/



















































































Автор: Ластовский Р.П.
Теги: органическая химия химия химическая промышленность химические реакции
Год: 1967




Текст
Редакционная коллегия
Р. 1!. Ластовский (гл. редактор), Е. А. Божевольнов,
А. В. Бромберг, В. Г. Брудзь, В М. Дзиомко,
И. А. Красавин, Г. И. Михайлов, Г. А. Певцов
СОДЕРЖАНИЕ
7-Азаоктагидропирроколин. А. А. Пономарев, И М. Скворцов . . 5
2-, 3-, 4-н-Амил-и 2-, 3-, 4-(5-нонил)-пиридин. Ю. И. Чумаков,
В. М. Ледовских............................................... 7
2-Амииотолуол-4-диметилсульфамид. Г. А. Тимохин, Б. И. Киссин 12
2-Антраценсульфохлорид. Н. И. Чернова, Б. М. Болотин, В. Г.Брудзь 15
1-Ацил-2,3-дигидроиндолы. А. К. Шейнкман, Л. П. Махно .... 17
5-Бром-2-нитрофуран. 3. Н. Назарова, Б. Н. Новиков............. 20
и-Бутил- и изобутилацегат. О. Н. Карпов, Р. М. Быстрова ... 23
/п/итгг-Бутилкарбазат. £. А. Макарова, Г. Н. Кошелева.......... 26
6-Гептилпиридазинон-З. А. А. Пономарев, В. А. Седавкина .... 29
Д-Глюкозамии хлоргидрит. Е. П. Крысин, Р. Е. Тарасова . ... 31
Диаммонийная соль о-сульфобензойпой кислоты. Е. Я. Яровенко,
М. Ф. Кондрашова............................................. 34
O-N-Диацетиляндоксил. В. М. Островская, И. А. Гоикер .... 36
2,3-Дигидроиндол. А. Н. Кост, Л. П. Махно, А. К. Шейнкман,
Г. А. Маркус................................................. 39
N, N'- Дика рбокс и мети лэтиленд нами ио-бис-мети л фосфиновая кисло-
та. Р. П. Ластовский, В. В. Сидоренко, А. В. Лапшина .... 42
3,3'-Диметилспиро-[бензтиазол ин-2.2'-(2'II-1'-бе изопираны)]. В. А.
Иншакова, И. А. Драпкина. В. Г. Брудзь, И. П. Плитина . . 44
2,5-Диметокси-2,5-дигидрофурфуриламияоэтанол, А. А. Пономарев,
И. А. Маркушина, И. С. Монахова.............................. 43
1,5-Ди (2'-окси-3', о', 6'-трихлорфенил)-3-ацетилформазан. В. М. Дзи-
омко, В. М. Островская...................................... 50
Индоксилац-тат. В. М. Островская, И. А. Горкер .............• 54
М-Карбоксиметилдиэгилентриамино-М',М"-бис-изопропилфогфииовая
кислота. Р. П. Ластовский, В. В. Сидоренко, Н. В. Лапшина.
Т. П. Коноплева ............................................ 57
7-Кетоноундекановая кислота. А. А. Пономарев, В. А. Седавкина . 59
2-Метил-З, 4, 5, 6-бис-инденопирилий перхлорат. Ю. А. Жданов,
Г. Н. Дорофеенко. В. А. Полчков .............. 63
З-Метил-1,2-дигидропирролизнн. А. А. Пономарев, И. М. Скворцов 65
3-Метил-5-диметиламинометил-1,2-дигидропирролизин. А. А. Поно-
марев, Л. Н. Астахова, В. И. Симонцев ........... 10
2-Метил-1,6-диоксаспиро-(4,4)-нонан. А. А. Пономарев, И. А. Мар-
кушина ...................................................... 72
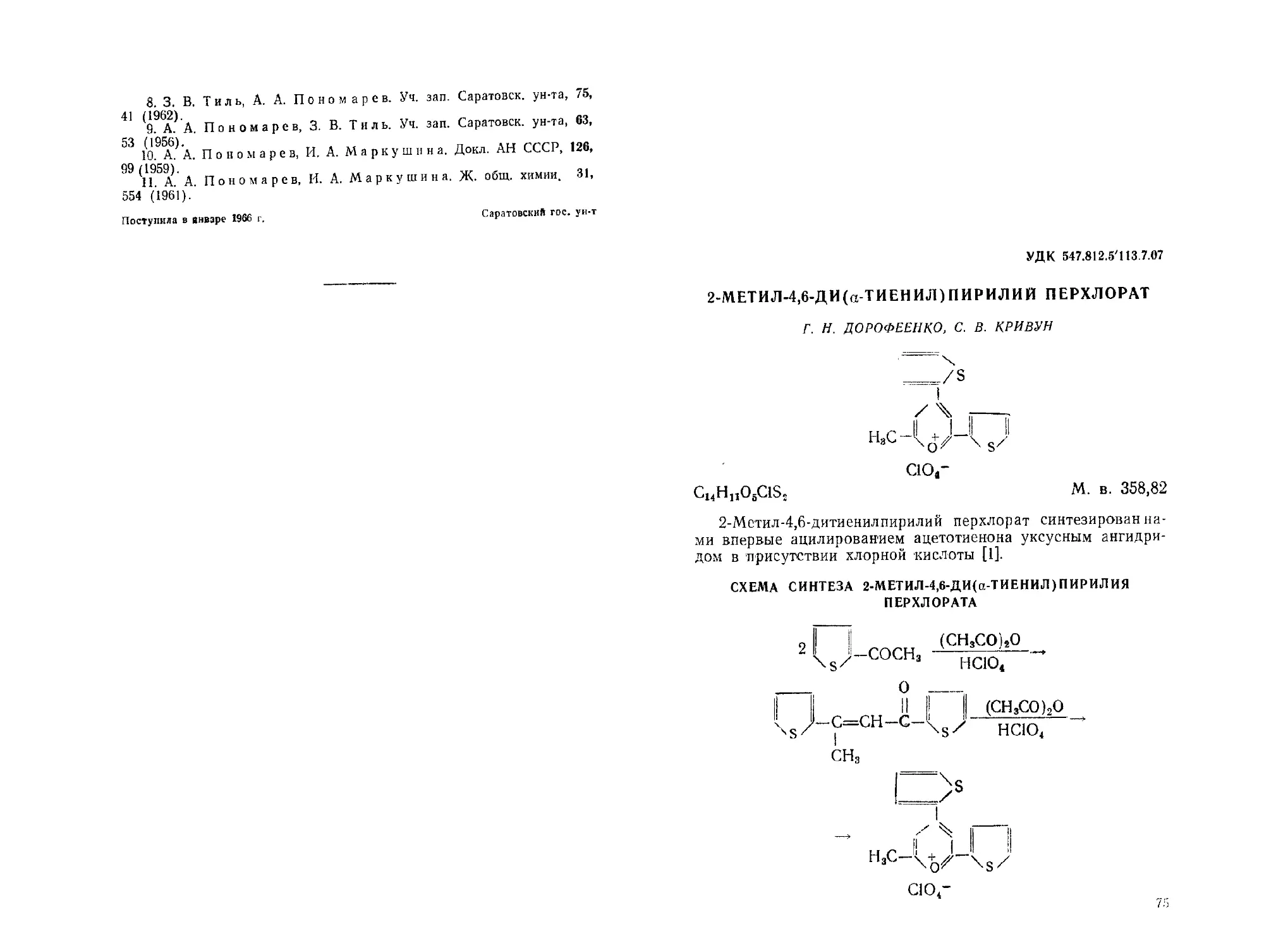
2-Метил-4,6-ди (а-тиенил) пирилий перхлорат Г. Н. Дорофеенко,
С. В. Крову н................................................ 75
2-Метил-2.4-диэтил-1,6-диоксас11иро-(4,4)-ио„ан. А. А. Пономарев,
В. А. Седавкина .............................................. 77
Метилеи йодистый. А. Л. Лифиц, Е. П. Агеев, В. П. Каролинский,
М. И. Ильичева............................................... 80
N-Метил-З-оксипиперидин. А. А. Пономарев, Н. И. Мартемьянова 82
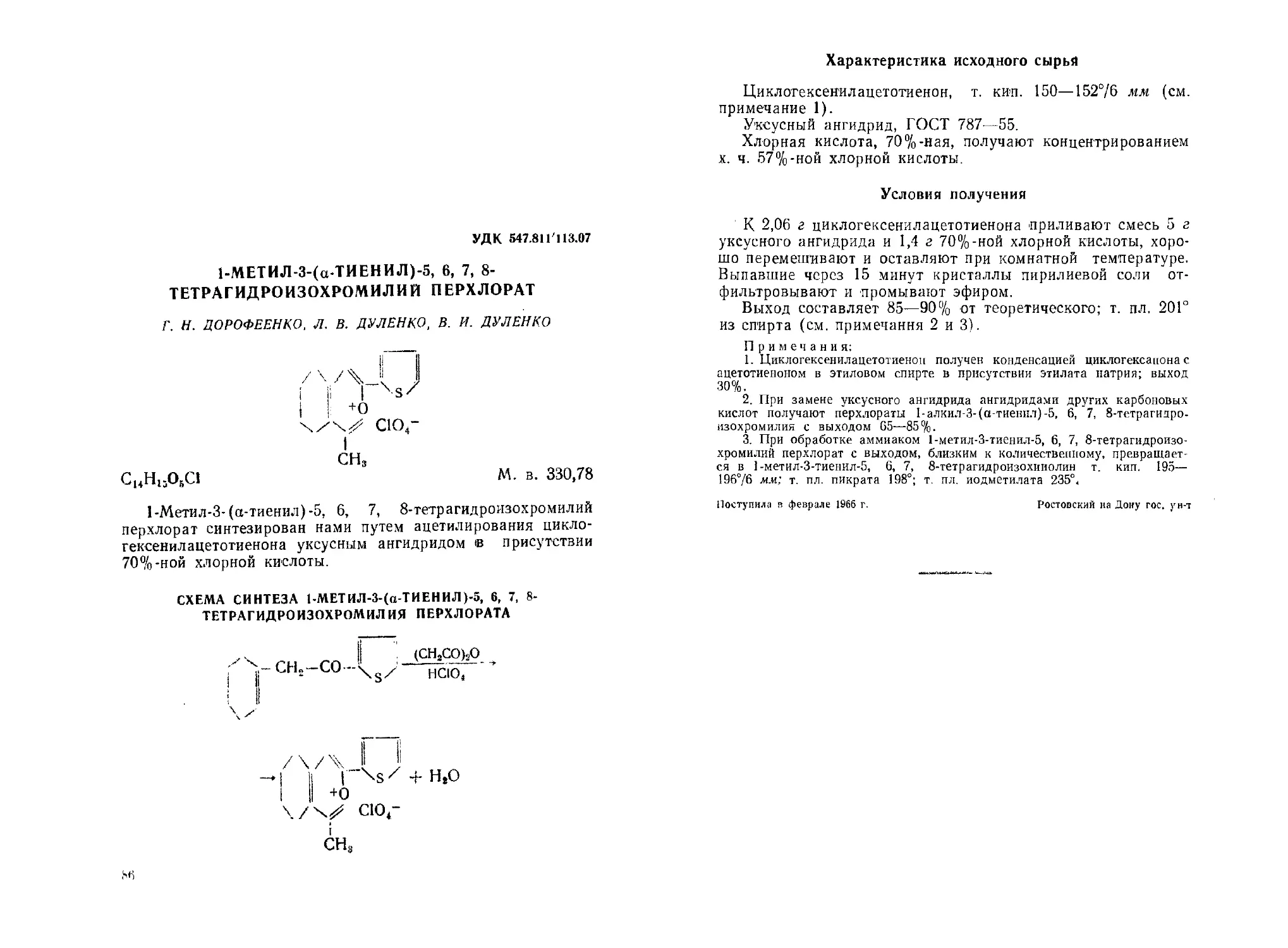
1-Метил-3-(а-тиенил)-5, 6, 7, 8-тетрагидроизохромилий перхлорат.
Г. Н. Дорофеенко, Л. В. Дуленко. В. И. Дуленко............... 86
2-Метил-1,6,8-триоксадиспиро-(4,1 4,2)-тридекаи. А. А. Пономарев,
И. А. Маркушина................................................ 88
!-Метил-3-фенил-5,6,7,8-тетрагидроизохромилий перхлорат и 1-ме-
тил-З-фенил-5, Ь, 7, 8-тетраг идроизохинолии. В. И. Дуленко, Г. 11.
Дорофеенко.................................................. 91
3-Метил-5-формил-!,2-дигидропирролизнн. А. А. Пономарев, Л. Н.
Астахова .................................................... 94
N-Метил-i-хлорпиперидии. А. А. Пономарев, И. И. Мартемьянова 97
2-Метокси-7 метил-1,6-диоксаспиро-(4,4)-ионсн-3. А. А. Пономарев,
И. А. Маркушина.............................................. 99
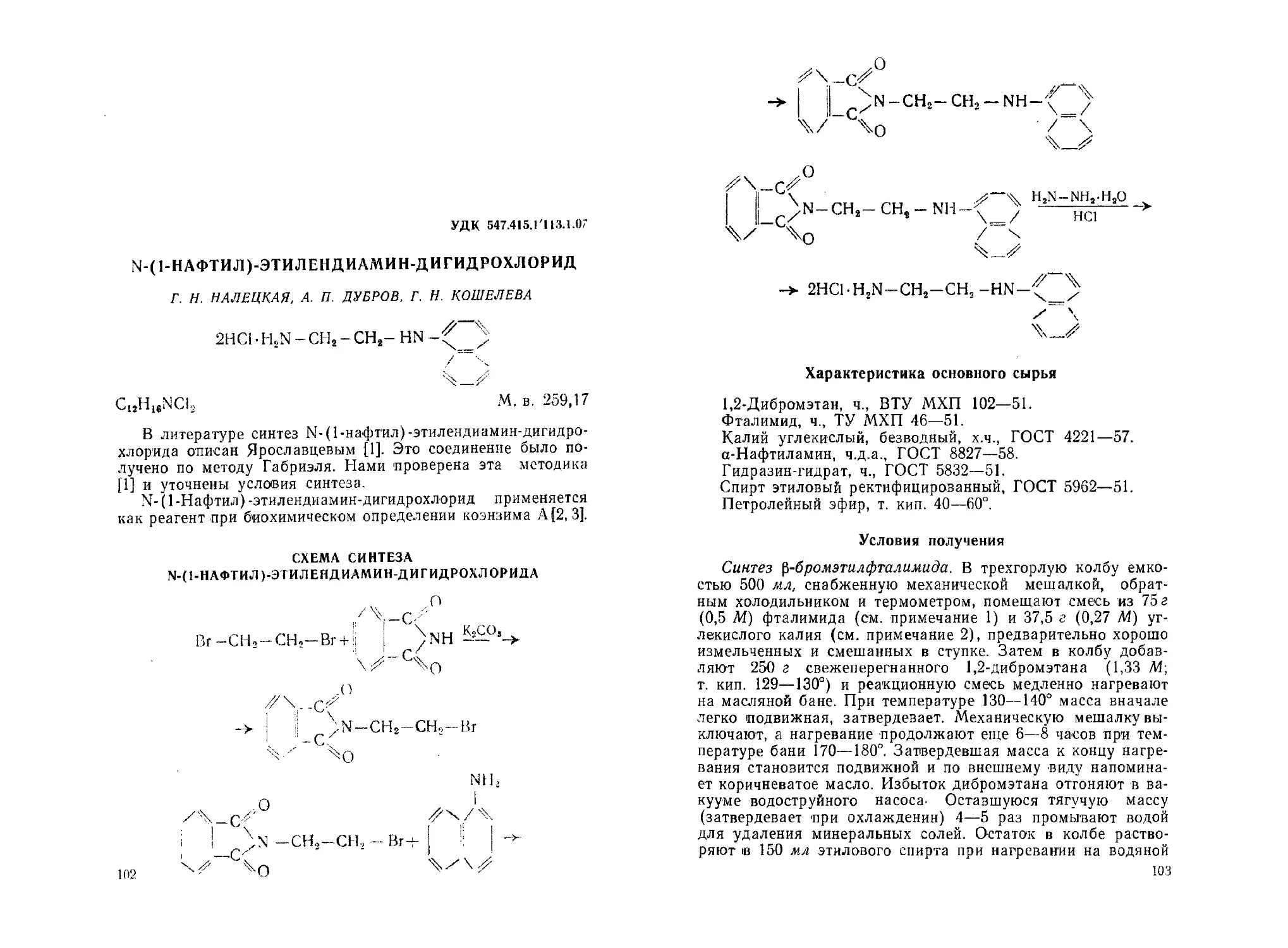
М-(1-Нафтил)-этилендиамин-дигидрохлорид. Г. Н. Налецкая, А. П.
Дубров, Г. Н. Кошелева..................................... 102
4-Нитро-4'-диэтиламиностильбен. В. Г. Брудзь, И, И. Чернова, Д. А.
Драпкина, Б. М. Болотин..................................... 106
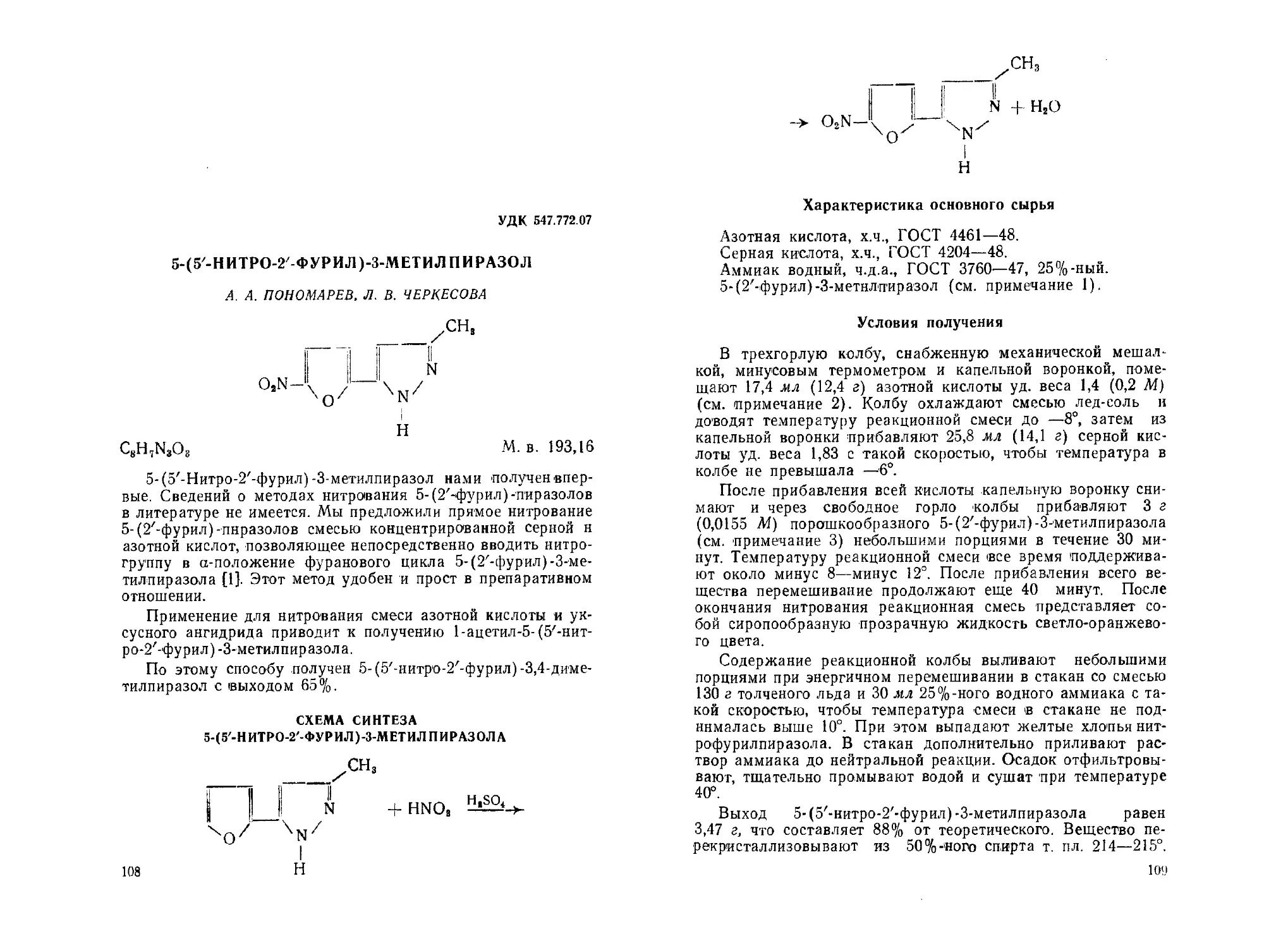
5-(5'-Ннтро-2'-фурил)-3-метилпиразол. А. А. Пономарев, Л. В. Чер-
кесова ....................................................... 108
(.5-Нитро-2-фуроил) ацетон. А. А. Пономарев, Л. В. Черкесова ... 111
N-Оксиимид малеиновой кислоты. В. С. Иванов, В. К. Смирнова,
Цао Юн....................................................... ИЗ
2-(3-Оксипропил)-5-(3-оксибутил)-фуран. А. А. Пономарев, И. А.
М /ркушина ... . ....................................... 116
8-Оксихннолин-(2-азо-2')-1'-оксинафталин. И. А. Красавин,
В. М. Дзиомко, И. И. Дударева, Ю. П. Радин.................. 119
Селеномочевииа. А. Б. Лундин.................................. 122
1-(а-Тетрагидрофу рил)-3-амннобутан и 1 -(5-метил-2-пнрролидил)
пропанол-3. А. А. Пономарев, А. П. Кривенько, М. В. Норицина 126
1-(а-Тетрагидрофурил)-3-мстчлпентанол-3 н 2-метил-2-этил-1,6-диок-
саспиро-(4,4) понан. А. А. Пономарев, 3. В. Тиль............ 130
N-Тетрагидрофурфурилэтилендиамин. А. А. Пономарев,
И. М. Скворцов.............................................. 133
N, N, N', N'-Тетраметил-!,3-пропаидиамин. В. М. Дзиомко, 3. С.
Сиденко..................................................... 135
Тиациклооктанон-5. А. А. Пономарев, И. С. Монахова........... 137
Тимоловый синий. Е. Я. Яровенко М. Ф. Кондрашова.............. 140
гг-Толуолсульфохлорид. Б. И. Киссин, Е. Н. Куракин, Г. А. Тимо-
хин ........................................................ 143
2, 3, 5-Трийодбензойная кислота. Г. Н. Налецкая, А. П. Дубров,
Г. И. Кошелева............................................. 146
2, 4, 6-Трнметилпирилий перхлорат и 2, 4, 6-триметилпиридии.
Г. Н. Дорофеенко, С. В. Кривун ................ 149
2, 4, 6-Трифенилпирилий перхлорат и 2, 4, 6-трифенилпиридин.
Г. Н. Дорофеенко, С. В. Кривун.............................. 152
1-Феиил-3-метил-4-аминопиразолон 5-хлоргидрат. Р. Б. Журин,
В. Н. Ивина ................................................ 155
9-Формил-3,4-беизакриднн. Б. С. Танасеичук, И. Я- Постовский . 158
Фураи-2-карбоновая кислота. 3. В. Назарова. В. Н. Новиков . . . 160
З-(а-Фурил) гексанон-5. Л. А. Пономарев, В. А. Седавкина .... 162
1-(а-Фурнл)-5-метил-3-аминогексаи. А. А. Пономарев, Н.П. Маслен-
никова .......................... 165
1-(а-Фурил)-3-метиламииопропан. А. А. Пономарев, М. В. Норицина,
И. М. Скворцов................................................ 167
1-(а-Фурил)-5-метилгексанон-3. А. А. Пономарев, 3. В. Тиль . . . 169
N-Фурфурилпропилендиамин-!,3. А. А. Пономарев. Н. В. Маслен-
никова ..................................................... 172
N-Фурфурилэтилендиамин. А. А. Пономарев, И. М. Скворцов . . . 174
(2-Хлорэтил)-триметиламмонийхлорид Г. Н. Налецкая, А. П Дубров,
Г. Н. Кошелева............................................. 176
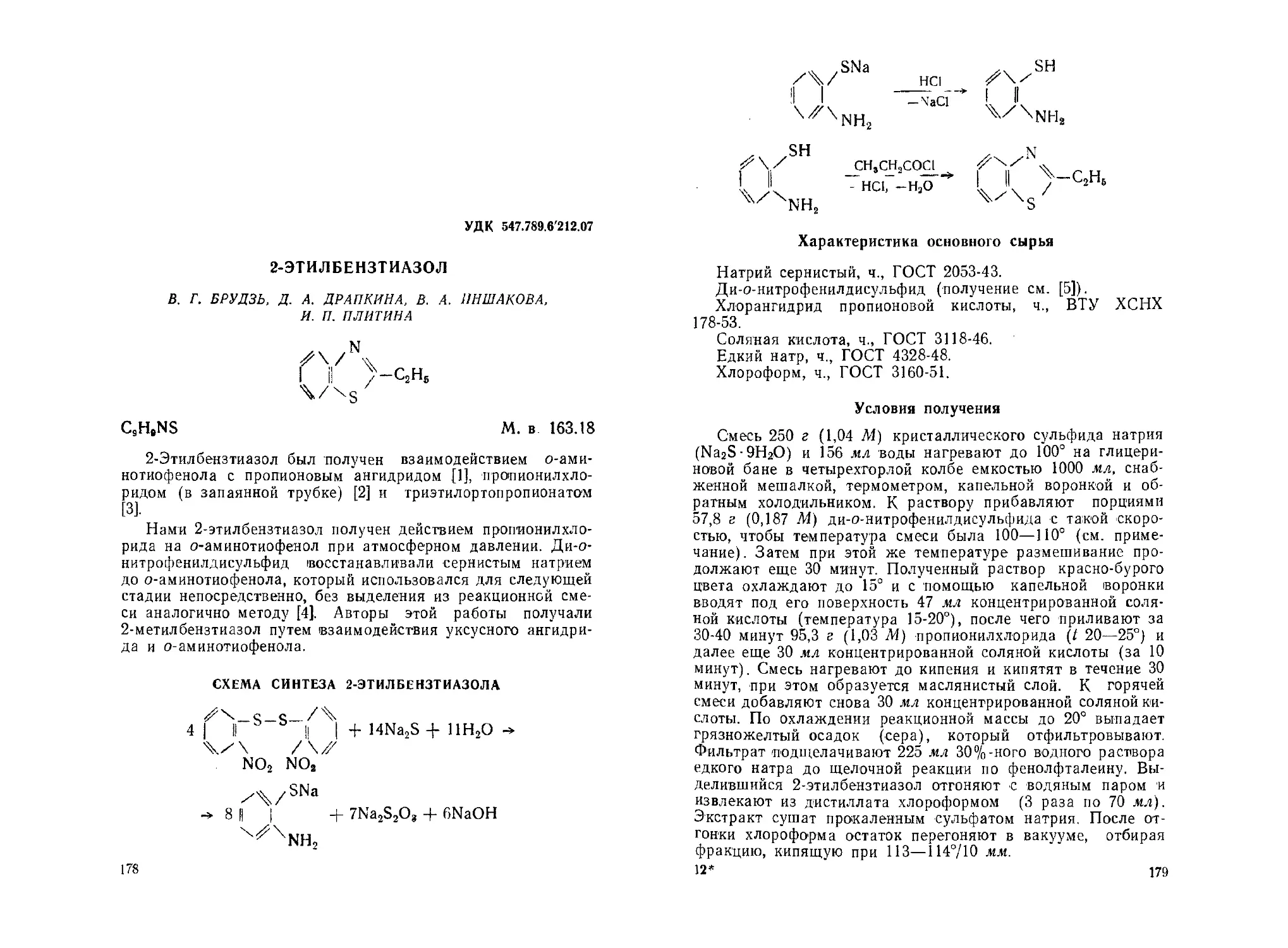
2-Этилбензтиазол. В. Г. Брудзь, Д. А. Драпкина, В. А. Иншакова,
И. П. Платина................................... ...... 178
Алфавитный перечень соединений, описанных в настоящем сборнике 181
4
УДК 547.834.2.07
7-АЗАОКТАГИД РОПИРРОКОЛ ИН
1,4-Диазабицикл о-(4.3.0)-нонан
.4. 4. ПОНОМАРЕВ, И. М. СКВОРЦОВ
c7hun2
М. в. 126,20
7-Азаоктагидропирроколины долгое время были трудно-
доступными соединениями; их получали восстановлением
алюмогидридом лития пролин-ацил-лактамов, в которых аци-
лом являются остатки а-аминокислот [1].
Нами предложен метод каталитического синтеза 7-азаок-
тагидропирроколинов путем дегидратации доступных N-тет-
рагидрофурфурилэтилендиаминов [2].
СХЕМА СИНТЕЗА 7-АЗАОКТАГИДРОПИРРОКОЛИНА
'-CH,-NH-CH2-CH,-NH2
О
-Н2О
ZrO,
NH
Характеристика основного сырья
N-Тетрагидрофурфурилэтилендиамин (см. соответствую-
щую статью в этом сборнике).
5
Условия получения
Через реактор (см. примечание 1), предварительно нагре-
тый в токе азота до температуры 300—305°, пропускают в
течение 2 часов 15 минут 16 г (0,111 М) N-тетрагидрофурфу-
рилэтилендиамина. Скорость тока азота через реактор долж-
на быть в пределах 800—1000 мл в час и поддерживаться на
этом уровне до прекращения выделения катализата из реак-
тора. После окончания процесса в приемнике собирается 12,9 г
катализата в виде желто-зеленой жидкости. К. ней добав-
ляют 20 мл эфира, а затем через обратный холодильник твер-
дое едкое кали до насыщения выделяющегося водного слоя.
Эфирный раствор отделяют и сушат твердым едким кали. По-
сле фильтрации раствора через пористый стеклянный фильтр
№ 3 и отгонки эфира остаток перегоняют в вакууме из кол-
бы с колонкой Видмера, отбирая фракцию с т. кип. 68—
70°/Ю мм. Получают 3,54 г (25,3%) 7-азаоктагидропирроко-
лина в виде подвижной жидкости с сильным запахом. Веще-
ство растворимо в воде, спирте и эфире; /гу)—1,4940;
<77°— 0,9679 (см. примечание 2).
Примечания:
1. Дегидратацию N-тетрагидрофурфурилэгилеидиамина проводят на
установке, описанной на стр. 65 этого сборника.
2. Аналогичным i _ . .... __ ______~
рил)-этилеидиамина получают З-метил-7-азаоктагидропирроколин с выхо-
дом до 30%, т. кип. 70—73710 мм; л‘р—1,4875; —0,9452.
путем дегидратацией Ы-(а-метнлтетрагидрофурфу-
ЛИТЕРАТУРА
1. A. Stoll, A. Hofmann, Th. Ре t г z i 1 k a. Helv. chlm. Aera
31, 1544, (1951).
2. А. А. Пономарев, И. M. Скворцов. Докл. АН СССР, 148,
860 (1963).
Поступила в январе I96tr г Саратовский Гос. уп—т
УДК 547.821.4.07
2-, 3-, 4-н-АМИЛ- И
2-, 3-, 4-(5-НОНИЛ)-ПИРИДИН
Ю. И. ЧУМАКОВ, В. М. ЛЕДОВСКИХ
II +CH2-CH3-CH2-CH..-CHg
C,oH,5N
2-, 3- или 4-изомер
М. в. 149,23
, zCH»-CH,-CH,-CHa
/ % /
II +СН
\N/- xCH2-CH3-CHa-CH8
CuHjjN
2-, 3- или 4-изомер
М. в. 205,34
Изомерные 2-, 3-, 4- н. амил- и 2-, 3- или 4- (Б'-нонмл) -пи-
ридин могут найти применение в качестве комплексообра-
зующих и хелатообразующих реагентов, а также в качестве
ингибиторов коррозии и селективных экстрагентов.
Обычно для получения 2-, 3- и 4- н. амилпиридина, а
также 2-, 3- и 4-(5'-н. нонил)-пиридина используют взаимо-
действие соответствующих изомерных метилпиридинов с га-
лоидными алкилами в присутствии амида натрия или калия
[1—3]. Недостатком этого метода являются низкие, плохо
воспроизводимые выходы продуктов и неудобства, связан-
ные с опасным в обращении амидом натрия в жидком аммиа-
ке.
В последнее время благодаря реакциям изомерных ме-
тилпиридинов с 1,3-бутадиеном стали легко доступны соот-
ветствующие им 2-, 3-, 4-(З'-пентенил)-пиридин и 2-, 3- или
4-(5'-н. нонадиен-2', 7'-ил)-пиридин [4—6].
Мы нашли, что изомерные пентенил- и нонадиенилпириди-
ны легко гидрируются в соответствующие н. амил- или 5'-но-
нилпиридины.
В качестве катализатора используется никель Ренея или
другой катализатор гидрирования в условиях, исключающих
возможность гидрирования пиридинового цикла.
СХЕМА СИНТЕЗА ИЗОМЕРНЫХ АМИЛ-И НОНИЛПИРИДИНОВ
/ Ч н
II +СН3-СН2-СН=СН-СН3 4^
2-,3- или 4-(3'-пентенил)-пиридин
/ Ч
-- II 4-CH8-CH2-CH2-CH2-CHs
\n^
2-,3- или 4-н.амилпиридин
СН2-СН =СН-СН4
II +СН -“f--.
хсн2-сн=сн-сн3
2,-3- или 4-(5'-нонадиенил-(2',7')-пиридин
/ /СНг-СНа-СНа-СНз
- II +СН
Xn/' хСН2-СНа-СН3--СНз
2-, 3- или 4-(5-нонил)-лиридин
Характеристика основного сырья
Изомерные пентенил- и нонадиенилпиридины (см. приме-
чание 1).
Водород, ГОСТ 3022—45.
Условия получения
Во вращающийся автоклав из нержавеющей стали поме-
щают раствор 0,125 М овежеперегнанного изомерного (3'-
пентенил)- или (5/-нонадиенил-2\7/)-пиридина в 50—60 мл
метилового спирта и 2 чайные ложки никеля Ренея. Надав-
ливают в автоклав азот до 2—3 атмосфер и выпускают его
для удаления воздуха из автоклава. Затем надавливают во-
дород до 30—40 атмосфер и приводят автоклав во вращение
8
при комнатной температуре до поглощения расчетного коли-
чества водорода. Если гидрирование не начинается, то авто-
клав нагревают до 80—90° (см. примечание 2). Гидрирование
занимает около 2 часов, после чего автоклав охлаждают, вы-
пускают избыточное давление водорода и продукт гидриро-
вания отсасывают в колбу Бунзена. Стенки автоклава обмы-
вают 25 мл метанола и присоединяют их к реакционной мас-
се после гидрирования. Никель Ренея отфильтровывают и
промывают на фильтре 15—20 мл метанола, которым пред-
варительно смывают склянку Бунзена. Метанол отгоняют при
атмосферном давлении, а остаток перегоняют в вакууме, по-
лучая с выходом, близким к количественному, соответствую-
щий изомер н. амил- или (б'-нонил) -пиридина (см. таблицу
1). Константы соединений по литературным данным приве-
дены в примечании 3.
Свойства изомерных амил- и нонилпиридинов
Название Температура кипения, °С[мм rf? Температу- ра плавле- ния пикрата, °C (испр.)
2-h. Амилпиридин 82,5-84,5/8-9 0,8881 1,4834 71,8-72,3
2-(5'-Нонил)-пиридин 81-83/0,5 0,8944 1,4912 93,5 -94,5
3-н. Амилпиридин 54,5-55/1 0,9070 1,4926 79,5-80
3-(5'-нонил)-пиридин 83-87/0,5 0,9257 1,5065
4-н. Амилпиридин 51,5-52/1 0,9077 1,4925 107,8-108
4-(5'Нонил)-пиридин 95-97,5/2 0,8911 1,4880 120,2-120,8
П р п м е ч а н и я:
1. 2-, 3-, 4-(3'-пентенил)-пиридин и 2-, 3-, 4-(5'-нонадиенил)-пиридин
получены согласно [5].
2. Более подробно гидрирование аналогичных продуктов описано
нами ранее на примере 2-винилпиридина [7].
3. По литературным данным изомерные пентенил- и нонадиенилпи-
ридины имеют следующие константы:
2-н.-амилпиридин—т. кип. 206,5—207°; т. пл. пнкрата 73°; d4°—0,9175;
—0,9016; т, пл. хлорплатината 160" [1];
3-н. амилпиридин—т. кип. 110—112° при 20 мм; т. пл. пикрата
78,8—79,2° [2];
4-н. амилпиридин — т. кип. 229—230°; т. пл. пикрата 102—103°;
dt°—0,9203; d420—0,9031; т. пл. хлорплатината 203° [I];
4-(5°-нонил)-пиридин — т. кип. 265—267°; т. пл. пикрата 114—116° [1].
ЛИТЕРАТУРА
1. А. Е. Ч и ч и б а б и н. Bull. Soc. Chirn. France, (о), 3. 1607 (1936!.
2. A. Miller, R. Levine. J. Organ. Chem. 22, 168, (1957).
9
3. E. H a r d e g g е г. Е. N ikies. Helv. Chim. Acta, 39, 505, (1956).
4. R. Wegler. G. Pieper. Ber., 83, 6 (1950).
5. Ю. И. Чумаков, В. M. Ледовских. Авт. свид. 158575; Билл,
изобр., № 22, 12 (1063).
6. Ю. И. Чумаков, В. М. Ледовских. Методы получения хи-
мических реактивов и препаратов, вып. 7, М., ИРЕА, 1963, стр. 38.
7. Ю. И. Чумаков. Методы получения химических реактивов и пре-
паратов, вып. 7, М„ ИРЕА, 1963, стр. 33.
Поступила в декабос 19с5 г. Киевский политехнический институт
УДК 547.541.52.07
2-AM И Н ОТО Л У О Л-4-Д И М ЕТ И Л С У Л Ь Ф А М И Д
Г. А. ТИМОХИН, Б. И. КИССИН
СНЭ
I
SOaN(CH3).2
C9HI4O2N2S М. в. 214,28
2-Аминотолуол-4-диметилсульфамид применяется в синте-
зе красителей и других препаратов. Он может быть получен
из п-толуолсульфохлорида или из о-нитротолуола. В первом
случае нитруют п-толуолсульфохлорид [1], во втором — суль-
фохлорируют о-нитротолуол [2, 3]. В обоих случаях первым
промежуточным продуктом является 2-нитротолуол-4-сульфо-
хлорид, который в дальнейшем подвергается амидированию
диметиламином, образуя 2-нитротолуол-4-диметилсульфамид.
Последний превращается в конечный продукт восстановле-
нием.
Для практики наиболее интересным представляется син-
тез 2-аминотолуол-4-диметилсульфамида из о-нитротолуола.
Восстановление промежуточно получаемого 2-нитротолуол-4-
диметилсульфамида сернистыми щелочами, в частности ди-
сульфидом натрия, до настоящего времени не описано в ли-
тературе.
СХЕМА СИНТЕЗА 2-АМИНОТОЛУОЛ-4-ДИМЕТИЛСУЛБФАМИДА
СН8 СНз
Г J
| rN0“ + 2HOSOsCl — \ rN°5 -J- H2SO, 4- HC1
4/ 4/
I
SO,Cl
CH, CH,
I I
A~N°a +2NH(CH,), fVN0? + (CH3),NH-HC1
4/ w
I I
SO2C1 4 SO2N(CHs),
ch3 CH3
I I
i^\-NO2 +Nags5 H2o _ ( X|-NH2 +NajSa08
4/ \/
I I
SOsN(CHs)2 SO8N(CHs),
Характеристика основного сырья
2-Нитротолуол, технический, ГОСТ 10206—62.
Кислота хлорсульфоновая, ч„ ТУ НКХП 385—41 или тех-
ническая, ГОСТ 2124—43, 1 сорт.
Диметиламин (раствор), ч„ ТУ МХП 2645—51 или техни-
ческий, ГОСТ 9967—62. о
Дисульфид натрия, водный раствор, содержащий при 50°
330—380 г/л Na2S2.
Условия получения
Синтез 2-нитротолуол-4-сульфохлорида. В четырехгорлую
колбу емкостью I л, снабженную мешалкой, термометром, об-
ратным холодильником и водно-глицериновой баней, вносят
740 г (6,35 /И) хлорсульфоновой кислоты и при температуре
не выше 40° приливают через капельную воронку 206,1 г
(1,5 714) о-нитротолуола. Массу размешивают 1 час при тем-
пературе 35—40°. Затем в течение 30—40 минут нагревают
ее до 50°, далее температура самопроизвольно доходит до
60—65°, после чего в течение 2 часов температуру смеси под-
нимают до 100—105°, размешивают в течение 2 часов и затем
охлаждают до 20—25°. Охлажденную сульфохлоридную мас-
су медленно, при энергичном размешивании, выливают на
2900 мл воды, имеющей температуру 0—2°. Образовавшуюся
суспензию 2-нитротолуол-4-сульфохлорида, имеющую темпе-
ратуру не выше 10°, размешивают 1 час и фильтруют. Осадок
на фильтре промывают двумя порциями по 300 мл холодной
(5—10°) воды и отжимают (см. примечание 1).
Выход 2-нитротолуол-4-сульфохлорида равен 296 г, что
составляет 83,6% от теоретического. Температура затвердева-
ния продукта 26,5—27°.
12
Получение 2-нитротолуол-4-ди.метилсульфамида. В четы-
рехгорлую колбу емкостью 1 л, снабженную мешалкой, тер-
мометром, обратным холодильником и водяной баней, вносят
90,2 г (2 М) диметиламина в виде 20—30%-ного водного
раствора и в течение 40 минут при размешивании прибавля-
ют 117,75 г (0,5 М) 2-нитротолуол-4-сульфохлорида. При этом
температура реакционной массы самопроизвольно поднима-
ется до 50—55°. После того, 'как температура начнет снижать-
ся, массу размешивают без охлаждения еще 1 час, а затем
охлаждают до 20°. Образовавшийся 2-нитротолуол-4-диметил-
сульфамид отфильтровывают и промывают на фильтре пятью
порциями (по 150 мл) воды, имеющей комнатную температу-
ру, отжимают и сушат при 35—40°.
Выход 2-нитротолуол-4-диметилсульфамида равен 111,1 г,
что составляет 91% от теоретического. Температура плавле-
ния вещества 86—87° (см. примечание 2).
Получение 2-аминотолуол-4-диметилсульфамида. В четы-
рехгорлую колбу емкостью 500 мл, снабженную мешалкой,
термометром, обратным холодильником и водяной баней, вно-
сят 100 мл горячей воды и при температуре 96—98° и разме-
шивании загружают 48,85 г (0,20 М) 2-нитротолуол-4-диме-
тилсульфамида. К полученной эмульсии при температуре
98—100° в течение 4—5 часов приливают через капельную
воронку 28,6 г (0,26 М) дисульфида натрия в виде водного
раствора, содержащего при 50° 330—380 г/л ЫагБг. Далее
массу размешивают при 98—.100° до полного восстановления
2-нитротолуол-4-диметилсульфамида. Конец восстановления
определяют хроматографией на бумаге (см. примечание 3).
По окончании восстановления массу охлаждают до 20° и при
этой температуре размешивают еще I час. Суспензию 2-ами-
нотолуол-4-д'иметилсульфамида фильтруют, осадок на фильт-
ре отмывают водой от щелочи, отжимают и сушат при тем-
пературе 70—80°.
Выход 2-аминотолуол-4-диметилсульфамида 38,14 г, что
составляет 89% от теоретического. Температура плавления
167—169° (см. примечание 4).
Общий выход 2-аминотолуол-4-диметилсульфамида по
всем стадиям синтеза составляет 66,7% от теоретического по
о-нитротолуолу.
Примечания:
1. Полученный 2-нитротолуол-4-сульфохлорид, ввиду его повышен-
ной способности к гидролизу, следует хранить при температуре не выше
20° и возможно быстрее перерабатывать в диметилсульфамид.
2. По литературным данным [4], т. пл. 2-нитротолуол-4-диметилсуль-
фамида 92—94°.
3. Методика определения конца восстановления состоит в следующем.
В мерный цилиндр емкостью 250 мл наливают чистого бензола до тех
пор, пока его слой по высоте цилиндра не будет равен 10—15 мм. На
полоску' фильтровальной бумаги шириной несколько меньшей, чем внут-
ренний диаметр цилиндра, с помощью стеклянной палочки наносят реакци-
13
онную массу в виде поперечной равномерной черты, на расстоянии 20—
25 мм от конца. Этим концом вниз бумагу опускают в цилиндр так, что-
бы она погрузилась в бензол, однако черта реакционной массы должна
находиться на 5—10 мм над поверхностью бензола. Цилиндр закрывают
часовым стеклом. Через 15—20 минут бумажную полоску вынимают и
дают бензолу с нее испариться. Отсутствие лимонно-желтой окраски вы-
ше нанесенной поперечной черты является признаком полноты восстанов-
ления 2-нитротолуол-4-диметилсульф амида.
4, По литературным данным [4], т. пл. 2-амннОтОлуол-4-диметнлсуль-
фамида 172—174°.
ЛИТЕРАТУРА
1. F. Re verdln, Р, С repie их., Вег., 34, 2992 (1901).
2. Герм. пат. 89997; Frdl., 4, 39.
3. BIOS, 1153, 40.
4. V Pet row, О. Stephenson. A. M. Wild J. Pharm. and
Pharmacol., 12, 705-19 (1960); C. A. 55, 8337 (1961).
Поступила в мае 1966 г. ЦЗЛ
УДК 547.672.2.07
2-АНТРАЦЕНСУЛЬФОХЛОРИД
Н. И. ЧЕРНОВА, Б. М. БОЛОТИН, В. Г. БРУДЗЬ
SO2C1
I I! I I
C,4H9C1O2S М-в. 276,74
В литературе описан единственный способ получения
2-антраценсульфохлорида нагреванием натриевой соли 2-ан-
траценсульфокислоты с пятихлористым фосфором и хлор-
окисью фосфора в присутствии уксусной кислоты [1}.
Нами разработан метод получения 2-антраценсульфохло-
рида по описанному в [2] общему способу получения хлоран-
гидридов кислот. В качестве хлорирующего агента использо-
ван хлористый тионил, а катализирующей добавкой служил
диметилформамид.
СХЕМА СИНТЕЗА 2-АНТРАЦЕНСУЛЬФОХЛОРИДА
SOsNa
I II ' I | +SOCU А
SO3C1
+SO24-NaCl
Характеристика основного сырья
11^2-Антраценсульфокислоты натриевая соль, ч., ВТУ РУ
15
Диметилформамид, ч., ВТУ РУ 1193—56.
Хлористый тионил, ч., ВТУ МХП 3591—52.
Условия получения
В фарфоровый стакан емкостью 200 мл, снабженный ме-
ханической мешалкой, термометром и капельной воронкой,
помещают 100 мл днметилформамида и при размешивании
присыпают 10 г (0,035 Л4) натриевой соли 2-антраценсульфо-
кислоты. Продолжая перемешивание, в течение 5—10 минут
по каплям прибавляют 11,2 мл (0,16 М) хлористого тиснила;
температура смеси при этом ие должна превышать 40°. По
окончании прибавления хлористого тионила реакционную
массу перемешивают еще 10 минут, осадок отфильтровыва-
ют, а фильтрат выливают в 400 мл ледяной воды при тща-
тельном перемешивании. Выпавший осадок желтого пвета от-
фильтровывают и промывают спиртом.
Выход 2-антраценсульфохлорида равен 8,42 г, что состав-
ляет 85% от теоретического; температура плавления веще-
ства после перекристаллизации последовательно из толуола
и хлороформа равна 142,5—143,0°; по литературным данным
122° [1].
Найдено, %; С 60,57; 60,36; Н-3,43; 3,10; С1—12,65; 12,47.
S—11,75; 11,96;
Ci4H9C102S. Вычислено, %: С-60,76; Н-3,27; С1—12,81; S-11,58;
ЛИТЕРАТУРА
1. W. Н е f f t е г. Вег., 28. 2258 (1895).
2. Н. Н. Bosshard, R. Могу, М. Schmid, Н. Zolinger
Helv. chim. acta, 42, 1653 (1959).
Поступила в апреле 1966 i. И PEA
УДК 547.753.07
1 -А Ц И Л-2,3-Д И Г И Д РО И Н Д О Л Ы
А. К. ШЕЙНКМАН, Л. П. МАХНО
1 -Ацетил-2,3-дигидроиндол
1-Бутирил-2,3-дигидроиндол
I
СОСНэ
C10HnNO М.в. 161,20
СОСН2СНаСН3
Cj2Hi5NO М.в. 189,25
1 -Капронил-2,3-дигидроиндол
I
СО(СН3)4СН3
ChHI9NO м.в. 217,31
1 -Хлорацетил-2,3-дигидроиндол
/\________________
I !l I
СОСНгС1
C!0H10NOC1 м.в. 195,64
Различные 1-ацил-2,3-дигидроиндолы (1-ацилиндолины)
являются удобными исходными соединениями для синтеза
разнообразных производных индола, замещенных в бензоль-
ном ядре. Кроме того, l-ацилиндолины могут использовать-
ся как полупродукты в фармацевтической и анилино-красоч-
ной промышленностях.
В литературе описано получение только простейшего пред-
ставителя этого класса соединений 1-ацетилиндолина [1], ос-
тальные 1 -ацил-2,3-дигидроиндолы получены нами впервые.
Обычными методами получения различных ариламидов
является ацилирование аминов ангидридами и хлорангидри-
дами кислот в присутствии соды либо в среде ацилируемого
амина. Однако эти способы не совсем удобны при ацилиро-
2 Зак. 266
17
вании индолинов, так как при этом часто протекают побоч-
ные реакции, что приводит к загрязнению получаемых
1-ацилиндолинов. Для их синтеза мы разработали простой и
удобный способ ацилирования на холоду 1-ацилпиридиние-
выми солями, дающий с почти количественным выходом чи-
стые кристаллические 1-ацилиндолины. При этом нет необ-
ходимости выделять свободные 1-ацилпиридиниевые соли.
Достаточно, чтобы они образовывались в ходе реакции в ка-
честве промежуточных соединений. Интересно, что подобная
реакция 1-ацилпиридиниевых солей с 1-алкил 2,3-дигидроин-
долами приводит к получению 1-алкил-5-(пиридил-4)-2,3-ди-
гидроиндолов [2], а не к продуктам ацилирования.
СХЕМА СИНТЕЗА 1-АЦИЛ-2,3-ДИГИДРОИНДОЛОВ
где R: СН8; С3Н7; С9Нц; СН2С1
Характеристика основного сырья
Индолин, свежеперегнанный, т. кип. 94—9578 мм.
Ацетил хлористый, ч, ГОСТ 5829—51.
Бутирил хлористый, т. кип. 100—102°.
Капронил хлористый, т. кип. 152—153°.
Хлорангидрид хлоруксусной кислоты, свежеперегнанный.
Пиридин, ч., ГОСТ 1625—61, абсолютированный (см. при-
мечание).
Условия получения
Синтез 1-ацетил-2,3-дигидроиндола. В трехгорлую круг-
лодонную колбу с обратным холодильником, защищенным
хлоркальцевой трубкой, мешалкой и капельной воронкой, по-
мещают 40,8 г (0,34 А1) индолина, 26,86 г (0,4 М) пиридина
и при интенсивном помешивании и охлаждении в ледяной
бане постепенно добавляют 31,4 г (30,4 М) хлористого аце-
тила за 0,5—1 час. Выпавшие кристаллы промывают водой
и кристаллизуют из горячей воды. Выход 1-а цетил-2,3-ди-
гидроиидола в виде слегка розоватых кристаллов равен 49 г,
что составляет 89,5% от теоретического; т. пл. 102—103°. По
литературным данным, т. пл. вещества 105° [1]. При хромато-
графировании вещества в нефиксированном тонком слое оки-
си алюминия в системе растворителей бензол: гексан: хлоро-
форм (6:1:30) обнаруживается одно пятно Rf 0,41, свиде-
тельствующее о чистоте полученного соединения.
18
Получение 1-бутирил-2,3-дигидроиндола. 1 -Бутйр ил-2,3-ди-
гидроиндол получают по методике, аналогичной вышеописан-
ной. При взаимодействии 17,7 г (0,15 М) индолина с 16 г
(0,15 М) хлористого бутирила в присутствии 15 г (0,15 Л4)
пиридина получено 25,8 г 1-бутирил-2,3-дигидроиндола, что
составляет 91 % от теоретического выхода. По внешнему ви-
ду это белые иглы с т. пл. 85—86° (нз воды); Rf 0,60 (бен-
зол: гексан: хлороформ соответственно 6:1:30).
Найдено, %: С-76,56; Н—8,51; N—7,50.
C1SH16NO. Вычислено, %: С-76,19; Н-8,1; N-7,40.
Получение 1-капронил-2,3-дигидроиндола. Как описано
выше, при взаимодействии 9 г (0,075 М) индолина, 12 г
(0,075 М) хлористого капронила и 8 г (0,075 А1) пиридина в
растворе петролейного эфира получают 12,7 г 1-капронил-
2,3-дигидроиндола, что составляет 83,8% от теоретического
выхода. По внешнему виду это розоватые кристаллы с т. пл.
62—63° (из ацетона); Rf 0,70.
Найдено, И; С—77,12; Н—8,71; N—6,56.
C14H19NO. Вычислено, %: С- 77,42; Н —8,7; N—6,46.
Получение Пхлорацетил-З^-дигидроиндола. К раствору
5,8 г (0,05 А1) индолина в безводном петролейном эфире до-
бавляют 4 г (0,05 А1) абсолютного пиридина и при интенсив-
ном перемешивании и охлаждении до —5°----------10° ледяной
баней с солью постелено добавляют 5,6 г (0,05 М) хлораце-
тилхлорида. Выпавшие кристаллы отделяют, сушат и пере-
кристаллизовывают из ацетона.
Выход 1-хлорацетил-2,3-дигидроиндола равен 9,4 г, что со-
ставляет 96,7% от теоретического; т. пл. 133—134°; Rf 0,54
(бензол; гексан: хлороформ соответственно 6:1:30).
Найдено, %: С—61,76; 61,75; Н—5,55; 5,36: С1—18,34; 18,81;
N-7,13; 7,15.
Вычислено, %•. С—61,38; Н—5,11; С1 —18,15; N—7,15.
Примечание.
Пиридин тщательно высушивают перед употреблением плавленым
едким кали с последующим кипячением над окисью кальция и затем пе-
регоняют на колонке.
ЛИТЕРАТУРА
1. Словарь орг. соед., т. 1—3, М., ИЛ, 1949.
2. А. Н. Кост, А. К. Шейнкман. Методы получения хим. реак-
тивов н препаратов, вып. 11. М., ИРЕА, 1964, стр. 8.
Поступила в мае 196И г. Донецкий филиал ПРЕД
2*
УДК 547.722
5-БРОМ-2-НИТРОФУРАН
3. Н. НАЗАРОВА, Б. Н. НОВИКОВ
|l II
Вт-Il JI-NO,
ч0 7
C4HaNO8Br М.в.191,97
5-Бром-2-нитрофуран (БНФ) впервые получен [1] взаимо-
действием 5-бромпирослизевОй кислоты с азотной кислотой в
уксусном ангидриде. При наших многочисленных попытках
воспроизвести эту методику выход не превышал 5—7%. При-
менив увеличенное по сравнению с указанной методикой ко-
личество азотной кислоты, мы получили БНФ с выходом 47%
[2]. Почти одновременно появилось сообщение [3], авторы ко-
торого применили увеличенное количество азотной кислоты и
более длительное перемешивание, причем выход продукта
был повышен до 56%.
Мы значительно упростили предложенную ранее [2] мето-
дику, заменив двукратное прибавление азотной кислоты од-
нократным и сократив до 1 часа время прибавления 5-бром-
пирослизевой кислоты. Нами установлено, что низкая темпе-
ратура в конце реакции нежелательна; на последней стадии
реакционная масса выдерживалась при температуре около
0°. Способ выделения продукта не отличался от описанного
ранее [1—3].
СХЕМА СИНТЕЗА 5-БРОМ-2-НИТРОФУРАНА
HNO3 ,
Вт—» II—СООН (СНз)го _>
4 0х
20
Характеристика основного сырья
5-Бромпирослизевая кислота, 'получают бромированием
пирослизе'вой кислоты [4],
Уксусный ангидрид, ч.д.а., ГОСТ 5815—52.
Азотная кислота (<7= 1,5), получают перегонкой смеси
ч.д.а. нитрата калия, ГОСТ 4217—55 и концентрированной
серной кислоты (d=l,84) в соотношении 2:1.
Условия получения
В трехгорлую колбу на 1 л, снабженную мешалкой, ка-
пельной воронкой и термометром, помещают 250 мл уксусно-
го ангидрида, охлаждают до —10° (лед с солью) и по кап-
лям при перемешивании добавляют 250 г азотной кислоты
(J=l,5). Скорость прибавления регулируется таким обра-
зом, чтобы температура в колбе поддерживалась околс
—10°. Отдельно растворяют 60 г 5-бромпирослизевой кисло-
ты в 300 мл уксусного ангидрида при нагревании. Раствор
охлаждают до комнатной температуры и добавляют его из
капельной воронки при энергичном перемешивании в течение
1 часа к нитрующей смеси, поддерживая температуру реак-
ционной массы —10°. Перемешивание продолжают при той
же температуре еще 1 час, затем в течение 2 часов постепен-
но повышают температуру до 0° и перемешивают еще 1 час
при 0°. Раствор выливают на лед, добавляют в смесь NaCl
до насыщения и пять раз экстрагируют эфиром [2]. Эфирные
вытяжки объединяют и перегоняют с паром. После отгонки
эфира отгоняется с паром БНФ, сначала в виде масла, а за-
тем в виде кристаллов, затвердевающих в холодильнике. Пе-
регонку ведут до тех пор, пока дестиллат не станет проз-
рачным. Погон охлаждают льдом с солью, кристаллы БНФ
отфильтровывают и сушат в эксикаторе над хлористым каль-
цием.
Выход равен 28 г, что составляет 47% от теоретического;
т. пл. 48°. По литературным данным, т. пл. вещества 48° [1].
Примечания:
1. Не рекомендуется применять смесь серной и азотной кислот (2:1),
так как попадание в погон серной кислоты, перегоняющейся в виде азео-
тропа с азотной, снижает выход БНФ.
2. БНФ при попадании на кожу вызывает покраснение и боль, как
при ожоге. На следующий день на пораженном месте появляются вол-
дыри, заживающие через 5—7 дней. При длительной работе с БНФ не-
обходимо чаше протирать руки ватой, смоченной в спирте.
21
ЛИТЕРАТУРА
1. J. J. Rlnkes. Rec. trav. Chim. 50, 981 (1931); Zbl, 1932, 1965.
2. 3. H. Назарова, В. H. Новиков. Ж. общей химии, 31, 263
(1961).
3. В. R. Balaguer. An. Real. acad. Farmac., 27, 47 (1961); РЖхии,
5Ж 209 (1962).
4. 3. И. Назарова, И. Г. Г а х. Ж. орган, химии, 30, 2322 (1960).
Поступила в феврале 1956 г. Ростовский на Дову гос ун т
УДК [66.062.612.142 166.062.612.143:547.07
н-БУТИЛ- И ИЗОБУТИЛАЦЕТАТ
О. Н. КАРПОВ, Р. М. БЫСТРОВА
СН3СН2СНаСН,ОССН3 СН3СН-СН2ОССП3
П I !!
свн12о.
СН3
о
М.в. 116,162
н-Бутил- и изобутилацетат очень широко используются
в качестве растворителей и в ряде синтезов. Как известно,
синтез сложных эфиров проводится в присутствии минераль-
ных кислот в качестве катализаторов, используемых только
однократно, что приводит к большому расходу воды при про-
мывании.
При применении катионитов этот недостаток исключает-
ся; кроме того, в меньшей степени идут побочные процессы,
в частности, образование непредельных соединений [I].
СХЕМА СИНТЕЗА н-БУТИЛ- И ИЗОБУТИЛАЦЕТАТА
сн3соон+с4н9он сн3соос:4нв-! н2о.
Характеристика основного сырья
Бутиловый спирт, ч., ГОСТ 6006—51 или 5980—51.
Изобутиловый спирт, ч., ГОСТ 6016—51.
Уксусная кислота, ч., ГОСТ 61—51.
Катионит КУ-2, ГОСТ 5696—51.
Условия получения
Синтез веществ проводят в трехгорлой колбе с механиче-
ской мешалкой, термометром и обратным холодильником с
водоотделительной насадкой. Нагревание производят на гли-
23
Цериновой или масляной бане, температуру которой поддер-
живают контактным термометром.
В колбу загружают 17 г катионита в Н-форме (см. при-
мечание 1), 74,86 г (1,01 М) и- или изобутилового спирта,
60,06 г (1 Л1) уксусной кислоты и нагревают- По достиже-
нии 98° начинает выделяться вода (воду, собравшуюся во
флорентине, сливают с таким расчетом, чтобы к концу синте-
за она оставалась заполненной водой почти полностью). Пе-
ред завершением реакции температура повышается для н-бу-
тилацетата ~ до 115°, для изобутилацетата —110°. Реакцию
ведут до выделения примерно 98—99% воды (17,7—17,8 мл)
так как ,кроме реакции этерификации три накоплении эфира,
идет дегидратация сложного эфира с образованием непре-
дельных соединений и кислоты. Синтез веществ с н-бутйло-
вым спиртом заканчивается за 45—50 минут, а с изо-бутило-
вым спиртом за 60—62 минуты (см. примечание 2). Затем
реакционную смесь охлаждают, отделяют катализат от ка-
тионита декантацией, пропускают через колонку с анионитом
АВ—17 или АВ—16 в ОН-форме, либо нейтрализуют 18—
20%-ным раствором соды до слабощелочной реакции и про-
мывкой водой до нейтральной реакции. Далее катализат под-
вергают разгонке на ректификационной колонке с эффектив-
ностью 10 теоретических тарелок.
Первую фракцию н-бутилацетата собирают до 122° с вы-
ходом 8,9—9 г, она состоит примерно из 51—52% спирта и
48—49% бутилацетата. Вторую фракцию, представляющую
собой эфир, собирают при 122 — 1277745 мм. Его выход со-
ставляет 106,8—106,9 г (или 92% от теоретического по кис-
лоте) с содержанием бутилацетата не менее 99% и кислот-
ностью не более 0,05%.
Первую фракцию изобутилацетата собирают до 110° с
выходом 6,5—8 г, она состоит также примерно из 51—52%
спирта и 48—49% изобут.илацетата. Вторую фракцию, пред-
ставляющую собой эфир, собирают при 110—1157745 мм.
Его выход составляет 108—110,3 г (или 93—95% от теорети-
ческого по кислоте) с содержанием изобутилацетата не ме-
нее 99,0% и кислотностью не более 0,05%.
Первые фракции с учетом содержания спирта и эфира
снова используются в синтезе или повторной разгонке, по-
следнее менее желательно.
Примечания:
1. При расчете количество катионита лучше выражать не в про-
центах по отношению к спирту или кислоте, а в граммах на грамм-моль
гидроксильных или карбоксильных групп. Эти данные для сравнения бо-
лее реальны и не меняются в зависимости от колебания молекулярно-
го веса кислоты или спирта. В данном случае необходимо 17 г катиони-
та КУ-2 на 1 грамм-моль гидроксильных (17 г) или карбоксильных
(45 г) групп. Если синтез вести с избытком 0,25 М спирта, то достаточно
24
уже 10 г катионита и реакция закапчивается примерно в два раза быст-
рее [2]. Следует также отметить, что первые два-три синтеза протекают
несколько дольше.
2. Необходимо заметить, что скорость образования эфиров из спиртов
изо-строения заметно медленнее, то есть продолжительность реакции за-
висит от прямой и обратной скорости диффузии внутри эериа ионооб-
ценной смолы реагирующих веществ и образовавшихся продуктов. Это
наблюдалось также нами иа примере образования н-амил- и изо-амилаце-
тата [3], н-бутил- и изобутил-, н-амил- и изоамилсалицилатов [4].
ЛИТЕРАТУРА
1. Н. Г. Полянский. Успехи химии, 31, 1046 (1962).
2. О. Н. Карпов, Л. Г. Федосюк, Р. М. Быстрова. Тезисы
докл. Донецкого Гос. у-та. «К новым успехам советской науки». ДГУ, До-
нецк, 1966 г., стр. 239.
3. О. Н. Карпов, Р. М. Быстрова, Л. Г. Федосюк. Химиче-
ские реактивы и препараты, вып. 29, М„ ИРЕА, 1966 г., стр. 329.
4. О. Н. Карпов, Р. М. Быстрова. Ж. прикл. химии, № 6 (1967).
Поступила в апреле 1966 г. Донецкий филиал ИРЕА
УДК 547.288.07
mpem-БУТИЛКАРБАЗАТ
/«ре/и-Бутилоксикарбонилгидразин
Е. А. МАКАРОВА, Г. Н. КОШЕЛЕВА
СН3
СН3—С—О—СО —NH—NHa
I
сн3
C6HlaNaO2 М.в. 132,16
трет-Бутилкарбазат употребляется при азидном методе
синтеза пептидов.
Нами проверена, уточнена и значительно упрощена мето-
дика получения этого препарата, описанная в литературе [11-
СХЕМА СИНТЕЗА ире/и-БУТИЛКАРБАЗАТА
(СН3)3С—OH-|-Na -> (CHs).CONa-M/2 Н.
(CH3)3CONa4-COS -> (CH3)3COCO-SNa
(CH3)8CO-CO-SNa+CH3J -> (CH8),CO-CO-S-CH,+NaJ
(CH3)3CO-CO-S-CHs+HaN-NH3 ->
(CHa)sCO-CONH-NH2+CH3SH
Характеристика основного сырья
грет-Бутиловый спирт, ч., ВТУ ХСНХ 355—60.
Натрий металлический, ч., ТУ МХП 1664—50.
Метил иодистый, ч., ТУ МХП 49—51.
Гидразин-гидрат, ч., ГОСТ 5832—51.
26
Условия получения
Синтез трет-бутил-8-метилтиокарбоната. В круглодонную
колбу емкостью 3 л, снабженную мешалкой с ртутным затво-
ром, обратным холодильником с хлор.кальциевой трубкой,
термометром и трубкой для ввода газа, помещают 500 мл
(5,5 М) трет-бутилового спирта, 1000 мл сухого бензола и
35 г (1,5 М) мелко нарезанного металлического натрия и
оставляют на ночь. За это время натрий растворяется полно-
стью. На следующий день добавляют еще 1 л бензола и через
реакционную смесь пропускают при энергичном перемешива-
нии быстрый ток сероокиси углерода (см. примечание). Тем-
пература реакционной смеси самопроизвольно поднимается
до 38—40°. Сероокись углерода продолжают пропускать око-
ло двух часов до тех пор, пока температура не начнет падать
и не понизится до 34°.
После этого заменяют газовводную трубку на капельную
воронку, быстро, в течение 15—20 минут, прибавляют ПО мл
(250 е>, 1,7 М) йодистого метила и перемешивают 8 часов
при комнатной температуре.
К полученной массе приливают 600 мл воды, верхний слой
отделяют, а нижний экстрагируют бензолом три раза по
200 мл. Объединенный органический слой высушивают пота-
шом. Бензол и избыток трет-бутилового спирта отгоняют с
дефлегматором при атмосферном давлении до температуры
85°, а остаток перегоняют в вакууме. Собирают фракцию
60—64720 мм.
Выход трет-бутил-Б-метилтиокарбоната в виде желтова-
той жидкости составляет 150 г (67% от теоретического).
Получение трет-бутилкарбазата. В круглодонную колбу
емкостью 2 л, снабженную мешалкой с ртутным затвором,
обратным холодильником, верхний конец которого соединен с
промывной склянкой, наполненной 20%-ным раствором едко-
го натра, помещают 700 г (4,7 М) трет-бутил-5-метилтиокар-
боната и 260 г (5,2 М) гидразин-гидрата. Нагревают при пе-
ремешивании в течение 7 часов при температуре бани внача-
ле 80—90°, а затем—105—110°. Выделение меркаптана вна-
чале бурное к концу этого времени прекращается. Затем ре-
акционную массу охлаждают, разбавляют 800 мл хлористо-
го метилена, отделяют верхний водный слой, а органический
слой высушивают сернокислым магнием и растворитель от-
гоняют в вакууме при температуре бани не выше 50°.
Выход белого кристаллического трет-бутилкарбазата ра-
вен 500—510 г, что составляет 91—93% от теоретического;
т. пл. 39—40°.
По литературным данным, после перекристаллизации из
петролейного эфира т. пл. продукта 41—42°.
Примечание.
Сероокись углерода получают, добавляя при перемешивании к рас-
твору 3 кг серной кислоты в 1 л воды раствор 230 г роданистого ам-
мония в 230 мл воды. Полученный газ очищают, пропуская через колон-
ки с серной кислотой и хлористым кальцием.
ЛИТЕРАТУРА
1. L. Carpin о. J. Amer. Chem. Soc.. 82, 2725 (1960).
Поступила в мае 1966 г.
Институт химии
природных соединений АН СССР
УДК 547.852.2.07
6-ГЕПТИЛПИРИДАЗИНОН-З
А. А. ПОНОМАРЕВ, В. А. СЕДАВКИНА
СпНмН,0
М. в. 196,29
Из известных в настоящее время методов синтеза 6-ал-
килпиридазинонов-3 наилучшие результаты дает взаимодей-
ствие у-кетокарбоновых кислот с гидразин-гидратом [1—8].
Нами разработан способ синтеза этиловых эфиров у-ке-
токарбоновых кислот из фурфурола через фурилалкилкарби-
нолы [9, 10]; при кипячении эфиров с гидразин-гидратом 6-ал-
килпиридазиноны-3 мы получили с количественными выхода-
ми.
СХЕМА СИНТЕЗА 6-ГЕПТИЛПИРИДАЗИНОНА-З
С,Н1Г1—С—СН2—CHS—СООСгН6 HaN--Ha_ I II
II * N
о O^N7
H
Характеристика основного сырья
Этиловый эфир у-кетоноундекановой кислоты, т. кип. 138—
13975 мм; По—1,4392; d420—0,9419 [10].
Гидразин-гидрат, ч., ГОСТ 5832—51.
29
Условия получения
Растворяют 8 г этилового эфира у-кетоиоундекановой кис-
лоты и 1,6 г гидразин-гидрата в 30 мл этилового спирта и
нагревают на водяной бане с обратным холодильником в те-
чение двух часов. Спирт отгоняют при пониженном давле-
нии, а остаток перекристаллизовывают из петролейного эфи-
ра.
Выход 6-гептилпиридазинона-З количественный. По внеш-
нему виду вещество представляет собой бесцветные кристал-
лы с т. пл. 56—57° (см. примечания 1,2).
П р и м е ч а н и я:
!. Аналогичным путем с количественными выходами получают:
6-амилпиридазинон-З из 8 г этилового эфира у-кетонононановой кисло-
ты и 2 г гидразин-гидрата с т. пл. 37—38°.
6-гексилпиридазинон-З из 5 г этилового эфира у-кетоиодекановой кис-
лоты и 1,2 г гидраэии-гидрата с т. пл. 55—56“ и т. кип. 140—143°/1,5 мм;
6-изогексилпиридазинон-З из 5 а этилового эфира у-кетопоиэодекаиовой
кислоты с т. пл. 54—55° и т. кип. 158—162°/4,5 мм;
6-октилпиридазинон-З из 13 г этилового эфира у-кетонодеКаиовок
кислоты и 3 г гидразин-гидрата, т. пл. 62—63°;
6-нонилпиридазинон-З из 7 г этилового эфира у-кетонотридекановой
кислоты и 1,6 а гидразин-гидрата с т. пл. 54—55";
6-децилпиридазинон-З из 8 г этилового эфира у-кетонотетрадекановой
кислоты и 2,4 г гидразин-гидрата; т. пл. 52—53,
6-традецилпиридазинон-З из 5 г этилового эфира у-кетоногептадекапо-
вой кислоты и 1 г гидразин-гидрата с т. пл. 61—63°.
2. Свободные у-кетонокарбоновые кислоты образуют 6-алкилпирндази-
ноны-3 с выходами до 96% в тех же условиях.
ЛИТЕРАТУРА
1. Е. Fischer, Ann., 233, 147 (1886).
2. W. Ove re nd, L Wiggins. J. Chem. Soc., 3500 (1950).
3. H. V. Pe ch maun. Ber.’ 33. 3223 (19 0).
4. Th. Curtius. J. Pract. chem. (2) 50, 508 (1894).
5. S. Gabriel, Colman. Ber., 32, 395 (1899).
6. Th. Curtius, Ber., 29, 778 (1896).
7. В. В. Ф e о ф и л а к т о в, H. К. Семенов. Ж. общ. химии 23,
849 (1953).
8. Н. Japp. Y. К 1 i п g е in a n n. J. Chem. Soc., 57, 662 (1890).
9. А. А. Пономарев, В. А. Седавкина. Ж. общ. химии, 32,
2540 (1962).
10. А. А. Пономарев, В. А. Седавкнна. Ж. общ. химии, 32,
984 (1961).
Поступила в январе 1966 г.
Саратовский гос.уии
УДК 547.455.623'233.1.07
Д-ГЛЮКОЗАМИН ХЛОРГИДРАТ
Е. П. КРЫСИН, Р. Е. ТАРАСОВА
н н онн п
1 I I I //
но-сн2-с-с-с-с-с--н
пн
ОН ОНН NH3C1
CeHuO6NCl М.В. 215,63
Получение Д-солянокислого глюкозамина кислотным гид-
ролизом хитина, выделенного из раковин омаров или панци-
рей крабов, описано в литературе [1—4].
Нами получена промышленная партия хроматографически
чистого хлоргидрата Д-глюкозамина из кормовой крабовой
муки, являющейся отходом рыбной промышленности.
В основу разработанной технологии положен метод полу-
чения Д-глюкозамина хлоргидрата из панцирей крабов [3]
через стадию выделения хитина {4}.
СХЕМА СИНТЕЗА Д-ГЛЮКОЗАМИН ХЛОРГИДРАТА
Н Н ОНН
НС1 I I I I л
Хитин —НО-СН2-С С-С-С-СПщ-СН3СООН
I I I I
ОН ОНН NH3C1
Условия получения
Выделение хитина из крабовой муки. В трехлитровую
круглодонную колбу, снабженную мешалкой, помещают 500г
кормовой крабовой мукн и 1 л воды. Массу кипятят 2 часа
при перемешивании, водный раствор сливают, а к осадку ос-
торожно, во избежание сильиого вспучивания массы от вы-
31
Являющегося углекислого газа, приливают 1 л воды н 150 мл
концентрированной соляной кислоты. Массу перемешивают
при комнатной температуре в течение 2 часов и оставляют на
ночь. Затем фильтруют, раствор сливают, а осадок промыва-
ют водой до нейтральной реакции промывных вод. Отмытый
от кислоты осадок снова загружают в колбу, прибавляют
раствор 50 г едкого натра в 1 л воды и кипятят при разме-
шивании 2 часа.
Окрашенный в темный цвет щелочной раствор, в который
переходят белковые вещества паицырей, отфильтровывают,
осадок промывают водой до нейтральной реакции и вновь
заливают разбавленной соляной кислотой (1 л воды+150 мл
концентрированной соляной кислоты), перемешивают 1 час
при комнатной температуре, отфильтровывают на вороике
Бюхнера, отмывают водой от кислоты и снова обрабатыва-
ют щелочью, как указано выше. Такую обработку осадка
кислотой и щелочью проводят трижды (см. примечание 1).
Полученную легкую массу хитина кремово-сероватого
цвета отфильтровывают, промывают водой, хорошо отжима-
ют и сушат при 100°.
Выход хитина равен 140 г.
Кислотный гидролиз хитина. Хитин загружают в кругло-
донную колбу, снабженную мешалкой и обратным холодиль-
ником, приливают 1,4 л концентрированной соляной кислоты
и смесь нагревают на кипящей водяной бане при перемеши-
вании в течение 2,5 часа. Почти весь осадок при этом раство-
ряется и масса приобретает темный цвет. После кипячения
в колбу приливают 1,4 л воды, добавляют 50 г активирован-
ного угля, перемешивают 15—20 минут прн температуре 60°
и фильтруют от угля на воронке Бюхнера (см. примечание 2).
Полученный осветленный раствор хлоргидрата Д-глюко-
замина заливают в круглодоиную колбу, снабженную насад-
кой Вюрца и прямым холодильником, и помещают ее в во-
дяную баню. Раствор упаривают в вакууме при остаточном
давлении 10—20 мм и температуре водяной бани 60—70° до
образования густой кашицы.
К упаренному осадку хлоргидрата Д-глюкозамина при-
ливают 80 мл этанола, смесь хорошо размешивают, помеща-
ют в холодильник и оставляют кристаллизоваться на ночь.
Образовавшийся осадок отфильтровывают на воронке Бюх-
нера, промывают 20 мл этанола, хорошо отжимают и сушат
в вакуум-эксикаторе над хлористым кальцием.
Выход хлоргидрата Д-глюкозамина равен 85—90 г, что
составляет 61—64% тот веса хнтниа и 17—18% от веса взя-
той крабовой муки; («)д =+84 (с= 1, вода) (см. примеча-
ние 3).
По литературным данным, (®о= + 100)—> + 72,5° (с=1,
вода).
32
Примечания:
1. После третьей обработки щелочной раствор приобретает светлую
окраску и необходимость в дальнейшей обработке осадка щелочью отпа-
дает.
2. Если фильтрат имеет темный цвет, то обесцвечивание повторяют
до тех пор, пока цвет раствора не станет светло-желтым.
3. При нанесении на электрофореграмму раствора 50 мкг хлоргидра-
та Д-глюкозамина в 30%-ной уксусной кислоте и проявлении нингидри-
ном или раствором азотнокислого серебра примесей у вещества не обна-
ружено.
ЛИТЕРАТУРА
1. G. Ledderhose. Вег., 9, 1200 (1876).
2. М. L. Wolfrom, М. 1. Cron. J. Amer, Chem. Soc., 74, 1715
(1952).
3. M. Stacey, I. M. Webber. Metbads in Carbohydrate Chemi*
stry, 1. 228 (1962).
4. Г. В. Лазурьевский, И. В. Терентьева, А. А. Ш а и ш у-
р и н. Практические работы по химии природных соединений, М„ изд.
«Высшая школа», 1961, стр. 61.
Поступила в июле 1966 г, ЦЗЛ
3
Зак. 266
УДК 547.583.2'021'139.07
ДИАММОНИЙНАЯ СОЛЬ о-СУЛЬФОБЕНЗОЙНОй
КИСЛОТЫ
Е. Я. ЯРОВЕНКО, М. Ф. КОНДРАШОВА
C,H12N2O6S
—COONHt
-SOjNH4
М. в. 236,24
Днаммоннйная соль о-сульфобензойной кислоты может
быть применена для получения сульфофталеиновых индика-
торов.
По литературным данным, ее получают нейтрализацией
моноаммоннйной соли о-сульфобензойной кислоты водным
раствором аммиака [1].
Нами эта соль получена окислением дитиосалициловой
кислоты (дитиосалициловая кислота является промежуточ-
ным продуктом при получении о-сульфобензойной кислоты)
перекисью водорода <в присутствии молибденовокислого ам-
мония в качестве катализатора.
СХЕМА СИНТЕЗА ДИАММОНИИНОИ СОЛИ
о-СУЛЬФОБЕНЗОИНОИ КИСЛОТЫ
соон соон
I I
I I I I + 5Н2О2 4- 4NH1OH -*
f^-COONH4
-* 2 +8Н,0
^J-SOaNH,
34
Характеристика основного сырья
Аммиак водный, ч., ГОСТ 9—57.
Перекись водорода, медицин., ГОСТ 177—55.
Дитносалицнловая кислота, ч., т. пл. 290—292° [2].
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную мешал-
кой, термометром и капельной воронкой, загружают 74 мл
(0,65 jW) 30%-ной перекиси водорода н 0,8 г молибденово-
кислого аммония.
Реакционную смесь нагревают до 45—50° н при этой тем-
пературе в течение часа прибавляют 40 г (0,13 А4) дитиоса-
лнцнловой кислоты, растворенной в 37 мл водного аммиака.
При окислении необходимо поддерживать pH 6—6,5 (по уни-
версальной индикаторной бумажке). После ввода в реакцию
всей дитносалициловой кислоты реакционную смеСь выдер-
живают прн 50° до полного исчезновения перекиси водорода
(по йод-крахмальной бумажке). Затем температуру подни-
мают до 60—70°, прибавляют еще 28 мл (0,25 Л4) перекиси
водорода и нагревают при 70° до полного ее исчезновения.
После этого в реакционную массу добавляют 0,2 г активиро-
ванного угля и снова нагревают при 70° в течение 30 минут.
Затем уголь отфильтровывают, фильтрат упаривают до ’/3
первоначального объема, охлаждают, выпавший продукт от-
фильтровывают н сушат при 70—80° до постоянного веса.
Получают 51 г днаммонийной соли о-сульфобензойной ки-
слоты, что составляет 82,5% от теоретического выхода, счи-
тая на дитиосалицнловую кислоту; т. пл. 246—246,5°.
Найдено, %: N—11,77; 11,71.
C7H1sN2O5S. Вычислено, %: N—11,85.
ЛИТЕРАТУРА
1. Re. Don me. J. Amer. Chem. Soc., 11, 74, 337 (1889).
2. A. J о n e s. J. Amer., Chem. Soc., 16, 366 (1894).
Поступила s ноябре J966 г. ИРЕА
3*
УДК 547.755.07
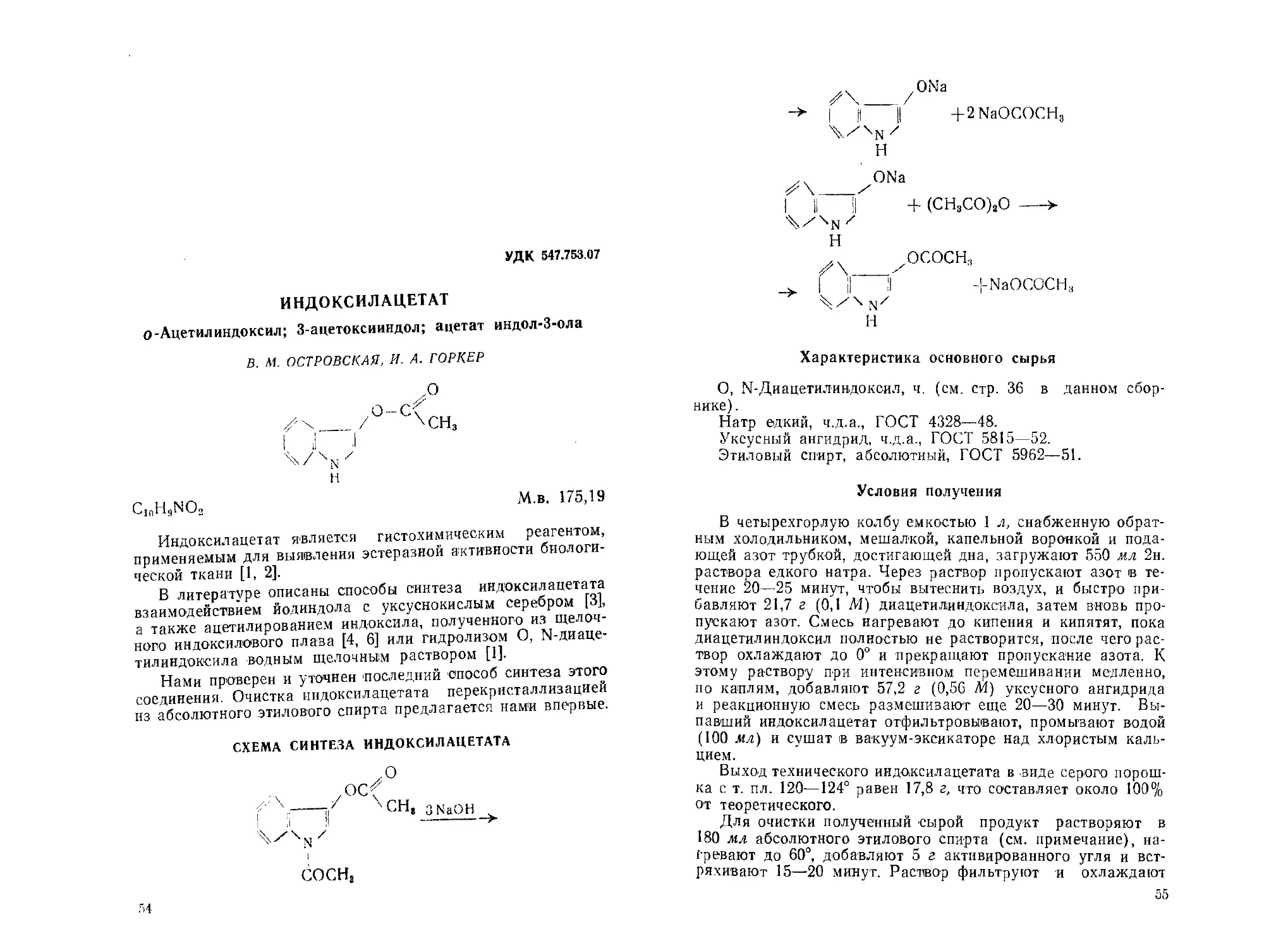
О,N-ДИАЦЕТИЛ ИНДОКСИЛ
1-Ацетил-З-ацетоксииндол; ацетат 1-ацетил-индол-З-ола
В. М. ОСТРОВСКАЯ, И. А. ГОРКЕР
ососн.
сосн3
C12HltNO3
М. в. 217,22
О, N-Диацетилиндоксил применяют для -синтеза иадокси-
лацетата, индоксилпропионата, индоксилбутирата [1], 1-аце-
тилиндоксила [2].
Диацетилиндоксил получают при действии уксусного ан-
гидрида на индоксиловую кислоту [3]; на N-ацетилиндоксил
или о-ацетилиндоксил [4]; на соли фенилглицин-о-карбоновой
кислоты [5]; на фенилглицин-о-карбоновую кислоту в присут-
ствии пиколина [6, 7]; на фенилглицин-о-карбоновую кислоту
в присутствии безводного уксуснокислого натрия [8]; на N-
ацетилфенилглицин-о-карбоновую кислоту в присутствии све-
жеплавленного уксуснокислого натрия [2]; на монокалиевую
соль фенилглицин-о-карбоновой кислоты в присутствии уксус-
нокислого калия (1].
Нами проверен последний способ получения диацетилин-
доксила.
СХЕМА СИНТЕЗА О,N-ДИАЦЕТИЛИНДОКСИЛА
,, ,соон
I II
+ С1СН2СООН + 2КОН
36
^соок ^.^соон
1'1 II + С1СН2СООК -* I II + КС1
Ч/XNH, 4/xNHCH2COOK
А/С00Н
| II + 3(CHSCO)3O -
'Ч / \ NHCHjCOOK
ососн3
+ СО, + CHsCOOK 4- зсн,срон
I .о
z\
\сн
3
Характеристика основного сырья
Антраниловая кислота, ч., ГОСТ 1718—48.
Монохлоруксусная кислота, ч., ГОСТ 5836—51.
Кали едкое, ч.д.а., ГОСТ 4203—48.
Калий уксуснокислый, ч.д.а., ГОСТ 5820—51.
Уксусный ангидрид, ч.д.а., ГОСТ 5815—52, свежеперегнан-
ный.
Этиловый спирт, гидролизный, ГОСТ 8314—57.
Эфир диэтиловый, ч., ГУФП 25/IV—41 г.
Активированный уголь, технический, ГОСТ 6213—53.
Условия получения
Синтез монокалиевой соли фенилглицин-о-карбоновой
кислоты.
Суспензию НО г (0,8 AI) антраниловой кислоты в 160 мл
воды и раствор 78 г (0,82 М) монохлоруксусной кислоты в
160 мл воды раздельно обрабатывают 30%-ным раствором
едкого кали (по 160 мл) до тех пор, пока не будут получены
прозрачные щелочные по фенолфталеину растворы. Затем
их охлаждают до 5°, смешивают, обрабатывают 8 г акти-
вированного угля и фильтруют на воронке Бюхнера через бу-
мажный фильтр. Фильтрат выдерживают 48 часов при тем-
пературе 50°. Образующуюся суспензию охлаждают до ком-
натной температуры, осадок отфильтровывают на воронке
Бюхнера, промывают водой (ЗХ§0 мл) и высушивают в су-
шильном шкафу при 70°.
Выход монокалиевой соли фенилглиции-о-карбоновой кис-
лоты равен 182 г, что составляет 97% от теоретического; т.
пл. (разл.) 257—263° (в запаянном капилляре).
37
Синтез О, N-диацетилиндоксила. Смесь уксуснокислого ка-
лия 150 г (1,41 М) и 910 г (8,66 М) уксусного ангидрида ки-
пятят в трехгорлой колбе емкостью 4 л, снабженной обрат-
ным холодильником и мешалкой с глицериновым затвором.
Затем быстро и осторожно добавляют 150 г (0,64 М) мелко-
растертой монокалиевой соли фенилглицин-о-карбоновой
кислоты. Полученный раствор кипятят 15—20 минут, охлаж-
дают до 10°, добавляют 900 мл охлажденной до 0° дистилли-
рованной воды и, продолжая охлаждение, размешивают в те-
чение 30 минут- Образовавшуюся суспензию диацетилиндок-
сила отфильтровывают, промывают 50%-ной уксусной кисло-
той (200 мл>), 50%-ным этиловым спиртом (дважды по
150 мл) н сушат на воздухе. Выход технического диацета.тан-
доксила в виде желтых кристаллов с т. пл. 77—78° равен
100 г, что составляет 72% от теоретического. Его перекристал-
лизовывают из 1100 мл кипящего этилового спирта с 10 г ак-
тивированного угля. Фильтрат охлаждают до —50°, выпавший
осадок отфильтровывают м сушат на воздухе. Продукт (84 г,
т. пл. 78,5—79,5°) помещают в коническую колбу емкостью
1,5 л с обратным холодильником, нагревают с 800 мл диэти-
лового эфира и 8 а активированного угля при осторожном
встряхивании на водяной бане, предварительно нагретой до
50°. Затем фильтруют, фильтрат охлаждают до —50°, выпав-
ший осадок отфильтровывают и сушат иа воздухе.
Выход чистого О,М-диацетилиндоксила равен 70 г, что со-
ставляет 50% от теоретического; т. пл. 80—82°. По литератур-
ным данным, т. пл. вещества 80—82° [1]; 83—84° (из этано-
ла) [1J; 81—82° (из воды) [3]; 82° (из этанола) [4. 5]; 83° [9].
ЛИТЕРАТУРА
1. S. J. Holt, in .General, cytochemical methods', v. 1, Acad. Press
ing., New York and London, 1958, p. 375-398.
2. K. D. Nenitzescu, D. Ratleanu. Ber., 91, 6, 1141 (1958).
3. Герм. пат. 133146 (1902); Fdl., 6, 554.
4. D. Vor lander, B. Drescher. Ber.. 34, 1858 (1901).
5. Герм. пат. 113240 (1900)'; Fdl , 5, 940 (1901).
6. A. Lawson, D. H. Mlles. J. Chem. Soc., 1959, 2865.
7. A. Lawson, D. H. Miles. J. Chem. Soc, 1960, 1945.
8. S. J. Holt, A. E. Kellie, D. O. O’S n 11 i v a n, P. W. Sad-
ler. J. Chem. Soc., 1958, 1217.
9. S. J. Holt, P. W. Sadler. Proc. Roy. Soc., 1488, 933, 481
(1958).
Поступила в мае 1966 г.
И FEA
УДК 547.754.07
2,3-ДИГИДРОИНДОЛ
Индолин
А. Н. КОСТ, Л. П. МАХНО, А. К. ШЕИНКМАН, Г. А. МАРКУС
//\_____.
C8H9N
М. в. 119,16
Восстановление различных производных индола в индо-
лины цинком в соляной кислоте известно очень давно, одна-
ко с самим индолом реакция обычно идет с очень низкими
выходами вследствие значительного смолообразования [1—3].
Венцинг улучшил процесс восстановления, проведя реакцию
в спиртовой среде [4], однако и этот способ давал хорошие
результаты только с 2-метилиндолом [4, 5, 6]. Индол хорошо
восстанавливается водородом с никелем-Ренея при 100° и
под давлением 100 атмосфер [7].
Мы проверили и несколько улучшили способ восстанов-
ления индола цинком с соляной кислотой в среде бутанола,
предложенный лабораторией органического синтеза МГУ.
Способ оказался очень удобным для получения сравнитель-
но чистого индолина и с успехом был воспроизведен в про-
изводственных условиях.
СХЕМА СИНТЕЗА 2,3-ДИГИДРОИНДОЛА
S\_______
| ||___|| + Zn + 2С1 - | || I Н-Zi
I ZnCle
Ч /4-Nx’
н
39
Характеристика основного сырья
Индол, технический, температура плавления 46—49°.
Бутиловый спирт, ч., ГОСТ 6006—51.
Цинковая !пыль, ТУ МХП 112—40.
Соляная кислота, ч., концентрированная, ГОСТ 3118—46.
Условия получения
В двухгорлую колбу, снабженную обратным холодильни-
ком и капельной воронкой, загружают 100 г амальгамирован-
ной цинковой пыли (см. примечание 1), 150 мл разведенной
1:1 соляной кислоты и сразу начинают добавлять раствор
36 г (0,31 М) индола в 250 мл бутилового спирта.
Реакционную смесь кипятят 2—2,5 часа, добавляя посте-
пенно 200 мл концентрированной соляной кислоты и кипяче-
ние продолжают до растворения цинковой пыли.
Охлажденную реакционную смесь разделяют в делитель-
ной воронке. Бутанольный слой, содержащий индол, промы-
вают небольшими порциями 5°/о-ной соляной кислоты по 25—
35 мл до полного извлечения индолнна (см. примечание 2).
Кислотные вытяжки присоединяют к водному слою, со-
держащему индолин, несколько раз промывают эфиром и
подщелачивают до полного растворения осадка. Из получен-
ного раствора эфиром или бензолом экстрагируют индолин.
Экстракт сушат поташом, отгоняют растворитель и перего-
няют в вакууме.
Получают 20,7 г индолина, что составляет 57,5% от тео-
ретического выхода; температура кипения 94—95°/7 мм. По
внешнему виду это слегка желтоватая маслянистая жид-
кость, Rf 0,81; «о-1,5905 (см. примечание 3).
Опытами, проведенными с неамальгамированной цинковой
пылью, получен индолин с 42—27%-ным выходом от теоре-
тического.
Примечания:
1. Амальгамированный цинк получают путем перемешивания в тече-
ние 5—10 минут цинковой пыли в растворе 7 г сулемы в 150 мл воды, в
которую предварительно добавляют 5 мл концентрированной соляной кис-
лоты.
2. Контроль полноты извлечения осуществляют с помощью хромато-
графирования в нефиксированном топком слое окиси алюминия в систе-
ме растворителей бензол:гексан:хлороформ соответственно 6:1:30; Rf ин-
долнна 0,80; Rf индола 0,70; активность окиси алюминия 2 степени по
Брокману.
3. По аналогичной методике получены с небольшими выходами: 2-ме-
тилиндолин с температурой кипения 70—72°/8 мм н —1,5661; 2,3-ди-
метилнндолин с т. кип. 125—134720 мм и Пд—1,5612; 2-этил-З-пропилин-
долин с т. кип. 130—132716 мм и Лр—1,5670.
40
ЛИТЕРАТУРА
1. 0. Jackon. Вег., 14,883 (1881).
2. A. Pictet. Вег., 19, 1065 (1886).
3. Е. Fischer. LSbigs Ann. Chem., 236, 123 (1886).
4. M. Wenzing. L6bigs Ann. Chem., 239, 242 (1887).
5.1. V. Braun. Ber., 37, 4729 (1904).
6. I. V. Braun. Ber., 45, 1265 (1912).
7. F. King, I. Barltrop, R. Wally. J. Chem. Soc.. 1945 277.
Постудила в мае 1966 г- Московский университет
им. М. В. Ломоносова,
Донецкий филиал ИРЕА
Фенольный завод
УДК 547.415.1'118-32.07
М,\'-ДИКАРБОКСИМЕТИЛЭТИЛЕНДИАМИНО-
БИС-МЕТИЛФОСФИНОВАЯ КИСЛОТА
Р. П. ЛАСТОВСКИЙ, В. В. СИДОРЕНКО, А. В. ЛАПШИНА
О О
II И
(НО)2 Р —CH, —N -СНа-СН, -N -СН2- Р(ОН)2
I I
НООСН2С СН,СООН
C8H18OleN2P2 М. в. 363,18
N, Nz-Дикарбоксиметилэтилендиамино-бис-метилфосфиво-
вая кислота была синтезирована в ИНЭОС АН СССР [1, 2].
Метод основан на взаимодействии этилендиамино-бис-ме-
тилфосфиновой ’кислоты с цианистым натрием и формальде-
гидом в присутствии щелочи. Продолжительность реакции по
указанному методу составляют 40 часов. Выделение кисло-
ты проводится через свинцовую соль с последующим разло-
жением ее сероводородом.
Нами найден способ получения N, Х'-дикарбоксиметил-
этилен диамино-бис-метилфосфиновой кислоты путем взаимо-
действия этилендиамино-бис-метилфосфиновой кислоты с мо-
нохлоруксусной кислотой в присутствии щелочи. Продолжи-
тельность реакции 18 часов.
СХЕМА СИНТЕЗА
М,М'-ДИКАРБОКСИМЕТИЛЭТИЛЕНДИАМИНО-
БИС-МЕТИЛФОСФИНОВОЙ КИСЛОТЫ
(НО)аР—СН2—NH—СН2—СН2—NH—СН3—Р(ОН)2 +
+ 2С1СНаСООН 2NaOH
42
о о
I! II
(HO)sP-CH2-N-CHs-CH2-N- СН.2 Р(ОН)„
СН2СООН снасоон
Характеристика основного сырья
Этилендиамино-бис-метилфосфииовая кислота, т. пл. 276°,
получена по прописи [1].
Монохлоруксусная кислота, ч., ГОСТ 5836—51.
Натр едкий, ч.д.а., ГОСТ 4328—48.
Условия получения
К смеси из 6,4 г (0,026 М) этилендиамино-бис-метилфос-
финовой кислоты; 15 мл воды и 5 мл 42%-ного раствора ед-
кого натра добавляют по каплям при -перемешивании 5 г
(0,053 Л-1) монохлоруксусной кислоты, растворенной в 2,5 мл
воды, после этого снова периодически прибавляют раствор
едкого натра до pH реакционной смеси 10—И.
Реакционная масса перемешивается 18 часов при 30°, при-
чем процесс ведется при pH 10—11. Затем при 10—20° кре-
акционной массе прибавляют концентрированную соляную
кислоту до pH 1. Раствор упаривают в вакууме до неболь-
шого объема при температуре не выше 40°. Выпавший через
несколько часов осадок (10 г) технического продукта про-
мывают 100 мл 70%-ного спирта и перекристаллизовывают
из воды.
Полученное вещество промывают водой до отрицатель-
ной реакции на ион хлора. После высушивания вещества при
80° до постоянного веса получают 4 г N1, N'-дикарбоксиметил-
зтилендиамино-бис-метилфосфиновой кислоты, что составля
ет 42,5%, считая на исходную этилендиамино-бис-метилфос-
финовую кислоту; т. пл- 218°.
Найдено, >4: С—25,8; Н—5,0; Р—17, 0; N 7,8.
C8HJ8O1->NaP!1. Вычислено, %: С-23,3; Н-5,0; Р-17,0; N-7,7 [2].
ЛИТЕРАТУРА
1. М. И. К а б а ч н и к, Т. Я. Медведь, Г. К. Козлова, В. С. Ба-
ла бу ха, Е. Л. Миронова, Л. И. Тихонова. Изв. СССР, Отд.
хим наук, 651 (I960).
2. Р. П. Ластовский, М. И. Кабачник, В. В. Сидоренко,
Н. В. Лапшина. Авт. свид. 185911; Бюлл. Изобр. 18 (1966).
Поступила в декабре 1966 г.
ИРЕЛ
УДК 547.814.07
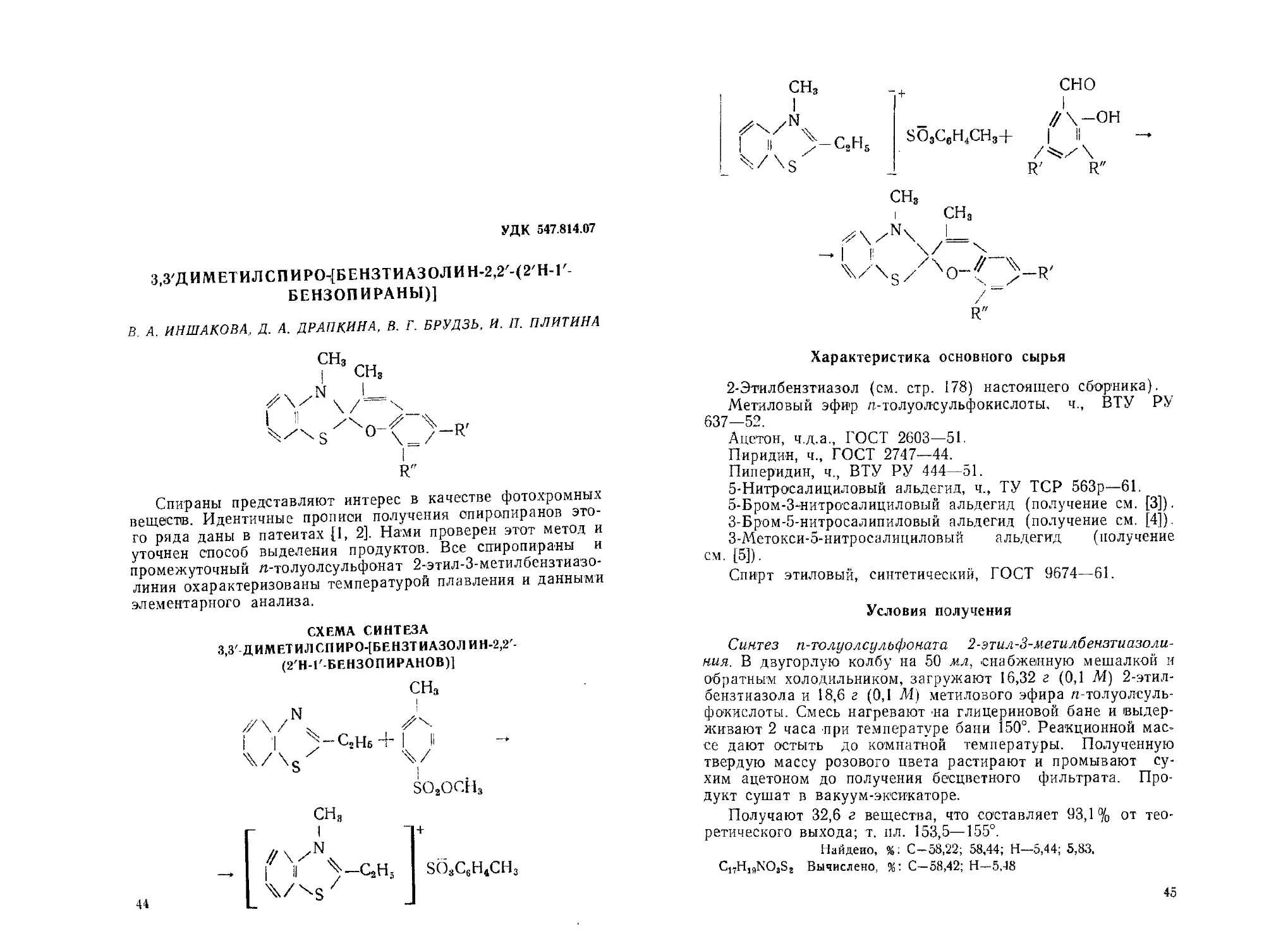
3,3'ДИМЕТИЛСПИР0т[БЕНЗТИА30Л HH-2,2'-(2'H-l'-
БЕНЗОПИРАНЫ)]
В. А. ИНШАКОВА, Д. 4. ДРАПКИНА, В. Г. БРУДЗЬ, И. П. ПЛИГИНА
Спираны представляют интерес в качестве фотохромных
веществ. Идентичные прописи получения спиропиранов это-
го ряда даны в патентах {I, 2]. Нами проверен этот метод и
уточнен способ выделения продуктов. Все спиропираны и
промежуточный п-толуолсульфонат 2-этил-З-метилбензтиазо-
линия охарактеризованы температурой плавления и данными
элементарного анализа.
СХЕМА СИНТЕЗА
З,3'-ДИМЕТИЛСПИРО-[БЕНЗТИАЗОЛ ИН-2,2'-
(2'Н-Г-БЕНЗОПИРАНОВ)]
сн3
Zv
—Сгц6 + | |
%/
SO2OCHs
сн3
//\/\
I II ^-СгН,
V\sz
SO4C6H4CH.
СНз
сно
Д-он
SO3C6H4CH3+ | II
R' R"
Характеристика основного сырья
2-Этилбензтиазол (см. стр. 178) настоящего сборника).
Метиловый эфир п-толуолсульфокислоты. ч., ВТУ РУ
637—52.
Ацетон, ч.д.а., ГОСТ 2603—51.
Пиридин, ч., ГОСТ 2747—44.
Пиперидин, ч., ВТУ РУ 444—51.
5-Нитросалициловый альдегид, ч., ТУ TCP 563р—61.
5-Бром-З-нитросалициловый альдегид (получение см. [3]).
З-Бром-5-нитросалипиловый альдегид (получение см. [4]).
З-Метокси-5-нитросалициловый альдегид (получение
см. [5]).
Спирт этиловый, синтетический, ГОСТ 9674—61.
Условия получения
Синтез п-толуолсульфоната 2-этил-З-метилбензтиазоли-
ния. В двугорлую колбу на 50 мл, снабженную мешалкой и
обратным холодильником, загружают 16,32 г (0,1 М) 2-этил-
бензтиазола и 18,6 г (0,1 М) метилового эфира n-толуолсуль-
фокислоты. Смесь нагревают на глицериновой бане и выдер-
живают 2 часа при температуре бани 150°. Реакционной мас-
се дают остыть до комнатной температуры. Полученную
твердую массу розового цвета растирают и промывают су-
хим ацетоном до получения бесцветного фильтрата. Про-
дукт сушат в вакуум-эксикаторе.
Получают 32,6 г вещества, что составляет 93,1% от тео-
ретического выхода; т. пл. 153,5—155°.
Найдено, %: С-58,22; 58,44; Н—5,44; 5,83,
C17HleNO,S, Вычислено, %: С—58,42; Н—5,-18
45
Получение 3,3 -диметилспиро-[бензтиазол-2,2'-(2'Н-1'-бен-
зопиранов)]. В двугорлую колбу на 50 мл, снабженную ме-
шалкой и обратным холодильником, загружают 25 мл пири-
дина (высушенного над щелочью и перегнанного) и при раз-
мешивании последовательно прибавляют 5,2 г (0,015 М)
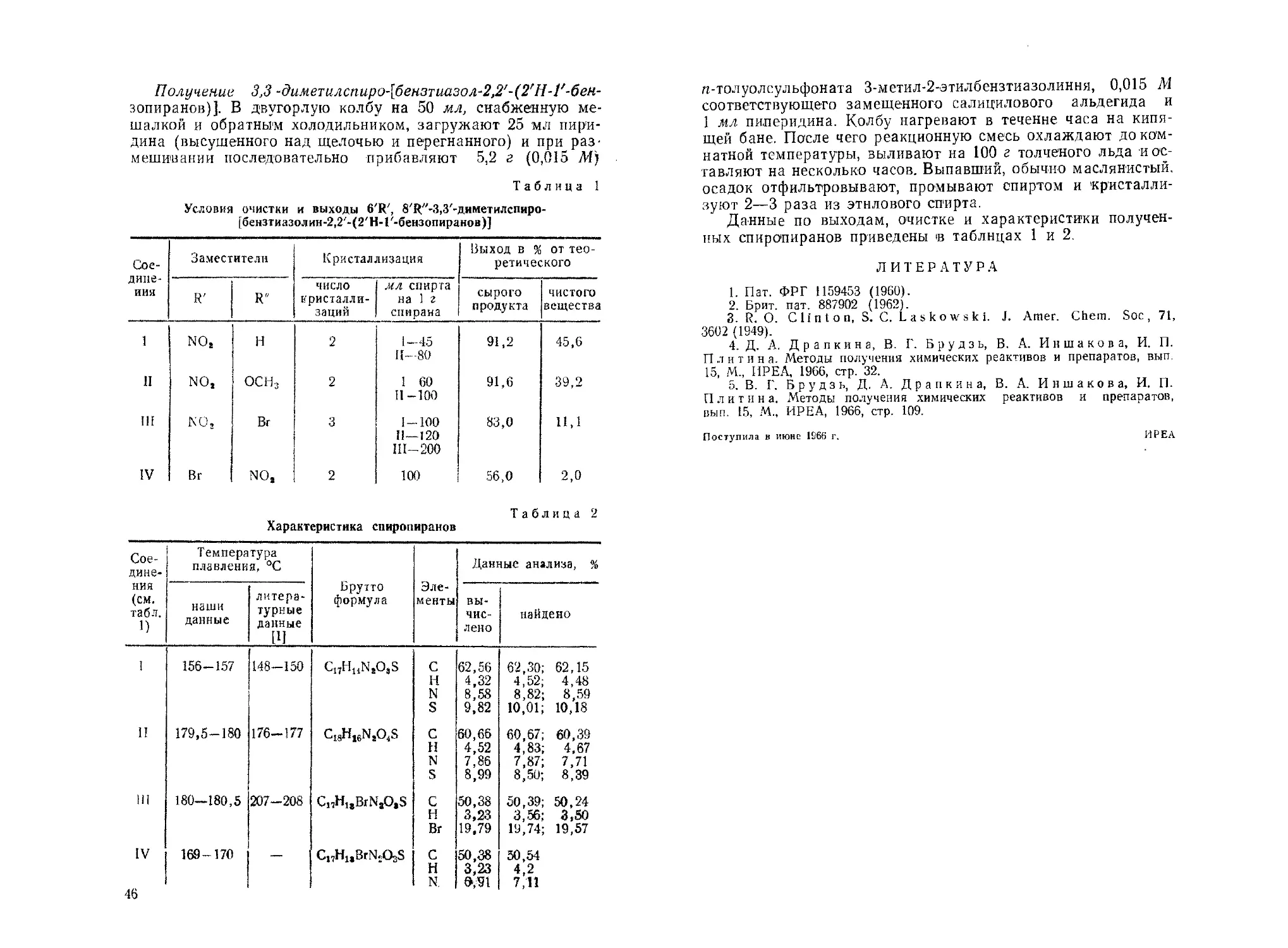
Таблица 1
Условия очистки и выходы 6'R', 8'Ц"-3,3'-диметилспиро-
[бензтиазолин-2,2'-(2'Н-Г-бензопиранов)]
Сое- дине- ния Заместители Кристаллизация Выход в % от тео- ретического
R' R" число кристалли- заций мл спирта на 1 г спирана сырого продукта ЧИСТОГО вещества
1 NO, Н 2 1-45 И—80 91,2 45,6
П NO, осн. 2 I 60 II-100 91,6 39,2
III КО; Вт 3 1-100 11—120 Ш-200 83,0 Н,1
IV Вт NO, 2 100 56,0 2,0
Характеристика спиропиранов
Таблица 2
Сое- дине- ния (см. табл. 1) Температура плавления, °C Брутто формула Эле- менты Данные анализа,
наши данные литера- турные данные [1] вы- чис- лено найдено
I 156-157 148-150 Ci,H14N,O,S с 62,56 62,30; 62,15
н 4,32 4,52; 4,48
N 8,58 8,82; 8,59
S 9,82 10,01; 10,18
II 179,5-180 176-177 cI8h16n,04s с 60,66 60,67; 60,39
н 4,52 4,83; 4,67
N 7.86 7,87; 7,71
S 8,99 8,50; 8,39
III 180—180,5 207-208 C„HuBxNAS с 50,38 50,39; 50,24
н 3,23 3,56; 3,50
Вт 19,79 19,74; 19,57
IV 169-170 — Cl7HltBrN.Q>S С 50,38 50,54
н 3,23 4,2
N. вда 7,11
46
н-толуолсульфоната З-метил-2-этилбензтиазолиння, 0,015 Л1
соответствующего замещенного салицилового альдегида и
1 мл пиперидина. Колбу нагревают в течение часа на кипя-
щей бане. После чего реакционную смесь охлаждают до ком-
натной температуры, выливают на 100 г толченого льда и ос-
тавляют на несколько часов. Выпавший, обычно маслянистый,
осадок отфильтровывают, промывают спиртом и кристалли-
зуют 2—3 раза из этилового спирта.
Данные по выходам, очистке и характеристики получен-
ных спиропиранов приведены в таблицах 1 и 2.
ЛИТЕРАТУРА
1. Пат. ФРГ Ц59453 (1960).
2. Брит. пат. 887902 (1962).
3. R. О. Clinton, S. С. Laskowski. J. Amer. Chem. Soc, 71,
3602 (1949).
4. Д. А. Драпкина, В. Г. Брудзь, В. А. И ишаков а, И. П.
П л и т и н а. Методы получения химических реактивов и препаратов, вып.
15, М., ПРЕД, 1966, стр. 32.
5. В. Г. Брудзь, Д. А. Драпкина, В. А. Иншакова, И. П.
П л и т и н а. Методы получения химических реактивов и препаратов,
вып. 15, М„ ИРЕА, 1966, стр. 109.
Поступила в июне 1966 г. ИРЕА
УДК 547.722.3.07
2,5-ДИМЕТОКСИ-2,5-
ДИГИДРОФУРФУРИЛАМИНОЭТАНОЛ
А. А. ПОНОМАРЕВ, И. А. МАРКУШИНА. И. С. МОНАХОВА
СН8О-\ /х
0 СН2—NH—CHa—CHgOH
C9H„NO4 м. в. 203,24
2,5-Диметокси-2,5-дигидрофурфуриламиноэтанол в,первые
описан нами в статье [1], где приведены данные о его синте-
зе и свойствах.
СХЕМА СИНТЕЗА
2.5-ДИМЕТОКСИ-2.5-ДИГИДРОФУРФУРИЛ АМИНОЭТАНОЛА
{ J—СН,- N—СН,—СН2ОСОСНа
О ' 1
СОСНз
сн„он_
(NHjBr)
СН3О-
j=—;хосн3
ЧО/Х CH2-N-CH2CH2OCOCH3
СОСНз
NaOH
--- /ОСНз
СН3О . /(
О 'CHo—NH— СН2-СН2ОН
Характеристика основного сырья
Ацетат N-ацетофурфуриламиноэтанола; т. кип. 152—
15372 мм, nDw —1,4918;. d^—1,1580 {1, 2}.
Метанол, ч„ ГОСТ 6995—54.
Аммоний бромистый, ч., ГОСТ 4454—48.
48
Условия получения
Растворяют 50 г (0,22 М) свежеприготовленного ацетата
N-ацегофурфурилами'НОЭтанола и 5 г (0,051 Л1) бромистого
аммония в 230 мл метилового спирта, помещают в электроли-
зер (см. примечание I) и охлаждают до —14° (см. примеча-
ние 2). Электролизер проводят в течение 8 часов при силе тока
3,5—2,3 а и напряжении 6—11 в. По окончании электролиза
слегка желтоватый раствор переносят из электролизера в
колбу и прибавляют к нему метилат натрия (см. примечание
3). На водяной бане при уменьшенном давлении отгоняют
метанол и аммиак. К кашеобразному остатку прибавляют
250 мл. 3 н. раствора едкого натра. Смесь переносят в дву-
горлую колбу, снабженную механической мешалкой и обрат-
ным шариковым холодильником, и кипятят на песчаной бане
20 часов. Затем содержимое колбы экстрагируют хлорофор-
мом (3 раза порциями но 50—60 мл) и сушат над прокален-
ным поташом. Хлороформ отгоняют на водяной бане при
уменьшенном давлении, остаток перегоняют в вакууме.
Собирают фракцию с т. кип. 120—122°/1,5 мм; nD' —-
1,4750; d420— 1,1340; т. пл. 2,4-динитрофенилгидразона 190—
191° (с разлож.).
Выход 2,5-дим етокси-2,5-дигидрофурфурил аминоэтанол а
равен 24,8 г, что составляет 55% теоретического из расчета
на ацетат N-ацетофурфур’ИЛамипоэтанола.
Примечания:
1. Описание электролизера см. в статье «2-Метокси-7-.мети.т-1,6-диок-
саспиро-(4,4)-нонен-3" данного сборника.
2. В качестве охлаждающей смеси используют бутиловый или изо-
бутиловый спирт, в который добавляют твердую углекислоту до дости-
жения необходимой температуры в элекролизере.
3. Метилат натрия, необходимый для связывания свободного брома,
образующегося в процессе электролиза, приготовляют из 1,2 г металли-
ческого натрия и 20 мл метанола.
ЛИТЕРАТУРА
1. А. А. Пономарев, И. А. Маркушина. Ж. общ. химии, 30,
976 (1960); Уч. зап. Саратовск. уи-та, 71, 135 (1959).
2. А. А. Пономарев, В. Плетенева, В. А. Седавкииа,
Л. Барская. Ж. общ. химии, 24, 718 (1954).
Поступила в январе 1966 г. Саратовский гос. ун.-т.
1 Зак. 266
УДК 547.556.9.07
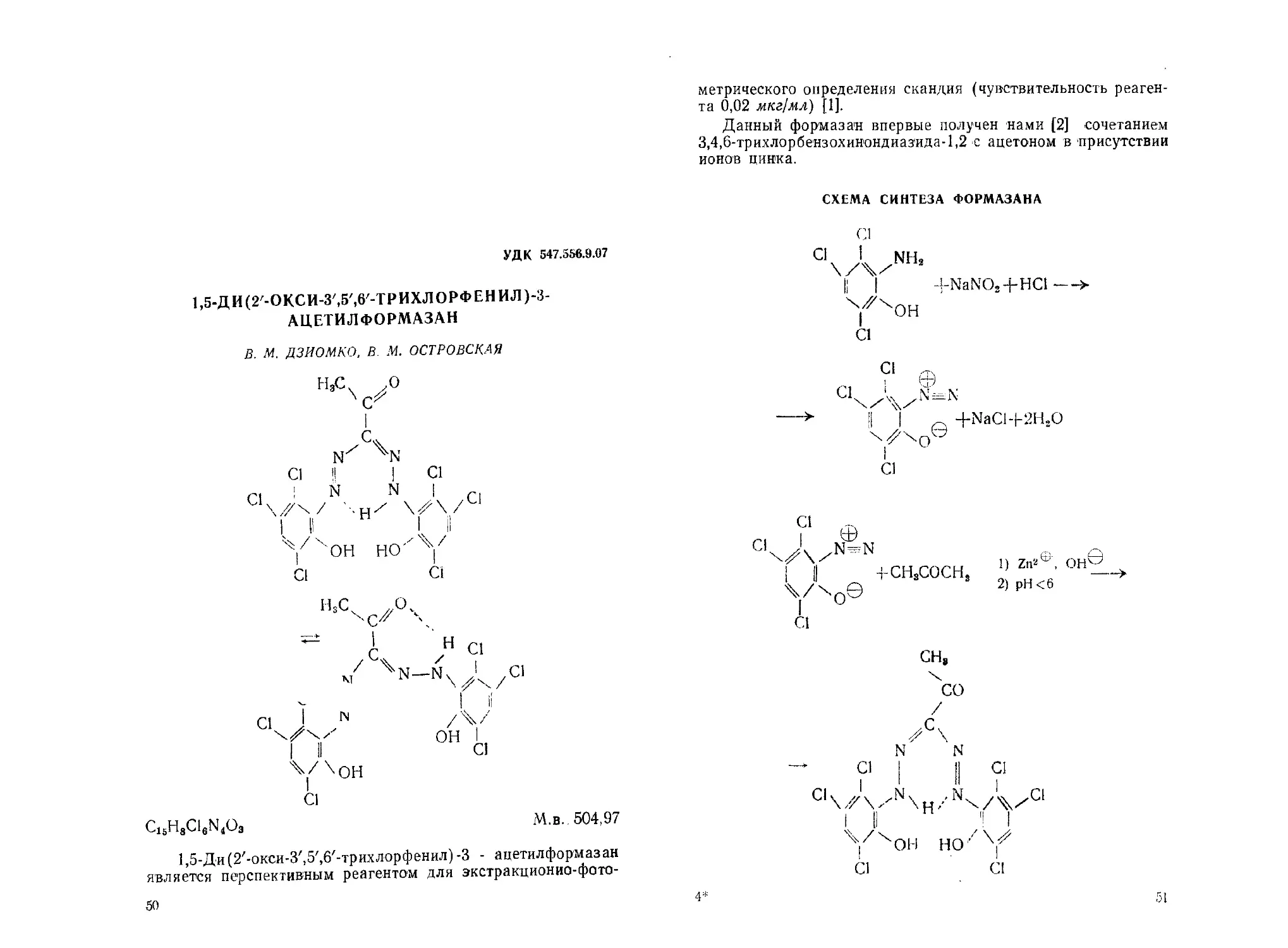
1,5^ДИ(2'-ОКСИ-3',5',6'-ТРИХЛОРФЕНИЛ)-3-
АЦЕТИЛФОРМАЗАН
В. М. ДЗИОМКО, В М. ОСТРОВСКАЯ
C16H8C16N4O3
М.в. 504,97
1,5-Ди(2''-окси-3/,5/,6,-трихлорфенил)-3 - ацетилформазан
является перспективным реагентом для экстракционио-фото-
50
метрического определения скандия (чувствительность реаген-
та 0,02 мкг/мл) [1].
Данный формазан впервые получен нами [2] сочетанием
3,4,6-трихлорбензохин'ондиаз'ида-1,2 -с ацетоном в присутствии
ионов цинка.
СХЕМА СИНТЕЗА ФОРМАЗАНА
-i-NaNO2+HCl--->
1) Zn2^, ОН©
2) pH<6
4*
51
Характеристика основного сырья
3,5,6-Трихлор-2-аминофенол, 87%-ный, т. пл. 122°
Соляная кислота, ч., ГОСТ 3118—46.
Натрий азотистокислый, ч., ГОСТ 4197—48.
Цинк сернокислый, х.ч., ГОСТ 417—48.
Кали едкое, ч.д.а., ГОСТ 4203—48.
Ацетон, ч.д.а., ГОСТ 2603—63.
Диметилформамид, ч., ВТУ РУ 1193—56 (дополнительно
перегнанный).
Условия получения
Синтез 3,4,6-трихлорбензохинондиазида-1,2. В трехгорлую
колбу емкостью 150 мл, снабженную термометром от —10° до
+ 50°, лопастной мешалкой, капельной воронкой, помещают
11,8 г (0,048 М) 3,4,6-трихлор-2-аминофенола и 50 мл воды.
Смесь размешивают, добавляют к ней 6,9 мл концентриро-
ванной соляной кислоты и охлаждают до —2Э. К полученной
суспензии по каплям, в течение 25 минут, прибавляют рас-
твор 3,5 г (0,05 М) азотистокислого натрия в 5 мл воды и
лают трехчасовую выдержку при температуре минус 5—0°.
Выпавший осадок отфильтровывают на воронке Бюхнера,
промывают на фильтре водой до нейтральной реакции, су-
шат на воздухе в течение 16 часов и оставляют до следую-
щего дня.
Получение и очистка формазана. Раствор 25 г ZnSOr/IbO
в 1250 мл воды смешивают в конической колбе с раствором
50 г едкого кали в 250 мл воды, охлаждают до 0°, перелива-
ют в фарфоровый стакан емкостью 4 л. При размешивании
этой смеси рамной мешалкой добавляют к ней свежеприго-
товленный раствор 3,4,6-трихлорбснзохинондиазида-1,2 в
50лл ацетона. Реакционную смесь размешивают 15 минут,
подкисляют до pH 1 добавлением 125 мл концентрированной
соляной кислоты, предварительно охлажденной до 5°. Через
5 минут размешивание прекращают, выпавшему осадку дают
отстояться около 20 минут, затем его отфильтровывают на
воронке Бюхнера, промывают на фильтре смесью воды
(50 мл) и ацетона (50 мл) и водой до нейтральной реакции.
Продукт сушат в сушильном шкафу при температуре не вы-
ше 70°. Выход технического формазана равен 7,4 г, что сос-
тавляет 61% от теоретического; т. пл. 176—178° (разл.).
Для очистки растворяют полученный продукт в 200 мл
диметилформамида при температуре бани 80° в конической
колбе емкостью 500 мл. Раствор фильтруют и оставляют в
холодильном шкафу на несколько часов (обычно на ночь).
Выпавшие кристаллы отфильтровывают на стеклянном пори-
стом фильтре № 4, промывают смесью 50 мл дистиллирован-
52
ной воды и 2,5 мл 5%-ной соляной кислоты, затем 50 мл дис-
тиллированной воды и сушат при 70°. Вес продукта равен
3,6 г (см. примечание). По внешнему виду вещество пред-
ставляет собой мелкокристаллический порошок коричневого
цвета с т. пл. 198—200° (разл.). Затем препарат перекри-
сталлизовывают из кипящего бензола (400 мл) и получают
3,3 г формазана, что составляет 27% от теоретического выхо-
да; т. пл. 202—203° (разл., см. примечание 2).
1,5-Ди(2/-окси-3/, 5', б'-трихлорфенил)-3-ацетилформазан
растворим в ацетоне (с кирпично-красной окраской), в вод-
ном ацетоне (с фиолетовой), в четыреххлористом углероде и
бензоле (с малиновой), в этилацетате и диоксане (с крас-
ной), хорошо растворим также в тетрагидрофуране (с корич-
невой окраской), в концентрированной серной кислоте (с си-
ней); в воде не растворим.
Примечания:
1. Дополнительное количество сырого продукта (около 2,7 г) можно
возвратить при добавлении к фильтру (после перекристаллизации из ди-
метилформамида) 200 мл дистиллированной воды я 2,5 мл 5%-ной соля-
ной кислоты.
2. После трехкратной перекристаллизации из четыреххлористого уг-
лерода температура плавления не меняется.
ЛИТЕРАТУРА
I. В. И. Дзномко, В. М. Островская. Химические реактивы
и препараты, вып. 28, М., ИРЕА, 1965, стр. 201.
2. В. М. Дзиомко, В. М. Островская. Ж. общ. химии, 35, 3,
502 (1965).
Поступила в мае 1966 г.
ИРЕА
УДК 547.753.07
ИНДОКСИЛАЦЕТАТ
О-Ацетилиндоксил; 3-ацетоксииндол; ацетат индол-3-ола
В. М. ОСТРОВСКАЯ, И. А. ГОРКЕР
CjflH9NO2
М.в. 175,19
Индоксилацетат является гистохимическим реагентом,
применяемым для выявления эстеразной активности биологи-
ческой ткани [1, 2].
В литературе описаны способы синтеза индоксилацетата
взаимодействием йодиндола с уксуснокислым серебром [3],
а также ацетилированием индоксила, полученного из щелоч-
ного индоксилового плава [4, 6] или гидролизом О, N-диаце-
тилиндоксила водным щелочным раствором [1].
Нами проверен и уточнен последний способ синтеза этого
соединения. Очистка индоксилацетата перекристаллизацией
из абсолютного этилового спирта предлагается нами впервые.
СХЕМА СИНТЕЗА ИНДОКСИЛ АЦЕТАТА
СН, зкаон
СОСИ
54
/0Na
-> | || || +2NaOCOCH3
H
,. ONa
^'\____x
I II ll + (CH3CO)SO ——>
H
^OCOCH,
. I ir~l -|-NaOCOCH3
h
Характеристика основного сырья
О, N-Диацетилиндоксил, ч. (см. стр. 36 в данном сбор-
нике) .
Натр едкий, ч.д.а., ГОСТ 4328—48.
Уксусный ангидрид, ч.д.а., ГОСТ 5815—52.
Этиловый спирт, абсолютный, ГОСТ 5962—51.
Условия получения
В четырехгорлую колбу емкостью 1 л, снабженную обрат-
ным холодильником, мешалкой, капельной воронкой и пода-
ющей азот трубкой, достигающей дна, загружают 550 мл 2н.
раствора едкого натра. Через раствор пропускают азот в те-
чение 20—25 минут, чтобы вытеснить воздух, и быстро при-
бавляют 21,7 г (0,1 Л1) диацетилиндоксила, затем вновь про-
пускают азот. Смесь нагревают до кипения и кипятят, пока
диацетилиндоксил полностью не растворится, после чего рас-
твор охлаждают до 0° и прекращают пропускание азота. К
этому раствору при интенсивном перемешивании медленно,
по каплям, добавляют 57,2 г (0,56 Л4) уксусного ангидрида
и реакционную смесь размешивают еще 20—30 минут. Вы-
павший индоксилацетат отфильтровывают, промывают водой
(100 мл) и сушат в вакуум-эксикаторе над хлористым каль-
цием.
Выход технического индоксил ацетата в виде серого порош-
ка с т. пл. 120—124° равен 17,8 г, что составляет около 100%
от теоретического.
Для очистки полученный сырой продукт растворяют в
180 мл абсолютного этилового спирта (см. примечание), на-
гревают до 60°, добавляют 5 г активированного угля и вст-
ряхивают 15—20 минут. Раствор фильтруют и охлаждают
55
до —50° на бане из ацетона с сухим льдом. Выпавший оса-
док отфильтровывают, промывают 10 мл абсолютного этило-
вого спирта, охлажденного до —50°, и сушат в вакуум-экси-
каторе над концентрированной серной кислотой. Перекрис-
таллизацию повторяют еще раз без активированного угля.
Получают индоксилацетат в виде белых кристаллов с т. пл.
127,5—128,5° (после повторной перекристаллизации темпе-
ратура плавления не меняется).
Выход чистого продукта равен 9,2 г, что составляет 52%
от теоретического.
По литературным данным, температура плавления индок-
силацетата 126° [3,5]; 126—127° [6]; 127,5° [7]; 128° [8]; 126—
128° [4]; 129° [3].
Примечание.
Продажный абсолютный этиловый спирт (1 л) обрабатывают 7 г ме-
таллического натрия, 23 г диэтилфталата и перегоняют. Выход 0,9 л.
ЛИТЕРАТУРА
1. S. J. Holt in .General cytochemical methods,” v, 1, Acgd. Press,
ing., New York, and London, 1958, p. 375.
2. T. В a r k a, P. J. Anderson. Histochemistry theory, practice,
bibliography, New York, Evanston, London, 1963, p. 383—385.
3. R. D. Arnold, W. M. Nutter, M. L. Stepp. J. Organ.
Chem. 24, 1, 117 (1959).
4. R. J. Barrnett, A. M Seligman, Science, 114,579,(1951);
C. A, 46, 2604 f (195/).
5. D. V or la nd er, B. Drescher. Ber., 34, 1858 (1901).
6 D. Vorlander, B. Drescher. Ber, 52, 327 (1919).
7. G. Spencer. J. Soc. Chem. Ind., 50. 63 (1931).
8. A. Etienne. Bull. Soc. Chim. France, 1948, 651.
Поступила в мае 1966 г.
ИРЕА
УДК 547.415.1'118-32.07
N-КАРБОКСИМЕТИЛДИЭТИЛ EHTPHAMHHO-N',N"-
-БИС-ИЗОПРОПИЛФОСФИНОВАЯ КИСЛОТА
Р. П. ЛАСТОВСКИИ, В. В. СИДОРЕНКО, Н. В. ЛАПШИНА,
Т. П. КОНОПЛЕВА
О
'1 1
N= CH3CH2NHC(CH3),P(OH)2 -2Н,0
। h
СН2
1
СООН
C12H29N3O8P2-2H3O М.в. 441,35
М-Карбоксиметилдиэтилентриамино-М/,М/,'-бис- изопропил-
фосфиновая кислота синтезирована и предложена ИРЕА в
качестве нового комплексующего агента. Синтез вещества
осуществлен путем взаимодействия Nz, N''-диэтилентриамино-
бис-изопропилфосфиновой кислоты [1] с монохлоруксусной
кислотой в щелочной среде.
СХЕМА СИНТЕЗА N-КАРБОКСИМЕТИЛДИЭТИЛ EHTPHAMHHO-N',N"-
-БИС-И30ПР0ПИЛФ0СФИН0В0И КИСЛОТЫ
О
II
HN= СН3СН2МНС(СН3)8Р(ОН)г f+С1СН,СООН->
о
II
-> N = CH,CH2NHC(CH3)2P(OH),
I ь
сн,
соон
57
Характеристика основного сырья
Диэтилентриамино-бис-изопропилфосфнновая кислота, по-
лучена по методу ИНЭОС АН СССР [2] т. пл. 231°.
Монохлоруксусная кислота, ч., ГОСТ 5836—51.
Едкий натр, ч.д.а., ГОСТ 4328—48.
Соляная кислота, ч., ГОСТ 3118—46.
Условия получения
К смеси 3,8 г (0,01 М) диэтилентриамино-бис-изопропил-
фосфиновой кислоты и 15 мл 42%-ного раствора едкого нат-
ра прибавляют по каплям при перемешивании 2,83 г (0,03 М)
монохлорукс.усной кислоты, растворенной в 5 мл воды, пос-
ле чего периодически добавляют раствор едкого натра до
pH смеси 10—11.
Затем реакционную массу перемешивают при температу-
ре 34—45° в течение 24 часов и обрабатывают 30 мл
концентрированной соляной кислотой. Выпавший хлористый
натрий отфильтровывают, фильтрат упаривают в вакууме до
сиропообразного состояния и свободную кислоту осаждают
спиртом. Белый аморфный осадок отфильтровывают, триж-
ды промывают метанолом порциями по 30 мл, кипятят 20 ми-
нут с обратным холодильником с 50 мл этилового спирта,от-
фильтровывают и сушат в вакууме над пятиокисью фосфора.
По внешнему виду полученная кислота представляет со-
бой белый мелкокристаллический порошок, растворимый в
воде и нерастворимый в органических растворителях.
Выход равен 1,8 г, что составляет 40% от теоретическо-
го, считая на диэтилентрнамино-бис-изопропилфосфиновую
кислоту.
Найдено, %: С-32,58; Н -7,63; N—476; Р—13.77.
C12H28N3OsP-j-2Н,О. Вычислено, Н: С—32,6; Н -75; N—9.5; Р—14,0.
ЛИТЕРАТУРА
1. М. И. К а б а ч н и к, Т. Я. Медведь. Изв. АН СССР, отд. хим.
наук 1960, стр. 651.
2. Т. Я. Медведь, М. В. Рудоми н о, Е. А. Миронова,
В. С. Балабуха. М. И. К а б а ч и и к. Изв. АН СССР, сер. хим., 1967,
№ 2.
Поступила л ноябре 1966 г.
ПРЕД
УДК 547.295.71.07
•(-КЕТОНОУНДЕКАНОВАЯ КИСЛОТА
Л. А. ПОНОМАРЕВ, В. А. СЕДАВКИНА
С6Н13--СН2-С-СН9-СНо-СООН
II
о
СпН20О3 М.в. 200,27
Одним из удобных методов синтеза у-кетонокарбоновых
кислот является расщепление цикла в фурилкарбинолах под
действием водных или спиртовых растворов хлористого во-
дорода. Этот метод, применяемый для получения левулино-
вой [1, 2] и ряда алкиллевулиновых кислот и их этиловых
эфиров [3—6], распространен нами на высшие у-кетонокарбо-
новые кислоты н их этиловые эфиры.
Исходные фурилалкилкарбинолы получены известными
методами при взаимодействии фурфурола с алкилмагнийга-
логенидами [7, 8]. Выходы достигают 92%.
Расщепление цикла в фурилалкилкарбинолах осуществля-
лось нагреванием их до 80° в течение 3 часов с 0,4%-ным
раствором хлористого водорода в абсолютном этиловом спир-
те. При этом получаются этиловые эфиры у-кетонокарбоновых
кислот с выходом до 62%.
Омыление эфиров проводилось нагреванием их со спир-
товым раствором калиевой щелочи на водяной бане; выходы
количественные.
СХЕМА СИНТЕЗА 7-КЕТОНОУНДЕКАНОВОЙ КИСЛОТЫ
I Lch-c,hi3 —>
Чо I
он
59
c2HrOH CjHjsCHj — C—CHS CH2COOC2HS —>
0
-> C,Hls-CH,-C-CHg-CH2-COOCsH5
о
Характеристика основного сырья
Фурфурол, ч., ГОСТ 10930—64; т. кип. 162°; —1,5261;
^42о __1 J 5g
Гексилбромид, ч., МРТУ 6-09-188—63.
Магний в виде порошка или стружек.
Спирт этиловый, безводный, ГОСТ 5962—ol.
Хлористый водород.
Условия получения
Синтез гексилфурилкарбинола. В трехгорлой колбе, снаб-
женной механической мешалкой, делительной воронкой и
обратным холодильником с хлоркальциевой трубкой, готовят
обычным способом н-гексилматнийбромид из 7,4 г магния и
50 г н-гексилбромида в 150 мл абсолютного эфира. Затем
при постоянном перемешивании приливают из делительной
воронки раствор 24 г свежеперегнанного фурфурола в 30 мл
абсолютного эфира с такой скоростью, чтобы не наблюда-
лось заметного разогревания реакционной массы. После при-
бавления всего фурфурола смесь в течение 2 часов нагрева-
ют на водяной бане до легкого кипения эфира и затем вы-
ливают в сосуд с колотым льдом. Эфирный слой декантиру-
ют, а водный слой разлагают насыщенным раствором хлори-
стого аммония и экстрагируют эфиром. Объединенные эфир-
ные вытяжки промывают свежеприготовленным раствором
бисульфита натрия (для удаления непрореагировавшего фур-
фурола), потом насыщенным раствором соды и дистиллиро-
ванной водой (по 50 мл) и сушат прокаленным сульфатом
натрия. Остаток после удаления эфира перегоняют в вакуу-
ме при 4 мм, отбирая фракцию с т. кип. 107—108°.
Выход гексилфурилкарбинола равен 28,6 г, что составля-
ет 63% от теоретического; tin—1,4726; d4Z0—0,9663 (см.
примечание 1).
Получение этилового эфира у-кетоноундекановой кислоты.
К 20 г гексилфурилкарбинола приливают 80 мл 0,4%-ного
раствора хлористого водорода в безводном спирте и смесь
нагревают на водяной бане в течение 3 часов. Затем отгоня-
ют 2/з спирта, а остаток выливают в насыщенный раствор по-
60
таша. Маслянистый слой тщательно (5 раз по 25 мл) экст-
рагируют эфиром. Объединенный эфирный экстракт сушат
прокаленным поташом, эфир отгоняют, а остаток перегоня-
ют в вакууме при 5 мм, отбирая фракцию с г. кип. 138—139°.
Выход этилового эфира у-кетоноундекановой кислоты ра-
вен 10,8 г, что составляет 43,2% от теоретического; Ло°—
1,4392; — 0,9419 (см. примечание 2).
Получение у-кетоноундекановой кислоты. Нагревают 9 г
этилового эфира у-кетоноундекановой кислоты в течение 1 ча-
са на водяной бане с 10%-ным раствором калиевой щелочи в
метиловом спирте. Спирт частично отгоняют, а остаток выли-
вают в воду, кипятят 2—3 минуты с животным углем, филь-
труют и по охлаждении подкисляют соляной кислотой до кис-
лой реакции по конго. Образовавшийся осадок отфильтровы-
вают и перекристаллизовывают из петролейного эфира. Вы-
ход у-кетоноундекановой кислоты равен 6,9 г, что составляет
97,5% от теоретического. По внешнему виду у-кетоноундека-
новая кислота представляет собой бесцветные кристаллы с
т. пл. 78—79°; т. пл. семикарбазона 129—130°.
По литературным данным [9], т. пл. вещества 77° (см.
примечание 3).
Примечания:
1. Аналогичным путем из фурфурола и соответствующих алкилмаг-
нийбромидов получены:
амилфурилкарбинол с т. кип, 98—9974 мм; rip 1,4690; dj20—0,9760;
выход 68%;
гептилфурилкарбинол с т. кип. 118—119/4 мм; «д —1,4719;
№ — 0,9529; выход 71,5%;
октилфурилкарбинол с т. кип. 128—13074 мм; «д —1,4715; d,=c —
0,9375; выход 69,1 %;
20
вторичнооктилфурилкарбинол с т. кии. 114—11774 мм; nD —1,4615;
А?0 —0,9397; выход 51,3%;
нонилфурилкарбинол с т. кип. 128—13072 мм; пд —1,4680;
rf4a0 — 0.9256; выход 83%;
додецилфурилкарбинол с т. кип. 159—16071 мм; т. пл. 39—40°; вы-
ход 88,7%.
2. Аналогичным путем из соответствующих алкилфурилкарбинолов
получены:
этиловый эфир у-кетонодекановой кислоты с т. кип. 134—13874 мм;
По —1,4358; d420—0,9437; выход 50,5%;
этиловый эфир у-кетонододекановой кислоты с т. кип. 145—14674 мм;
Чд—1,4413: о!,» —0,9384; выход 43%;
этиловый эфир у-кетонотридекановой кислоты т. кип. 153—15474 мм:
«д’—1,4430; (Т420 — 0,9327; выход 46,1%;
этиловый эфир у-кетонотетрадекановой кислоты с т. кип. 158—
16072 мм; Яд —1,4489; %-—0,9286; выход 60,1%;
этиловый эфир у-кетоногептадекановой кислоты с т. кип. 177—
179°/1 мм: т. пл. 29—307 выход 62%;
61
этиловый эфир 4-кетоно-6-метилдодекановой кислоты с т. кип. 140—
141,574 мм; —1,4429; с/Л’—0,9325; выход 43,8%.
3. Аналогичным путем с количественными выходами получены:
у-кетонодекановая кислота с т. пл. 65—66°; фенилгидразон с т. пл
79—80° (из спирта);
у-кетонододекановая кислота с т. пл. 78°; семикарбазон с т. пл. 128’,
по литературным данным [9], т. пл. кислоты 79°;
у-кетонотридекановая кислота с т. пл. 84—85°; семикарбазон с т. пл.
132°; по литературным данным [9], т. пл. кислоты 87°;
у-кетонотетрадекановая кислота с т. пл. 85—86°; семикарбазон с
т. пл. 160° (с разложением); по литературным данным [10], т. пл. кис-
лоты 87°.
ЛИТЕРАТУРА
1. А. М. Беркенгейм, Т. Ф. Данкова. Ж. общ. химии, 9, 924
(1939).
2. J. Н а с h i h a m а, М. 1 гл о t о. J. Soc. Chim. Japan. 45, 406(1942);
С. А., 44, 8859 (1950).
3. В. ф. Кучеров. Ж. общ. химии, 20, 1885 (1950).
4. М. И. Ушаков, В. Ф. Кучеров. Ж- общ. химии, 14, 1082
(1944).
5. J. Moffat, G. Newberry, W. We bast er. J. Chera. Soc.
1946, 451.
6. M. M. Кацнельсон, M. E. Кондакова. Докл. АН СССР
17, 363 (1937).
7. А. А. Пономарев, В. А. Седавкина. Ж. общ. химии, 31,
984 (1961).
8. А. А. Пономарев, В. А. Седавкина. Ж- общ. химии, 32,
2540 (1962).
9. Н. Kes kin. Rev. faculte Set univ. Jnstaubul, 15a, 54 (1950);
C. A., 45, 2904 (1951).
10. R. Lukes, A. Zobacova. Coll. Czech. Chem. Comm., 24, 3189,
(1959).
Поступила в январе J966 г.
Саратовский гос. ун«т
УДК 547.812.5'113.7.07
2-МЕТИЛ-3,4,5,6-БИС-ИНД ЕНОПИРИЛ ИЙ ПЕРХЛОРАТ
Ю. А. ЖДАНОВ, Г. Н. ДОРОФЕЕНКО, В. А. ПАЛЧКОВ
сн:
CjqH [jClOg
М.в. 370,79
2-Метил-3,4, 5,6-бис-инденопирилий перхлорат синтезиро-
ван нами впервые ацетилированием 2- (инденил-1) -инданона-1
уксусным ангидридом в присутствии 67%-ной хлорной кисло-
ты.
СХЕМА СИНТЕЗА 2-МЕТИЛ-3.4,
5,6-БИС-ИНДЕНОПИРИЛИЯ ПЕРХЛОРАТА
(СН,СО)2О
63
Характеристика основного сырья
2-(Инденил-1)-инданон-1, т. пл. 142—143° (см. примеча-
ние 1).
Уксусный ангидрид, ч., ГОСТ 787—55.
Хлорная кислота, 67%-ная, получают концентрированием
х. ч. 57%-ной хлорной кислоты.
Условия получения
В трехгорлую колбу емкостью 50 мл, снабженную меха-
нической мешалкой, капельной воронкой и обратным холо-
дильником, помещают 4,92 г (0,02 М) инденилинданона-1 и
16 мл уксусного ангидрида. Смесь нагревают до растворе-
ния кетона, и через капельную воронку при перемешивании
по каплям добавляют 2 мл 67%-ной хлорной кислоты. Про-
исходит сильное вскипание раствора и выпадение пирилие-
вой соли. Реакционную смесь слегка подогревают на горел-
ке в течение 2—3 минут, охлаждают, после чего осадок от-
фильтровывают, промывают эфиром и высушивают на возду-
хе.
Выход вещества равен 6,4 г, что составляет 87% от тео-
ретического; после перекристаллизации из уксусной кислоты
г. пл. продукта 280° (см. примечание 2).
Примечания:
1. 2-(Инденил-1)-инданон-1 получают следующим образом. В 25 мл
этилового спирта растворяют 0,75 г металлического натрия. К получен-
ному раствору этилата натрия прибавляют 19,8 г а-инданона. Смесь ос-
тавляют прн комнатной температуре на 2 суток. Из раствора медленно
выкристаллизовывается продукт. Осадок отфильтровывают на воронке
Бюхнера и промывают 15 мл этилового спирта.
2-(Ияденил-])-инданои-1 получается в виде светло-желтых кристал-
лов с температурой плавления 142,5—143°,
Выход равен 10 г, что составляет 54% от теоретического.
, 2. Прн замене уксусного ангидрида ангидридами других кислот по-
лучают 2-алкил-3,4,5,6-бис-инденопирилий перхлораты с выходом
80—90%.
Поступила в феврале 1965 г.
Ростовскик-ла-Дону гос. ун-т
УДК 547.741.07
3-МЕТИЛ-1,2-ДИГИДРОПИРРОЛ ИЗИН
Л. А. ПОНОМАРЕВ, И. М. СКВОРЦОВ
| N I
I
сн,
CgHnN М.в. 121,18
3-Метил-1,2-дигидропирролизии является наиболее до-
ступным представителем группы соединений ряда 1,2-дигид-
ропирролизина, получаемых по общему методу, разработан-
ному на кафедре органической химии Саратовского универ-
ситета [1—3].
Как было подтверждено нами совместно с Хоркиным [3]
и Астаховой [4], 1,2-дигидропирролизины, являющиеся N,a-
замещенными циклическими гомологами пиррола, 'могут быть
введены в большинство реакций, хорошо разработанных для
пиррола [5, 6]. В связи с этим 1,2-дигидропирролизины пред-
ставляют интерес для тонкого органического синтеза и полу-
чения потенциально биологически-активных соединений.
В литературе описан способ приготовления 1,2-дигидро-
пирролизинов [7] восстановлением 1-оксо-1,2-дигидропирроли-
зинов [8, 9], являющихся труднодоступными веществами.
Предлагаемый нами способ синтеза 1,2-дигидропирролизи-
нов отличается простотой, а исходные соединения—фура-
новые амины [10, 11], получаемые на базе фурфурола, явля-
ются легкодоступными продуктами.
Применяемая аппаратура
Реакция дегидратации проводится в проточной установке
обычного типа. Реактором служит стеклянная (пирекс) труб-
ка с внутренним диаметром 14 мм. Длина заполненной ката-
5 Зак. 266 65
лизатором части равна 24 см. Слой катализатора з трубке
фиксируется двумя тампонами из стеклянной ваты. Для ра-
боты берется 35 см? (насыпной объем) катализатора. Угол
наклона реактора к горизонтальной плоскости составляет
примерно 20°. Реактор помещают в цилиндрическую печь ти-
па Т-40/270 с электрическим обогревом. Температура обо-
грева регулируется ЛАТРом и измеряется непосредственно у
поверхности реактора в средней его части стеклянным термо-
метром на 500° (ГОСТ 215—57).
К входному концу реактора присоединяется градуирован-
ная бюретка для подачи амина (амин 'в бюретке находится
под азотом) и ввод для азота. На выходном конце крепится
обратный холодильник с приемником для катализатора и от-
водом через верхний конец холодильника отходящих газов
в вытяжной шкаф.
Скорость азота, пропускаемого через реактор, замеряется
реометром.
КАТАЛИЗАТОР (AlsO„ZrO2)
Характеристика основного сырья
Алюминий сернокислый, ч., ГОСТ 3758—47.
Цирконий сернокислый, ч., МПТУ 2443—50.
Аммиак водный, ч.д.а., ГОСТ 3760—47, 25%-ный раствор.
Вода дистиллированная, ГОСТ 6709—53.
Условия получения катализатора
Раствор 522,3 г (0,783 М) кристаллогидрата сернокислого
алюминия А12(5О4)з • I8H3O в 2500 мл дистиллированной во-
ды сливают с раствором 58,25 г (0,164 М) кристаллогидра-
та сульфата циркония (см. примечание 1) в 400 мл дистилли-
рованной воды. Затем к 1100 мл 10%-ного водного раствора
аммиака приливают при перемешивании подогретый до 70—
80° раствор сернокислых солей алюминия и циркония. Вы-
делившемуся осадку дают возможность осесть и жидкость над
осадком сливают сифоном (проба на полноту осаждения).
Осадок многократно промывают дистиллированной водой до
отрицательной реакции на сульфат-ион. Осадок отсасывают
на воронке Бюхнера так, чтобы получить слой толщиной в
1—1,5 см, переносят на стекло, разрезают ланцетом на куби-
ки с ребром 1—1,5 см и сушат в сушильном шкафу при 80—
90°, а затем при 120°. Высушенные кубики прокаливают при
700° в течение 3 часов. Размельченный до гранул с диамет-
ром примерно 3 мм катализатор используют для наполнения
трубки. Выход катализатора равен 61 г, что составляет 61 %
от теоретического.
66
Регенерация катализатора. Поверхность катализатора по-
сле дегидратации тетрагидрофурановых аминов становится
желтоватой или кремовой, а после дегидратации фурановых
аминов — серой или коричневой. Катализатор (НО—120 с .и3)
регенерируется в трубке из термостойкого стекла с диамет-
ром 28 мм, помещенной в печь типа Т-50/600 с регулируемым
обогревом. Температура регенерации контролируется термо-
парой в стеклянном кармане, введенном в слой катализато-
ра. Через трубку пропускается слабый ток воздуха и темпе-
ратура поддерживается в интервале 450—550° (см. приме-
чание 2). Сжигание органических соединений проводится до
тех пор, пока поверхность катализатора не станет совершен-
но белой (ом. примечание 3).
СХЕМА СИНТЕЗА 3-МЕТИЛ-1,2-ДИГИДРОПИРРОЛИЗИНА
|! || —ПаО | I |
'< J'-CH2-CH2-CH-CH3 А1А.2Ю,-* I /N- ..х
О I I
сн3
Характеристика основного сырья
1-(а-Фурил)-3-аминобутан, т. кип. 85718 мм; —1,4763;
0,9629 [10].
Условия получения
Реактор нагревают до 300—305° и одновременно пропус-
кают через него азот со скоростью 800—1000 мл в час (см.
примечание 4). По достижении заданной температуры и пре-
кращении выделения воды из катализатора прибавляют в
течение 3 часов 18,4 г (0,132 Л4) 1-(а-фурил)-3-аминобутана
со скоростью 1 капля за 10 секунд. Во время опыта скорость то-
ка азота поддерживают в интервале 800 -1000 мл в час. По-
сле окончания прибавления амина пропускание азота через
систему продолжают до прекращения выделения катализата
из трубки. Катализат (15,3 г) состоит из водного и органи-
ческого слоев. Органический слой — зеленоватая флуорес-
цирующая жидкость.
В полученный катализат добавляют 20—25 мл эфира, по-
сле чего в смесь через обратный холодильник вводят твердое
едкое кали до насыщения водного слоя. Эфирный раствор от-
деляют от водного и сушат твердым едким кали. Эфир от-
гоняют, а остаток подвергают разгонке в вакууме из колбы
с елочным дефлегматором длиной 14 см, собирая фракцию
с т. кип. 73—76716,5 мм.
5* 67
Получают 10,2 з 3-метил-1,2-дигидропирролизина, что со-
ставляет 63% от теоретического выхода; Пр —1,5130. За-
тем его перегоняют из колбы с колонкой Видмера и полу-
чают вещество со следующими константами: т. кип. 73—
74716 мм; «р—1,5139; d420 — 0,9684 (см. примечание 5).
Примечания:
1. Необходимо использован, свежеприготовленный раствор сульфа-
та циркония, так как при длительном стоянии из него начинают выде-
ляться основные соли сульфата циркония. Раствор сульфата циркония
готовят следующим образом: 87,6 г сульфата циркония, ч., содержащего
33,5% ZrCh, обрабатывают 400 мл дистиллированной воды, отфильтро-
вывают раствор сульфата циркония от двуокиси циркония при помощи
воронки Бюхнера, а затем еще раз — через складчатый фильтр.
2. При сжигании необходимо тщательно следить за температурой,
так как возможно сильное перегревание катализатора в связи с бурным
выгоранием органических веществ.
3. После регенерации катализатор активнее, чем свежеприготовлен-
ный.
4. Для дегидратации применяют технический азот из баллона через
редуктор.
5. Аналогичным путем были синтезированы:
/,2-дигидропирролизип с выходом 31%, т. кип. 69—70715 мм;
По—1,5308; с7°—1,0056;
3-изобутил-1,2-дигидропирролизим с выходом 54%, т. кип. 97—98°/.«л;
По —1,5001; d?’—0,9340;
2,3- тетраметилен-1,2-дигидропирролизин, с выходом 60%, т. ьип.
117—118710 мм. К™ —1,5365; dr0--1,0175;
I -этил-3-метил-1,2-дигидропирролизин, с выходом 64%, т. кип. 83—
85710 мм; По —1,5000; d420 — 0,9319;
1-изопропил-3-метил-1,2-дегидропарролизак, с выходом 53%. т. кип.
91—93710 мм; tip —1,4968; 7“— 0,9254 из соответствующих фурано-
вых аминов [10, 11]).
Для получения достаточно чистых 1,2-дигидропирролизи-
на и 1-этил-3-метил-1,2-дигидропирролизина необходима до-
полнительная очистка их от основных примесей. Для этого
вещества растворяют в пятикратном по объему количестве
эфира и быстро промывают раствором 5 %-ной серной кисло-
ты до кислой реакции промывных вод, затем водой, раство-
ром едкого кали, после чего эфирный раствор сушат твердыж
едким кали и далее поступают обычным образом.
ЛИТЕРАТУРА
1. А. А. Пономарев, И. М. Скворцов. Ж. общ. химии, 97 32
(1962).
2. А. А. Пономарев, И. М. Скворцов. Докл. АН СССР, 148,
860 (1963).
3. А. А. Пономарев, И. М. Скворцов, А. А. Хоркин. Ж.
общ. химии, 33, 2687 (1963).
4. А. А. Пономарев, 14. М. Скворцов, Л. Н. Астахова.
Докл. АН СССР, 155, 861 (1964).
5. Г. Фишер, И. Г. Орт. Химия пиррола. Л., ОНТИ, 1937.
68
6. Гетероциклические соединения, т. 1, М., ИЛ, 1953, стр. 219.
7. J. Petterson. J. В rase h, Р. Drenchko. j. organ. Ghent.,
27, 1652 (1962).
8. G. С 1 e in о, T. Melrose. J. Chem. Soc., 1942, 42-1.
9. R. Adams, S. Miyano, D. Fles. J. Amer. Chem. Soc., 82,1466
(1960).
10. А. А. Пономарев, H. П. Масленникова, А. П. Кри-
венько. Ж. общ. химии, 31, 958 (1961).
11. А. А. Пономарев, А. П. Кривенько, М. В. Но рицина.
Ж. общ. химии, 33. 1778 (1963).
Поступила в январе 1966 г. Са}атовский гос. ун-т
УДК 547.741.07
3-МЕТИЛ-5-ДИМЕТИЛАМИНОМЕТИЛ-
1,2-ДИГИДРОПИРРОЛИЗИН
А. А. ПОНОМАРЕВ, Л. Н. АСТАХОВА, В. И. СИМОНЦЕВ
CtIH,8N2
I N I
СН3х i * I
;n-ch. сн3
сн/
М.в. 178,27
В литературе нет каких-либо сведений о реакции Манни-
ха в ряду 1,2-дигидропирролизина. Принимая во внимание
структуру 1,2-дигидропирролизинов, являющихся N, «-заме-
тенными циклическими гомологами пиррола, мы распрост-
ранили метод, предложенный для синтеза пиррольных ос-
нований Манниха [1], на соединения ряда 1,2-дигидропирро-
лизина.
З-Метил-5-диметнламинометил-1,2-дигидропирролизин, по-
лученный нами в этих условиях из 3-метил-1,2-дигидропирро-
лизина, может быть использован в качестве промежуточного
продукта при синтезе потенциально физиологически-активных
веществ.
СХЕМА СИНТЕЗА
3-МЕТИЛ-5-ДИМЕТИЛАМИНОМЕТИЛ-1.2- ДИГИДРОПИРРОЛИЗИНА
+CH4O+(CH3)2NH -> i N !
ч/
СН3. I i
> -СН2 СНз
сн/
70
Характеристика основного сырья
3-Метил-1,2-дигидропирролизин, т. кип. 63710 жж;
«о —1,5139; <7420—0,9684 (см. стр. 65 в этом сборнике).
Диметиламин солянокислый, ч., ВТУ МХП 2885—51.
Формалин, техн. (36—40%-ный).
Условия получения
К 10 г (0,0825 Л4) свежеперегнанного 3-метил-1,2-дигид-
ропирролизина, помещенного в трехгорлую колбу емкостью
100 мл с мешалкой, капельной воронкой и обратным холо-
дильником, при перемешивании равномерно прибавляют рас-
твор 7,1 г (0,087 Л4) солянокислого диметиламина в 7,3 г
(0,087 М) 36%-ного формалина с такой скоростью, чтобы
температура в колбе не превышала 60°. По окончании при-
бавления, которое длится около 30 минут, реакционную смесь
перемешивают в течение 2 часов и оставляют на ночь. На
следующий день ее обрабатывают 20 мл 25%-ного раствора
едкого натра. Выделившееся красноватое масло экстрагируют
эфиром, соединенные эфирные вытяжки промывают водой
два раза по 20 мл и сушат прокаленным сульфатом натрия,
ффир отгоняют на водяной бане и остаток перегоняют в ва-
кууме, собирая фракцию с т. кип. 104—1067Ю жж;
по 1,5080; d420 — 0,9526.
Выход З-метил-5-диметиламинометил-1,2-дигидропирроли-
зина равен 9,2 г, что составляет 62,5% теоретического (см.
примечание 1). Т. пл. пикрата 102—103° (из метанола).
Примечание.
Аналогичным путем синтезированы из соответствующих 1,2-дигидро-
пирролизинов и вторичных аминов [2]:
5-диметиламинометил-1,2-дигидропирролизин с т. кип. 98—9978 л.и;
«О —1,5150; 0,9712);
3-метил-5-(Ы-морфолин.ометил)-1,2-дигидрогшрролизин с т.
156—158710 мм; По —1,5285; d420—1,0540;
3-метил-5-(Ы-пиперидинометил)-1,2-дигидропирролизин с т.
112-11375 ялт; Яд0 — 1,5260; — 0,9898;
3-метил-5-(М-гексаметилениминометил)-1,2-дигидропирролизин с
140—14172 мм; п'о —1,5288; </420 — 0,9972;
2,3-тетраметилен-5-диметиламинометил-1,2-дигидропирролизин с
148-150710 мм; п™ —1,5272; <4» — 0,9933.
кип.
кип.
кип.
кип.
ЛИТЕРАТУРА
1. W. Herz, К. Dittmer, S. Crlstol. J. Amer. Chem. Soc.
69,1698 (1947).
2. А. А. Пономарев. Л. H. Act ахова, В. И. С и м о и ц e в. ХГС
№ 1, 81 (1965).
Поступила в январе 1966 г.
Саратовский гос. ун-т
71
УДК 547,642.07
2-МЕТИЛ-1,6-ДИОКСАСПИРО-(4,4)-НОНАН
.4. А. ПОНОМАРЕВ, И. А. МАРКУШИНА
I I/O—г СН3
\о '
C8HIt0.2 М. в. 142,19
Известно несколько способов получения спиранов группы
1,6-диоксаспиро- (4,4) -нонана. Впервые 1,6-диоксаспиро- (4,4) -
нонан и его диметил- и диэтилгомологи получены [1, 2] из бу-
тиролактона, валеролактона и капролактона. В небольших
количествах 1,6-диоксаспиро-(4,4)-нонан образуется при гид-
рировании 0- (фурил-2)-акролеина в присутствии никеля Ре-
нея [3]. При гидрировании соответствующих у-фурилалкано-
лов с никелем на кизельгуре были получены [4] 1,6-диокса-
спиро-(4,4)-нонан и 2-метил-1,6-диоксаспиро-(4,4)-нонан. Гид-
рированием различных у-фурилалканолов синтезированы
многочисленные представители алкил- и арилпроизводных
1,6-диоксаспиро-(4,4)-нонана, но выхода их в большинстве
случаев невысокие и в значительной мере определяются струк-
турными особенностями исходных фурановых спиртов [5—9].
Нами разработан новый метод получения 1,6-диоксаспи-
ро-(4,4)-нонана и его гомологов электролитическим метоксили-
рованием у-фурилалканолов с последующим гидрировани-
ем образующихся 2-метокси-1,6-диоксаспиро-(4,4)-ноненов-3
[10, 11]. Этот метод позволяет получать спираны с хорошими
выходами (60—75%) независимо от строения исходного
у-фурилалканола.
СХЕМА СИНТЕЗА 2-МЕТИЛ-1,6-ДИОКСАСПИРО-(4,4)-НОНАНА
СН3О-
о----j-CH3 2Н2
72
%-сн.он
Характеристика основного сырья
2-Метокси-7-метил-1,6-диоксаспиро- (4,4) -нонен-3 [10, 11],
т. кип. 72—73°/5 мм; «о — 1,4572; <7г°— 1,0587.
Метанол, ГОСТ 6995—54.
Водород, ГОСТ 3022— 45.
Никель на кизельгуре — промышленный катализатор,
предварительно восстановленный в токе водорода при 220°.
Условия получения
В стальной вращающийся автоклав емкостью 150 мл по-
мещают 20 г (0,12 М) 2-метокси-7-метил-1,6-диоксаспиро-
(4,4)-нонена-3 (см. примечание 1), 20 мл метанола и 2 а ни-
келя на кизельгуре. Начальное давление водорода 90 атмос-
фер. Гидрирование проводят при температуре 100° и закан-
чивают по поглощении 5,8 л водорода, необходимых на вос-
становление одной двойной связи и отщепление метоксильной
группы. По окончании гидрирования катализатор отфильтро-
вывают и промывают на фильтре метанолом. Метанол отго-
няют на водяной бане, остаток перегоняют при атмосферном
давлении, собирая фракцию с т. кип. 162—164°; Пр —1,4427;
rf420 — 0,9911.
Выход 2-метил-1,6-диоксаспиро-(4,4)-нонана равен 12 г,
что составляет 72% от теоретического (см. примечание 2).
Примечания:
1. 2-Метокси-7-метил-1,6-диоксаспиро-(4,4)-нонеи-3 получают электро-
литическим метоксилированием 1-(а-фурил)-бутанола-3 (см. соответству-
ющую статью в этом сборнике).
2. Аналогичным путем получают:
1,6- диоксаспиро-(4,4)-нонан из Ь(а-фурил)-пропанола-3 с выходом
72%;
2-изобутил-1,6-диоксаспиро-(4,4)-нонан из 1-(а-фурил)-5-метилгекса-
нола-3 с выходом 70%;
2,2-диметил-1,6-диоксаспиро-(4,4)-нонан из 1-(а-фурил)-3-метилбута-
нола-3 с выходом 66% [10, 11].
ЛИТЕРАТУРА
1. R. Fittig. Ann, 256, 50 (1890).
2. R. Fittig. Ann., 267, 186 (1892).
3. H. Burdick, H. Adkins. J. Amer. Chem. Soc., 56, 438 (1934).
4. K. Alexander, L. Hafner, L. S c h n i e p p. J. Amer. Chem.
Soc., 73, 2725 (1951).
5. А. А. Пономарев, 3. В. Тиль, И. А. Маркушина,
К. М. Са п у н а р. Докл. АН СССР, 93, 297 (1953); Ж. общ. химии, 27,
ПО (1957).
6. А. А. П о н о м а р е в, 3. В. Тиль, А. Д. Петехонова,
В. Н. Решетов. Ж. общ. химии. 27, 1075 (1957).
7. А. А. Пономарев, В. А. Се давки иа. Уч. зап. Саратовск.
ун-та, 71, 143 (1959).
73
8. 3. В. Тиль, А. А. Пономарев. Уч. зап. Саратовск. ун-та, 75,
41 (1962).
9. А. А. Пономарев, 3. В. Тиль. Уч. зап. Саратовск. ун-та, 63,
53 (1956).
10. А. А. Пономарев, И. А. Маркушина. Докл. АН СССР, 126,
99 (1959).
11. А. А. Пономарев, И. А. Маркушина. Ж. общ. химии 31,
554 (1961).
Поступила в внваре 1966 г.
Саратовский гос. ун-т
УДК 547.812.5'113.7.07
2-МЕТИЛ-4,6-ДИ(а ТИЕНИЛ)ПИРИЛИЙ ПЕРХЛОРАТ
Г. Н. ДОРОФЕЕНКО, С. В. КРИВУН
С14Н nO5ClS2
М. в. 358,82
2-Мстил-4,6-дитиенилпирилий перхлорат синтезирован на-
ми впервые ацилированием ацетотиенона уксусным ангидри-
дом в присутствии хлорной кислоты [I].
СХЕМА СИНТЕЗА 2-МЕТИЛ-4,6-ДИ(а-ТИЕНИЛ) ПИРИЛИЯ
ПЕРХЛОРАТА
(СНзСО)аО
НС1О4
(СН3СО)2О
нсю4
СНз
сю4-
Характеристика основного сырья
2-Ацетотиенон, т. кип. 209—213° [2].
Уксусный ангидрид, ГОСТ 787—55.
Хлорная кислота, 70%-ная, получают концентрированием
х.ч. 57%-ной хлорной кислоты.
Условия получения
В двугорлую круглодонную колбу, снабженную капельной
воронкой и обратным холодильником, помещают 2,52 г
(0,02 М) 2-ацетотиенона и 6 мл (0,06 М) уксусного ангидри-
да. К полученному раствору прибавляют по каплям 1 мл
(0,012 М) 70%-ной хлорной кислоты и смесь выдерживают
сутки при комнатной температуре. Уже через несколько ча-
сов начинается кристаллизация пирилпевой соли. По оконча-
нии реакции к смеси приливают 5—7 мл изопропилового
спирта, осадок пирилиевой соли отфильтровывают, промыва-
ют смесью спирта и эфира (1:3), затем эфиром и высушива-
ют на воздухе.
Выход 2-Метил-4,6-ди(а-тиенил) пирилия перхлората ра-
вен 1,5 г, что составляет 41% от теоретического. После пере-
кристаллизации из воды с добавлением 1—2 капель хлорной
кислоты получают соль в виде светло-коричневых кристаллов
с т. пл. 254° (см. примечание 1 и 2).
Примечания:
I. 2-Метил-4,6-ди(а-тиенил)пирнлий перхлорат получается по выше-
приведенной методике при ацилировании тиофена. Из 1,68 г (0,02 М}
тиофена, 6 мл (0,012 М) уксусного ангидрида и 1 мл (0,012 Л1) 70%-ной
хлорной кислоты образуется 1,32 г (36%) пирилиевой соли.
2. Аналогичным путем при ацилировании п-метоксиацстофеноиа и
п-этоксиацетофенона получают 2-метил-4,6-ди-(п-метоксифенил)-пирилий
перхлорат (45%) и 2-метил-4,6-ди-(п-этоксифенил)-пирилий перхлорат
(41%).
ЛИТЕРАТУРА
1. Г. Н. Дорофеенко, С. В. Кривун. Украинский хим. ж„ 29,
1058 (1963).
2. Г. Н. Дорофеенко, В. И. Дуленко. Методы получения хим.
реактивов и препаратов, сб. 2, М., ИРЕА, 1961, стр. 75.
Поступила в апреле 1965 г.
Ростовский на Дону гос. ун-т
УДК 547.642.07
2-МЕТИЛ-2,4-ДИЭТИЛ-1,е-ДИОКСАСПИРО-(4,4)- НОНАН
А. А. ПОНОМАРЕВ, В. А. СЕДАВКИНА
С12Н,2О2
М. в. 198,30
Впервые 1,6-диоксаспиро-(4,4)-нонан и его диметил- и ди-
этилгомологи были получены из бутиролактона, валеролакто-
на и -капролактона [1, 2].
1,6-Диоксаспиро-(4,4)-нонан был обнаружен в продуктах
гидрирования фурилакролеина [3, 4]. Затем было показано,
что гомологи 1,6-диоксаспиро-(4,4)-нонана образуются при
гидрировании фурфурилиденацетона, фурфурилиденбутанола
в присутствии никеля Ренея [5], а также при гидрировании
1-(а-фурил)-пропанола-3 и 1-(а-фурил) бутанола-3 в присут-
ствии никеля на кизельгуре [6]. В дальнейшем было установ-
лено, что и при гидрировании третичных у-фурилалканолов с
никелем Ренея и никелем на кизельгуре, наряду с тетрагид-
рофурановыми спиртами, образуются гомологи 1,6-диоксаспи-
ро-(4,4)-нонана, причем с более высокими выходами [7].
Нами установлено, что при гидрировании третичных у-фу-
рилалканолов с алкильными заместителями у первого отцик-
ла углеродного атома боковой цепи выходы гомологов
1,6-диоксаспиро-(4,4)-нонана значительно повышаются, осо-
бенно в присутствии никеля на кизельгуре. В ряде случаев
гомологи 1,6-диоксаспиро-(4,4)-нонана являются главными
продуктами реакции [8]. Гидрирование у-фурилалканолов
осуществлялось под давлением 100—140 атмосфер при 120—
140°.
Исходные у-фурилалканы получены взаимодействием
насыщенных фурановых кетонов с алкилмагнийгалогенида-
ми в условиях магнийорганического синтеза.
77
СХЕМА СИНТЕЗА
2-МЕТИЛ-2,4-ДИЭТИЛ4,6-ДИОКСАСПИРО-(4,4)-НОНАНА
\OJ-CH-CH2-CO-CH3 -CAHiff£g-f-
С3Н5
( он
J „ Lch-ch2-c-c2h5-^
и I I
с2н6 сн3
i ко—/СНз
н2мк I И :\г„
'О/\____I
С2н6
Характеристика основного сырья
З-(а-Фурил) гексанон-5, т. кип. 108—110°/20 мм; пр —
1,4669; rf42’0 — 0,9879 [8].
Этил бромистый, х.ч., ТУ МХП 80—47, т. кип. 38—39°.
Никель на кизельгуре — промышленный катализатор,
предварительно восстановленный в токе водорода при 220°.
Получение 5-метил-3-(а-фурил)-гептанола-5. К. реактиву
Гриньяра, приготовленному из 12 г магния, 52 г бромистого
этила в 150 мл абсолютного эфира и охлажденному ледяной
водой До 5°, медленно при энергичном перемешивании реак-
ционной смеси вводят по каплям из делительной воронки
раствор 67 г З-(а-фурил) гексанона-5 в 70 мл абсолютного
эфира. После прибавления -всего кетона реакционную массу
нагревают в течение 30 минут на водяной бане, охлаждают
и разлагают образовавшийся магниевый алкоголят уксусной
кислотой со льдом. Экстрагируют эфиром, эфирный экстракт
сушат прокаленным сульфатом натрия, эфир отгоняют, а ос-
таток перегоняют в вакууме.
Выход 5-метил-3(а-фурил) гептанола-5 равен 61 г, что со-
ставляет 77,1 %.
По внешнему виду вещество представляет собой бесцвет-
ную жидкость с т. кип. 99—100°/7 мм; я* —1,4754; <74м—
0,9659 (см. примечание 1).
Получение 2-метил-2,4-диэтил-1,6-диок.саспиро-(4,4)-нона-
на. Во вращающийся автоклав емкостью 150 мл помещают
35 г 5-метил-З-(а-фурил) гептанола-5, растворенного в 35 мл
абсолютного спирта, добавляют 3 г порошкообразного нике-
78
ля на кизельгуре, предварительно восстановленного в токе
водорода при температуре 220°, и гидрируют при температу-
ре 140° и начальном давлении водорода 100—120 атмосфер
до прекращения поглощения водорода (6690 мл). Затем ав-
токлав разгружают, катализатор отфильтровывают, отгоня-
ют растворитель при нагревании на водяной бане и несколь-
ко уменьшенном давлении, а остаток перегоняют в вакууме.
Фракция с т. кип. 100—104720 мм представляет собой
2-метил-2,4-диэтил-1,6-диоксаспиро-(4,4)-нонан, «о —1,4452;
d420 — 0,9315.
Фракция с т. кип. 140—142720 мм является 5-метил-З-(а-
тетрагидрофурил) -гептанолом-5.
Выход 2-метил-2,4-диэтил-1,6-диоксаспиро-(4,4) -нонана
равен 23,3 г, что составляет 66% от теоретического; выход
5-метил-З-(а-тетрагидрофурил)гептанола-5—7,1 г (20%) (см.
примечание 2).
Примечания:
1. Аналогичным путем получают:
5-метил-3-(а-фурил)октанол-5 из 10 г магния, 50 г бромистого про-
пила, 41 г 3-(а-фурил)-гексанона-5 и 200 мл абсолютного эфира; т. кип.
97—9872 мм;П^ 1,4765; d.w 0,9566; выход 40 г (77%);
2£-диметил-3-(а-фурил)гептанол-5 из 12,6 г магния, 58 г бромисто-
го этила, 66 г 2-метил-З-(а-фурил) гексаиона-5 и 400 мл абсолютного эфи-
ра; т. кип. 109—111/°7 мм; «д —1,4755; А20 — 0,9590; выход 66,4 г
(86,2%);
2,5-диметил-3-(а-фурил)октанон-5 из 6 а магния, 30 г бромистого
пропила, 37 г 2-метил-3-(а-фурил)гексанона-5 и 150 мл абсолютного эфи-
ра; т. кип. 108—11072 мм; —1,4745; с/,2’— 0,9488; выход 43,7 г
(95%).
2. Гидрирование 24 г 2,5-диметил-З-(а-фурил)-гептанола-5 в присут-
ствии 2,5 г никеля Ренея в тех же условиях привело к образованию
2-метил-2-этил-4-изопропил-1,6-диоксаспиро-(4,4)-ноиана с т. кип. НО—
112720 мм; —1,4485; d420 — 0,9295: выход 20 г (78%).
Л И Г Е Р А Т У Р Л
1. R. Fittig. Лпп., 256, 50 (1890).
2. R. Fittig. Ann., 267, 186 (1892).
3. Н. В п г d i с k, Н. A d к i n s. J. Amc-r, Chem., Soc., 56, 438 (1934).
4. M. F a г 1 о v, H. Burdick, Н. Adkins. J. Amer. Chem. Soc.,
56, 2438 (1934).
5. A. Hinz, G. Meyer, G. S c h u с к i n g. Ber., 768, 676 (1943).
6. K. Alexa d er, L. Hafner, L. Schniepp. J. Amer. Chem
Soc., 73, 2725 (1951).
7.3. В. Тиль, А. А. Пономарев, А. Д. Пешехонова,
В. П. Решетов. Ж- общ. химии, 27, 1369 (1957).
8. А. А. Пономарев, В. А. Седавкина. Уч. зап, Саратовск.
ун-та, 71, 143 (1959).
Поступила в январе 196о г. Саратовский гос. ун-т.
УДК 547.224'211.07
МЕТИЛЕН ЙОДИСТЫЙ
А. Л. ЛИФШИЦ, Е. П. АГЕЕВ, В. П. КАРОЛИНСКИИ,
М. И. ИЛЬИЧЕВА
CHaJ2 М. в. 267,83
Метилен йодистый применяется в минералогическом ана-
лизе для разделения минералов по удельному весу, а также
для определения показателя преломления в микроскопии.
Метилен йодистый может быть получен нагреванием
хлороформа или йодоформа с концентрированной йодистово-
дородной кислотой или йодом в запаянных трубках [1], элек-
тролизом иодуксусной кислоты [2], действием йодистого ка-
лия на хлористый метилен в запаянных трубках [3].
Все эти методы сложны и не могут быть воспроизведены
в промышленном масштабе.
Промышленное применение нашел способ получения йоди-
стого метилена путем восстановления йодоформа мышьяко-
вистокислым натрием [4, 5, 6J. Однако производство вещества
по этому методу сопряжено с существенными трудностями
из-за применения ядовитого мышьяковистого ангидрида и
йодоформа, являющегося дорогостоящим сырьем.
В основу разработанного нами синтеза йодистого метиле-
на положена реакция взаимодействия хлористого метилена и
йодистого натрия в ацетоне под давлением [3].
СХЕМА СИНТЕЗА ЙОДИСТОГО МЕТИЛЕНА
СН,С12 + 2NaJ -> СН J3 + 2NaCI
Характеристика основного сырья
Метилен хлористый, технический, ТУ МХП 3105—52.
Натрий йодистый, безводный (отходы от производства).
Ацетон, технический, ГОСТ 2768—60.
80
Условия получения
В автоклав емкостью 30 л, снабженный мешалкой и паро-
вой рубашкой, загружают 10 л ацетона и 12 кг технического
безводного йодистого натрия, раствор нагревают при разме-
шивании до 30—40° и затем к нему добавляют 3 кг хлористо-
го метилена (см. примечание 1). Автоклав закрывают и даль-
нейший нагрев его производят при работающей мешалке та-
ким образом, чтобы давление в автоклаве в течение первых
4 часов было равно 5 атмосферам, а в течении последующих
4 часов—10 атмосферам (см. примечание 2). После этого
нагрев прекращают и пуском воды в рубашку охлаждают ав-
токлав до комнатной температуры. Когда давление опустит-
ся до нормального, автоклав открывают и содержимое аппа-
рата вакуумом переводят в перегонный куб, где и производят
отгонку 3,5—4 л избыточного ацетона, используемого в по-
следующих синтезах.
Остаток после отгонки ацетона выливают на воду (20 л),
отделяют нижний слой технического йодистого метилена,
промывают его водой 2—3 раза порциями по 5 л, отделяют
от воды и сушат в течение 24 часов с помощью прокаленного
хлористого кальция.
Высушенный продукт перегоняют под вакуумом, собирая
основную фракцию при остаточном давлении 10—20 мм.
Выход йодистого метилена равен 4,45 кг, что составляет
47% от теоретического; температура застывания 5,6°.
Первые фракции, содержащие хлорйодметан, загружают
в последующих синтезах вместо хлористого метилена.
Общий выход препарата равен 4,7—4,8 кг, что составляет
50% от теоретического.
По литературным данным, т. пл. йодистого метилена 4,5—
6° [7].
Примечания:
1. Хлористый метилен и ацетон предварительно просушивают хлори-
стым кальцием.
2. При проведении реакции при давлении в автоклаве ниже 5 атмос-
фер процесс идет в сторону образования хлорйодметана. Ступенчатый
подъем давления в автоклаве способствует сдвигу реакции в сторону об-
разования йодистого метилена.
ЛИТЕРАТУРА
1. W. Н. Perkin. J. Chem. Soc., 69, 1173 (1896).
2. J u 1 i u s S m i d t, K. Th. Widmann. Ber., 42. 1889 (1909).
3 W. H. Perkin, Scarborough. J. Chem. Soc., 119, 1400 (1921).
4. V. Auger. Coinpt. rend., 145, 810 (1907).
5. A. Gutmann. Ber., 52, 212 (1919).
6. Синтезы органических препаратов, т. 1 М„ ГХП, 1949, стр. 222.
7. Справочник химика, т. 2, Л., 1963, стр. 790.
Поступила в августе 1966 г.
6 Зак. 266
ЦЗЛ
81
УДК 547.823.07
N-МЕТИЛ-З-ОКСИПИПЕРИДИН
А. А. ПОНОМАРЕВ, Н. И. МАРТЕМЬЯНОВА
( Y0H
сн3
CeH13N0 М. в. 115,17
Один из путей получения N-алкил-З-оксипиперидияоз со-
стоит в расщеплении тетрагидрофуранового кольца газооб-
разными галоидоводородами с последующим замыканием
продуктов реакции в цикл [1—6]. Выход на каждой стадии
около 70%. Исходным сырьем является фурфурол или тетра-
гидрофурфуриловый спирт [6].
Другой путь заключается в сульфировании пиридина с
последующим щелочным плавлением пиридинсульфокислоты,
восстановлении образовавшегося с 90—95%-ным выходом
3-оксипиридина до 3-оксипиперидина (выход 80%) [7—9] и
алкилировании последнего до N-алкил-З-оксипиперидина
(выход 35%) [5]. Несмотря на высокие выходы в первых трех
стадиях, общий выход продукта по этому методу невелик.
В основу метода получения N-метил-З-оксипиперпдина на-
ми положены данные, приведенные в работах [1, 5]. Уточне-
ние оптимальной температуры реакции и условий выделения
продуктов позволило довести выход до 75—78% от теорети-
ческого.
СХЕМА СИНТЕЗА N-МЕТИЛ-З-ОКСИПИПЕРИДИНА
__nh2-ch,_
Nf — R, 6О-УО0
-CHS-*
82
HBr
CHjCOOH
ch2-ch,-ch,-ch-ch,-nh '
। i !
Br OCOCHs CHS
NaOH^
-OH
I
CHS
Характеристика основного сырья
Фурфурол, ч., свежеперегнанный, т. кип. 16Г/760 мм,
ГОСТ 10930—64.
Водород электролитический.
Метиламин солянокислый, ч., ТУ ОРУ 3—58.
Никель Ренея, приготовлен по [10].
Бензол криоскопический, ГОСТ 5955—51.
Бром, ч., ГОСТ 4109—48.
Получение тетрагидрофурфурилметиламина. В стальной
вращающийся автоклав емкостью 600 мл загружают 76,8 г
(66 мл, 0,8 Л1) свежеперегнанного фурфурола и 24,8 г (0,8 Л4)
метиламина в 130 мл метанола (см. примечание 1) и 5—7 г
никеля Ренея-
После предварительной «промывки» водородом в автоклав
подают водород под давлением 100—120 атмосфер. Вначале
реакцию проводят при температуре 60—70°. При этой темпе-
ратуре в течение 1 часа поглощается почти половина рассчи-
танного количества водорода. Затем в автоклав подкачивают
водород до первоначального давления и температуру повы-
шают до 90°; реакцию проводят до прекращения поглощения
водорода. Гидрогенизат фильтруют от катализатора. Метанол
отгоняют на водяной бане при уменьшенном давлении. Оста-
ток перегоняют в вакууме, собирая фракцию с т. кип. 40—
42°/9 мм, представляющую собой тетрагидрофурфурилметил-
амин.
Выход равен 62—65 г, что составляет 65—70% от теоре-
тического; Яд—1,4440.
Из отогнанного растворителя получают дополнительно
8—10 г вещества с теми же константами (см. примечание2).
Общий выход тетрагидрофурфурилметиламина составля-
ет 75—80% от теоретического.
По литературным данным, т. кип. вещества 155—156°;
4079 мм; d421 —0,928; Лд —1,4436; т. пл. пикрата 129°.
Получение N-метил-З-оксипиперидина. В трехгорлую кол-
бу на 500 мл, снабженную обратным холодильником, термо-
метром и барботером, помещают 78 г (0,68 М) тетрагидро-
фурфурилметиламина и 45 мл ледяной уксусной кислоты.
6* 83
Смесь нагревают до 100° и при температуре 1Q0—110° в те-
чение 3—4 часов пропускают 113,4 г (1,4 Л4) бромистого во-
дорода (см. примечание 3). Затем к реакционной смеси осто-
рожно при охлаждении прибавляют раствор 250 г (6,2 Л4) ед-
кого натра в 300 мл воды и смесь -перегоняют с водяным па-
ром. Дистиллят при охлаждении насыщают твердой щелочью
и выделившееся масло отделяют. Водный слой экстрагируют
эфиром и сушат углекислым калием. Эфир -отгоняют, а оста-
ток перегоняют в вакууме, собирая две фракции: первую с
т. кип. до 80°/15 мм, представляющую собой смесь тетрагид-
рофурфурилметиламина и N-метил-З-оксипиперидина (см.
примечание 4), и вторую с т. кип. 80—82715 мм, представля-
ющую собой чистый N-метил-З-О'КСипиперидин. Выход его ра-
вен 43 г, что составляет 68—72% теоретического; п1 2° 1,4733.
По литературным данным, т. кип. вещества 80—82715 мм
[5], т. кип. 68—7078 мм [8]; п” —1,4695 [5}; —1,4733
[8]; Д16 -0,9639 [5].
Примечания:
1. Мети ламин получают взаимодействием 50 %-кого раствора соляно-
кислого метиламина (61 г, 0,9 М в 60 мл воды) с 50%-ным раствором
щелочи. Выделяющийся газ пропускают в предварительно взвешенный
Дрексель, содержащий 130 мл метанола. Вес поглощенного метиламина
определяется по разности [11].
2. Отогнанный метанол при охлаждении подкисляют концентрирован-
ной соляной кислотой до pH 4—5, после чего спирт отгоняют; остаток
растворяют в небольшом количестве воды и подщелачивают твердой ще-
лочью при охлаждении. Выделившееся масло экстрагируют эфиром, эфир
отгоняют и остаток перегоняют в вакууме.
3. Бромистый водород получают [12] действием 248 г (78 мл, 1,55 М)
брома на 121 г (150 мл, 1,55 44) криоскопического бензола. Перед ре-
акционной колбой ставят U-образную ловушку, одно колено которой на-
полнено антраценом, а другое—бромным железом для улавливания иепро-
реагировавшего брома и бензола. Реакционную смесь после пропускания
бромистого водорода можно оставлять на ночь.
4. Первая фракция в количестве 16 г возвращается для расщепления.
Выход рассчитан па прореагировавший тетрагидрофурфурилметнламин.
ЛИТЕРАТУРА
1. R. Paul, S. Tchelitc’heff. Compt. rend, 221, 560 (1945).
2. Англ пат-. 598390 (1948).
' : 3. Пат. США 2489546 (1949)-.
-4. Пат: США 2477937 (1949).
5. J-. Н. Biel, Н. F г i е m а п Helen. J. Amer. chem. Soc., 74 1485
(1952). .
84
6. Англ. пат. 820(503 (1959).
7. Chen-Weng Као, J. Chem. Eng. China, 15, 80 (1948).
8. Пат. США 2802007 (1957).
9. Англ. пат. 804123 (1958).
10. Сб. «Синтезы органических препаратов», вып. 3, М., ИЛ, 1952,
стр. 338.
11. Сб. «Синтезы органических препаратов», вып. 6. М., ИЛ, 1956.
стр. 54.
12. Ю. В. Карякин. Чистые химические реактивы, Л., Химтеорет,
1955 стр. 244.
Поступила в январе 1966 г.
Саратовский гос. ун-т
УДК 547.811'113.07
1-МЕТИЛ-3-(а-ТИЕНИЛ)-5, 6, 7, 8-
ТЕТРАГИДРОИЗОХРОМИЛИЙ ПЕРХЛОРАТ
Г. Н. ДОРОФЕЕЙKG, Л. В. ДУ ЛЕН КО, В. И. ДУЛЕНКО
C14Hl5OsCI
М. в. 330,78
1 -Метил-3-(а-тиенил)-5, 6, 7, 8-тетрагидроизохромилий
перхлорат синтезирован нами путем ацетилирования цикло-
гексенилацетотиенона уксусным ангидридом в присутствии
70%-ной хлорной кислоты.
СХЕМА СИНТЕЗА 1-МЕТИЛ-3-(а-ТИЕНИЛ)-5, 6, 7, 8-
ТЕТРАГИДРОИЗОХРОМИЛ ИЯ ПЕРХЛОРАТА
(СН2СО)2О
нею,
Характеристика исходного сырья
Циклогексенил ацетотиенон, т. кип. 150—15276 мм (см.
примечание 1).
Уксусный ангидрид, ГОСТ 787—55.
Хлорная кислота, 70%-ная, получают концентрированием
к. ч. 57%-ной хлорной кислоты.
Условия получения
К 2,06 г циклогексенилацетотиенона приливают смесь 5 г
уксусного ангидрида и 1,4 а 70%-ной хлорной кислоты, хоро-
шо перемешивают и оставляют при комнатной температуре.
Выпавшие через 15 минут кристаллы пирилиевой соли от-
фильтровывают и промывают эфиром.
Выход составляет 85—90% от теоретического; т. пл. 201°
из спирта (см. примечания 2 и 3).
Примечания:
1. Циклогексенилацетотиенон получен конденсацией циклогексанона с
ацетотиеноном в этиловом спирте в присутствии этилата натрия; выход
30%.
2. При замене уксусного ангидрида ангидридами других карбоновых
кислот получают перхлораты 1-алкил-3-(а-тиеннл)-5, 6, 7, 8-тетрагипро-
изохромилия с выходом G5—85%.
3. При обработке аммиаком 1-метил-3-тиенил-5, 6, 7, 8-тетрагидроизо-
хромилий перхлорат с выходом, близким к количественному, превращает-
ся в 1-метил-3-тиенил-5, 6, 7, 8-тетрагидроизохииолин т. кип. 195—
196°/6 мм; т. пл. пикрата 198°; т. пл. иодметилата 235°,
Поступила в феврале 1966 г. Ростовский на Дону гос. ун-т
УДК 547.642107
2-МЕТИЛ-1, 6, 8-ТРИОКСАДИСПИРО-(4, 1, 4, 2)-
ТРИДЕКАН
.4. А. ПОНОМАРЕВ, И. А. МАРКУШИНА
---I/O— J-CH
---1
М. в. 198,26
2-Метил-1, 6, 8-триоксадиспиро-(4, 1, 4, 2)-тридекан полу-
чен нами впервые [1] электролитическим интрамолекулярным
метоксилированием 2-(3-оксипропил)-5-(3-оксибутил)-фурана
с последующим гидрированием образующегося 2-метил-1, 6, 8-
триоксадиспиро-(4, 1, 4, 2)-тридецена-12.
СХЕМА СИНТЕЗА 2-МЕТИЛ-1, 6, 8-ТР14ОКСАДИСПИРО-
-(4, 1, 4, 2)-ТР14ДЕКАНА)
НОСН.(СН,)г-11 |1-(СН2)г-СН(ОН)-СН!
СИ,ОН
'(NH4B0
---/| Т н.
— о/хо/х—I
—\i |/О—|~сн3
Характеристика основного сырья
2-(З-Оксипропил)-5-(3-оксибутил)-фуран [!}, т. кип. 155—
15671 мм; п$ — 1,4986.
88
Метанол, ч., ГОСТ 6995—54.
Аммоний бромистый, ч., ГОСТ 4454—48.
Водород, ГОСТ 3022—45.
Получение 2-метил-1, 6, 8-триоксадиспиро-(4, 1, 4, 2)-три-
децена-12. В 220 мл метилового спирта растворяют 55 г
(0,27 М) свежеперегнанного 2-(3-оксипропил)-5-(3-оксибу-
тил)-фурана (см. примечание 1) и 5 г (0,051 Л4) бромистого
аммония. Раствор помещают в электролизер (см. примеча-
ние 2) и охлаждают до —15° (см. примечание 3). Электролиз
проводят при силе тока 4,5—3,0 а и напряжении 9—11 в в те-
чение 6 часов. После окончания электролиза раствор перено-
сят из электролизера в колбу и обрабатывают метилатом
натрия (см. примечание 4). Метанол и аммиак отгоняют на
водяной бане при уменьшенном давлении. Выпавший осадок
бромистого натрия отфильтровывают (см. примечание 5) и
на фильтре промывают несколько раз эфиром. Эфир отгоня-
ют на водяной бане, остаток перегоняют в вакууме, собирая
фракцию с т. кип, 100—103°/2,5 мм; — 1,4792; с/.ф0—
1,094.
Выход 2-метил-1, 6, 8-триоксадиспиро-(4, 1, 4, 2)-тридеце-
на-12 равен 37 г, что составляет 68% теоретического по ве-
ществу и 69% по току.
Получение 2-метил-1, 6, 8-триоксадиспиро-(4, 1, 4, 2)-триде-
кана. В стальной вращающийся автоклав емкостью 150 мл
помещают 27 г (0,13 Л4) 2-метил-1, 6, 8-триоксадиспиро-(4, 1,
4, 2)-тридецена-12, 35 мл метанола и 2 г никеля Ренея [2].
Гидрирование проводят при комнатной температуре и на-
чальном давлении водорода 100 атмосфер и заканчивают по-
сле поглощения 3,8 л водорода. Катализат выгружают из ав-
токлава, никель Ренея отфильтровывают и промывают на
фильтре метанолом. Метанол отгоняют на водяной бане, ос-
таток перегоняют в вакууме при 2,5 мм, собирая фракцию с
т. кип. 84—88°; 4° —1,4621; с?420— 1,059.
Выход 2-метил-1, 6, 8-триоксадиспиро-(4, 1, 4, 2)-тридекана
равен 23 г, что составляет 85% теоретического (см. примеча-
ние 6).
Примечания:
1. Методику получения 2-(3-оксипропил)-5-(3-оксибутил-)фурана
см. в этом сборнике, а также в [1].
2. Описание электролизера и условия проведения электролиза см. в
статье «2-Метокси-7-метил-1,6-диоксаспиро-(4,4)-нонен-3» этого сборника,
а также в [3, 4].
3. В качестве охлаждающей смеси используют бутиловый или изо-
бутиловый спирт, в который добавляют твердую углекислоту до дости-
жения необходимой температуры в электролизере.
4. Метилат натрия приготовляют исходя из 1,2 г металлического
натрия и 20 мл метанола. Прибавление метилата натрия необходимо для
связывания свободного брома, образующегося в процессе электролиза.
5. Бромистый натрий отфильтровывают на шоттовском фильтре № 3.
6. Аналогичным путем получают:
89
1, 6, 8-триоксадиспиро-(4, 1, 4, 2)-тридецен-12 с т. кип. 89—91°/2 мм
из 2,5-фурандипропаиола-З; Лд —1,4895; d^— 1,1442; выход 70% по
веществу и 64,5% по току;
1, 6, 8-триоксадиспиро-(4, 1, 4, 2)-тридекан с т. кип. 66—67°/2,5 мм\
—1,4696; — 1,1020; выход 78% [1].
ЛИТЕРАТУРА
1. А. А. Пономарев, И. А. Маркушина. Ж. общ. химии, 33,
3955 (1963).
2. Сб. «Синтезы органических препаратов», вып. 3, М., ИЛ, 1952,
стр. 338.
3. А. А. Пономарев, И. А. Маркушина, Ж. общ. химии, 30,
976 (1960).
4. А. А. Пономарев, И. А. Маркушина. Уч. зап. Саратовск.
ун-та, 71, 135 (1959).
Поступила в январе 1966 г. Саратовский гос. yu-’i
УДК 547.811'113.7.07
1-МЕТИЛ-З-ФЕН ИЛ-5,6,7,8-
ТЕТРАГИДРОИЗОХРОМИЛИЙ ПЕРХЛОРАТ И
1-МЕТИЛ-З-ФЕН ИЛ-5,6,7,8-ТЕТРАГИДРОИЗОХИНОЛ ИН
В. И. ДУЛЕНКО, Г. Н. ДОРОФЕЕНКО
I II +6
4 7 V С1О4-
сн3
сн3
С1,Н„О6С1 М. в. 324,76 CltH,,N М. в. 211,30
1-Метил-3-фенил-5, 6, 7, 8-тетрагидроизохромилий перхло-
рат синтезирован нами впервые ацетилированием циклогек-
сенилацетофенона уксусным ангидридом в присутствии 70% -
ной хлорной кислоты [1].
При обработке пирилиевой соли аммиаком получен 1-ме-
тил-З-фенил-5, 6, 7, 8-тетрагидроизохинолин.
СХЕМА СИНТЕЗА 1-МЕТИЛ-3-ФЕНИЛ-5,6,7,8-
ТЕТРАГИДРОИЗОХРОМИЛИЯ ПЕРХЛОРАТА
И 1-МЕТИЛ-3-ФЕНИЛ-5,6,7,8-ТЕТРАГИДРОИЗОХИНОЛ ИНА
»-СНгСО-СвН, (СН3СО)3О
. нсю«
! С1О4-
сна
91
I C104-
CH3
+ 2NH3->
ааЯ
| | + NH4C1O4 + H2O
ch3
Характеристика исходного сырья
Циклогексенилацетофенон, т. кип. 172—176718 мм (см.
примечание 1).
Уксусный ангидрид, ч.д.а., ГОСТ 787—55.
Хлорная кислота, 70%-ная, получают концентрированием
х.ч. 57%-ной хлорной кислоты.
Условия получения
Синтез 1-метил-3-фенил-5, 6, 7, 8-тетрагидроизохромилия
перхлората. К 6 г циклогексенилацетофенона 'приливают све-
жеприготовленную при охлаждении смесь 15 мл уксусного
ангидрида и 4,3 г 70%-ной хлорной кислоты, энергично пере-
мешивают и оставляют при комнатной температуре. Через
15 минут, когда реакционная смесь охладится и выделятся
кристаллы пирилиевой соли, добавляют 20—30 мл эфира,
осадок отфильтровывают и тщательно промывают смесью
ацетона и эфира (1:4).
Выход 1-метил-3-фенил-5, 6, 7, 8-тетрагидроизохромилия
перхлората с т. пл. 198—200° (из спирта) равен 8,75 г, что
составляет 90% от теоретичезкого.
Получение 1-метил-3-фенил-5, 6, 7, 8-тетрагидроизохиноли-
на. В суспензию 10 г перхлората 1-метил-3-фенил-5,6,7,8-тет-
рагидроизохромилия в н. пропиловом спирте пропускают до
насыщения сухой газообразный аммиак, смесь в течение не-
скольких минут слабо подогревают на водяной бане. Выде-
ляющийся при охлаждении перхлорат аммония отфильтро-
вывают, а фильтрат разбавляют 100 мл воды. 1-Метил-З-фе-
нил-5, 6, 7, 8-тетратидроизохинолин выделяется в виде легко
кристаллизующегося масла. Продукт отфильтровывают, про-
мывают водой, высушивают на воздухе и перекристаллизо-
вывают из петролейного эфира.
Выход количественный; т. пл. 76—77°; т. кип. 19377 мм;
т. пл. пикрата 175—176° (из спирта), т. пл. хлоргидрата
192—194°, т. пл. йодметилата 210—211° (из н. пропанола).
Примечания:
1. Циклогексенилацетофенон получен щелочной конденсацией смеси
циклогексанона и ацетофенона [2].
92
2. При замене уксусного ангидрида ангидридами других карбоновых
кислот получены перхлораты 1-алкнл-3-фенил-5, 6, 7, 8-тетрагПдроизохро-
милия с выходом 70—80% от теоретического.
ЛИТЕРАТУРА
1. Г. Н. Дорофееико, В. И. Дулевко. .Ж. общей химии, 32,
3445 (1962).
2. М. D. Farrow, G. A. R. Коп. J. Cbeni. Soc., 128, 2128 (1926).
Поступила в феврале 1966 г. Ростовский на Дону гос. ун—т.
УДК 547.741.07
3-МЕТИЛ-5-ФОРМИЛ-1, 2-ДИГИДРОПИРРОЛИЗИН
.4. А. ПОНОМАРЕВ. Л Н. АСТАХОВА
I I
СНО СН8
СвН1(ЬЮ
М. в. 149,19
3-Метил-5-формил-1,2-дигидропирролизин в литературе
не описан. Впервые он синтезирован нами [1] путем форми-
лирования З-метил-1, 2-дигидропирролизина диметилформа-
мидом и хлорокисью фосфора в условиях, предложенных
Джемсом и Снайдером [2] для получения индол-3-альдегида.
3-Метил-5-формил-1, 2-дигидропирролизин представляет ин-
терес как промежуточный продукт при синтезе физиологиче-
ски-активных веществ.
СХЕМА СИНТЕЗА 3-МЕТИЛ-5-ФОРМИЛ-1.2-
ДИГИДРОПИРРОЛИЗИНА
II I + HCON(CHs)2
I
сн3
Характеристика исходного сырья
З-Метил-I,2-дигидропирролизин, т. кип. 63°/10 мм;
rig — 1,5139; с?4М — 0,9684 (см. стр. 65 в этом сборнике).
N, N-Диметилформамид, ч., ВТУ РУ—1193—55.
Хлорокись фосфора, ч., ВТУ 166—51.
Условия получения
К 68 мл (64,6 г, 0,883 М) свежеперегнанного диметилформ-
амида (см. примечание 1), помещенного в трехгорлую кол-
бу емкостью 0,5 л, снабженную мешалкой, капельной 'ворон-
кой и осушительной трубкой с безводным сернокислым каль-
цием, при охлаждении смесью льда с солью и перемешива-
нии прибавляют в течение 20 минут 20 мл (33,55 г, 0,22 М)
свежеперегнанной хлорокиси фосфора (см. примечание 2).
Затем к смеси, окрашенной обычно в розовый цвет, добавля-
ют по каплям в течение 40 минут раствор 24,24 г, (0,2 AJ)
З-метил-1,2-дигидропирролизина в 24 мл (22,83 г, 0,31 Мj
дмметилформамида (см. примечание 3). После тщательного
перемешивания в течение 30 минут температуру реакционной
смеси поднимают до 35° и перемешивают при этой темпера-
туре 1 час. Затем в колбу вносят 120 г колотого льда, осу-
шительную трубку заменяют обратным холодильником и при
энергичном перемешивании добавляют раствор 88 г едкого
натра (2,2 М) в 240 мл воды (см. примечание 4). Содержи-
мое колбы быстро нагревают до кипения, охлаждают до
комнатной температуры и оставляют на ночь. На следующий
день образовавшийся неорганический осадок отсасывают на
воронке Бюхнера и промывают два раза эфиром по 10 мл.
Выделившееся масло из фильтрата экстрагируют эфиром,
эфирные вытяжки сушат прокаленным сульфатом магния.
Эфир отгоняют на водяной бане, остаток перегоняют в ваку-
уме, «обирая фракцию с т. кип. 96—97°/4 мм.
Выход 3-метил-5-формил-1,2-дигидропирролизина равен
21,4 г, что составляет 71,7% от теоретического; ng—1,5712;
dt20— 1,0845.
Т. пл. азина 123—124° (из 50%-кого спирта).
Примечания:
о1- Использован свежеперегнанный диметилформамид с т. кип. 151 — 2 3 4 5 *
2. Использована свежеперегнанная хлорокись фосфора с т. кип. 106°.
3. Необходимо следить, чтобы на этой стадии температура не превы-
шала 10°.
4. Одну треть раствора прибавляют медленно по каплям, остальные
Две трети быстро. При этом происходит сильное разогревание реакцион-
ной смеси и на поверхности образуется коричневое масло.
5. Аналогичным путем синтезированы из 1,2-дигидропирролизина
^•формил-1,2-дигидропирролизин с т. кип. 108—10974,5 мм; ng—1,5902;
95
di,w—1,1325) и из 3-изобутил-1,2-дигидропирролизина З-изобутил-5-фор-
мал-1,2-дигидропирролизин с т. кип. 128,5—129,5 /5 мм; п1^ —1,5408;
d420—1,0179).
ЛИТЕРАТУРА
1. Л. Н. А с т а х о в а, И. М. С к в о р ц о в, А. А. П о и о м а р е в.
Ж. общ. химии, 34, 2410 (1964). А. А. Пономарев, И. М. Скворцов,
Л. Н. Астахова. Докл. АН СССР, 155, 861 (1964).
2. Сб. «Синтезы органических препаратов», вып. 11, М., ИЛ, 1961.
стр. 30.
Поступила в январе 1966 г, Саратовский гос. уи-т
УДК 547.822.3.413.07
N-МЕТИЛ-З-ХЛОРПИ ПЕРИДИИ
А. А. ПОНОМАРЕВ, Н. И. МАРТЕМЬЯНОВА
C6H1SNC1
М. в. 133,62
N-Метил-З-хлорпиперидин получают с выходом около 55%
действием избытка хлористого тионила в хлороформе на N-
метил-З-оксипиперидин [1, 2].
Ниже предлагается детально разработанная методика по-
лучения N-метил-З-хлорпиперидина с выходом 60%.
N-метил-З-хлорпиперидин является исходным веществом
для синтеза ряда антиспазматических и антиязвенных препа-
ратов [3].
СХЕМА СИНТЕЗА N-МЕТИЛ-З-ХЛОРПИПЕРИДИНА
SOCIa
NaOH
+ NaCi 4- Н3(.)
Зак. 266
97
Характеристика основного сырья
N-Метил-З-оксипиперидин, свежеперегнанный, т. кип. 80—
82°/15 мм; Пр—1,4733 (см. соответствующую статью в этом
сборнике).
Хлороформ, х. ч., ГОСТ 3160—51.
Хлористый тионил, ч., ВТУ МХП 3591—52.
Условия получения
В трехгорлую полулитровую колбу, снабженную механи-
ческой мешалкой, термометром, капельной воронкой и обрат-
ным холодильником, помещают 75 мл (123 г, 1 А1) хлорис-
того тионила в 100 мл хлороформа. Колбу охлаждают сме-
сью льда с солью до температуры 0—5° и при этой темпера-
туре в течение 2 часов добавляют что каплям 57,5 г (0,5 А1)
N-метил-З-оксипиперидина в 50 мл хлороформа. При этом на-
блюдается сильное разогревание (см. примечание). После при-
бавления всего N-метил-З-оксипиперидина температуру под-
нимают до 60° и смесь кипятят в течение 3 часов. Цвет смеси
к концу нагревания становится темно-красным. Хлороформ
и избыток хлористого тионила отгоняют при уменьшенном
давлении на -водяной бане. Остаток в виде темной густой
массы осторожно и при охлаждении выливают в 50 мл воды
и при 0° прибавляют избыток щелочи (2 Л1) в виде 40%-кого
раствора. Выделившееся масло экстрагируют 6 раз эфиром
порциями по 50 мл. Эфирные вытяжки сушат прокаленным
сульфатом натрия, эфир отгоняют и остаток перегоняют в
вакууме, собирая фракцию с т. кип. 52716 мм, представляю-
щую собой N-метил-З-хлорпиперидин с —1,4676.
Выход вещества равен 40,5 г, что составляет 60% от тео-
ретического.
По литературным данным [1], т. кип. продукта 52—
53°/16—17 мм; /1д—1,4676; т. пл. гидрохлорида 187—188°
п т. пл. пикрата 168—169°.
Примечание: температура реакционной смеси не должна быть
выше 10°.
ЛИТЕРАТУРА
1. Е. Brain, F. Р. Doyle, М. D. Me nt а. .1. Chem. Soc., 633
(1961).
2. R. G. F u s о n, C. L, Z i r k e. J. Amer. Chem. Soc., 70, 2760 (1948)
3. Англ. пат. 804123, № 5 (1958).
Поступила в январе 1966 г.
Саратовский гос. ун-т.
УДК 547.642.07
2-МЕТОКСИ-7-МЕТИЛ-1,6-ДИОКСАСПИРО-(4.4)-
-НОНЕН-3
А. А. ПОНОМАРЕВ. И. А. МАРКУШИНА
™ о—-сн3
ch3o-;oa_J
С,НИО. М. в. 170,21
2-Метокси-7-метил 1,6-диоксаспиро- (4,4)-нонен-З впервые
описан нами в статье [1], где приведены общие данные о его
синтезе и свойствах. Ниже приводится препаративный ме-
тод получения его из 1-(а-фурил)-бутанола-3 путем интрамо-
лекулярного электролитического метоксилирования.
СХЕМА СИНТЕЗА
2-МЕТОКСИ-7-МЕТИЛ-1,6-ДИОКСАСПИРО-(4,4)-НОНЕНА-3
Jl-CH.-CHt-CH(OH) -СН3
'О• * ’ 7 (NH4Br)
О—сн3
^ch3o-<oA_J
Характеристика основного сырья
1-(а-Фурил)-бутанол-3 [2, 3], т. кип. 126—128755 мм;
п™ —1,4743.
Метанол, ГОСТ 6995—54.
Аммоний бромистый, х.ч., ГОСТ 4454—48.
Аппаратура
Электролизер состоит из никелевого катода — двух ци-
линдров, вставленных один в другой (внешний — диаметром
7» уо
46X49 мм и высотой 230 мм, внутренний — диаметром
26\27 мм и высотой 148 мм), и угольного анода (диаметр
18 мм, высота 250 мм). Плотность тока на аноде 2 а! дм- (см.
рисунок).
Электролизер:
1 — анод, 2 — газоотводная трубка,
3 — термометр, 4 — крышка сосуда,
5 — сосуд для охлаждения смеси;
6 — охлаждающая смесь, 7 — катод
Охлаждающая баня — сосуд с двойными стенками, снаб-
женный теплоизоляцией, или сосуд Дьюара [4].
Условия получения
Растворяют 58 г (0,4 М) 1-(а-фурил)-:бутанола-3 и 5 г
(0,051 М) бромистого аммония в 230 мл метилового спирта.
Раствор помещают в электролизер, охлаждают до —12° и
подвергают электролизу в течение 9 часов. Сила тока 4,0—
3,5 а, напряжение 9—11 в. По мере протекания электролиза
электропроводность раствора падает, поэтому для поддержа-
ния необходимой силы тока подаваемое на электролизер
напряжение увеличивают до 25—28 в и ведут электролиз до
тех пор, пока ток при указанном напряжении не упадет до
1,5 а. Во время электролиза температуру в электролизере
поддерживают на заданном уровне (ем. примечание 1).
По окончании электролиза раствор переносят в колбу и
прибавляют к нему метилат натрия (см. примечание 2). От-
гоняют метанол и аммиак на водяной бане при уменьшенном
давлении. Выпавший в осадок бромистый натрий отфильтро-
вывают (см. примечание 3) и промывают на фильтре не-
сколько раз эфиром. Эфир отгоняют на водяной бане, оста-
ток перегоняют в вакууме (см. примечание 4), собирая фрак-
цию с т. кип. 72—7475 мм; п% —1,4572; — 1,0587.
Выход 2-метокси-7-метил-1,б-диоксаспиро-(4,4)-нонена-3
равен 53,8 г, что составляет 75% теоретического по веществу
и 76% по току (см. примечание 5).
100
Примечания:
1. Для охлаждения используют бутиловый или изобутиловый спирт,
в который добавляют твердую углекислоту до достижения необходимой
температуры в электролизере. В дальнейшем температуру поддерживают
на заданном уровне периодическим добавлением твердой углекислоты.
Метилат натрия приготовляют из 1,2 г металлического натрия и
метанола. Прибавление метилата натрия необходимо для связы-
свободного брома, образующегося в процессе электролиза.
Бромистый натрий отфильтровывают на шоттовском фильтре № 3.
Вакуумную перегонку проводят из колбы с елочным дефлегмато-
2.
20 мл
вания
3.
4.
ром высотой 8—10 см.
5. Аналогичным путем получают: 2-метокси-1,6-ди.оксаспиро-(414)-но-
нен-3 из 1-(а-фурил)-пропаиола-3 с выходом 77% по веществу и 83% по
току; т. кип. 83—84°/Ю мм; n'j^— 1,4650; dt№— 1,1093; 2-метокси-7-изо-
бутил-1,6-диоксаспиро-(4,4)-нонен-3 нз 1-(а-фурил)-5-метилгексанола-3 с
выходом 86% по веществу и 80% по току; т. кип. 81—8272 мм;
1,4518; №—1,0015;
2-метокси-7,7-диметил-!,6-диоксаспиро-(4,4)-нонен-3 из 1-(а-фурнл)-3-
метнлбутаиола-3 с выходом 59% по веществу и 56% по току; т. кип.
75—7777 мм; tig —1,4518, №—1,0267;
2-метокси-7-метил-7-этил-1,6-диоксаспиро-(4,4)-нонен-3 из 1- (а-фурил) -
3-метилпентаиола-3 с выходом 68% по веществу и 60% по току; т. кип.
92—9376 мм; п д—1,4563; № — 1,0204 [1].
ЛИТЕРАТУРА
1. А. А. Пономарев, И. А. Маркушина. Докл. АП СССР, 126,
99 (1959); Ж. общ. химии, 31, 554 (1961).
2. A. D и П I о р, F. Peters. The Furane, 1953.
3. А. А. Пономарев. Синтезы и реакции фурановых веществ, Са-
ратов, СГУ, 1960, стр. 108.
4. А. А. Пономарев, И. А. Маркушина. Ж. общ. химии, 30,
976 (1960).
Поступила в январе 1966 г.
Саратовский гос. ун-т
УДК 547.415.1'113.1.07
\-(1-НАФТИЛ)-ЭТИЛЕНДИАМИН-ДИГИДРОХЛОРИД
Г. Н. НАЛЕЦКАЯ, А. П. ДУБРОВ, Г. Н. КОШЕЛЕВА
2HCM-LN—СН2-СН2- HN
C12H18NCI2 М.в. 259,17
В литературе синтез N-(l-нафтил)-этилендиамин-дигидро-
хлорида описан Ярославцевым [1]. Это соединение было по-
лучено по методу Габриэля. Нами проверена эта методика
[1] и уточнены условия синтеза.
N- (1 -Нафтил) -этилендиамин-дигидрохлорид применяется
как реагент при биохимическом определении коэнзима А [2,3].
СХЕМА СИНТЕЗА
N-fl-НАФТИЛ )-ЭТИЛЕНДИАМИН-ДИГИДРОХЛОРИДА
I J /N-CH2-CH2 —NH-
w/“40
-> 2HC1-H2N—СН2—СНа -hn-^
Характеристика основного сырья
1,2-Дибромэтан, ч., ВТУ МХП 102—51.
Фталимид, ч., ТУ МХП 46—51.
Калий углекислый, безводный, х.ч., ГОСТ 4221—57.
а-Нафтиламин, ч.д.а., ГОСТ 8827—58.
Гидразин-гидрат, ч., ГОСТ 5832—51.
Спирт этиловый ректифицированный, ГОСТ 5962—51.
Петролейный эфир, т. кип. 40—60°.
Условия получения
Синтез ^-бромэтилфталимида. В трехгорлую колбу емко-
стью 500 мл, снабженную механической мешалкой, обрат-
ным холодильником и термометром, помещают смесь из 75 а
(0,5 М) фталимида (см. примечание 1) и 37,5 г (0,27 М) уг-
лекислого калия (см. примечание 2), предварительно хорошо
измельченных и смешанных в ступке. Затем в колбу добав-
ляют 250 г свежеперегнанного 1,2-дибромэтана (1,33 М;
т. кип. 129—130°) и реакционную смесь медленно нагревают
на масляной бане. При температуре 130—140° масса вначале
легко подвижная, затвердевает. Механическую мешалку вы-
ключают, а нагревание продолжают еще 6—8 часов при тем-
пературе бани 170—180°. Затвердевшая масса к концу нагре-
вания становится подвижной и по внешнему виду напомина-
ет коричневатое масло. Избыток дибромэтана отгоняют в ва-
кууме водоструйного насоса- Оставшуюся тягучую массу
(затвердевает при охлаждении) 4—5 раз промывают водой
для удаления минеральных солей. Остаток в колбе раство-
ряют щ 150 мл этилового спирта при нагревании на водяной
юз
бане в течение 30—40 минут. Горячий спиртовой раствор
фильтруют через воронку Бюхиера. При охлаждении спирто-
вого раствора выпадают кристаллы, слабо окрашенные в ко-
ричневый цвет.
Выход равен 100—120 г, что составляет 78— 100% от тео-
ретического. Эти кристаллы представляют собой смесь £-
бромэтилфталимида и дифталимидэтана. Их разделение про-
водят в аппарате Сокслета петролейным эфиром в течение
10—12 часов.
В петролейный эфир переходит только р-бромэтилфтали-
мид, который после перекристаллизации из горячего 75°/о-но-
го спирта и обесцвечивания небольшим количеством активи-
рованного угля получают в виде игольчатых бесцветных
кристаллов с т. пл. 82—83,5°.
Выход чистого p-бромэтилфталимида равен 70 г, что со-
ставляет 54 % от теоретического.
Получение N-(l-нафтиламино)-этилфталимида. Смесь из
51 г (0,2 М) p-бромэтилфталимида и 76,5 г а-нафтиламина
(0,53 М) тщательно растирают в ступке, переносят в колбу
и медленно нагревают иа масляной 'бане до 160°.
Образовавшуюся темно-коричневую тягучую массу про-
мывают большим количеством горячей воды (1000 мл), а за-
тем окончательно 50 мл теплого этилового спирта. Остаток
в колбе вымывают горячим этиловым спиртом.
По охлаждении спиртового раствора аморфный порошок
отфильтровывают, сушат и перекристаллизовывают из спир-
та и обесцвечивают активированным углем. По внешнему ви-
ду полученный продукт представляет собой золотисто-желтые
кристаллы с т. пл. 158°.
Выход N-(l-нафтиламино)-этилфталимида равен 51,7 г,
что составляет 82% от теоретического.
Получение N -(1-на.фтил)-этилендиамин-дигидрохлорида.
Навеску 51,7 г (0,16 М) р-(1-нафтиламино)-этилфталимида
суспензируют в 200 мл этилового спирта, затем прибавляют
6,5 г (0,06 М) 50%-ного гидразин-гидрата.
Реакционную смесь кипятят на водяной бане с обрат-
ным холодильником в течение 3 часов. К концу первого ча-
са нагревания в растворе появляются белые хлопья. Через
1,5—2 часа вся масса затвердевает. После трехчасового на-
гревания реакционной смеси образовавшийся белый осадок
подвергают гидролизу добавлением 100 мл 50%-ной соляной
кислоты (см. примечание 3). Затем нагревание продолжают
еще 1 час, после чего нерастворимый фталилгидразид от-
фильтровывают под вакуумом, промывают водой, а филь-
трат концентрируют путем отгона 150 мл спирта. По охлаж-
дении фильтрата снова выпадают кристаллы фталилгидрази-
да, которые отфильтровывают.
104
К остывшему фильтрату прибавляют крепкий раствор ед-
кого кали до явно щелочной реакции (по универсальной ин-
дикаторной бумажке).
Амин экстрагируют сухим эфиром, эфирный экстракт вы-
сушивают над твердым едким кали и переводят в колбу. По-
сле пропускания сухого хлористого водорода в эфирный рас-
твор амина выпадает белый хлопьевидный осадок N-(l-наф-
тил)-этилендиамин-дигидрохлорида. Его собирают на филь-
тре, быстро отжимают между листами фильтровальной бу-
маги и высушивают в вакуум-эксикаторе. Выход сырого про-
дукта равен 15 г (29% от теоретического).
После перекристаллизации из 6 н. соляной кислоты с ак-
тивированным углем получают белые кристаллы N-(l-наф-
тил)-этилендиамин-дигидрохлорида с т. пл. 182—188°.
Выход чистого продукта равен 12,8 г, что составляет 30%
теоретического. После нескольких перекристаллизаций из 6н.
соляной кислоты температура плавления веществ не изменя-
лась.
По литературным данным, т. пл. препарата 182—188° [1].
Примечания:
1. При отсутствии продажного фталимида его можно синтезировать
из фталевого ангидрида и концентрированного раствора аммиака пли из
фталевого ангидрида и углекислого аммония [4].
2. Углекислый калий обязательно нужно хорошо высушить в сушиль-
ном шкафу при 105°. Если углекислый калий не досушен, то выход
|3-бромэтплфталимида резко снижается.
3. Гидролиз р-(1-нафтиламино)-этилфталимида проводят по методу
[5].
ЛИТЕРАТУРА
1. Л. Ярославцев. Ж. общ. химии, 20, 2274—8 (1950).
2. D. Novell!. Methods for determination of coenzyme A In: Met-
hods of Biochemical analysis, v. Il, изд. Glick. Нью-Йорк—Лондон, 1955.
стр. 189.
3. A. P. Гусев a, M. Г. Б о p и x и н а. Биохимия, 23, вып. 2 M„ АН
СССР, (1958).
4. Сб. «Синтезы органических препаратов», вып. 1, М„ ИЛ, 1949,
стр. 448.
5. Н. R. Inga, R. Manske. J. Chem. Soc., 2348 (1926).
Поступила и декабре 1965 г. Ин-т химии природных соединений АН СССР
Биолого-почвенный факультет МГУ
УДК 547.829.07
4-НИТРО-4-ДИЭТИЛАМИНОСТИЛББЕН
В. Г. БРУДЗЬ, Н. И. ЧЕРНОВА, Д. А. ДРАПКИНА, Б. М. БОЛОТИН
O2N-^_^-CH=CH-^_^-N(CsH6),
C18H20NsO2 м. в. 296,37
4-Нитро-4'-диэтиламиностильбен обладает люминесцент-
ными свойствами.
В литературе это соединение не описано. Нами оно было
получено аналогично 4-нитро-4'-диметиламиностильбену [1].
СХЕМА СИНТЕЗА 4-НИТРО-4'-ДИЭТИЛАМИНОСТИЛЬБЕНА
ON2-^_^-CH,COOH + (C^N-^^-CHO
- O2N-^^-CH=CH -^_'^-N(C,HB)g + Н,0 + СОг
Характеристика основного сырья
п-Нитрофенилуксусная кислота, ч., ВТУ ХСНХ 259—60.
N, N-n-Диэтиламинобензальдегид, ч., СТУ 79—517х—60.
Пиперидин, ч., ВТУ РУ 444—51.
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную механи-
ческой мешалкой, термометром и обратным холодильником,
загружают 120 г (0,66 М) гс-нитрофенилуксусной кислоты,
117 г (0,66 М) n-диэтиламинобензальдегида и 30 мл пипери-
дина. Колбу помещают в глицериновую баню и при постоян-
ном перемешивании реакционную смесь нагревают. При 100°
начинается интенсивное выделение углекислого газа (ско-
106
рость перемешивания необходимо снизить), затем темпера-
туру поднимают до 130°, выдерживают 1 час 20 минут, (до
прекращения выделения углекислоты). Затем массу охлаж-
дают до 70—80° и выливают в 100 мл этилового спирта при
перемешивании. По охлаждении выделившиеся кристаллы
отфильтровывают, отжимают, промывают 100 мл горячего
бензола, охлаждают бензольный раствор, осадок отфильтро-
вывают и сушат при комнатной температуре.
Высушенный продукт имеет темно-красный цвет, не лю-
минесцирует в кристаллическом состоянии, но интенсивно
люминесцирует оранжево-красным цветом в бензольном рас-
творе. После перекристаллизации из бензола т. пл. вещества
182—182,5°
Выход 4-нитро-4'-диэтиламиностильбена равен 28 г, что
составляет 14% от теоретического.
Найдено, %: С-72,63—72,41; Н—7,01-6,96; N—9,21—9,39.
CisH.yNjO,. Вычислено, %: С—72,94; Н—6,80; N—9,49.
ЛИТЕРАТУРА
1. Р. Pfeiffer. Вег., 48, 1796 (1915).
Поступила в апреле 1966 г. ИРЕА
УДК 547.772.07
5-(5z-HHTPO-2 -ФУРИЛ)-3-МЕТИЛ ПИРАЗОЛ
А. А. ПОНОМАРЕВ. Л. В. ЧЕРКЕСОВА
CSH,N3O3
М. в. 193,16
5-(5'-Нитро-2/-фурил) -3-метилпиразол нами получен впер-
вые. Сведений о методах нитрования б-^'-фурилрпиразолов
в литературе не имеется. Мы предложили прямое нитрование
5-(2/-фурил)-пнразолов смесью концентрированной серной н
азотной кислот, позволяющее непосредственно вводить нитро-
группу в a-положение фуранового цикла 5-(2'-фурил)-3-ме-
тилпиразола [1]. Этот метод удобен и прост в препаративном
отношении.
Применение для нитрования смеси азотной кислоты и ук-
сусного ангидрида приводит к получению 1-ацетил-5-(5''-нит-
ро-2'-фурил) -3-метилпиразола.
По этому способу получен 5-(5'-нитро-2'-фурил)-3,4-диме-
тилпиразол с выходом 65%.
СХЕМА СИНТЕЗА
5-(5'-НИТРО-2'-ФУРИЛ)-3-МЕТИЛ ПИРАЗОЛА
ХСН3
I I |1 N + HNO3
н
108
сн3
+ Hs0
Характеристика основного сырья
Азотная кислота, х.ч., ГОСТ 4461—48.
Серная кислота, х.ч., ГОСТ 4204—48.
Аммиак водный, ч.д.а., ГОСТ 3760—47, 25%-ный.
5-(2'-фурил)-3-метнлп'иразол (см. примечание 1).
Условия получения
В трехгорлую колбу, снабженную механической мешал-
кой, минусовым термометром и капельной воронкой, поме-
щают 17,4 мл (12,4 г) азотной кислоты уд. веса 1,4 (0,2 М)
(см. примечание 2). Колбу охлаждают смесью лед-соль и
доводят температуру реакционной смеси до —8°, затем из
капельной воронки прибавляют 25,8 мл (14,1 г) серной кис-
лоты уд. веса 1,83 с такой скоростью, чтобы температура в
колбе не превышала —6°.
После прибавления всей кислоты капельную воронку сни-
мают и через свободное горло колбы прибавляют 3 г
(0,0155 М) порошкообразного 5- (2'-фурил)-3-метилпиразола
(см. примечание 3) небольшими порциями в течение 30 ми-
нут. Температуру реакционной смеси все время поддержива-
ют около минус 8—минус 12°. После прибавления всего ве-
щества перемешивание продолжают еще 40 минут. После
окончания нитрования реакционная смесь представляет со-
бой сиропообразную прозрачную жидкость светло-оранжево-
го цвета.
Содержание реакционной колбы выливают небольшими
порциями при энергичном перемешивании в стакан со смесью
130 г толченого льда и 30 мл 25%-ного водного аммиака с та-
кой скоростью, чтобы температура смеси в стакане не под-
нималась выше 10°. При этом выпадают желтые хлопья нит-
рофурилпиразола. В стакан дополнительно приливают рас-
твор аммиака до нейтральной реакции. Осадок отфильтровы-
вают, тщательно промывают водой и сушат при температуре
40°.
Выход 5- (5'-нитро-2'-фурил)-3-метилпиразола равен
3,47 г, что составляет 88% от теоретического. Вещество пе-
рекристаллизовывают из 50%-нопо спирта т. пл. 214—215°.
109
N'-Ацетильное производное имеет (из 50%-ного спирта)
т. пл. 176—177° (см. примечание 4).
Примечания:
1. 5-(2'-фурил)-3-метилпиразол с т. пл. 89—90° (из петролейного эфи-
ра) может быть получен по [1, 2].
2. При применении более концентрированной азотной кислоты выход
нитропродукта сильно снижается.
3. Вещество предварительно растирают в ступке.
4. N-Ацетилыюе производное получают действием уксусного ангидри-
да на 5-(5'-нитро-2'-фурнл)-3-метилпнразол.
ЛИТЕРАТУРА
1. А. А. Пономарев, Л. В. Черкесова. Ж. общ. химии, 33,
3946 (1963).
2. И. И. Гранберг, А. И. Кост. Д. В. С н б и р я к о в а. Ж- общ.
химии, 30, 2920 (1960).
Пэстушма 8 яшире 1966 г. Саратовский гос. уи-т
УДК 547.722.07
(5-НИТРО-2-ФУРОИЛ) АЦЕТОН
А. А. ПОНОМАРЕВ, Л. В. ЧЕРКЕСОВА
o2n-
JI—СОСН2СОСН3
хо7
C8H7NO6
М. в. 197,14
(5-Нитро-2-фуроил) ацетон получен нами впервые [1].
Синтез его был осуществлен ацилированием 5-нитроаце-
тилфурана уксусным ангидридом в присутствии трехфторис-
того бора. Неоднократные попытки получить этот дикетон
конденсацией этилового эфира 5-нитро-2-фуранкарбоновой
кислоты с ацетоном в присутствии этилата натрия не приве-
ли к положительным результатам.
СХЕМА СИНТЕЗА (5-НИТРО-2-ФУРОИЛ)АЦЕТОНА
(СН3СО)2О
o2n -I сосн,----------------->
\0/ BF,
- OeN -II l|-COCH,COCHs
4 о /
Характеристика основного сырья
Уксусная кислота, ч., ГОСТ 61—51.
Трехфтористый бор (см. примечание 1).
Уксусный ангидрид, ч., ГОСТ 5815—51.
Уксуснокислый натрий, ч.д.а., ГОСТ 199—52.
5-Нитроацетилфуран, получен по [2].
111
Условии получения
В трехгорлую колбу, помещенную в ледяную баню и
снабженную механической мешалкой, вводом для газообраз-
ного трехфтористого бора и выводной трубкой, защищенной
хлоркальциевой трубкой, загружают 12,5 г (12,0 мл) ледя-
ной уксусной кислоты. При энергичном ’перемешивании через
кислоту пропускают трехфтористый бор с такой скоростью,
при которой происходит его полное поглощение. По мере на-
сыщения кислоты подачу газа постепенно уменьшают и за-
канчивают после превращения содержимого реакционной
колбы в твердую массу (на это требуется около 2,5 часа). За-
тем трубку для ввода газа заменяют капельной воронкой, че-
рез которую приливают’ раствор 8,4 г (0,054 М) 5-нитро-2-
ацетилфурана в 16,5 г (15,2 дм) уксусного ангидрида. Пере-
мешивание продолжают еще 30 минут и далее реакционную
смесь выдерживают 12—14 часов при комнатной температу-
ре, после чего прибавляют к ней 140 мл 13%-ного водного
раствора ацетата натрия и кипятят с обратным холодильни-
ком 45 минут для разложения комплексов фтористого бора.
Выпавшие после охлаждения темно-коричневые кристал-
лы отфильтровывают, тщательно промывают водой и сушат.
Выход сырого (5-нитро-2-фуроил)ацетона равен 7,8 г, что
составляет 73% от теоретического (из метанола) т. пл. 112—
113° (см. примечание 2).
Примечания:
1. Трехфтористый бор готовят следующим образом: 50 a NH4HF2 сме-
шивают в ступке с 42 г Н3ВО3, при этом образуется жидкая кашица, ко-
торую в чашке упаривают, а затем осторожно прокаливают. Получен-
ную соль помещают в колбу Вюрца и из капельной воронки порциями
прибавляют 20%-нын олеум. Фтористый бор выделяется вначале бурно
без нагревания, затем колбу осторожно нагревают.
2. (5-Нитро-2-фуроил) ацетон использован для получения 5-(5'-нитро-
2'-фурил)-3-метилпиразола [1].
ЛИТЕРАТУРА
1. А. А. Пономарев, Л. В. Черкесова. Ж. общ. химии, 33,
3946. (1963).
2. Н. О. Салдобол, С. А. Гил л ер. Изв. АН Латв. ССР, 10, 101
(1958).
Поступила в январе iS66r.
Сараюнский гос. мьт
УДК 547.462.3.07
N-ОКСИИМИД МАЛЕИНОВОЙ КИСЛОТЫ
N-Оксимальимид
В. С. ИВАНОВ, В. К. СМИРНОВА, ЦАО ЮН
сн - с€
11 /N—ОН
СН - с<
C4H3NO3 М. в. 113,07
N-Оксимальимид, аналогично другим N-замещенным ими-
дам малеиновой кислоты [1], может представить потенциаль-
ный интерес в качестве мономера, вулканизующего агента
или активного вещества в сельскохозяйственной химии.
Сведения о получении N-ок'Симальимида в литературе от-
сутствуют. Попытка получить этот имид в одну стадию, исхо-
дя из -солянокислого гидроксиламина, по методу, описанному
для N-оксифталимида [2, 3], не увенчалась успехом: полу-
ченный таким способом продукт оказался тригидратом моно-
натриевой соли малеиновой кислоты [4].
В связи с этим синтез N-оксимальимида осуществлялся в
две стадии через N-оксималеаминовую кислоту, аналогично
описанному для получения других N-замещенных имидов ма-
леиновой кислоты [1].
Нами усовершенствована методика получения N-оксима-
леаминовой кислоты [5] и подобраны условия ее дегидрата-
ции [4].
СХЕМА СИНТЕЗА N -ОКСИМАЛЬИМИДА
О
сн - cf С СН - CON нон
II +H2N-OH--> ||
сн - с£ сн - соон
3 Иаь. 266 113
о
CH - CONHOH — H3O CH - cf
II -----„ li )n-oh
CH - COOH CH - c<
О
Характеристика основного сырья
Гидроксиламин солянокислый, ч.д.а., ГОСТ 5856—51.
Малеиновый ангидрид, ч., ГОСТ 5854—51.
Условия получения
Непосредственно перед началом синтеза готовят чистый
гидроксиламин из солянокислого гидроксилалгина [6].
В литровую круглодонную колбу с мешалкой, обратным
холодильником и капельной воронкой помещают 70 г тща-
тельно измельченного солянокислого гидроксиламина,
0,01—0,02 г фенолфталеина и 100 мл н-бутилового спирта,
перемешивают в течение 10 минут и прикапывают бутилат
натрия с такой скоростью, чтобы раствор оставался бесцвет-
ным. Во избежание загустевания бутилата капельную ворон-
ку снабжают рубашкой, через которую от термостата пропу-
кают горячую воду с температурой 90°. Бутилат натрия по-
лучают растворением 23 г металлического натрия в 300 мл
н-бутилового спирта.
Нейтрализация заканчивается в течение 2—3 часов. Обра-
зовавшийся хлористый натрий отфильтровывают. Из филь-
трата вымораживают кристаллический гидроксиламин при
10—15°, отфильтровывают на воронке Бюхнера, промывают
абсолютным эфиром и хранят в холодильнике.
Более удобный вариант приготовления чистого гидроксил-
амина заключается в замене н-бутилового спирта на метило-
вый. В этом случае не требуется нагревать капельную ворон-
ку при прикапывании метилата натрия. Отфильтрованный
от хлористого натрия метанольный раствор гидроксиламина
может быть непосредственно использован в первой стадии
синтеза.
Получение N-оксималеаминовой кислоты. В литровую круг-
лодонную колбу с мешалкой, обратным холодильником и ка-
пельной воронкой помещают 67 г размельченного малеиново-
го ангидрида и 130 мл диоксана. После растворения малеи-
нового ангидрида прикапывают раствор 22,5 г кристалличе-
ского гидроксиламина и 260 мл дноксана или метанольный
раствор гидроксиламина, получаемый по второму варианту,
перемешивают в течение 1 часа. Белый осадок N-оксималеа-
миновой кислоты отфильтровывают на воронке Бюхнера.
Ш
Выход кислоты равен 60 г; т. пл. 122—128° (с разложени-
ем).
Получение П-оксимальимида. Растворяют 44,3 г ЛАоксима-
леаминовой кислоты в 110 мл диметилформамида и помеща-
ют в круглодонную колбу с мешалкой, охлаждаемую проточ-
ной холодной водой. При сильном перемешивании через боко-
вой тубус в колбу медленно всыпают 57 г пятиокиси фосфо-
ра. Охлаждение колбы необходимо, так как происходит силь-
ное разогревание смеси. После получасового перемешивания
из реакционной вязкой массы при 50—60°/5 мм отгоняется
диметилформамид. Обычно удается отогнать только половину
взятого на реакцию диметилформамида. Оставшуюся в колбе
густую массу выливают в ледяную воду. Выпавший порош-
кообразный продукт отфильтровывают на воронке Бюхнера и
высушивают.
Выход сырого продукта ргИвен 7 г, что составляет 18%,
считая на кислоту. Полученный N'-окоималыимид перекрис-
таллизовывают с активированным углем из метанола до по-
лучения совершенно белого вещества с т. пл. 148—151° (с
разложением).
Примечание. При определении температуры плавления N-окси-
малеаминовой кислоты и N-оксимальимида капилляры с веществом поме-
щают в термостатированную жидкость за 10° до температуры плавления.
ЛИТЕРАТУРА
1. В. С. Иванов, В. К. Смирнова, Т. И. Сидорова,
И. И. М и г у н о в а, А. М. А б р а м о в а, Е. Г. Б а ш и н а, Т. Р. С е п п е и.
Методы получения химических реактивов и препаратов, вып. 15, ЛЁ,
ИРЕА, 1966, стр. 85.
2. W. R. Orndorf, D. S. Pratt. Amer. Chem. J., 47, 89 (1912).
3. M. A. St ol berg, W. A. Mosher, T. Wagner—hnregg.
J. Amer. Chem. Soc., 79, 2615 (1915).
4. В. С. Иванов, В. К. Смирнова, А Е. Семенова, Цао
Юн. Ж. орган, химии, 1. 1705 (1965).
5. G. Е. Endres, J. Epstein. J. Org. Chem, 24, 1497 (1959).
6. Сб. «Неорганический синтез», т. 1, М., ИЛ, 1952, стр. 87.
Поступила в январе ISS6 г.
Ленинградский гос, ун-1
УДК 547.722.07
2-(3-ОКСИПРОПИЛ)-5-(3-ОКСИБУТИЛ)-ФУРАН
А. А. ПОНОМАРЕВ, И. А. МАРКУШИНА
НОСН2 - СН, - СНа —И II- СН,-СН,-СН(ОН)-СН,
xoz
СцН]8О2
М. в. 198,26
Известные из литературы способы получения 2,5-фуранди-
пропионового альдегида и 2,5-бис-(3-оксобутил)-фурана, ос-
нованные на реакции конденсации фура,на с а, р-ненасышен-
ными карбонильными соединениями в присутствии кислых
катализаторов, дают низкие выходы [1].
Нами разработан новый способ получения некоторых ке-
тоальдегидов фуранового ряда, являющихся исходными ве-
ществами для синтеза а,а'-фурандиалканолов-3 [2]. Предла-
гаемый способ заключается в конденсации различных фура-
новых кетоиов с акролеином в присутствии уксусной кисло-
ты. Выходы фурановых кетоно-альдегидов достигают 43—
55%. При гидрировании фурановых кетоно-альдегидов над
медно-хромовым катализатором получаются соответствую-
щие а, а'-фурандиалканолы-З с выходом 70—80%.
СХЕМА СИНТЕЗА 2-(3-ОКСИПРОПИЛ)-5-(3-ОКСИБУТИЛ)-ФУРАНА
хо
СН, - СН, - со - сн,+ сн, = сн - сно
— OHC - CH, - СН2 - Il-СН,—сн,—со — сн,
xoz
116
- СН2-СН»-СН (ОН)-СН,
-> НОСНа - СН2- СН2 -
XOZ
Характеристика основного сырья
1-(а-фурил)-бутанон-3, т. кип. 86—87°/12 мм; —
— 1,4710 [3].
Акролеин, ч., ТУ МХП 2802—54.
Уксусная кислота ледяная, х.ч., ГОСТ 61—51.
Условия получения
Синтез 2-(3-оксопропил)-5-(3-оксобутил)-фурана. Смесь
53 мл (54,9 г, 0,4 М) 1-(а-фурил)-бутанона-3, 28 мл (23,5 г,
0,42 М) свежеперегнанного акролеина, стабилизированно-
го 1,6 г гидрохинона, 50 мл воды и 5 мл ледяной уксусной
кислоты помещают в автоклав емкостью 150 мл и нагрева-
ют при перемешивании в течение 4,5 часов при температуре
130°. После охлаждения реакционную смесь выливают в де-
лительную воронку, отделяют масло, а водный слой экстра-
гируют 2—3 раза небольшими порциями эфира (см. приме-
чание 1). Масло дважды промывают водой, добавляют к не-
му эфирные вытяжки и сушат над прокаленным сульфатом
магния. На водяной бане отгоняют эфир, остаток перегоняют
в вакууме, собирая фракцию с т. кип. 134—13872 мм;
«20—1,4922; rf420—1,090.
Выход 2-(3-оксопропил)-5-(3-оксобутил)-фурана равен
35 г, что составляет 43% от теоретического.
Получение 2-(3-оксипропил)-5-(3-оксибутил)-фурана. По-
мещают 120 г (0,62 М) 2-(3-оксопропил)-5-(3-оксобутил) -
фурана, 150 мл безводного спирта и 15 г медно-хромового
катализатора (см. примечание 2) в стальной вращающийся
автоклав емкостью 610 мл. Начальное давление водорода
110 атмосфер. Гидрирование проводят при температуре 120°
и заканчивают по поглощении 29,4 л водорода- По окончании
гидрирования катализат переносят в колбу и дают катализа-
тору осесть. Затем катализатор отфильтровывают, спирт от-
гоняют на водяной бане и остаток перегоняют в вакууме, со-
бирая фракцию с т. кип. 156—157°/1 мм; 1,4986;
1,068.
Выход 2-(3-оксипропил)-5-(3-оксибутил)-фурана равен
98, что составляет 80% от теоретического (см. примеча-
ние 3),
Примечания:
1. Экстрагирование проводят в вытяжном шкафу.
2, Медио-хромовый катализатор приготовляют по прописн [4].
117
3. Аналогичным способом получены:
2-(3-ок.сопропил)-5-(3-фенил-3-оксопропил)фуран (т. пл. 53—59° из
петролейного эфира) с выходом 56%;
2-(3-оксипропил)-5-(3-фенил-3-оксипропил)-фуран (т. кип 210—
21271 мм); «р —1,5464; <А25 — 1,115) с выходом 69% [3].
ЛИТЕРАТУРА
1. С. М. Шерл ин. А. Я. Берлин, Г. А. Серебренникова,
Ф. Е. Рабинович. Ж. общ. хим., 8, 7 (1938).
2. А. А. Пономарев, И. А. Маркушина. Ж. общ. химии, 33,
3955 (1963).
3. А. А. Пономарев. Синтезы и реакции фурановых веществ, Са-
ратов, СГУ, 1960, стр. 102.
4. Новые методы препаративной органической химии. М.—Л., ИЛ,
1950.
Поступила в январе I9S6 г. Саратовский гос. ун-т
УДК 547.831.
8-ОКСИХИНОЛИН-(2-АЗО-2)-1'-ОКСИНАФТАЛИН
2-[(1-Окси-2-нафтил)азо]-8-хинолинол
И. А. КРАСАВИН, В. М. ДЗИОМКО, Н. И. ДУДАРЕВА, Ю. П. РАДИН
М. в. 315,34
В литературе 8-оксихинолин-(2-азо-2/)-Г-оксинафталин не
описан. Он получен нами при взаимодействии 8-окси-2-гидра-
зинохинолина с 1,2-нафтохиноном в среде ледяной уксусной
кислоты.
Чистое вещество кристаллизуется из этанола в блестящих
темно-красных иглах с т. пл. 215—215,5°. Оно растворимо в
ацетоне, хлороформе и уксусной кислоте, при нагревании
растворяется также в этаноле, бензоле и немного — в гепта-
не, но нерастворимо в кипящей воде. С щелочами образует
хорошо растворимые в воде соли фиолетового цвета.
8-Оксихинолин-(2-азо-2')-Г-оксинафталин, в молекуле ко-
торого сочетаются хелатообразующие группировки 8-оксихи-
нолина и 1-(2-пиридилазо)-2-нафтола (ПАН’а), образует эк-
страгирующиеся комплексы с ионами многих металлов.
119
СХЕМА СИНТЕЗА
8-ОКСИХИ НОЛ И Н-(2-АЗО-2')-1'-ОКСИ НАФТ АЛ ИНА
'W'\n^\nHNH2
он
Условия получения
В широкогорлой конической колбе емкостью 250 мл с ме-
шалкой растворяют 4,38 г (0,025 М) 8-окси-2-гидразинохино-
лина (см. примечание 1) в 50 мл ледяной уксусной кислоты
при непродолжительном подогревании теплой водой. Раствор
охлаждают до 20° (см. примечание 2) и к нему при интенсив-
ном размешивании прибавляют за 15—20 минут 3,95 г
(0,025 М) измельченного 1,2-нафтохинона хорошего качества
(см. примечания 3 и 4) небольшими порциями с такой ско-
ростью, чтобы он успевал растворяться. Реакционная масса
окрашивается в красный цвет и вскоре начинает выпадать
осадок азосоединения. Когда добавление хинона окончено,
частицы его стряхивают со стенок колбы и смывают 10 мл
ледяной уксусной кислоты (см. примечание 5). Образовав-
шуюся суспензию перемешивают в течение 1 часа при ком-
натной температуре, выдерживают 1 час на водяной бане при
50° и охлаждают льдом.
Затем реакционную массу оставляют при комнатной тем-
пературе, чтобы замерзшая уксусная кислота оттаяла. Оса-
док отсасывают, смывая его из колбы фильтратом, отжимают
досуха и хорошо промывают водой (см. примечание 6). Ве-
щество вместе с фильтром переносят в 2-литровый стакан,
добавляют 1200 мл нагретого до 70—80° 0,5 н. раствора ед
кого кали (х.ч.), используя часть его для ополаскивания во
роики, и размешивают до полного растворения осадка.
120
Темно-фиолетовый раствор фильтруют и фильтрат под-
кисляют 10%-ной уксусной кислотой до pH 5—6, причем вы-
деляются красные хлопья. Чтобы получить хорошо фильтру-
ющийся продукт, нагревают стакан с суспензией 1 —1,5 часа
на кипящей водяной бане, затем светло-красный осадок от-
сасывают, многократно промывают на фильтре горячей во-
дой и высушивают в вакуум-эксикаторе.
Выход азосоединения 3,1—3,5 г (39,3—44,4% от теорети-
ческого); т. пл. 211—211,5°.
Для очистки вещество растворяют в 1200—1500 мл кипя-
щего этанола, раствор фильтруют и охлаждают до выделе-
ния игольчатых кристаллов, а затем выдерживают 2—3 часа
в бане с холодильной смесью (лед с солью).
Кристаллы отсасывают, промывают небольшим количест-
вом этанола, высушивают и получают 2,6—2,9 г (33—36,8%)
темно-красных игл с т. пл. 214,5—215°; повторная перекри-
сталлизация дает вещество с т. пл. 215—215,5° (ом. примеча-
ние 7).
Примечания:
1. Применялся перекристаллизованный из бензола 8-окси-2-гидрази-
нохинолин с т. пл. 179—180“, полученный по описанному нами ранее ме-
тоду [1].
2. При более низкой температуре вещество может выпасть в осадок.
3. Применялся 1,2-нафтохннон с т. пл. 144—146“ (с разл.), получен-
ный по методу [2] и тщательно промытый водой для удаления ионов же-
леза. Следует обратить особое внимание на качество применяемого хи-
нона, так как от пего зависит успех синтеза.
4. Хинон нельзя растирать в ступке, так как он сильно электризует-
ся. Поэтому его следует раскрошить шпателем и добавлять небольшими
кусочками.
5. Общий объем уксусной кислоты должен быть равен 60 мл. Если
количество кислоты уменьшить, то выход продукта повышается, но очи-
стка его становится затруднительной.
6. Для промывки вещества и приготовления растворов следует при-
менять дистиллированную воду.
7. Чистый образец в виде темно-красных игл с т. пл, 215—215,5° был
приготовлен перекристаллизацией из этанола и проанализирован.
Найдено, %: С—71,99; 72,04; Н—4,10; 3,94; N—13,41; 12,87.
C)eHiaN,O2. Вычислено, %: С-72,37; Н—4,16; N-13,33.
ЛИТЕРАТУРА
1. И. А. Красавин, В. М. Дзиомко, Н. И. Мирошкина.
Методы получения химических реактивов и препаратов, вып. 14, М.,
ИРЕА, 1966, стр. 86.
2. Синтезы органических препаратов, сб. 2, М„ ИЛ, 1949, стр. 353.
Поступила в феврале I9G7 г.
ИРЕА
УДК 547.495.2'123.07
СЕЛЕНОМОЧЕВИНА
Селенокарбамид
А. Б. ЛУНДИН
HSN - С - NH,
Se
CH4NaSe М. в. 123,01
Селеномочевина применяется в качестве исходного веще-
ства при синтезе ряда селенсодержащих гетероциклических
соединений (например, производных 2-селенобарбитуровой
кислоты [1] и 2-аминоселеназолов [2]).
Было найдено [3], что селеномочевина образуется при
взаимодействии газообразного селеноводорода с растворен-
ным в эфире цианамидом в присутствии малых количеств
аммиака. Существуют модификации этого метода, основан-
ные, например, на взаимодействии селеноводорода с водным
раствором цианамида [4].
В качестве исходного вещества для синтеза везде исполь-
зуется цианамид; однако, его получение из доступных реак-
тивов связано со значительными трудностями из-за неста-
бильности цианамида и его склонности к полимеризации с
образованием меламина. Поэтому нами был применен метод
получения селеномочевины без промежуточно го выделения
цианамида. Главным требованием при этом было получение
чистых растворов цианамида, лишенных примесей исходных
веществ. Поэтому из описанных в литературе способов полу-
чения цианамида [5—8] была выбрана методика, основанная
на взаимодействии окиси ртути и спиртового раствора тно-
мочевины [6] (см. примечание 1). Обработка полученного
спиртового раствора цианамида газообразным селеноводоро-
дом в аммиачной или щелочной среде приводит к образова-
нию селеномочевины.
122
СХЕМА СИНТЕЗА СЕЛЕНОМОЧЕВИНЫ
H3N - С - NH, + HgO — H2N - С N + HgS -ф Н2О
HSN - С N + H2Se — H2N - С - NH2
I,
Se
Характеристика исходного сырья
Тиомочевина, ч., ГОСТ 6344—52.
Окись ртути желтая, ч., ГОСТ 5230—50.
Селем, ч., ГОСТ 5455—50.
Спирт этиловый ректифицированный, ГОСТ 8314—57.
Условия получения
Синтез спиртового раствора цианамида. В толстостенный
стеклянный сосуд емкостью 1 л заливают 500 мл этилово-
го спирта, после чего загружают 76 а (1 Л1) тиомочевины,
238 г (1,1 At) окиси ртути и керамические шарики диамет-
ром 10—15 мм (см. примечание 2). Сосуд интенсивно встря-
хивают в течение 2 часом, после чего жидкость отфильтровы-
вают от твердой фазы (см. примечание 3). Прозрачная, слег-
ка желтоватая жидкость представляет собой раствор циана-
мида в спирте.
Получение селеноводорода. Селеноводород получают на-
греванием смеси минерального масла с порошкообразным
селеном.
В круглодонную колбу из термостойкого стекла емкостью
2 л, снабженную обратным холодильником и отстойником-
расширителем после него (см. примечание 4), помещают
120—150 г порошкообразного селена и 1 л минерального ма-
сла. При нагреве смеси до 200—250° получают устойчивый и
равномерный ток селеноводорода в течение 4—5 часов.
Получение селеномочевины и ее выделение. Спиртовый
раствор цианамида заливают в колбу Эрленмейера емкостью
I л. После добавки небольшого количества твердой щелочи
(около 0,1 г), применяемой в качестве катализатора, осуще-
ствляют барботаж газообразного селеноводорода через рас-
твор в течение 5 часов (см. примечание 5). Раствор быстро
мутнеет и окрашивается в красный цвет, а затем, видимо,
вследствие коагуляции грубодисперсной взвеси селена, вновь
становится прозрачным, сохраняя интенсивную красную окра-
ску. После 12-часового стояния раствор кипятят в колбе с
обратным холодильником до полного прекращения выделе-
123
ния газа. Поглощение газа осуществляют в склянке Тищен-
ко, заполненной 25%-ной щелочью. После очистки активиро-
ванным углем, фильтрации и охлаждения раствора часть се-
леномочевины выкристаллизовывается в виде хорошо обра-
зованных игл. Маточник упаривают и из него повторно про-
изводят кристаллизацию продукта. Обе порции селеномоче-
вины смешивают и очищают перекристализацией из этило-
вого спирта.
Выход сел еномочевины равен 25 г, что составляет 20% от
теоретического, считая на тиомочевину (см. примечание 6);
т. пл. 202—204° (разл.); 200° (разл.) [9].
Обрабатывая маточник после перекристаллизации, мож-
но дополнительно получить около 15 г селеномочевины.
Общий выход продукта составляет около 35%.
Примечания:
1. Нами было найдено, что введение в зону реакции массивных ша-
риков для постоянного обновления покрываемой сульфидом ртути по-
верхности окиси ртути позволяет значительно ускорить процесс отщеп-
ления сероводорода от тиомочевины и избежать применения больших
избытков окиси ртути.
2. Керамические шарики вводят для постоянного обновления покры-
ваемой сульфидом ртути поверхности окиси ртути. Без них даже при
большом избытке окиси ртути и многочасовом встряхивании провести
процесс количественно не удается.
3. Прежде чем начать обработку спиртового раствора цианамида се-
леноводородом, необходимо убедиться в отсутствии непрореагировавшей
тиомочевины, для чего в прозрачный фильтрат после отделения твердой
фазы, состоящей из сульфида ртути и ее окиси, добавляют небольшое
количество свежей окиси ртути. Если ее окраска после встряхивания ос-
тается неизменной, то можно считать, что непрореагнровавшая тиомоче-
вина в растворе отсутствует.
4. От генератора селеноводорода к барботеру газ подается через
обратный холодильник и отстойник-расширитель, в качестве которого мо-
жет быть применена колба Вюрца. Обратный холодильник вводят для
очистки газа от продуктов пиролиза минерального масла, образование
которых не исключено.
Следует помнить, что селеноводород очень ядовит. Поэтому вся ус-
тановка перед пуском обязательно должна быть проверена на герметич-
ность. Поглощение непрореагировавшего селеноводорода осуществляют в
двух последовательно соединенных склянках Тищенко, из которых вторая
служит индикатором полноты поглощения газа в первой склянке (уже
первые порции газа окрашивают щелочь в желтоватый цвет, видимо,
из-за разложения селеноводорода с образованием золя элементарного се-
лена).
5. Контроль за поглощением газа осуществляют периодическим
взвешиванием колбы с раствором цианамида. Барботаж селеноводорода
производится до достижения колбой постоянного веса.
В связи с тем, что для взвешивания колбы необходимо неоднократ-
но выключать обогрев генератора селеноводорода, что связано с падени-
ем давления в ием ниже атмосферного, необходимо между отстойником
и колбой для поглощения установить гидрозатвор, обеспечивающий под-
сос воздуха в систему при разряжении, еще недостаточном для засасы-
вания реакционной смеси через отстойник и обратный холодильник в
колбу для получения селеноводорода.
124
6. Окись ртути и селен из пепрореагировавшего селеноводорода мо-
гут быть почти полностью регенерированы. Первая нз сульфида ртути,
второй — из щелочного раствора, слитого из склянок Тищенко.
ЛИТЕРАТУРА
1. Н. О. Mauther, Ed. М. Clayton. J. Amer. Chem. Soc., 81, 6270
(1959).
2. H. J. Backer, H. Bos. Rec. trav. chim., 62, 580 — 4 (1943).
3. Verneuil., A. ch., 6, 9, 294.
4. Герм. пат. 607382 (1934).
5. «Препаративная органическая химия», перевод с польского, под
общ. ред. Н. С. Вульфсона, М., Госхимиздат, 1959, стр. 549.
6. Baumann. Вег., 6, 1376 (1873).
7. О. К э й д з и р о, И. X и д э о, О. С у с у м у. Японский пат., 957,
(1959).
8. О. Liebknecht. Angew. Cheffl., 45, 584 — 585. 10/9 (1932).
9. «Справочник химика», т. 2, 720, Л.—М., Госхимиздат (1951).
Поступила в июне 1965 г. Уральский политехнический
институт
УДК [547.7224-547.742:547.07
1-(а-ТЕТРАГИДРОФУРИЛ)-3-АМИНОБУТАН
И 1-(5'-МЕТИЛ-2-ПИРРОЛИДИЛ)ПРОПАНОЛ-3
/1. Л. ПОНОМАРЕВ, А. П. КРИВЕНЬКО, Af. В. НОРИЦИНА
•—СН4- СН, - СН - сн,
о7 I
NH,
1-СН2~СНг-СН,ОН
CHS N
н
C8Hj7ON М. в 143,‘23
Ранее были предложены два способа получения 1-(а-тет-
рагидрофурил)-3-аминобута1на. Первый — заключается в гид-
рировании N-ацетильного производного 1-(а-фурил)-3-амино-
бутана под давлением в присутствии никеля Ренея и после-
дующем омылении образующегося 1- (а-тетрагидрофурил)-М-
ацетил-З-аминобутана до свободного амина. Выход составля-
ет 50% ’в расчете на исходный фурановый амин. Недостаток
этого способа заключается в трудоемкости и длительности
стадии омыления [1].
Второй способ — прямое гидрирование 1-(а-фурил)-3-
аминобутана в среде абсолютного этилового спирта над ни-
келем Ренея при температуре 150°. Выход равен 63%, причем
образуются значительные количества смолообраэных продук-
тов и веществ неустановленного строения {2].
В препаративном отношении более удобен способ гидро-
генизации 1-(а-фурил)-3-аминобутана в водном солянокис-
лом растворе. В этих условиях в результате интрамолекуляр-
126
ной циклизации продуктов расщепления фуранового цикла и
их гидрогенизации наряду с 1-(а-тетрагидрофурил)-3-амино-
бутаном образуется также Ь^'-метил-З'-пирролидил) -пропа-
нол-3, представляющий самостоятельный интерес.
Способ применим для получения и других тетрагидрофу-
рановых аминов и а-пирролидилалканолов-3 (3—5].
Ь(а-Тетрагидрофурил)-3-аминобутан и его гомологи
представляют интерес как промежуточные продукты при по-
лучении соответствующих пирролизидинов путем интрамо-
лекулярной дегидратации [1, 6].
СХЕМА СИНТЕЗА
1-(а-ТЕТРАГИДРОФУРИЛ)-3-АМИНОБУТАНА И
1(-5'-МЕТИЛ-2'-ПИРРОЛИДИЛ) ПРОПАНОЛА-3
2Н,
СН, — СН2 —СН — СН,-------►
xoz I н+ нон
NH,
I l-CHj-CHo-CH - сн, +
"О7 |
nh2
I- СН, - СН, - СН2ОН
CH.^N
н
Характеристика основного сырья
1-(а-фурил)-3-аминобутан, получен по [1]; т. кип. 85° при
18 мм; п™ —1,4760; d420 — 0,9640.
Водород, ГОСТ 3022—45.
Соляная кислота, х.ч., ГОСТ 3118—46, 10%-ный раствор.
Никель Ренея, приготовлен по [7].
Условия получения
В стальной вращающийся автоклав емкостью 250 мл за-
гружают раствор 28 г (0,2 М) 1-(а-фурил)-3-аминобутана в
70 мл 10%-ной соляной кислоты и 2,8 г никеля Ренея, pH
раствора 4—5 (см. примечание 1). Начальное давление водо-
рода равно 100 атмосферам, температура опыта 100°. Реак-
127
ция заканчивается через 8 часов, после поглощения 10 л
(0,4 М) водорода, рассчитанного для насыщения двух двой-
ных связей. По охлаждении автоклав разгружают, гидроге-
низат освобождают от катализатора фильтрованием и нейт-
рализуют избытком твердого едкого кали. Выделившееся ма-
сло отделяют, водный слой экстрагируют эфиром три раза
порциями по 30 мл. Эфирные вытяжки соединяют с маслом
и сушат твердым едким кали. Эфир отгоняют, а остаток пе-
регоняют в вакууме из колбы с елочным дефлегматором вы-
сотой 20 см. При температуре 96—100718 мм собирают 1-(а-
тетрагидрофурил)-3-аминобутан, после чего давление осто-
рожно понижают до 5 мм и при 113—117° перегоняют 1-(5'-
метил-2'-пирролидил)-пропанол-3.
Выход 1-(а-тетрагидрофурил)-3-аминобутана равен 15 г,
что составляет 60% от теоретического; т. кип. 95—96718мм
(после второй перегонки); Яд —1,4570; af420—0,9253; т. кип.
ацетильного производного 139—14172 мм; —1,4741;
d<?° — 1,0086.
Выход 1-(5/-метил-2'-пироллидил) пропанола-3 равен 4 г,
что составляет 15% от теоретического; т. кип. 113—11475>Ы1
(после второй перегонки); при стоянии вещество застывает в
бесцветную аморфную массу с т. пл. 20—21° (см. примеча-
ние 2).
Данный способ применим для получения и других тетра-
гидрофурановых аминов и а-пирролидилалканолов-3 [3, 5]
(см. примечание 3).
Примечания:
1. Растворение сопровождается выделением тепла, поэтому сосуд с
амином помещают в баню с холодной водой, а затем осторожно при пе-
ремешивании приливают соляную кислоту. Последние капли кислоты при-
бавляют под контролем универсальной индикаторной бумажки, так как
при этом резко изменяется pH раствора.
2. Т. пл. определялась на шарике термометра [8].
3. Аналогичным путем получают:
из 1-(а-фурил)-1-этил-3-аминобутана —1-( а-тетрагидрофурил)-1-этил-3-
аминобутан с выходом 42%; т. кип. 109718 леи; Ид 1,4630 и 1-(5'-метил-3-
этил-2-пирролидил)пропанол-3 с выходом 36%; т. кип. 136—137712 мм;
т, пл. 46—48°;
из 1-(а-фурил)-1-изопропил-3-аминобутапа — 1-(а-тетрагидрофурил)-
1-изопропил-З-аминобутан. с выходом 27%; т. кип. 117—118714 мм;
Ид — 1,4635 и 1-(5'-метил-3'-изопропил-2'-пирролидил)пропанол-3 с выхо-
дом 49%; т. кип. 132—13478 мм; т. пл. 23—257
ЛИТЕРАТУРА
1. А. А. Пономарев, Н. П. Масленникова. Н. В. А л а к и-
на, А. П. Кривенько. Докл. АН СССР, 131, 355 (1960).
2. А. А. Пономарев, Н. П. Масленникова, А. П. Кри-
венько. Ж. общ. химии, 31, 9581 (1961).
3. А. А. Пономарев, А. П. Кривенько, М. В. Но рицин а.
Ж. общ. химии, 33, 1778 (1963).
128
4. А. А. Пономарев, M. В. Норицина А. П. Кривенько.
Докл. АН СССР, 156, 1, 102 (1964).
5. А. А. Пономарев, А. П. Кривенько, М. В. Норицина.
Авт. свнд. 158277; Бюлл. нзобр., № 21 (1963).
6. А. А. Пономарев, И. М. Скворцов. Ж. общ. химии, 32, 97
(1962).
7. Сб. «Синтезы органических препаратов», вып. 3. М„ ИЛ, 1952,
стр. 338.
8. С. В ай бель. Идентификация органических соединений, 1957,
стр. 314.
Поступила в январе 1966 г. Саратовский гос. ун-т
9 Зак. 266
УДК [547.722 + 547.642:547.07
1-(а-ТЕТРАГИДРОФУРИЛ)-3-МЕТИЛ ПЕНТАНОЛ-3
И 2-МЕТИЛ-2-ЭТИЛ-1,6-ДИОКСАПИРО-(4,4)-НОНАН
А. А. ПОНОМАРЕВ, 3. В. ТИЛЬ
ОН
I
СН^СНа-С-С2Н6
сн8
С10Н20О2 М. в. 172,26
С10Н18О3 М. в. 170,25
Третичные фурановые спирты, содержащие гидроксил при
третьем углеродном атоме боковой цепи (у-фурилалканолы),
легко получаются при взаимодействии предельных фурано-
вых кетонов с гриньяровскими реагентами [1—3].
Гидрирование третичных фурановых спиртов осуществля-
ется под давлением 100—140 атмосфер, при 120—140° в при-
сутствии никеля Ренея или никеля на кизельгуре. При этом,
так же как и в случае первичных и вторичных спиртов [4],
наряду с тетрагидрофурановыми спиртами, образуются гомо-
логи 1,6-диоксаспиро- (4,4) -нонана. Механизм этой реакции из-
ложен в работе [5].
При использовании никеля на кизельгуре выходы спира-
на выше, чем в присутствии никеля Ренея, а наличие замес-
тителей при первом от цикла атоме углерода боковой цепи
способствует реакции циклизации {6].
Другими способами 1-(а-тетрагидрофурил)-3-метилпента-
нол-3 и 2-метил-2-этил-1,6-диоксаспиро-(4,4)-нонан получены
не были.
130
СХЕМА СИНТЕЗА
1-(а-ТЕТРАГИДР0ФУРИЛ)-3-МЕТИЛ ПЕНТАНОЛА-3
и 2-МЕТИЛ-2-ЭТИЛ-1,6-ДИОКСАСПИРО-(4,4)-НОНАНА
__ ОН
I! I н,
i СН2—CHg—С—С8Н5 —
ХОZ 1
CHS
---- ОН |---1 /СН3
I /°-|\
' '-сн2сн2-с-с8н6 + L I хс2н5
О | о -------
сн3
Характеристика основного сырья
1-(а-Фурил)-З-метилпентанол-З, т. кип. 81—8372 мм;
Яд—1,4787; — 0,9942 (примечание 1).
Никель Ренея [7].
Условия получения
Гидрирование проводят во вращающемся стальном авто-
клаве емкостью 150 мл. Заданную температуру поддержи-
вают постоянной с помощью электронного регулирующего
милливольтметра типа ЭПВ-01.
В автоклав загружают 35 г (0,21 Л1) 1-(а-фурил)-3-метил-
пентанола-3, 35 мл абсолютного этилового спирта и 3,5 г ни-
келя Ренея. Начальное давление водорода равно 60—100 ат-
мосфер, температура 120°. Гидрирование обычно заканчива-
ется после поглощения около 1,5 моля водорода на 1 моль
вещества. По окончании гидрирования гидротенизат отделя-
ют от катализатора фильтрованием, растворитель отгоняют
на водяной бане, а остаток перегоняют под вакуумом. Сна-
чала собирают широкую фракцию с т. кип. 72—112710 мм,
а затем при температуре 114—1157Ю мм получают 23,4 г
(65,3% от теоретического выхода) 1-(а-тетрагидрофурил)-
З-метиЛ'пентанола-З.
Третичный спирт — жидкость с Яд—1,4608, <Л20—
0,9629, хорошо растворяется в этиловом спирте, эфире, бен-
золе; не растворяется в воде.
При повторной перегонке первой фракции получают 6,2 г
(17,5% от теоретического выхода) 2-метил-2-этил-1,6-диокса-
спиро-(4,4)-нонана (см. примечание 2). По внешнему виду это
бесцветная подвижная жидкость с т. кип. 102—104745 мм;
«о —1,4443; <Л20 — 0,9614, хорошо растворяется в обычных
9* 131
органических растворителях, не растворяется в воде (см. при-
мечание 3).
Примечания:
1. 1 (а-фурил)-З-метилпентанол-З получают из 1-(а-фурил) бутанона-3
и этилма! нийбромида с выходом 72% [1, 2].
2. Выход спирана при гидрировании 1-(а-фурил)-З-метилпептаиола-З
в аналогичных условиях в присутствии никеля на кизельгуре может до-
стигать 28—30%, но при этом соответственно понижается выход тетра-
гидрофуранового спирта.
3. Аналогичным путем при гидрировании 1-(а-фурил)-3,5-диметил-
гексанола-3, 1- (а-фурил)-З-метилгсптанола-З, 1-(а-фурил)-3-бутаиола-3,
1-(а-фурил)-3, 4, 4-триметилпентанола-З, 3-(а-фурил)-5-метилгептанола-5,
3-(а-фурил)-2,5-диметилгептанола-5 и других третичных фурановых спир-
тов получаются соответствующие третичные тетрагидрофурановые спирты
п гомологи 1,6-диоксаспиро-(4,4)-ионана [2, 6].
ЛИТЕРАТУРА
1. J. Kaslwagl. Bull. Chem. Soc. Japan. 2. 310 (1927).
2. А. А. П о и о м a p e в, 3. В. Тиль, А. Д. П e ш e x о н о в a,
В. П. Решетов. Ж. общ. химии, 27, 1369 (1957).
3. А. А. Пономарев, 3. В. Тиль. Ж. общ. химии, 27, 1075 (1957).
4. 3. В. Тиль, И. А. Маркушина, К. С а п у н а р, А. А. Пон о-
марев. Ж. общ. химии, 27, ПО (1957).
5. А. А. Пономарев, В. А. Афанасьев, II. И. Курочкин.
Ж. общ. химии, 23, 1426 (1953).
6. А. А. Пономарев, В. А. Седавкина, 3. В. Тиль. Ж- общ.
химии, 33, 1303 (1963).
7. Сб. «Синтезы органических препаратов», вып. 3, М., ИЛ, 1952,
стр. 338.
Поступила в январе 1966 г.
Саратовский гос. ун-т
УДК 547.722.07
N-ТЕТРАГИДРОФУРФУРИЛЭТИЛЕНДИАМИН
А. А. ПОНОМАРЕВ. И. М. СКВОРЦОВ
I '
I J-ch2-nh-ch2-ch2-nh2
о
C7HieN2O М. в. 144,21
Диамины тетрагидрофуранового ряда почти не изучены.
Между тем они представляют 'интерес для тонкого органиче-
ского синтеза, в частности, они могут быть использованы для
получения 7-азаоктагидропирроколинов (1J.
Простым и доступным способом приготовления N-тетра-
гидрофурфурилэтилендиаминов является каталитическое гид-
рирование N'-фурфурилэтилендиаминов, обеспечивающее по-
лучение конечных продуктов с выходами около 80%.
Другим менее удобным путем синтеза N-тет.рагидрофурфу-
рилэтилсндиамина является конденсация тетрагидрофурфу-
рил амина с этиленимином 1.2], но выход вещества по этому
методу невысокий.
СХЕМА СИНТЕЗА N-ТЕТРАГИДРОФУРФУРИЛЭТИЛЕНДИАМИНА
1П!
\ J1—CHS—NH—СН2—СН,—Nil, —2—►
\q/ ‘ Nl
' J —СН2—NH—СН2—СН.—NH,
Oz
Характеристика основного сырья
N'-Фурфурилэтилендиамин (см. стр. 174 «N-Фурфурилэти-
лендмамин» в данном сборнике).
Водород электролитический.
133
Условия получения
Во вращающийся автоклав емкостью 150 мл загружают
раствор 25 г (0,178 М) N'-фурфурилэтилендиамина в 63 мл
абсолютного спирта и 5 г никеля Ренея [3]. Начальное дав-
ление водорода равно 100 атмосферам. Гидрирование прово-
дят при 150° до прекращения поглощения водорода. Затем
автоклав разгружают, катализат отфильтровывают от нике-
ля, спирт отгоняют, остаток перегоняют в вакууме из колбы
с колонкой Видмера, отбирая фракцию с т. кип. 106—1077
10 мм.
Выход N-тетрагидрофурфурилэталендиамина равен 21,7 г,
что составляет 84,4% от теоретического. Вещество раствори-
мо в воде, спирте и эфире; п2$ —1,4750; с^20— 0,9888 (см.
примечание).
Примечание.
Аналогичным путем из М-(а-метнлфурфурнл)-этилсндиамнна получа-
ют N- (а-метилтетрагидрофурфурил)-этилендиамин с выходом 79,1% [11;
т. кип. 105—106710 мм; Лд — 1,4662; сб20—0,9524.
Л И Т Е Р А Т У Р А
1. А. А. Пономарев И. М. Скворцов. Докл. АН СССР, 148,
860 (1963).
2. А. А. Пономарев, И. М. Скворцов, Н. П. Масленни-
кова. Ж. общ. химии, 33, ИЗО (1963).
3. Сб. «Синтезы органических препаратов», вып. 3, М., ИЛ, 1952,
стр. 338.
Поступила в январе 1966 г.
Саратовский гос. ун-т
УДК 547.233.4'213.07
N, N, N'; 1\’-ТЕТРАМЕТИЛ-1.3-ПРОПЛНДИАМИН
в. м. дзиомко, 3. С. СИДЕНКО
(CH3)2N-(CH2)3-N(CH3)2
C7HisN2 М. в. 130,23
По литературным данным, N, N, N', ЬГ-тетраметил-1,3-
пропандиамин может быть получен многочасовым нагревани-
ем 1,3-дибромпропана со спиртовым раствором диметилами-
на [1, 2] (без указания выхода), перегонкой гидроокиси N, N,
N, N', N', №-гексаметил-1,3-пропандиаммония [3], перегон-
кой дихлорида 1, 1, 5, 5,-тетраметил-1, 5-диазония циклоок-
тана со щелочью [1] (с незначительными выходами), гидри-
рованием N, N, N', N'-тетраметил- 1,3-пропандиамина в при-
сутствии платинового катализатора с выходом 69% [4], а так-
же в качестве продукта расщепления при нагревании четвер-
тичного производного спермина [5] и при доказательстве
строения алкалоидов [6].
Однако все перечисленные выше методы синтеза N, N,
N', N'-тетраметил-1,3-пропандиамина являются довольно
трудоемкими и, по-видимому, не обеспечивают достаточно
устойчивого выхода продукта.
Известная же реакция Эшвайлера—Кларка [7, 8], которая
специально применяется для получения N-метилированпых
аминов (или диаминов) путем взаимодействия первичных
или вторичных аминов (или диаминов) с формальдегидом и
муравьиной кислотой, до сих пор не была применена к 1,3-
пропандиамину.
Нами осуществлен синтез N, N, N', N'-тетраметил-!,3-про-
пандиаммна метилированием по методу Эшвайлера-Кларка.
СХЕМА СИ HTE3AN,N,N'N'-TETPAMETHJI-1,3-ПРОПАНД КАМИНА
NHs-(CH2)3-NH2 (CH3)3N—(СН2)3—N (СН3)а
135
Характеристика основного сырья
1,3-Пропандиамин синтезирован по прописи [9}.
Формальдегид, 40%-ный раствор, ГОСТ 1625—64.
Муравьиная кислота, ч., 85%-ная, ГОСТ 5848—60.
Едкий натр, ч., ГОСТ 4328—48.
Соляная кислота, ч., ГОСТ 3118—46.
Едкое кали, ч., ГОСТ 4203—48.
Эфир этиловый, ГОСТ 6265—52.
Условия получения
В трехгорлую колбу, снабженную механической мешал-
кой, капельной воронкой и термометром, загружают 37 г
(0,5 А4) 1,3-пропандиамина и медленно ’при эффективном пе-
ремешивании приливают из капельной воронки 240 мл 85 % -
ной муравьиной кислоты, поддерживая в колбе температуру
минус 5° смесью льда с солью. После добавления всего ко-
личества муравьиной кислоты реакционную массу нагрева-
ют до 60° и в течение 30 минут приливают 165 мл 40%-ного
формальдегида. С момента выделения углекислого газа (по-
явление пузырьков) раствор нагревают на водяной бане 18
часов.
Затем содержимое колбы охлаждают, добавляют 200 мл
концентрированной соляной 'кислоты и избыточный объем во-
ды (320 мл) отгоняют. Остаток охлаждают льдом, прибавля-
ют 200 мл 50 %-кого раствора едкого натра и экстрагируют
эфиром. Эфирный экстракт сушат едким кали, эфир отгоня-
ют, а остаток перегоняют в вакууме и получают 32 г N, N,
N', N'-тетраметил- 1,3-пропандиамина, что составляет 49% от
теоретического выхода; т. кип. 37°/9 мм; т. пл. дипикрата 207°.
По литературным данным, т. кип. вещества 143—147°/
755 мм (1]; ; 1447760 мм [2]; т. пл. дипикрата 205 fl]; 207° [4].
ЛИТЕРАТУРА
1. L. К п о г г. Р. Roth. Вег., 39, 1428 (1906).
2. Н. Clarke. J. Chem. Soc., 103. 1699 (1913).
3. J. V. Braun. Ann.. 386, 295 (1911).
4. C. Mannich. K. Handke, K. Roth. Ber,, 69, 2112 (1936).
5, H. W. Dudley, O. Rosenheim, W. W. S t a r 1 i n g. Biochem.
J., 20, 1086. (1926).
6 К Wiesner, D. M. Mac —Donald. C. Bankiewlez. J.
Amer. Chem. Soc. 75. 6348 (1953).
7. W. Eschwei 1 er. Ber., 38, 880 (1905).
8. H. Clarke. H. Gillespie, S. Weisshaus. J. Amer. Chem.
Soc. 55, 4571 (1933).
9. A. H. К ост. Вест. МГУ, № 2. 141 (1947).
Поступила в июле 19^'6 г.
136
ИРЕА
УДК 547.518'122.1.07
ТИАЦИКЛООКТАНОН-5
А. А. ПОНОМАРЕВ, И. С. МОНАХОВА
C,H12OS М. в. 144,22
Тиациклооктанон-5 впервые получен циклизацией по Дик-
ману диэтилового эфира у, у/-т'иа-бис-масляной кислоты с по-
следующим гидролизом и декарбоксилированием; выход сы-
рого продукта 31—45% 111-
Синтез тиациклооктанона-5 осуществлен нами из 1,6-диок-
саспиро-(4,4)-нонана. Соединения группы 1,6-диоксаспиро-(4,
4)-нонана [2, 3] стали доступными после разработки нового
способа их синтеза посредством электролитического меток-
силирования у-фурилалканолов с последующим гидрирова-
нием образующихся 2-метокси-1,6-диоксаспиро-(4,4)-ноне-
нов-3 [4].
1,6-Диоксаспиро-(4,4)-нонан при действии концентрирован-
ных йодистоводородной или бромистоводородной кислот [3]
расщепляется с образованием 1,7-дийод- или дибромгепта-
нона-4. При обработке последних гидросульфидом натрия в
безводном этиловом или метиловом спирте образуется тиа-
циклооктанон-5 с выходом 42—54 % соответственно [5].
137
СХЕМА СИНТЕЗА ТИАЦИКЛООКТАНОНА-5
+ 2НВг -
— ВгСН2-СН2-СН2-СО-СН2-СН2-СН2Вг+Н2О
ВгСН3—СН,-СН2—СО—СН2—СН2—CH2Br + 2NaSH -
-{-2NaBr + H2S
Характеристика основного сырья
1,6-Диоксасииро- (4,4)-нонан, бесцветная летучая жид-
кость, т. кип. 82—83/60 мм; Пд —1,4481; получают по про-
писи {4}.
Гидросульфид натрия приготовляют насыщением раство-
ра этилата натрия сероводородом со скоростью 2 пузырька
в секунду [6].
Бромистоводородная кислота, ч.д.а., ГОСТ 2062—43.
Условия получения
Синтез 1,7-дибромгептанона-4. Кипятят 38 г 1,6-диоксас-
П'Иро-(4,4)-нонана и 220 мл бромистоводородной кислоты на
песчаной бане в течение 3 часов. Образовавшийся темный
тяжелый слой отделяют, промывают водой, растворяют в
эфире. Эфирный раствор сушат хлористым кальцием, эфир
отгоняют, остаток перегоняют в вакууме.
Выход 1,7-дибромтептанона-4 равен 46 г, что составляет
57% теоретического; т- кип. 137—13873 мм. По литератур-
ным данным [3], т. кип. вещества 14275 мм (см. примеча-
ние).
Получение тиациклооктанона-5. В 250 мл кипящего без-
водного этилового спирта, помещенного в четырехгорлую
колбу емкостью 1 л, снабженную мешалкой, обратным хо-
лодильником и двумя капельными воронками, прибавляют в
течение 1 часа одновременно раствор 15 г (0,055 Af) 1,7-ди-
бромгептанона-4 в 200 мл спирта и раствор гидросульфида
натрия (3 г, 0,13 Af металлического натрия в 200 мл спирта).
После прибавления реагентов смесь кипятят еще 5 часов, от-
гоняют спирт, остаток разбавляют эфиром. Эфирный раствор
138
фильтруют, промывают водой и сушат безводным сульфатом
натрия. Эфир отгоняют, а остаток перекристаллизовывают из
петролейного эфира и возгоняют.
Выход тиациклооктанона-5 равен 4,32 г, что составляет
54,4% теоретического.
По внешнему виду вещество представляет собой бесцвет-
ные кристаллы с т. пл. 53—54°. По литературным данным,
т. пл. 53,2—54,2° [1] (см. примечание).
Примечание.
Аналогичным путем получают 1,7-дибромоктанон-4 из 2-метил-1,6-ди-
оксаспиро-(4,4)-нонана; выход 44%; т. кип. 113—117/1 мм и 2-метилтиа-
циклооктанон-5 из 1,7-дибромоктанона-4; выход 36%; т. кип. 108—109°/
1,5 мм-, №— 1,0834; Чд--1,5213 [5].
ЛИТЕРАТУРА
1. Н. J. Leonard, Т. L. Brown, Т. W. М i 11 i g a n, J Amer, Chem.
Soc., 81, 504, (1959); С. С. О ver berg er, A. L u s 1. J. Amer. Chem.
Soc., 81. 506 (1959).
2. А. А. Пономарев. Уч. зап. Саратовского ун-та,71, 111,(1959);
М. F а г 1 о v, Н. Burdick, Н. Adkins. J. Amer. Chem. Soc., 56,
2498 (1914); A. Hinz, G. Meger, G. Schuking Вег.. 76B, b76 (1934);
K. Alexander L. Hafner, G. Smith, L. Schniepp. J. Amer.
Chem. Soc,, 72, 5506 (1950); А. А. Пономарев, В. А. Седавкина.
Уч. зап. Саратовского ун-та 71, 143 (1959); А. А. Пономарев, 3. В.
Тиль. Уч. зап. Саратовского ун-та, 75, 41 ( (1962); А. А. Пономарев,
3. В. Тиль, И. А. Маркушина, К. С а п у н а р. Докл. АН СССР, 93,
297 (1953); Ж. общ. химии, 27, НО (1957); А. А. Пономарев, В. А.
Афанасьев, К. И. Курочкин. Докл. АН СССР, 87, 983 (1952); Ж.
общ. химии, 23, 1426 (1953); А. А. Пономарев, 3. В. Тиль, А. Д.
Пешехонова, В. П. Решетов. Ж. общ. химии 27, 1075 (1957),
3. Н. Burdick, Н. Adkins. J. Amer. Soc., 56, 433 (1934).
4. А. А. Пономарев, И. А. Маркушина. Докл. АН СССР,
126, 99 (1959); Ж. общ. химии, 31, 554 (1961).
5. А. А. Пономарев, И. С. Монахова., Ж. общ. химии 34,
1244 (1964).
6. В. Д. Хиккинботтом. Реакции органических соединений, Гонти,
М., 1939, стр. 157.
Поступила в январе 1966 г.
Саратовский гос. ун-г
УДК 547.563.162.1.07
ТИМОЛОВЫЙ СИНИЙ
Т имолсульфофталеин
£. Я. Я РОВЕН КО, М. Ф. КОНДРАШОВА
СН(СН3)2 СН(СН,)а
Prso,H
"W
C£7H30O5S-l/2H3O
М. в. 474,24
Тимоловый синий является кислотио-основным индикато-
ром с переходом окрасок от красной к желтой при pH 1,2—
2,8 и от желтой к синей при pH 8—9,6.
Применяется для определения концентрации водородных
ионов, для неводных титрований и как сырье для получения
бромтимолового синего.
Тимоловый синий получают конденсацией тимола с
о-сульфобензойной кислотой, ее дихлорангидридом, ангидри-
дом или моноаммонийной солью в присутствии хлористого
цинка [1], хлорокиси фосфора, пятиокиси фосфора [2], смеси
хлористого цинка и хлорокиси фосфора [3].
Нами предложен способ конденсации о-сульфобензойной
кислоты с тимолом в присутствии смеси хлорной кислоты и
хлорокиси фосфора, что дает увеличение выхода индикато-
ра и улучшает его качество.
140
СХЕМА СИНТЕЗА ТИМОЛОВОГО СИНЕГО
соон он
||/4rso’H +2 ГАСН'СНЛ
х// сн3/х/
СН(СН3)а СН(СН3),
_но\Л А/°
А/\сАх А
СН3 1 СН3 +2Н,0
/A_SO3II
\/
Характеристика основного сырья
о-Сульфобензойиая кислота, ч., МРТУ 6—09—811—63.
Тимол, ч„ ТУ ГКХ 1633—61.
Хлорокись фосфора, ч„ ВТУ 166—51.
Хлорная кислота, ч., ТУ МХПОРУ 87—57.
Уксусная кислота, ч., ГОСТ 61—51.
Условия получения
В трехгорлую колбу емкостью 2 л, снабженную механи-
ческой мешалкой, термометром, капельной воронкой и хлор-
кальциевой трубкой, загружают 101 г (0,5 7И) безводной
о-сульфобензойной кислоты, 23 мл 58%-ной хлорной кислоты
и 25 мл хлорокиси фосфора, перемешивают и при 60° выдер-
живают в течение 30 минут. Затем прибавляют 150 г (1 М)
тимола, поднимают температуру до 90° и из капельной ворон-
ки в течение 1,5 часа прибавляют 50 мл хлорокиси фосфора.
После прибавления всей хлорокиси фосфора реакционную
массу выдерживают еще 30 минут до ее полного загусте-
ния.
К полученному плаву прибавляют 200 мл горячей воды и
при температуре 85—90° нагревают в течение часа, затем ох-
лаждают до 20—25°, выдерживают при этой температуре в
течение 30 минут, после чего отделяют декантацией водный
слой от плава.
К плаву прибавляют 200 мл уксусной кислоты, нагревают
до 85—90° и поддерживают эту температуру в течение часа.
Выливают раствор в фарфоровый стакан и оставляют стоять
141
24—26 часов, при этом тимоловый синий выпадает в осадок.
Его отфильтровывают, промывают водой и сушат при 90—
100° до постоянного веса.
Выход сухого продукта равен 165—167 г, что составляет
70% от теоретического, считая на о-сульфобензойную кисло-
ту. Содержание основного вещества в полученном тимоло-
вом синем, определенное спектроскопическим методом, равно
85—90%.
Для получения тимолового синего в химически чистом со-
стоянии его перекристаллизовывают из этилового спирта.
Найдено, %: С-67,85; 67,61; Н 7,09; 7,08; S-6,32; 6,11.
CjjHjoOjS. Вычислено, %: С—68,19; Н—7,01; S—6,74.
ЛИТЕРАТУРА
1. N. К.. Orndorff, F. К. Cornwell..!. Chem. Soc., 48, 981 (1926).
2.1. Chrzaszczewscki, M. К о s 1 n s k 1 C. A., 52,200361 (1958).
3. Г. И. Михайлов, Г. H. Кошелева. Авт. свид. 104125; Бюлл.
изобр., № 8, 17 (1956).
Поступила в ноябре 1966 г.
ИРЕА
УДК 547.541.513'533'023.07
п -ТОЛУОЛСУЛЬФОХЛОРИД
5. И. КИССИН, Е. Н. КУРАКИН, Г. А. ТИМОХИН
СНз
I
SO2C1
C7H7C1O2S М. в. 190,64
п-Толуолсульфохлорид—широко известный препарат,
применяемый в синтезе лекарственных веществ, красителей
и др. Его получают из толуола двумя способами: через п-то-
луолсульфокислоту с последующей обработкой ее хлорсуль-
фоновой кислотой fl} или непосредственной обработкой то-
луола хлорсульфоновой кислотой [2].
Второй способ проще, но приводит к получению смеси о-
и n-изомеров, обычно с преобладанием о-толуолсульфохло-
рида. Из этой смеси «'изомер выкристаллизовывают при ох-
лаждении.
Как установил Гардинг [3], реакция взаимодействия то-
луола с хлорсульфоновой кислотой идет через образование
сульфокислоты, которая затем с избытком хлорсульфоновой
кислоты переходит в сульфохлорид (в условиях обратимой
реакции).
Позднее было показано на примере сульфохлорирования
антрахинона [4], что добавка солей металла, в частности
сульфата натрия в определенном количестве, повышает выход
сульфохлорида. Та’ же закономерность наблюдается и при
сульфохлорировании толуола с целью получения л-толуол-
сульфохлорида [5].
Как установлено одним из авторов настоящего сообще-
ния [6], положительное влияние добавок неорганической соли
143
при сульфохлорировании толуола имеет место лишь при
строго определенных соотношениях реагирующих веществ.
Позднее мы установили, что оптимальное количество вводи-
мой неорганической соли на моль толуола зависит от темпе-
ратуры реакции и избытка хлорсульфоновой кислоты.
СХЕМА СИНТЕЗА П -ТОЛУОЛСУЛ ЬФОХЛОРИДА
Н3С-^ +2C1SO3H -> Нзс-^ ^-SOaCl+HaSO4 +НС1
Характеристика основного сырья
Толуол, ч., ГОСТ 5789—51.
Кислота хлорсульфоновая, ч., ТУ НКХП 385—41 или тех-
ническая, ГОСТ 2124—43, 1 сорт.
Натрий сернокислый, безводный, ч., ГОСТ 4166—48.
Условия получения
В четырехгорлую колбу емкостью 500 мл; снабженную ме-
шалкой с гидрозатвором, термометром, вертикальной отвод-
ной трубкой длиной 70—80 см, капельной воронкой и водяной
баней, вносят 92,1 г (1 А4) безводного толуола и 14,2 г
(0,1 /И) растертого в ступке сульфата натрия. Массу в колбе
охлаждают до 10° и при непрерывном и энергичном размеши-
вании приливают из капельной воронки в течение 5—6 часов
303 г (2,6 М) хлорсульфоновой кислоты. Температура реак-
ционной массы во время прибавления хлорсульфоновой кис-
лоты регулируется внешним охлаждением (баня с ледяной
водой) и не должна превышать 15—20°. По окончании прили-
вания хлорсульфоновой кислоты массу размешивают еще 10
часов при температуре 18—20°, после чего ее тонкой струей
выливают на 1400 мл охлажденной до 4—5° воды при непре-
рывном размешивании. Температура образующейся суспен-
зии n-толуолсульфохлорида не должна подниматься выше
10°. Суспензию размешивают 30 минут при температуре 10—
12° и фильтруют под вакуумом через двойной бумажный
фильтр. Осадок на фильтре многократно (12—15 раз) про-
мывают нагретой до 30° водой, порциями по 100 мл.
Высушенный в вакуум-эксикаторе над хлористым кальци-
ем п-толуолсульфохлорид имеет температуру кристаллизации
64—64,5° и содержит около 5% о-толуолсульфохлорида (см.
примечание).
Выход n-толуолсульфохлорида равен 152—153 г, что со-
ставляет около 80% от теоретического.
Для получения чистого препарата п-толуолсульфохлорид
очищают перекристаллизацией из бензола, из смеси хлоро-
форма и петролейного эфира или другими способами [7, 8].
144
Примечание. По литературным данным [9], температура засты-
вания чистого п-толуолсульфохлорида 66,7°.
ЛИТЕРАТУРА
1. К. Heumann, К ое с h 1 i n. Ber., 15, 1118 (1882). В. M. Род и-
онов. Bull. [4] 39, 306 (1926). С. 1926 1, 2925. А. В. Kulkarni,
R. С. Shah. J. Indian Chem. Soc., Ind. and News Ed., 17, 131
(1924); C. A. 49, 6861 (1955).
2. Герм. пат. 98030; Frdl. 4, 1261.
3. L. Harding. J. Chem. Soc, 119, 1261 (1921).
4. В. В. Козлов. Ж. прикл. химии. 20, 887 (1947).
5. И. Or ня но в, А. 3 а г о р о в а. Ж- прикл. химии, 29, 8, 1299
(1956).
6. Б. И. К и с с и н, К. А. Померанце в. Хим. наука н промышлен-
ность, 2, 6, 809 (1957).
7. Австрийск. пат. 170857; С. А. 46, 10203 е (1952).
8. S. W. Pelletier. Chemistry and Industry, 1034 (1953); С. A 48,
l!378f (1954).
9. A. F. H о 11 e m a n. P. С a 1 a n d. Ber., 44, 2505 (1911).
Поступила в мае 1966 г. ЦЗЛ
Ю Зак. 266
УДК 547.581.2'115.1.07
2, 3, 5-ТРИЙОДБЕНЗОЙНАЯ КИСЛОТА
Г. И. НЛЛЕЦКЛЯ. А. П. ДУБРОВ, Г. Н. КОШЕЛЕВА
СООН
C7H3O2J3 М. в. 499,81
2, 3, 5-Трийодбензойная кислота применяется в качестве
антигетероауксина в биохимии.
Единственный описанный в литературе метод получения
2, 3, 5-трийодбензойной кислоты основан на ряде последова-
тельных реакций с использованием в качестве исходного про-
дукта антраниловой кислоты [1—3]. йодированием антрани-
ловой кислоты хлористым йодом получают 2-амино-3,5-дийод-
бензойную кислоту. Диазотированием и последующей обра-
боткой раствором йодистого калия ее превращают в 2, 3, 5-
трийодбензойную кислоту.
Нами проверены и уточнены литературные данные.
СХЕМА СИНТЕЗА 2, 3, 5-ТРИЙОДБЕНЗОИНОИ КИСЛОТЫ
СООН соон
/^-NH2 \Z + 2JC1 NH2 X 2HC1
соон соон
I I
li^-NHs ||/^-N = NHSO4
H’so‘ j/xz/Xj
146
соон
J
J
Характеристика основного сырья
Антраниловая кислота, и., ТУ МХП 1713—48.
Натрий азотистокислый, х.ч., ГОСТ 4197—48.
Калий йодистый, х.ч., ГОСТ 4232—48.
Условия получения
Синтез 2-амино-3,5-дийодбензойной кислоты [3J. К раство-
ру 13,7 г (0,1 Л4) антраниловой кислоты в 300 мл 7%-ной со-
ляной кислоты при температуре 15—20° прибавляют порция-
ми 48,8 г (0,3 Л4) хлористого йода (4]. Общий объем реакци-
онной смеси доводят 7%-ной соляной кислотой до 500 мл.
Смесь механически перемешивают 48 часов, а затем из-
быток галоида удаляют при помощи бисульфита.
Образовавшийся в результате реакции осадок дийодкис-
лоты растворяют в горячем водном растворе аммиака, рас-
твор фильтруют, дийодкислоту осаждают из фильтрата ук-
сусной кислотой.
Температура плавления неочищенной 2-амино-3,5-дийод-
бензойной кислоты 231,5—232,5° (с разложением).
Выход равен 37,6 г, что составляет 96,5% от теоретичес-
кого. ,
Очистка кислоты достигается перекристаллизацией ее ам-
мониевой соли из воды и последующей перекристаллизацией
свободной кислоты из спирта. Температура плавления очи-
щенной 2-амино-3,5-дийодбензойной кислоты — 232—234°.
Однако применять 2-амино-3,5-дийодбензойную кислоту
для получения 2, 3, 5-трийодбензойной кислоты можно и без
дальнейшей очистки.
Получение 2, 3, 5-трийодбензойной кислоты. Растворяют в
150 мл концентрированной серной кислоты 38,9 г неочищен-
ной 2-амино-3,5-дийодбензойной кислоты и раствор при ох-
лаждении и перемешивании быстро выливают на 13 г порош-
кообразного нитрита натрия.
Смесь перемешивают 2 часа при 0° и затем выливают в
сосуд, содержащий 500 г измельченного льда; образовавшие-
ся окислы азота удаляют продуванием воздуха, затем рас-
твор фильтруют и фильтрат вливают в охлажденный кон-
центрированный раствор 50 г йодистого калия в воде. Затем
реакционную смесь нагревают до прекращения выделения
10" 147
азота, удаляют избыток йода тиосульфатом и, при охлажде-
нии, выделяют 47,5 г (95% от теоретического выхода) неочи-
щенной 2, 3, 5-трийодбензойной кислоты с т. пл. 224—225,5°.
Вещество перекристаллизовывают, растворяя в разбав-
ленной щелочи и осаждая разбавленной соляной кислотой,
фильтруют, осадок растворяют в спирте или ацетоне, кипя-
тят с небольшим количеством активированного угЛя и добав-
ляют иоду до начала кристаллизации, затем охлаждают. Тем-
пература плавления чистой 2, 3, 5-трийодбснзойной кислоты
225—226°. По литературным данным, т. пл. препарата 233—
234° {3].
ЛИТЕРАТУРА
1. Wheeler, Johns. Amer. Chem. 7., 43, 407 ('910).
2. С. 1. Klemme. J. H. Hunter. J. Organ. Chem., 5, 509 (1940).,
3. S. C. Oliver, W. P. Com ba. Rec. trav. chiin., 69, 22 (1950),
4. Руководство по препаративной неорганической химии, M., ИЛ,
1956, стр. 157.
Поступила в декабре 1965 г. Биолого-почвенный факультет МГУ
Институт химии природных
соединений АН СССР
УДК [547.811'113.7 + 547.822.3:547.07
2, 4, 6-ТРИМЕТИЛПИРИЛИЙ ПЕРХЛОРАТ
и 2,4,6-ТРИМЕТИЛПИРИДИН
Г. Н. ДОРОФЕЕНКО. С. В. КРИВУН
СН3
I
HsC-^+JI-CH3
С1О4
C8Hu06Cl М. в. 222,62
СН3
// \
H-c-kJ-CH=
C8HnN М. в. 121,18
Перхлорат 2, 4, 6-триметилпирилия может быть получен
ацетилированием окиси мезитила [1, 2] диацетонового спирта
[3], тр-т. бутанола {2—4] или изобутилена [2—5] уксусным
ангид[идом в присутствии эквимолекулярных количеств хлор-
ной гаслоты. Это же соединение получается с невысоким вы-
ходом при кислотной конденсации ацетилацетона и ацетона
[6].
Прилагаемый нами способ [7] заключается в ацилирова-
нии легкодоступного ацетона уксусным ангидридом в присут-
ствии «Бимолекулярных количеств 70%-ной хлорной кисло-
ты. Реакция проводится в одну стадию при комнатной тем-
ператур и позволяет легко получать 'весьма чистую пирилие-
вую есть в значительных количествах.
При обработке перхлората 2, 4, 6-триметилпирилия ам-
миакод получают 2, 4, 6-триметилпиридин (коллидин) с поч-
ти кол чественным выходом.
CXEIA СИНТЕЗА ПЕРХЛОРАТА 2, 4. 6-ТРИМЕТИЛПИРИЛИЯ
и 2, 4, 6 ТРИМЕТИЛ ПИРИДИНА
СН3СОСН, + О—C-СН..
НС1О4 149
СНз
I сн,со+
СН3СОСН=С-СН8 —!----->
сн3
I
+ нею
-> СН3-С-СНаСОСН3 I || ru
I сн*
I J-J Г' ч0 __
СН2СОСН3 Н’С С104
СН8
Характеристика основного сырья
Ацетон, ч.д.а., ГОСТ 2603—63.
Уксусный ангидрид, ч.д.а., ГОСТ 787—55.
Хлорная кислота, 70%-ная, получают концентрированием
х.ч., 57%-ной хлорной кислоты.
Условия получения
Синтез 2, 4, 6-триметилпирилия перхлората. К 57 мл
(0,56 М) уксусного ангидрида, помещенного в трехгорлую
колбу емкостью 250 мл, снабженную мешалкой, обратным хо-
лодильником и капельной воронкой, при перемешивании и
охлаждении (см. примечание 1) приливают по каплям 14,5 г
(0,1 М) 70%-ной хлорной кислоты (см. примечание 2). Пос-
ле прибавления кислоты охлаждение снимают и смесь вы-
держивают 15—20 минут при комнатной температуре.
К полученному ацетилперхлорату при перемешивании
приливают 14,5 мл (11,6 г, 0,2 Л4) ацетона. Реакционная
смесь при этом самопроизвольно разогревается до 40—50° и
через 2—3 часа начинается обильная кристаллизация. По ис-
течении 15—20 часов выпавший бесцветный кристаллический
осадок пирилиевой соли отфильтровывают, промывают эфи-
ром и высушивают.
Выход перхлората 2, 4, 6-триметилпирилия равен 9 а, что
составляет 40,5% от теоретического; т. пл. 245° (см. примеча-
ние 3 и 4).
Получение 2, 4, 6-триметилпиридина. К 5,6 г 2, 4, 6-три-
.метилпирилий перхлората приливают при встряхивании и
охлаждении 25 мл 25%-ного водного аммиака. После раство-
рения всего кристаллического продукта выделяется 2, 4, 6-
150
тр'иметилпиридин (коллидин), который экстрагируют эфиром
и перегоняют, собирая фракцию с т. кип. 170—172°.
Выход вещества равен 2,9 г, что составляет 96% от тео-
ретического.
Примечания:
1. Для охлаждения реакционный сосуд помещают в баню со льдом.
2. Возможно использование и менее концентрированной хлорной кис-
•Оты, например, 57%-ной, однако в этом случае необходимо добавлять
избыток уксусного ангидрида в расчете на воду хлорной кислоты.
I 3. Если к фильтрату прилить 300 мл эфира и смесь оставить на 2 —
3 уаса, то выпадает дополнительно 1—1,5 г менее чистого 2, 4, 6-триме-
тифтнрилий перхлората.
I 4. Полученную пирилиевую соль можно пускать в работу без допол-
нительной очистки. При необходимости продукт кристаллизуют из не-
большого количества воды, в которую предварительно добавляют немно-
го активированного угля и 1—2 капли хлорной кислоты.
ЛИТЕРАТУРА
1.0. Diels, К. Alder. Бег., 60, 722 (1927).
2. Р. F. Р г a i 11, A. L. W h i t е а г. Proc. J. Chem, Soc., 1959, 312.
3. Р. F. Р г a i 11, A. L. W h 11 е а г. J. Chem. Soc., 1961, 3573.
4. Н. И. Ш у й к и н, И. Ф. Бельский, А. Т. Балабан, К. Д. Н е-
\ницеску. Изв. АН СССР, ОХН, 1961, 491.
I 5. А. Т. В а 1 а b а п, С. D. N е п 11 z е s с и. Libigs. Ann. Chem., 625
4 (1959).
6. С. В. Кривун, Ж. В Шиян, Г. Н. Дорофеенко. Ж. орган,
химии, 34, 167 (1964).
7. Г. Н. Дорофеенко, С. В. Кривун. Укр. хим. ж. 29, 1058
(1963).
Поступила в феврале 1966 г.
Ростовский на Дону гос. ун-т
УДК 547.811'113.7+547.822.3:54/.07
2, 4, 6-ТРИФЕНИЛПИРИЛИИ ПЕРХЛОРАТ
И 2, 4, 6-ТРИФЕНИЛПИРИДИН
Г. Н. ДОРОФЕЕНКО, С. В. КРИВУН
С.Н5
/ Ч
н5св-А^-свн8
сю4
С33Н„О6С1 М. в. 408,83
/Ч
H5C6J4J-CeHs
C,3H17N М.в. 307,39
Для синтеза 2, 4, 6-трифенилпирилиевых -солей наиболее/
пригодны способы, основанные на конденсации ароматичес-/
ких альдегидов с жирноароматическими кетонами. В каче^
стве конденсирующих агентов применяют BF3 [1] и серную
кислоту [2]. j
Описано получение трифенилпирилий перхлората в две
стадии [3]: щелочной конденсацией бензальдегида и ацетофе-
нона сначала получают бензилиденбисацетофенон, который
затем при кипячении с трифенилметилперхлоратом в ледяной
уксусной кислоте превращают в конечный продукт.
Применение хлорной кислоты в качестве конденсирующе-
го агента в реакции конденсации бензальдегида с ацетофе-
ноном позволило нам упростить процесс и получить трифе-
нилпирилий перхлорат высокой степени чистоты и с хорошим
выходом [4]. Перхлорат трифенилпирилия получается при не-
продолжительном кипячении исходных веществ в среде то-
луола.
При кипячении полученной пирилиевой соли с ацетатом
аммония в уксусной кислоте получают 2, 4, 6-трифенилпири-
ДИН.
152
СХЕМА СИНТЕЗА 2, 4, 6-ТРИФЕНИЛПИРИЛИЯ ПЕРХЛОРАТА
И 2, 4, 6-ТРИФЕНИЛПИРИДИНА
СН3
I
сно
нсю4
сна
сю4-
CHaCOONH,
Характеристика исходного сырья
Ацетофенон, ч., ТУ ОРУ 28—55.
Бензальдегид, ч., ГОСТ 157—51.
Хлорная кислота, 70%-ная, получают концентрированием
х.ч. 57%-ной хлорной кислоты.
Толуол, ч.д.а., ГОСТ 5789—51.
Условия получения
Синтез 2, 4, 6-трифенилпирилия перхлората. К раствору
4,8 г (0,04 М) ацетофенона в 10 мл толуола приливают 2,1 г
(0,02 А1) свежеперегнанного бензальдегида и 3,5 а 70%-ной
хлорной кислоты (см. примечание 1). Смесь кипятят в колбе
с обратным холодильником в течение 1 часа. В процессе ре-
акции выделяется желтый кристаллический продукт, который
появляется сначала на стенках колбы, а затем во всей массе
(см. примечание 2). После охлаждения к реакционной смеси
приливают 100 мл эфира и отсасывают образовавшийся кри-
сталлический 2, 4, 6-трифенилпирилий перхлорат, его промы-
вают на фильтре смесью эфира и ацетона (5:1), а затем эфи-
ром и сушат.
Выход равен 4,0 г, что составляет 49% от теоретического.
Пирилиевую соль кристаллизуют из уксусной кислоты, в ко-
торую предварительно добавляют 1—2 капли хлорной кисло-
ты; т. пл. 273°.
153
Получение 2, 4, 6-трифенилпиридина. Смесь 4 г 2, 4, 6-три-
фенилпирилий перхлората, 7—8 г уксуснокислого аммония в
30 JM4 уксусной кислоты кипятят 30 минут в колбе емкостью
100 мл с обратным холодильником. Появляющаяся вначале
флуоресценция исчезает. Реакционную смесь охлаждают и
выливают в 150 мл воды. При стоянии выпадает 2, 4, 6-три-
фенилпиридин, который отфильтровывают, несколько раз
промывают водой и высушивают.
Выход равен 2,7 г, что составляет 90% от теоретического.
Продукт очищают перекристаллизацией из спирта; т. пл. 137°
(см. примечание 3).
Примечания;
1. Возможно применение продажной 57%-нон хлорной кислоты, одна-
ко в этом случае выход конечного продукта снижается.
2. Если при кипении появляются толчки, то во избежание выброса
реакционную колбу необходимо встряхивать.
3. Этим способом получены: 2,6-дифенил-4-(п-нитрофенил)-пирилий
перхлорат-, 2,6-дифенил-4-(м-нитрофенил)-пирилий перхлорат; 2,6-дина-
фтил-4-фенилгшри.лий перхлорат и др.
ЛИТЕРАТУРА
1. W. Dovey, R. Robinson. J, Chem. Soc, 1935, 1389.
2. R. Wlzinger, S. Los Inger. P. Ulrich. Helv. chim. Acta,
39, 5 (1956).
3. M. Siemiatlcki, R. Fugnitto. Bull. Soc. Chim. France, 1961
538.
4. Г. H. Дорофеенко, С. В. Кривун. Ж- орган, химии, 34, 105
(1964).
Поступила в феврале 1966 г.
Ростовский на Дону гос. ун-т
УДК 547.775'113.1.07
1-ФЕНИЛ -3-МЕТИЛ-4-АМИН0ПИРА30Л0Н-5
ХЛОРГИДРАТ
Р. Б. ЖУРИН, В. Н. ИВИНА
Н3С-С--CH-NH2-HC1-H2O
N С=О
\Nz
i
\ 1
Ч/
c10h12cin3o-h2o
М. в. 243,69
1-Фенил-3-метил-4-аминопиразолон-5 хлоргидрат может
применяться в качестве проявляющего вещества при обра-
ботке фотоматериалов [1].
Его получают восстановлением 1-фенил-3-метил-4-изонит-
розопиразолона-5 хлористым оловом [2] или каталитическим
гидрированием водородом >в присутствии скелетного никеля
{3].
Нами был использован последний метод, позволяющий
выделить амин в виде соли, устойчивой к окислению.
СХЕМА СИНТЕЗА
1-ФЕНИЛ-3-МЕТИЛ-4-АМИНОПИРАЗОЛОНА-5 ХЛОРГИДРАТА
Н3С—С СН2 НаС—С—C=NOH
II I НС1 1
N С=О NaNOj —N С=О
\NZ \nz
HSC—С—C—NOH
II I
N C=0
скелетный
никель
H8C-C—CH-NH,
II I
> N C=0
\NZ
HSC—C—ch-nh2 HsC-C—CH-NH2-HC1
II I II
N C=0 + HCt -> N C=O
\Nz \Nz
I I
II I li
4/ %/
Характеристика основного сырья
1-Фенил-3-метилпнразолон-5, ГОСТ 9593—61.
Нитрит натрия ч., ГОСТ 6194—52.
Соляная кислота техн, оинтет. ГОСТ 857—57.
Водород, ГОСТ 3022—61.
Спирт метиловый ч., ГОСТ 6995—54.
Никель скелетный.
Условия получения
Синтез 1 -фенил-3-метил-4-изонитрозопиразолона-5. В че-
тырехгорлой колбе емкостью 1,5 л, снабженной мешалкой,
капельной воронкой, термометром и трубкой для отвода вы-
деляющихся газов, растворяют 75 г (0,43 А4) фенилметилпи-
разолона в 250 мл метанола и 750 мл 15%-ной соляной кис-
лоты. К (полученному раствору прибавляют по каплям при
интенсивном перемешивании раствор 33 г (0,48 М) нитрита
натрия в 75 мл воды с такой скоростью, чтобы температура
реакционной массы не поднималась выше 15° (см. примеча-
ние 1) и чтобы не было заметного вспенивания. В этих усло-
виях реакционную массу выдерживают при размешивании
1 час, проверяя наличие азотистой кислоты по йодокрахмаль-
ной бумаге. Выпавший оранжевый осадок отфильтровывают,
промывают 500 мл воды до нейтральной реакции фильтрата
на конго и сушат в сушильном шкафу при 70—80° (см. при-
мечание 2). Получают 83—85 г технического изонитрозопро-
дукта, что составляет 95—97% от теоретического выхода,
считая на исходный фенилметилпиразолон; т. пл. 151—153°.
156
Получение 1-фенил-3-метил-4-аминопиразолона-5, хлор-
гидрата. В автоклав емкостью 1,35 л с 'мешалкой загружают
60 г (0,29 Л1) изонитрозопроизводного и 750 мл метанола
(см. примечание 3). К полученной суспензии при размешива-
нии добавляют 9 г скелетного никеля. Автоклав закрывают,
промывают водородом, наполняют водородом до давления
50 атмосфер и перемешивают без нагревания в течение 1,5
часов. Во время гидрирования температура в автоклаве по-
вышается до 32—35°. После поглощения рассчитанного коли-
чества водорода (примерно 2 моля/моль исходного вещест-
ва) содержимое автоклава размешивают еще 0,5 часа. По
окончании выдержки спускают давление, автоклав открыва-
ют и быстро переносят реакционную массу в колбу, содержа-
щую 45 мл концентрированной соляной кислоты. Затем ката-
лизатор отфильтровывают, метанол отгоняют под производ-
ственным вакуумом в токе азота (объем отгона 560—
580 .ил). Густой остаток охлаждают при перемешивании до
0—5° и выдерживают при этой температуре 1,5 часа. Выпав-
ший кристаллический желтоватый осадок отфильтровывают
и сушат на фильтре.
Выход технического 1-фснил-3-метил-4-аминопиразоло-
на-5, хлоргидрата составляет 52—54 г. Его промывают (см.
примечание 4) 3 раза по 200 мл смесью этиловый спирт—
бензол (1:1 по объему), после чего осадок отфильтровывают
и сушат в вакууме. Получают 42—44 г вещества, что состав-
ляет 55—58%, считая на исходный фенилметилпиразолон.
Содержание основного вещества в продукте равно 94—97%
(определяется йодометрически); при нагревании постепенно
разлагается.
Примечания:
1. При повышении температуры выше 15° происходит значительное
осмолснис реакционной массы.
2. Выше 85° продукт заметно возгоняется.
3. В метаноле техническом (ГОСТ 2222—60) реакция гидрирования
проходит неудовлетворительно.
4. Тщательная промывка смесью указанною состава необходима в
случае применения этого препарата в качестве проявляющего вещества.
ЛИТЕРАТУРА
1. М. С. X а й к и н, Г. В. Д е р с т у г а н о в, И. И. Левкоев,
В. А. Кухт ин, Д. Б. Ш а м и л ь с к а я. Труды НИКФИ, 37, 17 (1960);
РЖХим, 1962, 4Л421.
2. L. Knorr. Liebigs. Ann. Chem., 238, 185, 189 (1887).
3. Пат. ФРГ 1047618; Zbl„ 1959, 10452; Англ. пат. 833113- Ch. А., 55,
5207 (1961); Чешек, пат. 102347; РЖХим, 1963, 12Н143.
Поступила в июне 1966 г.
НИОПиК
УДК 547.835.07
9-ФОРМИЛ-З, 4-БЕНЗАКРИДИН
3, 4-Бенз-9-акридиловый альдегид
Б. С. ТАНАСЕИЧУК, И. Я. ПОСТОВСКИЙ
CI8HnNO
М. в. 257,29
Синтез 9-формил-3,4-бензакридина был ранее осущест-
влен [1] в несколько стадий: сначала из 9-метил-3,4-бензакри-
дина получен сульфометилат, затем с п-нитрозодиметилани-
лином приготовлен нитрон, а из нитрона — альдегид. Сум-
марный выход 9-формил-3,4-бензакридина из 9-метил-3,4-
бензакридина составил 32,5%.
Предложенная нами методика заключается в получении
9-формил-3,4-бензакридина в одну стадию; 9-метил-3,4-бенз-
акридин сплавляют с двуокисью селена, затем извлекают
альдегид из плава бензолом.
СХЕМА СИНТЕЗА 9-ФОРМИЛ-З,4-БЕНЗАКРИДИНА
SeO2
158
Характеристика основного сырья
(см. примечание 1)
9-Метил-3,4-бензакридин, т- пл. 124—125° (1].
Двуокись селена, ч„ МРТУ 6—09—1553—64.
Условия получения
В круглодонную колбу емкостью 50 мл помещают 4,86 г
9-метил-3,4-бензакридина, 2,24 г двуокиси селена н нагрева-
ют эту смесь при температуре бани 155—160° (сплав Вуда)
в течение 30 минут. Затем плав обрабатывают 500 мл бензо-
ла, бензольный раствор отфильтровывают от металлическо-
го селена и пропускают через колонку, наполненную окисью
алюминия. Бензольный элюат концентрируют до объема 30—
50 мл и охлаждают. Выпавшие кристаллы 9-формил-3,4-бенз-
акридина отфильтровывают.
Выход вещества равен 3,65—4,4 г, что составляет 72—
86% от теоретического; т. пл. 148—149° (см. примечание 2).
Примечания:
1, Особое внимание должно быть обращено на чистоту исходных ме-
тилбензакридииов.
2. Аналогичным способом из 9-метил-1,2-бензакридииа (т. пл. 144—
145°) [3] получают 9-формил-1,2-бензакридин.
Выход 40—60%.
ЛИТЕРАТУРА
1. А. Е. Поран-Кошиц, Г. С. Тер-Саркисян. Изв. АН СССР,
1951, 601,
2. Б. М. Михайлов, Г. С. Тер-Саркисян. Изв. АН СССР,
1954, 656.
3. А. Е. П о р а й-К о ш н ц, Г. С. Тер-Саркисян. Изв. АН СССР,
1951, 771.
Поступила в январе 1966 г.
Уральский политехнический институт
УДК 547.725.07
ФУРАН-2-КАРБОНОВАЯ КИСЛОТА
Пирослизевая кислота
3. В. НАЗАРОВА, В. И. НОВИКОВ
; j-COOH
CSH4O3 М. в. 112,0g
В литературе описано несколько способов получения пи-
рослизевой кислоты. Окисление фурфурола проводят перман-
ганатом калия [1, 2], кислородом в присутствии катализато-
ра Cu2O/Ag2O [3], воздухом в присутствии CuO/Ag2O [4], пе-
рекисью водорода с добавкой пиридина [5], гипобромитом
натрия [6] и другими окислителями. Однако большинство из
этих методов либо сложны и длительны, либо дают недоста-
точно чистую кислоту. Самым удобным и простым является
метод Салчинкина и Лайковой [6]. Нами установлено, что
для получения воспроизводимых результатов по этому спо-
собу следует применять избыток щелочи.
Кроме того, большое значение имеет, насколько быстро
вводится в реакцию раствор гипобромита; (необходимо начи-
нать прибавление фурфурола немедлен© после растворения
последней порции брома. Качество фурфурола не оказывает
влияния на выход кислоты, поэтому мы использовали фур-
фурол без предварительной перегонки. При соблюдении ука-
занных условий пирослизевая кислота получается с выходом
около 75%.
СХЕМА СИНТЕЗА ПИРОСЛИЗЕВОЙ КИСЛОТЫ
СНО NaOBr
1-СООН
160
Характеристика основного сырья
Фурфурол, ч.д.а., СТ ГОХП 27—1831.
Натр едкий, ч„ ГОСТ 4328—48.
Бром, х.ч., ГОСТ 4109—48.
Условия получения
В круглодонную колбу ю длинным горлом на 1 л налива-
ют раствор 135 г едкого натра в 500 мл дистиллированной
воды, охлаждают в бане до 0° (лед с солью) и при переме-
шивании добавляют 52 мл (1 Л1) брома порциями по 5—
7 мл. Очередную порцию брома прибавляют после того, как
предыдущая полностью прореагирует со щелочью. Немед-
ленно после растворения последней порции брома начинают
вводить фурфурол. Последний в количестве 96 г (1 Л4) при-
бавляют порциями по 3—4 мл при энергичном встряхивании
реакционной массы, поддерживая при этом температуру в
колбе от минус 5 до минус 10°. Прибавление фурфурола за-
канчивается в течение 1 часа. Смесь выдерживают в бане
еще 30 минут, после чего подкисляют предварительно охлаж-
денной разбавленной (1:1) соляной кислотой до кислой реак-
ции по Конго. Выпавшие белые кристаллы кислоты отсасы-
вают и переносят в колбу для перекристаллизации. Осадок
растворяют в 300 мл кипящей воды, добавляют 6 г активи-
рованного угля, кипятят раствор несколько минут и отфиль-
тровывают на воронке для горячего фильтрования. При ох-
лаждении фильтрата пирослизевая кислота выпадает в виде
белых игл. Через несколько часов кислоту отфильтровывают
и сушат при 60—70°.
Выход кислоты равен 83—85 г, что составляет 74—75%
от теоретического. Из маточника экстракцией эфиром с пос-
ледующим его удалением можно выделить дополнительно
6—7 г кислоты.
Общий выход кислоты равен 89 г (79,4%); т. пл. 132—133°.
По литературным данным, т. пл. вещества 133—134° [3].
ЛИТЕРАТУРА
1. ГО. К. Юрьев. Практические работы по органической химии,
вып. 1 и II, М„ МГУ, 1961. стр. 213.
2. А. А. Пономарев. Синтезы и реакции фурановых соединений,
Саратов, СГУ, 1960, стр. 158.
3. Сб. «Синтезы органических препаратов», вып. 8, М„ ИЛ, 1958,
гтр. 71. ; i.,
4. В. И. Лутков а, Н. Н. Ш м а г и н а. Пластические массы, вып.
4, I960, стр. 56.
5. С. П. Коршунов, Л. И. Верещагин. Ж. прикл. химии, 36,
1157 (1963).
6. А, П. Сал чинкпн, Л. Б. Л а п к о в а. Ж. прикл. химии, 29, 141
(1956).
Поступила в феврале 1966 г.
И Зак. 2S6
Ростовский на Дону гос. ун-т
161
УДК 547.722.2.07
З-(а-ФУРИЛ) ГЕКСАНОН-5
А. А. ПОНОМАРЕВ, В. А. СЕДАВКИНА
II II—СН- CH.-CO-CHS
\о/ I
с.2нЕ
С10Н14О2 М. В.. 166,21
Насыщенные фурановые кетоны, содержащие алкильные
заместители у первого от цикла углеродного атома боковой
цепи, могут быть получены путем присоединения магнийорга-
нических реагентов в положение 1,4-сопряженной системы
двойных связей фурфурилиденацетона.
З-(а-Фурил) гексанон-5 и некоторые другие кетоны подоб-
ного типа были синтезированы ранее [1]. Недостаток этого
метода заключается в необходимости применения двухкрат-
ного избытка магнийорганического реагента и больших ко-
личеств абсолютного эфира [2].
Нами осуществлен синтез 3-(а-фурил)гексанона-5 и 2-ме-
тил-З-(а-фурил) гексанона-5 из фурфурилиденацетона и этил-
магнийбромида и изопропилмагнийбромида с применением
лишь 20—40%-ного избытка магнийорганического реагента
при охлаждении реакционной смеси до 5°. Разложение маг-
ниевого комплекса осуществлялось разбавленной уксусной
кислотой со льдом [3, 4].
СХЕМА СИНТЕЗА 3-(а-ФУРИЛ)ГЕКСАНОНА-5
l4OJ-CH=CH-C=O+ C2H5MgBr -
СНз
^0Д-СН-СН==С-СНз-~ \0J-CH-CH,-C-CH3
162 С2Н5 OMgBr С8Н6 о
Характеристика основного сырья
Фурфурилиденацетон, свежеперегнанный, т. кип. 112—-
115°/10 мм [5].
Бромистый этил, х. ч., ТУ МХП 80—47, т. кип. 38—39°.
Магний, стружка или порошок.
Этиловый эфир, абсолютный, ГОСТ 6265—52.
Условия получения
В четырехгорлой колбе емкостью 1 л, снабженной меха-
нической мешалкой, обратным холодильником, делительной
воронкой и термометром, приготовляют этилмагнийбромид
из 24 г магния в виде стружек, 120 г бромистого этила и
250 мл абсолютного эфира. К полученному реактиву, охлаж-
денному до 5°, при энергичном перемешивании приливают из
делительной воронки раствор 80 г свежеперегнанного фур-
фурилиденацетона в 150 мл абсолютного эфира с такой ско-
ростью, чтобы температура реакционной смеси не поднима-
лась выше 10°. Затем реакционную смесь нагревают в тече-
ние 4 часов на водяной бане, охлаждают и переносят в колбу,
содержащую 500 а толчоного льда. Образовавшийся эфирный
слой декантируют, а водный слой, содержащий гидроокись
магния, обрабатывают уксусной кислотой, после чего экстра-
гируют тремя порциями эфира по 50 мл. Объединенные
эфирные вытяжки промывают насыщенным раствором соды
до нейтральной реакции, затем дистиллированной водой и су-
шат прокаленным сульфатом натрия. Эфир отгоняют на во-
дяной бане, а остаток перегоняют в вакууме и получают 3-(а-
фурил)-гексанон-5 с т. кип. 108—110720 мм; —1,4669
(см. примечание 1); <Л20— 0,9879.
Выход равен 62 г, что составляет 63,4% от теоретическо-
го.
Полученный кетон представляет собой слегка желтоватую
жидкость с приятным запахом. Хорошо растворим в обычных
органических растворителях, нерастворим в воде, т. пл. семи-
карбазона З-(а-фурил)гексанона-5— 112° (см. примечание 2).
Примечания:
1. Иногда показатель преломления кетона оказывается несколько за-
вышенным из-за примеси ненасыщенного фуранового спирта, получающе-
гося в результате присоединения магнийорганического реагента по карбо-
нильной группе. В этом случае необходима повторная перегонка продукта.
2. Аналогичным путем получают: нз 18 г магния, 87 г бромистого изо-
пропила и 80 г фурфурилиденацетона 2-метил-3-(а-фурил)гексанон-5 с
т. кип. 112—113°/18 мм-, Пд—1,4698; </420 —0,9829.
Выход равен 65,3 г, что составляет 64% от теоретического.
И’ 163
ЛИТЕРАТУРА
1. N. Maxim. Bull. Soc. Chim. France. (4), 49, 887 (1931).
2. В. В. Челинцев, 3. В. Тиль. Уч. зап., Саратовск. ун-та, 497
(1955).
3. А. А. Пономарев, В. А. Сед а в кина. Уч. зап. Саратовск.
ун-та, 71, 143 (1959).
4. А. А. Пономарев. Синтезы и реакции фурановых веществ, Са-
ратов, СГУ, 1960. стр. 130.
5. Сб. «Синтезы органических препаратов», вып. 1, М„ ИЛ, 1949,
стр. 451.
Поступила в январе 1566 г. Саратовский гос. у-пт
УДК 547.722.6.07
1-(а-ФУРИЛ)-5-МЕТИЛ-3-АМИНОГЕКСАН
А. А. ПОНОМАРЕВ, Н. П. МАСЛЕННИКОВА
\О/-СН2-СН2-СН-СН2-СН-СН3
NH3 СН3
CnHleON М. в. 181,27
Первичные амины фуранового ряда, содержащие амино-
группу в боковой цепи, могут быть получены восстановитель-
ным аминированием предельных и непредельных альдегидов
и кетонов в присутствии аммиака и никеля Ренея.
Таким путем синтезированы фурфуриламин [1] и несколь-
ко фурановых аминов из а, [3-непредельных фурановых кето-
нов [2]. Подробное изучение данного метода позволило нам
установить общий характер этой реакции в ряду фурана, в
результате чего разнообразные предельные и непредельные
фурановые кетоны могут быть превращены в первичные ами-
ны [3, 4]. Фурановый цикл в этих условиях не гидрируется.
СХЕМА СИНТЕЗА
1-(а-ФУРИЛ)-5-МЕТИЛ-3-АМИНОГЕКСАНА
1-СН=СН-СО-СН2-СН-СН» -N1’ Re- нз
| NH3(CH3OH)
СИ,
\о/—сн2-сн3-сн-сн2-сн-сн3
• NH3 СН3
165
Характеристика основного сырья
фурфурил'иде'нметилиз°бу™лкетон> т- кип- чэ—
12176,5 мм: п®—1,5520; синтезирован по [5].
Никель Ренея, приготовлен по [6].
Водород, электролитический.
Аммиак жидкий, в баллонах.
Метиловый спирт, х.ч., ГОСТ 6995—54, т. кип. 65°.
Условия получения
Во вращающийся автоклав емкостью 150 мл помещают
30 г свежеперегнанното фурфурилиденметилизобутилкетона,
70 мл насыщенного аммиаком прн 0° метилового спирта и
3,5 г никеля Ренея. Реакцию проводят при 80—100° и на-
чальном давлении водорода 100—120 атмосфер до прекра-
щения поглощения водорода (3—4 часа). Гидрогенизат осво-
бождают от катализатора фильтрованием, отгоняют метило-
вый спирт и аммиак. Остаток разбавляют эфиром и сушат
плавленым едким кали. Эфир отгоняют и остаток перегоня-
ют в вакууме.
Выход 1-(а-фурил)-5-метил-3-аминогексана равен 22,5 г,
что составляет 74% от теоретического, т. кип. НО—11 Г/Ю мм\
п£~ 1,4710 ; <А20 —0,9250.
Примечание.
Аналогичным методом были получены 1-(а-фурил)-1-аминоэтан,
1-(а-фурил)-2-аминоциклогексан, 1-(а-фурил)-5-аминогексан, ].( а -фурил)-
7-метил-5-аминооктан, 1-(а-фурил-6-фенил-5-аминопеитан [3—5].
ЛИТЕРАТУРА
1. J. Robinson, Н Snyder. Organic Sintheses. 23, 70(1943).
2. Z. Zafiriadis, P. Mastagli. Q. R. 236, 295 (1953).
3. А. А. По но м ap e в, H. П. Масленникова, H. В. Алакн-
н а, А. П. Кривенько. Докл. АН СССР, 131, 6 (1960).
4. А. А. Пономарев, Н. П. Масленникова, А. П. Кривень-
ко. Ж. орган, химии, 31, 958 (1961).
5. А. А. Пономарев. Синтезы и реакции фурановых веществ. Са-
ратов, СГУ, 1960, стр. 66.
6. Сб. «Синтезы органических препаратов», вып. 3, М., ИЛ., 1952,
стр. 338.
Поступила в январе 1966 г.
Саратовский гос. ун-т
УДК 547.722.6.07
1-(а-ФУРИЛ)-3-МЕТИЛАМИНОПРОПАН
Л. Л. ПОНОМАРЕВ, М. В. НОРИЦИНА, И. М. СКВОРЦОВ
Г!
\ Л-сн2-сн,-сн2
NH-CH.
CSH13ON М. в. 139,19
1- (а-Фурил) -3-метиламинопропан и его гомологи представ-
ляют интерес как полупродукты для тонкого органического
синтеза. Эти амины легко могут быть получены методом вос-
становительного аминирования фурановых а, ^-непредельных
и предельных альдегидов и кетонов в присутствии никеля Ре-
нея с использованием в качестве аминной компоненты метил-
амина.
СХЕМА СИНТЕЗА
1-(а-ФУРИЛ)-3-МЕТИЛАМИНОПРОПАНА
____„ О ___,
|| ' || 2Н„ NiRe :
\ /-СН=СН-СН /-СН2-СН2-СН, !
+ H2O NH-СН»
Характеристика основного сырья
2-Фурилакролеин, ч., ТУ 79-П-891-63, т. кип. 98—10075 л.и,
т. пл. 52—54° [1].
Метиламин солянокислый, ч., МРТУ 6-89-1385-64. ТУ ОРУ
3—58.
Натр едкий, технический.
Водород, ГОСТ 3022—45.
167
Условия получения
В стальной вращающийся автоклав емкостью 600 мл по-
мещают 61 г (0,5 Л4) фурилакролеина, раствор 25 а (0,8 А4)
метиламина в 120 мл метанола (см. примечание 2) и 6 а ни-
келя Ренея [2]. Начальное давление водорода составляет 80
атмосфер, температура 40—50°. За 2 часа поглощается рас-
считанное количество водорода. По охлаждении автоклав раз-
гружают и гидрогенизат освобождают от катализатора
фильтрованием. Метиловый спирт и избыток метиламина от-
гоняют в вакууме. Остаток растворяют в 100 мл 17%-нойсо-
ляной кислоты и экстрагируют эфиром органические приме-
си нейтрального характера. Затем к водному раствору соли
амина прибавляют твердый едкий натр (около 25 а) до вы-
деления амина из раствора. Амин отделяют, водный слой эк-
страгируют эфиром, эфирные вытяжки соединяют с амином
и сушат твердым едким кали в течение суток. Затем эфир
отгоняют, а остаток перегоняют в вакууме. Получают 35,1 г
1-(а-фурил)-3-метиламинопропана с т. кип. 83—84°/18 мм;
п™ —1,4713; выход составляет 50,5% от теоретического.
Температура кипения ацетильного производного 138—
139'75 мм, Яд—1,4932 (см. примечание 2).
Примечания:
1. Метиламин получают из солянокислого метиламина и щелочи по
методу, описанному в [3].
2. Аналогичным путем получены:
из фурфурилиденацетона 1-(а-фурил)-3-метиламинобутан с выходом
47% от теоретического; т. кип. 73—7578 мм; nffl —1,4711;
из 1-(а-фурил)-1-изопропил-бутаноиа-З 1-(а-фурил)-1-изопропил-3-ме-
тиламинобутан с выходом 71% от теоретического; т. кип. НО—111718 мм;
20
nD —1,4668. Перечисленные амины получены впервые.
ЛИТЕРАТУРА
1. А. А. Пономарев. Синтезы и реакции фурановых веществ, Са-
ратов, СГУ, 1960, стр. 51.
2. Сб. «Синтезы органических препаратов», вып. 3, М., ИЛ, 1952,
стр. 338.
3. Сб. «Синтезы органических препаратов», вып. 6, М„ ИЛ, 1956,
стр. 51.
Поступила в январе 1966 г. Саратовский гос. ун-т
УДК 547.722.2.07
1-(а-ФУРИЛ )-5-МЕТИ ЛГЕКСАНОН-3
А. А. ПОНОМАРЕВ, 3. В. ТИЛЬ
J-CH2-CH2-CO-CH2-CH-(CHS)2
СцНЛвО2 м. в. 180,24
Одним из общих методов получения предельных фурано-
вых кетонов является восстановление соответствующих а,
P-непредельных кетонов амальгамой натрия [1, 2] или по-
средством гидрирования в жидкой фазе при комнатной тем-
пературе на платиновом катализаторе [3—6]. В связи с мед-
ленным протеканием процесса эти методы мало удовлетвори-
тельны. При использовании никелевого катализатора гидри-
рование этиленовой связи при комнатной температуре и
обычном давлении также протекает медленно [7, 8].
Нами было найдено [9], что а, p-непредельные фурановые
альдегиды и кетоны могут быть быстро и с хорошими выхо-
дами превращены в соответствующие предельные соединения,
если гидрирование вести с никелем Ренея при комнатной тем-
пературе под давлением (от 20 атмосфер и выше). Для того,
чтобы исключить возможное в этом случае гидрирование
карбонильной группы с образованием спирта, в автоклав вво-
дится рассчитанное количество водорода. В 'качестве раство-
рителя используется безводный этиловый спирт, взятый не
менее, чем в равном объеме с веществом. Никель Ренея вво-
дится в количестве от 5 до 10% к весу непредельного соеди-
нения.
Метод проверен на гидрировании различных мононепре-
дельных и диеновых фурановых кетонов; выходы предельных
кетонов 72—87% от теоретического.
Получаемые таким образом предельные фурановые кето-
ны могут служить ценным исходным материалом для синтеза
169
третичных спиртов, алкенов и алканов как фуранового, так и
тетрагидрофуранового рядов.
1-(а-Фурил)-5-метилгексанон-3 ранее в литературе опи-
сан не был.
СХЕМА СИНТЕЗА
1-(а-ФУРИЛ)-5-МЕТИЛ ГЕКСАНОНА-3
J-CH=CH-CO-CH2-CH-(CH3)2 -
—J-CH3-CH3-CO-CH2-CH-(CH3)2
Характеристика основного сырья
Фурфурилиденметилизобутилкетон, т. кип. 115—11673 .«.и;
и^° —1,5528; семикарбазон с т. пл. 175—176° (см. примеча-
ние 1).
Никель Ренея, приготовляют обычным методом [10].
Условия получения
Раствор 20 а (0,11 М) фурфурилиденметилизобутилкето-
на в 75 мл абсолютного спирта и 2 г никеля Ренея помещают
в стальной вращающийся автоклав емкостью 150 мл. После
предварительной «промывки» водородом в автоклав вво-
дят под давлением 60 атмосфер 2800 мл водорода, что соот-
ветствует гидрированию одной двойной связи во взятой на-
веске. Гидрирование заканчивают через 1 час, при этом дав-
ление в автоклаве падает до нуля. Гидрогенизат отфильтро-
вывают от катализатора. На водяной бане отгоняют раство-
ритель, а остаток перегоняют в вакууме, отбирая фракцию с
т. кип. 111—112°/9 мм. Кетон — жидкость приятного запаха,
4° - 1,4630; — 0,9706.
Выход 1-(а-фурил)-5-метилгексанона-3 равен 15,35 а, что
соответствует 76% от теоретического (см. примечание 2).
Примечания:
1. Фурфурилиденметилиэобутилкетон получают конденсацией фурфу-
рола с метилизобутилкетоном в спирто-водной среде в присутствии ед-
кого натра [7].
2. Аналогичным путем получают:
фурилпропионовый альдегид из фурилакролеина;
фурилбутанон из фурфурилиденацетона;
фурилпентанон-3 из фурфурилидепметилэтилкетона;
1-(а-фурил)-гексанон-5 из фурилгексадиенона-5 [9].
170
ЛИТЕРАТУРА
1. J. Kasiwagi. Bull. Chem. Soc. Japan, 1, 90 (1926).
2. Harries, Kaiser. Ber,, 32, 1320 (1899).
3. K. Alder, C. Smidt. C. A. 37, 4702 (1943).
4. Q. Bapgellini, E. Martegiani; Zbl., 1913, 1, 296.
5. А. А. Пономарев, 3. В. Тиль, В. В. Зеленкова. Уч. зап.
Саратовск. ун-та, 34, 78 (1954).
6. А. А. Пономарев, В. В. Зеленкова. Ж. общ. химии, сб. П
1115 (1953).
7. Н. Wienhaus, Н. Leongardi. Zbl., 1930, 1, 224.
8. Н. Burdick. Н. Adkins. J. Amer. Chem. Soc., 56, 440 (1934).
9. А. А. Пономарев, 3. В. Тиль. Ж- общ. химии, 27, 1075 (1957).
10. Сб. «Синтезы органических препаратов», вып. 3, М., ИЛ, 1952,
стр. 338.
Поступила в январе 1966 i. Саратовский гос. ун т
УДК 547.722.6.07
N-ФУРФУРИЛ ПРОПИЛЕНДИАМИН-1,3
А. А. ПОНОМАРЕВ, Н. Н. МАСЛЕННИКОВА
I /—СН2—NH—СН2—СН2—СН2—NH2
4OZ
C8H14N2O М. в. 154,21
М-Фурфурилпропилендиамин-1,3 получен только по ниже-
описанному методу путем цианэтилирования фурфуриламина
и гидрированием образующегося аминонитрила [1].
М-Фурфурилпропилендиамин-1,3 может найти применение
в синтезе азотистых гетероциклов и 'потенциально биологиче-
ски-активных веществ.
СХЕМА СИНТЕЗА N-ФУРФУРИЛ ПРОПИЛ ЕНДИАМИНА-1,3
'-ch2-nh2
о
I J!-ch2-nh-ch2-ch2-cn
oz
NiRetH,
NH3(CH3OH)
< '-ch,-nh-ch3-ch2-ch2-nh.
о
Характеристика основного сырья
Фурфуриламин, ч., ТУ TCP 713-р-62, т. кип. 74—75°/60 мм,
п™ —1,4835 (2].
172
Акрилонитрил, ч., ТУ РУ 797—53, т. кип. 77,3°.
Никель Ренея, получен по [3].
Водород электролитический.
Аммиак жидкий, в баллонах.
Условия получения
Синтез П-(2-фурфуриламино)пропионитрила. К 60 а фур-
фуриламина с добавкой 0,7 г гидрохинона при температуре
не выше 20° в течение 75 минут добавляют 33,4 г акрилонит-
рила. После этого смесь перемешивают 4 часа, а затем на-
гревают при 100° 1 час. После 12-часового стояния при ком-
натной температуре полученный продукт подвергают фрак-
ционированной перегонке в вакууме.
Выход И-(2-фурфуриламино) пропионитрила равен 67 а,
что составляет 72% от теоретического; т. кип. 139710 яя;
п™ —1,4929; d420 — 1,0759.
Получение N-фурфурилпропилендиамина-!,3. Во враща-
ющийся автоклав емкостью 150 .ил загружают 46 мл мети-
лового Спирта, насыщенного аммиаком при 0°, 20,9 a N-(2-
фурфуриламино)|пропиопитрила и 6 а никеля Ренея. Началь-
ное давление водорода равно НО атмосферам, температура
120°. Гидрирование ведут до поглощения рассчитанного ко-
личества водорода. Катализат фильтруют, спирт отгоняют и
остаток перегоняют в вакууме.
Выход N-фурфурилпропилендиамина-!,3 равен 17 г, что
составляет 79,4% от теоретического: т. кип. 116710 яя;
—1,4985; d420 — 1,0266.
ЛИТЕРАТУРА
I. А. А. Пономарев, И. М. Скворцов, Н. П. Масленнико-
ва. Ж. общ. химии, 33, ИЗО (1963).
2. А. А. Пономарев, Н. П. Масленникова, Н. В. А л а к и и а,
А. П. Кривенько. Докл. АН СССР, 131, 6 (1960).
3. Сб. «Синтезы органических препаратов», вып. 3, М., ИЛ, 1952,
стр. 338.
Поступила в январе 1966 г.
Саратовский гос ун-т
УДК 547 722.6,07
N-ФУРФУРИЛЭТИЛ ЕНДИАМИН
А. А. ПОНОМАРЕВ, И. М. СКВОРЦОВ
ч z-CH2-NH-CH2-CH,-NH,
XOZ
C7HnNaO М. в. 140,18
Многие амины фуранового ряда являются ценными полу-
продуктами для тонкого органического синтеза [1—7].
Были предложены четыре возможных пути синтеза заме-
щенных алифатических диаминов, в которых замещающей
группой, помимо других, может быть и фурил [8].
Впервые N-фурфурилэтилендиамин синтезирован [9] с вы-
ходом 52%.
Нами предлагается улучшенная методика синтеза этого
вещества [10].
СХЕМА СИНТЕЗА N -ФУРФУРИЛЭТИЛЕНДИАМИНА
( f //° Н2
I Д. + H2N—СН,—СН2—NH2 -й-’
\ / — СП
-(Т
ii и
-* LcH2-NH-CH2-CH2-NH2
Характеристика основного сырья
Этилендиамин, ч., (гидратная форма).
Фурфурол технический.
Водород электролитический.
174
Условия получения
Во вращающийся стальной автоклав емкостью 780 мл за-
гружают раствор 87 г (1,11 Л4) этилендиамина в 200 мл ме-
танола, а затем при перемешивании туда же добавляют 34 г
(0,354 М) фурфурола (см. примечание 1) и 8 г никеля Ренея
[11]. Начальное давление водорода равно 100 атмосферам,
температура гидрирования 80°. После прекращения поглоще-
ния водорода автоклав разгружают- Катализат фильтруют,
метиловый спирт и избыток этилендиамина тщательно отго-
няют из колбы Вюрца на водяной бане при пониженном дав-
лении (см. примечание 2). К остатку добавляют примерно
100 мл эфира, при этом выделяется коричневый осадок.
Смесь сушат едким кали, затем эфирный раствор отфильтро-
вывают от щелочи и осадка. Эфир отгоняют и осадок пере-
гоняют в вакууме из колбы с колонкой Видмера, отбирая
фракцию с т. кип. 103—103,5710 мм.
Выход N-фурфурилэтилендиамина равен 33,8 г, что сос-
тавляет 68,2% от теоретического. Вещество представляет со-
бой бесцветную умеренной вязкости жидкость со слабым
аминным запахом, растворимую в воде, спирте, эфире;
пг° —1,5029; d^a— 1,0470 (см. примечание 3).
Примечания:
1. Применяют свежеперегнанный фурфурол с т. кип. 161’.
2. Полученный метанольный раствор этилендиамина пригоден для
повторного использования в этом синтезе.
3. Анало1ичным путем получают из метплфурфурола н этилендиамина
\-(а-метилфурфурил)-этилеидиамип [10] с выходом 73%: т. кип.
111,5710 мм; —1,4997; d420 — 1,0194.
ЛИТЕРАТУРА
I. А. П. Терентьев, Р. А. Грачева. Ж- общ. химии, 28, 1167
(1958).
2. А. П. Терентьев. Р. А. Грачева, В. А. Дорохов. Ж. общ.
химии, 29, 3474 (1959).
3. А. П. Терентьев, Р. А. Грачева, О. П. Ill к у р к о. Ж. общ.
химии, 30, 3711 (1960).
4. И. Ф. Бельский, Н. И. Ш у й к и н. Докл. АН СССР, 137, 331
(1961).
5. И. ф. Бельский. Ж- общ. химии, 32, 2908 (1962).
li С. Wilson. J. Amer. Chem. Soc., 67, 093, (1945).
7. А. А. Пономарев, И. M. Скворцов. Ж. общ. химии, 32, 97
(1962).
8. S. Lester. В. William. Пат. США 286SS33; РЖхим, 1961;
№ 2, 2Л181.
9. R. Graf. Пат. США 2817757; Chem. Abstrs., 37, 5988 (1943).
10. А. А. Пономарев. И. М. Скворцов, Н. П. М а с .т е и н и к о-
в а. Ж. общ. химии, 33, ИЗО (1963).
11. Сб. ^Синтезы органических препаратов», вып. 3, М„ ИЛ, 1952,
стр. 338.
Поступила в январе 1966 г. Саратовский гос. уи-т
175
УДК 547.233.4'113.1.07
(2-ХЛОРЭТИЛ)-ТРИМЕТИЛАММОНИЙХЛОРИД
Г. Н. НАЛЕЦКАЯ, А. П. ДУБРОВ, Г. Н. КОШЕЛЕВА
C1H2C-H2C-N-(CH3)3
Cl
C5H13NC12 М. в. 158,07
(2-Хлорэтил)-триметиламмонийхлорид применяется в ка-
честве антигиббериллина в биохимических 'исследованиях.
В литературе описано получение этого вещества взаимо-
действием холинхлорида с хлористым тионилом [1]. Холин-
хлорид может быть синтезирован из этиленхлоргидрина и
спиртового раствора триметиламина [2, 3].
Нами была воспроизведена литературная пропись и уточ-
нены некоторые условия синтеза.
СХЕМА СИНТЕЗА
(2-ХЛОРЭТИЛ)-ТРИМЕТИЛ АММОНИИХЛОРИДА
HOH2C-CH2Ci + (CH3).,N HOH2C-CH2-N—(СН3)з
Cl
НОН2С-СНа—N-(CH3) + SOC12 -*
Cl
С1Н2С-СН3-Й-(СН3)з + so2 + на
Cl
Характеристика основного сырья
Этиленхлоргидрин, ч., ТУ ОРУ 66—56.
Триметиламин солянокислый, ч., ВТУ МХП 2939—51.
Хлористый тионил, ч., ВТУ МХП 3591—62.
176
Условия получения
Синтез холинхлорида. Холинхлорид получают взаимодей-
ствием 33%-ного абсолютного спиртового раствора триме-
тиламина с этиленхлоргидрином (см. примечание).
Реакцию проводят в запаянных ампулах при температу-
ре бани 80—90° в течение 2 часов. В реакцию вводят эквимо-
лекулярные количества реагентов: 266 г (1,48 Л4, считая на
триметиламин) 33%-ного абсолютного спиртового раствора
триметиламина и 120 г (1,48 М) этиленхлортидрина.
Каждую ампулу заполняют реакционной смесью на поло-
вину ее объема.
По окончании реакции .ампулы вскрывают, этиловый спирт
удаляют на водяной бане в вакууме водоструйного насоса и
получают кристаллический холинхлорид, который пригоден
для следующей реакции без предварительной очистки.
Выход холинхлорида равен 132,7 г, что составляет 63%
от теоретического.
Получение (2-хлорэтил)-триметиламмонийхлорида. В
круглодонную двугорлую колбу, снабженную обратным хо-
лодильником и капельной воронкой, помещают 132,7 г
(0,095 М) холинхлорида и к нему добавляют по каплям 141 г,
(84,9 мл 1,1 А4) свежеперегнанного хлористого тионила. Ре-
акция сопровождается бурным выделением сернистого газа
и хлористого водорода.
После прибавления всего количества хлористого тионила,.
реакционную смесь кипятят в течение 1 часа, после чего из-
быточный хлористый тионил отгоняют от реакционного про-
дукта, который дважды перекристаллизовывают из смеси ме-
танол-эфир. Осадок выпадает при охлаждении раствора в
бане метанол-сухой лед. (2-Хлорэтил)-триметиламмонийхло-
рид по внешнему виду представляет собой кристаллическое
вещество белого цвета, сильно гигроскопичное, хорошо рас-
творимое в воде.
Выход равен 112,5 г, что составляет 75% от теоретическо-
го.
Найдено, % : N -8,88.
C,H13NCla. Вычислено, W: N—8,86.
Примечание.
Получение спиртового раствора триметиламина из солянокислого
триметиламина см. [2].
ЛИТЕРАТУРА
1. S. L. Fries, W, J. McCawille. J. Amer. Chem. Soc., 76, 8,
226 ) (19’>4)
2. Сб. «Синтезы орг. препаратов», вып. 1, М„ ИЛ, 1949, стр. 401.
3 К. А. Н о f m а п п. Chem., Вег., 44, 17о6 (1911).
Поступила в декабре 1961 г. Институт химии природных соединений АН СССР
Биолого-почвенный факультет МГУ
177
12 Чди,
УДК 547.789.6'212.07
2-ЭТИЛБЕНЗТИАЗОЛ
В. Г. БРУДЗЬ, Д. А. ДРАПКИНА, В. А. ПНШАКОВА,
И. П. ПЛ ИТ ИН А
C9H,NS М. в 163.18
2-Этилбензтиазол был получен взаимодействием о-ами-
нотиофенола с пропионовым ангидридом [1], пропионилхло-
ридом (в запаянной трубке) [2] и триэтилортопропионатом
[3].
Нами 2-этилбензтиазол получен действием пропионилхло-
рида на о-аминотиофенол при атмосферном давлении. Ди-о-
нитрофенилдисульфид восстанавливали сернистым натрием
до о-аминотиофенола, который использовался для следующей
стадии непосредственно, без выделения из реакционной сме-
си аналогично методу [4]. Авторы этой работы получали
2-метилбензтиазол путем взаимодействия уксусного ангидри-
да и о-аминотиофенола.
СХЕМА СИНТЕЗА 2-ЭТИЛБЕНЗТИАЗОЛА
4 | II Ц | + 14Na2S + 11Н2О
/\//
no2 no2
/4/SNa
-Э 8 || I + 7Na2S2O, + 6NaOH
178
SNa
NH2
hci
— NaCl
/\/SH
_сн,сн3соа
“hci, -h3o
^X/.N
I II >-c2h6
W\sz
Характеристика основного сырья
Натрий сернистый, ч., ГОСТ 2053-43.
Ди-о-нитрофенилдисульфид (получение см. [5]).
Хлорангидрид пропионовой кислоты, ч., ВТУ ХСНХ
178-53.
Соляная кислота, ч., ГОСТ 3118-46.
Едкий натр, ч., ГОСТ 4328-48.
Хлороформ, ч., ГОСТ 3160-51.
Условия получения
Смесь 250 г (1,04 М) кристаллического сульфида натрия
(Na2S-9H2O) и 156 мл воды нагревают до 100° на глицери-
новой бане в четырехгорлой колбе емкостью 1000 мл, снаб-
женной мешалкой, термометром, капельной воронкой и об-
ратным холодильником. К раствору прибавляют порциями
57,8 г (0,187 Л1) ди-о-нитрофенилдисульфида с такой скоро-
стью, чтобы температура смеси была 100—110° (см. приме-
чание). Затем при этой же температуре размешивание про-
должают еще 30 минут. Полученный раствор красно-бурого
цвета охлаждают до 15° и с помощью капельной воронки
вводят под его поверхность 47 мл концентрированной соля-
ной кислоты (температура 15-20°), после чего приливают за
30-40 минут 95,3 г (1,03 Л1) пропионилхлорида (/ 20—25°) и
далее еще 30 мл концентрированной соляной кислоты (за 10
минут). Смесь нагревают до кипения и кипятят в течение 30
минут, при этом образуется маслянистый слой. К горячей
смеси добавляют снова 30 мл концентрированной соляной ки-
слоты. По охлаждении реакционной массы до 20° выпадает
грязножелтый осадок (сера), который отфильтровывают.
Фильтрат подщелачивают 225 мл 30%-ного водного раствора
едкого натра до щелочной реакции по фенолфталеину. Вы-
делившийся 2-этилбензтиазол отгоняют с водяным паром и
извлекают из дистиллята хлороформом (3 раза по 70 мл).
Экстракт сушат прокаленным сульфатом натрия. После от-
гонки хлороформа остаток перегоняют в вакууме, отбирая
фракцию, кипящую при ИЗ—114°/10 мм.
12* 179
Выход 2-этилбензтиазола равен 33,2 г, что составляет
54,3% от теоретического, считая на ди-о-нитрофенилдисуль-
фид; Яд— 1,6030.
По литературным данным, т. кип. вещества 252°/760 мм
[2, 6, 7], 132°/18 мм [3]; =1,60048 [6, 7].
Примечание.
При слишком быстром добавлении дн-о-ннтрофенилдисульфида про-
исходит сильный разогрев реакционной массы и возможен выброс. Для
дайной загрузки время прибавления составляло 30 минут.
ЛИТЕРАТУРА
1. F. М. Н a m е г, R. J. Rathbone. В. S. Winton. J. Chem.
Soc., 1£47, 954.
2. A. W. Hofmann. Ber., 13, 21, (1880).
3. Q. L. Jenkins. A. M. Knevel, Ch. S. Davis. J. Org, Chem.,
26, 274 (1961)
4. M. В. Дейч мейстер, H. H. Свешников. Журнал «Кино-
техника». Научно-техн. сб„ (4) 117 (1963).
5. Син. орг. препаратов, 1, М., Госхимиздат, 1949, стр. 200,
6. J, Metzger, Н. Plank. Bull. soc. chim France, 1956. 1692.
7. J. M e t z g e г. С. C h e г r i e r. Compt. rend.. 2^6, 898 (1948).
Поступила в июне 1S66 г. ИРЕА
АЛФАВИТНЫЙ ПЕРЕЧЕНЬ СОЕДИНЕНИЙ,
ОПИСАННЫХ В НАСТОЯЩЕМ СБОРНИКЕ
7-Азаоктагидропирроколнн......................................... о
6-Амилииридазннон-З............................................ 30
2-, 3- или 4-н-Амилпиридин...................................... 87
Амилфурилкарбинол................................................ 81
2-Амипо-3,5-дийодоензойная кислота............................ 147
2-Аминотолуол-4-диметилсульфамид................................. 11
2-Антраценсульфохлорнд......................... . -........... 15
Ацетат 1-ацетилиндол-З-ола..................................... 36
1-Ацетил-З-ацетоксиандол........................................ 36
1-Ацетил-2,3-дигидроиндол........................................ 18
3,4-Беиз-9-акридиловый альдегид ..............................
Бор трехфористый............................................
Бромистый водород.............................................
б'-Бром-8'-нитро-3,3' - диметилспнро-(бензтиазол-2,2 - (’'H-l'-бенэо-
пиран)] ......................................................
js-Бромэтилфталимид ..........................................
5-Бром-2-нитрофуран.......................... ................
н-Бутилацетат.................................................
тргт-Бутилкарбазат ...........................................
лгретп-Бутнл-Б-метилтиокарбонат ..............................
ягрют-Бутилоксикарбоинлгидразин . . ..........................
1-Бутирил-2,3-дигидронндол....................................
158
112
81
46
1.03
20
23
26
27
26
19
Вторичнооктилфурилкарбинол . . ............................... 61
6-Гекснлпирндазинои-З.......................................... 30
Гексилфурилкарбинол ........................................... 60
6-Гептилпиридазинон-З.......................................... 29
Гептилфурилкарбинол............................................ 61
Д-Глюкозамин хлоргндрат........................................ 31
6-Децилпиридазинон-З............................................ 30
1,4-Диазабнцикло (4, 3, 0) нонан............................... 60
Диаммоннйпая соль о-сульфобензойной кислоты.................... 34
ОДЧ-Диацетилиндоксил............................................ 36
1,7-Дибромгептанон-4............................................ ’38
1,7-Дибромоктанон-4............................................. 139
2,3-Дигидропндол.................................................. 39
1,2-Дигндропирролизин............................................. 68
N, Ы-ДикарбокстГметнлэтиленднамино-бис-метнлфосфиповая кислота 42
181
5-Диметиламииометпа-1,2-дигидропирролизин.......................... 71
2,2-Диметил-1,6-диоксаспйро (4,4) нонан............................ 72
2,3-Диметилиндолии................................................. 40
3.3'-Диметилспиро-[беизтиазолин- ,2'-(2Н-Г-бензопираны)] .... 44
2,5-Диметилс)1иро-3-(а-фурил)-гептанол-5........................... 79
2.5-Диметил-3-(я-фурил)-октанол-;>................................. 79
2,5-Днметокси-2,5-ди1 идрофурфуриламиноэтаиол...................... 48
2,!’-Динафтил-4-фенилпирилий перхлорат........................... 154
1,6-Диоксаспиро-(4,4)-ионан........................................ 73
1,5-Ди-(2'окси-3',5',6'-трихлорфенил)-3-ацетилформазан............. 50
2,6-Дифеинл-4-(л-нитрофенил)-иирилий перхлорат................... 154
2,6-Дифенил-4-(п-нигрофенил)-пирилий перхлорат . . •........... 154
Додецилфурилкарбинол .............................................. G1
Изобутиланетат ................................................
З-Изобутил-1,2-дигидропирролизин.................................
2-Изобутил-1,б-диоксасииро-(4.4)-ионан.........................
Изобу 1Ил-5-формил- ,2-дигидропирролизин.......................
6-Изогексилпиридазипоп-З.......................................
6-Изопропил-з-метил-1,2-дигидропирролизии......................
2-(Инденил-1) индаион-1........................................
Индоксилацетат.................................................
1-Каппонил-2,3-дигидроиндол....................................
N- К арбоксиметил диэтил ентриамино-Ь1'1М"-бис-изопропилфосфино-
вая кислота..................................... ..............
(-Кетонодекановая кислота.....................................
f-Кет шододекановая кислота ..................................
-f-Кетоиотетрадекановая кислота................................
•f-Кетонотридекановая кислота..................................
у-Кетоиоуидекаиовая кислота ...................................
23
68
73
96
30
68
64
54
19
57
62
62
62
62
59
З-Метил-7 азаоктагидропирроколин.............. .................
2-Метил-3,4,5,6,-бис-инденопириллий перхлорат...................
3-Метил-5-(гексаметилениминометил)-1,2-дигидропирролизин . . . .
З-Метил-1,2-дигидропирролизин...................................
3-Метил-5-димегиламинметил-1,2-дигидропирролнзии................
2-Метил-4,6-ди-(п-метоксифепил) пирилий перхлорат...............
2-Метил-1,6-диоксаспиро-(4Д)-ноиаи..............................
2-Метил-4,6-ди-(«-тиенил)пирилий перхлорат......................
2-Метил-2,4-диэтил-1,6-диоксаспиро-( 1,4)-нонап.................
2-Метил-4,6-ди-(п-этоксифенил) пирилий перхлорат................
Метилен йодистый................................................
1-(5-Метил-3-изопропил-2-пирролидил)-пропанол-3.................
2-Метилиндолин..................................................
3-Метил-5-(М-морфолинометил)-1,2-дигидроппрролизип .............
N-Метил-1-оксипиперидии ........................................
3-Метил-5-(Ч-пиперидинометил)-1,2-дигидропирролизин.............
1-(5-Метил-2-пирролидил) пропанол-3.............................
2-Метил-4,6-ди-(я-тиенил) пирилий перхлорат.....................
1-Метил-3-(я-тиенил)-5,6,7,8-тетрагидроизохромилий перхлорат . . .
М-(я-Метилтетрагидрофурфурил) этилеидиамин......................
2-Метил-1,6,8-триокса дисп иро-(4,1,4,2)-три декан..............
2-Метил-1,6,8-триоксадиспиро-(4,1,4,2)-тридецен.................
2-Метилтиациклооктанон-5........................................
1-Метил-3-фенил-5,6,7,к-тетрагидроизохромилий перхлорат.........
1-Метил-З-фенил 5, 6, 7, 8-тетрагидроизохинолшт.................
З-Метил-5-формил-1,2-дигидропирролизин..........................
2-Метил-3-(а-фурил) гексанол-5......................... . . . .
6
63
71
65
70
76
72
75
77
76
80
128
40
71
82
76
126
75
86
134
88
89
139
91
91
94
163
182
б-Метил-З-(а-фурил) гептанол-5.............................. . 78
5-Метил-3-(я-фурил) октанол-5...................................... 79
М-(я-Метилфурфурни) этилендиамин................................... 175
N-Метил-З-хлорпиперидии ........................................... 97
2-Метил-2-этил-4-изопропил-1,6-диоксаспиро-(4,4)-нонан............. 79
2-Метил-2-этнл-1,6-диоксаспиро-(4,4)-нонан ........................ 130
1-(5-Метил-3-этил-2-пирро 1идил) пропанол-3........................ 128
2-Метокси-7,-диметил-1,6-диоксасииро-(4,4) нонен-3................. 101
2-Метокси-1,6-диоьсаспиро (4,4) нонен-3 .......................... 101
2-Метокси-7-изобутил-1,6-лиоксаспиро-(4>4)-нонен-3................. 101
2-Метокси-7-метил-1,6-диоксаспиро-(4,4)нонен-3................ 99
2-Метокси-7-метил-7-этил-1,6-диоксаспиро-(4,4)-нонен-3............. 101
М-(1-Нафтиламино)-этилфталимид..................................... 104
Ь1-(1-Нафтил)-этилендиамнн-дигилрохлорид........................... 102
6'-Нитро-8'-бром-3,3'-диметилспиро-[бензтиазол-2,2'(2'Н-Г-бензопи-
ран)]........................................................... 46
6'-Нитро-3,3'-диметилспиро-[бензтиазол-2,2'-(2'Н-Г-бензопиран)| . . 46
2-Нитро-4-лиэтиламиностильбен..................................... 106
2-Нитроюлуол-4-диметилсульфамид.................................... 13
6'-Нитро-Ь'-метокси-3,3'-диметилпсиро [бензтназол-2,2'-(2'Н-Г-беи-
зопиран)]....................................................... 46
2-Нитротолуол-4-сульфохлорид....................................... 12
5-(5'-Нитро-2'-фурил)-3-ме>илпиразол.............................. 108
(5-Нитро-2-фуроил) ацетон......................................... 111
2-3- или 4-(5'-Нонадиенил 2',7') пиридин............................ 8
Нонилфурилкарбинол................................................. 61
6-Нонилпиридазннои-З............................................... 30
2-3 или 4-(У-Нонил) пиридин......................................... 7
N-Оксиимид малеиновой кислоты...................................... ИЗ
N-Окси-малеаминовая кислота....................................... 114
2-[(1-Окси-2-нафтил)азо]-8-хинолинол.............................. 119
2-(.-'>-Оксинропил)-5-(3-оксиоутил) фуран......................... 116
8-Оксихииолип-(2 азо-2) 1'-оксинафталин........................... 119
2-(3-< >ксолропил)-5-(3-оксобутил) фуран.......................... 117
2-(3-Ок’иприпи i)-5 (З-фенил-З-оксопропил) г'уран................. 118
2-(3-Оксипронил)-5-(3-фенил-3-оксипропил) фуран .................. 118
6 о-О^тилпиридазинон-3............................................. 30
Октилфурилкарбинол ................................................ 61
2-,3- или 4-(3'-Пентенил) пиридил................................... 8
Пирослизевая кислота............................................. 160
Селенкарбамид .................................................... 122
Селеимочевина .................................................... 122
2,3-Тетраметилеи-1,2-дигидропирролизин............................. 68
2,3-Тетраметилеи-5-диметиламинометил-1,2-дигидропирролизин ... 71
1-(а-Тетрагидрофурил)-3-амннобутан................................ 126
1-(я-Тетрагидрофурил)-1-изопропил-3-аминобутаи ................... 128
1-(а-Тетрагидрофурил)-3-метилпентанол-3........................... 130
1-(а-Тетрагидрофурил)-1-этил-3-аминобутан......................... 128
N-Тетрагидрофурилэтилендиамин..................................... 133
Тетрагилрофурфурилметиламии ....................................... S3
N, N, N', М'-Тетраме1ил-1,3-пропаидиамип......................... 135
Тиацикоокта нон-5................................................. 138
Тимоловый синий................................................... 141
п-Толуолсульфонат-2-этил-З-метилбензтиазолнпий.................... 45
Толуолсульфохлорид 143
183
б-Трндецилпиридазинои-3 ........ ................................ 36
1,6,8-Тридиоксаспйро-(4,1,4,2)-трндекан..............................90
2,3,-5-ТрийодбензоЙная кислота .................... -............. 146
2,4,6-Триметилпнриллий перхлорат................................... 149
2,4,6-ТримеТИЛпиридин ............................................. 149
1,6,8-Триоксадиспиро-(4,1,4,2)-тридецен ....................... 90
2,4,6-Трифенилпиридин.............................................. 152
2,1,9-Трифеиилпиридий перхлорат..................................... 152
3,4,6-1рихлорбензохиноидиазид-1,2............................ . . 52
Феиилглнцин-о-карбоиовой кислоты монокалиевая соль
1-Фенил-3-метил-4-амииопиразолон-5 хлоргидрат . . . .
1-Фенил-3-метил-4-изоннтропиразолон;5..............
9-Формил-1,2-беизакридин ..........................
9-Формил-3,4-бензакридии...........................
5-Формил-1,2-дигидропирролизин.....................
Фуран-2-карбэновая кислота ........................
I-(а-Фурил )-5-аминогексаи ........................
1-(а Фурил)-2-аминоциклогексаи ....................
1-(а-Фурил)-1-аминоэтан............................
Фурилбутанон.......................................
I-,а-Фурил) гексанон-5......................... .. .
З-(а-Фурил) гексанои-5........................Л • -
1-(а-Фурил)-1-изопропил-3-метиламинобутан .С...
1-(а-Фурил)-3-метиламинобутан . ...................
1-(а-Фурил)-5-метиламиногексан . . ................
1-(аФурил)-7-метил-5-аминооктаи................. -
1 -(а-Фурил )-3-метиламинопропаи ...........
1 -(а-Фурил )-5-метил гексаион-3 . . ..............
1.(а-фурил )-6-феиол-5-амииопентан........... . . .
Фурилпентаион-З-хлоргндрат .............
Фурилпропноновый альдегид..........................
1Ч-(2-Фурфуриламиио) пропионитрил..................
М-Фурфурилпропилеидиамин-1,3 .......................
N-Фурфурилэтилендиамии.............................
. 37
155
156
159
158
95.
160
166
166
166
170
17*1
162
168
1,65
165
166
167
.169
16f
I7C
|7(.
173
J72
174
1-Хлорацетил-2,3-дигидроиидол......................1.’ . . . . 19
Холинхлорид .... ...................................... 177
(2-Хлоратил) трнметиламмоний хлорид........................... 176
Этилбензтиазол .................................... . .......
I -Э т ил -3-метил- > ,2-дигндропиррол изин....................
2-ЭгилтЗ-пропилнндолин................. . . . ..............
Этиловый эфир у-кетоногептадекановъй кислоты...................
Этиловый эфир у-кетонодекановой кислоты .......................
Этилоный эфир -j-кетонододекановой кислоты.....................
Этиловый эфир у-кетоно-6-мегил додекановой кислоты.............
Э1 иловый эфир у-ке7онотетрадекановой кислоты..................
Этиловый эфир у-кетонотриаекаиовой кислоты.....................
Этиловый эфир у-кетоноундекаиовой кислоты........................
178
68
40
61
6!
61.
62
Редактор Б. Г. Козлов
Техн, редактор А. И. Пирожкова
Корректор Н. Р. Казарина
Сдано в набор 8.4.1967 г. Подписано к печ. 21. II. 1967 г.
Формат бум. 60 x901/1# Печ. л. 11,5 Уч.-изд. л. 8,7
Л—56599 Тираж 1250 экз. Зак. 266 Цена 70 коп
Типография ВАХЗ
60
Замеченные опечатки
Q. О- Строка Написано Следует читать
20 3 .сверху Б. Н. Новиков В. Н. Новиков
HNO, HNO3
(СН,)3О (СН3СО)3О
22 2 сверху 1932, 1965. 1932, 1, 65. .
2 снизу Ж. орган Ж. общ.
39 2-я формула ... +2СГ- ...|2НС!^
49 6 сверху Электролизер Электролиз
\Л/ \А/
51 3-я формула | || + СН.СОСН, 2 | II + СН,СОСН3
\/\ | Ч/\ 1
60 2 сверху ...— СООС3Н3 ...— СООН
80 3 , А. Л. Лифшиц А. Л. Лифиц
94 2-я формула + HCON(CH2)2 + HCON (СН,),
101 .17’сверху -1,4518 —1,4570
105 и (29% (35%
106 2-я формула on2 O,N
128 13 и 14 снизу З-этил-2-пирролидил 3'-этил-2'-пирролидил
130 3 сверху 1,6-диоксапиро 1,6-диоксаспиро
151 5 снизу Ж. орган. Ж. общ.
154 3 , Ж. орган. Ж. общ.
172 NiRe,H2 > NlRe, Н,
З’Я-формула NH,(CH,OH) NH,(CH,OH)
173 6 сверху Синтез N- Синтез 0-
13 Выходом N- Выходом 0-
18 N-(2- 0-(2-
76 3-я формула - (СНг) + -(СН,),4-