/
















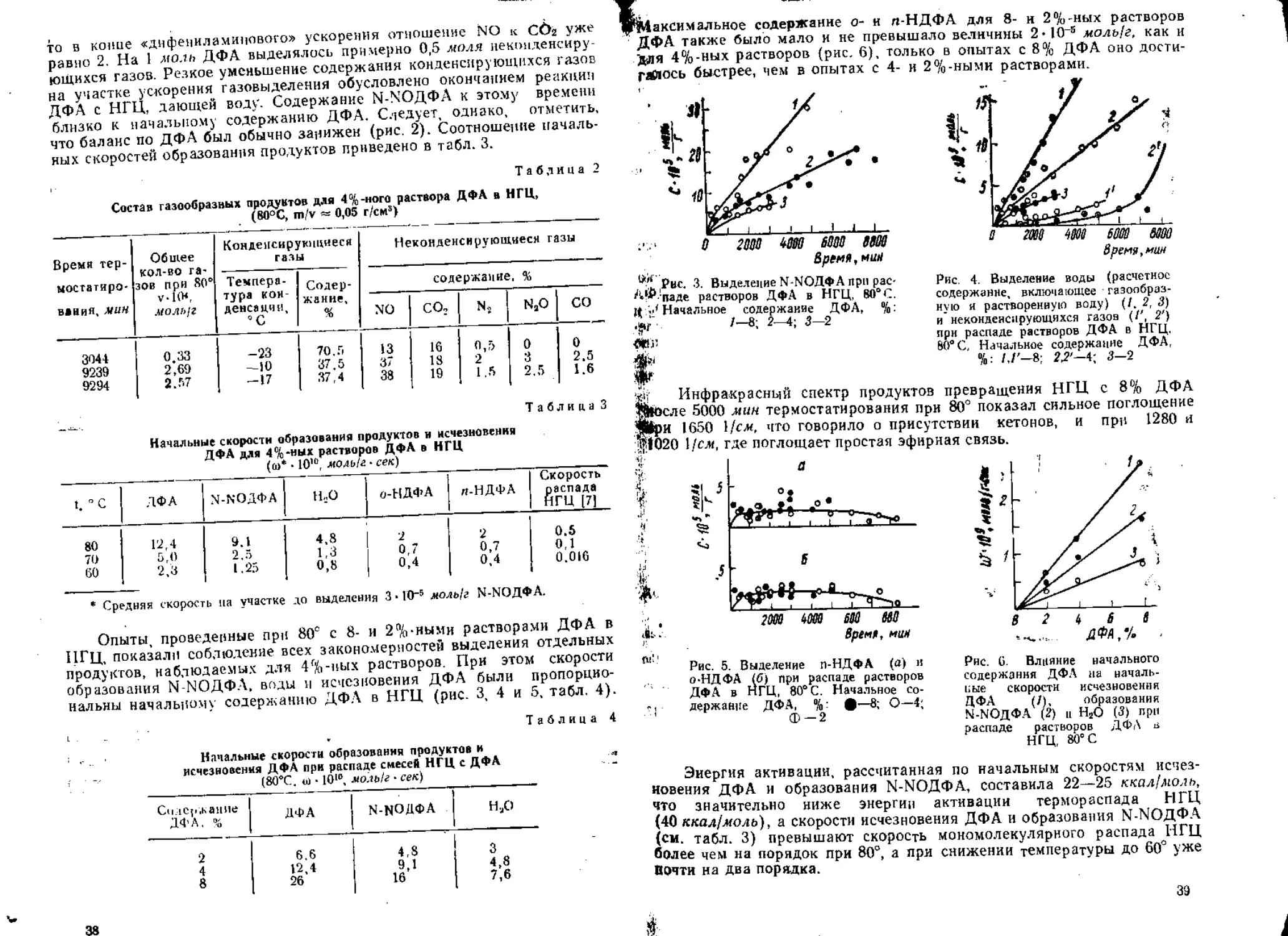

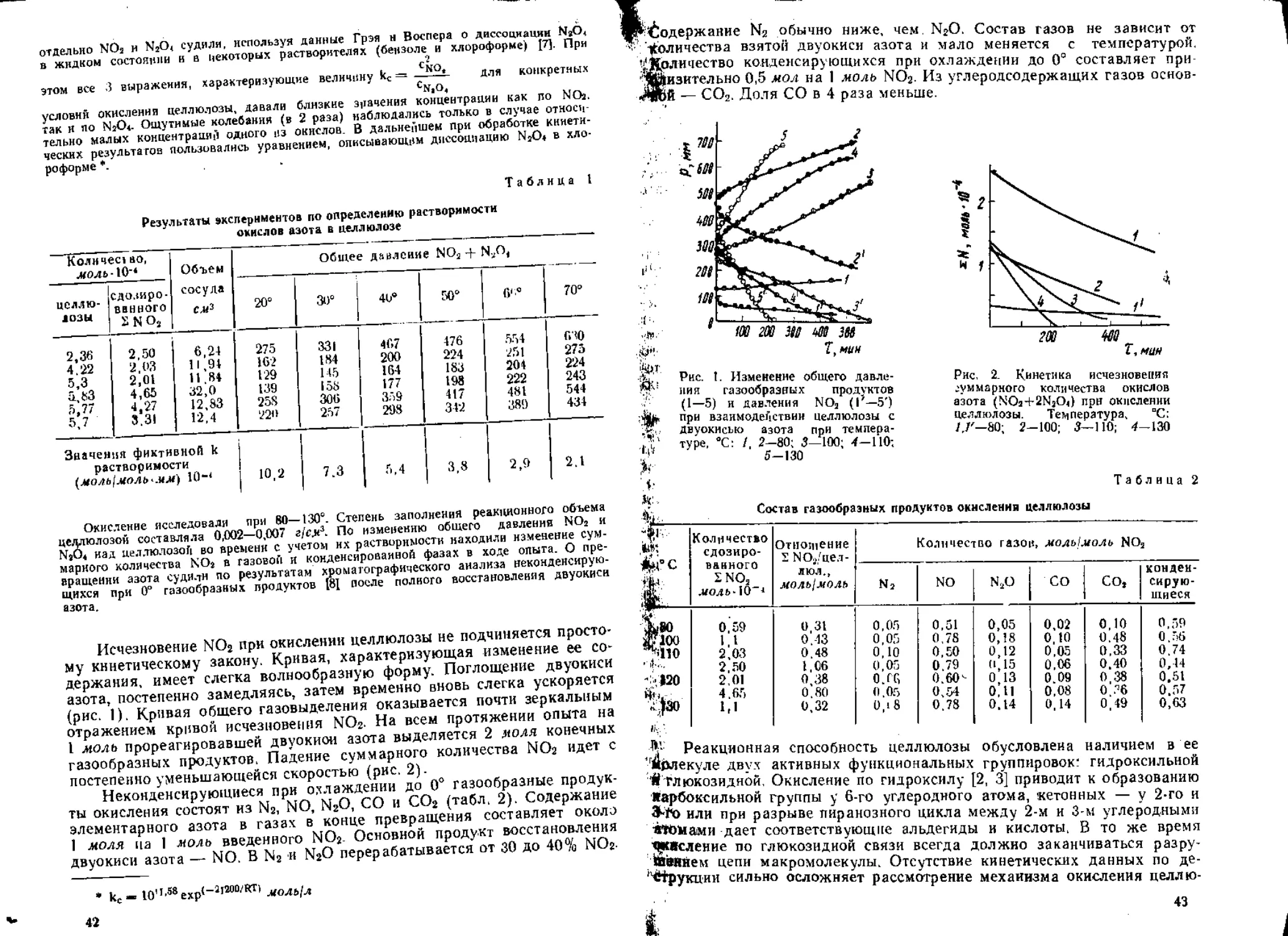




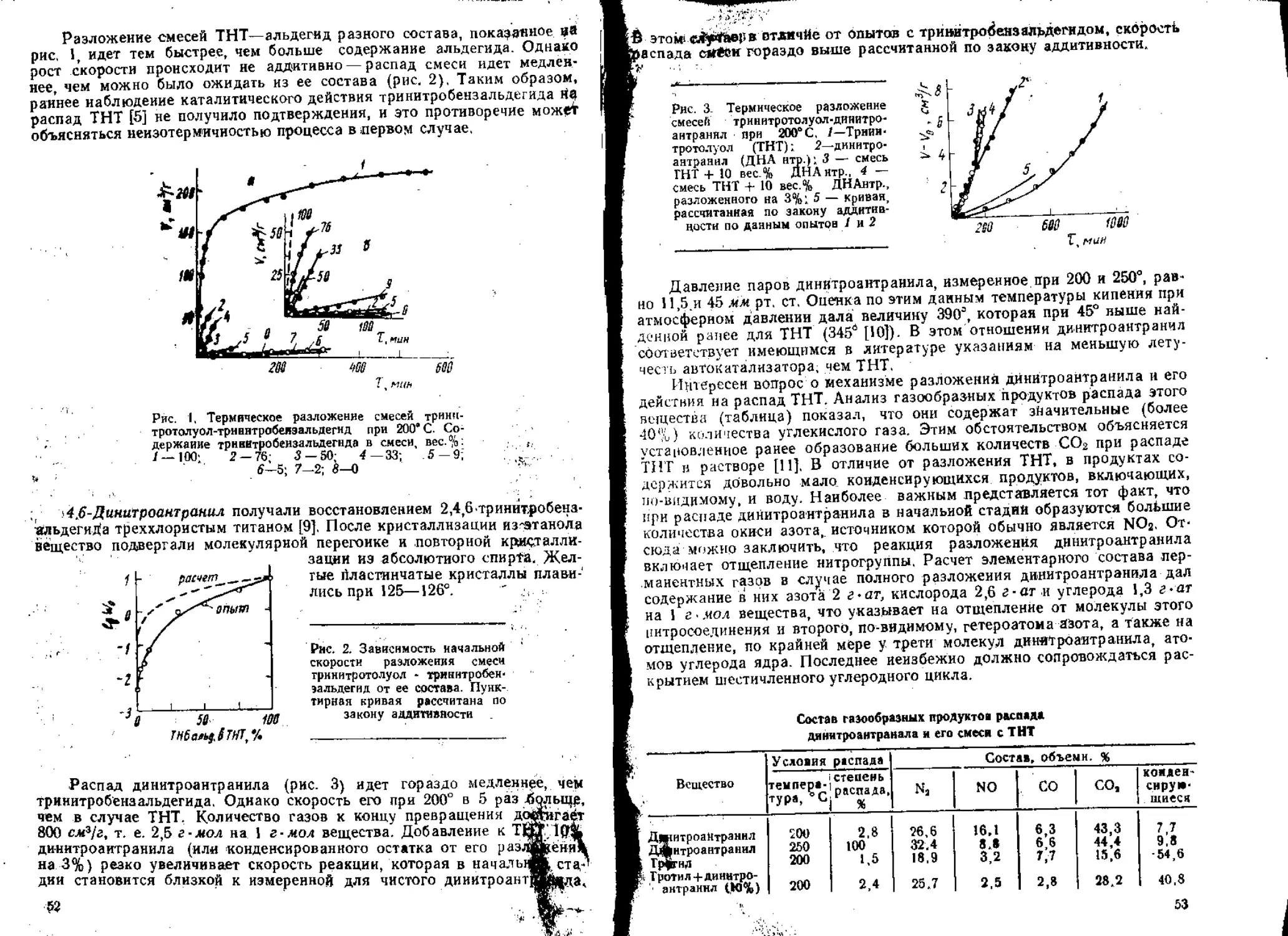

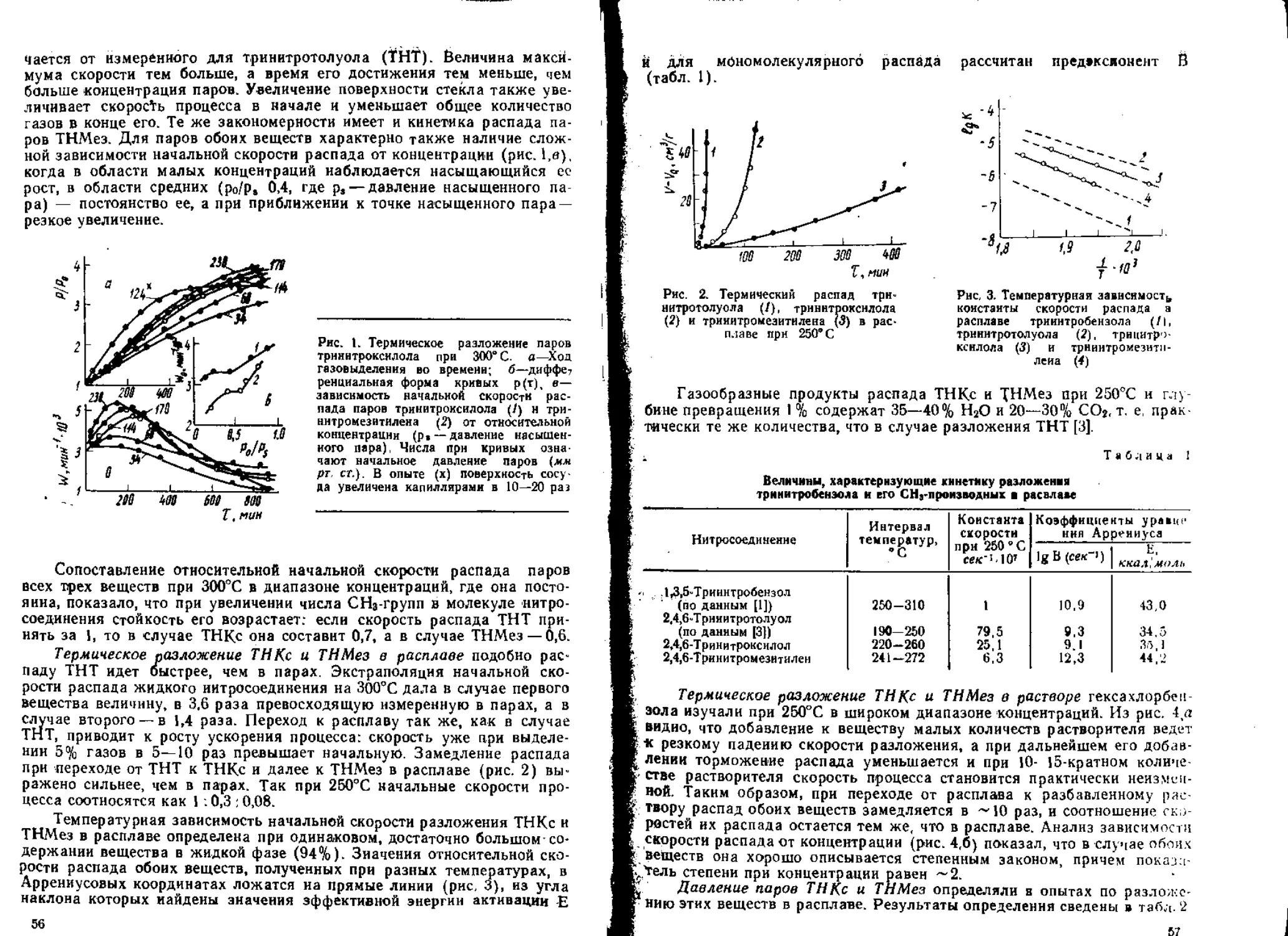


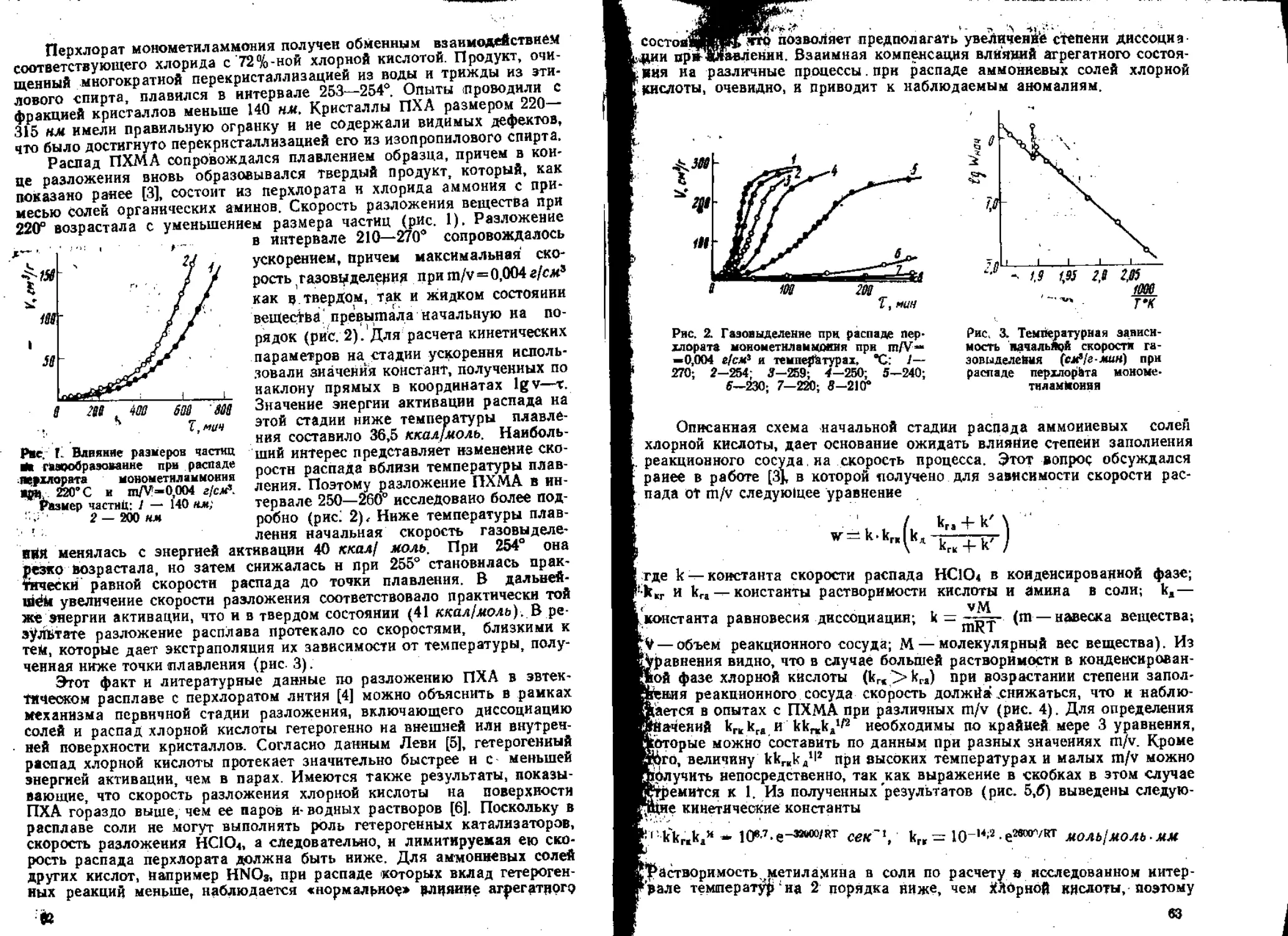


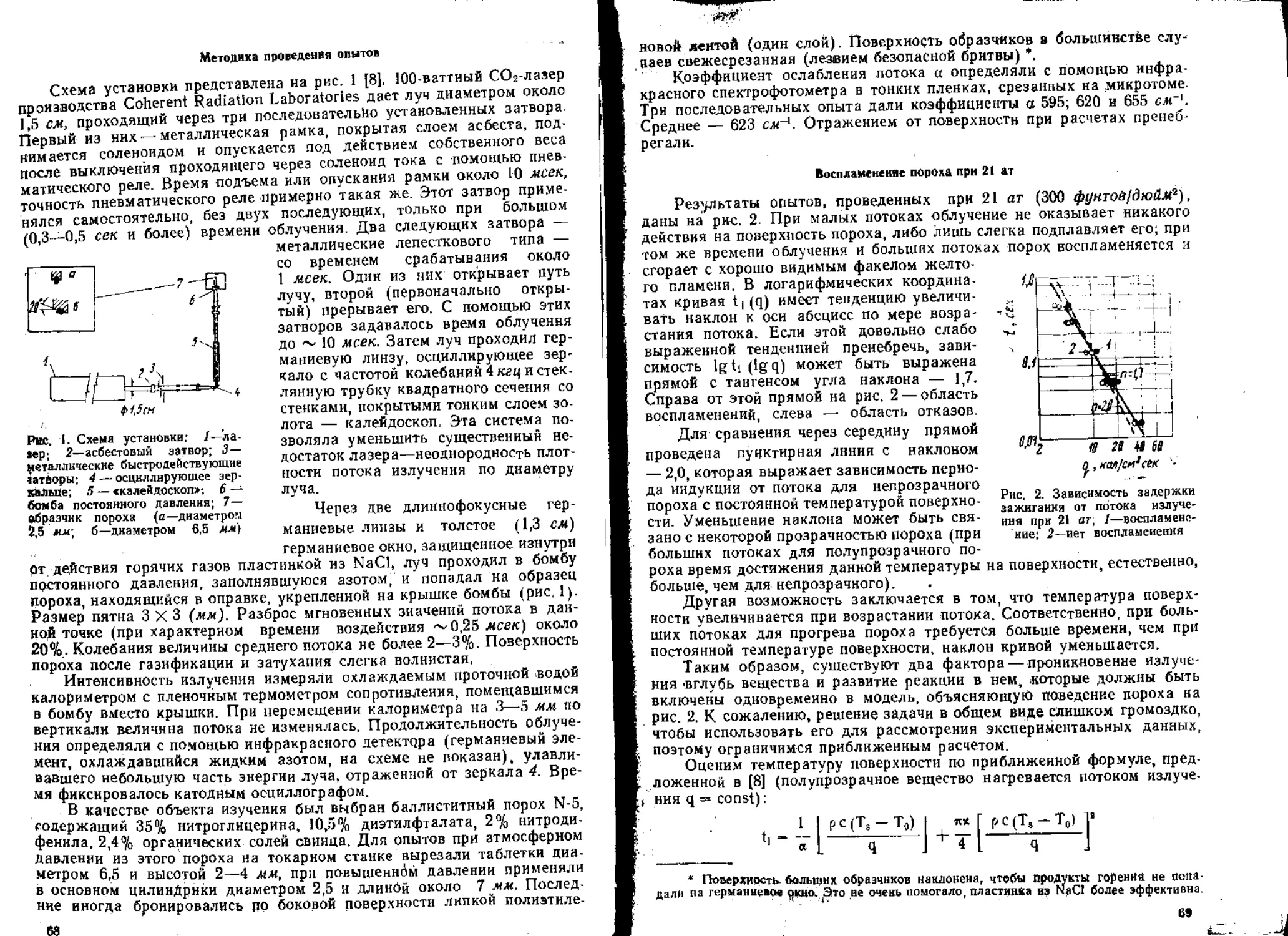
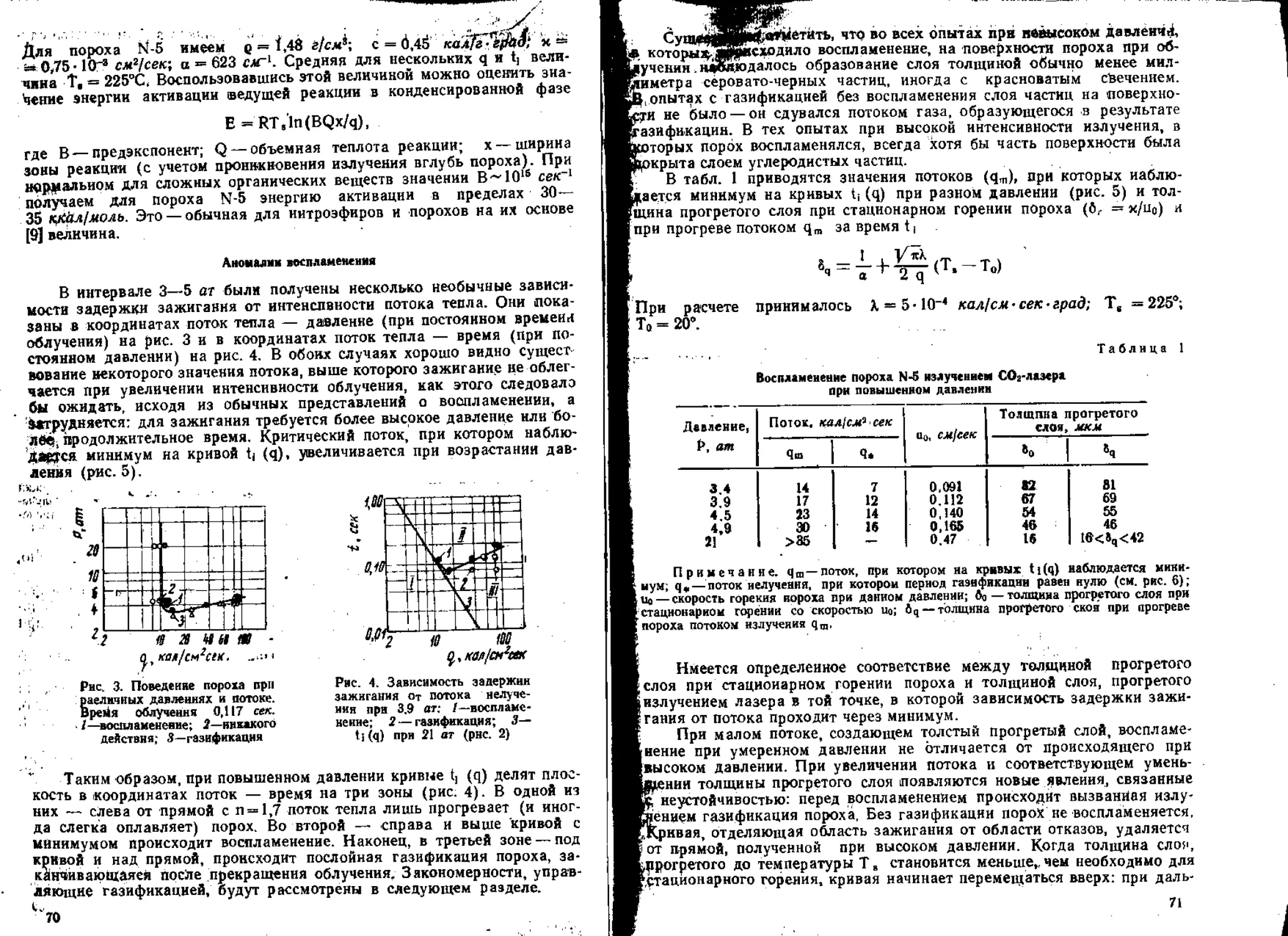
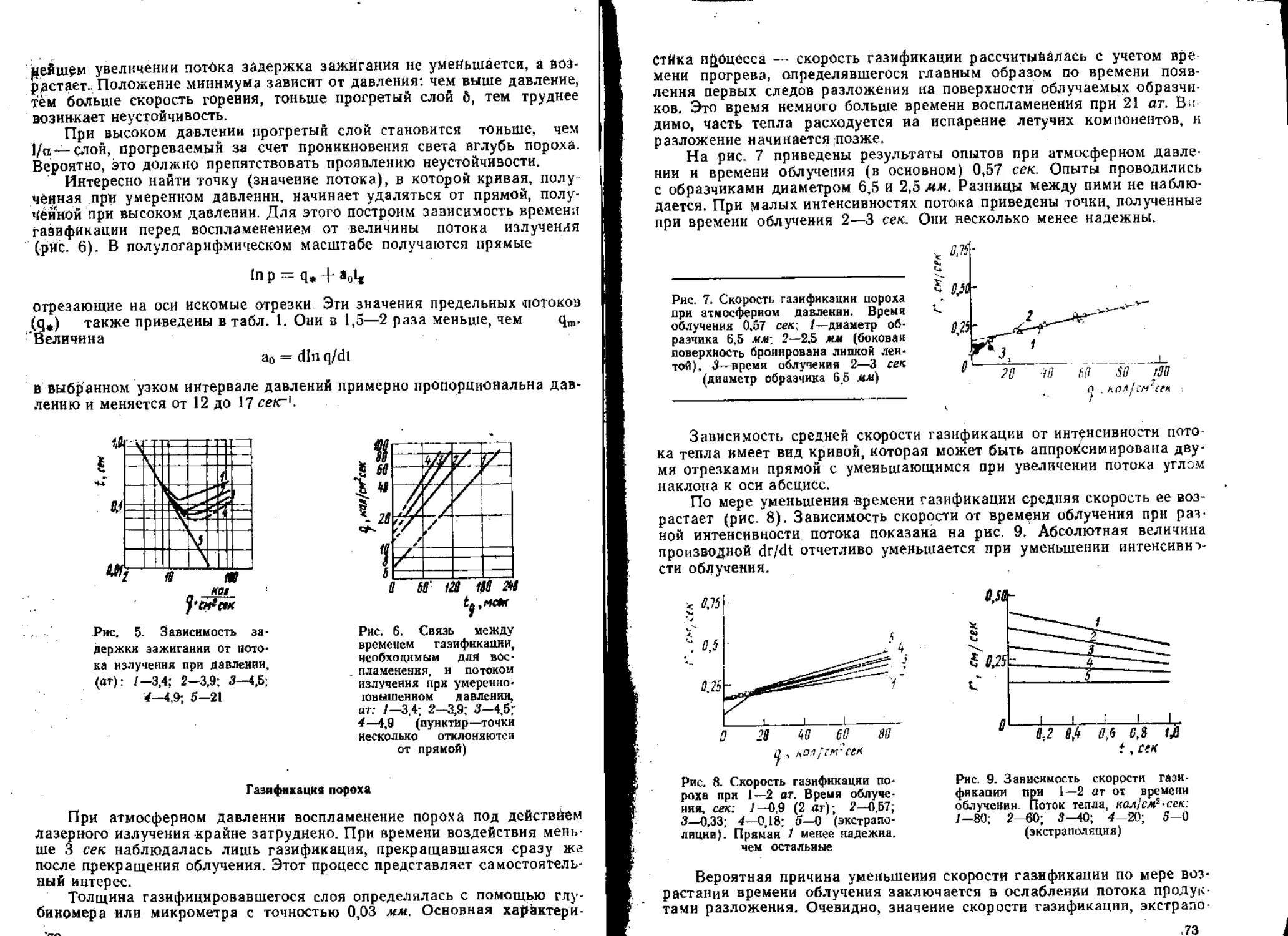













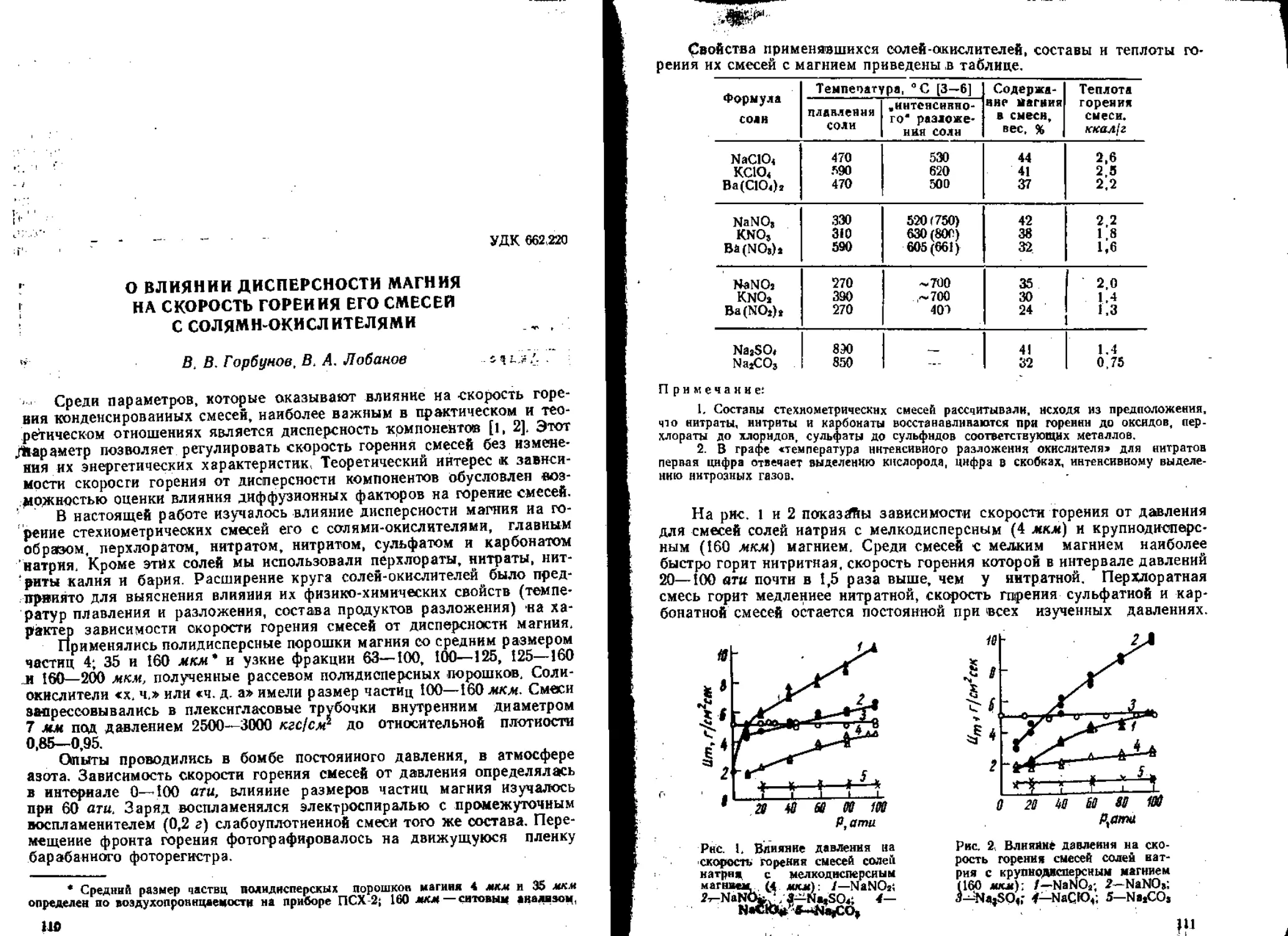

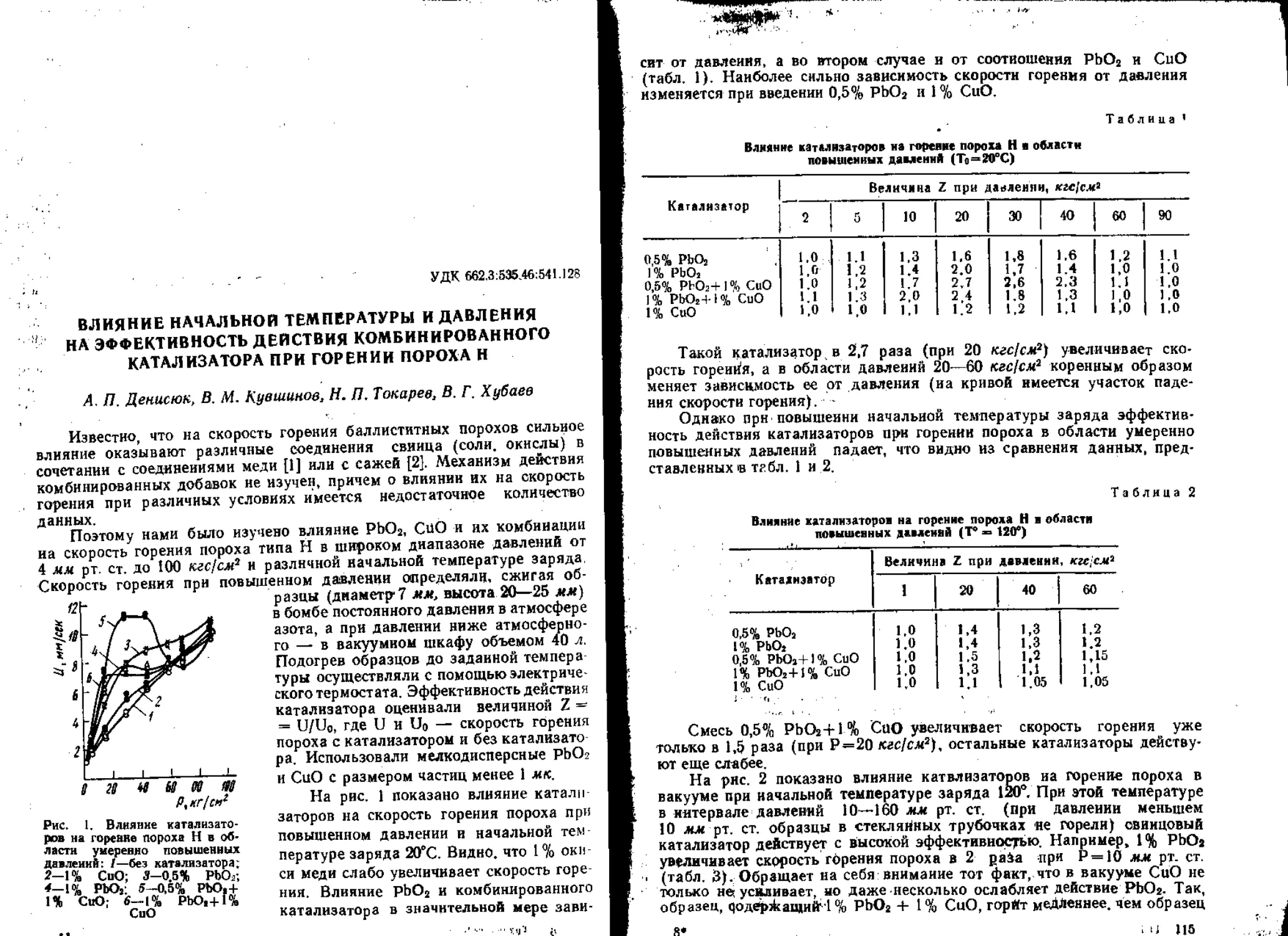
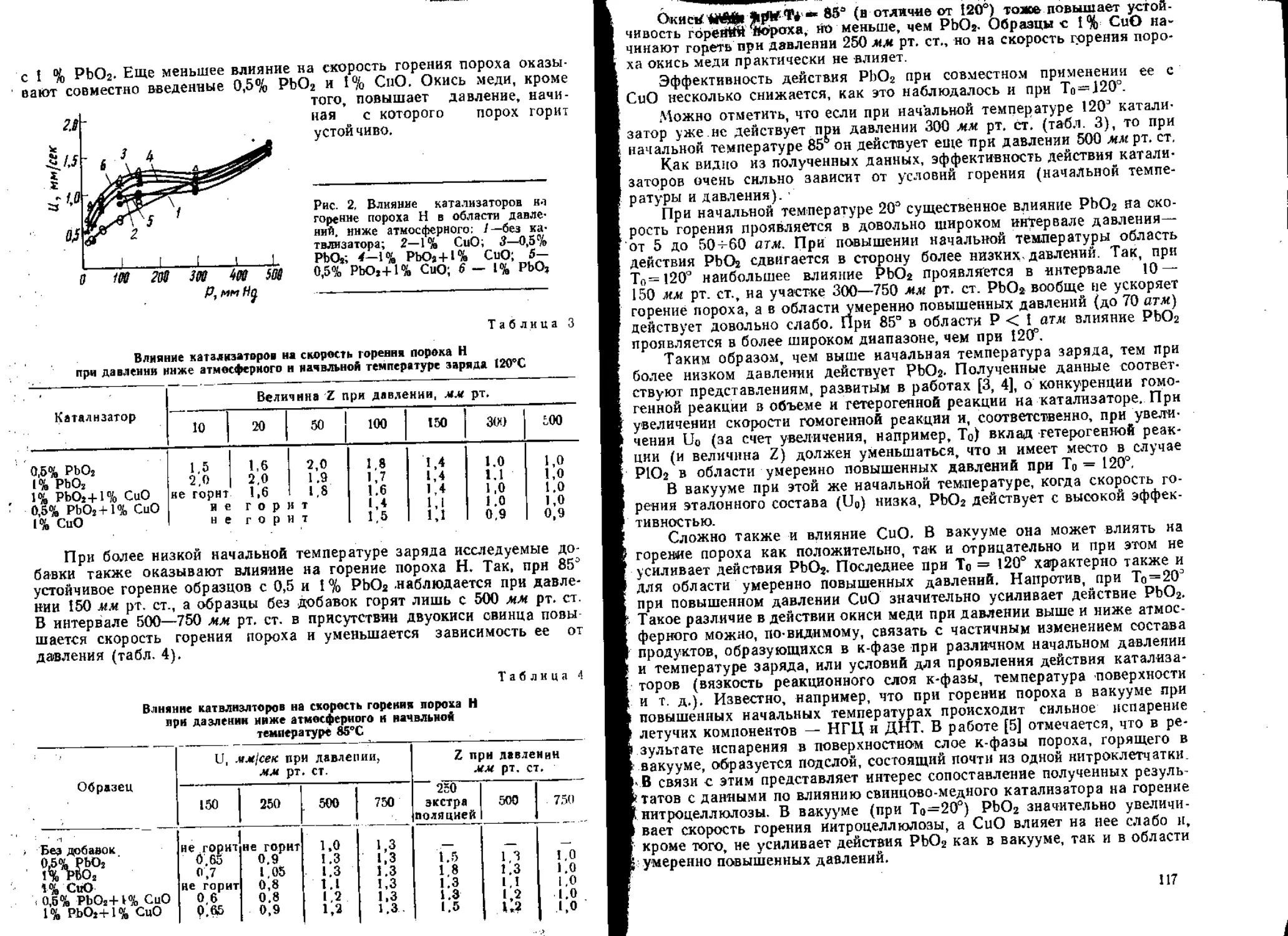















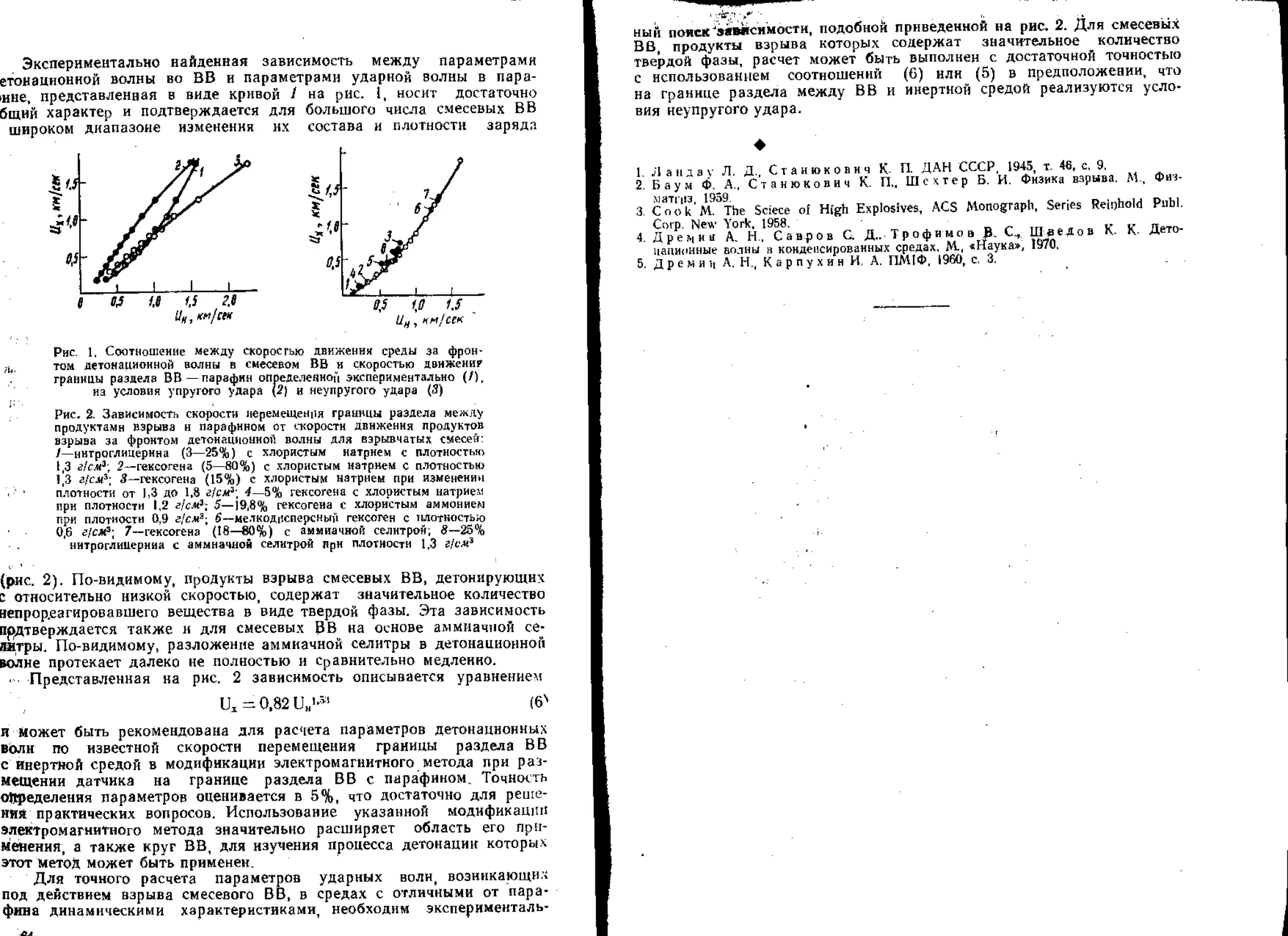
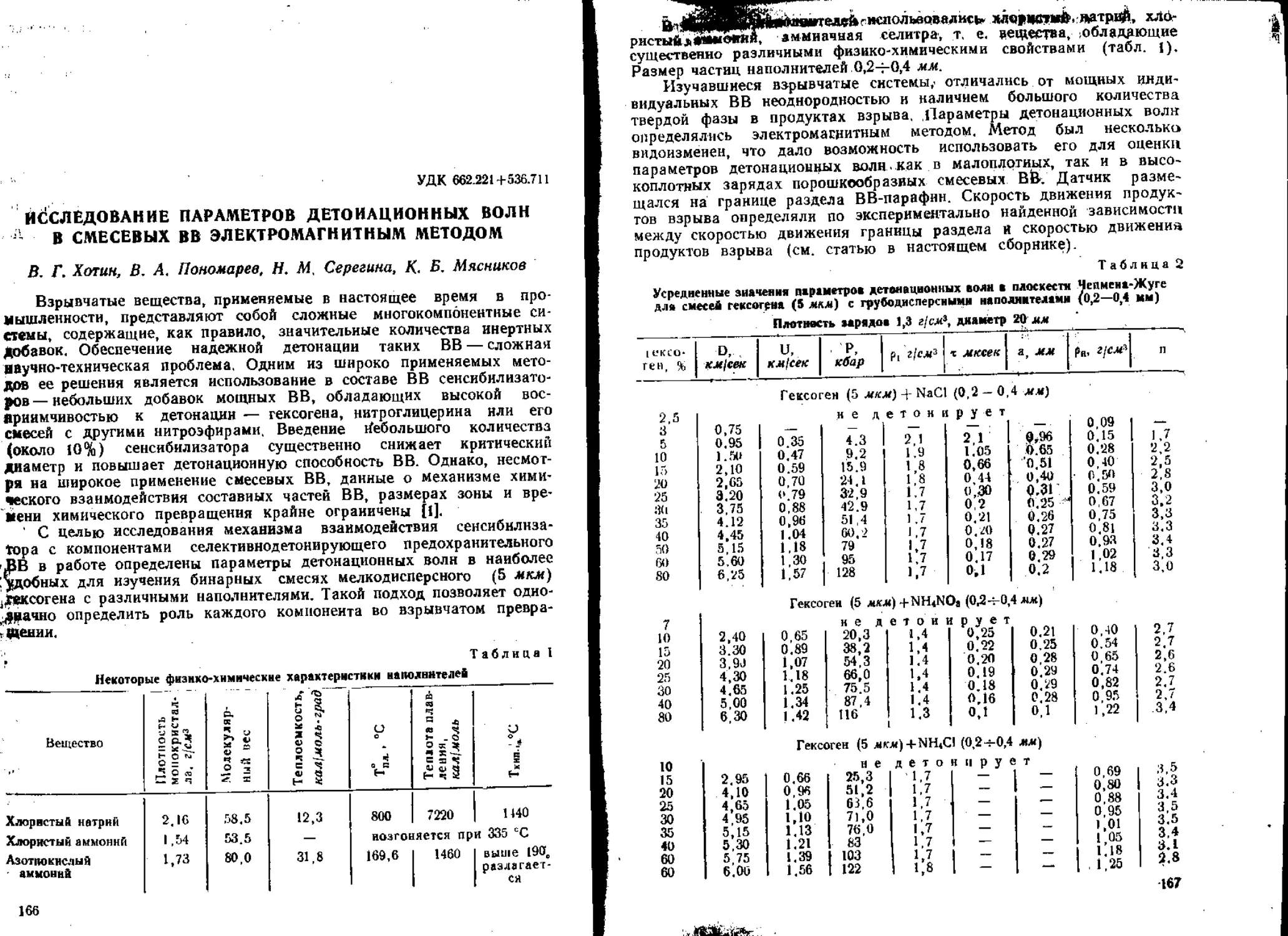
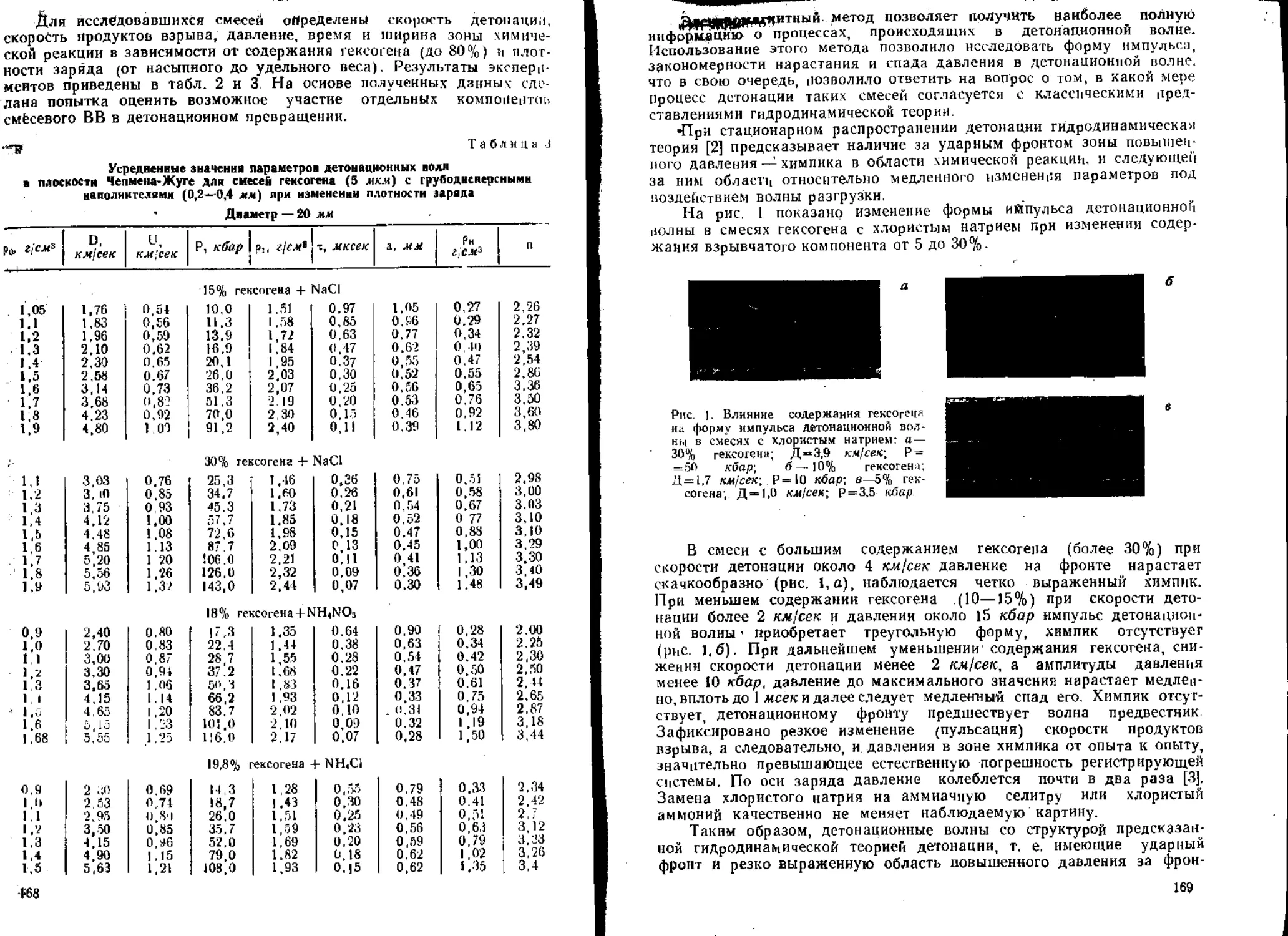



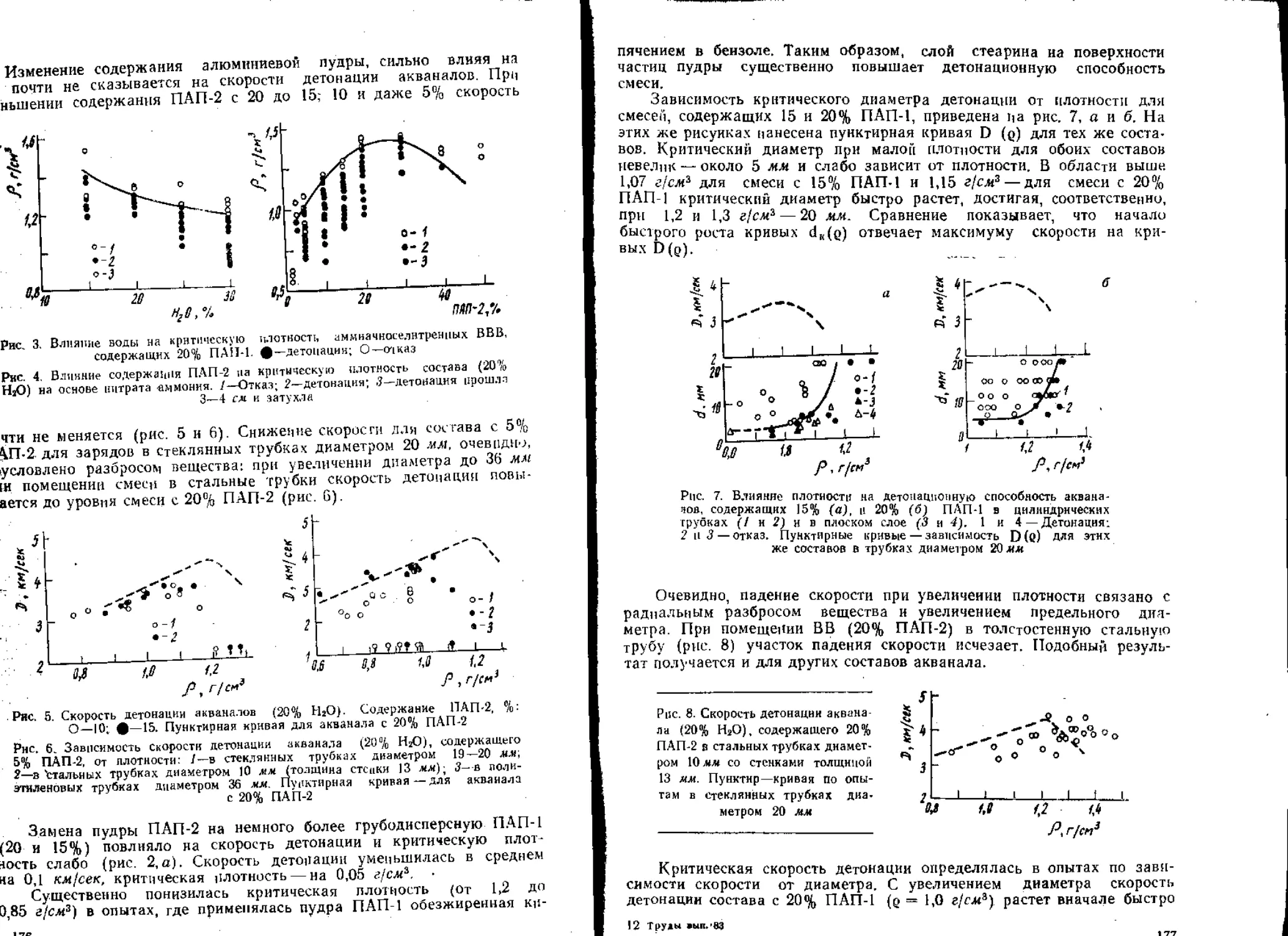












Текст
ВОПРОСЫ ТЕОРИИ
ВЗРЫВЧАТЫХ
ВЕЩЕСТВ
Министерство высшего и среднего специального образования СССР
Московский ордена Ленина и ордена Трудового Красного Знамени
химико-технологический институт имени Д. И. Менделеева
ТРУДЫ ИНСТИТУТА, ВЫПУСК LXXX1II
К ВОПРОСЫ ТЕОРИИ
I ВЗРЫВЧАТЫХ
Ш ВЕЩЕСТВ
' Сборник статей
\Ж '
аОвХ
МОСКВА —1974
SL006
Ндукова С13л1отека J
С-деського yiilceptHieiy
1м. I. 1. Мечникова |
У ПРЕДИСЛОВИЕ.
,ч
f Настоящий сборник посвящен 10-летию со дня смерти одного из
4'. ведущих специалистов в области теория взрывчатых веществ лауреата
^'Государственной премии профессора Константина Константиновича
г Андреева. В сборник вошли работы, выполненные в последнее время
ч учениками К- К. Андреева, главным образом, сотрудниками МХТИ
.им. Д. И. Менделеева, одной из кафедр которого он заведовал более
f 20 лет. Первоначально предполагалось, 4То сборник должен явиться
j творческим отчетом за это десятилетие, который отражал бы дальней-
( Шее развитие научных исследований в направлениях, р зарабатывавших-
ся или начавших разрабатываться профессором К. К- Андреевым. Одна-
ч. ко ограниченный объем издания не позволил в полной мере осуществить
> Этот план.
и- В связи с этим возникла необходимость предварить изложение
оригинальных статей кратким обзором работ, выполненных учениками ;>
и сотрудниками К К. Андреева в указанный период и опубл вкованных
|в периодической печати. Первая часть обзора.шк^ящена вопросам тео-
Жрни взрывчатых веществ, вторая—проблемам химии и технологии нит-
® росоединений. В конце обзора приведен полный список упоминавшихся
в нем оригинальных работ с указанием Названия работы и ссылкой иа
йямтературный источник. В обзор не вошли работы, помещенные в пре-
яОыдущем сборнике, вышедшем в 1967 году в издательстве <Высшая
жшкола». который в основной редакции был подготовлен к публикации
кК К. Андреевым.
Сборник состоит из трех разделов. Первый раздел включает ра-
боты по термическому разложению интроэфиров, ароматических ннтро-
Соединений и солей ряда перхлората аммония. Во второй раздел вошли
исследования по воспламенению и горению различных взрывчатых ве-
Й1еств, смесей и порохов. В третьем разделе помещены статьи по воз-
буждению взрыва ударом и детонации индивидуальных и смесевых
V взрывчатых веществ.
Редколлегия
1
ИССЛЕДОВАНИЯ В ОБЛАСТИ ТЕОРИИ И ТЕХНОЛОГИИ
ВЗРЫВЧАТЫХ ВЕЩЕСТВ
(Краткий обзор)
£, Ю. Орлова
I. ИЗУЧЕНИЕ ТЕРМИН ЕСКОГО РАЗЛОЖЕНИЯ, ГОРЕНИЯ
И ДЕТОНАЦИИ ВЗРЫВЧАТЫХ ВЕЩЕСТВ
Исследование свойств взрывчатых веществ (ВВ) и прежде всегз
их способности к самопроизвольному разложению, начатое профессо-
ром К. К. Андреевым, еще в конце 40-х гг., имеет непосредственное
отношение к решению теоретических и практических проблем химиче-
ской стойкости при хранении, переработке и использовании ВВ, а так-
же поведения их в ряде видов применения.
1. Термическое разложение нитроэфиров
В продолжение и развитие работ по проблеме реакционной способ-
ности нитроэфиров Б. С. Светлов, Б. А: Лурье, В. П. Шелапутина и
другие для детального выяснения кинетики и механизма разложения
нитроэфиров в конденсированной фазе разработали и применили целый
ряд новых методик, дополнивших манометрический способ. Примени-
тельно к решению этой проблемы были использованы хроматографи-
ческий метод анализа газообразных и конденсированных продуктов,
колориметрическое определение двуокиси азота, химические способы
определения малых количеств спиртов, альдегидов, карбоновых и азот-
содержащих кислот, нитритов. Применялась также инфракрасная спек-
троскопия.
- Были изучены кинетика и механизм [I—6], промежуточные и конеч-
ные продукты [3, 7—9] основных химических превращений, протекаю-
щих при термическом распаде большого числа нитроэфиров разного
строения. При этом было установлено, что химико-кинетические осо-
бенности разложения нитратов определяются протеканием четырех ос-
новных процессов: 1) гомолитического обратимого распада с ртрывом
NOa, параметры уравнения Аррениуса для которого Е = 38—42 ккал[моль
и В - 1015— 1018 сек"1 [1, 4, 6]; 2) реакций окисления с участием обра-
зовавшихся окислов азота н алкоксираднкалов с органическими проме-
5
1
«уточными продуктами и исходным веществом [10—13]; 3) гидролиза
[14—16] и 4) непосредственного взаимодействия нитроэфира с выделяю-
щимися органическими кислотами (переэтерификация) [17]. Вклад каж-
дого из указанных процессов определенным образом зависит от хими-
ческого строения нитроэфнра и условий его превращения. В частности,
различия в скорости первичного распада и гидролиза обусловлены ин-
дуктивным влиянием заместителей, входящих в молекулу нитроэфира
[4], а скорость гидролиза и переэтерификации его определяются -концен-
трацией и силой кислоты [15, 16]. На этой основе была проведена коли-
чественная классификация изучавшихся нитратов [6], которая позволяет
по особенностям химического строения предсказывать нх реакционную
способность в различных условиях.
Так, если нитратные группы располагаются в молекуле у рядом
стоящих атомов углерода, то эти вещества способны к развитию рез-
кого ускорения распада, аналогичного тому, как это было установлено
для глицеринтринитрата. В этом случае в продуктах накапливаются
высшие окислы азота и азотсодержащие кислоты. Кинетическое изуче-
ние кислотного гидролиза [14—16] позволило установить, что ускорение
распада действительно обязано развитию этой реакции, которая проте-
кает по автокаталитическому закону с малой энергией активации (20—
25 ккал/моль). Это обстоятельство и обусловливает ограниченную хими-
ческую стойкость нитратов типа глицеринтрииитрата.
Развитие распада остальных нитратов, которые, как установлено, в
меньшей степени склонны к гидролизу, определяется главным образом
быстрыми окислительными процессами. В ходе их восстанавливаются
ие только двуокись, но и окись азота, и основными азотсодержащим.!
•продуктами являются Na и N2O. В этом случае распад идет лишь с
небольшим ускорением [1], обусловленным преимущественно переэтери-
фикацией исходного нитроэфира образующейся карбоновой кислотой
[17]. Превращение нитроэфиров в присутствии восстанавливающих аген-
тов может быть рассмотрено при использовании данных работы [18].
2. Термическое разложение ароматических нитросоедннений
Начатое по инициативе и .под руководством К. К. Андреева систе-
матическое исследование термического разложения ароматических нит-
росоединений, перспективных с точки зрения поисков термостойких В В,
было продолжено в направлении выяснения механизма распада ВВ это-
го класса соединений и влияния на него химической структуры и агре-
гатного состояния. Для этих целей широко использовался хроматогра-
фический анализ газообразных продуктов по разработанной на кафед-
ре и усовершенствованной применительно к изучавшимся системам ме-
тодике, который часто дополнялся инфракрасной спектроскопией и фи-
зико-химическими методами анализа.
•г Изучение распада в парах 22 ароматических полинитросоединений !,
bj'молекуле которых имеются наиболее часто встречающиеся замести-
тели (NO2, СНз, NH2j F, Cl, Вг) [20—22], показало в целом одинаковые
кинетические особенности протекания процесса (мономолекулярность
1 ’.Для всех изучавшихся ароматических иитрососдине;шй мокофеннльного строе-
НИЗ.посоециальио разработанной манометрической методике были измерены давления
насыщенных ларов при разных температурах (19). По этим данным для многих нитро-
соединений впервые были определены температура кипения и скрытая теплота испа-
рения. .
6
тачальной стадии, небольшое ускорение, описыйаейое уравненйеМ автд-
Ждталитической реакции, протекание быстрой реакции на стенке и спе-
цифическое влияние концентрации паров), скорость которого, однако,
зависит от химической структуры нитросоединения. Она, как правило,
возрастает при усложнении структуры молекулы вещества. Установлено,
что механизм первичного распада нитропроизводных бензола [23] и их
м- и п-замещенных [21] идет через разрыв связи С—NO2 (Е = 50 —
65 ккал/моль), в то время как в случае о-замещенных происходит внут-
римолекулярное окисление заместителя, при котором эта связь сохра-
няется (Е^40 ккал/моль}.
Разложение нитросоединений в расплаве существенно облегчается
f межмолекулярными взаимодействиями [24, 25], сопровождающимися
1 протонным или электронным обменом, что в итоге обусловливает изме-
нение механизма начальной реакции разложения: в случае нитросоеди-
ъдаений, включающих водородсодержащий заместитель, происходит меж-
^молекулярное окисление последнего кислородом МО2-групп других мо-
4|Лекул. Впервые развитые представления о внутри- и межмолекулярном
{^цутях разложения одного из наиболее широко известных вторичных ВВ—
* тринитротолуола позволили установить основные реакции, определяю-
'iщие протекание этого процесса [26].
I Проведено также кинетическое изучение термического распада три-
* нитрофлороглюцина — последнего члена ряда ОН-замещенных трннитро-
^.бензола [27]. Установлено, что разложение этого ВВ имеет те же особен-
^'.ностн, что разложение пикриновой и стифниновой кислот, распад кото-
й&ых был исследован К. К. Андреевым. Найдено, что скорость процесса
возрастает при введении в молекулу нитросоеди пен и я каждой ОН-группы
среднем в 101 2 раз.
,’U Изучение кинетики и продуктов распада гексанитродифенил сульфи-
да и гексанитродифенилсульфона [28] показало, что начальная стадия
^разложения этих ВВ состоит не в отщеплении NO2-rpynn, а в-разрыве
связей С—S, сопровождающемся образованием гексаннтродифенила. На
Примере первого вещества [29] экспериментально подтверждена гипотеза
£р,роли примесей в искажении линейной зависимости Ig к(1/Т) три тер-
мическом распаде ВВ ниже точки плавления, высказанная К. К- Ан-
^реевым еще в 1960 году. Нацкновании этой гипотезы выведено уравне-
ние, описывающее температурный рост начальной скорости распада
’^рердого вещества, содержащего ожижающую примесь, вплоть до точки
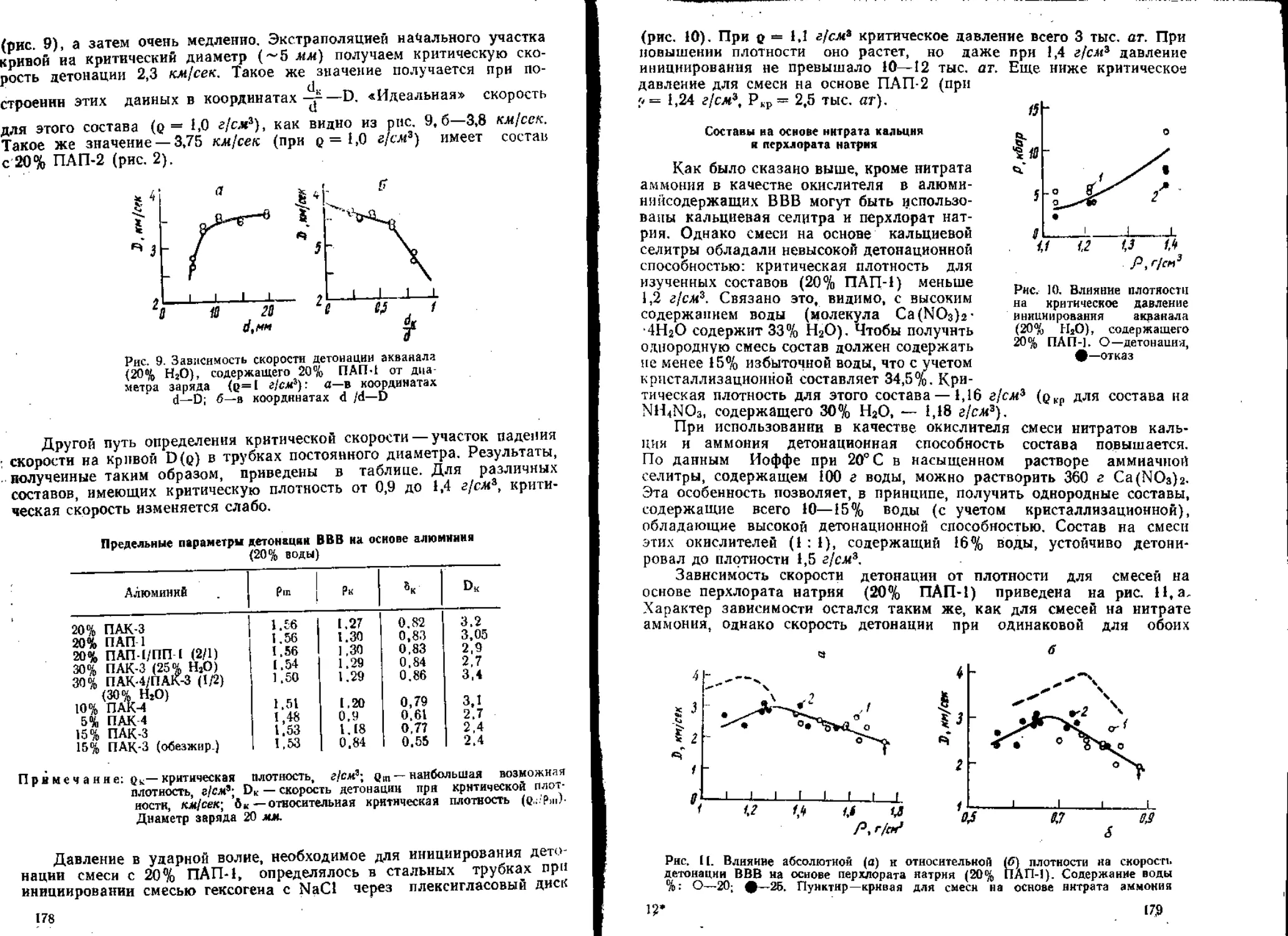
.^давления.
Продолжением сравнительного исследования кинетики разложения
у^икратов и стифнатов аммония, калия и свинца, проведенного К- К. Ан-
реевым, явились работы по распаду пикратов металлов первой и второй
групп периодической системы в условиях линейного нагрева (метод
ДТА) [30], а затем в изотермических условиях [31]. Было найдено, что
.кинетические параметры распада соли коррелируются с химической при-
родой катиона. Эти исследования значительно расширили представле-
ния л реакционной способности взрывчатых солей, включающих органи-
ческий анион, и могут служить экспериментальной основой для дальней-
шего развития теории горения ВВ.
1 3. Термическое разложение перхлората аммония
j Начатое К. К- Андреевым исследование термического разложения
перхлората аммония было продолжено Б. С. Светловым и В. А. Коро-
баном. Оно показало, что продукты превращения соли могут оказывать
7
различное по знаку влияние па распад [32]. При этом било обнаружено
накопление хлорной кислоты и окислов хлора в ходе реакции [33]. Иссле-
дование влияния каждого из продуктов впервые, на реальной основе,
позволило сделать вывод о химической природе самоускорения процесса,
а также дать обоснованную трактовку явления торможения распада во
дой, предусматривающую ионизацию первичного продукта распада пер-
хлората—хлорной кислоты [32, 34]. Эти исследования в период отказа
от «электронной» гипотезы разложения впервые в нашей стране пока-
зали необходимость обратиться к механизму протонного перехода, они
явились в его разработке значительным шагом вперед по сравнению с
зарубежными исследованиями этого времени и послужили толчком к даль-
нейшим исследованиям ведущих в данной области лабораторий страны.
Последующая детализация механизма распада перхлората аммония, в
частности, установление роли его взаимодействия с промежуточньнми
продуктами разложения — ок ислам и хлора и азота [34], диффузии и
разложения хлорной кислоты [35, 36] не только позволила установить
химическую схему распада перхлората и выделить в ней кинетически
ведущие стадии, но и помогла понять сложное поведение этого вещества
в практически важных условиях.
4. Стационарное горение
За истекшие десять лет были проведены фундаментальные иссле-
дования в области горения ВВ. Была освоена методика измерения тем-
пературы в зоне горения с использованием сверхтонких термопар
[37, 38], и с ее помощью установлен механизм горения тетрила [37] при
давлении до 50 ат. Показано, что реакция, определяющая скорость го-
рения, идет в конденсированной фазе. Опыты по влиянию разбавите-
лей — воды, углекислого аммония и Др. — на скорость горения ВВ [39]
показали, что гексоген горит аналогично тетрилу. В случае пироксилина
закон изменения скорости при разбавлении иной и, вероятно, обуслов-
лен ведущей реакцией в газовой фазе.
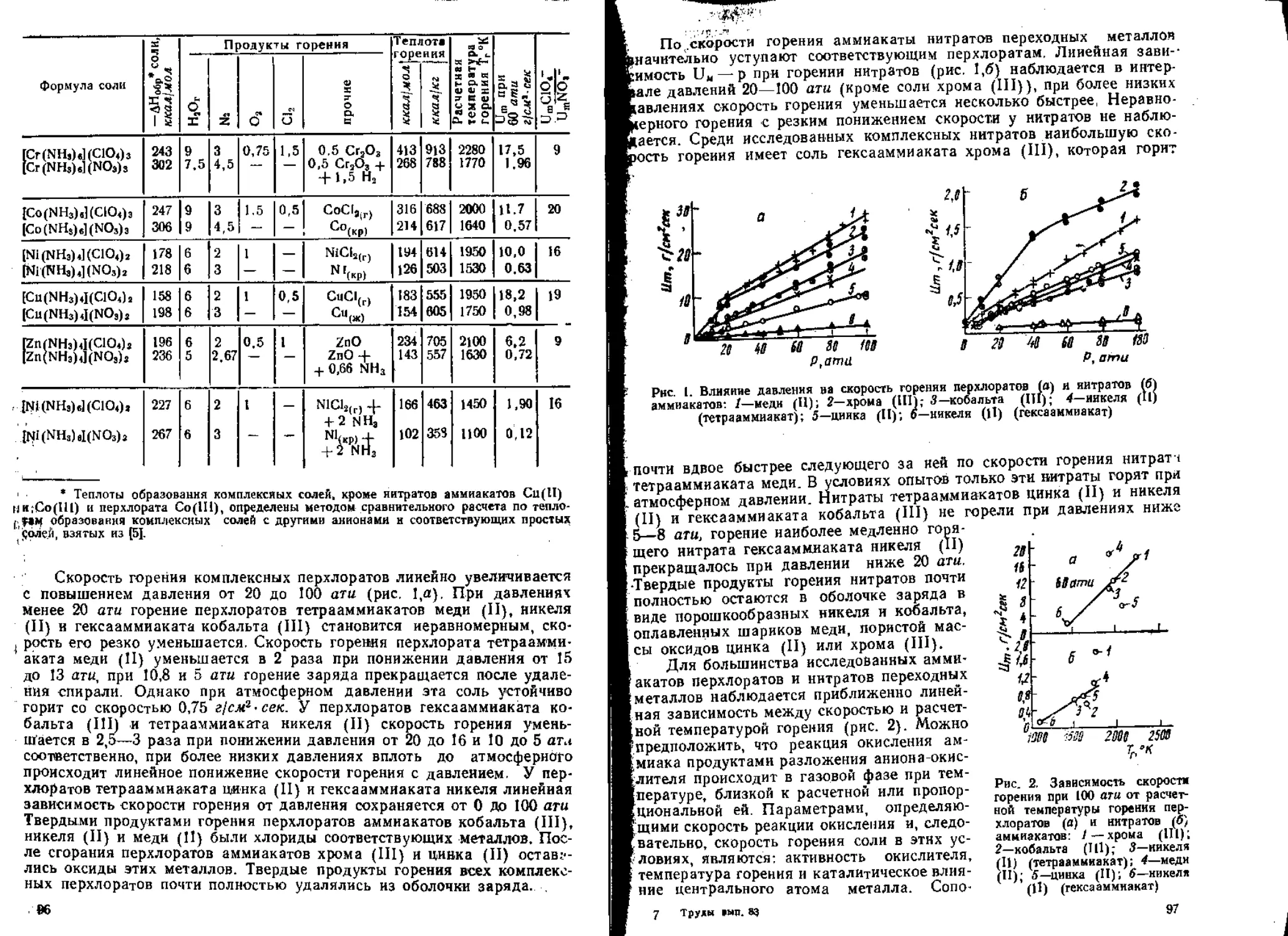
Большой цикл работ [40—49] посвящен выяснению связи между ско-
ростью горения и строением взрывчатых соединений. Начало этому на-
правлению было заложено в последних/работах К- К- Андреева, где
он указывал, что реакционная способность продуктов, образующихся
при разложении ВВ, может оказывать решающее влияние на скорость
горения. Исследование взрывчатых соединений различного строения по-
казало, что, действительно, изменение реакционной способности групп,
содержащих окислитель, приводит к изменению скорости горения на
порядок и болсс. Отмечено [44—46], что если активность окислителя
выразить в виде окислительного потенциала, то видно, что скорость горе-
ния резко растет при увеличении последнего. Обнаруженная зависимость
в общем закономерна, поскольку* горение — типичная реакция окисле-
лия-востановлсння, а окислительный потенциал дает возможность судить
Об относительной активности окислителей. Реакционная способность го-
рючей части молекулы ВВ влияет на скорость горения значительно сла-
бее. С развиваемыми взглядами согласуются представления о роли ме-
талла как 'возможного катализатора окислительных реакций при горении
быстрогорящнх солей взрывчатых кислот [40].
При изучении горения ВВ различного состава и строения — солей
нитроароматических кислот [40], органических перхлоратов [41 — 48], ам-
мониевых солей кислородсодержащих кислот [46], органических нитри-
8
тов [49, 50], нитро- и нитрозоаминов [51] и солей диазония [52] — было
установлено, что многие из этих соединений обладают высокой скоро-
стью горения, необычной зависимостью ее от давления, обнаруживают
различные формы неустойчивости горения.
Получены сведения [53], что при высоком (сотни атмосфер) давле-
нии скорость горения ряда нитросоединений определяется реакциями,
идущими в пламени при температуре, близкой к максимальной темпе-
ратуре горения. Термодинамический расчет температуры в сочетании с
измеренными в [54, 50] н приведенными в более ранних работах скоро-
стями горения дал энергию активации ведущей стадии горения О- п
С-нитросоединений около 30 ккал/моль. Аналогичные расчеты, проведен-
ные для N-нитросоединений [51], дали величину 16 ккал/моль, которая
близка к энергии активации восстановления в пламени N2O — основного
окислителя, образующегося при термическом разложении N-нитроами
нов. Кроме того, сравнение по скоростям горения этих ВВ с О- и С-нит-
росоединениями, которые имеют ту же температуру горения, показал,©,
что спи различаются так же, как смеси горючих газов с окислителями
NgO и NO<>.
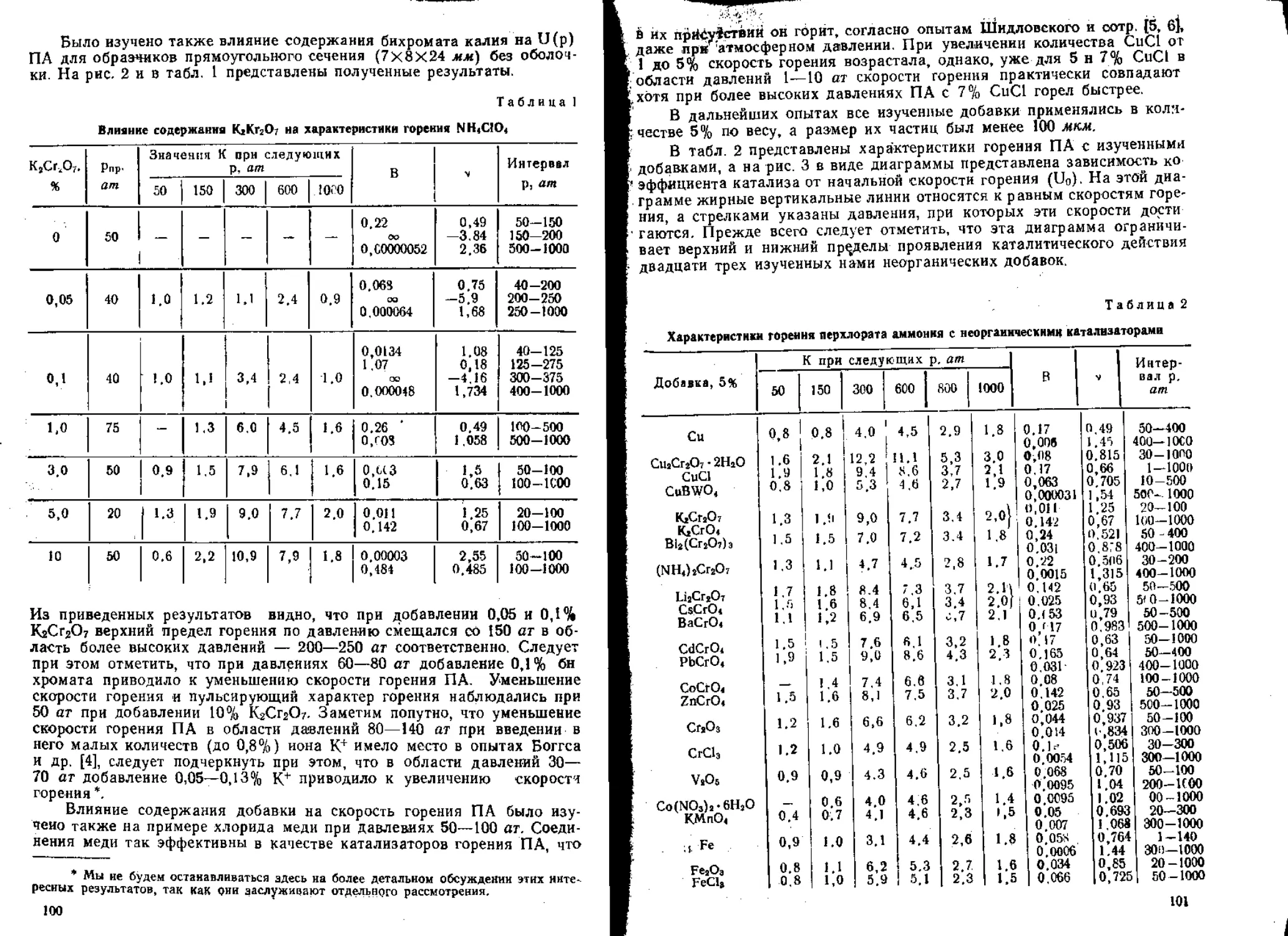
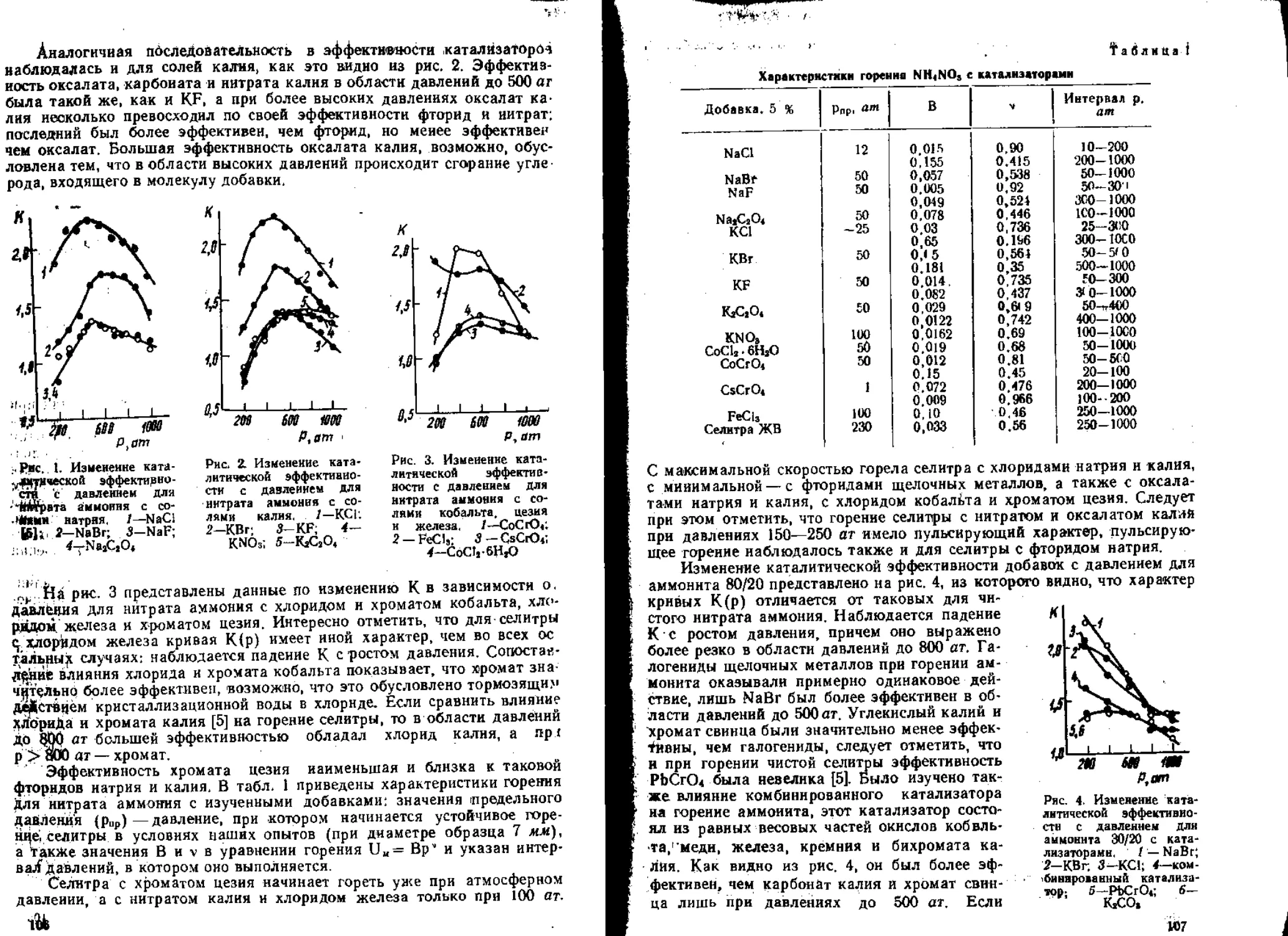
В работах [55, 56] было изучено влияние окиси железа на горение
некоторых органических перхлоратов и смесей на основе перхлоратов
калия и аммония. Показано, что влияние этого катализатора тем силь-
нее, чем ниже скорость горения соответствующего состава.
Во всех перечисленных работах изучалось в основном влияние дав-
ления на скорость горения. Влиянию начальной температуры, изучать
которое в общем труднее, посвящена только одна работа [57].
5. Предельные условия и устойчивость горения ВВ
Помимо скорости горения в технических приложениях теории зна-
чительную роль играют условия возможности и устойчивости горения.
Изучение этих вопросов было начато К- К. Андреевым еще в 1934 году,
когда он обнаружил существование критического диаметра горения ВВ,
Впоследствии было установлено, что некоторые ВВ ведут себя в этом
отношении не совсем обычно, Одни из них (например, тэн, стифниновая
кислота, эритриттетранитрат, нитрат гликолевой кислоты, нитрат и пер-
хлорат аммония, ди- и тринитрофеноляты свинца и др.) начинают гореть
только при высоком давлении (от нескольких ат для пикрата свинца до
1,5 тысяч ат для нитрата аммония). Другие (нитроглицерин, маннит-
гексанитрат, тринитро флороглюцин, некоторые органические перхлора-
ты) при повышении давления внезапно затухают, в некотором интервале
давлений не горят, а затем начинают гореть вновь.
Было высказано предположение [08], что эти особенности горения
связаны с его тепловой неустойчивостью, теоретически предсказанной в
1942 г. Я. Б. Зельдовичем, и показано, что в тех случаях, когда есть
возможность рассчитать критерий Зельдовича (Z), определяющий сте-
пень неустойчивости, случаям затухания в соответствии с теорией отве-
чает Z > Z*-
В работах [58, 59] были определены критический диаметр и предель-
ная скорость горения ряда ВВ. Оказалось, что предельные условия го-
рения легколетучих ВВ (метилнитрат) не отличаются от предсказывае-
мых теорией Зельдовича для газов: произведение критического диаметра
на скорость горения trudK = 0,01 г/см • сек, при увеличении диаметра заря-
да скорость растет в -^1,5 раза. Пределы горючести труднолетучих ВВ
9
^ебрйя, развитая для газов, не описывает: UMd(t = 0,l—0,4 г/см • сек.
при увеличении диаметра скорость остается постоянной.
Теория критического диаметра горения конденсированных веществ
рассмотрена в [60], Показано, что при Z> 1, UMd][> (Z—1)/г, где
r = (lTi/dT0 [Т(—температура поверхности, То—-начальная температу-
ра), Для многих веществ, горящих в конденсированной фазе, (Z—1)/г^
10, что дает качественное согласие с опытом.
В работе [61] теория Зельдовича была применена для объяснения
'результатов, полученных при разбавлении быстрогорящих веществ гру-
бодасперсным инертным порошком: скорость горения смеси почти не ме-
нялась вплоть до содержания порошка в ней более 50%, после чего
быстро падала. При 70—80% порошка горение затухало. Было выска-
зано предположение, что инертный разбавитель начинает влиять на горе-
ние, когда расстояние между частицами порошка приближается к вели-
чине критического диаметра горения ВВ.
Вопросы гидродинамической неустойчивости горения получили от-
ражение в работах [49] и [62], В первой нз них показано, что скорость
горения жидких ВВ на пределе устойчивости растет лишь до —-50 ат,
а затем остается практически постоянной. Предельное значение ско-
рости ~0,5 см!сек. Во второй — указаны некоторые отклонения от пре-
дельного значения критерия Андреева, полученного А. Д. Марголиным
и С, В. Чуйко, для перхлората аммония и смесевых составов. Показано
также, что взрыв пироксилина при поджигании и самовоспламенении
происходит, когда гидродинамическое сопротивление оттоку газов дости-
гает некоторого предела.
6. Воспламенение
При изучении зажигания ВВ и сравнении развитой главным обра-
зом в последние 10 лет теории зажигания с опытом основную трудность
представляет определение потока тепла, падающего на поверхность за-
ряда. Наиболее распространенным источником воспламенения является
горячий газ, обычно получаемый в результате сгорания заряда воспла-
менителя.
В качестве основного прибора для изучения зажигания была приме-
нена по предложению К- К. Андреева манометрическая бомба [63 , 64].
Развитие этого метода шло по двум направлениям. Для манометриче-
ской бомбы определялась зависимость потока тепла на поверхность за-
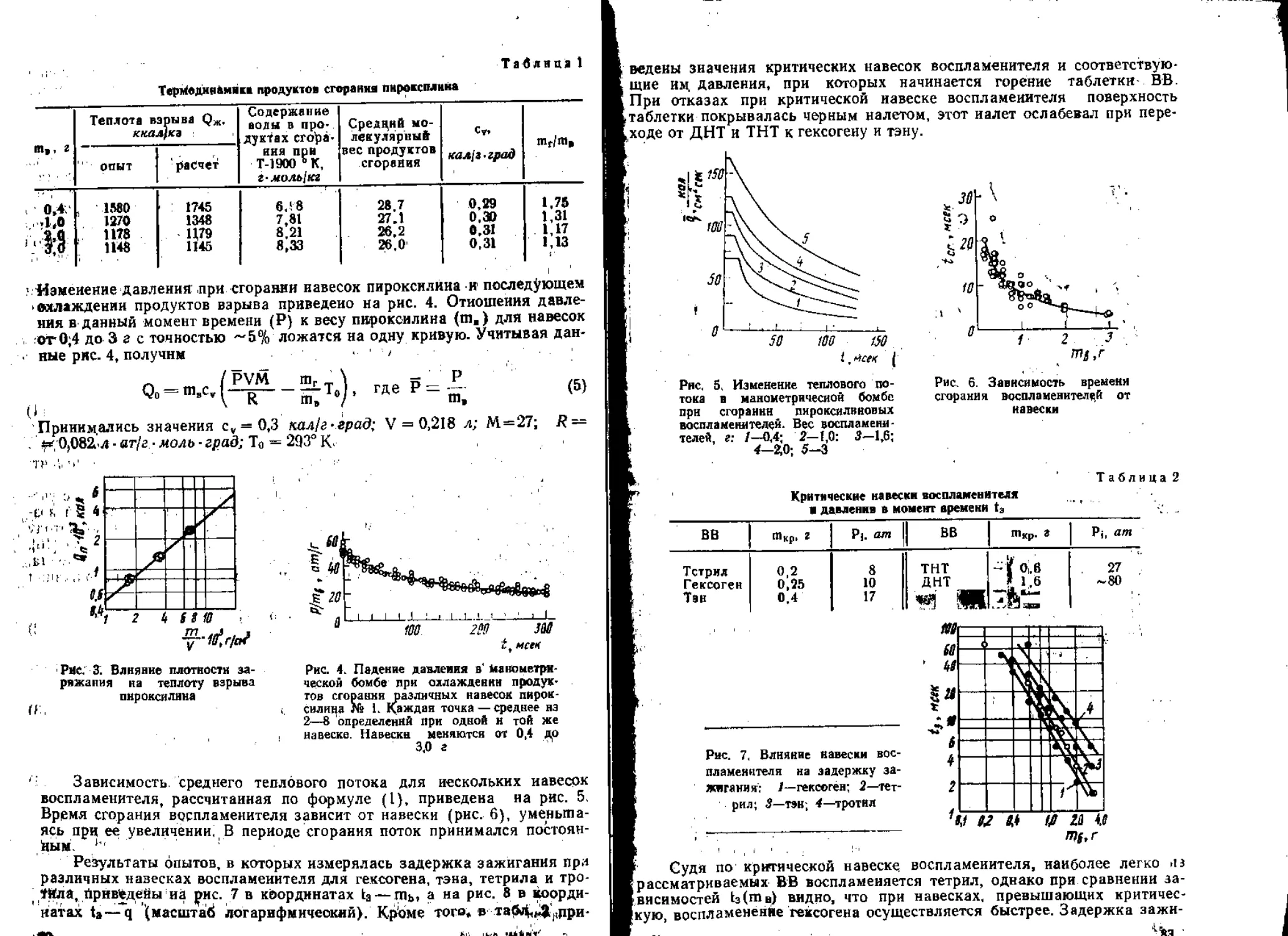
ряда от навески воспламенителя и времени. Определялась задержка
зажигания при данной навеске и построенная по этим дацным кривая в
координатах поток — задержка сравнивалась с полученной расчетом.
Такой подход реализован в статье, опубликованной в настоящем
сборнике. Теория зажигания сопоставлена с опытом в случае плавящихся
и испаряющихся вторичных ВВ. Оказалось, что при воспламенении тет-
рила и тэна проявляется тепловая неустойчивость их горения: задержка
зажигания заметно больше, чем предсказываемая теорией.
.Более подробно влияние неустойчивости было изучено в [65], где в
качестве источника воспламенения был применен углекислотный лазер
с хорошо измеренной до 100 кал1см2~сек плотностью потока, а в каче-
стве воспламеняемого ВВ — баллиститный порох. Оказалось, что при
высоком давлении увеличение потока может приводить к росту задержки
.зажигания, обусловленному тепловой неустойчивостью тонкого прогре-
того слоя. Установлено стабилизирующее влияние слоя твердых ча-
10
£Н1Ц — продуктов газификации пороха; еслй частицы срываются с по-
верхности потоком газа, воспламенение не происходит. Закономерности
газификации дают полезные сведения о процессах тепловыделения при
горении пороха.
Второе направление заключалось в моделировании процессов, про
исходящих при воспламенении ВВ в практических условиях, главным
образом при выгорании предохранительных ВВ в шпурах. Большую
роль играет здесь окружающий ВВ уголь, существенно повышающий
устойчивость горения и воспламеняемость аммиачноселитренвых ВВ.
Проведение опытов в манометрической бомбе в окружении угля позво-
лило получить результаты, оценить склонность к выгоранию предохра-
нительных ВВ, отечественных и зарубежных, и предложить пути сниже-
ния воспламеняемости [66, 67]. Комплексный подход к. оценке характе-
ристик ВВ, обусловливающих выгорание, с учетом детонационных
свойств, предложенный К. К. Андреевым, развит в работах [68, 69].
В [70] на основе изучения устойчивости горения и воспламеняемости
аммначноселцтренных ВВ разработан новый инициирующий состав для
патронов беспламенного взрывания.
7. Детонация
Одним из наиболее интересных результатов изучения детонационных
процессов было обнаружение возможности распространения в зарядах
ВВ высокой плотности — литых и прессованных — своеобразного режи-
ма распространения, не вполне правильно названного в одних работах
детонацией с малой скоростью, в других — ударным горением. В этом
случае слабая ударная волна, распространяющаяся практически со ско-
ростью звука в заряде, приводит к сжатию и воспламенению ВВ, а воз-
никающее за этим горение сопровождается значительным повышением
давления за фронтом ударной волны [71—74]. Такой комплекс из двух
’волн возникает и может распространяться при инициировании ударной
волной с постоянной скоростью на значительное расстояние. Ввиду боль-
шой протяженности комплекса для стационарного распространения не
обходимо наличие прочной массивной оболочки. В ее отсутствие процесс
затухает.
Полученные результаты оказались полезными при объяснении опы-
1ов по переходу горения в детонацию зарядов в стальных толстостен-
'ных трубках [75], Американскими и советскими исследователями было
установлено, что в ряде случаев рассмотренный низкоскоростной режим
'Является промежуточной стадией процесса перехода.
Большая серия работ посвящена решению проблемы обеспечения
устойчивости детонации смесевых ВВ, применяющихся в промышлен-
ности в разнообразных условиях. В работах [69, 76] изучено влияние
состава ВВ на возможность возникновения отказа детонации в случаях,
когда на детонирующий заряд оказывает действие взрыв рядом распо-
ложенного заряда, что наблюдается при короткозамедленном взрыва-
нии в угольных шахтах. Показано, что сильная ударная волна в среде
умеренной прочности в некоторых случаях вызывает детонацию смесе-
вого ВВ с повышенной скоростью [77].
В работах [78—82] исследованы некоторые пути, позволяющие
существенно повысить детонационную способность предохранительных
ВВ за счет выбора мощного сенсибилизатора детонации. Показана
принципиальная возможность создания индивидуального предохрани-
тельного feB , свободного от недостатков, присущих взрывчатым
смесям.
Решение практических задач потребовало развития теории детона-
ции взрывчатых смесей. В цикле работ [83—87] использован введенный
в практику научных исследований А. Н. Дреминым электромагнитный
метод оценки параметров детонационных и ударных волн. Исследована
структура детонационных волн, получены количественные данные об их
параметрах в системах, содержащих мощные сенсибилизаторы, а также
в промышленных аммонитах [84]. Оказалось, что строение детона-
ционной волны и структура гидродинамических течений за ее фрон-
том в реальных составах существенно отличаются от предсказанных
теорией.
В работе [88] показано, что возбуждение детонации в порошкооб-
разных ВВ слабым взрывным импульсом неизбежно связано с возник-
новением переходных процессов. Поэтому соответствие между детона-
ционной способностью ВВ и величиной минимального инициирующего
импульса, вытекающее из принципа Харитона, в подобных случаях на
практике це наблюдается.
В двух работах [82, 89] изучалась детонация новых, не исследовав-
шихся ранее в этом отношении веществ — солей хлорсодержащих нит-
роароматических кислот [82] и эфиров азотистой кислоты [89],
Работа со смесями, сенсибилизированными мощными индивидуаль-
ными ВВ, привела к необходимости изучения перехода их горения во
взрыв и детонацию. В работе [90] был получен при этом интересный ре-
зультат: увеличение содержания нитроглицерина в смеси с аммонитом
6Ж.В от 10 до 25% существенно уменьшило склонность к переходу горе-
ния смеси во взрыв (она взрывалась так же, как без нитроглицерина)--
очевидно, сказалась повышенная уплотняемость и малая газопроницае-
мость богатой жидкостью смеси,
Зарубежный опыт испытания промышленных ВВ по результатам
командировок в ЧССР и ГДР обобщен в работах [91, 92].
8. Чувствительность ВВ к удару
В отношении механизма возбуждения взрыва при ударе К- К< Ан-
дреев был последователем Н. А. Холево, идеи которого по поводу тече-
ния вещества, как основной причины возникновения взрыва при ударе
нашли в работах К, К. Андреева целый ряд ярких подтверждений. Они,
в частности, находятся в серьезном противоречии с предложенной в 40-е
годы методикой определения чувствительности к удару, зафиксирован-
ной у нас в качестве ГОСТ 4545—48. Доказательству ошибочности этой
методики посвящена работа [93], в которой рассматриваются также неко-
торые иные методы испытания.
Вместе с тем копер, предназначенный для изучения чувствитель-
ности к удару и являющийся в известной степени абсолютным прибором,
может быть использован для получения весьма полезной информации,
В работе [93] с его помощью было обнаружено, что энергия, необходи-
мая для возбуждения взрыва ударом, может быть определена с высокой
точностью без разброса результатов, столь характерного, как всегда
считалось, для опытов по определению чувствительности ВВ, и линейно
зависит от навески испытуемого ВВ. По этой зависимости удалось опре-
делить предельную скорость «истечения», при которой происходит вос-
пламенение ВВ ударом. Она оказалась около 400 м!сек.
12
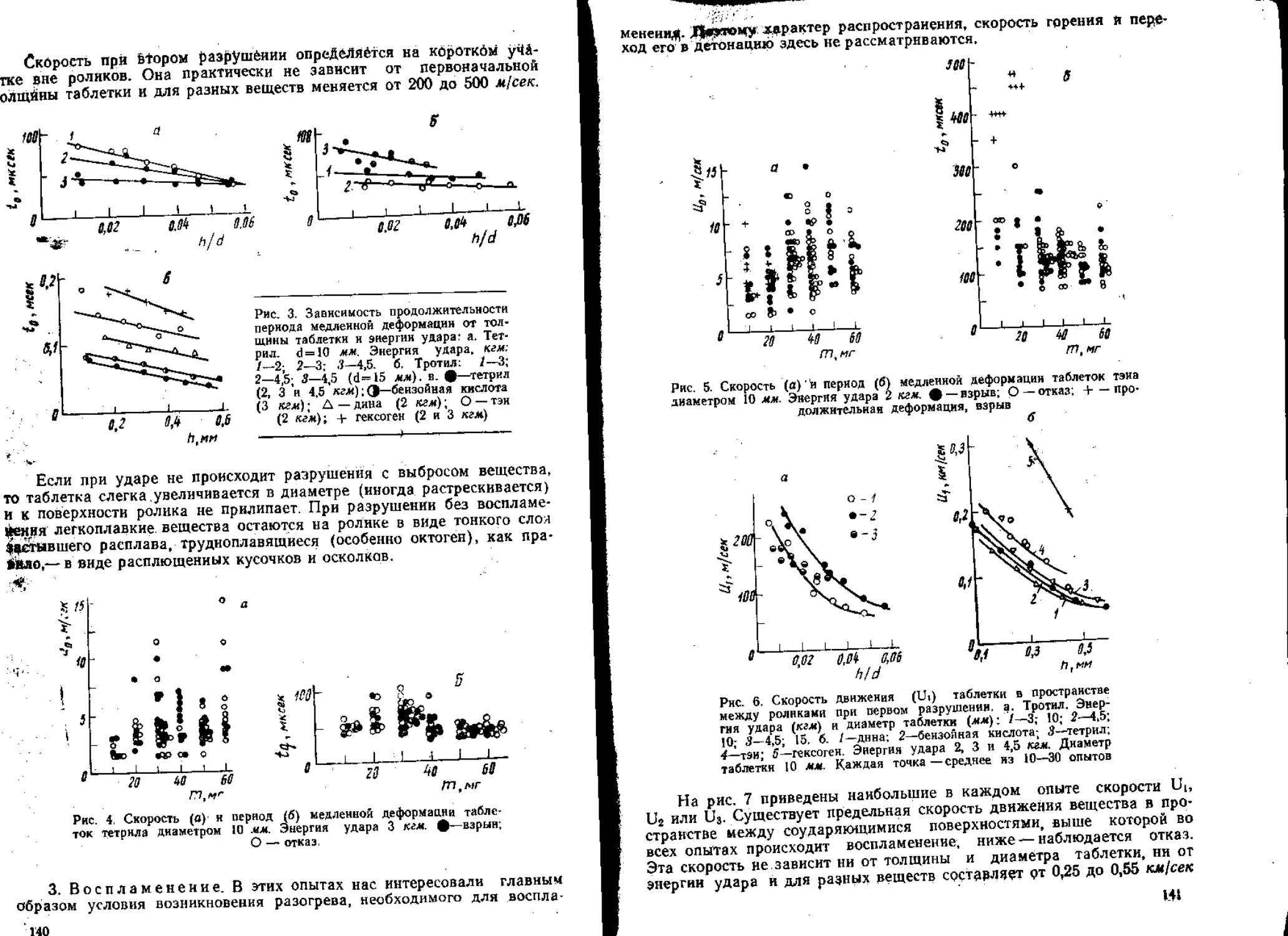
В связи с этим были проведены непосредственные измерения ско-
рости движения вещества в пространстве между соударяющимися по-
верхностями при ударе на копре [94, 95], Установлено, что вначале про-
исходит медленная деформация со скоростью порядка нескольких м/сек.
Затем таблетка разрушается, и осколки ее выбрасываются из простран-
ства между роликами со скоростью порядка сотен м/сек. Критическая
скорость, необходимая для взрыва, в случае разных ВВ составляет от
0,25 до 0,55 км/сек.
II. ВОПРОСЫ ХИМИИ И ТЕХНОЛОГИИ НИТРОСОЕДИНЕНИИ
Основное направление работ по химии и технологии нитросоедине-
ний связано с изучением процесса нитрования и является логичным про-
должением исследований, начатых А. Г. Горстом и Е, Ю. Орловой в
конце 30-х годов.
Большое внимание в работах последнего времени уделено природе
нитрующего агента и кинетике нитрования сер но-азотными кислотными
смесями [96, 97], представляющими наибольший интерес для промыш-
ленности. В качестве объекта для исследования выбирались как моно-,
так и динитросоединения. Рассматривалось также взаимодействие меж-
ду полностью пронитрованными соединениями и кислотными сме-
сями [98].
Наряду с процессом нитрования серно-азотными кислотными сме-
сями широко изучались различные реакции солен нитрония [99, 100],
азотиой кислоты [101—103], уксусно-азотных кислотных смесей [103] с
органическими соединениями.
В ходе этих работ были усовершенствованы методики проведения
эксперимента [100, 104], анализа реакционных смесей [105, 106], исполь-
зованы новые методы обработки получаемых результатов [107].
Значительное внимание уделено также изучению взаимодействия
нитросоединений с нуклеофильными агентами. Так, впервые в СССР
были проведены систематические исследования по замещению активи-
рованных групп (ОН, NOa и др.) в ароматических полинитросоединениях.
Особая роль отводилась получению галоид- и аминопроизводных три-
нитробензола, представляющих интерес в качестве термостойких ВВ.
Были не только разработаны препаративные методы синтеза указанных
соединений [Ю8—110], но и выяснен механизм процесса, в частности,
установлены роль катализатора [109, III] и строение промежуточного
продукта [112]; обнаружено также замещение «неактивированной» нит-
рогруппы при реакции с РОС1з [109].
Специфическое влияние нитрогрупп на реакционную способность
метильной и карбоксильной групп в полинитросоединениях подробно
прослежено на примере реакций Вильсмайера [113—116] и Шмидта [117].
1. Лурье Б. А., Светлов Б. С. О некоторых особенностях термического разложе-
ния нмтрозфиров в конденсированной фазе. Иэв. вузов, химия и хим. технология,
1967, т. 10, с. 1308.
2. Лурье Б, А., Светлов Б. С. Об особенностях термического распада некоторых
нитратов многоатомных спиртов в конденсированной фазе. Кинетика и катализ,
1968, т. 9, с. 183.
3. Л у р ь е Б, А-, С в е т л о в Б. С. О механизме термического распада некоторых нит-
ратов многоатомных спиртов в конденсированной фазе. Труды МХТИ нм. Д. И, Мен-
делеева, 1968, вып. 58, с. [89,
19
4. Афанасьев А. Г., Светлов Б. С. О термическом разложении полипнтратов
гомологического ряда нитроглицерина. Труды МХТИ нм. Д. И. Менделеева, 1968,
вып. 58, с. 185.
5, Светлов Б. С., Дурье Б. А., Дубнова С. Л. Об оценке скорости термиче.
ского разложения нитроэфнров в конденсированной фазе. Кинетика и катализ, 1968,
т. 9, с. 1163.
6. Светлов Б. С. Кинетика и механизм термического разложения нитроэфиров в
конденсированной фазе. 2-й Всесоюзный симпозиум по горению и взрыву. Рефераты
докладов, Ереван, 1969, с. 223.
7. Светлов Б. С., Лурье Б. А., Корнилова Г. Е. О термическом разложении
нитратов целлюлозы, Труды МХТИ им. Д. И. Менделеева, 1969, вып. 62, с. 62.
8. Лурье Б. А., Светлов Б. С. Термический распад 1,4-бутиленгликольдинитрата
в конденсированной фазе. Ж- физ. химии, 1969, т. 43, с. 2737.
Э.Светлов Б. С., Лурье Б. А., Мясникова С. И., Херсонский Н, С
Особенности термического распада органических нитритов в конденсированной фазе.
Кинетика и катализ, 1974, т. 15, с. 516.
10. Светлов Б. С., Лурье Б. А., Корнилова Г. Е. Взаимодействие декана с
двуокисью азота в жидкой фазе. Кинетика и катализ, 1972, т. 13, с. 1146.
19.
И. Светлов Б, С., Лурье Б. А., Корнилова Г. Е. Исследование превращении
двуокиси азота при взаимодействии с различными органическими соединениями в
конденсированной фазе. Горение и взрыв. Материалы 3-го Всесоюзного симпозиума,
М., «Наука», 1972, с. 780.
12. Светлов Б. С., Л у р ь е Б, А., С магии В, В., фе до ни на Л. М., Глухо-
ва И. А. Взаимодействие двуокиси азота с карбоновыми кислотами в жидкой фазе.
Труды МХТИ им, Д- И. Менделеева, 1974, вып. 80, с. 29.
13. Светлов Б. С., Л у р ь е Б. А., Корнилова Г. Е. Взаимодействие дибутилоаого
эфира с двуокисью азота в конденсированной фазе. Изв. вузов, химия и хим. техно-
логия, 1974, т. 17, с. 239.
14. Светлов Б, С., Шел а пути и а В. П. Изучение кинетики кислотного гидролиза
некоторых полнинтратов многоатомных спиртов Ж- физ. химии, 1966, т, 40, с. 2883.
15. Светлов Б. С., Ш е л а и у т и н а В. П., Город кона Л. Б. О влиянии строения
нитроэфнров на скорость их кислотного гидролиза. Кинетика и катализ, 1972, т. 13,
16. Св е т л о в Б. С., Шелапутипа В. П., Семенова Н. С. О тормозящей роли
воды при кислотном гидролизе глицеринтринитрата Кинетика и катализ, 1973, т. 14,
с. 589.
17. С в е т л о в Б. С., Лурье Б. А., С м а г и и В. В. О взаимодействии нитроэфирон с
органическими кислотами. Труды МХТИ им. Д. И. Менделеева, 1973, вып. 74, с. 34-
18. Лурье Б. А., С в е т л о в Б. С., Ч е р и ы щ о в А. Н. О взаимодействии водных рас-
творов солей азотной кислоты с алюминием. Труды МХТИ им. Д. И. Менделеева,
1973, вып. 75, с. 142.
Максимов Ю. Я. Давление паров ароматических дитросоединений при разных
температурах. Ж- физ. химии, 1968, т. 42, с. 2921.
Максимов Ю. Я., Дубовицкий В. Ф. Кинетика термического распада изо.
меров дивитробензола в парах. Докл. АН СССР, 1966, т- 170, с. 371.
Максимов К>. Я. Сравнительное изучение термического распада изомеров моно-
нитротолуола в парах. Ж- физ. химии, 1969, т. 43, с. 725.
Максимов Ю. Я. Термическое разложение ароматических полиннтросоединений
в парах. Ж. физ. химии, 1972, т. 46, с. 1726.
Максимов К>. Я., Егорычева Г. И. Исследование состава продуктов терми-
ческого разложения паров тринитробейэола. Кинетика и катализ, 1971, т. 12, с. 821.
Максимов К>. Я. О влиянии агрегатного состояния пар—жидкость на скорость
термического распада ароматических полиинтросоедннений. Ж- физ. химии, 1971,
т. 54, с. 793.
25. Ле йч ец к о А. А., Максимов Ю. Я- Автоассоциация ароматических нитро-
соединений. Ж. физ. химии, 1973, т. 47, с. 1265.
26. С а и р а в о в и ч В. Ф., Максимов Ю. Я., Маркелова М, Е. Исследование
состава газообразных продуктов термического распада жидкого 2,4,6-тринитротолу-
ола. Труды МХТИ им. Д. И. Менделеева, 1973, вып. 75, с. 147.
27. Максимов Ю. Я., Сапранович В, Ф., Павленко В, Г. Термическое раз-
ложение тринитрофлороглюцина. Изв. вузов, химия н хим. технология, 1974, т. 17,
с. 1585.
28. Короба и В. А., Максимов Ю. Я- Об особенностях термического распада гек-
саннтродифенилсульфнда и гексанитродифеннлсульфона. Изв. вузов, химия и хим.
технология, 1967, т. 11, с. 1032,
20.
21.
22.
23.
24.
29. Максимов Ю. Я. Об аномалии температурной зависимости скорости распада
взрывчатых веществ ниже точки плавления. Ж. физ. химии, 1967, т. 41, с. 1193.
30. Е л и ]i к о в а С. М., Максимов Ю. Я., Орлова Е. Ю. О поведении пикратов
щелочных и щелочноземельных металлов при нагревании с равномерно возрастаю-
щей температурой (при ДТА). Изв. вузов, химия и хим технология, 1971, т. 14,
с. 176.
31 Максимов Ю. Я., Воронцов Е. Д., Павленко В. Г. Термическое разло-
жение пикратов щелочных и щелочноземельных металлов. Кинетика и катализ. 1973,
. т. 14, с. 1139.
32. С в е т л о н Б. С., К о р о б а и В, А. О торможении термического разложения пер-
хлората аммония продуктами распада, Кинетика и катализ, 1967, т. 8, с. 456.
7 33. Светлой Б. С., Короба н В. А, О механизме термического разложения пер-
А хлората аммония. Физика горения и взрыва, 1970, т. 6, с. 12.
Ж 34. Кор обан В. А., Чу гун кн и В. М., Кудрявцева А. И., Светлов Б. С.
J Влияние некоторых продуктов па разложение перхлората аммония. Труды МХТИ
w им. Д. И. Менделеева, 1970, вып, 67, с. 50.
* 35. Короб ан В. А., Светлов Б. С., Чугункин В. М. Реакции пнзкогемпера-
* турного превращения перхлората аммония, Горение и взрыв. Материалы 3-го Все-
i союзного симпозиума, М., «Наука», 1972, с. 741.
? 36. Коробан В, А., Чугункин В. М.г Лобода В. И. Термическое разложенце
хлорнокислых солей метилзамещенных ионов аммония. Горение и взрыв. Материалы
fS 3-го Всесоюзного симпозиума, М., «Наука», 1972, с. 775.
$ 37. М а р г о л и и А. Д., Фогельзанг А. Е. О горении тетрила, физика горения и
л взрыва, 1966, № 2, с. 10.
^38. Денисюк А. П., Фогельзанг А. Е. Температурные профили при горении
баллиститного пороха с аномальной зависимостью скорости горения от давления.
Ф 1 Изв. вузов, химия и хим. технология, 1971, тч 14, с. 861.
>. 39. А и и и к о в В. Э,, К о и д р и к о в Б. Н., Полякова Н. А. Влияние воды, угле-
кислого аммония и некоторых других добавок иа горение ВВ. Физика горения н
• . взрыва 1969, т. 5, с. 60.
Светлов Б. С., Фогельзанг А. Е. О горении быстрогорящих ВВ- Физики
горения и взрыва, 1969, т. 5, с. 67.
»-41. Ф от е л ь з а н г- А. Е., Светлов Б. С., Алёшин В. Д., Опрышхо В. С.
№ ' О влиянии строения взрывчатых веществ на скорость их горения, 2-й Всесоюзный
симпозиум по горению и взрыву. Рефераты докладов Ереван, 1969, с. 32.
А.‘42. Алешин В, Д_, Светлов Б. С., Фогельаанг А. Е. О горении некоторых
X- органических перхлоратов. Докл. АН СССР, 1969, т. 185, с. 856.
Wr®. Фогельзанг А. Е., Светлов Б. С. О связи строения взрывчатых веществ со
Г- ' скоростью их горения. Докл. АН СССР, 1970, г. 192, с. 1322.
. 44, Фогельзанг А. Е., Светлов Б. С., Аджемян В. Я. Исследование влияния
на скорость горения реакционной способности окисляющей и горючей частей моле-
|>| кулы ВВ. Материалы IX Всесоюзной конференции по актуальным вопросам испа-
рения, горения и газовой динамики дисперсных систем, Одесса, ОГУ, 1970, с. 50.
,4& Фогельзанг А. Е., Светлов Б. С., Аджемяв В. Я. О роли химических
факторов при горении конденсированных систем. Материалы X Всесоюзной конфе-
ренции по актуальным вопросам испарения, горения и газовой динамики дисперсных
систем, Одесса, ОГУ, 1971, с. 51.
46. Фогельзанг А, Е., А д ж е м я н В, Я., Светлов Б, С, Влияние природы окис-
лителя содержащегося во взрывчатом соединении, на скорость его горения. Докл.
АИ СССР. 1971, т. 199, с. 1296.
47. Фогельзанг А. Е,, Светлов Б. С., Опрышхо В. С. Аджемян В. Я.
Исследование горения органических перхлоратов. Физика горения и взрыва, 1972,
т. 8, с. 257.
48. Фогельзанг А. Е., Аджемян В. Я., Светлов Б. С, О роли реакционной
способности окисляющей группы при горении взрывчатых соединений. Горение и
взрыв. Материалы 3-го Всесоюзного симпозиума, М., «Наука», 1972, с. 58.
49. Ко ндрнков Б. Н., Сидорова!. Т. Горение эфиров азотной и азотистой кислот.
Горение и взрыв. Материалы 3-го Всесоюзного симпозиума, М., «Наука», 1972, с. 58.
50. Кондрнков Б. Н., Сидорова Т. Т. О горении эфиров азотистой кислоты.
Дохл, АН СССР, 1971, г. 197, с. 125.
51. -ф от е л ь з а н г А. Е., Светлов Б. С., Аджемян В. Я., Колясов С. М.,
Сергиенко О. И. О горении нитро- н ннтроэоамниов. Докл. АН СССР, 1974,
т. 216, с. 603.
Фогельзанг А. Е., Аджемян В. Я., Светлов Б. С. Исследование горения
Л Солей диазония, физика горения и взрыва, 1974, г. 10, № 3, с. 449.
53. Кондриков Б. Н.. Райкова В, М., Самсонов Б. С. О кинетике реакций
горения нитросоедннений при высоком давлении. Физика горения и взрыва, 1973.
т. 9, с, 84.
54, Кондриков Б. Н., Свиридов Е. М. Горение ароматических ннтросоединснпй.
физика горения и взрыва, 1971, т. 7, с. 204.
55. Авдюнии В. И., Бахман Н. Н., Никифоров В. С.. Кинин Ю. С., Фо-
гельзанг Л. Е. Влияние окиси железа на скорость горения смесей с различными
перхлоратами. Изп. вузов, химия н хнм. технология, 1971, т. 14, с. 666.
56. В a k h ш a n N. N.. N i к i Го г о v V. S.. AvdyuninV.L, FogefsangA. Е.,
Ki ch in G. S, Catalylic effect of Ferrous Oxide on Burning Rale of Condensed
Mixtures, Combustion and Flame, 1974, v. 22, p. 77.
57. Фо ге ль з a i! г A. E,, Светлов Б, С., Аджемяп В. Я., Кол ясов С. №..
Гандельман М. Б, Исследование записимости скорости горения взрывчатых
соединений От начальной температуры. Материалы XI Всесоюзной конференции по
актуальным вопросам испарения, горения и газовой динамики дисперсных систем,
Одесса, ОГУ, 1972, с. 38.
58. Кондрикон Б. Н, Об устойчивости горения взрывчатых веществ. Физика горения
и взрыва, 1969, т, 5, с. 51.
59. Лнников В. Э., Кондриков Б. И. О зависимости скорости горения взрыв-
чатых веществ от диаметра заряда. Физика горения и взрыва, 1968, т. 4, с. 350,
60. Кондриков Б. Н., Новожилов Б. В, О критическом диаметре горения кон-
денсированных веществ, физика горения и взрыва, 1974, т- Ю, № 4, с. 354.
61. Алешин В, Д., Светлов Б. С,, Фогельзанг А, Е. Об особенности гореция
смесей содержащих быстрогорящее взрывчатое вещество, физика горения и взрыва.
1970, т'_ 6, с. 432.
62. Б а б а й ц ев И. В., Горбунов В. В„ Ко н д р ико в Б, Н,, Пономарев В, А.
Об условиях возникновения ускоренного горения пористых зарядов при постоянном
давлении. Материалы IX Всесоюзной конференции по актуальным вопросам испа-
рения, горения и газовой динамики дисперсных систем, Одесса, ОГУ, 1970, с. 41.
63. Рогожников В. М„ Лушкин В. П. Определение воспламеняемости ВВ с по-
мощью манометрической бомбы. Взрывное дело, М., «Недра», 1970, № 68/25, с. 134.
64, Кондриков Б. Н„ Козак Г. Д._ Лушкин В. П. Об одном методе определе-
ния воспламеняемости ВВ. ВарывнОе дело. М., «Недра», 1970, № 68/25, с. 139,
65. Kondrikov В, N. Ohiemiller Т., Summerfield М.‘, С a very L. Н.
Ignition Criterion am! Selfhealing of Propellant Subjected to Ontense RadiaHv Heat
Fluxes. The 1970, Technical Meeting ol the Eastern Section of the Combustion
Institute, Atlanta Ga, 1970,
66, Кондриков Б. H., Лушкин В. П. О некоторых путях снижения воспламеняе-
мости предохранительных ВВ. Инф. выпуск ИГД им, А. А. Скочинского, № 152,
М., 1965 с. 18,
67. Кондриков Б. Н„ Козак Г. Д„ Лушкии В. П. Изучение воспламеняемости
предохранительных ВВ в присутствии угля. Взрывное дело, М., «Недра», 1973,
№ 72/29, с. 217.
68. Андреев К. К., Хотип В. Г. О возможных путях предотвращения выгорании
ВВ при взрывных работах в угольных шахтах. Взрывное дело. М., «Недра», 1966,
№ 60/17, с. 20.
09. Chotjn W, G., Кг i ger G. Е. О некоторых проблемах обеспечения безопасности
взрывных работ в угольных шахтах, связанных с предотвращением отказов дето
нации и выгорания промышленных ВВ. bleue Bergbautechnik, 1971, N 9, s, /01.
70. Райкова В. М, Инициирующий состав для патронов беспламенного взрывания.
Материалы конференции по повышению безопасности взрывных работ в угольных
шахтах ИГД мм. А. А, Скочинского, №.. 1973, с. 39,
71, Бабайцев И. В., Кондрикон Б. Н., Т ы ш е в и ч В. Ф, О детонации с малой
скоростью литых зарядов тротила и тэна. Физика горения и взрыва, 1964, № 4, с, 601.
72, Б а б а й ц е в И, В., Кондриков Б. Н., Паукова 3. В., Тышеви ч В. Ф,
О детонации литых взрывчатых веществ с малой скоростью, физика горения и взры-
ва 1969, т. 5, с. 326.
73. Б а б а й ц е в И. В., К о и д р и к о в Б, Н., Тышевнч В. ф, О детонации с ма-
лой скоростью литых зарядов ВВ. Взрывное дело, М., «Недра», 1970, № 68/25, с, 215.
74. Babajfsev 1. V. Kondrikov В, N., Tishevich V. F. Low Velocity Pro-
cesses in High Density Charges. Abstracts of Papers Presented at the 3 rd Inter-
national Collonqum Gasdynamics of Explosions, Marceifle, France, September, 1971.
75. Афонина Л. В., Бабайцев И. В,, Кондриков Б, И. Метод оценки склон-
ности ВВ к переходу горения в детонацию. Взрывное дело, М., «Недра», 1970,
с. 149.
ТЯИК'гин В. Г., Кригер Г. Э. Влияние состава ВВ, условий заряжания и свойств
, Т*Тжружаю(цей среды на Возможность возникновения отказа детонации шпурового
, . Наряда под Действием взрыва соседнего, Сб. Отказы детоиапии ВВ на открытых
11 разработках. Киев, «Наукова думка», 1972, с. 14.
77. Богдановская Е. И,, Хотин В. Г, К вопросу о влиянии оболочки на процесс
, распространения детонации в зарядах порошкообразных ВВ. Сб. Использование
'взрыва в народном хозяйстве, т. 1, Киев, «Наукова думка», 1970, с. 49,
78. Хотин В. Г.г Хотина Л. Д., Кригер Г, Э,, Шаталова Н. А. Исследование
детонационной способности аммиачноселитренных ВВ, сенсибилизированных гексо-
/' геном. Взрывное дело, «Недра», М., 1970, № 68/25, с. 235.
79. Хотин В. Г., Сабса й А. А, К оценке полноты взрывчатого превращения селек-
. тивнодетонирующих ВВ. Взрывное дело, М., «Недра», 1973, № 72/29, с. 15.
8^1 Андреев К. К., К о н д р н к о в Б. Н., Рыбаков Е. А. О влиянии начальной
температуры н вязкости нитроглицерина на восприимчивость к детонации смесн его
с хлористым натрием. Взрывное дело, М., «Недра», 1970, № 68/25, с, 222.
Я^.Бабайцев И. В., Панарин Ю. Н,, Тышевнч В. Ф. Давление детонации
;•/ смесей ВВ с инертной добавкой. Взрывное дело, М., «Недра», 1973, № 72/29, с. 20.
5р. Андреев К. К., Хот ни В. Г., Рыбаков Е. А. О новом способе построения
предохранительных ВВ. Взрывное дело, М, «Недра», 1966, № 60/17, с. 96.
83. Хотнн В. Г., Пономарев В. А. К вопросу о схеме гидродинамических течений
у за фронтом детонационной волны в конденсированном взрывчатом веществе. Ма-
териалы XI Всесоюзной конференции по вопросам испарения, горения и газовой
динамики дисперсных систем, Одесса, ОГУ, 1972, с. 169.
8Ц Хотин В. Г., Пономарев В, А. Опёнка параметров детонационной волны в
аммоните 1—19. Материалы конференции по способам повышения безопасности
взрывных работ в угольных шахтах, ИГД им. А. А. Скочинского, М,, 1972, с. 38.
8у. Хотин В. Г,, Пономарев В. А, К вопросу о структуре детонационных волн
В малоплотных зарядах конденсированных взрывчатых веществ. Физика горения н
1взрыва, 1973, т. 9, с, 304.
86. Хотин В. Г., Пономарев В. А. Параметры детонационных волн в смесях гек-
ебгена с наполнителями. Материалы конференции по повышению безопасности взрыв-
ных работ в угольных шахтах, ИГД нм. А. А. Скочинского, М., 1973, с, 4.
81?,.Хотин В, Г., Пономарев В, А. О строении детонационного фронта смесевых
КВ, содержащих сенсибилизатор, при малой плотности заряда. Труды МХТИ им.
ЕД. И. Менделеева, 1973, вып. 73,
>8|(,Хотин В. Г., Батневский П, Ю., Пономарев В, А. О некоторых особен-
’ с^люстях инициирования детонации вторичных ВВ слабым взрывным импульсом. Сб.
-'Тцгказы детонации ВВ на открытых разработках, Киев «Наукова думка», 1972,
“ J Ю7.
ДО Бабайцев И. В., Кондриков Б, Н., Сидорова Т. Т., Тышевнч В. ф.
, ,/f. О детонационной способности некоторых эфиров азотной и азотистой кислот. Горе-
k- jHjre и взрыв. Материалы 3-го Всесоюзного симпозиума, М., «Наука», 1972, с. 470.
Лбт ин В. Г., Кондриков Б. Н., Свиридов Е, М, Исследование’склонности
• ,К, пере ход у горения во взрыв аммиачноселитренных ВВ сенснбнлизнрованных гексоге-
? -нОм н жидкими нптроэфирами, Взрывное дело, М., «Недра» 1970, № 68/25, с. 243.
*1;,.Ко н д р н к о в Б. Н. Методы испытания промышленных бВ Чехословацкой Со-
.. циалистической Республики. Взрывное дело, М>, «Недра», 1970, М 68/25, с. 180.
921. Хотин В. Г. Методы испытания промышленных ВВ в’ГДР. Взрывное дело, М.,
, «Недра», 1973, № 72/29, с. 225.
93. Кондриков Б, Н О некоторых методах определенна чувствительности ВВ к
, удару, Взрывное дело, М,, «Недра», 1970, № 68/25, с. 168.
94. Кондриков Б. Н., Чубаров В. Д. О возникновении взрыва при ударе, физика
горения и взрыва, 1970, т. 6, с. 318.
95. Kondrikov В, N., Tchubarov V. D, Deformation, Destruction and Ignition
of the Layer of Solid Under Impact. Abstracts of Papers Presented at the XV-th
International Symposium on Combustion, Tokio, 1974.
Шутов Г. M., Чиркова Р. Г., Орлова Е. Ю, Реакции нитроний-попа. IV.
Исследование реакционной способности мононитротолуолов по отношению к серно-
азотиым кислотным смесям. Труды МХТИ нм. Д. И, Менделеева, 1973, вып. 75, с. 40.
Шутов Г. М, Жаворонков Н А., Орлова Е. Ю. Зависимость скорости
нитрования от концентрации азотной кислоты прн оптимальной кислотности среды.
Труды МХТИ им. Д. И. Менделеева, 1973, вып. 74, с. 38.
Шутов Г. М., Улька О, Г., Светлов Б. С., Орлова Е, Ю. Реакции нитро-
ннй-нона, V. Исследование устойчивости растворов нитроглицерина и тринитрофе-
нола в смесях серной и азотной кислот. Труды МХТИ им. Д, И. Менделеева, 1973,
вып, 75, с. 138.
2 Труды вып, S3
96.
97.
98.
17
HjVKOBa б)Йл1отвка I
16
99. Шутов Г. М., Берг В. К-, Жил и п В. Ф., Орлова Е. Ю. Реакции нитроний-
нона I. Количественное определение нмтроннй-иона е среде органических раствори-
телей. Ж. анал. химии, 1972, т. 37, с. 731.
100. Шутов Г. М., Берг В. К., Орлова Е. Ю. Реакции нитроний-ион а. III. Терми-
ческое разложение перхлората нитрония а нитрометане, Ж. фиэ.-хнмик, 1972, т. Л
с. 2012. !
101. Шутов Г. М., Пастухова Р. Д, У л ь к о О. Г., Орлова Е. Ю. О взанми
действия 2,4- ди нитро толуол а с азотной кислотой в четыреххлористом угле^-Ьй*
Труды МХТИ им. Д. И. Менделеева, 1972, выл. 74, с. 40. ч.
102. Ж и л И и В. Ф., Збарский В. Л., Ш у т о в Г. М., О р л о в а Е. Ю. О мето ди#
исследования кинетики быстрых химических реакций. Ж. физ. химии, 1966, т. Я
с. 504. 7
103. Збарский В. Л., Шутов Г. М., Ж и л и и В. Ф., Орлова Е. Ю. О некотоР1Л
особенностях нитрования в ряду дифениламина. Ж. орг. химии, 1965, т. 1, с, 125Л
104. 3 б а р с к и й В. Л., Ш у т о в Г. М. Жилиц В. Ф., Орлова Е. Ю. Авт.' свЩ
СССР № 172338, 1965; Б. И., 1965, № 13. J
105. Шутов Г. М, Берг В. К.. Орлова Е. Ю. Авт. свид. СССР № 260951 МЯ
- Б. И., 1970, № 4. : / j
106. Шутов Г. М., Берг В. К., Орлова Е. Ю. Реакции нитроннй-иона. II. Utaei
кочастотное титрование смесей серной и азотной кислот. Ж. анал. химии,
т. 37, с- 807. \ 1
10/. Шутов Г. М., Орлова Е. Ю. К методике кинетического исследования реа^м
со сложной стехиометрией. Ж. физ. химии, 1971, т. 45, с. 832.
108. Orlova Е. У., Shutov G. М., Zbarsky V. L., Ztlin V. F. NucIedM
substitution by a halogen in aromatic nitro compounds. Tetrahedron, 1964, у.Д
s, 441. ’tl
109. Збарский В. Л., Шутов Г. М., Жилиц В. Ф., Чиркова Р. Г., От
в а Е. Ю. Нуклеофильное замещение галоидом в ароматических нитросое ди невИ
1 1 IV. Замещение галоидом, катализируемое диметилформамндом. Ж орг. хн|М
1971, т. 7, с. 310.
НО, ШРу т о в Г. М-, Жилин В. Ф., Збарский В. Л.,Орлова Е. Ю. Авт. СфИ
'СССР № 169504, 1964; Б. И, 1965, Ks 7.
III. Шутов Г. М., Збарский В. Л., Ж и л и и В. Ф., Орлова Е. Ю, НуклеофЩ
1,1 щ?е замещение на галоид в ароматических иитросоединеииях II. О каталитичвЙШ
Действии пиридина в реакциях полипитронроизводных бензола и фенола с 1М
. окисью фосфора. Ж. общ. химии, 1965. т. 35, с. 1358. <з‘Я
Г12; Шутов Г. М., Збарский В. Л., Жилин В. Ф., Чиркова Р. Г., ОрД|
в а Е. Ю. Нуклеофильное замещение на галоид в ароматических нитросоединмиИ
Ш. О промежуточном образовании катиона арил-пириднния в катализируемых*#
риднном реакциях замещениях ОН- и NO2-rpynn галоидом. Ж. общ. химии, IM
т.'37, с. 783.
ИЗ. Збарский В. Л., Шутов Г. М., Жилин В. Ф., Орлова Е. Ю. О peakna
онной способности активированной метильной группы в иитросоединеииях. 1. Нош
реакция тринитротолуола. Ж. орг. химии, 1968, т. 4, с. 1970. ,
Ц4. Збарский В. Л., Шутов Е М., Жилин В. Ф., Беланова Л. Н., Орла
ва Е. Ю. Авт. свид. СССР ЛЬ 216682, 1968; Б. И., 1968, ЛЬ 15.
115. Збарский В. Л., Борисенко А. А., Орлова Е, Ю. О реакционной сИО
собности активированной метильной группы в иитросоединеииях. II. Ароматич»
ские соединения в реакции Вильсмаейра. Ж. орг. химии, 1970, т. 6, с. 520. '
116. Збарский В. Л., Шутов Г. М. Жилин В. Ф., Орлова Е’. Ю. Авт. свид
ЛЬ 215924, 1968; Б. И., 1968, № 14.
117. Збарский В. Л. С он ис М. А., Орлова Е. Ю. Динитро феи и лкарбонова;
кислота в реакции Шмидта. Ж. прикл. химии, 1971, т. 44, с. 2378.
РАЗДЕЛ ПЕРВЫЙ
й
I УДК 541.64 + 541.127+ 547.26''! 17
ТЕРМИЧЕСКИЙ РАСПАД ДИЭТИЛЕНГЛИКОЛЬДИНИТРАТА
| В КОНДЕНСИРОВАННОЙ ФАЗЕ
I Б, А. Лурье, Б, С. Светлов
। Диэтилен гликольди нитрат (ДЭГДН) по кинетическим закоиомерио-
. стам термического распада является типичным представителем иитро-
эфиров второй группы, которые разлагаются в замкнутом объеме без
резкого ускорения [1—3]. В настоящей работе для выяснения механизма
его разложения исследуются природа и характер накопления основных
продуктов распада, способность ДЭГДН к комплексообразованию с NOj
и HNOa, а также кинетика и газообразные продукты окисления его дву-
окисью азота.
Распад и окисление ДЭГДН изучались манометрическим методом
в сочетании с хроматографическим определением выделяющихся газов
и функциональным анализом соединений, образующихся в конденсиро-
ванной фазе. Методика проведения эксперимента и анализов описана
ранее [1, 4]. О комплексообразовании судили по изменению температуры
затвердевания смесей ДЭГДН с NOa и HNO3, а также по коэффициенту
распределения азотной кислоты в композиции иитроэфир—вода при 30эС.
Содержание кислоты в нитрате определяли титрованием 0,02 N спирто-
вым раствором КОН в нитрометане, а в водной фазе - 0,1 или 1 N вод-
ным растгором NaOH-
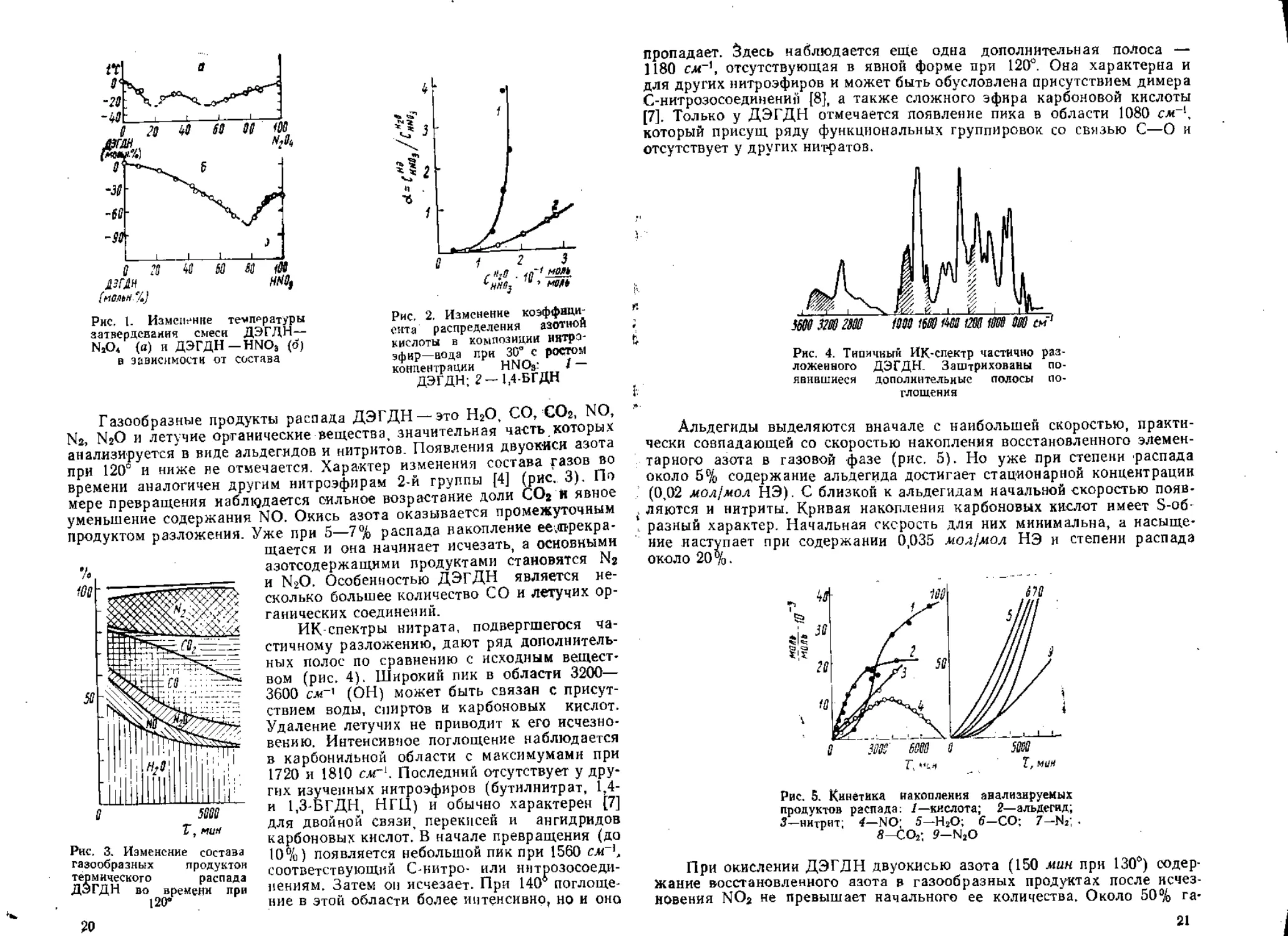
Диаграмма температура затвердевания—состав для смеси ДЭГДН—
N2O4 характеризуется максимумом при концентрации N2O4 30—35 мол.%
(рис. 1), что свидетельствует об образовании химического соединения—
2ДЭГДН-Ь12О4 с температурой затвердевания — 16°. Аналогичная диа-
грамма для смеси с HNOa не имеет максимума. Но растворимость HNO3
в композиции ДЭГДН—вода по сравнению с другими нитроэфирами
(1,4-бутиленгликольдинитратом и нитроглицерином), не содержащими
группировки простого эфира, явно выше и отношение с™0’/сщо‘
не является величиной постоянной, а растет с увеличением концентрации
HNO3 в воде (рис. 2). По-видимому, это указывает на образование по
кислороду простого эфира [5], комплекса между нитратом и кислотой,
который, судя по диаграмме затвердевания, должен легко диссоцииро-
вать [6].
2* 19-
ff w 60 SO iOS
ДЗГДН HHOt
(mbmh7.)
Рис, 2. Изменение коэффищг
ента распределения азотной
кислоты в композиции иятро-
эфир—вода при 30° с ростом
концентрации HNO^: / —
ДЭГДН; 2 — 1,4-БГДН
Рис, 1. Изменение температуры
затвердевания смеси ДЭГДН—
N5O4 (а) и ДЭГДН—HNO3 (о)
в зависимости от состава
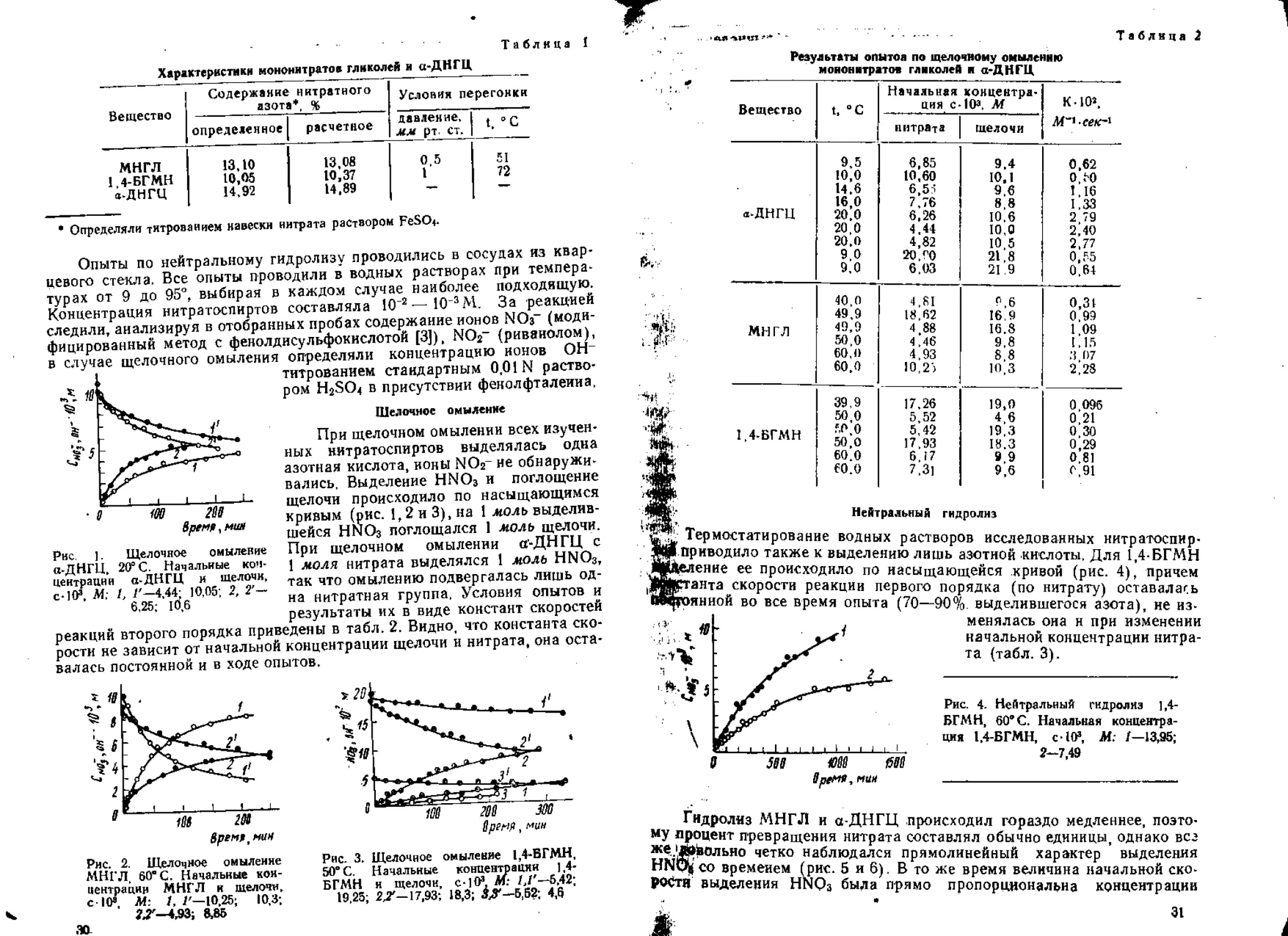
Газообразные продукты распада ДЭГДН —это Н2О, СО, СО2, NO,
Ns, N2O и летучие органические вещества, значительная часть которых
анализируется в виде альдегидов и нитритов. Появления двуокиси азота
при 120° и ниже не отмечается. Характер изменения состава газов во
времени аналогичен другим нитроэфирам 2-й группы [4] (рис. 3). По
мере превращения наблюдается сильное возрастание доли СО2 й явное
уменьшение содержания NO. Окись азота оказывается промежуточным
продуктом разложения. " ~ “'"'°'
Рнс, 3. Изменение состава
газообразных продуктов
термического распада
ДЭГДН во времени при
120*
Уже при 5—7% распада накопление еедарекра-
щается и она начинает исчезать, а основными
азотсодержащими продуктами становятся N2
и N2O. Особенностью ДЭГДН является не-
сколько большее количество СО и летучих ор-
ганических соединений.
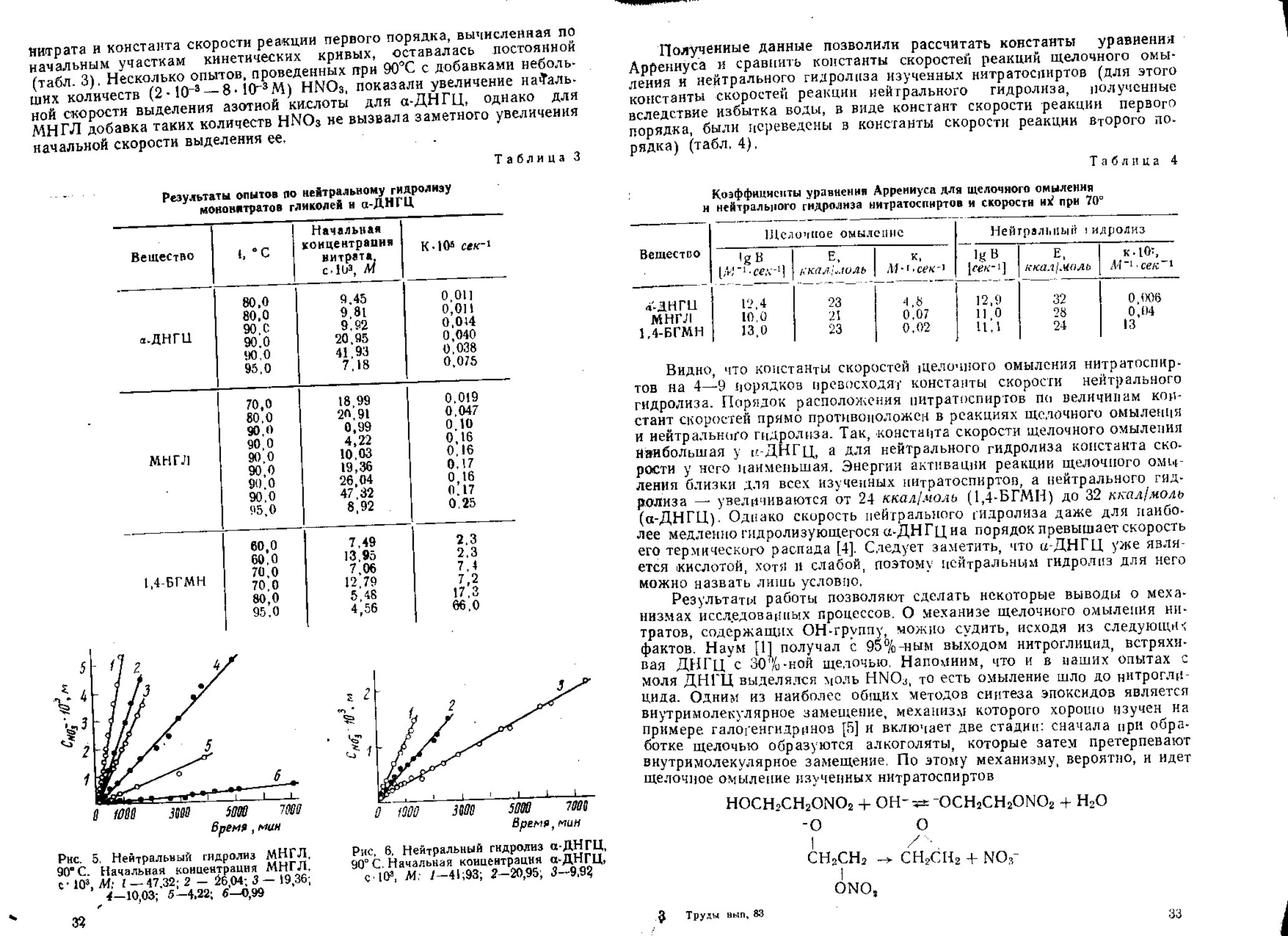
ИК-спектры нитрата, подвергшегося ча-
стичному разложению, дают ряд дополнитель-
ных полос по сравнению с исходным вещест-
вом (рис. 4). Широкий лик в области 3200—
3600 ежг’ (ОН) может быть связан с присут-
ствием воды, спиртов и карбоновых кислот.
Удаление летучих не приводит к его исчезно-
вению. Интенсивное поглощение наблюдается
в карбонильной области с максимумами при
1720 и 1810 слг1. Последний отсутствует у дру-
гих изученных нитроэфиров (бутилнитрат, 1,4-
и 1,3-БГДН, НГЦ) и обычно характерен [7]
для двойной связи, перекисей и ангидридов
карбоновых кислот. В начале превращения (до
10%) появляется небольшой пик при 1560 слг1,
соответствующий С-нитро- или ннтрозосоеди-
иениям. Затем он исчезает. При 140® поглоще-
ние в этой области более интенсивно, но и оно
пропадает. §десь наблюдается еще одна дополнительная полоса —
1180 см~\ отсутствующая в явной форме при 120° Она характерна и
для других нитроэфиров и может быть обусловлена присутствием димера
С-нитроэосоединений [8], а также сложного эфира карбоновой кислоты
[7]. Только у ДЭГДН отмечается появление пика в области 1080 см-1,
который присущ ряду функциональных группировок со связью С—О и
отсутствует у других нитратов.
Рис. 4. Типичный ЙК-спектр частично раз-
ложенного ДЭГДН. Заштрихованы по-
явившиеся дополнительные полосы по-
глощения
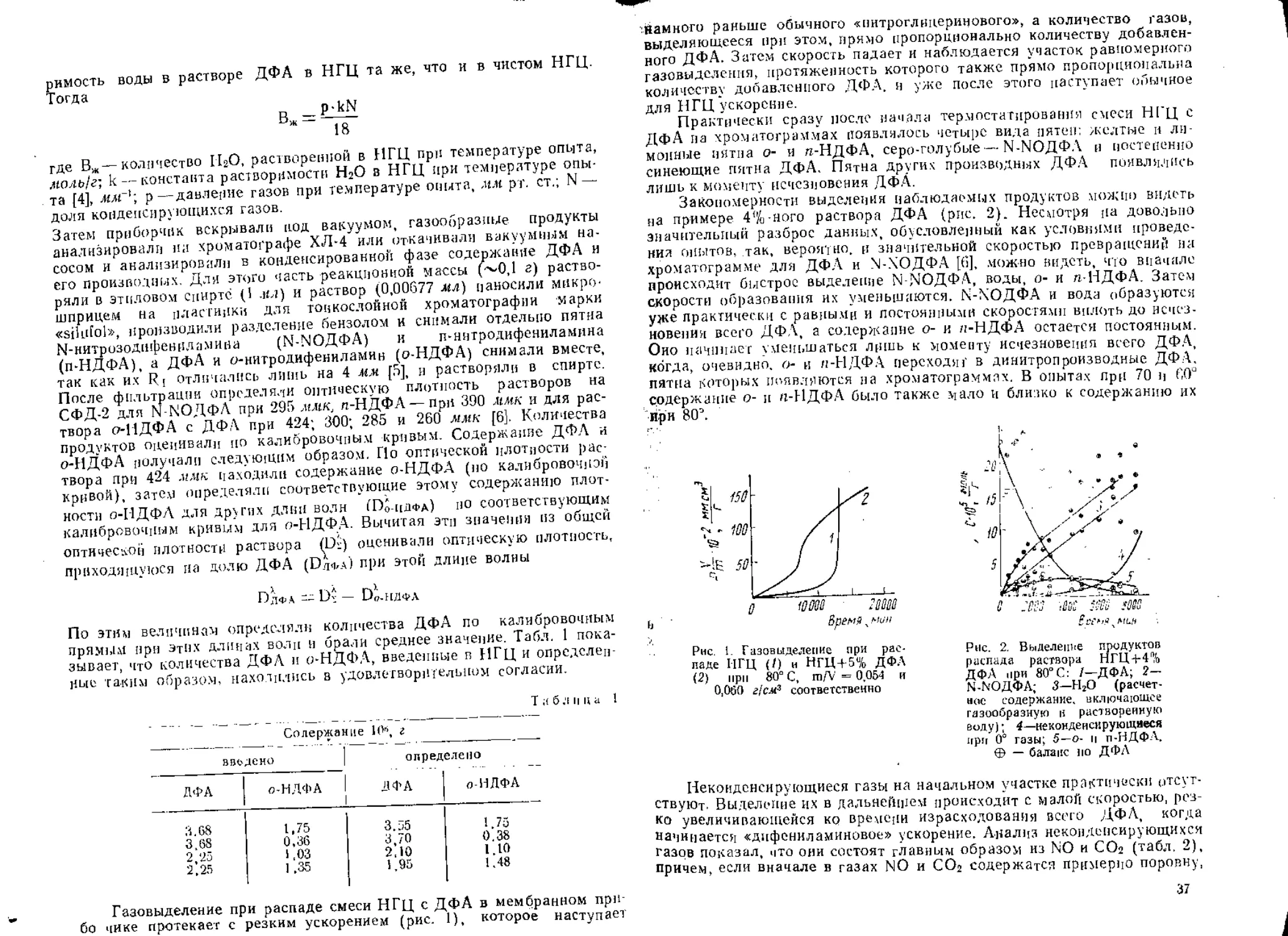
Альдегиды выделяются вначале с наибольшей скоростью, практи-
чески совпадающей со скоростью накопления восстановленного элемен-
тарного азота в газовой фазе (рис, 5). Но уже при степени распада
около 5% содержание альдегида достигает стационарной концентрации
(0,02 жол/мол НЭ). С близкой к альдегидам начальной скоростью появ-
{ ляются и нитриты. Кривая накопления карбоновых кислот имеет S-об-
, разный характер. Начальная скорость для них минимальна, а насыще-
ние наступает при содержании 0,035 лол/яол НЭ и степени распада
около 20%.
Рис. 5. Кинетика накопления анализируемых
продуктов распада: /—кислота; 2—альдегид;
3—нитрит; 4—NO; 5—Н2О; 6—СО; 7—N2; .
S-COj; 9—NaO
При окислении ДЭГДН двуокисью азота (150 мин при 130°) содер-
жание восстановленного азота в газообразных продуктах после исчез-
новения NOa не превышает начального ее количества. Около 50% га-
20
21
ЗОВ — это конденсирующиеся при охлаждении до 0°, остальное (%) —
8-N2; з—N2O; 19—NO; 15—СО и 5—СО2. Окисление при 70° в течение
300 минут приводит к образованию из неконденсирующихся газообразных
продуктов только 88% NO и 12% N2. Хотя полного восстановления NOa
здесь не происходит, суммарное количество элементарного азота, анали-
зируемого в газовой и конденсированной фазах (в виде сильной кислоты
и нитрита), меньше начального содержания двуокиси азота.
Обсуждение результатов
Близость начальных скоростей и величины энергии активации раз-
ложения ДЭГДН [1—3] к другим нитроэфирам 2-й группы как ди-, так
н мононитратам указывает на то, что термический распад его также
начинается с отрывом ИО2-группы. Величина константы скорости раз-
d(SNra34-HHTpHT'lra4
ложеиия, рассчитанная как --------------------- при 12U равна
1,8-10-7 сек-1. Аналогичные значения получаются для 1,4-БГДН и бутил-
нитрата,
O2NOCH2CH2OCH2CH2ONO2 -+ЭНССН2ОСН2СН2ОН 4-2NO2 (1)
Кинетические данные по окислению ДЭГДН двуокисью азота (80
120°1 [11 позволяют оценить константы скорости этой реакции по зави-
’ d (£ NO2ra3+ Е NO
симости начальной скорости исчезновения NO2 ---------т
от ее концентрации в конденсированной фазе. Последняя определяется
на основании общего количества сдозированных окислов азота и на-
чального их содержания в газовой фазе SNO2jbiu = SNOs—2NO2ra3.
Изменение коистаиты скорости окисления с температурой описывается
уравнением
. .«66 { 185С0
k = lOM.expl----—
которое при 120° дает значение 2- Ю-4 л]молъ • сек, Полученные величи-
ны констант обеспечивают практическое отсутствие NO2 в газах при тем-
пературах ниже 120° даже без учета других реакций с появляющимися
продуктами разложения, Ориентировочная оценка, исходя из условия,
что скорость распада равна скорости окисления нитроэфира, дает мак-
симально возможное давление NO2 в газе при 120°—3, а при 100° —
0,5 мм рт. ст,
Таким образом, среди конкурирующих процессов восстановления
двуокиси азота основным, особенйо на начальных этапах распада, яв-
ляется взаимодействие NO2 непосредственно с ДЭГДН по группировке
простого эфира, которое протекает, по-видимому, подобно другим соеди-
нениям этого класса [6, 9], через стадию образования и разрушения про-
межуточного комплекса. Это иллюстрируется составом продуктов раз-
ложения — аналогично окислению простых эфиров ДЭГДН дает вна-
чале преимущественно нитрит и альдегид, а в противоположность
1,4-БГДН, имеет меньшее количество элементарного азота в газах а
большее содержание летучих орг анических веществ, Соотношение между
начальными скоростями выделения первичных продуктов распада
(Н2О, EN„3, нитрит, альдегид) показывает, что окисление ДЭГДН
л!моль-сек
должно идти в значительной степени путем абстракции водорода с па-
раллельным выделением нитрита, Это можно представить как окисление
по а-углеродному атому простого эфира с образованием а-нитритзаме-
щенного продукта, легко гидролизующегося в ацеталь с последующим
Превращением ее в альдегид и спирт [9, 10]
O2NOCH2CH2OCH2CH2ONO2 + no2 O2NOCH2CH2OH 4-
4-OHCCH2ONO2 4-NO (2)
Быстрое прекращение накопления альдегида соответствует его боль-
шой склонности к окислению, протекающему обычно с высоким выхо-
дам карбоновой кислоты [II]
It
J5 ЯСНО 4- NO2 -+ RCOOH 4- NO (3)
мьдегиды, образующиеся при разрыве связи С—О—С в ДЭГДН, долж-
йн иметь в молекуле максимум 2 углеродных атома. Здесь можно ожи-
дать появления глиоксаля и формальдегида, окисление которых, как из-
жстно, сопровождается выделением значительных количеств СО [12],
ч ОНССНО 4- NO2 2СО + Н2О 4- NO (4)
Це зная точно концентрацию альдегидов, окисляющихся до СО, ио полз-
ая, что она близка к количеству анализируемого легко летучего альде-
№да, можно рассчитать величину к,-- / [ Альд ]лмуч [NO,] =
Й7-10-2 л/лоль сек, d т /
Превращение альдегида в кислоту многостадийный процесс [11] и
Мним из его промежуточных или побочных продуктов, по-видимому, мо-
жет быть ангидрид, который в условиях распада ДЭГДН способен на-
пиливаться из-за сильной конкуренции в отношении гидролиза и окис-
ления со стороны других соединений. Действительно, появление ангид-
мда отмечается при взаимодействии NO2 с октиловым альдегидом [13]
^перекисью бензоила [14].
2RCHO 4- 2NO2 -+ (RCO)2O 4- Н2О 4- 2NO (5)
)1; Карбоновая кислота тоже оказывается активным продуктом и по-
этому также прекращает накапливаться, достигнув некоторой стацио-
ЦДРной концентрации, Ее окисление идет с эквивалентным выходом СО2,
но заметно медленнее альдегида [13, 15],
RCOOH 4- NO2 -+ СО2 4- R’ 4- HNO3 (6)
: г
Основной путь ее исчезновения, вероятно, непосредственная реакция с
нитроэфиром путем переэтерификации [16].
XCH2OCH2CH2ONO2 4- RCOOH XCH2OCH2CH2OOCR 4-HNO3
2HNO3 + NO-л 3NO2 + H2O________________________________
XCH2OCH2CH2ONO2 4- RCOOH 4- 0.5NO -
- XCH2OCH2CH2OOCR 4- 1,5NO2 + 0,5H2O (7)
Получается, что скорость разрушения нитроэфира прямо пропорцио-
нальна концентрации кислоты, причем не только для ДЭГДН, но и для
1,4-БГДН. Значение константы скорости этой реакции для ДЭГДН рав-
Ног 1,5.10-* л1моль . сек.
23
Специальные исследования показали, что ангидриды карбоновых
кислот окисляются двуокисью азота довольно быстро с выделением
большого количества СО2
(RCO)2O I \'О2 -« R' 4- 2С0а 4- NO (8)
Сложные эфиры реагируют с NO2 явно медленнее, и выход углекислого
газа при этом сравнительно невелик. Поэтому можно сказать, что основ
ным источником СО2 при распаде ДЭГДН будут карбоновые кислоты и
их ангидриды.
Для выяснения роли отдельных реакций в наблюдаемых химичес-
ких превращениях необходимо оценить количества накапливающегося
спирта, ангидрида, сложного эфира, а также удельный вес разрывов
ДЭГДН посередине. Этому должно помочь подведение баланса по эле-
ментам, содержащимся, с одной стороны, в исходной нитратной группе,
а с другой, — в появляющихся газообразных продуктах и основных функ-
циональных группировках образующихся конденсированных соедине-
ний. В качестве таких типичных функциональных группировок нами
принимаются альдегид (А), ангидрид (Б), карбоновая кислота (К),
спирт (В), сложный эфир (Э) и нитрит (Г), Балансовое уравнение бу-
дет выглядеть следующим образом
по водороду — Н 4- А 4- К 4- ЗВ 4- 2Э 4- 2Г — 4х = 2п;
по кислороду — О 4- А 4- 2К. 4- В 4- 2Э 4- 2Г 4- ЗБ — х = Зп;
по углероду — С 4- А 4- К 4- В 4- 2Э 4- Г 4- 2Б — 2х = п; ,
по азоту — N 4- Г = п,
где Н, О, С, N — содержание этих элементов в газах; х — количество раз-
рывов связи С—О—С; п —количество нитратных групп, вступивших в
реакцию. По отношениям Н/С, Н/О, H/N, О/С, O/N, С/N можно полу-
чить выражения, позволяющие рассчитать содержания неаиализируемых
продуктов. Совместное решение уравнений, описывающих отношения
Н/С и Н/N, приводит к выражению В—х—Б = -—(I)- Аналогич-
но из отношений О/N и С/N получаем Б4-х = 2N—О 4- С 4- Г — К (II).
Суммирование (1) и (II) дает в результате равенство, описывающее
о 5N2O+3C-Н+2Г-2К ,ттп п
количество спирта В =----------1--------- (III). Расчет из отно-
шений Н/С и С/N в сочетании с выражением (II) позволяет получить
уравнение, характеризующее количество сложного эфира Э 4- 2Б =
_ -5N Н - С - 20 । 2Г - 4К -2А
4 1
Оцениваемое по балансу число разрывов, даже при максимально
возможном содержании ангидрида, оказывается близким к количеству
оторвавшегося элементарного азота (рис, 6). Поэтому можно считать,
что быстрое восстановление двуокиси азота тут же сопровождается раз-
рушением связи С—О—С. Наблюдаемое увеличение скорости разрывов
должно быть обусловлено повышением стационарной концентрации N02.
Из полученных результатов следует, что термическое разложение
ДЭГДН уже вскоре после начала превращения идет в основном через
24
переэтерификацию нитрата образующейся карбоновой кислотой с па-
раллельным окислением группировки простого эфира, заканчиваю-
щимся ее разрывом,
Рис, 6. Кривые накопления
спиртов (7); SN (2); раз-
рывов связи С—О—С (3);
сложных эфиров (4), полу-
ченные на основании мате-
риального и кинетического
балансов
Балансовые расчеты показали, что главным конечным продуктом
разложения является спирт и что сложный эфир накапливается гораз-
до медленнее, чем требуется, исходя из скорости взаимодействия кар-
тоновой кислоты с нитратом, Это находится в прямом соответствии со
способностью эфиров карбоновых кислот к гидролизу [17], которая ока-
зывается явно выше, чем у нитроэфира [18],
н-
RCOOCH2CH2OCH2X + Н2О ХСН2ОСН2СН2ОН 4- RCOOH (9)
►
'i. Ркисление же спирта, по-видимому, следует рассматривать как быстрое
Обратимое превращение его в нитрит [19] с последующим более медлен-
•Ифйым окислением нитрита до альдегида [20].
чг ROH + NO2-+RCHO + Н2О 4-NO (10)
V На рассматриваемом начальном этапе распада кинетическая оценка
, , отдельных химических стадий очень трудна, так как процесс еще не ста-
ционарен и удельный вес реакций меняется. Тенденция к стационарно-
dN dH
СТи намечается только, когда и стремятся к некоторым по-
стоянным значениям. При этом основными конечными продуктами раз-
ложения должны стать этиленгликоль, N2, СО2 и Н2О с небольшим со-
держанием N2O и СО,
С соответствующими допущениями можно оценить роль отдельных
^ёакций уже и на исследуемом этапе превращения, используя кинети-
ческие балансовые уравнения для случаев
ЦА] = 0> dNO = 0 „ dNO. = 0
d -t d x dt d -t
Уравнение, описывающее скорость накопления спирта, которое учи-
тывает образование его при самопроизвольном распаде нитроэфира,
при разрушении ДЭГДН по связи простого эфира и за счет гидролиза
сложного эфира
77 = V к, (НЭ] 4- (к2 [НЭ] [NO,] - £1) + (kT [НЭ] [К,] - —) (V)
□ \ d't/y dv
25
коЗйоляет сосчитать количество сложного эфирй.
d3 2^-(VI)
dr dx 2d? dr dr
Сопоставляя значения Эч-2Б из выражения (IV) и Э из уравне-
ния (VI), можно определить содержание ангидрида, который, судя по
этим данным, накапливается в ощутимых количествах только на более
поздних стадиях превращения*.
Скорости окисления альдегида в кислоту и ангидрид рассчитыва-
d(AJ о
ются из условия -д ' — когда
—кДНЭ] + HHSINOs] + kidBINOJ k^AlNOJ + 4A][NOa] (VII)
2 d?
d[KJ n
н ——- = 0, когда
d x
kJAINOaJ + k9[3IK] = MKINOJ + к7[НЭ][К] (VIII),
Причем скорость гидролиза сложного эфира равна
ri 3
к9[Э1К] = ЫНЭ1К] - (IX)
d т
d3
Задавшись на основании уравнения (VI)-= I • 10^ моль/л ~ сек,
получаем,что d т
MAffNOJ = ke[K][NOd + — 3 )0_fi 4- I • 10_fi = 4 • 10 6 моль/л. сек
d r
и kb -8‘ 10-3 л/моль • сек
Для условия 2 = 0, которое реализуется в конце рассматри-
d ?
ваемого участка превращения, имеем следующее уравнение
к,[НЭ] + — к7[НЭ][К] + 2 -^11 = 1 d-° - + к, [A][NO2] +
2 d? 2 dt
+ 2кз[А][МО2] + 1- ^2? + ka(H3][NO2] + kjoPINOJ, (X)..
2 d ?
где — = kJA^y4HNO2k —^ketKlNOJ + kelBINOj]
d ? d ?
Решение системы уравнений (Vll) и (X) позволяет оценить вели-
чины кз и кю, которые оказываются соответственно равными 2 • КУ"3 и
5 • 10ч л/моль сек.
* Такой способ нахождения количества ангидрида может давать значительную
ошибку, так как здесь оперируют разницей между двумя сравнительно небольшими
величинами, каждая из которых получается' с использованием еще нескольких экспе-
риментальных величин.
dC6t . .......
По зависимости • от концентрации ангидрида иа этапе, когда
. dt
содержание карбоновой кислоты фактически стационарно, можно опре-
делить кв (та 2 • КУ-3 л] моль • сек). Аналогичная зависимость -г от кон-
d -с
центрации карбоновой кислоты на начальном участке разложения дает
возможность получить значение к6(та 1 КГ3 л[моль • сек).
;<Т1олагая, что восстановление до N2 и N2O происходит через
эование и последующее превращение С-нитрозосоедииений
R' lNO-RNO; RNO + 3NO - R- + N2 + 2NO2
RNO —R'CH = NOH; R'CH = NOH + RONO -
- N2O + R'CHO + ROH
обра-
(12)
dNO n
условия ---- = 0 имеем следующее равенство
d т
Т + 4- + WA][NO2] +.2kJ[AINOJ] + МНЭР4О«1 +
2 de 2 de
+ kjolBHNOi] = 4 + ^2 + -Ьк7[НЭ][К]
ат d т 2
(XI)
ropoe вполне удовлетворительно сходится для имеющихся эксперимеи-
ых данных и кинетических констант. В момент времени,
dNO п d(CO+CO,) d(N,+ NtO)
=0, - - —J—sЧТО говорит в пользу
d т---------------de-dт
зтезы об образовании N2 и N2O через реакцию NO с алкильными
калами. Но так как при окислении соединений с двумя углеродными
МЙмами превращение последнего углерода в СО или СО2 не сопровож-
дается обязательным появлением алкильного радикала, то у ДЭГДН на
td <с0 + СОг),
,них этапах разложения -----может заметно превышать
»+N,O) dT
! d т
ф
Г Светлов Б. С. Теория ВВ (сборник статей), М., Оборонена, 1963, с. 278,
X Лурье Б, А., Светлов Б. С. Иав. вузов, химия и хим. технология, 1967, т. 10,
№ 12, с. 1308,
3. Светлов Б С., Л у р ь е Б. А., Д у б и о в а С. Л. Кинетика и катализ 1968, т. 9,
МД с. 1163.
4. Лур>еБ.А. Светлов Б, С. ЖФХ, 1969, т. 43, № И, с. 2737.
5- Кузнецов В. И, Успехи химии, 1954, т. 23, с. 654; Вдовенко В. М., Л и нов-
„ СДИ й А А.. К у з и и а М. Г. ЖИХ. 1957, т. 2, с. 975,
6. Шехтер Г. И, Успехи химии, 1966, т. 35, № 10, с 17; R lib [ n R., S i si ег Н. Н.,
Shecter Н. J. Amer. Chem. Soc., 1952, v, 74, p, 877.
7. Наканиси К- Инфракрасные спектры и строение органических соединений. М,,
<Мир>, 1965.
8. Mackor A., de Boer Th. J. Rec. Trav. Chem., 1970,. 89, 151.
9. Светлов Б. С., Лурье Б. А., Корнилова Г Е, Изв. вузов химия и хим.
^технология, 1974, т. 17, № 2, с, 239,
10. Б е к к е р Г. Введение н электронную теорию органических реакций. М., «Мир»,
1965, с. 156, 165, 181.
II. Я с и и и к и й Б. Г., Зайцев А. П. ЖОХ, 1966, т. И, № 6, с. 1002; Ogata Y,,
Tezuka Н., Sawakj Y. Tetrahedron, 1967, 23, 1007.
12. Pollard F. H., Wyatt R. M. H. Tran. Far. Soc., 1949, v. 45, p. 760; T h o-
m a s J. H. Tran. Far. Soc., 1953, v. 49, p. 630.
13. Светлов Б. С., Лурье Б. А., Корнилова Г. E. «Горение н взрыв».
Материалы 3 Всесоюзного симпозиума, М., «Наука», 1972, с. 780.
14. G i 11 G. В., W i 1 1 i a m s G. Н. J. Chem. Soc., 1956, v, 10, р. 5756.
15. Panday R. N., Barton D. J, Phys. Chem., 1970, p. 3459; Светлов Б, C.:
Лурье Б. А., Смагян В. В., федоннна Л. М., Глухова И. А. Труды
МХТИ им. Д, И. Менделеева, 1974, вып. 80, с. 29.
16. Светлов Б. С., Лурье Б. А., С маг ин В. В. Труды МХТИ им. Д. И. Менде-
леева, 1973, вып, 74, с. 34.
17. Pat a j S. The Chemistry of carboxylic acids and esters. 1969, p. 505.
|8. Светлов Б. С., Ш e л a n у т и и а В. П., Г о р о д к о в а Л. Б. Кинетика и ката-
лиз, 1972 т. 13 № 4, с. 880.
19, G г а у Р.’ Y о i f е. J. Chem. Soc., 1951, р. 1412.
20. LangenBeck, Richter М. Chem. Вег., 1956, v. 89, s. 202,
1
УДК 547.42:117.5:542.951.92:541.454:542.938
ЩЕЛОЧНОЕ ОМЫЛЕНИЕ И НЕЙТРАЛЬНЫЙ ГИДРОЛИЗ
НЕКОТОРЫХ МОНОНИТРАТОВ ГЛИКОЛЕЙ
И а-ДИНИТРАТА ГЛИЦЕРИНА
Б. С. Светлов, В. П. Шелапутина, Я. Г. Кравчинская
Ui. Полинитраты органических спиртов имеют широкое практическое
Изменение. Процессы их термического распада, гидролиза и омыления
Ёдбжны и включают превращения нитратов, содержащих ОН-группы как
гшрмежуточных продуктов, образующихся при этом. Целью настоящей
работы было изучение щелочного омыления и нейтрального гидролиза
цонбнитратов этиленгликоля и 1,4-бутиленгликоля, а также а-ди нитрат а
глицерина.
Моно нитрат этиленгликоля (МНГЛ) получали нитрованием этилен-
рлйколн 99%-ной азотной кислотой при 0°С [1], после выдержки реакци-
онную массу нейтрализовали содой и производили эфирную вытяжку.
МНГЛ, полученный после отгонки эфира, растворяли в небольшом коли-
честве воды, нерастворившийся динитрат этиленгликоля отделяли, воду
замораживали, а полученный МНГЛ перегоняли под вакуумом.
1,4-бутиленгликольмононитрат (1,4-БГМН) получали нитрованием
1,4-бутиленгликоля 67%-ной азотной кислотой [2J, слив вели при —15°,
затем выдерживали при 0° 19 ч и нейтрализовали массу содой. Выделив-
шийся слой динитрата бутиленгликоля отделяли, а из оставшейся мас-
сы производили эфирную вытяжку. После отгонки эфира 1,4-БГМН пе-
регоняли под вакуумом.
а-динитрат глицерина (ц-ДНГЦ) также получали нитрованием гли-
церина азотной кислотой [1] и очищали следующим образом. ДНГЦ рас-
творяли в 13-кратном количестве воды, нерастворившийся нитроглице-
рин отделяли и вводили кристаллическую затравку а-ДНГЦ, при охла-
ждении до 0° медленно выпадали .кристаллы гидрата а-ДНГЦ, которые
отфильтровывали и сушили на воздухе.
Характеристики полученных мононитратов и а-ДНГЦ представле-
ны в табл. 1.
29
J
Таблица I
Характер нети кн мононитратов гликолей и а-ДНГЦ
Вещество Содержание нитратного азота*, % Условии перегонки
определенное расчетное давление, мм рт. ст. t. °C
МНГЛ 13,10 13,08 0,5 51
1,4-БГМН 10,05 10,37 1 72
а-ДНГЦ 14,92 14,89 -* —
• Определяли титрованием навески нитрата раствором FeSO*.
Опыты по нейтральному гидролизу проводились в сосудах из квар-
цевого стекла. Все опыты проводили в водных растворах при темпера-
турах от 9 до 95°, выбирая в каждом случае наиболее подходящую.
Концентрация нитратоспиртов составляла 10~2— 10-3М. За реакцией
следили, анализируя в отобранных пробах содержание ионов МОз~ (моди-
фицированный метод с фенолдисульфокислотой [3]), NO2_ (риванолом),
в случае щелочного омыления определяли концентрацию ионов ОН-
титрованием стандартным 0,01 N раство-
ром H2SO4 в присутствии фенолфталеина,
Щелочное омыление
При щелочном омылении всех изучен-
ных нитратоспиртов выделялась одна
азотная кислота, ионы NO2 не обнаружи-
вались, Выделение HNO3 и поглощение
щелочи происходило по насыщающимся
кривым (рис. 1,2 и 3), на 1 люль выделив-
шейся HNO3 поглощался 1 люль щелочи.
Рис. 1.
а-ДНГЦ,
центрацнн
Щел очное омылевие
2(РС. Начальные кон-
а-ДНГЦ и щелочи,
с-]£Р, М: Л Г—4,44; 10,05; 2, 2'~
6,25; 10,6
При щелочном омылении а'-ДНГЦ с
1 моля нитрата выделялся 1 моль HNO3,
так что омылению подвергалась лишь од-
на нитратная группа, Условия опытов и
результаты их в виде констант скоростей
реакций второго порядка приведены в табл. 2. Видно, что константа ско-
рости не зависит от начальной концентрации щелочи н нитрата, она оста-
валась постоянной и в ходе опытов.
Рис. 2. Щелочное омыление
МНГЛ, 60° С. Начальные кон-
центрации МНГЛ к щелочи,
с 10», М: I, /'—10.25; 103;
22'—4,93; 8,85
30
Рнс. 3. Щелочное омыление 1,4-БГМН,
50° С. Начальные концентрации 1,4-
БГМН и щелочи, с-10*. М; /Д'—5,42;
19,25; 2.Z-17,93; 18,3; ЗЛ-5,52; 4,6
Таблица 2
1
• > All
Результаты опытоа по щелочному омылению
мононитратов гликолей и а-ДНГЦ
4 * — • Вещество t, °C Начальная концентра- ция с-103, Af К-10». ЛГ"1-сект1
нитрата щелочи
а-ДНГЦ С-. 9,5 10,0 14.6 16,0 20,0 20,0 20,0 9,0 9,0 6,85 10,60 6,55 7,76 6,26 4,44 4,82 20,90 6,03 9.4 10,1 9,6 8.8 10,6 10,0 10,5 21,8 21.9 0,62 0,60 1.16 1.33 2,79 2,40 2,77 0,55 0.64
МНГЛ ,ь 40.0 49,9 49,9 50.0 60,0 60.0 4.81 18,62 4 88 4,46 4,93 10,25 9.6 16,9 16.8 9.8 8,8 10,3 0,3V 0.99 1,09 1.15 3 07 2,28
w- 'W 1,4-БГМН 39,9 50 0 f,0 0 50,0 60.0 60.0 17,26 5,52 5,42 17,93 6.17 7.31 19,0 4.6 19.3 18.3 9,9 9.6 0,096 0,21 0,30 0,29 0,81 С.91
Нейтральный гидролиз
Термостатирование водных растворов исследованных нитратоспир-
приводило также к выделению лишь азотной кислоты, Для 1,4-БГМН
елеяие ее происходило по насыщающейся кривой (рис. 4), причем
рганта скорости реакции первого порядка (по нитрату) оставалась
роянной во все время опыта (70—90%. выделившегося азота), не из-
менялась она н при изменении
начальной концентрации нитра-
та (табл. 3).
Рис. 4. Нейтральный гидролиз 1,4-
БГМН, 60° С. Начальная концентра-
ция 1,4-БГМН, с-10», AL I—13,95;
2—7,49
Гидролиз МНГЛ и а-ДНГЦ происходил гораздо медленнее, поэто-
му процент превращения нитрата составлял обычно единицы, однако вез
ж*.уЙЛольяо четко наблюдался прямолинейный характер выделения
HNOfc со временем (рис. 5 и 6). В то же время величина начальной ско-
рости выделения HNOa была прямо пропорциональна концентрации
31
Нитрата и константа скорости реакции первого порядка, вычисленная по
начальным участкам кинетических кривых, оставалась постоянной
(табл. 3), Несколько опытов, проведенных при 90°С с добавками неболь-
ших количеств (2- ICh3 — 8-10'3М) HNOs, показали увеличение началь-
ной скорости выделения азотной кислоты для а-ДНГЦ, однако для
МНГЛ добавка таких количеств HNO3 не вызвала заметного увеличения
начальной скорости выделения ее.
Таблица 3
Результаты опытов по нейтральному гидролизу
мононятратов гликолей и а-ДНГЦ
Вещество 1, “С Начальная концентрация нитрата с-Юэ, М К - 10в сек-1
а-ДНГИ 80,0 80,0 90,С 90.0 90,0 95,0 9.45 9,81 9,92 20,95 41,93 7,18 0,011 0,011 0,014 0,040 0,038 0,075
МНГЛ 70,0 80,0 90,0 90,0 90,0 90,0 90.0 90,0 95,0 18,99 20,91 0,99 4,22 10,03 19,36 26,04 47,32 8,92 0,019 0,047 0 10 О’, 16 0,16 0,17 016 0.17 0,25
1,4-БГМН 60,0 60,0 70,0 70,0 80,0 95,0 7,49 13,95 7,06 12,79 5,48 4,56 2.3 2.3 7,4 7,2 17,3 66,0
Рнс. 5, Нейтральный гидролиз МНГЛ,
90“ С. Начальная концентрация МНГЛ,
С -10’, М: I — 47.32; 2 — 2604; 3 — 19 36;
4—10,03; 5—4,22; 5—0,99
Рис, 6, Нейтральный гидролиз а-ДНГЦ,
90° С. Начальная концентрация а-ДНГЦ,
elO3, М,- /—41;93; 2-20,95; 3-9,9?
3?
Полученные данные позволили рассчитать константы уравнения
Аррениуса и сравнить константы скоростей реакций щелочного омы*
ления и нейтрального гидролиза изученных нитратоспиртов (для этого
константы скоростей реакции нейтрального гидролиза, полученные
вследствие избытка воды, в виде констант скорости реакции первого
порядка, были переведены в константы скорости реакции Второго по-
рядка) (табл. 4),
Таблица 4
Коэффициенты уравнения Аррениуса для щелочного омыления
и нейтрального гидролиза нитратоспиртов и скорости ик при 70°
Щелочное омыление Нейтральный i идролиз
Вещество 1g в Е, 1g в Е, к • Ют,
"* - тех-1] к калило ль Д1 г ,cw 1 |ВД-!] к кал 1 моль АГ1 сек'1
а'-днгн 12,4 23 4, 8 12,9 32 0.W6
МНГЛ 10,0 21 0,07 11,0 28 0,04
1,4-БГМН 13,0 23 0,02 11,1 24 13
Видно, что константы скоростей щелочного омыления ннтратоспир*
тов на 4—9 порядков превосходят константы скорости нейтрального
гидролиза. Порядок расположения нитратоспиртов по величинам кон-
стант скоростей прямо противоположен в реакциях щелочного омыления
и нейтрального гидролиза. Так, константа скорости щелочного омыления
Наибольшая у u-ДНГц, а для нейтрального гидролиза константа ско*
рости у него наименьшая. Энергии активации реакции щелочного омы-
ления близки для всех изученных нитратоспиртов, а нейтрального гид*
релиза — увеличиваются от 24 ккал/моль (1,4-БГМН) до 32 ккал/,ноль
(а-ДНГЦ). Однако скорость нейтрального гидролиза даже для наибо-
лее медленно гидролизующегося а*ДНГцна порядок превышает скорость
его термического распада [4]. Следует заметить, что u-ДНГЦ уже явля-
ется (Кислотой, хотя и слабой, поэтому нейтральным гидролиз для него
можно назвать лишь условно,
Результаты работы позволяют сделать некоторые выводы о меха-
низмах исследованных процессов. О механизе щелочного омыления ни-
тратов, содержащих ОН-группу, можно судить, исходя из следующих
фактов. Наум [11 получал с 95%-ным выходом нитроглицид, встряхи-
вая ДНГЦ с 30%-НОЙ ще.ючыо, Напомним, что и в наших опытах с
моля ДНГЦ выделялся моль HNOj, то есть омыление шло до нитроглц-
цида. Одним из наиболее общих методов синтеза эпоксидов является
внутримолекулярное замещение, механизм которого хорошо изучен на
примере галогенгидринов [5] и включает две стадии: сначала при обра-
ботке щелочью образуются алкоголяты, которые затем претерпевают
внутримолекулярное замещение, По этому механизму, вероятно, и идет
щелочное омыление изученных нитратоспиртов
HOCH2CHaONO2 + OH-^OCH2CH2ONO2 + Н2о
-о о
1 /
СН2СН2 CH2CHZ + NO,-
ONO,
5 Труды вып, 83
Тем более, что влияние строения согласуется с предложенным меха-
низмом: чем более кислым является нитрат, тем быстрее проходит эта
реакция. Кроме того, наблюдаемые величины констант скоростей зна-
чительно (на 2—5 порядков) превышают соответствующие константы
для этил- и метилнитратов (по значениям для растворов в 60%-ном
спирте, где реакция идет быстрее, чем в воде), щелочное омыление кото-
рых идет по механизму главным образом реакции бимолекулярного
нуклеофильного замещения [6].
Прямо противоположное влияние строения на величины констант
скоростей нейтрального гидролиза нитратоспиртов может свидетельство-
вать в пользу того, что он осуществляется скорее всего по механизму
реакции бимолекулярного нуклеофильного замещения. Кстати, пара-
метры уравнения Аррениуса для нейтрального гидролиза МНГЛ и ме-
тилнитрата [7], гидролизующегося по механизму бимолекулярного нукле-
офильного замещения [6], практически равны. Значительное увеличение
константы скорости для 1,4-БГДН по сравнению с другими мононитра-
тами свидетельствует о том, что в данном случае ОН-группа имеет воз-
можность влиять на реакцию, очевидно, образуя циклическое соединение,
как это имеет место при щелочном омылении мононитратов гликолей,
УДК 662.232:547.622
fl' C,
1*'..
О ВЗАИМОДЕЙСТВИИ НИТРОГЛИЦЕРИНА
Й С ДИФЕНИЛАМИНОМ
I. Наум Ф. Нитроглицерин, М — Л., ОНТИ, Госхимтехяздат, 1934, с. 196,
2, Еременко Л. Г., Королев А. М. Изв. АН СССР, 1966, № 8, с. 1436.
3, С в с т л о в Б. С., Шел ал у тип а В. П, ЖФХ, 1966, т, 40, № 11, с. 2883.
4. А ф а я а с ь е в А. Г,, Л у р ь е Б. А., С в е т л о в Б. С. Сб. «Теория ВВ», М., «Высшая
школа», J967, с. 67.
5. W i п s t е i п S., L u с a s Н, J, J. Amer Chem. Soc., 1939, v. 61, p. 1576.
6. В a k e г J. W., E a s t у D, M. J. Chem. Soc, 1952, p. 1193.
7. -M с К i i) 1 e y-M с К e e J S., M о e 1 w у n-H и g h e s E. A. Trans. Far, Soc., 1952.
v, 48, p. 247.
В. С. Светлов, В. П. Шелапутина, Л, Д. Максимова, -- -
К В. П. Евтушенков
ф 1
;,,г, Дифениламин (ДФА) до сих пор является одним из наиболее по-
-Иигярных стабилизаторов химической стойкости нитроэфиров. Однако
Вдиеханизме его действия в литературе имеются противоречивые сведе-
.дая, говорящие как о возможности непосредственного взаимодействия
Jpt*A с нитроэфирами [1], так и, наоборот, о том, что он только связы-
вает окислы азота, выделяющиеся при распаде [2], В настоящей работе
Мы попытались выяснить этот вопрос, количественно проследив за про-
дуктами химического превращения смеси нитроглицерина (НГЦ)
с ДФА.
\ В работе использовали НГЦ, перекристаллизованный из 80%-ной
Потной кислоты и отмытый от последней, и ДФА с tn/l 52,7°С, перекри-
сталлизованный из этилового спирта. Опыты проводили при 60; 70 и 80°,
когда скорость распада НГЦ незначительна. Двух-, четырех- и восьми-
процентные растворы ДФА в НГЦ предварительно вакуумировали от
летучих примесей в стеклянных мембранных приборчиках [3] в течение
трех часов при остаточном давлении, меньшем IO"-3 мм рт. ст. Отноше-
ние навески вещества к объему реакционного сосуда составляло
0,05 г)смг. Через определенные промежутки времени термостатиро-
вания измеряли давления в реакционном объеме при температуре опыта
и при 0°, по которым вычисляли количество воды *, находящейся в га-
зовой фазе при температуре опыта. Процентное содержание конденси-
рующихся газов при этом получается таким же, как и при измерении
Давления газов при —20°, когда упругость водяных паров близка к
Нулю, Кроме того, определяли содержание растворенной воды, прини-
мая, что неконденсирующие газы нерастворимы в НГЦ, а раство-
* По опытам А. Г. Афанасьева, проводгшшего анализ газов распада 2 %-пых
Йвюгворов ДФА в НГЦ при 12Щ, количества конденсирующихся газов и воды совпадают.
римость воды в растворе ДФА в НГЦ та же, что и в чистом НГЦ.
Тогда
О _P-kN
Ол — ----
18
где Вж— количество Ц2О, растворенной в НГЦ при температуре опьла,
моль)г; к — константа растворимости Н^О в НГЦ при температуре опы-
та (4], лиг’; р—давление газов при температуре опыта, мм рг. ст.; N —
доля конденсирующихся газов.
Затем приборчик вскрывали вод вакуумом, газообразные продукты
анализировали на хроматографе ХЛ-4 или откачивали вакуумным на-
сосом и анализировали в конденсированной фазе содержание ДФА и
его производных. Дли этого часть реакционной массы р-0,1 г) раство-
ряли в этиловом спирте (1 .ил) и раствор (0,00677 мл) наносили микро-
шприцем на пластинки для тонкослойной хроматографии марки
«silufol», производили разделение бензолом и снимали отдельно пятна
N-нитрозодпфенпл амина (N-МОДФА) и п-нитродифениламина
(п-НДФА), а ДФА и о-нитродифениламнн (о-НДФА) снимали вместе,
так как их R[ отличались лишь на 4 леи [5], и растворяли в спирте.
После фильтрации определяли оптическую плотность растворов на
СФД-2 для N-КОД ФА при 295 ммк, п-НДФА —при 390 ммк и для рас-
твора о-НДФА с ДФА при 424; 300; 285 и 260 лшк [6]. Количества
продуктов оценивали но калибровочным кривым. Содержание ДФА и
о-НДФА получали следующим образом. По оптической плотности рас-
твора при 424 мяк находили содержание о-НДФА (но калибровочной
кривой), затем определяли соответствующие этому содержанию плот-
ности о-НДФА для других длин волн (Оо.цдфд) но соответствующим
калибровочным кривым для о-НДФА. Вычитая эти значения из общей
оптической плотности раствора (Dr) оценивали оптическую плотность,
приходящуюся на долю ДФА (D.t<i,a) при этой длине волны
ПдТФА =- D-; — Db-НЛФА
По этим величинам определяли количества ДФА по калибровочным
прямым при этих длинах воли и брали среднее значение. Табл. 1 пока-
зывает, что количества ДФА и о-НДФА, введенные в ЦГЦ и определен-
ные таким образом, находились в удовлетворительном согласии.
Т И б Л II ц а Содержание И*>, г введено | определено
ДФА о-НДФА 21 ФА о-НДФА
3.68 1,75 0,36 3.55 1.75
3.68 3,70 0.38
2,-25 1,03 2,10 1.Ю
2.25 1 .35 1 95 1.48
Гааовыделение при распаде смеси НГц с ДФА в мембранном при-
бо чике протекает с резким ускорением (рис. 1), которое наступает
Намного раньше обычного «нитроглицеринового», а количество газов,
выделяющееся при этом, прямо пропорционально количеству добавлен-
ного ДФА. Затем скорость падает и наблюдается участок равномерного
газовыдслення, протяженность которого также прямо пропорциональна
количеству добавленного ДФА. я уже после этого наступает обычное
для НГЦ ускорение.
Практически сразу после начала термостагировання смеси НГЦ с
ДФА па хроматограммах появлялось четыре вида пятен; желтые н ли-
монные пятна о- и n-НДФА, серо-голубые—N-МОДФА и постепенно
синеющие пятна ДФА. Пятна других производных ДФА появлялись
лишь к моменту исчезновения ДФА.
Закономерности выделения наблюдаемых продуктов можно видеть
на примере 4%-ного раствора ДФА (рис. 2). Несмотря ца довольно
значительный разброс данных, обусловленный как условиями проведе-
ния опытов, так, вероятно, и значительной скоростью превращений на
хроматограмме для ДФА и NJ-ХОДФА [б], можно видеть, что вначале
происходит быстрое выделение N-М'ОДФА, воды, о- и n-НДфА, Затем
скорости образования их уменьшаются. N-МОДФА и вода образуются
уже практически с равными и постоянными скоростями вплоть до исчез-
новения всего ДФА, а содержание о- и л-НДФА остается постоянным.
Ойо начинает уменьшаться лщпь к моменту исчезновения всего ДФА,
когда, очевидно, о- и н-НДФА переходя г в динитропроизводные ДФА.
пятна которых появляются на хроматограммах. В опытах при 70 и С0°
содержание о- и и-НДФА было также мало и близко к содержанию их
при 80'.
Рис. i. Газовыделение при рас-
паде НГЦ (Г) и НГЦ+5% ДФА
(2) при 80° С, го Д'1 - 0,054 и
0,060 г/с.и3 соответственно
Рис. 2. Выделение продуктов
распада раствора НГЦ+4%
ДФА при 80°С: /—ДФА; 2—
N-NO ДФА; 3—Н2О (расчет-
ное содержание, включающее
газообразную н растворенную
воду); 4—не конденсирующиеся
при 0° газы; 5—о- и п-НДФЛ.
ф — баланс по ДФЛ
Некондснсирующиеся газы на начальном участке практически отсут-
ствуют. Выделение их в дальнейшем происходит с малой скоростью, рез-
ко увеличивающейся ко времени израсходования всего ДФЛ, когда
начинается «дифениламиновое» ускорение. Анализ некондсшсирующихся
газов показал, что они состоят главным образом из NO и СОа (табл. 2),
причем, если вначале в газах NO и СО2 содержатся примерно поровну,
37
то в копие «днфеяиламинового» ускорения отношение NO к СОа уже
равно 2, На 1 моль ДФА выделялось примерно 0,5 моля неконденсиру-
ющихся газов. Резкое уменьшение содержания конденсирующихся газов
на участке ускорения газовыделения обусловлено окончанием реакции
ДФА с НГЦ, дающей воду. Содержание МЛ'ОДФА к этому времени
близко к начальному содержанию ДФА. Следует, однако, отметить,
что баланс по ДФА был обычно занижен (рис. 2). Соотношение началь-
ных скоростей образования продуктов приведено в табл. 3.
, Таблица?
Состав газообразных продуктов для 4%-ного раствора ДФА в НГЦ,
(80°С, m/v = 0,05 г/см3)
Время тер- ыостатиро- вания, мин Общее кол-во га- зов при 80° v-l(W, моль/г Конденсирующиеся газы Неконденсирующиеся газы
Темпера- тура кон- денсации, °C Содер- жание, % содержание, %
NO СО; Ns N»O СО
3044 0.33 -23 70.5 13 16 0,5 0 0
9239 2,69 -10 37.5 37 18 2 3 2.5
9294 2.67 -17 37,4 38 19 1.5 2,5 1.6
Таблица 3
Начальные скорости образования продуктов н исчезновения
ДФА для 4%-ных растворов ДФА в НГЦ
(о>* 1010, лоль/с сек)
t. ° С ДФА N-МОДФА Н,,0 о-НДФА п-НДФА Скорость распада НГЦ [7]
80 12,4 9.1 4,8 2 2 0.5
70 5,0 2.5 1,3 0.7 0,7 0,1
60 2,3 1.25 0,8 0,4 0,4 0.016
* Средняя скорость на участке до выделения 3-ГО-5 моль/г М-ИОДФА.
Опыты, проведенные при 80° с 8- и 2%-иыми растворами ДФА в
НГЦ, показали соблюдение всех закономерностей выделения отдельных
продуктов, наблюдаемых для 4%-ных растворов. При этом скорости
образования Ы-ЬЮДФА, воды и исчезновения ДФА были пропорцио-
нальны начальному содержанию ДФА в НГЦ (рис. 3, 4 и 5, табл. 4).
L Т а б л и ц а 4
Начальные скорости образования продуктов и
исчезновения ДФА при распаде смесей НГЦ с ДФА
(80°С. то 1010, мом!г сек)
Содержание ДФА, % ДФА N-НОДФА НаО
2 6.6 4,8 3
4 12.4 9,1 4,8
8 26 16 7,6
^Максимальное содержание о- н n-НДФА для 8- и 2%-ных растворов
ДФА также было мало и не превышало величины 2 • 10-в моль/г, как и
ДЛЯ 4%-ных растворов (рис. 6), только в опытах с 8% ДФА оно дости-
галось быстрее, чем в опытах с 4- и 2%-ными растворами.
время, мин
Рве. 3. Выделение N-ЫОДФА при рас-
АФ-паде растворов ДФА в НГЦ, 80°С.
Ц У Начальное содержание ДФА, %:
. /-—8; 2—4; 3—2
время, мин
Рис. 4. Выделение воды (расчетное
содержание, включающее газообраз-
ную и растворенную воду) (/. 2, 3)
и некондеисирующихся газов (/', 2')
при распаде растворов ДФА в НГЦ,
80° С, Начальное содержание ДФА,
%: /,/’-8; 2.2'—4; 3-2
Инфракрасный спектр продуктов превращения НГЦ с 8% ДФА
Юсле 5000 мин термостатирования при 80° показал сильное поглощение
и'" n-л п... 1280
WpH 1650 1/сл, что говорило о присутствии кетонов,
$1020 l/сл, где поглощает простая эфирная связь.
•V .
и при
2008 4000 вез ввз
время, мин
Рис. 5. Выделение n-НДФА (а) н
о-НДФА (б) при распаде растворов
ДФА в НГЦ, 80° С. Начальное со-
держание ДФА, %: ф—8; О—4;
ф —2
Рис. 6. Влияние начального
содержания ДФА на началь-
ные скорости исчезновения
ДФА (/), образования
N-ЫОДФА (2) ц НаО (3) при
распаде растворов ДФА а
НГЦ, 80° С
Энергия активации, рассчитанная по начальным скоростям исчез-
ШЛО П/fiA II -___ хт ктлт-я С
_______________________________ , В-« 11* гъ оь | Л 31 XL ‘1
новения ДФА и образования М-МОДФА, составила 22—25 ккал/моль,
что значительно ниже энергии активации термораспада НГЦ
(40 ккал/моль), а скорости исчезновения ДФА и образования N-МОДФА
(см. табл. 3) превышают скорость мономолекулярного распада НГЦ
более чем на порядок при 80°, а при снижении температуры до 60° уже
Почти на два порядка.
39
Полученные результаты доказывают, что при достаточно низких
температурах идет главным образом реакция взаимодействия ДфА с
НГЦ. Об этом говорят, как низкая энергия активации процесса, так и
прямая пропорциональность скоростей образования продуктов содер
жаиию ДФА, а главное, величина скорости превращения ДФА, в де-
сятки и сотни раз превышающая скорость распада НГЦ.
В опытах, проведенных до полного исчезновения ДФА (4% ДФА,
70 и 80°), он на 80—90% превращался в N-МОДфА, расчетное количе-
ство воды к этому моменту близко к содержанию N-ХОДФА, в продук-
тах присутствует кетон. Это позволяет представить уравнение главной
реакции исчезновения ДФА следующим образом
R2CHONO2 + HN(CGH5)a-* RaC = О + ON—N(C6H5)2 + H2O
Вероятно, благодаря распаду таких кетонов и образуются некоиденси
рующиеся газы.
Образование на начальной стадии о- и н-НДфА с суммарной ско-
ростью, примерно вполовину меньшей скорости образования N-МОДфА
(табл. 2), напоминает щелочное омыление НГЦ, при котором HNOa и
HNO3 отщепляются в соотношении 2: 1 [8], если считать, что отщепле
ние HNO2 дает N-.МОДФА, а отщепление HNO3 — соответственно
о- и п-НДФА, Вполне вероятно, что выделение продуктов реакции (на-
пример, воды) тормозит образование о- и н-НДфА, способствуя образо-
ванию лишь Ц^ОДФА.
Опыты показывают, что реакция взаимодействия НГЦ с ДФА имеет
первый порядок но ДфА. Если она первого порядка и по НГЦ, то урав-
нение для скорости исчезновения ДФА будет следующим
с1[ЛфД1 -
- -=3,G • 1°те «г - [НГЦ].[ДФА], моль л-сек
1. At и г а ии г 11. Bull. Soc. Chitn. lf/.'х,, .. 1#^.
2. F г e у A F.xplosrvsloflc, в. 1967, № 5, s. 97.
3. Андреев К. К., Глазкова А. П, Маурина H Д„ Светлов Г>. С, ЖфХ,
1938, т. 32, с. 1726.
4. Г о р <5 у и о в В. В. Сб. «Теория ВВ», М., Оборонена, 1963, с. 219, ,
5, Jacobsson О. «Chemical problems connected with the stability of explosives.
Slokgolm». 1967, p. 146.
6. Maurer R., Willy A, Ducts ch; A, «Chemical problems connected with the
Maoiiiiy of explosives*. Tyringe, 1970, p. 14,
7. Афанасьев А. Г., Светлов В. С., Лурье Б. А. Сб. «Теория ВВ», М., «Выс-
шая школа», Пин, с. 67,
8. Lowry Т. М., В г о w u n i п g К. С., Farmery, J. W. J, Chem Soc., 1920, v. 117,
№ 69, p. 552.
УД К 541 -124 + 541.127+54G. 174 + 547.458.8!
КИНЕТИКА ОКИСЛЕНИЯ ЦЕЛЛЮЛОЗЫ
ДВУОКИСЬЮ АЗОТА
Б. С. Светлов Б. Л. Лурье, Г. Е, Корнилова
Химическое превращение, целлюлозы под действием двуокиси азота
изучено довольно подробно [1—3]. но кинетика процесса и Природа вос-
станавливающихся азотсодержащих продуктов фактически нс известны.
В настоящей работе скорость взаимодействуй оценивали по исчезновению дву-
окиси азота из газовой фазы в замкнутом реакционном объеме. Конструкция при-
бора, в котором совмещались стеклянный компенсационный манометр [4] и оптическая
кювета, позволяла одновременно контролировать общее давление газообразных продук-
та!? и давление МО2 гг измеряемое по оптической плотности газов с помощью
ФЭК •М, соединяемого с кговётой гибкими стеклянными световодами.
Исследовалась хлопковая целлюлоза (ГОСТ 595—55, сорт К), предварительно из-
мельченная на голандере, многократно промытая дистиллированной водой п тщательно
высушенная от остатков влаги. Количественную дозировку окислов азота осуществляли
‘ перемораживаннсм в вакууме из специального дозировочного объема, снабженного стек.
' лянным компенсационным манометром [5].
Для расчета кинетических констант необходимо знать растворимость NO2 и Ns О,
. в целлюлозе. Ее оценивали на основании специальной серин опытов при относительна
невысоких температурах (20—70°С), когда доля растворенных окислов ощутима. Зная
сдознрованное количество ЬЮ2 и N2O( п их общее давление над целлюлозой с учетом
..известного равновесия .между ними в газе [б], легко по разности определить суммарное
^.доличество адсорбированных окислов азота (ИОг + З^гО^, Принимая зависимость сум-
/ маркой концентрации NOi, в целлюлозе от общего давления прямолинейной, находим
.некоторую фиктивную константу растворимости \ -=1222?
. которой с температурой описывается уравнением > Рцбы
(табл. 1), изменение
моль
ex i> I-- |----
V RT 1 моль-мм
Это уравнение позволяет оцепить общее количество растворенных* окислов азота
при более высоких температурах в условиях окисления целлюлозы. О концентрации
* Непосредственное определение констант растворимости NO2 и N2O< путем ре-
шения методом наименьших квадратов системы лпвейных уравнений N0> =
=®= ^NO, *Pno, + ^N,o, *PN,O, оказалось невозможным, так как из-за эксперимен-
тального разброса kN0. иногда получалось отрицательным и искомые величины хаоти-
чески менялись с температурой.
41
отдельно NOs и NaO( судили, используя данные Грэя н Веспера о диссоциации NaO(
в жидком состоянии и в некоторых растворителях (бензоле и хлороформе) J7). При
этом все 3 выражения, характеризующие величину kc = —N2* для конкретных
cN(o,
условий окисления целлюлозы, давали близкие значения концентрации как по NOa.
так н no NsO4. Ощутимые колебания (в 2 раза) наблюдались только в случае относи-
тельно малых концентраций одного из окнслов. В дальнейшем при обработке кинети-
ческих результатов пользовались уравнением, описывающим диссоциацию NjO« в хло-
роформе *.
Таблица I
Результаты экспериментов по определению растворимости
окнслов азота в целлюлозе
Количео во, моль 10-‘ Объем сосуда слР Общее давление NOa + NUO,
целлю- лозы сдозиро- венного S N О2 20° зо° 40е 50° 6‘.° 70°
2,36 4.22 5,3 а, 83 5,77 5,7 2,50 2,03 2,01 4,65 4,27 3.31 6,24 11,94 11.84 32,0 12,83 12,4 275 162 129 139 258 22» 331 184 115 158 306 257 407 200 164 177 359 298 476 224 183 198 417 342 554 251 204 222 481 389 640 273 224 243 544 434
Значения фиктивной к растворимости (мом1моль-.им) 10“‘ 10,2 7.3 5.4 3,8 2,9 2.1
объема
NOj и
при 80—130°. Степень заполнения реакционного
- — ---------- «*«=>»> давления
Окисление исследовали i.r.. __ ______
целлюлозой составляла 0,002—0,007 г{см\ По изменению общего
NjO4 над целлюлозой во времени с учетом их растворимости находили изменение сум-
марного количества NOS в газовой и конденсированной фазах в ходе опыта. О пре-
вращении азота суди.тн по результатам хроматографического анализа неконденсирую-
щихся при 0° газообразных продуктов (8( после полного восстановления двуокиси
азота.
Исчезновение NO2 при окислении целлюлозы не подчиняется просто-
му кинетическому закону. Кривая, характеризующая изменение ее со-
держания, имеет слегка волнообразную форму. Поглощение двуокиси
азота, постепенно замедляясь, затем временно вновь слегка ускоряется
(рис. !) Кривая общего газовыделения оказывается почти зеркальным
отражением кривой исчезновения NO2. На всем протяжении опыта на
I моль прореагировавшей двуокиси азота выделяется 2 моля конечных
газообразных продуктов. Падение суммарного количества NO2 идет с
постепенно уменьшающейся скоростью (рис. 2).
Неконденсирующиеся при охлаждении до 0° газообразные продук-
ты окисления состоят из N2, NO, N2O, СО и СО2 (табл. 2). Содержание
элементарного азота в газах в конце превращения составляет около
I моля на I моль введенного NO2. Основной продукт восстановления
двуокиси азота — NO. В N2 и N2O перерабатывается от 30 до 40% NO2.
• kc— i0’i,S8exp<-iiaoD/Rn МОЛЬ]Л
42
Ж . ”
^^^одержание N2 обычно ниже, чем. N2O. Состав газов не зависит от
'количества взятой двуокиси азота и мало меняется с температурой.
Вгличество конденсирующихся при охлаждении до 0° составляет прн-
изительно 0,5 мол на I моль NO2. Из углеродсодержащих газов основ-
ft — СО2. Доля СО в 4 раза меньше.
ЩрТ
V Рис. I. Изменение общего давле-
ния газообразных продуктов
(' (1—5) и давления NOa (Р—5')
ПРН взаимодействии целлюлозы с
двуокисью азота при темпера-
.Т1 туре, °C: /, 2-80; 5—100; 4-110:
1- 5—130
Рис, 2. Кинетика исчезновения
суммарного количества окнслов
азота (N0a+2N2O() при окислении
целлюлозы. Температура, °C:
1,1'—80; 2—100; 3—110; 4-130
Таблица 2
jfrj. Состав газообразных продуктов окисления целлюлозы
^i°c Количество сдозиро- ванного SNOa моль-10-' Отношение I NOj/це.т- люл., МО ЛЬ [МОЛЬ Количество газон, моль/моль NOa
ы2 NO NaO СО Со$ конден- сирую- щиеся
0,59 0,31 0,05 0,51 0,05 0,02 0,10 0,59
Ж ЛОО ’110 1,1 0.-13 0,05 0.78 0,18 0,10 0.48 0 55
2,03 0.48 0,10 0,50 0,12 0,05 0,33 0,74
2,50 1,06 0,05 0.79 0,15 0,06 0.40 0 14
2,01 0,38 о. ос. 0.60 < 0 13 0.09 0 38 0.51
Н<ь . 4.65 0.80 0.05 0.54 о.и 0,08 0 36 0'57
М 0,32 0,18 0.78 0.14 0,14 0,49 0,63
Реакционная способность целлюлозы обусловлена наличием а ее
’/молекуле двух активных функциональных группировок: гидроксильной
K-глюкозидной, Окисление по гидроксилу [2, 3] приводит к образованию
карбоксильной группы у 6-го углеродного атома, кетонных — у 2-го и
ЭИч> или при разрыве пиранозного цикла между 2-м и 3-м углеродными
аТОМами-дает соответствующие альдегиды и кислоты. В то же время
ЧЖКсление по глюкозидной связи всегда должно заканчиваться разру-
швЯйем цепи макромолекулы. Отсутствие кинетических данных по де-
'^+рукцим сильно осложняет рассмотрение механизма окисления целлю-
43
Лозы, так как трудно оценить вклад глюкозидной группировки, которая
способна разрушаться и за счет гидролиза [2, 3], Судя по прочности окис-
ленных целлюлоз [3], следует полагать, что реакция по глюкозидной
связи идет гораздо медленнее, чем по гидроксилу. Косвенные подтвер-
ждения этому видны из представленных экспериментальных данных.
Можно рассмотреть, как минимум, 2 вида окислительпо-деструктив
лых превращения по глюкозидной связи; первое с разрушением целлю-
лозного звена и параллельным выделением окислов углерода и второе—
с разрывом цепи без распада пиранозного цикла. Во всех этих гипоте-
тических случаях обязательным должно быть образование алкильных
радикалов, которые, как известно, активно способны перерабатывал!,
NO в Ns и N2O [9], Поэтому по количеству N2 и N2O (а оно относи-
тельно невелико — 10—20% в общем количестве азотсодержащих газов)
можно как-то судить о степени деструкции. .Мерой протекания деструк-
тивного процесса может служить и количество углсродсолержащих га-
зов, причем в первую очередь СО, так как она образуется обычно при
окислении органических соединений с минимальным углеродным скеле-
том [10]. Содержание СО мало и не превышает 10% от количества вос-
становленного \'Ой. СО2 же, выделяющаяся преимуществеио за едет
окисления карбоновых кислот [11], в случае целлюлозы способна образо-
вываться (от 6-го углеродного атома) н без разрушения пиранозного
никла. Поэтому, исходя из относительных количеств N2, N2O и СО, де-
струкция должна идти в 5-— 10 раз медленнее окисления по гид-
роксилу,
Анализ газообразных продуктов позволяет оценить и элементар-
ный состав получающих-ся конденсированных соединений. Если допус-
тить, что газы, конденсирующиеся при охлаждении до 0°, это Н2О, то
па 1 .ноль прореагировавшей двуокиси азота целлюлозное звено теряет
0,5С, 1Н и 0,50. Па один атом азота в газообразных продуктах прихо
дится 2 атома кислорода, из которых 0,5 (в СО и СО2) принадлежали
целлюлозе. Следовательно, 0,5 атома кислорода от NO2 в результате
окисления переходят в целлюлозное звено. Поскольку там каждый угле-
род уже имеет при себе кислород, то должно получаться 0,5 моля па
1 моль ЫО2 функциональных группировок, содержащих 2 кислорода на
углероде, по-видимому, в виде кислоты —(СООН). Это вполне согла-
суется с известным образованием карбоксильных групп по 6-му угле-
родному атому при взаимодействии целлюлозы с NO2 [1- 3].
Судя по .многочисленным литературным данным, окисление целлю-
лозы протекает преимущественно через образование и последующее
разрушение нитрита [1, 12], то есть процесс идет подобно взаимодейст-
вию со спиртами: ROH + N2O4 RONO + HNO3. Но целлюлоза эте-
рифицируется медленнее спиртов в силу особенностей ее химической
структуры [3, 13], а так же, очевидно, из-за худшей растворимости окис-
лов азота в ней.
Предлагавшиеся ранее механизмы дальнейшего превращения ни-
трита целлюлозы [1—3,12] в карбоновые кислоты во многом носят еще
дискуссионный характер. Полученные в настоящей работе кинетические
данные позволяют исключить из рассмотрения схемы, основанные на
самопроизвольном распаде нитрита, так как его скорость не способна
обеспечить наблюдаемых скоростей окисления. В наиболее благопри-
ятных условиях, когда нитрит практически не накапливается if
Йрасп ‘ ^-иитр == к,,к* - С2 ХЮ), (где С(1ИТр —— С;‘ NO,), НСОбхОДПМО, ЧГОбЫ
йрасп нитрита на 4 порядка превышала известные значения для случая
44
его гомолитического разложения в конденсированной фазе [14] (для де-
нмлнитпита при 80° kf>acil = 1,3- 10~9 сект1).
’ По-видимому, основным путем превращения нитрита в условиях
окисления целлюлозы будет реакция типа реакции Маурера [15], кото-
рая, судя по имеющимся скоростям взаимодействия децил нитрита с NO2
[14] (при 8(Г к — 7 - Ю-5 л/лоль сек), способна протекать довольно
быстро
rch2ono rc/N0H 22.°. RCOOH
ONO
Рассматривать разрушение нитрита под действием N20g, как рас-
пад на RO -т- NO, вероятно, ошибочно, гак как именно такой путь ре-
акции должен ограничиваться с одной стороны ее обратимостью, а с
другой - образованием стойкого нитрата.
Измеряемая скорость исчезновения NO2 при реакции ее с целлюло-
зой складывается из скорости этерификации целлюлозы и окисления
образующегося нитрита
w — k't' t-IIS, - CrjjO, 4" k2cHi)Ij,-<n»o4
По Данным Кеньона [16] и Ермоленко [1], количество накапливаю-
щегося нитрита ио сравнению с прореагировавшей двуокисью азота
мало. Рост концентрации нитрита заметен только в начальный .момент
превращения. Поэтому скорость этерификации выше скорости окисле-
ния нитрита только сначала, а затем па основном участке взаимодейст-
вия они близки к, c[[C<]-cN10i = к-.-с^.р-схщ,, откуда k(/k2 = с.^/с^.
Из данных Кеньона следует, что при 25° константа скорости окисления
нитрита целлюлозы выше константы скорости его образования почти ца
Цррядок. С повышением температуры концентрация нитрита падает [1].
В связи с этим должно возрастать отношение k2/kb то есть реакция
Окисления нитрита должна иметь более высокий температурный коэф-
фициент, чем первичная реакция этерификации целлюлозы четырех-
окисью дзота. Так как для обеих последовательных стадий близки ско-
рости и эквивалентный расход NO2, то скорость исчезновения целлю-
лозы при окислении будет составлять половину от величины, измеряе-
мой по падению суммарной концентрация двуокиси азота. Она, по-
видимому, подобно взаимодействию со спиртами [15, 17], должна быть
пропорциональна концентрации N2O^. Константу скорости окисления
Целлюлозы (табл. 3) рассчитывали, как для реакции 2-го порядка
1 W
к= пгтегж’ттт~т—1 . где [Цел.]—это число молей целлюлозных звеньев
2 |NsOJ[uen.] L
,, Таблица 3
Кинетика окисления целлюлозы двуокисью азота
°C 80 100 110 №.130 Колн чество, моль -10~> Начальная ско- рость исчезнове- нии ХМО3 моль/л-сек- J0-* Концентрация в целлюлозе NO2, N 3О4, моль/л-10'2 Константа скоро- с, и окисления, л1моль-сек Н0ж
целлюлозы S NO3
2,03 1,86 2,56 2,0 3,4 2,15 0,59 1,1 11 1,1 3,66 0.44 1,56 3,33 3.33 8 25 3 3 9 5.5 Н 3,5 11,0 1,0 0,91 0,92 1,78 5.9 20,8
в объеме ее навески. Значения скоростей брали по начальным участ-
кам кинетических кривых (рис. 2). когда роль вторичных процессов
относительно невелика. Изменение константы скорости с температурой
описывается уравнением
к —~ 106-7 ехр7 R т f.i/чо.Н’ ‘ссь'
Для выбранных условий окисления проведена оценка роли диф-
фузии. При этом рассматривается случай диффузии, сопровождаемой
химической реакцией [181, для которого можно записать условие
d2c
k-c = где к -константа скорости окисления; D — коэффициент
диффузии; с — концентрация N2O4 в целлюлозе; х — расстояние вдоль
поперечного сечения волокна. Решение его сводится к решению урав-
нения
сх — Are ' D + А,-е D
При условии, что при х = 0, с = со. а при х = -х-, (где d— диаметр во-
dc Л „ 2
локва целлюлозы) = 0, получаем 2 уравнения, которые позволяют
вывести следующее окончательное выражение*;
сд /с0 = 2 е а Ъ ,'е } D ф 1
Из пего легко убедиться, что в широком диапазоне значений коэффи-
циентов диффузии газа по полимерным материалам от 10~5 до
10“9 см2[сек [19] и при реальном сечении волокна целлюлозы (около
20 л(клг), сильное изменение концентрации NO2 и N2O< по толщине за
счет химической реакции для полученных значений констант скоростей
нереально, то есть, представленные кинетические данные определяются
химическими закономерностями, а не диффузией.
M.
' 11. С в e т л о в Б. С.. Лурье Б- А., С м а г и и В. В., федонииа Л. М,, Глухо-
в а И. А. Труды МХТИ им, Д. И. Менделеева, 1974, вып. 80, с. 29.
; 12. Каверзнева Е. Д„ С а лов а А. С. Изв, АН СССР, отд. хим. наук, 1959, № 2
с. 344.
Гетце К, Производство вискозных волокон. М,, «Химия», 1972.
Светлов Б, С., Лурье Б. А., Мясникова С. И„ Херсонский Н. С.
Кинетика и катализ 1974, № 5.
LangenbeekW,, Richter М. Chem. Вег,, 1956, v. 89, р, 202.
М с G г е е Р. A., F о w 1 е г W. F., Т а у 1 о г J, Е, W„ U n г u h С. С,, К е п у о п W. О.
J. Amer. Chem. Soc., 1947, v, 69, p. 355.
F a i r 1 i e A. M,, С a r b e г г у J. J., T г e а с у J. C, J. Amer. Chem, Soc., 1953, v. 75,
p, 3786; S 11 v e г w о о d R., T h о m a s J. H. Tran. Far. Soc., 1967, v. 63, p. 2476.
Ба ту вер Л. M., Поз ин M. E. Математические методы в химической техиоло-
логии. М., «Химия», 1968,
Р е пт л и н г ер С. А. Успехи химии, 1951, т. 20, № 2, с. 213.
15.
16
Л17.
Й.Ч8.
€ i9-
T
e* .
ы-
1. Ермоленко И. И. Спектроскопия в химии окисленных целлюлоз, Минск, Изд.
АН БССР, 1959.
2. Никитин В. И, Химия древесины и целлюлозы, Гослесбумиздат, 1964.
3. Роговин 3. А. Химия целлюлозы. М., «Химия», 1972.
4. Светлов Б. С., Лурье Б. А., Корнилова Г. Е. Кинетика и катализ, 1972,
т. 13, вып. 5, с. ] 146.
5, Горбунов В. В, Теория ВВ (сборник статей), М., Оборонгиз, 1963, с. 219.
6. Термодинамические и переносные свойства химически реагирующих газовых систем.
Часть 1, Минск, «Наука и техника», 1967.
7, Gray Р. Rathbone Р. J, Chem. Soc., 1958, р. 355(1; V о s р е г A, J. J, Chem
Soc.. А, 1970, р. 219].
8. Лурье Б, А, Светлов Б. С. Труды МХТИ им, Д, 14. Менделеева, 1967.
т. 53, с. 36.
9. Donarnma L. D., Carmody D, J. J. Org. Chem., 1957, v. 22, p. 635; Stra-
usz О. P., Gu an in g H. E. Canad. J. Chem., 1963, v. 41, p. 1207.
10. Pollard F. H., Wyatt R, M. H. Tran. Far. Soc., 1949, v. 45, p. 760; T ho-
rn a s J. H. Tran. Far- Soc,, 1953, v. 49, p. 630.
r
w
Ml.;
Ии
ПЙГ--.
I*-'.. ‘
“«яя пассматриваемой задачи предложен В. А. Коробаном.
Газообразные продукты распада ТНБ в расплаве (m/v ~ 0,1 г/сл3)
Шри 300°, как видно из рис. 2, содержат главным образом СО2 (до 40%)
'Wn2 (25—30%), содержание Н2О, СО и органических летучих веществ
составляет около 10% каждого, а относительные количества NO и N2O
.'долы (2—5% и менее 1% соответственно). Интересно, что в области
i ручейных степеней распада (0,3—1,8%) содержание СО2, Н2О и орга-
нческих летучих возрастает в ходе разложения ТНБ, а содержание NO
?N2 падает. Эти результаты существенно отличаются от аналогичных
ДОных, полученных для разложения ТНБ в парах [2]. Из таблицы видно,
fo при той же степени распада в случае разложения ТНБ в парах в
азах содержится в 20 раз больше NO, втрое меньше N2 и в ~ 10 раз
Меньше СО2.
УДК 662.237:541,124—13
ИЗУЧЕНИЕ СОСТАВА ГАЗООБРАЗНЫХ ПРОДУКТОВ
РАСПАДА ЖИДКОГО ТРИНИТРОБЕНЗОЛА
Ю. Я, Максимов, С. Б. Сорочкин, Б. Ф, Санранович
Исследование кинетики и продуктов термического разложения 1,3.5-
тринитробеизола (ТНБ) в парах [1, 2], а также распада его под дей-
ствием электронного удара [3] в целом показало чрезвычайно сложную
картину прекращения этого вещества. Достаточно надежно было лишь
установлено, что первичной стадией его является отщепление NOa-групп.
Кинетические данные о распаде жидкого ТИБ [4], а также сведения о
высокой склонности его к авто ассоциации в расплаве [5] указывают па
возможное изменение механизма начального распада ТНБ при переходе
от газообразного к жидкому состоянию,
В настоящей работе приведены данные о составе газообразных про-
дуктов термическою разложения ТНБ з расплаве (и, частично, в рас-
творе) при 280—300°С и малых степенях превращения (^2%).
Термический распад ТНБ проводили в сосудах объемом 5 мл, снабженных стек-
лянным манометром типа Бурдона. Для отбора газов па анализ к сосуду предвари-
тельно припаивали трубку с внутренней стеклянной перегородкой, выдерживающей
перепад давлений в 1—1,5 аг и разбивающейся от удара падающего стеклянного бойка
(pitc, I), Хроматографический .-щализ газов проводили в два приема: двухатомные
газы Ns, NO и СО разделяли па колонке с порапаком «Q», охлаждаемой до —70°, а
сумма этик газов, NsO, СО, н НаО (в виде С2Н3) давали отдельные пики на хромато-
грамме при разделении их на колонке с твердым носителем с нанесенным на него ДЮШ-
тилсульфоксидом [6]. Ввиду того, что часть NO необратимо поглощается порапаком
«Q», снимали калибровочную кривую на искусственных смесях NO-l-Nj и по ней опре-
деляли истинное содержание NO в анализируемой газовой смеси. Для перевода воды
в ацетилен к трубке с перегородкой припаивали отросток (рис. 1), в котором содер-
жалось определенное количество свежеизмельчеиного CaCj. Превращение 1ДО в СзНг
осуществляли многократным вымораживанием ее на карбид жидким азотом при одно-
временном нагревании сосуда с навеской ТНБ до 130°. В связи с тем, чго, к;зк было
установйепо, СаСэ может поглощать ч.теть СОа. проводили параллельный опыт по
распаду ТНБ и анализу газов без применения карбида, и содержание СОг в анализи-
руемой смеси газов определяли по ди иным этого опыта.
О содержании в газообразных продуктах органических летучих веществ судили
по количеству конденсирующихся при комнатной температуре веществ, из которого
вычитали количества имеющихся в газовой фазе ТНБ и воды.
ТНБ, применявшийся в работе, очищали трехкратной кристаллизацией из хлоро-
форма п, для освобождения его от возможной примеси тринитробецзойноц кислоты,
кристаллизовали из аммнянИогс, водип-лцегойового раствора. Точка плавления ТНБ
Имля павна 122,5— 123,0°.
Рис. 1. Схема при-
бора для изучения
кинетики газообразо-
вания при распаде
тринитробенаола с
приспособлением для
отбора газов на ана-
лиз и перевода воды
в ацетилен. /—Стек-
лянный прибор, вклю-
чающий сосуд с ве-
ществом и манометр
типа Бурдона-, 2—
стеклянная перего-
родка с лежащим на
ней бойком; 3—боко-
вой отросток с карч
бидом кальция
Степень раеподй, %
Рис. 2. Изменение состава
газообразных продуктов в
ходе распада тринитробен-
зол а s расплаве. Содержа-
ние данного газа равно вы-
соте относящейся к нему
площадки
'
Состав газообразных продуктов распада ТНБ в расплаве,
4 растворе и а парах (глубина превращения 1%)
и Условия распада >К. & Состав продуктов, %
NO N,0 СО СОЭ Н,0 органич. летучие
Л ь. Йасрлаа, 300°С 3,1 0.7 26,3 11.1 36,8 ИЛ 10.6
Раствор в гексахлорбен- .юзоле, 280°С Лары, 350°С [2] ? 19,7 12.8 15.3 32,8 16,5 2.9
67,5 — 7,8 16,9 3.9 3.9
Интересны в этом отношении результаты анализа газов при распаде
ТНБ в растворе, когда уменьшена возможность межмолекуляриых взаи
(Содействий в жидком веществе, В качестве растворителя был взят
гексахлорбензол, который в выбранных условиях обладает достаточной
устойчивостью и хорошей растворяющей способностью, инертен по отно
шенИю к ТНБ и продуктам его распада и имеет относительно низкую
таругость паров (т. кип. 320°). Опыты при 280° показали, что в 50%-пых
растворах этого растворителя ТНБ разлагается вдвое медленнее.
•#емв расплаве. Состав газообразных продуктов в этом случае (таблица)
8 целом занимает промежуточное положение между найденным при рас-
4* Труды «ЫП, в?
49
паде ТНБ в расплаве и в парах. Сказанное в первую очередь относится
к одному из главных продуктов разложения ТНБ —к NO, содержание
которого при переходе от расплава увеличилось более чем в 6 раз. Со-
держание азота, углекислого газа и органических летучих стало меньше,
а окиси углерода больше.
Изучение состава газообразных продуктов при малых степенях раз-
ложения ТНБ и отсутствие данных о кинетике его расходования не по-
зволили провести балансовый расчет процесса по элементам. На осно-
вании полученных результатов представляется возможным оценить
лишь скорость накопления последних в газовой фазе. Из рис. 3, где
показана кинетика роста во времени количества каждого из элементов
Ррс. 3,; Кинетика накопления в
газовой фазе кислорода (О),
азота (N), углерода (С), водо-
рода (Н) и суммы газов (V) в
процентах к общему их количе-
ству при разложении тринитро-
бензола в расплаве прн 300° С
(отнесенного к имеющемуся в молекуле ТНБ), видно, что скорости
накопления в газах азота и кислорода практически одинаковы. Следует
отметить,1 2 что приведенная на этом графике кривая суммарного газовы-
делеиия (отнесенная к общему количеству газов, выделяющихся яри рас-
паде ТНБ) имеет почти тот же угол наклона, что кривые, относящиеся
к этим элементам. Накопление же в газах углерода и особенно водо-
рода идет с заметно меньшей скоростью.
Выводы
1. Установлено, что при распаде тринитробензола в расплаве газо-
образные продукты имеют существенно иной состав, чем при распаде
в парах, а в случае превращения его в растворе состав газов занимает
промежуточное положение. Эти данные указывают на изменение меха-
низма начального распада тринитробензола при переходе от паров к
расплаву, которое, по-видимому, обусловлено межмолекулярными вза-
имодействиями в расплаве.
2. Показано, что суммарная скорость газовыделения при распаде
жидкого тринитробензола, по крайней мере, на начальной стадии его
практически равна скорости накопления в газах наиболее важных эле-
ментов N и О, входящих в молекулу тринитробензола, и может харак-
теризовать кинетику разложения этого взрывчатого вещества.
1. Максимов Ю. Я. ЖФХ, 1972, т. 46, с. 1726.
2. Максимов Ю. Я-, Егорычева Г. И. Кинетика и катализ, 1971, т. 12, с. 821.
3. Meyerson S., V а п d е г Haar R., Fields E. J. Org. Chem., 1972, v. 37, p. 4114.
4. Максимов Ю.Я. ЖФХ, 1971, т. 45, с. 793.
5. ЛейченкоА. А., Максимов Ю. Я. ЖФХ. 1973 т. 47, с. 1265.
6. Л у р ье Б. А., С в е т л о с Е>. С. Труды МХТИ им. Д. Н. Менделеева 1968, вып. 58.
с. 189. »•••••
11 . УДК 547.546:542.922:541.124
О РОЛИ НЕКОТОРЫХ КОНДЕНСИРОВАННЫХ
ПРОДУКТОВ В ПРОЦЕССЕ ТЕРМИЧЕСКОГО
' * РАСП АДА ТРИ НИТРОТОЛУОЛ А
Ю, Я. Максимов, В, Ф. Сапранович, Н. В. Полякова
Ранние работы по термическому распаду 2,4,6-тринитротолуола
'НТ) показали, что процесс его имеет автокаталитическую природу,
«чем автокатализатором является конденсированное вещество [J], об-
дающее малой летучестью [2]. Позднее из конденсированных продук-
в было выделено вязкое бурое вещество, растворимое в ацетоне и
4Т н разлагающееся быстрее исходного ТНТ, добавка которого суще-
венно ускоряет распад последнего [3]. Проведенный в недавнее время
оматографический анализ конденсированной фазы прн распаде ТНТ
5] показал, что в ней содержатся главным образом 4,6-динитроантра-
1Л и 2,4,6-триннтробензальдегид, добавка которого в количестве 25%
1 Весу к ТНТ вызывала сильное разложение двухграммовых навесок
о при 200°С.
В настоящей работе рассмотрен вопрос о стойкости этих двух ве-
ф£ств и влиянии их на разложение ТНТ.
2.., Термическое разложение веществ изучали манометрическим мето-
Жшв современном его варианте [3], а хроматографический анализ газов
9вводил и по методике [6, 7].
ТХ 2,4,6-Тринитробензальдегид получали конденсацией п-нитрозодиме-
Ва с ТНТ [8]*. После кристаллизации из этаноло-бензольной
ггинчатые кристаллы слабо-розового цвета плавились при 119°.
ческое разложение тринитробензальдегида при 200° (рис. 1)
?нь большой скоростью, которая в начале процесса в 10э 4 5 раз
ем у ТНТ, и в целом уменьшается во времени. Общее коли-
юв, выделяющихся при распаде альдегида в этих условиях,
ьше, чем в случае ТНТ н составляет лишь около 200 см&[г,
кол на 1 S'мол вещества, что может говорить о стадийности
азложения тринитробензальдегида.
ры благодарят Г. М. Шутова н В. Л. 3барского за консультации, значи-
чивщне проведение синтеза.
#•4* 51
Разложение смесей ТНТ-—альдегид разного состава, показанное «в
рис, 1, идет тем быстрее, чем больше содержание альдегида. Однако
рост скорости происходит не аддитивно — распад смеси идет медлен-
нее, чем можно было ожидать из ее состава (рис. 2), Таким образом,
раннее наблюдение каталитического действия тринитробензальдегида на
распад ТНТ [5] не получило подтверждения, и это противоречие мож^
объясняться неиэотермичиостью процесса в .первом случае,
Рис. I, Термическое разложение смесей тринн-
тротолуол-трннятробеяэальдегнд при 200* С. Со-
держание трннйтробензальдегида в смеси, вес.%:
/—100; 2 — 76; 3—50; 4— 33; .5-9;
5-5; 7-2; 8-0
4.6-Дини.троантранил получали восстановлением 2,4,6 тринитробена-
альдегида треххлористым титаном [9], После кристаллизации изгэтанола
вещество подвергали молекулярной перегонке и .повторной кристалли-
зации иэ абсолютного спирта. Жел-
тые йластинчатые кристаллы плави-'
лись при 125—126°. ' .
Рис. 2. Зависимость начальной
скорости разложения смеси
тринитротолуол - триннтробей-
эальдегид от ее состава. Пунк-
тирная кривая рассчитана по
закону аддитивности
Распад дянитроантранила (рис. 3) идет гораздо .медленнее, чем
тринитробенаальдегида, Однако скорость его при 200° в 5 раз Z»
чем в случае ТНТ. Количество газов к концу превращения д
800 сж3/г, т. е. 2,5 г-мол на 1 г-мол вещества. Добавление к Т
динитроаитранила (или конденсированного остатка от его раз
на 3%) резко увеличивает скорость реакции, которая в начали
дни становится близкой к измеренной для чистого диинтроант
&
льцрз,
, СТ Д’"
да
В этой са^ибаерв отличйс от опытов с тринитробензальдегидом, скорость
распада смеси гораздо выше рассчитанной по закону аддитивности,
V
Рис. 3. Термическое разложение
смесей тринитротолуол-динитро-
антранял при 200°С, /—Трини-
тротолуол (ТНТ); 2—динитро-
антранил (ДНА нтр.) i 3 — смесь
ТНТ + 10 вес.% ДНАитр., 4 —
смесь ТНТ + 10 вес.% ДНАитр.,
разложенного на 3%; 5 — кривая,
рассчитанная по закону аддитив-
ности по данным опытов 1 и 2
Давление паров динитроаитранила, измеренное.при 200 и 250°, рав-
но 11,5.и 45 мм рт. ст. Оценка по этим данным температуры кипения при
атмосферном давлении дала величину 390s, которая при 45° выше най-
денной ранее для ТНТ (345d [10]). В этом отношении динитроантранил
соответствует имеющимся в литературе указаниям на меньшую лету-
честь автокатализатора, чем ТНТ.
Интересен вопрос о механизме разложения дннитроантранила и его
действия на распад ТНТ. Анализ газообразных продуктов распада этого
вещества (таблица) показал, что они содержат значительные (более
40%) количества углекислого газа. Этим обстоятельством объясняется
установленное ранее образование больших количеств СО2 при распаде
ТИТ н растворе [11]. В отличие от разложения ТНТ, в продуктах со-
держится довольно мало конденсирующихся продуктов, включающих,
по-видцмому, и воду. Наиболее важным представляется тот факт, что
при распаде динитроаитранила в начальной стадий образуются большие
количества окиси азота,, источником которой обычно является NOj, От-
сюда можно заключить, что реакция разложения динитроаитранила
включает отщепление нитрогруппы, Расчет элементарного состава пер-
манентных газов в случае полного разложения динитроаитранила дал
содержание в них азота 2 г-ат, кислорода 2,6 г-ат и углерода 1,3 г-от
на 1 г. мол вещества, что указывает на отщепление от молекулы этого
цитросоединения и второго, по-видимому, гетероатома азота, а также на
отщепление, по крайней мере у трети молекул динитроаитранила, ато-
мов углерода ядра. Последнее неизбежно должно сопровождаться рас-
крытием шестичленного углеродного цикла.
Состав газообразных продуктов распад*
дянитроантранала н его смеси с ТНТ
Вещество Условия распада Состав, объем». %
темпера- тура, °C степень распада, % Na NO СО СО, конден- сируй- шиеся
ДпнитроаЧтраннл 200 2.8 26,6 16,1 6,3 43,3 7,7
Дп нтро а нт ра нил 250 100 32.4 8 Л 6 6 44,4 9.8
200 1,5 18.9 3,2 7,7 15,6 54 6
Тротил+динатро- ' актраинл (,!<]%) 200 2.4 25.7 2.5 2,8 28.2 40,8
53
Добавка к ТНТ 10% динитроантранила, как видно из таблицы, 4||-
дет к увеличению вдвое содержания в газах СОг и заметному росту
содержания азота. Эти данные в совокупности с результатами кинети-
ческих опытов позволяют предположить, что каталитическое действие
динитроантранила на распад ТНТ состоит во взаимодействии ТНТ с
первичным продуктом разложения динитроантранила, обладающим, по-
видимому, высокой реакционной способностью, которое приводит к уско-
ренному его превращению.
Выводы
1. Синтезированы 2,4,6-три нитробензол ьдегид и 4,6-диинтроантра-
нил, являющиеся основными промежуточными конденсированными про-
дуктами термического разложения ТНТ. Распад смесей ТНТ с первым
веществом идет медленнее, а со вторым — гораздо быстрее, чем можно
ожидать по закону аддитивности. Для второго вещества оценена точка
кипения, которая выше, чем у ТНТ. Эти данные согласуются с наблю-
дениями предыдущих исследований и позволяют приписать роль авто-
катализатора в данном процессе 4,6-дииитроант1ранилу.
2. Анализ продуктов распада динитроантранила показал, что раз-
ложение этого вещества включает отрыв NOj-групп и частичное разру-
шение 6-членного углеродного цикла. Найденные в продуктах большие
количества СОа объясняют происхождение этого газа при изучавшемся
ранее разложении ТНТ в растворе.
♦ ...............................
1. Рогинский С. 3., М а г и д А. М, ЖФХ, 1931, г. 2, с, 263.
2. Robertson A. Trans. Faraday Soc,, 1948. 44, 677.
3. Андреев К. К. Термическое разложение и горение взрывчатых веществ, «Нау-
ка», 1966.
4. Rogers R. Analyt. Chem., 1967, N 6, р. 730.
5. Da co ns J., Adolph H., К a m 1 e t M. J. Rhys. Chem,. 1970, v. 74, p. 3066.
6. Л у p ь e Б. А.. С в e т л о в Б. С. Труды МХТИ им. Д, И. Менделеева, 1968, вып.
5В, с. 180.
7. Максимов Ю. Я., Сорочкин С. Б., Сапранович В. Ф. Труды МХТИ ни.
Д, И. Менделеева. 1974, вып. 83, с. 48,
8. Sachs F-, EverdingW, Berichte, 1902. v. 35, p, 1236.
9. S p 1 i 11 e r J., С a 1 v i о M. J. Org. Chem., 1955, v. 20, p, 1086.
10. Максимов Ю, Я. ЖФХ, 1968, т. 42, с. 2921.
11. Сапранович В. ф., Максимов Ю. Я-, Маркелова М. Е. Труды МХТИ
им. Д, И. Менделеева, 1973, вып. 75, с. 147.
\УДК 547.54б/.525.5.-542.92:541.127-3
ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ И ДАВЛЕНИЕ ПАРОВ
ТРИНИТРОПРОИЗВОДНЫХ м-КСИЛОЛА И МЕЗИТИЛЕНА
Ю. Я. Максимов, И. В. Полякова, В. Ф. Сапранович
Исследование кинетики и механизма термического распада 2,4,6-три-
нитротолуол а [1—3] показало, что относительно низкая стойкость этого
ароматического нитроа>едннения обусловлена наличием в его молекуле
легкоокисляющейся -метильной группы.' Интересны в этом отношении
триннтропроизводиые л-ксилола и мезитнлена, молекулы которых вклю-
чают соответственно две и три метильные группы. Данные Р. Роберт-
сона [4], показывающие увеличение стойкости в ряду СН3-замещенных
тринитробензола, получены более 50 лет назад по несовершенной мето-
дике и нуждаются в уточнении. Целесообразно также рассмотреть воп-
рос о механизме термического распада этих двух нитросоединений.
Термическое разложение иитросоединений изучали манометричес-
ким методом с применением манометров типа Бурдона. Этим же мето-
дом измеряли давление насыщенного пара веществ при экстраполяции
давления, растущего в сосуде с веществом при его разложении, на время,
соответствующее началу реакции [б]. Хроматографический анализ газов
проводили по методике, разработанной в работе [6] и впоследствии усо-
вершенствованной [7].
2,4,6-Тршштроксилол (ТНКс) после кристаллизации нз толуола
плавился при 182,5—183°С. По литературным данным точка плавления
его равна 182,5—183,5’С [8].
2,4,6-Тринитромезитилен (ТНМез), перекристаллизованный из бен-
зола, имел точку плаалеиия 234,5 — 235°С, которая по литературным дан-
ным равна 235° С [9].
Термическое разложение ТНКс и ТНМез в парах, изучавшееся прн
300°С (рис. 1), характеризуется теми же особенностями кинетики, что
и распад большого числа других ранее изученных ароматических нитро-
соединений [10]. Как видно из рис. 1, превращение паров ТНКс идет с
увеличивающейся, во времени скоростью, которая может превышать
начальное ее значение в единицы раз. Общее количество газов к концу
реакции достигает 4 е^мол на 1 глол вещества и практически не отлч-
55
чается от измеренного для тринитротолуола (ТГНТ). Величина макси-
мума скорости тем больше, а время его достижения тем меньше, чем
больше концентрация паров. Увеличение поверхности стекла также уве-
личивает скорость процесса в начале и уменьшает общее количество
газов в конце его. Те же закономерности имеет и кинетика распада па-
ров ТНМез. Для паров обоих веществ характерно также наличие слож-
ной зависимости начальной скорости распада от концентрации (рис. 1,в),
когда в области малых концентраций наблюдается насыщающийся ее
рост, в области средних (ро/р» 0,4, где р, — давление насыщенного па-
ра) — постоянство ее, а при приближении к точке насыщенного пара-
резкое увеличение.
Рис. 1. Термическое разложение паров
трнннтрокснлола при 300° С. а—Ход
газовыделения во времени; 6—диффе?
ренциальная форма кривых р(т), в—
зависимость начальной скорости рас-
пада паров трнннтрокснлола (/) н три-
ннтромезнтилена (2) от относительной
концентрации (Р« — давление насыщен-
ного пара), Числа при кривых озна-
чают начальное давление паров (мм
рт ст.}. В опыте (х) поверхность сосу-
да увеличена капиллярами в 10—20 раз
Сопоставление относительной начальной скорости распада паров
всех трех веществ при 300°С в диапазоне концентраций, где она посто-
янна, показало, что при увеличении числа СНз-групп в молекуле нитро-
соединения стойкость его возрастает: если скорость распада ТНТ при-
нять за 1, то в случае ТНКс она составит 0,7, а в случае ТНМез — 0,6.
Термическое разложение ТНКс и ТНМез в расплаве подобно рас-
паду ТНТ идет быстрее, чем в парах. Экстраполяция начальной ско-
рости распада жидкого иитросоединения на 300°С дала в случае первого
вещества величину, в 3,6 раза превосходящую измеренную в парах, а в
случае второго — в 1,4 раза. Переход к расплаву так же, как в случае
ТНТ, приводит к росту ускорения процесса: скорость уже при выделе-
нии 5% газов в 5—10 раз превышает начальную. Замедление распада
при переходе от ТНТ к ТНКс и далее к ТНМез в расплаве (рис. 2) вы-
ражено сильнее, чем в парах. Так при 250°С начальные скорости про-
цесса соотносятся как 1 ; 0,3 ; 0,08.
Температурная зависимость начальной скорости разложения ТНКс и
ТНМез в расплаве определена при одинаковом, достаточно большом-со-
держании вещества в жидкой фазе (94%). Значения относительной ско-
рости распада обоих веществ, полученных при разных температурах, в
Аррениусовых координатах ложатся на прямые линии (рис, 3), из угла
наклона которых найдены значения эффективной энергии активации Е
56
и для мбномолекулярного распада
(табл. I).
рассчитан предвксионент В
Рис. 2. Термический распад три-
нитротолуола (/), трнннтрокснлола
(2) и трииитромезитнлена (3) в рас-
плаве при 250’С
Рис, 3. Температурная зависимость
коистаиты скорости распада а
расплаве тринитробензола (/1,
тринитротолуола (2), трнцитро-
кснлола (3) и трниитромезитп-
леиа (4)
Газообразные продукты распада ТНКс н ТНМез при 250°С и глу-
бине превращения 1 % содержат 35—40% Н2О и 20—30% СО2, т. е, прак-
тически те же количества, что в случае разложения ТНТ [3].
Величины, характеризующие кинетику разложения
три нитробензол а и его СНгпроизводных и рас вл аве
Таблица 1
Нит росоеди менне Интервал температур, °C Константа скорости при 250 ° С сек' МО1 Коэффициенты уравце ння Аррениуса
1g В (сек-1) Е, ккал', моль
. ,1Д5-Триннтробензол (по данным [1]) 250-310 1 10.9 43,0
2,4,6-Т рн и и т рото лу ол (по данным [ЗВ 190-250 79,5 9.3 34,5
2,4,6-Т р и н и троке и лол 220-260 25,1 9.1 3.->, 1
2,4,6-Т ри н и т ро м езн т и ле и 241-272 6.3 12,3 44,2
Термическое разложение ТНКс и ТНМез в растворе гексахлорбеп-
зола изучали при 250°С в широком диапазоне концентраций. Из рис. 4,<т
видно, что добавление к веществу малых количеств растворителя ведет
к резкому падению скорости разложения, а при дальнейшем его добав-
лении торможение распада уменьшается и при 10- 15-кратном количе-
стве растворителя скорость процесса становится практически неизмен-
ной. Таким образом, при переходе от расплава к разбавленному рас-
твору распад обоих веществ замедляется в ~10 раз, и соотношение ско-
ростей их распада остается тем же, что в расплаве. Анализ зависимости
скорости распада от концентрации (рис. 4,6) показал, что в случае обоих
веществ она хорошо описывается степенным законом, причем показа-
тель степени при концентрации равен ~2.
Давление паров ТНКс и ТНМез определяли в опытах по разложе-
нию этих веществ в расплаве. Результаты определения сведены в табл. 2
Г>7
Н нанесены на график в координатах tg р (1/Т) (рис. 5). Йа полученных
пряйых определены предэкснонент в уравнении р = А-ехр(—A/RT) я
теплота испарения X. Экстраполяцией этой зависимости на атмосферное
давление найдены значения температуры кипения, и по известным вели-
чинам рассчитаны значения отношения Трутона (табл. 3). Из таблицы
видно, что точка кипения ТНКс лежит выше, чем ТНТ, в то время как
ТНМез кипит при заметно меньшей температуре. Отношение Трутона
у обоих веществ мало и в случае ТНМез близко к нормальному его
значению (21).
Рис. 5. Температурная зависи-
мость давления паров тринитро-
ксилола (1) и трннитромезитиле-
на (2). Для сравнения приведена
аналогичная зависимость для три-
нитротолуола (3) н трннитробен-
зола (4) по данным [5]
Рис. 4. Термический распад тринитро-
ксилола (1) и тривнтромезитнлеиа (2)
в растворе гексахлорбензола при 250° С.
в —Зависимость начальной скорости
чмоцесса от соотношения компонентов:
^—логарифмическая зависимость удель-
н01 скорости распада от концентрации
1 (Вес.%)
Та блица 2
Давление паров триннтроксвяола к тринитромезйтилена
1 Ннтросоединекие Темпера- тура, °C Давление паров, мм рт. ст. Темпера- тура, °C а идеи не паров, мм рт. ст
Трвцнтроксилол 210 t 31 250 78
230 46 260 99
240 60
Три ннтром езити лен 240 112 263 191
1 246 13') 272 235
1 250 145 281 290
Полученные данные показывают общность кинетических особенно-
стей распада ТНКс и ТНМез, с одной стороны, н ТНТ—с другой. В со-
вокупности с одинаковым составом газообразных продуктов на ранних
стадияз! иренращеннявсех трех веществ это' позволяет считать „что
«
Таблицаi
Величины, характеризующие кипение ТНКс и ТНМез
Ннтросоедииение IgA (для р в мм) ккал [моль Тит. °C ^/^КИП*
Тринитротолуол (по данным [5]) 9.11 17,5 345 28,3
Тринитроксилол 7.75 14,0 356 22,3
Тринитроиезнтилен 7,67 13,2 330 21,9
[ механизм первичной реакции нх разложения одинаков м состоит в окис-
гении СНз’Групп. В свете развитых ранее представлений об одиовремеи-
Ьм протекании процесса по внутри- и межмолекулярному механизмам
: [3] интересен вопрос о их соотношении в ряду рассматриваемых веществ,
к В первую очередь следует рассмотреть влияние агрегатного состояния
г йа скорость их разложения. Так, отношение скоростей распада wM/wn
С при 300° закономерно уменьшается при переходе от ТНТ (7 по данным
г [3]) к ТНКс (3,6) и далее к ТНМез (1,4). С этой величиной коррели-
| руется и отношение Трутона, характеризующее степень ассоциации жид-
Е.ростей, что указывает на уменьшение роли меж молекулярных взанмо-
|t действий при распаде с увеличением числа CHj-групп в молекуле нитро-
Е' соединения.
Еч При переходе к растворам, вследствие подавления межмолекуляр-
Е' ных взаимодействий, скорость распада нитросоедипений становится на
№ порядок меньше, чем в расплаве и, как показал расчет, даже меньше,
I чем в парах. Последнее в общем не кажется удивительным, поскольку
Ескорость химических реакций в газовой фазе обычно больше, чем в жид-
в? кой. Важной особенностью разложения изучавшихся веществ является
г. пропорциональная зависимость константы скорости ийчального их рас-
Е пада от квадрата концентрации раствора (в случае ТНТ показатель
г,Степени при концентрации достигает 3 [3]). Это обстоятельство указывает
К па немоиомолекуляркость разложения рассматрцваемых нитросоедине-
iLlptfi в расплаве и делает необходимым вестн расчет предэкспонента в
L уравнении Аррениуса не для мономолекулярной реакции, как это дела-
к дось ранее, а для бимолекулярного распада. Такой расчет дал вели-
f ЗДну В в случае ТНТ 10е-9*, в случае ТНКс 10е7 и в случае ТНМез
Е10*'9 л • мол-1 • сект’.
к Одной из наиболее интересных закономерностей распада в ряду
Е метильных замещенных производных тринитробензола является замед-
гфецие его с увеличением числа СН3-групп в молекуле*. Оно каче-
ственно сходно с уменьшением скорости превращения, наблюдающимся
к,* ряду хлор производных три нитробензол а [1]. Однако это сходство, по-
г ййднмому, формально и не может быть объяснено с точки зрения элек-
Е тронных представлений, поскольку СН3-группа и атом С1 обладают про-
к ТИвопсложными донорно-акцепторными свойствами. Наиболее вероят-
к ным, по крайней мере, в случае СН3-замещенных, представляется объ-
£ яснение этого явления действием стерического фактора. Спектральные
Исследования [12] показали, что наличие в молекуле ароматического
• Интересно отметить, что в этом ряду веществ уменьшается также обнаруженная
недавно [11] способность нитросоединения тормозить процесс радикальной поли мери-
?ацин.
59
нитросоединения СМз-групп обусловливает поворот Г4ба-групп относи-
тельно плоскости бензольного ядра, Это может приводить к затрудне-
нию образования циклического активированного комплекса, растущему
с увеличением числа СН3-групп в молекуле,
Таким образом, увеличение стойкости в ряду моио-, ди- и триметил-
трннитробензолов происходит не только вследствие уменьшения склон-
ности нитросоединения к авто ассоциации в расплаве и доли межмоле-
кулярной составляющей разложения, но и за счет уменьшения скорости
внутримолекулярного процесса, которое обусловлено особенностями
строения молекул этих соединений.
1,. А и д р е е в К, К. Термическое разложение и горение взрывчатых веществ, М.,
«Наука», 1966, с. 57,
2. Rogers R. Analyt. Chem., 1967, N 6, p. 730; Da con» J„ Adolph H., K»m-
• let ,4. J, Phys. Chem,, 1970, v. 74, p. 3035.
3. С а п p а н о в к ч В. Ф,, Максимов Ю. Я,, Маркелова М. Е. Труды МХТИ
! им. Д. И. Менделеева, 1973, вып. 75, с. 147.
,4 Robertson R. J, Chem. Soc., 1921, v. 119, p. 1.
5. Максимов Ю. Я. ЖФХ, 1968, т. 42, с. 3921.
*6. Лурье В. А., Светлов Б, С. Труды МХТИ им. Д, И.-Менделеева, 1968, вып.
58, с. 189.
.7. Максимов Ю. Я., С ороч к и и С. Б., Сапранович В. ф, Труды МХТИ
им. Д. И. Менделеева, 1974, вып. 83, с. 48.
8. DesvergnesL. Ann. Chim. anal, app 1., 1920, (2), 2, 278.
9. de !,a n'g e P. Rec, tray, chim., 1926, 45, 57.
10. Максимов Ю. Я. ЖФХ, 1972, т, 46, с. 1726.
Н. Urbanski Т., Buzniak J. Roczn. Chem., 1971, 45 1841.
12. F г a nek R., W i 11 i a m s о п S. J. org. Chem., 1966, v. 31, p. 2420.
УДК 541.1247:662.232,
О ВЛИЯНИИ НЕКОТОРЫХ ФАКТОРОВ
НА РАЗЛОЖЕНИЕ ПЕРХЛОРАТОВ
АММОНИЕВЫХ ОСНОВАНИИ
В. А. Коробан,, Г, И. Смирнова, Б. С, Светлов
Как известно, условия термического разложения, которые включа-
ют как основной фактор степень заполнения реакционного сосуда, ока
зывают часто существенно^ влияние на начальную скорость разложения
взрывчатых веществ [1, 2]. В этих случаях для определения кннетиче
ских параметров стараются выбирать такие условия, при которых ее
влияние незначительно, Однако для ряда веществ и, в Частности, для
перхлоратов аммониевых оснований, влияние степени заполнения реак-
ционного сосуда (m/v) связано с механизмом распада [3], и выяснение
влияния m/v на кинетические характеристики необходимо не только с
практической точки зрения, но и для получения количественных данных
по отдельным процессам, протекающим на первой стадии разложения.
Ранее была предпринята попытка по влиянию m/v при различных тем-
пературах оценить значения энергий активаций и энтальпий этих про
цессов для перхлоратов метилзамещенных ионов аммония. В данной
.работе на примере перхлоратов аммония и моиометиламмоння влияние
m/v изучено более подробно, что, кроме указанных параметров, позво-
лило рассчитать абсолютные значения некоторых констант,-
Другим важным фактором, который оказывает влияние на началь-
ную скорость разложения, является агрегатное состояние. Возрастание
скорости разложения при плавлении вещества, наблюдаемое в боль
шиястве случаев, представляется настолько естественным, что часто это
правило распространяют иа такие объекты, на которых по тем или иным
причинам влияние этого фактора не изучено и может быть прямо про-
тивоположным. Так, например, имеются данные по разложению перхло-
, рата аммония (ПХА) в жидкой эвтектике с перхлоратом лнтия, которые
показывают, что начальная скорость распада ПХА в этом случае ниже,
чем в кристаллах [4]. Нами исследовано влияние агрегатного состояния
для близкого аналога ПХА —перхлората монометиламмоиия (ПХМА),
температура плавления которого достаточно низка (253°С), чтобы нс
следовать его разложение и в расплаве, причем В отсутствии добавик
других легкоплавких солей,
61
Перхлорат монометил аммония получен обменным взаимодействием
соответствующего хлорида с 72%-ной хлорной кислотой. Продукт, очи-
щенный многократной перекристаллизацией из воды и трижды из эти-
лового спирта, плавился в интервале 253—254°. Опыты проводили с
фракцией кристаллов меньше 140 нм. Кристаллы ПХА размером 220—
315 нм имели правильную огранку и не содержали видимых дефектов,
что было достигнуто перекристаллизацией его из изопропилового спирта.
Распад ПХМА сопровождался плавлением образца, причем в кон-
це разложения вновь образовывался твердый продукт, который, как
показано ранее [3], состоит из перхлората и хлорида аммония с при-
месью солей органических аминов. Скорость разложения вещества при
220° возрастала с уменьшением размера частиц (рис. 1). Разложение
в интервале 210—270° сопровождалось
ускорением, причем максимальная ско-
рость газовыделеция при m/v=0,004 г(смя
как в твердом, так и жидком состоянии
вещес+ьа превышала начальную на по-
рядок (рис. 2). Для расчета кинетических
параметров на стадии ускорения исполь-
зовали значения констант, полученных по
наклону прямых в координатах lg v—т.
Значение энергии активации распада на
этой стадии ниже температуры плавле-
ния составило 36,5 ккал/моль. Наиболь-
£
168-
I
S6-
6
Рве. Г. Г
Вк гмаообраэоваине при распаде
перхлората монометилам моим я
«№. ЙО’С и m/V—0,004 г/см\
Размер частиЦ: 1 — 140 нм;
2 — 200 мл
/06 400 §08 §06
1, МЦЧ
Блеяние размеров частиц Ший интерес представляет изменение СКО-
ростн распада вблизи температуры плав-
ления. Поэтому разложение ПХМА в ин-
тервале 250—260° исследовано более под-
робно (рис! 2)< Ниже температуры плав-
ления начальная скорость газовыделе-
НИЯ менялась с энергией активации 40 ккал/ моль. При 254° она
резко возрастала, но затем снижалась н при 255° становилась прак-
тически равной скорости распада до точки плавления. В дальней-
шем увеличение скорости разложения соответствовало практически той
же энергии активации, что и в твердом состоянии (41 ккал/моль). В ре-
зультате разложение расплава протекало со скоростями, близкими к
тем, которые дает экстраполяция их зависимости от температуры, полу-
ченная ниже точки плавления (рис. 3).
Этот факт и литературные данные по разложению ПХА в эвтек-
тическом расплаве с перхлоратом лнтия [4] можно объяснить в рамках
механизма первичной стадии разложения, включающего диссоциацию
солей и распад хлорной кислоты гетерогенно на внешней или внутрен-
ней поверхности кристаллов. Согласно данным Леви [5], гетерогенный
распад хлорной кислоты протекает значительно быстрее и с меньшей
энергией активации, чем в парах. Имеются также результаты, показы-
вающие, что скорость разложения хлорной кислоты на поверхности
ПХА гораздо выше, чем ее паров и- водных растворов [6]. Поскольку в
расплаве соли не могут выполнять роль гетерогенных катализаторов,
скорость разложения НСЮ4, а следовательно, и лимитируемая ею ско-
рость распада перхлората должна быть ниже. Для аммониевых солей
других кислот, например HNO3, при распаде которых вклад гетероген-
ных реакций меньше, наблюдается «нормальное» рлцяиие агрегатного
состоямИБЯЗГято позволяет предполагать увеличение степени диссоциа-
..дии apB'ISfявлении. Взаимная компенсация влияний агрегатного состоя-
. кия на различные процессы. при распаде аммониевых солей хлорной
кислоты, очевидно, и приводит к наблюдаемым аномалиям.
Рис. 2. Гаэовыделение при распаде пер-
хлората моиометнламцмия при m/V—
—0,004 e/cx<s и температурах, “С; 1—
Z70-, 2-254; 8-259; 4-250; 5—240;
5—230; 7—220; 8-210“
Рис, 3. Температурная зависи-
мость начальной скорости га-
эовыделейия (erf/г мин) при
распаде перхлората мономе-
тнл аммония
Описанная схема начальной стадии распада аммониевых солен
хлорной кислоты, дает основание ожидать влияние степени заполнения
реакционного сосуда, на скорость процесса. Этот вопрос; обсуждался
ранее в работе [3J, в которой получено для зависимости скорости рас-
пада ОТ m/v следующее уравнение
- Ь V h. k” + k' 'I
,r=k4k--i£+id
где k —константа скорости распада
‘•к»,, и кга — константы растворимости
^константа равновесия диссоциация;
НС1О4 в
КИСЛОТЫ
конденсированной фазе;
и амина в соли; кд —
(т—навеска вещества;
vM
mRT
к =
► — объем реакционного сосуда; М — молекулярный вес вещества). Из
равнения видно, что в случае большей растворимости в конденсирован-
ой фазе хлорной кислоты (кгк^>кга) при возрастании степени запол-
онил реакционного сосуда скорость должйа .снижаться, что и наблю-
дается в опытах с ПХМА. при различных m/v (рис. 4). Для определения
Качений krk кгЯ и kk^kнеобходимы по крайней мере 3 уравнения,
вторые можно составить по данным при разных значениях m/v. Кроме
бго, величину kk„k/I1 при высоких температурах и малых m/v можно
рлучить непосредственно, так как выражение в скобках в этом случае
тремится к 1. Из полученных результатов (рис. 5,6) выведены следую-
щие кинетические константы
'r’kkrtka* * l(A7.e-3SWK4RT сек'1, krr = 10-Н;2 •eMMP/RT моль’моль-мм
растворимость метиламина в соли по расчету в исследованном интер-
вале температурка 2 порядка ниже, чем кдбрной кислоты, поэтому
«3
точность определения была мала и не позволила надежно определить
температурную зависимость этой величины. На основании полученных
данных можно рассчитать значение энергии активации разложения хлор-
ной кислоты в конденсированной фазе, если известна теплота диссоциа-
ции соли на газообразные амин и хлорную кислоту. При теплоте дис-
социации, близкой 60 ккал/моль [3], энергия активации разложения хлор-
60
ной кислоты должна составлять около 32------+ 26 = 28 ккал/моль-
Значение констант скорости распада хлорной кислоты из полученных
данных рассчитать нельзя, так как отсутствуют надежные данные для
константы диссоциации этой соли. Гораздо больше может дать анало-
гичное исследование распада перхлората аммония.
Рис. 4. Влияние m/V на гаэо-
выделение при распаде пер-
хлората монометиламмоиия
при 210* С. Величины m/V,
г/сл"; /—0,004; 2-0,03; 3-0,1
Рис. 5. Температурные зависимости на-
чальных скоростей гаэовы деления пер-
хлората аммония (а) и перхлората
моиометиламмоиня (б) при различных
m/v. Величины m/v, г/см3: /—01, 2 —
0,03 (а), 0,04 (б); 5—0,004; 4 m/v->0
Характер газовыделення на начальных этапах разложения ПХА
отличался от того, что наблюдалось при распаде ПХМА (рис. 6), по-
этому при определении начальной скорости разложения пришлось
учесть некоторые обстоятельства, на которых мы остановимся ниже.
Г, мин
Рис. 6. Г а зовы деление в начале
распада перхлората аммония (/)
и перхлората монометнламиония
(3) прн 220* и m/v—0,1 г/см*
При выводе зависимости скорости разло-
жения аммониевых солей от степени за-
полнения реакционного сосуда предпола-
галось, что равновесие между твердым
веществом и газовой фазой устанавлива-
ется достаточно быстро и поэтому не вно-
сит существенного вклада в кинетику га-
зовыделения. Однако, если превращение
вещества определяется распадом НС1О<
на внутренних дефектах кристаллов, вре-
мя достижения этого равновесия будет
зависеть от диффузии продуктов диссоци-
ации по твердому веществу и может быть
довольно продолжительным. Особенно
ярко это проявляется при разложении
перхлората аммония. Так, газовыделение
>44
течение определенного промежутка времени чрезвычайно мало
связано, главным образом, с удалением нз кристаллов примесей
Ютворителя. Затем скорость разложения резко возрастает, и да-
е распад протекает согласно закону объемного роста реакционных
нтров. «Индукционный период» разложения зависит от температуры,
-ичем значение энергии активации в этом случае Составляет
ккал/моль. Для оценки влияния степени заполнения реакционного
суда скорость определяли сразу после стадии «индукционного перио-
й» в интервале степеней разложения 1—2 смг/г (0,2—0,3%). На осво-
ении зависимостей скоростей распада на этом участке от температуры
ля разных m/v (рис. 5,а) получено следующее значение растворимости
нслоты в перхлорате аммония
кгК = 10 ”14’9 • e-rwWRT моль [моль мм
'астворимость аммиака при этом на 1—2 порядка ниже. Найденное
о этим же результатам значение скорости при m/v-+0 составляет
кш ^о = к-кгккд'4 5 ~ Ю7-7-е 300W,'RT сек'1
то соответствует начальной скорости распада ПХА в условиях удале-
ия продуктов [8]. Из данных для km о, кгГ и найденной в работе [9]
онстанты диссоциации можно определить скорость распада хлорной
ислоты в конденсированной фазе
к __ ]07,7 -10,5+14,9 . е -(30000 -29000 +26000)/RT _ ] Q12 . е - 27000/RTCgK “ 1
е значение всего на порядок превышает то, которое получено при ис-
педовании распада НС1О4 в адсорбированном на кристаллах ПХА со-
гоя ни и [6], что вполне можно объяснить зависимостью константы от
оицентрации кислоты *. Энергия активации распада хлорной кислоты в
арах и ее водных растворах гораздо выше и составляет 45 ккал/моль-
начительное снижение энергии активации распада НСЮ4 в перхлорате
ммония может быть обусловлено, как уже отмечалось выше, гетеро-
нностью процесса.
Сравнение распада перхлоратов метиламмония и аммония дает
изкие значения скоростей разложения, а также аналогичный харак-
э -влияния степени заполнения реакционного сосуда. Поэтому с пол-
IM основанием можно заключить, что механизмы начальной стадии
зложения этих солей одинаков. Отличие в характере начальных участ-
в кинетических кривых, которое можно объяснить разными зонами
зникновения реакционных ядер, связано, очевидно, с различной реак-
юнной способностью катионов, Так как ион аммония окисляется
уднее, то для развития процесса разложения перхлората аммония
обходима высокая концентрация окислов хлора, которая достигается
шь при разложении НС1О4 в дефектах кристаллической решетки, т. е.
том случае, если выход окислов в газовую фазу затруднен. Ион метил-
4моний окисляется продуктами распада хлорной кислоты быстро. В ре-
ультате процесс
окисления способен конкурировать с реакциями
окис-
юв хлора в газовой фазе, и распад начинается с поверхности, где кон-
центрация НС1О4 максимальна,
* Концентрация НС1О4 в этих опытах была выше приблизительно на 5 порядков,
5 Труды, ВЫП. 83 65
। ;
L С И e т Я о в Б. С. Теория взрывчатых веществ. Сб. статей, М., «Оборонгиз», 1963,
с. 184.
2. Максимов Ю. Я. Труды МХТИ нм. Д. И. Менделеева, 1968, вып. 53, с. 73, 86.
3. КоробанВ. А., Чугункин В. М., Л о б о д а В. И. Материалы 3-го Всесоюзного
симпозиума по горению и взрыву, М., «Иаука», 1972, с. 775.
4 So Ту т о si F., R й nfcs М. Comb. and. Flame., 1966, v. 10 p. 398.
5. Levy J. B. J. Phys. Chem., 1962, v. 86, p. 1092.
6. Коробан В. А., Светлов Б. С., Чугункин В. М. Материалы 3-го Всесоюз-
ного симпозиума по горению и взрыву, М., <Иаука», 1972, с. 741.
7 Вес,налов Г. Н., Шидловский А, А., ЖФХ, 1968, т. 47, с. 2623.
8. Маиеллс Г. Б., Рубцов Ю. И. ЖФХ, 1968, т. 12, с. 770.
9. Inam 1 S. И., Rosser W. A, Wise Н. J. Phys, Chem., 1968, v. 67, р. 1077.
РАЗДЕЛ ВТОРОЙ
УДК 662.2
ВОСПЛАМЕНЕНИЕ И ГАЗИФИКАЦИЯ БАЛЛИСТИТНОГО
ПОРОХА ПОД ДЕЙСТВИЕМ ИЗЛУЧЕНИЯ СОгЛАЗЕРА
Б, Н, Кендриков, Т. Дж, Олемиллер, М, Самерфилд
Приближенная теория зажигания [1-8] позволяет предсказать усло-
вия, необходимые для возникновения горения конденсированных ве-
ществ при действии источника тепла достаточной интенсивности. Неко-
торые из этих предсказаний проверены на опыте. Так, в работах [2, 4]
установлено, что рассчитанные теоретически зависимости задержки и
количества тепла, необходимого для воспламенения пироксилина, полн-
винилиитрата и баллиститиого пороха Н, от потока тепла удовлетво-
рительно согласуются с опытом. Аналогичные результаты получены в
ряде других работ.
Вместе с тем,’ некоторые выводы теории до спх пор не подвергались
экспериментальной проверке, Одним из иих является предсказанный
' Зельдовичем и Либровичем [5, 6] рост задержки воспламенения при уве-
личении потока тепла сверх некоторого критического значения. Другие
допросы, например принудительная газификация под действием лучис-
той энергии, хотя и затрагивались (9, 10], ио далеко йе исчерпаны.
В связи с этим представляло интерес изучить воздействие источника
учистой энергии значительной мощности на баллиститный порох, явля-
ющийся сложным, но достаточно распространеиным объектом такого
юда исследований [3, 4, 11]*.
В качестве источника излучения использовался 100-ваттный угле-
дслотиый лазер, дающий поток до 90 кал/см2 • век. с длиной волны
0,6 мкм. Это излучение соответствует энергии кванта света 2-10-’3 эрг
т. е. ~ 3 ккал/моль), что на порядок величины меньше, чем энергия
тобой химической связи в молекулах соединений, входящих в состав
юроха. Очевидно, фотохимическими эффектами можно пренебречь. Мо-
нохроматический характер излучения позволяет надежно определить
[Коэффициент поглощения, необходимый для последующих расчетов и
Трудноопределимый в случае обычных дуговых нагревателей.
* Работа выполнена в декабре 1969—январе 1970 гг. во время пребывания Б. И.
Крндрикова в научной командировке в Принстонском университете (США).
5*
67
Рве. 1. Схема установки: I—ла-
Ир; 2- асбестовый затвор; 3—
Металлические быстродействующие
затворы; 4 — осциллирующее зер-
кВЛьЦе; 5 — «калейдоскоп»-, 6 —
бомба постоянного давления; 7—
абраачик пороха (а—диаметро.’.!
2,5 мм; б—диаметром 6,5 жж)
Методика проведения опытов
Схема установки представлена на рис. 1 [8]. 100-ваттный СОз-лазер
производства Coherent Radiation Laboratories дает луч диаметром около
1,5 см, проходящий через три последовательно установленных затвора.
Первый из них — металлическая рамка, покрытая слоем асбеста, под-
нимается соленоидом и опускается под действием собственного веса
после выключения проходящего через соленоид тока с помощью пнев-
матического реле. Время подъема или опускания рамки около 10 мсек,
точность пневматического реле примерно такая же. Этот затвор приме-
нялся самостоятельно, без двух последующих, только при большом
(0,3-—0,5 сек и более) времени облучения. Два следующих затвора —
металлические лепесткового типа —
со временем срабатывания около
1 мсек. Один из них открывает путь
лучу, второй (первоначально откры-
тый) прерывает его. С помощью этих
затворов задавалось время облучения
до «к 10 мсек. Затем луч проходил гер-
маниевую линзу, осциллирующее зер-
кало с частотой колебаний 4 кгц и стек-
лянную трубку квадратного сечения со
стенками, покрытыми тонким слоем зо-
лота — калейдоскоп. Эта система по-
зволяла уменьшить существенный не-
достаток лазера—неоднородность плот-
ности потока излучения по диаметру
луча.
Через две длиннофокусные гер-
маниевые линзы и толстое (1,3 см)
германиевое окно, защищенное изнутри
От действия горячих газов пластинкой из NaCl, луч проходил в бомбу
постоянного давления, заполнявшуюся азотом, и попадал на образец
пороха, находящийся в оправке, укрепленной на крышке бомбы (рис. 1).
Размер пятна 3x3 (мм). Разброс мгновенных значений потока в дан-
ной точке (при характерном времени воздействия ^0,25 мсек) около
20%. Колебания величины среднего потока не более 2—3%. Поверхность
пороха после газификации и затухания слегка волнистая.
Интенсивность излучения измеряли охлаждаемым проточной водой
калориметром с пленочным термометром сопротивления, помещавшимся
в бомбу вместо крышки. При перемещении калориметра на 3—-5 мм по
вертикали величина потока не изменялась. Продолжительность облуче-
ния определяли с помощью инфракрасного детектора (германиевый эле-
мент, охлаждавшийся жидким азотом, на схеме не показан), улавли-
вавшего небольшую часть энергии луча, отраженной от зеркала 4. Вре-
мя фиксировалось катодным осциллографом.
В качестве объекта изучения был выбран баллиститный порох N-5,
содержащий 35% нитроглицерина, 10,5% диэтилфталата, 2% нитроди-
фенила, 2,4% органических солей свинца. Для опытов при атмосферном
давлении из этого пороха на токарном станке вырезали таблетки диа-
метром 6,5 и высотой 2—4 мм, прн повышеннбм давлении применяли
в основном цилиндрики диаметром 2,5 и длинбй около 7 л.«. Послед-
ние иногда бронировались по боковой поверхности липкой полиэтиле-
68
Л ' ,.ьЧ . .......
новой лентой (один слой). Поверхность образчиков в большинстве слу-
чаев свежесрезанная (лезвием безопасной бритвы) *.
Коэффициент ослабления потока а определяли с помощью инфра-
красного спектрофотометра в тонких пленках, срезанных на микротоме.
Три последовательных опыта дали коэффициенты а 595; 620 и 655 см~1.
Среднее — 623 см~1. Отражением от поверхности при расчетах пренеб-
регали.
Воспламенение пороха прн 21 ат
Результаты опытов, проведенных при 21 аг (300 фунтов[дюйл&),
даны на рис. 2. При малых потоках облучение не оказывает никакого
действия на поверхность пороха, либо лишь слегка подплавляет его; при
том же времени облучения и больших потоках порох воспламеняется и
сгорает с хорошо видимым факелом желто-
го пламени. В логарифмических координа-
тах кривая t j (q) имеет тенденцию увеличи-
вать наклон к оси абсцисс по мере возра-
стания потока. Если этой довольно слабо
выраженной тенденцией пренебречь, зави-
симость Igti (Igq) может быть выражена
прямой с тангенсом угла наклона — 1,7.
Справа от этой прямой на рис. 2 — область
воспламенений, слева — область отказов.
Для сравнения через середину прямой
проведена пунктирная линия с наклоном
— 2,0, которая выражает зависимость перио-
да индукции от потока для непрозрачного
пороха с постоянной температурой поверхно-
сти. Уменьшение наклона может быть свя-
зано с некоторой прозрачностью пороха (при
больших потоках для полупрозрачного по-
Рис. 2. Зависимость задержки
зажигания от потока излуче-
ния при 21 аг; 1—воспламене-
ние; 2—нет воспламенения
роха время достижения данной температуры на поверхности, естественно,
больше, чем для непрозрачного).
Другая возможность заключается в том, что температура поверх-
ности увеличивается при возрастании потока. Соответственно, при боль-
ших потоках для прогрева пороха требуется больше времени, чем при
постоянной температуре поверхности, наклон кривой уменьшается.
Таким образом, существуют два фактора— проникновение излуче-
ния 'вглубь вещества и развитие реакции в нем, которые должны быть
включены одновременно в модель, объясняющую поведение пороха на
рис. 2. К сожалению, решение задачи в общем виде слишком громоздко,
[ чтобы использовать его для рассмотрения экспериментальных данных,
Г поэтому ограничимся приближенным расчетом.
г Оценим температуру поверхности по приближенной формуле, пред-
ь ложенной в [8] (полупрозрачное вещество нагревается потоком излуче-
L ния q = const):
РС(Т3-ТО) ]•
q
* Поверхность, больших образчиков наклонена, чтобы продукты горения не попа-
дали на германиевое {децп. Это не очень помогало, пластинка из NaCl более эффективна.
69
Для пороха т4-5 имеем с = 1,48 с = 6,45 кал[г- ZfkiS; н =*
5» 0,75-10’3 см2!сек\ а = 623 см-1. Средняя для нескольких q и tj вели-
чина Т, = 225°С, Воспользовавшись этой величиной можно оценить зна-
чение энергии активации ведущей реакции в конденсированной фазе
Е = RTsln(BQx/q),
где В — предэкспонент; Q — объемная теплота реакции; х — ширина
зоны реакции (с учетом проникновения излучения вглубь пороха). При
нормальном для сложных органических веществ значении В~ 1016 сек-1
получаем для пороха N-5 энергию активации в пределах 30—
35 кКал/моль. Это — обычная для нитрозфиров и порохов на их основе
[9] величина.
Аномалии воспламенения
В интервале 3—5 ат были получены несколько необычные зависи-
мости задержки зажигания от интенсивности потока тепла. Они пока-
заны в координатах поток тепла — давление (при постоянном временя
облучения) на рис. 3 и в координатах поток тепла — время (при по-
стоянном давлении) на рис. 4. В обоих случаях хорошо видно сущест
вование некоторого значения потока, выше которого зажигание не облег-
чается при увеличении интенсивности облучения, как этого следовало
бы ожидать, исходя из обычных представлений о воспламенении, а
Затрудняется: для зажигания требуется более высокое давление или бо-
ЛбЗ; продолжительное время. Критический поток, при котором наблю-
увеличивается при возрастании дав-
дается минимум на кривой tj (q),
лення (рис. 5).
Рис. 3. Поведение пороха при
раелнчных давлениях и потоке.
Врейя облучения 0,117 сек.
/—воспламенение; 2—никакого
действия; 3—газификация
Рис. 4. Зависимость задержки
зажигания от потока нелуче-
иин при 3,9 ат: /—воспламе-
нение; 2 — газификация; 3—
t; (ч) "ри 21 ат (рнс. 2)
Таким образом, при повышенном давлении кривые tj (q) делят плос-
кость в координатах поток — время на три зоны (рис. 4). В одной из
них — слева от прямой с п = 1,7 поток тепла лишь прогревает (и иног-
да слегка оплавляет) порох. Во второй — справа и выше кривой с
минимумом происходит воспламенение. Наконец, в третьей зоне — под
кривой и над прямой, происходит послойная газификация пороха, за-
канчивающаяся пойте прекращения облучения. Закономерности, управ-
ляющие газификацией, будут рассмотрены в следующем разделе.
'"70
। с*
ш которые
мучении.
обметить, что во всех опытах при невысоком давлении,
(сходило воспламенение, на поверхности пороха при об-
далось образование слоя толщиной обычно менее мил-
Бшметра серовато-черных частиц, иногда с красноватым стечением.
Шопытах с газификацией без воспламенения слоя частиц на поверхно-
Ееги не было — он сдувался потоком газа, образующегося в результате
(газификация. В тех опытах при высокой интенсивности излучения, в
которых порох воспламенялся, всегда хотя бы часть поверхности была
ыркрыта слоем углеродистых частиц.
Г В табл. 1 приводятся значения потоков (qm), при которых иаблю-
шается минимум на кривых t, (q) при разном давлении (рис. 5) и тол-
идина прогретого слоя при стационарном горении пороха (бг = k/uq) и
[при прогреве потоком qm за время t| .
за время 11
!’ = т+тг(т--т«>
[При расчете принималось X —5-10~4 кал/см-сек-град; Tg =225°;
То = 20°.
Воспламенение пороха N-5 излучением СОг-лазера
при повышенном давлении
Таблица 1
Давление, Р, ат Поток, калием? сек цй, см/сек Толщина прогретого слоя, мем
Чш Ч* »ч
3.4 14 7 0,091 82 81
3.9 17 12 0.112 67 69
4.5 23 14 0,140 54 55
4,9 30 16 0,165 46 46
21 >35 — 0.47 1« 16<»q<42
Примечание, q^—поток, при котором на кривых ti(q) наблюдается мини-
мум: Ч* — поток иелучення, при котором период газнфикации равен нулю (см. рис. 6);
,Ио — скорость горе кия пороха при данном давлении; до —толщина прогретого слоя при
'Стационарном горении со скоростью uo; dq—толщина прогретого скоя при прогреве
; пороха потоком излучения
| Имеется определенное соответствие между толщиной прогретого
[слоя при стационарном горении пороха и толщиной слоя, прогретого
[излучением лазера в той точке, в которой зависимость задержки зажи-
мания от потока проходит через минимум.
е При малом потоке, создающем толстый прогретый слой, воспламе-
Ьнение при умеренном давлении не отличается от происходящего при
(высоком давлении. При увеличении потока и соответствующем умень-
шении толщины прогретого слоя появляются новые явления, связанные
к неустойчивостью: перед воспламенением происходит вызванная нзлу-
Еяенцем газификация пороха, Без газификации порох не воспламеняется,
ГКрнвая, отделяющая область зажигания от области отказов, удаляется
вот прямой, полученной при высоком давлении. Когда толщина слоя,
ьррогретого до температуры Тв становится меньше,, чем необходимо для
F,стационарного горения, кривая начинает перемещаться вверх: при даль-
71
нейшем увеличении потока задержка зажигания не уменьшается, а воз-
растает. Положение минимума зависит от давления: чем выше давление,
тём больше скорость горения, тоньше прогретый слой б, тем труднее
возникает неустойчивость.
При высоком давлении прогретый слой становится тоньше, чем
j/а — слой, прогреваемый за счет проникновения света вглубь пороха.
Вероятно, это должно препятствовать проявлению неустойчивости.
Интересно найти точку (значение потока), в которой кривая, полу-
ченная при умеренном давлении, начинает удаляться от прямой, полу-
ченной при высоком давлении. Для этого построим зависимость времени
газификации перед воспламенением от величины потока излучения
(рйс. 6). В полулогарифмическом масштабе получаются прямые
In р = q* + «0*1
отрезающие на оси искомые отрезки. Эти значения предельных потоков
(q«) также приведены в табл. 1. Они в 1,5—2 раза меньше, чем qro.
'Величина
а0 = dlnq/dl
в выбранном узком интервале давлений примерно пропорциональна дав-
лению и меняется от 12 до 17 сек~'.
Рис. 5. Зависимость за-
держки зажигания от пото-
ка излучения при давлении,
(ат): /—3,4; 2—3,9; 3—4,5;
¥—4,9; 5-21
Ряс. 6. Связь между
временем газификации,
необходимым для вос-
пламенения, и потоком
излучения при умеренно-
ювышеииом давлении,
ат: 7—3,4; 2—3,9; 5-4.5;
4—4,9 (пунктир—точки
несколько отклоняются
от прямой)
Газификация пороха
При атмосферном давлении воспламенение пороха под действием
лазерного излучения крайне затруднено. При времени воздействия мень-
ше 3 сек наблюдалась лишь газификация, прекращавшаяся сразу же
после прекращения облучения. Этот процесс представляет самостоятель-
ный интерес.
Толщина газифицировавшегося слоя определялась с помощью глу-
биномера или микрометра с точностью 0,03 мм. Основная харёктери-
СтИка процесса — скорость газификации рассчитывалась с учетом вре-
мени прогрева, определявшегося главным образом по времени появ-
ления первых следов разложения на поверхности облучаемых образчи-
ков. Это время немного больше времени воспламенения при 21 аг. Ви-
димо, часть тепла расходуется иа испарение летучих компонентов, и
разложение начинается .позже.
На рис. 7 приведены результаты опытов при атмосферном давле-
нии и времени облучения (в основном) 0,57 сек. Опыты проводились
с образчиками диаметром 6,5 и 2,5 мм. Разницы между ними не наблю-
дается. При малых интенсивностях потока приведены точки, полученные
при времени облучения 2—3 сек. Они несколько менее надежны.
Рис. 7. Скорость газификации пороха
при атмосферном давлении. Время
облучения 0,57 сек: /—диаметр об-
разчика 6,5 juju; 2—2,5 мм (боковая
поверхность бронирована липкой лен-
той), 5—время облучения 2—3 сек
(диаметр образчика 65 лл<)
Зависимость средней скорости газификации от интенсивности пото-
ка тепла имеет вид кривой, которая может быть аппроксимирована дву-
мя отрезками прямой с уменьшающимся при увеличении потока углом
наклона к оси абсцисс.
По мере уменьшения времени газификации средняя скорость ее воз-
растает (рис. 8). Зависимость скорости от времени облучения при раз-
ной интенсивности потока показана на рис. 9. Абсолютная величина
производной dr/dt отчетливо уменьшается при уменьшении интенсивно-
сти облучения.
Рис. 8. Скорость газификации по-
роха при 1—2 ат. Время облуче-
ния, сек: /—0,9 (2 аг); 2—0,57;
3—0,33; 4—0,18; 5—0 (экстрапо-
ляция). Прямая / менее надежна,
чем остальные
Рис. 9. Зависимость скорости гази-
фикации при 1—2 ат от времени
облучении. Поток тепла, кал/см^-сек:
/—80; 2—60; 3-40; 4-20; 5-0
(экстраполяция)
Вероятная причина уменьшения скорости газификации по мере воз-
растания времени облучения заключается в ослаблении потока продук-
тами разложения. Очевидно, значение скорости газификации, экстрапо-
73
Лфобаййбс на время о^лученйя равное нулю, даст истйнНу||£виЙ^6ёТь
прн данном потоке (правильнее было бы использовать нулевое время
газификации, а не облучения, ио в данном случае эта разница на резуль-
тате совершенно не сказывается).
Зависимость скорости газификации от интенсивности потока для
разных времен облучения приведена на рис. 8. Начальный участок
этбй зависимости получен по данным для времени облучения 0,57 и 2—
3 сек (рис. 7).
Уравнения прямых
ш — rQ = a+bq,
(1)
написанные для массовой скорости
прн q < 15 кал/см2 сек
при q > 15 кал/см3 • сек
газификации (г/см2 • сек):
m = 0,11 + 15- 10"3q
m = 0,256 + 5,32- 10“3q
Уместно сравнить этот результат с обычным уравнением баланса
энергии на поверхности конденсированного вещества
rpcp(Ts-То) = q ф qg + r?Qs
где q — поток лучистой энергии извне; qg — поток тепла из газовой фавн;
Qe —теплота реакции в конденсированной фазе.
Отсюда массовая скорость газификации
cp(T»-T0)-Q, ] ‘q
вжи >
21,1 • ’ я qg h 1
а” cp(Ts — Т.) — Qs ’ CP(TS-TU)~QS
Легко нейти отсюда основные характеристики реакций, протекаю-
щих в газовой и конденсированной фазах
Че — |, ’ Qs — ср (Т. То) —
о b
Для значений а и Ь, полученных экспериментально, и Т6—То = consl =
= 220° при q < 15 кал/см2 • сек; qg = 7,3 кал/см2 - сек; Qs = 32 кал/г;
при q > 15 кал/см2 • сек; Ts= 280°. qg= 48 кал/см2 • сек; Q, = —66 кал/г.
Основной результат заключается в том, что, начиная с некоторого
не слишком большого потока энергии извне, обычная экзотермическая
реакция в конденсированной фазе сменяется эндотермической. По-види-
мому, продукты газификации компонентов пороха не успевают суще-
ственно прореагировать в конденсированной фазе, и реакция проходит
над ее поверхностью. Поток тепла из газовой фазы qg) соответственно
возрастает (хотя суммарное тепловыделение уменьшается).
Обсуждение результатов
При определенных условиях увеличение потока излучения может
приводить к росту задержки зажигания: поток тепла увеличивается —
условия воспламенения ухудшаются. Близость толщины зоны прогревз
в точке минимума кривой t( (q) к толщине прогретого слоя при стацио-
: 74
и ар Пороха (тайл, i) позволяет прйнЛёчь для в4ьяснёЯЙ Я
этого-теорию устойчивости горения Зельдовича [5]. В работе
[5] поюзано, что существует предельный поток тепла (qz) и отвечающая
ему предельная толщина прогретого слоя (б'}, при которых устойчивое
горение пороха невозможно. При б < б' градиент температуры у по-
верхности слишком велик, температура поверхности падает, реакция
прекращается. При потоке тепла от воспламенителя q > q' воспламене-
ние не происходит.
Согласно Либровичу [6] воспламенение возможно и при q > q', если
возникшая под действием потока тепла газификация пороха может
ослабить поток и довести его до q = qz.
Коэффициент Ослабления потока можно получить из рис. 6. При
данном давлении
d lnq/dt g = const *
В соответствии с законом Бугера ослабление потока экспоненциально
связано с поглощающей излучение массой вещества. Отношение плот-
ности потока, падающего на поверхность пороха, к плотности потока,
проходящего через окно камеры
q/q0 = e~a’1 (2)
где «| — коэффициент ослабления потока продуктами разложения по-
роха; х — толщина слоя продуктов. Если предположить, что толщина
слоя продуктов пропорциональна скорости газификации
dx = rdt = (а + bq)dt
то из выражения (2) получим
t
lnq0 = Inq + ai№ In q + си £ (a + bq)dt
или
“ q(a + bq) "*>dt при: t = 0, Ч = <Ы t = t„, q-q.
Интегрируя, находим
Пoлoживq = q*, t = tg, рассчитаем величину .< <
din q0
dlg = «t(a + bq0) = a, r0
; Это совпадает с результатом, полученным на рис. 6, поскольку скорости
газификации (г0) прн больших потоках слабо зависит от интенсивно-
сти излучения (рис. 8), а при повышении давления эта зависимость
’ имеет тенденцию уменьшаться (рис. 10).
Приняв г0 — 0,3 см/сек, dinq0/dt ~ 15 сект1, получим ctt = 50 см'1
^Коэффициент ослабления потока слоем частиц, накапливающихся на
поверхности горения, приблизительно в 12 раз меньше, чем исходного
порюха (очевидно, главным образом, в результате уменьшения плот-
ности вещества).
75
. При' атмосферном давлении поглощение гораздо слабее, вероятно,
вследствие того, что слой частиц уносится с поверхности потоком газа.
Величину dlnq0/dt можно получить, разделив величину dr/dt при раз-
ных q, полученную на рнс. 9, на Ъ/q = dr/dq (из рис. 8) и на q. Полу-
ченная величина не зависит от q и в среднем равна 0,73 сек'1. При
г = 0,3 см[сек это дает си = 2,5 с.и !.
Рис. 10. Скорость газификации при
умеренно повышенном давлении
(по опытам без воспламенения),
аг.- /-3,9; 2-4.5; Л-4,9; 4—
нуль линия при 1—2 аг (см.
рнс. 7). Время облучения 0,05—
0,12 сек.
Вероятная причина столь сильного уменьшения а, — унос слоя
частиц с поверхности потоком газа, образующимся при разложении
пороха. Согласно [12], сила, приводящая к уносу частиц с поверхности
Ьорящ^го вещества, пропорциональна массовой скорости газообразо-
вания н обратно пропорциональна давлению
F ~ 21
Р
Для того, чтобы сорвать частицы с поверхности, сила F должна достичь
критического значения F, = const, следовательно, в критических усло-
виях, при q — q*
in а + bq*
— =---------“ const
P P
ИЛИ
qt = Ap-B (3)
Если в координатах p —q$ провести прямую через критические зна-
чения потока/ отвечающие началу газификации, предшествующей вос-
пламенению, она имеет вид (3)
I q* — 5,3р — 10
Величина В = 10 кал}см2 • сек действительно близка к значению q2,
полученному из опытов по стационарной газификации при 1—2 ат
(7,3 кал)см1 - сек).
Величина q становится равной нулю при 2 ат. Это значит, что при
Р < 2 ат зажигание пороха N-5 невозможно ни при каком потоке.
Действительно получить воспламенение этого пороха облучением
при 1—2 ат не удалось. Порох М-9, дающий ввиду лучшего кислород-
ного баланса н меньшего содержания неорганических веществ менее
обильный конденсированный остаток, не загорается даже при 6,5 ат.
Л7б
Устойчивость его горения так мала, что и при 21 ат интенсивное облу-
чение приводило к затуханию.
Авторы [9], изучая горение пороха Н под действием излучения рас-
каленной графитовой пластины, при потоках до ~ 12 кал/см2 • сек, нашли,
что зависимость скорости газификации от интенсивности излучения име-
ет вид (1)
m ~ 0,095 + 21 • Ю"3 q, г/см2 • сек
Этот результат близок к полученному в данной работе для началь-
ного участка кривой r(q) *.
Характеристики химической реакции в конденсированной и газовой
фазах для обоих порохов сравниваются в табл. 2 **.
Несколько большее тепловыделение в газовой фазе для пороха N-5
возможно связано с наличием в его составе соединений свинца.
Следует отметить, что термопарные измерения дают яное распреде-
ление тепловых эффектов (и потоков) между зонами. Согласно Зенину
и Нефедовой [13] при комнатной температуре и атмосферном давлении
в конденсированной фазе выделяется 65, в газовой — 8 кал/г (рассмат-^
ривается только та часть тепла, которая возвращается к к-фазу). Сум-
марное тепловыделение примерно такое же как по опытам с облучением,
но распределяется оно иначе: в конденсированной фазе выделяется го-
раздо больше тепла, чем в газовой. Какой из двух методов дает более
правильные результаты, пока не ясно. Можно предположить, например,
что часть тепла qg/Шо генерируется вблизи от поверхности (например,
в слое частиц, волокон и нитей, получающихся при разложении и обуг-
ливании нитроклетчатки), а остальное получается из более удаленных
слоев газовой фазы, что фиксируется как тепло газофазных реакций
помощью термопарной методики.
Та бл и и 1 2 ТаблицаЗ
Сравнение характеристик Газификация перхлората аммония
газификации и горения порохов (с добавлением хромита меди)
Н, [9] и N-5 снеговым излучением [10]
Характеристики н N 5 Содержание хромита мелн, % 0 0,5 3
а, г^см-еек 0 1 95 0,11 a, г/см"1-сек 0,0266 0.0532 0,08’5
b-103, г1кал qg, кал/ем*-сек 21 4,5 15 7.3 b-IO3, кал]г коэффициент .1,37 5,32 1.67
Чг/П1(,*, кал/г Qs. кал!г 47 66 отражения 0,63 0,3 1 0,21
33 32 • кал'ем? сек qg/a, кал/г 7,2 271 6,8 128 4 0 47 •
* 1Лс=а-массоаая скорость без облучения. горения Qsr кал: г -90 4-60 +130
Аналогичным образом можно рассмотреть также данные [10] по
газификации перхлората аммония под действием .излучения электроду-
говой установки. Результаты расчетов с использованием коэффициентов
отражения, приведенных в работе [10], даны в табл. 3.
* В работе [91 членам уравнения приписывается иное значение: член, соответству-
ющий qg принимается равным скорости выделения тепла н к-фазе; Qs считается теп-
лотой реакции, протекающей в зоне подогрева к.фазы.
** Скорость горения пороха N-5 при повышении давления до 5 ат увеличивается
в 2,5 раза. Скорость газификации для больших потоков при этом почти не меняется
(рис. 10).
77
Интересно отметить, что введение хромита меди не только увеличИ
вает скорость газификации, ио и сильно влияет на характер реакций в
газовой и конденсированной фазах. При этом тепловыделение в газо-
вой фазе (qg/a, где а ?= т0) уменьшается, меняясь примерно обратно
пропорционально скорости газификации (поток тепла из газовой фазы
меняется мало, а скорость сильно растет).
В то же время количество тепла, выделившегося в конденсированной
фазе, при введении катализатора увеличивается. Можно думать, что
катализатор при разложении перхлората аммония, индуцированном по-
током излучения, а возможно и при горении его, действует в кондеиси-
рОЙДЙНОЙ фазе, ________swb. •_ ГЯЛгхм.._-
7 * Воспламенен не пороха М-9 7 ,
Дыли проведены также опыты с порохом М-9 (58% нитроглицерина,
1% этилнитрата, 1,5% KNO3), кислородный баланс и теплота взрыва
которого выше, чем N-5. Оказалось, что этот порох воспламеняется при
облученцц гораздо труднее. При 6,5 аг зажечь его не удалось, при 20—
Й лг горение возникало, но лишь в узкой области значений времен*:
облучедня и потока. В координатах lg q— Igt условиям воспламенения
отвечает полоса шириной 10—20 кал/см2 • сек. Слева от нее область про-
грева, справа — область газификации. Наклон левой (малые потоки)
таниды области воспламенения к оси абсцисс соответствует п = 2. Пра-
вдуграница, отвечающая большим потокам, несколько менее опреде-
ленна. Можно полагать, что причиной затухания при больших потоках
найдется неустойчивость горения в результате уменьшения толщины про-
гретого слоя при ускоренной газификации. Более отчетливый, чем для
N-5, характер явления, вероятно, связан с меньшим количеством остатка
на поверхности пороха М-9 и меньшей устойчивостью горения,
Г .1
.1. Зельдович Я. Б. ЖЭТФ, 1939, т. 9, № 12; Докл. АН СССР, 1963, т. 150, № 2.
2. А в ер с о и А. Э„ Барзыкнн В. В., Мержанов А. Г. Докл, АН СССР, 1968,
т. 178, № 1; ФГВ, 1968, т. 4, № 1; Роз е н б а и д В. И., Барзыкнн В. В., Мер-
жанов А. Г. ФГВ, 1968, т. 4, № 2.
3. Price Е. W., Bradley Н. Н., Hightower J. Т., Fleming R. D. А1АА
Preprint 1964, № 64 -120; Price Е. W., Bradley H. H., Deh or it у G. L.,
Ibirlcu M. M,, AIAA Journal, 1966, v, 4, p. 7,
4г Ковальский А. А., Хлевной С. С., Михеев В. Ф. ФГВ, 1967, т. 3, № 4;
, 1968.Т.4, № 1.
5. Зельдович Я. Б. ЖЭТФ, 1942, т. 12, в. И—12.
6. ЛибровичВ. Б. ПМТФ, 1963, т. 6, с.-74.
7. Baer A. D., Ryan N. W. AIAA Journal, 1968, v. 6, N 5; 1965 v. 3 № 5- 1966,
V. 4, № 8.
8. Ohlemiller T. J., Summerfield M. AMS, Report № 876, Princeton Uni-*
versify, 1968; Ракетная техника и космонавтика, 1968, т. в, № 5 с. 134
91 Ковальский А. А., Конев Э. В., Красильников Б. В. ФГВ, 1967, т 3 с.4.
IO. Levy J. В., Friedman R. 8th Symposium on Combustion N. V. 1962, p 663.
П.Похил П. ф., Белов M. M. Сб. «Физика взрыва», № 5, Изд. АН СССР, 1956,
с. 104.
12. Мержанов А. Г., Хайкии Б. Н. Докл. АН СССР, 1967, т. 173, М 6,
13. Зеинн А. А., Нефедова О. И. ФГВ, 1967, т. 3, № 1.
УДК 662.2
ВОСПЛАМЕНЕНИЕ И ТЕПЛООБМЕН
В МАНОМЕТРИЧЕСКОЙ БОМБЕ
Б. И. Кендриков, Г. Д, Козак, А. Д, Черных
Воспламенение вторичных взрывчатых веществ (ВВ) — один из
основных процессов, рассматриваемых в теории их горения и взрыва.
Загорание и взрыв при производстве и снаряжении ВВ в результате
нагревания или механических воздействий, возникновение преждевре-
менных взрывов артиллерийских снарядов в результате перегрузок, воз-
никающих при выстреле, выгорание предохранительных ВВ в угольных
шахтах при отказе детонации — во всех этих и многих других случаях
взрывное превращение начинается с воспламенения. В работах Андре-
ева, Беляева и Лейпунского качественно рассматривались пути возник-
новения устойчивого горения вторичных ВВ, необходимость достаточно
толстого прогретого слоя н др. [1—4]. Позднее на основе теории тепло-
вого взрыва был развит количественный подход к зажиганию [5—7]. Эти
теоретические работы были выполнены в основном в течение последних
10—15 лет. Одновременно проводилось экспериментальное изучение за-
жигания конденсированных веществ горячим газом, нагретым телом и
излучением [8—10], Объектом исследования служили смесевые или бал-
листитиые пороха либо их компоненты.
Обычные вторичные ВВ почти не изучались. Между тем, их изуче-
ние представляется интересным как в теоретическом плане, позволяя
рассмотреть применимость теории к зажиганию веществ, способных пла-
виться и испаряться, так и, особенно, в практическом — для обеспече-
ния безопасности при производстве и применении. Вторичные ВВ вызы-
вают интерес еще и потому, что при изучении предельных условий их
горения и затухания отчетливо проявляются эффекты, предсказанные
теорией тепловой неустойчивости горения [11]. Насколько существенна
она при воспламенении пока не известно'.
Для изучения воспламенения был применен предложенный Андре-
евым и сотр. метод с использованием манометрической бомбы [12]. Удоб-
ство метода заключается в его простоте. Кроме того, он позволяет моде-
лировать поведение ВВ в условиях их практического применения.
Методика проведения опытов
Воспламеняемость ВВ определяли в манометрической бомбе объе-
мом.218 мл с тензометрической записью давления во времени. Применя-
ли следующую аппаратуру: тензодатчики заводского изготовления с соб-
ственной частотой колебаний 5 кгц, тензометрическую станцию УТС 1
ВТ-12 с несущей частотой 35 кгц, магнитоэлектрический осциллограф
тр
Н-105. (Вибраторы ТИП V — собственная частота колебаний — 2 кгц).
Для нанесения меток времени использовали прибор П-104.
Скорость движения пленки при записи составляла 1000 мм[сек, эо
всех опытах применяли максимально возможный масштаб давления.
В качестве воспламенителя использовали пироксилин Na 1 (13,42%
азота), навески которого набивали в гильзы из папиросной бумаги и
прошивали вдоль нихромовой проволокой толщиной 0,1 мм. Испытуе-
мые вещества прессовали в виде плоских таблеток диаметром 36 и тол-
щиной 1,1 —1,2 мм до плотности 0,90—0,96 от максимально возможной,
Таблетки приклеивали к поверхности диска из плексигласа диаметром
40 и толщиной 3—5 мм с помощью раствора полиметилметакрилата и
дихлорэтане. Края шашки н боковую поверхность покрывали перхлор-
виниловым лаком,
Основные характеристики зажигания, определяемые в опытах
Основным параметром, с помощью которого оценивали воспламе-
няемость ВВ в манометрической бомбе, наряду с критической навеской
воспламенителя служило изменение давления в бомбе,
Центральным пунктом является вопрос о задержке зажигания прн
навесках воспламенителя больше критической.
. .. Кривая Р (t) при воспламенении и горении образца имеет три ха-
рактерных экстремума (рис. 1): максимум давления за счет сгорания
воспламенителя, последующее падение давления, минимум и, наконец,
максимум, который по времени близок к моменту окончания горения
таблетки ВВ,
Момент возникновения горения ВВ определяли, сравнивая кривые
P(tj 'в опытах по зажиганию и в холостых опытах с одними воспламе-
нителями' (рис. 1), Построение разности давлений ДР в этих опытах
от времени давало зависимость, показанную на рис. 2. Очевидно, в'мо-
мцнт tj < tmjn начинается горение образца. Задержка зажигания, таким
образом, будет складываться из времени сгорания воспламенителя и
времени tj, Как оказалось, возможны два случая: кривая 1 — воспламе-
нение происходит в периоде охлаждения продуктов сгорания воспла-
менителя; 2—воспламенение происходит в периоде сгорания воспла-
менителя.
. Рис. 1. Сравнение манометриче-
ских кривых в опытах по зажи-
ганию (!, 2) и в холостых опытах
(3) без шашки испытуемого ВВ.
1—зажигание в периоде охлаж-
дения; ,2—зажигание в периоде
сгорания воспламенителя
Рис. 2. Зависимость разности давле-
ний в опытах по зажиганию и в хо-
лостых опытах и определение момен-
та возникновения горения. Тетрнл,
навеска воспламенителя та =0,4 г
«V
ТакЗй подход к определению момента зажигания позволяет одновре-
менно определить еще одну важную характеристику — давление в мо-
мент воспламенения Р;.
Тепловой поток прн сгорании воспламенителей
Как показывает анализ имеющегося, литературного материала по
зажиганию, наиболее сложной и трудоемкой является часть, касающая
ся определения условий теплообмена и построения меняющегося во вре-
мени теплового потока. Первые работы по изучению! закономерностей
тепЛообмена в манометрической бомбе относятся к,'20-м гоЛам и связаны
с именем Мюраура [13]. Позднее вопрос теплбббмёна при высокой
температуре и давлении примеинтельйо к ракетной камере был подроб-
но рассмотрен в [14].
Теоретический расчет коэффициента теплообмена связан с почти
неизбежными погрешностями, экспериментальное определение — со зна-
чительными текническими трудностями.
Оценка тепловых потоков, развиваемых воспламенителями, может
быть осуществлена следующим образом. ) :
Тепловой поток за время t равен
—__Qn Q#
4 “~St
(1)
где Qn — теплота взрыва воспламенителя (при воде парообразной); Qo—
количество тепла, содержащееся в продуктах сгорания в данный момент*,
S — поверхность стенок бомбы (222 см2}.
Теплоту взрыва и состав продуктов сгорания воспламенителя с уче-
том бумажной оболочки, вес которой ~3% от веса пироксилина, и воз-
духа, содержащегося в бомбе, определяли расчетный путем по методу
[15] и экспериментально в стандартной калориметрической установке [16].
Результаты опытов приведены на рис. 3. Зависимость количества тепла,
выделяющегося прн сгорании пироксилина № 1, от плотности заряжания
выражается соотношением
Q* = 0,89-103
VvJ
(2)
В табл. 1 приведены результаты расчетов и сравнение их с опытом,
лота взрыва при воде парообразной
Qn = (Q> — 10,52’ пн, о) ш,
Теп-
СВ)
Количество тепла, содержащееся в продуктах сгорания’
с PVM
Qo = cvm,. (Т - То) - V R - - cvmrT.
(4)
где с¥ —теплоемкость газа при постоянном объеме; mr”m, + 0,3 —
масса газа в бомбе; Р, Т, М — давление, температура и средний молеку-
лярный вес газа; R — газовая постоянная; V = 218 см3— объем бомбы.
* Специальными опытами по резкому сбросу давления при простреле мембраны
было показано, что при 'сгорании пироксилина конденсаций воды на стенках бомбы
'За время 0,1—ОД «ей‘ие происходит.
а Труды »ып. 83
Таблица 1
Термодинамика продуктов сгорания пироксилина
ш,, г Теплота взрыва рж, ккал)кз : Содержание волн в про- дуктах сгора- ния пои Т-1900 ЬК, г-мом/кг Средний мо- лекулярный вес продуктов сгорания Су» кал/з-град mr/m.
опыт расчет
0,4 , 1580 1745 6,(8 28,7 0,29 1,75
ЛЛ 1270 1348 7,81 27.1 0,30 1,31
СТО;. .. 1178 Ц79 8,21 26,2 0,31 1,17
1148 1145 8,33 26,0 0,31 М3
’/Изменение давления при сгорании навесок пироксилина и последующем
‘Охлаждении продуктов взрыва приведено на рис. 4. Отношения давле-
ния в данный момент времени (Р) к весу пироксилина (шя) для навесок
:Ог0;4 до 3 г с точностью ~5% ложатся на одну кривую. Учитывая дан-
ные рнс. 4, получим 1 ?
где ₽ = £ (5)
0 \ к тв / т»
Принимались значения с¥ = 0,3 кал[г‘ёрад; V = 0,218 л; М = 27; R.=
. 0,082 л «г/г моль град; То = 293° К.
РИе.З. Влияние плотности за-
ряжания на теплоту взрыва
пироксилина
К'.,
Рис. 4. Падение давления в' йа но метри-
ческой бомбе при охлаждении продук-
тов сгорании различных навесок пирок-
силина № 1. Каждая точка — среднее из
2—8 определений при одной н той же
навеске. Навески меняются от 0,4 до
3,0 г
Ч. Зависимость среднего теплового потока для нескольких навесок
воспламенителя, рассчитанная по формуле (I), приведена на рис. 5.
Время сгорания воспламенителя зависит от навески (рис. 6), уменьша-
ясь при ее увеличении, В периоде сгорания поток принимался постоян-
ным. ь'
Результаты опытов, в которых измерялась задержка зажигания при
различных навесках воспламенителя для гексогена, тэна, тетрила и тро-
Тйла, йрнв’едены на рис. 7 в координатах t3— тпь, а на рис. 8 в коорди-
натах ta— q (масштаб логарифмический). Кроме того» в табл,,Д ;цри-
ведены значения критических навесок воспламенителя и соответствую-
щие им давления, при которых начинается горение таблетки- ВВ.
При отказах при критической навеске воспламенителя поверхность
таблетки покрывалась черным налетом, этот налет ослабевал при пере-
воде от ДНТ и ТНТ к гексогену и тэну.
Рнс, 5, Изменение теплового по-
тока в манометричесиой бомбе
при сгорании пироксилиновых
воспламенителей. Вес воспламени-
телей, г: /—0,4; 2—1,0: 3—1,6;
4—2,0; 5-3
Рис. 6. Зависимость времени
сгорания воспламенителей от
навески
Таблица 2
Критические навески воспламенителя
и давлении в момент времени t3
F Судя по критической навеске воспламенителя, наиболее легко из
рассматриваемых ВВ воспламеняется тетрил, однако при сравнении за-
висимостей h(mb) видно, что при навесках, превышающих критичес-
кую, воспламенение гексогена осуществляется быстрее. Задержка зажн-
Гания существенным образом зависит от теплового потока (навески вос-
пламенителя), уменьшаясь при его росте по закону (рис. 8)
ta = Aq-n
С ростом навески воспламенителя увеличива-
ется давление, при, котором происходит зажи-
гание. На рис. 9 приведены усредненные зна-
чения давлений в момент воспламенения Р[
при различных значениях гп в -Видно, что эти
давления близки для всех веществ при соот-
Рнс, 8, Влияние потока тепла на задержку зажигания.
Точки — результаты опытов; прямые — расчет по [71);
а—гексоген; б—тэн; в—тетрил; г—тротил
ветствующих навесках и, вместе с тем, при небольших воспламенителях
мало отличаются от максимальных значений давления.
Рис. 9. Изменение давления я
момент возникновения горения
с ростом навески воспламени-
теля. /—гексоген; 2—тетрил;
3~-тэн; 4—тротил. Прямая —
максимальное давление вос-
пламенителя без шашки ВВ
И
6 3 приведены величины максимальных давлений, развйваё-
ых воспламенителями в присутствии и в отсутствие таблетки тетрила
бомбе (опыты с тетрилом чередовались с холостыми), а также раз-
зсть между этими значениями. Видно, что до навески 1,4 г эта разность,
пределах ошибки опыта, несущественна, а начинай с навески 1,6 а
аксимальное давление в бомбе с помещенной в нее таблеткой тетрила
иле, чем в холостом опыте. Это говорит о том, что зажигание таблетки
1й воспламенителе 1,6 г и более происходит уже в. периоде его сгора-
[Я, отход кривой горения воспламенителя в пустой бомбе от кривой
рения ВВ в этом случае наблюдается еще до достижения максималь-
но давления (рис. 1)., Аналогичная картина наблюдается и для дру-
[х ВВ.
Таблиц* 3
Максимальные значения давлений при сгорания 1ЯСЯламеИИтелеЙ
в опытах по зажиганию тетрила
шв, г Опыты по воспламе- нению Холостые опыты А Р, ат
Рщ, ЧИСЛО опытов Ргп, ат ЧИСЛО опытов
0,2 ! 11,3+1.5 3 11,34:0.2 2 0
°,4 19,7±1.8 10 It),2± 1.3 8 0,5
0,6 28,8±3,7 и 29,9±2.5 6 --1,1
0,8 37.8 + 0.4 2 38,0+1,2 3 -0,2
1,0 48,24-2,5 4 47,4± 1,1 3 0.8
1,2 58,3± 1,8 2 56,5 2 1,8
1.4 70,5± 1,0 2 70,5 2 0
1.6 81,4±О,9 3 78 5 2 2.»
2,0 103.0 + 2.0 , 3 92,0 2 11,0
3.0 160±1,5 ' 3 147,5 + 2,5 2 12,5
Обсуждение результата»
На рис. 8 результаты опытов по воспламенению тетрила, тэиа, гек-
ена и тротила сравниваются с теоретической зависимостью ta(q),
утроенной по формулам [7]
(«)
(7)
Г
?'<!, (? — теплопроводность, теплоемкость и плетиесть вещества; В и
•—предэкспоиенциальный множитель и энергия активации; Q — теп-
»та реакции в к-фазе; Ts н То — температура поверхности ВВ и на-
ильная температура.
Значения теплофизических и кинетических констант, применявших-
я при расчете, приведены в табл. 4.
*5
Значения теплофизяческнх н кинетических констант ВВ [17]
ВВ Е, ккал! моль 1g В (сек“’) Q, кал! г 1-10*, калием •сею3 С, кал}г*град Рпл, кал]г р*, г ‘СМ3
Гегсотен 41,0 18,5 500 4.0 0.24 29,3 1,63
Тетрил 38,4 15,4 350 2,55 0,23 19,5 1.66
Тэн 39,0 15,6 500 4,0 0,4 17,7 1,67
Тротил 34,4 11.4 500 5,0 0,35 22,3 1,60
* Средние значения по результатам опытов.
Сравнение результатов расчета и опыта (рис, 8) показывает, что
качественное согласие между теорией и опытом имеется во всех слу-
чаях: расчетные кривые близки к экспериментальным точкам, задержка
зажигания уменьшается при увеличении потока и угол наклона расчет-
ной прямой отвечает темпу уменьшения задержки, полученному экспе-
риментально, В случае тротила и гексогена прямые на значительных
участках графика совпадают с точками, В случае тэна и тетрила расхож-
дение между теорией и опытом несколько больше: задержка зажигания,
полученнай на опыте, в 2—3 раза превышает полученную расчетом. Со-
отношение между расчетными и опытными значениями периодов индук-
ции зажигания для разных ВВ получается разным. Можно думать, что
эта разница обусловлена не только неточностью физических и кинети-
ческих характеристик ВВ, но и другими, более существенными причи-
нами.
На рис. 10 приведены задержки зажигания (средние из 2—14 опы-
тов} при данной навеске в зависимости от температуры кипения ВВ при
давлении Р|. Значения Тк найдены расчетом по данным [3]. В полуло-
гарифмическом масштабе точки для каждого вещества ложатся на пря-
мую вида
(п t3 = А — ВТК
Такое выражение легко получить из уравнения (7), предположив,
что поток пропорционален давлению, а давление экспоненциально свя-
зано с температурой кипения. При этом
2,3 RT?»<
где Qr>- теплота испарения; Ток—температура кипения вещества при
а+м'осферном давлении.
Кривые, соединяющие точки на прямых, относятся к опытам с оди-
наковыми воспламенителями. Минимумы этих кривых приходятся ни
гексоген, правые ветви поднимаются к тротилу, левые — к тетрилу 1‘
тэну. Подъем правой ветви кривой не дает существенно новой инфор-
мации: температура кипения тротила выше, чем гексогена, и задержка
зажигания при той же навеске воспламенителя соответственно больше.
Характер левых ветвей необычен: более низкая температура поверхно-
сти тетрила и тэна должна была бы достигаться раньше, чем гексогена,
8бх
к, еслиЯрГифй тй происходило воспламенение, Ьадёржка была бы
меньше, в она — больше. , ; /'
, На- рис. 11 приведено отношение толщины прогретого слоя (б „)
при нагреве до температуры кипения в манометрической бомбе к тол-
щииё прогретого слоя при стационарном горении (б =х/и). Значение
рассчитывали по формуле
8» = (Т« —ТЛ
В * X к
4 Чк
*де Ци — средний тепловой поток за время достижения Тк, Это отноше-
1ие для гексогена'в 2-3 раза больше, чем для тетрила и тэна, Соответ-
ртвенно, в случае гексогена устойчивость воспламенения больше.
Рис. 10, Занисимость вре-
мени воспламенения гексо-
гена (1), тетрила (2), тэиа
(J) и тротила (4) от тем-
пературы кипения. Числа
у кривых — вес воспламе-
нителя, г
Рис. 11, Влияние давления на от-
ношение толщин прогретого слоя
прн воспламенении и при стацио-
нарном горении: /—гексоген; 2—
тетрил; 3—тэн;
: Сопоставление рис, 8 и 11 позволяет предположить, что завышен-
ные задержки в случае тетрила и тэна связаны с меньшей устойчивостью
горения этих ВВ по сравнению с гексогеном. Тепловая устойчивость
Горения по измерениям критического диаметра тэна и тетрила ниже,
нем гексогена: прн 10 аг произведение критического диаметра на ско-
рость горения (гп • dK ) для них: 0,18; 0,12 и 0,06 г/см • сек соответственна
известно, что чем ниже устойчивость горения, тем больше Пэ-
тому прогрев тэна или тетрила до температуры кипения приводит не к
Горению, а к газификации вещества [10]. Газификация может привести к
Уменьшению потока тепла на поверхность ВВ [18], градиент температуры
У поверхности станет меньше, прогретый слой увеличится, и произой-
дет воспламенение.
I. Андреев К. К. Сб. статей по теории ВВ М., Оборонно, 1940, с, 39,
!. Андреев К. К, Костин И. Д. ДАН СССР, 1946, т. М, с. 231.
3. Беляев А. Ф. Горение, детонация и работа взрыва конденсированных снетем.
М„ <Наука», 1968.
17
4j Аристова 31..Й., Лейнунсккй О. И. 05. Физика взрыва, I9flflt, М>2. ДАН, •
СССР. 1946, т. Н с. 507; ЖФХ, 1946, т. 20, с. 1391.
А Зельдович Я-Б. ДАН СССР, 1963, т. 150, № 2, с. 283.
6. Аверсов А. Э., Барэыкин В. В., Мержанов А. Г. ДАН СССР, 1968,
т. 178, № 1.
7. Аверсов А. Э. БараыкннВ. В., Мержанов А. Г. ФГВ 1968, т. 4, № I,
с. 20.
8. Штейнберг А. С., Улыбни В. Б., Барзыкии В. В., Мержанов А. Г.
ИФЖ, 1966, т. 10, № 4, с. 482.
9. Роэенбанд В. И., Барзыкии В. В., Мержанов А. Г. ФГВ, 1968, т. 4,
№ 2, с. 171.
10. Кендриков Б. И., Олемнллер Т. Дж., Саммерфилд. Труды МХТИ
им. Д, И. Менделеева, 1974, вып. 83, с. 67.
11. Кондрнков Б. Н. ФГВ, 1969, 5, J* 1 51.
12. Андреев К- К-> Рогожников В. М. Сб. Теория ВВ М., 1967, с. 288.
13. Mur а о ur Н. Mem, Art. Franc. 1924, 3, № 1 339; 1925 4, № 2, 455- 478; Ми-
гаоиг И., Aunis, Comptes rendus, 1933, 196, 404; 1933, 196, 478.
14. Зельдович Я- Б., Рнвин М. А., Франк-Ка менецкий Д. А. Импульс ре-
активной силы пороховых ракет. М., Оборонгиз, 1963.
15. Андреев К. К., Б е л я е в А. Ф. Теория ВВ, М. Обороигнз, I960.
16. Гольбнндер А. И. Лабораторные работы по курсу теории ВВ, М., Росвуэиздат,
1963.
17. Андреев К. К. Термическое разложение н горение ВВ, М„ «Наука» 1966.
18. Л нбр ович В. Б. ПМТФ, 1963, № 6, с. 74. ;
УДК 662,2
О ГОРЕНИИ ЖИДКИХ ВЗРЫВЧАТЫХ СМЕСЕЙ
1
В. Э. Анников, Б. Н. Кендриков, В. М. Райкова, Л. Г. Спиженкова
Жидкие взрывчатые вещества находят все большее применение и
промышленности: при торпедировании нефтяных и газовых скважин,
при взрывном бурении, при обработке металлов взрывом. Преимуще
ствах их — высокая и не зависящая от диаметра заряда скорость дето-
нации, простота транспортировки, погрузки и выгрузки, заряжания шпу-
ров и скважин. Значительный интерес представляют жидкие взрывчатые
смеси на основе нитроэфиров, которые можно получить на имеющемся
оборудовании с хорошим выходом на широкой сырьевой базе.
Во всех случаях для надежного, безопасного и эффективного при-
менения этих смесей необходимо знать характер их горения — скорость,
устойчивость горения и связь их с основными физико-химическими и
взрывчатыми характеристиками. В настоящей работе изучалось горе-
ние смесей распространенных жидких нитроэфиров — мет,и л нитрата,
нитроглицерина и нитрогликоля с различными органическими раство-
рителями.
Нитроэфиры получали этерификацией соответствующих спиртов сер-
ноазотными смесями, промывали холодной водой, водным раствором
соды и снова водей. Метилнитрат перегоняли под атмосферным давле-
нием при 65°С. Остальные В В использовали без перегонки. Характери-
стики их приведены в таблице. Теплоты взрыва смесей оценивали по
методу Авакяна.
Растворитель Квалификация Плотность, г!см$ Ткнп. ° С
Гексан X, ч. 0.66 69,0
Этанол гидролизный 0,79 78.5
Метанол X. ч. 0 79 64.6
Уксусный альдегид ч.д, а, 0.78 21
Ацетон перегнанный 0.79 56,5
Бензальдегид перегнанный 1.05 179,5
Нитробензол «чистейший» (ГДР) 1,20 210.9
дпыты проводили в йомЙе постоянного давления в атмосфере азота
Жидкость наливали в кварцевые трубочки диаметром 7—8 мм, длиной
35 мм, со стенками толщиной 1—1,5 мм. Воспламенение осуществляли
свернутой в виде спирали нихромовой проволочкой. Скорость горения
измеряли с помощью фоторегистра ФР-11 (при малой скорости горе-
ния — с помощью секундомера).
Смеси на основе метилнитрата
Нормальное горение метилнитрата изучали Андреев и Пуркалн
[I]. Скорость горения в интервале давлений 0,5 —1,8 ат описывается
уравнением
u„— 0,140 р, г-!см2 -сек
При давлении 1,8 ат (им= 0,25 г/см2-сек) горение переходит иа турбу-
лентны# пульсирующий режим.
Зависимость скорости горения смесей метилнитрата с метанолом
от давления приведена на рис. 1. При увеличении содержания метанола
скорбеть горения уменьшается, повышается давление перехода горения
на турбулентный режим и изменяется характер зависимости и(р) нт
участке нормального горения. При содержании метанола более 10% на-
клон кривых н(р) в области высоких давлений уменьшается, при 40%
наблюдается плато, а при 45 и 50% — снижение скорости горения при
увеличении давления. При атмос-
ферном давлении было получено
горение для смесей, содержащих
5 — 40% метанола, смеси с боль-
шим содержанием спирта под-
жечь не удалось.
Рис, 1. Зависимость скорости горе- Рис. 2, Зависимость скорости горения
и ня смесей метилнитрата с метано* смесей метилнитрата с ацетоном от
лом от давления. Содержание мета- давления. Содержание ацетона, %:
иола, %: /-0; 2—5* 3—10; 4—15; 7—30; 2—40. Пунктирная кривая —
5—20; 5—30; 7—35; S—40; 9—45; 40% метанола
10- 50
На рис, 2 приведены результаты опытов для смесей метилнитрата
с ацетоном (30 и 40%). Пунктирная кривая — для смеси, содержащей
40% метанола, теплота взрыва которой близка к теплоте взрыва смеси
с 40% ацетона. До 100 ат при умеренном давлении скорость горения
смесей с метанолом выше, чем с ацетоном при одном и том же содержа*
«9
((ии лЯН^Ятеля: крнвай для смеси с 40% метанола по4ти совпадав1», С
кривейдля 30% ацетона. При давлениях выше 100- ат кривая для мета-
нола приближается к кривой для смеси с 40% ацетона, и при высоком
давлении скорости для смесей с одинаковой теплотой взрыва совпадают
Ускоряющее влияние метанола при умеренном давлении еще от-
четливее видно при сравнении со смесями метилнитрата с гексаном
(рис. 3). Состав с 40% метанола, теплота взрыва которого 666 иал!г
при давлении 5—30 ат горит не только быстрее смеси с 30% гексана
(Qv= 690 кал/г), но и смеси с 19% гексана (Qv = 910 кал/г). При дав-
лении 140 ат кривые для смесей с 40% метанола и 30% гексана пересе-
каются, и при высоком давлении скорости горения приходят в качест-
венное соответствие с теплотами взрыва.
Смеси на еснове нитроглнколя и нитроглицерина
В случае труднолетучих нитроэфиров (нитрогликоль и нитроглице-
рин) во избежание «выкипания» растворителя из прогретого слоя при-
менялись высококипящие растворители — нитробензол и бензальдегид,
На рис. 4 приведены данные для смесей нитрогликоля с нитробен-
золом-и нитроглицерина с бензальдегидом. До 100 ат зависимости ско-
рости горения от давления имеют вид прямых с тангенсом угла наклона
в логарифмических координатах близким к единице. Для этих смесей,
особенно на основе нитроглицерина, характерна значительная неустой-
чивость горения. Давление, при котором они начинают гореть, и крити-
ческая скорость тем выше, чем больше содержание растворителя.
Рис. 3. Зависимость скорости горе-
ния смесей метилнитрата с гексаном
от давления. Содержание гексана,
%: /—6; 2—19; 3-30. Кривые без
точек соответствуют смесям метил-
нитрата с метанолом, %; /'—10;
2*—40; 3'—50
Рис. 4. Зависимость скорости
горения смесей нитроглицери-
на с бензальдегидом и ннтро-
Гликоля с нитробензолом от
давления. Содержание раство-
рителя, %: бензальдегид —
1—20; 2—30; 3—40- нитробен-
зол — 4—30- 5—40; 6—50;
7—70; 8—80. Сплошная пря-
мая—13%, пунктирная кри-
вая—40% метанола
На том же рисунке -пунктирной линией изображена, зависимости
ц(р) для смеси метилнитрата с 40% метанола, теплота взрыва которой
(666 кал/г) меньше, чем любой другой из приведенных на рнс. 4 смесей.
Характерно, что для всех кривых имеется область давлений (правая
91
Граница которой лежит тем дальше от атмосферного давления, мем
больше растворителя в смеси), где скорость горения смеси с метанолом
выше, чем скорость горения смеси с нитрогликолем и нитроглицерином.
Иными -словами, у всех растворов на основе нитрогликоля (с теплотой
взрыва 700—1100 кал/г) имеется участок на кривой и(р), иа котором
скорость горения ниже, чем у низкокалорийной смеси с метанолом.
По мере повышения давления скорость горения постепенно приходит в
соответствие с теплотой взрыва.
Обсуждение результатов
В работе [2] показано, что скорость горения О—С-нитросоедине-
иий при высоком (300 ат) давлении определяется температурой пламени
В координатах 1/Tf—сим, где Tf—температура пламени, с =
|Л(Тг — Те)3/Т4(, данные
напрямую, которая
ля большинства изученных веществ ложатся
ает энергию активации ведущей реакция
27,9 ккал/моль. Если вместо обратной темпе-
ратуры пламени использовать обратную теп-
лоту взрыва нитросоединений, то получится
картина, изображенная на рис. 5. С использо-
ванием средней теплоемкости продуктов взры-
ва порохов, состоящих преимущественно из
О—С-иитросоединеннй, cv = 0,333 кал/г • град и
величины у = cp/cv= 1,24 — средней для тех
же порохов, легко по наклону прямой um—1/QT
найти эффективную энергию активации. Она
получается практически такой же —
27,4 ккал/моль.
Попытаемся применить аналогичный под-
ход и к нашим растворам. Наибольшее дав-
ление, для которого имеется достаточное чис-
ло данных — 100 ат. Это несколько ниже, чем
давление, взятое в [2], но для жидких ВВ нор-
мальные скорости горения при более высоком
давлении ввиду турбулизации получить труд-
но. Данные, полученные в настоящей работе,
в координатах 1/QV — иы. Точки для раз-
W / f,f Z
“г
Рис, 5. Зависимость скоро-
сти горения О, С-нитро-
сое ди нений при 300 ат от
теплоты взрыва
представлены иа рис. 6
ных растворов: метилнитрата в метаноле, гексане и ацетоне, иитро-
гликоля в нитробензоле, ложатся вблизи прямой, угол наклона которой
дает энергию активации 28 ккал/моль, что находится в хорошем соот-
ветствии с величиной, полученной в [2] для чистых нитросоединений при
высоком давлении. Существенного влияния природы растворителя н
нитроэфира при этом давлении не наблюдается. Можно только отметить,
что нитрогликоль дает растворы, горящие несколько медленнее. Если
сопоставить данные, полученные в настоящей работе для смесей с дан-
ными Для чистых нитроэфиров (белые точки и пунктирная прямая нт
рис, 6), то окажется, что нитроэфиры горят чуть медленнее, а точки для
системы нитрогликоль—нитробензол находятся ближе к последней
прямой.
Данные для изученных в работе смесей при давлении 10 ат приве-
дены на рис. 7. При этом давлении оказывается, что скорость горения
чистых нитроэфиров и растворов нитрогликоля в нитробензоле раза з
2 меньше, чем смесей метилнитрата с метанолом и гексаном. Растворы
метилнитрата в метаноле горят гораздо быстрее и разница увеличи-
вается при увеличении содержания метанола. Из чистых нитроэфиров
дигликольди нитрат и гидроксиэтилнитрат ложатся на прямую для си-
стемы метилнитрат—гексан. Данные для смесей с ацетоном лежат ме-
жду прямыми для смесей со спиртом и гексаном.
Рис, 6, Зависимость скорости
горения нитроэфнров и их
смесей с растворителями при
100 аг от теплоты взрыва.
Смеси метилнитрата ® — с ме-
танолом и этанолом; ф — с
гексаном; w - с ацетоном;
-I---йитрогликоль и его сме-
си с нитробензолом; О—
нитроэфиры (сверху вниз):
тэн; 1,3- и 1,2-пропилевглн-
ко льдн нитраты; 1,4-бутвлеи-
гликольди нитрат; этнлнитрат
Рис. 7. Зависимость скорости горе-
ния ннтроэфиров и их смесей с рас-
творителями при 10 ат от тепло-
ты взрыва. Смеси метилнитрата:
/—с метанолом и этанолом; 2—
с гексаном, ацетоном и ацеталь-
дегидом (+); 3 —4) —нитрогли-
коль и его смеси с нитробензо-
лом; О—нитроэфиры без добавок
Легко видеть, что при умеренном давлении метанол гораздо актив-
нее иных растворителей. Вероятная причина этого заключается в высо-
кой скорости взаимодействия двуокиси азота, образующейся при рас-
паде нитрата, с гидроксильной группой спирта. По данным [3] между
спиртом и NO2 уже при комнатной температуре идет быстрая реакция
СН3ОН + 2NO2 CH3ONO + HNO3
При горении реакция, вероятно, идет глубже — с образованием метокси-
ла, гидроксила и других активных продуктов.
Например,
CH3OH|-NO2- (CH3ONO+ ОН) -> СН3О- + NO + -ОН
СН3О- +NO-»CH2O + HNO
сн2о + • ОН -* СО + Н2О + Н’
HNO |- NO -> N2 + НО2'
При повышении давления (и, соответственно, температуры ведущей зо-
ны) эти реакции подавляются, например, в результате рекомбинации
активных центров, и активность спирта падает.
93
Особая роль спирта проверялась опытами с этанолом. По теплоте
Взрыва растворы метилнитрата в этаноле близки к растворам в мета-
ноле, В частности, смесь с 40% этанола в точности соответствует смеси
С 45% метанола. Результаты опытов (рис, 8} показывают, что смесь с
этанолом горит так же, как смесь с метиловым спиртом: скорости для
обоих смесей совпадают.
В целом зависимость скорости горения от давления для смесей ме-
тилнитрата со спиртами похожа на полученную ранее для органических
нитритов [4]. Вблизи 100 ат наблюдалось падение скорости горения при
повышении давления. Кривые для этилнитрата и смесей метилнитрата
со спиртами совершенно аналогичны друг другу (рис. 8). Ни один из
других изученных до сих пор растворителей такой картины не давал.
Рис. 8. Зависимость скорости горе-
ния метилнитрата с метаиолом и
этанолом от давления. 1—<0% мета-
нола; 2—40% этанола (О); 45%
метанола (ф); 3—50% метанола
Рис, 9, Зависимость скорости
горения смесей метилнитрата
с ацетальдегидом и метанолом
ст давления. 1—10% метано-
ла ; 2—11,7% ацетальдегида;
3—20% метанола
В опытах с нитритами было обнаружено сильное угнетающее влия-
ацетальдегида. Оно объяснялось тем, что ацетальдегид, в отличие
^^формальдегида, является ловушкой атомарного водорода — основ-
ного активного Центра реакции горения. Концентрация атомов водорода
J^йен'ьшается и скорость «падает. Были проведены опыты с растворами
Fацетальдегида в метилнитрате (рис, 4). Введение 12% ацетальдегида
уменьшило скорость горения сильнее, чем соответствующее ему количе-
ство метанола (10%), Смесь, содержащую 30% ацетальдегида, поджечь
,ре. удалось. . . .. ............._
♦ ... с t ;
I. Андреев К. Пуркалн М. М. ДАН СССР, 1945, т. 50, с. 281.
2. Кондриков Б. Н., Райкова В. М., Самсонов Б. С. ФГВ, 1973, № 1 с. 84.
3. F a i г 1 е А. М., Carberry J, J., Т г е а с у J. С, J, Am, Chem/ Soc. 1953 ’ v, 78,
р. 3786,
4. Кондриков Б, Н.. Сидорова Т. Т. ДАН СССР, 1971, т. 197, № I, с, 125,
УДК 536.46+ 662.222
О ГОРЕНИИ ПЕРХЛОРАТОВ И НИТРАТОВ
АММИАКАТОВ ПЕРЕХОДНЫХ МЕТАЛЛОВ
В. В. Горбунов, Л. Ф. Ш магии
Ранее было показано, что скорость горения нитратов аммиакатов
меди (И) и никеля (11) зависит от соотношения горючего (NH3) и окис-
лителя (NOa-) в молекуле соли [1]. При одном и том же катионе,
[CufNHajiJ2*, скорость горения комплексных солей определяется приро-
дой аииона-окислителя и убывает в ряду: ВгОа-, СЮз-, СЮ*-, МпОч-,
NO3“, 1Оа“ и NOa“ р]. В настоящей работе изучалось влияние централь-
ного атома металла на скорость горения аммиакатов перхлоратов и
нитратов хрома (111), кобальта (III), никеля (II), меди (11) и цинка
(11), в молекулах которых соотношение между количествам групп горю
чего и окислителя было равным двум (для гекса аммиак ат а никеля
(И) 6Г4Нз/2Лн = 3). Общая формула исследованных комплексных
солей
[Me^fNH^KAH-Jn,
где Мё+ — ион переходного металла; Ан~—СЮ*- или NOs- и Х = 1
или 6.
Соли были получены по методикам, описанным в [3, 4] и анализиро-
вались на содержание металла и аммиака. Содержание основного про-
дукта в полученных солях было 98—99%.
Скорость горения солей определялась при давлениях до 100 ати в
атмосфере азота. Навеску соли помещали в трубку из органического
стекла с внутренним диаметром 7 мм (в случае быстрогорящих пер-
хлоратов, - - 4 мм} и уплотняли под давлением 2500—3000 nscjCM1. Отно-
сительна^ 'плотность зарядов была 0,85—0,95. Воспламенение заряда
осуществлялось с помощью электроспирали, для определения скорости
горения применялся фоторегистр.
. Результаты расчетов теплоты и температуры горения исследован-
ных солей ио предполагаемым брутто-уравнениям реакции горения при-
ведены в таблице,
»5
Формула соли —ДНобр* соли, ккал 1 мол Продукты горения Теплоте Расчетная температура горения ТГ°К Um при 60 ати г/см1-сек ишС1ОГ UmNO,'
О Г « Z О сч и прочие u p rorlvm ккал 1кг =
[Cr(NHs)d(C104)3 [CrfNHehHNCbh 243 302 9 7.5 3 4,5 0,75 1,5 0,5 СГдО3 0,5 Сг3О3 + + 1,5 Н3 413 268 913 788 2280 1770 17,5 1,96 9
[Co(NH3),](CIO4)3 (Co(NH3)e](NO3)3 247 306 «сю 3 4,5 1.5 0,5 CoCISjrj Со(кр) 316 214 688 617 2000 1640 11.7 0,57 20
[Ni(NH3)4](C104)3 (Mi (NH3)4](\O3)2 178 218 6 6 2 3 1 — NiCi3(r) NffKpJ 194 126 614 503 1950 1530 10,0 0,63 16
[Cu(NH3)4I(C104)3 [Cu(NH3)4I(NO3)s 158 198 6 6 2 3 1 0,5 Cud(r) Си(ж) 183 154 555 605 1950 1750 18,2 0,98 19
[Zn(NH3)4I(C104)3 Ы\Н3),](ГЮ,Ь 196 236 6 5 2 2,67 0,5 1 ZnO ZnO + + 0,66 NHa 234 143 705 557 2100 1630 6,2 0,72 9
[Ni(NH3)<>](C104)» №(Nhs).I(no3)» 227 267 6 6 2 3 1 — NlGlS(r, 4- + 2 NHa +Ш 166 102 463 358 1450 1100 1,90 0,12 16
1 * Теплоты образования комплексных солей, кроме нитратов аммиакатов Си(И)
ии,*Со(111) и перхлората Со(111), определены методом сравнительного расчета по тепло-
[;У»М образования комплексных солей с другими анионами и соответствующих простых
'<;алей, взятых из [5].
Скорость горения комплексных перхлоратов линейно увеличивается
с повышением давления от 20 до 100 ати (рис, 1,а), При давлениях
Менее 20 аги горение перхлоратов тетр а аммиакатов меди (II), никеля
(II) и гексааммиаката кобальта (III) становится неравномерным, ско-
рость его резко уменьшается. Скорость горения перхлората тетраамми-
аката меди (II) уменьшается в 2 раза при понижении давления от 15
до 13 ати, при 10,8 и 5 аги горение заряда прекращается после удале-
ния спирали. Однако при атмосферном давлении эта соль устойчиво
горит со скоростью 0,75 г^м^сек. У перхлоратов гехсааммиаката ко-
бальта (III) и тетрааммиаката никеля (И) скорость горения умень-
шается в 2,5—3 раза при понижении давления от 20 до 16 и 10 до 5 ат л
соответственно, при более низких давлениях вплоть до атмосферного
происходит линейное понижение скорости горения с давлением. У пер-
хлоратов тетрааммиаката цинка (II) и гексааммиаката никеля линейная
зависимость скорости горения от давления сохраняется от 0 до 100 ати
Твердыми продуктами горения перхлоратов аммиакатов кобальта (III),
никеля (П) и меди (11) были хлориды соответствующих металлов. Пос-
ле сгорания перхлоратов аммиакатов хрома (III) и цинка (И) остава-
лись оксиды этих металлов. Твердые продукты горения всех комплекс-
ных перхлоратов почти полностью удалялись из оболочки заряда. ,
06
По.скорости горения аммиакаты нитратов переходных металлов
значительно уступают соответствующим перхлоратам. Линейная зави-
симость Uu — р при горении нитратов (рис, 1,6) наблюдается в интер-
вале давлений 20—100 ати (кроме соли хрома (III)), при более низких
Давлениях скорость горения уменьшается несколько быстрее, Неравно-
мерного горения с резким понижением скорости у нитратов не наблю-
цается. Среди исследованных комплексных нитратов наибольшую ско-
рость горения имеет соль гексааммиаката хрома (III), которая горит
Р.агтш
в й ад юз
Р, ати
Рнс. I. Влияние давления ва скорость горении перхлоратов (а) и нитратов (б)
аммиакатов: /—меди (Н); 2— хрома (III); 3—кобальта (Ш); 4—никеля (II)
(тетрааммиакат); 5—цинка (II); о—никели (П) (гексааммиакат)
г почти вдвое быстрее следующего за ней по скорости горения нитрат *
тетра аммиакат а меди. В условиях опытов только эти нитраты горят при
.атмосферном давлении. Нитраты тетрааммиакатов цинка (II) и никеля
(II) и гексааммиаката кобальта (III) не горели при давлениях ниже
5—8 ати, горение наиболее медленно горя-
щего нитрата гексааммиаката никеля (II)
прекращалось при давлении ниже 20 ати.
•Твердые продукты горения нитратов почти
полностью остаются в оболочке заряда в
виде порошкообразных никеля и кобальта,
оплавленных шариков меди, пористой мас-
сы оксидов цинка (II) или хрома (III).
• Для большинства исследованных амми-
акатов перхлоратов и нитратов переходных
! металлов наблюдается приближенно линей-
ная зависимость между скоростью и расчет-
ной температурой горения (рис. 2). Можно
^предположить, что реакция окисления ам-
^миака продуктами разложения аниона-окис-
елителя происходит в газовой фазе при тем-
[пературе, близкой к расчетной или пропор-
[циональной ей. Параметрами, определяю-
.щими скорость реакции окисления и, следо-
вательно, скорость горения соли в этнх ус-
ловиях, являются: активность окислителя,
температура горения и каталитическое влия-
ние центрального атома металла. Сопо-
а
ЬВати
26-
0 -
12-
8 ~
4 -
4 1
7 Трулы »ып. 83
"•2В~
V-
J,8-
5
д-4
2В69 2503
Т^К
Рис. 2, Зависимость скорости
горения при 100 ати от расчет-
ной температуры горении пер-
хлоратов (а) и нитратов (б)
аммиакатов: / — хрома (III);
2—кобальта (П1); 3—никеля
(II) (тетрааммиакат); 4—меди
(П); 5—цинка (II); 6—никеля
(11) (гекса аммиакат)
97
£
т
ставление скоростей горения перхлоратов и нитратов, ррдчиняющих-
ся линейной зависимости Uu — Тг, показывает, что при одной и той же
температуре горения нитраты должны гореть приблизительно в 10 раз
медленнее перхлоратов. Это объясняется более высокой окислитель»
активностью продуктов разложения перхлоратного аниона. При одно-
именном комплексном катионе перхлораты имеют несколько более высо-
кую температуру горения и соотношение скоростей горения перхлорат/
нитрат увеличивается до 16—20. Таким образом, при замене в аммиа-
кате аниона нитрата на перхлорат скорость горения увеличивается в
10 раз, вследствие увеличения активности окислителя и в 1,5 — 2 раза,
вследствие возрастания температуры горения соли.
Нарушение линейной зависимости иы — ТР у аммиакатов перхло-
ратов меди (11) и цинка (11) и нитрата хрома (111), по-вндимому, можно
объяснить влиянием центрального атома металла на скорость реакции
окисления аммиака. Хорошо известно, что наилучшим катализа+ором
горения перхлората аммония являются соединения меди (1), а соедине-
ния хрома оказывают такое же действие йа горение аммиачной селитры
[6]. Соответственно этому аммиакаты перхлората меди ^Н) и нитрата
хрома (111) горят почти в 2 раза быстрее других комплексных солей с
тем же анионом и близких к ним по температуре горения. Не вполне
понятны причины сравнительно малой скорости горения аммиаката пер-
хлората цинка (II), возможно, что оксид цинка, образующийся при го
рении соли в условиях наших опытов, ингибирует реакцию окисления
аммидоа.
Резюме: Скорость горения большинства изученных аммиакатов пер-
хлоратов переходных металлов в 16—20 раз выше скорости горения со-
ответствующих им нитратов. Различие скоростей горения в первую оче-
редь обусловлено более высокой окислительной активностью продуктов
разложения иона-перхлората. Температура горения и каталитическая
активность центрального атома металла оказывают более слабое влия-
ние на скорость горения изученных комплексных солей.
1. Горбунов В, В., Шндловский А. А., Шмагин Л. Ф. ФГВ 1971 № 4
с. 607—609. ’ ’
2. Г о р б у я о в В. В., Ш м а г и и Л. Ф. ФГВ, 1972, № 4, с. 523—526.
3. Gmelins. Handbuch der anorg. Chem., В. 58, 1964, В. 60, T. В„ 1959.
4. Неорганические синтезы. М,, И низ дат, 1952, ч. 111, с. 145—146.
5. W a g m а п D. D., Е v а п s W. Н. а. о., Technical Note 270—4, Washington, 19б9.
6. Андреев К. К. Термическое разложение н горение взрывчатых веществ. М..
«Неука», 1966, с. 152.
УДК 662.221+536.48
О ВЛИЯНИИ НЕОРГАНИЧЕСКИХ КАТАЛИЗАТОРОВ
НА ГОРЕНИЕ ПЕРХЛОРАТА АММОНИЯ
А. П. Глазкова
Впервые влияние ряда неорганических катализаторов на горение
перхлората аммония было изучено Фридманом и др. [1] в интервале дав-
лений до 340 ат, а в работе (2] — до 1000 ат. В настоящей работе влия-
ние катализаторов на горение перхлората аммония (ПА) было изучено
по той же методике [2]- более детально. В качестве добавок исследованы
соли шести- и трехвалентного хрома, соединения железа и меди, пяти-
окись ванадия и др. За критерий эффективности действия изученных
добавок принят коэффициент катализа К, равный отношению массовых
скоростей горения катализированного и чистого ПА. Поскольку опыты
по горению катализированного ПА проводились в плексигласовых труб-
ках, а при горении чистого ПА при давлениях выше 500 ат ведущим
является распространение пламени по поверхности раздела плекси-
глас— ПА, очень важным является во-
прос, какие значения скоростей горения
ПА брать для сравнения — в плексигла-
совых трубках или без оболочки, которую
мы приняли за истинную скорость горе-
ния чистого ПА [3].
Для определения влияния материа1-
ла оболочки на скорость горения ПА, ка-
тализированного 5% КаСгаОт, были по-
ставлены опыты, результаты которых
представлены на рис, I. Из рисунка вид-
но, что в данном случае материал обо-
лочки не влияет на скорость горения. Не
было обнаружено такого влияния и при
использовании в качестве оболочки плек-
сигласовых трубок, покрытых перхлор-
внниловым лаком. Исходя из этих дан-
ных, при расчете значений К брались ис-
тинные скорости горения; для ПА из опы-
тов без оболочки, для ПА с катализато-
рами—в плексигласовых трубках.
Рис. 1. Влияние оболочки иа
скорость горения перхлората
аммония с 5% би хромата ка-
лия. /—В плексигласовых труб-
ках [2]; 2—образцы прямо-
угольного сечения 7X8X24 мм
без оболочки; 3—в стеклянных
трубках, плотность образчиков
1,3—1,4
99
I
Было изучено также влияние содержания бихромата калия на U(p)
ПА для образчиков прямоугольного сечения (7X8X24 мм) без оболоч-
ки. На рис. 2 и в табл. 1 представлены полученные результаты.
Таблица I
Влияние содержания KiKr2O7 на характеристики горения NH4C!O4
К/ХО,. % Рпр. ат Значения К прн следующих р, ат В Интервал р, ат
50 150 300 600 1000
0 50 — — —* 0,22 0,00000052 0,49 -3,84 2,36 50-150 150-200 500-1000
0,05 40 1.0 1.2 1.1 2.4 0.9 0,063 0,000064 0,75 -5,9 1,68 40 -200 200-250 250-1000
0,1 40 1.0 1,1 3,4 2,4 1.0 0,0134 1.07 0.000048 1.08 0,18 -4,16 1,734 40-125 125-275 300—375 400-1000
1,0 75 — 1,3 6,0 4.5 1.6 0.26 ' о.гоз 0.49 1.058 100-500 500-1000
3,0 50 0,9 1.5 7,9 6.1 1.6 0,613 0,15 1,6 0.63 50-100 100-1 000
5,0 20 J !-3 1.9 9.0 7.7 2.0 0,011 0,142 1,25 0,67 20-100 100-1000
10 50 0.6 2,2 10,9 7,9 1.8 0,00003 0,484 2,55 0,485 50-100 100-1000
Из приведенных результатов видно, что при добавлении 0,05 и 0,1%
КлСгдО? верхний предел горения по давлению смещался со 150 аг в об-
ласть более высоких давлений — 200—250 ат соответственно. Следует
при этом отметить, что при давлениях 60—80 ат добавление 0,1 % бн
хромата приводило к уменьшению скорости горения ПА. Уменьшение
скорости горения и пульсирующий характер горения наблюдались при
50 ат при добавлении 10% КзСгаО?. Заметим попутно, что уменьшение
скорости горения ПА в области давлений 80—140 ат при введении в
него малых количеств (до 0,8%) иона К+ имело место в опытах Боггса
и др. [4], следует подчеркнуть при этом, что в области давлений 30—
70 ат добавление 0,05—0,13% К" приводило к увеличению скорости
горения *.
Влияние содержания добавки на скорость горения ПА было изу-
чено также на примере хлорида меди при давлениях 50—100 ат. Соеди-
нения меди так эффективны в качестве катализаторов горения ПА, что
* Мы не будем останавливаться здесь на более детальном обсуждении этих ните-,
ресных результатов, так как они заслуживают отдельного рассмотрения.
100
В Их присутствии он гбрйт, согласно опытам Шидловского и оотр. (5, 61,
даже при атмосферном давлении. При увеличении количества CuCl от
1 до 5% скорость горения возрастала, однако, уже для 5 н 7% CuCl в
, области давлений 1—10 ат скорости горения практически совпадают
хотя при более высоких давлениях ПА с 7% CuCl горел быстрее.
В дальнейших опытах все изученные добавки применялись в коли-
честве 5% по весу, а размер их частиц был менее 100 мкм.
В табл. 2 представлены характеристики горения ПА с изученными
добавками, а на рис. 3 в виде диаграммы представлена зависимость ко
эффициента катализа от начальной скорости горения (Uo). На этой диа-
грамме жирные вертикальные линии относятся к равным скоростям горе-
ния, а стрелками указаны давления, при которых эти скорости дости
гаются. Прежде всего ^следует отметить, что эта диаграмма ограничи-
вает верхний и нижний прф(елы проявления каталитического действия
двадцати трех изученных нами неорганических добавок.
Таблица 2
Характеристики горения перхлората аммония с неорганическими катализаторами
Добавка, 5% 50 К при следующих p. am R Интер- вал p, am
150 300 600 800 1000
Си 0,8 0.8 4,0 4,5 2.9 1.8 0.17 0,006 0,49 1.45 50-400 400—JOCO
CuaCr^U? * jiHaU 1,6 2.1 12,2 11.1 5,3 3.0 0,08 0,815 30-1000
CuCl 1.9 1,8 9.4 8.6 3,7 2,1 0,17 0,66 1—100(1
CuBWO4 0.8 1,0 5.3 4.6 2,7 1.9 0,063 0,000031 0.705 1,54 10—500 50C-. 1000
КаСГаО? 1.3 1.9 9,0 7.7 3,4 2,o| i>,011 1.25 20-100
KaCrO# 1.5 0,142 0,67 100—1000
BlitCrjOjJa 1.5 7.0 7.2 3.4 1.8 0,24 O.03i 0.521 0,878 50 - 400 400—1000
(Mll<)2Cr2O; 1.3 1.1 4.7 4,5 2,8 1.7 0.22 0.0015 0,5(16 1,315 30 -200 400-1000
Li^Cr^Or 1,7 1.8 JI.4 7 .3 3.7 2.Ц 0.142 0,65 50—500
CsCrO4 1.-5 1.1 1.6 8.4 6,1 3.4 2,Of 0.025 0,93 5< 0-1000
BaCrOt 6.9 6.5 c,7 2.1 0.(53 0 JI? 0,79 0.983 50—500 500-1000
CdCrOi 1.5 I. 5 7.6 6 1 3,2 1.8 0.17 0,63 50—1000
PbCrO, 1,9 1.5 9,0 8.6 4.3 2.3 0.165 0.031- 0,64 0,923 50—400 400-1000
CoCrO, — 1.4 7.4 6,6 3.1 1,8 0.08 0,74 100-1000
ZnCrO* 1.5 1.6 8,1 7.5 3.7 2,0 0,142 0.025 0,65 0,93 50—500 500—1000
CfjOs 1,2 1.6 6,6 6.2 3.2 1,8 0,044 0.014 0,937 I-.834 50-100 300—1000
CrCla 1.2 1.0 4,9 4.9 2.5 1.6 O.k 0,0054 0,506 1,115 30-300 300—1000
VjOa 0.9 0,9 4.3 4.6 2,5 1,6 0,068 0,0095 0,70 1.04 50-100 200—K00
Co(NO3)a • 6H2O 0.6 4.0 4.6 2,5 1.4 0,0095 1.02 00-1000
KMnO« 0.4 0.7 4.1 4.6 2,3 1,5 0.05 0.007 0,693 1,068 20-300 300-1000
. ;j. Fe . 0,9 1.0 3.1 4.4 2,6 1.8 0.05S 0.0006 0,764 1.44 1-140 эон—1000
FfiaOg 0.8 1.1 6,2 5.3 2.7 1.6 0,034 0,85 20-1000
FeCls 0,8 1,0 5.9 5.1 2.3 1.5 0.066 0,725 50—1000
101
' Как. было сказано нами ранее [3], кривую U(p) для Г1А можно
разбить на три характерные области: от 50 до 150 ат — область устой-
чивого горения с линейной зависимостью U(р); от 150 до 500 от — пере-
ходная область, характеризующаяся неустойчивым, пульсирующим горе-
нием, и, наконец, от 500 до 1000 ат — область, где скорость горения рас
тет с давлением по закону, в котором v значительно больше 1. При этом
реакция, определяющая скорость горения, протекает в конденсирован-
ной фазе в первой области и в газовой —при давлениях выше 500 ат
[7], нестабильное горение в переходной области обусловлено автоинги-
бироваиием процесса горения водой [8] и взаимодействием различных
стадий горения.
Рис. 2. Влияние содержания
К1Сг(От (числа у кривых, %)
иа зависимость скорости горе-
ния перхлората аммония от
давления (логарифмические
координаты)
Z/e > rJct^UK
Рис. 3. Зависимость каталити-
ческой эффективности от на-
чальной скорости перхлората
аммония с неорганически мн
катализаторами. 1—CujCrjO?-
-2HjO; 2- IJzCrrOr; 5-
Co(NO»)a-6HaO; 4 —KMnOt;
5—Fe
Если исключить из рассмотрения часть переходной области от 169
до 300 ат, в которой значение v отрицательно, тогда полученную диа-
SpMMy можно разбить на две области по давлению: от 50 до 150 ат,
je наблюдается слабый рост каталитической эффективности [К] с уве-
: лмчением начальной скорости горения и 300—1000 ат, в которой имеет
iMecro падение К с ростом Uo.
, В’области давлений 50—150 аг в интервале скоростей горения от
14 Др 2,4 г!см'г-с верхняя границ^ катализа описывается уравнением:
Sj = Т.ббив0’3®7, а нижняя — К = O^Uo9’*®. В области давлений 500 —
1000 ат в интервале скоростей от 1,3 до 7,5 г!с.м?-с уравнение катализа
йа верхнем пределе К = 13,6U0_0,76, а на нижнем — К. = 21,5Uo~o>e3,
Как в первой, так и во второй областях верхний предел проявления
катализа ограничен дигндратом бихромата меди, а нижний—перманга-
натом калия, азотнокислым кобальтом и железом. Разница между наи-
более и наименее эффективными добавками в первой области составляет
4 между Li-jC^O? и КМпО< при 50 аг и 3,7 при 150 аг между дигндратом
бихромата меди и азотнокислым кобальтом. Во второй области наиболее
..(CujCrsO?• 2Н2О) и наименее (Fe) эффективные добавки различаются
в 3,4 раза при 300 ат и в 2 раза при 1000 ат.
Ранее было установлено [9], что каталитические эффекты наиболее
ре?ко проявляются вблизи предельных условий горения н для веществ,
1ГЛ
$ способных Йёз катализаторов к горению или для медленйюгорящях
Вистем. Анализ результатов, приведенных на диаграмме рис. 3 показы-
Кает, что в случае ПА сама по себе скорость горения не является опре-
Кёляющей для эффективности каталитического действия. Большую роль
[проявлении максимальной эффективности добавки играет давление,
^Ьн котором начальная скорость достигается. Так, при 50 и 500, 150 и
ю ат ПА горит с одинаковой скоростью (пунктирные вертикальные
каин на диаграмме), однако при 50 ат бихромат лития увеличивал
Июрость горения в 2 раза, а при 600 ат Си2Сг2О7 • 2Н2О— в II раз. Если
^Еять один и тот же катализатор — дигидрат бихромата меди, то при
Юаг К было равно 2,1, а при 720 аг—8,2. Это свидетельствует о том,
важна не сама по себе величина скоростй горения, а те ведущие
^Кадции, за счет протекания которых она достигается. В области дав-
^Ккий 50—150 ат эти реакции протекают в конденсированной фазе и
^Крапление к ПА наиболее эффективных катализаторов — бихроматоз
Ктия, калия и меди приводило к уменьшению В н увеличению v в
мвнении горения, т. е. ведущая реакция в этом случае переходила из
Ишенси ров энной фазы в газовую. Подтверждением этого вывода могут
^вдкить также результаты опытов по влиянию плотности на скорость
фения ПА с 5% 1^Сг2О7. Как видно из рис. 1, скорость горения ката
Визированного ПА от плотности не зависела, т. е. реакция, опредёляю-
Кщя скорость горения протекала в газовой фазе, в то время как для
Йстого ПА картина была обратной [7]. В области высоких давлений наи
tutee эффективные добавки, напротив, уменьшали долю реакций, про-
Н^КИющих в газовой фазе, так, днгидрат бихромата меди снижал зиаче-
»е у-^в 3 раза (с 2,36 до 0,815).
К* Один из возможных путей увеличения скорости; горения катализа-
фами, особенно в переходной области (160—500 ат) — ускорение рзс-
дда хлорной кислоты [10], а также моно- и полигидратов ее. Влияние
Каталитических добавок на термический распад перхлорной кислоты з
арах было изучено Шоймоши и его сотр. [11}< наиболее эффективным
анализатором была окись хрома, особенно, если к ней добавлялась для
Кеиления электропроводности окись цинка. Как видно из табл. 2, при
фении окись хрома менее эффективна, чем соединения шестивалентного
дома. Вопрос об отсутствии корреляции между действием добавок на
гемическое разложение и горение ПА был рассмотрен ками ранее [2].
К^тому же выводу пришли позже Шоймоши и его1 сотр. [12]. Отсутствие
ИЙрреляции обусловлено тем, что при горении ведущими являются, по-
ЙХимому, другие реакции, к тому же следует учитывать и условия их
Нфотекания, в частности, влияние давления на них [13].
Что касается химического аспекта влияния катализаторов, то уско
ревие ими процесса горения обусловлено тем, что они могут вступать
Каю взаимодействие как с самим ПА, так и с продуктами его распада с
Образованием в качестве активных промежуточных соединений, в част-
ВОсти, в случае меди ее соединений с аммиаком и окислами хлора или
долее реакционноспособных перхлоратов, например, перхлората меди
Так, эффективность CuCl как катализатора горения ПА при атмосфер^
Гном давлении объясняется Шидловским [14] образованием малоустой-
fчнвого перхлората меди. По данным Горбунова и др. [15], скорость горе-
к ния перхлората тетрамнна меди [Cu(NH3)J(C10<)2 при 100 ат состав-
f-ляла 27 г.!см2-с, чистый ПА горит при этом давлении в 23 раза мед-
Г леннее.
103
j
Хотя полученные экспериментальные дайные не позволяют пока сЛё-
лать заключения о механизме влияния изученных добавок на химиче-
ские реакции, протекающие при горении, однако они показывают, что
регулирование скорости горения с помощью катализаторов может быть
осуществлено в широких пределах,
Для выяснения механизма влияния катализаторов при горении ВВ
целесообразно было бы изучать кинетику окисления углерода, водо-
рода, аммиака и простейших углеводородов различными окислами азот i
и хлрра в чистом виде и в присутствии катализаторов. Необходимо
также определить влияние катализаторов на состав продуктов горения.
К сожалению, такие данные пока практически отсутствуют,
Для низких давлений — до 140 ат имеются данные Леви и Фрид-
мана [16] по влиянию хромита меди на состав продуктов горения ПА
Добавление хромита меди не влияло на содержание NO2 и резко умень-
шало количество NjO, таким образом, катализатор ускорял, по их мне-
нию, превращение N2O в азот, т- е. скорость горения лимитировалась
газофазными реакциями. Однако прежде, чем в продуктах горения по-
явятся окислы азота, аммиак, из которого они получаются, должен
пройти различные стадии окисления и первые из этих стадий проте-
кают, по-иашему мнению, в конденсированной фазе, где их и ускоряют
катализаторы.
I. Friedman R„ Nugent R. G., R u m b e 1 К. E. Scurlock A. C, Vi Sympo-
sium (International) on Combustion. Reinhold. 1957, p. 612.
2. Г л a 3 к о в а А, П, ФГВ, 1966, Ni I, c, 59.
3. ' Глазкова А. П. ПМТФ, 1963, № 5, c. 121.
4. Boggs T. L., Petersen E. E., Watt D. M. J. Comb, and Flame 1972, v. 19
n 1, p. 131.
5. Шидловский А. А, Шмагнн Л. Ф. Нзв. вузов, 1, p. 131. Хим. и хим. тех-
нология, 1962, т. 5, № 4, с. 529.
6. Шидловский А. А., Шмагии Л. Ф., Буланова В. В. Там же. 1964 №5
с, 862,
7. GlaskovaA. Р, Explosivstoffe, 1970, № 4, s. 89,
8, Г л д з к о в а А. П. ФГВ, 1968, № 3, с. 314.
9. Глазкова А. П., Андреев О, К., 3-й Всесоюзный симпозиум по горению и
взрыву. М., «Наука», 1972, с. 78.
10. J а с о b s Р. W. М., Russel-Jones А, XI Symposium (International) on Combu-
stion. The Combustion Institute, 1967, p, 447.
11. Solymosi F,, Borcsok S„ Lazar E, Comb, and Flame, 1968, 12, n. 4, p. 398.
12. SOlymosi F., Gera L,, Borcsok S, XIII Symposium (International) on
Combustion. The Combustion Institute 1971.
13. Беляев А. Ф., Кондрашкой Ю. A., JI у к а nr e и я Г, В., Парфенов A, K.
Научно-технические проблемы горения н взрыва. 1965, вып. 1,25.
14. Ш и д л о в с к и й А. А. Сб. «Теория взрывчатых веществ». М., «Высшая школа».
1967, с, 364.
15. Горбунов В. В., III м а г и н Л, ф. ФГВ, 1972, № 4, с. 523.
16. Levy J. В., Friedman R, VI] I Symposium (International) on Combustion
Williams and Wilkins, 1962, p. 663.
УДК 662.221+536.4t
О МЕХАНИЗМЕ ВЛИЯНИЯ ГАЛОГЕНИДОВ
ЩЕЛОЧНЫХ МЕТАЛЛОВ НА ГОРЕНИЕ НИТРАТА
АММОНИЯ И АММОНИТА
А. П. Глазкова, О. К. Андреев
Изучение влияния катализаторов на горение нитрата аммония при
атмосферном давлении [I, 2], в условиях бомбы переменного давление
[3—4] и постоянного давления [5] показало, что наиболее эффективными
добавками являются соли шестивалентного хрома и галогениды щелоч-
ных металлов. Они же наиболее эффективны как катализаторы, и для
смесей аммиачной селитры с углем [4, 6], тротилом [5] и нитрогуанидином
[7]. При горении аммонита 80/20 было установлено также, что фторид
калия был более эффективен, чем фторид натрия [8], а для солей калия,
например, эффективность зависела также и от природы кислотного ос-
татка [9], в частности, наиболее эффективным катализатором горения
аммонита был бихромат калия [5].
В настоящей работе проведено более детальное исследование алия
ния щелочных солей на горение нитрата аммония и его стехиометриче-
скую смесь с тротилом. Изучались соли галоидоводородных кислот, ща-
велевой, угольной и азотной. Кроме того, в качестве добавок были при-
менены не изученные ранее соединения кобальта, цезия, свинца и железа
Опыты проводились в бомбе постоянного давления на 1000 ат по фото-
графической методике [10]. За критерий эффективности действия изучае-
мых добавок принят коэффициент катализа — К. равный отношению мас-
совых скоростей горения катализированного и чистого составов. По-
скольку чистая селитра не способна к говению, результаты сравнивались
со скоростью горения селитры ЖВ [5]. Добавки вводились в количестве
пяти весовых процентов, размер частиц всех компонентов был менее
100 мкм,
На рис. 1 представлены данные К(р) для нитрата аммония с солями
натрия; на кривых имеется максимум в области давлений -^500 ат, з
ряду солей галоидоводородных кислот наблюдается падение эффектив-
ности действия добавок от хлорида к бромиду и фториду натрия. Мак-
симальное увеличение скорости горения при 500 ат составляло 2,3. Окса-
лат натрия оказывал на горение селитры примерно такое же влияние,
как и фторид натрия.
105
> S
Аналогичная последовательность в эффективности катализаторов
наблюдалась и для солей калия, как это видно из рис. 2. Эффектив-
ность оксалата, карбоната и нитрата калия в области давлений до 500 аг
была такой же, как и KF, а при более высоких давлениях оксалат ка-
лия несколько превосходил по своей эффективности фторид и нитрат;
последний был более эффективен, чем фторид, но меиее эффективен
чем оксалат. Большая эффективность оксалата калия, возможно, обус-
ловлена тем, что в области высоких давлений происходит сгорание угле-
рода, входящего в молекулу добавки.
Рис.. I. Изменение ката-
y^umt ческой эффектирио-
' стя. 'с давлением для
-*ЙАтрата аммония с со-
-ММмн натрия, 1—NaCl
Й1, 2-NaBr; 3-NaF;
Рис. 2 Изменение ката-
литической эффективно-
сти с давлением для
нитрата аммония с со-
лями калия, 1—KCi:
2—КВг; FKF, 4-
KNO3; 5—KsCjO,
Рис. 3. Изменение ката-
литической эффектив-
ности с давлением для
нитрата аммония с со-
лями кобальта, цезия
и железа. /--СоСгОъ
2 -РеС19: 3-CsCrOc
4-CoCIj-SHtO
Йд рис. 3 представлены данные по изменению К в зависимости о.
давления для нитрата аммония с хлоридом н хроматом кобальта, хло-
рмдом железа и хроматом цезия. Интересно отметить, что для селитры
хлоридом железа кривая К(р) имеет иной характер, чем во всех ос
тальных случаях: наблюдается падение К с ростом давления. Сопостае-
Л^нйе влияния хлорида и хромата кобальта показывает, что хромат зна-
чительно более эффективен, возможно, что это обусловлено тормозящим
действием кристаллизационной воды в хлориде. Если сравнить влияние
длориДа и хромата калия [5] на горение селитры, то в области давлений
до QtyQ ат большей эффективностью обладал хлорид калия, а пр г
р > 800 ат— хромат.
Эффективность хромата цезия наименьшая и близка к таковой
фторидов натрия и калия. В табл. 1 приведены характеристики горения
для нитрата аммония с изученными добавками: значения предельного
давления (рир)—давление, при котором начинается устойчивое горе-
нде. селитры в условиях наших опытов (при диаметре образца 7 мм),
а также значения В и v в уравнении горения U„= Вр4 и указан интер-
вал Давлений, в котором оно выполняется.
Селитра с хроматом цезия начинает гореть уже при атмосферном
давлении, а с нитратом калия и хлоридом железа только при 100 ат.
1U
Таблица!
Характеристики горение NH^NO, с катализаторами
Добавка. 5 % Pnp. am В Интервал p. am
NaCl 12 0,015 0,90 10-200
0,155 0.415 200-1000
NaBr 50 0,057 0,538 50—1000
NaF 50 0,005 0,92 50- 30'i
0,049 0,524 300-1000
50 0,078 0,446 ICO-IOOO
KC1 —25 0,03 0,736 25-300
0,65 0.196 300-10C0
KBr 50 0,< 5 0,564 50—5/0
0.181 0,35 500-1000
KF 50 0,014. 0,735 50-300
0.082 0,437 3f 0-1000
KsCiOt 50 0,029 0,619 50—400
0,0122 0,742 400-1000
KNOS 100 0,0162 0.69 100-1000
CoCk. 6HjO 50 0.019 0.68 50-1000
СоСгОч 50 0,012 0.81 50-5C0
0,15 0.45 20-100
CsCrO4 1 0,072 0,476 200-1000
0.009 0,966 100--200
FeCi3 100 0,10 0,46 250—1000
Селитра ЖВ < 230 0,033 0.56 250-1000
С максимальной скоростью горела селитра с хлоридами натрия и калия,
с минимальной — с фторидами щелочных металлов, а также с оксала-
тами натрия и калня, с хлоридом кобалйта и хроматом цезия. Следует
при этом отметить, что горение селитры с нитратом и оксалатом калия
при давлениях 150—250 ат имело пульсирующий характер, пульсирую-
щее горение наблюдалось также и для селитры с фторидом натрия.
Изменение каталитической эффективности добавок с давлением для
аммонита 80/20 представлено на рис. 4, из которого видно, что характер
кривых К(р) отличается от таковых для чи-
стого нитрата аммония. Наблюдается падение
К с ростом давления, причем оно выражено
более резко в области давлений до 800 ат. Га-
логениды щелочных металлов при горении ам-
монита оказывали примерно одинаковое дей-
ствие, лишь NaBr был более эффективен в об-
ласти давлений до 500 ат. Углекислый калий и
Хромат свиица были значительно менее эффек-
тивны, чем галогениды, следует отметить, что
и прн горении чистой селитры эффективность
РЬСгОч была невелика [5], Было изучено так-
Рис. 4. Изменение ката-
литической эффективно-
сти с давлением дли
аммонита 30/20 с ката-
лизаторами, / — NaBr;
2—КВ г. 3—КС1; 4—ком-
>бинированный катализа-
тор; 5—PbCrOj; 6—
К1СО»
’ же влияние комбинированного катализатора
на горение аммонита, этот катализатор состо-
ял из равных весовых частей окнслов кобвль-
та.медн, железа, кремния и бихромата ка-
лия. Как видно из рис. 4, он был более эф-
фективен, чем карбонат калия и хромат свин-
ца лишь при давлениях до 500 ат. Если
MJ7
Провести сравнение с данными, полученными ранее [5], то все добавки,
изученные для аммонита в данной работе, уступают по своей эффектив-
ности бнхромату калия.
Характеристики горения аммонита 80/20 с изученными добавками
приведены в табл. 2,
• Таблица 2
Характеристики горения аммонита 80/20 с катализаторами
Добавка, б % рпр, ат В Интервал pt ат
NaBr 70 0.032 0,77 100-1000
КВт 75 0,625 0.80 80—1000
КС! 25—30 0,026 0,79 25-1000
КаСОэ 50 0,011 0,91 50—1000
РЬСгО, 60 0.0175 0,85 100-950
Комбинированный
катализатор 75 0,0125 0,893 75—1000
Аммонит 80/20 [10] 150 0.023 |,ю 150-1000
Из табл, 2 следует, что наиболее сильное понижение нижнего пре-
дела горения по давлению (в 5 раз) наблюдалось при добавлении к
аммониту хлорида калия, углекислый калий и хромат свинца снижали
рпр до 50 и 60 ат соответственно, а остальные добавки примерно вдвое.
. , Механизм влияния добавок типа хроматов щелочных металлов на
горение нитрата аммония уже обсуждался нами ранее [4], Рассмотрим
возможный механизм ускорения горения нитрата аммония и смесей на
его основе галогенидами щелочных металлов, например NaCl.
Первичной ступенью распада нитрата аммония и смесей на его осно-
ве при горении является, как уже предполагалось нами ранее, диссоциа-
ция: NH<NO3 т* NH3 + HNO3. Эта реакция имеет место, по-видимому, в
расплавленном слое конденсированной фазы, причем расплав должен
быть кислым вследствие лучшей растворимости азотной кислоты и боль-
шей летучести аммиака. Поскольку хлориды хорошо растворяются в рас-
плаве селитры, то в конденсированной фазе может протекать реакция:
HNO3 + NaCl = NaNO3 НС1. Причем эта реакция более вероятна,
чем реакция NaCl с аммиаком с образованием NH<C1, как это предпо-
лагалось Розианом [11] в случае термического распада, поскольку ам-
миак, вследствие, большей летучести, скорее будет находиться в газовой
фазе. Затемйтм, кстати, что наше предположение находится в соответствии
с экспериментальными данными Ван деи Берга и сотр, [12], когда спек-
троскопически было установлено, что при добавлении к селитре хлори-
дов немедленно наблюдалось образование HCJ.
Образовавшийся нитрат натрия (или калия и т, п.) быстрее окис-
ляет аммиак, чем азотная кислота. Значительно большую эффектив-
ность KNO3 как окислителя можно продемонстрировать следующим со-
поставлением: при атмосферном давлении стехиометрическая смесь
нитрата аммония с углем горит со скоростью 0,02 г/сж2-сек, а бессерный
порох, состоящий из 85% KNO3 и 15% угля, горит, по данным Беляева
и сотр, [13], со скоростью 0,35 г/см2-сек, т. е. в 18 раз большей, В даль-
нейшем образовавшийся нитрат может распадаться предположительно
МР
по реакции: 2NaNOa->Na2O + 2NO2 + 0,5О2 и тогда регенерация ката-
лизатора может протекать по реакции
Na2O + 2НС1 = 2NaCl + Н2О.
Образовавшаяся при распаде нитрата двуокись азота реагирует
затем с аммиаком, восстанавливаясь в конечном итоге до элементарного
азота. Очень важно при этом, что бы образовывались именно NO2 или
N2O, а не NO, поскольку, как было установлено Уайтом и др. [14], дву-
окись азота значительно легче окисляет аммиак, чем окись. Энергия
активации окисления аммиака составляет 27 500 кал/моль для NO2 [14]
и 54 700 кал/моль— при окислении его NO [15]. Таким образом, роль
катализаторов при горении нитрата аммония и смесей на его основе
заключается в ускорении реакции окисления аммиака.
1. Т а у 1 о г J. Ind. Chemist 1948, 24, р. 289.
2. Шид л о вский А. А. Изв. вузов. Хим. и хим. технология, 1958, № 3, с. 105.
3. Андреев К. К., Глазкова А. П. ДАН СССР, 1952, 86, № 4, с. 801.
4. Г л а з к о в а А. П. Автореф. канд. днсс, МХТИ им. Д. И. Менделеева. М., 1952.
5. G1 a s к о v а А. Р. Explosifs, 1966, и. 1, s. 5.
6. Андреев К. К-, Глазкова А.П. Сб. «Теория взрывчатых веществ»,М., «Выс-
шая школа», 1967, с. 314.
7. Г л а з к о в а А. П. ФГВ, 1971, № 2, с. 211.
8. Глазкова А. П., А н д р е е в О. К- ФГВ, 1969, № 3, с. 434.
9. Glaskova А. Р., Andreev О. К. Chimie et Industrie. Genie chimique, 1970,
103, n. 14, 1735.
10. Глазкова A. П., Терешкин И. А. ЖФХ, 1961, т. 35, №7, с. 1622.
11 Р о з м а и Б. Ю. ЖНХ, 1961, т. 6, вып. 4, с. 783.
12, Корег J. Н., Jansen О- G., van den Berg Р. J. Explosivstoffe, 1970, п. 8
s. 181.
13. Беляев А. Ф., M а знев С. Ф. ДАН СССР, 1960, т. 131, с. 887.
14 Rosser W. A., In, Wise Н. J. Chem. Phys. 1956, v. 26, п. 5, р. 1078.
15. Wise Н„F resh М. Е. J. Chem. Phys. 1954, v. 22, n.,8, p. 1463.
УДК 662,220
О ВЛИЯНИИ ДИСПЕРСНОСТИ МАГНИЯ
г НА СКОРОСТЬ ГОРЕНИЯ ЕГО СМЕСЕЙ
С СОЛЯМН-ОКИСЛИТЕЛЯМИ
” В, В. Горбунов, В. А. Лобанов ли'?,'--'
Среди параметров, которые оказывают влияние на скорость горе-
ния конденсированных смесей, наиболее важным в практическом и тео-
ретическом отношениях является дисперсность компонентов £1, 2], Этот
/Параметр позволяет регулировать скорость горения смесей без измене-
ния их энергетических характеристик, Теоретический интерес к зависи-
мости скорости горения от дисперсности компонентов обусловлен воз-
можностью оценки влияния диффузионных факторов на горение смесей.
В настоящей работе изучалось влияние дисперсности магния иа го-
рение стехиометрических смесей его с солями-окислителями, главным
образом, перхлоратом, нитратом, нитритом, сульфатом и карбонатом
натрия. Кроме этйх солей мы использовали перхлораты, нитраты, нит-
риты калия и бария. Расширение круга солей-окислителей было пред-
принято для выяснения влияния их физико-химических свойств (темпе-
ратур плавления и разложения, состава продуктов разложения) на ха-
рактер зависимости скорости горения смесей от дисперсности магния.
Применялись полидисперсные порошки магния ео средним размером
частиц 4; 35 и 160 мкм* и узкие фракции 63—100, 100—125, 125—160
л 160—200 мкм, полученные рассевом поли дисперсных порошков. Соли-
окислители <х, ч,» или «ч. д. а» имели размер частиц 100—160 мкм. Смеси
запрессовывались в плексигласовые трубочки внутренним диаметром
7 мм под давлением 2500- 3000 кгс!см* до относительной плотности
0,65—0,95.
Опыты проводились в бомбе постоянного давления, в атмосфере
азота. Зависимость скорости горения смесей от давления определялась
в интервале 0—-100 ати, влияние размеров частиц магния изучалось
при 60 ати. Заряд воспламенялся электроспиралью с промежуточным
воспламенителем (0,2 г) слабоуплотиенной смеси того же состава. Пере-
мещение фронта горения фотографировалось на движущуюся пленку
барабанного фоторегистра.
* Средний размер частиц полидисперскых порошков магния 4 мкм и 35 мкм
определен по воздухопроницаемости на приборе ПСХ-2; 160 мкм — ситовым анализом.
НО
Свойства применявшихся солей-окислителей, составы и теплоты го-
рения их смесей с магнием приведены в таблице.
Формула соли Температура, °C [3—6] Содержа- ине магния в смеси, вес, % Теплота горения смеси. ккал! г
плавления соли „интеясннио- го* разложе- ния солн
NaC10< 470 530 44 2,6
KC1O4 590 620 41 2,5
Ba (CIO,) 2 470 500 37 2,2
NaNO, 330 520(750) 42 2,2
KNO, 3i0 630(800) 38 1.8
Ba(NOj)i 590 605(661) 32 1,6
NaNOs 270 -ТОО 35 1 2,0
KNO» 390 —700 30 1.4
Ba (NO»)» 270 409 24 1,3
NajSO# 890 . 41 1.4
Na»CO3 850 --- 32 0,75
Примечание:
1. Составы стехиометрических смесей рассчитывали, исходя из предположения,
чю нитраты, нитриты и карбонаты восстанавливаются при горении до оксидов, пер-
хлораты до хлоридов, сульфаты ДО сульфидов соответствующих металлов.
2. В графе «температура интенсивного разложения окислителя» для нитратов
первая цифра отвечает выделению кислорода, цифра в скобках, интенсивному выделе-
нию нитроаных газов.
На рис. 1 и 2 показгЛы зависимости скорости горения от давления
для смесей солей натрия с мелкодисперсным (4 мкм) и крупнодисперс-
ным (160 лкл) магнием. Среди смесей с мелким магнием наиболее
быстро горит нитритная, скорость горения которой в интервале давлений
20—100 ати почти в 1,5 раза выше, чем у нитратной. Перхлоратная
смесь горит медленнее нитратной, скорость гдрения сульфатной и кар-
бонатной смесей остается постоянной при всех изученных давлениях.
Рис. 2, Влияние давления на ско-
рость горения смесей солей нат-
рия с крупнодисперсным магнием
(160 мкм); !—NaNOa; 2—NaNO»;
4—NaCIO,; 5—Na*COs
Рнс. L Влияя не давления на
скорость горения смесей солей
натрия с мелкодисперсным
магнием (4 мкм)-. /—NaNOii
Ж
При использовании крупного магния (160 лпслг) значительно воз-
растает скорость горения нитратной смеси и зависимость ее от давления.
Скорости горения нитритной и перхлоратной смесей, напротив, уменьша-
ются почти в два раза. Сульфатные и карбонатные смеси с крупным маг-
нием горят с той же скоростью, что и с мелким магнием.
Наблюдавшееся разнообразное влияние полидисперсных порошков
магния на скорость горения его смесей с солями натрия потребовало бо-
лее детального изучения вопроса с использованием узких фракций по-
рошка магния и других солей-окислителей.
Для перхлоратных и нитратных смесей (рис. 3) наблюдалось слож-
ное влияние дисперсности магния с минимумом скорости горения при
размерах частиц 40—70 мкм и максимумом при 120—160 мкм. Характер
этой зависимости оставался одинаковым у смеси магния с NaClO+ при
20, 60 и 100 ати. Соотношение скоростей горения Стах/иш]Ппри 50 ати
для смесей магния с КС1О4 и NaCIO4 около 2,7, у нитратных смесей
2,0—2,7 и увеличивалось в ряду солей К, Na, Ва. Горение перхлоратных
и нитратных смесей в области минимума скорости сопровождалось ред-
кой пульсацией, которая ослабевала при увеличении размеров частиц
мапиия. ___ f
Рис. 3. Влияние размеров
частиц магния на скорость
горения. при 60 ати его
смесей с перхлоратами и
нитратами; I—КСЮ<; 2—
NaClQ,; 3-Ва(ЫОз)2; 4—
NaNOa; 5-КЫО3
Рис. 4. Влияние размеров
частиц магния на скорость
горения при 60 ати его
смесей с нитритами, суль-
фатами и карбонатами: /—
Ba(NO2)2. 2—NaNOj. 3—
KNOj, 4 -Na2SO4. 5—
Na2COa
Скорость горения смесей магния с KNO2 или NaNO2 уменьшалась с
увеличением размеров частиц магния (рис. 4). Напротив, для смеси с
Ba(NO2)2 наблюдались минимум и максимум скорости горения подобно
смесям с нитратами и перхлоратами. Скорость горения смесей магния
с Na2SO4 или Na2COa оставалась постоянной при всех изученных раз-
мерах частиц магния (4—180 мкм). Пульсации, характерные для пер-
хлоратных и нитратных смесей, отсутствовали при горении смесей маг-
ния с KNOa, NaNO2, Na2SO4 и Na2CO3.
По влиянию дисперсности магния на скорость горения его смесей
с изученными солями-окислителями, последние можно разделить иа две
группы. KNO2, NaNO2, Na2SO4 и Na2COs — скорость горения их смесей
монотонно убывает или остается постоянной при увеличении размеров
частиц магния; NaClO+, КСЮ4, Ва(СЮ4)2, NaNOa, KNO„ Ba(NO3)2 и
П2
Ba(NO2)2 — при горении смесей магния с этими солями наблюдаются
минимум и максимум скорости горения.
Сравнение температур «интенсивного» разложения солей (таблица),
показало, что соли первой группы разлагаются при температурах выше
точки плавления магния (660s). При горении смесей с этими солями
окисляется расплавленный магний и поэтому первоначальный размер
его частиц оказывает слабое влияние (или не влияет) на скорость
горения. «Интенсивное» разложение солей второй группы (перхлоратов
и нитрита бария) происходит при температурах ниже точки плавления
магния. Ниже последней идет также первая стадия разложения нитра-
тов, в ходе которой образуются нитриты и выделяется кислород. При
горении смесей солей второй группы с магнием твердые частицы послед-
него окисляются по поверхности газообразными продуктами разложе-
ния солей. Это обусловливает сложное влияние дисперсности магния
на горение смесей. Появление максимума скорости совпадает с ослаб-
лением пульсаций. Возлюжно эго происходит вследствие стабилизации
фронта' горения крупными частицами магния. При дальнейшем увели-
чении их размеров преобладает уменьшение поверхности контакта окис-
лителя и горючего, которое приводит к падению скорости горения.
РЕЗЮМЕ1. Влияние размеров частиц магния на скорость горения
его смесей с солями-окислителями существенно зависит от условий окис-
ления магния в процессе горения, возникающих в зависимости от соот-
ношения температур плавления магния и «интенсивного» разложения
окислителя.
]. Бахман Н. Н., Беляев А, Ф. Гетерогенное горение конденсированных систем,
№.. «Наука», 1967.
2. Ш и д л о в с к н й А, *А. Основы пиротехники, М, «Машиностроение», 1973.
3. Сарвер С. Химия ракетных топлив, М., «Мир», 1969,
4. Gordon S., Campbell С„ Analyt, Chem, 1955, 27, s, 1102,
5. Freetn a n E. S„ J. Phys. Chem,, 1956, v. 60, p. 1487.
6. Верят У, Д., Машнрев В, П. и др, Термодинамические свойства иеорга*
ничееких веществ, №., Атомиздат, 1965.
g Труди, вин, ее
УДК 662,3:535.46:541.128
ВЛИЯНИЕ НАЧАЛЬНОЙ ТЕМПЕРАТУРЫ И ДАВЛЕНИЯ
НА ЭФФЕКТИВНОСТЬ ДЕЙСТВИЯ КОМБИНИРОВАННОГО
КАТАЛИЗАТОРА ПРИ ГОРЕНИИ ПОРОХА Н
Л, П. Денисюк, В. М. Кувшинов, Н. П. Токарев, В. Г, Хубаев
Известно, что на скорость горения баллиститных порохов сильное
влияние оказывают различные соединения свинца (соли, окнслы) в
сочетании с соединениями меди [1] или с сажей [2]. Механизм действия
комбинированных добавок не изучен, причем о влиянии их на скорость
горения при различных условиях имеется недостаточное количество
данных.
Поэтому нами было изучено влияние РЬО2, СиО и их комбинации
иа скорость горения пороха типа Н в широком диапазоне давлений от
4 мм рт. ст. до 100 кгс!см^ и различной начальной температуре заряда,
Скорость горения при повышенном давлении определяли, сжигая об-
разцы (диаметр-7 мм, высота 20—25 мм)
в бомбе постоянного давления в атмосфере
азота, а при давлении ниже атмосферно-
го — в вакуумном шкафу объемом 40 л.
Подогрев образцов до заданной темпера
туры осуществляли с помощью электриче-
ского термостата. Эффективность действия
катализатора оценивали величиной Z =
= и/и0, где U и Uo — скорость горения
пороха с катализатором и без катализато
ра. Использовали мелкодисперсные РЬО
и СиО с размером частиц менее 1 мк.
' На рис. 1 показано влияние катали
Рис. 1, Влияние катализато-
ров на горение пороха Н в об-
ласти уверенно повышенных
Давлений: !—без катализатора;
2—1 % СиО; 3-0.5% РЬО.-,
4—1% PbOj; 5—0,5% РЬО,+
1% СиО; 5—1% РЬО,+ 1%
СиО
заторов на скорость горения пороха при
повышенном давлении и начальной тем
пературезаряда 20°С. Видно, что 1% оки-
си меди слабо увеличивает скорость горе-
ния. Влияние РЬО2 и комбинированного
катализатора в значительной мере зави-
сит от давления, а во втором случае и от соотношения РЬО2 и СиО
(табл, 1), Наиболее сильно зависимость скорости горения от давления
изменяется при введении 0,5% РЬО2 и 1 % СиО,
Таблица1
Влияние катализаторов на горение пороха Н в области
повышенных давлений (То=20°С)
Катализатор Величина Z при давлении, кге/сж’
2 5 10 20 30 40 60 90
0,5% РЬОц 1.0 . 1,1 1.3 1,6 1,8 1.6 1.2 1,1
1% РЬО5 1.0 1.2 1.4 2.0 1,7 1,4 1,0 1,0
0,5% РЬОг+1% СиО 1,0 1.2 1.7 2,7 2,6 2,3 1,1 1.0
1% PbOj+1% СиО 1,1 1.3 2,0 2 4 1.8 1.3 1.0 1,0
1% СиО 1.0 1 0 1.1 1,2 1.2 1.1 1,0 1.0
2,7
раза (при 20
кгс[см2)
Такой катализатор в
увеличивает
ско-
рость гореийя, а в области давлений 2*0—60 кгс]см2 коренным образом
меняет зависимость ее от давления (иа кривой имеется участок паде-
ния скорости горения). -
Однако прн повышении начальной температуры заряда эффектив-
ность действия катализаторов при горении пороха в области умеренно
повышенных давлений падает, что видно из сравнения данных, пред-
ставленных в тгбл. 1 и 2,
Таблица 2
Влияние катализаторов на горение пороха Н и области
повышенных давлений (Т° » 120°)
Катализатор Величина Z при давлении, кгс^см1
1 20 40 60
0,5% РЬОа 1.0 1,4 1,3 1.2
1% PbOi 1.0 1,4 1,3 1.2
0,5% PbOi+1% CuO 1.0 1.5 1.2 1,15
1% PbOj+1% CuO 1.0 1,3 1,1 1.1
1% CuO 1,0 1.1 1,05 1,05
Смесь 0,5% РЬОг+1%
скорость горения уже
СпО увеличивает скорость горения уже
только в 1,5 раза (при Р=20 кгс/см2), остальные катализаторы действу-
ют еще слабее.
На ряс, 2 показано влияние катвлиэаторов на горение пороха в
вакууме при начальной температуре заряда 120°. При этой температуре
в интервале давлений 10—160 мм рт, ст, (при давлении меньшем
10 жл рт, ст, образцы в стеклянных трубочках не горели) свинцовый
катализатор действует с высокой эффективностью. Например, 1% РЬОа
увеличивает скорость гбрения пороха в 2 раза при Р = 10 мм рт. ст.
. (табл, 3). Обращает на себя: внимание тот факт, что в вакууме СиО не
только не усиливает, ио даже-несколько ослабляет действие РЬО2. Так,
образец, цодерзкащий ! % РЬО2 + 1% СиО, горйт медленнее, чем образец
я*
U 115
с ! % РЬО2. Еще меньшее влияние на скорость горения пороха оказы-
вают совместно введенные 0,5% РЬО2 и 1% СпО. Окись меди, кроме
того, повышает давление, начи-
ная с которого порох горит
устойчиво.
Рис. 2, Влияние катализаторов на
горение пороха Н в области давле-
ний, ниже атмосферного; 7—без ка-
тализатора; 2—1% СпО; 3—0,5%
РЬО»; 4—1% PbOa+1% CuO; 5-
0,5% PbOj+1% CuO; 6 — 1% PbO3
Таблица 3
Влияние катализаторов на скорость горения порока Н
при давлении ниже атмосферного н начальной температуре заряда 120’С
Катализатор Величина Z при давлении, мм рт.
10 20 50 100 150 300 500
0,5% РЬО2 1,5 1,6 2,0 1.8 1,4 1.0 1,0
1% РЬО2 2.0 2.0 1.9 1,7 1,4 1.1 1,0
, 1% PbOi+1% CuO не горит 1,6 1.8 1.6 1,4 1.0 1.0
0,5% PbO2+l% CuO Я £ гор И т 1,4 1,1 1.0 1,0
1% CuO н е гор н т 1.5 U 0,9 0,9
При более низкой начальной температуре заряда исследуемые до-
бавки также оказывают влияние на горение пороха Н. Так, прн 85J
устойчивое горение образцов с 0,5 и f% РЬО2 .наблюдается при давле-
нии 150 мм рт. ст., а образцы без добавок горят лишь с 500 мм рт. ст.
В интервале 500—750 мм рт. ст. в присутствии двуокиси свинца новы
шается скорость горения пороха и уменьшается зависимость ее от
давления (табл. 4).
Таблица 4
Влияние катвлнзлтеров на скорость горения пороха Н
при да зленин ниже атмосферного н начальной
температуре 85°С
Образец U, мм/сек при давлении, мм рт. ст. Z при давлении мм рт, ст*
150 250 500 750 250 экстра полицией 500 750
Бед добавок не горит не горит 1.0 1,3
0,65 0,7 0.9 1,05 1.3 1.3 1,3 1.3 1.5 1.8 1.3 1.3 1.0 1.0
1% СпО не горит 0,8 1.1 1,3 1.3 1,1 1.0
0,5% PbOj+1% CuO 0,6 0,8 1.2 1,3 1.3 L2 •1.0
1% PbOi+1% СцО 9.65 0,9 1,2 1,3. 1.5 U2 1,0
Cbrwa jWTx85° (н отличие от 120°) тоже повышает устой-
чивость горёййй пороха, но меньше, чем РЬО2. Образцы с 1% CuO на-
чинают гореть при давлении 250 мм рт. ст., но на скорость горения поро-
ха окись меди практически не влияет.
Эффективность действия РЬО2 при совместном применении ее с
CuO несколько снижается, как это наблюдалось и при То = 120°.
Можно отметить, что если при начальной температуре 120° катали-
затор уже.не действует при давлении 300 мм рт. ст. (табл. 3), то при
начальной температуре 85ьон действует еще при давлении 500 ллгрт. ст,
Как видно из полученных данных, эффективность действия катали-
заторов очень сильно зависит от условий горения (начальной темпе-
ратуры и давления). ’
При начальной температуре 20° существенное влияние РЬОг иа ско-
рость горения проявляется в довольно широком интервале давления—
от 5 до 50-J-60 атм. При повышении начальной температуры область
действия РЬОг сдвигается в сторону более низких, давлений. Так, при
То=120° наибольшее влияние РЬО2 проявляется в интервале 10 —
150 мм рт. ст., на участке 300—750 мм рт. ст. РЬО2 вообще не ускоряет
горение пороха, а в области умеренно повышенных давлений (до 70 атм)
действует довольно слабо. При 85° в области Р < 1 атм влияние РЬО2
проявляется в более широком диапазоне, чем при 120^.
Таким образом, чем выше начальная температура заряда, тем при
более низком давлении действует РЬО2. Полученные данные соответ-
ствуют представлениям, развитым в работах [3, 4], о конкуренции гомо-
генной реакции в объеме и гетерогенной реакции на катализаторе. При
увеличении скорости гомогенной реакции и, соответственно, при увели-
чении и0 (за счет увеличения, например, То) вклад гетерогенной реак-
ции (и величина Z) должен уменьшаться, что и имеет место в случае
Р1О2 в области умеренно повышенных давлений при То = 120°,
В вакууме при этой же начальной температуре, когда скорость го-
рения эталонного состава (Uo) низка, РЬО2 действует с высокой эффек-
тивностью.
Сложно также и влияние СнО. В вакууме она может влиять на
горение пороха как положительно, так и отрицательно и при этом не
усиливает действия PbOj. Последнее при То = 120° характерно также и
для области умеренно повышенных давлений. Напротив, при То = 20°
при повышенном давлении CuO значительно усиливает действие РЬО2.
Такое различие в действии окиси меди при давлении выше и ниже атмос-
ферного можно, по-видимому, связать с частичным изменением состава
продуктов, образующихся в к-фазе при различном начальном давлении
и температуре заряда, или условий для проявления действия катализа-
торов (вязкость реакционного слоя к-фазы, температура поверхности
и т. д.). Известно, например, что при горении пороха в вакууме при
повышенных начальных температурах происходит сильное испарение
летучих компонентов — НГЦ и ДНТ. В работе [5] отмечается, что в ре-
зультате испарения в поверхностном слое к-фазы пороха, горящего в
вакууме, образуется подслой, состоящий почти из одной нитроклетчатки.
В связи с этим представляет интерес сопоставление полученных резуль-
татов с данными по влиянию свинцово-медного катализатора на горение
нитроцеллюлозы. В вакууме (при То=2О°) РЬО2 значительно увеличи-
вает скорость горения нитроцеллюлозы, a CuO влияет на нее слабо и,
кроме того, не усиливает действия РЬО2 как в вакууме, так и в области
умеренно повышенных давлений.
117
' Непонятным является io; что при горении пороха 9 дакууме при
120р С60 повышает давление, с которого начинается устойчивое горение,
а в случае нитроцеллюлозы (правда, при То=20°) она, наоборот, сни-
жает это давление.
Выводы
1. Исследовано влияние СиО, РЬО2 и их сочетания на горение по-
роха Н в интервале давлений 10 мм рт, ст.—100 кгс/см? и различной
начальной температуре заряда.
2. Показано, что:
а) в области умеренно повышенных давлений при Т(| = 20°С окись
меди значительно повышает влияние РЬО2 как на скорость горения, так
и на зависимость U(P), хотя сама СиО скорость горения увеличивает
мало; при увеличении начальной температуры заряда до 120°С эффек-
тивность действия РЬО2 понижается, и СиО не усиливает ее;
б) в вакууме при То=120эС влияние РЬО2 на скорость горения про-
является в узкой области от 10 до 200 мм рт. ст. (Zmai = 2,0); СиО дей-
ствует слабее (Zm„ = 1,5), а, будучи введенной в порох совместно с РЬО2,
понижает эффективность действия последней.
Г. Патент США № 3138499; патент США № 3102834.
2. Йрекел Р. Р. Ракетная техника и космонавтика, 1965, 2, с. 238.
X НикифоровВ. С., Бахман Н. Н.'ФГВ, 1969, J& 2, с. 270.
4. Б а х м а н Н. Н. и др. ДАН СССР. 1972, т. 202, № 5, С 1107.
6. Але KCja ядро в В. В , X л е в и о й С. С. ФГВ, 1970, № 4, с. 238.
1. I-
1 ......... -
,ет '< iwr"' • ' . ;
УДК 662.311.1+536.463
ГОРЕНИЕ КОНДЕНСИРОВАННЫХ СМЕСЕЙ
С РАЗЛИЧНЫМИ ОКИСЛИТЕЛЯМИ
Л. Е. Фогельзанг, С. Af.. Каляеве. Б. С. Светлов
В работах [1, 2] показано, что изменение реакционной способности
Окислителя, содержащегося а молекуле взрывчатого соединения, оказы-
вает очень сильное влияние на скорость его горения. В случае газов
[3—5] также можно проследить подобную зависимость. Одновременно
для конденсированных смесей неоднократно отмечалась независимость
Скорости и возможности горения состава от теплоты реакции. Однако в
Настоящее время нельзя определенно сказать, будет ли для гетерогенных
tMecefi, так же" как для индивидуальных веществ н газовых смесей, на-
блюдаться корреляция между скоростью горения и активностью окисли-
теля, так как изучались главным образом смеси с окислителями С17+ и
S5' [6, 7]. В этой связи представляло интерес исследовать горение соста-
вов с различными окислителями, но с одним горючим.
f В качестве горючего использовали полиметил мет акрил ат (размер
кастиц около 3 мкм). Окислителями служили калиевые соли кислород-
содержащих кислот — КаСг3От, KNO3, KJO<, K.JO3. KCIO4, КС1О3,
КМпОх, КВгОз с размерами частиц 60—100 мкм. Смеси готовили, пере-
мешивая компоненты .резиновой пробкой на кальке в течение 20—30 мин.
Наряды прессовали в трубки из оргстекла с внутренним диаметром 4
1ли 7 мм под давлением 4000—5000 ат. Сжигание, образцов проводили
I бомбе постоянного давления. Скорость и характер горения фиксиро-
вали барабанным фоторегисгром. При давлении 40 ат для каждого окис-
кителя, изменяя соотношение компонентов в смеси, определяли макси-
мальную скорость, которую можно получить с ним. Расхождение между
результатами (4—5 опытов) не превышало 10%. Коэффициент избытка
кислителя а рассчитывали, исходя из того, что KJO4, KJO3, KCIO4, КСЮз,
|ВгО3 восстанавливаются до КН al, a К3СГ2О7, КМпОц KNO3 восста-
ивливаются до Сг2О3, MnO2, N2 и оксида калия соответственно.
f Остановимся сначала на горении составов, окислитель в которых
|№дадает наивысшей положительной валентностью. Смеси с нитратом и
119
бихроматом калия при 40 ат оказались неспособными к горению прй
изменении а от 0,3 до 2 (d3ap= 7 мм}. Наибольшую скорость ,горения
удалось получить с перманганатом калия (рис. 1). Несколько меньшая
скорость достигается с перхлоратом калия. Составы с перйодатом калия
горят медленнее составов с двумя другими окислителями. Отметим очень
резкий максимум на кривой U„(a) для смесей с перманганатом калия.
Зависимости UM(a) для составов с йодатом, -броматом и хлоратом
приведены на рис. 2. С наименьшей скоростью горят смеси с KJO3, Со-
ставы с хлоратом и броматом горят в 5—7 раз быстрее,
Рие. 1. Зависимость скорости го-
рения смесей полиметил метакри-
лата от состава при 40 ат с окис-
лителями; /—перманганат калия;
2-1перхлорат калия; 3—перйодат
калия
Рис. 2. Зависимости скорости го-
рении смесей поди метил метакри -
лата от состава при 40 ат с окис-
лителями; /—бромат калия; 2—
хлорат калия; 3—ио дат калия
Продуктами превращения всех окислителей, за исключением пер-
манганата калия, были белые шлаки, которые хорошо растворялись в
воде, причем растворы имели нейтральную реакцию, что указывает на
образование галогенидов калия, В опытах с КМпОх наблюдалось обра-
зование бурых нерастворимых в воде продуктов, вероятно, двуокиси
марганца.
В табл. [ приведены максимальные скорости горения, которые уда-
лось получить для смесей полиметилметакрилата с различными окисли-
телями. Видно, что и„ возрастает в ряду KJO3, KJO4, КСЮ4, КМпО4,
КСЮ3, КВгОз, Однако, если для индивидуальных веществ изменение
строения группы, содержащей окислитель, оказывает очень сильное
влияние на величину скорости горения [2], то в случае смесей роль окис-
лителя существенно слабее.
Зависимости UM(P) для смесей, имеющих состав, отвечающий аигаах
при 40 ат, приведены в табл. 2. Отметим, что последовательность в ско-
ростях горения смесей с различными окислителями, отмеченная при
40 ат, сохраняется до 50 ат'. Выше 50 ат состав с К-Юч начинает гореть
быстрее смесей с КСЮ< и КМпО<. Кроме того, смесь с перйодатом калия
отличается от всех остальных составов очень высоким значением v в
выражении U = BP ' для скорости горения.
Сопоставление максимальных скоростей горения смесей полиметил-
метакрилата с различными окислителями (табл. I) показывает, что, дей-
ствительно, никакой корреляции между величиной скорости и теплотой
120 '
' I'b • '“н Таблица)
Максимальные скорости горения смесей по ли мети л мет акрилата
с различными окислителями яри 40 ат
Окислитель ’“мах г! см2 се« Qp, ккал)/сг (Н3Огаз) расчет ко {81 тг,»К (расчет по ЭВМ) Стандартный окисли- тельный потенциал Еи соответствующей пары (в) (9]
КаСгдО? 0,3-2 не горит ЗЮ* Сг3+/Сг8+ —0,20
KNO3 0,3—2 не горит 430* 1650 NOras /NOa- “0• 15
Na/KO3- 4-0,25
KJO., 1 0,64 420 975 I-/IO3- +0,26
KJO4 0,75 1,9 520 1250 I-/IO4- 4 0,37
КСЮ4 0,57 2,4 84! 2000 С1-/С1О,- +0,51
КМпО4 0.81 2,7 230 900** МпОа/МпОг+"Л7
КС1Оз 0,75 3,1 890 2400 С1-/С1О3- +0,62
КВгОз 1 4,9 870 2050 Br/BrOa- +0,61
* Расчет прн а=1. Для смеси с КаСгзО, — конечные продукты КгСОз и СгаОз,
** Расчет по [8J,
_ Таблица 2
Зависимость скорости горения от давления дли смесей
полиметилметакрилата с различными окислителями
Окислитель а смеси Интервал давлений (ат) Зависимость Uv (Р) 1г1см2-сек}
КЮз 1 20-70 70 -100 U = 2,1-10"= Р°’э U =2,9.10-3 р'-38
KJO4 0,7.5 25-40 40—100 U = 2,6-1(Г4 Р'3-4 V = 1,4-10—2 Р[’33
КСЮ, 0,57 15—100 U =0,17 Р°;7
КМпО, 0,81 1100 U = 0,35 Р0’55
КСЮ, 0.75 5-100 U =0,24 Р°’7
КВгОз 1 1-100 U =0,25 + 0,5 Р0’65
горения, а также и его температурой не существует. Попытаемся по-
, этому сопоставить полученную последовательность в скоростях горения
, смесей с изменением активности окислителя, входящего в их состав,
за меру которой, как к в работе [2], примем окислительный потенциал
(Ео). Однако Ео сильно зависит от реакции среды, причем при измене-
нии последней меняется не только абсолютное, но и относительное зна-
чение Ео- В рассматриваемом случае для сравнения реакционной спо-
собности окислителей, вероятно, следует исходить из окислительных
1 потенциалов в нейтральной среде, поскольку все выбранные соединения,
121
Противоположность солям аммония, образованы при взаимодействий
слоты с сильным основанием. Как видно из табл, I, наблюдается
овлетворительное совпадение между возрастанием скорости горения
ееей и величиной окислительного потенциала. Есть только одно исклю-
Нйе _ tMetn с КВгОз горят значительно быстрее смесей с КС1О3,
;нако известно [10], что несмотря на близкие окислительные потен -
[алы пар СГ/С1О3~ и Вг’/ВгОз окисление броматом идет быстрее,
го, возможно, связано с тем, что в случае хлора окисление затруднено
-за меньшей величины Ер на начальной стадии. Действительно, если
юследить восстановление хлора и брома
0,49
I 1
_ 1,36 „ 0,40 ,* (1,66 ______ 0,33 I _ 6,36_______
Cl —С1_, - ~----------СЮ —C10t------------------- С1О3 —:-----СЮ4 ,
„ - 1-07 „ 0,45 „ 0,51 „ л _
Вг «------Вга--------ВгО ----------ВгО3 ,
> видно, что Ео для нары ВгО7ВгОэ_ больше Ео пары С1О7СЮз" В слу-
ie восстановления перйодата начальная стадия идет с очень высоким
этенциалом _
г . i2 10~ . 10з- юг.
днако затем наблюдается образование сравнительно инертного йодата,
й*тому при горении смеси с перйодатом калия можно было бы ожн-
вть, что восстановление Р+ также как в случае перйодата аммония,
становится на l5*. Однако тепловой эффект реакции при восстанов-
ении перйодата калия до I5*, в отличие от перйодата аммония, близок
нулю, что делает горение по данному пути практически невозможным,
(озтому для смесей с перйодатом калия конечным продуктом реакции
акже должен быть йодид.
' Таким образом, в области относительно низких давлений для сме-
ей полиметилметакрилата с различными окислителями, так же как и
дя индивидуальных веществ, наблюдается корреляция между скоро-
тью горения и реакционной способностью входящего в состав окисли-
еля,. выраженной в виде окислительного потенциала, однако в этом
лучае влияние активности окислителя значительно слабее.
Ф
1., Фогельзанг А. Е. С в е т л о в Б. С. Докл. АН СССР, 1970, т. 192, с. 1322.
2. Фогельзанг А. Е., Аджемян В. Я. Светлов Б. С. Докл. АН СССР,
1971 т. 199, с. 1296.
1 McD .Gummings G. А., Н al 1 A. R. X Symposium on Combustion. 1965, p. 1365.
< Combourien J., Moreau G., Moreau R, Pearson G. S. AJAA Journal.
1970, v. 3, p. 594.
5. dPafrit er W. G., Wolf ha rd H. G. IV Symposium on Combustion. 1953, p. 420.
б.Бахман H. H., Беляев А. Ф. Горение гетерогенных конденсированных си-
едем, М., «Наука», 1967.
7. Андреев К- К. Термическое разложение и горение взрывчатых веществ. М.,
«Наука», 1966.
8. А ядреев К. К., Беляев А. Ф. Теория взрывчатых веществ. М, Оборонгиэ, 1960.
9. Латимер В. Э. Окислительные состояния элементов в водных растворах. ИЛ.,
М., 1954.
10. Ullmans Encyklopfidie der techniSchen Chemie, 1953, В. 4, S. 727.
122-
Л- Г,, .я 1 г; -
УДК 662.6)2 +622.615
ИССЛЕДОВАНИЕ ЙРОФИЛЯ ТЕМПЕРАТУРЫ
В ЗОНЕ ГОРЕНИЯ СМЕСЕВЫХ СИСТЕМ
Я. Н. Бахман, В, А. Зайцев, Ю, С. Кичин,
С. М, Каляеве, А. Е. Фогельзанг, В. Н. Чусов
При исследовании механизма горения большой интерес представ-
ляет профиль температуры Т(х) в зоне горения. В работах [1—5]
с помощью тонких термопар измерен профиль температуры в зоне го-
рения перхлората аммония (ПХА) и смесей на его основе. В работе
[2] определена температура поверхности (Тп) горящих зарядов ПХА+
сополимер бутадиена и акриловой кислоты. Разброс результатов очень
велик. Средние . значения Т„ при р = 1—70 атм лежат в пределах
510-^-660“С. Какая-либо Зависимость Т„ от давления, дисперсности
ПХА и наличия катализаторов не прослеживается.
В работе [4] значение Т„ для стехиометрической смеси ПХА + поли-
формальдегид возрастало от 630 до 680° С при увеличении давления
от 25 до 45 атм. Добавки алюминия слабо (иа ~20°С) увеличивали
Т„. Градиент температуры в газовой фазе у поверхности заряда
(dT/dx)„ = (pn возрастал с ростом давления (для состава без алюми-
ния— от 1,5* 105 до 2,5 • 105 градам при увеличении р от 25 до 45 атм).
Добавки алюминия увеличивали фп.
В работе [5] значения Т„ и срв для смеси ПХА-Ьполиметилметакри-
лат (ПММА) а = 0,61 возрастали с ростом давления (соответственно
от 400 до 700° Си от 1,4* 10*'до 5,5-105 град (см при увеличении р от 5
;до 60 атм). При добавке 1 % Ёе2О3 величина Тп практически не меня-
>лась, а фп уменьшалась.
Вопрос о точности и достоверности термопарных измерений в лите-
ратуре рассматривался главным образом качественно. Количественное
^рассмотрение некоторых погрешностей проведено Зениным .[6],
:• В качестве источников погрешностей рассматривались: конечная
^скорость теплообмена между термопарой и газом [6], теплоотвод
>в концы термопары [6], катализ на поверхности термопары [7], осаж-
дение сажи на термопаре [8]. При термопарных измерениях, как пра-
вило, предполагается, что поверхность горящего заряда является
123
1доской; отклонения от этого условия также ведут к ошибкам Еще
эдин источник ошибок — усреднение температуры по толщине термо-
тары. Указанные погрешности приводят к тому, что профиль темпера-
туры, записанный термопарой, всегда отличается от истинного. В опре-
деленных условиях эти отличия приобретают принципиальный
характер.
Цель данной работы — попытаться связать результаты термо-
парных измерений с общими представлениями о горении смесевых
систем [9] и более внимательно проанализировать ошибки измерений,
связанные с тепловой инерцией термопар.
Методика проведения опытов
Было исследовано распределение температуры в зоне горения
смесей ПХА с ПММА, параформом (ПФ) и серой. В качестве ката-
лизатора использовалась закись меди. Квалификация веществ: ПХА
и ПММА—техн., сера—серный цвет, ПФ и Сп2О—ч.; ПХА фракции
100—400 мкм сушился и затем измельчался и а вибромельнице, ПФ
и Си2О измельчались в агатовой ступке. Размер частиц ПХА состав-
лял ~ 12,5й, ~ 18,5s и ~302 мкм. Для ПММА исследовались фракции
~32 и 250—400 мкм (ситовой размер). Для частиц ПФ и Си2О сред-
ний микроскопный размер составлял 8,9 и 0,8 мкм соответственно
(количества измельченных ПФ и Си2О были недостаточны для изме-
рений на ПСХ-4).
. Порошкообразные компоненты смешивались на кальке резиновой
пробкой (обернутой в кальку) в течение 20—30 мин.
Термопары сваривались (под микроскопом) из проволочек 80%
W+20% Re и 95% W-|-5% Re диаметром 25 мкм. Сварка производилась
разрядом конденсатора емкостью 800—1000 мкф, заряженного до
20—40 в.’ Одним электродом служила молибденовая пластина, дру-
гим— вольфрамовый стержень. Затем термопара прокатывалась в
ленту толщиной b = 7—8 мкм и шириной ~70 мкм. Спай термопары
после прокатки имел форму полудиска, сопряженного с лентой; наи-
большая ширина термопары в этой точке составляла /2*100—130 мкм.
Опыты проводились с П-образными термопарами, предложенными
Зениным [6].
При приготовлении зарядов в оболочку из плексигласа (с внут-
ренним диаметром 7 мм и наружным 10 мм) насыпалась половина
навески смеси. Фигурным пуансоном (трапецевидной формы) делалось
углубление, в которое помещалась термопара после чего в оболочку
всыпалась вторая половина навески смеси. С^месь подпрессовывалась
сначала трапецевидным, а затем обычным пуансоном. Оболочку ста-
вили на Поддон с прорезями, в которые пропускались концы термо-
пары. Прессование проводилось при давлении рпрес =2000 атм.
Опыты при р I ата проводились в вакуумной камере объемом
40 л. Время горения т измерялось секундомером. Вычислялась средняя
скорость горения U = h/т, где h — высота заряда. Опыты при р>1 ата
1 В частности, в случае, когда на поверхности заряда имеется расплавленный
слой, термопара при выходе на поверхность еще некоторое время «тянет» за собой
пленку жидкости [8].
® Размер частиц d вычислялся ио формуле d = 6/р5уЛ. где SyJ — удельная по-
верхность порошка, измеренная с помощью прибора ПСХ-4.
124
проводились в бомбе объемом 3 л. Скорость горения измерялась фото-
регистром.
Сигнал с термопары через усилитель постоянного тока подавался
на шлейфовый осциллограф Н-700. При исследовании зависимости
U(T0) использовался медный блок (сечением 35X35 мм и высотой
120 мм с осевым каналом глубиной 110 лш) с окнами для фотогра-
фирования. Температура в канале контролировалась с помощью двух
хромель-копелевых термопар. Регулирование температуры осуществля-
лось с помощью потенциометра ЭПВ-01 (с точностью ± 1,5%). Время
выдержки заряда в термостате составляло 15—20 мин. Усреднение
результатов проводилось по 5—7 опытам При каждом р и То, '
Профиль температуры в конденсированной фазе (к-фазе)
На рис. 1 показан характерный вид записи сигнала термопары на
осциллограмме. Значение температуры поверхности заряда Тп опреде-
лялось двумя методами: 1) по излому записи на осциллограмме (при
достаточно низких давлениях на осциллограмме имелось небольшое
плато); 2) на графике lg(Т—То), х: в этих координатах опытные точки
группировались около прямой с двумя (рис. 2), а для смеси ПХА +
ПФ— с тремя изломами — соответственно при Т = Т**, Т* и Т„ (где
Т^<Т^ <Т„). Значения Т„, определенные обоими методами, практиче-
ски совпадали.
Рнс. 1. Характерный вид записи
сигнала термопары на осцилло-
грамме смесь ПХД (ъ 18,5 мкм)
+ ПММА (~3 мкм), а—0,75,
р«1 ата)
X, мк
Рис. 2. Зависимость lg (Т—То) = £(х)
для смеси ПХА (~18,5 лекм)+
ПММА (~3 мкм), а=0,75, р=1 ата
Значения Тп (см. табл. I, 2) существенно возрастали при увели-
чении давления (и соответствующем увеличении скорости горения).
Однако при введении катализатора (при p = consl) значения Т„ прак-
тически ие менялись, несмотря на значительной'(в 1,7—1,95 раза) уве-
личение скорости горения. При р=1 ата величины Т„ для некатализи-
рованных смесей ПХА + ПФ, ПХА + S, ПХА + ПММА были близки
друг к другу (нужно отметить, что для этих смесей была близка и ско-
рость горения).
Если в к-фазе при Т<Т„ отсутствуют зоны выделения или погло-
щения тепла и если температуропроводность а к-фазы не зависит от
125
температуры, то профиль Т(х) должен описываться михельсоновской
волной прогрева:
- dT U
-~=-(Т-Т0) (I)
dx а Г1 ' Т=Т0 + (Тп-Т0)е‘ ! т-т0 и : пт — Т = а х (2) 1 а 1 о 3
т. е. в координатах !g(T—То), х профиль температуры — это прямая
с тангенсом угла наклона к оси х, равном U/a.
Напротив, наличие изломов при T = TS (рис. 2) и Т = Т,Л свидетель-
ствует о том, что прн этих температурах в к-фазе происходит поглоще-
ние или выделение тепла или достаточно резко меняются теплофизи-
ческие свойства (см. следующий раздел). Излом при Т = ТЛ, видимо,
'сйнзан с фазовым переходом ПХА Торторомб.)-^ПХА (кубич.), отклоне-
ния Т, от табличного значения 240° С, видимо, связаны с погрешностя-
ОШи термопарных измерений в к-фазе (анализ этих погрешностей в лите-
"рвтуре практически отсутствует).
. '! '•'Таблица 1
Профиль температуры прн горении смесей ПХА+горючее
§ ' £ * Э X о □ * н О О * К и О (Фгавт)| k Ю S град(см * * * J5 о О н" (Тт.д>гЮ-‘. градам 11а, мкм О 1 | 3-10», 1 Ijzpad
Смесь ПХА ( ~ 30 мкм) + ПФ, а = 1
:.хз 0,32 180 300 460 7 6 690
,0.5 0.47 160 265 48 > 19 5 580 —
0,7 0,64 140 260 520 14 4 590 18 '52 1000
4.0 0,82 140 255 58') 41 7 710 27 17 930
Смесь 95% |ПХА( ~ 30 мкм} 4- ПФ, а = 1J 4-5% Cu3O
0.3 0,55 155 310 480 13 6 550
0,5 0.87 125 245 480 38 10 700 15 60 1080
1,24 130 285 530 40 5 680 - — —
W) ,1,55 125 260 580 j 29 6 650 — —
Смесь ПХА 10 мкм) — S, а = 0,75
1.0 *0.74 | - | 265 I 645 | - | _ | — |
Смезь ПХА ( — 18,5 мкм) — ПИМА ( ~ 3 лт/сн), я ='>,75
0,34,о;35 255 510 21 12 680 21 100, 1490 1,6
6,68 0.70 — 220 590 56 9 860 28 43 | 1510 1,5
J.0 -1,д. — 200 580 49 .7 830 26 32 1430 1, 1
11,6 5,7 , — 730 24 6 850 41 32 • 1170 1.7
21,5 6,9 — .760 17 5 890 58 ' i7l 1740 -
Смесь ПХА ( ~ 12 лкм} 4 ПММА (~ 3 мям}, и = 2
0,28(0,20 — — I 510 9 12 I 5901 46 । 1501 1560 1.6
1 1,0 0;50 — - — 630 56 5 1060 55 16 1830 I 1,8
1 ,0 Jb.O ф 1 650 .37 , 8, 1 8401 58 2,0
Расчет волны прогрев* в среде с фаэовымв переходам!
Пусть njpi Т = Т„ скачкообразно изменяется q (а значит и «и»),
с, X, а выделяется (или поглощается) теплота Q кал/г. Форму волны
прогрева найдем, решая уравнение теплопроводности
<РТ dT Л
А Т7 “ Р 1;с -Г" = О
dx1 dx
О)
отдельно для области 1 (Т0^Т <Т*)и области 2 (Т*<Т <ТП) при
/ dT\
граничных условиях =0, T|i_0=Tn. Условия сшивки решений
X О&
при Т-Т ф запишем в виде
Q(U[ = CtU = 02^2
/ dT \ / dT \ J
4dx )гт,~"чах aU^P1UQli
(4)
(5)
Отсюда для профиля температуры получим: в области I
(6)
в области 2
с, Qr2 Г Qll'l
f=T<-(T,-To)-t--I-+ Te-T. + (T.-Te)^ + f- -е^Г (7)
va L сз Ла
Скачок градиента температуры прн,Т «• Г* равен
dT\ /6Т\ 1
dx Д кахд Jt*
13, /К. \ c t 1
₽iu -r + hr-1 (8>
Л2 \ Л2 / Л1 J
При Qt2>0 (в принятых обозначениях это означает, что при переходе
от (Т„ —dT) KfT^ + dT) тепло поглощается) и /.[ > осуществляется
скачок, показанный на рис. 3,а, а при Q1?<;0 и < fa— осуществля-
ется скачок, показанный на рис. 3, б.
Оценим скачок градиента температуры при фазовом переходе
ПХА (орторомб.)-►ПХА (кубич.), приняв Qi -» 1£&г/см\ q2 = 1,76 а/сл3;
T* = 24(FC; Qi2=2,46 ккал/моль [11]; Ai = 1,08* 10~3 кал/см-сек-град;
Ci=0,3l кал/г-град (средние значения в интервале 50—24<^С, осно-
ванные на данных работы [10]). Приняв ад — 6* 10-4 смг/сек [3] и
с2—0,365 [12], получим ?.2 = 3,85• 10-4 кал/см- сек-град. Подставляя эти
Значения в (8), получим, что данный случай соответствует рис. 3, л.
При этом величина — (^3 ) ] равна 16,9 • 10* (pi и) град /см.
LX- Х /1 \ и* /1] Т,=24О°С "
При скорости горения и = 1 мм/сек это дает 3,3-10* град /см.
127
Профиль температуры в газовой фазе
В газовой фазе наблюдались два участка, где градиент темпера-
туры был максимален *. Можно думать, что первый максимум (<ртах),
связан с прохождением термопары через пламя продуктов газифика-
ции ПХА (NH3+HCIO4), а второй (фтиЬ расположен в зоне пламени
горючее-окислитель.
Рис. 3. Скачок градиента температуры в волне
прогрева при переходе через зону фазового
перехода: а—при Qu>0; <5—при Qia<0
Расстояние** h] от поверхности заряда до первого максимума для
• всех смесей в изученном интервале давления лежало в пределах
h] =4—12 мк. Систематической зависимости hi от а и р не прослежи-
вается (табл. 1). Зиачеиие температуры Ti, при которой лежит (фтах)г,
для некатализированных смесей при р^1 ата возрастало с ростом
давления, а при дальнейшем увеличении п оставалось примерно по-
стоянным (для катализированной смеси ПХА+ПФ+СпаО значение
Tj возрастало лишь при р^0,5 ага).
Величина (<pm„) 1 для некатализированных смесей при р^1 ата
резко возрастала с ростом давления. При дальнейшем увеличении р
(для смесей ПХА+ПММА) величина (<pmaili начинала быстро сни-
жаться. Для катализированной смеси ПХА + ПФ+СпаО величина
(<Pmax)i возрастала лишь при р^0,7 ата, а затем снижалась.
Поскольку первый максимум (<praai)i расположен очень близко
к поверхности заряда можно думать, что снижение величины (ч1,,^);
при р>1 ата (а для смеси ПХА + ПФ+Си2О — при р>0,7 ста) явля-
ется методическим эффектом, связанным с1 тепловой инерцией термо-
пары (см. следующий раздел).
Расстояние ha от поверхности заряда до второго максимума резко
уменьшалось с ростом давления при р^1 ата, а затем оставалось
Примерно постоянным (для смеси ПХАПММА, а=0,75) или даже
’ • Отметим, что в к-фазе, вблизи Т = Тп также имеется четкий максимум гра-
диента температуры, о два ко этот максимум связан с чисто методическими эффек-
тами. Мы видим, что при Т«ТЛ на осциллограмме, как правило, имеется излом,
а при низких давлениях — плато, т. е. ?п = 0 (это . связано с изменениями условий
теплообмена при переходе термопары из к-фазы ,н г-фаэу). Между тем при
Т < Тл везде Ф > О, что и приводит к появлению максимума градиента температуры
в к-фазе вблизи поверхности заряда.
** Значения ht; ht; Т,; Та брались для середины участков с максимальным гра-
диентом температуры, . , J
возрастало (для смеси ПХА+ПММА, а = 2). Температура Т2, отвечаю-
щая второму максимуму, сравнительно слабо зависела от давления. Ве-
личина (фтах)г возрастала с ростом давления (особенно для смеси
ПХА + ПММА, а = 0,75).
Значения температурного коэффициента скорости горения
Р = d In UI dT0 лежат в пределах (1,1—2) • Ю~3 \!град и слабо зависят
от а и давления.
Таблица 2
Профиль температур в г-фазе для смеси ПХА
(~12,5 мкм) + ПММА (250—400 мкм), а = 0,75
р, ата и, мм1сек Тп, °C I (?max^i 'Ю * । град/см h1; мкм Ti, РС (<ртах)з • Ю_« град}см h3, МКМ т„ °C
0.34 0,30 4 S0 9 5 520 26 115 1200
1,0 0.70 | 570 19 8 710 31 43 1380
В табл. 2 приведены результаты опытов для смеси ПХА + ПММА,
а = 0,75 с крупнодисперсным ПММА. Из сравнения табл. 2 и I следует,
что значительное увеличение размера частиц горючего не приводит
к заметным изменениям величин ht; h2 и (<pmax)2, однако значения Тв;
Т(; Та и (tpmax)i заметно снижаются.
Максимальный градиент температуры, который может быть записан термопарой
Рассмотрим простой случай, когда при выходе на поверхность
заряда (в момент времени t=0) термопара, имеющая температуру
Тт | t—о = Тп, попадает в газ с постоянной температурой Тга». Прогрев
термопары описывается уравнением
dT
ftCTST = Птв(Тгаа — Tr) (9)
где pt, ст, ST, Пт — плотность, теплоемкость, площадь и периметр по-
перечного сечения термопары, Й—коэффициент теплоотдачи от газа
к термопаре. Из (9) получим следующее выражение туры, который записывает термопара для градиента темпера-
dl\ 1 dTT “ ~ dx' “ U dt е где CD * — - " 1 *. cL. Рт Ставка 00) 2Ь(
?т “ ртс Дкви ’ иэив b + I
Из (10) следует, что при любом профиле температуры газа термопара
в’ принципе не может записать градиент температуры выше, чем еру*.
Проведем численную оценку <рт* для пламени NH34-HCIO4, при-
няв в первом приближении, что температура пламени NH34-HCIO4
р Труды вип. 83 129
в смесях ПХА + ПММА такая же, как и для чистого ПХА (Тгаз —
== Тпха), а массовая скорость потока газа в зоне пламени NH3 +
-H'NCIO равна m0Kf<U (где mok—весовая доля ПХА в смеси, a pU —
марсовая скорость горения смеси ПХА + ПММА) *.
. Nu Хгаэ
При вычислении 8 = -д------- воспользуемся зависимостью [ТЗ]
Ojks
Nu = A‘Pr"« Rem. Примем b=7 мкм\ /=150 мкм-, tj=4- 10_4 г!см-сек
•fiH Так как данные для прямоугольных профилей при малых Re
в [13] отсутствуют, примем для рассматриваемого диапазона Re зна-
чения А = 0,99; m = 0,305, рекомендуемые в [13] для круглых прово-
лочек. Примем >.гаа = 2-10~4 кал/см • сек• град [15] Рг=1, РЛ—
~ 0,75 кал[см3 • град (среднее значение для чистого W при Т = 1000—
2400°К [16]). При вычислении разности (Тпха — Тп) при р=1 ата
примем ТПха=967°С [17] и Тп=550°С (среднее значение, основанное
на результатах наших опытов и данных работ [2, 18—20]. Отсюда
Тпха — Тп)~417°С. Такое же значение (Тпха — Тп) примем и при
ря»0,3 ата, так как значения Тпха при р<1 ата в литературе отсут-
ствуют. При р=70 атм значение (Тпха—Т„)~987—750—237° С [17,
19, 21, 22]; при р = 11 агл интерполяцией получим значение
(Тпха-Т„)~391оС.
Значения ф/, рассчитанные из (10), приведены в табл. 3. Вели
чйна <рт* существенно уменьшается с ростом р и U.
Таблица 3
Значение предельного градиента температуры, который может быть записан
термопарой толщиной 7 мкм
". О р, ата U, мм]сек кал1см*сек-град. (ТПХА Тт*, град]см
0,75 0,34 0,35 0,105 417 500-10*
1 0,98 0,144 417 244-10*
11,6 5,7 0,245 391 67-104
2 0.28 0,20 0,0933 417 780-10*
1 0,52 0,1247 417 400.10*
Н 5,0 0,249 391 78 10*
Сравнивая табл. 1 и 3, отметим, что при р < 1 ата (фти) t < (рт ♦.
Однако при р>1 ата величина (фтах)i становится сравнимой с ф7*.
Это означает, что профиль температуры, записанный термопарой, уже
не передает реальный профиль Т(х) пламени NH34-HC1O<, а отражает
Лишь тепловую инерцию термопары. Возможно, именно с этим связана
обнаруженная в наших опытах независимость hi от давления, а также
падение (фт11)( с ростом давления в области высоких давлений.
• Это соответствует допущению, что при x^ht частицы ПММА еще не успевают
существенно газифицироваться (и тем более, сгореть). На самом деле, температура
пламени NH3+HCIOa в смесях ПХА + горючее— в зависимости от природы горю-
чего, а и давления может быть как выше (за счет подвода тепла от диффузион-
ного пламени), так и ниже (в результате затрат тепл* на газификацию горючего),
чем для чистого ПХА [9]. Величина ТПХА уменьшается при уменьшении давления.
Чтобы осуществить устойчивое горение ПХА при р^1 ата, необходимо либо подо-
греть его, либо ввести добавку достаточно активного горючего (или катализатора),
либо применить горючую оболочку.
Рассмотрим теперь более сложный случай, когда температура
газа возрастает по экспоненциальному закону:
т„я = Т„ + —(е _ 1
ТПож р \
00
(это соответствует михельсоиовской волне прогрева с граничными
/dTraj \
условиями Тга_, I Тп; I ~ j = (<рг„)„. Подставляя (11) в
(9) и решая полученное уравнение при граничном условии TTL-o =
dTr
Тп, получим выражения для Тт и <рт — Выражение для <рг
О X
имеет вид
/ гпм р Цсгаз \ / 4» \
j я ехр —1----------х — ехр — —~—— х
_, , ____L______V ртст^аквЦ 7 9
* ’ ” Шок р UCr„/kr„ - 4 в/ft (VUU ( '
Величину (фг1а)п, которая относится к газу и заранее неизвестна,
определим из (12) через величину (<pmai)i = (dT/dx)^,, записанную
термопарой. Из принятых граничных условий ясно, что при
х — 0, Тп = Тгаз, но по мере увеличения х разность (Тгаэ —Тт) воз-
((П* \
= 0. Это означает, что
dx /х=о
бесконечно тонкая термопара в момент выхода на поверхность заряда
должна записать горизонтальную «ступеньку»1, что дает возможность
определить координату поверхности и величину непосредственно
на осциллограмме. Нужно подчеркнуть, что градиент температуры
(?»)(» • который записывает термопара вблизи поверхности, резко зани-
жен по отношению к реальному градиенту температуры в газе, и его
нельзя непосредственно использовать в расчетах. Значение (<ргЭЭ)п мо-
жет быть вычислено из (12) или аналогичного выражения.
Так же, как и в предыдущем разделе, были сделаны оценки для
пламени NH3 + НС1О4 в смесях ПХА+ПММА (принято сгаз ~
= 0,3 кал/г-град, см. [24]). Оказалось, что При р^1 ата рассматри-
ваемая термопара с удовлетворительной точностью записывает про-
филь Т(х).При х=?=Ь| термопара заиижаеттемпературу при р~0,3 ата -
на 1—2,о%, а при р = 1 ата—на '*-10%. При х < hi ошибка в опреде-
лении Тгаз уменьшается (и при х = 0 обращается в нуль). Что каса-
ется градиента температуры, то при х = hi рассматриваемая термо-
пара занижает его при р ~ 0,3 ата—на 0,25—0,5%, а при р = 1 ата —
иа 2—5%а. Однако при уменьшении х ошибка в определении градиен-
та температуры возрастает и при х = 0 становится равной 100%
(см. выше).
* Для реальной термопары «ступенька» не будет вполне горизонтальной, так
Как тер мот ара имеет конечную толщину, и ее температура будет отставать от тем-
пературы конденсированной, фазы. Наличие «ступеньки» на термопарных профилях
и возможные причины ее появления обсуждались в [6, 23] и др.
* Однако, в реальных условиях ошибки в величине Т и dT/dx могут быть
существенно выше, так как .толщина термопары становится сравнимой с h[ (ата
ошибка здесь йе раосйатр ив елась)- г
а»
131
При р>1 ата рассматриваемая термопара (толщиной • 7 лкж),
;ак уже отмечалось, не может обеспечить достоверную запись про-
филя Т(х), и поэтому вычисление погрешностей теряет смысл.
Баланс тепла при горении
В работах [1, 4, 5] и др. по термопарным профилям вычислялся
юток тепла из г-фазы к поверхности заряда Хгаа (<ргаз)п • Затем вы-
шсля'лся поток тепла, выделяющегося в к-фазе.
ЧКф=рис(Т„-Т0) —Хгм((ргаз)„ (13)
где pUc(TJ( — То)—полный поток тепла, необходимый для нагрева
к-ф^ЗЫ от температуры То до температуры Тп.
Такой метод имеет следующий принципиальный недостаток.
В данном варианте уравнение (13) не является балансовым. Действи-
тельно, при сведении баланса каждое слагаемое должно измеряться
независимо, и их сумма сравнивается с заранее известной вличиной.
Именно так сводился баланс в работе [25] (величина <Ьф измерялась
с помощью импульсной калориметрической установки). Что же каса-
ется вычисления Яиф из (13), то такой метод можно рассматривать
лишь как качественную опенку д1гф. При этом зависимость величины
вычисленной из (13), от различных параметров во многих случаях
#6жет быть связана с чисто методическими эффектами. В частности,
ёйи с помощью данной термопары измерять профиль Т (х) при все
более и более высокой скорости горения (например, при увеличении
давления), термопара будет все сильнее и сильнее занижать поток
тепла Хгаз (?г.,3)а »' и рассчитанная из (13) доля тепла, выделяющаяся
в. к-фазе, будет стремиться к 100%, что является чисто формальным
результатом.
Сравним теперь поток из г-фазы ХРаз(<ргаз)п, измеренный в наших
ойытах, и полный расчетный поток тепла, необходимый для нагрева
й " газификации смеси ПХА + ПММА: qs = (1 — mOK) р U (Qr)a •+
qU(Qo,,)!!, где для суммарной теплоты газификации ПММА
(Qr)s примем значение (Qr)s = 380 кал/г [2б]( а суммарную теплоту
газификации ПХА (Qow)s запишем в виде:
(Qok)e = С] (240—То) + Qi2+c2 (Тп—240) +QMc-
Здесь сь с2 — теплоемкости орторомбической и кубической модифика-
ции ПХА, Q]2 — теплота фазового перехода ПХА (орторомб.)->ПХА
(кубич.); Q ДИС£,—теплота диссоциации NH4CIO4 (тв.) —* МНз(г) +
+НСЮ4 (г). Примем С] = 0,309 кал/г-град (в интервале 15—240° С)
[12], с2 = 0,365 кал/г-град. (Т>24О°С) [12], Qt2 = 2,46 ккал/моль [11],
Qwe = 58 ± 2 ккал/моль (при Т = 520—620'К, т. е. для кубической
модификации) [27],
Из табл. 4 видно, что при а = 0,75 (т. е. в восстановительной сре-
де) поток тепла из г-фазы при р^1 ата близок к , т. е. в этом
случае тепловыделение в к-фазе близко к нулю. При а = 2 (когда
термопара находится в окислительной среде) величина Хгаэ (<ргаз)п
при р = 1 ата значительно превышала q . Возможно в данном случае
термопара - завышала реальный градиент температуры в газе из-за
тепловыделения, связанного с окислением термопары.
В соответствии с замечанием, сделанным выше, сведения баланса
тепла при р> 1 ата в рассматриваемых условиях не имеет смысла.
132
Таблица 4
Теплота газификации смеси и поток тепла из г-фазы
a рФ ата U, CMjceK (Qok's , кал) г qs. кал1см2сек i ^газ 1?гзз)|ц । кал1см^‘ сек
0.75 0,34 0,035 655 34,8 38
0 63 0,072 680 74,1 104
1’ 0,093 685 101 93
2 0,28 0.020 049 21 5 18
В заключение отметим, что при достаточно низких давлениях
(р^0,7 ата) введение катализатора в смесь ПХА 4-ПФ увеличивало
примерно во столько же рость горения (см. табл. 1). раз, во сколько увеличивалась CKO-
Р. ата 0.3 0,5 О.7
Z = Uc катал/Нб£з катал 1,72 1,85 1.94
с катал/(Ттах)! без катал 1,86 2,0 2,86
Отсюда можно
давлениях действие
сделать вывод, что по крайней мере при
катализатора протекает в газовой фазе.
низких
Ф
1. Боболев В. К., Глазкова А. П., Зениц А. А., ЛеАпуискнЙ О, И.
ДАН СССР. 1963, т. 151, №3. с. 604
2. S a b a d е)) A. J., Wen og rad J., Summerfield M. AIAA J., 1965
v, 3, №9n. 1580,
3. Wise H. Ina mi S, H., Me Culley L. Combustion and Flame. [967,
v. 11, № 6, p. 483.
4. 3 e и и н А. А. Глазкова А. П-, Л e й п у н с к и й О. И„ Боболев В, К.
ФГВ. 1968. т. 4, № 3 с. 299.
5. Лейпунский О. И., Зе пин А. А., Пучков В. М. Сб. Горение и взрыв.
М., «Наука», 1972, с. 74,
6. 3 е н и н А. А. ПМТФ. 1963. 5.
7. Klein R., Mentser М.. Elbe G., Lewis В. J. Pbys. Coll. Chem.. 1950,
v. 54 № 6, p. 877.
8. Алексеев Ю. И.. Королев В. Л., КвяжицкиЙ В. П. сб. Горение
и взрыв. М., «Наука», с. >28,
9. Бахман Н. Н., Беляев А. Ф. Горенке гетерогенных конденсированных
систем М., «Наука», 1967.
10. R о s s е г W. A., I n a m i S. Н., W i s е Н, AIAA J., 1966, v. 4, № 4. p, 663.
11. Acheson R. J., Jacobs P. W. M.. AIAA, Paper, 1969. № 69 p. 447.
12. Jacobs P, W. M., Whitehead H. M. Chem. Rev., 1969, v. 69, p. 551.
13, Кутателадзе С. С., Борншанскнй В, M. Справочник по теплопередаче,
Л.-М.. Госэнергоиздат. >959.
14, Boisson J., Quentin Р. M&m des Poudres. 1961, 43, 301.
15. Friedman R. AIAA J.. 1967, v. 5, №7, p. 1217.
16. Филиппов Л. П., Юрчак P. П. IIФЖ, 1971 т. 21, № 3, с. 561.
17. Levy J, В.. Friedman R. 8-ih Symposium (lot) on Combuslion The Williams
and Wilkins Company, Baltimore. 1962, p. 663.
18. P о w I i и g J., S m i t h W. A. W. Combuslion and Flame, 1962, v. 6, № 3. p. 173.
19. SelzerH. 11-th Symposium (Int.) on Combustion. The Combustion Institub.'.
Pittsburgh, 1967, p. 439.
20. Horton M, D„ YoungbefgL, AIAA J., 1970, v. 8, № >0, p. 1738.
21, Powling J., Smith W. A. W. 10-lh Symposium (ini) on Combuslion The
Combustion Institute. 1965, p. 1373,
22. Arden E. A., Powling J., Smith W- A, W. Combustion and Flame, 1962,
v. 6, Jfti, p. 21. ___ ___
133
3 а р к о В. . Е С у х н н я и А. И., ХлевнойС. С. ФГВ, 1970, т. A, N* 4, с. 565,
!4. Guirao С., Williams F. A. AIAA J,, 1971, v. 9, №7, р. 1345,
25. Алекса идров В. В., Болдырева А. В., Болдырев В. В., Тух-
таев Р. К- ФГВ 1973, т. 9, №1, с. 140.
26. Дмитриев Б, М,, К ° 4 е т ° в О. А., Улыбни В. Б., Штейнберг А. С.
I Всесоюзный симпозиум по горению и азрыву, тезисы докладов, М., «Наука»,
1968 с. 22.
27\ Iп a m [ S. Н., Rosser W, Wise Н. J. Phys, Chem., 1963 v. 67, №5,
. p. 1077.
Раздел третий
УДК 662.24
ИЗМЕРЕН НЕ СКОРОСТИ ДВИЖЕИ ИЯ ВЕЩЕСТВА
ПРИ УДАРЕ ИА КОПРЕ
7 ' Б. Н. Кондриков, В. Д. Чубаров
При ударе по слою вещества, способного к экзотермическому пре-
вращению, в результате диссипации механической энергии происходит
разогрев н может возникнуть воспламенение. Неоднократно высказы-
валось предположение, что источником разогрева, приводящего к вос-
пламенению, является движение вещества относительно соударяю-
щейся поверхности.
Первые расчеты разогрева при движении тела в реагирующей
жидкости провел Харитон [1]. Он показал, что адиабатическое сжатие
жидкости при ударе и разогрев вне пограничного слоя не могут быть
причиной воспламенения, и что такой причиной является работа про-
тив сил вязкости в пограничном слое. Величина разогрева при дви-
жении тела в жидкости дается простой формулой
где U — скорость движения жидкости относительно поверхности тела;
с — теплоемкость. Разогрев пропорционален квадрату скорости. Из
(1) следует также, что разогрев не зависит от вязкости.
При экспериментальной проверке Харитон столкнулся с тем, что
возникновение взрыва существенно облегчается присутствием в жид-
кости пузырьков газа [2]. Вопрос о роли пузырьков был подробно
рассмотрен Боуденом [3].
Важность перемещения твердого вещества при ударе, приводя-
щем к воспламенению, была показана работами Холево [4] и Андреева
[5]. Было установлено, что воспламенение происходит только в том
f случае, если вещество деформируется и движется с достаточно боль-
'шой скоростью (течет).
; Ридил и Робертсон [6] измерили время от начала удара до возник-
новения взрыва (0,2—0,4 мсек) и оценили среднюю скорость
iтечения (103 см/сек). Они рассчитали разогрев при течении расплава
Между частицами твердого вещества с этой скоростью и получили
136
около 1500°. При этом, однако, не была учтена роль плавления [3],
ограничивающего разогрев. В случае низкомолекулярных органиче-
ских веществ температура плавления низка, и разогрев, необходимый
для воспламенения, возникнуть не может. Ридил и Робертсон, Козлов
и Болховитинов [6—8] предположили, что воспламенение может быть
результатом повышения температуры плавления ВВ под действием
высокого давления, развивающегося при ударе. Обстоятельные изме-
рения показали, что среднее давление при ударе составляет несколько
тысяч атмосфер и что существует критическое давление, ниже кото-
рого горение не возникает. Величина критического давления недоста-
точна для повышения температуры плавления до температуры вспыш-
ки. Предполагалось [8], что давление в центральной части ударника
в 2—3 раза выше среднего, и температура плавления при ударе близ-
ка к температуре воспламенения. Вопрос о скорости движения, харак-
терной для удара по тонкому слою органического вещества и необхо-
димой для его воспламенения, до сих пор оставался открытым.
Бриджмен, изучая действие гидростатического давления на органиче-
ские вещества, обнаружил, что «если начальная толщина диска
велика, прн возрастании давления возникает неустойчивое состояние,
и большая часть вещества диска выбрасывается в стороны с огром-
ной силой; Этот процесс сопровождается местным разогревом» [9].
В книге [8] указывается, что время разрушения по порядку вели-
чины составляет 10-5 сек и что, следовательно, скорость движения
вещества при разрушении -^lO5 см{сек.
В работе [10] установлено, что воспламенение Органических ве-
ществ при ударе не происходит, если затраты энергии на деформацию
и разрушение ниже некоторой предельной величины. В предположе-
нии, что энергия расходуется на скольжение вещества по слоям рас-
плава на поверхности ударника и наковальни и переходит в кинети-
ческую энергию осколков разрушенной таблетки, расчет дал для кри-
тической скорости, приводящей к воспламенению дины и тэна, -около
^00 м^сек *.
Необходимость прямого измерения скорости совершенно очевидна.
Методика проведении опытов
Опыты проводили, измеряя скорость движения таблетки веще-
ства, находящейся между плоскими торцами цилиндров (роликов)
из закаленной стали диаметром 19 и высотой 20—25 мм. Плоские
поверхности роликов полированные. Таблетки прессовались при дав-
лении 2500 кг/см2 до плотности 0,95 от плотности монокристалла. Диа-
метр таблеток 10 или 15 мм, толщина от 0,1 до 0,6 мм.
Скорость перемещения границы слоя вещества определяли с
помощью скоростного фоторегнстра ЖФР-3. Ось вращения зеркала
располагалась параллельно торцам цилиндров. Боковая поверхность
таблетки проецировалась на пленку в натуральную величину (объек-
тив «Юпитер-9» с переходным кольцом, расстояние от объектива до
объекта съемки 28 см). Скорость развертки во всех опытах 200 Mjcea.
* В |10 и 8[ было показано также, что разрушение таблетки, сопровождающееся
воспламенением и взрывом, происходит и при статическом сжатии на гидравличе-
ском прессе, причем не только прн подъеме давления, но н прн резком сбросе его
после окончании прессования.
136
Удар ио верхнему цйлйнДру производили паДаюЩйм грузов Мас-
сой 10 кг. Высота падения 20—45 см. Коэффициент восстановления
копра 0,70±0,02. В каждой серии при идентичных условиях проводили
по 10 опытов.
Груз при ударе замыкал контакт импульсной лампы-вспышки
ИФК-120, которая была установлена позади цилиндров с таблет-
кой и использовалась для подсветки. Длительность свечения импульс-
ной лампы 1 — 1,2 мсек. Съемка велась в проходящем свете, на пленку
падала тень от таблетки, вырезавшая в засвеченной части кадра
полосу, ширина которой равнялась диаметру таблетки. При ударе
диаметр таблетки увеличивался, полоса расширялась. Наклон границы
светлой полосы на пленке к оси времени характеризовал скорость
перемещения боковой поверхности слоя вещества в пространстве меж-
ду соударяющимися поверхностями и на небольшом расстоянии
(~2—3 иж) вне его.
В ряде случаев съемка производилась с помощью скоростного
фоторегистра СФР в режиме лупы времени с частотой 60 000 кадров/се к.
Остальные детали проведения опыта оставались без изменения.
Измерение давления при ударе производилось проволочным тен-
зометрическим датчиком сопротивлением 100 ом, наклеенным на ци-
линдрическую поверхность стального ролика диаметром 10 мм, уста-
навливаемого под цилиндр с таблеткой испытуемого вещества. Тензо-
датчик через усилитель постоянного тока подключали к катодному
осциллографу ORION-KTS тип 2780-С. Тарировку датчика осуществ-
ляли статическим давлением на гидравлическом прессе.
Подробно изучалось поведение при ударе шести Органических
веществ с различной температурой плавления: динитроксиэтилнитр-
амин (дина, tu/l = 52,5°), тринитротолуол (ТНТ, 1пд=60,2°), бензойная
кислота (1ил = 122°), тринитрофенилметилнитрамин (тетрил, tUJ) = 130’),
пентаэритриттетранитрат (тэн, tM~ 141° и циклотриметилентрнцигра-
мин (гексоген, (пл-202°). Изучались также, некоторые другие веще-
ства: тринитрофенол, три нитроксилол, гексанитродифениламиЩ :фкто-
ген н др.
Общая картина процесса при ударе по таблетке реакционноспо-
собного вещества состоит в следующем. Вначале давление растет
примерно пропорционально времени, и происходит относительно мед-
ленная (обычная до 10 м[сек) деформация таблетки. В конце периода
медленной деформации диаметр таблетки на 5—20% больше, чем
исходный.
Затем внезапно таблетка разрушается. Зазор между соударяю-
щимися поверхностями заполняется быстро расширяющимся вещест-
вом, скорость движения которого в десятки и сотии раз больше, чем
предшествующей медленной деформации. Давление резко падает.
С боковой поверхности таблетки в окружающую среду выбрасываются
тонкие расширяющиеся по мере движения струи вещества. Струи
следуют Друг за другом, приaesif скорость их становится все меньше.
После падения давление начинает расти вновь.
При разрушении из зазора выбрасывается только часть вещества,
и обычно расходуется лишь небольшая доля энергии удара. Поэтому
вслед за первым разрушением нередко происходит второе, скорость
которого обычно больше, чем первого (соответственно более высокому
давлению). Иногда наблюдается и третье разрушение. Следы второго
137
третьего разрушений, а также следы струй, истекающих в окружай-
те пространство, замерялись на периферии кадра (вне роликов),
В тех опытах, в которых наблюдалось воспламенение (или взрыв),
xejit на периферии кадра, вне роликов, резко обрывался. Больше раз-
ушений не происходило. Как правило, скорость движения, измерен-
ия по наклону следа на пленке, в случае взрыва была больше, чем
случае отказа. Воспламенение происходило по-разному: в одних
тучаях можно было заметить дым, залах продуктов разложения,
следы горения на небольшой части поверхности роликов]
других — сильный звук, пламя, ожог по всей поверхности торца.
В самых общих чертах поведение таблетки вещества при ударе
ндно из рис. 1, на котором приведены несколько фотографий и схема-
дщёрки изображена типичная картина, получающаяся иа пленке в ре-
оде развертки.
n n а а а п о
JU Q о
ft
,и
1£
__-ft
з /
I б О I 61
О п В п ввв
[ПГ-
а о о
Рис 1. Типичйые фоторегнстрограммы и схе-
матический рисунок (а), Показывающие пове-
дение таблетки при ударе, а. /—Ролики; 2—
таблетка; 3—струи вещества, истекающие в
окружающее пространство; d— диаметр таб-
летки; и0 и t0 — скорость и продолжитель-
ность медленной деформации, U,, Uj и Ua—
скорости последовательных разрушений,
б. Днна. Ь—0,17 мм, d=10 мм, энергия удара
2 кгм, отказ, в. Тэн. h—0,15 мм, d-10 мм,
энергии удара 2' кгм, взрыв. Скорость раз-
Результаты опытов
fil, L,Медленная деформация. Скорость медленной деформа
Ии слабо зависит от энергии удара (имеется тенденция к возрастанию
Порасти при увеличении высоты падения груза) и растет при увели-
ецт,‘толщины таблетки (рис. 2). Для легкоплавких веществ (tnjl <
^ДЗО0) период медленной деформации меняется от 0,05 до 0,2 мсек,.
П^рдабо уменьшается при увеличении энергии удара и толщины таб-
етцд (рнс. 3). Величина относительной деформации перед разруше-
чём \ («о = До/d) для разных веществ меняется в пределах 5—20%
; ;рдабо растет при увеличении толщины таблетки. Средние значения
Ja, to и ео для различных веществ приведены в таблице.
Для легкоплавких веществ не замечено никакой связи между
арактернстнками медленной деформации (LJ0, to, ео) и вероятностью
оспЛаменения; точки, отвечающие опытам со взрывами и отказами,
координатах масса таблетки — свойство располагаются совершение
беспорядочно (рис, 4).
Для^йцеств с более высокой температурой. плавления наблюда-
лись небощяно большие периоды медленной деформации — до 0,4—-
0,5 мсек, сравнимые со временем достижения максимума давления при
холостом ударе. Затем происходило разрушение и, как правило,—
взрыв (рис, 5). Значения Uo в опытах со взрывом ие отличались от
получаемых в опытах с отказами.
Рис, 2. Зависимость скорост» медленной деформации от толщины
таблетки (энергия удара 2, 3 и 4,5 кгм). а. 1—Дина; 2—тетрил;
3—гексоген; 4—тэн (О) и бензойная кислота (♦). б. Тротил.
/—Энергий удара (кам) и диаметр таблетки (мм): /—3; 10; 2—4,5;
10; 3-4,5; 15
Характеристики поведения некоторых веществ пря ударе,
(днаметр таблетки 10 мм, толщина 0,1—0,6 мм)
Вещество Энергия удара, кгм U,. м/сек to. м/сек *0. % U., км/сек
Дина 2 2,7 0,1 6 0,55
Тетрил 2; 3; 4,5 3,5 0.05 4 0,3
Тротил Бензойная 3; 4,5 в 0,04 в —
кислота 3 6 0,06 9
Тэн 2 6 0,12 17 0,25
Гексоген 2; 3 4,2 0.16 13 0,35
Примечание: По — средняя скорость медленной деформации; to — период мед-
ленной деформации; — относительное увеличение диаметра
таблетки в конце периода медленной деформации; U*—критиче-
ская скорость разрушения, необходимая для взрыва.
2. Разрушение, Зависимость скорости первого разрушения от
толщины слоя (h) показано на рис, 6. Приведены только те скорости,
на которые не оказывает влияния воспламенение вещества, В случае
днны, тротила и тетрила воспламенения при первом разрушении не
происходило. Для них приведены все значения Uj. В случае тэна и гек-
согена даются значения иг только в тех опытах, в которых момент вос-
пламенения (резкий обрыв следа на периферии кадра) далек от первого
разрушения. Для всех веществ скорость при разрушении в отличие
от скорости медленной деформации существенно уменьшается при уве-
личении толщины таблетки. Можно отметить, что при повышении тем-
пературы плавления вещества скорость имеет тенденцию увеличи-
ваться;
139
Скбрость при втором разрушении определяется на коротком уйй-
тке дне роликов. Она практически не зависит от первоначальной
олщйиы таблетки и для разных веществ меняется от 200 до 500 м!сек.
Рис. 3. Зависимость продолжительности
периода медленной деформации от тол-
щины таблетки и энергии удара: а. Тет-
рил. d = 10 мм. Энергия удара, кгм:
/--2, 2—3; 3—4,5. б. Тротил: 1—3;
2—4,5; 3—4,5 (d=15 мм). в. ф—тетрил
(2, 3 н 4,5 кгм); Q—бензойная кислота
(3 кгм); Д — дина (2 кгм); О — тэи
(2 кгм); + гексоген (2 и 3 кгм.)
--------------------->-----,----------
Если при ударе не происходит разрушения с выбросом вещества,
то таблетка слегка увеличивается в диаметре (иногда растрескивается)
и к поверхности ролика не прилипает. При разрушении без воспламе-
нения легкоплавкие вещества остаются на ролике в виде тонкого слоя
^стывшего расплава, трудиоплавящиеся (особенно октоген), как пра-
*Йло,“ в виде расплющенных кусочков и осколков.
Рис. 4, Скорость (а) и
ток тетрила диаметром
период (б) медленной деформации табле-
10 .«ж Энергия удара 3 кгм. ф—взрын;
О — отказ,
3, Воспламенение. В этих опытах нас интересовали главным
образом условия возникновения разогрева, необходимого для воспла-
• 140
менеии#. >ПЙи*>му'. характер распространения, скорость горения й пере-
ход его в детонацию здесь не рассматриваются.
Рис. 5. Скорость (а)' и период (б) медленной деформации таблеток тэиа
диаметром 10 мм. Энергия удара 2 кгм. ф —взрыв; О —отказ; + - про-
Рис. 6. Скорость движения (Ut) таблетки в пространстве
между роликами при первом разрушении, а. Тротил. Энер-
гия удара (кгж) и диаметр таблетки (мм): 1—3,- 10; 2—4,5;
10; 5—4,5; 15, б. /—днна; 2—бензойная кислота; 5—тетрил;
4—тэи; 5—гексоген. Энергия удара 2, 3 и 4,5 кгм. Диаметр
таблетки 10 мм. Каждая точка — среднее из 10—30 опытов
На рис. 7 приведены наибольшие в каждом опыте скорости Ult
Us или U3. Существует предельная скорость движения вещества в про-
странстве между соударяющимися поверхностями, выше которой во
всех опытах происходит воспламенение, ниже —наблюдается отказ.
Эта скорость не зависит ни от толщины и диаметра таблетки, ни от
энергии удара и для разных веществ составляет от 0,25 до 0,55 км!сек
Hi
'fJ&Mirika). В опытах -с гексогеном на нее не повлияла также флегма-
изация вещества (рнс. 7, г и д). Для чистого1 гексогена и с добавлё
1ием 5,5—6% флегматизатора она примерно одинакова —350 м!сек.
ГП,МГ
4
ГП, мг
_i__I__L—I___1___I__I__I__I__L.
10 20 38 40 Я 60 ft 08 90 ftt
m, мг
Pfec. 7. Наибольшие в каждом опыте скорости движения при воспламенении (черные
точки) и отказе (светлые точки) в зависимости от массы таблетки, а. Дина. Энергия
удара 2'кгм. d—10 мм. б. Тэн. Энергия удара 2 кгм, d=10 мм. в. Тетрил. d-Ю мм.
Энергия удара, кгм: Д —2; □—3; О — 4Д г. Гексоген, d = 10 мм. Энергия удара,
У*-’ Q —2; Д — 3. д. Гексоген флегматизнрованиый. О — энергия удара 3 кгм.
®“Юж.А—энергия удара 4,5 кгм. d—15 мм. е. Октоген. d"10 мм. Энергия
'J i удара, кгм: 0—2; Д—3
Ск^^ЖЙвЧЖВиЖеиия вёщества " при Взрыве мо!кет быть гораздо
больше, чем при отказе, очевидно, в результате ускорения за счет
воздействия продуктов горения и взрыва. В некоторых опытах наблю-
дались скорости до 1,5—2 км/сек.
Обсуждение результатов
I. Медленная деформация. При ударе по слою твердого
вещества, находящегося между жесткими поверхностями, начальной
стадией процесса является рост давления. Слой предварительно спрес-
сованного высокоплотного вещества вначале деформируется упруго.
Однако прочность низкомолекулярных веществ мала (обычно десятки
кг/сж2), н после небольшого участка упругой деформации вещество
начинает «течь». Происходит своеобразная пластическая деформация,
прн которой увеличиваются одновременно давление, плотность и проч-
ность вещества.
Когда давление достигает величины [8]
/ d \
р* "Ц1 + з/зъ) , , (2)
происходит разрушение. Величина предела прочности cf зависит
от температуры плавления вещества [8]
<Г1 = 5(ТГО—То), кг/см2 (3)
где Тш— температура плавления; То—начальная температура; of—
предел прочности в кг/см* [8].
Разогрев и зажигание при трении рассмотрены в [11].
Удельная работа деформации
Ac^r-Ad (4)
где т —среднее касательное напряжение сдвига. Согласно [8, 11],
- _ Тш —Т
Т~2]/3 Тт —То
где Т — температура разогрева; То — начальная температура.
/ X • t0 \’ft
Разогрев слоя толщиной й ~“ j (X, с, Q — коэффициент
теплопроводности, удельная теплоемкость и плотность) происходит за
счет работы трения (4)
(Т.-То)/ДТ- I+5-I0-’ (5)
Прн скольжении по поверхности стали можно принять коэффи-
циент теплового насыщения (й,.с,р) 1,3- 10т эрг/(смРград • сек4*).
Тогда величина в правой части (5) при Uo — 5 -101 см/сек, Ad~O,l см
получается равной 2. Температура разогрева приближается к темпера-
туре плавления. В случае многих ВВ температура плавления гораздо
ниже температуры воспламенения. Поэтому воспламенение на стадии
медленной деформации маловероятно.
Этот вывод соответствует результатам опытов: в случае легко-
плавких веществ взрыв возникает не на стадии медленной деформа-
МЗ
1
[И, в лишь (с большей или меиыцей— до 0,3 -мсск-задержиой) после
эрушения, И судя по рис, 7, только в том случае, если: скорость
зрушення достигает критической величины (U„),
2, Разрушение, Можно предположить, что скорость разруше-
1Я определяется упругой энергией запасенной на первой стадии уда-
। Считая что вся упругая энергия таблетки перешла в кинетиче-
(Р,-=)г p-U?
уэнергию осколков, получим —или U|=(Pi—о) /
qE)1'1, где Е — модуль упругости. Разность (Р] — о) отражает тот
акт, что разрушение возможно только, если Р, > о ~ Oi,
На рис. 8 результаты опытов с тетрилом и тэяом (рис. 6) пред-
авлеиы в координатах Р,—U,, Значения Pt рассчитаны по формуле
!) с? учетом увеличения диаметра таблетки при медленной дефор-
зпни. На ту же прямую ложатся точки для бензойной кислоты,
равнение прямой рис, 8:
U, = 32(Р] — 0,6), м)сек\ (Р,, кбар) (6)
кспериментальные значения ст; для тетрила 0,53, для тэна 0,60 кбар
J. Коэффициент перед скобкой—это (рЕ1Г,/’. Он равен 32 мЦсек-квар),
yin принять q — 1,6-103 кг/м3, а Е = 64 кбар (это отвечает скорости
1ука в ВВ 2 км/сек). Эти величины очень близки к реальным, В pa-
ne [12] аналогичные результаты были получены для янтарной кис-
эты и кристаллогидрата щавелевой кислоты.
Заметим, что в случае тонких таблеток средние скорости ниже,
jm .следует из формулы (6), Для дины и тротила разброс результатов
эльше, ' ' "
/сек.
и может быть получено приближенное выражение; U|C^35 Pi,
Рис. 8. Зависимость скорости разрушения (Ui) тзна
(О, энергия удара 2 жал); тетрила (ф, энергия удара
2, 3 и 4,5 кгм и бензойной кислоты (С|, энергия удара
3 кгм) от давления в момент разрушения (Pi), <1=10кя
Рис. 9. Зависимость давления разрушения Pj от
h/d для таблеток флегматизированлого гексогена диа-
метром 15 мм. Кривая проведена по формуле (2) для
- таблеток диаметром 10 мм
S
б ’
4
S,B2 ЗМ
h/d
2
Зависимость давления Р, от h/d для таблеток флегматизирован-
юго гексогена диаметром 15 мм приведена на рис, 9, Кривая прове-
яна по формуле (2) при ст, = 340 кг/см2 [13] для таблеток диаметром
0 мм. '
144
3. В осд>л вменение. При большой скорости, возникающей
в результате разрушения, разогрев более интенсивен, и температура
в тонком слое вещества у поверхности скольжения быстро приближа-
ется к температуре плавления, Достижению температуры плавления
способствует уменьшение последней при снижении давления: при раз-
рушении таблетки давление падает, и слой, температура которого была
ниже Тга, оказывается нагретым до Т^ТП(. Внешнее трение на поверх-
ности скольжения переходит во внутреннее —в тонком слое расплава
вблизи поверхности.
Обычно полагают [3, 7, 8], что температура слоя расплава не
может быть выше температуры плавления. При большой скорости
движения слоя это не так,
Составим уравнение баланса тепла для плоского слоя расплава
толщиной 6, ограниченного с обеих сторон веществом с температурой
плавления T,j . 1
tjU
—• dx — Q| + Q?,
где Qi = fi.c.Q.dT — расход тепла на прогрев жидкости; Q2 = Q'.jj.dfi =
2X-Nu(T —Т„,)
(7)
~------Ги--------расход тепла на прогрев и плавление твердого
вещества; Q'= Q,tl+c(Tm—То) — сумма теплот нагревай плавления;
Т — средняя температура слоя жидкости, Nu — критерий Нуссельта,
т) — коэффициент вязкости. ’ ’ ‘
В безразмерных переменных уравнение имеет вид
&ede = z(i-9)d£
при условии: £=1, 0=0,
2X-Nli 2‘X-NuQ,
(Т—Т.,,) —разогрев; % = ' с;' ;- — безразмёрны и пара-
метр; —— — толщина слоя. Решение уравнения (7) при % = const
zM = 4in(i-0)+e]
Предельное значение безразмерного разогрева 0 равно 1. Такой разо-
грев достигается при толщине слоя In g > 2/%. Наибольший разогрев
(при 0=1)
ДТ - Т — Тт - 2K,Nu
При U = 3-10* см/сек; р = 0,1 г см Iсек, А, = 1,5- 10х эрг/см-сек, град,
Nu = 6 получим АТ = ’500°. Максимальная температура в центре слоя,
естественно, еще выше. Наименее определенной величиной является
здесь коэффициент вязкости расплава при высоких температуре и дав-
лении. Не учтена также зависимость р от температуры. Поэтому полу-
ченную величину следует рассматривать как ориентировочную, пока-
зывающую, что разогрев в жидком слое возможен н, в принципе, мо-
жет быть достаточен для воспламенения.
Можно полагать, что в случае относительно легкоплавких веществ
реализуется механизм, согласующийся с наблюдениями английских
исследователей [14]. При разрушении и истечении вещества с большой
скоростью оно плавится, слой расплава нагрет до высокой темпера-
туры и через некоторое время воспламеняется. Это, в частности, отве-
10 Труды вып. 83
taer опытам, в которых воспламенений происходит после разрушения
: некоторой задержкой (диНа, тетрил, тэн). В ряде опытов с тэном,
ексогеном, октогеном воспламенение происходило в момент первого
нарушения: скорость движении фронта вещества намного превышала
)бычную скорость разрушения (6), достигая 1,5—2 кж/сек. Можно
1редволожмть, что в этих опытах в самом начале разрушения веще-
ство быстро нагревается до температуры воспламенения.
Тот факт, что критическая скорость, необходимая для воспламе-
нения, в случае флегматизироваиного и не флегм аудированного гек-
гогена практически одинаков^, позволяет предположить, что толщина
2Л0Я, нагретого до температуры воспламенения, больше, чем толщина
Моя флегматизатора.
Г-'-
1. Харитон Ю, Б. Сборник статей по теории взрывчатых веществ, под ред.
К К. Андреева и Ю. Б. Харитона, М., Оборонгнз, 1940, с. 177.
^Зельдович Я. Б. ЖЭТФ, 1942, т. 12, вып. 11—12, с. 498.
гЗйВоуден ф„ Иоффе А. Возбуждение и развитие взрыва в твердых н жидких
,, веществах, М.. ИЛ, 1955.
£ Холево Н. А- Сб. «Теория взрывчатых веществ», под ред. К. К, Андреева,
Ф. Беляева, А. И. Гольбнндера, А. Г. Горста, М., Оборонгнз,
V 1963, с. 5.
^ Андреев К. К. Там, же, с. 37.
,№Rideal Е. К\ Robertson A. J. В,, Proc. Roy. Soc. (London). 1948,
v. А 195, р. 135,
'^Болховитинов Л. Г. ДАН СССР; 1959, т. 125, № 3, 318.
'^Афанасьев Г. Т., Боболев В. К. Инициирование твердых взрывчатых
( йеществ ударом, М., «Наука», 1968.
ft Бриджмен П, В. Новейшие работы в области высоких давлений, М,, ГИНЛ,
1948.
JQ, К о н д р и к о в Б. Н., Ч у б а р о в В. Д. ФГВ, 1970, т, 6, № 3.
IK Амосов' А. П_, Бостанджнян С. А., Козлов В. С. ФГВ, 1972, т. 8,
:: № з, с, 362.
К, А ф а н а с ь е в Г. Т., Боболев В. К-, Д У б о в н к А. В. ПМТФ, 1971, № 3.
IS‘K а р п у х и н И. А., Боболев В. К. ФГВ, 1967, 3, № 4, с. 485.
14. Heavens S. N., Field J. Е. Combustion and Flame, 1972, p. 473.
- - --- . УДК 662.2
ДЕТОНАЦИЯ РАСТВОРОВ
НА ОСНОВЕ ЖИДКИХ ИИТРОЭФИРОВ
5. Н. Кондриков, В. М. Райкова
Детонация жидких взрывчатых смесей на осцове ннтроэфиров
изучалась в работах [1, 2J. Определялись критический диаметр дето-
нации в стеклянных трубках и критическая концентрация раствори-
теля при данном диаметре.
В работе [1J приведены данные Р. X. Курбангалиной по критиче-
ским диаметрам детонации смесей нитрогликоля с метанолом, хлоро-
формом и бромоформом. Авторы пришли к выводу, что химическая
природа растворителя не влияет на детонационную способность рас-
твора. Растворитель в значительной своей части испаряется и раз-
брызгивается без разложения, и реакция химического взаимодействия
его с продуктами распада ийтрогликоля происходит иа более поздних
стадиях процесса. В детонационной волне молекулы растворителя
играют роль изолирующей преграды, препятствующей прямой переда-
че реакции к соседним молекулам ВВ. Критический диаметр детона-
ций растворов определяется содержанием растворителя, выраженным
в мольных долях, т. е. зависит от молекулярного веса добавки.
В работе [2] изучалась детонация смесей метилнитрата с раство-
рителями различной плотности в стеклянных трубках диаметром
10 мм. Было установлено, что предельное содержание растворителя
при детонации с малой скоростью возрастает при увеличении плотно-
сти растворителя. При плотности 0,78 г/см3 (этанол, бензол) оно равно
10%, а при плотности 1,5—1,6 г/см3 (CHCI3, CCU) —40 — 50%, Опреде-
ление критического содержания растворителя, при детонации с боль-
шой скоростью дало неожиданный, результат: для хлороформа (плот-
ность 1,5 г/см3) оно равнялось 24%, а для ССЦ (плотность 1,6 г/см3)—
57%. Было высказано предположение, что такое различие в детона-
ционной способности смесей связано с величиной дипольного момента
растворителей: для СС1<он равен нулю, а для хлороформа:—1,06(?ебая.
Почему дипольный момент должен влиять на детонационную способ-
ность и именно так, а не иначе, авторы не пояснили.
10* 147
Задача настоящей работы заключалась в том, чтобы изучить
влияние природы растворителей на детонационную способность смесей
несколько более подробно, Опыты проводились с метилннтратой и
нитрогликолем, В качестве растворителей применяли метанол, толуол,
нитробензол, хлороформ, бромоформ, фреон,СС1<(2 от 0,8 до 2,9 а/см3)
(табл. 1), Для того чтобы получить более полную картину влияния
растворителей, опыты проводились как в стеклянных, так и в толсто-
стенных стальных трубках *.
Таблица 1
Характеристики растворителей
Растворитель Квалификация Плотность, г;с.ч^
Метанол ч, д, а. 0.79
Толуол ч, д, а, 0,87
Нитробензол чистейший (ГДР) 1,20
Хлороформ чистый (перегнан в интервале 60—61°С) 1,49
Четыреххлористый углерод технический (перегнан в интервале 76—77°С) 1,59
Фреон 114В2 (CjFfBr3) технический 2.18
Бромоформ ч, д. а. 2,89
Методика проведения опытов
Ннтрогликоль и метилнитрат получали обычным способом, Ме-
трл^итрат перегоняли при атмосферном давлении (температура кипе-
ния 65°С)..Нитрогликоль применяли без перегонки.
Характеристики растворителей приведены в табл. 1,
. Компоненты взвешивали на технических весах. Ошибка в содер-
жании растворителя по весу не более 0,5% абс,
.Опыты проводили в стеклянных и в стальных трубках. Толщина
ртецок стеклянных трубок 1 —1,5 мм, длина 10—12 см, внутренний
диаметр 10 мм, В ряде случаев применялись трубки диаметром 13—14
и 7 мм, эти опыты оговорены особо, В качестве свидетелей использо-
вали алюминиевые пластины. Внутренний диаметр всех стальных
Трубок 10 мм, толщина стейок 10 или 12,5 мм, длина трубок 15 см.
Для съемки процесса в стальных трубках по всей высоте были про-
сверлены 'отверстия диаметром 2 мм, расстояние между отверстиями
10—15 мм. Отверстия снаружи заклеивались прозрачной полиэтиле-
новой пленкой.
Заряды н трубках диаметром 10 мм инициировались с помощью
тетриловой шашкн весом 1,5 г, диаметром 10 мм и плотностью 1,62—
1,64 а/см3, для стеклянных трубок диаметром 13—14 мм использовали
шашку весом 2 г и диаметром 13 мм. Смеси в стеклянных трубках
диаметром меньше 10 мм инициировались непосредственно электро-
детонатором № 8,
• В проведении опытов участвовала Г, В, Филиппова,
148
?1ыЯИИИН®^*^°^ЬШо11 скоростью умесей в стеклянных труиках
.чч у'мЩДЯВус^нл етел ь обычно разрывало на -куски. Бели детонация
проходила с малой скоростью, то на свидетеле оставались следы дето-
нации.
Съемка процесса осуществлялась с помощью прибора ЖФР-3,
Все исследованные смеси представляли собой гомогенные раство-
ры. Смеси нитрогликоля с ССЦ и фреоном из-за плохой растворимости
этих веществ в нитроглнколе не испытывали.
1, Опыты в стеклянных трубках.
Результаты опытов в стеклянных трубках приведены на рис. 1
в виде гистограммы. Каждому опыту соответствует точка на горизон-
тальной прямой. По оси абсцисс отложено весовое содержание раство-
рителя. Сверху вниз располагаются
растворители в порядке возрастания
плотности.
На рисунке нанесены опыты в
трубках диаметром 10 мм, при ини-
циировании одной тетриловой шаш-
кой весом 1,5 г. Для смесей нитро-
гликоля с нитробензолом (30—40%),
кроме того, приведены опыты в
трубках- диаметром 13—14 мм, а
для смесей метилнитрата с хлоро-
формом и СС]< — в трубках диа-
метром 7 мм, (В данном случае из-
менение диаметра на результат не
влияло.)
Для смесей метилнитрата с ме-
танолом и нитробензолом детона-
ции с большой скоростью не было
Рис. 1, Детонация смесей метилни-
трата (МН) н нитрогликоля (НГЛ)
с органическими растворителями и
стеклянных трубках внутренним
Тиа метром 10 мм. 1 — большая ско-
рость; 2—малая скорость; 5—отказ
получено даже при содержании
10% растворителя. Смесь с 10% метанола детонировала со скоростью
2,14 км/сек, с 10% нитробензола — 2,21 и 2,39 км/сек и с 20% нитро-
бензола— 2,08 км/сек. В отличие от метилнитрата нитрогликоль
в смеси с 20% нитробензола детонировал с большой скоростью —
6,81 км/сек. Кроме того, была получена детонация для смеси с 10%
и 15% толуола. Причем в одном опыте для смеси с 15% толуола
в конце заряда как на свидетеле так и на пленке обнаружено затуха-
ние процесса.
Результаты опытов со смесями метилнитрата с СНС13 и CClt при
инициировании тетриловой шашкой весом 1,5 г согласуются с данны-
ми, приведенными в работе [2]. Детонация с малой скоростью прекра-
щается при содержании 50—60% растворителя. Детонация с большой
скоростью для СС14 наблюдается при 50%, тогда как в случае
СНС1з — при содержании не более 30% растворителя. Замена метил-
нитрата на нитроглнколь в смесях, содержащих хлороформ, привела
к повышению детонационной способности. Смесь нитроглнколя с
CHC1S детонировала точно так же как смесь метилнитрата с СС14:
при 50% добавки с большой скоростью, при 60%—с малой.
Для смесей метилнитрата с фреоном и бромоформом, а также
для смесей нитрогликоля с бромоформом детонации с малой ско-
ростью получено ие было. Эти смеси либо детонировали с большой
149
бкбростЬйУ, лнйо йе1 ДётойироЬали совсем: Йетонацня ьбрывйлась,
пройдя по заряду 2—4 см.
2.’0пыты в стальных трубках.
Результаты опытов в стальных трубках приведены на рис. 2.
При детонации смесей с большой* скоростью стальные трубки дробило
на куски, причем число осколков уменьшалось при снижении теплоты
взрыва смеси. Так, при взрыве нитрогликоля с 20% толуола получено
17 крупных и около 20 мелких осколков, а в случае смеси с 85% бро-
моформа трубку разорвало пополам.
Рис. 2. Детонация смесей мётилни-
трата (МН) и нитроглнколн (НГЛ)
с органическими растворителями в
стальных трубках внутренним диа-
метром 10 мм со стенками толщиной
10—12,5 мм. ф — Большая скорость;
(>— малая скорость; О — отказ
При детонации с малой скоростью свечение фронта видно плохо,
либо не заметно совсем. Трубка обычно не разрушается (при малом
содержании растворителя разрывало верхнюю часть трубки). В боль-
шинстве случаев наблюдалось лишь небольшое (на 2—3 мм) увеличе-
iime внутреннего диаметра трубки. В ,некоторых случаях (смеси нитро-
ИВДколя с 40—50% нитробензола) после опыта трубки оставались без
Шмейения, и о взрыве можно было судить по сильному звуку и отсут-
ствию следов жидкости на стенках трубки и камеры.
Таблица 2
Переход от детонации с малой скоростью к детонации с большой
при увеличении плотности растворителя в смеси с метил нитратом *
; Содержание раствори- lii1 теля, вес. % Плотность растворите- ля, Плотность смеси, .г'см* Скорость детонации, KMietfi
, > шреформ роыоформ
1']<| СО — IJ9 1,31 малая малая
-jti'. 1 1 " Гь.4;. 36 'И.?- 4 1,56 1,33 малая МАЛАЯ
32 'J 8 1,65 1,36 малая 2,17 5,51 5,60 5,57 5,62
12 1.74
• Теплота взрыва смесей 860—870 ккал!кг.
150
^ддцрофориоц нбро^оформои и мс
гнлццтт?^ЕдаЖ®^^₽иом и CCl*,. детонации , с,, малой скоростью
получеи^йс рыло. Для смесей обоих нитроэфиров' с метанолом,
а также для смесей метилнитрата с фреоном и бромоформом она была
подучена в узком (10%) интервале составов. Для толуола этот интер-
вал больше—20%, Смеси нитрогликоля с нитробензолом, начиная
с 35% растворителя, детонировали с малой скоростью, процесс сопро-
вождался сильным звуком и свечением во всем объеме камеры. Отказ
детонации был получен только для смеси с 75% нитробензола.
Для смесей метилнитрата с нитробензолом (так же, как для сме-
сей на основе нитрогликоля) при содержании, 30% растворителя была
получена детонация с большой, а при 35%»—с малой скоростью.
Обсуждение результатов
На рис. 3 приведена зависимость большой скорости детонации
от теплоты взрыва смеси для растворов иа основе -метилнитрата. Ре-
зультаты опытов в стеклянной и стальной оболочках приведены
вместе, так как влияния оболочки на величину скорости детонации
обнаружено не было.
ш №1 лш moi им юр
Qy'KKWtlir.
Phc, 3. Зависимость скорости детонации от теп-
лоты взрыва для смесей ив основе метилнитрата,
7--метанол; 2—нитробензол; 3— хлороформ; 4—
СС1<; 5—фреон; ,б—бромоформ; 7—смесь ХЖюо-
форма с бромоформом; 8—без растворителя [3]
Теплоты взрыва, были рассчитаны приближенно по методу
Г, А, Авакяна, При атом в отличие от некоторых предшествующих
работ принималось*, что растворитель реагирует в детонационной волне
(в смесях, содержащих галоидуглеводороды, при детонации образу-
ются галоидоводороды).
Скорость ^гонации для всех смесей линейно растет при увели-
чении теплоты взрыва:
Оболочка не влияет на величину скорости, ио влияет на предель-
ные условия детонации; для одной и той же пары интроэфир-раство-
ритель предельное содержание добавки в стальных трубках выше, чем
в стеклянных. Смеси на основе нитрогликоля в стеклянных трубках
оказались более детонационноспособными, чем на основе метил-
нитрата, В стальных трубках разница между нитрозфирами почти
незаметна.
\ В одинаковых условиях для смесей на основе одного и того же
нитроэфира предельное содержание зависит от природы растворителя.
Дак, для смесей на основе иитрогликоля в стальных трубках крити-
ческие содерм^а^ие растворителя по весу увеличивается в 3,5 раза,
151
ёсЛИ эамейить метанол на бромоформ. В стеклянных трубках разница
еще больше. Различие в предельных содержаниях растворителей со-
храняется и в том случае, когда они выражены в объемных или
в мольных процентах. Однозначная связь между критическим диа-
метром и мольной долей получилась в работе [1], видимо, случайно.
По нашим данным для детонации с большой скоростью в стеклянных
трубках диаметром 10 мм для смесей на основе нитрогликоля полу-
чились следующие предельные содержания растворителей в мольных
долях: тдлуол—0,3, нитробензол — 0,3, хлороформ—0,6, бромоформ-
0,6. Заметим, что в этой области концентраций критический диаметр
сильно зависит от содержания растворителя.
Из рис. 1 и 2 видно также, что предположение авторов [2] о важ-
ности дипольного момента растворителя (дипольный момент хлоро-
форма велик, поэтому предельная концентрация его мала) необосно-
ван). а) Если заменить в смесях с хлороформом метилнитрат на ниг-
рогликоль, то предельное содержание хлороформа для детонации
с большой скоростью в стеклянных трубках резко возрастает (от 25
до 55%)- б) Дипольный момент бромоформа (0,99 дебая) почти такой
же, как у хлороформа, а предельное содержание в смеси с метилни-
тратом (65%) выше даже, чем СС14. в) Различие между хлороформом
и СС14 исчезает при проведении опытов в стальных трубках: смеси
с обоими растворителями детонируют с большой скоростью при
содержании 70% растворителя и не детонируют при содержании 80%.
На рис. 4 приведена зависимость теплоты взрыва смеси в предель-
ных условиях для детонации с большой скоростью в стеклянных
трубках от плотности. Видно, что при повышении плотности критиче-
ская теплота резко падает. При изменении плотности на 25% (от 1,2
до 1,5 г/см3) Q* меняется почти вдвое (от 1200 до 700 ккал/кг), При
высокой плотности эта зависимость становится более слабой. Не-
смотря на приближенный характер расчета теплоты взрыва, примене-
ние в качестве взрывчатой основы моно- и динитрата и существенно
различную природу растворителей, разброс результатов не слишком
велик.
Рис. 4. Зависимость критической
теплоты взрыва от плотности смеси
для детонации смесей метилнитрата
(МН) и ннтротлцколя (НГЛ) с ор-
ianiru-cKHMH растворителями в стек-
лянных труокдх внутренним диамет-
ром 10 мм: 1 —метанол; 2—толуол;
3—нитробензол: 4—хлороформ; 4'—
хлороформ, бромоформ (80/20)’; 5—
СС14; б—фреон; 7—бромоформ (ини-
циатор—шашка тетрила весом 1,5г)
Были проведены также опыты со смесями метилнитрата с раство-
рами бромоформа в хлороформе. Эти данные приведены в табл. 2.
Видно, что при одном и том же содержании растворителя и почти
постоянной теплоте взрыва частичная замена хлороформа на бромо-
форм, сопровождающаяся увеличением плотности, приводит к повы-
132
шеи и юЙЙЙЙийонкой способности смеси; детонация с большой ско-
ростью возникает только при плотности смеси q^1,36 г! см1 2.
Предельная теплота взрыва для смесей в стальных трубках ниже,
чем в стеклянных. При большой плотности — для растворов с бромо-
формом детонация с большой скоростью может быть получена при
теплоте взрыва всего 200 ккал]кг.
Полученная в данной работе зависимость предельной теплоты
взрыва от плотности связана, вероятно, с повышением температуры
взрыва и разогрева в ударной волне при использовании в качестве
компонентов смеси тяжелых галоидуглеводородов. Этот вопрос будет
рассмотрен в дальнейшем.
1. Апин А. Я., Велина Н. Ф. сб. «Взрывное дело», 63/20, 5967, М., «Недра», с. 5.
2. Kusakabe М, М., Fujiwara S, 5-th Symposium on Detonation, 1970, p, 203.
УДК 662.22(4 "36.7(1
ИССЛЕДОВАНИЕ УДАРНОЙ АДИАБАТЫ
АММИАЧНОЙ СЕЛИТРЫ
В. Г. Хотин, С. П. Бачурин, В. А. Пономарев
Аммиачная селитра — основной компонент смесевых ВВ, нашел- '
их широкое применение как в мирной промышленности, так и в воен-
эм деле. Знание закономерностей поведения аммиачной селитры
ри сжатии ее ударными волнами могло бы объяснить многие особен-
>сти процесса возбуждения и распространения детонации в смесевых
В, интересующие практику.
Ввиду того, что теоретический расчет уравнения состояния кондеи-
зрованных веществ при тех высоких давлениях и температурах,
оторые реализуются в детонационной волне, в настоящее время не-
элможеи, решающее значение приобретают экспериментальные методы
сслвдования ударной адиабаты твердых тел [1]. В случае же вещестп,
пособных изменять свое агрегатное состояние при сжатии, как, на-
ример, аммиачная селитра, за счет химического разложения, экспери-
бнтальное исследование оказывается единственно возможным спосо-
ом получения характеристики вещества, сжатого ударной волной
олыпой интенсивности.
Сложность методов и малая точность получавшихся результатов,
идимо, отчасти могут объяснить то, что динамические характеристики
олыпинства компонентов смесевых ВВ до настоящего времени доста-
очно подробно не изучались. Положение улучшилось после того, как
лабораторной практике стал широко использоваться электромагнит-
:ый метод, позволивший сравнительно просто и с достаточной точ-
,остью определять параметры ударных и детонационных воли [2].
1 работе (3] определена ударная адиабата аммиачной селитры с плот-
юстью 0,86 г]см? и размером частиц 0,5—0,2 мм.
В нашей работе ударная адиабата аммиачной селитры определе-
ta при насыпной плотности 0,8 г/см3, умеренно повышенной 1,2 ejcM3
t близкой к удельному весу 1,7 г/см3, т. е. в интервале, соответствую-
цем возможному диапазону изменения плотности аммначноселитреи-
|ых ВВ в процессе их применения. Кристаллическая аммиачная
«Литра ГОСТ 37-61-65 ч.д. а. сушилась 24 ч при 80° С и измельчалась
154
в мельнице в течение 1,0 ч. для опытов нсполь-
япнмлЗрДрИИик'- получавшаяся рассевом на ситах с размером
частиц, <1^0,06 мм. Параметры ударных волн определялись элек-
тромагнитным методом.
В первой серии опытов источником ударной волны служили дето-
нирующие заряды, находившиеся в непосредственном контакте с тор-
цом заряда аммиачной селитры. Диаметр зарядов 20 мм. Определя-
лась скорость движения границы раздела между ВВ и аммиачной
селитрой, а также скорость движения среды за фронтом ударной
волны, в следующих друг за другом опытах, на удалении 5; 10; 15;
20; 25 и 30 мм от границы раздела. Скорость ударной волны опреде-
лялась в тех же опытах на базе 10 мм, в зарядах малой плотности
при малой скорости ударной волны
с помощью второго датчика, после-
довательно соединенного с первым,
или же, в зарядах повышенной плот-
ности при большей скорости удар-
ной волны с помощью одного дат-
чика, запрессованного в заряд, и
имевшего вторую горизонтальную
площадку (рис. 1, а, б>, тЛе. опреде-
лялась скорость ударной волны в
сечении, расположенном на 5 мм да-
лее чем то, в котором определялась
скорость среды. На большем удале-
нии от границы раздела, чем это
указано выше, параметры ударной
волны не определялись, так как
из-за влияния боковых волн разгрузки
Рис. I. Схема размещения датчиков
в зарядах аммиачной селитры с ма-
лой (а) и повышенной (б) плот-
ностью
наблюдалось резкое изменение
закона ее затухания.
Для того, чтобы определить ударную адиабату в достаточно ши-
роком диапазоне изменения параметров ударных волн, для их возбуж-
дения использовали заряды разной мощности: шашки гексогена
с плотностью 1,5 г[см\ детонировавшие со скоростью 7000 м сек, нли
же менее мощные заряды из смеси гексогена с аммиачной селитрой
30/70 с плотностью 1,3 sjCM3, детонировавшие со скоростью 4650 м!сек.
Изменение скорости ударных волн и скорости движения среды за
фронтом в зарядах селитры с плотностью 0,8; 1,2 и 1,7 г!см3 показано
на рис. 2, 3 и 4. Опыты с ударными вдлнами разной интенсивности
сопоставлены друг с другом, что позволило установить закон их
затухания (табл. 1).
Таблица 1
Плотность заряда аммиачной селитры, г[см3 Закон изменении параметров ударной волны по длиае заряда
скорость волны скорость среды за фронтом ВОЛНЫ
0,8 „ „ -0,М1 D = Doe с-, с,
1,2 D = D^'28' U = Uee"W;
1.7 D = D^ U = Uoe-0,761 ;
155
6 зарядах аммиачной селитры повышенной плотности 1,2 и
gfcM? ско'рость ударной волны и скорость движения среды моно-
ао убывают по мере удаления от границы раздела с ВВ. В зарядах
ыпной плотности 0,8 е/слР наблюдался иной тип зависимости. На
стке глубиной до 5 'мм скорость движения среды даже несколько
растала, по сравнению со скоростью перемещения границы раздела,
Рис. 2. Изменение параметров удар-
ных волн в зарядах аммиачной се-
литры с плотностью 0,8 г/см3 по мере
ударения от границы раздела с дето-
нирующим зарядом гексогена (J)
или 30%-иой смеси гексогена с ам-
миачной селитрой (2)
лишь затем падала. На этом же участке резко менялась форма
.пульса ударной волны и закон нарастания давления на ее фронте,
1 сопоставления осциллограмм видно, что в малоплотном заряде
Йтот момент на фронте волны первоначально треугольного профиля
Рис, 3. Изменение парамет-
ров ударных волн в заря-
дах аммиачной селитры с
плотностью 1,2 г/см3 по
мере удаления от границы
раздела с детонирующим
зарядом гексогена
Рис. 4. Изменение параметров
ударных волн в заряде ам-
миачной селитры с плотностью
1,7 г/см3 по мере удаления от
границы раздела с детонирую-
щим зарядом гексогена (/}
или 30%-ной смеси гексогена
с аммиачной селитрой (2)
юзннкает химпик (рис, 5), по мере распространения волны увеличи-
шется завал фронта, давление достигает максимального значения не
мгновенно, а спустя 0,1 мксек. В опытах с зарядами большей плот-
ности (1,7 г(см?) эти эффекты отсутствуют, ударная волна сохраняет
треугольный профиль с разрывом на фронте,
С зарядами нысыпной плотности (0,8 г/см?) проведена вторая
серия опытов, в которой определялось соотношение между скоростью
распространения волны и скоростью движения среды за фронтом,
причем профиль волны был треугольным, а давление на фронте нара-
стало скачком. Ударные волны разной интенсивности получали, меняя
характеристики активного заряда. Использовались смеси гексогена
с аммиачной селитрой, детонировавшие со скоростью от 2500 до
7000 м/сек. Между детонирующим зарядом и зарядом аммиачной
селитры помещали пластинку из плексигласа толщиной 5,3 лмь
В опыте определяли скорость движения границы раздела между плек-
сигласом и аммиачной селитрой и среднюю скорость ударной волны
в селитре на базе 10 мм. Затем с использованием соотношений, при-
веденных в табл. 1, вычисляли начальную скорость ударной волны.
Рис. 5. Изменение формы импульса ударной волны
по мере удалении от границы раздела с детонирую-
щим разрядом 30% ‘Ной смеси гексогена с аммиач-
ной селитрой в зарядах аммиачной селитрц с плот-
ностью 0.8 в/слг (а) и 1,7 г/см3 (б). Цифры у рисун-
ков—расстояние, прейденное ударной волной
В координатах D—U ударные адиабаты аммиачной селитры, опре-
деленные в первой серии опытов, аппроксимируются прямыми (табл. 2}.
Ударная адиабата для селитры с плотностью 1,7 г/см? легла несколько
выше (рис. 6), чем рассчитанная теоретически (3, 4]. Относительная
ошибка теоретического расчета может быть оценена в 17—20%, Экспе-
риментальная адиабата для селитры с плотностью 0,8 д/г.'з3 и с плот-
ностью 0,86 г/см\ определенная в работе (3], достаточно хорошо сов-
падают.
При различной начальной плотности ударные адиабаты аммиачной
селитры представляю^ семейство расходящихся прямых, описываемое
обидим уравнением D = Co+W. Коэффициенты Со и Л в интервале
плотности от 0,8 до 1,7 г/см? меняются пропорционально плотности
и могут быть вычислены из соотношений
Со = 2,44qo—1,28 (1)
Л = O,78qo + 0,68 (2)
Со тем болСМе,чем выше плотность аммиачной селитры
Таблица 2
Плотность аммиачной селитры, г/см3 Ударная адиабата Примечание
0.8 D = 0,48+1,3511 D - 0,4 +2,0 U для волны с разрывом на фронте.
0,86 D -0,84 + 1,42 U согласно [3|
1,2 D= 1,75+1,47 U
1.7 D -2,85+2,0 U
1,73 D - 2,20+1,96 U теоретический расчет согласно (3, 4]
Тангенс угла наклона адиабаты X уменьшается пропорционально
ййепию плотности аммиачнойселитры. Однако величина этого коэф-
Циентя определяется, по-вйдимому, не плотностью селитры, а зако-
йгерностью нарастания давления на фронте ударной волны. В удар-
ах волнах, распространяющихся в зарядах с плотностью близкой
6; Ударные адиабаты
ЯМмйачной селитры. Плот-
jBpCTb, г/см3: /—1,7; 2—
Ш [3, 41; 3-1,2; 4-0,86
[3J; 5—0,8
к удельному весу, да_вление па фронте на-
растает скачком и коэффициент X максима-
лен и равен 2,0. В ударных волнах, распро-
страняющихся по зарядам с меньшей плот-
ностью, давление на фронте растет относи-
тельно медленно. Завал фронта тем больше,
чем меньше плотность. Коэффициент X
уменьшается и при плотности 0,8 г/см?
равен 1,3.
Изменение закона нарастания давления
путем посылки в порошкообразный заряд
заранее сформировавшейся волны треуголь-
ного профиля, давление на фронте которой
меняется скачком, как это было сделано,
например, во второй серии опытов, ведет к
увеличению коэффициента X до 2,0, даже
в случае заряда селитры с плотностью
0,8 г/см3. Наклон адиабаты при малой
плотности в этом случае совпадает с на-
сДрНОм адиабаты при плотности, близкой к удельному весу, хотя
№Э'а уменьшения коэффициента Со скорость распространения ударной
l&tmt, при равном значении скорости движения среды за фронтом,
в Зйгряде малой плотности почти в 2 раза ниже (рис. 7).
На рис, 8 адиабаты аммиачной селитры с разной начальной плот-
ностью изображены в координатах Р—V. Взаимное положение ади-
абат отличается от предсказанного теорией (1] для пористых тел.
том случае, если давленыеиа фронте волны нарастает скачком,
ударная адиабата пористого вещества ложится правее сплошного,
и тем дальше, чем выше пористость, Во всем диапазоне давлений
от to до 80 Юбар при уплотнении пористого вещества плотность, рав-
ная удельному весу, не достигается. Это означает, что изначальная
пористость, вещества сохраняется даже при воздействии ударных
воли с достаточно высоким давлением, ’полного схлопывания пор не
происходит. Если же закон нарастания давления на фронте волны
иной, давление возрастает относительно медленно, как, например, на
фронте самопроизвольно распространяющихся по пористой аммиачной
селитре ударных волн, то достигается плотность при сжатии пористого
вещества даже большая, чем при сжатии сплошного.
Рис. 7. Ударные адиабаты
аммиачной селитры: 1—с
плотностью 1,7 г/с ле5; 2—с
плотностью 0,8 zjcM?, в ус*
ловиях действия ударной
аолпы треугольного профи*
ля; 3 — с плотностью
0,8 г/сле5, в условиях дейст-
вия самопроизвольно зату-
хающей ударной волны
Рис, 8. Ударные адиабаты ам-
миачной селитры. Плотность,
г/см5; 7-0,8; 2-1,2; 3—1,7;
4—0,8, в условиях действия
ударной волны треугольного
профиля
Оказывается, что конечное состояние малоплотного заряда ам-
миачной селитры, сжатого ударной волной, зависит от скорости нара-
стания давления на ее фронте. При равной амплитуде, конечная плот-
ность выше, если давление на фронте нарастает медленно.
Это обстоятельство может быть обусловлено малой скоростью
релаксационных процессов, протекающих в процессе уплотнения амми-
ачной селитры ударной волной, механизм которых, возможно, состоит
в следующем. Тепловой разогрев за счет трения между частичками
порошкообразного вещества при сжатии неизбежно вызовет разложе-
ние аммиачной селитры в местах концентрации напряжений, что долж-
но облегчить процесс уплотнения. Однако скорость разложения селит-
ры будет определяться сравнительно медленным процессом тепло-
передачи. Влияние разложения, не существенное при быстром нараста-
нии давления, проявляется лишь в ударных волнах с относительно
медленным нарастанием давления, как это имело место в опытах.
Таким образом, в работе определены ударные адиабаты порошко-
образной аммиачной селитры с размером частиц 0,1—0,8 мм при изме-
нении плртности от 0,8 до 1,7 г/см3 для случая самопроизвольного рас-
остр анемия по веществу затухающих ударных волн. Определен закон
гухания ударных волн в зарядах разной плотности. Показано, что
печное состояние сжатого ударной волной пористого вещества зави-
г от скорости нарастания давления на ее фронте, что связано, внди-
, с процессом разложения аммиачной селитры в ударной волне,
отекающим относительно медленно.
Зельдович Я- Б., Райзер Ю. П. Физика ударных волн и высокотемператур-
ных гидродинамических явлений. AL, «Наука», 1966.
Веретенников В. А., Дремин А. Н., Шведов К. К. ФГВ,, 1969, т. 5, с. 3.
Д р е м и н А. Н., Шведов К- К-. А в д о н и н О. С. ФГВ, 1970, т. 6, с. 4,
Афанасенков А, Н,, Богомолов В. М., Воскобойников И. М.
ПМ1Ф, 1969, с. 4.
УДК 662.221+536.711+531.66
О ВЫЧИСЛЕНИИ ПАРАМЕТРОВ УДАРНЫХ ВОЛН
В ИНЕРТНЫХ СРЕДАХ, ГРАНИЧАЩИХ С ЗАРЯДОМ
СМЕСЕВОГО ВЗРЫВЧАТОГО ВЕЩЕСТВА
В. Г. Хотин, В. А. Пономарев, К. В. Мясников
Задача о вычислении начальных параметров ударных волн
в инертных средах, граничащих с детонирующим зарядом ВВ, одна
из основных задач физики взрыва. Эта задача имеет большое практи-
ческое значение, так как позволяет установить количественную связь
между параметрами детонационной волны, параметрами ударной
волны в среде и .разрушающим действием взрыва. Решение задачи,
применительно к взрывчатому веществу, состояние продуктов взрыва
которого описывается уравнением Ландау-Станюковича PV8 = const
fl], дано в [2]. Имеется приближенное решение [3], рассматривающее
процесс взаимодействия: детонационной волны с инертной средой, как
абсолютно упругое соударение двух тел.
В практике лабораторных исследований приходится решать и
противоположную задачу, а именно: вычислять параметры детонаци-
онной волны или определять уравнение состояния продуктов взрыва
конденсированного ВВ по известным параметрам ударной волны в
инертной среде, граничащей с зарядом [2].
Например, в тех случаях, когда разместить датчик, используемый
в электромагнитном методе для определения скорости движения про-
дуктов взрыва в детонационной волне, непосредственно в заряде ВВ
нельзя, можно расположить его на границе раздела ВВ с инертной
средой, ударная адиабата которой известна, определить скорость пере-
мещения границы раздела, а затем вычислить параметры детонаци-
онной волны в ВВ. В работе [4] указано, что при размещении датчика
на границе раздела тротила с парафином на записи хорошо виден
химпнк на фронте волны. Точность расчета параметров детонационных
волн будет зависеть от справедливости допущений, сделанных при
выводе соотношений, описывающих закономерности изменения пара-
метров ударной волны на границе раздела двух сред. Можно было
полагать, что соотношения, полученные в [2, 3], могут быть исполь-
зованы лишь для расчета параметров детонационных волн достаточно
мощных индивидуальных ВВ. Возможность же использования метода
для оценки параметров детонационных волн в смесевых ВВ, приме-
няемых в промышленности, с низкой скоростью детонации следовало
изучить.
1 1 Труды вып. 83
161
В качестве объекта, удобного для исследования, была выбрана
1есь мелкодисперсного гексогена (средний размер частиц 5 лкл<)
хлористым натрием (0,4—0,2 мм). Меняя содержание гексогена,
эжно было в широких пределах изменять параметры детонационных
щи. 5%-ная смесь при плотности заряда 1,3 г/см? детонирует со ско-
)стью 950 MjceK, скорость детонации 80%-ной смеси 6250 м!сел.
дектромагннтным методом определялась скорость движения границы
щдела ВВ-парафин (Ux) и, с использованием соотношений извест-
ях из [2, 3], рассчитывалась скорость движения среды за фронтом
гтонационной волны в плоскости Чепмена-Жугс (U„). Парафин
арки МРТУ 6-09-6039-69 вакуумировался. Ударная адиабата пара-
нга йри начальной температуре 20° С в области давлений от 5 до
)к6ар айпрОКсимйроваЛась прямой Dx =2,ll -f-2,51 Us, излома адиа-
<ты в области давлений 60; кбар,. отмеченного в- работе [5J, не наблю-
алось. Полученные расчетом; значения., Uh [ Сравнивались с опреде-
знными экспериментально в параллельных опытах (таблица).
>держа- ДОгексо- Йа в сме- -fff. » Экспериментальные данные Расчетные Относи- тельная ошибка расчета %
DH, км>сек . UH, км/сек Uл, км/сек 0,17 0,26 0.36 0,46 0.55 0,65 0,75 0,85 1.04. 1.24 1,75 i парафина UH [2], км!сек UH [3J. км/сек
.ЙО! .у ЛМО Вн 15 L .25 Я с 35 40 «Г •1:lso Б'' *' *' При 0,95 1.50 2,10 2,65 3.20 3,75 4,12 4.45 5,15 , 5,60 6,25 расчете удар 0.35 0,47 0,59 0,70 0.79 0.88 0,96 1 ,1)4 1,38 1.30 1,57 пая 1 адиабат 0.17 0,28 0,35 0.43 0.48 0.55 0.64 0.71 0,86 1,04 1,55 экстраполи poi 0,24 . 0,31) 0.36 0,43 0,48 0,55 0,63 0.71 0,85 1,03 1,51 залась. 30 36 39 39 38 38 34 , 32 27 21» 4
^"Юба метода расчета в исследовавшемся диапазоне изменения
(йбйметров детонационных волн дают практически совпадающие ре-
тШтйты, однако относительная ошибка расчета, по сравнению с
шййриментом, велика, достигает 40% и уменьшается при переходе
Г'^йвласть детонационных волн с высокими параметрами. Причиной
цЬйь большого расхождения экспериментальных и расчетных резуль-
гОДуй служит, по-видимому, различие в составе продуктов взрыва
ЛёЛеДованных взрывчатых смесей, содержащих значительное количе-
ЯДОЭДердой фазы, в сравнении с гомогенной моделью, положенной
4бвюву расчета. Ввиду малой сжимаемости твердой фазы, при отно-
:rfrtUbHo большом (до 95%) ее содержании, изменение внутренней
Уйе|Угйи продуктов взрыва в момент выхода детонационной волны
на1’Границу раздела с парафином оказывается незначительно, и про-
ittitt Взаимодействия продуктов взрыва с инертной средой можег
5ытьрассмотрен как абсолютно неупругий удар. Необходимые огра-
Щйительные условия: система замкнута; влияние внешних сил отсуг-
^твует; осуществляется прямой центральный удар;—в'нащем случае
выполняются.
Wjf • . , ... •.
Потери Акинетической энергии в системе соударяющихся тел при
неупругом ударе могут быть вычислены из соотношения
mt • tn,
Л- o7m (у> - (l -кг) о
2(111! 4-шг)
где к — коэффициент носстановления, в случае неупругого удара
равен 0; Vt, V2— скорость тел до удара, Va = 0, Vi = UH; trit, m2 —
массы взаимодействующих тел.
Для нашего случая (!) перепишется как
A WK =
иц • т,
—3 - .из
2(mtTma)
(2>
Изменение внутренней энергии системы может быть определено, как
сумма изменения внутренней энергии продуктов взрыва и парафина
в момент удара
AW\= пт, • Ac( + т2-Ае2 (3)
где Де( — изменение внутренней энергии продуктов Взрыва; Дег —
изменение внутренней энергии среды (парафина). Для гомогенной
среды, какой является парафин, Де2 численно равно UX2I2, а масса
взаимодействующих тел Определится, как mj = 4„-— масса
продуктов взрыва; fns = pw-Dx—то же для парафина.
После подстановки в уравнение (2) получим
иг.
Ряи’Он 2 Рои-Da'
* I I J_, '--- А А
Рои-Г) н +pMDx " pMDx r’
(4)
Для взрывчатых смесей, продукты взрыва которых содержат болыпое
количество несжимаемой твердой фазы, вторым слагаемым в правой
части уравнения (4), ввиду малости величины Деь можно пренебречь
и расчет скорости движения границы раздела продуктов взрыва с
инертной средой производить по более простому соотношению
f Рон ' DH 4” Рох * Ох
На рис. I представлена зависимость между скоростью движения
продуктов взрыва (UH) и скоростью, движения границы раздела
ВВ-парафин (Ux), полученная экспериментально (кривая /) и путем
расчета в предположении, что удар упругий (кривая 2, акустическое
приближение [3]) и неупругий (кривая 3). В последнем случае, сов-
падение с экспериментом в области ударных волн малой интенсив-
ности лучшее. Первоначальное предположение о том, что процесс
взаимодействия продуктов взрыва смесевого ВВ, содержащих значи-
тельное количество твердой фазы, с инертной, средой носит не упругий
характер подтверждается. Условия упругого соударения реализуются,
видимо, только в области детонационных волн с весьма высокими
параметрами, т. е. при детонации мощных индивидуальных ВВ с высо-
кой теплотой взрыва и высокой детонационной способностью. В более
широком диапазоне наблюдаются промежуточные, между упругим и
яеунругим ударом, состояния.
11- V. х 163
Экспериментально найденная зависимость между параметрами
етонанионной волны во ВВ и параметрами ударной волны в пара-
>ине, представленная в виде кривой / на рис. 1, носит достаточно
бщий характер и подтверждается для большого числа смесевых ВВ
широком диапазоне изменения их состава и плотности заряда
Рис. 1, Соотношение между скоростью движения среды за фрон-
я„ том детонационной волны в смесевом ВВ и скоростью движение
границы раздела ВВ—парафин определенной экспериментально (/),
на условия упругого удара (2) и неупругого удара (3)
Рис. 2. Зависимость скорости перемещения границы раздела между
продуктами взрыва и парафином от скорости движения продуктов
взрыва за фронтом детонационной волны для взрывчатых смесей:
/—нитроглицерина (3—25%) с хлористым натрием с плотностью
1,3 г/см3; 2—гексогена (5—80%) с хлористым натрием с плотностью
1,3 г/см3; 3—гексогена (15%) с хлористым натрием при изменении
. плотности от 1,3 до 1,8 г/см3; 4—5% гексогена с хлористым натрием
при плотности 1,2 г/см3; 5—19,8% гексогена с хлористым аммонием
при плотности 0,9 г/см3; 5—мелкодисперсный гексоген с плотностью
0,6 г/сж3; 7—гексогена (18—80%) с аммиачной селитрой; 8—25%
нитроглицерина с аммиачной селитрой При плотности 1,3 г/см3
(рис. 2). По-видимому, продукты взрыва смесевых ВВ, детонирующих
с относительно низкой скоростью, содержат значительное количество
непрор.еа тировавшего вещества в виде твердой фазы. Эта зависимость
подтверждается также и для смесевых (JB на основе аммиачной се-
литры. По-видимому, разложение аммиачной селитры в детонационной
волне протекает далеко не полностью и сравнительно медленно.
Представленная на рис. 2 зависимость описывается уравнением
0,821V*1 (6s
и может быть рекомендована для расчета параметров детонационных
волн по известной скорости перемещения границы раздела В В
с инертной средой в модификации электромагнитного метода при раз-
мещении датчика на границе раздела ВВ с парафином. Точность
определения параметров оценивается в 5%, что достаточно для реше-
ния практических вопросов. Использование указанной модификации
электромагнитного метода значительно расширяет область его при-
менения, а также круг ВВ, для изучения процесса детонации которых
этот метод может быть применен.
Для точного расчета параметров ударных волн, возникающих
под действием взрыва смесевого ВВ, в средах с отличными от пара-
фина динамическими характеристиками, необходим эксперименталь-
If III" । ---
,
ный поиск "мвясимости, подобной приведенной на рис. 2. Для смесеных
ВВ, продукты взрыва которых содержат значительное количество
твердой фазы, расчет может быть выполнен с достаточной точностью
с использованием соотношений (6) нли (5) в предположении, что
на границе раздела между ВВ и инертной средой реализуются усло-
вия неупругого удара.
1. Ландау Л. Д., Станюкович К. П. ДАН СССР, 1945, т. 46, с, 9,
2. Баум Ф, А., Станюкович К. П„ Ill ех тер Б. И. Физика взрыва. М., Физ-
матгцз, 1959.
3. Cook М. The Sciece of High Explosives, ACS Monograph, Series Reinhold Publ.
Corp. New York, 1958,
4. Д p e м и и A. H., Саар os С. Д.. Трофимов В. С., [Ц.^еДов К, К. Дето-
национные волны в конденсированных средах, М, «Наука», 1970,
5. Д р е м и ii А. Н., К а р п у х и и И, А. ПМ1Ф, I960, с, 3.
УДК 662.221+536.711
ИССЛЕДОВАНИЕ ПАРАМЕТРОВ ДЕТОНАЦИОННЫХ ВОЛН
В СМЕСЕВЫХ ВВ ЭЛЕКТРОМАГНИТНЫМ МЕТОДОМ
В. Г. Хотин, В. А. Пономарев, Н. М, Серегина, К. В. Мясников
Взрывчатые вещества, применяемые в настоящее время в про-
мышленности, представляют собой сложные многокомпонентные си-
стемы, содержащие, как правило, значительные количества инертных
Добавок. Обеспечение надежной детонации таких ВВ — сложная
научно-техническая проблема. Одним из широко применяемых мето-
дов ее решения является использование в составе В В сенсибилизато-
ров— небольших добавок мощных ВВ, обладающих высокой вос-
яриимчивостью к детонации — гексогена, нитроглицерина или его
смесей с другими нитроэфирами, Введение Небольшого количества
(около 10%) сенсибилизатора существенно снижает критический
диаметр и повышает детонационную способность В В. Однако, несмот-
ря на широкое применение смесевых ВВ, данные о механизме хими-
ческого взаимодействия составных частей ВВ, размерах зоны и вре-
мени химического превращения крайне ограничены [!]•
' С целью исследования механизма взаимодействия сенсибилиза-
тора с компонентами селективнодетонирующего предохранительного
>ВВ в работе определены параметры детонационных волн в наиболее
.’удобных для изучения бинарных смесях мелкодисперсного (5 мкм)
,Гексогена с различными наполнителями. Такой подход позволяет одио-
Щачно определить роль каждого компонента во взрывчатом превра-
щении.
• Таблица I
Некоторые физико-химические характеристики наполнителей
Вещество Плотность | монокристал- ла, г [см3 । Молекуляр- ный нес i Теплоемкость, кал[ моль-град О О > а н f- Теплота плав- ления, кал! моль 1 ТкиЯ..;*с
Хлористый натрий 2.16 58.5 12,3 800 7220 1440
Хлористый аммоний 1,54 53.5 — возгоняется при 335 СС
Азотнокислый аммоний 1,73 80,0 31,8 169,6 1460 выше 190, разлагает- ся
166
0 ijilMPBwpwi^iniMiTTncrft "д"г'д- xjie>HCTMft. H»aTpH#, хлбс
рнстыйлЗЗ«5йяя, аммиачная селитра, т. е. вещества, обладающие
существенно различными физико-химическими свойствами (табл. i).
Размер частиц наполнителей 0,2-?0,4 мм.
Изучавшиеся взрывчатые системы,- отличались от мощных инди-
видуальных ВВ неоднородностью н наличием большого количества
твердой фазы в продуктах взрыва, Параметры детонационных волн
определялись электромагнитным методом. Метод был несколько»
видоизменен, что дало возможность использовать его для оценки
параметров детонационных волн>.как в малоплотиых, так и в высо-
коплотных зарядах порошкообразных смесевых Bfe. Датчик разме-
щался на границе раздела ВВ-парафнн. Скорость движения продук-
тов взрыва определяли по экспериментально найденной зависимости
между скоростью движения границы раздела и скоростью движение
продуктов взрыва (см. статью в настоящем сборнике).
Таблица 2
Усредненные значения параметров детонационных волн в плоскости Чеимена-Жуге
для смесей гексогрня (5 жкж) с грубодислерснымн на полните л а мн (0,2—0,4 мм)
Плотность аарядов 1,3 г/см3, диаметр 20 мм
L СКСО* ген, % D, км1сек и, км!сек Р, кбар р( г/сл3 т мксек ar мм ря, г/сле3 S п
Гексоген (5 мкм) + NaCl (0,2 — 0,4 мм)
2,5 и е д етонирует
3 0,75 — — — — . 0,09
5 0.95 0.35 4.3 2,1 2 1 0,96 0.15 1,7
10 1.50 0.47 ?.2 1,9 1.05 <>.65 0.28 2,2
15 2,10 0.59 15.9 1 8 0,66 0.51 0,40 2,5
20 2,65 0,70 24.1 1,8 0 44 1 0,40 0.50 2,8
25 Э.20 0.79 3’2,9 1,7 0,30 0.31 0.59 3,0
3(1 . 3,75 0,88 42.9 1,7 0,2 0.25 ’ 0,67 3,2
35 4.12 0,96 51,4 1,7 0.21 0.26 0,75 3,3
40 4,45 1.04 60.2 1,7 0.20 0.27 0.81 3.3
50 5,15 1,18 79 0,18 0.27 0,9.4 3.4
60 5,60 1,30 95 1,7 0.17 0.29 1,02 3,3
80 6,25 1,57 128 1.7 0,1 0.2 1.18 3,0
Гексоген (5 лкл) 4-NHiNOa (0,2щ0,4 жж)
7 не детонирует
10 2,40 0,65 20,3 1,4 0,25 0.21 0,40 2,7
15 3.30 0.89 38,2 1,4 0.22 0,25 0,54 2.7
20 3.9J 1,07 54,3 1.4 0.20 0 28 0,65 2,6
25 4,30 1.18 66,0 1.4 0.19 0.29 0.74 2.6
30 4.65 1.25 75.5 1.4 0.18 0.29 0,82 2.7
40 5,00 1.34 87,4 1.4 0,16 0.28 0,95 2,7
80 6,30 1.42 116 1.3 0.1 0 1 ] ,22 -3,4
Гексоген (5 жхж)+МНчС1 (0,2 4-0,4 мм)
10 15 20 25 2.95 4,10 4,65 0.66 0,96 1.05 н е 25,3 51,2 63,6 детонирует 0,69 0,80 0,88 3,5 з.з 3.4
1,7 1,7 1,7 1 1 1 1 1 1
30 4,95 1,10 71,0 1,7 — — 0,95 3,5 л К
35 5,15 1.13 76,0 1.7 —- — 1.01 3.5
40 5,30 1.21 83 1,7 — 1 05 3,4 О I
60 5,75 1.39 ЮЗ 1.7 — — — 1’18 Зе 1 2.8 167
60 6.00 1.56 122 1,8 — , 1,25
Для исследовавшихся смесей определены скорость детонации,
скорость продуктов взрыва, давление, время и ширина зоны химиче-
ской реакции в зависимости от содержания гексогена (до 80%) и плот-
ности заряда (от насыпного до удельного веса). Результаты экспери-
ментов приведены в табл. 2 и 3, На основе полученных данных- сде-
лана попытка оценить возможное участие отдельных компонентен,
смёсевого ВВ в детонационном превращении.
Таблица j
Усредненные значения параметрон детонационных волн
а плоскости Чепмена-Жуге дли смесей гексогена (5 лк.ч) с грубодиснерснымн
наполнителями (0,2—0,4 лл) при изменении плотности заряда
Диаметр — 20 мм
ро, г/см3 D, км/сек км'сек Р, кбар Pi, Z/CMt •с, мксек а, мм Рн г/е.и3 п
15% гексогена + NaCl
1,05 1,76 0,54 10,0 1,51 0.97 1.05 0,27 2,26
1.1 1,83 0,56 11.3 1.58 0,85 0,96 0.29 2.27
1,2 1,96 0,59 13.9 1,72 0,63 0,77 0,34 2,32
1.3 2.10 0.62 16.9 1,84 0.47 0,62 0,40 2,39
1.4 2,30 0,65 20,1 1,95 0,37 0,55 0,47 2,54
1.5 2,58 0.67 26,0 2,03 0,30 0,52 0,55 2,86
1.6 3,14 0.73 36,2 2,07 0,25 0,56 0,65 3,36
1.7 3.68 0,82 51,3 2,19 0,20 0,53 0,76 3,50
1,8 4,23 0,92 70.0 2,30 0.15 0,46 0,92 3,60
1.9 4.80 1,00 91.2 2,40 0,11 0,39 1,12 3,80
30% гексогена + NaCl
1,1 3,03 0,76 25,3 1,46 0,36 0,75 0,51 2.98
1,2 3, 10 0.85 34.7 1.60 0,26 0.61 0.58 3,00
1.3 3,75 0,93 45.3 1,73 0,21 0,54 0.67 3,03
1.4 4.12 1,00 57,7 1.85 0.18 0,52 0 77 3,10
1,5 4,48 1.08 72,6 1,98 0,15 0.47 0,88 3,10
1,6 4 85 1,13 87,7 2,09 0 13 0.45 1,00 3,29
1.7 5.20 1 20 !06,0 2,21 о,л 0 41 1,13 3.30
1.8 5.56 1,26 126,0 2,32 0,09 о:зб 1,30 3 40
1,9 5,93 1.32 143,0 2.44 0 07 0,30 1.48 3,49
18% гексогена+ NH«NO3
0.9 2,40 0,80 17,3 1.35 0,64 0,90 0,28 2,00
1.0 2,70 0,83 22,4 1.44 0,38 0,63 0,34 2,25
1.1 3,00 0,87 28,7 1,55 0,28 0,54 0,42 2,30
1.2 3,30 0.94 37,2 1,68 0,22 0,47 0.50 2,50
1.3 3,65 1,06 50,4 1,83 0.16 0.37 0.61 2,44
1. । 4,15 1,14 66,2 1,93 0,12 0.33 0,75 2,65
1.5 4,6.5 1,20 83,7 2,02 0,10 . (1,31 0.94 2,87
1.6 5,15 1 ,23 101,0 2,10 0,09 0,32 1 .19 3,18
1,68 5,55 1,25 116,0 2,17 0,07 0.28 1,50 3,44
19,8% гексогена NH4Ci
0,9 2 ;;о 0,69 14,3 1,28 0,55 0,79 0,33 2,34
i,i> 2,53 0,74 18,7 1.43 0.30 0,48 0.41 2.42
1,1 2.95 1) 8'1 26,0 1,51 0.25 0,49 0,51 2,7
1 .'1 3,50 0.85 35,7 1,59 0.23 0,56 0,6,1 3,12
1,3 4.15 0,96 52,0 1,69 0,20 0,59 0,79 3.33
1.4 4.90 1,15 79.0 1.82 0,18 0,62 1,02 3,26
1,5 5,63 1.21 108.0 1,93 0.J5 0,62 1.35 3,4
1-68
.метод позволяет получить наиболее полную
инфпр^япию' о' процессах, происходящих в детонационной волне.
Использование этого метода позволило исследовать форму импульса,
закономерности нарастания и спада давления в детонационной волне,
что в свою очередь, позволило ответить на вопрос о том, в какой мере
процесс детонации таких смесей согласуется с классическими пред-
ставлениями гидродинамической теории.
При стационарном распространении детонации гидродинамическая
теория [2] предсказывает наличие за ударным фронтом зоны повышен-
ного давления — химпика в области химической реакции, и следующей
за ним области относительно медленного изменения параметров под
воздействием волны разгрузки,
На рис, 1 показано изменение формы ийпульса детонационной
волны в смесях гексогена с хлористым натрием при изменении содер-
жания взрывчатого компонента от 5 до 30%.
Рис. 1. Влияние содержания гексогеця
на форму импульса детонационной вол-
ны в смесях с хлористым натрием: а—
30% гексогена; Д”Э,9 гсч/сек; Р =
=50 кбар, 6—10% гексогена;
Д=1,7 км/сек; Р=Ю кбар; в—5% гек-
согена; Д=1,0 км/сек; Р=3,5 кбар
В смеси с большим содержанием гексогена (более 30%) при
скорости детонации около 4 км/сек давление на фронте нарастает
скачкообразно (рис. I, а), наблюдается четко выраженный химпнк.
При меньшем содержании гексогена (10—15%) при скорости дето-
нации более 2 км/сек и давлении около 15 кбар импульс детонацион-
ной волны - приобретает треугольную форму, химпик отсутствует
(рис. 1,(5). При дальнейшем уменьшении'содержания гексогена, сни-
жении скорости детонации менее 2 км/сек, а амплитуды давления
менее 10 кбар, давление до максимального значения нарастает медлен-
но, вплоть до 1 мсек и далее следует медленный спад его, Химпик отсут-
ствует, детонационному фронту предшествует волна предвестник,
Зафиксировано резкое изменение (пульсация) скорости продуктов
взрыва, а следовательно, и давления в зоне химпика от опыта к опыту,
значительно превышающее естественную погрешность регистрирующей
системы. По оси заряда давление колеблется почти в два раза [3].
Замена хлористого натрия на аммиачную селитру или хлористый
аммоний качественно не меняет наблюдаемую картину.
Таким образом, детонационные волны со структурой предсказан-
ной гидродинамической теорией детонации, т. е, имеющие ударный
фронт и резко выраженную область повышенного давления за фрон-
169
oii — химпик, наблюдаются лишь о ’смесях с боль'шой теплотой
зрыва и малым временем химической реакции. По мере уменьшения
епЛоты взрыва системы картина резко меняется. Давление на фронте
етонацибяйой волны нарастает медленно, исчезает химпик, фронт
етонациоййой волны приобретает сложную структуру. В смесях с 5,
0 ’и 15% гексогена с хлористым натрием детонационной волне пред-
тествует волна-предвестник, по-видимому, связанная с опережающим
асйрбстрансинем звуковой волны в кристаллах наполнителя. *
а б
Гекаты,% Гекаты,%
Рис, 2. Влияние состава на параметры
детонационных волн для смесей гекса-
гена с наполнителями; a—NaCl; б—
NH4CJ; в—NHfNOs. Плотность зарядов
1,3 г/сл?
й На рис. 2 показана зависимость параметров детонационных волн
Ьрсорости детонации, скорости продуктов взрыва и давления) от со-
ржания гексогена во взрывчатых смесях. Видно, что в смесях
|»:дммиачиой селитрой и хлористым аммонием, при прочих равных
ЙЬювиях, параметры значительно, выше, чем в смеси с хлористым
|Йтрием. Однако, если сопоставить скорость детонации составов при
ЙВиой номинальной плотности гексогена *, т. е. плотности гексогена,
Исполняющего поры между частицами наполнителя, то можно видеть
3), что экспериментальные точки с ошибкой не более 6—8%
пкатся на одну прямую. Аналогичная картина ‘ наблюдается и для
йвиснмости давления от номинальной плотности сенсибилизатора.
Каким образом пропадает различие между «активным» наполните-
ММ—аммиачной селитрой и заведомо инертным, в отношении теплоты
Ьэрыва, хлористым натрием.
* Номинальная плотность (ри) рассчитывалась из соотношения;
т*
’ рн“т-?о(1 -*) ’
где у— плотность добавки; ро— плотность заряда; х— содержание гексогена в вес,
частях,
4ттЖ№шёдует вывод о том, что возможность участия наполни-
теля (определяется размером его частиц. Грубодисперсный наполнитель
с размером частиц 0,2-*-0,4 мм суще-
ственного влияния на параметры де-
тонационной волны не оказывает.
Влияние начальной плотности
заряда на параметры детонацион-
ных волн было изучено на примере
смесей хлористого натрия с 15 и
30% гексогена, хлористого аммония
с 19,8% гексогена и аммиачной се-
литры с 18% гексогена. Количест-
во гексогена в двух последних сме-
сях было выбрано таким, чтобы его
объемное содержание сооответство-
вало 15% смеси с хлористым натри-
ем, при этом при равном размере
частиц (0,2-^0,4 мм) соотношение по-
верхности сенсибилизатора и напол-
нителя в смесях должно быть по-
Рис. 3. Зависимость скорости детонации
и давления от номинальной плотности
гексогена для его смесей с наполните-
лями: /—NaCl; 2—NH,C1; 3-NH,NO3
(номинальная плотность гексогена ме-
няется за счет изменения содержания
гексогена' в сйеси)
СТОЯННЫМ. .1
С увеличением начальной плотности зарядов параметры детона-
ционных волн для всех изученных смесей непрерывно возрастают во
всем изученном интервале изменения плотности от насыпной до удель-
ного веса (рнс, 4). При приблизительно одинаковом содержании
гексогена скорость детонации и давление намного выше для смесей
с хлористым аммонием и аммиачной селитре^ чем с хлористым
натрием, a б
ДД Ц О U 1Л «Л V 1,4 . W « 1,6
Рв^/ем* Рв г/см3 Р^г/см3
Рис. 4. Влияние начальной плотности заряда на параметры детонационных волн для
смесей гексогена с наполнителями (0,2—0,4 мм): а—15% гексогена с NaCl; 6—30%
гексогена с NaCl; а—19,8% гексогена с NH^CI; г—18% гексогена с NH<NO3
fee ли построить зависимость параметров детонационных волн от
оминальной плотности гексогена, то и в этом случае скорость дето-
ации и давление определяются в основном номинальной плотностью
енсибнлизатора и в меньшей степени типом наполнителя (рис. 5).
Рис. 5. Зависимость скорости детонации
и давления от номинальной плотности
гексогена для смесей его с наполнителями:
1—15% гексогена с NaCl; 2—18% гексоге-
на с NH4NO3; 3—19,8% гексогена с МН4С1;
4—30% гексогена с NaCl
Таким образом, оказалось, что параметры детонационных волн
j смёсях, .содержащих мощный сенсибилизатор, определяются одним
фактором — плотностью сенсибилизатора, заполняющего поры между
истицами наполнителя. Возможность же участия наполнителя в хи-
мическом превращении в детонационной волне определяется его дис-
персностью.
ш ф
' i'/j
К'Дремин А. Н., Шведов К. К, Веретенников В. А. Сб. «Взрывное де лол,
№ 52/9, Госгортекиздат, 1963.
!. Зельдович Я- Б., Компанеец А. С. Теория детонации. М., Гостехиэдат, 1955.
1 X о т и и В. Г., Пономарев В. А. ФГВ, 1973, т. 9, с. 2.
УДК 662.2
ИЗУЧЕНИЕ ДЕТОНАЦИИ АЛЮМИНИЙСОДЕРЖАЩИХ
ВОДОИАПОЛНЕННЫХ ВВ
В. Э. Анников, Б. Н. Кондриков, Л. П. Парфенов,
Н. П. Смагин, Л. М. Шабалина
В настоящее время одним из наиболее перспективных .видов
взрывчатых веществ для горной и других отраслей промышленности
являются текучие и пластичные водонаполненные ВВ (ВВВ), содер-
жащие алюминий. Эти составы обладают наибольшей из всех извест-
ных промышленных ВВ теплотой взрыва —до 2000'ккал/кг, а хорошая
пластичность и малая опасность при механических воздействиях поз-
воляют использовать их для механизированного заряжания шпуров
и скважин.
Основной недостаток существующих ВВВ — относительно низкая
детонационная способность и малая чувствительность к инициирую-
щему импульсу; При содержании воды, обеспечивающем достаточную
пластичность—10—20%, они детонируют только в скважинах диамет-
ром более 5 см от промежуточного детонатора (заряд динамита, пен-
толита или скального аммонита), что удорожает производство взрыв-
ных работ и делает невозможным применение таких ВВ в целом
ряде областей — обработка металлов взрывом, шпуровые заряды и др.
Недавно были опубликованы работы сотрудников Горного Бюро
США [I, 2], в которых описан способ изготовления алюминий содер-
жащих ВВВ, обладающих высокой детонационной способностью.
Высокая детонационная способность этих составов обеспечивается
присутствием в них алюминиевой пудры с высокой удельной поверх-
ностью, обеспечивающей большую поверхность горения в детонацион-
ной волне, и наличием большого количества мельчайших пузырьков
воздуха, обеспечивающих необходимый разогрев за счет адиабати-
ческого сжатия в ударной волне.
В настоящей работе изучали детонационную способность и другие
физико-химические и взрывчатые свойства водонаполненных ВВ, ана-
логичных приведенным в {2], на основе различных окислителей и раз-
ных марок алюминиевой пудры.
pg
Была изучена возможность использования в качестве окислителей
более распространенных неорганических нитратов н перхлоратов,
залось, что высокую детонационную способность ВВВ можно обес-
йть, используя в качестве окислителей соли, достаточно хорошо
творимые в воде (рис. 1): нитраты аммония и кальция и перхло-
натрия. Другие перхлораты (аммония и калия) и нитраты (калия
атрия) могут быть использованы в качестве добавок: применение
этих окислителей самих по себе да-
т/г
дД, Растворимость некоторых нитра-
тов н перхлоратов в воде [3]
вало составы с высокой плотностью
и низкой детонационной способно-
стью.
Из отечественных сортов алю-
миниевой пудры высокую детонаци-
онную способность водонаполненных
аммоналов (ак в ан а лов) обеспечи-
вают пудры ПАП-1 н ПАП-21, обла-
дающие большой удельной поверх-
ностью (2,5 н 3 м5/г соответственно),
с тбнкодйёперсным алюминием ма-
рок ГГГГ-1, ПА-4 и др. результаты по-
лучились гораздо хуже.
В качестве загустителей алю-
минийсодержащих ВВВ были опро-
шены поливиниловый спирт, натровая н аммониевая соли карбок-
(ШйЛцеллюлозы (КМЦ), оксиэтилцеллюлоза, гуаргам, полиакрил-
а также некоторые флокулянты, используемые для обогащения
«Комета», «Ока», «Метас». Удовлетворительные результаты по-
веты с гуаргамом, полиакриламидом, аммониевой солью КМЦ и
Ызтилцеллюлозой. Основная часть опытов проводилась с использо-
юём гуаргама н полиакриламида. .
При разработке технологии изготовления учитывалось то обстоя-
Ььство, что она должна обеспечить необходимую степень разрых-
Йия состава, которая осуществляется за счет воздуха, вносимого
ЙНе с частичками алюминиевой пудры. Этого можно достичь, еслп
кцть алюминиевую пудру в жидкий, достаточно желатинированный
itoop окислителя. Вязкость раствора и прочность структуры жела-
Яы должны в этом случае обеспечить необходимую степень аэра-
I,' состава при изготовлении и высокую стабильность .при хранении,
ходе работы опробовано несколько разновидностей техиологи-
NKftt приемов изготовления ВВВ. Наиболее простой и хорошо
производимый в производственных условиях заключается в
/гПорошкообразный загуститель смешивают с окислителем и зали-
иОТ' гррячей водой. После желатинизации к полученной вязкой сус-
тзни при комнатной температуре (15—25?) присыпают алюминиевую
гГДру и перемешивают до получения однородной массы. Плотность
гстава зависит от времени перемешивания на последней стадии
иготовления ВВВ и увеличивается при продолжительном переметив-
ший, что позволяет в известных пределах регулировать ее.
ПА °®означенне этнх сортов алюминиевой пудры соответственно — ПАК-3
К4
Стуу^гт^ра и реологические свойства могут изменяться в широких
пределах: система получается от свободно льющейся до резнноподоб-
ной. Это зависит от количества води и алюминия, природы и количе-
ства загустителя, применения сшивающих агентов.
Составы снаряжали в стеклянные трубки диаметром 20 мм со
стенками толщиной 1—1,5 мм или в стальные трубки диаметром 10мм
со стенками толщиной 13 мм. Скорость детонации определяли с по-
мощью скоростного фоторегистра ЖФР, критический диаметр —
методом телескопических зарядов.
Составы ha основе аммиачной селитры
Скорость детонации состава, содержащего 20% алюминиевой пуд-
ры ПАП—2, приведена на pnic, 2,6. С увеличением плотности заряда от
0,9 до 1,2 г(см3 скорость линейно растет от 3,5 до 4,4 км/сек, а затем
уменьшается. На участке роста скорости выполняется соотношение
D = 3,75q, км/сек (q—в г/сж3)
При q= 1,35 а/сж3 детонация в зарядах диаметром 20 мм затухает
(достигается критическая плотность).
У5, рег/см*
Рис. 2. Скорость детонации акванала, содержащего 20% Н*О и 20% тонкодцс-
персного алюминия*, а—ПАП-t; б—ПАП-2
Изменение содержания воды в акванале сильно влияет на крити-
ческую плотность. Увеличение содержания воды от 15 до 30% (20%
ПАП-1) снизило QKp с 145 до 1,17 г/см3 (рис. 3). Влияние содержания
алюминиевой пудры ПАП-2 на критическую плотность составов, со-
держащих 20% воды, показано на рис. 4. Минимальное содержание
алюминиевой пудры, необходимое для обеспечения устойчивой дето-
нации ВВВ, —3% (Qkp= 0,95 г/сж3). Смеси, содержащие 1,5% ПАП-2,
не детонировали даже при плотности 0,55 г/см3. При увеличении содер-
жания ПАП-2 до 30% критическая плотность увеличивается до
1,41 г/см3, а при 40% ПАП-2 она снижается до 1,30 г/см3. Состав,
содержащий 50% ПАП-2, оказалось очень трудно получить достаточно
однородным. Смесь перемешивалась с большим трудом и получить
заряды с плотностью меньшей 1,29 г/см3 не удалось, а в заряде
с такой н большей плотностью детонация от ЭД-8 не возбуждалась
(рнс. 4). 1 *
1 Во всех случаях, кроме специально указанных,—заряд в стеклянной обо-
дочке, содержание воды — 20%, инициирование электродетриатором ЭД-8.
175
Изменение содержания алюминиевой пудры, сильно влияя на
почти не сказывается на скорости детонации акваналов. Прн
ньшении содержания ПАП-2 с 20 до 15; 10 и даже 5% скорость
Рис. 3, Влияние воды на критическую плотность аммиачноселитренных ВВВ,
содержащих 20% ПАП-1. ф—детонации; О— отказ
Рис. 4. Влияние содержании ПЛП-2 на критическую плотность состава (20%
HjO) на основе нитрата аммония. /—Отказ; 2— детонация; детонация прошла
3—4 гл и затухла
чтн не меняется (рис. 5 и 6). Снижение скорости для состава с 5%
4П-2 для зарядов в стеклянных трубках диаметром 20 лсм, очевидно,
условлено разбросом вещества: при увеличении диаметра до 36 мм
[Н помещении смеси в стальные трубки скорость детонации повы-
ается до уровня смеси с 20% ПАП-2 (рис. 6).
Рис. 5. Скорость детонации акваналов (20% НгО). Содержание ПАП-2, %:
О—10; ф—15. Пунктирная кривая для акванала с 20% ПАП-2
Рис. 6. Зависимость Скорости детонации акванала (20% НгО), содержащего
5% ПАП-2, от плотности: /—в стеклянных трубках диаметром 19—20 .я.я,
2—в 'стальных трубках диаметром 10 лл (толщина стсцки 13 л.ч), <?-в поли-
этиленовых трубках диаметром 36 мм. Пхиктирная кривая—для акванала
с 20% ПАП-2
Замена пудры ПАП-2 на немного более грубодисперсную ПАП-1
(20 и 15%) повлияло на скорость детонации и критическую плот-
ность слабо (рис. 2,а). Скорость детонации уменьшилась в среднем
на 0,1 км{сек, критическая плотность — на 0,05 г{см\
Существенно понизилась критическая плотность (от 1,2 до
0,85 в/сл3) в опытах, где применялась пудра ПАП-1 обезжиренная кц-
пячением в бензоле. Таким образом, слой стеарина на поверхности
частиц пудры существенно повышает детонационную способность
смеси.
Зависимость критического диаметра детонации от плотности для
смесей, содержащих 15 и 20% ПАП-1, приведена па рис. 7, а и б. На
этих же рисунках нанесена пунктирная кривая D (q) для тех же соста-
вов. Критический диаметр при малой плотности для обоих составов
невелик — около 5 мм и слабо зависит от плотности. В области выше.
1,07 г/см2 для смеси с 15% ПАП-1 и 1,15 г/см2 — для смеси с 20%
ПАП-1 критический диаметр быстро растет, достигая, соответственно,
при 1,2 и 1,3 г/см2 — 20 мм. Сравнение показывает, что начало
быстрого роста кривых dH(Q) отвечает максимуму скорости на кри-
вых D(q).
чов, содержащих 15% (а), и 20% (б) ПАП-1 в цилиндрических
трубках (f и 2) и в плоском слое (3 и 4). 1 и 4 — Детонация:
2 и 3 — отказ. Пунктирные кривые — зависимость D (в) ДЛЯ этих
же составов в трубках диаметром 20 мм
Очевидно, падение скорости при увеличении плотности связано с
радиальным разбросом вещества и увеличением предельного диа-
метра. При помещении ВВ (20% ПАП-2) в толстостенную стальную
трубу (рис. 8) участок падения скорости исчезает. Подобный резуль-
тат получается и для других составов акванала.
Рис. 8. Скорость детонации аквана-
ла (20% Н2О), содержащего 20%
ПАП-2 в стальных трубках диамет-
ром 10 мм со стенками толщиной
13 мм. Пунктир—кривая по опы-
там в стеклянных трубках диа-
метром 20 мм
| л о о
< °О о ° о 4
2>---1---1---1__I___I___i__I
W Ц
У3, г/сп3
Критическая скорость детонации определялась в опытах по зави-
симости скорости от диаметра. С увеличением диаметра скорость
детонации состава с 20% ПАП-1 (q = 1,0 е/с-и3) растет вначале быстро
12 Труды аып.’ВЗ
(рис. 9), а затем очень медленно. Экстраполяцией начального участка
кривой иа критический диаметр (~5 мм) получаем критическую ско-
рость детонации 2,3 км/сек. Такое же значение получается при по-
строении этих данных в координатах —D. «Идеальная» скорость
для этого состава (р = 1,0 zjcM^), как видно из рис. 9,6—3,8 км]сск.
Такое же значение — 3,75 км{сек. (при у =1,0 г/см3) имеет состав
с 20% ПАП-2 (рис. 2).
Рис. 9. Зависимость скорости детонации акванала
(20% НгО), содержащего 20% ПАП-1 от диа-
метра заряда (р= I г/см3): а—в координатах
d -D; б—в координатах d /d—D
Другой путь определения критической скорости — участок падения
скорости на кривой D(q) в трубках постоянного диаметра. Результаты,
полученные таким образом, приведены в таблице. Для различных
составов, имеющих критическую плотность от 0,9 до 1,4 zjcM3, крити-
ческая скорость изменяется слабо.
Предельные параметры детонации ВВВ на основе алюминия
(20% воды)
Алюминий рш Рх V D.
20% ПАК-3 1.56 1.27 0.82 3.2
20% ПАП 1 1.56 1.30 0,83 3,05
20% ПАП-1/ПП I (2/1) 1,56 1,30 0,83 2,9
30% ПАК-3 (25% Н2О) 30% ПАК-4/ПАК-3 (1/2) 1.54 1.50 1.29 1.29 0.84 0,86 2,7 3.4
(30% Н,О) 10% ПАКЧ 1.51 1,20 0,79 3,1
5% ПАК-4 1,48 0,9 0.61 2.7
15% ПАК-3 1.53 1.18 0,77 2,4
15% ПАК-3 (обезжир.) 1,53 0.84 0.55 2.4
П р и м е ч а н н е; критическая плотность, г1см?\ — наибольшая возможная
плотность, г/см9; DK — скорость детонации прн критической плот-
ности, /си/сек; 6К—относительная критическая плотность (с,;.'р>и)-
Диаметр заряда 20 леи.
Давление в ударной волне, необходимое для инициирования дето-
нации смеси с 20% ПАП-1, определялось в стальных трубках при
инициировании смесью гексогена с NaCl через плексигласовый диск
178
(рис. 10). При q = 1,1 г}см* критическое давление всего 3 тыс. ат. При
при 1,4 a/cjfi3 давление
Еще ниже критическое
повышении плотности оно растет, но даже
инициирования не превышало 10—12 тыс. ат.
давление для смеси на основе ПАП-2 (при
р = 1,24 г!см\ Ркр = 2,5 тыс. ат).
давление
акванала
на критическое
инициирования
(20% Н2О), содержащего
20% ПАП-1. О—детонация,
ф—отказ
Составы на основе нитрата кальция
я перхлората натрия
Как было сказано выше, кроме нитрата
аммония в качестве окислителя в алюми-
нийсодержащих ВВВ могут быть использо-
ваны кальциевая селитра и перхлорат нат-
рия. Однако смеси на основе кальциевой
селитры обладали невысокой детонационной
способностью: критическая плотность для
изученных составов (20% ПАП-1) меньше
1,2 г/сл3. Связано это, видимо, с высоким
содержанием воды (молекула Са(МОз)з-
4Н2О содержит 33% Н2О). Чтобы получить
однородную смесь состав должен содержать
не менее 15% избыточной воды, что с учетом
кристаллизационной составляет 34,5%. Кри-
тическая плотность для этого состава — 1,16 г!см3 (qk„ для состава на
МНчМОз, содержащего 30% Н2О, — 1,18 г/с-и3).
При использовании в качестве окислителя смеси нитратов каль-
ция и аммония детонационная способность состава повышается.
По данным Иоффе при 20° С в насыщенном растворе аммиачной
селитры, содержащем 100 г воды, можно растворить 360 г Ca(NOa)a.
Эта особенность позволяет, в принципе, получить однородные составы,
содержащие всего 10—15% воды (с учетом кристаллизационной),
обладающие высокой детонационной способностью. Состав на смеси
этих окислителей (1 : 1), содержащий 16% воды, устойчиво детони-
ровал до плотности 1,5 г!см3.
Зависимость скорости детонации от плотности для смесей на
основе перхлората натрия (20% ПАП-1) приведена на рис. 11, а.
Характер зависимости остался таким же, как для смесей на нитрате
аммония, однако скорость детонации при одинаковой для обоих
Рис. 1 [. Влияние абсолютной (а) и относительной (б) плотности на скорость
детонации ВВВ на основе перхлората натрия (20% ПАП-1). Содержание воды
%: 0—20; ф—25. Пунктир—кривая для смеси на основе нитрата аммония
1?* 17р
ставов теплоте взрыва (~ 1400 ккал/кг) заметно меньше, а крити-
ская плотность — больше. Состав, содержащий 20% воды, сохра-
(ет детонационную способность до плотности 1,7 г! см3. Учет более
лсокой плотности перхлората натрия — переход к относительной плот-
>сти (рис. 11,6)—уменьшает разницу в бкр между смесями перхло-
зта натрия и нитрата аммония.
Характер влияния дисперсности и содержания алюминия и воды
пя смесей на основе перхлората натрия такой же, как и для смесей
аммиачной селитрой. Увеличение содержания воды, так же как и
иеньшение содержания алюминия, в составе уменьшает критическую
дотность (рис. 12). При 30% воды (20% ПАП-1) ри,=1,20 г/см3, а
рн 40% воды в стеклянных трубках при q = 1,20 г':см3 получено два
отказа. Увеличение содержания
• ем «в* Ф
I ** 1 -I- 1_1 г
Рис. 12. Влияние воды на критическую
плотность составов на основе перхлората
натрин (20% ПАП-1), ф—детонация;
О—отказ
алюминия и применение более
тонкодисперсной пудры (ПАП-2)
так же, как для смесей на оспо'
ве нитрата аммония, увеличивает
критическую плотность.
Максимум скорости детона-
ции для состава с перхлоратом
натрия расположен левее, чем
для состава на аммиачной селит-
ре. Заметим, что больше и крити-
ческий диаметр; 9 мм для соста-
ва с NaClO+ по сравнению с
5 мм для состава на аммиачной
селитре.
Влияние начальной температуры
Детонационная способность ВВВ при низкой температуре изуча-
лась для составов, содержащих 20% ПАП-1. В качестве окислителей
были использованы нитраты аммония и кальция и перхлорат натрия.
Заряды в стеклянных трубках из смеси на основе нитрата аммо-
ния, содержащей 20% воды, устойчиво детонировали, если температура
заряда была выше —6° С. При температуре -—8° С детонация не воз-
буждалась даже для зарядов в стальной оболочке при инициировании
дополнительным детонатором. Не удалось повысить детонационную
способность этого состава при пониженной температуре введением
в смесь 5% хлористого аммония, существенно понижающего темпера-
туру затвердевания раствора аммиачной селитры (с —16° до —29° С).
Составы на основе смеси аммиачной и кальциевой селитры (1:1)
(t3 = —2(°С) и на основе перхлората натрия (t3 =—36°С), содер-
жащие 20% воды, также не детонировали при температурах ниже 0°С.
Результаты этих опытов, а также тот факт, что составы на основе
окислителей, плохо растворимых в воде, обладают малой детонацион-
ной способностью, дают основания полагать, что существенную роль
в обеспечении распространения детонации в составах, сенсибилизиро-
ванных тонкодисперсным алюминием, играет именно окислитель, рас-
творенный в воде. Нерастворенная в воде часть окислителя, так же
как и сама вода, видимо, более инертны и не успевают существенно
прореагировать во фронте детонационной волны,
вместе с тем, в работе (2] показано, что для подобных составов
суммарное количество инертной массы (вода и NaCl) не должно пре-
вышать 60%. При большем содержании инертной массы возбудить
детонацию смеси не удается.
Вероятно, именно этим можно объяснить тот факт, что при пони-
жении температуры детонационная способность ВВВ уменьшается.
Смеси, содержащие 20% воды, на основе аммиачной селитры чистой
и с добавками хлористого аммония пли кальциевой селитры (1 : 1),
смеси на основе одной кальциевой селитры и на основе перхлората
натрия не детонировали ниже -—5° С даже в стальной оболочке и при
инициировании дополнительным детонатором. С понижением темпера-
туры растворимость окислителя
массы (вода и нерастворившаяся
и при 0—5°С, видимо, достигает кри-
тического значения. Расчет показы-
уменьшается, количество инертной
часть окислителя) увеличивается
вает, что при температурах ниже
—5°С для всех этих составов содер-
жание инертной массы составляет
55—60%,
Основываясь на этом предполо-
жении, было рассчитано количество
инертной массы для составов, сенси-
билизированных 20% алюминия.
При расчете были нспользовацы дан-
ные по растворимости окислителей
при температуре затвердевания их
растворов: 36° для NaClO*; 28° для
Ca(NO3)2; 16° для NH4NO3. Ре-
зультаты расчета приведены на
рис. 13.
С увеличением количества воды
Рис. 13. Влияние содержания воды
На содержание инертного вещества
(20% алюминия)
в составе увеличивается содер-
жание растворенного в ней окислителя; при 37% воды для NaClO+
и 45% — для нитратов аммония и кальция окислитель полностью
растворен в воде, содержание инертной массы в этих случаях мини-
мальное. При большем содержании воды количество инертной массы
увеличивается. Отсюда следует парадоксальный вывод: чтобы полу-
чить состав с высокой детонационной способностью при низкой тем-
пературе, содержание воды в составе необходимо увеличить (от 20%
до 35—40%).
21
Окислипиль
-25 6 ijS i,t
W Са[М& MfjB i
Рис. 14, Влияние содержания-воды на детона-
ционную способность ВВВ при пониженной
температуре
181
Йа рис. 14 представлены результаты опытов с составами, содер-
|Щими 37—40% воды. Все составы устойчиво детонировали от
жтродетонатора в стеклянных трубках диаметром 20 мм вплоть до
гвердевания состава. Интересно, что для смесей на перхлорат?
грия и кальциевой селитре, растворы которых склонны к переохлаж-
нию, детонация была устойчивой вплоть до —52е и —38° С соответ-
чике.
Van Doi ah R. W„ Mason С. M., Forshey D. R. «Development of slarry
explosives for use in potentially flammable gas atmospheres». Rept. of Investigi-
iis 7195. US Dept, of the Interior, Bureau of Mines. 1968.
Mason С. M-, Lisoto E. C., Ruhe С. T., Van Dolah R. W, «Further
development of nonincedjve water gel explosives, Rept. N35 on Intern, conf, of
safety in mines research, Tokyo, 1969.
Справочник по растворимости, М.-Л., Изд. АН СССР, 1961—1962.
УДК 662.221
О ПОИСКЕ НОВЫХ НАПРАВЛЕНИЯ
В РАЗВИТИИ ПРЕДОХРАНИТЕЛЬНЫХ ВВ
В. Г. Хотин, Г. jM. Шутов, В. Л. Збарский
Современные предохранительные взрывчатые вещества — доста-
точно сложные многокомпонентные системы, теплота взрыва кото-
рых ограничена за счет введения ингибиторов воспламенения метано-
воздушной среды — преимущественно щелочных солей галоидово до род-
ных кислот. Эффективность ингибиторов зависит от их содержания п,
кроме того, от их удельной поверхности [1, 2J.
Добавка значительного количества тонкодисперсной соли в состав
смесового ВВ не является удовлетворительным решением, так как
существенно снижает детонационную способность. Кроме того, по тер-
модинамическим законам, при длительном хранении, особенно при
избыточном содержании влаги, частицы иргибятора укрупняются
и теряют свою эффективность. Поэтому находят компромиссные реше-
ния, позволяющие обеспечить удовлетворительную детонационную спо-
собность при некотором снижении предохранительных свойств,— при-
меняют сравнительно грубодисперсный ингибитор или выносят его
в оболочку на периферию заряда.
Известен другой путь — получение ингибитора (хлористого калия)
в мелкодисперсном состоянии в момент взрыва за счет реакции между
компонентами смесевого ВВ — хлористым аммонием и азотнокислым
калием. Но и этот способ имеет недостатки: составы, построенные
с использованием, этого принципа, маломощны и, кроме того, нет уве-
ренности в том, что реакция образования хлористого калия осуществ-
ляется до конца.
Следует отметить возможность образования пламегасителя при
взрыве хлоратных и перхлоратных ВВ, однако эти пути, по крайней
мере в нашей стране, практического применения не нашли.
В последние годы, наряду со стремлением повысить предохрани-
тельные свойства В В, не меньшее внимание стали уделять другой сто-
роне проблемы, а именно, вопросу предотвращения выгорания. К пре-
дохранительным ВВ стали предъявлять дополнительные требования.
183
любы исключить возможность отказа детонации, создающего пред-
осылку выгорания, оказалось необходимым обеспечить высокую дето-
ационную способность ВВ в уплотненном состоянии [3]. Однако всем
помянутым выше составам присущи недостатки смесевых ВВ, и в
ервую очередь, резкое снижение детонационной способности при
овышении плотности.
Указанные недостатки могут быть легко устранены, если атомы,
оставляющие ингибитор, ввести в молекулу индивидуального ВВ.
»та мысль принадлежит лроф. К. К- Андрееву, по инициативе которого
ыли разработаны методы синтеза ароматических полинитрохлорфе-
олов и анилинов, калиевые соли которых удовлетворяют поставлен-
ым требованиям [4—6].
Из предложенных соединений наибольший интерес представляют
алиевые соли 2,4,6-тринитрохлорфенола и 212'14,4/,6,6'-гексаяитро-
-хлордифениламииа. 2,4,6-Тринитрохлорфенол был получен из стиф-
иновой кислоты, при обработке последней хлорокисью фосфора в
:рисутствин пиридина и воды. Пиридин является катализатором нук-
[еофильного замещения оксигруппы на хлор, а вода препятствует за-
(ещению второй оксигруппы в стнфниновой кислоте.
2,2',4,4',6,б'-Гексанитро-3-хлордифениламин был получен конденса-
Лей тринитрохлорфенола с n-нитроанилином, донитровываннем полу-
еииого соединения до 2,2',4,4'6,6'-гексанитро-3-оксидифениламнна.
(амещение оксигруппы на хлор проводилось также прн действия хлор-
кйси фосфора в присутствии пиридина. Одно цз ВВ этого типа —
лорпикрат калия (калиевая соль 2,4,6-тринитрохлорфенола) было
гами изучено *.
Хлорпикрат калия представляет собой твердое вещество ярко-
келтого цвета нерастворимое в метаноле, ацетоне, бензоле, толуоле
[ хорошо растворимое в диоксане и диметилформамиде. Раствори-
<ость в воде при 15° составляет 8,15 г/л. Удельный вес, определенный
(пикнометре с толуолом, 1,88 cIcm*. 25% по весу в молекуле хлор-
шкрата калня приходится па атомы пламегасителя.
Некоторые взрывчатые характеристики хлорпикрата калня, по-
рченные путем расчета и определенные экспериментально, приведены
[Иже.
Расчетные данные *
Кислородный баланс, %.......................—29,2
Объем газообразных продуктов взрыва, л/кг 600
Теплота взрыва, ккал! иг . . , . . . ; 755
Температура взрыва, °К......................... 3080
Экспериментальные данные
Гемпература, вспышки, СС ........ . 290
Минимальное расстояние, на котором луч огня огнепровод-
ного шпура еще не вызывает горения вещества, см . , 7
4увствительнбсть к истиранию в фарфоровой ступке взрывов не дает
* В экспериментальной части работы принимал участие ииж. Е. А. Рыбаков.
184
Количество (%) взрывов при испытании на чувствитёЛьйостЪ
к удару (груз 10 кг, высота падения 25 см, навеска 0,02 г)
в приборчику № 1................................. 52
в приборчике № 2................................. 12
Минимальный заряд гремучей ртути, вызывающий детона-
цию, г................................................. 0,45
Критический диаметр детонации при плотности 1 г/слт3, мм 3,5
Скорость детонации в заряде диаметром 7 мм, м/сек
при плотности 1,4 г/см*............................5120
при плотности 1,8 г/см3.......................... 6540
Температуру вспышки определяли, нагревая в бане со сплавом Вуда
навеску ВВ 0,1 г, помещенную в стеклянную пробирку. Начальная
температура бани 100s С, в последующем возрастала со скоростью
2W/muh.
Предельный инициирующий заряд гремучей ртути, по отношению
к хлорликрату калия, был определен при испытании на свинцовых
пластинах (толщина 5,2 мм) электродетонаторов, содержавших в каче-
стве вторичного заряда 0,8 г хлорпикрата калия, запрессованного под
давлением 500 кг/см2, н переменное количество гремучей ртути, запрес-
сованной под давлением 300 кг/см2. Предельный заряд гремучей ртути,
начиная с которого диаметр отверстия в пластине остается неизмен-
ным, составил 0,35 г, что находится в одном ряду с данными Велера
для многих вторичных ВВ, Критический диаметр детонации хлор-
пикрата калия был определен при плотности Г г/см3 в конических
зарядах с тонкой бумажной оболочкой. Скорость детонации опреде-
лялась с помощью СФР в зарядах повышенной плотности диаметром
7 мм и длиной 5 см, составленных из отдельных шашек высотой 0,5 мм,
заключенных в целлофановую оболочку.
Удовлетворительные взрывчатые характеристики делают возмож-
ным использование хлорпикрата калия и в качестве составной части
предохранительного ВВ обычного типа на основе аммиачной селитры
и самостоятельно, в частности, в предохранительных средствах взры-
вания, Последняя возможность была изучена.
Проведено сравнительное испытание на антигризутность электро-
детонаторов, содержащих в качестве вторичного заряда по I г хлор-
пикрата калия и тетрила, и электродетонаторов заводского производ-
ства ЭД-8-56 (таблица):
Тип электродетонатора Число взрывов метано-воз- душной смеси к числу испы- таний % взрывав метано-воз* душвой смеси
ЭД-8-56 (гремучертутнотетрило- вый) 6,6 100
Экспериментальные: гремучертутнохлорпикратный 0,5 0
а а и дотетр и лов ы й 2/2 100
аз и дох лор пи кратный 0,'8 0
При изготовлении экспериментальных электро детонаторов заряд
вторичного ВВ во всех случаях запрессовывался давлением 500 кг/см2.
185
рпикрат калйя Прессовали в два приема для получения более реб-
ерной плотности. Заряд инициирующего ВВ (0,5 г) запрессовы-
н вместе с медной чашечкой под давлением 300 кг/см2. Хлорви-
овую пробку электровоспламенителя цангой не обжимали.
Установка для испытания детонаторов в отношении возможности
пламенения метано-воздушной смеси представляла цилиндриче-
й сосуд длиной 60 см и диаметром 25 см (емкость 30,5 л)г установ-
ный горизонтально (рис, 1), Пропеллер, вал которого был пропу-
I через сальник в дне сосуда, предназначался для перемешивания
•ано-воздушной смеси и для удаления продуктов взрыва после
(та. Второй торец сосуда после опыта закрывался стальной крыш-
г. Штуцер 1 в верхней части сосуда использовался для заполнения
уда метаном и отбора пробы газовой смеси до и после опыта,
pei штуцер 2 вводился электродетонатор. Оба штуцера имели гер-
газирующие прокладки. Через штуцер 3 в нижней части сосуд сооб-
лся с атмосферой.
Рис. I. Схема установки для оценки возможности воспламенения метано'
воздушной смеси взрывом электродетоиатора
и Рис. 2, График изменения величины обжатия крешера в бриаантометре
у ..Кдста тротиловыми шашками при увеличении содержания флегм ати за тора
, i тротиле при инициировании тетриловым детонатором (/) и хлорпи-
кратным (2)
th-
I- С помощью пипетки Зигера контролировали концентрацию метана
>К0₽уде до и после опыта. Первоначальная концентрация составляла
—11% и в течение получаса практически не менялась. При воспламе-
ейии метано-воздушной смеси анализ показывал отсутствие метана
пробе. Если воспламенения не возникало, концентрация метана
менялась незначительно (на 2—3%) за счет разбавления продуктами
зрыва детонатора. Результаты замера концентрации метана исполь-
овались для оценки результатов опыта.
Электродетонаторы, содержавшие в качестве вторичного заряда
г хлорпикрата калия, не вызывали воспламенения метано-воздушной
;неси при использовании в качестве инициирующего заряда как гре-
<учей ртути, так и азида свинца. Напротив, при испытании детона-
Юров ЭД-8-56 и экспериментальных азидотетриловых вероятность
юспламенения метана составила 100%,
Была определена способность гремучертутнохлорпикратных и
тетриловых электро детонаторов вызывать взрыв метано-воздушной
:иеси при различных навесках вторичного заряда. Предельный заряд
хлорпикрата калия оказался более 1,5 г и около 0,5 г в случае тетрила.
186
При косвенном методе определения инициирующей способности
детонаторов с весом вторичного заряда 1 г, вынос свинца, при про-
бивании свинцовой пластинки толщинок 4 мм хлорпнкратным кап-
сулем, оказался 0,7—1 г, т. е, почти в 3 раза меньше, чем при взрыве
детонатора ЭД-8-56 (2,8—2,7 г), хотя диаметры пробитых отверстий
практически не отличались (10,5—11,5 мм в обоих случаях).
При прямом методе, детонаторами с весом вторичного заряда
1 г инициировались шашки тротила (диаметр 25 мм, высота 42 мм и
вес 20 г), флегматизированного тальком, О возбуждении детонации
в тротиле судили по величине обжатия крешера взрывом шашки на
бризантометре Каста. Предполагали, что капсюль-детонатор с большей
инициирующей способностью будет способен вызвать детонацию тро-
тила с большим содержанием флегматизатОра. Оказалось, что ини-
циирующая способность детонаторов, снаряженных хлорпикратом
калия, не намного уступает инициирующей способности детонаторов,
содержащих в качестве вторичного заряда тетрил (рис. 2). Отказы
наблюдались при содержании флегматизатора более 30%,
Дополнительно была проверена способность различных типов де-
тонаторов возбуждать детонацию аммонита ПЖВ-20. Бризантность
ПЖВ-20 при инициировании экспериментальными электродетонатора-
ми, снаряженными хлорпикратом калия, не уступала бризантности
ПЖВ-20, инициированного промышленными детонаторами ЭД-8-56.
Обжатие свинцового столбика 14,4 и 14,2 jhjw соответственно.
Таким образом, опыт подтвердил возможность практического
использования нового типа предохранительного ВВ, Электро детона-
торы, в которых в качестве вторичного заряда была применена калие-
вая соль тринитрохлорфенола, практически не уступая по инициирую-
щей способности детонаторам, снаряженным тетрилом, не воспламе-
няют при взрыве метано-воздушную смесь, которую безотказно вос-
пламеняют стандартные гремучертутнотетриловые детонаторы ЭД-8-56.
I. Андреев К- К., Беляев А, Ф. Теория взрывчатых веществ, М.. Оборонгнз, I960.
2, Дубнов Л. В. Предохранительные взрывчатые вещества в горной промышлен-
ности, И., Углетехиэдат. 1953,
3. Андреев К. К., X о т и н В. Г. Доклад № 76 на X Международной конференции
по безопасности работ в горной промышленности, Варшава-Польша, 1961.
4. Андреев К. К., Рыбаков Е. А.. Хотин В. Г. Сб. Взрывное дело, М., <Недра»,
1966. № 60/17.
5, Шутов Г. М., Збарский В. Л., Жилин В, Ф,, Орлова Е. Ю. Ж. общ. хи-
мии, 1965, т. 35, с. 1358.
6. Збарский В. Л,, Шутов Г. М-, Жилин В. Ф., Орлова Е, JO. Ж- орг.
хи^ив, 1965, т- 1> с. 1237.
СОДЕРЖАНИЕ
СТР.
Предисловие
Е, Ю. Орлова. Исследования в области теории и технологии взрывчатых
веществ (краткий обзор) ..............................................5
Раздел первый
Б. А. Лурье, Б. С. Светлов. Термический распад диэтилеигликольдииитрата
в конденсированной фазе ......................................... . . .19
Б. С. Светлов, В. П. Шелапутина, Л. Г. Кравчинская. Щелочное омыление
и нейтральный гидролиз некоторых моиоиитратов гликолей и сс-дииитрата
глицерина ....... ............................. 29
Б. С. Светлов, В. П. Шелапутина, Л. Д. Максимова, В. П. Евтушенков.
О взаимодействии нитроглицерина с дифениламином.......................35
Б. С. Светлов, Б. А. Лурье, Г. Е. Корнилова. Кинетика окисления целлюлозы
двуокисью азота...........................................................41
Ю. Я. Максимов, С. Б. Сорочкин, В. Ф. Сапранович. Изучение состава газооб-
разных продуктов распада жидкого трииитробеизола..........................48
Ю. Я. Максимов, В. Ф. Сапранович, Н. В. Полякова. О роли некоторых кон-
денсированных продуктов в процессе термического распада тринитро-
толуола ..................................................................51
Ю. Я. Максимов, Н. В. Полякова, В. Ф. Сапранович. Термическое разложение
и давление паров трииитропроизводиых /г-ксилола и мезитилеиа ... 55
В. А. Коробан, Т. И. Смирнова, Б. С. Светлов. О влиянии некоторых факторов
иа разложение перхлоратов аммониевых оснований............................61
Раздел второй
Б. Н. Кондриков, Т. Олемиллер, М. Саммерфилд. Воспламенение и газификация
баллиститиого пороха под действием излучения СО2—лазера ... 67
Б. Н. Кондриков, Г. Д. Козак, А. Л. Черных. Воспламенение и теплообмен
в м аиометрической бомбе........................79
В. Э. Анников, Б. Н. Кондриков, В, М. Райкова, Л. Г. Спиженкова. О горении
жидких взрывчатых смесей........................ 89
В. В. Горбунов, Л. Ф. Шмагин. О горении перхлоратов и нитратов аммиакатов
переходных металлов .............. 95
А. П. Глазкова. О влиянии неорганических катализаторов иа горение перхло-
рата аммония................................... 99
j А. П. Глазкова, О. К Андреев. О механизме влияния галогенидов щелочных
>' металлов на горение нитрата аммония и аммонита...........................105
В. В. Горбунов, В. А. Лобанов. О влиянии дисперсности магнии иа скорость
Горения его смесей с солями-окислителями...........................110
А. П. .Д^^юк, В. М. Кувшинов, Н. П. Токарев, В. Г. Хубаев. Влияние иачаль-
иЙй^емпературы и давления иа эффективность действия комбинироваи-
иогонкатализатора при горении пороха Н........................... 114
Л. Е. Фогельван?, С. м Колясов, Б. С. Светлов. Горение конденсированных
смесей -р Различными окислителями................................ 119
И. Н. Бахман, Зайцев, Ю. С. Кичин, С. М. Колясов, А. Е. Фогельзанг,
В. Н. ^«>^;Исследоваиие профиля температуры в зоне горения
смесевых ...........................................123
189 ’'Л'П.й.,
Раздел третий
Б. Н, Кондриков, В. Д. Чубаров. Измерение скорости движения вещества
при ударе иа копре............................................... 135
Б. Н. Кондриков, В. М. Райкова. Детонация растворов иа основе жидких
нитроэфиров...................................................... 147
В. Г. Хотин, С. Я. Бачурин, В. А. Пономарев, Исследование ударной адиабаты
аммиачной селитры .................................................154
В. Г. Хотин, В. А. Пономарев, К. Б. Мясников. О вычислении параметров
ударных волн в инертных средах, граничащих с зарядом смесевого взрыв-
чатого вещества.................................................. .161
В. Г. Хотин, В. А. Пономарев, Я. М. Серегина, К. Б. Мясников. Исследование
параметров детонационных воли в смесевых ВВ электромагнитным
методом .........................; ...........................166
В. Э. Анников, Б. Я. Кондриков, Л. П. Парфенов, Я. П. С маг ин, Л. М. Шаба-
< лина. Изучение детонации алюмииийсодержащих водонаполиениых В В 173
В. Г. Хотин, Г. М. Шутов, В. Л. Збарский. О поиске новых направлений в раз-
витии предохранительных ВВ............................................. 183 ,
*. 1 ’f . ' ? ' . а
’ Ч / < S. » ' +
......................................................... , , '
‘ ‘ ' Г V-. . г
Труды МХТИ им. Д. И. Менделеева
Выпуск LXXXIH
Редакционная коллегия:
Б. С. Светлов (предс.), Б. Н. Кокдриков,
Ю. Я- Максимов (огв. ред.)
Корректоры
В. А, Евдокимова, Н. М. Ткач
Л 24776 от 24.11,75 г.______________________Объем 12 п. л.
Зака. 1112 Цена 1 руб. 20 коп. Тираж 1000
Типография МХТИ им. Д. И. Менделеева
ПРАВИЛА ДЛЯ АВТОРОВ
Издательский отдел МХТИ имени Д. И. Менделеева просит авторов, сдающих
статьи в труды института, придерживаться приведенных ниже правил,
1. Материал должен быть изложен кратко и тщательно отредактирован,
2. Статьи сдаются в издательский отдел в одном экземпляре. Объем статьи не
должен превышать 5 страниц машинописного текста, напечатанного через 2 интервала,
включая литературу и таблицы. Формулы должны быть вписаны четко черными чер-
нилами.
1 Статья должна содержать не более 5 рисунков. Рисунки могут быть выпол-
нены иа миллиметровой бумаге карандашом.
Все надписи с рисунков желательно сносить в подрнсуночные подписи я заменять
их цифрами. Размеры рисунков не должны превышать 9x12 см.
Подписи к рисункам должны быть напечатаны на отдельном листе.
4. На первой странице рукописи даются название статьи, затем инициалы и
фамилии авторов. Далее идет текст. В верхнем правом углу первой страницы ставится
индекс по универсальной десятичной классификации (УДК). Статья должна быть под-
писана всеми авторами. Если заведующий кафедрой не является автором, то на статье
должна быть его виза.
5. Обозначения величин и их размерность должны быть общепринятыми. Пред-
почтительна система СИ.
6. Литература приводится в койне статьи.
Для журналов: Ткач Н. М. ЖАХ, 1973, т. 25, вып. 7, с. 276—279.
Для книг; Лебедева В. Ф. Термохимия, М., <Химня», 1973.
7. Во избежание ошибок при наборе, в тех случаях, когда написание строчных
и прописных букв одинаково, автору следует сделать разметку (к. К, с. С, р, Р и
т. д.). Кроме того, следует указать индексы и показатели степени к буквам следую-
щнм образом — с , С^. Также необходимо четко различать написание
а — 2 ~ tn
букв I. J не, I. Греческие буквы следует обвести красным карандашом.
8. Статьи, представленные без учета данных правил, приниматься не будут.
9. В трудах института не печатаются работы, полностью или частично опублико-
ванные в других журналах.
10. К статье прилагается автореферат.