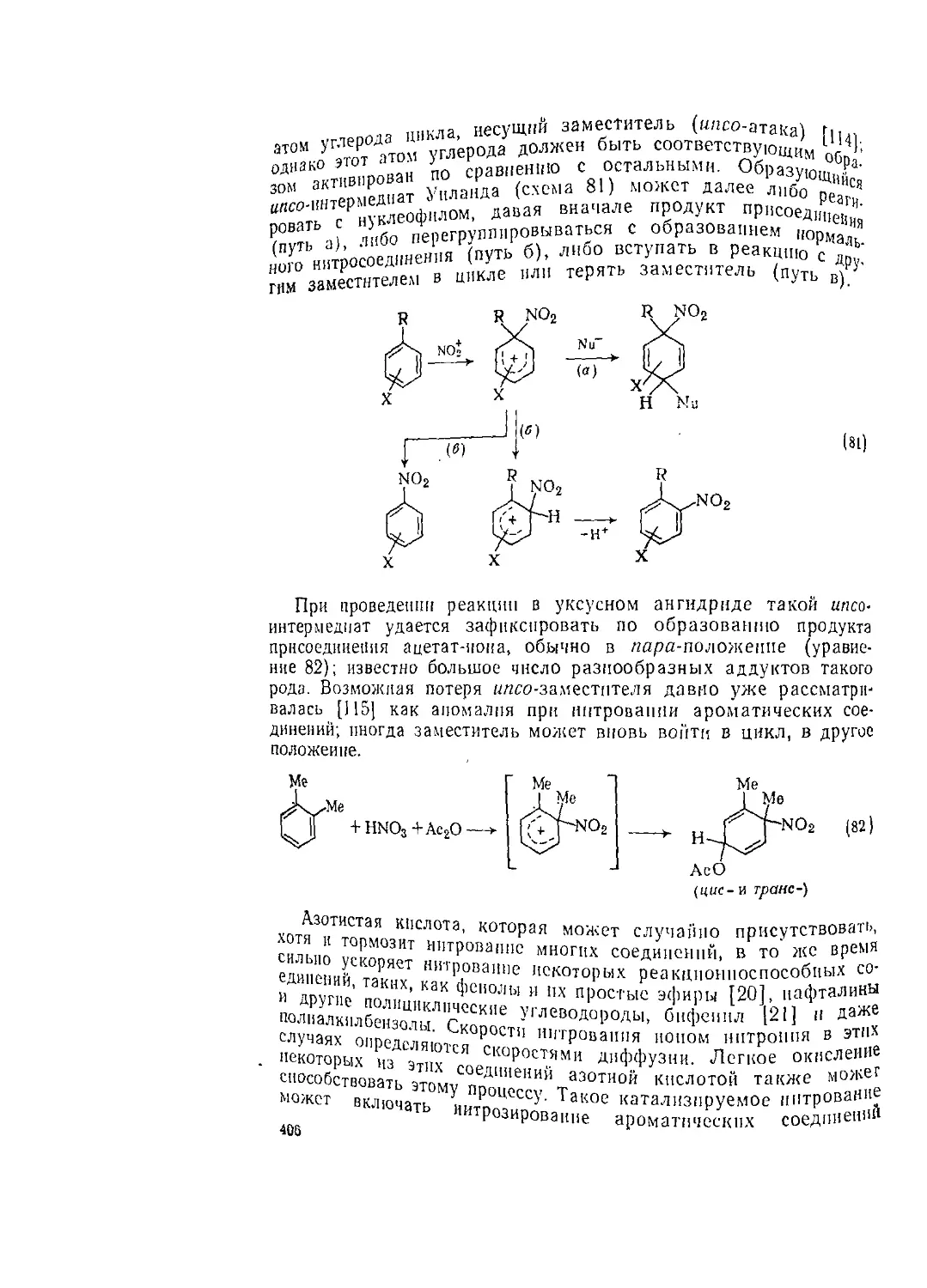
Различная способность к миграции наблюдается пр’!В1^Нне 80);
«ой перегруппировке оксимов и их производи I. СР ым цик.
при этом образуются бициклические амины с р Р веде.
°м (17) и (18). Аналогичные результаты а^,Д|е 81)
перегруппировки Шмидта в случае кето! Р „ ката.
“алкенов (уравнение 82; схема применима и к сп ip /
13пРУемон кислотами Льюиса реакции алк~"г "тенне хлора в
^огеиидами азота (уравнение 83) [69]. °™"" Овкой яв-
Рисутствии ионов серебра с последующей пер Р- р 84) [70].
U тся е1пе одним примером реакции типа Г (ур перегруппп-
Хкеач"Я °Т аТ0Ма й30Та К >'гЛерОдУ 4РподХдействием основания
(п^е 'етвертичных аммониевых солен под Д превращение
"вРегруппировка Стивенса) (уравнение 8о). ^то 1
начинается с образования плпда (в качестве активирующей груп-
пы X могут выступать COR, CO2R, фенил).
R R R
\| основание \1 - А \ I
^N—СНА -------->- /N-CHX ----> /N—СНХ (85)
Выяснение механизма этой реакции шло непростым путем; исто-
рия этого вопроса исследована в детальных обзорах [71, 72].
Для многих [I, 2]-процессов показано протекание стадий диссо-
циации — рекомбинации, как это изображено в уравнении 86; на
основании изучения методом ХИДПЯ предполагается, что реакция
идет в клетке растворителя с участием радикальной пары [73].
С процессом образования илидов аллиламмония часто конку-
рирует [2,3]-сигматропная перегруппировка; это направление ре-
акции лежит в основе одного из вариантов перегруппировки
зо
Соммле —Хаузера 171], применимого к бензиламмониевым сетям
(уравнение 87).
NaOH
Н2О, ОХ'
Me2N—CHCOPh
С-П
Р11^ ^Ме
Na Nil,
NHj
Me2N—CHCOPh
С- II
Pli^ ^Ме
Me2N<yHCOPh
С—и (86)
Ph ЧМе
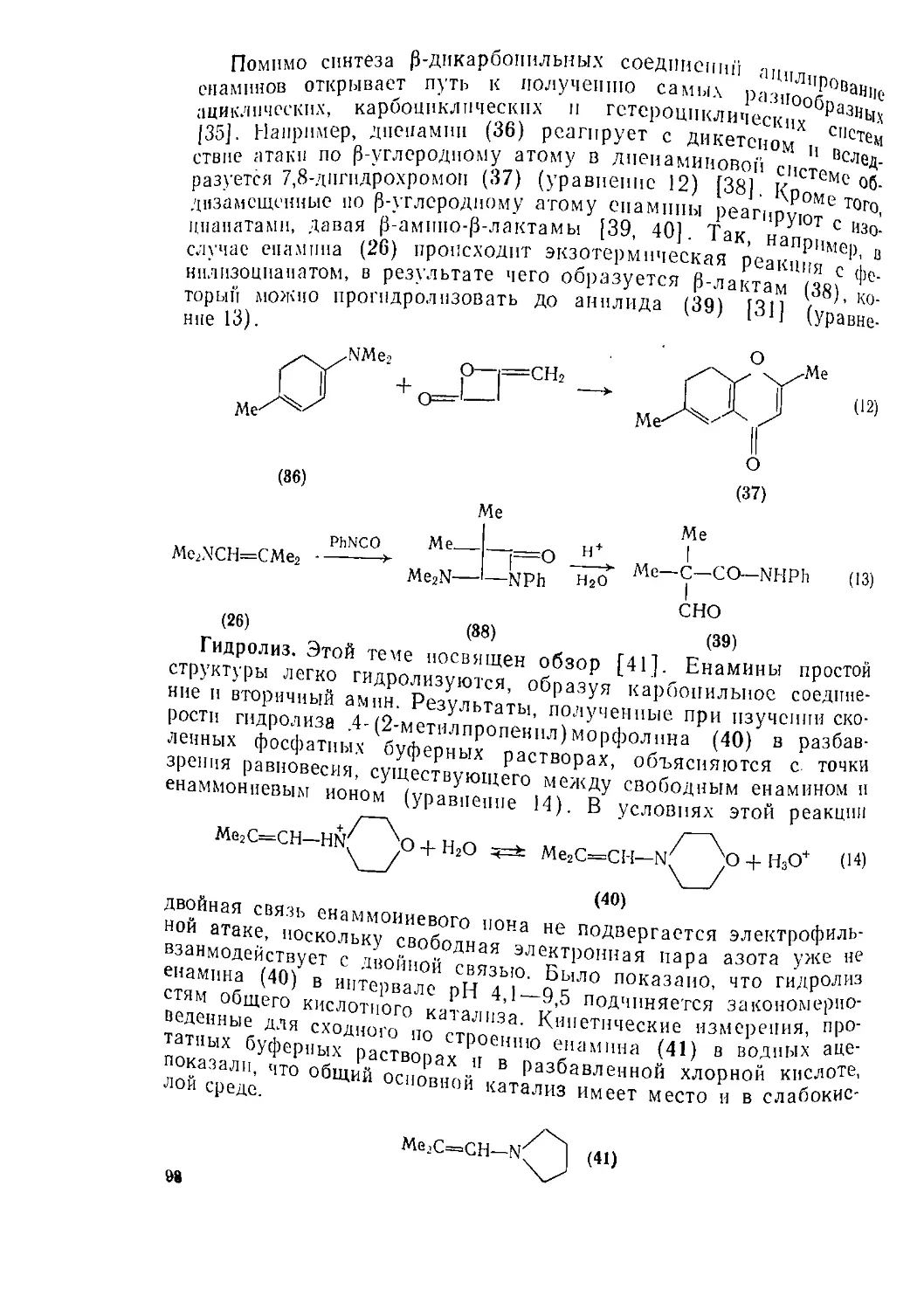
6.1.1.7. Получение циклических аминов [1, 74]
Многие методы, которые были рассмотрены в предыдущих раз-
делах, находят применение и в синтезе циклических азотсодержа-
щих соединений. Ниже приведены некоторые примеры использова-
ния этих методов, однако основное внимание уделено специаль-
ным методам синтеза циклических аминов, включая синтез малых
циклов.
(1) Реакции замещения
Образование циклических аминов из 1,4-, 1,5- и 1,6-дигалоген-
алканов и аммиака или аминов, как правило, протекает гладко;
однако в том случае, когда продуктом реакции является вторич-
ный амин, может происходить дальнейшее алкилирование.
Для получения азаадамантанового скелета было использовано
трпгалогеисодержащее соединение (уравнение 88) [1].
Можно использовать также подходящие галогенамнны, как
это было сделано Габриэлем, осуществившим в 1888 г. синтез
азиридина. Различия относительных скоростей циклизации в за-
висимости от размера цикла можно оценить из данных для гало-
генаминов (уравнение 89; R = Н) [75] (см. также табл, на с. 32).
Ч—fjiH
СН2(сН2)псНут-Вг
R-NH R-N
> H.Z > HZ V (89)
<СНЛ (ау„
3!
О
2
3
4
Относительная скорость
циклизация
0,014
800
14
0,02
(азиридин)
(азетидин)
(пирролидин)
(пиперидин)
(азациклогептан)
Кпочевымп интермедиатами в синтезе часто являются р-амн-
ноеппрты, однако OH-rpvnna является довольно невыгодной «ухо-
дящей» группой, что делает непосредственное замещение затруд-
нительным. В основе метода получения азиридинов, разработан-
ного Бенкером, лежит превращение ОН-группы в сложиоэфирную
действием серной кислоты н последующая циклизация в присут-
ствии основания. p-Ампноспирты легко получаются из оксиранов
(см. разд. 6.2.3), однако описаны и способы превращения оксира-
нов в азиридины [76].
Нуклеофильное замещение галогена на азот протекает по ме-
ханизму S»2, поэтому замыкание цикла — стереоспецифический
процесс. Одни из вариантов этого метода (на примере синтеза
азетпдиновой циклической системы) включает использование 1,3-
днбромпропапа и толуолсульфонамида; при этом образуется
N-толуолсульфоннльное производное (19), которое может быть
также получено из 3-амннопропанола (уравнение 90) [I, 74].
Внутримолекулярное замещение уходящей группы при азоте при-
водит к азиридинам (уравнение 91) [74], Если'активирующей
карбонильной группой является карбонил амидной группировки, то
образуются а-лактамы [77] (уравнение 92), однако в этом случае
не удается осуществить полное восстановление карбонильной груп-
пы с помощью L1AIH4 (хотя этот метод восстановления имеет общий
характер); в результате реакции получают р-ампноспирт.
|№ — NH
С5НцОП
характер); в результате реакции получают
j NHa TsCl, пиридин I------NHTs Na
---\ <S °C '------v EtOH
'OH \0Ts
r—NTs
(90)
/R2
R'-CO—x ,NZ
\Z \x
О
, II
R'CHjCNHR2
основание
------->- R'CO—.
(19)
/R2
-у COR'
NR2
(91)
трет-BuOC.l, трет- BuO~ Na‘
(92)
=0
NR2
(2) Методы восстановления
азп?ид11ИыСС(емЮхпеяНИ" азнР"иов шдрвдами металлов получают
32
зывается каталитическое гидрирование, как, например для пре-
вращения пирролов и пиридинов в пирролидины и пиперидины
При восстановлении LiA1H4 некоторые оксимы превращаются в
азиридины [78] (уравнение 93); используется и другой вариант
получения азиридинов, заключающийся в обработке иодметилата
диметилгпдразопа кетона реактивом Гриньяра [79], «-Хлориро-
вание нитрилов [80] или иминов [81, 82] с последующим восста-
новлением также позволяет получать азиридины (уравнение 94),
к этому же результату приводит восстановительная циклизация
Й-(р-бромалкил)ампдофосфатов, получаемых из алкенов [83].
PhCH2
PhClZ ОН
LIAlU,
PhcH, u
:NH
(93)
Ph H
R1
LIAIH<
R'CHCsN
R‘CCH=NR2
I
Cl
(3) Реакции радикала аминия
Известная реакция Гофмана—Лефлера [23] позволяет по-
лучать пирролидины путем циклизации N-галогенампнов, проис-
ходящей при неактивировапном б-углеродном атоме (уравнение
95). Реакцию проводили в присутствии различных кислот; для
инициирования использовали также нагревание, облучение или
химические инициаторы. Описанным методом был синтезирован
ряд соединений, включая алкалоиды пирролидинового ряда. Кон-
курирующим направлением реакции является циклизация по
е-углеродиому атому, причем иногда этот путь может стать, пре-
обладающим, если его протеканию способствуют конформацион-
ные факторы [84] (уравнение 96).
(94)
(96)
Радикалы аминия, образующиеся из
зоамипов, могут также присоединяться
2 Зак. Ю5
N-галоген- или N-нитро-
к двойным связям по
33
подробности
разд. 6.6.1,
RHVTOH- и межмолекулярному механизмам (уравнение 97). Основ-
S ссылки па работы нескольких групп, ведущих исследования
В данном направлении, можно найти в статье [85], ппппп«.,».
механизмов описанных реакций рассматриваются в
/Н \
RN + .С—С.
I I
RNH—С—С—CI
I I
(97)
(4) Каталитическое аминирование
Прямое аминирование неактнвированных алкенов в присутст-
вии палладия разработано Хегедусом, который применил эту реак-
цию для замыкания цикла, используя даже первичные алкил- и
арпламнпы. Замыкание цикла происходит при углеродном атоме
двойной связи, несущем наибольшее число заместителей. Проме-
жуточно образующиеся палладпйорганические соединения или ал-
кены подвергаются затем гидрированию (уравнения 98 99)
[86].
IPdCl. (cHaCN)2,
ТГФ
--------------->-
!. NEt3
(смесь изомеров)
Аминирование в аллильное положение, катализируемое палла-
дием, Трост применил в синтезе алкалоидов [87]. Построение ме-
зембринового скелета (уравнение 100) включает реакции обсуж-
давшиеся в разд. 6.1.1.2; на последней стадии осуществляется ка-
тализируемая циклизация по аллильному центру уществляется ка
____ 1.I.IAIH.
-СНО i.Tsci,
J-PhCHO,
бензол
2,NaBH4
з. Ас2О,
НС104
"ОАе
з.МвСН
ОАс Ы1А1Н.
(ЮО)
NH J11
34
(5) Циклоприсоединение
Реакции циклоприсоединения дают возможность получать са-
мые разнообразные циклические амины. Присоединение карбенов
к иминам даст азиридины [88] (уравнение 101). Хорошо известно
прямое присоединение нитренов к алкенам [89] (см., например,
уравнение 113, с. 43], а термически или фотохимически иниции-
руемое отщепление азота из трназолннов (20) нередко приводит к
азиридинам с достаточным выходом [90] (уравнение 102).
(20)
Превращения алкенов в азиридины были рассмотрены в разд,
б.1.1.3, однако днхлора.ткплбораны способны стереоспецнфически
превращать промежуточно получаемые р-иодазиды во вторичные
амины (ср. уравнение 58), а затем путем внутримолекулярного
замещения атома иода в азиридины, в которых сохраняется сте-
реохимия исходного алкена (уравнение 103) [91].
1. r’bci,
г.Основание
К- Н
Г103)
Особый интерес представляет внутримолекулярное присоеди-
нение нитренов к двойным связям. Эти превращения часто более
эффективны, чем межмолекулярные присоединения н позволяют
получать напряженные полициклические амины [92]. Ниже для
сравнения изображено образование азабпцнк.тобутана этим мето-
дом, а также присоединением метиленового звена к азприну с по-
мощью ди.метилсульфоиийметплнда [93] или обычным нуклео-
фильным замещением [94]; во всех случаях образуется одно и то
Же соединение (уравнение 104).
Присоединение изоцианатов к алкенам широко использовали
в синтезе [J-лактамов [95]. Предпочтение отдается N-хлорсульфо-
ннлизоцнапату (21) благодаря его высокой реакционной способ-
ности даже в случае простейших незамещенных алкенов, региос
пецифичностн и легкости удаления М-хлорсульфонпльиой группы
[96] после завершения циклизации. N-Хлорсульфоннлпзоцнанат
2*
35
5нг5Мег. ТГФ R=Ph
(1Н)
NaOH (водн.). A
облагает свойствами электрофила, содержащего в одной молекуле
разные реакционные центры [97]; это проявляется в его способно-
сти взаимодействовать с л-спстемоп, создавая положительный
заряд при углеродном атоме, затем карбокатионный центр атаку-
ется атомом азота, несущим отрицательный заряд. В условиях
кинетического контроля происходит 1,2-прпсоедпненпе (уравнение
105), однако в определенных условиях N-хлорсульфоиил-р-лакта-
мы могут снова раскрываться, образуя диполярные частицы и
способствуя, таким образом, 1,4-прпсоед11иепню к ациклическим
или циклическим диенам (например, уравнение 106) [98]. Осу-
ществлено 1,6-прпсоеднненпе к циклическим триенам и тетраепам;
в тех случаях, когда возможны скелетные перегруппировки карбе-
ниевого иона, подобные перегруппировки с последующим замы-
канием цикла также приводили к пяти- и шестичленпым цикли-
ческим структурам [96]. Присоединение кетенов к иминам дает
Р-лактамы.
использования других азотсодержащих
' >азом за-
?HX:oR“Z'Mep°B использования других азотсодержащих
мешенных азапик?ИЛ°В ДЛЯ П0ЛУ'1е11НЯ определенным образом за-
лают СИИТС311ПОВЯТ1>В’ И° Л11ШЬ 11еМ110г11е из этих реагентов позво-
стемы. Исключение '"исходные» незамещенные циклические си-
м является тозилцнапид, который легко при*
36
соединяется к циклопситадисну, давая при последующем гидро-
лизе первичного аддукта соединение (22; п= I) [99].
Присоединение углеродных мостиков к легко получаемым азот-
содержащим циклам иллюстрируется на примерах присоедине-
ния двухуглеродных фрагментов [65, 100) (уравнение 107) и
трехуглеродного фрагмента [101] (уравнение 108). Последний
синтез дополняет классический подход, основанный на реакции
Робинсона — Шеифа. В этом подходе к помощью двукратной ре-
акции Маппиха осуществляется «биогенетически похожий» син-
тез троиииопа (23; /1 = 2, R = Me) и исевдонельтьерина (23;
п = 3, R = Me) [102] (уравнение 109).
При использовании многих из приведенных выше реакций не-
обходимы дальнейшие операции, например восстановление на по-
следней стадии, и ясно, что выбранные примеры лишь наглядно
демонстрируют большое разнообразие методов, находящих приме-
нение в синтезе циклических аминов.
6.1.1.8. Разделение хиральных аминов
Многие природные вещества выделены в виде оптически чис-
тых форм, однако в лабораторной практике необходимость разде-
ления рацемических смесей на энантиомерные компоненты стала
очевидной уже давно. Общие сведения об оптической активности,
Разделении антиподов и их чистоте можно найти в гл. 1 тома 1 и
в обзорах [103]. Некоторое представление о методах, применяе-
мых для разделения хиральных аминов, дацо в этом разделе, а
вопросы асимметрии самого атома азота более подробно рассмот-
рены в следующем разделе.
37
й основе классического способа растепления хпральных ами-
В ° ®Рпкпия между рацемическим амином и оптически ак-
пмой^щгкхгой11 (ypaBiieinieP 110), в результате которой образуют-
с^дпастереомерные соли, различающиеся по свойствам. На про-
гмЛ.Лмин! „ 1(+)-Амии • (+)-Кислота}
{ (^) Амин J 'Кислота «—*- ц_).дМцц . (Ц-)-Кислота} >
тяжешш последних ста лет для этой цели использовали многие
оптически активные кислоты [7, 103], осооенио часто - винную
кислоту п ее производные, а также такие сильные сульфокисло-
ты как камфорсульфокислоту-10. В свою очередь для разделения
оптически активных кислот применяют оптически чистые амины,
как природные (например, хинин, бруцин), так и синтетические
(например, 1-феннлэтиламин). Наряду с образованием солен
имеется и’другой способ расщепления, заключающийся в получе-
нии диастереомеров с ковалентной связью п их разделении. Этот
метод применим в том случае, если регенерация свободного амина
протекает достаточно легко и эффективно, как, например, в слу-
чае диастереомерных уретанов, получаемых из рацемических ами-
нов и оптически активных ментнлхлорформиатов. Стандартным
приемом разделения диастереомеров продолжает оставаться дроб-
ная кристаллизация, однако часто для разделения диастереомеров
с ковалентной связью более удобными могут оказаться хромато-
графические методы. Адсорбционная хроматография иа колонках
с лактозой пли ацетатом целлюлозы была использована для час-
тичного разделения аминов.
Многие ферменты обладают способностью распознавать хи-
ральность, п часто происходит абсолютное различение энантио-
мерных молекул «гостя» молекулами биокатализатора, выступаю-
щего в роли «хозяина». Это используют для удаления из смеси
одного энантиомера, что позволяет выделить оставшийся энантио-
мер. В последнее время привлекли внимание синтетические хи-
ральные макроциклы, способные различать хиральность; несом-
ненно, что перспективы использования подобных соединений очень
широки. Крам с сотр. осуществили синтез различных макроцик-
лических краун-эфпров, в структуру которых включены хпраль-
ные бпнафтнльиые фрагменты. Распределение первичной алкпл-
аммонпевой соли^ между водной фазой и органической фазой,
содержащей такой хиральный комплексон, который играет роль
«хозяина», приводит к избирательному комплексообразованию,
степень которого можно оценить путем сравнения коэффициентов
распределения обоих энантиомеров. Этот принцип лег в основу
разделения на антиподы солей сложных эфиров аминокислот ме-
/КНДКОСТНО" 11 распределительной хроматографии; в по-
М сл7чае хнральиый комплексон ковалентно связан с силн-
чест«₽”е^"опол,1ст11родьнь,м сорбентом [104]. Используя в ка-
пронзвочипр с'.’Г ”сточников хпральных центров углеводы и их
’ ддарт с сотр. синтезировали хпральпые компл&к-
38
соны типа (24) и показали, что энантиомеры гексафторфосфатов
рацемических аминов комплексуются с этими макроциклическими
эфирами по-разному. В этой же работе [105], а также в разд.
6.1.2.2 описаны спектроскопические методы контроля за распреде-
лением энантиомеров и определения их абсолютной конфигура-
ции.
Кинетические методы разделения диастереомеров основаны иа
разнице энергий не основных, а переходных состояний. Компо-
ненты рацемического амина могут реагировать с хиральным реа-
гентом, например с ( + )-камфорсульфоннлхлорндом-10, с различ-
ными скоростями, это позволяет выделить оптически обогащенный
исходный амин в том случае, если реакция прекращена незадолго
до ее полного завершения. Однако при этом редко удается достичь
высокой степени оптической чистоты. Асимметрические синтезы с
использованием ахиральных субстратов и хиральных реагентов во
многих случаях дали возможность получать оптически активные
амины. Обычно в подобных реакциях обогащение смеси одним из
энантиомеров бывает небольшим, но может достигать и 100%
(см. уравнение 68).
6.1.2. СВОЙСТВА АМИНОВ
6.1.2.1. Структура и стереохимия простейших аминов
Атом азота в аммиаке н в простейших аминах находится в со-
стоянии др3-гнбрнднзацпп, поэтому естественно, что расположение
трех двухэлектронных связен п свободной электронной пары в про-
странстве приближается к геометрии тетраэдра. Это наводит на
мысль об аналогии с углеродным атомом с его способностью вы-
зывать появление оптической активности. Однако выделение двух
энантиомеров (25) и (27) с тремя различными алкильными заме-
стителями невозможно вследствие легкости взаимных превраще-
ний этих энантиомеров (уравнение 111). Инверсия протекает че-
рез переходное состояние (26), в котором атом азота находится в
состоянии зр’-гибриднзацнн, а свободная электронная пара зани-
мает рг-орбнталь.
Для аммиака (скорость взаимного превращения составляет
около 2-10н с"!) и простейших аминов свободная энергия ак-
тивации этого процесса мала (табл. 6.1.1). Если же вместо
3»
(25) (26)
(27)
(111)
свободной электронной пары имеется a-связь, как в случае четвер-
тичных аммониевых солей, например (28), и N-оксидов, конфи-
гурация при атоме азота характеризуется значительной устойчи’
Таблица 6.1.1. Барьеры инверсии у азота в ациклических
и циклических аминах [108]
Соединение К AG*, 3 кДж/моль Температура,
NH, (30) ~ 25 6 —
HNMe2 (31) — ~ 18,4 ° —
NF, (32) — ~ 251 r —
(33) Me 28,5 -125
(34) Cl 38,5 —87
Onr (35) Me 35,2 -98
(36) Cl 43,1 -68
r-NR (37) Me 42,7 -69
1 1 (38) Cl 56,1 -20
(39) Et 81,2 108
l/NR (40) трет-Ви 71,2 52
(41) Ph 49,0 -40
(42) CO2Me 29,7 -138
l/NEt (43) — 43,1 -65
Me'—is. ,NC1 (44) — 111,8 д 80
°\ .NBu-грет (45) — 138,2 120
Me—N
/NCH,Pli (46) 114,3 д 70
Me — 2H,Ph
° ПО данным м|<кроволнойоп"Гп«твоск,1"Ы:! 1?аст“°р"телях; 6 ПО данным ИК-спектроскопни;
вым исследования кинетики эиимердаации? npil6jIlllKeHHO рассчитаииаи величина; Адо дан-
40
востыо. В этих случаях взаимопревращения энантиомеров путем
пиганлпого обмена аналогичны ......1 -
Уг«рм. Если .ее
по паппииоп izMl ятшптол ____ ' 11
......ыическои системы,
разделение энантиомеров
- -Г П 3Hd
лигандного обмена аналогичны соответствующи
не, например (29), являются элементами бицик
инверсия исключается и возможно
[1, Ю6].
СН2СН2Ме
N—СН2СНМе,,
I
Me ОН
(28) (2S)
Стереохимическое поведение подобных «статических» систем
изучено достаточно хорошо, и а последнее десятилетие предметом
все более пристального внимания исследователей стала динамиче-
ская стереохимия соединении азота. С помощью метода динамите-
ской спектроскопия ЯМР можно измерять энергии активации в ин-
тервале 20—105 кДж/моль (~5—25 ккал/моль); классические
кинетические методы применимы для измерения более высоких
энергий. Влияние напряжения и электронных факторов выяснено,
в общем, достаточно хорошо. Ниже показано, что при наличии
подходящих заместителей энергия активации процесса инверсии
может достигать значения ~100 кДж/моль, допускающего разде-
ление инвертомеров физическими методами при комнатной темпе-
ратуре.
Характер влияния искажения валентных углов становится по-
нятным при рассмотрении примеров, приведенных в табл. 6.1.1,
например соединений (33), (35), (37) и (39), для которых мак-
симальная разница энергий активации составляет 53 кДж/моль.
В ходе инверсии валентный угол связей С—N—С в переходном
состоянии должен стать равным 120°. В случае ациклических
аминов или циклических аминов с достаточнс большим размером
цикла, например (33), этот переход совершается относительно
легко, ио все более затрудняется по мере уменьшения размера
цикла. Хотя в малых циклах напряжение несомненно существует (
и способствует увеличению энергии основного состояния с тетра-
эдрической гибридизацией, в плоском переходном состоянии
влияние напряжения еще более значительно. В результате этого
происходит общее увеличение энергии активации, которое может
быть очень существенным, как, например, в азиридинах (3 ) и
(44). Вследствие легкости измерения температуры, при которой
происходит усреднение сигналов в спектрах ЯМР энантиомерных
азиридинов, эта циклическая структура была использована в
честве эталона сравнения при изучении влияния замес и . •
Увеличение объема заместителей при атоме азота
Анческон системе) и вызванное этим их пространствен
41
оттаскивание приводит к дестабилизации основного состояния с тет-
паэдоической гибридизацией. Это отталкивание может несколько
уменьшиться в плоском переходном состоянии, приводя к ослаб-
лению напряжения в цикле и снижению барьера активации.
Были получены чанные, показывающие, что в некоторых случаях,
например’(23; R = трет-Ви, п = 3) [ПО], происходит существен-
ное уплощение пирамидальной структуры при атоме азота.
Гораздо сильнее энергия активации уменьшается при сопряже-
нии свободной электронной пары азота с соседней п-системой,
как в случаях (41), (42) и (43). Подобное сопряжение более вы-
ражено в переходном состоянии, в котором свободная электрон-
ная пара занимает р-орбпталь, а не в основном состоянии, где
гораздо сильнее проявляется s-характер. Сопряжением можно
объяснить, например, низкий барьер активации для процесса ин-
версии при атоме азота в амидах, для которых эта величина ле-
жит ниже уровня чувствительности методов измерения, основан-
ных на ЯМР-спектроскоппи.
Присоединение электроотрицательных атомов к атому азота
увеличивает s-характер свободной электронной пары. Это в свою
очередь приводит к дестабилизации переходного состояния по
сравнению с основным состоянием и к возрастанию энергетиче-
ского барьера. Предположили, что причиной подобного изменения
энергетических характеристик при замещении гетероатомамп яв-
ляются н другие факторы, включая взаимное отталкивание сво-
бодных электронных пар. Однако для пас в данном случае суще-
ственно то, что барьер активации действительно возрастает, как
это следует из ряда примеров, приведенных в табл. 6.1.1, при
замещении алкильной группы атомом хлора. Действительно, зна-
чительная дестабилизация переходного состояния в сравнении с
основным состоянием происходит при одновременном воздействии
двух факторов — напряжения цикла и «гетерозамещеиня». При
этом возникает возможность разделять пнвертомеры азота [на-
пример, в случае (44) методом газожидкостной хроматографии],
хотя в данном случае они являются диастереомерами, а не энан-
тиомерами. При асимметрическом хлорировании азиридина (47)
с помощью оптически активных хлорирующих агентов при низ-
ких температурах была получена смесь двух N-хлорзамещениых
энантиомеров в неравных количествах (уравнение 112), обладаю-
щая оптической активностью. Однако при О °C в течение несколь-
ких суток происходит полная рацемизация этой смеси [III]. При-
соединение нитрена к стиролу в условиях кинетического контроля
протекает стереопзбирательно (уравнение 113) и приводит только
к си«-ипвертомеру (48) в результате сильного притяжения между
фенильной н N-фталимидной группами в переходном состоянии.
Соединение (48) сохраняет свою стереохимическую чистоту при
температурах ниже О °C, обращение конфигурации при азоте про-
исходит лишь при повышении температуры. Таким образом, усло-
вия термодинамического контроля способствуют цолному переходу
42
(ИЗ)
в форму диастереомерного анти-инвертомера (49), в котором от-
сутствуют невыгодные пространственные взаимодействия [112].
Гетероатом, который способствует повышению энергетическо-
го барьера в структурах (44) и (48), является экзоцнклическим.
Однако высокие значения энергии активации наблюдаются также
и для соединении, содержащих эндоцпклпческий гетероатом, та-
ких, как оксазиридпны (45) и диазирпдины (46) (см. табл. 6.1.1).
Суммарное влияние нескольких гетероатомов наиболее сильно
проявилось в NF3 (32); энергия активации для него оценивается
в 251 кДж/моль.
Данные для других напряженных азабициклнческнх систем
пока еще довольно ограничены. В настоящее время проявляется
значительный интерес к изучению состава смесей двух инвертоме-
ров, которые не равноценны энергетически, то есть не являются
нзоэиергетическпми. Причины необычно высоких значений барье-
ров активации в 7-азанорборнанах и 7-азанорборнадиенах (50,
51) не ясны. В этих соединениях валентный угол между связями
С—N—С такой же, как в азетидинах (38), но при этом барьер
инверсии выше на 30—40 кДж/моль. Интересно отметить, что до-
статочно низкая скорость взаимопревращения (50) и (51) позво-
ляет обнаружить, что эти два соединения ведут себя по-разному
в реакции сольволиза, протекающей в присутствии ионов серебра
[114] (уравнение 114). Направление реакции определяется кон-
фигурацией уходящего от азота атома хлора; эта реакция являет-
ся ярким и в то же время редким примером стереоэлектронного
контроля процесса сольволиза в химии азота в сравнении с бес-
численным множеством подобных случаев в химии соединении
углерода.
Заслуживает обсуждения проблема вращения вокруг связи
азот — углерод в ациклических аминах и циклических аминах с
большим размером цикла, а также возможность инверсии Циклов-
Барьер инверсии в простейших алкилампиах обычно выше, чем
барьер вращения вокруг связен (для метиламина ,
8,4 кДж/моль, соответственно), и анализ таких случа ым
вает затруднения. С энергетической точки зрения наиболее выг д
43
Cl
способом достижения эквивалентного пространственного рас-
положения трех групп R в группировке R3C—N (например, трех
метильных радикалов в трет-бутилыюй группе) обычно является
простое вращение вокруг связей, которое протекает независимо
от более медленного процесса инверсии. Однако в пространствен-
но затрудненных трпалкпламннах, для которых скорости этих двух
процессов могут быть примерно одинаковыми пли энергия актива-
ции процесса инверсии выше барьера активации вращения вокруг
связей, инверсия больше не является независимым процессом. Для
подобных случаев Бушвеллер интерпретировал эквивалентное
распределение трех групп R в пространстве как результат сопря-
женного протекания процессов вращения вокруг связи R3C—N
и инверсии при азоте [115].
Вопрос о вращении вокруг валентных связей не возникает в
том случае, когда азот является элементом циклической системы,
по крайней мере в уже рассматривавшихся 3-, 4-, 5- и 7-членных
азациклах. В этих системах имеют место процессы искажения
цикла (ring-puckering) или псевдовращепия (пли оба этих процес-
са), но они протекают с такой большой скоростью, что не мешают
изучению инверсии методом динамической ЯМР-спектроскоппп.
Циклическая система пиперидина, напротив, привлекла к себе
больше внимания, чем все остальные азамопоциклические соедине-
ния вместе взятые. Этот интерес объясняется широким распрост-
ранением этой системы в природных веществах, легкостью полу-
лучения и сходством с циклогексаном. Для нее характерно равно-
весие междУ двумя возможными кресловидиыми конформациями
' 1 11 I )> в которых группа R занимает соответственно аксиаль-
ное или экваториальное положение.
инверсии цикла происходит взаимное превраще-
ние аксиального и экваториального конформеров, причем для N-
ХХПвТнПИ₽а (52*53; R = Me> предпочтительным является
экваториальное расположение метильной группы. В ходе критичв-
44
ского рассмотрения методов, используемых в конформационном
анализе (включая работы по протоиированию в условиях кине-
тического контроля, которое позволяет «фиксировать» конформа-
ционные состояния в форме четвертичных аммониевых солей),
Робинсон высказал предположение, что конформационная энергия
систем недооценивалась [116]. Несмотря на наличие обще-
принятой точки зрения на пространственную геометрию цикличе-
ской системы пиперидина и значения барьера активации инверсии
цикла (около 44—50 кДж/моль), по-прежнему спорным остается
вопрос, находится ли связь N—Н в пиперидине (52^53; R = Н)
предпочтительно в экваториальном положении [117, 119] или нет
[118а]. Анет, используя метод высокопольиой ЯМР-спектроско-
пии, обнаружил медленную инверсию при атоме азота в пипери-
дине при очень низких температурах [119]; инверсия при азоте
оказывается быстрым процессом по сравнению с инверсией цикла
как для пиперидина, так п для азациклооктапа (54), для которо-
го принята конформация «ванна-кресло» (boat-chair), присущая
самому цпклооктаиу. Энергия инверсии цикла, приводящей ко вто-
рому возможному конформеру «ванна-кресло», для (54) составля-
ет 33,5—37,5 кДж/моль; заместитель при азоте занимает исклю-
чительно экваториальное положение [1186]. Соотношения между
экваториальными и аксиальными изомерами различных аминоцик-
логексанов определены в работе [120]; в этой же работе обсужда-
ется возможность применения спектроскопии ЯМР 13С для такого
рода исследований.
6.1.2.2, Спектроскопические свойства аминов
Исчерпывающее описание спектроскопии аминогруппы являет-
ся слишком обширной задачей и в рамках данной книги представ-
ляется затруднительным. В этом разделе изложены спектральные
характеристики, имеющие наибольшее аналитическое значение
Для идентификации аминогруппы, и отмечены некоторые послед
яче достижения в этой области.
(1) ИК-Спектроскопия [121]
Характеристические валентные колебания евязе’'п^Н перв1«
f!blx алкпламшюв в растворе наблюдаются при 33
Антисимметричные колебания) и при 3320-зло
45
/симметричные колебания), причем взаимное расположение ЭТ11Х
полос связано эмпирически выведенным соотношением Vs
345 53 + 0,876vas. Валентные колебания связей NH ариламииов сме-
щены в более высокочастотную область. Вторичную аминогруппу
характеризует наличие одной полосы в области 3360—3310 см-i.
Необходимо подчеркнуть, что при измерении ИК-спектров аминов
следует использовать неполярные растворители (дп-, три- и тет-
рахлорметаиы, сероуглерод), поскольку за счет ассоциации, вызы-
ваемой образованием водородных связей в полярных растворите-
лях, особенно в конденсированной фазе, происходит уширение
указанных полос и смещение их в область более низких частот
(до 100 см-1). Дополнительным подтверждением структуры пер-
вичного амина может служить наличие полосы деформационных
колебаний связи N—Н в области ~ 1560—1650 см-1. Однако
идентификация третичных аминов и попытки анализа валентных
колебаний С—N в низкочастотной области спектра редко дают
удовлетворительные результаты при использовании обычных прие-
мов ИК-спектроскошш.
(2) ПМР-Спектроскопия [122]
Сигналы протонов при углеродном атоме, непосредственно
связанном с электроотрицательным атомом азота, обнаругкива-
ются в характеристической области слабых полей. Так, для N-ме-
тильных групп простых алкиламинов, как правило, они лежат в
интервале б 2,0—2,5. Сигналы протонов N-метплеповой и N-метп-
новой групп располагаются в еще менее сильных полях (при-
мерно до б 3,1). Однако на значение химического сдвига существен-
ное влияние может оказывать наличие ненасыщенных заместите-
лей или других гетероатомов при углеродном атоме метиленовой
или метиковой группы; так, например резонансный сигнал метиле-
новой группы в бензиламине наблюдается при 6 4. Попытки пред-
сказать характер влияния нескольких заместителей с помощью
принципов аддитивности ие всегда успешны; часто существенное
значение имеет зависимость химического сдвига от стереохимиче-
ских факторов, особенно для циклических систем, в которых влия-
ние относительно удаленных групп может быть значительным.
Для большей достоверности отнесения сигналов обычно проводят
сравнение с соединениями, имеющими близкое строение. Простой
способ отнесения сигнала протонов при углеродном атоме в «-по-
ложении к атому азота основан на иротоинрованпи азота, в ре-
зультате чего происходит заметное смещение сигнала в сторону
слабых полей иа 0,5—1,0 млн-1. Протоиироваиие достигается
при использовании в качестве растворителя трпфторуксусной ки-
слоты; иногда достаточно добавления нескольких капель этой ки-
слоты к раствору амина в CDCU (при условии удовлетворитель-
ной растворимости образующейся соли). Протоиироваиие являет-
ся полезным приемом, позволяющим избежать перекрывания си-
46
риалов в спектре и оцепить число протонов H-C-N в исследуе-
мой молекуле.
Еше более значительного упрощения сложных спектров аминов
можно добиться за счет использования парамагнитных комплексных
соединений редкоземельных элементов в качестве «сдвигающих
реагентов» (шпфт-реагенты) [124, 125]. Свободные электрон-
ные пары многих функциональных групп вовлекаются в образова-
ние координационной связи с комплексами, например с трис(ди-
нпвалоплметанатоевроппем Eu(dpm)3 (55). Вторичное магнитное
поле, обусловленное магнитным моментом парамагнитного иона
вызывает изменения химических сдвигов соседних ядер (псевдо-
контактные сдвиги). Если не учитывать возможные взаимодей-
ствия через валентные связи (контактные сдвиги), то можно при-
нять, что такие изменения зависят от удаленности этих ядер от
поиа металла. Кроме того, изменения в спектре определяются кон-
центрацией шпфт-реагента. Это часто позволяет разделить пере-
крывающиеся сигналы и осуществить их отнесение, а также упро-
стить спектры второго порядка до спектров первого порядка. Как
правило, для сигналов первичных аминов наблюдаются смещения
того же порядка, что и для любых других функциональных групп.
В аминах с большей степенью замещения (вторичные и третичные
амины) из-за пространственных затруднений возможно снижение
эффективности комплексования, вследствие чего смещения сигна-
лов происходят в меньшей степени. При повышенных концентра-
циях шнфт-реагентов наблюдается уширение сигналов, в резуль-
тате чего теряется информация о спин-спиновом расщеплении.
Шифт-реагеиты успешно применялись для изучения стереохимиче-
ских п конформационных проблем, несмотря на возможность
нарушения положения равновесия в конформационно подвижной
системе за счет присутствия этих комплексующих соединений.
(55) (56)
Оба энантиомера хирального амина, например а-фенилэтил-
амина (56), имеют идентичные спектры ПМР. Однако при превра-
Щевип рацемического амина в смесь двух диастереомерных солей
(см. уравнение 110) энаитпотопные протоны (например, Нх)
становятся диастереотопными и часто наблюдаются в спектре в
Впде раздельных сигналов. Это позволяет количественно оцени-
вать оптическую чистоту энантиомеров путем интегрирования
"МР-спектров. В подобных случаях различие в химических сдвн-
Гах является результатом внутримолекулярного взаимодействия,
47
пптко па спектры хиральных молекул могут влиять и воздейст.
вия межмолекуляриого характера. Процесс комплексообразования
с амином может быть обратимым, а время существования дпасте-
пеомерпых комплексов очень коротким, и тем не менее удается
зафиксировать влияние усреднения во времени па спектр ПМР.
Дтя получения раздельных сигналов от эпантпотопиых протонов
в ряде соединений Пиркл использовал хнральиыс растворители
[123]. Гораздо более значительных различии химических сдвигов
энаитпотопных протонов можно достичь С ПОМОЩЬЮ ПОДХОДЯЩИХ
оптически активных шифт-реагеитов; этот прием получил широкое
распространение [124, 125]. Так, оптически активный комплекс
европия (57) позволяет получать для сигналов метильной группы
в энантиомерах (56) разницу в химических сдвигах до 0,18 млн-1,
а для сигналов метшювых протонов Нх до 0,92 млн-1.
Значение химического сдвига сигнала протона группы NH
в аминах колеблется в интервале б 0,5—5,0; положение этого
сигнала в значительной степени зависит от природы раствори-
теля, концентрации и температуры, поскольку эти факторы влия-
ют на образование межмолекулярных водородных связей. Вслед-
ствие этого точное значение химического сдвига протона амино-
группы в указанном интервале имеет очень ограниченное значение
для идентификации. Быстрый обмен протонов па дейтерий, про-
исходящий в растворе амина в DoO, приводит к исчезновению из
спектра сигнала протона группы NH (сигнал DOH появляется при
б к. 5). Интегрирование спектра позволяет обнаружить исчезнове-
ние резонансного сигнала NH, даже если этот сигнал ие был раз-
решен в сложной картине спектра. Сигналы группы NH, как пра-
вило, бывают уширенными из-за влияния квадрупольного момента
ядер l4N, способствующего более быстрой спин-решеточной ре-
лаксации. Это уширение препятствует выявлению сппп-спипового
взаимодействия ядер 'Н, ’Н и >Н, ИМ.
(3) Спектроскопия ЯМР 3 * * * * * * * * * 13С
Огромные возможности метода ЯМР-спектроскопип с импульс-
ным преобразованием Фурье способствуют все более широкому
его применению в органической химии. Этот метод оказывается
особенно полезным в случае ядер 13С. Ранее использованию пре-
имуществ ядер |3С— более широкая резонансная область и как
следствие лучшее разрешение сигналов (резонансные сигналы
атомов углерода в органических соединениях наблюдаются в ин-
тервале —-200 мли~')—препятствовали низкая чувствительность
регистрации ядер 13С, равная '/ел чувствительности *Н, в соче-
™ С всвысоким естественным содержанием этого изотопа
(1,1 /о). Облучение образца в постоянном магнитном поле им-
пульсом мощности, покрывающим весь набор частот для данного
типа ядер, возбуждает одновременно все ядра; при релаксации
образца Поглощенная энергия выделяется в виде сложной волно-
48
вой формы, которая может быть преобразована с помощью ком-
шиотера в тот же набор частот, что и в обычном экспери-
менте при медленном прохождении всего интервала частот. Нако-
пление сигналов в спектре путем многократного повторения
импульсов, чередующихся через короткие интервалы времени до-
статочные для релаксации, осуществляют до тех пор, пока конеч-
ное преобразование не приведет к приемлемому соотношению
спгнал/шум (для этого в случае ядер 13С могут потребоваться
сотни и тысячи импульсов) п получению удовлетворительных
спектров при естественном содержании изотопа 13С даже для
разбавленных растворов. Обычная процедура шумовой развязки
от протонов упрощает спектры за счет исключения всех взаимо-
действии ядер 13С и >Н и приводит, как правило, к одному сиг-
налу для каждого углеродного атома и к увеличению интенсивно-
сти сигналов за счет проявления ядерпого эффекта Оверхаузера.
Спектрометры П усовершенствованные методики съемки спектров,
позволяющие получать информацию о спип-спиповом взаимодей-
ствии (то есть о расщеплении резонансных сигналов и значениях
констант спип-спинового взаимодействия ядер 13С и 'Н) рассмат-
риваются в превосходно написанных руководствах [126]. Следует
отметить, что количественное интегрирование сигналов в спектре
ЯМР |3С возможно только в том случае, если ядериый эффект
Оверхаузера не имеет места и если задержка между импульсами
превышает время сппи-решеточной релаксации наблюдаемых
ядер (+). Однако на практике такие случаи очень редки.
Спектры ПМР углеводородов, содержащих функциональные
группы, позволяют получать определенную информацию о протонах
при углеродных атомах, соседних с функциональной группой.
В противоположность этому с помощью спектроскопии ЯМР
|3С можно легко зафиксировать влияние заместителей в углерод-
ной цепи даже для S-углеродного атома. Введение аминогруппы
в пентан в положение 1 приводит к следующим смещениям сиг-
налов ядер 13С (в млн-1) относительно их положения в спектре
пентана: +28,8(a), +И,7(₽), — 4,8(у), +0,5(6) («+» означает
смещение в слабое поле). Подобная чувствительность к замещению
н тот факт, что эффекты замещения в спектрах ЯМР |3С имеютпоч-
тп аддитивный характер, дали возможность Джерассп вывести кор-
реляционные формулы, которые позволяют с высоком степенью
достоверности предсказать химические сдвиги сигналов углерода
Для неизвестного ациклического алифатического амина [12/J.
первый взгляд смещение сигнала у-углеродного атома в спль о
ноле может показаться неожиданным, но эта особенность я .
ся общим свойством циклических и ациклических стрхкт^р
случае, если в углеводород введена метильная группа ’ птель.
Полярный заместитель. Считают, что это смещен! .. впем
нои степени обусловлено пространственным 1’4-взаш £ Очередь
связанных заместителей в гош-ротамере, я 3 яминах 11271.
ает возможность изучать поворотную изомерш
- 49
,п „поь-тпк! ЯМР 13С для простых циклических амп-
ОП,1еаИнюТгДЖсоед11неи»11 сложного строения, например алкалои-
пов 11 МНО1 .t дметОда спектроскопии ЯМР |3С демонстрирует
пример взятой из работы Уэикерта, Жано и сотр [128]. Ими был
ПО v‘ien спектр виидолииина, в котором наблюдались отдельные
сигнато от каждого из 21 углеродного атома, и на основании от-
несения всех линий в спектре для ............а вместо структуры
(58) (принятой ранее для всего семейства алкалоидов) была
была
предложена структура (59). Применение шпфт-реагеитов в спек-
троскопии ЯМР 13С не играет такой роли, как при анализе спек-
тров ПМР, поскольку спектрам ЯМР 13С в гораздо меньшей
степени свойственно перекрывание сигналов. Однако шифт-реа-
генты с успехом применялись для отнесения линий в спектрах и
решения других задач, например для выяснения предпочтитель-
ной ориентации свободных электронных пар в молекулах азаби-
цнклов [113, 129].
(4) Спектроскопия ЯМР на ядрах азота
Спектроскопия ЯМР на ядрах азота для большинства хими-
ков-органиков является сравнительно мало известным методом.
Здесь будут отмечены лишь характерные особенности метода,
подробнее с этим методом читатель может ознакомиться в более
авторитетных источниках [130]. Естественное содержание изото-
пов 14N п 15N составляет соответственно 99,64 н 0,36%, причем
оба типа ядер примерно в 1000 раз менее чувствительны к воз-
буждению, чем ядра 44. Чаще используется спектроскопия ЯМР
на ядрах 14N, однако в последнее время увеличивается число ра-
бот, выполненных с применением спектроскопии ЯМР l5N. Это
связано с внедрением в практику импульсной ЯМР-спектроскопнп,
которая делает возможной съемку спектров при естественном со-
держании изотопа и не требует предварительного введения метки
N, являющегося достаточно дорогостоящей процедурой. Ядра
имеют электрический квадрупольный момент, небольшие
релаксации являются причиной характерного ушпренпЯ
Ппп, »,, (т”п1,чнои является ширина сигналов в 20—1000 Гц).
"°, V - «-окоразреиюииых спектров возможно в основном
при использовании изотопа ,bN. для которого характерна ширина
50
резонансных сигналов ~1 Гц. Интересной особенностью упоошя
ющей анализ ЯМР-сиектров на ядрах азота, является то ^тп
химические сдвиги могут быть измерены как от сигналов >«N ток
„ от сигналов N. Эти величины взаимозаменяемы поскоя™
„е существует экспериментальной разницы в константах экоанн
рОваш1Я азота для обоих изотопов. Приводившиеся ранее данный
0 различии химических сдвигов для изотопов азота являются
ошибочными. Химические сдвиги сигналов азота в органнч™
соединениях располагаются в области ~800 млн-'. Их
Значительную часть сведении о структуре органических ими
нов получают уже при использовании спектроскопии ПМР и ямо
,3С; применение спектроскопии ЯМР на ядрах азота ~
явные преимущества при изучении азидов, азосоедпнений тоиазо
лннов И т. д. Однако иногда очень полезной оказывается’возмож-
иость идентифицировать первичные, вторичные и третичные ами-
ны по значениям химических сдвигов атомов азота. Очень значи-
тельное смещение сигналов происходит при протонировании азота-
данные об изменении химических сдвигов под влиянием протони-
рованпя нашли применение при изучении таутомерии и в конфор-
мационном анализе. Данные об изменении констант 'Н, 15Х'-спнн-
спинового взаимодействия использовались для решения разнооб-
разных задач, в частности для изучения инверсии при атоме
азота [131]. Была изучена возможность использования спин-спн-
нового взаимодействия ядер 5 * * * * * * * 13С и 1SN для оценки гибридизации
]132]. Многие сведения о характере спектров ЯМР l5N получены
при исследовании соединений, обогащенных изотопом 15N. Иллю-
страцией возможности использования спектроскопии ЯМР 15N
(при естественном их содержании) и получаемых при этом спек-
тральных характеристик (химических сдвигов ядер 15N и кон-
стант спин-спинового взаимодействия ядер >Н и 15N) для изуче-
ния нуклеозидов н нуклеотидов служит работа Робертса [133],
в которой описывается также практическая сторона этого метода.
(5) Масс-спектроскопия [/34]
В масс-спектрах аминов пик молекулярного иона (М+) редко
имеет высокую интенсивность, но в обычных условиях съемки
спектров (при ионизующем напряжении 70 эВ) он, как правило,
вполне различим. Считается общепринятым, что при электронном
Ударе происходит потеря одного из неспаренных электронов азота.
Величина М+ особенно полезна для идентификации моноамннов
(практически для любых соединений, содержащих нечетное число
втомов азота), поскольку поп М+ имеет в таком случае нечетное
значение tn[e в отличие от четных значений М+ для соединении,
“Держащих атомы Н, С, О, S, С1 и т. д. Кроме того,, точ-
"°е определение относительной молекулярной массы
г° ио,,а позволяет однозначно установить молекуляр .
'-"У аиал1гзируемого соединения,
51
Mnnfionee интенсивный нон в спектре первичного алкнламнна
пПпХтся Обычно в результате a-разрыва, преимущественно
?гптя .I не исключительно) за счет элиминирования самого боль-
П10ГО ачкнльного фрагмента (уравнение 115). Нередко происходят
пазоыв’ы В-, Y-. б- " более удаленных углерод-углеродных связей,
особенно в'случае первичных аминов с высшим алкильным ра-
дикалом- при этом возникают серин ионов с понижающейся ин-
тенсивностью которые отличаются па 14 единиц массы, например,
mle 30 (основной пик), 44, 58, 72 и т. д. (уравнение 115,
R> = R’ = H).
R1
г ~+
R^CH^CH^C^-NIU
5 ? И2
У Р <*
-К3СН2С112
R1
\=nh2
R?
(115)
Аналогичная фрагментация может происходить во вторичных и
третичных аминах. При этом обычно обнаруживаются ионы, име-
ющие четное значение отношения заряда к массе и образующиеся
в результате всех возможных «-разрывов, причем, как правило,
более интенсивными являются ноны, возникающие путем элими-
нирования более тяжелых радикалов. Далее может происходить
перегруппировка, сопровождающаяся потерей алкеновых фраг-
ментов (например, уравнение 116).
ch2ch2r’ н
R’^-tf-CH2CH2R’-^R’cH=A( R*CH=i/
R’ R* V r2
(116)
«-Разрывы характерны и для фрагментации циклических ами-
нов, например пиперидина (уравнение 117) и циклоалкиламннов,
с
HN* HNt HN*
М~ 1 м*
наиболее
интенсивными часто оказываются пики
1 а-Н. Гомоли-
для которых
м—1, образующиеся в результате потери радикала а-Н. Гомоли-
тический разрыв одной из связей цикла приводит к сохранению
пяямопЛп’,’0Г<> *рагме1|та в молекуле, а характер замещения и
метании определя|От разнообразие путей дальнейшей фраг-
алкил^н i’v пвключа|ощих перегруппировку н элиминирование
зи ™ РаДикалов и (или) алкеновых фрагментов [134]. В свя-
и поири '.’.„Л™ ВСе больи1ее значение приобретает накопление
вс^О0озо^стаСю^пррТр°4МеТрИЧеСК°^ 1И1Ф°РмаЦИИ с помощью ЭВМ.
Р Щ е значение приобретает представление данных
62
масс-спектров в строго определенном виде. С помощью ЭВМ часто
удается идентифицировать ннкн молекулярных ионов, а данные
спектров высокого разрешения дают возможность установить
состав каждого иона (помимо его массы в относительной интен-
сивности). Специальные программы для ЭВМ позволяют выво-
дить структуры на основании данных масс-спектров низкого раз-
решения. Эффективность этих программ может быть значительно
повышена за счет привлечения данных ПМР-спектров. Дже-
рассн реализовал на практике многообещающий подход к одно-
значному отнесению структуры любого неизвестного амина, за-
ключающийся в объединении данных, полученных с помощью
таких мощных методов, как масс-спектрометрия и спектроскопия
ЯМР |3С и их анализе с помощью ЭВМ [135]. Значение этого
подхода становится очевидным, если напомнить, что молекуляр-
ной формуле C21H45N отвечает более 38-Ю6 структурных изо-
меров.
(6) УФ-Спектроскопия
Интерес к спектрам, наблюдаемым в УФ- н видимой областях
спектра, обусловлен главным образом возможностью определения
абсолютной конфигурации хиральных аминов по данным спектров
кругового дихроизма (КД). Выступая в роли монодентатных ли-
гандов в медных комплексах, хиральные амины могут вызывать
появление в области между 400 п 800 нм двух эффектов Коттона
противоположного знака. Для определения абсолютной конфигу-
рации необходимо лишь знание знаков эффектов Коттона, и этот
метод позволяет получать те же результаты, что и применение
других комплексов, например лантаноидных шифт-реагентов.
Сформулированы правила, связывающие абсолютную конфигура-
цию N-салпцилнденовых производных хиральных аминов с харак-
тером спектров кругового дихроизма [137], хотя аналогичные
правила для N-нитрозоамниов были пересмотрены [138].
6.1.2.3. Основность аминов
Наличие свободной электронной пары у атома азота придает
аИ||ногруппе основные свойства. В соответствии с теорией Брея-
птеда основание является акцептором протона; образующуюся
Ротоипроваипую форму амина (нон аммония) называют сопря-
^""Ой кислотой (см. уравнение 118). Согласно определению
т амнн может образовывать связь с кислотами « ьюп ,
сП(.,.Ль с любыми частицами, имеющими вакантную ор • >
эдектп1^'10 ПР,11,ЯТ>> участие в создании связи с нсП°^ь яется
Частш°Н110" па1’Ь1 основания. Таким образом, ПРОТО ’ ебе
ЧаиКп М "Римером кислот Льюиса, но именно он привлек к
Шшее внимание.
R3N : + АН RjNH + A
(па)
53
Равновесие кислота — основание устанавливается довольно
г Лпо Тпои обсуждении основности следует рассмотреть поло. ।
я ение равновесия в данной системе. Оно определяется разностью
AfiniBHX энергий AG0 основания н сопряженной кислоты. На
относительную устойчивость этих двух частиц влияют три основ,
ш х Фактора- электронные факторы, природа растворителя „
ётоуктурные особенности, которые будут рассмотрены ниже.
В таяние электронных факторов на основность можно оценить I
с помощью данных об основности в газовой фазе, полученных
рядом методов, таких, например, как масс-спектрометрия пли
ионный циклотронный резонанс. Эти методы позволяют изучать
иои-молекулярные взаимодействия (уравнение 119) ц рассчиты-
вать AG° по уравнению 120, где GB — основность в газовой фазе.
(119) '
(120)
GG° = GB (A) - GB (B)
Результаты, полученные при изучении большого числа алкил-
аминов, были использованы для количественной оценки влияния
алкильных групп на основность самих аминов. Порядок возра- '
стапия этого влияния согласуется с увеличением электронодонор-
ного «индуктивного эффекта» алкильных групп, который оказы-
вает сравнительно более сильное стабилизующее воздействие на
протонированные формы аминов, чем на свободные амины. В соот-
ветствии с этим наблюдается увеличение основности, например,
в следующем ряду:
NH3 < MeNH2 < EtNH; < h-BuNH2 < Me2NH < Me3N < Et3N < h-Bu3N
Понятие «индуктивный эффект» достаточно хорошо обоснова-
но экспериментально и является очень полезным для химиков-
органиков, однако стало очевидным, что алкильные группы спо-
собны стабилизовать не только положительные, но и отрицатель-
ные ионы. Это следует из характера кислотности спиртов в газо-
вой фазе [139, 140] (ВиОН > ЕЮН > МеОН > Н3О) и влияния
алкильных заместителей на увеличение силы кислоты в растворе,
тот эффект можно представить как результат делокализации
положительного или отрицательного заряда в молекуле вслед-
ствие поляризации различных связен. Как п следует ожидать,
Г-Ру°0ТРШ1аТеЛЬНЬ,е ат0МЬ| П0НИжают основность, например,
ch3ch2nh2 > fch2ch2nh2 > f2chch2nh2 > F3CCH2NH2
ценка основности в газовой фазе относительно аммиака
Дала возможность сравнить
(табл. 6.1.2); важно отметит
Йн4 + В NH3 + ВН
AG° = OB (NH3) — GB (В)
циклические и ациклические амины
'->> что для аминов, имеющих аиало-
54
Таблица 6.1.2. Основность некоторых аминов a .
----------------------------------------<«юорц фазе
Сослимние А0°' кДж'моль Соединение I
N I - кДж/иоль
-101,7
-103,0
—84,2
-84,6
—75,4
-75,0
-46,9
-64,9
-28,1
-47,3
—54,4
гкчные заместители, значения AG° близки (отрицательные значе-
ния указывают на большую силу основания). Отклонение, наблю-
даемое в случае азиридина, находит объяснение в рамках теории
гибридизации: орбиталь свободной электронной пары в азиридине
носит более выраженный s-характер, чем в диметиламине, и
поэтому менее вероятно ее участие в образовании связи с пр то
ном. Для бензиламинов, по-видимому, экспериментальных данны
недостаточно; для трех первичных аминов, представле ы
табл. 6.1.2, наблюдается удовлетворительная корреляция ме ду
характером гибридизации 0-углеродного атома >> значениi .
Закономерности, выявленные для основности разлп н .
иов в газовой фазе, привлекают своей простотон " ’
Большинство экспериментов в органической химии су ‘ 0.
ся в растворах, п в этих случаях изменение основноет
гДа описываться приблизительно такими же_ зак • Р вимНг,'
как и вслучае газовой фазы (например, BiisN > ,,~п>.но 2 4-дини-
получепныг; для растворов в хлорбензоле относг льно 2Д дини.
рофенола). Однако часто эти выводы не носят основности-
та\ в бензоле наблюдается следующий «°Рад°км °™ в, я-
> Bu3N > BuNH2. В течение многих лет Хх рас-
тпп°СОбы1' пнтеРес к закономерностям, существу ю ‘ свОбОдная
3iienrX' В Этом слУчае основным параметром • вь1раЖаемая
ок,Ргия пРот°1И1рованпя основания в воде Ди ( -н г’дО^НгО) =
сопряженной введены
!п/?7 1пКа]. Значения р/(а для простейших аминов I
“W 6.1.3,
55
Таблица 6.1.3.
гт/пяжемных алкиламинам (ПгО, -<> С)
Соединение
R3N
R2NH
RNH,
Nils
10,85 9,80
11,09 10,73
10,80 10,66
9,25
Отсутствие четкой закономерности в поведении алкиламинов
объясняли по-разному. Влияние пространственных факторов на
стадии протонпрованпя можно не учитывать, и долгое время при-
знавалась важность эффектов сольватации, протекающей в раз-
личной степени. Наличие данных по основности в газовой фазе,
подтверждающих закономерное повышение силы основания под
воздействием электронных факторов при увеличении числа ал-
кильных заместителей, служит теперь надежным критерием при
сравнении. Недавно Ауэ применил эти данные в сочетании с из-
вестными термодинамическими параметрами в водных растворах
для всестороннего анализа дифференциальной сольватации [142].
В ряду алкиламинов теплоты гидратации обычно закономерно
понижаются с увеличением размеров молекул. Это влияние
алкильных заместителей, называемое гидрофобными эффектами,
изучено недостаточно, однако предполагают, что подобные эффек-
ты почти полностью отсутствуют в нейтральных и протонирован-
ных аминах, находящихся в водной системе. Считают, что в
растворе важным фактором является влияние на сопряженную
кислоту ослабления взаимодействия между растворителем п прото-
нированным амином при делокализации заряда в ноне. К тем же
выводам приходят при интерпретации этого явления с точки зре-
ния электростатической сольватации (считают, что энергия соль-
ватации и ионный объем связаны обратной зависимостью) и соль-
ватации с участием специфических водородных связен (при этом
каждая специфическая водородная связь ослабляется вследствие
делокализации положительного заряда в ионе). Таким образом,
в тех случаях, когда усиление поляризуемости вследствие увели-
чения числа алкильных заместителей приводит к стабилизации
иона аммония за счет делокализации заряда, сольватация нона
должна происходить менее экзотермпчпо, способствуя ослаблению
стабилизующего влияния заместителей по сравнению с тем, что
'новеесчммАп,.прГаЗОВ01' фа3е- ПоэтомУ в ряду алифатических ами-
степенно оепХ * ВяЛ‘1Я1",е увели',е1шя степени алкилирования по-
ряда в тех слмиВ еТ/" может Фактически приводить к обращению
Р Д в тех случаях (например, Me3N в табл. 6.1.3), когда эффект
56
шепьшеппя стабилизации при сольватации сильнее, чем внутри-
Пекулярное стабилизующее влияние алкильных заместителей
И наоборот, В тех случаях, когда индуктивные эффекты могут вы-
звать дестабилизацию иона аммония, ион будет обладать повы-
шенной плотностью заряда на атоме азота и лучше сольватиро-
ваться; здесь вновь наблюдается противодействие электронным
оЛЛектам. Важность сольватации можно подчеркнуть тем, что
изменение свободной энергии при переходе ионов аммония из
тазовой фазы в водный раствор может составлять до 25—
110 кДж/м0ЛЬ (пРнмеРно аналогично изменению AG° за счет
электронных эффектов алкильных заместителей в газовой фазе).
Для более подробного и систематического знакомства с термоди-
намическим аспектом данной проблемы н уяснения природы эф-
фектов сольватации читателю следует обратиться к работам
[140-142].
В данном разделе не будет дублироваться обсуждение явле-
ния постепенного понижения основности при переходе от алкил-
ампнов к ариламинам (см. разд. 6 3.2) и амидам (см. разд. 9.4.2).
Понижение основности по мере усиления s-характера азота (на-
пример, в пиридине и нитрилах) также освещается в соответст-
вующих разделах.
Как уже отмечалось, данные, говорящие о каком-либо влиянии
пространственных факторов на перенос протона в газовой фазе,
отсутствуют, по при описании взаимодействия аминов с кислота-
ми Льюиса (уравнение 121) становится важным учет объема за-
местителей Этот фактор впервые был рассмотрен в классическом
исследования Брауна.
R3N + ВМе3 R3N->BMe3
(121)
Сравнение теплот диссоциации комплексов аминов с кислота-
ми Льюиса и теплот диссоциации соответствующих аммониевых
ионов позволяет достаточно точно оценить энергию пространствен-
ного напряжения, которое наблюдается у аминов, содержащих
заместители различного объема (табл. 6.1.4). „
Измерение констант равновесия дает сведения о сооственной
основности ряда ам инов, для которых пространственные факторы
не меняются, а изменение заместителей происходит на большем
Удалении от атома азота. Были изучены [Ю6] многие ДРУ™
комплексы аминов, например комплексы с ионами »еоталл°в'Х'
лексы с лантаноидными элементами (см. разд. • )> '
нами ц иолнпнтросоедпненпямп, включая пикрин У ме>
^еакцпя образования пикратов лежит в основе клас
да идентификации аминов. , ,, межмоле-
куг,?,11Ног11Уппы обладают способностью к в у р . ‘.цпональ-
НЫм?Н°В ооооииацип друг с другом или с ДРУГ1|‘' , связ11 (амин
группами. Оба возможных типа водородной связи I
57
ТлЛшиа 61.4. Энергия пространственного напряжения
для аддуктов аминов и три метилбора 1140]
Энергия
пространственного
напряжения,
кДж/мо.ть
Амин
Энергия
простра11сгаен
напряжения °
КДж/моль'
Me«NH
12,1
18,1
29,3
Et2NH зод
TpeT-BuNHj 33 5
Et3N 71,2
выступает как донор водорода или как акцептор) иллюстрирова-
ны формулами (60) п (61). Термином «водородная связь» в каж-
'4N—Н--Х X
/ /|
(60) (61)
дом случае принято обозначать более слабую из двух связей
с водородом. Образование водородных связей происходит в твер-
дом состоянии, в жидкой фазе, в растворе, а иногда даже в
в газовой фазе. По прочности водородная связь (~8—
40 кДж/моль) является промежуточной между ковалентными и
ван-дер-ваальсовыми связями. Особая важность этого типа связи
была продемонстрирована в ходе обсуждения основности в вод-
ном растворе. Определенное влияние водородных связен па физи-
ческие свойства выражается в том, что температуры кипения пер-
вичных аминов выше, чем температуры кипения углеводородов
приблизительно той же молекулярной массы, хотя в случае тре-
тичных аминов этот эффект, естественно, исчезает. Были изучены
спектроскопические проявления водородной связи; эти наблюде-
ния лежат в основе способов ее обнаружения и изучения. Про-
блеме водородной связи посвящены краткие обзоры [1431 и об-
ширные монографии [144].
6.1.2.4. Физиологическая активность аминов
В основе физиологической активности аминов [145—146], пе'
сомненно, лежит их способность образовывать водородные, кова-
лентные и ионные связи, которые определяют способность аминов
приближаться к рецепторному сайту в организме, реагировать с
58
... и образовывать, нарушать или модифицировать химические
'"язи. Обсуждение важности водородных связей в аминокисло-
тах белках н в каталитических процессах можно найти в ссыл-
ках упомянутых выше, ц в работах, приведенных в томе 10 на-
стоящего издания. Перенос аминов из водного раствора к рецеп-
торным сайтам регулируется прочностью связей с растворителем.
Ионные связи играют определенную роль при взаимодействии
лекарственного препарата с рецептором. Наиболее активные сое-
динения ионизованы или способны к ионизации при физиологиче-
ских значениях pH; к ним относится ряд азотистых оснований,
для которых характерны очень тонкие вариации р/(а при измене-
НИИ структуры и т. д. В том случае, когда соли четвертичных ам-
мониевых оснований полностью ионизованы (см. разд. 6.1.3.2),
проницаемость клеточной мембраны для этих соединений пони-
жается, однако при наличии в равновесной смеси даже неболь-
шого количества свободного амина проникновение через мембрану
может иметь место. Большой интерес представляет выяснение
возможности передачи заряда в процессах взаимодействия лекар-
ственного препарата с рецептором. Сложные ненасыщенные ами-
ны рассматривались как доноры, а некоторые соли ненасыщенных
четвертичных аммониевых оснований как акцепторы, но эти ис-
следования, по-видимому, еще не достигли того уровня, когда
могут быть сформулированы сколько-нибудь убедительные кор-
реляции между структурой и биологической активностью [145].
Обсуждавшиеся выше взаимодействия являются обратимыми,
обычно за исключением случаев образования новой ковалентной
связи, например при замещении водорода у атома азота в первич-
ном или вторичном амине. Действующие таким образом лекар-
ственные препараты, по существу, более токсичны; так, например,
ярп алкилировании атома азота в аминокислотах образуются так
называемые азотистые иприты.
В процессах вторичного метаболизма в растениях возникают
алкалоиды — весьма многочисленная группа соединений, содер-
жащих по крайней мере один атом азота, часто входящий в ци-
клическую систему. Примеры подобных соединений были приве-
дены в разделе 6.1.1, а многие классы алкалоидов, в основе кото-
рых лежат структуры пиридина, хинолина, индола и т. д., рас-
смотрены в томе 8. Почти все алкалоиды обладают тем или
иным фармакологическим действием, спектр их свойств от
чрезвычайной токсичности и способности вызывать психические
Расстройства до пригодности для использования в качестве лекар-
ственных препаратов [148]. Выяснено, что ряд аминов, получен-
Н(Ь1Х чисто синтетическим путем или путем последовательной моди-
у1каиии структуры алкалоидов, обладает полезными для челове-
а терапевтическими свойствами; эти соединения могут ыть
"'пользованы для лечения заболеваний, вызываемых "араз»т»РГ
""пчи в организме человека животными, насекомыми, rpi
59
„ бяктеоиями. Прочие важные азотсодержащие соединения
йда*—гл-224" »>• '«"««'it
(СМ гл. 23.5) И родственные соединения, рассматрпвак)н'“
в томе 10. Здесь стоит упомянуть наличие в организме человек
биологически активных аминов, образующихся путем декарбо
кодирования а-аминокпслот (то есть, в конечном итоге, из бед.
ков) при действии ряда ферментов [146]. При изучении этих
превращений возникают трудности, поскольку в амии превращает-
ся лишь небольшая часть белка. Однако несмотря иа низкую
текущую концентрацию этих аминов, установлена их необычайно
важная роль, в частности, для функционирования тканей мышц
и мозга. Были подробно изучены возрастные изменения содержа-
ния аминов и установлены соответствия между аномально высо-
ким или низким содержанием аминов и определенными патологи-
ческими состояниями. Эти корреляции нередко имеют большое
значение для диагностики. В организме человека обнаружены
амины, являющиеся производными ароматических аминокислот
(обычно модифицированные путем гидроксилирования, 0- и
N-метилирования, ацилирования и т. д.), например, (62) — (64).
В тканях организма человека обнаружены также менее хорошо
изученные алкиламнны, например этнламнп, нзопентнламин, «-бу-
тил- и изобутиламины, а также путресцин (66) и кадаверин (67)
(из орнитина и лизина), присутствие которых обычно связано
с самым крайним патологическим состоянием, смертью.
H2N(CH2)nNHj
(62) R1 = Н, I?2 — Н; допамин (65) серотошш
(63) Rl = Me, R2 = OH; адреналин
(64) R' = H, R2 = OH; норадреналин
(66) «=4
(67) я=5
Биологически активные амины имеют определенное значение
для развития различных опухолевых тканей. Существует связь
между метаболизмом аминов в ткани мозга и болезнью Паркин
сона, обусловленной взаимозависимым недостатком (62), (64)
н (65), который может быть следствием нарушения механизмов
накопления. В клетках крови и тканей организма происходи1
временное накопление природных аминов, которые могут вклю-
чаться в молекулы белков организма, зачастую с сохранением их
физиологического действия. Основной путь выведения аминов и3
организма заключается в ферментативном окислительном дезам»’
Д° С00тветствУюи1Их карбоновых кислот. Вещества,
пё =1"С ЭТ0Т ир011есс' нашли применение в качестве тера-
ких препаратов для лечения психических заболевании.
6.1.3. РЕАКЦИИ АМИНОВ
6.1.З.1. Нуклеофильные свойства аминов [8,10]
Химическое поведение аминов в значительной степени оппепо
ляется наличием свободной электронной пары на атоме азота и
обусловленной этим способностью атаковать электрофильны»
центры. В принципе небезосновательно ожидать корреляции меж"
ду нуклеофильным характером атома азота и его основностью
хотя нуклеофильность является кинетическим понятием а основ’
ность рассматривалась (см. разд. 6.1.2.3) с точки зрения термо-
динамики протонированпя. В том случае, когда для ряда аминов
пространственные (но не электронные) факторы постоянны, экс-
периментально определенные значения основности могут’быть
использованы для оценки нуклеофильности, но, как обычно, пер-
востепенное значение имеют пространственные факторы. Ниже
рассматриваются только алкиламины. По своему поведению арил-
амины и амиды, например, резко от них отличаются; в этих со-
единениях в конкурентную реакцию с электрофильной частицей
может вступить ароматическое кольцо н карбонильный кислород,
соответственно.
(1) Алкилирование
Примеры нуклеофильного замещения аминов под действием
алкилгалогенндов, алкилсульфонатов и других агентов отмечались
выше при рассмотрении синтеза аминов. Характерная для этих
процессов кинетика второго порядка, стереохимический результат
реакции и чувствительность к пространственным факторам согла-
суются с механизмом S№. Скорость реакции определяется нуклео-
фильностью амина, легкостью ухода замещаемой группы и поляр-
ностью растворителя (поскольку заряд возникает в переходном
состоянии). То обстоятельство, что амины обладают основными
свойствами, может осложнять ход реакции вследствие конкури-
рующих межмолекулярпых реакций отщепления в случае развет-
вленных алкилгалогенндов (уравнение 122), особенно когда в
Р-положепин к уходящей группе имеется активирующая функция,
например фенильная группа.
RsN + ^'CHClbX —> R3NH Х"+/С=СН2
Обычно простые алкены не подвергаются нуклеофильной ата
ке. однако описаны условия катализа, способствующего пр
иению к алкенам, диенам н стиролам (см. разд. • • • •
При действии аминов на алкены, содержащие груп ’ |1е п0
^,е|<тропоакцепторнымп свойствами, происходит пр i пе1ШЯ
Михаэлю, поскольку при образовании промежут i
(122)
создаются условия для лучшей стабилизации отрицательного
ряда (уравнение 123) [1]. Относительная реакционная
R
RNH2 + сн2=сп-с=о
R
I
CH2—СП—С—О
rnh2
за.
способ.
R
CH2_CH2-Lo (123)
RNH
ность при fl-углеродном атоме в алкенах, замещенных подобньи
образом, соответствует реакционной способности самих замести'
телей в реакции с аминами: а,р-пеиасыщенные альдегиды > кет„'
ны > нитрилы > сложные эфиры > амиды > кислоты. Пр0.
дуктом реакции является основание Манниха (см. уравнение 123)
Использование таких оснований Манниха (образующихся в соот-
ветствии с уравнением 51) в качестве исходных соединений в син-
тезе а,₽-непредельных карбонильных соединений демонстрирует
фактически возможности реакции, обратной аминированию алке-
на [24г] (уравнение 124, а также уравнение 7). Присоединение
аминов к простейшим алкинам иллюстрируют уравнения 45 и 46
Нуклеофильному присоединению к активированным алкинам по-
священ обзор [46].
0 сг Г ° 1 0
|| + KCN. Н2О II нем II
PhCCH2CH2NHMe ---------->- l_PhCCH=CH2J -----► PhCCH2CH2CN (124)
кипячение i ' 1
Атака амина по карбонильной группе альдегидов и кетонов
описывается уравнением 125. Промежуточно образующиеся 1-ами-
ноеппрты (68) не всегда удается выделить (см. разд. 6.2.3),одна-
ко детально изучен механизм их образования [149]. Вопросы
общего кислотно-основного катализа в случае таких сложных
реакций, протекающих в водной среде, рассматриваются в об-
зоре Дженкса [150]. В том случае, когда в реакции принимают
участие первичные амины, дегидратация промежуточно образую-
щегося соединения (68) в условиях кислотного катализа приводит
к иминам, в случае вторичных аминов образуются соответствую-
щие имипиевые соли. Значение этих реакций, представляющих
собой первую стадию при восстановительном алкилировании и
в реакциях Манниха, было уже отмечено выше.
R'R2NH +
;с=о
I R2=H
R'R2N—С—ОН ч=*
I
(126)
(68)
„Следует отметить, что существует аналогия между кето-еноль-
ной н нмии-епампшюй таутомерией (уравнение 126).
(126)
62
Епамнны МОЖНО получить ПЧ ВТОрнчпЫХ амИНов и ПОДХОДЯЩИХ
карбонильных соединении в присутствии кислотного катализатов?
например п-толуолсульфокислоты, с азеотропной отгонкой воды
Значение енаминов в синтезе органических соединений рассмот-
рено в разд. 6.2.1.
(2) Ацилирование
Ацилирование аминов путем нуклеофильной атаки по карбо-
нильной группе, содержащей потенциальную уходящею группу
схематически изображено уравнением 127. Эта реакция дает воз-
RNHa + С—О
+ I
RNII2—С—О'
X
I
RX'H—С=О + НХ
(127)
нежность получать вторичные и третичные амиды из первичных
и вторичных аминов. Третичные амины обычно не реакционноспо-
собны, однако в реакциях с хлорангпдрнда.ми кислот образуют
солеобразные продукты присоединения. По реакционной способно-
сти производные карбоновых кислот располагаются в обыч-
ной последовательности: RCOC1 > (RCO)2O > RCOOR >
RCONH2 > RCOOH, причем в отличие от энергичной реакции
аминов с хлорангидрпдамн кислот для реакции солей аминов с
карбоновыми кислотами необходима перегонка. Первичные амины
реагируют со сложными эфирами быстрее, чем аммиак, а в ряду
аминов, для которых стерпческий фактор остается постоянным,
скорость реакции возрастает с увеличением основности амина. Од-
нако пространственные факторы вновь приобретают значение в
случае вторичных п разветвленных аминов, а также в реакциях со
сложными эфирами, имеющими объемистые алкильные замести-
тели; в подобных случаях скорости реакций замедляются. Резуль-
таты исследования Механизмов реакций для ряда карбонильных
соединений приведены в обзоре [10], аминолпзу сложных эфиров
посвящена работа [151]. Более детальное обсуждение этойреак-
цип имеется в ч. 9, однако отдельные моменты, важные в препара-
тивном отношении, следует отметить уже здесь. При использова-
нии таких ацнлгалогенпдов, которые являются относительно сла-
быми ацилирующими агентами, или при ацилировании аминов с
пониженной нуклеофильностью часто необходимо связывать НС1,
чтобы сместить равновесие в сторону образования амида. В реак-
циях со сложными эфирами использовали металлические соли
алкпламидов. Нередко оказывается удобным применять гидрид
натрия в дпполярпых апротонных растворителях [152] (см., на-
пример, уравнение 128). При добавлении амина к смеси сложного
эфира и трехбромнстого бора реакция гладко протекает в мягких
условиях [153].
О
N.H l\ II / \ /.001
--->- \—CNH—( ) <128)
AejSO \/ \ /
63
Ргкомепдуемой обычно перегонки соли амина н карбоцом
• юты можно избежать при использовании Т1С14 или SiCl4
..up 1154 1551. Для создания амидной связи в пептидном
спйтТз" применяли многие другие методы активации карбоксилы
110ЙпХПм1ШФ0ваи»е (амидный обмен) имеет ограниченное пре-
павативное значение, но может быть использовано в реакциях
форчплпроваппя [10] (уравнение 129). Используя гомогенный
катализатор, карбонил рутения, при атмосферном давлении [157J,
удается избежать повышенных давлений, необходимых для фор-
матирования с помощью оксида углерода.
McONa
PhCHjNIh + Alc-NCHO ---> PhCH2NHCMO + Me2NH (129)
При взаимодействии аминов с фосгеном образуются карбамо-
илхлорпды RsNCOCl, однако в случае первичных аминов проис-
ходит отщепление НС1, приводящее к изоцианатам. Изоцианаты
реагируют с аминами, давая производные мочевины (уравнение
130). Эффективными ацилирующими агентами являются кетены;
наиболее часто используется легко получаемый простейший ке-
тен [1, 10] (уравнение 131).
О
RJNH + R2N=C=O —> R|N—С—NHR2 (130)
RNH2 + СН2=С=О —>
О
II
RNHCCHa
(131)
(3) Сульфирование
Амины весьма энергично реагируют с триоксидом серы; одна-
ко в растворе наблюдается более умеренное протекание реакции,
при этом образуются производные сульфамицовой кислоты (урав-
нение 132). Соединения цвиттерионной природы, получаемые из
третичных аминов (уравнение 133), используются как сульфирую-
щие агенты; они образуются также в реакции N-оксидов третичных
аминов (см. разд. 6.1.3.3) с диоксидом серы.
R'R!NH+SO3 —> R1R2NSO3H (R1, R2 = Н, Aik) (132)
R3N + SO3 —> RsN—SO3 (133)
Сульфонилхлорпды реагируют с аминами, давая сульфоиамн-
6 ’ эта Реакц11я лежит в основе классического
пичн^гх ,111тпотРГа’ 11спользуемого для различения первичных, вто-
*онн Xлппи₽Л ичных аМ||НОВ- При действии избытка беизолсуль-
R = Н) РотоПьШ пПДрв“Ч1,ь1й ам,ш получают сульфонамид (69;
щоонч ы °Z „°бра3у£Т волоРаствориму|о соль (70). В случае
реакция приводит к нейтральным сульфонами-
64
дам (69), а третичные амины не реагируют. Препаративное зна-
чение разделения аминов но Гинзбергу иногда преувеличивается.
Na*
PhSO2cl NaOH
R1R2NH ~NaOlI " R'R2N~SO2PI1 - R|=H > R2NSO2Ph (134)
(69) (70)
(4) Галогенирование
Галогенирование аминов но атому азота является окислитель-
ным процессом, требующим источника положительно заряженного
галогена. Реакция ооычно осуществляется с помощью гипогало-
гепита натрия пли соответствующей кислоты; нередко амин в виде
гидрохлорида добавляют к водному раствору гипогалогенита. При
взаимодействии первичных аминов с гипохлоритом или гипобро-
мнтом образуются N-галоген- (71; R1 = Н) или Ы.К'-дигалогенами-
ны (72); в случае вторичных аминов получают N-хлорпроизвод-
ные даже при проведении реакции на холоду.
NaOX NaOX
R'R2NH ------> R'R2NX - > R2NX2 (135)
К* = И
X = галоген (71) (72)
Хорошие результаты дает применение раствора хлорновати-
стой кислоты в эфире или трет-бутилгнпохлорита [7]; предвари-
тельно можно определить концентрации обоих реагентов, чтобы
обеспечить монохлорпроваиие первичных аминов. N-Хлорирование
с последующим дегидрохлорированием действием основания при-
водит к превращению амина в кетон [7] (уравнение 136) (в дан-
ном случае в прегненолон); элиминирование в мягких условиях
[158], имеющее преимущества перед альтернативными способами
элиминирования (по Коупу пли Гофману) дает алкены (уравне-
ние 137),
-"с\ NaOBt
Vhnhci ----*
RZ
Me.
;chnh2
IIOCI, Et2O
NajSO^
,NH2
r2c;
XCH3
Me.
XC=NH
RZ
HOCI. Et2O
Na2SO,
A'C12
r2c/
XCH3
и2о
CuCI
ДУ.сО
R2C=CH2
(136)
(137)
Получение галогенпроизводных (71) нередко удается осуще-
ствить непосредственным взаимодействием с галогенами в буфер-
ном водном растворе. Третичные амины при взаимодействии с
молекулярным бромом (или с хлором) в ССЬ превращаются в га-
логениды четвертичного N-галогеиаммония (73). Однако в присут-
ствии воды происходит элиминирование, а гидролиз образующейся
3 Зак. 105 65
пмшшевоп соли (74) приводит к карбонильному соеД1|11С1111ю
и вторичному амину (75) (уравнение 138).
(CH3)3N (СПз)зЙ-Вг (СНзЙ=СН2 ->
(73) (74)
—> (СНз)А'Н + HCI ю (138)
(75)
N-Xiop- пли М-бромсукцннимиды в эфирном растворе могут
действовать па амины как галогенирующие агенты, по-внднмому,
путем прямого обмена галогена (такой обмен был доказан в слу-
чае днметнламнна и N-хлорсукцпннмнда [159]). Ранее отмеча-
лось, какое большое значение имеют реакционноспособные N-хлор-
прои’зводпые для реакции аминирования, в реакции Гофмана —
Лефлера п в реакции сольволиза, катализируемой нонами сереб-
ра. Эти производные являются источником ионов и радикалов
азота (см. гл. 6.6); подробному рассмотрению методов получения
и реакционной способности N-хлор- и N-бромамниов посвящен
обзор [23].
(5) Нитрозирование
Нитрозированне аминов происходит путем нуклеофильной ата-
ки свободной электронной пары атома азота на соединения, явля-
ющиеся источником иона N0+, например ON—N02, ON—0Н2 или
ON—Hal. Так, например, нитрозирование можно осуществлять,
действуя нитрозилгалогенидом в инертных растворителях типа
эфира, однако более распространенный метод нитрозировання
включает генерирование водной азотистой кислоты HNO2 нз соли
этой кислоты сильной кислотой в водном или уксуснокислом рас-
творе. Сама HNO2 в молекулярной форме не реагирует непосред-
ственно с аминами. В равновесии находится целый ряд частиц;
большинство работ, посвященных детальному изучению механиз-
ма этой реакции, было выполнено на примере нитрозировання
ароматических аминов [10]. Для эффективного протекания нитро-
зирования необходимо наличие свободного амина; при работе
с алкиламинами pH реакционной смеси должен быть не ниже
трех, в то время как ароматические амины, которые (являясь
более слабыми основаниями) содержат в равновесной смеси до-
статочное количество свободного амина могут реагировать и при
более низких значениях pH.
Вторичные амины реагируют гладко (и обратимо), давая
N-нитрозонроизводные, не обладающие основными свойствами н,
как правило, легко выделяющиеся из раствора. Как было пока-
зано выше (см. уравнение 22), подобные интермедиаты, образую-
щиеся при действии алкнлнптритов, являются ключевыми соеди-
нениями при проведении а-алкилировапия вторичных аминов.
Гретнчные амины не реагируют с азотистой кислотой, и с помощью
этой реакции их можно отличить от первичных п вторичных ами-
66
нов. Однако в действительности реакция происходит легко лишь
в том случае, когда присутствует свободный амин (то есть в том
случае, когда значение pH не очень низкое). Промежуточно обра-
зующееся N-ннтрозосоединенне претерпевает расщепление; пре-
имущественно происходит отрыв небольших по размеру алкиль-
ных групп и бензильной группы, при этом образуются вторичные
амины, нитрозируемые избытком реагента (уравнение 139).
\ ONX
nchr2 -----* chr2 —> n=cr2--------*
/ /\ / -O=ci<2
NO
)N—NO
(139)
Наиболее интересны первичные амины, поскольку они реаги-
руют со всевозможными нитрознрующими агентами (уравнение
140). Промежуточно образующееся N-нитрозосОедннениё имеет в
этом случае водород при атоме азота, поэтому возможно новое
направление реакции, приводящее к неустойчивому алкилдиазо-
нневому иону (76). Быстрый выброс молекулы азота сопровож-
дается образованием карбенневых ионов, реакционная способ-
ность и перегруппировки которых [160, 161J подробно рассмотре-
ны в гл. 2.7. Некоторые алкилдиазониевые солн, содержащие
водород при углеродном атоме в a-положении к аминогруппе, пре-
вращаются в нейтральные дназоалканы [162] (см. разд. 6.5.3).
ONX +
RNH2 ---->- RNH2—NO -----> RNHN=O —►
-н+
-—► RN=NOH —> R\'=N —► Продукты дезаминирования (140)
(76)
Для ннтрознровання применяли также N2O4. Применение это-
го реагента при низких температурах позволяет проводить дез-
аминирование первичных аминов, содержащих первичные или
вторичные алкильные заместители, в более мягких условиях, при-
чем вероятность протекания перегруппировки или реакций отще-
пления уменьшается, а выходы возрастают [163]. Арилдиазоние-
вые соли гораздо более устойчивы по сравнению с алкильными
аналогами вследствие резонансного взаимодействия между аро-
матическим кольцом н диазогруппой. Они могут подвергаться
нуклеофильной атаке первичными и вторичными аминами, обра-
зуя трназепы (уравнение 141).
ArN=N + HNR, —> Ar.V=N—NR2 (141)
6.1.3.2, Основные свойства, соли четвертичных
аммониевых оснований и катализ
Протоиироваиие аминов, приводящее к образованию солей,
а также основность по отношению к кислотам Льюиса обсужда-
лись в разд. 6.1.2.3. При алкилировании аминов получаются
з» 67
„ртвептичные аммониевые солп, однако из-за крайней чувствптздь,
ногти этой реакции к пространственным факторам в случае В1ХС
X аииламшюв и разветвленных трналкнламипов она протекает
не всегда легко. В подобных случаях полезным оказывается пр,,
ененне реакционно-способных эфиров трнфторметансульфок„е,
лоты [164], а образованию продуктов реакции, несущих заряд
в значительной степени способствует использование днполярнцх
апротонных растворителей. Несмотря на пониженную пуклеофиль-
ность, пространственно затрудненные амины сохраняют способ-
ность’ связывать протоны (то есть связывать кислоту, образую-
шуюся при алкилировании аминов). Это было удачно использова-
но при проведении реакции исчерпывающей кватернизации
(образование четвертичной соли). Эта реакция иллюстрируется
на примере метилирования чрезвычайно слабого основания (77;
рЛа = 2,6) в присутствии основания (78), не обладающего нуклео-
фильными свойствами (p/G = 11,25) (уравнение 142) [165].
(77) (78)
Примеры напряженных азнрндинневых и азетидипиевых солей
приведены в разд. 6.1.3.6, а вопросу стереоизбпрательности в про-
цессе кватернизации третичных аминов посвящен обзор [166].
Изучен обмен алкильных групп в четвертичных солях [167]; уда-
ление алкильных групп, приводящее к нейтральным аминам, рас-
сматривалось в разд. 6.1.1.2.
Начиная с 1851 г., для получения алкенов стали применять
четвертичные аммониевые соли (расщепление по Гофману) [160,
168]. Расщепление проводят путем нагревания четвертичного
аммониевого основания (обычно получаемого из галогенида дей-
ствием оксида серебра) примерно до 100—150 °C в водном или
спиртовом растворе; использование смеси безводных ДМСО н
ТГФ позволяет успешно провести реакцию н при комнатной тем-
пературе [169]. Четвертичные аммониевые основания полностью
ионизованы и являются столь же сильными основаниями, как
гидроксид натрия; иногда используют алкоксиды тетраалкнламмо-
ния пли солп с другими аннонами основного характера (см., на-
пример, уравнение 159). Классической областью применения этой
реакции было установление структуры неизвестных аминов
с предварительным «исчерпывающим метилированием» (напри-
мер, по уравнению 143).
Реакция протекает по механизму £2; в случае простых соеди-
нений происходит отщепление наиболее кислого н (или) наиболее
доступного атома водорода (правила отщепления по Гофману)
с образованием алкена с наименьшим числом алкильных заме-
ьв
стптелеп (уравнение 144). Показано, что из трео-изомера (79)
образуется траис-а-метилстильбеп (а из эритро-изомера — цис-
а-метилстпльбен); эти результаты согласуются с механизмом
акга-элимпнпроваиия (уравнение 145), который обычно н наблю-
дается.
103%)
♦ -ОН л|----* СН2=СН2 + EtNPr2
(CH3CH2)2N(CH2CH2CH3)2 ----- (1%) (144)
1--->• СН3СН=СН2 + PrNEt.
Ph Me (*NMe3
/СГТ’РЬ
ЕЮ H H
(79)
(145)
Заместители при 0-углеродном атоме, способные к сопряже-
нию, п конформационные факторы влияют па регио- н стереоспеци-
фичность элиминирования (схема 146). Наличие фенильной груп-
пы в (80) и (81) придает Р-водородному атому более кислый ха-
рактер и, кроме того, способствует образованию более устойчивой
двойной связи в продукте реакции. В соединении (80) происходит
онти-элпмиипроваипе, но оно невозможно в соединении (81),
в котором должно осуществляться син-элимпнироваипе через
переходное состояние, для которого в свою очередь характерно
коплаиариое расположение отщепляющихся групп. Различие в ско-
ростях реакций для (80) и (81) (133: 1) свидетельствует о суще-
ственном преобладании ««тп-элпминнровапия над смя-элпмннпро-
ваппем и в то же время ясно показывает, что сии-элимпнпрова-
ппе ие является запрещенным процессом в том случае, когда
ант«-элпмп1шровапие невозможно. Необходимость копланарности
заместителей (двугранный угол, равный 0 пли 180° между отще-
пляющимися группами) подтверждается результатом, полученным
в случае жесткой системы (уравнение 147), для которой наблю-
дается исключительно счи-элнминировапне.
09
При действии сильного основания возможно удаление а-водо-
оодного атома вместо ^-водородного атома в ионе четвертичного
аммониевого основания; обычно при этом получают плпд (82)
[1701 Предположили, что плиды являются промежуточными со-
единениями в ходе расщепления по Гофману, но играют очень
незначительную роль в реакциях, осуществляемых в обычных ус-
ловиях. Например, замена |3-Н на D в (81) приводит не к
DCH2NMe2, как следовало бы ожидать в случае механизма,
включающего илид, а к Me3N в соответствии с нормальным би-
молекулярным син-элпмииированнем. Однако образование плпдов
является основным процессом в тех случаях, когда галогениды
четвертичных аммониевых оснований обрабатывают сильными
металлорганическпми основаниями, например фениллптием (ср.
с перегруппировкой Стивенса, разд. 6.1.1.6); стереоспецифическое
син-элиминированне протекает в соответствии с уравнением 148
[168].
СН3
CH,NR2
(148)
Когда имеются факторы, создающие значительные препятствия
для атаки по |3-водородпому атому, или когда подлежащие отще-
плению группы с большим трудом занимают коплапариое распо-
ложение, происходит конкурирующая реакция образования про-
дуктов замещения (уравнение 149). Если необходимо направить
реакцию в сторону замещения, то лучших результатов можно
ожидать при использовании явно выраженных нуклеофилов, а не
сильных оснований. В соответствии с этим при пиролизе галоге-
нидов четвертичных аммониевых оснований получаются алкплга-
логенпды, а не алкены. С успехом применяют и многие дру-
гие нуклеофилы, но, вероятно, наиболее широко пспольэуют карб-
анионы, обеспечивающие образование повой углерод-углеродной
Me
Io
R—N—Me ~Х
Me
-> RNMe2 + МеХ
(149)
ТО
Амины широко используют в органическом синтезе в качестве
оснований (например, в реакциях дегидрогалогенирования). Для
связывания кислот, выделяющихся в ходе таких реакций, как
этерификация или образование амидов, обычно применяют тре-
тичные амины. Большой успех был достигнут при использовании
тетраалкнламмоииевых солей в качестве катализаторов фазового
переноса при проведении реакций в двухфазных системах. К реак-
циям, которые включают эту форму катализа, относятся генери-
рование днгалогенкарбенов [172, 173], окисление аминов гипохло-
ритом [171], многие реакции нуклеофильного замещения и синтез
изоцнапндов из первичных аминов по Гофману. Другие случаи
применения этого типа реакций приведены в обзорах [172, 173],
в которых обсуждаются и механизмы этих реакций; приводится
также оценка эффективности применения различных солей чет-
вертичных алкнламмониевых (и фосфониевых) оснований
[174].
Простое объяснение описанных выше реакций иллюстрируется
схемой (150), в которой показано превращение алкилбромида
RBr в производное RX (где, например, Х = цианид, азид, тиофе-
ноксид). Анион X- переносится ионом четвертичного аммониевого
основания Q+ из водной фазы в органическую, которая содержит
алкилбромид. Подвергшийся замещению бромид-ион возвращает-
ся затем в водную фазу, чтобы вновь принять участие в реакцион-
ном цикле. После завершения реакции RX может быть выделен
из органической фазы с помощью минимального числа операций.
Значение этого подхода заключается в возможности успешного
проведения в мягких условиях реакций между такими соедине-
ниями, как неорганические солп п нейтральные органические
молекулы, причем очевидно, что отпадает необходимость в высо-
ких температурах и в специальных растворителях (которые под-
ходили бы для обоих компонентов). Не возникают трудности и
при выделении продукта реакции.
Водная фаза Na+ X’ + Q+ Br’ Q+ X + \та+ Вг
------------------1!-----------------— (I50)
Органическая RX + Q+ Br <— Q+ X + RBr
фаза
Q+ = mou четвертичного аммониевого основания
Дальнейшее развитие этого метода применительно к реакции
замещения атома брома в алкнлбромидах на цпаннд-нон связано
с применением гетерогенного катализатора (полистирол, с кото-
рым связана соль четвертичного аммониевого основания), Посколь-
ку катализатор не смешивается ни с водной, ни с органической
фазой, этот метод получил название «трехфазного катализа»
[175].
71
6.1.3.3. Окисление аминов
Длины легко окисляются. Возможны многочисленные варцаи
ты проведения этого процесса, и продолжают появляться всё
новые и новые реагенты, обладающие большей избирательностью
нлн повышающие эффективность синтезов. Основные методы
окисления аминов описаны в этом разделе, а более подробны^
сведения можно найти в работах [1, 10].
[О] |OJ |О)
RNH2 ---г [RNHOH] ----* 1RNO] ---> RNO2 (15|)
[О,
r2nh —> r2noh (152)
|OJ
R3N > P-\’->0 (153)
Уравнения 151 —153 в обобщенном виде представляют «при-
соединение кислорода» к аминам, например, при действии пер-
окснкислот п пероксида водорода. Гпдрокснлампн и нптрозопро-
изводные являются промежуточными соединениями при получе-
нии алкилнитросоеднненпй (с.м, гл. 7.2) из первичных аминов;
дналкилгидроксилампны п N-окснды образуются соответственно
из вторичных и третичных аминов (см. гл. 6.4). N-Оксиды могут
быть снова превращены в исходный амин действием восстанови-
телей, например трпфенилфосфина.
Термический распад аминоксидов (элиминирование по Коупу
[160]) происходит очень легко; ио характеру протекания его мож-
но сравнить с реакциями элиминирования, описанными в
разд. 6.1.3.2. Распад подчиняется закономерностям механизма £,
(уравнение 154); циклическое пятпчленпое переходное состояние
должно быть плоским и лишь в немногих случаях конформацион-
ные ограничения препятствуют успешному и стереоспецифичному
протеканию син-элимипирования, приводящего к алкенам с высо-
ким выходом. Условия реакции являются мягкими; при использо-
вании в качестве растворителя диметилсульфокспда и тетрагид-
рофурана температура реакции может быть понижена до комнат-
ной. В случае бензил- и аллиламппов, не содержащих атомов
водорода при p-углеродном атоме, происходит перегруппировка
Мензенгеймера [176] (уравнение 155), которая очень похожа па
перегруппировку нлидов азота по Стивенсу (см. разд. 6.1.1.6).
Ряд данных свидетельствует в пользу несинхронного [1,2]-сдвига,
PhCH—ЙМе2
Н2С*) 07 ---► PWCH=CH2+Me2NOH
PhCH2—NMe2 —PIiCH2ONMc2 (155>
О’
72
однако термическая перегруппировка N-оксидов N-аллиламинов
[177] (уравнение 156) связана с протеканием синхронных [2,3]-
спгматропных процессов.
н
Me2NO Me
(.156)
Хотя описанные выше реакции окисления обычно можно осу-
ществить в соответствующих условиях, в случае первичных и вто-
ричных аминов часто сталкиваются с затруднениями, если угле-
родный атом, расположенный рядом с атомом азота, связан с во-
дородом. Удаленпе всех атомов водорода, например, при действии
перманганата в нейтральном или щелочном растворе может при-
вести к иминам, енаминам, нитрилам (уравнения 157 и 158) или
их производным.
R*=H
R'R2CHNH2 —> R'R2C=NH --------> R‘CsN (157)
RCH2CH2Nr2 —> RCH=CHNR2 (158)
Способы окисления весьма разнообразны: диоксид марганца [178]
используют для превращения третичных N’-метнламинов в N-фор-
мильные производные и первичных аминов в альдегиды; превра-
щение аминов в альдегиды и кетоны осуществляют с помощью
сульфоннлнероксндов при низких температурах [179], гипохлори-
та (в присутствии катализаторов фазового переноса) [171J и
солен золота п палладия [180]. Тетраоксид рутения избирательно
окисляет метиленовые звенья, находящиеся в а-положенин к тре-
тичному атому азота, с образованием карбонильной функции; эта
реакция лежит в основе часто используемого метода деграда-
ции [181].
Леонард с сотр. использовали соли ртути [24в] для окисления
третичных аминов в соли нмпння (которые в условиях реакции
могут находиться в равновесии с таутомерной енампнной фор-
мой). Реакция протекает через стадию ангп-элпмннпрования в
промежуточно образующемся комплексе типа (83). Обычно обра-
зуется импнпевая соль с наибольшим числом заместителей, часто
ее можно выделить в виде перхлората. Эти соединения являются
электрофильными агентами, находящими применение в синтезе.
На примере синтеза производного азацпклоионана показана ну-
клеофильная атака нмпнпевых солен (уравнение 159); синтез
включает стадии кватерпнзацпп и направленного элиминирования
по Гофману. Гидролиз нмпнпевых солей (уравнение 160) при-
водит к удалению алкильных заместителей в амине; эта реак-
ция обратна реакции восстановительного алкилирования (см.
разд. 6.1.1.2) и является ключевой стадиен в интересном синтезе
алкалоида спартеина (84), в котором дважды используется реак-
ция Манниха (уравнение 161).
73
1.Реакция
Манниха
2 Восстановление
(161)
Окисление первичных аминов тетраацетатом свинца протекает,
по-видимому, через промежуточное образование нитренов [182]
(см. разд. 6.6.4). Этим методом из N-аминофталнмида и был по-
лучен нитрен, фигурирующий в уравнении 113.
6.1.3.4. Кислотные свойства аминов
Основность аминов довольно подробно обсуждалась в
разд. 6.1.2.3, однако аминогруппа первичных н вторичных аминов
способна также отдавать протон, проявляя тем самым свойства
очень слабой кислоты. Изучены [183] ароматические амины, ими-
ды и аналогичные соединения (в тех случаях, когда сопряженное
основание стабилизуется путем взаимодействия с соседней л-сис-
темой), однако для алкилампиов простейшего строения, по-видн-
мому, таких данных очень мало. Значения рКа Для аммиака И
диэтпламниа составляют соответственно 34 и 36, что соответствует
кислотности протонов метильной группы толуола. Слабая кислот-
ность аминов означает, что сопряженные нм основания обладают
значительной силой; действительно, алкнлампды щелочных метал-
74
лов являются сильнейшими из известных гетероатомных основа-
ний. Однако металлалкилы превосходят их по основности и это
обстоятельство позволяет получать металлические соли аминов
(уравнение 162).
RjNH + MR2 r'n- M+ + r2h (162)
Эти основания представляют значительный интерес для син-
теза, особенно в тех случаях, когда одна или, лучше, две алкиль-
ные группы R являются вторичными или третичными и, таким
образом, экранируют анионный центр на азоте, препятствуя при-
ближению электрофильных частиц, за исключением частиц, ми-
нимальных по размеру, например протона *. От использования
простых диалкнлампдов перешли к таким соединениям, как диизо-
пропиламнд лития (85), дицнклогексиламнд лития (86), 2,2,6,6-
тетраметилпиперндид лития (Li—ТМП) (87) и 1,1,3,3-тетраметил-
М-(1-этилцпклогекспл)бутиламид лития (88).. Все эти вещества
(85) (86) (87) (88)
получаются довольно легко, хотя из-за пространственных затруд-
нений синтез соединения (88) путем реакции исходного амина с
метиллптием продолжается в течение нескольких часов. Эти
(и им подобные) соединения представляют собой эффективные
основания, не обладающие нуклеофильными свойствами и способ-
ные избирательно отрывать протоны от субстратов, которым свой-
ственна чрезвычайная чувствительность к нуклеофильной атаке
обычными основаниями. Ненуклсофнльные основания имеют еще
одно преимущество: они обладают высокой растворимостью
Даже в неполярных растворителях. Критический разбор особых
достоинств различных иеиуклеофпльных оснований имеется в об-
зоре [184]; ниже приведены примеры, ясно демонстрирующие
преимущества оснований этого типа.
Ценность аринов как промежуточных соединений в органиче-
ском синтезе снижается в тех случаях, когда атака арниов осно-
ваниями, используемыми для их получения, происходит гораздо
быстрее, чем другими реагентами, вводимыми в реакционную
среду. При попытках осуществить реакцию, изображенную
.. * Подобные основания были названы протонными гарпунами
0 011,1 напоминают скорее жертву гарпуна, кита, устройство поло Р
I 1 о таково, что он может поглощать планктон для удовлетворения своего ч\до
нциото аппетита, по ие может заглатывать крупных рыб.
75
n vmBiieiiiiii 163, С помощью амида натрия u о-бромаппзола полу-
ирны лишь продукты осмолеиия. При действии псиуклеоф11ЛЬ||щ
оснований па бензилхлориды могут образоваться оеизилкарбспы;
их присоединение к алкенам или алкинам приводит соответствен’
но к циклопропанам пли циклопропенам с хорошими выходами,
причем реакция не осложняется конкурирующим процессом бен-
зилирования основания. В уравнении 164 представлен пример
превращения, которое, как и следовало ожидать, не удается осу-
ществить с помощью традиционных оснований (реактивы Гринь-
яра, алкоксиды, алкиллптий); однако при использовании нену-
клеофильных оснований реакция протекает успешно с выходами
до 90%. Эти основания имеют еще одно очевидное преимущество,
заключающееся в том, что в процессе обработки реакционной
смеси амин может быть легко отделен от нейтральных продуктов
реакции и регенерирован.
,ОМе ,ОМе
Ы-ТМП. LICsCPh / \
е \—С1 ---------------------> е 2—CsCPh (163)
(8О»/о)
CO2Et
основание _ PhCOCl I
Et,CHCO2Et *• Et2CCO2Et------------------->- PhCOCEb (164)
Повышенная основность солей аминов с успехом используется
в реакциях анионной полимеризации алкенов и в процессе обмена
водорода па дейтерий. Металлические соли аминов сравнимы с
3-аминопроппламидом калия (89), необычным «сверхоснова-
ннем» (поскольку в качестве противоиона выступает ион калия),
которое является, по-впдимому, внутренне сольватированным
[185]. Последнее обстоятельство исключает снижение активности
за счет ассоциации, которое наблюдается в случае многих других
сильных оснований, поэтому реакционная способность (89) в реак-
циях изомеризации (при использовании в качестве растворителя
сухого исходного диамина) в 1О5—1О6 раз выше, чем реакцион-
ная способность трет-бутокепда калия в ДМСО и примерно равна
реакционной способности диметиламида лития в гексаметнлтрн-
амидофосфате [186]. Другим интересным примером «внутренне
сольватированного» основания является соединение (90), образу-
ющееся при действии гидрида калия на моноаза-18-краун-б-эфнр
[187]. Образование тесной ионной пары в этом соединении невоз-
можно из-за координации иона калия в полости макроцикла.
Краун-эфпр (90) используется для проведения реакций 1,2-элп-
минпрованпя н обнаруживает высокую растворимость в углеводо-
родах типа толуола.
В растворах щелочных металлов (как правило, натрий пли
лнтнн) в жидком аммиаке имеются сольватированные электроны,
тн реагенты являются эффективными восстанавливающими сис-
76
к*
(89)
темями многоцелевого назначения, которые особенно часто при-
меняли для восстановления сопряженных молекул (восстановле-
ние по Берчу) и которые сыграли громадную роль в сннтетнче-
ских исследованиях [188].
R = H, Aik, AlkO
Циклогексадиены-1,4 образуются из производных бензола в
условиях кинетического (а не термодинамического) контроля,
обусловленных присутствием спирта, который препятствует созда-
нию концентрации а.иид-нонов, достаточной для протекания ката-
лизируемой изомеризации в диен-1,3 н, следовательно, восстано-
влению до циклогексенов. Электронодонорные заместители (R =
алкнл, алкокси) в ароматическом кольце оказываются в положе-
ниях, не подвергшихся восстановлению (см. уравнение 165),
а электропоакцепторные группы оказываются у атомов углерода,
подвергшихся восстановлению. Таким образом, когда R в (91)
является алкоксигруппой, гидролиз простого эфира енола (92)
водной уксусной кислотой приводит к р,у-непредельному кетону
(действие минеральной кислоты вызывает смещение двойной свя-
зи и образование сопряженной системы). Преимущество системы
литий — алкиламины заключается в возможности работать при
комнатной температуре (и более высоких температурах), но эти
системы действуют более энергично и с меньшей из иратель
КОСТЬЮ.
77
6.1 3.5. Введение защитных групп в амины
и их удаление [4,7,189, 190]
Аминогруппу нередко требуется защищать, если она М0Жм
быть затронута в условиях реакции, проведение которой цеобх^
днмо для модификации функциональных групп в каких-либо дпу
гих частях сложной молекулы. Необходимым требованием к лю
бой защитной группе является легкость н избирательность
введения и удаления, причем, желательно, в мягких условиях
Основной интерес к защите аминогруппы связан с важностью
этой процедуры для пептидного синтеза (см. гл. 23.6), но мы це
намерены связывать методы защиты аминогруппы только с этой
частной областью. Ниже рассмотрены примеры защитных групп
относящихся к различным классам органических соединений'
включая, по мере возможности, новые методы защиты. Мы не
будем подробно останавливаться на реакциях, которые были опи-
саны в других разделах этой главы.
(1) Амиды
Простейшей реакцией, служащей целям защиты аминогруппы,
является, вероятно, превращение в амид; обратным процессом
является гидролиз. Широко используются простые ацилпропзвод-
ные (формильные, ацетильные, бензоильные); галогенацетильные
группы, подобные трпфторацетильной, расщепляются в настоль-
ко мягких щелочных условиях, что регенерацию свободного ами-
на можно осуществить с помощью ионообменных смол полнампн-
ной природы [189]. Другие защитные группы (и методы их удале-
ния) были упомянуты в связи с рассмотрением синтеза аминов;
это циклические имиды, арилсульфонильпые, трифторметилсульфо-
ннльные н фосфорильные группы (см. уравнения 8—12)./1-Толуол-
сульфонильная (тозильная) группа была использована в реак-
ции, описываемой уравнением 90. Эта группа может быть избира-
тельно введена в амины в присутствии ОН-группы с помощью пер-
хлората М-метил-Ы-тозилпирролидиния [191]. Новые методы уда-
ления сульфонильных защитных групп связаны с использованием
действия света [192], раствора серной кислоты в уксусной кис-
лоте [193] пли бис(2-метокснэтокси)алюмогндрида натрия [194].
О
II
C1CH2CNHR
о-диамннобепзол
о
(166)
78
Интересные методы удаления защитных групп, включающие
внутримолекулярное участие соседних групп, были применены
в случае ацильных защитных групп [189j, таких, как а-хлораце-
тпльная группа (уравнение 166); защитная группа (93) (получае-
мая из 94) «приводит к амидам с лабильностью сложноэфирной
группы» [195] (уравнение 167). у 1
RNH2
(93)
CONHR
CHzOCOPh
(94)
CHjONa.
СНзОИ. 20‘С
(167)
(2) Уретаны
Бензилоксикарбоннльная и трег-бутилоксикарбонцльная груп-
пы могут быть введены в амины с помощью соответствующих
хлорформиатов или азпдоформнатов (уравнение 168).
О Q
II у
ZCOR3 ||
R'R2NH ~цп „п. > R'R’N'COR3 (168)
НВг, НОАс
R2 = Н, Aik; Z = CI, N3; R3 = Ph, PhCH2, трег-Ви
Для удаления этих защитных групп применяли различные спо-
собы. Гидролиз происходит в более мягких условиях, чем гидро-
лиз амидов (см. разд. 6.1.1.5); можно применить также восстано-
вительное расщепление (см. разд. 6.1.1.2). Для введения защит-
ной группы рекомендуется также применение аллнлхлорформиата
[196]; удаление аллплокснкарбонильной группы осуществляется
с помощью системы Ni(CO)4 — MesNCHzCH’NMe, ДМФ
Н2О. Уретан (95) устойчив к действию кислот п оснований; спо-
соб регенерации амина из подобных соединений приведен в урав-
нении 169 [197]. 9-Флуоренилметпльная группа может быть
О
(169)
0 CH2OCNHR
9-аптрил—СЩСО ХО2. ДМФ
RNH2 . ГУ-*
CHSS- »* -20 °C, Д.ЧФ
(95)
79
удалена путем растворения защищенного амина в жидком амМ11_
'[190] а-Бромбутилокснкарбонпльные производные устойчцвь, '
инертных растворителях, таких, как бензол; преимуществом это,°
защитной группы является чувствительность к обычному сольво'
визу. Удаление защитной группы сольволизом в этаноле объясни
ется соучастием соседней группы (уравнение 170) [190].
О О
О СН2Вг
II I
RNHCx /СМе,
О О о
Eton +
—> rnh=c;
rnh2 + о=с:
(ПО)
Очевидно, что при превращении вторичных аминов в амиды не
остается свободных NH-rpynn, а первичные амины, как правило,
дают амиды или уретаны, которые хотя и являются менее реак-
ционноспособными, но все еще обладают свободной NH-группой.
Интересный вариант защиты аминогруппы, который (подобно об-
разованию имида) связан с образованием двух новых связей с
атомами углерода, представлен в уравнении 171.
(3) Имины
Аналогичный результат получают в случае иминов, образу-
ющихся из первичных аминов и бензальдегида или других альде-
гидов. Арнлиденовые производные, стабилизованные за счет обра-
зования водородной связи, более устойчивы в условиях восстано-
вительного удаления защитной группы, чем обычные бензилидено-
вые производные (уравнение 172) [189, 198].
(4) Алкильные и другие группы
Бензплгалогениды реагируют с аминами, давая моно- (или
ди-) бензильные производные. Бензильная группа может быть
удалена каталитическим гидрогенолизом (см. разд. 6.1.1.2), а
также другими методами, например действием натрия в жидком
аммиаке. Бензоильная защитная группа может быть восстаповле-
80
„ Лрнзплъпую (метод восстановления — гм „„
бывает еще одни путь к получению бензил ам^о’Г’кпо25’’ ЧТ0
т превращение лежит в основе восстановитец, пг“’ роме того>
Гения бензоильной группы. Аллилхлорформиа'т может^ы?.УДЭ’
„ользоваи для удаления не только бензильных защитных rnv*40’
" и простых алкильных групп (см. уравнения 20 2П йЙРРУ ’
Тпцфенилметнльного (тритильного) заместителя
легко осуществляется действием трнтплхлорида и основан"7
пример пиридина; эту защитную группу можно удалить мягким
кислотным гидролизом или каталитическим гидрогенолизом Вве
дение в ароматические кольца тритильной группы заместителей’
повышающих устойчивость карбениевого иона, еще более увечи’
чивает чувствительность этой группы к кислоте, а большой объем
этой защитной группы может также способствовать уменьшению
уязвимости других заместителей, находящихся в а-положеиии к
аминогруппе, по отношению к нежелательным воздействиям [189].
6.1.3.6. Реакции азиридинов
Многие реакции циклических аминов (с небольшим размером
цикла) и циклических N-алкпламинов аналогичны уже описан-
ным реакциям вторичных и третичных аминов. Однако напряже-
ние, существующее в малых циклах, в частности в азиридинах
(энергия напряжения 113 кДж/моль), приводит к гораздо более
легкому раскрытию цикла путем расщепления связей С—N и,
иногда, С—С. Ниже описаны некоторые реакции азиридинов,
протекающие с сохранением и раскрытием цикла. Поведение ази-
ридинов обычно заметно изменяется при наличии у атома азота
заместителей (таких, как ацил н фенил), которые могут вступать
в сопряжение со свободными парами электронов; однако такие
амиды п ариламнны здесь подробно рассматриваться не будут.
Имеющиеся полные обзоры, посвященные азиридинам [75] и цик-
лическим аминам [74], содержат более подробное описание при-
веденных ниже реакций.
Более выраженный s-характер свободной пары электронов
атома азота в N-алкилазиридинах (см. с. 55) не приводит к
сколько-нибудь существенным качественным изменениям характе-
ра нуклеофильности. С протонами и кислотами Льюиса азпрндп-
пы образуют четвертичные аммониевые соли; однако эти соли,
как правило, являются реакционноспособными по отношению
нуклеофильным агентам, и их можно выделить лишь в тех <
аях, когда используются растворители и противоионы р
Рат, тстрафторборат и т. д ) со слабыми нуклеофильными сво^
генчт"1’ Сол“ а311Рпдиния могут быть синтезированы ₽ 173);
о.,„ЭТ1,Лам1,11°в с помощью перхлората серебра (>Р синте-
зе ЯВля10тся обычными промежуточными соедини ы в
рСа. *ех слУчаях> когда азот играет роль У430,1'85.1,^ в^обзоре
‘ Щиях замещения (например, уравнение 174) I
61
[200] обобщены работы в области
ЗЭРЯД' AgClO,
r2nch2ch2ci —77—*
малых гетероциклов,
песУЩ11.х
-ст
(174)
N-Алкилироваиие вторичных азиридинов с помощью алкилга-
логенидов п аналогичных соединений требует присутствия основа-
ния (часто используется избыток триэтнламина пли неорганиче-
ского основания, например карбоната калия) для быстрого н не-
обратимого связывания выделяющегося галогецоводорода пли
других продуктов кислотной природы. Таким образом исключает-
ся' катализируемое кислотой раскрытие цикла. Нуклеофильное
присоединение вторичных азиридинов к альдегидам и кетонам
происходит обычным образом, а присоединение по Михаэлю к
а,p-непредельным карбонильным соединениям и нитрилам, а так-
же к стиролам осуществляется в отсутствие кислоты или основа-
ния (уравнение 175). В реакциях раскрытия оксиранового цикла
(уравнение 176) азиридины ведут себя как нуклеофильные части-
цы, причем азиридиновый цикл сохраняется, однако в некоторых
случаях реакция может осложняться полимеризацией.
(175)
(176)
Реакции ацилирования, сульфонилирования и фосфорилирова-
ния при атоме азота обычно осуществляют действием соответ-
ствующего хлорангндрпда (или заменяющего его реагента) при
низких температурах и в присутствии основания. N-Галогеназн-
риднны получают в обычных условиях (см. разд. 6.1.3.1); их ис-
пользовали в синтезе 1,Г-дпазнридлнов (уравнение 177), вводя в
реакцию с литиевыми производными азиридинов (ср. разд. 6.1.3.4).
итрозирование азиридинов действием нитрозилхлорлда при ииз-
62
ких температурах приводит к ожидаемым N-иитрозопропзводным
которые устойчивы только при низких температурах и быстро’
разлагаются даже при температурах ниже комнатной, претео.щ
вая стереоспецифическое превращение в соответствующие алкены
(уравнение 178). Аналогичное стереоспецифичное дезаминирова-
ние можно осуществить с помощью дпфторамина. Р
R R
NOCl.Etp
Н -78°С
R R ,
R R
н'-Л/’-'н
N—NO
(178)
J Нн
Во многих случаях можно провести модификацию заместите-
лей, не затрагивая азиридиновый цикл, который устойчив к
реагентам типа LiAlH.(. Даже избирательное каталитическое гид-
рирование ненасыщенных заместителей может происходить без на-
рушения целостности азиридинового цикла, который, однако, мож-
но раскрыть при использовании соответствующего’ катализатора
в особых условиях (уравнение 179). При гидрогенолизе С-алкил-
азирпдинов наблюдается преимущественное расщепление связи
между атомом азота и наименее замещенным углеродным атомом
цикла; в С-арилпроизводных разрывается связь между атомом
азота и углеродным атомом, несущим арильный заместитель.
Н2. катализатор I I
(179)
NR
H NHR
кетонной группе (заместитель) осуще-
циклпческой структуры; $к2-замещение
наличии хороших уходящих групп про-
Реакции Гриньяра по
ствляются без нарушения
у углеродных атомов при
исходит также, как правило, с сохранением цикла. Незамещен-
ный азиридиновый цикл раскрывается нуклеофильными агента-
ми, однако эта реакция протекает медленно (уравнение 180).
В тех случаях, когда при атоме азота имеется ацильная пли дру-
гая группа, способная стабилизовать возникающий отрицатель-
ный заряд, реакция происходит быстрее. Заметное влияние кис-
лого катализатора на повышение скорости расщепления связи
С—N можно уяснить, обратившись к уравнению 181. Так, рас-
крытие цикла в (96) под действием амина в отсутствие катализа-
тора (уравнение 182) (которое иллюстрирует также обращение
конфигурации при атоме углерода, подвергшемся нуклеофильной
атаке) можно противопоставить быстрой реакции азиридинов с
водной кислотой при обычных температурах и носящей характер
взрывного процесса полимеризации, происходящей при оораоотке
незамещенного азиридина кислотой. В связи с этим часто 1,ео *
«"МЫ особые меры предосторожности, чтобы исключить услов;
оторые могут способствовать полимеризации при пров
Реакции с азиридинами.
83
H;NEt. H2o
120°C, 16дней
II Me NIIEt
/—^41
EtNH Me
(182)
NEt
(96)
Раскрытие цикла протекает в основном по механизму Sn2
(раскрытие незамещенного азиридина водой полностью отвечает
этому механизму), однако не исключается и некоторый вклад
5м1-механизма, в том случае, когда расположение алкильных или
других заместителей способствует стабилизации образующегося
карбениевого иона. Описание реакции раскрытия азиридинового
цикла с точки зрения «чистых» SnI- и 5н2-механпзмов не дает
удовлетворительного объяснения соотношения двух возможных
продуктов реакции, получаемых при раскрытии несимметрично
замещенных азиридинов, однако обычно легко решить, какой из
двух углеродных атомов цикла «более чувствителен к Sn 1-атаке»
(например, бензильный углерод или углерод, несущий большое
число алкильных заместителей), а какой — «более чувствителен
к 5м2-атаке» (наименее замещенный углерод). Можно ожидать,
что изменение полярности растворителя или нуклеофильности
атакующих групп будет обычным образом влиять на характер
разрыва и образования связей в переходном состоянии; во многих
случаях можно удовлетворительно объяснить результаты реак-
ш'г использовании сильного нуклеофильного агента
(NCS-) 5»2-замеи1ение происходит при первичном углеродном
атоме (уравнение 183). Если разрыв связи опережает образование
новой связи, атаке подвергается наиболее «5^1-чувствительный»
углеродный атом (уравнение 184).
Me Me
Ph
Me
IISCN | +
-----» NCSCH2CNH3 SCN
I
Me
Ph
ИС1, u2o 1 t
---------> C1CI1CH2NH3 СГ
(183)
(184)
84
Раскрытие цикла в азиридинах под действием воды, галоген-
,.ул<ащкх кислот и других реагентов не имеет большого нрена-
С° tBiioro значения, так как 0-амнноспнрты или 0-галогенамины
Рат1 сами являются исходными соединениями в синтезах азири-
,,aCQB (см. разд. 6.1.1.7). Больший интерес представляет раскры-
Д11" азнрнДн,,0В0Г0 чикла аминами, в результате чего получают
диамины, а также использование других нуклеофильных аген-
тов (уравнение 185).
R2COCI
О
1
C1CH2CH2NRCR2
(185)
Азиридины могут претерпевать многие перегруппировки, при-
водящие к расширению цикла [74, 75, 201]; отдельные примеры
образования 4-, 5-, 6- и 7-членных циклов приведены в уравне-
ниях 186—189. Во всех случаях основным фактором, способству-
ющим перегруппировке, является уменьшение напряжения цикла.
Расширение азиридинового цикла с образованием азетндннона
(уравнение 186) не относится к типичным перегруппировкам
[202], однако перегруппировки впннлазиридинов, приводящие к
3-пирролннам (уравнение 187) могут происходить при нагревании
[203] или в условиях катализа нуклеофильными реагентами, на-
пример иодпд-ноном [204]. Сольволиз N-хлоразирпдинов [205]
COON а
Ви-трет
(186)
SOC12, 25 °C
NaH, ТГФ
представлен в уравнении 188; были тщательно исследованы ката-
лизируемые нонами серебра реакции конфигурационно устойчи-
вых N-хлоразиридинов [70]. К термическим электроциклическим
реакциям относится перегруппировка Коупа (уравнение 189) и
заключительная стадия в уравнении 190 [206]. Предыдущие ста-
Д|1И этой схемы демонстрируют сохранение азиридинового цикла
в ходе осуществления реакций с использованием реаген
85
потенциально спосооных раскрывать цикл. Однако эти рСаГе
принимают участие в более быстрых реакциях, проиСходЯЩ11"Т«
других частях молекулы.
(189)
ЛИТЕРАТУРА
1. «Meihodicum Chimicum», vol. 6, «С—N Compounds», ed F. Zymalkowski,
Academic, New York, 1975, pp. 439—597.
2. «The Chemistry of the Amino Group», ed. S. Patai, Interscience, London,
1968.
3. «Methoden der Organischen Chemie (Houben-Weyl)», vol. XI/I, 4th edn.,
E. Muller, Thieme, Stuttgart, 1957.
4. /. T. Harrison and S. Harrison, «Compendium of Organic Synthetic Methods»,
Wiley, New York, vol. 1 (1971) and vol. 2 (1974).
5. C. A. Buehler and D. E. Pearson, «Survey of Organic Synthesis», Wiley-In-
terscience, New York, 1970.
6. H. Goldwhite, in «Rodd’s Chemistry ol Carbon Compounds», 2nd edn.,
_ e,d- Coffey- Elsevier, Amsterdam 1965, vol. lb, chapter 6.
7. L. F. Fieser and M. Fieser, «Reagents for Organic Synthesis», Wiley, New
York, 1967—1977, vol.s. 1—6 [Физер Л. Физер M. Реагенты для органиче-
ского синтеза. Т. 1—6. М., Мир, 1970—1975.]
о. Р. A. S. Smith, «The Chemistry of Open-Chain Nitrogen Compounds», «Ben-
jamin, New York, 1965, vol. I, chanter 2.
9. M. S. Gibson, в cc. 2, chapter 2, p 37.
10. В. C Challis and A. R. Butler, в cc. 2, chapter 6, p. 277. , „
* 1969 ^42 ’ Ш^'а1таЬе' T' pupta’ and T- P- Pan- Bul1- c,lcirL Soc’ Ja|’ ’
!?' S F Hennion and R. S Hanzel, J. Amer. Clicm. Soc., 1960, 82, 4908
7 7Snt£a!s,a’ S 'P Murahashi, and I. Morltani, Tetrahedron Letters,
919 ’ Gibson and R- W'. Bradshaw, Angew. Chem. Internal. Edn., I9b .
Ik V Mukaiyama and T. Taguchi, Tetrahedron Letters, 1970, 3411.
. B. Hendrickson and R. Bergeron, Tetrahedron Letters, 1973, 3839.
86
17
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
Л. Zwierzak and J. B. Piotrowicz, Angew. Chem. Internal Edn 1977 16 107
H. Zimmer, M. Jayawant, and P Outsell, J. Org. Chem., 197o’, 33, 2826
У. Ohsluro, M. Komatsu, and T. Agawa, Synthesis 1971 89
/. D. Hobson and J. G. McCluskey, J. Chem, Soc. ’(C) 1967 2015
Я. A- ?^0П' R C Sc'mUr’ L BumS’ and J' B- P^e< Tetrahedron Letters
1977, 1507- ’
D. Seebach and D. Enders, Angew. Chem. Internal. Edn 1975 14 15
P. Kovacic, M. K. Lowely, and K. IR. Pield, Chem. Rev 1970 70 639 '
H. 0. House, «Modern Synthetic Reactions», Beniamin, Menlo Park Califnr
nia, 1972, (a) p. 71; (6) p. 81; (в) p. 401; (?) p. 655. ’ Г
H. C. Brown and P. Heim, J. Org. Chem., 1973, 38, 912.
R, F. Borch, Tetrahedron Letters, 1968, 61.
A. Rahman, A. Basha, N. Waheed, and S. Ahmed, Tetrahedron Letters 1976
219.
IV. Umino, T. Iwakuma, and N. Itoh, Tetrahedron Letters 1976 763
J. A. Secrist III and M. HZ. Logue, J. Org. Chem., 1972, 37, 335.'
E. J. Corey, K. C. Nicolaou, R. D. Balanson, and Y. Machida Synthesis 1975
590.
J. F. Knifton, J. Org. Chem., 1975, 40, 519.
M. Rabinovitz, in «The Chemistry of the Cyano Group», ed. Z. Rappoport
Interscience, London, 1970, p. 307.
N, M. Yoon and H. C. Brown, J. Amer. Chem. Soc., 1968 90, 2927
R. F. Borch, J. Org. Chem., 1969, 34, 627.
S. Yamada, K. Tomioka, and K. Koga, Tetrahedron Letters, 1976, 61.
R. Rausser, L. Weber, E. B. Hershberg, and E. P. Oliveto, J. Org. Chem 1966
31. 1342, 1346.
J. F. Archer, D R. Boyd, W. R. Jackson, M. F. Grundon, and It7. A. Khan
J. Chem. Soc. (C), 1971, 2560.
R. F, Borch and A. I. Hassid, J. Org. Chem., 1972, 37, 1673.
B. L. Sondengam, J. H. Hemo, and G. Charles, Tetrahedron Letters, 1973,
261.
S. H. Pine and B. L. Sanchez, J. Org. Chem., 1971, 36, 829.
H. 117, Gibson, Chem. Rev., 1969, 69, 673.
M. P. Cooke Jr. and R. M. Parlman, J. Org. Chem., 1975, 40, 531.
J. P. Kulney, G. B. Fuller, R. Greenhouse, and I. Itoh, Synth. Comm., 1974,
- 1 gg
J. Schlott, J. C. Falk, and K- W. Narducy, J. Org. Chem., 1972, 37, 4243.
Ц7. Kruse and R. F. Kleinschmidt, J. Amer. Chem. Soc., 1961, 83, 213.
/. Miller and R. Tanaka, in «Selective Organic Transformations»,
B. S. Thyagarajan, Wiley-Interscience, New York, 1970, vol. 1, p. 143.
Hassner, Accounts Chem. Res., 1971, 4, 9 и приведенные там ссылки.
Hasser, R. P. Hoblitt, С. H. Heathcock, J. E. Kropp, and M. Lorber,
J. Amer. Chem. Soc., 1970, 92, 1326.
С. H. Heathcock, Angew. Chem. Internal. Edn., 1969,-8. 134.
H. C. Brown and J. T. Kurek, J. Amer. Chem. Soc., 1969, 91, o647-
A. F. M. Iqbal, Helv Chim. Acla, 1971. 54, 1440.
S. J. de Solms, J. Org. Chem., 1976, 41, 2650.
G. Kinast and L.-F. Tietze Angew. Chem. Internal. Edn., 1976. 15, 239.
N. L. Holy and Y. F. Wang, J. Amer. Chem. Soc., 1977, 99, 944.
N. Yoshimura, I. Moritani, T. Sliimamura, and S.-I. Murahashi, J. Amer.
Chem. Soc., 1973, 95, 3038. „ „ „ . ,„„r rhom
M. И7. Rathke, N. Inoue, K- R. Varma, and H. C. Brown, J. Amer. Che .
Soc., 1966, 88, 2870. u ,
Y. Tamura, J. Minamikawa, and M. Ikeda, Synthesis, 1У77, „
H. C. Brown M. M. Midland, and A. B. Levy, J. Amer. Chem. Soc., 19/3,
95, 2394.
R. H. Mueller, Tetrahedron Leiters, 1976. 2925.
G. Alvernhe and A. Laurent, Tetrahedron Letters, 1973 I0W.
E. J. Corey R. J. McCaully, ami II. S. Saclidev, J. Amer. Chem. 00c,
92, 2476.
4,
7?.
С.
S.
ed.
Л.
A.
₽ л navis and P A. Mancinelli, J. Org. Chem., 1977, 42, 398.
G.ATs^l4hashi, S. Iriuchijinia, and K. Manila, Tetrahedron Letters,
^O'Connor Quart. Rev , 1970, 24. 553.
L A Carpino and D. E. Barr, 5. Org. Chem., 1966, 31, 764.
D V. Banthorpe, в co. 2, chapter 10, p. 58o.
P G Gassman and R. L. Cnjberg, J- Amer Chem Soc., 1969, 91, 2047
C Kaiser and J. Weinstock, Org. Synth., 1971, 51, 48.
p Kovacic, M. K. Lowely, and P. D. Roslcos, Tetrahedron, 1970, 26, 529
p G Gassman, Accounts Chem. Res., 1970, 3, 26.
A R Lepleti and A. G. Giunianini. in «Mechanisms of Molecular Minratin™,
ed. B. S. Thvagarajan, Wiley-Intcrsciencc, New York, 1971, vol. 3. b
S H Pine, Org. Reactions, 1970, 18, 403.
F. D. Ollis, M. Rey, I. 0. Sutherland, and G. L. Class, J. C. S. Chem. Comm
1975, 543.
R. Livingtone, in «Rodd’s Chemistry of Carbon Compounds», 2nd edn
ed. S. Coffey, Elsevier, Amsterdam, 1973, vol. IVA.
О. C. Denner and G. E. Ha/n, «Ethylenimine and Other Aziridines», Academic
New York, 1969.
1. Shahak, К Utah, and J. Blum, Tetrahedron Letters, 1976, 4003.
/. Lengyel and J. C. Sheehan, Angew. Chem. Internal. Edn., 1968, 7, 25.
K. Kotera and K. Kitahonoki, Org. Synth., 1968, 48, 20.
G. Alvernhe, S. Arsenyiadis, R. Chaabouni, and A. Laurent, Tetrahedron Lei-
ter 1975 355
K. Ichimura and M. Ohta, Bull. Chem. Soc. Japan, 1967, 40, 432.
IV. DeKimpe, R. Verge, L. DeBuyck, and N. Schamp, Synth. Comm., 1975, 5,
269.
L. Duhamel and S.-Y. Valnot, Tetrahedron Letters, 1974, 3167.
A. Zwierzak and K. Osowska, Angew. Chem. Internal. Edn., 1976, 15, 302.
R. M. Duperyre and A. Rassat, Tetrahedron Letters, 1973, 2699.
R. A. Perry, S. C. Chen, В. C. Menon K. Hanaya and Y. L. Chow, Canad.
J. Chem, 1976, 54, 2385.
L. S. Hegedus, G. F. Allen, and E. L. Waterman, J. Amer. Chem. Soc, 1976,
98, 2674.
В. M. Trost and J. P. Genet, J. Amer. Chem. Soc, 1976, 98, 8516.
H. Yamanaka 1. Kikui, K. Teramura and T. Ando J. Org. Chem, 1976, 41,
3794.
«Nitrenes», ed. W. Lwowski, Wiley, New York, 1970.
P. Scheiner. in «Selective Organic Transformations», ed. В. C. Thyagarajan,
Wdey-Interscience, New York, 1970 vol 1, p 327
A. B. Levy and H. C. Brown, J. Amer. Chem. Soc. 1973, 95, 4067.
7 P°rt°gl‘ese and D. T. Sepp, Tetrahedron, 1973, 29, 2253. „
, G Hartmann and D. A. Robertson, J. Amer. Chem. Soc, 1972, 94, 2758.
W. Funke, Angew. Chem. Internat. Edn, 1969, 8, 70.
A. S. Isaacs, Chem. Soc. Rev, 1976, 5, 181.
I л Klassen and A. Hassner, Chem. Rev, 1976, 76, 389. . и
iqrt апЧлгм' Allen lr-- and M- J- Broadhurst, J. Amer. Chem. Soc.,
ijii, Уо, 4oO<5.
/ r ^mass ,a'\d Р,- Tweddle, J. C. S. Perkin I, 1977, 874.
s 1Э73.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
эд f г IYYI— ГТ И’ J' ‘ с, Ь. Perkin I, 19//, 8/4.
100 А р м„ Хап Le"sen, J. Org. Chem. 1974, 39, 564.
’ G R' “j IJ'™’ J- Or« Chem> 1975' 4°’ 255L t Edn
1974 13’ fI0C/'"!^’ a"d M. R. Hoffman, Angew. Chem. Internal. Edn.,
816^ C°Pe' Dryden, and C. F. Flowed, Org. Syntli. Coll. Vol. 4, 1963,
^'^^'t^science^^w York^Tg^^voI.^e'^^lbT^
W. D yCur°i8saDanAd p - n Cra"1’ J' Anicr- Chem. Soc, 1976, 98, 3038.
Comm., 1975,’835 U,t!'er> Л F- s‘°ddart, and G. II. Sones, J. C. S. Chem.
103.
104.
105.
88
106 / IF Smith, в cc. 2, р. 161.
;07; L. МJackman and F. A. Cotton, «Dynamic N. М. R. Spectroscopy», Acade-
mic New Уогк, ty/o. rj
108. J. в. lambert, «Topics inI Stereochemistry», ed. N. L. Allinger and E. L. Eliel
Wilcy-Intcrscience, New York, 1971, vol. 6, p. 19. ’
109 J M. Lehn, Fortschr. Chem. Forsch., 1970, 15, 311
110. ! R- Wiseman, H. O. Krabbenhoft, and R. E. Lee, J. Org. Chem., 1977, 42,
HI. R. Annunziata, R. Fornasier, and F. Montanari, J. C. S. Chem. Comm., 1972
112. R- s. Atkinson and J. R. Malpass, J. C. S. Perkin I, 1977, 2242
[13. EMorishima and K. Yosliikawa, J. Amer. Chem. Soc., 1975 97 2950
[14 V. Rautenstrauch, .1. C. S. Chem. Comm., 1969, 1122.
115 С. H. Bushweller, W. G. Anderson, P. E. Stevenson and J IF O'Neil I
Amer. Chem. Soc., 1975, 97, 4338. ' '
116. P. J- Crowley, M. J. T. Robinson, and M. G. Ward, Tetrahedron 1977 33
915. ' ’ ’
117. I. D. Blackburne, A. R. Ratritsky, and У. Takeuchi, Accounts Chem Res [975
8, 300.
118a. Z. B. Lambert and S. I. Featherman, Chem. Rev., 1975, 75. 611.
[186.7. B. Lambert and S. A. Khan, J. Org. Chem., 1975, 40, 369.
119. F. A. L. Anet and I. Yavari, J. Amer. Chem. Soc., 1977, 99, 2794.
[20. FL Booth and M. L. Jozefowitz, J. C. S. Perkin II, 1976, 895.
121. L. I. Bellamy, «Infra-Red Spectra of Complex Molecules», 3rd edn., Chapman
and Hall, London, 1975.
122. L. M. Jackman and S. Sternhell, «Applications of NMR Spectroscopy in
Organic Chemistry», 2nd edn., Pergamon, Oxford, 1969.
123. IF. H. Pirkle and S. D. Beare, J. Amer. Chem. Soc., 1969, 91, 5150.
124. В. C. Mayo, Chem. Soc. Rev., 1973, 2, 49.
125. A. F. Cockerill, G. L. O. Davies, R. C. Harden, and D. M. Rackham, Chem.
Rev., 1973, 73, 553.
G. C. Levy and G. L. Nelson, «1SC Nuclear Magnetic Resonance for Organic
Chemists», Wiley-Interscience, New York, 1972; J. B. Slathers, <13C NMR
Spectroscopy», Academic, New York, 1972.
Fl. Eggert and C. Djerassi, J. Amer. Chem. Soc., 1973, 95, 3710.
A. Ahond, M.-M. Janot, N. Langlois, G. Lukacs, P. Potier, P. Rasoanaivo,
M. Sangare, N. Neuss, M. Plat, J. Le Men, E. IF. Hagaman, and E. Wenkert,
J. Amer. Chem., Soc., 1974, 96, 633.
K. Jankowski, J. Org. Chem., 1976, 41, 3321.
«15N NMR», ed. M. Witanowski and G. A. Webb, Plenum, Pondon, 1973.
H. Paulsen and IF. Greve, Chem. Ber., 1970, 103, 486.
J. M. Schulman and T. Venanzi, J. Amer. Chem. Soc., 1976, 98, 6739.
К Markowski G. R. Sullivan, and J. D. Roberts, J, Amer. Chem. Soc., 1977,
99, 714. , ,
H. Budzikiewicz, C. Djerassi, and D. H. Williams, «Mass Spectrometry ol
Organic Compounds», Holden-Day, San Francisco, 1967.
R. E. Carhart and C. Djerassi, J. C. S. Perkin II, 1973, 1753.
F. Rerek and G. Snatzke, Angew. Chem. Internal. Edn 19/,a, 14, 109
II. E. Smith J R. Neergaard, Ё. P. Burrows, and F.-M. Chen, J. Amer. Chem.
Soc., 1974, 96. 2908. „ , . , T , h . , p)
B. Liberek J. Ciarkowski K. Plucinska, and K. Stachowiak, Tetrahedron Let
tors, 1976, 1407.
R- IF. Taft and L. S. Levitt, J. Org. Chem., 1977, 42, 916.
R. IF. Taft, in «Proton Transfer Reactions», ed E. Caldin and Got ,
Chapman and Hall, London. 1975, chapter 2.
E- M. Arnett, в cc. 140, chapter 3. т r. royg 98,
Л H. Aue, IE M. Webb, and M. T. Bowers, J. Amer. Chem. So .,
L C. Speakman. «The Hydrogen Bond and Other Intermokcular Forces ,
Chemical Society, London, 1975.
126.
127.
128.
131.
132.
133.
134.
135.
136.
137.
138.
139.
140.
141.
142.
143.
89
«The Hydrogen Bond» ed- P- Schuster. G. Zandet, «ml C.
^Encyclopedia oi Pharmacology and Therapeutics», secti r
vol 1 «Structure-Activity Relationships», ed. C. J. Cavallito, Perganton
fF4rS and K. Eysell, «Biologically Active Amines Found in Man» p„
gamon. Oxford, 1969.
r r F Banks в cc. 2, chapter 9, p. 499.
r A koan «An Introduction to the Alkaloids», Blackwell, Oxford tear
° M |ay“r’, B. Pinsky, A. Sclionbrunn, and W. Washtien, J. Amer. Cfiem
Soc. 1974, 96, 7998.
Ц7 P lencks Chem. Rev., 1972, 72, 70o.
T D Singh and R. It7. Taft. J. Amer. Chem. Soc., 1975, 97, 3867.
В Singh Tetrahedron Letters, 1971, 321.
И Yazawa К Tanaka, and К. Kariyone, Tetrahedron Letters, 1974,3995
T H. Chan and L. T. L. Wong, J. Org. Chem., 1969, 34, 2766.
J O Wilson and H. Weingarten. Canad. J. Chem.. 1970, 48, 983.
S Yamada Y. Kasai, and T. Shoioiri, Tetrahedron Letters, 1973, 1595.
j' I Byerley G. L. Rempel, N. Takebe, and B. R. James, J. C. S. Chem
Comm., 1971, 1482.
J. A. Tonnis, P. Donndelinger, С. K. Daniels, and Kovacic, J. C. S. Chem.
Comm., 1975, 560.
T. Higuchl and J. Hasegawa, J. Phys. Chem., 1965, 69, 796.
E. H. White and D. J, Woodcock, в cc. 2, chapter 8, p. 407.
C. J. Collins, Accounts Chem. Res., 1971, 4, 315.
Al. Regilz, Synthesis, 1972, 351.
D. H. R. Barton and S. C. Narang, J. C. S. Perkin I, 1977, 1114.
J. Burden and V. C. R. McLoughlin, Tetrahedron, 1965, 21, I.
H. Z, Sommer, H. I. Lipp, and L. L. Jackson, J. Org. Chem., 1971, 36, 824.
A. T. Bottini, in «Selective Organic Transformations», ed. B. S. Thyagarajan,
Wiley-Interscience, New York, 1970, vol. I, p. 89.
T. H. Lane and J. L. Speier, J. Org. Chem., 1976, 41, 2714.
J. L. Coke, in «Selective Organic Transformations», cd. B. S. Thyagarajan
Wiley-Interscience, New York, 1972, vol. 2, p. 269.
J. E. Hofmann, T. J. Wallace, and A. Schriesheitn, J. Amer. Chem. Soc., 1964,
86, 1561
/ Zugarauescu and M. Petrovanu, «N-Ylid Chemistry», McGraw-Hill, New
York, 1976. J
',70 Г V Loe, and П H- Freedman, Tetrahedron Letters, 1976, 1641.
И, , n “ehmlow, Angew. Chem. Internal. Edn., 1974 13, 170
173. J. Dockx, Synthesis, 1973, 441.
174. A. W. Harriott and D. Picker, J. Amer. Chem. Soc. 1975, 97, 2345.
17Г о л '^7 J J- Лтег- chem- Soc- 1976, 98, 6270.
„L ; (,°,h,nsl?n^ in «Mechanisms of Molecular Migrations», ed. B. S. Tltya-
177 I &W' ey’ nn7Cience’ Ncw York> 1969. vol. 2, p. 249.
78 A I k ' t?da- a'ld Y- ,пои«е- J- c- s- Chem. Comm., 1973, 848.
ITO d и и У?’ Synthesis, 1976, 133.
inn м г „'[man, J. Amer. Chem. Soc., 1976 98 6702
181’ О' ВеНом"с ap‘l T'S'-Hc11’ 1 Org- Chtm-, '976, 41, 2742.
Chem ?OT6 il 27ВД m’ Morlacchi- N- and V. Tortorella, J. Org.
183 T Quart- Rcv., 1971, 25, 407
'84. R. A MO^so°n and С Лтсг Chem- Soc- l97fl' 98’ 3399' s 6RI
582. ' " and C' Dougherty, J. Amer. Chem. Soc., 1973, 95, 581.
। Й^Г?Г^ С5'“тС','™72СОзГ1'4975' 222
У' °Vol^’ l970er(lS6tre'ra!ledrOn LettCrS’ 1977’ 32t
Y. W0Jmn„, B cc 2> chap(er (j ^66g
144.
145.
146.
147.
148.
149.
150.
151.
152.
153.
154.
155.
156.
157.
158.
159.
160.
161.
162.
163.
164.
165.
166.
167.
168.
169.
170.
185.
186.
187.
'89. v: Woiman,
90
190.
191.
192.
193.
194.
195.
196.
197.
198.
199.
200.
201.
202.
203.
204.
205.
206.
I Л. Carpino, Accounts Chem. Res., 1973, 6, 191
K. Kamata. S. Kosuda, and Y. Ban, j, c. S. Chem Comm., 1972,
A. Abad, D. Meiller, J. P. Pele, and C. Porlella, Tetrahedron Letters 1971
4555. ’ 1
P. D. Carpenter and M. Lennon, J. C. S. Chem. Comm 1973 664
£ H. Gold and E. Babad, J. Org. Chem., 1972 37 2208
B. F. Cain, J. Org. Chem., 1976, 41, 2029.
E. J. Corey and J. VY. Suggs, J. Org. Chem., 1973 38, 3223
A. Kornblum and A. Scoll, J. Org. Chem., 1977, 42, 399
B. G. Ramsay and R. !. Sloodley, J. Chem. Soc. (Q 1971 3864
C. F. Hammer S R Heller, and 7. H. Craig, Tetrahedron’ 1972 28 239
D. R- Crist and N. J. Leonard, Angew. Chem. Internal. Edn 1969 R 069
H. VY. Heine, in «Mechanisms of Molecular Migrations» ed В S Thvaat'
rajan, Wiley-Interscience, New York, 1971, vol. 3 p. 145 У g
7. A. Deyrup and S. C Clough, J. Amer. Chem. Soc., 1969 91 4590
R S. Atkinson and C. W. Rees, J. Chem. Soc. (C) 1969 778 ’
P. Scheiner, J. Org. Chem., 1967, 32, 2628.
D. C. Harwell and C. W. Rees, J. C. S. Chem. Comm., 1969, 1428.
E. Vogel, IV. Prelzer, and IV. A. Boll. Tetrahedron Letters, 1965, 3613.
6.2. ПОЛИФУНКЦИОНАЛЬНЫЕ АМИНЫ
ДЖ. М. 3. ГЛАДЫХ, Д. ХАРТЛИ (Allen and Handbury’s Research Lid., Ware
Herljordschire}
6.2.1. НЕНАСЫЩЕННЫЕ АМИНЫ
6.2.1.1. Енамины
В 1954 г. в практику органического синтеза был введен до-
вольно общий метод препаративного алкилирования п ацилирова-
ния карбонильных соединении с помощью третичных енаминов
(1) [1, 2]. Начиная с этого времени было выполнено значитель-
ное количество работ по химии енаминов; проблеме енаминов по-
священы обзоры [3—7].
(1) Методы получения
Из альдегидов и кетонов. Наиболее общим методом получения
енаминов является конденсация альдегидов и кетонов со вторич-
ными аминами (уравнение 1). Были описаны многочисленные
модификации этого метода. Для повышения скорости реакции бен-
зол может быть заменен более высококипящимн растворителя-
ми— толуолом или ксилолом, применение которых также способ-
ствует азеотропном)' удалению воды [8, 9]. Могут быть также
использованы такие кислые катализаторы, как п-тол}олс\льфо
кислота [10]. Для удаления воды используют н осушающие аген
ТЬ|> например карбид кальция [И] пли молекулярные сита 1
R3 R4 R3
R'R-'NH + О=С—CH—R5 r'R2N—С=С—R' + H2O (О
(О
91
Источником амина в описываемой реакции служат „е то
ап,,с амины. Например, циклогексанон при кипячении с ‘ °
стметилтрпампдофосфатом (3) в течение 40 мин превращает*"’
значительной степени в 1-двметнлампноцпклогексеи (2)
Тетракис(днметиламино)титан (4) гладко превращает 3-мети ’
бутанон-2 в енамин (5) [14]; этот метод особенно пригоден 2
синтеза енаминов из пространственно затрудненных кетонов „
тля синтеза ендпампнов из N.N-диалкнлацетамидов [15].
NMe2
NMe2
Mc2N—Р=О
I
NMe2
NMe2
I
Me2N—Ti—NMe2
I
NMe2
(2) (3)
NMe2
)
CH2=C—CHMe2
(4)
Rs
R\* ।
\j=C—CHR4R5
R2/
X'
Из иминиевых солей. Иминиевые соли типа (6) реагируют с
основаниями давая енамины (1), и, следовательноц-методы их по-
лучения взаимосвязаны (см. также разд. 6.2.5.1). Алкилированию
иминов с помощью алкнлгалогеиидов посвящен обзор [16].
На приведенном ниже примере показано, что алкилирование
имина (7) приводит к пмнниевой соли (8), которая при действии
основания превращается в енамин (9) [17] (уравнение 2).
аминов через енамииов из циклических третичных
ботан Леонардом и сотг,°"'°е полУчеШ1е иминиевых солей разра-
' Р’ в результате систематического изучения
окисляющего действия ацетата ртути п„
показано на примере превращения х« ,L. e"t,llle этого метоп,
дегидрохинолизидии (11) прн дей °а,^ади«а (|0) в Д1^а
водной уксусной кислоте [18J (уравнеш е 3) Ртути в 5%-ной
Из лактамов. Пяти- и шестичленные циклические N-алкиллак-
тамЫ, например (12), прн взаимодействии с реактивами Гринья-
ра дают пирролины (13а) и пииеридеины (136) [4]. Эта реак-
ция открывает возможность синтеза циклических енаминов, кото-
рые нельзя получить другими способами (уравнение 4).
/ RMgX z—(СН2)п
°=\ /---------------” 'М > (4)
NMe NMe
(12) (13а) n = 1
(136) л = 2
Частичное восстановление ароматических аминов. При исполь-
зовании восстанавливающей системы литии — н-нропиламин джу-
лолидпи (14) восстанавливается до енамина тетрагндроджулоли-
дина (15), а I-метил-1,2,3,4-тетрагидрохпнолин (16) в тех же усло-
виях превращается в смесь енаминов (17) и (18) [19] (урав-
нения 5, 6).
(2) Структура и свойства
енамина можно представить две мезомервые
свободная электронная пара атома азота мо-
с л-электронами двойной связи. Для электро-
BUd.uOmnOCT.’i: по атому азота
рвом случае образуется енам.мо-
--------------------------- /901
93
Структура. Для
формы, в которых
W перекрываться u .. ~„«.„nPTn
фильнои атаки открываются две в з.
или ПО Р-углеродиому атому; в пер*”--- /20).
Ччевая соль (19), во втором — нмичне
к 1954 г. появилось сообщение [1] о том, что метилппПн
енамина (21) с последующим кислотным гидролизом при₽в°0Вд^
ПРИВОДНТ
R
(20)
моноалкплкетону. Очевидно, что эта реакция включает промежу-
точное образование енамина (23) из имннпевого попа (22), кото-
рый далее не алкилируется. Образование изомерного енамина
(24) исключается по пространственным причинам [8], так как в
этом случае имело бы место ощутимое взаимодействие между ме-
тильной группой и метиленовой группой, соседней с атомом азо-
та, при условии сохранения перекрывания между свободной элек-
тронной парой азота и двойной связью. Было высказано предпо-
ложение [20], что метильная группа в (23) должна иметь акси-
альную ориентацию, поскольку в альтернативной конформации
метильная группа была бы почти копланарной с метиленовой
группой, соседней с атомом азота, и в значительной степени пре-
пятствовала бы атаке по этому центру. Позднее аксиальная
ориентация метильной группы в (23) была однозначно доказа-
(21) (22) (23)
Спектроскопия.
в ИК-спектрах
1630—1660
сдвиг
94
(24)
Валентные колебания двойной связи енамшю»
см-1 loo ЯД1Яе^ся в ВИде интенсивной полосы пр
па 20—50 гм-1 Эта полоса обнаруживает пшеохромнь
в том случае, когда протонировав!
енаминов по 0-углеродиому атому приводит к обпЯзо«я
ветствующих имничевых солей. Указанное смевши ИЮ с00т’
ся [22-24] для определения положения двой^й Ис|,ользует-
телыю атома азота. Это смещение не наблюдает а СВЯЗИ Отиоси-
ие лротопируются при Р-углеродном атоме ’ есЛИ ена“ииы
планарности и электронного перекрывания в А 0ТсутстВия ко-
В УФ-спектре наличие двойной связи сопп„ еме енамина.
азота, вызывает батохромный сдвиг полосы поХиТ С атол,о‘!
телыю ее положения для насыщенных аминов ; ’Я о?ТНОСИ'
е 3000). Вследствие этого максимум попюшрД "акс 215 нм-
расположен прн 230 +10 нм, а их коэсЬЛииие ДЛЯ енам,1НОв
Р(е 5000-9000) обнаруживает гиперхромны? эффект
особенности спектров могут быть объяснены с точки ™ b Э™
крывания свободной электронной пары азота и пере’
двойной связи. Р а и ^-электронов
Спектры протонного магнитного резонанса енямчип
родным атомом также имеют характерный вид. Положение™^
ческого сдвига а-винильного протона отражает степень Хект
вания между свободной электронной парой атома азота Тдвой-’
нои связью [26]: чем меньше степень перекрывания, тем в более
сильном поле наблюдается резонансный сигнал протона Няппи
мер, сигнал вннильного протона енамина (21) расположен ппй
б 4,17 (в млн ; в более слабом поле относительно тетраметнт
силана). Этот сигнал смещается в слабое поле в область 6 4 55-
4,62 в случае аналогичных енаминов - производных морфолина и
пиперидина, но наблюдается при 6 5,24 в случае N-феннлена.мина
(25), в котором свободная электронная пара атома азота вовле-
кается в сопряжение с фенильной группой, так что степень пере-
крывания с двойной связью енамина оказывается очень незначи-
тельной.
(25)
Свойства. Степень проявления енаминами ос я.япкпль'ных
видимому, зависит от наличия или отсутствия а- и iР -
групп. а-Алкильные заместители повышают ° ’енамп-
алкильные группы понижают ее [27]. Стевенв лыл0 устаиовле-
иов была измерена для растворов в хлороформ, , ( ОСНОва-
ио, что енамины являются в 10-30 раз ол енаМ1|На иро-
ниями, чем исходные вторичные аМ11нь';/пппт01111р0ВаНная форма
зонируется в ‘первую очередь, н эта_^прото^ у, дат
может перегруппировываться в Ср-прото1 Р в аминную
Рая затем легко гидролизуется, превращаясь
„ карбонильную составляющие. При алкплирова.......епампновп,,
хпорнафто.хиноном образуется пурпурная окраска; эта реакция
ла предложена как удобная качественная проба на енамины [28]
(3) Реакции
Алкилирование. В прнципе алкилирование енаминов молод
осуществляться как по углероду, так н по азоту, но на практике
как правило, протекает С-алкплпрование. Легкость алкилирова-
ния определяется структурой енамина п, в частности, его основ-
ностью и легкостью образования тригонального атома в переход-
ном состоянии, а также природой алкилирующего агента и раст.
верителя.
Енамины, производные ацетальдегида или моиозамещенных
ацетальдегидов, обычно не алкилируются [29], поскольку в усло-
виях алкилирования осуществляется их самоконденсация. Однако
в тех случаях, когда енамины содержат объемистые заместители,
продукты С-алкилнровашгя могут быть получены с хорошими вы-
ходами [30]. Например, енамин (26) первоначально N-алкнлиру-
ется бензилбромпдом, но дальнейшее нагревание с последующим
гидролизом приводит к продукту С-алкилпрования (27), вероят-
но, за счет межмолекулярной реакции (уравнение 8) [31].
Me
I. л |
Me2NCH=CMe2 + PhCH2Br -----------> PhCH2CCH=O (8)
2, I ндролиз ।
Ме
(26) (27)
Прямое С-алкилирование происходит при действии аллил- и
пропаргилгалогепндов, а также алкилгалогенидов на енамины,
получаемые из циклических кетонов; реакция чувствительна к по-
лярности растворителя [32].
Алкилирование енамина обладает преимуществами по сравне-
нию с непосредственным алкилированием кетона, поскольку в
первом случае главным продуктом реакции обычно является мо-
ноалкпльное производное [8, 32]; однако при использовании силь-
ного органического основания и избытка алкилгалогепида преоб-
ладает процесс диалкилироваиия [32]. Так, к примеру, реакция
енамина (21) с аллилбромпдом (2 моль) в присутствии дпцпкло-
гекенлэтиламина с последующим гидролизом образующегося
алкилированного енамина приводит к 2,6-диаллилцнклогексаноиу
(28) с почти количественным выходом (уравнение 9).
О
сн2=снсн-.
(21) —>
•СН2СГ1=СН2
(9)
(28)
96
В диенаминах, например (29), имеются т.
«еиия для электрофильной атзки; атсп, Яя три возможных поло
в р. и 6-положениях. Однако реакции ироХ," атомы
редь при P-Углеродном атоме. Так, иапр^Т В ”ервУ’° °че
дпенамина (29) с метилиодидом в ДМфР в случае реакции
кристаллическая N-метиламмониевая соль тчы ™ 1|) °®Разхется
одиако нагревание смеси енамина (241 „ °' с вь1ходом ’929f>-
шении 1:1, или соли (30), в дМф\ ’ Лв соотно-
ла, 70 ч) при последующем гидролизе" дает тпп, Паян11ая аМву-
реакции - С-алкилпрованный дикетои /з > Один пР°Дукт
экспериментальных данных четко доказывает ' Ряд дРУ™х
в результате диссоциации соли (30) с послеХш пР°НСХОдит
ваяием. сдующим С-алкилиро-
о
о
о-
(29)
Арилирование.
лом, в результате
получают 2-арилкетон (32) [34].
Ме
(31)
Енамин (21) реагирует с 2,4-динитрохлорбензо-
чего после гидролиза с очень высоким выходом
О
(21)+ С1
>—no2
(Ю)
;—NO2
no2
NO2
(32)
Ацилирование. Ацилированию енаминов посвящен обзор [35].
Енамины ацилируются по углероду или азоту, но обычно проис-
ходит С-ацнлирование. Поскольку енамины являются более силь-
ными основаниями, чем ацилированные енамины, половина взято-
го количества енамина превращается в соль, однако потерю ена-
мина можно предотвратить путем добавления в реакционную
смесь органического основания, например трнэтпламина [36].
Ацилирование енаминов, полученных из альдегидов, протекает
нормально в том случае, если енамин добавляют к хлорангидрн
ДУ кислоты [37]; добавление енамина (33) к ацетплхлорид} в
эфире приводит к образованию пмпнпевон соли (34), при гидрол
Зе которой получают кетоальдегид (35) с выходом оо /0.
С Г СОМе СОМе
ofV.CH=cMe3 Ме_,С—СНО (П)
(38)
4 Зак. log
(34)
(35)
97
нтма й-дикарбонильных соединений ацнлироВаН)1с
Помимо с11НтеаРТР путь К получению самых разнообразных
енаминов ««Р’^пбоциклпческцх в гетероциклических СИСТем
ациклических, кароо ц реагирует с дикетсном и Вслед.
|35]. Например, Д 'атому в дненаминовои системе об-
ствие атаки по р Р ,37) (уравнение 12) (38]. Кроме того
разуется 7<8-'ll,ri'0T гРлсроДиому атому енамины реагируют с ИЗо’_
тизамсшеиные 1 ₽ . д ла’КтаМы [39, 40]. Так, например, 0
цианатами, ДаваЯ Р поонсходит экзотермическая реакция с фе.
случае енамина (2b) ронс^ образуется р-лактам (38), К0-
Ш1ЛПЗоиианатом,"В ™ анилида (39) [31] (уравне-
торын можно прогпдролизовать до
вне 13).
Me-
(86)
NMe2 _
р—|=СН2
(12)
Me
I
Me—С—СО—NHPh
I
сно
(39)
Енамины
Me
PhNCO Me____________
Ме2ХСН=СМе2-------->- I
Me2N-----NPh
|=O
на
Н20
(13)
простои
соедине-
(26) (88)
Гидролиз, Этой теме посвящен обзор [41]. ......
структуры легко гидролизуются, образуя карбонильное
нне и вторичный амин. Результаты, полученные при изучении ско-
рости гидролиза .4-(2-метилпропеннл) морфолина (40) в разбав-
ленных фосфатных буферных растворах, объясняются с точки
зрения равновесия, существующего между свободным енамином и
енаммонневым ионом (уравнение 14). В условиях этой реакции
Ме2С=СН—HNy у + Н2О Ме2С=СН—+ Н3О+ (14)
(40)
двойная связь енаммоииевого иона не подвергается электрофиль-
ной атаке, поскольку свободная электронная пара азота уже не
взаимодействует с двойной связью. Было показано, что гидролИЗ
енамина (40) в интервале pH 4,1—9,5 подчиняется закономерно
стям общего кислотного катализа. Кинетические измерения, про-
веденные для сходного по строению енамина (41) в водных аце-
татных буферных растворах н в разбавленной хлорной кислоте,
казали, что общий основной катализ имеет место и в слабокис
лои среде.
Галогенирование. Бромирование енамина (42) протекает изби-
рательно [42] н приводит к монобромеиамину, при гидролизе ко-
торого образуется а-бромкетоп (43). При прямом бромировании
кетона, синтетического предшественника енамина (42), происхо-
дит замещение как в а-положении к кетогрунне, так и в аромати-
ческом кольце. При действии на еиамииы N-бром- и N-хлорсук-
цинимндов получают р-галогененамины [43].
ОМе ОМе
(42) (43)
Реакции с галогенцианами. Енамин (21) вступает в реакцию
с хлорцианом (I экв) в присутствии трпэтилампна (1 экв) в
диоксане, в результате чего после гидролиза промежуточно обра-
зующегося цианенамнна получают 2-цианоииклогексанон (44) с
хорошим выходом [44].
CN
(21)
(44)
Реакции с альдегидами. Получаемые из циклических кетонов
енамины реагируют с алифатическими и ароматическими альдеги-
дами, давая с хорошими выходами 2-моноалкилиденовые произ-
водные кетонов [45]. По-впдимому, первой стадией реакции яв-
ляется образование диполярного иона (46), который после пере-
носа протона теряет молекул)' воды, превращаясь в диенамин
(47), а последний при гидролизе дает непредельный кетон (48)
(уравнение 17).
О'
4*
99
„иклоприсоедииения. Енамины — производные 11ИКЛи.
Реакции никло Р И]1 е аз11ДаМ1| образуют производна
веских кетонов- leiiaMiii| (49) реагирует с «-ннтрофеинлаэп-
Тма3давая трназолин (50), который ирн действии кислоты 11ре.
^р щаетм <51) (УР‘”ие,8> I46’'
(13)
NOj
(48)
(50) (51)
Получаемые из циклогексанона енамины в условиях хорошо
известной реакции [47, 48] присоединяют дпхлоркарбеи, давая
аддукт состава 1 : 1. Енамин (49) реагирует таким образом с
дихлоркарбеном в интервале температур от —10 до —20 °C; в
результате реакции образуется устойчивый аддукт (52), при тер-
мическом разложении и гидролизе которого получают кетон (53)
(уравнение 19).
(53)
Алкилсульфоннлхлориды, имеющие атом водорода в «-положе-
нии, также присоединяются к енаминам, являющимся производ-
ными альдегидов и циклических кетонов, с образованием цикли-
ческих сульфонов. Например, при взаимодействии енамина (49)
С ''ета|^У;|ьФО|1НЛХЛОР!!дОм получают сульфон (54) [49] (урав-
SOs
(49) + MeSOsCl
+-
ен
(54)
npH?inManMviiacT^e реа.КцИИ’ Енамины н диенамины могут
ных 2 ,п Фотох11М11',есК11Х реакциях; примеры
Р 1 щений приведены в уравнениях 21—23 150—521.
100
(20)
также
подоб-
(a) R=H, n=4
(6) R=H, n=5
(в) R=Me, n=4
(21)
(22)
(23)
Восстановление. Двойная связь енаминов легко восстанавли-
вается с помощью борогидрида натрия или алюмогидрида лития;
при этом получают насыщенные амины [53].
6.2.1.2. Аллиламины
В этом разделе рассматриваются Методы получения, свойства
и реакции простых аминов, содержащих аллильную группировку,
например аллилампна (55), который является характерным пред-
ставителем этого класса соединений.
(1) Методы получения
При реакции аллилхлорпда (56) с водным раствором аммиака
или аминами в присутствии хлорида медп(1) образуются аллил-
амины с выходами 36—90% [54]. Аллиламин может быть полу-
чен также (выход 64%) обработкой аллилхлорпда (56) гуаниди-
ном с последующим щелочным гидролизом (уравнение 24) с вы-
делением продукта реакции перегонкой [55].
NH
|| NaOH
СН2=СНСН2С1 + NHz—С—NH2 -------> CH2=CHCH2NH2 (24)
(56) (55)
Аллиламины могут быть получены гидролизом аллилпзотпо-
цианатов соляной кислотой [56]. Сходный метод получения вклю-
чает перегруппировку аллилтпоцианатов в изотиоцианаты и
гидролиз образующегося продукта реакции (уравнение 25). Этот
Ю1
п.ставтяет практический интерес, поскольку позволяет
метол пР^с2аВ„.3.1МС1Цеиныс аллиламины, с трудом получаем,,,с
синтезировать а Для сш,теза аллпламипов применяют
другими ме' о ское восстановление акрилонитрила, наир,,.
ТТН1 свиниовом катоде [58], и катализируемое иаллад„ем
ититрование аминов с помощью простых п сложных аллиловых
эфиров (уравнение 26) |59).
NH2
н+ I
R'R2C=CHCH,SCN —> R'R2CCH=CH2 -----> R'R2CCH=CH2
Pd
Et2NH -f- MeCO2CH2CH=CH2 Et2NCH2CH=CH2
(2) Свойства
Наличие аллильной двойной связи снижает основность аминов
[60] (р/Са Для аллнламина 9,49, для дпаллиламина 9,29 и для
трпаллпламина 8,31). Эти данные получены при измерении основ-
ности в воде; значения относительной ......-..-
гранс-аллиламинов были определены потенциометрическим ти-
трованием в нитробензоле |61].
Данные ИК-спектра аллнламина
(25)
(26)
основности у-замещенных
показывают, что влияние
орбитали свободной электронной пары при азоте, изменяющей
индукционный эффект аллильной группы, на положение и интен-
сивность полосы поглощения аминогруппы крайне незначительно
при условии сохранения хр3-гибрпдизации атома азота (62].
В спектре ЯМР олефиновые протоны аллнламина имеют следу-
ющие химические сдвиги (6): 5,95 (Н‘), 5,03 (Н2) и
5,14 (Н3) [63].
н3, ,ch2nh2
>=< (55)
Н!' н'
(3) Реакции
По отношению к алкилирующим и ацилирующим агентам, а
В РеаД'1!1И образования оснований Шиффа с альдегидами
nnnBulo ° проявляет себя как типичный амин. Лзометино-
на с ппмпп1СВЯЗЬ В 0с,|0ван11ЯХ Шиффа может быть восстановле-
активированной хлоридом
ни-
при
— с помощью алюминиевой фольги
ртути (II) (уравнение 27) (64]
келя Ренея (уравнение '
этом сохраняется.
;> или водородом в присутствии
28) (65]; аллильная двойная связь
СН;СОМе
+ (55)
А1
HgCI2
Me
ch2chnhch2ch=ch2
(27)
102
оно
•СНгЫНСНгСН=СН!
(28)
Для олефиновой связи не характерны какие-либо особые хи-
мические свойства, хотя в определенных случаях как аминогруп-
па, так и двойная связь могут вступать в химическую реакцию;
например, аллплампн (57) реагирует с димедоном, давая енамн-
нокетоп (58), при облучении которого получается трицикличе-
ский кетон (59) [66] (уравнение 29).
(И)
О
MeNHCH2CH=CH2 +
(58)
(29)
Было показано, что комплексные соединения аллиламинов с
палладием реагируют с нуклеофильными атомами углерода, а
продукты реакции можно восстановить до сложных у-амнноэфи-
ров или кетонов с высоким выходом [67]. В уравнении 30 приве-
ден пример такой реакции; в качестве нуклеофильного агента
используется анион дпэтилмалонового эфира.
MeaNCH2CH=CH2
(EtO2C)2CH Cl
I / -Д'
Л
Me Me
(30)
Перегруппировки типа перегруппировки Клайзена для
аллиламинов не так характерны, как для соответствующих про-
стых аллиловых эфиров. Однако примеры перегруппировки N-
аллпланилппа (60) [68], катализируемой кислотой Льюиса, и
термической перегруппировки оксазина (61) [60] показали, что
перегруппировки этого типа могут быть использованы в органи-
ческом синтезе (уравнения 31 и 32).
nhch2ch=ch2
(60)
A, ZnCh
ксилол
•ОН2СМ«СН2
nh2
(31)
103
6.2.1.3. Амины алленового ряда
(1) Методы получения
В литературе имеется довольно мало сведений об аминах
алленового ряда. Однако были синтезированы такие тетраамнно-
аллены, как (64), путем взаимодействия 1,3-днампно-1,3-дпхлор-
аллил-катпоиа (62) (в виде соли с перхлорат-анионом) с вторич-
ным амином и последующего превращения продукта реакции (63)
в амин (64) при обработке н-бутиллитием или избытком амида
натрия в безводном жидком аммиаке [70] (уравнение 33).
Cl CI Me2NH г NMe2 н.Вии,
Me2N/S/^NMe2 Me ^NMez
в сю; н cio;
(62) (63)
Me2N< xNMe2
—+ V,=C=C(f (88)
Me2br д^Ме2
(64)
Аллены с первичной аминогруппой в боковой цепи, например
(66), были получены исходя из спиртов (65), содержащих одно-
временно двойную и тройную связи (уравнение 34) [71], а также
восстановлением азидов аналогичного строения (67) с помощью
LiAlH4 [72] (уравнение 35). Реакции же цинк- и магнийоргани-
ческих производных 1-бромбутина-2 с простыми геж-амшюэфира-
ми и с алъдпминами были использованы в синтезе третичных
(68) и вторичных а-аллеиаминов (69) [73].
CHsCCRl(OH)C(R2)=CHR3 —> CHsCCR'=CR2CH(OAc)R3
(65)
104
—> CH2=C=CR'CHR2CH(OH)R3 ----------------►
ri-MuC0H,SO2Cl
г-.т 1. Фталимид калия
CH2^C=»CR'CHR2CHR3OSO2CliH<Me----------------
2. n2Hj
—► CH2=C=CR'CHR2CH(NH2)R3
(66)
R=IC„H. R’ — Me; R'=IV=R3—Me
(84)
C№=CCR'=CR2CHR’N3 ------
(07)
R । Me, R2 —H;
Me R
I I
CH2-C=C—CHNEt2
(68) R — H или Ph
* CH2=C=CR'CHR2CHR3NH2 (35)
R' — R2 = Me, R3 —H
Me R
I I
CH2=C=C—CHNHEt
(69) R a= Ph или Me2CH
(2) Свойства
Амины алленового ряда, например (64), представляют собой
жидкости, которые можно перегонять. Они не разлагаются при
хранении в отсутствие воздуха и влаги, однако самопроизвольно
реагируют с воздухом.
(3) Реакции
Алленамии (70) реагирует с водой, давая амид (71); его ана-
лог (64) при реакции с фенилцнанатом и суспензией серы в сме-
си дихлорметана и эфира образует соли (72а) и (726), выделен-
ные в виде перхлоратов, а с диоксидом углерода дает бетаин
(73) [73].
Et2N. ,NEt2 Н20 Et2N\
)С=С=С/ -------► ;C=CHCONEt2 (36)
Et2N/ 'NEt2 Et2NZ
(70) (71)
Me2N NMe2 Me2N NMe2
(72a) R = CN (73)
(726) R = SH
6.2.1.4. Ииамины [74,75]
Название «пнамин» введено, чтобы подчеркнуть аналогию
между К,К-дизамешенными 1-ампноалкннами-1 (74) и соответ-
ствующими соединениями олефинового ряда, известными как
«енамины».
zR2
R'—С^С—N' (74)
\R3
(1) Методы получения
Первый представитель пнампнов (75) был получен в 1958 г.
случайно в результате пропаргиловой перегруппировки [76]
(уравнение 37).
105
(75)
Впоследствии было показано, что пропаргиламины можно полу-
чить в мягких условиях и с хорошим выходом и затем изомери-
зовать в инамнны в присутствии основания, например гидроксида
калия в дпметилсульфокспде [77]. Хотя первоначально эта реак-
ция применялась исключительно к N-арилзамещенным производ-
ным, позднее она была распространена п на N-алкильные произ-
водные за счет использования особых катализаторов, таких, как
амид натрия на оксиде алюминия [78].
В настоящее время наряду с изомеризацией для получения
пнампнов применяют реакции элиминирования и замещения. Так,
например, отщепление галогена в а,р-дигалогеиенамипах (76) дей-
ствием бутнллптия [79] или отщепление хлористого водорода в
1,1-дпхлорампнах (77) с помощью М.М-дизамещениых амидов ли-
тия [80] приводит к инамипам. Синтезу инаминов, исходя из а-
хлоренаминов, посвящен обзор [81].
С1 С1
I I
R'C=C.\R-’R3
(76)
I I +
R'CH2C—NR2R3 R'CH2C=NR2R3
Cl cr -
(77)
Элиминирование метантнола в N,S-ацеталях кетена (78) [80] или
сероводорода в фенилтиоацетампдах (79) [82] при действии ами-
дов лития пли натрия также позволяет получать ииамины (урав-
нения 38, 39).
SMe
(78)
Ph—СН=С—N
(38)
S
Ph—СН2—С—NR2R3 —> (74)
(79)
(39)
Присоединение дихлоркарбепа к енамину (80) с последующим
Дегидрохлорированием под действием трет-бутоксида калия в
лнметплсульфоксиде приводит к алкенпнамипу (81) [83] (уравне-
__лабораторный способ синтеза инаминов (74; R2 —
' = Et) заключается но взаимодействии дпфторалкепа (82) с
диэтнламидом лития [84]. Последний реагент замещает также
106
Меч
J)C=CHNMe;
Me/
(80)
Me Me
Tp<?r-BuOK
CHCIm
rper-BuOK
ДМС0
CH2
II
Me—C—CsC-NMe2
(81)
(40)
атом галогена в фенилхлорацетилене (83), давая инамин (84) с
высоким выходом [85]. Кроме того, реакция 1-галогеналкинов с
алифатическими третичными аминами приводит к солям четвер-
тичных аммониевых оснований (85), которые декватернизацией
избытком третичного амина могут быть превращены в инамин
(86) [85, 86].
RCH=CF2 Ph—CsC- С! Ph—С=С—NEt2
(82) (83) (84)
R3N
R>OeC—X -----> R'CsC—NR| X'
R'C=C—NR22 (41)
(85) (86)
В реакции с дналкиламидамн лития [87] вместо соответству-
ющих галогенпропзводных как предшественников инаминов (74)
могут быть применены простые эфиры ацетиленового ряда, напри-
мер (87).
EtOsCOEt (87)
(2) Свойства [75]
Инамины представляют собой, как правило, бесцветные жид-
кости с запахом, напоминающим запах аминов. Обычно они
устойчивы к действию повышенных температур и безопасны в ра-
боте, особенно третичные С-замещенные соединения. В полярных
растворителях инамины обладают повышенной реакционной спо-
собностью; их реакционная способность зависит от характера за-
местителей при азоте. Как и следовало ожидать, N-арпл- и N-
трифторметилпроизводные являются соответственно наиболее
устойчивыми и наименее реакционноспособными соединениями.
В ИК-спектрах несимметрично замещенных инаминов наблю-
дается сильная полоса поглощения при 2220 см~‘, характерная
почти для всех дизамещенных производных.
(3) Реакции
Реакционную способность инаминов можно объяснить с точки
зрения двух резонансных структур (88) и (89), хотя, по-видимо-
му, первая из них преобладает. В результате электрофильной
107
атаки на ииамипы возникают структуры кетенпмпппя, энерГеТп.
чески менее выгодные, чем пмнипсвыс соли, получаемые из ена.
мпнов [88]. Следовательно, можно ожидать, что ппамнпы буДут
реагировать с монофункциональными нуклеофильными агентами
хуже, чем енамины, причем реакция будет осуществляться пре.
имущественно путем присоединения в ос- и р-положення по отно-
шению к атому азота.
—C=C=N;
—C^C—N'
(88) (89)
Нуклеофильное присоединение. Катализируемая кислотой гид-
ратация инампнов (74) с образованием амидов (R'CH2CONR2R3)
является, по-видимому, одной из первых изученных реакций
инаминов [89]. Гидратация ииаминов протекает настолько лег-
ко, что инамины были использованы в качестве дегидратирующих
агентов в синтезах ангидридов кислот, амидов и пептидов [90
91].
Аналогичным образом спирты и амины также присоединяются
к инамииам (уравнения 42 [90] и 43 [89]).
ZR!
Р.'-С=С->/
(74) 4R3
Eton xNR’R3 н2о,н+
-----R'CH=c( ---------------► R'CIACOOEt (42)
X)Et
HNEtj /NEt2
(74) -------► r'ch—c;
MSIR2R8
(43)
Ацилирование. Ацилгалогениды, такие, как фосген, тиофосген и
тионилхлорид, реагируют с инампиами (74), давая аддукты типа
(90) практически с количественным выходом [92]. Продукты при-
соединения термически устойчивы и могут быть перегнаны в ва-
кууме без разложения. Однако в этих соединениях имеются два
реакционноспособных атома хлора, которые могут взаимодей-
ствовать с бифункциональными агентами, давая гетероцикличе-
ские соединения, например (91) [93] (уравнение 44).
О R' NR2R3
II I I
G1—С—С=С—Cl +
(91)
(44)
(90)
Реакции циклоприсоединения. Инамииы присоединяются к раз-
личным активированным двойным связям с образованием цикли-
ческих продуктов. Например, они реагируют с 1,2-дифепилмети-
ленциклопропеиами, давая 3-амннофульвены (92), которые нельзя
108
получить обычными методами [94]. При реакции инамина с ди-
метиловым эфиром ацетилендпкарбоновой кислоты образуется за
мешенный анилин (93) [90].
(92) (93)
Реакция инаминов с кетенами, например ннамина (95) с кете-
ном (94), протекает легко, по-внднмому, через стадию образова-
ния промежуточного цвиттерионного соединения (96) [95] и при-
водит к цнклобутенону (97) с почти количественным выходом
(уравнение 45). Однако незамещенный кетен реагирует с инами-
ном (98), давая в качестве главного продукта реакции аллен
(99). Предполагают, что в этом случае циклизация промежуточ-
ного цвиттериона затрагивает и атом кислорода, в результате
чего образуется оксет (100), который быстро перегруппировы-
вается в аллен (99) (уравнение 46).
Ph2c=C=O + Phc^CNEtz —>
(98)
(46)
(100)
,Ме
—>- сн:=с=<\
\cONEt2
(99)
Кетенимпны (101а) и (1016) также вступают в реакцию ци-
клоприсоединения к ниаминам (95) и (98), давая -аш ’
ны (102) [96[ (уравнение 47); однако эти реакции протекаю
более медленно, чем аналогичные реакции цпклоприсоединен
кетенами [95].
Циклоприсоединение ни амина
представляет особый интерес,
(98) к 5-метплциклогексенону
поскольку гидролиз продукта
109
(95) или
Г R=O=C—NPh
„,, + В-С_С-НРЬ - [ ,(r_l_c_.a
(IOIa)
(1016)
R
циклоприсоединения в контролируемых условиях приводит к кис-
лоте (103) с тремя асимметричными центрами, которые имеют
лишь показанную ниже относительную конфигурацию (уравне-
ние 48) [97].
(103)
Реакции циклоприсоединения инаминов к хинонам можно
инициировать как фотохимическим способом, так и термически.
Отдельные примеры подобных реакций приведены в уравнениях
49 и 50 [98].
(49)
ацетиленового
6.2.1.5. Амины ацетиленового ряда
В этом разделе рассмотрены другие амины
^п»^0ТЛ'1Ча'°"1"еся по структуре от инаминов. Некоторых
аспектов этой темы касаются обзоры [99, 100].
но
(Г) Методы получения
Пропаргиламивы (105) могут быть получены из галогенацети-
лспов типа (104) действием избытка аммиака или соответству-
ющего амина (101] (уравнение 51), при использовании гексаме-
тилентетрамина (102] или по реакции с литиевыми производными
вторичных аминов [103]. Реакция Манниха с монозамещенными
ацетиленами в присутствии медного катализатора является наи-
более распространенным методом синтеза третичных пропаргил-
аминов (105) [101, 104]. Пропаргиламин простейшего строения
легко получается в условиях синтеза Габриэля (уравнение 52)
[105].
R2R3\H
R'C^CCH2Br --------> R'C=CCH2NR2R8 (51)
(104) (105)
Бутин-З-амин (106) был синтезирован с помощью реакции аце-
тиленидов металлов с галогенаминами [106] (уравнение 53), а
соединения с более длинной алкильной цепью, например (107),
могут быть получены путем восстановления соответствующего
амида ацетиленового ряда алюмогидридом лития (107] (уравне-
ние 54).
CHsCNa + BrCH2CH2NH2 —> CtesC—CH2CH2NH2 (53)
(106)
Me Me
| LiAIH, I
MeCs=C—CH2CHCONR‘R2----------* MeCsCCH2CHCH2NR'R2 (54)
(107)
(2) Свойства
Для экспериментально измеренных значений рХа в случае
третичных ацетиленовых аминов была установлена связь с кон-
стантой а в уравнении Тафта [108]. Значение рКа для пропаргил-
ампна составляет 8,15.
Были сняты ИК- и КР-спектры некоторых ацетиленовых ами-
нов и диаминов; полоса поглощения в о6ласт^ 600—650 см 1
была отнесена к деформационным колебаниям С = Сг1 [109]. изу-
чен также характер масс-сиектрометрической фрагментации неко-
торых вторичных и третичных ацетпленамннов [НО].
Отдельные ацетиленовые амины обладают значительно»
биологической активностью; особого внимания заслуживают иар-
ГИЛИН (108) и оксотреморип (109). Паргилин является ннгиинтором
ill
ыоиоампиооксидазы и эффективно снижает кровяное давле.
Нне хотя побочные действия и ограничивают его применение
Оксотреморпи вызывает судороги в организме животных и ис-
пользуется в лабораторных исследованиях для выявления лекар-
ственных препаратов, пригодных для лечения болезни Паркин-
сона.
Me
РЬСнД-СНг-С^СН Q)N-CHs-teC-CH2-N^J
(108) (109)
(3) Реакции
В этом разделе основное внимание уделено перегруппировкам
н реакциям циклизации ацетиленовых аминов, а также превраще-
ниям, затрагивающим обе функциональные группы.
Прототропная перегруппировка вторичных пропаргплампнов
(ПО) приводит к иминам (111) [111], в то время как третичные
амины, например (112), в присутствии калия или амида калия на
оксиде алюминия [112] перегруппировываются в ипамины (113).
CHsC—СН2—NHR —> СН2=СН— CH=NHR (55)
(НО) (111)
СН=С—СН2—NEt2 —>- Me—CsC—NEt2 (56)
(112) (113)
Перегруппировка диацетиленамина (114), катализируемая
трег-бутокспдом калия, является более сложным процессом п
приводит к трем основным продуктам реакции (уравнение 57)
[113].
/СеСРЬ
MeN^ —>
^CsCPh
(114)
Ph Ph
Реакции циклизации с участием ацетиленовых аминов широко
используются в синтезе гетероциклических соединений, например
натпм°ЛтТ!НЯ/ТИазолинов (,15) по Реакции с бензоилизотиоциа-
(Уравнение 58). Аналогичным образом циклизация
ропаргиламипов под действием полнфосфорной кислоты
112
легла в основу нового подхода к
„зопавннов (117) [115] (уравнения
Me
I PhCONCS
H2N—С—CsCH -----------
I
Me
(116) и
(58)
синтезу изохинолинов
59, 60).
Реакциям циклизации ацетиленовых аминов посвящен обзор
[116].
Региоспецифичная гидратация пропаргиламинов (118) дей-
ствием серной кислоты, содержащей сульфат ртути(II), позво-
ляет легко получать амины типа (119), которые являются замас-
кированными винилкетонамн (120). Эта реакция была использо-
вана для получения ключевого промежуточного соединения (121)
в синтезе эстрона [104].
RC=C—CH2NEt2 —> RCOCH2CH2NEt2 —» RCOCH=CH2 (61)
(118) (119) (120)
IIPNE',
jOO
(121)
6.2.2. ДИАМИНЫ И ПОЛИАМИНЫ
6.2.2.1. 1,1-Диамины [117]
Эти соединения, структура которых представлена общей фор-
мулой (122), называют также гел-диаминамн или амнналями.
Rs
R'R-’N—С—NR’R4
I
R"
(122)
113
(1) Методы получения
1,1-Дпампвы могут быть получены реакцией метилендцгя
нидов с аммиаком пли аминами [118] и конденсацией альдеЛ°Ге'
или кетонов с вторичными аминами [119]. В качестве nrv/l,fl0B
веществ —.....рП»пП1>,^тп„ ,........ Мых
лотовый
62, 63).
были использованы соединения, содержащие Дихлорма-
фрагмент [120], и фспилглиоксаль [121] (уравнения
HNRSRI
О
(62)
PhCOCHO + HNR3R* —> PhCOCH(NR3R’)2
(63)
Реакция карбиноламинов (см. разд. 6.2.3.1) с первичными
или вторичными аминами также является хорошим методом по-
лучения 1,1-диаминов [47]. Так, несимметричные 1,1-диамины
(123) могут быть синтезированы [122] реакцией формальдегида
с дициклогексилампном и другим вторичным амином (уравне-
ние 64). Аналогичным путем получены [123] трициклические
соединения (124) (уравнение 65).
(C6H,,),2NH + HCHO + R3R’NH —> (C6H,,)2NCH2NR3R< (64)
(123)
(66)
(124)
Этилендиамины, например Ы,Ы'-дифенилэтилендиамин (125), так-
же реагируют с альдегидами, давая имидазолины (126) (уравне-
ние 66), которые обычно выделяют в кристаллическом виде
(128)
(66)
(67)
114
9) Эта реакция была предложена для идентификации альде-
' [124]. Было показано, что реакция фенолов с олефинами
ia,гример (|27Б Двойная связь которых обогащена электронами’
также приводит к имидазолинам (128) [125| (уравнение 67L
(2) Свойства
Устойчивость 1,1-диаминов определяется их
также свойствами веществ, с которыми они находятсяа
те. Например, метилендиамин H2NCH2NH2 не был rL™ КОНтак'
бодном состоянии, однако жидкий тетпя«рТ„ Д 1ен в сво-
Me2NCH2NMe2 устойчив в водноГрёетГо^
пали, такие, как (124) и (126) — кпистяпп икл,,ческие амн-
у’стойчпвые в щелочной (но не в’ кислой) средё^Некотооые^™3’
...
ные 1,1-диамины, имеющие водородный атом е
[126], и вторичные 1,1-днамины RNHCH2NHR II171
ке разлагаются на амин и имин. 1
... жно перегонять при атмосферном давлении. Однако третич-
их мол, „ио1А„пш ..------- в положении 2
] при перегон-
(3) Реакции
Хаит и Вагнер [127] изучили действие кислоты на 1,1-диами-
ны и образование солей 1,1-диаминов. Они пришли к выводу,что
большинство ампналей разлагаются при действии кислот в при-
сутствии или в отсутствие воды или спиртов. И хотя можно по-
лучить соли некоторых более устойчивых 1,1-диаминов, как пра-
вило, эти соли недостаточно устойчивы при хранении вследствие
разложения до карбениевого иона, стабилизованного наличием
атома азота:
R—N—СН2
I
R—Й=СН2
I
В работе [128]
(129).
приведены данные о гидролизе имидазолинов
NMe
NMe
(129)
В присутствии кислот спирты превращают 1,Ьдпамнны в аце-
тали (131), однако при использовании мягко девствующих ката-
лизаторов, например гидрохлорида пиридина, можно выделить
фостой 1,1-аминоэфир (130) [117].
RJNCH.NRJ + R,0H C5"5N-HCI> r=och2nrJ R?0CH’0R’ (68)
(130) (,31)
115
1 1 .Диамины легко расщепляются в условиях каталитическое
пигоогенолиза и при действии дпбораиа (уравнения 69, 70)ог°
также при восстановлении металлами, например цинком, рас'’ '*
Гяемымн в щелочи [117]. Хлорапгидрпды кислот, галогены, гал£
геново дороды, метплхлорметиловыи эфир и метплхлорметилсуль
фид также расщепляют 1,1-диамины, давая а-галогена
R‘R2NCH2C1 (см. разд. 6.2.5.1).
Ми ИЦ
(69)
(70)
Алкилирование 1,1-диаминов протекает медленно; это говорит
о том, что они обладают менее сильными нуклеофильными свой-
ствами, чем простые амины. Однако из них тем не менее могут
быть получены соли моночетвертичных аммониевых оснований,
например (132а) [129]. Получена также и соль (1326); согласно
Me2NCH2NMe3 ВГ Me3NCH2NMes 2Г
(132а) (1326)
литературным данным [130] она устойчива при хранении в атмо-
сфере сухого азота. Однако, как правило, эти соли неустойчивы,
что свидетельствует о том, что появлявшиеся ранее в литературе
сведения о получении этих соединений были ошибочными [130].
6.2.2.2. 1,2-Диамины
В этом разделе рассматриваются в основном синтез, свойства
и реакции замещенных этилендиаминов (133) и, в частности, са-
могб этилендиамина (133; R1 = R2 = R3 = R4 = Н),
Rk ,RS
^NCH2CH2N(^ (183)
(7) Методы получения
Хотя реакция аммиака с 1,2-дигалогепэтаном при комнатной
^99 ПрИ нагРеваиии под давлением приводит к эти-
скопыги . ’ эт0т метоД ие совсем удовлетворителен, по-
У ет происходить дальнейшее алкилирование с образо-
116
пем пиперазина и линейных полимеров [131]. В основе более
, ачного метода получеш.я этилеидиамина лежит синтез ,юГаб-
пиэлю (уравнение 71). По этому методу дибромэтаи
фталимидом калия, а образующийся
переводят в этилендиамин [132].
О
нагревают
замещенный фталимид
ВгСН2СН2Вг
кислота,
щелочь
NH2NH2.H2O
(133)
(71)
Еще один вариант синтеза основан на реакции 1,2-дихлорэта-
на с мочевиной при повышенных температурах, приводящей к
бис(мочевине) NHzCONHCHoCHzNHCONHs, которая гидролизу-
ется в щелочных условиях до этилендиамина [133]. Кроме того
удовлетворительными методами синтеза этилендиаминов (133)
являются также перегруппировка Курциуса диазидов янтарной
кислоты и восстановление а-нитроаминов и а-аминонитрилов [134].
(2) Аминоенамины
1,2-Бис(диалкиламино)этилены (аминоенампны) (135) полу-
чают из третичных 2,2-дихлорэтиламииов (134) реакцией с ами-
дами лития с последующим элиминированием амина при нагрева-
нии [135] (уравнение 72).
L1AIH, L,NR2
CUCCONR? -----------► CI0CHCH2NRI --------
(134)
nr?
hcch2nr]
NR?
(72)
(13Я)
(3) Свойства
Сведения о свойствах этилендиамииа содержатся в обзоре
[136]. Во многих отношениях его физические свойства напоми-
нают свойства воды, однако этплендиампн является^ сильным
основанием с двумя константами диссоциации, pAi 10,0 и
Р'Н 7,0 (при 20 °C).
117
- „„леп-шампп как полярный растворитель сра,
Безводны» эг11Л?’ 4 11ел применение как растворитель
с жидким ^'"Литоватя слабых кислот [137]. ЛЛ1‘
потеипиометрп еск в газовой фазе было обнаруЖеНо
Пр» 1,3>'чи “" тнпендиампиа, объединенных водородным,, СВя.’
чТО для МОЛ^на значительная энергия напряжения. Это уиа31>).
зями, характерназ! водородная связь стремится к линейной
вает на то что
геометрии I1 „ /..) этпленднашша в интервале темпера-
освдяет1,9Д [139].
ТУРКак7ам этилендпампн, так в Ы=PR^
лендпа^иштетрауксуснаЯ]^'^|''°1с".;1.1Л0В образу1ОТ хелаты [140].
“ ФоГгмент 1 2-диамина является элементом структуры некото-
оы лекарственных препаратов, в частности, входит в состав
^т»гистами!1пых препаратов, родственных пириламину (неоан-
терган) (136). __
\—N—СНг—(f /—ОМе (136)
'N । \=/
CH2CH2NMe2
(4) Реакции
Наиболее важными реакциями этилепднамина, отличающими
его от обычных аминов, являются реакции, в которых принимают
участие обе аминогруппы и образуются циклические соединения..
В качестве примера ниже приведены реакции с диэтилкарбона-
том (137) [141], сероуглеродом (138) [142], 1,3-дикетонами (139)
[143], пирокатехином (140) [144], сложными эфирами 2-галоген-
карбоновых кислот (141) [145] и дигалогенпронзводнымн (142)
[146] (уравнения 73—78).
ЕЮ.
(133) + V=O
ею/
(137)
NH
NH
(133) + cs2
(188)
(133) + R‘COCH2COR2
(75)
(189)
118
(142)
Так же как и другие а,о>-дпамнны, этилендиамин реагирует с
альдегидами, образуя альдимины (143), и с адипиновой кисло-
той, давая полиамиды типа найлона [147].
RCH=\CH2CH2N=CHR (143)
(5) Пиперазин
Основанием для рассмотрения этого циклического 1,2-диами-
на (144а) и его производных в специальном разделе служит зна-
чительный объем сведений, касающихся химии этих соединений.
(144а) R'=R2 = H
(1446) R1 = R-
Получеиие. га-Нитрозоанилин может вступать в конденсацию
с этплендпбромидом, а образующееся ди (л-нитрозофепнлпропз-
водное (1446) расщепляется в щелочной среде, превращаясь в
«-нитрозофенол н пиперазин. Этаполамин HOCH2CH2NH2 или
М-(2-гндрокспэтил) этилен диамин HOCH2CH2NHCH2CH2NH2 мож-
но превратить в пиперазин с помощью процесса цпклодегпдрата-
Чии. Пиперазин можно получить также восстановлением 2,5-дпке-
топиперазина натрием в спирте [148].
Свойства. Пиперазин (144а) представляет собой гигроскопич-
ное вещество, которое образует гексагпдрат п легко растворимо
в воде. Константы диссоциации этого соединения в водном ра-
створе составляют 9,8 (pKi) и 5,7 (рКг)-
Пиперазиновое ядро входит в состав самых разнообразных
соединений, проявляющих биологическую активность [150]. Неко-
торые препараты интересные с медицинской точки зрения, пред-
ъявлены формулами (145) - (150). Сам пиперазин и некоторые
119
его производные являются аптпге.пьмннтиыми препаратами, на-
пример дпэтилкарбамазян (гетразан) (145), применяющийся прп
лечения филариоза. Пипсразииовый цикл является элементом
структуры ряда транквилизаторов, таких, например, как npoiii
водное фепотиазпна — трпфторперазин (стелазии) (146) и
тиотиксен (наван) (147). Пиперазиновый цикл присутствует так-
же в структуре гидроксизина (атаракс) (148), родственного Цик.
лизину (валопд) (149) — препарату, который применяется ддя
облегчения приступов морской болезни. Относительно недавно в
качестве препарата для снижения повышенного кровяного давле-
ния был предложен празосин (гиповаз) (150).
(149) (150)
Реакции. При действии хлорангидрндов или ангидридов кислот
или сложных эфиров пиперазины дают моно- пли диацплпроиз-
водные в зависимости от относительной концентрации реагентов.
Аналогично, несмотря па легкость образования диалкилпипера-
зинов, при взаимодействии 2 моль пиперазина с 1 моль алкилга-
логепида получают моноалкилпроизводные пиперазина с выхода-
ми 50—60% [151]. Однако легкость получения и очистки моно-
CICOOEI
------>• EtOCON
NH
конц. I1CI
-----------
RH.il
(79)
120
«пиперазинов часто делает оправданным применены» -гг,
gon схемы синтеза моноалкилпроизводшй (урав^Х'
[152].
6.2.2.3. 1,3-Диамины
(1) Методы получения
Для синтеза 1,3-дпамииопропаиа (пропандиамин-131 (кп
м0Гут быть использованы методы аналогичные методам получе-
„я этнлеидпамина: а) реакция 1,3-дигалогенпропанов с аммиа
ом, хотя предпочтительнее использовать синтез Габриэля с фтали-
мидом калия (уравнение 80); б) перегруппировка Курциуса диа-
Вг(СН2)3Вг
—>- H2N(CH2)3NH2
(151)
(80)
зида глутаровой кислоты ЫзСО(СН2)зСОКтз пли перегруппировка
Гофмана диамида глутаровой кислоты H2NCO(CH2) 3CONH2;
в) восстановление [З-аминонптрплов [131]. Пропандпа.мнн-1,3 по-
лучают также с помощью модификации последнего метода—ка-
талитическим гидрированием акрилонитрила над никелем Ренея
при высоком давлении в присутствии аммиака [153].
(2) Свойства
Значения констант диссоциации пропандиамниа-1,3 в водном
растворе при 20 °C составляют 10,6 (р/С) и 8,65 (рКл). В ин-
тервале температур 298—318 К дипольный момент (ц) пропан-
диампна-1,3 равен 1,94 Д [154].
З-Аминопроппламид калия, «сверхоснование», легко получае-
мое из гидрида калия и избытка пропандиампна-1,3, проявляет
исключительную активность в прототропных реакциях [155]. Под
воздействием этого реагента, например, происходит чрезвычайно
быстрый процесс миграции тройной связи из середины углеродной
Цепи в концевое положение; эта миграция получила название
«ацетиленовой молнии» (уравнение 81).
I. КН, (151) rerw (81)
H(CH2)mCsC(CH2)„H —>• H(CH2)m+„C^CH ( )
Данные ЯМР-спектроскопии свидетельствуют о том, чт -1
Ротонироваппые формы некоторых 1,3-дпаминов еущест .
ЭД6 Циклических соединении с водородной связью [
121
п ... масс-сиектрометрпческом распаде 1,3-дпаминов под дей.
П ч ектрониоп, удара характеристическим процессом ЯВЛя.
нейтральной молекулы амина 113 Мо.
ЛеККил^даамнношй фрагмент - обычный элемент струну.
; комических препаратов, оказывающих воздействие на
е й г .ьцхю нервную систему, таких, например, как антндепрес-
еаит нминрамнн (тофрапил) (152).
(152)
N
CH..CH?CH2NMe2
(3) Реакции
Химические свойства пропандпамнна-1,3 (151) аналогичны
свойствам этилеидиамина. Так, его можно моиоацилировать и
алкилировать способами, аналогичными способам, описанным
выше (см. разд. 6.2.2.2). Он реагирует, например, с альдегидами,
карбонатами и 1.2-днкетопамп, давая соответственно циклические
соединения (153) — (155) (уравнения 82 [159], 83 [160], 84
[158]).
HN-,
RCHO + (151) —> к—( \ (82)
НХ—
. (153)
/ОЕ1 HN—,
(151) + O=C( —> О=( ) (83)
Х»Е1 HN~^
(154)
Ph\zO Ph. „У—\
(151) + Y —> Y \ (84)
PbY'4) Ph/4N_/
(155)
При нагревании гидрохлорида пропаиднамина-1,3 с ацетатом
натрия при 170 200 °C образуется тетрагидропнрпмндип (156),
однако при 270 С продуктом реакции является М.М'-днацетилпро-
папдиамин (157) [158].
N-,
) MeCONH(CH2)3NHCOMe
HN-7
(156) (157)
122
в.2.2.4. Полиамииы
(1) Гексаметилентетрамин (гексамин) (158)
Получение. Амин (158) легко получается при упаривании
'.алыегида и концентрированного раствоояУ Л? fMeCH
Сойства и структура. Амин (158) РредстТля “Х Г621'
„,авкое белое кристаллическое вещество, возгоняется ,щи
”аНИи в вакууме. Его структура была установлена мет , ре‘
еновской дпффракцни. Как и 1,1-дпамины, амин Н58
очень слабыми основными и нуклеофильными свойств,
станта его диссоциации (рК.) в видном разоре^при'’25°°С
равна 6,3.
Препарат, состоящий из гексамина и миндальной кислоты не
пользуется при лечении заболеваний мочевыводящих путей
Реакции. Вследствие слабой нуклеофильности гексамин подвей
гается однократному алкилированию, и то с трудом В качееХ
алкилирующих агентов могут выступать только реакционной
собные соединения типа бензил-, аллил- и фенацилгалогеинлов
В результате реакции образуются гексаминневые соли. Подобные
соли являются ключевыми соединениями в синтезе первичных
аминов (163) по Делепину (уравнение 85) и в синтезе альдегидов
(162) по Соммле (уравнение 86). В основе реакции Делепина
[163] лежит катализируемый кислотами алкоголиз соли (159)
позволяющий вывести формальдегид из реакционной среды в виде
относительно инертного ацеталя формальдегида. В реакции
Соммле [164] формальдимин (160), образовавшийся при гидро-
лизе соли (159) в слабокислой среде, дегидрирует амин (161) до
имина, при гидролизе которого получается альдегид.
Раствор гексаметилентетрамина в уксусной кислоте использо-
вали в качестве мягкого окисляющего агента для превращения
СН2К
(168) (‘59)
[rCHjNHj + CH2=Nh1 RCH2NH3Ci+3NH4CI +6CHjOEtj2 (85)
(16‘) (160) (163)
г
рсН=1ун + M₽NH2j ------>- ECHO + 3NH3 + MeNH2 + 5CH2O (86)
J (162)
123
аминов в альдегиды пли кетоны [165]. В реакции Даффа i16R,
гексамин реагирует с активированным фенолом в ирисутс-ги
глнцерпнборной кислоты, в результате чего с низкими выходам,,
образуются о- или (если орто-положення заняты) п-гидрок "
бензальдегиды. Было установлено, что различные ароматические
соединения, даже простые углеводороды, при нагревании с тек-
самином и трифторуксусиои кислотой превращаются в альдегиды
причем в случае активированных ароматических производных вы-
ходы выше [167].
Гексамин (158) реагирует также с дымящей азотной кислотой
лучше всего в присутствии нитрата аммония и уксусного ангщр
рида, давая взрывчатое вещество циклонит (гексоген) (164) [168].
O2N-N^N—no2
1^ J (164)
N—NO2
(2) Линейные и макроциклические полиамины
Установлено, что конденсация первичных п вторичных диами-
нов с электрофильными агентами приводит к линейным полиме-
рам. Например, при взаимодействии пиперазина с альдегидами
[169] и с метпленбпс(акриламидом) [170] образуются соответст-
венно полимеры (165) и (166). Однако недавно стал возмож-
ным синтез разнообразных макроциклических полпаминов.
(165)
О О
II II
NCH2CH2—С—NHCH2NH—С—СН2СН2-
(166)
Основные типы полученных макроциклических соединений
представлены формулами (167) — (172) [171].
, HN.
СН2Г >СН,1
2л^ЫН \ 2/'
(166)
Циклические полнамииы обычно получают с помощью реакций
конденсации, в которых ион переходного металла выступает в
роли матрицы, удерживающей реагенты в положении, благо-
приятном для образования продукта реакции. Это явление полу-
чило название координирующего матричного эффекта.
Прямая конденсация с участием карбонильных соединений и
комплексов металл — амин дает удовлетворительные результаты
124
(172а) (1726)
(1726)
лишь в случае соединений никеля (II) и меди(П). Диссоциация
возникающих комплексов происходит с таким трудом, что во мно-
гих случаях получение макроциклов, не содержащих металла,
представляет сложную проблему. Тем не менее из некоторых ком-
плексов никеля(II) были регенерированы циклические полиами-
ны, например, с помощью агентов, еще сильнее связывающих ме-
таллы, таких, как циаппд-пон в водном растворе (уравне-
ние 87). Циклические полиампны были получены также прямой
конденсацией диаминов с карбонильными соединениями. После
выделения свободного амина могут быть получены его комплек-
сы с ионами других металлов.
NiL2t + 4CN —► Ni(CN);" + L
L =лиганд
Для макробициклнческнх диаминов типа (172) характерны
три конфигурации (172а) —(172в) [172]. ои/-о«/-Изомер
(1726; k = I = т = 9) при растворении в 50%-ной дейтеротрн-
фторуксусной кислоте превращается в /n-in-изомер, по достиже-
нии равновесия смесь содержит примерно 40% изомера (172а) н
около 60% изомера (1726). Галогенид-ионы взаимодействуют с
формой (172а), в результате чего образуется новын катион, в ко-
вром галогенид-нон заключен в полость молекулы бпцнклнческо-
Го амина. Этот процесс включает диффузию галогеннд-пона в по-
^ость бициклического амина и получил название катапиноза.
Радующиеся ионные пары называются катапинатнымн ионами.
^Ругие примеры комплексных ионов и ионов включения приведе-
ны в разд. 6.2.4.4.
н мУг8ТО1)Ые линейные полнамины. например
.«=«•
125
(87)
Avhkiuiii: установлено [173], что эти соединения связаны с фа|[.
торами, стимулирующими в ингибирующими процесс деления
клеток.
6.2.3. АМИНОСПИРТЫ
6.2.3.1. 1,1-Аминоспирты
Эти соединения, являющиеся а-гидроксиамннамн с общей фор.
мулон (173), называют также карбпноламннами.
R3
R'R2N—С—ОН (173)
I
R«
(1) Методы получения
Аммиак и амины присоединяются к низкомолекулярным аль-
дегидам, образуя 1,1-аминоспирты. Механизм конденсации фор-
мальдегида п аммиака, приводящей к получению амипометанола,
изучен с помощью спектрофотометрии. Однако продукт реакции
неустойчив и легко отщепляет молекулу воды, давая гексамети-
лентетрамин (158) [174]. В случае первичных и вторичных ами-
нов формальдегид действует в кислой среде как метилирующий
агент, однако в присутствии щелочи легко образуется 1,1-амино-
спирт. При конденсации других алифатических альдегидов с
аммиаком получают 1,1-ампноспирты, которые можно выделить в
кристаллическом состоянии, однако во многих случаях эти «аль-
дегндаммпаки» представляют собой, вероятно, гидраты 2,4,6-
трпалкнлгексагидро-1,3,5-трназинов [175].
Кетоны (например, ацетон) могут вступать в конденсацию с
аммиаком п аминами, образуя сложную смесь продуктов реак-
ции; однако известны примеры выделения карбннолампнов и в
случае кетонов. Например, циклопропанон реагирует с аминами,
давая 1,1-ампноспирты с хорошими выходами [176]. В 1,1-амнпо-
спирты могут быть превращены также некоторые полуацетали;
например, при взаимодействии бициклического полуацеталя (174)
с метиламином получают аминоеппрт (175) (уравнение 88) [177].
(174) (175)
Гидратация азометпнов часто приводит к 1,1-аминоспнртам,
если продукты реакции стабилизованы за счет межмолекулярных
водородных связей. Этот процесс особенно типичен для гетеро-
циклических систем [178] и при образовании пеевдооснованпн 113
солен четвертичных оснований [179].
126
(2) Свойства
Соединения, образующиеся при взаимодействии формальдеги-
да первичных аминов, как правило, представляют собой бес-
цветные реакционноспособные жидкости, однако при пропускании
тока аммиака через эфирныи раствор ацетальдегида получают
кристаллическое вещество, которое существует не в форме про-
стого мономера, а, скорее всего, в виде трнгндрата триметилгек-
Сагидро-1,3,5-трназнна |180]. Это соединение и .ipvrue представи-
тели класса «альдегидаммиаков» |RCH(OH)NH2]„ обычно
неустойчивы во влажном воздухе; однако нх стабильность увеличи-
вается по мере возрастания величины заместителя R. 1,1-Амино-
спирты, полученные из альдегидов и аммиака или первичных ами-
нов, даже при хранении склонны терять воду н тримеризоваться
Однако отдельные 1,1-амнноспирты, способные к образованию
внутримолекулярных водородных связей, например (176) [181]
И (177) [182], согласно литературным данным, обладают значи-
тельной устойчивостью.
R1
I
CH2CCO2R3 * * * * 8
I
NHCH(OH)R2
НО
НО
(176)
О2\—
О
S
>—сн.\н—С
I
он
(177)
N
разбавленными кис-
(3) Реакции
«Альдегндаммиаки» легко гидролизуются
лотами; при этом происходит регенерация компонентов, образо-
вавших эти соединения. Вследствие этого для очистки альдегидов
иногда получают их аддукты с аммиаком. Дегидратация этих
аддуктов в мягких условиях приводит не к алкн.тнденнмнну
RCH = NH, а к 2,4,6-трпалкнлгексагидро-1,3,5-триазинам [174].
Аддукты, образованные высшими альдегидами и первичными
аминами, в меньшей степени проявляют склонность к полимери-
зации; в ЭТ|1Х СЛуЧаях выделяют соответствующие основания
Шиффа. Механизмы образования карбниоламинов и их дегидра-
тации рассматриваются в обзоре (183].
1.1-Амнноспи рты, как правило, очень неустойчивы, ооразова
|111е производных обычного типа для них не характерно, хотяI
описано N-ацилпрованне с помощью кетена [l/о]. Однако J1'-
фильные агенты могут замещать гидроксильную группу, и
«ымн реакциями 1,1-амнносппртов являются реакции с пер -
«ыми н вторичными аминами, приводящие соответственно
аминам и 1,1-аминоэфирам (см. разд. 6.2.2 и о. . )•
127
6.2.3.2. 1,2-Аминоспирты
Этот раздел посвящен описанию этанолами
лы (178) и включает рассмотрение химии
(179), представляющих собой особую группу 1,2
R' Rs
НО—С—С—NR’R®
I I
R2 R
Лг R3
НО—С—С.~NRr’Rs
Н Н
нов общей форму,
арплэтаполаминов
-амииосппртов.
(178)
(179)
(1) Методы получения
1,2-Ампноспирты, в частности 2-аминоэтаполы R5RGNCH2CH2OH
н 2-аминопропанолы R5R6NCH2CH2(ОН)Ме обычно получают пу-
тем раскрытия цикла в легко получаемых этилен- и пропиленок-
сидах. Скорость реакции аминов с эпоксидом (180) и направле-
ние раскрытия цикла определяются характером пространственных
и электронных факторов в нуклеофильном агенте и замести-
телях R1 и R2, а также константой диэлектрической проницае-
мости и температурой растворителя. Как правило, атака аминами
начинается у наименее пространственно затрудненного центра, в
результате чего происходит гранс-анти-пернпланарное раскрытие
цикла по механизму SN2, как, например, при образовании соеди-
нения (181). В некоторых случаях, особенно когда заместители
R1 и R2 способны стабилизовать частичный положительный заряд
при наиболее пространственно затрудненном атоме углерода, мо-
жет происходить конкурирующая реакция, в результате которой
образуется продукт (182). Протеканию этого процесса способ-
ствует повышение температуры реакции.
QU «аномальный» . г, «нормальный»
I путь 1\ \ / \ путь I
R‘R2C-CH2 <------------ \/ \ -----------------> RIR2C-СН2 (89)
NRSR6 R’ NR5R“
(182) (180) (181)
В результате изучения реакции этаиоламина с эпоксидами
[184] было показано, что гидроксильная группа может способ-
ствовать раскрытию цикла по механизму, изображенному в фор-
муле (183). Этот «пуш-пульный» механизм, при котором груп-
па ОН выступает в роли слабого электрофила, обусловливаюше-
Г2 R1
128
уклеофпльного раскрыт.^ эпок™ '!<i y,,eJ",4eHIie
₽0 Наряду с ямпнолпаом эпоксидов в ряде случаев ботее vnr>6
|1ЫМ может оказаться другой вариант раскрытия эпоХдов Л'
чаюшийся в использовании в качестве нуклеофил™ ;,-^’
а азид-аипопа с последующим восстановлением азиднойф,2'
In,, алюмогидридом лития или дибораном [18f,| (уравнение 90)'
О Н2 X НО R5
„'.v-z.». ,ч
R N3 NH,
R1in,y~—V1"1 R
R2 R4
Считают, что катализируемый кислотой гидролиз азиридинов
с препаративной точки зрения ие представляет большого интере-
са [186], однако этот метод применяли для получения отдельных
1,2-амнноспиртов [187, 188]. Данные кинетических исследований
показывают, что разрыв связи при углеродном атоме, предпочти-
тельно образующем карбокатионный центр, происходит по меха-
низму SN1. Однако это допущение не полностью объясняет стерео-
избирательность, наблюдаемую при кислотном гидролизе бицик-
лического соединения (184) (уравнение 91), поэтому был предло-
жен механизм, промежуточный между SN1 п Зм2 [189].
Ph. ,sJJH
f '1 0,2h.H2SO4 ч
I J so-/. дмсо-н2оУ
(91)
(184) 957.
Методы получения 1,2-аминоспиртов, исходя из эпоксидов или
азиридинов, приводят обычно к транс-\,2-аминоспиртам или сме-
сям цис- и транс-изомеров. Поэтому заслуживает внимания иссле-
дование Шарплесса и сотр. [190], направленное на разраоотку
стерео- и регпоизбирательных синтезов цис- 1,2-аминоспиртов. ти
авторы обнаружили, что хлорамин Т реагирует с олефинами в
присутствии каталитических количеств тетраокснда ОСМ‘1Я с ’
Разовапнем сульфамидных производных цис- 1’2'aMIIBJ: Р ея
°,следующее удаление сульфамидной группы осу
«ствием натрия в жидком аммиаке (уравнение 92).
он он
Na. ХНз (92)
NHTs NH2
МеЖут₽УГ0М ва1’1|анте этого метода [191], K0T°P“eB™fi реагентвза-
иц01'-°'|1!ое образование сульфамида, пмпд восстановлении
Им°ДеиСТвует Непосредственно с олефинами, а прн восста
6 3“к. 105
TsNCI Na+
"1% 0,0.,
продуктов реакции получают /(/«.МЛамшюснирты с высоким,,
выходами (уравнение 93).
„ '• OsCi|' rp.-pBuNI^Caju, ]>1|С!1(О1!)С!1 Ni!UlJ.r
PhCH=CH; 1
Апробированные методы
Ний в этаполамнны приведен
превращения карбонильных
ы в уравнениях 94—100.
Вг
соедини-
ВГ2. СНС1з I
R’CCTCHaR3 —----------R’COCllR
NRSR“
1
R’COCllR3
R2MgHr
HO NR6R“
I I
R'R2C—CHR3
(94)
|n2. Pt
OH NR5R6
R'CH—CHR3 (95)
Br OH Br OH NR5R“
I NaBHi I I RSR6NH I I
R'COCHR3 --------> R'CH—CHR3-----------y R'CH—CHR3 (96)
NOH OH NH2
пептилпптрит II Ha, PI I I
{'COCHaR3 ----------+ R'COCR3----------► R'CH—CHR3 (97)
OH OH
HCN 1 LiAIH, I
R'CHO ------* R'CHCN--------> R'CHCHaNHj (98)
OH
Ha, Pd, RSNHa I
R'COCHO -----------> R'CHCHaNHR5 (99)
OH
112. PI I
R'COCN----------> R'CHCHaNHa (Ю0)
Использование этих методов в синтезе арплэтаиоламниов
обсуждается в работах [192, 1931.
Область применения цнангидринового синтеза (уравнение 98)
ограничивается альдегидами, однако введение в практику органи-
ческого синтеза триметплсилилцнаиида позволило распространить
этот метод и на некоторые кетоны [194]. В присутствии катали-
тических количеств иодида цинка образование а-силилоксипитри-
лов (185) протекает быстро даже при комнатной температуре и
1,2-аминоспирты (186) получают с хорошим общим выходом
(67—98%) (уравнение 101).
ОН
1. LIAlIl., |
------> R1—С—R2
2. Н* |
CH.N1I2 <101>
(186)
OSiMe;
MejSiCN
R'COR2 ---------->
Ziilj
R1—С—R2
CN
(186)
130
В настоящее время для получения 1,2-аминоспиртов испочь-
зу|0Т «замаскированные» а-аминокарбапионы. Зеебах и Эндере
[195] установили, что нитрозирование вторичных метиламинов
позволяет вводить атом лития к углеродному атому, находящему-
ся в а-положеиии к азоту. Образовавшийся карбанион легко
реагирует с карбонильными соединениями, давая аддукты (187),
которые превращаются в 1,2-амииоспнрты с высоким выходом
(уравнение 102).
NO NO
EtONO |
MeNHR5 ------->- MeNR5
(изо-Рг)г№ Li* |
-------------> Li+ ’CH2NR6
R1R2CO
H2O
OH NO OH
I I H+ |
—► R'R2CCH2NR5 -------> R'R2CCH2NHR5 (102)
(187)
Разработана методика проведения описанной последователь-
ности реакций в одном реакционном сосуде, сводящая к миниму-
му контакт с потенциально канцерогенными N-нитрозоамннами
[196]. Другой возможный вариант, позволяющий исключить рабо-
ту с N-нитрозосоедипениями, состоит в использовании литий-
производпого N-(дифенилметплеи)метиламина (188) (197] (урав-
нение 103).
ОН ОН
R1R2CO | И+ I
LiCH2N=CPh2 ------->- R'R2CCHN=CPh2 --->- R>R2CCH2NH2 (103)
(188)
В реакциях с кетонами и альдегидами металлические произ-
водные изоцианпдов (189) также могут выступать в роли «замас-
кированных» а-аминокарбаиионов (198]. Для того чтобы избе-
жать образования побочных продуктов, анион (190) превращают
в ацетат и при последующем гидролизе получают этаноламнн
(191) с высоким общим выходом (уравнение 104).
Li Li+ ’О \’С
I RIR-’CO | I AcCl
R’R’CNC --------> R'R2C—CR3R< --------*
(189) (190)
г AcO NC I OH NH2
—» LR'R2C—CR3R‘J —R‘R!C—CR3R’ (KM)
(191)
Реакция металлических производных пиридинов (192) и ме-
таллических производных 2-алкппамииов (194) с карбонильными
соединениями лежит в основе специальных методов получения
1,2-амииоспиртов, для структуры которых характерно наличие
пиперидинового цикла (например. 193) [199] или ацетиленового
Фрагмента (195) (200], соответственно.
5*
131
(105)
nr-'R’ h° “r5r‘
I R1R2CO I I
Li(iH_C=CR --------” R'R’C-CHteCR
(194) (>95)
(106)
Синтезы, включающие восстановление аминокислот „ли их
сложных эфиров алюмогидридом лития [201] (уравнение 107),
или действие реактивов Гриньяра [202] на сложные эфиры ами-
нокислот (уравнение 108) также могут быть использованы для
получения ’1,2-амниоспиртов (196) и (197), соответственно.
R3
ЫАЦ-Ц
R8
I
R2OOC—С—NR'RS
I
R*
R'MgBr
HOCH2—C—NR5R5
I
R
(196)
R' R8
I I
HOC—C—NR5R8
I I
R‘ R*
(Ю7)
(108)
(197)
(2) Свойства
1,2-Ампноспирты с низкой молекулярной массой в большин-
стве своем являются вязкими жидкостями, обладающими силыю-
основнымп свойствами. Они растворимы в воде и нс отщепляют
аммиак. В случае благоприятной стереохимии они образуют
координационные соединения с солями меди и кобальта [203,
204].
Основные свойства 1,2-амипоспнртов, отличающие их от дру-
гих алканоламинов, обусловлены образованием водородной связи
между гидроксильной и аминогруппами. Вследствие электроотри-
цательного характера гидроксильной группы понижается основ-
ность аминной формы (198), однако водородная связь способ-
ствует стабилизации протонированного иона (199). Влияние во-
дородной связи на реакционную способность и спектральные
особенности отдельных функциональных групп наиболее четко изло-
жено в статье Рапопорта и Масамупе [205], посвященной стерео-
химическому отнесению цис- и транс-изомеров 10-гидроксикодеи-
на (200). Данные, полученные при изучении растворимости, ИК-
спектров и рКа, были использованы для объяснения легкости
окисления цис-изомера и его деградации по Гофману и для
132
объяснения устойчивости к гидрогенолизу
ной группы в транс-изомере.
бензильной гидроксиль-
Фрагмеит 1,2-ампноспирта в виде этаноламина, холина или
серина является структурным элементом глицеринфосфатов (201)
и сфингозинов (202), относящихся к различным классам фосфати-
дов. Эти соединения, в состав которых входит как липофильная,
так и гидрофильная группы, представляют собой, вероятно, пере-
ходный тип между водорастворимым белком и неполярными
липидами п играют определенную роль в разнообразных физиоло-
гических процессах наряду с их основной функцией в метаболизме
жиров.
CH=CH(CH2)i2Me
неон
I
RCONHCH
I
CH2OR2
(202)
R2 = CH2CH2NMe3
, У кефалины
R1 = CH2CH2NMe3 )
R1 = CH2CHNH2; лецитин
I
СО2Н
RCO = остаток жирной кислоты
CHSOCOR
I
RCOOCH О
I II
ch2opor>
I
он
(201)
R1 = CH2CH,NH2
Входящий в состав фосфатидов в качестве основания холин
выполняет также специфические функции как донор метильной
группы в процессе превращения гомоцистеина в незаменимую
аминокислоту метионин и как предшественник ацетилхолина
AcOCH3CH2N+Me3, являющегося .химическим медиатором некото-
рых механизмов нервной регуляции в организме человека, таких,
как парасимпатические процессы в периферической нервной си-
стеме.
Фрагмент 1,2-амппосппрта входит также в состав двух других
физиологически важных соединений: адреналина (203) и нор-
адреналина (204)—главных медиаторов симпатической нервной
системы. Терапевтическая ценность этих катехппампнов признана
уже давно; история их открытия, получения и первых попыток
133
,тпх соединений интересно описана Хартунгом
синтеза аналогов этих с 30 лет наметился прогресс в
’ ... соединений, которые оказывают то же действие,
медиаторы, или изменяют его характер. Для
.I.",-, активности важно наличие 1,2-амино-
г Лпагмента'"'однако вариация заместителя при атоме
спиртового фрагмен , |а пЮВОЛИЛа получить широко приме-
клинической практике терапевтические средства, напри-
(205) -- (208).
R\
CH(OH)CH2NHR2
[206]. Однако лишь
области поиска соед
что и эти природные
проявления биологической
азота пли в остатке
няемые в к....
мер соединения
но-
(203) R^OH, R2 = Me
(204) R’ = OH, R2 = H
(205) R' = OH, R2 = uao-Pr; изопреналин (меднгалер);
бронхорасширяющее средство
(206) R’ — HOCHo, R2 = Tper-Bu; сальбутамол (веитолин);
бронхорасширяюшее средство
(207) R’=H2NCO, R2 = CH(Me)CH2CH2Ph; лабсталол
(трандейт): антигипертензивное средство
НО.
CH(OH)CH2NHBu-rper
но/
(208); тербуталин (бриканил); броихорасширяющее средство
(3) Реакции
Химические реакции 1,2-амииоспиртов можно рассмотреть на
примере свойств простейшего представителя этого класса соеди-
нений, этаноламина. Для него характерны реакции по обеим
функциональным группам, хотя во многих случаях на скорость
реакции влияет наличие внутримолекулярной водородной связи,
римером служит замедление скорости алкилирования амино-
р ппы | и/]. Следовательно, особенность химического поведения
те ,а,п"1'0СП"рТОВ. связаиа с возможностью проведения пзбира-
тзкже nenvfК1-11И fKaK П° амии0'- так 11 по спиртовой группе, а
Ю1Ш1Х обе (hvu" образова|"1Я Циклических соединений, затрагива-
ющих обе функциональные группы.
ствляется"₽п,Впппе> ЗТОМа азота в 1,2-ампноспиртах легко осуиге-
использоПанипРэквпммьиогоИО?аИ11Й (|1апр"меР’ ппрпдииа) при
гидроксильная группа поп° к“Л|,чества алкилирующего агента;
он группы на алкгД. Р "е затРагивается. Замещение
создания ка рбен1?еКвогоИГРУППУ °бЫ'"10 требУет предварительного
группу. Имеются aamuJ,0II\i "а УглеР°Дном атоме, несущем ОН-
к > деются данные о N-алкилировании этаноламина при ки-
пдченпп с производными четвертичных оснований (208], однако
белее удачным способом моно-Гй-алкилпровання является восста-
новительное алкилирование и присутствии альдегида или кетона,
как например, при почти количественном превращении этанол-
амн’на в N-изопропилпроизводное [209].
Так же легко осуществляется катализируемое основанием
ацилирование атома азота; гидроксильная группа при этом не
затрагивается. Для избирательного ацилирования гидроксильной
группы обычно необходимо предварительно защитить атом азота.
Наиболее простой способ защиты аминогруппы заключается в
протоинроваиип с помощью минеральной кислоты, а последующее
ацилирование гидроксильной группы осуществляют хлорангидри-
до.м кислоты. Аналогичный метод был использован в синтезе
триалкилсплилоксипроизводных гидрохлорида эфедрина в безвод-
ных условиях [210] (уравнение 109).
ОН Me RaSiO Me
I I t RsSiCl. C5H5N I I t
PhCH—CHNH^Me Cl’ -----------> PhCH— CHNH2Me СГ (109)
Для замещения гидроксильной функции галогеном или алко-
кепгруппой используются условия, в которых углеродный атом,
несущий гидроксильную группу, образует карбенпевын нон (см.
разд. 6.2.4 и 6.2.5). Для превращения этаноламнна в 2-бромэтил-
амин необходимо нагревание с 48%-ной бромистоводородной
кислотой |211], тогда как арплэтаноламины можно превратить в
хлорамины (209) с помощью хлористого водорода на холоду
[212] (уравнение 110).
ОН С1
I * по I *
ArCIJCHjNHs СГ ---> ArCHCHsNHj СГ (110)
(209)
Предварительное образование карбениевого иона лежит также
в основе метода Венк.тера (уравнение 111), позволяющего пре-
вращать 1,2-амивоспирты в азиридины путем перегонки их О-
сульфатов в присутствии щелочи. Замыкание цикла облегчается
при использовании 1,2-дизамещенных этаноламинов; при этом по-
вышаются и выходы азиридинов (186,213].
он oso; nh
I H2SO, I + "ОН / \
RCHCH:NH2 ------RCHCHjNHa ------► RHC---СНг (HI)
Карбенпевые ионы, получаемые из арплэтаноламинов, нашли
особое применение в синтезе 4-арилтетрагидропзохпнолпнов с по-
мощью катализируемой кислотами циклизации N-бензилпропзвод-
ных (уравнение 112) (214].
Аг
НО Аг |
СО- - ОС»
135
гпглых условиях арилэтаиоламипы могут вступать в
В силь"° " 'обусловленные «окснамииовым распадом»
побочные речки . 0>ъяснеш1я этого распада было предложи
(ураВНемехЯнпзма однако последние данные подтвердили Промс.
нодва механп , енамина (211) и не было обнаружено ни-
,21"1|2|5>-
он он
ArCH-CHR3
(2Ю)
/R3
АгСН=С
OH NR5R°
ArCH—CHRS -
-> ArCHiCOR3 (113)
(211)
При обработке иодной кислотой 1,2-аминоспирты с первичной
или вторичной аминогруппой могут также расщепляться по
углерод-углеродной связи в соответствии с реакцией, аналогич-
ной реакции расщепления а-глпколей [216]. Соединения с третич-
ной аминогруппой не реагируют с перйодатом, по расщепляются
при действии тетраацетата свинца (уравнение 114) по механиз-
му, включающему образование иминиевого попа (212) [217].
PhCH(OH)CH2N( \ —> PhCHO + Tch2=N ')1 (114)
'—' L \—/J(212)
Окисление перманганатом калия было использовано как метод
превращения протонированных 1,2-ампноспиртов (213) в амино-
кислоты (уравнение 115), однако выходы были ниже, чем при
использовании соответствующих N-ацилпроизводных [218].
R3 । R3
R“R5NCCH2OH 7-^^- R«R5N-C—СООН
R< ' '
(213)
Гоф^Ваа|1уРобнчн»Япп[2пМИНОСПИртОВ c ,,оследУ1Ощим распадом по
чаях (см. 1|апппмрп'В0ДИТ К Э||оксиДам, Однако в некоторых слу-
углеродио’й связи f2P19]1>aB"e"lie И6) П₽ОИСХ°ДИТ разрыв углерод-
(115)
(116)
ВСН(0Н)-у; ОН^ОСН2СН2НЗше
Me /
Термине i .
еапии с помощью хлооТп1т<,СеТа1,Иа’ ПОлУчаемого при кватерни-
136 ц та НатРия, используется также как
метод обнаружения этаиоламипов [220J. Образующийся в этой
реакции ацетальдегид можно определить с помощью раствора, со-
держащего морфолин и нитропруссид натрия (уравнение 1’17).
CICH2CO2Na „ д
R2NCH2CH2OH ---------► R2NCH2CHzOH ---->- R2NHCH2CC2H + СНзСНО
СНгСО; (117)
При действии нитрозирующих агентов на 1,2-аминоспирты мо-
жет происходить перегруппировка, аналогичная пинаколиновой
перегруппировке [221, 222] (уравнение 118).
R2 R3
| | I. нмо2
R'-c-c-r4
но nh2
г R2 R3 -1 R2
I I I
R'—С—С—R4 —► R1—С—С—R3
I ‘ 8 1
L НО J OR4
(118)
Частным примером этой перегруппировки является расшире-
ние цикла 1-аминометилциклоалканолов (перегруппировка Тиф-
феио — Демьянова), широко используемая для превращения цик-
лических спиртов или кетонов в циклические же продукты, цикл
которых содержит на один атом углерода больше (уравне-
ние 119).
W/0H HHO2 W п
/ У ------>- ( У=О (119)
'У \ch2nh2 '----------'
Кроме образования оксазолинов и морфолинов соответственно
из карбонильных соединений и этпленоксида (см. разд. 6.2.4) пер-
вичные и вторичные 1,2-амииоспирты способны реагировать с ря-
дом реагентов, давая циклические соединения; типичные примеры
приведены в уравнениях 120—123 [223—226].
137
(i.2.3.3. 1,3-Аминоспирты
(1) Методы получения
„ . .„„„тп R'R2C(OH)CH2CH2NR3R4 обычно получаю,
| З-Лмнноспирты К к DIDirnfH ГН ND1D1 ikj"1
.«mm, ft-ампнокетонов R K CUL112С112М R !R4 2271
HpeiXuecTBeiu'io алюмогидрндом лития (228]. Другой метод по-
биения I 3-амнпоспп|>тов действием реактивов Гриньяра нередко
' зет низкие выходы, что обусловлено енолизацпен кетона и по-
следующим образованием соли. Несмотря на это осложнение,
реакция в ряде случаев находит применение [229]; лучшие ре-
зультаты дает использование литийорганических соединений
1230] Для получения 1,3-ампноспнртов в реакциях с реактивами
Гриньяра и с восстанавливающими агентами гидриднои природы
можно исходить также из 0-а.минокислот и их сложных эфиров
[231].
Поскольку ненасыщенные цианоэфнры (215) легко получают-
ся по реакции Кпевеиагеля (232], восстановление этих соедине-
ний алюмогидрндом лития может оказаться наилучшим способом
получения некоторых 1,3-амнпоспнртов (уравнение 124) [231].
ZCN
RCH=CZ —> RCH2—CH— ch2nh2
^•CChEt I
CH2OH
(124)
(215)
Еще один общий метод получения 1,3-ам|шосппртов состоит в
катализируемом основаниями алкилировании ампиосоединенпй
или их N-ацилпропзводных действием 1,3-галогеигидринов в кон-
тролируемых условиях с последующим удалением N-защитной
Э™ мет°Д позволяет получать производные пиперидина
(216) с хорошим выходом (233] (уравнение 125).
Me
.Me
NCH2CH2CH2OH
(125)
(216)
оказалось возмож-
аммнака или ямкип» емое основанием присоединение
лени у а е, ад ™°ВЫМ СП"рТаМ при повышенном дав-
₽-Щ.каХилыи: ^-вов^оио^ аминирование
снг=снсн2он Mc2NCH2CH2CH2OH
(Месо)2сн2
♦ Me2NCH(Me)CH2CI 1(01 l)Me
(126)
(127)
138
(2) Свойства
Сходство свойств этого класса соединений и 1,2-амииоспиртов
заклю,,аетСЯ в том, что и в данном случае внутримолекулярная
водородная связь увеличивает устойчивость и растворимость и
понижает основность. Значение рКа З-гидроксипроииламина прн
25 °C составляет 9,96.
(3) Реакции
Как и для 1,2-амииоспнртов, для 1,3-амииоспиртов характер-
ной химической особенностью является возможность избиратель-
ного ацилирования или алкилирования как но атому азота, так
и по атому кислорода. Гидрокси- и аминогруппы 1,3-амииоспир-
тов с первичной пли вторичной аминогруппой могут, кроме того,
вступать в реакции циклизации с образованием производных 1,3-
оксазина. Так, например, конденсация с альдегидами и некоторы-
ми кетонами приводит к тетрагидро-1,3-оксазинам (217) [236].
(217)
6.2.4. ПРОСТЫЕ АМИНОЭФИРЫ
6.2.4.1. 1,1-Аминоэфиры [117,237]
Эти простые эфиры карбииоламинов изображают общей фор-
мулой (218).
R3
R'RWCOR5 (218)
(/) Методы получения
1,1- Амииосппрты, полученные in situ из шиффовых оснований
RCH = NR или методом, описанным в разд. 6.2.3, реагируют со
спиртами, образуя простые 1,1-ампноэфвры. Наилучшие резуль-
таты этот метод дает в случае относительно стабильных карбннол-
аминов, полученных, например, из формальдегида и вторичных
аминов (уравнение 128). Альтернативный метод получения 1,1-
аминоэфиров (219) состоит во взаимодействии вторичных аминов
с простыми 1,1-галогенэфирами. например с C1CH?OR [5, 238].
EtOH
EtjNH + HCHO —> EtiNCHjOH -----------► EbN’CH.OEt (128)
(2)9)
139
(2) Свойства
Ничише 1,1-ампноэфпры представляют собой жидко-
..... которые можно перегонять без разложения, часто даже
поп атмосферном давлении. Они легко растворяются в органиче-
ских растворителях и не растворяются в воде. Точно измерить
основность этих аминов невозможно, поскольку они очень легко
гидролизуются разбавленными кислотами, однако ва основании
рассчитанных для них значений р/G, можно предположить, что
индуктивный эффект простои эфирной группы понижает основ-
ность па 2—3 единицы по сравнению с основностью соответству-
ющих алкпламннов.
(3) Реакции
1,1-Амнноэфиры более устойчивы, чем исходные карбинолами-
11Ы, однако химическое поведение обоих классов соединений весь-
ма схоже. Опп проявляют себя как электрофильные агенты по
отношению к ряду соединений: реакции этого типа приводят к за-
мещению алкоксигруппы. Например, при действии хлористого во-
дорода пли галогенангндрндов карбоновых кислот 1,1-амнпоэфп-
ры превращаются в а-галогенампиы R'R’NC(R3R4)Hal (см.
разд. 6.2.5.1). В условиях каталитического гидрогенолиза или при
действии алюмогндрида лития происходит восстановительное
расщепление 1,1-аминоэфиров до соответствующих аминов
R‘R2NCHR3R4 [239].
Простые 1,1-амнноэфиры получили широкое распространение
как аминометилирующие реагенты (уравнение 129) [240], причем
особенно легко происходит замещение в пара-положение фенолов,
даже при комнатной температуре и в отсутствие обычно добав-
ляемой кислоты (уравнение 130). Кроме того, в отличие от обыч-
ной реакции Мапниха простой амнноэфпр (219) реагирует с
хлормагниевым енолятом (220), давая р-аминокетон (221) [241]
(уравнение 131). 3 ’ 1 1
140
Высокая реакционная способность 1,1-аминоэфиоов
теплю К реактивам Гриньяра позволила разработать
способ "ff'Zi ,'аЛО,с,,адов и ацетиленов соответственно
амины (222) {242] и аминоспирты ацетиленового
1243]. В модификации этои методики для получения И а
'он (225) |244] используют реакцию а-бромамидов (224)
эфирами в присутствии магния. '
PI1CHNR] + R-Max
OBu
no OTHO-
простой
(i-аминоами-
амино-
PhCHNRj
1
R2
(222)
NR'R2
НО(СН2)4СНС=СН
(132)
,NR'R2
(224)
CHsCMgBr + ( ;o
BrC(Me)2CONH2 + О
NCH»OBu
(223)
(225)
(134)
(133)
(4) Оксазолидины [245]
Характерными представителями простых циклических 1,1-ами-
иоэфиров являются оксазолидины (226).
Получение. Общий метод получения циклических простых эфи-
ров карбпполамнпов (226) из 1,2-аыиноспиртов и альдегидов или
кетонов показан в уравнении 135. Однако только кетоны легко
конденсируются с первичными 0-гидроксиалкиламииами (227;
R1 = Н).
R3 NR‘
R3R2CO + HOCH2CH2NHR1 —* X I (135)
“Н2° R2/ \ X
О
(227) (226)
Получение оксазолидинов восстановлением оксазолинов, на-
пример (228), сопровождается осложнениями вследствие легкости
сверхвосстаповлення до ампноеппртов (231) (схема 136). Однако
продукты восстановления (230) солей четвертичных аммониевых
оснований (229) борогидрпдом натрия не находятся в равновесии
со своей ациклической формой и могут быть выделены /246].
Соль четвертичного основания (229) реагирует с двумя экви-
валентами реактива Гриньяра в эфире; при этом в результате
Раскрытия цикла образуется аминоспирт (232). Этот процесс,
включающий образование комплекса между магнием и атомом
кислорода оксазолидппового цикла, может быть подавлен р
Проведении реакции в гексаметплтрпампдофосфате (г<.к
141
R’ClhNllC(Me)jCH’OH
R’
\—NMe
о( УСМе2
HjNClMehCHzOU
R'CO.H----------------
(231)
fNaBH4.
EtOH
R’
о' )с.Ме2
Mel
MeNO2
(228)
R?MgHaI,
EtzO
R;C-V
I
,/Me
'4C(Me)2CH2OH
(230)
NaBlli,
ЕЮН
R1
NMe
О\^СМе2 (136)
(229)
RSMgHal,
ГМФТА
R1 R2
V-NMe
о; )сМе2
(232)
этом растворителе арильные, бен-
(233)
фосфортрнамид, ГМФТА). В этом растворителе арильные, бен-
зильные и алкенильные (но не алифатические) реактивы Гринья-
ра взаимодействуют с солью (229), образуя оксазолпдпны (233)
[246]. _
Свойства. Оксазолндины представляют собой жидкие или твер-
дые вещества, которые можно перегонять. Они не поглощают
в ближней ультрафиолетовой области, за исключением тех слу-
чаев, когда в структуре присутствуют хромофорные заместители.
Но в ИК-спектрах оксазолпдинов наблюдается характеристиче-
ский триплет полос поглощения в области 1200—1080 см-1.
Для ПМР-спектров оксазолпдинов типа (230) 1247] характер-
но наличиесигналов в следующих областях: б 4,82—3,90 (OCHN),
Зр7як,ш4и2 v0™2)- 6 2,19—2,0! (NMe) и 6 [,18—0,95 (СМе2).
н пГипвм ’ СТ0|,ч,,п°сть оксазолидпиов в условиях гидролиза,
и этаноп’ямш|МПКа’ <~>кеазол11-'1Ш|, полученный из цпклопептанопа
зуются киспп^хГНДР°’1!1пУеТея водой- Вее оксазолндины гидроли-
|И1Н. Описаны птп/° '2-амнносппртов и карбонильных соедине-
пропзвотные окг^г™Ше -СТойчнвые гидрохлориды н N-ацильные
путь окислительно[245’ Оксазолпдинь. можно подверг-
137) 12451- ппп Раешеп'лепшо в аминокислоты (уравнение
138- см. также гзгГрдяГ "НХ Реагс11тов Гриньяра (уравнение
1 1-101 или при восстановлении образуются
замешенные амипоспирты (231). Однако все возрастающее эиаче
соединения
) 12491.
пне они приобретают именно как промежуточные сеед
при синтезе альде! идов или кетонов из оксаэолинов (228)
/—°, КМпО,
—Ph --------► HO2CCH2NHMe + PhCOOH
NMe
(137)
/—0
—Ph + MeCH=CHMgBr
NMe
Ph
MeCH=CHCHNMeCH2CH2OH
(138)
(5) Тетрагидро-1;3-оксазины
Методы получения, свойства и реакции тетрагпдро-1,3-оксази-
цов (234) почти такие же, как и в случае оксазолидннов; этн сведе-
ния изложены в обзорах [236, 250, 251],
Тетрагпдро-1,3-оксазпны (234) приобрели большое значение
в последние несколько лет как промежуточные соединения в
чрезвычайно широко распространенном методе синтеза карбоно-
вых кислот из дпгидро-1,3-оксазннов (235) [246].
(234) (235)
6.2.4.2. 1,2-Аминоэфиры
(1) Методы получения
Общим методом получения простых 1,2-ампноэфнров
R’OCH2CH2NR2R3 является алкилирование соответствующих
1,2-амипоспиртов (см. разд. 6.2.3.2) дпалкилсульфатом в сильно-
кислом растворе (уравнение 139) [252], 2-Метоксиэтиламин (236)
можно также получить с очень высоким выходом по реакции эти-
ленимипа (237) с метанолом в присутствии тре.хфтористого бора
[253] (уравнение 140).
Me2SO4. 6н. HCI. АсОН
НОС.Ч2СН2МН2 --------------------> МеОСН2СН.\Н2 (139)
(236)
\Н BF-.
/ \ 4- МеОН -------- (236) (140)
(237)
2-Метокснэтиламин (236) был синтезирован и нз монометило-
вого эфира этиленгликоля (238) при действии аммиака и водоро-
да в присутствии никелевого катализатора при атмосферно*
из
Г9541 а также реакцией тозилата (239) с бис (бензол-
даВжП,Т„(.пом пития и последующим взаимодействием с тнофе.
SSSKV--.
„ом 2±!> Ме—(/ y-SO2OCH2CH2OMe --------------.
НОСНгСНгОМе ме Х^/
(239)
<238)
—» (PhS)2NCH2CH2OMe -------” (236)
(141)
(2) Морфолины [251,252]
Получаемые из этиленоксида и 1,2-аминоспнртов диэтанол-
амины (240) (см. разд. 6.2.3.2) легко циклизуются в кислой среде,
образуя тетрагидро-1,4-оксазины (241) с высоким выходом. При
нагревании с концентрированными минеральными кислотами дп.
этаиоламин превращается в морфолин (241а), тогда как при на-
гревании гидрохлорида (2406) в метаноле образуется диметиль-
ный аналог (2416). Морфолин (241а) и его N-замещеииые про-
изводные легко получаются также при взаимодействии
2,2-дихлордиэтилового эфира (242а) пли дисульфопата дпэтилен-
гликоля (2426) с аммиаком или аминами.
О
н7 HO(CH2)2NHCR2CH2OH —> СХ' NH к (142)
(240а) R = H (241а) R = Н
(2406) R = Me (2416) R = Me
КНз ХСН2СН2ОСН2СН2Х > (241а) (143)
(242а) X = CI
(2426) X = OSO2Ph
прос-
(3) Свойства
> эффекта алкоксигруипы основность
ниже основности соответствующих алкил-
например, для 2-мегоксиэгиламииа (236) рХа = 9,61, а
Из-за индуктивного
тых 1,2-амииоэфнров i
аминов; г-------
ДЛЯ морфолина (241а) рХа = 8,33 (прн 20°С),' '
г1ПЗПП1Р я пiirh о типп л..... ,, '
простые амииоэфиры растворимы
Низшие алифатические
в воде.
пскотооых ”«тТаЯ группа является элементом структуры
ион и седативнойTBeHIIblX пРепаРатов, обладающих аитпгпстамин-
ных Д11фепиапамчаКТ"А1ЮеТЬ10' в чг,гтиости, соединений, родствен-
вый никл иаЩ)о1ееУчтсто'|1Лп'!',') (243)' КрОме Т°Г°’ M0p(!>0JI!’110'
мент в некоторых пХ, |1сп°льзуют как третичноаминнын ФРаг'
нервную систему- f„i₽ варатах’ воздействующих па центральную
У- является также важным элементом струк-
144
тупы недавно предложенного
валян) (244).
Ph2CHOCH2CH2NMe2
(243)
.\П
(244)
(ви-
(4) Реакции
Реакции 1,2-амнноэфиров обусловлены наличием простой
эфирной и аминогрупп. Эти соединения устойчивы. Так, морфолин
не гидролизуется водой при 200 С, 10%-ным водным гидроксидом
натрия или 11 и. соляной кислотой, а также не окисляется пер"
манганатом калия. Однако при действии пероксида водорода он
превращается в 4-гидрокспморфолпн. Морфолиновый цикл рас-
крывается бромцнаиом, а при распаде солн четвертичного основа-
ния (245) по Гофману образуется простой виниловый эфир (246).
О\ /NMe> I —» О\__________/NMe2 (144)
(24Б) (246)
Морфолин обладает также свойствами хорошего нуклеофила,
и вследствие этого его предпочитают всем другим вторичным
аминам в таких реакциях, как реакция Маннпха (уравнение 145)
[256], и модификация Киндлера реакции Внльгеродта (уравне-
ние 146) [257].
с/ ^йн2 СГ + МеСОМе + НСНО —> с/ ^ЫСН2СН2СОМе • HCI (145)
(146)
СН2СООН
6.2.4.3. 1,3-Аминоэфиры
(I) Методы получения
З-Метокснпропнлампп можно получить прямым
пнем З-гпдроксппроппламнпа диметилсульфатом в к । Д ₽
творе [258| а также восстановлением З-метоксппроппопптрп
MeOCH.CH^CN в условиях каталитического «рова.шя над
никелем Ренея в присутствии аммиака [259] (вых Д /о)
Действием натрия в спирте [260].
145
1,3
женин
легко
, л л,„пи содержащие гидроксильную группу в поло-
’^'(247) представляют собой важную группу соединений,
полЙмЫхР пз эпихлоргидрина (уравнение 147) [261].
/ \ кон / \ R2NH2
R.OH + C1CH2^^ — R'0CH>----------------*
он
R'OCH2CHCH2NHR2
(247)
(U7)
(2) Свойства и реакции
Как п в случае простых 1,2-аминоэфиров, свойства и реакции
1,3-амипоэфпров складываются из свойств, присущих алкокси- и
аминогруппам.
2-Гидроксианалоги (247) и особенно те соединения, в которых R
является арильной группой, имеют значение как терапевтические
средства. Подобные соединения подавляют действие адреналина
на рецепторы организма, в частности на адренорецепторы сердца.
На основе этих веществ был создан ряд лекарственных препара-
тов для лечения ангины, сердечной аритмии, а в последнее время
и для лечения гипертонии. Наиболее широко используемыми пре-
паратами являются пропранолол (индерал) (248а) и окспрено-
лол (тразпкор) (2486).
ROCH2CH(OH)CH2NHCHMes
(248а)
(2486)
6.2.4.4. Макроциклические аминоэфиры [262—264]
(/) Методы получения
ДоВатРеТХт?^,1пЭфИрЬ' ТППа (255> МОГУТ быть получены после-
Реакция дпампппР7!МспИ’ приведеН11ЫХ в Уравнении 148 [265].
бавлеппом пасттпр249 С д11ХЛора|1П1дридом (250) в сильно раз-
становленпп алюмогито'щт‘Т К Л1,амидУ <251). который при вос-
Дает тетраоксадпамйнР(2521 В К1|пящем тетрагпдрофуране
хлорангпдридом , ’’ г’10 соед||1|ение конденсируют с ди-
дмеск„н.диампд. (253) воестТ^.’’’.'^.^Р^тате этого б|1ЦПК-
ноборапа) (254), кОТо1,,ц;ОССТаиавлива1О'г Дпбораиом до бнс(ами-
ляпой кислотой ппевпа’п^Л.?"010 очеРедь. при гидролизе 6 И. со-
ОбщиП кыход кощчХТпХт ДПГ1,дР°хд°РИд Диамина (255).
пательпости реакций гп Ролукта ПР” проведении этой иоследо-
составляет около 25% (уравнение 148).
(254)
Gh.HCI
Г 0 0 1
(255)
(148)
Макроциклические аминоэфиры получают также, не прибегая
к сильному разбавлению реагентов, используемых обычно для
циклизации [266]. Описано получение макротрпцпклнческого
амииоэфира (256) [267] н макротетрациклпческого амииоэфпра
(257) [268], а также соединений типа (258) [271].
147
(2) Свойства
. гптктсоиым свойством макробнцнклнческпх амино.
, На" игшетсяРнх способность образовывать соединения включе,
эфиров двоя ’°” иахад11ТСЯ в центральной полости
макооб цик«ческой системы, такой, например, как (256). Для
“того нового класса комплексов металлов было предложено на-
ЗВ7оеХюш|Те'Т1?255Г9является очень сильным комплексующим
агентом- в его водном растворе медленно растворяется осадок
сульфата бария, а в бензольном растворе - перманганат калия.
Изменения, происходящие в растворах соединения (255), напри-
мер в хлороформе, к которым была добавлена соль металла,
можно проследить с помощью ЯМР-спектроскопни. Так, тиоциа-
нат калия растворяется за несколько секунд, тогда как растворе-
ние хлорида бария требует нескольких дней. Комплекс амииоэфи-
ра (255) с калием более устойчив, чем комплексы с натрием пли
рубидием; комплексы с литием н цезием крайне неустойчивы
[270].
Соединения типа (258) также образуют криптаты, например,
с КВг, CsBr и ВаВг2 [271). Описаны крпптат-апиопы, схемати-
чески представленные формулой (259) [272]. Липин, соединяющие
атомы азота в (259), изображают полиметнленовые цепи, и хотя в
роли криптандов выступают полициклические тетрампны, этот при-
мер включен в данный раздел для общности изложения. Еще один
пример крпптат-анпона сходного строения приведен в разде-
ле 6.2.2.4. Аииои X- удерживается в полости молекулы тетрапрото-
нированного лиганда водородными связями. На сегодняшний день,
по-видимому, наиболее устойчивым анионным комплексом является
хлоридныи комплекс. Изучение подобных соединений должно пред-
ставлять интерес в связи с развертыванием исследований по изу-
чению связывания и транспорта анионов в биологических мем-
бранах.
в.2.5. ГАЛОГЕНАМИНЫ (С-ГАЛОГЕНЗАМЕЩЕННЫЕ)
6.2.5.1. а-Галогенамины
(1) Методы получения
а-Галогенамнны (261) „огут быТь
способами: действием галогена
реакцией галогеповодородов с
й-дпа л кил аминоэфира.мп (262 \
'(2621С /МШ,алям’1 (260) или 1
лд (УРавне1111я 149, 150)
Можно предположить, ,1Т0 прпппй „
ется присоединение катиона (нЛп, даеи ЭТ|1Х реакции явля-
И8 катиона (например, хлорония или протона)
получены [273] следующими
। па ампиали ([, 1-дпамппы) (260),
: аминалямп (260) пли простыми
п взаимодействием ацплгалогсшн
простыми а-дналкпламппоэфнрами
R3 R3
I llalx I
R.R2N-СИ-NR'R2 ----------- R'R2N-CH-Hal + R.R2NX (I49)
<260) (261)
X = Hal, H, COR6
Г Ha!X R3
R'R’N-CH-OR’ R'R2N-CH-HaI + R<OX (150)
(262) (261)
X = H, COR6
к свободной паре электронов у атома азота. Образующийся при
этом аддукт затем распадается, давая более устойчивые фрагмен-
ты, в том числе и а-галогеиампн (261).
’Из всех ранее упомянутых методов наилучшнм методом полу-
чения а-галогенамннов является взаимодействие ацилгалогенпдов
с ампиалями (260). Исходные соединения доступны, распад про-
дукта присоединения протекает количественно н сразу же вы-
падают солеобразные а-галогеиамнны в аналитически чистом со-
стоянии. Прн увеличении объема заместителей при атоме азота
выходы уменьшаются вследствие повышения растворимости а-га.то-
генамииов в органическом растворителе.
Ампиали (260) расщепляются фторангндридами карбоновых
кислот лишь после повышения реакционной способности фторан-
гидрпдов добавлением в реакционную среду эфирата трехфторис-
того бора.
(2) Свойства
а-Галогенампиы нерастворимы почти во всех инертных рас-
творителях [273]. Эти соединения правильнее всего изображать
в виде галогенпдных солей карбенневых ионов (263), стабилизо-
ванных за счет наличия атома азота [274].
(263)
На Г
С этим способом написания согласуются также и другие фак-
ты (уравнения 151). Так, а-галогенамины образуют в ацетонитри-
ле кристаллические перхлораты с перхлоратом серебра, при соль-
волизе безводной синильной кислотой получают замещенные амп-
ноацетоиитрилы (264), а взаимодействие с реактивами Гриньяра
в эфире приводит к третичным аминам (265).
Фторметнлдпалкнламнпы CH2FNR‘R2 [275] представляют
собой нпзкокипящпе жидкости, физические свойства которых о
лее всего согласуются с ковалентным характером связи угле
Род —фтор. Однако их реакционная способность напоминает ре-
акЦИоипую способность хлоридов, бромидов Н 11ОД11Д0В, нмеющи-
и°нное строение.
149
(261)
R\ /R’
xn-ch;
R3/ XCN
(264)
ft
IICM R'MgBy
*---- (263) -------*
^АвСЮ,
R'\+ /”
>\=cz cio;
Rs/ 'I?1
R'\ /R3
;n—chz
R2/ \R<
(265)
(151)
Поп контакте с водой а-галогенамппы разлагаются, образуя
эквпмольные количества соли соответствующего амина и альде.
гида.
(3) Реакции
а-Галогенамнны обладают свойствами мощнейших амипометн-
лпрующпх агентов, реагирующих с большинством анионных н
нуклеофильных центров; многочисленные реакции этого типа
систематизированы ниже. Для общности изложения сс-галогепами-
ны во всех случаях изображены в форме пммопиевой соли [см.
(263)1.
Образование связи С—С [276]. Реакция с цианидами. а-Гало-
генампны реагируют с цианидами или цианистым водородом, об-
разуя а-амипонитрилы. Например, хлорид N-(метоксикарбоиилме-
тилен)пипериднния (266), легко получаемый из дихлоруксусной
кислоты, вступает в реакцию с безводной синильной кислотой,
давая пнпернднпоинаноацетат (267) (уравнение 152; см. также
уравнение 151).
CN
С* исх /—\ I
N=CHCO;Me СГ -----> <__/N-CH-CChMe (152)
(266) (267)
Реакции с соединениями с активной метиленовой группой.
Введение диалкиламипомстнлыюй группы в соединения, содержа
щне активные метиленовые звенья, можно осуществить по реак-
ции металлических солей этих соединений с а-галогеиамииами.
пппично^0?-0пКрЬ1ВаеТ Путь к получению диалкиламппометпльпых
тпимп „"]Х Р'Д11Кетоиов 11 Р-кетоэфпров, которые обычными ме-
Напонмрн Н1!^!'’ет’1Л1’РУ10Тся с трудом или вовсе не реагируют,
реагппхют с об 0РмеТ11ЛП|1пеР'1Д1ш и натрпйдпэтилэтилмалонат
этплмалоиовой кислХ"Э<11ИРа П1'Пер"Д11“0ЫеТ“Л'
с октавной метиленовой группой.
I5Q
CH2-C(CO2Et)2
(268)
Механизм образования азетидннона (271) также можно рас-
сматривать как процесс, включающий промежуточное образова-
ть а-галогеиампиа (уравнение 153). Получающиеся при взаи-
модействии шиффовых основании (269) и соответствующих гало-
геиангпдрилов карбоновых кислот аддукты (270) циклизуются в
присутствии трнэтпламина с образованием азетидинонов (271)
[278].
R'-
_р<снгСОС1 Frir2CH—R’ R‘R3CI-1—N—г?3"]
I I СГ
L r4ch2—со r*ch2—со J
(270)
RJ/
(269)
Et3N
(153)
R2
R'—I—\_R3
R’~----1=0
(271)
металлорганическими соединениями. Замещенные
синтезировать реакцией а-галогенамииов с реактн-
Реакции с
амины можно _ , __
вамп Гриньяра (уравнение 154) [274, 279—281] (см. также урав-
ние 151).
R3
R1\t RMgX R1\ I
;n=c( сг ----------> ;nchr
R2Z \R3 R2/
(154)
В результате реакции аллнлмагнийгалогенидов с а-галоген-
аминами с почти количественным выходом получают уф-ненасы-
щениые амины (272), которые превращаются в 3-хлорамины прн
присоединении хлористого водорода (уравнение 155) [276].
R\ + /н
:у=сх
R'/ XR3 СГ
СН2=СНСПгМвВг
R3
| HCI
R'R2NCHCH2CH==CH, —>-
(272)
R3 Cl
—> RiR2NCHCH2CHMe (155)
Реакция дибромалкпламииов (273) (получаемых присоедине-
нием брома к енаминам) в форме пммоппевои соли с реактивами
I риньяра приводит к а-разветвленным р-бромалкиламинам (2/4),
которые реагируют с еще одной молекулой ал кил маги и пором ида.
Образующийся реактив Гриньяра (275) гидролизуется затем до
амина (276) [282].
151
(274)
R3
Н2О I
---* RCH2CHNR‘R«
(276)
(166)
Br R3
?r ZR' RSMgBr I I R3MgBr
RUcH=< Br- RC1I—CH—NRlR2 —
XR2
(273)
BrMg R3
__, RCH—CHNR'R:
(275)
Литийорганические соединения реагируют с а-галогенампнами
анаюгичным образом, давая а-амшюалкилпроизводные; два за-
служивающих внимания примера показаны в уравнениях 157, 158.
Реакция жидкого диметилфторметнлампна с трихлорметиллц-
тием (278) при —110 °C приводит к диметил (₽,₽,р-трихлорэтил)-
амину (277), который выделяют в виде солянокислой солп с вы-
ходом 70%. В результате обработки основания подпетым метилом
получают соль четвертичного основания (279) с выходом 90%
[275].
CIsCLi (278) Mel ♦
Me2NCH2F --------1- Cl»CCH2NMe2 -----> CI3CCH2NMe3 Г (157)
(277) (279)
При взаимодействии 2,2-дифеиил-1-хлорэтена (280) с «-бутил-
литием при —110 °C образуется карбеноид (281), который реаги-
рует с а-галогенампном, давая амин (282) [283].
CI
, н-ВиЫ | R1R-’N=CHR3 СГ
РЪ2С=СНС1 ------->- Ph2C--=C—L1 --
(280) (281)
Cl R’
—>- Ph2C=C—CH—NR'R2
(282)
Реакции с олефинами. Реакция а-метилстпрола (283) с суспен-
зией диметилхлорметиламнна (284) в ацетонитриле при компат-
отиошеншИ([284]ВОДИТ * и30меР0в (285> » <286’ в С°‘
Me
1
phC=CH2 + Me2N=CH2 CI’
(283) (284)
В аналогичном превращении (vo
Щеииых коричных кислот (287)
ется Декарбоксилированием
хлориды замещенных аллил’
152
(158)
CHj Me
® I
PhCCH2CH2NMe2 + PhC=ClICH2NMe2 (159)
(285) (286)
авнеине 160) реакция заме-
с а-галогепаминами сопровожда-
в результате чего образуются гидро-
аминов (288) [285].
Ме2Й=СН2 СГ + Me2N—О
CH=CHCO2H
(287)
CO2H
•CH—CHCH2NHMe2 2СГ ••
—> Me2N
CH=CHCH2NHMe2 СГ
(160)
(288)
а-Галогепампны могут реагировать также с диенами по ме-
ханизму циклоприсоединения типа реакции Дильса — Альдера;
примером служит реакция с 2,3-диметилбутадиеном (уравнение
161) [286].
Me Me
.Me
N=CH2 Br" + CH2=C—C=CH2
—Me
Br' (161)
Реакции с енаминами. В результате присоединения 1-морфо-
лино-2-метилпропена (289) к N-.хлорметнлморфолину (290) (сус-
пензия в ацетонитриле) получается прозрачный раствор, из кото-
рого при добавлении эфира осаждается соль (291). При гидро-
лизе последней образуется 2,2-диметил-З-морфолпнопропаналь
(292) [287] (уравнение 162).
Cl' CH2=N' О (2901
ме2с=сн—n; ;о
(289)
/—\ С' */ \ ’он
°\ )n—СН2—СМе2—CH=N О
(291)
/~\ /Н
о N—СН2—СМе2—С
(162)
(292)
Реакции с диазосоединениями. а-Галогенамины энергично
реагируют с дпазосоедпиеппями, превращаясь в производные | -
хлорэтилампиа (293) [288].
Rs
R'R2NCII 4Ial + CH2N2
(261)
R3
RiR’NCHCHsCI
(293)
(163)
153
амины могут рсо* 1 0TCVTCTBIIe катализатора (уравнение 164)
Д^ваГироизводпыебепзнлампиа.
(261) |- Аг" —* iVR-NCHAr (164)
Г„«гезы через u.utdu фосфонин. а-Галогенампны реагируют с
С Л .Дп-мими- ион этом за счет формального а-элпмн-
^фовХ^азуютея аллены [289]. Этплпдеитрпфеиилфосфоран
Ж днметил-а-хлорбензилампн образуют фосфониевую соль
295 которая отщепляет «-протон в результате перевлидирова-
ния н затем претерпевает р-элиминироваиие с образованием фе-
нилаллена (296) с 60%-ным выходом (уравнение 165).
СГ Me NMe2
|PhCH=X.'l«l сГ + I I 12941
PhsP=CHMe--------------- P'i3P—CH CHPh
(294) (295>
Me NMej
I I
Ph3P=C—CHPh
(165)
CH2—C—CHPh
(296)
Образование связи C—N. Взаимодействие а-галогепамииов с
азиридинами приводит к образованию мопочетвертпчных имнда-
золидиниевых солей [290]. Например, N-(р-феиилэтпл) азиридпи
(297) реагирует с М-хлорметплшшерпдипом (298), давая азнрп-
диниевую соль, которая перегруппировывается в устойчивое мо-
ночетвертичиое соединение (299) (уравнение 166).
____ СГ CH2=nQ
\ / |288i ~ \ / /—\
PhCH,CH2N--------------- phCHaCHjNCHjN^ СГ —>
(297)
СН2СН;С1
РЬСНгСНг—\—CHgN^
PhCHzCHjN7 'n
СГ (166)
(299)
Образование связи С—О. Реакция а-галотепамимов с этилен-
оксидом, эпихлоргидрином, стиролоксидом и оксетаном сопровож-
дается раскрытием никла, а образующиеся р- пли у-хлоралкоксн-
метилампны циклизуются в соли оксазолидипия (300; л = 1) пли
тетрагидрооксазнния (300; п = 2) [2911.
п<'е2^₽а3°ВанИ2 СВЯЗИ С— Р. N-Хлорметилпиперндин (298) pearli-
р. с трнэтилфосфитом, при этом с выделением этил хлор н да обра’
154
о
Cl- C1L=N^)O + —ЧСНЛ„ [o^NCH2OCH2(CH2)aCI
(290)
(CH2)„
(300)
зуется диэтиловый эфир пиперидииометилфосфоновой кислоты
(301) [292].
О
СГ СН2=Х\ ^+(Е1О)3Р —> (EtO)2PCHA'^^ + EtCl (168)
(298) (301)
Образование связи С—S. Взаимодействие соли хлорметилди-
этпламина (302) с тираном в ацетонитриле приводит к получе-
нию гигроскопичного хлорида М.М-диэтилтиазолидиния (303а).
Спиртовый раствор этого соединения реагирует с 70%-ной пер-
хлорной кислотой, давая устойчивый на воздухе перхлорат (3036)
]293].
S * .V .—.
/ \ + СГ CHP=NEf2 —> RsCHjNEts —> \’Et2 X’ (169)
I/ cl- \z
(302) (303a) X = Cl
(3036) X = C1O4
сс-Галогеиамнны реагируют с ксаптогената.мп калия (304) в
апротонных растворителях; при этом с хорошими выходами по-
лучают дпалкилампнометилтпокарбонаты, например (305) [294].
S S
ROCS’ К++ (302) —» ROC—SCH.NEh (170)
(304) (305)
6.2.5.2. р-Галогенамины
(7) Методы получения [295]
Р-Галогенамины R'R2NCH2CH2Ha! (306) обычно получают в
инертном растворителе из соответствующих 2-ампносппртов при
Действии: а) тионплхлорида [296] 'или хлористого водорода.
б) тиоиилбромнда 1296] пли бромпстоводороднои кислоты i-s/j.
в) пептахлорида фосфора пли тригалогенпда фосфора в безвод-
оом хлороформе [298]. Фторпропзводные (306, На! )
155
в этаноле [298] 1(2о
(8вв) R'R’N | X- ’
нагреванием вторичного амина с 1-бром-2-фторЭтаном
r'R2NCH2CH2OH + их (17|)
(307)
Способы получения р-галогенаминов из а-галогенамшюв пред,
ставлены в уравнениях 157, 158, 163.
(2) Свойства
б-Галогеиамины обладают основными свойствами и образуют
кпнстатлическпе соли, иногда обладающие гигроскопичностью.
В Гатогеиамииы в форме свободных оснований умеренно раство-
римы в воде, однако, например, в водном ацетоне они растворя-
ются с образованием этнлеппмшшевых солей (307), которые в
свою очередь реагируют с водой с образованием 1,2-аминоспиртов
[299]. Ионы этнлениминня реагируют также со свободным хлор-
амином. давая соли пиперазпння (308) [299]. Фторпроизводные
(306; Hal — F) в водном ацетоне разлагаются очень медленно
или вовсе не разлагаются [300].
(307) + (80S) —у R'R2N^ ^NR'R2 2Х" (172)
(308)
0-Галогенамнны (306) обладают ярко выраженными фармако-
логическими свойствами [295]; онн ингибируют действие норадре-
налина н адреналина на специфические адренорецепторы. Одним
из таких так называемых а-адренергических блокирующих препа-
ратов является фенокснбеизамин (дибеизилнн) (309), который
применялся для лечения гипертонии и нормализации кроветока
в периферической сосудистой системе.
PhOCH2CH(Me)NCH2CH2Cl (309)
CH2Ph
(3) Реакции
К™ уже отмечалось выше, ₽-галогенамины (306) легко цикли-
явтяютея °®Разованнем этиленимнниевых ионов (307), которые
В ' 196Q г л 'ежуточиыми соединениями в реакциях замещения,
этих оеакпийПр f1,K0BaH 0030Р [301], посвященный применению
этих реакции в медицинской химии.
ным PpeI?eHTOMMa(NIi)aKU1n Р'га-10геиам"на (306) с нуклеофилы
и галогенидщону ’и LТ’,ОДЯи1ая к ам.щу NuCH2CH2NR'R*
в реакциях с паавртП°„0ЪЯСНЯет изменен1'й, которые наблюдаются
можно объяснить оГш еИ|1ЫМ|1 гал°ге»амннамп. Такие изменения
единений, котооые млгр°Ва'1Нем ЦИКЛ1|ческпх промежуточных со-
-,т РаскРываться двумя различными сносо-
100
бамп. приводя к двум изомерам. Например, р-хлорамин (3101
иреврашаегся в ион этилеииминия (311) (уравнение 173)- ней со
'вльпая частина может атаковать этот ион как по а-С (хрщщ
„не 174), так и по р-С (уравнение 175). '
R3
ci-ch-ch2nr‘r2
• fl л 1
(310)
R2
R*
fl/ \,/R2
R3 । N''p>
f 11,11
Nu-CHjr-CH-NR'Ri
(174»
R3
R3
Nu—CH—CH2—NR'R2
(1751
Очевидно, что когда R = H, в обоих случаях должно образо-
ваться одно п то же соединение. Как правило, когда R =# Н, со-
отношение образующихся изомеров определяется особенностями
строения р-галогенамина и нуклеофильного реагента. При синте-
зе антигистаминного препарата прометазина (уравнение 176)
в смеси преобладает более активный изомер (312) [302].
С1СН(Ме)СН2ХМе2
S S
-СОЗ <оо
N N
I I
МеСНСН2\Ме2 CH>CH(Me)NMe,
(312)
(176)
Взаимопревращения 2-хлорметнлпнрролидннов (314) и 3-хлор-
ииперидппов (315) (уравнение 177) объясняются промежуточным
образованием общего для них бициклического катиона (313).
Кроме того, обе циклические системы (314) и (315) при прове-
дении реакций нуклеофильного замещения (уравнение 178) [303
307] превращаются в одно и то же производное (316).
Ьч * R --у Лсн2 (177) \—/ СзСн-ci
(зи) (313) <S*S)
157
(316a) X“CN
(3166) х-он
ойпяются очень активными алкилирующими
р-Га.тогеиамИны „сак1(Нп при атомах кислорода, серы,
агентами; ^''ньв уравнениях 179-184 [308-313Ь
азота н углерода прнвме У г
Ph,CHO’ Na+ + ClCH.CHiNAtc;
(317)
Р-Гллогеиямииы
pb2CHOCH2CH2NMe2
(179)
PhCH(OH)CO.Ag + BrCH5CH2NMe3 Вг ►
—> phCH(OH)CO2CH2CH2NMe3 Br’ (180)
'SH
(317) ’—** HSCHjCHsNMea (181)
Ph2CHCOXU + ClCH2CH2N’^> Ph2CHCONHCH2CH2NQ) (182)
CH2CH2NMe2
CH2CH2NMe2
knh2 I
Ph2CHCH2CH2OEi + (317) -----* Ph2CCH2CH2OEt (184)
При взаимодействии с магнием в тетрагндрофуране (3-галоген-
амины не образуют реактивов Гриньяра. Например, 2-диметил-
амнноэтилхлорид превращается в этилен (уравнение 185). Кроме
того, в результате вторичной реакции между N-хлормагцпйдиме-
тиламином (318) и р-галогенамином (317) (уравнение 186) обра-
зуется диамин (319) [314]. Однако морфолииоэтплхлорнд (320)
димеризуется с образованием дисинропиперазинпевой соли
[31а].
(317) +Mg —> Me2NMgCl + СН2=СН2
(318)
(317)+ (318) —+ Me2NCH2CH2NMe2 + MgCU
(319)
(321)
(185)
(186)
(187)
(З-'i)
138
6.2.5.3. у-Га.югснамины
(I) Методы получения
Первоначально гидробромид З-бромнропиламина Br(CH2)3NH>-
IfBr был синтезнровац 110 методу Габриэля путем расщеп-
ления фталимида (322) бромистоводородиой кислотой; этот метод
(322)
псе еше широко применяют для получения у-галогенаминов с пер-
вичной аминогруппой [316].
Третичные амины аналогичного строения (323), как правило,
получают путем взаимодействия 1,3-дигалогенпропанов с вторич-
ными аминами в инертном растворителе [317] или действием
тионилгалогенидов иа З-дналкнламинопропанолы, например (324)
[318] (уравнения 188, 189).
С1(СН2)3Вг+ RIR2NH —> C1(CH2)3NR'R2 (188)
(323)
SOC12
HO(CH2)3NR1R2 --------► (323) (189)
(324)
у-Галогенамииы можно также получить из а-галогенамннов
(уравнение 155).
(2) Свойства
Как основания у-галогенампны образуют кристаллические
соли, которые иногда обладают гигроскопичностью. у-Га.тоген-
амины в виде свободных оснований умеренно растворимы в воде.
В очень разбавленных растворах у-бромпропн.тди.метиламии
(325) претерпевает внутримолекулярную циклизацию, давая кри-
сталлическую соль (326) (уравнение 190) [319]. Наблюдалась
__NMe2 Вг'
Br(CH2)3NMe2—> | |
(190)
(325)
(326) —е Вг(СН2)3
(326)
г Me
I*
—N—(СН2)3—
I
L Me
NMe.- жВг’
(191)
(327)
159
„ПШ1ЯЯ перегруппировка солп (326) в линейный поли-
также медлен * Р, '|91) [3!9]. Другие у-бромнропиламины,
мер (327) (}|>< . Ц N.N-дибутилнроизводиые, также об-
так"е ^иетвёптпчпые соли в результате внутримолекулярной
разуют четвер сОедПнеипя, по-впдимому, более устойчивы
циклизации, но л.
(320]. Реакции
Реакциям у-галогенаминов посвящен опубликованный в г969 г.
обзор [301], в котором особое внимание уделено применению
этих реакций в медицинской химии.
Циклические соли типа (326) играют, по-впдимому, определен-
ную роль в реакциях замещения в у-галогена1иипах. Например,
при нуклеофильном замещении в За-хлортропане (328а) За-кон-
фигурация сохраняется н в продукте реакции (329). Этот факт
может быть объяснен образованием циклического промежуточного
соединения (3286) (уравнение 192) [32/].
По отношению к нуклеофильным центрам у-галогенампны
являются эффективными алкилирующими агентами (уравнения
193-/97) [322-325].
(H2N)2C=S H’N\ Cl
C1(CH2)3NMC! ------>- \—S(CH2)3NMe;
IW
НО'
---► HS(CH2)3NMe2 (193)
(339)
(194)
(CH2)3NMe2
. Na _ <3301 I ,rnc.
Ph2CO 777* PhjC-O' 2Na+ —♦ Ph?C(CH2)3NMe2 (195>
Nr»3 112•)
ОИ
PhoCO | ,
El2N(CH2)3MgCI -> Ph,C(CH2)3NEt2 <196'
(331)
uO^CaX
N ^^NX^(CH2)3NMe2
160
п отличие от р-галогенаминов у-галогенамины реагируют с
,,аПшем нормальным образом, давая реактивы Гриньяра, напри-
мер (331). Эта реакция была использована в синтезе б-гидрокси-
аминов (уравнение 196J [324] н для аминоалкнлирования гетеро-
dIV ___,,,tv оПАППИАИПЙ (УПяпиаипр 1Q7\ fQORl 1
/331). Эта реакция
мер /________,„„о юйс
циклических соединений (уравнение 197) [3251'.'
ЛИТЕРАТУРА
I о. stork, R. Terrell, and 1. Szmuszkovicz, J. Amer. Chem. Soc., 1954 76
2029- ' ' '
о 0. stork and H. K. Landesman, J. Amer. Soc., 1956, 78, 5128.
3 J, Szmuszkovicz, Adv. Org. Chem. (963), 4, 1.
4' K. Blaha and 0. Cervinka, Adv. Heterocyclic Chem., 1966, 6, 147.
5' «Enamines: Synthesis, Structure and Reactions», ed. A. G. Cook, Marcel
Dekker, New York, 1969
6a. M E. Kuehne. Synthesis, 1970, 510.
6б'. P. IT. Hickmoll and H. Suschitzky, Chem. and Ind. (London), 1970, 1188
7. S. F. Dyke, «Chemistry of Enamines», Cambridge University Press. 1973.
8. G. Stork, A. Brizzolara, H. Landesman, I. Szmuszkouiicz, and R. Terrell
J. Amer. Chem. Soc., 1963, 85, 207.
9. R. Fusco, G. Bianchelli, and S. Rossi, Gazzetta, 1961, 91, 825.
10. M. E. Herr and F. If. Heyl, J. Amer. Chem. Soc., 1953, 75, 5927.
11. I. L. Johnson, M. E. Herr, J. C. Babcock, A. E. Fonken, J. E. Stafford, and
F. If. Heyl, J. Amer. Chem. Soc., 1956, 78, 430.
12. K. Taguchi and F. H. Weslheimer, J. Org. Cliem., 1971, 36, 1570.
13. R. S. Monson, D. N. Priest, and J. C. Ullrey, Tetrahedron Letters, 1972, 929.
14. It. Weingarten and If. A. White, J. Org. Chem., 1966, 31, 4041.
15. H. Weingarten and W. /1. White, J. Amer. Cliem. Soc., 1966, 88, 850.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
I. Coerdeler in «Methoden der Organischen Chemie» (Houben-Weyl), Thieme,
Stuttgart, vol. XI/2, pp. 616—618.
G. N. Walker and D. Alkalay, J. Org. Chem., 1967, 32, 2213.
N. J. Leonard, A. S. Hay, R. W. Fulmer, and V. If. Gash, J. Amer. Chem.
Soc., 1955, 77, 439.
A'. J. Leonard, С. K. Steinhardt, and C. Lee, J. Org. Chem., 1962, 27, 4027.
If R. N. Williamson, Tetrahedron, 1958, 3, 314.
F. Johnson and A. Whitehead, Tetrahedron Letters, 1964, 3825.
G. Opilz, H. Hellmann, and H. If. Schubert, Annalen, 1959, 623. 112.
У . I. Leonard and V. W. Gash, J. Amer. Chem. Soc., 1954, 76, 2781.
У J. Leonard and A. G. Cook, J. Amer. Chem. Soc., 1959, 81, 5627.
У . /. Leonard and D. M. Locke, J. Amer. Chem. Soc., 1955, 77, 437.
If. D. Gurowilz and M. A. Joseph, J. Org. Chem., 1967, 32, 3289.
R- L. Hinman, Tetrahedron, 1968, 24, 185.
У . B. Henbest and P. Slade, J. Chem. Soc., 1960, 1555.
G. Opitz and H. Mildenberger, Angew. Chem., 1960, 72, 169; Annalen, 1961,
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
6
T. J. Curphey and J. C. Hung, Chem. Comm., 1967, 510.
К- C. Brannock and R. D. Burpitt, J. Org. Chem., 1961, 26, 3576.
, • Opdz, H. Mildenberger, and H. Suhr, Annalen, 1961, 649, 47.
U- K. Pandil, If. A. Zwart Voorspuij, and P. Houdewind, Tetrahedron Let-
‘"s. 1972, 1997.
n' £ Kuehne, J. Amer. Chem. Soc., 1962, 84, 837.
e’ Kickmott, Chem. and Ind. (London), 1974, 731.
S. Iliiilig, E. Benzing and E. / icke, Chem. Ber.. 1957, 90, 2833.
R о a,l(l Yoshizawa, J. Org. Chem., 1967, 32. 404.
Mp Millwanp J. Chem. Soc,. 1960, 26. 1069 84 4988
Perelmana and S. A. Mizsak, J. Amer. Cliem. Soc., 1962, 84, 49
Opdz and J. Roch, Angew. Chem., 1963, 75, 167.
Зак, 105
161
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
F 1 Staminas, in «Enamines: Synthesis, Structure, and
i A G Cook Marcel Dekker, New York, 1969, pp. JOI—i 13. Keactl°ns>,
M £ Kuehne,.}. Amer. Chem. Soc., 1961, 83, 1492.
e' I Ниппа and M V. Lessard, J. Amer. Chem. Soc., 1968, 90, 2439
Al E Kuehne, J. Amer. Chem. Soc., 1959, 81, 5400.
L Birkofer S. M. Kim, and H. D. Engels, Chem. Ber., 1962, 95, 1495
D. Pocar, G. Bianchetti, and P. Dalia Croce, Gazzetta, 1965, 95, 1220
Л1. Ohno, Tetrahedron Letters, 1963, 1753.
! Wokinsky, D. Chan, and R. Novak, Chem. and Ind. (London) logs
G Stork and I. Borowitz, J. Amer. Chem. Soc., 1962, 84, 313. ’ ’ 67 68 69 70 71 72 * * * * * * * *°-
E. Elkik and P. Vaudescal, Compt. rend., 1967, 264, 1779.
LA Paquette Tetrahedron Letters, 1963, 2027.
О L. Chapman and G. L. Eton, J. Amer. Chem. Soc., 1968, 90, 5329.
I «7. Daly and B. Witkop, J. Org. Chem., 1962, 27, 4104.
N M. Morlyan, A. G. Muradyan. L. O. Esayan, and Sli. O. Badanuan Ar
myan. khim. Zliur., 1975, 28. 75 (Chem. Abs., 1975, 83, 9062) [H. M. Морлян
А Г Мурадян, Л. О. Эсаян, 111. О. Баданян—Арм. хим. жури, 1975 28
75]. ’ ’
М. Olomucki and Р. Hebrard, Tetrahedron Letters, 1969, 13.
M. T. Leffler, Org. Synth. Coll. vol. II, 1943, 24.
R. H. De Wolfe and W. G. Young, in «The Chemistry of Alkenes», cd. S. Pa-
tai, Interseience, London, 1964, 724.
У. D. Smirnov, L. I. Saltykova, and A. P. Tomilov, Zhur. priklad. Khim
1970, 43, 1620 (Chem. Abs., 1970, 73, 87351) |7O. Д. Смирнов, Л. И. Сал-
тыкова, А. П. Томилов — /КПХ, 1970, 43, 1620].
59. К. Takahashi, A. Miyake, and G. Hata, Bull. Chem. Soc. Japan, 1972, 45, 230
(Chem. Abs., 1972, 76, 98 670).
60. I. IE. Smith, in «The Chemistry of the Amino Group», ed. S. Patai, Intersci-
ence, London, 1968, 178.
61. R. I. Kruglikova, G. R. Kalinina, and Z. F. Abramova, Reakts. spos. org.
Soedinenii, 1972. 9, 79 (Chem. Abs., 1972, 77, 100 551) fP. И. Кругликова,
Г. P. Калинина, 3. Ф. Абрамова — Реакционная способность органических
соединений, 1972, 9, 79].
62. Р. J. Krueger and D. W. Smith, Canad. J. Chem., 1967, 45, 1605.
63. O. Yamamoto, M. Yanagisawa, H. Tomita, T. Ogawa, Y. Senga, K. Hayandzu,
and F. Taka/ Analyt. Chem., 1972, 44, 2180.
64. Chinoin Gyogyszer-es Vegyeszeti Fermekck Gyara Rt„ Brit. Pat. 1 242 109
(1971) (Chem. Abs., 1972, 76, 34 015).
65. IE. Himmele, IE. Aquila, H. Siegel, A. Amann and H. Giertz, Ger. Offen.,
2 157 454 (1973) (Chem. Abs., 1973, 79, 42 248).
66. У. Tamura, 11. Ishihashi, M. Hirai, Y. Kila and M. Ikeda, J. Org. Chem.,
1975, 40, 2702.
67. R. A. Holton and R. A. Kjonaas, J. Amer. Chem. Soc. 1977, 99, 4177.
68. N. Takamatsu, S. Inoue, and Y. Kishi, Tetrahedron Letters, 1971, 4661.
69. R. E. Ireland and A. K. Willard, J. Org. Chem., 1974, 39, 421.
70. H. G. Viehe, Z. Janousek, R. Compiler and D. Lach Angew. Chem. Internal.
Edn., 1973, 12, 566.
71. J. P. Dulcere, B. Ragonnet, M. Sanlelli and M. Bertrand Compt. rend., 1972,
0-7Л O-7C ’ ’ 1
72.
73.
74.
75.
76.
77.
78.
79.
80.
Л P Dulcere, M. SanteW, and M. Bertrand, Compt. rend., 1970, 271, 585.
C. Niverl and L. Miginiac, Compt. rend., 1971, 272, 1996.
„ "4 V‘e,,e- Ange« Chem. Internal. Edn., 1967, 6, 767.
n. G. Viehe, in «Chemistry of Acetylenes»,ed H G Viehe Marcel Dekker,
New York, 1969, pp 861—912. ’ •
H. E . Zaugg. L. R. Swett, and G. R. Slone, J. Org. Chem., 1958, 23, 1389.
1. L. Dumont, Compt rend., 1965, 261, 1710.
A./. Hubert and H. G. Viehe, J. Chem. Soc. (C) 1968 228.
о u a'u ,, Barbara. Bull. Soc. ehim. France, 1965, 2787.
5' 584^ C’ ^a^eul>’ a'}d W. G. Viehe, Angew. Chem. Internal. Edn., 19чб,
162
81.
82.
83.
84.
85
8G
87.
88
91.
Ghosez and I. Marchand-Brynaert in «Iminlum Salts in Organic Che-
niistry», part 1, ed. H. Bohme and Ц. G. Viehe, Wiley, London. 1976, vol. 9.
p. 421.
A. Ilalleux, //. Reimlinger, and H. G. Viehe, Tetrahedron Letters 1970 3141
Г968' Т^ОЭ5, E' Ri ‘“PS’ A' N' КиГ‘г’ AngeW C U 1,,’1ег"а*'’ Ed"
D. R. Strobach, J. Org. Chem , 1971, 36. 1438.
II. G. Viehe and M. Reinstein, Angew. Chem. Internal. Edn. 1964 3, 506
fl. G. Viehe, S. I. Miller, and 1. I. Dickstein. Angew. Chem ’internet Edn
1964, 3, 582.
P. P. Monlijn, E. Harryvan, and L. Brandsma, Rec Trav chim 1964 83
1211. ’ ’ ’
M. E. Kuehne and P. J. Sheeran, J. Org. Chem., 1968, 33, 4406
lz. Wolf and F. Kowitz, Annatcn. I960, 638, 33.
fl. G. Viehe, R. Fuks, and M. Reinstein, Angew. Chem. Internal. Edn 1964
3. 58E
F. Weygand, W. Konig, R. Buyfe, and H. G. Viehe, Chem. Ber. J 965 98,
3632.
92. R. Buyle and H. G. Viehe, Tetrahedron. 1968, 24, 3987.
93. R. Buyle and H. G. Viehe. Tetrahedron, 1969, 25, 3453.
94. T. Eicher and T. Pfister, Tetrahedron Letters, 1972 , 3969.
95. M. Delaunois and L. Ghosez, /Angew. Chem. Internal. Edn.. 1969, 8, 72.
96. L. Chosez and C. de Perez, Angew. Chem. Internal. Edn., 1971. 10, 184.
97. J. Ficini and A. M- Touzin, Tetrahedron Letters, 1974. 1447.
98. M- E. Kuehne and H. Linde, J. Org. Chem., 1972, 37, 4031.
99. R. A. Raphael, «Acetylenic Compounds in Organic Synthesis», Butterworths,
London, 1955, pp. 61—64.
100. M. Olomucki, Ann. Chim. (Italy), 1960, 5, 845.
101. D. Z. Simon, R. L. Salvador, and G. Champagne, J. Medicin. Chem., 1970.
13, 1249.
102. Y. Besace, A. Marszak-Fleury, and I. Marszak, Bull. Soc. chim. France 1971,
1468.
103.
104.
105.
106.
107.
108.
109.
110.
Hl.
112.
E. V. Ermilova, N. E. Khlebnikova, L. A. Remizova, and 1. A. Favorskaya,
Zhur. org. Khim., 1974, 10. 2290 (Chem. Abs. 1975. 82. 72 493) [E. В. Ерми-
лова, H. С. Хлебникова, Л. А. Ремизова, И. А. Фаворская — ЖОХ. 1974, 10,
2290]
G. H. Doubglas, L M. H. Graves. D. Hartley, G. A. Hughes. B. J- McLough-
lin, J. Siddall, and H. Smith, J. Chem. Soc., 1963, 5072.
C. Glacet and R. Coupe, Bull. Soc. chim. France, 1963, 2464; modified by
L. Brandsma, «Preparative Acetylenic Chemistry». Elsevier, Amsterdam, 1971,
pp. 165—166.
/. L. Dumont. W. Chodkiewicz. and P. Cadiol, Bull. Soc. chim. France, 1967,
588.
R. S. Gyuli-Kevkhyan, L. S. Papoyan, and G. T. Tatevosyan, Armyan. khim.
Zhur., 1973, 26, 837 (Chem. Abs.\ 1974, 80. 81951) [P. С. Гюли-Кевхян.
Л. С. Папоян, F. T. Татевосян — Арм. хим. журн., 1973, 26, 837].
/. Koshkina. L. ,4. Remizova, E. Г. Ermilova, and I. .4. Favorskaya
Reakts. spos. org. Soedinenii, 1970, 7, 944 (Chem. Abs.. 1971, 75. 19 604)
\И. Л1. Кошкина, Л. А. Ремизова, E. В. Ермилова. H. А. Фаворская —
Реакционная способность органических соединении, 1970, 7, 944].
Z. Alaune, Z Talaikyte, and V. Mozolis, Liet. TSR Mokslu Akad. Darb..
Ser. B., 1967. 67 (Chem. Abs., 1968, 68, 64090).
C. Bogenioft Y. Svensson, and B. Karlen, Acta Ptiarm. Suec., 1973, 10. 21o
(Chem. Abs., 1973, 79. 77618).
D. A. Ben-Efraim, Tetrahedron, 1973, 29. 4111.
H. G. Viehe and A. 1. Hubert, U. S. Pat.. 3 439 038/1969 (Chem. Abs.. 1969,
71. 13 131)
P.'l. Carral! and Soon Bin Neoh, J. Amer. Chem. Soc 1975 97, 3255.
I- N. Azerbaev L. A. Tsoi. L. T. Kalkabaei-a. *2 H л-o'
gelerotsikl. Soedinenii 1971. 199 (Chem. abs., 19/4, 80. 133a’2) [//. H. Азер-
6*
163
115.
116.
117.
118.
119.
120.
121.
122.
123.
124.
125.
126.
127.
128.
129.
130.
131.
132.
133.
134.
135.
136.
137.
138.
139.
140.
141.
142.
143
144.
146.
147.
148.
149.
баев Л Я. Uou. •'!. T. Кабаева. " " Амксеева ~ Х,,м,,я гстс,Роцпк.Ц|.
dnd * D- J- C' S- P^kin I, 1973,
₽58f«fe, and H. G. Viehe, in «Chemistry of Acetylenes», ed. II. G. Viehe,
Marce^Dekker, New York,J9^9, Nitrogen Compounds», Benjamin, New Yorki
^wloncsMd H F- Phalen, J. Amer. Chem. Soc., 1925, 47, 1343.
П Blanche ti DPocar, and R. Stradi, Gazzetta. 1970, 100, 726.
T Kaop E Lender, and E. Ziegler, Monatch., 1968, 99, 990.
11 B6hm’eandY. S Sadanandam, Arch. Pharm. 1973. 306, 227.
0. Zinner and W. Kliegel. Chem. Ber., 1967, 100, 2515.
H. R. Hensel, Chem Ber., 1966, 99, 868.
H 117 Wanzlick and IV. Lochel, Chem. Ber., 1953, 86, 1 163.
L Hocker, H. Giesecke, and R. Merten, Angew. Chem., 1976, 88, 151.
C. Mannich and H. Davidsen, Ber. 1936, 69, 2106.
117. C. Hunt and E. C. Wagner, J. Org. Chem., 1951, 16, 1792.
T. H. Fife and J. E. C. Hutchins, J. Amer. Chem. Soc., 1976, 98, 2536.
H. Bohme and W. Lehners, Annalen, 1955, 595, 169.
H. Bohine, M. Dahne, IV. Lehners, and E. Ritter, Annalen, 1969, 723, 34.
«Chemistry oi Carbon Compounds», ed. E. H. Rodd, Elsevier, Amsterdam,
1951, vol. IA. p. 697.
H. G. Ing and R. H F. Manske, J. Chem. Soc., 1926, 2348.
USSR Pat., 176303 (Chem. Abs., 1966, 64, II 084) \E. P. Чертой. А. П. Тор-
ба Авт. свид. СССР № 176 303].
R. B. Wagner and H. D, Zook, «Synthetic Organic Chemistry», Chapman and
Hall, London, 1953, p. 691.
A. Ilalleux and H. G. Viehe, !. Chem. Soc. (C), 1968, 1726.
С. H. Shih, Petrochem., 1973, 12 (3), 49 (Chem. Abs. 1974, 80, 3010).
M. L. Moss, J. H. Elliott, and R. T. Hall, Analyt. Chem., 1948, 20, 784.
R. Yamdagni and P. Kebarle, J. Amer. Cliem. Soc., 1973 95, 3504
₽. Trunel, Ann. Chim. (France), 1939, 12, 93 (Chem. Abs., 1939, 33, 8066).
«Chelating Agents and Metal Chelates», ed. F. P. Dwyer and D. P. Mellor,
Academic, London. 1964.
A. P. iV. Franchimont and E. A. Rlobbie, Rec. Trav. chim, 1888, 7, 258;
J Chem. Soc., 1889, 124.
C' Cr °’ Ede"s- and ' Allan, Org. Synth. Coll. Vol. Ill,
6. Schwarzenbach and K. Lutz, Helv. Chim. Acta 1940 23, 1139
«Cheims ry of Carbon Compounds», ed. E. H. Rodd,' El sevier, Amsterdam,
19o4, vol. П1А, p. 466.
p'fnl“r,i"7rl!^ e" A1artcl1- J- Amer- Chem. Soc., 1950 72, 4301.
p (Л“"£ rF. Bergstrom, J. Amer. Chem., 1931, 53, 1846.
Res Ini-. V' im “mV.» r!lara' a"d T- Morisita, Bull. Inst. Phys. Chem.
Res. Tokyo, 1940, 19, 1448 (Chem. Abs., 1943, 37, 6900)
№9emvol.r5;VB, pp 13n38,C?3m4rndW' ed' E' H' R°dd' E,SeVier’ Ams,erdanb
15, №8S’ <Piperazine>>' Kirk-Othmer Encyci. Chem. Techuol, 2nd Edn., 1968,
150.
151.
152.
153.
154.
155.
156.
M. Popescu, Farmacia (Bucarest), 1972, 30, 535
54 (C1Z’ Abs 1974ra80m27 2^).M' СЖ'П' I973' 54'
W L McEwen o’ »' HUr"n' arld E J' De"/o,‘, S. Kushner, L. M. Braneone.
и II at 3260759 “'ЧЛ Subbar,,1. Org. Chem., 1948, 13, 134.
я srejbft; IS: 8-g ”5’
!.' Hi^and W^S ALiVa]mnl'‘trhS' Лтег' Chem' Soe ’ 1975' 97’ 891-
rrine ana 117. 5. Li, J, Qrg. Chem., 1975, 40, 1795.
164
162.
J 63.
164.
165.
166.
167.
168.
169.
170.
,Г7 G Eckhardt, /(. J. Goebel, G. Tondorj, and S Geonerhcn м
|57' trometry 1974, 9, 1073. ueonechea, Org. Mass Spec-
[58 T. Haga and RMajimaBer., 1903, 36, 333; J. Chem. Soc. 1903 291
159. f959',’volyivk par|2°9n5. C°mp0Unds^ ed- E Rodd, Elsevier,'Amsterdam,
ГКО E. Fischer and H. Koch, Annalen, 1886, 232, 222- J Chem Snr 1яяк сот
6L 1- Га/ter «Formaldehyde», 3rd edn., Reinhold, New York ИЭб^п' 5п’
-- F. Meissner, E. Schwiedessen, and D. F. Othmer, Ind. Eng. Chem 1954 46 724
A. T. Bottini, V. Dev and 1 Klinck, Org. Synth. Coll. Vol V W3 ill
S. J- Angyal, Org. Reactions, 1954, 8, 197. ’
S. J- Angyal, G. B. Barlin, and P. C. Wailes, J. Chem. Soc. 1953 1740
L N. Ferguson, Chem. Rev., 1946, 38, 230. ' ' U’
W. E. Smith, J. Org. Chem , 1972, 37, 3972.
Г. E. Bachmann and J. C. Sheehan, J. Amer. Chem. Soc., 1949 71 1842
IF. T. Forsee, Jr. and С. B. Pollard, J. Amer. Chem. Soc.' 1935’ 57’ 2363
G. E. Hulse, U. S. Pat. 2759913 (1956) (Chem. Abs., 1956, 50 17’533)
171. J. J- Christensen, J. O. Hill, and R. M. Izatt, Science, 1971 '174 459’
172. С. H. Park and 11. E. Simmons, J. Amer. Chem. Soc., 1968, 90, 2431.
173. J. C. Allen, C. J. Smith, M. C. Curry, and I. M. Gaugas Nature 1977
267, 623.
174. A. Kawasaki and У. Ogata, Mem. Fac. Eng. Nagoya Univ., 1967 19 1
(Chem. Abs., 1968, 69, 58 669).
175. A. T. Nielsen, R. L. Atkins, D. W. Moore, R. Scott, D. Mallory, and J. M. La-
Berge, J. Org. Chem., 1973, 38, 3288.
N. J. Turro and W. B. Hammond, Tetrahedron Letters, 1967, 3085.
H. 0. Krabbenhoft, J. R. Wiseman, and С. B. Quinn, J. Amer. Chem. Soc
1974, 96, 258.
176.
177.
178. A. Albert, Adv. Heterocyclic Chem., 1976, 20, 117.
179. D. Beke, Adv. Heterocyclic Chem., 1963, 1, 167.
180. E. W. Lund, Acta Chem. Scand., 1958, 12, 1768.
181. A. Hajos and P. Soliar, Experientia, 1965, 20, 434.
182. J. M. I. Gladych, Chem. and Ind. (London), 1966, 31.
183. P. У. Sollenberger, and R. B. Martin, in «The Chemistry of the Amino Group»,
ed. S. Patai, Interscience, London, 1968, p. 349.
184. D. R. Barfield, Seng-neon Gan, and R. H. Smithers, J. C. S. Perkin I. 1977,
666.
185.
186.
187.
188.
189.
190.
191.
192.
193.
194.
195.
196.
197.
D. D. Miller F -L. Hsu, K- N. Salman, and P. H. Patil, J. Medicin. Chem.,
1976, 19, I80'.
P. E. Fanta, in «The Chemistry of Heterocyclic Compounds with Three and
Four Members Rings», ed. A. Weissberger, Interscience, New York, 1964,
vol. I, p. 524. _
FL M. Kissman D. S. Tarbell, and J. Williams, J. Amer. Chem. Soc.. 19o3, 7o,
2959.
S. Eguchi, and У. Ishii, Bull. Chem. Soc. Japan, 1963, 36, 1434. ,
G. Berti, G. Camlci, B. Macchia, F. Macchia, and L. Monti, Tetrahedron
Letters 1972 2591
К. B. Sharpless, A. O. Chong, and K. Oshima, J. Org. Chem. 1976, 41 177.
К. B. Sharpless, D. IF. Patrick, L. K- Truesdale, and S. A. Biller, J. Amer.
Chem. Soc., 1975 97, 2305. , . „
R. T. Britain, D. lack, and A. C. Ritchie, in «Advances m Drug Research
ed. N. J. Harper and A. B. Simmonds, Academic, London, 1970, »ol. 6,
R. ^Howe, A. F. Crowther, J. S. Stephenson, B. S. Rao. and L. H. South,
J Medicin. Chem. 1968, 11, 1000. 1974 39 914.
D. A. Evans, G. L. Carroll, and l.. К- Tr«esrfa/e J. 0ro. -. - Wein-
D. Seebach and D. Enders, «New Synthetic Methods», g
heim, 1975, vol. 2, p. 66. „ „ nQ ,0Q„
D. Seebach and D. Enders, Chem. Ber.. 197o, 108, 12J3. lnternat Fdn.,
T. Kauffmann E. Koppelmann, and II. Berg, Angew. Chem. Internal. Ldn..,
1970, 9, 163. ’
165
108. D. Hoppe, «1
p. 17.
199. /. Sant,
200.
201.
202. F
203.
204.
205.
206.
207.
208.
209.
210.
211.
212.
213.
214.
215.
216.
217.
218.
219.
220.
221.
222.
223.
224.
225.
226.
227.
228.
229.
230.
231.
232.
233.
234.
235.
- .. .... <Ncw Synthetic Methods», Verlag Chemie, Weinheim, 1975, vol. 2,
' it- D. Nobles Vacik. and Af. V. About Enein, J. Pharm. Sei., i971i w_
936. f nnfj p Mercier Synthesis, 1977, 183.
, Л. Epselern and F Метет Ilelv. Chiin. Acta, 1948, 31, 2088.
. p. Rarrer and P.Portmmn i
p Beltz eche and Л. Elulicfi, l i iy>u ,
ш H eber and E. Levip Annalen, 1932 aOO 14.
"• 11 ~ rr ; Г W J С11СШ. SOC., 1У32, 2oOl.
H CRapojport"andS. ’ ^ne^.A^r. S°C’ l955’ ?7' 4330'
1 ^DiUonand^ ManfsM.’S. Amer.’Chem. Soc., 1975, 97, 5417.
8 H n g and W. Barron, Chem. Ber. 1957, 90, 395, 403
£ Л1 Hancock and .1. C. Cope. Org. Synth., 1946 26 38.
4 H Beckett, D. C. Taylor, and I. IK Gorrocl, J. Pharmacol., 1975, 27, 588.
О Kamtn and C. S. Marvel, Org. Synth. Coll. Vol. I, 1941, 25.
8 Racldin and I. Enemark, J. Medicin. Chem, 1969, 12, f089.
«.Methoden der Organischen Chemie (Houben-Weyf)», Thieme, Stuttgart, 1958,
vol 11/111 228.
T. J. Schwan. 0. S. Lougheed, and S. E. Burrotis, J. Heterocyclic Chem., 1974,
11 807
s.’/l. Fine and I. Shreiner, J. Org. Chem., 1974, 39, 1009.
E. L. Jackson. Org. Reactions, 1944, 2, 341.
J. B. Aylward. Quart. Rev., 1971, 25, 407.
7. H. Billman and E. E Parker, J. Amer. Chem. Soc., 1944, 66, 538.
A C. Cope and E. R. Trumbull, Org. Reactions, I960, 11, 355.
M. J. Rosen, Analyt. Chem., 1955, 27, 114.
C. J. Collins, Accounts Chem. Res., 1971, 4, 315.
D. V. Bantliorpe, in «Chemistry oi the Amino Group», ed. S. Patai, Intersci-
ence, London, 1968, 585.
S. Raines and C. A. Kovacs. J. Medicin. Chem., 1968, 11, 854.
M. E. Open and D. Swern, Chem. Rev., 1967, 67, 197’.
J. A. Deyrup and C. L. Moyer, J. Org. Chem., 1969, 34, 175.
H. Witte and ll’< Seeliger, Angew. Chem. Internal Edn., 1972, 1 1, 287.
F. F.Blicke, Org. Reactions, 1942, 1, 303.
K. Hayes and G. Drake, J. Org. Chem., 1950 15 873
R. Baltcly and J. W. Billinghurst, J Org. Chem. 1965, 30, 4330.
П M ?,ar,r,on’ G- H- ,ial1- ' L- H- F- Ridley, R. G. W. Spicketl, and
D. K. Vallance, J. Medicin. Chem., 1965, 8, 836.
.4, Durnov, 0. Messwarb, and H. H. Frey, Chem. Ber., 1950, 83, 445.
u. Jones, Org. Reactions, 1967, 15, 204.
1940?'7^«№’ U' Salvador~ Md S. C. Laskowski, J. Amer. Chem. Soc.,
11, 00OO,
O. Hromatka, Ber., 1942, 75, 131.
ear 7 Srk‘la and F' Keil- BeK 1929, 62B, 1142
237. H Heterocyclic Chem., 1963, 2, 311.
238 MtUlcrr,',1965yV%VI/3’PP™eme,
1060. ,a"' On’ L' 0,rardei’ and A. Boucherle, Bull. Soc. chim. France, 1954,
24(3 H. HMnan^ and С Cm?’ rl'em' Ber" I957. 90’ I5'
241. M. Charl>eftiierMoGr'izePHZ'pCh-^ ,Ber" 1956’ 89' 8k
242. A95T ^StewaH ^^‘1957 5^73^’ B' Тс,10“ЬаГ’ ComP1' rend '
?4Л' ? Glacel and'D. CouiuEr i' Amer- Chem. Soc., 1955, 77, 1098.
244. 1 Canceill, J. Jaeyues ‘nri R rP ' r<ind'. 1968. 267- 1624.
2«- 53,а5Ь9СОГПР‘' rend- 1959' 248' 331 L
247- > C. Nordin, J Het'ero^c^Chem'.’^t'^ 987'
166
248.
249.
250.
251.
252
253.
254.
255.
256.
257.
258.
259.
260.
261.
262.
263.
264.
265.
266.
267.
268.
269.
270.
271.
272.
273.
274.
275.
276.
277
278.
279.
280.
281.
282.
283.
284.
285.
286
287
288.
289.
290.
291.
292
293
J LUini and ll. Normant, Bull. Soc. chim. France 1957 1454
д J. Meyers and E. D. Mihelich, Angew. Chem 1976 88 321
‘Hek'r0CytliC CM'Pom.ds». ed. R. C. Llumltl0, W.ley,
^R Ramage E. H Rodd, and j'.K- Lundquist, in «Chemistry of the Car-
bon Compounds», ed, E, H, Rodd, Elsevier, Amsterdam, I960 vol. IVC
p. 1465. ’ ’
Brit Pat. 1070307/1967 (Chem. Abs., 1967, 67, 53 671).
U. Harder, E. Pfeil, and K. F. Zenner, Chem. Ber 1964 97 5Ю
й/й1960Г54^ пТж"' and W' LeViS' U- S’ Pat-' 2928877/1960 (Chem.
T. Mukaiyama and T. Taguchi, Tetrahedron Letters, 1970 3411
Г. F. Blicke, Org. Reactions, 1942, 1, 303.
M. Carmack and M. A Spielman, Org. Reactions. 1946. 3. 83
Brit. Pat. 1070307/1967 (Chem. Abs, 1967, 67, 53 671).
L L. Boguslavskaya, L. Kh. Vinograd. and Y. B. SMeinberg zhur priklad
Khim., 1961, 34, 1392 (Chem. Abs., 1961, 55. 23332) [Я. Л. Богуславская
Л. X. Виноград, Ю. Б. Штейнберг — ЖПХ, 1961. 34, 1392].
A. N. Kost and V. G. Yashunskii, Doklady Akad. Nauk SSSR. 1952. 83. 93
(Chem. Abs, 1953, 47, 2696) [A. H. Кост, В. Г. Яшунский — ДАН СССР
1952, 83, 93].
A. F. Crowther and L. Н. Smith. J. Medicin. Chem, 1968, 11, 1009.
J. M. Lehn, «Structure and Bonding», Springer, Berlin, 1973. vol. 16. p. 1.
B. Dietrich, J. M. Lehn, and J. P. Sauvage, Chem. L’nseret Zeit, 1973. 7, 120.
G. №. Gokel and H. D. Durst, Synthesis, 1976. 168.
B. Deitrich, I. M. Lehn, and J. P. Sauvage, Tetrahedron Letters. 1969, 2885.
/. E. Richman and T. I. Atkins, J Amer. Chem. Soc, 1974. 96, 2268.
/. Cheney. I. M. Lehn, J, P. Sauvage, and Л1. E. Stubbs, J. C S. Chem. Comm.,
1972, 1100.
I. M. Lehn, J. Simon, and J. Wagner, Angew. Chem. Internal. Edn, 1973,
12, 578.
B. Dietrich, J. M. Lehn, and J. P. Sauvage, Tetrahedron Letters. 1969. 2889.
M. R. Trnter and C. J. Pedersen, Endeavour. 1971. 30. 142.
E. Graf and J. M. Lehn. J. Amer. Chem. Soc, 1975. 97. 5022.
E. Graf and 1. M. Lehn. J. Amer. Chem. Soc, 1976. 98. 6403.
H. Bohme and K. Hartke. Chem. Ber., 1960. 93, 1305 и цитированные там
работы.
г> - Г-. « Z-. Т? L/_10X7 ОЛ OTlflQ
н.
н.
н.
н.
G.
Н.
Н.
92, 1608. „„
Н. Bohme and Н. Ellenberg. Chem. Ber, 1959. 92 2976.
A. Kirrmann E. Elkik and P. Vaudescal. Compt. rend, 1966. 262, 1268.
H. Bohme and №. Stammberger. Arch. Pharm.. 1972_ 30a зва.
H. Bdhme and №. Fresenius Arch Pharm., 1972. 30a, 601.
H. Bohme and №. Fresenius, Arch. Pharm, 1972, 30a 610.
H. Bdhme K. Hartke and A. Miiller. Chem. Ber 1963. 96, 607
H. Bdhme, K. Osmers. and P. Wagner. Tetrahedron Letter., 1972. 278a.
W. Bdhme, E Mundlos, №. Lehners, and О. E. Herboth. Chem. Ber, i.ar,
90, 2008.
J. Bestmann, Angew. Chem, 1965. 77. 651.
Bilme and H. Orth, Clietn. Ber, 1966 99. 2842.
H. Bdhme and P. Wagner, Chem. Ber, 1969, 10--bal
H. Bdhme. L. Koch, and E. Kohler Chem Ber 196 95. 1849.
//. Bdhme and G. Ddhler, Chem. Ber., 1970, 103, 3058.
Вогте, Е. Mundlos, and О. Е. Herboth, Chem. Ber., 1957, 90, 2003.
Bohme and M. Hilp, Chem. Ber., 1970, 103, 104.
Gross and E. Hoft Angew. Chem. Internal. Edn., 1967. 6, 335.
Bohme E Mundlos, and G. Keitzer. Chem. Ber.. 1958. 91, 656.
Bohme. S. Efel, and K. Hartke. Chem. Ber, 1965. 98, 1463. 4 K. Bose.
Spiegelman. and M. S. Manhas. Tetrahedron Letters. 1971 3167.
Bohme. №. Lehners, and G. Keitzer, Chem. Ber. 19o8. 91. 340.
Bdhme, H. Ellenberg. О. E. Herboth. and №. Lehners. Chem. Ber., 19o9,
Н.
II.
Н.
167
294. //. Bokme anil D. И. Oito, .Arch. Pharm., 1967, 647
295. I. D. P. Graham, in «Progress in Medicinal Chcimstrv» r-
- чл,..г 1 ппЛт. Builirworilw, 1962, vol. 2, p 132 ’ о. p
296. ...................................................... A
297.
298.
299.
300.
301.
302.
303.
304.
305.
306.
307.
308.
309.
310.
311.
312.
313.
314.
316.
317.
318.
319.
320.
321.
322.
323.
324.
325.
eigmw * — * iZ
//“£7'7 Ь&Я* f. ВЯМЪ S, cw
s/s 'ai?S f- «" /a"lcs- •' Chem Soc- 1953’ 1865-
^F Aden and N B. Chapman. J- Chem. Soc., 1960. 1482.
V J' C'”m- m ’Д4П21°3-
Af Mioraue and l. P. Darios, Chim. 1 her.. 1969, 4 363.
P Charpentier. Conipt, rend., 1947, 225, 306; P. Charpentier and p. p)ltcro^
n“c fuson end C. L Zirkle. J. Amer. Chem. Soc., 1948, 70, 2760.
E, G. Brain, F. P. Doyle, and M. D. Mehta. ,1. Chem. Soc., 1961, 633.
R H Reitsema, J. Amer. Cliem. Soc., 1949, 71, 2041.
/ H Biel E. P. Sprengeler, H. A. Leiser, J. Horner, A. Drukker, and
H L Friedman. J. Amer. Cliem. Soc., 1955, 77, 2250; J. H. Biel, W. K. Horn
and H. .4. Leiser. ibid., 1959. 81. 2527.
R. Paul and S. Tchelitchefj. Bull. Soc. chim. France, 1958, 736.
T. F. Dankova, A'. A. Preobrazhenskii, and M. A. Miropolskaya, Zhur. ob-
schchet Khim., 1951, 21, 570 {Chem. Abs., 1951, 45, 8484) [7, Ф. Данкова,
H. А. Преображенский. M. А. Миропольская — ЖОХ, 1951, 21, 570].
H. Horensiein and H. Pdhlicke, Ber., 1938, 7l, 1644,
IF. O. Foye and D. H. Kay, J. Pharm. Sci., 1962, 51, 1098.
M. Bockmiihl and G. Ehrhait, Annalen, 1949, 561, 52.
С. P. Hullrer, C. D/erassi. IF. L. Beears, R. L. Mayer, and C. R. Scholz,
J. Amer. Chem. Soc., 1946, 68, 1999.
A'. Sperber, Л1 Sherlock, and D. Papa, J. Amer. Chem. Soc., 1953, 75, Ц22.
H. Gurien. J. Org. Chem., 1963. 28, 878.
1. P. Mason and H. IF. Block, J. Amer. Chem. Soc., 1940, 62, 1443.
S. Gabriel and 1. Weiner. Ber., 1888, 21, 2669.
Swiss Pat. 221596/1942 (Chem. Abs’, 1949, 43, 666)
R. C F.lderfield. L. C. Craig. Il7 M. Lauer. R. T. Arnold, IF. /. Gensler,
, ‘’nr'u H- Be",brH. H- R- Mighton, J. Tinker, J. Galbreath, A. D. Hol-
G- Goldman, J. T. Maynard, and hl. Picus, J. Amer. Chem. Soc., 1946,
oe, idlb.
C F. Gibbs and C. S. .Marvel, J. Amer. Chem. Soc., 1935 57, 1137.
C. F Gibbs andC. S Marvel, J. Amer. Chem. Soc., 1934, 56, 725.
lAmw/’ Яе.п-,Г- R' Lewis- l- W' Schulenberg. and M. I. Unset,
7 Am er. Chem. Soc., 1957, 79, 6337.
Fi ^'Lli8^r' Helv' Cllim- Ac,a- 1954, 37, 472.
A' Mww hi ППсн- ' Ei Casefl' lr' J- Or(?- Chem-, 1965, 30, 3959.
A. Marxer, Helv. Chim. Acta, 1941, 24 209E.
arper, U. Salzmann. and F. Hofer. Helv. Chim. Acta, 1971, 54, 2507.
6.3. АРОМАТИЧЕСКИЕ АМИНЫ
p. ДЖ. ЛИНДСЕИ (IC1 Organics Division, Blackley, Manchester)
6.3.1. МЕТОДЫ ПОЛУЧЕНИЯ АРОМАТИЧЕСКИХ АМИНОВ
6.3.1.1. Первичные ароматические амины
включа°отЬ1’восст'ГноХш1р ДЛЯ с“”'1еэа ароматических аминов,
в ряду ароматическиуНПе пптРосоеД»ненпй, реакции замещения
перегруппировки II пе- галоге|1,1Р°113водпых н гпдрокснсоедпненпи.
» рс/кс, прямое аминирование.
168
(1) Восстановление
Первичные ароматические амины наиболее часто получают
восстановлением соответствующих ароматических нитросоедиие-
пнй. Восстановление можно вести в кислых или основных усло-
виях или прямым воздействием водорода в присутствии катализа-
тора, Выбор восстанавливающего агента в значительной степени
определяется природой других групп, присутствующих в молеку-
ле. Как правило, предпочитают восстанавливающие агенты кислой
природы, основные агенты обычно стараются не применять. Ка-
талитическое восстановление применяют в особенности в случае
таких соединений, как н-нитроацетаннлид, содержащий группу,
которая может гидролизоваться в кислом растворе. Варианты
методов синтеза ароматических аминов путем восстановления нит-
росоединений систематизированы ниже (уравнения 1—9).
—NO2 + 6TiCl3 + 6НС1 —► —N'H2 + 6TiCl< + 2Н2О (1)
---NO2 + 3Fe + 6HCI —► ууу—NH2 + 3FeCl3 + 2H2O (2)
ЛА—jy=N + 2Na2S2O4 + 4Н2О —>- 2 —NH2 + 4NaHSO, (3)
dNO2 /NH2
NaOH. S fl \
—Et ------► --Et (4)
(69%)
ею2с—yyy—no2 e102Hi 25 ;c>- Eto2c——NH’ (5)
(91—100%)
Me_AA__N0 PdA Me—ЛЛ—XH2-HC1 (6)
me \ /.«и: CHCls \_/
(89%)
169
Повоино часто для восстановления используют комбинацию
кислоты. Обычно применяют олово [1], железо Илн
инк и соляную кислоту, наиболее удобно проводить восстановлю
ние раствора пптросоедпиення в спирте [2]. Типичном методикой
п я табораторных условий является методика Адамса „ ДжонСо.
„я 131- классический метод Вешана [4] - восстановление желе-
том в уксусной кислоте. Метод восстановления хлоридом или
сульфатом титана (Ш) в соответствующей кислоте лежит в осно-
ве количественного определения ннтрогрупп.
В промышленных масштабах анилин * * обычно готовят вос-
становлением нитробензола действием железа в небольшом коли-
честве соляной кислоты; общий выход на стадиях нитрования и
восстановления превышает 98%. Образующийся хлорид желе-
за(П) в значительной степени гидролизуется до гидроксида и сво-
бодной минеральной кислоты, которая вновь вступает в реакцию.
Восстановление нитросоединения в кислом растворе протекает
через стадии промежуточного образования нитрозо- и гидроксил-
алинопропзводпых (уравнение 10). В щелочном растворе реак-
ция протекает по тем же направлениям (уравнение 11), однако
в этих условиях промежуточно образующиеся нитрозо- и гидро-
ксиламннопронзводпые конденсируются с образованием азокси-
соедннений (1), которые при дальнейшем восстановлении и окис-
лении могут быть превращены в гидразосоединення (2) и азосо-
едпнеиия (3). При восстановлении азо- и гидразосоеднненнй с
помощью гидросульфита натрия получают амины с хорошим
ходом.
ДгМО2 —> АгХ’О —>- ArNHOH —> ArNHj
О
ArNO +ArNHOH —> ArN'=NAr —> ArNH—NHAr —> ArN=NAr
<>) (2) (3)
бензола1''1/ ± П°ЛуЧеН C выходом 65% при нагревании нитро-
Оппсано тяк-кРДЫМ едким иатР°м в изопропиловом спирте [5].
смесыс гиппокгн0ОССТановление ароматических нитросоединений
творе [6) Р ДЭ иат')ПЯ ” сеРы в метанолыю-ацетоновом рас-
каталитХскоГ?вдр!нРХиТнГ1ГН8Ь ™тросое№Не11 ий является
гладко и приводит к 8’ ’ восстаиовленпе протекает
Дом. В качестве катя оно ветствУющему амину с высоким выхо-
и палладий на угле 1Ч1аТ<1/>0В пр11ме!,ЯЛ!< никель Реиея, платину
соединений, азидов нитпчп/'аТаЛИТ"ЧССКОе п°сстаиовление нитро-
д ’ ”ИТРПЛОВ ,1Л” оксимов в растворителе, содер-
* Впервые синтез анилин» 9
осуществлен Н. Н. Зииииым в 1849^ ,УПЛЯМИПа 11 -«-аминобензойиой кислоты был
получения" ,еульф"яом аммония. В%астояС1Х"ОВЛе'тСМ с00™атстеующих иптро-
еппп, % М|'!1°п Ряда апт|>ах1Н1П11з ™я,11 время этот метод применяют для
гении. — Прии ред, и юстичного восстановления полппитро-
170
вы-
(10)
(II)
жащем небольшое количество хлороформа, приводит к гидрохло-
ридам амшюв [Ю]. Интересным примером каталитического вос-
становления является использование природного газа 1111 Так
при восстановлении нитробензола с помощью природного'газа’
содержащего 93 А метана, при повышенной температуре и в пои-
сутствни Cr-Ni-Co-катализатора выход анилина составляет 90%
Восстановление нитробензола в парах осуществляли с помощью
водяного газа в присутствии угля как катализатора [121
Анилин н алкиланплииы можно получить высокотемператур-
ным дегидрированием-восстановлением иптроциклогексана в при-
сутствии ряда катализаторов [13]. 1
Вместо методов прямого гидрирования можно с успехом при-
менять восстановление питросоедпнеиий действием борогпдрида
натрия п платинового или палладиевого катализатора.
Для избирательного восстановления нитрогруппы в присут-
ствии других функциональных групп разработано несколько мето-
дов. Так, метанольные растворы додекакарбонилтрижелеза
Fe3(CO)i2 избирательно восстанавливают ароматические ннтросо-
единения в амины с высоким выходом при наличии самых разнооб-
разных функциональных групп [14, 15] (уравнение 12).
бензол,
кипячение
ArNO2 + Fe3(CO)|2 + СН3ОН ------->- ArNH;
(12)
При использовании этого метода получают только амин; не про-
исходит образования азо- пли азоксипропзводных, не затрагива-
ются щелоче- и кнслотолабпльные группы и остаются без изме-
нений карбонильная группа и двойная связь. Этим методом нз со-
ответствующих иитросоедииенип были получены анилин (77%),
о- и п-хлораиилины (83%, 86%),и-толуидпн (73%),л- и п-фенилен-
диамииы (95%, 63%), о-броманилин (86%) и п-аминофенол (66%).
Если нитрососдинеиие содержит ненасыщенную боковую цепь или
альдегидную группу в ароматическом ядре, восстановления этих
групп можно избежать, применяя в качестве восстанавливающего
агента сульфат железа(П). Если присутствует только альдегидная
группа, то можно применить хлорид олова(П). Гидразпигпдрат в
присутствии небольшого количества никеля Ренея избирательно
восстанавливает ароматические питросоединения до аминов с очень
хорошим выходом [16].
(2) Замещение галогена
Атом галогена в ароматическом ядре инертен в условиях
нуклеофильного замещения, и поэтому при действии аммиака на
ароматическое галогеипроизводпое трудно получить амин. Есте-
ственно поэтому, что этот метод очень ограниченно применяется
в синтезе ароматических аминов. Однако атом галогена в аро.ма
тическом ядре оказывается гораздо более реакциоиноспосооных,
если в орто- и пара-положепнях. находятся нитроза.местптелп.
171
в этих случаях реакция
нами протекает легко и
хлорпропзводиых с аммиаком или ами-
приводит к первичным, вторичным или
ТР7"Нпро"зводпь1е бензола удается превратить в анилин пр„
действии амидов натрия или калия в жидком аммиаке при Ш13.
кой температуре (уравнение 13).
NH3 (жпдк.)
ArX + NaNH2---------------- ArNH2 + NaX
(13)
Замещение амидами металлов может протекать по одному из
двух направлений или по обоим направлениям одновременно
[17 18]. Первый путь —это обычное нуклеофильное замещение
(уравнение 14).
/н
. Л~~\ /NH2
—X"
•х + nh2
ад (14)
Второй путь включает образованиедегидробензола (уравнения 15,
16). С помощью галогенпроизводиых бензола, меченных 14С,
было показано, что аминогруппа оказывается частично у мечен-
ного углерода, а частично у углеродного атома в орто-положенпп,
в результате чего образуется смесь анилинов. Для объяснения
этих результатов было постулировано промежуточное образова-
ние дегидробензола (4).
пара- или жета-Замещенные галогеипронзводные дают смесь
анилинов, а орто-замещепиый галогенид — только Л1ета-замещеи-
иыи анилин.
I, Рал°ген0113Фталип реагирует исключительно через промежу-
таламии°овРа3°ВаНИС дег11ДРонаФталина’ Давая смесь 1- и 2-наф-
с вмным^м^1"10 также полУчить при нагревании хлорбензола
давлением (ЕХ.щ "iT"™" °КС"Да ПрИ 200°СП°Д
_______ н 1е механизм этого процесса не ясен.
2\=/ С1 + 2!4нз + Си2О —> —NH2 + 2CuC1+Н2О (W)
(80%)
172
(3) Замещение гидроксильной группы
Методы превращения фенолов в соответствующие анилины то
есТь методы замещения гидроксильной группы в ароматическом
кольце на аминогруппу в том же положении, весьма немногочис-
ленны п имеют ограниченное применение. При соблюдении соот-
ветствующих условии такая замена происходит в случае фенолов
склонных к таутомерным превращениям, например в случае наф-
толов и резорцинов. Область применения хороню известной реак-
ции фенолов с сульфитом аммония (реакция Бухерера) (уравне-
ние 18) ограничена, в общем, нафтолами и гидрокси- и ампнофе-
ноламп. Для фенолов, активированных наличием электроноакцеп-
торных заместителей, например для 2,4-дпнитрофенола, также
характерно замещение гидроксильной группы [19], Поэтому в
ряду производных бензола для получения амина гидроксильную
группу следует, как правило, превратить в другую функциональ-
ную группу.
ОН ЩИЩЗОз
nh2
(95%)
(18)
Фенол можно превратить в сложный эфир днэтплфосфорной
кислоты (уравнение 19); реакция этого эфира с амидом калия и
раствором металлического калия в жидком аммиаке позволяет
получить анилин [20] (уравнение 20).
ArOH + NaOH+ (ЕЮ)2РОС1 —> ArOPO(OEt)2 (19)
(80-90%)
ЫНз (жидк.)
ArOPO(OEt)2 + KNH2 + К ----------->- ArNH2 (20)
(70%)
Другой достаточно общий метод превращения ароматических
гидрокспсоедннеиий в соответствующие амины [21] изображен
Уравнением 21. Так, например, получаемый из фенола и гидрида
173
ЩеночЯТ натрия реагирует с 2-феиил-4-хлор.
натрия в Д»м,'«с ‘!звзя 2-фснпл-4-фенокспхнпазолин (6). Прп
хпназолшюм РГ (6\ перСГрупппровывается с образованием
„атревапчп хппазол 'Лепота (7). гидролизом которого мож.
2-3-Д|1Феи,'-’'!^ ,и с выходом 7! %
но получить аншюв табе анилин готовят непосредственно
в промыш ие
осуществляют в паровой фазе прп
Sff «•’“«»»«
SiO2 — AhOs; выход анилина 93 /о-
(4) Перегруппировки
Перегруппировка Гофмана. Перегруппировка Гофмана (уравне-
ние 22) [23, 24] состоит в превращении амида при обработке
бромом в щелочном растворе или гнпобромнтом щелочного ме-
талла в амни, содержащий на один углеродный атом меньше.
При проведении этого процесса в промышленности используют
хлор. Эта реакция и родственные перегруппировки Курцпуса к
Лоссена протекают по общему механизму [25], включающему
образование изоцианата (9) в результате миграции арильной
группы к атому азота с пониженной электронной плотностью в
промежуточно образующемся ацилннтрене (8). Гидролиз изоциа-
ната (9) приводит к амину (уравнение 23).
Вг2. “ОН н2о
ArCONHj ----------9- ArNCO -----> ArNH2 (22)
О О
II II
Ar-C-NH2 Ar-C-N3
I ВгО~,~ОН__________ д
(Гофман) I (Курциус)
+ + (23)
° -он ° _
Ar C-NHOAc Ar^C^j; >- Ar—№=0=0 Ar»H2
(»)
Перегруппировка Гофмана была успешно использована для
превращения бензамида [26], нафтамида и их гомологов в соот-
ветствующие амины с высоким выходом. Так, например, салицнл-
амид при действии щелочного раствора гипохлорита превращает-
ся в о-амшгафенол с выходом 72%.
Перегруппировка Курциуса. В обзоре [27] детально рассмот-
рено расщепление азидов карбоновых кислот по Курц»УсУ
(уравнения 24 и 23) и проведено сравнение эффективности этой
реакции и перегруппировки Гофмана, которой она родственна.
174
фактически обе реакции служат методами превращения
иеских карбоновых кислот или их сложных эфиров
аромати-
в амины.
АгСОгН
ArCOCl
ArCO2R
ArCONHXH,
MONO
ArCON,
-n2
н2о
_> АгМСО ------> ArNHj (24)
Превращение карбоновых кислот проводят через стадию поо
межуточного образования хлорангидрида кислоты, а превращение
сложных эфиров через стадию образования гидразида. Обра-
зовавшийся ацилазид (уравнение 24) теряет молекулу азота и
перегруппировывается в арилпзоцпаиат. Далее протекают те же
превращения, что и при перегруппировке Гофмана (уравнение 22)
На практике реакция Курцпуса дает хорошие выходы аминов
Перегруппировка Курцпуса не имеет каких-либо существенных
преимуществ перед реакцией Гофмана, но ее предпочитают в
случае ароматических карбоновых кислот, содержащих в качестве
заместителей активные атомы галогена.
Перегруппировка Лоссеиа. Общим для перегруппировки Лоссе-
на [28] (уравнения 25 и 23), которую претерпевают соли щелоч-
ных металлов гидроксамовых кислот и их производных, и реак-
ций Гофмана и Курпиуса является образование изоцианата в ка-
честве первичного продукта реакции при перегруппировке ацил-
нптреиа. При гидролизе изоцианата образуется амин. Практиче-
ски реакция проводится не со свободными гидроксамовыми кис-
лотами, а с нх О-ацильными производными, что позволяет полу-
чить более высокие выходы целевого продукта. В препаративном
плане перегруппировка Лоссеиа имеет меньшее значение, чем
реакции Гофмана и Курцпуса, и находит ограниченное примене-
ние в органическом синтезе.
_______нго________
I
ArCO—NK—ОСОСНз —► ArN=C=O + СН3СО2К АгХН- (25)
Реакция Шмидта. Реакция Шмидта [29] (уравнение 26) это
реакция между эквимольиыми количествами азотистоводородной
кислоты и карбонильного соединения в присутствии сильной ми-
неральной кислоты. Эта реакция является удобным методом пре
вращения карбоновых кислот в амины, а ее преимущество по
сравнению с реакциями Гофмана и Курцпуса заключается в о -
иостадиппости процесса. Механизм реакции Шмидта в рс> ,
аналогичен механизму реакции КурииУса 11 включает о ра ‘
ацнлазпда в качестве промежуточного соединения. р ДУ Р •
тпческих соединений эта реакция дает хорошие °“*°д ’ 'дом
пример, бензойная кислота превращается в аи1 выходы
85% |30]. Заметное влияние на скорость реакции и выходы
аминов оказывают положение и тин замс^Т"™ ота да£т л-толс-
скои карбоновой кислоте. Так, ,/-толуиловая кислота да ?
7П<>/ в то время как льтолуиловая кислота —
ниш С выходом /и/0, ' »
лишь 24°/о .«-толуидина-
ArCO2H + HN, —ArNH2 + N, + CO, (26)
пР„егр¥иПировка Бекмана. Перегруппировке Бекмана (уравие-
„ 2П псвХ обзор [31]. При действии на кетоксим реаген-
Z кислой природы, таких как пентахлорид фосфора, дифосфор,
пентаоксид серная кислота, триоксид серы, тноиплхлорид или
тпХорпд бора, образуется амид, который можно гидролизом
пеоевести в амин. Так, например, ацетофенон превращается
в анп.
1Л11Н с хорошим выходом (уравнение 28).
Лг2С=хон __> ArCONHAr —> ArNHj + ЛгСО2Н (27)
НО, О? гидролиз
4C=NPh —► /С—NHPh --------►
ph\
XC=NOH
PhNHs + МеСО2Н
(28)
Наиболее интересной особенностью реакции Бекмана является
то, что определяющим для миграции алкильного или арильного
заместителя является не нх природа, а стереохимическое распо-
ложение. Мигрирующей группой всегда является арильный пли
алкильный заместитель в кетоксиме, находящийся в анти-положе-
нии к гидроксильной группе (уравнение 29).
R'\
R-’/
НО,
V=N
R2/ \R1
(29)
(5) Прямое аминирование
Этот метод имеет ограниченное значение для синтеза. Прямое
аминирование бензольного ядра протекает удовлетворительно
лишь в крайне жестких условиях. Одни из методов включает
реакцию бензола с безводным аммиаком при высоких температу-
рах 11 давлениях в присутствии катализатора, состоящего пз соли
молибдена, вольфрама или хрома и оксида меди или никеля [32]
(уравнение 30).
+ NH3
MoSs, CuO
30 атм, 400 °C, 3 чХ
(30)
(100 %)
6.З.1.2. Вторичные и третичные ароматические амины
(1) N-Ллкил- и N ,П-диалкилариламины
р!,'. У У/У'У/'Действии „а ввили,, алкллгллогеиида сб-
USУ,««»•
Дастся в третичный амин (например,
enaBiieiiue 31). В качестве алкилирующих агентов могут быть ис-
’тьзовапы также алкилсульфаты и сульфонаты, такие, ка ди-
"етилсульфат пли этил-п-толуолсульфонат. Часто образуется
есь вторичного и третичного аминов, которую можно р!зд=
помощью реакции с ацетаигпдридом или бевзолсульфокилхлори
юм- Вторичны)! амин реагирует с образованием амида или суль-
фонамида- которые не обладают основными свойствами а тое-
’„чиый амии неивступает в реакцию, и его можно извлечь мине-
ральной кислотой.
NH2 + Mel —>
Mel
•NHMe -----
NMe2
(31)
Алкилирование первичных ароматических аминов можно осу-
ществить с помощью триалкилортоформиатов в присутствии сер-
ной кислоты [33, 34]. Первичным продуктом реакции является
^алкилформанплид, который легко гидролизуется с образова-
нием N-алкиланилииа, Таким образом, например, N-этил-л-хлор-
анилпн можно с хорошим выходом получить из л-хлоранилина,
триэтилортоформиата и серной кислоты (уравнение 32).
NHi Et—N—СНО NHEt
(80—86%) (87—92%)
N-Алкилироваппе можно также проводить в присутствии ще-
лочных металлов [35]. При растворении натрия в анилине обра-
зуется анилид натрия; образование анилида можно катализиро-
вать добавлением металлов или их оксидов. При введении в такой
раствор этилена под давлением получается смесь N-этилаинлинз
(86%) и Ы.Ы-диэтилаиплииа (9°/о), алкилирования ароматиче-
ского ядра при этом не происходит. Эта реакция применима
в случае толуидинов, феиплеидиампнов и нафтнламииов.
Реакция анилина с первичными алифатическими спиртами
также может приводить к N-алкилпропзводным. Так, например,
если нагревать сульфат анилина с метаиолом под давлением, о
разуется смесь моно- и дпалкилаиплшюв (уравнение 33). Неак-
можно проводить также в присутствии никеля Ренея |3б,
I или кислотного катализатора [38] (уравнения 34 и )
ArNH2 -р МеОН —>
NH- + RO1I
ArNHMe + Н2О
NI Ренея
кипячение, 16 ч
ArNMej
МеОП
NHR
(82%)
(33)
(34)
R=Et, Рг, Ви
177
Nib + McOH
SIO2, Л1гОз
3 ч, 280 °C, давление,
(98%)
(35)
Эта реакция лежит в основе промышленного способа получе-
ния N-алкил- и Ы.Ы-дпалкпланилшюв: анилин и его сульфат или
гидрохлорид нагревают с нужным спиртом под давлением прн
температурах до 170-180 °C. В качестве катализатора использу-
ют порошок меди пли хлорид кальция. Обычно выбирают усло-
вия, позволяющие получить преимущественно вторичный или тре-
тичный амин, которые очищают перегонкой.
Особые методы получения вторичных аминов. Для получения
вторичных аминов существует несколько особых методов. Для
этого необходимо защитить одну из связей N—И в первичном ами-
не, чтобы сохранить ее при проведении дальнейших реакций.
Приемы защиты аминогруппы рассмотрены в обзорах [39—42].
Удобным и широко распространенным методом защиты амино-
группы является превращение амина в амид или замещенный
амид. Наиболее часто для зашиты аминогруппы в ходе реакций
алкилирования используют ацетильную группу, более устойчи-
вую по сравнению с формильной группой [39]. Ацетильные произ-
водные первичных амшюв получают ацилированием с помощью
хлораигидрнда пли ангидрида кислоты. Ацетанилид может давать
натриевое производное, которое легко реагирует с алкплгалоге-
нидом; получаемый при этом N-алкплацетанилид в кислых или
щелочных условиях гидролизуется до вторичного амина (уравне-
ние 36).
Mel КОН
АгКНСОМе —> ArNCOMe Na+ ——-----> ArN(Me)COMc ----> ArNHMe (36)
Т0ЛУ°Л (90%)
Введение защитной группы по одной из N — Н-связей в пер-
вичном амине может сопровождаться увеличением реакционной
способности другой связи; это наблюдается, например, в случае
превращения первичного амина во вторичный путем алкилирова-
ния бензол- пли Д-толуолсульфонильиых производных. Гидролиз
178
Восстановительное алкилирование Кои
ароматического амина с альдегидом или "СацИЯ ^винного
шая конденсация с кетоном приводят к Ши*ж^иЛегко пРотекаю-
ИН1 аннлу, который можно восстановить у Основаии>о (11)
водородом в присутствии никеля Ренея пп 8 кислоте или
вторичный амин [43] (уравнение 38) ' Р ЭТОм образуется
RCHO + ArNH, рсн=ж.
КСпр.Х'НАг
(38)
В случае низших алифатических альдегидов типа формальде-
гида получают сложную смесь продуктов реакции, при восстанов-
им которой образуется смесь первичных, вторичных и третич-
ных аминов.
Разработан простой метод восстановительного алкилирования,
дают111”1 хорошие результаты [44]. Первичные или вторичные
ароматические амины можно алкилировать или арнлировать дей-
ствием алифатических и ароматических альдегидов в присутствии
аниона гндридотетракарбонилжелеза MHFe(CO)4. С препаратив-
ной точки зрения преимуществом этого метода является возмож-
ность получения, главным образом, моно- или диалкилпронз-
водных из первичных аминов просто за счет изменения мольного
соотношения амина, альдегида и MHFe(CO)4 (см., например,
уравнения 39 и 40).
1:2:2* ft \
NH2 + HCHO —
(39)
NHMo (65%)
Me_0_NH2 + HCHO Me-Q^-NMe, (80%) (Я
•Соотношение ArNH,, НСНО и MHFc(CO)4.
Прочие методы. Моноалкиланнлнны можно ‘''|Н^31’Р^0м
путем восстановления N-алкплфенплгидрокснлами ю Р
в присутствии палладия на угле [45] (уравнение )•
ArNIIOH — ArNROH ArNHR <41’
Интересный общин метод синтеза [46] 8 бен-
алкил- или арилдпхлорбораиов с °РгаИ'1,,е“'еся соединение
зольном растворе. Промежуточно образу щается в со-
(12) в условиях щелочного гидролиза легко nPeBP|QQO/ /уравке-
ответствующ|й) вторичный амин с выходами 84- 1W/о (.1
пне 42)
NaOH. 132_^ ArRNII (42)
RBC12 + ArN3 —> ArRNBCh
(12)
179
аплоятических углеводородов с галогепамииами пьп
± невысоки, хотя в некоторых случаях удается добиться вЬ1сР0.
«у выходов Так, например, реакция N-хлордиметиламииа [491
КЙЛ ВЫХидм1. _____„ nirnwiTiltia м ППТПГШРТЙгдо rxnl
“ 90% бя—
хлорида алюминия и ^нитрометана при 0°С
(43)
(90%)
ОН
KsFe(CN)e. КОН, Н2О /
-----------------** \ /------NHMe
(93%)
(44)
При действии трикалийгексацианоферрата на третичные аро-
матические амины происходит N-деметилпрование с образованием
вторичных аминов [50] (уравнение 44).
(2) N-Арил- и И,И-диариламины
Реакция первичного ароматического амина с арплгалогенндом
протекает с трудом, и ее осуществление требует специальных
условий. Так, дифениламин н его гомологи можно получить по
реакции Ульмана, которая заключается в нагревании анилина
или ацетанилида с бромбеизолом в нитробензоле в присутствии
меди или иодида меди и карбоната калия (уравнение 45).
Си
PhNHCOMe + PhBr + К2СО3 --> Ph2NH + СОг + MeCO.K + KBr (45)
(60%)
Реакция дифениламина с нодбензолом в нитробензоле в ана-
логичных условиях или в присутствии солей лития приводит к
трнфеннлампну с высоким выходом [51, 52] (уравнение 46). Ди-
фениламин можно также получить по реакции анилина с фено-
лом в присутствии хлорида цинка или трнхлорнда сурьмы (урав- 47 * * * *
2Ph2NH + 2РЫ + К2СО3 2PlisN + СОг + 2KI +Н2О
(85%)
ОкЛгт , г,!.,,,, ZnCi2 , 260’С
PhOH + PhNH2 -----------------> Ph2NII + H2O
(46)
(47)
испо^Ллпяи!63" диФеинламниа и его производных может быть
мена) 1531 н еРС| (1УппиРовка нмпдатов (перегруппировка Чеп-
Хр1д1м1АпЛЛ1,Ме^ПрИ взапмодействнн бензанилида с пента-
Ф ф ра образуется имндонлхлорид (13), обработка
180
агового замещенным фенолятом натрия приводит к ,
S)P При нагревании последнего происходит перегруппировка^
JX, щелочной гидролиз которого дает вторичный ами? (>рав
пение 48)•
ArNHCOPh + PCIs —> ArN=CCIPh -----—т г дг^=с/°Аг
^Ph
(13) (14)
Аг\ “он
—> NCOPh -----> ArNHAr1
Аг'/
Технический способ получения дифениламина состоит в нагре-
вании смеси анилина и его гидрохлорида чаще всего в присут-
ствии хлорида алюминия [54] под давлением и при высокой тем-
пературе (уравнение 49).
PhNHs + PhNH2-HCl + AlCl3 ph2NH (49
(95%)
6.3.1.3. Нафтиламины
(1) а-Нафтиламин (1-аминонафталин)
а-Нафтпламин впервые получил Н. Н. Зинии [48] при восста-
новлении 1-нптронафталина сульфидом аммония. Обычными ме-
тодами получения а-нафтнламина являются: восстановление в
растворе кислоты действием железа, цинка или хлорида олова
(II) и каталитическое гидрирование. Этими методами а-нафтпл-
амин удается получать с хорошими выходами. Описано [55] по-
лучение а-нафтиламина с высоким выходом при восстановлении
1-нитронафталина дисульфидом натрия в водном растворе в при-
сутствии небольшого количества натриевой соли ароматической
сульфо- или карбоновой кислоты (уравнение 50). а-Нафтпламнн
можно также получить нагреванием а-нафтола с комплексом хло-
рида цинка с аммиаком (уравнение 51).
NO» №
(81%)
181
(2) fr-Нафтиламин (2-аминонафталин')
п„я получения р-нафтиламина обычно не используют восста-
„пвлеиие поскольку 2-питропафталин труднодоступен. Практиче-
скор значение имеет лишь одни метод — аминирование легкодо.
степного В-нафтола. Этот метод известен под названием реакции
Бухерера [56-58]. р-Нафтол нагревают иод давлением с раство-
ром сульфита аммония и аммиаком при laU—lol) С, прн этом
происходит почти количественное превращение в нафтплампн
(уравнение 52). Механизм реакции Бухерера [59] представлен
уравнением 53.
•ОН (NH.|l2SO3, 150 Dc. nh2
давление, 8 ч
(52)
OH ffso;
so;
(15)
О
-USO
(53)
NH
NHa
(54)
so;
(16)
Кетоформа (15) р-пафтола н пмннопроизводное (16) стабили-
зованы за счет присоединения бпсульфит-поиа. Как а-, так п
р-нафтиламип были получены нагреванием соответствующих наф-
тойных кислот с гидроксиламином и полпфосфорион кислотой
[60[ (уравнение 54).
/СООН
MH2OH-HC1
ПФК, 160 °C. кон'
(82%)
6.3.1.4. Полифункциональные ароматические амииы
(/) Ароматические амины,
содержащие алкильные заместители в кольце
метпплГ'п™1’1 Н КС||Лилины обычно получают с помощью общих
нов Так Н(1е»1Ь3^еМЫХ для синтсза первичных ароматических ами-
гановлепие о-, л1- н /г-ннтротолуола, например, дей-
182
стВпеи железа в разбавленной соляной кислоте позпо
"„тветствуюшип изомер толуидина. позволяет полуЧиТь
При реакции ароматических аминов с апКРип
аН1Ш1да алюмиппя происходит алкилпроваине зппПР”Сутетвин
,ра [61, 62[. Каталитическое действие анилида я „ Р матическ°Ео
йыть значительно усилено введением хлорида ал1пм'°МИ'!,)Я Мож«
катализаторов Фриделя — Крафтса таких Я ИЛ(* ДРУ-
s3(IV), тетрахлориды титана и кремния,’ трифтоп°Л0‘
риД цинка. Так, при нагревании анилина, алюминия R ИЛИ хло'
синяя и этилена под давлением образуется Р й Хл0Ри-1а а.тю-
(гравяение 55). Реакция протекает через порт»™, ЭТНЛапИли»
зованпе анилида алюминия. Не Наблюдается заме нМЬНОе °бра'
положение или N-алкплпровання. Аналогичным пйпДТ В пара'
превращается в г^етнл-б-этнланилнп с выходом 87 о?’™5'"'
лундин дает 4-метил-2,6-д11этплаи11лиН с выходом 95°/ П'™'
В ряду ксилидинов происходит подобным же образом r ®ЩеИПе
условиях а-нафтиламнн превращается в 1 о ™ же
с выходом 90%. Р Я В 1 амино’2'Этилнафта.1нн
nh2
nh2
.. .,r. Ek xEt
. Л1. Л1С1з >5^
j + 2СН2=СН2 ------------► Т Г
(55)
(96%)
Ароматические амины, алкилированные в ядро,
также из соответствующих гидроксисоедииеиий
промежуточного превращения в диэтплфосфаты
Me Мс
Л-Он ИЩСОЬРОС, Jv/OPO(OE‘b ,. !(ЭТ1г, К г
L JL 2. NaOH *” L 1 2. КНз I
^'Ме
Me
получают
через стадию
(уравнение 56).
Me
X/NH;
(56)
Me
(78%)
Применяются также реакции Гофмана и ШмД^о'вой
при гофмаиовскоп перегруппировке аМ1,ла аРперегрупппровка
кислоты получают п-толуидпн с выходом 98/о, Р к с0.
Шмидта в случае о-, м- и «-толуиловых кис Р
°тветству|о|цим толуидинам с выходами 4о, 24 о
(2) Диамины и триамины
плсстаиовлеипсм о-нитро-
О'Фенплеидиамни обычно получают ®°,с „иртового раствора
Сплина действием цинковой пыли и - пе о-иитроаиплин'
едког° натра. Каталитическое палладия на Ф>'ллеР0'
’присутствии палладия на угле [64] । высоким выход •
Лй земле [65] также приводит к дпамн»)
57 58). Если необходимо сохранить ацетильнук
(уравнения , „ в первиЧ„ых ароматических аминов
'ij'iiue использовать каталитическое гидрирование (уравнение 59).
NHa NaOH. кипячение
3Zn + н»о
NO.*
nh2
(85%)
•NH,
NH,
(57)
NH»
NO,
Н2. Pd/C f |Г
70—90 °C. давление
(85—90%)
(58)
NHCOMe
Н2, Pt. спирт
(59)
л-Фенилендпамппы можно получить восстановлением легко-
доступных .ч-динитробензолов с помощью железа в соляной кис-
лоте' Например [66], восстановлением в этаноле 2,4-динитрото-
луол превращается в 2,4-дпампнотолуол (уравнение GO).
EtOH. Fc, HCl
(74%)
(60)
л-Фепилендиамин получают восстановлением п-нитроанилнна
или восстановлением n-амипоазобензола гидросульфитом натрия
пли хлоридом олова (II) в соляной кислоте. льТолунднп можно
превратить в 2,5-диампнотолуол аналогичным образом (уравне-
ние 61).
Me Me
пьм г, , )/ \ ... К1ОН А \ Na2S2O4
PnN2Cl + —nh2 ------>- Ph—N=N—/ X)—nh, —--------*
(17)
Me
—> H2\— NHj+PhNHs (61)
В ряду три-, тетра- и пентааминобензолов нх устойчивость по-
степенно падает. Эти соединения можно получить восстановле-
на соответствующих иитросоедпнений. В другом варианте ами<
184
„rnviuiy вводят непосредственно, через пппм».
"зосоединения [67] как в синтезе,2,4,Р5-тетХмТ°^ПОЛучение
„.фенилеидпамииа (уравнение 62). ‘Раамцнобензола из
1. *N2ceH4so- ВД
NH2
(62)
H:N nh2
(85%)
(3) Аминофенолы
Аминофенолы обычно получают восстановлением ™
юШНх нитрофенолов чаще всего действием с^ьфата
вводном аммиаке. Используют также и каталитической (П)
ванне, п-аминофенол можно получить гидрированием нитробензо'
ла [68] в гетерогенной системе в присутствии серной кислоты н
платинового или палладиевого катализатора (уравнение 63?
/ \ Н2. Pd г—х '
\___/ N°2 H2SO4, 60-120 «с* Н0 \ /------NH2
(73%)
Полинитрофенолы при восстановлении превращаются в почи-
аминофеиолы; например, прн восстановлении аммониевой соли
2,4-дпнптропафтола-1 (18) гидросульфитом натрия получают
2,4-диаминонафтол-1 (уравнение 64).
о- nh;
no2
+ 6Na2S2O4 + 8НоО —->
(63)
NO2
(18)
ОН
nh2
+ HNaHSO3 + Na(NH4)SOs
(64)
NH2
Другой метод введения аминогруппы в фенольные соединения
ключастся в проведении реакции сочетания с солью арилдиазо-
ппп " Последующем восстановлении образующегося гидроксиазо-
л.\ 1<3!!О-'11!ОГ°, например (19), гидросульфитом натрия пли хлорн-
noilvOJ'OBa(I1) [69—72] Например, 2- и 4-ампноиафтолы-1 можно
ств1ЧПТЬ/13 нафтола-1 с выходами 72—75 и 8- 85 А, с0°
1Н0 (уравнение 65). Но этим способом нельзя получит
185
t аналогичного строения, так
сочетаются С солями дпазопия в щепа-положение,
он
л-ампнофеполы пли ампяовафтолы
как фенолы < тл««г
ОН
1. n,c6h4so;
2. №011
он
NaaSgO.)
пли SnCI2
(65)
N=NC6H(SO3Na
nh2
(19)
Для превращения гидроксибензойиых кислот в аминофенолы
можно воспользоваться реакцией Шмидта. Так, л-гндроксибепзой-
ная кислота превращается в лпамннофеиол с выходом 80%. Одна-
ко салициловая кислота в условиях перегруппировки Шмидта
дает только 2! % о-аминофеиола; перегруппировка Гофмана в слу-
чае салициламида позволяет получить о-амннофенол с выходом
70%.
(4) Нитроамины
Непосредственное действие азотной кислоты на анилин дает
неудовлетворительные результаты, поскольку аминогруппа чув-
ствительна к окислению. Однако аминогруппу легко защитить
ацетилированием. Так, нитрование ацетанилида с последующим
гидролизом приводит преимущественно к п-нцтроанилину (урав-
нение 66). Если необходимо получить о-ннтроанилип, то пара-
положение можно блокировать сульфированием (уравнение 67).
(66)
IS6
(56%)
(67)
Иптроамины можно получить также в результате замещения
£”На аммиаком в активированных нитроароматических гало
См"' изпример, о- и n-нитроанилины могут быть получены
ге пней о- и п-хлориптробепзолов с аммиаком. Подобным ««
С РеаКЦЙЯ 2’4-д,шнтР°-1-хл°Рбензола с аммиаком в прнД
°ВРНИ ацетата аммония приводит к 2,4-динитроанилину [731
/уравнение 68).
и zno2 ,no2
МеСО2МН< .—(
Cl + 2NHs |70 oC 6q 1 O2N--'J
оЛ
(68)
(68-76%)
л-Ннтроапнлии и его аналоги обычно получают частичным
восстановлением .н-дииптробензола гидросульфпдом аммония или
натрия [74]. Используют также сульфид натрия, сульфид аммо-
ния и хлорид олова (II) в соляной кислоте. Эта реакция примени-
ма и к дннптрофенолам; например, 2-амнно-4-нитрофенол получен
частичным восстановлением 2,4-динитрофенола [75] (уравне-
ние 69).
(58-61%)
.и- и н-Нитроаинлины были получены с помощью реакции Гоф-
мана из соответствующих N-бромиптробензамидов (уравнение 70);
о-, л- н n-ннтроанилпны можно получить с хорошим выходом из
соответствующих иптробензойиых кислот по реакции Шмидта.
,CONHBr /NHj
O2N-Q O.N-0 (70)
(70%)
Достаточно подробно описано [76] получение нитронафтил
амилов. Прямое нитрование а- н р-нафтиламина удается осущест
В|,Ть обычно лишь при низких температурах в концентрированн i
серной кислоте, прп этом образуются 5- и в-нптропроп^однь^
Ацетил-а-нафтиламин нитруется как правило, в по
Nils
(71)
(60%)
187
и 4 в то время как N-ацетал-р-нафтиламин дает преимущест
НО Лпитролропзводиое. 4-Нитро-а-нафтиламип может быть „оХ
Чен прямым аминированием нитроиафталина с помощью гидро^'
спламииа [77] (уравнение 71).
(5) Галогенсмины
Наличие аминогруппы активирует орто- и нара-положення в
анилине таким образом, что по этим положениям очень легко осу.
ществляются реакции с бромом И хлором. Так, взаимодействие
анилина с бромом приводит к 2,4,6-трпзамещенному производному
с количественным выходом; эта реакция используется для обна-
ружения анилина (уравнение 72). Трихлорлроизводное можно
получить с высоким выходом обработкой анилина безводным хло-
ристым водородом и хлором [78] (уравнение 73). Большая часть
других ароматических аминов бронируется по всем имеющимся
в молекуле орто- и нора-положениям по отношению к аминогруп-
пе; например, толуидины дают дибромпроизводные с высоким
выходом.
NH2
-V ЗВгз
н2о
(100%)
(73)
степени галогенирования необходимо дезак-
Для уменьшения степени галогенирования необходимо дезак-
тивировать аминогруппу с помощью ацетилирования. При этом
понижается электронная плотность как в ароматическом ядре,
так и на атоме азота (по сравнению с анилином), поскольку
свободная электронная пара азота вовлекается в сопряжение с
амидной связью (20). Несмотря на дезактивирующий эффект
(20), степень активации ароматического ядра все еще достаточна
для избирательного галогенирования в орто- и пара-положеииЯ-
Ацетильная группа легко удаляется в условиях кислотного или
щелочного гидролиза, при этом регенерируется амии. Так, гало-
1в8
получают
р,„1Рованием ацетанилида с последующим гидролизом
ге^Роге(!анилии (уравнение 74).
(20)
(80%)
(74)
о-Галогепаиилппы можно синтезировать из анилина, защи-
тив предварительно /гара-положение сульфированием (уравне-
ние 75), затем сульфогруппу удаляют гидролизом. Другой вари-
ант синтеза о-хлоранилипа состоит в восстановлении о-нитрохлор-
бензола активированным железом [79] (уравнение 76).
При действии безводного хлорида олова(II) на иитрооензол
происходит одновременное восстановление и хлорирование, в ре-
зультате чего образуется /i-хлорацетанплид с количественным вы-
ходом [80] (уравнение 77).
—NO2 + Ac2O+ SnCl2-2H2O —> Cl—NHAc (77)
(100%)
Для получения галогепамипов используют реакции Гоф''31|а
Шмидта. Например, льбромапплпп получают из - р ' а.
** -“°;”"™' ..........................................—
189
п ши 1811 ’ реакцию проводят в присутствии нитрометана,
2? служит источником гпдрокснлампна (уравнение 78).
CHsN'Qa. ПФК Л~\
c|_ZA-COiII -Koil7l^
(80%)
KOTO
(78
коп, из °C
Гатогсннафтнламнны обычно получают восстановлением гало-
гснннтронафталинов железом в соляной или серной кислоте
В качестве восстанавливающих агентов можно также использо-
вать гидросульфит натрия или водород в присутствии никеля
Ренея.
(6) Аминобензолсульфокислоты
п-Аминобензолсульфокпслоту (сульфаниловую кислоту) полу-
чают нагреванием равных частей анилина н концентрированной
серной кислоты до 180°C. Механизм реакции включает миграцию
сульфогруппы от атома азота в ароматическое ядро (уравне-
ние 79).
HjSO)
ISO °C
NHSO3H
-Н2О
НОз S---—NHj
~ (79)
Из производных этого типа наиболее трудно получить о-ами-
нобензолсульфокпслоту (ортанпловую кислоту). Нанлучшнй ме-
тод ее синтеза состоит в сульфировании n-броманилпна с после-
дующим удалением брома восстановлением (уравнение 80).
-ч-Аминобензолсульфокислоту (метаниловую кислоту) синтезиру-
ют восстановлением лмштробеизолсульфокнслоты, получаемой
сульфированием нитробензола дымящей серной кислотой при
60—70 °C (уравнение 81).
NO2
no2
nh2
восстановление
(81)
SO3H ^^SO3H
мн [821: ппямым Lm к синтезируют несколькими метода-
Бухерера [831" исхли^1’'^'30^а'Шем 1!аФт|1ламинов; по реакции
талнисульфокислот с "3 наФт,олсУльФокпслот; нитрованием наф-
последующим восстановлением продукта
пеакчии и аминированием галогеннафталинсульфокислот. Приме-
ром таких превращении является последовательность реакций
включающая стадии нитрования и восстановления (уравнение 82).
6.3.2. СВОЙСТВА АРОМАТИЧЕСКИХ АМИНОВ
6.3.2.1. Основность [85,86]
Основность ароматического амина определяется доступностью
свободной электронной пары на атоме азота для протонирования.
В случае анилина она понижена за счет резонансной стабилиза-
ции, возникающей вследствие взаимодействия свободной пары
электронов с делокализованными л-орбиталями ароматического
кольца:
В результате этого анилин оказывается более ела ю’
пнем по сравнению с аммиаком или алифатпчески» и - •
Структуры дифениламина и трифениламина открывают
ше возможностей для резонансной стабилизации i, Ф вообще
еще более слабое основание, чем анилин, а трпфе i • м пред.
нельзя считать основанием. Исходя из строения ’’’’ случае,
сказать, что резонансная стабилизация максим . ' е п‘о от.
если аминогруппа может занимать к0,“анар' действительности,
ношению к ароматическому кольцу. ОдНа значительной сте-
итя резонансная стабилизация и происход 0Го кольца
печи, полная копланарность аминогруппы и Р
“С достигается. ароматическое ядро
Введение заместителей при азоте и. f могут вызывать
Изменяет основность анилина. Эти зам,,т и’_д/. (мезомерные)
т/ или —1- (индуктивные) эффекты и +А -
19J
п вследствие этого они повышают или понижают
эффекты, стабилизации свободной пары электр0110
степень 1овательно, ароматические амины могут зиачи.
атома азота- • ' от друга ио основности.
тельно отличаться ДР) нп ме тс же 311ачС1111я рК
N-McT,kial "Г ‘"'“же Хотя метильная группа вызывает „ояв-
caw а""Тп, ио -o 4-/-эффскта, он не настолько значителен, чтобы
ление небо.iwiio + ь резонансную стабилизацию. В случае
существенно • .1Г1о.1ат11ческнх амнион следует также при.
^”аа^ние * реох!1м!1ЧССКпе факторы. Между N-алкиль-
"^заместителями и водородными атомами в орто-иоложен11ях
аоомэтического кольца могут возникать пространственные взан.
действия в результате чего понижается степень планарНОСТИ
молекХ Этот эффект становится все более выраженным при
увеличении числа и объема N-алкильных заместителей и прнво-
пнт к существенному снижению степени резонансной стабилиза-
ции и повышению силы основания. Эти взаимоотношения поясня-
ют значения рКа для N-алкиланнлинов, приведенные ниже:
PhNHj 4,58 PhNEt2 6,56
PhX'HMe 4,85 PhNHCHMe2 5,77
PhNMet, 5,06 PhN(CHMe2)2 7,37
PhNHE 5,П
Влияние заместителей в цикле па основность ароматических
аминов зависит не только от мезомерного и индуктивного эффек-
тов, но и от положения заместителей (табл. 6.3.1). Метильная
группа оказывает небольшой положительный индуктивный эф-
фект, и можно ожидать, что введение такого заместителя в аро-
матическое кольцо вызовет небольшое повышение основности.
Это и наблюдается в действительности для мета- и пара-заме-
щепных анилинов, по не для орто-изомера. Этот аномальный
«орто-эффект» не ограничивается только метильной группой, но
наблюдается и для других заместителей. Причины этого явления
понятны пока не полностью.
Таблица 6.3.1. Основность замещенных анилинов (p7fa)
Заместитель Р^а
орто-изомера лета-изомера пара-изомера
S!4’ 4,45 4,72 5,10 nb 2,64 3,50 3,98 ОСН 4.74 4,30 5,65 NH 3 4’52 4,21 5,34 NH2 V4 4,98 6,16
Нитрогруппа вызывает появление сильного —1- п —М-эфФ614"
тов, и поэтому нитроаиилнны являются гораздо более слабыми
192
„пями, чем анилин. В случае onro- и „„
ос"ов»я стабилизация амина еще более ycini^TИЗомеров
на"с" аЛЬиейшему понижению основности.* 5 ’ ТСЯ’ что
Д"ТК '°\* /”А " °\. /=ч
N—< >—NH2 «-► ф:=А_♦
\=/ -п/ \ /~NH*
резо-
приво-
Галогениды как заместители вызывают сильный _/
с1,тельно слабый J-M-эффект поэтому хлоранилины более°сла-
бые основания, чем анилин. В случае лара-изомера +М-эффект
направлен против +Л4-эффекта аминогруппы, что приводит ?не
ольшому усилению основности Гидроксильная, алкокситьная и
аминогруппы вызывают сильный +/-эффект, и при введении в
лйра-положение эти „группы обусловливают увеличение основно-
сти в гораздо большей степени.
В ряду нафтиламинов заместители, находящиеся в том же
кольце, что и ам1шогруппа, вызывают те же эффекты, что
и в ряду анилинов. Если заместители находятся в соседнем коль-
це то эти эффекты выражены гораздо слабее.
6.3.2.2. Спектроскопические свойства
(1) ИК-Спектры [87]
Для ИК-спектров первичных ароматических аминов характер-
но присутствие двух полос поглощения в области 3500—3300 см-1,
обусловленных симметричными и несимметричными валентными
колебаниями связей N—Н. В спектрах первичных аминов обна-
руживается поглощение в области 1650—1590 см-1 за счет де-
формационных колебаний связей N—Н. Наличие водородной свя-
зи приводит к смещению полос поглощения в область более низ-
ких частот. В спектрах вторичных аминов наблюдается только
полоса валентных колебаний N—Н при 3500—3300 см-1, а по-
лоса деформационных колебаний N—Н, как правило, слишком
слаба и не проявляется. Третичные амины не имеют поглощения
в области валентных колебаний N—Н н поэтому их крайне труд-
но идентифицировать с помощью ИК-спектров. Частоты поглоще-
ния ароматических аминов в ИК-областп спектра (в см
приведены ниже: .
&еМаЬминКы0Леба,,ИЯ N~H 3500-3300 (ср.)-
Вгоричиые амины 3500-3300 (=₽•)
>т»чиые амины Поглощение отс5тствуе
5еР»иХ“аачшны,ИЬ,е КОлеба"НЯ N“H 1650-1590 (с.-ср.)
Вт°Ричные амины [63о-15о0 (ел.)____
иди в п°тосы, смещающиеся в область низких частот при
Г, ; 'чае твердых образцов,
°лна полоса.
1 Зак. Jos
193
(2) Спектры Я MP
о „пч-гнх классов органических соединении, из спект-
Как И ДЛЯ Д1 ’>“• амина можно извлечь цепную информа.
ра ЯМР apo'iaT"2Le' резонанс протонов NH-группы ароматиче-
цшо о его струной ’ задается в виде единичных узких спгиа.
ского амина ои' 9 е_4 7 ,1Т0 типично для быстрого химического
в ° ^мов водорода’аминогруппы. При разбавлении образца
обмена ат0“0в -1ТРеПеМ сигналы смещаются в более сильное
”''пГ Сигналы протонов группы N-CH алифатических замести-
поле. Сигналь Р д несколько смещены в более слабое поле
телец при aiu. п010женнем в спектрах аналогичных алкил-
амннов8’в случае ‘ацетилирования или протонирования N-алкил-
ап ламинов эти сигналы заметно (примерно на 0,5 млн-') сдай-
Хтея в более слабое поле. Для химических сдвигов (в млн-')
аооматпческих протонов характерно значительное смещение в
сторону сильных полей по сравнению с их положением в спектре
бензола:
о-Н Л4-Н п«Н
Ph\’H2 —0,75 -0,24 -0,63
PhNMe2 -0,60 —0,10 -0,62
Сигналы ароматических протонов также смещаются в слабое
поле прн ацетилировании (о-Н---------1-0,8, лг-Н---(-0,3, п-Н —
4-0,5 млн-1) или протоипрованнн (о-Н и п-Н---------(-1,0 и л-Н —
—(-0,5 млн-') ароматического амина. Не менее информативны
в спектры ЯМР 13С; как и следовало ожидать, наличие амино-
заместителей вызывает смещение резонансных сигналов орто- и
пара-углеродных атомов в сильное поле по сравнению с положе-
нием линий в спектре незамещенного бензола (табл. 6.3.2).
Таблица 6.3.2. Данные спектров ЯМР '3С
для углеродных атомов ароматического кольца
ароматических аминов С0Н5Х
Химические сдвиги ядер 13CS
Анилин (X = NH2) N,N-Диметил авилин С-1 147,9 (+19,2) Орто 116,3 (-12,4) мета 130,0 (+1,3) пара 119,2 (-9,5)
(Х = .\'Ме2) 101,0 (-f-22,b) 113,1 (—15,6) 129,7 (+1,0) 117,2 (-11,5)
Мд+Диэтиланилин (X = NEts) Ацетанилид 148,6 (+19,9) IQA О/i < . . 113,4 (-15,3) 130,1 (+1,4) 116,5 (-12,2)
(X = NHCOMe) (оУ.о 11Д) 118,8 (-9,9) 128,9 (+0,2) 123,1 (-5,6)
а Химические сдвиги (п „
терлзуют смещение сигиц-юв от'пгпп-лг^Т1,10С!Пе‘'ь‘ю (шкала 6); цифры в скобках харак
иложения сигнала углерода в спектра бензола.
194
(3) УФ-Спектры [58]
для УФ-спектра бензола характерны две интенсивные полосы
поглощения при 80 и 200 им и более слабая полоса поглоще-
ния при 260 нм. При замещении бензола характер всех трех по-
лос значительно изменяется. Введение полярных заместителей
имеющих свободные электронные пары, например аминогруппы
приводит к явно выраженному смещению полос поглощения н
значительному увеличению их интенсивности. Так, например,
в случае анилина полоса при 200 им (первичная, или 'TQ-nonoca)
смещается до 230 нм (е 8600), а полоса при 260 нм (вторичная,
Илп бензольная полоса) смешается до 280 нм (е 1430). Анало-
гичные изменения наблюдаются в УФ-снектре диметиланилина
[Хмакс В спирте 251 нм (е 14000) и 299 нм (е 2100)]. Как н сле-
довало ожидать, эти изменения сильно подавляются при протони-
ровании, и УФ-спектр катиона анилииия |/.„акс 203 нм (е 7500)
н 254 нм (е 160)] напоминает спектр бензола. Эти измене-
ния УФ-спектров ароматических аминов в растворах кислот мо-
гут быть использованы как тест на наличие структуры сопряжен-
ного ароматического амина.
6.3.2.3. Физиологические свойства
Ароматические амины широко используются в качестве полу-
продуктов при производстве красителей, фармацевтических пре-
паратов и в резиновой промышленности. Некоторые из них очень
токсичны, другие токсичны лишь в незначительной степени, мно-
гие ароматические амины раздражают кожные покровы и явля-
ются сенсибилизаторами. Особым токсикологическим свойством
ароматических аминов является их способность образовывать мет-
гемоглобин [89], продукт окисления гемоглобина, который дей-
ствует угнетающим образом на центральную нервную систему
человека. Обычно амин проникает в организм человека через
кожу или при вдыхании паров. В последнее время амины стали
предметом интенсивного изучения в связи с возможностью про-
являенпя ими канцерогенных свойств. Уже в 1895 г. были заре-
гистрированы случаи рака мочевого пузыря у рабочих предприя-
тий по производству красителей, где в качестве исходного сырья
использовался анилин. Однако в настоящее время считают, что
предполагавшаяся ранее канцерогенность анилина на самом деле
обусловлена примесями. Известно лишь ограниченное число кан-
церогенных ароматических аминов с одним ароматическим ядром;
главную опасность представляют ароматические амины с двумя
и более циклами. Так, {J-нафтплампн (21), бензидин (22) п4-ами-
нобифенил (23) являются сильными канцерогенами, вызывающи-
ми образование раковых опухолей у человека. Использование
этих веществ в промышленности в настоящее время запрещено.
Применение других аминов сходной структуры находится под
7» 19*
очень строгим
я-нафт1,лаы1,в’
дин (26).
NH2
контролем; к этой группе веществ отноСЯТСя
о-толпдпп (24). днхпорбензидпн (25) и дпа11Нз11
(21)
HsN-
nh,
(22)
H2N-
—NH2
Me Ме
(24)
H2N—(
(23)
h2n—'Г~\—/~Ч-
nh2
СЕ ХС1
(25)
—NH2
MeO'
OMe
(26)
Бензидин представляет особый интерес, поскольку это соеди-
нение в течение многих лет использовали как реагент для обна-
ружения крови; для этой же пели применяли о-толпдпн и дпанп-
зидин Недавно для этих определений был предложен 3,3', 5,5'-
тетраметилбензидин (27) [90], который, по-впдимому, безопасен
и может быть использован вместо описанных выше реагентов.
(27)
Считают [91], что имеется убедительное доказательство того,
что ароматический амин канцерогенен в том случае, когда амино-
группа находится в таком положении полиядерноп ароматической
системы, которое эквивалентно положению 2 в нафталине или
пара-положению по отношению к бифенильной связи. Дальней-
шее введение заместителей в ароматические амины может повы-
шать или понижать канцерогенную активность. Так, усиление кан-
церогенных свойств наблюдается в том случае, когда ароматиче-
ское кольцо песет метильные пли метоксильные заместители, в
то время как гидроксильные, карбоксильные пли сульфогруппы
понижают канцерогенность амина.
6.3.3. РЕАКЦИИ АРОМАТИЧЕСКИХ АМИНОВ
6.З.З.1. Реакции с азотистой кислотой
Реакции, происходящие при действии азотистой кислоты на
первичные, вторичные п третичные амины, различны [92]. Пер-
вичные ароматические амины образуют соли дпазония. Из вто-
196
ричных ароматических аминов получают N-нитрозамины
время как третичные ароматические амины нитрозиоуются
ро с образованием С-нитрозосоединений.
В то
в яд-
(1) Первичные ароматические амины
При действии азотистой кислоты на первичные ароматические
амины в присутствии минеральных кислот получают соли диазо-
ния. Обычно реакция протекает быстро и с количественным вы-
ходом- Анилин превращается в бензолдиазонийхлорнд [931 диа-
зотирование толуидинов [94, 95] и нафтиламинов также протека-
ет удовлетворительно (уравнения 83—85).
NHa • НС! + NaNO; + НС1 —> NJ СГ + NaCl + 2Н2О (83)
(85)
Если заместители находятся в орто- или пара-положении, диа-
зотирование осуществляется с большим трудом. Однако возмож-
но диазотирование ароматических аминов, имеющих в ядре один
из следующих заместителей: галоген, нитрогруппу, гидроксил или
альдегидную группу. Так, например, успешно диазотируются ни-
троанилины [96] и амипофенолы [97] (уравнения 86, 87).
—nh2 + hno2 + hbf4
’BF4 + 2НгО
(86)
2HO---f \—NH2 + 3H2SO4 + 2NaNO2
(87)
При наличии нескольких орто- или пара-заместителей основ-
ность амина может понизиться до такой степени, что диазотиро-
вание в нормальных условиях становится невозможным. Так,
диазотирование 2,4,6-триброманилина и 2,4-динитроаиилина мож-
но осуществить только прп высокой концентрации кислоты в рас-
творе. Слабооспбвиые ароматические амины, например 2,6-дпиод_-
4-нитроанилпн [98], лучше всего диазотируются в смеси серной
и фосфорной кислот (уравнение 88).
Представляет интерес диазотирование соединений, содержа-
щих одновременно ароматическую и алифатическую аминогруп-
пы. Можно провести диазотирование ароматической аминогруппы,
не затрагивая алифатическую аминогруппу. Таким образом,
-197
0-N—'£^-NIl24-NaNO2 °2N \=^/ N* 'OWl (88;
наппимер можно осуществить диазотирование п-амшюбензиламина
1Г991 а последующее восстановление фосфиновой кислотой приво-
дпт к бензиламину (уравнение 89).
Н2И_/”Л—CH..NHJ —► +Ns—уЗ/—CHlNH2 —* —СНЖ
4=7 (89)
Реакции солей дпазонпя можно разбить на две группы: реак-
ции замещения диазогруппы, сопровождающиеся отщеплением
молекулы азота, и реакции с сохранением азота. В определенных
условиях дпазонпевая группа может быть замещена гидроксиль-
ной группой, водородом, галогеном, нптрпльной или арильной
группами (схема 90).
PhOH
Ph—Ph
PhCN
Си
ЕЮН
KCN
разб. HjSOj,
кипячение
EtOHv кони. H2SO4
-iPhN? X’
NaNO2. A
CuCl. HCI
(CuBr, ИВг; KU НВГ4, Д)
PhH
PliCl (Br, I, F)
(90)
К наиболее важным реакциям второго типа относятся реакции
с фенолами, нафтолами и первичными, вторичными и третичными
аминами (схема 91).
Ph-X=N-C6H4NR2-n
R
I
Ph—N—N=N—ph
PhX'R2
PhOH
PhNHR
PhNj X' —
PhNHj
Ph—N=N—CoH.OH-rt
(91)
PhNH—N=N—Ph
Восстановление солей диазония сульфитом натрия [100] при-
водит к натриевой соли гидразинсульфокислоты (28), которая
при кипячении с соляной кислотой превращается в фенилгидра-
зин (уравнение 92).
PhNj X' —» PhNHNHSO3Na —► PhNHNHj (92)
(28)
(2) Вторичные ароматические амины
кислотойЧНоЛпДрОМаТм'1еСкие амш1ы> взаимодействуя с азотистой
поевпашаетса УюТ ^’Питрозопроизводпые. Так, N-метиланилин
Втооичные япп нитР030'^'мет,1ланнл1ш [101] (уравнение 93).
р матические амины можно ннтрозировать также
198
действием нитрозилхлорида в смеси уксусной кислоты и уксус-
ного ангидрида; примером служит получение метилового эфира
3.п-(Н-иитрозацетамидо)фенилпропионовой кислоты (29) из ме
тидового эфира 3-(п-ацетамидофенил)пропионовой кислоты 11021
(уравнение 94).
// \ NaN02, ИС1 г,—\ /NO
V —NHMe -------- . // \\ /
\ / о—iu»c
(93)
МеО2С(СН2)г
—NHAc
NOCI, Ас2О
АсОН.КОАс
/-У ли
МеО2С(СН2)2—{94)
\=/ \Ае
(29)
Если ароматический нитрозамин вводят в реакцию с раствором
хлористого водорода в смеси абсолютного спирта и диэтилового
эфира, нитрозогруппа мигрирует в пара-положение ароматическо-
го кольца (см., например, уравнение 95). Эта реакция известна
как перегруппировка Фишера — Хеппа, Так, \-нитрозодифенил-
амин перегруппировывается с образованием п-иитрозоднфенил-
амина [103], а Ы-иптрозо-М-метилаинлин дает п-нитрозо-Х-метил-
анилин (уравнение 95). Нитрозогруппа всегда перемещается в
пара-положение [104], и если это положение блокировано, пере-
группировка не происходит, В ряду производных нафталина N-
нитрозо-1-нафтнламин дает 4-нитрозопроизводное (30), в то
время как Й-иитрозо-2-нафтиламин превращается в 1-ннтрозо-
производное (31) [105] (уравнения 96, 97).
Легко видеть, что перегруппировка Фишера Хеппа откры-
вает удобный путь для введения ннтрозогруппы в ароматическое
кольцо вторичных ароматических аминов. Прямое ннтрознрование
вторичных ароматических аминов осуществляется с помошыо
твердого нитрита натрия или нптрознлсерной кислоты (lUoj.
199
Пп„ восстановлен.... N-niiTpo30Coe.nineniii> цинковой пы.,1Ь1
,баРв енпоП кислоте образуются замещенные гидразины; uanpH
разбавлено зо-М-мстпланпд|Ща ,юлуча10Га-метилфени,,,.,,^
знн’ равнение 98). В более жестких условиях регенерн-
исходный амин (уравнение 99).
,__, .NO
—NZ
РУется
Zn, разб. llOAc
20—80 °C
Ж
•Nf
(98)
'NO Sti или Zn, 1 (CI
-Me
•NHMe + NH3
(99)
(3) Третичные ароматические амины
В случае третичных ароматических аминов происходит нитро-
зирование ароматического кольца с образованием С-питрозосоеди-
ненпй. Например, диметиланнлпн превращается в п-нитрозодиме-
тнланнлнн [107] (уравнение 100), а З-бром-М.М-дпметиланилин
дает 3-бром-4-н11трозо-М,М-диметнлан11лпн. Замещение обычно
происходит в пара-положение, однако, если это положение
занято, получается орто-нитрозосоедпнение. Наличие заместителей
в орто-положении дезактивирует ароматическое кольцо вследствие
нарушения резонансной стабилизации из-за пространственных
препятствий. В результате этого нитрозирование становится не-
возможным. Так, например, М,М-дпметил-2,6-диметпланилин не
реагирует с азотистой кислотой в условиях, обычно применяемых
для питрознрованпя.
NMe2
води. НС1, NaNOg, 8 °C
-> ON
—NMe 2
(100)
При окислении п-иптрозоднметплапилпна получают соответст-
вующее ннтросоедипеиие (32), при восстановлении — N.N-днме-
тилпропзводпое н-фепиленднамина (33), а при гидролизе горячей
Щелочью — и-нитрозофенол (34) (уравнение 101). Таким образом,
нптрознрование третичных ароматических аминов представляет
определенный интерес для синтетической практики.
НО——NO
(34)
ТмаОН; /JOI)
гидролиз '
(33)
200
6.3.3.2. Ацилирование
Общим свойством первичных и вторичных ароматических ами-
нов является способность к превращению в замещенные амиды
при взаимодействии с ацилирующими агентами. У третичных
ароматических аминов прн атоме азота нет водорода, способного
к замещению, поэтому они не образуют N-ацильных производ-
ных.
Наиболее часто для ацетилирования первичных ароматических
аминов используют уксусный ангидрид; примером служит полу-
чение N-ацетил-п-анизидина (35) [108] (уравнение 102). Хлор-
ангидриды карбоновых кислот, тиоуксусная кислота и карбоно-
вые кислоты [109] также реагируют с ароматическими аминами,
давая замещенные амиды (уравнение 103). Нафтиламииы реаги-
руют с анетплхлорндом в присутствии ацетата натрия, образуя
N-ацетпльные производные (36) [ПО] (уравнение 104).
// \ (МеСОЦО f,—К
МеО —NH2 _МеСОгИ > МеО—NHCOMe (102)
(35)
f, \ MeCOSH Z \ RCOjH
// \\—NHCOMe «------- </ \—NH2 -------
\__/ \ / -H2O
| RCOCI
-ArNHj-HCI
NH2 NHCOMe
(36)
Превращение амина в амид или замешенный амид является
удобным способом защиты аминогруппы, что позволяет синтези-
ровать, например, моноалкнлпронзводные. Метилирование ацета-
нилида метилподцдом приводит к N-метилпроизводному (37),
Щелочной гидролиз которого дает N-метиланплпн (уравнение 105).
Me
Mel 1 КОН
PhNHCOMe ----->- PhNCOMe ---► PhNHMe (105)
(37)
Наличие галогена или иитрогруппы в ароматическом ядре
замедляет процесс ацетилирования, и для ацетилирования таких
соединений требуется присутствие катализатора, например кон-
центрированной серной кислоты. Так, 2,4,6-триброманнлнн не
ацетилируется уксусным ангидридом, однако в присутствии ката-
литического количества концентрированной серной кислоты процесс
ацетилирования протекает быстро |Ш| (уравнение 106).
201
Поп обработке ароматических аминов избытком уксусного
ангидрида могут образоваться диацетилпроизводные. Этой реак-
ции способствует наличие в ароматическом ядре в орто-положении
к аминогруппе заместителей типа галогена, нитро- или алькиль-
ной групп. Так, при нагревании анилина с избытком укусного
ангидрида образуется смесь 1Ч,1Ч-диацетилаиилина и ацетанили-
да [112], а о-толупдпн в аналогичных условиях дает диацетил-
о-толуидпн (38) [113] (уравнение 107).
чти MpHC—М—CCiMq
(38)
При взаимодействии ацетанилида с хлорноватистой кислотой
получают N-хлорацетанилпд, который превращается в смесь о- и
п-хлорацетанилидов при обработке хлористым водородом в ле-
дяной уксусной кислоте (уравнение 108). Эта реакция известна
под названием перегруппировки Ортона [114]. Перегруппировка
приводит главным образом к и-хлорпроизводным; катализатора-
ми могут быть карбоновые кислоты и бензоилпероксид. Механиз-
му этой реакции посвящен обзор [115].
В некоторых случаях происходит С-ацил ирование, а не N-ацп-
хлопипя1 о.,,лаК’ ХЛоРацетилиРование ацетанилида в присутствии
Р миния приводит к ацилированию ароматического
NHCOMO+C1CH2COC1 X ClCH2CO-HQ>-NHCOMe
С=/ NEt2 + C3F7COCI —> CSF,CO——NEt2
(109)
(ПО)
(89)
202
кольца (уравнение 109), N.N-диэтилаиилии, реагируя с гепта*топ
бутирплхлорпдом, тоже превращается в нротукт С Яп,Л„п Ф Р’
(39) [Н6] (уравнение 110). фодукт L-ацилироваиия
При взаимодействии диметилапилииа с (Ьосгеипм nr,n
кетон Михлера (40) (уравнение 111), который может быть испочь”
зоваи в синтезе трифепилметановых красителей. Примером
таких синтезов служит получение кристаллического фиолетового
(41) (уравнение 112). * "-ниию
2^ %----NMez + COCla
'—co
NMe;
dll)
(И2)
6.3.3.3. Алкилирование и арилирование
аминов
алкил-
Для N-алкилпроваиия первичных ароматических
удобно использовать реакцию амина с соответствующим
галогенидом [117] или алкилсульфатом. При этом обычно образу-
ется смесь вторичного и третичного аминов. Так, например,
реакция метилиоднда с анилином приводит к последовательному
образованию вторичного и третичного аминов (уравнение ИЗ), а
а-нафтиламин при взаимодействии с этплбромпдом в спирте
образует N-этилпроизводное, которое выделяют в виде свободного
амина после обработки основанием (уравнение 114). Иногда
$ Ч—NII2 Ме'>- \—NH.Me ------► / S—N’.'le2 (ИЗ)
N'HEt
NH2
I. EtBr
2. кон
N-алкилнровзпие проводят в присутствии щелочных металлов.
Реакция анилина П других первичных ароматических аминов с
первичными алифатическими спиртами, часто в присутствии ката
лизаторов, также является методом N-алкплпрованпя. Первич
ароматические амины можно проалкилировать депствием трпа.
кплортоформиатов в присутствии серной кислоты.
(114)
203
, „ин присоединяется к ненасыщенным углеводородам, обра.
Ан"Ля 'с™е первичных продуктов реакции замещенные ащ1л„,
ЗУЯШЯ) Протек ивио реакции сносооствует присутствие СоеДп.
вы ।1 , ,,х метаччов Так, анилин реагирует с этиленом и
пенни шело i пь д д соответствующие N-замещенные произ.
ПРпныее1(уравиение 115). Интересно отметить, что прн реаКЦ11и
водные 1>к в присутствии алюминия алкилирование цдет
Хе ароматических аминов к тронной углерод-углеродной
связ ацетиленов дает енамины; в качестве примера приведено
образование енамина (42) прн реакции N-метпланилипа с дИме.
толовым Эфиром ацетиленднкарбоновои кислоты (уравнение ц6)
-----мн. NHEt
nh2
NHCHMe2
CH2=CHMe
CH2=CH2
(115)
NHMe
CCO2Me
III
CCO2Me
x /Me
/ \ /H
J ^C=CZ
MeO2Cz ^COjMe
(116)
(42)
Реакция между первичным ароматическим амином и арнлга-
логенпдом происходит с трудом вследствие низкой реакционной
способности арплгалогенидов в реакциях нуклеофильного заме-
щения. Однако реакцию между анилином и хлор- или бромбензо-
лом можно осуществить в присутствии йодида меди. Эта реакция
известна как реакция Ульмана, и с ее помощью из анилина или
ацетанилида можно получить дифениламин (уравнение 117). При
взаимодействии анилина с фенолом в присутствии хлорида цинка
или трнхлорида сурьмы также образуется дифениламин. Дифе-
ниламин можно синтезировать, кроме того, реакцией анилина с
его гидрохлоридом под давлением и при высокой температуре в
присутствии хлорида алюминия.
PhNHCOMe + PhBr + K2COS Ph2NH + СО2 + МеСОЖ + КВг (117)
В отличие от неактнвированных галогенбензолов 2,4-динитро-
хлорбензол легко реагирует как с первичными, так и со вторич-
ными ароматическими аминами. Например, реакция анилина с
(уравиеннеХП<8)^еНЗ°ЛОМ ПР1|ВОДИТ к диннтроднфениламнну (43)
гои??РИ Реакц11и третичного ароматического амина с алкилгало-
ИЛИ алкилсУльФатом образуется соль четвертичного аммо-
запия °сиова1шя (44) [121] (уравнение 119), Прямая кватерни-
рвичных или вторичных ароматических аминов проис-
2С4
(118)
ходит не во всех случаях, хотя описана реакция 4-метоксианилина
с „етилподндом в присутствии 2,6-диметилппридина, приводящая
К иодиду 4-метоксифеиилтриметпламмоиия (45) с хооошим rL.
ходом (уравнение 120). Кватернизацию первичных историчных
ароматических аминов (в виде нх солей) можно также тегко
осуществить с помощью диазометана [122]. В качестве примера
приведено образование тетрафторбората Диметплдифениламмония
(46) (уравнение 121).
PhR!R2N + MeI —> PhR*R2NMe I"
(46)
Наличие заместителей в ароматическом ядре замедляет про-
цесс кватернизации [123], причем этот эффект проявляется в наи-
большей степени, когда заместители находятся в орто-положенип.
Кватернизация ЫД’-диалкпланилинов протекает достаточно
легко, однако реакция алкилгалогенпдов с алкплдиариламннамп
или с триариламинами, которые приводили бы к солям четвер-
тичных аммониевых оснований, практически неизвестны. Однако
имеются данные [124], что при взаимодействии трпфенила.мина с
третичными оксоииевымп солями образуются четвертичные аммо-
ниевые соли, как, например, в случае получения тетрафторбората
метилтрифеипламмоння (47) (уравнение 122).
Ph3N + Ме3О+ BF? PhsNMe BFJ (122)
(47)
При нагревании гидрохлориды N-алкпл- н К,М-дналкнлани-
линов и четвертичные аммониевые соли перегруппировываются в
С-алкилпрованиые анилины. Эту реакцию называют перегрхппи
ровкой Гофмана - Марциуса '[125]. Алкильная группа входит
преимущественно в лара-положение ароматического кольца, аie
°но занято, то в орто-положение. В качестве примера при
205
„пиоовка хлорида фенплтриметнламмония (48) (yp
перегруппировка 1_ реа,.цпп был предложеп Хь
пение Ud). ‘ • 19Д_|27) Эту перегруппировку можно провп
'['261 т^"®"тем нагревания Ы-алкнларнламипа с хлор11даМ11
ДП алюминия [127].
HNMe, СГ
цппка или
NMe3 Cl
NH.Me СГ
J. .Me
NH3 СГ
Me\ /Me
(123)
Me
Me
Me
(48)
PhNHiR X' —»- PhNH2 + RX (124)
RX —> R+ + X‘ (125)
R+4-PhNH2 —> RCeHiNHj + H* (126)
RC8H4NH2 + H+ —► RC6H<NH3 (127)
6 3.3.4. Образование изоцианатов
и производных мочевины
Реакция первичного ароматического амина с фосгеном (1 экв)
приводит к арилкарбамоилхлориду, который при нагревании
теряет молекулу хлористого водорода, превращаясь в арилизо-
цианат. Так, например, я-интроанплпн дает я-нитрофенилпзо-
цианат [128] (уравнение 128) через промежуточное образование
п-иптрофенплкарбамоилхлорида (49). Диамины превращаются в
дппзоцианаты аналогичным образом. Реакция первичных арома-
тических аминов с тиофосгеном приводит к арилизотиоцпанатам;
пример такого превращения — получение п-хлорфенплнзотпоциа-
пата [129, 130] (уравнение 129). Фенплизотиоцнанат синтезируют
аналогичным способом из анилина и тнофосгена.
NH2 NHCOC1 NCO
rnri /х осторожное I
г п \ нагревание
О ППБХГ П| —--------------------->- П) <128)
N°2 NO2 NOj
(49)
csci2 z—x
С|-< )—ncs
•—nh2
(129)
оксипа^кя пияНп^Па C ХЛ0Р°Ф0РМ°М и спиртовым раствором гидр-
ванпи с сеоой ппг>ИоВ0ЛИ1 к Фенилиз°Чиаииду, который при нагре-
Р р ращается в фенилизотиоциапат (уравнения 130,
206
131). При реакции апплппа с сероуглеродом и гидроксидом амм
ния получают фенилдитиокарбамат аммония (5of пТн об?
которого нитратом свинца образуется фенилнзотиоцнаиат
пение 132).
PhNH2 + СНС13 + ЗКОН —> PHNC + 3KC1+31W
нагревание
PhNC + S------------ phNQs
PhNHs + CSj + NH<OH —_Ha0 > PhNHCS2N'H( PbtN°a)2>
(50)
—► PhNCS + NH,XO3 + HNO3 + PbS
о-
при обработке
-----; (урав-
(130)
(131)
(132)
Феннлизоцианат реагирует с первичными и вторичными ами-
нами, давая производные мочевины (51), а замещенные тиомо-
чевины (52) получают при взаимодействии аминов с фенил изотио-
цианатом [131] (уравнение 133). Реакция анилина с избытком
фосгена приводит к N.N-дифенилмочевине (53). N.N-Днфенил-
тиомочевина (54) образуется при нагревании анилина со спир-
товым раствором сероуглерода и твердым гидроксидом калия
(уравнение 134).
PhNCS PhNCO
PhNHCSNHR *-------- RNH2 -------> PhMHCOXHR (133)
(52) (51)
CS2. 2KOH COCI2
(PhNH)2C=S <—, _2Hz0 2PhNH2 (PhXH)2C=O (134)
(54) (53)
6.3.3.5. Окисление
Окисление анилина является очень сложным процессом, в хо-
де которого в зависимости от природы окислителя возможно об-
разование фенилгндрокспламина, нитрозобензола, нитробензола,
азобензола, азоксибензола, я-бензохинона пли анилинового чер-
ного. Различные способы окисления анилина рассмотрены в обзо-
ре [132].
Основные реакции первичных ароматических аминов с окис-
ляющими агентами протекают по двум принципиально разным
направлениям. Окисляющий агент может или отдавать кислород
молекуле ариламина, пли отнимать водород из аминогруппы.
Реакции первого типа приводят к образованию гидроксиламинов,
нитрозо- пли нитробензолов, в то время как реакции второго типа
дают азобензолы, азоксибензолы и хиноны.
При использовании окисляющих агентов первого типа первич-
ным продуктом реакции, обнаруживаемым в смеси, является
арилгпдрокспламин, а дальнейшее окисление приводит к нптро-
зобензолу или, в более жестких условиях, к нитробензолу (урав-
нение 135).
ArNH2 J21. ArNHOH АЛО АЛО, (135)
207
„ nnnvuatoT, используя в качестве окислителей
Нитроз0^,1,,C;,lauSep)alOiipI,> 2-амино-5-нитро.
перокепкисзо™ Haul? ; ^,111;]Ш1а [134] кислотой Каро (11еро.
толуола [133] пл1'-';П „отучают соответствующие нптрозосое-
ксомопосерпоп кисли;, Более глубокое окисление нитро,
дпчеипя (уравнения 1 , азотной кислоты приводит к
зосоедппеиии. например, с
нитро производным.
(136)
(137)
Для окисления ариламинов до нитросоединений можно с успе-
хом применить перокепкарбоиовые кислоты [135]; по данным
[136, 137], особенно подходящим реагентом является раствор
пероксида водорода в трпфторуксусной кислоте. При окислении
анилина этим реагентом получают нитробензол, а 2,4-динитро-
аннлпн превращается в 1,2,4-тринитробензол (уравнение 138).
Можно также использовать раствор пероксида водорода в ледя-
ной уксусной кислоте [138, 139]; в этих условиях пз и-толундина
получают и-ннтротолуол (уравнение 139), а 2,4,6-трпхлоранилпн
дает соответствующие нитрозо- и нитропроизводные (уравне-
ние 140).
Окислители второго типа (отнимающие водород от аминогруп-
пы) не приводят к образованию нитрозо- или нитросоедииений.
ак, натриевая соль пероксоборной кислоты в уксусной кислоте
окисляет первичные амины до азосоедипеиий [140]; например, из
Х=Вг,С1,Ме
(141)
208
NHCOMe
2
„„ 'MeCONH
\ra ВОз
H3BO3
z^*yxNHCOMe
N=NA>I 042)
(55)
J азоксибензолу [142, 143] (уравнение 143). P " пРив°лит
H2O2. MeCN
(143)
Азокспсоедпнення образуются также при обработке первичных
ариламннов пероксидом водорода в водной уксусной кнспоте а
В присутствии небольшого количества хлорида меди(П)’ выде-
ляют азопроизводпое [144]. В качестве примеров приведены
окисление льнитроанплппа до 3,3'-динитроазокснбензола (56) ч
3,3'-динитроазобензола (57) (уравнение 144).
МеСОзН
МеСОзН
СиС12
(57)
Реакция анилина с кислородом в присутствии катализатора
™риднн — хлорид меди(1) приводит к азобензолу [145, 146]
сравнение 145), который образуется также в результате реак-
1,11 анилина и нитробензола в присутствии порошкообразного
»оксида натрия или металлического натрия [147] (уравнение
а„.,' Г"чохлорит натрия в водном растворе окисляет полифтор-
П1.,ИНЬ| Д° азосоединений [148] (уравнение 147). Описаны и др>-
чсгсоо™ГеНТЬ1 для окисления ароматических аминов в азо- на
ацСТ1тЛ"Нен"я: Диоксид свинца в уксусной кислоте [141 ], Р
[152| СВ11Н14а [150], феинлиодацстат [151] и перок )- Ф
—NH2
CuCI. О2. С5П5М
—2НгО
(Н5)
209
R = F, H, Me
Под действием концентрированной серной кислоты происходит
перегруппировка азокспбензолов с образованием гидроксиазобен-
золов.' Эта реакция носит название перегруппировки Валдаха
[153]. Так, например, сам азокспбензол превращается в 4-гидр-
оксиазобензол (58) (уравнение 148).
О
H2SO4
N=N
—ОН (148)
(58)
При действии диоксида марганца на анилин конечным про-
дуктом реакции является n-беизохинои. Гомологи «-бензохинона
можно получить, исходя из толуидинов и ксилидинов. Окисление
н-диамино- и л-аминогидроксибензолов дихроматом калия в сер-
ной кислоте также приводит к n-бензохинону; в этих случаях
реакция протекает через промежуточное образование л-бензохн-
нондиимина (59) и /г-бензохинонимипа (60), соответственно (урав-
нение 149).
NH2 NH о О он
(59) (60)
Окисление а-нафтиламина хромовой кислотой [154] приводит
к , -нафтохинону, а прп окислении как а-, так и р-пафтиламина
перманганатом калия получается фталевая кислота. Окисление
ir1)! м'Да1М1!На На воздУхе на свету приводит к днбензофеиазину
'll0! (Уравнение 150), который образуется также при окис-
(61)
210
нятрня ВДИ "РИ -P~
^Сп.чиые ароматические амины, например диметиланилии
₽агпрУ»от с пероксидом водорода с образованием N-оксидов (voa'
Р не 15D- Известно также [156, 157], что при обработке моно^
Тналкиланилииов сильными окислителями происходит отщепле
„е алкильных групп, а взаимодействие метил- или диметичани-
н1„а с диоксидом марганца приводит к окислению г- '
группы с образованием соответствующего форманилида
(уравнение 152).
~ I
•NMe2
диоксидом марганца приводит к окислению
метильной
i [158]
•NMe2
(01, н2о2
(151)
•NRMe
MnOj
NRCHO
(152)
R = H, Me
6.3.3.6. Реакции с альдегидами и кетонами
Общей реакцией первичных ароматических аминов с альдеги-
дами является образование оснований Шиффа [159]. Лучше все-
го эта реакция протекает с ароматическими альдегидами. Так,
например, при реакции анилина с бензальдегидом образуется
бензилиденанплин (62) [160] (уравнение 153). Основания Шиффа
легко гидролизуются до свободных аминов, поэтому их образова-
ние является методом защиты аминогруппы. Производные анили-
на, замещенные в ядро, такие, как толуидины или ннтроанилины,
реагируют аналогичным образом. Эти реакции обратимы и не
требуют присутствия катализатора.
ОН
PhNH2+PhCHO PhCHNHPh «=* PhCH=\Ph + Н2О (153)
(62)
Шиффовы основания, образовавшиеся при взаимодействий
первичных ароматических аминов с алифатическими альдегидами,
как правило, неустойчивы и легко полимеризуются. Например,
продуктом реакции п-толундина и формальдегида является сое-
динение (63), которое может полимеризоваться или реагировать
с еЩе одной молекулой «-толуидина [161] (уравнение 1э4).
n-MeC6H4NH2 + НСНО —> n-MeC6H4N=CH2 —> Полимеры (Ю4)
(63)
п-толупднн
(MeC6H4NH)2CH2
211
Рткиия между первичными ароматическими амилами и Кет
вами и о сходит гораздо труднее; ирп нагревании беизоф^
“ Z ион 160 °C реакции не наблюдается. Однако в пп,
сутсти катализатора (хлорид Цинка [162] или иод) основа,^
Ш'п£ нагрева'гии^первпчного ароматического амина, в котороч
ОРТО-ПОюжевие не замешено, с глицерином, копцептрвровацад
сепной кислотой н окислителем, например нитробензолом, в ре.
зультате сложной последовательности реакции ооразуется хино-
лин. Эта реакция известна как синтез Скраупа [163]. Считают,
что глицерин превращается в акролеин (64), который затем реа^
гирует с анилином или ариламином (уравнение 155).
ОНС
^СН2
./СН, ~-н!° ’
NH
СН;=СН-СНО
(64)
сн
^сн
I
/СП1
Реакция п-хлораннлнна с глицерином в присутствии л-нитробен-
золсульфокислоты как окислителя [164], приводящая к 6-хлор»
хинолину, является еще одним примером синтеза Скраупа (урав-
нение 156). Нафтиламины реагируют аналогичным образом, при-
чем из а-нафтиламина образуется 7,8-бепзохинолпн, а из 0-наф-
тиламнна— 5,6-бензохинолни. Если вместо альдегидов исполь-
зуют кетоны, то образуются замещенные хинолины. В качестве
примера приведено получение 2,4-диметилхинолина (уравнение
1о7) [165]. К,реакциям того же типа относится получение индола
прн взаимодействии первичного ароматического амина с сс-гало-
incc?T0H0M’ напРимеР- реакция бромацетофенопа с анилином
![166] приводит к 2-фенилиндолу (уравнение 158).
+ МеСОСН=СНМе
(1ST)
212
+ PhCOCH2Br
NH2
Ph
NH
(158)
Гомологи хинолина можно синтезировать из первичных арил-
енов по реакции Дебиера - Миллера при нагревании аромати-
ческого амина и альдегида с соляной или серной кислотой. На-
пример, анилли реагирует с паральдегидом с образованием 2-ме-
тилхинолина (уравнение 159).
Родственной реакцией ароматических аминов с карбонильны-
ми соединениями является реакция, известная как синтез Фрид-
лендера. Ее используют для получения хинолинов, замещенных
в гетероциклическом ядре. Реакция заключается в конденсации
(в щелочной среде) о-аминобензальдегида или о-аминофенилкето-
иа с альдегидами, кетонами или эфирами кетокислот, содержащи-
ми фрагмент—СОСН2— (уравнение 160). Например, о-амино-
бензальдегид и этиловый эфир ацетоуксусной кислоты образуют
этиловый эфир 2-метилхинолин-З-карбоновой кислоты (65) (урав-
нение 161).
(161)
CH2CO2Et
I
СОМе
(65)
1,3- Дикарбонильиые соединения конденсируются с первичны-
ми ароматическими аминами в присутствии серной кислоты; при
этом первоначально образуется аннл, который далее превращаег-
213
(163)
MeCIIO-|- PhCH=CH2
P,h
(67)
v.u.nniiH 11671 Так, реакция «-толуидина с а цетила цетоном
поивод Т 2,4 6 трпмети.пхинолцну (66) [168] (уравнение 162),
аРи , . neariiDveT с ацетальдегидом и стиролом в смеси ледяной
уксусной п "ервой кислот, образуя 2-мет11л-4-фенпл-1,2,3,4-тет-
рагидрохинолпн (67) (уравнение 163).
6.3.3.7. Галогенирование, нитрование и сульфирование
(1) Галогенирование
Наличие аминогруппы в ароматических аминах активирует
орто- п пара-положения настолько сильно, что анилин крайне
легко реагирует с бромом или хлором, давая 2,4,6-тригалоген‘1
производные с высоким выходом (уравнение 164).
С1 Вг
Аналоги анилина реагируют столь же легко; толуидины дают
дибромпронзводные. Бромирование анилина или его аналогов с
образованием 2,4,6-трцзамещенных производных фактически
протекает настолько легко, что этот процесс трудно регулировать.
Одиако при переходе к N-ацетцламинам процесс галогенирования
поддается контролю. Например, бромирование незамещенного
n-толуидина приводит к 2,6-дибром-п-толуидину, но если толуидин
предварительно ацетилируют, в результате бромирования полу-
чается Ы-ацетнл-2-бром-п-толуидин, ацетильную группу в котором
можно легко удалить гидролизом и выйти к 2-бромтолуидНну
(уравнение 165). Направленное галогенирование ацетилпроцзвод-
ных первичных ароматических аминов можно осуществить с помо-
щью хлорноватистой кислоты. Например, п-хлорацетанилпд удает-
ся получить при обработке ацетанилида хлорной известью в
уксусной кислоте [170] (уравнение 166). Для достижения нужно-
го характера замещения можно также блокировать одно нз поло-
жении ароматического кольца. Например, хлорирование сульфа-
мида приводит к 4-амино-3,5-дихлорбеизолсульфоиам11дУ
( ) [171], который в условиях гидролиза превращается в 2,6-дП-
214
\.ЮР'
палплии [172] (уравнение 167). Аналогичным обвячом
''"тезировать и 2,6-димброманилин. ДимегиланилииРоеагип^"0
6Р МОМ в уксусной кислоте, образуя 4-бромдиметиланилинУ од‘
f,P я присутствии серной кислоты И СОЛИ СРПпАг., Н| од
Гбромпроизводное М (уравнение 168) И Серебра ^У^ся
3 Е NHCOMe NHCOMe
Br2
50—55 °C
(165)
Me
NH,
•Вг +
И или “Он
Br
Me
•NHCOMe
'—NHCOMe
(166)
Me
хлорная известь, AcOH
Введение иода в ядро ароматического амина можно осущест-
вить прямой реакцией с иодом; выделяющийся в ходе реакции
йодистый водород удобно удалять с помощью бикарбоната нат-
рия [174, 175] (уравнение 169). Более удачный метод синтеза
состоит во взаимодействии ароматического амина с .монохлоридом
иода. Реакцией и-нитроаннлнна с монохлорндом нода в хлоро-
форме или ледяной уксусной кислоте был получен 2,6-дииод-4-ни-
троанилин [176] (уравнение 170).
N-Хлорариламины — крайне нестойкие соединения. N-Алор-
аинлнн не был получен, однако М,М-дихлораннлнн можно син-
’сзировать действием хлорноватистой кислоты на анилин в ДИ-
NHz NHz
+ Iz + NaHCOs —►
I +NaI+HzO + COz (169)
SIS
(170)
„„„ qA,,n0 поп —20 °C, хотя выделить его не удается из-за
₽го'"оЛивостн. Наличие электроиоакценториых заместителей
! АПОХ атическом ядре повышает устойчивость N N-дихлораминов,
В neuTivioo-N N-т.лхлоранплпн устойчив при обычных темпера-
тепах Поп действии соляной кислоты или хлористого водорода
вчЛпое на N N-дихлоранплин происходит реакция, аналогичная
пеоегруппировке N-хлорацетаиилида (перегруппировке Ортона),
в результате чего образуется 2,4-дпхлораннлпи и некоторое коли-
чество 2 6-дихлоранилнна (уравнение 171). Прн восстановлении
N М-дихлоранн-ита получают азобензол и N-фенилбензохннон-
днпмнн г1-С6Н5П = С6Н4=МН.
nci2 nh2 nh2
A hci Ачс1 с,чАгС|
U —" ч) + ч J °
С1
(2) Нитрование
Прямая обработка анилина азотной кислотой не дает удов-
летворительных результатов, поскольку аминогруппа чувствитель-
на к окислению. Однако аминогруппу можно легко защитить аце-
тилированием. Примером нитрования защищенных арнлампнов
является нитрование ацетанилида и 2-ацетамидоиафталпна [177]
(уравнения 172, 173), Нитрование П.Ы-диметил-п-ТОлуидпна
смесью азотной н серной кислот приводит с высокими выходами
к орто- (69) или лета-замещениому (70) продукту в зависимости
от условий проведения реакции [178] (уравнение 174).
низкая
кислотное 11,
нонишенная
КИСЛО!HQC гь
(6S)
(174)
восстановлении нитроанилинов металлами
При ...... г-—металлами в kiiciot--
образуются соответствующие диамины. При частичном восстанов-
лен 2,4-динитроаннлина гидроксидом аммония и сероводородом
получают 1,2-диамино-4-ннтробепзол [179] (уравнение 175) а вос-
становление о-иитроаиилина цпнковои вылью в присутствии гид-
роксида натрия [180] приводит к о-фенилендиа.мину.’Однако при
кипячении о- и п-нитроаннлинов с водным раствором гидроксида
натрия происходит реакция замещения, приводящая к нитрофе-
иолам (уравнение 176).
NaOH
кипячение
Zn, NaOH
(175)
(176)
При взаимодействии анилина с азотной кислотой получают
нитрат анилина, реакция которого с уксусным агнидридом дает
фенилнитрамин (N-нитроанилин) [181]. Наиболее интересной реак-
цией арилнитраминов является их перегруппировка [182, 183] в
присутствии водного раствора сильной кислоты или хлористого
водорода в органических растворителях. В результате перегруп-
пировки образуются ннтроариламнны. Так, например, фенилнитр-
амин превращается главным образом в о-нитроаннлнн, побочно
образуется небольшое количество я-изомера (уравнение 177).
NO,
(3) Сульфирование
Реакция анилина с серной кислотой при высокой температуре
"Р'1в°Дит к п-амннобензолсульфокнслоте. Реакш я "Р° пеое-
Рез промежуточное образование N-производного, Р
ппвыяается [184] в сульфокислоту, замещенную в аро
ГРУППИ^ВХ Примером такой реакции является перегруппироа.
тическое ядр ; Р Р _тол дина (7 ) в 4-амино-З-метилбензол-
ка кислого сульф ( равнение 178). Если «ара-положение за.
сульфокислоту и ' т0 образуется о-аминосульфокислота;
пято, какn-ор а можеТ быть удален восстановлением
в данном случае , натром, в результате получают о-амино-
ХлХкислоту (уравнение 179). Если а-нафтилампн натре-
вал с концентрированной серной кислотой при высокой темпе-
name то основным продуктом реакции является 1-аминонафта-
линсул'ьфокислота-4. Бензол- и аминонафталинсульфокислоты ши-
роко используют для синтеза красителей.
6.3.3.8. Реакции аминофенолов
и N-фенилгидроксиламина
сближенности гидроксильной и аминогрупп в
о-амп-
Вследствие
пофеиолах последние отличаются по химическому поведению от
мета- и пара-замещенных аналогов. Так, о-аминофенолы легко
реагируют с рядом соединений, образуя гетероциклические струк-
туры.
При нагревании о-амииофеиолов с карбоновыми кислотами,
хлорангидридами и ангидридами кислот н родственными соеди-
нениями образуются 2-замещенные бснзоксазолы (схема 180).
Например, реакция о-амннофенола с уксусной кислотой приводит
к 2-метилбеизоксазолу (72) [186], а в реакции с нитрилами в
кислой среде образуются 2-замещенные бензоксазолы (73) [187].
ри действии фосгена получают 2-п1дрокспбензоксазол (74).
е кцпя о-аминофенола с гидрохлоридом гпдроксиламина и
и б™™ хл0Ральг1,дРат°м В присутствии ацетата натрия приводит
к оензоксазолальдокспму-2 (75) [188].
£>'ам,,1|ОФеиола с фосфорной кислотой при высокой
Р У ре в присутствии хлорида цинка дает феиоксазип (76)
21В
[189] (уравнение 181). Пикрилхлорид реагирует с о-аминофе-
иолом в нейтральной среде, давая 2'-п1дрокси-2,4,6-тринитроди-
феииламин (77), прн обработке которого основанием получают
2,4-дпнитрофеноксазин (78) [190] (уравнение 182).
о-Лминофепол легко реагирует с хлораигпдрндо.и хлоруксус-
110й кислоты, образуя N-.хлорацетильиое производное (79), кото-
Рое в присутствии основания может циклизоваться в 2,3-днгндро-
ч-оксо-1,4-бензоксазпи (80) (уравнение 183). Производное бенз-
(79) (8°>
219
(184)
CCO2R
о
CCO2R
,„а почгчается также прп реакции о-аминофеиола с эфипОм
ацетилендикарбоновой кислоты [191] (уравнение 184).
Пои окислении анилина кислотой Каро или пероксидом водо.
попа образуется N-фенилгидроксиламин. Вероятно, он получается
в результате перегруппировки оксида амина (81) возникающего
при непосредственном присоединении кислорода (уравнение 185).
О'
PhNH2 + PhNHj —> PhNHOH
(81)
(185)
N-ФенилгидроксиламИН перегруппировывается в присутствии
разбавленной серной кислоты, давая в качестве главного продукта
реакции 4-амннофенол. Эта реакция носит название перегруп-
пировки Бамбергера [192]. Примером этой перегруппировки слу-
жит превращение о-хлорфеннлгидроксиламина в 4-амнно-З-хлор-
фенол (уравнение 186). Если в лара-положенни ароматического
кольца находится метильный заместитель, то в результате пере-
группировки может образоваться хинол (82) (уравнение 187).
о-
химическому поведению
м-
своему
6.3.3.9. Реакции ароматических диаминов
и фенилгидразина
и п-Фенилендиамнны заметно различаются по
о-пнамиш, ' г,„, ----- Наибольший интерес представляют
рядом соединенийЛЬпГ °ПИ сг|особиы конденсироваться с целым
(Уравнения 188, 189)бРаЗУЯ вещества гетероциклической природы
ам1ш^м||'образуаотся xiZt б0111и1Ьг'т соединений с фениленди-
амин и глиоксяпь ппо ' 10КСаЛ1,11ы t193]; например, о-феииленди-
и глиоксаль превращаются в хпиоксалип (83) Реакция о-фе-
220
(188)
нялендиамина с дицианом приводит к диаминохиноксалину (84),
прн гидролизе которого разбавленной кислотой получается соот-
ветствующее дигндрокснпронзводное [194], Реакция с фенантра-
хиноном используется для идентификации о-фенплендиамина,
поскольку продукт реакции (85) дает интенсивное красно-корич-
невое окрашивание с серной кислотой. Реакция 1,2-диаминонафта-
лнна с глиоксалем приводит к 5,6-бензохиноксалину (86) (уравне-
ние 189).
(86)
В результате взаимодействия о-феипленднамина с ацилирую-
щими агентами получают бензимидазол [195] (уравнение 190),
сам незамещенный бензимидазол (87) образуется при реакции
О’феииленднамнпа с муравьиной кислотой. К замещенным бензи-
мидазолам (88) приводит взаимодействие о-феинленднамнна с
нитрилами пли альдегидами в присутствии ацетата меди(11)
Азотистая кислота реагирует с о-фсннлсиднампном в разбав-
1нои кислоте с образованием беизотрпазола (90) [ 1 (УР
821
некие 191); реакция протекает через промежуточное образовав
монодпазоПпевоП соли (89).
•- -N?
UNO?, AcOII
•NH2
Nib.
N
' V
(191)
NH
(89) (90)
Окисление о-феииленднамина водным раствором хлорцда
жетеза(Ш) приводит к 2,3-Д11аминофеназпну (91); для этой
реакции характерно появление темно-красного окрашивания
(уравнение 192). При окислении о-фенилендиамина молекуляр-
ным кислородом в присутствии хлорида меди (II) и пиридина
происходит раскрытие цикла с образованием 1,6-днцианогексадие-
на-2,4 (92) [198] (уравнение 193). Аналогичным образом реаги-
рует п 1,2-диаминонафталин.
2| J > [ 1 [ I (192)
(91)
CuCI2
пиридин
(92)
(193)
В случае м-феиплендпаминов реакции замыкания цикла не
протекают, и обе аминогруппы можно ацилировать п т. д. При
избытке азотистой кислоты происходит диазотирование обеих
аминогрупп; в конечном итоге образуется основный коричневый
(93), один из первых известных азокрасителей (уравнение 194).
(93)
мннобензоловД по п и' вступают в реакции, типичные для 1,4-диа-
при действии’ нт Д'1Я ”"х хаРактерны и реакции окисления. Так,
кислоте обтзспт КС"'У маРганца или дихромата калия в серкой
кислоте образуется и-беизохинои (уравнение 195).
222
(195)
NH, ОжЛ=\
w2so4 и=ж=/ Q
При восстановлении хлористого
„аТпия или хлоридом олова (И) в соляной /Л°"ИЯ с>7,ьФитом
Йлгидразин. Он может быть также ц’0“Те образуется
хлорамина с апплпном или реакцией аиилииа с г»ТДеНСацией
О.сульфокислотои [199]. Наиболее важной
разпна является реакция с карбонильными фсН1!'1,1'Д-
моносахаридами, в результате которой образую™ДИ/еНиями и
30ны или озазоны. Эту реакцию используют дня™ феН1™ра-
альдегидов и кетонов. Особенно часто для этой ,,Р Д ТИфикации
2,4-дннитрофенилгидразин; в этом скчае пор "Равняют
образуются кристаллические производные (уравнение *1 Жественно
/NO2 '
/ /\О2
H2N-
02N-
•nhnh2+o=c; —o2n.
\ —Н2О
—ХНК=С^ (196)
Под действием азотистой кислоты фенилгидразины превраща-
ются в нитрозосоединения (94), которые перегруппировываются
в фенплазиды (уравнение 197). При обработке концентрирован-
ной соляной кислотой при 200 °C происходит перегруппировка
фенилгидразина в и-фенилендиамин [200] (уравнение'
реакция имеет определенную аналогию с бензидиновой
ппровкой (см. ниже).
водн. НС1
PhNHNH2 ----——-
NaNO2
198). Эта
перегруп-
> PhN,
—пгО
(197)
(94)
Z \ конц. НС1
(' х)—nhnh2--------------> нлг-
\ / 200 °C
Если нитробензол или азобензол восстанавливать
пылью в водном гидроксиде натрия, образуется N.N'-дифенил-
гндразин или гидразобензол. Из реакций гидразобензола наиболь-
шее значение имеет его катализируемая кислотами перегруппи-
ровка в бензидин (4,4'-дцаминоби'фенил) (95) [201]
пне 199). Обычно реакцию проводят в разбавленной
кислоте, в растворе серной кислоты в спирте или в
Z—\ н+
>—XH2
(198)
цинковой
(уравне-
СОЛЯНОЙ
растворе
R—!
•NH—NH-
N'H:
(199)
(95)
223
n Xпористого водорода в толуоле Пооочпо образуется »еко.
С' Г количество дпфепилппа (2,4'-днамшюонфенпла). Г11дразо.
бензолы замешенные в кольцо, вреврашаются в соединения (9в),
называемые семндипам» (уравнение 200ф |Jt
R——NH—NH—/ / •-------►
(96)
(200)
ЛИТЕРАТУРА
1 R Schroter, in «Methoden der Organischcn Chemie» (Houben-Wcyl), ed.
E. Muller, Georg Thieme Verlag, Stuttgart, 1957, Vol. 11/1, p. 341.
2 R IV. IVesl, J. Chem Soc., 1925, 127, 494.
3i R. Adams and J. R. Johnson, «Laboratory Experiments in Organic Chemistry»,
Macmillan, New York, 4th edn., 1949, p. 319.
4. H. Koopman, Rec. Trav. chim., 1961, 80, 1075.
5. N. S. Kozlov and M. N. Tovshlein, J. Org. Chem. (U. S. S. R.), 1967, 3, 132
[H. С. Козлов, M. H. Товштейн — ЖОХ, 1967, 3, № I, 138].
6. L. Legradi, Chem. and Ind. (London), 1965, 1496.
7. R. L Augustine «Catalytic Hydrogenation», Marcel Dekker, New York, 1965,
pp. 91-102.
8. P. N. Rt/lander, «Catalytic Hydrogenalion over Platinum Metals», Academic,
New York, 1967, pp. 168-203.
9. R, Adams and F. L. Cohen, Org. Synth. Coll. Vol. 1, 1941, 235.
10. L. A. Secrist and M. II7. Logue, J. Org Chem., 1972, 37, 335.
If. I'. Kharkov, S. U. P. 240885/1972 [В. Харьков. Авт. свид. СССР № 240 885
(1972)].
12. J. Harast, U. S. P. 3041377/1962 (Chem. Abs., 1963, 58, 474)
13. Atlas Chem Jnds. Ltd., U S. P. 3427355/1969 (Chem. Abs., 1969, 70, 67 862).
14. J. M. Landsburg, L. Katz, and C. Olsen, J Org. Chem., 1972, 37, 930.
15. H. Alper and J. T. Edward, Canad. J Chem. 1970, 48, 1543.
16. D. Balcom and A. Furst, J. Amer. Chem. Soc’., 1953, 75, 4334.
17. H. Heaney Chem. Rev., 1962, 62, 81.
18. J. F. Bunnett, Quart. Rev., 1958, 12, 1.
19. E. К Spencer and G. F. Wright, Canad. J. Res., 1946 24B, 204.
20. R. A Rossi and J. F. Bunnett, J. Org. Chem., 1972, 37, 3570.
21. R. A. Scherrer and H. R. Beatty, J Org. Chem., 1972, 37, 1681.
on УЧС0?Л‘ lnc’ U S P- 3578714/1971 (Chem., Abs., 1970, 73, 35 024).
n, r ,, Wallls and F- La,w< OrE Reactions, 1946, 3, 267.
24. E. Magnten and R. Baltzly, J. Org. Chem., 1958, 23, 2029.
20. p. Sykes Guidebook to Mechanism in Organic Chemistry», Longmans,
London, 1965, p. 94.
9,' p ?• saoser,. and ®’- B- Renfrow, J. Amer. Chem. Soc. 1937, 59, 121.
Ой и , V , Org Reactions, 1946, 3, 337.
oo и nc '« Cl’em Rev., 1943, 33, 209.
on Г Org- Reactions, 1946, 3, 307,
al /' r' nand l ^-L'Jttlelon, J. Chem. Soc., 1943 421.
32 Unfv₽r«M°m □“"л 'Г ZA HMt- Org' Reactions, 1960. 11, 1.
32 1527). 1 ° Produds Co- u- S. P. 2948755/1960 (Chem. Abs. 1960, 55,
224
3'1
Organic Chemistry», ed.
ly/о, p. 4<3.
Amer. Chem. Soc., 1973, 95,
„»» gas й s J: № «л&ь.’й na i™
S '- °' A®., Л.*,*
з6 Го- *се and E- ' Kohn- U- S- p' 28'3124/1957 (Chem. Abs., 1958, 52,
, p 6G R>ce and E- ~ Ko!1"’ JAAl^er- Chem- s°c-. 1955 77 4059
31 Chin-Nippon Rika Co., Jap. P. 124687/1972. ’
39 f'MiK Georg Thieme Verlag, S^uRgTrh^y^vol"1'!?/! ’ped'
л. Ы»°* the Amino 6rou₽> 4 s
/ПС 'tf' Barton, in «Protective Groups ind
4 ’ 1' F W. McOmie, Plenum, London and New York,
J/ Д Carpino, Accounts Chem. Res., 1973, 6, 191.’
i IF S Emerson. Org Reactions, 1948» 4, 174.
n n' P Boldrini, Synthesis, 1974, 733.
ля 6 E. Utzinger, Annalen, 1944, 556, 50.
46 H C. Brown, M. M. Midland, and A, B. Levy, J,
2394-
,7 p Kovacic, in «Friedel Crafts and Related Reactions», ed G A Olah ’nb.
4 science New York, 1964, vol. 3, chapter 44. ’ lnter’
<8 Zinin, J. prakt. Chem., 1842, 27, 140.
49 H. Bock and K. L. Kompa, Chem. Ber., 1966, 99, 1361,
50: T. D. Perrine, J. Org. Chem., 1951, 16, 1303.
51. F. D. Hager, Org. Synth. Coll. Vol. 1, 1941, 544.
52 0. Neunhoeffer and P. Heitmann, Chem. Ber., 1961, 94, 2511.
53 !, IV. Schulenberg and S, Archer, Org. Reactions, 1965, 14, 1.
54. Koppers Co. Inc., U. S. P. 2645662/1953 (Chem. Abs., 1953, 47, 12 421).
55 0. 4. Timokhin and B. Kissin, S: U. P. 255767/1972 ГГ. A. Tuwxuh
Б. И. Kuccun. Авт. св»д. СССР № 255 767 (1972)].
56. N. L Drake, Org. Reactions, 1942, 1, 105.
57. И7. A. Cowdrey, J. Chem. Soc., 1946, 1046.
58. H. Seeboth, Angew. Chem. Internal. Edn., 1967, 6, 307.
59. A. Rieche and H. Seeboth, Annalen, I960, 638, 57, 66.
60.7/. R. Snyder, С. T. Elston, and D. B. Kellom, J. Amer. Chem. Soc., 1953, 75,
2014.
61. R. Stroh, Angew. Chem., 1956, 68, 387; 1957, 69, 124.
62. 0. G. Ecke, 1. P. Napolitano, A. H. Filbey, and A. J. Kolka, J. Org. Chem.,
1975, 22, 639
63. L. H. Briggs, G. C. De Ath, and S. B. Ellis, J. Chem. Soc., 1942, 61.
64. U. О. P. Ltd., U. S. P. 3230259/1966 (Chem. Abs., 1966, 64, 11 126).
65. Monsanto Chemicals. U. S. P. 3576876/1971 (Chem. Abs.. 1967, 67, 99 833).
66. 8. 4. Mahood and P. V. L. Schaffner, Org. Synth. Coll. Vol. 2. 1943, 160.
'7 P- Raggli and R. Fischer, Helv. Chim. Acta, 1945, 28, 1270.
R G. Benner, C. A. P„ 002354/1970.
C F. Fieser, Org. Synth. Coll. Vol. 2, 1943, 39.
"Beyer and G. Wolter, Chem. Ber., 1952, 85, 1077.
, “fcocq, Bull. Soc. chim. France, 1951, 183.
г „ anii M- Fieser, J. Amer. Chem. Soc., .1935, 57, 49L
- " сю UUCI v.. i . п. /1Ш-П, wig. oyiuu. O»*<. • “>--
» Hodgson, J. Chem. Soc., 1945, 590, 663, 794.
H Hartman and H. L. Silloway, Org. Synth. Coll. Vol. 3, 19м, 8-.
н. Hodgson and D. E. Hathway J Soc. Dyers and Colourists, 1947, 63. , .
67.
68.
69.
70.
71.
72.
7^ r V ‘,сг,г' и,ш JVi- fieser, j. amer. Lueui. oul, jsuu,
7a 5 B- Wells and C. F. H. Allen, Org. Synth. Coll. Vol. 2, 1943, 221.
- ' J. Chem. Soc., 1945, 590, 663, 794.
„ y. Hartman and H. L. Silloway, Org. Synth. Coll Vol. 3, 19м,
H. Hodgson and D. E. Hathway J. Soc. Dyers and Colourish 1947. 6 .
С- 0. Price and Voong-sing Tail, Org. Synth. Coll. \ oh 3, 19o5 664
«.?790W and L P N°P°lita"0’ 6' S- P- 2675909/1954 ,Che
r l-HaHet and C. A. Dornfeld, J. Amer. Chem. Soc 1944, 66, 1781.
G » о K-iewiet and H. Stephen, J. Chem. Soc, 1931, °-- „
°- s- Bachman and J. E. Goldmacher, J. Org. Chem., 1964, 29, -b/o.
8 Зак. 105 226
75.
76.
77
78.
79
80
81
82 N DvnMson, «Naphthalene Compounds», Arnold, London, Ю58, p|)
83. J- Pri,lit Cl,c,n" 19CM (ii)'69'49; N' L °rake' O,'e' Rcacti°4
£ ^Tsm^'in^^e^istry of the Amino Group», cd. S. Pntaj, ln|er.
8G p.'Tyk^'^G^ Mechanism in Organic Chemistry», Longmans
87 L°A °BeS, «flic Infra-Red Spectra of Complex Molecules», Methuen, Lon-
88 cV^Vao.^Hraviolet and Visible Spectroscopy», 3rd edn., Butterworths,
89 Л'^PaRjo^hidusirial Hygiene and Toxicology», Interscience, 1963, vol. Ц,
90 V.^R^Holland, В. C. Saunders. F. L. Rose, and A. £. Walpole, Tetrahedron,
91. L7H.3^eSrger and E. K. Weisburger, Chem. and Eng. News, 1966, 43,
92.
93.
94.
95.
96.
97.
98.
99.
103.
104.
С. K. Ingold and E. D. Hughes, Nalurc, 1950, 166. 642.
0 H. Coleman, Org Svnth. Coll. Vol. 1, 194 1, 442
H. T. Clarke and R. R. Read, Org. Synth. Coll. Vol. 1, 1941, 514.
L. A. Bigelow, Org. Synth. Coll. Vol. I, 1941, 136.
E B. Starkey, J. Amer. Chem. Soc., 1937, 59, 1479.
F. B. Dains and F. Eberly, Org. Synth. Coll. Vol. 2, 1943, 355.
R. B. Sandin and T. L. Cairns. Org. Synth. Coll. Vol. 2, 1943, 604.
A'. Kornblum and D. C. Iffland, J. Amer. Chem. Soc., 1949, 71, 2137.
100. L. Thompson, J. Soc. Dyers and Colourists, 1921, 37, 7.
101. IF. IF. Hartman and L. I. Roll, Org. Synth. Coll. Vol. 2, 1943, 460.
102. IF. A. Skinner, H. F. Gram, and B. R. Baker, J. Org. Chem., 1960, 25, 777.
O. Fischer and E. Hepp, Ber., 1886, 19, 2991,
С. K. Ingold, «Structure and Mechanism in Organic Chemistry», Bell, Lon-
don, 1955. p. 613.
P. IF. Heber and H. Rauscher, Annalen, 1942, 550, 182.
Monsanto Chemicals. U. S. P., 3504034/1967.
R. Adams and GJl. Coleman, Org. Synth. Coll. Vol. I, 1941, 214.
109. Л BMln^er-andG. IF. Anderson, J. "AmerBchem. Soc’.. 1952, 74, 5514.
ЖОХ, 1951. 21, 1503] . ...
105.
106.
107.
108. P. E. Fanta and D. S. Tarbel'l’, Org. Synth Coll. Vol. 3,’ 1955,’ 661,
109. J. Biodinger-and G. IF. Anderson, J. Amer Chem. Soc. 1952 74, 5514.
ПО. В. I. Ardashev, J. Gen. Chem. (U. S.S. R), 1951, 21, 1503 Г А. И. Ардашев —
ЖОХ. 1951. 21, 1503]. 1
111. A. E. Smith and К. J. P. Orton, J. Chem. Soc., 1908 1242.
112. I I. Sudborougli. J. Chem. Soc., 1901, 533
• Л г n Wilkinson and I. L. Finar, J. Cliem. Soc., 1946 115.
114. F. D. Chattaway and К. I. p. Orton, J. Chem. Soc., 1899, 1046; T. S. Ste-
V\mn an iao Watts, «Selected Molecular Rearrangements», Van Nostrand,
l У / <j, p. i у J.
115. Cm. cc. 104. pp. 604 п сл.
ll6’ IQKO !zvest Akad- Nauk S. S.S. R„ 1969, 112 (Chem. Abs.,
и™’ah’Jrrn 9 [Л M- Платошкин, Ю. А. Чебурков, И. Л. Кнунянц -
117 А /Л СССР, Сгр хим., 1969. № 1, 112].
118 IF I HHkinhot/' Сг- and D- P“rdie- J' Chem. Soc., 1943, 58.
J ’£• Jf- nicRinbottotn J, Chem Soc IQS9 9A4R
1 КГ u imissSm ’
S *• ьВтл7/4 vл1 изд- cfc.'s.v®
iB j « у ‘ "*“• 31
25 А °rg' Chcm 1968. 3:i. 2554.
' MoL Migr .Tgn, 3,l237' A' Marti,,s’ Bcr - 187U 742i c- GrlHot, Meeh.
226
Jon G M ......“ v.i.v»,. I non i/uz.
111. A. H. Cook, A. C. Davis, I. Heilbron, and G. H. Thomas, J. Chem. Soc, 1949,
132.
133-
134-
гм cc 104, PP- 615 и сл.
126- i Peillu and W. J. Hickinbottom, J. Chem. Soc. 1920 103
'g' ‘gTshriner, W. H. Horne and R. F. B. Coz. Org. Synth. Coll., Vol. 2. 1943,
fl53M. Dyson, Org. Synth. Coll. Vol. 1, 1941, 165.
л M Dyson and H. 1. George, J. Chem. Soc., [924 1702
u- л н n«r.,'c / r> tr ’г,' .
M^J/edayatu/ZaA, Bull. Soc. chim. France, 1972, 2957.
H J Page and B. R. Heasman, J. Chem. Soc, 1923 3241
W. !. Mi/s, S. E. Hoekstra, R. M. Ulmann, and E. Havinga, Rec Trav chim
1958 77, 746.
„ I D:Ans and A. Kneip, Ber, 1915, 48, И44.
or IF D. Emmons and A. F. Ferris, J. Amer. Chem. Soc, 1953 75, 4623
a? IF. D. Emmons, J. Amer. Chem. Soc, 1954, 76, 3470.
И8 R. R- Holmes and R. P. Bayer, J. Amer. Chem. Soc, I960, 82, 3454
39 IF. D. Emmons, J. Amer. Chem. Soc, 1957, 79, 5528.
140 S. M. Mehta and M. V. Vakilwala, J. Amer. Chem. Soc, 1952, 74. 563
Hl’ P. Santurri, F. Robbins, and R. Stabbings, Org. Synth, I960, 40. 18.
[42 G. B. Payne, P. H. Deming, and P. H. Williams, J. Org. Chem. 1961 26,
' 659.
143 F. D. Popp and A. Catala, Tetrahedron Letters, 1962, 1071.
144. E. Pfeil and К. H. Schmidt, Annalen, 1964, 675, 36.
145.
146.
147.
148.
149.
150.
151.
152.
153.
О A. Russel, Е. G. Janzen, H.-D. Becker, and F. I. Smentowski, J. Amer
Chem. Soc., 1962. 84, 2562.
L. Horner and ! Dehnert, Chem. Ber.. 1963, 96. 786.
Af. Martynoff, Compl. rend., 1948, 227, 1371.
I. Burden, С. I. Morton, and D. F. Thomas, J. Chem. Soc., 1965, 262L
S. Goldschmidt, Ber., 1920, 53, 28.
К. H. Pausacker and I. G. Scroggie, J. Chem Soc., 1954, 4003.
К. H. Pausacker, J. Chem. Soc., 1953, 1989.
M. K. Seikel, J. Amer. Chem. Soc., 1940, 62, 1214.
O. Wallach and L. Belli, Ber., 1880, 13, 525; E. Buncel, Accounts Chem. Res.,
1975, 8, 132.
К. P. Griffin and IF. D. Peterson, Org. Synth. Coll. Vol. 2, 1943, 242.
G. R. Clemo and E. C. Dawson, J. Chem. Soc., 1939, 1114.
154.
155. .. . .
156. /. Г. Edward, J. Chem. Soc., 1954, 1464.
157. S. F. D. Orr, P. Sims, and D. Manson, J. Chem. Soc., 1956. 1337.
158. H. B. Henbest and A. Thomas, J. Chem. Soc., 1957, 3032.
159. H. Schiff, Annalen, 1864, 3, 343; K. Dey, J. Sci. Ind. Res. India. 1974, 33, 76
160. L. A. Bigelow and H. Eatough, Org. Synth. Coll. Vol. 1, 1941. 80.
161. /. G. Miller and E. C. Wagner, J. Amer. Chem. Soc., 1932, c>4, 3698.
162. G. Reddelien, Annalen, 1921, 388, 165.
163. R. H. F. Manske and M. Knlka. Org. Reactions, 1953, 7, o9.
164. W. p, Utermohlen, Jr., J. Org. Chem., 1943, 8, 544.
165. K. N. Campbell and i. I. Schaffner, J. Amer. Chem. Soc., I94o. 67. 86.
A. Combes, Bull. Soc. chim. France, 1888, 49. 89.
£ Roberts and E. E. Turner J. Chem. Soc., 1927, 183-
K.-D. Hesse, Annalen, 1970, 741, 117.
'I7- 7. Jones and KIP. Orton, J. Chem. Soc, 1909. 10o6.
м. K. Seikel, Org. Synth. Coll. Vol. 3, 1955, 262.
47- K. Seikel, J. Amer. Chem. Soc. 1940, 62, 1214.
ivt. w. г. ulerffiOfiien, Jr., J. urg. Miem., c? aa
165. K. N. Campbell and i. I. Schaffner, J. Amer. Chem. Soc, I94o. 67. 86.
166. F. G. Mann and F. Brown, J. Chem. Soc, 1948. 847, 858.
167. • " • - - • - . ,n on
168.
169.
170.
I7l.
^>ri>tei, o. Miner, слеш. joc. . jt.,.
17a о % Corvin, J. Chem. Soc, 1953, 1237.
17- uo Brewster, Org. Synth. Coll. Vol. 2, 1943, 347. 63 43s.
Г Militzer, E. SnMi, and E. Evans, J. Amer Chem Soc, 1941,
177 w B-Sandin et al., Org. Synth. Coll. Vol. 2, 1943. 3 438
178 и У' Hartman and LA. Smith, Org. Synth. Colh \ol. 2.
179 и n' Hodgson and A. Kershaw, J. Chem. Soc, 13, 1955, ,42.
1Z9‘ £ P. Griffin and IF. D. Peterson, Org. Synlh. Coll. 4
8*
227
180. E. L. Martin, Org. Synth. Coll. Vol. 2, 1943, 501.
181. E. Bamberger. Ber.. 1895, 28, 399.
182. A. H. Lamberton, Quart, Rev., 1951, 5, 75.
183. £ D. Hughes and C. K. Ingold, Quart. Rev., 1952, 6, 34.
184. 0. Illuminati, J. Amer. Chem. Soc., 1956. 78, 2603
185. C. F. H. AHen and !. A. Van Allan, Org. Synth. Coll. Vol 3 iqv,
186. IP. Theilacker, J. prakt. Chem., 1939, 153, 54. ' ’
187. £ L. Hollies and E. C. Wagner, J. Org. Chem., 1944, 9, 31.
188. K. Dickore, K. Susse, and К-D. Bode, Annalen, 1970, 733, 70
К G. Gruzdev, Zhur. org. Khim.. 1971, 7, 1085 (Chem. Abs 1071 -re „„
[В. Г. Груздев — Ж0Х, 1971, 7, 1085]. ’ 1J71' 75 • 63277)
G. S. Turpin, J. Chem. Soc., 1891, 723.
У. Iwanami, J. Chem. Soc. Japan. 1964, 82, 634.
E. Bamberger, Ber., 1894, 27, 1347
O. Hinsberg, Annalen. 1887, 237, 327.
O. Hinsberg and E. Schwantes, Ber., 1903 36, 4039
J. B. Wright, Chem. Rev., 1951, 48, 397.
R. Weidenhagen and G. Train, Ber.’, 1942, 75, 1936.
J. B. Wright, J. Amer. Chem. Soc. 1949, 71, 2035
H. H. Sister, Ц. S. A/iuja, and ,V. L. Smith J. Ore Chem ion, ,
R Gosl and A. Meuason, Chem. Ber.. 1959, 92 2521 ® ’ ^6. 1819.
I. Thiele and L. H. Wheeler, Ber., 1895, 28, 1538.
1 r.on Л<-» Л Cl
189.
190.
191.
192.
193.
194.
195.
196.
197.
198.
199.
201. D. V. "Santhorpe and Ё. D. Hughes, J. Chem. Soc., 19G2, 3308.
6.4. ПРОИЗВОДНЫЕ ГИДРОКСИЛАМИ HA
Дж. С. РОБЕРТС (University of Stirling)
6.4.1. МЕТОДЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ГИДРОКСИЛАМИНА
6.4.1.1. Номенклатура и вводные замечания
Со времени открытия гидроксиламииа и его производных но-
менклатура этих соединений неоднократно менялась. Так, до
1937 г. в Chemical Abstracts N-замещенные гпдрокенламины
попадали в раздел бета-гидроксиламинов, а 0-замеш.енные —в
раздел альфа-гидроксиламинов. Этот способ номенклатуры был
заменен системой, согласно которой О-замещенные производные
рассматривались как алкокси (или арплокси) амины, а N-замещен-
ные производные — как алкил (или арил) гпдрокенламины; N,O-
днзамещепные называли алкоксиалкиламинами. Начиная с 1972 г.
в Chemical Abstracts для всех гидроксиламинов используют
номенклатуру соответствующих аминов. Так, (CH3)2NOH описы-
ется как М-гидрокси-И-метилметанамин. Однако в оригиналь-
ных публикациях и обзорах [I] большинство авторов придержи-
вается простой системы, в основу которой положено название
родоначального соединения, гидроксиламииа. Так, упомянутое
®1™е соединение в соответствии с этим принципом называют
N JN-дпметилгндроксиламином, а C2H5(CH3)NOCH2C6H5 — N-ме-
7?,_'Лтнлгг'°енз"лг’1ДР01<силамин°м, причем N-замсстптели пред-
-101 и-заместателям в соответствии с положением этих букв
тинском алфавите. В данной главе использована именно эта
система номенклатуры.
228
Ациклические гидроксиламины можно п,
горни В соответствии с характером .замещ™Л"Ть на три кате-
городе или при замещении у ЭТих атомов™ °Р" а30те 11 ве-
ществу имеются два типа моиозамещеншЛ Ремеи1’0' По су-
несуших заместитель или при азоте (1) ,,,. гидРокси.'|аминов,
Диалогично, для дизамешенных гидроксилаLuJ₽H якис“'1оРаде (2)’
могут быть при азоте (3), илп оба атоМа 1 " t°6a зам^ителя
м0гут нести по заместителю (4). Очевидно что ЙПз2?СЛ°Р°А~
один тип ациклических тризамещеиных Жен ЛИшь
Аналогично, в соответствии с характером замешекЛаМ',НОВ <5)-
гидроксиламины делят на четыре подктассиид Циклические
.................... --------------- ------- Основные
отдельных раз-
методы получения этих соединений
изложены
в
делах.
RNHOH H2NOR R2NOH RMHOR r2nor
(1) (2) (3) (4) (5)
6.4.1.2. N-Монозамещенные ациклические
гидроксиламины
Несомненно, что наиболее распространенным и эффективным
способом получения соединении типа (1) является восстановление
азотсодержащих соединений, находящихся в более высокой сте-
пени окисления, чем сам гпдроксиламнн. Например, довольно
широко распространено восстановление нптросоедпнений и окси-
мов, в меньшей степени — иитрозосоединений. В случае восста-
новления нптросоедпнений наиболее часто используют цинк в
водном растворе хлорида аммония [2]. Выходы в этой реакции
как для N-алифатических, так и для N-ароматпческпх гндрокенл-
ампнов могут довольно значительно колебаться, н поэтому в пос-
ледние годы было предложено много других способов восстанов-
ления. к ним относятся каталитическое гидрирование [3], электро-
химическое восстановление [4], применение диборана 1 ],
амальгамы алюминия [6] и гидросульфида натрия в присутствии
хлорида кальция [7]. Однако для большинства этих методов
2сн°вная проблема заключается в подавлении образования
оочных продуктов реакции, таких, как амины н лнЦершяе -
«си- (10), азо. (Ц) и глдразопронзводные (12), кот Р
л °т пРи конденсации продуктов реакции, находят
игых степенях окисления (уравнение !)•
229
R—NOs
|H|
A0 R—NHOII R—NHj
(Hl
r-N=N-R r-N=N-R -----> R—NH—NH—R
O'
(10) (**) (12>
Более общим методом восстановительного синтеза соединений
типа (1) является превращение оксимов в гидрокспламины с
помощью диборана [8] или цпаноборогндрнда натрия в кислой
среде [9]. Было показано [10], что незатрудненные оксимы
реагируют с избытком литийорганических реагентов с образова-
нием N-алкилгндрокспламннов (уравнение 2).
Rk
T=NOH
R2/
R'
R3Li I
----> R2—C-NLi—OLi
R’
I
R2—C—NHOH
I
R3
Непосредственное моноалкнлированне гндроксиламина низко-
молекулярными алкилирующими агентами не относится к удоб-
ным препаративным методам получения гидроксиламинов (1) в
связи с трудностями, возникающими в результате дальнейшего
алкилирования (уравнение 3). Однако следует отметить, что
подходящим образом активированные производные бензола,
например (13; X = С1, Вт или ОМе), могут быть использованы
для прямого превращения гидроксиламина в N-арилгндроксил-
анин [11].
X'
RX RX рх , PY
NHjOH > RNHOH ----* R2NOH -—> R3N—О' ------> R3N—OR (3)
(13)
Разработан ряд
возникающих при i
. приемов, позволяющих избежать трудностей,
попытке осуществить контролируемое моноал-
килироваиие. К этим приемам относятся алкилирование калиевой
с и Г1,ДРокснл амин-М,О-дисульфокпслоты с последующим
мепип1пЫМ. ГИДРОЛИЗОМ [12] и гидролиз нитрона [13] или нзо-
поп«ип.Ат°КСаЗИрИДИ1м (Уравнения 4, 5). Исходные нитроны
прямым N-алкцлированием соответствующих оксимов,
щим методом синтеза изомерных оксазпрндннов является
230
оК11слеч-е соответствующих иминов иерокснкнслотами (см разд
6,41'9) кон R
-O3S-NH-°S°3’ 2K++RX - -O3S-N—OSO; 2K+ r£,h,.oh {4)
O’ + О
\ ,/U H3O* f НзО* \ / \
/ 4 —" 4— ;cCA-r (5)
6.4.1.3. О-Замещенные ациклические гидроксиламины
Для получения О-замещенных гндроксиламинов (2) разрабо-
тано два основных метода. Первый из них заключается в нуклео-
фильном замещении „уходящей группы при атоме азота алкокси-
нли арилоксигруппои (уравнение 6). Обычно в этой реакции
используют хлорамин (Х = С1) [15] и гндроксиламнн-О-сульфо-
кислоту (X = OSO3H) [16]. Другой метод состоит в алкилиро-
вании N-ацилзамещенных производных гидроксиламииа с после-
дующим кислотным гидролизом в мягких условиях (уравнение 7).
N-Ацильные группы могут быть различными: R1 = алкил или
арил (гидроксамовая кислота) [17] и R! = OEt (N-гндроксиуре-
тан или карбамат) [18].
H2N—X + 'OR —► HjNOR (6)
О о
|| R2X. "ОН II Н3О+ ♦
R'CNHOH ----------->- R'CNHOR2 ---> H3NOR2 (7)
Вместо N-ацильной группы
группа: при О-алкилироваиии
может быть использована имнно-
илн О-арилировании оксима [19]
или алкилгидроксимата [20] получают соединения (14), лабиль-
ные в условиях гидролиза. Аналогичная последовательность
алкилирования и гидролиза была применена в случае солей гидр-
оксиламин-М^-дисульфокислоты (15) [21] и N-гидроксифтал-
имида (16) [22].
О
(14) (16) (16>
6.4.1.4. N.N-Дизамещенные ациклические
гидроксиламины [23]
гиппоксн л аминов N.N-i11'
В отличие от N-монозамешенных г др х моЖно пол)-
замещенные производные (3) в некотор • • - ксиламнна, 1,л1
Ч11ть или прямым диалкилированнем Р
231
моиоаткилированнем N-монозамещенного гпдрокснламина, ocuG
но в тех случаях, когда вводимая алкильная группа (пли груПП1Д
достаточно объемиста [24]. Для получения ЫЛ-ДизамещениЬ1'
гпдроксиламшюв с успехом применили восстановление иитроцо*
В условиях каталитического гидрирования над платиной J25J
с помощью алюмогпдрпда лития [26] В некоторых случаях мо-
жет быть использовано окисление М.М-дпзамещеиных аминов
в контролируемых условиях действием пероксида водороДа
в присутствии катализатора, например вольфрамата или молиб*
дата натрия [27]. Одним из наиболее эффективных методов
получения гидрокснламинов типа (3) является распад по Коупу
суть которого состоит в выделении летучего олефина из оксида
амина (17) про нагревании, при этом образуется нужный гидро-
кспламнп. Часто эта реакция протекает гладко н дает хорошие
выходы [28]. Наконец, следует отметить, что некоторые N.N-ди-
замещенные гпдроксиламины можно получить путем присоедине-
ния реактивов Гриньяра к алифатическим и ароматическим нит-
розо- [29] и ннтросоедпнеипям J30].
R’ оЛ
\Л/ Н
сн/сн2
(17)
6.4.1.5. 1^,0-Дизамещенные ациклические
гидроксиламины
Основным методом получения этих соединений (4) является
диалкилированне или ступенчатое моноалкилирование N-гидр-
окснуретаиа с последующим кислотным гидролизом [31] (уравне-
ние 8). Та же последовательность реакций может быть использо-
вана и для ступенчатого алкилирования N-гидроксимочевииы (18)
и М-гидрокси-М'-фенилмочевины (19) [32].
ООО
Il R'x II R2x II н3о+ *.
EIOCNHOH ->- EtOCNHOR1 -> EtOCNR2OR' -► R2NH2OR‘ (8)
О О
V II
H2N—С—NHOH CeHsNH—С—NHOH
(18) (19)
6.4.1.6. М,М,О-Тризамещенные ациклические
гидроксиламины
Для синтеза этих соединений (5) использовали ряд методов,
но ни один из них не является универсальным. К этим методам
относятся алкилирование и диалкилированне М,О-замеш.енных и
232
о.3амещениых гидрокспламннов coot™-
подала лежит способность большннХЗ^'1»0- В основе nDvrn
(как устойчивых, так „ неустойчивы Л "ИТР°к™Диых
радикалами алкильной или ар™,,,"*' Реаг“Ровать с
в целом может быть представлен Природы- Этот руГими
9W метод обсуждается ^а3д,б.4.5У3Р еН,1ем более щ'ТрХо
R • + R—NO
o.
(9)
R?N—OR
Еше один метод синтеза производных (5), пригодный только
для введения О-аллильиой группы (и замещенных О-аллильных
групп) пли О-бензильнои группы, это термическая перегруппи-
ровка соответствующим образом замещенных оксидов аминов
(20; R3 = аллил или бензил) (перегруппировка Мейзенгеймера)
(уравнение 10). Эта реакция послужила предметом интенсивного
изучения; полученные данные обсуждаются в разд. 6.4.4.3.
R’\ R\
N—О' —> XN—OR3 (10)
R2/jl3 R2/
(20)
Наконец, следует отметить, что трп-трет-бутилгндрокснламин
(21) является основным побочным продуктом, образующимся при
взаимодействии 2-метил-2-нитропропана с натрием в 1,2-днмет-
оксипропане. Главным продуктом этой реакции является ди-трет-
бутилнитрокснд (22) [33].
трет-Втк трет-Ви^
л’—OBu-трет N—О»
трет-Ви^ трет-Ви
(21) (22)
6.4.1.7. М,1\1-Дизамещенные циклические
гндроксиламины [34]
Некоторые из методов получения N.N-днзамещенных ацикли-
ческих гидрокспламннов (6) применимы также и для синтеза
Циклических аналогов. К этим методам относятся восстановление
Циклических нитронов [35], окисление циклических аминов [27J
\ х (23)
4Z 'Et
<J + R--> </~R (И)
"О—N+ НО—N
233
о
о
(| j] ,+ h2noii — > I
N
)
OH
(12)
„ пиролитическое отщепление этилена из этилзамещенных оксидов
пикшческнх аминов, таких, как (23) [36]. Для получения заме-
тенных циклических гидроксиламшюв используют реакцию при-
соединения реактивов Гриньяра пли лптииорганпческих соедине-
нии а также других агентов карбанионного характера (уравне-
ние’11) [37]. К прочим методам синтеза относится присоединение
гидроксиламииа к сопряженным диенонам [38] (уравнение 12)
и восстановление нптрокспльных радикалов в мягких условиях,
например, феннлгпдразпном [39].
6.4.1.8. М,М,О-Тризамещенные циклические
гидроксиламины (7)
По существу имеется только два метода синтеза гидроксил-
ампнов этого типа (7). Первый состоит в О-алкплировании
М.М-дизамещенного циклического гидроксиламииа, а второй
основан на легко протекающей рекомбинации циклического иит-
рокснда с каким-нибудь другим радикалом (см. уравнение 9),
6.4.1.9. N.O-Ди- и М,М,О-тризамещенные
циклические гидроксиламины (8) и (9)
Типичными примерами циклических гндроксиламинов (8) и (9),
в которых как атом азота, так и атом кислорода входят в состав
цикла, являются наиболее распространенные циклические систе-
мы: оксазирпднны (24), 1,2-оксазетидины (25), тетрагидро-1,2-ок-
сазолы (нзоксазолндипы) (26) и тетрагпдро-1,2-оксазииы (27).
_м~ N’~ N-
и о Q>
,24> (25) (26) (27)
Имеется несколько путей синтеза оксазиридипов (24) [40].
наиболее широко используют окисление альднминов или кети-
минов (основании Шиффа) с помощью пероксикислот (уравне-
1лпРОпДе11"я этой Реакции применяют и другие
елн ’ р'оскольку энергетический барьер процесса
(см ПРс /о°Де азота для ЭТ|!Х соединений достаточно высок
актив,,мхД' „ возможно получение устойчивых оптически
ок азиридинов с помощью оптически активных перокси-
234
кислот [42], таких, как (+)-пероксикамфОпная
лением хиральных оснований Шиффа [43] кисл°та, или икис-
*-г-1свч4со3ц (R')IR
„„
о
Другой ДОВОЛЬНО подробно изученный способ по ,
зиридпнов заключается в фотохимической п 'ГНМ окса’
нитронов [44]. Приведенные в работе |441 пяи ntperPVnnl1poBKe
что процесс замыкания цикла подчиняется пп1е «оказывают,
симметрии и происходит дисротаторно как пп^аМ оРб|'тальиой
превращения (28) в (29) (уравнение 14) Поско На пРимеРг
родом и кислородом образуется о-связь л-ообитян?’ Между угле‘
регнбридизуется в зр3-гибридизованную орбэтапьатома,азота
электронной пары, в результате чего получается оке! °ДИ0Й
с той же самой конфигурацией, что н в исходном нит |гДИН
мошыо хиральных оксазпридинов Бойд с сото р“Не' С по-
эте фотохимическое превращение обратимо К ЧТ°
верейным методам синтеза оксазпридинов относятся реакцияТ
тона или альдегида с хлорамином (X = СП нтн г,™
О-сульфокислотой (X = OSO3H) в ^рнсутТт^’^^^аХ'^ав'
нение 15) и озонолиз алкена в присутствии амина (уравнение 16)
Необходимо отметить, что некоторые нитрены присоединяются
о двойной связи карбонильной группы с образованием оксазирн-
динов [46], а нитроны типа (30) реагируют с п-толуолсульфонил-
лоридом в водном растворе щелочи [47], как показано в
^Равнении 17.
235
OTs
(SO}
Известные 1,2-оксазетпдпны (25) представлены в значительной
степени их фторпропзводными [48]. Примером этих соединений
мГжпт з,3,4(4-тетрафтор-2-трпфторметпл-1,2-оксазетидцн (31), ко.
топый образуется в. результате термического присоединения ни-
трозотрпфторметана к тетрафторэтилеиу [49]. Соединение (32),
явтяюшееся одним из немногих примеров иефторированных 1,2-
оксазетидинов, было получено путем обработки гидробромида
ациклического гидроксиламииа (33) тр^т-бутоксидом калия [50].
Было обнаружено, что пиролиз необычно устойчивого нитрозооле-
фнна (34) в вакууме приводит к термически лабильному 4Z/-1.2-
оксазету (35), который разлагается при нагревании с образова-
нием ди (трет-бутил) кетона и цианистого водорода [51].
[51].
F
(31)
CF3
I
трет-Ви
I
Г1
трет-Ви
трет-Ви
тргт-Ви
трет-Ви
трет-Ви—!___
I II
О—N
(32)
(33) (34)
(35)
Основной метод получения тетрагидро-1,2-оксазолов (26) [52]
заключается в 1,3-дпполяриом присоединении нитрона к олефину
(уравнение 18). По поводу механизма этой реакции высказыва-
лось два предположения — о согласованном процессе и о ступен-
чатом бирадикальном процессе. Совокупность экспериментальных
данных подтверждает первое предположение [53]. Хорошо
известны многие примеры этой реакции как меж-, так и внутри-
молекулярного характера в основном благодаря блестящим рабо-
там Хьюсгена [54] и Ле Беля [55]. В случае несимметричных
олефинов межмолекулярная реакция, как правило, отличается
высокой регпо- и стереоспецифичностью. В ряде работ Тартаков-
ский и др. [56] показали также, что нитроновые эфиры типа (36)
легко вступают в реакцию 1,3-дцполярного присоединения
к олефинам, в результате чего образуются 2-метоксш130ксазолп-
TTWU1.I 1 4
(18)
286
Eme один метод прямого получен»»
заключается в диалкилироваинн N JL,TTparibWU.^(.., ,
j^nfipoMuponana; при этом получается мНурета|,а Действием
производное (37). Аналогичным образом
Дана может быть получен М-это1/°М’ нсх°Дя из 1 л ное
gs. <*» от.«. - «Ьгя>»»йй;
ные с заместителями в цикле удаетея’ н Другне ' >^к
циклические гидроксиламины путем рсв* ’ть в незамещенные
ааиия в присутствии водной кислоты. Р За « Декарбохсилиро
О
О
" xNCOzEt
,ОМе
X'CO2Et
Ar'
(36) (37) (38)
Вероятно, наиболее универсальный метод синтеза тетрагндро-
1,2-оксазииов (27) основан на относительно легко протекающей
реакции Днльса — Альдера между' дненом н иитрозосоединением,
активированным наличием сильной электроноакцепторной груп-
пировки при азоте (см. уравнение 19) (58]. В некоторых случаях
получаемые этим способом 3,6-днгидро-1,2-оксазнны (39) можно
превратить в тетрагидро-1,2-оксазины путем удаления активирую-
щей группы действием основания и каталитического гидрирования
эндоциклической двойной связи в мягких условиях. Аналогичным
образом нптрозобензол вступает в реакцию [4+2]-циклоприсо-
единения с циклопентадиеном, превращаясь в соединение (40)
[59], и в реакцию [6 + 2]-циклоприсоединения с цнклогепта-
триеном, давая соединение (41) [60].
С+1х~|
о
NX
)
-О
(19)
X = CF3, C(Me)2CI, C(Me)2CN,
Ph
(39)
n-O2NC6H4, и-С1С5Н4
/ Ph
и т. д.
(41)
Эшенмозер и др. [61] показали, что сопр.’’жеу”Ь'ц3 6-хлорни-
розония (42), которые можно генерировать
тронов (43), вступают с олефинами в реакцию' ' ' •
иного типа. Получающиеся циклические дегнд„ ,
можно превратить пли в а.р-ненасьшюни >! • _ОКСазннов (45),
Ретро-реакции Дильса — Альдера 5,6-дип ДР
пли в у-лактоиы (47) (схема 20). 237
(40)
попы HllT-
— Альдера
соли <44>
льдегнды (46 по
О
6.4.2. ФИЗИЧЕСКИЕ СВОЙСТВА ПРОИЗВОДНЫХ ГИДРОКСИЛАМИНА
6.4.2.1. Структура и стереохимические свойства
Большое число экспериментальных [62] и теоретических иссле-
довании [63] посвящено изучению энергии активации конформа-
ционных изменений в ациклических п циклических гпдроксилами-
иах. В сущности, важны два конформационных изменения,
а именно инверсия при атоме азота и вращение вокруг связи
N О- В случае трех-, четырех- и пятпчлеиных циклических
гндроксиламииов этот второй процесс не может являться ско-
ростьопределяющим, поскольку вращение вокруг связи N—О не-
возможно в силу ограничений, накладываемых геометрией этих
циклических систем. Для многих из этих циклических систем,
в частности для замещенных оксазнридинов, были измерены
барьеры инверсии при атоме азота [42, 62], которые характери-
зуются наибольшими значениями энергии активации процесса
инверсии (105 134 кДж/моль), В случае немногих тетрагидР0'
236
„гчзолоп, для которых были выполнены ПОЛОЛ..,.,
о11,' ческие барьеры процесса ипве„сшГ , измеРения,
мб-67 кДж/моль) [64]. Как уже отмечалось, высоки^зна
ергпн активации для оксазиридииов подтверждаются тем “и
яиеиия можно получить в виде устойчивых оптически акт
“Л изомеров [42, 43]. В ряду тетрагидро-1,2-оксазоло> у=к
"азделить ипвертомеры (48) и (49) [65]. Другие ЭКЗоцикличХе
Кектроотрппательные заместители при азоте (хлор, Сепа
поод) также повышают барьер инверсии [62],
Ллычно связывают с дестабилизацией плоского
ояния за счет взаимодействия
валентных электронов.
ОМе
МеО2С
и кис-
и это явление
переходного со-
между иеподеленными парами
ОМе
МеО2С X
МеО2С
С\
(48)
CN
(49)
В ациклических или шестичленных циклических гидроксиламн-
нах скорость конформационных изменений может в принципе
определяться или инверсией, или торсионными взаимодействиями.
Увеличивается число теоретических [66] и экспериментальных
данных [62, 67], подтверждающих, что процесс инверсии имеет
более высокий активационный барьер (~50—54 кДж/моль), чем
энергетический барьер процесса вращения (33—38 кДж/моль).
Что же касается геометрии ациклических гндроксиламинов,
то теоретические расчеты [63, 66] указывают на то, что ротамер
(50), изображенный в ньюменовской проекции, имеет наиболее
устойчивую конформацию в том случае, когда отталкивание не-
поделенных электронных пар минимально. Этот ротамер является
наиболее выгодным, и его структура находится в полном соответ-
ствии с результатами измерений дипольных моментов [68] и с
данными изучения М,М,О-триметил-, М.О-дпметпл- и О-метил-
гндроксиламинов методом электронографии [69]. Интересно от-
метить, что эта предпочтительная геометрия частично находит
свое отражение в кристаллической структуре К-(л-карбокснбен-
зил)тетрагпдро-1,2-о ксазинз (51), для которой характерен
(50)
(51)
439
..„„„ij угол при связи N—о, равный 67°, что значительно
^'’ыве “ем Соответствующий угол в циклогексане иахоДЯщем’°
конформации нормального кресла (т. е. 55 ) [70]. В упомяну.
той выше работе [69] методом электронографии оыло показав
, ИИ связи N—О в трех гидроксиламииах составляют соот’
;” SX 3 <±0.В). 148,8 (±0.8) « 146,4 (±0.3) „“J
тишь немного больше, чем значение 145 пм для гидрохлорида
N-метчлгидроксилампиа (данные рентгеновского анализа) [71]
6 4.2.2. Спектроскопические и кислотно-основные
свойства
В УФ-, ИК- и ЯМР-спектрах гидроксиламинов обычно не
удается обнаружить особенностей, которые были бы характерны
только для гидроксиламиииой группы. На основании изучения
ИК-спектров большого числа гидроксилампиов выявлено, что
полоса поглощения при 900—1000 см~' связана с валентными
колебаниями связи N—О [72].
Низшие гпдрокспламины обладают типичным «аминным»
запахом и представляют собой жидкости или твердые вещества.
Как можно предвидеть, замещение при атоме кислорода приводит
к понижению точки кипения: например, для N-метилгидрокснл-
амина т. кип. 115°С, а для О-метилгидроксиламина т, кип. 48°С.
Гндроксилампны обладают основными свойствами и образуют
соли с минеральными кислотами (например, гидрохлориды и
гидробромиды), можно также получить оксалаты н пикраты гид-
роксиламинов. Во многих случаях эти соли гораздо более устой-
чивы, чем свободные гндроксилампны, и гпдрокспламины пред-
почитают хранить в виде солей. Увеличение степени замещения
при атоме азота н кислорода понижает основность, как и в слу-
чае производных гидразина. Для простых гидроксиламинов [73],
а_ также для тетрагидро-1,2-оксазола и тетрагидро-1,2-оксазина
[74] значения рКа соответствующих сопряженных кислот обычно
находятся в интервале 4—6. N-Моио- и М,М-дизамещенные гндр-
оксиламины обладают также свойствами очень слабых кислот,
однако из-за неустойчивости этих соединений в сильнощелочной
среде измерить их кислотность невозможно; эти значения должны
лежать приблизительно в интервале 12__13.
6.4.3. РЕАКЦИИ ПРОИЗВОДНЫХ ГИДРОКСИЛАМИНА
6,4.3.1. Нуклеофильные свойства
слепчет" LL хами'1еского поведения гидроксилампиов в целом
веакиночной дослать короткое введение относительно
бенностч по С°б,!0Ст" незамеще„„ого гидрокспламниа, в осо-
давио было vptI°ше!"”° к карбонильным соединениям. Не так
у иовлеио, что гидроксилам,,,, относится к ряду
240
"У^ЙнуЮ реакционную способность^ сравнению "₽дпВЛЯ,°Т
"Хулами «ли ионами, обладающими сходнымизначениями
ТО есть для них характерно положительное отклонение п
SJa Бреистеда. Общим структурным признакомэтихреакцион
□способных нуклеофильных частиц является то, что нуклСофиль’
"ый ие»тр Расположен Рядом с атомом- имеющим неподе=ю
тронную пару; это явление получило название а-эффет
751. Хотя для объяснения этого эффекта был предложен мд
Пресных гипотез, включая внутримолекулярный катализ
дифференциальную сольватацию, стабилизацию переходной
состояния и отталкивание свободных электронных пар, приводя-
щее к расщеплению энергетических уровней, по-видимому, одно-
значного решения этой проблемы не существует. Реальное
объяснение должно, по-видимому, учитывать ряд факторов Не-
давно Либман и Поллак [76] подвергли критике эти гипотезы
обратив внимание на тот факт, что a-эффект наблюдается лишь
в случае субстратов с л-связями. Они высказали предположение,
что увеличение скорости реакции может быть обусловлено боль-
шей стабильностью шестиэлектронного ароматического переход-
ного состояния соответственно для О- и N-атакн [(52) и (53)]
карбонильной группы.
"ДкЭ ©О
oQ ®o
oO ©c
®c
(S3)
как нуклеофильного агента
(52)
Использование гидроксиламина
в реакциях с карбонильными соединениями может оказаться
довольно сложным, так как атака может происходить как азот-
ным, так и кислородным центром. В случае реакций с такими
соединениями, как ацетон, ацетилацетон и этиловый эфир ацето-
уксусной кислоты, для обнаружения и идентификации промежу-
точно образующихся соединений Коцнвера и др. [77] использо-
вали метод проточной ЯМР-спектроскопии. Применение этого
метода позволило установить строение тетраэдрического проме-
Жутиого соединения (54), образующегося при реакции гидроксил-
амина с ацетоном, и соединений (55) И (56), образующихся соо.
ветственно в реакциях с ацетил ацетоном и этиловым эфир
ацетоуксусной кислоты. Если карбонильная группа находится
® сопряжении с двойной связью, как в случае 4-метнлпент ‘
Х11си мезитила), то присоединение гидроксплампна в
Л°ВЦЯХ приводит К СЛОЖНОЙ смеси продуктов Реа
п 11Им 1,3 главных продуктов реакции является нзокс ’
^агают, что он образуется в результате шжлнзацш 0-а;™«л
Лроксиламкна (58), который получается присоеднн
. „ inc топочного атома гидроксилампнп. Однако 11еп..г,
Михаэлю ' ‘ Р ;,1еШ1Я Н-мсгнлгпдрокснламнна к этиловой
ХГкоршшои кислоты при pH 8,5 не является присоед11Не
по Михаэлю; показано, что вначале образуется аддукт (59), к"е
тоЛ затем циклизуется в конечное соединение нзоксазольной
™нроды (60) [79]. Из приведенных примеров следует, что меха!
Z присоединения гидроксиламина к сопряженным карбониль-
н м соединениям нельзя считать твердо установленным, и неко.
тонне гипотезы, сформулированные ранее, необходимо проверив,
с помощью современных аналитических и спектроскопических Ме.
спектроскопических Ме.
тодов.
Ме
I
HOHN—С—ОН
I
Ме
HONH О
Me\l II
Сх /С—OEt
но/ хсй2
(54)
\/
-О'
N
H2N
(55)
О
II
PhCH=CHCONHMe
(57) (58) (59) (60)
N-Алкнл- и N-арнлгндроксплампны легко реагируют с аль-
дегидами и несколько труднее с кетонами, давая нитроны, а О-мо-
нозамещенные образуют О-замещенные окснмы. Эти реакции
находят применение в синтетической практике. Реакции некото-
рых N,N- и N.O-дизамещенных гидроксилампиов с альдегидами
приводят к карбиноламинам и 1,1-дигидроксиламинопроизводным
[80, 81]. Ацилирование N-моиозамещенных гидрокснламннов
действием хлорангндридов и ангидридов карбоновых кислот
обычно происходит по атому азота, при этом образуются гидро-
ксамовые кислоты (уравнение 21). В более жестких условиях
последние ацилируются дальше до М.О-днацилгидрокснламинов.
Известно два основных’ исключения из этого общего правила.
О-Ацнлироваипе становится предпочтительным направлением
реакции, во-первых, для пространственно затрудненных гидро-
скиламинов (например, R'-тритнл) [82] и, во-вторых, при исполь-
зовании классических реакционноспособных ацилирующих агентов,
таких, как zi-нптрофецилбеизоат и ацетилнмидазол [83]. Инте-
ресно, однако, отметить, что Перкинсу и Хорду [84] при ацили-
ровании П-трет-бутилгидрокснламина этнлхлорформиатом удалось
получить соединение (61) с выходом до 30%. В то же время по
данным Хауса и др., [85] обработка N-трет-бутилгидрокснламина
ацетангидридом в отсутствие пиридина приводит к продукту
-ацетилирования. Для получения N-алкнл-О-бензоилгидрокснл-
амина иаиоолее подходящим методом является реакция первич-
ного амина с пероксидом бензоила [86] (уравнение 22),
242
он
I
трет-Hu—N—CO2Et
R'NIIOH + R2COC1
О О
(21)
(22)
(61)
НО О
I «
R'N—CR2
О
RNH2 + Р11СООСР11 —> RNIIOCPh
Как уже отмечалось в разд. 6.4.1.2 и 6.4.1.4, алкилирование
ппрокснламнна может быть очень сложным процессом, который
"Учительной степени определяется пространственными особен
остями исходного гидроксиламииа и алкилирующего агента
Несомненно, что первоначально алкилирование происходит по
азоту и в случае незамещенного гидроксиламииа не наблюдается
значительного a-эффекта. Этот факт служит еще одним подтвер-
ждением того, что при любом объяснении a-эффекта необходимо
учитывать взаимодействие с л-связыо в субстрате.
6.4.3.2. Окисление и восстановление
Окисление N-моно- и N.N-дизамещенных гидроксиламинов
протекает крайне легко п под действием целого ряда окислите-
лей [87]. В препаративных целях окислительное превращение
N-монозамешенных гидроксиламинов в С-нитрозопронзводные
лучше всего проводить с помощью реактива Фетизона (карбонат
серебра, нанесенный на целит) [88] или дпэтилового эфира
азодикарбоновой кислоты [89], поскольку в обычных условиях
оба этих реагента не вызывают дальнейшего окисления до С-ни-
тропронзводных. Третичные нитрозопроизводные получают или в
виде интенсивно окрашенного мономера, или в виде бесцветного
димера; первичные и вторичные нитрозосоединенпя, как правило,
Довольно быстро таутомеризуются в соответствующие оксимы.
Однако если в гидроксиламине вблизи реакционного центра име-
ется двойная связь, как, например, в (62; п = 2 или 3), происходит
Циклизация с образованием соответствующего пяти- пли шести-
членного циклического гидроксиламииа (63) [85^90]. Считают,
что механизм этой циклизации включает цепной радикальный
процесс, начинающийся с образования моиоалкнлнитроксида ( )
Ароматические гидроксиламины особенно склонны к конденсации
с образовавшимися иитрозосоединенпямп, что приводит к азо
кснпРоизводным (см. уравнение 1).
(СН2)«
R2C\ /CHCHs
N
(СН2)«
R.C^ ХСН
:nhch2
l
O'
(64)
243
(CH2)n
хсн
HONH С PI2
ОН
(63)
(62)
v„TO rtoice сильные окислители превращают N-монозамеп,
HHP гпдрокенламины в С-пнтросоедпиеиня [91], реакции J’’
имеют сколько-нибудь существенного значения ДЛя
т,,па " го синтеза. Окисление ЧЫ-днзамещеиных гидроксилам
нов может приводить к образованию производных двух “»•
зав спмостн от природы заместителей при углеродных атом"?
соединенных с атомом азота. В том случае, если хотя бы од,,*'
,в заместителей является атом водорода, как, например, в
Х = Н) продуктом реакции является нитрон (66), а в случае
соединений типа (65; X = алкил или арил) ооразуется соответст-
вующий иитрокенд-радикал (67) (уравнение 23). Более подробно
реакции этого типа рассмотрены в разд. 6.4.5.1.
f /ОН
:с-ь/
+ /°'
C=NZ
\ (23)
I /°-
;c—N
(67) (65) (66)
Большая часть попыток окисления 0-монозамеш.енных гидро-
ксиламинов приводила к расщеплению связи N—О, к перегруппи-
ровкам или к обоим типам превращений одновременно. Однако
имеются данные, что окисление О-метнлгидрокснламииа тетрааце-
татом свинца в присутствии тетраметилэтилена приводит к мето-
ксиазирндпну (68), промежуточно в этой реакции образуется ме-
токсинптрен (69). В результате более глубокого изучения этой
реакции и в ходе других попыток генерирования О-нитренов
Кери и Хейес [92] пришли к заключению, что механизм образо-
вания алкоксиазнридинов в реакциях окисления тетраацетатом
свинца не обязательно предполагает промежуточное генерирова-
ние О-нитрена. Более удовлетворительно механизм реакции мо-
жет быть представлен как электрофильное присоединение евн-
нецоргаиического промежуточного соединения (70) или нона
О-иитрения (71) к двойной связи олефина.
I
ОМе
N—ОМе
RONHPb(OAc)3 RONH
(68) (69) (70) (71)
Основанием для
этого вывода
мере
---------------------------- послужили в значительной
результаты нестереоспецифической реакции цис- и транс-изомеров
утена . Заканчивая перечисление реакций с участием ионов
’WeHM, следует заметить, что Гассмаи и Хартман [93] принзу-
и ,^лЛТаН0ЛИЗа ряда (пнперндиннл-!)бензоатов (72) пришли
ram moT-'''"'' выводу: в ходе превращений, связанных с разры-
плення „Лял„егче Реализуется переходное состояние для расше-
бои связи N—О с последующим генерированием иона
244
„ чем переходное состояние для
iii|TPeI1’го же тина, но более прочной.
с»»3" т° о
ионизации
связи
С-О,
\j—о с—
(72)
Восстановление гидрокспламннов можно осуществить пей
г1ВиеМ ряда восстанавливающих агентов [1]. Обычно восс анов’
е приводит к расщеплению связи N-0 с образованийчТ
„ В случае циклических гидрокспламннов, таких как (26 н
,27) н их производных восстановительное расщепление с по
,ошыо цинка в уксусной кислоте [94] или методами каталитщ
ческого гидрогенолиза [95] приводит к ациклическим амино-
спиртам.
6.4.3.3. Перегруппировки
Можно предвидеть, что наиболее склонными к перегруппиров-
кам гидроксиламинамп будут оксазнрндины, поскольку для них
характерно наибольшее напряжение цикла. К изученным пере-
группировкам относятся термические [96] и фотохимические
превращения [45] в нитроны, а также термически и фотохими-
чески инициируемые перегруппировки в амиды.
Достаточно давно известны две перегруппировки N-арил-
гидрокснламннов, а именно перегруппировки Бамбергера и Штиг-
лица. Первая заключается в обработке N-фенплгидроксиламина
водной минеральной кислотой, в результате чего образуется п-
аминофенол. Полагают [97], что механизм этой реакции вклю-
чает генерирование иона нптрения (73), который может атако-
вать вода и другие нуклеофилы, такие, как хлорид, спирты, фе-
нол и даже бензол (если в качестве кислого катализатора
используется трифторметансульфокнслота [98]); при этом полу-
чают о- и n-замещенные анилины.
(73)
Область применения перегруппировки Штиглица [82]
на N-моиоалкилгндрокснлампнами, у которых атом угл Р в
тпиеДСТву с атомом азота полностью замещен, как, р
х₽1пп"ЛГИдрокс'1лаМ1111е (74)- Обработка этого соеднненг1
' Ридом фосфора вызывает миграцию одной из фени- ..)П)ацня
4ТОМУ азота с *об ованнсм пмпНа (75). Считают, что миграция
г 245
является внутримолекулярным процессом, протекающим Так,
изображено в формуле (76).
Ph3CNHOH Ph2C=NPh Ph2C—№Г CI
О—Р^
(74) (75) <76>
Еще одна перегруппировка, обнаруживающая близкое сходство
с хорошо известной бензидиновой перегруппировкой, была откпц.
та Шерадским и Салемником [99], которые пытались превратить
карбамат (77) в N.O-дизамещениое производное (78) при дейст
вии трет-бутоксида калия и о-нитрофторбензола. Вместо ожидае-
мото соединения было выделено производное бифенила (79); авто-
ры пришли к заключению, что его получение объясняется проте-
канием перегруппировки бензидинового типа через промежуточное
образование соединения (80). Движущей силой этой перегруппи-
ровки является расщепление слабой связи N—О в промежуточ-
ном соединении (78) и образование связи С—С.
(79) (80)
Три очень интересные перегруппировки креминйоргаиических
производных гидроксиламнпа наблюдали Вест с сотр. [100].
В литиевой соли аниона (82), образующейся из N.O-днзамещенио-
го гидроксиламнна (81), происходит внутримолекулярный аннон0-
иднын 1,2-сдвиг О-трпметплсилилыюй группы от кислорода к азо-
ту с образованием аниона (83) (уравнение 24). В большинстве
случаев для соединений с различными группами при атоме крем-
ния равновесие между двумя анионами сдвинуто в сторону анно-
на типа (83). Соответствующий соединению (81) радикал (84),
лученный пз (81) путем отрыва атома водорода с помощь10
тРет утоксилыюго радикала, перегруппировывается подобным »е
р зом в радикал (85). В обоих случаях выигрыш энергии за
246
трканнЯ перегруппировки должен превышать
1 пРоТ , т е разницу энергий связей Si—О и Si—N (443
кДя^пль соответственно). В случае третьей перегруппи-
1 318 т’рцс(органосилил)гидроксиламина (86) приводит
"пвк11 п"рОЛ'(87) Предполагают, что механизм этой реакции мо-
г.илоксанУ ’ прОцесс, изображаемый формулой (88) и заклю-
кеТ вклт°чат^ внедрении кремнийалкильной группы по связи
^1ЙсЯере110се алкильной группы с кремния на азот.
Me3SiNOSiMe;
(82)
(Me3Si)3N-О-
(85)
Me О—SiMe:
I I
MejSiN—SiMea
(87)
H-BuLl
Me3SiNHOSiMe3
(81)
Me3SiNOSiMe3
(84)
(Me3Si)3N0‘ Li+ (24)
(83)
(MesSiJjNOSiMes
(86)
Me3Si-N----O—SiMe3
SiMe2
Me
(88)
6.4.4. ОКСИДЫ АМИНОВ [101]
6.4.4.1. Методы получения
Легко и с высокими выходами оксиды аминов (89) получают
из соответствующих третичных аминов при действии ряда окисли
телей, включая пероксид водорода [102], различные пероксикис-
лоты [ЮЗ, 104] и озон [105]. Для получения оксидов низкомоле-
кулярных симметричных аминов в некоторых случаях с усп •
может быть применен давно известный метод исчерп в
алкилирования гидроксиламнна (см. уравнение 3).
R3N— О R3NOH X"
(89) (9°)
6Д.4.2. Физические свойства
б ** кристаллические
Оксиды аминов обычно представляют со массой чрезвычанно
Щества; соединения с низкой молекуляр v411T1)jBaH полярну
'“роскопичны. Как п следовало ожидать, У 247
ковалентную природу связи N О, большая часть оксидов амии
очеш хорошо растворима в воде и ограниченно раствор, .м*"""0»
' J органических растворителях. Оксиды аминов проя ' «е-
свойства слабых основании, образуя с кислотами устоО’
кристаллические соли ЫЛЫ-тризамещешюго гидроксилам^'
(90). Значения рК. сопряженных кислот оксидов аминов На °Я“’
ся обычно в интервале 4—5 [106].
Результаты исследования структуры оксидов аминов раз1„
ны.ми спектроскопическими методами свидетельствуют о тетп'аэл'
рическом расположении четырех заместителей вокруг атома азота
и о ковалентной природе связи N—О. Так, методом электроногра
фин [107] показано, что длина связи N —О в оксиде трпмети/
амина составляет 136 пм. Эти данные согласуются с наличием в
ИК-спектре поглощения в области 950—970 см-1, относимого к ва-
лентным колебаниям связи N—О [108]. Другим подтверждением
тетраэдрического расположения заместителей вокруг атома азота
является возможность разделения оксидов несимметрично заме-
щенных аминов на энантиомеры путем образования диастереомер-
ных солей, последующего их разделения и регенерации оксидов
аминов. Наконец, у оксидов простых аминов был обнаружен зна-
чительный дипольный момент (~5Д) [108], что согласуется
с общим характером связей в этих соединениях.
6.4.4.3. Реакции
Известно много методов регенерации исходных третичных ами-
нов из их оксидов. В качестве реагентов обычно используют сое-
динения фосфора (111), включая трифенилфосфин [109] и трихло-
рид фосфора [110]. Общие принципы, лежащие в основе этой
реакции, были с успехом использованы в альтернативном методе
мягкого окисления борорганического соединения, промежуточно
образующегося при реакции гидроборирования [111]. Присоеди-
нение дигидрата триметнламииокспда к триалкилборану протека-
ет в соответствии с уравнением 25. Оксиды аминов реагируют так-
же с эффективными алкилирующими агентами, в результате чего
образуются соли тетразамещенного гндроксиламмоиия (91). При
обработке подобных производных пиридпн-М-окснда (92) гид-
роксидом натрия образуются ароматические альдегиды [U2]-
R
RaB-Q-^Mej ---->- R2B—OR+Me3N И
R3nor
(91)
iCoh
(ЛуО^Н-АГ
(92)
248
човании ацилирующих агентов, например ацетангид-
и 11С'10‘" шуточно образующиеся соли ацнлокснаминов (93)
,\а, пр0М«аается выделить, они претерпевают реакцию Поло-
"е(|И] Эта реакция может протекать ио различным на-
-кого ' зависимости от структуры оксида амина и природы
1,а8Ле1111Я4!₽го агента. Если один из заместителей R при атоме
ц11Л»РУ‘° ставлен метильной группой, то продуктами реакции
’,ота преЛ,,цлированный вторичный амин (94) и формальдегид.
L, 1ЯютсЯ о механизм этой реакции включает промежуточное
читают, чт оШ)Ой или радикальной пары. При реакции триме-
„браЗ°ваН!!е с трнфторуксусным ангидридом можно выделить
тцлаМ1|Н0КС яТ N^-диметилформальдиммопня (95) [114]. Для
1рпфт°Рацет‘1 „чес'ких и гетероциклических аминов нормальным
рейдов цИК,'? по-./...»-! является ацнлоксилнрование в
направлением реакции
(уравнение 26) [115].
О
4 II
RsN—ОСМе OjCMe
(93)
Ядро
О
R2NCMe
(94)
АсаО
CHj=NMez OtCCF,
(95)
+N—О'
N хОАе
(26)
Как отмечалось
пенным реакциям
Мейзенгеймера
в разд,
оксидов
-. изу-
амннов относятся перегруппировка
6.4.1.6 и 6.4.1.7, к двум наиболее
мейзенгеймера и элиминирование по Коупу. Перегруппировка
Мейзенгеймера [116] может происходить в двух различных фор-
мах в зависимости от природы заместителей в аминоксиде. При
наличии бензильного заместителя, как в (96), термическое воз-
действие вызывает [1,2]-сдвиг с образованием триалкилгидроксил-
щина (97). Эта реакция была изучена довольно подробно [Ш[.
В настоящее время методом ХИДПЯ в опытах по захват) ра-
дикалов окончательно установлено, что эта реакция протека
путем гомолитического разрыва связи N—бензил с образование
радикальной пары (98), которая затем рекомбинирует. -
«рмолиза оксида амина с аллильным заместителем, ка
“е««н (99), имеет место [3,2]-сигматропная пеРегРрпДХСя со-
W которой в результате согласованного пРоцесса P,.e\aHii3Ma
«менне (100). Блестящим доказательством такого
’Мается почти полное сохранение хиральности Р i^jgf От-
№ке оптически активного аминоксида (1° ) ® . гндрокснл-
чадось, однако, что первоначально образу может лретер-
пр»? МОЖет оказаться термически неустойчив у примере
"е»«ь дальнейшую перегруппировку, как показано Н
₽eBPaiUeulw (103) в (104) н (105) [119].
249
Me3N—О
СНгРЬ
(96)
Мех CHa
Me3NOCIhPli
(1’liCU. О-NMe,)
(97)
(98)
Me
Me. I
X)—O—CH—СI I=CH3
O’ CH.
(99)
Me
I
CH
Ме5Й/ XCH
I #
° CH\ph
(101)
(100)
Ph
I
Me2N—О—Clk
хсн=сн.
(Ю2)
N—
(103) (104) (105)
Успешное протекание реакции Коупа [28], происходящей при
нагревании, определяется наличием по крайней мере одного
^-водородного атома, расположенного таким образом, что воз-
можно образование плоского пятпчленного циклического переход-
ного состояния (106) (уравнение 27).
Н Го
R2Cj (^Nr2
(106)
R2C=CH2 + HONR2
(27)
Были получены многочисленные доказательства г<ггс-характера
элиминирования, в частности, в тех случаях, когда важную роль
играют пространственные затруднения. Это положение пояснено
на примере соединения (107), которое легко претерпевает внутри-
молекулярную реакцию с образованием соединения (108), в то
Г10П1Я пак его диастеРе°мер (109) В этих условиях инертен
Ио аналогичным причинам происходит гладкое расщепле-
ние двух циклических аминоксидов (ПО) и (111) в противопо-
"Х ,!Ч®с-'ТИЧЛенномУ аналогу, который также инертен-
Ш ии [121] приведены данные о том как различные УсЛ
250
н1я эксперимента могут влиять на изменение направле-
1|ПРовС'ас1а элиминирования в сторону образования побочных
проЦСССлЫ11но считаемых нежелательными. Например, в изо-
Впо,тУ1‘тоВ’-°системе оксид третичного амина (112) может окис-
11ров»11Н°и продуктов реакции, диалкилгидроксиламин (114), с
’ Д оД||Н " цитрона, который в свою очередь может вступать в
’La30Bal,l!oNi 21-циклоприсоединения с олефином (113), давая
In—ме
(107)
ОН
I
N—Ме
Me
♦ I
Л-0
L-Ме
(1°8) (щ)
(111)
к
(ИО)
О'
д
Ci2H2sNMe2 ' *- C1DH21CH=CH2 + HONMe2
(112) (ИЗ)
(И4)
(28)
С10Н21 (
о
(ИЗ)
О'
I
h2cZ‘xch3
амин-
годы
6.4.5. НИТРОКСИДЫ
6.4.5.1. Введение. Методы получения
Изучение иитроксидов (нитроксилов, аминилоксндов нлн
охсплов) началось сравнительно недавно и за п„ав1еннях.
претерпело экспоненциальный рост в различныхмическиеаспек-
Изучались теоретические, спектроскопически • п пменения
ты данной проблемы, а также возможность шир п химии
этих соединений в молекулярной биологии, Ф знач|1ТелЬный
высокомолекулярных соединений. В с^'шв предшествующее
вклад в ускорение изучения ннтроксидов вн ‘ Хьн0 короткий
Развитие ЭПР-спектроскопнп. За^ этот относительно^^
период опубликовано несколько обзорных публикации
посвященных Химии ннтроксидных радикало . тур в ко-
’“Детельствуют о большом разнообразии типов стр.
°Рые входит нптроксидпый фрагмент. 251
Сообщение Пилот» » Шверина [123] о получении перво,
1111Троксида. порфирсксида (115), относится к 1901 г., а в
дующие годы Виланд [124] отметил образование некоторых Ди
арилпитроксидов (116). однако большая часть синтетических метщ
дов в химии ннтроксндов была разработана лишь в последней
время Окисление М.М-Дналкилгндроксиламинов (117) в ни е
оксиды (118) легко осуществляется с помощью ряда мягких окис-
лптелей, таких, как оксид серебра, диоксид свинца, трнкалийгек-
саниапоферрат и кислород. По данным Лебедева [125], который
„отучил нитрокспд (120), более общий метод состоит в окислении
вторичного амина (119). В качестве окислителей используют пер.
оксид водорода в присутствии четвертичного аммониевого основа-
ния солн ванадия, молибдена или вольфрама, или пероксикислоты
[126].
С помощью этих двух методов можно было бы, казалось, синте-
зировать множество ннтроксндов, однако, как показано в
разд. 6.4.5.2, общей предпосылкой устойчивости алифатического
нитрокснда является наличие максимального числа заместителей
при соседнем углеродном атоме. Сфера применения этих методов
окисления ограничена доступностью соответствующих гидроксил-
аминов и (или) вторичных аминов. Тем не менее тот факт,
что двукратное присоединение аммиака к форону (121) по Михаэ-
лю приводит к 2,2,6,6-тетраметпл-4-оксопиперидину (122) с доста-
точным выходом, указывает на возможность получения огромного
числа ннтроксндов путем окисления с последующей модификацией
карбонильной группы. Было показано [127], что кетоны обычно
реагируют с 2-амшю-2-метилпропанолом, давая 2,2-дизамеш.ениые
4,4-диметилоксазолидипы (123), которые могут быть превращены
в соответствующие ннтроксиды при действии пероксикислот
(уравнение 29).
Еще один способ получения ннтроксндов, применяемый доста-
точно успешно, заключается в присоединении соответствующим
образом замещенного реактива Гриньяра или литийоргаиического
соединения к нитрозо- или иитросоедииеипям [128] и к интронам
[12J] (уравнения 30, 31). В случае реакции с шггрозосоедпнеиня-
252
(121) (122)
Я'\=0 + н0\ —/NH*
/\
RCOjH
(29)
я„ и нитронами первоначально в качестве промежуточного сое™
нения образуются гпдрокенламины, которые окисляются киепппп
ДОМ воздуха до соответствующих нитроксидов. В реакции с нити»’
соединениями ооразуется соль (124), разложение которой приво'
дит к нитроксиду. Эти процессы протекают в случае хопошп
известного ди (трет-бутил) нитроксида (22), образование которого
из 2-метил-2-нитропропана в присутствии металлического иатрня
как было показано, протекает путем атаки радикала трет-бутита’
образовавшегося из анион-радикала (125), на второй аннон-ради-
кал с образованием соли (124; R = трет-бутил) [33)
R2MgX (R2Li) R'NO2 > ^N—0 • R2// R2MgX (R2LI) -< R'NO (30)
^C=N—Ви-трет / 1 R2MgX (R2Li) —\—Ви-трет (31)
0‘ z 1 1 R 0.
R2NO2 трет-Ви—NO2
(124) (125)
Другой основной путь препаративного получения
заключается в присоединении алкильного радика.
Мнению или нитрону (уравнения 32, 33).
• R1 + R2NO —> R1—Nr—R2
о •
(33)
253
Оба эти процесса нашли свое применение в методе спиновой
ювушкп (СМ. разд. 6.4.5.4), В то же время они представлю"
собой интересный способ синтеза ряда ннтроксндов, которые труд,
но было бы получить другими методами. Показательный np11MeD
этой реакции — фотохимическое превращение бициклического дп.
мера (126) в бициклический нитрокснд (128), которое проте.
кает через стадию образования дпннтрозосоедниення (127)
как установили Рассат и Рен [130] (уравнение 34). Успешное
илий9
(34)
(U6) (127) (И8)
протекание этой реакции легко понять в свете появившейся ранее
работы Бура с сотр. [131], которые показали, что нитрозомономе-
ры претерпевают фотохимически инициируемый гомолиз связи
С—N, в результате чего генерируются алкильные радикалы, кото-
рые взаимодействуют с непрореагировавшнм мономером, обра-
зуя ицтроксиды. Другим примером внутримолекулярного образо-
вания нитроксида может служить превращение нптронитрозопро-
взводного кариофиллена (129) в трициклический нитрокснд
(130) при реакции с иодом. Полагают, что это превращение про-
исходит через соединение (131) [132]. Позднее было показано, что
эта внутримолекулярная реакция может быть использована как
общий метод получения пяти- и шестичленпых моно- и бицикличе-
ских ннтроксндов [90, 133] (уравнение 35). Реакция нитронов с
алкильными радикалами имеет гораздо меньшее значение [134].
254
6.4.5.2. Структура, стереохимия
и спектроскопические свойства
Электронная устойчивость, присущая нитроксильному рапикт-
, может быть легко объяснена при рассмотрении частичной
диаграммы энергии молекулярных орбиталей: «сгичнои
Считают, что характер связей при атоме азота приближается к
состоянию зр2-гибрндизацни, а a-связь образуется за счет пе-
рекрывания одной из этих гибридных связей с 2р-орбнталью кис-
лорода- Перекрывание 2рг-орбпталей азота и кислорода приво-
дит к заполнению л-орбиталн, а неспаренный электрон распола-
гается на л*-аитисвязывающей орбитали. Все это в совокупности
приводит к образованию двухцентровой трехэлектронной связи.
Используя валентные связи (черточки), ннтроксидную группу
можно приближенно представить двумя мезомерными структура-
ми (132) и (133).
R—N—R <-> R—N—R
(132) (133)
На первый взгляд может показаться удивительным, что нит-
роксидная группа термодинамически устойчива и обнаруживает
очень слабую тенденцию к димеризации, за исключением особых
случаев, отмеченных ниже. Такое поведение становится понятным
при рассмотрении электронной конфигурации нптроксидной груп-
пы; ясно, что проигрыш в энергии делокализации не может быть
компенсирован небольшим энергетическим выигрышем, возникаю-
щим в результате образования слабой связи О—О. Другая точка
зрения на этот феномен предполагает учет двойной квартетнои
гипотезы Линне [135], согласно которой димеризация лишь увели-
чивает межэлектронное отталкивание, но не приводит к увеличе
пню числа связывающих электронов.
Как отмечалось ранее, общим критерием устойчивости дна.-
’илнитроксидов является наличие максимально возможного чнсл
заместителей при углеродных атомах по соседству с атомо азота.
ественно, что этот пространственный фактор своди п ''0
У тенденцию к димеризации. В том случае, когда у
255
с азотом атома углерода имеется хотя оы один водород.., -
стптель (134; R = Bu-tpct), происходит диспропорции,,’’ Зак'е-
до соответствующего нитрона (135) и гпдрокспламипа
(уравнение 36). Эта бимолекулярная реакция была ц3у,
вольно подробно [136]; предполагают, что происходит обо”8
образование димера, вероятно, через (137) и (иди) (138) "
дующим отрывом 0-водородпого атома и распад д0 ц.
гидрокенламнна. Подобный процесс образования нитрона'
гетически невыгоден в случае бициклического иитроксила /ЭНе₽’
который довольно устойчив [137], но димеризуется в твер 13й'
стоянии. Из данных рентгеновского анализа этого дца РЯОм
димера следует наличие фрагмента (140), в котором ;
О лежат в вершинах прямоугольника [138].
(136)
-а до-
атЧмое
с после.
•'"трона и
) г,.-
(139)’
со-
'Магнитного
атомы N п
О-
(134)
(36)
(137)
тионитроксиды (питрсульфищ’>)СПОСОбЯЬ1Х к обРазоваН1"° димеров,
"О генерировать лишь теЙпп ’ Г°раЗЯ0 Менес Ус™йчивы. Их мож-
б"с (диалкиламино) дисул^ид^у^в^
N-S-S-N
Д или hv
2( ]N—S.
(37)
В то же время известии „
™па (»41), имеющие рХопо'100"™1’"0
о,г р в°Д°Родньш атом
256
устойчивые пптрбкепды
[134]. Стабильность по-
дойного радикала определяется двумя
„ими особенностями, препятствующим,фактоРами: простоя»
... диспропорционирования и тРЫ 1 ” бпмолекуляпнг.С раНсгвен-
ю орбитали атома Ха™00р,,е,Паа11я
^ектрон, не вполне соответствует той АОдеРЛащей иесиа» Н
легкое расщепление. Идеализир0Ваш“аяПра ко™рой np0HtA Р ₽Й
гоприятстпрощая гомолитическому Х1н?М«Трия «олекуты L
показана в формуле (142) [140] Р ВУ ₽'в°А°РОдного '
(141)
(142)
В бициклическом нитроксиде (139) связь С—Н в голове мостика
располагается практически под прямым углом к орбитали с
неспаренным электроном. На устойчивость блокированных диал-
кплнитроксидов могут влиять и другие факторы, о чем свидетель-
ствует легкость, с которой связь С—N может претерпевать гомо-
литический разрыв в соединениях (143) [141] и (144) [142].
В первом случае первичными продуктами распада являются
2-метил-2-нитрозопропан и гндроксиламин (145). Их образование
может быть объяснено относительно более высокой стабильно-
стью выбрасываемого аллильного радикала (имеет место процесс,
обратный улавливанию спина).
(143) (144) (145)
В случае диарил- и арилалкилиитроксидов, изученных
стером с сотр. [143], неустойчивость определяется_ АРУ Р1ЛЬ.
вой. В результате перекрывания между нитроксид_ Ф
пой группами, носящего характер сопряжения, пр ’ чесК|)Х
кализация спиновой плотности, как изоораж „„>,,auHH пеали-
формах соединения (146). Вследствие этой дес екаюЩей через
зуется другом тип бимолекулярной реакции Р нЫЙ амИн
®““еР (147). Затем может происходить Рас" ц ,га0й-полоЖения
U48) и п-хинониминоксид (149). Блокирование фаКТ0.
или введение орто-заместителей (или налит тся „з состоя-
РОв), в результате чего иптроксндная гРУ”"а °“Вподобных нитро-
,я сопряжения, может повышать стабн.
хси.тов.
® Эек, 106
257
Многие спектроскопические и стереохимические особенности
нитрокспдов согласуются с общей картиной молекулярных орбита-
лей пптроксидной группы. Так, валентные колебания связи N—О
характеризуются поглощением примерно при 1350 см-', что сог-
ласуется с кратностью связи, равной 1,5 [144]. Две полосы при
230 нм (е « 2500) и 410—250 нм (е » 10) в УФ-спектре на осно-
вании характера смещения этих полос под влиянием растворителя
были отнесены соответственно к л—л*- и п—л*-переходам [145],
Поглощение при 410—450 нм является причиной характерной
оранжево-красной окраски многих нитроксидов. В ряде случаев
при исследовании нитроксидов методом рентгеновской дифракции
были выявлены две важные особенности [146]. Было найдено,
что длина связи N—О зависит от конкретной структуры и состав-
ляет около 126—131 пм. Можно считать, что это согласуется
с двухцеитровым трехэлектронным характером связи. Кроме того,
из большинства рентгеновских спектров следует, что связь N—О
не лежит в плоскости CNC, а образует с ней угол 20—30°, что
указывает на пирамидальную геометрию при атоме азота. Хотя
эти параметры и относятся к твердому состоянию, есть основания
считать, что в растворе сохраняется та же геометрия [140]; ряд
расчетов ab initio [147] в большей или меньшей степени согласу-
ется с таким толкованием. Считают, однако, что инверсия при ато-
ме азота — очень быстрый процесс, причем энергия неплоской
формы лишь на ~4—8 кДж/моль ниже энергии плоской конфор-
мации. Как отмечает Рассат [148], нитроксиды во многих отноше-
ниях сходны с карбонильной группой, находящейся в п—л‘-воз-
бужденном состоянии. Действительно, эта аналогия вполне прием-
лема с точки зрения картины молекулярных орбиталей. Из нее
следует, что единственным различием между ними является то,
что в ннтроксиде в основном состоянии обе пары несвязывающих
ектронов расположены при атоме кислорода. В соответствии с
э ои частичной аналогией находится тот факт, что некоторые из
ппглЧеНИЫХ 0ПтичесК|1 активных нитроксидов в спектрах Kpyroj
вою дихроизма обнаруживают эффект Коттона (отвечающий
,.лтД°Л°Сг П0ГЛ01йения с низкой экстинкцией) с тем зна-
, рыи был предсказан с помощью хорошо известного К
258
существенно уточненного правила октантов для карбонильной
группы [149].
Эти п другие спектральные характеристики ннтроксндов важ-
ны, однако наиболее информативной спектральной характеристи-
кой ннтроксндов является спектр ЭПР, который в своем простей-
шей форме представляет триплет сигналов с интенсивностями
1:1:1- Он возникает в результате сверхтонкого взаимодействия
неспаренного электрона с ядром азота l4N (/=±1). Обычно
константа сверхтонкого взаимодействия (вк) равна 1,4 —1,7 мГ
для дналкилнитроксидов и 0,9—1,1 мТ для диарилнитроксидов.
В случае иитроксидов, атом азота которых расположен рядом с
ненасыщенной группой, таких, как ацил- и имниилинтроксиды,
наблюдаются и более низкие значения константы сверхтонкого
взаимодействия. Другие виды сверхтонкого расщепления иа ядрах
15N и 13С (при их естественном изотопном содержании) и на уда-
ленных ядрах ‘Н наблюдаются при большом усилении и высоком
разрешении. Как на а^, так и на g-фактор (значение которого
обычно составляет около 2,0060) влияет полярность растворителя,
причем с возрастанием полярности растворителя увеличиваются
значения ан и понижаются значения g-фактора. Параметр а>. про-
порционален Z-фактору Косовера [150]. Это становится понятным,
если учесть, что полярные растворители стабилизуют днполяриую
форму (132), увеличивая тем самым спиновую плотность иа атоме
азота. Мнения, однако, значительно расходятся по вопросу о рас-
пределении относительной спиновой плотности на атомах азота и
кислорода.
6.4.5.3. Реакции
Реакции иитроксидов могут быть разбиты иа две группы в со-
ответствии с тем, затрагивает ли реакция радикальный центр или
нет. К наиболее важным реакциям, не затрагивающим радикаль-
ный центр, относятся реакции 2,2,6,6-тетраметпл-4-оксопиперндвн-
1-оксила (150), химия которого разрабатывалась, главным обра-
зом, Розанцевым (Москва) и Рассатом (Гренобль).
С этим соединением можно провести практически любой тип
реакций, в которые обычно вступает кетогруппа, в результате
чего были получены его многочисленные функциональные произ^-
водиые. Успешное осуществление этих реакций в значительной
степени способствовало расширению возможностей исследовате-
лей, занятых изучением спиновой метки (см. ниже). Небольшая
часть этих реакций приведена на схеме 38 [151].
Одиако в условиях некоторых реакций происходит исчезнове-
ние радикального центра. Примеры двух подобных реакции, а
именно бимолекулярное диспропорционирование и разрыв связи
С—N, уже приводились. К другим реакциям такого типа относят-
ся: присоединение радикала к атому кислорода, реакция с кисло-
тами, одиоэлектроиноеокисление и восстановление ’’
Не слишком неожиданным является тот факт, что н р
9* 359
захватывают другие свободные радикалы, образуя трнзамещенные
гндроксилампны (151). Эта реакция не была изучена очень под-
робно. Примером является образование соединения (145), проис-
ходящее вслед за термическим разложением (143) [14)}. Несом-
ненно, что такие процессы рекомбинации исполыуют для стабили-
зации полимеров путем добавок нитроксидов, действующих как
агенты обрыва цепи. Восстановление пптроксндных радикалов Д°
днзамещенных гидроксилампиов (152) является относительно
легко протекающей реакцией, которую можно провести с помощь10
ряда мягких восстанавливающих агентов. Наиболее удобным 113
260
них следует, видимо, считать фенилгидразин [391. Известны так
я(е методы обратного превращения нитроксидов во вторичные
амины.
+ е-, + Н+ л
r2n ОН > R2N—о . ----> r2n=
I <“>
1 I
(39)
R2N—OR R2N—ОН
(151) (154)
Как можно было предполагать, нитроксиды в п->- .-^-возбуж-
денном состоянии гораздо легче отрывают атом водорода чем в
основном состоянии [152]. Эта способность нитроксидов исполь-
зована в интересном примере фотолиза нптроксида стероидной
природы (155), в результате которого образовались соединения
(156) и (157) с функциональными группами, удаленными от ради-
кального центра.
(156) (157)
Нитроксиды можно также окислять, например, с помощью
хлора, брома и л-хлорпербензопноп кислоты. При этом образуется
соль иммопнйокспда (153), которая, будучи генерирована in
situ, способна вызывать окисление вторичных спиртов до кетонов
[154]. Хотя растворы нитроксидов в органических растворителях
можно промывать холодными разбавленными водными раствора-
мч кислот без значительной потери парамагнитных свойств,
нитроксиды являются слабыми основаниями п способны иротони-
роваться (и образовывать комплексы с кислотами Льюиса), пре-
вращаясь в катион-радикалы (154), которые диспропорционируют
До (153) н соли гидроксиламмоппя (158) [155].
R2NH-OH (158)
261
ti.4.5.4. Спиновая ловушка и спиновая метка
Как уже было отмечено, два препаративных метода получен,,
ннтроксндов заключаются во взаимодействий алкильных ради1!И’
лов с нитронами пли пнтрозосоедииеипя.мп. Однако эти реакиа’
паипп гораздо более широкое применение в химии свободны,
радикалов вообще вследствие тон центральной роли, которую 0“„
играют в так называемом методе спиновой ловушки [156] Этот
метод, открытый в 1968 г. тремя группами исследователей [1571
является новым способом изучения многих свободнорадикальных
реакций. В основе метода лежит легкость, с которой соответствую-
щим образом замещенные нитроны п трет-ннтрозосоедппения, по-
добные (159) и 2-метил-2-нптрозопропану, захватывают коротко^
живущие радикалы самых различных типов, образуя значитель-
но более долгоживущие нптроксиды. Это явление позволяет
регистрировать спектры ЭПР вновь образующихся иитроксидов-
на основе полученных данных, включающих значение g-фактора и
констант сверхтонкого взаимодействия (в частности, aN и ан),
зачастую можно идентифицировать захваченный радикал. Этот
метод может быть использован во многих других случаях (напри-
мер, при проведении химических, фотохимических, электрохими-
ческих реакций и радиолиза) для обнаружения свободных ради-
калов и установления их структуры. Кроме того, дальнейшее усо-
вершенствование этого метода связано с использованием новых
нитронов, таких, как (160; R = трет-Ви), которые интронным кон-
цом улавливают радикалы с радикальным центром на С-атоме и
служат донором фенольного водородного атома при взаимодейст-
вии с О-радикалами [158], а также с использованием [2Нз]-2-ме-
тил-2-нитрозопропана, который сводит к минимуму сверхтонкое
расщепление в трет-бутилыюй группе в нитроксидах, полученных
па его основе [159]. Работающим в этой области следует, однако,
помнить сделанные в свое время Форрестером и Хэпберном [160]
предостережения относительно того, что сигналы, соответствую-
щие нптроксидам, могут наблюдаться в спектре ЭПР и в тех слу-
чаях, когда свободные радикалы и не являются интермедиатами
в реакции.
PhCH—N—Зи-трет
О'
(159)
—CH=N—Bu-трет
О'
(160)
одной из главных научных дисциплин,
влияние оказало введение в практику
Вне всякого сомнения,
на которую благотворное
КПП-1ЛТ1ЛП г, •• опадали DDCAClinv о nyun-------/
Эта - 111111 Н1,тР°ксидов, является молекулярная биология.
finn^un”10 С пР1,менеи||ем метода спиновой метки для изучения
ческих структур на молекулярном уровне. С момента по-
262
зления первой публикации Стоуна и др. [161] в )965
подобных исследовании колоссально расширилась. Теория этого
метода п его приложения неоднократно рассматривались (162?
В рамках этой короткой главы невозможно хотя бы поверх ,осто
по охватить этот эффективным и привлекательный метод кото
рый за последние годы позволил получить чрезвычайно полезную
информацию по таким фундаментальным вопросам, как ctdvktv-
„ функция некоторых ферментов и молекулярное движение’в
биологических мембранах. В сущности, метод спиновой метки
основан па том* что нитроксид (иногда называемый не спиновой
меткой, а спиновым зондом), который включен в биологическую
систему с помощью ковалентного или нековалентного взаимодей-
ствия, может посредством спектра ЭПР дать значительную ин-
формацию о своем окружении. Это позволяет получить сведения
о нескольких параметрах, поскольку значения On и g-фактора для
ннтрокспда анизотропны в том смысле, что они меняются с из-
менением ориентации радикала относительно приложенного поля
и с изменением полярности растворителя. На основании значе-
ний ан, g-фактора и изменения формы сигналов в спектре ЭПР
можно сделать вывод о локальной полярности, вязкости н ско-
рости молекулярного вращения. В свою очередь, эта информация
в сочетании с данными о структуре включенного в систему нит-
роксида может быть использована для оценки таких параметров
фермента, как полярность центров связывания, геометрия актив-
ного центра, времена вращательной корреляции н динамические
свойства биологических мембран.
Совсем недавно был разработан другой столь же мощный
метод, связанный с нитроксидными спиновыми метками. Он ос-
нован на использовании влияния, которое парамагнитный нит-
роксид оказывает на ЯМР-спектр биологической структуры. В
данном случае радикал уменьшает времена релаксации и тем
самым уширяет линии резонанса близко расположенных магнит-
ных ядер. Результаты измерения времен спин-решеточной и спнн-
спииовой релаксации в сочетании с измерением времен корре-
ляции позволяет рассчитывать расстояние, которое отделяет не-
спаренный электрон от возбуждаемого ядра. На этом основе
возникает эффективный способ исследования геометрии центра
связывания фермента.
ЛИТЕРАТУРА
’ A A. S. Smith, in «Open-chain Nitorgen Compounds», Benjamin. New York,
1966, vol. II. chapter 8; B. Zeeh and H. Metzger, in «Methoden der Оrgan-
schen Chemie (Houben-Weyl)», Georg Thieme Verlag Stuiigart 1971,
™1. Ю/l, pp. 1091—1279; S. R Sandler and W.Karo in «Organ.e Fl nctic> a!
Group Preparations», vol. 12-III of «Organic Chemistry», ed. A. T. Blomqm.t
о nn „ Wasserman, Academic, New York, 1972. v и Clark
2- E. Bamberger, Ber., 1894, 27, 1548; R. Bonnett, R.F- C.Broj:-n
1- 0. Sutherland and A Todd, J. Client. Soc., 19o9, 2094,
F. Pazos, J. Org. Chem., 1969, 34, 2269.
263
25.
26.
27.
28.
29.
30.
34.
85.
36.
37.
38
39
264
. ! п ytcnmm Annalen, 1957, 606, 24; К. Brand and J c,„.
3. i. Hor™' OT.-‘ ML Scheinbaum, J. Org. Chem, 1970, 35, 279(1 “''
Ber., 1922. 55. 87o AL J. Amer. Chem. Soc., 1951 73
4. ' Soc chim France, 1966, 1858, 1867. °'’ 5675.
5. /Л Feue^R- S. Bartlett, B. F. Vincent, and R. S. Anderson, J. Org. Chem
1965, 30, 2880. L Baumgarten. J. Amer. Chem. Soc toe,
6- №,"1139; A Ca№r A/?: Forrester, and S. P. Hepburn, Org. Syn^
7 ^p'd Haworth. and A. Lapworth, J. Chem. Soc., 1921, 768
8 H Feuer B. F. Vincent, and R. S. Bartlett, J. Org Chem 1965, 30, 2877
a J? F Borch M D- Bernstein, and H. D. Durst, J. Amer. Chem. Soc. 1971
93, 2897; H'СК House and L. F. Lee, J. Org. Chem., 1976, 41, 863; С././аЦ’
10. HynGheRW<e^Jr., R. C. McLane, and С. I. Phillips, Tetrahedron Letters, 197s
233.
II ll? D. Emmons. J. Amer. Chem. Soc., 1957, 79, 5739.
12 IV Traube and A. P. Schulz. Ber., 1923, 56, 1856.
13. L. IV. Jones and M. C. Sneed, J. Amer. Chem. Soc., 1917, 39, 674; A. C. Cope
and A. C. Haven. Jr., ibid., 1950, 72, 4896.
14 A H Beckett K. Haya, G. R. lanes, and P. H. Morgan, Tetrahedron, 1975
' 31, 1531.
15. P. Truitt, L. M. Long, and M. Mattison, J. Amer. Chem. Soc., 1948, 70, 2829;
И7. Theilacker and K. Ebke, Angew. Chem., 1956, 68, 303; L. A. Paquette
J. Amer. Chem. Soc., 1962, 84, 4987.
16. C. L. Bumgardner and G. L. Lilly, Chem. and Ind. (London), 1962, 559.
17. 0. L. Brady and F. H. Pdakin, J. Chem. Soc., 1930, 226.
18. L. A. Carpino, C. A. Giza, ond B. A. Carpino, J. Amer. Chem. Soc., 1959, 81,
955; T. Sheradsky, J. Heterocyclic Chem., 1967, 4, 413.
19. B. J. R. Nicolaus, G. Pagani, and E. Tesla, Helv. Chim. Acta, 1962, 45, 358.
20. 0. Zinner, Arch. Pharm. (Weinheim), 1970, 303, 317; У. Tamura, J. Minami-
kawa, K. Sumoto. S. Fujii, and M. Ikeda, J. Org. Chem., 1973, 38, 1239.
21. A. F. McKay, D. L. Carmaise, G. Y. Paris and S. - Gelbtum, Canad. J. Chem.,
1960, 38, 349.
22. IV. B, Lutz, J. Org. Chem., 1971, 36, 3835; E. Grochowski and J. Jurczak,
Synthesis, 1976, 682.
23. S. Wawzonek and J. V. Kempf, Org. Prep. Proced. Internal., 1972, 4, 135.
24. G. E. Utzinger and F. A. Regenass, Helv. Chim. Ac(a, 1954, 37, 1885.
n J. T liesing and H. Mayer, Chem. Ber. 1956, 89, 2159.
0. Exner, Coll. Czech. Chem. Comm., 1955, 20, 202.
O. L. Lebedeo and S. N. Kazarnovskii, Trudy Khim. i khim. Tekhnol, 1959,
2, 649 (Chem. Abs., 1962, 56, 15479) [О. Л. Лебедев, С. H. Казарновский-
1рулы по химии и хим. технологии, 1959, 2, 649].
u (i,C?peJand Е- R- Trumbull, Org. Reactions, 1960, 11, 317.
H. Wieland and К Roth, Ber., 1920, 53, 210
G. O. Buckley, J. Chem. Soc., 1947, 1492; S. IFawzonefc and J. V. Kempf,
J. Org. Chem., 1973, 38, 2763.
31 S’ T- Mai°r and E. E. Fleck, J. Amer. Chem. Soc., 1928, 50, 1479; E. Testa.
766 ' ° Rlc°laus. L. Mariani, and G. Pagani, Helv. Chim. Acta, 1963, 46.
A' $re“lz,ktamP and p- Messinger, Chem. Ber., 1967, 100, 3463.
Chem E- Gelblum, and W. G. Hodgson, J. Amer.
and A He,'86, 639, 646; A K. Hoffmann, A. M. Feldman, E. Gelblum
and A. Henderson Org. Synth, 1968, 48, 62.
A PFmi a'ld M K- El'Hawari, Heterocycles, 1974, 2, 669
J Flies?, g and W ! HhThom^. J- Chem. Soc., 1965, 1224.
! Hain!r8nr,ddAVM,rrfnbc''^ Cllcm' Ber- 1959> 92• 1748.
F г о У Л- Macaluso, Chem. Rev., 1964 64 473
T D Leland1'! T’wu' 'КЧга''^™< 1964, 20, 131.
e and J. F. W. Keana, J. Org. Chem, 1975, 40, 3145.
32.
33.
•10.
41.
42.
43.
44.
45.
it? Rundel, in «Methoden der Organischen Chemie .
Thieme Verlag, Stuttgart, 1968, vol. 10/4, pp 449_4™иЬ?п'УеУ1)», Georg
r Hamer, in «1,2-Cycloaddition Reactions» ed G A ’ nt’ S ^uller and
New York, 1967, chapter 11, p. 103. ' ea' A' 0Iah, Interscience,
r. A Tolstikov, U. M. lemitev, V. P. Jurjev F в
^7°2U’6T33rahedrOn LCHCrS’ ,97'' 2807; Л P’ S'Chimann
Dd \ ВВУоЪа'1. /5%.
^7б'б0бтРЬе11’ N' L' Th°mpSOn‘ and B- ^mngs, J C. S. Perkin^H,
c. Belzecki and D. Mostowicz, J. Org. Chem 1975 40 307c. m n, ,
II /. Moretti, G. Torre, G. D. Andreotti, G. Boeelli and’p s^abotM
J c. s. Chem. Comm., 1976, 60. ’ ' bgarabotto,
N. R. Beutoniere and G. W. Griffin, in «Organic Photochemistry
ed. 0. L. Chapman, Marcel Dekker, New York, 1973 vol 3 n m-’
J. S. Splitter T.-M. Su H. Ono, and M. Calvin, J. Amer.’Chem Soc 1971
93, 4075; M. Lamchen, in «Mechanisms of Molecular Migrations»
ed. B. S. Thyagarajan, Interscience, New York, 1968 vol Ip]
I. В forgo, D. R. Boyd, R. M. Campbell, and D. C.’ Neill J C S Chem
Comm., 1976, 162. ’
46. T. Hiyama, H. Taguchi, S. Fujita, and H. Nozaki, Bull. Chem. Soc Japan
1972, 45, 1863. ' ’
47. J. P. Alazard, B. Khemis, and X. Lusinchi, Tetrahedron Letters, 1972, 4795.
48. L. L. Muller and J. Hamer, in «1,2-Cycloaddition Reactions», ed. G. A. Olah,
Interscience, New York, 1967, chapter III, p. 257.
49 D. A. Barr and R. N. Haszeldine, J. Chem. Soc., 1955, 1881, 2532; ibid
1956, 3416.
50. J. E. Baldwin, А. K. Bhatnager, S. C. Choi, and T. J. Shortridge, J. Amer.
Chem. Soc., 1971, 93, 4082.
51. K. Wieser and A. Berndt, Angew Client. Internet. Edn., 1975, 14, 70.
52. У. Takeuchi and F. Furusaki, Adv. Heterocyclic Cliem., 1977, 21, 207; см.
также D. St. C. Black, R. F. Crozier, and V. C. Davis, Synthesis, 1975, 205.
53. В. M. Benjamin and C. J. Collins, J. Amer. Chem. Soc., 1973, 95, 6145.
54. R. Huisgen, J. Org. Chem., 1968, 33, 2291.
55. N. A. LeBel and E. Banned, J. Amer. Chem. Soc., 1970, 92, 5278.
56. V. A. Tartakovskii, Z. Ta. Lapshina, I. A Savost'yanova, and S. S. Novi-
kov, Zltur. Org. Khim., 1968, 4, 236 (Chem. Abs., 1968, 68,9o734)
[В. А. Тартаковский, 3. Я. Лапшина, И. А. Савостьянова, С. С. пови-
ков —ЖОХ, 1968, 4, 236]. „ , „
57. Н. King, J. Chem. Soc., 1942, 432; F. G. Riddell and D. A. R. Williams,
62.
Tetrahedron, 1974, 30, 1083.
58. G. Kresze and J. Firl, Forschr. Chem. I---........ F -mh
and G. Kresze, Angew. Chem. Internal. Edn., 1976, 15, 7oU.
59. G. Kresze and G. Schulz, Tetrahedron, 1961, 12, 7.
60. P. Burns and Ж A. Waters, J. Chem. Soc. (C), 1969, 27.
61. U. M. Kempe, T. K. Das Gupta, K. Blatt, P. Gygax DnPel,xn^.da о Wx
Moser, Helv. Chim. Acta, 1972. 55, 2187; T K. Das Gupta, D.
U. M. Kempe, and A. Eschenmoser, ibid., p. 2198, P. JS ,
Gupta, and A. Eschentnoser, ibid., p. 2205. c„„rtrosconv» ed E. F. Moo-
/. O. Sutherland, in «Annual Reports on NMR Spectroscopy»,
ney, Academic, London, 1971, vol. 4, p. 71. „ ,дц. цу /у ptnk,
L. Pedersen and K. Morokunia, J. Chem. ‘B'S-. 1 c ' u/o’/b / 11. Tel, and
D. C. Pan, and L. C. Allen, ibid., R^oni W':f. Hehre, and
I. G. Csizmadia, Canad. J. Chem., 1973 51, 24_ „г гаг(ц /. 0. Csizma-
f. A. Pople, J. Amer. Chem Soc., 1972 94 237 F^Bern , jb.
dia, A. Mangini, H. B. Schlegel, M. H. Whangbo, and 5. / ,
97. 2209. „.„m Comm 1968. 1403;
f- G. Riddell .1. M. I.ehn, and Ch ’
D. L. Griffith and B. L. Olson, ibid., 1968, 1682.
Forsch.,< 1969, 11, 245; H. Nitsch
63.
64.
266
Letters, 1975,
Haynes, and
1 Fsclienruoser, Helv. Chim Acta, i960, 52, 1823.
65 . K. Mid er ,976 4) ,7J8
67 ?' C°RiddellMand E. S. Turner, J. C. S. Perkin II, в печати
68 R. Л L Aines, Л- R- Kalr!lzk‘J' S Sab0- and ' L SParrow- J- C. S. Perki,, ц
so л97ЯялИп M. R. Todd, E. G. Riddell, and E. S. Turner, частное сообше,,,,
70 F G. Riddell. P. Murray-Rust, and J. Murray-Rust, Tetrahedron 1974. 3q
7, l^oft and В Jerslev. Acta Chem. Scand., 1967. 21, 1383.
79 M Davies and .V. .-1. Spiers. J. Chem. Soc., 1959, 3971.
73 J Mollin F Kasparek, and 1. Lasovsky, Chem. Zvesti, 1975 29, 39.
74 H J Brass J О Edwards, and Л'. J. Fina. J. C S. Perkin II, 1972, 726
75 R F. Hudson, in «Chemical Reactivity and Reaction Paths», ed G. Klopman
Wiley, New York, 1974, chapter 5; N. I. Fina and I. O. Edwards, Internal, j'.
Chem Kinetics. 1973, 5, I. „ „
76 J F Liebrrian and R. M. Pollack, J. Org. Chem., 1973, 38, 3444.
77 . Al. Cocivera, A. Effio, H. E. Chen, and S. Vaisli, J. Amer. Chem. Soc., 1976
98, 7362; M. Coclvera and K- W. Woo, ibid., p. 7366; M. Cocivera and 'A.
fio ibid., p. 737L
78 A Belly, R. Jacquler, F. Petrus, and J. Verducci, Bull Soc. chim. France 1972
330- A. Belly, F. Petrus, and I. Verducchi, ibid., 1973, 1395.
79 K. R- Fountain. R. Erwin, 7. Early, and H. Kehl, Tetrahedron
’ 3027.
80 . G. Zinner and W. Kliegel, Chem. Ber.. 1966, 99, 895.
81 . G. Zinner. IV'. Kliegel, and W. Ritter, Chem. Ber., 1966, 99, 1285.
82 . /. Stieglitz and P. N. Leech, J. Amer. Chem. Soc., 1914, 36, 272.
83. №. P Jencks, J. Amer. Chem. Soc., 1958, 80, 4581, 4585.
84. M. J. Perkins and P. Ward, J. C. S. Chem Comm., 1973, 883.
85. H. O. House, D. T. Manning, D. G. Melillo, L. F. Lee, O. R.
В. E. Wilkes, J. Org. Chem., 1976, 41, 855.
86. G. Zinner, Arch. Pharm. (Weinheim). 1970, 303, 488.
H. Metzger and H. Meier, in «Methoden der Organischen Chemie (Houben*
Weyl)», Georg Thieme Verlag, Stuttgart, 1971, band 10/1, pp. 960—968.
/. A. Maassen and Th. /. de Boer, Rec. Trav. chim., 1971, 90, 373.
E. C. Taylor and F. Yoneda, Chem Comm., 1967, 199.
W. B. Motherwell and I. S. Roberts. J. C. S. Chem. Comm., 1972, 328;
H. O. House and L. F. Lee, J. Org. Chem., 1976, 41, 863.
H. G. Padeken, O. von Schickh, and A. Segnitz, in «Methoden der Organischen
Chemie (Houben-Weyl)», Georg Thieme Verlag, Stuttgart, 1971, band 10/1»
p. 106; см. также co 87, pp. 848—849.
92. F. A. Carey and L. J. Hayes, J. Org. Chem. 1973, 38, 3107; S. J. Brois,
J. Amer. Chem. Soc., 1970, 92, 1079.
93. P. G. Gassman and G. D. Hartman, J. C. S. Chem Comm., 1972, 853.
94. G. Kresze and O. Korpiun, Tetrahedron, 1966 22, 2493
95. R. Hmsgen, H. Hauck, R. Grashey, and H. Seidl Chem. Ber., 1968, 101,
2568.
96. M. Lafnchen, in «Mechanisms of Molecular Migrations» ed B. S. Thyagara-
Jan, Intcrscience, New York, 1968 vol I n 1
т and c- K- Ingold, Quart. Rev., 1952, 6, 34.
an т c,kamcl ?’ K- ^‘lo- and T- 0Ma- J- Amer. Chem. Soc., 1975, 97, 7184..
inn D , n,!'1 °-Snto,",'cZe’Te,ral’ef,ron Letters 1971 645. ,
100. P. Boudfouk and R. West, J. Amer. Chem. Soc., (971 93, 5901; R. West and
fdyB°bid/OI<k’703d'’ 1973' 95’ 39831 3987; CM1 ТаКЖе S1 ™1е and D' °' SM“'
\0\. H Freytag in «Methoden der Organischen Chemie (Houben-Weyl)», Georg
StUttsart’ 19581 band 4/2. P- 190; D- Swern, Chem. Rev^
; м i 1 'i4; г гетеРо>1|,1'лическнх N-оксндах ем: Л. R. Katritzky and
London "fev' <<ChemistrY of lhe Heterocyclic N-oxides», Academic Press,
102. Л. A. Oswald and D. L. Guertin, J, Org. Chem., 1963, 28, 651.
266
87.
88.
89.
90.
91.
103.
104.
105.
106.
107.
108.
109.
R Koster and Y. Morita, Annalen, 1967, ’704 '70^
H. C. Hedgecock, Jr., J. Org. Chem., 1975, 40, 1776. ’
1 CN Х Л rVVX*' indTM0^’^11’ 2 3«3-
; B. Henbest and MJ. W^tratjord, J. Chem. Soe 1%4 7'11 ’958’ 4 5 *°57'
P32NA gVCSe" Md" 1938' *®’ 48 i^'m. Abslr., 1938,
m’ IP. Lister and L E. Sutton, Trans. Faraday Soc 19ЭД v. ao-
G Tsoucaris, J. Chim. phys., 1961, 58, 619. " 3 ' 3j’ 49j'
F Howard, Jr. and №. F. Olszewski, .1. Amer. Chem Snr lorn 01 ,
fi Hamana, J Pharm. Soc. Japan, 1955, 75, 121 (Che™. Al ® и я 171
d Roster and 1. Morita, Annalen, 1967, 704 70' 6 № 7, i 8 7^'
llb H. C. Hedgecock, Jr., J. Org. Chem., 1975, 40, 1776. ' W' КиЬМа and
]12. №. Feely, W L. Lehn and V Boekelheide, J. Org. Chem. 1975 22 m-
113 G. A. Russell and G. J. Mtkol, m «Mechanisms of Molecular Mia oh °
1 ed B. S. Thyagarajan, Interscience, New York, 1968 vol m Г °П5>1
F. Kajfez, D. Kolbah, H. Hoeman, and M. Stromflr, Te?rahed?on Letters
114 A. Ahotid, A. Cave, C. Kan-Fan, H.-P, Husson, J. de Rostolan and P Pnfi^r
tfWr N-Lanslois’F- b
115. V- J- Traynelis, Л. I. Oallagher, and R. F. Martella, J. Org. Chem., 1961 26,
4365. ’ ’
116. R- A. №. Johnstone, in «Mechanisms of Molecular Migrations» ed В S Thva-
garajan, Interscience, New York, 1969, vol. 2, p. 249. ’ ’
117. A. R. Lepley, P. M. Cook, and G. F. Willard, J. Amer. Chem Soc 1970 92
1101; J. P. Lorand, R. №. Grant, P. A. Samuel, E. M. O’Connell, J. Zaro
!. Pilotte, and R. IF. Wallace, J. Org. Chem., 1973, 38, 1813.
118. M. Moriwaki, Y. Yamamoto, J. Oda, and Y. Inouye, J. Org. Chem., 1976, 41.
300; У. Yamamoto, J. Oda, and Y. Inonye, ibid., p. 303.
119. V. Rantenstrauch, Helv. Chim. Acta, 1973, 56, 2492.
120. A. C. Cope and N. A. LeBel, J. Amer. Chem. Soc., i960, 82, 4656.
121. R. G. Laughlin, J. Amer. Chem. Soc., 1973, 95, 3295.
122. A. R. Forrester, J. M. Hay, and R. H. Thomson, «Organic Chemistry oi
Stable Free Radicals», Academic, London, 1968, chapter 5; E. G. Rozantsev,
«Free Nitroxyl Radicals», Plenum, New York, 1970; H. G. Aurich and
№. Weiss, Topics Current Chem., 1975, 59, 65; E. G. Rozantsev and V.D.Shol-
le. Synthesis, 1971, 190, 401.
123. P. Piloty and B. G. Schwerin, Ber., 1901, 34, 1870, 2354.
124. H. Wieland and F. Kogi, Ber., 1922, 55, 1798.
125. O. L. Lebedev and S. N. Kazarnovskii, Trudy Khim. i khim. Tekhnol., 1959,
126.
127.
' 128.
129.
130.
J3I.
2, 649 [О. л. Лебедев, С. H. Казарновский — Труды по химии и хим. техно-
логин, 1959, 2, 649].
Е. J. Rauckman, 0. М. Rosen, and М. В. Abou-Donia, Synth. Comm., 197о,
5, 409; G. Sosnovsky and M. Konieczny, Z. Naturlorsch., 1976, 31b, 1376.
J. F. №. Keana S. B. Keana, and D. Beetham, J. Amer. Chem. Soc., 1967, 89,
3055. PJ ori,
Y. Brunei, H. Lemaire, and A. Rassat, Bull. Soc. chim. France, 1964, 1895.
T. D. Lee and J. F. №. Keana, J. Org. Chem., 1976, 41, 3237.
A. Rassal and P. Rey, Chem. Comm., 1971, II6I. ,Q7,
J- A. Maassen fl. Hiltenhausen, and Th. J. de Boer, Tetrahedron \/nrrm
3213; Th. A. J. №. Wafer, A. Mackor, Th. J. de Boer, and J. D. №. van Voorst.
Tetrahedron, 1967, 23, 4021. _ . r. Comm
D. M. Hawley, J. S. Roberts, G. Ferguson, and A. L. Porte, Client.
1967, 942. „ 107„ i9R7
IF. B. Motherwell and J. S. Roberts, Tetrahedron Letters P972,^28
Jwamiu a and N. Itiartioto, Bull. Cheni. Soc. •’< P -
Л F. I.iniiett, J. Amer. Chem. Soc., 1961, 83, 2643. 7J g3 ^55-.
D. 1. Bowman, T. Gillan, K. U. Ingold, J. Amer. Chem. Soc., IJ/^ ,
K. Adamic, D. F. Bowman, T. Gillan, and K. U. Ingold, id , 1
ino Hupeyre and A. Rassat, J. Amer. Chem. Soc., . • 27 392.
138- -4. Сарино!,/ в Chion, and J. Lnjzerowicz, Acta Cryst., 1971, в L
’ 267
132.
133.
134.
135.
136.
139. Г С. Dane,, and O. D. Ne^irk J. An,er. Chem. Soc., 1976, 98, 516; g
r'c^iljeFin'Kofin '» SUreoctanisTry», ed. N L. Allinger and E.L Eii.
IS **• *.%
Ио О IV.CMamde: 4072.'
143 A R Forrester and S. P. Hepburn L Chem. Soc. (C)- l97°. I277; ibid., 1971
3322; A. Calder, A. R. Forrester and S P Hepburn, J. C. S. Perkm 1,1973,456
г Morat and A Rassal, Tetrahedron, 1972, 28, 73o.
11 P Briere H. Lemaire, and A. Rassat. Tetrahedron Letters, 1964, 1775.
46 ! Laizeroaicz-Bonneteau. in «Spin Label,ng. Theory and Applications»
ed L J. Berliner, Academic, New York, 1976. p. 239.
147 A U7 Salotto and L. Burnelle, J. Chem. Phys., 1970, 53, 333; T. D. Davis
RE Chrisloffersen, and G. M. Maggiora, J. Amer. Chem. Soc.. 1975, 97, 1347
148. A. Rassal, Colloqucs Internationau.x du Centro National de la Recherche Sci-
entifique. 1966, no. 164, p. 427.
149. /. S. Roberts and C. Thomson, J. C. S. Perkin II, 1972, 2129; R. Ramasseul,
A Rassat, and P. Rey, Tetrahedron, 1974, 30, 265; A. Collet, J. Jacques,
В Chion. and J. Lajzerowicz, ibid., 1975, 31, 2243.
150. R. Briere, H. Lemaire, and A. Rassal, Bull. Soc. chim. France, 1965, 3273.
151. B. J. Gaffney, in «Spin Labeling, Theory and Applications», ed. L. J. Berliner,
Academic, New York, 1976, p. 183.
152. J. F. W. Keana, R. J. Dinerstein, and F. Bailis, J. Org. Chem., 1971, 36, 209.
153. J. A. Nelson. S'. Chon, and T. A. Spencer, J. Amer. Chem. Soc., 1975, 97, 648.
154. J. A. Celia, J. A. Kelley, and E. F. Kenehan, J. Org. Chem, 1975, 40, 1860;
B. Ganem, ibid . p. 1998.
155. V. Malatesfa and K. U. Ingold, J. Amer. Chem. Soc., 1973, 95, 6404.
156. Af. /. Perkins, in «Essays on Free Radical Chemistry», Chemical Society Pub-
lication, 1970, no. 24, p. 97; E. G. Janzen, Accounts Chem. Res., 1971, 4, 31.
157. C. Lagercranfz and S. Forschult, Nature, 1968, 218, 1247; G. R. Chalfont,
M J. Perkins, and A. Horsfield, J. Amer. Chem.- Soc., 1968, 90, 7141;
E. G. Janzen and B. J. Blackburn, ibid., p. 5909.
158. J. G. Pacifici and H. L. Browning, Jr., J. Amer. Chem. Soc., 1970, 92, 5231.
159. R. J. Holman and M. J. Perkins, J. Chem. Soc. (C) 1971, 2324.
160. A. R. Forrester and S. P. Hepburn, J. Chem. Soc. (C), 1971, 701.
161. T. J. Stone, T. Buckman, P. L. Nordio, and H. M. McConnell, proc. Nat. Acad.
Sci, U. S. A., 1965, 54, 1010.
162. О. H. Griffith and A. S. Waggoner, Accounts Chem Res., 1969, 2, 17;
п. M. McConnell and B. G. McFarland, Quart. Rev. Biophys., 1970, 3, 91;
«Spin Labeling, Theory and Applications», ed. L. J. Berliner, Academic, New
York, 1976.
6.5. ПРОИЗВОДНЫЕ ГИДРАЗИНА
И РОДСТВЕННЫЕ СОЕДИНЕНИЯ
Р. С. АТКИИСОН {University of Leicester)
6.5.1. АЛИФАТИЧЕСКИЕ, АЦИКЛИЧЕСКИЕ
И ЦИКЛИЧЕСКИЕ ГИДРАЗИНЫ, АРИЛГИДРАЗИНЫ
И ПОЛИФУНКЦИОНАЛЬНЫЕ СИСТЕМЫ
6.5.1.1. Методы получения [1—5]
(1) Алкилирование
Самого гпДРазина имеет препаративное зиа-
гидоазинн ЛУЧЭе' ~огда необходимо получить монозамешеппые
гидразины и может быть применен избыток гидразина. Тетраал-
2р8
„гидразины могут быть получены при использовании избытка
1\к1|Л11рУ,оШе10 агеита- Однако при использовании эквимольных
а'о'[ичсств гидразинов н алкилирующих агентов обычно получают
^и'зкпе выходы продуктов реакции вследствие трудности контроля
числа входящих алкильных групп, а также региоспецифичности
пк'плироваиия. Тем не менее при алкилировании монозамещенных
гидразинов 1,1-дизамещеиные могут быть получены с приемлемыми
выходами (уравнение 1) [6]. Замещение можно направлять в опре-
деленное положение, используя временные защитные группы (урав-
нения 2, 3) [7].
СН2С1 + MeNHNlh
ch2nnh2
I
Me
(1)
(92%)
В присутствии цианоборогпдрида натрия происходит восста
новительное алкилирование гидразинов альдегидами с о разова
нием тетраалкилгидразинов (уравнения 4, 5) [8].
NH NaBH.CN
| + ОНС(СН2)3СНО ------
NH
(S241
h2nn;
NaBHsCN / \.VM.
;nnh2 + ch2o-------------- Лх-,е-
(4)
(5)
(40%)
Внутримолекулярное а-1КПЛ,,Ро8ан1‘е ® ^основных Условиях
Ровки солеи гидразинпя происходит в с обычно высту-
(Уравнение 6). В качестве мнгрнрхющи. Р. , п реакция
пают замещенные бензильная или аллпльна Р:
nl),,„T 11 2]-миграцию в промежуточно образующемся ам,
включай I , j ] ^дцздметенные гидразины превращаются
м'Ттп замешенные через амниофоефораны, причем выход ’
аж Д стадии составляет 85% и выше (уравнение 7) Ц0]. 'а
•Л
MeiN
Vl-IJ’h
кон
•Л»-301> °C
► Me?NNJlCH.>Ph
(80 %)
/R' Etsx ♦ /R'
phsPBrs + U..N\ -----* Р1’зРХ'Н\п2
Ph3P=NN
KOH, ЕЮН
* /R1
XT KT'
zR1
R3NHbZ
\R2
С помощью оптически чистого амина было показано [11], что
при превращении аминов в гидразины через дназотаты (уравне-
ние 8) замещение на 53% происходит только путем инверсии.
Ph t.cicojEt Ph rn
,}с-МН2 зков" Д-N^N-O’ К+ NH2NH-C, (в)
Me/ 2 зк°Ви-и^л, у \/zMe
H H H
(40%)
Алкилирование гидразина 1,2-димезилатами приводит к N-
аминоазирндпнам (уравнение 9); их получают также путем гид-
разпнолнза фталимидоазиридинов, образующихся при присоеди-
нении фталпмидонптрена к олефинам (уравнение 10) [12].
MeCHOSO2Me
I + h2n-nh2
MeCHOSO2Me
(мезо)
Me
(55%)
(52%)
(93%)
270
(2) Восстановление соединений, содержащих связь N—N
Восстановление связи C=N в гидразонах (уравнение 11) и
,„1Нах можно осуществить с помощью каталитического гидриро-
вания, действием алюмогндрнда лития, борогидрида натрия и
водородом, выделяющимся при растворении различных металлов.
,\зосоедипения также восстанавливаются целым рядом реагентов
(см. РаЗД- 6-5-21- с,!"тез. 1.2-днзамещеиных гидразинов для полу-
чения азосоедпиеинй). Аналогичным образом, легко получаемые
нптрознрованием вторичных аминов N-нитрозосоединения при вос-
становлении превращаются в 1,1-дизамещенпые гидразины (урав-
нение 12) или, реже, при действии реактивов Гриньяра —в 1 ’ °
тризамещенные гидразины (уравнение 13) [13].
КаВН4, EtOH
MeNNH2 + R‘R2CO —► MeNN=CR'R2 ------------»-
I I
COMe COMe
1,1,2-
1. H2o. HCI
—> MeNNHCHR'R2 -----—>- MeNHNHCHR’R2 (11)
। 2. NaOH
COMe
Me
I
PhCH2—N—NO
HCI. EIOH, Hj-катол
(или Zn, HO Ac)
Me
I
PhCHz-N-NH2
(76%)
(12)
NHBu-rper
Me2N—NO + rper-BuMgBr —> MeNCHjBu-rper (13)
(3) Образование связи
Для аминирования первичных, вторичных и третичных аминов
используют хлорамин NH2C1 и гндрокснламин-О-сульфокислоту
(2) (уравнение 14). Более удобно применение О-мезнтоилгидрок-
силамина (3) [14] (уравнение 15) п О-(2,4-динптрофенил)гнд-
роксплампиа [15], поскольку эти соединения более устойчивы,
чем хлорамин, и требуют менее основных условий, чем (2). Ами-
нирование с помощью N-метплгндрокспламин-О-сульфокислоты
протекает менее успешно (уравнение 16) [16].
+ McNHOSOsII
N—NHMe
(25%)
Тоехчленные циклические гидразины (дпазирндниы) были по-
печены Шмитцем с сотр. [17] при реакции иминов с хлорамином
Посчедуюший мягкий гидролиз приводит к ациклическому гид.’
пазину чанная процедура в целом соответствует аминированию
амина из которого был получен исходный имин (уравнение 17)
(64%) (81%)
Мочевины и сульфонамиды превращаются в гидразины в ре-
зультате реакций, включающих образование связи N—N и напо-
минающих перегруппировку Фаворского (уравнения 18, 19). Бла-
годаря этому возникает возможность аминирования хиральпого
амина после перевода его в производное мочевины. При этом об-
разуется соответствующий гидразин с полным сохранением конфи-
гурации [18]. В некоторых случаях были выделены промежуточно
образующиеся диазприднноны (4) и тиадпаз11рпд11П-1,1-диокси>
ды (5).
[SO2 -I
Et2N--NHj -~"а^СОз>- Et2NNH2 (19)
(5) (79%; данные
титрования)
(4) Циклоприсоединение азодикарбоксилата
иие^ктивипоп3 цнкличес,!|1х гидразинов используют присоедпне-
Zy“^n~ а3°СОеДИ"е'”"* к "° Дильсу-Аль-
(20)
272
„йс-Лзосоединения (6) — /gj яи„
горными группами, - ?”8''Р»»аниые
очнако они часто нестабильны
я7й, обычно путем -
гидразина.
___ электроноакцеп*
I, являются особенно хорошими диенофилами,
нестабильны ц должны быть генерированы in
соответствующего циклического
окисления
о Лгь N-y О о nA II ;s о
(6) (7)
R
(«)
Делались попытки создания таких заместителей которые бы
активировали азогруппу и легко удалялись в мягких условиях
приводя к свооодному гидразину. К ним относятся, например дп-
бензил- и бпс (2,2,2-трнхлорэтил) азодикарбоксилаты (10) и (II)
и сложный эфир азодифосфоновой кислоты (12) [19] Эфир (12)
в отличие от алкиловых сложных эфиров карбоновых кистот мож-
но гидролизовать без нагревания в присутствии основания (урав-
нение 21).
ro2cn==nco2r
(ю) н-сн2рц
(11) r = ch2cci8
(PhO)2PN=NP(OPh)2 +
(12)
rp'NPtOPh^
""^'NP(OPh)2
О
(68%)
l.H2,Pd/C
2.2И.НС1
3.NaHCO3
(21)
(5) Арилгидразины
Для получения арплгпдразпиов разработаны специальные ме-
Т°ДЫ. Распространенным способом их получения является хими-
ческое ИЛИ электролитическое восстановление солей арендиазония
Однако из-за низкой устойчивости эти соли предварн-
ельно превращают в более устойчивые соли арнлазосульфокис
оты (13), которые могут быть восстановлены в более жестких
)сл°виях (уравнение 22).
Лг%7 + hso; —>• ArN=\'so; ---------- atKHNHs (22)
(13)
о о
273
Лучшим способом получения днарнлгпдразппов является ВОс
,2. Р,ше соответствующих азососдииснии или прнсоелш^...
к Рипона (см: разд. 6.5.2). Используют также
;ПР замещение уходящих групп (обычно галогена)
вировапиых ароматических ядрах (уравнение 23) [21].
Рнсоедпцени
нуклео’
в актц.
Однако
О
О 85%)
(23)
замещение галогена в неактивпрованных ароматических ядрах с
помощью реакционноспособного, но опасного в обращении гид-
разида натрия может приводить к смеси изомеров вследствие
промежуточного образования аринов. Для превращения неакти-
внрованных ароматических йодидов в арилгпдразины был пред-
ложен медьсодержащий снлнлпрованнып реагент (14) [22].
Некоторые фенолы могут быть превращены в соответствую-
щие гидразины с помощью реакции Бухерера (уравнение 25); из-
вестен также вариант реакции Чичибабпна, в котором использу-
ется гидразин (уравнение 26).
6.5.1.2. Структура и стереохимия [23]
(пирамитаВ гидРа311На* характеризуются тетраэдрической
роды заместптрпп"'ЛП плоск°й геометрией в зависимости от при-
нимальной эпепгнп" П₽к азоге- в бипланариых гидразинах мн-
делениые элек^щшТп^61 К0"Ф°Рмаи1|я- в которой две непо-
ции (16) с ₽Ь1 °РТ010»алы1Ы (15). Для копформа-
ми) существуе? 1иеп2’“ электРоинь,ми иар'ами (и заместителя-
рг тическпи барьер вращения вокруг связи
N; экспериментально определенное значение AG* для этот
N1' „пя составляет кДж/моль. дя зТО[о
баРьСР
>2-4
(15) (1в)
В случае ациклических бипирамидальных гидразинов простой
„,,ет отталкивания электронных пар показывает, что наименьшей
энергией обладает конформация (17). Однако имеющиеся экспе
рпмептальные данные н многие теоретические расчеты подтверж-
дают представление о том, что в конформации С MllUUUo Hi
энергией (18) двугранный угол
вен ~ 90° [24].
в конформации с минимальной
между электронными парами ра-
(17) («)
Для бипирамидальных гидразинов можно представить два ти-
па процессов вращения: переход (19) (20), в ходе которого
реализуется лишь одно заслоненное взаимодействие заместите-
лей (с н Ь), и переход (19)(21), где неизбежно происходит
переход в заслоненное положение обеих пар заместителей и сво-
бодных электронных пар. Предполагают, что первый процесс
характеризуется более низким энергетическим барьером.
с с XX
(21) (19) (20)
(27)
Кроме вращения вокруг связи N—N бппирамидальные гидра-
зины могут претерпевать инверсию при обоих атомах азота. По-
скольку электроотрицательные .группы при азоте в амине увели-
чивают барьер инверсии (см. разд. 6.1.2), то неудивительно, что
Для гидразинов характерны несколько более высокие значения
барьера инверсии, чем в случае аминов. Для гидразинов недавно
было точно доказано наличие двух типов барьеров инверсии J.
а) превращение (22) в (23), в котором при инверсии заместите-
ли не оказываются В заслоненном положении в переходпол
стоянии («пепересекающаяся» инверсия, non-passing шес
275
/991 в (24), npn котором необходим переход За
б) превращение (-^ за^01!С1ПШе подожеипе («пересекающая^
месТ1ПСТ’ой«гп8 ’н^он).
„„версия. рв«млК
d
Ь
d
(28)
(23) (22) (24)
Предполагают (26], что именно различие в отталкивании меж*
ду пеподеленными электронными парами азота является главным
фактором, отличающим высоко- и нпзкоэпергетические пути
инверсии для бнпнрамндального гидразина. По нпзкоэпергетиче*
скому пути (уравнение 29) превращение не связано с переходом
электронных пар в заслоненное положение в отличие от высоко-
энергетического пути (уравнение 30).
видов N-инверсии было нсполь*
Существование двух различных видов N-инвсрсии было нсполь*
зоваио для объяснения поведения некоторых ациклических и циК*
лцческих гидразинов во время снятия ЯМР-спектров при различ-
ных температурах [27, 28]. Барьеры процесса «непересекающей-
ся» инверсии при азоте составляют 28—34 кДж/моль, а барьеры
«пересекающейся» инверсии имеют значения 42—50 кДж/моль.
11Пг,т,аЬ'Г11Ке-'1И'0 трудно бывает провести различие между «ие-
пппп<.рлп10Ще"СЯ> ,1НВеРсией ПР>1 азоте и более ннзкоэпергетическнм
гоуппипоп враще”ия [('9)-'- (20)]. При включении гидразннной
ограничение „Л цш<л"ческУ’° систему происходит некоторое
тывать кон<ЬопмМ°/КНОСТ” вРащен»я; однако при этом надо учп-
систем Тяк^ лпаа11И9,1ИЬ1е энеРгетнческие минимумы циклических
как было показано’7Дп^еТ'1ЛГеКСаГИДроп,’Р',дазина’ ДлЯ К0ТОР0Г0’
устойчивой является кп?.*°ЩЬЮ СПе«тР°«<олнп ЯМР *3С, наиболее
НИИ температуры замел *°Рмация <25), прежде всего прп понпже-
зультате этого в спектр щ> "1,011QCC инверсии цикла [28]. В ре-
лов равновесной смеси (2f>?-£*выделяются из сигна-
женпи температуры rmn 1(28). При дальнейшем пони;
ературы процесс взаимоиерехода (27) (28). через (26)
„иедляется n ,Mi“ i"lau1’-' времени, необходимого для съемки
спектра ЯМР. В получается спектр ^замороженной» смеси
? i(28). w™ иренмЯцестР.ино присутствующим конформером
„се еше является (25). Содержание (26), через который устанав-
ливаете4 равновесие между (27) и (28), слишком мало, чтобы его
можно было обнаружить.
(28)
Гидразины принимают плоскую конформацию при одном из
атомов азота, если в результате замещения прв этом атоме про-
исходит эффективная делокализация иеподелеинон электронной
пары. Значения барьеров вращения вокруг связи N—N в этих
плоскопира.мндальпых гидразинах занимают промежуточное по-
ложение между значениями энергетических барьеров для бнпнра-
мидальных и бнпланарных гидразинов (63—71 кДж/моль) и мо-
гут быть измерены с помощью ЯМР-спектроскопин. Например, из
ЯМР-спектров днбензпларнлгпдразина (29) следует, что его бен-
зильные группы эквивалентны.
NO2
PhCH2v
^NNH—( \—NO2 (29)
PhCHC '='
Однако бензильные протоны Нд и Нв каждой бензильной группы
не эквивалентны, их магнитное окружение изменяется в резуль-
тате процессов вращения и инверсии при N-1 (топомернзация)
(схема 32).
Процесс вращения имеет более высокий энергетический барьер,
11 Именно этот барьер может быть вычислен на основании дан-
пЬ1х о температуре, при которой происходит слияние сигналов
277
, „пых протонов, в на основании разницы их хнмИЧе
бензилы!^ ' , „ „„рамидальпо-планпрпых гндря31|„ах '*
сдвигов ЛО- переход неноделенны.х электронных иар в '
избежно вклю ает перс > объяснЯет более высокие з11а
Л0,,е’Х* энергетических барьеров, че^
чте"б11Н1>рамнда.1ьчых гидразинов [29]. Полезным методом п₽1|
‘уме и, конформационных проблем ii xiunni гидразинов оказы.
ет фотоэлектронная спектроскопия [30].
6.5.1.3. Реакции [2, 5, 31, 32]
Снта пиразинов как основании зависит манным образом от
„нъкнноиного II резонансного эффектов заместителей. Сам гид-
разни (рКэ 7,95)—более слабое основание, чем аммиак (рКа
9 94)- основности монопротопированного гидразина N2HJ край-
не мала н близка к основности воды. В отличие от аминов заме-
щение атомов водорода алкильными группами в гидразинах по-
нижает основность, однако гидразин протонпруется по более ал-
килированному азоту. Резонансная делокализация пеподеленной
электронной пары в феннлгпдразпие (р/<а 5,27) значительно по-
нижает основность, по не в такой степени, как в гидразидах
(например, в MeCONHNH2 (рКа 3,24)]. Нередко простые алкил-
гпдразнны медленно поглощают диоксид углерода (п воду) из
воздуха, образуя солп кислоты NH2NHCO2H. Солп алкилгндразн-
нов с минеральными кислотами часто являются кристаллическими
соединениями и могут быть использованы для идентификации или
хранения.
(1) Нуклеофильные свойства
К наиболее характерным реакциям гидразинов относятся реак-
ции, в которых гидразины проявляют себя как нуклеофильные
агенты. Гидразин и многие его производные обаружнвают ано-
!альпо высокую нуклеофильную активность по отношению к угле-
родным центрам с 5р2-гибриднзациеГ[, связанным с соответствую-
уходящими группами. Такая реакционная способность
(“-эффект [33]) характерна и для других нуклеофильных агентов
с электронными парами на электроотрицательных атомах рядом с
нуклеофильным центром. Количественно эту способность можно
rwnnnr™?’, путем ЕРав,[еи11я скоростей реакции гидразинов со
пА-Р ¥г,ЯМ' реакцин аминов, обладающих близкими значениями
„тп 7!, П°Ка еще нет полного объяснения a-эффекта (возмож-
пагт\птп11пааПреДеЛЯеТСЯ несколькимн факторами, зависящими от
акнииУ Ро,< /7° нУклеоФИЛ1>ного агента н осуществляемой ре-
обпазовапне 40™°"^™' ,Ю’В|,Д11МОМУ, в реакциях, для которых
в переходном состоянии" Л 3,,а'”,тель,,ой степени происходит уже
3"ча уменьшается и а-эфф1ктМеРе уМеНьшеш,я основпостп гидра-
278
(2) Алкилирование
Алк11-’"Ф1’ва1’"е гидразинов обычно протекает по более зам₽
шс1111ому аюмУ азота (уравнение 33). Если N-заместитель обла-
дает электроиоакиепториыми свойствами или если алкилирующий
яепт имеет большой объем, атаке могут подвергаться и другие
атомы азота. Прн алкилировании эпоксидами (уравнения 34 35) и
азиридинами также, как правило, реагирует замещенный’ атом
азота. Алкоксиды гидразиння, получаемые из 1,1-диза мешенных
гидразинов, превращаются в амннимиды (30) [34]. Арилирование
также происходит по наиболее замещенному атому азота (уравне-
ние 36).
PhNHNHj + 2Ме) —> PhNMe2 Г
NH2
(33)
MejNNHn + RHC---CH2
\)
RCHCH,NMe,
I 'I
O' NH;
r'nh
! I +
r‘nh
,1 ]
r'nh oh
RCHCH2NMe2
I I
OH "NH
(30)
r2cho
----
(3) Ацилирование
Ацилирование несимметричных гидразинов происходит менее
предсказуемым образом, чем алкилирование, и в значительной
степени зависит от условий проведения реакции. Осложняющим
обстоятельством является то, что образование продукта реакции
может контролироваться кинетическими или термодинамически-
ми факторами. Однако нз-за электроноакцепторной природы
ацильной группы моноацплпрованпе осуществить проще, чем мо-
ноалкплировапие. Ацилирование моноалкилгпдразннов обычно
приводит к смеси 1- и 2-моноацплпропзводных. моноарвлгндразн-
ны ацилируются в положение 2. Однако были разработаны усло-
вия ацетилирования метплгндразпна, позволяющие получать
аиетил-1-метплгидразин с очень высоким выходом [35J. Срав
пенне различных N-защптных групп было проведено на пример»,
получения 1-арил-1-ароплгпдразннов. Найдено, что нанлучшеи за-
щитной группой является N-ацетпльпая группа (уравнение )
279
Как нуклеофильные агенты гидразины реагируют с сул
136’ ?фосфорчлгалогепндамп, с другими ацилирующими аУГен^
мн т как кетены и изоцианаты а также с имидо,^
аамп. давая амндразопы (уравнение 38) [37]. ₽«
ArNHNH2 * ArNHNHCOMe
ArtCOCI
COAr1 NH2
I hci, Eton |
—> ArNNHCOMe ----------> ArNCOAr1'
Cl R NHMe R
I I I I
PhC=NMe + H2NNMe —> PhC=N—NMe
(37)
(38)
(4) Реакции с соединениями,
содержащими группы C = O и C=C
Активированные алкены реагируют С гидразинами (присоеди-
нение по Михаэлю) (уравнения 39, 40). Как и при алкилировании,
со второй молекулой акрилонитрила предпочтительно реагирует
наиболее замещенный атом азота. Аналогичная реакция присоеди-
нения в случае ацетиленов приводит к ппразолам (уравнение 41);
региопзбирательность присоединения согласуется с первоначаль-
ным присоединением метилированного атома азота к тройной свя-
зи с последующей циклизацией.
NH2NH2 + CH2=CHCN —♦ NH2NHCH2CH2CN —> NH2N(CH2CH2CN)2 (39)
+ PhCOCsCCOPh
/N—C=CHCOPh
XjUcOPh
к
HtOH
NHC=CHCOPh
I
COPh
(40)
MeNHNH2 + PhC-sCCOMe
(41)
(90%) (10%)
и его производных
(ЗП —m? ,,звсст,,ая конденсация гидразина
ине TPrnLC альдеГ1|Дами и кетонами происходит через образова-
ние 42)₽аКп ГСК0Г0 пР°межУточиого соединения (35) (уравне-
действпю к ,е., аЛЛ"3уеМ0СТЬ’ 3 также устойчивость гидразонов к
НХ широкому Jenn0 срав"сн,”°' например, С иминами привела к
кетонов. Озазоны (™?ова'"”° для идентификации альдегидов и
на в а-дпкетопов ’ явля,01ц,1еся производными фенилгидразИ-
леводов Другие пи-.,/ а'г,!дРокс,|кетоиов, применяют в химии уг-
280 ' Р Функциональных групп гидразонов лежат
основе получения ряда гетероциклических соединений, содержа
их связи N—N [38].
(35)
+ 2PHNHNH?
(36)
(43;
(5) Перегруппировки
Бензидиновая перегруппировка заключается в превращении
1,2-диарилгидразинов в кислых условиях в бензидины (37), ди-
фенилины (38), о-бензидины (39) н о- и /г-семидины (40) и (41).
Кроме этого иногда получают небольшие количества продуктов
диспропорционирования (амины, азосоединеиия).
(41) (4°)
Много усилий было затрачено на изучение механизма бензн-
Аиновой перегруппировки. Реакция является внутримолекулярной
281
ДВУМЯ параллельными путями: один путь-это реа„
и протекает двум . пторой _ реакция второго порядка отн*
пня перво Предложено несколько механизмов, которце
ситмыю in I- !01-)е другпе да11)1„,е. ЭТц механизмы подробпо
учитывают эти । у Перегруппировка происходит также
обс’’ЖДХт1пи в отсутствие кислоты, однако в этом случае 2
"Ротекает менее гладко н образуется большее количество нродук.
представляют собой Днполярные ионы, вкл,очаю.
о ож тельио заряженный азот, связанный с азотом, несущИм
(ятезьный заряд. Амннимнды склонны к перегруппировкам
Р (1)]; химия этих соединений рассматривается в обзоре [40].
(6) Окисление
Различные ионы и радикалы, промежуточно образующиеся
поп окислении гидразинов в азосоедннення и амннонитрены, пред-
ставтены па схеме 45. Эти промежуточные продукты были выде-
лены нт идентифицированы спектроскопически [41]. Окисление
до азосоединенпп происходит под действием ряда реагентов, вклю-
чая диоксид марганца, оксид ртути(П), пероксид водорода,
гипохлориты и днэтпловый эфир азоднкарбоновои кислоты. Если
гидразин легко доступен, его окисление представляет собой удоб-
ный путь получения азосоединений.
-е~ — R2 . — е~
~ " => R'N—NR2 5=»
I +Е‘
гндразил-
радикал
—N=NR2
'N—1
R'N—NR1
гидразин ион-радикал
гидразнння
(45)
-Ri
ИОН
диазенля
азосоедннснне
(диазен)
R‘N=N
а.мииош'трец (аза.мин
или диазен с разде-
лепными зарядами)
При окислении гидразинов, особенно 1,1-днзамещенных гид-
разинов (42], часто постулируется промежуточное образование
амиионитренов. Однако амннонитрены не удается выделить и
даже наблюдать спектроскопически, и в ряде случаев не ясно,
действительно ли они являются промежуточными соединениями-
Прн проведении реакции окисления различных 1,1-днзамещенных
гидразинов в присутствии олефинов (уравнение 46) Рнс с сотр.
1(43] выделили азиридины с очень высокими выходами. Этот факт
убедительно доказывает промежуточное образование аминопнтре-
282
„ Окисляюшим агентом всегда является тетраацетат свиииа
’’IaC), а замешенный атом азота гидразина входит в гетероТик-
',‘ческую систему.
\ \ / РЬЮАс),
^nnh2 + ------->
К остальным продуктам, которые обычно образуются при окис-
пенин ДРУГИХ М-Днзамещенных гидразинов [44], относятся соот-
ветствующие амины, тетразены или соединения, возникающие в
результате выделения молекулярного азота (уравнение 47).
Окисление аллилгидразпнов (42) оксидом ртути(II) приводит к
аллильным азосоединениям (43) в результате'[3,2]-сигматропной
перегруппировки промежуточно образующегося диазена (амино-
нитрена) [45J (уравнение 48).
R'—R2 + N2 «—- R'RjNNH2 —>• R\HR2 (47)
I
R‘R2NN=NNR’R’
(42)
(43)
Амннонитрены не удается выделить, однако в некоторых слу-
чаях были получены протонированные формы амннонитренов —
ионы диазения. Наиболее хорошо изучен ион 1,1-днметилдиазе-
нпя, полученный окислением гидразина бромом в кислой среде и
выделенный в виде соли хлорной кислоты (44). Это соединение
претерпевает ряд интересных превращений [46], включая димери-
зацию в тетразен, происходящую при нейтрализации кислого рас-
твора, присоединение к бутадиену по Дильсу — Альдеру, электро-
фильное присоединение к стиролу и реакцию циклоприсоединения
с норборненом, приводящую к азиридину (схема 49).
4
NNMe,
a
Me2N=NHCIO4 OH-4- Me2NN=NNMe2
(44)
I PhCH=CH2
- ~ HBr ' ' I
(43)
Me2i$HNHCH2CHPh
Br
Br
283
Алкилированием азосоеднпепий [41] был получен ряд д
солей дназения, включая соединение (4о). >
I. AgBH<
CHa=»CHCHjN=NBur/>er _
С1Ь—CHCH2N===NBu-rpej.
Bu-rper
(45)
Использование гидразина в качестве восстанавливающего
агента [47] предполагает наличие одного из трех условий'
а) присутствия основания, как в случае восстановления карбо^
пильных соединении по Кчжнеру — Вольфу; б) использования
диспергированных металлов в качестве катализаторов при вос-
становлении, например, ароматических соединений; в) примене-
ния гидразина как источника динмпна (см. разд. 6.5.2.4).
6.5.1.4. Гидразил-радикалы [48]
При действии брома тетраарилгидразпны окисляются (урав-
нение 51) до катнон-радикалов (радикал-ионы гидразиния;
см. уравнение 45). Изучение катнон-радикалов тетраалкилгидра-
зипа, полученных электролитическим окислением гидразинов или с
помощью сурьмяной соли (46), методом ЭПР приводит к выводу
[49], что ни для одного из атомов азота не характерна плоская
геометрия. В то же время считают, что катион-радикал (47),
полученный окислением гидразина тетраацетатом свинца, имеет
плоскую геометрию при обоих атомах азота [50].
AraN—NAra + Bra -—> AraN—NAra Вг" (S1)
(”'
N SbClj
(46) (47)
Наиболее известным гидразильиым радикалом (см. уравнение
45) является дпфенилпикрилгндразил (ДФПГ) (48). Этот ра-
дикал удается выделить, и он не реагирует с кислородом. Однако
до последнего времени алкилгидразнлы были почти неизвестны,
едавпо в продуктах фотолиза ди-трег-бутилпероксида в присут-
(50)
R-rper-Bu
284
с1В1ш 1,Ьдналкилгидразинов методом ЭПР
„ужены 1.’1’диаЛК,’ЛГ"дРа3ил-радикалы а 'С"ектроско»нн обна-
У'тойчнвыи радикал (49) образуется пп., от»осительно бой!
троваиин солн дназення (50) [51] (R Рре^Д1°воме7'Рическом "тн-
Другой класс устойчивых гиюаш»» 8
(51) —(53) [52]. Все они обладают сход.нымнТаВЛЯК1Т веРДазилы
явностями, и нх устойчивость анатогнч аТтСТ"РУК1урным« «со
' устойчивости ДФПГ
N-N'
\ /
N—N.
;n—nc;
.//
(51) (52) (53)
6.5.2. АЗОСОЕДИНЕНИЯ (ДИАЗЕНЫ)
6.5.2.1. Методы получения [1—3, 30, 31]
(1) Окисление гидразинов
Синтезы 1,2-замещенных гидразинов можно одновременно
рассматривать как синтезы соответствующих азосоединеннй. Не-
обходимое для получения азосоедннення окисление обычно про-
водят с помощью оксида ртути(II), брома, гипохлоритов или со-
лей меди н воздуха. Для получения несимметричных азосоедине-
иий используют приведенные ниже методы (уравнения 53, 54)
ТбЗ].
Ме
Меу /Ме LIAID< | /Ме Нз0+
V=N—N=C---------------► Ph—С—NH—N=C' ----------►
Ph/ \ph I 'Ph
D
Me Me
I MejCO I I. Hs. Pt
—> Ph—C—NHNH2 -------► Ph—C—NHN=CMe, - ~
| | 2- HgO
D 6
Me
—> Ph—C—N=NCHMe2 (53)
I
D
NaH NaH
EtO2CNHNHCO,Et ------------> EtO2CNNHCO2Et —*
CH2=CHCH2OS02Ph I
CH2=CHCH2
. HgO
—► EtO2CNNCO2Et CH«=CHCH2\HNHMe 1
CH2=CHCH2
CH«CHCH,N=X>
235
(2) Перегруппировки, приводящие
к образованию связи N=N
Поскольку несимметрично замещенные мочевины (54}
легко получить из изоцианатов и аминов, широко распп0^ М0}|<1|°
их превращение в несимметричные азосоединения через nnPai1e"0
точное образование гидразинов. Омэ, Шмитц и сотр поел °Ме'кУ-
использовать N.N'-дналкилсульфонамиды (55) [54,55]. Р ДЛО!К"Л1,
R'NHCONHR2 + трет-BuOCl + грег-ВнОН_>
(54)
— ► [R’NHNHR2] —R>N=NR2
R‘NHSO2NHR2 + NaOCI + NaOH —> [R'NHNHR2] —к R'N=Np2 <M)
(55) K
Для дихлоразоалканов, легко получаемых из соответствующих
азинов, характерно замещение галогена прн действии ряда нук-
леофильных агентов [32], включая Н“ (LiAlH4) и D~ (LiAlD,)
(уравнение 57) [56]. Алкилирование по Фриделю — Крафтсу
приводит к трет-алкилазоалканам (56) [57] (уравнение 58).
Симметричные трет-алкнлазоалкаиы (57) получают также
средственно из грег-алкиламннов (уравнение 59).
Heno-
Rk /R3 С|2 ZR3
XC=N-N=c/ -------► ;с—N=N—(/ ----*
R2/ XR1 R27^ Cl R<
R' /R3
—-+ 'C—N=N—
R2/Z I |\r4
Y Y
Y = CN, N3, SPh н др.
R'\ mc13 R\ /*’
C—N=N—C +R3Al (Ar3Al) ---->- V—N=N—C'
R2/k /V7 R2/l IXr2
Cl R(Ar) R(Ar)
Me2C-NH2 + JFS -°C' п”р“д'1]1
(57)
(58)
(59)
(56)
Me.C—X=N—CMe2
(57)
разоиов в азоалкапы°Д(уп ирсвРаще""я более легкодоступных гид-
под действием основанпГТбТ п~63)’ вкл,очая "зомернзашио
имеющие а-водоооаипп ’ Однако многие азосоедннення,
(или света) таутомеп,,™ dT0MbI’ под воздействием температуры
сужает применнмостьРданнпгА В б°Лее УСТ0Й',,|ВЫе гидразоны, что
° данного метода.
286
/*2
phNH—N=C
XCH2R'
R2
12. ROH, NaOAc |
-> PhN=N—c—OR
I
CH2R'
r<
XC=N—NHR3
r2/
РЬ(ОЛс),
R2—C—N=NR3
I
OAc
R2 ♦ . R2
I 1. PhNMej Вгз |
R'CH2C=N—NHSO2Ar ----------->- R'CH=C—N=NSO2Ar
KOH
R'R2C=N-NHMe ---> R'R2CHN=NMe
(60)
(61)
(62)
(63)
6.5.2.2. Циклические азосоединеиня
Простейшим представителем этого класса соединений является
диазнрин (58; R' = R2 = H), неожиданно оказавшийся несрав-
ненно более устойчивым, чем изомерный диазометан (для кото-
рого первоначально предполагалась циклическая структура). Ди-
азирины, как правило, получают окислением дпазнрндинов (уравне-
ние 64); окислителем обычно служит оксид ртути(Н). В отличие
от диазоалканов диазирины умеренно устойчивы к действию кис-
лот. Напряжение в цикле проявляется в способности этих со-
единений реагировать с реактивами Гриньяра [59]. Подобно ди-
азоалканам для дназнринов характерно термическое или фото-
химическое разложение до карбенов (уравнение 65) [60].
R1
R2
HgO
Me
(58)
I. RMgBr
2. H2O
(64)
N
XII
Ph N
(65)
Известны очень немногие дназетнны (59), однако пиразолпны
легко получаются в результате 1,3-днполярного присоединения
Дназоалканов к активированным олефинам (уравнение 66) [60].
(66)
287
Ря1 тетпагитроппрпдазннов был получен присоединением Д|16
р, 3 Хгтнкарбокснлатам "° Д,К"”СУ “ ЛльлерУ с по
4ЮВ„пим восстановлением. гидролизом, декарооксилированне^'
S ЖМ (уравнение 67). При иснользованпн 1,2-бнс(₽.Тоз “
° "кс карбонпл)дпазена (уравнение 68) (61] удается цзбеж
применен»я жестких условий, необходимых для гидролиза кар6а.
" н HR
Vb rA/Nco^
г NCO2R И К
R
H
''kco2(ch2)2ts
NCO2(CH2)2Ts
l.KOEt,
EtOH,20aC
2.CF3CO2H *
+ NCOjK
RO2CN^
CO2(CH2)2Ts
I' +
TsfCH^C7 Fe(CO),
---Продукты (68)
6.5.2.3. Ароматические азосоединения
Специальные методы [3] получения соединений этого класса
показаны на примере реакций, приведенных ниже (уравнения
69—81). Некоторые из этих реакций чрезвычайно важны для про-
мышленности, производящей красители.
Агк; + АгХ —► ArN=NArX W
X = NR'R2, ОН, OR и т. д.
R'R2CH2 + ArNJ —► R‘R2CHN=NAr (70>
R1, R2 = CO2R и t. д.
ArNO+Ar'NHj —► ArN=NAr' <71'
Cl Me
ArNHNHs + MeCoinCO.Et —> ArN=NC=CHCO2Et (72)
ArMgBr 4- Ar'N? СГ ZnCI2 —► ArN=NAr' + MgBrCl + ZnCh (73>
ArNO2 + Zn + NaOH —> ArN=NAr (74)
ArNO2 + NaBH4 ArN=NAr (75'
ArNHNHAr'+ EtO2CN=NCO2Et —> ArN=NAr'+ EtO3CNHNHCO2Et (76)
288
ArNO + Ph3As NAr1 —>- ArN==NAr' + PlhAsO
1. Cu, aiiciu,,
Л1Х BF< -----------------* ArN=NAr
(77)
(78)
(79)
(80)
(81)
•NO2
SNHAr
► ArN==NAr X = MnO2, NaOCl, IF6 или Cu» H2O:
ArN] X‘ + AgCsCPli —► ArN=NCs«CPh 4. AgX
ElOH
NaOH
6.5.2.4. Монозамещенные диазены и диимины
Получение и изучение монозамещенных диазеиов RN=NH
начато только в 1965 г. в основном Косовером с сотр. [62]. Эти
соединения чувствительны к воздействию кислорода и оснований
„ устойчивы только в разбавленных растворах. Дназеннльный во-
дород обладает слабокислыми свойствами (р/(а « 18), а ди.
азеннл-анион является хорошим нуклеофилом и присоединяется
к карбонильным группам (уравнение 82). Считают, что монозаме-
щенные диазены выступают в качестве промежуточных соедине-
ний при восстановлении карбонильных соединений по Кижнеру —
Вольфу.
“ОН
Me3CN=NH + СН2О ---->- Me3CN=NCH>OH (82)
Сам динмин HN=NH оказался ценным реагентом для осуще-
ствления ч«с-прнсоедпнения водорода к ненасыщенным системам,
обычно к связи С=С. Его получают in situ окислением гидрази-
на метаперподатом натрия [63], солями меди (II) и пероксидом
водорода, а также через N-хлордипзопропнламин или термолизом
производного антрацена (60) (уравнение 83) [64].
(60)
6.5.2.5. ijuc-транс-Изомерия
Синтез ациклических и ароматических азосоеднненни неизмен-
»° приводит к получению более устойчивого транс-изомера. Для
ароматических азосоедпненпй цпс-пзомер обычно может быть по
путем фотоизомернзащш [65]; для удаления TPa^"paQ’
Рисутствующего в возникающей равновесной смеси, Kal\F^B'^|X’
льзуют методы хроматографии. В обычных Ц1
10 Зак. юз 289
конечно, преобладает цас-пзомср, однако т
дзосоедниениях. ’() 'быЛ получен Овербергером ccorp.^j
Iг-дназациклоонте \ > цис-2-Азопропан (62) является 0Д11,^
при фотолизе /( 6- 3 с|1_|х а,)11фат1|чес,{нХ цнс-азосоединенпй, ко-
из немногих а11’ т. Предполагают, что цис-„-азоиропап og.
торые УДал0С" ’'Ь'-„е«но "3 тиадиазнрнДпн-М-диоксида в Усдо.
разуется неносредстве " п0 методу Омэ н др„ поскольку
ВНЯХ термического роз. Д( ОМ |]р|11,од,1т только к транс-ъъо-
окисление ’"^^"^.'„рборпан (63) также достаточно устойчив
Г^<!Хт1вь№лен167!.
(61)
(63)
Результаты измерения энтальпии переноса растворителя пока-
зывают, что термическая ipc-транс-изомеризация азобензола
протекает через инверсию при атоме азота, а не по механизму,
включающему вращение вокруг связи N=N [68].
6.5.2.6. Термическое и фотохимическое разложение
Некоторые азосоедннення широко используют как источники
получения свободных радикалов при нагревании (или фотолизе)
[69]. Выброс молекулы азота может происходить путем согласо-
ванного разрыва двух связей при азогруппировке (уравнение 86)
или через диазенильяый радикал (уравнение 86).
R'N=NR2 —> . pi + n2 + . R2
R‘N=NR2 ч=±: ,ri + . N=NR2 —n2 + . R2
(85)
(86)
дов [70?С"обпячов1ИННОГО Mexa,,ll3Ma был использован ряд мето-
женни фепнлазотпижви диазенильных радикалов (64) прн разло-
ХИДПЯ [711 ЧЯмв?о плметаиа наблюдалось с помощью метода
аллильную в (6611 1 алк11ЛЬ11°й группы R [метильной в (65) на
8 газовой фазе хотя начительио влияет на скорость разложения
зей R-Н различаются9 няР'№liaMH для аналогичных свя-
имел место согласпНои ~ кДж/моль. Если бы в данном случае
290 'ласованиыи процесс, то следовало бы ожидать
тельного увеличения скорости реакции для (66) по сравне-
нию С (65)- ph№NCPhj —► PhN=N• + Ph3C• (87)
(64)
Me№=NCH2CH=CHj CH2=CHCH2N=NCH2CH=CH2
(65) (66)
Было высказано предположение, что в растворе согласованный
механизм более вероятепв том случае, когда оба генерируемых
радикала одинаково устойчивы. Для того чтобы выяснить, какой
|13 процессов [(85) или (86)] реализуется, использовали внутри-
молекулярное улавливание дпазенильных радикалов, рацемиза-
цию хиральных азосоединений, исследовали влияние давления и
вязкости на скорость реакции и кинетические изотопные эффекты
[72]. Из-за более низких значений энергий основного состояния
(29—33 кДж/моль) для транс-азосоедннений по сравнению с
((uc-соедпненнями свободная энергия активации при разложении
цис-соеднненнй имеет более низкие значения [73]. Полагают, что
термическое н фотохимическое разложение пиразолинов (уравне-
ние 88) протекает через соответствующий бирадикал (67), точная
структура которого изучается [60]. На основании данных по изу-
чению влияния заместителей сделан вывод, что в переходном со-
стоянии происходит разрыв связен N—С при обоих атомах азота.
Образование бирадикала (68) при облучении исходного азосоеди-
нения в стекловидной матрице при 5,5 К [74] доказано методом
+ СН3СН=СН2
(67)
(88)
(66)
(89)
(67)
—► Продукты
(90)
(70)
291
Ю»
„пегоппп (уравнение 89). Однако термическое разло.
ЭПР-спектроскопп (Полшюв м протекать и другим сп
жение некоторых ш и об ого 1,3-динолярного присоединения
бом, а именно »} приводит к продуктам, образующие
Так, »агРев.а™ насяду с продуктами, получаемыми более обыч.
из карбена (70), 1р лУ! ад11кала (уравНенпе 90).
ВЫМ путем nP"/^ v азота (деазетнровавие) из многих дРу.
Отшепленне мол ннй приводнт к углеводородам в р«.
гнх цпкл'1Ч1СомбннацП1| промежуточно образующегося бирадика-
зультате Рек0“°1П пя,ложенпе было, в частности, использовано
ла. Фотохжьшческ Р ,, с напряженным циклом (71) —(74)
Св=еН91-94) Наиболее эффектным из этих синтезе-
ется получение призмана (71).
В ЯВЛЯ-
(72) (100%)
(92)
(74) (30%)
[651 часто nnnUf°e Разл°жение ациклических азосоедннений
меризаиии с по СХ°ДИТ путем Фоюкатализуемой транс-> цпе-изо-
ного иас-соединр11ияУ1ОШ"М теРмичес1<нм разложением нестабиль-
чески устойчив пабл1л яРадика‘"ы- °ДНако если quc-нзомер терми-
налов ИЗ транс-изомераД [75]Я не”осРедствеиное образование ради-
которое не аклю^е/стял16" рЯЛ пРпмеР°в деазетирования [76],
стадии промежуточного образования биради-
калов (последние не удается зафиксировать). Эти процессы пред-
*тавляют собой реакции циклореверсии, контролируемые прави-
лами орбитальной симметрии (например, уравнения 95—97)
(97)
Описанные процессы отщепления азота протекают стереоспе-
цифично; продукты замыкания цикла не образуются. Более того,
отщепление азота обычно происходит быстро по сравнению с этим
процессом в случае модельных соединении, разложение которых
включает образование радикалов. Так, (76) — восстановленный
аналог (75) необходимо нагреть до 100°С для того, чтобы прои-
зошло выделение азота. Реакция цнклореверсин нашла примене-
ние в синтезе дигидропнрпдинов [77] (уравнение 98).
CO2Et
^NCO,Et
/
‘NCO2Et
tmj3 .
м г
COjEt
I.MeOH,
KOH
2.Me ОН, Г
HgO
(9S)
sea
nwfinoc азота из Д3-1,3,4-тиадиазолинового цикла в услпо
ях термолиза приводит к илиду (77) (обратное 1,3-диполяр1
циклоприсоединение) и затем к тиирану (78) [78]. Рнад
(77) (78)
6.5.2.7. Прочие реакции азосоединений
Среди реакций азодикарбовнльных производных, являющихся
сильнейшими диенофилами, особенно распространены реакции
циклоприсоединения (уравнение 100) [79]. С кетенами они реаги-
руют по схеме [24-2]-циклоприсоединения (уравнение 101). При-
соединение аналогичного типа является, видимо, первой стадией
при фотохимическом присоединении азобензолов к дпкетеиу
(уравнение 102). Для обеих этих реакций предполагается, что реа-
гирующей частицей является именно цнс-азосоеднпеиие.
ваге’’т У||,1Ве1)саль1!()й природы, днэтиловый эфир азодикар-
К1,сло™’ в Реакциях с алкенами может выступать и
3irmoitCTa\e ено(1”1ла- Это было использовано для введения гидра-
1801 Vctmmim"1 При С''’ 1,11 Р|1М1< днпового цикла (уравнение 103)
пениям 1811 .’’Ротекает присоединение карбанионов к азосоеди-
является'nL( p;‘B’^"ie |04)- Обычно наблюдаемой реакцией
зованнсм Х 'пё'Ь''' "ере'-’Ос ^тронов к связи N=N с обра-
оедипеппй при последующем гидролизе (урав'
(63%)
BrMg MgBr
PhN=NPh + 2RMgX —> PhN—Npli + R—R
(79)
H20
(105)
PhNHNHPh
Можно осуществить присоединение одного электрона к связи
N=N, в результате чего образуется анион-радпкал азобензола
(80) (уравнение 106). Известны также примеры присоединения
карбена [82] и нитрена [83] (уравнения 107, 108) к связи N=N.
дмео
PhN=NPh + PhNHNHPh + трет-ВиОК ------> [PhN=NPh]‘ К (Н.6)
Под действием многих окислителей азосоедннення превраща-
ются в азоксипроизводпые (см. разд. 6.5.2.8), однако азины по-
ручают с помощью реакций отщепления водорода под действием
Радикалов (уравнение [09). Алкилирование атомов азота aaoipynnu
295
,п1,м диазсппя [84]; для осуществления этой
приводит к с '• ” эффективные алкилирующие агенты (урав£
акции иеооход
пае 1'0)-
рю zPil
\HN=NCH' +-СС!з —>
Me' \Ме
р1|\ /Ph
C=N—N=CZ
Me/ \Me
(109)
phN=NPh + MeOSOzF —>
ph\ +
N=NPh ’OSO2F
Me'
<110)
Соли N-алкоксидназения (81) образуются из N-нитрозосоедиие-
НЯЙ при алкнлирова.... реагентом Меервейиа (уравнение 1Ц)
Эти соли легко претерпевают депротонпрованпе с образованием
дпполярных частиц (82), которые вступают в реакцию циклопри-
соединения с азометпновой связью гетероциклических соединений
и оснований Шиффа и также вызывают дегидрирование (уравне-
ния 112, 113).
Ph, Ph,
Ч—NO + Et8O+ 'BF4 —> /N^NOEt 'BF, (111)
Me' Me'
(81)
-EtOH ,
-2H >
(112)
Продукты
роль
Ионы азокарбения ^,С—N=N— играют определенную
собетвтот06’06 дихл0Раз0алкаН0в [32], а наличие связи N=N спо-
лпзе &83) ®^]®спеН11ю Уводящей брознльной группы при сольво-
6.5.2.8. Азоксисоединения [3, 5]
нения^Ц—1 методов получения азоксисоеднненшй (урав‘
случаях, когда В отсУтствии регпоспецифичности в тех
ные о том что nv естителп ие идентичны. Однако имеются Дан'
исленне азосоединепий такими окислителями,
г„гг.бутнлгидроперокснд — гексакарбонил
молибдена — дипивалонлметан, протекает
[86].
молибдена и оксид
региоизбирательно
। л)
ArNO + Ar'NHOH —> ArN=NAr' + ArN=NAf*
A- A-
(115)
ArN=NAr' + RCO3H —> ArN=NArl + Ar\'=NAr' (116)
O’ o-
Алкилпрованне тозплатов ннтрозогндрокснламнна с помощью
реактивов Гриньяра (уравнение 118) и реакция X.N-дихлоралкил-
амииов с алкил- или арплнптрозосоедниениямн (уравнение 119)
[87] лежат в основе двух подходов к региоизбнрательному синте-
зу азоксисоедпненпй. Еще одни метод синтеза соединений этого
класса, разработанный Моссом с сотр. [88], заключается в алки-
лировании дназотатов [89] (уравнение 120).
ArN=NOTs + RMgCl —> Ar.\=NR (118)
А- А-
R‘NC12 + R!NO —R‘N=XR! (119)
О'
Х = НО-, CuCN, CuCI, KI
Me _ ,, _ ,, Me
KON=NEt + CI-CT -------► ,C—N=NEt I120)
I
Для азокснсоеднненнй, выделяемых из природных источников,
обычно характерно проявление физиологической активности; на-
пример, элайомицин (84) обнаруживает как канцерогенные свои-
СТВа. так и свойства антибиотика. Д-Дигндровлайомицнн был
297
получен 12-стадшшым синтезом, включающим алкилипов
азотата, с оВщпм выходом 8% (уравнение 121). 1 Bail»e ди.
Н Н
г-
\+ 'ъ
/
О
СН2ОМе м©
'7-сс"юн
н н
м
«-CgHij
EtOjCNH Me .-С0'» К ON=Jj Me
МеОСНг^г/ 2- NA_________>. МеОСНг<^_с/ Л-С8нп1
/ 3' KGBa~BVem / ^7].j "гмфд"
н он н отгп
х-С8Н|7
_ Me
’° Р’А-/
/ / \>н
МеО / *
Н ОТГП
"-С8Нп
_ /N=N
° сн2«$
/ 2 Р
МеО /
Н
Me
/
он
МеОН >
"н7 г
Термодинамически более устойчивые транс-азоксисоединения
в условиях фотолиза [90, 91] превращаются в равновесную смесь,
содержащую цнс-изомеры и изомеры, образующиеся в результате
миграции кислорода. Например, фотолиз (85) приводит к четырем
возможным изомерам [92] (уравнение 122).
”О\ + /Ме
^N=-=NZ —►
Ph/
(87)
(SB)
Ph\
N=N
+ (85)
(122)
ным o6naznr,PO'i',KTOB мигРации кислорода связано с промежуточ-
да эту Рнеоб>аНИвМ 01!садиазиРиДчна (86). Известны примеры, ког-
других азокс'цЧгАУЮ ,1ИКЛ„ИЧескУ<о систему выделяли при фотолизе
превращаются в^Хс'нзом^' X°™ некотоРые Ч^-азоксиалкаиы
(87) не ияпмВп.,о,₽ кс 113°меры даже при комнатной температуре,
изомера азоксибензоТа "в В кипящем бензоле. Превращение цчс-
туре происходит быстоо „J/’awc'H30MeP ПРИ комнатной темпера-
чем в этаноле ПпеХ’,,",? В ГеПтане пР«мерио в 10 раз быстрее,
ветствующие азоксиппл„1е "е цнклическнх азосоединеппй в соот-
значительной теомич^А-ВОЛ"Ые М0Жет приводить к появлению
О" теРмическои устойчивости [91]. В то время как
298 lie
диазен (88) не
согласованной
175 °C.
удается выделить вследствие
фрагментации, его N-оксид
быстро протекающей
устойчив вплоть до
(86)
R-yzjwza-Bu, Л-Ви, «за-Рг
(88)
• Диарилазоксисоедииеиия в условиях перегруппировки Валла-
ха, проводимой в сильной кислоте, превращаются в н-гидроксиазо-
соединения (89) [95]. Известей также фотохимический вариант
этой перегруппировки (уравнение 124), который включает мигра-
цию кислорода азоксигруппы в орто-положение более удаленного
ароматического кольца, как было показано с помощью метки 15N.
Me
I
C„H13CHN=N
I V0’
C6H13CHN=N^
'CH,
EtsCO"
Me O'
—> C6H13(!)HN=NCH2Me
+
(125)
Азоксигруппа может стабилизовать положительный заряд, не-
спаренный электрон или отрицательный заряд на углеродном
атоме, связанном с неокисленным атомом азота. Протоны прн
Углеродном атоме рядом с окисленным атомом азота обладают
достаточной кислотностью и могут быть удалены с помощью
^рет-алкоксидов (уравнение 125) [96].
6.S.2.9. Азодиоксисоедииения
Эти соединения представляют собой димеры нптрозосоедине-
чий (см. разд. 7.1). Ациклические азодпокспсоедпнення регенери-
руют мономеры при переходе в раствор при комнатной темпера-
туре или при нагревании [97]. Димеры нптрозоалканов получают
при окислении бензальднмпнов с помощью перуксуснон кислоты
(Уравнение 126) или прямым окислением аминов. Синтезированы
299
, Р полициклические производные, например (90) „
некоторые пол ц аЗОД11Оксиформе даже при нагревании
которые остаются г О 1 о
/ \
RN=CHPl’ + МеСОзН —> LRN-----CHPhJ —> 0/ \R
01)
[98]
(126)
6.5.3. ДИАЗОСОЕДИНЕНИЯ [20, 99]
Диазосоединення обладают формальным и химическим сход-
ством с азидами (см. разд. 6.5.4):
/С—N=N «-» /C=N—N —N—N=N ч—>• — N=N=N
Подобно азидам они выделяют молекулу азота с такой лег-
костью, что низкомолекулярные представители этой группы соеди-
нений в чистом виде представляют известную опасность. Соли
диазония (92) формально получаются из диазосоедннепнй путем
протонировання или алкилирования по углероду, эти соли могут
быть также получены нитрозированием амииов (уравнение 127).
Однако алифатические диазопиевые солп выделить не удается
[100], так как они спонтанно разлагаются с выделением моле-
кулы азота под влиянием нуклеофильной атаки или в результате
образования карбенневого иона [101]. Многие реакции диазосо-
единений протекают путем протонировання и последующего раз-
ложения диазониевых частиц. Ароматические диазонневые соли
ArN +Х- значительно более устойчивы (см. разд. 6.3.3). Спект-
роскопические свойства азидов и диазосоединений также сходны.
Характеристическое поглощение диазосоедииеиий Хмакс 2040—
2120 см-1, для азидов Хмакс 2140—2240 см-1. Алкил-, арил- и
ацилазиды бесцветны, диазосоединения окрашены в желтый нли
красный цвет.
^-NH, 027)
(92)
6.5.3.1. Методы получения [102]
виде раствопя°иНЬ!^ усЛ0в|1ях диазометан обычно используют в
нитрозоуретаны (qv/43101 при Действии основания на Ы-алкил-Ы-
розоуретаны (93) или дру,.]1е ДОступныс соед!шеннн (94) или
300
,, которые более устойчивы и не обладают
-Ч’люуретанон. [2Н2]-Диазометан получают
уХни этого метода [103].
канцерогенностью
С помочи.И1 моди-
(128)
NO
MeNCO2Et + NaOEt —> CH2N2 + EtOH + Na2CO3
(93)
(129)
(94)
NO
| D2O, (CH2OMe)2
MeNCONHj + KOD > CD2N2 + KCN'O + HOD (130)
(95)
Менее трудоемким способом получения диазометана является,
возможно, действие хлороформа п основания (т. е. дихлоркарбе-
на) на гидразин. В последнее время выходы в этой реакции были
увеличены за счет использования каталитического количества
18-крауна-6 в качестве катализатора фазового переноса [104].
Использованию и работе с большими количествами диазо.метана
препятствует опасность этого вещества, однако диазометан можно
генерировать и расходовать/^ situ в том случае, если сопряженное
основание кислоты, подвергающейся метилированию, одновремен-
но используется для разложения исходного соединения (уравне-
ние 131). Этим способом удается избежать возникновения значи-
тельных концентраций диазометана. Естественно, разложение
N-нитрозоуретанов основаниями используется не только как метод
получения диазометана (уравнение 132).
NO
| Z—X медленно
MeNCONH2 + NaO—( /—N°2 -------------*
НО—V у—NO2 + CH2N2 + N’aCNO
(131)
быстро
ON R
NOCI I I______
------>- EtO2CN—CHSO.Ar
R
E102CNHCHSO2Ar
R
ДД ,\=N=CSO:Ar (132)
301
нием ....-
зуюгщьхея дп<ь
свести к минимуму
П единения могут быть получены умеренным нагрева.
Дназосоед пс1" то/ИЛГИДразонов, причем разложение обра.
•'"1Т"С оосоединенпн за счет образования карбенов удаеТся
...........У если диазосоединсиия можно удалять пере-
;о“н\оп по мере нх образования /УР^^\с,УДе„J^,,^°^?»afor1I4.
ное разложение о-нг ,
днт под действием оспонанпя
AdCHO+ NH2NHTs
>--- л л и.ггппбензолсульфеинлтпдразонов (96)
ное Разлояу>шелунигрооензо(у)ав1е|111е
BuLI
AdCH=NNHTs ------►
нронсхо-
Li
I 70—120 °C
—> AdCH=NNTs , 10 MM pT. "•* AdCHN2 (133)
Ad = адамаитил
NO2
r.—\ Ar\ Et3N
/ "—SCI + /C=NNH2 --------►
NO2
—> Al\=NNHS—(Л ^C=N=N (134)
r/ (96) Rz
Окисление гидразонов до дназосоедпнешш обычно проводят
с помощью оксида ртути(П) (уравнение 135); используют оксиды
и других металлов.
О Ph О Ph
11 1 Hg0 11 1
PhC—C=NNH2 -----* PhC—CN2 (135)
(87%)
Для синтеза а-карбоннлдназосоеднпений разработано большое
число методов, имеющих препаративное значение; так, для полу-
чения эфиров а-диазокарбоновых кислот и а-диазокетонов исполь-
зуют нитрозирование соответствующих аминов. Активирующий
эффект карбонильной группы способствует отщеплению протона
в диазониевой соли еще до выброса молекулы азота (уравнение
136). Вместо азотистой кислоты рекомендуется применять изопен-
тилнитрит [106].
+ NaNO2
EtO2CCH2NH3 Cl' ------> [EtO2CCH2N2] СГ —> EtO2CCHN2 (136)
(80%)
При нитрозированин енамина (97) образование диазосоедине-
пня сопровождается присоединением нуклеофильного агента
(уравнение 137). Еще проще а-диазокетоны получаются из соот-
ветствующих тознлгидразопов при действии основания; основани-
ем может служить даже гидразин, используемый для получения
тозилгпдразоиа (уравнение 138). Аналогичную чувствительность
к действию оснований проявляют дназосоедппення. несущие заме-
стители Р=О и S=O (уравнение 139).
302
(60%)
TsNHN О Ns О
II II МеОН || ||
RC—Р(ОМ.е)2 + NaBH, ------* RC—P(OMe)j
(137)
(138)
Для исследований с применением фотоаффинной метки был
довольно просто получен 17-ди азоацетат эстрадиола (98) с по-
мощью тозплгидразона глиоксиловой кислоты, хотя и с низким
выходом (уравнение 140). Одним из способов введения диазоке-
тофрагмента в кольцо D (99) является обработка оксима хлор-
амином (уравнение 141). Этот метод первоначально был предло-
жен Форстером [107].
ОН
303
Prwuin переноса диазогруппы [102] особенно полезны в c„v
Ч1е „ревращения активной метнлеиовон группы в дпазофуик^
,с пользованием тозилазида и основания (уравнение 142f°
{g>Lt++ TSN3 — (3C=/TS [G^jN^HTs
—* O^2 *—* ON2
(142)
Известен ряд модификаций метода переноса диазогруппы.
К ним относится использование полимерносвязанного тозилазида
[108], который в отличие от тозилазида не взрывается при ударе.
Обработка реакционной смеси в этом случае сводится к фильтро-
ванию и упариванию. В другой модификации используют двух-
фазную систем}' и соль четвертичного аммониевого основания
в качестве катализатора фазового переноса (уравнение 143).
N2
R<N X". CII2CI2 II
МеСОСНгСОгВи-трет + TsN3 ------—— ------> МеСОССОгВи-трет (143)
о н. NaUH
(92%)
Одним из недостатков реакции переноса диазогруппы является
необходимость наличия двух активирующих заместителей (С=О,
Р=О, S=O и т. д.) при метиленовой группе. Моноактпвированные
метиленовые группы могут вступать в реакцию в том случае,
если активирующей является формильная группа. При осуще-
ствлении реакции переноса формильная группа удаляется (пере-
нос дпазогруппы с деформилнрованпем) (уравнение 144) [109].
Аналогичным образом, сложные эфиры и амиды диазоацетоуксус-
нои кислоты можно расщепить действием основания в условиях
переноса дпазогруппы в том же растворителе, который был ис-
пользован для их получения (уравнение 145) [110].
МеСОСВД —- MeCOCHR — N“‘ EWH>- McCocr 4- TsNCHO (1*9
(90%; R == СН2СН=СМе2)
14eCOCHsCOX-p TsN3 MeJcOX JcOX + MeCO2K d«)
X = QAlk, NEl2, — \
304
Превращение простых Диазосоединений в их
так,ке использовать в синтезе, посколькх L Р°ИЗвоДные мож-
обнаруж»вает неожиданную устойчивость aL™™ диазоФуикция
тана хлорангидрндамн кислот известно как п₽п? аИие диазоме-
цнп Арндта - Эистерта. Вследствие кислотное™ * СТадия Реак'
тоНа диазоацетатов или диазометилкетонов п Иового ПР°-
использованы для получения сложных эфипон -0»1111 МОгУт быть
карбоновых кислот или а-диазо-В-гидрокспкрЛ'диаз°-₽-гидрокси-
едпнения образующегося карбаниона к ал Л путем «рисо-
(уравиення 146, 147) [111]. Введение функционалын^*1 КеТонам
„вложение можно осуществить также с помо,Т„ * грУпп в это
диазосоединения (100) (уравнение 148) Kmn»» сеРебряной соли
на (101) с 1 моль бензальдегида (реакция Лап,яиТЯ ДИаз°кето-
прнводит к эпоксиду (102) [Ц2] (уравнение U9) избиРательно
R N2
МеОН - ЯСНО | ||
N:CHCO2Et + КОН -------[N2=CCO2Et] ----------► HOCHCCO2Et (146)
НО N2
ТГФ, — 78 »С | )
N2CHCO2Et + Ме2СО + BuLi ----------->• Ме2С—CCO2Et (147)
(76%)
n2 I N2 i n2
II II R1 f
Et02CCH+Ag20 —> LEtO2CCAgJ ------------► EtO2CCR (148)
(100) (66 %; R = аллил)
“он
C1CH2COCHN2 + PhCHO -------► Ph——COCHN2 (149)
О
(101) (102) (69%)
6.5.3.2. Реакции
Термолиз или фотолиз диазосоедпнений является известным
способом получения карбенов (см. разд. 2.3.4). Разложение дн-
азосоединений в присутствии солей некоторых металлов приводит к
образованию карбеноидов — частиц, в которых карбен и соль ме-
талла связаны друг с другом в комплекс; реакционная способ-
ность карбеноидов отличается от реакционной способности карбе-
нов [ЦЗ].
(1) Реакции циклоприсоединения
с°едпи>е3'“ЛЬТате PeaKllii*i 1,3-липо л яркого присоединения- диазо-
енин к алкенам получают пнразолины (уравнения 150, 151).
305
Объяснение региоселективное™ и реакционной способное?.,
пн ^соединений в этих реакциях циклоприсоединения было полу
Д £ пр^льтате рассмотрения двух наиболее важных взацн„
в?«° " немо. „;
Еых коэффициентов внутри этой пары [114].
(tso)
Во многих случаях выделенный пиразолин является таутомер-
ной формой первоначально образовавшегося продукта (уравнения
152, 153)
N2CHCO2Et + [Цо
(64%)
(152)
(153)
Выделяемые пиразолипы, в которых сохраняется связь N=N,
имеют особое значение как источники 1,3-триметиленовых бира-
дикалов, а следовательно, и циклопропанов, образующихся при от-
щеплении молекулы азота [60]. Если двойная связь алкена об-
ладает значительной электронной асимметрией, первоначально
образующиеся пиразолипы крайне неустойчивы и превращаются
в замещенные алкены путем отщепления молекулы азота (урав-
306
ga»H°e
яЛ1<1И|аМ
равнения
значение имеет также 1,3-диполярное присоединение
и двойным связям, включающим другие гетероаюмы
(91%)
S=C=NCO2R + CH2N2
N—х
—NHCO2R
s
(156)
Ароматичность пир азольного цикла нередко способствует
и 51-снгматропной перегруппировке первоначально образующегося
соединения (уравнение 157),
СО2Ме
+ II
СО2Ме
МеО2С
Me—
(38%)
СО2Ме
(157)
NCOMe
Равновесие, устанавливающееся между диазоимином и триазолом
(103) (которые являются продуктами 1,3-диполярного присоеди-
нения азидов к ацетиленам), может быть истолковано как внут-
римолекулярное 1,3-днполярное присоединение к связи C=N
[116]. Регноселективность циклоприсоединения к адамантантиону
в значительной степени определяется выбранным растворителем
(уравнение 158); в петролейпом эфире выходы соединении А и Б
составляют соответственно 87 и 13%, в метаноле — 22 и 78%.
(ЮЗ)
307
(2) Перегру1ЧшР0ВКЛ Вольфа [JJ7]
Прпегоуппировка Вольфа (уравнение 159) представляет собой
превращение диазокарбонпльиых соединении в кетены цри д
Хи света или температуры. Ее можно также катализировать со
лями металлов. Нередки случаи, когда образующиеся кетены не
выделяют; первичные продукты превращаются in situ в друГце
продукты реакции, как, например, при превращении карбоновых
кислот в их гомологи по Арндту —Эистерту (уравнение 160).
П1
Д, ftv или катализатор 4
— N2
(159)
R2C4
RCOCI + CH2N2
д rR\
rcochn* lr/c=c=o
LHZ
R\
—> ,CHCO2CH2Ph
(160)
Конкретный механизм перегруппировки Вольфа определяется
условиями проведения реакции, а также особенностями субстрата.
Во многих случаях стадии миграции и отщепления молекулы азо-
та являются согласованными, однако, как было показано недавно,
некоторые фотохимически инициируемые случаи перегруппировки
Вольфа протекают (по крайней мере, частично) через промежу-
точное образование оксирена (104) (уравнение 161) [118]. Пере-
группировку Вольфа широко применяют для сужения циклов
(уравнение 162).
О N2 о
1 II ftv II ..
PhC—CPh —-> PhC-CPh
• — Кг *
C = I3C
(104)
Ph\
Y~C=0 (161)
P1Z* ‘
ftv>300 HM
TpeT-BuOzCNHNHa
NHCChBu-rper
NHCO—j—j'^Me
o=l—NH
(162)
Образованию карбена
соединению к двойной
собствует добавление
308
(карбеноида) п внутримолекулярному при-
связи в ходе перегруппировки Вольфа спо-
солей металлов (в частности, меди). Лег-
_ получения диазокетонов делает их особенно пр,!ВЛекаТРп.
< " го^и зрения синтеза напряженных циклических систем
||UM" в состав многих веществ природного и 11Р,’
8^’^дения, например (105) (уравнение 163) (119]. Р Р Д °
Л|> о
(163)
(105) (30%)
(3) Нуклеофильные реакции
Диазосоединения чувствительны к действию кислот; в основе
использования диазометана в качестве реагента для метилирова-
ния карбоновых кислот лежит стадия предварительного протони-
рования с образованием неустойчивой соли дпазония (106) (урав-
нение 164). Диазометаном можно также метилировать фенолы,
енолы и даже спирты (в присутствии кислотного катализатора)
Известны примеры алкилирования гидропероксидов (уравнение
165) [120].
н+ + rco2h
CH2N2 CH3N2 * RCO.Me (164)
(106)
Me2CPhCO2H + N2=CR' R2 —► Me2CPhCO2CHR'R2 (165)
Циклизация диазокарбонил-Л’-ппразолинов в присутствии
кислот была использована Моором и сотр. [121] в качестве мето-
да получения диазабпцикло[3.2.0] гептенонов и 1,2-диазепинонов
(уравнение 166). Аналогичным образом, эфират трехфторпстого
бора катализирует реакции внутримолекулярного внедрения дп-
ДЗокетонов (107), в то время как в присутствии медного катализа-
тора в результате внутримолекулярного присоединения карбена
шразуется циклопропан (уравнение 167).
1рехфтористый бор превращает также упомянутые ранее а-ди-
М-гидрОКсикетоны (108) в ацетилены с хорошим выходом
Уравнение 168) [122]. Реакции солей диазония, генерируемых в
ит°ВИЯХ Нротоиироваппя, не всегда, однако, связаны с отшепле-
' м Молекулы азота; иногда наблюдается атака по атому азота,
соответствует реакции сочетания ароматических дпазониевых
(Уравнение 169) [123].
|Рпи,еакЦпи триалкилборанов со многими дпазосоедпненпямп, со-
»поив1Ш,"М" Функциональные группы, позволяют спнтезпрова
воХ ’ Мегилы, нитрилы, сложные эфиры и их а-деитеропро з-
В11я П\тем последующего протоиолиза (дейтеролпза) (>Р
1,и—172) [124].
309
OH n2
I II
R'CH—CCOR2+ BF3-OEt2 —» R’C^CCOR2 (168)
(108)
I. — N2
R3B + N2CHX — - - > RCH2X (I70)
2. I12U
X = OHO. CO2R, CN, COR
D
R3B + N2CHX ; n Ыг> RCHX U7I>
2. P2O
О No и PJ+
II II fl f
BEt3 + CC~C'') ТГО.НгО» ^C—CH- (172)
\CH2)4> <(CH3)4-
(98%)
310
^клеофи^ное присоединение диазосоедш.ений лежит в осно
реакций с карбенпевыми ионами (уравнение 173) с ?ало
86 ямн (УРаВНеНИе } (ЭТ° Х°РОШИИ мет°Д получения гельдига-
ГС" Хов), с синглетным кислородом и альдегидами. Последняя
•'“акция позволяет синтезировать озониды (уравнение 175) [125].
(173)
(174)
X == галоген
R!C-N2+‘O2 —*
ph3C C10J + CH2N2 Et20 * lPh3CCH2N2] •—► Ph;C=CHPh
R2C=N2 + X2 —> Г R2CN2 X -| -—-> R2CX2
I I I ~N2
Lx J
(175)
Пербензойная кислота реагирует с дифенилдиазометаном, да-
вая бензофенон с высоким выходом; в данном случае атака угле-
рода при диазогруппе направлена на электроположительный атом
кислорода, а не на кислый протон (уравнение 176) [126].
X) т,
РЬС^( / Н Ph
PhCO,H + Ph2C=N2---*• \< /~^ч1 + ---*• PhaCO + PhCO2H
°-q _CTN=N "N2 zl7»)
| U I176/
Ph
Нуклеофильная атака дназосоединений по карбонильной груп-
пе альдегида или кетона приводит к эпоксидам или к высшим
гомологам исходных соединений (уравнение 177). Эта реакция
редко представляет интерес с препаративной точки зрения, по-
скольку обычно происходит дальнейшая гомологизация образовав-
шихся кетонов, в результате чего возникает смесь веществ. Более
А
R\ /O'
X
R2/ \ch2n; r2-
R1
ch2n2 -f-
O О
R'CCH2R2 + R2CCH2R'
Me Me
Me Me
=0 + N2CHCO2Et
BF3-OEt2
=0
(177)
(178)
CO.Et
(96%)
311
того, в тех случаях, когда кетон несимметричен, не всегд
добиться региоспецифичпости на стадии миграции. Однак УДав1ся
ренне цикла в ряде а-моно- и а,а-дизамещеиных цикло° РасшИ-
нов и циклогексанонов при обработке этилдиазоацетатом ,Та”0-
сутствин трехфтористого бора протекает с высокой региос В При'
ностыо путем миграции менее замещенного а-углепо™ СеледтИв.
(уравнение 178) [127]. Рудного aT01fa
Внутримолекулярные реакции диазоалканов с ка
соединениями также протекают более успешно (ур
[128], даже в случае сложных эфиров (уравнение
.0
Рб°ннльНЫЫи
авнение 179)
180) [129/9)
. •fcH2)2CH=N2
CO2Et d
(180)
(4) Электрофильные реакции
Диазоалканы довольно инертны по отношению к основаниям,
и многие из отмеченных выше методов получения включают ис-
пользование оснований. Однако сильные нуклеофильные
обычно вступают в реакцию, атакуя концевой атом азота
иення 181 — 184).
О
„II
RCCHN2 4. Li А1Н4
Ph2CN2 + r/,ej..BuMgC1
R2CN2 4. p3p ____
агенты
(урав-
(181)
(182)
(183)
(184)
312
О
II
—>- RCCH=NNH2
—f Ph3C=MNHBu-rpcr
1?2C=N—N=PPh3
(109)
Среди прочих реакций диазосоедииений
Фотохимическая изомеризация а-д11азоП,Ици пРеДставляет
:,ого света, приводящая к Диазнр^Х)"?
Д)Я относится к серии ЭлеКТроцИКЛ][130]. Эта ’
с„стеМы оксазиридин — нитрон, а ™ Реакций, включав
-карбошм-илид н другие. Н ~ аз°метннилил
А; ^ПОК'
СИД
6.5.4. АЗИДЫ [131]
Аналогия между азидами и диазосоединенн«..„ й
ранее (см. разд. 6.5.3). Азиды легко обнаруживают ЫЛа 0Тмечена
вескими методами (vM1KC 2140—2240 см"1) аюТся спектроско-
6.5.4.1. Методы получения [132—134]
Наиболее распространенным методом синтеза алкилазидов
является нуклеофильное замещение. Значительная нуклеофиль-
ность азидной группы может быть усилена за счет применения ди-
полярных апротонных растворителей, таких, как ДМСО, ГМФТА
и ДМФ. С успехом применяется катализ фазового переноса.
К обычно используемым уходящим группам относятся галогениды,
тознлаты, мезилаты и т. д. При синтезе 2'-азидо-2'-дезоксиурндииа
1110) происходит замещение не совсем обычной уходящей'группы
[135]. При проведении этой реакции часто наблюдается некоторая
избирательность; например, при обработке тримезилата (111)азид-
ионом (1 экв) получают (112) (уравнение 186) [136].
LiN3, ГМФТА
150"С
“W
MsO
(til)
NriN:1
ГМФТА
(18»)
(18в)
С'
Ц11Л П°М011Ч110 нуклеофильного замещения можно получить также
’ сУльфоппл- н другие азиды (уравнения 187—191).
_ MeCN
C1CN + NaN3 --------> N3CN + NaCl
о О
’’Per-BuOCCl 4- n; H2N=C(NMe)2 --C-Ia'—+ rper-BuOCNs
(187)
(188)
313
MeCN
CIS0-C1 + NaN3
дмф
IC1 + NaN3 — :
MeCN
NO?BF; + LiN3 —' ~
N3SO,C1 + NaCl
IN3 + NaCl
NO2N3 + L1BF4
(189)
(190)
(191|
Наблюдается нуклеофильное замещение как при азидной гВУп
не, так и с помощью азидной группы осооеш,ю при использовании
п-толуолсульфонплазида (уравнения 192-194) [137].
N3Ts
-
(27%)
(W2;
N3
МсОСН2СН2ОМе I
RC(CO2Et)2 + N3Ts ------------> RC(CO2Et)2 (193)
RNH2+N»SO2CF3 —> RN3 +H2\’SO2CF3 (1M)
Присоединение азида пода (получаемого в виде раствора в
полярных растворителях) к алкенам протекает регио- и стерео
специфично [138]. Образующиеся транс-|3-иодалкплазнды могут
быть превращены в ряд соединений, включая а-азидовннплкето-
иы (113) (уравнение 195).
/ \
рь со н
1N3
МеСЫ
NaN3
ДМФ
(ИЗ)
(195)
Взаимодействие алкенов с азидом ртути(II) и демеркурирова-
нпе^выглядит как присоединение азотнстоводородной кислоты к
двойной связи (уравнение 196) [139]. Комбинированным воздей-
ствием диацетоксииодбензола и трнметилсплнлазнда с последую-
щим хроматографированием иа силикагеле алкены превращают
в а-азндокетоны (уравнение 197) [140].
+ Hg(Ns)2
Na ВИ,
Ns
-C-C-
HgN3
(196)
+ Phl(OAc)2 + Me3SiNs
CH2ci2
(197)
(48%)
314
азид-
наирав-
, но
(198)
(199)
Ar'N’a (200)
дпнлазнлы обычно получают нитрозированием арилгидпазин™
Soie 198) '»"• Реакцией азида с солями диазония (уравне
O'PJiq9) Если имеется только арилгалогенид, то иожпо п„»
1,1,6 последовательность реакций, изображенных уравнением 200
н'^ая в качестве заместителя в ароматическом “ e
группа проявляет себя как электроиоакцепторная
"’’дальнейшее замещение в орто- и лара-положения.
'1Яе ArNHNH, + HNOj —> ArN3
ArNj X'+NaN3 —> ArNa
Ar'MgX ArSOjN—N=NAr'
Ar‘A
6.5.4.2. Структура и устойчивость
Отшепленне молекулы азота из органических азидов можно
осуществить фотохимическим или термическим путем (уравнение
201). Образующиеся соединения могут быть идентифицированы на
основании их реакций как нитрены (114) (см. разд. 6.6.4) [141],
однако часто отщепление азота протекает синхронно с процессами
миграции других связей. Это означает, что свободный ннтрен
не образуется, как в случае перегруппировки Курциуса (уравне-
ние 202).
А или hv ••
R—N3 --------->- N, + R—N:
(114)
(201)
R-/
R—N=C=O
(202)
О"
Термическая устойчивость различных азидов колеблется в ши-
роких пределах. При термическом разложении азидов константа
скорости достигает значения ~10“3 мин-' в случае алкилазпдов
при температуре около 190 °C, а для алифатических ацилазндов
при температуре немного выше комнатной. Эта разница в устой-
чивости может быть объяснена резонансной стабилизацией
в ацилазидах, которая понижает степень двоесвязанности между
N-1 и N-2, благоприятствуя тем самым выбросу молекулы азота
(Уравнение 203).
О о- О
11 1 С <• -
/ \n—N=N \<=N=N (203)
Термическая устойчивость азидов уменьшается в следующем
Ряду: алкнлазиды > арилазнды > азндоформиаты, сульфонилази-
> ацилазиды. Однако известно много случаев, когда выбросу
скулы азота способствует участие некоторых других фу
етгя°ЛиКуле' в Результате чего температура разложения пон
ся' Наиболее вероятно, что фотохимическое разложение аз! д
315
ячота приводит к образованию настоящего нтре.
с отшеплениеы азота Р и01,,10Щ.110Т в длинноволновой области
на- Арнл- и ДХ? возможным проведение их фотолиза в пр|,.
“S'- других хромофоров.
6.5.4.3. Реакции [142—146]
Исчерпывающему рассмотрению нитренов, которые часто вы-
ступают в роли промежуточных соединении, образующихся При
фотохимическом распаде азидов, посвящен разд. 6.G.4. Считают
что разложение арплазидов приводит к образованию синглетного
нитрена, который находится в равновесии с азирином (М5)
В присутствии дпэтплампна получают азиридин (116), который
путем электроциклпческого раскрытия цикла и таутомерного пре-
переходпт в азепнн (117) [147]. Обычно при раскрытии
азиридинах образуется также некоторое количество
вращения
цикла в
Продукты реакции обоих типов наблюдаются при фотолизе ази-
добензо [6] тиофена (118) [148]; аналогичные направления реак-
ции характерны для фотолиза о-азидобензоатов (119) в спиртах
(13%)
(следы)
316
СО;Ме
Sv. ROH
(119)
Ч I Н
N^xoi?
(206)
другие центры молекулы могут 3d>rhB.
с арильным кольцом за захват нитрена L „ ДХл КОнкУРироеаГь
[V молекулы азота. Это проявляется в боле? бегвовать выбро-
гемператур, необходимых для разложениег (уравХи^ 207"Т Х
317
Ароматические азиды могут быть использованы
Ароматические азиды могут быть использованы в качестве
фоточувствительных защитных групп (уравнение 215). При
лизе генерируется свободная кислота с выходом 60—70%
ОМе ОМе
фото-
1150].
+ PhCO2H
(215)
азида,
N3 СН2—OCOPh
Межмолекуляриая реакция нитрена, генерированного из
может наблюдаться лишь в том случае, когда ароматическое коль-
цо несет заместители с сильно выраженными электроноакцептор-
ными свойствами (уравнения 216, 217) [151].
л \ 132 °с
Ns—< х)—CN + PhNMe2 ---------->
NMe,
!—~CN + Me2N-
I—NH——CN (216)
(25%)
(3, 4%)
Me0\A/Ns МеО. N N
f II + RCN
NV"
N'
:n
ОМе
МеО R
О;
Л
Me
N N
/
MeNVNpin
0 R
теРмолиза или фотолиза арилазидов в прнсут-
оазуются'нр \К"СЛ0Т может приводить к продуктам, которые об-
Р нз нитрена, а в результате превращений протониро-
318
„гП азида или иона питрсния (уравнения 218 219) flW
’““поведеиие диазониевои соли и карбеш.евого пока при ката
(ср'руемом кислотами разложении диазосоединений; разд 6 3^
Результаты измерения термодинамических параметров процес-
са термолиза вннплазндов также указывают на то, что в ходе
реакции не образуются свободные нитрены (уравнение 220), и,
следовательно, эта реакция аналогична перегруппировке Курциу-
са [153]. В противоположность азнрпнам, которые, как предпола-
гают, образуются в ходе разложения азидов, но выделить которые
не удается, устойчивые азирины получают из вннплазндов [154].
Огщепление молекулы азота нз азидной группы иногда вызывает
распад всей молекулы, особенно в том случае, когда это способ-
ствует отщеплению еще одной молекулы азота (уравнения 221 —
223).
ZCN
PhN=c;
\ph
(220)
NPh
(221)
(85%)
(222)
(79%)
319
R
2 /*=С=0 (223)
NC
ц=Вй-трет
Ппклопропплазиды стереоспецифпчески распадаются до 0Ле.
ф„на п нитрила (уравнение 224); наблюдается также образование
азетина (уравнение 225) [155].
[ Me
(85%)
(224)
Ph
Cl
(55%)
(225)
Продукты перегруппировки при термическом разложении мно-
гих азндохинонов и азидосодержащих гетероциклических систем
были изучены Моором с сотр. [156]. Некоторые примеры этих
превращений приведены в уравнениях 226—229.
(229)
(1) Реакции циклоприсоединения
Азиды относятся к многочисленному семейству 1,3-диполярных
агентов, которые вступают в реакцию циклоприсоединения с ал-
кенами. Очень легко реагируют алкены с напряженными двойны-
ми связями, давая трназолины, которые часто оказываются не-
устойчивыми. Сопряжение с фенильными и сложноэфнрными
группами также увеличивает реакционную способность двойной
связи (уравнения 230—232).
МеО2с Н
+ PhN3 —>
о’я
со2ме (в1%)
PhCH=CH2 + PhN3 ->
(231)
PhN 4N
Ph A-'
H
Особенно реакционноспособными являются енамины и простые
эфиры енолов, а перегруппировки промежуточно образующихся
триазолпнов приводят к сужению цикла (уравнения 233, 234)
1*57]. И наоборот, присоединение к экзоциклической двойной свя-
зи может приводить к образованию продуктов расширения цикла
(Уравнение 235) [1581. .
Распад триазолннов может включать промежуточное образо-
вание бетаина (120), который в результате перегруппировки Ва
(1291~^МеервеЙ11а превращается в имины (121) пли “''J
(*22) (уравнение 236) [159]. Конечно, образование азиридин
11 Зак. 105
R = CN, SO,Ar
азидов и алкенов может происходить через нитрен, однако воз-
можно альтернативное направление реакции, которое включает
циклоприсоединение, приводящее к трпазолину, с последующим
выбросом молекулы азота [160].
MesC=CH2 + NaCN —>
Ме2С—СН2-1
NC—1>/ \
N -I
1
NC-N
1,
ме/ \:н2Ме
(121) (48%)
Ме2С—СН2Ы2
I
‘N—CN
(120)
(122) (34%)
(238)
включая дегидробеизоч' р!соедниение азидов к ацетиленам,
(уравнения 237—2301' I водит к более устойчивым триазолам
322 отя в слУчае (123) методом ЯМР можно
обнаружнть существование равновесия между триазолом и а-ди.
азоимндатом
Примеры циклоприсоединения к кратным связям, включающим
гетероатомы, приведены в уравнениях 240 и 241; азнд-тетразоль-
ная изомерия [161] (уравнение 242) более удовлетворительно
описывается как электроциклическая реакция.
N=N
C4H8N3 + ArSO2N=C=S -► C4HgN S (241)
Y
NSO-jAT
•N3.
( I И
<N----N
В тех случаях, когда механизм реакции исследовался, было
установлено, что эти реакции циклоприсоединения являются син-
хронными. Это примеры [4-, + 2л]-циклопр11Соединсн11я, являюше-
гося супраповерхиостным по отношению к каждому к0МП0"е”-•
Как и в случае диазосоединений, регпонзбнрательность пр i
нения к алкенам (уравнение 243) может быть о ъясн
предсказана [114].
(242)
R'N.
R2 /YY />
+xv
N N
(2431
R2
323
11*
„ п „«иное циклоприсоединение азидов к енаминам чрезвы-
1,3.'Д Лнюспецпфпчпо для самых разнообразных азидов и ена-
чаино в реаКцПю. Во всех случаях концевой атом азо-
таазндной группы образует связь с углеродным атомом, несущим
азот в енамине [163].
(2) Восстановление
Алкены удается стереоизбирателыю превратить в азиридины
путем присоединения азида пода с последующим восстановлением
LiAlH (уравнение 244). Восстановление азидов до аминов, ко-
торое можно проводить также и каталитически, может быть ис-
пользовано в синтезе. Избирательное восстановление азидной
группы в присутствии двойных связей и карбонильных групп [163]
можно осуществить с помощью катализатора Линдлара. Альтер-
нативный метод превращения азидов в амины заключается в об-
разовании фосфииимпиа и последующей обработке его аммиаком
(уравнение 245) [164].
Н Me
Хс=с
/
Me
м hie
N3 & NH
,ы \ uaih4 / \
rf,C—C -----* Hwy—(244)
Me H
Me
H
n5-h2c o Th Ph3l,
ph3p=N—H2C о Th
NlljOH
RO
RO
(245)
HjN-HjC о Th
Th=тимин
RO
Азиды могут подвергаться нуклеофильной и электрофильной
атаке, хотя они обладают основными н нуклеофильными свой-
ствами в меньшей степени, чем дназосоедннения. Концентриро-
ванные кислоты вызывают отщепление молекулы азота и пере-
группировку алкилазндов с образованием альднмпнов и кетими-
нов (уравнение 246); арилазнды превращаются в анилины с
заместителями в цикле (уравнение 247). Вероятно, образование
(124) [165] включает нуклеофильную атаку триметилсилилазида
по карбонильной группе ангидрида (уравнение 248).
Txv H2SO4 ]]2()
РадНМз ---► Ns+PhCH=NPh ----* PhCHO+lhNPli (246)
U1SO, Л--\ +
CtH6Na ---г ио——NHS HSO~ + N2 (2V)
324
/ \ 0 ' Mc2SIN3
\ / 2. Elon
О
NH
(248)
О
(124) (69%)
Дцилазиды в присутствии кислот перегруппировываются в изо
цианаты, а изоцианаты в этих условиях претерпевают гидполиз
и декарбоксилирование. При проведении реакции Шмидта ази™
получают in situ из карбоновых кислот, минеральных кислот и
азотистоводороднон кислоты; общим результатом реакции явзяет-
ся превращение кислоты в амин, сопровождающееся потевей
одного углеродного атома (уравнение 249). 1
H2SO4, HN3
rco2h ----------►
RNH3 + CO2 (249)
Азиды не особенно чувствительны к действию нуклеофильных
агентов; р-галогенэтилазиды при действии спиртового раствора
щелочи превращаются в винилазиды. Однако нуклеофильные
углеродные центры атакуют концевой атом азота в азидах, в ре-
зультате чего образуются триазены (уравнение 250) пли триазолы
(реакция Димрота; уравнение 251) [166].
Ar‘SO2N3 + Ar2MgX —> Ar‘SO2N=NNHAr2 (250)
R\ ,CO,Et
[RN=C=O]
“OEt
R'COCH2CO2E1 + R2N3 ------->- R1COCHCO2Et
N=N—NR2
R2N' 'N (251)
N
Арилсульфонилазиды широко используют для перенесения
диазогруппы на активированную метиленовую группу (см.
разд. 6.5.3.1). Реакции этих азидов с различными замещенными
индолами изучены Бейли с сотр. [167]. Для ацил- и активирован-
ных арнлазидов характерно замещение азидной группы (уравне-
ние 252) (а не атака но азидной группе) нуклеофильными аген-
тами; способность ацилазндов выступать в качестве ацилирующих
агентов использована как метод создания пептидных связен
(Уравнение 253) [168].
О
О'
R3CN3 + R'R2NH
R‘
Cl' R3
R3CNHR'R2
R2CONHCHCO2H + II3NCHCO2H
О
R3CNR‘R2+HN3 (252)
Ns
О
»зР(0Р1^ r2CONHCHCONHCHCO2H
ДМФ, EtsN (253)
R1
Rs
325
6 55 СИСТЕМЫ, СОДЕРЖАЩИЕ ПОЛИАЗАФРАГМЕНТЫ
свободных электронных пар при трех или более
Нал,,ч1,е “° „ота находящихся в состоянии 5Д3-гибридИзае
смежных атомах азо , дестаб1)ЛИзующее влияние. Цепи Из
дни. маэыва”’, пятп атомов азота обладают устойчивостью, до.
7реХ’ Чепй оя выд леиня соответствующих соединений, лишь втом
статочной ,для выде атома аз0Та находятся в состоянии
случае, когда два i подобных соединениях присутствует
^"±,^aN=NTpynn..poBKa. Извинений этого типа лучше
аМ"п"и«чены трчазеиы. Наиболее известный способ их получения
во взаимодействии ароматических диазонневых солей
Гамшами (уравнение 254) [169]. В основе других методов полу-
«ет пежит реакция диазонневых солеи с анионами амидов
(тавиёние 255) и присоединение реактивов Гриньяра (уравнение
256) или триалкилалюминия к азидам. N-Оксиды триазенов легко
получают путем сочетания диазонневых солеи с гидроксиламина-
ми (уравнение 257).
1 + ArNH2
(254)
(125)
-30 °C
PhNj Cl' + (Me3Si)2NNa ---► PhN=NN(SiMe3)2
s s
Г jT Me + RMgX —> -Il —Me
ArN2* + R2NOH —► ArN=N—NR2
(255)
(256)
(257)
ЩпзоиДЗРявля1°тся алкилирующими агентами
давать /я ’ эт0 свойство в сочетании со способностью
пппчинлй" Л ние1!Ые производные при действии кислот может быть
кислого ппотпняРОГе11И°СТ" МНОгих из этчх соединений. Наличие
чировкой вызыВаеГпхГреаРзлХшарТОИе рЯД°М С тРназеиовой гру"'
ствви оснований н тпп^ени до диазосоеди11еи1Ш в присут-
ниями в реакциях прприп™ являются промежуточными соедине-
отсутствии кислого прОтоичД",а3°гРУППЬ1 (см' УРавнеине 142). При
санной выше реакции 1а’ "еобхоДнмого Для протекания опи-
дентиого аниона TDin4PnJ°/KeT пР011СХ°Дить алкилирование амби-
а трпазена (уравнение 259).
326
PhN-NNHBu + ArCOjH
ArCO2Bu
(258)
ArN=NNHAr*
ArN—N—NAr1
RX
zAr' Ar
—► ArN=NN + ;NN=NAr>
XR r/
(259)
g3 данных спектров ЯМР следует что 1 3
м в растворе часто находятся в виде смё™“ещеннь1е триазе-
Спектры ЯМР, снятые при низких температурк и™ Sp°B ll7!l-
сгвуют о наличии ограниченного вращения свидетель-
у—N-связи. Термическое разложение фенилз™ ®°Круг простой
нов приводит к фенильным радикалам уравнешТосп^' трназе-
ложение изучено методом ХИДПЯ в спектрах ПМР0): ЭТ0 Раз'
спектрах ЯМР ,3С продуктов реакции (бХл ^Л.Также в
Л1,н) [173]. В этих условиях распад трназена приводит . 1 аНН’
катиону через промежуточное разложение диа3пнио -феНИЛ'
Триазены образуют устойчивые комплексна Д'азониевои соли,
гимн металлами [174]. соединения со мно-
PhNHN=NPh > Ph . +. NHPh + n,
(260)
Известно лишь несколько примеров замещенных триазанов,
например (126). Неустойчивость трех атомов азота, связанных
о-связямп, продемонстрирована на примере электроциклического
раскрытия цикла в (127), в результате чего образуется более
устойчивый илид (128) [175]. По-видимому, более устойчивыми
являются соли триазанпя (129) [176] илн бетанны триазання
(130) [177].
CO2Et
PI1NH2 + Et2OCN=NCO2Et —> PhNHNNHCO2Et (261)
(126)
327
.—> H2Nv + / v t
HNZ 'Nil + NHsOSOjNa —> N' Ж 2M,
V_/ H2n/ V_/ \Nh2 3 (263)
(129)
NH2
I -
RJNNHг+ R'——NCONHR’ R]N—NCONHR2 + R.CHo
(130)
Тетразены значительно более многочисленны, чем тетразаньг,
что также является результатом стабилизующего влияния нена-
сыщенности этих систем. Тетразены-1 получают азосочетанием
даазониевых солей с гидразинами (уравнение 265). Тетразены-2
обычно синтезируют окислением 1,1-дизамещенных гидразинов
(уравнения 266, 267); с формальной точки зрения тетразены-2
являются продуктами димеризации ампнопитреиов. Незамещен-
ный тетразен-2 (131) был выделен при протолизе сполна силили-
рованного производного (132) (уравнение 268) [179].
R
I
ArNj + RNHNH2 —► ArN=NNNH2
(Me3Si)2NN=NN(SiMe3)2
СГ3СО2Н. CHjC12, -78 °C
------------------> NH2N=NNH2
(132)
(131)
(265)
(266)
(267)
(268)
При нагревании или в условиях фотолиза тетразены разлага-
\°Тйп1 С °бРазоваи|1ем азота и аминорадикалов (уравнение 269)
1180). Подобно триазенам они образуют комплексные соединения
с металлами, например (133) и (134) (уравнения 270, 271) [18И-
(269)
Me2NN=NNMe:
N2 + 2Me2N •
CqF3N3 Njp2
Nil6
C0Fs-nx xN_Cops
N==Jl
(270)
(133)
MeN3 + Fe2(CO)9 ------к
n=n
WJjNMe
Fe(CO)3
(134)
(271)
иные тетразаиы имеют большое число замес-
синТезИРоваv ичй'' Г'ол1 " растворе гексафенил-
Вс.. w......... радикалы
уста-
цсе v>m.---,
«пгелей, например (135) и (13g) большое чисто ,
тегразан обратимо диссоциирует и ’ В Р”'”"’"-- мес’
(уравнение 274). Структура диполярной 1"1Дрази-'1Ь«ые Da ‘
„озлена методом рентгеновской дифр акади Д П37> была
(272)
(135) (90%)
СО.Ме СО2Ме
80 «С | I
► PhN—N—N—NPt,
I I
CO2Me СО-Me
+ MeO2CN=NCO2Me
(273)
Ph Ph
I I
Ph2NN— NNPh2
(138) (69%)
(274)
2Ph,.V—NPh
(275)
(137)
Некоторые пентазадиены (138) были получены азосочетанием
Диазонневых солей с первичными аминами 1184]. Их разложение
протекает со взрывом.
Me
2ArN£ + MeNHi - °"''’ ArN=NNN=NAr (2/6>
(138)
1. Е ЛИТЕРАТУРА
Chemie (НоиЬепЛ^7б»-
3 j?6®. vol. 2. U ’ “01>en Chain Nitrogen Compounds», Benjamin, New York,
<lenile, New Yoi b''VoJo' '^<г/'0’ «Organic Functional Group Preparations», Аса-
и,к> 1Уоо, Vol. 1.
329.
7.
8.
9.
10.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
i hob 7 Chem.» 1968» 8, 41.
л p r Stowell, in «The Chemistry of Hydrazo Д,л
1- J Г Timberlake end /. ^.^lersc’ience, New York. 1975. part 1. ’ Аго «hj
«ftTs M ^ler' 1 r‘ McLe“n' °"d L'
^Тс^^Я^^зГ'1963’85'214'’-
'K^'T^bedZlZs2^,
D. M. Maytum and В R. 1. Л« .
H. Zimmer andG. Smg^ * Qrg Cllclll1975, 40, 1213.
0 feta V * ЛШег. U- Hom- - Joos‘ L Scl,reiber- a,,d A- Cschenmoser>
П 1975’ 4°’
G. AFCHuang.T. °Okamoto,M. Maeda, and Y. Kautazoe, Tetrahedron Letters,
J,9'j; and R Turley, Z. Nalurforsch., 1973. 28b, 829.
E Schmitz, «Dreiringe mit zwei Heteroatomen», Springer-Verlag, Berlin, 1967,
fH.9E. Baumgarten, P. У.-А. Chen, H. IV. Taylor, and D.-R. Hwang, J. Org.
Chem.. 1976, 41, 3805.
] L Meisel, Tetrahedron Leflers, 1974, 3847.
«. Zollinger, «Diazo and Azo Chemistry», Interscience, New York, 1961,
p. Ю9
F. Yoneda and T. Nagamatsu, Synthesis, 1975, 177.
F. D. King and D. R. M. Walton, Synthesis, 1975, 738.
y. Shvo, in «The Chemistry of the Hydrazo. Azo and Azoxy Groups»,
ed. S. Patai, Interscience, New York, 1975, part 2.
24. S. F. Nelsen and J. M. Buschek, J. Amer. Chem. Soc., 1973, 95, 2011.
25. R. А. У. tones, A. R. Katritzky, K. A. F. Record, and R. Scattergood, J. C. S.
Perkin 11. 1974, 406.
26. G. R. Weisman and S. F. Nelsen, J. Amer. Chem. Soc., 1976, 98, 3281.
27. Cm. cc. 23, p. 1043 и сл.
28. G. R. Weisman and S. F. Nelsen, J. Amer. Cliem. Soc., 1976, 98, 7007.
29. M. J. S. Dewar and U7. B. Jennings, J. Amer. Chem. Soc., 1973, 95, 1562.
30. S. F. Nelsen and J. M. Buschek, J. Amer. Chem. Soc., 1974, 96, 6982.
31. C. G. Overberger, J. P. Anselme, and J. G. Lombardino, «Organic Compounds
with N—N Bonds», Ronald Press, New York, 1966.
32 j C’ "7“c Chemistry of the Hydrazo, Azo and Azoxy Groups»,
11 , Pa a lnterscience, New York, 1975, part 2.
'„лпеси s' D' Dats,n- Amer. Chem. Soc., 1972, 94, 6998; F. Fillipini
34 Я л’ 12l.S Chem' Comm- W2’ 522.
35 N P p £ ’cJ cOr^ Chem' 1968' 33- l374-
36 AR »cC’arSti,n"wek л>гй’?’ Cre^ J' Огй- Chem., 1976, 41, 2733.
37 J-'chem. Soc (Bl'i969°||85A- W’ Я 8arnes- L- E- Sutton' and C- Ainsworth,
'* 1973, 38, 1344^ S’ J°l'nson. %- A. Abgotl, and M. J. Madden, J. Org. Chem.,
[О. П. ШвХа“ ваПН АотЛ Ar,^nm- Buss- Chem. Rev., 1972, 41, 833
R. A. Cox end'e\НшАр.Tf-Vw. химии, 1972, 41, 1788].
Groups», ed. S Patai Ini'pi-Pr Cliemistry of the Hydrazo, Azo and Azoxy
«7- !. McKiltip eTs № Y°^ !975' 2art 2.
1973, 73, 255. ' ' Culbertson, and S. Wawzonek, Chem. Rev.,
°- -j1- temal^in ^Nitrcnm'5^' Chem- Soc., 1974, 96, 1788.
P- 345. * ed- W. Lwowski, lnterscience, New York, 1970,
/. Anderson T I r„i i .
(Q, 1970, 576.' ’ ‘ tuis 1 D1 c- Harwell, and C. W. f^ees, J. Chem- Sop.
38.
39.
40.
41.
42.
43.
330
93, 788.
..........'-г. Chem.
ранние работы.
Stable
Amer.
2745.
n Aulward, Quart Rev., 1971, 25, 418.
44. ! r Raidwin, ! £• Brown, and G. lidfle, J. Amer c
45. L "ц Urry, 2- L. E. Gatbel, !. C. Duggan, and S. S Tseng’s д71, !
46. IQ73 95, 4338 и приведенные там ссылки на Г»,™,4, Amer-
ff№- Berio, ond S ”0<>,0n' Chem. Rev. ?965 65 T" ' -
47- j' R Forrester, J. M. Hay and R. H. Thomson, «Organic Chemist t
48. a Radicals*. Academic, London, 1968. g C Chemistry of
„ Tr ».»;», V“’ ° ’ «»«. C
. с Д. Neugebauer and H. Iaeger Tetrahedron Letters, 1976, 2083
c, $ F. Nelsen and R. T. Landis, J Amer. Chem. Soc., 1973 95 644
И F A Neugebauer, Angew. Chem. Internal. Edn., 1973 12 455
« В H. Al-Sader and R. !. Crawford, Canad. J. Chem., 1970 48
0 J Crawford and K. Takagi, J. Amer. Chem. Soc., 1972 94 74(p-.'
P S Engel, Tetrahedron Letters, 1974, 2301.
« J W Timberlake, M. L. Hodges, and K. Betterton, Synthesis 1972
приведенные там ссылки.
56 P. L. Grizzle, D. V. Miller, and S. E. Scheppele, J. Org. Chem 1975 40
1902.
к? IP Dulsmann, H.-D. Beckhaus, and C, RUchat Tetrahedron Letters 1974 2H5
« J Buckingham, Quart. Rev., 1969, 23, 37. ' У
59 E. Schmitz, Adv. Heterocyclic Chem., 1963, 2. 122.
И K. Mackenzie, In «The Chemistry of the Hydrazo, Azo and Azoxv Groups»
ed. S. Patai, Interscience, New York, 1975, part 1. . и ,
61 S. Masamune, N. Nakamura, and J. Spadaro, J. Amer. Chem. Soc., 1975 97
918.
632 и
62. E. M. Kosower, Accounts Chem. Res., 1971, 4. 193.
63. I. M. Hoffman and R. H. Schlessinger, Chem. Comm., 1971, 1245.
64 S. Hunig, H. R. Miiller, and IF. Thier, Angew. Chem. Internal. Edn. 1965
4, 271.
65. P. S. Engel and C. Steel, Accounts Chem. Res.. 1973, 6. 275.
66. C. G. Overberger, M. S. Chi, D. G. Pucci, and J. A. Barry, Tetrahedron Let-
ters, 1972, 4565.
67. P. S. Engel, R. A. Melaugh, M. A. Page, S. Szilagyi, and J. IF. Timberlake,
J. Amer. Chem. Soc., 1976, 98, 1971.
68. P. Haberfield P. M Block, and IL S. Lux, J. Amer. Chem. Soc., 1975, 97,
5804.
69.
70.
71.
72.
73.
74.
75.
76.
79.
80.
81.
82.
83.
84.
P. S. Engel and P. D. Bartlett, J. Amer. Chem. Soc. 1970, 92. 5883; J. IF Tim-
berlake and A. IF. Garner, J. Org. Chem., 1976, 41, 1666.
A. IF. Garner, J. IF .Timberlake, P. S. Engel, and R. .4. Melaugh, J. Amer.
Chem. Soc., 1975, 97, 7377.
H. Fischer and J. Bargon, Accounts Chem. Res., 1969, 2, 110.
8. E. Scheppele, P. L. Grizzle, and D. IF. Miller, J. Amer. Chem. Soc., 1975,
97, 6165.
P. S. Engel and D. L Bishop, J. Amer. Chem. Soc., 1975. 97,6754.
S. L. Buchwalter and G. L. Gloss, J. Amer. Chem. Soc., 1975 97, 38o7.
N. A. Porter and M. O. Funk, J. C. S. Chem. Comm.. 1973. 263
- LP. Snyder and D. N. Harpp, J. Amer. Chem Soc.. 197t> 98. 7821.
”. О. M. Stout, T. Takaya, and A. I. Meyers, J. Org. Chem 1975 40, 563.
^8. R. M. Kellogg, M. Noteboom, and I. K. Kaiser, J. Org. Chem..
P- Fahr andH. Lind, Angew. Chem. Internal. Edn. 1966 5, 372; L Hamer.
«1,4-Cycloaddition Reactions», Academic. London, 19bT p.
E. C. Taylor and F. Sowinski, J. Org. Chem.. 1974, 39. 907
Я a. Jzydore and S McLean, J. Amer. Chem. Soc 197o. 00“-
E “
Л SUS «4 .. S,Г c S. М» r * * "
an- Tetrahedron Letters, 1973, 2889.
331
L Org. Chem., 1976, 41, ]ilfi
Л/ nnH P I г A l4b-
• J- Amer.
1267.
1975, 40,
J 1 c R Flunn J. Amer. Chem. Soc., 1972, 94, 5891.
85 E. L. Allred and С K. > tyn^ j Qrg chem, 1974. 39j 407
86. Al. A. Johnson ^,fnzSa^d p, Kovacic, J. Org. Chem., 1976, 41, 175,
88 R A^Mo^and G. M. Love, J- Amer. Chem. Soc., 1973, 95, 3070; Synth^,
89. R.A. Moss, Ac“Uc*s7^™' and'O- Buchardl, Chem. Rev., 1970, 70, 231
90. G. C. Spence E.C. lay or a SocI973i 95, 5518.
91. S.-B. KheeandH.'H.Jac.A Amer. Chem. Soc., 1972, 94, 250.
81 S«>-» ««'
IJ “ fe«"-«»S
99. «Diazoalkane». ed. M. Regitz Thieme Stuttgart 1977.
100. W. Kirmse, Angew. Chem. Internal. Edn., 1976 15, 2ol.
101' C. J. Collins, Accounts Chem. Res., 1971, 4, 315.
102 M Regitz, Synthesis, 1972, 351.
103* S. M. Hecht and J. W. Kozarich, Tetrahedron Letters, 1972, 1501.
104 H Ledon, Synthesis, 1974, 347.
105. T. Sasaki, S. Eguchi, I. H. Ryu, and У. Hirako, Telrahedron Leiters, 1974,
2011.
106. N. Tahamura, T. Mizoguchi, K. Koga, and S. Yamada, Tetrahedron Lellers,
1971, 4495.
107. M. P. Cava and B. R. Vogt, J. Org. Chem., 1965, 30, 3775.
108. W. R. Roush, D. Feiiler, and J. Rebek, Tetrahedron Letters, 1974, 1391.
109, M. Regitz and F. Menz, Chem. Ber., 1968, 101, 2622.
110. Af. Regilz, I. Hocker, and A. Liedhegener, Org. Prep. Proc. Inlernat., 1969,
1, 99.
111. E. Wenkert and C. A. McPherson, J. Amer. Chem. Soc., 1972, 94, 8084;
U. Schollkopf and H. Frasnelli, Angew. Chem., 1970 82, 291.
112. N. F. Woolsey and M. H. Khalil, J. Org. Chem., 1973, 38, 4216.
113. «Carbene Chemislry», ed. W. Kirmse, 2nd edn., Academic, New
p. 85.
V.l' П N-H°uk, J. Amer. Chem. Soc., 1972, 94, 8953.
r м Coms‘ns' F: %- L>ean, and L. E. Houghton, J. C. S. Perkin 1,
116. Л4 Regi/z and G. Himbert, Telrahedron Letters, 1970, 2823' R. d. uarmm,
117 И M»"» ’ 4 u' S“PJa,' and Л Iohnson, J. Org. Chem., 1970, 35, 3444.
Chem. Internal. Edn., 1975, 14, 32.
95 124- s’ A ' K',Ogi’ and °. P, Strausz, J. Amer. Chem. Soc., 1973,
119. / Mein’waidand G Vwohi A Sam"‘es; J- c- S- Pcrk"‘ L '972. 2623'
120 H Kronl С R nk n.Wahl Chem. and Ind. (London), 1965, 425.
121. A. Nabeua 'р\вС,1п' and L-pohlenburg, Tetrahedron, 1970, 26, 3279.
122. E. Wenkert at^C^^MDh A' M°ore’ J- OrK- Chem-. 197°. 35’ 2015'
123. 1. St. Purek and m Arh^0'1' Synlh- Comm., 1972, 2, 331.
124. J. Hoor and 6 '/ M ?®li:z,Jetrahedron Letlers, 1970, 2651.
D. Af. Gunn and H Rnr°rr‘s0,l’Clt'™d- 3. Chem., 1970, 48, 868; J. Hooz,
О-P. Higlei and R № м'< Chem- 197'. 49, 2371.
126. R. Clinch E D dFuria and'h^i/' Am^' Chem' Soc 197t. 98’ ^30 * 32 33°-
27- H. I Liu and S. P Ma mLMera,\ah J- OrS- Chem., 1971, 36, 3774.
28- c. D. GulscAe and// J 7„ 5’,S’’n II Comm., 1975 , 5, 125.
30 R a’ cUleit ancl 1 ElRendngdSi rr’ 4 Cheilt' I974’ 39- 324-
30. R A. Franick, G. Lowe mid' /' o' ?' Pl;rk"1 •' 1973. 1658.
32 8C onV5lry of th« AzMo Gronn *7’ C' S' Pcrkin •> 1972- 2034-
mio\iSo,'?er a,!fi 47- Karo <Пг»^' c’ Pa.tai- Interscicncc, New York, 1971.
, New York, 1968, vol. 2’ П1С Ри11с41°па1 Group Preparations», Acade-
York, 1971,
1974, 66.
E. Harmon,
332
144.
145.
146.
147.
148.
149.
158.
159.
160.
161.
162.
163.
,33. S- S°!ar- and °' E- P°tanSky’ Chem. Internal. Edn
7 Dori and R. F. Zlolo, Chem. Rev., 1973, 73, 247.
1 P- 4- Ve'lieHdm’ °- WaSneG and J. G. Moffatt J Oro rn
135-7- Org. Chem., 1971, 36,
- a Kuzuhara and S. Emoto Tetrahedron Letters 1973 cnci
138. f jWeininger, S. Kohen, S. Mataka, G. Koga aid I P . ,
137 ciiem 1974,39,1591. ' ana !--p- Anselme, j. Org
. Д Ha’ssner, Accounts Chem. Res., 1971, 4, 9.
!зд IE Galle and A. Hassner, J. Amer. Chem. Soc., 1972 94
art / Elirenlreund and E. Zbiral, Annalen, 1973, 290. ' ' 3°'
41 «Nitrenes», ed. W. Lwowski, Interscience, New York 1970- w t. „ s-
«• A- ds ^riaC- M- Thomps°'1’ R- E- Wilde- ond S.-y'cC7org“’
1975 40, 2608. ’ ' « c-m-m.,
142 J. H. Boyer and F- C' Canler- Chem. Rev. 1954, 54, 1.
143 E Leiber, R. E. Minnis, and C. N. R. Kao, Chem. Rev 1965 65 477
1 К Dehnicke, Angew. Chem. fnternat. Edn., 1967, 6, 240 " '
A. Schonberg, «Preparative Organic Photochemistry», Springer Berlin 195Я
G l'Abbe. Chem. Rev., 1969, 69, 345. > P g ’ erhn’ 1918
R. J. Sandberg, D. W. Gillespie, and B. A. DeGraff, J. Amer. Chem Soc
1975, 97, 6193.
R, K. Smalley, W. A. Strachan, and H. Suschitzky, Synthesis 1974 503
B. Iddon. M. W. Pickering, H. Suschitzky and D. S. Taylor, j. C S Perkin f
1975, 1686.
150 D. H. R. Barton, P. G. Sanimes, and G. G. Weingarten, J. Chem. Soc (C)
’ 1971, 721.
151 R A. Abramovitch, S. R. Cltalland, and E. F. Scriven. J. Org. Chem. 1972
37, 2705.
152. R. J. Sandberg and К. B. Sloan, J. Org. Chem., 1973. 38, 2052.
153. G. I'Abbb and G. Mathys, J. Org. Chem., 1974, 39, 1778.
154 M. Komatsu, S. Ichijima, Y. Ohshiro, and T. Agawa, J. Org. Chem., 1973, 38,
4341.
155. A. Hassner, A. B. Levy, E. E. McEntire, and J. E. Galle, J. Org. Chem., 1974,
39, 585; G. Szeimies, U. Siefken, and R. Rinck, Angew. Chem. Internal. Edn.,
1973, 12, 161. „ r
156. D. S. Pearce, M. Locke, and H. W. Moore, J. Amer. Chem. Soc., 1975, 97,
6181, 6187 и более раиппе работы.
157. S.-I. Yamada, У. Hamada, К- Ninomii/a, and T. Shioirl, Tetrahedron Letters,
1976, 4749.
R. A. Wohl, J. Org. Chem., 1973, 38. 3862.
M. E. Hermes and F. D. Marsh, J. Org. Chem., 1972, 37, 2966, 2969.
P. P. Nicholas, J. Org. Chem., 1975, 40, 3396.
M. Tisler, Synthesis, 1973, 123. Q7- s)q
M. K. Meilahn, B. Cox, and M. E. Munk, J. Org. Chem., 1 0, , •
E._7. Corey, К. C. Nicholaou, R. D. Balanson, and У. Machida, Synthesis,
L. Greene, G. Л. Heavner, and R. L. Lelsinger, J. Org.
AhD''\t7arre^0/.1W59MaCA4tllfl«, and S. S. Washburne, J. Org. Chem.,
713 о
166. G. l’Abbe, G. Mathys, and S. Tappet, J. Org. Chem., 1975, 18 1973
167. A. S. Bailey, A. I Buckley and I. F. Seager, J. C. S. Perkin t,
ICO l80?,11 Оолсе раипие работы. IQ7, 549. p Shioiri, K-
168. У. S. Klausner and M. Bodanszky, Synthesis, 197 .
,ro "‘‘'l/а, and S. Yamada, .1. Amer. Chem. Soc., 1972 9^, o-u •
69. E. V. Brown and J. J. Duffy, J. Org. Chem 967, 32 112^
17l' 0 r-' Wl,ile aml H- Sclierrer’ Tetrahedron Te**crjg73 6' 293.
170 u Curci antl К Lucchini, Spectroscopy Lexers 197. t> S()c ,968,
172' W. P. Marullo, С. B. Mayfield, and E. H. Wagener, J. A'”cr-
90, 510.
1975. 590.
47. S. Mungall, G. L
164.
165.
1975,
1602,
Nino-
333
,t u T Satuvere, Ind- chim. beige, 1971, 36, 1070
с Lippn,aa. T. Comm. 1971. 1315.
74 S D. Robson and : G it Д1 j Raiue, C. ft. Rees, and R. C. Slorr, J. c .
« < R. Challand S- ? uu • ' s-
,7B Fta 1969, 725, 1
77 E- Sch’,l,‘2' T L Gilehnst, and С. V Rees. Chem Comm., 1971, 800-
да P. < ^’^p' p Glover, and K. D. Vaughan, J. C. S. Perkin , 1975, 1232
5. ^ег№Л-нЕда1/еГ „nd H. Rachhnber, Angew. Chem, Internal. Edn., 1975'
l79' l77' \ а к 11 Insold J Amer. Chem. Soc., 1973, 95, 3228.
180. J. R. Roberts and K. U-J g Q д j c s. Dahon_ 1972
181. I. dMeit-SmM M- G™'1967, |243.
31. Dekker and O.R.K™*.'± and I. T. Sharp, J. C. S. Perkin I, 1975, 1065
JO J E Wefd^ner E Ruhr, M- I- RRhter, and К. H. Koch, Angew. Chem'.
]nMa^ and M- Matrka' Chcm' Pr™ysI'
1973, 23. 133 (Chem. Abstr., 1973, 79, 31 bus).
6,6. АЗОТОРГАНИЧЕСКИЕ ИОНЫ И РАДИКАЛЫ,
НИТРЕНЫ И РОДСТВЕННЫЕ ЧАСТИЦЫ
7. Л. ДЖИЛКРИСТ (University of Liverpool)
Путем формального удаления заместителей или валентных
электронов из амина R3N могут быть получены азотсодержащие
промежуточные продукты различных типов. Наиболее важные из
них приведены в табл. 6.6.1.
Внутри каждой из этих групп могут существовать соединения
с различной электронной структурой. Так, аминнльные радикалы
могли бы обладать о-электронным основным состоянием (1) или
л-электронпым основным состоянием (2), если принять, что они
имеют нелинейную структуру. Как нитрениевые ионы, так и ни-
трены потенциально могут существовать в синглетном состоянии,
для которого характерно наличие шести валентных электронов и
вакантной орбитали, или в триплетном состоянии с двумя неспа-
ренными электронами. Приведены возможные синглетные п три-
плетные структуры для нптреипевых попов (3) и (4) и для ни-
тренов (5) и (6).
а .ища , . , Промежуточно образующиеся азотсодержащие соединения
Название Формула Число электронов на налентной орбитали
Амидные анионы R2N- . . ———
Радикалы аминия3 8
Аминильные радикалы r»n: 7
Нитрениевые ионы RsN • 7
Нитрены R2N+ 6
RN 6
« н *
”"“е ««ммониПил» или <аи№ки°§’£а"ен тсрм|щ «ацивий», однако 1UPAC рекомендуй
Внутри отдельных групп структура промежуточных соединений
может оказывать существенное влияние на нх свойства. Понятно
что синглетный нитрен, содержащий электронные пары и вакант-
ную орбиталь, должен отличаться по своему поведению от три-
плетных бирадикальных частиц. Данные о природе электронного
основного состояния некоторых азотсодержащих промежуточных
соединений получают в ходе спектроскопических исследований,
особенно в результате исследований, проводимых при низкой тем-
пературе для относительно долго живущих частиц.
Из полученной таким способом информации не всегда можно
было сделать однозначный вывод о химических свойствах неустой-
чивых промежуточных соединений, которые генерируют в раство-
рах. Можно установить типы реакционной способности, которые
соответствуют отдельным типам промежуточных соединений. Од-
нако иногда одному типу реакционной способности может отве-
чать более одного типа промежуточных соединений, что затруд-
няет строгую классификацию. Это положение хорошо иллюстри-
рует химия N-хлорампнов и родственных соединений; в принципе
N-хлорамины способны выступать в роли предшественников не-
скольких различных типов промежуточных азотсодержащих со-
единений. Конкретные условия реакции и природа заместителей
в N-хлорампне играют очень важную роль при определении типа
генерируемого промежуточного соединения.
6.6.1. N-ГАЛОГЕНАМИНЫ И КАТИОНЫ НИТРЕННЯ
Химии N-бром- н N-хлораминов посвящен обзор [1].
6.6.1.1. Методы получения N-галогенаминов
Большая часть методов получения N-галогенамшюв вклю^^^
Галогенирование вторичных аминов. Напооле Р„,01311п^ются
^-хлорам||ны. Вторичные алкнламииы лучше всего ло юте
водным раствором гипохлорита пли трет-бутилгппохлоритом Р
335
го 31 Моноалкплампиы могут быть превра
юик1«темпеРатУРахД0.Р„ датирования обычным образ0м
щены в пР^тю-Пих количеств хлорирующего агента. дЛя
с полотьюлкилампиов используют обработку хлОром
„0,1V4euiui N’N’X бРкарбоиата |2]. Аналогичные методы 11р„Ме.
вводном растворе о иь * 11Л11НОВ; однако вследствие тенДС11Ц11||
ияют для сиите.3аПрпегг,упш1ровываться в производные, хлорнро-
ЭТ11х соединении перс р М.ХЛОрапнлинов следует проводить
ванные в кольцо, и - > Склонность к перегруппировке понцжа-
пр11 низких «^Р^^троиоакцепторных групп. Некоторые N.N-
ется при пали 11 J ы К-дихлор-4-1111трофеииланплин, были
дихлораипл! 1иы, '• Т‘ОЙ,111В’Ь1Х твердых веществ [4]. N-Хлорсук-
выделены в вид > такжс являются эффективными агента-
шпишпд и j Р ‘ j-,--] Для хлорирования 2-нитроаннлина
МИ N-хлорпрова! i [ Ь некоторые N-хлорамины получают
° .А, X .1— = Р). N-Бром » N-под-
ашпиХли получены из вторичных аминов реакцией с соответ-
ствующпм галогеном [ 1 ].
6.6.1.2. Реакции с олефинами
Связь N—С1 может расщепляться несколькими различными
способами (уравнения 1—4).
R2NC1 —> r2n. + ci«
R2NC1 —> RzNHCl —> R2NHt + Cl •
R2NC1 —> R2N+ + cr
R2NCI —> R2N' + C1+
(1)
(2)
(3)
(4)
Таким образом, N-хлорамины являются возможными источни-
ками по крайней мере четырех различных типов промежуточных
азотсодержащих соединений. Частицы электрофильного пли ра-
дикального характера, генерируемые в ходе подобных реакций,
могут присоединяться к простым олефинам, так что существует
несколько схем взаимодействия алкенов с N-хлорамннами.
Присоединение простых N-галогенамшюв к алкенам (соотно-
шение реагентов 1 . 1) с предварительным гомолизом (уравнение
), если, в лучшем случае, и происходит, то весьма неэффективно,
поскольку известно, что амннильные радикалы, генерируемые н
dD?'Mc К д\Т0^мн’ присоединяются к олефинам с трудом (см.
m.vw иднако Реакция происходит при использовании
текаю? реакции а’,алогичных соединений. Наиболее успешно про-
в этом случае ‘1'алогенамин°в с олефинами в кислой среде,
тают облучению Гх'1Ы<',1.РаСТВОрЫ' содеРжащие хлорамины, подвер-
солей жетеза(П\ ' ‘ еакцию можно инициировать и с помощью
[9, Ю]. Йспользоваи” аналоги]]НЬ1х восстанавливающих агентов
в синтезе ограничен" ''® ',еаки-1,й фотохимического присоединения
335 едствие протекания конкурирующей реак-
р,₽..»»» 2>" ">™ »<ф«« ж
f1 c\ +
IIе1"1 R2NHC1 —> R2NH4-C1+
(5)
Присоединение радикала амииня протекает по цепному меха-
,1 (уравнение 6). В результате реакции с хорОШИМИ выУХОдаМ1)
" П’чают аддукты состава 1:1 с сопряженными олефинами
" ? умеренно дезактивированными олефинами, однако в стучае
Фотохимического инициирования реакции с простыми алкенами
с процессом присоединения эффективно конкурирует реакция
пирования. ,
V/ RjNH + R2NHC1
,С С 4~ R2\'h;
хло-
С—С
r2nh:
(б>
Типичные примеры присоединения приведены в табл. 6.6.2.
Реакции, инициируемые солями железа (II), успешно применимы
для самых разнообразных олефинов, поскольку в этом случае
ампниевые радикалы генерируются быстрее, чем происходит элек-
трофильное хлорирование олефина. Стадия инициирования приве-
дена в уравнении 7.
R2NHC1 + Fe2+ —> R2NH: + FeCl2* (7)
В табл. 6.6.2 включены также типичные примеры присоедине-
ния, катализируемого железом (11). Реакции присоединения чрез-
вычайно региоселективны, однако их стереонзбирательность низка.
Реакция N-хлордналкнламннов с олефинами может происхо-
дить также и в нейтральной среде в присутствии солей Cu(I),
Fe(II) или Ti(III) [9, 10], причем выходы аддуктов достаточно
Таблица 6.6.2. Присоединение ^-хлораминов R:NCla к олефинам в кислых средах
Олефин Способ инициирования Аддукт Выход, % Литера- тура
СНг=СМе. /iv R„NCH2CMe2Cl 10“2° И CH2=CHCH=CH2 ftv R;nCH2CH=CHCH2C1 40-60 8 СНг=СМеС1 hv R2NCH2CMeCh 80-90 P ^=CHCH2C1 ,v R,NCH2CHC1CH2C1 80-90 [8 cH2=CHPh Fe2+ RNCH-CHCJPh 60 lluaJ
Ви; 1<2=-(СН2)5- и т. д.
337
„ , ппотекают, по-вндпмому, через образование ами.
высок». Реакции про« комплексов с металлом. Радикальный
пильных Рад"° ния следует из того факта, что оксид а30та
механизм ПР‘е°,д" чород могут ингибировать промежуточно об-
(уравнение »1 к ' Реакция с кислородом лежит в основе рас.
разуюшиеся радпк g 1е„1)я а.ДИалкиламинокетонов (урав.
пространенного
пенне 9).
faXCl + CH!=CHPh
R2NCH2CHPh —> RzNCHsCHClPh
| NO
(8)
RjNCHaCPh
II
NOH
FeSO(, O2
RjNCl + CH-=CHR2--------->• R]NCH2COR3
(9)
Известны также примеры внутримолекулярного присоединения
N-хлорамннов. Эти реакции применяют в синтезе циклических
третичных аминов (уравнения 10 [12] и И [13]).
Имеются данные о том, что реакция внутримолекулярной цик-
лизации N-хлорамина (7) катализируется попами серебра, причем
в качестве промежуточного продукта реакции предполагались
ионы ннтрення [3]. Эти результаты и их интерпретация были
поставлены под сомнение в работе, появившейся позже; по-видн-
мому, с имеющимися данными в большей степени согласуется
радикальный механизм [14]. Хлорамин (8) циклизуется в при-
сутствии кислот Льюиса [15] (которые могут способствовать об-
разованию ионов иитреиия), а также в типичных условиях, ис-
пользуемых для генерации радикалов аминия [16].
ОМе
338
Следует отмстить, что присоединение хлораминов к пп ж
„жпо применять в синтезе. Промежуточными спа Ф”Нам
□тих реакциях являются радикалы аминпя и чмиии *е'1иями
" в виде комплекса с металлом. Роль свободны*™Hue Ради‘
Пикалов и ионов ннтрення в этих процессах стпогп ,МИН,1ЛЬ1ШХ
^„а. Другие примеры присоединения радикалов аминияТоле’
фииам приведены в разделе 6.6.3. нния к оле-
6.6.1.З. Реакции с ароматическими соединениями
N-Хлордиалкиламнны аминируют бензол, алкилбепзолы и атк-
оксибензолы в условиях, сходных с условиями, используемыми
для присоединения к олефинам [17]. Промежуточными соедине-
ниями являются, вероятно, радикалы аминия, а стадия иницииро-
вания та же самая, что и при присоединении к олефинам.
В случае бензола предполагается цепной радикальный механизм
(уравнения 12, 13).
Н NHR2
К • Д + R2NHC1
Н С1
(8)
(13)
В результате отщепления НС1 от диена (9) образуется аро-
матическое соединение. В случае анизола и бензолов, активиро-
ванных аналогичным образом, диэтиламиногруппа входит глав-
ным образом в /гара-положеиие, однако для алкилбензолов велика
Доля лгета-замещепня. Так, взаимодействие толуола и N-хлордпме
тиламина в концентрированной серной кислоте приводит к смей
2- (9%), 3- (53%) и 4-(38%) диметнламинотолуолов. Вуслс,,
фотохимической реакции хлорирование алкплоензол ' е
цепь может с успехом конкурировать с замещенп
['§] Известны также реакции внутримолекулярного
н«я; пример такой реакции приведен в уравнении
339
6.6.1.4. Хлорирование связей С—Н.
Реакция Гофмана Лефлера
процесс
'Уравне’
Инициируемая светом пли солями железа(II) реакция Пп0,
.J х \'-хпордиалк11ламииов с насыщенными алнфаТ||
мпРсоелипеннямп лежит в основе метода нх хлорнроД
"юшегося заметной избирательностью (17, 20]. В об1Цем ;
пеакния представлена уравнением 15. Наиболее вероятным Ме^
I’u’vom этой реакции считается ценной радикальный
причем переносчиком цени являются радикалы амппия
пня 16 и 17). +
R'r:r>CH + RJNHC1 —> R'R2R3CC1 + R<NH2
R]NH: + R!H —> RjNH + • R2
•R2 + R]NHC1 —> R2C1 + R]NHi
(15)
06)
(W)
Высокая избирательность характерна как для монохлорпро-
вання, так и для замещения при углеродном атоме рядом с кон-
цевым атомом в алкильной цепи ((со — I)-углеродный атом]. Из-
бирательность атаки в это положение возрастает при использова-
нии объемистых диалкпламииов. Примеры реакций, в которых
получены высокие выходы конечных продуктов с высокой сте-
пенью избирательности замещения в определенное положение,
приведены в табл. 6.6.3.
При использовании N-хлордиалкнламинов с алкильными груп-
пами, содержащими цепь из четырех п более углеродных атомов,
реакция может происходить также и внутримолекулярно. Внутри-
молекулярное хлорирование с последующей циклизацией приводит
к производным пирролидина или пиперидина (уравнения 18,19).
Реакция в общем виде известна как перегруппировка Гофмана —
Лефлера * [I, 21].
С1
J,,,-, Tiv + основание / \
Me(CHj)3NHBu ---> C1(CH2)<NHjBu --------->- / J) (18)
NBu
С) (79%)
Me(CH2)5NHPr MeCHCl(CH2)4NH2Pr -i”"8,
(19)
Me NPr
(60-65%)
осущестьтеиию\1е.пя°КО пРименяется в синтезе, однако иногда ее
является распад хмпя n<)C()'IK,je реакции. Возможным процессом
с Расщеплением nomi?™3 (Уравнение 20), связанный, вероятно
—_------_ Р ежуточно образующегося радикала амннпя
34Q 1 Же 9еаки-ией Гофмана—Лефлера—Фраптага,— Прим. Ps^‘
Таблица 6.6.3. Хлорирование алифатическ^
, , „ ♦ “веских соединений
действием R2NHC1 (17, 20)
соединепне Хлорамин
Избиратель-
кость, %
90
Ь6
68
64
Иродукт реакции
ЙЖЯЙГ
asaae _
Лефлера не может
Исходное
ГГлГНоЬСНгОИ
СН^СО2Ме
J даьсн.о
(22]. Перегруппировка Гофмана - Лёфлера не
овать с внутримолекулярным арилированием [19] ми СК°
г..„„,,папным присоединением к двойным rQa,n„ r<»i °«>‘ри-
(u3o-Pr)2NCI
Me2NCl
Me2NCl
(U3o-Pr)2NCl
молекулярным присоединением к двойным связям [23]
коац'Н250’
ч J+C1X/'4'Me
N
(20)
(25—30%)
6.6.1.5. Перегруппировки N-хлораминов.
Промежуточное образование ионов нитрения
Помимо реакции Гофмана — Лёфлера основным типом пере-
группировок N-хлоралкиламинов является перестройка скелета в
результате формальной миграции связи к атому азота с понижен-
ной электронной плотностью [3]. Подобные реакции катализи-
руются солями серебра и хлоридом алюминия; в некоторых слу-
чаях перегруппировки происходят в метанольном растворе без
катализатора. Наиболее давно открытой перегруппировкой этого
типа является перегруппировка Штиглица (уравнение 21); другие
примеры приведены в уравнениях 22—25.
н2о +
PhsCNHCI -----► [Ph2C=NHPh] —> Ph.CO + PhNH. (21)
RC1N
Ag*, MeCN, 0 °C
MeOH
2O°C
RX—
|J
(38-65%)
(10-15%)
AIC13, CHjCh
Bu3CNC12 ------------*
Cl Cl
I I
Bu2C—NBu
(22) (24]
(23) [25]
(24) ]26]
(25) [Si.
341
- mo«« зрения возможно, что все эти peaR.,u
С ф0Рмал,,’7еТеролнт11ческим разрывом связи N-CJ с Ц01и
инициируются гет I жуточных продуктов нитреН11е °’
разоваиием в_ ка может протекать синхронно с миграций
йотов R2N+. 1етер случаях в результате перегрупПИр0 "
связи, поскольку во все и011 карбеиия. Системь" в
образуется отстие при гетеролизе маловероятно, на.
которых анхпмеР ди^ не реагируют <: ионами серебра в Ме.
прнмер „Гкомнатной температуре [27, 28]. син- и аяги-Инвер-
таноле при ПОдВергаются сольволизу с различными ско-
Т0МерЫи и лают различные продукты, что указывает на отсутствие
₽°СТ»пг поомежуточного соединения [28]. При проведении пере-
пт,ловки хлорамина (12) под действием ионов серебра было
отмечено влияние «тяжелого атома» [3, 29]. В большинстве рас-
таопнтелей главным продуктом является перегруппированный
Гмин (13) однако в присутствии хлороформа или бромоформа
преобладает неперегруппированный амин (14). Это объясняется
превращением синглетного иона ннтрення в основное триплетное
состояние под воздействием растворителей, содержащих тяжелый
атом; затем триплетный нон вырывает атом водорода с образо-
ванием (14). Аналогичные перегруппировки наблюдались при ди-
азотировании гидразина (15) и при генерировании тозилгндроксил-
амина (16) в гексане. В обоих случаях предполагаются механиз-
мы с участием нона нитрения [30].
NX
12) х=С1
К Х-Н
15 X=NH,
(1в) X=OTs
аивлипя^. °ГгаВная грУппа перегруппировок связана с N-хлор-
при иагрев’аипи°т?РЫХ ХЛор мпгР»РУет к атому углерода цикла
проходить И ПОЛе"КЦНЯ катал,|31|руется кислотой [5], но может
катализируемые “’,лСотойМп,'°ИОВ Серебра !31Ь Перегруппировки,
обладанием опт изг, ° ’ пр|,водят к °- и н-хлоранилннам с пре-
Мехаинзм этих ппЛ“яРа о : п не менее 2:П-
^^^x’^-N-xaopan^i,^’1е"звестен. Для перегруппировки
342 в 8 С111фте в буферном растворе и при его
pvtctbhh и для их сольволиза, катализируемого ионямм .
нН предполагается промежуточное образование ионов НитпР ра
3 ,’полиз приводит к включению молекулы оаств^п нитРепия.
реакции Так, М-трет-бутил-Н-хлоранилин₽U7(Рр™Я В
i’S™ 26). ес,,хцйн:
* ано, реакция может быть использована для получен °КИ'
Рексадиенов (уравнение 27; R = трет-Ви),
’ЛОКИ-
ИЯ цикло-
Образование ионов арилнитрения характерно и для других
типов реакций. Ионы диарилнптренпя (22) были обнаружены не-
посредственно как долгоживущие промежуточные соединения при
электрохимическом окислении некоторых диарилампнов [32],
(22)
Предполагают, что попы арилнитрения являются промежуточ-
ными соединениями в реакциях арилгидроксиламннов, катализи-
руемых кислотами [33]. Они могут также возникать при разло-
жении арилазидов; предположено их промежуточное ооразова i
в процессе метаболической активации канцерогенных амин в L !•
6.6.1.6. Прочие реакции N-хлораминов
N-Хлорапилпиы реагируют с диметилсульфидом и
Лиалкилсульфпдами с образованием азасульфонпе “5 'иров£а
обработке этих солей основанием происходит Р 0ВЕРа ле-
^ммле-Хаузера (уравнение 28) [35]. 9™пер^ р) аРЛК11Л11р0.
тв основе дающего хорошие результаты м соединений
BdI1™ анилинов. Использование в качестве исходных
343
аШП11роВаш.ых сульфидов позволило распространить ЭТо,
«^дополучение ..иД0л°В.
*SR2
nijq/ \cH2R3 NUR'
cixiiR1 K । I
I 1 ОСОПЗ.П.С /v"|'"SR2
A Qcr -------------------> Qf (28)
м n„Tiin-N-x iopann.nui выступает в роли предшественника фс.
н.ппптреиа (см. разд. 6.6.4). К-Хлор-г-ш.троаиплшг при обработ-
КР основанием быстро реагирует с образованием беизофуроксана
(гпавнение 29) [6]. В случае монохлоралкиламинов реакция с
основанием может сопровождаться дегидрохлорированием [36],
а некоторые дихлоралкиламнпы при действии основания перегруп-
пировываются в а-ампнокстоны [37]. Эта реакция очень напоми-
нает перегруппировку Небера и, вероятно, протекает через проме-
жуточное образование азирппа (уравнение 30).
При нагревании дпхлорамппы могут также превращаться в
азосоединения, а прн взаимодействии с нцтрозоалканамп пли ннт-
розоаренами — в азоксисоедипения [38]. N-Хлордиалкнламины
сохраняют в некоторой степени основные и нуклеофильные свой-
ства, присущие исходным аминам; они могут образовывать соли
[39] и вступать в реакции с электрофильными агентами, такими,
как кетены и аф-ненасыщенные карбонильные соединения [40].
R>R2N' + HJ
6.6.2. АЗОТОРГАНИЧЕСКИЕ АНИОНЫ
. Первичные 11 вторичные амины способны в принципе выступать
в Роли кислот (уравнение 31).
R'R2NH R1R2N- + н+ (31)
мере11ыЧХчаен'1'яЬпК'ВЛп10ТСЯ 0ЧеН1’ слабь,ми кислотами. Точно из-
главным обпазеш / ДЛЯ °т,1осите-пьно небольшого числа аминов,
приведены в таб» бк? о,1Лам1”10В 11 анилинов; эти значения
в водном раствопе тот'4' 311аче,1ис рХа незамещенного анилина
является еще ботее гтчгТ” п₽имеР110 27 [41]. Циклогексиламин
зи е слаб0() кислотой с рХа » 35 [42].
Таблица 6.6.4. Значения р/<а дифениламин- . .ыи, ,
и анилинов* |4/а] we Ar NHAr2
дифениламина
Анилины
РЛ,
24-ДцвитрО"
фНитро-
2-НитрО'
З-Нитро-
[4езамеше1111ь1е
Ф,Метил-
13,84
15,67
17,91
19,53
22,44
22,95
4-Иитро-
4-Циано-
З-Циаио-
З-Хлор-
18.91
22,68
24,64
25,63
а ОпреД®лены в ИОДНОМ днметнлсульфокснде.
Кислотность алкпламииов в газовой фазе, определенная мето
дом спектроскопии ионного циклотронного резонанса, понижается
в ряду Et2NH > трет-ВиСН2КН2 трет-BuNH, > Ен
/«30-PrNH2 > PrNH2 > EtNH2 > Ме.\Н2 > \’Н3' [43]
этот ряд В очень слабой степени отражает порядок кислотности
перечисленных соединений в растворе, где сильны эффекты соль-
ватации и образования ионных пар. С достаточным основанием
можно ожидать, что кислотность первичных аминов в растворе
будет подчиняться тем же закономерностям, что и кислотность
спиртов, то есть анионы объемистых первичных аминов будут
обладать основными свойствами в большей степени, чем их' не-
затрудненные аналоги.
В синтетической органической химии амнд-анионы играют
важную роль в качестве сильных оснований. Системы цпклогек-
силампд цезия — циклогексилампи и циклогекспла.мид лития —
циклогексиламии широко используют при измерении равновесных
значений рКа слабых карбоновых кислот [44]. Были измерены
значения рКа столь слабых кислот, как толуол (рЛ'а 40,9). С той
же целью использовали систему N-метнланилид лития— К-метнл-
анплин [45]. В синтетической химии в качестве основания наиболее
часто используют анионы, получаемые из диалкнламинов, особен-
но литиевые соли этих анионов. Как реагенты эти анноны харак-
теризуются несколькими привлекательными особенностями: они
легко генерируются и определяются, растворимы в большинстве
органических растворителей; структуру дналкиламида можно по-
добрать таким образом, что его можно будет использовать для
Решения определенной химической задачи. Одно из наиболее на-
глядных проявлений влияния структуры на свойства это п
жение нуклеофильности по мере увеличения объема заместитаде’
ПР« этом основность может меняться мало или Да_ -
ваться. Более тонкие эффекты проявляются в сп°^ соеди-
иых диалкилам идов вырывать отдельные виды типов
нения, которое может образовывать карбанионы ^скольких тип^
Напоолее удобным методом получения Д • ме1Н11цтня с
шляется реакция бутиллптия, фенпллития пли “ет-‘
345
(1) Диэтиламид лития
Диэтиламид лития — сил
пользуется
анионных
33 [48]).
для генерирован
перегруппировок
0
Ph
пос основание, которое широко нс-
я енолят-апионов и для проведения
эпоксидов (уравнения 32 [47] и
l1nk*2>- PhCOCH2Ph , ,
(70%) ’
Et
LINEtj / \ I
------> <. CHOH (33)
—H
Et
Это соединение представляет собой эффективный реагент для
генерирования карбанионов в а-положешш к сульфоксидной груп-
пе [49]. Оно вызывает также распад 2-фе1шлоксатиоланов до
олефинов (уравнение 34) [50]. Диэтиламид лития обладает до-
статочно сильными основными свойствами для того, чтобы гене-
рировать дегидробензол из хлорбензола, и достаточно нуклеофи-
лен, чтобы присоединяться к дегидробензолу (уравнение 35).
(2) Дииэопропиламид лития
свойствами™0аяНвляе±ТеТ весьма слабыми нуклеофильными
енолятов лития и мипг наилУчшим реагентом для генерирования
зования диизоппоиит«п.Хп другнх карбанионов. Примером нсполь-
жить perHocneuH<h.r’uPnL)Ad лития для алкилирования может слу-
₽-дикетонов (уоавнеииЛ1ЛЛЛ'11Л,’роваШ|е енолятов циклических
менение для введения „„3® „ [511' Этот реагент нашел важное при*
34а ития в “'Положение к карбоксильной труп-
а я карбоновых кислотах [52] и их сложны*
рдения Г~™ТИЯ в Р-ДикетонТ^! 1т’ и для
т’’нтрно>>-2,4,6 может быть превращен в трилн°иев0₽ 1 ’ ?аК’ Ге"’
:"Ь, X"»р»»у«ст„но ;Ers
род О
о
трет-ВиО
о
1. LIN(Pr-u30)a
2. ^\/Вг
трет-ВиО
(36)
(~100%)
Динзопропнламнд лития (и другие диалкиламиды лития) об
разуют комплексы состава 1:1с гексаметилтриамидофосфатом
[55]. Этот комплекс, по-видимому, является более эффективным
основанием, чем сам днизопропиламид лития.
(3) Дициклогексиламид лития
Это соединение является еще более эффективным основанием,
чем дпизопроппламид лития [56]. В качестве примера использо-
вания этого реагента можно привести генерирование из метило-
вого эфира бромуксусной кислоты карбаниона, который вступает
в реакцию типа реакции Дарзана с циклогексаноном (уравнение
37) [57].
О + ВгСН2СО2Ме
LIN(CtHuh
-78 °C
СО-Ме
(93%)
(37)
осно-
этого
(4) 2,2,6,6-Тетраметилпиперидид лития
Для этого амида характерно сочетание свойств сильного
вавпя с крайне низкой нуклеофильностью [56]. Вследствие
ОН способен генерировать дегидробензол из хлорбензола анало-
гично днэтилампду лития, однако последующее его присоединение
« Дегпдробензолу не имеет места; вместо него могут присоединять-
ся другие нуклеофильные агенты, присутствующие в Реак“||ОНИОЙ
среде. 2,2,6,6-Тетраметилппперидид лития реагирует та’®‘ „
зилхлорндом, образуя фенилкарбеи, который может )•
ле»> олефинами [56, 58]. При действии этого реагента
а З-хлорццклогексене происходит 1,1-отщепление £
>2,6,6-Тетраметнлпиперидид лития — прекрасное о
енерирова1шя карбанионов, стабилизованных иал> • Р
ЕЩе одНИм преимуществом тетра.метилпнперидида литня по^
нню с другими основаниями лнпшамидноп Р Р
М7
К отсутствие обычно протекающей побочной реакцН11;
С'",Та итоида пз а-положепия па субстрат. Этот процесс ’
и0'а поскольку оба ct-углеродиых атома являются трет11Ч11ЫМ1 )0’
же„ поско. j с более 1III3K0(I нуклеофильностью м0'],
АМть из пространственно затрудненных аминов, таких к‘°
?Уб?г>1л-тргт-пеитиламип [56] и дп-^т-бутнламнн [60].' ак
6.6.3. АЗОТОРГАНИЧЕСКИЕ РАДИКАЛЫ
Исследованы два класса свободных радикалов азота: ней-
твальиые аминильные радикалы R2N- [Ы-63] и катион-радика.
лы амннпя RsNt. [61, 62, 64].
6.6.З.1. Аминильные радикалы
Многие алкил- и арплзамещенные аминильные радикалы луч-
ше всего генерировать термолизом пли фотолизом тетразенов
r2NN = NNR2 [29, 65]. Энергия активации процесса распада
тетраметилтетразепа составляет около 150 кДж/моль. Обратимый
термический распад некоторых тетраарилгидразинов Ar2NNAr2
позволяет получать дпарнлампнпльные радикалы. Исходными со-
единеньями для получения некоторых амннильных радикалов слу-
жат амины. Так, у-облучеине анилина в присутствии адаманта-
новой матрицы приводит к фепиламинильному радикалу [66].
Импульсный фотолиз арплампнов также приводит к арилами-
ннльным радикалам [67]. Некоторые диалкиламипильные радика-
лы, например азетпдпн-1-илы1ый радикал (23), генерируют путем
отрыва радикала из исходного амина [68], хотя чаще под дей-
ствием радикалов происходит отрыв водорода при а-углеродном
атоме. Альтернативный подход к получению радикалов этого типа
!(24)О[69]В Фотолизе сложных эфиров пероксикислот, например
COaOBibypej
г—N. N ,.
I I /
(23)
Диметиламинильные ради
трет-бутоксильиых радикалов
получения дифеиилампииль
тенированпи стабильного и
этнлфосфита 1711 Но.
лы генерируют из N Л рь,е ЦИКЛ11'
Мно?„еРСпш1лТн,‘РРЗМ,,,,ОВ li-
able частицы однако Раднкя'1ы пп
радикалов. Важ'ш '"меется
зывается пространств^™№
348
(24)
радикалы были получены при реакции
•’юз с (Me2N)3P [70]. Интересный путь
иных радикалов заключается в деоксп-
нптроксида Ph2NO- с помощью три-
пые циклические диалкиламинорадика-
Радикалы представляют собой неустойчи-
иесколько типов более устойчивых
предотвращения димеризации ока-
экраиироваине радикального центра
^„‘‘электронодоиорпых и электроноакцепторных заместителей'
поедполагается, что димеризация этих частиц не вызывает зна'
1ьного увеличения устойчивости [73]. т зна’
Ч1!Т Л$и-трет
(C6C15)2N« t/w-Bu—NH
—XCOPh
4Bu-rper
(26)
(27)
(25)
Данные ЭПР-спектроскоппи для алкиламинильных радикалов
обычно свидетельствуют в пользу л-структуры, то есть структуры
в которой неспаренный электрон располагается на р-орбнталн.'Та-
ким образом, днметплампнпльный радикал лучше всего изобра-
зить как структуру с д/Агпбридпзацпей (28).' Эта структура со-
гласуется и с тем фактом, что радикал является более слабым
основанием, чем диметпламин (значение рКг сопряженной ему
кислоты равно 7,0). Неподеленная электронная пара располагает-
ся на орбитали с менее выраженным р-характером [74]. Значе-
ние рЛа кислоты, сопряженной фенпламинпльному радикалу,
также равно 7,0 [67]. Для арнламинильного радикала возможно
существование о-осиовного состояния, если заместители в кольце
обладают электроотрицательностью, достаточной для делокали-
зации свободной электронной пары на азоте. Однако данные ЭПР
тех ариламинильных радикалов, спектры которых расшифрованы,
говорят в пользу л-основного состояния. Атом азота в простых
ариламинильных радикалах находится в валентном состоянии,
приблизительно описываемом состоянием $р2-гибридизацип, при-
чем неспарепный электрон находится на р-орбитали, перекрыва-
ющейся с ароматической системой. Это приводит к существенной
делокализации, особенно в системах, для которых характерна
плоская геометрия. Даже для ппрролильного радикала (29) ха-
рактерна л-структура, несмотря на то, что ароматический сек-
рет сохранился бы и в а-структуре. [75].
(28)
ГлотрИНИЛЬНЬ1е радикалы имеют характеристические спскатРтворе.
,н 111 ния. Продолжительно живущий радикал (26 Р ₽н погЛО.
в гексане дает розовое окрашивание с макси )
325 » 410 нм [76 • Генерируемые в условиях ,)Мпу
щенияпрн 32Р (Ьеинламппильные радикалы имеют максиму*
ного Ф°тол11'ХЛо8 и 400 НМ, Основное поглощение 1-цаф™\
:£» =s w”?r
, х стабильных аминпльиых радикалов является днмеризаВДя
Тетраарплгндразииы склонны к обратимой диссоциации, причем
легкость юмашт определяется природой и положением зам.
стптетей Этектронодоиорные заместители в пара-положенин спо.
еобствуют диссоциации, в то время как электроноакцепторные за.
местнтели оказывают противоположное действие. Заместители, по.
водимому скорее стабилизуют гидразины, чем дестабнлизуют
радикалы: Пространственно затрудненные радикалы (26) также
быстро рекомбинируют с образованием гидразинов, однако при
комнатной температуре реакция обратима 178]. Продолжительно
живущие дпарпламиинльные радикалы не реагируют с кислоро-
дом или иодом, однако взаимодействуют с монооксидом азота и
трифенилметильным радикалом. Неустойчивые диалкпламиниль-
ные радикалы легко реагируют с кислородом, давая ннтрокспды;
так, 2,2,6,6-тетраметнлпипер>и)1л превращается в иитрокспл (30).
При взаимодействии равновесной смеси тетраарилгидразинов и
днариламиннльных радикалов с пероксидом водорода образуются
нитрокснлы (79|.
N
I
О.
Диалкиламинильные радикалы могут димеризоваться, а также
днспропорщюнировать, как показано для диметилампнильного
радикала (уравнение 38).
2Me2N.
Me2Nl I + MeN=CH2
(38)
тивного участия
-зногл Рп?„ЧЛ°’ алк1,ламин»лы1ые радикалы не принимают ак-
оатикяплп и7„"» ° межм°лекулярных процессах отрыва других
иы выпивать яш1РеаКЦИЯХ пРпсоеД||иен|1я. Эти радикалы способ-
чае соединений J’’ акт"11ПРОва[|иые атомы водорода [80]; в слу-
конкурирует днспрмопп"о1РОВа1",ЫМ’' аТ0м31,“ с эт,)м процессом
Раметилпиперадщьппй . ,,Прован,1е- Исключением является тет-
грет-алкпльиые замест1тетн11Ка'"’ "ме101Ц11Г1 У атома азота Л"ШЬ
Да из углеводородных Л ' " способ?п вырывать атомы водоро-
терна пзбирателыХ рвс1в0Рителм- Ддя этого радикала харак-
действня атомов бпомя де 1СТВ,,Я> сравнимая с избирательностью
рыва водорода пз поп» "ЛИ .псР°кс,1д,1ых радикалов (скорости or-
350 первично,,, вторичной алкильной п бсизилЫЮ«
„ относятся как 0,0005:0,02:1) rg11 ы
CeP°B присоединения простых аминилы ых ЛС™0 ,!ем"°го
? ам В растворе диметиламинильпые рад„кяРа"ИКалов « оле.
Z Я К простым алкенам [82], одпако ХХС "Рисое^-
пппсоедпнеш.и к этилену в газовой фазе дан"«е об
J присоединяются также к стиооте г Эти ради-
phC(Me) (NMe2)CH2NMe2 с низким выходом [84Г о” аддУкг
i скорости присоединения к замещенным а-мети гт2 °“те;,ь’
Даются уравнением Гаммета с положительным Р°лам опи-
Со,69± 0,03), что свидетельствует о ХХ ЗНачен,,е« Р
„кала- В этом, вероятно, состоит причи1[а песнДи^™ ра’
Скала присоединяться к простым алкенам. В п р ,C vTeT? ЭТОГО
цинка он образует комплекс, проявляющий уХнТ М°‘
Сильные свойства (обнаруживает некоторые чепты п элек'
даям). Присоединение полученного комплекса к ЛЛ раяикала
гекает более эффективно [84]. «олефинам про-
Прн 130 °C происходит дпсротаторное раскпыти»
,^-2,3-дифенплазирлдпнил-радикале (уравнение 39) [6g] а
N,
130 °C ? Ph4.N Ph
Ph Н
(39)
Ph’
6.6.3.2. Радикалы аминия
Наиболее важным методом генерирования радикалов диалкил-
аяиния R‘R2NH; является их образование из хлорамина в кис-
лой среде (см. разд. 6.6.1). Фотолиз N-ннтрозодиалкплампнов
в кислоте также приводит к радикалам аминия (уравнение 40)
[85]. Радикалы аминия можно также генерировать путем одно-
мектропного окисления дифениламииов. Опп могут выступать
в качестве промежуточных соединений в ходе катализируемой
кислотами перегруппировки нитраминов (уравнение 41) [86].
R'R2NNO hV' Н-> R'R’NHt+NO W
Ph—N—NO2 4- НА Ph—NH—NO2 + A'
I
Me Me
| <4!>
N-Мстнл-о- и -п-ннтроаннлнны <— PhNH-J-’NOs
Me
1879 г.
Соли аминия впервые были "?л^““етнл-«-Фе11'1Ле”^Х'сут-
№Р. При обработке N,N,N\N'-TeTpa^^cTepa) в „рисут
Р°МОМ ОНП получили синюю соль ( перхл°Рат 3
етв«ч Избытка хлорной кислоты образуется
п-пзека обусловлена устойчивым радикалом амнпия (31) ()
560 л 606 им) [8'1-
MezN—< NMe, (31)
Аналогичные соли были получены из других п-фепиленднам
НОВ 1881 Эти соли устойчивы в растворе в узком интервале зна-
““ pH (3,5- 6), поскольку в более кислой среде происходи,
, Х дальнейшее протонпрованпе, а в основной среде - депротон„.
пование. Их применяют в качестве антиоксидантов и окислитель,
по-восстановительиых индикаторов.
Другие стабильные радикалы амшшя были получены Вилан-
дом с сотр. при реакции триариламипов с хлором пли бромом.
Соли Ar3N- X- интенсивно окрашены (Ph3N: имеет хмаи
640 нм в ацетонитриле), а некоторые из них достаточно устойчи-
вы в твердом состоянии при низких температурах. Устойчивость
этих радикалов увеличивается прп наличии электроподонорных
заместителей в мета- и яяря-положеннях, что следует из умень-
шения значений констант сверхтонкого взаимодействия при атоме
азота в спектрах ЭПР этих радикалов [89]. Соли, получаемые
из трис (я-бромфенил) амина, приобрели значение в качестве реа-
гентов одноэлектронного окисления и были использованы для
генерирования других катнон-радикалов [90]. Этот радикал
является также эффективным катализатором оксигеинровавия
эргостернлацетата [91].
Для генерирования радикалов трпариламинпя из третичных
аминов помимо галогенов можно применять н многие другие
окислители. Эффективным методом является анодное окисление,
которое было использовано для генерирования менее устойчивых
радикалов ампния из третичных алкпламшюв [92]. К другим
методам генерирования радикалов трналкилампиня относятся
окисление амина трикалнйгексациаиоферратом [93], диоксидом
хлора [94] и N-хлорбензотриазолом [96], фотолиз комплекса
амин—хлор в кислой среде [96] и восстановление оксида амина
действием солей железа(П) [97].
ии»?ГЛяаС110 Расчетным данным радикал амнпия Me->NH должен
иметь плоскую структуру; исследование методом ЭПР подтвер-
рХа;ГЛ:'В0;1 [aN = 19 Гс Тл) для MesNH'] [98].
дальнею геометр’ ,,ахолящпеся в голове мостика, имеют пирами-
увеличивается гт*0’ ОЛ'1,ако с увеличением подвижности системы
триал=шП1я^“ К УПЛО'-‘С"1110 [961- Для радикал
пример, радикал (₽UxTC₽ll° nor(10Weilne в видимой области; на
с У.мс 465 нм [94] неустойчивая частица красного цвета
+ •
352
радикалы ампния могут вступать в большее число ,
нейтральные аминильные радикалы, вероятно Л Веакций-
"е «тоофильностн соединений первого типаР РеакциипД°ЛЬШеЙ
’/Йлампния, полученных из хлораминов, описаны в разГбб?
£" '-алы ампния, полученные из нитрозамннов т»™
РапЛефииам и отрывают водород аналогичным образом ПпЮТСЯ
Снейпе к олефинам происходит довольно успению при ^оТоб
разуются аддукты состава 1.1с высокими выходами [85 99]
^.{соединение протекает с высокой региоселективносАю пои
том промежуточно образуется наиболее устойчивый углеродный
радикал, который затем рекомоиинрует с NO (уравнение 42)
Av Н+ МеОН У° N0H
BtiCH=CH2 + Me2NNO > BuCHCH2NMe2 —> BuCCH2NMe2 (42)
2Me2NHt —> Me2NH2 + MeNH=CH2 (43)
в отсутствие других нуклеофилов радикалы диалкиламиния
распадаются в результате бимолекулярной реакции саморазложе-
ния; возможный тип такого превращения показан в уравнении43.
В спльнокислых средах устойчивость радикалов ампния увеличи-
вается по мере увеличения кислотности растворителя [100].
6.6.4. НИТРЕНЫ
Предположение о существовании промежуточных соединений
одновалентного азота было впервые выдвинуто в XIX столетии
для объяснения протекания перегруппировок Лоссена н Куринуса.
Эти промежуточные соединения, обычно называемые в настоящее
время нитренами [101], были обнаружены и исследованы с по-
мощью современных физических методов, н их нередко включают
в схемы реакций. Хотя нет сомнения, что нитрены могут существо-
вать в твердых матрицах прп низких температурах, доказатель-
ства того, что они в качестве самостоятельных промежуточных
соединений принимают участие в реакциях, протекающих в рас-
творах, иногда не вполне строги. Осложняющим обстоятельством
является то, что в реакциях могут участвовать как синглетные,
так и триплетные нитрены, хотя эти частицы обладают различ-
ив характером реакционной способности. Синглетное состояние
присуще молекуле со спаренными спинами и низкоэнергетическон
св°бод||ой орбиталью, а триплетное состояние — бирадикал.
Доказательство промежуточного участия нитренов в реакш ж ,
Дотекающих в растворах, иногда основано на данных
л *х 11сследонаин|"|, но чаще па изучении природы в " как
Р дуктов реакции. Некоторые реакции рассматрнв Н||Х
является п1П"чиые для ''''тренов. Наиболее важн^^ для
Синг,™ Реакция внедрения по связи С Н, „ р' J (упав-
нитренов и для немногих частиц иной пР'1Р°Дреакцпя,
в КС|С 44), |. отрыв водорода--типичная радикальная^р^акп^,'
Рую вступают триплетные нитрены и в р У
‘2 Зак. 105 3
fB случае нитренов), как правило, происходит reIlep„p0B
ЙЙ-ГРУП® (уравнение 45).
RN + 4C-H —> RNHC^
К +/| |\ «4)
С—н
RN- + /C-H RNH ---------> RNH2 №)
К другим реакциям, в которых могут принимать участие нит-
рены относится присоединение к двойным связям и другим 11у.
клеофильным агентам, перегруппировки (особенно 1 ,2-Miirpamnj
заместителей), распад п димеризация. Само по себе выделение
продуктов любого из этих типов реакций не является достаточ-
ным доказательством промежуточного образования нитренов,
поскольку предшественники витренов или другие промежуточные
соединения нередко могут давать те же продукты. Примером
являются 1,2-сдвиги, которые могут протекать синхронно с выбро-
сом азота из азидов (уравнение 46), и образование азиридинов
из азидов и олефинов, которое может происходить через промежу-
точное образование неустойчивого триазолпиа (уравнение 47).
R1
2 Пф
R—С—N—N=N >-
R3
RN3 + У=(
^2
\C=NR1 + n2
(46)
NR
Именно поэтому необходима осмотрительность в том случае,
когда промежуточное образование нитренов предполагается про-
сто на основании характера получаемых продуктов реакции. Хо-
Ров'1™ доказательством участия нитренов является образование
внедрения; в случае их отсутствия общие выводы сле-
дует делать осторожно.
6.6.4.1. Методы генерирования
вескийe*oToxnm11,°')rai"IllIeCKlle а3||Ды могут претерпевать терми-
Можпо считать что*'"1 " катал,|о31|РУемый кислотой распад [102]-
дов идет с теРммческпй и фотохимический распад аз11‘
соединений (уравнепис^д?3""^ |П1тРе|10в ка|< промежуточных
354 ,ие 48), в случае же реакции, каталпзирУе
- кислотой, этого сказать нельзя, посколи™ „„„„
«°11 стадия протонировання азида. У Р паду пРедше-
RN3 —> RN + n,
(48)
Органические азиды обычно разлагаются в растворе при тем
„ературах 130-200 С. Это происходит в случае простых жткил
арил-, впил- и сульфонилазидов. Однако цианазид N3CN разла-
гается при 40 50 С [103], а ферроценилазид —при 70—8о°С
1104]. Арнлазнды с нитрогруппои или подобными заместителями
‘ орто-положении разлагаются при температурах ниж»
[00 °C [105]. в то же время триметнлсилилазид разлагается лишь
очень медленно в растворе при 200°C.
Маловероятно, чтобы алкилннтрены были промежуточными
соединениями в процессе термического разложения простых ал-
килазидов [105, 106]. Межмолекулярные реакции почти никогда
не наблюдаются; основным типом реакции является 1,2-сдвнг
водорода или алкильной группы. Даже в случае разложения ази-
дов с азидной группой, находящейся в голове мостика, для кото-
рых 1,2-сдвиг алкильной группы должен привести к сильно напря-
женному имину, протекает именно эта перегруппировка, а не от-
рыв водорода или внедрение по связи [107]. Возможно, что при
разложении некоторых алкилазидов побочной реакцией и являет-
ся образование нитрена; например, при термолизе бифенилазнда
(33) можно выделить продукт внутримолекулярного внедрения по
связи (уравнение 49) [108].
'СМеЛ’з нагревание
хМе
\Ме
%
(49)
(33) (6,5%)
Для некоторых других классов азидов промежуточное образо-
вание нитренов в ходе их термолиза доказано более строго, по-
скольку часто удается выделить продукты межмолекулярноп
реакции. Арил-, алкокснкарбонил-, сульфонил- и иианазиды оора
зуют при термическом разложении продукты межмолекулярноп
Реакции. Скорость разложения мета- н иаро-замешенных Р
азидов не зависит от природы заместителя и от полярности р
творптеля, что и следовало ожидать, если промежуточными сое-
•шнепнямп являются арплпнтрепы [109]. Более легко Р
11’,е термолиза многих орто-замещенных арилазпдо , 4tq61J
обусловлено соучастием заместителей н У^^Х^ные™™
этих реакциях принимали участие нитрены. О ов (34)
ростн термического разложения 2-замешенпых ф J
(в Декалине, 161 °C) приведены ниже [110]:
12»
355
к
*OT11
(34)
Ns
н
NsPh
NOs
COMe
COPh
CH2OH
6680
537
254
45
0.8
соучаствующие группы должны находиться в со-
Впдно, что соу п0скольку гидроксиметильпая группа не уве.
пряжении с никл 1 Ш1. Реакции могут происходить путед
личивает скороеi приВодяЩеи к образованию аро.
синхронно» Перес Р скнх соеДШ1еиий (уравнение 50). Аиа-
матическпх iciep протекающее термическое разложение
логично, довольнс легко с Рсоучастием двойиой СВЯ311. так цто
Удойное образование винплнитрепов маловероятно.
)Y + N2 (50)
"'X '
Прн нагревании ацилазидов происходит перегруппировка Кур-
циуса; имеются убедительные данные, подтверждающие, что эта
реакция протекает по согласованному механизму. Разложение
алкоксикарбонилазидоа происходит с образованием нитрена, при-
чем для данного азида скорость разложения не зависит от харак-
тера среды [111].
Нитрены образуются также при фотолизе различных азидов,
овым важным направлением в фотохимическом разложенца
стало использование триплетных сенсибилизаторов, применение
ячп°?Ь1;' позволяет прямо получать триплетные нитрены. Алкил-
мрхяи«Л° ДИМ0МУ’ Разлагаются в растворе по согласованному
нериоовать ЛХ43'™51 иитРе,10в I106, П2], хотя их и удается ге-
характеоня я рдОИ |',атР|1це ПРП 4 К. Для трналкилсилилазидов
ДругиеГадесы я7ИЧйаЯ согласова""а* перегруппировка [ИЗ],
рены, дают Ш1тре"ы°и ЙПРИ терм0Лизе которых образуются нит-
Ацилазиды так-vp п ’ В услОвнях фотохимического разложения,
нитренов; одновпемРаЗЛа1'аЮГСЯ с пРомежУточиым образованием
ложение, приводя!порН° С Этим пРо,,сходит и согласованное раз-
форнлазиды (ГО РпыперегрУ,1П|,Ровке Курцпуса fill], а Фос‘
8 фосфорилнптре||Ь| иг, Усл°ниях фотолиза превращаются
ЦИи при фотолизе ' 1*°казэно, что выходы продуктов реак-
Увеличить, проВОдЯ nP?.°T°phIX аР||Лазидов можно существенно
Для получения iinrnJI'0 прн высоких температурах [Но]-
Р ’ W«e йет°лы аналог3 МОгут быть использованы п некого-
ЛВДы- например с\-ль*Им Разложению азидов. Некоторые
858 ^ьфИмиды (35) [116j и амиинмиды (36) [1И1>
могут давать нитрены при фотолизе Ими
в форме циклических валеитн.« Мидоилазиди «
фотолизе дают имидонлнитрены (уХ°Мер°В’ 1
логично генерируют авдл. и ‘ 'WWne 5|) г л'ов'
фотОЛИЗом диоксазолонов или оксад^Х^- jePX*2М
* сравнение 52J
R3N—NX
(36)
RaS—NX
(35)
N—N
R'c\ zN
NR2
hv
N—О
Ar—\=q
X
hv
или нагревание
II + ^2
NR2
(51)
Лг.х ."
|| + СО2
X
Реакции 1,1-элиминирования как метод получения нитренов
нашли незначительное применение. Феннлпптрен был получен
из Ы-литнй-Ы-хлоранплина (119] н путем термолиза К’,О-бнс(три-
метилснлил)фенилгидроксиламнна PhN (SiMe3)OSiMe3 [120].
Реакция М-(п-нитробепзолсульфоп11Локси) карбаматов с основанием
является хорошим способом получения алкокенкарбопилиитренов
(уравнение 53) [111]. Реакция некоторых арепсу.тьфонплгпд-
разинов с основаниями приводит к соответствующим аминоннтре-
иам (уравнение 54) [121].
ROCONHOSOjAr ] -С' ' > ROCONOSOsAr —> ROCOX + ArSOJ (53)
R\ rRl\+ 3 основание
JN-NHSOsR3 >=NH 'SO2R
R’-/ 1 y
(52)
(54)
Дезоксигенирование нитро- и нитрозос° д быть объяснено
Дит к продуктам, образование которых чт0 наиболее
промежуточным участием нитренов. net ’ . ’ ар0Матическнх
важными из этих реакции являются ре 0Сн0Ве фосфо-
Пнтро- и иитрозосоединений с реагент.л п₽акиця ароматпче-
Ра(Ш) [122]. Типичным случаем ?’влве^, ₽п»п комнатной плп
ского нптрозосоедипення с триэтилфоефн 1 реагируют при
более низких температурах; пнтросо д рова1,ця и при Ра>
нагревании. В результате реакции дезок, од|1нзковые продук-
Ложеиин соответствующих азидов вь,а • тов внедрения слуя*>
а выделение в обоих случаях продуктов ,,
Хорошим доказательством того, что ннтре сОПоставДенне ДВ)*
Промежуточны мп соединениями. Тда' ЬНЬ]Ч производных (Р
Управлений Реакции в ряду З-бифеи'-^^^пя в характере
в°Дящих к карбазолам) выявило некоторые
„„ „подукюв реакции. При этом, однако, п„„
раСПреДТо Промежуточные соединения, образующиеся нз азн^
заН°’нГт0осоединенпй, не всегда имеют одинаковую реакционна
" Хть [123]. Алифатические нитрозосоединения при ВЗа
сп? °™,, с трнэтплфосфитом, по-видимому, не дают „итре-
Де И241 хотя при дезоксигенировании нитрозоалкина (з7)
образуется’нитрен (уравнение 55) [125].
р(ОЕ1)з •• *•
rper-BuC=CNO ----------” r/wr-ButeCN *-> rpw-BuCCsN (55)
(37)
в счучае ароматических нитросоединенпй используют другие
дезоксигенирующие агенты, в частности соли железа (II), кото.
рые, вообще говоря, не столь эффективны, как реагенты на осно-
ве трехвалентпого фосфора. Для некоторых субстратов можно
с успехом использовать и дисиланы [126].
В принципе окисление Ь1Н2-группы позволяет получать нитре-
ны, однако этот подход имеет ограниченное практическое значе-
ние. Наиболее важными реакциями этого типа являются реакции
1,1-дизамещенных гидразинов, прп окислении которых получаются
продукты, свидетельствующие о промежуточном образовании
аминоиитренов (уравнение 56) [121, 127]. К наиболее часто ис-
пользуемым окислителям относятся оксид ртути(II) и ацетат
свинца (IV).
R2NNH2 + HgO —> R2NN + Hg + Н2О (5)
О промежуточном образовании нитренов в ходе этих реакций
окисления свидетельствует главным образом природа образую-
щихся продуктов, особенно если эти же продукты образуются и
нз других источников нитренов. В некоторых случаях возможно
промежуточное образование комплексов металл — нитрен или
ионов ннтрення. Иногда выделяют продукты перегруппировки пли
разложения, которые могут образовываться в результате согласо-
ванных процессов, как в случае перегруппировок прп разложении
азидов. О-Замещенные гидроксиламины также пытались использо-
вать в качестве источника алкоксиннтренов, однако п в этом
случае промежуточными соединениями являются скорее всего,
ионы нитреипя, а не нитрены [128].
,,гп?азложе|и'е аз,||Р||Днн°в обычно включает стадию разрыва
ДУглеР°ДН°й связи; исключением является образование
нм5™Т1°29]ТРе1'а ПРП фотолизе Фталимидоазиридинов (уравне-
368
б.в.4.2. Структура
Основные сведения о структуре нитренов мОЖН„ „
визируя спектры ЭПР, полученные в матрице нпи Л17Ч1!Ть,
а1,пааЛХах ИЗО] • Спектры ЭПР X ₽” "Ts №
"лРкЛ арил-, сульфонил- и алкоксикарбонилнитреноГи''для
цуаионитреиа. Из них следует, что все эти типы нитренов им^?
триплетное основное состояние. В алкил- „ сульфонилнитренТх
плотность спина иеспареиного электрона локализована „реимуще-
ственио на атоме азота, однако в арилиитренах она в значитель-
ной мере делокализована на ароматическом кольце причем
степень делокализации увеличивается при наличии пара-заместите-
лей в бензольном цикле или при наличии конденсированных цик-
лов. Так, для нафтил- н антрилнитренов характерна значительная
делокализация, и эти нитрены имеют довольно продолжительное
время жизни (порядка нескольких секунд при комнатной темпе-
ратуре). Для алкоксикарбонилнитренов также характерна подоб-
ная делокализация. Цпанонитрен является единственным в своем
роде радикалом вследствие его симметрии; он также обнаружи-
вает значительную степень делокализации, а его основное состоя-
ние лучше всего изображается формулой линейного бирадикала
•N=C=N- [ЮЗ].
Для спектров поглощения арилнитренов характерны максиму-
мы в области 370—390, 310—370 н 220—260 нм [131]. Расчеты
предсказывают для ацилпптренов триплетное основное состоя-
ние [132]. Для ампионитренов также предсказывали триплетное
основное состояние, хотя все наблюдаемые для них реакции ти-
пичны для синглетных частиц.
6.6.4.3. Реакции
Как следует из спектров ЭПР, многие нитрены существуют
В триплетном основном состоянии. Однако большая часть мето-
дов генерирования нитренов, за исключением фотолиза в присут-
ствии сенсибилизаторов, приводит, вероятно, первоначально к син-
глетным частицам в соответствии с принципом сохранения спина.
Это означает, что при проведении реакции в растворе могут реа
лизоваться как синглетные, так и триплетные нитрены; степень
Участия триплетных частиц определяется относительными ск Р
стами захвата синглетной частицы и ее распада до трип._ _
°Дио осложнение состоит в том, что, когда синглетное и р
Предшественник —> Синглетный TP1'nJieT1’bli'
нитрен нитрен
Продукты реакции
359
Лпичкп энергетически, вибрационно возбужден»,,?
Сп,ипет"может вновь превращаться в синглет. Эти взапмоотноше'
тр пппрпнм в уравнении 58.
Н11ЯКак показано ранее, реакции триплетных нитренов тнпнч„ы
пня радикальных частиц. Синглетные нитрены способны прИ|1„,
1РХстне в согласованных одностадийных) реакциях, по.
этому ихреакции с С-Н- нс С=С-связямн полностью стерео™,
„ектнвны — сохраняется конфигурация тетраэдрического центра
ню, олефина соответственно. Формально продукты внедрения и
ПП соединения могли бы быть также получены через ряд стадий
в результате радикальных реакций триплетного нитрена, однако
совершенно не обязательно, чтобы эти процессы были стереосе-
лективны. Стереоселектпвность (или ее отсутствие) при образо-
вании продуктов внедрения или присоединения часто принимается
за меру степени участия синглетного пли триплетного нитренов.
На практике ситуация часто еще более упрощается за счет того,
что процессы внедрения и присоединения, если они происходят,
полностью стереоселективны. Это указывает па то, что в реак-
циях не принимают участия триплетные нитрены.
Следовательно, природу аддуктов, образующихся в ходе реак-
ции нитрена, можно в некоторой степени контролировать. При
фотолизе в присутствии сенсибилизаторов будут, вероятно, обра-
зовываться продукты превращения триплетного нитрена, образо-
вание продуктов внедрения или продуктов реакции с олефинами
нлн другими нуклеофильными агентами маловероятно. Если
в реакции используется предшественник синглетного нитрена, то
могут образоваться продукты как синглетной, так и триплетной
природы, хотя время жизни синглета можно значительно увели-
чить подбором подходящих растворителей, таких, как гексафтор-
бензол или дихлорметан [134]. В реакциях с алкокси- и амино-
нитренами получают продукты, которые должны были образовать-
ся нз синглетных промежуточных соединений.
ся из синглетных
(1) Реакции внедрения
С-Н при тетраэдрическом углеродном центре,
Эта реакция синглетных нитренов происходит главным обра-
с“ связямн С-Н при тетраэдрическом углеродном центре,
зям г р<Йукть| внедрения могут образовываться также и по свя-
пХппй ,другнх Т11пов 11 "° связям Х-Н. Это одна из немногих
при нея’ктпвиппЬ1е ПОЗВОЛЯ10Т вводить функциональную группу
цпально кп1й. ва1'"ом Углеродном центре, и поэтому она потен-
снстем (пггем'ш в.аж”а как метод создания гетероциклических
субстратов с п 'утримолекУляРИ°го внедрения), для связывания
системах 1Т0Р||Ь1ми сайтами (центрами) в биологических
ные центры в стероидахВ[еИ4]''Я фу||Кц||0||алЬ||Ь|х групп в °тДален'
карбенов можно ож1щ''дГ°фма’1а с СОТР- лля Реакций внедрения
А ть> 4Т0 переходное состояние в случае со-
woQ
„er-BuCO
EtOCO
(EtO)sRO
fbeSOi
Таблица 6.6,5. Селективность реакции
внедрения нитренов RN по связи С-Н
______________________
Относительная реакционная слосоОкость я „
па С—Н-связь сть “ пересчете
гласованного внедрения нитрена по связи С—Н будет почти ли
ценным, причем атака водородных атомов нитреном происходит
вдоль оси связи С Н [136]. Показано, что в случае некоторых
„«тренов, например сульфонилнитренов [137] и алкоксикарбонил-
«итренов [111], реакция полностью стереоселективна. По-види-
мому, продукты внедрения не в каждом случае образуются путем
отрыва и рекомбинации триплетных нитренов. При внедрений по
„еактнвнрованпым связям С—Н выходы обычно умеренные (25—
60%), хотя внедрение фосфорплнитрена (EtOJaPON: в циклогек-
сан происходит с выходом 88% [114].
Селективность внедрения по связям С—Н различного типа
определяется природой нитрена и в меньшей степени природой
субстрата и условиями проведения реакции. Обычно легкость
внедрения уменьшается в ряду: третичные > вторичные > пер-
вичные связи С—Н (см. табл. 6.6.5).
Реакции внедрения наблюдаются не для всех нитренов. В слу-
чае алкил-, арил- и амнноинтренов эти реакции происходят очень
редко. Например, феннлиитрен внедряется по связям С—Н опти-
чески активного 2-фенилбутана, однако оптическая активность
в продукте реакции сохраняется лишь частично, и первичный ки-
нетический изотопный эффект (йн/^о = 4,15) в большей степе-
ни согласуется с механизмом отрыва — рекомбинант, триплет-
ного нитрена, чем с непосредственным внедрением [138]. Однако
внутримолекулярные реакции внедрения сопровождаются полным
сохранением конфигурации (уравнение 59) [139].
•/Ме Ме
нагревание
(59)
N3^ NH Et
Часто наблюдаемое образование карбазолов из
тренов можно формально рассматривать как внедрен_ че.
' Л1- в Действительности реакция происходит, по- „ обра-
Реэ синглетное состояние, однако это сложный процесс Р
ванне карбазола включает более чем один м вап1|анта.ми
прямым внедрением возможными альтернативны. Р
361
„„кяоприсоедппеппе к ароматическому кольцу и ц
„цтреиа с последующей перегруппировкой (ур^.
внедрение по связи С—Н______________
ЯВЛЯЮТСЯ
заиия
ние 60)
(60)
(2) Реакции отщепления
Отрыв водорода от молекул растворителя относится к обыч-
ным реакциям триплетных нитренов. Типичным конечным продук-
том является амин, образование которого можно объяснить осу-
ществлением актов последовательного отрыва (см. уравнение 45),
хотя, учитывая явно низкую активность аминильных радикалов
в реакциях отрыва водорода, эффективное протекание второй ста-
дии представляется неожиданным.
Эта реакция происходит в случае триплетных арилннтренов.
Фенилнитрен, например, может превращаться в анилин с выхо-
дом выше 50%. Сульфонилнитрены могут давать сульфонамиды,
однако в ароматических растворителях выходы выше, чем в али-
фатических. Это наводит на мысль, что в данном случае меха-
низм реакции не исчерпывается простым радикальным отрывом.
Возможно, он включает перегруппировку промежуточно образую-
щегося соединения, получаемого в результате присоединения нит-
рена к ароматическому кольцу [105].
Вместо отрыва второго атома водорода может происходить
рекомбинация промежуточно образующегося радикала RNH с
углеводородным радикалом, полученным в результате первого
отрыва. Рекомбинация приводит к продукту формального внедре-
ния Сравнение 61). По-виднмому, этот процесс происходит в не-
рачительной степени в случае триплетных арилннтренов и явля-
ется основной реакцией в случае цианоннтренов [103].
RN" + R«R»R<CH —У + > RWHCR^’R* (61>
(3) Присоединение к кратным связям
чепияРИнекоторыхе тиХеН°В К 0;ietl)IIHa-M ~ удобный способ полу-
нитренов представляет г а?ир”Д11нов- Присоединение сппглетньь
яет собой согласованную реакцию, для кот0'
рой характерно сохранение стереохимии игхп„
дукте реакции, в то время как присоединен^0 ОлеФииа в 11Во
нов (менее типичная реакция) приводи" гР"™етиых ии"^
потере исходной стереохимии. Пример" * ^с™Чной или полной
к олефинам (уравнение 62), иротекаюш/гп ДИНеиия Греков
выходами, приведены в табл. 6.6.6. Щ 0 с препаративным ®
.. 1 , NR
(62)
Стереоселективность присоединения этоксикарбонилнитрена
к алкенам использована Львовским с сотр. для определения соот-
ношения синглетных н триплетных нитренов в ходе их генерации-
чем выше концентрация присутствующего олефина, тем скорее
происходит захват синглетной частицы и более стереоселективным
оказывается присоединение [III].
Фталимидоннтрен является типичным представителем группы
гетероциклических N-нитренов, которые вступают в реакции при-
соединения к олефинам. Он представляет особый интерес для
синтеза, поскольку круг олефинов, которые дают аддукты с хоро-
шими выходами, очень широк. Фталимидную группу можно уда-
лить реакцией с гидразином: образующиеся при этом N-амнноазн-
ридины сами являются ценными синтетическими интермедиата-
ми [141]. Присоединение к олефинам при низких температурах
приводит к пространственно менее выгодному син-нивертомеру,
который прп нагревании превращается в равновесную смесь син-
Тиблица 6.6.6. Присоединение нитренов RN к олефинах
R Типичные олефины Литература
С№ Алкены, циклооктатетраеи 1103]
EtOCO 6 Алкены, 1,3-двены 1111]
rper-BuCO 6 Алкены [111, Ша]
C6FSB Алкены, стильбен 11116]
О Алкены, стирол, стильбен, сложные эфиры а, p-непредельных кислот, 1111В]
О 1,3-днены
Ph——Р1( в N —-—- Алкены [1!!г]
Триплетный; 6 еииглегны» и триплетный; ’ сивметаый.
363
лппм ill-’] Дрхгпе типы нитренов, включая арпл- „
обычно не присоединяются к олефинам с обра£.
Ф°Н Sbwuob, хотя азиридины можно получить „а С00!Тв^
стюшнх азидов н олефинов через промежуточное образовав
Тр1'ич°вестио несколько примеров присоединения нитренов к трой.
и,,м связям. Этоксикарбоивлиитрсн присоединяется к дЙфеннл.
ппе итену п сложным эфирам ацетилепкарбоновых кислот с обра.
зованием оксазолов (уравнение 63) и к нитрилам с образованием
пксадиазолов [III]. Нельзя сказать с определенностью, образуют-
ся ли эти аддукты путем 1,3-днполярного присоединения'ад»
путем присоединения с образованием трехчленных циклов с по-
следующей перегруппировкой. Фталимндоинтрен присоединяется
к некоторым алкинам, давая 2Я-азпрнны с низкими выходами
(уравнение 64); в этом случае, вероятно, промежуточно образу-
ются Ш-азирины (38) [127]. о
E1OCON + RCsCR —> —0Et
R N
(63)
Г NX Д N q
/\ \/R
XN + RCsCR —> Lr——Rj —> R—"------\ (64)
(38) 'X
X “» фталнмлдо
(4) Реакции с ароматическими субстратами
В результате реакции нитренов с ароматическими соединения-
ми бензольной природы образуются два типа продуктов: азепппы
и продукты замещения. Азепппы могут получаться путем цикло-
присоединения нитрена к ароматической системе с последующей
валентной таутомеризацией. Образование продуктов замещения
может протекать по нескольким механизмам: в результате элек-
трофильного замещения синглетного нитрена или радикального
замещения триплетного нитрена, путем прямого внедрения, по
пи™1™ отрша~ рекомбинации ароматических С—Н-связей или
луитя ~еРегРУипировкв промежуточно образующегося циклоад-
пп««7, /Т обРазон- продукты замещения могут получаться или
прямым (а) ИЛИ косвенным (б) путем (уравнение 65).
'NUR
NR
(65)
364
Дзспипы были выделены в результате реакции
„пииенпи ряда бензола и нитренов RN ароматических
$.0Лп |Ш1. SO“R l,43l. COR 1441 dra/м \=CN [1031
Группы (МО). Прн R = C6fJ [14>] азёпишГл !’451 ИаРиль;
цнровачы в качестве промежуточных соединений’“"Я "ден™Фи-
“ ‘тих систем были выделены также продукты Z' Д я миогих
„ова-вя полагать, что они образуют^ Хжл
пым образом (путь «б» в уравнении 05). Трите™.?7 к°свеи'
“Х“мГ|И8]аТаК0ВаТЬ ПР(,"ЗВ0Д'1Ь,е f~a "о РадикХ^
м ряд реакции внутримолекулярного замещения яли,.
обшей формулы (39) лежит в о’снове удоб.шх метХ
некоторых гетероциклических соединении [1491 Реакция и!
является прямым замещением при орго-углеродном атоме как
можно заключить на основании относительного распоюжевия
заместителей в исходных соединениях н продуктах реакции (упав-
ненне 66); вероятно, продукты реакции возникают нз промежу
точно образующихся сппродненпльных соединений. '
(39) X = S, О, NR, СН2, СО
Циклоаддукты получаются прн взаимодействии ароматических
гетероциклических соединений с повышенной электронной плот-
ностью, таких, как фураны, пиррол и тиофены, с этокснкарбонил-
питреиом [111] и фталнмндоннтреном [150], хотя эти аддукты
неустойчивы и самопроизвольно изомеризуются. Однако цикло-
аддукт бензофураиа с фталнмндоннтреном удалось выделить
[150].
(5) Реакции с нуклеофильными агентами
Наиболее электрофильные нитрены реагируют с нуклеофиль-
ными агентами различных типов. В случае синглетного нитрена
происходит промежуточное образование нлида (уравнение 67);
в некоторых случаях этот илпд может быть выделен, однако чаще
происходит его спонтанная перегруппировка. Например, наблю-
даемое внедрение этокснкарбоннлнитрена по связям О Н и
N—Н может происходить путем промежуточного образования
нлида.
RN-f-tY —> RN—Y —> Продукты реакции <67)
К нуклеофильным агентам, которые образуют более у
вые 11лиды с нитренами, относятся диалкплсульфнды [11 Ь
тилсульфокснд [127, 151] и трнфеннларспн v
этих реакций не могут быть истолкованы однознач , -
некоторые предшественники нитренов (например, аз д )
гУг реагировать с нуклеофильными агентами.
365
(6) Перегруппировки и фрагментация
Маловероятно, чтобы хорошо известные скелетные переГруп.
„„ппвк. такие, как перегруппировки Гофмана, Курцпуса, А.
LX п Штпглина, которые, как полагали ранее, протекают с об-
пазованием нитренов, в действительности происходили 11утем
Межуточного образования нитренов. Во всех случаях имеющиеся
памые подтверждают механизм, в соответствии с которым мпгра.
пня связи"происходит синхронно с выбросом уходящей группы.
Окислительная перегруппировка гетероциклических N-амино-
соединений, которая может включать образование N-ннтрепов,
служит удобным способом получения некоторых гетероцик-
лов 127]. Примером такой перегруппировки является образова-
ние 1.2,3-беизотрназ1и1ов при окислении 1-аминоиндазолов (урав-
нение 68) [153].
R «
РЬ(ОАс),
\ ---------->- I
ЧА
N—NH2 N
Аминонитрены аллильной природы склонны к [2,3]-сигматроп-
ным сдвигам (уравнение 69) [154] .
N-V
(68)
(69)
SN z
При окислении других гетероциклических N-аминосоедипешш
происходит раскрытие цикла и фрагментация [127]. Обычно
в этих реакциях происходит отщепление одной пли более молекул
азота. Окисление 1-аминобензотрназола представляет собой эф-
фективный способ генерирования дегидробензола (уравнение 70).
Аналогичные способы разработаны для получения многих дру-
гих аринов, включая 1,8-дегндронафталнн, а также олефинов и
ацетиленов. Окисление 2-аминобензотриазола является примером
другого типа расщепления, для которого характерен выброс лишь
одной молекулы азота и раскрытие обоих колец (уравнение 71)-
> П11
nnh2 ЧЧ
JnNH2 PbfoAc)^ |^CN +
'N *
В различных vcnoRHciv
типы перегпуппнпАЧ * аР1|лпитрены претерпевают различные
или Дезоксигенипова|1|1<?,ГЛе'50д1!ого скелета. Фотолиз арилазидоь
₽ ание производных нитробензола в присутствии
2N2
(70)
- (71)
, ,,.jx нуклеофильных агентов, напримео втг.п.,,.
с"л приводит К ЗН-азепинам (40) с выходамиР“чиых алкилами-
"rf" значение [118, 155] ? Вероятиен "та рХ^я П₽ак'
Т6пазование бициклического азнрииа (уравнение 791 Ючае!
°^Р .еТся нуклеофилом. Возможно, что в пяти, ’ КОТ0Рии
:; ;Х.« . '«' “хд ~
обиружшакпе». ее»,, „ »риеуте,.™с“'‘“
нуклеофилы, которые могут с ними взаимодействовать. е
(40)
При высоких температурах в газовой фазе происходит суже-
ние никла в феннлннтрене, приводящее к образованию цианоци-
клопентадиена [156]. Эта реакция может включать промежуточ-
ное образование карбена (41) (уравнение 73). Диалогичные'про-
цессы сужения цикла происходят в случае гетероциклических
нитренов, таких, как 2-пнрндил- л пнразниилнитреиы. Реакция
происходит тем легче, чем выше электрофильность нитренов; по-
видимому, это прямой процесс, не связанный с промежуточным
образованием карбена.
внедрение
(41)
(7) Димеризация
Димеризация нитренов маловероятна вследствие малой про-
должительности жизни этих частиц в растворе и их высокой
реакционной способности по отношению к другим субстратам.
Продуктами разложения арилазидов часто оказываются азобен-
золы. Возможно, что димеризация арилнитреиов происходит
вследствие их низкой электрофильности и относительной устойчи-
вости. Более высокие выходы азобензолов наблюдаются при фо-
толизе азидов в присутствии сенсибилизаторов, поэтому их вето
ятными предшественниками являются триплетные нитрены I J-
1.
2.
3.
4.
5.
е.
Литература
Р. Kovacic, М. К. Lowery, and К. П7- Field, Chem. Rev. |9>0; 7М 5д
Р Stroh, in «Methoden der Organischen Chemie (Houben-WejD»,
8. G. Gassman, Accounts Chem. Res., 1970, 3, 26.
£ Goldschmidt and L. Sirohmenger, Ber., 1922, 5aB, JW. £ A Sf_
ЧЛ Timpe, Adv. heterocyclic Chem., 1974, 17, f Зд 29, 3390;
Я. S. Neale, R. G. Schepers, and M. R- »alsh .1. Org. cnem
D- F. Paul and P. Haberfield, J. Org. Chem L7b. ♦
A Chapman and L. Dyall, Austral J. them, 19'6, 2»,
8 7
9.
10.
!0а.
Chem., 1974, 52, 2123.
, w Snndermeuer Z. Naturforsch. B, 1969, 24, 774.
7- 19711 *i ?- p.'3273.'e' 1 0Г& ChCm" 19671 321 32«;
R.S .Neale and A'. L c^s‘ 1975, 8, 165.
F. 4,l'"'sfl1 К D J Rawlinson, in «Advances in Free Radical Chemistry»
G. Sosnovskyaml i £ondon, 1972, vol. 4, p. 203. ryi'
ed. 6. N. W'll'a™s Л[ сёсеге, Tetrahedron Letters, 1966, 3163.
A '/HSC',. R. Ga a a роЦ.па Chimica e Industrie, 1965, 47, 736.
11 F. Mt/usa, л (J°‘‘ 1 , лНл_с iQ7f) Q7
12: «. Heusler. Tdr?l!c%^(^et|et).ahXon Letters, 1974, 2191.
l3' О £. ^dwards.^'Bernat'h. 1. Dixon, J. M. Palon, and D. Vo,idle, Canad. J.
- &R Furstoss and B. Waegell, Tetrahedron Letters, 1972, 993.
RMss"g^ Espositch P. Teissier, Waegell, Bull. Soc. chink France,
1974, 2485,
17 F. Minisci, Synthesis, 1973, I.
R R 9 Neale and E. Gross, J. Amer. Chem. Soc., 1967, 89, 6579.
19 P. S. Anderson. G. F. Lundell. J. L. Cias, and F. M. Robinson, Tetrahedron
20.
21.
22.
23.
24.
25.
N. С. Ьепо, It7. E. Billups, R. Fishbein, C. Pierson, R. Whalen, and J. C. IVp-
ckoll, J. Amer. Chem. Soc., 1971, 93, 438; N. C. Deno, R. Fishbein, and
I. C.'Wuckofl, ibid., p. 2065.
At. £. IVolff. Chem. Rev., 1963, 63. 55.
G. Adam and K. Schreiber, Tetrahedron, 1966, 22, 3581; £. Leete and
A. R. Hargens, Tetrahedron Letters, 1966, 4901.
J.-M. Surziir, L. Stella, and P. Tordo, Bull. Soc. chim. France, 1975, 1429.
H. H. Wasserman, H. IV. Adickes, and O. Espejo de Ochoa, J. Amer. Chem.
Soc., 1971, 93, 5586.
D. C. Harwell and C. It7. Rees, Chem. Comm., 1969, 1428.
26. R. E. White, M. B. Nazareno, M. R. Gleissner, and P. Kovacic, J. Org. Chem.,
1973, 38, 3902.
27. О. E. Edwards, D. H. Paskovich, and A. H. Reddoch, Canad. J. Chem., 1973,
51, 978.
28. V. Rautenstrauch, Chem. Comm., 1969, 1122.
29. P. G. Gassman, F. Uneyama, and 1. L. Hahnield, J. Amer. Chem. Soc., 1977,
99, 647.
30. P. G. Gassman and K. Shudo, J. Amer. Chem. Soc., 1971, 93, 5899; P. G. Gas-
sman and G. D. Hartman, ibid., 1973, 95, 449.
311 fov?' a.aSssan’ G- Cafnpbell, and R. C.' Frederick, J. Amer. Chem. Soc.,
39 lid > , I; f,,G, Gassman and G. A. Campbell, ibid. 1972, 94, 3891.
2' ihirfS IO7s° m ™V' D' Parker- J' Amer- Chem. Soc., 1974, 96, 1234; D. Serve,
шт., У/, 432.
34 Dks"dbOner\S^ld°And T',ohla' J' Amer Chem' Sor' l975’ 97’ 7I84'
4' p r r n r’ 2rg' Chem- 1976, 41, 3820
Chem. SocSi'974 «Адет D' P Gilberl- and B- C“e. J- Amer
H. Ruschig W Fritsch 7 s' h' G' D' Gruetzmaclier. ibid, 5487.
88,883. S' ' c"' L Schmidt-Thome, and W. Haede, Cheni. Ber, 1955,
v'. Nelson'Ts^ria n^and p' ^elersen- J- Chem. Soc, 1960, 82, 459.
39. C. Allenslein and Go heau 7™^' Chem" 1976' 41’ l751'
« D. Scholz and H G m Z'.anor«- Chem, 1963, 322, 145,
'la O' i J' "h™. E 1967m44' ЗоТ 5lZ
4ia. u. Dolman and p ,i<J4.
6. Halelinger and A. Sbeitwie^ci '''‘T' 19671 451 L
E l- Brauman and L. g Bmp 1’ 4 Km' Bcr ’ 1968. ”»• 6?2-
3?lV( , ' ' ’ J1 Amer' Chem- Soc, 1969, 91, 2126; 1971, 93,
^‘reitwieser J и цп
«'Т’гл0/,'ё9'67’89' ^"‘A°streih Ciul,ari"' an<t J- A Brauman, J. Amer.
bld1’ 631 A- Streitwieser ипЛл Streitwieser, E. Ciuffarin, and J. H. Hammons,
1 E Scannon, ibid, 1973, 95, 6273.
36.
37.
38.
43.
44.
368
47
48.
49.
50.
51.
52.
53.
54.
55.
60.
61.
62.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
- pl,el and G. Rilterbusch, Annalen, 1967, 704 15
Л n Weyl)». 1958, 1 1/2, 187; U. Sehbllkopf,
II применении этих оснований см. также: М Fleser „ '1 , <Р "°'
^'«Reagents for Organic Synthesis», WileyAnieS“%ew Yort
д'с.'соре, P. * Trumbull, and E. R. Trumbull, J. Amer. Chem. Soc„ 1958.
d°'p^Thummel and B. Rickborn, J. Org. Chem., 1971 36 1365
и M. Trosl and J. L. Sinton J. Amer. Chem. Soc., 1975, 97,' 4018
Я Jones, P. Temple, E. J. Thomas, and G. H. Whitham, J. c. S.'Perkin I
1Q74 433. ’
r, Slork and R. L. Danhetser, J. Org. Chem., 1973, 38, 1775
P. L. Creger J. Amcr_ Chem. Soc., 1970, 92, 1396; В. M. Trosl and Y. Ta-
таги, ibid., 197o, "Л “pzo.
до Ц7. Rathke and D. /•. Sullivan, J. Amer. Chem. Soc., 1973 95 3050
p T. Howarth, G. P. Murphy, and T. M. Harris, J. Amer. Cheni. Soc., 1969
p’l. Herrmann, G. R. Kieczykowski, and R, H. Schlessinger, Tetrahedron
Letters, 1973, 2433.
56 R. A. Olofson and С. M. Dougherty, J. Amer. Chem. Soc., 1973 95, 581 582
57' H. Taguchi, H. Yamamoto, and H. Nozaki, J. Amer. Chem. Soc., 1974 96,
' 3010.
58. R. A. Olofson, K. D. Lotts, and G. N. Barber, Tetrahedron Leiters, 1976, 3381.
59 R, Kow and M. 117. Ralhke, J. Amer Chem. Soc., 1973, 95, 2715; D. S. Matte-
' son and R. J. Moody, ibid., 1977, 99, 3196.
T. G. Back and D. H. R. Barton. J. C. S. Perkin 1, 1977, 924.
S. F. Nelsen, in «Free Radicals», ed. J. K. Kochi, Wiley, New York, 1973,
vol. 11, p. 527.
A. R. Forrester, J. M. Hay, and R. H. Thomson, «Organic Chemislry of Stable
Free Radicals», Academic Press, London, 1968.
63. W. C. Dane", and F. A. Neugebauer, Angew. Chem. Internal. Edn., 1975, 14,
783.
64. A. J. Bard, A. Ledwith, and H. J. Shine, Adv. Phys. Org. Chem., 1976, 13,
155.
65. IF. C. Danen and T. T. Kensler, J. Amer, Chem. Soc., 1970, 92, 5235; S. F. Nel-
sen, R. T. Landis, L. H. Kielile, and T. H. Leung. Ibid., 1972, 94, 1610.
66. R. V. Lloyd and D. E. Wood, Mol. Phys., 1971, 20, 735.
67. E. J. Land and G. Porter, Trans. Faraday Soc., 1963, 59, 2027.
° W. C. Danen and T. T. Kensler, Tetrahedron Letters, 1971,2247. _
S. Sustmann, R. Sustmann and C. Ruchardt, Chem. Ber.. 197o. 108. Io27.
R. IF. Dennis and В. P. Roberts, J, Organometal. Chem., 1972 43, C2.
/• I. G. Cadogan and A. G. Rowley, J. C. S. Perkin 11. 1974 1030.
A. T. Balaban, Rev. Ronmaine Chim., 1971, 16, 725; R. Istraioiu. I. Pascaru,
apdw' n' Ba,aban- 7- Naturforsch. (B), 1973, 28 543 ,
R- V. Baldock P. Hudson A. R. Katritzky, and F. Soti, J. C. S. Perkin ,
1974, 1422.
R «7. Fessenden and P. Neta, J. Phys. Chem, 1972, 76, 2857.
70^' Bvleib. P- M. Horowitz, and T. S. Lee. J. Amer. Chem. Soc,
I94,8; A- Samimi and P. Neta, J. Phys. Chem, 1973, 77, 16-9.
P ‘-Land and G. Porter, J. Chem. Soc, 1961, 3540.
n Cauguls, J. Cognard, and D. Serve, Tetrahedron Letters, 19/1,
^riiler- t- R- C Barclay, and K. U. Ingold, J. Amer. chem. Soc,
n.'4; Neugebauer and P. H. H. Fisher, Z. Natu.rJ°rs?nRR^R|396C /. Michejda
and w^y ?nd A. Waters, J. Chem. Soc. (C) 1966, 813, C.
1 R i/Ph !,0SS' J- Amer.’Chem. Soc, 1970 92, 6_. • 95 3228.
В «• Roberts and K. U. Ingold, J. Amer. Chem Soc 19/3,
R- Cowley and IF. A. Waters, J. Chem. Soc., 1961, 122».
1973, 95,
4645.
1975, 97,
369
oo .1 Good
oo- , и,
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
1О0.
101.
102.
103.
104.
105.
106.
, r j Thimne J. Chem. Soc. (B), 1967, 684.
- od and J. L.J- J Campbell, J. Amer. Chem. Soc, 1976 ns
C j. Mlche/da and ,rL-retraliedro» Letters, 1977, 577.’ ’ 6?28:
C. 1. htichejda am D 1973. 6, 354.
7.L CbS?-} A и c Mile and A. Fenliman, J. Org. Chem, 1976, 41 3.Rr
IV. Ц. ^!1! e'!!' p'sc iitberl, and S. Granick, J. Amer. Chem. Soc, 193c? §?
Gmick’ibid-194316511747-
CM JC-,67MP„n2rf4R IV Tail J. Amer. Chem. Soc, 1967, 89, 5172.
F A Bell A Ledwilh, and D. C. Sherrington, J. Chem. Soc. (B), 1969, 27,9;
й H.^Bmlon, &. 'beclerc, P. D. Magnus, and I. D. Menzies, J. C. S. Che,,,
“etohn4/P. Л Hintz, J. Amer Chem Soc 1972, 94, 7114; J. R. Lind.
П, 1976, 1172.
G 7. Davis, M. Л1. Demek, and D. H. Rosenblatt, J. Amer. Chem. Soc, 1972,
Г’/от/И'/ 5",M md A 5' Sacld- J- C- S- Perkin l976> 741-
jp c Dane» and R. C. Rickard, J. Amer. Chem. Soc, 1975, 97, 2303.
J p ' Ferris R. D. Gerwe, and G. R. Gapski, J. Amer. Chem. Soc, 1967, 89
5270- J Org Chem , 1968, 33, 3493; J. R. Lindsay Smith, R. О. C. Norman
and A. G. Rowley, J. C. S. Perkin I, 1972, 228.
IV. C. Danen and R. C. Rickard, J. Amer. Chem. Soc, 1972, 94, 3254.
T. Mojelsky and У. L. Chow, J. Amer. Chem. Soc, 1974, 96, 4549.
I'. Malatesla and K.. U. Ingold, J. Amer. Chem. Soc, 1973, 95, 6400.
«Nitrenes», ed. W. Lwowski, 1 nterscience, New York, 1970.
«The Chemistry of the Azido Group», ed. S. Patai, Interscience, London, 1971.
A. G. Anaslassiou. H. E. Simmons, and F. D. Marsh, в cc. 101, p. 305.
R. A. Abramovilch, С. I. Azogu, and R. G. Sutherland, Chem. Comm, 1971,
134.
R. A. Abramovilch and E. P. Kyba, в cc. 102, p. 221.
R. A. Abramovilch, in «Essays on Free-radical Chemistry», The Chemical
Society, London, 1970, p. 323.
107. H. Quast and P. Eckert, Annalen, 1974, 1727; 7. O. Reed and W. Lwowski
J. Org. Chem, 1971, 36, 2864.
mo м Ai Abram°vilch and E. p. Kyba, J. Amer. Chem. Soc, 1974, 96, 480.
109. M. Appt and R. Huisgen, Chem. Ber, 1959, 92, 2961; P. A S. Smith and
... , 7-' 7°J' Лтег Chem. s°c, 1962, 84, 480.
1 иЛ Di,ali.and L E- K™iL J- Chem. Soc. (B), 1968, 976.
111. W. Lwowski, В cc. 101, p. 185. ' "
*1 16 p Ra and Lwowski, J. Org. Chem, 1976, 41, 96.
6'154L Abramov‘lch- S- R- Thailand, and Y. Yamada, J. Org. Chem, 1975, 40,
Soc.J^с/мздГзтб L Liilchrist, D. C. Harwell, and C. IF. Rees, J. Chem.
Ill R. Mhrhf0Har“ydaJndSRUc’ ™rakedron Letters, 1974, 2945.
movitcli and E P^Kuhn' ,Re?rclon‘ Tetrahedron, 1970, 26, 1379; R. A. Abra-
D. Berry M I MrGBb, i 1 Ат,ег/Chem- Soc, 1971, 93, 1537; R. E. Banks,
113. D. R. Parker and L H a"d 9’ L Moore- J- Chem. Soc. (C), 1970, 1017.
1.14. R. Breslow, A Feir'inn eTi,1' A,uer' Chem- Soc- 1976. 98’ 6|8'
J- M. Lindley t M MrPnhhF’ ner,".’an’ J' Amcr- Chcm- Soc-, I974- 96’ 5?37'
Letters. 1976,’4513,' McRobb'4, O- Melh-Cohn, and H. Suschitzky, Tetrahedron
M. Naslasi, ^“.’'strub Ctem' Рсч-, 1977, 77, 409.
<ipr’,BLM- Culbertson 'and s wSlreil,t’ Tetrahedron Letters, 1976, 4719,
С A B «- lot n Chem. Rev, 1973, 73, 255.
iVrlWe an(i n о к- •
• R- Dimmel, J. Лтсг. Che,n Soc , 1072| 94| 860l),
115.
116.
117.
118.
119.
370
120-
|2l.
122.
123.
124.
|25.
126.
127.
128.
Л and E. Stanton, J. Chem. Soc^ (C) 1971, 988.
130.
131.
132.
133.
134.
135.
136.
F. p. Tsui, Y. // Chang, T. M. Vogel, and G. Zun, j. Org С|1ад
338L letnal, в cc 101, p. 345. ' '
i 'TSi. Vi *
Abramovitch, J. Court, and E. P. Kyba, Tetrahedm.Ч Иetters.‘973,761.
_c Molte and H. G. Viehe, Chimia (Switz.), 1975, 29 515 eFS' 1972, <1059-
p.p Tsui. T. M. Vogel, and G. Zon, J, Org. Chem, 1975 40 7fii
□ v Ioffe and M. A. Kuznetsov, Russ. Chem. Rev, 1972 41 ’ 131 гк я и A
t Л1 А- Кузнецов - Усп. химии, 1972, 41, 241]; T. L ' Gilchrist с W Ир°Ф'
ЧЛ*.< « p“"
I andon 1У» *• 'Ul J‘
C j Brois. J. Amer. Chain. Soc, 1970, 92, 1079; L. J Hayes F P Rm-
leu and C. Trindle, J. Org. Chem, 1972, 37, 3924; В. V Ioffe аги1'рВуП&
Zbur.org. Khim, 1972, 8, 1548 [8. В. Иоффе^.ТКоролева ^Z.
Opf, XliM-i 1У/Д °’
n Ц7. Jones, J. C S. Chem, Comm,, 1972, 884; T. L. Gilchrist C W Ppp*
. i . .. T /"1л „„л <? !Г\ 1 f\y । ОЙЯ ’ ' ” '
Up ‘Wasserman, Progr. Phys. Org. Chem, 1971, 8, 319.
r S. Berry, в cc. 101, p. 13.
p. F. Alewood, Р. M. Kazmaier, and A. Rauk, J. Amer. Chem.
5466; J. F. Harrison and G. Shalhoub, ibid, 1975, 97, 4172.
N, C. Baird and R. F. Barr, Canad. J. Chem, 1973, 51, 3303.
R. C. Belloli and V. A. LaBahn, J. Org, Chem, 1975, 40, 1972.
J. R. Knowles, Accounts Chem. Res, 1972, 5, 155.
R, C. Dobson, D. M. Hayes, and R. Hoffmann, J. Amer. Chem.
6188. О данных, подтверждающих триангулярное переходное
cc. 111.
Soc., 1973, 95,
Soc, 1971, 93,
состояние, см.
137 D. S. Breslow, E. I. Edwards, E. C. Linsay, and H. Omura, J. Amer. Chem.
Soc, 1976, 98, 4268.
138. J. H. Hall, J. U7. Hill, and J. M. Fargher, J. Amer. Chem. Soc, 1968, 90, 5313.
139. G. Smolinsky and В. I. Feuer, J. Amer. Chem. Soc, 1964, 86, 3085.
140. R. J. Sundberg, D. 117. Gillespie, and B. A. DeGraff, J. Amer. Chem. Soc,
141.
142.
143.
144.
145.
146.
147.
148.
149.
150.
151.
1975, 97, 6193.
D. Felix, R. K- Muller, U. Horn, R. loos, 1. Schreiber, and A. Eschenmoser,
Helv. Chim. Acta, 1972, 55, 1276.
R. S. Atkinson and R. Martin, J. C. S. Chem. Comm, 1974, 386; R. S. Atkin-
son and J. R. Malpass, ibid, 1975, 555.
R. A. Abramovitch, T. D. Bailey T. Takaya, and У. Uma, J. Org. Chem, 1974,
39, 340.
M. Georgarakis, H, J. Rosenkranz and H. Schmid, Helv. Chim. Acta, 1971,
54, 819.
D- If. Jones, J. C. S. Chem. Comm, 1973, 67.
L. Krbecheck and H. Takimoto, J. Org. Chem, 1968, 33, 4286.
R- A. Abramovitch, S. R. Chai land and E. F. V. Scriven, J. Amer. Chem. Soc,
1972, 94, 1374.
R- A. Abramovitch, G. N. Knaus, and V. Uma, J. Org. Chem, 1974, 39, 1101.
n7' Cadogan, Accounts Chem. Res, 1972, 5, 303.
£ W'. Jones, J. C. S. Perkin I, 1972, 225, 2728; см. также cc. 129 .
R E- Banks and A. Prakash, J. C. S. Perkin I, 1974, 136o; P. D. Kenne.ell
152 Ta‘Jlor- Chem. Soc. Rev, 1975, 4, 189.
153 я7'.?' Cadogan and I. Gosney, J. C. S. Perkin I 1974, 460, 466
3 £ Adger, S. Bradbury, M. Keating, C. U7 Rees, R. C. Store. ana
154 jFniiliamS' J-Cs- Perk’in ’• ,975- 3L , Л Soc 1971 93, 788.
155 D c, Bald™in, J. E. Brown, and G. Hofle, J. Amer. Chem. Soc, 19/ , *
513Л RU'rl‘"^r8’ S- R- Suter- and M- Brcnner,’ J. 1974'. »«. 7491,
156 С Тр//Л' DeGruff, D. If. Gillespie, and R. J-e8 {258 R. Harder
aJn^ and C- Wentrup. J. Amer. Chem. Soc, 1976, 98, 12os, n.
157 , A h Weilir4P, ibid, 1259. o ,Q7| q3 <05i,
' Re,ser and L. J, Leyshon, J. Amer. Cliem Soc, 197 , ।
371
ЧАСТЬ 7.
НИТРО и НИТРОЗОСОЕДИНЕНИЯ
7.1. НИТРОЗОСОЕДИНЕНИЯ
Р. ДЖ- КУМБС (The City University, London)
Химии этого класса соединений посвящено несколько общих
обзоров [1]. Две важные особенности нитрозосоединений (1) при-
дают методам их получения и свойствам особую сложность. Пер-
вая из них состоит в склонности многих нитрозосоедш1сщ|й обра-
зовывать биснитрозосоединения (уравнение 1), которые могут
обладать цис- (2) или транс- (3) конфигурацией относительно
связи N=N. Вторая особенность — это тенденция алифатических
иитрозосоединений, содержащих атом водорода при а-углеродном
атоме, к изомеризации с образованием соответствующих оксимов
(4) (уравнение 2).
хО +/R R\+ t/O-
R-N^ ч=± N=Nz + N=Nz (О
‘си No- -o' Nr
(') (2) (3)
^CH-N=O —> ^C=N-OH (2)
(4)
кртЛГ-х ,ДВУХ СВ0Йствах нам не Раз придется вспоминать в соот-
ветствующих местах данного раздела.
и7 *пЛЕЛ0ДЬ1 ПолуЧЕНИЯ АЛИФАТИЧЕСКИХ
АРОМАТИЧЕСКИХ НИТРОЗОСОЕДИНЕНИИ [1д]
( ) кисление аминов и ^-замещенных гидроксиламинов
быть окислены до\нХг>Гг.Л!'3аМеЩеннь'е гпдрокенламины могут
до нитрозосоедииений (уравнение 3).
_ RNHj —> RNHOH —> RNO (3)
градиционным реагентом „„
веских аминов в иитпло ДЛЯ пРевРаЩения первичных ароматп-
372 Рассоединения (выход 35—70%) является
«,ралпзованная мопопероксосерная кислота H2SO.
||е" \ дтя получения котором используют кош »„ 'Кислота
КаР° ’ оцелоту и пероксисульфат аммония [21 ^1ри1>ованную
первичных алифатических аминов в котош.х Н° окис-
х»»™ »=™«р=*=»е»«о к тр«,z;
п‘ , ко выходы образующихся при этом соответствующих лР Да’
0Д осоединенин невысоки (~4%). Если амнХуТа РВ
С1Р рвичпым или вторичным атомом углерода, то окн слей не этим
е г нтом может привести в зависимости от кислотносп среды
бок первичным или вторичным бисиитрозосоединеииям тибо
изомерным оксимам (см. уравнение 2). Вторичные аром’а™че°
ск1,е амины также можно превратить в ннтрозосоединения ис-
пользуя методы деградации; так, например, из N-бензиланплнна
образуется нптрозобензол.
Некоторые 2,6-дигалогенпропзводные первичных ароматнче-
скпх аминов были превращены в ннтрозосоединения окислением
перокспкислотамп, причем оказалось, что для предотвращения
образования азоксн- и (или) азосоедпнений [3, 4] необходимо,
чтобы заместители в положении 2 создавали определенные стери-
ческне препятствия. Первичные алифатические амины, в которых
аминогруппа присоединена к первичному или вторичному атому
углерода, превращаются в биснитрозосоедннеиия [5]. Такое же
превращение могут испытывать и некоторые вторичные амины,
способные отщеплять алкильный заместитель в виде карбониль-
ного соединения. Считают, что подобное окисление аминов про-
исходит через N-замещеиные гпдрокспламины, которые в отдель-
ных случаях удалось даже выделить, и включает, по-видимому,
нуклеофильную атаку амина по атому кислорода гидроксильной
группы пероксикпслоты. Лучший метод получения алифатических
первичных п вторичных бисннтрозосоедпненпй (выходы 30—
№%) (6] состоит в использовании азометинов и оксазпридинов
В качестве промежуточных соединений (уравнение 4), причем на
каждой стадии окисления используют алифатическую пероксикнс-
1ОТУ. Ароматические азометпны (основания Шиффа), а также
алифатические и ароматические нитроны (уравнение^5) также
можно окислить перокспкислотамп до иитрозосоединений.
О
+ Et2CO —> RN=CEt2 RN^CEh (RNOh + Et-CO W
O’
RiN=CR2 МеС°з1Х (RiNO)2 + R22C=O
п°ДУчКаИ|оЛеШ'е Ы-замеЩенпых гидроксилахгииов^ которые обыч^^
тивиым восста!!°влением нптросоединеили,• * г!!др0Кснл-
аминон мет°Дом, несмотря на склонность не Р нитрозосо-
единенг ЛИСПропоРц-1!О!1!!Ровать ПР!1 хра!!С!’ии н пп.В11димому, лишь
Не, ограничением которого может быть,
(5)
доступность необходимых предшествепнпкоп. Можно ,|Гпп
большой набор эффективных окислителей. !]pn ol()itVlc, ™лЬз01)а
пых или вторичных М-алкилгпдроксиламниов могут ок"ер°ич
ватьея изомерные оксимы. Для получения или Фитйческн ?>•
эососдпиешш в качестве окислителен используют вол 1,,тРо-
творы галогенов, хромовую кислоту, оксид ртути(II) Ые Pac-
кислоту и карбонат серебра на целите в дихлормстане Г71Н0д11У»
производных ароматического ряда эффективными оказ- ' 3
манганат в кислой среде, тетраацетат свинца » хлопЛНСЬ ПеР-
за (III). При перйодатном окислении М-метплгидг,г;Д‘ Же-’е-
образуется 1;нс-бис(11птрозомстан) (2; R = CH3) Г8а1 КцНЛам,1»а
особо чувствительных соединении подходящим окислнтрп СЛу,!|с
ется диэтиловый эфир азодикарбоновон кислоты- 0) ЛеМ я®ля'
использование для синтеза бнс(нитрозоцнклопропан’а) (яг|Ил его
ленпе гидроксиламинов - лучший способ получения
ннтрозосоедииений. у я т₽етпчивд
(2) Восстановление нитросоединений
Это восстановление редко псплчгяи,^»
ных целей, так как трудно остановить ппонрг/ЛЯ препаРат«в-
ДНП. Необходимо работать со слабыми п Р <С Иа нУЖи°Й ста-
мами в почти нейтралы^^хС°растворЛагВТлСяТаэтоВ"1ТеЛЬНЬ1М11 —
валн гидроксилами», сутьфид натт1’яД ,? целп пспольз°-
метанольном растворе щеточи Ml Л«и \ОКСНД олова(П) в
ваются до 1штРозош1^обс1Тзо£]в Д^Р^ЛЫ восстанавлк-
^^’трвмо-зекулярного» бнсннтрозосоедщ^ц гроб!,Феннл—
"О—n=n—су
(5)
N=0"
0
Ph H
NO
(6)
COPh
Известны примеры внутримолекулярных окислительно-восстано-
вительных реакций, например образование о-нитрозобепзофено-
на 110а] (уравнение 6):
ако2
TsCl
снон
I
Ph
мер преврРатиьЖо.}\°Жп.? осУЩествить и фотохимически, напри-
лоту. Может покатят, р°бензальДегид в 2-ннтрозобензойную кис-
ны обязательно нахппнг’ ЧТ° НЙТР0- 11 альдегидная группы доля*-
ко в подобную реакпп,пС» В орто-положенпн друг к другу, оД”а‘
374 У Реакц,,ю встУпает и 4-ннтробензалвдегнд [Ю»]-
,||1трозохлорсоед|1пе11ия можно получить обработкой п ,
пм пнтросоедипеппи эфирным раствором хчооиЛа ей
^'(уравнение 7). Прн взаимодействии некоторых замешенТ’
^поалкенов с хлористым водородом (уравнен^ 8) обпХтЫХ
а-нитР° , 2.дихлорсоедниения. 1 "“Разуются
нитро30’ ’
/ \ HCI
-N02 —> ( /=Ж Na+-----------
М<ч
;с=сн2 + нс1 ——
o2nz -Нг0
(7)
03%)
NO
Me—С—СН2С1
I
С1
(34—45%)
(3) Нитрозирование по атому углерода
Замещение водорода в алифатических соединениях. Прямое
замещение атома водорода на нитрозогруппу возможно прн
наличии электронооттягнвающпх ненасыщенных групп, связанных
с тем атомом углерода, по которому должно пройти замещение
(уравнение 9) [11]. В качестве нитрозирующих средств обычно
используют азотистую кислоту или ее эфиры, причем в последнем
случае реакция проводится в сильнокислой или сильнощелочной
среде. Активирующими атом водорода группами могут служить
карбонильная, алкокспкарбонильная, имино-, нптро-, циано- или
арильная группы; сложные эфиры монокарбоновых кислот недо-
статочно реакционноспособны. Если реакция проходит по метиль-
ной пли метиленовой группе, то первоначально образовавшееся
иитрозосоедпненпе быстро перегруппировывается в оксим. Сами
иитрозосоединепия образуются лишь в немногих случаях: при
замещении у третичного углеродного центра, при котором невоз-
можна таутомерия, пли прп проведении реакции в условиях,
близких к нейтральным, например при использовании оксидов
азота (главным образом, Ы20з) в эфире (уравнение 10) пли
этнлнитрита и ацетнлхлорида. Нптрозопроизводные а-монозаме-
Щенных Р-кетоэфпров, малоновых эфиров и кислот настолько
склонны к образованию оксимов, что, если не соблюдаются мяг-
К||е условия питрозироваипя, часто происходит элиминирование
алкоксикарбоппльной группы, причем в этих и некоторых других
«Учаях реакции расщепления С-С-связей становятся суще-
„Венными ['2а). Если нитрозирование может проходить по
б °Жениям, то реакция обычно направляется пзбпра .
' е К0Р°ткий менее разветвленный радикал. Изопро • •
3-мет’ Тако’ дает как б11с(3-метпл-3-ннтрозобутанон-2), _
««Рида 7КС''М1,'ЮОуТа1,°"’2 ПР" деГ1СТвШ1 ЭТХНстсРя разработать
₽l’Ad (уравнение 11) [126] . Возможно, удастся ра р
375
такие Условия реакции в
зосоедпнения до того,
/Н ,няо2" \г/‘
X-------- Ах
которых можно будет выделять
они I
------------- отделять и,,_
подвергнутся таутомеризацШ]
X = COR, C(R) = NH, NO2, CN, Ar
Ity
NO
phCOCH.COPh ----->- PhCOCHCOPh
(50—60%)
MeaCHCOMe
EtONO
MeCOCI
• i° )
.Me2CCOMe/2 + Me2CHCOCHNOH (!!|
Замещение водорода в ароматических соединениях. Прямое
электрофильное нитрозирование действием азотистой кислоты воз
можно для тех ароматических соединений, которые активированы
наличием сильных электронодонорпых заместителей, и в препара-
тпвных целях ограничивается лишь фенолами, нафтолами, неко-
торыми простыми эфирами фенолов, вторичными, третичными и
некоторыми первичными анилинами и нафтилампнами. Замещение
в этих соединениях протекает в лара-положение, если оно не за-
нято, и в случае фенолов происходит по <45Е2-механизму с про-
межуточным образованием цпклогексадиенона (6), причем стади-
ей, определяющей скорость реакции, является отщепление про-
тона [13а].
/=\/н
°~O\N0 (б)
Именно этим объясняется тот факт, что, хотя участвующий
в этой реакции электрофил гораздо менее активен, чем электро-
фил в реакции нитрования, где лимитирующей стадией является
электрофильное присоединение, значения параметра р для обоих
процессов близки, и это указывает на их одинаковую избиратель-
ность. Оиа была продемонстрирована при изучении орто-нптрози-
рованпя лара-замещепных фенолов [1.36]. В случае ди- п тригид-
рокенбеизолов возможно дниитрозпровапие; например, из резорцина
пониоРил'Ми вь,ходом получают 2,4-диинтрозорезорцпн (УРав’
niw»2»J' Н|'тР03||Р0ваи”е простых эфиров фенолов часто сопро-
паблтилотп'10^'"101' Реакцией — разрывом эфирной связи. Иногда
п-коезолз ’ °^”сле1,ие пптрозогрупиы; например, нитрозирование
чить опто-нпт'ппэЛ"Т К 4'мет11Л-2-нитрофенолу [14]. Чтобы облег-
солн тяжелых '‘рова””е /гаРа’заме|Цб11пых фенолов, используют
вывать комплексыТасЛЛп°В’ напримсР медп(П), способные образо-
та [161 пспол! -ivin-p °’н”тР030Фе»°лам11 [15]. В реакции Бауди
и соли медиПп смес1> гидроксиламнна, пероксида водорода
в воде при 25°C- ат^ЛИ амм|,11°пентацпаиоферрата (II) иатР'|Я
’ реагент является эффективным орго-11!1ТР°
1
л,ним агентом фенолов, даже если пара-пом^,.
;йрУ Нитрозофеиолы образуются и непосредствен,^"е заНя'
г°' °’v соединений путем окисления — нитпочип.,п° И3 аР°мати-
’есК"Х, тех же реагентов (уравнение 13) НИТрозиРования с приме-
пением юл /
0Н ON- он
-* но~O-N0
но
(12)
Сц2+ или Nag [Fc(CN)5NH31
NHaOH, НаОа
(13)
Нитрознрование первичных ароматических аминов обычно чя.
трагивает аминогруппу, приводя к ее диазотированию, но некото-
рые амины, не содержащие электроноакцепторных заместителей
например а-пафтиламин [17], ннтрозируются в положение 4 ядра'
если в качестве нптрозпрующего агента используют нитрозилсер-
ную кислоту. Подобная же реакция протекает в некоторых слу-
чаях (например, для 2,3-дпметиланнлипа) и при использовании
обычного реагента, азотистой кислоты. Сходные N-алкплзамещен-
ные вторичные амины также можно непосредственно пронитрози-
ровать ннтрознлсерной кислотой, нитритом натрия в дымящей
серной кислоте или в насыщенных хлористым водородом спирте
или уксусной кислоте. В обычных условиях реакции, однако, об-
разуются N-нитрозамнны, которые можно ввести в перегруппи-
ровку Фишера — Хеппа, приводящую к С-нптрозоарнламинам
(см. разд. 7.4.3). Обычно Ы.Ы-дпалкпланилины легко нитрозпру-
ются в пара-положение, хотя наличие объемистого заместителя,
например в случае Ы-трет-бутил-Ы-метиланплпна, может замед-
лить реакцию [18]. На основании низкого значения параметрар
можно было бы ожидать непосредственного нптрознровапия н ме-
нее реакционноспособных ароматических соединений, чему, види-
мо, препятствует неустойчивость азотистой кислоты; имеется лишь
одно сообщение о непосредственном нитрозированпи бензола и
толуола [19]. Ни одна из этих реакций не дает удовлетворитель-
ных выходов.
Нитрование некоторых реакционноспособных ароматических
соединений, катализируемое азотистой кислотой и протекающее,
как принято считать, путем нитрозированпи и окисления, Х0Р°
1,10 известный процесс. Он реализуется, когда скорость^ Реак™
с ионом интропня значительно снижена из-за его малой„
Winn и когда скорость реакции определяется диффузией-
Реакции в случае фенола и анизола дают почти "ск‘'"°ч”д^^1е
Н||1тросоедипен11я [20]. В то же время накапливаютс А
У азывающие на то, что по крайней мере |,е,'°т р чамещение.
Р цессов не включают нормальное электрофил аТ11Ческ1,х
“°’первых, каталитическое нитрование некоторых ароматическ
пп„,,еп нафталина и бифенила [21), „Потп
соединен»», папр ожидат)>> исходя „э Г1Х реакцИоЙ
•1егче/'е™ м отношению к другим электрофилам. Во-в '"oli
еп0С° веское нитрование Ы.Ы-ДНметялапилина происходит в“ '
каталитическое п саМ0 и11ТрозИров;ише не идет [22а1 п.
К,1Х уМп-Х’ 6Ы1 предложен механизм с переносом электрод’
этого их ая (УТочное образование катпон-радпкала [2261
включаюпшн промер 1ИИЗМ )1Меет более общий харак ,[
ВП0Л*’Х,? Срочаиия, катализируемых азотистой кислой
АЛ нигоо зирование мегаллорганических соединении. Реакция ал.
t.„„п иипртттных соединений с нитрозирующими агентами Пр„.
Хт кпп'трозосоедннеииям путем замещения металла п связан-
ного с ним лиганда. Арнлртутьгалогеииды или -ацетаты реаги.
«уют с нптрозплморидом, N2O3 или N2O4, давая пнтрозоареиы;
ппп реакции дпфенплртути с иитрознлхлоридом образуется не-
большое количество нитрозобензола. Диалкилртутпые соединения
реагируют с NO или иитрознлхлоридом при УФ-облучепии или
нагревании; реакция в данном случае протекает по механизму,
вктючающему промежуточное образование алкильных радика-
лов [23]. Взаимодействие производных бис(алкнн-1-ил)ртути
с иитрознлхлоридом при —78 °C приводит к крайне неустойчивым
1-нитрозоалкинам, перегруппировывающимся прп нагревании
в 2-оксоалканнитрилы (ацплциаииды) (уравнение 14) [24]. Аро-
матические реактивы Гриньяра реагируют с иитрознлхлоридом и
N2O4 с образованием нитрозосоеднпенип; аналогично ведут себя
алифатические реактивы Гриньяра в реакциях с нптрозплхлорп-
дом и NO. В случае алифатических соединений реакция протекает
через промежуточное образование N-иитрозогндроксиламинов,
которые разлагаются в кислой среде (уравнение 15). Взаимодей-
ствие трпалкилалюминия с иитрознлхлоридом приводит к бнс(ни-
трозоалканам); олово- и креминйоргапическне заместители в аро-
матических соединениях вытесняются нитрозирующими агентами
(уравнение 16). Обработка днфторацетатов арплталлпя пропил-
нитритом н хлористым водородом в хлороформе при 25°C
иптрозоарены, как правило, с высоким выходом [26].
(Dr_ri ,, х°с1 л
tRC==C)2llg РС-°!0 RC0CN
С\ N'OCl / \ /ОН fi+ i- / у "1
/~М8С| ——> f у/ —> ГАЛ__мо1
дает
(14)
(15)
(16)
МеО Х0С‘ . ______
SiiMe, -----> МеО—# \ч—N0 + Me3SnC!
(80%)
боновых киЛотТк Г₽упп' ^|,’гРознроваине ароматических кар-
чую группу/актиВ11п™Ти атом углерода, несущий карбокснль-
378 ₽ Г1° отношению к электрофильному Реа
, соседней гидроксильной группой, сопровождаете, „
ге«гУпяа1)цсм; алкокеигруина гораздо менее активнТ декаРбок-
с11лпР°ватак, например, 2-гидроксииафтойиая , t ЭТОМ отно’
^"‘позонафтол-г. Цпклоалкаикарбоновые кис поты н'°Та Дает
1-«" Р е карбоновые кислоты также декарбоксилирМотг, п’Развет'
’•’е1 лпании; если в качестве нптрозпрующего агент, ри Нит’
Р,,гр«илсер!<У!0 кислоту, образующееся иитрозосоединен^пепТ
’ ппиро«ь!ВаетСЯ В °КСНМ- ПеР°ксикислоты, за исключением X
гр’заметенных, превращаются в иитрозосоединения пр действий
озплхлорпда (уравнение 17) [27]. р Действии
NOCI
R2CHCO3H > RjCHN'O + СО2 + О2
(17)
(4) Превращения алкилнитритов
чпС-Бис(иитрозоалканы) (2) можно получить из алкилнитри-
тов при их кратковременном термическом разложении (200—
]000°С), обычно при пониженном давлении [28]. Первоначально
образующиеся алкоксильные радикалы распадаются с образова-
нием наиболее объемистого из возможных алкильных радикалов,
который затем соединяется с оксидом азота (уравнение 18). Так,
бути.типтрит дает ч«с-бис(1-нитрозопропан), а из этил-, изопро-
пил- п трет-бутплнитрптов образуется цис-бпс(нитрозометан) (2;
R = Me). Условия реакции исключают возможность таутомериза-
ции и позволяют, если потребуется, «уловить» мономер.
д
R2CR'ONO ---► R2CR'O« + NO
------->- • Rl + NO —f R’N’O (18)
—r-co
R2 = H, Aik
ацетнлперокспдом при 73°C полу-
R1 = Aik,
При обработке бутил-2-ннтрнта
чают транс-бис(ннтрозометан) (3; R = Me) [29].
Фотолиз (<330 нм) алкилнитритов в газово1"1 фазе протекает
по упомянутому выше механизму, включающему распад алкок-
сильных радикалов (ср. уравнение 18); в зависимости от условий
Реакции могут образоваться цис- или транс-димеры. Из тРет'^У'
пинитрпта, например, образуется транс-бпс(нптрозометан) [30].
Нитриты, содержащие чстырехутлеродную цепь, прн облучении
(>330 нм) в инертных растворителях подвергаются перегруппи-
ровке Бартона с образованием бпс (нитрозоспиртов) |см.
Разд. 7.3.1 (3)]. ..... проходящие с распадом алкокспльных
Радикалов, сопровождаются существенными побочными проце
Сами, особенно в случае вторичных п третичных нитритов^ пряк.
u Фторированные ацплпнтрпты, которые образхютс Р сооГ.
в ' безв°Дных серебряных, ртутных или св!,нцов \ 'вых К!)С.
ют ТвУ|0щ,,х алифатических или ароматических Р анг!)д-
с иитрозидхлорндом пли при взаимодействии NA®
Р"дам» карбоновых кислот, легко подвергаются термин
379
-юбоксилировашно в газовой фазе н дают с хороцц1М11 8
де Р.ит»озосоедш1еиня (уравнение 19) [311.
Д8Ш‘ c9p,COiAg — C,F;COONO -> CSF, • + NO C3F7No
(5) Реакции с участием свободных радикалов
Помимо уже рассмотренных выше случаев, алкильные рад
1Ы можно термически пли фотохимически генерировать ,13 * * ‘
°ов иолндов н бромидов, а их последующая реакция сОк
до* азота приводит к нптрозосоединеппям. Именно эта реакс'а
послужила основой для использования оксида азота как ингХ
тора свободнорадикальных цепных процессов. Таким путем щ0)к,
но получить мономерный трифторпитрозометап (уравнение 20)-
подобное замещение осуществимо и у sp -гибридизованного атома
углерода (уравнение 21) [31].
CF3I —> -CF3 -----► CF3NO
ftv
CF2=CFI + NO cf2=cfno
(20)
(21)
(6) Присоединение к алкенам
Использование присоединения ннтрознлхлорида к алкенам [23)
в значительной степени способствовало ранним работам по раз-
делению и идентификации терпенов. Нетерминальные или терми-
нальные алкены неаллильного типа реагируют при низких темпе-
ратурах с нитрозилхлоридом, который можно генерировать in situ
из пентплнптрита, давая su^-хлорнитрозоаддукты, образование
которых можно было ожидать на основании механизма полярною
присоединения с участием наиболее устойчивого карбевиевого
иона (уравнение 22).
R
(22)
Г R I С1
RiC=CHR + NOC1 —► Lr2C—CH—NOj СГ —> R2C— CH—NO
В случае менее реакционноспособных соединений реакция
ускоряется добавлением кислот Льюиса, а также N2O4. При при-
соединении пнтрозилхлорида к хлорэтиленам с использованием
„ ал,°м,|1111я как катализатора правила ориентация,
линч “бл'одаются, в то время как присоединение к А9-окта-
к нообопнЛРОДС' 0'Н"трозо'9'хлоРдекалии [34а], а присоединение
к норборнадпену, цис-димеру соединения (7).
/Л /'СЛ <7>
z--^-^Z-'NO
[346], объясняют»»6!?'1 пРедложен четырехцептровын механизм
3S0 тереохимпю продукта реакции; нет данных,
е указывали бы иа нуклеофильное уЧастир и„
^'’Рщдиировку- в зависимости от условий реактив. СКелетнУО
^'давать продукты цис- или траиС.Присоедн ' ЦИК оОГ<;ксен
цо*е ..цепне некоторых цис- или транс-ахдуХв'п^Л „Взаим°-
"рС М ИТЬ что такое присоединение может бшь обп "Ред'
"'’'“’иеские исследования, согласующиеся с полярным^ ЫМ Ки’
^'.скИочают свободнорадикального процесса ₽[35? пХанизмом-
“когда иитрозогруппа связана с первичным ипи СЛу'
• ^.Ненасыщенные альдегиды, кетоны, кислоты “и*:
1ры 11 их производные образуют а-нитрозоаддукты, хотя в этих
случаях часто необходимо применение кислот Льюиса как ката-
шзаторов.
’ N,O, присоединяется к алкенам, давая внц-иитрозонитриты
(например, уравнение 23), хотя данные о структуре продуктов
п ориентации присоединения противоречивы. Предполагали что
первоначально происходит электрофильное нитрование, нитрозн-
ровапне или свободнорадикальная атака, однако более поздние
данные [36] согласуются с участием N2O3 как электрофильного
шпрозпрующего агента, причем продуктом реакции 2-метилпро-
пепа является 2-метил-2-нитрито-1-ннтрозопропаи (уравнение 23).
ONO
м2о3 |
Ме2С=СН2 -------► Ме2ССН2МО
(23)
Действие N2O4 иа алкены может привести к ге.и-нптронитри-
там по свободнорадикальному механизму; в отсутствие слабо-
основных растворителей этот реагент осуществляет электрофиль-
ное нитрозирование, приводящее к ге.ч-иитрозонитратам. Нитро-
зилсериая кислота и нптрозплформнат [37] присоединяются к
алкенам; гидролиз образующегося в последнем случае нитрозоал-
кплформиата открывает путь к бис (гидроксинитрозоалканам).
(7) Галогенирование оксимов
Реакция альдоксимов или кетокспмов с бромом или хлором
в кислой или щелочной среде или с иитрознлхлоридом дает гем-
галогеннитрозосоедпиеиия, вероятно, по механизму присоедине
впя — отщепления (уравнение 24), хотя имеются данные 0 т°“’
что необходимо наличие водорода при а-углеродном атоме. ®
Ц|1Я с алкилгипохлорптамп в растворе трпхлорфторметана Р
кает В мягких условиях [38]. В случае альдоксима таутомериза-
Ц||Я продукта и дальнейшее галогенирование могут npi
к Ы-дигалогеи-! -нитрозосоединению.
Hal
PaC=NOH + X—Hal —> R2C-N.
Х)Н
,На!
r2c;
X _цх
(24)
381
(8) Фотохимическое внедрение нитрозогруппы
а шшческая реакция (380 420 им) алканов в гачл.,
фоТОХ ' ' ' , азота в присутствии иеоолыиого количества v?06
фазе с оксидо вашп0 6пс(пптрозоалкапов) (уравнение 25) &
?РгХ"о предложенному механизму атом, хлора отрывает Д'
о• ткана и образующийся алкильпыи радикал затем реа? '
Pver с NO При облучении смеси алкана, хлора и NO в СРО" '
п е ни ( Vi. находящейся в газовой фазе, продуктом реакц„н
яв яется’ гс.н-пптрозохлорсоедиисние (см., например, ypaB^
пне 26), которое можно восстановительно дегалогенировать д0
оксима.
/«V
RH + Cl2 + NO ------► [RNOh 4- HCI
i + NO + ’ACh <CD'\No + 2HCI
(25)
(26)
Оксим образуется также и в том случае, если алкан, например
циклогексан,' насытить хлористым водородом и затем облучить
в присутствии NO. Если реагентом служит нитрозплхлорид, то
могут образовываться оксимы и гел1-нитрозохлорсоединения. При
использовании же избытка NO (3:1) или при удалении образую-
щегося хлористого водорода током азота получаются также и
бис(нитрозосоединения). К недостаткам метода следует отнести
его пеизбирательность по отношению к ациклическим алканам;
например, в случае гептана образуется смесь различных изомер-
ных бис(нитрозогептанов).
Альтернативная методика [40] фотохимического нитрозирова-
ния алканов, содержащих легко удаляемый первичный или вто-
ричный атом водорода, предусматривает участие трет-бутоксиль-
ного радикала, образующегося при фотолизе трет-бутилнитрита.
Облучение при 400 нм и 0—5 °C дает транс-бис (нитрозосоедине-
ния) с хорошим выходом (уравнение 27). При более низких тем-
г, kv ни ио
rper-BuOXO _NO> 7-per-BuO- ------> rper-BuOH + R • ------►
rper-BuOH + [RNO]2
(27)
пппп?Л?,Х МОГУТ быть выделены «(пс-изомеры. Найдены условия
ньши 1тл.Я ЭТ0П РеакцИ|1 применительно к соединениям с третич-
побочных реакцгйЛеРОДа’ ГЛе возможно более легкое протекание
7.1.2. свойства нитрозосоединении
Почти Структура
ствовать в в1|де!с?ип1°'СОедННе"ия диамагнитны, они могут суше-
пых димеров [ 1В1 ц,„ 'г"1 зеле,1ь,х мономеров (1) или бесцвет-
382 11С(|,|,тРозосоединения) имеют структуру
i2'C, более устойчивы, чем соответствующие мономе™ Л МИ'
,|С !кю1Х заместителей, как, например, в 2-бвом 9 ,? ' али'
ч1,е Т<1 „„трозотрифторметапе, ведет к стабилт- ,итР°зопропа-
«е "1’ к - более ’устойчивым" фор^^м®щ гХозо^"^^;
ктральиых характеристик с характеристиками виутриХкч
ярШх димерных систем существующих в цис-конф,,гур^ии как
рпмер, 2,2 -динитрозоб,,феипл (5). Известно, что ц"сфОрМы
огда оказываются первичными продуктами реакции (сМ
зд 7.1.D и могут образовываться из транс-изомеров см
азД. 7.1.3 (6)]. Самопроизвольное превращение цис-изомеров
в граяс-изомеры может осложняться образованием оксимов одна-
ко эксперименты, предпринятые с целью определить, происходят
при этом ,1лп нет процессы «захвата» „ли перекрестные реакции
показали, что изомеризация в ацетонитриле цис-бис (нитрозоме-
тана) и цис-бнс (нитрозоцпклогексаиа) протекает исключительно
путем диссоциации и рекомбинации свободных мономерных
форм [42]. Структуры (2) и (3) согласуются с химическими
и физическими данными, они были надежно подтверждены рент-
генографическими исследованиями [43]. Длины связей [N—N
(127—131 пм), N—О (130—135 пм)] указывают на то, что крат-
ность этих связен больше единицы. В транс-бпс(1-нитрозонзобу-
тане) фрагмент О—N—N—О плоский, углы у атомов азота близ-
ки к 120°. Первичные и вторичные бис (нитрозоалканы) дают бес-
цветные растворы, а некоторые — даже бесцветные расплавы,
причем для появления голубой окраски, присущей мономерам,
необходимо дальнейшее нагревание; при быстром охлаждении
мономеры вновь димеризуются. Третичные бис (нитрозоалканы),
однако, диссоциируют намного легче. Имеются данные структур-
ных исследований различных мономерных нптрозосоединений
в газовой фазе; сообщается, что угол между связями С N О
составляет ~ 120° [44].
Бис(ннтрозоарены) могут находиться в цис- или транс-конфи-
Урацпи; бпс (нитрозобензолу) отвечает первая из них а для
бчс(о-нитрозотолуола) известны обе формы [4а]. эти Д1- Р
обычно быстро диссоциируют в растворе, этот процесс уск р
пРи наличии в пара-положепни молекулы электронодонорных за-
местителей. н-Иоднитрозобензол — мономер зеленого С—N"
? кристаллическом состоянии, а меньшая лл"”а тельного
1'28 пм) позволяет предположить существоваип и
заимодействия за счет сопряжения между нптр Р'ю5ого типа
CTafil4eCK"M КОЛЬИ°М' Наличие орго-заместител исклю-
илизуст д„Мер, „ бис(о-ноднптрозооензол) • бис(нит-
Ч11тель>,0 в таком виде. Люттке [1в] предположил, что
ОЙО
< любое сопряжение между л-электропамц с.
розоарспах) мат1]1]ССКОго кольца ослабляет связь
O'N'е7 устойчивость димера. Вследствие этого эффекта и
уменьшает и образуется, а в цкс-копфигурацнц в р“Нс-
бис(ннтрозоо °Jibua выведены из плоскости О—N-N’J0 °’0'
пояжение” в достаточной мере снижено, что н позволяет обра^
Р Л тнмеру орто-Заместители увеличивают устойчивость дТ/
"Г ас X фенильные кольца выйти из плоскости снс^
&/-N-0 а подача электронов по цепи сопряжения от вд„„
заместителей’ препятствует димеризации. По-видимому, раано/ ;
ное превращение несимметричных оис(пптрозосоедниеипй) вС1!М.
метричные происходит слишком быстро, чтооы нх можно было
выделить.
(2) Физические и спектроскопические свойства
Чмс-Б||с(иптрозомета1|) имеет температуру плавления 93,5—
95°C, а транс-изомер плавится при 122 °C. трйнс-Изомер раство-
рим в воде и спирте, умеренно — в эфире, нерастворим в пентане.
гранс-Б11с(2-метпл-2-111|Трозопропан) быстро образует пары моно-
мера при плавлении (83—84°C). Мономерный интрозотрпхлорме-
тан — жидкость интенсивного голубого цвета, кипит с разложе-
нием при 57—58°С, а нптрозотрнфторметан— темно-синий газ
(т. кип. —84 °C). Бледно-зеленый цис-бис (иитрозобензол) в рас-
плаве (т. пл. 68—69 °C) становится изумрудным (т. кип. 57—
58°С при 18 мм рт. ст.). Температуры плавления бпс(о-, м- и п-
нитрозотолуолов) составляют соответственно 72—75, 53—53,5 и
48,5°C. транс-Б||с(нптрозоалкапы) значительно более летучи,
чем цис-изомеры; их разделение можно осуществить дробной воз-
гонкой. Это указывает на то, что димеры могут существовать и
в газовой фазе [46].
Характерная окраска мономерных нитрозосоедипенш"! обус-
ловлена малоинтенсивной полосой поглощения при 630—790 нм
(е ж 45—60) (п->л*-переход). Дополнительные полосы поглоще-
ния при 270-290 нм (е « 80) и <220 нм (е « 5000), наблюдае-
мые для алифатических соединений, полностью перекрываются
тш>аг»1МН |10гл0щення. соответствующими л — л*-переходу арома-
чяяпгиУ ПРОПЗВОД,1Ь1Х- Положение длинноволнового максимума
мольная яТбД^еСТИТеЛеЙ в алкилЫ1ОН пли арильной группе, и
пени дпмепп1™Ц11°пНаЯ спос°бность может служить мерой сге-
дается потопа Ш' спек~рах транс-бис (пптрозоалкапов) набЛ|О-
(е» 10 000) ппйП0ГЛ07йеШьп’ соответствующая л—л*-переходу
растворителя |1М’ пол°жение которой зависит от
цис-димеров устой,>Г»₽а алк1,льной группы. Полоса поглощения
на всегда при бопой'и*^ только в водных растворах, расположив-
кругового дихроизмя SИ“аХ воли (265—271 нм). Спектры
вторичных нптрозосорПпН 63?—790 нм для оптически активны*
од ишенпй, полученных окислением амино
«казаться пол'ёзп^^
11011 II окру>ке,,1|я хромофора |47]. Ределснии ориеи-
‘‘'“частоты валентных колебаний связи N=O для ,
„пзоалкапов н мономерных нитрозоаренов лежа/ “ОНОМеР«ь1х
111,1/Хасти 1539—1G21 н 1488—1513* см- дТя т^ТВеТСТВен-
перво интенсивное поглощение (е « 600-1500^ ирГнТ*
io (алифатические соединения) и 1253-1299 «-> (аоом1т~
скпе соединения), которое приписывают N-O-валеитиымколе’
алиям- У цмс-димеров эти полосы замещены дублетами и сме’
;„ы в область больших частот: 1323-1344 и 1330-149П р/ i
соаииепи»), 1389 „ И09 '(X™™
уединения).
Данные спектров 11МР позволяют определить степень дез-
,краиировання протонов пптрозогруппой: для протонов CH,-rnvn
„ы в траяс-бнс(а-ннтрозотолуоле) б 5,38 млн-‘; для СН3-группы
в транс- н чис-бпс(ннтрозометане) соответственно 4,00 и
4 20 млн—1 и Для СНз-группы в гра«с-бпс(2-метил-2-нитро’зоппо-
пане) 1,60 млн-1 (для СН3-группы мономера б 1,25 млн-1).
Смешение в сторону слабых полей сигналов о-, м- и п-протонов
цикла в нптрозобензоле по сравнению с бензолом (для раство-
ров в гексане) составляет соответственно 0,56, 0,26 и0,31мли-‘.
Для некоторых мономерных пара-замещенных нитрозобензолов,
имеющих электронодонорные заместители, с помощью ЯМР-спек-
троскопии можно определить барьеры затрудненного вращения
вокруг связи С—N [48] (для п-нптрозоанизола ДО* составляет
42 кДж/моль). Была проведена корреляция химических сдвигов
”N н 15N с положением длинноволновой полосы поглоще-
ния [49а]; при изучении равновесия между нптрозосоедпиениями
п оксимами [496] также использовали значения химических
сдвигов 14N.
В масс-спектрах нптрозосоедннепнй, в том числе иногда и для
димеров, присутствуют пики молекулярных ионов, а также ионов,
обусловленных характеристической фрагментацией; перегруппи-
ровки скелета довольно редки [5, 50а]. Спектр нитрозобензола
содержит интенсивный пик молекулярного попа, который распа-
дается с последовательными выбросами NO (давая пик с те //,
Ю07о) и С,Н2 (т/е 51; 93%). Как следует из масс-спектров, об-
щей особенностью является легкое отщепление NO [эОб].
(3) Изомеризация в оксимы
Таутомерная перегруппировка в оксимы, Хпесс
ПеРвичных и втори иных нптрозоалкапов,— ''^^"''ХншГида
Сы- Уравнение 2). Опа может происходить при • • ацни
Е?ре"пи’ ее скорость может превышать по-видимому,
Вй/?еризация нитрозометана в парах вы ’ оаСТВОрители,
Утрпмолекулярнып перенос водорода. По. р Р
13 Зак. 105
К11сюты или основания, а также NO уекорЯ1ОТ ,
Г11ЛЬ" R случае катализа под действием оснований суЩестаР '
заШ,Ю по Вадимов', является отрыв водорода из а-положе я
НЫ\Хпс1пе. Скорость перегруппировки вторичных б11с(1|
К НХе 1Н ), каталнзпруемон хлористым водородом, „е 3aBIJ
зосоедннепш , с10ты. Это позволяет предположить, что ГЛ
°" Г'отХляюЩеп скорость реакции, является дпссоц,,ац11я *
Д" 'Ха 511- подтверждением этому служит тот факт, ш
«Хозотолсол после превращения в траяс-двмер становится Чрез.
3 устойчивым. в кислой среде может происходить Пряь,Ое
“евращеипе первичных бис (нптрозосоедпненип) в алкнлиденац11Л.
™лоа31шы (уравнение 28) [51]. Оксимы могут образовываться „ в
результате фрагментации некоторых третичных нитрозосоеднненнй
(например, уравнение 29) [52].
(RCH2NO)2 RCH=NNHCOR + Н2О
hoch2cr2no —> r2c=noh + СН2О
(28)
(29)
(4) Таутомерия нитрозофенолов и нитрозоанилинов
Ннтрозирование фенола пли взаимодействие гидроксиламииа
(1 экв) с бензохиноном дают один и тот же продукт, кото-
рый можно получить в двух кристаллических формах. Химиче-
ское поведение этого продукта указывает на то, что он может
реагировать либо как /г-ннтрозофенол (8), либо как оксим бензо-
хинона (9). Судя по спектральным данным, n-ннтрозофенол суще-
ствует, главным образом, в виде таутомерного оксима (9), в то
время как о-питрозофенол — это именно нитрозосоединевие, суще-
ственную роль в стабилизации которого играет внутримолекуляр-
ная водородная связь. Следует отметить, что таутомерное равно-
весие нптрозофенолов и -нафтолов очень сильно зависит от при-
роды растворителя и осложняется возможностью ионизации
путем отрыва протона [53].
В случае п-нитрозоаннлинов (вещества зеленого или голубого
цвета) имеется ряд данных, подтверждающих существенный
"±Л"^1,Л1,ЫХ СТ₽УКТУР в резонансные структуры (10) этих
imcwuL „ Н1 г1, В частностч, относятся высокая мольная абсорб-
моменти щon 'пСТЬ дл1|нноволн°вой полосы, высокие дипольные
( > Д для и-ннтрозо-М,М-димет11ланплина), а также
386
sLisz»”'ука,ива" - ’г«
(5) Внутримолекулярные взаимодействия
Попытки получения о-динитрозобензолов приводили к бее
цветным или желтым мономерам, имеющим строение бензофурок-
санов (Па и 116). С помощью ”0-, ИС- „ >Н-ЯМР-спектоо
ск0Ш|ц было изучено равновесие 11ат±11б (R = H) (AG* ~
~63 кДж/моль) [55]. При высоких температурах такое равно-
весие для бензофуроксанов устанавливается быстро (дан-
ные ЯМР); по некоторым косвенным данным, эти равновесные
превращения происходят с участием о-дннитрозосоединення (урав-
нение 30). Полагают, что простые фуроксаны также изомеризу-
ются с участием динитрозоалкенов [56]. Индивидуальные изомеры
О-
(На)
(Пб)
(30)
(11) можно различить при низких температурах. В равновесной
смеси 5- и 6-нитробензофуроксанов (11, R = NO2) преобладает
6-иитроизомер, его содержание при 31 °C составляет 70%; в слу-
чае 4-нитробензофуроксана равновесие смещено в сторону 4-нитро-
изомера [57]. Нптробензодифуроксан и -бензотрнфуроксан, для
которых также возможны подобные структурные взаимопревраще-
ния, образуют устойчивые кристаллические комплексы с рядом
ароматических молекул, обогащенных электронами.
7.1.3. РЕАКЦИИ НИТРОЗОСОЕДИНЕНИИ
(1) Окисление
Ароматические и алифатические ннтрозосоединения обычно
можно окислить до соответствующих нптросоедпнений целым
Рядом реагентов; этим объясняются низкие выходы нитрозосоеди-
неиий, когда их получают окислением аминов. Некоторые третич-
ные соединения чрезвычайно легко окисляются на воздухе. Для
окисления используют азотную кислоту, щелочной раствор пер-
оксида водорода, перманганат, оксид хрома (VI), кислоту Каро-
гипохлорит, озон, перуксусную и трнфторперуксусную кисло-
ТЫ- Для окисления ароматических производных
более сильные пероксикнслоты; окислению способствуе
13* 387
.iv чяместнтолбИ в лара-положенпи и ...
э-1СКтрОП„едполагают, что стадией, определяющей скорость р^'
группе. HPef',.VK.ieot|,ильная атака атомом азота нитрозопХ ®
группы «ОТЫ [58].
(2) Восстановление
Многие мягкие восстановители превращают Ш1трозоаре1ш
в гидроксплампны. Гидроксплампны затем к0ндевспру10ТСя
® „нтпозосоедапеппем, образуя азоксисоедппеппя, которые обыЧ11и
являются основными продуктами реакции. Спиртовые расТВОрЬ1
шеяочей борогпдрпд натрия и арсенит могут применяться как
восстанавливающие агенты; восстановление хлористым водородов
может сопровождаться «ара-замещснпем. Для защиты С-ннтрозо-
группы от восстановления и конденсации используют аромати-
ческие сульфиповые кислоты, образующие невосстанавлпвающиеся
далее гидроксплампны [59]. Прп более энергичном восстанов-
лении с использованием системы металл кислота [60], катали-
тического гидрирования [61], никеля Ренея или дитпоипта натрия
образуются амины; из интрозобепзола может образоваться азо
бензол, вероятно, путем восстановления до анилина и последую-
щей конденсации с неизмененным нитрозобензолом. Менее рас-
пространено гидрирование алифатических нптрозосоедцненцй.
1(ис-Бпс(нитрозометан) количественно восстанавливается до
метиламина, из бис(нптрозоциклогексана) получают цпклогексил-
амин. Бис(нитрозоалканы) могут восстанавливаться до азокси-
соединений, гидразинов и аминов непосредственно, минуя стадию
перехода в мономеры. Гидразобензол восстанавливает нитрозо-
бензол до фенилгндроксиламнна. При восстановлении гелинптро-
зохлорсоединеннй гидридами металлов или прп гидрировании
образуются соответствующие оксимы; тот же процесс, правда,
с невысоким выходом, происходит н прн фотохимическом восста-
новлении в эфире или циклогексане [62].
Нитрозосоединения дезоксигенируются соединениями трех-
валентного фосфора с нуклеофильным атомом фосфора, приводя
к самым разнообразным продуктам [63]. Ароматические нитрозо-
в с’ РеагиРУют с трпфенилфосфипом или трпэтплфосфитом
мешеинпгА ДЭВаЯ азоксибензолы (уравнение 31); в случае пеза-
нптрозобензола реакция идет с низким выходом.
ArNO + (ЕЮ)3Р —+ ArN=NAr (31>
I
В °"
в отсутстш!е°Дпазстппп,Ц!1ЯХ’ ос°бенно при избытке реагента или
нмцдаты [иаипимепР/гХх! ;ф1ГУт также образовываться фосфор-
П₽" проведХ^О):Р—NAr] ИЛИ продукты их гидролиза,
золов невысоки в „вк без растворителя выходы азоксибеи
388 ’ ольиюм количестве образуются амины. При-
поая продуктов этих реакций и сравнение с результатами
°'олиза и пиролиза арилазидов привел,, к общепринятой
; °, астояшее время выводу, что реакция протекает с участием
>ТУчГГ/ОсмОАРаб6)О,ЦИХСЯ apM,!,iTpe'10B <Аг№) "йавие
н»я 32 '”И • '•
ArNO+(EfO)3P » ArN——P(OEf)a —► (E1O)SPO + ArN * (32)
О
ArNs + ArNO —» ArN=NAr
। и»)
O'
ArN S-f-(EtO)aP —>. (E10)aP=NAr (34)
Особенно убедительным доказательством промежуточного
образования нитренов было получение 2-амиио-3//-азепинов (12)
прп реакции нитрозобензола с трифенилфосфином в бензоле
в присутствии диалкнламннов (уравнение 35). Предполагают,
что ароматические иитрозосоедииения с электроноакцепторными
заместителями также генерируют нитрен, которые дает продукты
замещения с используемыми в качестве растворителя аромати-
ческими соединениями с электронодонорньшн заместителями при
условии, что в данном растворителе иитрозосоедииения сущест-
вуют, главным образом, в мономерной форме (см., например,
уравнение 36) [64а].
Л—\ N. N-диметиланилвв
NC----\ Э—3'0 + P(0Et)s —----------------------
NMe2
> /—NO +Азоксисоединенне (17%) + Амия (7%) (36)
(18% о-, 8% л-)
Нитрен, образующийся из интрозопентафторбензола, можно уло-
вить в виде N-арнлазеппна — продукта реакции с аренами со
слабыми электронодонорцычш заместителями; например, из
анизола образуется 2-метокси-1-пеитафторфенил-1//-азепнн (13)
[646].
/ОМе
J^^N-CcF5 03)
3S9
« ,4a приведенные данные, свидетельствующие
HeCM°Xe ов нельзя полностью исключить и согласований
участив нптреио , на аТ0М азота а предшественник
отшепление фосф установления привело к синтезу p®.
развитие этих соединений. В качестве примеров М0Р
тетеропнклпческ елму|о ц| заци,0 2-ннтрозобиарИло“
"Р'1вес™ ,,и 2-(о-ннтрозофенил)пиридина в пиридо[1,2-б] ин.
“ кяпбазолы Пр|) обработке нитрозопентафторбензола
....... азнр11Дин
алкену
привести
в карбазолы и
Даг°тко^трнХ ф’осфитГв “среде алкена ‘ образуется
"а с™стереоспецифического присоединения нитрена
(уравнение 38).
к
'NO
P(OEt)3
эф«р, 0 °C
(~100%)
(37)
e
(18%)
Н\ Z1 (ЕГО)зР „
CgFjNO + )=\ _юОс' >' CeFsN
Me Me
(38)
Реакции нитрозоалканов с производными трехвалентного
фосфора изучены в меньшей степени. 2-Нитрозо-2-хлорпропан при
взаимодействии с триэтнлфосфптом дает производное оксима
диэтилизопропплнденамииофосфат (EtO)2P(O)ON=CMe2. гем-
Галогениитрозоциклоалканы при комнатной температуре обра-
зуют промежуточные соединения, которые подвергаются пере-
группировке Бекмана с расширением цикла (уравнение 39). Было
показано, что реакция мономерных монофункциональных алифати-
ческих и алициклических иитрозосоединений с триэтнлфосфптом
не идет через нитрены, а приводит к азометинам с миграцией
алкильной или, предпочтительнее, арильной группы (уравне-
ние 40) |66]. Последующий гидролиз азометннов может привести
к амину и кетону. Ннтрозосоединения удается дезоксигенировать
также пзоцпаиидами; при этом, вероятно, через стадию образо-
вания нитренов получаются карбоднпмиды (уравнение 41).
О
.Cl
NH
'NO
'-' СГ -РЬзРО
р ГМА P(0Et>3
KsCNO ------> R2C=NR + OP(OEt)s
CPaNO rp“. MeNC
CFaNj ------>- CFsN=C=NMe
(39)
(40)
(41)
(3) Реакции с ненасыщенными
Взаимодействие нитрозоперфторсоединений
ыМ11 алкенами может привести к оксазетидииям фтоР3амещеН-
ЫравИенне 42), через ион-радикальнь.е ’ <’4)
,я [67] и к сополимерам, образующие я Х,У ч"ь,е соедине-
яизкнх температурах. В случае алкенов неРим^ЩеСТВенно пРа
ванных заместителей, но содержащих а-метиленоДДМ фтоРиР0'
рода, возможен дальнейший гидролиз до пХ„ ?‘,Ы юдв'
роксиламинов. Из нефторированных соединений Щ ,ЫХ гид‘
0Рбычио не образуются [68] хотя из интроХТта °Газетидии«
ксиэтилена был получен 3,3,4,4-гетраметокси-2-*енил о Трамет°-
Д11н. Реакции ароматических нитрозосоединений г п,336™’
алкенами обычно приводят только к азоксиаоеня? различиыми
становления нитрозосоединенпй [69]. ПАоплалкД.. Д СЧеТ ВОс‘
ны [68], например (15) [уравнение 43], формально заХт''ТР°'
щепления двойной связи в алкене; в случае реакции со г рас'
был обнаружен неустойчивый промежуточный продукт'^ИбГ
который распадается с образованием бензплиден-Л'-фенилнитрона
Аддукты (17) и (18) идентифицированы как продукты взаимо-
действия нитрозотрифторметана со стиролом „ трифторстиротом
соответственно. 3 н цщи.юм,
F2CCXY -I
I + CF3N0: —>
CF3NO. J
f2c—cxy
—► I 1
CF3N—О
(14)
PhNO + Ph2C«CH2
CF3NO + F2C=CXY
Ph
. CF3
I /Ph
/NPh
О
(16)
(17)
+ CF3NO
O'
I
-> Ph2C=NPh
(15)
' T F
Ph— — —F
CF3-N—N-CF,
I I
O' О
(18)
(42)
(43)
Многообразие продуктов реакции ароматических питрозосоедине-
н,|й с алкенами, имеющими а-метпленовые атомы водорода,
можно объяснить протеканием реакций, показанных на схеме
44 |70]. Первая стадия — это присоединение алкена, приводящее
('9). Если в ароматическом цикле содержатся «нейтральные8
11 электроноакцепторные группы, реализуется, главным обр ,
веДУЩий к нитронам и ароматическим азокспсоедш
м- При наличии электронодонорных заместителей
391
О
термические реакции тут в основном по пути М)
алхеииларилампиы и «.^-ненасыщенные нитроны Hi °брадШТс
зультате последующих реакций образуют сложную сме°^Ы в Ре’
(19)
Алифатические мономерные нитрозосоедпненпя
в роли диенофилов, если при а-углеродном атоме у
жнтся электроиоакцепторнын заместитель [71]; в
реакции образуются цнклоаддукты (например, 20),
ОТПИНТОПО О П — . - t П ' . — .
выступают
них со дер -
результате
* jniи; псдuj?11л!vр, }у имеющие
структуру 3,6-днгпдро-!,2-оксазина (уравнение 45). гел-Нитрозо-
цианосоединення дают устойчивые аддукты. гелг-Нптрозохлор-
соедииення образуют менее устойчивые продукты, вероятно,
РД,ТВ’,еГОГ0' ЧТ0 ХЛо₽нл—хорошая уходящая группа; однако
с тп«?аКЦНЮ ПРОВОД11'Г1> в спирте, удается получить гетероциклы
выходом„ (уравнение 45). При взаимодействии нптро-
J оедпненли с перфторалкенами образуются устойчивые
/С1
RiC +сн2=спсн=сн2 —> Г ?'
Е10Н
L о
(20)
+ R2C(OEt)s
'* О
(45)
392
рь, (уравиеиие 4b).
(46)
Несимметрично замещенные диены иногда дают смеси изоме-
ров, а из I-арнлбутадиенов образуется исключительно тот изо-
мер. в котором кислород присоединен к бензильному атому
углерода. Электронные факторы в диене не оказывают, как пра-
вило, влияния ни на ориентацию, ни на скорость присоединения;
наличие же электропоакцепториых заместителей в нитрозосо-
единенни заметно ускоряет реакцию. Бнс(л-нитронитрозобеизол)
присоединяется к 2,3-диметилбутадпену, давая производное
тетрагндропирпдазпн-М.М'-диоксида (21). В обзоре [72] приведе-
ны данные о диенофильной природе некоторых С-ннтрозосо-
единений, например ннтрознлциаипда и нитрознлкарбонильных
соединений.
(21)
Реакцию перфтораллена с нитрозотрифторметаном можно
провести в контролируемых условиях и получить соответствующий
оксазстидни: при взаимодействии тетраметилаллена с нитрозо-
бензолом образуется соединение (22) [73]. Сам ацетилен инер-
тен в реакциях присоединения, но различные алкины реагируют
с интрозобепзолом, давая зачастую сложные продукты так,
например, из толана (PhC = CPh) получают нитрон (23р при
реакции дегидробензола с интрозобепзолом получают ср hhj
карбазол (24).
С1!г=
Ме Мс
1 I /
=С—С—С—Nz
J I \
N Ме
Ph
ОН
О'
О’ !
ph-N=C-C=N-Ph
* I I *
Ph Ph
(22)
(23)
(I)
Реакция присоединения и конденсации
,..пямнны легко можно получить, подм
^-Днарилгидрокс ого реакт„ва Гриньяра „а apoVa.
ствовав и’бытк^гоедпнеппе в проведя затем гидролиз; моЖе1
^четкое ннтрозосоедю __ ВОССтаиовлеиие гидроксила мина Дз
пройти К побочная реаки g оматнчСском кольце содерЖаТся
вторичного амина «-1 заместители, гидроксиламин ВЬ1де.
еХые электро»одо»ор«^тся втор11ЧИЫЙ ам)1Н н азобеиз^
лить не Удается; ,ьейиллития с иптрозобензолом являются
471 1741
- X
Ароматические ннтрозосоедпнения реагируют с соединениями
с активной метиленовой группой в присутствии основного катали-
затора (пиридин, Na^COa). Первоначально образующийся аддукт,
предположительно имеющий структуру (25), может отщеплять
воду, давая анпл (26) (реакция Эрлиха — Закса) [75], или
окисляться в нитрон (уравнение 48). Трудно предсказать, какая
из реакций будет преобладать, хотя, по некоторым данным,
алифатические соединения с активной метиленовой группой дают,
главным образом, анилы, а активированные толуолы — нитроны,
которые могут перегруппировываться в бензанилиды. Если ме-
тиленова'я группа активирована цианогруппой, реакции могут
осложняться протеканием дополнительных процессов.
)СН2 + ArNO
C=NAr
(25)
I
(26)
(48)
О'
I
:c=NAr
бензилХоХиашГв^ реагируют с некоторыми
нитроны [761- чти -it пР11сУтствпн сильных оснований, образуя
ниевых солей (n₽Bu, и П??ДУКТЬ1 получают и из N-бензплпириДП-
ния можеГи~ЯцрТройке) (уравнение 49) [77]. Эта реак-
илиды серы, наппнмеп ' >е3 пРомежУ'го,1иое образование илидов,
Диненнямн давая ни РеагиРУют с ароматическими нитрозосое-
напротив, при ветки, ? !’Ы ^Равнение 50). Илиды фосфора,
неиие 51), что напп».,КТ'к>зосоеД1111с1111ЯМ|1 дают анилы (урав-
‘ нает реакцию Виттига. Взаимодействие
тпочобс>13()Ла с фенилгидразонами ароматических япипл
и'ётонов приводит к соответствующим нитронам [78а]. Д°В
,__, О’
+ // \ , основание I __.
Ar'CHaN^ 2 + ArNO ---------—► Ar'CH=NAr + (,
О’
’ ’ I
RaC—SMe2 + ArNO —>• R2C=NAr + Me2S j
Ph2C—PPh3 + ArNO —> Ph2C=NAr + Ph3PO (
Многие диазоалканы легко реагируют с нитрозосоединеииями
давая нитроны, вероятно, путем электрофильной атаки атомом азота
нитрозогруппы по атому углерода диазоалкана (например, уравне-
ние 52). В случае диазометана обычно образуется бис(нитрон)
(уравнение 52), хотя при наличии объемистой алкильной или ариль-
ной группы можно получить и метиленнитрон [786]. С нитрозотрп-
фторметаном диазометан образует сополимер. При реакции арома-
тических иитрозосоединений с азотистоводородной кислотой с хоро-
шим выходом получают арнлазпды. Возможно, реакция протекает
через образование неустойчивой частицы ArN(O)=NH; электро-
иоакцепторные заместители ускоряют реакцию.
°‘ Г 0 1
I Ph2CN2 СН2\2 | I
C6F6N=CPh2 «-------- C6F5NO -------> C6FSN=CH- (52)
+ u + J2
В присутствии изопропоксида алюминия бензальдегид реа-
гирует с нитрозобензолом, давая гидроксамовую кислоту
PhCON(Ph)OH; кетоны инертны, а тпокетоны дают аинлы.
Первичные ароматические амины реагируют с ароматическими
нитрозосоединениями с образованием азосоединеннй (уравне-
ние 53); иногда в качестве побочных продуктов образуются
азоксисоедпненпя.
ArNO + Ar>NH2 Ar.N=NAr! + H2O (53)
С алифатическими аминами, однако, реакция не идет столь легко.
Некоторые ннтрозосоединения реагируют с дифениламином
в среде концентрированной серной кислоты; при этом происходит
замещение амина в пара-положение и образуются ^ярко-синие
хиноннмпиы. Эту реакцию можно использовать для оонаружения
вчтрозосоединений. При конденсации \’,№-днхлораминов с нитро
зосоединенпями могут образовываться несимметрично замещенные
азоксиалканы и алкилазоксиарены [79]. Взаимоденствне арома-
тических иитрозосоединений с гидроксиламином в ше-’о °, Р
РРчводит к сын-диазотатам (уравнение 54), а с ’ к1’
^арилгидроксиламинами - к азоксисоединениям. Ьслн; в у
Реакцию ввести ArNO и Ar'NHOH, образуются как неснмметр
395
,е азокспсоедипеиия, что указывает на
так „ СИММИР ' равновесие между реагирую,Ц11м®
бистро устанавливаюш ннтрозоарепов с несимметричны^,
гоедипеиия""- Ко1'Ддразниам11 приводит к триазец-Ы-окспду,
Хамешевпьши ДО, ок1,слеш1Я промежуточно образующей,
предположительн , J ,ураВНе)1пе 55). Вызывает сомнение
N-гидрокспиропзвод ого ^обензола с фенилгидразином. Сооб-
сообщение , N. рпдрокспдпарнлтрназена как главного
шалосъ о уоаввепие 55), однако по другим данным,
продукт;'РеаХ„даРмиии, количественно, азобензол,
образуются дифен
ArNO + NHzOB
(54)
Ph\
XN—NH2 + PhNO
Me/
O’
Ph. I
>—N=N—Ph
(55)
Ароматические иитрозосоедииения подвергаются самоконден-
сацпи в присутствии концентрированной серной кислоты. Если
пара-положение свободно, образуются п-интрозодиарилгидроксил-
ампны (уравнение 56), если же оно занято, то продуктами
являются феназины и феназинокспды наряду с небольшими коли-
чествами М,М-диарилгндроксиламинов (уравнение 57). Нитрозо-
соединения могут конденсироваться с фенолами, давая хиноидные
системы; реакция п-иитрозофеиола с фенолом — основа пробы
Либермана на нитрозосоединения (см. разд. 7.4.3).
(5) Реакции захвата радикалов
не11иями0Л"олпРаДИКаЛЫ РеаГ11РУют с мономерными интрозосоеди-
Обычно 04eniPwm- 11"1Рокснлы,ые свободные радикалы; они
тифицпповат/е ИЧНВЫ " МОгУт быть легко обнаружены и идеи-
ловушки) 1801С QOMOUlb10 ЭПР-спектроскопин (метод спиновой
2-1штрозопропай оС° еН1Ю Ш||РО1<ое применение нашли 2-метил-
тилнитрозобензол- е т"‘п’3‘Н|1ТРоз°бута11Ои-2 и 2,4,6-три-трет-бу-
арильные, алкок™... .,!'Х "омощью удалось уловить алкильные.
лнп реакций пьоилл1остш\Ло1ЛЬИЬ,е И Т[||,-'1ьные радикалы. Этот
ного радикала обп-иь,₽’,’°Ва" пР|1ме1’°м захвата ........
ЗЭ6 ’ “Роющегося при фотолизе N-бромсукциннмида
„ш-нпе 58). В общем случае реакция с пя
^полностью замещенным гидроксиламииам;1^ирХр" XT"
к „тяиа п метильного радикала образуется тп, m„J ’ птру‘
3°Ми Особенно хорошей ловушкой для гидпокси-п ,™ЛГИДР°КС11Л-
как ингибиторы полимеризации. используют
трет-ВиХО
(58)
Ароматические ннтрозосоединения реагируют с NO, давая
нитраты диазония или продукты их разложения; первоначально,
по-впдимому, образуется динитрозогпдроксплампн (27), неустой-
чивый аналог которого был действительно выделен при реакции
нитрозотрифторметаиа с NO при —100 °C. Другие алифатические
ннтрозосоединения образуют ннтросоедпненпя, нитраты и следы
нитритов, вероятно, по той же схеме [81].
zONO
Аг>/ (27)
\no
(6) Фотохимические реакции [82]
При облучении ароматических и алифатических нитрозосоеди-
иенин обычно образуются нитроксиды. Для алифатических нитро-
соединении этот процесс происходит при /. > 660 нм (красный
свет), для питрозобензола требуется применение УФ-облученпя.
Полагают, что в реакцию вступают мономерные молекулы (урав-
нение 59) [83а]; при облучении красным светом могут образо-
ваться смешанные арплалкилиптроксилы (уравнение 60), нитро-
зобеизол при этом не затрагивается. При УФ-облучении аро-
матических иитрозосоединений может также происходить их
восстановление до азокспсоедпнений; нптрозоцпклогенсан дает
главным образом изомерный оксим [836].
RNO I?NO* R2N0‘ (59)
. трет- Bik
PhNO + трег-BuNO * + <^r-Bu)2XO • (60)
1 >000 нм ph/
Облучение при 320—360 нм транс-бнс (пптрозоалкапов) в «стек-
лах» 2-метилтетрагидрофураиа при —198°С приводит п и,
йиацпи до мономера [83в]. Если повышать температуру можно
10 п темноте образуются цис-бис (иитрозоалканы), и.
397
, —25°C. Эта методика была с успех
храш-ть ло Tt‘XP\jv ряду питрозоалканов, за исключением Тр“
применена к ' Н11ТрОЗодифенилметана. При облучен,,'
Аих pbX ”птрозотрифторметана образуется димер N-Hl1TpH"
красным светом < Р 6]). возможн0, „ при этом происходит
тоампна ОР гя41 фотохимическое разложение некоторых
фотодиссоциация i р объяснить протеканием реакции ф',то.
нитрозоалканов 62). Фотолиз ге.и-нитрозохлорсоеди
злиминирован. яДр-ней(уравнение 63). во Х0Рд
НеННИи неясенР Предполагают, что в случае Ьнитрозо-Ьхлор.
Sгексана [85а] и некоторых других соединении [856] может
“'Ходить первичное расщепление связи С-N; фотоокисление
X 2-нитрозопропана также включает первичную фотодиссо-
ш.аци.о. в присутствии избытка кислорода образуется трет-бутил-
нитрат [85в].
CF3NO
hv
----—> (CFshNONO
>600 нм
(61)
NO
I /IV
Ме2ССН2СОМе ———* Ме2С=СНСОМе (62)
vvu n М
С м МС
| hv Ме\ I
’С—NO -------->- ,C=NOH + МеСОС=.\ОН (63)
' >600 им, /
МеОН Et
ароматп-
в случае
действием
(7) Реакции замещения
в ароматических нитрозосоединениях
Электронов кцепториый эффект нитрозогруппы в
ческих нитрозосоединеннях в большей степени, чем
нитрогруппы, облегчает обмен napa-заместптелей под .
нуклеофильных агентов. Так, п-иитрозодпметпланилин гидроли-
зуется водной щелочью до монооксцма хинона и диметиланнлина,
тогда как 2,4-динитроанилин реагирует очень медленно. Эту
реакцию можно рассматривать как важный метод получения
чистых вторичных аминов. Простые эфиры п-нитрозофеиола
расщепляются горячей щелочью, и алкоксильную группу можно
upr™™Tb Иа аР[,лаМ11Н0ГРУппу при действии первичного аромати-
ческого амина в присутствии кислоты.
в н.,™^ ®ЫЛ° ожидать, что электрофильное замещение
как при бпг>м?Ле приведет к Л(ета-продуктам, но на самом деле
нитровании де’йием’ N ДетыреххлоР,,стом углероде, так и при
ристом углепопр в пРисутствпи РгО6 в четыреххло-
показано что бпомРаЗУЮТСЯ продУКТЬ1 лара-замещения. Выло
стадией, лимнтиоуюшрйТ11"6 ~ аатокаталитическая реакция, а
бромистого водопола СК0Р°сть реакции, является присоединение
быстрое замещеиир . , рааован['ем гидроксиламина; затем идег
за8 трофильным бромом в гсара-прложенпе и
7.1.4. НИТРОЗОЛОВЫЕ КИСЛОТЫ
«.Нитрозооксимы (28), называемые также нитрозоловыми
„„слотами, недостаточно стабильны, чтобы их можно было выде
л ть; сравнительно устойчивы их соли с металлами. Например'
калиевую соль этилнитрозоловои кислоты (28; R = Me) синее
кристаллическое соединение, можно хранить при О °C. Нит’розоло-
вые кислоты можно получить из оксимов гидроксамовых кислот
(29) путем их диспропорционирования пли перйодатного окисле-
№Я. ыпи Юн
R—СХ
\nHOH
(29)
^»°H
R—C
\no
(28)
Известен трифенилметиловый эфир этилнитрозоловои кислоты
[87]; однако попытки получения бензиловых эфиров нитрозоло-
вых кислот приводили к несимметрично замещенным 1,2,4-окса-
диазол-4-окСПдам, например (30) (уравнение 64).
NOK
R—С'' + АгСН2Х —>
Чю
о
(30)
(64)
7.2. НИТРОСОЕДИНЕНИЯ
Химия нитросоединений, RNO2, очень многогранна; мы реко-
мендуем читателю обратиться к специальным обзорам. В этом
разделе рассмотрены .тишь наиболее важные методы получения
и свойства этого класса соединений.
7.2.1. МЕТОДЫ ПОЛУЧЕНИЯ АЛИФАТИЧЕСКИХ
И АРОМАТИЧЕСКИХ НИТРОСОЕДИНЕНИИ [88]
(1) Прямое нитрование [<W]
Реакции у насыщенного атома углерода. Проведение этой ре
а*нии в жидкой или газовой фазе [90] имеет ПР°МЫШ“!
значение для получения простых иптроалканов, однако
Роторной практике из-за обилия образующихся продуктов
ходимости применять жесткие условия она не полу i • й
'° Распространения. Реакцию обычно 1’P0B2^Ir впемя кон-
с СТеме, используя 50-70%-ную HNO3 (400-/00 С, время
399
n 9 с) или N20„ (250—600 С, 14 мин). При yMepeiI
такТа '’rvn'ix (300 -500°C) не происходит скелетная перегр“х
температурах н1|Трование сопровождается окислением; 0^"'
"пр°ХсЯ кислородсодержаитие продукты и нитросоеД1„1еР я
зуюпикся К У частности, алкены и нитроалканы с ме1,
С"о> ™’’углерода. Эти реакции нитрования
д ка ьиый характер [91]; их можно с успехом применять Для
нитрования алифатических боковых цепей в ж»риоаромат»ческнх
“'пЕГ' гетеролитпческое нитрование азотной кислотой
иногда в полифосфориой кислоте, можно тем ие менее использо-
вать в лабораторной практике для нитрования соединений с ак-
тивной метиленовой группой; при этом в мягких условиях можно
получить иитросоединснпя с высоким выходом. В качестве при-
мера приведем получение диэтилнитромалоната из диэтил.мало-
ната (уравнение 65).
СЖСОДДЙ O2NCH(CO2Et)2 (65)
(92%)
Образующийся при этом нптроэфнр разлагается в присутствии
азотистой кислоты, поэтому ее удаляют добавлением мочевины.
Легкость, с какой индандиои-1,3 подвергается нитрованию, а
образующийся а-нитрокетон — щелочному гидролизу, обеспечи-
вает возможность создания удобного способа синтеза первичных
нитроалканов [92] (уравнение 66). Нитрование соединений с ак-
тивной метиленовой группой действием N2O4 в четыреххлорпстом
углероде. или хлороформе, иногда с добавкой сульфата меди,
протекает с участием свободных радикалов; нптросоедпнений
образуются с умеренным выходом (~50%). Диэтнлмалоиат,
например, окисляется в указанных условиях до 2-оксопропандпо-
вой кислоты (мезоксалевая кислота). Нитрование, по-видимому
электрофильное, алканов и циклоалканов солями нитрония
в инертных- растворителях протекает с низкими выходами [93]
иеакиия обычно идет медленно, при 25 °C выход нитрометана из
зетана составляет лишь 0,1%, хотя в среде суперкислоты он
„ тп, ра3 ВЬ1П1е- Нитрование 2-метилпропана смесью азотной
тросоедицений°Т ПРИ 5°°С такжс ИРИВОД‘” к низким выходам ни-
о
о
со;
+ rch2no2
со; (в6)
зоваиием анионов ^с-оотп1’1!- pCai "I1-101 с алкилнитратами с обра-
,вд ^отвстствуЮЩ11х питросоедпнеиий; флуорен,
лаеТ калиевое производное 9-нитрофлуореиа (уравие.
:::s
EtOK
+ EtONO2 -------
(67)
no; kh
(>70%)
псоедпненпя, образующиеся из соединений такого типа,
неустойчивы, и при подкислении солей с целью выделения
'1аСТ°ных нитросоедпнений может происходить их частичное раз-
"сХ0Д Было найдено, что З-метнлбутилннтрат в сочетании
•10ilie' бутилатом калия в тетрагндрофураие при
с ' ямнппм калия в жидком аммиаке
е1це проще,
с успехом
разд.
я.ш-дпнптроалкапов
лидировав условия
его расщепления,
ине 69) [95а, в].
i —50 °C, пли,
' с амидом калия в жидком аммиаке, может быть
.Ли„' использован для этой цели (95а] (см. также
(3) ]. Эта реакция является общим методом синтеза
|Д‘ V (уравнение 68) (956]. Можно также, моди-
реакции, получить мононитрокетои и продукт
эфир ы-ннтрокарбоновой кислоты (уравне-
В реакциях с арплацетоиитриламии
мн формально происходит замещение не вод( Р )да’ а „л|[
(уравнение 70). Нитрование мопозамеше > ых ма.тоновы^^
ацетоуксусных эфиров нитратом ацетон^'А^уравнение 71).
гвип избытка гидрида натрия дает a-ни р ф Р
PhCH2CO2Et рьсн-ыо;
(80%)
ono2
I ’Nall
PCI I(CO2E1)2 + Me2C—CN ->
(70)
(71)
r+ Ц- (EtO)2CO
NO2
RCIICO2Et
(-50%)
При реакции пропилнптрата с
кг1рбоповой кислоты образуется
дианионом
иитроалкан.
псразветвленной
содержащий на
401
меньше, чем исходная кислота (уравце.
0Дпн атом углерода
пне 72) |96] I. ргомо2
г(„да-рг)2м^ RCHLiCO2Li ----;----">• RCH2NO2 (7?.
RCH2CO2H K -' 2. H : —CO2 1’2)
(45-68%)
n ,,u v «Агибридизованного атома углерода в алифатиче-
п^няду Дм получения нитроалкенов в ряду стероидов исполь-
CKOM ряду- *м ;еиов с холодным эфирным раствором азотной
зовал''р ^з холестерилацетата, например, с выходом 72% полу-
чей 6-ннтрохолестерилацетат. Аналогичные реакции замещения
протекаю/в случае триарилэтиленов (уравнение 73) [97] „ не-
которых дезактивированных эфиров коричной кислоты. Нитро-
алкены можно также с хорошим выходом получить путем
лвухстадийного процесса (уравнение 74): сначала происходит
присоединение N2O4 в присутствии кислорода по двойной связи,
приводящее к смеси вш(-динитросоединении, 0-интронитритов и
g-нитратов, а затем каталнзуемое основаниями отщепление при-
водит к нитроалкену [98].
IN U2
HNO3 I
Аг2С=СНАг--------> Аг2С=САг (73)
К2о4, О2
20 °C
El3N
------------у.
комн. темп.
(74)
X = NO2, ONO или ONOs
Подобным способом было проведено нитрование холестерилацета-
та. Можно модифицировать стадию присоединения и получить
вщрдипнтросоедннения с выходом ~40%, тогда как обычно
продуктами этой реакции являются p-нптроиптраты и -нитраты и
продукты их гидролиза. Присоединение происходит с участием
свободных радикалов, и оно нестереоспецифично. В присутствии
кода образуются р-пптроалкилнодиды, которые при дегидрогало-
генировании действием ацетата натрия дают иптроалкены. N2OS
присоединяется к алкенам в днхлорметане при —25°C с образо-
ванием р-нптропитратов. Взаимодействие алкенов с тетрафторбо-
к Тпп И”ТРОНИЯ в безводном ацетонитриле при —15 °C приводит
ацетаХ7а(уравТн11е75)°[9И93]е К°Т°РЫХ полУча,от производные
R2C=CHR + NO$ "BF, —-|120 > R2C(NHCOCH3)CH(NO2)R (75)
ре (15—50%)
—20~— ЗО^начипае01"0'' К||СЛ0Т0Й в уксусном ангидриде при
между собой и обт J,nLn0CJle Т0Г0, как Регенты прореагируют
Р< - тся а|1ет||лнитрат. Обычно главным про-
авчяется p-пнтроацетокспсоединение няп«я«
1'’<'ами п р-ннтроалкенамп. В некоторых случаях^аб Р'нитро’
соединение- Это привело к предположению что'Т™^
па нт через циклическое переходное состояние flOOal РАаК11НЯ
< J “данные, например образование 3- и 4-нитр оциХ°Т*°
Жг'к1огексена, заставляют предположить, что иноП?7
Р°ЛИ00ТГТ ИГРаТЬ ПР°МеЖуТ0ЧИ0 обРазуЮЩийся УкТрТе’-
РВЫЙИОН [1UUOJ.
Под действием как ацетнлнптрата, так и нитрилхлорида слож
J эфиры енолов превращаются в нитрокетоны (уравнения76 77\
' дашнческне винилокснсилаиы в мягких условиях реагируют
, геграфторборатом нитрония [)01] с образованием а-нитрокето-
нов (уравнение 78). Ннтрнлхлорнд присоединяется также к терми-
иачьным алкенам по свободнорадикальному механизму, давая
|.нптро-2-хлоралкапы. Взаимодействие алкенов с нитритом сере-
бра и подом приводит к Р-ннтроподалканам, и в этом случае
реакция имеет свободнорадикальный характер. Эта реакция
позволяет избирательно получать нитроалкены и нитроалканы
в мягких условиях; она нашла применение в ряду стероидных
а.чкеиов [102].
Ме
I
МеСН=С—ОАс
OSiMe3
Me—С=СН2 + NO? "BF4 —>- MeCOCH2NOs (78)
(45%)
Замещение
Реакции у ароматического атома углерода [103].
атома водорода в ароматическом ядре на нитрогруппу долгое
время служило объектом внимания теоретиков, так как, во-пер-
вых, введение нптрогруппы значительно дезактивирует ядро
к Дальнейшему замещению и, во-вторых, оказ; юсь, что проведе-
ние реакции в резко различающихся условиях дает очень близкие
соотношения изомерных нптросоедпнений. Эта реакция инее
гР°мадное значение для получения ароматических нптросоедине-
Нчй. Данные о роли иона нитрония NOn" как эффективного'
Фпла при самых разных условиях и о влиянии заместите ‘ х
^ьнеишего замещения в замещенных ароматических соединения
1»огокраТ110 обсуждались [103а]. Реакция протекает по схеда
о,. ‘ В|1С||ие 79) с участием интермедиата )> иланда ( вт0.
Рой стаю°ЩеЙ являегся перваЯ СТаД11Я’ \7,“ы?'пЬрКо°страиственных
стаДЧи не снижена из-за очень сильных пр 1
Вз<1|1МоДейсТвнй. В случае соединений с достаточно высокой р
403
' ,, i/i z к()постьо1и)еделяю1цсч"| стадией может a,
Ц1'0'",0'1Саиш1ая°л''Ф'1>-'3|,й “(,110П ",,ТРОИ"Я " М°ЛеКУл сУбсТр^
также
та [104].
/тту.Н в
АгН+^^Д^Х^ ^ВН + ArNOj (?9)
(31)
Иля нитрования можно использовать азотную кислоту или ее
пастворь/ в водных кислотах (особое место здесь зан11Маег
сепная кислота), когда пои нитрония образуется в результате
оавновесной реакции (уравнение 80). Концентрацию кислоты
и температуру можно изменять в широких пределах —от водной
Н+ 4. HNO3 Ч=* FfeNOs НгО + NO,
(80)
НЫОз при о °C до дымящей азотной кислоты в олеуме при на-
гревании — в зависимости от реакционной способности субстрата.
На практике скорость нитрования максимальна в 90%-ной серной
киелоте прн низкой концентрации азотной кислоты; если же ско-
рость реакции все еще низка, используют олеум, который спо-
собствует дегидратации азотной кислоты. Существенным ослож-
няющим фактором является очень низкая растворимость боль-
шинства субстратов в этой среде, поэтому результаты кинетических
измерений, проведенных в гомогенных условиях, ие всегда можно
использовать для препаративных синтезов. С дополнительной
сложностью приходится сталкиваться прн работе с субстратами,
обладающими основными свойствами, так как в реакцию могут
вступать их протонированные или непротоннроваииые формы
(последние могут присутствовать в очень малых количествах).
Анилин, например, нитруется как сопряженная кислота, а 4-нпт-
ро-2-хлорап11лин реагирует как свободное основание. Вопреки
распространенному мнению, соотношение изомерных продуктов
реакции с одним в тем же электрофилом может меняться с из-
менением^ кислотности даже в случае субстратов, способность
взапмоденствпя которых со средой мала. Это было показано на
примере толуола и о-ксилола с поправкой на «исо-замещенпе
(см. ниже) [105].
Нитрование азотной кислотой в инертных органических
(-уксус,)ая кислота, сульфолан, нитрометан и
ннтрппс т рпСтьп| УГЛСР°Д) представляет большой теоретический
скопогт! иЛпЛ315 СК0Р0СТЬ Реакции вполне может сравниться со
использоваппеРэтойа"11Я "°"а Н,1ТРОН||Я- Однако препаративное
ционноспособшши Реакцпи ограничено лишь относительно реак-
ХОТ» ЧТ
кислоту. Азотная «пепп реагента можно добавить серную
образуя ацетвлннтппт ?Та РеагиРУет с уксусным ангидридом,
Считалось, что в этой (®раствоРе) мощный нитрующий агент,
среде присутствует некий, отличный от
•404
„„трония, Эффективный электрофил, с нал„ц, о
а-ш высокое соотношение орто- „ пара-нзХ'? К0Т0р0Г0
спя31'1® йх субстратов. Для них был пРедлоТен'ХбРЫйВ1УС:У1аС
яеК"замеШе"ИЯ с первоначальной атакой на заместптеч, ИЗМ
Ле учитывали, что для такого механизма' замХи’ °Днако
”Стпебуется участия какого-то иного электроф|1”аЩ“Иняр вовсе
Яе тр„,я По данным работы (226), ион ннтпониа ’-а Не иона
Дется’ активным агентом прн нитровании ацетХ^атоГи
’ «пайпен мере в случае некоторых соединений „ ’
^шлида [106], анизола [107] „ бнфеиила Д’, ^Ример
одиыости прибегать к осооому механизму зависимого от среды
„^.замещения. Результаты в данном конкретном случае оное
"няются сочетанием каталитического эффекта азотистой кисло
ты [20, 21], взаимодействия со средой (включая образование
водородных связен) 11 ио, 107] н, возможно, для бифенила про-
теканием предварительной реакции по замещающей группе. Для
нитрования реакционноспособных соединений можно применять
ацетплнитрат, полученный нз ацетилхлорпда и нитрата серебра
в инертном растворителе. Считается, что нитрование концентри-
рованных растворов ароматических соединений в четыреххлорис-
том углероде действием N2O5 прн низкой температуре происходит
в результате обычной, хотя и автокаталитической, реакции с ио-
ном нитрония. Было найдено, что при низкой концентрации суб-
страта протекает бимолекулярная реакция с участием \;О5 и
субстрата [109], хотя имеются серьезные доводы в пользу
влияния растворителя на этот процесс. Можно полагать, что
беизоплнитрат выступает в роли источника N2O5, хотя в присут-
ствии пероксидов реакция может происходить и по свободно-
радикальному механизму [ПО]. Использование различных солен
и эфиров азотной кислоты в различных растворителях также
приводило к необычному соотношению изомеров; применение
этих реагентов для некоторых соединений традиционно связывалось
с высоким орто: пара-соотношением. Метод, позволяющий полу-
чить нитротолуолы из толуола с соотношением орто- и пара-изо-
меров, равным 0,68 («нормальное» значение составляет ~ 1,6),
основан на использовании азотной кислоты и безводной суль-
фатированной ионообменной смолы на основе полистирола [Ill],
Простой метод нитрования, осуществляемып в мягких условиях
в отсутствие кислот, состоит в использовании устойчивых солеи
""трония; механизм этой реакции служил предметом многочис-
ленных теоретических споров [104, 112]. Тетрафтороорат нитро-
"ия, например, нитрует в сульфолане простые ароматнч
1'Ле|!0Дороды, в том числе и цианиды, с хорошим выходом да
ычное соотношение изомерных продуктов. Можно псп . ,
кже ряд доступных реагентов в сочетании с к, Р •
еакцнн Фриделя — Крафтса [1126, 113]. „„тпованнем
ПомнМО участия в реакциях делротонпроваппя яТаКовать
ОТОРЬ1Х речь шла выше, электрофил способен т '
405
цикла, несущий заместитель (шгсо-атака) г,,.,
этом углерода1 а дмжен быть соответствующий Щ ;
однако этот ато. у В||ен11ю с остальными. Образуют,,?
зом актпВ"Р°ГЛт Уиланда (схема 81) может далее либо в
“^^нуклеофилом, лааая вначале ПРОДУКТ "Р^еди^
ровать с вук w „егруппПровываться с образованием ц— 'ия
(nry0TbH«po оедннения W б), либо вступать в пеа.„
заместителем в цикле или терять заместитель
атом углерода должен
Уиланда («ема _80 ^ожм далее_ либо ре^8
1 нормаль
пать в реакцию с
(путь
Дру.
в).
Н Nu
(81)
При проведении реакции в уксусном ангидриде такой unco-
интермедиат удается зафиксировать по образованию продукта
присоединения ацетат-ноиа, обычно в пара-положепие (уравне-
ние 82); известно большое число разнообразных аддуктов такого
рода. Возможная потеря и/гсо-заместптеля давно уже рассматри-
валась [115] как аномалия прн нитровании ароматических сое-
динений; иногда заместитель может вновь войти в цикл, в другое
положение.
Me
АсО
(цис-и транс-)
(82)
хота3иТИСТаЯ к11СЛ0Та’ которая может случайно присутствовать,
сильно нитрование многих соединений, в то же время
единений таких НИ1Р0Ваи11е некоторых реакционноспособных со-
и другие пол ;фс,,олы И 11Х п1)осгыс эфиры [20], нафталины
полиалкплбензоты 'ск^"6 углеводоР°ЛЬ|> бифенил [21] и Даже
случаях определяются Р°СТ" и,|тРовап11я ионом нитрония в этих
. некоторых вд этих Я СКОРОСТ„ЯМИ Диффузии. Легкое окисление
способствовать этомуСпеДЦИеИИ^ азотиой кислотой также может
может включать ииЛ°ЦСССу’ Уакое катализируемое нитрование
40s Р03иР°ванпе ароматических соединений
пслеДУ1°и11,м окислением образующихся
с П°пЧЛ например, действием N2O4 'll 16) Почт Р°ЗОСОадинений,
'3 реакции образуются нитросоединеиия с иео’бычнымЗУЛЬТаТе
Р1ием изомеров; скорость реакции также отчичае?гя пт С°°ТН0’
S катализ может осуществляться и другим путем как°иЯВДае’
И°п в случае иона М,М-дпметпланилииия |29я1 в ’ кЭК’ напРи’
протонированных аминов или четвертичных амТониХ
солей представляет собой особый случай, его следует рассХри
ать отдельно. И в этой реакции возможно ипсо-замещенне ми
нером чему служит питрозодеподирование - окисление 2 4-ди-
10Д-3,5-диметнланпзола [117]. В настоящее время ясно’ что
нитрование указанных выше типов соединений по опубликован
ИЫМ препаративным методикам включает, в той или иной степе-
ни, катализируемые азотистой кислотой превращения.
Для нитрования аминов и фенолов использовали тетранитро-
метан в щелочной среде [118а]; родственное соединение, три-
яитрофторметан, превращает толуол в а,2,4,6-тетранитро-
толуол [1186].
(2) Реакции с нитритами металлов [88s]
Реакция нитритов лития, натрия или калия с алкилбромида-
ми или -иодидами (но не с алкнлхлоридами) в диметилформ-
амиде или диметилсульфоксиде приводит обычно к смеси алкил-
нптритов п нитроалканов; основной компонент смеси — нитро-
алкаиы (~60%). Для того чтобы предотвратить последующее
нитрозирование образовавшегося нптроалкана, необходимо ис-
пользовать днполяриып апротонный растворитель; обычно до-
бавляют также мочевину и агент, разрушающий алкилнитрит.
Из трет-бутил- и цпклогекснлгалогенндов, однако, образуются
продукты отщепления; это не позволяет использовать указанную
реакцию для синтеза питростерондов. Особенно успешные резуль-
таты эта реакция дает в случае эфиров а-галогенкарбоновых
кислот (даже а-хлоркарбоновых кислот), выходы эфиров а-ннтро-
кислот составляют 70—90% (уравнение 83). В случае же соеди-
нений типа RCH2X, где R —очень сильная электроноакцепторная
группа, реакция не приводит к желаемому результату, что, ве-
роятно, связано с питрозироваиием продукта. При взаимодей-
ствии хлорацетата калия с водным раствором нитрита натрия
образуется нитрометан, что говорит о чрезвычайной легкости
самопроизвольного декарбоксилирования образующенся ни р
Уксусной кислоты; в случае же высших гомологов выходы' *Р
низки (119]. Нитрит серебра реагирует с алкилгал0Г“'^‘
(Реакция В. Мейера) при 25°C и дает более высоки: вых .Д^
первичных питросоедпнеиий (~80%); этот метод о выше
сообразно использовать применительно к упохп < ' прнг0(
RCH,xP Д« прочих
нитрит натрия (см. также разд. 7,o.l(l)J-
407
реакции нитрита серебра с эфирами а-галогспо|<целот
использовать а-нодкислоты (уравнение 84). С iiiitdi т 6xoAnM
лов реагируют и илкилнитраты, давая интроалкаиы [12о/<П Мета.1°
Вг NO2
I NaNOj I
MftCCOjEf -------* MejCCOjEt
« .i
(СНгСОгЕ!-f-AgNO2 ---* OzNCH2CO2Et
(77%)
(83)
(84)
(3) Превращение оксимов в алифатические
нитросоединении
Окисление оксимов в иитрососдинення
действием перокенкнелоты, образующейся
ангидрида и пероксида водорода. Реакцию
можно осуществить
из трифторуксусного
приводят в кипящем
ацетонитриле в' присутствии буферного раствора и небольшого
количества мочевины (уравнение 85).
мочевины (уравнение 85).
С1-зСО.-ОП
MeCN
N02
(85)
(62%)
что первоначальным продуктом реакции является
ннтросоединення, которая затем таутомеризуется.
Полагают,
аци-фор.ма
Для получения а-пптрокарбоиильных соединений по этой схеме
нет необходимости в использовании буферного раствора, раство-
рителем может быть трнфторуксусная кислота пли хлороформ.
По-вндимому, на эти реакции окисления влияют стернческие фак-
торы, в случае пространственно затрудненных соединений предпоч-
тительиее проводить бромирование N-бромсукцнпимидом (NEC)
on Р0мкитР03опРо||ЗВОДн°то, затем окисление до бромпитросо-
Нет Honfivnn восстановнтельиое дебромирование (уравнение 86).
в слччяр ' "МОстп выделять промежуточные соединения, однако
не дает еповпптпИМ°В И аР0Мат|,ческпх кетокенмов эта реакция
[•211 кототя ,рИтельнЬ1Х результатов. Аналогичная методика
пенни’ (4О_8ро/ \ вспеЧ||вает более высокие выходы ннтросоедп-
рование (хлоп в пЛСаМЫХ ра31|ЫХ кетокенмов, включает хлори-
нктрозоеоеднневия обРаз> иощегося хлор-
ине в щелочной спепе М И’ нако,,еЧ> каталитическое гидрпрова-
способом осуществляюДЛЯ Удале|||,я атома хлора. Аналогичным
сима до гел!-н||Т(>озош,т„„С111'Тез нитростероидов: нитрование ок-
ге-'|-Д||ннтрососд|1пСН||!1 I, СОед,1|1е1>ня, последующее окисление до
лсиие до MOHoiuiTiiocTonnn ЗЭТ?М стеРеосГ!счифпческос восстанов-
ив 1 "да (Уравнение 87) [122]. геж-Дниптро-
,„Р1ШЯ можно получить также из оксим™
<оОваиием действием N2O<. °ксимов 'Фи
С1,1,1 NBC /NO
r2c=noh 5_10„с> 1ЪС(
\1г
окислении
HNO3
Ч2О2 (30%.HL16)
/N°! NaBII,
> R2C -> r2cHNq
xBr
(Ю-48%)
Н№з /N0 И2О2 pt
RjO=NOH * ЪС\ыО2 HN°3* R2C(N°2>2 R2CHN'O2
(86)
(87)
(4) Превращение первичных аминов в нитросоединения*
Эффективным окислителем, превращающим первичные амины
с третичным алкильным радикалом в нитросоедннения (вы-
ход 70—83%), служит раствор перманганата калия в водном
ацетоне при контролируемом значении pH. Амины, содержащие
первичную или вторичную алкильную группу, удовлетворительно
окисляются перуксусной (но не пероксптрнфторуксусной) кисло-
той, и, например, циклогекснлампн можно окислить до ннтро-
цпклогексаиа с выходом 70% [123]. Пербеизопная и льхлор-
перокснбензойпая кислоты также применяются для этой цели,
особенно для окисления стероидных аминов. В некоторых слу-
чаях прн такого рода окислении образуются бнс(ннтрозосоедине-
вия). Озонолиз первичных алкиламинов в безводных условиях
также приводит к соответствующим ннтросоедпненням [124].
Перокситрпфторуксусная кислота окисляет замещенные ани-
лины в производные нитробензола с почти количественным
выходом [125]; при этом удается избежать образования азокси-
бензолов, появляющихся при использовании менее кислых реаген-
тов. По-видпмому, стерические препятствия, создаваемые объ-
емистыми орто-заместителями, не влияют па реакцию. Альтер-
нативный способ получения нитроареиов состоит в замене
диазониевон группы диазотированного первичного ароматического
амина на ннтрогруппу. Это замещение проходит только в ней-
тральном и щелочном растворе [126], необходимо очень тщатель-
но соблюдать условия реакции, и тем не менее этот метод дает
количественные выходы о- и п-днпитробензолов.
(5) Прочие методы
Имеется большое число различных «е\оД1,к>1кот°Р“Д.М°*ны
'юиользовать для синтеза пптроалканов |88а, б]. i 1 - (
1,(|жцо получить с удовлетворительными выходами (
' См. также разд. 7.1.1 (1) п 7.1.3.(1).
409
, ,„.РОТНОМ растворителе при 270-300 С ацплц||Тра.
нагревании в "" Р 0Чередь готовят т situ из галогенангнДри.
тов, которые в № к11слэт разл „„ способами; реаКЩ1я
дов нлн анг Р механизму 112/Ь Терминальные нитро-
ходит но радик^' сУВ|>|ходамн 60-9о% при восстановлен,
алканы образуют g ,др11Д0М натрия в этаноле [128];
р.питроалкп.iiiTp 1|а1ОТ 113 алкенов-1, оксидов азота и
ннгроалкилпптра ||Ы образующиеся из альдегидов [см.
кислорода, а-п ^жн0 легК0 „ эффективно восстановить [129]
разд. UH»)’ „ нптроалканов действием борогндрнда натрйя
. 2“п”.«»"«тр»ле «Р» 0-S-C и рНЗ-6 |ур’ав.
w В оптимальных условиях реакции удается в значи-
НеН"цпй8 степени подавить процесс образования вторичного
Т®ЛЛ ° тя (32) Этот метод дает возможность удлинять углерод.
X щпь в-Ннтроацетаты можно восстановить непосредственно
^соответствующие нптроалканы действием борогндрнда натрия
в пимеппсульфоксиде при комнатной температуре; эту реакцию
можно использовать как одну из стадии в методе удлинения це-
пи проводимом без выделения продуктов отдельных стадии (130].
При электролизе трчалкилборанов RsB в растворе нитрометана
в присутствии галогенида тетраалкиламмопия гладко нс хоро-
шими выходами образуются высшие нптроалканы RCH2NO2 (131].
Некоторые ароматические
[132] реакцией пирилиевых солей с нитрометаном в щелочной
среде (уравнение 89).
NaBH4
pH 3-€
нитрососдпиения можно получить
R'C(NO2)CH2R2
R2CH2CHR’NO2 + R2CHCHR’NO2
(32)
R
(88)
R!CH=CR‘NO2
R
(89)
О'
МеХО2
грет-ВиОК, rper-BuOIl * L J1
NO2
(6) Алифатические нитросоединения
с другими функциональными центрами
Перво^ачаХлвпп М0Я1И0 получить [133] по схеме, включающей
последующее п РИСОед|1нение NO2X к подходящему алкену и
лиз ₽ аХксшжтпоае'1Ие НХ 7.2.1 fl)]. Мягкий пиро-
этот метод с успехом б^Н0В (24°‘~600°с) Дает а-иптроалкены,
лена. Р-Нитроспнпт. бЫЛ нспользован для получения ннтроэти-
алканов с атьлегипа’ гладко образующиеся при реакции интро-
не легко превратить в кстоиам" I™- РДЗД- 7.2.3(10)], мож-
ВДнпя с фталевым ||тРоалкены дегидратацией путем иагре-
4,0 1г1|Дрчдом или превращением в 0-цитро-
с последующим отщеплением уксусНпй
3<“ е90). УКСусН0Й кислоты
|VPaB ЛсС1 кнг
„,С1ЗДКО1 М(
^аЛ°ма’трпвает превращение гидроксилиой\(метаХлЬ°Г80%)
лр1Ху лучшую уходящую группу, и последующее ееФот,ЙЛ0-
If под действием триэтиламина (134]. Ц-Нитроспирты пХ
"„не из бензальдегида, часто подвергаются самопроизвоАьнХ
з^пленвю. Получение галогеннитросоединений рассмотрено
j обзоре Н35] -
(7) Алифатические ди- и полинитросоединения
Характерные особенности реакций, приводящих к образованию
этнх соединений, описаны достаточно подробно |136]. Обычно
,!Я m получения требуются лишь незначительные модификации
тех реакций, которые обсуждены выше [см. разд. 7.2.Ц1) —
72.1(5)]. гел<-Динитросоединения удобнее всего получать из
гел-ннтронитрозосоединений, которые образуются при нитровании
оксимов; для окисления иитрозогруппы можно использовать
окислители (уравнение 91). гельДинитросоединения образуются
|)37] также при реакции первичных или вторичных нитроалканов
с нитритом серебра и нитритами металлов в щелочной или ней-
тральной среде (уравнение 92), однако в том случае, когда R'
или R2 — электроноакцепторная группа, реакция не идет. Методы
синтеза трннитрометильных соединений рассмотрены в обзо-
ре [138].
NOH NO
II HMDs I
Et—С—CO2Et ------->- Et—С—CO2Et
I
NO2
(91)
R\
'C=no; + 2Ag+ + no;
(92)
NO.
Et-C-CO.E:
I
\Os
7.2.2, СВОЙСТВА НИТРОСОЕДИНЕНИЯ [
(1) Алифатические нитросоеди rSO2 —Уме‘
Общие характеристики. Простые ”нт^нь ограниченно рас^
ренно токсичные бесцветные жидк ’ при 20 СС). Он1’ .,еть
коримые в воде (10% для можно
мт высоким,, температурами Кш ольнЫмп момеН^ мйогими
^ атмосферном давлении) ’’ смеШиваются « ряте-
W<02 т. кип. 102 °C, и = 3,5Д. Они важИЫМн расп’Р^
Руническими растворителями и я ' ,етан обладав
л*м’ Для некоторых полимеров. Нитро.' 41!
cKoii постоянной (37) [139] н может применяться
проведения различных ионных реа Ка
быть представлена в виде резонансного
Н1элек1рн'|ес..-
растворитель для
Ннтрогруппа может
рида (33).
' + /°‘
_— N
(33)
нандены следующие значения длин св
нм (N—О); угол О—N—О равен 127°; Сц“'
! вокруг
имеется интенсивная
15 ООО), однако в ближ-
Для нитрометана
147 пм (С—N), 122 - - . ... , ,
тема С—NO» — плоская с низким барьером вращения сок
связи С—N. Основность иитросоедииеиип очень низка (р/(,
«—11); они сполна протоиируются лишь в таких силыюкислых
средах, как олеум. В еще более кислых условиях наблюдалось
растепление протонированных нитроалканов с образованием
карбенневых ионов [140]. С донорами протона ннтрогруппа
образует лишь слабую водородную связь.
В ” электронном спектре нитроалканов
полоса п->л*-перехода при '—210 им (е «
ней ультрафиолетовой области спектра (270—280 нм) поглощение,
обусловленное п->л*-переходом, очень слабое (е « 20) [141|.
В ИК-спектре присутствуют две интенсивные полосы, соответ-
ствующие симметричным и антисимметричным валентным ко-
лебаниям связей N—О; в большинстве случаев частоты, соот-
ветствующие этим полосам, являются характеристичными для
первичных, вторичных и третичных алкилиитросоединеипй: для
первичных алкилнптросоедмиеиий 1382 + 6 и 1554 + 6 см-', для
вторичных 1370 + 4 и 1550+ 3 см-1 и для третичных 1349 + 5
и 1539 ± 5 см-1. На положении низкочастотной полосы
сильнее, чем какие-либо другие факторы, сказывается сопряже-
ние; известны примеры поглощения вне указанных пределов.
В спектрах ПМР сигнал протона группировки CHNO2 постоянно
находится при 6 4,38 — 4,28 млн-'. В масс-спектрах нитросоеди-
нении (за исключением MeNO2) пик молекулярного иона или
очень невелик, или отсутствует вообще. Главными процессами
фрагментации являются отщепление NO2 и последующий распад
ак1Ь110Г° Радикала> или отщепление двух атомов кислорода
0Tnienj>3puBiiao"i4Mr>^)Pai'MeH'ra’ экв11валеитного нитрилу (142а,б|-
алкена — «я, г О’ 11 последующая фрагментация возникающего
тйчных иитпйр°ЛеС суш.сствен11ая особенность масс-спектров тре-
точное обпзаопл^111!СПИИ i‘42al’ для которых возможно иромежг-
Таутомевия'и"е шест1,1|лениой циклической структуры [142в]-
ричные н11тпоЛ/Ч“'НИТросоединения [143]. Первичные и вто-
с оц»-11Итросоепнп11Ы находятся в таутомерном равновесп
такж4 ЙХХдХ?" (34) (УР*"е 93), называемым'
Последнее название 10 !1ями 11Л”> чаще, нитроновыми кислота-
название особенно применимо для обозначения >>
таУ
на’
ляет
равно
„ГОДНЫХ, СООТВеТСТВуЮЩИХ СЛОЖНЫХ Эфиров Делпп
^"Сросоединения до так называемого нит понят Рирова‘
* Нный процесс, для которого характерен общий”3 (35)^
'С,,3 Между кислотностью нитроалкана н скопп, ОСНОвной
< соблюдается соотношение линейности свободных10 И°Н‘!за’
111 „бное соотношение существует между силой С энергии:
'Сетью ионизации. Из-за низкой скорости иОНИЗ°яТЗНИЯ и
5ны называют псевдокислотами Из двух таутомеЦИоИ0 ИИТР“'
Сомер гораздо более устойчив (см. уравнение 93) пр,“ко °’
нон температуре соотношение ICH2=NO2H)/1СН3.\0,1 сосТав
.рЮ-’; для 2-нитроиропана соответствхющее значХ
3-Ю'3. Превращение аци-нитросоединеиия в нитросоед,
Lне обычно протекает достаточно медленно, так что концен-
трацию аци-формы удается определить титрованием бромом.
Таутомеризация каталкзуется и кислотами, аналогично снолиза-
mi» альдегидов и кетонов. Реакции ннтросоединеннй, катализуе-
мЫе кислотой (см. разд. 7.2.3), могут проходить через промежу-
точное образование аци-формы нитросоединення.
R\ _ н*
СН\О2 з=ь /С—N.
R2.
R2-
(35)
/О'
(93)
RlZ XOII
(34)
Бесцветные нитронаты натрия и калия — наиболее часто
получаемые соли; они образуются при действии водного раствора
щелочи или спиртового раствора алкоголята на нитроалкан.
Серебряные соля обычно получают из этих солей щелочных
металлов путем метатезиса. Соли нитроновых кислот взрыво-
опасны в отличие от самих мононнтросоединеннй. Кинетически
контролируемый продукт протонирования ннтронат-иона — <щи-
нитросоединение- судя по спектральным данным, можно преДп°'
пожить, что отрицательный заряд в ннтронат-ионе cocPMOTO4eJ
главным образом, на кислороде. Протонирование ПР^С ’ А'
собой наиболее удобный и широко используемый мет • -
!ШЯ а,<«-нитросоедииений. Типичная методика в клюяолРкнсл0.
’’птроа.ткана водной щелочью, а затем избытком • ____________
ты пр,, о—5 °с Другой путь получения
присоединение по Михаэлю карбанионов
например, уравнение 94). Кроме того,
начальные продукты окисления оксимов
Ph
I i:t2o
PhMgBr + PhCH=C—NOs-------------
оци-нитросоединений
к я-ннтроалкенам (см.,
оЧи-нитросоедннення-
[см. разд. /.2.1(0))-
ph о-
РЮСнСС
\эн
(94)
4,3
„ЧШ1РШ1Я лучше растворимы в воде, более пи„
Sfes^5“-“*5‘5
МпХе в том числе жирноароматические соединения, устой^
ДР риение более длительного периода. Например, аЧи-нптрОцИкл
додекан (т. пл. 84—86 °C) устойчив в течение нескольких Дней
С хлоридом железа(III) они дают красное окрашивание. Хо"я
существует возможность чис-транс-изомерии ач+ннтросоединея
ннй индивидуальные изомеры не были обнаружены [144].
уф.Спектры ачп-нитросоедпненни, нх солеи и эфиров оВДь
сходны между собой; они характеризуются сильным поглощением
при 220—230 нм (л->л>'-переход, в» 10 000) (для анионов на
~10 нм выше); сопряжение смещает полосу поглощения в об-
ласть больших длин волн. В ИК-спектре поглощение С=М-связп
лежит при 1610—1680 см-1 (как и для оксимов). Для нитро-
натов натрия (но не для эфиров) наблюдается смещение в об-
ласть меньших частот (1587—1605 см-1). В спектрах аци-ннтро-
соединений появляется широкая полоса валентных колебаний
О—Н при 2500 — 3000 см-1, характерная для ассоциированных
слабых кислот. На основании ПМР-спектров нптропат-нонов
[145] их можно идентифицировать в щелочных растворах нитро-
соединений (для CH2NC>2Na+ б 5,83 млн-1, для CH3CHNOi Na1
б 1,73 и 5,14 млн-1). В эфирах нитроновых кислот сигнал вп-
нильного протона в соединениях типа RCH=NO2Et находится при
6 ~ 7,0, если R = арил, и при ~6,0, если R = алкил; в каждом
из этих случаев в спектре смеси цис- и транс-изомеров наблю-
дается два пика.
При восстановлении третичных иптроалканов металлическим
натрием в инертном растворителе [ср. разд. 7.2.2(2)] образуются
анион-радикалы, распадающиеся на третичные алкильные ради-
калы и нитрит-ион. Подобное восстановление 2-метил-2-ннтропро-
пана приводит к соли трет-BuNO; Na+, при добавлении к которой
воды образуется ДП-трет-бутилпнтроксил, rper-Bu2NO- [146].
замещенные нитросоединеиия. Известны ди-, три- и тетраипт-
«нмГДНЬ1’ ^еРВЫ11 113 Н||х нестоек, а остальные — высококнпяшие
давлении’ гг7мг>Ы? можно перегонять, лучше при пониженном
воде Z Т> К/п' 126°CJ- 01111 менее растворимы в
ОДи-фопмы в пР°МеТаН' 9 Увеличеиием числа ннтрогрупп доля
тана),РувелпчивзетгаС1101! СМеС" В03Растает (4% ДЛЯ динитроме-
[для CH2(NO У Пй-Я также сила нптросоедпнений как кисл
иие числа woornvn’ РаШ1° 3’6 11 ^я CH(NO,)3]. Увеличе-
разделению хаоактепи соедш,еи11Ях этого типа ведет к большему
1332 ±5 и 1575 +19 ,полос в ИК-спектрах (гел-дшштр0?
±М- В иЙпектпа" '’М-тринитро-: 1302 + 5 и 1600 ±
Рах соответствующих анионов отсутствуй
„_ые полосы, которые можно было бы <««
»11<РНИЯ связи C=N или NO2-rpynn, что ука-нТ™' За «et
лоГЛ°^ делокализацию заряда; в УФ-спектпе ян Т На ЗНачи‘
^соединен111"' наблюдается характерное поглощение08 ге^-
e”nTPu°M Данные о физических свойствах этих сое ™ "J” 320~
380 Нв'обзоре [136]. Тетранитрометан имеет низкую ли^ приве-
де о-ю постоянную (~2), с алкенами и ароматийр ТРИ'
Мнениями он образует молекулярные комплексы. Все n0T
росоединемпя чрезвычайно взрывоопасны, „ с ними надо об'
щаться очень осторожно. ад0 00
Р а-Ннтроалкены обладают лакрнматорным действием- это
светло-желтые или бесцветные жидкости (для нитроэтилена т кип
Ч8°С), легко, а иногда и бурно полимеризующихся в присутствии
„снований с образованием короткоцепных (20-30 звеньев поли-
меров. В их УФ-спектрах поглощение прн 220-250 нм, обуслов-
ленное л->л*-переходами, маскирует поглощение, вызванное
n-^-переходами. В ИК-спектрах частоты основных полос
лежат в более низкой области вследствие сопряжения (1353 ±6 и
1424±4 см-1 для моноалкилнитроэтиленов; 1346+9 и 1515±4 см+
для ди- и триалкнлпроизводных). В спектре ПМР нитроэтилена
[147] присутствуют сигналы трех протонов, химические сдвиги ко-
торых приведены в формуле (36).
ОА\ /Н (6, 55)
(36)
(7, 12) W \Н (5, 87)
Галогеннитроалканы также представляют собой высококипя-
шие жидкости; трнхлорннтрометан (хлорпикрин) — бесцветная
жидкость, т. кип. 112 °C, сильный лакриматор.
а-Ннтрокетоны, вероятно, могут существовать в трех тауто-
мерных формах (уравнение 95). По данным спектров ПМР,
ациклические а-иитрокетоны в нейтральной среде обычно
существуют в кето-форме (37), тогда как циклические соедине-
ния, например 2-нитроциклогексанон, в мягких условиях, особен-
н° в апротонных растворителях, представляют собоп смеси тау-
томера (37) и одного из енольных производных [9об, о].
Структурные и спектральные особенности этих соединении опн-
са«ы в обзоре [149]. 0-
R’COCHR2NO2 ч=+ R'C=CR2NO2 ч=* R'COCRUl^ <95)
ОН
(37)
а-Нитропроизводиые первичных алифатических^ аминов,
Р вило, неустойчивы и разлагаются при комнатнон • Р
м’»110-2-иитроэтан, например, нс удается nePerH3J’3. фОрмуЛы
р.п^яоиитролы - нейтральные соединения ш,тРмосоедп-
;£(№)№,; они проявляют характерпучо для ивтр^с(1)]>
111 склонность к обратимой димеризации [см. р
1 415
как
йпервломитрол) бесцветное твердое вешил
Бпс(1-мет"лэтил »с Пссвдопнтролы можно п0„ТВ° t
едким заг,а- п01щ|10 взаимодействием N2O4 с кето|ХУ'"1Т|‘
по PeanKU’c"oB№ реакции опп мОГУт ОК1,СЛЯТЬСЯ до гед-д *аЧ
однако в УСЛпГРВ2О„итролы можно также получить нитрозпп ₽0'
4rSZ-OB R‘R2CHNO2. Применен,,/03^,
нием ВТ^ ч чае альдоксимов осложняется таутомеризац,1еРй
П°Хо я пе вичиого псевдопитрола RCH (NO) NO2 в ннт
ча10щсг°ся Р )=NOH, которая образуется также при нитпп
,<,1СЛ0Т „и первичного питроалкана. АцетонитроловаяР к”с р
Е Ж.»»» »‘с <раз“-)» «д«»«о S?
гяатся при храпении. Наличие двойной связи в нитроловой Мс
тоте обусловливает существование^ геометрических изомеров
которые образуют два типа солеи. Геометрические нзоМеры
некоторых ароматических нитроловых кислот удалось идеитифп.
цировать отдельно от соответствующих псевдоиптролов и щ
димеров [150].
(2) Ароматические нитросоединения
Физические и спектроскопические свойства. Ароматические
нитросоединеиия — бесцветные пли светло-желтые высококппящпе
жидкости (для нитробензола т. кип. 209 °C) или ипзкОплавкие
твердые вещества, часто перегоняющиеся с паром и обладающие
характерным запахом. Они плохо растворимы в воде и многие
из них чрезвычайно токсичны. Нитробензол (диэлектрическая
постоянная 35) используют как полярный растворитель, в част-
ности, для проведения реакции Фриделя — Крафтса. Арома-
тические полинптросоединения — твердые вещества, они часто
взрываются при нагревании. Тринитротолуол — хорошо извест-
ное взрывчатое вещество (тол, тротил); лабораторные образцы
такого рода соединений обычно хранят под водой.
Молекула нитробензола имеет плоскую форму со слегка
удлиненным бензольным кольцом (длины связей С-2—С-3 и
С-5—С-6 составляют 146 пи), но геометрия ннтрогруппы в нитро-
ензоле почти не отличается от таковой в нитрометане [151]. Ни-
трогруппа в нитромезитилеие (1,3,5-триметпл-2-нитробсизол) повер-
нута на 66 относительно плоскости кольца которое н само значи-
тельно искажено [151].
с\тгт|ХТСПеКТрах аРоматическпх нитросоединенпй обычно при-
В оХи 250-ЗПВП1аЯ (е~ 600°) '""рокая полоса поглощения
женне и интопр ЗМ М> ОиУСловлеипая л->п*-иереходом. Поло-
от растворите™'В"0СТЬ ЭТ0Й полосы поглощения сильно завися:
кольце. Интенсивное °Т приРоды заместителей в ароматпческо
Факторы заставляют™ ПОЛОСЫ снижается, если простраиствснн
ческого кочьиа (1 я "н1РОгРУппу выйти из плоскости aP0”afi
416 ольца (например, для нитромезитилепа е « 800). В бо-
ЯЛВ11ЯОВОЛЧОВОИ Области (-330 1ш) м
’перегиба- Жет наблюдаться
^ияето встречающееся утверждение о том что пг,о
„и валентных колебании ароматической интенсивные
л0'1()тПё лежат в узком интервале частот кажется 1Руппы Е ИК-
с’е«олы<У на частоту колебаний нитрогруппы замР°иНИТе''’ЬНЬШ’
«оМ"'от заместители в цикле; более Ln" 06 Влияние
1357 и 1487-1555 см-' [152]. Сигналы о-, . и “ппо™ЧеНИЯ
Спектре ПМР нитробензола смещены относительно бензолаеоот-
ветственно на 0,93, 0,21 и 0,33 млн в сторону слабых полей
В масс-спектрах ароматических нитросоединений [153] в отличие
оТ спектров нитроалканов обычно присутствует пик моад”яп
ого иона; основной пик в спектре соответствует иону, полуХ
ному при отщеплении NO2. В значительной мере под действием
электронного удара происходит образование изомерного фенил
нитрита PhONO и отщепление от него NO. Аналогично ведут себя
я- и n-нитротолуол, однако для о-ннтротолуола основной пик
соответствует иону, полученному при отщеплении НО- за счет
циклического взаимодействия заместителей. Полагают, что пре-
вращение в нитриты — это существенная часть .процесса фраг-
ментации ароматических нитросоединений. Спектры отрицатель-
но заряженных ионов привлекли к себе внимание в последнее
время [154].
Образование комплексов. Многие ароматические ди- и поли-
ннтросоедннения, в частности 1,3,5-тринитробензол и родственные
соединения, образуют довольно устойчивые кристаллические
комплексы с обогащенными электронами ароматическими угле-
водородами (наирнмер, с нафталином), ароматическими аминами
и фенолами. Эти комплексы ярко окрашены и поглощают в ближ-
ней УФ- или видимой области спектра. Комплексы, в особенности
с пикриновой кислотой (2,4,6-трннитрофенол), часто используют
для выделения, очистки и идентификации ароматических углеводо-
родов; эти углеводороды можно регенерировать обработкой
комплекса горячей водой. Структура окрашенного в красный
Цвет кристаллического комплекса n-иоданнлпна u 1,3,5-триннтро-
бензола такова, что расстояние между ароматическими кольцами
обеих молекул >300 пм. Такие комплексы называют комплекса-
ми с переносом заряда или, по- Малликену, донорно-акцепторными
комплексами, В связи с тем, однако, что ван-дер ваальсо
взаимодействия играют главную роль в стабилизации ком
ЭТого типа, для них было предложено более подходящее иазва
«"о-молекулярные комплексы [155]. Устойчивость этих «охш
ексов возрастает с увеличением числа алкнльп ix • )1ем
11 Циклов в ароматическом углеводороде, а так ' н01екулу
большего числа электроиоакцепторпых заместителей ,пения
Рематического интросоединения. Обычно темпер„ -—тыо. По-вн-
;iiJ.,ITC11C,,BII()CTb окраски увеличиваются со сгЛ ‘ ’ ,1езав11снмыми
Мсл’У, эти комплексы ие являются кинетически пезаш
417
И Зак. Ю5
медиатамп в каких-либо последующих реакциях еоетавля,0
™?’Х KXue"^комплексов, образующихся с перечисленным,.
В ЭТЛИА „лпннми соединениями, ди- и триннтробепзоЛ
выше Ma6of а 5 ^Нитробензол и родственные соединения
в частности Г алкокСпл-, азид-, сульфит- или ц^
аг11руют ясиг"71нфатичесК11МИ аминами с образованием анионных
110НаМппексов называемых комплексами Мейзенгеймера (урав
р.мшплексов g к0Т0рых оба взаимодействующих компонента
и"я А „ЧДзио Такие комплексы часто можно выделить в виде
СВЯЗапр'ииих солей щелочных металлов; они надежно охарактерц.
°Х™ спектроскопией ПМР. Возможно, что в растворах реаген-
тов присутствуют молекулярные комплексы, находящиеся ,
состоянии быстрого обратимого равновесия и предшествующ,,е
комнтексаы Мейзенгеймера. В случае 2-замещенного 1,3,5-три-
нитпобензола вначале может образоваться термодинамически
менее остойчивый комплекс Мейзенгеймера, который затем пере-
гпуппнровывается в более стабильный (уравнение 97). Добавле-
ние кислоты к аннону Мейзенгеймера по аналогии с алифати-
ческими производными могло бы приводить к образованию аци-
формы нитросоединения. В кислой среде, однако, эти комплексы
обычно быстро распадаются, образуя полннитроароматнческпе
соединения; в некоторых случаях удалось идентифицировать и
аци-нитросоединения (например, 38) различной степени неустой-
чивости [157]. Если молекула ароматического нптросоедипения
содержит уходящую группу, может пройти нуклеофильное аро-
матическое замещение [см. разд. 7.2.3 (15)]. При взаимодействии
ароматических ннтросоедпнений с кетонами, содержащими «-ме-
тиленовую группу, в водном растворе в присутствии основания
появляется яркая окраска (реакция Яновского) [158]. Аналогич-
ная реакция была использована как проба на соединения с ак-
тивной метиленовой группой, в том числе на 17-кетостероиды.
Продукту взаимодействия 1,3-динптробепзола с ацетоном при-
писывают строение о-комплекса (39). Те же соединения в
ствнн этокснда натрия в спирте дают анион (40); это
реакции Циммермана [159].
o2n no2 _0Et
прнсут-
прнмер
H OEt
NO2
N02
NO2
(96)
418
Возможны и другие типы взаимодействия полинитросоедине-
инй С основаниями [160]. 1,3-Дциитробеизол и 1,3,5 тринитоо-
бензол обменивают атомы водорода никла в присутствии основа
Н11Я, возможно, через промежуточное образование карбаниона
например (41), однако, если такие анионы н образуются, кон-
центрация их очень мала и они не могут быть причиной
окрашивания растворов. Возможен также отрыв протона нз ме-
тильной или замещенной метильной группы, находящейся в
активированном положении в ароматическом нитросоедннении.
Из-за высокого сродства ароматических нитросоединений
к электрону может произойти перенос, электрона от основания-
донора, приводящий к анион-радикалу, который можно
идентифицировать с помощью ЭПР [161]. Такие анноны обра-
зуются из нитротолуолов и 1,3-динптробензола, но не нз три-
нитросоедпнеиий, дающих устойчивые a-комплексы. Анион-ради-
калы ароматических нитросоединений можно генерировать
электрохимически, фото.титическм, действием щелочных метал-
лов и реакцией с дитиопптом натрия.
7.2.3. РЕАКЦИИ НИТРОСОЕДИНЕНИЙ (88г]
Реакции первичных и вторичных интроалканов тесно связаны
с реакциями соответствующих оци-иитросоединении [J43];
с подтверждением этого не раз мы встретимся прн дальнейшем
рассмотрении материала.
(1) Термические реакции
Термическое разложение нитроалканов прн темперэтхре
выше 300 °C может идти по двум направлениям [162]. Возможно
отщепление азотистой кислоты, приводящее к алкену, и для всех
нитроалканов за исключением нитрометана эта реакция пр
ладает при температурах <450°C. Другой путь — это гох •
ческнй разрыв С—N-связи с образованием продуктов фрагме
тации. При первом процессе, который имеет [1631.
энергию активации, может происходить цнс-отщеп. i J в
Ациклические а-пнтрокетоны разлагаются ПРП Д.1 ов,ен1[я)
спирте и дают кстокснм (формальный ”Р0Д>'^ разрыва
и карбоновую кислоту, образующуюся в результат р е
14* 419
NOH
BuCOCMe + BuCO2Bu + BuCO2H + PrCH(OBu)2 (9gj
ннтросоедпнений, которые
происходит ----
no2
I BuOll
BuCOCHMe —
Ппполиз ароматических ннтросоедннении, которые легко
' "L приводит к появлению свободных ароматических ра.
Чкалов ’[165а]. Если нитробензол подвергнуть пиролизу при
йПП°С (20 с) получающиеся при этом продукты образуются из
фенильного й фепоксильного радикалов Фенильный радикал
может образоваться из нитробензола [1656], а феиоксильный
ладит появляется, вероятно, из промежуточно образующегося
нитрита что напоминает процессы фрагментации при масс-
спектрометрии [153]. Были изучены реакции ароматических
радикалов, генерируемых таким образом, а также реакции
радикалов, образующихся из нитрометана и ннтроадаманта-
на [165а].
(2) Фотохимические реакции
Фотохимические реакции ннтросоедпнений подробно освещены
в литературе [166]. В случае простых ннтроалкаиов первичной
реакцией, по-видимому, является фотодиссоциация (уравне-
ние 99); спектр ЭПР МО2 был зарегистрирован при фотолизе
нитроэтана. Соответствующий нитрит RONO, обнаруженный среди
продуктов, мог образовываться путем рекомбинации радикалов,
хотя возможна и согласованная перегруппировка. Образование
этилена при фотолизе нитроэтапа происходит, вероятно, благодаря
Диспропорционированию радикала Е1- или фотоциклоэлиминиро-
ванию. Для объяснения образования нитрозобензола и «-нитро-
фенола при фотолизе нитробензола было выдвинуто предположе-
ние о разрыве N—О-связп (уравнения 100, 100а), хотя, по-види-
мому, нельзя исключить и другие возможные пути реакции, вклю-
чающие первоначальную фотодиссоциацию.
(99)
(ЮО)
(100а)
hv
RNO, ---> R. + NO2
hv
PhNO2 --> PhNO + О •
O- + PhNO2 —► HO——NO2
(Е)-*(2)-°|ютопз™°» П0ВедеН11е нитроалкеиов. Известны примеры
шей по триплетипт'3аЦН" !1,1тРгостиролов в растворе, проходя-
нии же в твердой фазе^дди''ЗНу ^167^ <У1’авче”ие 101); прноблуче-
блюдалось также никJ’Р'"1ГГРОСТИР°Л образует димер (42). На-
личным алкенам, npHBOnJJ,^eill’HeHHe •₽-1|1,тРостпр°ла к раз-
нилышя группы находятся,.6 " аддУктам, Е которых нитро- и Фс"
420 транс-положеинн относительно ДРУГ
^Подвергаются перегруппировке под так’
f0K минокетопы (уравнение 103). iZХт,
через промежуточное образование нитритов. Реакция МОжет
'Д „ по другому пути, приводящему к ароматическим альдеги
и оксидам нитрилов, вероятно, через дРугое возбужденное
состояние. Эта перегруппировка не ограничивается лишь 1 арил-
о.нитропропенамн-1, она легко протекает и в случае неароматиче-
rKIix соединении, для которых невозможна конкурирующая реак-
цПЯ ведущая к нарушению сопряжения [168]. Эта реакция де.
конъюгации состоит во внутримолекулярном отрыве 7-водорода
к образовании |3,?-ненасыщенных нптросоедпнений.
(101)
NO2 NOH
| hv(>290 нм) II
PhCH=CMe ------------>“ PhCCOMe
ацетон
(102)
(ЮЗ)
фото-
обра-
Ароматические нитросоединеиия могут подвергаться
восстановлению в присутствии доноров водорода [169] с
зованием смеси продуктов, соотношение между которыми зави ;
от длины волны; главными продуктами реакции обычно являю
гидроксиламины (уравнение 104).
hv (366 нм) « , । г-п
PhNO, ------------> PhNHOH + МегСО
изо-РгОН
(104)
о-Нитротолуол проявляет фотохромизм за счет B^Tp”.M°]ITpo.
лярного отрыва водорода и образования ярко-ci i дНОму
соединения (уравнение 105). Это приводит также к водородное
обмену в метильной группе под действием облучен; .
421
Еще одной фотохимической реакцией ароматнчесхпу
соединений является их фотоипклопрпсосдииснпе к *
приводящее к продуктам, имеющим некоторое сходствоаЛКенам.
дуктамп озонолиза (см., например, уравнение 106). пре С п₽°-
что в этой и подобных реакциях образуются промеж ЭГа101,
аддукты, например (43), вероятно, в результате ступ Т°ЧН!>1е
присоединения; соединения этого типа были выделены^по,ГЧа'г°го
ции нитробензола с циклогексеном. ПР" Реак-
hv
PhNOj + МегС=СНМе ►
Me Me
O^NPh
—Ь MeiCO + MeCHO + PliNHCOMe + I I
KhN\/U
Me Me
(106)
(43)
Реакции циклоприсоединения происходят и с алкинами; извест-
ны также внутримолекулярные реакции ароматических нитрогрупп
с ненасыщенными участками молекулы, хотя в этих случаях дол-
жен образовываться циклический интермедиат другой природы
(уравнение 107). Ранее уже упоминались реакции внутримолеку-
лярного фотохимического окисления — восстановления ннтробенз-
альдегндов [см. разд. 7.1.1 (2)].
О
II
-> Ph (107)
R N+
I
O’
необы'1ная\еакиипиип!>,НД^’рова”11ых реакций была подмечена
иий в реакциях ,?” ° ?СОбиость ароматических нитросоедпие-
гидрокспл-ноном питяпи Ь”0Г0 замец1е”11я [170] с аминами,
422 Р Д ном и цианид-попом. В частности, пукле-
жильное замещение может происходить в „„„
Группе (уравнения 108-110), причем, по-видимому ° ГвДГ7’
£енно« состоянии именно жета-иоложение оказываете’» , УЖ’
ванным для нуклеофильной атаки. вается активнро-
/МО2
hv
'801-Г
(108)
МеО—(
+ МеОН
АОг
i»O—/хх
(109)
(ПО)
а-Иоднитроалканы претерпевают фотохимическое разложе-
ние в растворе через промежуточный а-иитроалкильный радикал,
давая а-нитроалкены с выходом 70—80% [171].
(3) Восстановление и гидрирование
Синтетическая значимость ннтросоедпнений, особенно арома-
тических ннтросоедпнений, обусловлена легкостью их восстанов-
ления в амины, которые находят широкое применение в синтезе
(см. также разд. 6.3.1.1). В общем случае восстановление нптро-
соединений может дать целый набор возможных продуктов [172а],
как показано на схеме 111, которая первоначально была выведена
на основании результатов электрохимических исследований. Обзор
по электрохимическому восстановлению приведен в [1726]. Вос-
становление в кислой среде обычно препятствует образованию
азоксн- [RN=N(O)R], азо- (RN=NR) и гидразо- (RNHNHR)
соединений, возникающих в щелочной среде за счет реакций кон-
денсации, в которых участвуют промежуточно образующиеся
иитрозосоедииения и амины или гпдрокенламины. В случае пер-
вичных и вторичных иитроалканов существенной может оказаться
таутомеризация ннтрозосоедииенпя в оксим, который может
Далее восстановиться до амина. Ранее уже упоминалось [см.
RNO2 —>- RNO —> RNHO11 -------*
|rno
RNO
(111)
RN_NR — RN=NR — RNHNHR
азоксн-
сосДиненне
азосое-
динеине
гидразо-
соединение
423
ЧТО получение ИПТрОЗОСОСДППСППП путем ВОСГт
разД' 7' „1ПЧИО трудно осуществимо. СТа-
НОТосстаиовлеиие нитроалканов хлоридом олова (II) дает
„Лоро силамииы, при действии хлорида хрома (II) образуй*
1 Л1731 При восстановлении а-пптроалкепов хлоридом олп
°я7т nSatOT а-хлороксимы. Хлорид титана (III) Ив
®аствОре восстанавливает первичные и вторичные нитроалкаиы
по ишшов (вероятно, через оксимы), которые затем гидролизу^
w а дегппов или кетонов с общим выходом 45—90%
Поедварптелыюе превращение интросоедипения в соль а<(«-форм^
лает лучшие результаты. На основе этой реакции может быть соз-
дан метод превращения иитрогруппы в карбонильную группу, т. е
метод обращения «полярности» атома углерода, ранее свя-
занного с нптрогруппой [см. уравнение 166 разд. 7.2.3(16)].
Аналогично ведет себя хлорид ванадия(Ш) [176]. Каталитиче-
ское гидрирование ннтроалкаиов можно остановить на стадии
образования гидроксиламнна; подходящими реагентами для полу-
чения алкил- и арнлгпдроксилампнов из соответствующих нитро-
соединений являются также шшковая пыль в нейтральной среде
или амальгама алюминия.
Для восстановления питросоедпнеиий в азоксн- или азосоеди-
нения обычно используют алкоголяты натрия. Восстановление
ароматических нитросоедииений борогидридом натрия [176, 177]
также дает азокси- или азосоединеиия, а при действии алюмогид-
рида лития обычно образуются азосоединения. Восстановление
гидразином и его производными в присутствии рутения или нике-
ля Ренея приводит к гидразо- или азокснсосдиненпям; гидразо-
соедииепия получают при обработке ароматических нитросоедп-
нений гидразином в присутствии палладиевого катализатора в
щелочных условиях. Азокси- или гидразобензолы могут также
быть основными продуктами при восстановлении гидроксидом
железа(П), а действие станнита натрия в зависимости от условии
реакции может приводить к азоксп-, азо- или гпдразосоедннениям.
Известно много методов восстановления нитросоедииений до
^I7R1°B ^СПеш,ю применяется каталитическое гидрирование
|178] в различных условиях, однако восстановление иитрогруппы
может происходить медленнее, чем конкурирующее гидрирование
пп связи' В последнее время используют гомогенные реакции
лбпе1,ех°А(1Ых металлов как катализаторов, которые
восетЯ1я1ппСОбО“ избпРателыюстыо [179]. Алюмогпдрид лития
Tvoe ппп _вагШ1Троалка”ь’ до аМш10В при комнатной темпера-
имушествепгто реакШ1Я не идет, что позволяет осуществить пре-
иитроцпклоалкагтССТаПОВЛен11е ДРУГИХ групп, однако в случае
леппе борогидрПдомВн3атпЖ11Ь' пеРегРУпш1Ровкп [ 180]. Восстанов-
затора дает лиип я рия в присутствии палладиевого катали-
единений пропсхо\птМИИЫ' Эффективное восстановление питросо-
водными в ‘от оТ."агРсваппи с гидразином п его пронз;
НЛп в присутствии катализатора (палладии
гле). Широко использовались железные опилки „ „
" ,слот [181а], а также олово и иногда цинк в соляной"^™"
хотя в спльнокпслой среде могут протекать побочные ’
снижающие выход. Для восстановления ароматич"ко
единений можно применять хлориды титана (Ц[) [1816 xnZ?m
„ олова (И); столь же эффективны дитионит натрия t бисульЙ
натрия, хотя последний реагирует медленнее. Целый ряд реаген
тов, близких по своей природе, был использован для избивав
„ого восстановления ароматических полинитросоединений до нит^
роамннов [182а]. К ним относятся сульфиды щелочных металл
сульфид аммония и полпсульфнд натрия; лучшим реагентом явля-
ется, по-вндпмому, гидросульфид натрия в метаноле [1826] Из-
вестны методики гидрирования, использующие перенос водорода
от доноров водорода в присутствий металлических катализаторов
[183]; с этим связана способность нитробензола, используемого
как растворитель в реакции Скраупа, выступать в роли дегидри-
рующего агента. Восстановление ароматических нитросоединений
карбонилами железа было проведено в присутствии катализаторов
фазового переноса [184].
(4) Дезоксигенирование соединениями
трехвалентного фосфора [63]
Ароматические ннтросоединеппя реагируют с соединениями
фосфора(Ш) медленнее,чем соответствующие нитрозосоединення.
Считают, что происходит ступенчатое дезоксигенирование нитро-
группы сначала в нитрозогруппу (уравнение 112), которая при
последующем дезоксигенировании превращается в нитрен [см.
разд 7.1.3 (2)]. Поскольку питросоедииения более доступны, чем
ннтрозосоедпнения, этот подход имеет широкие синтетические воз-
можности. Лучшим реагентом обычно является триэтилфосфит;
при его реакции с нитробензолом и простыми о-алкилнитробензо-
лами в качестве основных продуктов образуются соответствующие
триэтилфосфоримпдаты (Е1О)зР=МАг, а также другие продукты
превращения нитрена. 2-Нптробифепплы можно перевести в кар-
базолы; по-вндпмому, предпочтительной является 1,5-цнклпзацня
(уравнение 113). Некоторые о-нптростнролы или -стпльоены наря-
ду с другими продуктами дают индолы (уравнение )• ₽
реакции трянс-2-нитростильбсна в качестве промежуточного соеди-
нения был выделен 1-гпдроксн-2-фенилпндол; это позволяет пр
положить, что нитрен, по всей видимости, не является ннт Р-
Диатом в этих реакциях. а-Нитростпльбен дает индол' с
тельпо меньшим выходом, а-ннтростпрол не образует г ’ ’
Р'Нптростнрола получают с низким выходом фенил ц т
ArNO2 + R3P —>
-ЩРО
АгХО
(112)
425
При реакции 2-о-нитрофенилпиридина замыкание цикла провд.
ходит по обогащенному электронами атому азота цикла; это ха-
рактерно для синтезов таких гетероциклов, как индазолы, трпазо-
лы и родственные им системы (уравнения 115—117). Кажущаяся
«шестичлеппая» циклизация прн образовании феиотиазииов из
2-нитроднарнлсульфндов на самом деле иллюзия (уравнение 118);
такие спиродиенильные интермедиаты участвуют также и в про-
цессах дезоксигенирования других нитросоединенпй, для которых
протекание перегруппировки очевидно [185]. Фосфорсодержащий
нуклеофильный реагент может иногда даже заместить активиро-
ванную нптрогруппу, вместо того .п„,„„
(186]. Восстановление замещенных нитробензолов
различных трнс(диалк»лампно) фосфинов в присутствии
амина приводит к замещенным ЗН-азепннам [187].
чтобы дезоксигенировать ее
действием
избытка
(117)
валентиогоИЛоесС*опЯРп^ш’'’ шгтРоалкапов с соединениями трех-
КИХ температупах" 2 Н₽,< СТЬ'п ннтРоалкапы ие реагируют при «пз-
I Прах. 2-Нптро-2-хлорпропап и еея-галогеипитроциК’
-пканы по своему поведению схожи со споим,,
’"Лем. разд. 7.1.3 (2)). Известно также фо=^^^
генирование в присутствии триэтилфосфита [188] ское Дезокси-
(5) Окисление
Ароматические питросоединения инертны к окислению »„
роалкапы окисляются на воздухе в щелочном растворе (уравнеи?
19). Получающиеся продукты образуются вИрезультатеРСвйбол
„□радикальной цепной реакции, Друг„е окислители также даюй
соединения (уравнение 120). Соли нитроновых кислот моХ
окислить в соответствующие карбонильные соединения действием
перманганата в нейтральном С"*™ нош — u bl,LM
гпрпо ricni
среде [*“9] или пероксисульфата'
озонолиз солеи нитроновых кислот протекает быстро и глалхп
____л™..-г I/ L’lnfintf tl IklJLK.f nnfllHtio.Twr,.. /..._ [190]
приводит к карбонильным соединениям (уравнение 121)
О2. “ОН
Ме2СНМО2 _„с > Ме2СО + XOJ (Н9)
O2N NOj
Na2O2 • | |
Me2CHNO2 — > [Me2CNO2] —>. Me2C—CMe2 (120)
’OMe i. Оз
R,R2CHN°2 R1R2C=NO2 R'RZCO (121)
(6) Реакции с кислотами и основаниями
Ароматические питросоединения, в общем, инертны к действию
сильных кислот. Однако пптроалканы, имеющие водород при
а-углеродиом атоме, гидролизуются с различными скоростями под
действием горячих концентрированных минеральных кислот [191],
давая соответствующую кислоту или кетон (уравнения 122. 123);
в первом случае (уравнение 122) можно выделить гидроксамовую
кислоту. Скоростьопределяющей стадией этих реакции является
образование аци-пптроеоедииеиий [см. разд. 7.2.3(12)]. а-Нитро-
кетоиы, содержащие группировку—COCH(NO2)—, превращаются
с высоким выходом в карбоновую и гидроксамовую кислоты
(уравнение 124) [192], однако некоторые циклические а-нитроке-
ТОПЫ стероидного ряда дают М-гидроксппмиДЫ (уравнение Шэ)
[193].
RCH2NO2 " ' А >- RCONHOH * RCO2H + NH2OH (122)
R2CHNO2 —Н ' Д->- RsCO + NsO
R2
R'CoinNOa —> R'CO2H+ R2CO.\'HOH
—COCHNO, —> — CON(OH)CO—
(123)
(124)
(125)
427
я вторичные иптроалканы реагируют с OC1W,
Пе1* первоначально соли [см. также разд. 7.2.2(1)1, к^
Н”ЯМ11’ nrv? оеагнровать дальше при стоянии или при uarpeBaHI°
торые могут Реа р нитрометана превращается в Сол'
(уравнение 126). Другие иерВ11ЧНЬ1е
HHTpojKC CHOI (аиь1 м конденсироваться под дей.
Р°аЛК оснований давая пзоксазолы (уравнение 127); в слД^е
"етичных нитроалканов с активированной нитрогруппой происхо-
пит отщепление азотистой кислоты (уравнение 128). а-Нптрокето-
„ь пасшепляются в присутствии основании даже в мягких уело-
пнях прячем в случае циклического ннтрокетона^ образуется 0.
ттпокнслота по механизму ретрокляизеиовскоп конденсации
(194) Гидролиз а-ннтроалкенов в слабокпслои и щелочной средах
ппиводит К нитроалкаиу и карбонильному соединению по ретро-
реакции Генри [см. разд. 7.2.3(10), уравнение 145]. Взаимодей-
ствие ароматических нитросоединений с основаниями рассмотрено
в разделе 7.2.2 (2).
CH2NO2 к+ -----> -o2n=chco2- 2К1
V/
-он /=\
RCH2NO2 ----► R—
N
(126)
(127)
N02
I МеО"
MC2CCH2CI -------► Me2C=CHCl
(128)
(7) Алкилирование, арилирование и ацилирование [/95]
Реакции алкилирования нитросоединений ограничены лишь
теми алифатическими соединениями, которые способны образовы-
вать карбанионы; алкилирование обычно проходит по атому кис-
лорода, приводя к эфирам а/<1/-нитросоедппепий [196]. Дпазоме-
тан подходящий реагент для алкилирования нитросоединений,
представляющих собой довольно сильные кислоты (p/G < ~8),
и для аци-пптротаутоыеров менее кислых соединений. Успешно
протекает^ реакция алкплиодидов с серебряными солями иитро-
лниенни> име1°ших дополнительные электроотрицательные за-
Иза11М°Действне алкилгалогенндов с солями щелочных
веооятпп ЯИТРОСОеД1111еп1111 в кипящем спирте хотя и протекает,
пения ппивоП™?’ея<Ута'1'1ЫМ оСРазоваиием эфира а^и-иитросоедИ-
Щему’пзРалкнлг'1пбЫ,11,° К каРбонильному соединению, возникаю-
зовать ка“СОбТ'1Да' " °КС”Му- Эту реакцию можно исполь-
129) (1971- лУчеш,я карбонильных соединении (уравнение
более устойчивых ” альтсРпатпвпый подход — разложение
среде. ' Пиров жщ-питросоедпнений в слабоосновпон
Ме2С=\-О; Ха+ +/'=Х\ ,,
V/ Вг —>• Me2C=NOH +
(129)
428
Солп цптроалкапов реагируют с тетп-щ ,
оксоипя при О С с образованием эфнро^ад н°иРаТ0М фиалки,-
"01(ТН количественным выходом (уравненне То Х'СоеДннений с
ры могут существовать в виде цис- и транс-S ' Этн Ди-
So быстро разлагаются при комнатной темпепат^°В\0НИ Довод”,
тых н11Троновых кислот обычно представл^ЩЛ Эфиры прос.
нпзкоплавкие твердые вещества [таутомепи, ” Жидкостч или
описана также в разд. 7.2.2(1)]_ а>т°мерия нитросоедИКен^
R'\ . R1
)c=NO2- Na++R3iO*-BF. \г_ы„ 3
R?/ / С-ЬОЛ + RjO (I30)
фепнлпрованне тозилатом дпфеннлнодоиия в диметитфоом
амиде проходит по а-углеродному атому н приводит к а-феиилнит-
роалканам. Сообщалось о С-алкплированнп с участием промежу-
точно ооразующихся аннон-радикалов [198] или четвертичных
оснований Маннпха. Соответствующим образом активированную
ннтрогруппу удается также заместить аци-нптро-анноном, вновь
по анион-радпкальному механизму [1986, 199]. При использова-
нии некоторых замещенных беизнлгалогенидов возможно как О-,
так и С-алкплировапне в зависимости от уходящей группы в гало-
гениде. Предполагают, что и в данном случае С-алкплнрованне —
анион-радпкальный процесс.
Трудности осуществления С-алкплировапня не позволяют в
полной мере использовать анионы ачи-пптросоедпнений в синтезе.
Однако в настоящее время из первичных иптроалканов стало воз-
можным получать диапионы (44) [200а] при действии н-бутпл-
лития при —90 °C. Эти анионы реагируют как нуклеофилы с эфи-
рами, ангидридами и галогеиапгидрндами карбоновых кислот,
а также с алкнлгалогеипдамн (уравнение 131), альдегидами н
кетонами, давая продукты ацилирования пли С-алкплирования.
Если в активированном а-положенпн находится только один атом
водорода, как в 2-иптропропане, или если в р-положении нитро-
соединения имеется дополнительная активирующая группа, как
CH2NO2 ---------->
тгф, гмхтл,
90 0
- 1. R2Br
R>C=f< 2Li+ ———* IWCHNO: (131)
\0- 2- ЩЛ
(44)
(45)
Е - Е
Н\1 Д/° RCHCH2NO2
R^^ \0-
(132)
429
о „бпазуется а.р-дпдепротоиировапный 'Тоо
в 2'аР45У’ХТ0рОь'йа'реагируеТ' с эдектрофидом (Е) в ₽-положеН11е
&Йге'|32) !200б); !ЫХ „ вторпчных нитроалКанов с
Пр'1 в3а,,МОХХных условиях первоначально происходи
нНаМ1”’аМя'анпе по кислороду [201] (уравнение 133).
алкилирование R3 Q.
К I I
2 „зг,,х,0 _> r]NCOCHOM=CR| + RsNCOCH—N==CR’ (I33)
rJNGssCR’+R^HN^
Известны реакции С- и О-ацилпроваппя нитроалканов. Так,
нптроалкапы образуют с ацилирующими агентами не.
3 вые смешанные ангидриды нитроновых кислот, а первт-
£" тросоедипення могут за счет дальнейшей реакции ангндрн.
па н троновой кислоты превращаться в производные карбоновых
ш гидроксамовых кислот [202а]. Удается выделить триацилгнд-
поксиламины, образующиеся в результате реакции некоторых пер-
вичных нитроалканов с уксусным ангидридом н ацетатом натрия
при ~80°С (уравнение 134) [2026]. При взаимодействии первич-
ных нитроалканов с метокснмагннйметнлкарбонатом (ММК) про-
исходит С-карбоксилпрование, что представляет собой метод син-
теза а-лнтрокарбоновых кислот и их эфиров (уравнение 13о) [203].
АсгО
RCH2NO2 -Т-уугТ*' RCO.VAcOAc (134)
СООН
1. MeOMgOCO2Me, ТГФ |
RCH!N02 Тнс]--------------- RCHN02 (135)
оснований
а прн а-углерод-
м монохлорпропз-
......»й катализируемый кислотами
стадией в котором является пре-
“ "J ] геохимическом хлорнро-
ие только по а-углеродиому атому.
(8) Галогенирование
Нитроалканы легко галогенируются в присутствии
с последовательным замещением атомов водород
ном атоме; реакцию можно ограничить синтезе
водных. Известен и более медленный ----
процесс, скоростьопределяющей стадией в
вращение в ацн-нитросоединсние. При фо'
ваини замещение проходит ~
(9) Реакция Генри и родственные реакции [204]
₽ Ре11Рп состоит в катализируемом основаниями прпсо-
ш/ппГт замеще»иых и незамещенных первичных н вторичных
чесчим К алиФат11ческпм альдегидам н кетонам и аромати-
ческим альдегидам (уравнение 136).
R2C0 + RjCHNOs
ОН
основание |
► R2CCR2NO2
(136)
430
ностп с нитрометаном (уравнения 137, 138) [205]; эта
ные циклизации также нашли применение в химии
яжлектнвиымн катализаторами этой реакции
* алкоголяты, карбонаты и бикарбоиа^^Х^ Г1«Рокси-
Эпоксид кальция, этоксид алюминия, эток™НЫХ металлов
'я ашюнообмепные смолы, третичные амины Л™'” и алк>ми:
Щелочность среды следует тщательно коитоолипг Т амм°ния.
дат пройти альдольная конденсация или реак^^к' иначе «°-
8 случае альдегидов, а также катализируемо Канниццарп0
женке нптроалканов [см. разд. 7.2.3(6)]в сл^ва,!И7М11 Разло-
роалканов в присутствии основания (1 экв) обпЯЛРВИчных нит‘
нитро-таутомера продукта реакции и подкис™? “Ль ач“'
смесь для выделения этого продукта следует остоп.РеаКЦИОН^’ю
жанне протекания реакции Нефа [см. разд 7 2з7|о\Г° В° "збе'
нежелательна. В том случае, если образуется' тор™ ВС’1Н оиа
алкан, который не может давать соли, щелочносп №ро’
растает по мере протекания реакции, что приводит КР^ ра В03'
выходам; для повышения выхода выделяющееся оСноваш1еИХМ
зывают, например, диоксидом углерода. Образующийся" ™
реакции р-гпдроксинитроалкан может отщеплять воду и даГа?к
а-интроалкен. В случае ароматических альдегидов бывает трудно
провести реакцию без того, чтобы не прошло отщепление хотя
обычно оно происходит под действием сильных кислот. Первичные
нитроалканы могут реагировать с двумя молекулами карбониль-
ного соединения, так что соотношение реагентов надо подбирать
очень тщательно. Особенно широко эти реакции применялись в
химии углеводов.
~ Дпальдегиды могут ^вступать в реакции циклизации, в особен-
'т" и родствен-
сахаров.
431
(10) Присоединение по Михаэлю [204,206]
Первичные и вторичные иптроалканы в присутствии огг
катализатора могут реагировать с а.р-ненасыщеиными елп "Ог°
ГПАЛГГ..РИПЙМ», цианидами и 'К,1Ь1М||
------------------ и аналоги,,
ПОНЧ" --- - - • • ---•••«« ДО ДЛ..
(уравнение 139) с хорошими выходами. Присоединение Эл’°
........... ” ........ ИИТроад^1
ЛЛт _ П<
--нД
ФОСфц.
1 Могут
1 Могут
ирнсоеди-
х кондеи-
’ такими,
КИСЛОТ и
катализатора могут реагировать
эфирами, карбонильными соединениями, цианидами ц
ны.ми соединениями, давая продукты присоединения по
________- vnnninrnrii пмУЛПами Пп.,---
пойти п дальше, если в реакцию вводили первичный*нитпоМ°>Кет
В качестве основного катализатора обычно используют аЛКан-
натрия или диэтиламип в спирте; применение третичных лТ0КС11Д
нов дает более высокие выходы [207]. Продукты реакци Ф0Г'''"
затем отщеплять азотистую кислоту [208]. а-Ннтроалке "
сами по себе служить слабыми акцепторами в реакциях п'Ы
пения ио Михаэлю; известно большое число примеров их
сации с соединениями с активной метиленовой группой
как ацетплацетон, эфиры ацетоуксусной ц малоновой '
другими соединениями (например, уравнение 140) и
анион -ОЕ( может легко присоединяться к а-пптроалкеняТ'?^
уравнение 143), вместо него следует Р йенам (
ванне.
. алкенам
использовать другое
(см.
осно-
no2
r2chno2 + сн2=снл —> r2cch2ch2a
а = СО2Ме, СОМе, СПО, SO2Me и т. д.
(139)
CII(CO2Et):
«RC6h,<!;hch2no2
«RC6h,ch=chno2 + CH2(CO2Et)2 —>
уЙЯТ.’ЙК игг -........................... —’
тации продукта конденсации и как РезУльтат Дегидра-
Уравнение 142). К а-ннтроалкевя ПОследУ'°шего присоединения
осноиГ11е 1|УклеоФилы, в том чиспрМ пр11соедш1яются по Михаэлю
ания, а также реактивы Гриш яп"Р/ТЫ’ амины и Родственные
No2 No Р 1 Ра (например, уравнения 143,
МеС=СНг+М^НСН20Ме _ NaOEt |,°!
МеСНСНгССНгОМе
NOa
[Me2c=CHNO2] -McN0^.
RO’ Na+ + PhClI==CHMn M^C(CI12NO2)2
“ ™0< - "«МСЫО,- N„.
(140)
„описанных выше типа ре-
пи, а именно присоединение
алифатический"" наиоодее Удой-
... - ~ к,х ди‘ и яолиннтросоединеннн
— "•° ™па присоединение может
и последующего
(Ml)
(142)
(143)
педииение воды к питроалкенам в елабокислой или
М41- Пр „ой среде приводит к p-нитроспиртам, которые могут
1 «бо1ДеЛ° " петрореакцип Генри (уравнение 145). Метилиддиме-
с;01веРгат1” „ия дает иитроциклопропаны {209], причем вначале
;1Йл,,ф°* присоединение по Михаэлю (уравнение 146). Присо-
лпо»с1‘оД1,Т .ппловых эфиров енолов в условиях кислотного ката-
Лшенне си уЩесТВить непосредственный синтез 1,4-дикето-
л,13а(уПр°а3внение 147) {2101-
цОб иг
R‘CH=CHNO2 + R2MgX —> R'R2CHCH2NO2 (IM)
он
, / H2° \ I / \
/=Чо2 /С-СНЧо2 /=O+-CH2NO2 (ц5)
О R
II I
Me2S=CH2 + RCH=CNO2 —>.
+ СН2=С
(146)
(147)
OSiMe3
Первичные и
акцию Манниха
формальдегидом
зом (например,
изводное ’'
про-
(11) Реакция Манниха
вторичные алифатические амины вступают в ре-
с первичными и вторичными нитроалкаиамн и
[204]. Реакцию можно проводить обычным обра-
уравнение 148), можно также использовать про-
N-(гидроксиметил) амина (например, уравнение 149),
гел-диамин [например, (R2N)2CH2] или гидроксиметильное
изводное нитроалкана (уравнение 150).
RjNH-t- СН2О + RfCHNOj —> R]C(NOj)CH2NR'2 + Н2О
RCH2NO»4- C5HioNCHsOH —> C5HioNCH2CHRN02 + H20
R’NH2-|-HOCH2CR?NO2 —> R’NHCH2CR?NO2 + H2O
Нитромета......-
(148)
(149)
(150)
«„прометай н нитроэтап могут конденсироваться ы
обычно
Мгндроксиметпл)амина, высшие первичны Р аминов
Реагируют лишь с 1 экв амина. В случае р ‘ 0Ваипе основного
Реакция протекает с трудом, требуется и ся гетероиик-
катализатора. В этих реакциях могут °°Ра п!.е1“1СТВпи первнч-
лическпе системы [211]. Например, при ® 1 убытком форм'
иого иитроалкана с 2 экв первичного амина на (уравне-
альдегида получают производное гексагпдр Н0(11еН11Н 1:1: 3,
ине 15)). еслп те же реагенты берутся . „ азн11 (уравнение
образуется З.б-диалкил-б-иитротетрагндро-!, пр01!ЗВ0Дпые
152). Гетероциклические соединения, в 1
„..ртища и гексагпдроппрпмидпиа, получают пп,
тетра"™ш1 перчаииш-х нптроалкаиов с формальдс1.11до ₽» взаи.
моде11(-1В1 “ ач.
миаком-
R,CH1NO2 + 2R-’NH’ + CH2° (НЗбЫТ0К)
^'\/—NR!
—н2о
R'\y—°
R’NH. + R'CH2NO2 + 3CH2O _„2O ” /< )
О2У '—mrj >'«)
(12) Реакция Нефа
Выделение обсуждения этой реакции из разд. 7.2.3(6) сделано
произвольно, однако это удобно. Реакция Нефа, происходящая
при добавлении соли шрлннтроформы первичного или вторичного
нитроалкаиа к избытку холодной разбавленной минеральной кис-
лоты [191], приводит соответственно к альдегиду или кетону с
выделением N,O. В случае особо устойчивых нитросоедииений
(например, п-нитрофеинлнитрометана) они являются основными
продуктами реакции в результате протонировання аниона и тау-
томеризации. Реакция Нефа обычно протекает гладко, выходы
продуктов умеренные, и она находит применение в синтезе (урав-
нение 153).
н* н+
RsC=X’O2- ----> R2C=NO2H —* R2CO + '/2N2O + l/2H2O (153)
Н2О
Первичные нитроалканы в этих условиях дают альдегиды, в то
время как при действии горячих концентрированных растворов
кислот образуются гидроксамовые или карбоновые кислоты [см.
разд. 7.2.3 (6)]. Эфиры простых аци-нитросоедннеинп весьма сход-
ны с солями как по составу продуктов, образующихся из них в
результате реакции Нефа, так и по характеру его изменения,
в зависимости от условий реакции, хотя ясно, что ац/г-пптросоеди-
сопрп-Не СЛуЖаТ 11нтеРмел11атамп в этих реакциях [191]. Эфиры,
сор ржащие’ 11апР11меР> а-оксогруппу, гидролизуются до нитро-
аии-нптп!1Я " СП11Рта- Очевидно, что и реакция Нефа, и гидролиз
оовапнпй с?едниен’’й проходят через стадию образования протоки-
терме и тпФТЫ' R2CN+(OH)2; имеются также данные, что ин-
ние R2C(OH)NOPenKU"" -НеФа является гидроксннитрозосоедиие
Различные СХВМы ^Х°ОЪРяс,1еНия реакции были предложены Д
безводный ХЛПп„ - ]• ^сли в качестве реагента использова
ния, а НХ гиппом.Ь1И В0ДоР°Д> образуются а-хлорпитрозосоеди
к ожидаемым nnoav» катаЛ1131!РУемый ионами серебра, привод
Иногда особен!™ реакщ1» Нефа [213]. цпе
а'<“-иитросоед„1,еш,й * 'вторичных соединений, РаЗЛ^ЕД0.
434 Р1 PH 2—4 приводит к оксимам и nLV л
яду с нормальными продуктами реакции; очевидно
рОлаМ "яр 'а„Пе происходит ио различным путям [214]; вооб-
„ рх °бр нефа сопровождается образованием иитроловых кис-
, pW1'11"'1 штполон как побочных продуктов. Подробно реакции
1Л. и цсевДо1 ...„“j и их эфиров в условиях кислотного катализа
Л°‘ ,-rnnCOCAllHLU ttxQl
jgS%b. в р ’
(13) аци-Нитросоединения и их эфиры [/«]
Многие реакции первичных и вторичных и«тр0а.1канов „„„
яые в разделе 7.2.3, протекают с участием аци-интпотатт м
„ли обычного аниона ацк-интросоедииеиия (питрона? ипн) пРа
становление аци-интросос.тинеиий иод действием большою
реагентов, например йодистого водорода, протекает ^ „7’
личным выходом приводит к соответствующим оксимам Так
протекает восстановление и эфиров соответствующих нитроиовш
кислот. а-Галоген-а^й-иитросоединения очень 'реакционноспособ
„ы, попытки выделить их не увенчались успехом. Атом гатогеиа
в этих соединениях весьма склонен к нуклеофильному замещению
Считают, что реакции соответствующих а-хлорнитроа тканов
являющиеся, по-видимому, обычными реакциями замещения про-
текают через аци-нптротаутомер. Таким процессом является
реакция Меера — замещение галогена слабонуклеофильным нит-
рит-ионом.
Ранее уже отмечалось диспропорционирование эфиров щи-
нитросоединении in situ с образованием оксимов и карбонильных
соединений [см. разд. 7.2.3(7)], и в случае простых по строению
сложных эфиров его можно использовать для синтеза оксимов.
Эфиры аци-нптросоединенпн, которые можно генерировать in situ,
вступают
дают с
пне 154).
в реакции 1,3-диполярного присоединения к алкенам и
хорошим выходом 2-алкоксиизоксазолпдины (уравне-
ОД' КО,
°2N\ +/0' СН2С12
;c=n( + сн2=сн2 Me°-N\
O2n/ -ОМе ' о'
(73%)
(154)
(14) Замещение ароматических нитрос
. „„ нуклеофильное заме-
Активирующее влияние нитрогруппы Г’я1огена, хорошо ма-
шине орто- или пара-заместителя, особен ‘ тсЯ сильнее в»
вестно [215, 216]. Эта активация ооычно ск зм прнСоеди-
°Р7о-заместителе; общепринятым считает • • ПпомежуточнЫМ
нения — отщепления (например, УРа®неЛ"йзе'нгеймера) (46^-
образованием а-комилекса (комплекса А 435
(46J
разнообразными цУклр„
ами НО- AlkO- pho°J
банионямк. rwtrr»_ •
Галогены могут быть замещены самыми
Хми в том числе аминогруппой, нон ............. , ,
Hal- RS-, N3- и NCS-, а также карбанионами. Отиоситель
мая реакционная способность различных галогенидов зависит 0,
природы самого галогенида, нуклеофильного агента, растворителя
и температуры. Помимо галогена «уходящими» группами могут
быть нитро-, алкокси-, арцлокси-, сульфонатная или другая слож-
ноэфирная группы, а также амино- и аммонийная группы. Извест-
но много реакций, в которых и атом водорода замещается
нуклеофилами. Недавно прн изучении реакций нуклеофильного
ароматического замещения была установлена способность арилга-
логенидов захватывать электроны от подходящего донора и склон-
ность образующихся анион-радпкалов выбрасывать галогенид-
ион, давая арил-радикал, то есть в некоторых случаях может про-
исходить «нуклеофильное» замещение по механизму SRNI [217],
Хорошо известен перенос электрона к ароматическим ннтросоеди-
нениям [см. разд. 7.2.2 (2)]; побочные реакции, протекающие 8
результате этого процесса, могут, вероятно, сопутствовать нор-
мальному замещению. По-видимому, пока пет данных, подтверж-
дающих предполагаемое образование [218] аннон-радикалов или
молекулярных комплексов [см. разд. 7.2.2 (2)] [155] в качестве
предшественников а-комплексов. Протекание реакции по схеме
присоединение — отщепление возможно не только при замещении
в производных ароматических ннтросоедпнений [219], такой же
ход реакции часто наблюдается и в азотистых гетероциклах,
с которыми их часто сравнивают. Обмен галогена на металл
в орто-нитрогалогенбепзолах также протекает с большей
легкостью.
Прн нагревании ароматического ннтросоедпнений со спирто-
вым раствором цианида калия происходит отщепление пптрогруп-
м» □ ввеДе|Ще карбоксильной группы в орто-положение к прежне-
пеякииГпп™10 <Реакц,,я Рихтера) (уравнение 156); обычно эта
лежпо ч«тРГКаеТ С невысок,,м выходом. В настоящее время на-
о-нитоозпбепДЛН10’гоот н'!теРмед,1атом 8 ЭТ011 реакции является
взаимодействии'22° ’ А|!алоги'и»>|й процесс происходит п при
ванную нитоогпмпКОТОрЬ1Х питР°беизолов, содержащих активиро-
растворителе пр^и"Щ0^На'ШД'И°ПОМ В Д1,П0ЛЯРН0М апротонном
положение к при этом "Уклеофнл входит в орто
группой; таким пчтрмР ППе’ которая замещается гидрокспльи
утем “ожпо получать о-циаиофеполы (уравнение
436
|221]. Перегруппировка Сманлса (уравнение 1^
'5/) .обой внутримолекулярную реакцию нук ieo«hJ„ } представ-
<ского замещения; эта реакция обычно осущест8иХ° аР°Ма‘
^ароматическом кольце активирующей нитрогрупПь| Р" Налн’
/ \.—ХО2 4- KCN
С1-~\ / (изб.)
кипячение
48%-ный водный EtOH
+ N;
со2н
(156)
/CN
/ \ KCN z—/
phcO—№2 "дм«Г* PhCO—
(55-60%)
• I I ‘on I I
HY—С—С—X—Ar --->- НХ—С—C—Y—Ar
II II
X = so2 или О; Y = O или NH
Ароматические нитросоединеиия вступают в обычные реакции
электрофильного замещения, давая в основном продукты мета-
замещения. Эти реакции приходится, как правило, проводить в
жестких условиях из-за сильного дезактивирующего эффекта нит-
рогруппы. Так, для нитрования нитробензола в водной серной
кислоте найдены следующие парциальные факторы скорости'
f„= 1 -10-8, Л, = 16- Ю-8, /л = 0,7. ю-8.
Влияние иитрогруппы на реакции ароматического свободно-
радикального алкилирования и арилироваиня, как правило, незна-
чительно; обычно образуются продукты орто- п «ара-замещения.
Однако бензильный и некоторые другие радикалы могут ата-
ковать пнтрогруппу [222]; нитробензол может служить эффектив-
ной спиновой ловушкой для некоторых радикалов (уравнение 159)
[223]. Нптрогруппа в значительной мере дезактивирует молекулу
по отношению к действию радикала •CH2NO2, генерируемого из
нитрометана и ацетата марганца(П), тогда как при взаимодей-
ствии этого радикала с бензолом феннлнптрометан получается
с хорошим выходом [224]. Свободнорадикальное галогенирование
питросоедпнеиий может привести к замене нитрогруппы на
галоген.
PhNO2 +
(159)
(15) Прочие реакции
[225]
кото-
также
437
Взаимодействие нитросоедииений с реактивами рР||Яьяра
^Рвопачалыю дает соль диалкил-а({И-1П1тросоеднпеи1 я
рои могут быть получены дпалкплпптроксиды Iе
разд-
КХаСь. [226] прп реакции с ароматическим,,
НИЯМИ-. R'\ + /0 RIMgX' R'\
RWgX + R!N°2 ~* R2/\0MgX R’-/N °MeX ('«О)
,т „пирны МОГУТ выступать в роли диенофилов в пея^,
дильс!- Альдера [204]. Например, имеющая общий xapa!J
Д Д, между циклопептадпеном и нитроалкенами приводи ₽
?Х»»”"122 |)гептенаи-2 (»— 161).
(161)
а-Нитроалкеиы являются также дпполярофилами в реакциях
циклоприсоединения [227]; например, прп взаимодействии с окси-
дами ароматических нитрилов образуются производные 1,2-окса-
зола (уравнение 162). а-Нптроалкпны, неустойчивые при комнат-
ной температуре,—исключительно реакционноспособные диено-
филы [228]. ph
, \/н
RhCsN—О' + СН2=СН\О! —> N" Y (162)
V xno2
Хотя ароматические ннтросоедпиения не вступают в обычные
реакции ацилирования, те из них, которые имеют алкильный за-
меститель в орто- пли пара-положении, могут ацилироваться по
а-положешпо этой активированной алкильной группы (уравнение
163); они также вступают в реакцию Гепрп. В обзоре [229] рас-
смотрена синтетическая значимость реакций, включающих взаи-
модействие ароматической пптрогруппы с орто-боковыми цепями,
особенно в связи с синтезом гетероциклов.
O2N—АА—СНз -ААА —сн2сОСООЕ1 (|б3!
\—./ NaOht \ /
1«,ю.е')А!,1'1ССКОе '^'Циклоприсоединение прп взаимодействии нит-
ип„НЭ?Л0В' имеющих электроноакиепториые группы, с высоко-
и п "'е"11ЫМ".аА“ протекает при комнатной температуре
нпеР!06) [2301 Ш’Л'!оксазолиди11ам (уравнение 164, ср. уравне-
ArNCJ,
— Ar—N
(164)
438
(16) Синтез природных соединений
q оТ раздел завершается рассмотрением дВух „„„
„пые иллюстрируют, как специфические свойства и реакций>
использовать в синтезе природных соедииеивй пР°Группы
^ синтезов (уравнение 165), приводящий К Х 1ерВЬ,Й из
s„“«,
2. Гидролиз
I. Присоединение
N'aBH,
МеОН
HsO'PrjNH
CHCI3
(81%)
(165)
(50%) (высокий выход)
фолипа) к а-нитро алкену по Михаэлю и отщепление азотистой
кислоты от р-пнтрокарбош1льного соединения [231]. Во втором
синтезе (уравнение 166) для получения цис-жасмона (47) [232]
используется способность атома углерода, несущего нитрогруппу,
образовывать аннон; для этого первоначально алкплгалогеннд
переводят в нптросоединеппе, затем группу CHNO2 превращают
в карбонильную, чтобы активировать соседнюю метиленовую
группу в реакции циклизации.
xx^CH2NO2
СН21
NaNO2
Д.МСО
МсСОСН=СН2
usoPrjN’H
7.3. НИТРИТЫ И НИТРАТЫ [233]
7.3.1. ЭФИРЫ АЗОТИСТОЙ КИСЛОТЫ
(1) Методы получения
Алкилиитриты R—О—N=O могут образовываться пр„ Вз
действ,спиртов с азотистом кислотой или ее производя" '
Спирты необычайно быстро этернфицируются азотистой К|1СЛ0*
В разбавленном водном растворе при О С (уравнение 167). С
ная модификация этой методики состоит в использования вас",
шейного водного раствора нитрита натрия, содержащего сульфат
алюминия. Метил- н этилиитриты - газы, они могут самопрощ.
вочьно выделяться при переэтерификации спирта более высоко-
кппяшим нитритом, например З-метилбутплинтритом (т. Кип
99°С). Как реагенты можно также использовать пары оксидов
азота (содержащие N2O4, NO и N2O3), нитрозилхлорнд в при-
сутствии пиридина (особенно для стероидов [234]) и тетрафтор-
борат питрозония.
ВОДИ. H2SO4
ROH + NaNO2 -------»- RONO (167)
При реакции нитрита серебра с алкплгалогепидами образу-
ются как интроалкаиы [см. разд. 7.2.1 (2)], так п алкилиитриты;
выход последних возрастает при переходе от первичных к вторич-
ным и третичным галогенидам [235]. Обе реакции протекают с
обращением конфигурации, и увеличение карбенпй-иониого ха-
рактера замещения коррелирует с образованием нитритов. При-
соединение ннтрозилфторида к некоторым перфторкетонам приво-
дит к перфторалкилиитритам.
(2) Свойства
Простые алкилиитриты— летучие, бесцветные, приятно пахну-
щие, но высокотокснчпые соединения, смешиваются с обычными
органическими растворителями, нерастворимы в воде. Метил- И
этилнитриты—газы (т. кип.— 16 н 18°С, соответственно). Нит-
риты более летучи, чем соответствующие спирты или изомерные
интроалкаиы. Их можно хранить при О °C; при комнатной темпе-
ратуре они медленно разлагаются с выделением оксидов азота;
иереюпка при температурах выше 25°С также обычно сопровож-
дается разложением. Цнклопропилиптриты намного менее устой-
явь* И подвеРгаются гомолитическому разложению с раскрытием
цикла в интервале температур —80—j-20 °C [2361.
вчет°аияиито^а11"»Ы,'1 спеитРос|(опических исследований, сУШе££
34 кДж/мопь 11ЫИ йаРьеР вращения вокруг связи RO—N (Д^
S-цис- н ХлЛ'п меТ||Л11||ТР||та), допускающий существование
связности ЭТОЙ свРя°знТР0В’ вероят"°’ благодаря заметноп двое-
11 связи (длина связи О— N 137 им). В случае пер-
440 '
метилиитрита
х алкилнитритов присутствуют как S-4UC т
nf причем первая из них преобладает ? ак и Странг
'К1, возможно, вследствие гиперконъюгации- втопМеТ”ЛНИтРита
g ты существуют, главным образом, в виде ™Р”чн,ые
" Le алкилнптриты - почти исключительно ₽анс‘Ф°Рм, тре-
т" уф-спектрах алкилнитритов обнаруживает" ^“-ротамеры.
BJ.ie (4—5 полос), соответствующее «^*.п₽по ОЯ!иое погло-
нм (е « 20-80) [238, 239], которое S в °^ТИ
мости ОТ соотношения цис- и транс-изомеров вР>п Зависи‘
” сп а также п°глощеш1е, соответствующее Раа,'овесной
“| С20 нм (в « 2000). ИК-Спектрь, харХе^с?^П₽И
полос поглощения при 751—814 см- (Vo N Рс У,зся наличием
Г1653-1681 см- (с может быть afc) и 16 ГХ^0'
с может быть дублет) [238]. Последние две полос? пп° “
8(а,от валентным колебаниям связи N=O соответственно "uT
,, 5-трпнс-форм. Частоты деформационных колебаний связей ON
„ N=O в группировке O-N=O расположены при 565-62? и
617-691 см-1, соответственно. Проведено также отнесение полос
в спектрах комбинационного рассеяния. В спектрах ПМР некого
рых простых алкилнитритов RONO (s-цис- и s-транс-) ‘химические
сдвиги «-протонов составляют: при R = Me д 3,7 и 4,7; R = Е( 6
4,0 н 5,1; R = н-Рг 6 3,9 и 5,0; R = изо-Рг 6 4,9 и 5,7 [237а] Дан-
ных по масс-спектрометрии нитритов крайне мало. Известно лишь,
что пики молекулярных попов в спектрах почти незаметны, при-
сутствует интенсивный пик с m/е == 30(NO+); обнаружена тен-
денция к образованию фрагментов путем расщепления связей, на-
ходящихся в 0-положении к кислороду [240].
Известны эфиры гппоазотистой кислоты HON=NOH; невысо-
кий дипольный момент позволяет предположить, что они облада-
ют транс-конфигурацией (48). Они образуются при взаимодействии
гипопптрпта серебра с алкилгалогенпдамн. При выдерживании
этих соединении при 25 °C, при нагревании или в присутствии
воды при температурах выше 40 °C они разлагаются с выделением
азота по механизму, предполагающему участие алкоксильных
радикалов (уравнение 168). При восстановлении образуется толь-
ко спирт, что подтверждает О-алкпльпую структуру.
zOEt
.N=\z
Е1СИ
(48)
—> EtOH + MeCHO + Ni
(168)
(3) Реакции
Легкость образования алкилнитритов c0„n.0CJaB'1p приводящего
"х гидролиза в кислой (но не в основной) Р ’ ет псПользо-
к спирту и азотистой кислоте. Это свойство’ , как источник
Вать алкилнптриты (обычно З-метилбути.и Р
”, кислоты вместо нитрита натрия. Их можно тяю,,
азотистой ki НевОдного диазотирования аромат,,че "Рк-
”еГтТде нитрит натрия непригоден. Взаимодействие аЛк™
“ оеакшюпноспособным фенолом дает ннтрозофеиол, при ’а
Р Хл опами также происходит нитрозирование. В пос^Цйй
С X оеакпшо обычно проводят в присутствии этилата иДНе“
СЛ?ка!ПЯ (дтя соединений с активной метиленовой группой)
, 'Л I”». Р“. 7.1.1 (3) I;
ных соединений (например, для алкилбензола) в качестве Ос '
вання используют амид натрия. Прп взаимодействий с реакт,,Ва
м„ Гриньяра промежуточно образующееся пнтрозосоединенпе Мр
Жет реагировать дальше, давая дналкплгпдроксплампн.
NOK
К+ "OEt II
EIONO +PhCH2CO2Et --------> PhCCO2Et + EtOII (1Й)
Скорости катализируемого основаниями гидролиза алкнлннт-
рнтов быстро убывают по мере замещения прп а-углеродном атоме.
Исследования, проведенные с использованием оптически активного
н меченого октил-2-нитрита, показали, что преимущественно раз-
рывается связь О—N, а не С—О. Если водород прп Р-углеродном
атоме активирован, может наблюдаться отщепление азотистой
кислоты (уравнение 170) [241].
ONO
I F.ljN
RCHCH2NO2 ------► RCH=CHNO2
(170)
Окисление нитритов в нитраты можно осуществить действием
N2Os [242] и этилгидроперокснда; прн действии пероксида водо-
рода образуются продукты гидролиза. Ллкплнптрнты легко вос-
станавливаются в спирты, хотя в некоторых случаях, возможно,
происходит сольволиз, а не восстановление. В качестве эффектив-
ного восстанавливающего агента можно использовать гидразин в
присутствии металлических катализаторов или алюмогндрид ли-
Голо Каталитическое гидрирование этплннтрита над никелем
' и—130 С) дает этпламип, этанол и аммиак' при более высо-
ят,,темпеРатУРах (>300°C) продуктами реакции являются этил-
нр, АИН°ГДа' аЦстО"итРНл. Полагают [243], что оба эти соеди-
хотя впоТп"1ка10т вслеД за изомеризацией в питроалкан [120],
Пои попит ВОзМ0>К1Ю> чт0 опп образуются из этанола и аммиака,
тов соелинр|111яГе11ер1'роваТ11 иптРены дезоксигенированием нитрн-
ветствуюшпр МИ т',ехвале1'тпого фосфора получали лишь соо
В X? L ”РТЫ' возм™»°. с Образованием гппош.тритов
ПреХХ ЖУТОЧНЬ,Х сосД'шешш.
в нитроаткаиь, X ?,.,.теРХ1,1ческой изомеризации алкилпитрнт
Р алкднь, ир„ 100-130°C [243] кажется маловероятным; ПО
ванпи а.
гида
НИИ
„„„шея данным, первичный процесс тепы,,,
"ме1Х а образовании алкоксильиых радикаРа3ло>кения
с0С ‘’образования иитрозосоединений пр“м п" Ш Чта S
сТСплИзе нитритов, то эти вопросы уже пасем Р°Цессе или при
t разд. 7.1-1 (4)]. КонкурирующаяУреакцИЯСМ„а^ва;1ись ранее
‘ омическом разложении нитритов, состоит в дТпппКающая "Ри
гер'-. алкоксильиых радикалов с образование . Р рциониРО-
пли кетона. Октанол-2, полученный в резуитяРТа и альде-
113 оптически активного 1-метилгепт1[1Нитп„тТ? ЭТ0Й реак‘
т это согласуется с разрывом O-N-связи (2441 ж Опт"чески
' е’ разложение алкилпитрптов в растворе пооиехпОто;",ТИч!>
‘емеханизму и приводит к алкоксщ^ьп. раХа ^ П°
распадаться [245], днспропорцпопировать, образовыва’т отоб|,Ь1м
соединения [см. разд. 7.1.1 (4)] „л„ отрывать водород о Xi
растворителя. Фотохимическая реакция в растворе в пР Сутст
№ кислорода (Х> 300 им) может дать алкнлнитраты, kotoZ
яе поглощают при Л > 310 нм и не вступают в дальнейшие реак
шт [246]. Если же алкильная цепь содержит более трех атомов
углерода, удается наблюдать процесс внутримолекулярного отры
ва водорода в алкоксилыюм радикале, приводящий в конечном
итоге к 4-ш1трозос11прту (49), хотя сам бутилннтрит дает этот
продукт с низким выходом. Указанный процесс (уравнение 171)
известный как реакция Бартона [247], явился особенно удачным’
приложением фотохимии к органическому синтезу. На стадии от-
рыва водорода требуется, чтобы реализовалось шестичленное
переходное состояние; цпклоалкнлнптрнты реагируют по тому же
пути, если щестнчленное переходное состояние стерпчески воз-
можно. Именно благодаря таким стсрически.м требованиям реак-
ция может протекать и с участием атомов боковой цепи н даже
первичных С—Н-связей.
O=N
/IV. N2. tj .
>330 им, s Г)
c»ll. ; |
-NO ” I J
ли _ 0N\ ОН
no х. ।
(49)
Реакция Бартона широко применялась. для в®®^Н"ЯСтероидов,
нальных групп в обычно неактивные участки . помощью ак-
В частности В СН3-группы в положениях 1» и। • тако|.. реак.
спальных стероидных нитритов. Первым р‘ „.йа-прегнан-гор-
ЧИи было превращение 2Ор-пптрита Зр- 1 ан.зв 206-дпола
ола (50) в 3-ацетат 18-гидрокс1ШМПпо-_ <* ПР ствие стереохн-
51) с выходом 34% [248] (уравнение “ото;,а при фотолизе
Мпческпх требований к реакции отщеплен” р х 20, 15, И 1,ЛП
стероидов, содержащих группу ONO в полi ' стерондов с груп-
может атаковаться положение 18, а в' сЛ.уча®п010жеипе 19 [скь
"ой ONO при С-2, С-4, С-6, С-8 пли С-Н-пол
Формулу (52)].
443
(50)
И»)
Предпочтительным является 1,3-днаксиальное расположение связи
С—Н и нитритной группы; важное значение для протекания реак-
ции могут иметь конформационные эффекты, например неблаго-
приятные днаксиальиые взаимодействия. Известно также, что кон-
фигурация атома углерода, несущего нитритную группу, может
изменяться в ходе реакции. Использование экваториальных нитри-
тов может привести к введению в цикл функциональных групп
(уравнение 173). В отсутствие стернческнх препятствий нитрозо-
соединения могут димеризоваться, при кипячении в пропаноле-2
образуются оксимы. Алкильный радикал может вступать в реак-
ции внутримолекулярного присоединения или подвергаться пере-
группировкам в противовес реакции с оксидом азота (уравнение
171) в силу тех или иных структурных особенностей (см., напри-
мер, уравнение 174) [249].
оеакпиш r3q циклогексплнитрнта, который не может встунать 8
рнт и нпклопплп3’ пр”водпт к цнклогексаполу, а цнклобутилнит-
розоатьаегилЕ НЛНИТР11Т Дают продукты раскрытия цикла
В’случай иД“’ог °Т°рЫе МОГ>'Т Реагировать и дальше [250 ;
тона степеохнмиирли'11,ИТ|1ИТа 11 Диклооктилпнтрита реакция Бар
ки возможна. Фотолизом соответствуют11* а
444
янтрптов В присутствии ацетата
йИГЮ двойную связь (уравнение 175) (ЯР ,УЛается ввес
1 „„трита приводит к оксиму тетпяг J' ф'<го,Лести Ш-
реакции образуются прен^ще^;”аР°фУРФуро,1а '7,T,e“f
“ е пятичлениыи цикл (уравнение 176) ,Но пР°ДУкты т 25 С1;
' 1и-№ржа-
Н .
7.3.2. ЭФИРЫ АЗОТНОЙ КИСЛОТЫ [252]
(1) Методы получения
Общими методами получения алкплнптратов RONO.- являются
непосредственная этерификация спиртов, реакция алкилгалогени-
дов с нитратом серебра п сольволиз алкилгалогенндов в присут-
ствии нитрата ртути. Для этерификации можно использовать ды-
мящую азотную кислоту саму по себе или в уксусном ангидриде,
а также смесь азотной и серной кислот (О—10’С) (уравне-
ние 177). Если не принять необходимых мер предосторожности,
эти реакции могут пойти очень бурно, особенно в случае легко
окисляющихся спиртов; обычно добавляют мочевину, чтобы свести
к минимуму образование азотистой кислоты. Механизм образова-
ния нитратов включает, по-видимому, О-ннтрованпе ионом нитро-
ния. Алкнлнптраты могут также образовываться при алкоголизе
тетрафторбората нитрония и ацилпптратов. Столь же эффективна
11 реакция спиртов с N2Os 1120].
МеОН + HNO3 —HaS0< >- MeONOs (6&—80%)
мелко пзме.’11'ченнь1М<
Алкнлгалогениды могут реагировать !ле в гетерогенной
нитратом серебра в инертном раствор! если в каЧеСТ
реакцию можно провести и в гомоген 19531. Для пол1'ч^,п.
растворителя использовать ацетонитрп- поль30вать алкил р
первичных п вторичных нитратов лучше 1Д08 и галогенид
*Щды п -йодиды, в случае третичных < 445
„ бензильного рядов можно применять и алкиЛХл
аллильного и ое> моЖйО получпТь в мягких условиях пр„ >•
ды- '-,ХрфоР«оатов с и,праТО“ сеРебРа 0 п"РиДйи^ а^'
ция алкплхлорф Р смешанный ангидрид разлагается '
‘‘УХ#»’™™”’' 1781 |2Я|-
ROCOCI + - IKOCOOSO.1 RONO. + од
и 1тпаты С аллильным, бензильным, первичными „ втОр11чн
Нптра™ радикалам» можно подучить с хорошими выхода*'
аЛК" Л,и . Соответствующих бромидов с нитратом ртутит*"
ПРИ ре^тлксиэтане' в большинстве случаев этот метод дае“
’2 * * * * *'даМ X пезутьт’аты [255]. Алкплбромпды или -иодиды удД
'1аИвпапт Рв нитРа™ Действием 98%-ной азотной кислоты
превратить г ного ангидрида.
аЗОНе°которые полпметнлпрованные арены дают бепзилиптрад
поп обработке дымящей азотной кислотой. При нитрован,,,, тп.
при °0Раи" «-ксилолов были выделены нитраты, производные
гндроксиц, клогексадпенона (53) [256]. Азотная кислота может
присоединяться к некоторым алкенам с образованием нитрата
Снейпе 179), так же ведут себя нитраты бромония и „ода-
НИЯ 1257а]- при окислении алкенов и циклопропанов нитратом
таллия (III) в пентане при 20°C могут образовываться 1,2- и
1,3-динитраты (уравнение 180) [2576].
С1 С1
\=Ц /Не
XZ У
(53) X, Y = Me пли Cl
-20 °C
Ме2С=СН2 + НМОз ------* Ме3СОХОг
ONO2
TI(NO3)3 I
С,И17СН=СИг -------->- CjHnCHCHjONOj
пентан
(85%)
(179)
(180)
(2) Свойства
Чевие как взрыв"1ят>шХ многоатомных спиртов имеют большое зна-
иий (например, ннтпП11пВЛЩеСТВа' ^же 1!3 1(х тривиальных иазва-
О-иитросоедипения Пп ' 10Л03а’ нитроглицерин) следует, что это
ческнм свойствам ааипТ1'1' алкилиитраты напоминают по ф»зи'
Рат, т, кИп. 64 6ОС- чти |,ТР11ТЬ!- но они менее летучи (метилинт-
лом о1ш часто обра а™ЛЙ,,тРат, т. кип. 88°C). С водой и этано-
446 азеотропные смеси. Следы кислот увели-
.,от склонность нитратов к разложению, „ пш, „
’" просто при хранении при комнатной те-,1,Рт1Грсвании «ли
М£ и‘взрыв. Продуктами такого разложенияРг> Г Может про-
С оксиды азота, альдегид „ли кетон, соответ ° PM0M явля’
uv спирту- и вода. Типичные длины связей ,, ующии исход-
"пиведены в формуле (54); имеются данные о то«ЛЫ В ""тратах
«fc. °-N0’ "
с
118°
О
(54)
в УФ-спектрах алкилнитратов наблюдается сильное погтоше
пне при <220 нм и плечо при260-290нм (е « 10); интенсивность
поглощения при 260—290 нм возрастает с увеличением числа ал-
кильных заместителей при а-углеродном атоме [259]. В ИК-спект-
рах присутствуют частоты симметричных и антисимметричных
колебаний соответственно при 1272—1282 и 1626—1634 см-1; про-
ведена корреляция между положением второй полосы и значе-
нием о*. Симметричным колебаниям в спектре вторичных алкил-
питратов соответствует дублет [260]. Влияние нитратной группы
на химические сдвиги протонов в спектрах ПМР можно проиллю-
стрировать на примере этилнптрата: 6(СН2) 4,49 млн-1;
б(СНз) 1,39 млн-1. В масс-спектрах простых нитратов никогда
не наблюдались пики попов, отвечавших исходным молекулам
[261]; характеристические пики соответствуют ионам с т/е
30(NO+), 46 (NOt) и 76 (CH2ONO2), их могут маскировать
пики ионов, образующихся за счет фрагментации алкильной цепи,
если она содержит более четырех атомов углерода.
(3) Реакции
При нагревании нитратов (>150°C) в контролируемых уело
виях образуются алкилиитриты с выходом до 75%. По-вндпмо^у,
сначала происходит гомолитическое расщепление, а затем в а
Действие алкоксилыюго радикала с оксидом азота, B031™i, ' ет
счет восстановления N2O4. Избыток оксида азота способ У
эт°му превращению, тогда как избыток ЬШ ториоз!
Алкплннтраты медленно реагируют е основав № £ ата.
Южин три направления (схема 181). <а) i y 0 сродного
Ка на атомы С пли N; (б) удаление водорода °т “ РаиНОму
ома с образованием карбонильного соединения п •
механизму [262[ it (в) отрыв
с образованием алкена.
Н 11
I I
~С—С—В + No3'
II
водорода от p-углеродиого аТвда
Н И
-С-С-СГ++BNOa
I I
н н
I I
В: + — С—С—0\02
I I
—с—с=о + no; + +вн
Процессы (а) и (в) могут проходить с промежуточным обра-
зованием карбениевого иона. Первичные нитраты претерпевают
главным образом сольволиз, однако бензплпптраты, в особенности
те пз них, которые содержат электропоакцепторные заместители,
более склонны к реакции (б), приводящей к бензальдегидам.
Вторичные нитраты даже в отсутствие активирующих заместите-
лей татже в значительной мере дают карбонильные соединения.
В случае третичных нитратов (например, ПЬдиметилэтплнитрат)
образуются алкены по реакции (в). Если а-углеродный атом свя-
зан с карбонильной группой, то под действием ацетата натрия
(основание) образуется карбонильное соединение, что является
удобным способом синтеза а-дикетонов [263]. Аналогично, если
₽-углеродный атом связан с соответствующей группой, реакция
отщепления, ведущая к алкену, облегчается н может стать
главной.
М07РвПд„МРтвпе0фИ*Ы’ в том Ч1,сле -CN> ~SCN> R0'
иптроалканы) а также° п^Т последнем слУчае образуются
довольно неактивно П1 то Р иые’ вторичные п третичные амины
тах с образованием nn'^^'5"07 (,1,тРа™У'о группу в алкплнптра-
лина может ппойп! -1 Р°ЛУКТОВ замеЩепия. Однако в случае анп-
п спирт. НуклрпгЫ,?аКа по азотУ 11 образоваться фенилнитрамии
случае, если алкпчьнп М°,->т Реагировать по атому азота и в ™
трет-алкилнитраты я11Я 1Р^ппа в Длкилннтрате третичная, поэтому
ний с активной метипо>Я10Т^Я нптРУ1ощцми агентами для соедппе-
с Длкилпнтратами могу80” гРУППов- Эти соединения при реакции
однако путем подбопя 1О41ИЛИ алк!1Л|1Р°ватъся, или нитроваться.
Удается из кетонов пптп п°ДхЗДящнх основания и растворителя
сульфонатов, сульфо110пРатоп’ ампД°в, эфиров карбоновых кислот,
448 11 толУ°лов получить соли пптросоедиис"
|сМ„ например, уравнение (68) в р
< ^агентом для такого рода лревращеннй Д 7'21 (1>]. Полез-
Снаигидрппа Me2C(CN)ONO2 [204]; его можноТ-ИИТ₽ат аие‘
Д-™ |,0ЛУ,,ен,1Я ""трампнов из пеовичЛ,
„мипов; высвобождающийся анион ацетоиадангидп ” 8Т0₽ичн“х
М а анетои п инаппд-поп. ид₽ииа распадает-
СЯ Известно, что алкилиитраты как правило, довоч,^
.....— ... во-’ыю устойчивы
кислотой, содер.
^превращаются
' Из не-
ки сло-
серной
к действию кислот, но при кипячении с уксусной кХS
ей каталитические количества серной кпстоты й’
* ацетаты [265], а с муравьиной кислотой-в *™"₽неВ₽а1
.“ и „р,. обработке W«oft"
та>ш Образуются бензальдегиды. В концентрированной
кислоте происходит сольволиз и нитраты превращав в
нитрония NO? и спирты, которые могут далее окисляйся По™
му алкилиитраты используются как нитрующие агенты в ™
кислых растворах, хотя практические преимущества этого меХ
„„видимому, невелики. Некоторые бензнлннтраты с большим чис!
„ом алкильных заместителей при действии кислот превращаются
„ ароматические питросоединения, вероятно, по схеме включа
юшей сольволиз, окисление п ннтродекарбокснлированне. Силь-
ные кислоты Льюиса могут вызвать разложение нитратов со взры-
вом.
Восстановление алкнлнптратов различными реагентами [хло-
рид желеэа(П), алюмогпдрнд лития, гидразин, цинк и уксусная
кислота], а также электролитическое восстановление и каталити-
ческое гидрирование приводят к спиртам с хорошим, иногда с ко-
личественным, выходом. Легкость восстановительного расщепле-
ния в мягких условиях наряду с инертностью по отношению к ос-
нованиям п окислителям позволили использовать О-нитрогруппу
для защиты гидроксильной функции в синтетических превраще-
ниях [266] в химии стероидов и в химии углеводов.
О фотолизе алкнлнптратов мало что известно; возможно, ос-
новной начальный процесс состоит в образовании алкоксилыюго
радикала.
7.4. НИТРОЗАМИНЫ
Сведения об алифатических ннтрозампна-х содержатся в обзо-
ра [267а] и в более общих обзорах [2676].
7.4.1. МЕТОДЫ ПОЛУЧЕНИЯ N-НИТРОЗОСОЕДМНЕНИЙ
тцч?рбь1‘1110 ПР“ нагревании вторичных а-1ПФа™че^ уК™снокис-
, тСКПХ а',,,,,ов с азотистой кислотой в водно. ( • riins\_NO.
В р„ Растворе образуются вторичные нитрозами электро-
фп/абокпсл°й среде такая реакция ,пРотека яинН0М, однако в
с111ьн°Г0 ВЗапмоДенетвия N2O3 со свободным ’ ов'одородяых
‘1ьн°кислой среде, особенно в присутствии галогенов 16
16 3«к. 105
пезкцпя становится обратимой [268]. Для Ш1трозирова
кислот. Реакция^ ш |()р||д, дающин высокие выходы
пспольз.мот Т N2O1 „ тетрафторборат ннтрозоп'
розамннов I- -1, озогруппы на вторичные амины в отсутст’ '
пср<’,,0СХет служить N-нптрозо-З-ннтрокарбазол (55). 5 ТВИе
gjlClOT М0АС1 v.v'
n-TYN°’
N
I
NO
Сообщения о возможности реакции с оксидом азота, по-вцднмому,
ошибочны, в то же время N2O3 п N2O4 — эффективные нитрозщ
пующне агенты в центральных и даже щелочных водных раство-
рах' реакция протекает по свободнорадикальному механизму с
участием N2O4 и NO [270а], Было показано, что дпэтиламин
питрозируется нитритом натрия в присутствии формальдегида пли
хлораля при pH 6,4—11 [2706]; постулируемый механизм пред-
ставлен уравнением (182).
+ NO-
Et2NH + CH2=O ------>• Et2N=CH2 < — Et2NCH2ONO —»
-liO'
—► Et2NNO + CH2=O (182)
N-Ннтрозогндразпны RN(NO)NH2 также обычно получают
ннтрознрованием чрезвычайно реакционноспособных N-алкплгпд-
разнпов азотистой кислотой; из М.М'-диалкплгпдразпнов получа-
ют ЫЛ'-дннитрозосоедннення [271]. Лучшим методом получения
М-нитрозогпдроксилампнов оказалось нитрозирование алкнлнит-
рнтом в щелочной среде, хотя они получаются и в слабокислом
растворе. В случае О-алкилгндрокспламннов RONH2 реакция в
кислом растворе протекает путем электрофильного нптрознрова-
ния по азоту, а при реакции с N-метилгидроксилампиом MeNHOH
происходит О-нптрозирование и затем миграция пптрозонневогэ
нона, причем эта стадия может стать скоростьопределяюшей в
сплыюкислых средах [272].
Первичные ароматические нптрозамины представляют собой
таутомерные формы арнлдпазогидроксидов ArN=NOH. Нптрозп-
рованне первичных ароматических аминов обычно дает соответ-
11°"Ь1 Д|,а30Ш1я (см. разд. 6.5.3 и 6.3.3). В качестве на-
ш'т пР0Д1’кта реакции метиламина с иитрознлхлоридом при
ческий ™”nePaTi'Pax был идентифицирован первичный алнфати-
ванием м,РЛ3аМ‘Ш [277а^ он Раадагается при -25 °C с образо-
ннтрозпрованн^т31'3' Не|<0Т0РЬ1е третичные амины могут прп
нильныеРсоедине1П1яВ|?ЩаТЬСЯ 80 ВТ0Р11Ч11ые нптрозамины н карб -
.той среде э?а оёяю, >равне,,,,е 183> t273ab однако в сильнок с
иый амин ппн L,”. 1 пРотекает медленно. Смешанный трети
•ствип тетрапптрометапа в присутствии ппрнд11
450
па в этаноле превращается во вторичный
пым пнтрозпрующпм средством для третий, Р аМИ"’ эФФект"в
З-гидропероксл-г-нитронроиан (уравнение 184“*[2Пб]°В СЛуЖИТ И
R’NCHR‘ R'n(NO)chr= r,*=cr2
Н20
-Rj-CO
—UNO
—> r'nh2 —r,nno
Et3N + Me2C(NO2)OOH Et2NNo + MeCHO S
Нитрозампны образуются при nacinenneu,.,, „ ,
дов питрозплфторпдом а также при восстановлен!™"трамииов
алюминием в щелочной среде, при реакции дналквлкарбамои °
хлоридов с нитритом серебра (уравнение 185) и при реакши
N-хлордиалкнламипов с NO в присутствии ионов железа (11) и!и
медн(1).
MeCN
Me2NCOCl + AgNO2 ------► [Me,NCOONO] —> Me2\’NO + CO, (185)
Алифатические и ароматические N-нитрозогпдроксиламины
RN(OH)NO можно получить пря взаимодействии реактивов
Гриньяра с оксидом азота. Имеется сообщение о том, что реак-
цию оксида азота с любым соединением, содержащим водород
при активированном к иптрозированию атоме углерода, в присут-
ствии сильного основания можно с успехом применить для полу-
чения ннтрозогпдрокспламинов [274]. В случае соединений с
активной метиленовой группой может происходить и дизамещеиие.
Известны еще две старые, но до конца пе исследованные реак-
ции — это получение N-нптрозофеиилгидроксилампна из нитробен-
зола и гидроксиламина в присутствии этокспда натрия и протека-
ющее с высоким выходом превращение N-нитрозоанилидов в
N-иптрозогидроксиламины под действием пероксида водорода.
Было предложено получать эти соединения обработкой арплдна-
зонпевых соединений раствором хлорида медп(Н) и сульфата же-
леза (II), насыщенным оксидом азота [275].
7.4.2. СВОЙСТВА N-НИТРОЗОСОЕДИНЕНИЙ
Алифатические вторичные нитрозампны Р,К—КО представ-
ляют собой вязкие жидкости желтого цвета или нпзкоплавкие
твердые вещества, устойчивые при комнатной температуре в тече-
ние нескольких недель и растворимые в воде ( е2 ,
т кип 154 °C- EUNNO, т. кип. 175 °C). Ароматические нитрозами-
ны нерастворимы в воде и разлагаются при перегонке при атмо-
сферном давлении (К-метнл-М-иптрозоапплни, т. пл. ’
т. кип. 128 °C при 19 мм рт. ст.). Продукты термического разло-
жения диарилпнтрозамипов образуются за счет радик ‘ *
[276], в присутствии кислорода получаются в иов
С-нптродиариламины. N-Ннтрозопроизводные первич
15*
пптив в высшей степени неустойчивы; судя по данным у<к
напротив, » метилнитрозамнп образуется при низких
оятХ По ранним литературным данным, нрн нпзких темв^
?vnax образуются твердые соединения, которые, ио-вадД*'
являются первичными ароматическими интрозаминами. НекОто '>;
Хада»л- » триазолплиптрозамины, »апр»мер (56), МоЖно Рв“
делить в кристаллическом состоянии 12776]; они устойЧ11вы “
комнатной температуре.
N—N
Ri—/ —NHNO
NR2
(56)
Интерес к нитрозаминам возрос в последнее время в связи
с выяснением их роли как канцерогенов, присутствующих в объек-
тах окружающей среды [278а]. Полагают, что ферментативное
а-гндрокснлпрование активирует нитрозампны. Образующиеся
при этом продукты, расщепляясь, дают дназогидроксиды, которые
способны алкилировать нуклеиновые кислоты; важную роль в
этом процессе, по-впднмому, играет поп дназоипя. Предполагают
также, что некоторые нитрозампны могут выступать как перенос-
чики нитрозогруппы в реакциях транс-нптрозпроваппя, а также
активировать электрофильные реакции. а-Гпдрокснннтрозамнны
не удается выделить из-за их быстрого разложения, но а-ацето-
ксиннтрозамины были получены [2786]. Они действуют как мута-
гены без мнкросо.мальной активации и поэтому чрезвычайно
опасны.
Несимметричные нитрозампны могут существовать в виде сме-
си плоских геометрических (А)- и (Z)-изомеров (57) и (58).Для
N-иитрозоэтилметиламина, например, отношение (£):(Z) = 4 :1,
^279сЯ] ^'ннтР030 ^'метиланнлииа обнаружен только (Е)-изомер
Ek Ме.
n-ч. ;n—Nx
Ме^ ХО
(57) (£) (58) (Z)
Предполагается поэтому, что каноническая форма R2N=N—О"
тв^ТпДеС°МЫЙ Вклад в СТРУК'ГУРУ этих соединений; это под-
<11 данвь,мп ИК-спектроскопии, дипольными моментами
60~кЛ«/ Н баРьеРами вращения вокруг связи N—N (76—
тнлам„*о™. В зг1в,1еимостп от окружения). В N-пптрозодиме-
124 пм- угол К'3 мВЯл составляет 134 нм, связи N—О—
меров диалкилнит Равен 114° [2796]. Равновесную смесь изо-
шш, 1|ротекающейРчепМИН°В м,о!кно подвергнуть фотоизомернза-
ние, в фотостаингшяп^3 ” возбужденное синглетное состоя-
[279в]. Рное состояние с преобладанием (Z)-изомера
462
п уф-спектрах интрозамннов постояnun
иецне при Л < 400 нм с максимумом, имеющим то^6™ П0|'Л0
"рн 340-385 нм (е« 100), обусловленным Т„0^^руктУру,
"Р также поглощение вблизи 235 нм (е« 700(0 №рехадом-
^„•-перехода [239]. В случае нитрозопро^звод^ых“Г
веских аминов поглощение, обусловленное л-*п*-пепехою^
является при больших длинах воли, а цри наличии Хмистых'
заместителе», препятствующих сопряжению, полоса состою
ствуюшая п-*п -переходу, как и следовало ожидать вновь сме
шается в область меньших длин волн. Длинноволновое поплоше’
ние (п-+л*-переход) зависит от природы растворителя- этот
факт связывали с межмолекулярным дипольным взаимодействием
На основании хнроптических свойств N-ннтрозохромофора можно
определять абсолютную конфигурацию исходных аминов; суще-
ствует правило секторов для этого хромофора [280]. В ИК-
спектрах нитрозамипов имеются интенсивные полосы прн 1421 —
1476 см-1 (vn-o), 1352 1367 н 1266—1290 см-1; частота валент-
ных колебаний группы N—О ниже, чем группы N=O |см.
разд. 7.3.1 (2)], что согласуется с приведенными выше доводами
об их структуре.
Существование (£)- п (Z)-изомеров доказано с помощью
ПМР-спектров [279а]. Например, в спектре днметилнитрозамина
присутствуют два сигнала равной интенсивности прн 6 3,76 и
2,96, которые относят к протонам метильных групп, находящихся
соответственно в транс- и цис-положении по отношению к кисло-
роду. Эти сигналы не сливаются вплоть до 180 °C вследствие вы-
сокого барьера вращения вокруг N—N-связн. В масс-спектрах
этих соединений [50, 281] обнаружены пики молекулярных ионов;
от молекулярного иона алифатических н ароматических соедине-
ний прежде всего отщепляется молекула NO. В случае алифати-
ческих нитрозаминов, однако, существенную роль играет н отрыв
ОН, происходящий, по-видимому, в результате перегруппировки
типа перегруппировки Мак-Лаффертн. Запись спектров нитроз-
аминов, в особенности ароматических нитрозаминов, следует про-
водить с большой тщательностью, так как в ионном источнике
масс-спектрометра нитрозамнны в значительной мере разлагают-
ся до аминов.
М-Арил-К-ннтрозогидрокснламины ArN(OH)NO представляют
собой, как правило, твердые, умеренно устойчивые вещества.
Так, например, N-ннтрозофенплгидроксиламин плавится при
59°С и разлагается прн 75°C; его устойчивая аммониевая соль —
это аналитический реагент купферон. Образование солен тако
типа указывает на кислотные свойства А-ннтрозогпдрокснлвмн
с незамещенной гидроксильной группой, для N-нитрозо- ф
гидроксиламииа, например, РА'а = 4,42. Простые И-алкнл-А-нит-
розогидрокснламины RN(OH)NO существуют лишь в водном
Растворе, М-интрозо-М.О-диалкплпропзводные КЩикр
более устойчивы (для ^ннтрозо-^О-днметилгадроксиламива
03
кп’С пин 30 им рт. СТ.). УФ-Спектры N-нитрозогидроксил
Т- К"пп сходны СО спектрами иитрозамипов, за исключением Т0Г01
а”п отсутствует малопнтенснвпая длинноволновая полоса.
Нитпозогпдразпны обычно малоустойчивы; известны ,1Х ал-
кппьпые п арильные производные различных степенен замещения.
7.4.3. РЕАКЦИИ N-НИТРОЗОСОЕДИНЕНИЙ
Простые нптрозамины можно восстановить до соответствующих
гидразинов цинком в уксусной кислоте, но в случае ннтрозампнов,
содержащих заместители, большие, чем пропильная группа, основ-
ными продуктами становятся вторичные амины (уравнение 186).
R2NNH2 <— R2NNO —> R2NH + N2 (186)
Для получения гидразинов в качестве восстановителя исполь-
зуют алюмогидрид лития; прп тщательном соблюдении определен-
ных условий реакции его можно применять п для высших нитро-
заминов. Эффективно идет восстановление прп использовании
такого реагента, как цинк — гидроксид аммония — карбонат аммо-
ния [282], а также прп каталитическом гидрировании над плати-
новым, палладиевым или родиевым катализаторами с добавками,
например, сульфата магния, которые сводят к минимуму образо-
вание вторичных аминов. Для преимущественного образования
вторичных аминов используют более энергичные восстановители,
такие, как амальгама натрия в подкисленном спирте, а также гид-
рирование над никелем Ренея [283] или палладиевым катализа-
тором с добавкой основания. Попытки дезоксигенировать ннтроз-
амины соединениями трехвалентного фосфора оказались безус-
пешными, а при восстановлении литионитом натрия пли литием
в жидком аммиаке промежуточно образуются дпалкилампноннт-
реиы (азамины), которые затем превращаются в тетразены или
биалкилы (уравнение 187) [284].
цг/
CH—N—СН
I
NO
Na2S2O4 Ме\ /Ме
------>- ,СН—N—CHZ
R*/ I \ri
N:
Me, Me
TH-CHZ
(187)
враща1отся''вТ|н|И азотно’1 кислоты вторичные нптрозамины
КО И с Хинь 1Р ,ИНЫ; эт,° жс "ревращеиие
и и ОТЛИЧНЫМ ВЫХОДОМ •
ситрифгоруксусной кислоты
водорода п трифторуксусиого
ХарЬаствоТах№еД'1НеМЯ’ обРа«е‘
пре-
. .,— > можно просто, глад-
90%) осуществить действием перок-
нолучасмой смешением пероксида
1 ангидрида в дпхлорметаие [285]
С011р0В0Ждаться ЭЛектро-
иитраминов можно исполь-
- ион нитрония в сильнокис-
454
парофазпом фотолизе N-нитрозодиалкиламииов первоиа-
образуются оксид азота и диалкиламино-раднкал (уравие-
нпе'188)-
(rCH2)2NNO —(Rch2)*n * * (RCH2)2 NH + RCH=NCH2R (188)
в то Же время пптрозамииы довольно устойчивы к фотолизу
кой фазе в отсутствие сильных кислот, а в присутствии хло-
8 *того водорода в метанольном или водном растворе основными
₽"од\ктамн являются ампдоксимы наряду с небольшим количе-
п₽ У альдегида и амина [286а]. Первичным процессом в этих
'стовпях является генерирование оксида азота и аминиевого
ндпкала R2NH : [2866] с последующим образованием HNO и
альдамина (уравнение 189); параллельно может также происхо-
дить восстановление до исходного амина.
NOH
/IV, НС1 II
Bu2NNO ~_hno' х [BuN=CHPr] —>• BuNHCPr + Bu2NH (189)
PrCHO
В присутствии кислот происходит фотолитическое присоединение
N-нитрозаминов к алкенам (уравнение 190) [287а], причем иит-
розогруппа обычно присоединяется к наименее замещенному
атому углерода. Аналогичное 1,4-прнсоедннение наблюдается и
в случае симметричных сопряженных диенов [2876]. Если воз-
можно внутримолекулярное присоединение, пятнчлениые циклы
образуются предпочтительнее, чем шестпчлениые; это открывает
Удобный путь синтеза пятичлеипых азациклических соединений
[288а]. Может происходить также и разрыв С—С-связи (уравне-
чпе 191). Если фотоприсоединение проводят в атмосфере кисло-
Рода, реакция полностью направляется па образование 1-амнно-
нитратоалкапов с отличными выходами [2886], хотя в случае
инов может наблюдаться перегруппировка, а нитраты аллило-
1Х сп,|ртов неустойчивы.
Bu2NNO + [ j] М £ (190)
(/NNO + Me2C=CMe2 —Me2C=NOH + Ме2СО + ( ^NH (191)
П+ \—/
ЯИя^цд де'1СТВП11 диметилсульфата, тетрафторбората триэтплоксо-
ХоДИт С) алкнлиоДидов в присутствии перхлората серебра пропс-
^'К+^’Д^олнроваипе вторичных интрозамннов, приводящее к
Ходом я Можно также провести, хотя и с низким вы-
11289а]'- JlKllJ,,1P°Baniie се-углеродиого атома в диметнлнптрозампне
1 в Настоящее время кислотность атомов водорода прн
•455
„ИПМ атоме хорошо изучена [289о]. Обработка N-нитро-
а и ИНОВ некоторыми ацилирующими агентами приво-
3O'N’ Хшешио пптрозогруппы на ацильную (уравнение 192)
В присутствии уксусного ангидрида N-нитрозо-а-аминокис-
[29UJ. в Щ - с образованием спдноиов (уравнение 193)
Zo Соответствующие нитрилы циклизуются в сидноиимины уЖе
Z действии хлористого водорода. В присутствии оснований не-
котовые соединения этого типа претерпевают 1,2-сдвиг ннтрозо-
Дуппы с образованием оксимов; по-видимому, эта реакция про-
текает с участием карбаниона как промежуточного соединения
NO
I но «с
PbNMe + AcoO ---—
(192)
zA
RX XCO2H
Ас2О
(193)
нитрозами-
А с
I
PhNMe
(65%)
IvA-P
RC<CZ
II
О
Катализируемые основаниями реакции некоторых
нов находят применение в синтезе. В определенных случаях гид-
рид натрия вызывает элиминирование HNO с образованием имина
[например, ArN=CR2 нз ArN(NO)CHR'], хотя возможны и дру-
гие направления реакции [293]. В некоторых случаях, например
(59), в присутствии оснований образуются диазоалканы (уравне-
ние 194). Водные минеральные кислоты медленно гидролизуют
нитрозамины до аминов; обычно для удаления образующейся азо-
тистой кислоты используют мочевину или ионы медн(1) или же-
леза(П) (уравнение 195). При этом замещенные анилины могут
подвергаться перегруппировке Фишера — Хеппа, приводящей к
соответствующему/г-нитрозамипу (уравнение 196).
NO
MeNCMe2CHjCOMe - °с"°ва'")|:
(59)
„ , НС1, Н2О
R2NNO--------—
RNNO
Мс2С=СНСОМе + CH2N2 (194)
(195)
RjNH + NOCI
RNH
(196)
NO
[1то эта реакция представляет собой
ч,‘Денптрозирования п илтрозпрованпя
Долгое время считали .,т
межмолекулярный процесс N-
456
„апо, однако лоздиее было доказано (2941
’Zpo«K“ " денитрознроваиия^д^1 4J> что Процессы
„'отекаюшие реакции, которым поДВергае^ь,'ь'е
^.витрозамкч. Детали внутримолекулярной мигпя°Т°ИИрс,ВаннЬ1й
сИдии (уравнение 197), остаются невыясненный кочевой
реак^ денитрозирования включает атаку П°-^ЮМу
интрозамин. Высвобождающаяся в ф,1Ла Па прото
мроваиня азотистая кислота обеспечивает возМХ°ЦеСсе ™тгр0.
алення реакции Либермана, которая го° °ЖН0СТь осущест.
мгрозосоедниения с фенолом в ко/щеНтр„РХХй В ИагРевании
те. При этом возникает Красное окрашЦнК “ сеРн°й кисло-
бое при разоавленпп н подщелачивании Р/0ДЯЩее в голу.
Згу пробу дают все нитрозампны и нитплтк. (yPas«enne 198)
С-яитрозосоединения. 1 ы> а также некоторые
(197)
(198)
При нагревании вторичных *-иитрозамщ^
субстратом в органическом растворите реакция идет по
зпрованпе [295а]. Полагают, что в э ДоЖНР было бы ожи-
свободнораднкальиому механизму [295 ]. предшеству-
дать, что в гетеролптических условиях эт°’р^“’в PJ4ae вро-
ет деиитрозирование, однако было устано . вшегося\ связи
тонированного М-иитрозо-М-метнлаиплина ( у ‘ также при
с исследованием перегруппировки ф,|ШСРа ~/ тиомочевнну ре-
иереносе нитрозогруппы от этого соединения азотистой
акция идет непосредственно, минуя стадию о ра лам)1Н также
кислоты [295в]. Протонированный N-иитрозод Ф гр,,ппу на
способен непосредственно переносить сц01° , процессом
^-метнлаиилнц; в случае других нуклеофилов « кислоты
V°>KeT конкурировать реакция с участием а связана-,
[2956]. С возможностью протекания таких*р
ск°рее всего, канцерогенность этих соединении. пРПГ|1пуют при
Некоторые простые вторичные иитрозамипы Р в эфн*
«пакой температуре с фениллптием или тРет'^ „боаботке водой
Р®’ давая N'-алкнллитпевые соли, которые р Пз0МетинпмнНЫ
(9чг?т^нолом подвергаются элиминированию P,u,-rei<cariiAP0'
96]. Обычно при стоянии эти имины образуют^ ре- , fi
TeWiH!b), их мкпо также уловить н по продуктам р
|,Мп соединениями (уравнение 199).
4S7
(КСНг)А:\'°
CIIjR
RCH=N—NPh
R
(199)
r пЛшрм случае, по-видимому, нуклеофильная атака по нитрозо-
rnvnne и введение лития к а-углеродному атому - конкурирую-
пне процессы. Последний преобладает прн использовании в ка-
честве реагента днпзопроинлампда лития [297а]; образующийся
я-татионнтрозамин может далее вступать в реакции с диоксидом
углерода бензилбромпдом, ацетальдегидом или бензофеноном
'(уравнение 200), давая соединения, которые можно затем легко
дённтрозировать [2976]. Реакция лптнноргаинческих соединений
с пеенолизующимнся нитрилами дает 1,2,3-триазолы, так, на-
пример, (60) образуется из N-иитрозодиметиламина и бензони-
трила [298]. При взаимодействии алифатических и ациклических
ннтрозаминов с различными реактивами Гриньяра, взятыми в из-
бытке, образуются трнзамещенные гидразины за счет алкнлиро-
вання по а-углеродному атому и нитрозогруппе [299].
R\ (иэо-PrhNLi R\ R\
N—N'O ------->- NO —> zN—NO (200)
X X X
/ ' Xu ' XE
E = CO2H, CH2Ph, CH(OH)Me, C(OH)Ph2
(60)
Выше уже отмечалась термическая неустойчивость N-нитрозо-
гидрокснла.мииов; главными продуктами их разложения являются,
а°'видимому, соответствующие С-иитрозосоедпнения. Катализируе-
nnnnvv5.’TOTaM11 Разложе||ие может приводить к разнообразным
ЭМ’ напР11меР' 113 ОЛ'-диметил-П-нитрозогндроксиламина
ние mwi? Melail0-’1> формальдегид и азот. Алкилнрова-
дяет oki-iiiun 'н'1тРозог11Лроксиламннов, например, метнлподндом,
(62)- пт, ° а Воз,М0Ж|1ых О-алкильных производных, (61) н
работке аткоксиХГТг‘''-олсУльФонилхлорида и последующей об-
новление N пито™ обРазУстся лишь изомер типа (62). Восста-
амин или искоъ»,»Г|’дР0КС1|ламинов обычно дает соответствуюшии
3o-N,O-Air3Tii4rH4nn„?l;i'>OKCHJlaMHH’ хотя восстановление N-интро-
(63). Главное а'ЛЮМОГ11ДР11Дом лития приводит к
нов — эт0 их 1ССК°м поведении N-нитрозогидрокснлами-
** способность образовывать комплексы с металлами,
1HI1OH выступает как бидеитатиый лиганд, как, например,
»р<а!
В (64)'
Ph—N—N
NO
I
OR2
Ri—N=NORJ
О’
EtNOEt
nh2
о:
'О
(61) <в2> (63) (64)
м
Пои нагревании N-алкнл-М-ннтрозогидразииы легко отщепля-
олекулу N2O с образованием аминов; М,№'-диалкил-.\’-нитро-
^пдпазпиы разлагаются более медленно и дают азосоединения.
В кислой среде М-арил-М-нитрозогндразины перегруппировыва-
ются в арилазпды [300] двумя путями; при одном нз них, не-
равном, происходит случайное перераспределение меченого ато-
ма азота и, вероятно, образование циклического интермедиата
(65Ь Г zNl
I Ar—iZ || ArNHNHNO
L Nd
(65) (86)
Главный путь перегруппировки состоит во внутримолекулярном
1.2-сдвпге, приводящем, предположительно, к промежуточному
\'-кнтрозосоед)1ненню (66). В случае М,М'-дналкил-М-нитрозогид-
разннов удается наблюдать катализируемую кислотами N-*N'-
миграцию нптрозогруппы прежде, чем пройдет гидролиз до исход-
ного гидразина. N-Замещенные N-нптрозогидразнны могут реаги-
ровать с альдегидами и кетонами, давая нптрозогндразоны по
незамещенной аминогруппе. М-Алкнл-Ы-ннтрозогидразины могут
также подвергаться N'-алкилированию и N'-ацилированию, а при
действии этилнитрнта в щелочной среде образуются нзоалканди-
азотаты (уравнение 201). Имеется лишь небольшое число данных
° восстановлении [301] и окислении нитрозогидразпнов; при вос-
становлении, по-видимому, происходит разрыв N—N-связи.
NO
I EtONO . „
McNNH2 -------> MeN=N—О’ Na++ NSO (201)
NaOMe
7.3. НИТРАМИНЫ
UieiTnrtBlI4HblM алифатическим нитраминам R—NH—МОг посвя-
3°P [302], о них говорится и в более общих обзорах [303].
7.5.1. МЕТОДЫ ПОЛУЧЕНИЯ НИТРАМИНОВ
тать^кя'06 ,|||1ТР0Ваипе аминов, протекающее, как принято счи-
зыва’етг К °°Ь1ч||ая реакция с участием иона нитрония, редко ока-
я ^спешным, хотя некоторые малоосновные первичные и
459
₽ -шины например (67) и (68), могут образовать Нитр.
^0Р’’чНЛпХе ароматических соединении как, например, при
амин. В сл>'^ Р офепплнчтрамниа из 2-нитро-, 4-нитро- „
получении 'образованию нитрамина обычно сопутствует
2,4-д1ШИтроаиплшк , 1 троваН))Я в кольцо, которые, возмож-
обра3°ВХют ,з нитрамина [304, 305]. Для получения перв,1ч-
ВОЗХ, шов широкое применение нашел подход, основанный
!1ЫХ нитрампне> Рстп амШ10Группы введением легко удаляемой
на снпжснш ос бодае част0 „спользуют метод, включающий
ацильнои зашит • 1 е уретана (уравнение 202); сходным об-
(ура»»»»е 203).
1 ----- HN(CH2CF3)2
(68)
no2
MeNCOjEt
HN(CH2CO2H)2
(67)
NH3
MeNHNOj (202)
ClCOjEt _ HNOs
MeNH2--------- MeNHCO2Et
MeNHCONH2 + HNO3 —> MeN(NO2)2
(203)
Алкилированием нитроуретанов можно получить необходимые про-
межуточные Ы-алкил-М-нитроуретаны. Для получения вторичных
нитраминов в качестве исходных веществ можно использовать
М,М-дизамещеииые амиды или сульфонамиды, так как некоторые
ацильные и сульфонильные группы замещаются ннтрогруппой при
действии нитрующей смеси (см., например, уравнения 204, 205)
[306]; с этим процессом конкурирует окислительное дезалкилиро-
вание, которое становится преобладающим в случае М,М-диалкил-
уретанов (уравнение 206).
АсгО
Me2NCONH2 + HNO3 ---------> Me2NNO2 + СО2 + NH4NO3 (204
(СГ3СО)2О
R2NSO2Ph + HNO3 -----—c > R2NNO2 (~ 100%) (205)
NO2
, HNOs I
Me2NCO2Et -------> MeNCO2Et (206)
или
... -30 до 0 °C или с NjOi прп—80°С —
прямой путь получения вторичных (но не первичных) нитраминов.
попвОВаНИе (у’^'ДИХЛ0Рам1,,,0В в избытке уксусного ангидрида
Реакция аминов c N2Os в четыреххлористом углероде
эфире при температурах от —Г" """
-- ulV|,nHjr
Нитрование М,Н-днхлорамнлов l „uCvhul у1^унги»и аи.нмг1**-
приводит к N-хлорннтраминам, которые можно восстановить Д°
первичных нитраминов (уравнение 207) [307].
Н0С1
RNH2 -------*- PNC’-.
Наличие хлорид-нона за
пример, хлорида цинка
460
HNOs I NaHSOa
—RNNO2 ---------------> RNHNO2 (2У7)
ЛС2О
счет введения в реакционную смесь, на"
или гидрохлорида амина ускоряет непо-
цеп-
ценное образование нитраминов при нитровании вторичных
<ов в уксусном ангидриде. Выход нитрамина при реакции ни
а)|" пиметнламмоння с уксусным ангидридом возрастает с 5 по
СТаппн' введении хлорид-пона; полагают, что реакция носит
Sfxapa^P (УРав"с,,,1Я 208-210)[308].
НС1 + HNO3 + 3/2Ас2О AcOCl 4-V2N2O3 4-2АсОН
AcOCI 4- R2NH —> R2NCI 4- AcOI-I
r2NC1 4-HNO3 4-Ac2O •—»- R2NNO2 + AcOCI 4-AcOH
(208)
(209)
(210)
Лучшим методом получения первичных нитраминов является
п|1Трованне первичных аминов действием нитрата ацетонцианги-
дрина в щелочных условиях (выход ^-50%) (уравнение 211); этот
реагент применим также и для нитрования диалкиламинов с не-
разветвленнымп алкильными радикалами [2646]. В прошлом в
качестве реагентов использовали другие простые алкилннтраты;
успешная модификация этой методики состоит в предварительном
превращении первичного ароматического амина в его литиевое
производное [309]. Окисление арендиазотатов, обычно трикалнй-
гексацианоферратом, представляет собой общий метод превраще-
ния первичных ароматических аминов в нитрамины (уравне-
ние 212). Выше уже упоминалось об окислении нитрозаминов
в нитрамины; для получения нитраминов можно также воспользо-
ваться реакцией, аналогичной приведенной в уравнении (185),
только вместо нитрита серебра взять нитрат.
ONO2
I ~он
EtNH2 4- Me2CCN -----> EtNHNOj 4-Me2C(OH)CN (211)
PhNH2
hno2
“OH I- K3Fe(CN)e
PhNj ------>- PhN20‘ ------------->- PhNHNO2
A 2. Il+
(80%)
(212)
7.5.2. СВОЙСТВА НИТРАМИНОВ
Простые нитрамины RNHNO2 и R2NNO2 представляют
как правило,
(MeNHNO2,
I- пл. 55 °C-
phNHNO2 т’
•*-> •• II v I . I, 1 IHOIIIHV IIVU4II u> - - . . - р — .
1мС1?.°1,Я1°тся в воде обладают слабокнелыми свойствами
№NHNO2> р/(а 6,1; PhNHNO2, рКа 3,75). Мопоалкилнптрамины
е1|ее летучи, чем вторичные нитрамины, при нагревании они мо-
N (~>РаЗЛаГаться (из метилпитрампна образуются метанол, N.N- и
-^Диметилнитрамнны и N2O), однако их можно перегнать
BaioTn°HH>KeilliOM Давлении. Арилнитрамины при нагревании взры
групп,?’ осоЕ’сино если в молекуле присутствуют и другие нитро
’ °ии неустойчивы и иа свету. Предполагают, ч
461
собой,
жидкости или нпзкоплавкие твердые вещества
. пл. 38 °C- EtNHNO2, т. пл. 6 °C; Me2NNO2,
Et2NNO2, т. кип. 48 °C при 0,5 мм рт. ст.,
пл. 46 °C). Низшие первичные нитрамины легко
пззчожспне первичных ароматических нитраминов в
термическое |> • |Очаст гомолитический разрыв связи N—П
растворе ПР» J R_N=NO2H [310]; в результате образуют.
а'"'-Ф°;Х ‘г^роксила и арилдиазотата. Некоторые вторичные
ся Р ^кие 'нитрамины подвергаются термической пере-
арОппшовке (уравнение 213) [276]. Известны также примеры
Хох мичсскойперегруппировки, однако их механизмы на»™,.
НИ? Огромный практический интерес к этой области
обусловлен тем, что нитрамины можно использовать как
чатые вещества и реактивные топлива [312].
no2
неясны
химии
взрыв-
CgMe иN
(n-MeC6H4)2NNO2 80O---^ «-MeCANH——Me
(213)
Амбидентный анион (69), образующийся при депротоинрова-
ннп первичного нитрамина, можно проалкнлпровать по азоту пли
по кислороду. Соответствующие соли щелочных металлов N-алки-
лируются низшими алкилгалогенидами, что создает еще одну
возможность получения вторичных нитраминов (уравнение 214).
Серебряные соли алкилируются по кислороду (уравнение 215);
при этом образуется смесь геометрических изомеров N.O-дпал-
кнл-ач«-нитрамннов, представляющих собой жидкости с более
низкими температурами кипения [313], чем для N-алкилпптрамп-
нов. При реакции нитраминов с диазометаном может происходить
N- ц О-метнлнрованне [314а]. Методы N-алкилнроваиия были
использованы для получения замещенных вторичных N-амппоме-
тилиитраминов [3146].
Г - 4/°'1
I R—N—N К—N=N
L \т \г.
(69)
(E1NNO2)' Na+ + Mel —> EfN(Me)NO2 + Nal (214)
(EtNNO2)’ Ag* + Mel —> EfN=NOOMe + Agl (215)
четыре ато-
длины свя-
такие же, как у С-ннтросоедипеиий.
..............С—N и N-N (для
В УФ-спектрах нитраминов на-
н 7000 для пенвшшиу "рИ/<320 "М (*«»«» 225-250 нм, в » 5523
В щелочном паствопр ™Р.ПЧН“Х нитраминов, соответственно).
в сторону больших длин волн за счет
Как в первичных, так и во вторичных нитраминах
"К1т •у™“,
зеи МО2-группы примерно такие же, как у ~
?вяз~?Яэ™К°ТОРОС УК0'’°,1С,"1е « С-...................
угол С—N—N c????TBy^'Jlr’»меР,ю 15%-ной двоесвязпости),
блюдается погчошешш ЯСГ 1 1, УФ-спектрах нитраминов на-
н 7000 для пе'рвпчп • Т1 < 320 ПМ 225~250 "*’• Е « 5500
В щелочном раствоое мя торпчных нитраминов, соответственно),
нов несколько смешяетг? о'МуМ ПОГЛОЩС"11Я первичных иптрамп-
462 ' в ст°рону больших длин волн за счет
„и,нця аниона. В ИК-спектре полоса поглощения снимет-
обРа3° валентных колебании нитрогруппы расположена при
р||Ч1)Ы 1304 см'1, асимметричные валентные колебания проявля-
|2^пИ 1550—1630 см для первичных и при 1500—1530 см-1
ютсЯ „7'пичных нитраминов. Диалкил-аци-нитрамины характе-
1лЯ ‘га полосами поглощения при 1544-1564, 1232-1252 и
Р'«10оО7 СМ-’-
некоторых нитраминов сняты спектры ПМР; химические
г, ппотонов при а-углеродном атоме составляют (6) 399
Д*2). 3,63 (CH3CH2NHNO2) 3,45 [ (CH3)2NNO2]Дез-
паниРУюШПЙ эФФект гРУППь| ~N(NO2)— схож с влиянием кис-
пода в О-алкнлпронзводных. Благодаря существованию взаимо-
действия между ,4N иитрогруппы и а-СН-протонами сигналы этих
лоотонов расщеплены или уширены [315]. В масс-спектрах прос-
тых нитраминов отмечаются пики молекулярных ионов [316]; в
спучае первичных нитраминов присутствуют также интенсивные
пики ионов с т/е 46 (NO2+) н 30 (NO+). Все эти ноны отсутству-
ют в спектрах простых вторичных нитраминов, которые более
охотно отщепляют радикал NO2*; к гетероциклическим системам
это не относится.
7.5.3. РЕАКЦИИ НИТРАМИНОВ
Первичные интрамины разлагаются водными кислотами. При
этом в результате превращении промежуточного карбениевого
попа (уравнение 216) образуются алкены, спирты и другие продук-
ты реакции с нуклеофилами; так, например, при взаимодействии
с соляной кислотой образуются алкилхлорпды. Для разложения
некоторых вторичных нитраминов требуются гораздо более кис-
лые условия [317], например 95%-ная серная кислота. При этом
происходит отщепление азотистой кислоты; в некоторой степени
протекает п гидролиз, приводящий в конечном итоге к нитроза-
мнну (уравнение 217).
RNHNO2 + Н+ —> if + N2O + Н2О (216)
।—► HNO2 + / \=N—/ / •---*• / \=О + CgHuNHj
(C»H11)2NNO2— \\—' '—' (217)
I—> (C„Hu)2NH —> (C0Hn)2NNO
Первичные или вторичные N-ннтроаннлпны при нагревании с
Рпз явленной кислотой подвергаются нптрамппнои перегрупппров
Дают главным образом соответствующие о-нитроарилампны
с неболыинм количеством пара-изомера. Ориентация пр
ф„]ов заметно отличается от таковой в случае перегруппировки
г 'ера ~ Хенна (см. разд. 7.4.3). Механизм нитрамншюи пер -
0Дной"РОВК" Не выяснсн. Существуют две точки зрения.
из них [318а] в пптрамшиюй перегруппировке уча у
463
I! так называемый кульбитиый механизм
о-ннтрптоиптермеднат
Хьюза (уравнение 2'8). °
PhX'H2-< — Ph—NH2—О—N=O
phNHNU* \q-
NHa
i ~*UL (218)
Ч/р-ю-N=O S“x^NO2
H
Согчасно второму механизму, предложенному Уайтом [3186],
происходит гомолитический разрыв связи н последующая реком-
бинация частиц в клетке растворителя (уравнение 219). Врядля
можно ожидать, что во всех случаях реализуется единый меха-
низм [318в].
PI1NHNO2 —* PhNH2NO2 —> [PhNHa NO2] —f
Н
клетка
(219)
Нитрамины обычно относительно инертны к действию основа-
ний, даже к оксиду натрия в кипящем этаноле, хотя первичные
нитрамины образуют соли, а некоторые соединения, в особеннос-
ти содержащие электроотрицательные заместители, разлагаются
прн нагревании (уравнение 220) [319]. Вторичные нитрамины
медленно разлагаются горячей щелочью до альдегида или кетона
п первичного амина с промежуточным образованием имина (урав-
нение 221).
R'R!CHNHNO2 ----------> R’R2C=O + N2 + Н2О (22°)
основание 1 1
R‘CH!NR!NO2 —NOJ 4- R'CHO 4-R2NI-I2 (221)
обычно
пвпвоммТкЗппптР°Ва""Я соле“ черничных нитраминов
точного соепине, Дукта“ Разложения предполагаемого промежу-
наряду°сСнеболь1пнм*~ аи,1ЛИИТрам|,иа (уравнение 222) [320]
действии меТ11лхлоо<1,щ''‘"ЧеСТВОм Г'1'ац11ЛЫ|ого производного; при
значительных количёствах^ПгN,ацплпр°чзводиоо образуется в
НОВ гипохлоритом иЛп' ОбРаботка первичных арплпптрами-
ArNClNO2, которые "РПВОД,1Т к N -хлорннтрамипам
тагов с хлором Эти также ПР" реакции арепдиазо-
4м Р Jr" «W'eKHa легко гидролизуются до аминов;
,||С Я также М.М-диметилнитрамин и N,0 диметилни^” Мета‘
рплнитрамина в этих условиях - катион бензолдиаздня™' Э Н3
R.COCI + R’NNO; Ag+ _> R’COOR2 + зд + AgC| (229.
Первичные нитрамины реагируют и с формальдегидом обоа
,уя продукты присоединения или конденсации в зависимости-
ловий реакции (уравнения 223, 224) [321]. ИМ0СТИ от
ХО2
н2о I
O2NNH(CH2)2NHNO2 4- СН2О ---> 02NNH(CH2)2NCH2OH
no2 no2
90%-пая H2SO. I I
rnhno2 + CH2O--------——> rnch2nr
(223)
(224)
Ы-(Хлорметил)нитрамины [322] являются производными гнд-
рокспметилнитраминов, атом хлора в них можно заместить на
различные функциональные группы. Некоторые первичные ннтр-
аиияы могут также вступать в реакцию Манниха и образовывать
различные продукты в зависимости от используемого амина
[323а]; реагировать могут оба атома водорода аминогруппы
(уравнение 225). Основания Манниха легко разлагаются прн дей-
ствии оснований на соли нитраминов, давая амины и формальде-
гид, хотя их удается стабилизовать в кислой среде [3236].
Г NO: I
2R'NHNO2 + CH2O + R2NH2 —► irXLCHAR1 J2 (225)
Нитрамины устойчивы к окислению; они подвергаются ступен-
чатому восстановлению. Так, при осторожном восстановлении
М-метил-М-фенилнитрамппа цинком получают нитрозамин, вос-
становление первичных нитраминов в щелочной среде (цинк и
аммиак, алюминий в щелочи или амальгама натрия) дает изоди
азотаты, которые в случае алифатических нитраминов ,“°ПТ
ст. превращаться в диазоалканы. При избытке щелочи
зиавлпвающего агента могут образовываться гидра _
Ц|,н''0Пле"Ие вторичных нитраминов в кислой сред . pHb[ ₽’
h °ы в уксусной кислоте, дает соответствуют)’ р пер.
8пчныхрое кол,,чество исходных аминов. При в°^”аях обра3уются
:оотв^теар°Л1аТ1|ческ,|Х нитраминов в этих.уел 'ятпаН1|Ртра-м11на,
Наппп»ТС1Ву10щие Дназоииевые ионы; в случае ж Нце нитр-
ам"иовСоб,°бра3уеТСЯ мета,,ол- Эперг1'чн0^ ВВЯЗИ „ образованию
aMlI1° обычно приводит к разрыву N-N-связн
в ” аммиака.
465
ЛИТЕРАТУРА
la p <1 S. Smith. «Open-chain Nitrogen Compounds», Benjamin, New
l96“’ ^м'пЬг^and H D. Springall, «The Organic Chemistry of nji-
,6 {'h T'nMV Sidgewick)». 3rd edn., Clarendon Press, Oxford, 1966, p 339°^»
Liittke, Quart. Rev., 1958, 12, 321. P' "9-
J0 ®' GH GBotler in «The Chemistry on the Nitro and Nitroso Gro
lr ed H. F<Xr,' Interscicnce, New York, 1969, part. l. p. 215. [Дж, г
В ku.: Химия нитро- и нитрозогрупп. Т. 1. Пер. с англ. М., Мир, 1972
5 Т^Sandler and IF. Karo, «Organic Functional Group Preparations», дса.
Hnmir Press New York, 1971, vol. 2, p. 383.
2 IF. D. Langley, Org. Synth. Coll Vol. Ill, 1955 334.
3 у Yost and H R Gutmann, J. Chem. Soc. (C), 1970, 2497.
J L Di Nunno, S. Florio, and PE. Todesco J. Chem Soc. (Cl, 1970, |433
r' A H Beckett G. R. Jones, and R. T. Coutls, Tetrahedron, 1976, 32, 1267
6 IF D Emmons J. Amer. Chem. Soc., 1957, 79, 6522, 5739.
7 I A Maassen and Th. J. de Boer, Rec. Trav. chim., 1971, 90, 373.
8a T Emery and J. B. Neilands, J. Amer. Chem. Soc., 1960, 82, 4903.
86. R. Stammer, I. B. F. N. Engberts, and Th. I. de Boer, Rec. Trav. Chim,, 1970,
89, 169.
9. /. Meisenheimer and E. Hesse, Chem. Ber., 1919, 52, 1161; /. Meisenheimer
and E. Patzig, ibid., 1906, 39, 2526.
10a. W. B. Dickinson. J. Amer. Chem. Soc., 1964, 86, 3580.
106.6. W. Wubbels, R. R. Hautala, and R. L. Letsinger, Tetrahedron Letters, 1970
1689.
11. 0. Touster, Org. Reactions, 1953, 7, 327—378.
12a. M. M. Rogic, J. Vitrone, and M. D. Swerdloff, J. Amer. Chem. Soc., 1971, 99,
1156.
126 I. G. Aston and M. G. Mayberry, J. Amer. Chem. Soc., 1935, 57, 1888.
13a.В. C. Challis and A. J. Lawson, J. Chem. Soc. (B), 1971, 770.
136.В. C. Challis and R. J. Higgins, J. C. S. Perkin II, 1972, 2365.
14. H. H, Hodgson and E. A. C. Crouch. 1 Chem. Soc., 1943, 221.
15 G. Cronheitn, J. Org. Chem., 1947, 12, 1, 7.
,c 0. Baudisch and S. H. Smith, Naturwiss., 1939. 27. 769; K. Maruyama, I. Ta-
mmoto, and R. Goto, J. Org. Chem., 1967 32, 2516.
L Blangey, Helv. Chim. Acta, 1938, 21, 1579.
C55"an Hoek’ p- E- Made, and В. M. Wepster, Rec. Trav. chim., 1958,
В. C. Challis, KJ, Higgins, and A. I. Lawson, J. C. S. Perkin II, 1972, 1831.
. G. Hoggett, R. в. Moodie, and K. Schofield, Chem. Comm., 1969, 605.
“ Wor Tetrahedron Letters, 1972, 1755.
225 / и m Lne,J' D' Mills’ and J- w- Ridd, J. C. S. Chem. Comm., 1976, 19.
226.1 H. Raid, частное сообщение.
E Rohs!!!, a'll.i O’Connor, J. Org. Chem., 1964, 29, 2012.
C REahom ! )nM-,Tedder, Proc. Chem. Soc., 1963, 13. 7,
870 ’ ' D' lenkms, and D.-. R. M. Walton, J. C. S. Perkin 1, 19?4.
Jnf Danfortli, and A. McKillop, J. Org. Chem., 1973, 38, 2088.
m. uuoes j. Org Chem., 1959 24 295
M. 5 CKhmas!h l-i ^hem’ Soc» 1956’ 167°' 1957
22,37. ’ T- H' Meltzer, and IF. Nudenberg, J. Org. Chem., I957’
C. W^Taytor TTF-,Dc‘umani, J. Amer. Chem. Soc., 1948, 70, 1516.
С. E- о4!\\ЛВг'с^т1 R. L. Wear, J. Org. Chem., 1962, 27 1№>
pQC-, I960, 139g ' Ha^eldine, Proc. Chem. Soc., 1959, 369; J. cb * *
' ^-К^а^н'^ !L,S- I^ss. Chem. Rev., 1968, 37,
. “С n. с. Зефир0в _ycn XHM1Ill> 196g> 37) 1243].
16.
17.
18.
19.
20.
21.
23. j
24. ,
25. i
26.
27.
28.
29.
30.
31.
32.
33.
466
62.
63.
64.
65.
66.
67.
68.
69.
3« {9ме8М074.У’ C' M'!"tWa,d' Л Л'- Ba/ier. HI, J. Amer, chem Soc.
C Me‘’'Wald a"d T' N' BUker' 111 ’ 1 ЛтСГ Soc,
, r fiuinr' H. G. Haulhat, and W. Pritzkow J ura|,( rh m
з [g Park and D. L.H. Williams, J. C. S. PerkhH 1976 828 4' 2®' Ж
,7 н C Hamann and D. Swern, Tetrahedron Letters 1966 44ni
н Diekmann and W. Liillke, Angew. Chem. Internal Edn W.8 7
эд E Midler and H. Metzger, Chem. Ber., 1957, 90, 1179 ’ 9ВД' 71 387‘ 388
,0 A. Mackor and Th J de Boer, Rec. Trav. chim., 1970 89 15. >5q
I I F. Farther and I. W. Lmnett, J. Chem. Soc. (A) 1967 1928 Я n и
court J. Mol. Structure, 1971, 9, 221. ’ ’ 1 R- D- Har-
,1 TH. A. J. 117. Wafer and Th. J. de Boer, Rec. Trav. chim 1972 or «к
43 R. Dietrich and D. C. Hodgkin, J. Chem. Soc., 1961 3686 ’ ' 65
44 I'. Hanyu and J. E. Boggs, J. Chem. Phys., 1965. 43, 3454
45 D. A. Dieterich, I. C. Paul, and D. У. Curtin. J. Amer. Chem Soc 1974 96
6372. M. Azonlay, B. Stymne, and G. Wettermark. Tetrahedron 1976 32 2961
J6. L. Batt, B. G. Gowenlock, and J. Trotman, J. Cliem. Soc. I960 2222
47 Л'. D. Vietmeyer and C. Djerassi, J. Org. Chem., 1970, 35, 3591 '
48. I. C. Calder and P. J. Carratt, Tetrahedron, 1969, 25, 4023.
49a. L.-O. Andersson, J. (Banus) Mason, and W. van Bronsti-iik J Chem Snr
(Л), 1970, 296. '
496. M. Wilanowski, L. Slefanidk, H. Fanuszewski, S. Szymanski and G.A. Webb
Tetrahedron, 1973, 29, 2833.
50а. J. Collin, Bull. Soc. roy. Sci. Liege, 1954, 23, 201.
506. G. Schroll. R. G. Cooks, P. Klenimensen, and S.-O. Lawesson Arkiv Kemi
1968, 28, 413.
51. R- Hohn, H. Schaefer, H. Hiibner, M. Wahren, W. Pritzkow, G. Lauterbach.
P. Fuide. and P. Herrmann, Tetrahedron Letters, 1965, 2581.
52. IF. Pritzkow and IF. Rosler, Annalen, 1967, 703, 66.
53. V. V. Erschov and G. A. Nikiforov, Russ. Chem. Rov., 1966, 35, 817 [В. В. Ер-
шов, Г. А. Никифоров — Усп. химии, 1966. 35, 1953].
54. С. Romming and Н. J. Talberg, Acta Chem. Scand., 1973, 2246.
55. F. A. L. Anet and I. Yavari, Org. Magn. Resonance, 1976. 8, 158.
56. F. B. Mallon/ and A. Cammarata, J. Amer. Chem. Soc., 1966, 88, 61.
57. R. K. Harris, A. R. Katritzky, S. Oksne, A. S. Bailey, and №. G. Paterson.
J. Chem. Soc., 196.3, 197.
58. К. M. Ibne-Rasa, C. G. Lauro, and J. 0. Edwards, J. Amer. Chem. Soc., 1963,
85, 1165.
A. Darchen and C. Moinet, J. C. S. Chem. Comm., 1976, 820.,
Thieme Veriag, Stuttgart, 1957, Band Xl/1, p. 490.
61- P- N. Rylander, «Catalytic Hydrogenation over Platinum Metals», Academic
Press, New York, 1967, p. 164. o . D ..
A- Schonberg, «Preparative Organic Photochemistry», Springer, Berlin,
I. I. G. Cadogan, Quart. Rev., 1968, 22. 222; /. H. Boyer, in «Nitrenes»,
°d. \V. Lwowski, Interscience, New York, 1970, P- 163
R- A. Abramovilch, S. R. Clialland, and E. F. V. Screen, J. Org. them.,
2705; J. Amer. Chem. Soc., 1972, 94, 1374. rh 1975 40.
A. Abramovilch, S. R. Clialland, and У. Yamada, J. Org.
B. Sklarz and M. K. Sultan, Tetrahedron Letters, 1972, 1319; R- -4. Abramo
vCa' Co“rt. and E. P. Kyba, ibid., 4059. Vasil'eva M. F. Le-
F A Ginsberg. A. N. Medvedev, L. L. Martynova, M. N- s> 1965
b^a,S. S. Dubov, and A. Ya. Yakubovich.EGemOem. (U.S.b.K,,
о ' '9l7 [В А. Гинзберг и др. — ЖОХ, 196э, Зэ, 1-- J- , |1ес)гоп 1965, 2L
273f Cr'№"- N- Г- Hep/inger, and В. L. Shapiro. Tetrahedron.
! Hamer and A. Macaluso, Tetrahedron Letters, 1963, 381.
59. A. Darchen and C. Moinet J. C. S. Chem. Coram., 1976, 820.
60. R. Schroter, in «Metlioderi der Organischen Chemie» (Houben-Weyl). Georg
467
74.
75.
, n R Penner Tetrahedron, 1971, 27, 6201.
70, G. T. Knight arid ' Л ' in «1,4-Cycloaddition Reactions», ed J u
71, /. Hamer and hrnaa. 4]g. s Needlem(m H
Academic Press, 5. G ^resze and j, Firl, Tetrahedron 1'чсй“«е
Kuo, Chem. Rev, ,9%%’ pranCoiie, M. Van Meerssche g’
a~ s“-15'°
»s- J- K' .fS 8ЛВй«
1563
' p- 'Ci **/. them'Ber.. I95S. SI 14,4.
% F Krlhnke Ange^v Chem internet Edn 1963, 2, 380.
7R Fr F Baldwin A. K. Qureshi, and B. Sklarz, Chem. Comm., 1968, 373.
7ЯЛ r F Baldwin A. K- Qureshi, and B. Srlarz, J. Chem. Soe. (C), 1969 iq73
V. Kelson S P. Kovaeic. J. C S Chem. Comm., 1975, 312.
ЯП E G Janzen Accounts Chem, Res, 1971 4, 31.
81 M. I. Christie, J. S. Frost, and M. A. Voisey, Trans. Faraday Soc, 1965, 61,
674
82a I. G. Calvert and I. N. Pitts, lr„ «Photochemistry», Wiley, New York, 1966,
pi 475.
826. H. A. Morrison, в cc Ir. p. 165.
83ai/. A. Maassen, H. Hlttenhausen, and Th. J. de Boer, Tetrahedron Letters,
1971,3213.
836. A. Mackor and Th. J. de Boer, Rec. Trav. chim, 1970, 89, 164.
83b A Mackor Til. A. J. IF. Wafer, and Th. J. de Boer, Tetrahedron Letters, 1967,
2757.
84. A. H. Dinwoodie and R. N. Haszeldihe, J. Chem. Soc, 1965, 1675.
85a. B. G. Gowenlock. J. Pfab, and G. Kresze. J. C. S. Perkin II, 1974, 511.
856.73. G. Rowenlock, G. Kresze, and J. Pfab, Tetrahedron Letters, 1972, 593.
86b.F. J. G. Broekhoven, Th. A. S. M. Bolsman, and Th. J. de Boer, Rec. Trav.
chim., 96, 12; 7. Pfab, J C. S. Chem. Comm, 1976, 297, 616.
86. P. W. Robertson, T. R. Hitchings, and G. M. Will, J. Chem. Soc, 1950, 808.
87. D. F. Wiemer and N. J. Leonard, J. Org. Chem, 1976, 41, 2985.
88a.S, R. Sandler and IF. Karo, «Organic Functional Group Preparations», Aca-
demic Press. New York, 1968, vol. 1, p. 411.
886. H. O. Larsen, в cc. Ir, p. 301.
88b. N. Kornblum, Org. Reactions 1962 12,101.
ВВг.См. cc. la, p. 391.
89. A. V. Topchiev, «Nitration ol Hydrocarbons and other Organic Compounds»,
Pergamon, London, 1959 [A. S. Топчиев — Нитрование углеводородов И лру-
9П л“dPo‘"Г»ски^ ™едН|1е|'1:й- М„ Изд. АН СССР, 1956].
п о °'!d К Ya- Shtern, Russ. Chem. Rev. 1976, 45, 721 [А. П. Бал-
91 С, Я' Штерн-Уеп. химии. 1976 45, 14281.
MacmnhnSN' ^ГС? [?adical Reactions in Preparative Organic Chemistry»,
macm llan, New York, 1964 n 213
Cten"* '? “"“«Ч fellSA' l”’f. W# Д1
p. 160. ’ A’ U b' SymP°siuin Series No. 22, A. C. S, Washington, ! J71’
а’з/622Л<?ИгГ' Л' M' IIul1’ S- Oolden, and R. L. Reitz J. Org. Chem, 1968. 33.
gijg pa ' ®
95в. H. Feuer and P A Anderson, J. Org. Chem, 1971, 36, 140-
36- P- E. P/elle, andp Org' Chem- 1966, 31. 3152.
97- M P. Buu-Hot j p Tetrahedron Letters, 1970, 699. e
1964. 1842. ’ Sabatkier, and P. Jacquignon, Bull. Soc. chim- Fran 125
125.
D<nes, J, Org. Chem, 1971, 36, 3641.
92,
93.
94.
98.
99.
468
, f G Bardwell and E. W Garbisch, J. Amer. Chcm s™ lo,„ „
^4 A Griswold and P. S. Starcher, J. Org. Chem кЛ S’- 82’ 3588
Ю б-Я/, Shvarts, V. N. Yarovenlto, M. M. Krayushkin ie'
|0L V V. Sevost’yanova Bull. Acad. Sci. 11. S. S. R?1976 25 and
R H Яровепко, M. M. Краюшкин, С. С. Новиков В n И/ Шварц,
Изв. ЛН СССР, Сер. хим. 1976, 1674]. ’ Севотянова —
д' Hassner, !. Е. Kropp, and G. J. Kent, J. Org. Chem I960 qi or„„
£, I G. Hoggetl, R. B. Moodie, J. R. Penton, and К Schofield
03 Aromatic Reactivity», Cambridge University Press, 1971. N lral,on and
,тл p B. D. de La Mare and J. II. Ridd, «Aromatic'Substitution- MUr <•
^Halogenation», Butterworths, London, 1959. ' Nltration and
miB IP. M. Weaver, in «The Chemistry of the Nitro and Nitroso г
3 ed. H. Feuer, Interscience, New York, 1970, part. 2, p i ry Ji.OrEUps*'
Химия нитро- п нитрозогрупп. T. 2. М, Мир, 1973. См с 51 Р' В Кн :
J Н. Ridd, Accounts Chem. Res., 1971, 4, 248.
j. IF. Barnett, R. B. Moodie, K. Schofield, and I. B. Weston J c S Pnrtinii
1975, 648. ' ' Lralnlt-
R, B. Moodie, K. Schofield, and P. N. Thomas, частное сообщение
j. IF. Barnett, R. B. Moodie, K- Schofield, J. B. Weston R. G Coombes
I. G. Golding, and G. D. Tobin, J. C. S. Perkin If, 1977, 248.'
R. G. Coombes and I. G. Golding, Tetrahedron Letters, 1976, 771.
V. Gold, E. D. Hughes, С. K- Ingold, and G. H. Williams J. Chem Soc
1950, 2452.
M. E. Kurz, L. T. A. Yang,
1973, 38, 2271.
0. L. Wright, J. Teipel. and
112a.G. A. Olah. Accounts Chem. Res., 1971, 4, 240.
104.
105.
106.
107.
108,
109.
110.
Е.
Р. Zahora, and R. C. Adams, J. Org. Chem.,
Thoennes, J. Org. Chem., 1965. 30, 1301.
D.
1126. G. A. Olah and S. J. Kuhn, in «Friedel-Crafts and Related Reactions»,
ed. G. A. Olah, Wiley-fnterscience. New York, 1964, vol. 3, p. 1393.
113. G. A. Olah, in «Industrial and Laboratory Nitrations», ed. L F. Albright and
C. Hanson, Л. C. S. Symposium Series, No. 22, A. C. S., Washington, 1976,
p. 1.
114. S. R. Hartshorn, Chem. Soc. Rev., 1974, 3, 167; R. B. Moodie and К Scho-
field, Accounts Chem. Res., 1976. 9. 287.
115. D. V. Rightingale, Chem. Rev., 1947, 40, 117.
116. T. G. Bonner R. A. Hancock G. Yousif, and E. R. Rolle, J. Chem. Soc. (B),
1969, 1237.
117. K. Olsson, Acta Chem. Scand. (B), 1975, 29, 405.
118a. T. C. Brilice, M. !. Gregory and S. L. Walters, J. Amer. Chem. Soc.. 1968.
90, 1612
1’86. Л4. E. Sitzmann, L. A. Kaplan, and I. Angres, J. Org. Chem., 1977, 42.
563.
119.
120.
121.
122
123
124.
125.
126.
127
128
129
130.
131.
132.
133.
134.
135.
W. Treibs and H. Reinheckel, Chem. Ber., 1954, 87, 341.
0. В Bachman and N. W. Cannon. J. Org. Chem., 1969, 34, 41-1.
M. IF. Barnes and J. M. Patterson, J. Org. Chem., 1976. 41. 73 .
' ,R Bid/, E. R. H. Jones, and G. D. Meakins J. Chem. Soc., i960. 2WI.
IP- D. Emmons, J. Amer. Chem. Soc., 1957, 79, 5o28
A. Keinan and Y. Mazur, J. Org. Chem.. 1977. 42, 844.
IP- D. Emmons. J. Amer. Chem. Soc., 1954. 76. 3470. ]96()
A- R Ward, С. Ь. Johnson, and J. G. Hawkins, J. '4009?
6- B. Bachman and T. F. Biermann, J. Org. Chem., ,-74'
Л 4' lGarkin and K- L Kreuz- J- Or~ C,hem"
Л- I. Meyers and J. C. Sircar. J. Org. Chem. l9o7-!‘, 2810
0- B. Bachman and R. J. Maleski. J. Org. Chem .1972n(h \|s ,976. 616.
Takahashi. M. Tokuda. M. Hoti, and A. Suzukt Sxntn . • |634.
Dunroth G. Brauninger and G. Neubauer O'em. Ber., .
I № and J- D X°se- Ql,art- Rev- l9A7' ' 3.075 40 <’138: H. H. Baer and
L Melton and J. E. McMurry. J. Org Chem.. 197o. 40,
G Л Ze 9mr^- ibid.. 1976,'41, 3474. eBrll/iov, and S. S. Novikov,
б- Л. Schvekhgeimer, V. A, Smimyagui, R- Sadl1
О v ы/Т j^^Borgaidt, and W. L. Reed. Chem. Rev.. 1964. M
£ В Kaplan and IL Shechter, J. Amer. Chem. Soc., 1961, 83, 3535. ' '9'
Л. А. Капмш, вег. 103в, c_^221. ]958
G A^OMand T. E. Kiovslty, J. Amer. Chem. Soc., 1968, 90, 6461.
ff T Aptin°M. EischepD. Becher, H. Budzikiewicz. and C. Dferassi, j. Л|11и
nor. vheM m'"Nibbling andTh. J- de Boer, in «Some Newer Physical Methods
,426' sYructural Chemistry», ed. R. Bonnett and J. G. Davis. United Trade S
i 9G7 ) 47 '
142n ^^Larkiiis', F. E. Saaljeld, and L. Kaplan, Org. Mass. Spectrometry, J969
143
144.
145.
146.
147.
148.
136.
137.
138
139.
140.
141.
142a.
4. H. P- Pao- B c n
i, R. i • „и""- .«о
Chem. Soc.. 1965, 87, 4888_
in
London, 1967, p.
47.
2, 213.
A. T. Нильсен, в
A A. Criswold^and P.'S. Starcher, J. Org. Chem.. 1965, 30, 1687.
A. К Hoffmann, A. M. Feldman, E. Gelblum, and W. G. Hodgson, J. Amer.
Chem. Soc., 1964, 86. 639.
/?. R. Eraser, Canad. J. Chem., 1960, 38, 2226.
T Simmons R. F. Love, and К. I.. Kreuz, J. Org. Chem., 1966 , 31, 2400,
3152.
cc. I г, с. 260.
149. Cm. cc. 143, p. 406.
150. L. I. Khmel’nitskii, 0. V. Lebedev, V. I. Slovetskii, and S. S. Novikov, Bull.
Acad. Sci. U. S. S. R., 1961, 627 [Л. И. Хмельницкий. О. В. Лебедев,
В. И. Словацкий, С. С. Новиков — 1\зтз. АН СССР, Сер. хим.. 1961, 678].
151. Л Trotter, Acta Cryst., 1959. 12, 605, 884.
152. L. I. Bellamy, «The Infrared Spectra of Complex Molecules», Chapman and
Hall, London, 1975, vol. 1, p. 335.
153. I. H. Beynon, R. A. Saunders, and A. E. Williams, Ind. chim. beige. 1964.29.
311; /. H. Beynon, M. Bertrand, and R. G. Cooks J, Amer. Chem. Soc., 1973.
95, 1739.
S. R. Robinson. С. H. J. Wells, R. B. Turner, and J. E. J. Todd J. C. S. Per-
kin II, 1976, 1363.
D. V. Banthorpe, Chem. Rev., 1970, 70, 295.
M. J. Strauss, Chem. Rev.. 1970. 70, 667; T. N. Hall and С. F. Poranski,
в cc. 103b. p, 329 [7. H. Холл, Ч. Ф. Поранский. мл., в сс ЮЗв с 254].
С. Moberg and О. Wennerstrom, Acta Chem. Scand., 1971, 25, 2355. _ ..
154.
155.
156.
157. . . ______ _____
158. M. R. Crampton, in «Advances in Physical Organic Chemistry», ed. V. Gold,
Acadennc Press, New York, 1969, vol. 7, p. 238.
mo ₽ F°ster,and R- K. Mackie, Tetrahedron, 1962, 18, 1131.
6?' A or1’ Л R- Norris' ai,d К- E- Russell, Quart. Rev., 1068, 22, 123.
Ri.h, D°rrenler,- L M- Ha,J- and R- " Thomson (Organic Chemistry of
162 Gm Vе Ra<!lca's», Academic Press. New York, 1968, p. 268.
' 37МВПЦ!г"‘ма'цВ' Ma’,el,'s' and F. I. Dubovitskii. Russ. Chem. Rev., 1968,
37 1443] ' Иа31Ш' Г' Б- Манелис, Ф. И. Дубовицкий — Усп. химии,
164. P'lE'Mo°le7andnk т он"’ Tetrallcdron, 1963, 19, Suppl. 1, 23.
165а. Е К Г eMs a J\ - Se‘d- ™га|^гоп Letters; 1976, 2283.
1656. C. W Hand Cd м M^er,SO11' Adv. Free-Radical Chem., 1975, 5, 101.
166. Cm. cc 82a „ 477 md C- DiPietro. J. Org. Chem.. 1977. 42,
H. J. Kulm Wtrn ’<A' Momson. 0 cc. lr, c. 165. _ mn.
1976, 824 ’ K' S!raalmann, and D. Scliulte-Frohlinde, J. C. S. Chem. Comm.
169 tMS' .^6-°44?7.P' J' M°,es' S' T' Reil1- and к- т- Т0!!1™. Tetrahedron Let-
170'. ?. Cornel,«T'end E''и;, С1’1'-'ГП'- l975. 5 A 49. , rhem,
1976, 47, 1. ' Chem. Rev., 1975, 75, 353; Pure Appl- Chc
- - . 1 —' ••• hi r inaitai \-/i I
Academic Press, New York, 1969 vol. 7 p. 238
₽ ncipinr n IZ 14--L- -c I ’ ’____
1968,
841.
167.
168.
470
r л В. M. Bolsman and Th. J. de Boer, Tetrahedron m,-
171- L к 60, S. 360. ’ lelrahedron, 1975, 31, l019.
>'2j! H Lund, in «Organic Electrochemistry», ed M M n
l'26 New York, 1973, p. 316. Barzer, Marcel Dekker
/ R. Hanson, Synthesis, 1974, 1.
Ar'r'zalinic1 C Li лл. ГТ ~ ~ 1 r\rr , —
174-
175.
176.
177.
178.
179.
180.
1816. T.-L. Ho and С. M. Wong, Synthesis, 1974, 45.
182a. H. K. Porter, Org Reactions, 1973, 20, 455.
1826J. P. Idoux, J. Chem. Soc. (C), 1970, 435.
183.
/ £ McMurray, Accounts Chem. Res., 1974 7 281
g Kirchhoff, Tetrahedron Letters. 1976, 2533 ’
B. Loubinoux, J. J. Chanot, and P. Caubere, J. Organometa„ic cheffl 19^
R ' 0. Hutchins, D. IF. Lamson, L. Rua, C. Milewski „„а a и
J Org. Chem., 1971, 36, 803. muewsltt, and B. Maryanoff,
Cm. cc. 61. |). 168.
J. F. Rniflon, J. Org. Chem., 1976, 41, 1200.
H. I. Barber and E. Lunt, J. Chem. Soc., 1960, 1187
D. C. Owsley and !. J. Bloomfield, Synthesis, ’1977, 118
H. Imai, Т. Nishiguchi, and К. Fukuzumi, Chem. Letters, 1976, 655- I D Ent-
wistle, A. E. Jackson, R. A. W. Johnstone, and R. P. Telford j. C S Perkin 1
1977, 443; H. Imai, T. Nishiguchi, and R. Fukuzumi, J. Org.’ Chem.' 1977, 42’
431. ’ ’ ’
184. H. des Abbayes and H. Alper, J. Amer. Chem. Soc., 1977, 99, 98.
185. /. /. G. Gadogan, D. S. B. Grace, R. R. R. Lin, and B. S. Tait J C S Chem
Comm., 1972, 520.
186. J. I. G. Cadogan, D. I. Sears, and D. M. Smith, J. Chem. Soc. (C) 1969
1314.
187. F. R. Atherton and R. W. Lambert, J. C. S. Perkin 1, 1973, 1079.
188. R. J. Sundberg, В. P. Das, and R. 11. Smith, J. Amer. Chem. Soc., 1969, 91,
658.
F. Freeman and D. K. Lin, J. Org. Chem., 1971, 36, 1335.
/. E. McMurry, /. Melton, and H. Padgett, J. Org. Chem., 1974, 39, 259.
N. Kornbliim and R. A. Brown, J. Amer. Chem. Soc., 1965, 87, 1742.
7. Simmons and К. L. Kreuz, J. Org. Chem,, 1968, 33, 836.
A. Hassner and J. Larkin, J. Amer. Chem. Soc,, 1963, 85, 2181.
4. S. Matlack and D. S. Breslow, J. Org. Chem., 1967, 32, 1995.
К /. Erashko, S. A. DShevelev, and A. A. Fainzil'berg, Russ. Chem. Rev.,
1966, 35, 719 [5. И. Ерашко, С. А. Шевелев, А. А. Файнзильоерг — Ъсп.
химии, 1966, 35, 1740]. o£> OCQ1
N. Kornblum and R. A. Brown, J. Amer. Chem. Soc., 1964 86, 2b81.
W. B. Hass and M. L. Bender, Org. Synth. Coll. Vol. IV, 1963, 3•
См., например: V. Kornblum, R. E. Michel, and R. C Kerber J. Amer. C
Soc., I960, 88, 5662; G. A. Russel and W. C. Danen, ibid.,5bt>3.
189.
190.
191.
192.
193.
194.
195.
Lin, J. Org. Chem., 1971, 36, 1335.
ion а 'к'ччч, rtiigtw. v^iicui. пне»
N- Rornblum, S. D. Boyd, and F. .
... 5783; N. Rornblum and S. D. Boyd, ibid., 5_7_8L^ 505. R Hen.
'”ng, F. Lehr, and D. Seebach, Helv. Chim. Acta,JpJ^^'-fctrahetiron Letters,
>977, 1 161.
196.
197.
198a
mor 7,"'"’ '-"''O 00, .juijo v. /I. Лиооег u.»w n. « .., ._ .
nn6'^’ Kornbliim, Angew. Chem. Internal. Edn., 1975, 14, TH. Q 92
>90- N. Rornblum, S. D. Boyd, and F. W. Stnchal, J. Amer. Chem. Soc. 1970, ,
enn л^З; N. Rornblum and S. D. Boyd, ibid., 5784
Oa. D. Seebach and F Lehr, Angew. Chem. Internal. Edn. .
snnr Eehr, and D. Seebach, Helv. Chim. Acta, .- • “
2006. 0. Seebach, R. Henning, F. Lehr, and I. Gonnermann,
2№ Bonenjant, and C. Barbara, Tetrahedron. Letters, 1972, 41.
202a A. McRillop and R. L. Robylecki, Tetrahedron, 1974 30, 136o.
203 н Stecmitz and F. A. Norris, J. Org. Chem., 1. . g3 28 215.
204 n £' Fi"kbeiner and G. IF. Wagner, J. Org. Chem.. DM.
205 p' St Fa‘r and E. Utbas, в cc. 103, p. 75. „. 3 nfl. „ ,7Q
206 P n' Eichtenthater, Angew. Chem. Internal. Eclr., . tjons 1959, 10. 179.
207 D ?• B^smann, D. Ginsberg, and R. %73 3597.
208 m' o' White and A1. M. Balzer, Tetrahedron Leiters. DfaHger and U - A 5 >
M- C. Rtoetzet, J. Amer. Chem. Soc., 1948, 70, 3571, W. K«
‘etrahedron Letters, 1967, 1177. 47J
209. /
210. "
211.
212.
213.
211
215.
216.
217.
218.
219.
220.
221.
222.
i- «я H Sftechter J. Org. Chem., 1968, 33, 1164.
Xashtta, T Yanami, and A. Yoshikoshi, J. Amer. Chem. Soc., 1976, 98
S Tsun а^П1Ге/-!««^ l971’ 27' 323-
S°C" 19571 7<l’ 25‘5'
r/j de Boer and I. P. Dirkx, в cc. 1 r, p. 487 [7. Дж. де Бур, Ц, п „
B cc Ir c 371] 11 приведенные гам ссылки. P ,
j Miller', Aromatic Nucleophilic Substitution», Elsevier, Barking, esscj
lJ^F Bunnell, Accounts Chem. Res., 1972, 5, 139.
$ Л1 Shein L V. Bryukhovetskaya, and 1. M. Ivanova, Bull. Acad с.<
USSR 1973, 22, 1543; L. V. Bryukhovetskaya, L. V. Mironova and
S Л1 Shein ibid., 1550 [С. Л1. Шейн, Л. В. Брюховецкая, T. М. Иванова-
Изв АН СССР Сер. хнм., 1973, 1594; Л. В. Брюховецкая, Л. В. Мироно-
ва, С. М. Шейн, там же, с. 1601]; A. Abe and У. Ikegaml, Bull. Chem. Soc.
Japan, 1976, 49, 3227.
P. Buck, Angew. Chem. Internat. Edn., 1969, 8, 120.
M. Rosenblum, J. Amer. Chem. Soc., 1960, 82, 3796.
J. H. Gorvin, Chem. Comm., 1971, 1120.
R. A. Jackson and IV. A. Walers, J. Chem. Soc., 1960, 1653.
223. E. G. Janzen and J. L. Gerlock, J. Amer. Chem. Soc., 1969, 91, 3108.
224. Al. E. Kurz and R. T. Y. Often, J. C. S. Chem. Comm., 1976, 968.
225. G. D. Buckley, J. Chem. Soc., 1947, 1492; S. Wawzonek and /. V. Rernpf
J. Org. Chem., 1973, 38, 2763.
226. У. Yost, H. R. Gutmann, and С. C. Muscoplat, J. Chem. Soc. (C), 1971, 2119.
227. G. A. Sftvekhgeimer, A. Baraitski, and M. Grzegozek, Synthesis, 1976, 612.
228. V. Jager and H. G. Viehe, Angew. Chem. Internat. Edn., 1969, 8, 273.
229. P. N. Preston and G. Tennant, Chem. Rev., 1972, 72, 627.
230. J. Leitich, Angew. Chem. Internat. Edn., 1976, 15, 372.
231. I. 97. Patterson and I. E. McMurry, Chem. Comm., 1971, 488.
232. J. E. McMurry and J. Melton, J. Org. Chem., 1973, 38, 4367.
233. Cm. cc. la, p. 455; 16. p. 83.
234. D. 11. R. Barton, J. M. Beaton, L. E. Geller, and M. M. Pecliet. J. Amer.
Chem. Soc., 1961, 83, 4076.
235. N Kornbluin, R. A. Smiley, R. K. Blackwood, and D. C. JHland, J. Amer.
Chem. Soc., 1955, 77, 6269,
236 li- DePuy, H. L. lanes, and D. 11. Gibson, J. Amer. Chem. Soc., 1972, 94,
239.
240.
241.
212.
243.
244.
2376 ? J- Mol. Spectroscopy, 1964, 13, 305.
238 с V°8oVn' J- C- S- chcm- Comm., 1973. 450.
RNHn Ru.and P- Bftaskar, Ref. 1(d), pp. 139, 146, 147, and 156,
R N , m mdu!- !under> J- Chem. Soc., 1954, 691.
L D\ r lmd ‘nr o'11 { Л H- MalHnson, J- Chem. Soc., 1955, 4172.
WK Seder 'tC‘cs‘n'rlM- SoC’ roy Sci- Li7KC 1953. 22- 285'
G r n ' J- Or8- Chem., 1963, 28, 125.
P Neogl andT^Ol N'^' Connun- J- Org. Chem,, 1969, 34, 4121.
R. XornbX Ler°®?n"’ J Chem' Soc- 1916’ 701i 1917’ 899-
P. E Car n -h "f r r r etnt J Amcr Chem. Soc., 1949. 7I’ 22G- II
1973, 2019 Л ’ S- G' Gowenlock, and C. A. F. Johnson, J. C. S. Perkin 17
M^wX^Adv °phnf' Г’ r°w,nle,J- J- Amer. Chem. Soc., 1962, 84, 2711.
"ions, in «Modern Хн ет'' 964' 2' 2G3; 77 V- Blackburn and С. I-
^strand Reinhold, Lndom^"^?^ Syiltl,<isis>>. ed- C- J' Timm°"S'
Chem. Soc.,7эб0,'8J2, 2640Л' l; 'Getter, and M. M. Pecliet, J Amer-
'™^'aZ: аЛХг®: юа48^п"“' p- o,,l'cl0’ D',h R'Bar
246.
247.
248.
249
472
p Kabasakalian. and E. R. Townley, J Org. Chem., 1962 27 941 я
250- ypghovic and T. Sriuc, Tetrahedron Leiters, 1976, 561 ’ ’ 2J 8-
251a.7. Rie!le and N. A. Moore, J. Org. Chem., 1972 37 414 „ „
-M. Surzur, Bull. Soc. chim. France. 1973, 2393. ’ 4 3’ M 'P Bertrand
^THoueR'nan and L^W. \£° CarfaSydrale chem^'g^
plhS^^CLeOn' 1 ° MarkS' md ° EmnWS’ J Chem.’
g 4 Mortimer, J. Org. Chem., 1962, 27, 1876.
A McKillop and M. E. Ford, Tetrahedron, 1974, 30 2467
« n\ Suzuki, K. Ishizaki, S. Maruyama, and T. Hanajusa, J. C. S. Chem. Comm
1975, 51. ’’
пе?! / W. Lown and Л. V. Joshua, Canad. J. Chem., 1977, 55, 508.
m t п.юПоНр ntifl P J. Bertsch .T Orcr Phom At лто^
D ‘aJscroggiu, 1. M. Riveros, and E. B. Wilson, J. Pl]..s )974 w
1376.
у M. Csizmadia, S. A. Houlden, G. I. Koves, 1. M. Boggs and / G Csizma
dla. J Org Chem 1973, 38 2281 ' ' ™
R. A. C. Carrington. Speclrochim. Acta. 1960, 16, 1279.
R. T. M Fraser and N. C. Paid, J. Chem. Soc. (B), 1968. 659, 1407.
E. Buncel and A. N. Bourns. Canad. J. Chem.. 1960. 38, 2457.
N. Kornblum and H. IF. Frazier, J. Amer. Chem. Soc., 1966. 88, 865.
Й7 D. Emmons and J. P. Freeman, J. Amer. Chem. Soc., 1955. 77, (a) p 4391
(6) p. 4387.
Ross, E. R. Coburn, and M. Finkelstein, J. Org. Chem, 1968, 33. 585.
R. Barton, M. J. Day, R. H. Hesse, and M. M. Pechet. J. C. S. Perkin I,
2252.
Fridman, F. M. Mukhametshin, and S. S. Novikov, Russ. Chem. Rev.,
40, 34 [Л. Л. Фридман, Ф. Л1. Мухаметшин, С. С. Новиков — Усп.
1975, 51.
„,7 / №. Lown and Л. V. Joshua, Canad. J. Chem., 1977, 55 508
J. Ouellette anil R. J. Bertsch, J. Org. Chem., 1976. 41’. 2782
2*g n n r^nnitin I Л4 Riveros nod F R r z-i---- .
259.
260.
261.
262.
263.
264.
265.
266.
S. P.
D. H.
1975.
267a.A. L.
1971,
химии. 1971, 40, 64].
2676.См. cc. la, p. 455; cc. 16, p. 592.
268. 7. H. Ridd, Quart. Rev., 1961, 15. 418.
269. R. E. I.yle, J. E. Saavedra, and G. G. Lyle, Synthesis. 1976, 462.
270a. B. <?. Challis and S. A. Kyrtopoidos. J C. S. Chem. Comm., 1976, 877.
2706 .7. A'. Keefer and P. P. Roller. Science, 1973, 181, 1245.
271. J. R. Perroll, G. Stedman, and N. Uysal, J.C. S. Perkin 11, 1977, 274.
272. T. D. B. Morgan G. Stedman and M. N. Hughes, J. Chem. Soc. (B), 6 ,
344. , ,
273a. G. E. Hein, J. Chem. F.duc. 1963, 40, 181; P. A. S. Smith and R. N. Loeppky,
J. Amer. Chem. Soe., 1967. 89, 1147. , , , . F.n ,q7(1
2736. B. Franck J. Conrad and P. Misbach, Angew. Chem. Internal. Edn., 197 ,
9, 892.
274. Cm. cc. la, p. 501.
™ n Miniski and R. Galli Chimica et Industrie, 1964, 46, 423.
277 г ^е1ге1' Chem- Ber- 1971 104’ 808 „ ,nrn oa 1541
277a. E. Mailer, H. Haiss. and № Rundcl, Chem. Ber., 1960. 93, 1541.
27«f n' Miiller and II. Haiss, Chem. Ber.. 1963. 96, 570.
2786 r' r' Magee and J. M. Barnes, Adv. Cancer Res. 1967, . sinskey, and
^L E. Baldwin, S. E. Branz, R. F. Gomez. PL Kratt, A.
279т r n Tannenbaum, Tetrahedron Letters. 1976 333. , j cpein 1974.
79a'„D- Cooney, S. K. Brownstein, and I. Г ApSimon. Canad. a.
52 ЗООй
279n r' ^acleinacher and R. Stolevik. Acta Chem. Scand.. 1969. 23, Comm.,
mJ- Michejda, N. E. Davidson, and L. K. Keefer, J.c.o.
280 r97R- 633
281 S a'id K. Prater, Tetrahedron. 1976 32 847 lrv i973, 7, 431;
lw .ts- н- " Jaffe. and F. Kaplan. Org. Mass Spe t
282. В 7' ^/’Lrnon and J. D. Cooney. Canad. J. Ch™-,g70 '1088.
°- T. Haye, and T. S. Stevens, J. Chem. Soc. (C), l»/«.
473
D Enders. T. Hassel. R. Pieler, B. Renger, and D. Seebach, Synth,
!976, 548. G ^„„tbardino, and R. C lliskey, .1. д|11ег fl
* .exAViS* »
285. IP. 0- an™ j. M. Lavanish. Tetrahedron Leiters, 19G4
286a' у L ChowSid., 1964, 2333.
2866 у L Chen,' J.'Org. Chem., 1967, 32
2873 M P. Lau ACI. Cessna, Y. L. Chow, and R. W. Yip, J. Amer. Chem.
1971, 93, 3808, .... „ ....
2876.1. .
l«is,
Soc
1378.'
1221;
296.
297.
298.
299.
300.
2IOq;
i. Soc.,
CtoK0°C. A Colon, and D. W. L. Chang, Canad. J. Chem., 1970, 48,
288a Г6!' C/ЮИ, R. 4- «• C. Menon, and S_ C Chan Telrahedron Letters,
288 1971 1545 T L Chow. R. 4. Perry, and В. C. Menon, ibid., 1971, 1549
9«ял К S Pillau and Г. L. Chow, J.C. S. Perkin II, 19/7, 93.
LK. Keefer and С. H. Fodor, J. Amer. Chem. Soc., 1970, 92, 5747.
2896 R. R- Frazer and I- K. Kg, J. Amer. Chem. Soc., 1976 98, 5895.
9Qfi E Yu Belyaev, M. S. Tovbis, G. <4. Sitboch, and Л. V. El tsov, J. Org. Chem
' (US. S. R.), 1976, 12, 462 [E. Ю. Беляев, AL С. Тоабис. Г. А. Субоч.
Л. В. Ельцов — Ж. орг. хим., 1976, 12, 466].
291 И7 Baker and IP. D. Ollis, Quart. Rev., 1957, 11, 15; F. Fl. C. Stewart, Chem.
' Rev., 1964, 64, 129.
292. H. U. Daeniker, Helv. Chim. Acta, 1964, 47, 33,
293 I E. Baldwin, D. H. R. Barton, N. J. A. Gutteridge, and R. J. Martin,
J. Chem. Soc. (C), 1971, 2184.
294. D. L. H. Williams, Tetrahedron, 1975, 31, 1343.
295a. C L. Bumgardner, K- S. McCallum, and J. P. Freeman, J. Amer. Chem. Soc.,
1961, 83, 4417
2956. В. C. Challis and AL R. Osborne. J. C. S. Perkin II, 1973, 1526
295b. 0. L. H. Williams, J. C. S. Chem. Comm., 1975, 375.
P. R. Farina and H. Tieckehnann. J. Org. Chem., 1973, 38, 4259.
D. Seebach and D. Enders (a) Angew Chem. Internal. Edn., 1972, 11. 301;
(6) ibid., 1975. 14, 15; Chem. Ber., 1975, 108, 1293.
D. Seebach and D. Enders. Angew. Chem. Internal. Edn., 1972, 11, 1102.
P. R. Farina and H. Tieckehnann, J. Org. Chem., 1975, 40, 1070.
E. Yu. Belyaev. M. S. Tovbis, G. N. Kuznetsova and A. V. El'tsov, J. Org.
Chem. (U.S.S. R.), 1976, 12, 405 [E. Ю Беляев AL С Товбис, Г. И. Куз-
нецова A. В. Ельцов -Ж. орг. хим., 1976, 12, 412].
A. I- Graefe, J. Org. Chem., 1958 23, 1230
ДА Гл™,"' Р- /vsl'in- and S- S- Novikov, Russ. Chem. Rev„ 1969, 38,
1448] Фридман, В. П. Ившин, С. С. Новиков — Усп. химии, 1969, 38,
сМ467С1-’м Р/ 4n5; uG- F Vri^hl в СС. 1г, р. 613 [Дж. Ф. Райт, в се. 1г,
Quart Rev' 1951 5^75 АЬГЕПЗ Sammlun§- 1912- |8’ ',59; А- Н- Lamberton.
Т. ^sh^and'f' Н J С' S' Chem' C(,,nm’ ,972' 641'
ed L F Albrinht ' я'ДА’ In «,ndustrial and Laboratory Nitrations»,
Washington lArl. p ЮЗ.' HanS°n’ A’ C S’ ^“Posinm Series No. 22, A.C.S.,
307. GHNRnn°stnart J1IAmer- C1’em- Soc- 1955' 77’ 2453'
308. G. F. WrtfM B L "r Д До WriRht, Canad. J. Res. (B), 1948, 26, 284.
E. J. Winters D В 833 1Дж. Ф. Райт, в cc. 1г, с. 467].
В. N. Biddle' f> s Ln^ni and C- Desai, J. Org. Chem., 1965, 30, 2471.
,1968, 31 j /• Davtds°n. S. H. Harvey, and A. F. Singleton, Chem.
Wiley.tnte^^c\M^,%s™ °’ Moiccular Migrations», ed. B. S. Thyagarajan,
C Urbanski «Chemi.r k' 197L 3. I>. 109.
Oxlord, 1964. ’ Ty and Technology o! Explosives», Perganton Press’
301.
302.
303.
304,
305.
309.
310.
311.
312.
474
)/ /.uinbertim and H. M. Yasul, j Ch,„„ c
.110' utltcrhalt and D. Thainer Arch. Pharm ior?" 1969 3q7
Unterl.ult and D. Tlianter, SynU,. "f™4’,.М 307 731 ' ®7'
3!^ //' Lumberion, I. O. Sutherland E W6'
315- L (B), 1968, 6; P. Hampson and А м№’ ancl H. м V ,
л g. Farndner and G. A. Webb, Tetrahedron^ iS' Chun. Comm^ In3 Chem.
S Bulusu, T. Axenrod, and
j/«• -Q i*»3ss 5
,17 c. Halstead and A. 11. Lamberton J Clmm c p c r°metry, |g70,
18a s' Brownslein, C. A. Bunton, and E. D Huah’ l952' 1886
N- While and R- Klink, J. Ore Chem Chem- Soc fqr.k
З н. s. White, and A. Fentiman, ibid., 1976 41 4',Jfi970' 35’ 965;
,,o„ n V. Banl/iorpc and !. G. Winter j C s’o ’Л ' ^‘4
j'g.'/.'Barro/l, M. I. Gillibrand, and A.' N. Lar^bert'o'1’i1912' J259-
£. H. White and C. A. Aujdermarsh Jr. j AmPVr Chem. Soc., 1951 iom
H79. en Chem- Soc , J?)6J 83,,fi
0. Woodcock, J. Chem. Soc., 1949 1635- uz r „ ’ ' '
E. L. Jenner, N. W. MacNatlghton'and C p' Mnv,!mmn’ #’' I- Horton
1950, 72, 3132; L. Goodman, ibid., 1953 75 3019 J' Amer ch™ Soc’
/. Major and !. Denkstein, Coll. Czech chem r
323a. С. C. Bombardier! and A. Taurine. Canad J Chm lor’'1^?' 3|' 2547-
3236.4. H. Lamberton, C. Lindley, P. G Ouston a?d’ J r' 923
Soc., 1949, 1641, ’ u™ 1- C- Speakman, J. chem
320-
321.
322.
ЧАСТЬ 8.
ИМИНЫ, НИТРОНЬЬ НИТРИЛЫ И ИЗОЦИАНИДЫ
8.1. АЗОМЕТИНЫ
ДД. ТЭННАНТ (University о} Edinburgh)
8.1.1. ВВЕДЕНИЕ
В настоящее время двойная связь углерод—азот как функцио-
нальная группа по своему значению в органической химии сопер-
ничает с двойной связью углерод кислород, а в некоторых
отношениях и превосходит ее. Различным аспектам химии азометн-
нов посвящено свыше 300 обзоров и учебников, В данном разде-
ле сделаны попытки представить исчерпывающую картину химии
соединений, содержащих двойную связь углерод—азот. Обсужде-
ние, однако, ограничится лишь ациклическими азометпнами и
циклическими структурами, в которых двойная связь угле-
род—азот не является составной частью ароматической системы.
Более того, и о циклических азометпнах речь будет идти лишь
постольку, поскольку их свойства будут определяться их ацикли-
ческим компонентом.
8.1.1.1. Общие методы создания С=1М-связи [2—4]
(1) Конденсация аминов с карбонильными
соединениями [2—5]
ппл^АГСНЧеСКИ*’ метод введения в молекулу углерод-азотной
с oawmuiT1311 состопт в конденсации альдегидов и кетонов (1)
миак" амины" '°ед1,неи,|ям11> содержащими аминогруппу (2) (ам-
ление элеме1’1товДволп1Л,аМИлЫ " гидРазииы)’. происходит отщеп-
нов (4) (огиАпа. и, й, °°Разование соответствующих азомети-
Процессы этого тяпаШц"^Фа’ 0КС|1МЫ’ гидРаз01ш) (уравнение 1).
нежности применения раньше представляли интерес из-за воз-
Дегидов и кетонов уДЛЯ °'1ИСТКИ, характеристики и защиты аль-
карбопильными соепни1и1ЮЕЛено . что конденсация аминов с
Д1,ю в целом пяпр „М" пРедставляет собой решающую ста-
жем. ниже); это CTHMvnuJ'0I,ecc0[i' катализируемых ферментами
476 стимулИрйвало в последнее время интерес к иссле-
о.гизма этой конденсации. На основании данных кине-
.пНю ,деХД едованпй 15. 7) предложен двухстаднйный меха-
'‘рск«х ,,ие 1) образования азометпнов (и обратной реак-
f". (уравне пКЛЮчаю1Пий образование тетраэдрического
к гнДР „арбииоламнна (3), который в некоторых случаях
1едната.’пить (например, при взаимодействии хлораля с ги-
.С1иЯ 8 см также ссылку [76]), а в других — обнару-
100КС»лама,иппЯх с помощью проточной спектроскопии ЯМР [8].
Y-n-r3
R2/ \н
|11,змих ПЪ’)
цнн И* -
ннтер!ме.
'<амнном; см- такЖе ССЫЛку 17бВ.
5ь% растворах ° ‘
К'\
ХС=О + R3NH2
Rz/
(1)
(2)
(1)
R‘\ R,\°H /н
XC=NR3 + Н2О V-.V
Rz/ RiZ \R3
(4) (3)
Ri, R2 = H, Aik, Ar: R3 = A!k, Ar, OH, OR. NHR, Hal
Образование азометпнов, как правило, катализируется кислотами.
Скорость конденсации обычно тем выше, чем больше нуклеофиль-
ность аминного компонента, она сильно зависит от значения pH,
оптимальные условия —pH 4. В нейтральной среде скоростьопре-
деляющей стадией является дегидратация карбииоламнна, тогда
как в кислой среде — его образование. При конденсации с гид-
роксиламином или гидразином равновесный процесс (см. уравне-
ние 1) в значительной мере смещен вправо и прямо приводит к
образованию оксимов и гидразонов. В реакциях конденсации кар-
бонильных соединений с аммиаком и аминами, напротив, равно-
весие сильно смещено влево; для получения имина или основания
Шиффа необходимо вести азеотропную отгонку воды. На практи
ке из-за склонности карбиноламинного интермедиата к полные
рнзации получить имин при реакции альдегида с аммиаком i.
солями аммония не удается. „ „
Альдегиды и диалкилкетопы гладко кондеиснру₽
иымп аминами, давая соответствующие основания ФФ .
как арндкетоны обладают относительно низкой Реакди , и
ибпостыо, в этом случае требуется большее врем Р . т
вменение катализаторов (протонные K,IC]™™’TbZ есд„ прово-
пнт ’ ИнеР™ость арилкетонов удается преод . ’ в жиД.
,гь реакцию с производным амина и щелочного' ‘ ’ g л п0.
* аммиак С помощью этого метода из бензофеио а №
его анпл с выходом 50%. Основания Шиффа,
Формальдегида или алифатических альде ' ’ 0На идет п0
к полимеризации, причем в последнем с. у
, 47 (
,M11..ml)n. В TO же время шиффовы Ос1!
„молы'01' к .„‘ьтегидов в кетонов устойчивы. Обра’’
111В- из арО‘'1!|Т"Чт'")\а из альдегидов п кегопов Ч;|СТО приводит
тЛХапИ'^^рёо^омеров (см. разд. 8.1Л.2), Вдто£
ваНЛсИ С««- “ «7“ выделить порознь из-за низкого эмерГетиче.
редко Удае,переходов.
„ вза1П'' конденсация легкодоступных у. Вд)
Внутрпмолекуляриа H))fi (7; «=1 пли 2) представляе1
. --------- \ ---- ( шестичленных циклических
iii путь 22'1" ил„ 2) [9]. Примерами такого
X основании I , и представленные уравнения-
ыизации служа» к
НГ1Я
b„—.
к смеси син
очень i
ского барьера
б-аминокарбоппльных
собой общий путь синтеза пяти- ц
шиффовых с-”"”’1
рода цпк:.____
ми 2 и 3.
(СНЛСОзН
(СНг)л«Н8
(6)
Н2С—СН2
Ас“ л г/ \сн 1
------> АгС,^ /(СНгрг
о nh2
(7)
Ar s= м-РОС-Н» п = 1
Ar (2)
N
(8)
нлн 2
r'ch=ch\cor’
+
r:cik
\vo2
R’-
основание Р!СН—СН2
-----------I I
R3CH COR2
''noh
Ni, M2
Zn.HC)
•R2
(31
R'—'
N
Основания Шиффа выделяют также как промежуточные со-
единения в ряде важных, хорошо известных синтетических про-
цессов, в том числе при гомологизации аминов и при синтезе
альдегидов. Так, образование азометинов — ключевая стадия в
гидрогеиолитнческом алкилировании аммиака и первичных ами-
нов альдегидами и кетонами над никелевым или платиновым ка-
тализатором [10], представляющем собой общий метод синтеза
пДкяпё1Х|1п “™РИч,1Ь1Х алкиламииов. В родственной реакции
пегниами „ „2 амм»ак и первичные амины алкилируются аль-
служит и пллЛ°НаМП В пРисУтс,1'впп муравьиной кислоты, которая
вайлера - Кларм°ВГ1Т0еЛ1 И " расТв0Р”т?лем- в модификации Эш-
та заменена гЛЛ /’ ’ Реакции Леикарта муравьиная кисло-
полагаемым» hm-J° Ф°РмальДеГ11Да и муравьиной кислоты. Пред-
иии аминов из я Рмелчатамн при восстановительном образова-
ла ZZ ТсобХКСё::ИАОВ (УРав"е-е 4) являются имои.
пенных кетонов [191 1РекомеилУется Для аминирования затруД-
ними соединениями и'|,,,,Иевые катионы являются промежуточ-
сндролпза, используем» РсакД,|ЯХ последовательного окисления "
в альдегиды взаимо»е&Х ДЛЯ пРевРаш-е11ия арнлметилгалогениде11
пня Соммле [131/ МпС™ем с гексаиетилентетрамнном (реак’
npoMexv/L Г“Ж"° “«Делить основания Шиффа, являю
альдегидов путем впг,.-," соедниеш|ями в эффективных синтезах
478 ановления Иитрнлиевых солей триэтплси-
1141 ИЛИ восстановительного аминирования карбоцовы>
с1‘1СПользова11ием лития в метиламине |15). '
MC3SIN3
:с^о----------
R30.
। ,OR3 HN3
„/ X0R:
N3 ыАщ, [R3O4 мн
Г/
R'
R2
-«ЮН
lT
R\
,C=NH
R’v л'н2
R
R
В настоящее время образованию (и гидро™™!
Ш11ффа стали придавать особое значение. Было
„О способность пиридоксаль-5-фосфата выступать KodX' n 6,1
ферментативных превращениях а-аминокислот (напримеп пт
рацемизации, а- и р-декарбоксилироваиин [17], ретроальдатьХ
распаде, а.р-элнминпровании и трансиминировании Г18П ™
,,ена пониженной электронной плотностью в a-положении амино’
кислоты вследствие образования основания Шиффа. Примеры та-
кого рода процессов а-декарбоксилирование и а,В-элиминирова-
ние —приведены па схеме 5. г
(йуСНО)
У
Н—С—X
I
Н—С—СО2Н
N=CHpy
-со2,
-Н’
(5)
н—C-Qc
г-1
с—СО2Н
N=CHpy
с-со2н
N=CHpy
встут 11 ВтоР|,чных аминов (перхлораты и
11мш,,‘Ют в Реакцшо с альдегидами и кетонам
иссые соли (уравнение 6); этот метод >
фторбораты) легко
,н [2, 19, 20], давая
особенно применим
479
кпуиномасштабного синтеза этих универсальных ci))ITeTll
Д , г ш гермедиатов. Имшшсвые соли - ключевые интермедиат;;
пп а >юалкилировании соединении с активным атомом вод^11
Л вза одейетвпем с альдегидом пли кетоном и вторичным а£„.
ном в кислой среде (реакция Манниха [21]); реакция Иаход,„
широкое применение в синтезе [22].
R\ R3\*/11
х Ж X"
R;/ R</ \1 R2/ XR’
(Ю) О’)
Коп тет ания [2, 23, 24] альдегидов п кетонов с
амином (образующимся in situ из соли прн действии
Н О-з:>мешснным1> гидрокспламинами, приводящая к
C=0 +
R'-
R3
X"
(6)
гидроксил-
основания)
оксимам и
О-замещенным оксимам, обычно протекает легче, чем конденса-
ция аминов с карбонильными соединениями. Окснмнрование по-
добно образованию шиффовых оснований катализируется кисло-
тами, но его обычно проводят в буферном растворе (например,
в фосфатном буферном растворе) при pH ~ 4, чтобы обеспечить
оптимальную скорость реакции. Окснмнрование стерически за-
трудненных кетонов рекомендуют проводить в основной среде.
Вместо гидроксиламииа для оксимирования альдегидов и кето-
нов можно использовать дпнатрневую соль гпдрокснламиндпсуль-
фоновой кислоты HON(SO,r Na+js, которую можно легко приго-
товить in situ пропусканием диоксида серы в водный раствор ни-
трита натрия и бисульфита натрия.
Из альдегидов и кетонов образуются обе стереоизомерные (сип
и анти) формы оксимов (см. разд. 8.1.1.2), преобладает более
термодинамически устойчивая анти-форма. В отличие от син- и
анти-форм шиффовых оснований (см. выше) син- и анти-изомер-
ные оксимы могут существовать порознь; часто удается провести
окснмнрование в контролируемых условиях, так чтобы отдельно
получить каждый изомер.
Альдегиды ц кетоны конденсируются с гидразином, давая не-
замещенные по атому азота гидразоны или азины, причем азины
часто являются основным или единственным продуктом. Для об-
гтпп^аИ11Я Ы-"еза“адеш1ых гидразонов необходимо полное отсут-
К11СЛ0Т 11 ИСпОльз°ванне большого избытка гидразина. N-He-
пиен'^Т'"’10 Г||дРазоиЬ1 могут быть получены и обменной реаК'
в клп1поНа С 1идРа31|ном- Гидразоны бензальдегидов, содержащих
Т1пя'мпппЛ^иКТрОНОДОИОРиую ГРУППУ (например, гидразон п-диме-
са'мопоо1пкс13аЛЬЛеГНДа) отл«чаются особой неустойчивостью и
ЗОНЫ бр1Па’п|Ь-Ю Д|1СПР(,ПОРЦ|10Нируют В ЭЗШ1Ы, ТОГДЯ КЯК ГПДрЗ’
устойчивы (’||япп7'Д01! с злектроноакцепторнымп заместителями
К самопроизвольном^;’ 1'|д₽а3011 'г-иптробензальдегида не способе
реакцинРс гидпазпип образова11шо азина). АлкиларилкетонЫ пр
в присутствии У,,, „ °М Е ОтсУтствпе кислоты дают гидразоны.
с. оты азины. Диарнлкетоны слабо реагирую
,тализаторы дегидратации). емпературы,
' Моно- [25] и несимметричные дизамещеииые гидпазИии
г„руют с альдегидами и кетонами однозначным образом реалию
асто проводят в кислом среде и выходы соответствую^ х гид„
а30нов высоки. Аналогично реагируют «-толуолсульфонилгвдр
а;Ш1(тознлгидразин) n-MeC6H4SO2NHNH2, семикарбХ
NH,CONHNH2 II тпосемнкарбазид NH2CS\HNH2. ₽-Галоге₽нкето
11Ы и альдегиды (12а) или основания Маиниха (126) реагиоуют
с гидразином и моиозамещеннымн гидразинами (13) давая цик-
лические гидразоны (Д2-пиразолины [26]), например (14а) и
(146) (уравнения 7). Аналогичные реакции с гидроксиламином
ведут к Д2-изоксазолпиам [27].
R\
zR2
r1—с—сн2—с/ +h2n—NHR5
II |\R3
О R’
(12)
(a) R2, R3 = Me; R4 = Cl; R5 = H;
N\ /
R5N R’
(13) (14)
(6) R2, R3 = H; R* = NMes; R5 = Ph
(?)
Альдегидный пли кетонный компонент в рассмотренных выше
реакциях конденсации с успехом может быть заменен «замаски-
рованным» карбонильным реагентом (гидрат альдегида нлн ке-
тона, ацетат, ацеталь, тиоацеталь, простой эфир енола, дисуль-
фид и гелрдигалогенпроизводное), в особенности если исходное
карбонильное соединение неустойчиво. Так, дпэтилацетали гладко
реагируют с аминами и дают соответствующие основания Шнффа
с хорошим выходом, а при реакции простого эфира енола с гид-
рохлоридом гидрокспламнна в присутствии эфирата BF3 получа-
ют^ соответствующий оксим. Использование ге.и-дпхлор- нлигел-
Аибромпронзводиых особенно оправдано в тех случаях, когда ис-
ходные карбонильные соединения слишком вяло вступают в кон-
•Waunio, как, например, в случае ди арил кетонов. Так, дихлорид
(*а) при нагревании конденсируется с этплкарбаматом (16). Да’
®ая бензофенони.мин (17) (уравнение 8). Аналогично реагируют
1н??Ч1,Ые амины с дннодметаном, давая нминневые со.
)-*(19)] (уравнение 9).
Ph\ гага- р1\
\ci2 + NH2CO2Et ---------~► C=N’H + EtOH
pi/ -с°2 pi/
(15) (16) (17)
\ С"212 > / \\=СН2 Г
\ / — 111 \ /
(18) (19)
'G Зак. |05
(8)
(9)
431
,„nro псимеров конденсации первичных амидов с
Известно IKM’O Р ^„водящей к ацн.-пшннам [28]. В то
альдегидами и м- вступают такие реагенты, как этил-
Же вре«я- в хпорами1) NH2C1 п сульфипампды RSONH2.
карбамат - этого т1и1а приведена конденсация
в качестве (20) — с суд,,
рсакниоииосиос 01ЯЩая к карбпноламииу (22), который
фпнампдам» (_ ') • в алК11Л1|дСнсульф1ИИ1Мид (23) при дей-
можно дсп‘д) „„г.иппда или фосфорилхлорида в пиридине; вы-
'3»-7№> 1291 <”»
неп,1е 10)- 0 он о г _ о
Р г В F*C\ I II ЬС\ II
г,с\..о + >«Д-« - • . с>-я-5-’
F.c/ F3C/ F*c (10)
(20) (21) (22) (23)
[2, 4]
(2) Реакции иминного обмена
Возможность синтеза азометннов, основанного на реакции
иминного обмена, обусловлена равновесной природой реакций кон-
денсации аминов с карбонильными соединениями (ср. уравне-
ние I). Так, анилы R2C = NAr реагируют с гидроксиламином
или гидразинами и дают в результате иминного обмена соответ-
ствующие оксимы и гидразоны; в свою очередь, при реакции ок-
симов с гидразинами получают гидразоны. Равновесие часто мож-
но сместить в нужном направлении, особенно если аминный ком-
понент, подлежащий замещению, более летуч чем вновь вступаю-
щим. Отсюда следует, что имины R2C=NH будут реагировать с
ампносоединеииями с преимущественным отщеплением аммиака и
образованием других азометннов (оснований Шиффа, оксимов,
в гп«3|0Н°В ' ^Т° пРевРаш.ение особенно целесообразно применять
нов кпЛимннов- являющихся производными затрудненных кето-
иую конденсацию6Д0М° С ТРУДОМ будут встУпать в непосредствен-
котопомТлин и»пяН0 получ”ть и таким траисимипнроваиием, при
щий в состав я/ 01nlJlblibiii компонент замещает другой, входя-
Регенерация кетоиовТнНчапКПрНМер°М ЭТ°Г° типа Реакцпй служ|1Т
в из оксимов при действии формальдегида.
'ЦШг конденсации по метиленовой группе [2—4]
Ароматические ни
мУю основаниями тр°зввоеД|1иепия (24) вступают в катализируе-
леновой группой (25- у|1сац’110 с соединениями с активной мети-
ли) (Реакция Эолитгя a * ~ электроноакцепторные заместите-
соедивеиця (производна гКСа^’ образующиеся промежуточные
тировать в азометины Д7УидР0ксиламииа) (26) можно дегпдра-
482 11лп окислить в нитроны (28) (урав-
„енпе И)- Эта реакция каталнзуется различными основаниями
NaOH, К2СО.? пиперидин); в нее вступают различные соеХ
„я с активной метиленовой группой, в том числе ареиметилциа-
)ДЫ, малоновые эфиры, fl-кетоэфиры, Р-дикетоны, флуорены цик-
топентадиены, а также активированные замещением в ядре ме-
'тнларены (например, 2,4-дпнитротолуол). При использовании для
конденсации сильноосновных катализаторов образование азомети-
нов преобладает над образованием нитронов. Следует, однако, от-
метить, что реакция Эрлиха — Закса имеет ограниченное приме-
нение из-за относительной малодоступности исходных ннтрозоаре-
нов н низких выходов вследствие конкурирующего процесса обра-
зования интронов.
ArN=O + п2с
основание
(24) (25)
-* Аг.\’—СН
I х
он
(26)
-н2о | (01
(11)
/Х
ArN=C'
(27)
* /Х
ArN=(/
о-
т
При реакции соединений с активной метиленовой группой (29)
с различными ннтрозирующими агентами (NaNCh. Н+; N0C1;
RONO, NaOR или НС1; N2O3) при 0—50°C (или ниже) образуют-
ся неустойчивые нитрозоалканы (30), превращающиеся обычно
в оксимы (31) [2, 24, 30] (уравнение 12). Следует избегать из-
бытка иитрозирующего агента, иначе может произойти дезокси-
мирование в соответствующее карбонильное соединение.
NaNO2. И+
;сн2------------»-
VZ или
Yz RONO, NaOR
(29)
х. Y = СНО, COR, COZR, CN,
NO2
C=X0H
(12)
Соединения с метиленовой группой, активированной находящейся
"° соседству одной сложиоэфнрион группой, не вст> па”т в Ре„а.
|1тРозпровання, тогда как соответствующие монокето-,
10- или моноинтрометнленовые соединения гладко ннтр> 1 р
я. Так, нитрозированпе первичных нитропарафинов в bi '
(нитронаты)—лучший синтетический способ получени . £ '
ьх кислот (уравнение 12; X = NO„ Y = H или алкшлГ. Алкнль
'Руппы, активированные пнтроарильиым заместит.
16* 483
,„.,„п,.,отся Соединения с активной метиленовой rpV1.
X оксимировать и N-иитрозамииамн в отсутствие ката-
"^топо Было показано |31], что при реакции циклогексанона
т? е иитрознлхлоридом в спиртовых растворителях в присущ,
пш хлористого водорода образуются 2-алкокси-З-оксимииоцикло-
М21 а в отсутствие хлористого водорода — эфиры 6-окси.
««» <м> У
иитролиза углерод-углероднон
связи (уравнение 13).
О
OR
NOCI.
HCI
к Oil
NOCI
КОИ
^NOU
(32)
(33)
CH=NOH
I
(CH2)4 (I3j
co2r
(34)
Оксимы получают также с высоким выходом [2, 24] при об-
лучении насыщенных алифатических или циклоалифатических уг-
леводородов прп 325—600 нм в присутствии хлора и оксида азо-
та или нитрозилхлорида и хлористого водорода. Фотонптрозиро-
вапне — свободнорадикальный процесс; из-за отсутствия специ-
фичности основная область его приложения — цнклоалкапы. Так,
например, фотонитрозирование циклогексана в цнклогексанонок-
сим является ключевой стадией прн производстве капролактама.
Тем не менее фотонитрозирование метильных групп представляет
собой практически важный путь получения альдоксимов [32].
Внутримолекулярное фотонитрозирование метильных групп, при-
водящее к альдоксимам, служит основой реакции Бартона [33].
Этот элегантный и широко применяемый метод позволяет вводить
функциональные группы в инертные ангулярпые метильные заме-
стители в стероидах и родственных соединениях путем внутримо-
лекулярного нитрозироваиия, осуществляемого при фотолизе дол-
жным образом расположенных нитритных групп. Так, удобный
метод синтеза [33] ацетата 1,2-дегидроальдостероиа включает на
ключевой стадии специфическую функционализацию (окепмпро-
ванне) метильной группы при С-13 путем фотолиза II ₽-нитрита
н Р-П1Дроксипрегнадиеи-1,4-диоца-3,20 (35) (уравнение (4),
ы
ONO
(14)
(35)
Арендиазонпевые соли
J"0 С СОединеннЯм'‘ с акти’вной метил ен'мо'й" группой "в форме
(37), как правило, вступают в конден-
0ЙЧ11вые промежуточные азосоедннення (39), которые таутоме'
„зуются в соответствующие гидразоны (40) (уравнение 15) [2
25 34, 35]. Заместители, активирующие метиленовую группу мо
гут быть самыми различными (X н Y; уравнение 15)- их можно
расположить в следующий ряд по степени активации метиленовой
группы к реакции сочетания:
NOi > СПО > COR > CN > CO2Et > СО.\’Н2 > СО2Н > SO2R > SOR > Ph
Заместителями, находящимися прн метиленовой группе и обеспе-
чивающими успешную реакцию сочетания, могут также быть ге-
тероарпльные остатки, четвертичная аммониевая (пиридиниевая,
+NRs) и сульфоипевая группы. В реакцию сочетания могут всту-
пать также енамины и простые эфиры енолов. Если один из за-
местителей прн метиленовой группе — карбоксил, может происхо-
дить самопроизвольное декарбоксилирование; продуктом реакции
в этом случае является монозамещенный гидразон (40; Х = Н).
X
, + J к=н
Ar—+ CR------:
I
Y
(37) (38)
Ar—N=N—СН
Y
(39)
(40)
п.
Y Y В р
Ar—N=N—С—R > ArNHN=C + BY
Й^ \
(41) (42)
способа синтеза
„„ ___мьзования алкн-
.метиленовых компонентов (38;
варианте сочетания (реакция
удобного <
счет испо.
Область
гидразонов
лированных
R = алкил ...„
Яппа — Клингемана) .—, <•,,
(41; R = аЛ|{ИЛ ||ЛН |Л) (в некоторых случаях его удаетсявы-
Делить) превращается в алкилированный пли арилированнын Р
азон (42) путем сольволитического отщепления ацильного за-
местителя. Не было ни одного случая, когда бы при реакции
Яппа — Клингемана происходило сольволитпческое
Ччапогруппы. Ожидаемый порядок предпочтительного отщеплен,
анильных заместителей таков,- СНО > RCO > АгСО. ГнДРаз°
чы, продукты реакции Яппа — Клингемана, используются
честве субстратов в синтезе индолов по Фишеру.
применения этого очень
значительно расширена за
и арнлпрованных i.~.
или арил). В данном г..,.
1 136 3711 промежуточное азосоединенне
485
z4) Дегидрирование (окисление) аминосоединений
( “ в азометины [2]
Дегидрирование ампиов [3 4] редко используют как метод прак.
мирского синтеза оснований Шиффа, так как трудно предотвра.
™ть пеоеокнсленпе. Однако имеется несколько сообщений [3, 4]
п пегпдрированни первичных или вторичных алкиламинов над
«..«левым платиновым или хромовым катализаторами пли при
действии серы или селена; соответствующие азометины были по-
лечены с приемлемыми выходами [3, 4].
Использование таких окислителей, как диоксид марганца или
перманганат калия, также дает неудовлетворительные результа-
ты Однако окисление вторичных алкиламинов перокспкислотамп
при низких температурах (О °C) дает соответствующие шиффовы
основания с выходами до 80%. трет-Бутплпероксид также окис-
ляет вторичные алкпламины по свободнорадикальному механиз-
му, шиффовы основания образуются с выходом ~60%.
Реагенты, осуществляющие отрыв гидрид-пона (фторбораты
дназоппя, тритилперхлорат), окисляют третичные амины (43) до
иминиевых солей (44) (уравнение 16) с отличными выходами;
единственное ограничение состоит в том, что образующаяся имн-
нпевая соль должна быть более слабым акцептором гидрида, чем
применявшийся реагент [19а, 20]. Этот метод особенно подходит
для синтеза иминиевых солей из пространственно затрудненных
третичных аминов.
Rk /R3 Pl>3c+ сю; R\ + /R3
\'-cir —-----------* сю;
\R< R2/ \R4
(43) (44)
Моно- и дпзамещеппые гидроксиламипы окисляются [2, 24] в
оксимы под действием различных окислителей [хиноны, галогены,
гипохлорпстая кислота, перокснкислоты, кислород в присутствии
солей меди(П), оксид ртути, хлорид железа (III), перманганат ка-
лия, дихромат калия]. При окислении дизамещенпых гидроксил-
амииов в качестве побочных продуктов часто выделяют нитрозо-
алканы. Анодное окисление [38] монозамещенных гидроксилами-
нов также приводит к оксимам.
птГлп?СТОР°Л<||ОМ окислении гидразинов бромом, хлоридом же-
гитоачош.‘т °КСИДОМ Ртути(П) наряду с азинами образуются
пив ппсме-Дт’ веР0ЯТП0’ за счет возникновения и таутомериза-
кхема 17) \Х'’Ь!Х кат1|01ШЬ1х азосоединепнй [ (45) — (46)—*(47)]
симметрично личлЛИ',СН"Я’ 11олУча'ои111еся при анодном окислении
если к этому естьХТктуп1и1о1ДРаЗП,’ОВ |38]’ таУтомеР|13УютсЯ’
руктурпые предпосылки, образуя гидразоны.
Мс2\ NHMc —— rMe2N=NMej —>- Me2NH—N==CH2 <17)
(45) L ®r” J Br"
’ {47)
(16)
(46)
486
(5) Реакции отщепления, приводящие к азометинам [2, 3]
термическое или катализируемое основаниями отшрп
Истратов (48), образующихся при атаке эл^офи!
рентами (при галогенировании, нитроз,,ровании , Z' ании’
сульфоиилированни) по атому азота в первичных и вторичных
алкпламннах, является общим (и не таким уж необычным) мето
дом введения углерод-азотной двойной связи в молекулу (упав
пение 18). Так, галогенирование первичных и вторичных алкит-
аминов трег-бутплгппохлоритом дает N-хлорамины, которые под-
основание нли нагревание R'\
C-N( ----------------—--------> \=N
-нх
(48)
(18)
,R3
(49)
вергаются самопроизвольном}' пли катализируемому основаниями
отщеплению хлористого водорода, приводящему с хорошими вы-
ходами к соответствующему имину или шиффову основанию. Эта
методика представляет собой лучший метод получения [9] Д'-пир-
ролина и Д'-пиперидепнов и является одной из стадий асимметри-
ческого синтеза аминов, включающего трансаминирование [39].
Полагают, что окислительное превращение первичных п вторич-
ных аминов в соответствующие основания Шиффа с высоким
(~90%) выходом прп действии аренсульфонилперокспдов в этил-
ацетате при —78 °C происходит путем N-сульфонилнроваиия п по-
следующего отщепления [40]. Образование связи C=N за счет
подобного отщепления использовано в синтезе in situ N-ацилпмп-
иов [41[ и синтезе ацилгпдразопов из алкилгалогенндов [42].
Катализируемое основаниями превращение о-аиетиларенсульфоги-
дроксамовых кислот в оксимы [2, 24] представляет собой родст-
венный процесс.
Образование С=М-связи в результате отщепления „широко
используется как общий метод синтеза иминиевых солей |2, 9,
19, 20] (см., например, уравнения 19, 20). Считают [43], что
обычное а-амндоалкнлирование ароматических соединении, оле
фипов И соединений с активным атомом водорода действием
М-(а-хлоралкил)ампдов п кислой среде происходит с образова-
нием в качестве реакционноспособного промежуточного с
ПИя ацилимиииевого катиона.
Ilg(OAc)2
II2O, AcOIl
(19)
487
(RCH2)3N
I
(RCWsN-Br Br
I_______
|c.(NO2),
(RCH2)3N-NO2 Br'
I_________________
| NO* -bf4
(RCH2)3N—NO "BF,
_______I
(RCH2)2N=CHR X’ (20)
. HHal * SO2 +
(RCHs)sN—OH Hal' -*--- (RCH2)3N—О ► (RCH2)3N—OS02'
Двойную связь углерод—азот можно ввести в молекулу п с
помощью реакций фрагментации. Так, илпды азота, образующиеся
из азиридинов и карбенов, самопроизвольно распадаются на ал-
кены ц азометины [44), а фотолитическое отщепление азота [45]
из нмннотетразолов представляет собой путь синтеза импиоазпрп-
динов. Общим методом синтеза шиффовых оснований является
фотолитический отрыв диоксида углерода от N-нцтрозо-а-амино-
кислот [46], а термическое отщепление диоксида углерода из
1,2-оксазнниевых солей, приводящее к нмндатам,— ключевая ста-
дия в элегантном синтезе [47] циклических диеновых сложных
эфиров. Для синтеза импнпевых солей обычно используют реак-
ции фрагментации различных четвертичных аммониевых соеди-
нении {19а, 20] и распад у-ампногалогеиидов и -тозилатов п
а-амннокетоксимов по Гробу [48].
8.1.1.2. Геометрическая (сцн-аяти)изомерия,
прототропная таутомерия и молекулярные
перегруппировки азометинов.
(1) Геометрическая (син-анти) изомерия [4,49]
и н^Г113нейОпостиТ^Ь',ОЙ жесткос™ углерод-азотной двойной связи
азометины ПппЛы РУКТур’ содержащих эту связь, N-замещенные
альнГмоту? 1ФСеТВиЬ' °с,,ова11ая- оксимы и гидразоны) потенци-
мерных формах (501 иУкц8 пВуХ РазЛ|™1Ь|Х геометрически изо-
термодинамически foLo'/’ - случ„ае N-замещсппых альднмипов
R2 = Н, X = ал.,.,„ лее Устойчивой оказывается структура (50,
жен по ту же стопонч” арилЧв которой атом водорода располо-
жите; ее обозначает °Т ДВ0^Н0|”( связи, что и заместитель при
Х = алк.1Л мКоатвеХт-ИЗОМер- Стр™а (5,; Г =
В случае оксимов н г,,™™ ает менее устойчивому сия-пзомер)•
188 Дразопов более устойчивые структуры (50;
„,_Ц; X = OR>NR2)> атом водорода в которых нахо™.
R; сТОропы двойной связи, что и заместитель при азоте „Т™1
* ^изомерами; «яти-изомер изображен формулой (51-R2—Вц’
'°^OR,NR2). В случае N-замещенных кетиминов, кеток ’
„празонов кетонов обозначение конфигураций следует oroBaJ
aIk особо. Конфигурация (син или анти) азометпнов может
6ить однозначно установлена с помощью спектроскопии ЯМР [50]
R'\c/R2 r'x_/R’
II
N.
N
(50)
R2
(51)
RJ
Способность альдоксимов и кетоксимов существовать в cm- и
вни-изомерных формах была выявлена уже довольно давно; это
один из классических примеров геометрической изомерии. Суще-
ствование сан- и анта-форм у шиффовых оснований и гидразонов
было однозначно показано лишь недавно с помощью ЯМР-спект-
роскопии, которая вообще оказалась мощным инструментом изу-
чения син-анти-пзомерип азометпнов. Ранее публиковавшиеся со-
общения о раздельном существовании и выделении сан- н анти-
форм шиффовых основании и гидразонов необоснованы; скорее
всего речь
морфизм и
ЯМР было
альдпмииы
чнтельно в
или арил)
Х = алкил .... _________,—
перегруппировки антн-изомера при низких температурах (от —70
До—100 °C), при которых скорость термическом реверсии относи-
тельно низка и за ее ходом можно проследить с помощью ЯМР.
Втоже время, по данным исследований С-арнл-К'-алкнлальдимн
нов методом ЯМР [52], оказалось, что прн комнатной температу-
ре присутствует менее устойчивый сия-изомер; его равновесная
Концентрация довольно значительна. Кроме того, прн комнат
температуре равновесие устанавливается достаточно *|е_‘ ’
это позволяет проводить съемку спектров ЯМР ПРН 113М® .,HRPncHH.
температурах и тем самым оценить скорость син ? ЙС.
•Алкил- и N-арилкетпмпны в твердом состоянии с> п’яствоое
ключительно в более устойчивой изомерной форме, а Р СКРО.
Устанавливается равновесие между син- и анти'$ р’ ПОмОщью
ямп инвсРснп п в этом случае можно опреде. пысокоэнерге-
т^р’спектроскопнн при различных температура.. быЛ0 п0.
lecKiie барьеры син анти-инверсии в имида .. ывает на
' 33110 [53], снижаются при протоннроваиии, э
48.
шла не об изомерии, а о таких явлениях, как поли-
термо- и фотохромизм. С помощью спектроскопии
отчетливо показано, что многие N-алкил- н N-арил-
в твердом состоянии и в растворе существуют нсклю-
более устойчивой анти-форме (50; R2 = H,X = алкил
[51]. Менее устойчивый сан-изомер (51; R" = Н,
или арил) часто удается получить лишь путем фото-
тп чт0 отталкивание пеподелеипой электронной [1г1ры в зНач„т
ной степени определяет конфигурационную стабильность азо^
Т"НК'1к еже отмечалось, оксимы обладают достаточно высоко т
гоифигурацнонноп устойчивостью, что обеспечивает раздельно
ev „ествовапве в возможность выделения син- и анти-1130
В пастворе же быстро устанавливается равновесие между син.
и Лги-формамп; в этом случае также можно определить скорость
процесса с помощью ЯМР при различных температурах. Напро.
tub конфигурационная стабильность простых эфиров оксимов ца.
столько высока, что спектры ЯМР их растворов заметно не Ме.
няются при изменении температуры. В то же время, cuu-auru-изо-
меризашпо этих эфиров оксимов можно осуществить путем прото-
пирования или облучения. Уместно в связи с этим упомянуть, что
простые тиоэфиры оксимов взаимопревращаются в 10" раз быст-
рее, чем их кислородные аналоги, что было установлено на осно-
вании спектров ЯМР при разных температурах [54].
При синтезе гидразонов обычно получают более устойчивый
изомер, который в растворе находится в равновесии со своим
конфигурационным партнером. Скорости син я* анти-взаимопре-
вращений гидразонов часто можно определить довольно легко г
помощью ЯМР-спектроскопии. Напротив, спектры ЯМР N-гало-
генкетнмщюв не меняются с изменением температуры; это нахо-
дится в соответствии с их конфигурационной устойчивостью.
Часто удается выделить син- и анти-формы этих азометннов.
Достижения последнего времени [55] в области изучения син-
анти-пзомерпзацпи азометннов относились, главным образом, к
выяснению топкого механизма, по которому происходит это взаимо-
превращение. Было предложено два механизма: внеплоскост-
ной (put-of-plane) «вращательный» процесс, включающий дипо-
лярное переходное состояние, и механизм плоскостного «бокового
сдвига» с линейным переходным состоянием. Параметры актива-
ции син 5=ь анти-взаимопревращения для N-алкилкетпшиюв [56]
и -гидразонов [57] согласуются с последним. Однако, по-впднмо-
му, факторы, определяющие, по какому механизму пройдет изо-
меризация, очень тонко сбалансированы: механизм боковогосдви-
мр/тЗМСНЯеТСЯ ИЛ вРа™ы,ый при изменении природы яара-за-
X^М1хпопа?ХекСафторацетоннминах [58]. Расчет по
TDQHOR uunnn 3 свидетельствует о том, что в случае ни-
тронов инверсия проходит по вращательному механ изму.
(2) Прототропная таутомерия
Двойной связью прототропной таутомерии соединении с
1124] [(уравнеииеУ2ПР°?Ч9\130Т/!:ОТНОСЯТСЯ; <а) иитрозо-окепмная
[60, 61[ [ 52 «Д1 (5у2)Л 53)’ Х = °Ь б) азо-гидразонная
'[(52)«(53), X = CR 1 Tai 11 азометин-азометпннаЯ
490 21 у соединений, содержащих иоДвиЖ’
и11Й атом водорода прн а-углеродном атоме г™
зометиниой группировке), возможны еще чва ("° “™°Шению к
г,1Яразо11-епгидразнниая 60, 61 [(52)«=>(54) х а. ™Утомерии.
Хамиипая [4] [(52) = (54); X = CR2] Д =?R] И азо“е-
.... триадпых прототропных систем наблюдается ст₽^.ДР°Гей'
ядок увеличения скорости превращения: 'азометинХ«нн Z
азо-гидразои < пнтрозо-окенм; он соответствует <
„осу протона к более электроотрицательному центру (гД'1пГ
Следствием легкости С —О-прототропного сдвига является то V™
первичные и вторичные нитрозоалканы перегруппировываются к
оксимы, как только образуются; этим пользуются для потения
оксимов путем нптрознрования по углероду (см. уравнение 12)
Неустойчивость азоалканов, образующихся [34, 35] как первнч
ные продукты при сочетании соединений с активной метиленовой
группой с солями арепдиазонпя (по сравнению с устойчивостью
соответствующих гидразонов) также показывает легкость
C-^N-[1,3]-прототропного сдвига. В то же время, по данным
спектроскопических исследований [60], в растворе имеется не-
большое количество азо-таутомера (53, X = NR) в равновесии с
гидразоном (52; X = NR). Превращение гидразона в азо-тауто-
мер можно также провести в присутствии оснований [61]. Равно-
весие гидразон — енгндразин [(52) = (54); X = NR] также значи-
тельно смещено в сторону образования гидразона [61]. хотя моно-
гидразопы p-кетоэфиров п p-кетонитрнлов в растворах [62] пол-
ностью или частично перегруппировываются в енгидразины, без-
условно, в результате стабилизации за счет сопряжения.
R2e4
,C—N—X
z=r2ch
z.
(21)
H
(53)
II
H
(54)
Триадная прототропия типа
H
(52)
азометпн-азометин [(52) = (53),
X = CR2] протекает довольно вяло; обычно она ускоряется прн
нагревании в присутствии сильного основания, например t, • а
в EtOH [63]. Было показано, что происходящий при этом
[1,3]-протонный сдвиг осуществляется ступенчато путем пеР®™са
протона с присущей этому процессу полно» стереоспецифич
[64] с одной плоскости промежуточно образующегося аз ..
"ого аниона [65] на другую. Стереоспецифичность "PtBPa“e
азометпн-азометин делает эту систему привлекательно! к ’
изучения ферментативного трансаминирования ада
•ходящего при участии пирпдоксаль-5-фосфата lb L пного
Как можно было ожидать для случая г /«у--(54)•
сдвига, азометнн-енаминная прототропия тип Н
X = CR2] гораздо легче осуществима, чем
Щая азометин-азометипная прототропия. Подвпж. структурой
Mbl такова, что хотя в твердом состоянии )CTOi
491
омывается азометпппая форма {4, 9, 66], в растворе, особенно
в полярных растворителях, таких, как днметнлсульфоксид [67],
PvuiecTHyer равновесие азометин— енамин, которое легко обпару.
жить физическими и химическими методами [4]. В определении
положения равновесия азометин — енамин важную роль играют
н эффекты заместителей [68].
(3) Молекулярные перегруппировки
Катализируемое кислотами превращение оксимов (55) в ами-
ды (56) п (57) (перегруппировка Бекмана [49, 69]; уравнение 22),
несомненно, является наиболее известной и наиболее тщательно
исследованной перегруппировкой азометпнов.
R[\ катализатор
XC=N -------------->- R'CONHR2 + R’CONHR1 (22)
Rj/ \)H
(55) (56) .(57)
К числу обычных катализаторов, которые вызывают это превра-
щение, относятся пентахлорид фосфора в эфире, концентрирован-
ная серная кислота, тиоинлхлорпд, полифосфорная кислота и
аренсульфоннлхлорнды. Бекмановская перегруппировка эффектив-
но протекает и в слабо основных условиях прп нагревании окси-
ма в гексаметплтрнамидофосфате [70]. Известны н другие мето-
дики, позволяющие в мягких условиях и с высоким выходом про-
вести эту перегруппировку: кипячение оксима в ксилоле в при-
сутствии силикагеля [71] п реакция с карбоновыми кислотами и
присутствии азоэтнлформиата н трифепялфоефнпа при 0°С в ап-
ротонных растворителях (тетрагидрофураи) пли в слабокислой
среде [72]. Перегруппировка Бекмана широко использовалась для
превращения циклических кетокепмов в соответствующие лактамы
с расширением цикла. Прп этом нельзя не упомянуть о промыш-
ленно важном превращении оксима циклогексанона в капролак-
там, а также о расширении цикла прп переходе от кетокепмов в
составе кольца А стероидов (58) к А-азастероидам (59) (урав-
нение 23). Перегруппировку Бекмана использовали также для
синтеза а-аминокислот и пептидов [73].
он й
(58)
(59)
(23)
манВОвскоНйИщпРеа±аХ [49’ 69! по исследованию механизма бек-
группа смешаетсяУрШ1₽°ВК"11 было П0каза110, что мигрирующая
тронной плотности чскпп°еи электР011Н°й нарой (увеличение элек-
ти ускоряет перегруппировку, введение электроно-
492
L.nenTopHblx заместителей замедляет ее) .. М|,
’ утрпмолекулярчо (отсутствие перекрест. ых ПР*ЦИЯ "Расходит
е конфигУРашш у мигрирующего центра) „ РД<‘КЦ|,И; сохраНе.
предпочтительная миграция .руППЬ11 находящей»ереос(1еиифичио
. „ К гидроксильном группе), Тознлаты оксиме» “"’'“положе-
"отся В имидоилтознлаты самопроизвольно » ЛерегруппиР°вы-
лязатора в отличие от исходных оксимов, инертны/?1*”* ката’
' |Ях, Это свидетельствует о том, что функцией ЭТ1|Х По-
является превращение гидроксильной группы OKcHMaTM3aTOpa
уходящую группу. Эти наблюдения в сочетании с 6о1₽в ЛучШую
исследованиями механизма перегруппировки БекЗТ 1,оздними
данные о том, что оксимы, содержащие в а-поюжХ ',/ Также
группировки (например, гидроксильную или аминогруппу?10^
собны превращаться в нитрилы, привели к выводу о mLT
„ости механизма этой перегруппировки (схема 24) '«Новмал.няа'
перегруппировка может быть представлена как согласованний
процесс, включающий непосредственное превращение оксима (60)
в цмндат (62) через мостиковое промежуточное соединение (61)
Образование продукта перегруппировки, амида (66), объясняется
ионизацией нмпдата (62) в нптрплпевый ион (65) с последующей
его гидратацией. Альтернативная схема («аномальная» бек’манов-
ская перегруппировка) состоит в образовании нитрилневого иона
за счет фрагментации н рекомбинации [(60)-► (63) + (64)-<-(65)].
Промежуточно образующиеся ннтрилиевый и карбениевый ионы
при «нормальной» и «аномальной» перегруппировках
можно «уловить» соответствующими нуклеофилами [76].
Бекмана
R* -R2 R\ -
/C=N -----------> R1—d=N -----» \=N~R2
₽? \)X X0*
(60) (61) _____I®.2)
j
(R^ + R-C=N —> R’-CSN-R2 <-r'~C=N-R2
(63) (64) (65) I*
II
ri-C-NH-R-
(66)
осуществить И Ф°‘
Векмаиовскую перегруппировку по-вндчм°-му’7’}ер®о-
’охимически. Однако эти процессы вклюн^^^ н1,трОн
начальную перегруппировку в соотв образование
т°Рый, как полагают, через проиежу 1П1<ровк». llMU°
Ридина дает наблюдаемые продукты -
надежные доказательства фотохимическом циклизации ,111тр
в о№' прид-пы [78]. Показано, что это высокостереоспециф
ПЫЙ процесс [79]; в результате перегруппировки могут обра3одь.
ваться амиды ]78, 80]. Нитроны перегруппировываются в амид',
[781 и ПР» действии ацилирующих aieiiroe. Эти превращения
можно объяснить [1,3]-сдвигом в промежуточно образующее
N-ацилоксопиевом попе [81]. Расширение цикла при получении
А-азастероидов нз стероидных интронов, содержащих эту группщ
ровку в кольце А, катализируемое л-толуолсульфонплхлоридом в
пиридине в присутствии небольшого количества воды, протекает
с высоким выходом, его можно рекомендовать [82] вместо бек-
мановской перегруппировки. Из других перегруппировок азометн-
нов в последнее время привлекли внимание перегруппировки Не-
бера п Чемпена [49]. Первая из них состоит в катализируемом
основаниями превращении тозплатов оксимов, четвертичных со-
лей гидразонов плп N-хлорпмшюв в а-амииокетоиы через проме-
жуточные азпрпиы, которые удается выделить. Перегруппировка
Чепмена — это термически индуцируемая реакция внутримолеку-
лярного нуклеофильного ароматического замещения, используе-
мая для превращения Ы-арплбепзпмпдатов в N-ароилдпфеннл-
ампны. Перегруппировка Чепмена нашла некоторое применение
в синтезе дпарилампнов, необходимых как исходные соединения
в синтезе акридина. Типичная перегруппировка гидразонов — син-
тез индолов но Фишеру [83], который широко применялся для
синтеза природных соединений, в частности, алкалоидов.
8.1.2. АЛИФАТИЧЕСКИЕ, АЛИЦИКЛИЧЕСКИЕ И СОДЕРЖАЩИЕ
АРОМАТИЧЕСКИЕ ЗАМЕСТИТЕЛИ АЗОМЕТИНЫ
8.1.2.1. Специфические методы синтеза
простых азометинов [2—4]
(1) Из субстратов, содержащих кратные связи
Двойные связи, активированные элсктроиоакцепторными груп-
пами, расщепляются прн реакции с первичными аминами, давая
азометппы (уравнение 25). Аналогично реагируют вторичные
амины, приводя к имннпевым солям. В результате родствен-
ого процесса при гидразпнолнзе производных стирола гидрази-
runn ,,атР||я в присутствии гидразина получают соответствующие
дразоиы. Восстановление иптроалкенов комплексными гпдрпда-
/СОМе
ш/с==с\ + R3NH2
ХСОМе
NHR3
R'x | /СОМе
,с—С 7
Н
R'\ /СОМе
C=NR3 -у н2СХ
(25)
•СОМе
494
)ipI1 температурах ниже О °C с иослел^
дает N-пела метенные имины, а пои 1, Щим Вдгк|<м гндпо„
установлении [38] нитроалкенов в ?езуль*Р'ЭЛиРУем°м эл Р
него процесса образуются соответствсющ^ ХмЧеТЫ₽ехэл«трон.
реактивы Гриньяра реагируют с имидоитхп
меШеи1,сМ атома хлора, давая с хорОщим/Х°Ри^ми (67) сза.
яЫе азометины (68). Так же реа ’ИРУЮТ и аВХ^,£'за“ец№
меры неустойчивых имидовых кислот (70)] (схема 26) * (таути'
С1
R'~C=NAr
(67)
О
II
R’—С—
(69)
R2
R2MgX j
------► R'-C=NAr
(68)
| R2MgX
OH
(26)
(70)
Замещение атома хлора в промежуточно образующихся имндоил-
хлорндах на водород с образованием азометпнов, которые можно
выделить, составляет ключевую стадию в ряде хорошо известных
синтетических реакций. Так, восстановление нитрилов действием
хлорида олова(II) и хлористого водорода в эфирном растворе
(восстановление по Стефену [84]) с последующим кислотным
гидролизом до альдегидов (это удобный метод синтеза альдеги-
дов) включает первоначальное образование и восстановление ими-
доилхлорпда в комплексную соль имина н олова. Свободный имин
часто удается выделить нз этого комплекса осторожной ооработ-
кой аммиаком или трпэтнламнном. Синтез альдегидов по Зонну —
Мюллеру [85] из анилидов также включает в себя восстановле-
ние до азометпна. Имины являются промежуточными соединения
Mil в синтезе ароматических альдегидов реакцией акт,1ВИР°
аренов (фенолы н простые эфиры фенолов) с иПаН!}™'’ /»|Г|
Дом и хлористым водородом в присутствии кислот . ГяТТеОМЗ-
нли ZnCh) как катализаторов (синтез альдегидов "°
НУ [86]). Восстановление четвертичных солеи i Р арРНВОднт
вами Гриньяра или оксимов хлоридом титана! )
к соответствующим иминам с хорошими выходак'1',.ктам11 в целом
Азометины являются также конечными Р д- льн0) цикло-
Ряде реакций, которые включают (хотя бы Ф Р 2"). Так,
присоединение п ретроцпклоприсоедпненпе ( р нами [9] (71;
альдегиды н кетоны гладко реагируют с нзониана-
= 8—0), сульфурдипмпдами [2] (71; - Г21 („ЧцНофосфорава-
тами [88] (71, X = С=0) п фосфпна.мииамн [2Н « ЫЧ(74). Вме-
ми) (71, X = РР1Ъ), давая соответствующие t
ст° Ф°ефииамнпов можно использовать анион
оказались пригодными для синтеза S-
в тех процессах. когор > = gph> R2 apuJb Ш щ N.T1I0.
арплтпоальдокеамоо (7 ' ! = sphi х = pph;i) [89]. АзометИвд
а^Ф°±пцгь’.. взаимодействием фосфораиов с ..
1 ‘'“"'cuiiibi
арилазпдами и
о
ЕЮ\|| -
Р—NR
ею/
(76)
Простые ацетилены присоединяют амины при высоких темпе-
ратурах и давлениях, давая енамины, которые таутомеризуются
в азометпны. Эти реакции протекают гораздо легче, если троп-
ная связь активирована электропоакцепториымп заместителями.
Реакции присоединения к нитрилам также приводят к разно-
образным азометикам (79) (уравнение 28): к пмпдоилхлоридам
(Х = С1), пмндатам (X = OR), амидинам (X = NR2), ампдок-
сима.м (Х = Х:НОН), а также к основаниям Шиффа (X = Н, ал-
кил, арил). Здесь мы обсудим лишь реакции присоединения к
нитрилам, приводящим к шиффовым основаниям. В последую-
щих разделах речь пойдет в о других реакциях присоединения.
X
R1—C=N + R2—X •—> Ri—c=NR2
(28)
(77) (78) (79)
Простейший способ превращения нитрила в азометнп (79)—
присоединение водорода с образованием альдимппа (79; R‘>
Х = Н). На практике каталитическое восстановление нитрилов
[90] трудно контролировать, так как образующиеся имины в°с'
стаиавлпваются далее до аминов, в связи с чем этот метод име
ет незначительное препаративное значение. Однако восстановле-
ние нитрилов комплексными гидридами металлов [90] удается
довольно надежно контролировать и получить хорошие выходи'
ммниов. Особенно эффективно эта реакция протекает, если про00
Т' Н”ЗКОЙ температуре н использовать модифшшрован’
0Рдн[,е uT’nv!npiiMe₽ тр11ЭТОКС1'ал’°М0Г11дрид натрия.
присоедииеппиУ п^аХ мстод°в синтеза кетиминов [91] состоит
комплексам им' активов Гриньяра к нитрилам, приводящем
Хь гндпоX ' И3 1ЮТ°РЫХ в ««™х условиях (чтобы избе-
ги Дролиза до кетона) действием хлористого водорода ил»
496 1
„лппго аммиака можно получить свобод.„ч
входом- Присоединение как алифатических, так иТ С Хорошим
* S.BOB Гриньяра с последующим осто ожвым
мплекса им.п.а при ипзкои температуре приводит РКР(^еииеи
[ощему кетимину с выходом -70%. Пространственно затоТ
ё,п.ь.е кетимины относительно устойчивы к гидролизу 77
10HiIIo выделить обработкой реакционной смеси водой АлкиЛ
зриллитпевые реагенты также легко присоединяются кв7Хм
давая кетимины с высокими выходами. Реакцию нитрилов с Л-
активам.. Гриньяра можно использовать для синтеза [91 „икни
ческпх азометннов (уравнение 29) - Д'-пирролинов (82 п = п
„ Д'-пиперидеинов (82; п = 2). Комплексы иминов с реактивами
Гриньяра, являющиеся первичными продуктами реакции нитрилоз
с соединениями Гриньяра, можно также использовать для полу-
чения азометннов, содержащих функциональные группы. Так
комплексы, образующиеся при взаимодействии бензонитрила с
реактивами Гриньяра, реагируют с хпральными эфирами сульфи-
иовон кислоты и дают хнральные сульфпиамиды с оптической
чистотой, превышающей 95%.
।—(СНг)л ArMgBr ।—(СН2)„
СК CN с1/ ^kAr
N Аг
I
MgBr
(81)
,—(СН2)„
(29)
Аг
(82)
Производные ацетонитрила, содержащие хотя бы одни а-водо-
| родный атом, в присутствии сильных оснований подвергаются ди-
мерпзацнн альдольного типа н дают нминоннтрилы [91, 92]; ка-
тализаторами служат амид натрия или алкоксиды натрия. Внут-
римолекулярная конденсация а,й)-дпнптрнлов дает циклические
'‘''’'"Ойитрцлы (реакция Циглера — Торпа [92]).
(?) Процессы, включающие [/,2]-С —► N-миграции [93 95].
РЬве«ен РЯд реакций, в которых образование кетиминов яв
кил [^„Результатом [1,2]-миграцни заместителя (водорода ал-
этот'01' "J"1 аРилыюй группы) от углерода к азоту. ПР
Ш1ЯМ11пПа МОГУТ катализироваться как кислотами, так и
М1,Ч р ’г °Ип МогУт происходить также при термических и ф -
лоацЯу11Х В03действиях в отсутствие катализатора. ы
зующ. ЭТ,,Х Реакций происходит последующее РазДОЖ/н,'ео°Х
чнтепрХСЯ азоМетпиов п возникает смесь продукт •1ТЬ
'« XЭТим реакциям обусловлен скорее стР“';’’Д|,,д некоторых
^Учаях ЭМ’ Чем возможностям., их "впМЬЗОВваН”Яое?Руппи5овк..
КетиМ1.нЫ "ако> образующиеся в результате Р ДдаЛ.
орилач., ж,,° выделить в препаративных кол'" кет1Мцны,
Л4311Дь. под действием кислот при нагревании дают кети.
,, которых приводит к аминам п карбонилы^
кислотный гидролаз ко i катаЛнз»руемая кислыми пере.
соединениям. В ™ *азпдов (S3) представляет сооои удобныГ|
гРУ"п,1₽0\«аЦ,Хл^Че"ких азометпнов (84) с расширением цНКЯа
Кавиев’шЗО)"11
(83)
HaSO, , <
CHCls
N
(84) (80%) (85) (2%)
>=NH
(30)
Аналогичные перегруппировки алкплазпдов катализируются кис-
лотами Льюиса (например, SbCi5 и А)С)3). Согласно данным,
полученным при изучении кинетики и механизма этой реакции, j
перегруппировку вступает не свободный азид (86), а сопряженная
кислота (87) (ион ампиодиазония); миграция идет синхронно
с отщеплением азота, а не происходит через стадию образования
нптренпевого иона (уравнение 3)).
н+ ТЛ Г+
K3C-N3—~>R2C—NH—N2—->R2C=NHR -y^>R2C=NR (31)
(86) (87) (88)
Трпарилметнлазпды довольно инертны в условиях кислотного
катализа, тогда как алкилазиды дают продукты расщепления азо-
метина (амин и карбонильное соединение). Аналогичные C->N
сдвиги (перегруппировки Штиглица [95, 96]) происходят при об-
работке трет-алкилгпдроксиламипов пентахлоридом фосфора в
инертном растворителе или при обработке трет-алкнл-М-галогеи-
аминов основаниями (уравнение 32). Реакции этого типа могут
быть использованы для препаративного синтеза кетиминов. Дан-
ные [96] об участии интермедиатов типа ионов пптреиия в пере-
группировках Штиглица противоречивы.
А r-К'ГЧтт PCljy основание- ZQ01
АгзСХЮН --------> Аг2С=Х'Аг -«--------- Ar3CNHCI (32)
\7 ?
термической1*1 пепегоуппьпСре';1е т'р11аР,1-лме™лазнды подвергаются
бензофенонов с пПР₽м Р ВКе При 200-300°С, образуя аниль.
в таких перегруппнпов’ЛМЫМИ ВЫхоДаМ11- Склонность к миграция
арильных остаткахР нЛ* "е завнснт от природы заместителей #
применение, -jaK кя леДствие этого они имеют ограничен!'01-’
лов образуются СМРг,, елУчае несимметричцых трпарилалкплази-
арильных групп. изомеров за счет миграции любой и3
Аналогичную теп
трет-алкилазиды Обси'!,еСК'10 пеРегРУ"пировку претерпевают "
ступенчатому, через пппм»СЯ Вопрос’ 110 какому механизму-'
498 Р е/КУточио образующийся нитрен, "Л1
рпгласовашюму — происходит термическая
слазил03' Д°каза,,° |J7j участие нитпе'!,^ГруппнР^вка а„
“ступенчатом процессе. Аналогично указ 10 ,!«^РмЛ„й;
протекает термическая фрагм^Гл^
„ов (легкодоступных в результате никпп Д Ш2,3-трна, ” “
к алкенам), также приводящая к кетиминам" ВозТ"^ аз«Дов
здаания этой реакции в синтезе определяется уД п?Ность «еполь
депия. Так, при относительно низких теХ^Т ЯМИ ее аРове-
сохраняется стереохимия заместителей в той» УРаХ (40~90сС)
образуется лишь один кетимин, а не смесь !' ,'Эад,;|,овс« цикле и
лее высокой температуре. Термолиз триазо XIT08' Как ”₽и <”>
например, для синтеза циклического азоме’т, ЫЛ нсп°льзоваи,
наше .33). ’ на’ пнРролина (урав-
СН=СН2 50 °C
Прп фотолизе трст-алкилазпды также дают N-замещенные
кетимины; с препаративной точки зрения эти перегруппировки
удобнее, чем реакции, катализируемые кислотами, так как дают
более чистые продукты. В отличие от термической перегруппиров-
ки азидов, для которой надежно доказано образование в качестве
промежуточного соединения синглетного нитрена, механизм фото-
химического процесса строго не установлен. Имеющиеся данные
свидетельствуют об участии как синглетного, так и триплетного
нитрена. Было показано [93], что предпочтительность миграции
топ или иной группы прп фотоперегруппировке определяется кон-
формацией основного состояния азида. Это означает, что реали-
зуется согласованный механизм без участия нитрена.
С-►N-Сдвиги, приводящие к образованию N-замешенных кет-
вмпнов, происходят также прп окислении трет-алкиламйнов тетра-
ацетатом свинца [98].
8.1.2.2. Реакции простых азометииов [4, 99,
(1) Реакции с электрофильными реаген
г„„пые иминпевые соли
1 Шиффовы основания образуют усто1 с хлорной KHC,1C>™S''.'
ПЭа] с протонными кислотами^ (напР' •’а быстрого гидро._
лУЧше их получать в безводной СР®Д н еЖН0 определено ср
который происходит в ВОДНОЙ сред • аМ11. В зависимо
, 1Ие солей гидразонов с протонными к _ ягь ^р2-гибриди
’Р’Фоды заместителей (которые м°2,д я по атому азота ' нх
3°та) гидразоны могут протонпроватьсочеНЬ мала, н.
Pinny или аминогруппы. Основност растворах.
лч легко гидролизуются в разбавлен
499
ПОП присоединение галогенов (хлор, 6pl)Mi n .
Описан0 |Н>И "I ,ерод-азот в N-арплальднц.......ах
1 2-типу к лвопнои е iie>6biJ}„ „сследовапы. Продукту реак.а’
ко "Р^^^плнна о бР°ыоы В ’•етыреххлорнстом углер0'де ПЦ1‘"
бензилиден б оы„да N-бромпмшшя PliCII-=N(Br)Ph R
li0%1031 х<,ором в В°Л
JIPaJ. Хл° ирова кпслота) или в безводной среде (хл0‘Й
(ра3б^;п|"в "» ‘к формальному замещению водорода нмнд11льР0°й
форм) привод! *е‘м г1,ДрОКспмонлхлорндов. Полагают, ЧТо э ”
ГР-Хы образуются В результате промежуточного образован,,^
ПР Д^оТво. ЗВОЛЬНОЙ таутомеризации _ неустойчивых гещ-интр030.
1 Д Рктов которые являются устойчивыми Конечными прОдУк.
ДДп щи аналогичном хлорировании кетокспмов. Подобным же
пЛтзом с кетокспмамн реагирует ором, давая гельбромнитрозо-
производные. Гидразоны обладают заметной реакционной способ-
костью ПО отношению к галогенам [2о, 61]. Так, реакция N-неза-
мешеипых гидразонов с подом [Ю4] приводит к окислению их
‘ алкенов, по-впдпмому, через промежуточное образование
N-иод- и С-иоддпазонпп-катнонов. Реакция с хлором и с бромом
[104] приводит к сложной смеси продуктов в результате фрагмен-
тации. сопутствующей галогенированию. N-Моноарплгидразоны
альдегидов устойчивы к окислению, хлорируются и бромируются
по пмидильному атому углерода с образованием гндразоноилга-
логенпдов [25, 61]. Хлорирование может протекать и дальше;
атака по атому азота приводит к неустойчивым N-хлорпропзвод-
ным, которые склонны перегруппировываться в изомеры с атомом
хлора в цикле. Днннтрофеннлгидразопы алифатических кетонов
подобно самим исходным кетонам подвергаются бромированию
по атому углерода в а-положеннн к азометнповой функции п
дают продукты, дегпдробромнровапием и последующим гидроли-
зом которых можно получать аф-ненасыщепные кетоны. Удобный
способ регенерации альдегидов н кетонов из их п-толуолсульфо-
нплгпдразонов с высоким выходом (80—90%) состоит в обработке
этих гидразонов N-бромсукцннпмндом в ацетоне при комнатной
температуре в присутствии метанола [105]. Полагают, что при
этом сначала бро.мируется NH-группа, а затем происходит
мХт аТака ыетан°лом с отщеплением брома, приводящая к проме-
Твощтрпя^ ПРОСТО:'!У азоэфнру; последующая атака молекулы рас-
homv На азоэФпР приводит к ацеталю п затем к карбониль-
ному соединению.
агентами1!^67 ы"эТерес Реакц,1я азометннов с ннтрозирующнмн
хлоридом ппн а?’еще1|,1ые кетимины реагируют с нитрозпл-
1.2-Ln11Tpo3o C"" пТеМПераТуре (~30°С). Давая ожидаемые
по крайней мрпр п ДДУКТЫ, которые довольно неустойчивы, хотя
N Арнлальдпмииы од,,ом случае [106] они были выделены,
высокими выходами (80™алиЧ’ напроТ|1В> удается превратить с
кетоны и аренппя,™, 9и/о) в соответствующие альдегиды и
ювые соли при действии фторбората (пли
500
и‘Ксифторзптимоиата) нитро юиня нпи
ой температуре I Ю7| Механизм реакции вкТ1Т”°Й "ли «оиижеи
ное N-иитрозироиаиие, Последующую ник™* ЮЧает "ервонач*^
нЫй оксадиазе111днни1! и распад на продукт п“.И*° “ "Джутом.
такпм образом, может служи, ь и удобным " Э,а Р^нния
кропания, и методом регенерации альдеги "оВ“н СИОсобо“ лиазо-’
ветствуютпх иминов. Дезокснмирование оке* , Ке1оиов Иа «ют-
1|Ых соединении при ннтрозпровапии также им^ ,до ка₽6еииль-
„|)е, поскольку позволяет вводить функций бо-'1ьшое зиаче-
алкильные заместители с помощьюфотооксимип HUe 'Руииы в
мер, при реакции Бартона [33] (см с 44ЛР°тания’как.на"ри-
симов с нитрозплхлорндом в пиридине' ипи_%ггР^Ки-ия ок'
ветствующий нитримин, который легко гидоопи^ дает соот'
чении с водой и дает карбонильное соединение ? 7. ПрИ кипя'
90%) |Ю8). Проврашонае “““ №-
нитрозированпи хорошо известно для кетиминов не₽И
„.yw.ua ат«,м«. (103). Д.„ ..............
боппльные соединения можно также использовать фторбХ
нптрозоння или фторборат нитрония при О'С [1091 Оксимы vm
ется превратить в исходные карбонильные соединения и проио
обработкой азотистой кислотой [103]. Все Указанные реакции*^
зокснмнровання, по-видимому, инициируются атакой по атому
азота в оксиме. В то же время многие алифатические кетокснмы
реагируют с N2O4 по имидпльиому этому углерода, давая гем-
нитроннтрозосоеднненпя (псевдонитролы). Однако и в этих слу-
чаях, вероятно, С-нптрозироваиие есть результат [1,2]-.\’-*С-сдви-
га в первоначально образовавшемся N-нптрозоинтермедиате
[103]. Гидразоны также легко реагируют с ннтрозирующнмн аген-
тами [61, 25]. Так, N-незамещенные (или N-моноацилированные)
гидразоны прн действии нптрит-нона в слабокислой среде (напри-
мер, в уксусной кислоте) превращаются в соответствующее кар-
бонильное соединение и азотистоводородиую кислоту, тогда как в
сильнокпслой среде образуются амиды, продукты перегруппиров-
ки через иминодиазонневый катион (перегруппировка Шмидта).
При проведении реакции N-ацетилгидразонов с нитрозплхлорндом
или N2O4 в присутствии ацетата натрия при —5°C образующийся
иминодиазонневый нон «улавливается» анетат-ионом и дает гем-
азидоацетат, который можно выделить [НО]. Ннтрозированне
N-мопоалкпл- пли -арилгндразонов нитритом натрия в уксусно
кислоте или алкилпнтритамн в апротонной СРВДД
N-нитрозогидразоны, которые в случае арильных пр i
склонны к формальному [1, 3]-N-^C-сдввгу ннтрозогрхпп
после таутомеризации соответствующие азооксимы | , ’в
Прямое алкилирование N-алкилальдпмино солн
идет по атому азота и дает соответствуюши • постраиствен-
[4, 19а, 100]. Подобные процессы чувствите.Ямистые алкильные
ным затруднениям. Так, имины, имеющие Ю1СЯ лишь с
группы или N-арильные заместители, i • •
rnviOM- В этом случае необходимы более активные алкилнр,,,
прзгенты например трналкплоксонпевые соли R3O+ Х- К 111111
вследствие своего амбидентного характера [102] могут
гаться прямому О- или N-алкплпрованвю в последнем случ°^
разуются интроны [см. разд. 8.1.6.1 (2)]. Ооычио ocymc„Л °6'
алкилирование серебряных солеи оксимов, которое проходит
дючвтелыю ио кислороду; при этом исходная конфигурация оЛ'
ма сохраняется и в О-эфире. Прн обработке оксимов алкипгап"’
геппдамн или алкнлеульфатамп в присутствии щелочи алкилип
ванне и арнлнрование происходят преимущественно по кислорщ
тогда как в отсутствие щелочи (и в темноте) образуются нитрон?'
Прн действии диазометана алифатические альдоксимы превращ»'
ются в смесь О- и N-метнльных производных, тогда как арнлац'
доксимы дают прн реакции с диазометаном только О-эфирьь Г»
разоны тоже легко реагируют с алкилирующими агентами [25
61, 112]. В случае N-иезамещенных гидразонов параллельно
с алкилированием аминного азота происходит их диспропорцио-
нирование в азины. N-Алкилнроваине N-мопозамещенных гндразо-
нов можно провести внутримолекулярно (уравнение 34); это об-
щий метод синтеза А2-ппразолш1ов. Интересно отметить, что, если
в результате такой циклизации может образоваться шести- или
семичленное кольцо, реакция предпочтительно протекает по пер-
вому пути, включающему внутримолекулярное алкилирование по
иминному атому азота (уравнение 35). М.Ы-Днзамещенные гидра-
зоны реагируют с алкплгалогенидами, образуя четвертичные ам-
мониевые соли [112].
R'CH—।
I
R2CH 1
I
X 1
<89)
Х = С1, ОН, OR, SH, SAr, NR2, NR3
axzx
x основание
^NNHR 4^'V^NNHR X'
(91) (92)
лотАациыорндам!!,,иФя(1>ОВЫХ основа1ШЙ I4. IOOI ангидридами кис-
азота и приводит к ' “''ЛЦ11а“|1лам" начинается с атаки по атому
ной связи углепоп—-.Р соадш,еН1|ю ацилирующего агента по двою
синтезе природных ел°Т' РеаКдНИ этого типа нашли применение»
линов с гомофт-први еД1,,|е|,|1и- Так, реакция 3,4-днгидроизохпп0'
синтезе алкалоидов Т' а|1Г11ДР|,дам11 явилась ключевой стадиен
основаниям? X X пР°табсРбернна [ИЗ]. Катал изпрУ^’
НИИ электроноакцептоп1и ,^11еТ"ЛХ"'10р"Д0В (имеющих в “-поло*
502 р труппу и по крайней мере одни аТ0‘
CR3
II
N
I
NHR*
R'\ ,R3
~-\/N
NR'
(90)
(34)
(35)
пола) с N-арилальднмниами начинается г »,.
’° «азота и приводит к р-лактамам, которые •— Ирован“и
<1ТПМ Y ..a I а г a iidti О mi т и п л / . _ *
g^o реагируют
АГ. /Н
nV Я30»« u * I r'
310 L для ХИМИИ пени,
"T“oec - с аиил
CHC12
c=o -
I
Cl
,N
РГ
м0ГУТЛ?ед£та'вить
-------------1МЫ
ми нормальным
ArCH-cci2
PhN—cq I'36)
(93)
(91)
циллииа (уравнение 36) HiK™81
i- и сульфоиилгалогеиидаг b °КСИ|
An ZH
IW 'c<
Л
Ph + C'
II
О
(95)"
(96)
образом и дают О-ацнльные или О-сульфонильиые производные
|)02]. Считают, однако, что реакция стероидных кетоксимов с ук-
сусным ангидридом в пиридине, применяемая для синтеза трудно-
доступных еиамидов (выход 70—90%), протекает по радикально-
му механизму и начинается с ацилирования по азоту |П5].
N-Моноза.мешенные гидразоны [61] ацилируются по $р3-атому
азота, давая N-ацильиые производные, которые из-за высокой
чувствительности к гидролизу следует получать в безводных ус-
ловиях (например, использовать хлорид цинка в присутствии пи-
ридина). Аналогично можно получить N-сульфонильные производ-
ные гидразонов. Ацилцнаниды присоединяются к двойной связи
углерод—азот в гидразонах, давая 1,2-аддукты, подобные тем,
которые образуются из шиффовых оснований.
(2) Реакции с нуклеофилами
По аналогии с двойной связью углерод—кислород, двойную
связь углерод—азот можно представить как резонансным гибрид
канонических форм (97) и (98).
(97) (98)
Биполярный характер двойной связи углерод—азот объясняет
тот факт, что, как уже отмечалось, электрофилы реагируют пре-
'мущественно пли исключительно по атому азота в азок
сюда следует, что нуклеофильные агенты 10
обстет""ы по чмпднльному атому углерода; показ , соответ,
стк,°"Т "меН|’° так- Более того, пминиевые соли I J повы.
ш .с Резонансными структурами (99) и (,0°) ' ,.,к,1еофиль-
по,- 10н Реакционной способностью по отношению
10,1 атаке.
Шенной
(99)
(100)
503
Пиролиз азометннов [4, 76, 99, 100] до исходных аМ1
карбонильных соединении представляет сооон проетейн.пй ,11
кар о‘л„,„,,1Пп атаки по связи C=N. Эта реакция ofin е’
dS* образования азомепша [см. разд. 8.1.1.1 (1)], ’,10д1р^^
ся закономерностям общего основного катализа. Как н ре^ ’’
образования азометннов (основании Шиффа, оксимов, г,1дКц ’
нов) гидролиз азометннов - двухстаднннын процесс; он ВК.Р '
ет присоединение молекулы воды к двойной связи углерод-,' [
с образованием карбнноламннного интермедиата, котопщ
затем распадается на амин и карбонильное соединение. Скоро "
гидротпза азометннов, как н скорость реакции нх образования за
висит от pH. При низких pH скоростьопределяющен стадией’ЯВ-
ляется разложение карбшюламнна, тогда как при проведении ре.
акции в условиях, близких к нейтральным, скоростьопределяющен
стадией становится образование карбшюламнна (т. е. гидратация
С=Ы-связп). Гидразоны [61, 99в] (за исключением семнкарба-
зонов), особенно N-арнлгпдразопы, относительно устойчивы к гид-
ролизу. Однако нмпниевые соли R2C=N+R2 Х~ [9] чрезвычайно
быстро гидролизуются водой, и получать нх надо в строго безвод-
ных условиях. Легкость гидролиза нмннневых солен была исполь-
зована для синтеза вторичных аминов пз первичных аминов,
включающего превращение первичного амина в альдпмин
R1CH=NR2, затем алкилирование (см. выше) в имннневую
соль R1CH=N+R2R3 X- и гидролиз, приводящий ко вторичному
амину R2NHR3. Поскольку гидролиз оснований Шиффа является
составной частью ряда катализируемых ферментами реакций, в
последние полтора десятилетня механизм гидролитического рас-
щепления двойной связи углерод—азот подробно исследовался;
условия реакции моделировали условия in vivo и in vitro [76, J16].
Гидролиз иминов — ключевая стадия в синтезах альдегидов по
Соммле [13], Стефену [84], Зонну — Мюллеру [85] и Гаттерману
[86] (см. выше).
Алкоксиды присоединяются к основаниям Шиффа, давая, как
*! слеД?вало 0>К1|дать> соответствующие а-алкокснамнносоедпнеивя
]/б, 99а, 100]. Присоединение этого типа лежит в основе ключе-
вой стадии элегантного стереоспецифического синтеза (без виде-
=,,ПроыежутоЧ|1ЫХ соединений) производных пешщил-™13'
.... ^,АМ0Гут быть использованы для получения новых произвол-
noLU ГОСПОр1,На (УРавнен'1е 37) [117]. Он включает N-хлор»'
липового пяпя"ЛнппОр"рОван"е легкодоступных амидов пенни'1' •
в присутс^и, г ° действием трет-бутилгипохлорита в ме™!1,0,^
которые присоеХшот"1’11 °°С’ !!11||поля|цее к ацплимпнам ( ’
нениой а-стопоны » танОл с менее пространственно затрр
Ациклические I Val0T аддукт (104> с "Уж"ой стереохкк»
с™ соли метиле! им Jl'^4eSK'Le [?] соли пмнння, в час™ь1.
шенной реаднон пй""ИЯ H-’C=N+R2 Х~ [19а], обладают по «
алкоксидов, что '"осоС’11остью к I ^-присоединению спирт
ш открывает щ11р0К11е возможности для спит^
□и1?
I C1 H
PbOCH?CONH-T—WBuoci, РЮСНцСОК ’
Л"'
(101)
(102)
PhOCH2CON
(ЮЗ)
QMe
Me on PhOCH2CONH-4—<1
(104)
а-алкоксиаминосоедннений. Алкоксиды легко присоединяются и к
двойной связи углерод—азот в гидразонах [61]. Фенолы не реа-
гируют с простыми азометинами, а при реакции с имнниевыми со-
лями происходит электрофильная атака бензольного кольца, при
которой образуются орто- и пара-аминоалкилированные фенолы.
Сероводород легко реагирует с N-замещеннымн кетиминами
при низких температурах (—40—О °C) [4, 76, 99а, 100); первич-
ные аддукты быстро вступают в дальнейшую реакцию, образуя
гм-днтиолы. N-Арилальдимипы легко присоединяют тиолы (в ви-
де соответствующих тнолятов) [4, 76, 99а, 100] и дают ожидае-
мые 1,2-аддукты, при диспропорционировании которых в конечном
итоге образуется амин (продукт восстановления имина) и
дисульфид. Тиоляты легко присоединяются также к двойной уг-
лерод—азотной связи в гидразонах [61] и имнниевых солях
Взаимодействие шиффовых оснований с первичными аминами
[4, 99а, 101] приводит к аддуктам, склонным к разложению, в
результате которого образуются новый имин н первичный амин,
так что суммарно этот процесс выглядит как иминный обмен
1С«. разд. 8.1.1.1(2)]. Скорость иминного обмена увеличивается с
ростом основности первичного амина. Амид натрия [101] PearKPJ
ет с альдиминами (105), давая амидины (106), что формаль
выглядит как замена имидильного атома водорода (уравнение3»).
NH2
PhCH=NPh NaN"2' phC=NPh + NH. (38)
(105) (106)
Иминневые соли также гладко амнналей
«ые н третичные амины с оОРаз°в®“ [19а]. растворе
^лписшй) или их четвертичных М11 в эфирном рас »
и,,я (107) реагируют обычно с хл р ,„тНОй темпера1)Р - Рцэд
в течение нескольких часов при ‘ 0, /уравнение
ДназнрВДш(ы (Ю9) с выходами 40 ' А 50э
лппмяпьного циклоприсоединения имеет пиша,,
Эта реакция Ф Р(1д ' использовании самого хлораминаР?ц?
0бЛп6погоР ему реагента, гпдроксиламнн-О-сульфокислоты) ,м"
подобного ею р 1|П0В в реакцию вступают разнообрази,"
основания - производные альдегидов н ациклических
^SSecKHX кетонов. Во многих случаях шиффово основание
OH IO превратить в дназпридш. <n situ с выходом 50—80%, пр^
Хя реакцию карбонильного соединения с аммиаком или перв^.
ньш .ив вторичным амином в присутствии гидрокснламин-О-суль.
Локисзоты пли N-замешенион гидрокснлампн-О-сульфокислоты
По видимому, дназнриднн образуется в результате первоначально-'
го нук геофильного присоединения по двойной связи углерод-азот
п последующего замыкания цикла с. одновременным отщеп-
лением В отличие от оснований Шиффа оксимы [102] реагируют
с хлорамином или гидрокснламнн-О-сульфокнслотон, давая д11а.
зоалканы (реакция Форстера).
(39)
(109)
Присоединение цианистого водорода к шиффовым основаниям
протекает легко [4, 76, 99а, 100]. Эта реакция представляет со-
бой удобный способ синтеза а-амннонитрилов, гидролиз которых
дает а-аминокпслоты (синтез Штрекера [119]). Эта реакция
(аналогичная реакции образования циангидринов из карбониль-
ных соединений) обычно проводится с использованием безводного
цианистого водорода в инертном растворителе (эфир или бензол).
Более удобным оказалось, однако, использование цианида натрия
в фосфатном буферном растворе; выходы продуктов при этом
выше. Этот реагент служит также для превращения оксимов в
соответствующие а-гндроксиламнпопнтрилы [996, 100, 102].
Было предложено [120| использовать в синтезе Штрекера не
цианистый водород, а гораздо более безопасный триметилсилил-
цианнд. Этот реагент присоединяется к шиффовым основаниям в
присутствии таких катализаторов, как хлорид алюминия(Ш).
».„»™Л|ВКа "л" тРис(ацетилацетонато) алюминий, давая
ют и иг'- ’тР11Мет”лсилилам|1ны, которые, не выделяя, гидролизу-
(80—9 о!?а“ среде в “’аМ|1»оинтрнлы с высоким выходом
силплин-м ипя р" К11слот"ом гидролизе этих аддуктов триметил-
снлилцпапип п Л"° Сра3у ПОлУ,|11ть а-ампнокислоту. Триметил-
оксимы в а гтппРПСуТСТВ"" 1,од,|да mniKa(II) также превращает
вольно Инер?мыРе KCX™I10H''Tp,1J,b' (120)' Г11ДР а зоны, обычно до-
тируют с цианистомИ™110 нуклеофильных реагентов, легко Реа'
«-цианогпдразинп с vr, ОД°рОдОМ " ацетонциангидрином И Да10Г
6й6 рошим выходом. Ациклические [19] п Ц|1К*
.«не I9! нминиевые соли экзотеп1;!щнп n
водородом И В некоторых случаях с водным^’'01 С ЦИаНи'
с' "атР"я> давая с отл”,(”ьши выходами S16*" ЦИа’
1Н^ “ '1иа-1К1маминоиит-
Шиффовы основания (ПО), не СодерЖаЩ)(е
.„.положении к двойной связи углерод-а30т аг™Ов Города
® м1, Гриньяра и алифатическими или арОмаТИ’‘?ют с Реа«-
“аннческими соединениями так же, как и капбп ими литий-
•;;; „Р„ ™p«»Sf обр.з,»™„, Ор»
"•чают вторичные амины (112) с выходами 60~Х / ' П°'
(0). Реакдкя протекает лучше с ароматическими ачь^мииТ"6
может считаться общим методом синтеза вторичных аминов о”
мко он имеет ряд недостатков. На присоединение метапоогаии'
чадах соединении оказывают влияние пространственные зато™'
цепня, создаваемые заместителями (например, N-Tper-6vT«^
группой) В шиффовых основаниях; стерический эффект замес™
телен в металлорганпческом реагенте, по-внднмому, ие прояви’
ется. Кроме того, в некоторых случаях (особенно пои пеакнии г
реактивами Гриньяра) выходы вторичных аминов снижены из зя
параллельно протекающей восстановительной димеризации шиж
фова основания [(110)^(113)]. Область применения этого мТ-
года синтеза аминов удалось расширить [121] за счет использо-
вания в качестве шиффова основания N-алкнлнденаренсульфен-
амндов (НО; R3 = SAr). При реакции этих субстратов с литий-
оргаиическпми соединениями и последующем гидролизе подучают
первичные втор- и трет-алкиламины (112; R3 = H) с выхода-
ми 40—80%.
RK
,C=NR3
(112; R3 = Н) с выхода-
Rk /R3
,C-N'
R3/Z XMgX(Li)
(Ш)
R4MgX или R4Li
(HO)
|r<MsX
zR1
R\
XC—NHR’
(40)
RK
R2/f fXR2
R3HN NHR3
(113)
К аф-непасыщенным альдиминам
присоедипя|отся в положения 1,4, и i
Гриньяра
гтвЛР 1р2]- Наличие должным
). алкокспка]
R*
(112)
‘ (,|1> К™%рИу»«
..................................- П0СЛе Геп1ды (,а,6) <)'равне'
“кидаемые ^-замещенные насЫШ'е11Н?'е„,аЛВДпасположенного заме
ние 41) [122]. Наличие должным °бРаз<\ р Ппы), сП0С')бТ,е
жителя (например, алкокспкарбонильнот I. Гр11НЬяра в ходе
к°орд|пшроваться с атомом мел.ал.'!п Способствует выс0 ’цоо]
акого 1,4-лрпсоедннення [ср- (,|0^ поодемонстрнР°ва
Реоселекгнвностп этого процесса, что р 507
образованием хиралыюю р-замещеипого альдегида из х
шппиото предшественника [(114)->-(115)->(| (в) j. "Ра-'1Ьного
Ациклические [19| и циклические [9, 19] иминневые соли реа-
гируют с реактивами Гриньяра и с алкил- и ариллнтиевыми
реагентами нормальным образом и дают соответствующие третич-
ные амины. В связи с этим уместно упомянуть о том, что присое-
динение дитианиллитпя (118) к солям альднмииия (117) н после-
дующий гидролиз аддукта (119) представляет собой элегантный
способ синтеза «-аннпоальдегндов (уравнение 42) [19а].
+ /—3
rch=nr2 + ( ]
R
S, ।
)—CIINRj
сно
и 2 о I j 1 -
---> RCHNR2 (42)
(120)
(47) (118) (П9)
™ДЛгД|>КЛ,1МЫ I'02’ Пмеюшие а-водородных атомов) реагп-
и\зшт ото? Реактива Гриньяра или литийорганичсского реагента
ми Говн амины- взаимодействие кетокснмов с реактпва-
гидраРзоиы Р(П1ппО,Те1<аеТ б0Лее сложно 11021- М.М-Дизамещенные
лорганическнми соед'шенпящГт'^''’^ а™М°В) Ре?™РУ,от с меТаЛ’
однако 1,2-пР11соеди11Р ир п Же’ как 11 и'"ФФ0ВЫ основания,
пнями [6Ц. N-Xnnn.m, .е Реагента осложнено побочными реак-
ных соединений с хппДЫ (легк°Лоступные по реакции карбонпль-
обменивают атом xnrJ, М1,11ом) ПРП действии реактивов Гриньяра
бой удобный способ г,,,,!'3 рад|,кал реагента, что представляет со-
вы основания, не сно R3a шпФФ0ВЬ1х оснований [101]. ШпфФ0'
“водородных атомов Сп°Р 1,Ые К енол1|зацни, т. е. не имеющие
соединение по двойной т^'1а|от в реакцию Реформатского; при-
^фично. ОбразX .и!"13” У™ерод, — азот протекает стере0'
лить при низкой темцрмСЯ эрУтР°'Р'амяноэфпры можно виде-
«мопроизвольпо цХ ’ "УРе (-10ОС)> в других условиях они
oc'iinKTa!,1OB 1,231- Щелочи^51’ ЧТ0 ОткРывает путь к синтезу
взинямп, содержат,,.,. 'е мета,,ль1 реагируют с шнффовыМ”
60а 1 Л|1шь арильные заместители, дав*1*1
N С-бис(металл) производные, которые можно далее алки-шоо
‘ать или ацилировать сначала ио углероду, а затем по азоту и
получить смесь продуктов С- и N-алкилирования, -ацилирования
‘ алкилирования - ацилирования [4, 100, 124]. 1
Азометнны (шиффовы основания, оксимы и гидразоны) с
д.алкильпыми заместителями, содержащими по крайней мере один
свободный а-водородный атом, проявляют особую реакционную
способность по отношению к металлорганическим и подобным
реагентам вследствие возможности отрыва этого а-водородного
атома (это сходно с образованием енолята). Так, енолизующиеся
альднмииы н кетимины подвергаются С-алкилированию при дей-
ствии реактивов Гриньяра; реакция протекает через гриньяров-
СКИН комплекс енаминпого таутомера, после гидролиза которого
с высоким выходом получают соответствующий а-алкилированный
альдегид или кетон. Этот процесс был использован для синтеза
хпральных а-алкилированных циклогексанонов с высокой (85%)
оптической чистотой, исходя из хиралыюго исходного имина
[125]. а-Алкилнрованные кетоксимы (121; Y == ОН) и четвертич-
ные соли гидразонов (121; Y — +NRj) циклизуются в азирины
(122) при взаимодействии с реактивами Гриньяра (перегруппи-
ровка Небера; см. с. 494); дальнейшая реакция приводит к азири-
динам (123) с хорошими выходами (уравнение 43) [126].
RIMrX R'\ R'\ /R8
/СН CR3 * —у R3 * /\ (43)
R2/ <!>Y *г/V R!ZX\H R'
(121) (122) (123)
В настоящее время мезомерные азааллильные анноны, которые
генерируются из а-С- п N-алкилированных азометпнов прн дей-
ствии сильных оснований, используют как промежуточные соеди-
нения в различных синтезах. Так, шиффовы основания (124),
имеющие а-С-заместптель, обычно гладко металлируются диалкил-
амидамн лития, например динзопроппламндом лития, в эфире
или тетрагидрофуране при —70 Н-------30 °C и дают литиевые соли
азааллильпых анионов (125) (уравнение 44) [127]. На амбидент-
ный характер этих анионов указывает тот факт, что они ацили-
руются как но углероду, так и по азоту. Их можно использовать
как карбанионные эквиваленты формн.т.метнленового фрагмента,
что позволяет при «непосредственной альдольнон конденсации»
с альдегидами и кетонами получать с высоким выходом (~9О/о)
группировку а,р-ненасыщенного альдегида (128) в составе различ-
ных, в том числе и природных соединений [127]. Первоначально
образующиеся а,^-ненасыщенные имины легко превращаются в
свободные альдегиды путем перегонки с паром в присутствии
Щавелевой кислоты. 1-Азааллильные анионы легко алкилируются,
11 эта методика алкилирования иминов была использована (исхо
дя из хпральных иминов в качестве субстратов) для спите.
509
хиральных «-алкилированных кетонов [128]
[129] с высокой степенью оптической чистоты.
u а-амтюкислот
UN(Pr-z»o)t | Li+ >0=0
ТГФ, -70 °с Ck-/Nx
•СХ /
С н,0
О.
Li'
•С'
ОН 1
(127)
N.
(125)
(126)
Н30’
;с=с—сно
(128)
(44)
Н
А
Конденсация а-С,0-дплптийоксимов с альдегидами и кетона-
ми дает р-гндрокспокспмы с выходами 50—70%. Эти соединения
легко циклизуются под действием холодной концентрированной
серной кислоты, что открывает путь к синтезу Д2-изоксазолинов
[130|. N-Незамещенные гидразоны, не способные к енолнзацпн,
гладко металлируются амидами щелочных металлов в жидком
аммиаке и дают М,М-дианноны, из которых в зависимости от ко-
личества используемого алкилирующего агента можно получать
моно- п диалкильные производные [61]. Жесткая обработка
N-незамещенных гидразонов сильными основаниями (например,
трег-бутоксидом калия) в диметилсульфокснде приводит к отщеп-
лению азота и образованию соответствующего углеводорода
(восстановление по Вольфу—Квжнеру [131]). В N-моиозамещен-
ных гидразонах ароматических альдегидов протон NH-группы об-
ладает слабокнслымп свойствами. Металлирование этих гидразо-
нов действием амидов щелочных металлов в жидком аммиаке
дает амбидентные диазааллнльные моноанцоны, которые можно
проалкплпровать по концевому атому азота, это представляет со-
бой удобный способ синтеза Ы,№дпзамещенных гидразонов [61].
N-Монозамещепные гидразоны, содержащие в а-положенни пер-
ы" 11Л" ВТ0Р|1ЧНЬИ”1 алкильный заместитель, реагируют с ами-
моно- м®таллов днпзопропиламндом лития, давая
чппуются по В В1'Яе с00тветствУ1ощнх солей), которые алки-
Большой * °ТУ U уМеР?л>'- ответственно [ 132].
поведение " теоРет"'1еский интерес представляет
Реакциях с снльннмиЛп^°”ИЛГПЛраЗОНОВ альДетндов и кетонов в
МОСТИ от Условий мпг основаниями [133], при которых в завпеи-
кены, как претерпевшие ооразопаться циклопропаны и (или) ал-
(схема 45). Направченч’ № 11 Не претерпевшие перегруппировку
вания, так и от ппйпп„^ реакШ111 зависит как от природы осио-
этплеигликоль) и Растворптедя. И в протонных- (обычно
510 ’ р т ,1,их (обычно Д11МСТ11Л0ВЫЙ эфир Д11’
,т1(ле11глпколя или N-метилпирролидон) п,
сильного основания (обычно алкоксид наРтп^°рителях действие
тонирование тозилтидразова (129) в aHI)0JP " , В1^^вает мпр^
тозилат-иои, превращается в диазоалкан (13п ’/ КОторий. теряя
ляюшая стадия) (реакция Бамфорда— Стивенса <иЖ?СТЬ0ПРеде-
тельная судьба которого определяется поипопп” ^]), оконча-
В отсутствие источника протонов („ли есл п Растворителя
„сходит медленно) отщепляется азот и обпяП?°ТОНИроваиче про-'
который внедряется по соседней углевоз ггДУ ся каРбен (132)
циклопропан (134) или претерпевая и о, р°ДНОи СВязи. давая
дает неперегруппированный алкен (133) в родНый сдвиг
гелях протоиироваиие протекает быствее “ "ротонны* раствори-
и разложение диазоалкаиа (131) поиволйт ? °™егмение азота,
(карбокатиону) (135), который стабилизуется КпарбенИевомУ иону
R'\
'CH-CH=NNHSO2Ar
R2Z
2Li+
c—CH=NNSO2Ar
(129) (138)
I
V
;сн—CH=NNSO2Ar —>- Vh—CH=N=N )c=
R2/ R2/ RiZ
(130) (131) (is»;
J
*)сн-сн2
R2Z
(135)
I
*'CH-ch2R2
(136)
j-n-
R,c'4^CHir
(137)
R-\ ..
Vh-ch
R2/
R!\
'c=CH2
R.’Z
(133)
„ потери протона, приводящих к перегруппированному алке-
р0Во'<7\ В, хоты продуктов реакции Бамфорда-Стивеиса в пр0-
‘ „чгтпопитепях часто довольно низки, поэтому для получе-
тонных рас 1 опа)1ОВ> так и неперегрупиированных алкенов
пеакш.ю стедует проводить в апротонных растворителях.
Р SfioTKn тознлгидразонов, имеющих по крайней мере
„•шп свободный а-водородный атом, алкнллптпевым реагентом в
чппотошюм растворителе (обычно в эфире) при комнатной тем-
прпатуое (133] позволяет с высокими выходами получать непере-
го-ш1иповаш1ые алкены. Такой вариант приведенной выше
схемы имеет большое значение в синтезе. Образование алкенов
в' этом случае .можно объяснить тем, что вначале происходит де-
протоннрование с образованием днаннона (138) (о его образова-
Н1И1 можно судить по характерным реакциям, в которые он всту-
пает), который распадается с отщеплением тозвлат-пона и азота;
прото’нированпе образовавшегося внннлыюго карбаниона (139)
приводит к неперегрупппрованпому алкену (133) [133]. Синтети-
ческая ценность этой реакции тознлгидразонов значительно рас-
ширилась после того, как было показано, что промежуточно обра-
зующийся карбанион (139) можно ввести в реакцию с различны-
ми электрофилами (кетонами, алкплгалогенидамп, диоксидом
углерода, реагентами Вильсмайера), в результате которой можно
получить алкены, а,0-неиасыщенные спирты, альдегиды и карбо-
новые кислоты [134]. Было показано, что регпоспецпфпчпость
превращения тознлгидразонов а,₽-пепасыщенпых карбонильных
соединений в диены под действием алкпллитпевых соединении
[135] зависит как от природы растворителя, так и от стереохимии
тозплгпдразона. Алкилирование тознлгидразонов альдегидов ал-
кпллнтпевымп реагентами приводит к образованию углеводородов
с выходами 40—60%. В целом этот процесс соответствует восста-
новительному алкилированию исходного карбонильного соедине-
ния [136], Пиролиз солей щелочных металлов тознлгидразонов в
жидкой пли паровой фазе — хорошо известные методы генерирова-
ния карбенов in situ [137]. Использование этих методов для син-
теза напряженных циклических систем (за счет внедрения или
каРбевов) проиллюстрировано уравпенпя-
явчортро " П39]. Последнее из приведенных превращений
степного пр,1мсРом элегантного синтеза [6]парациклофана (недо-
цнклоЛяппГуг11МП сп°собами), наименьшего из известных пара-
му иска-< ’ В K0T0P0il носкость бензольного кольца, по-впднмо-
‘j> л\сг| а,
альдегидов Н1||Р°крНпе ^,М-диметнлгндразонов а-алкплнрованпых
температуре (—78°ПВ днизопРоп”ламидом лития при низкой
ствующпх 1-ачятн^>п Гдадко приводит к литиевым солям соответ-
теза полифуикш.овялк,','.?”0®- Было показано [140], ЧТО ДЛЯ С1И1-
нменно эти соедине,шя"ЫХ молекУл лучше всего использовать
образующиеся иГ "С.еноля™ ¥» азааллнлыгые анионы,
512 НМ,1НОВ <см- выше). Они могут быть легко по-
Ml1’X да углерода. Эти соединения обладают ™С',0₽ода'
" ; °Хй способностью; реакции с ю' участий ВЬ"ие"Н0Й ре‘
большей региоснецифичиостью и приводят к иродуХ™/
“ешения (а не полизамещення, как в слу1^ X” ° 3
свойства, а также легкость регенерации карбонильною соедине
н,1Я нз образующеюся в результате реакции гидразона (см инже)’
делают их идеальными реагентами для непрямого введения Хнк
Зональных групп в альдегиды н кетоны. Так, азааллильные ани-
оны, образующиеся из диметилгндразонов хиральных альдегидов
и кетонов, подвергаются энантиоселективному алкилированию с
более высоким общим выходом и большей оптической чистотой”
чем в случае анионов, производных иминов [141]. Было показано
[142], что азааллильные анионы из N.N-диметилгидразонов аль-
дегидов образуются с высокой степенью стереонзбирательности и
что стереохимия их не изменяется при алкилировании нли аци-
лировании.
(46)
весьма
а,^-не-
путем 1
выхо-
N, N-диметнлгндразонов
высоким выходом (ча-
Включение атома азота
остроумно использовано в
гндразона в состав цикла было
j____ ______,_____ _ элегантном синтезе хиральных
насыщенных альдегидов (которые трудно получить
с высоким химическим (60- 80%) н оптическим (60-SUA)
Дами [143], Азааллильные анионы из
альдегидов и кетонов можно превратить с —--- спрдине-
сто превышающим 90%) в ₽- и у-гидрокспкароонпльны едннй
Н11я> а также в 1,4- и 1,5-дикарбоннльные соеди р бннсону
н1*?!?3 является улучшением метода никелирован 1 значитМЬН0
I 44]. Более того, область применения этон Р „зонов альде-
Расшприлась за счет того, что анионы д|,"®]т1, ))Х гладкого
Д°в и кетонов можно использовать 1|4щ эквиваленты ани-
Ревращенпя в а-тиометильные производные к_ , HV10 функпи-
‘евЬ1х ионов. Это позволяет проводить пхк. , „аудирование)
ализацпю (например, алкоксилнрованне, 5)3
Зак. 105
„,^-Риае к карбонильной группе (и отличие от обычной
В.,^фи.,ь!'о>1 функивопализан.п» с высоким выходом (90—
97%протоннроваШ1е а-СЛ’-Диалк,,лироваииых азометинов (144)
д евро то, привести либо к 1-азааллилыюму аниону
Т л б2азааллилы.ому аниону (146) (уравнение 48). НаУ
.п .ктнкс денротоппрованне преимущественно привод,, г к -
Хп-ьвому аннону из-за его большей устойчивости
1-аза-
[14GJ.
н
LiN(Pr-U30)E I LlN(Pr-Kw),
—-----------/Ns, / -------------
ТГФ,-70°С П СС, ТГФ,-70С
(144) Н
(145) (146)
2-Азааллнльные анионы (148) можно легко получить [65, 147]
депротонированием N-алкилпрованных оснований Шиффа (147),
не имеющих а-водородиого атома, при низкой температуре
(—70°С) действием днизопропиламнда лития в тетрагидрофуране
(уравнение 49), или путем конротаториого раскрытия цикла лег-
кодоступных N-лнтнпазпрндинов (149). Как уже отмечалось, ме-
зомерные структуры типа (148) принимают участие в каталнзуе-
мой основаниями азометин-азометинной прототропии [см. разд.
8.1.1.2(2)]; интерес к ним обусловлен также и тем, что они могут
участвовать в реакциях циклоприсоединения (см. с. 524 сл.).
Н R1
I I
I'N^R2
(147)
LiN(Pr-U3tf)2
ТГф,-70°С
(49)
ты ^НИглНяЫлЛТ0Г° ТИПа М0ГуТ вь:гтУпаТь как нуклеофильные
давая (аишлЛ1аП1РУ10Т\ С альдеГ|1Дамп и кетонами [147,
действием ,,л ,денам»н°)спирты, которые
(действием тионнлхлорида в пн„
2-азабутТдТевовТзЯ'?й яКмТЛ0ТЫ^ ЧТ° откРывает ПУТЬ к синтезу
’ р аминосинртов, соответственно (схема 50).
514
типа могут
выступать как нуклеофильные
а ген-
148],
можно дегидратировать
в пиридине) ил,, гидролизовать (дей-
1. r2C=O
2.Hfi0
R\ Ph
/С—ch2n=c/ 2^
R ' \>h
OH ia
SOClj,
пиридин
R\ Al
/C=C\ Ph
К XN=C(
xPh
ch2nh2
OH
(50)
Альдиминиевые соли, не содержащие а-С- и N-алкильных заме-
стителей со свободными атомами водорода (150; R1 = н R2
R3¥=Me или первичной или вторичной алкильной группе) депро’
тонируются при действии трпэтилампна в таких растворитетях
как дпхлорметан пли ацетонитрил, давая продукты (152), кото-
рые можно рассматривать (хотя бы формально) как результат
самосочетанпя амииокарбенного интермедиата (151) (схема 51)
[19а]. Однако карбены типа (151) не удалось «поймать» с по-
мощью различных типичных «карбенофнлов» (например, трифе-
нилфосфнпа, тетрацианэтилена, циклогексена); это свидетельству-
ет против того, что онп существуют как самостоятельные частицы
и принимают участие в этих превращениях [19а]. а-С-Алкилиро-
ванные иминиевые соли, имеющие хотя бы один свободный атом
водорода [(150; R1 =Ме)], депротонируются основаниями в ена-
мины (153) [19а]. И наоборот, иминиевые соли, несущие N-ме-
тильную, первичную пли вторичную алкильную группы и лишен-
ные а-С-водородных атомов [(150; R’=#Me или первичной или
вторичной алкильной группе, R2, R3=Me)] под действием осно-
ваний, например феннллития, превращаются в неустойчивые илн-
Ды (154). Эти нлнды склонны реагировать с исходными соедине-
ниями, давая конденсированные иминиевые солп (155). которые
при гидролизе распадаются на кетон (156) и 1,2-диамин (157)
[19а]. . .
Двойная связь углерод—азот подобно двойной связи угле-
род—кислород легко вступает в реакции конденсации альдольно-
го типа [4, 100, 101]. Шиффовы основания и N-замещенные арил-
альднмины, в частности, гладко реагируют с соединениями с ак
тивиой метиленовой группой в самых разных условиях, давая ад
Дукты (160), склонные к отщеплению амина, что приводит к соот-
ветствующему алкену (уравнение 52). Эти процессы если " “Х
преимущества по сравнению с реакциями конденсаци < Р '
пых соединений, то очень незначительные В качает рР= ,це)
приведена реакция N-метилбеизальдимпна (15S. Аг . ’
с 2-нитроацетофенопом (159; X = COPh V = NOJ в
уксусного ангидрида, приводящая к (бенз*',л1 псакц‘ии этого
нону (iGj; ArJph xLcOPh, Y=NO2). -Многие реакции
’ 515
17*
Ph.
Ph
PhC=NR2R3 PtlC NR R R3R2N/ \nR2R3
BtsN.
CH2CI2
R2, RS^Me.
пврВ' или втор-А1к
R'\ +
V=NR2R3
PhZ
RleMe
ElaN, MeCN
(150)
Ph,
XC—NR2RS
H2C^
(153)
RSssI^saMe,
Ri^Me,
пере- или втор-Мк
(51)
Ri + Л1е
><
Ph' 'CH2
(154)
Ri +/Me
XC=NZ /R'
pi/ \ch2—c;
|\ph
NMe2
(155)
R\ /R’
;c=o + MeNHCH2C'
PhZ PPh
NMe2
(156) (157)
типа приводят к довольно устойчивым аддуктам, которые, будучи
р-фупкцнонализировапиыми аминами, являются важными проме-
жуточными соединениями в различных синтезах. Так, в частности,
этиловые эфиры арилуксусных кислот (159; X = Ph, Y = CO2Et)
присоединяются к N-замсщенным бензальдимипам в присутствии
основания или кислоты Лыонса (например, А1С1з), давая (3-ами-
ноэфиры (160; X = Ph, Y = CO2Et), которые могут циклизовать-
ся в производные (3-лактамов (114]. Аналогично, p-кетоэфиры в
условиях кислотного катализа присоединяются к бензилиденаин-
лам; образующиеся при этом продукты можно использовать в ка-
честве исходных соединений при синтезе хинолинов [149].
Аг\с/Н АгСН—NHR
II +ХСН2У —> X—CH—Y ------------—* АгСН=С;
пк —RN112
(52)
(,68> Я6») (160) (16!)
мпмЕт,?’ <l63\ c "лидами серы (162), стабплнзованны-
тод синтеза R ГРУ”1Ю"’ та'<а<е Представляет собой важный ме-
р-аминоалкенилкарбоннльных соединений (166)
516
пение 53) [150]. Эти реакции протекают по схеме, вклю-
(УРав первопачальиое нуклеофильное присоединение с после-
чаЮ'яе,м замыканнем цикла и затем раскрытие цикла в промежу-
дУ|йШ1 образовавшемся азиридиновом производном (165). Это под-
точно ается тем, что подобно тому, как из карбонильных соеди-
чверЖД бразуются оксираны, при реакции анилов с метилидом
1'еИ етилоксосульфония получаются азиридины [151] с умеренны-
„невыходами.
‘CH COR ~
(162)
ArN=CHAr
(163)
Me2S-^CH-COR
ArN—CHAr
(164)
"С.
Fr COR
(165)
ArNH- COR
\ /
c=c
/ \
Ar H
(166)
(53)
а,р-Неласыщениые альдпмнпы подобно соответствующим кар-
бонильным соединениям также вступают в реакцию 1,^присоеди-
нения по Михаэлю с карбанионами, получаемыми из соединений
с активной метиленовой группой. Показано [122], что хиральные
аф-неиасыщеиные альдимпны в присутствии трет-бутоксида калия
присоединяются к диэтилмалонату, приводя к соответствующим
хпральным аддуктам (асимметрическое присоединение по Михаэ-
лю) с умеренным химическим (~50%) и оптическим выходами
(50-80%). Пол агают [152], что реакция пмпнпевых солей с сое-
динениями, содержащими активную метиленовую группу, явля-
ется ключевой стадией в реакции Мапнпха — широко используе-
мом методе аминоалкилирования СН-кислот реакцией с карбо-
нильным соединением (обычно формальдегидом) в присутствии
соли вторичного амина. Недостатками этого метода являются
большая продолжительность реакции, трудность оптимизации
выходов, неудовлетворительная воспроизводимость результатов и
осложнения за счет побочных реакций. При введении в реакцию
конденсации с соединениями с активной метиленовой группой за-
ранее полученной пминиевой соли удается преодолеть практиче-
ские сложности, присущие этой реакции, и значительно расширить
область ее применения [19а]. Для этой цели особенно подходя-
щей оказывается дешевая и легко получаемая устойчивая имин е
вая соль, хлорид N,N-диметил(метилен)аммония СНг^-У ег •
Этот реагент гладко превращает альдегиды и кетоны (2 , >
в соответствующие дмпиоалкильпые производные с высоким: вы о-
дом (50—90%) [153]. Примером легко протекающей реак i
517
сечей с енаминами служит элегантный биомпметпче.
"М "•'"синтез'0 ни котип а (170) [154], включающий ооразоВание
4 w.iTpoiiiipnAnHa (167) и его реакцию с ацетатом
%е'пч-ДМифролшшя (168) с последующим окислением кисло-
родом воздуха (уравнение 54).
(467) (168) (‘в») (™)
(3) Реакции циклоприсоединения [/55]
Циклоприсоединение к двойной связи углерод—азот хотя н
изучено не столь широко, как присоединение к углерод-углеродной
двойной связи, постоянно привлекает внимание. Отсутствие ин-
тереса к реакциям этого типа было обусловлено относительно
меньше устойчивостью связи C=N по сравнению с С=С (на-
пример, в реакциях сольволиза) и склонностью способных к ено-
лнзации азометинов участвовать в реакциях циклоприсоединения
как енамины, а не как нмнны [156].
Реакции [1 + 2]-циклоприсоединения карбенов и карбеноидов
к азометпнам в настоящее время хорошо известны [155, 157];
они представляют собой удобный метод синтеза азиридинов. Про-
стейшим примером циклоприсоединения этого типа служит реак-
ция бензилиденанилпна (171) с дихлоркарбепом, приводящая к
1,3-дифенил-2,2-дихлоразиридину (172) с хорошим выходом (55%)
(уравнение 55).
(172)
(55)
(171)
Р°Да Успешное применение нашел катализ
невипу₽мыйР1еН0Са [118Ь БЫЛО показано> что дихлоркарбен (ге-
тнвен'по отНпшрХ^°Р0ф0рМа 11 основания) в 1,65 раза более ак-
логичной vnenm11^ К двоин.ой связи углерод—азот, чем к ана-
также отметитГ т?еР°ЛН0И двойной связи [159]. Интересно
(тригалогенмети’л)Чтт1|ИГа^пГеНКарбе''Ь1’ ге|1еР"РУемые "3 Фепил
электронодефицитной двойип^0 реаГ11РУют с соединениями с
с имииофосгенамн Р\=сс1>И УглвРод-азотиой связью, например
как с обогащенным., 2 с образоваиием азиридинов, тогда
6 „ игроками шиффовыми основаниями типа
OJO
^ензплндепатинта (171) реакция це м„ет
разуются также при реакции шиффовых J 571 • Пиридины об-
Спммонса - Смита (дииодметап - цинк-мею аиии с Реагентом
разование амидов типа <«7в) при реХТ.Г: ''?.Р.а’ U6OJ. Об-
ВЫХ солеи (173) с трпхлорацетатомРцатрия .« их иминие-
результат присоединения карбена с обоачо^"0 объвд<ить как
„вевого интермедиата (174), который noXnra₽ С"иРОаз'фиди-
цикла иод действием трихлорацетат-ионя 7 гР ся Ра“рытию
волизу (уравнение 56) [155]. последук>щему со.ть-
(175)
(176)
В настоящее время хорошо известны реакции циклоприсоеди-
нения азометннов к большинству обычных кратных связей (на-
пример, С = С, C=N, С=Р, N=N, N=0, С=С), приводя-
щие к циклоаддуктам типа 1 : 1 (соединениям с четырехчленным
циклом) пли продуктам раскрытия цикла [155]. Реже всего из
перечисленных типов реакций проводят циклоприсоединение
к простым алкенам; в качестве примера можно привести синтез
азетинов (180) с выходом 35—70% при реакции N-аренсульфо-
Ннлбензальдиминов (177) с дпампналямп кетена (178) [155].
Объяснить эти реакции можно, исходя из предположения об уча-
стии неустойчивого промежуточного цнклоаддукта (179), который
легко отщепляет 1 моль амина и дает конечнын продукт (уравне-
ние 57).
PhCH=.NSO2Ar
(177)
+
адх
хс=сн2
ад/
(178)
Н NRj
Н----1—NR«
Ph----NSOjAr
н -*
(179)
Н—j=j—XRs
Ph——XSO,Ar
H
(ISO)
Азометины вступают также и в реакцию с алке|'®'"'
фонилимин (182) реагирует с алкенами (181) и Д
Так, суль-
выхода.ми
519
60—90%
ПОЛуЧСШ1Я
яппукты (183) [161], которые можно использовать Аля
^-ненасыщенных «-аминокислот (уравнение 58),
Н
(181) (182)
(183)
Соли мегилсниминия R,N+=CH2 X” не вступают в реакцию
циклоприсоединения с простыми алкенами, а скорее выступают в
doth электрофилов, давая карбениевые ионы, депротопированпе
которых дает смесь у,6- и р.у-неиасыщенных аминов [19а].
Азометины (как ациклические, так и циклические) легко всту-
пают в реакции [2 + 2]-циклоприсоединения с кетенами; это явля-
ется общим методом синтеза (З-лактамов, протекающего с высоким
выходом [88, 114, 155, 162]. Такого типа реакции циклоприсоеди-
нения азометпнов изучены особенно широко, так как они нашли
применение в синтезе пенициллинов [114, 162]. Так, взаимодей-
ствие дпфеяилкетеиа (184), например, с А2-тназолпнами (185) при-
водит к производным пенама (186) (уравнение 59).
(188) (186)
используемые в этих реакциях, обычно по-
(184)
Неустойчивые кетены,
лучают in situ действием оснований, например трнэтнлампна, на
замещенные ацетилхлориды или из ацплазпдов с помощью терми-
ческой или фотохимической перегруппировки Вольфа. В отдель-
ных случаях кетены генерируют in situ термическим раскрытием
цикла мезононных оксазолонов (187) (уравнение 60) [114], а циа-
теРмическим разложением азидокротонолактонов [163]
или 2,5-диазпдо-1,4-беизохиц<)нов [164].
’СК °
Аг.
>—R 3^-8
Лг/\ /
N—Me
(187)
Единственным i
Rl .R2
О $
Me/ \COR
(188)
Me Ar R1
। I I
RCOX——г—R2 (60)
O--=L-NR3
(189)
ределепных KeTen<nwJl\°M ?т01° мет°Да является склонность оп-
Б20 "0В (в незамещенного кетена и ал-
кИлзамешенных кетенов) реагировать в димерной <hnnM
типами, в результате, чего образуются ше?тичлениые а
(ппперидиндионы), а не соответствующие р-лактамЫ
циклоприсоединение азометннов к кетенам протекав « 62 •'
степени стереоселективно [165], то есть протекает согтДп ‘СШ“
процесс. Однако наблюдаемую стереоселектнвность равнымХТ
зом объясняет и двухстадийный механизм с участием дииотярного
промежуточного соединения (уравнение 61), который к тому же
надежно подтверждается данными исследования механизма П6Ч
165] образования р-лактамов из кетенов и азометннов. Кетены
присоединяются также к двойной связи углерод—азот в N N-ди-
адвлгидразоиах, давая Ц-(дпациламино)-р-лактамы [162].’
н
_NPh
PhCH=NPh PhCH=NPh ph_
P2C=C—O' ” Я—
R,c=c=o
(61)
Циклоприсоединению шиффовых оснований к простым изоци-
анатам (191) [155, 166, 167] предшествует (как и в случае цик-
лоприсоединения к кетенам, см. выше) обратимое образование
цвиттер-ионного интермедиата (192) (уравнение 62), который в
некоторых случаях удается выделить [166]. Ход дальнейшей
реакции определяется как устойчивостью цвиттер-ионного интер-
медиата (и, следовательно, главным образом, стерическимн н
электронными эффектами заместителей в молекуле азометина),
так и подчинением реакции кинетическому или термодинамическо-
му контролю. В целом наличие электронодонорных /гара-заместн-
телей в бензилиденовом фрагменте шиффова основания (190)
и проведение реакции при низких температурах (а следовательно,
(62)
521
„„•mi контроль реакции) способствуют непосредственному
кинетическш Р0 )е (192), приводящему к [2 + 2]-
замыканию ш. . на0б0р0т, электроноакцепторные пара-зз-
цпклоаддукт) , 7|Л(|ден0В0М фрагменте шиффова основания
лчТм относительно высокие температуры проведения реакции
ппрловатетьно термодинамический контроль) способствуют
(IL™d90MV |4 + 2] -циклоприсоединению цвиттер-ионного интер-
Тпилтя (192) к присутствующему в равновесной смеси шиффову
основанию (’90) или'изоцианату (191) „ образованию 2:1 или
1 -2-аддуктов (194) н (195) [166, 16/].
Реакции циклоприсоединения шиффовых основании к ацнлнзоциа-
иатам [168] и ацплизотпоцнаиатам [169], в общем, подчиняются
тем же закономерностям, но проявляют тенденцию к непосред-
ственному [4 + 2]-циклоприсоединению (в отличие от реакций по
Т1ПУ (2 + 2]- или 1,4-диполярного [4 + 2]-присоединения). Шиф-
фовы основания (196), способные к таутомеризации типа нмнн-
епамин, реагируют с изоцианатами, давая ациклические продукты
(199), для образования которых можно предположить [2 + 2]-
циклоприсоединепие изоцианата к енаминному таутомеру и по-
следующее раскрытие цикла в образующемся неустойчивом аддук-
те (198) (уравнение 63) [166, 167, I/O].
Me
£
Ph/ %JPh
(196)
R
I
N
£
II
О
r°
Ph---NR
NHPh
PhNH4 /Н
^C=CZ (63)
Ph/ ^CONHR
СН2
t
Ph/^NHPh
(197)
Реакция оснований Шиффа с арилидеифосфораиами, протекаю-
щая по типу реакции Виттнга, приводит к алкенам п фосфиними-
нам (уравнение 64) [171] ; этот результат объясняют формальным
[2 + 2J-циклоприсоединением двойной связи углерод—азот
к двойной связи углерод—фосфор с последующим ретроцикло-
присоединением.
Аг'\../и 'К./Н
(198)
Шиффа с арилпденфосфораиами, протекаю-
(199)
PPh3 N,
АНИС—CHR
I I
Ph3P NAr2
Ar'HC—CHR
Ph3P—NAr2
Ar'CH=CHR
Pb3P=NAr2
(64)
522
довольно редким примером [2 + 2] -циклоприсоедине,,,
7а по тройной углерод-углеродной связи слу^ я азомети-
„(дроизокииолина (200) с инамином (201) \пян,?еаКЦИгя 3’4‘Ди-
водяшая к неустойчивому азетииу (202) (этот Х'™ Со)’ Ири'
перегруппировке с расширением цикла, „0 moZS СКЛоне,!
чеН» восстановлением) [155]. ожет быть «уЛОв-
' (200)
Протеканием подобного процесса можно объяснить реакции пна-
мпна (201) с ациклическими шиффовыми основаниями или нмн-
ниевыми солями, приводящие через неустойчивые азетины (203)
или (205) к продуктам раскрытия цикла (204) или (206) (схе-
ма 65) [19а, 172].
Реакции [3 + 2]-циклоприсоединения дпполярны.х молекул
(1,3-диполи) (207) к различным соединениям с двойными (208)
(и тройными) связями с образованием пятичленных циклоаддук-
тов (209) (уравнение 66) [173, 174] имеют такое же большое зна-
чение, как и реакция Дильса — Альдера.
+ Y у
(207) х/ / \
+ —> Х\ /
(208) А=В Х-В
(66)
(209)
Потенциальные синтетические возможности 1,3-диполярного цпк
Лоприсоединеиия впервые в полной мере были раскрыты Р
Демонстрированы Хьюсгеном с сотр. [173 . Поскольку 1,3-днпо
ляриая молекула должна содержать один (или бол е) Р ’ ’
эта реакция приобрела особое значение для синтеза। >
нообразиых гетероциклических систем. Наибольше (210),
ют 1,3-диполн двух типов: а) алленил-пропаргил
1 523
.„лнкации Хыосгепа [173], относятся 1,3..
к которым- № стабилизацию п содержащие ()рто^
ноли, hmwuiJ“,^ связь ч б) тина аллильного аннона (211)
„..льну» двойную « • Хыосгепа, относятся 1,3-ДИПол '
к которым- ‘габилизашно, по не содержащие ортого11ал1).’
оеюшие oKTCTii ю с 0,,ям перцО1о типа относятся диазоалка-
ной двойноп связь Л(,1 ов (2J06) ш1трпл11М1111|>1 (-210В) и Вит-
ны (210а), овода Р как С1ОСДШ1С1ШП второго типа иредставле-
азометннов (нитроны) (211а), азометннимииамн
(2116) и азометин-илпдамн (211в).
h x=Y-Z x=Y=z
у = м Z = CR2
Z == О
Z = NR
Z = CR2
> - /К
X
X = CR; Y = N;
Y = N;
Y = N;
(210а)
(2106)
(210в)
(210г)
X = CR;
X = CR-
xz
(211a) X = CR2-, Y = NR; Z = O
(2116) X = CR2; Y = Z = NR
(211b) X = CR2; Y = NR; Z = CR2
Реакции 1,3-диполярного циклоприсоединения по двойной свя-
зи углерод—азот [155, 173, 174] менее исследованы, чем реакции
с двойной углерод-углеродной связью [175] (как и в случае
[2 +2]-циклоприсоединения, см. выше). Однако известны приме-
ры циклоприсоединения азометннов к 1,3-диполям обоих типов с
октетной стабилизацией (см. выше); полученные результаты не
вызывают сомнения. Подобно тому как циклоприсоединение диа-
зоалканов к алкенам дает Д'-пиразолины [175], шиффовы осно-
вания гладко реагируют с диазоалканами (например, с диазоме-
таноы) в присутствии каталитических количеств метанола или
воды и дают Д2-1,2,3-трвазолины (214) (уравнение 67) [173,
'йлектРоноак11ептоРные группы в шиффовом основании спо-
mvnnu^I Циклоприсоединению, тогда как электронодопориые
нии с кЯт??1|тЗЯТ еГ°' Так„ого РОда влияние заместителей в сочета-
высокая прн1пспСК1ж действ,|ем метанола и воды в наблюдаемая
триазолинов и ПтгуЙ"ЧН0СТЬ ^обРазованне исключительно 1,2,3-
приводящего к Д:-19 Т'п альтеРнат|'виого направления реакции,
ванному 1 3-диполяпи™ Иа3°Л11Ну соответствуют согласо-
через полярное пенехп-».£ Ш1клопРисоедииени1о, протекающему
симальная стабилизация 6 СОСТОЯ|1пе- в котором достигается мак-
молекулярных орбитатей гкчмпт пеРекРыва»ия высших занятых
мн молекулярными ппКи-г в Диполе с низшими свободны-
174] рными орбиталями (НСМО) в диполярофиле [155,
521
,Ar
ArN \ CIlaNa Г1, АГ\
< *—X----- ArCH=NAr - - 2 2, / Аг
(67)
N
(212) (213) 4
’ (214)
Подпой противоположностью реакциям 1 3-дниопяп,
„соединения, наблюдаемым для шиффовых ад^аииГ ЦИКЛ°'
ся реакции пмиииевых солеи [как ациклических >1 ' вляют’
лических] с дназоалканами (обычно с диазометаном/^п" ЦИК‘
щие к солям азиридиниев (217) [9, 19, 176J; эти оеакви рИЕОДЯ'
считать наиболее общим методом синтеза азирид11ииеаых“
(уравнение 68). Реакция протекает гладко и с высоки J о ей
при О °C в инертных растворителях. По-видимому^первона"
„дет нуклеофильное присоединение к карбиминиевой двойной
связи, а затем замыкание цикла с потерей азота [196 1761 Пои
соединение дназоалкапа к имнниевым солям, хотя п дает главным
образом цыс-азиридиниевую соль, не вполне стереоспецифично
(обычно выделяют и небольшое количество транс-продукта) т. е
в качестве промежуточного продукта может выступать катион ди-
азоння (216) пли образующийся из него карбениёвый ион, у кото-
рых отсутствует связь C=N и обусловленная ею стереохимия ис-
ходной карбиминиевой соли; это также -------------------
тый механизм данной реакции.
R'\r/Ri Rk„^-R2
|| CHeN2
r3/+\r4
подтверждает ступеича-
Р‘ .Р£
Cf
(215)
R3/ VR4
(216)
(217)
(68)
Х= BF4,C1O4
Реакция циклической нмииневон соли с дназометаном, приво-
дящая к спироазпридиниевой соли, и последующая фрагментация
возникающего нлида иллюстрируют (уравнение 69) общий метод
введения в молекулу экзоцпклпческой двойной связи; особое
значение эта реакция имеет в химии стероидов [19а].
'-Г
R;
525
о ОКСИДОВ нитрилов по ДВОЙНОЙ связи уГл
Присоединение ок зд0 детчС1 чем По двойной связи yr"®'
род—азот, ПР°ТХ'° илЯст собой общий метод синтеза Д<124'
род-кислород. чреда с liblcoKIlM ВЫХОДОМ (уравнение 70)
охсадиазол»»08 1/й 'омпоне|„ (219) в циклоприсоединении ЭТОГо
174]. Азометидавы) (ескпм N-алкил- или N-арилальдимином
типа может 0|‘ть 3 аЛ1(11Л „Л|| арил) или входить в состав Ш1К.
„л11 -кетимидам I присоединения оксидов нитрилов (2|8) к
ла, как в слу nr\ приводящего к конденсированным
ДМиразо.и.нам (221.Х 7[) [174]. в такую
структурам । . зд0 Менее реакционноспособная двойная
SbX- (219; Х-ОН),
сели ее сначала активировать путем образования координацией-
Того соединения с кислотой Льюиса (например, с эфвратом трнф.
бооа)' это открывает возможность синтеза недоступных
ZX способами производных 4-М-гидрокси-Д2-!,2,4-окса диазо-
лпна (220; Х = ОН). Таким же образом присоединяются к окси-
дам нитрилов в /^-изоксазолины (221; Х = О), давая сочленен-
ные структуры [(222); Х = О)] [174].
R'\
XC=N
ArCsN-0
(218)
Аг-
R'-
(219)
---j—R2
\Ar3
(221)
ArCsN-0
(218)
Ar-
(70)
(71)
/X
(---NZ
I J/R1
(X /Xd!
О K
(220)
R1
o I
' ------i—R2
-x’x.
X K
(222)
1,3-Диполяриое Циклоприсоединение азометпнов к иптрилими-
нам является, по-видимому, наиболее удобным общим методом
синтеза Д2-1,2,4-триазолииов [155, 173, 174, 177]; необходимые
нппя?Т°Г° 1'итР|1лп1'|,и1ы обычно генерируют in situ действием ос-
9 1 nt-'L "а г||дРазоно|,лгалоге||иды или термолизом подходящих
вним^ни^^пУп51^’ тетРазолов„ (схема 72) Обращает на себя
иость (в отгшч^р Х1,М|,чсский выход и высокая региоспецифпч-
Диазоажанам; см. выищ)^11"1' 11ВКЛОПР1|Соел11не1|11я азометпнов к
обладает бопьшей°Нл1" ТеХ Же уСловипх означает, что связь C=N
С=О. Оксимы также ппХОфиЛ’’11оГ| акт"вностыо, чем связ17
алдукты, которые в с "р,1соел-и|1я1отсЯ к ивтрплимииам, давая
вольной дегидратации5 Пп альдокснм°в подвергаются самопроиз-
пропзводным 1,2 4-тшп’запк1а°/АЯ с °'|С|1Ь хорошими выходами к
’ ‘рназолов (уравнение 73).
02(5 '
Ск
ph/C'N\NHPh
Et3N;
-HC1
phN—№CPh+ \=N
Me/ 'Рг-изо
'''NPh
uao-PrN-
Me
(72)
R'O\-NPh
Me
(84%)
(73)
Известно довольно мало примеров циклоприсоединения азоме-
тинов к октетно-стабилизованным 1,3-диполям, лишенным орто-
гональной двойной связи [155, 173]. Этот тип циклоприсоедине-
ния азометинов иллюстрируется взаимодействием N-иминов и N-
илидов 3,4-дигидроизохинолина (223; X = NR или CRa) с N-a.i-
кил- или N-арилальдиминами (224; R2 = алкил или арил), приво-
дящим с высокими выходами к конденсированным 1,2,4-триазоли-
динам » имидазолпдпнам (225; X = NR или CR?) (уравнение 74)
[155, 173].
(223) (224) (223)
(74)
К тому же типу превращений относится п интересное [3 + 2]-
циклоприсоедииецие азометинов к тиазприднннмннаы (образую-
щимся in situ путем пиролиза имнвотнатрназола), которое с хоро-
шим выходом приводит к нмидо-1,2,4-тиадиазолидпнам (уравнение
'°) [178]. Циклоприсоединение идет рсгиоселектнвно; это означает,
527
что в гиазирлдвивмипе реакция предпочтительнее
нолю S—С—N, чем по диполю N—Г—м иДет
СС)< PhCH2N~s
PhCH2N у
по ди.
NTs
r'CH=nR; r‘-|—
PhCHjN^A^zS - ^PhClltv /S
(75)
"NTs
NTs
может
В реакциях [3 + 2]-циклоприсоединения С=М-связь
служить не только двухатомным фрагментом, ио и входить как
компонент в тре.хатомпый фрагмент. К этой категории реакций
относятся реакции 1,3-диполярного циклоприсоединения с уча-
стием азометннимннов и азометин-илидов (некоторые из них уже
обсуждались выше); подробнее о них и о родственных реакциях
циклоприсоединения N-окспдов азометннов (цитринов) речь пой-
дет несколько ниже [(см. разд. 8.1.6.2(3)). Реакции 1,3-дианион-
ного циклоприсоединения [65, 147, 174) легкодоступных 2-азаал-
лильных анионов (см. с. 514) также можно отнести к категории
[3 -г 2] -циклоприсоединения, в котором производное азометина
выступает как трехатомный компонент. 2-Азааллильные анноны
способны участвовать в самых разнообразных реакциях «1,3-анио-
иофильного»циклоприсоединения с различными' типами связей
O-aaaahnTuu °’ N’ (X = C, N). Циклоприсоединение
тркярт 'гЛ , ЫХ ан,|онов к двойной углерод-углеродной связи про-
нообопна\иои^Л11ШЬ в слУчае напряженных систем (например,
приводит к степ <Н° ПРО11СХОД!|,Г региоселективно, поскольку оно
пифическое иис-ти^п Ме"ее ВЫГОДНОМУ продукту; это стереоспе-
лильноме аннону т пр!1соед,,неИ|!е но отношению как к азаал-
(226) присоединяется к 2 atаЛКеНУ 1И7]' Так’ Цтаяс-сшльбен
пому in situ kohiwt.J 2 азааллилыюму аниону (227) (получен-
Дифенилазнридина) ' орным раскрытием цикла 1М-литио-цис-2,3-
2286) с выходом 73°/ (ЛЭЯ тетРаФеН||лиирролидин (228а или
специфичность может [147]. Наблюдаемая стерео-
го Ms 4-i2s) процесса t • ЯС|!еиа в рамках как согласованио-
Ром принимает участие ’..'„t “ двУхстадпйиого механизма, в кото-
пение, прострацственное гт аЛ''!°1)Га|!1|‘'ескОе промежуточное соеди-
с maX которого сохраняется благодаря
лексообразующего агенте',, !оскольку, однако, добавление комп-
•?’|РЯс°ВДинепия, это свн-тетОВЛ1,Яет 1!а стереоспецифичность ши<-
pta ,нК0Г° т«Р^Апата"°Г^С1Тт п₽от”“ участия металлор-
реакции. двата и подтверждает схему согласованной
oat.
(227)
H Li+ Pb
Arc-
Ph N
H
(226) (228a)
или
H
(2286)
(76)
1,3-Анионофильное присоединение сопряженных диенов к 2-аза
аллильным анионам приводит к продуктам [3 + 2]-циклоприсо-
единения, а не [4 + 3]-циклоприсоединения, которое также могло
бы иметь место [147]. Так, бутадиен-1,3 реагирует с 2-азааллиль-
иым анионом (229), давая производное пирролидина (230), а не
тетрагидроазепиновое производное (231) (схема 77) [147].’в от-
личие от альдегидов и кетонов, которые дают ациклические про-
дукты (Р-гндроксимины) (см. с. 514), шиффовы основания присо-
единяются к 2-азааллпльным анионам и приводят к ожидаемым
[3 + 2]-циклоаддуктам с умеренными выходами. Так, бензилидеи-
анплин реагирует с 2-азааллильным анионом (229), давая произ-
водное имидазолидина (232) с выходом 50% (схема 77) [147].
В реакциях 1,3-аииоиофилыюго циклоприсоединения гетерокуму-
ленов (изоцианаты, изотиоцианаты, карбодиимиды) вследствие их
высокой реакционной способности генерирование 2-азааллильного
аниона следует проводить в отсутствие амина или амида лнтня
раскрытием цикла N-литпоазиридинов [147]. В таких условиях
изоцианаты и изотиоцианаты реагируют с 2-азааллильными анио-
нами, давая с хорошими выходами производные имидазолидина
(233; Х = О или S) [147]. В подобные реакции циклоприсоедине-
ния вступают п карбодиимиды, тогда как в случае азосоединений
образуются производные 1,2,4-триазолидина (234) [147].
—пуст атом водорода, способ-
пльный анион за счет
2-азааллильному анио^^Ь
Алнины и нитрилы, у которых отсутствует
Н|'и Дезактивировать 2-азааллпльный
ваяЯ’лзе!К° ^"соединяются к 2-^^....... „
А 'Плрролпны или Д3-пмкдазолины (^оо,
529
некоторых случаях окисляются in
пли имидазолы [147].
(уравнение 78), которые в
в соответствующие пирролы
situ
Н Li + Ph I.ArCaX
2.НгО
Xph
(235)
X= CPh,N
(236)
(78)
Двойная связь углерод—азот способна выступать н в рОли
диенофила и как составная часть диена в реакциях [4 4- 2]-цикло-
присоединения 1,3-дпенов с диенофилами (реакции Днльса —
Альдера). В общем случае, однако, наличие двойной связи угле-
род—азот в диене пли в диенофиле приводит к резкому снижению
реакционной способности данного соединения в реакции [4 4-2]-
цпклопрнсоедпиения; этим и объясняется столь незначительное
число примеров реакций с участием производных азометпнов
[155, 179). Что касается диенофильной активности, то лишь азо-
метины, содержащие сильные электроноакцепторные заместители
у атомов углерода и азота С=Ы-связи, обладают значительной
реакционной способностью. Так, N-аренсульфонилпмины (238) —
производные хлораля и его фторированного аналога — присоеди-
няются к 1,3-диенам (237) и дают ожидаемые [4 4-2]-циклоад-
дукты (производные тетрагидропиридина) (239) (уравнение 79)
(79)
(237) (238)
Х = С1, F
м "аЛ"Ч!1е электР°1,оакцепториой группы в диене также, по-впди-
ому, способствует протеканию реакции. Это е
вИ I;ц™лопР1)соадннен))я дигидроизохинолииа
14nuv* Py пентадИен-2,4-овой кислоты (241)
1,4-циклоаддукту (242) ____ ' ’’
вывартся » v самопроизвольно перегруг
вывается в сопряженный изомер (243) (уравнение 80) [155].
Это видно из примера
<а (240) к этило-
14-цпклоЯ7п™'т,',"/1>л,оС^’’'’ииии кислоты (241), приводящего к
вывается в clnnLnuub К°™РЫИ, самопроизвольно перегруппиро-
(243)
530
,2>.Циклоприсоединение шиффовых оснований k
,и&» 1М5>- » ,и,
"° „ем никла циклобутеиов (244), позволяет осуществив Р
Йгпый рсгио- И стереоспецифический синтез тетрагидрои1охино-
внов <246>й ИНТереС ДЛЯ ХИМИИ алкалоидов
лравнение 81) [180]. Легкость этих превращений может быть
обусловлена как наличием электроноакцепторной группы (циано
группа) в диене, так и выигрышем в энергии вследствие арома-
ArCH=NCHgPh
(245)
(81)
Важность оттягивания электронов в диеновом компоненте осо-
бенно ярко проявляется в том, что реакционная способность гете-
родиенов-1,3 в реакциях [4 + 2]-циклоприсоединения гораздо
выше, чем у аналогичных гомодиенов-1,3. Так, ацил- и тноацнл-
изоцианаты и ацилизотиоцианаты легко подвергаются 1,4-цикло-
присоединеншо в реакциях с простыми шиффовыми основаниями,
давая с отличными выходами 1,3,5-оксадпазпны и 1,3,5-тиадиа-
зины (249) (уравнение 82) [155, 168, 169, 181]. В отличие от от-
носительно инертных простых азометинов импниевые соли вообще
11 метпленимнниевые соли, в частности, очень легко присоединя-
ются к 1,3-диенам по реакции Дильса — Альдера (уравнение 83)
[19, 155, 179]. Производные азометинов могут выступать н в роли
Диенового компонента в реакции Дильса — Альдера, что видно
’*3 [4 + 2]-циклоприсоединения енаминов (253) к N-ацнлиминам
(254), открывающего путь к синтезу 5,6-дпгидро-4//-1,3-оксазинов
(255) (уравнение 84) [182].
(247) (248) (249)
X, Y = O, S; R2, R3, R1 = Alk, Аг
(82)
531
[4-j-2]-Циклоприсоединением промежуточно образующихся
N-ацпл- п N-тиоацплпминиевых солей (257; X = О или S) также
объясняется образование 5,6-дигидро-4//-1,3-оксазинов и -тиази-
нов (258; X = О или S) в результате катализируемой хлоридом
олова (IV) реакции N-хлорметиламндов или N-хлорметилтпоами-
дов (256; Х=О или S) с алкенами (уравнение 85). Наблюдае-
мые в этих реакциях высокая региоспецифичность и цис-стерео-
специфичность (см. уравнение 85) можно объяснить протеканием
согласованного [,44s + n2s]-процесса с участием шестичленного
циклического переходного состояния. В других случаях, особенно
при реакциях алкенов с избыточной электронной плотностью
(например, а-метплстирола), в ступенчатом процессе участвует
карбений-ионный интермедиат [19а, 182].
м’4п’?игиДР9ИЗОХШ|олвна с N-фенилмалепмидом
(ур^вненнеаК8ЦбТ Л£'?игидр?изохи11ОЛ|1на с N-фенилмалеимндом
86) 11831 11 образование аддуктов 2 : I продуктов
(см. также^111^!1'51 "ЗО1115аиатов к шиффовым основаниямЧ18*1
механизм [4 4-21 ни ХСМ'>' можно объяснить, если привлечь
полям содеРж1± жТРИСОеДИНения к потенциальным 1,4-Д«'
содержащим фрагмент имипиевого катиона.
(4) Окисление [4, 185]
Двойная связь углерод—азот подобно двойной связи углерод—
углерод подвергается атаке разнообразными окислителями: от
пероксикислот и озона до типичных металлсодержащих окислите-
лей, таких, как оксид ртути(II) и тетраацетат свинца. Окисли-
тельные превращения азометинов представляют значительный
интерес по двум причинам. Во-первых, возможность использова-
ния азометинов (иминов, оксимов, гидразонов) как предшествен-
ников карбонильных соединений и как их эффективных синтети-
ческих эквивалентов (см. с. 509) зависит от метода последующей
регенерации исходного карбонильного соединения, для чего часто
применяют и окисление. Во-вторых, окисление азометинов может
оказаться синтетически важным и само по себе, поскольку оно
может привести к продуктам, недоступным иными методами.
В дальнейшем обсуждении окисления азометинов мы рассмотрим
оба эти аспекта.
Неконтролируемое окисление [4] шиффовых оснований пер-
оксикислотами приводит к расщеплению двопной связи углерод
азот и к образованию карбонильного соединения и нитрозосоедн-
Нения. В то же время окисление пероксикислотами при низкой
температуре (0°С) представляет собой отличный путь синтеза
оксазнрпдцпов (261) (уравнение 87) [4, 118, 185]. При использо-
вании таких пероксикислот, как перуксусная кислота или .«-хлор-
вербензойиая кислота, выходы составляют 40 90 /о. Данный м
Тод применим для окисления алифатических, циклоалифатических
и ароматических N-алкнл- и N-арнлальдиминов u -ReT,,M™° J
Ребуемые шиффовы основания можно получать in situ из к р
иХН°Г0 соеД”"ення и амина. Образование оксаз"р"'1"'“’н„ю
ффовых оснований аналогично хорошо известному 0
ieil°B в оксираны под действием перокспкислот.
633
nnouecc протекает медленнее, что позволяет провеет,,
.ибпоатмьное окисление действием пероксшшслот двойной сцяз„
гЖ-азот в присутствии двойной связи углерод-угле.
йпн 11851 По поводу того, происходит ли окисление шифф0Вых
оснований в оксазирпдппы под действием пероксикислот по согла.
гованному механизму или ступенчато, не имелось единого мне-
ния П85] По-видимому, этот вопрос уже решен. Показано, что
образование оксазпридинов не протекает стереоспецпфично; это
предполагает ступенчатый механизм, включающим электрофиль-
ную атаку пероксикислотой на шиффово основание и последую-
щее замыкание цикла в образующемся интермедиате (260).
R1
\=N
(259)
R1 II
в4со3н \ /
----->- С—N
R2C> R3
b-CR4
ik
О ’Н
(260)
R1
(87)
R2 О
(261)
R‘,R2= H, Aik, Ar; К3=А1к'нли Ar
Катализируемая кислотами перегруппировка оксазирндипов
(263), образующихся при окислении циклических имидатов (262)
пероксикислотами при низких температурах, представляет собой
удобный метод синтеза циклических гидроксамовых кислот.
Было показано, что окисление шиффовых оснований в оксазп-
ридины действием пероксикпслот и последующая катализируемая
основаниями перегруппировка (уравнение 89) представляют со-
ои модель ферментативного окислительного дезаминирования
в а'кет°кислоты, катализируемого пиридоксальпн-
Эта реакция сходна с хорошо известным перемеше-
межутоиш,Н°ИГСВЯЗ" в аллиловых спиртах, протекающим с про-
межуточным образованием оксирана.
\н-МН2 + _ R' \ |1 л<-С1СвН4СОзН
8,/ SAOT0 —
'i. ЩО*
(89)
534
й отличие от шиффовых оснований кетоксимы глачко окисляют
Вроксикпслотами (например, перокситрифторуксусной кислотой!
Соответствующие нитроалканы, а реакция может быть испол?
о вана для синтеза нитроалканов [188]. Гидразоны а^оматиче-
„х альдегидов под действием пероксикислот прев^шются
смешанные жирноароматические азоксисоединения (266) 1961
Хорошо известна способность гидразонов реагировать с молек^
дярным кислородом 125 185] при этом получаются желтые фе-
лилазогидропероксиды (267) (уравнение 90). 1
Me.
X—N=NAr
Ме^ I
ООН
(267)
02
Rl = R2 = Me
R'\
XC=N
Rs/
(265)
ArCH2N=NAr
I
O'
пероксикислота
R1=H. К2=Аг'
(90)
(266)
Первоначальным образованием гидропероксида можно также
объяснить окислительное расщепление шиффова основания из
2-амннопиридпна и нзобутиральдегида под действием трет-бут-
оксида калия в диметилсульфоксиде, которое приводит к ацетону
и 2-формамидопнридину с высоким выходом (80—90%). При этом
окислительном превращении п, как и при бактериальной люмине-
сценции, появляется голубое свечение, что согласуется с механиз-
мом, включающим образование и распад дноксетанового интер-
медиата (уравнение 91) [189].
(91)
способ
Окисление пероксидом натрия можно использовать как
Регенерации карбонильных соединений из тозилгндразонов с вы
ходом 40—80% [190]. Окисление алкил- и арплальдоксимов перо-
НатРия приводит к карбоновым кислотам с выхода»
80 /о, по-видимому, через соответствующие гндроксал
кислоты [191]. Гидроксамовые кислоты действительно образ)юте
Ри окислении альдоксимов пероксисернои кнелотон 1 .
11 окисление пероксикислотами, озонолиз [4, 11 ’ 1
шов (шиффовых оснований, оксимов, гидразонов)
535
л .ой птаки по двойной связи углерод азот и при-
с электрофильно „ ам)]ду (продукту его перегруппировки),
водит к оксазприд щепле|||)Я _ карбонильному и нитрозо-
а также * '|р^.етилг|щразонь1 альдегидов п кетонов расщепля-
СОеД,,"о вь < РХором перйодата натрия в очень мягких уСЛо.
ются водным I е исходные карбонильные соединения об-
В,,ЯХ S'c отт’н'выкш выходами (90—100%) [140, 144]. Этот
Р‘ХйСэффективный метод регенерации карбонильного соедине-
мягкий эффе синтетическую ценность диметилгндра.
х: ка ° 'впвалентов карбонильных соединений (см. 5f3).
рД однако нельзя применить для диметнлгидразонов <х,р-нена-
альдегидов из-за преимущественно протекающего эли-
ZZ Щ’!- Использование бе».
30 ""гидразонов как защитных групп в синтезе пенициллина об-
,-словтено возможностью последующей регенерации карбонильных
групп ПОД действием перйодата натрия [192]. Обработка гидра-
зонов, оксимов и семпкарбазонов кетонов ангидридом бензолсе-
левиновой кислоты в тетрагпдрофуране при 50—60 С предста-
вляет собой эффективный метод регенерации соответствующего
карбонильного соединения [193]. Полагают, что образование кар-
бонильного соединения в указанных условиях происходит в ре-
зультате [2,3]-сигматропного сдвига первоначально образующего-
ся производного селена с последующей ионной или радикальной
фрагментацией (уравнение 92) [193].
лР-N^f?) R р
► *ЧС=О +• ArH + PhSeNu + N2 (92>
PhSe-^O R RZ
NuH
Ангидрид бензолселениновой кислоты можно использовать Ji
в случае гидразонов аф-нецасыщениых карбонильных соединении,
хотя он неприменим для диметнлгидразонов н О-Метилокспмов-
Так, в частности, использование этого метода в сочетании с
дегидрированием л-ннтрофеинлгндразопов 3-кетостеропдов Де1*
ствием иода или п-пптробеизойиой кислоты в присутствии тр«у
холомС'7^>опоЛЯ’ протекаЮ1в»м в одну стадию и с высоким в-
синтеза М°ЖН0 ₽ассматрнвать как эффективный мФ Л
,р- насыщенных кетонов в кольце А стероидов [194]-
53В
Окисление азометннов окислителями иа основе
С cZZiirTAC)Ha[S?79^HPnKO “Примере
^8) Действием ТАС образуются альдегвд^'^оГа'ри^
а«'и (273) и соответствующее производное азобензола (272) 1961
/уравнение 93). Образование амина и производного азобензола
объясняют участием в этом процессе нитрена (271), образующего-
ся промежуточно при ионном распаде первоначально возникаю-
щего свинецорганического производного (269). Если в N-ариль-
ном кольце анила присутствует подходящий орто-заместитель
(гидроксильная или аминогруппа), окисление под действием ТАС
приводит к образованию гетероцикла (бензимидазола, бензокса-
зола) (274) [185, 195]. Тетраацетат свинца дегидрирует N-неза-
мещенные альдамины в соответствующие нитрилы. Поскольку не-
обходимые для этого альдимины можно получить реакцией аль-
дегида с аммиаком in situ, данную реакцию можно рассматривать
как удобный метод синтеза нитрилов из альдегидов [185, 195].
как удобный
ОАс
PhN-^HAr
I
РЬ(ОАс)3
(269)
I
РЬ(ОАс)4
Z = H
(268)
РЬ(ОАс)4
Z=NHR
или ОН
ArCHO + Ph—N
(270) (271)
------> phN=NPh
(272)
донор Н
-----> PhNH2
(273)
при-
при-
Реакцня оксимов с ТАС протекает неоднозначно и может
водить к самым разнообразным продуктам в зависимости от .
Роды субстрата, условий реакции и даже от соотношения суб-
страта и окислителя [98, 103, 185, 195, 197]. Одной нз при-
!1ИН интенсивного изучения окисления оксимов под действием
*АС было использование этих реакций для генерирования ряда
свободнорадикальных интермедиатов. При окислении оксимов 1AL
было обнаружено [195] п изучено с помощью ЭПР-спектроско-
нии не менее шести свободных радикалов, в том числе имино-
«сильиые радикалы R,C=N—О-. Одиако радикалы не являются
единственными интермедиатами, участвующими в образовании
XдуКтов окисления оксимов под действием ТАС. Возникнове
некоторых из этих продуктов можно объяснить параллельным
до^еканпем ионных процессов [185, 195]. Из-за наличия в •
Ча"мах импднльного водорода их реакции с ТАС резь.
ся от реакций кетоксимов. Ароматические а д
537
„P(IVT себя иначе, чем алифатические альдоксц.
в свою очередь, вед> ,.,еШ1И последних зависит как от конфи,
мы; более того хw субстрата, так и от температуры, при ко-
rvpauiin (с«« I'-’" степне. Так, прн окислении алифатических
торой проводится окис-теп^ ал)^ к2==Н) при _78ОС под
син-альдокспмов < . (Ш1Ш( выходами (~80% ) получают
ствпем j27s которые можно «уловить» путем их реак-
оксиды Н11ТР"'" ’ ] з-диполярофпламп. Окисление алифатн-
цпи С Разн°°“Р' в действием ТАС прн низких температурах
ческпх енн-а. Д ' цен[|Ь1й общий метод получения оксидов али-
нитрилов [195, 197]. Реакцию получения оксидов
И,Хов обычно проводят в присутствии третичного основания,
наппХ триэтиламнна, чтобы удалить уксусную кислоту (кото-
пляРоеагирует с оксидом пптрпла), также образующуюся в ре.
Хтате реакции. Окисление алифатических син-альдоксимов при
комнатной температуре действием ТАС или окисление алифати-
ческих антн-алъдоксимов при -78 °C или прн комнатном темпера-
Tvn₽ лает неустойчивые гел-нптрозоацетаты (279), которые удает-
ся₽выделнть с выходами 40—70% в виде димеров (280) [195, 197].
RL X R'\-/R2
Xf/ РЬ(ОАс), СНгСЪ
1 " I “-78°С
\эн Х>РЬ(ОАс)3
(275) (276)
СН2С12. -78 -+2П°С |
[ОАс 1 ОАс О’
R'CH—N=oJ —» R’CH—N=N—CHR1
I ‘ 1
O' OAc
(279) (280)
R' = AIk, R2 = H
R'CssN—0‘
(278)
(94)
В противоположность этому ароматические син- и анти-алъи-
оксимы (275; R1, R5 = Ar, Н) реагируют с тетраацетатом свинца
при 0—20°С и дают исходное карбонильное соединение наряду с
побочными продуктами, которые можно представить как ДИ-А’
оксиды альдазинов (282) или азокспангидрнды (283) (схема 95)-
Природа этих продуктов строго не установлена; имеющиеся в на'
%йо^еио«₽е?пЯ данные> по-видимому, подтверждают структур)
(2»2) (195 197]. Окисление алкил- и циклоалкплкетоксимов Де11‘
1АС ° 1,неРт,,ых растворителях прн низкой температуре
veSuLK ге'"'Ш1ТРОзОа,1етатам (284), выделяемым в виде не-
У х маслянистых жидкостей голубого цвета с выходам
538
75% [195, 197]. Они легко распадаются
аЯ соответствующий кетой; этим и объясняется оЛ Среде’ да'
„иа (вместо г^-нитрозоацетата) при окиспении °„разова«ие ке-
“«„лкетоксимов действием ТАС в уксусной кислоте^п" ВДкло‘
“.„магических кетокепмов под действием ТАС™ Окисление
Хжно, И помимо исходного карбонильного соедини?,еКЭ,еТ более
вероятно, возникает из промежуточного ад.нит"
<285) и a»-N."ch&2!
превращение оксимов при их окислении ТАС (схешХ™™?
ае установлены [98, 195, 197]. Однако общепризнано'ч
начально образующимся интермедиатом в большинстве „
является свинецорганическое производное оксима (2761
ЮШИЙ гетеролиз пли гомолиз которого и дает все
выделенных продуктов. Так, считают [98 195 1971
розоадетаты (279) или (284) (и, вероятно, образующиеся
карбонильные соединения) и ---- --- щ
счет непосредственной ионной перегрупп'ировки^ и
яяя в интермедиате (276) (хотя для образования к-нигро^оаде-
вероятно, возникает
-------------------------------- и ди-СЧ-оксидазина (286)
ои1?7азокснангидрид (287)] [195, 197)^Пути, по которым идет
Г.яП„,ошгр ni/еимлп ППП »”'---- '“j ТОЧНО
что перво-
случаев
, последу -
все разнообразие
|ГУ71 что гем-нит-
v_,___миразующиеся из них
карбонильные соединения) и нитрплокепды (278) возникают за
пня в интермедиате (276) (хотя
I u
Аг'СНО 4-Ar'CH=N+ или Ar'CH=N+
I I
О—N=CHAr' L О—№=CHAr,J
+
(281) (282) (2831
' I
Ar^C-N-o. (27в)
(277)
ОАс
R'\i
Y—\г=о
R2/
(284)
(95)
(R1, R2 = Alk)
(286)
539
iпожен и альтернативный механизм [197]). В образе,
тагов предложенj. 5) р Д||.К-оксидов азппов (2821
ва„Ш1 м0'ю'Ы'ХвД(283)] И (286) пли (287), как полагают [98
Й0К^,"Г'уЧгствуют иминокспльпые радикалы (277), образу,0[
1 ' 1 го оптическом распаде интермедиата (276),
щпеся при гомо. альдегидов и кетонов под действием
тшироХ --зг-алось [98, 185, 195, 198, 199]. N,N-Неза-
TAG также ш р R3 = H) дают при этом диазоалканы
схема 96) которые достаточно устойчивы (п их можно вы-
Нпптъ если в исходных гидразонах импдпльныи атом углерода
™зан с этектроноакиепторными группами [например, (288;
pi==R’ = CF R3 — Н)]. В противном случае диазоалканы уда-
ется «уловить»’ по продуктам реакции с избытком окислителя,
Ходящей к а^-дпацетатам (294), или по продуктам реакции
с уксусной кислотой, образующейся в качестве побочного продук-
та при которой получаются моноацетоксипроизводные (295).
Аткены образующиеся вследствие отщепления протона от проме-
жуточно образующихся карбениевых попов, участвующих в про-
цессе [(291)-»(295)], также были выделены как побочные про-
дукты. Таким образом, окисление 1Ч,М-незамещенных гидразонов
альдегидов и кетонов под действием ТАС может быть использо-
вано для превращения карбонильной группы в ацетокси- или гем-
днацетокенгруппу или для введения - ......
Процессы этого типа использовали
в молекулу двойном связи,
синтезе стероидов, например
в
R\ /R2
РЬ(ОАс)4
II -------------->
SlHR3
Rk /R2
N.
R3^.H
R'CsN—NR3
(290)
(288)
РЬ(ОАс)3
(289)
Rl=R3=,Ar
R'\- ♦
,C-N=N
R3=H
(291)
R'
]C—N=NR3
R3/ I
OAc
(292)
AcO.
C=N (96>
'NHAr
(293)
R'
уСНОАс
R2/
(295)
54U
R'\
/Web
рг/
(294)
О Ac
(296)
S gft -н> »»«""» ТЛС^^ЙКЙ^Г
епленпем уксусной кислоты из азоацетата (292; R* = Н) Поо
Шуточно образовавшегося в результате перегруппировки "ви-
„органического производного гидразона (289; R3 = ю [98 .“Л
98] В соответствии с этим механизмом, окисление N-монозаме-’
ленных гидразонов кетонов^ (288) под действием ТАС приводит
азоацетату (292) (который можно выделить) [201]; процесс его
образования аналогичен процессу образования гел-иитрозоацета-
тов из кетоксимов при окислении ТАС (см. выше). Отсутствие
поглощения в спектрах ЭПР [198], кинетические параметры [201]
и влияние заместителей [198] при окислении N-монозамещенных
гидразонов кетонов под действием ТАС находятся в полном соот-
ветствии с механизмом, включающим образование н гетеролптиче-
ское (а не гомолитическое) расщепление свинецорганнческого
производного гидразона (289).
Образование азоацетатов (298) при
(297) действием ТАС было использовано
токсицнклопропанов (299) (уравнение 97)
окислении пнразолпнов
в изящном синтезе аце-
[198].
R,YVR2 AeoTXf
N\ / \рз / \пз
NH N
(297) (298)
R1
(299)
•R2
(97)
, Окисление N-монозамещенных гидразонов альдегидов (288;
R’ = H) ТАС дает соответствующие N-ацетилгидразпды (296).
Образование этих соединений можно объяснить тем, что проме-
жуточно образующиеся нптрплпмины (290) «захватываются»
Уксусной кислотой и дают гидразпноплацетаты (293), которые
затем претерпевают [1,4]-ацетильный сдвиг (см. схему 96) [19о].
Промежуточный ннтрплнмнн (290) можно «поймать» с помощью
Реакции с подходящим 1,3-днполярофнлом; действительно, окисле-
но N-мопозамещсппых гидразонов альдегидов деиствпе.м ТАС
УЖнт важным способом генерирования in situ этих интересных
Реакционноспособных интермедиатов. Отсутствие поглощения
“ центрах ЭПР н кинетические параметры для процессов окис-
ТАсЯ К,-Мо|'озамещенпых гидразонов альдегидов под депствием
холо И в Этом случае согласуются с ионными процессами, пропс
°кисл₽М" при УчасТ|ш интермедиата (289; R2 = Н) [9 , ]•
тиоср? ие Действием ТАС N-моиозамещенных семпкарбазонов и
ИМ сое!,1Карба?опов альдегидов н кетонов, а также
Дарений, представляет значительный интерес для • 5
541
.азолов и 1,3,4-тнадпазолов (уравнение 98)
к систем (уравнение 99) (199]
РЬ(ОАс)д
:= о или s
/,3,4-оксадпазолов и
спроваипых е.'.'стем <
iR'x v
J1C=N X
й2'/ \ И ,,
д NH—C—NHR3
(300)
RTI
r2/S<^4nR3
(301)
" к°пдец.
(»8)
(303)
(302)
X
гидразо-
Окисление азометннов (оснований Шиффа, оксимов,
нов) различными окислителями на основе металлов с переменной
валентностью .хорошо известно [25, 102, 185], и в ряде случаев
его используют в синтезе. Так, окисление N.N-незамещенных гид-
разонов альдегидов и кетонов диоксидом марганца [185] и окси-
дом ртути [202] — хорошо известные способы синтеза диазоалка-
нов, а окисление оксимов гексацианоферратом (III) или оксидом
серебра — удобный метод генерирования устойчивых нминоксиль-
ных радикалов (например, ди-грет-бутилиминокснла) [203].
Интерес к изучению окисления азометннов под действием пере-
ходных металлов стимулировало все возрастающее значение этих
соединений (особенно оксимов и гидразонов) как синтетических
интермедиатов; как следствие этого, возникает необходимость в
новых реагентах, которые превращали бы азометины вновь в кар-
бонильные соединения эффективно и в мягких условиях. Так,
синтетическая ценность диметилгидразонов альдегидов и кетонов
(см. с. ЫЗ) определяется тем, что их можно окислить до карбо-
нильных соединений с выходом ~-100% в мягких условиях
няпп„11Н’ КОмнат11ая температура, pH 5) действием солей меди(П).
телУнп'6? ацетата меДИ(П) [145, 204]. Аналогично можно окислп-
алкил ™еНТР0Вать альдегиДЫ и кетоны (выход 80-95%) из
щих (Ьенппг, ’ИЛаЛЬДОКСИМОВ ||ЛИ -кетоксимов (пли соответствую-
ной (|- „„ТТ.ИЛ" сем1|карбазонов) путем кратковремеп-
комнатиой темп^пРаб0ТК1'лА,!траТ0м таллия (III) в метаноле при
соединений из Skchmob6 ю205 ' ДлЯ РегеиеРаЧ11И карбонильных
гидразонов с высоким’п?МеТИЛГЖазонов и «-толуолсульфонил
кобальта(ЦП [2061 чВых0Д()м (80—90%) рекомендуют фторид
регенерации капбп шЛ,Т0Т арсенал реагентов для окислительной
пополнился гексасЬтпп <пЫХ соед1|1|еш1й из азометннов недавно
лена ]208] пероксЖ?' УРЛ’1а [2071’ гексафторидом молиб-
следиий реагент особенно(тРиФС|||1ЛФ°сфип) палладием [209]; п
мов в мягких условиях ° пР||10ден Для дезоксимироваппя ока •
542
(5) Восстановление
Поведение двойной связи углсрод-азот по отношению к
тя вливающим агентам сходно с поведением двойной свя°и
Vнерод-кнсдород- Реагенты типа металл - донор „рОтОНа (на
мер, натрии, амальгама „атрия, магний или алюминий в эта'
е- «инк или алюминии в водном растворе щелочи; цинк в ™
“сИой кислоте) гладко восстанавливают шиффовы основания
ученные отдельно или in situ из альдегида „ли кетона и
амина) до соответствующих аминов [4, 100]. При действии ще
‘очных металлов в инертных растворителях, таких, как эфир или
толуол, возможна восстановительная димеризация по механизму
спаривания радикалов, приводящая к диаминосоединениям в ка-
честве главных продуктов (схема 100) [4, 100,210].
Na . - Na - .
R'C=N-CHR2 — R'C—N—CHR2 R-C-N-CHR2
Ar Ph Ar Ph Ar Ph
I--------------------' I nh4ci.
вода
Ar Ph Ar Ph Ar Ph
R'C—N—CHR2 nhsci R'CH—NH—CHR2 R'CH—NH—CHR2
1 - 7* I (100)
R'C—N—CHR2 В°дз R’CH—NH—CHR2
1 1 Ar Ph 1 1 Ar Ph
(60%) (20%)
Восстановление гидразонов [25] системой металл —донор
протона приводит к гидразинам. Шиффовы основания восстана-
вливаются до соответствующих аминов и при действии муравьи-
ной кислоты. Полагают, что такого рода восстановление азомети-
нов является ключевой стадиен синтетически важного восстано-
вительного алкилирования аммиака и аминов альдегидами и кето-
нами в присутствии муравьиной кислоты (реакция Лёйкарта) или
муравьиной кислоты в сочетании с формальдегидом (реакция
Эшвайлера — Кларка) [10,11].
Способность муравьиной кислоты восстанавливать пминневые
соли до аминов лежит в основе метода прямого восстановления
енаминов до аминов (уравнение 101) [196]. Особенно прпмеча-
ельио, что восстановление алкалоидных енаминов муравьиной
ислотой протекает в высшей степени стереоспецифпчно |19б].
НСО2Н
R\ /Rs
> xch-ch2n;
(101)
нсо;
543
шиффовых оснований муравьиной кислотой про.
Восстановленпе‘ ^дрцдного переноса, это было показано
исходит 110/'Х' |1ев0|| меткой [196]. Восстановление оксимов
в "^'^Дчорил - цинковой парой в тетрагпдрофуране Пр„
л?г"0Д« этермпчеекий прочесе; образующиеся при этом НМ1111Ь,
0 С „Ттиются до соответствующих карооппльных соедине-
ЛеГ-Кс Общим выходом 50-90%. Этот метод используют для цос.
" ! „ Аитетыюй регенерации карбонильных соединении из окси-
1?081 Х'юрнд титана (!!!) также позволяет провести глад-
кое превращение оксимов в имины, которые, в свою очередь,
моя но восстановить борогидридом натрия до первичных аминов.
Последовательное восстановление этого типа позволило осуще-
ствить успешное превращение оксима эритромицина в эрнтромп-
шпа.мин [211]. Хлорид титана (!!!) в 50%-ной водной уксусной
кпетоте используют для превращения п-толуолсулъфонплгпдразо-
пов (особенно ряда стероидов) в соответствующие кетоны при
комнатной температуре с высоким выходом (>90%) [212].
Двойная связь углерод—азот подобно двойной связи углерод—
кислород легко восстанавливается комплексными гидридами ме-
таллов [4, 100]. Восстановление такого рода, вероятно, представ-
ляет собой наиболее эффективный и удобный метод превраще-
ния азометинов в амины. Так, алюмогндрид лития в тетрагидро-
фурапе при комнатной температуре (или, в сложных случаях,
ври повышенной температуре) гладко восстанавливает основания
Шиффа во вторичные амины с высоким выходом (>90%). Столь
же эффективным восстановителем является и борогидрид натрия,
его даже следует предпочесть алюмогидрнду лития, поскольку он
инертен по отношению к большему числу растворителей, в кото-
рых проводится реакция, и более специфичен, вследствие чего
другие заместители, например хлор пли ннтрогруппа, восстана-
вливающиеся алюмогидридом лития, не затрагиваются борогидрп-
дом натрия. Еще более эффективным восстановителем является
иианоборогидрпд натрия NaBHjCN. Этот реагент довольно успеш-
но использовали при восстановительном алкилировании аминов
карбонильными соединениями [213]; применение его для восста-
новительного метилирования амшюв позволяет проводить эту
реакцию более эффективно и в более мягких условиях, чем в слу-
чае широко используемой реакции Эшвайлера — Кларка, в кото-
"Р",'1С"ЯеТСЯ Формальдегид [10, 11]. Так, реакция алифатп-
пегпппм'ли аРомаТ|1ЧесК|,х первичных аминов с водным формаль-
ноле п'ппвппР”СУТСТВ||И ччаноборогидрида натрия в водном мета-
ты с копошим 1 меТ|1Л||Рова||н°му амину высокой степени чисто-
заранее Р|пи генепппу°М '2!41 • ,ШиФФ°вы основания, полученные
двнений также эФл2ЛМЫе ‘П Sltu 113 амннов и карбонильных сое-
плексом гидрид щадочногл’0 В0ССтаНавливаются Д° амшюв 1<ом'
NaHFe(CO). [21ГЛ ого металла— карбонил железа, например
иминов (легко пол1ийП?ССТаН0ЛЛЬ|||1е N-замещенных а,а-дихлор-
1 получаемых а-С-хлорнроваиием иминов действием
044
„„кцпнимида) алюмогидрндом лития поелста», >
Ь'-хЛ°Р сшггеза азиридинов (выходы 80-90%Р) Со6ой
«етоД к восстановлению борогидридом натпи» „ 1иксимы
>’ЧИЭВТО было использовано для сшпеза
восстановления моиооксиминокетоиов борогРидпИп<>м° Пу'
01 слеДУ|0Щей РегеиеРацией карбонильной группы pm В
ci оксимы можно восстановить до спиртов С ВЫСОКИМ выходом
<90%) нагреванием с борогидридом натрия в водном пя 2
Л,»™ [2181 Циаиоборог!1дрид ,„гри,
„ТОМ восстановления оксимов в гидроксиламины [213
„,gi. Длюмогидрид лития восстанавливает арилгидразоны в соот-
%-ствуюшпе гидразины [25]. Борогидрид натрия, напротив не
Останавливает гидразоны, что, как н в случае оксимов ’(см
выше), было использовано в синтезе а-гидроксикетонов [2201
диалогично восстановлению по Вольфу - Кижнеру п-толуолсуль-
фоиплгндразоны альдегидов и кетонов превращаются в” алканы
„од действием алюмогидрида лития [25], или, лучше, цианобо-
рогпдрнда натрия [221] или борана [222]. Следует отметить, что
использование катехинборана позволяет осуществить восстанови-
тельное превращение n-толуолсульфонилгидразонов в углеводо-
роды в исключительно мягких условиях, высокоизбирательно
(другие функциональные группы, например двойная связь, не за-
трагиваются) п с высоким выходом (~97%) [222]. Шиффовы
основания восстанавливаются в растворе уксусной кислоты ком-
плексом дпметиламин— боран в таких условиях, в которых не
затрагиваются другие способные к восстановлению группировки
(например, хлор н нитрогруппа), и дают с высокими выходами
(80—97%) вторичные амины [4, 100]. Восстановление ацикличе-
ских и циклических имннневых солей алюмогидрндом лития, ба-
рогидридом натрия нли дпбораном дает третичные амины с хо-
рошими выходами [9, 196]. Борогпдридное восстановление ими-
яиевых солей, полученных in situ реакцией амидов с оксихлори-
дом фосфора, представляет собой новый способ синтеза аминов
(выходы 50—90%) [223].
Шиффовы основания гладко восстанавливаются до аминов
при гидрировании над платиновым, никелевым пли хромовым ка-
’’алпзаторами [4, 100, 224]. Так, анилы восстанавливаются до
коричных аминов практически с количественными выходами при
2Р11рова|111" "ад платиновым катализатором при 60 °C. N-Неза-
Щеннце пиццы R,C=NH дают первичные амины; выходы не-
Ш'С°К|1 "З'за Реакции конденсации исходного имина с образовав-
Ноп ? амИ||°м. Гидрирование смеси аммиака и первичных амн-
п», алкнл- ц арнлальдегндамн и -кетонами над нпкелевы ,
влеН1,а°ВЬ1М 11 палладиевым катализаторами включает восс^
иьш ,шиФФ°аых оснований in situ и является простым, но
с очень Ui"M методом синтеза первичных и вторичных а„1
соб с11нДЬ1С0К,1‘'"1 вых°Дами в мягких условиях. Этот це е
теза аминов применим п для полпфункциональны
Зак, юз 545
примера может служить получение а-амцц01(||
кул; в ка^еt r '||а) „ «-кетокислот (например, пировшюгра
(например, а- ,а;шт1|„еС1(0М гидрировании иминов набл'одае 1
К"С '0Т шетрическая индукция [225], хоральные амины подучав
СЯкопошн 1 оптическим выходом. Новый метод эффективного пре.
ппашешя шиффовых основании в амины с отличным выхода.
Si) в мягких условиях состоит в нх восстановлении алк11л.
( в присутствии катализаторов па основе переходных м«.
^Х напримеР, (Ph3P)3RhCl] [226]. Оксимы [Г02
9241 также легко восстанавливаются водородом над платиновым
папа лиевым катализатором до первичных аминов, хотя при
восстановлении над платиновым катализатором могут образовы-
ваться и производные гидроксиламнна. Гидразоны [25, 224] вос-
станавливаются не так легко, как оксимы, и для нх превращения
в гидразины лучше использовать никелевый или платиновый ка-
тализаторы.
Двойная связь углерод—азот восстанавливается электрохими-
чески [38, 100] гораздо легче, чем двойная связь углерод—кисло
род. Шиффовы основания из альдегидов и кетонов можно восста
новнть электрохимически до соответствующих аминов в широком
интервале pH, но восстановление азометннов этого типа ослож-
няется параллельно протекающим гидролизом шиффова основа
ния. Восстановительное алкилирование аминов альдегидами и ке-
тонами с промежуточным образованием шиффовых оснований
можно провести не только каталитическим гидрированием, но п
электрохимическим путем. Так, при контролируемом восстановле-
нии раствора циклогексанона в метиламине N-метилциклогексил-
амип образуется с высоким выходом [38]. Электрохимическое вос-
становление N-замещенных ароматических альдиминов и кетими-
нов происходит путем ступенчатого присоединения двух электронов
и вначале дает анион-радикал, при протонировании которого обра-
зуется промежуточны!! радикал, который может дальше либо вос-
становиться до вторичного амина, либо димеризоваться в 1,2-ди-
аминосоединение (уравнение 102) [38, 227, 228].
’hCH-MPh > PhCH-NPh PhCH—NHPh PhCH2NHPh
н+
| (102)
PhCH—NHPh
PhCH—NHPh
иминиевые соли [229?В'чпЛЬИО” дпмеРИзаццц подвергаются н
фовых оснований в пг1п!..-Г.еКТ|,П'',1'1||1|ес|<ое восстановление шиф'
54g ' твни Х11ральиых солей сопровождается
„метрической индукцией с образованием хипялкнпг
227]. Электрохимическое восстановление оксимов гМИ‘
"а проводить при кислых или нейтральных значениях pH оГ
2„ь) и затем «»™ 138). Гидразоны „к-ктрехн!
g восстанавливаются только в кислой среде; нрн этом проис-
‘однт РазРыв N-N-связи и образуется смесь первичных
нов [38].
Шиффовы основания обычно восстанавливаются до вторичных
аминов при фотолизе в спиртовых растворителях [230] По ана
логин с подобным фотовосстановлением карбонильных соедине-
ний первоначально полагали, что фотовосстановление шиффовых
оснований включает отрыв водорода имином в возбужденном со-
стоянии. Однако более поздние исследования показали [230], что
„мпн участвует в процессе не в возбужденном, а в основном со-
стоянии; этот процесс инициируется кетпльными радикалами,
генерируемыми из присутствующих в растворе примесей (меха-
низм так называемой «химической сенсибилизации»). Неспособ-
ность имина в возбужденном состоянии оторвать водород, в отли-
чие от поведения карбонильной группы в такого рода процессах,
связана, скорее, с исключительно низкой скоростью отрыва водо-
рода, чем с увеличенной скоростью безызлучательного распада
имина в возбужденном состоянии вследствие вращения вокруг
двойной связи углерод—азот. Имеются данные [230], что N-ацил-
импны в отличие от простых шиффовых оснований фотовосстанав-
лнваются в среде пропанола-2 за счет отрыва водорода (в про-
тивоположность механизму «химической сенсибилизации»), одна-
ко, по более поздним данным [231], фотовосстановление и про-
стых шиффовых оснований в пропаноле-2, по-видимому, также
происходит за счет отрыва водорода.
8.1.2.3. Циклические азометины с малым циклом
(азирины, азетины и родственные соединения)
В основе Д’-азирппов — циклических азометинов с наимень-
шим размером цикла — лежит структура 1 -азациклопропена
(304а). Эти соединения первоначально были известны лишь как
интермедиаты в перегруппировке Небера (см. с. 494). из)че
химии этих интересных гетероциклов было надолго отложено ч
стнчно из-за их относительной неустойчивости, но главнымi с। р
зом из-за отсутствия общих методов их синтеза. Прон
А-азиринов в настоящее время легкодоступны, поэтомх их г •
ческие свойства изучены весьма широко [230, 232 235],
за последние годы. Следующие гетерологн азпрпнов -а
ты (1-азациклобутены) (305)—относительно ' в по-
отя эти соединения еще не так доступны, как Д -a3i Р
18*
,„₽пиие годы они все в большей степени становятся объеКТОм
обстоятельного химического изучения.
X
(304а) X = cr2 (305)
(3046) X = NR
(304в) х = о
(304г) x = s
(1) Методы получения /М-азиринов [232, 233, 235],
Ь'-азетинов и родственных соединений
Азирины легко можно получить с высокими выходами при
термолизе или фотолизе винилазидов [232, 233, 235]. Этот метод
нашел широкое применение из-за доступности исходных винил-
азидов [235]. Випнлазпды (310) получают присоединением азида
иода к алкенам (306) с последующим отщеплением йодистого
водорода в присутствии основания, а также взаимодействием
оксиранов (307) с азпд-поном п последующей дегидратацией об-
разовавшихся азидоспиртов (309). Этот подход дополняет метод
с использованием азида иода, поскольку он дает региоизомерные
випнлазпды.
Rl\ ,R>
,C-CZ
R2/l Г'Н
I Na
(308)
основание
(310)
-И2О
R2//1 |\i
HO Na
(309)
Д'-
Термолиз винилазидов был
азирина (311) газофазным
использован для синтеза 2-фенил-
пиролизом а-азндостпрола (310а).
1310а) <31О (80%)
548
(104)
. „яко более удобным методом является Лптляи,
Поскольку его можно проводить при ИИЗФКИХ
’° Убавленном растворе; это приводит к более высоким It УР
фунтов, а ТаКЖ6 "°зп0ляет избе>кать разложения илн по“
"р Унт неустойчивых азиринов, что часто набтюлаетга „
g термолиза. При пиролизе терминальных виннлазидовнГоб'
уются 2-незамещенные азирины (вероятно, из-за их последую-
go термического разложения), в то время как при фотоли-
Зе при -50°C они получаются гладко н с хорошим выходом
Однако фотолитическая конверсия виннлазидов в Д'-азирииы
может быть снижена из-за образования кетениминов (313)
(уравнение 105).
из-за образования кетениминов (313)
EtO»C\ /Me hv
>=< —*
Aik' XN3
Aik CO2Et
EtO2C
+ ,C=C=NMe
Aik'
(105)
(3106) (312) (80%) (313) (20%)
Первый успешный синтез незамещенного азирина (315) уда-
лось осуществить с удовлетворительным выходом [326] путем
флеш-вакуумного пиролиза вннилазида (310в) прн 400°С, в то
время как фотолиз этого соединения привел к азирину лишь
с низким выходом [326]. Предполагали, что образование азирн-
нов из виннлазидов представляет собой ступенчатый процесс
(уравнение 106), включающий циклизацию промежуточно образу-
ющегося винилнитреиа (314) [232, 233, 235]. Этот ступенчатый
механизм с участием нитрена не согласуется, однако, с кинетиче-
скими параметрами для циклизации вннилазида в азирнн, т. е.
отщепление азота происходит в значительной степени по согласо-
ванному механизму [237].
(106)
А'-Азирины образуются
А -Азирины образуются также как конечные продукты Р
брмолизе и фотолизе некоторых пятичленных гетероциклов. ’
с,рол"3 ^’Фталнмндо-1,2,3-триазолов (316) ПР||^',Т * м0-
муУЮ1Ц"м З-фталимидо-А'-азпрннам (319) н I320)’ и
дУ’ ?6рез промежуточно образующиеся иминокарбе (
оЛ. азирин (318) (уравнение 107) [328]. Участием промежуточно
РазУющегося Д’-азприиа можно также объяснить о р
549
Д'-азприна при окислительном
да (321) с алкинами [239].
взанмодействи» N.ai
(316)
(318)
(319)
(320)
З-Ацил-Д’-азирнны (323) получают
фотоиндуцированном сужении цикла
ствием света с длиной волны 254 нм.
облучении светом с длиной волны
11233].
с высоким выходом при
изоксазолов (322) под ден-
Обратный процесс идет при
>300 нм (уравнение 108)
Ph
254 нм
Нч ,COPh
(108)
Ph , t
>300 КМ
о
(322)
Сужение цикла в изоксазолах,
.можно провгетн н г-------------
•МИ выходами (~20—ЗО°/о), чем' при фотохнмиче^™ BUcoKi's'
-]233, 240]. В ряде случаев Д’-азприны были п°л^\жосфи”01(С'1 t
выходом путем термического отщепления т(>пч>е1 ,ь(!сф0Р'|',|° 3!|.
из оксазафосфолов, образующихся при Реа1<и1’1’„„„.еза ^'а
оксидами нитрилов IWti ст—
Ph.
N
(323) (82%)
. приводящее к ацй^И
термическим путем, но с гораздо oO11ectf
чем при фотохимически '.„„.ы
- Ряде случаев Д'-азприны были получены
выходом путем ТОПч »<.-
оксидами нитрилов’ [гЗЗЬ^'от путь с|111|Ма “
550
„ проиллюстрирован синтезом спиро-Д>.азирива (уравнение
Й“й31'
<61°Z°) (84%)
В отличие от азпрпнов трехчленные циклические азометины
держание два гетероатома [дпазнрины (3046), оксазирины (304в)
я тиазирпны (304г), неизвестны. Предполагают, что диазипины
и тиазирпны могут участвовать как неустойчивые промежуточные
соединения в фотохимически индуцированных реакциях 1*3 дипо
лярного циклоприсоединения, в которые вступают некоторые ме-
зопонные гетероциклы. Так, возможно, что образование произвол
пых пиразола (327а) и лзотиазола (3276) при фотолизе сиднонов
(324а) и их тиаанологов (3246) в присутствии диметилового эфи-
ра ацетилепдикарбоиовой кислоты (ДМАД) происходит по урав-
нению НО. Отщепление диоксида углерода дает нестойкие диази-
рии (325а) и тназпрпн (3256), находящиеся в равновесии с соот-
ветствующими 1,3-диполями — нитрилимпном (326а) и нитрил-
сульфидом (3266), которые присоединяются к ДМАД и дают на-
блюдаемые продукты циклоприсоединения (327а и 3276) [241].
v R v МеО2С
* Ы X МеО»СС=ССОгМе
(325)
(324)
,СО2Ме
-COg
(а) Х= NR
(Ь) X=S
X.
(327)
(НО)
x^n^r
(326)
Основной общий способ синтеза л азетпнов-—ппролиз
Циклопропанов, протекающий с расширением Ц • ка0бенов
Доцпклопропаиы обычно получают прнсоед 105—125°С И
к азидоалкенам. Пиролиз протекает гладко пр i
регпоспецифпчно (уравнение 111).
CI С1
х 5 105-125°С
Ьк/\Х-----------------
R' Ч1Г
(328)
при 105—125 °C и
R
R2
N—I—R3
J-----С1
С1
(329)
не
R1
С1
С1
9
R"
R"
(330)
(ш)
551
К.|та визируемое основаниями расширение цикла [235] легко.
„Линых трихлорметилазиридппов (331) также приводит к д>.
азетпиам (332) с умеренным выходом.
С1»С\ zR2
R1-
KOBu-rper
Д.МСО
CI-
R2
N-----Н
1______.R‘
(112)
Cl
(332)
ряде случаев при фото-
(331)
Д’-Азетины были получены также в
химической элсктроциклизации циклических азаполиенов. Приме-
рами указанного типа процессов могут служить фотопревращение
производных дигидроазепина (333) в неустойчивые бициклические
азетины (334) [230] (уравнение ИЗ) и любопытный способ полу-
чения пиридина Дьюара (336) в результате фотоперегруппнровкч
пиридина (335) [243]. Продукт (336) переходит обратно в пири-
дин в течение 15 мин при комнатной температуре. Однако его
структуру удалось надежно установить
в относительно устойчивый азабпцнкло [2.2.0] гексен (337)
(уравнение 114).
ДТ
R'-
R'-
путем восстановления
[243)
:n
(333)
R‘=H, Me;
ln fi—i—N
R2
5v R\/xLn
(334)
Ц2 ~ ОМс, NMez
NaBH4 ---j—МН
#13)
(114)
N
(335) (336) (337)
я-!рРиип',еД'10ЖеННЬ1Х ведав11° практических способах синтеза Д1-
ние ключевой стадии используют фотоциклоприсоедине-
устойчивые^л'?06^1111611116/а'10Мат|1чеек11х "чтрнлов к алкенам дает
ленная 2 ^Рав,1ен^ ’’5) [244]. Термически дозво-
ненных поонТво-.пТт’’। ч’РеаКЦ“я РетР°Диенового распада сочле-
азетииы (341) по г- \ '^-оксазина (340) позволяет получать Д'-
нне 116) [2451Р г'‘°спецнФичес1<” и с хорошим выходом (уравне-
присоейнЕ алкенов ТзЗЭ)( П°ЛучаЮТ '2 + 21 ’Фотоцпкло-
1кенов (339) к 1,3-оксазинону-4 (338).
ArC==N
+
Ме\ Me
>=<
Me' \мр
Me
N——Me
Лг II I „
Ar।—
Me
(116)
552
225 «С
(341) (57%) <ие в“Д«леа)
(И6)
(2) Реакции к'-азиринов и Д'-азетинов [233]
Реакции, типичные для двойной связи углерод—азот в про-
стых азометинах (см. выше), в случае Д'-азиринов и Д'-азетинов
протекают иначе, так как цикл напряжен. Так, реакция Д'-азирн-
нов п Д^азетпнов с электрофильными и нуклеофильными реа-
гентами, характерная п для простых азометпнов (ем. выше);
часто сопровождается раскрытием цикла, иногда с последующей
перегруппировкой.
Низкая основность Д'-азиринов объясняется тем, что орбиталь
у атома азота, содержащего неподеленную электронную пару,
приобрела в значительной мере s-характер. В сильных кислотах
(например, в хлорной кислоте) протонирование возможно (343,
R',R2 = Me), но оно сопровождается раскрытием цикла с обра-
зованием имииокарбениевых ионов (344), которые удается
ВИТЬ» путем реакции с нитрилами, в результате чего образуются
производные имидазолина (346) (схема 117) [233] • Д Р
реагируют с карбоновыми кислотами (например, с оензонн
-1отой) и дают кетоамиды (347), по-видимому, с ^громеж [233],
образованием производных азиридинов (345;
При аналогичной реакции с сульфокислотами оорази 0
''Мины [например, (345; R3 = SO2Ar)] устойчивы н и“
Мелить [233]. Раскрытие цикла азиридиновых
45, R3 = H), образующихся при гидратации Р окетОноз
Гч1с?Тавляет собон удобный способ получени Небера (см.
с 12331> который аналогичен псРсгРУпп"Р°енпя. Таким об-
’ 494), но имеет более широкую область при. яЗИРНнОв часто
лоп°М’ С1!Нтез а-амннокетоиов путем сольволи получать
пзо>0ЛНЯе1 перегруппировку Небера, поскольку пспользо-
МеРные а-амииокетоны. Сольволиз азиринов если при-
ме? " пЛЯ получения а-аминоальдегидов (на р оЖ1[о получить
? ^незамещенные азирины), которые невозможно )
*Родством перегруппировки Небера, так как тозплаты
в превращаются главным образом в нитрил
553
(344)
(117)
I. MeCN
2. H20
Me
Ph Me
(346)
(345)
R3 —COPii | R3=H
I 1
0 0 0
PhCNHCHaCPh PhCCH2NH2
(347) (348)
Л'-Азнрнны реагируют
Л'-Азнрнны реагируют с хлорангндридамн и ангидридами
кислот и дают 2-хлор-Ы-ацнлазпридпны (351), которые можно
выделить и которые перегруппировываются при нагревании в по-
лярных растворителях в производные оксазола (354) через не-
устойчивые оксазолнновые интермедиаты (353) (схема 118) [233].
Разложение 2-хлор-Ы-ацилазиридииов (351) в более мягких усло-
виях может служить способом получения производных А2-оксазо-
лнна (355) [246]. Д’-Азнрины реагируют также с сульфопилга-
логеиндами в присутствии пиридина, ио в этих условиях N-суль-
фонилазирпдиповые интермедиаты (350) неустойчивы, происходит
самопроизвольное раскрытие цикла с образованием N-алкенил-
сульфоиамидов (352) [233].
Лпл^нкп?Пп»ИаЯ способ1'°с^ь Д’-азирппов по отношению к нуклео-
они ппоявРч^птНТаМ П° характеру сходна с той, которую
соединение uvtw Реакциях с электрофильными агентами. Прн-
азот приводит °Ф1'ЛЬН0Г0 реагента по двойной связи углерод—
"о которые склонны и? чДИНаМ’<. К0Т0Рые часто можно выделить,
ла и последующим 3 СВОе ’ ||еУстойчпвости к раскрытию цнк-
тов к Д^азирпнам кятяПГРУПП11')0ВКам (233]. Присоединение спир-
азпридцны (357) Эти изиРУется алкоксидами и дает 2-алкокси-
устойчнвы и попврпгп еД|1нення МОЖНО выделить, однако они не-
цнкла, приводящей к тяСЯРТДаЛ1,нейшей реакции с раскрытием
деталям а-аминокетонов (360) [233]
(351)
1. МеОН.20’С
2. NaHCO3
”R,=R2=M’ Me
R
N^\)
—J----L_OMe
Me Ph
(355)
(349)
Ri=.h| PhSOjCI,
кгвме|пиридин
(350)
(353)
(118)
ci\ /Me
Ph/ \fflSO2Ph
Ph
(352) (354)
реагируют л с аминосоединениями. В случае
конечными продуктами после обработки, вклю-
вляются амидины (361), образующиеся, веро-
------------------------------fS58)
А’-Азирины охотно
первичных аминов
чающей сильволнз, являются амидины о>и1л - (^а\
ятно, путем фрагментации неустойчивого аминоазнрнД!
МеО“
МеОН
R1 = H
R2 = Me
Me0\ A'll2
XC—
MeOZ | |\Me
Ph H
(360)
(356)
NHjX
R2 = CONH2
(119)
PhNH2 Ri = R2 = H
Ph
PhHN
(358)
C=\TPh
(361)
555
. л .пппнновые производные (359; X — NH2 пли ОН), ogD
|23шисся К реакции Д'-азпрппов с гидразином или гидр0КсР‘л.
3)”°Т мо о выделить. Они служат удобными предшСстве1111н.
ам"н°м. °'езах оц))клов (например, производных 1,2,4-Тр11.
камгол?] Очень легко Д'-азирины реагируют с карбаииоипы
мГ’осагснтами; присоединение происходит по двойной СВЯ311 уг.
МИ Рс^7зот £ случае реактивов Гриньяра нуклеофильная атака
пппХодит стереоспецифичио с менее пространственно затруднен-
ПР. Попоны азпршювого цикла (363), и после гидролиза полу,
чают производные азиридинов (362) (схема 120) [233]. а-Заме-
(пение наблюдаемое при реакции ациклических С-алкилированнщ
азометинов с реактивами Гриньяра (см. с. 509), не происходит
V Д'-азкрпнов. Первоначальная атака азиринового цикла карба-
иионамн образованными из соединений с активной метиленовой
группой,’ также идет по С-2, но возникающие при этом аддукты
(364) неустойчивы и превращаются в пирролы (ЗЬ8) [233]. Ана-
логично по отношению к Д'-азпринам ведут себя и реактивы Ре-
сЬооматского [248]. В отличие от столь сложно протекающей реак-
инн с карбанионами, полученными из соединений с активной ме-
тиленовой группой, реакция Д'-азнринов с дпметнлеульфопии-ме-
тилидом Приводит к производным любопытной циклическом си-
стемы 1-азабицикло[1.1.0]бутана (365), с хорошим выходом (60-
70%) [233,249].
(367)
(366)
656
В последнее время интерес к химии л<
„озрос. Это связано с их ярко выраженной'склон,'.°в 3,,аЧ1«ельно
оеакши! циклоприсоединения с самым,, Y Остью «ступать
Датами [230, 250, 25Ц. Особого вив ’ , ПЯР S-
а,от наиболее хорошо изученные реакции за^У^-
^псоедипения Д'-азиринов [230, 2511 ппи - "ого иикло-
^охимического раскрытия цикла из аз(ппнпи°МгВ РезУльтате
стойкие нитрнл-илиды, которые и участвуют » „ образуются не-
сениях [см. разд. 8.2.42(3)]. Далее будут пасХТЙШ‘'Х "Ре-
нии циклоприсоединения [250], в которых вероятио^^' реак'
участие неизмененный Д'-азириновый никл’в час ’ рииНмает
ск« разрешенные реакции [3+2]- и [л4 + 2 1 СТН0СТ"’ Термиче-
ния, в которых эти соединения выступают как
диенофилы. KdK дип<мярофилы или
Д'-Азприны могут также выступать и в по-,,,
«ого компонента _в_ термически^ реакциях 1,3-диполя“ n°ptoe-
[250]. Пожалуй,
присоединения служит реакция Д'-азиринов (371) с'нестойкими
азометнн-илпдами 'VTPU тв°пмн“
ского раскрытия
КИМ выходом к
ние 121) {250].
неизмененный Д'-азириновый цикл, в частности', ‘термиче-
t's которых эти соединения выступают как диполярофилы или
Д'-Азприны могут также выступать и в роли диполярофиль-
динения благодаря наличию в них двойной связи углерод—азот
(2501. Пожалуй, наиболее четким примером такого рода цикло-
I (370) (генерируемыми in situ путем термиче-
цикла в азиридинах (369) |, приводящая с высо-
1,3-диазабицикло [3.1.0] гексанам (372) (уравне-
CH2Ph
А
PhY\>H
""'/Ph
(369)
д
бензол
CH2Ph
Y-,н
Ph Ph
(370)
СН2РЬ
ph-~N Ph-, «LyPii
YY (mi
Ph~A——N
(372)
[250], в которых, по-внднмому. пронсхо-
К другим реакциям [250], в которых, д,’)Поляриых
Дит образование п раскрытие цикла J]e>,c „ д,.Я7,|П„’нов с дпазо-
Циклоаддуктов, относятся
алканами при комнатной j ,____
азидам, н образование карбодпимидов в р^л-'и'^ером участия Д'-
азиринов с оксидами нитрилов при 0 Д'- р _ дльде'ра служит
азирпнов как диенофилов в реакции "'’°'
их термическое взаимодействие сглра,'
приводящее к ЗН-азепинам ( . .
происходит отщепление оксида углерода
Дукта (375) с образованием 2-- —
пировывается в наблюдаемый продукт
1*,5]-водородного сдвига (уравнение
в аналогичные реакции Дильса —
не цил«<1 HV7V4V......
также реакции Д'-азиринов с диазо
‘ 11 температуре, приводящие к аллильным
азириноВ'‘ оии“-1и°«1,1,е карбодппмидов в результате реакции Д'-
оксцдами нитрилов при 0°С. Примером участия
—• Дильса — Альдер. ............
гвие с цнклопентадпенонами (373),
(377) [250]. Прп этом, по-ввдпмому.
да углерода из первичного цпклоад-
г 2Н-азепнца (376), который перегруп-
^’51-водоп*пМ В "а<’г'10-;ас--чыи (377) за счет допустимого
в Залоги,,, "°Г0 СД8нга (уравнение 122). Д'-Азирины вступают
ск',мн 1 з _ Ь1е реакцпн Дильса — Альдера и с другими циклнче-
Д1енами (например, с циклопентадненом) [250].
557
При взаимодействии тиобензоилизоцианата (378) с Д’-азпринамп
образуются термолабильные аддукты состава 1 : 1 (380), которые
легко подвергаются реакции расширения цикла с образованием
тиадиазепиионов (381); это служит иллюстрацией участия Д'-ази-
ринов в реакциях [4-г 2]-циклоприсоединения с гетеродиенами-1 3
(уравнение 123) [250],
Двойная связь углерод—азот в Д'-азиринах, как и в случае
простых азометннов, может быть легко восстановлена (см. с. 543).
Восстановление комплексными гидридами металлов (LiAlHi,
NaBH<) приводит к соответствующим азиридинам с отличными
выходами (80—100%) и в связи с легкой доступностью Д1-ази-
рннов может считаться удобным методом синтеза азириди-
нов [233]. Неожиданно оказалось, что каталитическое гидрирова-
ние над палладием на угле привело к разрыву о-связи N-1—С-3
в Д'-азирипах [233].
В литературе содержится мало сведений о химическом пове-
дении простых Д'-азетннов, вероятно, вследствие их неустойчиво-
сти (например, г-фенил-Д'-азетпн легко полимеризуется [252])-
В последнее время центральным объектом изучения стали более
стабильные 2-алкоксипропзводиые Д’-азетшюв (которые легко-
доступны путем О-алкилпроваиия азетпдинонов [253]); изучень
в основном реакции их циклоприсоединения. Циклоприсоединени
658
,п0ку.муленов к ^-алкокси-Д'-азетииам
Детыми азомстинамп ^м. с. 520 сл.); OHOlg»’< «х реакциям
‘ ааз I ’2- no-видимому, с лервойачаль ““ A"7 адлУ^ам
) ^полярных интермедиатов, которые ШеТ /бразо^«
(«молекулу гетерокумулеиа. Типичным ипИмР"₽ Соеди»я/от ВТо.
g 2-метоксп-3,3,4,4-тетраметил-Д-.а3етш"рим^ служит р™к.
д,5Ьфоиилнзоцианатом (уравнение 124) (2541 9 л с ^-толуол-
тины реагируют подобным образом и с другими Д^^-азе.
й) (например, с трет-бутнлциаиокетеном (25511 п ерокУмУлена-
„бразоваиием н последующим раскрытием „ии/о РВОИачаль«ым
„нтермедкате можно объяснить реакцию 2 Ли В ;,?1ПшяРком
диметнловым эфиром ацетилеидикарбоновой Л, И'Л'’азетин°в
Тате которой образуются 3-азагексатриены-1,3,5 (з^’ го резуль’
МеО SCW
Me
Me —I—
Me—j—n.
•К,о
,N'SO2Ar
Т
О
(385)
MeCeH4SOiN=C=O,
25 °О
MeCeH4SO2N=C=O,
25 °p __________;
(R'= R2= Me)
(382)
(124)
MeO2CC=CCO2Me
(R*= R2= H)
(384)
(386)
8.1.2.4. Сопряженные полиазаены одцн
Описано довольно большое число Энного п0-111®'13
|,л« больше углеродных атомов в цепи Р Ь1Х случаев, со
метены азотом. За исключением двух в 1 ы> „, наско.тьк
111|я этого типа систематически не пссл 559
вестно автору данного раздела, вс существует обзора, в ко,
была бы рассмотрена химия сопряженных полиазаенов Oxr °м
целиком эту весьма обширную область невозможно в рамКахаТить
пой главы, поэтому мы ограничимся рассмотрением двух нацбДан'
хорошо изученных классов сопряженных полиазаенов: соппя 6
ных 2,3-дпазабутадненов-1,3 (известных обычно как азины)
[99в, 256], перекрестно сопряженных моноциклпческих п-хк 7
диимннов (388) it сопряженных моноциклпческих о-хинондину11'
нов (389) [257,258]. *'
V=N w
R2/ XN=C/
(387) (388)
(J) Методы получения азинов [2, 256]
Симметрично замещенные альдазнны и кетазпны можно легко
н с высокими выходами (60—90%) синтезировать реакцией аль-
дегида пли кетона (или образованного из них альдпмина или
кетимина) в водно-спнртовом растворе с избытком гидразингнд-
рата, гидрохлорида или сульфата гидразина, или (в некоторых
случаях) с безводным гидразином в отсутствие растворителя.
Образование альдазинов или кетазинов (392) в этих условиях —
двухстадпнный процесс, включающий промежуточное образование
гидразона (391) (уравнение 125). С альдегидами н диалкнлкето-
намп реакция протекает гладко, и промежуточные гидразоны
обычно не выделяют, тогда как алкиларил- или диарплкетоны
реагируют медленно, и реакция часто может останавливаться на
о
Ч‘\ NH2NH2 ri—С—R2
С=О---------> yC=N\ -------------------
цг/ R2/ \\'И2
(390) (391)
О
R3—C-RI
R‘\
,C=N4
R2/ \\'=C
(392)
Z
\R2
r.X(125)
d2/ XN=C
(394)
(393)
W
гидразона- В этих случаях для получения азина иагреВа10г
гтзД"11 ^/подходящем растворителе в присутствии или в отс7т
кислоты; при этом происходит диспропорционирование и об-
азпп л молекула гидразина. При нагревании гидразона
раз)'» * 1)СХОД1)ОГо карбонильного соединения или с другим
г 'Сьным соединением можно получить симметричные (392)
,мметрнчные (393) азины.
<з1О0<1Д№С™° этого метода является катализируемая кисло-
\ оеакния диазиридииов (394) с альдегидами и кетонами
” „кающая с раскрытием цикла и приводящая к симметричным
”Р несимметричным азинам.
"''"взаимодействие 1,4- или~J 5-Дикетонов (395) с гидразином
..................... ... ........ семи-
'"взаимодействие 1,4- или 1,5-днкетонов (395) с г;—
аппит к замыканию цикла и образованию теста- или
Ценных циклических азинов (396) [2,256].
л = 2 или 3
N-Моноалкилгидразины (но не арильные производные) всту-
пают в катализируемую кислотами конденсацию с альдегидами
в кетонами; это общий способ синтеза ациклических азиниевых
солей [112]. Так, метилгидразин (398) гладко реагируют с аце-
тоном (397) в присутствии НС1 и хлорида олова (IV), приводя
к пентаметилазиниевой соли (399) (уравнение 127). Прн анало-
гичной конденсации N-моноалкилгидразинов (401) с (З-дикетонами
(400) образуются, как правило, эндоциклические азиниевые соли
(402) (уравнение 128) [112]; экзоцнклические азиниевые соли
(405) легко можно получить при реакции циклических гндразо-
ниевых солей (например, пиразолнниевых солей) (403) с кароо-
чильнымп соединениями (уравнение 129) [112].
Ме^
Ме/С=О + McNHNH;
SnCIi
HCI
SnCIi”
(127)
(397)
(398)
COR'
| HX
R3—C—R2 + r-<nhnh2 —!
COR'
(400) (401)
(399)
(128)
(402)
561
Н Н
(403) (‘,04)
SnCI,
(129)
(405)
конденсац. приводящих к азинам, со-
альдегид013 с фосфиназинамн (реакция
Другой тип реакции
СТ0ПТ дкцпи'вХиг™ ^56]' Польку фо’сфин-
/408) 1259] легко можно получить реакцией диазоалка-
(чио; (. j___ жлг<Н|1тям11 или катализнпуемпн пт,-
Другой тип
типа реакции
а^Ь4ОбГс'(Ьосфпнамв или фосфитами пли катализируемой осно-
В п,Z ’р а£и!й гидразонов (407) с дибромфосфппамн, кондеи-
гп шю фосфиназниов с альдегидами можно рассматривать как
пбгинй способ получения симметричных и несимметричных азинов
130) Азины являются также конечными продуктами кон-
денсация фосфора нов с диазоалканами [ (406)->(409)] [256]
и пиролитического разложения диазоалканов в инертных раство-
рителях [256].
R'\- *
;с-мг
R2/
(406)
R1\
;c=Nk
r2/ xnh2
(407)
R3CH=PPI>3
РЬзРВгз.
Et3N
(130)
R1
R2
(409)
/H
\R3
R3CHO
-Ph3P-O"
/C=N\
R2Z ^N=PPh3
(408)
Известен ряд препаративных окислительных и восстановитель-
ных методов синтеза азинов. Как альдазпиы, так и кетазпны
получаются с приемлемыми выходами при окислении N,N-ne3aMC-
«™иых гидразонов иодом или хлоридом железа(III) {256].
прягрнтпо ра3уются также при действии свободнорадикальных
неипр содеРжащие а-водородпые атомы (урав-
(413) . кптпппо ’ И,1теРес»Ь1е по структуре дп-М-окснды азинов
пнем азнноп ”евозможно получить непосредственным окпсле-
(35—40%) пути К° М°ГУТ ®ь,ть П0ЛУ1|ены с умеренным выходом
действием саУзлнии°К11СЛ,1ТеЛЬП0Й Л,1Меризацип оксимов (412) под
(Ш) щелочного МетХаллКа’СЬ1"О'16’’ 1"апР,,меР’ гексацнаиоферрата
еилла, М2О4) гипохлорита натрия, иода — «аР
боната калия] (260]. Ди-Л1-оксид бензальдазина (4131 обп
также при обработке фенилдназометана (414) оке, „ образУется
(уравнение 132) [260]. °™м азотаЩ)
RS или С|3С
(410)
(131)
K3Fe(CN)e
2PhCH=N\0H
PhCH=N
\j=CHPh
NO +
'-- 2PhCH—N2
(132)
(412)
(413) (414)
Азины получаются с хорошим выходом (70—80%) и при восста-
новительной конденсации нитрилов с гидразином в присутствии
никеля Ренея (уравнение 133) [256]. Контролируемое электро-
восстановление легкодоступных пиридазиновых производных изби-
рательно проходит по связи С-4—С-5; это является способом син-
теза соответствующих циклических азинов (4,5-дигидропирндази-
нов) [38]. Примером такого рода синтеза азинов может служить
двухэлектронное восстановление З-диметиламино-6-фенилпйрида-
зина (417) в кислой среде, приводящее к легко выделяемому
4,5-дигндропронзводному (418) (уравнение 134) (38].
(415) (416)
(417)
(134)
Циклоприсоединение с последующим ретроцнклопрнсоеднне-
нием может служить удобным способом введения в молекулу
Двойной связи углерод-азот (см. с. 495 сл.). Реакции такого типа
можно использовать и для получения характерного для азинов
2,3-диазабутадиенового-1,3 фрагмента. Наиболее яркнм^ приме-
ром этого подхода к синтезу азинов является элегантный общин
способ синтеза циклической системы 3,4-диазабнцнкло[4. . Jге
тадиена-2,4 (422) [261 [ (представляющей собой новый интерес-
"ый класс циклических азинов), включающий циклоприсоединение
инклопропенов к 1,2,4,5-тетразинам. Возможно, что вначале Р
х°Дит [42]-циклоприсоединение, приводящее к бнцпклнческ
563
(2) Реакции азинов [99в, 256]
Азины подобно простым азометииам реагируют как с электро-
Жильными так и с нуклеофильными реагентами. Их реакции
с электрофилами напоминают реакции шиффовых оснований,
но из-за наличия сопряженной системы связей им присущи неко-
торые специфические особенности. Азины достаточно основны и
дают азиниевые соли [112] с протонными кислотами. Так, при
взаимодействии ароматических альдазинов или кетазинов с хло-
ристым водородом в апротонных растворителях, таких, как бен-
зол или эфир, образуются легко выделяемые гидрохлориды; соот-
ветствующие гидробромиды и гидросульфаты образуются соответ
ственно при контакте с 48 %-ной бромистоводородной или концен-
трированной серной кислотой. Азиниевые соли, образующиеся из
алифатических альдазинов и кетазинов, можно выделить лишь
с большим трудом. В случае а-С-алкилироваиных азинов (423)
протоиироваиие безводными кислотами часто сопровождается ци-
клизацией в А2-пиразолииы (427) с хорошим выходом (50—70%)
(уравнение 136) [256]. Кроме того, возможна и циклизация
в пирролы (424) (синтез пирролов по Пилоты); этот процесс ана-
логичен синтезу индолов по Фишеру [99в].
(424)
(423)
BuBr v
В'-Н^-Ме
Me
Bun xMe
(428)
(136)
ci"
Me
Me н
/б
Me N=C
CI Me
(425)
564
MeC—CIL-.-H
JI
HN Me
(427)
(426)
Непосредственное алкилирование [61 цо]
алкилгалогенндов (например, метилиоди’та! ,! действием
Лпример, диметилсульфата) проходит 'усиеХ алкилсУЛьФатов
бензальдазина. Однако при использований ™ " х Т”' В СЛучае
борфторид трнэтилоксоипя или метиловый эфир как
кислоты, возможно алкилирование и других соединяй ИФ°Н°В0Й
случае алкилирование часто сопровождается - этом
Газолины (428) [112]. При реакции аминов с карболовым В
лотами или ацнлгалогеиидами образуются N-яиипгиД И к,ис‘
256], а в случае а-С-алкилнрованных азинов-МПЙНЫ [61,
„ (256, 262J. Пр» —60 С происходит „ериоси" „ф‘е
соединение хлора к кетазинам по нерадикальному цепному м₽хГ
ИИзму, приводящему к 1,4-днхлоразоалканам [61 256 2681 м2.
ИНЗМ 1,4-присоединения хлора к кетазинам (429- R = Мм
полагающий атаку хлорид-ионом с наименее пространственно1^'
трудненнои стороны карбениевого интермедиата, стабилизован'
кого азотом (уравнение 137), был выдвинут [263] на оснований
наблюдаемого влияния заместителей и стереоспецифичности
В отличие от кетазииов прн хлорировании альдазннов
К Н) происходит постадийное замещение имидитьных
водорода хлором [264, 265д].
Ph
C=N
N=C
Н
(430)
(429;
атомов
CI г,
ссц
R-H
Ph
Cl
Ph
(429)
R»Me | С?
сц.
Ph C=N
Cl 1
CI
(432)
CI
i-niMe
N=NZ ^Ph
Ph/*, /
JC
Me*^ I
Cl ч
(431)
(137)
себя
Так,
В реакциях с нуклеофильными реагентами азины ведут
подобно простым несопряженным шиффовым основаниям,
например, нуклеофильное присоединение часто приводит к насы-
щению лишь одной из двух углерод-азотных двойных связей и об-
разованию производных гидразонов. Катализируемый кислотой
гидролиз азинов [256] и азнниевых солей [112] приводит к рас-
щеплению на исходные карбонильный и гпдразинный компоненты.
Интересно, что при кислотном гидролизе моно-К'-оксидов азинов
образуются карбонильное соединение и диазоалкан [266]. а-С-Ал-
килированные азины под действием дпизопропиламида лития
в Тетрагидрофуране депротонируются, давая в зависимости от
565
атомов моно- плл днаниоиы, которые могут
важных синтетических превращений. Так,
анноны (434), образующиеся из моноалкн-
а-50°С Сдавая бисазпны (435),' гидролиз которых
1 А л А -» t I 1 «л гт tl П » < ( Д л(т 1 I UtX О И И А I» .. .
1 4-^карбонильным <—ч , —mv
числа а-водородных
подвергаться ряду _
моно-1-азаа.1.1»лм‘^ сдваиваются в присутствии хлорида
/’ /-„«„«„uu М.ЧН) гнлпп.пна иптлт.^
соединениям (436) (уравнение
лироваивых
меди(1) при
приводит к
138) [267].
Р\
/ ’
К LiN(Pr-aw);
тгФ
Ph
R2
R
(433)
Ph
CH2R'
Ы+ ' CHR'
(434)
CuT .
-50»С '
,3
R
Ph
R2
N=C
CHR3
CHR3
r2\
с
I 3
CHR3
CHR3
(138)
.2
R2 О
R (435)
Бис(азааллильиые) анионы, образующиеся из диалкиллрован-
ных кетазинов (437) при действии диизопропиламида лития в те-
трагидрофуране при комнатной температуре, самопроизвольно
циклизуются и дают после гидролиза пирролы (440), правда,
с умеренными выходами (17—52%) (уравнение 139) [268]. Фор-
мальное внутримолекулярное «.«'-сочетание в азиновой системе,
участвующей в этой реакции, можно объяснить циклизацией ди-
аниона дикетимина (439), возникающего путем перегруппировки
бпс(азааллильиого) аниона (438), сходной с перегруппировкой
Коупа [268].
RH2cv
4C=N.
(436)
перегруппировкой
д (uao-PrhNU
"~ТГФ, 20 °C?
(437)
г RHC—CHR
► Аг—с:
\ / Аг
N N' 2Ц+
(439)
н2о
RHC
Ar—cf
CHR
Sc—Ar
'N—N' 2Li
(438)
R R
Аг— —Аг
NH
(440)
(139)
566
реакции азинов с нуклеофильными пеягрц
,1ЫЙ центр которых находится на атоме азота Г’ 1!уклеофиль-
К продуктам гидролитического расщепления tL ривОдят обычно
Лизе азинов с хорошими выходами ( — 750/; а ’ При ГидРазино-
ствуюшне гидразоны. Гидразинолиз азинов ' °0разуются соответ-
для получения тех гидразонов, которые DaiI ° рек°Мендовать
лучить из-за их неустойчивости [2561. В от™, Ие удавалось по-
филов нуклеофильные агенты с нуклеофильном116 °Т этих нуклео-
углерода вступают, как правило, в реак,„<«л „ Це1,Тром на атоме
динения по одной из двух азометнновых- сХТ™ 1’2'пРисое-
аая устойчивые моноаддукты (гидразоны) с Zm В 33ине’ Да‘
Так, при действии цианид-иона образуемся аЛукт"^8??0^'
а при действий карбанионов из гриньяровскиГи™ ( 12э6Ь
ческнх соединений — (443) [99в 2561 хотя « -титнноргани-
может происходить и дальнейшее присоединен 1учае
дящее к бисаддуктам (445) (уравнение 140) Результатом’
реакции бензальдазпна (441; R1 = Н- R2 = ph> татом бурной
метилоксосульфоння в диметилсульфоксиде при Си2^ИД?“ ДИ'
пературе является также продукт 1 2-прнсоединения N бензити'
денаминоазиридпн (444) [269], ия’ 1ч-оензили-
Н CN
HCN
-<----
(442)
Me2SOCH2
R1 = H.
R2=Ph
(441)
I
N=CHPh
I
(444)
(140)
R1
R2
PhMgBr;
H2O
(443)
PhMgBr
H2O
R\ /R1
,c—мн—nh-c;
R:/1 I XR2
Ph Ph
(445)
Реакции циклоприсоединения азинов стали предметом
ннтен-
..... ..ао,1пи“ 16^'1^'256? 2701. Хоро-
спвиых исследований в последние годы [ > ’,KTVpy сопряжен-
ию известно, что введение атомов аз° способность в реакции
кого диена резко снижает его реэкцпонн) . Поэтомусреак-
Дильса — Альдера ([4 + 2] -циклоприсое' ц с СОпряжени«М1Г
Ниями Дильса — Альдера, которые, по • азинов, приходится
Диенами, должны были бы быть обычп - • способность азн-
встречаться крайне редко. Низкую Р^1’0'1^ „ с тем, что пз-за
»ОВ связывают с нх иеплоскостным стро
рпятствий «-г<«с-геометрия переходного состояния,
стсрнчсских "Реля пешного [42]-циклоприсоединения, недо-
необходимая дляу кл„чесК11Х азинов, для которых выпол-
стнжнма. “*,Р геометрические требования существования
„яются »е°°ХОД состояния, реакция Дильса — Альдера проте-
|4 + 2]-_перех°Ц уер, взаимодействие 3,4-дпазабицикло-
—’___________А-2,4 (446) с алкенами (хотя и напряженными),
[4 + 2] -циклоаддуктам (448), которые можно
такими, как • Р oi „пя ппиктам (448), котопые можно
ны-2,5, приводит к
выделить [271].
СО,Me
В
СО2Ме
(141)
(447)
СО2Ме
(448)
N
СО2Ме
(446)
Двойные связи углерод—азот в азииах, если п вступают, то
редко, в реакции (2 + 2]-циклоприсоединения, характерные для
простых азометинов [разд. 8.1.2.2(3)]. Преобладающим для ази-
нов является так называемое перекрестное (criss-cross) цикло-
присоединение, а не реакции [2 4-2]- и [4 4-2]-циклоприсоедине-
ния [61, 156, 256]. Бензальдазины, например, обычно реагируют
с двумя эквивалентами электронодефицитных алкенов (этилакри-
лат, малеиновый ангидрид, N-феннлмаленмнд), давая производ-
ные 1,5-диазабнцикло[3.3.0]октана [61, 156, 256]. Типичной для
такого циклоприсоединения является реакция незамещенного бен-
зальдазина (449) с метилакрилатом, приводящая к диметнловому
эфиру 2,6-дпфеиил-1,5-диазабицнкло [3.3.0] октапднкарбоновой-3,7
кислоты (451) (схема 142).
Ph
СИ2=СНСО2Ме
PhCH=Nx -----> МеОгС
xN=CHPh
—СОгМе (142)
(449> (451)
В отлнчцё °т этой вяло текущей реакции, для осуществления ко-
In₽ntne06XOfl"MO нагРеваиие при 125—130°С в течение 48 ч,
»апппмепТ1^Х вдклопР1,соеД|1нение электронодефнцитного азина,
ним э1еРктооиа^иГеКСа^Т°РаЦеТОНа (452)’ к алкенам, обогащен-
очень легко Эту ’ реакцию^ о" 2’метнлпропе11у (453), протекает
фотохимически п Р ц МО/КНО индуцировать термически или
продукта (455) °',е,!Ь
этого типа проводить при mJ 561’ Еслп Циклоприсоединение
стехиометрических количеств ™ , темпеРатуРе с использованием
trB азина и алкена, то можно выделить
с хорошим ВЫХОДОМ ЦИКЛОаддукпг
троением экзоциклнческих азометин,,» СТава ' : 1
Д в свою очередь, легко р6агИруС"1’0/ (454) [156 27?^
г«М алкеном И Дают симметрично т с Же caMbI^ 2731
ные циклоаддукты (455) или (456) [273] ' СИмметРично замещен”
(452)
(453)
Me Me
(453)
(143)
(455)
Me Me F3C CF3
(456)
Отсюда следует, что «перекрестное» циклоприсоединение азинов
на самом деле представляет собой сложение двух [3 + 2]-цикло-
присоединении. Последовательное введение в эту реакцию различ-
ных субстратов с кратными связями может стать потенциальным
способом синтеза различных азабнгетероццклов. Так, использова-
ние алкинов в прямом [156] и двухстадийном [273] «перекре-
стном» циклоприсоединении к азинам дает моно- и диненасыщен-
ные аналоги соединений типа (455). Более того, подобное цикло-
присоединение азинов не ограничивается лишь соединениями
с двойной углерод-углеродной связью, оно осуществимо н в слу-
чае соединений с двойной углерод-азотной связью. Примером слу-
жит реакция бензальдазина (449) с фенилпзоцианатом, приводя-
щая к производному 1,3,5,7-тетраазабицикло[3.3.0]октана (40/)
(Уравнение 144) [256].
(449)
(457)
569
nprm окисляются без разрыва связен прп пспользова
Азины легко ом 1сства пероксикислоты и дают соответ-
вин эквива.теитно|и аЗШЮВ с приемлемыми выходами (50—
ствпошие моно-in Дальнейшее окисление не приводит к
60%) 1В 9-,в’ , ’ (1!Х приходится получать обходным путем,
дп-М-окспдам аз 11СХОД„Т расщепление азина с образо-
сМ7 ^„схо дного карбонильного соединения и азота. К такому
ваннем исход „‘окисление азинов сильными окислителями,
такими как азотная кислота [256] или озон [256]. С высокими
такими, ко продукты фрагментации альдазинов и кетази-
новтожио получить при действии иодной кислоты в мягких усло-
вна (15 МИИ при комнатной температуре в уксусной кислоте или
дпметплсульфокснде); это является удобным способом регенера-
U11U карбонильного соединения [см. разд. 8.1.2.2 (4)] [275]. При
окнетеиин азинов в относительно мягких условиях действием
диоксида марганца [185, 256] происходило главным образом их
разложение в отличие от реакции с тетраацетатом свинца при
действии которого алифатические кетазнны (458), не имеющие
а-водородного атома, превращаются в азобисацетаты (459), кото-
рые можно выделить (уравнение 145) [195]. Аналогичное превра-
щение было описано в разделе, посвященном гидразонам (см
с. 540). Ароматические кетазнны устойчивы к действию тетрааце-
тата свинца; ароматические н алифатические альдазины (458.
R2 = Н) циклизуются прп действии этого реагента в производные
1,3,4-окса диазол а (461), по-видимому,
доилацетаты (460) [195].
RA
>C=NX /К1
02/ ’N=C
через промежуточные ими-
РЬ(ОАс).|
R1-
(458)
R2=H |рЬ(ОАс)4
R\
;c-n4 /R'
R2/| ^N-C(
OAc | \r2
OAc
(459)
(145)
R1-
О R>
N---N
(460) (46Г)
В зависимости0^0 ПРОСТЬ,М азометинам можно восстановить,
шение одной из ^ользУемого реагента происходит либо насы-
гидразонов лХД полное СВвоеЙ УГЛер°Д~ азот с образованием
гидразинов, которому Mn., BQCCTaii0BJIemie до соответствующих
и образование первпиш,°ЖйТ сопутствовать разрыв N—N-связи
9 х ам1шов. Амины обычно образуются при
„становлении азинов системой металл — доноп „„„
ss“«sux==-“==-;.=5
результаты дает использование трифенилфосфина (274] Подей
ствин трнфенплфосфина происходит восстановление и дн-N окси’
дов азинов в соответствующие азины, тогда как восстановление
сульфидом аммония приводит к разрыву N-N-связи с образова-
нием оксимов 1.274]. Лучше всего проводить восстановление азинов
до гидразинов действием алюмогидрида лития или гидрированием
над платиновыми катализаторами [99в]. Каталитическое гидриро-
вание или восстановление азиниевых солей комплексными гидрида-
ми металлов обычно останавливается на стадии образования
гидразонов [112]. Ход электрохимического восстановления азинов
зависит от pH среды [38]. В нейтральном растворе бензальдазин
подвергается шестиэлектронному восстановлению, сопровождаю-
щемуся расщеплением N—N-связи, с образованием бензиламина,
тогда как в кислой среде происходит двухэлектронное восстановле-
ние до бензилгидразона бензальдегида [38].
(•3) Методы получения бензохинондииминов [257, 258]
Основной метод синтеза п- и о-бензохинондииминов состоит в
окислении производных п- и о-фенилендиамина, обычно в инерт-
ном растворителе (эфир или бензол), при комнатной температуре,
а в случае менее реакционноспособных субстратов — при несколь-
ко более высокой температуре (например, 50—60°C). Из-за не-
устойчивости многих бензохинондиимннов выбор окислителей огра-
ничен: могут быть использованы лишь те реагенты, которые обес-
печивают быстрое и практически количественное окисление в мяг-
ких условиях. К ним относятся трикалийгексацианоферрат, оксид
серебра и тетраацетат свинца, причем последний часто дает наи-
лучшие результаты. Сульфат церия (IV) в серной кислоте также
успешно использовали для окисления n-феннлендиаминов в л-бен-
зохинондиимины [276]. n-Бензохинондиимины, содержащие N-неза-
мещенную импногруппу, весьма неустойчивы к влаге и свету н
легко полимеризуются. Поэтому N-незамещенные и N-монозаме-
Щенные /г-бензохинонднимины (463; R1 = Н, R3 = Н, алкил или
арил) обычно не выделяют из раствора, а сразу вводят в реакцию
после генерирования из соответствующих производных п-фенплен
Диамина [257].
Окисление производных п-фенилендиамииа ионом серебра в
незамещенные п-бензохииондиимины и сочетание пос. .
situ с подходящими субстратами с образованием кр •
®ит в основе процессов цветной фотографии. В слу ие
т₽ичиых М,М-дизамещенных п-фенплендиамин
571
R3 = R1 = H
(146)
(466) (467) (465)
протекает через радикал-катионные интермедиаты (464) н приво-
дит к неустойчивым п-бензохинонпминимнниевым солям (465)
(уравнение 146) |257, 276). Эти соединения обычно также не вы-
деляют из раствора и немедленно вводят в реакцию [276]. Сим-
метричные N.N'-днзамещенные п-бензохинондиимнны сравнительно
устойчивы, особенно это относится к М,М'-диацнл-п-бензохн-
нондпиминам и М.йГ-дисульфонил-п-бензохннондннминам, продук-
там окисления N N'-днацнл- или М.М'-дисульфонил-п-феннленДи-
аминов (462; R' = R3=COR или SO2R). В отличие от более
простых п-бензохннондннмннов эти соединения обычно представ-
ляют собой довольно устойчивые кристаллические вещества. Было
показано [276), что /г-бензохинопнминнминиевые соли (465) мож-
но нолучить т situ фотолизом Ы,М-дизамещенных п-азидоанили-
нов (466) в водном растворе, вероятно, с промежуточным образо-
ванием производных нитрена (467) [277].
(4) Реакции бензохинондииминов [257, 258]
х11ипипип.ЛЛЛУЧае хинонов’ иа реакционную способность бензо-
щению r ИуптА"СИЛЬИ°е ,влияние оказывает тенденция к возвра-
бензохинонтиимшш У'° бензоиднУ10 структуру. Эта тенденция у
реакциям нуклрон. В пРоявляется в повышенной склонности к
ления котовое плпто"°Г0 пРпсоеДинения и в легкости восстанов-
большее число да^ных3^ быстрее’ ч„ем в случае хинонов. Наи-
Нмннов получено РеакцИ0||||°й способности бензохинонди-
и ^y-4b4>oH1WnpoH3BOAHbi4xei[258]OTHOC1,TeJlbHO Устойчивых диациЛ’
реагентами [257 %5о\,зох‘,1101,ДНИМ|,иов (468) с нуклеофильными
нне; в результате после™!'»сегда происходит 1,4-прнсоедине-
С72 последуЮ1цеи ароматизации образуется заме-
,llioe в ядро производное п-фенилендиамина (470V »
1т провесе представляет собой замещение (УРавн2'е 47) За
дним или двумя исключениями реакций 1,2-присоединения к
миногруппе не наблюдалось. Примечательной особенност. ю
“Геофильного присоединения к п-беизохинондииминам являе?
я то, что благодаря легкости окисления образующихся моноза-
мешенных производных n-феиилендиамина удается осуществить
Замещение путем последовательного окисления и присоедине-
ния [(470)-> (471)(472)] (трн- и тетразамещение обычно не
происходит).
не
NHR
NR
NHR
уч
NHR
(147)
NR
NR
(470) (471) (472)
Кислотный или основной гидролиз [257] простых N-незаме-
щенных или N-алкилированных п-бензохинонднимпнов приводит
к дезаминированию в соответствующий п-бензохпнонпмин (урав-
нение 149; R = Н, Х = О); с процессами такого рода нуклеофиль-
ного 1,2-присоединения к бензохинондиимннам приходится встре-
чаться довольно редко.
NR
(468)
В случае п-беизохпнонпмнннминиевых солей (465) наяРавл
«“е г11дро.1Шза заВ11сит от рн. В нейтральных или К'шлых
Р х дезаминируется пмпногруппа, а в щелочны.
^еститель [1257, 258]. Окисление N.N-Днзамещенньх W
в Д11амшюв С последующими дезаминированием восСДанов-
ленирУЧа'още'|СЯ п-бсизохиноипмиипмпнпевои сол. реакЦии
ением хлоридом олова (II) или поднд-ионом (все реаки
S73
„ пином сосуде, без выделения промежуточных про-
нроводяия в> л оы > дсгавляет собой метод синтеза труд-
SSSSS» i........................ IWI.
™°Cniicvibtboiii^-n-BeiBoxiHioHAiiiiMiiHbi относительно устойчивы
n слов нях кислотного и основного гидролиза, однако прн дейст-
вии концентрированной соляной кислоты в уксусной кислоте ПЛI
X орнстого водорода в бензоле гладко протекает 1,4-присоедине
и образуются соответствующие хлорированные п-фениленди-
^мины (уРравненпе 147; R = COR или SO3R, X = Cl) [257, 258],
Подобным образом ведут себя в ИМИ'
нов (уравнение 150; R — COR пли SO2R, X С1) [2э7, 258].
(475) (476)
(150)
Диацнльные производные п- и о-бензохинондпиминов легко
подвергаются также 1,4-прпсоединению карбоновых кислот в от-
сутствие катализатора с образованием соединений (470) и (476)
(уравнения 147 и 150; R = COR, X = ОАс) [258].
Дисульфонилбензохннондинмпны в таких реакциях менее
реакционноспособны: присоединение карбоновых кислот к пара-
пропзводным происходит в присутствии кислого (эфират трехфто-
ристого бора) или основного (EtaN, NaOAc) катализатора и не
происходит вовсе в случае орто-производных [258]. При 1,4-при-
соеднненпи спиртов к диацил-n- и -о-бензохннондииминам (урав-
нения 147 и 150; R = COR, X — OEt) также необходимо приме-
нение эфирата трехфтористого бора как катализатора [258]. 1,4-
Присоединение алкан- и арентполов к бензохинондниминам, на-
против, идет в отсутствие катализатора (уравнение 147; R =
= SO2R, X = S-Alk пли S-Ar), однако при использовании три-
этиламииа как катализатора удается подавить возможную па-
раллельно протекающую реакцию восстановления бензохинонди-
имина до фенилендиампиа (уравнение 147; R = SO2Ph, X = Н)
ri сократить продолжительность целевой реакции [258].
еакции п- и о-бензохинондипмннов с аминами более сложны,
и характер их протекания зависит в заметной степени как от
рироды субстрата (бензохннондннмин), так п от природы амп-
а. [ретичиые ароматические амины (478) конденсируются с
NH
(477) (478)
(479)
574
м „езамешенными n-бензохинондинминамн (47
N „ожение арнламнна, образуя красители 479 (Л, в пара'
|10ЛЭта реакция сочетания имеет большое знамен,
фотограф'"1 1257]- При реакции Д'<сульфонил-п-бе™’“7ИОЙ
„п с первичными алифатическими аминами (напоим™ „онд,иими’
"мнном) происходит 1,2-присоединение к сульфоиилимиё к'бутил’
о = SO2R, X Мои н) |2о/, 258]. Однако первичные авилямиии
(например, анилин) и вторичные алкиламины (например чор
фолпн, пиперидин) больше склонны к реакциям последователь-
иго in situ 1,4-присоединения- окисления - 1,4-присоединения
давая 2-замешенные и 2,5-диза мешенные производные п-феннлен-
дпамина (уравнение 147, R SO2R, X = анилино или морфолино)
|257, 258]. Производные 2-амино- и 2,5-диамнно-п-фенплендиами-
нов [(470) и (472); R SO2R, X = Y = NH2], которые нельзя
получить непосредственно, легко можно синтезировать путем
контролируемого последовательного 1,4-присоединения азотисто-
водородной кислоты (уравнения 147 и 148; R = SO2R, X = Y =
= N3) и последующего восстановления гидросульфитом [258].
Приведенные выше примеры демонстрируют важность реакций
1,4-присоединения к п- и о-бензохинондииминам для введения
функциональных групп в доступные производные п- и о-фенилен-
дпамина с помощью нуклеофилов, содержащих кислород, серу
или азот. Такого рода 1,4-присоединение к п- и о-бензохинонди-
иминам не удается осуществить для реактивов Гриньяра (которые
более склонны вызывать восстановление, чем вступать в реакцию
присоединения), однако оно обычно гладко проходит в случае
карбанионов, полученных нз соединений с активной метиленовой
группой; образующиеся при этом продукты замещения в ядро
представляют интерес как исходные вещества в синтезе индолов
[257, 258]. Например, М.М'-дпбензолсульфонил-п-бензохннонди-
имин (468; R = SO2Ph) реагирует с ацетилацетоном в бензоле в
присутствии каталитического количества метоксида натрия, да-
вая ожидаемый 1,4-аддукт (т. е. продукт присоединения по Ми-
каэлю) с хорошим выходом [470; R = SO2Ph, X = СН (СОМе)2] -
•’Незамещенные п-бензохинонпшшпмпнневые соли (465), напро_-
Т|1В, конденсируются с соединениями с активно,, метиленовой
группой с участием „минного атома азота; образу*?""1®?’ KP,aSB_
рЛи находят применение в цветной фотографии [2о7. 2а J. •
„ |1аРенсульфонил-/1-бензохинондиимины (468; R = SO2-г) Р
[?,от также с активированными аренами (фенолами, про
(Мп Мп ФенолоВ, арнлампнамп) в присутствии кислот ” к.
Ь, BF3) как катализаторов и дают соответствуют. р .
’ аРил11роваипя (470; R = SO2Ar, X = Ar) [2t>7, 2а8].
3oxiruaKlln" циклоприсоединения к N.N'-диаренсульф •
Ч|'леПпНд"ИМШ1ам протекают с участием углерод-ун’е но-
^сРкоД;аЗОТНоП дп°йной связи; В° ЕСеМ °СТяаЛНапримёр, такие
олько-нибудь исключительного характера. Р
575
лтчом участвуют как 2л-компоиенты в пеак
^+2] ш^опрнсосдш1е.шя | (4SO) - (482) ] |2о8].
NSOaPb
NSOiPh И
[258].
•Me
-Ph
Ph
Pl,2c -X'z
NSO2Ph
•Me
(152)
Me
NSOjPh
(481)
NSO2PI1
(480)
NSO2Ph
(482)
Большой интерес представляет способность 1,4-диазабутадиен-
1 3-ового фрагмента в о-бензохпнондииминах выступать в роли
^-компонента в реакции [4 + 2]-циклоприсоединения к алкенам
[179 270, 280]. Этот довольно редкий случай участия аза-1,3-дце-
на в реакции Дильса— Альдера (см. с. 568) проиллюстрирован
взаимодействием Ю'-дибеизолсульфонил-о-бензохинондннмина
(483) с дпфенплкетеном (484), приводящим к тетрагидрохинокса-
линовому производному (485) [280].
SO2Ph
(483) (484)
SO2Ph
(485)
(153)
8.1.3. ИМИДОИЛГАЛОГЕНИДЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ
Имидовые кислоты (так называемые изоамиды)—гипотетиче-
ские соединения общей формулы (486; Х = ОН). Свободные имн-
довые кислоты, по-видимому, до сих пор не получены. Однако неко-
торые вещества, полученные при катализуемой кислотой гидрата-
ции нитрилов, ведут себя химически и спектроскопически как кис-
лые соли имидовой кислоты (487; Х = ОН, R2 = И), а не как солн
амидов |281]. В то же время хорошо известен ряд классов соеди-
нений производных имидовой кислоты; к ним относятся соот-
ветствующие галогенангидриды, сложные эфиры н амиды импдо-
пям К1Мй£’ <^ычно называемые соответственно пмидонлгалогени-
(486- х — ’моГУа11’ имила’гам« (486; X = OR) и амидинами
'.(ред’ *2'’ В данном разделе описаны имидоилгалогенидЫ
(486- pa п р°Л?тви,НЬ1е соединения — гидрокенмонлгалогепндь!
$ = ЫрГх Ня1Т 11265г1 - гндразононлгалогеннды (486;
< nr2, X — Hal) [265д] и фосгеннмины (486; X = R’ = Hal)
576
г0,- 282-284]. Имидаты
E5bX4 И8.Г6.
x\
V=NRS
R1/Z
(486)
X = Hal, OH, OR, NR2
и амидины рассмотрены в разде.
Х\ */R2
;c=nz
R1' \рз
(487)
8.1.З.1. Методы получения имидоилгалогенидов
и родственных соединений
(1) Галогенирование карбоксамида-
и тиокарбоксамидосоединений
Галогенирование (главным образом, хлорирование)' легкодо-
ступных амидов и тиоамидов и близких по структуре соединений,
таких как производные гидроксамовых кислот (488; X = О,’
p3 = OR) и гидразиды (488; X = О, R3 = NR2), широко ис-
пользуется для синтеза имидоил-, гидроксимоил- и гидразо-
ноилгалогенидов (в основном хлоридов) и фосгениминов. Так,
классический и до сих пор наиболее удобный метод синтеза
имидоилхлоридов (489; R' = R2 = Alk или Аг) с высокими вы-
ходами [264, 2656] состоит в нагревании вторичного алифа-
тического или ароматического амида (488; X = О, R1 = R2 = Aik
пли Аг) самого по себе или в растворе инертного растворителя
(эфир, толуол) с подходящими хлорирующими агентами, выбор
которых чрезвычайно широк (например, PCI5, PCU — Cl2, SOCh,
ArSO2Cl, СОС12, PhPCL, PhjP —CCI4). Первоначально образую-
щимися продуктами являются хлориминиевые соли (490; R3 = Н),
которые превращаются в имидоилхлориды прн нагревании или
при обработке основанием, например триэтиламином. Особенно
удобно проводить хлорирование тионилхлоридом, так как побоч-
ные продукты реакции газообразны. Использование фосгена
СОС12 для хлорирования алифатических амидов позволяет избе-
жать осложнений, связанных с а-хлорированием, которое возмож-
но при применении таких реагентов, как пентахлорпд фосфора,
Хлорированием под действием системы трифенилфосфпн четы-
реххлористый углерод дает отличные выходы имидоилхлоридов
(80—90%), однако продукт реакции обычно трудно отделить от
образующегося трпфенилфосфчноксида. Эту трудность удается
преодолеть, если использовать трифепплфосфпн, связанный с
Лпмериым носителем [285]. Хлорирование алифатических н ар
этических третичных амидов (488; R’ = R= = R = Ш
^помощью таких реагентов, как пентахлорид фосфора, оксалил
°рид, тпопплхлорнд пли фосген (панлучииш реагент), р
о|,,0ответствУ10Щнм хлоримпниевы.м солям (так назыв м.дл.
р"Дам амидов [9861 ) (490- R' = R2 = R3 = Alk или Аг). N Ал
к,1Льиые произЬдХ этих солей при -НО’С подвергаются
19 Зак. Ю5
prOMV отщеплению алкнлхлорнда (отщепление типа
пиролнгнческому с образованием (1мндонлхлорпдов (489) (284]
распада ио ьРа>“>п'о.. хюрироваипе тетразамещеппых мочевин
Подобным »e,H°%38Mx = O «ли S, R1 = NR2, R2 = R3==Alk
или тпомочевин • оксалнлхлорнда дает хлорформ.
или Аг) с помощью фосгену nr2) Такпе реагент^
аТ!~дРфосфора. дегидратируют алифатические и аромати-
ческне первичные амиды в нитрилы.
Ск
XC=NNR1
R,Z
(494)
Ск
XC=NOR3
(493)
Фосгенимины (491) и соответствующие фосгенимнниевые со-
ли (492)—реакционноспособные соединения, широко используе-
мые в самых разнообразных синтезах. Их легко получают с от-
личными выходами из амидов или тиоамидов под действием ряда
хлорирующих агентов. Так, фосгенимины (491, R2 = Aik пли Аг)
[265а, 282, 283] образуются при хлорировании (под действием
хлора или фосгена) днтиокарбаматов (488; X = S; R1 = S_ Na+;
R2 = Aik или Аг; R3 = H), которые либо предварительно синте-
зируют, либо получают in situ из первичных аминов и сероугле-
рода. Арилзаыещенные фосгенимины (491; R2 = Аг) можно полу-
чить хлорированием форманилидов (488; X = О, R1 = R3 = Н,
R2 = Аг) хлором в присутствии тиоиилхлорида [265а]. Фосген-
имннневые соли (492; R2 = R3 = Aik или Аг) образуются при хло-
рировании ткокарбамоилхлорндов (легко получаемых нз первич-
ных аминов и тнофосгеиа или хлорированием дпзамещеппых
тиоформамидов), тнурамдисульфидов, днзамещеиных дитпокарба-
«агсюпЛИ лчзамещеиных тпоформамидов (488; R’ = Н, Cl, SMe,
~ R3 ~ Aik пли Аг) действием хлора или
у Фу рилхлорида без добавок или в присутствии пеитахлорнда
578
Лосфора I282’ 2871' ВИ0№ ПР” эт°м обычно веема
М) Хлорирование гидроксамовых кислот м ВЬ1С0Ки (80—
y ^ O; R1 = Aik или Ar; R2 = Н; R3 = « ™Дразндов (488;
„Ы фосфора также используют для поеплпа’пентахлорн-
(433) Й 2Кг| .L
[265в] • ’ *
(2) Галогенирование азометинов
Галогенирование азометинов (495) с замещением им.
аТома водорода уже вкратце обсуждалось ранее км X?
8.12.2(1)] • Это наиболее эффективный препаративный 1етод син'
теза гидрокспмоилгалогенидов и гидразононлгалогенидоЕ> (урав-
нение 155, R — OR или NR2, X —Cl, Вг,1). Непосредственное
галогенирование альдпминов [264, 2656] действием галогенов
трудно контролировать, поэтому эта реакция не имеет большого
препаративного значения. Реакция с трет-бутилгнпохлорнтом прн
низкой температуре вполне пригодна для превращения альдими-
нов в нмндоилхлориды (уравнение 155; R> = R2 = Alk или дг,
Х=С1) [264, 2656]. Хлорирование формимидонлхлорндов in situ
действием сульфурплхлорида при 15—20 °C — удобный метод по-
лучения фосгениминов (уравнение 155; R1 = Х = С1) [265а].
Нх Хч
:C=NR2
'C=NR2 -
R1Z
(495)
Хлорирование алифатических
действием хлора в эфире или хлороформе или, предпочтительнее,
в концентрированной соляной кислоте представляет собой класси-
ческий метод синтеза гидрокснмоилхлоридов (уравнение 155;
496; Х = С1; R! = Aik или Ar; R2 = ОН) [24, 264, 265в, 288]; вы-
ходы могут достигать 90%. трет-Бутилгнпохлорпт в метаноле и
нитрозилхлорид также применяли как хлорирующие агенты; ис-
пользование брома приводит к соответствующим гидроксимоилбро-
Мндам (496; X = Br, R2 = ОН). При хлорировании и бромиро-
вании гидразонов альдегидов (495; R2 = NR2) выходы соответ-
ствующих гидразоноилхлоридов и -бромидов (496; X = С1 или Вг,
°2 ~ NR2) составляют 60—90%; этой! реакции, однако, может со-
путствовать галогенирование N-арильного заместителя [2о ,
265д, 289].
(155)
(496)
н ароматических альдоксимов
(3) Присоединение галогеноводородов к нитрилам
Нитрилы присоединяют галогеноводороды с образованием мо-
н°- и бисаддуктов, относительно природы которых не было едн-
||ОГо мнения. Особое внимание привлекли бисаддхкты, о р }
'Циеся при присоединении хлористого водорода к (
кольку, возможно, они являются промежуточными с д
19* 679
. п»аК|111Я’( на основе нитрилов, как синтез альде-
в таких важных Р и синтез кетонов по Гешу. В настоящее
гидов во ‘ • общепринято, что бпсаддукты хлористого
время, no-вид»^' деле ЯВЛЯ!0Тся хлоридами хлор,
водорода । «ии_„3 = Н) |0 природе этих соединений н их ро-
оеакипоаиоспособных интермедиатов подробнее см. 8
лн Й99 9НИ Присоединение хлористого водорода к комп-
p33t Пейтах тори та сурьмы с хлорцпаном дает хлорид фосгени-
л 5 mL ' R2= R3 = Н) (265а]. Присоединение хлористого
" < к оксидам нитрилов (497) с высоким выходом приводит
к гндроксимоил.хлорндам (498) (24, 265г]. Этот метод, однако,
имеет невысокую препаративную ценность, так как именно гидро-
+ hci С1\
R_c=\—О’ -------> ''C=NOH
(156)
(497)
(498)
кснмоплхлориды на практике используют как предшественники
для получения оксидов нитрилов.
(4) Прочие методы
Классический метод синтеза фосгениминов (500)—низкотем-
пературное хлорирование изотиоцианатов (499) в хлороформе (схе-
ма 157) [265а] (выходы часто превышают 90%). Он применим к
алкил-, циклоалкил- и арилизотноцнанатам. Эту реакцию прово-
дили и в промышленном масштабе, однако из-за возможности про-
текания побочных реакций она не нашла широкого применения.
Аналогичное превращение изоцианатов (499а) в фосгенимнны (500)
происходит менее легко: необходима обработка пенгахлоридом
фосфора в хлорбензоле прн 20—50°C [265а]. Арилизоциапаты бо-
лее склонны образовывать карбодинмпды, чем фосгенимнны. В то
же время более реакционноспособные сульфонилизоцианаты глад-
ко превращаются в соответствующие фосгенимнны при действии
хлора в бензольном растворе при 60 °C [265а]. Алифатические и
ароматические изоцианиды (501) дают фосгепнмнпы (500) с коли-
чественным выходом при обработке хлором или сульфурилхлорп-
«мг/ У°Р°Ф°Рме ПРИ комнатной температуре [265а]. Этот метод
циаНадоРве7сма”132Н5аЧ1е)ННе ВСЛадСТВие •’1егкой Д°стУпности изо-
Cl2. CHCls, RN=C=S — C12 ИЛИ SOC12
(499) SC'2 RN=C-О ₽CI5' PhCI —»• RN=CCI2 ч— (500) 2u °c RN=C (501) (157)
(499a) 580 R'C (502)
указанные превращения изоцианидов и п
ге11Метил) ртути с диэтиловым эфиро " Р\ак««и Фенил (тригалп
!290] или с фенилазидом (502; R °Д|»°»овой S"
фосгеном инам, могут иметь сходные механизмы Ь Приво^иё к
8.1.3.2. Реакции имидоилгалогенидов
и родственных соединений
В отношении точной структуры имидоилгалогенипов
ствепиых соединении первоначально не cv.no Д и род’
мнения [264]. Повышенная реактюпноспособноё??Ва’''0 единог°
РИДОВ и фосгенимпнов по отношению к иуктеоЛш.. 11мвдонлад°-
там на первый взгляд противоречит коватеити реаген’
типа (503) и (505), которые обычно отиоситХн^ин₽С„ТрУКТурам
лобных реакциях; нм должны были бы соответствср-т^ В П°'
нитрплиевые структуры (504) „ (506). Однако простые имидоил®
хлориды и фосгенимины представляют собой жидкости иди низко
плавкие твердые вещества, легко растворимые в неполярных сре-
дах, что совершенно не согласуется с ионными структурами (504)
и (506). В настоящее время общепринято, что имидойлхлориды и
фосгенимины — соединения с ковалентными структурами (503) и
(505), содержащими сильно поляризованные связи С—С1. Вслед-
ствие легкой ионизации этих связей возможен переход к нитрн-
лиевым соединениям (504) и (506), вероятным реакционноспо-
собным промежуточным соединениям в большинстве реакций
имидоилхлорндов и фосгенимпнов с нуклеофилами. Четвертичные
производные имидоилхлорндов и фосгенимпнов, как правило,
Ск
V—\’R2 R1—CssN—R2 СГ
R1Z
(503) (504)
Ск
Cl—CsN—R СГ
С1/ ж
(505) (506)
(507) (508)
Г /R1 Ck ,/R1
ci—с—n' /с=\ С1
I \r2 с/ xR2
Cl
(509) (S10)
«81
твердыми веществами с типичными свойствами солей
/ВЛ.Хтет.ио высокие температуры плавления, нерастворимость
«°Х явных средах). Полагают, что им соответствуют ионные
типы 1508) II (510), а не альтернативные ковалентные
структуры (507) и (509) (264, 286].
(1) Реакции с электрофильными реагентами
Атом азота в имидоплхлорпдах способен присоединять про-
тон с образованием хлорпмпниевой соли [R'C(C1)=N (Н) R2 Х~].
Реакция с галогеиоводородамп в инертных растворителях дает
соответствующие галогениды хлориминия, которые вновь превра-
щаются в’пмндоплхлориды при нагревании [264]. При исполь-
зовании достаточно мощных алкилирующих агентов, например
тетрафторбората трнэтплоксонпя, пмндоплхлориды можно проал-
кплпровать по атому азота; при этом образуются устойчивые хлор-
пминневые соли R'C(CI) = N(Et)R2 X". Внутримолекулярное аци-
лирование галогенпмннневых солей (512), полученных в резуль-
тате присоединения галогеноводорода к ы-циапоацнлгалогенидам
(511), использовали как общин метод построения цикла в синте-
зе гетероциклов различных типов (например, галогенппридинов,
галогенизохпиолинов, галогенхиназолинов, галогенбензотиазпно-
нов и тиазепинонов) (уравнение 158) [292].
,N X
X
;Ч\
С—X
II
о
(511)
с—X
II
о
(512)
О
(513)
(2) Реакции с нуклеофильными реагентами
Гладкое замещение имидильных атомов галогена в галоген-
азометинах — имидоилгалогенидах, гидроксимоилгалогенидах,
гидразоноилгалогенидах (514; R2 == Aik, Аг, OR или NR2) (схе-
ма 159) и фосгениминах (520) (схема 160)—прн действии самых
разнообразных нуклеофилов является характерной особенностью
этих соединений.
Особенно сильными электрофильными свойствами обладают
хлориминиевые соли R1C(C1)=NR2 X- и фосгениминиевые соли
С12С—NR2 X- из-за наличия положительно заряженного атома
азота, имиднльный атом хлора в этих соединениях обладает по-
вышенной реакционной способностью по отношению к иуклео-
582
0
ri-Л—NHR2
H2O
(ho-)
(515)
|h2S
|(H5-)
s
r|—C—NHR2
X\
y.=NR!
R'Z
(514)
I
R’o-
RfNH R’N,
(159)
(516)
R’O
,C=NR2
R1Z
(517)
R’S.
\=NR2
R'/
(518)
фильному замещению. В реакциях с нуклеофильными реагентами
хлоразометпны ведут себя аналогично галогенангидридам кислот
и являются поэтому важными синтетическими интермедиатами.
Более того, возможность последовательного нуклеофильного за-
мещения атомов хлора в фосгенимпнах и фосгениминиевых солях
придает этим соединениям особую ценность.
(Н0‘) С1\ R2S- R2S\
R'NHCOCl ->------- ;c=nr> ----------->- ;c=nr' —
cv cv
(521) (520) (523)
| |r!°* )«2s‘
R2<\ R’S,
r‘nhco2h ,c=nr' ;c=nr>
CK R2SZ
(524) (522) (528)
(160)
R2O. R2°\
R'NHCONHR1 XC=NR' C=NRl
r20/ R2SZ
(525) (526) <527)
Простейшим проявлением реакционной cn°E°^HOcT"
^огенндов (хлоридов) [101, 264, 2656, 293, 294], r''«P°KC'™B
галогенидов (хлоридов) [996, 101, 265г] п гпдразоноилгалогеннд
боридов „ бромидов) [265д, 295] по о7"™* Н>с ^б-
Ф лам является легкость их гидролиза в вод1 , ?о^самидОв,
ованпем соответствующих амидосоедипенни ( Р
J 583
запи-
гидразидов, соответственно) (515; R2 =
111Дроксамовых кислот схему 159) Хлоримпнийхлорнды
ли- Лг vK »**и‘ '
= А ’ г,п2 п- также энергично реагируют с водой, давая
R’C(Cn=NR2 u ые ам11ды [286]. При гидролизе род-
соответствуют.lieР хлорформамид!шиевых солеи (529) в
ствениых соедшеи > • могут образовываться либо моче-
симостн от природ с Рарбод1ШМцды (531) с высокими выхо-
ss (%*о%) д с™ «').
rc=r<=h + /R3 R'\ II /R
R'X’=C=NRS -*--------- R’/ ,c=\ N-C-N
С1/ \r4 R2/ xR4
(531) (s29)
H\>~|
(530; X = 0, S)
(161)
(532)
OR5
N—C—OR3
OR5
(533)
Фосгенпмпны устойчивы при действии холодной воды, но при
нагревании быстро реагируют с ней и дают симметрично заме-
щенные мочевины (525) с промежуточным образованием неустой-
чивых карбамоилхлоридов (521) и карбаминовых кислот (524)
(см. схему 160). В то же время сольволиз фосгениминов в среде
сильных кислот (например, в хлорсульфоиовой кислоте) пред-
ставляет собой ценный метод синтеза изоцианатов (схема 162)
[265а, 284].
RN=C=S
P2Ss
/С1
rn=c;
\ci
C1SO3H
|rnh3 сг
RN=C=NR
/С1
RN=(/
X)SO2Cl
|-SO2C12
RN=C=O
(162)
Реакции имидоилхлоридов и родственных соединений с серо-
водородом или ионом -SH можно использовать как эффективный
пн3 Г51дТ°¥ _’гп3а, т110каРб°ксампдосоед1!ненин. Импдоплхло-
м п ’ , (полученные отдельно или синтезированные
nearnnvioT3с 1р'!п вторичных карбоксамидов с фосгеном) гладко
вторичных Т1>пкяпН°Д0Р0Д0М " ла,от ВЫС0К|,е выходы (60—90%)
вторичных тиокарбоксамндов (516) [264, 2656, 296] (см. схему
584
«г«о₽п»луч«'«и«“"е>?'‘™тад™р,“’ "
R'N=Cs Ck
+ —> ,C=NR>
R2COC1 R2COZ
О S
R2—С—C-MHR
(163)
ГИД-
(514;
мина
Обработка гпдрокснмоилхлоридов (514- R2 = он Y <-к
росульфнд-ионом [996, 297], а гидразоноилбрДТов
p2=NR3, X — Br) сероводородом в присутствии трнэтила
[295] приводит аналогично к тиогидроксамовым кислотам и тио
гидразидам соответственно (516; R2 = ОН или NR2). При реак-
ции с сероводородом хлориды хлориминня R'C(C1)=NR-;C1" ппе
вращаются в третичные тиокарбоксамиды, а хлориды х’чорфопм-
амидиния (529)—в тиомочевины (530; X = S) [286] Реакция али-
фатических и ароматических фосгенимпнов с сульфидами 1NH Ч
(NH4)2S, P2S5] [265а, 284] (схема 162) может быть с успехом £
пользована для крупномасштабного синтеза изотиоцианатов вме-
сто тнофосгеннровання первичных аминов (см. с. 630).
Имндоилхлориды [101, 264, 2656, 293, 296], гидрокспмоилхло-
риды [101, 265г, 297] и гидразоноилхлориды и -бромиды [265д,
295, 298] (514; X = С1 или Вг) гладко реагируют с алкоксидами^
феноксидами, тиолятамн и тиофеиоксидами с образованием
О-алкнл- и О-арплпмидатов, -гидроксиматов, -гидразонатов (517)
и их тиоаналогов (518), соответственно (R2 = Aik, Аг, ОН или
NRs). Свободные фенолы при реакции с имндон.тхлоридами
склонны образовывать продукты замещения в бензольном цикле
(см. с. 594), Продукты монозамещения, образующиеся, как пра-
вило, при реакции хлориминпевых солей со спиртами, фенолами,
тиолами и тпофенолами [264, 2656, 286, 296], представляют со-
бой ценные интермедиаты в синтезе сложных эфиров и их моно-
и дитиоаналогов (схема 164). Хлорпминпйхлорид (534) гладко
реагирует с этанолом и этантиолом, давая соответствующие про-
изводные нмпдата (535) и тиоимндата (536), которые при обра-
ботке водой пли сероводородом с высокими выходами ПР]’®РД'
Фаются в этплпропиопат (537), изомерные монотпоэфнры (538)
11 (540) и дптпоэфир (541), соответственно. Однако конечными
Продуктами реакции хлориминпевых солеи (534) с алкоксидами,
феноксндами, тиолятамн и тиофеиоксидами являются ацетали
(539) ц тпоацеталп (542) амидов, образующиеся путем прпсоедн-
"еиия второй молекулы реагента к промежуточному у
(535) или тпопмидату (536). Аналогичная реакция хл0РФ°Ре’ [ны
(549?Bb‘x солей с алкоксидами приводит к ацеталямi • е
опт 1В РезУ-чьтате дальнейшей реакции образуются р -
Рт°эфиров (533) (см. схему 161) [286] (см. также ра д.
585
(534)
НОН
11.111
EtSH
О
II
Et—C—XEt
(537) X = О
(540) X = S
(538) X = O
(541) X = S
(535) X = О
(536) X = S
S
J
Et-C-XEt
(539) X = 0
(542) X = S
(164)
Имидаты, образующиеся при взаимодействии галогениминие-
вых солей со спиртами, могут подвергаться дезалкилированию с
образованием алкилгалогенида. Так, именно дезалкилированием
промежуточного имидата в присутствии фторнд-пона можно объ-
яснпть [299] способность фторированных алкплампнов выступать
в роли эффективных фторирующих агентов по отношению к спир-
товым субстратам (особенно к 11а-гидроксп-19-норстероидам).
Предполагаемый механизм 1299] этого интересного превращения
показан на схеме 165.
F
Et2N-C—CHC1F
F
•RQH у
Г F
Et,N=C
2 \
CHCIF
ROH^
PTR7P о
+ 1 II
Et2N=Cx EtjM-С-CHC1F
CHC1F
(165)
Было показано [300], что иминпевые соли CICH=NMe2 Cl"
или Cl2POCH=NMe2 С1~, образующиеся при реакции диметил-
формамида с пентахлоридом фосфора, являются эффективными
реагентами для превращения спиртов в алкилхлориды. Различная
реакционная способность атомов хлора в фосгениминах (520) по
тношению к нуклеофильным агентам позволяет осуществить нх
сипоепДОтВ«ппатА°е за'',с',1ение "ОД действием алкоксидов, фенок-
обшим метл / " ™Феноксидов, что, таким образом, является
5261« М СИНТе3а хлоРФ°Рмим,!Датов (522), имииокарбона-
тов (526) и их тиоаналогов (523) и (528), а также «смешанных»
586
ггпоДУктОВ’ папРпмеР (527) [265а, 2841 (г-
"{’ра в фосгенимпниевых солях обтяпк' м' Схему 1601 а
^/способностью. Так, они могут быть ? ПОвь‘шеиной ре/™МЬ|
свободных спиртов, феио^вб^^7амеЩеиыР;^-
Пр11 реакции со спиртами образуются X” "/,гоФеиолоаР Д’
/орформимидаты (544), которые самопо™“ ™ь1е Ч(-‘Тверт1ч/«
ются под действием гало ген ид-иона ГтХт ЛЬ”г° деза-™и№
(545) П алкил галогенид (546) (схе.ма 166) чРТКарбамоиЛхлорИд
формимидаты (544; R = Ph), образующиеся 1пиВерТ1'ЧНЫе ХЛ0Р-
колами, относительно устойчивы; 11Х можно * Р реакции с фе-
Зу до карбаматов (547) 11лп превратить с п™ Ве₽Г"№ гидроли-
„ого замещения и последующего гидп0Л1,„ °ЩЬЮ н-''к-’еофи.и-
(549) [287]. Дролиза в ими11ОкарбОцаты
СГ
Cl2C=NMe2
СГ С1
С k + R = Alfc |
^C=NMe2 ---------► О=С—NMe2 + RC1
ROH
(543)
н2о
j R = Ph
(544)
PhOH;
R = Ph
(545) (546)
(166)
О
II
PhO—C—NMe 2
(547)
PhO.
XC=NMe2 —>
Pho/
(548)
PhO.
XC=NMe
PhO/
(549)
Имидоил-, гидроксимоил- и гидразоноилгалогениды (550;
X = Cl, Y = CR2, О, NR) легко реагируют с карбоксилат-аннона-
мн (ацетатом, бензоатом) и дают неустойчивые ацнлнмидаты
[264], ацилгидроксиматы [265г] и ацилгидразонаты [265д, 295],
которые гладко перегруппировываются (если позволяет их струк-
тура) в N-ациламиды (552), О-ацилгидроксаматы (553) и N-ацил-
шдразиды (554).
Х\ рьсо; PhCOO. y=cr2
,C=NYH -------
'C=NYH
RZ
(550)
(551)
I y = NR
» v/C°Ph
R-C-\
x^HRs
(552)
(167)
Y = O
О
R—C—NHOCOPh
R
О
p—C—NH—N.
R \coph
(554)
587
(553)
„О кпсюты реагируют с хлорпмиппевымп [2656, 286|
Карбоновые hI1“°соаяМ)1 [287) в мягких условиях и даЮг
или фосгеннминпев • м|) вь|ХОдам„ (уравнение 168). Эти ре.
ацнлгалогенпьс )[|е под действием хлор-нона в реак-
акции вклкиак .тОчном соединении, что позволяет также
ционноспосооно J1 СТ|1Лф мам„да как эффективного катализа-
объЯС"'' еа1 инн карбоновых кислот с фосгеном или тнопилхлорн-
приводящей к ацнлхлорпдаы с высоким выходом (>90%).
С1
рлр!- + _/ - RCO,H .
Me,NCHO ------► Me2N=C Cl Ь
2 H
/ci
ч Го-cor
1CZ ---у RCOC1 + Me2NCHO (168)
ЧН
Не является неожиданным и то, что реакционноспособные ими-
дильные атомы галогена в имидонлхлоридах [101, 264, 2656, 293,
301], гндрокснмоилхлоридах [101, 265г] и гидразононлхлоридах
и -бромидах [265д, 295] легко замещаются прп действии аммиака
п первичных или вторичных алкиламинов и арпламинов. Эти ре-
акции с практической точки зрения дают более чистые продукты,
чем в случае аналогичного нуклеофильного замещения в нмида-
тах [см. разд. 8.1.4.2(2)], и в том случае, когда соответствующие
хлоразометины доступны, представляют собой наилучший способ
синтеза амидинов (519; R2 = Alk или Аг), амидокспмов (519;
R2 = ОН) и амидразопов (519; R2 = NR2) (см. схему 159). Со-
ответствующие реакции [101, 264, 2656, 293] имидоилхлорндов с
гпдрокспламииом и гидразином и их производными протекают
гладко и также приводят соответственно к ампдокспмам и амн-
дразонам. Легко осуществимо ступенчатое замещение атомов хло-
ра в фосгениминах (520) при действии первичных или вторичных
алкиламинов или арпламинов, что представляет собой удобный
,оскг1особ) синтеза хлорформамидннов (555) и гуанидинов
(55b) [265а, 284] (схема 169). В свою очередь, пиролиз гуаниди-
нов (о ), производных первичных аминов, дает соответствую-
рЦ1улг,КаР 0ДИИМИДЫ (559) [265а, 284], которые получают также
гипплхпл™ выходом ПРИ прямой реакции (имннофосгенпроваипе)
иминами^!?08 пеРВИЧ1,^х алкиламинов и ариламинов с фосген-
(5581 полуир! .СХе''1- 62) J265a, 284]. Производные гуанидина
чивьг’ они легкоЫпасшрКЦ1,еЙ Фосгенпминов с аммиаком, неустой-
замещенные цианаМИдЬЦ560)С[265ща2”высоким выходом моно-
588
(H2N)2C=N1V
(558)
R’=R’=H | NH,
R'NII—Сзй4
(560)
R2R3NH
ChC^NR1 ;
(520)
R2R3N.
,C=N
С1/ \R.
(555)
R3 = H | R2NH2
R2R3NH
R2R3N
\=NR
r2rsn/
(556)
(169)
(R2NH)2C=NR‘ -----> R!N=(;_^R1
<557) (559)
фосгениминиевые соли (543) реагируют аналогично давая п
реакции с аммиаком дизамещенные цианамиды (561) [2871 а
первичными аминами — тетразамещеиные гуанидины (262) [287]
cl" NHAr
NH, + ArNH2 |
Me2N—CsN Cl2C=NMe2 >- Me2N—C=\Ar (170)
(561) (543) (562)
Благодаря своему бисэлектрофпльному характеру и легкости вза-
имодействия с самыми разнообразными нуклеофильными реаген-
тами, нуклеофильный центр которых сосредоточен на атоме кисло-
рода, азота или серы, фосгенимины широко использовались для
замыкания кольца при синтезе гетероциклов. Эти реакции невоз-
можно подробно рассмотреть в данном разделе. Однако ценность
фосгенимпнов для синтеза гетероциклов достаточно ясно видна
уже на примере их реакций с такими бифункциональными нукле-
офилами, как гидразиды, тиогидразиды и амидразоны; это важ-
ный подход к синтезу [302] циклических систем 1,3,4-тнадназо-
лпя, 1,3,4-оксадназолия и 1,2,4-триазолия (уравнение 171) ме-
зоионных структур, привлекающих в настоящее время внимание
исследователей.
R1
I
,С=Х
+
nh2
Cl
Cl V
н
X-O,S,NR4
Имидпльиые атомы
местить группами CN и
галогена в галогеназометина)< можно*за-
„п, - „ Na (уравнение 172). Цианнд рвдам11
м Илн спиртовом растворе реагирует с i Д •
р- = АПч или .V), давая соответствующие пмпдоплипаииды
R-’=Alk пли Аг) [264]. Лналопгчно реагируют „ гндразо.
„омхлорнды (563; R-’ = NR2) [265д]. При действии цианида ме-
non 180 °C замещается лишь одни атом хлора в фосгеипминах
(563 R' = Cl) с образованием цнаноформпмндоилхлорпдов (564
R|=Jci) 1284]. Импдоплхлорпды [264, 303], П1Дроксимо11лхлорц-
пы П01 265г 303] и гпдразоноилхлорпды и -бромиды (563'
JaIIc Аг. ОН НЛП NRJ [265д, 295, 303] гладко реагируют с
азпд-иоиом, давая соответствующие азидоазометины (имидонл-
азиды) (565). Эти соединения могут существовать в равновесии
с соответствующими таутомерами с тетразольпым кольцом (566)
(азндоазометнн-тетразольная таутомерия). Тетразолы и в самом
деде часто являются устойчивыми конечными продуктами реак-
ций галогеназометннов с азпд-иоиом (синтез Брауна — Рудоль-
фа [264]). В отличие от цианпд-пона азнд-ион замещает оба
атома хлора в фосгеипминах (563; R1 = С1), давая через проме-
жуточный неустойчивый бнс(азпдо) продукт (565; R1 = N3) со.
ответствующий азпдотетразол (566; R1 =N3) [265а, 284].
С,\
;c=NR2
R,Z
(563)
-cn NC.
'C=NR2
R,/Z
(564)
n;
(172)
;c=nr2
R'Z
(565)
N
N/ 4N
II I
R'C-----NR2
(566)
реакциями галогеиазометииов с
к образованию
! ПО
и Гёшу (см. ни-
иепий, легко замещают импдпльные атомы галогена в галоген-
Пожалуй, наиболее важными г—
нуклеофилами являются реакции, приводящие ..
углерод-углеродной связи. К, ним относятся ацилирование
Вильсмейеру — Хааку — Арнольду, Гаттермаиу г. Гёш/ -•••
же). Карбанионы, образующиеся из металлорганнческпх соеди-
нений, легко замещают импдпльные атомы галогена в галоген-
азометинах и дают соответствующие алкилированные или арили-
ровапные азометины. Синтез шиффовых оснований путем реакции
имидоилхлорндов с реактивами Гриньяра уже рассматривался
ранее (см. с. 495) [2—4, 2G4, 2656]. При аналогичной реакции
гидроксвмоилхлоридов с реактивами Гриньяра [265г] получают
кетоксимы. Реактивы Гриньяра реагируют с хлоримпнпевыми со-
¥*—гп^гоя?^ Н) 1^64, 2656] и фосгениминпевыми солями 567;
па» К "Г, давая продукт полного замещения; это представ-
т со ои эффективный метод синтеза недоступных иным путем
590
уретпчных карбинаминов
(уравнение 173).
„ , R2MgBr
RjCH—NR) <-х-=н
(568) и (569) с выходами
Ск
/C=NR)
40-80%
R’MgBr
"х^сГ
(567)
(569)
Галогеназометины обычно реагируют такж₽
„слученными из соединений с активной мети™>С карба11ионами,
дают функционально-замещенные азометины яип°1°И гРУпп°й. и
Ш1 соединениями для синтеза самых разнообоазН^Щ"еся ценны-
Азометпны, получаемые реакцией [264 26561 и гетероцикл°в.
дев с соединениями с активной метиленовой гпуПппйД°И'1Галогени-
выделить; они нашли применение в синтезе “ожпо легко
Так, например, N-феиилбензимндонлхлорпд (570- Р°-рь?а i74,‘
руст с ацетоуксусным эфиром в присутствии этокса натпия"^
дает в качестве основного продукта азометин (572- R = рм
торый при нагревании легко циклизуется в производное хиноти
на (573). Соответствующие азометиновые интермедиаты няп™
ч (572; К-ОН ,™ NHPH). »6р8,у„щ„есЛ“ ” £
роксимоилхлорпдов [265г] и гпдразононлхлоридов (и бромидов)
[265д, 304] с соединениями с активной метиленовой
против, циклизуются самопроизвольно в пзоксазолы
разолы (575), соответственно.
Rlc-N’RJ
(568)
(173)
группой, на-
(574) и пн-
С1\
;c=n
Ph/ \r
(570)
MeCOCH2co2Et MeCOCHCO2Et
(571) |
-----------—> Ph—C=NR
R=NHPh
------->.
Ph CO2Ef
NPh
(572)
(575)
фостешшиниевые соли, например '''“^„'^^оедннениямн. У к0'
сутствии оснований легко кондененру о1Ноп пл» ДвУмягВ
торых метиленовая группа активиров - ])ЛИ у —С -
алкоксикарбоиилынымп гРУппа^\пп11Стого водород ° пр
COOR), и даютР за счет отщепления ^Хре1.амвноН11трпда ,
латаемого интермедиата (57?) ' 591
(543) (уравнение
175), в при-
,„е. y nnn №CN ИЛИ CO2R). Соединения этого типа
-эфиры (<>7 , ' который можно легко заместить и прнсут-
содержат атом ЛЫ1ЫХ групп, и поэтому являются ценны-
степи других Ф)111'41 ,„87] Пр|1 введении в реакции этого типа
М11 »1'ГеР'1СД',‘‘Тн„Ых метиленовых компонентов (576; X = CN
моноактивнроваииь. & качестве катализатора необходимо нсполь-
"Л"т сТлы ое основание, например бутиллитий [287]. В отличие
3 ™ших реакций конденсации, в которые вступают ци.
от традиш ош ы- алкокс11карбон1|лметпленовые группы, ено-
тХщиея карбонилметиленовые соединения реагируют с фос-
г Гийевыми солями по-другому; итогом реакции является за-
мешение атома кислорода карбонильной группы на хлор. Эти
реакции н их применение в синтезе рассмотрены ниже.
cr Me2N X
основание I I
С12С=Х:Ме2 + X-CH2-Y ---------->- ChC-CHY
(543) (576) (577)
основание
Me2N\ /X
;с=с;
(578)
До сих пор не обсуждался механизм (или механизмы) разно-
образных реакций нуклеофильного замещения галогеназометинов.
Однако интерес, который проявляется к механизмам сольволнти-
ческнх реакций винильных систем [298], позволяет считать, что
настало время решать вопрос и о механизме аналогичных про-
цессов у галогеназометинов. Большая часть данных о механиз-
мах реакций галогеназометнновых систем получена при исследо-
вании нмидоилхлоридов [264, 294, 301] п гпдразоноилгалогени-
дов [295, 298, 301]. Возможны четыре механизма, выбор между
которыми зависит от условий реакции, природы нуклеофильного
агента и структуры субстрата (схема 176). В случае субстратов,
не содержащих при атоме азота заместителей, к которым пепо-
Е"в“"° пР1|соеД,11,еи водород, наиболее обычный путь образо-
нпг. „пл°Д^КТа ЭТ0 Зм1-и°11изацпя до резонансно стабилизован-
ное те ivionilt ’то'111'1 г0 азакарбениевого (нитрнлиевого) нона с
->(585)]. Этот'XwTlW-l ВпГПР0АУКТ РеаКЦ,'И И579)-И58°)->
норных заместитрпжг^ *294^ облегчается наличием электронодо-
рителей с относительно™^6"^0”6™6’ ,|Спользовачием раство-
применеиием низких конпе»тпК°И „А1|ЭЛектРической постоянной и
что он протекает'степрлгпоТР^Ц11И нУКЛС0Фнлов. Было показано,
соединяется к субстрату ециФ1|ЧН0 [298, 305]; нуклеофил при-
мой электронной папе азота СГОРОИЬ,> противоположной понеделен-
Щ азота азометииа
592
X
#*Y
(581)
r'C^NYR2 tfc=NYR2
-H+ x
(579)
R’c^NYR2
(580)
-X
R2/H Nu
r'C^NY
r‘c-nyr2
X~
R'C^NYR2
(176)
(584)
Nu
(582)
(583)
Nu
—У R1 C=N YR2 4
(585)
Nu
Альтернативный SN2-npouecc присоединения — отшепления
|(579)_>(582)-► (585)] обычно возможен лишь при высоких кон-
центрациях нуклеофилов в растворителях с низкой диэлектриче-
ской постоянной н преимущественно в случае галогеназометннов
содержащих электроноакцепторные группы [298, 301]. Ьыло по
казано [298], что 3№-замещенне протекает стереоспецифнчно с
сохранением конфигурации, что вполне согласуется с мех •
присоединения — отщепления; такой же стереохимическш р У
m наблюдается и для сольволнтических процессов в В! '
системах. Следует, однако, отметить, что РезУдь”т ор„чными
ского изучения [301] реакций нмидонлхлоридо нвзча за.
аминами можно было объяснить в рамках ДРУГОГ° поомежх'-
мещення по типу SN2, а именно сольволиза с уч 1,583чЦ,
точной тесной ионной пары (Зк21Р-механнзм[(сДс0Дтвуюг
*(585)]. Протеканию реакции по этому ме- у т0Гда как
электронодонорпые заместители в галогена . > ПрП.
электроноакцепторпые заместители направляю а30та в
соединения — отщепления. Если заместитель Аода, то един-
алогеиазометпне содержит хотя бы один ат и умерен-
твенно осуществимое направление Реа£Ц!ДптШеПленпе в 1,3-ди-
концентрациях нуклеофила)—это EU '’ “ч [306] - важ-
„...ь О'чтрнл-илпд, интрилимин или оксПД и образовании
ПО ",1И интсрмедпат в дальнейшем пРевР показано I307!’
РОАУкта [(579)_>(581)_>(584)->(585)] Деде по механиз-
му Ф°сгснимины гидролизуются в нейтраль а как в
Му ^1 (через шщщлпсвыи поп) (ср. схему 17W,
“ея1 отщеплешь; по этому же механизму происходят „ р^Ийе-
замещения в фосгенпмпнах под действием вторичных РевкЧнн
[307]. ,НОв
(3) Реакции с соединениями, содержащими обогащение
электронами кратные связи 1е
гЯпогепазометннов имеют ряд особенностей и От.
Эти реакции гал реаКЦИЙ с нуклеофилами (см. ВЫШе)>
лпчаются ОТ оолев отделЬном разделе. Наиболее известны
поэтому они расеь рафТСа с использованием галогеназоме-
реавдии фР’'^трОф)П‘Ьцых реагентов. Так, например, имидоил-
тпнов как ЭЛекч очень легк0 реагИруют с активирован-
хлоридып 5ЬЬ к мП) простыми эфирами фенолов, третич-
ными а?е1И ' в присутствии кислот Льюиса (например,
нымп арилам , ) пр ц аэомеТ11НЫ
хлорида ™ выделять Гидролиз азометинов с хорошим выхо-
K0T°^:Z к SX’(589) (схема 177) [264]. Более реакци-
онноспособпые фосгешшшш (586; R1 = С1) реагируют даже_с не-
активированными аренами, например с бензолом (587, R -Н),
и дают имидоилхлориды (590), гидролиз которых приводит к ами-
дам (591) [265а, 284[.
(586) (587)
А1С1з
R1 = CI,
R2=H
Cl
I
PhC=NPh —> PhCONHPh
(590) (591)
(177)
Ri = Ph, А|Г,
R2=NMe( А,С'3
~ Крафтса идеальными Pe;
проме₽^,1'ЛЬНостп- Это свой'еВЬ’е C0J1" вслелствие их высокой
скими !УТ0'!НЫх соединений п™ обУСЛОвило их применение как
синтез ТМерами являются РРЯДе Важ1!ЫХ процессов. Классике-
чаю в Гг°Н0В по Гёшу [86 по Гаттермаиу и
стого во± Гене'>и150ван1ю хп ’ 28’’ Ж 30Э1' Э™ Реак“ии вКЛ,°‘
сацню г Р?Ла или Ч11трила Д рпм"1!|1йхлорида in sitlt 113 ц||а”"’
фенола и>ч 71ИВ||рОва1Шым -> л°ристого водорода и его конде*1'
или N.N-дпзз “м ареном (фенолом/ простым эфиром
Б94' Щеш,Ь1м арпламином) в присутствии хло-
,и цинка- Образующуюся С-арилнмнниевую
Спатам путем гидролиза в соответствующий Ыделяют и
"^ Область применения реакций этого тим v„Дегид или
кеТ0 ’ пясширпть за счет применения поостпй о. Удал°сь порази-
тель’^ных позиций!) стратегии; N,N’-дизамеше±”ИВаЯ ЭТо с с°'
8Родили в реакцию с хлорирующим агентомЩ(напримепРб°КСаМИД
S Фосфора- фосгеном или, что более удЯ
Й) in situ и получали раствор соответствующей' хлориХшХй
С или реагент Вильсмеиера _ Хаака - Арнольда Ж
2656, ЗЮ, 311]. Возможности использования реагентов ВХА
применять которые в полной мере начали примерно с I960 г’
иллюстрируются схемой 178, на которой приведены некоторые из
реакции наиболее часто используемого реагента, полученного из
днметплформамнда.
СНХ
CH=NMe2
I СГ
CH2NMe2
NaBH,
(594)
(593)
(595)
PhH
С1\ *
V=NMe2
н/
R‘ ,R3
Чс=с/
R2/ \l
R'\ /R3 Г1- н2о
С=СД * ---’
RL,/ \=NMe2
W
^СНО
(178)
(592)
(596)
(597)
NaBH,
R2 = NR2
H2O
R2=C!. H2O
R3=H
R3
C—CHCHO
R'\ /R3
/C=C\
R2/ ^CH2NMe2
(598)
Cl\ /Н
v—cz
R1/ \сно
(599)
(600)
Реагенты ВХА достаточно электрофильны п всТ^пав) в отсут-
Ц|||| Фриделя — 1<ра фтса (нмипоалквлирование ар чес/ве
твпе ооычных катализаторов. Образуюш ,,пЖНо выделить н
«уточных соединений имнннйхлориды ( 1 „ в ароматиче-
Ревратпть в условиях контролируемого сол • [[л|| восста-
кие альдегиды или тноальдсгпды (594; л — твеТцчные амины
Вить цри действии комплексных гидрид
„„повапне в сочетании с восстановлением леЙ
/595). Иш1НоаЛпК^педставляет собой удобный способ аминоалК1,.
ствием гплрнД0В преде
лнровапм аре"00' тов вХА возможно нмнноалкилнрование Не
С помощью pear ’ eHIlfl) но и алкенов [310, 311]. Мо.
только аРот'а™ мешенные алкены дают еннминиевые ннтерМед11.
ио-, Тр ода которых приводит с хорошими выходам,, к
аты (596), ГИДР°Л альдегидам (597), а восстановление комплек-
а р-ненасышенным оалк11Л„рованным алкенам (598); этот
сным гидридом 6 дополняет амнноалкнлнроваиие по мап.
процесс, таким о Р ’ые эфиры енолов и ацетали также реаг„.
ниху (см- с- ova в случае енаминов образуются проме-
руют с Peare‘,TaJ я (59б; R? = NR2), при гидролизе которых
ТХкя кетоальдегиды (599). В реакциях нмнноалкплнро.
образуются р ке метИлкетонов н ацетамидов (т. е. метил-
ВаН«пНнЛных соединений) участвуют нх енольные формы; эти
кар ЛЬсо оовожДдаются замещением карбонильной группы на
К Образующиеся хлорированные еннминиевые соединения
S' R1 =С1 R3 = Н) легко гидролизуются в З-хлоракролепиы
Si которые являясь реакционноспособными винилогами хлор-
ангидридов кислот, широко используются как реагенты, в осо-
бенностн при синтезе стероидов п гетероциклов [310].
Соединения с иным типом кратных связен (например, соеди-
нения, содержащие цианогруппу [312]) также подвергаются ими-
ноалкилированню с одновременным хлорированием при действия
реагентов ВХА (уравнение 179).
Ck J./R1 R3C.N с,\ СГ ,
;c=n; сг ---------► yc=Nx _ л/к
(179)
(601)
(602)
обпязпы моТ й товт Реакции фосгеннмппнсвых солей (главным
сти в сн’нтети™ ^87’ 3 открыло новые, неожиданные обла-
(прнсоелппрнчрСК011 °Рга"пчсскоН химии. Так, эти соли реагируют
нХ сХпят» ТР"рОва,Шем) с разнообразными иенасышен-
ЦнаТам£Р кет”, (ВКЛ10'1аЯ с”олы' е”амииь, „намины, нптрилы,
п^ХВ12Г'"М1ЙН) I287’ 3(3—315]; эти реакции
нов (603) с высок общ1ш способ синтеза полнхлорметиицпаш1'
1 высоким,, выходам,, (уравнение 180).
Cl + R’
X V
Cl
chcF^C
R1 R1
V V R
I I I
CI CI CI
(603) (94%)
(180)
СГ
I кпагодаря наличию «пуш-пульного» винями
£ эти цианины проявляют повышенную электр"^В°Г0 °Стова
1 ность. с химией этого нового класса соединяй* ЬНую спо’
' '.'.^ ознакомиться самостоятельно [287,313,314) читатель мо-
(4) Реакции отщепления
Отщепление в ряду галогеназометннов может происходит
„азных условиях; его протекание в значительной степени
ПТ природы входящих в их состав заместителей. Реакции
и* ‘ ‘ плгли оэллготпилп ____ “ц,и|
Ь В
зависит
«„ия'‘от галогеназометннов (имидонлхлорадов,' гидроксимои'!’
хлорилов и гидразопоилхлорпдов и -бромидов), катализируемые
основаниями и приводящие к 1,3-диполям (к оксидам нитрилов
иптрилиминам и нптрил-илидам), подробно рассмотрены в сле-
дующем разделе.
' Импдоплхлориды, содержащие вторичный С-алкнльный заме-
ститель (604; R1 = MesCH, R2=Ar), в присутствии триэтилами-
на превращаются в кетенпмииы (605) (схема 181) [264, 2656).
С^-Дпарнлпмидоилхлориды (604; R1 = R2 = ph) при обработке
кислотами Льюиса, например пентахлоридом сурьмы, дают соли
нптрилия с комплексным анионом (606) [264, 2656]. С-Арнл-N-
алкилимидоплхлорнды (604; R1 = Аг, R2 = Aik) при пиролизе
\
отщепляют алкплгалогенид и превращаются в соответствующие
нитрилы (607) [264, 2656]. Аналогичная пиролитическая фраг-
ментация пмидоилгалогенида, получаемого in situ из ацнламина
и галогенирующего агента, например пентахлорпда или пентабро-
мпда фосфора, представляет собой важнейшую стадию в дегра-
дации по Брауну [264], используемой при исследовании структу-
ры природных соединений (особенно алкалоидов). Реакция фос-
гениминов (604; R1 = С1) с трпфенплфосфином при 60—100 С или
с подпетым водородом приводит к соответствующему изоцианиду
(609) [265а, 284].
Ме^
V=C=N4
Me' vV
Et3N
R1=Mc2CH,
R2=Ar
(605)
CI\
r.=\R!
Rl/
(604)
PhsP
RI = CI
R!N=C:
(609)
SbCls
|r1=R2 = P11
PhfeNPh
sbci;
(606)
нагревание
Ri=Ar
R2=Alk
ArCN + AlkCl
(607) (60S)
(181)
597
8.,.4. ИМИДАТЫ П РОДСТВЕННЫЕ СОЕДИНЕНИЯ
Пптты (эфиры и.мндовой кислоты), нмнноапалогн сложные
Г Я также родственные им окспмппопронзводные (гндрок*
эфиров, а так а'зоиопропзводные (гидразонаты) - это соедцЛ
симаты) ‘ наличием алкокси- или арилоксиазоме
ния, характернзуюзи и-н = = Д(к |[Лп Дг, х Д(к зоне.
ГХЖ’-назометииовый фрагмент входит В ЦИКа °TQ
имидат может’ иметь полностью или частично эндоциклнческую
с ожтуру, К таким имндатам относятся: Д’-оксазолины (611;
J,T= I) Р5.6-дпп1дро-1,3-оксазнны (611; п = 2)и О-замещенные
тяктамы (612). Химия имидатов в целом [316, 317], а также
Д?оксазолииов [318], 5,6-днгпдро-1,3-оксазииов [182, 319] „ 0.
замещенных лактамов [320] была достаточно подробно рассмот-
рена сравнительно недавно. В данном разделе речь пойдет лишь
о наиболее важных особенностях химии имидатов и их тиоанало-
гов— тиоимидатов (610; SR2 вместо OR-).
r2o
,C=NX
(610)
O^N
R '
(611)
(612)
8.1.4.1. Методы получения имидатов, тиоимидатов
и родственных соединений
(1) Реакции присоединения к соединениям
с кратной связью
Классический общий способ синтеза имидатов (615) —это
синтез Линкера [316, 317]. Он состоит в присоединении первич-
ного или вторичного спирта к алифатическому или ароматическо-
му нитрилу (613) в инертном растворителе (например, в бензоле
или эфире) при низкой температуре (0—5°C) в присутствии кис-
лого катализатора, обычно хлористого водорода (схема 182).
ия, ,Д-'ет ст₽ого придерживаться этих условий, поскольку перво-
КЯ Н „ ра3у,ощаяся соль имидата (614) термически нестой-
имндата чтстл Р°Л1,3уеТСЯ даже Е присутствии следов влаги. Соль
но однако лягилП°ЛЬЗ"ЮТ в Дальней|инх превращениях; ее мож-
обработки ’суспензинРеВрЛТИТЬ В своб°Д11Ь1й имидат (615) путем
ком при низкой темПератупе°Ё<'ПоС11еРТИ0М РаствоР"теле аММ
Синтез Пиннепа мм 7 'J
лучения О-арилимип^Т бь|ТЬ с Успсх°м использован и для по-
если вводить в ' ' ‘Д ’ ? также S-алкил- и S-арилтиопмидатов,
венно [316 317] НрИЮ Фе,|0ЛЬ|, тиолы и тиофеполы, соответст
торы; с третичными ^аЛ°ВаЖ"у|° Р°ль играют стерическпе фак-
59g шртами и пространственно затрудне|,,1Ы>
СГ
R’O.
о
R2OH / \ О
R'z
(614)
(613)
R1
N'
(617)
основание
он он
Ме2ССН2СМе2
Me Me
(182)
R2O
XC=NH
R'/
(615)
нитрилами реакция
некоторые нитрилы, содержащие функциональные
пример циангидрины, г
образующихся импдатов ее не удается пронести’ГнитриламиТпот
не
Ме\| j
Ме (Э ХР'
(616)
идет. В этой реакции можно использовать
функциональные группы на-
однако из-за самопроизвольного распада
R PP UP опоа'гол пола,__ _
нпженной электронной плотностью (например, с трнхюрацето
нитрилом) и с а-аминонитрнламн. Использование в синтезе Пин-
кера 1,3-диолов или у,6-ненасыщенных спиртов приводит к 56
д||гпдро-1,3-оксазинам (616) [182, 319]. Реакция нитрилов с
эпоксидами в присутствии концентрированной серной кислоты прг-
низких температурах — это общин способ синтеза Д2-оксазолинок
(617) [318].
Реакция Пнннера всегда приводит к N-неза.мещенным имида-
там или тноимидатам.
OEt
EtgO+ ~BF4 + EtOH 1
RCsN ------------>- RC^NEt 'BF4 ----------> RC=XEt (183)
(618) (619) (620)
Однако при реакции нитрнлневых солей (619) (легко получае-
мых алкилированием нитрилов фторборатамн триалкнлоксония)
гидрокси- или меркаптосоединенпямп можно получить N-заме-
ч«1ные пмндаты, например (620) (уравнение 183) [316, 317].
мп- Р|1соеД||пеи11е спиртов к нитрилам, приводящее к имидатам,
j*110 осУи1ествпть и в присутствии основании, например при
25орЛь.ЗОваи11ч алкоксида натрия в соответствующем спирте прн
чесю, • 3,61- В случае простых алифатических н ароматн-
ЖениА- HiitP,1ji°b выходы невысоки, но в случае нитрилов с понн-
» - нейтронной плотностью (например, хлорацетонитрила,
обг>яЛ0оепз0нптРпла)> то есть именно тех нитрилов, которые не
вЬ1со2,1ОТ,л,1М1|дат0в в условиях синтеза Пнннера, выходы очень
рают ак 11 Для реакции Пнннера, стерическпе фактор
спппто /К11У10 роль в катализируемом основаниями прнсоедт
₽10в х нитрилам.
599
Ичипаты и тпоимидаты являются копейными продуктами „
соединения спиртов н тиолов к кетсинмппам (см. с. 640, 641) Р"'
изоциаипдам (см. с. 704). 11 к
(2) Реакции замещения производных азометинов
Реакции нуклеофильного замещения имидоилхлорндов
ствеиных соединений при действии алкоксидов, фенокспдов ?°Д'
лятов и тпофеиоксидов широко используются для получения'10'
кил- и арилимидатов и -тиопмпдатов. Применение этих пеяк ЭЛ'
подробно рассмотрено ранее (см. с. 585, 586). 1 акций
При нагревании пмидата со спиртом в присутствии катал
ческого количества соответствующего алкоксида происходит "™’
этерификация, если температура кипения вводимого в те П6ре’
спирта выше, чем образующегося [316]. Хотя для осуществл^'
этой реакции требуется наличие уже готового имидата Л6Я
рпфикация может найти применение, например, при
трет-бупьшшдатов, которые непосредственно получить це'
СЯ, ИЛИ при ............... - -
тов [316].
в реакцию
--.1пя
переэте-
при синтезе
синтезе имидатов путем переэтерификации' тио/мид”'
(3) Алкилирование амидов, тиоамидов
и родственных соединений *
алкилирующие агенты, такие, как тетрафторборат
атомам кислорода или
(184)
Мощные
триэтплоксоиия и метиловый эфир фторсульЛонпнпД‘^Г4'™1’0
...........................................................
торых по Пиннеру невозможен [316 317] С°еД"НеН,,Я' с,,итез ко'
f ? ♦ XEt
R-C-N у -1:130 ~BFt I
л Y ---------------- R—C=N—Y
Этот мето, м (62,) (622)
тов, гндразонатов и ихЬтппяиоп3°ВаН И для С|||1Теза гидроксима
роксамовых кислот, ГИДО 7п'‘аоЛ0ГОВ И3 гидроксамовых и тиогид-
(Уравнение 184; R = Aik ,,п„ « „ Т|,Ог1|Лразидов, соответственно
) 3'7Ь а также для ,и.Гт’ Х =° ил" S' Y = 0Н
। ° 1- Тиоамиды и тиогнчпяя G3a 1Шкл||,|ескпх имидатов (624)
® лПр“ действии алкилР Ы ЛСГК0 алкилнру ются по атому
1297,316,3)7]. “галогенидов в присутствии оснований
/ Л Е!д(Г ~ве,
нА
-------. (623)
См. также гл. g.g
(624)
(185)
600
О-Ллкилпровапие карбоксамидов действием алкилга тоген.,™
,.1Пжпо лишь в том случае, если их ппеппяп.^•1га;юге,,||Д°з
в0^ребряну.о соль [316, 321]. Однако Ха Перевести
’«лоне» 14 перегруппировке Чепмена, катализипуемпй1*™ имидат
(« с- 494) НС„„р, „ > ==в
серебра служит удобным методом синтеза имидатов, являющихся
рР0извоДНЬ.ми пространственно затрудненных нитрилов котовые
нельзя получить по методу Пиннера. Для непосредствённог^а
„ S-этнлирования алкиламадов „ -тиоамидов можно использо-
вать э™д?Л0[Й)91рМИрТ (CICOjEt) [316] и этилтиохлорформиат
(ClCOSEt) [322]. В то время как провести межмолекулярное
О-алкнлнроваппе амидов действием алкилгалогенидов довольно
трудно, внутримолекулярное О-алкнлирование легко осуществи-
мо; его использовали в самых разнообразных вариантах для син-
теза циклических имидатов — А2-оксазолннов [318, 323] и'об-ди-
г»дро-1,3-оксазинов (626; «= 1 или 2) [182, 319, 323] с выхода-
ми порядка 30—70 %•
(CH2)„/R2
HNZ
R'C^0 X
н2о
О R2 R»
II
R’C—О—С—(СН2)„—NH2 (186)
(625) (626)
(627)
X = Hal, OSOsC6II4CH3-n
(4) Реакция аминов с ортоэфирами
Ароматические первичные амины (это не относится к алифа-
тическим первичным аминам) реагируют с ортоэфирами (напри-
мер, с этплортоформпатом, этилортоацетатом) в присутствии
кислого катализатора (например, соляной кислоты, серной кис-
лоты, уксусной кислоты пли n-толуолсульфокислоты) и образуют
имидаты с хорошими выходами [316, 317]. В случае первичных
алифатических аминов продуктами реакции оказываются соответ-
ствующие амидины; они же образуются и из первичных аромати-
ческих аминов, если их реакцию с ортоэфпрами проводить в нейт-
ральных условиях. При реакции сульфамидов и гидразинов с ор-
тоэфирами образуются соответствующие гидразонаты [316, 317].
8.1.4.2. Реакции имидатов, тиоимидатов
и родственных соединений
Многие из реакции, в которые вступают имидаты и тиоимида-
ты, аналогичны реакциям, характерным для простых азометинов
(См- Разд. 8.1.2.2), и о них стоит лишь упомянуть. В других слу-
чаях наличие кислорода пли серы придает их поведению осооые
еРты; это заслуживает более полного обсуждения.
601
(/) с электрофильными реагентами
,„тпя с электрофильными реагентами напомпна,
РеакЩ....азометпнов и почти всегда протекают так,
ют реакции прост . атод, азота [3)6> 317]. Протопирование
что атака наир- ва111110 довольно устойчивых солей, ко-
по азоту nPBB0J gofi „ервнчпые продукты прп синтезе нми-
торые пР№птав'Х /См Выше). Алкилирование N-замещеиных
ДаТ0В1тов (628) дает четвертичные соли (629) [318]. Лцилирова-
"пеД, еул&онилироваиие [316, 317] N-иезамещеиных импдатов
также протекает обычным образом и приводит к N-ацил- и N-
сульфопилпмндатам.
/\ + Mel Xs
С1СН2—СНг
I
/NC1
Ph
(628)
>- [
°у X'
Ph
(630)
(галогены, гипохлорит натрия,
нмидаты
О
(631)
(187)
Ph
(629)
Га.тогенирующне агенты .
бутилгппобромит) превращают N-пезамещенпые
галогенпроизводные [316, 317], тогда как N-замещенные имндаты
(628) дают N-галогенпмнниевые соли (630), такие соли, получен-
ные из А2 *-оксазолинов, склонны к раскрытию цикла [(631)]
[318, 323].
трет-
в N-
(2) Реакции с нуклеофильными реагентами
Реакции импдатов и тнонмидатов с нуклеофильными реаген-
тами [316, 317] хотя и напоминают реакции простых азометпнов
[см. разд. 8.1.2.2(2)], однако несколько отличаются от них из-за
наличия заместителя в виде гетероатома.
Ациклические нмидаты (632; X = О) или их соли легко гид-
ролизуются при низких значениях pH до соответствующего слож-
ного эфира (633; Х = О) и амина (634) или соли амина (схема
188), тогда как при высоких значениях pH проггуктамп гидролиза
оказываются амид (636; X = О) и спирт (637) [316, 317]. Обыч-
ными продуктами гидролиза ациклических тнонмидатов (632;
X = s_) являются тиоэфир (633; X = S) и амин (634); однако в
кислой среде реакция может пойти в основном в сторону обра-
зования тиоамида (636; X = S) и спирта (637). Механизм гидро-
АгпЦо,",М"ДаТ°В ” т,1О|1Ы,|Датов был тщательно изучен [316, 324],
пик Д'п В связп с тем- что такие процессы напоминают иското-
эдрических'ни^пк превРащеипя' протекающие с участием тетра-
скихЧимКидатовТе(626^11?Л°В <С“' С’ 504)- Прп ‘«ДРОлизе циклпче-
азинов [182 3191 и их '°’(са?°Л|1Н0В I318] и 5,6-дигидро-1,3-оке-
обоазовзнпр' рппч-И их ,солеи) происходит раскрытие цикла и
ложных эфиров амипосииртов (627) (см. уравпе-
602
)Я6). Поскольку 5,6-дигндро-1,3-оксазниы „
'“'сположепием заместителей и известной стереохиХТТ*11™
go получить (например, по реакции ДнХ^Ха)*™
’ дролпз представляет собой регио- и стереоспецифический спо
об получения 3-амшюпропаиолов, важных в синтетическом от"
ошепии соединении Кроме того, легкость гидролитического рас-
Йления никла в ДМксазолипах и 5,6-дигидро-1,3-оксазинахРпо-
зволяет |,сп?оо=° ЛТс’1 п гетероциклы в различных синтезах
(см- ниже) [325, 326]. Взаимодействие имидатов и тиоимидатов
632; Х = О или S) с сероводородом приводит соответственно к
£оэфпрам и дитпоэфпрам (635) [316, 317]. Соли имидатов реа-
гируют в среде инертного растворителя (эфир, петролейный эфир)
со спиртами, взятыми в большом избытке, и дают простые или
сме1паниые ортоэфиры (638). Свободные же имидаты, напротив,
реагируют со спиртами иначе (см. выше). Превращение имидатов
в ортоэфпры с последующим восстановлением алюмогидридом
Я11^„СТ П ГПППЛПНЗЛМ ------- ' ,,ч
ЛПТПЯ II гидролизом Представляет собой удобный
датов к альдегидам (640) [316,317]. * ьн переход
от
нми-
KMgA /RS
RCH=NPh-----------► R2CH—NHPh R'C—NZ
\?s
(642) (643) (641)
| RMgX R4NHRS j
X=O, R1=H, R3=Ph
1 (188)
О s
II Il
R'C—XR2 R'C—XR2
(633) H.o R2X4 H2s (635)
+ ч------------ Y=NR3 -----------> +
R3NH2 R'/ r3nh2
(634) (632) (634)
II2O | R<OH. MCI
| x-o (
X
R'C—NHR3 + R2OH R'C(OR’)s
(636) (637) (638)
|liaih(
П2О
R'CHO *-------- R'CH(OR')s
(640) (639)
603
. нпр чпметение фрагмента, содержащего кислород
Ну-е* , дттах II тпонмидатах под действием аминов [316,
Ж 1н из важнейших общих методов синтеза амино-
3!7, 32/], 4 реакции аналогичны реакциям замещения
"на°в галогсназометннах при действии аминов (см. с. 588)
nfi нем приводят к тем же продуктам, хотя в некоторых слу-
е такой эффективностью. Так, реакция N-незамещенных
имидатов или тиоимидатов (632; Х=О
с di — R2 = Aik пли Аг, R3 = Н, Aik пли Аг) (или их со-
прМ с аммиаком, первичными или вторичными аминами, солями
(HI;R'-Alk OU Ar. Aik или Ar)
[293 316 317, 328]. Аналогично реагируют простые эфиры и тио-
эфнры лактимов [циклические имидаты и твоимидаты типа
(624)] [320]. Единственное осложнение, с которым приходится
сталкиваться прн реакции N-незамещениых имидатов и твоими-
датов с первичными аминами,— иминиын обмен между первона-
чально образовавшимся монозамещенным амидином и избытком
амина, приводящий к возникновению К’.К’-дпзамещ.еиного ами-
дина. Соли имидатов п тиоимидатов, как N-замещениых, так и
незамещенных (632; X = О или S, R1 — R2 — Aik или Аг, R3 = Н,
Aik или Аг), реагируют с гидразином и его моно- и днзамещен-
нымп производными (как симметричными, так и несимметричны-
ми). Эта реакция представляет собой общий способ синтеза
амндразонов (641; R1 = Aik или Аг, R4 = NRs, R3 = R5 = Н,
Aik или Аг) [316, 327, 329]. Реакция протекает очень гладко и
часто с высокими выходами. Исключение представляет взаимо-
действие N-незамещеиных имидатов с гидразином; прн этом
возможно образование побочного продукта, гидразндниа (1,4-ди-
гидро-1,2,4,5-тетразнн). Эту трудность удалось преодолеть, обраба-
тывая легкодоступные гидроподнды тиоимидатов безводным гид-
разином в инертной атмосфере; выходы гидроподидов N-незаме-
щенных амндразонов составляют примерно 80% [327]. Имидаты
и тиоимндаты реагируют, как правило, и с гидрокснламппом п
дают соответствующие амндоксимы (641; R' = Aik пли Аг,
R ~он> R3 = H, Aik или Аг, R5 = Н) [996, 330, 331].
Приведенные выше данные ясно показывают, насколько раз-
нообразны реакции имидатов и тиоимидатов с кислород-, серу-
и азотсодержащими нуклеофилами. Применение субстратов, со-
I'r-?5 должным образом расположенных нуклеофильных
пост1Р ппя ДЫ к"СЛ0Р°да> сеРы или азота), открывает возмож-
систем Этот м'1раТо'Я общего метода синтеза гетероциклических
жат обише мртппД "спользУется очень широко. Примерами слу-
XaaoZroB lvn^ С""Те^ А^оксазолниов, Д2-тназолииов и А1-
нмидазолинов (уравнение 189) [316, 317]
атомов Д подобно°"гплД<1Ти’ "С содсР)Каш.не «-С- и a-N-водородных
тветствующпм шиффовым основаниям (сМ-
604
Rs R'
R|Z '
K N
(189)
(645)
(646)
IP
R’—C—XU .NH
I + R5-cf
R2—C—NH» \qE|
Rl
(644)
v = O, S, NR
c. 508, 509), легко реагируют с реактивами Гриньяра и литийоога
„„ческп.мп соединениями; первоначально происходит 1 2-пРисХи
цепче по двойной связи углерод-азот. Так, при реакции [316 3171
й-арилпмндатов (632) с избытком реактива Гриньяра образуется
вторичный амин (см. схему 188). Сначала происходит ^-присо-
единение, затем в результате отщепления алкоксида возникает
11Ыцн (642), присоединение к которому второй молекулы реагента
и дает вторичный амин (643). Если использовать стехиометриче-
ское количество реагента, то имин можно выделить; гидролиз
имина позволяет получать альдегиды [316]. 1,2-Присоединение
лптпйоргаинческих соединений или арилмагнийгалогенидов к со-
лям ациклических тпоимпдатов [332], легкодоступным солям
Д2-оксазолинпя [325, 326] или 5,6-дигидро-1,3-оксазнния [333] и
последующий гидролиз также позволяют получать альдегиды и
кетоны с выходами 50—90%. Реакции конденсации [316] нмида-
тов и тпоимпдатов с соединениями с активной метиленовой груп-
пой, приводящие к енаминам, также можно объяснить, допустив,
что вначале происходит 1 ^-присоединение по двойной связи угле-
род—азот.
а-С-Алкилированные имидаты и тиоимидаты, подобно прос-
тым азометинам (см. с. 509—513), действием литийорганических
соединений или диалкнламидов лития можно перевести в карб-
анионы, являющиеся ценными синтетическими интермедиатами.
Особенно широкое применение нашли карбанионы, полученные
из 2-метнл-Д2-оксазолпиов (647) [325, 326] . Было показано, что
реакцией таких карбанионов с первичными и вторичными алкнл-
галогепидами можно получать моно- и дналкплпронзводные (вы-
ходы 93—99%) н что сольволиз алкилированных Д*-оксазолинов
(649) в контролируемых условиях дает гомологичную кислоту или
ее эфир (651; R-* =Н или Et) с высокими выходами (77—98%)
(схема 190) [325, 326] . Кроме того, использование карбонильного
соединения в качестве электрофильного реагента приводит к
’(^гидроксналкил)-Д2-оксазолинам (650), которые можно пре-
Ратитьлибо в а,6- и 6,у-ненасыщенные кислоты и эфпры (6э2)
‘Ли (653), либо в сложные эфиры р-гидроксикислот (654). Высо-
,В!»оды в последней реакции (80—87%) позволяют чепол
Зопать ее вместо реакции Реформатского [325, 326]. При пспо-
хирального Д’-оксазолпна [325, 334] как субстрата уда-
осуществить соответствующие асимметрические
605
етПМ достаточно высок (51—86%). 1>4.п
птдческпй выход пр” ^ОесКих соединений к хиральным 2-(алке11'.
°" ' „н»е лптппорганп <е оШ11н сольволиз приводят
Д’-оксазолпнам ис очейЬ высоким оптическим ВЫхо.
R'CH2CO2R2
(651)
I Н‘°Н'
Me
Me
КУ"-
(647)
(649)
(190)
ОН
Me, г О I ТЧ.-ная 112SO.,
V \_Ch2-C-CH2R2
R'
I
R2C112—C—CH2CO2Et
OH
(650) (654))
IH3O*
R\ ,R' R!CHk /Н
>=< + /c=c(
W VHjCOjR3 R,z xCO2R3
(652) (653)
(3) Реакции циклоприсоединения
13-ИдиполяпнпглН0НМ11ДатЫ ведут себя в Реакииях [2 +2]- и
1’155] Так ?о-д 9)и11КЛ01'Р"С0едпнеппя как простые азометины
алкокснф-:1актам^’^лопР1'^еД”исние импдатов к кетенам дает
стах азометпнов ,, , X (2 + 2]-циклоприсоединение про-
еДииенпе импдатов к О,”|сапо 1|551 |3 + 2]-циклопрнсо-
стых азометпнов Пп тР||л"М1Шам, которое, как и в случае про-
c. 526), ’ Р1,В0Дпт к производным 1,2,4 -трназола (см.
р (4) Окисление
MC'ibwe 1316], чем'Яг>ея['Датов " тнонмидатов известны гораздо
8 !'2'2 (‘1)). Окисление ПР°С™Х азометпнов [см. pa3*1-
60s мидатов пероксикпслотамп протекает
аналогично окислению простых шиффовых оснований (см с 534
535) и приводит к соответствующим оксазиридинам "з 16 336]'. ’
(5) Восстановление
Имидаты и тпопмндаты легко восстанавливаются до соответ
ствующи.х аминов при взаимодействии с системой металл—донов
протона (например, амальгама натрия или цинка в растворе кис-
лоты, натрии в спирте пли жидком аммиаке) или при катартиче-
ском гидрировании под давлением [316, 317]. Удобнее всего одна-
ко, провести восстановление имидатов до аминов действием алю-
могидрида лития или борогидрида натрия [316, 317) Простой
метод N-мопоалкплпровапня первичных ароматических аминов со-
стоит, таким образом, в кипячении амина с ортоэфиром с после-
дующим in situ восстановлением образующегося прн этом нмидата
борогпдрндом натрия [337, 338]. Электрохимическое восстановле-
ние имидатов в растворе кислоты дает амины с хорошими выхо-
дами.
8.1.5. АМИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ
Амидины (655; X = Aik пли Аг) [293, 328], амндоксимы
(655; Х = ОН) [996, 330, 331] и ампдразоны (655; X = NR2)
[329, 339] можно рассматривать как имино-, оксимиио- и гпдразо-
ноаналоги амидов, соответственно, пли, обобщенно, как аминоазо-
метпны. Однако если в ряду этих соединении возможна тауто-
мерия [340] (уравнение 191), то амндоксимы и ампдразоны мож-
но рассматривать также как пмпиопронзводные (656; X = ОН или
NR2) гидроксамовых кислот и гидразидов, соответственно.
NR2
R2R3NX R3 = n ||
C=NX ( R'-C-NH-X (191)
R1Z
(655) (656)
8.1.5.1. Методы получения амидинов
и родственных соединений
Амшюазометины получают присоединением аминосоеднпенпи
к субстратам, содержащим кратные связи (в основном к нитри-
лам), или с помощью реакции замещения соответствующих азоме-
тинов (большей частью галогепазометпиов, имидатов и тиопмида-
тов) под действием амшюсоедниепнй. Наибольшее распростране-
чие получил второй метод; он подробно рассмотрен в разделах,
посвященных реакциям галогеназометпиов (см. с. 588) и имидатов
11 тиоимидатов (см с 604), п его применение для синтеза амидн-
"ов [293, 328], амидокспмов [996, 330, 331] и амндразонов [329,
439] пе требует дополнительных пояснений. .
тезировать
п амп
m Присоединение амчносоединенид
к субстратам, содержащим кратные связи
„„пииенпя аминов к нитрилам, позволяющие син-
Реакции прпсоеди Зо8], амПдОксимы [996, 330, 331]
тезировать амиДО» I ’у1обиее рассмотреть в разделе, посвя-
п амидразоиы [о-• 'биост„ нитрилов (см. разд. 8.2.2.2).
ше1П10,м реакционно , сиосоо^ „ ампдоксимов 331]> ^1.
Спосооы синтеза4 |соед111|с1|)1я а.мнпосоединепий к оксидам
ванные па реаь , 0Ш1Сапы в разделах, в которых рассмат-
“XI!%"««=». спо«й«ть »™ оу&тр.™ |„.
разд. 8.24.2(2) и 8.2.5.2(2)].
(2) Прочие методы
Кажущийся возможным способ синтеза амидинов, амидокспмов
и ампдразопов (или, формально, имино-, окснмино- и гндразоно-
поопзводпых амидов) путем амипокарбонильнои конденсации
|см разд 8 1.1.1(1)1 в применении к амидам осуществить в обыч-
ных условиях не удается вследствие чрезвычайно низкой реакци-
онной способности карбонильной группы амида. Однако в неко-
торых особых случаях с помощью такой конденсации были полу-
чены амидины с препаративными выходами. Примерами могут
служить термическая конденсация вторичных амидов с арилизо-
цианатами [схема 192; (657) —► (658)] [328] и их реакция с амин-
ными комплексами титана, такими, как тетракпс (диметиламино)-
титан |(657)->(659)] [328]. В отличие от простых амидов гидро-
ксамовые кислоты легко конденсируются с арнламинами в полн-
фосфорноп кислоте при 110—115°C и обычно дают амидоксимы с
хорошими выходами |99б, 330, 331]. Более высокой реакционной
способностью тнокарбопнльной группы (по сравнению с карбо-
нильной группой) можно объяснить гладко протекающую конден-
сацию тноамидов с аммиаком и первичными и вторичными амина-
ми в присутствии хлорида ртути(II) (для связывания образующе-
гося сероводорода); это общий метод синтеза амидинов (660)
[328]. Аналогичная реакция тноамидов с гидроксилам ином и
разииом приводит соответственно к амидокспмам [996, 330,
и амидразоиам [329].
ArNCO,
150 °C
Х = О
гид-
331]
NAr
Я
R’-C-NHR2
(658)
X
, II
R —С—NHR2
(657)
X = o|ti(NMc2)4
RSNHRI R3R4N,
HgCig , C=NR2
X=S ’ R1Z
(660)
(192)
Me2bk
\=NR2
(659)
608
к числу менее общих методов синтеза амидинов относятся кон
денсаипя хлороформа со вторичными аминами в ирису?™ мето
КСПДЗ натрия как катализатора [341] и коидеисЦя пбвичных
аминов с этилортоформиатом в кислой среде [341] Амидины об
разуются и как конечные продукты самопроизвольного обложе-
ния ДМ,2,3-триазолинов - продуктов Циклоприсоединения би-
доВ к енаминам [342]. Таким путем был осуществлен синтез
циклического амидина с наименьшим (из известных) размером
цикла (уравнение 193) [343]. ' н р м
NR
R = SO2C6H4CH3, СОгВи-трет, СО2Е(
Каталитическое восстановление легкодоступных нитроловых кис-
лот (661; X = ОН) и гидразонов (661; X = NHAr) (получаемых
сочетанием а-нитроарилметанов с арендиазонневыми солями; реак-
ция Яппа — Клингемана, см. с. 485) представляет собой удобный
метод синтеза амидокснмов [996, 331] и амидразонов [329]
(662; Х = ОН или NHAr) (уравнение 194), хотя он связан с опре-
деленными ограничениями. Гуанидин [H2NC(NH2)=NH] получа-
ют нагреванием тиоцианата аммония [344].
O2Nx Н2 pt H2N,
Y=NX ---------> Y=XX . (194)
r/ r/
(661) (662)
8.1.5.2. Реакции амидинов и родственных соединений
Вследствие присутствия аминогруппы, заместителя с высокой
электронной плотностью, аминоазометины (амидины, амидоксимы
и амидразопы) обладают более высокой реакционной способ-
ностью по отношению к электрофильным агентам, чем другие азо-
метины. И наоборот, двойная связь углерод — азот в аминоазо-
метинах проявляет пониженную реакционную способность по
отношению к нуклеофильным агентам по сравнению с другими
азометинамп.
(1) Реакции с электрофильными реагентами
Амидпны относятся к числу сильнейших органических основа-
ний (если не считать четвертичные аммониевые основания), а к,
Уже простой амидин, например ацетампдпп, является более силь-
ным основанием, чем соответствующий алнфатическпп амин, и
Дает истинные соли даже со слабыми кислотами, например с
сусной кислотой. Протонпровапне происходит по атом) а
iiDOBKii п значительная основность амидинов
азометпиовой групп 1 бо’ьшей устойчнвостыо образующейся со-
может быть ооъяс ствие резонанса нона амндиния (урав-
„ряженноп кп ---ледст no„„of e3OHaHCHoft ста.
неипе 190)- ultl . КПРПоты обусловленном наличием треть-
б,.лизат.ей ^Ряжешиш кислоты, r?= r2 = r3 = R4 =Р Н)>
ХГХясш.ть исключительно высокую основность гуаннднна
(рЛ'= 13,65) [344].
NHR2 /NHR2
R'-cf R’-СГ
(195)
(663)
(664)
Такие реагенты, как 1,5-дназабицпкло[4.3.0] нонен-5 (665) н 1,8-ди-
азабицикло[5.4.0]упдецен-7 (666), являются мощными дегалогени-
рующими агентами, что можно объяснить наличием в их структу-
рах фрагмента амидина [345]. При действии этих реагентов де-
гидрогалогенирование идет быстро при низких температурах, что
позволяет синтезировать многие неустойчивые соединения, напри-
мер стнбабензол (668) (уравнение 196) [345].
(196)
[344] или
свободной
При нагревании амидинов [293, 328], гуанидинов
амндразонов [329], содержащих не менее одной
NH-группы, с алкплгалогенидамп (лучше всего с йодидами) или
алкилтозплатами происходит N-алкилпрованпе с преимуществен-
ным образованием N-алкнлпмннопроизводных. Амндоксимы (в ви-
де солей с металлами), напротив, алкилируются по атому кисло-
рода [996, 330].
Амидины [293, 328], гуанидины [344] (в том числе ампно-
гуанпдины [346]) и ампдразоны [329, 339] легко N-ацплируются
при действии карбоновых кислот, их ангидридов и хлорангидрп-
ппп . .... ... последую-
, г---------------------„„ действием
класст'ргЛЫ1ЫХ 1’еаг„ептов) представляет собой основу для ряда
K.naccmiprvnv „ пропзвод-
ампдппов
" 1>а,,1,Д1"10В 13441 ) и к производным пиразола, 1,2,4-триа-
, , , -оксадпазола и 1,2,4-трпазнна (в случае ампногуанидп-
610
дов. к-Лцилпроваипе амипоазометииов в сочетании с
щвм замыканием цикла (самопроизвольным пли под
JI f~\ П П 1111 -тт гч гт т t * • ,. ч
классических реакций гетероциклнзацпи, приводящих к
иым имидазола, пиридина и пиримидина (в случае
[328] и гуанидинов 3441 ) •• - ---- у
нов [346] и амидразопов [329, 339]). Все эти п»
н0 рассмотреть в данном разделе поэтомм »Р кции нев°змож-
теля к оригинальной литературе (см. также т 81 0ТСЬ1-’1аем чита-
Реакния N-пезамещенпых амидинов с
там», например с гипобромитом ИЛИ ГИПОХЛОритХР н=И [3471'
приводит к диазиридинам. Вполне вероятно что пп Т [3471'
Хт последовательное N-moho- „ -дибХ-М.^ванизаТм 'заТ
кан„е цикла сопровождающееся отщеплением бромп -X
(уравнение 197) [293]. Иногда удается выделить N-моно атоген
интермедиаты этих реакции. Электрофильная атака гипогалоге'
НПТОВ Э3 ГуанпД1,нов превращает нх в N-галогенХи.
дины [344]. Примерами электрофильной атаки по азоту гуаниди
иа являются также нитрозирование, и нитрование, приводящие
соответственно к N-нитрозо- и N-иитрогуанидпиу [344].
менее
(2) Реакции с нуклеофильными реагентами
Аминоазометины благодаря наличию аминогруппы
склонны подвергаться атаке нуклеофильными агентами, чем дру-
гие азометины, о которых уже шла речь ранее. Однако их реак-
ции с нуклеофилами аналогичны реакциям азометпнов с повышен-
ной электронной плотностью (т. е. импдатов и тнонмидатов) и по-
этому заслуживают лишь краткого упоминания.
Гидролиз амидинов [293] и гуанидинов [344] катализируется
как кислотами, так и основаниями и приводит в первом случае к
амидам (в случае N-незамещениых амидинов к аммониевым
солям кислот), а во втором — к мочевинам. Ампдоксимы [99 ,
330] также гидролизуются до амидов как в кислой, так и в основ
ной среде, в тех же условиях амидразоны [329] дают гидразиды.
Для получения тиоамидов использовали реакцию амидинов с сер
водородом в пиридине [293, 328], однако она идет при отноептель-
"о высокой температуре, и при этом часто обРазУюдс” ом
продуктов. Прп взаимодействии амидразопов с сероводородом
L гораздо «
чем имидоил галогениды (см. с. 588) или нмидаты (см. с.
20* 611
„лг,Ли„ вступать в обменные реакции с аминосоедииеиия-
ТаКМТак Й-’-Днзамсщспныс амидины [293] подвергаются аммо-
нолизу при реакции с хлоридом аммония в жидком аммиаке и да-
М^незамешениые амидины. Нагревание N-мопозамещенных
.ппн’иов 1293 328] с первичными аминами при высокой темпера-
тгпр приводит к №,№-дпзамешенным амидинам. Аналогичным
образом амидины могут реагировать с гидроксиламином (293,
3281 и с гидразинами [293, 328, 329] с образованием соответствен-
но ампдоксимов п амидразонов. Ампдоксимы [996] и амидразоны
[329] подвергаются аналогичным реакциям обмена.
(3) Реакции циклоприсоединения
По реакционной способности в реакциях циклоприсоединения
амииоазометины напоминают имндаты и тиоимидаты (см. с. 606).
Так, [2 4-2]-циклоприсоединение амидинов к кетенам дает амино-
азетидиноны (производные (3-лактамов) [155, 328], а при такого
же рода присоединении изоцианатов (670) к гуанидинам (669)
образуются 4,4-диамино-1,3-дназетидпноиы-2 (671) (уравнение
198) [328]. Известно также [3 + 2]-циклоприсоединение ами-
ноазометннов к нитрилимииам [155]. В случае амидинов этот
процесс приводит [155] к производным 1,2,4‘триазола, вероятно,
вследствие отщепления аминогруппы от первоначально образовав-
шегося А!-1,2,4-трназолина. Соответственно при [32]-цикло-
присоединении амидокепмов к нитрилимииам, приводящем к
1,2,4-трназол-4-Ы-окспдам, необходимо окисление in situ промежу-
точно образующихся 4-Ы-гидрокси-1,2,4-триазолинов [155].
Me2N
2 /C=N4 + R’N=C=O — Me2N—NR1
Ме2у/ XR‘ R2N—'=0 U 1
(669) (670) (671)
(4) Восстановление
Амииоазометины довольно устойчивы к восстановлению. Так,
амидины [293] инертны в реакции каталитического гидрирова-
ния, но их можно восстановить системой металл — донор протона
ХАа^аКтГУаИИДПИЫ 13441 по отношению к обо^м ти-
пам восстанавливающих агентов. При действии амальгамы нят-
амин1юУоХеОП XT6 [293] 11р°11СХ0Д»Т восста.ювите^ьно^ дез-
аминов а пнп электп^еННЫХ амидннов с образованием вторичных
щенпых ам?лLon JP, ИМ”ЧССК0М восс™’овлен1111 [38] N-цезамс-
внчные амины Поетп'п^Л°1! 11ЛП Щелочн°й среде образуются пер-
нировании происходит поомежу? ПР“ восстан°витель"ом дезами-
соединеиия могут быт. вРомежУточи°е образование иминов; эти
мо.уг быть Обнаружены при восстановлении амидинов
612
наТрием в этаноле [293], Этот процесс, включающий
in SltU. ПЫЛ 11(*ППЛ1^Лййи ...... иичсПОЩИИ
I1MIma т situ был использован для синтеза (с высоким
альдегидов- Амндоксимы можно каталитически (99б
электрохимически [38] восстановить до амидинов.
гидролиз
выходом)
330] или
8.1.6. ДИПОЛЯРНЫЕ СИСТЕМЫ
Д,.полярные системы, содержащие двойную связь угтеоод-
азот (диполярные азометииы), включают три'важных класса сое-
динении: N-окспды азометинов (называемые обычно нитронами)
для которых характерно наличие группировки типа (672) и их
аналоги, азометинимпны и азометпн-плиды, содержащие фрагмен-
ты (673) п (674). Эти структуры, способные подвергаться 1,3-ди-
поляриому циклоприсоединению, по классификации Хьюсгеиа
[173] относятся к октетиостабилнзоваппым 1,3-диполям, не содер-
жащим ортогональной двойной связи (см. с. 523, 524). ’ *
^C=N—О' \=N-N- \=,y_c/
/ I / 1 / | \
(672) (673) (674)
В отличие от относительно устойчивых нитронов, свойства и реак-
ции которых достаточно хорошо изучены [996, 348, 349], азоме-
тиинмипы п азометин-плпды—новые классы довольно неустойчи-
вых органических соединений, п их химические свойства (за исклю-
чением поведения в реакциях циклоприсоединения) исследованы
в гораздо меньшей степени [112, 173—175, 177, 350, 351]. Ниже
рассмотрены лишь такие представители этих трех классов дн-
поляриых азометинов, которые являются либо ациклическими
соединениями, либо циклическими соединениями с низкой сте-
пенью резонансной стабилизации. Поэтому здесь не описаны та-
кие гетероциклические ароматические структуры, содержащие
Фрагменты дпполярных азометинов [(672) —(674)], как N-окси-
ды [352], широко изученные N-нмппы [353] и N-нлпды [354],
бетаины [355] и мезоиониые соединения [302].
8.1.6.1, Методы получения N-оксидов азометинов (нитронов)
(I) Реакции отщепления в производных гидроксиламина
Элиминирование в производных гидроксиламииа, полученных
заранее или приготовленных in situ с помощью соответству ющ|.
Реакций конденсации, является ключевой стадиен в РЯДС м
синтеза интронов. По-видимому, самым простым и пкг„.
Щчм методом является использование реакции образования okcii-
Мов путем конденсации аминов с карбонильными сое i_ '
(«1. с. 480). Обычно взаимодействие N-алкил- или Nарнл ДР
°ксила»мина с алифатическим или ароматическим альдегид
643
ПРОВОДЯТ, как правило, в
по^ёт ошее отщепление воды приводит
рону (679), часто с хорошим выходом
отсутствие катализатора;
к соответствующему нит-
(80—90%) (схема 199
(679) (680) (67»)
Ха-е= Н,ОН, Hal, ON, C5H5N+Y; R3N’ Y'
(199)
у— 014) [996, 348, 349]. Альдегиды, как правило, гладко реаги-
руют при комнатной температуре в растворителях типа эфира или
этанола, тогда как для осуществления конденсации менее реакци-
онноспособных кетонов с N-замещсцнымн гидроксиламппами
обычно требуется нагревание. Образование интронов из простых
алифатических кетонов (например, из ацетона) и N-замещениых
гидрокспламннов осложняется склонностью нитрона вступать в
дальнейшую альдольную конденсацию. Эту трудность удается
преодолеть, если в качестве карбонильного компонента использо-
вать ацеталь или кетимин. Так, взаимодействие кетиминов с
N-метплгпдрокснламип-О-сульфокпслотой идет в мягких условиях
и дает кетонптропы с выходами 50—90% [356].
Конденсация гидрокспламннов с карбонильными соединениями
подвержена влиянию пространственных факторов (кетоны и
N-замещеиные гидроксплампны с объемистыми заместителями в
реакцию не вступают), что снижает ее ценность для синтеза ни-
тронов. Еще одним ограничением является относительно малая
доступность исходных производных гидроксиламииа. Однако гене-
рирование требуемого гидроксиламииа можно осуществлять
in situ из более доступного предшественника, например нитро- пли
интрозосоедипепня. Так, стандартным методом синтеза цикличе-
ских нитронов является самопроизвольная циклизация N-заме-
щепных гндрокспламииокарбоннльных соединений, образующихся
/„.Дп” ПРН ВОССтан°вле|1ии а.р-нитрокарбонильных соединений
( роальдегидов и, главным образом, нитрокетонов) под действи-
м цинка п хлорида аммония. С особым успехом этот метод был
ten льзовап для синтеза Д'-ппрролин-М-оксидов (683), важной в
6И
группы соеднненнй (уравнение
200)
(200)
„нтетпческом отношении
[996, 348, 349, 357].
o2n
(681)
(682)
(683)
Оксимпио (нитрозо) соединения, образующиеся n„u „
Бартона (см. с. 484), также были использованы как проме™"
иые соединения в синтезе полициклических нитоопов - 04
ипе 201) [82, 358]. Избирательное восстаиовление^с^о^дХ?’
вием цнанооорогпдрпда натрия, приводящее к N-замещеиным гиГ
рокснламинам, и последующая конденсация с форматьдегилом
позволяют получать метпленнитроны [359]; таким способом удает-
ся синтезировать различные соединения этого типа. '
Генерирование in situ N-замещенных гидрокспламннов (677)
(схема 199), которые могли бы в результате отщепления дать
производные нитронов (679), можно также легко осуществить пу-
тем катализируемой основанием конденсации иптрозоаренов с раз-
личными соединениями с активной метиленовой группой (676в-е),
в которых одни нз метиленовых заместителей является хорошей
уходящей группой (схема 199; R3=Ar). Иллюстрацией этого об-
щего подхода к синтезу нитронов может служить катализируемая
основанием конденсация иптрозоаренов с бензплгалогенидамн
(676в; R1 = Аг, R2 = Н), приводящая к СД'-Дпарплальдоннтро-
паы. По-влднмому, промежуточно образуются М-(аталогеналкил)-
гпдроксиламппы, отщепляющие затем галогеиоводород [996, 348,
349]. Аналогичным образом конденсируются с иптрозоареиами и
“л-дизамещепные ацетонитрилы (676г) (в присутствии метоксида
Натрия) и, отщепляя синильную кислоту, дают соответствующие
кетонптропы (679; R3 = Аг) [996, 348, 349, 360]. Область приме-
11е,И1я такого рода конденсаций удалось значительно расширить, а
,1Х эффективность повысить за счет использования в качестве ме
тчленового компонента N-метпленпирпдиипевых солеи (о7Ьд),
в качестве основания пиперидина, карбоната или гидроксида
личного металла (схема 199; R3 = Ar). Вероятным '’^^яется
м прп 9ТОМ вар11апте реакции — реакции Кройке
615
„„п.тшпй-бетанп (678). Высокая эффективность синтеза обуслов-
₽— что ппридииин-катион — одна из лучших уходящих
пп 1996 348 349]. Необходимые для синтеза пиридиниевые
coin можно легко получить путем кватернизации пиридина соеди-
нениями с подвижным атомом галогена, например фенацнлброми-
дом или хлорацетонитрнлом. Их можно получить также in situ
поп взаимодействии пиридина с соединениями с активной метиле-
новой группой в присутствии иода (реакция Ортолева — Книга).
Реакция Кронке может быть с успехом применена для синтеза
функционально замещенных нитронов. Так, конденсация нитрозо-
аренов с фенапилппридипийбромидом (686) в присутствии основа-
ния как катализатора протекает гладко и дает ацилнитроны
(687) с хорошими выходами (уравнение 202).
Вг'
дает ацилнитроны
O'
I
+ PhCOCH=NAr
ArNO
(202)
(686) (687)
Вместо метпленппрпдпипевых солей в реакции Кронке можно ис-
пользовать четвертичные метпленаммониевые солп (676е) и также
получать нитроны (679; R3 = Аг) с хорошими выходами. Нит-
роны образуются как конечные продукты и при реакции алифати-
ческих и ароматических иитрозосоединений с дпазоалкапами и
сульфонлйилндами ]99б, 348, 349, 361] (уравнение 203). По свое-
му механизму эти процессы сходны с реакцией Кронке. По-види-
мому, вначале образуется аддукт (690; X = No или SRs), который
затем превращается в интрон (692) либо непосредственно, либо
через новый интермедиат, оксазнрндин (691). Примером синтеза
нитронов по этой схеме может быть реакция третичных нитрозо-
алканов с диазометаном, приводящая с хорошим выходом к метп-
леинитропам (схема 203; R> = R2 = Н, X = N2, R3 = rper-Alk)
rI
г/6
r2/i.
X
(688)
R1
2/4"------->JR
R 7 V
(691)
л IN Г
"4 А-
(б90)
(689)
r!.
(203)
О"
(692)
НОЙ метилс11оХ“1НёрупёёйРе(б76 СХП—М11М" соед,,ненияы" с актив-
также приводит к нптоопям ки Н В пР1,сУтствпн оснований
616 Р°"аМ [см' также Разд. 8.1.1.1(3)]. Полака-
т что вначале образуется N-замешенный -
viH, R3 = Ar), который затем дегвдрируетгЯД1’°КС,<'Ламин <677:
J„eM нитрозоарена [996, 348, 349]. Х^метоп in‘Z“ "°Д
ка_ Закса) не нашел применения для синтез (реакция Эрли-
ренных (-50%) выходов продукта; конкурир У“е’
является дегидратация промежуточно образовав Р°Цессом
«ого гидроксиламииа (677; X = Н) с ofina™, шегося произвед-
шего анпла. Гораздо большую ценность дпяТинтХ™""^'0'
представляет окисление специально полученных N Л, нитронов
гпдрокспламннов, содержащих хотя бы одСХдор"hZCJ
действием пероксида водорода, трет-бутилгпдропероксида трика
лнйгексацнаиоферрата или перйодата натрия (996 348 ХоГ
Было показано что особенно эффективным реагентом для’осу.,
ствленпя подобного превращения служит тионилхлорид (3591
Окислительный способ синтеза нитронов особенно успешно приме-
ним для превращения циклических гпдроксилампнов (693) в цик-
лические нитроны (694); в этом случае наплучшпм окнетптетем
является желтый оксид ртути, выходы при этом очень высоки
(80—90%) [996, 348, 349, 359].
^\/Н н*°
НО—hlPh
(693)
Ph
(204)
(694)
(2) Алкилирование оксимов
Оксимы представляют собой амбидентные нуклеофилы. Она
могут подвергаться алкилированию по кислороду и давать про-
стые эфиры оксимов или по азоту, образуя нитроны. Направление
алкилирования зависит от структуры оксима, природы алкилирую-
щего агента и условий реакции. Использование простых алкили-
рующих средств—алкплгалогепидов плп -сульфатов — приводит
обычно к смеси обоих продуктов, хотя прн алкилировании сереб-
ряных солей оксимов в смеси может преобладать нитрон. Этот
метод широко используется для получения нитронов, так как про-
дукты реакции часто удается легко разделить. Тем не менее, не-
смотря на простоту выполнения этой реакции и, как правило,
высокие выходы нитронов, алкилирование оксимов нельзя все же
рассматривать как общий метод получения нитронов. Для синтеза
первого представителя четырехчленных циклических нитронов
(696) было использовано внутримолекулярное алкилирование ок
сима (695) [363].
Me Me
/С основание
TsO—' —Me
НО—N
(695)
Me Me
(205)
(696)
617
(S) Окисление азометинов пероксикислотами
((см7,с “eS
Перегруппировкой в тщательно контролируемых условиях (чтобы
избежать возможной перегруппировки оксазнридпиов в амиды
топ можно рассматривать как общин метод синтеза нитронов
гпяввение 206) (996, 348, 364]. Окисление обычно проводят при
низкой температуре (О’С), в качестве окислителен используют
пероксид водорода, перуксусную кислоту или, что более удобно,
л-хюрпероксибепзойную кислоту. Довольно часто оксазиридины
не выделяют а они самопроизвольно перегруппировываются в нит-
роны (365].’На стадии окисления выходы составляют 50—90%,
а на стадии перегруппировки 50—100%. Этот метод применим к
азометпнам, содержащим алкильные, циклоалкпльные п арильные
заместители. Недавно было предложено использовать силикагель
как катализатор
1[366].
R'\
C=W
R2/
перегруппировки оксазнридпиов в нитроны
перокси-
кислота
О
С——N—R3
(697)
(698)
R\
XC=N—R3 (206)
R!/ i-
(699)
8.1.6.2. Реакции N-оксидов азометинов (нитронов)
Структуру нитронов удобнее всего представить как резонанс-
ный гибрид трех канонических форм (700), (701) и (702), С точки
зрения реакционной способности наибольшее значение имеет
структура «растянутого карбонила» (700) [348]. Как видно, нит-
роны должны подвергаться электрофильной атаке по кислороду
и нуклеофильной атаке по азоту, что и наблюдается в действи-
тельности.
(7°0) pal) (7()2)
(207)
(1) Реакция с электрофильными реагентами
кислорода с образованием соотв.
(схема 208) [996, 349]. Алкилир,
му кислорода; его можно легко
алкилирующим агентом напппмпп ~ ’ ’ —г >uuFun v
НОМ растворителе (напрймер^в б^оле”^^2™' *
618
(НС1, НВг,
протонирование по атому
соответствующих солей (704; R4 = Н)
килиповаипе также происходит по ато-
осуществить, нагревая нитрон с
и использовании более
мошного алкилирующего средства, иапримео *Tnnftn „
оксония, образуется соль О-алкилирова.шогоНитрона (ЖХ
3491. Реакция нитронов с ацилиру10щими X'TJ™4> t"6’
,,РП с хлорангпдридами кислот, ангидридами (иапри-
рСЬ, РОСЬ) протекает более сложно может nponZT’ PCU>
группировка (особенно в случае N-алкилнитроновГво ат Пер®-’
S третичный амид [78, 996, 349]. Полагает X пн pZZ
пИровках такого рода вначале происходит координация Z X
„о атому кислорода нитрона и образование ацилоксиммониево со
ли (705). Переход от этого интермедиата к конечному продукту ocv
ществляется путем последовательных [ 1,2]-сдвигов авдлоксигрущ
пы и заместителя и затем сольволиза [(705)->(706)-^(708)->-
(709)->(7Ю) ]. Альтернативная схема образования амида вклю-
чает замыкание цикла в оксазиридиновое производное (706) пре-
вращение в (707) и последующую перегруппировку оксазиридииа
в амид (7Ю) [/о, ЗЬ4].
R'\ + zr3 R,X
X- T=N('
R2Z X
(704)
X’
R'\ */R3
xC=n'
R2/ XOCOR1
(705)
R'\ . zR3
R<COX
R1
R2/
(703)
OCOR’
(Znr3
(706)
r’\°cor
;c—N—R3 (208)
(708)
R1.
о о
/\ ,11 /R2
;c----NR3 -----> R'C-N'
R2Z \R3
(707) (710)
O(W
I ZK
r'-c-n;
* XR3
(709)
(2) Реакции с нуклеофильными реагентами
Будучи по своему характеру соединениями с «растянутым кар
бопплом» (см. выше), нитроны обычно вступают в реакции
1,3-прпсоедпнепня с нуклеофильными реагентами п образуют про
взводные N.N-дцзамсщенного гидроксиламнна, которые в
‘,0Сти от природы нуклеофильного агента либо могут ыть ’
и, либо распадаются на исходное карбонильное пр ,
(или его производное) и гпдроксиламии. Легкость гидр
лчческпх нитронов (711) до карбонильного компонента ( )
"РоизводногЦРгЦдрокснламЦна (715) (схема 209) в У”я0ЧныЦ
обп'ЮГО ил“ осиовного катализа можно объяснить ₽ кс112аЛкпл)-
Разованием неустойчивых производных N-(a-niAP
619
Rk + /R3 r<nh2
xc=v •----------------
R2/ 'O'
(711)
мер Д'-пирролнн-1-N-оксиды [9],устойчивы к гидролизу.
R'\?H' ZR3 ><3°
;с-<
R2/ ^ОН
(712)
«\Г>
C—N'
R2/Z ЧЗН
(713)
(209)
R'\
V=O + R3NHOH
Ri/
(714) (715)
R'\
(715) + ;C=NR<
R!Z
(716)
Подобным же образом ациклические нитроны разлагаются
при взаимодействии с аминосоедннениямп (первичными аминами,
гидроксиламином, гидразинами) и дают соответствующие азомети-
ны (716; R’= Aik, Аг, ОН, NR2) — основания Шиффа, оксимы,
гидразоны —п производные гидроксиламииа (715) [348, 349].
В отличие от реакций с аминосоедннениямп при взаимодействии
интронов (как ациклических, так и циклических) с нуклеофиль-
ными агентами, у которых нуклеофильный центр сосредоточен на
атоме углерода (циаппд-пон, металлорганические соединения, кар-
банионы соединений с активной метиленовой группой), образуют-
ся производные М,М-дизамещенпого гндрокспламина с выходами
60—80%. Эти соединения обычно довольно устойчивы, и цх мож-
но выделить [9, 996, 348, 349, 366]. Примерами процессов этого
типа могут служить превращения А'-пнрролин-М-окспдов (схе-
ма 210).
(718)
KCN, Н+
MeNO?
NaOEt
(210)
Ha
ют
620
(717)
I. R3MgX
12. H2O
(720)
I
O*
С7ерПш1ёск11ЛеИНфакторы',^пао1110ИНЬ1Х реагентов к
p факторы, поэтому взаимодейств.
(721)
интронам нлия-
ie с альдоицтро-
нами (или соответствующими 2-пезамРпг™
аналогами) протекает более эффективно "ю?™1' '«««квми
реагируют с карбанионами. Так, например в "ИТроны труднее
Гриньяра происходит не 1,3-присоединещ® СЛучае Реактивов
ронов до соответствующих шиффовых основамос.стан°вление нит-
прп 1,3-присоединенин реактивов Гриньяпа « Случающиеся
изводные гидроксиламииа легко окисляются , ЛОн"тРонам про-
зволяет использовать эту реакцию для синтА?’' ВЫШе)’ что п°-
альдонитронов, например (720)->(721 j а кетонитронов из
Атомы водорода в а-С-алкилзамешенныу
кислыми свойствами. Вследствие этого такие ронах обладают
работке сильными основаниями могут подвепгятДИНеНИЯ при об‘
превращений: конденсации, напоминающей альдоп АТ ,типам
числе даже и димеризации под действием основания) (в
[348], и [1, 3]-прототропному сдвигу (перегруппnLA21 °
[78]). Последний соответствует взаимопревращениям атьдониТ
ронов и кетонитронов под действием оснований (уравнение 212)
« сходен с рассмотренной ранее (см. с. 490) трнадной прототпо.
ппеи шиффовых оснований. риютро-
Ме Me Me Me
основание
20 °C
(723)
Me Me
(722)
(211)
PhCHO
KOH, EtOH,
кипячение
Me Me
CH=CHPh
(724)
C=N—CHaAr
I
O"
(725)
основание Al\ t
_-------->• xCH-N=CHAr (212)
‘ n/ I
К 0-
(726)
(3) Реакции циклоприсоединения
Нитроны представляют собой “^'АщАГортогомльной двой-
стаонлпзоваииых 1,3-диполеи, не содержат . P ш [34-2]-
пой связи [173] (см. с. 523, 524), они встУпа’°11 АиХрофилами.
Циклоприсоединения с самыми разнообра 1 выходом при
Реакции этого типа обычно протекают с (например, в то-
Чагревапии компонентов в пиертпом Раса® ‘ . различные гетеро-
луоле). С пх помощью можно сиитезпр ппугнмп способами.
Циклические системы, которые нельзя полу1
„ апкппп возросло еще более, когда стало ясно
Значение этой Р'икн“ дд ты образуются из нитронов, ко-
136-], что, п°с"> получены нз карбонильных соединений,
ГОрЫе\.,о1иСпе превращения этих аддуктов эквивалентны превраще-
послед)юшие преир осуществлены на исходных карбониль-
ном, которые могли^бь^осупц^правило, менее реакционноспо-
НЫХ соединения • 3-дцполярного циклоприсоединения, чем азо-
SX SSX1 |». P-Д. S.1.6.3(2)]. Более то„.
"до штропы более активны, чем кетонитроны с которыми
neaniPViOT лишь наиболее активные из днполярофилов. На лег-
кость 3 + 21 -циклоприсоединения оказывает влияние и стереохи-
мия интронов- так, цис-нитроны более реакционноспособны, чем
транс-изомеры. В последнем случае часто требуются жесткие ус-
ловия для проведения циклоприсоединения [174].
Способность нитронов к 1,3-диполярному циклоприсоедине-
нию—это единственный аспект химии нитронов, который в пос-
леднее время подвергся широкому изучению [173—175, 367, 368].
Из реакций этого типа наиболее подробно было исследовано взаи-
модействие с алкенами [175, 359, 367—369]. Ациклические и
циклические нитроны легко вступают в реакции [3 + 2]-циклопри-
соединения как с электронодефицитными алкенами, так н с алке-
нами с повышенной электронной плотностью, и дают изоксазоли-
дины, причем выходы часто близки к количественным. Высокая
реакционная способность в реакциях циклоприсоединения этого
типа видна из того факта, что даже циклогексен, весьма малоак-
тивный 1,3-дцполярофнл, вступает в эту реакцию. Циклоприсое-
динение с равным успехом проходит и с нитронами, полученными
in situ путем конденсации гпдрокснлампиов с карбонильными со-
единениями [370, 371]. Образование изоксазолндииов подчиняется
кинетическому контролю; эта реакция в
специфична, причем стереохимия алкена
аддукте, то есть это стереоспецифичное
(схема 213),
высшей степени стерео-
сохраняется и в цпкло-
цис-циклоприсоединенпе
622
п условиях кинетического контроля [3 + 91 „„
„симметрично замещенных алкенов к нитпои;лопр"СоеДииение
^специфично. Присоединение к нитрон м"Р°'Гекает высо-
1-дизамешенных алкенов с высокой электпони ?замещенных
"„’слегка обедненных электронам,, иеизмен.ю ппоИсх^°ТНОСТЬЮ
образом, что образуется связь между атомом кислорота н,™™
° наиболее пространственно затрудненным концов алкена вТ
"уктами реакции являются 5-моно- и 5,5-дИзамеще„, ые " о ^P0'
лпдпны. Регпоспецифичность циклоприсоединения, которую м„ж
„о объяснить в рамках теории молекулярных орбиталТ |Х
обусловлена тем, что НСМО диполя с наибольшим коэффициент
том У атома углерода взаимодействует с наименее замещенным
атомом углерода (концом) алкена с образованием связи. 13 + 21
Циклоприсоединение подобно [4 + 2] -циклоприсоединению (реак-
ции Дильса Альдера) обратимо. Отсюда следует, что высокая
стерео- и регпоспецифичность, наблюдаемая при реакции алкенов
с нитронами, сохраняется лить при условии кинетического конт-
роля реакции и исчезает в случае термодинамического контроля
Описание [3 + 2] -циклоприсоединения алкенов к интронам
(и других родственных реакций [3 + 2]-циклоприсоединения)
как одностадийного согласованного процесса, включающего стро-
го одновременное образование двух новых о-связей в высокоупо-
рядочениом переходном состоянии, полностью согласуется с на-
блюдаемыми кинетическими характеристиками процесса (относи-
тельно низкой энергией активации и высокой отрицательной
энтропией активации), а также с высокой стерео- и региоспецифнч-
ностыо [174]. Был предложен альтернативный бирадикальный ме-
ханизм [372], ио он отвергнут [373]; в частности, он не согласует-
ся с данными |4С-кинетического изотопного эффекта, который
изучался на примере образования 2,3,5-трифенилпзоксазолпднна
нз стирола и С,Ы-дифенилнитрона по реакции [3 + 2]-циклоприсо-
единения [374].
Было показано, что внутримолекулярное циклоприсоединение
алкенов к нитронам — эффективный метод синтеза [359, 369] . При
построении новых сочлененных и мостиковых систем можно было
вполне полагаться на стерео- и регпоспецифичность таких процес
сов, что и было остроумно использовано при синтезе сложных
структур, отвечающих природным соединениям; особенно показа
тельным примером служит синтез лнкоподиевого ал^ал,°"Лгд?
Дулина, осуществленный с высоким выходом (схема L ) L Б
Реакции [3 + 2]-циклоприсоединения нитронов не огра,
Ются лишь примерами использования двойной углер ду Р
связи в алкенах. Их можно также успешно ’ ерОд —
ь,мн связями углерод—углерод в алленах [367, J, У |13ОТцо-
« « углерод—сера1 р’геЛроку.^т В X
«арСод„„„„д,х а сероуглероде) КЦ (72S)
Слу ,ае продуктами являлись [3 + 2] „ I комнатной
Ма 215), которые обычно довольно устонч.
температуре, ио распадаются или перегруппировываются по-разно-
му при нагревании. Аллены (728а) дают нзоксазолнднпы (729а),
перегруппировывающиеся в соответствующие пнрролпднноны-З
(731а) при нагревании, вероятно, через цвиттер-ионные интерме-
диаты (730а). Термолиз циклоаддуктов [1,2,4-оксадназолидпно-
нов-5 (7296) и -тиоиов-5 (729в)], полученных из арнлизоциапатов
(7286) и арплизотиоциапатов (728в), напротив, приводит к амиди-
нам (732). Эту реакцию можно рассматривать как метод синтеза
амидинов из карбонильных соединении через нитроны. В случае
H;
YC=N\ +
0“
(727) I
(728)
Н
(730)
а,д
н
'N^'X
(729)
6,6
N 'S
H
(734)
ArN=C=O
(735)
(215)
. H
y—i=0
(731)
(a) X=Y=CR.
N NHR
(732)
(733)
•W X-Y-NAr; (e)x.Y-SY'°; W ’Y=S• to X-S,Y-NAr; '
624
апилизотиоциаиатов (728в), наряду с иикпп п
связи C=N происходит циклоприсоединение пГсвязи"^ П°
приводит к образованию в качестве побочных ппп™1 C=S- что
ствующих 1,4,2-оксатиазолидинов (729г) однако^ ” С00твет‘
присоединения ацнлизотпоцнанатов к ’нитропам пТ’ае цикдо'
единственными продуктами. Термолизом этих циклоал™™ ЯЮТСЯ
но получить тиоамиды (734) что, таким образом может XT’
методом их синтеза из карбонильных соединений че£зZ «
1,2,4-Оксадиазолидинонимин-5 (729д) образетш..»Р р ны'
нов и карбодиимидов (728д), при нагревании иодверХ’тся^Х
группировке в 1,2,4-триазолидиноны-5 (731 д) и распаду в амидины
(732) Образующиеся при [3 + 2]-циклоприсоединеНии\ероу лер0
да (728е) к нитронам 1,4,2-оксатназолидинтиопы-5 (729с) неустой
чивы и распадаются на имины (в случае СЛ’-диарилнитронов) или
тиоамиды (734) (если в качестве исходных использовали С N-ди-
алкилнитроны) [174, 367]. Нитроны легко присоединяются также
к двойной связи углерод—сера в тиокетонах и двойной связи
углерод—фосфор в фосфоранах, давая соответственно 1,4,2-ок-
сатиазолидины и 1,2,5-оксазафосфолидпны [174 , 367J. Нитроны
бурно реагируют с простыми алкинами; получающиеся А4-изокса-
золпны неустойчивы, они превращаются или в нлиды вследствие
раскрытия цикла, либо в Д3-изоксазолпны путем переноса водо-
рода. Однако Д4-изоксазолииы, получаемые при циклоприсоеди-
нении дпметилового эфира ацетилепдпкарбоиовой кислоты к нит-
ронам, достаточно устойчивы, и их можно выделить (см., напри-
мер, уравнение 216) [137, 367]. Для объяснения образования
₽-лактамов из нитронов и ацетиленидов меди было выдвинуто
предположение о перегруппировке первоначально возникающих
^’-изоксазолинов [376]; выход |3-лактамов 20—60%.
Ме\/---у _ МеО2ССэССО2Ме Ме'
Me'
(216)
Ме
(736)
О---—СО2Ме
(737)
(4) Окисление
Нитроны устойчивы к действию большинства обычных окпсли
леи. При озонолизе, однако, происходит окислительн Р
П ° соединения и нитросоединеипя [348 34VJ. о‘ аль-
до °дата [349] пли тетраацетат свинца I'85’ 1 J ' у (иапри-
меп Т^|1Ь| нлп С-2-иеза мешенные циклические Р ш их
0®Р> Д'-пирролпи-П-окспды) ДО гидроксамовых кисло
ЦеТНЛЬНЫх производных.
625
(5) Восстановление
восстановление нитронов может происходить таким образом,
X Хея П..6О N N-днзамещенные гидроксиламииы (путем
что образуйте СВЯзИ), либо шиффовы основания (путем де-
‘"грования), либо вторичные амины (когда реализуются
абп Типпавлепня). Восстановительная система металл - донор
то 1 "(например, цинк в уксусной кислоте) превращает нитро-
в соответствующие шиффовы основания; при действий щелоч-
m ..ртяпля в спирте возможно дальнейшее восстановление с
об аз ваннем вторичных аминов [996, 348, 349]. Комплексные
гидриды металлов (LiAlH4, NaBH4) восстанавливают нитроны в
N N-дпзамещепные гидроксплампны с хорошим выходом [996,
348 349] При каталитическом восстановлении над никелевым,
платиновым или палладиевым катализатором в зависимости от
условии реакции и применяемого катализатора образуются вто-
ричные амины, N,N-днзамещенные гидроксплампны или шиффовы
основания [996, 348, 349]. Дезоксигенирование нитронов в шиф-
фовы основания идет с высоким выходом (~90%) прп действии
фосфорсодержащих реагентов, например трифенилфосфина [996,
348, 349].
8.1.6.3. Азометинимины и азометин-илиды
(1) Методы получения
Устойчивые N-p-цнаноазометинпмпны можно легко получить
взаимодействием диазоалканов с анти-дпазоцнанидами (уравне-
ние 217) [173, 175, 177, 351, 377].
одним из следующих способов:
^нХ11^11рХ,.!Хе™11ИМИ,,1ь’ (74°) обычно получают in situ
солей nw дспРоточпроваиием гидразопневых
том числе и вХХ0ЖН0 П0ЛуЧИТЬ различными методами, в
(738- R = 2 4 ™УнХпМ°/еКуЛ/'’гНЬ1М алкилированием гидразонов
' К 23-динитрофенил) И2 точ 1то? ом
крытием цикла в легкодостепХ 73, 175’ 177’ 351Ь 7
под действием кислот / Д ТДПНах (742; R = Me)
гораздо более общего метола(пДеМа -218) ил" с помошыо
Да реакцией альдегидов н кетонов
626
(743) с 1,2-дизамещеннымн гидразш,ам1!
л0Т (схема 239) [112). При взаимодействии я? пр11сУтствип кис
с 1,2-днзамещенными гидразинами часто „Л №вдов « кетонов
вольная дег.гдратаи.и! и непосредственное T^X°MT самопрОнз!
„ЙЙНОВ (схема 219) [112, 173, 175 i77 35], Р зоваиие азометин-
ляются промежуточными соеД!1Нения ’ '"‘Ь Азометинимииы я“.
днях циклоприсоединения к азину гекса<Спа"ТКре7вь,х> Реак-
„х можно выделить. 4 1оРацегона (СМ. с. 568);
(747)
тому, как депротоиированне гидразониевых солей прн-
азометиниминам (см. выше), депротонирование п.чнние-
можно получить N-алкнлированием иминов
Вг'
... ^.\О2
Bt3N
-НВг
Подобно
водит к
вых солей, которые
N BrHsC-
(220)
627
ппрпииепий с подвижным атомом галогена, позволя-
ло,! действием соедин азоыет)1Н.|1ЛНДЬ1 (уравнение 220) [173-
ет/С.77РТоВ1 Применение этого метода ограничено из-за возмож-
17о, 177, Заир Р простых нмпниевых солеи в енамины.
IIOCth депрспонир используемым методом, пригодным для
Более оошиа< ,,-устойчивых азометин-плндов, является тсрмц-
получения ins» > фотохимическое днеротаторное раскры-
ЧТшиК°аР^^производных азиридина (схема 221) [174, 350, 351,
о, Не,,Ртойчивые азометин-илиды можно также получать
?n s<7« Термолизом соответствующих ДМДЗ-трназолинов [380].
A-' +f
U 4 поме N „ Мео2с N СОМе
Hs. ,К-/С°2Ме..... Н. / \ .Л Л-J Y , ,
221
кН Л1)
100«с Ц
МеО2С Н
(конротаторное
раскрытие)
н н
(диоротаторное
раскрытие)
МеО2С СО2Ме
(2) Реакции
и азометин-илиды — очень реакцнонноспособ-
Лзометинимины
ные 1,3-диполп. Характернейшей особенностью их химического по-
ведения является способность вступать в реакции [3 + 2] -цикло-
присоединения почти со всеми типами кратных связен. Подробно
эти реакции рассматриваются в других руководствах, здесь же
мы коснемся их лишь в общих чертах.
Циклоприсоединение к азометинпмпнам и азометпн-илпдам
происходит легче, чем к нитронам [см. разд. 8.1.6.2(3)], его мож-
но провести в инертном растворителе при комнатной (или повы-
шенной) температуре. Поскольку образующиеся гетероциклические
аддукты часто удается выделить с высоким выходом, реакции
[3 2]-циклоприсоединения к азометиниминам и азометин-илп-
дам (как п к нитронам) используют для получения разнообраз-
ных гетероциклических систем, некоторые из которых другим
путем получить невозможно. Более того, поскольку обычно цикло-
присоединение протекает и стерео-, и региоспецпфичио, с его по-
мощью можно получить гетероциклы с определенным расположе-
нием заместителей и заданной стереохимией.
Все алкены — простые (например, этилен), с повышенной
электронной плотностью (например, енамины, простые эфиры ено-
щ 11 электронодефицитные (например, акрилонитрил, дпэтпл-
мпнямМ’ Д’,эт,|Лмалеат) — гладко присоединяются к азометинп-
дают niin-nrniTT''”лидам " с высокими выходами (часто > 90
Т''“ ил" пирролидины (749; X = NR плп СШ)
Наблюл чём МеМа 222) 1173~175, 177, 351, 378, 381].
Фнчност! и кигр В ЭТПХ процессах высокие регно- п стереоспеци-
фичность и кинетические параметры (низкая энергия активации.
628
(222)
высокая отрицательная энтропия активации) объяснимы с точки
зрения согласованного механизма, в котором реализуется высоко-
упорядоченное переходное состояние. Внутримолекулярное цикло-
присоединение алкенов к азометпнпмпнам и азометин-нлидам бы-
ло использовано для регно- и стереоизбнрательного построения
конденсированных и мостиковых бициклических пиразолидннов
[359]. Циклоприсоединение азометпннмииов (748) по связи
C=N можно проиллюстрировать их взаимодействием с изоциана-
тами и карбодипмпдамп; при этом с отличными выходами обра-
зуются 1,2,4-триазолидпны (750) [173—175, 177, 382]. Азометин-
имины и азометин-нлпды легко присоединяются также и к двойной
связи углерод—кислород в альдегидах, давая 1,3,4-оксадиазоли-
дпны и оксазолидины (751, X = NR или CRs), которые нельзя по-
лучить другим путем [173—175, 177]. Реакции [3 2] -циклопри-
соединения азометинпмпиов и азометин-плпдов не ограничиваются
лишь соединениями с двойной связью, они легко проходят и с
тройной связью углерод — углерод в алкинах, давая пиразолпны и
ппрролины, соответственно (752; X = NR или CRz) [173 175, 7,
378, 379 , 383].
8.1.7. ГЕТЕРОКУМУЛЕНЫ
Гетерокумулены — соединения, содержащие двойную
Углерод—азот, представлены четырьмя важными классами
Чических соединении: изоцианатами (753), изотиоцианатами
КаРбодипмндамн (755) и кетепиминами (756).
RN=C—О RN=C=S RN=C=NR
(753) (754) (755)
л”о,СТемЬ| кумулированных двойных связен в
ф °т повышенной реакционной <
связь
орга-
(754),
обла-
r2c=c=nr
(756)
ПЫХ связен " ^^цд^ях^нуклсо-
, мвышепиои реакционной способностью в ]Реа* ' позв0Ляет
ьиого присоединения и циклопрпсоедпн •
62»
„ ,„,ctv важнейших органических Функциональных
от,,еСТ"гётеРОКу'улень. (754) - (756) нашли широкое примене
ГРуПП лпганическо^й химии как синтетические интермедиаты. Изо-
н1,е в органик i в ка„естве мономеров (например, толплен.
ш,а“а,™ на днфенплметандиизоцианат) для синтеза полиМерОв
яДИтакже как исходные соединения в синтезе лекарственных пре.’
ёаГатов (например, для лечения гипогликемии) и веществ, приМе.
няемых в сетьском хозяйстве (например, гербициды, инсектициды,
Л ипиды) Только за один 1974 г. мировое производство нзоциа-
?атов составило 10® т [384]. Изотиоцианаты по сравнению с дРу.
гимн гетерокумуленами, содержащими двойную связь углерод-
азот обладают пониженной реакционной способностью; они
встречаются в природе - входят в состав горчичных масел [385].
8.1.7.1. Методы получения изоцианатов,
изотиоцианатов, карбодиимидов
и кетениминов [386, 387, 389—394]
(1) Синтез из аминов
Метод, наиболее широко используемый для синтеза изоциана-
тов [344, 386, 387], состоит в нагревании гидрохлорида амина с
фосгеном в расплаве пли, что более удобно, в высококппяшем рас-
творителе, например о-днхлорбензоле. Промежуточными соедине-
ниями в этом синтезе являются карбамоилхлориды. Их обычно
не выделяют, однако можно показать, что они дают изоцианаты
при нагревании [388] или при обработке третичным амином
(уравнение 223; X = О). Выходы составляют 85—95%. Исполь-
зование соли амина, а ие свободного амина позволяет избежать
последующей реакции с образовавшимся изоцианатом. В промыш-
ленности изоцианаты получают реакцией аминов с фосгеном в па-
ровой фазе. В качестве ацилирующего агента при синтезе изоциа-
натов из первичных аминов вместо фосгена можно использовать
оксалилхлорнд.
+ Ск
RNH3 Cl' + Y=X —
X
II
RNH—С—С1 -нс7> RN=C=X (223)
(759) (760)
мопейсти.л. ----- как конечные продукты при взап-
X = S) 1344 чяспХЛОаИЛОВ ам11нов с тиофосгеном (уравнение 223;
шпх к пчтпЛч^, . •т’тн реакции в отличие от реакций, приводя-
скольку иЦтиофосгёнУДи обп асег° провод"ть а иодной среде, no-
устойчивы к гидролизу. образующиеся изотиоцианаты довольно
гремвпсСёпдрохлТрРбдоДв11ём?ДОВ ИСПОЛЬЗУ1ОТ продолжительное на-
X = NR2) [2, 390] р 71 ам,1,10в с фосгенпминамп (уравнение 223,
630
(757) (758)
Изотиоцианаты образуются
(2) Синтез из карбаматов ам1,р„
и их тиоаналогов ° ‘ МОчевин
ряд практически важных методов ГИИто,
„скован на реакциях отщепления, котопи,., гетеРокумуленов
маты, амиды, мочевины и их тиоаналоги ОднакперГаются каРба-
таких процессов а именно пиролиз карбаХОв ЖтеЙШИЙ из
50-70 °C
- ROSiMe3
—ROH
+ ROH
(224)
Ви-трег
I
RNCOC1
(764)
РеС1з'6Н2О AgNOa
83-1.0 °C ’ RN=C=O < MeCN
(762)
О
II
RNH-C-SR
(765)
пиролитическое расщепление
легко получаемого
п представляет со-
В то же время
N-трнметплсилилкарбамата (763) необратимо
бой хороший путь к изоцианатам [167]. Изоцианаты можно полу-
чить с хорошим выходом и при нагревании М-алкил-М-трет-бутпл-
карбамоплхлорпдов (764) (легко образующихся при обработке
N-алкил-трег-бутиламинов фосгеном) с гидратом хлорида желе-
за(Ш); при этом происходит отщепление изобутена и хлористого
водорода [344]. Под действием солей тяжелых металлов происхо-
дит отщепление тиолов из тиокарбаматов (765), также приводя-
щее к изоцианатам [344].
В результате сходного процесса (реакции Калуна) соли лвтио-
карбаматов [например, дитиокарбамат аммония (766; Д =д
под действием солей тяжелых металлов [например, HgCh.Ag з,
Pb(NO3)2], гипохлорита пли фосгена (последний наиболее эф
Фектпвеп) превращаются в изотиоцианаты (767) (схемаi )
1344, 389]. Этот метод, однако, имеет orPa,I",'e“'l°eVTf./каталп-
поскольку получение солей дптиокарбаматов (766) У . .....
зчруемоп основаниями конденсации сероуглерода с Р
амннами проходит успешно лишь в том случае, к __дг,
паточно осповпы. Методы синтеза изотиоцианатов у о , * д
1395, 396] путем пиролиза метплдитиокарбама лит1юка’рбаматов
(7в7пе)’ иолучаемых метилированием сол основаниями
; R = Аг, х = NHEt3), а также каталю. дптиокар-
сц1еплеппя S-(N-метилпиридиниевых) пр пппя1Жем
Шматов (768; R' — CjHiNMe), получаемых гетарилир
„Й,матов (766- X==NHEt3), имеют те же недостат-
содей дитиокароам.i i методы синтеза изотиоцианатов,
кн. Недавно были ть исполь3ования токсичных реагентов,
которые поз°одя1°яжелых металлов и фосген, и преодолеть огра-
таких, как сол т е н0стЬ10 солей дитиокарбаматов [397].
лпчения, связа! ные £ д У,ском разложении бис (металлирован-
Они основаны P м = Lj или MgBr), которые образу-
ют при действии литпйорганпческих соединений или реактивов
Гриньяра и сероуглерода на первичные амины.
S
II СОС12
RNH-C-S- Х+ ---------- RN=C=S
(766) <7в7>
д
(225)
(226)
S MS
I, I II
RNH-C-SR' RN-C-SM
(768) (769)
Карбодиимиды (771) можно получить путем обессеривания
(термического или под действием солей тяжелых металлов, напри-
мер, AgNO3 или HgCl2) S-алкил- или S-ацилизотиомочевин (770;
R3*== Aik или СО2Е() [344, 391], а также путем разложения
5-(Ы-метплпирндИнпевых) производных пзотномочевин (770;
R3 = С5Н4 NMe) под действием оснований [398].
NR2
II
R'NH-C—SR3 —-> R’X=C=NR2
(770) (771)
Аналогично первичным аминам, первичные алкил- п арилкар-
боксампды и -сульфонамиды прп реакции с фосгеном дают соот-
ветствующие ацплизоцпаиаты [344, 386] п сульфонплнзоциаиаты
[167, 391, 394]. Добавлением каталитических количеств изо-
цианата или заменой фосгена как ацилирующего агента оксалнл-
хлоридом удается повысить выходы продуктов и заметно увели-
чить скорость реакции [167, 344, 391—393].
а-Хлоримндонлхлорпды (773), получаемые хлорированием аце-
тамидов (772) или раскрытием цикла в днхлоразиридпнах (776),
при дехлорировании под действием йодида натрия превращаются
в кетенимины (774) с выходами 40—60% (схема 227) [293, 399].
1чпеИ7Иош\’Ы М0ЖН0 также ПОЛУЧИТЬ с более высокими выходами
Lunvnnn L П"'Т/4'-> лег”дРохлоР11Ровапня а-С-алкплированиых ими*
пгипт'/1п °1! ? ’ котоРь,е> в свою очередь, получают прп конт-
Р^'РКмом хлорировании производных ацетамида (772) [293,
b С хорошим выходом кетеипмпиы образуются и при дегнд-
632
«Г0”(772) ф«фоР1
P ? C1 Cl
II PC15 I I NPh
ph2CH-C-NHPh -------> Ph2C-C=NPh Dh / \
era ’«-co,
I V77ti)
pels I_____p2°5- CsHsN__ Nal, MejC0
Cl
I Et3N
Ph2CH—C=NPh -------> Ph2C=C=NPh
в Пи-
1227)
(775)
(774)
Кетениминиевые соли (778), обычно генерируемые in situ можно
выделить в виде относительно устойчивых тетрафторбопатов ппи
действии тетрафторбората серебра на а-хлороенамины (777)
R'\ ZC1
>=<
R2/ \\'Ме2
(777)
AgBF,
-60 °C
(228)
(778)
Для синтеза изоцианатов, изотиоцианатов и карбодиимидов
особенно удобными исходными соединениями являются мочевины
и тиомочевины. Так, например, взаимодействие 1,3-диалкил-
и 1,3-диарилмочевин (779) с фосгеном при комнатной температу-
ре (в случае дпалкилмочевин) или при 150 °C (для диарилмоче-
вин) является общим методом синтеза изоцианатов (782). Особый
интерес этот метод представляет для синтеза соединений, которые
ие удается получить прямым фосгенироваипем соответствующих
аминов [167, 394]. Промежуточными соединениями в этих синте-
зах являются аллофаноилхлориды (780), подвергающиеся само-
произвольному термическому разложению (схема 229). В случае
диалкилмочевии наряду с аллофаноилхлоридом может также об-
разовываться хлорформамидинийхлорид (781); в некоторых слу-
чаях это направление становится основным.
О О СОС1 СГ С1
RNH—С—NHR С0С-> RNH—С—NR + RNH=C—NHR
(779) (780) (781)
I (229)
д или
Д основание
RN=C=O RN=C=NR
(782) <783)
633
< „„псгпыЬопил-З-алкнлмочевин с фосгеном позволяет
Реакция -ар 'с ’фО1111;111зоц11а11аты 1391, 393, 394]. Арнлизотпо.
синтезировать ч и обработкои недорогих и легкодостуд.
Ц“ан^пип11омочевнн соляной кислотой или уксусным ангндрн,
ных 4 шпи иетевых продуктов образуется соль амина или его
S. 'bnoe производное 1389]. Лучшим препаративным общ11м
ом синтеза карбодинмидов является обессеривание 1,3 •диал-
М .in । З-днарилтиомочевин действием солеи тяжелых метал-
ш псутствпи дегидратирующих средств (пли с использованием
"нзеотроиной отгонки образующейся воды) [344, 390]. Обессе-
а, „ > з.1|,замещенных тпомочевни можно осуществить так-
^ГдХтввем хлора или гипохлорита [344, 390] или сильного ос-
новация например гидрида натрия; выходы в последнем случае
составляют 45—75% [390]. Карбодиимиды (783) - конечные
продукты формального дегидратирования 1,3-диалкнл- н 1,3-дн-
арилмочевин под действием различных хлорирующих агентов, на-
пример пеитахлорида фосфора, оксихлорида фосфора, п-толуол-
сульфохлорнда, фосгена [344, 390, 394]. Успешное протекание
процессов этого типа определяется образованием и дегидрохло-
рированием in situ (под действием оснований или в результате тер-
мической обработки) хлорформамидиннйхлоридов (781). Так,
наплучшим .методом синтеза такого важного реагента, как дицик-
лотексплкарбодиимид, является дегидратация 1,3-дициклогексил-
мочевины при комнатной температуре под действием фосгена в
присутствии пиридина пли, предпочтительнее, трпэтиламнна; выхо-
ды достигают 90% [390].
(3) Синтез из соединений с подвижным атомом галогена
Реакции замещения подвижного атома галогена под действи-
ем изоциановой кислоты, цианатов или тиоцианатов металлов в
ряде случаев позволяют осуществить синтез изоцианатов и изо-
тиоцианатов.
Классические методы синтеза ацилизоциаиатов [386, 391, 392]
и сульфонплпзоцианатов [391, 393] — взаимодействие ацил- и
сульфонилхлоридов с цианатом серебра; выходы продуктов,
правда, невысоки (5—8%). Наилучшим реагентом для синтеза
ацилизоциаиатов является легкодоступная изоциановая кислота,
при реакции которой с хлорангидрпдами и ангидридами кислот
в присутствии пиридина выходы ацилизоциаиатов достигают в не-
которых случаях 90% [167, 392, 393]. Алкилгалогепиды тоже мо-
™-.,т«Т'ПаТЬ В РеаК11ИИ замещения подобного рода, однако для
тл& nnf, алкнл,,зоц,|аиатов взаимодействие с изоциановой кисло-
новпйРЛ" П1’ОВОД1'ТЬ в жестК||х условиях. Реакцию с пзонна-
пзоинанатпп1 пл "ельзя рассматривать как общий метод синтеза
авилга1огеп1<пчСК0',ЬКУ °На склоина к полимеризации. Простые
циановой кист "BePT!lbl в реакции с цианатами металлов и пзо-
взанмодействие апш' ЛЯ С|,1|теза арилнзоцпаиатов используют
1 |- диазонневых солей с цианатом калия [386].
634
Один ИЗ наиболее ранних методов с
,ю1ня алкилгалогеиидов с тиоцианатам Н3от1’оцианатпп
С “калия и аммония [389]. Вслед^
^оннаиат-аииоиа при реакциях замещения этоХ™0'0 хаРа«еРа
еси алкнлизотиоцианата и алкилтиоцианата О1!а °б₽аз>10Тся
ц анаты легко изомеризуются в изотиоцНана™\ °ДНаК0 «-‘килтио-
можно осуществить, не выделяя тиоцианата а Л? изом^«ацию
полярном растворителе (например, в ацетонитоХ?Т рсакцню *
" ,н триэтиламнна, или просто увеличив впемй п или в «Рисут-
н"е тиоцианатов подавляется и Пр„ использова Z””- Об',аао’
тяжелого металла, например тиоцианата свинца кПТПп -0Ц1’аната
,|От для превращения ацнлхлоридов в соответствуюпш? Нсполь’
тиоцианаты, хотя и с низким выходом [169, 344 389 адТЛп®°’
синтеза алкоксикарбонилизотиоциапатов было пп’ет^= b Для
вать эквимольную смесь алкил- или арилоксикапбонпЛ^ Нагре'
.“„„«зотроитнагом "Р" 120— iso-с Сез X» ™ '
инертном растворителе-толуоле или ксилоле [169] Встетё™.^
высокой летучести хлорсилана, образующегося Как побочный поо
дукт, ЭТОТ метод особенно целесообразно применять в тех случаях
когда требуется выделить изотиоцианат. ’
(4) Синтез из соединений, содержащих кратные связи
Существует ряд методов, позволяющих создать гетерокумуле-
новые структуры изоцианатов, изотиоцианатов, карбодиимидов
или кетенимпнов на основе превращений кратных связей, уже при-
сутствующих в молекуле. Простейшими процессами этого типа
являются реакции присоединения некоторых производных изоциа-
натов или изотиоцианатов к двойной углерод-углеродиой связи.
Очевидно, что реагентом может быть изоциановая кислота HNCO;
она довольно вяло реагирует с простыми алкенами, одиако легко
присоединяется к двойной связи с повышенной электронной плот-
ностью в виниловых эфирах и дает соответствующие алкилизоциа-
наты [167, 344]. Реакция изоциановой кислоты с алкенами уско-
ряется в присутствии трет-бутил гипохлорита; при этом образуются
Р-хлоралкилизоцианаты [167]. При взаимодействии легкодоступ-
ного псевдогалогена, изоцианата иода INCO, с алкенами, проис-
ходит строго стереоспецифическое транс-присоединение; это общий
метод синтеза |3-иодалкилизоцианатов [386]. Эти соединения мож-
но использовать как исходные вещества для стереоспецнфическо
го синтеза 2-амииоспиртов и 1,2-диаминов [401]. Гетеролитиче
ское присоединение диродана к алкенам также пРоте а„
’’Ране-типу и приводит к смешанным аддуктам а-тиоциа Р
тиоццацатам [402]. „„ /гм
п Повышенная реакционная способность Ф0СГен11М''Н метоДОВ
сииД' обусловливает использование их в р натов
и Je3K гетеР°кумулеиов, особенно изоцианатов, в (784)
Карбодиимпдов. В сильнокислых условиях ф
635
платаются в изоцианаты (785; X = О) [167], однако
ГЛаТ ппРлаРпревраше.1НЯ можно использовать лишь для получе.
такого рода пре Р гидролизу, поскольку реакция
ння 1'30ци/нваТв°0Влн0Уй среде. Изотиоцианаты (785; X = S) более
проводится в вод Рпоэтому „х можно получать взаимоденст-
УСТОИЧ1'В. ршшинов < сульфидом натрия или аммония в водном
ВПе™,?е 265а 284]• выходы изотиоцианатов -80%. Для получе-
н^я увсIbSt лькых’к воде изотиоцианатов реакцию следует про-
водить с пентасульфидом фосфора в инертном растворителе (то-
луол пли хлорбензол) при повышенной температуре. Взаимодей-
ствие N-арилфосгепиминов с азиридинами в присутствии триэтил-
ам, на приводит к имндоилхлоридам (786), которые легко изоме-
пизуются при нагревании или в присутствии кислых катализато-
ров при 25—33°C и образуют карбодиимиды (787) с высокими
выходами [403].
RN=CCl2
(784)
I
RN=C=X
(785)
уС.—% Et3N.
R2Z
СС1(, 5 °C
Cl—C=NAr
R‘\l
C—CH2N=C=NAr
R2/
(786)
(787)
(230)
Склонность изоцианидов вступать в реакции внедрения была
использована для синтеза гетерокумулепов. Так, изоцпапиды окис-
ляются до изоцианатов диметилсульфокспдом в присутствии ка-
талитического количества брома, по-видимому, через неустойчи-
вый промежуточный изоцианодибромпд [167, 344, 386]. При реак-
ции изоцианидов с первичными аминами в присутствии оксида
серебра и каталитического количества хлорида палладия (II) при
комнатной или повышенной температуре образуются с высокими
выходами (72—94%) карбодиимнды (404]. Полагают [404], что
этот метод превосходит все имеющиеся и может быть исиользовап
для синтеза симметричных и несимметричных карбодиимидов, со-
держащих алкильные, арильные п алкенильные заместители.
Внедрением в комплекс изоциаиидов с металлом объясняют об-
разование карбодиимпдов при термической конденсации азидов с
пЗОЦко"НДао^\ .в присутствии неитакарбопила железа (выхо-
ды ьи ьи/о) [405]. Родственным процессом является реакция
кароеиов (генерируемых термолизом дназоалкаиов) с изоцианида-
ми, позволяющая синтезировать кетенимпиы [399].
lx бЫЛ° ПОказа"° ||а Г|Р||Мере образования двойной свя-
У Р д - азот при синтезе азометинов (см. с. 495, циклопри-
.636
соеД1|иенИе с последующим ретроциклопг,
Ыть С успехом использовано для тХТп°еД,,нение“ может
связи. Этот метод особенно пригоден д.1я *0 в МолскУлУ кратной
„.рованных двойных связей; известен уже ° еР°?"я ««еДуХ
Омеров синтезов такого рода [406]. Этот по1 РЯ/ И1’тереснЫУх
ван и в синтезе гетерокумуленов, содержащих °Д-был «спользо
дарод-азот. Так, фосфпнимины (789 зонную связь уг.
изоцианатами в реакцию типа реакции ВиттИгЯ Т « кетенамп и
ИМИНЫ [293, 399] и карбодиимнды [3901 соотвс-т образУЮт кетен-
яие 231; X — R2C или RN). Взаимодействие гЬпЛ™11"0 ^'Рав»е-
„зоцпанатамп или с карбодпимидами пвнКоп^Ф°ра"0в (794) с
(796) [399] (уравнение 232; X = О пли NR). К кете1’чминам
х=с=о
(788)
RN=C=X
(791)
RN=PPh3
(789)
PhN=C=X
(793)
R—N-j-pph
(790)
Ph3P = O
(792)
PhN=C=CPh:
(796)
1.231)
Ph2C=PPh4
(794)
Ph2C~+-PPh:
(795)
Ph3P=X
(797)
(232)
2
классиче-
(5) Перегруппировки
Изоцианаты являются первичными продуктами ряда
«их перегруппировок, в которых происходит ['^''С-^-сдвнг.
перегруппировок Гофмана, Лоссена, Шмидта и Курииуса [4U ].
Однако лишь перегруппировка Курцпуса может рассматриваться
как общий метод синтеза изоцианатов, поскольку условия про е
Деиия остальных процессов таковы, что изоцианат выд . 'л
можно, и он превращается прп сольволизе в амин i.’ ’
В случае перегруппировки КуРЧиуса [94] пзоцнана Т |д^
так как термическое или фотохимическое разлол Пола-
(800) проводится в апротонных условиях (УР3™®1 согласованный
гают, что прн термическом процессе Реал 'у „ механизм
'^реход групп и выброс азота [94]; альте₽ п цесС Промежу-
федполагает участие нитрена и ступенчат р ют протекание
очным образованием триплетного нитрена 1130щ|аиаты [94].
Фотохимического превращения ацплазндов которые
егкой д0Стуи„остью необходимых ацилаз ид ов 1 К11Слот
(7<ш\° П0ЛУЧ||ть питрозированием пеР5 * * 8'1'1^, с аЗОтистоводород-
nnii "лп реакцией ацилгалогеиидов ( ) етсЯ тот факт,
кислотой пли с азидом натрия, п ют с помощью
Г0 С||»тсз изоцианатов чаще всего осущес«'
,nmn Kvpmiyca (помимо реакции аминов с фосгеном)
леР^^н’^б]. э™ лучший лабораторный метод их синтеза.
1 ’ НМОг
RCONHNHj
(798)
RCOC1
(799)
NaN3
Д ИЛИ 11V
RCON3 --------RN=C=O
(800) (801) (81-86%)
(233)
В то же время из-за опасностей, связанных с использованием азо-
тистоводородной кислоты и азида натрия, а также ацплазцда, пе-
пегруппировку Kvpunyca, по крайней мере в ее классическом ви-
ле нельзя, к сожалению, использовать для промышленного произ-
водства изоцианатов. Были предприняты поиски новых вариантов
осуществления перегруппировки Курцпуса с помощью таких реа-
гентов, которые были бы эквивалентны азиду натрия и азотпсто-
водородиой кислоте, ио менее опасны. Заслуживают внимания
такие реагенты как триметилсвлилазпд MesSiNs и трибутилстан-
нилазнд H-BusSnN?,—устойчивые, безопасные в обращении сое-
динения [408, 409]. Их можно легко получить при реакции триме-
тилхлорснлана или трибутнлстанннлхлорида с азидом натрия в
водном растворе. Особым практическим достоинством является их
растворимость в инертных неполярных органических раствори-
телях, например в толуоле. Возможность использования этих
соединений для синтеза изоцианатов показана на примере их
экзотермической реакции с галогенаигидрндами и ангидридами
алифатических и ароматических кислот при комнатной темпера-
туре, образующиеся ацилазиды можно превращать in situ в изо-
цианаты с высоким выходом (>90°/о) [408, 409]. Описано также
применение устойчивого азида тетрабутиламмонпя для осуществ-
ления перегруппировки Курциуса в апротонной среде [410].
Перегруппировка Гофмана включает превращение N-галоген-
амидов в водно-щелочном растворе, и поэтому ее можно использо-
вать лишь для получения гидролитически устойчивых изоцианатов.
С приемлемыми выходами изоцианаты образуются при окислении
первичных амидов тетраацетатом свинца [98]. По аналогии с пе-
регруппировкой Гофмана прн перегруппировке Лоссеиа — прев-
ращении производных гидроксамовых кислот в изоцианаты — так-
же возможно получение лишь устойчивых к гидролизу изоциана-
тов. В некоторых случаях, однако, перегруппировку Лоссена можно
провести в апротонных условиях [167, 396, 411]. Перегруппиров-
ка типа перегруппировки Лоссена была применена также для син-
теза ранее неизвестного 2-тпенилнзоцнаната [412].
изотип1п??п^ЧеСК" 0СУществимых методов синтеза изоцианатов,
коытпп ппкл?’ "аРб°Ди»миДов и кетеииминов основан на рас'
поимев в ппв^4’РаГМе"Таци” ло”упных гетероциклов. Так, иа-
ДИТ пасшеплри nTBi"o трнфенилфосфниа (катализатор) происхо-
Р отепление 1,2,4-дитиазолннтпоиов-З, 5-имино-1,2,4-тиадИ-
638
азолпнов п 5-иминоизотиазолипов, привод,
к тиоацилизотиоцианатам, имидоилкарбодиими^ “ответственно
иМНнам с отличным выходом [4131. Изониаи^ "‘^«лоилкетен-
импды [390] и сульфоннлкарбодинмиды [4151 мпг4 4]; кМ°Ди-
чены путем термического распада 1 2 4 тпИЛ УТ бь,ть ПОЛУ-
1,5-дизамещенных тетразолов и 5-сульфо„’илиРм ан3°7’‘,л 1)0,)°в-3,5,
азолинов, соответственно; выходы продуктов о-Х о’3’4'™^11’
действием лнтпйорганических реагентов нзоксазоти’’'^^' П°Д
тиазолы и триазолы претерпевают раскрытие цикла и пЛСа3°ЛЫ’
ются в кетенпмины [416]. Ц ла и превраща-
8.1.7.2. Реакции изоцианатов, изотиоцианатов
карбодиимидов и кетениминов
Основными реакциями гетерокумуленов, содержащих двойнею
связь углерод азот, являются нуклеофильное присоединение н
циклоприсоединение. В данном разделе большее внимание будет
уделено реакциям нуклеофильного присоединения, в которые всту-
пают изоцианаты, изотиоцианаты, карбодиимиды и кетенпмины
поскольку реакции циклоприсоединения уже были описаны ранее’
(!) Реакции с нуклеофильными реагентами
К числу наиболее важных реакции нуклеофильного присоеди-
нения, которым подвергаются изоцианаты, изотиоцианаты, карбо-
диимиды н кетенпмины, относятся реакции с гидроксисоедииения-
ми. Изоцианаты (802а) [344, 386, 387] легко присоединяют воду
в нейтральной среде (в кислой или основной среде присоединение
идет еще быстрее [417]), образуя неустойчивые карбаминовые
кислоты (804а). Карбаминовые кислоты легко отщепляют диоксид
углерода, давая амины, которые, взаимодействуя с исходным изо-
цианатом, образуют производные мочевины. Производные моче-
вины часто оказываются устойчивыми конечными продуктами ре-
акции простых изоцианатов в водной среде, тогда как реакция
ацилпзоцпапатов [392, 393] и сульфоннлпзоцианатов [391, 418]
с водой останавливается на стадии образования «амина», и про-
дуктами реакции являются первичные карбоксамиды и сульфон-
амиды, соответственно. Так, например, хлорсульфонилизоцнанат
1391, 418] превращается в водной среде в сульфамоилхлорид
NH2SO2C1, являющийся превосходным реагентом для введения
сульфонамидной группы в молекулу. Изотиоцианаты [ > *’
которые, в общем, не так реакционноспособны по отн
‘уклеофнльпым агентам, как изоцианаты, устойчивы к
горячей воды, н для их превращения в производиi _ на.
в кислых или основных условиях требуется пр д • ' •
'Реванпе. Карбодинмидь, (802в) [349, 390], вапрот в,^егко гид
Ратпруются как в кислой, так и в основной ср. ' с пер.
водные мочевины (804в). Взаимодействие каРб°д^
°Кс>'ДОм водорода приводит к перокспкарбоксимндовым
639
, Olli которые являются эффективными реагентами для
(803в; R'= 1 <; ,н0 аренов в соответствующие оксиды
окисления а-,|ке1|ОВ ' ов ^802г) [293, 299] с водой, приводя-
1"9)' '’Тс'™ м 804г) (схема 234; R2 = H), протекает мед-
Щая к каР®"Кч„е10т быстрой гидратации высокореакционноспособ<
солей [400].
OR2
R'NH—С=Х
(803)
пенно в от.
них кетенимпнневых
О
II
R'NH—С—ХН
(804)
R2=»H
OCOR2
I
R'NH—C=X
(805)
R2OH
R2CO2H
NR2RS
II
R’NH-C—X
(808)
R2NHR2
X
R2M II
R1N=C=X ----► R'NH—C—R2
(234)
(802)
R2SH
(809)
S
II R2=H
R'NH-C—XH *-----
(808)
SR2
I
R'NH—C=X
(807)
(a) X = О; (б) X = S (b)X=NR; (r)X = CR2
Реакции изоцианатов, изотиоцианатов, карбодиимпдов и ке-
тениминов с сероводородом и гидросульфид-ионом протекают ана-
логично реакциям с водой и гидрокснд-ионом. Изоцианаты [387]
реагируют с сероводородом или гидросульфид-ионом с образова-
нием неустойчивых тиокарбаминовых кислот (808а), которые, как
и их кислородные аналоги (804а), распадаются на амины и дают
далее производные мочевины. Дитнокарбаминовые кислоты (8076;
R2 = H), образующиеся при действии гидросульфид-иона на изо-
тиоцианаты (8026) [389], можно выделить в виде калиевых со-
лей. Проведение реакции с несимметрично замещенными карбо-
диимидами (802в) [344, 390] позволяет осуществить синтез несим-
метрично дизамещениых тиомочевин (808в).
Присоединение первичных и вторичных спиртов и фенолов к
изоцианатам (802а) с образованием карбаматов (уретанов)
(803а; R2 = Aik или Ат) [167, 344, 386, 387] — один из важней-
ших промышленных процессов [384]. Использование в этой реак-
ции диизоциаиатов создает основу для синтеза полимеров, особен-
но полиуретанов. Скорость присоединения к изоцианатам увели-
чивается под действием кислот, оснований [417] и освещения
too ЯОЧ1 случаех вь1сокореак11иоиноспособиых ацилизоциаиатов
[зад, цуц] и сульфонилизоцианатов [391, 418] реакция протекает
640
экзодерм ичпо. Особый интерес вызывают капЛ '
из хлорсульфоиилизоцианата и третичные Рбматы’ п°лученные
нни температуры выше комнатной отшепляС11НрТОВ’ 11ри ||овыше-
И образуются (с выходами 40—90»/ 1 е,ся диоксид углерода
RNHSO.Cl [421]. Поскольку хлорсуиоиил^ЛЬфаМОИЛХЛО₽^
быть легко удалена, этот процесс можно Я грУппа может
тод превращения третичных спиртов в 1ПРт,(?’а1рИВаТ1' как ме'
позволяющий в отличие от реакции Ритги» г е карбинамины,
сильных кислот. Если реакцию х.торсульйюни™ ЙТЬ воздействия
днть с фенолами при повышенной тХощуре ПР°В0‘
единения по изоцианатной группе идет зямрн/Л вместо пРисо-
образование арилоксисульфонилизоцнаната [422^ °Со г°ГеНа И
»Ф™“" н фенолам»
получения тиокарбаматов (8036; R-’= Д1к или АН 1391 4231
в качестве реагента лучше всего использовать соответствующий
алкоксид или феиоксид. В отличие от простых изотиоцианатов
ацилизотиоцианаты реагируют со спиртами и фенолами экзотер-
мично 11оУ, 393), доказывая тем самым, что они обладают повы-
шенной реакционной способностью по отношению к нуклеофиль-
ной атаке. Карбодиимиды (802в) также ведут себя пассивно в
реакциях со свободными спиртами и фенолами, однако в присут-
ствии солей меди или при использовании алкоксидов и фенокси-
дов реакция идет быстро с образованием соответственно О-алкил-
и О-арилизомочевин (803в; R2 = Aik или Аг) с выходами 50—
100% [344, 390]. Гидрогенолизом О-арилнзомочевины, полученной
из фенола и карбодиимида, можно осуществить восстановление
исходного фенола [424]. Алкоксиды легко присоединяются к ке-
тениминам [293, 399] и кетениминиевым солям [400] и дают в
первом случае имидаты, а во втором — М,О-анетали кетена (812)
с высоким выходом (70—90%) (уравнение 235).
СГ
R1\ t/R3
;c=c=n;
NaOEl R'\ /0Е*
-----у XC=cf (235)
R'\ ZC1
ХС=С/
R2^ ^NR’R4
LR2Z XR<J R2z \WK'
<8I0) (811) (SI 2)
Реакции изоцианатов, изотиоцианатов, карбодиимндов и кетен-
иминов с тиолами и тиофенолами аналогичны их реакциям со
спиртами и фенолами и поэтому заслуживают лишь краткого
Упоминания. Тиолы, тиофенолы или их соли реагируют с просты-
ми изоцианатами [386], ацилизоцианатами [392, 393). сульфо-
«нлизоцианатами [391] (802а) и их тпоаналогамн (8026) |1Ь9,
389, зэз, 423] (см сх 234) и дают соответствующие тиокар-
®маты илн дитиокарбаматы [(807а) пли (8076), R — • ' •
илн RSO2, R2 = Aik пли Аг] обычно с хорошими выходами,
^рбодипмиды (802в) реагируют с тиолами ' т»0Фе”?^Мд|).С и°л„
Аг)°^з”4ием S-алкнл- н S-ариЛизотиомочевнн (80гв, R
*1 1И
641
Гетеоокумулепы, содержащие двойную снизь углерод—азот
„«...то реагируют с кислотами (например, с карбоновыми ИЛ||
AoccbopiiwMii кислотным) 110 связи О-Н и образуют очень реак-
щшшоспоЛбпые смешанные ангидриды, например (805). СоСД11.
нения этого типа, особенно полученные из карбодиимидов и кетен-
пминов можно использовать как реагенты в процессах, связанных
с образованием новых связен, в частности в реакциях, приводящих
к созданию связен С—N и Р-0 в пептидах п фосфатах. Менее
пригодны для этих целен смешанные ангидриды (805а) и (8056),
полученные нз карбоновых кислот и изоцианатов [344, 386, 387,'
417] и изотиоцианатов [344, 389J, поскольку они быстро распада-
ются па амиды или дпспропорционнруют с образованием смеси
ангидридов карбоновых и карбаминовых кислот. В то же время
хорошо известно [390, 425, 426|, что дпциклогексилкарбодппмид
(ДЦП\) способствует образованию пептидов (815) и фосфатов
(820) (например, нуклеотидов). Это легко объяснимо, если до-
пустить промежуточное участие смешанных ангидридов (814) и
(819). Соединения этого типа очень реакционноспособны, и их
обычно невозможно выделить из-за быстро протекающей пере-
группировки, O->-N-ацильной миграции, например (814)->-(817).
Недавно удалось получить устойчивую 0-ацилизомочевину типа
R2 R2
I
COCHNHR1
t
C6HnN—CONHC6Hn
(817)
(236)
R2 OCOCHNHR'
I дцгк I
R'NHCHCOjH --------> C6HnN=C—NHC0H„ —>
(813) (814)
R3
I
H2X'C1ICO2R4
R2 R3
R'NHCHCOX’HCHaW + C6H,,NHCONHC6Hii
(815) (816)
О 0 NCcH,,
и
V. ДЦГК II II R2OH
R'O—Р—ОН--------> R‘O—Р—О—С—NHCgHh -----------►
I I
ОН он
(818) (819)
О
—> R'O—Р—OR2 4- (816) (237)
I
ОН
(820)
642
(814) [4271, ее устойчивость приписывают 2-кпнаиг ,
стельно двойной связи углерод - азот, соответХюшиГ/ГГ
мер^подвергается самопроизвольному O-^N-nepXyацилыюй
'РУВозможность такой миграции снижает ценность ДЦГК как
конденсирующего агента при образовании пептидной и фосфо
эфнрнои связей; кроме того, продукт реакции обычно трудно от
делить от сопутствующей емудициклогексилмочевины Решитьобе
эти проблемы удается, используя связанный с полимерным носи-
телем несимметрично Дпзамещенный карбодиимид [428]. В тако-
го рода субстратах электронная плотность на атомах азота раз-
лична, что затрудняет 0->N-ацильную миграцию, а использова-
ние полимерного носителя исключает возможность загрязнения
продукта реакции мочевиной. Образованием реакционноспособных
интермедиатов типа (805в) можно также объяснить роль ДЦГК
при получении сложных эфиров карбоновых кислот (выход >90%)
[390] и простых эфиров фенолов [390], а также прн эффективном
синтезе диацилпероксидов из карбоновых кислот и пероксида во-
дорода [390]. Карбодпимиды на полимерном носителе можно с
успехом использовать для мягкой дегидратации кислот в ангид-
риды [429]. Как и следовало ожидать, тетрафторборат \-метил-
М.М'-Ди-трег-бутплкарбодппмпдия [rp<?r-BuN=C=N+(Me)Bu-rpeT
-bf4] оказался еще более активным реагентом, чем ДЦГК, в ре-
акциях образования связей С—N и Р—О [430]. Высокореакцнон-
носпособные смешанные ангидриды могут образовываться не
только из карбоднимидов, но и из кетенимвнов. Особое значение
приобретают кетеннмины, которые ооразуются in situ путем рас-
крытия цикла в нзоксазолиевых солях и могут быть использованы
в синтезе пептидов [426,431] и нуклеотидов [399].
ДЦГК способствует также образованию нлида (824) прн реак-
ции дцметилсульфоксида
„ . • Me,SO
C6H11N=C=NC6H11
(821)
с гидроксисоедпнениямн; эта стадия
O“SMe*
I
C6H„N=C-NHC6Hn
(822)
кон .
-(CjHnNHljCO r
21*
« r эпегантпом методе окисления гидроКСн
является Реп'аЮ“1еП ыВ „ кетоны (825) (метод Пфпцпера - Мо?
едниепп»; в альдегид удоб|П>|й мс10д введения метилтио.
фатта) H32J .ппыв орто-положенпе фенолов также включает ста-
метильной гр)ппы к ( )433] Приме1.1енис карбодиимцдОВ
Д’"0 об1'аЗОВ полимерным носителем, для окисления по ПфИцНе:
СВЯЗаХ**тгтУ позволяет устранить загрязнение продукта реак.
ру-ЛЮфФагту ев)1Ны; карбонильные соединения получа-
Ц"" "Ротлн>пым выходом (70—90%) [434]. Вместо карбодиими.
доТвСЯв окислении по Пфицнеру - Моффатту можно использовать
КСТНет’н|1чего9удивптельного в том, что изоцианаты, изотиоциа-
паты карбодиимнды и кетенимины реагируют с аминосоедине-
нням’п (аммиаком, первичными и вторичными аминами, гидро-
кетаминами гидразинами) по типу 1,2-нуклеофильного присоеди-
нения. Эти реакции обычно протекают с хорошим выходом. Так,
прп взаимодействии аммиака или аминов (первичных и вторич-
ных) с простыми изоцианатами (802а) [167, 344, 386, 387, 411|
образуются производные мочевины (806а); аналогично ведут себя
ашглнзоцианаты [392, 393] и сульфоннлизоцианаты [391]. По-
добным же образом, но более медленно, реагируют изотиоциана-
ты (8026), образуя производные тиомочевины (8066), а также
ацнлизотпоцнанаты [169, 393]. Карбодиимиды (802в) гладко при-
соединяют аммиак и первичные и вторичные амины с образова-
нием гуанидинов (806в) с высоким выходом [344, 390]. Продук-
тами присоединения аминов к кетениминам (802г) являются ами-
дины (806г) [293, 399]. Особенно высокую активность в реак-
циях этого типа проявляют кетениминиевые соли [400], дающие
с очень высоким выходом амидины.
Гетерокумулены с двойной связью углерод—азот реагируют
по типу, 1,2-прнсоединення и с нуклеофилами, у которых нуклео-
фильный центр сосредоточен на атоме углерода. Обычно эти
реакции проходят с цпаипд-ионом, металлорганическимн соедине-
ниями, с карбанионами, полученными из соединений с активной
°о’ их используют для превращения изоциана-
доч) 67’а44, 386, 3871’ изотиоцнанатов [169, 344, 389, 391, 392,
в2солепжпбп,ДИИжИД0В [344’ 3901 и кетениминов [399] (802а-г)
ны и енямииы ФУнкцнональные группы амиды, тиоамиды, амиди-
ны и енамины, соответственно (809а—г).
(2) Реакции циклоприсоединения
держащие лвой|тп°г0ВЬ1ЯСН'1ТЬ' встУпают ли гетерокумулены, co-
ll + 2] нЦ1клоприУсоедин3 * *ентЛеРОД7а3°Т (азакУмУлены)- в Реакц““
первичные продукты т-J ” С каРбенами и нитренами, поскольку
Шим превращениям IRR Иадс>еаКпЦИЙ легк0 подвергаются дальней-
Д°в с диазосоединениями и"’. 4351' Взаимодействие карбодинми-
и азидами, приводящее соответственн
К азиридинам и диазиридииам, можно Лоп
как пример |1 + 2)-Циклоприсоединения кЯпТ'?ЬН0 Рассматриватъ
кумуленовой системе [436]. карбена и нитрена к аза-
13 отличие от процессов [l + 21-nuvn™
[2+ 2]-, [3 + 2]- и [4 + 21'ци1о1рХдХ^Д?еНИЯ РеЭКиии
изотиоцианатам, карбодннмидам и кетенимии, изоцианатам,
[88, 399, 406]. Циклоприсоединение ХУмЛ нОВХОРТ
и С=О уже рассматривалось в разделе Уп\е °В К язям C==N
азометинам (см. с. 495 н 521); участие азакумг^Т0 г 1рОСТЫМ
в реакциях [4 + 2]-циклоприсоединения, в который“эта Феункции
выступала как 4я-компонент (гетеродиен) тчкХ система
лось выше (см с. 530). Реакции [3 + 2]-циклоприеоедине°ниТаза’
кумуленов описаны в разделах, посвященных нитронам (см с 621)
и азометиниминам и азометин-илидам (см с 628) Таким б
зом, единственный тип циклоприсоединения азакумупенов кото'
рый заслуживает здесь подробного разбора, это' [2+ 21’никло
присоединение по связи С=С и [4 + 2]-циклоприсоединение в ко-
тором азакумуленовая функция выступает в роли 2л-компонента
(диенофила).
К числу наиболее тщательно исследованных реакций [2 + 2]-
циклоприсоединения относятся реакции изоцианатов с соедине-
ниями с двойной углерод-углеродной связью. Простые алкены
не вступают в реакцию термического [2 + 2]-циклоприсоедине-
ния с изоцианатами, хотя недавно было описано фотохимиче-
ское [2 + 2]-циклоприсоединение фенилизоцианата к стиль-
бену [437]. Изоцианаты легко вступают в реакцию термиче-
ского [2 + 2]-циклоприсоединения с алкенами с повышенной
электронной плотностью (енамины, виниловые эфиры), что, таким
образом, можно рассматривать как метод синтеза р-лактамов (831)
(уравнение 239; R2 = NR2 или OR, R3 = Aik или Аг) [88, 114,
167, 170, 344]. Присутствие в молекуле изонианата электроноак-
цепторного заместителя у атома азота облегчает [2 +^(-цикло-
присоединение. Так, обладающий высокой реакционной способ-
ностью хлорсульфонилизоцпанат взаимодействует не только с
простыми алкенами (см. например, уравнение 239; =
R3 = SO2C1) 188, 114, 391, 418, 438], но и с 1,3-диенами [88 114
391, 418, 438—440], алленами ]П4, 438] и кетенами [391, 418]
по типу [2 + 2] -циклоприсоединения.
R1
3 . СИ2 . p'/Tl (239)
НВ,1ДО
(848) (829)
Присоединение, хлорсульфонилизоциаиата к двойные'
Р°Д—углерод происходит с высокой Рег’’°' р ]х данных
иостыо [441, 442]. Предположенный на основании этих д
„г довитый термически разрешенный [л2а + n2s]-процесс
ичоГ не согласуется е наблюдаемыми параметрами актнвацт,.
я[ „кжС влиянием полярных заместителе,. и растворителей на
рго скТоомь [442]. По-впдимому, реализуется либо двухстадий,
механизм, включающий образование цвиттер-ионного интер-
(830) 1438], либо согласованный процесс через высокопо-
• яр .^‘переходное состояние [442]. В пользу первого представле-
ния говорило успешное выделение [443] и «улавливание» [444]
дпполярного интермедиата, тогда как образование Р-лактамов
в качестве первичных продуктов [445] свидетельствует в пользу
второго. Таким образом, вопрос о механизме присоединения
хлорсульфоиилизоцианата к двойным связям углерод углерод
остается открытым [440]. При термическом [22]-циклоприсое-
динении карбодиимндов к кетенам, приводящем к 4-пмнноазетпди-
нонам-2 [446], удалось зафиксировать образование цвиттер-ион-'
ного интермедиата, что подтверждает ступенчатый механизм.
Те же продукты образуются н при термическом [2 -ф 2] -цикло-
присоединении изоцианатов к кетеппминам [447].
Примеры реакции [4 ф- 2]-циклоприсоединения, в которых аза-
кумулены выступают в роли 2л-компоиента, приведены ниже
(уравнения 240—242). Присоединение анилнзотиоцианатов (833)
к 1,3-дпенам (832) приводит к производным тетрагидропирндина
(834) [393]. При реакции кетенов (135) с карбодинмидами (836)
образуются 1,3-оксазпноны (837) [4066], а в результате электро-
циклизацпп ------- ---------------------
ноны (839;
[449].
гетеродпенилизоцианатов (838) получают 1,3-оксази-
X = CR) [448] н 1,3,5-оксадпазиноны (839; X = N)
646
8.2. НИТРИЛЫ И ИЗОЦИАНИДЫ
8-2.1. ВВЕДЕНИЕ
В основе структуры нитрилов RCN и и™,,,.
тройная связь углерод -азот, относящаяся К ч"ДДГ RNC лежит
функциональных групп в органической химии (Ат *лассических
ченпе тройном связи углерод-азот как Лункин™ я. Ь-Шое зна'
связано с легкостью ее введения в молекулу Ц" р< и ГРУ"ПЬ1
Но высокой реакционной способностью, об'усювтен "^•'1ючитель-
ним сочетанием ненасыщенности, поляризуемости и н₽У«иКаЛЬ-
чувствительности к стерпческвм факторам. В данном обзоре^сдТ
дана попытка возможно более полно описать соединения содержа'
щне тронную связь углерод-азот. В то же время требований к
объему обзора, с одной стороны, и разнообразие аспектов совпе
меннои химии нитрилов и изоцианидов [451], с другой, заставили
исключить из рассмотрения некоторые вопросы, связанные глав-
ным образом с реакционной способностью свободных радикалов
[452] и фотохимией [453] циано- и изоцианосоединений.
8.2.2. НИТРИЛЫ
8.2.2.1. Общие методы получения алифатических,
алициклических и ароматических нитрилов [309, 454—456]
(1) Реакции замещения
Один из классических методов синтеза алифатических нитри-
лов состоит в реакции нуклеофильного замещения алкилгалогени-
да [309, 454] пли тозилата [454, 455] цпанпд-поном. Реакцию с
Цианидом щелочного металла проводят в водно-спиртовой среде
при комнатной или повышенной температуре. Вследствие амби-
дентного характера цианпд-пона часто в небольшом количестве
образуется и соответствующий изоцианид, который становится
основным продуктом, если в качестве источника цианид-нона
используется цианид тяжелого металла (например, цианид сереб-
ра) (см. разд. 8.2.5.1). Образование изоцианпдов подавляется,
если реакцию проводить при повышенно!! температуре в подходя
Щем для этой цели растворителе, например в этиленгликоле, п
скольку в этих условиях происходит необратимая перегруппиро
«а изоцианида в нитрил [457, 458]. Как и следовало ожидать для
гипичного процесса нуклеофильного замещения, порядок р
ной способности алкплгалогенидов по отношению к ш,а ' ы„’ен.
ДУЮщий: F < Cl < Br < I, фториды совершенно инертны. ^ме^
ко повышенной реакционной способностью а.ткнл! Д' юЩег0
ьясппть каталитическое действие подпда натр, , рсакш1я
аимодействие алкил хлоридов с цианид-ионом.
«голого» цианид-
нитрила в мягких
455] Первичные алкилгалогениды даЮт
1|Дет медленно ,{01Ь образованно нитрилов ,13 вторц,, ,‘ И’
большие выхо-W 1 1 ,а;оге„,)доВ не столь эффективно „з-3;|
или гретпчиг х .лк алкенов. Если замещение циаН11
курени».। фотонных дпполярпых растворителях, таких,
проводить р а'Р ,ет11лформамнд или гексаметилгР11аМ||„0К
д.|Метилс>Л1>фоки.д, температуру и »родолжптелЬиЛ
фосфат, то дл к 110ВЬ1Шению выхода нитрила д‘„
реакции, ,п“ "Р.Т||151|0СТП метода синтеза нитрилов путем 3a
повышения И б предпринят поиск более эффект *
*еШТ'г тов и еловой реакции, в большей мере*™
"“юшп протекаю.» нуклеофильного замещения. В качестве
S^oiiiero агента был использован цианид тетраэтиламмОЙ|)я
[4591 Препм'шеством этого реагента по сравнению с Цианидами
„.елочных металлов является растворимость в органических рас.
творнтелях, например, в дохлорметане, ацетонитриле, диметил-
гслыЬокспде, превращение алкилгалогенндов в нитрилы происхо-
дит быстро (30—50 мин) при температуре <50 С с выходами
50—90%. Для улучшения условий, способствующих нуклеофиль-
ному замещению галогенидов пианпд-ионом применяют катализ
фазового переноса [460] или используют циаипд-пон в виде ком-
плекса с 18-крауном-6 [461] или аналогичным азамакроцякличе-
скнм полиэфиром [462] (так называемый «голый» цнанпд-ион).
В обоих случаях, особенно прн использовании
нона, наблюдалось сильное повышение выхода
условиях проведения реакции.
Ацплцианиды [454] можно легко получить
хлоридов пли -бромидов цианидом меди(1). Реакционную смесь
нагревают в ацетонитриле при 80 °C или в эфире при 35 °C
в присутствии йодида лития как катализатора [463]. Существует
еще один мягкий метод синтеза ацилциаиидов [454], заключаю-
щийся во взаимодействии ацилгалогенпдов с безводным цианис-
тым водородом в инертном растворителе в присутствии основания,
например пиридина. В отличие от ацилгалогенпдов сульфоннлга-
логениды не реагируют с цианид-ноном с образованием сульфо-
нплцианидов [464]; их можно получить реакцией сульфинатов
с хлорцпаном [464].
1„РнХаКЦИЯ> °®Ратная замещению галогена цианид-ионом,— заме-
капбянппня,,?'3 В галогенцианах (предпочтительно в хлорциане)
uiianornvnn.,' м°жет быть использована для введения
с активной мрти ' Кароанионы, образующиеся из соединении
дпиианом и даютЛекак°п ГруПпой’ Реа™РУЮт с хлорцианом пл»
ные. Так наппимпп К пРавило, соответствующие цнанопроизвод
малонового э<Ьипдиэ’Гилцианомалонат был получен из иатриЙ-
реакцип енаминов ХЛ0РЦиана. Использование в аналогично
ствить синтез В-кето„итпСЛеДу1?Ш'ий гиДРолпз позволяют осуше-
648 ₽ етонитРилов (уравнение 243) [454].
обработкой ацил-
CN
(243)
(840)
<>(сн,)я 0==A
2. 14,0’ J \Р<СН2),
(841)
Общий, казалось бы, метод синтеза алифатические и,
цодействнем реактивов Гриньяра с хлопииян™ н11тР‘Мов взаи-
производным, полученным из нерв?^ °РЫХ X
дольку реактивы Гриньяра со -орич^ми^^Хми^ П°’
ными радикалами склонны распадаться с образовав а;1киль’
[4541. Однако л„ш„
нитрилы НЗ реактивов Гриньяра с первичным и вторичным ал
кильным радикалом [454]. ричным ал-
Неактпвированные арилгалогениды в обычных условиях не
реагируют с цианидами металлов, однако замещение удается ocv
ществить, и часто с отличным выходом, при нагревании галогенида
с цианидом медн при высокой температуре (>200°С) в поисут-
ствии пиридина [454, 455]. Применение этих методов ограничено
из-за использования жестких условий. Гораздо большее значе-
ние имеют реакции замещения неактивированных арилбромидов
и -иодидов под действием цианид-иона в присутствии металличе-
ских катализаторов. Так, ароматические нитрилы удается полу-
чить с выходами 80—90% в мягких условиях при реакции арил-
бромидов или -иодидов с цианидом натрия или калия в присут-
ствии ацетата палладия [465] нли комплексов палладия [466]
или никеля [467] с трпфеннлфосфнном. Арнлцианнды. можно так-
же получить с выходами от 50 до 90% при кипячении арилтал-
лий(III)ацетатов нлн -перхлоратов с цианидом медн в ацетонит-
риле илн пиридине в течение 5—10 ч. Полагают, что эти превра-
щения протекают по ионному, а не по свободнорадикальному
механизму [468]. Интересно, что при аналогичной реакции арил-
таллпй(III)бис(трнфторацетатов) с избытком цианида калия в
водном растворе образуется комплексный ион, для превращения
которого в соответствующий нитрил (выходы 27—80%) требуется
облучение; по-видимому, это радикальный процесс [469]. Особен-
ностью этих методов синтеза нитрилов является то, что цпанид-
пон замещает таллий, ориентацию которого (и, следовательно, по-
ложение входящей цпаногруппы) можно контролировать [ J.
Стандартная методика синтеза ароматических нитрилов по реак
чип Зандмсйера состоит во взаимодействии солен арендна
с комплексом цпапнда калня и цианида меАИ;.Лц1а,гЛЮТ2Л] "
акция протекает по радикальному механизму риу, • Г -
Реактивы Гриньяра, полученные из арплгалоге; и , ше,
но соединениям с первичным алкильным радика. ( •
Р^Пфуют с хлорцианом и дают ароматические нитрн.
<49
„ки^гются также (правда, с низким выходом) при рс-
llnipii.'iiJ образ)к поенов с дициапом пли бромцнапом в
акции активном 1 в\ловнЯх реакции Фриделя -
к®с-™ 1451' 455] ‘ Разновидностью этого метода (синтез Губе-
, Ф Лт) является обработка активированных аренов (прос-
lia-Jh, ьd/еноты) трпхлорацетопптрнлом в присутствии хлори-
зГа«м“ниГи хлористого водорода, образующиеся трихлорме-
тнлкетимпны при обработке основанием дают нитрилы.
Авены можно н непосредственно превратить в ароматические
ннтрюы с помощью фотоиидуинрованного замещения нптрогрупп,
яткокспгрупп и водорода циавпд-ноном [4о4, 455, 470]. Исследо-
ван механизм этих процессов [471]. Они часто протекают с вы-
соким выходом, особенно если замещению подвергается нитро-
пли алкокснгруппа [472]. Фотозамещение водорода цпанид-ионом
происходит нецзбирательно и не очень эффективно, хотя исполь-
зование цнаннд-поиа, солюбилизированного в ацетонитриле до-
бавлением 18-кра\’иа-6, повышает выход нитрила [473]. Арома-
тические нитрилы образуются также и при электролизе аренов
в присутствии цпаппд-попа, хотя и в этом случае выходы непо-
стоянны п часто образуются смеси изомеров [454, 455, 470, 474],
(2) Реакции присоединения
Синтез некоторых цианосоедпнений часто можно осуществить
путем присоединения цианистого водорода или иона цианида к
кратным связям. Простые алкены обычно инертны в такого рода
реакциях даже в жестких условиях, однако присоединение циа-
нистого водорода к напряженной двойной связи в норборнене
протекает гладко в присутствии катализатора и приводит к соот-
ветствующему нитрилу с хорошим выходом [455]. Наличие в мо-
лекуле алкена как электроиодопориых, так и электроноакцептор-
ных заместителей способствует присоединению цианистого водо-
pi^- и111 Ф,аиНд‘,юна (454]. Так, простые виниловые эфиры (842,
R1 = п, R2 = OR) легко присоединяют цианистый водород в при-
сутствии основного катализатора (цианид щелочного металла пли
п'ФВДин) " дают соответствующие а-цианоаддукты (843; R1 = Н,
1849. DI!_ D> ь присоединение цианистого водорода к енаминам
' ’ < К 1хМе2) протекает настолько легко, что не требует
илтнай Лм’ каталнзатоРа. реакция завершается за 12 ч при ком-
цванистого'вопопоп- *ураВ11еНие 244>- Сопряженное присоединение
электионоакпептАп’411 1ЛН Ц||анил'иона к алкенам, активированным
ацильной нитпо Рп^ж замс‘стителями (алкоксикарбоппльной,
Р-инаноа^дукТам’(844)ФИ54ГНег ИЛН цианогРУГ111°"). приводит к
безводный цпаиистнй ваД54^’ можно осуществить, используя
еще проще обпабпт!.-А-ОЛОРОД в ||Р1|СУТСТВ1Ш Цианида калия, или
сусной кислоте. И цпанпдом "алия в водном спирте или ук-
650
CN и т.
R'\ (a)
-С СНз * с—-Г-ц
т/1 СНг
CN
(843)
и (или) R2 = OR или NR2; (б) Ri
ед R'\
CH-CH2CN (244J
(842> (844)
И (или) R. = г;зд COR,
В перечисленных условиях, однако, присоединению цИяиил
путствует большое число побочных реакций: гидролиз продукта'
образование димера и присоединение по Михаэлю продукта пе
аКшш к исходному ненасыщенному субстрату; реакция хапайте'
р||3уется низкой избирательностью [475]. Этих недостатков можно
избежать, если проводить присоединение цианистого водорода в
присутствии трпалкплалюминня как катализатора [454 4751
Однако триалкилпроизводные алюминия — пирофорные и потому
опасные соединения. Более приемлемым решением является ис-
пользование «голого» цианид-пона (то есть комплекса цианида
щелочного металла с краун-эфиром) вместе с ацетонциангидрином
Применение такого реагента позволяет осуществить стереоспеци-
фическое присоединение цнанид-иона к <х,р-ненасыщенным карбо-
нильным соединениям с высоким выходом (83—86%) [475].
Наиболее известным процессом, приводящим к цианосоедине-
ниям и основанным на присоединения цианистого водорода к крат-
ной связи, является, по-видимому, образование циангидринов
[309, 454, 476]. Он состоит в нуклеофильном присоединении циа-
нистого водорода к карбонильной группе альдегидов н кетонов.
Обычно эту реакцию проводят, обрабатывая карбонильное соеди-
нение (или его производное, например аддукт с бисульфитом)
цианидом натрия или калия в присутствии водного раствора сер-
ной, соляной или уксусной кислоты; циангидрины получают, как
правило, с высоким выходом (~80%). Для синтеза циангидринов
можно использовать также безводный цианистый водород в при-
сутствии основного катализатора (цианид, гидроксид или карбо-
нат щелочного металла). Присоединение цианистого водорода к
карбонильной группе — процесс обратимый, в случае альдегидов,
(845; R2 = Н) равновесие смещено в сторону образования продук-
та (847), тогда как для днарилкетонов оно смещено влево, т. е.
Циангидрины днарилкетонов настолько неустойчивы, что выделить
"X нельзя. При реакции n-бензохинонов с три.метнлсилилипанидам
8 присутствии цнанид-иона происходит эффективное пр р
лишь одной из двух карбонильных групп; образуют шея -р -
метилсилилциангидрнн гладко регенерирует кар ош. ж..
Не||,|е при обработке фторидом серебра в водном «трапщроф^
Ране при 25 °C [477]. Такой прием впервые позволил ос Щ
«зопрательную защиту одной из двух карбоннльны.
н°не, что открывает путь к еще более широком) испо.
инонов в синтетической практике.
651
R1'
R4 /О’ Н*
r:Z
(843)
(846)
R\ ZCN
/ОН R3R4NH
с/ -------------1
R-' XCN
(847)
(245)
(848)
и при
присут-
Образование циангидринов — промежуточная стадия
peaS «гидов н кетонов с цианистым водородом в
Е аммиака или первичных и вторичных аминов, приводящей
v „.ямпнонцтрнлам (848), классическим интермедиатам при син-
тезе а-ампнокислот (синтез Штрсккера) [454, 478]. Свободный
цианистый водород в синтезе Штреккера можно заменить циа-
нидом аммония или сочетанием хлорида аммония с цианидом
калия Еше более удобный вариант этого синтеза (реакция Кнёве-
нагеля — Бухерера) состоит в приготовлении in situ бисульфитно-
го производного карбонильного соединения и его непосредствен-
ной реакции с цианидом калия в присутствии соли аммония или
амина.
Цианистый водород и цпаннд-ион легко присоединяются так-
же и по двойной связи углерод—азот в азометинах (шиффовы
основания, оксимы, гидразоны), что приводит к а-амннированным
нитрилам с хорошими выходами [454]. В качестве реагента мож-
но использовать безводный цианистый водород или цианид нат-
рия в сочетании с бисульфитом натрия. Модификацией этого
метода [479] является обработка основания Шиффа или оксима
трпметилсплилцпанпдом в присутствии в качестве катализатора
кислоты Льюиса (например, AlCl3,ZnI2), последующий гидролиз
триметилсилпльного производного (850) в нейтральных условиях
приводит к а-аминоиитрнлам (851) (схема 246; X = Н, Aik, Аг или
ОН). Легкость присоединения цианистого водорода и цианпд-иона
по двойной связи углерод—азот была использована в изящных
методах превращения альдегидов и кетонов в гомологичные нитри-
лы. В одном нз них карбонильное соединение превращают в ал-
коксикарбонилгндразои (849, X = NHCO2Me), который, в свою
очередь, вводят в реакцию с цианистым водородом или цианидом
гч ПРИСУТСТВНИ хлорида аммония и получают цианоаддукг
(SM). Окисление этого аддукта бромом дает азосоединение (855),
пепятипяТ КОТ°Р°ГО П°Д Действием основания при низких тем-
выхоппРм гомологичному нитрилу (853) с высоким
щение апенгмпьЖлн° Более УД°биым способом является превра-
X = NHSOA’r\<n илг,|дРазона карбонильного соединения (849;
652
SiMes
MegSICN.
AJC/з или ZnCI2 ” у
X = U. OH ,C=NX
RzZ
HCN или
NH,C1.
KCN
x=nhco2m^
(850)
H2O
(849)
R,\
Rz/|~NHNHCOiMe
CN
(854)
X-NHSOjA,
KCN,
McOH или
AcOH
R‘\
XC—NHX
Rz/ I
K CN
(851) .
R'\
C—NHNHSOiAr
(852)
Brg
NaHCOj
R'\
C—N=N’CO2Me
R-/1
CN (246)
(855)
NaOMe. MeOH
и -C
R‘\
VhCN
R2/
(853)
Особым случаем образования нитрилов путем присоединения
цианида по двойной связи углерод —азот является реакция Рейс-
серта [454, 483, 484]. Она состоит во взаимодействии л-дефицнт-
ных азагетероароматических соединений (например, хинолина или
изохинолина, но не пиридина) с цианидом калия в присутствии
ацилгалогенида в двухфазной системе вода — дихлорметан. Обра-
зующийся N-ацил-а-цианоаддукт (857) обработкой пентахлоридом
фосфора можно превратить в ароматическое соединение, цнано-
замещенный гетероцикл (858) (уравнение 247); этот же аддукт
можно использовать и в других синтетических превращениях (см.
ниже). Гетерогенная среда, используемая в реакции Рейссерта,
часто обусловливает низкую степень конверсии. Выходы, однако,
во многих случаях можно резко повысить (от 20 до 70 ^), если
использовать катализатор фазового переноса, например проводить
Реакцию гетероциклического соединения с ацнлгалогенидом и
Цианидом калия в системе дихлорметан — вода в присутстви
хлорида бензилтриметил аммония.
P11COCI. KCN
С11,С12. Н2О
(856)
(j) Реакции отщепяения
и.„|б0.1ее распространенные .. зачастую наиболее удобные Ме-
ЛК создания тронной связи углерод-азот включают реакции
ею я К ним относится классически., метод дегидратации
” X шоа, который широко использовался для синтеза нитри-
7102 300 454 -186] Непосредственную дегидратацию альд-
(>ксим >В И нитрилы ча.це всего проводят действием таких реаген-
тов как пентаокод фосфора, триэтилфосфат или хлорангидридь.
(например, тнопилхлорил, ацетнлхлорид, арепсульфоннл-
х зори ты Дегидратацию под действием хлорангпдрндов удобнее
всего’проводить в диметплформампде. Одним из наиболее прос-
тых и эффективных методов превращения альдоксимов в нитрилы
явтяется нагревание при —100°C с уксусным ангидридом в при-
сутствии основания, например пиридина [454, 455]. Был изучен
ряд новых реагентов, позволяющих провести эффективную деги-
дратацию оксимов в мягких условиях. Так, при использовании
трпфторуксусного ангидрида превращение альдоксимов в нитрилы
идете высоким выходом (>90%) в мягких условиях [487, 488].
Ценным реагентом оказался и фенилизоцианат; он вызывает де-
гидратацию п декарбоксилирование а-гидроксиминокислот в ни-
трилы с высоким выходом (>90%), реакция протекает за 15 мин в
бензоле при комнатной температуре [489]. Крометого, для дегид-
ратации алкил- и арилальдокепмов в мягких условиях рекомен-
дуют также н-хлорфенилхлортионоформиат /;-CIC6H4OC( = S)OCl
[455] и фенплхлорсульфит PliOSOCl [490]; при действии
последнего на бензольные растворы альдоксимов в присутствии
триэтнлампна при комнатной температуре выходы нитрилов со-
ставляют 90—98%. Высокой степени превращения альдоксимов в
нитрилы в мягких условиях можно также добиться при использо-
вании трнфенплфосфпна, связанного с полимерным носителем, в
присутствия четыреххлористого углерода [491], цнапурхлорида и
пиридина прн комнатной температуре (эта методика особенно
пригодна для кислотолабильных субстратов) [492] или фосфо-
ннтрплхлорида в присутствии триэтнламина при комнатной тем-
пературе [493]. Альдоксимы можно просто и с хорошим выходом
превратить в нитрилы кипячением с ортоэфирами в присутствии
кислоты [494]. Превращение альдоксимов в нитрилы в основных
условиях можно осуществить обработкой дихлоркарбеном, гене-
рируемым в условиях катализа фазового переноса [456]. Нитри-
™ обРазуются И3 альД°КСИМ0В с очень хорошими выходами в
пеагентов°какШтпН°п НеНТ')аЛЬ11Ь,Х ровнях прн действии таких
I 1' дикап’бош п?ПХ °Рацет°Н11тР11л> Диипклогексплкарбодпимид и
1,1 -Дикарбонплдпнмпдазол [456]
ронВнепТоедс\СвХРеХ1Х ВЫ'"е методах той “ас™цей, 113 ™™-
окенм. В иекотооых р1™3^ТСЯ нитРпл, является О-замещеннып
выделять н затем вволитьЯв ТаКИе “нтеРыелиа™ целесообразно
затем вводить в реакцию отщепления. К этой кате-
654
горн» методов синтеза нитрилов относится „
мов [455[ и катализируемый основании ‘/Р°Лнз 0-ацилокси-
рнэтиламнн) распад 0-2,4-динитрофениловихИДРлОКСИД нат₽ия,
(которые можно легко получить из соотвеХ, Эфиров оксимов
. 0-2,4-ДННнтрофенилгндроксиламннаЬ “ ТСТ8Иощего альдегида
высоки (81—94%) [495]. Возможен! и дРЖ“ h“t₽«™b весьма
трплов, который состоит В образовании „ °ДХод " синтезУ ни-
мов ш situ. Так, нитрилы получают нагоев™ раТаЦии окси’
гидрохлоридом гидроксиламнна и формиатом и альдеги«>в с
рия в муравьиной пли уксусной кислоте [454 4m3atTaTW; иат-
иием с гидрохлоридом гидроксиламнна в диметн-,1 ?И иагрева'
средний метод, несмотря иа простоту, применив Р^ЭМИДе; П0‘
алифатических, алициклических и ароматических синтеза
„Л, .ь,хо11И. |.%| к
г||дов без выделения промежуточных продуктов относятся Л
реакция альдегидов с O.N-бис(трифторацетил)гидрокс, ламином
в пиридине [454, 455], с гидрокснламнн-О-сульфокнслотой 49/“
В водной среде при комнатной температуре илн при 65'С или
реакция с карбодпнмндамп в присутствии солей меди (11) и три.
этнламнна при комнатной температуре в течение 1—3 ч; послед-
ний метод позволяет получать нитрилы самой разнообразной
структуры с выходами >90% [498].
Структура кетоксимов обычно не позволяет осуществить обра-
зование тройной связи углерод-азот за счет реакции отщепле-
ния. Однако наличие a-заместителей, таких, как карбонильная,
амино-, гидрокси- пли алкоксигруппа, так называемые электроно-
донорные группы [499], способствует их распаду по Бекману [74.
309, 454, 499] с образованием нитрилов при нагревании или в
присутствии таких агентов, как пентахлорпд фосфора, тнонилхло-
рпд, полнфосфорная кислота. Нередко таким способом удается
получить в мягких условиях нитрилы с высоким выходом; пример
такого превращения приведен в уравнении 248 [499].
NOCOMe
80%-ный водный Е1ОН
(100%)
К методам .синтеза нитрилов, основанных на д®Г11ДраДаЦИв
альдоксимов, тесно примыкают методы раскрыт,
С-З-незамеще.шых изоксазолах [454, 500 501) и
золах под действием оснований [486, 502], в Р_ аоляты с хо-
ооразуются соответственно цианоеноляты н w Ф ксазолы ц
РОШИМИ выходами. Поскольку «-незамещенныерас-
1.2-бензпзоксазолы легко можно получить, Р аТрИВаТь как
кРЫТнем цикла под действием основании можно ’-711х11111тр11Лов.
общий метод синтеза алифатических и -4°
„ „UK .а В изоксазоле (859) под лещ-пшем щелочи
Раскрытие «м ‘ ы(. способ „олучеиия in situ неусгоичн-
(уравиенпе >' (861) (454], а аналогичное раскрытие
воп> ч^^'^^^епз^оксазолия (862) позволяет использовать
,Тс%стрТты »’качестве конденсирующих агентов в пептидном
, тезе (503] (уравнение 250).
NC. Н
С
II
С
н V>h
(860)
CN
I
сн2
сно
(861)
Промежуточное образование Д2-оксазолинов и раскрытие цикла
удовлетворительно объясняют те глубокие превращения кетонов,
которые в результате реакции с п-толуолсульфонилметилизоциани-
дом в основных условиях превращаются в нитрилы со вторичным
алкильным радикалом с хорошим выходом [456, 500],
Нитрилы можно также получить, и часто с хорошим выходом,
путем отщепления амина от гидразона или гидразониевой соли
(864) альдегида при нагревании или действии оснований (урав-
нение 251), а также путем раскрытия цикла в аналогичных по
строению циклических соединениях (866) (уравнение 252)
[61,309,454,455,486].
556
+ NaOMe
RCH=N—NMe3 X' ---------> RC=N + Me3N + NaX
(884) (865)
R’ R!
R> V Rs R1
\/ \ 210 - 230 °C I I
Y „ H3N-C-C-CN
R ¥
<Юв> (8OT)
(251)
(252)
,janorn,IIII,ie nP0Iieccu’ приводящие с хопош,
^включают растепление N-(anH™?° м вь,ходом к нитон
J'действием основания [504) 7 термХХ)'1/’4’Триазолов
"палкплидсвампно)- и Марилиденамино)™™^ фра1,ментапию
Йалл«1’ова,1ип N’N-Диметнлгидразонов LpS^’2 1505ф П₽и
,,0В (868J происходит отщепление амина и ofinl х альде’
‘в (870) с выходом 77—90% (схема 253) (506? 7<1Ние нитРи-
сравнению с другими методами растения 7PWCC“
" дко протекают при низкой температуре (-40 77|ХА,разонов
граты (гидразоны) нет необходимости получав снеди 77'
/можно синтезировать и подвергать распаду in situ S ^
метого, Карбахо»* н^Р^ов (871), полученные из идр’аз^ов
альдегидов (868; R = Н), имеющих в «-положении вторичный а°
пильный радикал, могут т sltu реагировать с алкилгалогенидамн
или карбонильными соединениями и давать трет-алкилднаниды
(870) (77-94%) пли 0-гидроксинитрилы (872) (83-90%) (5061
Действие основании на N-хлор- [454] и N-сульфоннларилальд-
импны [507] позволяет получать ароматические нитрилы с выхо-
дами 70— 100%.
, R3 t :
R\l LlN(Pr-«3o),
>-CH=N-NMe2 р^ФтА-бензол, ,>
-40 v -1O°C К
i rS
H2O K\ I
—>- 2/C-C=K
Rz
(«70)
, R Li
R\l
,C-C=fNrNMe.
R‘z V u
(869)
R3-h| ______
R
(868 >
R.
_^С=О
. CN OH
R\l I/
)c—c(
r2/ X
(872)
Нитрилы можно получить с приемлемыми выходами окислена
ей (дегидрированием) альдиминов или первичных ал^в‘1ам’'.
Действием таких окислителей, как гнпогалогениты 1 , 1>
1309, 454).
25”
обработка первичного а.1-
в присутствии кислоро-
г ' __________ buvnnOM
R* _Li+
;C—
r2/
(871)
с приемлемыми выходами окнслени-
* .... « 11,41 т о WHHOR
(253)
..vp»—..
гнпогалогениты |_оОУ,
ацетат свинца [98, 455] . Вполне вероятно, что в последнем, слу
чае вначале происходит рас'
г°ся свинецорганического с..,_
“бе возникающего при этом альдимина
!“] Более мягким методом является о'
«“ламина хлоридом меди(1) в пиридине =' “;"-енным выходом
Да, при этом получают нитрил, правда, с у н
(м0%) [508].
Rch2nh2 J'b(UAc)" бензол
кипячение
(878)
[RCH2NHPb(OAc)3 —* RCH NH1 —Нз
(874) (878) (254)
---<- RCsN
(876)
657
' „„„пишем окислении альдиминов, полученных ш S(7(l
Данные оо jcie„ ,1Ь,1ег|1Д013 с аммиаком, в нитрилы [309, 454,
путем KO.Liei сан )пя Q т0М. ,1т0 ,1М1„1Ь, по-впдимо.
45 1 иермеднагами в процессах окислительного превра.
МУ' ТХвпчных алкнламинов в нитрилы (ср. уравнение 254)
неппостХем случае [454, 455] кислород пропускают через рас-
В Р Лпег ,па и аммиака в метанольном растворе метоксида нат-
пняР “содержащем хлорид медн(II). В данном способе можно
Хчьзовать II другие окислители - иод, пероксид никеля или
тетраацетат свинца [454. 455]. Поскольку реакция проводится в
снтьноще точной среде, ее нельзя использовать для получения али-
Аттических нитрилов из-за чувствительности алифатических аль-
дегидов к щелочам. Нитрилы можно получить с хорошим выходом
окнетигельной фрагментацией азометинов. Так, ароматические
нигриты образуются с высоким выходом (70—90%) из арнлиден-
аткнламннов при действии динзопропнлперокспдикарбоната в
бензоле при 60°C [5091 - Ароматические нитрилы получают также
окислением гидразонов ароматических альдегидов оксидом рту-
ти (Н) в кипящем диглиме или 1,2-диметокснэтане; выходы дости-
гают 80% [510].
Простой и широко используемый метод образования тройной
связи углерод—азот состоит в отщеплении воды от первичных
карбоксамидов. Классический вариант этого метода [309, 454]
синтеза нитрилов заключается в нагревании амида с такими
реагентами, как пентоксид фосфора или хлорапгидрнд кислоты
(например, пентахлорнд фосфора, фосфорилхлорид, фосген, тио-
нплхлорид, метансульфонилхлорид или п-толуолсульфонплхло-
рид) в присутствии основания (например, пиридина или трпэтил-
ампна [455]). Особенно хорошие результаты дает применение
тноннлхлорнда в сочетании с дпметилформамидом [455]. Из-за
обратимости реакции первичных карбоксамидов с ангидридами
кислот эти ангидриды, как правило, нельзя использовать как деги-
дратирующие агенты при получении нитрилов из амидов [454|
Однако было показано [488], что прн действии трнфторуксусиоп
ангидрида в присутствии пиридина первичные карбоксамиды пре-
вращаются в нитрилы с высоким выходом (85—99%), в мяг
ких условиях (0 20°C). К числу реагентов, позволяющих осуще
ствпть дегидратацию алифатических и ароматических первичны.'
карооксамидов в соответствующие нитрилы с превосходными вы-
ходами и в мягких условиях в присутствии оснований (например,
трнэтиламина, пиридина, N-метплыорфолина), относятся дицнкло
гексилкарбоднимил [455] и хлорид тптана(1П [4561. С высоки-
дейстшп?^'ям б?-100'?’) ""ТРИЛЫ М0Жн0 получить [456] при
да и тпиэти амтР"Фе,11,лфосфина, четыреххлористого углеро-
лимеоным и'оснтрпв осоГ,е',но У-юбно использовать связанный с по
загрязнения пвотукт/Р1'*!'1"^^*1111’ это УСтРаняет возможность
карбоксамидыР поУппб, Зр1|'',е"11лФосФ«»окспДом [492]. Первичные
’ До но альдоксимам (см. выше), можно с успе-
658
am преврати™ в нитрилы реакцией с
ровнях [456] пли нагреванием с фосфоюирцд^’04 в ос»овиых
rnOPiiTeJI>l llJ111 в II,ieP11,0M PacTBopineie тя1 хлоРидом без пае
>100-С |45G|P ftjf. x,»pta,P“
is svis. p““ “
дЫ Эффективными методами синтеза иитриши
методы, основанные на отщеплении сероволоппДВЛЯЮТСЯ также
ио! действием хлорида ртутн(П) как к== Г. Гамидов
i«4i «л» Ре,™4
ЦШ1 амидов,-дпхлоркарбена [456] или трнфенЦ^иИ ,Драта’
„етаннн с четыреххлорпстым углеродом н тпизти-Л? Ф ,в С0‘
Бы,ю показано [511], что тноамнды в мягких ус1овияхН°М 456''
КПМ„ выходами (64-86%) превращаются в нитрилы при дейетТ’
эфирного раствора днэтилового эфира азоднкарбоновой кистоты
И трпфенплфоефнна при комнатной температуре «чиюты
В некоторых случаях кислоты удается ’перевести в нитриты
непосредственно, минуя стадию образования амидов или тиоами
дов [454]. Один из таких процессов состоит во взаимодействии
карбоновых кислот (877) с хлорсульфонилизоцнанатом в присут-
ствии днметнлформамнда; промежуточно образующийся смешан-
ный ангидрид (878) самопроизвольно отщепляет диоксид углерода
затем происходит отщепление хлорсульфоновой кислоты, приво-
дящее к нитрилам с хорошими выходами (уравнение 255) [454].
ciso2n=c=o
RCO2H --------->- RCOOCONHSO2C1
(877) (878)
ДМФ
•—> RCONHSOjC! -------’
(879)
Стадия отщепления в хлорсульфонамиде ,
мент в приведенной выше схеме — была использована в «сквоз-
ном» синтезе Р-кетонитрплов (883), состоящем во взаимодействии
алкилкетонов с хлорсульфонилизоцнанатом в присутствии диме-
Т11лформамида (уравнение 256) [512]. Эта реакция имеет общин
О
г>, Д ciso2x=c=о
К -C-CHR2R3 ----------->
-со2
(255)
RCX
(880)
(879) — ключевой мо-
0 R2 О
!l I I!
ri-C-C-C-NHSOjCI
I
R3
(882)
О R2
-c-i-cx
R’
(883)
ДМФ
(881)
(256)
659
„ ОР можно применять для получения нитрилов нз цикло-
чпкпл дпалкил-, пли арплалкплкетопои (по не диарнлкетонов);
"“Tn^wviii фрагментация гетероциклов может представить
пенный (в препаративном отношении) метод синтеза нитрилов.
Пп'вставляют интерес примеры термической [500 513| п фотохи-
м.неской фрагментации [500], а также распад 12,5-оксадназолов
и их N-оксндов под действием фосфитов [500]. В последнем
случае образуется смесь нитрила и его N-оксида (см., напри-
мер уравнение 257). Нитрилы образуются с высокими выходами
7йО_90%) и при окислительной фрагментации N-амино-!,2,4-три-
азолов (886) под действием тетраацетата свинца (уравнение 258)
[514].
о +
xN—О"
fcC CssN—О"
(257)
(884) (885)
N—N
N—NH2
Pb(OAc),
бензол, 0 °C
2RCN + N2
(258)
(886) (887)
(4) Перегруппировки
Нитрилы часто образуются с приемлемыми выходами в резуль-
тате молекулярных перегруппировок. Простейшим примером яв-
ляется превращение нзоцнаннда в нитрил [454, 457, 458J, кото-
рое протекает необратимо при температуре >150°C. Поэтому
те процессы, которые в других условиях приводили бы к изоциани-
дам, при повышенной температуре приводят к нитрилам. Примером
может служить получение ароматических нитрилов из арил-
изотноцианатов (888), протекающее, по-видимому, через промежу-
точное образование изоцианнда (889) под действием трифенил-
фосфита (уравнение 259) [454].
(PhO)3p
ArN=C=S -----------> [ArN=C,] —► ArCsN (259)
<888) (889) (890)
илиНИфотохимЯииЛрЯгип“Я также коченными продуктами термической
8 азидостипоп » перегруппировки азидов. Так, например.
[94] Пепегоуппипг? щается в Феиилацетонитрнл с выходом 74%
(9 1. Перегруппировки этого типа могут быть использованы для
660
пяноциклопропанов (892) (выходы 62—84%) путем тер-
•пте38 ц фотохимического сужения цикла в азидоциклобуте-
•Чмо
•»1 р р ,
I
р_ I —Д ИЛИ hv
(891)
(260)
R = COR, CN
4N
(892)
Превращение цикла в некоторых гетероциклических
год действием амида калия в жидком аммиаке можетпривести^
высоким выходом к нитрилам, содержащим функциональные Dvn
пы, хотя природа этих процессов зачастую до конца не раскоы
Та; примером этому может служить превращение 1,3-дибромизо-
хинолина (893) в о-цианобензилцианид (894) с выходом 97»Х
[516].
Br
(893)
Br
KNH2. жидк. NH3
(8M)
CHsCN
(261)
CN
8.2.2.2. Реакции алифатических, ациклических
и ароматических нитрилов [91, 281, 309, 51 j
Представление о тройной связи углерод—азот как: о Р^°на®то
ном гибриде [(895)** (896)] согласуется с Данными ° 0.
иуклеофильная атака направляется на атом угл р' , направ-
фильиые агенты присоединяются к aTfi°“yJa о'в результате та-
ление выражено слабее (уравнение 26-). ОД кодированный
кого присоединения образуется резонанс: естественно, под-
нитрплиевый катион [ (897) ** (898)], к°^. Р ’ остью, чем исход-
кергается нуклеофильной атаке с оольше ’ • способность к
«ый нитрил. Присущая тройной связи углер д ером придает
поляризации в сочетании с ненасыщенным • рядом замести-
Дианогруппе свойство активировать наход способность
Тель за счет оттягивания ею электронов и, Р катализуемому как
Подвергаться нуклеофильному присоедин । ’ кцИН некатализуе-
кислотами, так и основаниями, и вступа ..не1-|Ная циан07!1)™
‘,0го циклоприсоединения. Помимо это • ярепятствия,
здает незначительные пространст затрулнсни" Р
®«печивает полное отсутствие стер
ArvinecTBпенни присоединения потройной связи углерод азот. Уии-
ппй ‘характер цнапогрхнны как функционального заместителя
станет ясным из последующего рассмотрения реакции, в которые
вступают нитрилы,
♦ - х* + +
RCsN *-* RON ------> RC=N-X *->- RON-X (262)
(895) (896) (897) (898)
(!) Реакции с электрофильными реагентами
Нитрилы малоосповны и не образуют солеи с минеральными
кислотами в водной среде. В то же время наличие свободной
электронной пары на атоме азота позволяет осуществить протопи-
ровапне нитрилов в безводных условиях в апротонной среде под
действием достаточно сильных кислот. Процессы этого типа при-
влекли внимание исследователей, так как многочисленные пре-
вращения нитрилов в кислой среде протекают, вероятно, через
промежуточное образование протонированных форм. В апротон-
ной среде нитрилы реагируют с галогеноводородамн и при низкой
температуре (—50 4---5 °C) образуют аддукты с одной и двумя
молекулами галогеноводорода; при несколько более высокой тем-
пературе (0—20 °C) образуются димерные продукты [91, 281,
309, 517, 518]. Данные о природе всех этих аддуктов весьма про-
тиворечивы; в особенности это относится к мономерным бпеаддук-
там, электропроводность которых и спектральные характеристики
соответствуют как структуре нитрилиевой соли (RC^NH НХ~2),
так и структуре соли имидоилгалогеннда [RC(X)=NH2X-]. По-
видимому, однако, мономерные моноаддукты являются нитри-
лиевыми солями (900), а бисаддукты — солями пмидоилгалогени-
дов (901); выбор же между альтернативными структурами нмпдо-
плгалогенида (902) и нитрилиевой соли (903) для димерных ад-
дуктов еще предстоит сделать.
Основность нитрилов видна также и из их реакций с кислотами
Льюиса (А1С13, SbCls, SnCU) (схема 263) [91], приводящих к
комплексам нитрилов с металлами, например (904). При реакции
(904) с хлористым водородом при низкой температуре образуются
незамещенные нптрилиевие солп (905), а последующее превращу-
ние при >50°С дает соли имидоилхлорндов (906) [91; 518]. Кар-
боновые кислоты крайне вяло реагируют с нитрилами, для осуще-
ствления реакции требуются температуры >200°C; однако в при-
сутствии каталитического количества минеральной кислоты реак-
цию можно провести и при более низкой температуре Продуктами
ппп?.^РеаКЦ1’" являются «МИДЫ (908), образующиеся за счет само-
лимипгии'.’у'"' пеРС1 Р^ппнроакн первоначально возникающих О-аии-
чёский КПСЛ0Т (й907) [91’ 281' 309’ 5171- Некоторый каталити-
зьщает х^ри" железаUlbTl™ ° "
66.2
rc=nh x‘ RCsNH X’
(900)
| HX
их
- rcsn
(899)
R = Pli|snCI,
OCOR
(997)
X
RC=NH2 x-
(901)
(PhCNhSnCI,
(904)
I HCI
о
RC—NHCOR
(908)
(263)
X\
r/C=N\c/R
NH
(902)
(PhCssNH)2 SnCl2'
(905)
I HCI, И =C
или
RC=N. /R
XCZ
II
NH X
(903)
=\Н2Л$пС12,'
(906)
основность, нитрилы можно перевести
Несмотря на низкую основность, нитрилы можно иереосс.п
в N-алкильные [91, 309, 517, 518] и N-арильные |о18] производные
в различных кислых и основных условиях. Образующиеся ннтрт
лневые соли имеют большое значение как синтетическиещшт р
диаты. N-Алкнлпроваине можно осуществить нагре®®““ „ ю.
ла с трпалкилоксониевой солью (тетрафторборат Р р '
мииат или гексахлорантпмопат) [схема 264; (9^
обработкой комплекса нитрила с металлом (см. в 1(909)->-
вторичным или (лучше) третичным алк11Л''^ н11Трилов кар-
911)->(912)[ [91, 309, 517, 518]. Алкилирование ю тР«СТОГо и
Рениевыми ионами лежит в основе реакции п имЫШЛенного и
широко используемого метода, пригодного а следова-
лабораторпого получения вторичных кар ,ьтатегидролиза
тельпо, и вторичных аминов, образующихся Р г0 царбение-
карбоксамидои [309, 517, 520]. Необходимый * нЫХ субстра-
Ыи ион можно получить in situ из самых р 1Огенов), обычно
Ов (спцр10В1 простых эфиров, алкенов^ а, _’д5%-ной фосфорной
сильно кислых условиях, например, в еак11ця с ПРОСТ“МИ
^оте или в 90%-пой муравьиной кисло ,ер< с тгНГ'^к
11 Функционализированными нитрил- и приводи ,
иамц и а-аминонитрилами, идет при
„по Г опичвымн выходами к соответствующим вторичным
"рав™ (914) 'вероятно, через промежуточно образующийся кати-
„ Хвизия (913). Ряд факторов согласуется е представлением
пб v.астип ионов карбсния в реакции Риттера: образование про-
ектов скететной перегрсиппровкп, опыты с меченными дейтерием
сБедпнеипямч [521] и образование продуктов, типичных для ре.
акции Риттера при взаимодействии устойчивых карбенпевых ио-
нов (например, тритильного [520] и хлордифенилметильного
(5921) с нитрилами. Примером реакции Риттера можно считать
взанмотепствие алифатических или ароматических нитрилов с
альдегидами в 85—98%-ной серной кислоте в отсутствие или в
присутствии второго растворителя (например, муравьиной кисло-
ты, фосфорной кислоты, уксусного ангидрида или хлороформа)
при 30—50°C, приводящее к М.М'-алкилидеибпс(амиду) (915)
[90, 517]. В конденсациях этого типа особенно высокую эффек-
тивность обнаруживает формальдегид, в качестве катализатора
можно использовать метансульфокислоту.
♦ н2о
----* R'CONHBu-rper
(913) (914)
Ме3СОН
илн
Ме2С=СН2
'BF, R2 (264)
♦ EW* -BF4 R2CHO I
PhCs=\Et t—-— R'GsN ---------------> R'CONHCHNHCOR1
Rl = Pn
(910) (909) (915)
R=Me SbCI5
< - MeCI t
MeC=N—SbCl6 -------> MeCsNMe 'SbCle
(911) (912)
Самопроизвольное электрофильное замыкание цикла в проме-
жуточно образовавшихся нитрилиевых катионах, полученных в ре-
зультате реакции Риттера с диенами, гликолями и аналогичными
субстратами, лежит в основе общего метода гетероциклизацпи,
проходящей с высоким выходом и позволяющей получать Д'-пир-
ролины, Д2-имидазолнны, Д2-оксазолипы, Д2-тиазоЛнны, дпгпдро-
1,.3-оксазпиы и дпгидро-1,3-тиазины (523, 524]. Примерами реак-
ций этого типа могут служить катализируемые кислотами реакции
оксиранов, тииранов и азиридинов с нитрилами, приводящие со-
ответственно к Д2-оксазолииам, Д2-тиазолинам и Д2-пмидазолн-
нам (уравнение 265) [523, 624].
664
N-Арилнитрилиевые соли нельзя получить пеак„Ирй
арилгалогенидов с нитрилами. Они образуются при 0Е
нитрилов арендиазониевыми солями, разлагающимися при темпе
ратуре ниже 60 С; если реакция проводится при более высокой
температуре, то выходы снижаются из-за побочных реакций 191
517,518]. 11
Нитрилы обычно не удается непосредственно превратить в
N-ацилировапные производные. Синтез N-ацилнитрилиевых со-
лей (например, R‘C=N—COR-’ А1С1Г) проводят взаимодействием
ацилгалогенидов с комплексами нитрилов с металлами [91, 517,
518). Межмолекулярное ацилирование цианогруппы происходит
с трудом, однако внутримолекулярное прямое ацилирование про-
текает гладко и, как и в случае внутримолекулярного алкилиро-
вания нитрилов (см. выше), позволяет синтезировать большое
число гетероциклических соединений (см., например, сравнение
266) [623, 525].
CH2CN
СОС1
(2) Реакции с нуклеофильными реагентами
Реакции нитрилов с нуклеофильными агентами об“чно
кают в присутствии кислого или основного катализа' Р „ 1ЬН0
но эффективен кислый катализ, так как субстрат_ р ,сМ
превращается в высокоэлектрофильный нитрилиприсоеди-
УРавнение 262). Простейшим примером нуклеоф . хор0Шо
нения к тройной связи углерод —азот служи , ’карбокс-
вестная реакция гидратации нитрилов в Р зу и для их
амиды [91, 281, 309]. Нитрилы устойчивы к п'^ие' кислые или
Ревращения в амиды обычно необходимы у(ощеМу гидро-
“ровные условия, которые способствуют и }в в основных
ЛИ8У До кислоты. В случае алифатических нитрило
атппчя стадия, гидролиз амида в кислоту, может быть
условиях втора стадия. П|Дратац„я нитрила в амид,
ролее оыир > f 'и| „„...рн.цц, е целью получения амидов
?’^-ет проводить в концентрированной серпов или полифосфор.
оте при комнатной или повышенной температуре. Для
по'вр; ценпя нитрилов в амиды без последующего их гидролиза
« к еюты можно также использовать трифторнд оорав уксусной
,с оте 191] В тех случаях, когда гидратация тронной связи
v 'neooi-азот протекает особенно медленно, ее можно ускорить
испотьзованием более мощного нуклеофильного агента, гидроне-
поксид-ашюна ("ООН). Действительно, нитрилы гладко и с вы-
сокими выходами превращаются в амиды при обработке перокси-
том водорода в слабоосновпой среде (реакция Радзншевского)
[91 309] Первоначально образующиеся прн этом производные
перокеппмидовой кислоты RC(OOH)=NH могут использоваться
как окислители. Так, для эпоксидирования алкенов и для N-okhc-
леиня азагетероцнклов широко используют реакцию с пероксидом
водорода в присутствии ацетонитрила или, лучше, бензонитрила
[91, 309]. Нитрилы превращаются в амиды и при непродолжитель-
ном кипячении с гидроксидом калия в трет-бутпловом спирте; вы-
ходы достигают 85—95% [526]. Недавно предложены методы
гидратации нитрилов, которые позволяют отказаться от сильно-
кислых или спльнощелочных условий, используемых в классиче-
ских методиках; получение амидов идет в строго нейтральных
условиях. Один из таких методов, в котором как бы смоделиро-
вана способность ферментов превращать нитрилы в амиды путем
гидролиза в мягких и строго соблюдаемых нейтральных условиях,
состоит в использовании меркаптоэтанола как катализатора
[527]. Гидратация нитрилов в мягких нейтральных условиях мо-
жет ускоряться также и гомогенными катализаторами иа основе
комплексов переходных металлов [528]. Так, пептаамминохлоро-
рутений (!!!) хлорид [ (NH 5) 5RuC1]C12 катализирует гладкое пре-
вращение нитрилов в водной среде при комнатной температуре,
выход амидов 64 99%. Гидролиз ацилциаипдов водным раство-
ром кислоты протекает «нормально», давая а-кетокпелоты, тогда
как по отношению к другим нуклеофильным агентам ацилциани-
ды ведут себя необычно (см. ниже) [91]. Однако при кислотном
гидролизе продуктов реакции Рейссерта происходит их расщепле-
пАгип °бразоваШ1С гетероциклической карбоновой кислоты и аль-
ненир%К7?ТВтТСТВУ10ЩеГО 11СХОД1|°МУ ацилирующему агенту (урав-
лучения япьпр0 можно Рассматрнвать и как косвенный способ по-
лучения альдегидов из хлорангидридов кислот [483]
” 0CV '
НМ=1_Д Br
I + РИНО
N (267)
СООН
-^XxNCOPh Ас°н
(60%)
666
^„сое/ишенне сероводорода к 1)НТр
' ..ьфнд-чона позволяет получать с в "Р^Лствии
^амиды образуются также пр„ взаимХ^ вых°Дом тиоамип^'
„оводородом в диметилформамиде при 6ovTBHH НитРи-’ов с се'
। 1111а или грпэтиламина [9|]. С В "РИсУтствии
I 'реакции алифатических и ароматически,
,, фенолами и их тноаиалогами как в к|,с,,'1'‘.'ТР'Ш'в со спирта-
средах представляют собой общий метод „X’ак " « Ровных
имидатов и -тиоимндатов. Реакции присоед е„ия КИ/" 11 а1)и-’-
робно рассмотрены ранее (см. разд. 8 1 4) „ НР ’ТОго ™На гюд.
тельных пояснений. Взаимодействие спиртов Требуют Дополни-
проходит ие по типу присоединения к тройной ев« аии’1цнаниДами
з, скорее, по типу замещения цианогруины г пй? УМерод~азот,
„ых эфиров [91]. ЮШ1Ы с 00Раз°ваинем слож-
Реакшш присоединения аминосоединений к нптпи,
ляют осуществить успешные синтезы аминоазомеХ i П03В0'
|91, 281, 293, 309, 32 8], ампдоксимов [91 996 30SW ИДННОВ
дразонов (91, 309, 329, 339]) (см разд я’Д7 „ ’ 3311 и ами-
удобны, хотя и менее эффективны, чем методы, осиованны^иа
реакциях нуклеофильного замещения, которым подвергаются
имидоилгалогениды (см. с. 589) и имидаты (см. с 604) при деТ
ствии аминосоединений, В случае аммиака непосредственное при
соединение к тронной связи углерод-азот - обратимый процесс.
Реакция проходит лишь с нитрилами, содержащими электроно-
акцепторные заместители, например с трнхлорацетонптрилом
(916); в этом случае продуктом реакции является N.N-незаме-
щенный амидин (918) (схема 268; “
R1 =CC13, R2 = R3 = H).
R3C=n
(916)
R\
3/NH
R3/
(917)
NH
. II /R2
R-C-N< ,
R3
(918)
Me3NNH2 Cl ,
mpem - BuOK,
mpem -BuOH,
кипячение
R’-H
(268)
.NH
R-сф-
XN-NMej
(920)
nh2
t I :
R—C=NR
(919)
гцдрат Л1и1С|||[е аммиака по тройной
этому ац,1я’ каталпзуется как кислотами, так
"с'1оЛьзДЛЯ осУш-ес’гнления этой реакции г
|,РакТцк°Г!ать аммоиневую соль или а.
е N, N-незамещенные амидины
связи углерод-азот, как и
так п основаниями, по
ЛцШ1 вместо аммиака можно
аМ"Д поучат нагреванием
667
„ р тиоцианатом или алкилсульфоиатом аммония в рас.
НИТР" Лппботкой нитрилов хлоридом или бромидом аммония в
плаве, оОрао |2,5—)50°С иод давлением, а также обра.
?'1ЛК„Т>П1ТОИ'ЮВ амидом натрия или калия в жидком аммиаке,
00[ Д требуются более высокие температуры, в аммиаке под
"l'*' шем иногда с добавкой растворителя (например, бензола,
то to а или ксилола). Продуктами реакции нитрилов с амидами
метЛтов являются металлированные амидины, гидролиз которых
то амидинов следует проводить осторожно, чтобы не допустить
гидролиза амидина до амида. Непосредственное присоединение
аминов (917) по цпаногруппе, приводящее к N-замещеииым ами-
динам (918) подчиняется тем же требованиям, что и присоедц.
некие аммиака; для успешного осуществления этой реакции тре-
буется кислый пли основной катализатор. Однако взаимодействие
нитрилов с гидрохлоридами аминов редко приводит к удовлетво-
рнтельным выходам амидинов. Лучшим способом синтеза амиди-
нов является сплавление нитрила с арилсульфонатной солыо
амина. Этот метод особенно пригоден для синтеза N-замещенных
бензамидпнов (918; R1==Ar, R2 = Aik, R3 — Н или R1 = R2 =
= Rs = Аг) из первичных алкилампнов или диариламинов. Для
получения М.М-диалкцламндпнов (918; R2=R3 = A!k), исходя из
арилсульфонатов дпалкилампнов, этот метод не пригоден, так
как в условиях реакции проходит и дезалкилирование. Альтерна-
тивный метод, позволяющий получать N-замещенные амидины с
приемлемым выходом,— нагревание смеси нитрила и амина с хло-
ридом алюминия в инертном растворителе и гидролиз образующе-
гося комплекса амидина с хлоридом алюминия до свободного
амидина. При реакции нитрилов с металлированными аминами
также получают N-замещенные амидины. Металлирование можно
осуществить in situ, нагревая смесь нитрила и амина в инертном
растворителе с амидом натрия или калия. Более удобным мето-
дом является получение металлированных аминов обработкой
аминов гидридом натрия в диметилсульфоксиде и последующее
взаимодействие их с нитрилом в этом же растворителе при ком-
натной температуре. Присоединение аминов к нитрилам с об-
разованием N-замещенных амидинов можно осуществить также
н с помощью комплексов аминов с магнием, которые образуются
Ж»тпТ’”'’0ДеИСТВИИ ВТ°1’ИЧЧЬ1Х аминов с реактивами Гриньяра.
з„Ум чГпе5ЛЯрНОе пРисоединение аминогруппы к тройной свя-
пользуемого обшРтСТаВЛЯеТ осиову классического и широко ис-
[92 5231 Пиимеп Синтеза гетероциклических амидинов
уравнении (269) Р тероциклизадии этого типа [529] приведен в
NHj
(269)
668
внутримолекулярное присоединение амина к
В/аеТ самопроизвольно и в этом отношении ?Т₽МУ ’исто поо
ответствуюшнх межмолекулярных про™ 'Z™ ^aZaZ
олЫ”им трудом. Это указывает на то, что fi„ "Ходящих с
1ра11ствеппое расположение реагируй их г±*ГоТЯТН(* "Ро-
яльное присоединение к тройной связи °б;'егЧает "У-
В отличие от аммиака гидроксиламин ЛГЛерод~азот.
,ен „ способен присоединяться как к алифаХ?^0 Нукле*-
мат„ческим нитрилам при 60-80 °C в этанол п ’ Так и к 'W
лпзатора; это практически общий метод cnZJZ'1'"81’6 Ката-
кобенно пригодный для получения бензамидоигиЛ ?Мидоксимов,
(9)6)—»—* (919); R1 = Ar, R2 = ОН, R3 = “ИАок~ [схема (268);
„Л11 в сочетании с гидразидом натрия присоед СаМ по себе
аналогично и дает N-незамещенные амидразоны ? * нитрилам
(916)-*—*(919); R2 = NH2, R3 = Н] ПоисТп u [схсма (W
вых солей к алифатическим и ароматическим^*™® 'Идразини«-
ствии трет-бутокспда калия гладко приводит к aJL’Z В п₽исуг'
(амннимидам) (920), соединениям своеобразной crp^^S
Примером присоединения анионов к тройной связи умеоол'
азот может служить оощая реакция нитрилов с азнд-ионом в
результате которой образуются тетразолы, вероятно через про-
межуточное соединение — имидоилазид (уравнение 270) [523]
N’ R\
Ns II -
RC=N ---► RC—N—N=N —> V \\
н+ /=л
—* HN (270)
N
Нитрилы, ие содержащие атомов водорода в a-положении, на-
пример ароматические нитрилы, легко присоединяют карбанион-
ные реагенты по тройной связи углерод—азот с образованием
иминов, которые можно выделить или гидролизом превратить в
кетоны. Процессы этого типа с участием реактивов Гриньяра,ли-
тии°рганических соединений и карбанионов из соединений с
Дивной метиленовой группой (например, реакция Циглера —
. Рпа) были рассмотрены ранее при обсуждении синтеза иминов
1Разд. 81.2.1(1)], Результат присоединения реактивов Гриньяра
н,,аенз°Н11ТРилу, как и следовало ожидать, зависит от соогноше
ЛьипСу®стРата и реагента; реакцию можно контролировать i
бИняТЬ с хорошими выходами имины, кетоны или трети4 _
кИаамины [531]. Примером реакции присоединения каРоа”®
“•Литий0” СВЯЗи УглеР°Д—азот является также „^нтнйнитроз-
аминов %°,М!|ТНИ0В (оксимов и гидразонов) [53 ] общрего
ИеТо° '5331 с нитрилами. Эта реакция лежи изоксазо-
л°в и 1Со'оТе3а ”ят"членных гетероциклов (пир• ;в с карб-
•икоцом 73'тР),аз°-"ов)- КР°ме тОГ'?’ ре^Ме$ОСН,$Ме позво-
1 (метилтиометил)метнлсульфоксида МеЬ
пгтвпть НОВЫЙ синтез эфиров а-ацетилампиокиелот
щиаивЛЙ [9D веДУт себя в реакциях с реактивами
[531а). ЛИ"-' как обычные нитрилы: вместо присоединения
rp"llbXinunc происходит их расщепление с образованием кето-
"° ПоХ.,> и аиеталеи ацилциаиидов с литиидлтнапом протекает
норна.Хм образом, с ее помощью можно осуществить синтез
а-днкетоиов (уравнение 271) [5346].
R'
(R2O)2C-CN
(68%)
При взаимодействии нитрилов, содержащих а-водородный
атом, с карбанионными реагентами в большей степени происходит
а-депротоинрованпе, чем сопряженное присоединение к циано-
группе. Поскольку, однако, депротонпрование и присоединение
основания — конкурирующие процессы, направлением атаки мож-
но до некоторой степени управлять путем выбора основания и сре-
ды, в которой проводится реакция: в случае относительно мало-
ионизованных оснований в малополярных средах присоединение
преобладает над депротонировапием [91]. Карбанионы, получен-
ные а-металлированием нитрилов,— реакционноспособные интер-
медиаты. Они могут вступать в различные реакции, типичные для
карбанионов из соединений с активной метиленовой группой; их
можно алкилировать, ацилировать и нитрозпровать. Процессы
этого типа проиллюстрированы на примере феннлацетоиптрила
PhCH2CN
основание
RCHOH
PhCHCN
RCHO
PhCHCN
RONO
N=O
PhCHCN (272)
-Н2О
CHR
II
PhCCN
HCO2Et
2RX
CHO
PhCHCN
I
CN
I
PhCR,
NOH
II
PhCCN
670
Галогенирование нитрилов в а-положенне к к,
осуществить в стандартных условиях |3091 ц иаН01РУппе трудно
сокпм выходом проходит при обработке а’пиан°« ° Легко 11 с в“-
тЫреххлористым или четырехбромистым VZ„,окаРбанионов че-
эТокспкарбоиильную группу в «-положение [°35L Ввести
„егко И С высоким выходом путем металлироВанНи°вГруППе
‘ зоиропиламидом лития в тетрагнДрофуранеР Яа»ИЯ «»т₽ила №
с последующей обработкой этилхлорформиатом L 74 ~ ~78 с
этилкарбонатом [536]. Использование днизоппоП1памЛУЧШе’ ди’
Гтетрагидрофуране при -78 °C позволяет провес И мп»а ЛИТИЯ
рование алифатических первичных нитрилов, которое
ществить каким-либо другим способом [537]. При ое™ У’
винных алифатических нитрилов с избытком МетИПИоди ' ”и
метиллития в зависимости от соотношения основания и аткилХЛ
шего агента можно получить бис-а-метилнрованные нитриты со
ответствующие кетимины или кетоны (схема 273) [538] *
? ? 2MeI-
I II 4MeLl 94eLi
Ме2С—С—Me <-------- RCHaCN -------
4Mel,
4MeLl
R Me
I I
Me2C—C=NMe
R
I
Ме2С—CN
(273)
с иодидом меди
Легкодоступный по реакции цпанометиллитня
синтон, цианометилмедь CuCH2CN, при реакции с аллилгалоге-
нидами подвергается лишь моноалкплированию, это позволяет
осуществить синтез у,6-ненасыщенных нитрилов с высокими выхо-
дами [539]. Применение водной щелочи в присутствии 18-крау-
на-6 позволяет провести алкилирование фенил- и дифенилането-
нитрплов с выходами 75—85% [540].
а-Цпанокарбанионы в последнее время привлекли к себе вни-
мание благодаря способности выступать в роли «замаскирован-
ных ацильных карбанионов», т. е. реагентов, использование кото-
рых как бы обращает (umpolung [541]) нормальный электро-
фильный характер карбонильной группы [схема 2/4, ( - )
"*(922) --*(923)]. Существуют два метода, в которых исполь-
зуется эта способность а-цианокарбаипонов. В одном из них. [ -J
ЛК||Л’> циклоалкил- пли арилциангидрин переводят в bi •
зХ тетРагидропираниловый эфир (зашита НО-группы) [(924)],
чают металл»РУ'от действием дипзопропиламид V5Ke
Мо“ каРбанн°н защищенного циангидрина ( ..’’ емРэлекТро-
ф.. 0 вв°Дить в желаемые превращения под л• . Последую-
агеитов (например, провести алкилировати Послед *
фУн °бработка кислотой и основанием позвол • ‘ /923). Метод
пионализованному карбонильному соеди
r0\c^n
>/
(924)
t
| IICN
0
II
R-C-H---------
(921)
RO. _ zCN
0
RZ
(925)
О
II
R—C
(522)
(926)
О
II
------* R—С—E
(923)
(274)
можно улучшить, используя триметил силиловые эфиры циангид-
ринов [543]. Таким путем удается провести превращение бенз-
альдегидов в дпарилкетоны обходным путем (арилировапнем
карбаниона из манделонитрила) [544], а также превращение аль-
дегидов в 1,4-дикарбонильные соединения с высоким выходом
(70—85%) путем 1,4-присоединения к аф-ненасыщенным карбо-
нильным соединениям [545]. Последняя реакция имеет особое
значение [545], поскольку другие возможные эквиваленты ацил-
карбанпона, дитиан-анпоны, реагируют с аф-ненасыщенными со-
единениями по типу 1,2, а не 1,4-присоединения. Карбанионы ци-
ангидринов участвуют также в катализируемом цианид-ионом
присоединении альдегидов к аф-ненасыщениым карбонильным и
родственным соединениям в апротонных растворителях (напри-
мер, в диметилформамиде или диметилсульфоксиде) [546]. С
участием карбанионов циангидринов протекает также самопроиз-
вольный [2,3]-сигматропный сдвиг в аллиловых эфирах «-метал-
лированных циангидринов, приводящий к р.у-ненасыщенным ке-
тонам с выходами 36—90% (уравнение 275) [547],
Возможность реализации второго подхода к использованию
ТоЦе^°КарбаНИОИОВ как эквивалентов ацил-карбаниона опреде-
лрпиячJnT4HeM метода’ пригодного для мягкого окислительного
^92T17qa9a?_ "°“е функционализации карбаниона [(927)-*-
сукпипимиля лп*а 923 Эт° достигается применением N-бром-
X = SPh) nonvupu окислен11я а-феиилтиоироизводного (932;
ученного взаимодействием функционализированно-
679
г0 карбаниона (930) с феннлсульфенилхлоридом либо иепосред-
стненно, либо после превращении ,, промежуточный силнлкеЛи-
нмш. <93?;4J:54v8L^uXZT можлке',еиимина (93‘)
нитрил (932 х — О. который можно окислить до карбонильного
соединения (932) действием оксида серебра [548].
CN
I
R-CH2
(927)
Li+ "N(Pr-U3o)2
1 ГФ, —78 °C
CN
R—CH —
CN
R—CH—E
(928) (929)
R\ Ви-трет
Y=C=\\ |
Е/ ^-SiMej
(931)
bi+ "N(Pr-U30)2
ТГФ, -78 °C
CN
I SnC
—С—E ------
I (89
OOH
CN
I
R-C-E
I
OH
0
II
R—С—E
(933)
12
ИЛИ
PhSCI
CN
I
R—С—E
I
X
(932)
(276)
(934) (923)
Ag2O(X = l) |
NBS(,X = SPh)
Другой (и более' удобный) -
нии функционализированного карбаниона (930) реакцн '
лородом в гпдропероксид [933). Гидропероксид взапм д
с хлоридом олова (11) можно перевести в с00Т8е^Т8>Ю™ карбо.
гидрин (934), из которого действием основанияi J cnofQ6a
нильное соединение (923) [548]. Пренмушест . - нлтао. и
по сравнению с окислительным превраще Ф ‘ ПОЛуче-
смюднитрплов является возможность применен! г-<о] Длки-
чия диалкил-, а также диарил- и арнлэлкил ^ановнтель-
лпрованке а-цианокарбанионов с послед)ю „ етнлтриамидо-
ным децианированпем действием кал118 спиртов и адьде-
фосфате было использовано в синтезе в • ^ета1лйрование
™дов; выходы продуктов очень. вь,с“Пг1,зпида натрия в диме-
"Родуктов реакции Рсйссерта действием г др раз.ПОжениеу
плформамнде и последующими алкплир
22 Зак. Ю5
способ окисления состоит в превраще-
„птп,,я _ паилучшпй метод синтеза 1-алкилизохино-
м^ТГГособеииости промежуточных соединений в синтезе изо-
Х,ИПрХома?еакц"1й°иитр8>мов84]с' активированными ароматиче-
Пр слепи Р1ШЯМП (фенолами и их простыми эфирами) в усло-
виях реакцш, Фриделя - Крафтса может служить синтез ^ето-
О Гёшу (см с 594). Внутримолекулярное взаимодействие
иитонтневого катиона с бензольным кольцом происходит, по-ви-
п мому при реакции Бишлера - Напиральского, широко исполь-
зуемой’для получения 3,4-дигидроизохинолииов, применяемых в
синтезе изохинолииовых алкалоидов [523].
(3) Реакции циклоприсоединения [523]
Тройная связь углерод—азот в отличие от двойной связи угле-
род—азот [см. разд. 8.1.2.2(3)] с большим трудом вступает в ре-
акции циклоприсоединения. Так, например, для нитрилов неиз-
вестны, по-видимому, простые реакции [ I + 2] - (карбены, нитре-
ны) и [2 + 2] -циклоприсоединения. Хотя цианогруппа н выступает
в роли днполярофила в диполяриом [3+2]-циклоприсоединении
(см. с. 523), такие реакции проходят только с ароматическими
нитрилами, с нитрилами, активированными электроноакцепторны-
ми заместителями (например, с этилцианоформиатом EtO2CCN)
или с нитрилами, предварительно связанными в комплекс с кис-
лотой Льюиса (например, с трифторидом бора). Имеется большое
число примеров участия ароматических нитрилов в реакциях
[3 4-2]-циклоприсоединения с октетностабилизоваиными 1,3-дипо-
лями, содержащими ортогональную двойную связь (с оксидами
нитрилов, нитрилимииами, нитрнл-плндами, диазоалканамн и ази-
дами) [523, 550]. С помощью этих реакций можно получить раз-
нообразные пятпчлеиные гетероциклические системы—1,2,4-окса-
диазолы, 1,2,4-триазолы, имидазолы, 1,2,3-триазолы и тетразолы.
В то же время число примеров [3 + 2]-циклоприсоединения нит-
рилов к октетностабплизовапным 1,3-диполям, не имеющим та-
кой двойной связи (к нитронам, азометиниминам и азометин-или-
дам), сравнительно невелико [523]. В случае азометиниминов
процессы этого типа приводят к Д3-1,2,4-триазолинам (уравие-
NO,
NO2
(277)
стых интпимиз к I ос°ер’1нение (реакиия Дильса-Альдера) про-
температурах (200-400 "сь "1’°текает оче,1ь вяло. nP" высоких
лых катализ-,торов 15231 Ппп f ускоряется в присутствии кис-
ров 1,523]. Продукты этих реакций-производные
674
пиридина (938)—образуются за
, ,----- счет ароматизации первичных
“"г"7ялдуктов' (3,6-дигидропиридииов) (937) в крайне жестких
|1И „„ах проведения реакции (уравнение 278; R = Me, Et, Ph)
мсДОВ'1>1Л 1
[523].
= Me, Et, Ph)
(935) (936) (937)
Нитрилы, активированные электроноакцепторными заместителя-
ми, например трифторацетонитрил, вступают в реакцию [4 + 2)-
цпклоприсоедииения с 1,3-диенами в более мягких условиях [523].
Особенно высокой реакционной способностью в этих реакциях
обладает п-толуолсульфонилцианнд [551]. Было показано [551],
что при его взаимодействии с норборнадиеном (939) происходит
редкий тип [2 + 2 + 2]-гомосопряженной реакции Дильса —Аль-
дера, хотя и с низким выходом (18%) (уравнение 279).
(939)
jz-MeC6H4SO2CNk
80°С,Зч
(279)
(4) Восстановление [90, 309]
Тройная связь азот—углерод подобно двойной связи ^леР°Д
-азот [разд. 8.1.2.2 (5)] может быть восстановлена поддеис
самых различных агентов, включая системы м т - 1зат0.
тона, комплексные гидриды и водород в прпсутс_ ‘ 0 „
ров. Реагенты типа металл-донор протона, например эл<эв^_
соляная кислота, натрий и этанол или амальг перв11чных амн-
ион среде, способны восстанавливать ’«очными реакциями
нов. Однако восстановление осложняется реакцця нитрилов
и имеет ограниченное препаративное значе i ~ „о„ода с после-
с хлоридом олова(II) в присутствии хЛор|‘7 ™ на до альдегида
ДУющим гидролизом образовавшейся со j _ относптся к ре-
(снитез альдегидов по Стефену), строго P . т,)1ДОцЛгалоге-
акциям восстановительного дехлорирован! ‘ С11НТезу азоме-
«ида и была рассмотрена в разделе, носвяшешюл -
22*
405) Бы io показано, что восстановление ароилциа-
тпнов (см. С. D присутствии хлористого водорода
',,|ДОВПяет°пРолучнть с исоким выходом (50—78%) гидрохлориды
позволя . . rnrH \|Нз С1- [552], которые трудно полу.
арО"'-1кимЛтибо другим способом. Щелочные металлы в гексаме-
тчптонам дофосфате вызывают восстановительное децианнрованне
ш,=в в плсводороды [553], что позволяет осуществить уда-
„ шпилгпуипы после того, как а-цпаиокарбанионы сыграли
свою роль как нуклеофильные агенты (см. выше) [549].
Восстановлен по нитрилов алюмогидрндом лития в обычных
гечовиях дает первичные амины; вторичные амины, осложняющие
поонесс каталитического восстановления нитрилов (см. ниже)
[901 при этом не образуются. Частичное восстановление нитри-
лов \о иминов можно успешно осуществить с помощью алюмо-
гидрида лития прп низкой температуре; это можно рассматривать
как удобный метод синтеза альдегидов (если проводить in situ
гидролиз образующихся иминов) [90]. Восстановительное превра-
щение нитрилов в ароматические и алифатические альдегиды че-
рез имппы можно также провести с высоким выходом с исполь-
зованием трпэтоксналюмогидрида лития [90]. Производные
ацетонитрила, содержащие а-водородные атомы, устойчивы к вос-
становлению под действием алюмогидрида лития вследствие инерт-
ности промежуточно образующегося комплекса металла. Тем не
менее восстановление в таких случаях можно удовлетворительно
провести, если использовать алюмогидрид лития в сочетании с
трихлоридом алюминия [90]. Простые нитрилы не восстанавли-
ваются борогидридом натрия [90, 554], а более реакционноспособ-
ные ннтрнлневые соли восстанавливаются этим реагентом до пер-
вичных аминов [554]. Успешное восстановление нитрилов в амины
под действием диборана можно объяснить первоначальной коорди-
нации! восстанавливающего агента по атому азота и образованием
нитрилиевого комплекса, который уже более склонен к присоедине-
нию гидрнд-иопа [90]. В отличие от полного восстановления до
первичных аминов под действием борогидрцда натрия и диборана
восстановление алкил- н арилнитрилиевых солей триэтилсиланом
bt3Sil! происходит частично, образующиеся имины можно под-
вершуть гидролизу in situ н получить алифатические и ароматиче-
ские альдегиды с выходами 41—90% [554]
С=Г° вТ г=г УглеР°д-азОт в меньшей степени, чем связи
Однако ипчмлтпп подвергается каталитическому гидрированию,
лов в первичные атпи?”06 Кй7алптическое гидрирование нитри-
творе а-^Х^прГГб'^С ST™
ХноТ'в^уТтат^прщоещш образование вторичных
го амина, к промежуточное Я пР0Лукта реакции, первично-
тельного дезаминирования [90 '55Ы "дп°СЛедуюи1е|'° восстанови-
* Действительно, каталитиче-
t>7b
яв-
аминов [90],
• - - - - *1 Н с
катализатора; в
в нейтраль-
ное гидрирование нитрилов в присутствии .
ляется удобным способом синтеза Х рвичных аминов
Каталитическое гидрирование нитрилов Р”ЧнЫх г" •— '
использованием платинового или пачпя™ Ожно пРовестн ।
кислой среде продукт реакции - Хи “'° Ка“
„он - вторичный [90]. Каталитическое гидов™ Г
подобно восстановлению комплексными ₽ Ние НитРилов
Ти в контролируемых условиях н по пучить и И М0ЖН0 пР°вес-
ле гидролиза in situ - альдегиды. Таким иутеГб.’.f И3 Них ПОс’
синтез труднодоступного миндального альдегида А?СнУптг^
путем гидрирования нитрила миндальной кислоты н»л(°Н)СН0
Ренея в смльнокислои среде [556]. Ароматические Н1,келем
станавливаются (гидратированными электронам^ “ трилы яс-
ские альдегиды при облучении в водном Рраствопе ,„₽п°Ма™Че’
ходы составляют 40-75% [557]. растворе щелочи; вы-
8.2.3. ПОЛИЦИАНОСОЕДИНЕНИЯ
Накопление в одной молекуле высоко полярных и не создающих
пространственных затруднений цнаногрупп зачастую придает по-
лицианосоединенням уникальные физические и химические свой-
ства. Выделение полицианосоединеннп в особый класс было сде-
лано сравнительно недавно [558—560], это оправдывает вынесе-
ние их в отдельный раздел в рамках данного изложения.
К полицианосоединения.м можно отнести большое число струк-
тур: от полицианоалканов до полицпаноалкеиов и полицианоаро-
матическпх соединений. Несмотря на различие в структурах, у
них, однако, много общих химических характеристик, и именно на
основе этого родства решено рассматривать химию полицианосо-
единенпй с единых позиций. В данный обзор, естественно, вошел
не весь имеющийся обширный материал по данному вопросу
[558]; цель данного обзора состоит в том, чтобы на избранных
примерах обрисовать наиболее яркие особенности синтеза и реак-
ционной способности полицианосоедпнений. Там, где это необхо-
димо, речь пойдет о синтезе и реакционной способности моноциа-
ноалкенов и дицианосоединений.
8.2.3.1, Методы получения полицианосоединений
Методы, приводящие к полицнаносоедннениям, основаны, глав-
ным образом, на тех превращениях, которые применимы для
еза простых нитрилов (см. разд. 8.2.2.1). Они вкл'°чаелинения
"не цианогруппы путем реакций замещения или р ощью’
а также создание новых тройных связей углерод ® субстратов.
Реакций отщепления или перегруппировок. Вв д J
677
,,„v ..miKiroviniv » реакции конденсации или от-
уже. ^примёпяХ, ^частности для синтеза циаиоалкенов.
“ХГХ- циа’<ла меди(1) в диметилформамиде „р„
п п'с ' >опсходит гладкое замещение фтора в 1,3,5-трициапо-
гТб-трпфторбеизоле [561], а также одновременное замещение ал-
Йснгруппы н .хлора в 1-алкокс11-3,5-дпхлор-2,4,6-тр|1циаиооепзо-
ле 156’]' это реальный путь синтеза гексацпанобеизола. Обработ-
ка цианидом медп(1) при повышенной температуре (150 С) ис-
пользуется также для превращения цис- и транс- 1,2-диподэтнле-
нов в нитрилы малеиновой н фумаровой кислот, соответственно
[454] Лучшим методом синтеза интересного полнцпаноалкаиа
цианоформа (трицианометана) является реакция броммалоноди-
нптрнла с цианидом калия [558, 563]. Вообще же полпцпаноал-
каны чаще всего получают замещением галогена в галогеиоцпа-
нах действием а-циан'ометилеиовых соединений или образованных
из них карбанионов. Примерами превращений такого типа могут
служить взаимодействие ацетонитрила с хлорцианом при сильном
нагревании, приводящее с количественным выходом к малоиоди-
нитрплу [563], и синтезы трицнаиометанов RC(CN)3 [558, 563],
в частности гексацианоэтана [558], реакцией хлорциана с нат-
риймалоиодпиитриламн или натрийпентацианоэтанпдом Na~
-C2(CN)s, соответственно. Используя хлорциаи и гидрид натрия,
можно осуществить ступенчатое превращение циклопентадиенид-
иона (942) в трпцианопроизводное (943), которое обработкой
хлорцианом в присутствии трихлорида алюминия можно перевес-
ти в тетра-л пентацпаноцпклопентадиенид [(944) и (945)1 [5581
(уравнение 280).
CN
(942) (943)
стен
CN А1с|з>
CN
NC' CN NC 1 CN
NC' । CNA1C|3 ме-' ' CN
(280)
(944) (945)
Присоединение цианида к
вить синтез цианоалкенов
мер, акрилонитрил получают
И
кратным связям позволяет осущест-
их полицианоаналогов. Так, иапрп-
в промышленных масштабах присо-
в присутст-
и хлористого водорода
мнение цианид-иона к
и последующее окисление обра-
''<л,:д2ми азота представляет со-
полициано-п-бензохиноиов. Примером
-дпхлор-б.б-дициано-п-бензохииопа
ia (946) (уравнение 281;
(948;
(946,
рпимбмпли TI. - * MdLLlIl
дииенисм цианистого водорода к ацетилену пни 80 °C
вин хлорида меди(1), хлорида аммония и
(катализатор Ныоланда) [454] Присоедии
хлорированным п-беизохинщщм н пХ
бой'Хщ '1ианог1‘^о-но>,ов оксидам
ooh Общин метод синтеза полицпанп-и
(948)ТизСЛУ2 35 6 ,10ЛуЧе,,Ие 2-3-Д»хлор
X = Y == Cl)’ ’ HTCcPHTe3P'2'3C5H63OXmiOllL '^-иеии
х = Cl, Y = CN) '[454 ’558] Лор'3’6_Д1’и11ано''г-бензохииопа
678
О
X II
он
•CN
у XCN
ОН
(947)
о
(946)
Реакции присоединения к легкодоступному тетпяпи и
(ТЦЭ) (см. ниже) - еще один источник интевегнт Леиу
соединений. Так, реакция цианида натрия с избытком^Хциано'
этилена (949; R = CN) (во избежание образования ЯИИ™Т
калов, см. ниже) приводит к пентацианоэтаниду натрия (950)
[558]. Каталитическое восстановление ТЦЭ или, что более доб
но, восстановление действием иодистоводородной или меркапто'
уксусной кислоты - удобный источник получения тетрацианоэта-
на (951) [558, 559]. Окисление ТЦЭ водным раствором перокси-
да водорода — лучший способ синтеза тетрацианэтиленоксида
(952) [558]. Присоединение натрийброммалонодинитрила к ал-
килиден- или арилиденмалонодинитрплам, как полученным спе-
циально, так и приготовленным in situ из алифатических или
ароматических альдегидов или алифатических кетонов и малоно-
динитрила, в присутствии водного раствора иодида калия (реак-
ция Впдеквпста) представляет собой общий способ синтеза раз-
личных тетрацианоцнклопропаиов (953) [558,563].
Me Me
Na* \ /
Х'С\ . /CN NaCN NC\ /R N3t BrC(CNl2 /ч /CN
xr_Cz -«--------- XC=CZ ------------► X-_____
Nc/ \CN R=CN NC/ XR NC'^CN
(950) (949) (953)
HSCH2CO2H I НгО2(водн.1
R = CN R=CN
(282)
NC. /CN
/C—C\
NCZ I I XCN
H H
(951)
(952)
* ОТУЧИТЬ с помощью
Ненасыщенные полпцианосоедпненпя можно п • - создания но-
Реакцнй отщепления, к которым относятся Р свЯЗВ уГЛерод—
в°й цианогруппы за счет образования р ю связь угле-
азод, н процессы, позволяющие ввес™ „гщсгтствует циано-
Р°Д —углерод в субстраты, в которых у и примере синтеза
гРУппа. Оба этих подхода иллюстрированы на пр
обладающего уникальной структурой азаполн
„„„чиоацетнлена, о0Л * N) Его можно получить De;i
мкина (^^.„да с пентоксидом фосфора [558] (РЭТо
ацетпленднкарб01^ |)ТДр|1ла дегиДратацНеи амида, см с. 658) Р ’
мер образования J » И м газофазного пиролиза 4,5-д||Ц11а|'о л«-
с высоким выходец >можно легко получить реакцией ц £
дитиолоиа-г котор м с последу1ОЩС11 обработкой o6pa3oB*a
натрия с серо пк д)1МСркаптомаЛеодпнитрила фосгеном
SSeS) [500,558].
NC\C/S Na COCI2 NC 600—800 oC С
=° -----------* III (283).
NC S f
CN
(76%)
Полицианоалкены часто удобно получать с помощью классиче-
ских приемов галогенирования и последующего дегалогенирова-
ния или дегидрогалогенирования; типичным примером является
бромирование — дегпдробромнрование трпцианоэтана, приводящее
к трицианоэтплену [559]. Mono- или дпхлорированпе 1,2-дициано-
циклобутана пеитахлоридом фосфора и последующее дегидрохло-
рирование триэтнламином пли дехлорирование никелем Ренея
позволяет получить 1,2-дицианоциклобутен [564].
Для синтеза полицианоалкенов с успехом применяют также
и реакции конденсации с участием малоноднннтрнла или его про-
изводных. Классическим примером является конденсация альде-
гидов п кетонов (954; Х = О) с малонодннитрплом (955) по Кнё-
венагелю в присутствии основного катализатора (например, алк-
оксида металла, пиридина, пиперидина или амида натрия) или
уксусного ангидрида, позволяющая в ряде случаев получать с от-
'[4541Ь563В565]1ОМ нлиденмалонодинитРиль1 (956) (уравнение 284)
R‘\ ZCN
С=Х + CH2(CN)2 •—> ХС=С,
S
I
c
II
s
"CN
С.
(284)
реак (954) (955) (956)
кие, нТлишеньГ'^и однако’ '«смотря на широкое распростране-
которых карбоиппп?,Т0₽Ь1Х недостатков'- трудная доступность не-
затрудненных егбе-тпп* соединен< невозможность использования
сов, главным обдппм ' а также протекание побочных процес-
Реагировав|цего мт/₽еакции присоединения по Михаэлю непро-
[454, 465]. Общий J '0Д|1нитРила к аф-непредельиому нитрилу
трудности и получит! ,°Л’ КОТОРЬ1Й позволяет преодолеть все эти
ми (YS-.go^j 7 миденмалонопитрилы с высокими выХ°Дд]
— N~ М+) (легкоплг В К0Нденсации кетимата металла (95 >
680 тУпного в результате реакции нитрила
реактивом Гриньяра или литийорганическим гг.».
мя эквивалентами малоиодинитрила К уединением) с дву-
гексапдиоиа-1,4 (957) с малонодиит ри ,юм в Конденсачия цикло-
па приводит к бисаддукту (958) котопис? “РисУтствии (J-алани-
сукцииимидом или бромом в присутствии ,ри окисленни К-бром-
амина превращается в 7,7 8 8-тетпя..и-к, ' Иридина или т₽иэтил-
(959) (уравнение 285) [558, 563]. рацианохинодиметан (ТЦХМ)
(285)
Этот метод является общим методом синтеза технически важ-
ных полицианоалкеиов вследствие легкой доступности исходных
циклогександнонов-1,4, получаемых восстановлением по Берчу
1,4-диметоксибензолов. Конденсация альдегидов и кетонов с
цианалкнлиденфосфоранами [454] или цнаналкилфосфонатами
[566] по Виттиту, позволяющая получать <хф-ненасыщенные нит-
рилы с отличными выходами, неприменима для получения или-
денмалононитрилов, так как соответствующие дицианофосфорные
реагенты слишком устойчивы. Тетрацианоэтилен можно получить
окислением малоиодинитрила хлором или монохлоридом серы при
450 °C [558, 559]; более удобным методом синтеза служит авто-
конденсация диброммалонодинитрила в присутствии медного ка-
тализатора [558, 559]. Было показано, что при кипячении произ-
водных ацетонитрила (например, этилцианацетата) с тионнлхло-
ридом образуются соответствующие 1,2-дицианоэтилены с хоро-
шим выходом (65—70%) [567]. Однако при использовании этого
метода в случае малоиодинитрила тетрацианоэтилен был получен
с низким выходом [567]. Тетрацианоэтилен (962) можно полу-
чить также реакцией тетрациано- 1,4-дитиина (960) с цианидом ка-
лия с последующим окислением образовавшегося анион-радика-
ла хлором (уравнение 286) [558].
кек . -/ет
ксХ Хсы —
(960) (9«1) №
Дицианосоединения нередко оказываются конмными^^ме-
тами перегруппировок диазидопроизводных. К. о.-тазидобеи-
ром может служить пиролитическое превращен
золов (963) в цммис-1>4-дпцианобутадиены-1,3 (964) ()Рав
681
с| N'<\ /CN
ХС=С/ (286)
,п. лги лчя] К тому же результату приводит окисление
Ггда^чи’нобензолов' (966) тетраацетатом свинца [98, 454, 458]
пероксидом никеля (IV) [454, 458]; по-вндимому, ннтермед,,.
ат мп служат нитрены (965). Было показано, что такое превра-
щение можно осуществить и просто окислением 1,2-дпампнобензо-
м кислородом воздуха в присутствии хлорида медн(1) и пири-
дина [568]. Конротаторным замыканием никла в промежуточно
образующемся /{//с^г/г-дициано-о-хннодцметане^ (968), определяе-
мым необходимостью сохранения молекулярной симметрии, мож-
но объяснить стереоспецифическое образование граде-1,2-дпциа-
но-1,2-д11гидробеизоцпклобугепа (969) при вакуумном флеш-пиро-
лпзе 2,3-дпазпдонафталпна (967) [569].
Термолнтнческая перегруппировка 2,3-диазидо-1,4-нафтохино-
на (970) приводит к ожидаемому продукту раскрытия цикла,
о-фталонлнианнду (971), а также к необычному продукту —
2-аза-3-цпано-1,4-нафтохинону (973), по-видимому, с промежуточ-
ным образованием 2-азидо-2-цнаноиндандиона-1,3 (972), который
можно выделить [570].
682
I
О
О
(973)
(289)
8.2.3.2. Реакции полицианосоединений
Химические свойства лолициаиосоетине ”
столько реакциями самих циаиогрупп 'скопк*1 0ПРеДеляются не
нием электроиоакцепторных цнаногочпп и/, ° Совмес™ым влия-
местнтелей. Так, например, это приводит к °ведение Других за-
стабильности соседних анионных центров а резкому П0,,Ь!Шению
реакционной способности ненасыщенных ’coentuTJ “ повышению
нуклеофильного присоединения и циклоприсоединения P„KuHS,x
этн аспекты реакционной способности полиииан^™™ Именно
двинуты на первый план в приведенном ниже обзоре “ ВЫ'
(1) Реакции с электрофильными реагентами
Трицианометан (цианоформ) существует в таутомерной кетен-
имнннои форме (NC)2C=C=NH [558], поэтому он легко пеаги
рует с галогеноводородамн н дает 1-амино-1-галоген-2,2-дивдано-
этилены (NC) 2С=С (X) NH2; реакция, по-видимому, начинается
с протонирования и завершается присоединением галогенида
[558]. 1,2-Прнсоединенне хлора к тетрацианоэтилену и 1,6-при-
соединенпе хлора к тетрацианохинодиметану, напротив, катализи-
руются хлорпд-ионами. Полагают, что при этом вначале протекает
нуклеофильная атака хлорпд-ионом с образованием хлоркарбани-
она, а затем хлорирование этого карбаниона (т. е. хлор по отно-
шению к ТЦЭ и ТЦХМ ведет себя как нуклеофил, а не как
электрофил) [558]. Как ни странно, галогеноводороды (НС1, НВг,
HI) и бром легко присоединяются к днцианоацетилену, образуя
соответственно 1,2-дицнановинилгалогенпды (стереохимия продук-
тов не установлена) и 1,2-дибромфумародииитрил (выход 57%).
Поскольку маловероятно, чтобы к тройной углерод-углеродной
связи в дпцианоацетилене с крайне пониженном электронной
плотностью происходило электрофильное присоединение, объясне-
нием этих реакций, скорее всего, служит предположение о перво-
начальной нуклеофильной атаке галогенпд-ионом с образованием
карбанионного интермедиата, который далее подвергается прото-
ннрованию или галогенированию. Как и в случае ТЦЭ и. 1Ц- < ,
ярко выраженная пониженная электронная плотность тройной уг
лерод-углеродной связи в дицианоацетплене способствует, по- и
Днмому, тому, чтобы обычные электрофильные реагенты в
ли в роли нуклеофилов. . „ VVPVr-
Тетрацпаиэтнленоксид ведет себя обычно в реакш . - 'е_
иым ангидридом и ацетплхлоридом, давая соотв (558].
тат тетрацпанэтиленгликоля и 2-х.юртетрациано т
(2) Реакции с нуклеофильными реагентами *
Способность цпаногруппы актпвнроВ1ТЬ_^ойа”таке хорошоиз-
нУю углерод-углеродную связь к нуклеофи. ’ ‘ та 1егк0Сть. с
вестна. Простейшей иллюстрацией этого с. па3нообразные
которой акрилонитрил присоединяет с
лн (СПИРТЫ тиолы, ампносоединения, соединения с актив-
нуклеофилы (сшщ ,. служит основой использования
»««« tSZlf. как а Мо-
этою соеди к не1|асЫ1ЦСН11Ые полициапосоедпнеппя обладают
довало ожи'* ’ опной способностью к нуклеофильному прпсо-
П0ВЫ» пио в Рэтом отношении илпденмалоноиптрилы напоминают
кпрбошльиую группу [560]. Это служит еще одним доводом в
пользу существования аналогии между группировкой NC-C-CN
,, кислородом И, следовательно, между малоподинптрнлом
Н C(CN)2 и водой [560, 572]. Поэтому гидролитическое расщеп-
ление илидсималонопптрплов [974; X = C(CN)2] до карбонильных
соединений п малонодпнптрпла (с предварительным образовани-
ем гидратов) [560] протекает аналогично хорошо известному об-
ратимому процессу гидратации карбонильных соединений (урав-
нение 289).
, ОН
R'\ н2о R\|
,С=х з=ь ,С-ХН
R'\
V=O + H2X
(289)
(975)
(976) (977)
R2/
(974)
X = C(CN)2, О
Гидролиз тетрацианоэтилена (978) в нейтральной или слабо-
кислой среде, напротив, дает трнциаиовиниловый спирт (980;
Х = ОН), образование которого можно объяснить первоначальной
гидратацией и последующим отщеплением цианистого водорода
(уравнение 290) [558, 559]. Гидролиз тетрацнаноэтилена в ще-
лочной среде протекает более сложно и приводит к пентациано-
пропенид-иону (NC)2C=C(CN)C_(CN)2 за счет сопряженного при-
соединения первичного карбанионного аддукта к иепрореагн-
ровавшему тетрацианоэтилену [558, 559]. Прн обработке циано-
форма водой гидратируется лишь одна из трех цианогрупп что
вполне согласуется с представлением о том, что он реагирует в
таутомерной кетепиминной форме (982) (уравнение 291) [558].
ксЛ i1 /CN
С—с —HCN
NCZ \CN
(979)
х\ zCN
— )с=с(
х/ ^CN
(981)
NVC/X -
NC/C \NH2 *
(983)
- я-Ме2&1СцН,
ХС\ /CN нх
NC/ C\CN
(978)
NC.
CN
;c=c:
(980)
(290)
NC.
/C=C=NH
NC/
(982)
X = OH, OR, Nr2, сн2соме”
684
NC\
— ,CH—CNH2 (291)
NCZ
(984)
HX
X = OH
Повышенная реакционная способност.
отношению к нуклеофильной атаке вип,я Ц”анобензола по
„дна из цианогрупп замещается уже ппосто пп Т°/° факта’ 4Tfj
при комнатной температуре; выход пента, обРа6°тке водой
95% [573]. Спирты (иди. лучшей составляет
также реагируют с гексацианобензолом поп ко?? а.лко*<сиды}
туре с замещением одной циаиогруШ1Ы и обпяя» темпеРа'
эфиров пентацианофенола [562]. При нагреваний ТПЧ Пр°СТь,х
тамн в присутствии мочевины также происходит п„7п С° СПИр’
ное замещение двух из четырех циаиогрупи- мпж» последователь-
циановиннловые эфиры (980; X = OR) и вne3v?,?° '8Ыдел"ть,тРн-
реакции, ацетали дицпанокетена (981; X = OR? [553е «тЬНрйшеи
ты присоединяются к цианоформу в его кетениминной формой
дают 1-алкокси-1-амино-2,2-дицианоэтилены (983- X = OR) 155R1
Высокой склонностью к реакциям нуклеофильного присоединения
обладает тронная связь углерод-углерод в дицианоацетилене-
реакция его со спиртами и тиолами происходит очень четко ппи
комнатной температуре и приводит к смеси цис- и транс-изоме-
ров 1,2-дициаиовинилового эфира или 'тиоэфира, в которой обыч-
но преобладает цис-изомер [558]. Легкость протекания этих ре-
акций особенно заметна при сравнении условий их протекания с
условиями, необходимыми для присоединения тех же агентов к
диметиловому эфиру ацетилендикарбоновой кислоты (150—200°С),
что свидетельствует о повышенной реакционной способности трой-
ной связи углерод—углерод в дицианоацетилене к нуклеофильной
атаке. Интересными примерами взаимодействия полииианосоед.;-
неннй с серусодержащими нуклеофилами могут служить реакции
тетрацнанэтплеиоксида (986) с дпалкнл- и диарилсульфидами.
Эти реакции протекают с разрывом С—С- и С—О-связей и при-
водят с хорошими выходами к илидам серы (987) (выход 62—
85%); их можно использовать также как эффективный метод по-
лучения труднодоступного карбонилцианида (988) (уравнение
292) [558].
NC\ /°\ ZCN • -/СХ
RaS + —* RsS-C
NCZ 'CN XCN
(985) (986) (987)
Реакции полицианосоедпнений с азотсодержащими ну клеофн
ламп протекают так же, как с кислород- н серусодержа
единениями. Так, по аналогии с реакцией со спиртами ( _
ше), цианоформ (982) присоединяет пиперидин и д£>е СОО1В с
вующий амнналь [983; X = N(CH2)s] !558ф BW-
себя и тетрацнаиоэтилен (978)— при деиствш р двух циа-
Ричных аминов происходит последовательное з.. ^ •^9g0.
обРазоМ1,||е BHa4a'’’%?i'.(TP'-NR.>) [558.'559]. При
a~NR2), а затем аминалеп (981, i
685
.с\
(292)
(988)
. „шянохнноднметаиа с первичными и вторим-
взаимодействии wpaw провесТи ступенчатое замещение двух
,|ЫМП амниамн также M o9Ti,;iieii реаг11руеТ с аммиаком и с
цпааогрупи 1&о»1 • ‘ птнВРТСТВуЮщпс бжЦтрпцпановпннльпые) про-
гидразином, давая couiьс ?
взводные [558, 559].
(NC)2C==C(CN)—(NH),.-C(CN)=C(CN)2 и=1 или 2
Вследствие повышенной склонности к реакциям нуклеофильно-
го Внписоед неиия дицнаноацетнлен реагирует с аммиаком,
пепв ч ыми » вторичными аминами при комнатной (или понижен-
ной) температуре и дает смесь цис- и траяс-1,2-двциановинил-
амнносоеднненпй [558]. Реакция тетрацнапэтиленокспда с пири-
дином протекает так же, как и с сульфидами (см. уравне-
ние 292), и приводит к соответствующим пиридинии-илпдам; в этом
случае побочный продукт реакции, карбонилцнанид, выделить не
удается [558].
Дополнительным доказательством высокой реакционной спо-
собности полициаиоалкеиов по отношению к нуклеофильному при-
соединению служит их легко протекающее взаимодействие с карб-
анионными реагентами. В отличие от рассмотренных выше реак-
ций присоединения кислород-, серу- и азотсодержащих нуклео-
филов прп реакции с карбанионами обычно образуются устойчи-
вые 1,2-аддукты. Примером реакций этого типа может служить
сопряженное присоединение реактивов Гриньяра [560] и трихлор-
метаннд-пона СО. [574] к илнденмалоноиитрплам. Подобное
1,2-присоединение наблюдается и при взаимодействии тетрацнано-
этилена с еиолизующимися кетонами, например с ацетоном, ката-
лизируемом трнфторидом бора или ионами серебра [ (978)->(979)',
Х = СН2СОМе] [558, 559]. Сопряженное присоединение аниона
малоиодинитрила к ТЦХМ (959), напротив, сопровождается по-
следующим отщеплением цианид-иона, так что суммарный резуль-
тат реакции состоит в нуклеофильном замещении одной из циано-
групп карбанионным реагентом [558].
Выше уже обсуждалась склонность цианоалканов к а-депрото-
„п?~Вани,° См' с' 670 674); оно протекает с еще большей лег-
, с ИР11 наличии двух или большего числа цианогрупп, в ре-
аниоиа 6 Tnv° )Ееличнвается стабильность соответствующего карб-
натпийм™^а"РИМеР’ малон°Аинитрил легко превращается в
ле прн обваботкеТРНЛ С ПОМо1ЦЬ|° различных методов, в том чпс-
пщридом натш я вЭпТ. м°ЛЬНЫМ Г™0?™ ЭТ0КС!'^ н
ожидать, иатрнймалонодиннтппчФ°бСВДе [563]' КаК И слеДовало
ностыо и Может поетеппрпат1Р Л Г’а;1ает высокой нуклеофиль-
ная моно- н днгалоген1шпт Ь РазиооГ>разные превращения, вклю-
дналкилиропание (пои РЛрйрНе ^папРимеР, пРп действии брома),
(при реакции с этилхлопЛ^13'.'1 метилн°ДиДа) И ацилирование
число примеров, свидете^ст-пг*иатом) [563]. Известно большое
686 " },0Щих о том, что анион малонодП-
нитрила воупает в реакции конденсации с кяпг
ствеипымп пм соединениями [5631 Cv» . ™н“Шим и ии
эффект ипаногрупп в полициаиоалкаиах (ЛР,"?'Й стаб«ли3уЮщий
ме, тетрациапоэтаие) настолько велик ЧТп t Ример’ в Чиайофор-
устойчивы в виде соответствующих анионов соеДинения более
ствуюшпх свободных кислот. Эти соединен^ В 8ИДе Со°твет-
ваны в водном растворе, и их лучше всего R олностью ионизо-
в виде солей щелочных металлов или чртвй„тЬ1ДеЛЯТЬ и хРанить
солей [558, 572]. Наибольшей устойчивостью аммониевых
аноцпклопеитадиенид-ион (945). Он vctohoL Дает пенташ<-
400 °C и не протонируется даже в хлорной кнстоте В°ЗДухе да
но, соответствующая ему кислота сильнее хлорной' [558?°н1еЛЬ’
тря на исключительную устойчивость, полнциа. окарбанною’,
реагируют, правда, вяло, с электрофильными реагентам СХ
ми реакции такого типа могут служить бромирование и ’
рование трнцпанометанида серебра (NC)3C- Ag+ [5581 Искпю
чением является тетрацианоциклопентадиенид-пон который
будучи ароматическим соединением иебеизоидной структуры тегко’
подвергается электрофильному замещению (например, нитрова-
нию, галогенированию, ацилированию по Фридетю — КпагИтгН
(схема 293) [558]. р ф •’
Электрофильная способность полнцианоалкенов видна также
из того, что тетрацпаноэтплен (978) может реа™рОва ь, ов
вированнымп аренами (например, с простыми эфир Ф ‘ ]
Ы.М-диметпланиЛином) и даже с неактивированншш ^аренами
(бензолом, толуолом) в присутствии тР,,хлор“®нн^о1ХдныеК
катализатора, давая соответствующие трицпанови ^^^р Щ)ЯХ
например (980; X = n-McjNCeHj^ [эо8, 5 J. _ который
Удается выделить промежуточный аддукт ) исстедовано, .
Распадается по механизму Е\сВ; было “X Х'чпано-
каким именно образом происходит его пр 1 ‘ моЖет всту-
впнильный продукт [576]. Тетрацианохи рОваннымп аре-
пать в аналогичные реакции заметенп
ками [558].
687
(3) Реакции циклоприсоединения
- т. „оосты.х а В-пепасыщеппых нитрилов, таких, как
CnocOl)"?,„ и 2-дпциапоэтплсиы (например, фумародинитрил,
акрилонитрил . ,ц|1КЛОбутеи), выступать в роли диено-
маЛ0Д,,П,,;Ром;юЙ Р “кН»" классического [4 + 2] -циклопрн-
Ф1'"* Дильса-Альдера) хорошо известна [175,
кгт ^771 и ие будет подробно рассмотрена в данном разделе.
Гуммаопый электроиоакцепториып эффект цианогрупп в полициа-
шиткенах увеличивает эту способность до такой степени, что
по мцпаиоалкепы относятся к числу наиболее реакционноспособ-
ных партнеров в реакциях циклоприсоединения. Так, тетрацнано-
эттеи (990) хотя и пе реагирует с соединениями с неактнвиро-
ванной двойной связью углерод-углерод, ио с алкенами с повы-
шенной электронной плотностью (например, со стиролами, вини-
ловыми эфирами, внннлеульфпдамн и енаминами) легко вступа-
ет в реакции [2 + 2]-циклоприсоединения и образует производные
цик.тобутапа (992); реакция проводится при низких температурах
(0—30°С), выходы продуктов 80—94% (схема 294; X = Аг, OR,
SR, NR?) [175, 438, 558, 559, 578]. Отметим при этом, что [2+2]-
цпклопрпсоеднненпе фторалкенов к алленам протекает при тем-
пературах 100—200 °C [559]. Поскольку в соответствии с правила-
ми сохранения орбитальной симметрии согласованный процесс
термического [2 + 2]-циклоприсоединения алкенов запрещен, его
рассматривают как ступенчатый бирадикальный процесс [175,
438]. С предположением о синхронном механизме не согласуются
кинетические параметры и часто низкая стереоспецифичность ре-
акций [2+ 2]-циклоприсоединения тетрацианоэтнлена, хотя они
вполне отвечают возможному ступенчатому присоединению через
хсн=сн2
(989)
+
(NC)2C=C(CN)2
(990)
X—СН—СН2
(NC)2C—C(CN)2
(991)
X
NC---------CN
NC CN
(992)
X=OEt
MeOH
(294)
Mc2CO
(994) (91%)
ЕЮ
MeO—CH—CH2
(NC)sCH—C(CN)2
(993) (70%)
688
j 4-диполярныи интермедиат (991) 1430 -70
реакции отвечают и данные о влиянии „о,,™ °Ь ТакомУ ходу
среды на скорость [2 + 2] -циклоприсоедины Риих заместителей и
„у [438, 558, 578, 579, 581]. ТаКР Хр^ня к ™Рациаио?тиле.
и-метоксистирола к ТЦЭ в иитроМ?;^',М^,«'«лоприсоедиХе;
нуту, тогда как в четыреххлористом углеХ* Д За ОДНУ ««-
до конца и за месяц. Ступенчатый механизм РпрТ?ИЯ-Не Доходнт
соединения ТЦЭ к алкенам с повышенной Реакции циклопри-
стыо подтверждается еще и тем, что как бы ектР°ННой плотно-
1,4-диполярные интермедиаты (991) ’МОжип «,° Показано недавно,
растворителями [ (991 )-*(993) ] [579,582] ил? что"1” "оляРны“и
но, подходящими 1,4-диполярофилами н’аппи!ЩеСолееваж'
[(991)-*(994)] [583]. Ф И’ напРИмер ацетоном
Тетрацпаноэтилеи выступает и в попн „„ .
циях [4 + 2]-циклоприсоединения (реакция Дитьса"—ТД В реа,Г
он, по-видимому, наиболее реакционноспособный из ве«ЬДера);
филов. Согласованное циклоприсоединение к 1 3-диенам
(996)] протекает быстро и с высоким выходом ’(>90%)
натной или более низкой температуре (уравнение 295 R1- D2
= Н, Me или Ph, R3 = R4 = Hf Ц&, 5$, 559 578, 58^] ~ R
= H, Me или Ph, R3 = R4 = H)
(998) (997)
Тетрацпаноэтилеи настолько легко вступает в реакции цикло-
присоединения, что его часто используют как «ловушку» для не-
стойких промежуточных 1,3-дпенов [558, 585]. Наиболее показа-
тельным примером служит факт «поимки» неуловимого прежде
валентного изомера цпклооктатетраена (уравнение 296) [558].
CN
(296)
689
i 2 ппеновыи компонент пространственно затруднен ,Иц
Если же W-ai'etr пе может принять цисоидную коифор-
по структурным Р успе1|шого осуществления [4 + 2[-цпкло-
машпо, неоо, > J происходить дииолярпое [2 + 2[-ппклопри-
присоединения moa^tjPr2 = R3 = R4 = Мс)-(997) + (998'}].
соединение [I • . осОбепностп в полярных средах, оно может
В"еК°ТДинственным’направлением реакции. Еще одним примером
секционной способности ТЦЭ в реакциях циклоприсо-
еХшя может служить его взаимодействие с норбориадпеном
99 результате которого происходит редкий тип гомосопря-
«много [2 +2+ 2]-присоединения (уравнение 297) [558]. Дици-
яноапетитеи по способности выступать как 2л-компонент в реак-
циях [4 + 21 -циклоприсоединения и родственных реакциях сравним
С тетрациаиоэтиленом пли даже превосходит его [5о8]. Так, он
образует с высоким выходом цпклоаддукты с такими 1,3-диена-
МИ как бута диен-1,3 и цнклопентадпен, даже при О С, а с нор-
борнадпеиом (999) вступает в гомосопряженное [2 +2 + 2]-цик-
лоприсоединение.
К
(1000) (999)
NCC=CCN
(297)
Тетрацпанэтиленокспд (1002) также активно участвует в ре-
акциях циклоприсоединения; он легко присоединяется к простым
алкенам, труднее к алкинам, и дает 1,3-циклоаддукты [тетрагид-
рофураны (1004) и 2,5-дигидрофуракы (1005)], обычно с очень
высоким выходом (схема 298) [174, 558, 586]. 1,3-Диены и аллены
могут выступать и как моиоеновые компоненты в таких реак-
циях циклоприсоединения [(1002)->->-(1006)]. Даже бензол с те-
трациаиэтиленокепдом дает циклоаддукт (1007). Столь неожидан-
ное поведение тетрацианэтилеиоксида по сравнению с другими
оксиранами еще раз подчеркивает, к каким разительным измене-
ниям реакционной способности приводит сосредоточение несколь-
ЦоггЛп%’0ГРУПП В молек>'ле' Д° недавнего времени не было едп-
меа.. 'г'ЛгтТпИ0С"ТеЛЬ110 ПуТ" Реакиин " Т0ч»0Й природы интер-
сида 158f? 4171 ySIMr0 в„ц”клопР11соеЛинеиип тетраннанэтилепок
-р««"
лепоп и папнпр n 5°’ 58G^ к Двойной связи углерод—угле-
ются лишь с представпениемМ о'13ОТОПНОМ Эффекте 15881 согласу’
карбонил-илидпого иптермадиата ЖТ инклоприсоедииепип
зовапного I З-дппотя т„ДИаТа 11003^ (пример октетиостабили-
д ля’ л"шениого ортогональной двойной связи
[174]), образующегося в результате обратимой,
рапового цикла в тетрацианэтиленоксиде
раскрытия окси-
(4) Восстановление
Благодаря суммарному электроноакцепторному действию цна-
ногрупп полицпаноалкены проявляют высокое сродство к электро-
ну. Они могут выступать как л-акцепторы (л-кнслоты), спо-
собные к переносу заряда от соединении с повышенной электрон-
ной плотностью, л-доноров (л-оснований), например дненов, ал-
кенов с электроиодонорными заместителями, аренов [589]. Обра-
зующиеся при их взаимодействии интенсивно окрашенные комп-
лексы с переносом заряда (л-соли) в некоторых случаях удает-
ся выделить. Об образовании таких комплексов в начальных
стадиях реакций полицианоалкенов (например, в реакциях
[2 + 2]- и [4 + 2]-циклоприсоединения тетрацианоэтилена) мож-
но судить по появлению окраски после смешения реагентов. В ре-
зультате полного переноса заряда образуется устойчивый аннон-
радпкал, который часто удается выделить в виде относительно
устойчивой соли. Так, тетрацнаноэтилен (1008) и тетрацианохнно-
Диметан (1010) восстанавливаются металлами. иодидамп и тре-
тичными аминами до соответствующих анион-радпкалов (
(•ОН), дающих характерные спектры ЭПР и относительно .
чивых как в растворе, так и в виде металлических со. ,
РЫе легко можно выделить.
(NC)2C=C(CN)2
(1008)
(NC)2C=^—^=C(CN)2
(1010)
—> (NC)2C—C(CN)s
(1009)
(nc)2c-hQ-c(cx)’
(1011)
(299)
(300)
691
„ прприосом заряда, образованные ТЦХМ с таки-
Комплексы с перепосо Р валсп (ТТф) (I0I2> Х = S)
ГД0НХТсеХф>-львален (ТБеФ) (1012; X = Se) [591],
[590] п те1Ра ®' 6е особое внимание, поскольку по своим
привлекли к - )ческ1|м св0йствам они напоминают металлы,
оптическим п э. Н тцХМ—ТТФ обладает наиболее высо-
Так. например, • ))3 всех пзученпых органических соеди-
юнпй 'ТоХшшая в этом отношег.. с графитом [558, 592]. «Ор-
ган ескне металлы» этого типа уже используются в электронной
промышленности [593]. Быстро развиваются и продолжаются ис-
следоваппя в этом направлении, целью которых является улуч-
шение свойств таких комплексов либо путем сравнительно прос-
тых структурных изменений л-акцептора (ТЦХМ) [594] или
л-донора (например, Т8еФ) [595], либо путем создания новых
аналогов ТЦХМ [596]. Примером реализации второго подхода
может служить синтез интересного сернистого аналога (1013),
который, несмотря на формальное сходство с ТЦХМ [597], ока-
зался электроизолятором!
X X
s
.CN
NC.
ХС=С^ 'С=С^
NC/ у' X1N
(1013)
электрону у полнцианосоединений
свойства мощных окислителей. Так,
окисляет даже
к
X X
(1012)
Повышенное сродство
придает полицнанохннонам
например, 2,3,5,6-тетрацнано-п-бензохинон [558]
воду; хорошо известна способность 2,3-хлор-5,6-дицнано-п-бензо-
хннона выступать в роли дегидрирующего агента, особенно в ря-
ду стероидов [598].
8.2.4. ДИПОЛЯРНЫЕ СИСТЕМЫ
Тройная связь углерод—азот входит в качестве структурного
элемента (по Крайнев мере, в формальном смысле) в состав трех
важных и структурно весьма близких классов днполярных соеди-
нений. Это оксиды нитрилов (1014) [599], а также их азотные и
ип1«\Д!1Ь1е аналог"’ "итрилпмины (1015) [174] н нитрнл-илиды
нений. Это оксиды нитрилов (1014) [599]
(1016) [174].
R—C=N—О Ri—c=\_ NR2
О014) (1015)
Т0ИлимЛины°иК^ССИф1!КаиИН Хь10СгеНа [173] ,„пИ„липи,дШ,
1 3 лнпочями 1ТР11Л'И-',ИДЫ являются октетностабплизовапными
и (2101 н (211н'0Н?1И 0Ртогональ”Ую двойную связь [см. с. 523
выделить (5991 я котоРые оксиды нитрилов иногда удается
выделить [599], а иитрнлимнны п нитонл-нлплы iwwtiii как
реакционноспособные интермелпяти У известны как
воре. и'срмедпаты, существующие лишь в раст-
692
R1—C—N—C.R,2
(1016)
иптрнлокспды, HII-
8.2.4.1. Методы синтеза оксина
нитрилиминов и иитрил-ИЛвдовРИЛ08’
Наиболее общий метод синтеза нитрилоксил™
и инТрнл-илидов состоит в дегидрогалогенирован’ НИТрилиминов
х (см. разд. 8.1.3.1) галогеназометннов (ги1рокси ‘ егкодостУп-
гидразоноилбромндов или -хлоридов и Имидои„Пп„О1ихдоР"дов,
лизируемом основаниями. Дегидрогалогепированщобычно’ Ката‘
дят при низком температуре с использованием трИЭТ„ыпров°-
дегидрогалогенирование N-монозамещенных гидразонов™до в
или -бромидов - удобный метод синтеза нитрилиминов in X
например C.N-дифеиилнитрилимина (1018) (уравнение чпь
R=Ph, Х = С1, Y = NPh) [61, 174, 175, 177? 264,Р265д] Генерщ
рование нитрил-илпдов tn situ также гладко происходит при деги-
дрохлорировании имидоилхлоридов, катализируемом триэтилами-
ном (уравнение 301, R = Ph, X = Cl, Y = л-О2Х'С6Н4СН) [173—
175, 264]. Нитроловые кислоты (легко получаемые при нитрозиро-
вании первичных нитроалканов или при нитровании альдоксимов
действием оксидов азота) легко, а иногда и самопроизвольно от-
щепляют молекулу азотистой кислоты; это может служить реаль-
ным методом синтеза оксидов нитрилов (уравнение 301; X=NO2,
Y = О) [600]. Превращение гидразонов а-нитроальдегидов (их
можно получить путем диазосочетания первичных нптроалканов)
в натриевые соли под действием изопропоксида натрия в изопро-
пиловом спирте и последующий их пиролиз прн 80 °C позволяют
осуществить синтез нитрилиминов (уравнение 301; X = ХО»,
Y = PhN) [174, 175].
х\
,С=М.
(301)
R—C=N—Y
-HX
(1017) (l0!8>
Удобным способом получения оксидов адплнптрилов in situ
с высокими выходами является генерирование и самопронзволь
иын распад неустойчивых диазонневых производных, образх '
ся при нитрозпрованпп диазокарбонильных соединении н*”Р _
натрия в хлорной кислоте при 0°С (уравнение 1 • ’’
X==N2+,Y = O) [601]. Преимуществом этого методапо ?
ию с методом синтеза оксидов нитрилов путем де р ^мОче_
лЛаНия (см- вы'Де) является возможность 11СП°‘Ч продуктов.
ильных субстратов и получения щелочела • _ Р и npjf
0 ......трилов можно получить с высоким! _апианоферра-
та(1п?"И альдокс,,мов прн 0 °C подде'', ™1ещелочного металла
) или, предпочтительнее, гипобромпта
A.-iic'iiiTeibiioe образование оксидов нитрилов
,600]. “Х'Хсвинца удается осуществить лишь ®
иод действием т рщ 1 (см также с 538) N-Монозамещец.
сык.альдоьсима}.^1П^в действ|1„ теТраацетата свинца пре-
ные гидразе'нь ‘^тве||||0 в питрилпмнпы (уравнение 301; X = Н,
уР1Щмк ) 11741 Было показано, что дегпдроднмеры арплальдок-
л„ Гготовые изображают по-разному (см, с. 538 и схему 95):
11,60 как дп-К'-окснды альдазинов (282), либо как азокснангид-
пит! 1283)1 подвергаются термолизу в мягких условиях (кипя-
п ИЙ бензо! или хлороформ) и дают с хорошим выходом (—60%)
оксиды нитрилов [602]. Ценность этого метода особенно высока,
так как с его помощью можно получать оксиды нитрилов в со-
вершенно нейтральных условиях. Оксиды нитрилов получают так-
же с хорошим выходом при дегидратации нитроалканов действи-
ем фосфорплхлорпда или, что более удобно, фенилпзоцианата в
присутствии следов триэтиламина [600]. Этот метод используют
для синтеза оксидов нитрилов и из них путем циклоприсоедине-
ния (см. ниже) изоксазолов — исходных соединений в новом ме-
тоде построения коррпновой системы [603].
Используются также и другие методы получения оксидов нит-
рилов, нитрплпминов и ннтрил-нлпдов, основанные на превраще-
ниях различных гетероциклических систем. К ним относятся вос-
становительные, термические и фотохимические фрагментации
1,2,5-оксадпазолов (фуразанов) или их N-оксидов (фуроксанов),
рассмотренные выше (см. с. 660), которые хотя и не используют-
ся широко, но представляют потенциальную возможность синтеза
как нитрилов, так и их оксидов. Термическое или фотохимическое
отщепление диоксида углерода из соответствующих пятичленных
гетероциклических субстратов также представляет собой общий
метод получения in situ нестойких 1,3-диполей. Так, например,
термическое отщепление диоксида углерода из легкодоступных
1,3,4-оксатиазолонов-2 (1019; X = S) позволяет получить нптрил-
сульфнды (1022, X = S) [604, 605], которые хотя и невозможно
выделить, но удается «уловить» реакцией с подходящими диполя-
рофплами. К тем же соединениям приводит и фотофрагментация
1,о,2-оксатиазолпи-5-олятов (1021; X —S): предполагают, что при
этом образуется тиазириповый интермедиат (1020), подвергаю-
щимся далее обратимому раскрытию цикла [241, 606]. Еще одним
примером синтеза этих интересных тиааиалогов оксидов нитри-
D?=Mni!ieYCj2-c?ITb полУче''не т situ бензоиитрилсульфида (1022;
ЛОПОПЯ ’пт М За СЧеТ 0ТШ'еПЛе1Ч1Я двух молекул фтористого во-
ный спосоГ?ппч«3рЛ11М1'110СУЛьфур-'111фтоРида (1026) [607]. Удоб-
тоэлиминимвт Н1,ТРИЛ1’М"НОВ (1022) in situ состоит в фо-
азолнй-5-о£ятов)\102?КХ- \п?ТР°Да И3 С|,Д11ОНОВ (1,2,3-оксадн-
В неустойчивых nr.,., ,Л и последующем раскрытии цикл-
X = NR) [241] нР|?тпС^УТ°ЧНЫХ диазиРн,|ах (1020) (схема 302
скпм отщеплением азота’от'Тб М0ЖНО полУчит1, также термине-
а301а от 2,5-днзамещеиных тетразолов (1023)
R1 N
'у#' ''-X
О-----1=0
(1019)
O—P(OR4)3
(1024)
_n Phx M Cf
F i /CH-j-N=S^
H/k-^ F
(302)
(1026)
[173 175, 177]. Практическая ценность этого метода, однако,
весьма ограничена из-за довольно высоких температур, требуемых
для осуществления этого превращения (160°С в случае’2,5-днарил-
тетразолов и ~220°C для алкилзамещенных тетразолов). Ннт-
рил-илиды (1022; X = CR?) можно генерировать in situ путем
термического отщепления диоксида углерода из оксазолпнонов-5
(1019; X = CRj) [608] или отщепления фосфата (термического
пли фотохимического) из Д4-1,4,2-у5-оксазафосфолинов (1024)
[174]. Однако гораздо более общим методом получения in situ
неустойчивых нитрил-илидов является хорошо известное фотоли-
тическое раскрытие цикла в легкодоступных (см. разд. 8.1.2.3)
А’-азиринах (1020; X = CR$) [174, 230, 251]. Полагают, что обра-
зование нитрил-илидов в этом случае обусловлено раскрытием
Никла в /ьл*-синглетном возбужденном состоянии А’-азирина, их
Удается «уловить» и изучать в углеводородных матрицах при низ-
ких температурах или же использовать как субстраты в самых раз-
нообразных реакциях 1,3-диполярного циклоприсоединения [1/4,
230,251].
8.2.4.2. Реакции оксидов нитрилов, нитрилиминов
и нитрил-илидов _
Строение 1,3-диполей, содержащих троинУ^aH0H114ecKHX пре-
ззот, можно описать как резонансным г р а))больший вклад
Дельных структур, изображенных на схе:''® „ структуры (Ю27а)
® основное состояние вносят целиком ок можно объяснить
1 (10276); наличием структур (Ю-7в) i ( Щ)И нуклеофильно
Длонность этих соединений вступать в р
. I З-днполярного циклоприсоединения. Вклад
1,3.Присосдииешш .<арбиП1Ый характер, не столь у*
структура 13-диполях, содержащих тронную связь
отчетливо прй некоторых обстоятельствах она мо-
жетвыйпГнТ’первын план.
R-C^N-X
(1027а)
r—C=N=X R—C=N—X
(10276) (1027b)
(303)
R—C—N=X
(1029)
R—C=N—X
(1028)
Помимо реакций циклоприсоединения наиболее изучены реак-
ции довольно часто выделяемых оксидов нитрилов [609]. За иск-
лючением их отношения к гидролизу и восстановлению, эти окси-
ды мало чем напоминают по своим химическим свойствам исход-
ные нитрилы; они ближе к азидам и диазосоединениям.
(1) Реакции с электрофильными реагентами
1,3-Диполи, содержащие тройную связь углерод—азот, индиф-
ферентны по отношению к атаке электрофильными агентами.Так,
оксиды нитрилов инертны в реакциях галогенирования (бромиро-
вания, иодирования) [23] и, хотя они легко вступают в реакции
1,3-присоединения с галогеноводородами (HCI, НВг, HI), давая
соответствующие гндроксимонлгалогеииды (1030; X = С1, Вт, I),
эти реакции начинаются, как полагают [609], с атаки галогенид-
ноиом и лишь затем происходит протоипрование. Можно, одиако,
предложить и противоположный механизм, поскольку оксиды нит-
рилов, нптрнлпмины и иитрпл-плиды способны протонироваться,
давая иитрнлпевые соли [609]. Оксиды нитрилов реагируют и с
карбоновыми кислотами, приводя к ацнлгидроксаматам (1037).
Их образование можно объяснить тем, что в результате 1,3-при-
соедпнеиия образуются ацнлгидроксиматы (1033), которые затем
подвергаются перегруппировке типа перегруппировки Чепмена
(уравнение 304) [609]. Аналогично реагируют карбоновые кисло-
ты и с ннтрилимнпамн, приводя к неустойчивым ацнлгндразона-
там, которые подвергаются самопроизвольной перегруппировке
ниеМзе0б) И'.шГы соответствУ10Щие гидразиды (1038) (уравне-
\И ССТН0 несколько примеров 1,3-присоедпнення
пазуются О -inV* К .........трнлов, в результате которого об-
нитонлов п,и™ ‘гидрокс"МО!!лгалогениды (1032) [609]. Оксиды
Р руют также с ангидридами и хлорапгидридами
696
по типу 1,3-присоединения, давая соответственно бис-
кисДоТ ппцкснматы (1034) и О-ацилгидроксимоилхлорнды
» . . >.х Х\
A\C=NOH *------- R'-CsN-O -----► )c=!W
R1/Z
(1030)
(1031)
(1032)
|r2CO2H
OCOR2
I
Ri—C=NOH
(1033)
I
|(R2co)2o
OCOR2
Ri—(L=NOCOR2
(1034)
|R!Coci
CI
I
R1—C=NOCOR2
(1035)
(304)
r OH 1 О
I , I’
Lr'—c=nocor2J r'—c—nhocor2
(1036) (1037)
[OCOPh 1 О COPh
I II I
PhC=N—NHPh J —> Ph-C-NHXPh
(305)
(1038)
(2) Реакции с нуклеофильными реагентами
Способность оксидов нитрилов, нитрилиминов и нитрил-илидов
подвергаться нуклеофильному 1,3-присоединению может соперни-
чать с их склонностью (как 1,3-диполей) вступать в реакции 1,3-
диполярного циклоприсоединения. Однако легкость, с которой оп-
ределенные галогеназометины (гпдроксимонлхлориды, л-моиоза
мещенные гидразоноплгалогениды и N-етор-алкилпмидоилхлори
Ды) подвергаются нуклеофильному замещению, непосредст е
связана с промежуточным образованием соо^ве9с.т9®-1 поодукты
шюнноспособных 1,3-диполей [см. разд. 8.1 )]•
Реакций нуклеофильного присоединения к оксид _ в
идентичны продуктам, полученным из Г1’ДР°К91,2., ’так₽напри-
Результате реакций замещения [см. разд. 8; ‘ . п011 35-'С окси-
меР, при обработке разбавленной серной кп • вые кнслотЫ
нитрилов гидратируются, давая гидр • ОСУЛЬф11Д.нона
(’041) (схема 306) [23, 609], а при действ ни взрос
получаются тиогидроксамовые кислоты ( (тиолов и тио-
'Рчсоединение спиртов, фенолов и их Tiioai • . действием
Фенолов) к оксидам нитрилов происходи йТ соответст-
“11слот пли оснований как катализаторов Щ „ тноГИД.
Роке0 К гиДРоксиматам (1042; R2 — А ' г н0 реагируют со
Роксиматам (1043; R2 = Aik или Аг). Аналои
женоламп тиолами н тпофеиоламн н нптрилнмины
спиртами. JH ’O ш’нпе первичных и вторичных алкнламинов а
Яшиных арпламинов к оксидам нитрилов протекает гладко «
R-’
R'—C==NOH
(1049)
| R2MgX
NO2
К'—C=NOH
(1048)
CN
1
ri—C=NOH
(1047)
NO;
CN
OR2
I R2OH ♦ -
Ri—C=NOH •«------- R1—CssN—О
(1042) (1039)
OH 1
_ri—c=nohJ
(1040)
I
О
II
Ri—C—NHOH
SH 1
R>_ c=nohJ
(1044)
S
R1—C—NHOH
SR2
I
R’—C=NOH
(1043)
|RiNH «
nr!
R1— C=NOH
(1045)
(1041) (1046)
приводит к соответствующим амидоксимам (1045) [23, 609),
тогда как аналогичное присоединение аминов к пнтрплпмпнам
дает ампдразоиы [610]. Примером 1,3-присоеднненпя анионов
может служить реакция оксидов нитрилов с цианид-ноном, при-
водящая к циаиоокснматам (1047) [609], а также реакция с ннт-
рит-ионом с образованием интроловых кислот (1048) [609]. 1,3-
Прпсоеднненне реактивов Гриньяра к оксидам алифатических п
ароматических нитрилов приводит к кетокснмам (1049) [23, 609],
которые используют как исходные вещества в синтезе нзоксазолов.
(3) Реакции циклоприсоединения
Наиболее важными и наиболее изученными реакциями 1,3-ди-
полей, в состав которых входит тройная связь углерод—азот, яв-
ляются, пожалуй, реакции 1,3-диполярного циклоприсоединения
[173, 174], Оксиды нитрилов, питрплпмниы и нитрил-нлпды легко
вступают в реакции [32]-циклоприсоединения практически со
всеми типами кратной связи. Реакции этого рода, в которых при-
нимают участие соединения с двойной и тройной связью угле-
(,‘JS
оД_азот, уже рассмотрены аыше ГСм г -о
а реакции с другими соединениями, в котоп°26 и разд- 822
/я связь прн атоме углерода, пр!1ве^.°Рвд «РНсУтствуеЛп^’
о ц- 2] -циклоприсоединения оксидоп u а СХеме 307 р! рат'
К-ивмипов [173, .74] и ни^Х.ид <173 >74 аб1Ц.7
протекают с высокими выходами и служат n li73’ 174- 230 Д’
разнообразных пятичленных гетеродин^ *? ПОЛУче»”я ай
?“о"от СВ? «“"и /
X=O, N«, UKz), Л 1,3,4-диоксазолинов Л2 i ч < алКеНов (.051-
„ дг-оксазолинов и их тиоаналогов из капб™. ,4'оаддиазолииов
пильных субстратов, соответственно [(1052? и ™окарбо-
NR пли CR2], а также нзоксазолов, липазой 053); Х==О
кпнов [(1054); X = О, NR, CR2], ’ '"!ра-жтов и пирролов из
(307)
(1053)
(1052)
Процессы этого типа обычно характеризуются высокой стереоспе-
цифичностью [174] и регпоспецнфичностью [174]. Например,
циклоприсоединение монозамещеиного пли 1,1-дизамещенногоа,
, кеиа к оксиду нитрила обычно протекает таким о Раз°п'
1 аддукте сохраняется стереохимия исходного алкена ( ₽ -
' стереоспецифическое цис-прпсосдвиение), и обраа71 J |е ре.
ио- пли 5,5-днзамещенные Д2-изоксазолины, а „чоксазотины
пюизомеры — 4-моно- пли 4,4-днзамещенные
[174]. Для объяснения регпоспецпфичпостпгра.
цессов [3 + 2]-цнклоприсоедивеппя с успех вопрос 0
яично-орбитальный подход [174]- Несмотря » ’оляриого прп-
сиихрошюстн образования связен в процессе , ' опр||Соедпне-
^аднснпя спорен [372, 373]. реакции Р+ I ^^дов следует
1и|я оксидов нитрилов, HHTpiuwMHHOB н г протекающнмй
считать полностью согласованными ироне’)Ь1е Состоя,111Я-
’еРез высокоупорядоченпые циклические п Р - () рег11о-
с'10ианчем для такого вывода служат в параметры реак'
ЧЧфичпость процессов, а также кине высокие отрпна-
т’И~ог,1оснтсльио ипзкпе энергии активация
иные значения энтропии активации.
599
8.2.5. ИЗОЦИАНИДЫ
п метавнего времени изоциаииды RNC [451, 612] были от.
Д°л но мало изучены, не столько из-за того, что, как извест-
HOC?RH1 они обтадают крайне неприятным запахом, сколько из-за
Н0 подходящих общих препаративных методов их синтеза.
взпХбразному развитию химии изоцианидов; изоцианогруппа
стала одной из важных функциональных групп.
8.2.5.1. Методы получения изоцианидов [309, 612—615]
Несмотря на амбидентный характер, цианнд-нон алкилируется
преимущественно по углероду при действии алкнлгалогенпдов и
-сульфатов; при этом в основном образуются нитрилы, нзоциани-
ды являются минорными продуктами (см. с. 647). В то же время
«связывание» цианид-иона в комплекс с тяжелым металлом, таким
как серебро или медь, приводит к тому, что алкилирование идет
преимущественно по азоту. Таким образом, при реакции цианида
серебра (I) или цианида меди(1) с алкилиодпдом (1055) обра-
зуется нзоцнанидный комплекс (1056), который реакцией с циа-
нидом калия можно перевести в свободный изоцианид (1057)
(схема 308) [309, 612—615]. Выходы изоцианидов в этом класси-
ческом методе синтеза, однако, невелики, не выше 55%• Эффек-
тивность этого метода можно заметно повысить, если в качестве
пзоцнапируюшего агента использовать комплекс цианида серебра
с трифеннлметилфосфоннйгалогенидом, поскольку его можно
применять в гомогенной среде в апротонных растворителях [615].
Было показано [616], что алкилгалогеннды, в особенности алкпл-
иодпды, в некоторых случаях можно перевести в изоциаииды прак-
тически с количественным выходом, проводя реакцию с дпциано-
аргентатом тетраметиламмоння. Интересно отметить, что ацилпо-
ным выхпГппУ10Т С и1,а"ВДом «Ребра при —30°C и дают с отлпч-
выходом неизвестные ранее ацилнзоцианнды (RCONC) [617].
AgCN
RI -------► RNC-AgI
(|055) (1056)
OSO2Ar
RN=CH
(1062)
+ - СПС13
rnh2cci2 -«----------
основание
(1059)
A-SOjCl
RNHCHO
(1061)
700
RNH2
(1058)
основание |
---------* RNC
(1057)
RN=CHC1
(1060)
(308)
Et3P
RN=CCl.
(1064)
Et3P;X==S)
iWpt»o) |
Ь RN=c=X
(1063)
Другой классический метод синтеза
ная реакция Гофмана) [309, 612—6151 сосХГКп°В (кгРб™амин-
первичного амина с хлороформом в присутст»™ взаимадействии
вания, например спиртового раствора едкого Л„ СНЛЬ|10Г0 осно-
трег.бутокспда калия в трет-бутнловом спноте Х/Ли РаствоРн
цианидов в этом случае можно рассматривать кХ n2°BaHHe И30’
воначальной атаки амина (1058) дихлопкаЛР ультат пеР'
“ итермеи.ту <1059), », Итор““Р“
«.элиминирования и а-элимннированпя обпазуетЛДОватель,1Ь,х
51057). При термической реакции арнламинов?с трихлора“ом
натрия, приводящей к арилизоцианидам, по-видимому ппоХТ
аналогичные процессы [309, 612-615]. В своем классическом ви
Де карбиламинная реакция не носит общего характера и выхо-т
продуктов обычно невысоки. В присутствии катализаторов Фазо
вого переноса превращение аминов в изоцианиды идет в одну
стадию; выходы изоциаиидов в этом случае сравнимы с выходами
которые наблюдаются при использовании двухстадийной методи-
ки с участием формамидов, наиболее широко применяемой для
этой цели [618]. Недостатком этого метода, однако, является то,
что его нельзя использовать, если субстрат или продукт чувстви-
тельны к основаниям.
Изоцпаниды могут быть получены с хорошими выходами де-
гидратацией N-монозамещенных формамидов ^1061) действием
самых разнообразных реагентов (например, Р2О3, РС13, SOC12,
СОСЬ, ArSOaCI, POCI3) в присутствии основании (например, дн-
и триалкила.мпиов, пиридина, трет-бутоксила калия) [309, 612—
615]. Этот метод наиболее широко используют для синтеза изо-
цианидов. Образование пзоцианидов в указанных условиях мож-
но легко объяснить, предположив протекание а-элп.миннровання
в промежуточном формпмидате (1062) под действием основания
(на примере дегидратации под действием ареисульфонплхлорида).
Особенно удобным реагентом для крупномасштабных синтезов
изоциаиидов из N-моиозамещеиных формамидов является фосген
в сочетании с триэтпламином или пиридином; выход изоциаиидов
70—95%. В то же время опасность, связанная с необходимость
работы с фосгеном, делает этот метод непригодным „
иевных лабораторных синтезов пзоцианидов. Более уд •
мепее эффективным реагентом является фосфорплхл Р
сутствпи пиридина пли трет-бутоксила калия. ’ „омаТ11.
’’Рет-бутоксида калия особенно необходимо при ( Р пр„.
ческих изоциаиидов, образующихся с низким! „ _ лацпР суль-
мепении в качестве катализатора пиридина. Д' . действием
Фоиильиых производных Ы-етор-алкилформамн п_Ю°С слу-
Фосфорпл.хлорида в присутствии трпэтиламин Р Нидов (на-
Ж|,т удобным методом синтеза сульфопилме • пх большое
пример, п-толуолсульфоиплметнлнзоцианнда;. рместо фосгена
начепие как синтетические интермедиаты I Г к110 исполь-
Реакцпц дегидратации формамидов в пзоц ‘
„„(Ьосгеи Гперхлор(метил)формиат, С1СО2СС13] [620]
30ВапТеагснт Вильсмсйера, хлорид хлордиметилформам11ДИния
. <т_гнС1 С1~ который можно легко приготовить in situ из
пиметмформамида и тионилхлорида [621]; выходы „зоцианидов
оставляют 60-90%. с отличным выходом (89—91 /0) изоцианц.
ТЫ Обоазуются при дегидратации формамидов действием трифе-
питфосфпна и четыреххлористого углерода в присутствии три-
лтпламппа 1622]; использование трифеиилфосфпна и диэтилового
эфира азодпкарбоновой кислоты менее эффективно [623]. Изо-
цпашиы являются конечными продуктами восстановления изо-
цианатов или изотиоцианатов (1063; Х = О пли S) действием
триэтплфосфпта пли тризамещенных фосфинов; они образуются
также при восстановлении фосгенимпнов (1064) [309, 612—615].
Эти методы, однако, имеют ограниченное применение, поскольку
в условиях проведения реакции (повышенная температура) воз-
можна перегруппировка изоцианида в нитрил [457, 458].
8.2.5.2. Реакции изоцианидов
Изоциаиогруппа— единственная функциональная группа, со-
держащая двухвалентный углерод. Эту группу можно изобразить
как резонансный гибрид двух предельных канонических форм
(1065) и (1066), первая из которых указывает на ее двухвалент-
ный, карбенный характер, вторая — на возможность использова-
ния ее как нуклеофильного агента [624]:
N=C: -<-> —№=с
(1063) (1066)
Отсюда следует, что реакционная способность изоцианогруппы
,,СЛеД0В/ТелЬН°’ нзоинанпдов) определяется ее стремлением
хапяктрп^к е ^тоичивое ковалентное состояние; для изоциаиидов
гаются эпР^тпп<ьЦ11И в„недРсния’ а не присоединения; они подвер-
гнутся электрофильной, а не нуклеофильной атаке.
(1) Реакции с электрофильными реагентами
РУЮТР с галогенами (хлором^бтомТм ИаНИДЬ\ (1067) бУРн0 Реаги'
ратуре эти реакции становятся ’ ИОДОм^ "Ри низкой темпе-
дуктами являются соответств-1ошпГР°‘"ИрУеМЬ|1''"1’ Пр11 ЭТ0М пр°’
(1068; Х = С1 Вг ИЛИ п 1чпоЩоС |1МИ|1°карбонилгалогениды
реагируют цзовдаииды c l ’ 615’ 625]' СтОль же энергпчи<
НВг’ HI); проведение реакпшЛ??"1 гад2ге||0в0д0Р0даМ|1 <ИС1
ожидаемые аддукты —(Ьопмии РП ~ 5 С позволяет получит’,
водородных солей) (1069- X __1!д<г|,1ДГа-г,ог('|Ц|ды (в виде галогено-
по-впдпмому, устойчивы к пейсто ’ Вг' I) 625]. ИзоццанпДЫ,
10 с ацилхлоридами паппптпп |1Ю ajlKI,JIHPyiouinx агентов [309],
702 ’ Р "1в’ Реагируют охотно и дают а-кето-
„доилхлориды (1070) [309, 625]. Прн ВЯяик
ВЫХ кислот с изоцнанидами получа1Отр,’;Даи«одейсТВ11н карбоно-
формамиды (1072), это можно объяснить ппгДЫ Кис;10т (1073) и
зованнем ацилимидата (1071), который буХТЖут°ч'1ь,м
ру|Ощим агентом, реагирует со второй моле™ М°ЩНич а^и-
лоты, давая указанные продукты [309 roJ °ы каРб°новой кис-
переносом в аналогичном промежуточном,я,„ Но Сильным
можно объяснить протекание уникальной тоД™”"™ <1074)
денсацнп изоцианидов с альдегидами „лн к;т*компонентиой кон.
ми кислотами, в результате которой полх-чя™" 11 каРб°новы-
-................. (реакция Пассернни) [309, 612? g]g'ag2qOKC1,KaP®"
оксамиды (1075)
R3
I
R>NHCOCOCOR2
R4
(1075)
R3 OCOR2
:n-c—c=nr>
R4
(1076)
R5-
R\ R3 O COR2
n-c-c-nr‘
Rg/ I
R4
(1077)
R2co2h,
R3COR4,
RSNHR6
R2CO2H
R3COR4
, х2
R'NC -----> R'N=CX2
(309)
(1067) (1068)
I
r2CO2h MeCOCl
HX
COMe
[R'N=CHOCOR2] PhN=C—Cl R'N=CHX
(1071) (1070) (1069)
R’NHCHO + (R2CO)2O
(1072) (1073)
«скрепляющего узла»
и соединениями и
а-а.мннокпслот
Способность Изоциаппдов выступать в роли < так.ЖР поп
® таких многоцентровых конденсациях пР0!’в-’1Яе_п1,ири„яин ,f
взаиМОдействии пх с аминами, карбонильным! оТ1|1ЧНЫ-
каРбоновым1| кислотами. При этом в мягких усл . .I.iinvlir ПОТ
nn7^X?''laM" (80—100%) получают пР0ИЗ®°д”“еая конденсация»
*077) [612, 627, 628]. Такая «четырехкомпонентна вдсь иС.
°И11аццдов имеет большое значение в С1|И ’шегося высокой
ПЩ|ьзовать для нового синтеза пептидов, отлпчаюшеюс
/(|О
TII0 независимостью от природы присутст-
стереопзбирателыю > (.отору10 вводят в реакцию в качестве
вуюшей а-амичоки . - тем, что в одной реакции пр0ПСХ()
одного из ком лоне! > х ПСПТ(1ДНЫХ связей [612, 629].
днт образование дв>л
(2) Реакции с нуклеофильными реагентами
,, „„„аногруппа обладает преимущественно нуклеофильным
и ГАПОМ (см выше) II поэтому устойчива-к нуклеофильным
характером пзоциапнды довольно стабильны в ус-
ГоХ^шелоХо гидролиза [309, 615] п не реагируют с такими
hvk еофилами, как спирты и амины, даже в жестких условиях
И.яппимео они не изменяются при нагревании с аминами до
120°C) [625]. Однако, как и в случае нитрилов (см. с. 665), в ус-
ловиях кислотного катализа изоипаниды вступают во взаимодей-
ствие с нуклеофильными агентами [630]. Именно этим можно
объяснить легкий кислотный гидролиз изоциаиидов, приводящий
к формамидам (1080; X = О), из которых в свою очередь можно
получить первичные амины (1081) [309, 625]. Аналогично реаги-
руют изоцианиды и с сероводородом, давая тиоформамиды (1080,
X = S) (схема 310) [612].
В отличие от свободных пзоцианидов, инертных по отношению
к нуклеофилам, комплексы пзоцианидов с переходными металла-
ми гладко реагируют со спиртами и тиолами и дают соответст-
вующие формпмпдаты и тпоформимпдаты (1082; Х = О или S),
как правило, с отличными выходами (80—95%) [625, 631]. Реак-
ции присоединения этого типа легко осуществить, обрабатывая
изоцианид спиртом или тиолом в присутствии хлорида меди(1);
промежуточно образующийся комплекс карбен—металл удалось
обнаружить [631].
В условиях кислотного катализа идет присоединение аминов
к изоцианпдам. Так, прн взаимодействии пзоцианидов с гидро-
арпламинов получают соответствующие формамидины
( 1 ° ,= ^г’ R3 = Н) [625], Практически с количественным
„^х°дом т°Р^амидппы образуются прп нагревании изоциаипда с
6191 °ПпПР" в присутствии хлорида медн(1) [309,
СИЛЯМ,„,Р" ®заим°Деиств1‘и пзоцианидов с гидрохлоридом гпдрок-
ожипярмт ,отсУтствпе металлического катализатора получают
625 тпгпя фоРмамиД°кС"м (1083; R2 = ОН, R3 = Н) [309, 612,
Щенная мочевин* хлорида цинка образуется N-заме-
эффектнвны:ГХЙ^ио^\6’2;оТ1^,10р,,Д медп(1) является
анидам- Фопмяип^'зат0Рсш присоединения гидразинов к нзоци-
образуются(1083’ R2 = NMe2, R3 = Н) обычно
Ценность этой пеаЛ, , Выходом ( — 80%) [632]. Синтетическая
склонности нзо.ша,,, " нрисосдппеппя несколько снижена из-за
Катализируемая cn„JA0B образовывать смеси продуктов [633]
'тональными кислопол-' металлов реакция изоциаиидов с бпфупк-
704 " А’’ сеРУ- и азотсодержащими нуклеофилами
IlaO I 1. R3X ||
R2CHO -<--------- R'N=CR2 ---------->- R2—c_D3
2. H2o K K
(Ю86) (1085) (Ю87)
t R2M
(1079)
X
II
RlNH—CH
(1080)
l-hx ZnCb
i---- R'NC —------------>
ii+ NiijOH-Hci
(1078)
R2X11
XR2
I
R'N=CH
(1082)
R'NHCONHj
(1084)
(310)
1
R'NH2
(1081)
(например, с амииосппртами, ампиотполами, диаминами) приво-
дит к гетероциклизацип; ее можно использовать для получения
различных гетероциклических систем, в том числе оксазолинов,
тиазолннов, имидазолинов и 1,3-оксазннов [631]. Было показано,
что при такого рода гетероциклизацип фенилизоцнанпда с тносе-
микарбазидами в зависимости от природы заместителей в тносе-
микарбазиде образуются 1,2,4-триазолтионы-З или 2-амино-1,3,4-
тнадиазолы [633].
Изоциаииды, не содержащие а-водородного атома, гладко при-
соединяют лптпйорганнческие реагенты и реактивы Гриньяра и
дают металлоальднмины (1085) [634]. Эти соединения можно
использовать как интермедиаты в различных реакциях; например,
их можно превратить путем гидролиза соответствующих альдими-
иов в альдегиды (1086) с высоким выходом [634, 635] пли же
использовать как эквивалент ацил-карбапиона. Так, при их ал-
килировании и последующем гидролизе получают кетоны (1087),
также с высоким выходом (86%) [634, 636]. Было осуществлено
внутримолекулярное присоединение карбаниона к изоцпанпдной
группе, позволившее перейти от орто-метплированных арнлампнов
к индолам (1090) с высоким выходом (86—100%); реакция проте-
кает через стадии образования о-метиларплизоцпаипдов (1088)
и их металлирования, сопровождающегося циклизацией (схема
311) [637]. Было показано [637], что первоначально образующий-
ся лптийоргаипческпй реагент (1089) можно проалкилпровать, а
23 Зак. 105
703
и получить в результате 3-заме-
снова пром^лиров расширяет граниЦы данного метода
Ш1Д0ЛЫ , ‘обладающего большими потенциальными воз-
а затем
щениные
синтеза
(1091)
(1092) (Ю93)
Как и в случае нитрилов (см. с. 669—674), при взаимодейст-
вии изоцианидов, содержащих в a-положении атом водорода, с
анионными реагентами происходит а-депротонированпе, а не при-
соединение реагента к изоциаиогруппе [636, 638]. Так, изоциани-
ды, содержащие енльпокпелые а-водородные атомы, например
эфиры а-пзоцнанокарбоновых кислот или аренсульфонилметили-
зоцианиды (например, п-толуолсульфонилметплизоцпаннд) [636,
638], можно прометаллировать при действии таких оснований,
как трет-бутоксид калия или гидрид натрия — диметплсульфокспд.
Даже относительно слабо кислые изоцианиды, такие как метил-
изоцианид, удается превратить п а-изоциапокарбанионы при реак-
ции с бутиллитпем в тетрагидрофуране при —70°C [636, 638].
Прн реакции а-алкилированпых нзоцианидов, например, цикло-
гексилизоцианида, с литнйорганпческимн соединениями идет как
присоединение по изоциаиогруппе, так и а-депротоннрование; ме-
таллирование в таких случаях лучше всего проводить с помощью
сильного основания (но слабого нуклеофила) 2,2,6,6-тетраметил-
пиперидида лития [636, 638]. Способность пзоциапогруппы ста-
билизовать отрицательный заряд па соседнем атоме была обна-
ружена впервые лишь в 1968 г. [636, 638], несмотря на формаль-
ную аналогию между изоциано- и нптрильной группами В на-
стоящее время а-нзоцианокарбанионы очень широко используются
как интермедиаты. Подробное рассмотрение всех аспектов химии
а-металлироваипых нзоцианидов невозможно в рамках данного
“X" ”-ра'..’Т|й’ од
70Ь
в ^ВуА прекрасних обзорах по химии а-изоцианокарбаниомов
[636, 638]. Изоцианиды получаются из аминов и могут бить в них
снова превращены кислотным гидролизом (см. выше), поэтому
а-металлированные изоциаииды стали использовать как замаски-
рованные а-аминокарбанпоиы, которые в свою очередь пригодны
для нуклеофильного а-амипоалкилирования, например для удли-
нения цепи эфиров а-аминокислот. Для этой цели эфир «-амино-
кислоты (1094) обычным образом переводят в соответствующий
эфир а-пзоцианокислоты (1095), который превращают в а-изоциа-
нокарбанион (1096), затем алкилируют н подвергают сольволизу
действием спиртового раствора хлористого водорода (схема 312)
[636, 638].
NHi
R'CH— CO2R3
NC NC
1 NaH I R2X
-> R’CH—CO2R3 -j— R'C—CO2R3 -------------
(1095) (1096)
NC NH, СГ
R X I EtOH. HCl R \ I
Y—CO2RS -----------> Y—CO2R3
R2/ R2/
(1097) (1098)
(1094)
(312)
Нуклеофильное присоединение а-пзоцнанокарбаинонов (1100)
к соединениям с различными полярными кратными связями С^Х
(Х==С, О, S, N) [(1099)] происходит гладко и приводит к аддук-
там (1101), которые вследствие наличия по соседству реакционно-
способной пзоцианогруппы находятся в равновесии с карбаниона-
ми (1102) соответствующих пятичлеиных гетероциклов; протонн-
рованием этих карбанионов можно получить гетероциклы (1103)
(схема 313).
R* X
/С—с'
к2/| l>R-
NC X
(ИМ)
-JR'
(1100) (1099)
R*
r2/|
R Ny/X
С
(1102)
Эти соединения можно выделить,
присутствующих заместителей они
ствующне им арома......
Rl /R"
2/С—сх (313)
Г, Jr К
CH
(1103)
хотя иногда в зависимости от
....... менее устойчивы, чем соответ-
атпческис циклические системы [636, 638].
707
23*
Гетероциклизация этого типа часто происходит с высоким выхо-
лим что позволяет использовать ее для синтеза Д2-ппрролинов
(через Д'-изомеры) и пирролов из электронодефицнтпых алкенов
(636 638], Д2-пмидазолпнов и имидазолов из азометинов [636,
638 639], Д2-тиазолинов п тиазолов из тиокарбонпльных соедине-
ний’ 1636’638, 640] п Д2-оксазолинов и оксазолов из карбонильных
соединений [636, 638] (схема 313; X = С, N, S, О). Кроме того,
Д2-оксазо.чиннл-карбанионы, промежуточные соединения в приве-
денной схеме, в зависимости от природы заместителей можно под-
вергнуть тем или иным превращениям, так что суммарным итогом
является введение функциональных групп в исходный альдегид
или кетон. Так, например, оксазолинил-карбанионы (1107;
R1 = R2 = R3 = Ph, R4 = H), полученные при реакции аромати-
ческих альдегидов и кетонов с а-изоцианокарбанионами (1105),
неустойчивы и самопроизвольно распадаются при комнатной тем-
пературе (отщепление цианата лития протекает стереоспецифи-
чески) н образуют соответствующий олефин (1108) с высоким вы-
ходом (~70%). Этот процесс рассматривают как пример редкой
1,3-диполярной [4 4-2]-циклореверсии (схема 315) [636, 638].
Хотя эта реакция (карбонил-олефинированне) может быть успеш-
но использована лишь для синтеза арилзамещенных алкенов, ее
R
,/CH-NC
R
K-BuLi ,
ТГФ-70°С
R1 Li + R3-
R\_ 4/C о
2/C-NC -2----->
R
70b
(1104)
Rs
(1105)
R2n P-r4
CN O'
Ajl
(1106)
•R3
;с—о
I I т?4
С' Ы+
(1107)
Й-Mt
p’’
Ar SO,—c—CX
, .+ I K4
L1 K
CH
(1109)
R/R2-R3-Ph,
r4>h
-ыоск
32-.c=c^R'
I
nhcho
(1110)
Ph
/C=c
Pli
(1108)
ArSO.-c-c<R .Нзо4
-л->
CH Li+
(1110)
(315)
можно применять в тех случая
более общей реакцией Виттига. ОксазшпНельзя ^пользоваться
лученные из изоциаиидов с сильный 1 НИЛ’анио11Ь1 (1107) по
местителями, напр„мер с SO2Ar, "и 10^^'°^
превращаются в С-4-карбанио„ы ( 09) = SCl’Ar’ R2 = H)],
протона; последующее самопроизвольноеX™ 0Ь1стр°г° переноса
карбанионах приводит к гетеропентаме^ Р 'Т"е иикла в этих
протонировании которых получают алкенил™™ (Н,0)’ п₽и
Всю указанную последовательность реакцийЛ^рмамидь' (НИ),
зом, рассматривать как удобный метод капбоТЛТЭКИМ °бра’
метнленироваиия [636,638]. А карооиил-формиламино-
(3) Реакции Циклоприсоединения
Благодаря своему карбенному характеру (см. выше) изоциани-
ды легко реагируют с большинством соединений с обычными
кратными связями, давая различные трех-, четырех- и пяти™
ные циклоаддукты, которые формально можно рассматривать
как продукты взаимодействия субстрата (кратная связь) с реаген-
том (изоцианид) в соотношении 1:1, 1 :2 и 2: 1. Такого рода цик-
лоприсоединение присуще лишь изоцианпдам и происходит обыч-
но в отсутствие катализатора, хотя в некоторых случаях необхо-
димо наличие кислотного катализатора. Реакции пзоцианидов с
соединениями (1113), содержащими двойную связь прн атоме уг-
лерода, протекают обычно ступенчато (схема 316). Они начина-
ются с образования цвиттер-ионного интермедиата (1114), направ-
ление дальнейшего превращения которого, по-впдимому, опреде-
ляется природой исходного субстрата с двойной связью. Непосред-
ственная циклизация в трехчленный [ 1 + 2] -циклоаддукт (1115),
если и происходит, то весьма редко. Было показано, что цикло-
присоединение такого рода наблюдается при взаимодействии
циклогекспл- и арилизоцпапидов с тройной связью углерод—угле-
род в ииаминах и напряженных циклоалкинах; продуктами реак-
ции являются циклопропенимины; выходы от умеренных до от-
личных [644]. Прп взаимодействии пзоцианидов с днметиловым
эфиром ацетиленднкарбоиовой кислоты образуется смесь продук-
тов вследствие внедрения изоцнанида как по связи С= , так ип
связи С=О [612, 643]. Чаще же первоначально образовавшими
цвиттер-ионный интермедиат (1114) реагирует ли ° a™pOI’BQ.”.
лекулоп изоциаиида, либо со второй молекулой у Р
ной связью. Образующиеся при этом новые промежуточны цвпт_
__ /nicy I /111 ft 1 пнипизуются соответственно в {етырех
тер-ноны (1116) и (П18) в пятнчлеиный цпклоад-
танам, например [1117; R —к .„„....Лцопейнщптными кетонами
643]. Реакция же пзоцианидов с элемронодеЧ пц
/например гексафторацетоном) приводит с высоким выходом
Жи к пятпчленным (2:1) циклоаддуктам (пмнно-1,3-диок-
е— . «з™««=р «О»; - О’- х - °’m «Ч-
Подобным же образом изоциаииды внедряются по двойной связи
уперод-кислород в кетенах, приводя к имино-1,3-диоксоланам
Г641 643 645]. Интересно отметить, что с простыми кетонами
(например, с ацетоном) пзоцнанпды реагируют в присутствии
трифторида бора, образуя четырехчленные (1:2) циклоаддукты
(1 2-диимииооксетаны), например (1117; R2 = R3 = Me, X = О); с
выходами 40—96% [612, 641, 643]. Катализируемая кислотой Льюи-
са реакция изоцианидов с альдиминами, напротив, дает пятичлен-
ные (2:1) циклоаддукты (имииоимидазолидины) (1119; R2 ~ Ph,
R3==H, X = NR) [643], тогда как в отсутствие катализатора та
же реакция приводит к четырехчленным (1:2) циклоаддуктам
(2,3-динминоазетидннам) (1117; R2 = Ph, R3 — Н, X = NR) [642,
643].
X
II
R’NC + R2CR3
X
^С—'C=NR‘
X'
I t
;C—C=NR‘
(1113) (1114)
(Шб)
R’NC
X
II
R2CR3
R\
,C—X'
Rs/I
R‘N=C—C=NR’
(1116)
R\ X /R2
(316)
R'N=C+ X"
(1118)
R2\
,C—X
R\ x /R2
R3/ | 'R3
R’N=C-----X
(И19)
r’N=C—С=ж-
(ИП)
С 1,3-диполями, такими как азометнпимипы [646], азометип-
плиды [380) и нитрил-илиды [647], изоциаииды вступают в более
обычные реакции [1 + 3] -циклоприсоединения и дают с хороши-
ми выходами ожидаемые четырехчлеппые цнклоаддукты —ими-
иодназетпдииы, имппоазетпдипы и иминоазетииы, соответственно.
Примерами реакций [1 -R4]-циклоприсоединения пзоцнанидов
могут служить образование нмнио-Д2-оксазолпиов (1122) из N-
ацилиминов (1120) и циклогсксилизоцнанида (1121; R2 = цикло-
гексил) (уравнение 317) [641, 643], а также синтез импнооксазоло-
710
ПОВ и иминооксазолтионов (nosi
(ШД) ацилизоцианатами и ациХоХциана^^^г^^^»11®»
- „ “и PhCH,S О2^ап,атами (П23) (урав-
О илн S) [641,643]. ’ h или циклогексил,
N=CR‘
(F3C)2c^ \)
C=NR2
(1122)
N
R'C^ \C=Y
X—C=NR2
(И25)
нение 318); R1 ==
X = О, Y = "
(F8C)2C=N-CR‘ + C=NR2
II <
О
(1120)
N
(1121)
(317)
R'CZ
X
(1128)
+ C=.NR2
(1124)
(318)
2.
3.
4.
5.
6.
ЛИТЕРАТУРА
«The Chemistry of the Carbon-Nitrogen Double Bond» ed s pat„- t <
science, New York, 1970. ' °' Pata1' ,nter‘
S. Dayagi and Y. Degani, в cc. 1, chapter 2, pp. 61—147.
S'i S,i”1(k’ T (<9реП 2“1ln Nitr°gen Compounds», Benjamin, New York
1965, vol. I, chapter 7, pp. 327—336. ’
R. W. Layer, Chem. Rev., 1963, 63, 489.
R. L. Reeves, in «The Chemistry of the Carbonyl Group», ed. S Patai In-
terscience, New York, 1966, vol. 1, chapter 12, pp. 600—614; P. Y. Sollen-
berger and R. B. Marlin, in «The Chemistry of the Amino Group», ed. S. Pa-
tai, Interscience. New York, 1968, chapter 7, pp. 367—395.
T, C. Bruice and S. Benkovic, in «Bioorganic Mechanisms», Benjamin, New
York, 1966, vol. 2, chapter 8, pp. 181—300.
7a. R. K. Chaturvedi and E. H. Cordes, J. Amer. Chem. Soc., 1967, 89, 4631.
76. A. F. Cockerill and R. G. Harrison, in «The Chemistry of Double Bonded
Functional Groups», ed. S. Patai, Interscience, New York, 1977, Suppl. A,
part 1, chapter 4, pp. 288—306.
7b. A. F. Noels, J. N. Braham, A. J. Hubert, and P. Teyssie, J. Org. Chem., 1977,
42, 1527.
M. Cocivera, C. A. Fyfe, A. Effio, S. P. Vaish, and H. E. Chen, J. Amer.
Chem. Soc., 1976, 98, 1573.
K. Blaha and O. Cervinka, Adv. Heterocyclic Chem., 1966, 6, 147.
О. H. Wheeler, in «The Chemistry of the Carbonyl Group», ed. S. Patai, In-
terscience, New York, 1966, vol. 1, chapter 11, pp 529—532.
M. L. Moore, Org. Reactions, 1949, 5, 301.
E. P. Kyba and A. M. John, Tetrahedron Letters, 1977 2737.
S. J. Angyal, Org. Reactions, 1954, 8, 197 [С. Дж. Анжиол В сб Органи-
ческие реакции. Т. 8. Пер. с англ. М„ Издатинлит, 19э6. См. с. 263].
w. A. Bergin, and J. D. Adhins,
'u^hn^nd^E^aniz92^. Chem Internal. Edn., 1977 16, 328
P. S. Tobias and R. G. Kallen J Amer. Chem. Soc 197o 97, бэЗО, и.
mans, F. H. Westheimer, K. Nakaoka, ana’RKluger, ibid•• ^975 9/,
18. E. H. Abbott and A. E. Martel, J. Amer Chem. ^a’ ^^“istry»Part 1,
19a. H. Bohme and M. M in «Iminium Sa Its. Orgam c Che У
ed. H. Bohme and H. G. Viehe vol. 9 of «Advincei n org
ed E C Taylor, Interscience, New York, 1976, chapter л, pp-
711
8.
9.
• 10.
12.
13.
14.
15.
16.
17.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
lqe , g PaM in «Tl-е Enamines», ed. A. G. Cook, Dekker. New York. 1969,
2o Икс Enamines», ed. A. G. Cook. Dekker, New York. 1969,
' chapter 2. PP-79-81 )9J2 ( 303 |Ф. Ф. Блик. В сб.: Органические
21. F. F. ;СЙ°ПМ._ IIsAannunr, 1948. См. с. 400].
реакции, i- • c' (l «в |()7з 703
23 р'аТяпЫ' in «Open Cham Nitrogen Compounds», Benjamin, New York,
19би'?^2/ Cina₽«4ret8hoden63der68’Organischen Chemie (Hauben-Weyl)», ed.
J «" '» <
г8;"Гй/а5 AUHPiih'to and o'F. Fenton, J. Chem. Soc. (C), 1971, 80.
rJ^cVnilLdF L. Scott. Tetrahedron Letters, 1970, 2993.
К ^ug“rg8j SAlba2'auer,7F.4Kafig. and S Penninger, Annalen, 1977, 624.
О Toaster Org. Reactions, 1953, 7, 327 [О. Тоустер. В сб.: Органические
оеакшш т’ 7 Пер. с англ. М, Издатиилпт, 1956. См. с. 409].
М. М. Regie, /. Vitrone, and М. D. Swerdloff, J. Amer. Chem. Soc., 1977,
F'Juher and .4. E. Bottcher, Chem. Ber., 1975, 108, 1475.
D. H. R. Barton, N. K. Basu, M. /. Day, R. H. Hesse, M. M. Peerie!, and
A. N. Starratt, J.C. S. Perkin I, 1975, 2243.
E. Enders, in «Methoden der Organischen Chemie (Houben-Weyl)», ed.E. Mul-
ler, Thieme Verlag. Stuttgart, 1965, vol. 10, pari 3, pp. 490—522.
S. Л4. Parmerter, Org. Reactions, 1959, 10, 1 [С. M. Пармертер. В сб.: Орга-
нические реакции. Т. 10. Пер. с англ. М, Издатинлнт, 1963. См. с. 7].
Е. Enders, in «Methodcn der Organischen Chemie (Houben-Weyl)»,
ed. E. Muller, Thieme Verlag, Stuttgart, 1965, vol. 10, part 3, pp. 522—544.
R. R. Phillips, Org. Reactions, 1959, 10, 143 fP. P. Филлипс. В сб.: Орга-
нические реакции. Т. 10. Пер. с англ. М„ Издатинлит, 1963. См. с. 148].
Н. Lund, a cc. I, chapter 11, рр. 505—564.
S. Yamada. N. thota, and К. Acliiwa, Tetrahedron Letters 1976 1001.
R. V. Hoffman, J. Amer. Chem. Soc, 1976, 98, 6702.
R. K. Olsen and A. J. Kolar, Tetrahedron Letters, 1975, 3579.
Л В Hendrickson and D. D. Sternbach, J. Org. Chem, 1975, 40, 3450.
n и a*d У B' Marlin’ Org. Reactions, 1965, 14, 52 1Г. Цаугг,
см c 65]K' B C6Z Органнчес|'11е PeaKmni. T. 14. Пер. с англ. M, Мир, 1967.
w(°98, 6033Шапа1,е’ T- Matsubara, and A. Touch,, J. Amer. Chem. Soc,
H. Quast and L. Bieber, Angew Chem. Internat Edn 1975 14 428
У. L Chow. Tetrahedron Letters, 1965 2473 ’ ’
К B8BeXcker and E^n",os^H^ Chim. Acta, 1977, 60, 507.
Groups», ed. M. pa’tai’ Interscien^^1’0 v'st,ry °f Double-Bonded Functional
ter 8, pp. 694—696- C A flrMd ^e'v У?гк1 19771 Suppl. A, part 2, chap-
с. G. McCarty В ее I Cham ? 6 Ап№Л Ctem- Int<'rnal Edn. 1969, 8, 535.
- G. Ц7. Buchanan and ’в Гn? PP' 303~464-
51. M. Kobayashi, M. Yoshida ad Н м’ C?na?' J' Chcm- 19771 551 1437.
52. . Bjorgo, D. R. Boyd C G °rS' Chem, 1976. 41, 3322.
,, oCPerkin 11, 1974 ’108I ' alson< W. B. Jennings, and D. M. Jerind,
53. R M. Moriarty. C. L. ye!, !( r „
rT Soc’ 19701 921 6360 ’ ' ' Кат<,У' °nd P. U7, Whitehurst, J. Amer.
54. C.. Brown, В. T. GraySOn and R F и
Й t'Tn и работе- F- L Sm///1‘rfi°'l’Jetrahedrorl Letters, 1970,4925.
m 470 47I er and M- J- Perkins „t 'П .<OrSanfc Reaction Mechanisms»,
F?ennings s Л1 c, SC'CnCe- NCW York' 1973' chal)ter l3’
PMki" lb 1976, 150L'S',Oa',"'a’h О- »- Boyd, and R. M. Campbelt, J. C. S.
49.
50.
55.
56.
712
61.
62.
63.
64.
65.
66.
67.
57 У. Shvo and A. Nahlieli, Tetralmdmn i
58. G. E. Hall. 117. I. Middleton, and°J D ffX ? °’.42Z3'
93, 4778. ’ аПа Л Ц ™>eHs, J. Amer. chem. Soc 197)
S t1&&,•£££. Kfeрт. их.
s-
Y. P. Kitaev, В. I. Buzykin and T V t. ~
39, 441 [10. П. Китаев, Б. И Бузыкин / B^n R“SS' Chem' Rev- 1970'
1970,39,961]. " ьузыкин, T, В. Троепольская - Усп. химии,
H. Ahlbrecht and H. Henk, Chem. Ber. 1976 109 1516
P. A. S. Smith and С. V. Dang, J. Org. Chem 1976 41 901ч
M. D. Broadhurst and D. J. Cram, J. Amer. Chem^Soc' 2?ЭТ4 96 5R1 «
веденные там ссылки. ’’ “6, э81 и при-
R. М. Kellogg, Tetrahedron, 1976, 32, 2180
J Teysseyre I. Arriau A. Dargelos, I. Elguero, and A R Katritzku Bull
Soc. chim. beiges, 1976, 85, 39 (Chem. Abs., 1976, 85 77 3251 B '
... R. A. Clark and О. C. Parker, J. Amer. Chem.'Soc., 1971 28. 5481
68. H. Ahlbrecht, H. Hanisch, 47. Funk, and R. D. Kolas, Tetrahedron 1972 28 5481
69' M „ 1 'n «Mo'ectHar Rearrangements», ed. P. de Mavo, Interscience,'
New York, 1963, Part I, chapter 8, pp. 483—507.
70. R. S. Monson and В. M. Broline, Canad. J. Chem., 1973, 51, 942.
71. I. B. Chaltopadhyaya and A. V. Rama Rao, Tetrahedron, 1974 3(1, 2899
72. S. Bittner and S. Grinberg, J. C. S. Perkin I, 1976, 1708.
73. L. A. Cohen and B. Witkop, in «Molecular Rearrangements», ed. P. de Mayo,
Interscience, New York, 1964, Part II, chapter 15, pp. 989—990.
74. C. A. Grob and /. Ide, Helv. Chim. Acta, 1974, 57, 2571 и приведенные там
ссылки.
75. R. Т. Conley and S. Ghosh, in «Mechanisms of Molecular Migrations»,
ed. B. S. Thyagarajan. Interscience, New York, 1971, vol. 4. p. 197.
76. A. C. Huitric and S. £>. Netson, J. Org. Chem., 1969. 34, 1230: B. J. Gregory,
R. B. Moodie, and K- Schofield, J. Chem. Soc. (B). 1970, 338; У. Yukawa and
T. Ando, Chem. Comm., 1971, 1601.
77. G. Just and M. Cunninggham, Tetrahedron Letters, 1972, 1151; G. G. Spence,
E. C. Taylor, and O. Buchardt, Chem. Rev., 1970. 70, 240.
78. M. Lamchen, in «Mechanisms of Molecular Migrations», ed. B. S. Thyaga-
rajan Interscience. New York. 1968, vol. 1. pp. 1—60.
79 I S Splitter, T. M. Su, H. Ono, and M. Calvin, J. Amer. Chem. Soc., 1971,
93, 4075; D. R. Boyd and D. C. Neill, J. C. S. Chem. Comm., 1977. 51.
80 D St C Black and A. B. Boscacci', Austral. J. Chem., 1977. 30, П09.
81. S. Tamagaki, S. Kozuka, and S. Oae, Tetrahedron 1970 26 179u-
82. D. H. R. Barton, M. J. Day, R. H. Hesse, and M. M. Pechet, J. C. S. Perkin I,
1975, 1764. , ,O7.
I I Grandh“ra and V. I- Sorokin, Russ Chem. Rev., 1974. 43. 115
[И И Грандберг В. И. Сорокин — Усп. химии, 1974, 43, 266] и приве-
83.
84.
85.
""New York 1964, vol. Ш, part II, chapter 38,
7, 282. . KI
Heterocumulenes», Academic, New'
and R. K. Norris. J. C. S. Perkin I,
iroup», ed. Z. Rappoport,
87.
88.
89.
90.
оТ»и«». » Ы!- °’’"”™
ed. G. A. Olah, Interscience, N:
pp. 1191—1210.
J E. McMurry, Accounts Chem. Res., 1974
H Ulrich in «Cycloaddition Reactions oi
York, 1967, chapter 4. pp. 122-219.
J. Almag, D. H. R. Barton, P. D. Magnus,
M^RaMnovitz, in «The Chemistry of_ the Cy?no, Or
Interscience, 1W York, 1970, chapter 7, pp. 307-340.
713
ОI p C. Schaefer, in
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
113.
114.
115.
in «The Chemistry of, the Cvano.Group». ed. Z. Rappoport,
jri terscience, New York. J™’ cljn₽«Vhe’Chemistry of the Cyclic Enaminonitri-
£. C. Taylor ojxl A^MIoP. m 4 ><- ./ in Organic chemistry», ed.
les and o-Ammomtnles», \oi. /. 7q___«y4
E' Сл Tu;l0r' 1П'!е/?ап<1С£ p'VKi°ba 'in «The Chemistry of the Azido Group»,
ЛЛ Se SmftA' i97«Mo'«ular Rearrangements», ed. P. de Mayo, Interscience,
WKchem. Rev., 1970, 70, 664.
R. A Abramovitch ond E.'P. Kyba, J. Amer. Chem. Soc., 1974, 96, 480.
P V'/smiV^^VdpenChain’NHrogen Compounds», Benjamin, New York,
(a) 1965 vol.' 1, chapter 7. pp. 297-322; (6) 1966, vol. 2, chapter 8,
pp. 29—63; (a) 1966. vol. 2, chapter 9, pp. 149—171.
K. Harada, в cc. 1. chapter 6. pp. 255—298.
R. J. Morath and G. №. Stacy. в cc. 1, chapter 8, pp. 327—362
H Metzger, in «Melhoden der Organischen Chemie,
E. Muller. Thieme Verlag, Stuttgart, 1968, vol. 10, part
/. P. Freeman, Chem. Rev., 1973, 73, 283.
A. Pross and S. Sternhell, Austral. J. Chem., 1971, 24,
статьи этой серии.
G. Rosini, J. Org. Chem., 1974. 39, 3504.
M. Wiessler, Angew. Chem. Internal. Edn., 1974, 13, 743.
Л1. P. Doyle, №. Wierenga, and Л1. A. Zaleta, J. Org. Chem., 1972, 37,
(Houbcn-Weyl)», ed.
4, pp. 217—282.
1437 и предыдущие
1597.
108. C. R. Narayanan. P. S. Ramaswamy, and Л1. S. Wadia, Chem. and Ind. (Lon-
don), 1977, 454.
109. G. 4. Olah and T. L. Ho, Synthesis, 1976, 610.
110. R. H. McGirk, C. R Cyr, №. D. Ellis, and E. H. White, J. Org. Chem., 1974
39, 3851.
111. J. Buckingham, Tetrahedron Letters, 1970, 2341.
112. J. Elguero and C. Marzin, in «Iminium Salts in Organic Chemistry»,
ed. H. Bohme and H. G. Viehe, Part 1 of vol. 9 of the series «Advances in
Organic Chemistry», ed. E. C. Taylor, Interscience, New York, 1976, pp. 533 —
587.
116.
117.
118.
119.
120.
121.
122.
123.
124.
125.
714
M. Cushman, J. Qentry, and F. №. Dekow, J. Org. Chem., 1977, 42, 1111;
M. A. Haimova, N. Л1. Mollov, S. C. Ivanova A. I. Dimitrova and V. I. Og-
nyanov. Tetrahedron, 1977, 33, 331.
N. S. Isaacs, Chem. Soc. Rev., 1976, 5, 181.
»' {•/cCG/'Ze’Kobins°n. °- H- R- Barion, D. C. Harwell, and
I F Meer.’ u L 1975' 1237i «• B- B°aO T. K. lethnah.
19f75 748 Robmson' a,'d D- H- R. Barton, J. C. S. Chem. Comm.,
1977,C99?337B9Pa°0Te: md M' PoBack. Amer. Chem. Soc,
Soc. Гэт'з ^эз 2401 Urban. R D. G. Cooper, and F. L. lose, J. Amer. Chem.
У !JeterocytIll? Chem, 1963, 2, 104.
.Ogata and A Kawasaki, J. Chem. Soc. (B\ 1971 395
F °AnDavis ап^Р а'^о Nak?‘suBa*'a. Synthesis, ’1976,' 207.
8. Hashimoto NPNonnsMnaeS’ 42’ 398'
'/7n 2j°7 “ п1,11"еде"ние там ссылки " K°8a’ Tetrahedron Letters,
384L W3, 1668. M°reau' and A’- Gaudemar, Bull. Soc. chim. France. 1972,
J. G. Smith, } Wn and П Г r e-
L K- Whitesell and M. A wLsell A °rg, f;1’™'- 197 д 49 • 495-
wfiueseii, j. org. them., 1977, 42, 377.
126.
127.
128
129.
130.
131.
132.
133.
134.
135.
136.
137.
138.
139.
140.
141.
142.
143.
144.
145.
146.
147.
148.
149.
150.
151
152.
153.
154.
155.
156.
157.
158.
159.
160.
161.
162.
D.
E.
E.
A.
T.
T
1977, lib, 2659.
W S Kozlov, V. A. Serzhanina. and R. D. Sauls, Khim. geterotsikl. Soedine-
nii 1973 1082 (Chem. Abs.. 1973 , 79. 126279) [Я. С. Козлов. В. A. Cep-
' «» Л . ________ ICVTJ tAQOl
мипини, Г ЛА- '~гиуи1----- Ail.mi'i .......' V < 7-1 t>
A. !. Speziale, С. C. Tung. K. W. Ralls, and A. }ao, J. Amer. Chem. Soc..
1965, 87. 3460. L „ ....
E. !. Corey and M. Chaykovsky. J. Amer Chem. Soc.. 196o 87. 13a3. _ —
• 1» , r* -iL-iIa । лпе -л НлгпЯГПтП \P1X 1ПГК
R. Bortnik, У. Diab, and A I ,
денные там ссылки. ’ Tetrahedron, 1977 33 ,,,„
G. Wittig and H. Ret" An Ch ' ' 1279 ” "₽«»'•
A. I. Meyers, D. R. Williams and M n.,eLna ' Edn" 1968, 7, 7
98> и r n 8er' J' Amer' Soc., 1976
S. /. Yamada, T. Oguri and r Л ’
C. A. Park, C. F, Beam, E. M Kaiser RC 1S/Л™' Comm >976, 136
C R'Hauser. I. Heterocyclic Chem 1976 « 449°“Й°'1’ Л £ and
H. H. Szmant, A. Birke. and M. P Lau I A™ n
H. H. Szmant and С E Alciaturi I Or,’, A, er- Chem. Soc., 1977 99 logo.
F. E. Henoch, K. G. Hampton and cl h"™' '?77' 42' 1081
91, 676. p ' and C- * Iiauser- Amer. Chem., Soc., 1969.
R. H. Shapiro, Org. Reactions, 1976. 23, 405.
I. E. Stemke. A. R. Chamberlin, and F T Bond tj i j , .
^7rPnC-LaaSrHTB°Delens’ md H- LeUers' 1976'
1^77G99^3414'!' T ^rmnn^Amer. Chem. Soc..
E. Vede/s and W. T. Stolle, Tetrahedron Letters 1977 ич
IT J. Baron, M. R. De Camp, M. E Hendrick M P W / •
lCchaXSl’. J "-°™ 3nd R- A' Moss:
ai Br)ArWlbere' G' L BurSnaier. and P. Warner, J. Amer. Chem. Soc., 1971,
о ОI Z4 0,
V. V. Kane, A. D. Wolf, and M. /ones, J. Amer. Chem. Soc., 1974 96, 2643
E. /. Corey and D. Enders, Tetrahedron Letters, 1976, 3.
D. Enders and H. Eichenauer, Angew. Chem. Internal. Edn, 1976 15
549.
M. Newcomb and D. E. Bergbreiler, J.C, S. Chem. Comm., 1977, 486.
Enders and H. Eiclienauer, Tetrahedron Letters, 1977, 191.
/. Corey and D. Enders, Tetrahedron Letters. 1976, 11.
/. Corey and S. Knapp, Tetrahedron Letters. 1976, 4687.
Ahlbrecht and W. Farming, Chem. Ber.. 1977. 110,596.
Kaufftnann, Angew. Chem. Internal Edn., 1974. 13, 627-
Kauffmann, H. Berg, E. Koeppelmann, and D. Kuhlmann, Chem. Ber.,
n's ^Kozlov, V. A. Serzhanina, and R. D. Sauts, Khim. geterotsikl. Soedine-
nii, 1973, 1082 (Chem. Abs.. 1973 , 79. 126279) [Я. С. Козлов. В. А. Сер-
жанина, P Д. Сауц — Химия гетероцикл, соед., 1973. 1082]
А. !. Speziale, С. С. Tung. К. W. Rails, and A. Yao, J. Amer. Chem. Soc..
1965, 87. 3460. L „ ....
E. !. Corey and M. Chaykovsky. J. Amer Chem. Soc.. 196a 87, 13э3.
H. O. House, in «Modern Synthetic Reactions», Behjamm, Xen \ork, 1972,
2nd edn., chapter 10, pp. 655—660. .
G. Ktnast and L. F. Tietze, Angew. Chem. Internal. Edn.. 19<6. la, 239.
E. Leete, J. C.S. Chem. Comm., 1972. 1091.
!. P. Anselme, в cc. 1, chapter 7, pp. ^»9-o-»
А. Ж.ХП « «J
ed. S. Patai, Intcrscience, New York, 1977. auppi. n. p
pp. 612—617. . ,, . r.„,„ IQ74 48 2129 (Chem. Abs.,
Л4. Makosza and A. Kacprowicz, Roczniki Chem., 97 . .
1975 83,78 798). . „ « n0i„blln Nov. Khim. Karbenov
A. F. Khlebnikov, R. R. Kostikov and KA. ,st 1972, 197 (Chem.
Mater. Vses. Soveshch. Khim Ka^eno[1 p Костиков. К .4. Оглоблин.
Abs., 1975, 82, 138 993) 1ЛЛр1'±'КПервого' Всесоюзного совещания п-
Повое в химии карбенов. Матер, .
химии карбенов и их аналогов. А .. • ргапсе, 1972. 82э.
Р. Baret.H. Bii/fef, and J. L. ple'rJ;. ' s chem. Comm., 1976. 484.
O. Achmalowicz and M J. C is. cne,
A. K. Mukerjee and R. C. Srivastava. Synthesis.
715
165. F, Durarii
166.
167.
168.
169.
170.
171.
172.
173.
174.
180.
181.
182.
183.
184.
185.
. i i с.-пи I Amer Chem. Soc., 1976. 98,
r Moore. L. Hernandez, end A. S.ng, J-
Tlysenko M M Joullic, /. Athmu, and R. Rodebaugh. Tetrahedron Letters,
161 n.Tand L. Letters. .970, 245; /. Deeazes, J. L. Lt,
-
i N. N. Zobam, Syn^is 1974, 461.
Sed. A. G. Cook, Dekker, New York, 1969,
Seng Tetrahedron 1965. 21 1373.
ft. Fuks and H. 0. Viehe, Chem. Ber. 1970 103, 573.
R Huis gen Angew. Chctn. Internat. Edn., 1963, 2, 565.
G sSi: C De Micheli, and R. Gandolji in «The Chennstry of Double-
bonded Functional Groups», ed. S. Patai, Intersc.ence, New York, 1977,
Suppl. A, part 1, chapter 6, pp. 369—532. . .
175 Я Huisgen R. Grasheu, and J. Sauer, in «The Chemistry of Alkenes»,
ed s Patai Interscience, New York, 1964, vol. 1, chapter ll, pp. 739 953.
176 D. R. Crist'and N. J. Leonard, Angew. Chem. Internat. Edn., 1969, 8, 962.
177 ft Huisgen, Bull. Soc. chim. France, 1965, 3431.
178 <7 L’abbe, G. Verheist, S. Toppet, G. S. D. King, and !. Briers, J. Org.
Chem, 1976, 41,3403. л
179 . M. Lora-Tamayo and R. Madronero, in «1,4-Cycloaddition Reactions», ed.
J. Hamer. Academic, New York, 1967, pp. 127—142.
T. Kametani. T. Takahashi, /(, Ogasawara, and K. Fukumoto, Tetrahedron,
1974, 30, 1047.
R. Huisgen, M. Morikawa, D. S. Breslow, and R. Grashey, Chem. Ber, 1967,
100, 1602.
ft R. Schmidt, Synthesis, 1972, 333.
M. Morikawa and R. Huisgen, Chem. Ber, 1967, 100, 1616.
ft. E. Walrond and H. Suschitzky, J. C. S. Chem. Comm, 1973, 570.
P. Л1. Henry and G. L. Lange, in «The Chemistry of Double-bonded Func-
tional Groups», ed. S. Patai, Inlerscience, New York, 1977 Suppl A nart 2
chapter 11, pp 1067—1082.
D- St, C. Black, R. F. C, Brown, and A. M. Wade, Tetrahedron Letters, 1971,
4519.
S. E. Dinizo and D. S. Watt, J. Amer. Chem. Soc, 1975, 97, 6900.
ft J. Sandberg and R. A. Bukowick, J. Org. Chem, 1968 33 4098
F. McCapra and ft Wrigglesworth, Chem. Comm, 1969, 91. ’
ten ri k n'i 1 %A-,Oluh’ Synthesis, 1976, 611.
Ж м Ai T' L' Ho' Synthesis. 1976, 807.
AK± Mv
N.A. Usher, J. C.S Perkin’ 1 1977 1477 Г Pal!ad,m,lrl™. J- V. Turner, and
D.H. R. Barton, D. J. Jester, and S. V. Ley, J. C. S. Chem. Comm, 1977,
Perldn R Шганэд. C' C0U’ J' McGarrity, an!l D. A. Widdowson, J. C. S.
A. CaH^KCo^an^B^RlndMe'ali'dr Chem. Rev, 1973, 73, 93.
2723. ' °- Ч^опе, and C. Scolashco, Tetrahedron Letters, 1973,
R- N. Butler' Chem and Ind’ (н°п3°п)- 1972' 523-
200. ‘i>№ ^heS S 1968’ 437’
201 R3|V В ll П’ ‘ McOhie' a,,d P- L. Batten, J. chem. Soc. (C), 1970,
°. сХГ
186.
187.
188.
189.
190.
193.
194.
195
196.
197.
198
199
204.
205.
206.
207.
208.
209.
210.
211.
212.
213.
214.
215.
216.
217.
218.
219.
220.
221.
A.
Milewski, and В. E. Maryanojf. J. Amer. Chem. Soc.,
C. Yang, J. H. Chandler, and !. 0. Baker, Synthesis.
J. Shannon, J. Org. Chem.. 1977. 42. 2082; Atta-ur-
T.
222.
223. M. Ё. Ruehne and
P.
v>3. R. U. Ingold and S. Brownslein, J. Amer Chen, c
веденные там ссылки. chem- See , 1975, 97, 1817 и ппк
E- a"d. S^app' Tet^hedron Letters 197г, '
л. McKillop, J. D. Hunt, R. D. Naylor and FC 3^7',
Soc., 1971, 93, 4918 4‘ ’ and C- C Taylor, J Amer. Chem
G. A. Olah, J Welch, and M. Henninger Svnth»«m
G. A. Olah, J. Welch, and T. L. Ho J Amer Ch»’ c77, 308'
G. A. Olah. J. Welch, G. R. Surya Prakah, and T ТЛ 6717
808. ' L- ,io. Synthesis, 1976,
R- Maeda, I. Moritani, T. Hosokawa and S Murahoshi т > i.
1974, 797. muraliushi, Tetrahedron Letters,
R. N. Mehrolra and В. P. Ctrl, Synthess 1977 489
G. H. Timms and E. Wildsmilh, Tetrahedron letters 1971 ion
V Smthankar’ S 6 Chem. and Ind.
«• 99oB70rC''’ M' D- Berr,slein' and fl- °- Durst, J. Amer. Chem. Soc 1971
93, 2oy/.
R. F. Borch and A. I. Hassid, J. Org. Chem., 1972 37 1673
H. Alper, J. Org. Chem., 1972, 37, 3972; G. P. Boldrini, M Panunzio and
A. Umani-Ronchi, Synthesis, 1974, 733. °’ ana
N. De Rimpe, R. Verhe, L. de Buyck, and N. Schamp. Synthesis 1976 62
M. Nazir, W. Rreiser, and H. H. Inhofjen, Synthesis, 1977 466 ’ ’
R. H. Bell, Austral. J. Chem., 1970, 23, 1415.
C. Bernharl and C. G. UZermuth, Tetrahedron Letters. 1974, 2493.
T. Takeda, M. Ueda, and T. Mukaiyama, Chem. Letters. 1977. 245.
R. O. Hutchins, C.
1973, 95, 3662.
G. Г. Rabalka, D.
1977, 124.
"5, ". ". _______! P. j. Shannon, J. Org. Chem.. 1977. 42. 2082; Atta-ur-
Rahman, A. Basha, N. Waheed, and S. Ahmed, Tetrahedron Letters, 1976,
219.
224. R. L. Augustine, in «Catalytic Hydrogenation», Dekker, New York,
chapter 5, pp. 81 — 123.
225. R. Harada and R. Matsumoto, Bull. Chem. Soc. Japan, 197 , 44
E. Frainnet, P. Braquet, and F. Moulines, Compt. rend. (C), 1971. 272,
226. I. Ojinta, T. Rogure. and Y. Nagai, Tetrahedron Letters 1973 247o.
— L. Horner and D. H. Skaletz, Tetrahedron Letters. 1970 1103, 3679.
A. J. Fry and R G. Reed, J. Amer. Chem. Soc., 1969. 91. 6448
С. P. Andrieux and J. M. Saveant, Bull. Soc. chim France. 1968. 4671
A. Padua, Chem. Rev., 1977, 77, 37; A. C. Pratt. Chem So . R r 19/7, 6,63.
N. Toshima. R. Aoki, and H. Hirai. Bull. Chem. Soc. Japan 19/7.50 123o.
G. Smolinsky and C. A. Pryde. in «The Chemistry of the Az>do Group»,
ed. S. Patai Interscience. New York, 1971 chapter 10, pp. 5o5-58o.
F. W. Fowler, Adv. Heterocyclic Chem., 197!.13. 4o.
D. J. Anderson and A. Hassner, Synthesis, 1J7o,48J
G. L'abbe, Angew. Chem. Internal. Edn !97o 14, 77m
R. G. Ford. J. Amer. Chem. Soc.. 1977, 99. 2389.
G. L’abbe and G. Mathys, J. Org,Chem.. R £ J C S Perkin 1. 1973. 555.
T. L. Gilchrist, G E O^-.^d C W. R es and C. W. Rees, J.C.S. Per-
О. J. Anderson, T. L. Gilchrist, G. E. Gymer, ana
kin I. 1973, 550 , K koiuio J. Chem. Soc. (C), 1971,
T. Nishiwaki, T. Saito, S. Onomura and R-^ona0'
2644; H. Wamhoff. Chem Ber., 1972 °5.- 74“ , chim Acta. 1971. 54, 1275;
M. Marky. H. J. Hansen, and H. Schmid- №. L”971 2749; H. Gotthardt.
Fl. Gotthardt and F. Reiter. Tetrahed chapman and D. R. Eckrofh,
ibid., 1277; R. M. Moriarty. R- Org. Chem.. 1971 36,
ibid., 397; C. S. Angadiyavar and M. V- G g Chem Comm 1970.
1589- H. Rato T. Shiba, H. Yoshida, and o. ri,
1591.
219.
1965.
1068;
1435.
227.
228.
229.
230.
231.
232.
233.
234.
235.
236.
237.
238.
239.
240.
241.
717
1974
2051
1973
242.
243.
244.
245.
246.
247.
248.
249.
250.
251.
252.
1976,
1977,
253.
254.
255.
256.
257.
258.
259.
260.
261.
262.
263.
264.
265.
266.
267.
268.
269.
270.
271.
Letters, 1970, 1623
272. A'. Burger, IV. Then,
474-
„Edn JM77
, f г McEntire and J. F- Calle, ,1. Org. Chem.,
. Hafner, A. B.Levy, E. F. Мсьпег y Cliem Sq(, _ |g7|_
39. 585; A. R.'Rinck. Angew. Chem. Internal. Edn.,
q Szeimies, U.
12, 161. i n i Rausch J Amer. Chem. Soc., 1970, 92, 2178.
A' £ ^“b. Kim V' Chiang, and T. Hamada. J. C. S. Chem. Comm.,
7Л. ЛоеЛ. R- 4. Higgins, and II. F. Schuster, Tetrahedron Letters,
A'/to.sner, S. 5- Rnrhe. and J. Cheng-Fan 1. J. Amer. Chem. Soc., 1975, 97.
T^ishiwaki and T. Saito, J. Chem. Soc. (C), 1971, 2648; T. Nishiwaki and
n ^Zzka'and^Tanrent Tetrahedron Letters, 1977, 31.
t Tnortmann and D. A. Robertson J. Шег Chem. Soc., 1972, 94, 2758.
n Л /Inrferso'i and /1. Hassner, Synthesis 1975, 483.
4 Paclwa Accounts Chem. RCs., 1976, 9, 371.
p G Kostuanovskii I. Л4. Gella, and K. Khafizov, Izvest. Akad. Nauk
SSS R Ser khim.; 1971, 893 (Chem.- Abs., 1971, 75, 62 815) \P. Г. Ko-
стяновскпй, И. M. Гелла, X. Хафизов — Изв. АН СССР, Сер. хпм., 1971,
8931
D Н Aue and D. Thomas, J. Org. Chem., 1975, 40, 2360.
D. H. Aue and D. Thomas, J. Org. Chem., 1975, 40, 2356.
£>. H. Aue and D. Thomas, J. Org. Chem.. 1975, 40, 2552.
D. Kolbalt and D. Koruncev. in «Methoden der Organischen Chemie (Houben-
Weyl)», Thieme Verlag, Stuttgart, 1967, vol. 10, part 2, pp. 89—120.
К. T. Finley and L. K. J. Tong, в cc. 1. chapter 14, pp. 663—729.
R. Adams and IV. Reifschneider, Bull. Soc. chim. France, 1958, 23.
A. W. Johnson, in «Ylid Chemistry», Academic, New York, 1966, chapter 6,
217-247.
D. Kolbah and D. Koruncev, in «Methoden der Organischen Chemie (Houben-
Weyl)». Thieme Verlag, Stuttgart, 1967, vol. 10, part 2, pp. 121 —122.
A. Sfeigel, J. Sauer, D. A. Kleier, and G. Binsch, J. Amer. Chem Soc., 1972,
94, 2770.
1 Elguero, R. Jacquier, and C. Marzin, Bull. Soc. chim. France 1970, 4119.
D. S. Malament and J. E. McBride, J. Amer. Chem. Soc. 1970 92, 4593
R. Bonnett, в со. 1, chapter 13, pp. 597—662.
R- Girich in «The Chemistry of Imidoyl Halides», Plenum New York, 1968,
a) chapter 2 pp 13-54; (b) chapter 3, pp. 55-112; (в) chapter 4, pp. 113-
117, (1) chapter 6, pp. 157—172; (д) chapter 7, pp. 173—192.
v„i Л, , 1 п Cllaln Nitr°gen Compounds», Benjamin, New York,
\ui. z, cnapicr И, p. 317.
T. Kauffmann and M. Schonfelder, Annalen 1970 731 37
H Metzger апс^К^ее^У^кГТ1’ ?'trahetiron Letters, 1976, 3823.
59, 6337) S • L Naturforsch„ 1963, 18b, 336 (Chem. Abs., 1963,
' Lo.ra’Jan,a.'/0 and J. L. Soto, in «I 4-C
mer. Academic New York. 1967, chapter 7
' F Wittmar, G. Heinrichs, A Steigel
Letters 1970 1R94 'eigei,
n' a”d Л- Gigren- *»». Chem. Internal Edn., 1974, 13,
274 РЕАП J’s’ R R°Uh' H' S<Mckaneder, Angew. Chem. Internal
275 2-chapter 1F p э/к'” ^rogen Compounds», Benjamin, New York,
В J. &U-..«.
278 К г н°е1г"М mi1 K-'j^’/ong lnt0/nat- Edn» 1977. 1(i 266-
C. Broom and 1. g Corbett J c S P r’ Clem' Soc» I971, 93. 1347-
718 ' C'S- Perkln И. 1977, 1125.
Soto, in «1,4 - Cycload d it ion Reactions» cd. J. Ha-
?. pp. 179—204.
f, T. Troll, anil J. Saner, Tetrahedron
285.
286.
287.
288.
289.
290.
291.
Cook, and
292.
293.
294.
295.
296.
297.
298.
301.
302.
303.
304.
305.
306.
307.
308.
079. A. G. Pinkus and J. Tsuii J Or,,
280. W- Friedrichsen and H.'d Oeser Ch' 97o' 39, 497 •
Letters, 1974, 4373. ' ’ 'h’™' Her., 1975, 108, 3E Tetrah a
281. E. N. Zilbermun, Russ. Chem. Rev 1962 ,1 rr ’ °П
химии, 1962, 31, 1309]. ’’ 31' Gi'« [£. H. Зильберман - yen
282. E. Kahle, B. Anders, and G. Zumach Aiw™ ci,
649. ’ Chem. Internal. Edn. 1967 6
283. H Holtschmidt, Angew. Chem. Internal. Edn 1962 1 fine ’ '
284. E. Kuhle, Angew. Chem. Internal. Edn 1969 '8 on ’’ 632'
C. R. Harrison, P. Hodge, and W I Rogers'I'erm .
H. Eilingsfeld, M. Seefelder, and H Weidm'ger& гГ' 41',
Z. Janousek and H. G. Viehe, in «iminium Sahl m п”™ ’ 72' 836'
Part 1, ed. H. Bohme and H. G. Viehe vol 9 ol «^gan,C Che™str^
ррЬеТГз-У41’9Са- E' C- ТаУ,0Г' York,'ai976, 'SrapteVt
Y. H. Chiang, J. Org. Chem., 1971, 36, 2146
F. L. Scott, F. A Groeger, and A. F. Hegarty, J. Chem. Soc. (Bl 1971 un
D. Seyferlli and H. Shih, j. Amer. Chem. Soc., 1972, 94 2508
H. H. Gibson, J. R. Cast, J. Henderson, C. IV.’ Jones В F
J. B. Hunt, Tetrahedron Letters, 1971, 1825.
G. Simchen and G. Enlenmann, Angew. Chem. Internat. Edn., 1973, 12, 1 19
P. A. S. Smith, in «Open Chain Nitrogen Compounds», Beniamin New York
1965, vol. 1, chapter 4, pp. 137—208.
A. F. Hegarty, J. D. Cronin, and F. L. Scott, J. C. S. Perkin II, 1975. 429.
R. N. Butler and F. L. Scolt, Chem. and Ind. (London), 1970. 1216.
IV. Walter and K. D. Bode, Angew. Chem. Internat. Edn., 1966, 5, 447.
IV. Waller and E. Schaumann, Synthesis, 1971, 111.
A. F. Hegarly, M. T. McCoi mack, B. J. Hathaway, and L. Hulett, J. C. S. Per-
kin II, 1977, 1136.
E. J. Bailey, H. Fazakerlcy, Л4. E. Hill, G. E. Newall. G. H. Phillipps,
L. Stephenson, and A. Talley, Chem. Comm., 1970, 106.
A. G. Anderson, N. E. T. Owen, F. J. Freenor. and D. Erickson, Synthesis.
1976, 398. „ ,OJ_
R. Ta-Shma and Z. Rappoport, J. Amer. Chem. Soc.. 1977, 99, 1840
IV D Ollis and C. A. Ramsden, Adv. Heterocyclic Chem.. 1976. 19, 1.
M. E. C. Biffin J. Miller, and D. B. Paul, in «The Chemistry 01 the Azido
Group», ed S. Patai, Interscience, New York, 1971, chapter 2, pp. 57 190.
A. S. Shawali and H. M. Hassaneen, Tetrahedron 1973. 29 121
da. i972' «•
AD. ‘:PH№^1 RgT^ 'cUtry OI thej^onyl
ti^.’^^^^’mr^iSn^U^Ben^in, New York,
1965, vol. 1, chapter 5, pp. 209—231. ! ed H Bohme and
c. Jutz, m «Iminium Salts in Oigame Chenw( ed e. c. Tavlor,
H. G. Viehe, vol. 9 of «Advance in Organic Chemistry.
' ' New York, 1976, chapter 4 pp. 22o л-
I 1977, 33, 731; Synthesis, 1976,
299.
300.
309......................... -
1965, vol. 1, chapter 5, pp. 209—231. _ ,
310. C. Jntz, in «I "
I . _. ______
Interscience, > В7П-
311. D. Burn, Chem. and Ind. (London), l9".8'O.
312. J. Liebscher and H. Hartmann, Tetrahedron,
H. G. Viehe, Chem. and Ind. (London),1977. 386. j chem. Research (S),
M. Huys-Francotte, Z. Janousek and H. U. ’jbid I05; д. Elgav, and
1977, 100; E. Coffin, 7. Legrand, and fl,6 lg]. //. G. Viehe B- Le
H. G. Viehe, Angew. Chem. IniernaL Edm >.7 Q |bld189.
Clef, and A. Elgavi, ibid., 182; B. S e"t. Edn., 1976, 15 459.
D. Lloyd and H. McNab Ange* cl'e\\m‘dines and Imidates», ed. S. Pata,
D. G. Neilson, in «The Chemistry 385—489.
Interscience, New York. I97o, cbapt g] ]79
R. Roger and D. G. Neilson, Chem. Rex., ><o>,
313.
314.
315.
316.
317.
719
318.
319.
320.
321.
322.
323.
324.
325.
326.
327.
328.
329.
330. r
331. j
<
332.
333. '
334. .
I
335. .
336.
337. ,
338. .
339. '
340.
341. '
342.
343.
344.
345,
346.
347.
348.
349.
350.
351.
352.
(Houben-Weyl)»,
4, pp. 209—213.
(London), 1972,
353.
354.
I A Frump, Chem. Rev, 1971, 71, cheni19G3, 2, 311.
Z Eckstein ami F. Urb4'1^' G^anik Adv?Helerocyclic Chem, 1970, 12, 185.
«. £. Glushkov U,ul\. O- ^rfl'';c;lron Lettcrs. i976r 4073.
J. R. Pougny and P. S a J to siebenthall, J. Org. Chem., 1973, 38, 2242.
S. L. E M Flagg ami t R g< T
IF. Seehger, b. Auplernanr .lt'rIlaf, Edn., 1966, 5, 875.
and H. IIellmann, Ange • d Q L schmir, J. Amer. Chem. Soc.,
fivrart0™ McClelland, ibid.. 1974 96 3690; A. C Sallerthwait
aml’lV P lenoks, ibid.. 7018, 703Д. P. Deslongchamps, C. Lebreux, and
R. Tailiefer, Ca“dF \C^7he|idi7 Angew. Chem. Internal. Edn., 1976, 15, 270.
A. I. Meyers, in «Hetfro^cles in' Organic Synthesis», Wiley-Interscience, New
York. 1974. . . rmA eoo
к M Doule and F. Киггег, Synthesis, 1974, 5o3.
I A Gautier M Miocque and С. C. Farnoux, in «The Chemistry of Amidines
and Imitates», ed. S Patai, Interscience, New York, 1975, chapter 7, pp. 283-
^M Watson and D. G. Neilson, in «The Chemistry of Amidines and Imida-
tes> ed S Patai. Inlerscience. New York, 1975, chapter 10, pp. 491—545;
D. G. Neilson. R. Roger. J. IF. M. Heallie, and L. R. Newlands, Chem. Rev.,
1970, 70. 151.'
F. Eloy and R. Lenaers, Chem. Rev., 1962, 62, 155.
H. Metzger, in «Methoden der Organishen Chemie
ed. E. Muller, Thieme Verlag, Stuttgart, 1968, vol. 10, part
T. Yamaguchi У. Shimizu, and T. Suzuki, Chem. and Ind.
380.
G. R. Malone and A. I. Meyers, J. Org. Chem., 1974, 39, 623.
A. I. Meyers, G. Knaus, K. Kamata, and M. E. Ford, J. Amer. Chem. Soc.,
1976, 98, 567.
A. I. Meyers and С. E. Whitten, J. Amer. Chem. Soc., 1975, 97, 6266.
1. F. IF. Keana and T. D. Lee, J. Amer. Chem. Soc., 1975, 97, 1273.
R. A. Crochet and C. DeWitt Blanton, Synthesis, 1974, 55.
H. Lund, in «The Chemistry of Amidines and fmidatcs», ed. S Patai, Inter-
science, New York, 1975, chapter 5, pp. 241—253
A. L. Rusanov, Russ. Chem. Rev, 1974. 43, 795 ГЛ. А. Русанов — Усп. хи-
мии, 1974, 43. 1669].
<7. Fodor and В. A. Phillips, in «The Chemistry of Amidines and Imidates»,
с'Л Patab.Inlerscience, New York. 1975, chapter 2, pp. 85-155. ’
S. C. WicherinkI. w. Scheeren, and R. J. F. Nivard Synthesis 1977, 273.
i B°urE°‘s and F Texier, Compt. rend (C), 1977, 284, 509
2045 crandoli’ L- c- Crawley, and J. B. Komin, J. Org. Chem, 1975, 40,
1965, vSol.Srchapter^,Ppp Con'POU,llR’- Bonjamua, New York,
H. Oediger, F. Moller, and K. flier,'Synthesis 1972 591
M. WZGrays1on andAD <м‘',ГУ’ ?"Fa' Ch^T1- ln4crnat Edn- 1963, 2< 459-
G. RDelpiereand M in Le,mal^- Chem. Soc, 1976 98, 1278.
A.MneninXch"^ Rev, 1965, 19, 329, 7. Harner and
ed. e’mSc, Thie^Verhn зГн Or,ga™ach«n Chemie (Houben-Weyl)»,
R. M. fe/fogg/T’.'tXdmr^97S6U t2Ba2r 781968’ V°L '°' ₽art *• PP- 3,1
C. G^ Stuckwisch. Synthesis. 1973', 469
a. Katntzkt/ and 1 м т < .
N-Oxides», Academic, New YorkS<l «г’p'1 n<</hcmistl y of tlle Heterocyclic
Elsevier. Amsterdam 1967 ' ' " Ocluai, in «Aromatic Amine Oxi-
G. ЗигХ«АЛ'/НркГ°±Ис 1974' 17' 213.
Tetrahedron, 1978, 32, 2847 ’ P' and A. Lablaciie-Combier,
720
356.
357.
358.
359.
360.
361.
362.
363.
364. IP. Rundel, in
365.
366.
367
368.
369.
370.
371.
372.
373.
374.
375.
376.
377.
378.
379.
380.
381.
382.
383.
384.
385.
L.
R.
J.
A.
K.
R.
A.
A.
H.
A.
386.
387.
388.
389.
390.
391
392.
393.
394.
395.
396.
397.
398.
399
400
355. R- Dennis, A. R. Kalrilzky, and У .
1976, 15, 1. J' 13 ^uckg Angew. chem. Internal FHn
M. Abou-Gkarbia and M. M. Joullie Svnih„ 1
D. St. C. Black and А. в. Boscacci ^usfral ’1 cY’ 3>S'
H Suginome,T. Mizoguchi and T.'Masamune' 29,’2561.
Г. Angew Chem. internal, Edn., 1977 16 10 гкш ’’ 1976' 2365
M. Jawdosiuk, B. Oslrowska, and M Makn^„ ru
A. W. M. Kayen and T. J. de Boer Rec TraJ 197L 548
J. E. Baldwin, A. K. Qureshi, and В Sklarz \ rh '?77’ 96’ 8-
D. St. C. Black R. F. C. Brown, В. T. Dunstan andTsT*/??’ 1969> 1073-
Letters, 1974, 4283. "unstan, and S. Slernhell, Tetrahedron
ed. E. Muller, Thieme Verhg, Stuttgad ^Эбв^о! So^plrt (,НоиЬе";У,:У1)‘>
7. Bjoergo,D C. Boyd, D. C. Neill, and V ВJennings№ t51?472'
1977, 254; D. R. Boyd and D. C. Neill, ibid., 1308. n‘ngs’ iCS- p«<<m I,
n D<t,Ler a"d д Fn ®r ^еапа- J- Org- Chem., 1976, 41, 3237.
v' r t C' i,i aCk.\ r' r' Croz,ier\ and °- c- Davis, Synthesis, 1975 205
У. Takeuchi and F. Furusaki, Adv. Heterocyclic Chem 1977 21 *207
A. Padwa, Angew. Chem. Internet. Edn, 1976, 15 123." ' ’
C. Belzecki and I. Panfil, J. C. S. Chem. Comm. 1977 303
A. Vaselia, Helv Chim. Acta, 1977, 60, 426, 1273.
R. A. Firestone, J. Org. Chem, 1968, 33, 2285.
R. Huisgen, J. Org. Chem, 1968, 33, 2291.
В. M. Benjamin and C. J. Collins, J. Amer. Chem. Soc, 1973, 95, 6145
G. Cum, G. Sindona, and N. Uccella, J. C. S. Perkin I, 1976, 719.
' K. Ding and W. J. Irwin, J. C. S. Perkin I, 1976, 2382.
Huisgen, R. Fleischmann, and A. Eckell, Chem. Ber, 1977, 110, 500.
IP. Lown and В. E. Landberg, Canad. J. Chem, 1974, 52, 798.
Padwa, D. Dean, and T. Oine, J. Amer. Chem. Soc, 1975, 97, 2822.
Burger, F. Manz, and A. Braun, Synthesis, 1975, 250.
Huisgen and A. Eckell, Chem. Ber, 1977, HO, 522, 540.
Eckell and R. Huisgen, Chem. Ber, 1977, 110, 571.
Eckell and R. Huisgen, Chem. Ber, 1977, 110, 559.
J. Twilchett, Chem. Soc. Rev, 1974, 3, 209.
Kjaer, in «Organic Sulphur Compounds», ed. N. Kharasch, Pergamon,
Oxford, 1961, vol. 1, chapter 34, pp. 409—420.
I. H. Saunders and R. J. Slocombe, Chem. Rev, 1948, 43, 203.
R. G. Arnold, J. A. Nelson, and J. J. Verbanc, Chem. Rev, 1957, 57, 47.
R. Bacaloglu and C. A. Bunton, Tetrahedron, 1973, 29, 2721, 2725.
S. J. Assony, in «Organic Sulphur Compounds», ed. N. Kharasch, Pergamon,
Oxford, 1961, vol. 1, chapter 28, p. 326—338.
F. Kurzer and K. Douraghi-Zadeh, Chem. Rev, 1967, 67, 107; В. I. в°<*аг°£
Russ, Chem. Rev, 1965 34, 212 [5. В. Бочаров — ion. химии, 196a, 34,
488].
H. Ulrich, Chem. Rev., 1965, 65, 369. u n..^wnuau
K. A. Nuridzhanyati, Russ, Chem. Rev., 1970, 39, 130 [К- Л. УР
Усп. химии. 1970, 39, 259]. , „ ,nco л nn.
M. O. Lozinskii and P. S. Pelkis, Russ. Chem. Rev, 1968, 37, 363 [M.O. JIo
/7. С. /Уе.гмте — Усп. химии, 1968 37, 840].
H. Ulrich and A. A. R. Sayigh, Angew. Chem. Internal. Edn, 1966, 5, 704.
R. M. Ottenbrite, J. C. S Perkin I, 1972, 88. 1077 573.
T. Shibanuma, M. Shiono, and T. Mukaiyama Chem. . • „ 425;
S. Sakai, T. Fujinanii, and T. Aizawa, Bull. Chem. Soc, Japan, 19//, on,
T7Schiba2nmna, M. Shiono, and T. Mukaivama Chem Letters, 1977, 57o.
G. R. Krow, Angew. Chem. Internal. Edn, 1971 J , jn Organjc che-
L. Ghosez and !. Marchand-Brynaert, m «Im . ()1 ser|es
mistry», ed. H. Bohme and H. G. Viehe Part 1 0o . «
«Advances in Organic Chemistry», ed. E. C. Taylor, Intersoence,
1976. pp. 421—532.
721
jOl. A. Hassnei
402.
103.
404.
405.
406.
407.
408.
409.
410.
411.
412.
413.
414.
415.
416.
417.
418.
419.
420.
Edn.. 1968, 7. 172.
421.
422.
423.
424.
425.
426.
427.
428.
429.
430.
431.
432.
433.
434.
Д Hussner, R- P- H°blitt- C' ^athcnck’ Л £ Kr°PP’ a'U‘ M' L°rber‘ 1 Amer-
RKG. G°uy, lR7°Bome^and D. Lanigan, Chem. and Ind. (London), 1969,
O°A. Tonialia, T. J. Giaeobbe, and IV. A. Sprenger, J. Org. Chem., 1971, 36,
Г «о, T. Hirao, and T. Saegusa, J. Org. Chem., 1975 40, 2981
T Saegusa, Г. Ho, and T. Shimizu. J. Org. Chem., 1970 35, 3995.
H Ulrich in «Cycloaddition Reactions of Heterocumulenes» Academic, New
York 1967, (a) chapter 5, pp. 220—253; (6) chapter 6, PP- 254 26G-
P A S. Smith, in «Molecular Rearrangements», ed. P. de Mayo, Interscience,
M.-W York 1963 Part 1. chapter 8. pp. 528—558.
J H MacMillan and S. S. Washbcirne. J. Org. Chem., 1973, 38, 2982;
H R Kriclieldorf, Chem Ber., 1972, 105, 3958; Synthesis, 1972, 551.
H R Kriclieldorf and E. Leppert, Synthesis. 1976, 329.
,4. Brandstrom, B. Lamm, and 1. Palmertz, Acta Chem. Scand. (B), 1974, 28,
699.
L. Bauer and 0. Exner, Angew. Chem. Internat. Edn., 1974, 13, 380.
«7. C. McCarthy and L. E. Foss, J. Org. Chem., 1977, 42, 1508.
! Goerdeler J Haag, C. Lindner and R. Losch, Chem. Ber., 1974, 107, 502.
H. Wamhoff and K. Wald, Chem. Ber., 1977, 110, 1699.
G L’abbe E. Van Loock, R. Albert, S. Toppet, G. Verhelsf, and G. Smets
J. Amer. Chem. Soc.. 1974. 96, 3973.
D. J. Woodman, P. M. Stonebraker, and L. Weiler, J. Amer. Chem. Soc.,
1976. 98, 6036; U. Schollkopf and I. Hoppe, Annalen, 1974, 1655; A. I. Mey-
ers and G. N. Knaus, J. Amer. Chem. Soc., 1973, 95, 3408.
D. P. N. Satchell and R. S. Satchell, Chem. Soc. Rev., 1975, 4, 231.
R. Graf. Angew. Chem. Internat. " ' , *.
S. Krishnan, D. G. Kuhn, and G. A. Hamilton, Tetrahedron Letters, 1977,
1369.
S. P. McManus, H. S. Bruner, ll. D. Coble, and M. Ortiz J. Org. Chem.,
1977, 42, 1428.
! B. Hendrickson and I, I of fee, J. Amer. Chem. Soc, 1973, 95, 4083.
G. Lohaus, Chem. Ber, 1972, 105, 2791.
r' ,ra,!ef ?nd K ° B°de’ Angew- Chem. Internat. Edn. 1967, 6, 282.
E. \ owmkel and C. Wolff, Chem. Ber, 1974, 107, 907.
paG“7e' °' de Tar and R-'Silverstein, J. Amer. Chem. Soc,
1УОО, о», !Ulo, 1020,
л Sn KPitisn,:T afd M- Bodanszky, Synthesis, 1972 454
Chem. Sof^f977 99r201fOrmaCA’ °' Fer«uson- 'and P- !- Robert*, J- Amer-
N TakamatIuko^atlSUi i“t m lc'dkiza!ii~ Chtrr- Letters, 1977, 539; H. Ho,
>/“11 and ' Hhiktzakt, ibid. 1975 577
R C SHinurhandeF aFd ^hedron Letters, 1972, 3281.
£ Oleson and ^UMarinoi Tetrahedronil97^m26^<\TTg^5’ m
New York° 971” wlX2 Xme^' R' L, Augustine and' D- J- Trecker, Dekker,
Chem. Rev. 1967 67 9 rn’T r> ' PP' ?,46; pP^in and F. IT. Sweat,
L p. Marino к EPfitznera,Tv TTT Chem- l9G9- «, 343.
. N M. Weinshenker aid СM Shen T ? 1971. 27, 4181.
435. Cp. R. IE. Hoffmann and M Ы?’ rahcdIon Letters, 1972, 3285.
moto, H. Tokanou H иепшга^ппТй^'?^^' l976' ,09’ 2565; Л Уата'
G Labbe, С. C. Yu and S Ton G°,oh- J- C s- Pcrkin L I977- 1241.
475; A. 1. Hubert, A Feron T An|?e^ Chcm- Internat. Edn, 1977, 1«,
1976, 317, ,eron- R- and P. Teyss.e, Tetrahedron Letters,
П R,lbola and II. Sakurai I Г -< ri.
R. Compper, Aneew Chem i । ' Chem. Comm, 1972 362
F- !. Moricinl ^nd W C"У,1'11"",31- Edn-- 1969, 8, 312
L R. Malpass and N.'j\weddl^\°cSo ^1С1Т1 • 1971 - 36, 2841.
'c J C- s- Perkin 1, 1977, 874.
436.
437.
438.
439.
440.
722
441. W- J. Friedrich, Tetrahedron Letters 197] 99R1
442. F. Effenberger, P. Fischer, G. Prossel anrir ,
1987, 2002. °ssel’ and G. Kiefer, Chem, Ber 1971 )04
443, R. Gonipper and B. Wetzel, Tetrahedron i „н
444. T. J. Barton, R. J. Rogido, and J. С Clarduтм 9J? a529 ,
445. T. J. Barton and R. J, Rogido, Tetrahedron Inti Letters. 1970, 2081
446. Г. T. Brady and E. D. Dorsey iI nr„ eV 1972' 3901
L Hernandez, and Л Sing, J. Amer. Chem Soc 1976 98 3728 M°°re'
% 1973. 27!
„9 I. T. Kay and I. T. StreetingSynthes,? 1976 зГ ' '976' 718
450. «The Chemistry of the Cyano Group»', ed Z Rannonorf i . •
York, 1970. P Kappoport, Interscience, New
451. «Isonitrile Chemistry», ed. I. Ugi, Academic, New York 1971
4,52. H. D. Hartzler, в cc. 450, chapter 11, pp. 671-716
453. S. T. Reid, in «Photochemistry» ed. D Bryce-Smith n T .
Reports Chemical Society, London. 1977, Х8^chapte?б' " p^"
тома этой серии. r нсдшд^ш.ис
454. К. Friedrich, and К. Wallenfels, в cc. 450, chapter 2, pp. 67—122.
455. /. T. Harrison and S. Harrison, in «Compendium of Organic Synthetic Me-
thods», Interscience, New York, 1971, vol. 1, chapter 13, pp. 457—478.
456. /. 7. Harrison and S. Harrison, in «Compendium of Organic Synthetic Me-
thods», Interscience, New York, 1974, vol. 2, chapter 13, pp. 185—192.
457. К. M. Maloney and B. S. Rabinovitch, в cc. 451, chapter 3, pp. 41—61.
458. J. Casanova, в cc. 450, chapter 16, pp 885—946.
459. 6. Simchen and H. Kobler, Synthesis. 1975, 605; D. A. White and M. M. Bai-
zer, J. C S. Perkin I, 1973, 2230.
460.
461.
462.
463.
464.
465.
466.
467.
468.
469.
470.
471.
С. M. Starks and R. M. Owens, J Amer. Chem Soc., 1973, 95. 3613.
J. W. Zubrick, В. I, Dunbar, and H. D. Durst, Tetrahedron Letters, 1975. 71.
M. Cinquini, F. Montanari, and P. Tundo, J. C. S. Chem. Comm., 1975, 393.
!. F. Nor inant and C. Piechuck, Bull. Soc. chim. France, 1972, 2402.
F. P. Corson and R. G. Pews, J. Org. Chem.. 1971, 36, 1654; A. M. van Leu-
sen and 1. C. lagt. Tetrahedron Letters 1970. 967.
K. Takagi, T Okamoto, Y. Sakakibara, and S. Oka, Chem. Letters, 1973, 471.
A. Sekiya and N. Ishikawa, Chem Letters, 1975, 277.
L. Cassar, J. Organometallic Chem., 1973, 54, C-57.
S. Uemura, У. Ikeda and K. Ichikawa, Tetrahedron, 1972, 28, 30_o
E. C. Taylor, H. W. Allland. R. H. Danforth, G. McGillivray, and A. McKd-
lop, J. Amer. Chem. Soc., 1970, 92, 3520.
S. Nilsson, Acta Chem. Scand., 1973,27,329. 1075
J. Cornelisse, G. P. de Gunsl, and E. Havinga, Adv. Phys. Org. Chem., 1.7a,
472.
473.
474.
475.
476.
477,
478.
479.
480.
481.
482
A.'wfnno, С. Рас, and H. Sakurai. J C S Chem. Comm 1975, 553.
R. Beugelmans, M. T. Le Goff, J. Pussef, and G. Roussi, J.C.S. Chem.
Comm., 1976, 377 , rntTim 1079 441- L Eb er son,
VS. NHssonZdl. Rielz^ Che,? Scand. 1972, 26, 3870; K. Yoshida and
Уе^о^Ль^.^Н. Za/kow, Tetrahedron Letters. .977,
Y. l7Ogata and A. Kawasaki, in «The Chemistry,o'h‘pterCLrbppn'!-°90UP '
ed. J Zabisky, Interscience, New YorkA ^rL'^dale"} Amer. Chem. Soc. 1973.
D. A. Evans, J. M. Hoffman, and L. К Truesdale, J. Amer
ETaiiiades and A. Commeyras Tetrahedron 197-L 30. 2493.
/. Oft,na, S. Inaba, and K. Nakatsugawa Chem Letters
Г E. Ziegler and P. A. Wender 1 Org^hem 1977.
D. M. Orere and С B. Reese. J. C. S- Chei (London), 197-, -13-
S. Cacchi, L. Caglioti, and G. Paoluiu. Chem.
723
483.
484.
485.
486.
487.
488.
489.
490.
491.
492.
493.
494.
495.
496.
497.
498.
499.
502.
503.
504.
505.
F. D. Popp, M'l• HeterКМ СЬ Compounds» ed. S. Coffey, El-
S. F- Dyke\ 'П/™°1076 vol W chapter 27. pp. 424-426.
sevier. Amsterdamt 1976, vol 1< } .. Synlhesis, 1977> 497
Ty. Г&Га X'^g. R-Cbe-Rev.. 1966, 35, 9 [Ю. A. Hay-
Henrickson" K. W^Bai^md P. M. К echo, Tetrahedron Letters, 1976,
^Campagna /1. Carolli, and G. Casini, Tetrahedron Letters, 1977, 1813.
PTaus^fsS'SkaM 'synthesis, 1975, 502.
C R HarHsofp Hodge, and №. J. Rogers, Synthesis, 1977 4 L
r C/mkrabiirli and T M. Hotlen, J. C. S. Chem. Comm.. 1972, 1226.
G RosnflBaccolini, and S. Cacchi J. Org. Chem. 1973,38, 1060
M. MRogic, J. F van Peppen, К- P. Klein, a,id T. R. Denimtn, J. Org.
Af'T MUter and^M. Loudon, J. Org. Chem., 1975, 40, 126.
J. Liebscher and H. Hartmann, Z. Chem., 1975, 15, 15, 302 (Chem, Abs., 1976,
C ' Bizet'and J. Streifh, Tetrahedron Letters, 1974, 3187.
E. Vowinkel and J. Bartel, Chem. Ber., 1974, 107, 1221; E. Vowinkel, Angew.
Chem. Internal. Edn., 1974, 13, 351.
C. A. Grob and P. №. Schiess. Angew. Chem. Internal. Edn., 1967, 6, 9.
Cp. cc. 326, chapter 10, pp. 243—306.
A. De Munno. V. Bertini, and F. Lucchesini, J. C. S. Perkin II, 1977, 1121.
Л1. L. Casey, D. S. Kemp, K. G. Paul, and D. D. Cox, J. Org. Chem., 1973,
38, 2294.
D. S. Kemp, S. J. Wrobel, S. №. Wang, Z. Bernstein, and !. Rebek, Tetra-
hedron, 1974, 30, 3969 и предыдущие статьи этой серии.
Н. G. О. Becker, Н. Hubner, Н. J. Timpe, and М. Waliren, Tetrahedron, 1968,
24, 1031.
J. В. Lapat, R. J. Blade, A. 1. Boulton, 1. Epsztajn, A. R. Katritzky, !. Lewis,
P. Molina-Buendia, P. L. Nie, and C. A. Ramsden, Tetrahedron Letters, 1976,
2691.
506. T. Cuvigny, !. F. Le Borgne M. Larcheveque and H. dormant. Synthesis.
1976, 237, 238.
507. R. S Glass and R. C. Hoy, Tetrahedron Letters, 1976, 1777, 1781.
,i Kametani, K. Takahashi, T. Ohsawa, and M. lhara Synthesis 1977, 245.
509. H. Ohta and K. Tokumaru, Chem. Comm., 1970, 1601.
M1?' m Bri Mobbs, and H Suschilzky, Tetrahedron Letters, 1971, 361.
511. M. D. Dowle, J. C. S. Chem. Comm., 1977, 220.
Rio л Kasniussen arld A. Hassner, Synthesis, 1973, 682.
514 К kPkmfdf o' л' Org- Chem., 1973, 38, 1054.
' F Schronnf nnfi ^r,se'm<?/.,Tetrahedron Letters, 1970, 3851; К- K. Mayer,
... r a prl ’ d ' Sauer- lbl<r' 1972, 2899
s c B,4hrA Ch,etn' Ber 1973. '°6. 3544.
93, 298 ' M' Vm Di,k' md F ^log, Rec. Trav. chim., 1974,
Si’s f'e' 3"льбеР“п«-Усп. хим., I960, 29, 709.
519. P. МП Diamond, S^Dinizo T'w'f* I ₽P' 123~166'
520 l ^a“’I C Soa^ Comm.J97A 298eerfoe'’’ Haltiwan4er’ and
521. p, j, Beebu and S Rcac*ions. '969, 17, 213.
522. D. H. R. 'Barton P *d"m ' Austrab J- Chem., 1971 24, 809
1973,331. ' P- °' Ma^. ^d R. R. Young,'J. c s. Chem. Comm.,
3 g F '^PafP CMaS^ - 450. chapter 8, PP. 341 -42,.
syntg^rSChcm",966'6-95-
' lsler' Org. Chem., 1976, 41, 3769.
527.
528.
529.
530.
531.
532.
533.
C. Zervos and E-. H. Cordes, J. Or„ C],.m l0_, „„
M. A. Bennett and T. Yoshida, J. Am сЛ 36, 166L
rnond, B. Grant, G. M. Tom, and H Taube т,?ос,-I973' 951 303°- S. E Dia-
E. C. Taylor and !. L. LaMaltlna, J. Org Chem ‘\ч77Г4? !^ters' 1974. «25
R. F. Smith, L. L. Kinder, D. G. Walker LJT"n 42’ 1523'
J. Org. Chem., 1977, 42, 1862. ’ ' A' Buckl<!y. and !. M. Hammond
G. Alvernhe and A. Laurent, Tetrahedron Letters IQ71 ,nK,
C. F. Beam, R. S. Foote, and C. R Hauser I н * °57'
]83. ’ Л Heterocyclic Chem, 1972 , 9,
D. Seebach, D. Enders, R. Dach and p dm.
I879 K' uacn' and Pieter, Chem. Ber.. 1977, 110,
534a.K. Ogura and Tsuchihashi, J. Amer. Chem. Soc 1974 4fi mm
— I Kawamotc, SMuramatsu, anaI Y. Yura, Tetrahedron’ Letters 1974 4223
R. Seux, G. Morel, and A. Foucaud, Tetrahedron 1975 31 134s ' ' 4223'
!. P. Albarella, J. Org. Chem, 1977, 42, 2009. ’ 5'
D. S. Watt, Tetrahedron Letters, 1974, 707.
S. Brenner and M. Bovete, Tetrahedron, 1975, 31, 153
E. J. Corey and I. Kuwajirna, Tetrahedron Letter’s 1972 487
M. Makosza and M. Ludwikow, Angew. Chem. Internal. Edn 1974 13 665
D. Seebach and D. Enders, Angew. Chem. Internal. Edn, 1975 14’15'
G, Slork and L. Maldonado, J. Amer. Chem. Soc, 1971, 93, 5286^
" Deuchert, U. Herlenstein, and S. Hunig, Synthesis, i973. 777.
Kalir and D. Baiderman, Synthesis, 1973, 358.
Stork and L. Maldonado, J. Amer. Chem. Soc, 1974, 96, 5272.
Stetter, Angew. Chem. Internal. Edn, 1976, 15, 639.
Cazes and S. Julia, Tetrahedron Letters. 1974 , 2077.
5346.
535.
536.
537.
538.
539.
540.
541.
542.
543.
544.
545.
546.
547.
K.
A.
G.
H.
B.
548. S. /. Selikson and D. S. Watl, J. Org. Chem, 1975 , 40, 267 и предыдущие
статьи этой серии.
549. A. Debal, Т. Cuvigny, and М. Larcheveqtie, Synthesis, 1976, 391; Л!. Larche-
veque and T. Cuvigny, Tetrahedron Letters, 1975, 3851.
550. K. Bast, M, Christi, R. Huisgen, and IF. Mack, Chem. Ber, 1972. 105, 2825.
551. J. C. Jagt and A. M. van Leusen, Rec. Trav. chim, 1977, 96, 145.
552. C, G. Stuckwisch, J. Org. Chem, 1972, 37, 318.
553. T. Cuvigny, M. Larcheveqtie, and H. dormant, Bull. Soc. chim. France, 1973,
554.
555.
556.
557.
558.
559.
560.
561.
562.
563.
564.
565.
566.
567.
568.
569
670.
1174.
J. L. Fry, J. C. S. Chem. Comm, 1974, 45.
R. L. Augustine, in «Catalytic Hydrogenation», Dekker, New York, 1965,
chapter 5 pp. 96—100.
P. Tinapp, Chem. Ber, 1971, 104, 2266.
J. P. Ferris and F. R. Antonucci, J. Amer. Chem. Soc 1972, 94 8091.
E. Ciganek, W. I. Linn, and О. ll7. Webster, в cc. 450, Chapter 9, pp.
638
T. L. Cairns and В. C. McKusick, Angew Chem, 1961 73, .
K. Wallenfels. K. Friedrich. 1. Rieser, W. Ertel, and H. K. Thieme, Angew.
Chem. Internat. Edn, 1976, 15, 261.
K. Friedrich and S. Oeckl, Chem. Ber, 1970, 103 3951.
K. Friedrich and S. Oeckl, Chem. Ber., 1973, 106, 3796.
F. Freeman, Chem. Rev, 1969, 69, 591. ™ . H1 Chim. Acta, 1973,
D. Belins, K. von Bredow, H. Sauter, and C. D. Weis, neiv.
56» 3004. _ ,. • 1 n?R 70^
E. Campaigne and S. W. Schneller, Synthesis, • j Org. Chem,
S. E. Dinizo, R. IF. Freerksen, W. E Pubs , and D.S.
1976, 41, 2846; R. R. Wroble and D S. „• ,392 4.
C. !. Ireland and J. S. Pizey, J.C S_Chem_ Con , ')972 2, 181 (Chem.
H. Takahashi, T. Kajimoto, and J. Tsup, Synth. Comm,
Abs, 1972, 77, 139 494). „ <. Perkin I, 1974, 1260.
M. E. Peek, C. IF. Rees, and R. C //S^Amer Chem. Soc, 1975, 97,
D. S. Pearce, M. I. Locke, and И. W. Moore J. Amer.
6181; H. IF. Moore, Chem. Soc. Rev, 1973, 2,
725
573. К- I'-
574. "
575.
576.
577.
578.
579.
580.
581.
582.
583.
584.
585.
586.
587.
588.
589. ........
590. J. Ferraris, D. 0. Cowan, V. VPalatka, and /. H. Perlslein, J. Amer. Chem.
591.
592.
593.
„ , iQ40 к 79- P F. Butskus, Russ. Chem. Rev
57h H. A. ~ Or£ Reac -s, 1949, 5, 79,
1961. 30, 583 1П. 1 ter 5 np. 209-237.
572. «7. A. Sheppard псс АЗО chapt , I i06, 2361.
--- Z Friedrich and SOecU.wn-r, |977> 553.
К Nanjo, K. Suzuki. andM. S k; ,С ,970. |63Q
PE Lord^M"p Naan A DSlall, J. Chem. Soc. (B), 1971, 220; Z. Rap.
R. Hungen,’r.QSdi«g?a«'' °- Sle<’,er' A,1"t>W' Chcm' 1,,lernat- Edn- l974' 13'
??’ I1 ’uaii and R Ykman. J. Amer. Chem. Soc., 1975, 97, 800.
7' R Dombroski, M. L. Hallensleben, and IF. Regel, Tetrahedron Letters,
1 071 3K81
Й IP Hoffmann and W. Schafer, Angew. Chem. Internal. Edn., 1970, 9, 733.
₽' Sc/irnr and R Huisgen. J. C. S. Chem. Comm, 1975, 60.
H. Noerenberg, H. Kratzln, P. Boldt, and V. S. Sheldrick, Chem. Ber, 1977,
M к.' Newman and IF. X. Bajzer, J. Org. Chem, 1970, 35, 270.
H. C. van der Plas, in «Ring Transformations of Heterocycles», Academic,
New York, 1973, vol. 1, chapter 1, pp. 13—15.
H. Hamberger and R. Huisgen, Chem. Comm, 1971, 1190.
W. F. Bayne and E. I. Snvder, Tetrahedron Letters, 1970, 2263; VP. R. Dol-
bter and S. H. Dai, ibid, 4645.
L. R. Melby, в cc. 450, chapter 10, pp. 639—669.
Soc, 1973, 95, 948.
E. M. Engler and V. V. Patel, J. Amer. Chem. Soc, 1974, 96, 7376.
A. F, Garito and A. !. Heeger, Accounts Chem. Res, 1974, 7, 232.
S, Yoshitnura. Y. Holt, M. Yasuda, M. Murakami, S. Takahashi, and K. Hase-
gawa, Electron. Components Coni, 1975, 25, 77 (Chem. Abs, 1976, 85,
12669); Y Kisliimoto and F. Oda Japan Kokai Pat 7622092 (1976) (Chem.
Abs, 1976. 85, 39 875).
!. R. Angersen. R. A. Craven, !. E. Weidenborner, and E. M Engler J C. S.
Chem Comm. 1977, 526 . ’
7. R. Andersen, C. S. Jacobsen, G. Rindorf Ц. Soling and K. Bechgaard
J. C. S. Chem Comm, 1975, 883.
M. L. Kaplan, В. K. Teo, and J. Marshall, J. Org Chem. 1977 42,
1 000. b ’
N. F. Haley, J.C. S. Chem. Comm, 1977, 207,
D. Walker and J. D. Hieberl, Chem. Rev. 1967 67, 153
Berhn""^!."" and P' OrunanRer’ in Nitrile Oxides», Springer-Verlag,
См. cc. 599. chapter 3, pp. 31—61.
C. GrundnmnnaafdG''p. Syrnhesis.”973 ЛС‘“’ '793, 56’ 457’
iJ:W-pSoc’V,’GltMa^%-P
О-P. PolUm, Chem. Comm. ’1971 926 ’ G637' °' Travcrso- Barc°.
R. K. Howe, T. A Gruner' and I p г , ~
R К Н°Ше anl!t ibid/'1974, 39'9’62 E' C1’Cm" 1977’ 42’ '813;
Rr K- Howe and J. Jz ('ran? ICQ г'ь r*
H. Gotthardt. Chem. Ber“1972, lOs.US ’ С°П,т" ’973' 524'
W'. Steglich' Pmo ruber ^'ij^He^0'' Letlers- 1975. 1739.
".“F ал“' a“
1966, vel. 6, ...... .' ..’ t . Compowids,. Bvnjamln, .. . York,
594.
595.
596.
597
598.
599.
600
601
602
603.
601.
605
606.
607.
608
609.
610.
726
в cc. 450, chapter 15,
PP-„9—3,9,’
615. F. Millich, Chem. Rev., 1972, 72, 101.
620.
621.
622.
623.
624.
625.
626.
627.
628.
629.
630.
631.
632.
633.
634.
635.
636.
637.
638.
639.
640.
641
642.
643.
644
645.
646.
647.
CU Cm. cc. 599, chapter 5, pp. 85—139
612. P. D guarding, H. Kliimann, and I Ugi
pp, Ouo—ООО. ' 6‘i
613. P. Hoffmann, G. Gokel, D. Marquarding. and l. Vgi „ cc ,,, „ ,
pp. 9—39. б! в cc. 451, chapter 2>
fil4 /• Ugi, U, Fetzer U. Eholzer, H. Knupfer nn/l к
Internal. Edn., 1965, 4, 472. ’ ^'nermann, Angew. Chem.
61б' L. B. Engemyr, A. Marlinsen,' and J. Songslad rx.
A28, 255. sxongsiad, Acta Chem. Scand, 1974,
617. O. Hoefle and B. Lange, Angew. Chem. Internat Edn 1077 xa
618 W'- P- Weber, G. W. Gokel, and I. Ugi Angew Chen ’’ 1 , ’ 'Л62'
11, 530; W. P. Weber and G. W Gokel Tetrahedron & 1972'
G. W. Gokel R. P. Widera, and IF. P. 'Weber Or Syn h^’lCT
619. A M. van Leusen G. J. M. Boerma, R. B. Helmholdl tf slderxus'and
J. Strating, Tetrahedron Letters, 1972, 2367. menus, and
G. Skorna and I. Ugi, Angew. Chem. Internal., Edn. 1977 16 259
H. M. Walborsky and G. E. Niznik, J. Org. Chem., 1972, 37, 187.’
Я ^PP^' R' Kleinstuck, and K. D. Ziehn, Angew. Chem. Internat. Edn., 1971,
B. Beijer, E. von Hinrichs, and I. Ugi, Angew. Chem. Internat Edn 1972
11,929.
J. A. Green and P. T. Hoffmann, в cc. 451, chapter 1, pp. 1—7.
T. Saegusa and Y. Ito, в cc 451, chapter 4, pp. 65—92.
D. Mar guarding, G. Gokel, P. Hoffmann, and I. Ugi, 451, chapter 7, pp. 133—
143.
G. Gokel, G, Lttdke, and I. Ugi, в cc. 451, chapter 8, pp. 145—149.
/. Ugi, Angew. Chem Internat. Edn., 1962, 1, 8.
G. Gokel, P. Hoffmann, H. Kleimann, H. Klusacek, G. Ludke, D. Marquarding,
and I. Ugi, в cc. 451, chapter 9, pp. 201—215.
Y. Y. Liin and A. R. Stein, Canad. J. Chem., 1971, 49, 2455.
T. Saegusa and Y. Ito, Synthesis, 1975, 291.
P. lakobsen, Acta Chem. Scand., 1976, B30, 847.
S. Treppendahl and P. lakobsen, Acta Chem. Scand., 1977, B31, 264.
G. E. Niznik, IF. H. Morrison, and H. M. Walborsky, J. Org. Chem., 1974 , 39,
G. E Niznik IF H. Morrison, and H. M. Walborsky. Org. Synth., 1971, 51,
31.
D. Hoppe, Angew. Chem. Internat. Edn., 1974, 13, 789.
У. Но к. Kobayashi, and T. Saegusa, J. Amer. Chem. Soc, 1977. 99. ЗэЗ..
У. Зс/1бШгор/, Angew. Chem. Internat. Edn, 1977. Io, 3 .
A. M. van Leusen, J. Wildman, and О. H. Oldenael. J. Org. Chem, 1977,
U' Schdllkopf, P. H. Porsch, and E. Blume .Annalen, 1976, 2122, G. D. Hart
man and L. M. Weinstock, Synthesis, 1976 681.
H. !. Rabbe, в cc. 451, chapter 5, pp. 93—107.
G. IF. Gokel, в cc. 451, addendum, pp. 235—зао.
B. Zeeh, Synthesis, 1969, 65. internat Edn 1971, 10. 409.
A. Rrebs and H. Kimling, Angetw Chem. In
T. El Gomati, J. Firl, and I. Ugi. Chem Ber 1977, 1W>.
I. A. Deyrup, Tetrahedron Letters, 1J71. 21 j
K. Burger, J. Fehn, and E. Muller, Chem- В .,
Внимание!
ветственно
Согласно
как N- и
16
предметный указатель
международной практике вторичные и третичные
N.N-дизамещенные первичного амина по старшему
амины даны
радикалу.
соот-
Адамантан .»
производные 16. 31. 3U2, зи/
Адреналин 60. 133. 155
вор- 60. 133. 155
Азааллил-анноны 513 сл.. зоб
Азамины 454
320. 519.
Азепин 4.
гексагидро-, производные 40
днгйдро-. Производные 552
производные 316. 364 сл.. 389. 426,
теграгидро-, производные 529
Дзет
дпгидро-, производные
551-553. 558. 559 , 710
тетрагидро-, производные 32. 40, 55,
348, 558, 612, 710
Азиды
получение 22. 313 сл.,
реакции 18. 22. 122.
316 сл.. 354 сл.,
637. 638
спектры 50 . 300
структура 300. 315
фотохимия 366
Азины 560 сл.
N-оксидЫ 538 . 562 сл , 570. 571
Азиридин 31, 55
459
174. 175,
497 сл.,
179,
548,
557
523,
151,
304,
549,
производные
получение 31 сл.. 135, 282. 283. 321,
324, 362 сл., 509. 517. 518. 525, 545.
554 сл.
реакции 42, 81 сл., 129, 154 , 279 , 351.
358
структура 40, 41
Азирин 549
производные
получение 32 319 , 364 , 547 , 560
реакции 22. 35, 316, 341, 547, 550,
553 сл., 695
Азобензол
получение 207, 209 . 216, 388 . 537
структура 290
реакции 169 , 223
Азобензол {производные) 209, 2)0. 295
Азодикарбоковая кислота 294
Азодиоксисоединения 299, 300
Азоксибензол 207. 209, 210, 298
3.3-динитро- 209
Азокснсоединевня 207 сл., 296 сл 388
.395. 396. 424. 535
Азометип-плиды 613. 622, 628, 629 645 710
Азометинимины 613, 622. 626 сл. 645 710
Азометины 476 сл.
Азонорбориан 290
2-Азопропан 290
Азосоедннення 51. 271, 273, 281, 282 285
сл., 424 ’
Азотистой кислоты эфиры 445—449
Азотной кислоты эфиры 440—445
Акрилонитрил 102, 678, 683 688
Аланин 546
Аллеи, производные 104 105 154 393
Аллнламниы 55. 101 сл., 153'
Лллнлокснкарбонильная защита 79
«Альдегндоаммиаки» 127
Альдольная конденсация 431
Альдостерон, производные 484
Амид-анионы 334. 314 сл
АМ"Л"‘б6В 570i 588' 6Ш’ 607 СЛ-’ 624’ О67’
амино- 604 . 507
Амндоксимы 588. 604 . 607 сл., 667-669 704
Амидразовы 280, 588, 589 604
667, 669, 704 ’ ’ 607 сл.,
Амиды металлов 345 сл.
Аммнали 113 сл.
Ампнпднны 282
Аминильные радикалы 334. 338 339 ггя
Ампния радикалы 334, 337, 338’351 сл
Аминослирты 126 сл., )41, 143, ’i55 156
Аминоэфиры простые 139 сл. ’
Амины
алифатические ц сл,
ароматические 168 сл.
галоген- см. Галогенамнны
гидрокси- см. Амнноспнрты
диамины ИЗ сл.
оксиды 247 сл.
основность 53 сл.. 191
полиамины 123 сл.
получение 11 сл., 168 сл., 398 424 544 сл
607, 676, 677
реакции 61 сл.. i3l, 196 сл., 372, 373 600
спектры 45 сл., 193 сл.
структура 39 сл.
физиол. св-ва 58—60, 196, ]97
хиральные, разделение 37—39
циклические 31 сл.
эфиры см. Амнноэфнры
Аммиак
основность 54, 56. 74 , 278, 345
структура 39. 40
л-Апнзиднн 192, 201, 205
Анизол 339 , 377 . 389. 405
производные 76, 377, 385, 407, 423
Анилин
алкилирование 177, 183, 202 сл.
ацилирование 201
галогенирование 188, 2i4—216
диазотирование 197
конденсации 211 сл.. 223
окисление 207 сл., 220
основность 191. 192. 344
нитрование 186, 216. 217, 404. 448
получение 169 сл.
спектры 194, 195
сульфирование 190, 217
физиол. св-ва 195
фотохимия 348
Анилин (производные)
N-аллпл- 103
N-бевзнл- 373
бензилиден- 500, 518, 529
бром- 171, 189. 190. 218
4-бром-М,Ь1-бис(4-бромфснил)- 352
3-бром-N N димстил- 200
З-бром-Ы.Ы-ди метил-4-нитрозо- 200
N-трет-бутил-N-метил- 377
N-трет-бутил-N-хлор- 343
гидрокси- см. Фенол, амино-
N.N-диацетнл- 202
2.6-дибром- 215
2.6-дииод-4-иитро- 197, 2l5
2.3-диметил- 377
N.N-диметил- 407. 594
алкилирование 204
ацилирование 203
галогенирование 214
нитрование 378, 407
нитрозирование 200
основность 192
получение 160 , 203
спектры 194, 195
Н,Ы-димстил-4-«Итро- 200 . 386 , 39о
728
2.7, 398
ftK 21^^s
177. 192-194. 204
4‘ИТ°Л СМ Толуидины
Киил 192, 198. 201, 203, 204, 211. 345
4МмМеети“-2.6-АКэтил. 183
Й.м1п!л“м-’1НТР°3°- 198-200 , 451, 452 , 457
2-метил-6-этил- 183
метокси- СМ. Анизидии
натрий- 177
2-нитрО-
ОСНОВНОСТЬ 196
получение 186, 187
реакции 183. 208, 211. 217. 336
3-иитро- 68, 186, 187, 209 , 211
4-нитрО- 217
ОСНОВНОСТЬ 193. 340
получение 169. 186
реакции 184, 197. 206, 2|1, 215
N-нитро- 217. 461, 464
нитрозо- 119. 386
М-нитрозо-И-фенил- 199, 45/
4-нитрофснил-М,М - дихлор' 336
нитрохлор- 344, 404. 465
пептахлор- 216
2.6.М.М-тетрлметил- 200
2 4,6-трибром- 188. 197, 201, 214
2,4.6-трНхлор' 188. 208, 214
N-фенил- 188. 191, 394 , 395
2-хлоп- 171, 189, 192
3-хлор- 189, 192, 345
4-хлор- 171. 177, 190, 192, 193, 212
N-хлор- 215, 345
циано- 345
этил- 169, 177, 192
Анилиновый черный 207
Антрацен, производные 289, 359
Арндта >— Эйстортэ реакция 305, 308
Атаракс 120
Ацетальдегид 127
Ацетамидин 609
Ацетанилид
алкилирование 178. 180, 201, 204
ацилирование 202
галогенирование 188, 214
нитрование 186, 216, 405
спектры 194
Ацетанилид (производные) 189, 202, 209, 214
Ацетилен 21
дициано- 660, 683, 685, 696. 690
«Ацетиленовая молния» 121
Ацетилцитрат 403
Ацетильные защиты 78 "9. 178
Ацетон 126, 526
производные 449, 482, 490, 569
Ацетонитрил 678
производные 599, 667, 670. 671
Ацетонитроловая кислота 416
Ацетофенон 176
производные 212. 515
Бамбергера перегруппировка 220, 245
ьамфорда — Стпвенеа реакция 511, 512
“Ортона реакция 379, 443, 444, 484, 615
ЬаудцШа реаКцИЯ з7б
Бекмана
перегруппировка 29. 176. 492, 493
расщепление 144
ьеиадрил 144
Ьензалъдаэян 505, 507—509, 571
N-ОКСНД 563
Ьслзальдегид
4-амнло- 213
гидрокси- 124
4"ДИметнламшю- 480
Бензальдегид
имин 515
нитро- 374. 422, 480
Бензамид, 2-нитрозо- 436
Бензанилид 180
Бензидин 195. 196 223
производные 196, 281
Бензидиновая перегруппировка 223, 246, 281,
Бензиламин 46, 198
производные 198, 595
Бензилиденовая защита 211
Бензилоксикарбонильная защита 79
Бензилцианид. 2-цяаио- 661
Бензильная защита 80, 81
Бензоил изотиоцианат 1’12
Бензоильные защиты 78, 80
Бензойная кислота 175
амид 174
4-амино- 169. 170
гидрокси- 186
нитро- 187
2-нитрозо- 374
хлор- 189, 190
Бензол
аминирование 176, 180, 339
окисление 377
реакции с иминами 594
— — цианидами 690
спектры 195. 417
Бензол (производные)
амино см. Анилин
гексациано- 578. 685
дегидро- 172, 323. 346. 366
1,2-диамиНо- 183, 217. 220 сл.. 682
производные 217, 574 сл.
1 3-диамино- 171. 184. 185. 220. 222
1,4-диамино- 171, 184. 210. 220. 223
производные 200, 351, 571 сл.
1,2-дигидрокси- см. Пирокатехин
1.3-дигидрокси- см. Резорцин
дпннтро* 187, 409. 418. 419, 423
2,4-ЛИНИтро- 1-хлор- 187, 204
иоднитрозо- 383
литий- 394
метил-см. Толуол
метокси- см. Анизол
нитро- 217. 425
восстановление 169—171, 185, 223
галогенирование 189
нитрование 437
нитрозированне 451
пиролиз 419
получение 208
спектры 417
структура 416
сульфирование 190
физ. св-ва 416
Фотохимия 420
нитрозо- 373, 378, 383—385, 388-
3&9. 391 сл.
ннтрозопеитафтор- 389, 390
питронптрозо- 208. 393
1-нитро-2.4.6-трихлор- 208
1-нитро-З-фтор- 423
нитрохлор- 187
1-нитро-2-этил- 169
стиба- 610
тстраампно- 185
1,2.4-триамино- 169
2 4^6-три трет-бутил Ьнитрозо- 396
Г.з’5-тримстил-2-питро- 416
тринитро- 208, 417—419
2.4,6-трнфтор-1.3,5.-трнцнано- 678
хлор- 198
Бензолдиазонийгалогеипды 197, 1Уо
Бензолсульфокислота
2-амиио- 190, 192. 218
3-амино- 190, 192
4-амино- 190. 192, 217, 218
729
Бензонитрил l!’?Hq34^349ffl4!69
производные 5W. «м.
да
Берснда перегруппировка 6-1
388
388
Б';*Гнз^е3^.^%4. .383. см. также
Еиипклоатканы1 азапронаводные 35. 309.
Б| 1 550 5S7, 563 . 568. 569, 610
Башлсра - Наинральского реакция 6/4
азапроиэводные 35, 309.
реакция 674
Брауна
распад 597
реакция 15, 28
Брауна — Рудольфа синтез 590
Бриканнл 134
Бруцин 38
БУтаион-2
З-метнл- 92. 375
З-метпл-З-нмтрозо- 396
Бутиламии 54 , 55 . 60
N-6yTH.n-N-iHiTpo3o- 455
Ы-бутнл-Н-хлор- 337
N.N-днметил- 15, 54. 55
N-метпл- 55
трет-Бутнламин 345
Бутилннтриты 379
З-метнл- 440 . 441
Бутин, амино- 111
трет-Бутоксикарбопильная защита 79, 80
Бухерера реакция 173. 182, 274
Вагнера — Меераейна перегруппировка 321
Валлаха перегруппировка 210, 299
Валлоид 120
Вснталин 134
Вердазилы 285
Виволан 145
Вилоксазин 145
Внльгеродта — Киндлера реакция 145
Вильсмейера — Хаака — Арнольда реагенты
595. 596
Виндолении 50
Вминлазид 549
Виниловый спирт, цизнопроизводные 684,
. 685
Вольфа перегруппировка 308, 520
Вурстсра синий 351
Габриэля синтез 13. 28, 111
Гдлогенамины 148 ел.
«Гарпуны» протонные 75
Гаттермана синтез 494. 504 . 580. 594
Гексаметилентетрамин 14, 123, 124, 126
Гексоген 124
Геири
реакция 430, 431
ретро-реакция 428. 433
Гептан, нитрозопронзводные 341 382
гептаитрион-2,4.6 347
Гептилнитрит, 1-метил- 442, 443
Гетразап 120
Гёша синтез 580 . 594 . 674
Гидразндин 604
Гидразиды 17
Гидразил'радикалы 284 ^85
Гидразины
нитрозо- 450
основность 278
получение 268 сл., 450
реакции 17. 268. 278 сл., 358, 454 480 сл
структура 274—278 » -и. TW сл,
Гидразобензол 223, 388
Гидразонаты 598. 600. 601
тио- 600
Гидразоноилгалогеннды 577, 579, 582 сл
590. 591. 597, 697 **
Гидразоны 19, 476 сл.
Гидразосоединения 124
Гидроксамовые кислоты 175, 434, 594
Гидроксизин 120
Гидроксиламниы 228 сл.
нитрозо- 450. 451, 453, 454, 458
основность 240
получение 229 сл., 388 сл.. 480 сл.
реакции 218—220, 240 сл.. 372 сл., 476 сл
613 сл.
спектры 240
структура 238—240
Гидрокспматы 598, 600, 696, 697
тио- 600
Гидроксимоилгалогеииды 576 сл. 580 сл
590. 597, 697
Гинзберга синтез 13. 64
Гипоазотистая кислота, эфиры 441
Гиповаэ 120
Глицерин, ннтро- 446
Гомоцистеин 133
Гофмана — Лефлера реакция 33, 66, 340. 341
Гофмана — Маркиуса перегруппировка 205
Гофмана перегруппировка 29, 174, 175 637,
638
Гофмана реакция 71, 70!
Гроба реакция 488
Гуанидины 588. 589. 609—611
Губена — Фишера синтез 650
Дарзана реакция 305
Даффа реакция 124
Дебнера — Мюллера реакция 213
Дегидробензол см. Бензол, дегидро-
Декалнн, нитрозохлор- 380
Делепина реакция 14. 123
Джулолнднн 93
Диазення соли 283
Диазены см. Азосоединеиия
Диазет
дигидро-, производные 287
тетрагидро-, производные 612, 710
Ди азиридии
окса- 298
производные
получение 82. 272. 505. 610
реакции 287. 656
структура 40. 43
тиа- 272
Диазирин 287
производные 287, 313, 551
Диазония соли 300 сл., 484, 485
Диазосоединения 300 сл.
Дианизидин 196
Дибензи.ппп 156
Лиимии 289
Дикарбоиилдиимидаэол 654
Дильса — Альдера реакция 603. 674. 675*
688. 689
Димедон 103
Днмрота реакция 325
1,3,2-Диоксазол, тетра гидро-, производные
1.3.4-Диоксазол, дигидро-, производные 699
1.3-Диоксолан. Производные 710
1.2.4-Дитиазол, дигидро-, производные 638
1.3-Дитиаи, производные 508
1.4-Дитиин. тетрациано- 681
М-Днтиален, производные 680
Дифеиндрамип 144
Дифенилип 224. 281
Диииклогексилкарбодиимид G34, 842, 654,
658
Дициклогекеилмочевина 634
Диэтилкарбамазаи 120
Допамин 60
730
сл.
504
242, 438. 5)0,
Енамнву G3, 73, 91 сл'
Жасмон 439
Заидмейсра реакция 649
Защитные группы аминов 78
Зонна — Мюллера синтез 495,
Изобутиламин 60
Пзоксазол
дигидро-, производные 241,
Д 526, 625. 699
производные 428, 591, 655, 698, 699
тетрагндро-. производные 236 сл.. 435,
622 сл.
Изонитрососдинения 4)2—414
Изопавин, производное 1)3
Изопентиламин 60
Изопреналин 134
Изопропилами» 345
N-изопропил- 75, 289, 34), 346, 347
Изопропил метил кетон см. Бутанон-2,3-ме-
тил-
Изопропилнитрит 379, 441
Изотиазол
дигидро-, производные 639
производные 551
Изотиоцианаты 629 сл.
Изохинолин 653
днгидро-, производные ИЗ. 502, 523 , 527,
530. 532, 674
производные 582, 661, 665
тетрагидро-, производные 34, 531
Изоцианаты
получение 64, 175, 206, 584, 585, 630 сл.
реакции 17, 35. 580, 639 сл.
Изоцианиды 131, 580, 581, 600, 647, 700 сл.
Изоциановая кислота 634, 635
Имидазол
бензо-, производные 221, 537
дигидро-, производные 114, 115, 529, 504,
664. 705. 707, 708, 710
производные 221, 529. 610. 674, 70S
тетрагидро-, производные 527, 529
Нмидаты 576
получение 585, 586, 598 сл., 641. 704
реакции 586, 60) сл., 6)1, 667
тно- 598 сл,, 667. 704
Имидовые кислоты 576. 598
производные 576 сл.
Имидоилгалогсниды 576 сл., 611. 697, 702
Имнпрамин 122
Инамины 105 сл.
Индазол, производные 366. 426
Индандион-1,3 400
производные 682
Индерал 146
Индол 425, 705
днгидро-, производные 34
производные 2)2, 425, 485, 494
Ипсо-атака 406
Кадаверин 60
Калуца реакция 631
камфора, производные 38, 39
^анииццарро реакция 431
капролактам 484. 490
Карбазол, производные 361, 390. 393, 42о,
Карбодиимнды 629, 630, 632 сл., 654. 658
карбонилцианид 685
кариофиллен, производные 254
Катапиноз 125
Кетен, дицнало- 685
и!?НИм,,||Ы 597. 600, 629, 632 сл.
к^фалялы 1зз
Жнера — Вольфа восстановление 284, 289.
к 510
Кноп«.?На РетР° конденсация 428
^вснагелн — Бухерера реакция 652
» «о
Корриды даМН0Ь110й 222
Коуна
"^группировка 85
Расщепление 72 . 232 249 кг,
п’Крезол 376 ' 250
Криптаты 148
Кронке реакция 394 615 fii«
Ксилидины 183 210 ’ 51 6 6
Ксилолы 404, 406, 446
Курпиуса ^Регюппировка 174,
oW, Зоб, 366, 637 , 638
175, 315,
Лабеталол 134
^«остерин. производные 541
Лейкарта реакция 20, 478 543
Лецитин 133
Либермана реакция 396 457
Лизин 60
Лоссена перегруппировка 174. 17S, 353. 366.
vu /, 0-50
Луцидулин 623
233.
Мак-Лафферти перегруппировка 453
Малеиновая кислота, динитрил 678
Малоновая кислота
динитрил 678, 680. 681, 684, 686 сл.
диэфир, замещенные 400. 448
Манделонитрнл 672, 677
Манниха реакция 62. 433 , 434 . 480 . 517
Медигалер 134
Мезимбрин 34
Мезоксалевая кислота 400
Меера реакция 435
Мейера реакция 407, 435
Мейзенгеймера перегруппировка 72.
249, 435
Метан
диамино- 115
динитро- 4)4 _
нитро- 407. 41) сл.. 416. 4)9. 428, 433, 437
нитрозо- 374. 379, 383 сл., 388
нитрозотрифтор- 380, 383. 384 , 39). 393,
397. 398
нитрозотрихлор- 384
нитро(4-ннтрофеннл)- 434
ннтротрих.юр- 415
нитрофенил- 437
тетранитро- 40/, 414, 41э
тринитро- 407. 414, 41э
ЮЗ-695. 697
Метаннловая кислота см. Бензолсульфо-
кислота, 3-амино-
Метанол 54
амино- 126
Метиламин 54-W,
N N-диметН.т- г>4. эЬ. Я», w
4М
> «’
N-НИТрОЗО- 4э-
Метилнзоцяанпл
Метиликтрат «
Метилиитрнт
Метионин № а нитрил 6,.’. 6Т7
Миндальная ьисло 6--
Миндальный - д „е 280. «2
Михаэля лрнсоьд
Михлера кетон Л
Морфм'1" 44’ 9S 137, 144. 145. 153. 1»
производные
201. 210 СЛ.
амино-
379
Нава» 120
Нафталин 378
1-аМИИО- 3W
получение181. № 2М> 210 212
реакций 183. 1W. ™
фкзиол. св-ва 19о
2амиио-
получение 170. 173. >82
реакции 1/7, 181 1Л.
физиол. св-ва 19о
2-амико-1-аиетил- 188
1-амино 4-литро; 1»ь
аминоннтрозо- 199. ен
аминосулъфо- 190
1 амиио-2-этил- 183
ацетиламино- 187. 216
гидрокси- см. Нафтолы
дегидро- 172, 366
1,2 диамино- 221, 222
2.3-дказндо 682
нитро- 181. 182
сульфо но
этиламино- 203
Нафтиламнны см. Нафталин,
Нафшлннтрен 359
Нафтойные кислоты 174, 182,
Нафтол-1 181, 185
аминопроизводные 185, 186
2 4-динитро- 186
Нафтол-2 173, |82
1-нитрозо- 379
Нафтолсульфокислоты 190
1,4-Нафтохин 210
производные 682
Небера перегруппировка 344 , 494 , 509 , 547,
553
Неоантерган 118
Нефа реакция 431, 434, 435
Никотин 518
Нитроаминнля перегруппировка 463
Нитрамины 448, 454, 459—465
Нитраты 445—449
Нитрениевые ноны 334, 338, 339, 341—-
343
Нитрены 353 сл.
амино- 282, 283
образование 74. 174, 175. 316. 389
реакции 35 . 42, 359 сл.
структура 334 , 335, 359
Нитрилиевые соли 599, 661 сл.
Ннтрил-илиды 692 сл.. 710
Нитрияимины 692 сл.
Нитрилы 647 сл.
оксиды 538, 539 692 сл.
получение 73, 149, 378 536, 537. 647 сл
реакции 18. 25, 33 , 495 . 496, 579, 580 598
599. 6С-7. 608
Ннтримины 501
Нигриты 440—445
Нитроглицерин 446
Нитрозамнны 445 сл.
Нитрозоловые кислоты 399
Нитрозосоедннеиия 372 сл. 382 сл 424
425, 614 сл.
Нитроксяды 251 сл 350. 396 397
тио- 256
Ннтроловые кислоты 483 . 609 693
псевдо-Нитролы 415. 416. 601
Нитроновые кислоты 412—414
Нитроны 230, 393 сл., 482 613
питросоединения 18 20 3 74’ 399
535
Нитроцеллюлоза 446
Ннтросульфиды 256
Порборнадлен 380 , 528 , 675 , 690
7-аза-, производные 43
Норборнак
7-аза-, Производные 43
киано- 650
Норбориен 233 , 650
732
СЛ., 645
сл..
Озазоны 223, 280
1,3 5-Оксадиазин, производные 531, 646
1’,2’4-Оксадиазол
дигндро-. Производные 526
производные 674
1 2 5-Оксадиазол см. Фуразан
N-оксиды см. Фуроксан
1,3,4-Окса диазол
ангидро-, производные 699
производные 542 , 589. 610
тетрагидро-, производные 625, 629
^ди^дро-, производные 234, 236. 245, 391,
тетрагндро-. производные 236
1 2-Оксазин
’дигндро-. производные 237, 392. 393
тетрагндро-, производные 234, 237
1,3-Оксазин
бензо-, производные 220
дигидро-, производные
получение 103, 143, 237, 531, 532, 552, 598,
599. 601, 603, 664
реакции 602, 603, 605
производные 646, 705
тетрагидро-, производные
получение 139, 154, 433, 434
реакции ЮЗ, 143
1,4-Оксазнн см. Морфолин
Оксазнрндин, производные
получение 230 , 231, 234 , 235. 607
реакции 313, 373, 534, 618
структура 40, 43, 238, 239
Оксазмрнн. производные 551
1,2-Оксазол см. Изоксазол
1,3-Оксазол
бензо-, производные 218, 537
дигидро-, производные
получение 137, 554, 598, 599, 603, 604, 699»
705. 710
реакции 141, 601. 602. 605, 606, 708, 709
производные 364, 554. 708
тетрагидро-, производные 141—143, 154,
252, 664
Оксатиазолы, производные 625 , 694
Оксатяолан, производные 346
Оксетан, производные 154, 710
Оксиран 154
производные
получение 533, 534
реакции 32, 82, 128,
тетрациано- 679, 683,
Оксирен, производные
Оксотреморнн ill, Ц2
Оксирснолок 146
Орнитин 60
Ортаниловая кислота
Ортолева — Кинга реакция 616
Ортона перегруппировка 202
«Органические металлы» 692
Паргилин 111
Пассерннк реакция 703
Пенам, производные 520
Пенициллин 50.3—505, 520 536
Пентадиен, полициано- 678
Пентазадиены 329
Пеитен-4-илнитрит 445
Пентнламии 49
трет-Пентиламин, N-трет-бутил- 348
Переамидирование 64
Пикриновая кислота см. Фенол 2,4 6-трИ-
нитро-
Пннаколииовзя перегруппировка 137
Пиппера синтез 598
Пиперазин см, Пиразин, гексагидро-
пи22^ле,,п см' ПиР»ДИ!1. тетрагидро-
перидии см. Пиридин, гексагидро-
оензо-, производные И9
129, 346
88 5 686, 690
308
Бензолсульфо-
... .. см.
кислота, 2-амино-
3-метокси- 145
ЦРопилннтрит 44
• Полин. 3-амино
1 ipOliiiOHH mil п Я
Пиразин 367
гексагидро-, производные U9, 121, 156
Пиразол
дигидро-, производные
получение 287, 29J. 305, 306 48) 564
565, 629, 664. 699 ' ' ’
реакции 292, 309, 641
производные 280. 551, 591, 610. 669, 699
тетрагидро-, производные 625, 628, 629
Пиридазин
днгидро-, производные 553
гексагидро-, производные 276. 563
тетрагидро-, производные 288, 393
Пиридин
бициклический 552
гексагидро 32, 52, 58
производные
получение 132, 138. 150, 157 , 252 , 340
реакции 154, 253, 259 сл., 347, 350, 455
структура 44. 45
дигидро-, производные 293, 657
N-оксид, производные 245. 248 , 615, 620
производные 33, 132 , 367, 390 , 426, 582, 610
тетрагидро-, производные 93 487. 497.
530, 535, 646
Пириламин 118
Пиримидин 6Ю
гексагндро-, производные 433, 434
дигидро-, производные 274
тетрагндро-, производные 122
Пировиноградная кислота 546
Пирокатехин 118
Пиррол 349, 365
дигидро- 487
производные, получение 85, 93. 478 497
499, 529, 699, 708
N-оксид, производные 614
производные
получение 530, 556, 564. 566. 699
реакции 33
тетрагндро- 32, 55, 58
производные
основность 55
получение 33 , 340 , 529, 624 , 628
реакции 78, 93, 157
структура 40
Пирролидин см. Пиррол, тетрагндро-
Пирролин ем. Пиррол, дигидро-
Полназаены 559 сл.
Полицианосоединения 677 сл.
Полоновского реакция 249
Понцио реакция 416
Порфирексид 252
Празосин 120
Прегнан, производные 65. 443, 484
Призмам 292
Прометазин 157
Пропан
амино- см. Пропиламми
2-бром-2-цнтро- 383
2-гндроксн-2-нитро- 451
1,3-диамино- 76, 77. 121, 122
2-метил-2-нитрито-1 -нитрозо- 381
~-метил-2-нитро- 414
^-метнл-2-питрозо- 396. 398
2-нитро- 413 427. 429
Ьнятрозо- 379
2-нитрозо-2-хлор- 390
2-нитро-2-хлор- 426 „
•‘Ропанол. амнполроизводпые 32, 128, 139.
Пропаргиламины 106, 111—113
Цропаргилояая перегруппировка 105
“ропиламии 55, 345
Збром- 159. 160
З-гидроксл. isq, 145
1
- 55
-метокси- 145
Пропранол 146
Протоберберины 502
I севдонигролы 415, 416 501
Псеадопельтьерии 37
Путресцин 60
Пфицнера — Моффатта метод 644
Резорцин 376
Рейссерга реакция 653
реформатского реакция 508
Й реакция 14- 23, 663, 664
нихтера реакция 436
Робинсона аннелирование 513
Робинсона — Шепфа реакция 37
Салициловая кислота 174 186
Сальбутамол 134
Семидины 224. 281
Серин 133
Серотонин 60
Симмонса — Смита реагент 519
Син/антн-изомерия 488—496
Скраупа синтез 212, 425
Смайлса перегруппировка 437
Соммле реакция 123, 478, 604
Соммле — Хаузера перегруппировка 31. 343
Спартеин 73
Спермидин 125
Спермин 125
Стелазии 120
Стефена восстановление 495, 504, 675
Стивенса перегруппировка 29, 721
Стильбен, производные 69, 152, 351, 425
Стирол 21. 130, 283, 351, 391
производные 154, 420, 548, 660
Сукцинимиды, галоген- 336, 396
Сульфаниловая кислота см. Бензолсульфо-
кислота, 4-амино-
Сфингозин 133
Тербуталин 134
Тетразаны 120, 328
Тетразены 232, 328, 348
1,2.4,5-Тетразин
гексагидро-, производные 457
дигидро- 604
Тетразол, производные
получение 323, 452, 590, 669. 674
реакции 323, 357, 525, 527, 639, 694
Тиадиазепин, производные 558
|,3.5-Тиадназнн, производные 531
1,2,4-Тиадиазол
дигидро- производные 638
тетрагидро-, производные з2/
1,3,4-Тиадназол, производные 294, м2, ээ».
1,3-Тиаз?н. дигндро-, производные 532, 582.
664
Тиазлрин, производные з51
т Дегидро-, производные 112. о», о04.
705. 708
производные 155. '08
Тнаетриа"м.0Д»'г"»1>0-- производные 639
Тииран 155
производные оМ
Тиотиксен 120
Жлях-»—
- ж-675' ж
701 .. -й
тпчитьная защита 7о
т“ см Толям. тринитро-
Толан 393
о-Толидин 196
Толилеидпизоцианат
о Толуидин 183. 1S-. -3“
662
137
733
л-Толуидин 164, 192, 197
д-Толуидин
основность 1У«а
1К™°з’з“е 3«.»5.' 407
2-амино-5-нитро- 208
диамнно- 184
2?н?РГ>!а Ж. 405. 417. 421. 438
нитрозо- 383—385
З-нитро-6-ннтрозо- 208
тетранитро- 4IG
тпниитро- 416
л-Толуолсульфоннлниапиды и -изоцианиды
36, 675, 700. 701
д-Толуолсульфоннльная защита 78
Тофраинл 122
Тразикор 14б
Транзеит 134
«Трехфазный катализ» 71
Триазаиы 327
Триазепы 325—327. 396
1,2 3-Трназин, производные 366
1.2’4-Триаэии, производные 5аб, 610
1.3’5-Триазнн, гексагидро-, производные
127
126,
1.2.3-Трназол
бензо-, производные 222 , 323 , 366
дигидро-, производные 322, 526, 609
производные
получение 35, 100. 321, 323 , 354 , 426,
468. 524, 527. 669
реакции 307. 499, 549
тетрагндро-, производные 529
1,2,4-Триазол
днгидро-. производные 524, 612, 629, 674,
706
производные
получение 452, 589, 606, 610, 674
реакции 452. 657. 660
тетрагндро-, производные 629
Триалкилсидилазиды 355. 638
Тритильная защита 81
Трифториеразик 120
Тропам, производные 160
Тропин, производные 15. 37
Тротил ем. Толуол, тринитро-
Уксусная кислота, нитро- 428
Ульмана реакция 204
Уридин, 2'-дезоксн-313
Феназин, производные 210. 222
Феиантрахинон 22!
Феиилаэид 356 . 357
Феинлазотрцфенилметан 290
Фенилгидразин 198 , 223 , 396
производные 200. 223
Фенилгидроксиламин 220, 388
Фенилендиамины см. Бензол
Феиилизотиоцинат 206, 207
Феиилизоцнанат 207. 654 694
производные 200. 206 ’
Фемнлизоцианнд 207 705
Фенилпнтрамин 217
Феиилнитреи 357, 362, 367
Фенилнитрит 417
Фемоксаэин 218
диамино-
производные 219
Фсноксибензамин 156
Фенол 173, 174. 180, 198. 377 386
2-амиво- 186. 192 218 219
3-амино- 180. 192’
2-амиво 4-нитро- 187
4-амиво-З-хлор- 220
2,4-диннтро- 187
4-метнл-2-нитро- 376
4-иитро- 185 , 377
нитрозо 119, 200 . 377, 386
Фенол
пентациано- 685
2.4.G-1 рннитро- 417
Фенотиазип. производные 120, 426
Фсрроиеи. азидо- 355
Фиолетовый кристаллический 203
Фишера — Хенна перегруппировка 199 377
45G
Флуорен 400
9-Флуореиилметильная защита 79
Формальдегид
имины- 123, 477
конденсации 20, 23, 123, 126, 127, 139
433, 465
Формильная защита 78, 178
Форстера реакция 50G
Фосгенимнны 576 сл.
Фриделя — Крафтса реакции 594 . 595
Фридлендера синтез 213
Фталевая кислота 210
Фталевый альдегид 431
Фталимид, производные 74, 270, 358, 363 сл.
Фталонлцианид 682
Фульвален, селена- и тнапроизводпые 692
Фульвен, производные 108
Фумаровая кислота, динитрил 678, 683, 688
Фуразан, производные 660, 694
Фуран
днгидро-, производные G90
производные 3G5
тетрагндро-, производные 690
Фуроксан
бензо-, производные 384 , 387 , 388
производные 387
Фурфурол 445
Хинаэолин 174
производные 174, 582
Хинин 38
Хннодиметан, цнанопроизводные 681—683,
686, 687, 691, 692
Хиноксалин 220
производные 220, 576
Хинолизин, производные 93
Хинолин 212. 591. 653
производные 109. 212, 213, 653
тетрагидро-, производные 93
Хлораль 477, 530
Хлорамин Т 129
Хлорпикрин 415
Хлорсульфопилизоцианат 639, 645, 646
Холестерин производные 402
Холин 133
Хромой, производные 98
Хьюсгена классификация 524 сл.
Цефалоспорины 504
Цианазнд 355
Цианамиды 589
Цнангндрииный синтез 130
C-Цианиды см. Нитрилы
Цианины 596, 597
Цнанокснматы 698
Цианонитреи 359
Цианоформ 678, 693-695. 697
Циглера — Торпа реакция 497, 669
Цчклиэин 120
Циклобутан. 1,2-дициано- 680
Циклобутен, цианопроизводные 680, 682, 688
Циклобутилнитрит 444
Циклогексадиенон, нитрозо- 376
Циклогексан 382, 484
нитро- 375, 408, 409
нитрозо- 383, 388, 397
4-нитро-1-хлор- 375. 382, 398
Цнклогексанол 444
Циклогексанон 92, 484
2-нитро- 415
оксим 404. 484 492
2-циано- <К)
734
Циклогексен 129, 337, 403, 422
l-днметиламино- 92
Ццклогскссион. производные 109 403
Цнклогсксиламин 344 , 345, 388 , 409, 455, 546
N-мет алл- 75, 34б
N-цпклогекскл- 75, 114
Циклогсксилизоциаиид 706
Цнклогексилнитрит 444
Циклогептан, аза- 32
Циклогептилнитрит 444
циклододекан, нитро- 414
Циклонит 124
Циклоионан, азапроизводные 73
Циклооктан, аза- 45
Цнклооктатетраен 689
циклооктси 402
1,2-диаза- 290
циклооктилнитрит 444
Циклопентаднеп 438
циаиопронзводные 367, 678, 687
Циклопентанон 142
Циклопропан
нитрозо- 374
тетрацнанопролзводное 679
Циклопропанов 126
Циклопропилнитрит 444
Циммермана реакция 418
Чепмена перегруппировка 180, 494 , 600, 696
Чнчибабииа реакция 274
Шиффа основания 373, 476 сл.
Шмидта перегруппировка 29, 175 , 325 , 601,
637
Штиглица перегруппировка 245, 341, 366, 498
Штреккера синтез 506, 652
ЭлайомицНн 297
Эмде восстановление 14
Эпихлоргидрин 146, 164
Эргостерин, производные 352
Эритромицин, производные 544
Эрлиха — Закса реакция 391, 482, 618
Эстрадиол, производные 303
Эстрон ИЗ
Этан
амино- см. Этиламин
Ьамино-2-гидрокси- см. Этаноламик
1-амино-2-ннтро- 415
1,2-диамино- см, Этилендиамин
нитро- 414, 415. 420. 433
цианопроизводные 678—680 , 687
Этанол 54, 142
Этанолами» 119, 123, 134, 1зб
производные 128 сл, 144
Этиламин 47 , 54 , 56, 60 . 345
2-бром- 135
М,М-диметилтрнхлор- 152
N.N-диметилхлор- 158
2,2-дифтор- 54
N.N-диэтил- 54, 55. 56, 58
Ц-иэоиропил-2-фенил- 16
М-мстил-Ы -нитро- 462
М-метил-Гч'-нитрозо- 452
2-метокси- 143. 144
N-нитро- 461, 463
N-HHtpo-N-этил- 461
I-фенил- 38
фтор- 54, 74
N-хлор-Й-этил- 337
N-этил- 55, 56. 58, 203 , 345, 346, 460
Этилен, цианолроизводные 680. 681, 683 сл.,
688 сл.
Этиленгликоль, тетрациано- 083
Этилендиамин 116—118
производные 118, 119
Этиленднаминтетрауксусная кислота 118
Этиленоксид см. Оксиран
Этилнятрат 446—448
ЭтидиИТрнт 375, 379, 440—442, 459
Этокснкарбонилнитрен 361, 364, 365
Эфедрин, производные 135
Эшвайлера — Кларка реакция 20 . 543
Яновского реакция 418
Ялпа — Клингемаиа реакция 485, 609
ОБЩАЯ
ОРГАНИЧЕСКАЯ
ХИМИЯ
ТОМ 3
АЗОТСОДЕРЖАЩИЕ
СОЕДИНЕНИЯ
Редакторы:
М. И. ПАСТУШЕНКО
О. И. СЛУЦКИЙ
Художник
Н. В, НОСОВ
Технический редактор
Г. И. КОСАЧЕВА
Корректор
П. Б. ИВАНИЦКАЯ
ИБ № 1485
Сдано в наб. 15.03.82
Иодп. в печ. 30.07 82
Уч.-изд л’зо'та ТУ л' «Р.-ОТТ. 46.
Заказ •* 1О5.це"Г'к
Изд. Л', 2053.
Ордена «Звак ПоВДта»
ЮТ076 М?скваТ°? *Хнмия»-
<посква, Стромынка, 13.
Лголад7о“Спре”№ 2
1>1'До0ог0^р™Ж',еч0'>“»а
Ленипградското °б° ед1,',«Ме,’И
’Те»-<»чеекаякщ1^:,!",,я
">*3—лнграф^ромЛа°"п“"
"“-граф,,,, „ "x;eS™’
&в5т«.л.к,
вски1| проспект, 29.
Фотографировал Семенюченко Владимир
chem_vova@mail.univ.kiev.ua; vova2002@mail.ru
ОБЩАЯ
ОРГАНИЧЕСКАЯ
ХИМИЯ