/























































































































































































































































































































































































































































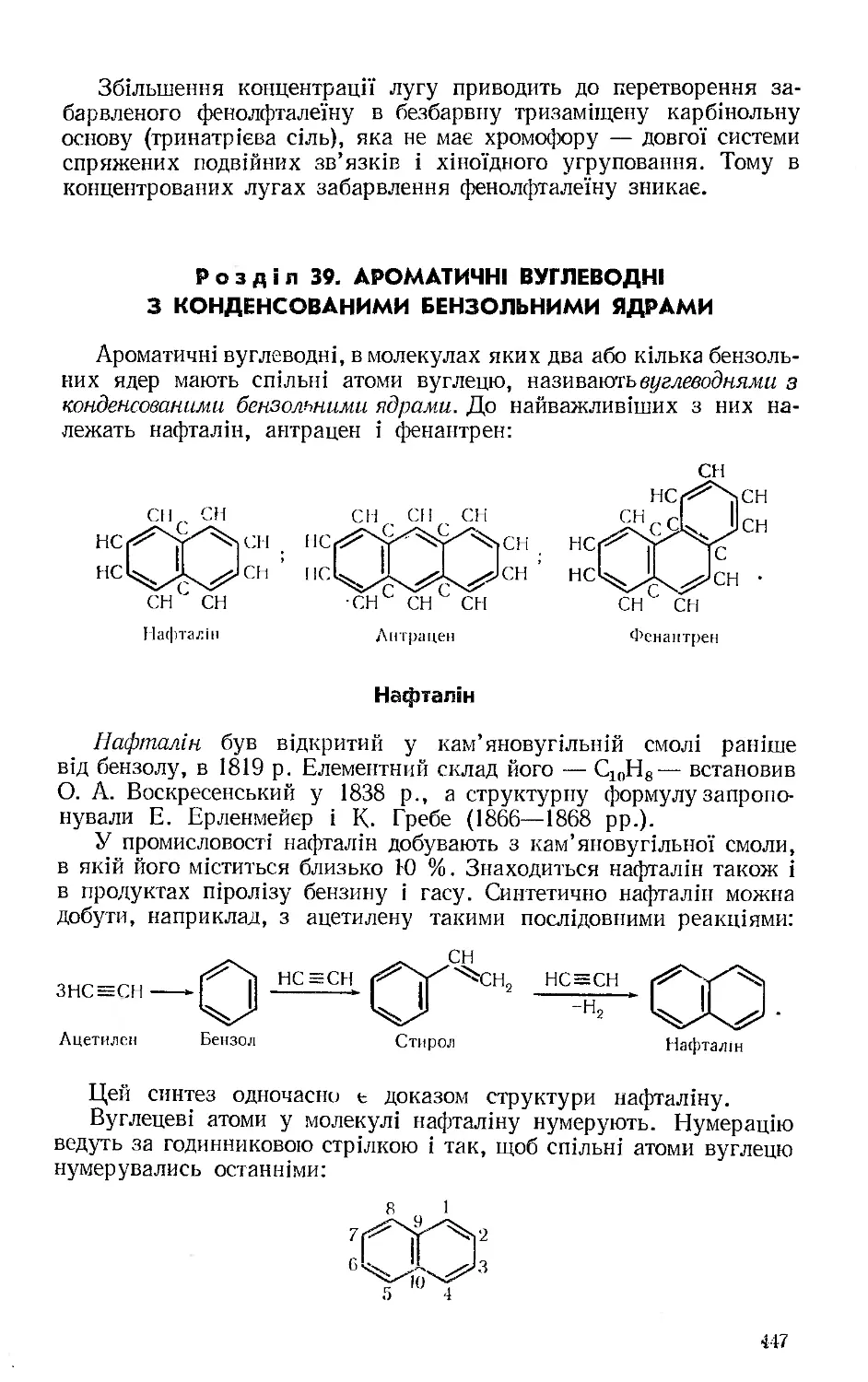








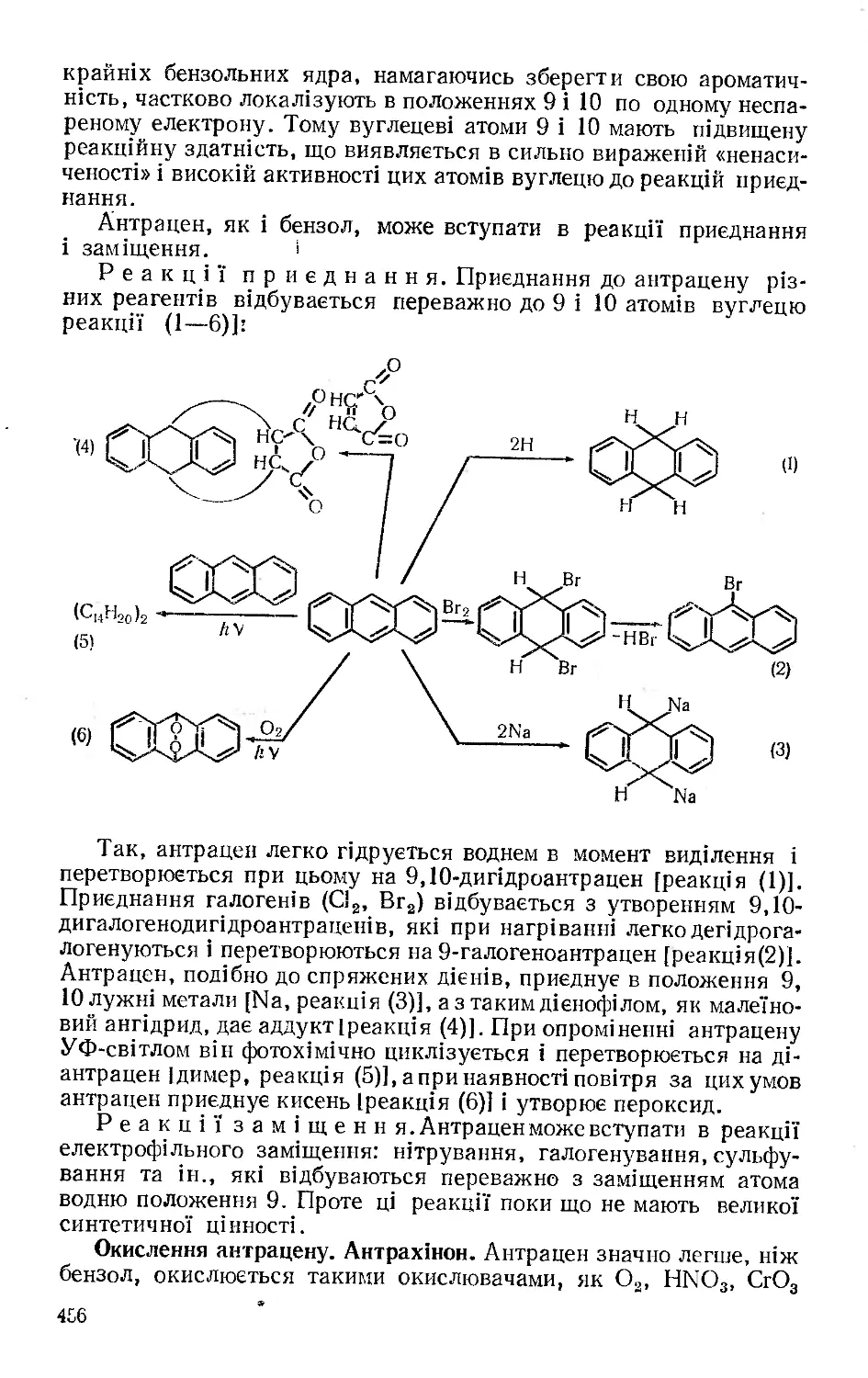



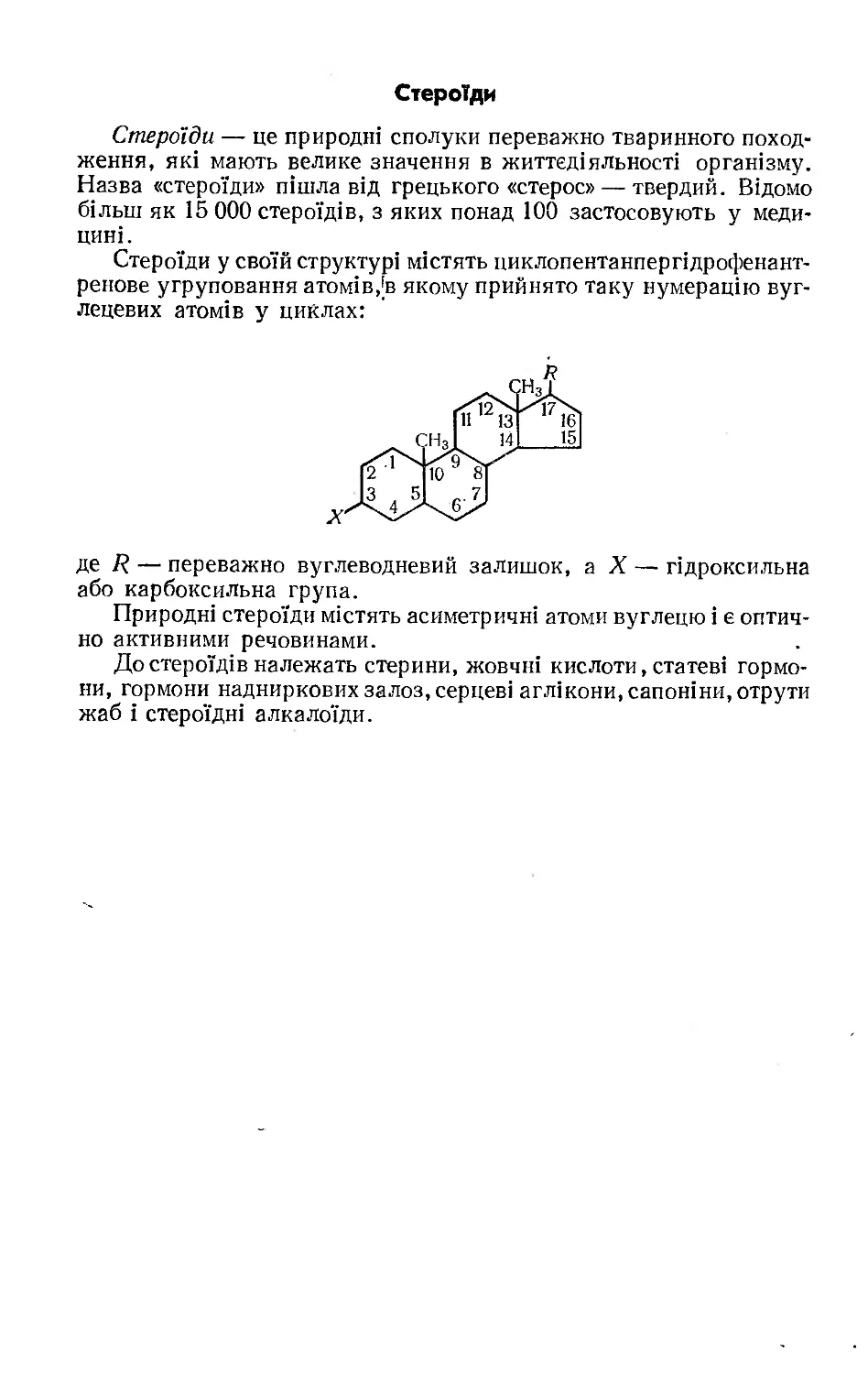



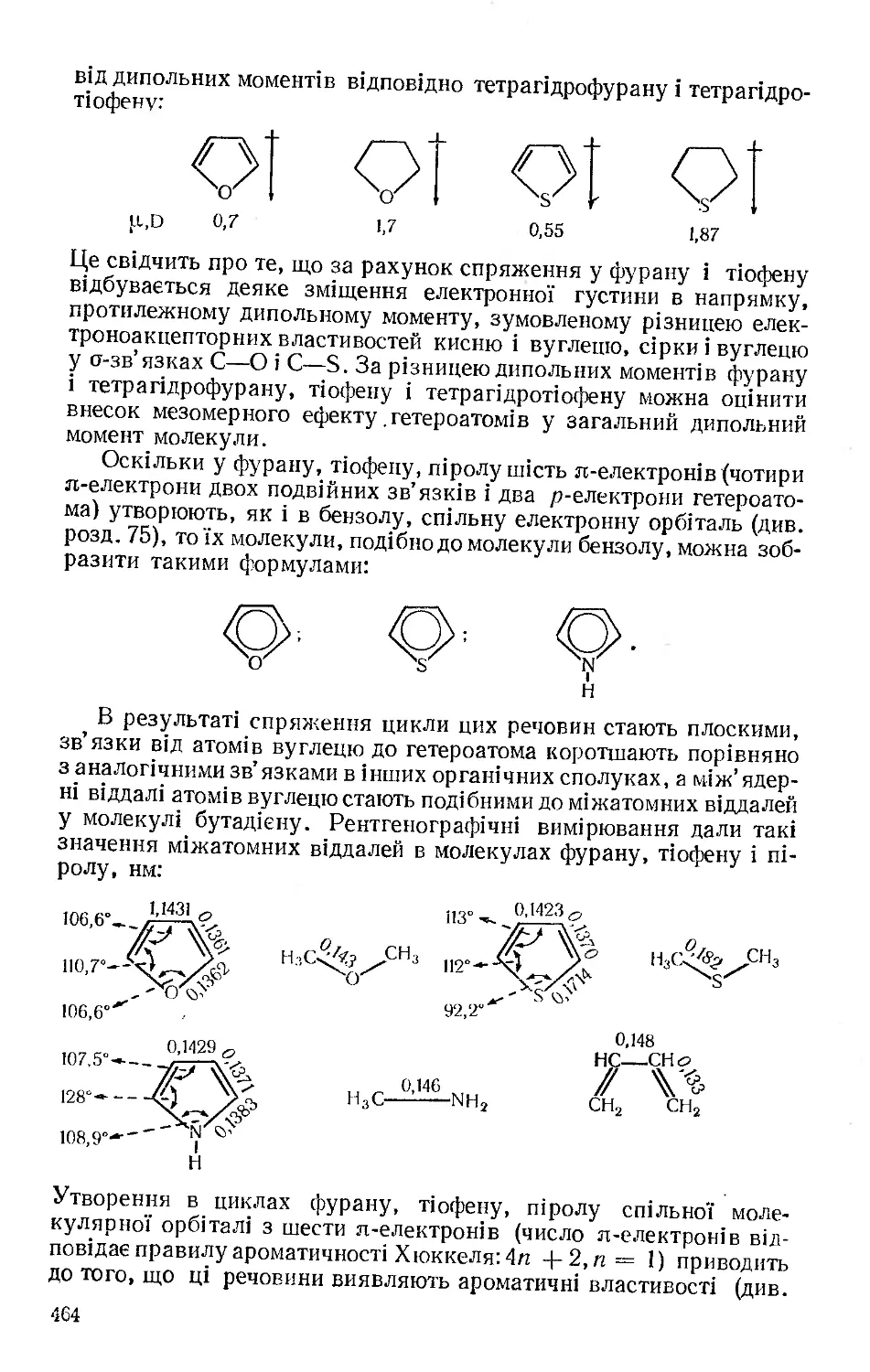


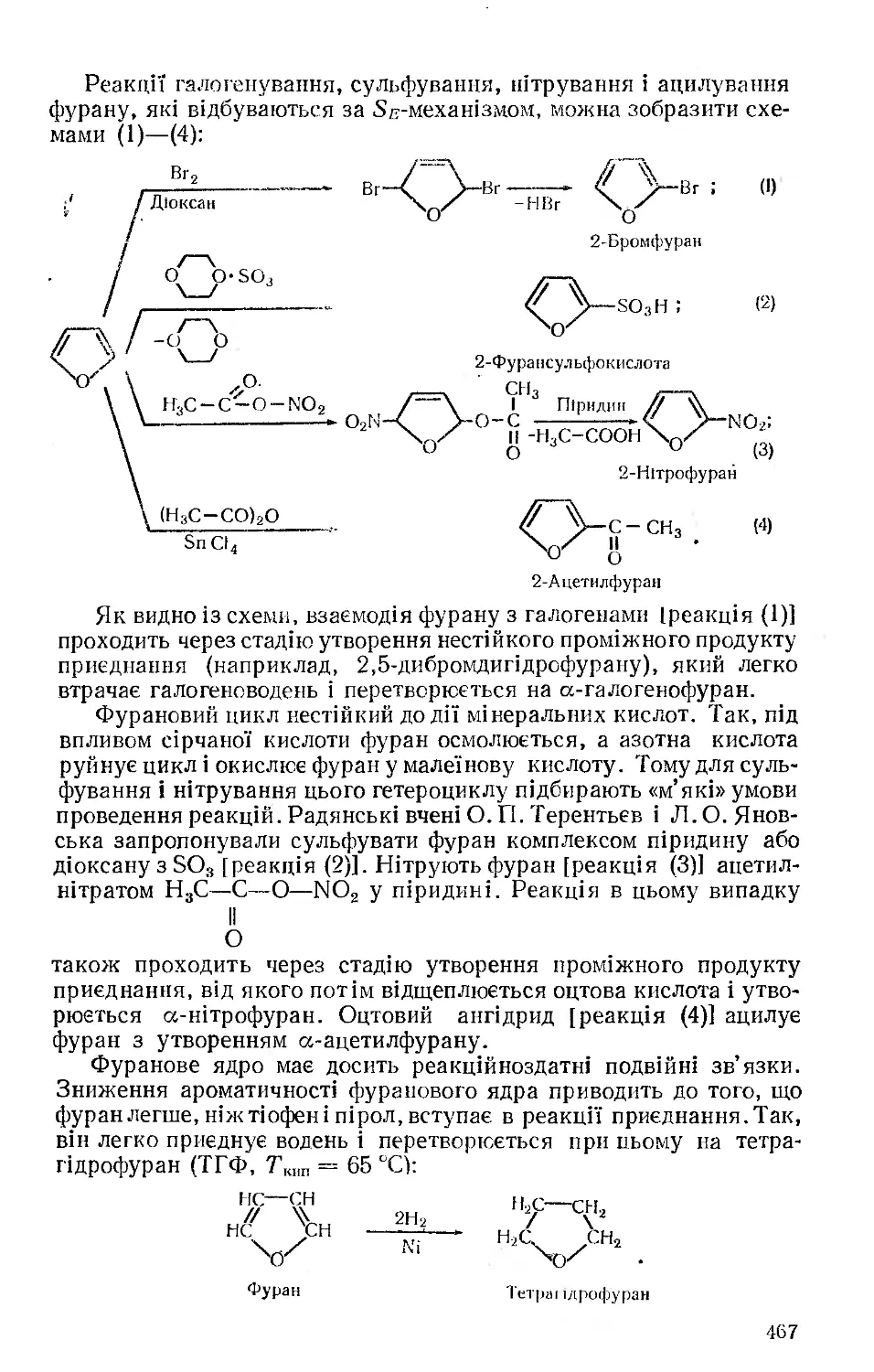







































Текст
Органічна ХІМІЯ
Допущено Міністерством народної освіти України як навчальний посібник для студентів природничих та природничо-географічних факультетів педагогічних інститутів
гч о о.
і з
і
ББК 24.2я73 Д66
УДК 547 (07)
Висвітлено найважливіші теоретичні положення органічної хімії, розглянуто основні класи органічних сполук, їх номенклатуру, ізомерію, хімічні властивості, методи добування, а також застосування в народному господарстві. Викладено теоретичні основи будови і реакційної здатності органічних сполук: електронна природа хімічного зв’язку, будова електронних оболонок і особливості хімічних зв’язків атома вуглецю, взаємний вплив атомів у молекулах, механізми органічних реакцій тощо.
Для студентів природничих та природничо-географічних факультетів педагогічних інститутів
Освещеньї важнейшие теоретические положення орга-нической химии, рассмотреньї основньїе классьі органиче-ских соединений, их номенклатура, изомерия, химические свойства, методьі получения, а также применение в народ-ном хозяйстве. Изложеньї теоретические основні строения и реакционной способности органических соединений: електронная природа химической связи, строение електронних оболочек и особенности химических связей атома углерода, взаимное влияние атомов в молекулах, механиз-мьі органических реакций и др.
Для студентов естественньїх и естественно-географи-ческих факультетов педагогических институтов
Рецензенти: канд. хім. наук Д. І. Шейко (Київський державний університет), канд. техн. наук Н. Д. Хижняк, канд. хім. наук Г. М. Дрмоленко (Харківський державний педагогічний інститут)
Редакційна група літератури з хімії і гірничої справи Редактор Л. Є. Канівець
1705000000-180
ДМ211(04)—92
©А. В. Домбровський, В. М. Найдан, 1992
І5ВИ 5-11-002504-5
ВСТУП
Органічна хімія — наука, що вивчає сполуки вуглецю з іншими елементами. Найчастіше вуглець утворює сполуки з такими елементами, як водень, кисень, азот, сірка, фосфор. Тому ці елементи були названі органогенами. На сьогоднішній день синтезовано сполуки вуглецю майже з усіма елементами періодичної системи елементів Д. І. Менделєєва (крім благородних газів).
Назви «органічна» хімія, «органічні» речовини походять від слова «організм». Ці назви ввів у науку в 1806 р. відомий шведський хімік Я. Берцеліус. На його думку, органічна хімія повинна вивчати речовини рослинного і тваринного походження, які він назвав органічними речовинами. Ці назви збереглись і до сьогоднішнього дня, хоча зміст їх істотно змінився. Я. Берцеліус (1827 р.) у своєму підручнику з хімії вперше виділив сполуки вуглецю і водню в окремий розділ. Проте під впливом ідеалістичної філософії, яка панувала в той час, Я. Берцеліус і багато його сучасників вважали, що утворення органічних речовин відбувається з участю потаємної і недоступної для пізнання «життєвої сили» (по-латинськи уіз уііаііз). Цей ідеалістичний і невірний погляд на утворення органічних речовин одержав назву віталізму. Суть життєвої сили ніхто не міг пояснити, але мали на увазі, що в основі її лежить божественне і надприродне начало. Віталістична теорія твердила, що хіміки можуть добувати органічні речовини тільки з готових природних органічних матеріалів, а на самому початку для створення цих речовин необхідна «життєва сила організму». Отже, ця теорія заперечувала можливість синтезу органічних речовин поза організмом рослин і тварин і тим самим ставилась перешкода розвитку хімічної науки, зокрема органічної хімії. Віталізм являє собою типовий приклад ненаукової теорії, яка виникла під впливом містики і ідеалістичних філософських систем, на яких виховувались західноєвропейські вчені минулого століття. Наука спростувала і відкинула погляди про існування «життєвої сили», нібито необхідної для створення органічних речовин. Ще при житті Берцеліуса почали з’являтись експериментальні роботи, в яких було показано, що органічні сполуки можна добувати з неорганічних речовин руками хіміків в лабораторії, шляхом синтезу. Учень Я. Берцеліуса німецький хімік Ф. Велер у 1824 р. з води і неорганічної речовини диціану
З
Добув досить поширену в рослинному світі щавлеву кислоту:
С=М /О
| +4НаО—+2ИН3.
СееМ Диціан хон /0 хон Щавлева кислота
Надзвичайно важливе значення для дальшого розвитку органічної хімії мав синтез сечовини (речовини тваринного походження), який був проведений також Ф. Велером у 1828 р. Упарюючи розчин неорганічної сполуки ціанату амонію, він уперше хімічним шляхом добув типовий продукт життєдіяльності тварин — сечовину і вказав шлях її синтезу з неорганічних речовин:
К—-2^ К—О—(№Цг50*, н4№-О—2ї?-.ванТ Н2И—С—М-І2.
Ціанід Ціанат калію Ціанат амонію II
калію О
Сечовина
Синтез сечовини наніс досить відчутний удар по віталізму, оскільки довів можливість синтетичного добування органічних речовин з неорганічних, і тому мав велике значення для подальшого розвитку органічної хімії. Після цього синтезу почався період розвитку синтетичної органічної хімії.
Розділ 1. ТЕОРІЯ ХІМІЧНОЇ БУДОВИ
□Теоретичні уявлення в органічній хімії формувались, змінювались і вдосконалювались у міру розвитку хімічної науки. До середини XIX ст. хімія Досягла настільки значних успіхів, що склалися умови для виникнення наукової теорії хімічної будови органічних сполук, одним з головних творців якої був видатний російський хімік О. М. Бутлеров.
До кінця 60-х років XIX ст. хіміки відкрили явище ізомерії, ввели поняття гомології і гомологічних рядів, атома, молекули, атомних мас, валентності елементів. У 1858 р. було встановлено, що вуглець чотиривалентний (А. Кекуле, А. Кольбе) і що його атоми можуть сполучатись між собою в довгі ланцюги (А. Кекуле, А. Ку-пер). А. Купер запропонував зображати зв’язки між атомами в хімічних сполуках за допомогою рисок (цим умовним позначенням хіміки користуються і тепер). Всі перелічені відкриття стали важливою передумовою для створення О. М. Бутлеровим теорії хімічної будови органічних сполук.
Теорія хімічної будови органічних сполук О. М. Бутлерова. Основна ідея теорії хімічної будови органічних сполук була сформульована О. М. Бутлеровим у 1861 р. Суть цієї теорії полягає ось у чому.
4
1. Атоми в молекулах органічних речовин сполучені між собою в певному порядку. Послідовність сполучення атомів у молекулі О. М. Бутлеров назвав хімічною будовою, або структурою, молекули.
2. Атоми в молекулах органічних речовин сполучені між собою відповідно до їх валентності. Вуглець у молекулах органічних сполук чотиривалентний.
3. Властивості молекул органічних речовин залежать від природи атомів, що входять до складу молекул, від кількості цих атомів і від порядку їх сполучення в молекулі, тобто від хімічної будови молекули.
4. Органічні речовини з однаковим якісним і кількісним складом, але з різною хімічною будовою — ізомери — мають різні хімічні властивості.
5. Знаючи властивості органічних речовин, можна встановити їх хімічну будову.
6. Реакційна здатність (хімічна активність) атомів, що входять до складу молекули органічної речовини, може змінюватись залежно від того, з якими іншими атомами вони сполучені в даній молекулі. Ця зміна хімічної активності атомів зумовлюється взаємним впливом безпосередньо сполучених (сусідніх) атомів, а також взаємним впливом атомів через інші атоми (посередній вплив).
Великим успіхом теорії хімічної будови було обгрунтування явища ізомерії, яке протягом майже сорока років не знаходило теоретичного пояснення. Наприклад, дві різні речовини—етанол і диметиловий ефір — мають одну і ту саму молекулярну формулу С2Н6О, але різну хімічну будову:
Н3С—СН2—ОН; Н3С—О—СН3.
Етанол Диметиловий ефір
Це зумовлює їх різні властивості. Так, етанол добре розчиняється у воді, за звичайних умов — рідка речовина, реагує з металічним натрієм, в той час як диметиловий ефір —газоподібна речовина, не розчиняється у воді і не взаємодіє з металічним натрієм.
Теорію хімічної будови О. М. Бутлеров та його учні, а потім і інші хіміки підтвердили, синтезувавши всі передбачені ізомери найпростіших органічних речовин. Так, були синтезовані ізобутан, ізобутилен, третинні спирти тощо.
Теорія хімічної будови органічних сполук набула дальшого розвитку завдяки дослідженням учнів О. М. Бутлерова—В. В. Мар-ковникова, О. М. Зайцева, Є. Є. Вагнера, які глибоко вивчили взаємний вплив атомів у молекулах органічних речовин, розробили нові методи синтезу різних класів органічних сполук. Ця теорія стала загальною теорією органічної хімії.
Вивчення будови органічних речовин. Для встановлення будови органічної речовини її насамперед слід очистити, а потім визначити якісний і кількісний склад, загальну формулу та молекулярну
5
масу. Після цього різними хімічними і фізичними методами визначають. які фрагменти, функціональні групи і зв’язки є в даній речовині, і на основі одержаних результатів роблять висновок про її будову.
Одним з найефективніших методів дослідження будови органічних речовин можна вважати оптичну спектроскопію. При проходженні ультрафіолетового (УФ) або інфрачервоного (ІЧ) світла крізь розчин органічної речовини відбувається його часткове або повне поглинання (це залежить від енергії пучка світла і будови органічної речовини). Іншими словами, оптична спектроскопія досліджує залежність інтенсивності поглинання світла від довжини хвилі (енергії) світла. Енергія, яку поглинула молекула, може викликати або перехід електрона з одного енергетичного рівня на інший, енергія якого вища (УФ-спектроскопія), або привести до коливання і
Рис. 1. УФ-спектр поглинання ацетону
Рис. 2.УФ-спектри фенолу
(1) і феноляту натрію (2)
обертання атомів (ІЧ-спектроскопія). Спектри поглинання в УФ- і видимій областях пов’язані з електронними переходами. Тому ці спектри називають ще електронними спектрами. В загальному спектрі електромагнітних хвиль вони знаходяться в інтервалі від 200 до 1000 нм*.
Залежність інтенсивності поглинання від довжини хвилі записують у вигляді спектральної кривої, яка характеризується двома величинами: положенням максимуму поглинання (в нанометрах) і його інтенсивністю (в одиницях е або 1§е). Наприклад, електронний спектр поглинання ацетону має вигляд, зображений на рис. 1, УФ-спектри фенолу і феноляту — на рис. 2. Порівнюючи спектри поглинання, зображені на рис. 1 і 2, легко помітити, що вони різняться між собою виглядом, положенням максимуму поглинання і інтенсивністю.
Різні класи органічних сполук поглинають УФ-світло різної довжини хвилі і з різною інтенсивністю. На основі цього роблять висновок про будову речовини. УФ-спектри автоматично записують сучасні спеціальні прилади — спектрофотометри.
* 1 нм (нанометр) = 10м.
6
В табл. 1 наведено дані про положення максимумів поглинання та інтенсивність поглинання УФ-світла ряду органічних речовин.
Таблиця 1. Поглинання УФ-світла деякими органічними речовинами
Сполука Характеристика поглинання
X,, нм (І£ е) X,, НМ (І£ Є)
Н2С=СН—сн=сн2 Бутадієн-1,3 217(4,32) •—•
Н3С—СІ Хлористий метил 150 173 (2,30)
Н3С—Вг Бромистий метил •— 204(2,30)
Н3С— 1 Йодистий метил 150—210 258 (3,50)
Н3С—ОН Метиловий спирт 150 183 (2,18)
Н3С— N11, Метиламін 173 213 (2,8)
/О н— хн Мурашиний альдегід (в пароподібному стані) 295(1) —
Н3С—СООН Оцтова кислота (в розчині спирту) 204(1,61) —
Інфрачервона (ІЧ) спектроскопія досліджує поглинання речовин в області від 200 до 5000см-1 (обернені сантиметри). У цих одиницях вимірюють хвильове число (або частоту) V, яке показує кількість хвиль, що вкладаються в одиниці довжини шляху променя світла. Існує таке співвідношення між хвильовим числом (частотою) і довжиною хвилі X: V = 1/Х. Інфрачервоні спектри дають найбільшу інформацію про структуру речовини. ІЧ-спектри називають також молекулярними спектрами.
Інфрачервоне світло не має достатньої енергії, щоб викликати перехід електронів з одного електронного рівня на інший. Але його енергії достатньо для переведення молекули з основного коливального стану в більш високий коливальний стан. Такі коливання пов’язують з окремими зв’язками або групами зв’язків молекули. Кожна група атомів поглинає інфрачервоне світло тільки при певній довжині хвилі. Це дозволяє на основі аналізу ІЧ-спект-ра зробити висновок про вуглецевий скелет, про наявність в молекулі функціональних груп. Одні й ті самі групи атомів у різних молекулах мають однакові частоти в ІЧ-спектрах. Такі частоти називають характеристичними. Характеристичні частоти ряду зв’язків і груп атомів наведено в табл. 2. Кожна органічна речовина має характерний набір смуг поглинання в інфрачервоному спектрі. Ряд смуг в області 1500—700 см-1 є своєрідним «відбитком пальців» речовини, за яким можна встановити її хімічну будову.
7
Таблиця 2. Деякі характеристичні частоти поглинання в ІЧ-спектрах
Зв’язок Сполука Частота, см’1 І нтенсивність і характер смуг
•—с—н 1 Алкани 2850—2960 Сильна
1 1 —с—с— 1 1 Алкани 2200 Сильна
=с-н 1 Алкени і арени 3010—3100 Середня
-С=С— Алкени 1620—1680 Перемінна
=с—н Алкіни 3300 Сильна, різка
— с==С— Алкіни 2100—2260 Перемінна
1 —с—о— і Спирти —С—ОН, прості ефіри 1 1 —С—О—С—, карбонові кислоти 1 1 —С—ОН, складні е.ііри —С—0—С— II ‘ні о о 1000—1300 Сильна
-0—н 1 Спирти —С—0—Н, феноли =С—0—Н 1 3590—3650 Перемінна, різка
\(2=О Альдегіди —С—Н II О 1720—1740 Сильна
\с=О Кетони —С—С—С— 1 II 1 О 1705—1725 Сильна
^с=о Кислоти —С—0—Н II О 1700—1750 Сильна
—С—ин2 1 Аміни 3300—3500 (подвійний пік) Середня
8
На рис. З зображено ІЧ-спектри ацетону НаС- С— СН3 і метил.
етилкетону Н3С—С—СНо—СН3, молекули яких відрізняються II О
Довжина хвилі
З Ц 5 5 6 7 8 5 /0 /2 /4
3600 2800 2000 2000 1800 1600 /400 1200 1000 800 Частота, саг' а Довжина хвилі
З 4 5 5 6 7 8 8/0/2/4
3600 2800 2000 2000 №00 1600 Ш00 1200 1000 800
Частота, см~* д
Рис. 3. ІЧ-спектри ацетону Н3С—С—СН3 (а) і метилетилкетону
Н3С—С—СН2—СН3 (б)
тільки на - СН2-групу атомів. З порівняння ІЧ-спектрів цих речовин видно, що поглинання світла, пов’язане з наявністю С—Н-і С=О-зв’язків, як для ацетону, так і для метилетилкетону відбувається при одній і тій самій частоті: для С—Н-зв’язку при частоті близько 2900 см-1, а для С=О-зв’язку при частоті 1720 см-1. Тому спектри ацетону і метилетилкетону в області від 4000 до 1250 см'1
9
дуже близькі. Смуги поглинання між 1250 і 675 см-1 пов’язані із зміною коливальної і обертальної енергії цілої молекули і характерні для кожної даної молекули. Цю частину спектрів називають областю «відбитків пальців».Вона надзвичайно важлива для визначення хімічної однорідності речовини.Порівнюючи спектри ацетону і метилетилкетону в області 1250—675 см-1, легко помітити їх істотну відмінність.
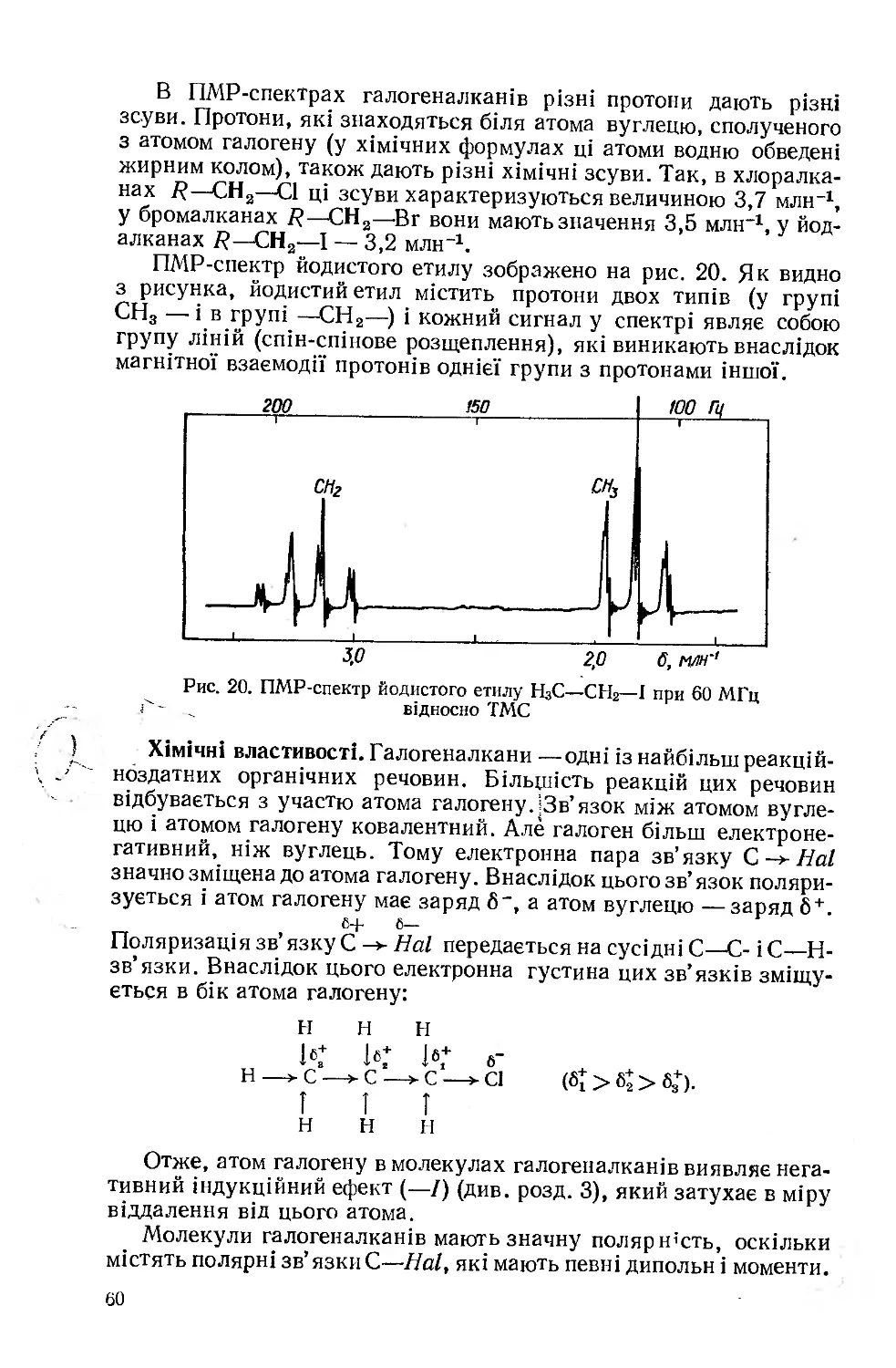
В останні роки для вивчення будови органічних речовин почали широко використовувати метод ядерного магнітного резонансу (ЯМР). Особливо добре відомий ядерний магнітний резонанс на протонах — протонний магнітний резонанс (ГІМР), принцип якого полягає ось у чому. Ядра Деяких атомів, у тому числі і водню (про-
Рис. 4. Спектр ядерного (протонного) магнітного резонансу етилового спирту Н3С—СН2—ОН при 60 МГц
(ТМС — сигнали протонів еталона — триметилсилану)
тони), мають магнітний момент. Якщо протон знаходиться в постійному магнітному полі, то його магнітний момент може бути напрямлений вздовж силових ліній магнітного поля або проти них. Оскільки орієнтація в напрямку цього поля більш вигідна, змінити його на протилежний можна тільки при наданні протонові додаткової енергії (А£). Якщо органічну речовину опромінювати радіохвилями із змінною частотою, то можна підібрати таке значення /IV, яке для конкретного протона буде дорівнювати Л£, тобто при
.... І . 1
цьому можлива зміна орієнтації магнітного моменту І спін-
змінюється на -!—І-). У цей час буде спостерігатись поглинання речовиною радіохвиль, що супроводиться появою відповідного піку поглинання. Змінюючи таким чином частоту в області всього спектра, можна одержати сигнали всіх протонів, які містяться в даній органічній речовині.
Сигнали різних протонів знаходяться один від одного на певній віддалі. Ця віддаль називається хімічним зсувом і позначається грецькою буквою б. Зсув вимірюється відносно сигналу еталона— тетраметилсилану (НаС)48і і виражається у відносних одиницях (мільйонних частках магнітного поля—млн"1), які називаються резо
10
нансною частотою. Число сигналів у спектрі ПМР показує, скільки типів протонів знаходиться в молекулі речовини, а хімічний зсув (положення сигналів) визначає, які це протони (аліфатичні, ароматичні, первинні, вторинні і т. д.). ПМР-спектр етилового спирту, який містить три різних типи протонів, показаний на рис. 4.
Сигнали ПМР багатьох протоновмісних груп розміщені в досить вузькому діапазоні значень 6, і, що особливо важливо, області поглинання протонів, які часто зустрічаються в органічних сполуках, як правило, не перекриваються. Наприклад, олефінові протони поглинають в інтервалі 4,0—6,5 млн-1, ацетиленові — в інтервалі 2.3—2,9 млн-1, ароматичні протони похідних бензолу—в інтервалі 6,5—8,3 млн-1, альдегідні —в інтервалі 9,0—10,5 млн-1. Значення хімічних зсувів протонів деяких протоновмісних органічних груп наведено в табл. 3.
Таблиця 3. Інтервали хімічних зсувів протонів
Протоновмісні груґін б, МЛН”1
—сн3 0,8—4,0
—сн2= 1,1—4,5
1 —сн— 1,4—6,3
=СН (олефіни) | 4,0—6,5
гзСН (ацетилени) 2.3—2,9
Ароматичні протони (монозаміщені 6,5—8,3
похідні бензолу)
—Сг" (альдегіди) ХН 9,0—10,5
—ОН (спирти) 2,0—4,5
—ОН (феноли) 4.5—9,0
—ОН (кислоти) 9,0—14,6
/№Н (аміни) 1,0—2,4
>ЬІН (аміди) 5,0—8,0
Розділ 2. ТЕОРІЯ НАПРЯМЛЕНИХ ВАЛЕНТНОСТЕЙ
Валентний стан і гібридизація атомних орбіталей (АО) вуглецю. Вуглець знаходиться у 11 періоді періодичної таблиці елементів Д. І. Менделєєва. Його порядковий номер 6. Отже, атом вуглецю
11
має шість електронів, які розміщуються на двох електронних шарах. В так званому основному стані атома вуглецю ці електрони перебувають на таких АО:
Як видно з електронної схеми, в такому стані вуглець має тільки два неспарених електрони. Тому в хімічних сполуках він мав би бути двовалентним елементом, оскільки валентність визначається кількістю неспарених електронів в атомі даного елемента. Проте в численних сполуках вуглець майже завжди чотиривалентний. У квантовій механіці чотиривалентність вуглецю пояснюють припущенням, згідно з яким вуглець при утворенні хімічних зв’язків переходить у збуджений стан, тобто в такий, коли один електрон з 28-підрівня (з затратою енергії) переходить на вакантну 2р-орбі-таль. Енергія, необхідна для такого переходу, порівняно невелика. У збудженому стані електрони атома вуглецю знаходяться на таких АО:
У такому стані вуглець має уже чотири неспарених електрони і його атоми можуть утворювати з іншими атомами чотири ковалентних зв’язки. З електронної будови вуглецю в збудженому стані випливає, що атом вуглецю повинен утворювати чотири ковалентних зв’язки за рахунок одного 2х-електрона і трьох 2р-електронів. На рисунку 8-орбіталь зображують у вигляді сферичної поверхні (рис. 5,6, а), а р-орбіталі — у вигляді об’ємних вісімок (рис. 6, б).
р-Орбіталі атома вуглецю орієнтовані в трьох взаємно перпендикулярних площинах (рис. 6, б). В електронних формулах їх позначають символами рх, ру, рг. Орбіталі чотирьох неспарених електронів атома вуглецю відрізняються між собою (одна сферична, а три —об’ємні вісімки, гантелеподібні). Це наводить на думку, що чотири зв’язки атома вуглецю з чотирма іншими однаковими атомами, наприклад з атомами водню в молекулі метану (СНД повинні бути нерівноцінними (один з них повинен відрізнятися від трьох інших), оскільки три р-орбіталі атома вуглецю можуть легко перекриватись з Іх-орбіталями трьох атомів водню, перекриття ж 2«-орбіталі атома вуглецю з І5-орбіталлю четвертого атома водню не може бути досить повним у зв’язку з просторовими умовами. В результаті цього мали б утворитися три міцні вуглець-водневі зв’язки і один—неміцний. Згідно з квантовою теорією, така молеку
12
ла утворитися не може. Ще в минулому столітті було беззаперечно доведено, що в симетрично побудованих молекулах типу С/?4, наприклад у СН4, СС14, С(СН3)4 тощо, всі чотири зв’язки атома вуглецю (всі його чотири валентності) рівноцінні і напрямлені до вершин правильного тетраедра. Рівноцінність зв’язків вуглецю в квантовій механіці (Л. Полінг, 193! р.) пояснюють гібридизацією валентних орбіталей в його атомах у процесі переходу їх у збуджений стан і утворення ними хімічних зв’язків, тобто змішуванням цих орбіталей і вирівнюванням їх за формою.
Форма гібридизованих електронних орбіталей визначається тенденцією до максимального, «лобового» перекриття, що зумовлює
Рис. 5. Схема орбіталі 1«-електрона:
а —сферична форма 15-орбіталі; б—зменшення електронної густини при збільшенні віддалі від ядра
Рис. 6. Схеми орбіталей електронів:
с—орбіталь 2з-електрона; б — орбіталі 2р-електронів, які розміщені в трьох взаємно перпендикулярних напрямках (2рх, 2Ру, 2Р2)
більший виграш в енергії при утворенні о-зв’язків, тобто більшу їх міцність. Гібридизація завжди супроводиться зміною форми електронної орбіталі. При цьому гібридизована електронна орбіталь асиметрична: має більш витягнуту форму по один бік від ядра, ніж по інший. Тому хімічні зв’ язки, що утворюються з участю гібридизованих орбіталей, більш міцні, ніж зв’язки з чистих негібриди-зованих електронних орбіталей.
8 /АГ ібридизація. В результаті гібридизації одного 2х-електрона з трьома 2р-електронами вуглець у молекулах органічних речовин утворює чотири рівноцінні гібридизовані орбіталі (рис. 7), які називаються <з-орбіталями. Таке вирівнювання орбіталей називається 8рР-гібридизацією'.
2&2рх2ру2рг
Гібридизація
(5р3)4.
13
При «/^-гібридизації в лінійну комбінацію з атомною 2«-орбітал-лю входить три атомні 2р-орбіталі. Можна уявити, що кожну з «р3-гібридизованих орбіталей складають на 25 % 2«-орбіталь і на 75 % —2р-орбіталь. Гібридизована «р3-орбіталь має форму несиметричної об’ємної вісімки, витягнутої по один бік від ядра (рис. 8). Така гібридизована форма орбіталей сприяє більшому перекриванню з електронними орбіталями інших атомів, ніж перекривання «чистих» негібридизованих 2«- і 2/?-орбіталей.
Рис. 7. Схема утворення чотирьох «р3-гібридизованих орбіталей вуглецю
Рис. 8. Конфігурація гібридизованої «р3-ор-біталі
Рис. 9. Будова молекули метану:
а — розміщення о-зв’язків у молекулі; б — тетраедрична модель молекули; в — кулестержнева модель молекули; а—масштабна модель молекули за Стюартом — БрІглебом
Гібридизовані електронні орбіталі внаслідок відштовхування електронів розміщуються в просторі на максимальній віддалі одна від одної. Експериментально доведено, що чотири ковалентних зв’язки вуглецю в молекулі метану напрямлені в просторі один відносно одного під кутом 109°28', тобто до вершин правильного тетраедра. Це означає, що осі чотирьох «/^-гібридизованих орбіталей вуглецю також напрямлені в просторі до вершин правильного тетраедра, тобто під кутом 109°28' одна відносно одної (рис. 9). «р3-Гібридизацію називають також тетраедричною гібридизацією, або першим валентним станом вуглецю.
14
Електронегативність вуглецю в 5р3-гібридизованому стані дорівнює 2,5. Перехід атома вуглецю з основного стану в збуджений і 5р3-гібридизація повинні здійснюватись з поглинанням 678 кДж/моль енергії. Ця енергія з надлишком компенсується енергією, яка виділяється при утворенні двох додаткових зв’язків.
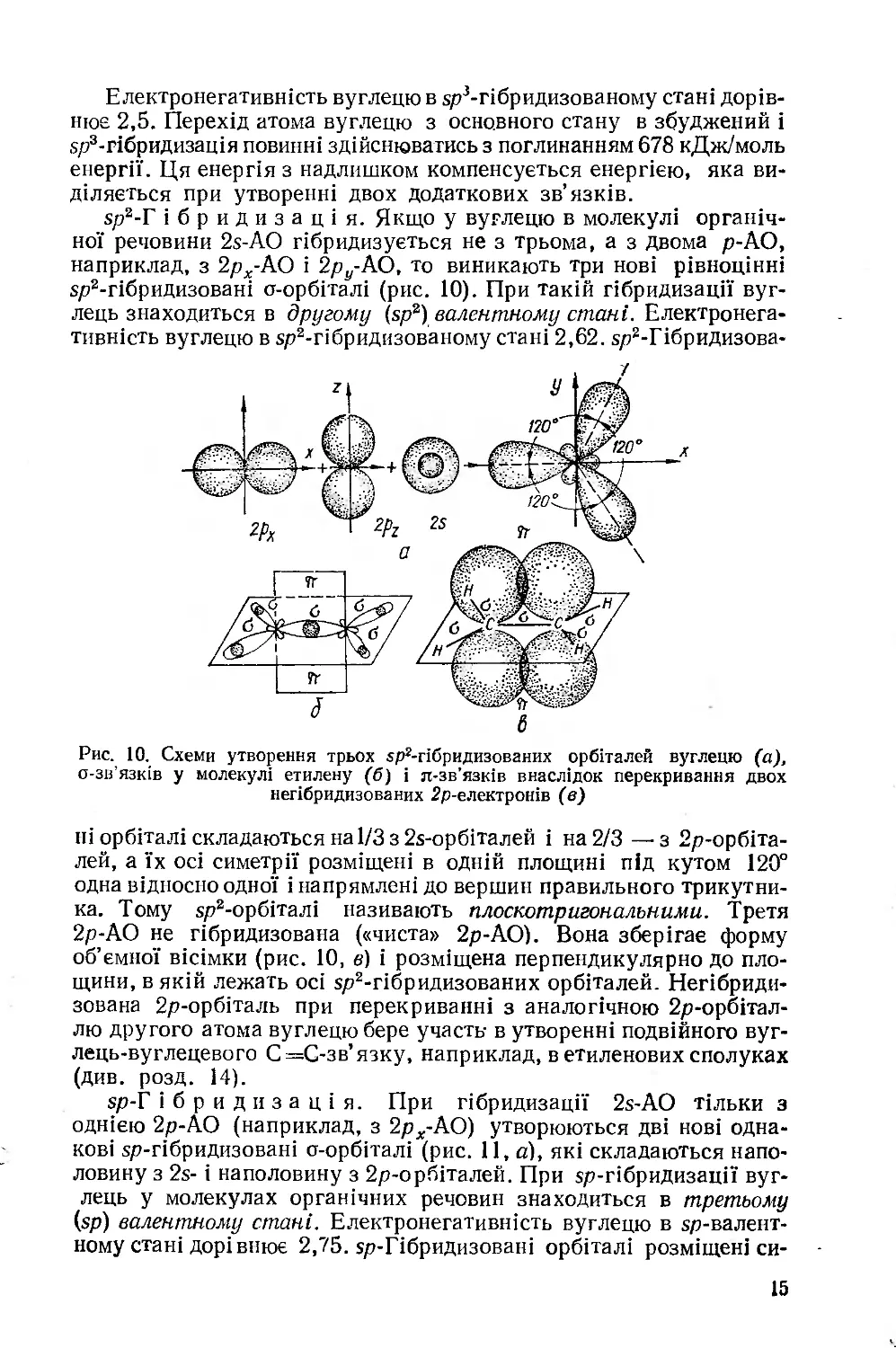
5р2-Г ібридизація. Якщо у вуглецю в молекулі органічної речовини 2х-АО гібридизується не з трьома, а з двома р-АО, наприклад, з 2рд.-АО і 2/^-АО, то виникають три нові рівноцінні 5р2-гібридизовані о-орбіталі (рис. 10). При такій гібридизації вуглець знаходиться в другому (зр2) валентному стані. Електронегативність вуглецю в 5р2-гібридизованому стані 2,62. 5р2-ГібриДизова-
Рис. 10. Схеми утворення трьох «р2-гібридизованих орбіталей вуглецю (а), о-зв’язків у молекулі етилену (б) і л-зв’язків внаслідок перекривання двох негібридизованих 2р-електронів (в)
ні орбіталі складаються на 1/3 з 25-орбіталей і на 2/3 —з 2р-орбіта-лей, а їх осі симетрії розміщені в одній площині під кутом 120° одна відносно одної і напрямлені до вершин правильного трикутника. Тому зр2-орбіталі називають плоскотр азональними. Третя 2р-АО не гібридизована («чиста» 2р-АО). Вона зберігає форму об’ємної вісімки (рис. 10, в) і розміщена перпендикулярно до площини, в якій лежать осі $р2-гібридизованих орбіталей. Негібриди-зована 2р-орбіталь при перекриванні з аналогічною 2р-орбітал-лю другого атома вуглецю бере участь- в утворенні подвійного вуг-лець-вуглецевого С=С-зв’язку, наприклад, в етиленових сполуках (див. розд. 14).
«рТ ібридизація. При гібридизації 25-АО тільки з однією 2р-АО (наприклад, з 2рх-АО) утворюються дві нові однакові хр-гібридизовані о-орбіталі (рис. 11, а), які складаються наполовину з 2«- і наполовину з 2р-орбіталей. При 5р-гібридизації вуглець у молекулах органічних речовин знаходиться в третьому (зр) валентному стані. Електронегативність вуглецю в 5р-валент-ному стані дорівнює 2,75. $р-Гібридизовані орбіталі розміщені си-
15
азональною. В третьому валентному
Рис. 11. Схема утворення двох «р-гібридизованих орбіталей вуглецю (а) та схематичне зображення зв’язків у молекулі ацетилену (б)
метрично, їх осі співпадають, але напрямлені в протилежні боки (під кутом 180°). «^-Гібридизацію називають лінійною, або дістані електронні орбіталі
2рй-АО і 2рг-АО вуглецю не гібридизовані. Вони зберігають форму об’ємних вісімок (рис. 11,6), розміщені в просторі одна відносно одної під прямим кутом і беруть участь в утворенні потрійного зв’язку —С = С—, наприклад, в ацетиленових сполуках (див. розд. 15).
Виходячи з вищесказаного, можна зробити висновок, що вуглець в органічних сполуках може знаходитись у трьох валентних станах, які характеризуються відповідно зр\ зр2- і зр-гібридизацією його орбіталей. Стан гібридизації атомів у молекулі даної сполуки можна визначити. Для цього підраховують кількість «груп» (атомів або груп атомів), які сполучені з тим атомом, гібридизацію якого хочуть встановити, а потім користуються таким правилом: чотири «групи» біля атома відповідав його зр2-гібридизації, три «групи» — зр2-гібридизації, дві «групи» — зр-гібридизації. Наприклад:
сі і
н
С = N - Н ;
Характеристика ковалентного зв’язку. Ковалентний зв’язок між атомами характеризується енергією, довжиною, полярністю, поляризованістю і напрямленістю в просторі.
Енергія, яка необхідна для розщеплення окремого зв’язку з утворенням Двох нейтральних атомів, називається енергією дисоціації зв’язку. Її виражають у джоулях на моль. Часто неможливо визначити енергію, необхідну для розриву окремого зв’язку. Тому замість енергії дисоціації зв’язку використовують середню величину, яку називають енергією зв’язку.
Довжина зв’язку —це та віддаль між ядрами атомів, що утворюють хімічний зв’язок, на якій сили притягання урівноважуються силами відштовхування.
Ковалентні зв’язки можуть бути полярними і неполярними. В неполярних ковалентних зв’язках область максимальної електронної густини (іншими словами,пара електронів) розташована на однаковій віддалі від ядер обох атомів. Наприклад, ковалентний 16
*
зв’язок у молекулі воднює неполярним. Переважна більшість ковалентних зв’язків має полярний характер. Це означає, що область максимальної електронної густини розташована не точно посередині між ядрами атомів, а в більшій або меншій мірі зміщена до одного з них. Напрям зміщення електронної густини зв’язку визначається за електронегативністю атомів.
Електронегативність характеризує здатність атомів даного елемента в сполуці відтягувати електрони від сусідніх атомів і визначає додаткову електронну спорідненість тих атомів, які вже заповнили вакантні орбіталі і після цього ще намагаються відтягнути спільні електрони на себе. Електронегативність атомів можна визначити за допомогою шкали електронегативностей, запропонованої Л. Полінгом:
Електронегативність атомів за Полінгом
Елемент к N8 Ьі Мо А1 8» н с 5
Електронегативність 0,8 0,9 1,о 1,2 1,5 1,8 2,1 2,5 2,62 2,75 2,5
Продовження
Елемент N СІ Вг І О р
Електронегативність 3,0 3,0 2,8 2,5 3,5 4,0
Найбільш електронегативним є фтор, тому в сполуках з іншими елементами електронна пара зв’язку завжди зміщена в його бік. Вуглець знаходиться посередині шкали і залежно від валентного стану може виявляти різну електронегативність. Більшість елементів, які входять до складу органічних речовин, виявляють більшу електронегативність, ніж він, тому атом вуглецю в цих речовинах часто несе частковий позитивний заряд.
Полярність різних зв’язків можна кількісно характеризувати різницею електронегативностей сполучених між собою атомів. Чим більша ця різниця, тим полярніший даний зв’язок, тим він більш активний у хімічних реакціях.
Внаслідок різної електронегативності атомів у хімічному зв’язку (в молекулі) порушується симетрія розподілу зарядів (електронної густини). Одні атоми, в результаті зміщення до них електронної густини, набувають часткового негативного заряду, інші атоми, внаслідок зміщення електронної густини від них, набувають часткового позитивного заряду. Ці часткові (неповні) заряди, величина яких становить частки заряду електрона, позначають грецькою буквою «дельта» (6 + і б-). Наприклад, ковалентний зв’язокміж
/Й. І?
< \
атомами вуглецю і водню (С—Н) полярний, оскільки вуглець більш електронегативний і електронна густина зміщена до його атома, що схематично показано стрілкою. В результаті на атомі вуглецю виникає частковий (менший від одиниці) заряд 6-, а на атомі водню — однаковий за абсолютною величиною, але протилежний за знаком заряд б+:
с—ф-н •
Полярними є також зв’язки
8+8+гх 8-8+/ч 8- 8+г\ 8- /-х 8- 8+8-/\ 8+
С V/ Ма>с-Г7*С!> С-Ь4-Вг, С-Н+-К, с-ь4*о, м-Чч-н.о+ч-н,
та ін. Частковий заряд 6 може мати різні значення, але вони завжди менші від одиниці. Наприклад, у зв’язку С—Н в молекулах насичених вуглеводнів значення такого заряду на атомах вуглецю і водню дорівнює 0,04, а в зв’язку С—СІ —0,25 заряду електрона.
Полярність зв’язку оцінюють, виходячи переважно з величини дипольного моменту молекули.
Дипольний момент рі молекули є добутком абсолютного значення заряду <7на віддаль/між центрами ваги всіх позитивних і негативних зарядів молекули: ц = ді. Дипольний момент вимірюють в дебаях* (О) або в кулон-метрах (Кл • м): Ю = 3,34 х X 10-3 Кл • м.
Дипольний момент —векторна величина. Його позначають символом +*. У хімічній літературі дипольний момент прийнято вважати напрямленим від позитивного кінця диполя до його негативного кінця. Для двохатомної молекули дипольний момент зв’язку е одночасно і дипольним моментом молекули. Дипольний момент молекули, яка має кілька полярних зв’язків, виражається векторною сумою Дипольних моментів окремих зв’язків: Наприклад:
Н СІ
Зв’язок тим полярніший, чим більше зміщена спільна пара електронів до одного з атомів, тобто чим більші часткові заряди атомів і чим більша довжина диполя /. Тому в молекулах, подібних за будовою, дипольний момент зростає із збільшенням різниці електро-негативностей атомів, які входять до їх складу. Наприклад, ди
* Цю одиницю названо на честь голландського фізика і фізикохіміка лауреата Нобелівської премії Петера Дебая (1884—1966).
18
польні моменти галогенопохідних метану Н3С—І, НОС—Вг,Н3С—СІ і Н3С—Р становлять відповідно 1,70; 1,82; 1,86 і 1,87С.
Важливою характеристикою хімічних зв’язків є їх полярність, яка часто визначає фізичні й хімічні властивості органічних речовин. Від наявності того чи іншого часткового заряду на атомі залежить його реакційна здатність. Полярність зв’язку в молекулах органічних речовин — нестала величина. Вона може змінюватись під впливом багатьох факторів: природи атакуючого даний зв’язок реаген-та, характеру розчинника тощо. Тому крім полярності, яка характеризує хімічні зв’язки в статичному стані, кожний зв’язок має ще й поляризованість. Поляризованість зв’язку — це його здатність змінювати свою полярність під дією зовнішнього електромагнітного поля.
Ковалентні зв’язки завжди мають певну напрямленість у просторі. Між напрямами ковалентних зв’язків атома утворюється валентний кут. Наприклад, у молекулі метану цей кут становить 109°28' (див. розд. 7), етилену —120° (див. розд. 14), ацетилену — 180° (див. розд. 15) і т. д.
Характеристика ковалентних зв’язків атома вуглецю. Елемент вуглець може утворювати ковалентні зв’язки з атомами багатьох інших елементів. В органічних сполуках найчастіше трапляються вуглець-вуглецеві і вуглець-водневі ковалентні зв’язки. Причому атоми вуглецю можуть утворювати між собою одинарні, подвійні і потрійні ковалентні зв’язки. Порівняння деяких параметрів у сполуках з такими зв’язками на прикладі етану, етилену і ацетилену наведено в табл. 4.
Таблиця 4. Характеристики (параметри) одинарних і кратних зв’язків у молекулах етану, етилену і ацетилену
Параметри Н.С-СН, н,с=сн2 н—с=с—н
Зв’язок С—С Валентний стан атома С зр3 зр2 вр
Частка в-хмари, % 25 33,3 50
Довжина зв’язку С—С, нм 0,154 0,133 0,120
Ковалентний радіус атома С, нм 0,077 0,067 0,060
Енергія зв’язку, кДж/моль 350,0 607,1 831
Відносна електронегативність атома С 2,5 2,62 2,75
Характеристичні частоти коливань 1000 1640 2200
в ІЧ-спектрах, см-1 Рефракція для лінії В, см3/(г-моль) 1,296 4,17 6,03
Зв’язок Н—С Довжина зв’язку С—Н, нм 0,1094 0,108 0,106
Енергія, кДж/моль 401,9 435,5 473
Дипольний момент, В 0,4 0,6 1,0
Зміна валентного стану атома вуглецю і збільшення кількості електронів на утворення зв’язку між атомами вуглецю приводить до того, що в ряду етан —етилен —ацетилен довжина зв’язків і
19
ковалентний радіус атома вуглецю зменшуються, Змінюється також і енергія зв’язків між атомами. Так, при утворенні другого зв’язку між атомами вуглецю загальна енергія подвійного зв’ язку не подвоюється, а тільки зростає на 257,1 кДж/моль. З цього випливає, що л-зв’язок слабший від о-зв’язку. На ще меншу величину зростає енергія зв’язку між вуглецевими атомами при виникненні потрійного зв’язку.
Молекули, які містять кратні зв’язки, поглинають світлові промені з більшою довжиною хвилі (мають нижчі частоти коливання), ніж молекули насичених сполук. л-Зв’язки легше поляризуються, ніж о-зв’язки. Мірою поляризованості зв’язків частково може бути рефракція зв’язку, яка збільшується в ряду етан—етилен —ацетилен.
Характфр зв’язку між атомами вуглецю впливає на властивості сполучених з ними атомів водню. В ряду етан—етилен—ацетилен енергія зв’язку С—Н зростає. В цьому ряду зростає також і кислотність атомів водню. Так, атоми водню в молекулі ацетилену і в групі СзеС—Н мають здатність заміщуватись на метали. Зміна хімічної активності зв’язків С—Н у молекулах цих сполук пояснюється тим, що в ряду етан—етилен—ацетилен зростає електронегативність вуглецю, що в свою чергу приводить до збільшення полярності зв’язку С—Н і відповідно до підвищення активності атома водню біля «^-гібридизованого атома вуглецю. Зростання електро-негативності атома вуглецю в ряду етан—етилен—ацетилен зумовлене збільшенням частки 2«-електронної орбіталі в гібридизованих орбіталях в порядку зрл < зр2 < зр. Оскільки енергія 2«-електро-нів в атомі вуглецю на 5,3 еВ нижча, ніж енергія 2р-електронів, то зростання частки 2«-електронної хмари в гібридизованій орбіталі буде сприяти сильнішому притяганню електронів.
Розділ 3. ТЕОРІЯ ЕЛЕКТРОННИХ ЗМІЩЕНЬ
В молекулах органічних речовин під впливом наявних у них різних за своєю природою атомів або груп атомів відбувається перерозподіл електронної густини хімічних зв’язків. У симетричній молекулі, яка складається з подібних за електронегативністю атомів (при умові, що молекула перебуває в статичному стані), електронна густина розподілена рівномірно.Якщо молекула побудована з різних за елекронегативністю атомів,то в цьому випадку відбувається зміщення електронної густини в бік атома або групи атомів з більшою електронегативністю. Наприклад, в молекулі хлористого. метилу атом хлору, як більш електронегативний, викликає зміщення електронної густини на себе:
НзС-ф—СІ .
У зв’язку з цим сг-зв’язокС—СІ стає полярним. На атомі хлору ви-20
никає негативний б_-заряд, а на атомі вуглецю — рівний за величиною, але протилежний за знаком позитивний б+-заряД.
Зміщення електронної густини в ковалентному зв’язку впливає не тільки на ті атоми, які безпосередньо сполучає дана електронна пара, але передається й на інші атоми. Розглянемо цей вплив на прикладі Н3С-групи:
Оскільки вуглець більш електронегативний, ніж водень, то електронна густина від атомів водню зміщується до атома вуглецю і на ньому виникає заряд 8~. Внаслідок цього електронна густина сусіднього з цим атомом вуглецю зв’язку відштовхується від нього, зміщується, тобто також поляризується. Здатність певної групи атомів або окремого атома викликати зміщення електронної густини вздовж о-зв’язків під впливом різних за своєю природою атомів або груп атомів за рахунок електростатичної індукції називається індукційним ефектом (/-ефектом). Цей ефект є найпростішою формою вияву взаємного впливу атомів у молекулі. Схематично індукційний ефект позначають прямою стрілкою яка напрямлена в бік більш електронегативного атома або групи атомів, що викликають зміщення електронної густини на себе.
Індукційний ефект може бути позитивним (-(-/) і негативним (—І). Якщо атом або група атомів викликають зміщення електронної густини від себе на сусідні зв’язки (відштовхують від себе електронну густин-у), то такий індукційний ефект називається позитивним. Протилежний ефект, коли атом або група атомів відтягують електронну густину сусідніх зв’язків на себе, називають негативним індукційним ефектом.
-(-/-Ефект виявляють: а) елементи менш електронегативні, ніж вуглець, наприклад лужні метали; б) групи атомів з повним негативним зарядом. У цьому випадку -(-/-ефект зменшується при збільшенні електронегативності атомів: ~;С_ >—-ЕГ:>—О:~; в) алкільні групи, причому -(-/-ефект їх зменшується в ряду
(Н3С)3С - >(Н3С)2СН-> Н3С-СН2->Н3С->Н-.
—/-Ефект виявляють елементи більш електронегативні, ніж вуглець, тобто галогени, азот, кисень та ін., а також групи атомів з повним позитивним зарядом на атомі, сполученому з атомом вуглецю, наприклад:
21
—/-Ефект мають групи з семіполярним зв’язком —С—N—О:-,
/ ц
О
а також групи з подвійними зв’язками: /С=О, С=И—7? та ін. —/-Ефект зменшується справа наліво в періодах і зверху вниз у групах періодичної системи елементів Д. І. Менделєєва, наприклад:
—Р > —О— > ^і\'; —Р > —СІ > —Вг.
Якщо замісник має повний заряд, —/-ефект збільшується із збільшенням електронегативності атома, який несе позитивний заряд:
-40:>">1Ч—; ;О:> )8: >\8е:. / / / । / । / ।
Для більш складних груп атомів -/-ефект залежить від природи атомів, які входять у цю групу. Наприклад, —/-ефект кисню зростає, якщо він сполучений з групою, що виявляє —/-ефект:
—О—С—СН, > — О—Н > — О—СН,.
II О
—/-Ефект залежить також від гібридизації атома. Наприклад, електронегативність атома вуглецю найвища в зр-гібридизованому стані, а найменша — в зр8-гібридизованому стані.
В міру віддалення від атома або групи атомів, які викликають часткове зміщення електронної густини, індукційний ефект у насичених системах швидко слабшає (зменшується, затухає)^ Наприклад:
с+"«+" в*' б"
Н3С -> СН2 -> СН3 ->- СІ (6+"' « б+" « 6+').
Величина заряду 6+' набагато більша, ніж 6+", а величина заряду 6+" набагато більша від б+"'. Вважають, шо індукційний ес]:ект у четвертому положенні відносно тієї групи, яка цей ефект викликає, практично вже не виявляється.
Переконливим прикладом прояву індукційного ефекту є зростання сили монохлороцтової кислоти порівняно з оцтовою кислотою:
/О /О
Н3С—; С1*-Н2С*-С^ .
хо—н І
О-^-Н
= 1,76 • 10~в Лд = 150 10-®
Як видно з порівняння констант дисоціації (/(д) цих кислот, хлороцтова кислота набагато сильніша від оцтової. Пояснюється
22
це тим, що атом хлору, маючи —/-ефект, викликає зміщення електронної густини сусідніх зв’язків на себе (схематично показано стрілками). Таке зміщення приводить до підвищення полярності зв’язку між атомами кисню і водню в групі О—Н. Тому атом водню цієї групи в молекулі хлороцтової кислоти легше відщеплюється у вигляді іона Н+. Отже, хлороцтова кислота дисоціює легше, ніж оцтова. А сила кислоти, як відомо, визначається ступенем її дисоціації.
У молекулах з подвійним, потрійним зв’язком атоми і групи атомів з позитивним та негативним індукційним ефектом викликають зміщення (поляризацію) л-електронної густини цих зв’язків. Наприклад:
н
і н-с
!
н
ї
сн=снг; н—с—с=сн.
н
Під впливом метальної групи, яка має +/-ефект, відбувається зміщення електронів л-зв’ язку в бік кінцевого незаміщеного ненаси-ченого вуглецевого атома і на ньому виникає надлишок електронної густини (заряд 6_).
Ненасичені сполуки, в молекулах яких чергуються прості і подвійні або потрійні зв’язки, як, наприклад, в бутадієні Н2С=СН—СН=СН2, У хімічних реакціях поводять себе своєрідно. Під час деяких реакцій приєднання такі подвійні зв’язки виявляють себе як єдине ціле. Так, бром приєднується переважно до першого і четвертого атомів вуглецю в молекулі бутадієну (див. розд. 16). У зв’язку з цим виникло уявлення, що такі подвійні зв’язки між собою сполучені (знаходяться в спряженому стані). Якщо в молекулі органічної сполуки є система спряжених подвійних зв’язків (див. розд. 16) або біля атома вуглецю з подвійним зв’ язком чи 'атома вуглецю ароматичного ядра знаходиться атом або група атомів з вільною електронною парою, то електронна густина л-зв’язків або окремого л-зв’язку і вільних р-електронів взаємно перекривається і стає спільною. У таких сполуках взаємний вплив здійснюється через л-зв’язки і через цю спільну електронну густину. Передача взаємного впливу через л-зв’язки називається ефектом спряження, або мезомернпм ефектом (М-ефект). Наприклад, у молекулах хлористого вінілу, вінілметилового ефіру, фенолу, аніліну та інших органічних сполук існує ефект спряження:
н2с = сн-сі: ;
Хлористий ВІНІЛ
Вінілметиловий ефір
н3с-р-сн=сн2;
Анілін
У молекулах цих речовин відбувається перекривання (спряження) вільних р-електронів атомів хлору, кисню, азоту з л-орбіталями
23
подвійного зв’язку і бензольного ядра. В результаті такого перекривання ці атоми сполучаються в молекулі з вуглецевим атомом не тільки а-зв’язком, а ще й додатковою л-електронною хмарою, яка утворилася в результаті спряження, і передача взаємного впливу в цих сполуках відбувається по системі спряжених зв’язків.
Спряження в молекулах органічних сполук зображують за допомогою зігнутих стрілок. Початок стрілки вказує, які р- або л-електрони перекриваються (беруть участь у спряженні), а кінець стрілки —зв’язок або атом, до якого зміщуються ці електрони. Залежно від того, які електрони —>р- чи л- —беруть участь у спряженні, розрізняють р, л-спряження, л, л-спряження і навіть о, л-спряження. У вищенаведених прикладах—молекулах хлористого вінілу, вінілметилового ефіру, фенолу, аніліну—відбувається р, л-спряження, оскільки р-електрони атомів хлору, кисню, азоту перекриваються з л-орбіталлю подвійного зв’язку або бензольного ядра. У молекулі бутадієну відбувається зустрічне л,л-спряження:
112^=СН^СН=СН2 .
На відміну від індукційного ефекту, при якому відбувається тільки зміна полярності о- або л-зв’язку, при спряженні електронна хмара частково зміщується в область сусіднього зв’язку.
Мезомерний ефект буває позитивним (4-М) і негативним (—М). Позитивним вважають такий ефект, при якому атом або група атомів викликає зміщення електронної густини до л-зв’язків. При негативному мезомерному ефекті атом або група атомів викликає зміщення електронної густини від л-зв’язків до себе, у свій бік.
4-М-Ефект виявляють атоми і групи атомів, які мають вільні електронні пари. Причому для елементів, розміщених в середині другого періоду періодичної системи, цей ефект тим більший, чим менший заряд ядра атома, який несе електронну пару. Тому 4-М-ефект атома азоту більший, ніж атомів кисню і фтору, заряди ядер яких відповідно становлять 7, 8 і 9:
б—>—Г:.
/ І
4-М-Ефект виявляють від’ємно заряджені атоми. їх ефект та-- -кож тим більший, чим менший заряд ядра, і більший, ніж ефект тих самих атомів у незарядженому стані. 4- М-Ефектзменшується при збільшенні електронегативності атома, який має вільну електронну пару, оскільки при цьому знижується тенденція віддавати цю пару, наприклад:
>с->-і4-> -о-
24
4-Л4-Ефект зменшується також при збільшенні об’єму атома, який несе електронну пару, наприклад:
Тривалентний фосфор практично не має ефекту спряження.
Ефект спряження зменшується в тому випадку, коли атом, який має вільну пару електронів, з’єднаний з групою, що відтягує електронну густину на себе:
Н2Й— > — N11—С—СІ І3.
При переході вниз у межах однієї групи періодичної системи ф-ЛІ-ефект падйє. Зменшення ефекту спряження у цьому випадку пояснюють більшою енергетичною вигідністю перекриття орбіталей, близьких за розмірами. Внаслідок цього 2/?-орбіталі краще перекриваються з 2р-орбіталями, гірше з Зр-орбіталями і ще гірше — з 4р-орбіталями.
—М-Ефект виявляють атоми, які мають вакантну низькорозта-шовану орбіталь (В, С+), і групи, які мають л-зв’язок між атомами з різною електронегативністю, якщо ця група в молекулі приєднана до атома вуглецю менш електронегативним атомом (^С =0, —-С^ІЧ, —М02, /С=1ЧН, /В02 і т. д.).ТІри цьому зміщення електронної густини відбувається в бік більш електронегативного атома:
>С = 0, -С=И , -М02', хС = NN. >8О2
і менш електронегативний атом має недостачу (дефіцит) електронів, тобто несе б "-заряд. Тому зміщення л-електронпої густини сусіднього з ним подвійного зв’язку чи ароматичного ядра відбувається в бік цього атома:
I-
Н2С = СН-С чн .
—ЇИ-Ефект збільшується із збільшенням електронегативності атома.'Він тим більший, чим більша різниця в електронегативностях атомів, що утворюють дану групу.
Групи, які мають подвійні або потрійні зв’язки між однаковими
спряження обох знаків.
25
Ефект спряження в міру віддалення л-зв’ язку від атома або групи атомів, які цей ефект викликають, зменшується (затухає). Вважають, що ефект спряження затухає приблизно через три подвійних зв’язки.
У процесі експериментальних досліджень ХІМІКИ помітили, що ефект, подібний до Л4-ефекту, виникає також при наявності ал-кільного радикала біля подвійного зв’язку або ароматичного ядра. Цей ефект напрямлений в бік подвійного зв’язку або до бензольного ядра і зображується зігнутою стрілкою:
Такий ефект називається гіперкон'югацією, надспряженням, а,п-спряженням, або ефектом Натана—Бекера. Надспряження алкільних радикалів зменшується в такій послідовності:
Н8С— > Н3С—СН2—> (Н3С)2СН—> (Н3С)3С—.
Лї-Ефект має значний вплив на фізико-хімічні властивості органічних сполук (див. розділи 34, 36).
Розділ 4. ІЗОМЕРІЯ
В органічній хімії часто трапляються сполуки з однаковими хімічним складом, загальною формулою та молекулярною масою, але з різними хімічною будовою і властивостями. Такі сполуки називають ізомерами, що в перекладі з грецької означає «рівний», «однаковий». Явище існування таких сполук називають ізомерією.
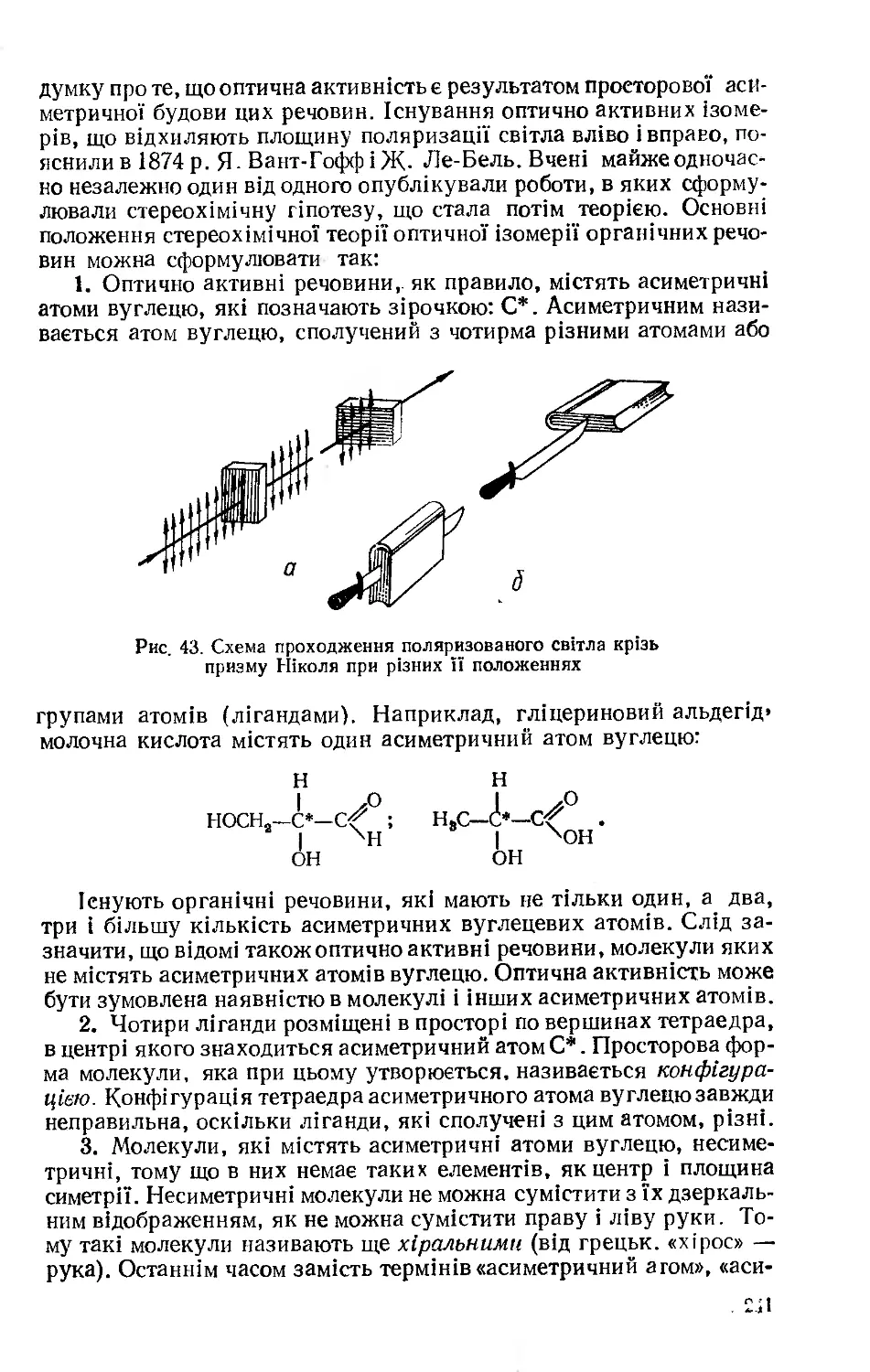
Одним з перших випадків ізомерії, який привернув до себе увагу, було відкриття ціанової НО—О=Н і гримучої НО—N=0 кислот. Ціанову кислоту відкрив у 1822 р. Ф. Велер. На рік пізніше Ю. Лібіх вивчив гримучу кислоту і, на своє велике здивування, встановив, що, незважаючй на різкі відмінності у властивостях, вона має такий самий склад, як і ціанова кислота. Через кілька років Я Берцеліус знайшов ще одну пару ізомерів — винні кислоти. Цей вчений і запропонував термін «ізомерія».
Чітке пояснення явища ізомерії дала теорія хімічної будови О. М. Бутлерова (див. розд. 1). Дальший розвиток цієї теорії дав змогу пояснити нові типи ізомерії (оптичну, геометричну), доповнити її стереохімічними уявленнями.
На сьогоднішній день усі типи ізомерії органічних сполук класифікують так: 1. Структурна ізомерія', а) ізомерія вуглецевого скелета; б) ізомерія положення; в) ізомерія взаємного положення; г) метамерія. 2. Просторова ізомерія: а) поворотна ізомерія (конформація); б) геометрична (цис-, транс-) ізомерія; в) оптична (дзеркальна) ізомерія. 3. Динамічна ізомерія (таутомерія).
26
Найпростішим типом ізомерії є структурна. Ця ізомерія зумовлює велику різноманітність органічних сполук.
1. Ізомерія вуглецевого скелета. Одним з видів структурної ізомерії є ізомерія, зумовлена різною будовою вуглецевого скелета. Хімічні сполуки, в яких вуглецеві атоми утворюють нерозгалужений ланцюг, називають сполуками нормальної будови. Сполуки з розгалуженим вуглецевим ланцюгом називають ізоспо-луками. Наприклад:
Н3С—СН2—СН2—СН3 і Н3С—СН—СН3; Н2С=СН—СН,—СН3 і Н„С=С—СН3'
Бутан | Бутилен |
сн3 сн3
Ізобутан Ізобутилен
2. Ізомерія положення. В органічних сполуках, крім ізомерії вуглецевого скелета, спостерігається ізомерія, зумовлена положенням функціональної групи, положенням певних хімічних зв’язків або положенням атомів-замісників. Наприклад:
Н2С=СН—СН,—СН3 і Н3С—СН=СН—СН3;
Бутен-і Бутен-2
Н3С—СН,—СН,—СН, і Н3С—СН,—СН—сн3.
0 4 4^40 4 0
СІ СІ
1-Хлорбутаи 2-Хлорбутан
3. Ізомерія взаємного положення. Різновидом структурної ізомерії є ізомерія взаємного положення. Вона зумовлена різним взаємним розміщенням окремих атомів або груп атомів у молекулах органічних сполук. Наприклад:
Н,С—СН,—СН—СН3 і Н3С—СН—СН—СН3
II II
СІ сі сі Сі
1,3- Дихлорбутан 2,3-Дихлорбутан
4. М е т а м е р і я (за О. М. Бутлеровим, «ізомерія нецільних структур») зумовлюється різним положенням у молекулах аліфатичних сполук таких атомів, як кисень, азот тощо, які називають гетероатомами. Наприклад:
Н3С—СН2—О—СН2—СН3 і Н3С—О—СН2—СН2—СН3.
Діетиловий ефір Метилпропіловий ефір
Інші види ізомерії будуть розглянуті далі, в конкретних класах органічних сполук.
Розділ 5. КЛАСИФІКАЦІЯ ОРГАНІЧНИХ РЕАКЦІЙ
Динаміка хімічних перетворень органічної речовини (субстрату), яка вступає в реакцію з якою-небудь іншою хімічною речовиною (реагентом) або піддається дії температури, опромінення та інших факторів, характеризується механізмом реакції. Механізм реакції це своєрідний шлях, який проходять вихідні реагенти, перетворюючись на продукти реакції, і являє собою послідовність простих
27
(елементарних) реакцій даного хімічного процесу. Елементарна реакція є одностадійною або охоплює декілька стадій. У ній не утворюється проміжний продукт (інтермедіат).
Реакції органічних речовин класифікують за напрямком перебігу та за їх механізмами.
Класифікація реакцій за напрямком. Реакції заміщення, або 8-реакції (лат. БиЬзШиііопе—заміщення),—це елементарні або багатостадійні реакції, в яких один атом або група атомів у молекулі заміщуються іншим атомом або групою атомів. Наприклад:
Н3С—СІ -р ОН" —> Н3С—ОН + СГ.
Реакції приєднання, або А-реакції (лат. Асііііопе — приєднання) такі хімічні реакції, в результаті яких з двох або трьох реагуючих молекул утворюється один продукт реакції. В результаті реакцій приєднання утворюються два нових хімічних зв’язки і зменшується кратність зв’язку в одного з реагентів. Наприклад:
Н2С=СН2 + Вг2 —> Вг—Н2С—СН2—Вг.
Реакції відщеплення, або Е-рєакції (лат. Еіітіпаге — відщеплення),— зворотні реакціям приєднання. Принципово важливими є реакції 1,2-відщеплення, або реакції р-елімінування. Наприклад-
0 а
Н3С—СН2—ОН —► Н2С=СН2 + Н2О.
Перегрупування — це реакції, у яких відбувається зміна тих частин молекули, яких реагенти безпосередньо не торкаються. Перегрупування супроводяться зміною порядку сполучення атомів у молекулі. Наприклад, ізомеризація 1-бромпропану в 2-бром-пропан:
Н3С—СН2—СН2—Вг —> Н3С—СН—СН,
Вг
або перегрупування оксиму циклогексанону в капролактам (перегрупування Бекмана):
Н2С—СН, ХСН2—сн2
/ \ Н2ЗО4 Н2С
Н2С С = Х-ОН-------
\ / н2с—сн2
Н2і
с=о.
’СН2—мн
Класифікація за механізмом реакції (за типом розриву хімічних зв’язків). Можливі два головних типи розриву ковалентних зв’язків: гемолітичний і гетеролітичний. Розглянемо їх на прикладі ковалентно побудованої органічної сполуки Р3С—X, де Р можуть бути, наприклад, вуглеводневі радикали або інші різноманітні частини органічної молекули, а X —атоми Н, СІ, Вг, І, ОН, МО2 або атоми вуглецю інших хімічних груп. Реакції розриву супроводяться в основному поглинанням теплоти і збільшенням об’єму
28
системи, а реакції утворення нових зв’язків — виділенням теплоти і зменшенням об’єму.
Гемолітичні реакції. Гемолітичними називають реакції, що супроводяться гемолітичним розривом хімічних зв’язків. При гемолітичному розриві зв’язку С—X у молекулах /?3С—X відбувається «симетричне» роз’єднання пари електронів ковалентного зв’язку так, що один електрон залишається біля атома вуглецю, а другий переходить до частинки X:
П Е
Я". С44------- Я: С + • X •
Я ’ ~~ /?
Частинки або атоми (X може бути атом), які утворилися при цьому і мають по одному неспареному електрону, називають вільними радикалами. Отже, радикал — це частина молекули, яка має неспарений електрон, але не має заряду, наприклад, радикал метил Н3С. У радикалах типу /?3С на валентній орбіталі вуглецевого атома є 7 електронів. Три з них належать групам /?, які з атомом вуглецю утворюють три ковалентних зв’язки, а чотири електрони — це «власні» вуглецеві, з яких три спарені з групами а один— неспарений. Він є р-електроном і знаходиться на зв’язуючій молекулярній орбіталі.
Алкільні радикали Н3С, Н3С—СН2та інші дуже нестійкі (корот-коживучі) і час їх існування становить тисячні частки секунди. Радикали триарилметанового ряду, наприклад трифенілметил (СеНв)3С, стабільні (довгоживучі), тому що в них відбувається спряження неспареного р-електрона з л-електронами фенільних ядер. Вільні радикали мають високу реакційну здатність, велику спорідненість до електрона і виявляють електрофільні властивості .
За напрямком перебігу гемолітичними можуть бути реакції заміщення (£я) і реакції приєднання (А^).
Гетеролітичні реакції. Гетеролітичними називають реакції, які супроводяться гетеролітичним розривом хімічних зв’язків. Гетеролітичний, або іонний, розрив о-зв’язку відбувається «несиметрично». При цьому в молекулі /?3С—X розри« о-зв’язку С—X може відбуватися з переходом пари електронів до атома вуглецю або до частинки X. Якщо пара електронів перейшла до X, то утворюється карбкатіон /?3С+, а якщо до вуглецевого атома, то утворюється карбаніон /?3С”:
/?3с(га-—А>3С++х~; /?3с :(х-^р3с~+ х+ .
Карбкатіон містить тривалентний атом вуглецю з вакантною р-орбіталлю і, подібно до вуглецю-в молекулі етилену, знаходиться в стані $р2-гібридизації.
29
Більшість алкільних карбкатіонів настільки нестійкі, що за звичайних умов їх існування не вдається виявити. Енергетична стійкість карбкатіонів знижується в ряду:
СН3
І + + +
Н3С—С+—СН3 > н3с—сн—сн3 > н3с—сн2 > Н3С.
Найстійкішими є третинні, найменш стійкими—первинні карбкатіони. Зміна стійкості карбкатіонів викликана тим, що в цих частинках має місце взаємний вплив атомів. Так, у третинного бутильного катіона три метальні групи, маючи позитивний індукційний ефект, викликають зміщення електронної густини зв’язків до третинного атома вуглецю, який несе позитивний заряд. Таким впливом ці групи зменшують величину позитивного заряду на даному вуглецевому атомі і підвищують стійкість третинного катіона, тобто стабілізують його. У вторинного і первинного карбкатіонів кількість алкільних груп біля атома вуглецю, який несе позитивний заряд, відповідно зменшується. Тому зменшується вплив цих груп на величину позитивного заряду вуглецю і знижується стійкість таких катіонів. Третинні карбкатіони у зв’язку з цим можна навіть виявити спектроскопічними методами. Досить стійкими є карбкатіони триарилметанового ряду Аг3С+ (див. розд. 38).
Карбкатіони є найсильнішнми електрофілами. Електрофіли (від лат. Еіесігоріїіііс —той, що любить електрони), або електро-фільні реагенти, — це частинки, які мають повний або частковий позитивний заряд (вакантну зв’ язуючу орбіталь або дефіцит електронів) і тому є акцепторами електронів. Молекули електрофільних реагентів у процесі реакції можуть також генерувати електрофільні частинки. До електрофільних реагентів крім карбкатіонів Д3С+ відносять катіони Н+, С1+, Вг+, І+, N0* та ін. Електрофіли вступають у реакції заміщення і приєднання (5ц- і Лц-реакції). Наприклад:
С„Н„ + N0+ —> С« Н5-МО2 + Н+;
Бензол ЕлектрофІл Нітробензол + 4-С1-
Н2С=СН2 + н+ —> Н3С—сн2-----> Н3С—СН2—СІ.
ЕлектрофІл’ Хлористий
. етил
Карбаніони — це органічні аніони, які мають парне число електронів або вільну пару електронів на тривалентному атомі вуглецю. В карбапіоні Д3С“ атому вуглецю належать п’ять валентних електронів, тобто на один електрон більше, ніж в «^-гібридизованому стані.
Карбаніони є одними з найсильніших нуклеофілів. Дуклеофми (від лат. ЬІисІеорЬіііс—той, що любить ядро), або нуклеофільні реагенти, —аніони або молекули з вільними парами електронів, які мають спорідненість до електрофілів або до позитивно зарядженого центра субстрату і намагаються з ним сполучитися. Нуклео-фільними реагентами є основи Льюїса, серед яких крім карбаніонів
ЗО
/?3С“ найчастіше трапляються аніони Н3СО-, С2НБО-, С6НБО-, НО-, СМ~, МН2, Н3ССОО-, ІВг-, СІ-, Р-та ін., нейтральні молекули з вільними парами електронів — Аііг —ЬШ2, ЬШ3, Н26:; Н3СОН, С2Н5ОН тощо.
Нуклеофіли найчастіше вступають у реакції заміщення (Зм-реакції). Наприклад:
Н3С—СН2—Вг + С2Н5О—На -> С2НБ—О—С2Н5 + Н’аВг.
Нуклєофіл Діетиловий ефір
Молекулярність і порядок реакції. У хімічній кінетиці розрізняють молекулярність і порядок реакції. Молекулярність відображує механізм елементарного акту реакції і показує число молекул, які беруть участь у найповільнішій стадії процесу, яка визначає швидкість усієї реакції. Відповідно до цього реакції називають мономолекулярними, бімолекулярними, тримолекулярними і т. д.
Мономолекулярні реакції — це реакції перетворення молекул одного виду, коли з вихідної речовини А утворюється продукт В або продукти В, С тощо без перебігу будь-яких проміжних стадій:
А -> В або А -► В + С +...
До мономолекулярних реакцій належать внутрішньомолеку-лярні перегрупування, наприклад, ізомеризація 2-бромпропану в 1-бромпропан, гетеролітична дисоціація трифенілхлорметану на трифенілметильний катіон і аніон хлору:
Н3С—СН—СН3-> Н3С—СН2—СН2—Вг;
Вг
(С„Н5)3С-С1 - (СсН5)3С Ч- СІ-.
Швидкість мономолекулярних реакцій описується кінетичним рівнянням V = /? [Л], де /г —константа швидкості реакції.
Бімолекулярні реакції — це процеси, які відбуваються при зіткненні молекул двох видів або двох реагуючих частинок А і В. Такі реакції можна зобразити схемою:
ЛТ-В^-С + Р.
В органічній хімії бімолекулярні реакції найбільш поширені. Швидкість бімолекулярної реакції виражається рівнянням:
V = ЧЛ] [В].
Отже, швидкість бімолекулярних реакцій пропорційна добутку концентрацій реагуючих речовин А і В. Бімолекулярними є, наприклад, реакції заміщення галогенів у первинних галогеналка-нах В—СН2—X (X—СІ, Вг, І) на групи ОН, СК' та ін.
Порядком хімічної реакції за даною сполукою називають ступінь, в якому концентрація цієї речовини входить у рівняння швидкості реакції. Загальний порядок реакції являє собою суму цих ступенів. Кінетичний порядок реакції за даним реагентом визначають експе-
31
риментально. Для цього встановлюють, як змінюється сумарна швидкість реакції при зміні концентрації даного реагента. Наприклад, якщо при підвищенні концентрації вдвоє швидкість реакції збільшиться у два рази, то це реакція першого порядку (2і) за даним реагентом. Якщо при збільшенні концентрації вдвоє швидкість реакції збільшується в чотири рази, то це реакція другого порядку (22) за даним реагентом. Розглянуті вище реакції ізомеризації 2-бромпропану в 1-бромпропан, гетеролітичної дисоціації трифе-нілхлорметану є реакціями першого порядку.
Порядок реакції позначають цифрою. Наприклад, ЗдЛ —реакція нуклеофільного заміщення першого порядку, 5л?2— реакція нуклеофільного заміщення Другого порядку і т. д. Дана номенклатура хімічних реакцій і відповідні скорочені позначення були запропоновані К- Інгольдом.
Розділ 6. КЛАСИФІКАЦІЯ ОРГАНІЧНИХ СПОЛУК
Сучасна класифікація органічних сполук базується на теорії хімічної будови. В основу класифікації покладено особливості будови вуглецевого ланцюга вуглеводнів, оскільки вони прості за складом. Крім того, сполук вуглецю та водню відомо найбільше і в більшості відомих органічних речовин вуглеводневі радикали складають головну частину молекули.
Залежно від будови вуглецевого ланцюга органічні сполуки поділяють на дві великі групи: нециклічні (ациклічні) і циклічні. Ациклічні сполуки називають ще сполуками жирного ряду, або аліфатичними. До них належать усі вуглеводні та їх похідні, молекули яких не містять кілець або циклів. У молекулах цих сполук є тільки так звані відкриті ланцюги вуглецевих атомів. Ациклічні сполуки поділяють на насичені вуглеводні гомологічного ряду метану і ненасичені сполуки, в молекулах яких між атомами вуглецю є кратні (подвійні або потрійні) зв’язки.
Циклічні сполуки поділяють на карбоциклічні, цикли яких складаються тільки з атомів вуглецю, і гетероциклічні, в цикли яких крім атомів вуглецю входять також інші атоми, найчастіше азоту, сірки, кисню. Карбоциклічні органічні сполуки в свою чергу поділяють на аліциклічні і ароматичні. До аліциклічних сполук належать усі карбоциклічні-вуглеводні та їх похідні, за винятком ароматичних сполук. До ароматичних сполук відносять бензол, нафталін, антрацен та інші.
Органічні сполуки залежно від складу і будови поділяють на класи. Класи органічних сполук одержують при заміні атомів водню в молекулах вуглеводнів на інші атоми або групи атомів, так звані функціональні групи. Функціональні групи і окремі атоми-замісники, введені в молекулу органічної сполуки замість атомів водню, визначають типові хімічні властивості сполук цих класів. Найважливіші класи органічних сполук та їх функціональні групи наведено в табл. 5.
32
Таблиця 5. Функціональні групи і класи органічних сполук
Функціональна група Клас сполук Загальна формула
Галогени Галогенопохідні К*НаІ (//а/ —Сі.Вг, 1, Г)
Гідроксильна —ОН Спирти к—ОН
Карбонільна —С— О Альдегіди ї а;
Кетони Я-С-7? 11 О
Карбоксильна —С—ОН II О Карбонові кислоти .О /?—с^ 4 ОН
Нітрогрупа —1\Ю2 Нітропохідні «-N0,
Аміногрупа —МНЯ Аміни /?-нн2
Сульфогрупа —8О3Н Сульфокислоти к— 8О3Н
Тіолова група —8—Н Тіоспирти (меркаптани) К—8—Н
* К — вуглеводневий радикал (залишок).
Відомо багато класів органічних сполук, і з розвитком науки кількість їх постійно збільшується. Поряд з речовинами, які мають одну функціональну групу, існують сполуки, молекули ЯКИХ МІСТЯТЬ кілька однакових або різних функціональних груп.
Вивчення органічної хімії починають, як правило, з сполук аліфатичного, або жирного, ряду і з найпростішого класу органічних речовин — вуглеводнів.
2 1-120
ЧАСТИНА І
Ациклічні сполуки аліфатичного ряду
Розділ 7. АЛКАНИ (НАСИЧЕНІ ВУГЛЕВОДНІ)
Сполуки, що складаються з двох елементів —вуглецю і водню, називають вуглеводнями. Залежно від того, як сполучені між собою атоми вуглецю в цих сполуках, існує кілька типів вуглеводнів. Найпростішими є вуглеводні з відкритим, незамкненим вуглець-вуглецевим ланцюгом. їх називають ациклічними. За характером зв’язків між атомами вуглецю ациклічні вуглеводні поділяють на насичені і ненасичені.
Г Насиченими називають такі вуглеводні, атоми вуглецю в молекулах яких сполучені між собою простими (одинарними)о-зв’язками. Всі інші одиниці валентності атомів вуглецю у цих сполуках зайняті (насичені) атомами водню. Атоми вуглецю в молекулах насичених вуглеводнів перебувають у першому валентному стані, тобто в стані $р3-гібридизації. Насичені вуглеводні називають ще алка-нами, або парафінами І Парафінами ці органічні сполуки називають тому, що довгий часта вважали малореакційноздатними (від лат. рагпш—мало і аНіпіз — має спорідненість). Стара назва насичених вуглеводнів —аліфатичні, або жирні, вуглеводні (від лат. аііїаііс — жирний). Ця назва походить від назви перших вивчених сполук, які колись відносили до цих речовин, —жирів.
\ Насичені вуглеводні утворюють ряд сполук (табл. 6) з загальною’ формулою СпН2п+2 (п — 1, 2, 3, 4, ...). Найпростішою сполукою цього ряду є метан СН4. Тому ряд цих сполук називають ще рядом метанових, вуглеводнів. Сполуки ряду метану мають подібні будову і властивості. Такий ряд сполук, представники якого мають близькі хімічні властивості і характеризуються закономірною зміною фізичних властивостей, мають однотипну структуру і відрізняються один від одного на одну або кілька —СН2-груп, називають гомологічним рядом (від грецьк. «гомос» — послідовний, подібність)7Як видно з даних табл. 6, кожний наступний вуглеводень даного'ряду відрізняється від попереднього на групу —СН2. Ця група називається гомологічною різницею, а окремі члени цього ряду — гомологами.
Назви перших чотирьох насичених вуглеводнів (метан, етан, пропан, бутан) виникли випадково. Наприклад, корінь слова «етан» пішов від латинського слова аеійег —ефір, тому що залишок етану —С2Н5 входить до складу медичного ефіру. Починаючи з С5Н12, назви алканів утворені від грецьких або латинських числів-
34
Таблиця 6. Фізичні властивості деяких насичених вуглеводнів нормальної будови
Назва Формула Тпл. °С Т °С 1 кип» 90 Густина
Метан СН, — 182,5 —161,5 0,4150 (при —164 °С)
Етан н3с—сн3 — 173,3 —88,6 0,5610 (при —100 °С)
. Пропан Н3С—сн2—сн3 —187,7 —42,7 0,5853 (при -44,5 °С)
Бутан Н3С—СН2—сн2—сн3 — 138,3 —0,5 0,6000 (при 0 °С)
Пентан Н3С—(СН2)3—СН3 — 129,7 +36,7 0,6260
Гексан Н3С—(СН2)4—СН., —95,3 68,7 0,660
Гептан Н3С—(СН2)3—СН3 —90,6 98,4 0,6838
И Октан Н3С-(СН2)6-СН3 —56,8 125,7 0,7025
іб Нонан і І3С-—(СН2)7—СН3 —53,5 150,8 0,7176
^о^Декан Н3С (СН2)8 СН3 —29,6 174,1 0,7301
а * ІІентадекан Н3С—(СН2)13—СН3 -|-10 270,5 0,7689
кіЕнкозан Н3С—(СН2)18—СН3 36,8 342,7 0,7780 (прн 37 °С)
Пентакозан Н3С—(СІ І,).3—сн3 53,7 400 0,8012
Т риаконтан Н3С (СН2)2Я СН3 65,8 446,4 0,8097
Гектан НдС—(СН2)98—СН3 115,2 — —
ників, які вказують кількість вуглецевих атомів у молекулі даного насиченого вуглеводню, з додаванням до цих назв суфіксу -ан. Так, вуглеводень С5Н12 має назву пентан (від грецьк. «пента» — п’ять), С6Н14 — гексан (від грецьк. «гекса» —шість), С7НІС —гептан (від грецьк. «гепта» — сім) і т. д.
Наведені в таблиці алкани мають нерозгалужені вуглець-вугле-цеві ланцюги. Такі вуглеводні називають нормальними. Існують вуглеводні і з розгалуженим вуглецевим ланцюгом. їх називають ізосполуками. Наприклад, ізобутан має таку будову:
н3с—сн—сн3.
сн3
Атоми вуглецю в молекулах органічних сполук поділяють на первинні, вторинні, третинні і четвертинні. Атом вуглецю, який безпосередньо сполучений тільки з одним сусіднім вуглецевим атомом, називають первинним-, атом вуглецю, сполучений з двома сусідніми атомами вуглецю, — вториннйм. Якщо атом вуглецю безпосередньо сполучений з трьома або чотирма С-атомами, то такі вуглецеві атоми називають відповідно третинними або четвертинними:
2*
35
Первинні атоми вуглецю в даній сполуці обведені колом, вторинний — квадратом, третинний— трикутником, четвертинний — колом, зображеним штриховою лінією.
Якщо від молекули насиченого вуглеводню відняти один атом водню, то утвориться частина молекули, яка називається одновалентним радикалом. Одновалентні радикали мають один неспарений електрон (див. розд. 5). Назва радикала утворюється з назви відповідного насиченого вуглеводню шляхом заміни в його назві суфікса -ан на -ил(-іл): Н3С—метил, Н3С—СН2—етил, Н3С—СН2—СН— первинний пропіл (неспарений електрон біля первинного атома вуглецю), Н3С—СН—СН3 — вторинний пропіл (неспарений електрон біля вторинного атома вуглецю), який називають ізопропілом, і т. д. Загальна формула одновалентних радикалів насичених вуглеводнів СпН2п+1—. Ці радикали називають алкілами*.
Якщо від молекули метану відняти два атоми водню, то утвориться двовалентний радикал —СН2—, який називають метиленом. Тривалентний радикал —СН— називають метином.
І
Номенклатура. Для назви органічних речовин комісією Міжнародної спілки теоретичної і прикладної хімії (ШРАС1 2) були розроблені правила систематичної (наукової) номенклатури. Згідно з цими правилами, назву вуглеводню дають таким чином.
1. У молекулі вуглеводню вибирають основний—найдовший і найскладніший (який має найбільше число відгалужень)— вуглецевий ланцюг:
і 2 з
Н3С—СН--СН—СН2—СН3 | |4 5 6
сн3 сн2—сн2—сн3.
2. Нумерують атоми вуглецю основного ланцюга. Нумерацію здійснюють послідовно з того кінця ланцюга, який дає радикалу найменший номер. Якщо існує кілька алкільних радикалів, то порівнюють величину цифр двох можливих послідовних нумерацій. Та нумерація, в якій першою зустрічається менша цифра, ніж в другій послідовній нумерації, вважається «меншою» і використовується для складання назви вуглеводню:
6 5 4 3 2 1
н3с—сн2—сн—сн2—сн—сн3.
сн3 сн3
2,4-Диметилгексан
Нумерація справа наліво буде «меншою», ніж нумерація зліва направо.
3. Називають вуглеводневі радикали, які утворюють бічні ланцюги. Перед назвою кожного радикала ставлять цифру, яка вказує номер вуглецевого атома головного ланцюга, біля якого зна
1 Вуглеводневі радикали раніше розглядали як залишки алкоголів (спиртів). Звідси і пішла назва — алкіли.
2 Від перших букв англійської назви спілки ШРАС — Іпіегпаііопаї Нпіоп оі Риге а псі Аррііей Сйеіпіїігу.
36
ходиться даний радикал. Цифру від назви відділяють дефісом. Назви алкільних радикалів перелічують в алфавітному порядку.Якщо вуглеводень має в своєму складі кілька однакових радикалів, то записують в порядку зростання номери вуглецевих атомів, біля яких стоять ці радикали. Цифри відділяють одну від одної комами. Після цифр записують префікси: ди- (якщо однакових радикалів два), три- (коли однакових радикалів три), тетра-, пента- і т. д. (якщо однакових радикалів відповідно чотири, п’ ять і т. д.). Префікси вказують, скільки однакових радикалів має даний вуглеводень. Після префікса ставлять назву радикала. У тому випадку, якщо два однакових радикали знаходяться біля одного вуглецевого атома, номер цього атома вуглецю ставиться у назві двічі.
4. Називають вуглеводень основного пронумерованого вуглецевого ланцюга, пам’ятаючи при цьому, що назви всіх насичених вуглеводнів мають суфікс -ан.
Наведений нижче приклад допоможе уяснити ці правила: сн3
І 2 3| 4 5 6 7
Н3С—СН—С—сн—сн,—сн—сн3.
III І
сн3 сн3 сн2 сн3
сн3
4 -Етил -2, З, 3, 6-тетрам етил гептан
Інколи алкільні радикали бічних ланцюгів розгалужені. В цьому випадку їх називають так, як відповідні насичені вуглеводні, тільки замість суфікса -ан вживають суфікс -ил.
Вуглецевий ланцюг розгалуженого радикала нумерують. Атом вуглецю цього радикала, сполучений з основним ланцюгом, одержує номер 1. Для зручності вуглецевий ланцюг розгалуженого радикала нумерують цифрами з штрихами і повну назву такого радикала беруть у дужки:
12 34 56 7 « 9 10
Н3С—СН—СН—СН„—СН—СН,—СН—СН2—СН—СН,.
II І І І
СН3 СН3 СН—сн3 сн2 сн3
СН— сн3 сн3
сн3
2'-Диметилпропіл)-7-етнл-2, 3, 9-триметилдекаи
Для кількох розгалужених радикалів зберігаються такі назви: Н3С—СН— ; Н3С—СН—СН2— ;
СН3 СН3
Ізопропіл Ізобутил
сн3
Н3С—СН,—СН—; Н,С—С—£/ ''
І І
сн3 сн3
втор-Бутил треш- Бутил
з;
Н3С—СН—СН2— СН2— ;
СН3 Ізопентил
сн3
Н..С— С—СН,—;
І
сн3
Неопентил
сн8 І
Н3С—СН2—С—;
СН3 трет-Пенти л
н3с—сн—сн2—сн2—сн2—.
сн3 Ізогексил
Крім систематичної для назви насичених вуглеводнів використовують ще раціональну номенклатуру. За цією номенклатурою насичені вуглеводні розглядають як похідні метану, в молекулі якого один або кілька атомів водню заміщені на радикали. Назву насиченого вуглеводню за раціональною номенклатурою утворюють таким чином: називають за ступенем складності всі радикали, які знаходяться біля атома вуглецю з найбільшою кількістю замісників (зазначаючи їх кількість, якщо вони однакові), а потім додають основу назви вуглеводню за цією номенклатурою — слово «метан». Наприклад:
СН3 СН3 СН3
І І І
Н3С—С—СН3; Н3С—С—СН2—СН2—СН3; Н3С—СН—СН—СН2—СН3.
І І І
СН3 СН3 СН3
Тетр а метил метан Триметилпропілметан Метил етил їзопропілметан
Раціональною номенклатурою користуються для назви порівняно простих вуглеводнів. Ця номенклатура не така вдосконалена і набагато менш зручна в користуванні порівняно з систематичною номенклатурою. За раціональною номенклатурою одна і та сама речовина може мати різні назви, що надзвичайно незручно. Крім того, за даною номенклатурою можна назвати далеко не всі насичені вуглеводні.
Ізомерія. Для насичених вуглеводнів характерна структурна ізомерія, пов’язана з розгалуженням їх вуглецевого ланцюга. Структурні ізомери відрізняються між собою тільки порядком розміщення атомів вуглецю в молекулі.
Структурна ізомерія насичених вуглеводнів починається з бутану. Бутан існує у вигляді двох, пентан —у вигляді трьох ізомерів;
Н3С—СН2—СН2—СН3; Н3С—СН—СН3;
-О
(Т'КИП = -Н,7°С)
сн3
І
Н3С—СН2—СН2—СН2—СН3; Н3С—СН—СН2—СН3; Н3С—С—СН3.
(Т — 36 °С) І І
1 ки" 1 сн3 сн3
(^=27,9 °С) (7КИП = 9,5 °С)
Гексан має п’ять структурних ізомерів, гептан — дев’ять, октан —18, нонан — 35, декан —75 і т. д. Чим більше в молекулі, насиченого вуглеводню атомів вуглецю, тим більше ізомерів він має. Для вуглеводню С13Н28 можливі 802 ізомери, для С14Н30 — уже 1858 ізомерів, дляС^Нза — 4347, для С20Н42 можливі 366 319 ізомерів і т. д.
Для насичених вуглеводнів крім структурної властива ще і поворотна ізомерія. Простий о-зв’язок не створює перешкод Для обертання атомних угруповань, які оточують його. Тому, наприклад, в молекулі етану внутрішнє обертання однієї групи —СН3 відносно другої не повинно викликати деформації о-зв’язку С—С і може бути вільним. Досліди показують, що при кімнатній температурі цей обертовий рух гальмується. Причиною гальмування є взаємодія не сполучених між собою атомів водню у двох СН3-групах, що приводить до відштовхування між цими атомами. Такий
Рис. 12. Поворотні ізомери етану: а — загальмований; б — заслонений
загальмований обертовий рух в молекулах етану та інших вуглеводнів і їх заміщених приводить до появи поворотних ізомерів.
При обертанні однієї метильної групи-в молекулі етану відносно другої навколо вуглець-вуглецевого зв’язку атоми цих груп можуть займати в просторі багато різних взаємних положень. Це легко простежити на моделі молекули етану. Такі структури молекули речовини з різним взаємним розміщенням атомів у просторі, які виникають при обертанні навколо простого вуглець-вуглецевого зв’язку, відносно якого визначається положення атомів у просторі, називають конформаціями (від лат. сопїогтіс — подібний), або поворотними ізомерами (коиформерами). З великої кількості конфор-мацій етану можна виділити дві крайні. Якщо.обертати в просторі одну СН3-групу відносно другої і дивитись на модель етану вздовж С—С-зв’язку,то можна помітити, що при одній з крайніх конформа-цій атоми водню однієї СН3-групи будуть «заслоняти», «затіняти» атоми водню другої СН3-групи. Таку конформацію називають заслоненою, або затіненою. При другій крайній конформації атоми водню однієї СН3-групи розміщені в проміжках між атомами водню другої СН3-групи. Таку конформацію називають загальмованою (рис. 12).
Для більш наочного зображення конформацій часто користуються іншою проекцією, де в центрі, на перетині трьох прямих, знаходиться атом вуглецю ближчої до нас метильної групи. Зв’язки
39
\мі
Рис. 13. Графік залежності потенціальної енергії Е від кута обертання <р в молекулі етану
0° 60е /20° 180° 240°500°560° у
Н СІ
І І н-с-с-н
І І
СІ н
Рис. 14. Конформації 1,2-ди-хлоретану:
а — транс-коиформація; б, в — гош-конформації
С—Н Другої метильної групи розміщені за площиною кола (на рис. 12 показані штриховими лініями). Такий спосіб зображення конформацій одержав назву формил Ньюмена.
Конформери, зображені на рис. 12, відрізняються один від одного взаємним поворотом в просторі С—Н-зв’язків однієї метильної групи і С—-Н-зв’язків другої метильної групи відносно зв’язку С—С. Це взаємне їх розміщення в просторі характеризують кутом повороту ф.
Від звичайних ізомерів конформери відрізняються тим, що їх, як правило, не можна виділити в індивідуальному стані. Атоми в молекулі знаходяться в безперервному русі і одна конформація легко переходить в іншу. Конформації, які виникають при вільному обертанні навколо о-зв’язків, мають різну стійкість. Як правило, молекула намагається набути такої конформації, при якій її потенціальна енергія була б мінімальною. Більш енергетично вигідною, а тому і більш стійкою є загальмована конформація, оскільки в ній атоми водню однієї метильної групи знаходяться на найбільшій віддалі від атомів водню другої метильної групи, і тому взаємне відштовхування між ними буде мінімальним. Запас внутрішньої енергії такої молекули зменшується. У зв’язку з цим при таких положеннях водневих атомів внутрішнє обертання навколо С—С-зв’язку якоюсь мірою гальмується. Звідси і назва конформації — загальмована. Для переходу молекули із загальмованої конформації в затінену необхідно затратити 12,6 кДж/моль енергії (рис. 13). Та
40
•СІ О Н
кий незначний енергетичний бар’єр, що знаходиться на рівні енергії теплового руху молекули, не дозволяє виділити окремі поворотні ізомери, але існування їх може бути зафіксоване деякими фізичним і методами, наприклад, електронографічним, спектроскопічним та іншими.
При збільшенні кількості вуглецевих атомів у ланцюгу зростає кількість можливих конформацій. Так, для бутану їх уже шість. Дизаміщені етану, наприклад, 1,2-дихлоретан СІ—СН2—СН2—СІ, мають конформацій більше, ніж етан: шрснс-конформацію, дві скошені конформації і затінені конформації (рис.14).Утранс-конфор-мації атоми хлору розміщені в просторі по різні боки від С—С-зв’яз-ку і найдалі один від одного (лат. ігапз — по той бік). Тому транс-конформація для дихлоретану є енергетично найвигіднішою і найбільш стійкою. В скошених кон-формаціях атоми хлору розміщені в просторі ближче, ніж у транс-конформаціях. Скошені конформації, в яких двогранний кут між окремими замісниками дорівнює 60°, називають гош-конформаціями (від старого французького слова, що означає «скручування»), гош-Конформації дихлоретану менш стійкі, ніж його транс-конформа-ції. Енергія молекули дихлоретану в еош-конформації більша від енергії в транс-конформації на 5,9кДж/моль. Максимальну енергію його молекули мають у заті
нених конформаціях (рис. 15). Період існування молекули дихлоретану в кожній з цих конформацій становить близько 10 -1° с.
Конформери дихлоретану відрізняються дипольними моментами. Так, шранс-конформер неполярний, а аош-конформери — полярні. Внаслідок цього 1,2-дихлоретан має середній дипольний момент (1,27 П при 25 °С) .При підвищенні температури кількість менш стійкої гош-конформації збільшується і дипольний момент дихлоретану підвищується. При 315 °С він уже становить 1,57 П.
Методи добування. Для добування насичених вуглеводнів використовують як синтетичні методи, так і природні джерела (газ, нафту, вугілля, віск, деревину, торф тощо). З синтетичних методів добування алканів найбільше значення мають такі:
1. Відновлення галогенопохідних насичених вуглеводнів воднем при наявності каталізатора, атомами водню в момент виділення або йодоводнем:
Н2 2Н
неї + Н3С—СН3 ч— Н3С—СН2—СІ----------> Н3С—СН3 + неї;
Ргї Хлористий етил (НСІ + 2п)
Н3С—СН,—І + НІ -> Н3С—СН3 + І2.
а б б г В б а
Рис. 15. Графік залежності потенціальної енергії молекули дихлоретану від будови поворотних ізомерів
41
З метою добування насичених вуглеводнів з їх галогенопохідних можна спочатку добути магнійорганічні сполуки, а потім діяти на них речовиною з активним атомом водню (див. розд. 9).
2. Гідрування ненасичених (етиленових або ацетиленових) вуглеводнів (див. розділи 14, 15).
За допомогою названих методів добувають насичені вуглеводні з такою самою кількістю вуглецевих атомів у молекулі і з такою самою будовою вуглецевого ланцюга, як у вихідній речовині.
3. Взаємодія галогенопохідних насичених вуглеводнів з металічним натрієм (реакція Вюрца):
Н3С—СН2—І + 2К’н + І—СН2—СН3 -> Н3С—СН2—СН2—СН3 + 2№І.
Бутан
Внаслідок цієї реакції з йодистого етилу, наприклад, можна синтезувати бутан.
П.П. Шоригін довів, що при дії на галогенопохідні насичених вуглеводнів металічного натрію як проміжні продукти утворюються натрійорганічні сполуки, які потім взаємодіють з галогеналкі-
лами:
Н..С—СН2—І + 2Ма Н,С—СН,—На -4- ИаІ;
Етилнатр ій
б-
с
Н3С—СН,— І + На'—СН,—СН, -> Н3С—СН2—СН2—СН3 + №1.
Якщо в реакцію з металічним натрієм вводити галогенопохідні різних вуглеводнів, то в результаті реакції утворюється суміш насичених вуглеводнів:
2Ка
Н3С—І + Н3С-СН2-І Н3С-СН3+Н8С-СН2-СН3+Н3С-СН2СН2-СН3
Етан Пропан ‘ Бутан
За допомогою реакції Вюрца із сполук з меншою кількістю вуглецевих атомів утворюються насичені вуглеводні з більшою кількістю атомів вуглецю.
4. Електроліз водних розчинів натрієвих або калієвих солей карбонових кислот (реакція Кольбе). Аніони карбонових кислот при електролізі переміщуються в розчині до анода і віддають йому електрон, перетворюючись при цьому на відповідний радикал з не-спареним електроном на атомі кисню. Такі радикали карбонових кислот нестійкі і відразу ж розкладаються на вуглекислий газ і вуглеводневий радикал. Вуглеводневі радикали взаємодіють між собою і утворюють насичений вуглеводень:
х° Г •' 'У
2Н3С-С* — 2е-*-2 Н3С^с'
чо~ [ 1 о-_
Ацетатний аніон Ацетатний радикал
2СО2 + 2Н3С- -* Н3С-СН3.
Метильний радикал Етан
На катоді при цьому утворюються водень і гідроксид відповідного лужного металу.
42
5. Сплавляння солей карбонових кислот з лугами (синтез Дю-ма). При сплавлянні солей карбонових кислот з їдкими лугами або з натронним вапном відбувається їх декарбоксилування і одночасно утворюються насичені вуглеводні, в яких на один атом вуглецю менше, ніжу вихідній солі. З ацетату натрію в цих умовах добувають метан:
-О І
+ №О|Н
О—№ І
-► СН4 N а2СО3.
У лабораторних умовах метан добувають при нагріванні ацетату натрію з натронним вапном (суміш №ОН 4- Са(ОН)2). Зручним методом добування метану в лабораторії є дія води на карбід алюмінію:
А14С3+ 12Н/)->ЗСН4 + 4А1(ОН)3.
У промислових масштабах насичені вуглеводні з кількістю вуглецевих атомів до Си включно можна виділити фракційною перегонкою природного газу, бензинової фракції нафти або з синтетичного бензину (синтину), який добувають з оксиду вуглецю (II) і водню при наявності залізокобальтового або кобальт-торій-марганцевого каталізатора:
-300 °С
пСО-|- (2«+ 1) Н2 * СпН2П+2 -|- пН2О.
Проте індивідуальні насичені вуглеводні виділити цим методом досить важко.
Поширення насичених вуглеводнів у природі. Насичені вуглеводні широко поширені в природі. Вони містяться в природному газі, нафті, вугіллі. Природний газ в основному складається з метану. Нафта являє собою суміш гомологів метану з числом вуглецевих атомів у молекулі відС^ ДоС30—С40. Метан міститься в газах кам’яновугільних шахт (рудниковий газ), входить до складу болотного газу. В болотах і болотистих місцевостях, де частина рослин покрита водою, целюлоза (СвН10О5)п, з якої восновному складаються рослини, піддається анаеробному бродінню, що відбувається під дією мікроорганізмів без доступу кисню повітря. В результаті такого бродіння утворюється болотний газ, який піднімається на поверхню боліт. Він складається переважно з метану:
(СвН10О8)п 4- пН2О —> ЗпСН4 4" ЗпСО2.
Вищі насичені вуглеводні з кількістю вуглецевих атомів від 27 до 37 входять до складу так званого озокериту.
н-НонакозанС29Н60 був виділений з листків капусти (1,5 г цього вуглеводню з 220 кг капусти). В шкірці яблук містяться насичені вуглеводні з кількістю атомів вуглецю С27 і С29. Бджолиний віск містить гептаконтан С2ТНБв і гентриаконтан С31НМ.
43
Фізичні властивості, фізичні властивості насичених вуглеводнів, як і інших органічних речовин, визначаються їхніми складом і будовою. Як видно з даних табл. 6, насичені вуглеводні з числом атомів вуглецю від 1 до 4 за звичайних умов — газоподібні речовини, починаючи з С-, і до С16 — рідини, а з числом вуглецевих атомів 16 і вище —-тверді речовини. Температури кипіння і плавлення послідовно підвищуються із збільшенням молекулярної маси вуглеводню. Тут чітко виявляється закон діалектики про перехід кількісних змін в якісні: збільшення молекули насиченого вуглеводню на групу —СН2—-приводить кожний раз до появи нової сполуки?^
; Густина насичених вуглеводнів менша від густини води і зростає із збільшенням молекулярної маси алкану. Газоподібні і тверді вуглеводні не мають запаху. Рідкі вуглеводні мають характерний запах бензину і гасу. '
Рис. 16. Будова молекули етану:
а. — розміщення а-зв’язків у молекулі; б— тетраедрична модель молекули; в — куле-стержнева модель молекули; г — масштабна модель молекули за Стюартом — Бріглебом
Насичені вуглеводні — сполуки неполярні і важко поляризуються. Вони практично не розчиняються у воді, але добре розчинні в більшості неполярних органічних розчинників. Рідкі насичені вуглеводні —добрі розчинники для багатьох органічних речовин.
В ІЧ-спектрах алканів валентні коливання зв’язку С—Н лежать в області 3000—2850 см-1, а їх деформаційні коливання — в області 1470—1380 см-1 (С—Н в метальній і метиленових групах).
Насичені вуглеводні поглинають УФ-промені в області з довжиною хвиль, меншою за 200 нм, і тому їх використовують як розчинники при спектроскопічних вимірюваннях.
ЯМР-спектри насичених вуглеводнів розшифровуються важко, оскільки хімічні зсуви по-різному розміщених протонів мають близькі значення (біля 0,5—2 млн-1).
£_ Хімічні властивості. Хімічні властивості насичених вуглеводнів зумовлені наявністю в їхніх молекулах атомів вуглецю, водню і зв’язків С—Н та С—С.
У молекулі найпростішого алкану—метану хімічні зв’язки утворюють 8 валентних електронів (4 електрони атома вуглецю
44
і 4 — атомів водню), які розміщені на чотирьох зв’язуючих молекулярних орбіталях.
Отже, в молекулі метану з чотирьох хр®-гібридизованих орбіталей атома вуглецю і х-орбіталей чотирьох атомів водню утворюються чотири яр®—я (С—Н) ковалентних зв’язки (див. рис. 9).
У молекулі етану, утвореній з двох вуглецевих тетраедрів, є один яр3—яр®(С—С) ковалентний зв’язок і шість яр®—я(С—Н)кова-лентних зв’язків (рис. 16). У розглянутих типах ковалентних зв’язків області найбільшої електронної густини знаходяться на лінії, яка з’єднує ядра атомів. Ці ковалентні зв’язки утворені локалізованими о-МО і називаються а-зв' язками. Важливою особливістю цих зв’язків є те, що електронна густина в них розподілена симетрично відносно осі, яка проходить через ядра атомів (циліндрична симетрія електронної густини). Завдяки цьому атоми або групи атомів, які сполучені цим зв’язком, можуть вільно обертатися
навколо осі зв’язку, не порушуючи перекривання орбіталей і не викликаючи деформації зв’язку. Кут між напрямками валентностей атомів вуглецю в молекулах алканів становить 109°28'. Тому в молекулах цих речовин навіть з прямим вуглецевим ланцюгом атоми вуглецю в дійсності розміщуються не по прямій. Цей ланцюг має зигзагоподібну форму, яка пов’язана із збереженням міжвалентних кутів атомів вуглецю (рис. 17). У молекулах алканів з досить довгим вуглецевим ланшого.м цей кут збільшений на 2° внаслідок відштовхування валентно не сполучених між собою атомів вуглецю.
Кожний хімічний зв’язок характеризується певною енергією. Експериментально встановлено, що енергія зв’язку С—Н у молекулі метану становить 422,9 кДж/моль, етану —401,9 кДж/моль, інших алканів —близько 419 кДж/моль. Енергія зв’язку С—С дорівнює 350 кДж/моль. Висока енергія зв’язків С—С і С—Н зумовлює низьку реакційну здатність насичених вуглеводнів при кімнатній температурі. Так, алкани не знебарвлюють бромної води, розчину перманганату калію, не взаємодіють з іонними реагентами (кислотами, лугами), не реагують з окислювачами, з активними металами. Тому, наприклад, металічний натрій можна зберігати в гасі, який є сумішшю насичених вуглеводнів. Навіть концентрована Н28О4, яка обвуглює багато органічних речовин, при кімнатній температурі не діє на алкани. Враховуючи порівняно малу реакційну здатність насичених вуглеводнів, їх у свій час назвали парафінами. Алкани не мають здатності приєднувати водень, галогени
45
та інші реагенти. Тому цей клас органічних речовин назвали насиченими вуглеводнями.
Хімічні реакції насичених вуглеводнів можуть відбуватися за рахунок розриву зв’язків С—С або С—Н. Розрив С—Н-зв’язків супроводиться відщепленням атомів водню з утвореннямненасиче-них сполук або подальшим заміщенням відщеплених атомів водню іншими атомами чи групами атомів.
Залежно від будови алкану і умов реакції в молекулах насичених вуглеводнів зв’язок С—Н може розриватися гемолітично:
н3сЦ-, Н->Н3С- Ч- • Н
І Метальний радикал
і гетеролітично:
Н3С:'|Н -> Н3С;- + Н+; Н3С| :Н -»Н3С+ + :Н’.
* Метальний Метальний
аніон катіон
При цьому можуть утворюватися вільні радикали, що мають неспарений електрон, але не мають електричного заряду, або карбкатіо-ни чи карбаніони, які мають відповідні електричні заряди. Вільні радикали утворюються як проміжні частинки в реакціях радикального механізму, а карбкатіони і карбаніони — в реакціях іонного механізму.
Внаслідок того, що зв’язки С—С неполярні, С—Н-зв’язки — малополярні і ці о-зв’язки мають низьку поляризованість, гетеролі-Тичний розрив о-зв’язків у молекулах алканів з утворенням іонів вимагає великої затрати енергії. Гемолітичне розщеплення цих зв’язків потребує менше енергії.Тому для насичених вуглеводнів більш характерними є реакції, які відбуваються за радикальним механізмом. Розщеплення о-зв’язкуС—С вимагає меншої затрати енергії, ніж розщеплення зв’язку С—Н, оскільки енергія С—С-зв’язку менша від енергії С—Н-зв’язку. Проте хімічні реакції частіше відбуваються з розщепленнямС—Н-зв’язків, оскільки вони більш доступні для реагентів.
к ції з а м і ш е н н я.\Взаємодія алканів з галогенами. Заміщення водневих атомів атомами галогенів —одна з найхарактерніших реакцій насичених вуглеводнів. Швидкість галогенування алканів різко зменшується в ряду: Е >С1 > Вг > І. Реакція з вільним фтором супроводиться вибухом і руйнуванням молекул вуглеводню, а продуктами реакції е вуглець і фтороводень. Отже, фтор викликає розрив (крекінг) вуглецевого ланцюга, оскільки реакція фторування екзотермічна і в процесі її виділяється більше теплоти (435,4 кДж/моль), ніж потрібно для розриву зв’язку С—С (350 кДж/моль):
Нзс—СН3 + ЗРг -> 2С + 6НР.
46
Розбавляння фтору азотом дає можливість добувати поліфтор-похідні алканів з досить високим виходом:
N.
СН4 + 4Р2 —> СР4 + 4НР.
Тетра-фтор-метан
Насичені вуглеводні активно вступають у реакцію з хлором. Хлорують алкани атомами або катіонами хлору, які більш реакцій-ноздатні, ніж молекулярний хлор. Дисоціація молекули хлору на атоми вимагає затрати 242,8 кДж/моль енергії. Така дисоціація хлору легко відбувається при звичайній температурі під дією УФ-світла, поглинання якого молекулою надає їй 293,0 кДж/моль енергії. Для термічної дисоціації молекули хлору на атоми необхідна температура близько 300 °С. Дисоціація молекули хлору на іони вимагає затрати 1130,2 кДж/моль. З наведених енергетичних даних видно, що насичені вуглеводні найлегше хлорувати на світлі.
Хлорування алканів відбувається з виділенням 108,8 кДж/моль теплоти і є менш екзотермічним процесом, ніж фторування. Фотохімічне хлорування насичених вуглеводнів проводять при розсіяному світлі, оскільки при прямому освітленні реакція відбувається з вибухом. При хлоруванні атоми водню алканів поступово заміщуються на хлор. В результаті утворюються хлоропохідні насичених вуглеводнів:
сі2 сі2 сі,
СН4 + СІ, ——> Н,С—СІ ——> СН,С1, > СНС13 ——> СС14.
4 г 2 —ПСІ 3 . —неї „ 2 2 —неї „ 3 —неї ,, 4
Хлористий Хлористий Хлоро- Чотири-
метил метилен форм хлори-
стий
вуглець
Хлорування насичених вуглеводнів на світлі, як встановлено роботами М. М. Семенова, відбувається за ланцюговим радикальним механізмом (5/?):
сі ч-.Сі і
Світло
(А ^478нм)
2С1- ;
Сі' + Нп-!СН3----------►Н : СІ + -СН3 ;
/Нг-^СН, ЛсН^СІ
н3с- +С1р-’СІ - Н3С : СІ + сі----і- н: Сі + Н3с- » І т.д
Під дією світла молекула хлору гемолітично розпадається на атоми, які далі реагують з молекулами алканів, наприклад з метаном, і викликають гемолітичний розрив зв’язку С—Н. В результаті утворюється молекула хлороводню і метильний радикал, який вступає в реакцію з молекулою хлору, утворюючи хлористий метил і атом хлору. Атом хлору, який утворився, реагує потім з іншою молекулою метану і т. д.
В процесі хімічної реакції відбувається зміна взаємного розміщення частинок у системі. Щоб відбулася взаємодія, дві частинки
47
Координата реакції
Рис. 18. Зміна потенціальної енергії системи А->-В->-С вздовж координати реакції:
1 — вихідний етап А; 2, 4 — перехідні стани; 3 — проміжний стан В; ' 5 — продукт реакції С
___________а-+сн3сі
Координата реакції
Рис. 19. Зміна потенціальної енергії реакції хлорування метану
повинні спочатку зіткнутись, для чого їм потрібен певний мінімум кінетичної енергії. Мінімальна кількість енергії, необхідна для перебігу реакції, називається енергією активації (Еакт).
Хімічні реакції часто проходять у кілька стадій, які можна розглядати як самостійні реакції зі своїми вихідними і кінцевими продуктами. Послідовність проміжних станів, через які проходить система в процесі елементарного акту, називають шляхом реакції (координатою реакції).
При зміні положення частинок в системі змінюється її потенціальна енергія. Крива зміни цієї енергії залежно від координати реакції зображена на рис. 18, мінімуми на якій відповідають стійким розміщенням частинок (вихідні і кінцеві продукти реакції). Для переходу від одного мінімуму, який відповідає вихідним речовинам, до другого, який відповідає кінцевим продуктам реакції, необхідно перейти через перехідний стан (енергетичний максимум), або активований комплекс, енергія якого вища від енергії вихідних і кінцевих речовин. Хлорування метану, як видно із схеми зміни потенціальних кривих (рис. 19), проходить через стадії утворення таких активованих комплексів.
Хлорування насичених вуглеводнів при наявності каталізаторів, таких як А1С13, відбувається за ланцюговим іонним електрофіль-ним механізмом (5е):
•СІ^СЇ: + А1СІ3 — [А1.С14]С1+:
_ ‘_2 2. _" ’_
1 :сі++н:!сн3 — н: Сі + сн3; А і_"-----------.і \
і----і-------сн3
і н3с+ + :сі: і Сі: — н3с: с і + \сг^-----
Молекула хлору при дії каталізатора розпадається гетеролі-тично з утворенням комплексної іонної пари [А1С14]"С1+, оскільки алюміній сполучений з електронегативними атомами хлору і має
48
потребу в електронах. У зв’язку з цим він відщеплює від молекули С12 атом хлору з парою електронів. При цьому одночасно утворюється катіон хлору, який потім взаємодіє з молекулою метану і гете-ролітично розриває зв’язок С—Н. Така взаємодія приводить до утворення метального карбкатіона. Останній далі вступає в реакцію з молекулою хлору і утворює хлористий метил та катіон хлору, який реагує з іншою молекулою метану. Такі іонні реакції, в яких проміжними частинками є позитивно заряджені іони, називають електрофільними.
Хлорування насичених вуглеводнів при наявності каталізаторів проводять при нагріванні реакційної суміші, оскільки дисоціація молекули хлору на іони вимагає значної затрати енергії.
Хлорування етану відбувається аналогічно хлоруванню метану. На першій стадії реакції утворюється хлористий етил, який може хлоруватися далі:
Світло, 25 °С
Н3С—СН3 4- С1„---------> Н,С—СН,—СІ + неї.
При хлоруванні гомологів метану, в молекулах яких крім первинних є вторинні, третинні вуглецеві атоми, з одного насиченого вуглеводню може утворюватись кілька ізомерних продуктів залежно від того, який атом водню заміститься. Так, пропан, н-бутан та ізобутан при хлоруванні на світлі на першій стадії реакції утворюють суміш двох ізомерних хлоропохідних:
Н3С—СН2—СН3 + С12 -> Н3С—СН2—СН2—СІ + Н3С—СН—СН8 + неї;
І -Хлорпропан (45 %) І
СІ
2-Хлорпропан (55 %)
Н3С—СН2—СН2—СН3 + С12 ->
-> Н3С—СН2—СН2—СН2 + Н3С—СН2—СН—СН3 + неї;
СІ СІ
1-Хлорбутан (28 %) 2-Хлорбутан (72%)
сн3 сн3 сн3
Н3С—СН—СН3 + С12 -> Н3С—СН—СН2—СІ + Н3С—С—СН3 4- неї.
2-Метил-1-хлорпропан |
(64 %) СІ
2-Метил-2-
хлорпропан (36 %)
Як видно з наведених схем реакцій, хлорування гомологів метану відбувається не вибірково (не селективно) відносно тих атомів, які знаходяться біля первинного, вторинного і третинного атомів вуглецю.
Алкани здатні бромуватися. В цьому легко переконатись, провівши такий дослід. Якщо до н-гексану в темноті добавити краплю брому, то забарвлення брому зберігається протягом багатьох днів. Під впливом сонячного світла забарвлення брому в н-гексані зникає через кілька хвилин, оскільки в молекулі гексану відбувається фотохімічне заміщення атома водню на бром:
Світло
СвНи + Вг2-------> Свн„-Вг + НВг.
49
Легкий видих повітря біля отвору посуду з реакційною сумішшю викликає появу білого туману, який свідчить про утворення в результаті реакції бромоводню.
Бромування алканів відбувається з виділенням 39,8 кДж/моль теплоти і є набагато менш екзотермічним процесом, ніж реакція хлорування. Тому швидкість реакції бромування значно менша порівняно з хлоруванням. Механізм бромування на світлі, як і хлорування, — ланцюговий, радикальний. Фотохімічне бромування гомологів метану значно селективніше, ніж хлорування, відносно тих атомів водню, які заміщуються:
Світло, 127 °С
Н3С—СН2—СН3 + Вг2----------> Н3С—СН2—СН2—Вг +
1 -Бромпропан (3 %)
+ Н3С—СН—СН3 + НВг;
Вг
2-Бромпропаи (97 %)
Світло, 127 °С
Н3С-СН2-СН2—СН3 + Вг2----------->- Н3С—СН2—СН2—СН2 +
Вг
1-Бромбутан (2 %)
+ Н3С—СН2—СН—СН3 + НВг; І Вг
2-Бромбутаи (98 %)
сн3 сн3
І Світло, 127 °С |
Н3С—СН—СН3 + Вг2-------------> Н3С—СН—СН2—Вг + Н3С—С—СН8 + НВг.
1-Бром-2-метил- |
пропан (1 %) Вг
2- Бром-2-м етил-пропаи (99 %)
Значне підвищення вибірковості заміщення атомів водню на бром пов’язане з повільнішим перебігом реакції бромування.
Реакція йодування алканів ендотермічна. Для того щоб відбулася взаємодія атомів йоду з алканом, необхідна досить висока енергія — 129 кДж/моль. Тому йод практично не реагує з насиченими вуглеводнями навіть при температурі 300 °С.
{Взаємодія алканів з азотною кислотою'} Насичені вуглеводні при звичайній температурі не взаємодіють з концентрованою азотною кислотою. При нагріванні ця кислота діє як окислювач. Якщо ж на алкани Діяти розбавленою азотною.кислотою при нагріванні (140 °С), то може відбуватися реакція нітрування, в результаті якої атоми водню в молекулах насичених вуглеводнів заміщуються на нітрогрупу:
/?— {Н+~НО!—N0, Я— N0. + Н2О.
Вперше реакцію нітрування насичених вуглеводнів здійснив у 1888 р. М. І. Коновалов, тому її називають реакцією Коновалава.
БО
Швидкість реакції нітрування алканів невелика і вихід нітросполук досить низький. Найкращі результати одержують при нітруванні алканів, що мають третинні вуглецеві атоми. Нітрування насичених вуглеводнів супроводиться окислювальними процесами. На нітрування витрачається тільки близько 40 % азотної кислоти. Решта кислоти діє як окислювач. Тому при нітруванні порядз нітросполуками утворюються різні кисневмісні сполуки — спирти, альдегіди, кетони, кислоти. Нітрування алканів супроводиться також розривом С—С-зв’язків у їхніх молекулах (крекінгом), що веде до утворення нітросполук з меншою кількістю вуглецевих атомів, ніж у вихідного алкану. Так, при нітруванні пропану утворюється 34% 1-нітропропану Н3С—СН2—СН2—МО2, 32 % 2-нітро-пропану Н3С—СН(Г\Ю2)—СН3, 26 % нітроетану Н3С—СН2—Г\Юа і 8 % нітрометану Н3С—Г\Ю2.
Азотну кислоту, як нітруючий реагент алканів, можна замінити оксидами азоту (П. П. Шоригін, О. В. Топчієв, А. І. Титов).
Реакція нітрування алканів — радикальний процес. Початковою стадією цієї реакції є взаємодія вуглеводню з оксидом азоту _ (IV), який за будовою є радикалом, і утворення вуглеводневих радикалів.
Останні з оксидом азоту (IV) дають нітросполуки:
Я— н + N0; ->/?• + НМО2;
НМО2 + НМ03 -> М2О4 + Н2О;
н2о4^±2но;:
/?• + но;->/?—мо2.
Оксид азоту (IV) міститься в азотній кислоті і утворюється також внаслідок окислення вуглеводнів азотною кислотою.
\ Сульфування насичених вуглеводнів! Алкани за звичайних умов стійкі до дії концентрованої Н28О4. ‘При високих температурах вона діє як окислювач, а при слабкому нагріванні димуча сірчана кислота сульфує насичені вуглеводні, тобто заміщує в їхніх молекулах атом водню на сульфогрупу:
/?— ;н + НО1; 8О3Н -> /?— 803Н + Н2О.
і і Алкансульфо-
кислота
! Сульфохлорування насичених вуглеводнів./Сульфохлорування ал-кайІЕГзДїйснюють сумішшю хлору і оксиду сірки (IV). В результаті реакції утворюються алкансульфохлориди — хлорангідриди сульфокислот насичених вуглеводнів:
Р _ н + 8О2 -р С12 -* /? — 8О2С1 + НС1.
Реакцію проводять на світлі або при наявності каталізаторів, які утворюють в реакційній суміші вільні радикали. Фотохімічне
51
сульфохлорування алканів має ланцюговий радикальний механізм (5а):
і Сі
К— Н + С1- -—>«'• ----->-/? —8О2——8О2С1 + СІ • і т. д.
—неї
Реакція сульфохлорування має широке практичне застосування в промисловості, оскільки її продукти—алкансульфохлориди при взаємодії з лугами або содою утворюють солі сульфокислот:
7? — 8О.2С1 + 2№ОН -> 7? — 8О3№ + №С1.
Солі алкансульфокислот з 12—18 і більшою кількістю вуглецевих атомів застосовують для виготовлення синтетичних миючих засобів. Для їх добування в реакцію сульфохлорування вводять важкі фракції перегонки нафти або синтину4
Реакції відщеплення водню. Каталітична дегідрогенізація алканів. При пропусканні насичених вуглеводнів над оксидом хрому (III) при температурі 300—400 °С від їхніх молекул відщеплюються атоми водню і утворюються ненасичені вуглеводні (О. О. Баландін):
Сг2Оз б*пН2п+2 * б’пН2п -р Н2.
Реакції відщеплення водню від органічних речовин називають реакціями дегідрогенізації. Д.(Кщюг(лйзащ я алканів має велике промислове значення, зокрема при виробництві синтетичних каучуків (див. розд. 16).
Реакції розщеплення. Крекінг алканів.' При нагріванні насичених вуглеводнів до температури понад"400 °С відбуваються перетворення, що супроводяться розривом С—С-зв’язків у молекулах. Залежно від температури, при якій ведуть процес, розрізняють реакції крекінгу, що відбуваються при температурах, нижчих за 650 °С, і реакції піролізу, які відбуваються при температурах понад 650 °С.
Крекінг (від англ. іо сгаск —розщеплювати) вперше здійснив О. О. Лєтній, а В. Г. Шухов у 1891 р. взяв перший патент на установку крекінга. Цей процес має широке промислове застосування. З допомогою крекінгу з висококиплячих нафтових фракцій (гас, солярове масло, мазут) одержують низькокиплячі і таким чином підвищують вихід бензину із сирої нафти майже в три рази. Основні процеси, які відбуваються при крекінгу, —це дегідрогенізація алканів і розрив С—С-зв’язківїх молекул. Одночасно відбуваються також реакції ізомеризації і циклізації молекул насичених вуглеводнів.
Крекінг здійснюють або термічно в металевих трубках при 500—700 °С і різному тиску, або каталітично при температурі 500 °С на алюмосилікатних каталізаторах.
62
Як приклад розглянемо крекінг бутану при атмосферному тиску і 600 °С, в результаті якого утворюється суміш метану, етану, етилену, пропілену, бутилену і водню:
Н3С—СН2—СН2—СН3 -> СН4 + Н2С=СН—СН3; (1)
Н3С—СН2—СН2—СН3 Н3С—СН3 + Н2С=СН2; (2)
Н3С—СН2—СН2—СН3 -> Н3С—СН=СН—СН3 + Н2. (3)
Встановлено, що з 100 моль бутану 48 розкладається за реакцією (1), 36 —за реакцією (2) і 16 —за реакцією (3).
Якщо термічний розклад алканів проводять без каталізаторів при температурі 700—800°С і вище, то такий процес називаютьпі-ролізом. При піролізі алканів відбуваються більш глибокі зміни в їхніх молекулах, аж до утворення вуглецю (сажі або коксу).
Окислення насичених вуглеводнів. Окислення алканів можна здійснювати як при високих, так і при невисоких температурах. Енергійне окислення цих речовин киснем повітря при високих температурах, яке приводить до повного руйнування молекул алканів і утворення СО2 і Н2О, називають горінням. Цей процес має велике значення, оскільки він відбувається в усіх двигунах внутрішнього згоряння і лежить в основі добування енергії з палива. Окислення насичених вуглеводнів проходить через стадію утворення гідро-пероксидів Ц —О—О—Н. У двигунах внутрішнього згоряння вуглеводні нормальної будови утворюють пероксиди, які викликають передчасне загоряння пари бензину без участі свічки запалювання. Це явище називають детонацією. Воно завдає великої шкоди, оскільки веде до передчасного зношування двигунів, а також не дає можливості повністю використати його потужність. Насичені вуглеводні з розгалуженою будовою не детонують. Особливо цінним як моторне паливо є ізооктан:
СН3 СН,
І І
Н3С—С—СН2—сн—сн3.
сн3
2. 2, 4-Триметилпентан (ізооктан)
Ізооктан був покладений в основу умовної шкали для оцінки моторних палив за «октановим числом».Ізооктану в цій шкалі було приписане октанове число 100, ан-гептану —0. Бензини порівнюють з цією шкалою, тобто з сумішшю ізооктану і н-гептану. Наприклад, бензин з октановим числом 72 горить так, як суміш, яка складається з 72 % ізооктану і 28 % н-гептану.
При звичайних температурах кисень повітря і такі окислювачі, як КМпО4, К2СгО4, К2Сг2О7, не окислюють насичені вуглеводні. При невисокій температурі (~150 °С) і наявності каталізаторів, наприклад солей марганцю, алкани можуть окислюватися з утворенням різних кисневмісних сполук: спиртів, кетонів, кислот, оксикислот, кетонокислот тощо. Контролюючи і регулюючи процес окислення, можна добувати тільки певний продукт. При каталітичному окисленні вищих парафінів нормальної будови (С12—С23)
53
у промисловості добувають вищі жирні кислоти і вищі жирні спирти. Вищі жирні кислоти використовують для добування синтетичних миючих засобів (див. розд. 18), а вищі жирні спирти —для добування пластифікаторів пластичних мас.
Окислення метану киснем повітря при нормальному тиску дає мурашиний альдегід, а якщо окислення метану вести при тиску 151,5 • 10Б Па і 400 °С, то головним продуктом такого процесу є метиловий спирт.
Ізомеризація алканів. Насичені вуглеводні нормальної будови при температурі 100 °С і наявності каталізатора (А1С13) ізомеризуються в алкани з розгалуженою будовою. Так, пентан в цих умовах ізомеризується в ізопентан. Цей процес застосовують у промисловості, оскільки з ізопентану синтезують ізопрен—вихідну сировину для добування синтетичного ізопренового каучуку (див. розд. 16). Вважають, що при ізомеризації каталізатор відщеплює від пентану гідрид-аніон Н” з утворенням вторинного карбонієвого іона, який потім перегруповується через проміжну циклопропа-нову похідну в катіон ізопентану, а останній, приєднуючи іон водню, перетворюється на ізопентан:
Н3С—СНа—СН—СН2—СІ І3-> Н: + Н3С-СН2—СН-СН2—СІІ8->
1 .. і
! Н • і- і _
+Н+ + +Н:
-> І г + Н3С—СН—СН—СН3 -> Н3С—СН—СН—сн3 >
сн2 сн3
-> н3с—сн2—сн—сн3.
СН3
.„Взаємний вплив атомів у молекулах алка-„н і в. Експериментальні дослідження показали, що реакційна здатність водню в молекулах насичених вуглеводнів залежить в основному від того, біля якого вуглецевого атома (первинного, вторинного чи третинного) він знаходиться, а не від природи алкану, до складу якого цей водень входить. Відмінність у реакційній здатності атомів водню в алканах пов’язана з тим, що в молекулах цих речовин спостерігається взаємний вплив атомів, який у першу чергу виявляється в різній активності таких атомів водню:
сн3 І
Найбільшу активність виявляє атом водню біля третинного атома вуглецю, оскільки три метильні групи, маючи позитивний індукційний ефект (див. розд. 3), викликають зміщення електрон
54
ної густини (у хімічних формулах це зміщення зображено стрілками) і цим самим збільшують полярність, а відповідно і активність зв’язку третинного атома вуглецю з воднем. На активність атома водню біля вторинного атома вуглецю впливають дві метильні групи. Тому полярність і активність цього С—Н-зв’язку менша порівняно з попереднім. Найменшу активність має атом водню, який знаходиться біля первинного вуглецевого атома.
За відносною реакційною здатністю атоми водню в молекулах насичених вуглеводнів можна розмістити в такий ряд:
Водень біля третинного атома вуглецю
Водень біля > вторинного атома вуглецю
Водень біля Водень у первинного > молекулі атома вуглецю метану.
Така послідовність підтверджується і значеннями енергії даних зв’язків. Так, енергія первинного С—Н-зв’язку становить 419 кДж/моль, вторинного С—Н-зв’язку — 393,6 кДж/моль, третинного С—Н-зв’язку — 372,6 кДж/моль.
Експериментально встановлено, що співвідношення між швидкостями заміщення (при 300 °С) водневих атомів на хлор біля первинного, вторинного і третинного атомів вуглецю становить 1 : 3,8 : 5,0, а відносних швидкостей бромування — 1 : 82 : 1600. Швидкості відриву атомів водню від цих вуглецевих атомів відносяться як 1 :2 : 10.
Напрямок перебігу хімічних реакцій залежить від стійкості проміжних частинок, які утворюються в процесі даної реакції. Оскільки здатність водневих атомів відщеплюватися зменшується в порядку
Третинний > Вторинний > Первинний > в СН4,
то здатність до утворення вільних радикалів повинна зменшуватися в такому ж порядку:
Третинний радикал > Вторинний радикал > Первинний радикал > > Метильний радикал.
Така послідовність здатності утворювати радикали відповідає зміні їх стійкості. Зміна стійкості радикалів зумовлена взаємодією неспареного електрона з С—Н-зв’язками сусідніх а-атомів вуглецю. Наприклад, третинний бутильний радикал більш стійкий, ніж вторинний пропільний, який, у свою чергу, стійкіший від етильно-го радикала:
Різна стійкість цих радикалів спричинюється тим, що в третинному бутильному радикалі неспарений електрон взаємодіє (знаходиться в спряженні) з дев’ятьма сусідніми С—Н-зв’язками, у вто
55
ринному пропільному радикалі таких сусідніх зв’язків шість, а в первинному етпльному радикалі—три. Тому в третинному радикалі неспарений електрон найбільш делокалізований сусідніми С—Н-зв’язками. Це зумовлює його підвищену стійкість. Делокалі-зація неспареного електрона у вторинних і первинних радикалах порівняно з третинними зменшується, що приводить ДО відповідного зменшення їх стійкості.
Встановлено, що чим стійкіший вільний радикал, тим легше він утворюється в процесі реакції. Отже, у хімічних реакціях з участю алканів найлегше будуть утворюватись третинні радикали, тому біля третинних атомів вуглецю в першу чергу будуть відбуватися реакції заміщення атомів водню. З цієї ж причини кількість продукту заміщення біля третинного атома вуглецю значно більша, ніж біля вторинного і первинного. Проте слід пам’ятати, що на вихід продукту реакції впливає також реакційна здатність самого реагенту: чим менша реакційна здатність реагенту, тим селективніше він діє.
Контрольні запитання і вправи
1. Зобразіть графічні формули всіх можливих ізомерів вуглеводнів складу С6Н12, СеН]4 і назвіть їх за міжнародною номенклатурою.
2. Якими зв’язками сполучені між собою атоми в молекулах насичених вуглеводнів? Яка їх електронна природа?
3. Назвіть найважливіші реакції насичених вуглеводнів. Зазначте їх механізм.
4. Порівняйте активність атомів водню в молекулах алканів біля первинного, вторинного і третинного атомів вуглецю та стійкість первинних, вторинних і третинних радикалів, катіонів.
5. Обчисліть масові частки (у процентах) 1- і 2-хлорбутану в суміші, яку добувають при радикальному хлоруванні бутану. Реакцію проводять при кімнатній температурі. Відносна реакційна здатність первинного, вторинного і третинного атомів водню відповідно 1:4: 5.
Розділ 8. ГАЛОГЕНОПОХІДНІ АЛКАНІВ
Галогенопохідними називають сполуки, які містять зв’язки вуглець — галоген. При заміщенні в молекулі насиченого вуглеводню одного атома водню на атом галогену одержують моногалогено-похідні алканів, або галоїдні алкіли, які описуються загальною формулою С„Н2п+1— X, де X — Р, СІ, Вг або І. Залежно від природи галогену розрізняють фтор-, хлор-, бром- і йодалкани.
Утворення, будова і номенклатура деяких монохлорпохідних алканів наведені в табл. 7.
Класифікація. Залежно від того, біля якого (первинного, вторинного чи третинного) атома вуглецю знаходиться галоген, монога-логенопохідні поділяють на галогеналкани з первинним, вторинним і третинним вуглеводневими радикалами. Наприклад:
Галогеналкани з первинними радикалами:
Н3С—СН,—Сі; Н,С—СН„—СН,—СІ; Н„С—СН„—СЕ,—СН,—СІ;
Н,С—СН—СН,—СІ;
І
СН3
56
Таблиця 7. Утворення і номенклатура деяких хлоралканів
Формула алкану Формула хлоралкану Номенклатура
систематична Історична
сн4 Н3С—СІ Хлорметан Хлористий метил
Н3С-СН3 Н3С—СН2—СІ Хлоретан Хлористий етил
Н8С-СН2-СН3 Н3С—СН2—СН2—СІ Н3С—СН—СН3 1 СІ 1-Хлорпропан 2-Хлбрпропан Хлористий пропіл Хлористий ізопропіл, або хлористий в/пор-пропіл
нас—сн2—сн2—сн3 сн3 Н3С—СН2—СН,— СН2—СІ Н3С—СН2—СН—сн3 СІ СН3 1 3 1-Хлор'утан 2-Хлорбутан Хлористий бутил Хлористий в/пор-бутил
Н3С—сн—сн3 Н3С—СН—СН2—СІ сн3 Н3С—С СНд СІ 2-Метил- 1-хлор пропан 2-Метил-2-хлорпропан Хлористий ізобутил Хлористий тре/п-бутил
Н3С—СН2—СНСН2—СН з Н3С—СН2—СН,—СН2—СН2—СІ Н3С-СН2—СН2-СН-СН3 СІ Н3С—СН2—сн—сн2—сн3 СІ 1-Хлорпентан 2-Хлорпентан З-Хлорпентан Хлористий аміл Хлористий втор-аміл
з вторинними радикалами:
Н3С—СН—СН3; Н3С—СН2—СН—СН3;
СІ СІ
з третинними радикалами:
Н3С—С(СН3)~- СН3; Н3С-С(СН3)_ СН2—СН3.
СІ сі
Номенклатура. Моногалогеналкани називають за історичною або за систематичною номенклатурою. За історичною номенклатурою назву галогеналкану утворюють з назви відповідного одновалентного радикала з додаванням до неї назви галогену. Наприклад, хлористий метил, хлористий етил, хлористий пропіл і т. д. (табл. 7). За систематичною номенклатурою назви галогеналканів утворюють від назв відповідних насичених вуглеводнів, додаючи до них назву галогену, і цифрою вказують його положення у вуглеводневому ланцюгу. Наприклад, 1-хлорбутан, 2-метил-1-хлорпро-пан і т. д. За систематичною номенклатурою назви галогенів перелічуються в алфавітному порядку поруч з назвами вуглеводневих радикалів.
Ізомерія. Галогеналканам властива структурна ізомерія, зумовлена будовою (розгалуженням) вуглецевого ланцюга і положенням атома галогену в ланцюзі, а також конформаційна ізомерія (див. розд. 7). Ізомери з різним положенням атома галогену властиві вже галогенопохідним пропану — 1-хлорпропан і 2-хлорпропан (табл. 7), а ізомери з розгалуженим вуглецевим ланцюгом — починаючи з галогенопохідних бутану.
СІ
Н3С—СН2—СН,—СН2—СІ; Н,С—СН—СН,—СІ; Н_С—С—СН3.
СН3 сн3
Методи добування. З синтетичних методів добування моногало-геналканів найбільше значення мають такі:
1. Взаємодія насичених вуглеводнів з галогенами (див. розд. 7). Оскільки безпосереднє йодування алканів йодом провести не вдається, то йодопохідні насичених вуглеводнів з одним атомом йоду можна добувати реакцією обміну між відповідною хлоропохідною і йодидом натрію в ацетоновому розчині (реакція Фінкельштейна):
Я — СІ + №1-> Я — 1 + №С1.
Фтористі алкіли, які не можна добути взаємодією алкану з фтором, добувають дією фториду срібла на йодисті алкіли:
Н3С—1 + АеР -> Н3С-Р + А6І.
2. Заміщення в молекулах спиртів гідроксильної групи на галоген. Це можна здійснити кількома способами:
58
а) взаємодією спиртів з галогеноводнями, яка відбувається за такою загальною схемою:
П — ОН-р НХ ^/? — % + Н2О (X —СІ, Вг, І).
Реакція спиртів з галогеноводнями оборотна. У зв’язку з цим, щоб вона відбулася практично до кінця, необхідно в реакцію вводити газоподібні галогеноводні і в реакційну суміш не вводити воду.
За допомогою цього методу можна отримати хлор-, бром- і йодалкани;
б) дією на спирти галоїдних сполук фосфору або сірки (див. розд. 9).
3. Приєднання галогеноводнів до етиленових вуглеводнів (див. розд. 14).
Фізичні властивості. Галогеналкани мають характерний запах і багато з них діють як наркотики. У воді галогенопохідні насичених вуглеводнів майже не розчинні, але легко розчиняються в органічних розчинниках, таких як спирт, ефір, тощо. Самі галогеналкани є добрими розчинниками для багатьох органічних речовин. Нижчі галогеналкани — газоподібні речовини, середні — безбарвні рідини, вищі —тверді речовини жовтого кольору. Найважливіші фізичні властивості галогеналканів наведені в табл. 8. З даних табл .8 видно, що із збільшенням вуглеводневого радикала і атомної маси галогену, який входить до складу молекули, температура кипіння галогеналканів підвищується. При збільшенні атомної маси галогенів зростає також і відносна густина цих речовин.
Таблиця 8. Фізичні властивості деяких моногалогеналканів /?—X
Галогеналкан /?—X т 1 кип °С Густина ,20 "4 Галогеналкан —X ^кип» °С Г усти-Иад2° 4
Н3С—Р —78,6 0,877* Н3С(СН2)2СН2—Р 32,5 0,776
Н3С—СІ —23,7 0,991** Н3С(СН2)2СН2—СІ 78,5 0,887
Н3С—Вг 3,5 1,732*** Н3С(СН2)2СН2—Вг 101,6 1,276
Н,С—І 42,5 2,279 Н3С(СН2)2СН2—І 130,4 1,615
Н3С—СН2—Р —37,1 0,816**** Н3С(СН2)3СН2-Р 62,8 0,791
Н3С—СН,—Сі 12,4 0,921*** Н3С(СН2)3СН2—СІ 108,2 0,878
Н3С— СН2—Вг 38,4 1,461 Н3С(СН2)3СН2—Вг 127,9 1,218
Н3С—СН2—І 72,3 1,936 Н3С(СН2)3СН2—І 154,2 1,517
НЯС—СН„—СН2—Р —2,5 0,778*** Н3С(СН2)4СН2—Р 93,2 0,800
Н3С—СН2—СН2—СІ 46,6 0,892 НЯС(СН2)4СН2—СІ 132,9 0,876
Н3С—СН„—СН2—Вг 71,0 1,351 Н3С(СН2)4СН,—Вг 156,0 1,176
Н3С—СН2—СН2—І 102,5 1,749 Н3С(СН2)4СН2—І 180,0 1,441
* —при—79 °С; ** — при —25 °С; *** —при 0сС; **** — при —37 °С; /^ — вуглеводневий радикал СПН.2П+1; X — Р, СІ, Вг, 1.
В ІЧ-спектрах галогеналканів смуга валентних коливань зв’язку С—Р знаходиться в інтервалі 1000—1350 см-1, зв’язку С—СІ — близько 600—800 см-1, зв’язків С—Вг і С—І — близько 500— 600 см-1.
59
В ПМР-спектрах галогеналканів різні протони дають різні зсуви. Протони, які знаходяться біля атома вуглецю, сполученого з атомом галогену (у хімічних формулах ці атоми водню обведені жирним колом), також дають різні хімічні зсуви. Так, в хлоралка-нах 7?—СН2—СІ ці зсуви характеризуються величиною 3,7 млн-1, у бромалканах К—СН2—Вг вони мають значення 3,5 млн-1, у йод-алканах Я—СН2—І — 3,2 млн-1.
ПМР-спектр йодистого етилу зображено на рис. 20. Як видно з рисунка, йодистий етил містить протони двох типів (у групі СН3 — і в групі —СН2—) і кожний сигнал у спектрі являє собою групу ліній (спін-спінове розщеплення), які виникають внаслідок магнітної взаємодії протонів однієї групи з протонами іншої.
Рис. 20. ПМР-спектр йодистого етилу Н3С—СН2—І при 60 МГц
Л- відносно ТМС
Хімічні властивості. Галогеналкани —одні із найбільш реакцій-ноздатних органічних речовин. Більшість реакцій цих речовин відбувається з участю атома галогену. ІЗв’язок між атомом вуглецю і атомом галогену ковалентний. Але галоген більш електронегативний, ніж вуглець. Тому електронна пара зв’язку С -> Наї значно зміщена до атома галогену. Внаслідок цього зв’язок поляризується і атом галогену має заряд б-, а атом вуглецю —заряд 6+.
6+ с—
Поляризація зв’ язку С —>- Наї передається на сусідні С—С- і С—Н-зв’язки. Внаслідок цього електронна густина цих зв’язків зміщується в бік атома галогену:
(6Х > > 63).
Отже, атом галогену в молекулах галогеналканів виявляє негативний індукційний ефект (—/) (див. розд. 3), який затухає в міру віддалення від цього атома.
Молекули галогеналканів мають значну полярність, оскільки містять полярні зв’язкиС—Наї, які мають певні дипольн і моменти.
60
У табл. 9 наведено деякі характеристики зв’язків С—Наї у молекулах галогеналканів.
Таблиця 9. Характеристика зв’язків С — Наї у молекулах галогеналканів
Зв’язок Елек троне гативи і сть галогену Радіус атома галогену, нм Довжина зв’язку 1, нм Енергія зв’язку (середнє значення), кДж/моль Поляризація зв’язку, СМ3 Дипольний момент И, О
С—Р 4,0 0,064 0,142 429,24 1,7 1,4
С—СІ 3,0 0,099 0,177 332,64 6,5 1,5
С—Вг 2,8 0,114 0,191 278,04 9,6 1,4
С—І 2,5 0,133 0,212 220,08 14,6 1,2
Як видно з наведених даних, збільшення радіуса атома галогену зумовлює зростання між’ядерної віддалі І зв’язку С—Наї. Внаслідок цього енергія зв’язку в ряду С—Е,С—С1,С—Вг, С—І зменшується. Найменшу енергію має зв’язок С—І, тому в хімічних реакціях він найактивніший. Отже, зв’язки С—Наї за хімічною активністю розміщуються в ряд:
С—І > С—Вг > С—СІ > С—Р.
Для галогенопохідних насичених вуглеводнів найхарактернішими є реакції заміщення галогену (5-реакції) і реакції відщеплення галогеноводнів (Е-реакції). Висока полярність зв’язків С—Наї зумовлює високу реакційну здатність галогеналканів.
{ Р е акції за мїще н н я. Галоген, сполучений з атомом вуглецю, є слабкою основою (див. розд. 9). Його у вигляді іона можна легко замінити на сильнішу основу, тобто на іншу нуклеофільну частинку. Тому для галогеналканів характерними є реакції нуклео-фільного заміщення (Зуу-реакції). При нуклеофільному заміщенні нуклеофіл атакує молекулу і надає для утворення нового зв’язку свої електрони. Електрони зв’язку, який розривається, відходять разом з іоном, який при цьому утворився. У загальному вигляді нуклеофільне заміщення галогену в галогеналканах можна зобразити такою схемою:
/?: X + :7 -> /?: 7 + :Х.
Нуклеофільним реагентом X- можуть бути іони ОН-, ОТ?-, СМ-, 7?СОО-, 8Н-, 87?-, 8СА'-, N07 тощо, а також нейтральні молекули, які мають вільну пару електронів: Н2б, ІЧН3, 7?—КІН2, 7?'—NN—Ц", (С6Н5)3Р та ін. Найсильнішим нуклеофілом є карба-ніои 7?3С~. Реакції нуклеофільного заміщення галогену в галоген-алкднах є одними з найважливіших органічних реакцій.
Швидкість заміщення атомів галогенів у молекулах галогеналканів досить різна і значною мірою залежить від будови вуглеводневого радикала, з яким сполучений атом галогену, а також від природи галогену, нуклеофілу і розчинника. Так, швидкість реак
61
ції бромистого метилу з гідроксидом натрію, в результаті якої утворюється метиловий спирт
Н3С—Вг + ОН- -> Н8С—ОН + Вг,
залежить від концентрації обох реагентів —[Н3СВг] і [ОН-] — згідно з рівнянням V = к [Н3СВг] [ОН-], де к —константа швидкості реакції. Для третинних галогеналканів швидкість реакції залежить тільки від концентрації галогеналкану і не залежить від концентрації [ОН-]. Наприклад, швидкість реакції
(Н3С)3С—Вг + ОН- -> (Н3С)3С—ОН + Вг-
залежить тільки від концентрації [(Н3С)3С—Вг] і виражається рівнянням V = [(Н3С)3С—Вг]. Отже, взаємодія бромистого метилу з гідроксидом натрію — це реакція нуклеофільного заміщення другого порядку (5 м 2), оскільки її швидкість залежить від концентрації обох реагуючих речовин, а взаємодія бромистого трет-бутилу з №ОН —реакція першого порядку (5л1), оскільки її швидкість зай£2<ить від концентрації тільки однієї речовини.
|Різний кінетичний порядок реакцій заміщення атома галогену бі’ля первинного і третинного атомів вуглецю пояснюється неоднаковим механізмом нуклеофільного заміщення. У молекулі первинного галогеналкану іон ОН~ атакує атом вуглецю галогеналкану з за рядом 6 + (недостача електронів). Причому іон ОН - у ході реакції розміщується найдалі від атома галогену, на якому є заряд 6~. Тому ОН- атакує молекулу галогеналкану (зв’язок С—Наї) з боку, протилежного від атома галогену (з затилля відносно зв’язку С—Наї). Реакція проходить через перехідний стан, в якому атом вуглецю частково сполучений з ОН-групою і, крім, того, цей же атом ще не втратив зв’язку з галогеном. Іншими словами, зв’язок С—ОН у цьому стані ще не повністю утворився, а зв’язок С—Наї ще не зовсім розірвався. В уявленнях теорії МО перехідний стан при нуклеофільному заміщенні виникає в результаті перекривання вищої зайнятої орбіталі нуклеофілу і нижчої вакантної орбіталі реагуючої молекули. Перехідний стан не є проміжною сполукою і не має ознак, характерних для хімічних сполук (стабільних значень між’ядерних віддалей і валентних кутів). Він має максимальну енергію (енергію активації) порівняно з енергією початкового і кінцевого станів системи. Зміна енергії в ході такої реакції показана на енергетичній кривій (рис. 21). У перехідному стані утворення зв’язку між атомом вуглецю і ОН-групою і розрив зв’язку С — Наї відбуваються одночасно (синхронно).
Якщо у реакцію вводять оптично активну галогенопохідну сполуку (див. розд. 23), то ОН-г^рупа займе в просторі не те положення, яке займав раніше га логе^. Молекула спирту який утворюється, має конфігурацію, протилежну конфігурації молекули галогенопохідної. Так, якщо атом галогену був розміщений у просторі біля атома вуглецю справа, то ОН-група в добутому спирті буде розміщуватись біля цього атома вуглецю зліва (рис. 22), якщо ж галоген
62
Рис. 21. Зміна потенціальної енергії реакції нуклеофільного заміщення, яка відбувається за механізмом 5д,2
Рис. 22. Схема взаємодії хлористого метилу з ОН~-аніоном: а — атака «з затилля»; б — перехідний стан; в — «фронтальна» атака
був біля атома вуглецю зліва, то ОН-група стане справа. Отже, Зл^-реакції відбуваються з повним обертанням конфігурації (див. Р оз^23).
реакційну здатність алкілгалогеніду в реакціях 8дг2 впливає будова алкільної групіщ Щоб зрозуміти вплив будови алкіл ьної групи на швидкість8м2^реакції, розглянемо, як буде змінюватись перехідний стан при переході від бромистого метилу до бромистого етилу, а потім до ізопропіл броміду
Рис. 23. Просторові перешкоди при 5д,2-реакціі
і трет-бутилброміду. В міру того, як атоми водню біля атома вуглецю, сполученого з галогеном, будуть замінюватись на метильні групи, що мають значно більший об’єм, ніж атоми водню, збільшуються просторові перешкоди біля цього атома вуглецю (рис. 23). Це, у свою чергу, утруднює підхід ОН-групи до атома вуглецю і тим самим збільшує енергію утворення перехідного стану. Енергія акти-
СЗ
вації стає вищою і реакція відбувається повільніше. Тому в міру збільшення кількості метильних або інших груп біля атома вуглецю, сполученого з галогеном, реакційна здатні сть галогену до 5№2-реакцій зменшується:
Сполука Н3С—Вг > НаС—ЗН,—Зг > (Н3С)2СН—Вг > ^Н3С)3С—Вг. ,
Відносна реак- 150 1
цінна здатність атома брому до заміщення на I-
0,01
0,001
Отже, у реакціях 5,у2 реакційна здатність галогеналканів зменшується у такому порядку:
Н3С—Наї > ПеРвинний > 3 галогеналкан
Вторинниі .Третинний галогеналкан галогеналкан.
Розглянуті вище кінетичні дані і стереохімія реакції заміщення атома галогену доводять 5м2-механізм.
Тепер 'розглянемо механізм нуклеофільного заміщення галогену, який знаходиться біля третинного вуглецевого атома. Гідроліз третинних галогеналкілів, наприклад трет-бутилбфВмІду, відбувається у дві стадії. На першій повільній стадії із третинного гало-геналкілу утворюється іонна пара—третинний карбонієвий катіон і аніон галогену (мономолекулярна реакція):
(Н3С)8С-Вг^ (НаС)3С+ + вс.
Карбонієвий іон на другій стадії швидко взаємодіє з іоном гідроксилу або з молекулою води, яка має здатність надавати пару електронів для заповнення вакантної орбіталі вуглецевого атома:
(Н3С)3С+ + ОН- (Н3С)3С — ОН;
(Н3С)3С+ + :б-Н^ [(Н3С)8С:О:Н]+ (Н3С)3С-ОН р Н*.
Н ! Н !
І 1
Процес утворення з галогеналкаиу карбонієвого катіона(перша стадія) відбувається повільно і визначає швидкість всього процесу, оскільки загальна швидкість реакції визначається швидкістю найповільнішої стадії. Тому весь процес заміщення галогену біля третинного атома вуглецю проходить відповідно до кінетичного рівняння першого порядку (5дг1). Зміна енергії в процесі реакції 5лі показана на енергетичній кривій (рис. 24).
Якщо в 5д4-реакцію вступає оптично активна галогенопохідна сполука, то продукт заміщення галогену наОН-групу являє собою суміш сполуки з протилежною конфігурацією і рацемату. Отже, -реакції відбуваються з частковою рацемізацією. Пояснюється це тим, що в результаті дисоціації третинної галогенопохідної утворюється карбонієвий іон, зв’язки в якому напрямлені до кутів правильного трикутника. Такий карбонієвий іон має плоску структуру. Нуклеофільний реагент (ОН-, Н2О) може атакувати цей
64
плоский іон з будь-якого боку і залежно від того, з якого боку відбувається атака, утворюється продукт з тією або іншою конфігурацією (з двох його можливих конфігурацій). Вище вже зазначалося, що стадією, яка визначає швидкість 5л/1-реакції, є стадія утворення карбонієвого іона. Тому реакційна здатність алкілгалоге-нідів до 5д4-реакцій залежить головним чином від стійкості цього карбонієвого іона і змінюється в такому ж порядку, як і стійкість таких іонів (див. розд. 5). Реакційна здатність галогеналканів до ЗдЛ-реакцій зменшується в такому порядку:
Рис 24. Зміна потенціальної енер тії реакції нуклеофільного замі щення, яка відбувається за меха пізмол
Третинний ^Вторинний .Первинний . __Наї
галогеналкан •'* галогеналкан •'* галогеналкан 3
Наприклад: (И3С)3С—Вг (И3С),СН—Вг Н3С—£Н2—Вг Н3С—Вг.
Відносна швид- 100 000 000 45 1,7 1,0
кість заміщення атома галогену на ОН-групу
Слід зазначити, що на механізм нуклеофільного заміщення галогену крім будови вуглеводневого радикала впливає також концентрація нуклеофільного реагенту, його природа, полярність розчинника. Висока концентрація нуклеофільного реагенту сприяє 5л'2-механізму, а низька концентрація — навпаки, 5л-1-механізму. Сильний нуклеофіл «виштовхує» атом галогену з молекули і тим ; самим сприяє 5л/2-реакції, а слабкий нуклеофіл «чекає», доки іону галогену не відійде від атома вуглецю, що сприяє 5л?1-реакції.
Вторинні алкілгалогеніди мають проміжну між первинними і третинними алкілгалогенідами структуру і тому можуть реагувати як за 5м2-, так і за 5л4-механізмом. Збільшення концентрації іонів ОН~ прискорює реакцію другого порядку і не впливає на швидкість реакції першого порядку. При високій концентрації ОН “-іонів реакція другого порядку відбувається настільки швидко, що практично весь вторинний алкілгалогенід реагує за 5дг2-механізмом. Аналогічно зменшення концентрації ОН~-іонів сповільнює реакцію другого порядку і не впливає на реакцію першого порядку.
Механізм реакції часто може визначатися також полярністю розчинника. Чим полярніший розчинник, тим більше він прискорює дисоціацію галогеналкану з утворенням карбонієвого іона і іона галогену. Отже, підвищення полярності розчинника прискорює швидкість 5дЛ-реакції. 5м2-Реакції відбуваються з утворенням між реагуючими речовинами перехідного стану. Перехідний стан
З 1.120
65
менш полярний, а тому і менш сольватований, ніж вихідні речовини, оскільки негативний заряду такому стані розподілений між групою ОН і галогеном. Тому підвищення полярності розчинника сповільнює 5д?2-реакцію.
Розглянемо деякі реакції нуклеофільного заміщення атомів
галогену в галогеналканах.
/7 При дії на галогеналканн водного розчину лугу, води або волового оксиду срібла утворюються спиртн:лр
сі Ч" і,
н3с—с\—І + К+ОН" н3с—оу-он + КІ.
і Етило^і}^ спирт
Реакція галогеналканів з водою є оборотною:
С+ 6 0+ б 'і
Я—Є} + НОН Я—ОН + Нбф
Взаємодію галогеналканів з водою називають гідролізом. Гідроліз ведуть при нагріванні та наявності лугів або карбонатів лужних металів і широко використовують Для добування спиртів.
Галогеналканн при взаємодії з алкоголятами утворюють прості ефіри: ' •' •
б* С д------- ’
Н3С—СН^—Вг + №0—СН2—СН3 -> Н3С—СН^-О—СН2—СН3 + ИаВо
Етилат натрію Діетиловий ефір
Солі органічних кислот при нагріванні з галогеналканами утворюють скдадні ефіри: о ,
б+ ' с" + _ І
Н3С—СІ^—І + Ая—О—С-СН3 Н3С—СН,-О—С-СН3 + А§ї.
' II II
О О
Ацетат срібла Оцтовоетнловий ефір
Взаємодією галогеналкілів з солями синильної кислоти добувають ціанисті алкіли (нітрили) та із^ціаніди (ізонітрили):
*-Н2С—СИ (5д,2-реакція)
> НЬгрил Оцтової КИСЛОТІ
б* _
НзС-І-І-К
1 !<.С—N С (5^1-реакція)
’ у Метіеіізоціаиід СЛа -є
Нуклеофільна частинка — іон СІМ", що атакує галогеналкіл, виявляє подвійну реакційну здатність (амбідентні властивості) і тому може приєднуватись до атома вуглецю галогеналкілу або атомом вуглецю, або атомом азоту. Якщо в реакційній суміші як проміжні частинки утворюються карбонієві іони (Х^І-механізм), то амбідентний СІМ "-іон приєднується до карбонієвого іона більш електронегативним атомом, тобто азотом. Продуктами такої взаємодії є ізонітрили. При 5м2-механізмі СМ~-іон взаємодіє з атомом вуглецю галогеналкілу менш електронегативним атомом,тобто вуглецем, і утворює нітрили.
(.6
При нагріванні галогеналканів з нітритом срібла утворюються : нітросполуки і ефіри азотистої кислоти — нітрити (див. розд. 12).
Галогеналкани в спиртовому або ацетоновому розчині можуть вступати в реакцію з солями галогеноводневих кислот .'-Таким чином можна замінити в молекулі галогеналкану атом одного галогену на інший. Якщо заміщують хлор на бром або йод, то реакцію потрібно проводити з солями лужних металів, оскільки хлориди цих металів розчиняються важче, ніж броміди і йодиди. При заміщенні галогену з більшою атомною масою на галоген з меншою атомною масою реакцію необхідно проводити з солями срібла, оскільки йодид срібла менш розчинний, ніж бромід і особливо хлорид:
Я—СҐ+ Маґ^> /?-($-
Я—І -}- А§С1 ч- Я—СІ + А§1.
Галогеналкани досить легко реагують з аміаком і амінами. При цьому можуть утворюватися первинні, вторинні або третинні аміни, а також солі амонійних основ (див. розд. 13). Наведемо схему реакції утворення тільки первинного аміну:
еі+б-" .. . і' +№, : '
Н3С-І + •^^С-НН2 + МН4І.
: І ( Йодистий ‘ ^Метиламін
. метадамоній '-'і/
З наведених прикладів видно, що реакції заміщення дають можливість на основі галогеналканів синтезувати найрізноманітніші класи органічних сполук.
Реакції відщеплення (дегідрогалогену-в а н н я) називають також реакціями елімінування, або Е-реак-ціями.
Поряд з реакцією нуклеофільного заміщення галогену дуже часто паралельно відбувається реакція дегідрогалогенування з утворенням ненасиченого етиленового вуглеводню. Наприклад, при дії на галогеналкани спиртового розчину лугу від них легко відщеплюються елементи галогеноводневої кислоти і утворюються ненасичені вуглеврдні:
Н є —нх !
Н3С—СН2—X + КОН —Н2С=СЙІ+ КХ + Н2О.
*.-
З наведеної схеми реакції видно, що в ході реакції дегідрогалогенування від одного вуглецевого атома відщеплюється галоген, а від сусіднього атома вуглецю (з Р-положення) відщеплюється водень. Якщо елементи галогеноводню можуть відщеплюватися в кількох напрямках, то водень відщеплюється від найменш гідро-генізованого Р-вуглецевого атома. Такий порядок відщеплення галогеноводнів відомий в органічній хімії як правило Зайцева, який вперше встановив і обгрунтував його. При відщепленні галогеноводнів від галогеналканів, згідно з правилом Зайцева, утворю-3* 67
ються етиленові вуглеводні, які мають найбільше число алкільних радикалів біля вуглецевих атомів з подвійним зв’язком:
—неї
Нзс—СН—сн2—СН3 + КОН -—> Н3С—СН=СН—СН3 + КС1 + Н2О; . І ' Р Бутен-2
І СІ
СН3 сн3
І - НВг |
Н3С—С—СН2— 6Н'3+ КОН ---> Нзс—С=СН—СН 3/+ КВг + Н2О.
І Спирт
За здатністю до реакцій відщеплення галогеноводнів галоген-алкани розміщуються в такий ряд: Я—І > £—Вг > £—СІ > К—Р.
Активність відщеплення галогеноводню визначається також і структурою вуглеводневого радикала.
Реакції відщеплення галогеноводнів, як і реакції нуклеофільного заміщення, можуть відбуватися за мономолекулярним (£1) і бімолекулярним (£2) механізмами. Швидкість мономолекулярних £1-реакцій визначається тільки концентрацією галогеналкану, а швидкість бімолекулярних £2-реакцій залежить як від концентрації галогеналкану, так і від концентрації дегідрогалогенуючого реагента, наприклад лугу.
В реакції £1-елімінування найлегше вступають третиннігало-геналкани, в реакції £2-елімінування—первинні галогеналкани. Така залежність механізму відщеплення від природи вуглеводневого радикала пов’ язана з відносною стійкістю алканів, які утворюються при £2-реакції, і стійкістю карбонієвих іонів, які утворюються на найповільнішій стадії £1-реакції.
Реакції, які відбуваються за £1-механізмом,двостадійні. На першій, повільній стадії, яка визначає швидкість процесу дегідро-галогенування, третинний галогеналкан дисоціює з утворенням карбонієвого іона, від якого на наступній стадії дуже швидко відщеплюється протон і утворюється етиленова сполука:
_ н,о (Н3С)3С—СІ
' * /д Повільно
(Н3С)3С++-СГ;
НзС
Швидко
сн2 II
Н3С —С +
СНз
Ізобутилен
У цьому випадку розчинник, наприклад вода, сприяє відщепленню іонів галогену від галогеналкану, а подальша дія ОН “-іонів лугу на зв’язок С—Н полегшує відщеплення протона. Електронна пара, яка вивільняється після відщеплення протона, використовується для утворення іншого зв’язку між атомами вуглецю. Отже, швидкість дегідрогалогенування третинних галогеналканів залежить тільки від концентрації галогеналкану.
68
При дегідрогалогенуванні за Е2-механізмом нуклеофільний реагент, наприклад іон ОН", атакує |3-вуглецевий атом галоген-алкану і відщеплює від нього протон. Одночасно з відщепленням протона відщеплюється від галогеналкану і аніон галогену. Цей процес відбувається без утворення проміжних сполук, але йде через перехідний стан, у якому нуклеофільний реагент утворює зв’язок з протоном в такій мірі, в якій відбувається відщеплення аніона галогену:
б- в-
НО~ + Н—СН2—СН2—X -* [НО - • -н. • -СН2—СН2- • -Х| ->
Перехідний стан
-> СН2 = СІ І2 + Н2О + X".
У цьому випадку, як і у випадку третинних галогеналканів, електронна пара, яка вивільняється після відщеплення протона, іде на утворення подвійного зв’язку між вуглецевими атомами:
і І—І І
НО" + И[:СН2-+ СІ І2|: X-> СН2=СН2 + Н2О + X".
Швидкість Е2-реакції пропорційна концентрації обох реагентів — і галогеналкану, і нуклеофільної сполуки.
Взаємодія галогеналканів з металами. При дії на галогеналкани металів галоген заміщується на атом металу.’Ця реакція єдіайваж-ливішим методом синтезу металоорганічних сполук — речовин, які містять зв’язок С—Ме. Так, металічний літій у безводному діетиловому ефірі утворює з^алогеналканами літійорганічні сполуки:
( Н3С—Вг'+ 2Іл'-*Н8С—СН2—Ш + ЕіВт'.
і, А Етиуі^і^ій
З металічним натрієм галогеналкани утворюють насичені вуглеводні. Проміжними продуктами при цьому є натрійорганічні сполуки (див. розд. 7). 'З металічним цинком галогеналкани утворюють цинкорганічні сполуки: ч
2Н3С—СНа—Вг + 22п (Н3С—СН2)22п 2пВг2.'
. ; - - Діет^л^инк
Особливо важливою є реакція галогеналканів з металічним магнієм, в результаті якої утворюються змішані магнійорганічні сполуки (див. розд. 20):
Ефір 7?—І + ----* —І.
Реакція активно відбувається в безводному ефірі.
Магнійорганічні сполуки були відкриті у 1901 р. французьким хіміком В. Гріньяром, тому їх називають реактивами Гріньяра. Ці реактиви знайшли широке застосування в органічному синтезі. В. Гріньяру за відкриття магнійорганічних сполук і розробку синтезів різноманітних органічних речовин на їх основі у 1912 р. було присуджено Нобелівську премію.
При взаємодії галогеналканів зі сплавом натрію і свинцю утворюються свинецьорга нічні сполуки* Найбільше значення серед них
має тетраетилсвинець (ТЕС), який у промисловості добувають з хлористого етилу:
140 °С
4С2Н6С1 + 4РЬі\а-> (С2Н5)4РЬ + 4КаС1 + ЗРЬ.
Тетраетил-свинець
Можливо, ця реакція відбувається з утворенням проміжного продукту етилнатрію. ТЕС використовують як антидетонатор для моторного палива, він міститься в етильованому бензині. ТЕС надзвичайно отруйний (уражає центральну нервову систему), що обмежує його застосування.
Контрольні запитання і вправи
1. Зобразіть графічні формули всіх можливих ізомерів речовин складу С4Н9С1, СвНиС1, С6Н13Вг та назвіть їх за міжнародною номенклатурою.
2. Які речовини утворюються під час взаємодії 1-бромбутану з водним розчином лугу; спиртовим розчином лугу; ціанідом калію; металічним натрієм; ацетатом срібла; оксидом срібла?
3. Розмістіть наведені нижче сполуки в порядку зміни реакційної здатності в реакціях 5д,2 і 5д,1:
а) 2-бром-2-метилбутан, 1-бромпентан, 2-бромпептан;
б) 1-бромбутан, 1-бром-2,2-диметилпропан, 1-бром-2-метилбутан, 1-бром-3-метилбутан.
4. Яка з наведених речовин — хлористий метил, хлористий бутил чи хлористий трет-бутил — буде енергійніше реагувати з йодидом калію в ацетоні, з гідроксидом калію в етанолі, з нітратом срібла у водному етанолі?
Розділ 9. АЛКАНОЛИ (СПИРТИ)
Спирти —• це похідні вуглеводнів, у молекулах яких один або кілька атомів водню заміщені на відповідну кількість гідроксильних груп —ОН. Спирти називають ще алкоголями (від арабського «алкоголь» — спирт).
Класифікація. Спирти класифікують за кількістю гідроксильних груп, які містяться в його молекулі, а також за характером вуглецевого атома (первинного, вторинного чи третинного), з яким сполучена гідроксильна група. Кількість гідроксильних груп у молекулі спирту називають його атомністю. За атомністю спирти поділяють на одно-, двох-, трьох- і багатоатомні. Одноатомні спирти мають одну гідроксильну групу, двохатомні —дві, трьохатомні — три гідроксильні групи і т. д. Спирти, які мають чотири і більше гідроксильних груп, називають багатоатомними. Прикладом одноатомного спирту є етиловий спирт Н8С—СН2—ОН, двохатомного— етиленгліколь НО—СН2—СН2—ОН, трьохатомного— гліцерин СН2(ОН)—СН(ОН)—СН2—ОН, багатоатомного—гексит СН2(ОН)—СН(ОН)—СН(ОН)—СН(ОН)—СН(ОН)—СН2—ОН.
Залежно від того, з яким вуглецевим атомом—первинним, вторинним чи третинним — сполучена гідроксильна група, розрізняють первинні, вторинні і третинні спирти.Наприклад, первинний бутиловий спирт Н3С—СН2—СН,—СН2—ОН, вторинний бутиловий спирт Н3С— СН2—СН(ОН)—СН3, третинний бутиловий спирт
70
(Н3С)3С—ОН. Ознайомлення з класом спиртів почнемо з найпростіших — насичених одноатомних спиртів, які мають загальну формулу СпН2п +1 —ОН.
Одноатомні спирти
Одноатомні спирти утворюють гомологічний ряд, який можна легко записати, користуючись гомологічним рядом насичених вуглеводнів, замінюючи в них один атом водню на гідроксильну групу (табл. 10).
Номенклатура. Одноатомні спирти називають за історичною, раціональною та систематичною номенклатурами (табл. 10). За історичною номенклатурою назва одноатомного спирту походить від назви радикала, з яким сполучена гідроксильна група. Наприклад, метиловий спирт, етиловий спирт, пропіловий спирт і т. д. За раціональною номенклатурою спирти розглядають як заміщені найпростішого із спиртів—метилового, який називають карбінолом. Наприклад, метилкарбінол, етилкарбінол і т. д. За систематичною номенклатурою назву спирту утворюють від назви відповідного насиченого вуглеводню, додаючи до неї суфікс -ол. Положення гідроксильної групи позначають номером атома вуглецю, біля якого вона розміщена. Вуглецевий ланцюг нумерують так, щоб атом вуглецю з гідроксильною групою мав найменше числове значення. Наприклад, бутанол-2 Н3С—СН—СН2—СН3, 2-метилпропанол-І
ОН
НУС—СН—СН2—ОН.
І сн3
Деякі спирти мають ще емпіричні назви, які пов’язані з історією відкриття або добування цього спирту з того чи іншого природного продукту. Наприклад, метиловий спирт часто називають деревним, оскільки його добувають сухою перегонкою деревини; етиловий спирт називають винним, оскільки його вперше було виявлено у виноградному вині і довгий час добували перегонкою вина. Звідси пішла і латинська назва цього спирту —Зрігііиз уіпі —«дух вина».
Ізомерія. Ізомерія спиртів може бути зумовлена розгалуженням вуглецевого ланцюга їх молекули, положенням гідроксильної групи в цьому ланцюгу, а також конформаційна (див. розд. 7). Перші два члени гомологічного ряду спиртів (табл. 10)— метиловий Н3С—ОН і етиловий Н3С—СН2—ОН—ізомерів не мають. Пропіловий спирт існує у вигляді двох ізомерів — первинного і вторинного (ізо-) пропілового спирту. Легко ПОМІТИТИ, що ці спирти відрізняються один від одного положенням гідроксильної групи в молекулі. У бутилового спирту чотири ізомери (табл. 10) — первинний і вторинний бутилові, ізобутиловий та третинний бутиловий спирти. Ізомери бутилового спирту різняться між собою уже як будовою вуглецевого ланцюга, так і положенням гідроксильної групи в молекулі.
71
Таблиця 10. Гомологічний ряд, номенклатура і фізичні властивості деяких одноатомних спиртів
Насичені вуглеводні Одноатомні спирти
Формула Назва Формула Назва т 1 КИП’ °С Густина .20
історична | раціональна ІЮПАК
сн4 Метан н3с—он Метиловий спирт Карбінол Метанол 64,7 0,792
н3с—сн3 Етан н3с—сн2—он Етиловий спирт Метилкарбінол Етанол 78,4 0,789
Н3С-СН2-СН3 Пропан н3с—сн2—сн2—он Пропіловнй спирт Етилкарбінол Пропанол-1 97,2 0,804
Н3С—СН(ОН)—сн3 Ізопропіловий спирт Диметилкарбі-нол Пропанол-2 82,3 0,785
Н3С-СН2-СН2-Сн3 Бутан 1 Н3С—СН2—СН2—СН2—Он Бутиловий спирт Пропілкарбінол Бутанол-1 117,1 0,809
1 Н3С—сн2—СН(ОН)—сн3 Вторинний бутиловий спирт Метилетилкар-бінол Бутанол-2 99,5 0,808
Нзс—СН—сн3 сн3 Ізобутан,, 2-метпл-пропан н3с—сн—сн2—он сн3 І зобутиловий спирт Ізопропілкарбі-нол 2-Метилпро-панол-1 107,3 0,801
СНз
' сн3—с—сн3 он Третинний бутиловий спирт Триметил карбінол 2-Метилпро-панол-2 82,8 0,788
Методи добування. Насичені одноатомні спирти можна добути такими методами.
1. Окисленням насичених вуглеводнів. Алкани при каталітичному окисленні можуть утворювати ряд кисневмісних сполук, у тому числі і спирти (див. розд. 7):
Н3С—(СНа)п—СН3 —> Н3С—(СНа)т—СНа—ОН, де т < п.
Цей метод має промислове значення для добування вищих жирних спиртів.
2. Гідролізом галогеналканів, який приводить до заміщення атома галогену на гідроксильну групу (див. розд. 8):
Я— СІ + он- -> Я—ОН + СІ".
3. Гідратацією етиленових вуглеводнів (див. розд. 14).
4. Відновленням альдегідів, кетонів, карбонових кислот і складних ефірів воднем при наявності каталізаторів. Альдегіди, карбонові кислоти і складні ефіри при цьому приєднують водень і утворюють первинні спирти, а кетони в аналогічних умовах —вторинні спирти (див. розд. 17). Як відновники цих речовин використовують також комплексні гідриди металів.
5. Взаємодією альдегідів, кетонів, складних ефірів з металоорганічними речовинами, головним чином магнійорганічними (реактивами Гріньяра) (див. розд. 17).
6. Лужним або кислотним гідролізом складних ефірів карбонових кислот (див. розд. 19).
Фізичні властивості. Насичені одноатомні спирти від Сі до С10 за звичайних умов—рідини. Вищі спирти, починаючи с Си,— тверді речовини (табл. 10). Всі спирти легші за воду. їх густина менша від одиниці. Нижчі спирти, від С, до С3, змішуються з водою в будь-яких співвідношеннях; спирти від С4 до С1о важко розчиняються у воді, а вищі спирти в ній практично не розчинні. Запах нижчих спиртів алкогольний, починаючи з С4 — неприємний. Вищі спирти запаху не мають. Температури кипіння спиртів підвищуються в міру збільшення їх молекулярної маси (табл. 10). Слід зазначити, що температури кипіння спиртів значно вищі, ніж температури кипіння відповідних насичених вуглеводнів та їх галогенопохідних з такою самою кількістю вуглецевих атомів, як і в спирту. Наприклад, етан кипить при —88,3 °С, бромистий етил С2Н5—Вг — при ф-38,0°С, а етиловий спирт — при +78,5°С, хоча молекулярна маса бромистого етилу вища, ніж етилового спирту. Таке підвищення температури кипіння спиртів пояснюється тим, що молекули спирту, як і молекули води, хоч і меншою мірою, є асоційованими і сполучені між собою водневими зв’язками:
е+ б~ б+ с* б' б+ еГ б+ б“ є* б~ -Н—О: -Н—О: -Н—О: -Н—О: -Н—О: -Н—О: -
II І І І І Я Я Я Я Я Я
Електронегативний атом кисню, який має ще й вільні електронні пари, однієї молекули спирту притягує атом водню гідро
73
ксилу другої молекули спирту, на якому є заряд 6+. Така міжмолекулярна взаємодія приводить до асоціації молекул спирту, що і є причиною високої температури кипіння цих речовин. Чим менше алкільних груп оточує гідроксил в молекулі спирту, тим легше виявляються водневі зв’язки і тим більшою мірою відбувається асоціація молекул спирту, а отже, тим вища температура кипіння спирту. Тому первинні спирти киплять при вищій температурі, ніж ізомерні їм вторинні, а третинні спирти з тієї ж причини мають ще нижчу температуру кипіння (табл. 10). Отже, спирти нормальної будови киплять при вищій температурі, ніж спирти з розгалуженим вуглецевим ланцюгом.
Енергія водневих зв’язків набагато менша, ніж енергія звичайного ковалентного зв’язку, і дорівнює 20,934 —41,868 кДж/моль, у той час як енергія звичайного зв’язку О—Н становить 456,36 кДж/моль. Під час кипіння всі водневі зв’язки руйнуються.
Молекули спиртів полярні і мають дипольні моменти. Наприклад, дипольний момент метилового спирту.дорівнює 1,690. Валентний кут, утворений зв’язками атома кисню в молекулі спирту, , становить 110°25'.
С Н
В ІЧ-спектрах зв’язок кисень — водень групи ОН дає характеристичну смугу поглинання. На це поглинання істотний вплив має водневий зв’язок. Наприклад, в газовій фазі, де водневого зв’язку практично немає, в ІЧ-спектрі етанолу є порівняно чітка смуга поглинання при 3700 см-1, яка свідчить про наявність неасоційованої гідроксильної групи (рис. 25, а). В ІЧ-спектрі 10 %-го розчину етанолу вСС14 (рис. 25, б) цю смугу важко розрізнити при 3640 см-1. Однак в цьому спектрі є відносно широка смуга при 3350 см-1, характерна для ОН-групи, сполученої водневим зв’язком. Зсув частоти спостерігається приблизно на 300 см-1, оскільки водневий зв’язок послаблює зв’язок О—Н, і тому поглинання відбувається при нижчих частотах. Розширення смуги зумовлене тим, що гідроксильні групи асоційовані в агрегати різного розміру і профілю. Внаслідок цього з’являються різні типи асоціацій і поглинання зумовлює спектр, який складається з тісно розміщених смуг, спричинених ОН-групами. В дуже розбавлених розчинах спиртів у неполярних розчинниках водневі зв’язки утворюються у надзвичайно малій кількості. В міру збільшення концентрації спирту все більше молекул стають асоційованими і інтенсивність смуги, зумовленої асоційованою гідроксильною групою, зростає за рахунок смуги неасоційованого гідроксилу. Частота смуги, пов’язана з асоціацією, може бути мірою міцності водневого зв’язку. Чим більше частота асоційованого гідроксилу зміщена в бік низьких частот відносно положення неасоційованої ОН-групи, тим міцніший водневий зв’язок.
У ПМР-спектрі етилового спирту (див. рис. 4, розд.1)легко помітити наявність сигналів протонів трьох типів (—ОН, —СН2— і —СН8). Хімічний зсув гідроксильного протона знаходиться в ін
Ї4
тервалі 2,0—4,5 млн*1. У випадку етилового спирту він є розширеним трпплетомза рахунок спін-спінової взаємодії з протонами сусідньої групи—СН2—і зміщенийубік слабшого магнітного поля.
Хімічний зсув гідроксильного протона сильно змінюється залежно від природи розчинника, зміни концентрації спирту ітемпе-
Довжина хвилі
3600 2800 2000 2000 1800 1600 1400 1200 1000 800
Частота, сг-г’
Довжина хвилі
3600 2800 2000 2000 Ш 1600 1400 1200 1000 800 Частота, сг-г'
б
Рис. 25. ІЧ-спектри етанолу в газовій фазі (а) і 10 %-го розчину етанолу в ССЦ (б)
ратури, оскільки залежить від ступеня молярної асоціації спирту за рахунок водневих зв’язків і від їх міцності. При збільшенні міцності водневого зв’язку резонансний сигнал з’являється при слабших магнітних полях (хімічний зсув має більше значення). Так, хімічні зсуви ОН-протонів спиртів у рідкому стані становлять 4—5 млн*1. При зменшенні ступеня утворення водневих зв’язків
75
за рахунок розбавляння спирту чотирихлористим вуглецем сигнал ОН-протона зсувається в бік сильнішого поля. Різниця цих зсувів для етилового спирту в рідкому стані і в сильно розбавленому розчині (у СС14) становить 3 мли-1.
і Хімічні властивості. Спирти є високореакційноздатнимисполу^ каїми завдяки .наявності в їх молекулах функціональної групи ОН» У молекула^ спиртів міститься полярні зв’язки О—Н і С—О, які легко вступають у різноманітні реакції. V ' Ч-
К ислоти о-основні властивості хімічних сполук. Уявлення про кислотні і основні властивості речовин складалися і вдосконалювалися в міру загального прогресу хімічних наук. У класичному визначенні за Оствальдом — Арреніусом (1880—1890 рр.) кислотами називають сполуки, які при електролітичній дисоціації у водному розчині утворюють іони водню Н+, а основами —сполуки, які в тих самих умовах утворюють іони гідроксилу ОН-. Таке визначення включає в групу кислот і основ порівняно невелику КІЛЬКІСТЬ ХІМІЧНИХ сполук. Більш загальне ПОНЯТТЯ кислот і основ, яке охоплює значно більшу кількість сполук і реакцій, було сформульоване в теорії Бренстеда — Лоурі (1923— 1934 рр.), згідно з якою кислоти — це донори протонів, тобто сполуки, здатні віддавати протони (протонізуватися), а основи—це акцептори протонів, тобто сполуки, що приєднують протони.
Паралельно з теорією Бренстеда—Лоурі виникла і розвинулась концепція узагальнених кислот і основ, запропонована Дж. Льюї-сом, у якій дається ще ширше поняття кислотно-основних властивостей хімічних сполук. За Дж. Льюїсом, кислоти — не сполуки, які є донорами протонів або акцепторами електронів. Кислотами Льюїса можуть бути будь-які молекули або іони, здатні приєднувати частинки з неспареними (вільними) парами електронів і утворювати з ними хімічний зв’язок. У кислотах Льюїса, в крайньому разі, один з атомів водню має вакантну електронну орбіта ль. Так, НС1, Н3С—СООН тощо самі по собі не є кислотами, кислотою буде протон Н+ (акцептор електронів), який вони генерують. До кислот Льюїса крім протоновмісних сполук відносять велику кількість апротоиних кислот, які не містять атомів водню, зокрема ВР3, А1С13, РеС1а, 8О3, СІ*, N0?, карбкатіони /?3С+, /?—С+=Ота ін. Кислоти Льюїса є електрофільними реагентами.
Основи Льюїса — це речовини, які є донорами електронів або акцепторами протонів. До них належать аніони, атоми або молекули,які мають хоча б одну вільну пару валентних електронів. До основ Льюїса відносять аніони ОН-, Р-, СІ-, Вг-, І-, Л/Ю-, С(ІН5О-, 7?СОО-, Д3Н, 7?—ОН, карбаніони 7?аС-, а також по’ двійні (С=С), потрійні (С=С) зв’язки, бензольне ядро [С2)} ’ які мають л-електрони, що легко збуджуються. Основи Льюїса є нуклеофільними реагентами.
76
Кислотність і основність — це відносні властивості, які виявляються тільки при наявності компонентів кислотно-основної взаємодії: сполука, яка потенціально здатна бути кислотою, стає нею тільки при наявності основи, і навпаки, сполука, потенціально здатна бути основою, стає нею тільки при наявності кислоти. Отже, кислота і основа є спряженими кислотно-основними парами. Залежно від типу реагента або розчинника одна і та сама речовина може виявляти як кислотні, так і основні властивості.
Теорія кислот і основ була розвинута головним чином в застосуванні до кисневмісних кислот (О—Н-кпслот) і основ. Проте положення цієї теорії придатні і до інших типів речовин, зокрема до сполук, які мають здатність дисоціювати з розривом МН-зв’язків (МН-кислоти), С—Н-зв’язків (С—Н-кислоти), зв’язків між атомами водню і галогену,і взагалі зв’язку будь-якого хімічного елемента з воднем.
Органічні сполуки, молекули яких містять групи О—Н, М— Н, 8—Н, мають значну кислотність за Бренстедом—Лоурі. Ступінь кислотності зв’язку за Бренстедом—Лоурі визначається головним чином характером атома, з яким сполучений водень, і, зокрема, здатністю цього атома утримувати електронну пару після того, як від нього «відійде» іон водню. Здатність атома утримувати електронну пару залежить від різних факторів, у тому числі від електро-негативності атомів і їх розмірів. Отже, в періодах таблиці Д. І. Менделєєва кислотність зростає із збільшенням електронега-тнвності атомів:
Кислотність: Н—СН3 < Н—МН2 < Н—ОН < Н—Г; Н—8Н < Н—СІ.
У групах кислотність зростає в міру збільшення розмірів атомів:
Кислотність: Н—Р < Н—Сі < Н—Вг < Н—І; Н—ОН < Н—8Н.
Ступінь основності певного хімічного зв’язку визначається в основному природою атома, який несе вільну пару електронів, його електронегатнвністю, розміром і зарядом. При цьому зберігається така залежність: чим сильніше атом утримує електронну пару, тим вона менш доступна для утворення зв’язку, тим слабкіші основні властивості зв’язку.
Дальшим розвитком вчення про кислотно-основні властивості стало введення поняття про жорсткі і м’які кислоти і основи (ЖМКО). Принципи теоріїЖМКО сформулював у 1963р.Р. Пірсон.
До жорстких кислот належать малі за розміром електроно-акцепторні частинки, які мають високий позитивний заряд і не мають у своїх валентних оболонках електронних пар. Для жорстких кислот характерні висока електронегативність і низька поля-ризованість. Наприклад, до жорстких кислот належать Н+, №+, К+, М§2+, Са2+, А13+, А1С13 та ін.
М’які кислоти мають електроноакцепторні атоми великих розмірів, але з невеликим позитивним зарядом, а у своїй валентній
77
електронній оболонці містять вільні пари р- або ^-електронів. М’які кислоти мають високу поляризованість і низьку електронегативність. До м’яких кислот належать Сн+, А^+, Н§+, Н§2+, І+, Вг + тощо.
До жорстких основ відносять сполуки з електронодонорними атомами, які мають високу електронегативність і низьку поляризованість. До жорстких основ належать ОН“, Е~, Н3С—СОО~, РОІ-, 8ОІ-, С1~, /?О", Н2О, Я— О—Н, Я2О. ЬІН3 та ін. Жорсткі основи міцно утримують свої валентні електрони.
Для м’яких основ характерні низька електронегативність, висока поляризованість, вони легко окислюються і порівняно слабко утримують валентні електрони. До м’яких основ належать І-, 8Сі\", Я8-, С1М-, СО, Н2С=СН, та ін.
Найміцніший зв’язок утворюється в результаті взаємодії між жорсткими кислотами і жорсткими основами. Більшість комбінацій між м’якими кислотами і м’якими основами нестійкі.
; К ис лоти о-основ н і властивості спиртів. Наявність у молекулах спиртів гідроксильної групи зумовлює їх кислотно-основні властивості. Оскільки атом кисню цієї групи більш електронегативний, ніж атоми вуглецю та водню, то електронна густина зміщується від цих атомів до кисню. Зв’язки О—Н і С—О поляризуються:
Н
|е+ 6. е+
Р—С —> О ч— н.
І н
Тому в хімічних реакціях спирти можуть віддавати протон і виявляти, таким чином, кислотні властивості. Крім того, на атомі кисню гідроксильної групи спирту є вільні пари електронів. За рахунок цих електронів (у реакціях виявляє активність тільки одна електронна пара) спирти можуть також приєднувати протон до атома кисню ОН-групи і виявляти при цьому основні властивості. Отже, спирти, згідно з теорією Бренстеда — Лоурі, матимуть кислотно-основні властивості.
Для спиртів існують реакції, які відбуваються як з участю атома водню гідроксильної трупи (кислотні властивості), так і з участю всієї гідроксильної групи (основні властивості). Отже, спирти мають амфотерні властивості. Але як кислотні, так і основні властивості в них виражені дуже слабко. Тому розчини спиртів у воді нейтральні і не змінюють забарвлення індикаторів. Константа іонізації для метилового спирту дорівнює приблизно 1 • 1017, для етилового спирту — 1 • 10-18, а для води — 1,8 • 10“1в, тобто спирти порівняно з водою мають слабкіші кислотні властивості.
Реакції з участю водню гідроксильної групи. Спирти —практично нейтральні речовини, але атом водню гідроксильної групи, у зв’язку з її високою полярністю, може виявляти кислотні властивості. Так, спирти реагують з активними металами. При ньому виді-
78
ляється водень гідроксильної групи, який заміщується в процесі реакції на метал. Продуктами такої взаємодії є металічні похідні, які називають алкоголятами-. '• '
27?—ОН2Иа2/? — Оі\а.+Н2. /А
Назву алкоголятів утворюють'з назви вуглеводневого радикала спирту, додаючи до неї суфікс -ат і назву металу. Наприклад, метилат натрію Н3С—О№, етилат натрію Н3С—СН2—О№ і Т. д.
Алкоголяти—тверді речовини, легко розкладаються водою, яка є сильнішою кислотою, ніж спирт:
7?—О№ 4- НОН 7?—ОН + ИаОН.
Спирти активно вступають у реакцію з магнійорганічними сполуками. При цьому утворюються насичені вуглеводні і галогенал-коголяти магнію:
Н3С—СН2—ОН + Н3С—М§—І —> СН4 -р Н3С—СН2—О—М§—І.
Йодетилат магнію
Цю реакцію використовують для визначення кількості гідроксильних груп у молекулі спирту, яку легко встановити, вимірявши об’єм насиченого газоподібного вуглеводню, наприклад метану (реакція Чугаєва —Церевітинова).
Кислотні властивості спиртів змінюються при введенні в їх молекулу інших атомів або груп атомів. Так, якщо в молекулу спирту ввести атом або групу атомів, що мають негативний індукційний ефект і викликають зміщення електронної густини сусідніх зв’язків на себе, то ці атоми або групи атомів збільшують полярність зв’язку О—Н і, отже, підвищують кислотні властивості спиртів:
СІ
СІ — 11,С — СІІ,*- О — І!; СІ — С — СН-і — О — Н С'Х
Тому ступінь дисоціації 2-хлоретанолу в 10 разів більший, ніж метанолу, а 2,2,2-трнхлоретанолу—ще в 10 разів підвищується порівняно з 2-хлоретанолом. З тієї ж причини спирт (Е3С)3С—О—Н стає настільки кислотним, що може витісняти вугільну кислоту з її солей.
Введення в молекулу спирту алкільних радикалів також впливає на його кислотні властивості. Алкільні радикали, маючи позитивний індукційний ефект, збільшують електронну густину на атомі кисню гідроксильної групи, що призводить до зниження стійкості аніонів 7?—СН2—О". У зв’язку з цим імовірність відщеплення протона від такого спирту зменшується, а відповідно зменшується і здатність спирту утворювати такого типу аніони. Ще склабкіше виражені кислотні властивості у вторинних і особливо
79
третинних спиртів (за рахунок позитивного індукційного впливу трьох радикалів):
СН3
І
Н3С->С-*О—н.
і сн3
За здатністю відщеплювати водень у вигляді протона спирти можна розмістити в такий ряд:
Діетиловий Первинні Вторинні Третинні спирт 'спирти '> спирти ^спирти.
У наведеному ряду кислотні властивості найсильніше виражені в метилового, найслабкіше — у третинних спиртів.
Спирти виявляють і основні властивості. Так, вони вступають у взаємодію з сильними кислотами. При цьому протон кислоти займає вільну електронну пару атома кисню групи —ОН і приєднується до нього координаційним зв’язком. Така взаємодія приводить до утворення позитивно зарядженого іона алкоксонію, який є аніоном кислоти і дає відповідну сіль оксонію:
І1 + н+Вг—>/?—О—Н -р Вг".
'’І_____| Н ; .
Основні властивості змінюються порівняно з кислотними в протилежному напрямку. Групи атомів, які збільшують електронну густину на атомі кисню групи —ОН, підвищують швидкість приєднання протона кислоти до атома кисню цієї групи і підсилюють таким чином основні властивості спирту. За здатністю приєднувати протон спирти можна розмістити в такий ряд:
Третинні Вторинні _ Первинні Метиловий спирти спирти спирти "> спирт.
У цьому ряду основні властивості найсильніше виражені в третинних, найслабкіше — в метилового спирту.
' Реакції нуклеофільного заміщення гідроксильної групи. Під час взаємодії спиртів з галогено-водневими кислотами або галогеноводнями, галогенідами фосфору абозтіонілхлоридом гідроксильна група в їх молекулах заміщується на галоген.
Реакція з галогеноводнями. Спирти легко реагують з галогеновед-нями і утворюють при цьому алкілгалогеніди. Ця реакція є одним з найважливіших методів добування алкілгалогенідів. У реакцію вводять сухий газоподібний галогеноводень, який пропускають крізь спирт, або ж спирт нагрівають з концентрованим розчином кислоти. Реакційна здатність спиртів до реакції заміщення ОН-групи на галоген зменшується в ряду:
Третинний Вторинний Первинний спирт '> спирт спирт.
80
Галогеноводні в реакції із спиртами також виявляють різну активність. Найактивнішим є йодоводень, найменш активним — хлороводень. Отже, за активністю галогеноводні можна розмістити в такий ряд:
НІ>НВг>НС1.
Так, найменш реакційноздатний галогеноводень НС1 реагує з первинними спиртами тільки при наявності ХпС12, а найбільш реакційноздатні третинні спирти (наприклад, тре/и-бутиловий спирт) легко реагують з концентрованою соляною кислотою і при кімнатній температурі швидко перетворюються на третишуі алкіл-хлориди:
/пСІ,
Я—ОН + НС1^=&7?—СІ + Н2О;
(Н3С)3С—ОН + НСІ^(Н3С)3С-С1 + Н2О.
Реакція спиртів з галогеповоднями каталізується кислотами. Навіть у тому випадку, коли водний розчин галогеноводню є сильною кислотою, добавляння до реакційної суміші сірчаної кислоти прискорює утворення алкілгалогеніду. Вважають, що каталітична дія кислот пов’язана з утворенням іона алкоксонію /?ОН2, який потім вступає в реакцію нуклеофільного заміщення з іоном галогену. Причому третинні і вторинні спирти реагують за ЗдЛ-меха-нізмом, а первинні спирти —-за 5#2-механізмом. Заміщення гідроксильної групи третинного і вторинного спирту на галоген відбувається в декілька стадій:
7?3С—ОН + Н+Х-«=і/?3С—ОН+ + X"; /?3С - ОІі+^=>/?3С+ + НВО; /?3С+ + Х-->/?3С—X.
(1)
(2)
(3)
Спочатку спирт приєднує іон водню і утворює іон алкоксонію (стадія 1), який далі дисоціює на воду і карбонієвнй іон (стадія 2— найповільніша). Карбонієвнй іон потім швидко приєднує іон галогену і утворює алкілгалогенід (стадія 3). Підтвердженням такого Злгі-механізму служить той факт, що при заміщенні ОН-групи в третинних і вторинних спиртах на галоген спостерігається перегрупування алкільних радикалів: галоген не завжди утворює зв’язок з тим атомом вуглецю, який спочатку був сполучений з гідроксильною групою. Наприклад:
СН3 СН3 / СН3 ч
І +нсі 1/11
Н3С—СН—СН—СН8------->Н3С—С—СН2—СН3І а не Н3С—СН—СН—СН3 І.
он сі \ сі /
3-М етилбутавол-2 2»Метил-2-хлорбутан
Наявність такого роду перегрупувань свідчить про утворення в процесі реакції заряджених проміжних частинок — карбонієвих іонів.
61
ПІД час реакції заміщення гідроксилу на галоген у первинних спиртів алкільні радикали перегрупувань не зазнають. Тому вважають, що первинні спирти реагують за 5л'2-механізмом, тобто через стадію утворення перехідного стану:
с-
х- + яон+-дх... Я • • • ОН2]->Х—Я + н2о.
Хлороводень і бромоводень легко вступають у реакцію в момент утворення, наприклад, якщо на суміш солі №С1 або І\Вг з спиртом діяти концентрованою Н28О4. При цьому спирт спочатку взаємодіє з сірчаною кислотою і утворює складний ефір цієї кислоти, який, наприклад, у випадку етилового спирту називається етилсірчаною кислотою:
Н3С—СН2ОН 4- НО8О3Н->Н3С—СН2—О—8О3Н 4- Н2О.
Етил сірчана кислота
Галогеноводнева сіль утворює з Н28О4 галогеноводень, який, взаємодіючи з ефіром сірчаної кислоти, дає галогеналкіл:
ИаСІ 4- Н28О4->НС14- КаН8О4; КВг 4- Н28О4->НВг 4- КН8О4;
НВг ПСІ
Н28О4 4- Н3С—СН„—Вг< Н3С—СН2—О—8О3Н------>Н,С— СН2— СІ 4- Н,8О4.
Взаємодія з галоїдними сполуками фосфору і з хлористим тіонілом. Галоїдні сполуки фосфору при дії на спирти легко заміщують їх гідроксильну групу на галоген:
ЗЯ—он 4- рсі3 -> зя—сі 4- н3РО3;
я—ОН 4- РС1В -> Я—сі 4- неї 4- РОС13.
При взаємодії РС13 з первинними спиртами як побічний продукт утворюється складний ефір фосфористої кислоти:
зя—он 4- РС13 -> (ЯО)3Р + ЗНС1.
Для реакції із спиртами можна брати не готові РВг3 і РІ3, а вводити в реакційну суміш окремо фосфор і галоген. У цьому випадку з спиртами реагують галогеніди фосфору в момент їх утворення:
2Р 4- ЗІ2 = 2РІ3; ЗЯ—ОН 4- РІ3 = ЗЯ—14- Н3РО3.
Хлоропохідні з високим виходом добувають при дії на спирти хлористого тіонілу 8ОС12:
Я—ОН 4- 8ОС12 -> Я—СІ 4- НС14- 8О2.
У цьому випадку утворюються газоподібні НС1 і 8О2, які легко відділяються від галогеналкану.
Вважають, що спирт спочатку взаємодіє з 8ОС12 і утворює ал-кілхлорсульфіт (І), який потім утворює внутрішню іонну пару (II), а вона далі розкладається із збереженням конфігурації:
:рО
Я - ОН + СІ - 8ОСІ ~НС| • Я ^8 = 0-* Я—^8 = О — Я - СІ + 8О2 .
:сі :с/
і 11“
82
Реакція етерифікації. Спирти вступають у реакцію з мінеральними і органічними кислотами. Продуктами такої взаємодії є відповідні складні ефіри. Якщо в реакцію із спиртом вступає багатоосновна кислота, то залежно від співвідношення кислоти і спирту, введених у реакцію, можуть утворюватися або кислі, або середні ефіри:
,О—Н , ,О—Н, 9Г н
С2Н5ОН + н-о-р/ ->с»нь4о-р/ ."-І-
А 2 , г II \о-II їЛо-н
- - ?. Л- о о
Етил фосфорна
кислота
(кислий ефір)
->(С2Н5О)3Р = О + Н2О;
Триетилфосфат (середній ефір)
,О ,0
тс—с-^ + по—с,н Дн3с—+ н2о.
хон 9 ' хо—с*н5.
Оцтовоетил ОЕИЙ ефір, етилацетат
Дегідратація спиртів. Дегідратація спиртів може відбуватися у двох напрямках: внутрішньомолекулярно і міжмолекулярно. Напрямок дегідратації залежить від природи спирту і умов проведення реакції.
При внутріпіньомолекулярній дегідратації спирту утворюється ненасичений етиленовий вуглеводень, в результаті міжмолекулярної дегідратації — простий ефір. Так, при нагріванні спиртів з такими водовідбирними речовинами, як концентрована Н28О4, Н3РО4, безводна щавлева кислота, оксид алюмінію тощо, утворюються ненасичені сполуки етиленового ряду.
Реакційна здатність спиртів до дегідратації, тобто до утворення з них етиленових сполук, змінюється в такому порядку:
Третинні Вторинні Первинні спирти спирти спирти.
Деякі третинні спирти дегідратуються настільки легко, що їх можна перегнати тільки у випадку, якщо запобігти попаданню в них навіть лабораторного повітря, яке містить у незначних кількостях пару кислот.
Дегідратація спиртів при наявності концентрованої Н28О4 залежно від температури, співвідношення об’ємів спирту і кислоти може відбуватися з утворенням різних продуктів. Наприклад, етиловий спирт при 105° С утворює з сірчаною кислотою кислий складний ефір —етилсірчану кислоту (реакція 1). При надлишку спирту і вищій температурі (130—140 °С) відбувається міжмолекулярна дегідратація, головним продуктом якої є діетиловий ефір (простий ефір; реакція 3). При температурі понад 160°С етилсірчана кислота розкладається з утворенням етилену (реакція 2):
сХ'ОН-
Н25О4
105°С —Н,О 160°С
—що +с2н,он
-Н,0 *
С2Н/)5О3Н;
НВС = СН2;
с2гі3—о—санв
(1)
(2)
(3)
63
О. М. Зайцев (1875 р.) встановив, що при відщепленні води від спиртів утворюються найбільш алкіловані етиленові вуглеводні (правило Зайцева). Наприклад, при дегідратації З-метилбутанолу-2 відщеплення води відбувається за рахунок гідроксилу і атома водню, який знаходиться біля сусіднього найменш гідрогенізованого Р-вуглецевого атома. В результаті утворюється 2-метилбутен-2:
сн3 он сн8
Відщеплення води від спиртів за правилом Зайцева пов’язане з утворенням у ході реакції проміжних найстійкіших карбкатіо-нів, які потім перетворюються на найбільш алкіловані етиленові вуглеводні, термодинамічно стійкіші від менш алкілованих алкенів.
Спирти легко дегідратуються і в газовій фазі над нагрітим оксидом алюмінію або пористою глиною. Наприклад, з етилового спирту за цим методом добувають етилен з високим виходом:
Н3С—СН2—ОН Ац°н го°°С^ = СН2.
Алкілування спиртів. Алкілуванням називають реакції введення в молекули органічних речовин алкільних радикалів. При введенні алкільного радикала в молекулу спирту замість атома водню ОН-групи добувають прості ефіри. Алкілувати спирти можна спиртами, як це відбувається при їх міжмолекулярній дегідратації:
---------- н+
/?—о—;н + н—оі—-> д—о—/? + н2о.
Можна алкілувати спирти галоїдними алкілами. Однак спирти є слабкими нуклеофілами і з галоїдними алкілами взаємодіють надзвичайно повільно. Якщо реакцію галоїдних алкілів проводити не з спиртами, а з алкоголятами, то прості ефіри утворюються швидко, оскільки алкоголят-іони — сильні нуклеофіли і алкілування в цьому випадку відбувається дуже активно:
6+ в- —і-
Н3С—І + С2Н6(Жа-*Н3С—О—С2Н8 + №1.
Метилетиловий ефір
Алкілувати алкоголяти можна і діалкілсульфатами:
' /?—СЖа + О—/?' + Д'О—8О3Ж
Взаємодія алкоголятів з алкілгалогенідами є загальним методом добування простих ефірів, відомим під назвою синтезу Вільям-сона.
Реакція алкоголятів з галогеналкілами відбувається за 5д>2-ме-ханізмом:
б- б-
/?—О" + Х-ЯЯ—О %]->/?—О—/?' +
84
Окислення спиртів. Спирти при 300—500 °С і наявності мідних та інших каталізаторів окислюються киснем повітря. Такі окислювачі, як КМпО4, хромова суміш, окислюють спирти вже при кімнатній температурі. Залежно від того, який це спирт —первинний, вторинний чи третинний, — при окисленні утворюються різні продукти.
Первинні спирти при окисленні дають альдегіди з такою ж кількістю вуглецевих атомів, як і в молекулі вихідного спирту. Альдегіди в цих умовах можуть окислюватись до карбонових кислот. Щоб запобігти дальшому окисленню, альдегіди необхідно швидко виводити з реакційної суміші:
/О—|Н /ОІЛ1 ЛО
Я—Н + [О]->Н2О +/?——->/?—С<^ .
ХН І \н 4 он
Первинний Альдегід Карбонова
спирт кислота
Окислення спиртів хромовою сумішшю супроводиться зміною оранжево-червоного забарвлення реакційної суміші (іони Сг2О7 ) на зелене (іони Сг3+):
.О
ЗН3С—СН2—ОН + К2Сг2О74- Н28О4-^ЗН3С-=-С^” 4- Сг2 (5О4)34-
. ' хн
Оцтовий
альдегід
4-К28О4 +7Н2О.
Первинні спирти можна також окислити до альдегідів тонко подрібненою міддю, нагрітою до 280—300 °С. У цих умовах від молекули спирту відщеплюється два атоми водню і в молекулі органічної речовини, яка при цьому- утворюється, з’являється подвійний зв’язок вуглець—кисень (—С=О). Таке перетворення
І спиртів називають дегідруванням'.
—
р с"
п 300сС
Вторинні спирти при окисленні, а також при дегідруванні перетворюються на кетони:
/?-сн-/? /?-С-/? 4- На.
ОН О
Вторинний Кетон
спирт
Наприклад, втор-пропіловий спирт у цих умовах перетворюється на кетон, який називають ацетоном:
ЗН3С—СН—СН3 4- 2КМпО4 -> Н3С—С—СН34-2МпО, 4- 2КОН 4- 2Н.О.
І II
ОН О
Пропанол-2 Диметилкетон
(ацетон)
Третинні спирти окислюються Досить важко з одночасним розривом вуглецевого ланцюга їх молекул і утворенням сумі ші карбонових кислот і кетонів. Таке окислення цих спиртів пов’язане з тим, що в умовах реакції окислення вони дегідратуються і перетворюються на етиленові вуглеводні, які при наявності сильного окислювача окислюються з розривом молекули за місцем подвійного С=С-зв’язку (див. розд. 14):
СН3 сн3
Н3С--СН2—С—СН3 кМп°а, Н25О4^ с_сн=[;_сн , н 0.
ОН сн, І /С
5Н3С—СН=С—СН3 4- 6КМпО4 + 9Н2$О4 -> 5Н3С—С" +
ХОН
Оцтова кислота
+ 5Н3С— С—СН3.
II О Диметилкетон (ацетон)
Отже, за продуктами окислення спиртів можна зробити висію, вок про будову спирту.
Ідентифікація спиртів. Спирти можна відрізнити від інших класів органічних речовин за їх кольоровою реакцією з оксидом хрому (VI) в середовищі сірчаної кислоти. Хромовий ангідрид СгО3 у водному розчині Н28О4 окислює первинні та вторинні спирти і протягом приблизно двох секунд прозорий оранжевий розчин реакційної суміші стає голубувато-зеленим і мутнішає. Третинні спирти цієї реакції не дають.
Для того щоб встановити, яким є даний спирт-—первинним, вторинним чи третинним, користуються пробою Лукаса. Ця проба грунтується на різній реакційній здатності цих спиртів відносно галогеноводнів. Спирти, молекули яких містять не більше шести вуглецевих атомів, розчиняються в реактиві Лукаса, який являє собою суміш концентрованої соляної кислоти і хлориду цинку. При розчиненні спирту в цьому реактиві утворюються відповідні алкілхлориди, про утворення яких свідчить помутніння розчину, оскільки алкілхлориди в реактиві Лукаса не розчиняються. Час, необхідний для появи помутніння реактиву Лукаса, є мірою реакційної здатності спирту. Третинні спирти реагують з реактивом Лукаса відразу ж, вторинні спирти — протягом 5 хв, а первинні спирти при кімнатній температурі з цим реактивом майже не взаємодіють.
Етиловий спирт і спирти з загальною формулою Н3С—СН—/?
ОН взаємодіють у лужному середовищі з йодом і утворюють йодоформ СНІ8 (йодоформна реакція). За здатністю давати йодоформну реакцію можна відрізнити дані спирти від інших спиртів.
66
Окремі представники. Метиловий спирт (метанол, карбінол, або деревний спирт) Н3С—ОН відомий з 1834 р. Він являє собою прозору рідину із специфічним запахом, який нагадує запах етилового спирту. Змішується з водою в усіх співвідношеннях. Основним промисловим методом його добування є гідрування оксиду вуглецю (II), яке відбувається під тиском при наявності каталізаторів (7пО, Сг2О3) і температурі 400 °С:
СО + 2Н. НоС—ОН.
1 а /ПО, *>
Раніше метиловий спирт добували сухою перегонкою деревини, тому його назвали деревним спиртом.
Метиловий спирт широко застосовується в промисловості для добування формальдегіду, органічних барвників, фармацевтичних препаратів та як розчинник лаків і політур. Його використовують як вихідну речовину для введення метильної групи в різні органічні сполуки.
При роботі з метиловим спиртом слід пам’ятати, що він надзвичайно отруйний. Попадання в організм кількох грамів (~10 мл) метилового спирту викликає сліпоту, а дещо більші кількості (~30 мл) — смерть. У зв’язку з цим метиловий спирт використовують тільки для технічних потреб. На склянці з речовиною слід обов’язково робити напис «Метанол — отрута».
Етиловий спирт (етанол) Н3С—СН2ОН — безбарвна рідина з характерним спиртовим запахом. З водою етанол змішується в усіх співвідношеннях, з нею він утворює азеотропну суміш складу: 95,57 % спирту і 4,43 % води. Дану суміш неможливо розділити фракційною перегонкою. В процесі очистки (ректифікації), яку проводять ретельною фракційною перегонкою, одержують 95 %-й спирт — ректифікат. Для добування безводного (абсолютного) спирту водний спирт нагрівають протягом тривалого часу з СаО, з безводним Си8О4 та іншими речовинами, які хімічно зв’язують наявну в спирті воду. Зневоднити етанол за допомогою безводного СаС12 не можна, оскільки спирт з хлоридом кальцію утворює комплекс складу СаС12 * 4С2Н5ОН.
Етиловий спирт — багатотоннажний продукт хімічної промисловості. За об’ємом виробництва він посідає перше місце серед інших органічних продуктів. Його добувають різними методами. Один із них —спиртове бродіння цукристих речовин, наприклад, глюкози, при наявності дріжджів:
СеН12О6 Др1ждж1^ 2И3С—СН2—ОН + 2СО2.
Глюкоза
Необхідна для цього процесу глюкоза або міститься в готовому вигляді, наприклад у виноградному соку, або її одержують гідролізом крохмалю. Спирт, добутий цим методом, називають винним, або харчовим. Крохмаль міститься в таких харчових продуктах, як пшениця, жито, ячмінь, кукурудза, рис, картопля. У наш час для виробництва етанолу для технічних потреб харчові продукти пов
87
ністю замінено нехарчовими. Так, крохмаль був замінений клітковиною (целюлозою), яка у великих кількостях міститься у відходах деревообробної, целюлозної та лісової промисловості (тирса, стружки, гілки, хмиз тощо). Клітковину спочатку гідролізують:
(СвН10О5)п -р /гН2О |5о „ і70°с яСвН12Об.
Клітковина В-Глюкоза
Добуту таким чином глюкозу піддають бродінню. Етиловий спирт, одержаний цим методом, називають гідролізним. Гідролізний метод добування етанолу вигідніший, оскільки дає можливість економити великі кількості харчових продуктів. 3 1т тирси можна добути До 200 кг етанолу. Проте гідролізний спирт містить домішки, в тому числі й отруйний метанол, тому його використовують тільки для технічних потреб.
Широкого застосування в промисловості набув метод добування етилового спирту шляхом гідратації етилену при наявності Н2ЗО4 або Н3РО4:
Н2С = СН2 + НОН н3с—сн2—он.
Етиловий спирт має широке застосування. Важко назвати галузь народного господарства, де б не застосовувався етанол. Перш за все, він використовується для добування синтетичного каучуку, оцтової кислоти, органічних барвників, діетилового ефіру, складних ефірів, пахучих речовин (фруктові есенції), фармацевтичних препаратів тощо. Етиловий спирт має антисептичні властивості і використовується в медицині, зокрема в хірургії для знезараження хірургічних інструментів, рук хірурга, операційного поля. Етанол—прекрасний розчинник. Для органічної хімії він має таке саме значення, як для неорганічної хімії —вода. Застосовують етиловий спирт також у парфюмерії, харчовій промисловості.
Етиловий спирт у невеликих дозах викликає сп’яніння, а у великих — зумовлює стан, близький до наркозу, з загальною нечутливістю, який може закінчуватись смертю. Отже, за дією на організм етиловий спирт є наркотиком і може викликати захворювання, відоме під назвою алкоголізм.
Вищі спирти. У природі трапляються цетиловий С16Н33—ОН і мірициловий С31Н63—ОН спирти. Перший з них у вигляді складно
ного ефіру пальмітинової кислоти С15Н31—Об складає
О—С1вН33 основну частину спермацету, який добувають з черепної порожнини китів і застосовують як високоякісне мастило для прецизійних інструментів, а також у парфюмерній та текстильній промисловості. Мірициловий спирт у вигляді ефіру пальмітинової кислоти
С15Н31—С<^ входить до складу бджолиного воску.
О с31н63
88
Двохатомні спирти
Спирти, молекули яких містять дві гідроксильні групи, називають двохатомними, або гліколями. Загальна формула двохатомних спиртів СпН2,г(ОН)2. Двохатомні спирти утворюють гомологічний ряд, який можна легко записати, користуючись гомологічним рядом насичених вуглеводнів, замінюючи в їх молекулах два атоми водню на гідроксильні групи.
Першим і найважливішим представником двохатомних спиртів є етиленгліколь СН2ОН—СН2ОН. Ттт = 197 °С. З нього виготовляють антифриз.
Стійкими є гліколі, в молекулах яких гідроксильні групи розміщені біля різних вуглецевих атомів. Якщо ж дві гідроксильні групи знаходяться біля одного вуглецевого атома ,то такі двохатомні спирти нестійкі, легко розкладаються, відщеплюючи за рахунок гідроксильних груп воду, і перетворюються на альдегіди або кетони:
।—, ОтН і
/ І
/? —СН І
А.іьдсі ід
------► /? — с — /?
-н,о II о
Кетон
Номенклатура. Залежно від взаємного положення гідроксильних груп розрізняють а-гліколі (у них гідроксильні групи розміщені біля сусідніх вуглецевих атомів, що стоять поряд, в положенні 1,2), р-гліколі (ОН-групи в них розміщені в положенні 1,3), у-гліколі (ОН-групи—в положенні 1,4), б-гліколі і т. д. Наприклад: а-гліколь —СН2ОН—СНОН—СН2—СН3, р-гліколь — СН2ОН—СН2—СНОН—СН3, у-гліколь—
СН2ОН—СН2—СН2^СН2ОН.
За раціональною номенклатурою назву а-гліколів утворюють від назви відповідного етиленового вуглеводню, до якої додають слово гліколь. Наприклад, етиленгліколь, пропіленгліколь і т.д.
За систематичною номенклатурою назву гліколів утворюють з назви насиченого вуглеводню, до якої додають суфікс -діол, вказуючи номери вуглецевих атомів, біля яких знаходяться гідроксильні групи. Наприклад, етиленгліколь СН2ОН—-СН2ОН за номенклатурою ІЮПАК—етандіол-1,2, а пропіленгліколь СН3—СНОН—СН2ОН —пропандіол-1,2.
Ізомерія. Ізомерія двохатомних спиртів залежить від будови вуглецевого ланцюга: СН2ОН—СН2—СНОН—СНУ і СН2ОН—СН—СН2—ОН, а також від положення гідроксильних
СН3
груп у молекулі спирту, наприклад, пропандіол-1,2 і пропан-діол-1,3.
89
Методи добування. Гліколі можна добути такими методами: 1. Гідролізом дигалогенопохідних насичених вуглеводнів:
Н3С—СН—СН, 2Н0Н Н,С—СН—СН, 4- 2НС1.
II II'
СІ СІ он он
2. Гідролізом галогеноспиртів:
СН,—СН, + НОН -> СН,—СН, + НС1.
11“ І ' І
СІ он он он
3. Окисленням етиленових вуглеводнів перманганатом калію або надмурашиною кислотою (див. розд. 14).
4. Гідратацією сс-оксидів:
н+
н2с— СН2 + нон--* СН2ОН—СН2ОН.
6
Оксид етилену
5. Бімолекулярним відновленням карбонільних сполук (див. розд. 17).
Хімічні властивості. Хімічні властивості гліколів аналогічні властивостям одноатомних спиртів і визначаються наявністю в їх молекулах двох гідроксильних груп. Причому в реакціях можуть брати участь одна або обидві гідроксильні групи. Однак внаслідок взаємного впливу однієї гідроксильної групи на другу (особливо у а-гліколів) кислотно-основні властивості гліколів дещо відмінні від аналогічних властивостей одноатомних спиртів. У зв’язку з тим що гідроксил має негативний індукційний ефект, то одна гідроксильна група відтягує електронну густину від другої подібно до того, як це робить атом галогену в молекулах галогензаміщених одноатомних спиртів. В результаті такого впливу кислотні властивості двох атомних спиртів порівняно з одноатомними підвищуються:
Н—О СН2 СН2 О Н.
Тому гл'іколі, на відміну від одноатомних спиртів, легко вступають у реакцію не тільки з лужними металами, але й з лугами і навіть з гідроксидами важких металів. З лужними металами, лугами гліколі утворюють повні і неповні алкоголяти (гліколяти):
СН2—ОН а СН,—ОН СН2—ОК’а
н2 + | <-±^-| ±^| * +Н2.
СН2—ОН СН2—ОН СН2—ОИа
З гідроксидами деяких важких металів, наприклад з гідроксидом міді, гліколі утворюють комплексні гліколяти. При цьому не розчинний у воді Си(ОН)2 в гліколі легко розчиняється:
Н
СН,—ОН СН,—О х ,:О—СН,
2 | +Си(ОН),->| ,Сиб | .
СН2—ОН ' СН2—О:'' 4 О—СН2
Н
80
Мідь у цьому комплексі утворює з атомами кисню два ковалентних зв’язки і два —координаційних. Реакція є якісною на двохатомні спирти.
Гліколі можуть утворювати повні і неповні прості і складні е ріри. Так, взаємодією неповного гліколяту лужного металу з галоїдними алкілами добувають неповні прості ефіри, а з повного гліколяту — повний простий ефір:
СН„—ОЬ’а СН2—О—С.,Н5
| + Сі — С2Н5-> НаСІ| }
сн2—ОН .. . ‘ сн2—он
Моноетиловий ефір етилен* гліколю, етилцелозольв
СН2—О№ СН2—О—С2Н5
| + 2С1 — С2Н3 -> 2ИаС1 + |
СН2—ОИа СН2—О—С2Н8
Діетиловий ефір етиле игл і колю
Метил- і етилцелозольви застосовують як розчинники у виробництві лаків, бездимного пороху (піроксиліну), ацетатного шовку ТОЩО.
З органічними і мінеральними кислотами двохатомні спирти утворюють Два ряди складних ефірів:
СН„—О—N0, СН2—ОН СН2—О—N0,
| ‘ НОМОа І 2ИОКО, І
"--ЩО | -2Н2О І
СН2—ОН СН2—ОН СН2—О—ЬЮ2
Мононітрат Дині трат
етиленгліколю етиленгліколю
Динітрат етиленгліколю —сильна вибухова речовина, яку використовують замість нітрогліцерину.
Окислення гліколів відбувається ступінчасто, з участю однієї або обох гідроксильних груп одночасно і з утворенням таких продуктів:
.О ,о ,о с^ с^ “до! Н.яїНг0"™.І " Е .0 /О*" сн2он 1 (і/ с<у СН2ОН \он хн Щавлева Гліоксаль Гліколе* кислота вий альдегід <ХОН хон Щавлева кислота сЧ° с^° 1 ронЕ 1 ч СН2ОН С" \н Гліколова Гліокса* кислота лева кислота
91
Двохатомні спирти вступають у реакцію дегідратації. Причому а-, 0- і у-гліколі, залежно від умов реакції, по-різному відщеплюють воду. Відщеплення води від гліколів може відбуватися внутрішньо- і міжмолекулярно. Наприклад:
НО—СН2—СН2—ОН -но-* 1Н2С=СН—ОН1 Ізомеризац^-> Н8С—С^° ; « * — і'іг’-’ 1 * 1 о \ т т
Вініловий спирт Оцтовий
альдегід
НО-СН2—СН2—СН2—ОН -н--о > Н2С=СН—СН2—ОН;
СН2-СН2-СН2-СН2 он он
Аліловий спирт н2с------сн„
І І •
н2с сн2
Тетрагідрофуран
У 1906 р. О. Є. Фаворський, переганяючи етиленгліколь з сірчаною кислотою, Добув циклічний простий ефір —діоксан;
/О'н по-к
І + І н,(- ,------^сн,
Д)НІ ІІО<
IX
Н 40 Н>С СІІо - X 21 | + 2Н2О.
Н,(’ >;Н'
О Діоксан
Діоксан — рідина, яка кипить при 101 °С; розчиняється у воді в будь-яких співвідношеннях, застосовується як розчинник і як напівпродукт у деяких синтезах.
При міжмолекулярному відщепленні води від гліколів можуть утворюватися оксіефіри (спиртоефіри), як наприклад, діетилен-гліколь:
но—сн2—сн2— іон ч- н; — о—сн2—сн2—он^щр но—сн2—сн2—о—сн2сн2—он.
Д іетиленгл і коль
Діетиленгліколь добувають також взаємодією етиленгліколю з оксидом етилену:
НО—СН2—СН2—ОН + ІІ2С—СН2 -> но—сн2сн2—о—сн2сн2—он. о
Діетиленгліколь—рідина з температурою кипіння 245,5 °С; застосовується як розчинник і для заповнення гідравлічних пристроїв, а також в текстильній промисловості.
Широкого застосування як добрий розчинник набув диметило-вийефір діетиленгліколю (диглім)
Н3С—О—СН2СН2—О—СН2СН2—О—СН3.
92
Етиленгліколь при нагріванні з оксидом етилену при наявності каталізаторів утворює в’язкі рідини —поліетиленгліколі:
НО—СНаСН2—ОН + Н2с—;СН2 о
нгс^—;СН2
->НО—СН2СН2—О—СН2СН2—ОН—2-------->
Д і етиленгліколь
-> НО—СН2СН2—О—СІ І2СН2—О—СН2СН2—ОН і т. д. -> (—СН2—СН2—О—)л. Т ри етил е нгл І коль Пол і етилен гл і кол ь
Полігліколі використовують як компоненти різних синтетичних миючих засобів.
Широкого застосування набули поліефіри етиленгліколю з двохосновними кислотами, які використовують у виробництві синтетичних волокон, наприклад лавсану (див. розд. 35).
Перегрупування 1,2-гліколів. 1,2-Гліколі при каталітичній дії кислот можуть перегруповуватися в кетони. Реакції цього типу подібні до перегрупувань карбонієвих іонів, які утворюються при дії кислот на одноатомні спирти. Так, при дії кислот, наприклад, на 2,3-диметилбутандіол-2,3 (пінакон) він перегруповується в кетон — 3,3-диметилбутанон-2 (пінаколін). Таке перегрупування називають пінакон-пінаколіновим. Пінакон-пінаколінове перегрупування відбувається в Декілька стадій. Спочатку протон кислоти приєднується до одного з гідроксилів двохатомного спирту і утворює оксонієву сполуку (І). Цим самим протон активує даний гідроксил до реакції відщеплення. На наступній стадії від оксонієвої сполуки (І) відщеплюється за рахунок активованого гідроксилу вода і утворюється карбонієвнй іон (II), який потім перегруповується шляхом переміщення сусідньої метильної групи (1,2-змі-щзння) в оксонієвий катіон (III). Катіон (III) на заключній стадії депротонізується і перетворюється на кінцевий продукт реакції — кетон:
СН3 СН3 + сн3 СН3 (срХсІ13
н3с - с—с- сн3^==± н3с - с—с - ,сн3 - - - н3с - с—с - сн3 II І І /-'I +
:он:он он+он2 «:он
2,3-Диметил-
бутандІол-2,3 । И
(пінакон)
СН3 СН3
—- Н3С-'С-С-СН3 .. ' НзС-С-С-СН, + н +
- 3 II І II І ‘
+он сн3 о сн3
ЛІ 3,3-Диметилбутанон-2 .(піиаколін)
Трьохатомні спирти
Трьохатомні спирти мають три гідроксильні групи, їх хімічний склад описується загальною формулою СпН2п-і(ОН)8. Стійкими є такі трьохатомні спирти, у молекулах яких гідроксильні групи
93
розміщені біля різних вуглецевих атомів. Якщо три гідроксильні групи знаходяться біля одного вуглецевого атома, то такі спирти нестійкі, легко розкладаються, відщеплюючи воду за рахунок гідроксильних груп, і перетворюються на карбонові кислоти:
+Н,о.
Карбонова кислота
Першим і найважливішим представником трьохатомних спиртів є гліцерин СН2ОН—СНОН—СН2ОН (Тшш =290 °С), який відкрив К. Шееле у 1779 р. За систематичною номенклатурою назву трьохатомних спиртів утворюють з назви відповідного насиченого вуглеводню, до якої додають суфікс -тріол, вказуючи номери атомів вуглецю, біля яких знаходяться гідроксильні групи. Так, гліцерин за цією номенклатурою має назву пропантріол-1,2,3.
Методи добування. Гліцерин добувають гідролізом (омиленням) жирів (див. розд. 21) і синтезом з пропілену. Синтетичний метод добування гліцерину з пропілену включає декілька стадій:
н2с=сн—сн8 н2с=сн—сн2сі
Пропілен Хлористий аліл
, н с_сн__сн он - — - ——>
Аліловий спирт
->СН2ОН—СНС1—СН2ОН + сн2сі—снон—сн2 ->
он
Моиохлоргідрини гліцерину
сн2он—СНОН—сн2он.
Хімічні властивості гліцерину подібні до властивостей етиленгліколю і зумовлені наявністю в його молекулі трьох гідроксильних груп. Завдяки взаємному впливу гідроксильних груп кислотні властивості гліцерину сильніші, ніж у одноатомних спиртів. Ступінь дисоціації гліцерину приблизно в 10 разів більший, ніж метилового спирту. Внаслідок цього гліцерин легко утворює гліцерати (алкоголяти) не тільки з лужними металами, а й з лугами та гідроксидами заліза, міді, кальцію, барію та інших металів. При цьому не розчинні у воді гідроксиди, наприклад гідроксид міді, розчиняються. Розчинення Си(ОН)2 відбувається з утворенням комплексної сполуки (гліцерату міді) характерного синього кольору:
СН2-ОН
2 СН - он + Си(@Н)л СНа-ОН
14
єн2-0—,--:о-єн2 сн-о:-""” и—~~о-сн і н і
СН2-ОН- но-сн2
Гліцерат міді
94
Ця реакція є якісною на гліцерин та інші багатоатомні спирти, за її допомогою можна відрізнити багатоатомні спирти від одноатомних, оскільки одноатомні спирти з Сп(ОН)2 не взаємодіють.
Слід зазначити, що властивість етиленгліколю, гліцерину та інших багатоатомних спиртів розчиняти Сп(ОН)2 пов’язана не тільки з підвищенням їх кислотності, а також із здатністю цих спиртів утворювати внутрішньокомплексні високостінкі сполуки.
Гліцерин може вступати в реакцію з галогеноводнями (НС1 і НВг). При цьому в його молекулі одна або дві гідроксильні групи заміщуються на галоген і утворюються галогеноспирти, які назива-ють^галогеногідринами гл іцерину.
СН2ОН—СНОН—СН2ОН —
->СН2СІ—СНОН—СН2ОН + СН2ОН—СНС1—ЄН2ОН — Монохлоргідриии гліцерину
СН2С1—СНС1—СН2ОН + СН2С1—СНОН—СН2С1.
Днхлоргідрини гліцерину
Органічні і мінеральні кислоти утворюють з гліцерином складні ефіри. Залежно від кількості введеної в реакцію кислоти утворюються повні або неповні складні ефіри. Наприклад, при достатній кількості НМО3 утворюється повний ефір азотної кислоти —тринітрат гліцерину, який часто не зовсім вірно називають нітрогліцерином:
СН,—ОН СН,—О—N0,
І І
сн—он ч- зноьго2 сн—о—но2 ч-зн2о.
сн2—он сн2—о—но2
Тринітрат гліцерину
Тринітрат гліцерину —вибухова речовина і використовується для виготовлення динаміту. Тринітрат гліцерину отруйний, але в малих кількостях він розширює кровоносні судини серця. Тому його використовують у медицині для лікування стенокардії.
Гліцерин може утворювати повні і неповні складні ефіри і з органічними кислотами, наприклад з оцтовою:
СН2—О—С—СН«
І
сн2—ОН | о
| СН—о—с—сн3
СН—ОН Ч-ЗНО—с—СН3-> | Ч-ЗН2О.
СН2—ОН О СН2—О—С—СН3
о
Гліцернитрнацетат
Взаємодія гліцерину з вищими карбоновими кислотами приводить до утворення складних ефірів, які називають жирами (див. розд. 21).
95
Гліцерин, ЯК І ІНШІ спирти, легко окислюється. Причому окисленню можуть піддаватися одна, дві або всі три гідроксильні групи. Первинні гідроксильні групи окислюються до альдегідних, а потім до карбоксильних груп. Вторинна ОН-група окислюється до кетонної. Склад продуктів окислення залежить від природи окислювача.
При окисленні гліцерину можна добути такі речовини:
с^° сн,—он ,0 С^ с^° ,О С^
1 1 - | хон | хон | ХОН
С=О ; 1
сн-он 1 сн—он ’ сн—он ’ с=о
1 СН2—ОН 1
сн2—он Діоксі- СН2—ОН Ьо 1 лО
Гліцериновий ацетон Гліцеринова с\
альдегід кислота хон хон
Тартроиова Мезоксалева
кислота кислота
Контрольні запитання і вправи
1. Зобразіть графічні формули всіх ізомерів спиртів складу С4Н8ОН, С5НПОН та" назвіть їх за міжнародною номенклатурою.
2. Які речовини утворюються під час дегідрування, окислення, дегідратації бутанолу-1, бутанолу-2, З-метилбутанолу-2, а також при взаємодії цих спиртів з металічним натрієм, бромоводнем, оцтовою кислотою, концентрованою сірчаною кислотою?
3. Порівняйте кислотно-основні властивості бутилових та ізобутилових спиртів.
4. У чому подібність і відмінність між властивостями одно-, двох- і трьохатомних спиртів?
5. Як за допомогою хімічних методів та ЯАЇР-, ІЧ-спектроскопії можна розрізнити етанол і йодистий етил, етанол і етиленгліколь, етанол і діетиловий ефір; етанол, ізопропіловий спирт і діізопропіловин ефір?
Розділ 10. ПРОСТІ ЕФІРИ
Простими ефірами називають органічні сполуки, у молекулах яких два вуглеводневих радикали сполучені між собою атомом кисню. Ці сполуки можна розглядати як продукти заміщення водню гідроксильної групи спиртів на вуглеводневий радикал. Загальна формула простих ефірів —О—Н або Ц—О—/?'. У першому випадку обидва радикали однакові, у другому — різні. Ефіри з різними радикалами називають змішаними. Прості ефіри утворюють гомологічний ряд, який можна написати, виходячи з загальної формули цих речовин. Для цього в загальній формулі необхідно замість Д записати відповідні вуглеводневі радикали.
Прості ефіри називають за назвою вуглеводневих радикалів, сполучених з атомом кисню, а за систематичною номенклатурою — за назвою насиченого вуглеводню, додаючи до неї назву відповідної алкоксильної групи Ц—О—. Гомологічний ряді назви деяких простих ефірів наведено в табл. 11.
96
Таблиця 11. Гомологічний ряд, номенклатура і фізичні властивості деяких простих ефірів
Формула Назва 1 КИП ’ °С Густина . 20 'Ц
історична | систематична
Н3С—0—сн8 Диметиловий*, або метиловий Метоксиме-тав —23,7 0,617**
НЯС—О—СН2СН3 Метилетиловий Метоксіетан 7,9 0,697
Н3С—О—СН2СН2СН3 Метилпропіловий Метоксипро-пан 39 0,738
Н,с—0—СН—СН3 1 сн, Метилізопропіловий 2-Метокси-пропав 32,5 0,735
Н3ССН2—о—СН2СН3 Діетиловий*, або етиловий Етоксіетан 34,6 0,714
Н3ССН2СН2 — о—СН2СН2СН3 Дипропіловиіі*, або пропіловий Пропокси-пропан 91 0,736
* Якщо радикали однакові, то префікс ди- (ді-) часто опускають.
** При 0®С.
Ізомерія простих ефірів залежить від ізомерії вуглеводневих радикалів, сполучених з атомом кисню (наприклад, метилпропіловий і метилізопропіловий), а також від положення атома кисню в молекулі ефіру (наприклад, діетиловий і метилпропіловий; табл. 11). Останній вид ізомерів називають метамерією.
Методи добування. Прості ефіри добувають із спиртів, відщеплюючи від них воду, і взаємодією галогеналканів з алкоголятами (див. розд. 9). Важливим методом Добування простих ефірів є також каталітичне приєднання спиртів до етиленових вуглеводнів. Так, діізопропіловий ефір у промисловості добувають приєднанням ізо-пропілового спирту до пропілену при наявності каталізатора ВР3:
є- б* с- в* вг
(Н3С)2СН—О—Н + Н2С=СН—СН, ——> (Н3С)2СН—О—СН (СН3)2.
Фізичні і хімічні властивості. Перші представники гомологічного ряду простих ефірів (диметиловий і метилетиловий) є газоподібними речовинами. Діетиловий ефір і наступні гомологи —безбарвні, легкорухливі рідини, легші за воду (табл. 11). Прості ефіри дуже леткі, мають характерний запах, температури кипіння їх нижчі, ніж температури кипіння тих спиртів, з яких ці ефіри синтезовані. Це пов’язано з тим, що між їх молекулами, на відміну від спиртів, не утворюються водневі зв’язки і молекули простих ефірів є мономерними.
В ІЧ-спектрах простих ефірів є інтенсивна смуга поглинання в області 1060—1300 см-1, характерна для ефірного зв’язку С—-О (тс-о).
4 1-120
97
У хімічному відношенні прості ефіри є досить стійкими і мало-реакційноздатними речовинами. Вони важко вступають у хімічні реакції. Так, прості ефіри надзвичайно важко гідролізуються, на них не діють концентровані луги, концентровані кислоти, за виключенням йодистоводневої, яка вже при кімнатній температурі розкладає їх з утворенням галогеналканів і води:
Н3С—СН2—О—СН2—СН3 4- 2НІ -> 2Н3С—СН2—І + Н2О.
На атомі кисню в молекулах простих ефірів є вільні пари електронів: К—Р—Д. Тому кисень у цих сполуках може приєднувати протони та інші електронодефіцитні реагенти і виявляє, таким чином, основні властивості:
(С2Н6)2 О + Н* [(С2НБ)2ОН]\
Основність ефіру, як і інших слабких основ, можна охарактеризувати величиною константи рівноваги Ко (константою основності) даної оборотної реакції. Для діетилового ефіру рД0 — —3,6 (рЛо = = -№).
Основні властивості прості ефіри виявляють під час взаємодії як з сильними протонними, так і з апротонними кислотами, з якими вони утворюють солеподібні сполуки з тривалентним позитивно за-
рядженим атомом кисню (оксонієвий кисень —О—). Ці сполуки на-
I
зивають оксонієвими (подібно до сполук амонію). Утворення оксоні-євих сполук відбувається таким чином, що атом кисню молекули простого ефіру віддає на утворення зв’язку з протоном пару електронів і утворює з ним координаційний (донорно-акцепторний) зв’язок:
ГС2НВ
>0—н |С2н/ Діетилоксонійсульфат
8О4Н- °4-
СГ.
Діетилоксонійхлорид
3 простих ефірів найбільше значення має діетиловий, який часто називають просто ефіром. Цей ефір був добутий ще у 1540 р. алхіміком В. Кордусомдією концентрованої Н2ВО4 на етанол і названий ним сірчаним ефіром. Етиловий ефір —безбарвна, рухлива, надзвичайно вогненебезпечна рідина із специфічним запахом, важко розчиняється в воді (у 100 масових частинах води при 20°С розчиняється 6,5 масових частин ефіру) і сам частково розчиняє воду (л-'1,2 %). Повністю безводний ефір називають абсолютним. З органічними розчинниками діетиловий ефір змішується в будь-яких співвідношеннях.
Діетиловий ефір застосовується у виробництві бездимного пороху, штучного шовку, колодію, в медицині —для наркозу. Цей ефір використовують як розчинник при синтезі магнійорганічних речовин (реактивів Гріньяра; див. розд. 20).
88
Відомі також і циклічні прості ефіри, прикладом яких є діоксан (див. розд. 9):
Н2с---СН2
°\ /°
Н2С—сн2
Основні властивості діоксану виражені сильніше, ніж діетилового ефіру: р/<о діоксану становить —3,2.
Прості ефіри при зберіганні на світлі окислюються киснем повітря і утворюють вибухонебезпечні гідропероксиди:
Н3С—СН2—О—СН2—СН3 + О2 -> Н3С—сн—о—сн2—сн3.
оон
Утворення пероксидних сполук було причиною багатьох нещасних випадків під час перегонки ефіру. Тому перед початком роботи з простим ефіром необхідно перевірити, чи містить даний ефір пероксидні сполуки. Для цього пробу ефіру перемішують з підкисленим розчином йодиду калію. Побуріння розчину свідчить про наявність пероксидів, які окислюють йодид водню До вільного йоду. Останній забарвлює ефір у бурий колір. Щоб очистити ефір від пероксидів, його перемішують з сульфатом заліза (II).
Розділ 11. ТІОСПИРТИ, ТІОЕФІРИ ТА ІНШІ СПОЛУКИ СІРКИ
Сірка є аналогом кисню. Тому органічні сполуки, молекули яких містять сірку, хімічно подібні До аналогічних кисневмісних сполук.
Тіоспирти. Тіоспирти (тіоли, або меркаптани) — це органічні сполуки, молекули яких містять сульфгідрильну групу—8Н. Загальна формула тіоспиртів /?—8—Н. Тіоспирти можна розглядати як похідні сірководню, в молекулі якого один атом водню заміщений на вуглеводневий радикал.
Тіоспирти називають за історичною та систематичною номенклатурами. За історичною номенклатурою назву тіоспирту утворюють з назви вуглеводневого радикала, до якої додають слово меркаптан, а за систематичною — з назви відповідного вуглеводню, до якої додають суфікс -тіол і вказують номер вуглецевого атома, біля якого знаходиться група—8—-Н. Наприклад, Н3С—8Н —метилмеркап-тан, або метантіол; Н3С—СН2—8Н — етилмеркаптан, або етантіол; Н3С—СН(8Н)—СН3—ізопропілмеркаптан, або пропантіол-2.
Тіоспирти добувають з галогеналканів, діючи на них гідросульфідом натрію, або взаємодією спиртів з сірководнем при наявності каталізатора:
Н3С—СН2— І + к— 511 -> Н3С—СН2—8Н + КІ;
Н3С—СІ І2—ОН + Н—5—11 Н3С—СН2—5Н + Н2О.
99
Фізичні та хімічні властивості тіоспир-т і в. Тіоспирти киплять при значно нижчих температурах, ніж відповідні спирти, що пояснюється меншою асоціацією їх молекул. Так, етантіол кипить при 37 °С, тоді як етиловий спирт — при 78,39 °С. Тіоспирти важко розчиняються у воді і легко — у багатьох органічних розчинниках. Нижчі тіоспирти —рідини, вищі — тверді речовини. Особливістю нижчих тіоспиртів є надзвичайно неприємний запах, за яким можна виявити меркаптан у повітрі в дуже невеликих кількостях — 1 частина меркаптану на 5 • 1010 частин повітря. Тому тіоспирти добавляють до природного газу, який не має запаху, як одорант1 для виявлення його витоку з газопроводів.
Хімічні властивості тіоспиртів визначаються наявністю в їх молекулах сульфгідрильних груп —8Н. Властивості цієї групи зумовлені сіркою, яка, на відміну від кисню, виявляє змінну валентність, має меншу електронегативність, більший атомний радіус. Збільшення атомного радіуса сірки порівняно з киснем приводить до того, що зв’язок між сіркою і воднем більш слабкий, ніж між киснем і воднем.Тому тіоспирти, на відміну від спиртів,мають сильніші кислотні властивості. За хімічними властивостями тіоспирти наближаються до слабких кислот. Але завдяки ф-7-ефекту алкільної групи 7? ->8—Н кислотні властивості тіоспиртів слабкіші, ніж сірководню. Так, для етантіолу р/<к = 10,9, а для сірководню — р/\’к = = 7,02.
Тіоспирти, виявляючи кислотні властивості, легко реагують з лугами, оксидами металів і утворюють солеподібні сполуки —іпіоля-ти, або меркаптиди:
Н,С—СН2—8Н + К’аОН -> Н8С—СН2—8К’а + Н,О.
Тіоспирти порівняно легко окислюються. Слабкі окислювачі окислюють їх у дисульфіди:
2С2Н4—8Н + 12-> С2Н5—8—8—С2Н5 +2НІ.
Діетилди сульфід
Дисульфіди легко відновлюються воднем і знову перетворюються на тіоспирти.
Сильні окислювачі (І\МпО4, концентрована НХО3) окислюють тіоспирти до сульфокислот;
Н3С—СН2—811Н3С—СН2—8^-ОН.
Етансульфокислота
Отже, при окисленні тіоспиртів, на відміну від спиртів, окислюється сірка, а не вуглець.
Тіоефіри (сульфіди). Тіоефіри є аналогами простих ефірів і мають загальну формулу А—8—Л. У молекулах тіоефірів атом сірки
1 Одорант (від лат. одогаїіо.і — запах.
100
сполучений з двома вуглеводневими радикалами, які можуть бути як однаковими, так і різними. Називають тіоефіри за історичною та систематичною номенклатурами. За історичною номенклатурою назву тіоефірів утворюють з назви вуглеводневих радикалів, до якої додають слово -сульфід. За систематичною номенклатурою назву тіоефірів утворюють з назви відповідного вуглеводню, додаючи до неї назву групи 7?—8 — (алкілтіо-). Наприклад, Н3С—8—СН3 — диметилсульфід, або метилтіометан; Н3С—8—СН2СН3 — метил-етилсульфід, або метилтіоетан; Н3С—СН2—8—СН2—СН3 — діетил-сульфід, або етилтіоетан.
Тіоефіри добувають взаємодією галогеналканів з сульфідом натрію або калію і меркаптидами лужних металів:
2Н3С—СН2—І + К25 -> Н3С—СН2—8—СН2—СН3 + 2 КІ!
Н3С—СН2—8—Ка + І—СН2—СН8 -> Н3С—СН2—8—СН2—СН3 + КаІ.
Тіоефіри — рідини з температурою кипіння, близькою до температури кипіння тіоспиртів з такою самою молекулярною масою. Наприклад, диметилсульфід кипить при 38 °С, а етилмеркаптан — при 37°С. Тіоефіри не розчиняються у воді, мають ефірний запах. За хімічними властивостями тіоефіри — нейтральні речовини і не взаємодіють з кислотами.
За рахунок вільних електронних пар атома сірки.тіоефіри виявляють нуклеофільні властивості і вступають у реакцію з галоген-алканами (особливо легко з йодистим метилом), утворюючи при цьому сульфоніеві (тіонієві) солі:
І с-
Н3С—8—СН3 + Н3С—1 ->
Н3С—8—СН3 І СН3
Йодистий триметилтіоній
Тіонієві солі аналогічні оксонієвим солям простих ефірів (див. розд. 11). Але сульфоніеві солі значно стійкіші порівняно з оксонієвими і утворюються легше, НІЖ оксонієві солі.
Тіоефіри, як встановлено роботами О. М. Зайцева (1866 р.), досить легко окислюються. Слабкі окислювачі, наприклад пероксид водню, окислюють їх до сульфоксидів:
Н3С—8—СН3 + Н2О2 -> Н3С—8—СН3 + Н2О.
І
о
Диметилсульфоксид
Сильні окислювачі (КМпО4, ННО3) окислюють тіоефіри і сульфоксиди до сульфонів:
О
Н3С—8—СН3 —нзс—8—СН3 ^^121 Н3С— 8—сн3.
?) і
Диметилсульфон
101
Деякі похідні тіоефірів знайшли практичне застосування. Так, диметилсульфоксид (Н3С)28 О (7\ип= 189 °С) широко застосовують як високополярний апротонннй розчинник (дипольний момент 3,9В).
Розділ 12. НІТРОСПОЛУКИ АЛІФАТИЧНОГО РЯДУ
Нітросполуки—-це похідні вуглеводнів, у молекулах яких міститься нітрогрупа — ЙЮ2, атом азоту якої сполучений з атомом вуглецю вуглеводневого радикала. Нітросполуки мають загальну формулу 7?—МО2.
Будову нітрогрупи і природу зв’язків атома азоту з атомами кисню в цій групі легко зрозуміти, розглянувши процес утворення нітросполук окисленням пітрозосполук:
я — N = О + :О: —> /?—Й=О.
ї І :О:“
3 наведеної схеми реакції видно, що атом азоту нітрозогрупи віддає на утворення зв’язку з киснем вільну пару електронів і заряджається при цьому позитивно, а кисень, приєднавши пару електронів, заряджається негативно. Внаслідок цього між атомами азоту і кисню утворюється семіполярний зв’язок. Отже, у нітрогрупі є і подвійний зв’язок , і семіполярний. Структуру цієї групи можна зобразити так:
.. +
або #—N=0, або Я—N=0.
»6: І ।
„ О о-
Проте встановлено, що віддалі N—О в нітрогрупі однакові X)
(0,120нм), кут N<5 дорівнює 130°, що свідчить про рівноцінність \0
обох атомів кисню в цій групі. Вирівнювання зв’язків у групі —ЬЮ2 відбувається за рахунок спряження поляризованого подвійного зв’язку —N =0 з вільною парою електронів другого атома кисню
4°
О"
і приводить до рівномірного розподілу негативного заряду між обома атомами кисню. Тому найправильніше зображати нітросполуки формулою з дробовими зарядами (І) або у вигляді двох граничних структур (II і III):
+
К-Ч-2-\о 2
І
+ л°
Д-І^
\о-
II
/°”
-ЧІ
102
Назву нітросполук за систематичною номенклатурою утворюють з назви відповідного насиченого вуглеводню, Додаючи до неї префікс нїтро-.Місцеположення нітрогрупи в молекулі зазначають цифрою. Наприклад: Н3С—І\Ю2 —нітрометан, Н3С—СН2—ЬЮ2 — нітроетан,Н3ССН2СН2—МО2—1-нітропропан, Н3С—СН(МО2)—СН3 —2-нітропропан і т. д.
В ІЧ-спектрах нітросполук спостерігаються інтенсивні смуги поглинання в області 1375 і 1580 см-1, які характеризують відповідно симетричні і антиснметрнчні коливання групи —МО2.
Нітросполуки аліфатичного ряду добувають нітруванням алканів (див. розд. 7), а також взаємодією йод- і бромалканів з нітритом срібла. Застосовуючи в останньому методі такі розчинники, як ди-метилформамід (ДМФА) або диметилсульфоксид (ДМСО), можна замінити в цій реакції дорогий нітрит срібла на дешеві нітрити натрію або калію:
МаВг 4- Я—Г\’О2 ’~дмсо або дмфа ^О2 А§Вг.
В результаті такої взаємодії відбувається нуклеофільне заміщення атома галогену в галогеналкані на нітрогрупу. Іон І\Ю2 має амбідентні властивості (див. розд. 8) і може сполучатися з атомом вуглецю алкільного радикала як атомом азоту, так і атомом кисню. Амбідентні нуклеофіли при Хд^-реакціях атакують галогеналкан місцем найбільшої нуклеофільності, а при Хл^І-реакці-ях — місцем найбільшої електронної густини (правило Корнблю-ма). Тому первинні і вторинні галогеналканн в цій реакції утворюють не тільки нітроалкани, вихід яких становить 50—60 %, а й побічні продукти — ефіри азотистої кислоти. Наприклад;
І->Н3С—СН2—МО2; (8д,2-реакція)
• Н3С—СН2—І + АБКО2 Нітроетан
->Н3С—СН2—О—N=0. (8д,1 -реакція)
ЕтнлнІтрит
Хлористі алкіли з нітритом срібла реагують дуже повільно, а бромистий трет-бутил у цих умовах відщеплює бромоводень і перетворюється на ізобутилен.
Нітрогрупа має сильний — /-ефект і спричинює рухливість атомів водню біля а-вуглецевого атома (а-водневих атомів):
*І +С°
і °’
Підвищена рухливість а-водневих атомів приводить До того, що первинні і вторинні нітросполуки існують у вигляді двох тауто-
103
мерних форм — нітроформи І (нормальні нітросполуки) і аци-нітроформи II (ізонітросполуки):
N8 011
неї
о-н
/? - СН = N
X
О
II
ІМаОН
неї
О-N3 /
/? - СН = N
X
О
III
Лфі-нітроформа характеризується кислотними властивостями і реагує з лугами, утворюючи солі (III). При цьому рівновага в суміші таутомерних форм зміщується вправо. При підкисленні відбувається зворотний процес: спочатку утворюється кисла бщц-нітро-форма, яка потім ізомеризується в нітроформу. Отже, нітросполуки аліфатичного ряду — це псевдокислоти, оскільки самі вони нейтральні, не мають електропровідності, але утворюють нейтральні солі лужних металів. Взаємодія нітросполук з лугами відбувається повільно, на відміну від реакції нейтралізації справжніх кислот, яка, як відомо, відбувається миттєво.
Характерною реакцією нітросполук є їх здатність до відновлення. Відновлення нітросполук здійснюють атомарним воднем, добуваючи при цьому первинні аміни:
/?—К02 + 6Н -> /?— N14 2 4- 2Н2О.
Нітроалкани використовують у техніці як розчинники, а також як вибухові речовини, в реактивній техніці, у гумовій промисловості як вулканізатори.
Розділ 13. АМІНИ АЛІФАТИЧНОГО РЯДУ
Аміни можна розглядати як продукти заміщення атомів водню в молекулі аміаку на вуглеводневі радикали. Залежно від того, скільки радикалів сполучено з атомом азоту в молекулі аміну (скільки атомів водню в молекулі аміаку заміщено на радикал), розрізняють аміни первинні Ц—МН2, вторинні /?2ПН і третинні
КН3; Н3С—МН2;
Аміак Метиламін
(первинний амін)
Н3С—МН—СН3;
Диметиламін (вторинний амін)
Н3С—ІЧ—сн3.
І сн3
Триметиламін (третинний амін)
Номенклатура. Назви аліфатичних амінів утворюють додаванням суфікса -амін до назви алкільної або алкільних груп, сполучених з атомом азоту. Складніші аміни називають за назвою відповідного насиченого вуглеводню, додаючи до неї префікс аміно- (або М-метиламіно-, М,М-диметиламіно-, И-етиламіно- і т. д.).
104
Наприклад:
Н3С—СН2СН„СН„—N11,;
Бутиламін, 1-амінобутан
Н8С—КН—СНСН2СН3;
СН3
Метил-в то р-бу тил амін, 2- (//-метил аміно)-бутан
сн3
Н3С—N1—СНСН3.
сн3
Димет нл- і зо п роп і л-амін, 2- (ЬІ, М-диме-тиламіио)-пропан
Ізомерія амінів залежить від положення аміногрупи у вуглевод
невому ланцюгу, від кільї з атомом азоту. Наприкла, Н3С—СН2—МН—СН2—СН3 Н3С—Ш—(СН)2—СН3, тр Н3С—НН-СН2—СН3 і про
Стереохімія азоту. Методом дифракції електронів встановлено, що в молекулах аліфатичних амінів, таких як метиламін, диметиламін, триметиламін та інших, середнє значення валентного кута ато-
ості і будови радикалів, сполучених „ ізомерами є діетиламін
і метилпропіламін
іметиламін (Н3С)3М, метилетиламін ііламін Н3С—СН2—СН2—ИН2.
Рис. 26. Конфігурації молекули метилетил-пропіламіну
ма азоту становить НОНІ0. Це свідчить про пі-
рамідальну конфігурацію
молекул таких амінів і про ^//-гібридизований стан їх атома
азоту.
Розглядаючи моделі молекул амінів, легко переконатися, що молекулу аміну, в якій атом азоту сполучений з трьома різними радикалами, неможливо сумістити з її дзеркальним відображенням, тобто вона є асиметричною. Тому такий амін, як, наприклад, метил-етилпропіламін, повинен існувати у двох енантіомерних формах 1 і II (рис. 26), кожна з яких (окремо від другої) повинна виявляти оптичну активність (див. розд. 23). Проте енантіомери такого типу не були виявлені. Вивчення ПМР-спектрів амінів показало, що енергетичний бар’єр для переходу однієї пірамідальної конфігурації в другу досить невеликий і навіть при кімнатній температурі молекули швидко переходять з конфігурації / в конфігурацію II, тому розділити їх і встановити оптичну активність неможливо. Очевидно, що вільна пара електронів на атомі азоту не може виступати в ролі четвертої групи, яка закріплює конфігурацію (див. розд. 23). У молекулах четвертинних солей амонію, які утворюються при взаємодії третинних амінів з галогеналкілами, з атомом азоту сполучені чотири алкільні групи і четвертинний атом азоту стає тетраедричним. Якщо біля такого атома азоту розміщені чотириріз-ні групи, то четвертинні солі виявляють оптичну активність і існують у вигляді конфігураційних енантіомерів. Наприклад, йодид метилалілфенілбензиламонію існує у вигляді двох оптично активних енантіомерних форм (рис. 27),
105
Дзеркало
I
Рис. 27. Оптично активні енантіомерні форми йодиду метилаліл-фенілбензила монію
' Методи добування. Аміни аліфатичного ряду добувають такими методами:
1. Відновленням нітроалканів (див. розд. 12).
2. Відновленням нітрилів воднем при наявності каталізаторів N1, Рі, Реї або натрієм у спирті. Продуктами такого відновлення є первинні аміни: ’
Н3С—СН2—+ 2Н2 ™ Н3С—СН2-СН2—№ІЙ.
Пропіонітрил Пропіламіи
3. З амідів карбонових кислот (див. розд. 19).
4. Взаємодією галогеналканів з аміаком (реакція Гофмана, 1849 р.). Галогеналкани з аміаком утворюють суміш солей різних (первинних, вторинних і третинних) амінів. Реакція може закінчуватися взаємодією третинного аміну з галогеналканом і утворенням повністю заміщеної амонійної сполуки, так званої четвертинної амонійної солі:
с+і б- । н
н3с—і + №3-> [н3с—ІЧН4І + н3с-йн2;
Йодид метил- Метиламін
амонію
- ./ Ю+ _ + N11,
н3с-кн2 + н3с-і,-Ч(н3едш2]+і----------мнзіє)і21кн
Йодид Диметиламін
диметиламрніЮ'
НзС - І
—. [(.н3]0)^нГі - » (ІІ3С).,К 1------- [,(.Нз:С)4К], І .
Йодид . . .
триметиламоніюі Триметиліаміні Йодид тетраметиламонію
(четвертинна амонійна сіль)
При надлишку аміаку утворюється суміш амінів, більшу частину якої складають первинні аміни. Добуту суміш розділяють, використовуючи різну реакційну здатність первинних, вторинних і третинних амінів.
Фізичні властивості. Метиламін, диметиламін і триметиламін— гази, інші нижчі аміни —рідини. Нижчі газоподібні і рідкі аміни мають аміачний запах і подібно до аміаку добре розчиняються у воді. Більш складні аміни — рідини з неприємним запахом зіпсованої риби.
106
В ІЧ-спектрах первинних і вторинних амінів спостерігається поглинання в області 3300—3500 см-1, яке відповідає валентним коливанням N —Н-зв’язків. Смуги поглинання, які відповідають валентним коливанням зв’язків N—С, розміщуються в області 1100 — 1300 смг1 і важко ідентифікуються.
' Хімічні властивості. Властивості амінів визначаються в першу чергу наявністю в їх молекулах аміногрупи.
Кислоти о-основні властивості амінів. Атом азоту аміногрупи має вільну пару електронів. Тому аміни, подібно до аміаку, виявляють основні властивості і вступають у різні реакції як нуклеофільні реагенти. Так, у водних розчинах аміни приєднують протон води і утворюють гідроксиди амонійних сполук:
Т\ІІ2 + НОН [7?—ІЧН3] + + ОН-.
У зв’язку з цим водні розчини амінів містять у надлишку іони ОН" і мають лужну реакцію. Константа рівноваги даної реакції Ко, яка називається константою основності, становить Ко = [/?—№Іо] [ОН*] „ . , ,,
— —^__мн і—• Проте основні властивості у аліфатичних амінів виражені сильніше, ніж у аміаку, оскільки під впливом -ф/-ефекту алкільних груп електронна густина на атомі азоту аміногрупи збільшується:
Н—Ь1—Н;. Н3С-*НН2! Н3С->ЙН-<-СН8,
І н
а це підсилює електронодонорний характер змінної групи. Тому аліфатичні аміни активніше і міцніше, ніж аміак, зв’язують протон, що є свідченням підвищення їх основних властивостей порівняно з аміаком. Кількісною мірою основності амінів є константа основності Ко, значення якої зростає при переході від аміаку до аліфатичних амінів:
К'Н8 Н3С- І\Н2 Н3С—МИ—СН3 (Н8С)3И
К?5 1,8 • 10~§ 43,8 • 10-5 51(2 . ю-5 5>з . 10-3,
Як видно із наведених значень Ко, основність послідовно зростає при переході від первинних амінів до вторинних, але зменшується при переході від первинних і вторинних амінів до третинних. Послаблення основності третинних амінів порівняно з первинними і вторинними викликана тим, що іони триалкіламонію у зв’язку з просторовими утрудненнями сольватуються гірше і тим самим є менш стабільними, ніж іони алкіл- і діалкіламонію.
Аміни виявляють основні властивості і в реакціях з мінеральними кислотами, з якими вони легко утворюють алкілзаміщені амонійні солі. Наприклад:
І 4
(Н3С)3М: + Н+СГ [(Н3С)3КН]+СГ, або (Н3С)3М • НС1.
Хлорид Солянокислий
тр имет ил амон ію тримет ил амін
107
Для первинних і вторинних амінів відомі також реакції з участю с- с+ водню, який знаходиться біля змінного азоту. Зв’язок N -<- Н полярний. Електрони цього зв’язку зміщені до більш електронегативного елемента — азоту. Внаслідок цього атом водню,сполучений з атомом азоту, має підвищену активність і виявляє слабкі кислотні властивості. Так, первинні і вторинні аліфатичні аміни взаємодіють з металоорганічними сполуками, з лужними металами і утворюють солі — алкіл- і діалкіламіди:
2/?— МІ2 + 2Кта2/?—МН—Кта + Н2;
2/?2К’ Н + 2№ -> 2/?2Н—№ + Н2.
Алкіл- і діалкіламіди легко гідролізуються водою:
Я—ЬІН—№ + НОН -> /?—І\'Н2 + ИаОН;
/?2М—№ + НОН -> /?2МІ + ИаОН.
Алкілування амінів. Аміни легко вступають у реакцію з галогеналканами, в результаті якої відбувається поступове заміщення атомів водню біля змінного азоту на алкільні радикали. Такого типу взаємодію було названо реакціями алкілування. Алкілуванням можна з первинних амінів добути вторинні, а з вторинних — третинні аміни (реакція Гофмана; див. розд. 13).
Ацилування амінів. Первинні і вторинні аміни взаємодіють з органічними(карбоновими) кислотами, ангідридами і гало-генангідридами карбонових кислот. При цьому атом водню аміногрупи заміщується на ацильний залишок органічної кислоти. Таку взаємодію називають реакцією ацилування:
С2Н5-Кх1'-+'ер-С-СНз-----►С2Н5-.\'И-С-СН3 + неї!
й О
Ацетилхлорид N-Ацетил етил а мін
.. (С2Н5)2МІН + Н3С-Є-(Я- С-СН3--►(С2Н5)2К,-С-СН, + СНзСООН.
1 О О о
Оцтовий ангідрид \’-АцетилдіетиламІн
Ацильні похідні амінів можна гідролізом знову перетворити на вихідні аміни. Реакція ацилування амінів використовується для розділення суміші первинних, вторинних і третинних амінів.
Взаємодія з азотистою кислотою. Первинні, вторинні і третинні аміни аліфатичного ряду відрізняються реакціями з азотистою кислотою. Вважають, що діючим реагентом у реакції амінів з азотистою кислотою є азотистий ангідрид П2О3, який утворюється з азотистої кислоти:
2НО—N=0 <=> 0=Н—О—N=0 + Н20.
Оскільки азотиста кислота у вільному стані не існує, то її добувають безпосередньо в реакційній суміші взаємодією водного розчину нітриту натрію з мінеральною кислотою.
108
Первинні аліфатичні аміни взаємодіють з азотистою кислотою з виділенням азоту і утворенням спиртів, алкенів та інших речовин. При цьому азотистий ангідрид взаємодіє спочатку з аміном і утворює М-нітрозалкіламін, який далі ізомеризується в тауто-мерний алкандіазогідрат. Останній у кислому середовищі утворює нестійку сіль діазонію, яка розкладається з виділенням азоту і перетворюється таким чином на карбонієвий іон. Карбонієві іони, взаємодіючи з водою, утворюють спирти, а також перетворюються на етиленові, циклічні вуглеводні, алкілгалогеніди. В усіх цих перетвореннях солі діазонію розкладаються з виділенням азоту:
' ІЛ', О№ ? СОКО V
а>-сн2-к:+г/ч - у-сн2-н _ — /г-сн^н-о-
Н ° Н О Нітрозалкіламін
Н"' _ +
---А>—СН2 —N — N = О - /? —СН_,—N = N-^О~ Н —----Нітрозалкіламін Алкандіазогідрат
+ + но)н +
— /?-сн2-№н — їм» + /?-сн2—► у —сн2—он + н ;
Іон солі алкандіазонію
/?-сн2 + сі —-/?-сн2-сі;
А? — СН2і--—» Етиленовий вуглеводень .
Наприклад, етиламін в умовах цієї реакції утворює етиловий спирт з виходом 60 %. Пропіламін за цих самих умов дає пропіло-вий спирт (7 %), ізопропіловий спирт (32 %), пропілен та інші продукти.
Вторинні аміни з азотистою кислотою утворюють И-нітроза-міни:
(С2Н5)2И|НТНО|-НО -> (С2Н6)2ГМ-М=О + Н2О.
М-Нітрозодіетиламін
И-Нітрозопохідні вторинних аліфатичних амінів при нагріванні з розбавленою соляною кислотою Гідролізуються з утворенням вторинного аміну і азотистої кислоти.
З третинними амінами на холоді азотиста кислота не реагує.
Діаміни. Сполуки, в молекулах яких є Дві аміногрупи, називають діамінами. Стійкими є тільки такі діаміни, в молекулах яких аміногрупи розміщені біля різних вуглецевих атомів. Тому мети-лендіамін у вільному стані не існує. Найпростішим з первинних діамінів є етилендіамін,або 1,2-діаміноетан Н2П—СН2—СН2—МН2. ” ” ’ (1,3-діамінопропан)
(1,4-діамінобутан) (1,5-діамінопентан) (1,6-діаміногексан) або путресцин (від
Гомологами його Н2М—(СН2)3—МН2, Н2П-(СН2)4-МН2, Н21Ч(СН2)5->Ш2, Н2М—(СН2)0—МН2 та ін. 1,4-Діамінобутан, лат. рпігезсеге — той. що розкладається, гниє), і 1,5-діамінопентан,
є триметилендіамін тетраметилендіамін пентаметилендіамін гексаметилендіамін
109
або кадаверин (від лат. сасіауегозиз —трупний), відомі під назвою «трупні отрути», які утворюються при гнитті білків (трупів тварин), зокрема при декарбоксилуванні амінокислоти орнітину. Кадаверин у невеликих кількостях знайдений у мухоморах, пивних дріжджах. Він виявлений також у сечі іекскрементах людей, хворих на холеру і цистинурію.
Нижчі діаміни розчинні у воді і є сильнішими основами, ніж моноаміни. Діаміни утворюють солі з двома еквівалентами кислот, можуть алкілуватися і ацилуватися з участю як однієї, так і обох аміногруп.
Найбільше практичне значення має гексаметилендіамін (Тпл = — 40—43 °С), який є одним з компонентів при добуванні синтетичного волокна найлон (див. розд. 22).
Аміноспирти. Аміноспирти — це біфункціональні сполуки, молекули яких містять одночасно як спиртову, так і аміногрупу. Аміногрупа може бути первинною, вторинною і третинною. Існують також четвертинні амонійні солі цих спиртів. Спиртова група в молекулах аміноспиртів може бути первинною, вторинною і третинною. Гомологічний ряд первинних аміноспиртів починається з аміноетилового спирту Н2Й—СН2СН2—ОН, який називають р-етаноламіном, або коламіном. За систематичною номенклатурою назви аміноспиртів утворюють з назв відповідних спиртів, Додаючи слово аміно- і зазначаючи номер вуглецевого атома, біля якого розміщена ця група. Наприклад: 2-амінопропанол-1 Н3С—СН—СН2—ОН.
ЬШ2
«-Аміноспирти добувають приєднанням аміаку до «-оксидів. Так, при взаємодії оксиду етилену з аміаком утворюється етанола-мін:
Н,С—СН, + Н—МЙ, —-> Н,С-СН2—ГШ,.
О-......; он
Оксид етилену Етаноламіи
Важливу біологічну активність виявляє гідроксид триметил-р-оксіетиламонію — холін, який можна синтезувати взаємодією гідроксиду триетиламонію з оксидом етилену:
Н2с—СН2 + [НН(СН8)3]+ ОН- —> [НО—СН2—СН2— Н(СН3)3]+ ОН".
\ / Хол ін
о
Холін —фізіологічно активна речовина, яка знижує тиск крові в організмі. При ацетилуванні холін перетворюється на ацетилхолін:
[НО—СН2—СН2—М(СН3)3[+ ОН- + Н3С—С—ОН —>
II о
—>ГН3С—С—О—СН2—СН2—М(СН3)Л+ОН-.
II
І о ] . -
Ацетилхолін
по
Ацетилхолін відіграє важливу роль у діяльності центральної нервової системи, оскільки бере участь у передачі нервового збудження. Ацетилхолін синтезується в закінченнях нервових КЛІТИН з холіну і оцтової кислоти під впливом ферменту холінестерази. Проникнення холіну в місце контакту нервового волокна і іннер-вуючої клітини приводить до її збудження (або гальмування). Після цього ацетилхолін за допомогою холінестерази гідролізується на холін і оцтову кислоту.
Холін є також складовою частиною важливих природних жироподібних речовин—фосфатидів (див. розд. 21).
Розділ 14. АЛКЕНИ (ЕТИЛЕНОВІ ВУГЛЕВОДНІ, ОЛЕФІНИ)
Вуглеводні, молекули яких мають на два атоми водню менше, ніж алкани (вуглеводні ряду метану), і характеризуються наявністю в молекулах подвійного вуглець-вуглецевого зв’язку С=С (етиленовий зв’язок), називають алкенами, етиленовими вуглеводнями, або олефінами. Хімічний склад етиленових вуглеводнів описується загальною формулою СпН2п. Алкени утворюють гомологічний ряд, який можна записати, виходячи з гомологічного ряду алканів. Для цього необхідно відняти від двох сусідніх вуглецевих атомів алкану два атоми водню (по одному від кожного атома вуглецю)./Б удова, номенклатура і фізичні константи деяких алкенів наведейТв табл. 12.
Номенклатура. Алкени і багато їх похідних називають олефінами, оскільки рідкі алкени є оліїстими речовинами (від лат. оіеиш —рослинна олія). За систематичною міжнародною номенклатурою назву алкенів утворюють з назв аналогічно побудованих насичених вуглеводнів з тією самою кількістю вуглецевих атомів шляхом заміни суфікса -ан на суфікс -єн. При цьому в номенклатурі алкена вказується номер вуглецевого атома, після якого розміщений подвійний зв’язок (табл. 12). За основний вибирають найдовший вуглецевий ланцюг, в який обов’язково повинен входити подвійний зв’язок. Основний ланцюг нумерують так, щоб атом вуглецю, який вказує положення подвійного зв’язку, одержав менший порядковий номер.
Для алкенів часто використовують також назви, які склалися для них історично (тривіальна номенклатура). Тривіальні назви алкенів утворюють з назв відповідних насичених вуглеводнів, замінюючи в них суфікс -ан на суфікс -илен (-ілен).
Крім того, найпростіші алкени називають за раціональною номенклатурою («етиленова система» в назві), за якою ці вуглеводні розглядають як похідні етилену, утворені заміщенням атомів водню в його молекулі відповідними алкільними радикалами (табл. 12).
Якщо від етиленового вуглеводню відняти один атом водню, ТО одержують одновалентні радикали, які містять подвійні зв’язки. Радикал етилену Н2С=-СН— називають вінілом (етенілом). Існує
111
Таблиця 12. Гомологічний ряд, номен
Насичені вуглеводні
Формула Назва Формула
н3с—сн3 н3с—сн„—сн3 Н3С— СН2— СН2— СІ І3 н3с—сн2—сн2—сн2—сн3 Етан Пропа її Бутан Пентан н2с=сн2 н2с=сн—сн, н2с=сн—сн2—сн2 Н. .11 ;с=с< НдС/ ХСН3 Н3СХ /Н ;с=с< їй хсн3 н2с=с—сн3 1 сн3 н2с=сн—сн2—сн2—сн3 н3с—сн=сн—сн2—сн3 н2с=с—сн2—сн3 1 СНз Н2С=СН—сн—сн3 сн3
* При —102 °С, * ** при —47 °С, *** при 0°С.
три радикали пропілену: пропеніл Н3С—СН=СН—, ізопропеніл Н3С—С=СН2 і аліл Н2С=СН—СН2—. Двовалентний радикал етилену Н2С=С<^ — вініліден.
Ізомерія. Алкенам властива структурна ізомерія, зумовлена будовою (розгалуженням) вуглецевого ланцюга і положенням подвійного зв’язку в ланцюгу. Тому кількість структурних ізомерів у алкенів більша, ніж в алканів з такою самою кількістю вуглецевих атомів у молекулі. Ізомерія в ряду алкенів, як і алканів, починається з вуглеводню, що містить чотири атоми вуглецю, тобто з буте-ну, ізомерами якого є бутен-1, бутен-2 і метилпропен (табл. 12). Крім структурної, в ряду алкенів має місце ще геометрична ізомерія, передбачена Я. Вант-Гоффом у 1874 р., яка є одним з видів просторової ізомерії (стереоізомерії). Стереоізомери мають однаковий порядок хімічних зв’язків, але різне розміщення атомів або груп атомів (лігандів) у просторі. Просторове розміщення лігандів у молекулах стереоізомерів називається конфігурацією.
Геометрична, або цис- трснс-ізомерія, — це такий вид просторової ізомерії, коли ліганди по-різному розміщені в просторі від-
112
клатура і фізичні властивості алкенів
Етиленові вуглеводні
Назва за номенклатурою . Тпл. °С ГКИП’ °С Густина ,20
систематичною | тривіальною | раціональною
Етен Етилен Етилен —169,4 — 103,9 0,596*
Пропен Пропілен Метилетилен — 185,2 —47,0 0,609**
Бутен-1 а-Бут лен Етилетилен —130,0 —5,0 0.668***
/(ИоБуіен-2 Р-Бутилен (псевдобути-лен) саж-Диметиле-тилен —139,0 3,7 0,724
трснс-Бутен-2 — — —105,6 1,0 0,635
Метилпропен Ізобутилен НЄСМТИ-ДиМЄТИЛ-етилен —140,3 —6,9 0,5942
Пентен-1 а-Амілен Пропілетилен —138,0 29,9 0,6405
Пентен-2 Р-Амілен Метил етил етилен — 139,0 36,9 0,6515
2-Метилбутен-1 у-Ізоамілеи несши-Метил-етилетилен —137,6 31,2 0,650
З-Метилбутен-1 а-Ізоамілен Ізопропілетилен —135,0 25,0 0,648
носно площини подвійного зв’язку. Даний вид ізомерії властивий етиленовим сполукам, у молекулах яких атоми вуглецю з подвійним зв’язком сполучені з лігандами, наприклад речовинам типу 7?—СН=СН—В (В можуть бути як однаковими, так і різними), і зумовлений відсутністю вільного обертання навколо зв’язку С=С, яке потребує розриву л-зв’язку і затрати 125—210 кДж/моль. У випадку, коли однакові 7? розміщуються в просторі по один бік від площини зв’язку С=С, утворюється і{пс-конфігурація (від лат. сіз — по цей бік). Якщо 7? розміщені по різні боки, — утворюється /пране-ізомер (від лат. Ігапз —через, по той бік). Наприклад:
цис-Бутеи -2
транс -Бутен -2
113
У складніших випадках, коли, наприклад, чотири ліганди різні, ццс-/п/шнс-позначєпня є умовними. Тому для сполук типу аЬ С=С сй за системою ШРАС запропонована Е—/-номенклатура. Для цього введено старшинство замісників. Замісник тим старший, чим більший атомний номер елемента, який безпосередньо сполучений з атомом вуглецю зв’язку С=С. Так, старшинство замісників зростає у ряду Н < С < N <С О < Р < Р <с 8 < СІ < Вг < І і т. д. Якщо атоми, сполучені з атомом вуглецю зв’язку С=С, однакові, то необхідно враховувати старшинство сполучених з ними замісників. Старшинство замісників зростає в ряду Н3С—< <Н3С-СН2—<(Н3С)2СН—.
У тому випадку, коли біля однакових атомів є різна кількість замісників, старшим вважається атом з більшою кількістю замісників, наприклад: (Н3С)3С— > (Н3С)2СН— > Н3С—СН2 — (ЗС > >2С>1С).
Якщо в сполуці типу аЬ С=С аі а старше, ніж Ь, а с старше, ніж^, то конфігурацію, в якій старші замісники а і с знаходяться в цпс-положенні, позначають буквою 7. (від нім. Хизатшеп — разом). Якщо старші замісники а і с знаходяться в транс-положенні, то таку конфігурацію позначають буквою Е (від нім. ЕпДюреп— напроти). Наприклад:
Н3С. .Н Н8С—СН2Ч .Н
)С=С< ; >С=С<
Н3С—Н2СХ 'СН2СН2СН3 Н3СХ ХСН2СН2СН3.
2-3-Метил гептен-З Е-3 - Метил гептен-3
(^«^-конфігурація) (тран с-конфігу рація)
Геометричні ізомери відрізняються за фізичними, хімічними і навіть фізіологічними властивостями. Молекули цпс-ізомерів мають центр і площину симетрії, а шранс-ізомери —тільки центр симетрії. Для визначення конфігурації геометричних ізомерів використовують фізичні параметри. Так, температури плавлення транс-ізомерів, як правило, вищі, ніж відповідних 4{ис-ізомерів. Цис-ізомери мають більшу густину, вищу температуру кипіння і вищий показник заломлення, ніж відповідні тромс-ізомери. У транс-ізомерів дипольний момент дорівнює нулю, а і}пс-ізомери мають певний дипольний момент. Помічено певні'відмінності також в ІЧ-спектрах цис- і шранс-ізомерів.
Методи добування. Ненасичені вуглеводні етиленового ряду в вільному стані в природі зустрічаються порівняно рідко, наприклад, в деяких нафтах.
З синтетичних методів добування алкенів найбільше значення мають такі:
1. Дегідратація спиртів, тобто відщеплення від них води (див. розд. 9).
2. Дегідрогалогенування галогеналканів (див. розд. 8).
3. Дегалогенування віцинальних дигалогенопохідних алканів. Віцинальні дигалогеналкани при нагріванні з подрібненим цинком або магнієм у відповідному розчиннику, наприклад у спирті, легко 114
перетворюються на етиленові вуглеводні. Так, з 1,2-диброметану в цих умовах утворюється етилен:
Н2С—СН2 -р 2п —> Н2С=СН2 + 2пВга.
Вг Вг
4. Дегідрування алканів (див.розд. 7).
5. Часткове гідрування ацетиленових вуглеводнів (див. розд. 15).
6. Каталітичний крекінг алканів (див. розд. 7).
Фізичні властивості,' Перші три представники гомологічного ряду етиленових вуглеводнів — гази, починаючи с СБН10 — рідини, а С]8Н36 і вищі алкени — тверді речовини (табл. 12). Із збільшенням молекулярної маси алкену підвищуються їх температури плавлення і кипіння. Алкени з вуглецевим ланцюгом нормальної будови киплять при вищій температурі, ніж їх ізомери, які мають розгалужену структуру. Переміщення подвійного зв’язку в центр молекули викликає підвищення температури кипіння і температури плавлення алкену. ~1
Для алкенів хартктерне вибіркове поглинання ІЧ-світла, тому ІЧ-спектри широко використовуються для встановлення будови етиленових сполук. В ІЧ-спектрах валентним коливанням С=С-зв’язків відповідає смуга поглинання в області 1680—1640см-1. Це коливання не є строго валентним, оскільки поряд з розтягуванням зв’язку С=С відбувається зміна валентних кутів Н—С=С. Смуга валентного коливання С=С-зв’язку в ІЧ-спектрах слабка, особливо в тих випадках, коли етиленовий зв’язок знаходиться в середині вуглецевого ланцюга молекули.
Частота валентних коливань =С—Н спостерігається при ЗОЮ— 3095 см-1, причому значення т^с-н визначається ступенем заміщення: для ==СН7? характерне коливання з V =3040—ЗОЮ см-1, для групи =СН2 з’являється коливання з частотою 3095—3075 см-1.
Інтенсивними і характерними для алкенів є смуги неплоских деформаційних коливань Н—С=С—Н, розміщені в області 1000— 800 см-1. Поглинання при 970—965 см-1 характерне для транс-ізомеру, при 670 см-1 —для цпс-ізомеру, а при 890 см-1 —для угруповання Н2С=С\\
В УФ-області спектра алкени поглинають при 180—200 нм.
Характерними для алкенів є ПМР-спектри. Протони алкенів у цих спектрах дають хімічні зсуви біля 4,5—6 млн-1.
Хімічні властивості. Хімічні властивості алкенів зумовлені в першу чергу наявністю в їх молекулах подвійного вуглець-вуглецевого зв’язку С=С, який являє собою комбінацію о- і л-зв’язків. Для описання електронної будови зв’язку С=С за теорією МО використовують один з прийомів —метод гібридизації. Атоми вуглецю, сполучені подвійним зв’язком, знаходяться в з/Лгібридизова-ному стані (див. розд. 2),
По
Утворення подвійного зв’язку між атомами вуглецю можна уявити собі на прикладі реакції дегідрогенізації насичених вуглеводнів, При відщепленні двох атомів водню, наприклад, від молекули етану утворюється етилен. В процесі відщеплення водню від кожного з вуглецевих атомів етану розпаровуються дві пари електронів і утворюється другий зв’язок:
її; ;'н
'А’- -2Н
Н2С-----СІ Ь------ Н2С - сн2,
Цей другий зв’язок за своєю природою не може бути повністю ідей -тичнпм а-зв’язку С—С в молекулі етану, оскільки в такому випадку вуглецевий атом повинен був би мати два електрони з чотирма однаковими квантовими числами, що привело б до порушення принципу Паулі. Розпаровані внаслідок відщеплення атомів водню електрони атомів вуглецю знаходяться на р-орбіталях, оскільки вони є більш зовнішніми, ніж х-орбіталі. р-Орбіталі, як відомо, мають форму об’ємних вісімок (гантелей) і розміщені в просторі в трьох взаємно перпендикулярних площинах рх, ру, рг (рис. 6, розд. 2). Взаємне перекриття двох р-атомних орбіталей (по одній від кожного з вуглецевих атомів) дає л-молекулярну орбіталь. Ковалентний зв’язок, утворений цією орбіталлю, називається л-зв’яз-ком. Інші зв’язки в молекулі етилену (С—С і С—Н) є сг-зв’язками. Вони утворені 5р2-гібрпдизованпми орбіталями атома вуглецю:
На утворення а-зв’язків
ТГ-зв’язку
При 8р2-гібридизації в лінійну комбінацію з 2х-АО вуглецю входять дві його 2р-АО, а третя негібридизована 2р-орбіталь іде на утворення л-зв’язку. 5р2-Гібридизовапі орбіталі атомів вуглецю в молекулі етилену, перекриваючись між собою, утворюють о-зв’я-зок С—С, а перекриття 8р2-гібридизованих орбіталей атомів вуглецю з І8-АО чотирьох атомів водню дає чотири о-зв’язки С—Н (рис. 28). Негібрндизовані («чисті») 2р-АОдвох атомів вуглецю спарюються (по одній від кожного атома) і утворюють л-зв’язок (рис. 29).
Відповідно до теорії МО ЛКАО при бічному перекриванні (латеральне перекриття) двох р-орбіталей утворюється зв’язуюча л-МО (рис. ЗО, «), енергія електронів якої нижча від енергії цих же електронів на ізольованих р-кЬ, і розпушуюча л*-МО (рис. ЗО, б), на якій за звичайних умов електронів немає. Електрони на зв’язуючій л-МО підсилюють притягання між атомами вуглецю, тому дов
116
жина зв’язку С=С зменшується порівняно із зв’язком С—С. Зв’язуюча л-МО, на відміну від орбіталей о-зв’язку, несиметрична відносно лінії зв’язку і має вузлову площину. В екстремальних умовах
Рис. 28. Будова молекули етилену:
а — розміщення С-зв’язків у молекулі; б — тетраедрична модель молекули; в — куле-стержнева модель молекули; г — масштабна модель молекули за Стюартом — Бріглебом
Рис. 29. Схема утворення л-зв’язку в молекулі етилену:
а —розміщення зт-зв’язку в просторі; б — бічне перекривання хмар р-електронів; в — розміщення електронної хмари л-зв'язку над площиною і під площиною атомних ядер
Рис. ЗО. л-Зв’язуюча (а) і л-розпушуюча (б) молекулярні орбіталі у молекулі етилену
енергія електронів на розпушуючій л*-МО значно вища, ніж на ізольованих р-АО, що зумовлює їх відштовхування і, відповідно, відсутність зв’язку.
Особливістю подвійних зв’язків є те, що навколо них немає вільного обертання. Оскільки електронна густина л-зв’язку розміще
ну
на в просторі в площині, перпендикулярній до ст-зв’язку, то л-зв’я-зокнедає можливості вільно обертатись одній частині молекули відносно другої, сполученої з першою С— С-зв’язком. А це приводить до появи у заміщених алкенів геометричних цис-транс-ізомерів.
Найпростішим і типовим представником сполук з подвійним С=С-зв’язком є етилен, молекула якого плоска, всі шість її атомів розміщені в просторі під кутами, близькими до 120° (див. рис. 28—ЗІ),
Зв’язки в молекулі етилену характеризуються такими параметрами: зв’язок С —С — довжина 0,134 нм, енергія 607,1 кДж/моль; зв’язок С—Н—довжина 0,108 нм, енергія 435,5 кДж/моль.
Істотною особливістю л-зв’язку молекули етилену та інших алкені в є те, що максимальна електронна густина його знаходиться вище і нижче від осі зв’язку (рис. 29, ЗО), а область перекривання 2/7-орбіталей дещо менша, ніж у зрг-орбіталей. Тому при утворенні другого зв’язку між атомами вуглецю загальна енергія подвійного зв’язку не подвоюється, а збільшується тільки на 257,1 кДж/моль. з цього випливає, що л-зв’язок слабкіший від о-зв’язку.
л-Електрони зв’язків С=С під дією різних факторів легко зміщуються до одного з атомів вуглецю і таким чином поляризуються. Підтвердженням цього є здатність алкенів поглинати світло в більш довгохвильовій частині спектра порівняно з поглинанням алканів, що свідчить про нижчі частоти коливання молекул алкенів, ніж алканів, і про більшу поляризованість С=С-зв’язків порівняно із а-зв’язками С—С.
Нагромадження електронів і поєднання о- і л-електрон і в" у зв’язку С=С збільшує загальну електронну густину цього зв’язку і надає йому певного мірою властивість основи Льюїса, або нуклео-фільність. Завдяки цьому етиленовий зв’язок виявляє високу реакційну здатність відносно частинок, що мають меншу електронну густину, —'радикалів і катіонів. Тому для етиленових сполук характерні реакції приєднання, реакційним центром у яких є л-зв’язок. ;
_;Р е а к ц і ї приєднання. Найважливішим типом хімічних перетворень етиленових сполук є реакції приєднання (А-реакції) за місцем подвійного зв’язку за рахунок розриву енергетично слабкішого л-зв’язку. л-Зв’язок є донором електронів, які легко утворюють о-зв’язки з реагентами, що приєднуються. До зв’язку С =С можуть приєднуватися водень, галогени, галогеноводні, вода, спирти та інші реагенти (аденди). Залежно від природи адендів, умов реакції (температура, природа розчинника тощо) А-реакції можуть відбуватися за іонним електрофільним (Аг) і радикальним (А/?) механізмами. Реакції приєднання нуклеофільних реагентів до етиленових сполук мало характерні. У процесі реакції приєднання атоми вуглецю подвійного зв’язку переходять із зр2- у 5р3-валентний стан.
Гідруеання.і Гідруванням називають реакції приєднання водню до ненасичених сподук. При гідруванні подвійний зв’язок С=С розривається і утворюється простий зв’язок С-С, а алкени при
118
цьому перетворюються на алкани. Гідрування є одним з найзручніших методів синтезу алканів. Практичне значення має гетерогенне гідрування алкенів молекулярним воднем при наявності каталізаторів, таких як Пі, Рі, Реї. Гідрування алкенів — екзотермічна реакція. Наприклад:
. Н2С=СН2 + Н2 Катал1зат°Р^ Н3С—СНа + 137 кДж.
Електрофільне приєднання,Цля алкенів надзвичайно характерними є реакції електрбфільного приєднання (Лг-реакції) до подвійного зв’язку полярних (іонних) реагентів Х+ £~' або реагентів, які легко поляризуються. Частинка Х+ є акцептором електронів (електрофілом) і починає процес приєднання до етиленового зв’язку.
Ая-реакції описуються двостадійним механізмом. На першій, повільній стадії електрофіл X+, який має вакантну орбіталь, взаємодіє з л-електронами етиленового зв’язку і утворює л-комплекс (І). Далі у л-комплексі внаслідок приєднання електрофілу за рахунок л-електронів утворюється о-зв’язок з одним з атомів вуглецю, л-Комплекс таким чином перетворюється на ст-комплекс (карбкатіон) (II). Рівноважний стан між л-комплексом (І) і о-комплексом (II) завершується другою, швидкою стадією — приєднанням протилежно зарядженого іона і утворенням кінцевого продукту реакції — аддукту (III). Приєднання протпвоіона 2“ відбувається за транс-механізмом, тобто з тильного боку відносно частинки X, яка вже приєдналася:
Гідрогалогенування» Гідрогалогенуванням називають реакцію приєднання ЇФНаї до ненасичених сполук.
Приєднання ЕШаї до етиленового зв’язку, як і приєднання інших протонних кислот, описується вищерозглянутим механізмом. У цьому випадку електрофілом є протон. Протон спочатку утворює з алкеном л-комплекс, а потім гетеролітнчно розриває л-зв’язок і за рахунок пари електронів утворює ст-зв’язок з атомом вуглецю. Таким чином л-комплекс перетворюється на о-комплекс. Останній дуже швидко приєднує аніон НаІ~ і утворює галогеналкан:
хС = С< + Н+НаГ
—с
На І
119
Швидкість приєднання галогеноводнів до алкенів зменшується в ряду НІ > НВг > НС1 > НЕ, тобто найлегше приєднується йодоводень, а найважче —фтороводень.
Правило Марковникова. Особливий інтерес являє приєднання протонних кислот до несиметричних етиленових сполук. Ще в 60-х роках минулого століття О. М. Бутлеров та інші вчені показали, що НІ, приєднуючись до пропілену, утворює йодистий ізопропіл. Реакція в цьому випадку відбувається за схемою:
Н3С—СН=СН2 + НІ —> Н8С—СН—СН8.
Перші узагальнення про порядок приєднання галогеноводнів до алкенів належать В. В. Марковникову, який сформулював таке правило (1869 р.): «...галоїд приєднується до найменш гідрогенізо-ваного вуглецю». Тепер відомо, що приєднання галогеноводнів та інших протонних кислот відбувається згідно з правилом Марковникова тільки в тому випадку, коли механізм приєднання гетеро-літичний (іонний).
Електронна інтерпретація правила Марковникова базується на тому, що під впливом електронних ефектів замісників подвійний зв’язок у молекулах несиметричних алкенів поляризується (статичний фактор). Наприклад, у молекулі пропілену під впливом ефекту, який виявляє метильна група, атом вуглецю метиленової е+ б-групи має частковий негативний заряд 6_: Н8С->СН=СН2. Застосування розрахункових квантово-механічних методів показало, що в молекулі пропілену розподіл електронної густини на атомах вуглецю (в одиницях заряду електрона) такий:
+0,02 +0,03 —0,05
Н3С—сн=сн2.
Полярність молекули пропілену підтверджує наявність у неї дипольного моменту, що дорівнює 0,36П.
Гідрогалогенування пропілену відбувається за вищеописаним механізмом електрофільного приєднання у дві стадії. Причому приєднання протона відбувається відповідно до поляризації подвійного зв’язку —до атома вуглецю з зарядом 6" (статичний фактор):
6+ б- + НаІ~
Н8С->СН=С«2 +Н+5±Н„С—СН=СН2^±Н3С—СН—СН, —> Н,С—сн—сн„
V І
Н+ Наї
Такий напрямок приєднання ННаї зумовлюється також утворенням на проміжній стадії втор-пропільного катіона, який стабілізується метальними групами і є стійкішим (див. розд. 5), ніж первинний пронільний катіон (динамічний фактор). Отже, правило Марковникова в застосуванні до полярних реакцій приєднання формулюється так: При гетеролітичному приєднанні полярних молекул до алкенів позитивна частинка реагенту приєднується до
120
атома вуглецю подвійного зв’язку з більшою електронною густиною, а негативна частинка приєднується до атома вуглецю з меншою електронною густиною.
За електрофільним механізмом, згідно з правилом Марковни-кова, крім галогеноводнів до етиленових сполук приєднується ще багато інших реагентів, напрямок приєднання яких показаний нижче на прикладі реакцій з пропіленом:
м+он
по Сі
ь-
-сн=сн..
н3с—сн—сн, І он
Н3С-СН-СН2
ОН СІ
гОї
НО8О3Н
Н3С-СН-СН
н3с-сн-сн,
О Г\Ю2
О8О3Н
Нзс-сн-сн» І І СІ !
Напрямок реакції приєднання галогеноводнів та інших прото-новмісних кислот до зв’язку —СН=СН— з однаковим ступенем гідрогенізації атомів вуглецю визначається емпіричним правилом Зайцева —Вагнера (1875 р.), згідно з яким «галоїд приєднується до того з ненасичених вуглецевих атомів, який знаходиться у зв’язку з метилом». Електронна інтерпретація цього правила грунтується на тому, що приєднання протонних кислот до подвійного зв’язку відбувається у напрямку утворення енергетично стійкішого карбкатіона (динамічнийфактор). Стабілізація карбкатіона зумовлена найбільшою кількістю а-водневих атомів, спряжених з вакантною орбіталлю на атомі вуглецю, тобто спряжених з позитивним зарядом. Наприклад, в реакції приєднання НІ до пентену-2 енергетично стійкішим буде проміжний карбкатіон (І), який стабілізують п’ять а-водневих атомів, а не карбкатіон (II), який стабілізують чотири а-водпевих атоми. Тому головним продуктом приєднання буде 2-йодпентан, а не його ізомер — 3-йодпентан:
•ні н3с-сн2-сн=сн-сн3—
і—* н,с-сн
► Н3С-СН2-СН2-СНСН3:
н
н
н3с4с-сн-с^сн -►н3с—сн2—сн-сн2—сн3.
V Т '
н н і.
121
Отже, напрямок приєднання галогеноводнів та інших полярних реагентів до несиметричних алкенів зумовлюється як поляризацією подвійного зв’язку (статичний фактор), так і стійкістю карбо-нієвих іонів (динамічний фактор), які утворюються на проміжній стадії реакції.
Якщо в молекулі алкену міститься сильний електронегативний ліганд, то приєднання полярних реагентів до таких алкенів відбувається проти правила Марковникова. Наприклад, проти правила Марковникова відбувається приєднання хлороводню трифторпропі-леном, який в результаті реакції утворює 1,1,1-трифтор-З-хлор-пропан:
б- с+
Р3С Ч— СН=СН2 + Н+СГ —> Р3С—СН2—СН2—СІ.
Проти правила Марковникова?; приєднується до пропілену та інших несиметричних алкенів бромоводень, якщо реакцію здійснюють при наявності кисню повітря, пероксидів або інших джерел вільних радикалів (пероксидний ефект Караіиа):
о2
Н3С—СН=СН2 + НВг-----> Н3С—СН2—СН2—Вг.
Пероксидний ефект пояснюється тим, що реакція приєднання НВг в цих умовах відбувається не за іонним, а за ланцюговим атомарно-радикальним механізмом. Спочатку радикал (наприклад кисень 'О—О’) гемолітично розщеплює молекулу НВг, утворюючи при цьому атомарний бром Вг‘, який потім приєднується до більш гідрогенізованого атома вуглецю з подвійним зв’язком. Новий складніший радикал, який виник при цьому, взаємодіє з наступною молекулою НВг, приєднує до вторинного атома вуглецю атом водню з одночасним утворенням нового атома брому Вг‘, який продовжує ланцюг реакції:
/?• + Н у;-- Вг —> /?—Н + Вг;
0+ 6-
Н8С—СН=СН2+Вг- —Н3С—СН—СН2Вг;
Н3С—СН—СН2Вг + Н Вг —> Н3С—СН2—СН2Вг + Вг* і т. д.
Пероксидний ефект спостерігається тільки у випадку приєднання бромоводню. Однак якщо приєднання НВг проводити при відсутності кисню, пероксидів та інших джерел вільних радикалів, то в цих умовах реакція відбувається за електрофільним механізмом і бромоводень приєднується до несиметричних алкенів за правилом Марковникова.
Приєднання до алкенів НР, НС1, НІ завжди відбувається за правилом Марковникова, тому що пероксиди не розкладають ці галогеноводні, оскільки гемолітичний розрив зв’язку в молекулах НР, НС1 вимагає великої затрати енергії (для НР — близько 618 кДж/моль). НІ має сильні відновні властивості і сам розкладає пероксиди; крім того, атоми йоду, які утворюються з НІ, мало реакційноздатні.
122
__ С.ЙДйшшДщД'іДратацією називають реакцію приєднання води. Гідратація етиленових сполук відбувається при наявності таких каталізаторів, як Н2$О4, Н3РО4 тощо. Реакція проходить через стадію приєднання мінеральних кислот до алкену з утворенням складного ефіру мінеральної кислоти, який при гідролізі дає спирт і відповідну мінеральну кислоту. Наприклад:
4-ноп
Н2С=СН2 + НО8О3Н —> Н3С—СН2—О8О3Н------> Н3С—СН2—ОН + Н2$О4.
Етилсірчана кислота
Гідратація етиленових сполук є одним з найважливіших методів добування спиртів.
^Галогенування. Алкени легко приєднують галогени з утворенням віцинальних дигалогеналканів:
Н,С=СН2 + СІ, —> СІ—СН2—СН2—СІ.
Алкени з алкільними радикалами біля атомів вуглецю з подвійним зв’язком приєднують галогени швидше, ніж етилен.
Реакційна здатність галогенів різко зменшується в ряду Р2 > >С!2 > Вг2 > І2. Під час взаємодії алкенів з фтором виділяється 452,5 кДж/моль. Ця енергія більша від енергії С—С-зв’язків, для розриву яких потрібно 350 кДж/моль. Тому фторування алкенів супроводиться загорянням реакційної суміші.
Йод з алкенами взаємодіє повільно на сонячному світлі.
Галогенування алкенів належить до реакцій електрофільного приєднання (Ле -реакцій).
Загальний механізм полярного приєднання галогенів до алкенів описується розглянутим вище двостадійним механізмом: спочатку катіон галогену утворює л-комплекс, який потім перетворюється на о-комплекс (карбкатіон), після цього відбувається шронс-приєд-нання до карбкатіона аніона галогену:
-с 8 + 8-
II + \\'^НаІ-На/л^.
-С “V
Поляризація галогену
Є,—Наї —С—Наї
І І ———*- І ,с- Наї—С
— Є'
І + '.НаІ НаГ
-Є.' а
- І -4
Л-Комплеке с-Комплекс>
Демонстраційний дослід знебарвлення бромної води при пропусканні крізь неї газоподібного етилену являє собою типову Ае-реакцію. Бромна вода (розчин брому у воді) — суміш розчинених речовин, серед яких під дією полярних молекул води виникають катіони Вг+. При взаємодії з етиленом вони спочатку притягуються до л-МО подвійного зв’язку, перекривають свої вільні р-АОз його л-молекулярною орбІталлю і утворюють л-комплекс (рис. 31). Цей л-комплекс перетворюється далі на о-комплекс (карбкатіон), який
123
г г
:Вг~-Вг:---:ІЇг -Вг‘-
• • «» «а
їг-Ломллекс
Рис. 31. Схема утворення л-комплексу з подвійним 38 *ЯЗКОМ
швидко приєднує аніон брому Вг і утворює кінцевий продукт реакції — 1,2-диброметан (бромистий етилен):
„ .. +
н.,б. +'вг 0 Вг [н,о:вг н.,о +
Ві + ] Вг- ’>
Н,С ІІ,С'- ІІ,С-Вг
‘II + Ві + —н'Ві^“ 1 + н,с н.с- сіі2
Вг~ Н,.С-Вг Ні — СН,
Алкілування. Алкілування алкенів являє собою, в принципі, приєднання алканів до етиленового зв’язку, яке відбувається в порівняно м’яких умовах при наявності кислотних протонних і апро-тонних каталізаторів (Н28О4, Н3РО4, НР, А1С13, ВР3 тощо). Алкілуванням алкенів синтезують вуглеводні переважно з розгалуженою структурою, які використовують як моторне паливо (наприклад, синтез авіаційного бензину). Так, 2,2,4-триметилпентан (ізооктан), який має високе октанове число, добувають алкілуванням ізобутилену ізобутаном:
І.....1 снз
є- е+ є— | (Н3С)2С=СН2 + н—Н2С—с—н
І І
............. сн3
сн3
Щ5О, н С_(1_СН СН_СНз_ ю °С | |
СН3 СН3
Каталітичне алкілування відбувається за механізмом електро-фільного приєднання.
Оксосинтез. Оксосинтезом, або гідроформілуванням, називають каталітичну реакцію приєднання до алкенів оксиду вуглецю (II) і водню (СО-]-Н2, водяний газ), в результаті якої утворюються переважно альдегіди. Реакцію проводять під тиском при нагріванні наявності карбонілів кобальту або нікелю (метод Реппе). З етилену, наприклад, цим методом добувають пропіоновий альдегід:
Н2С=СН2 СО + Н8125 сс/ 20,6 мг.’а~* Н3С—СН2—•
Пропіоновий альдегід
124
З гомологів етилену цим методом одержують суміш альдегідів з нормальним і розгалуженим вуглецевим ланцюгом:
О .О
Я-СН=СН, 4- СО + Н, —» /?-СН2—СН2—С< + І?—СН—С" .
сн3
/П олімеризаці я/Характерною особливістю багатьох ненасичених сполук є їх здатність вступати в реакцію полімеризації і утворювати полімери. Полімеризацією ненасичених сполук називають хімічну реакцію утворення полімеру(високомолекулярної сполуки) за рахунок з’єднання між собою великої кількості молекул ненасичених сполук (мономерів) ковалентними зв’язками, які виникають внаслідок розрив}' кратних зв’язків (л-зв’язків) у молекулах мономерів. Полімеризацію можна розглядати як своєрідне хімічне множення молекул мономерів. При полімеризації не виділяються побічні низькомолекулярні продукти. Полімеризація, наприклад, однозаміщених похідних етилену.може бути зображена загальною схемою:
лН2С=СН —> (—Н2С—СН—)„, 4 І
Мономер Полімер
деи — ступінь полімеризації, який може мати значення до кількох сотень тисяч одиниць. При значенні п = 2, 3, 4 ...10 сполуки називають олігомерами (від грец. «олігос» — небагато). Полімери знаходяться в генетичному(спорідненому) зв’язку з вихідними мономерами. Замісники 7? —атоми водню, хлору або групи —СН3, -СМ, С6Н5—, Н2С=СН—, —СООЛ№ та ін.
У хімії полімерів широко використовують також сумісну полімеризацію кількох різних мономерів, яку називають співполімеризацією.
Полімери, добуті методом полімеризації, називають переважно за назвою мономерів, до якої додають префікс полі-, що означає «багато». Наприклад, полімер, синтезований з етилену, називають поліетиленом, полімер пропілену — поліпропіленом і т. д.
За характером перебігу реакція полімеризації буває двох типів — ступінчаста і ланцюгова (лінійна). Ініціатором полімеризації можуть бути теплова енергія, тиск, опромінення та спеціальні хімічні реагенти.
За механізмом розрізняють іонну (катіонну і аніонну) та радикальну полімеризації.
Ступінчаста, або перервна, полімеризація була відкрита в 1873 р. О. М. Бутлеровим на прикладі «ущільнення» ізобутилену при нагріванні його з 20 %-м розчином Н2ЗО4. Ступінчаста полімеризація ізобутилену є типовим прикладом катіонної полімеризації, механізм якої описав ф. Уітмор в 1932 р. Ініціатором полімеризації в цьому випадку є протон кислоти, який приєднується до мономерного ізобутилену і утворює при цьому карбкатіон. Карбка-
125
тіон потім реагує з іншою молекулою мономеру, приєднуючись до атома вуглецю з подвійним зв’язком з більшою електронною густиною. В результаті утворюється димерний катіон, який, відщеплюючи протон з метиленової групи або однієї з метальних груп, може перетворитися на октен (І) або октен (II). Згідно з правилом Зайцева, утворюється переважно октен (І). Димерний катіон також може реагувати з третьою молекулою ізобутилену і утворювати тримериий карбкатіон, який далі може перетворюватися на тримери (III) і (IV) (триізобутилени):
8+ 8- +. +
(н3с)2с=сн2 + н—-(Нз,с)3с ,
8+ . + -»
(Н3о2с = сн2 + (н3с)аС+—- (н3с)2с - сна“ с (сн3)а;'
С-СН-С(СН3)3-^+ сн3
+(н3с:)2с =сна
-Н3С-С=СН-С(СН3)3
ЄН3 І
- Н2С = С -сн2- С(СН3).3
СНз і)
Днмери Ізобутилену
сн3
(Н3С)2С - СН2 -С-СНв-С(СНз)з-^ СН3,
(82%);
(18’4
СН3
І—.(н3с)2с ~ сі і-с-сн2-с(сн3)3; СНз ш
СНз
Н2С = С-СН2-С-СНа-С(СН3)3 .
СН3, СН3 Ц/
Тримери ізобутилену
За певних умов полімеризацію можна зупинити на необхідній стадії шляхом обриву реакційного ланцюга.
При гідруванні димерів І і II утворюється 2,2,4-триметилпентан (ізооктан). Аналогічно ступінчасто полімеризуються інші алкени з утворенням ненасичених сполук складнішої структури, гідруванням яких добувають цінне паливо і мастила.
‘ Ланцюгова полімеризація* Найпоширенішим типом полімеризації є ланцюгова, або лінійна, яка характеризується тим, що макромолекула утворюється в процесі однієї безперервної реакції внаслідок сполучення мономерів о-зв’язками за рахунок розриву л-зв’язків. У механізмі ланцюгової полімеризації розрізняють три стадії: 1) ініціювання і початок росту ланцюга; 2) ріст ланцюга; 3) обрив ланцюга полімеризації.
Полімеризація —екзотермічна реакція. На кожну ланку мономеру, що приєдналася, виділяється ~42 кДж.
Радикальна полімеризація. Радикальною називають полімеризацію, яка відбувається за радикальним механізмом. Радикальний
126
механізм полімеризації характеризується тим, що ініціаторами і проміжними частинками процесу є вільні радикали. Радикальна полімеризація відбувається під дією ініціаторів полімеризації, тобто речовин, які мають здатність генерувати в реакційному середовищі вільні радикали. Серед численних ініціаторів такої полімеризації часто застосовуються пероксидні сполуки, наприклад персульфат амонію, пероксид бензоїлу та інші, які вже при слабкому нагріванні розкладаються з утворенням частинок з неспаре-ними електронами (Р'):
ГШ4О8О2-
О—2О2ОМН4
(ГШ4)28О4 + 5О3 + -О';
О—С—СвН6 —> СО2 + свнБ- +свн5с-о-. І о
СвН6—С—О II о о
Пероксид бензоїлу
Особливо ефективним як ініціатор є 2-азо-2-метилпропаннітрил (азо-біс-ізобутиронітрил), який розпадається за схемою:
(Н3С)2С ЧдС(СН3)2 ІЧ2 + 2(Н8С)2С. СИ СМ СМ
Важливого значення набула полімеризація етилену в поліетилен (політен):
«Н2С=СН2 —> (-СН2-СН2-)«.
Етилен Поліетилен
Вперше полімеризацію етилену здійснив російський хімік Г. Г. Густавсон у 1884 р., який отримав полімер у рідкому стані з Ткнп — 128 °С і ступенем полімеризації п та 65. Полімеризація етилену може відбуватися під дією як радикальних, так і іонних ініціаторів. Радикальну полімеризацію проводять, нагріваючи етилен під тиском з киснем (молекули .О—О.— радикальні ініціатори). Процес полімеризації відбувається постадійно.
1. Ініціювання і початок росту ланцюга починається з приєднання радикальної частинки ініціатора до мономера і утворення початкового радикала:
і"....і Г'ї
/? 1 + Н2С-^-СН2 —> сн2—сн2.
Мономер Початковий радикал
2. Ріст ланцюга полягає в послідовному приєднанні молекул етилену до радикала, молекулярна маса якого послідовно зростає. Таке приєднання відбувається доти, доки ланцюг, який росте, збе= рігає радикальну будову:
Я—СН2—СН2 + «Н2С=СН2 —> /?—(СН2—СН2)„—СНа—СН2.
3. Обрив ланцюга (припинення полімеризації) може регулюватися спеціальними речовинами (інгібіторами полімеризації) або
127
відбуватися самовільно внаслідок сполучення двох радикалів чи з інших причин:
—(СН2—СН2)„-—СН2—СН2 + /?• > Я (СН2 СН2)„—СН2—сн2—7?;
—(СН2СН2)П—СН2СН2 -рСН2СН2 (СН2СН2),| 7? > /?—(СН2СН2)2„+2—р.
Поліетилен з молекулярною масою близько 20 000 складається з багатьох залишків етилену і насправді є алканом з дуже довгим вуглецевим ланцюгом.
іонна полімеризація ненасичепих мономерів відбувається при каталітичному ініціюванні цього процесу катіонами або аніонами. Тому іонну полімеризацію . поділяють на катіонну і аніонну.
Катіонна полімеризація, ініціюється сильними протонними і агіротонними кислотами, наприклад Н28О4, НС1, ВВ3, А1СІ3, ТіС14 та іншими, що перетворюють мономер на карбкатіон, під дією якого починається далі ріст ланцюга. Наприклад, низькотемпературна катіонна полімеризація ізобутилену під дією ВЕ3 відбувається у такі стадії:*
1. Початок росту ланцюга починається з приєднання каталізатора до мономеру і утворення карбкатіона:
с+б- + _
(Н3С),С=СІ І2 + ВГ3 —> Н3С-С-СН2 : ВР3
СН,
Ізобутилен
2. Ріст ланцюга відбувається шляхом приєднання молекул мономеру до карбкатіонів, молекулярна маса яких поступово зростає:
СН, б— СН,
І ' Г ; І Г3В—СН2—С+ + пН2С==С —>
сн3 сн3
сн, 1
—> В3В—СН2—с----
І сн3
СН3-1 сн3
І І
СН2—С— сн2—с* .
СН,- п—і сн3
Ланцюг росте доти, доки випадкове зіткнення з яким-небудь аніоном не обірве його.
Аніонна полімеризація широко застосовується з 1963 р., коли К. Ціглер добув (1952 р.)’комплексні металоорганічні сполуки, а потім разом з Д. Натта застосував їх як каталізатори полімеризації. Ініціатором полімеризації в цьому випадку є органічні аніони. Наприклад, комплекс з високополяризованим зв’язком [С2Н5]“[ТіС13] + утворюється під час взаємодії триетилалюмінію з тетрахлоридом титану:
АЦС^), + ЗТІС14 ЗС2Н8ТіС13 + А1С13.
128
Такі комплекси є високоактивними каталізаторами аніонної полімеризації, яка під їх впливом відбувається з високою швидкістю навіть при кімнатній температурі. Наприклад, аніонна полімеризація етилену при наявності 1 % (С2Н5)3А1 + ТіС14 (каталізатор Ціглера —Натта) легко відбувається в середовищі сухого бензину при невисокому тиску (15,15 • 10Б Па) і температурі 50—60°С без кисню повітря. В процесі такої полімеризації з етилену утворюється лінійний полімер (поліетилен) з молекулярною масою до 3 000 000. Процес полімеризації в цьому випадку також складається з трьох стадій.
1. Ініціювання ланцюга починається з приєднання першої мономерної молекули етилену до каталізатора і утворення поляризованої, але вже складнішої металоорганічної сполуки:
Н.С—СН. ТІЄЇ.
] і *— Ч"
—> Н3С—СН2—-СН2—СН2ТіС13.
Тб+ Г "1т6—
...СНа
2. Ріст ланцюга відбувається шляхом приєднання молекул мономеру до металоорганічних сполук, молекулярна маса яких поступово зростає:
Н3С—СН2—СН2—СП2ТіС13 + пН2С=СН2 —>
—> Н3С—СН„—(СН.СН.).—СН2—СН„ТіС13. V А ' А і’ І і А А О
3. Обрив ланцюга відбувається за рахунок відщеплення каталізатора:
НаС—СН2—(СН2СН2)П—СН2—СН2ТІС18 —>
—► Н8С—СН2(СН2СН2)„—СН=СН2-|-НТіС18.
Ріст ланцюга продовжується до повного використання мономер-ного етилену.
Аналогічно під впливом каталізатора Ціглера—Натта легко полі мер изуються і гомологи етилену, наприклад, пропілен:
(С,Н,)8АЬТІСІ4 пН2С=СН ---------------> (—Н2С—СН—)„.
сн3 Ін8
Пропілен Поліпропілен
Характерною особливістю каталізатора Ціглера — Натта є те, що під його дією в багатьох випадках утворюються стереорегулярні полімери. Такі полімери утворюються при полімеризації несиметричних ненасичених мономерів, наприклад пропілену. Стереохімічний контроль полімеризації пропілену, як і інших несиметричних мономерів, здійснюють модифікацією каталізатора. При полімеризації на каталізаторі, нанесеному на кристалічний носій, добувають ізотактичний поліпропілен, у якого всі метильні групи розміщені по один бік від площини макромолекулярного вуглецевого ланцюга '(рис. 32, а). Каталізатор, нанесений на інший кристалічний носій,
5 1-120
1’9
дає можливість сдержувгп п синдіетактичний поліпропілен, у'якого метильні групи регулярно розміщуються то по один, то по другий бік вуглецевого ланцюга (рис. 32, б). Якщо каталізатор Ціглера— Натта нанести на аморфний носі й або полімеризацію пропілену вести під впливом інших ініціаторів, то утворюється атактичний поліпропілен, у якого метильні групи розміщені без будь-якого порядку відносно площини вуглецевого ланцюга (рис. 32, в).
Рис. 33. Ізотактичнпй поліпропілен
Різна просторова будова полімеру зумовлює його різні властивості. Так, в молекулі атактичного поліпропілену вуглецеві ланцюги внаслідок нерегулярної будови не можуть щільно прилягати один до одного. Тому атактичний полімер аморфний, низькоплав-кий(7пл =80°С), що обмежує його практичне застосування. В молекулі стереорегулярного ізотактичного поліпропілену метильні групи розміщуються в просторі дуже тісно одна біля одної (рис. 33, а), полімерний ланцюг набуває форми спіралі, в якій зникають просторові утруднення метильних груп (рис. 33, б). Така спіральна будова робить ізотактичний поліпропілен високоплавким (Тпл = 170 °С); з нього виготовляють міцне волокно.
130
Реакції окислення/ Ненасичені сполуки легко окис-лібютьсязз місцем етиленового зв’язку. Окислювачами можуть бути О2 (повітря), О3,СиО, А§20. 1\МпО4, СгО3, ОзО4, Н2О2, надкислоти, наприклад надоцтова Н3С—С—О—О—Н та ін. Окислення вико-
II
О
ристовують для промислових і препаративних синтезів, а також для встановлення місцеположення подвійних зв’язків у молекулах не-насичених сполук. Цілеспрямованим окисленням алкенів у промисловості добувають двохатомні спирти, альдегіди, карбонові кислоти та інші кисневмісні сполуки.
Окислення без розриву С—С-зв’язків. При каталітичному окисленні алкенів киснем повітря розривається тільки л-зв’язок і за місцем розриву приєднується атом кисню. Продуктами такого окислення є тричленні циклічні прості ефіри, які називають оксиранами, епоксисполуками, або а-оксидами. Наприклад, при га-зофазному окисленні етилену киснем повітря над металічним сріблом добувають оксид етилену:
Д8, 250 °с
Н„С=СН, + О,------> Н.С----СН,.
“ “ ' * “ А А
Ряд окислювальних реагентів взаємодіє з алкенами у м’яких умовах. В результаті такого окислення відбувається розрив тільки л-зв’язку, за місцем якого приєднуються дві гідроксогрупи, з утворенням двохатомних спиртів — гліколів. Особливе значення має відкрита в 1898 р. Є. Є. Вагнером реакція окислення алкенів перманганатом калію (реакція Вагнера), яка відбувається по типу м’якого окислення. Під час реакції етилену з ЦМпО4 при кімнатій температурі утворюється етиленгліколь:
ЗН2С=СН2 + 2КМпО4 + 4Н2О=Н2С—СН2 + 2Мп02 ф- 2К.ОН.
НО ОН
Перманганат калію окислює ненасичені сполуки в нейтральному, лужному або кислому середовищі.
Окислення алкенів перманганатом калію відбувається за механізмом і{нс-циклоприєднання І\МпО4 до подвійного зв’язку з утворенням проміжного нестійкого циклічного складного ефіру (І), який далі гідролізується водою і перетворюється на гліколь:
нон
і і
— С — С— + МпО3 , НО он
Другий тип окислення — взаємодія алкенів з пероксидом водню в мурашиній кислоті, а також з надкислотами, такими як Н3С—С—О—О—-Н, В3С—С—О—О—Н, СсНБ—С—О—О—Н, та ін
131
тими, з утворенням а-оксидів алкенів (реакція Прилежаєва).При такому окисленні також утворюються двохатомні спирти — гліколі, але приєднання ОН-груп до алкену в цьому випадку відбувається у транс-положення.Транс-приєднання гідроксильних груп зумовлене тим, що проміжними продуктами при такому окисленні є прості ефіри оксирани (оксиди алкенів), які потім взаємодіють з водою за 5д'2-механізмом. Розщеплення оксиранового циклу відбувається при каталітичній дії протонів. Протони спочатку утворюють з оксиранами іони оксонію, а останні під дією молекул води піддаються нуклеофільному заміщенню (5№) і в місці атаки нуклеофілу при цьому відбувається обертання конфігурації, що спричинює тронс-приєднання гідроксогруп до алкену:
^С = СС + Н3С —С—О—О—Н II о
-СНзСООН
V с о он
Х І |
у* * С х : * С н +
і І
О/ он он
н
Іон оксонію
Алкени при наявності каталізаторів можуть окислюватися киснем повітря. Так, етилен при наявності паладій-мідного каталізатора перетворюється на оцтовий альдегід.
Для алкенів, наприклад для пропілену, відома також реакція каталітичного окислювального амонолізу, яка є економічно вигідним промисловим методом добування одного з найважливіших мономерів — акрилонітрилу, утворення якого відбувається за такою реакцією:
Н2С=СН—СН3 -}- 1,5О2 + ГШ3 —> Н2С=СН—СИ + ЗН2О.
Акрилонітрил
\ Окислення алкенів з розривом С—С-зв’язків. Більш енергійна дія окислювачів (нагрівання, збільшення концентрації окислювача) приводить до розриву молекули алкену за місцем подвійного зв’язку і утворення карбонільних сполук або карбонових кислот. Наприклад, при нагріванні пентену-2 з КМпО4 молекула алкену розщеплюється за місцем подвійного зв’язку з утворенням оцтової і пропіонової кислот:
кмпо4, X) £)
Н.С-СН=СН—СН2-СН3------------»- Н3С-С^ + Н3С-^СН2-С^ .
Пентен-2 3 нагрівання \ОН Т \0Н
Оцтова кислота Пропіонова кислота
Утворення в результаті такого окислення кислот, молекули яких містять відповідно два і три вуглецевих атоми, свідчить про
132
те, що подвійний зв’язок у молекулі пентену знаходився після другого С-атома.
Окислення такого типу використовують для встановлення місцеположення подвійного зв’язку в молекулі алкену, а отже, будови алкену.
Більшість алкенів, як встановив К. Гаррієс, навіть при низьких температурах реагують з озоном. При цьому озон приєднується до подвійного зв’язку з одночасним його розривом і утворенням циклічних пероксидних сполук, які називають озонідами. Озоніди, подібно до більшості сполук, які містять пероксидні зв’язки (—О—О—), можуть несподівано вибухати з великою силою. Тому озонування необхідно здійснювати з великою обережністю. Озоніди, як правило, не виділяють у чистому вигляді, а розкладають, гідролізуючи водою. При цьому утворюються карбонільні сполуки, за будовою яких можна встановити положення подвійного зв’язку в молекулі алкену. Наприклад:
НаС=СН—СН2—СН3 + О3 > Н2С------сн—сн2—сн3 >
Бутеи-1 \ /
4~Н2О
—> Н2С—О—СН—СН2—СН3 —- о > Н2С=О +О=СН—СНа—СН3;
II 22 Мурашиний Пропіоновий альдегід
О-----о альгегід
Н3С—СН=СН—СН3 + О3 Н3С—СН СН—СН3 —*-
Бутеи-2 \п /
'“'з
+Н,О
—> Нзс—СН—О—СН—СН3---------> 211,С—С^ і
[ | -н2о, \н
О------О Оцтовий
Н3С—С=СН—СН3 + О3 —> (Н3С)2С----СН—СН3 —>
!снз Х°зХ
2-Метил бутеи-2
--> (Н3С)2С-О-СН-СН3 (Н3С)2С=О + О=сн-сн3.
II —ПаО» А „
І Ацетон Оцтовий
, о----о альдегід
/Реакції заміщення. Для алкенів можливі реакції заміщення атомів водню, наприклад, на галогени. При цьому утворюються ненасичені галогенопохідні, які мають велике промислове значення. Так, при термічному хлоруванні (450—500°С) пропілену хлор не приєднується до подвійного зв’язку, а заміщує атом водню в метальній групі. В результаті утворюється хлористий аліл (З-хлорпропен-1, ТкИп = 45 °С). Реакція відбувається за радикальним механізмом заміщення (5^-реакція):
С1Ц" СІ—>2С1-;
Н2С=СН— СН3 + - сі —Н2С=СН—СН2 + неї; і н.с=сн—сн,
Н2С=СН—СН2 +СІ СІ—>Н2С=СН—СН2—С1 + СР-----------> 1 т. д.
Застосування алкенів. Етилен, пропілен та Ізобутилен широко використовуються в промисловому органічному синтезі:
133
Використання етилену, пропілену та ізобутилену в промисловому органічному синтезі
Синтетичні волокна
СН2С1-СН = СН2
Н3С-СН-СН2
(-СН-СН2-) І СНз
СН3-СН-СН ОН
С6Н5-СН(СН3)2
Н3С-СН=СН2
СіІ2 —
СН2ОН - сн он - СН2ОН
— Н2С = СН-СИ
І
Синтетичні каучуки, синтетичні волокна-
МН3,О2
Н2С—СН2
Н3С- НС\ о о—СН2 ♦ '
134
Контрольні запитання і вправи
1. Зобразіть графічні формули всіх ізомерів вуглеводнів, що мають склад С5Н10, і назвіть їх за міжнародною номенклатурою.
2. Якими зв’язками сполучені між собою атоми в молекулах етиленових вуглеводнів? Яка їх електронна природа?
3. Які речовини утворюються під час взаємодії етилену і пропілену з Вг2, НС1, Н2О при наявності концентрованої Н28О4 і з водним розчином КМпО4?
4. Які речовини утворюються під час взаємодії бутену-1 з НС1, НВг і НІ при наявності пероксидних сполук та без них?
5. Поясніть, чому утворення Вг—СН2—СН2—Сі у реакції етилену з Вг2 при наявності Сі" доводить, що реакція починається з електрофільної атаки алкену катіоном Вг+?
6. Поясніть, чому в розчині метилового спирту (Н3С—ОН) бром, приєднуючись до етилену, утворює не тільки 1,2-диброметан, а також і Вг—СН2—СН2—О—СН3.^
7. Розмістіть етилен, пропілен та ізобутилен у порядку зростання активності їх подвійного зв’язку до реакцій бромування.гідрогалогенування та гідратації. Напишіть схеми цих реакцій.
Розділ 15. АЛКІНИ (АЦЕТИЛЕНОВІ ВУГЛЕВОДНІ)
-Л Ненасичені вуглеводні, молекули яких містять між вуглецевими атомами потрійний зв’язок —С=С—, називають ацетиленовими вуглеводнями, або алкінами. Потрійний зв’язок —С=С— для ацетиленових, які подвійний ^>С=С<^дляетиленових вуглеводнів, ввів у органічну хімію Е. Ерленмейер у 1862 р. для збереження в цих сполуках чотиривалентності вуглецю.
Загальна формула ацетиленових вуглеводнів СпН2п—2. У молекулах алкінів на чотири атоми водню менше, ніж в алканах.і на два атоми водню менше порівняно з алкенами. Алкіни утворюють гомологічний ряд, який можна легко записати, виходячи з гомологіч-нихрядів алканів або алкенів. Для цього необхідно відняти’від двох сусідніх вуглецевих атомів алкану чотири атоми водню (по два від кожного, табл. 13), або, якщо виходити з гомологічного ряду алкенів, то потрібно відняти два атоми водню (по одному від тих атомів вуглецю, які сполучені між собою подвійним зв’язком). Першим представником гомологічного ряду алкінів є ацетилен Н—О=С—Н. Члени гомологічного ряду алкінів, як і члени рядів алканів і алкенів, відрізняються між собою на гомологічну різницю, групу —СН2—.
Номенклатура. За сучасною систематичною номенклатурою назви ацетиленових вуглеводнів утворюють від назв відповідних насичених вуглеводнів з тією самою кількістю вуглецевих атомів шляхом заміни в їх назві суфікса -ан на суфікс -ин (-ін). За основний вибирають найдовший вуглецевий ланцюг, в який обов’язково повинен входити потрійний зв’язок. Положення потрійного зв’язку в ланцюгу позначають цифрою, яка вказує, після якого вуглецевого атома розміщений цей зв’язок, наприклад, бутин-1, бутин-2 і т. д.
135
Таблиця 13. Гомологічний ряд, номенклатура та фізичні властивості алкінів
Насичені вуглеводні Ацетиленові вуглеводні
Формула Назва Формула Назва за номенклатурою Гпл. °с Гкип’ °С Густи- на
систематичною | раціональною
н3с-сн3 Етан нс=сн Етин Ацетилен —81,8 —83,6 0,565*
н3с—сн2—сн3 Пропан нс^с—сн3 Пропін Метил ацетилен —102,7 —23,3 0,670*
Н3С—сн2—сн2—сн, Бутан нс=с—сн,—сн3 Бутин-1 Етилацетилен —122,5 8,5 0,678**
н3с—с^с—сн3 Бутин-2 Диметил-ацетилен —32,3 27,0 0,691
н3с—сна—сн2—сн,—сн3 Пентан нс^с—сн,—сна—сн3 Пентин-1 Пропілаце-тил єн —98,0 39,7 0,691
н3с-с^с-сн2—сн3 Пентин-2 Метилетил-ацетилен —101,0 56,1 0,710
* При температурі кипіння.
** При 0 °С.
(табл. 13). Ланцюг нумерують так, щоб атом вуглецю, який вказує положення потрійного зв’язку, одержав менший порядковий номер
Інколи для назви ацетиленових вуглеводнів користуються раціональною номенклатурою. За цією номенклатурою алкіни розглядають як ацетилен, у якого один або обидва атоми водню заміщені на відповідні вуглеводневі радикали (алкіли). Наприклад, метил-ацетцлен, етилацетилен, диметилацетилен і т. д. (табл. 13).
Ізомерія. Для алкінів властива ізомерія, зумовлена положенням потрійного зв’язку (наприклад, бутин-І і бутин-2) та розгалуженням вуглецевого ланцюга:
Н—С=С—СНа—СН2—СН3 і н—с=с—сн—сн3.
Пенткн-1 • 1
СН3
ЗДАетії/! бутин-1
Методи добування. Ф. Велер у 1862 р. вперше добув ацетилен з карбіду кальцію, діючи на нього водою. Карбідний метод добування ацетилену (після того, як А. Муассан у 1892 р. розробив технологічний спосіб виробництва карбіду кальцію з вугілля і вапняку) набув широкого промислового застосування:
цзс, 3000 »с
СаСО3 ——> СаО--------------> СаС2 + СО;
СО‘! Карбід
кальцію
•С."н іон
ЧІІ + і
N с н іон
НС=СН + Са(ОН)2.
Проте карбідний метод добування ацетилену досить дорогий. У промислових масштабах ацетилен добувають окислювальним піролізом метану:
1500 °с
6СН4 4- О2--> 2НС=СН + 2СО + 10Н2.
Гомологи ацетилену добувають такими методами:
1. Дегідрогалогенуванням віцинальних дигалогеналкапів, які, в свою чергу, добувають галогенуванням алкенів. Наприклад:
Н3С—СН=СН2 + Вг2 —>Н3С—СН—СН2—с—~^агг—?ння-> н3С—С = СН.
Пропен І | Пропік
Вг Вг
2. Дегалогенуванням тетрагалогенопохідних алканів, нагріваючи їх з порошкоподібним цинком:
Н3С—СВг2—СНВг2 + 22п ► Н3С—С = СН + 22пВга.
3. Алкілуванням ацетиленідів лужних металів:
б- с+ б- с+
Н3С—С=С—На +1—СН3 — Н3С—СгС—СН3 + Иаі.
Метил ацетилен Ід Бутнн-2
натрію
137
Рис. 34. Перекривання р-орбіталей в молекулі ацетилену
Рис. 35. Будова молекули ацетилену:
а — тетраедрична модель МОЛЄКУЛИ; б — КуЛЄСТЄр>К-иева модель молекули; в —масштабна модель молекули за Стюартом .—
Бріглебом
а
\ Фізичні властивості. Ацетиленові вуглеводні від С2Н2 до С41 і;, заґзвичайних умов — газоподібні речовини (табл. 13), починаючи з С5Н8 — рідини, а С16Н30 і вищі алкіни —тверді речовини. Закономірності в зміні температур кипіння і плавлення в гомологічному ряду алкінів аналогічні змінам в рядах метану і етилену. Алкіни мають вищі температури кипіння, ніж подібні до них алкани і алкени з такою самою кількістю вуглецевих атомів у молекулі. Густина ацетиленових вуглеводнів також вища, ніж алканів і алкенів. Алкіни-1 мають невеликі дипольні моменти (0,75 — Ю), що свідчить про полярність їх молекул.
В ІЧ-спектрах ацетиленові вуглеводні мають характеристичні смуги поглинання валентних коливань зв’язку —С=С— при 2100—2300 см'1. УФ-світло алкіни поглинають в області до 200 нм.
В ПМР-спектрах протони групи —Сн^С—Н дають сигнали в області 2—3 млн-1._Д
Хімічні властивості. Хімічні властивості алкінів зумовлені наявністю в їх молекулах потрійного зв’язку —С=С—, який складається з одного о-і двохл-зв’язків. Найпростішим представником сполук з таким зв’язком є ацетилен. Утворення потрійного вуглець-вуглецевого зв’язку можна уявити на прикладі реакції дегідрогенізації етилену. При відщепленні водню від етилену в кожного з його вуглецевих атомів розпаровується по одному р-електрону, при спа-ренні яких утворюється другий л-зв’язок. Два л-зв’язки в молекулі ацетилену орієнтовані в просторі у взаємно перпендикулярних
138
площинах, оскільки р-електрони атома вуглецю, які утворюють ці зв’язки, орієнтовані в просторі у взаємно перпендикулярних площинах (рис. 34). Інші зв’язки в молекулі ацетилену є о-зв’язками (рис. 35). Вони утворені яр-гібридизованими орбіталями вуглецевих атомів: )
Гібридизація
На
На
утворення утворення
5ЇІ -зв’язку Л2-зв’язку
На утворення 0-зв’язків
Фізичними методами дослідження встановлено, що молекула ацетилену має лінійну будову, в ній усі чотири атоми розміщені на одній прямій (рис. 35). Вуглець в молекулі ацетилену знаходиться в яр-гібридизованому стані. Обидві яр-гібридизовані орбіталі кожного з них напрямлені в просторі у взаємно протилежні боки під
Рис. 36. Циліндрична л-електронна орбіталь молекули ацетилену
кутом 180° (див. рис. 11). я-Електронна густина потрійного зв’язку має циліндричну симетрію і є симетричною відносно лінії, яка сполучає ядра чотирьох атомів (рис. 36).
Молекула ацетилену характеризується такими параметрами: зв’язок С===С—довжина 0,12 нм, енергія 831 кДж/моль; зв’язок С—Н —-довжина 0,106 нм, енергія 473 кДж/моль; дипольний момент становить 1,00.
Між я-електронною густиною подвійного С=С- і потрійного С=С-зв’язків існує певна відмінність, я-Електронна густина зв’язку С =С легко змінюється, поляризується піддією різних факторів. я-Електронна густина зв’язку С=С значно важче піддається поляризації, оскільки довжина цього зв’язку менша порівняно із зв’язком С=С, електронегативність вуглецю в яр-гібридизованому стані порівняно з вуглецем в ярг-гібридизованому стані вища і досягає значення 2,75 (приблизно така, як брому). Тому електрони атомів вуглецю у яр-гібридизованому стані більш глибоко втягуються всередину молекули, більше сконцентровані між вуглецевими ядрами
139
і сильніше утримуються електронегативнішими вуглецевими атомами, ніж у зв’язку С=С. В результаті ацетиленові вуглеводні, незважаючи на більшу ненасиченість, виявляють меншу реакційну здатність до приєднання електрофільних реагентів, ніж етиленові. В той же час в алкінах зовнішні області ядер атомів вуглецю з потрійним зв’язком стають відносно збідненими електронною густиною. Тому вони виявляють більшу .схильність, ніж алкени, до реакцій приєднання нуклеофільних реагентів, таких як спирти, аміни, синильна кислота тощо.
Алкіни вступають у реакції приєднання, які можуть відбуватися за радикальним, електрофільним і нуклеофільним механізмами, а також в реакції заміщення атомів водню, розміщених біля атома вуглецю з потрійним зв’язком.
' Реакції приєднанн я/Приєднання різноманітних реагентів'до алкінів відбувається в основному в дві стадії. Спочатку утворюються сполуки етиленового ряду, а потім —насичені вуглеводні або їх похідні.
Приєднання водню (гідрування)^ Алкіни послідовно приєднують До зв’язку С==С 2 моль водню:-»'
+нг +н,
/?—с=с—р —> р—сн=сн—р —> р—сн2—сн2—р.
Гідрування алкінів, як і алкенів, відбувається при наявності таких каталізаторів, як Рі, Рб, Мі тощо. Потрійний зв’язок С=С гідрується активніше, ніж зв’язок С=С в молекулах алкенів. Використовуючи спеціальні реагенти, до потрійного С=С-зв’язку можна приєднати тільки два атоми водню і добувати таким чином алкени. Часткове відновлення ацетиленових вуглеводнів цікаве ще й тим, що, залежно від природи відновлюючого реагента і умов проведення реакції, гідрування алкінів можна здійснити стерео-напрямлено. Так, при відновленні алкінів натрієм у рідкому аміаку добувають транс-алкени, а при відновленні над каталізатором Ліндлара (Рд з домішкою РЬ)—^цс-алкени. Наприклад:
Н3С—С=С-СН3
Бутив-2
Н.С, ,н ка+кн, 3 \ /
-------> С=С І
Нх \сн3 тране-Вутен-2
Рд/РЬ
Н3С\ /СН, с«=с хн
цде-Бутен-2
{ Галогенуванням Алкіни можуть приєднувати за місцем потрійного С^С-зв’язку дві' молекули галогену. Приєднання, наприклад, хлору до ацетилену в газовій фазі відбувається дуже швидко і може супроводитись вибухом. Тому хлорування проводять у рідкій фазі, в середовищі тетрахлоретану при наявності каталізатора 8ЬС13.
140
При цьому спочатку утворюється транс-1,2-дихлоретилей, який потім з другою молекулою хлору утворює 1,1,2,2-тетрахлоретан. Галогенування алкінів, так .як і алкенів, може відбуватися як за радикальним, так і за електрофільним механізмом. Наприклад:
8ЬС1,
СІ: СІ =г=* СІ+ + :С1“! ' .
СІ СІ
НС=сн+СГ-*[нС=СН—Сі]„/С==С\( сн—сн.
1 Нх ХС1 І І
• - - • . СІ СІ
Гідрогалогенування. Галогеноводні приєднуються за місцем потрійного СзаС-зв’язку також у дві стадії. Спочатку утворюються вінілгалогеніди, які потім приєднують за правилом Марковникова другу молекулу галогеноводню і утворюють гемінальні дигало-геналкани. Підбираючи умови, можна зупинити реакцію на першій стадії.
Найбільше практичне значення має приєднання до ацетилену хлороводню, оскільки продуктом реакції є цінний мономер — хлористий вініл:
НдСІ, 4-НС1, 2пС1,
НС=СН + неї------> Н2С=СН-С1------------> Н3С—СНС12.
Приєднання галогеноводнів до гомологів ацетилену відбувається в т/щне-положення і за правилом Марковникова. Спочатку утворюється вінілгалогенід, який потім приєднує другу молекулу галогеноводню і дає гемінальну дигалогенопохідну:
б" 4-НС! 1
Н3С->С=СН + Н+СГ —> Н3С—С=СНа--------> Н3С—СС12—СН3.
9 х
Приєднання води (гідратація). Алкіни при наявності каталізаторів приєднують воду і утворюють карбонільні сполуки. Промислового значення набула реакція гідратації ацетилену, відкрита в 1881 р. М. Г. Кучеровим, який встановив, що приєднання води до ацетилену відбувається в середовищі 10 %-го розчину На8О4 при наявності 5 % сульфату ртуті. Продуктом реакції є оцтовий альдегід:
Ня8О41Н„5О4 >0
НСгСН + Н2О----------> Н3С—С" .
ХН
Гідратацію ацетилену називають реакцією Кучерова. Механізм цієї реакції ще не повністю вияснений. Вражають, що спочатку до зв’язку С^=С приєднується каталізатор і утворює з ним продукт приєднання (І). Продукт (І) далі гідролізується в енол —нестійкий вініловий спирт (її), який відразу ж ізомеризується (таутоме-ризується) в термодинамічно стійкішу сполуку (в карбонільну форму) — оцтовий альдегід (III):
141
НС = СН + Н§504 -> НС:
сн —-нкзо,
і І
Н.,С=СП— О—н
І...... і
ІІ
о о
Органічні сполуки, в молекулах яких ОН-група сполучена з атомом вуглецю з подвійним зв’язком, як наприклад, у вінілового спирту (II), називають енолами (єн означає наявність у молекулі подвійного С=С-зв’язку, ол -—ОН-групи спирту).
О. П. Ельтеков у 1877 р. сформулював правило, згідно з яким ненасичені спирти з групою ОН біля атома вуглецю з подвійним зв’язком є нестійкими і самовільно ізомеризуються в стійкі карбонільні сполуки. Це правило виконується переважно для простіших енольних систем.
Дослідження хіміків показали, що завжди при добуванні сполуки з енольною структурою одержують ізомерну їй сполуку з карбонільною структурою. Між енольною і карбонільною структурами існує рівновага:
[—С=С—О—н] —X —С—С=О.
Енольна | |
структура }]
Карбонільна
структура
Таке перегрупування енольної структури в карбонільну’вїдбу-вається особливо легко і тому рівновага цього процесу сильно зміщена в бік карбонільної структури. Причина такої ізомеризації полягає в тому, що внаслідок високої полярності зв’язку О—Н атом водню цієї групи енолу може легко відщеплюватись від атома кисню у вигляді іона. При зворотному приєднанні цей іон водню може приєднатися як до атома кисню, так і до атома вуглецю з подвійним зв’язком. Якщо Н+ приєднається до кисню, то дуже легко зможе знову відщепитися. Якщо цей іон приєднається ДО вуглецю, то вже залишиться сполученим з ним, оскільки полярність зв’язку С—Н досить мала. В результаті відбувається перегрупування, при якому сильніша кислота перетворюється на значно слабкішу:
І І
—С=С—О-Н 4=*
Сильніша кислота
II Г _____ II
—С=С—о] Н+—І—с—с=о.
І н
Значно слабкіша кислота
142
Структурні ізомери, між якими існує рівновага, називають таутомерами, а явище існування такої ізомерії називають таутомерією (див, розд. 24).
Гідратація гомологів ацетилену приводить до утворення кетонів. Приєднання води до алкінів-1 відбувається за правилом Марковникова. Наприклад, при гідратації проліну утворюється Диметилкетон (ацетон):
б- с+ 6-
Н8С->СнеСН + НОН
НЄ8О4
Н,8О4
'Н8С—С=СН21-----> Н3С—с—сн8.
ОН .1 о
Диметилкетон, ацетон
Алкіни можуть вступати також у реакції нуклеофільного приєднання. Так, ацетилен при наявності каталізаторів приєднує спирти, карбонові кислоти, синильну кислоту та інші нуклеофіль-ні реагенти. В результаті утворюються вінільні сполуки, які є цінними мономерами для добування полімерів, а також вихідними речовинами в органічному синтезі.
О. Є. Фаворський встановив, що ацетилен і його гомологи при наявності гідроксиду калію досить легко приєднують спирти і утворюють прості вінілові ефіри:
НС=СН + II—О—СІ Т2—СН8 —- Н2С=СН—О—СН2—СН8.
ВІнілеталовий ефір
При наявності кислотних каталізаторів до ацетилену приєднуються карбонові кислоти. Продуктами такого приєднання є складні вінілові ефіри:
НСнСН + Н—О-С—СН3 н2с=сн—О—С—СН3.
II II
О О
Оцтова кислота Вінілацетат
Ацетилен легко приєднує синильну кислоту і утворює важливий мономер — акрилонітрил:
НО=СН + НС1Ч н2С=СН—СМ.
Акрилонітрил
і Реакції заміщення. Зміна характеру гібридизації атома вуглецю в ряду етап, етилен, ацетилен приводить до зміни його електронегативності, величини атомного радіуса, а це, в свою чергу, —до зміни віддалей між атомами в молекулах, дипольних моментів зв’язків С—Н в молекулах цих сполук і кислотних властивостей цих зв’язків (табл. 14).
Як видно з даних табл. 14, електронегативність вуглецю в ряду етан, етилен, ацетилен зростає і в стані 5,о-гібридизації стає най-
143
Таблиця 14. Характер гібридизації і будови етану, етилену 1 ацетилену
Вуглеводень Гібридизація Елек? ренегат ив-ність вуглецю Атомний радіус вуглецю, нм Між'ядерна віддаль зв’язку С—Н, нм Дипольний момені зв’язку С— н, Н Константа кислотності зв'язку СН рК25 н к
Н3С—сн3 хр3 2,50 0,0722 0,1102 0,307 42
І ІгС=СНа «р2 2,62 0,074 0,1081 0,629 36,5
нс=сн їр 2,75 0,069 0,1064 1,05 25
6— с+
більшою. Внаслідок цього електрони зв’язку С-«- Н в молекулі ацетилену зміщуються до атома вуглецю більше, ніж в молекулах етану і етилену, що зумовлює зростання полярності цього зв’язку А це веде до зменшення між’ядерної віддалі, збільшення дипольного моменту С—Н-зв’язку і значного посилення здатності атома водню, який знаходиться біля хр-гібридизованого атома вуглецю, відриватись у вигляді протона.
В результаті підвищення полярності зв’язку між атомами вуглецю з потрійним зв’язком і водню, а також внаслідок того, що в аніоні А—Ск=С~ ступінь х-характеру орбіталі, яка несе не-поділену пару електронів, складає 50 %, ацетилен і алкіни-1 /?—С==СН мають кислотні властивості і відносно високу рівноважну кислотність:
Я—О=С—Н 5==а= Я—с=с- + Н*
В табл. 14 наводяться значення рА> для етану, етилену і ацетилену (чим менше значення р/(к, тим сильнішою є кислота). Для порівняння наводимо значення р/Ск' для води, що дорівнює 15,7. З величин р7(к цих речовин видно, що кислотні властивості С—Н-зв’язку в ацетилені, порівняно з етиленом і етаном, найвищі, але менші, ніж у води. Тому атоми водню в молекулах ацетилену і ал-кінів-1 легко заміщуються на атоми металу. В результаті утворюються солеподібні металічні похідні, які називаютьацетиленідами. Ацетилен і алкіни-1 утворюють ацетиленіди з калієм, натрієм, амідом натрію. Ацетиленіди лужних металів використовують для синтезу гомологів ацетилену:
Н—С=С—Н + 2К —* К—С=С—К + Н8.
Ацетиленіди лужних і лужно-земельних металів — стійкі речовини, не вибухають. Стійким є також ацетиленід кальцію СаС2, який називають карбідом кальцію. Вода, яка є сильнішою кислотою, ніж ацетилен, миттєво розкладає ацетиленіди цих металів на ацетилен і гідроксид відповідного металу:
Ка-С^С-Ма + 2НОН->Н-СвС-Н + 2№ОН.
144
Ацетилен і алкіни-1 утворюють з розчинами гідроксидів міді (І) або срібла не розчинні у воді ацетиленіди міді червоно-коричневого кольору і ацетиленіди срібла білого кольору:
Н—С=С—Н + 2 [Си (№Ч3)2] ОН > Си—С=С—ОД + 4НН3 Д 2Н2О;
Н—С=С—Н + 2 [А§ (№3)21 ОН > АЄ_С=С—Ай] + 4ИН3 Д 2Н2О.
Ацетиленіди срібла, міді та інших важких металів у сухому стані нестійкі і розкладаються з вибухом. Ацетиленіди важких металів, на відміну від ацетиленідів лужних і лужно-земельних металів, водою не розкладаються, а розкладаються тільки розбавленими кислотами. Реакцію утворення не розчинних у воді ацетиленідів міді і срібла використовують для якісного відкриття водню (ацетиленового водню) в групі С=С—Н.
/'Атом водню ацетилену і алкінів Д—С=С—Н має достатню кислотність і вступає в реакцію з алкілмагнійгалогенідамн (реактивами Гріньяра). При цьому виділяються алкани і ацетиленові магнійорганічні сполуки, які називають реактивами Іоцича. Ці реактиви широко використовуються в різноманітних синтезах:
д_С=С—Н + С2Н8—Ме— Вг —>/?—С=С—Вг + С2Нв.
Полімеризація ацетилену'. Для ацетилену можлива циклічна і лінійна полімеризація. М. Бертло в 1866 р. помітив, що при пропусканні ацетилену через нагріту до 500 °С скляну трубку три молекули ацетилену циклізуються в молекулу бензолу. Радянські хіміки М. Д. Зелінський і Б. О. Казанський (1924— 1930 рр.) вдосконалили цю реакцію. Пропускаючи ацетилен над нагрітим до 400 °С активованим вугіллям, вони добули бензол з кількісним виходом:
сн
СН Активоване
4 |йП вугілля НС< СН
. СН Н(Д. СН
% СН
Пізніше, в 1948 р., В. Реппе встановив, що при наявності карбонільних сполук нікелю ацетилен при 60—70 °С циклізується в бензол і одночасно з ним утворюється ~12% стиролу. Змінюючи склад каталізатора, можна провести циклізацію чотирьох молекул ацетилену і отримати його тетрамер — циклооктатетраєн-1,3,5,7.
НС=СН ССеН^р-МСС(Ж
СН
СН
’НС=СН
4-
сн==сн
145
Ю. Ньюленд встановив, що при наявності хлориду міді (І) і хлориду амонію, розчинених у соляній кислоті, ацетилен поліме-ризується лінійно: дві молекули ацетилену сполучаються і утворюють бутен-1-ин-З (вінілацетилен). Така димеризація має велике промислове значення, оскільки вінілацетилен легко приєднує за місцем потрійного зв’язку хлороводень (за правилом Марковникова) і утворює 2-хлорбутадієн-1,3 (хлоропрен) — важливий мономер для виробництва хлоропренового каучуку:
н'с-сн + н-с-сн С||СІ- Н.С-СН-С.СН
| і Вінілацетилен
—> н2с=сн—с=сн2.
І
СІ
Хлоропрен
Каталітичним окисленням можна здійснити окислювальну поліконденсацію ацетилену і поступово добути бутадіїн-1,3, гекса-тріїн-1,3,5 тощо, аж до карбіну (алотропна видозміна вуглецю) в його поліїновій модифікації:
о. +нс=сн+о2
2НС=СН ЗЗЩГ НС—С—С=СН--------Гщо-----” НС^С-С=С—С^СН і т. д. -»
Бутадіїн-1,3 Гексатріїн-1,3,5
— (_с^С-С^С-)„.
Карбін
Окисле н н я. Зв’язок С:^С окислюється дещо важче, ніж зв’язок С=С в алкенах. Це легко помітити за повільнішим знебарвленням розчину КМпО4 ацетиленом порівняно з аналогічною реакцією етилену. При окисленні ацетилену КМпО4 відбувається розрив л-зв’язків його молекули з утворенням оксалату калію:
ЗНС=нСН + 8КМпО4=ЗКО—С—С—ОК + 8МпО2 + 2КОН + 2Н2О.
Оксалат калію
Алкіни, подібно до алкенів, горять. Так, ацетилен на повітрі горить кіптявим полум’ям, а в струмені кисню згоряє повністю і створює при цьому температуру до 2800 °С, що використовують у техніці для автогенного зварювання металів.
Застосування алкінів. З алкінів велике промислове значення має тільки ацетилен, який є сировиною для виробництва багатьох органічних сполук. Основні напрямки промислового використання ацетилену показані на схемі:
146
Основні напрямки промислового використання ацетилену
Синтетичний СНСІ2— СНСІ2 каучук бутадієновий
Полімери'
Синтетичні волокна
Синтетичний каучук Штрильний
СН2 = СН-СІЧ
СН2=СНСІ
СІІ,-СН,-ОН СНСІ = СНС1
Синтетичний каучук бутадієновий
Автогенне зварювання металів
СН2 — СН - сн2 -—не=С - СН2ОН СН 2 = Сі1 - с СН
ОН ОН ОН хО
Н-Сх неї
'н нс|
но- сн2- с=с - сн2- он сн2=сн-с=сн2
СІ
Полімери
сн2=сн-о-с
о4 о
С4Н<)-ОН
СН2=СН—О—С4Н9
І
Полімери
° І Ь,О
но- сн2- сн2—сн2—сн2 - он
І
Синтетичний каучук бутадієновий Синтетичний каучук хлоропреновий
Контрольні запитання і вправи
1. Зобразіть графічні формули всіх ізомерів вуглеводнів складу С5Н8, С6Н10 та назвіть їх за міжнародною номенклатурою.
2. Які речовини утворюються під час взаємодії ацетилену, бутину-1 і бутину-2 з Вг2, НС1 і Н2О при наявності солей ртуті?
3. Як за допомогою спектральних методів та хімічних реакцій можна розрізнити такі речовини: я-пентан, пентен-1, пентен-2, пентин-1 і пентин-2?
Розділ 16. АЛКАДІЄНИ [ДІЄНОВІ ВУГЛЕВОДНІ)
Ненасичені вуглеводні з двома подвійними етиленовими зв’язками називають алкадіенами, або дієновими вуглеводнями. Алкадіє-ни ізомерні відповідним алкінам і мають таку саму загальну формулу — СпН2ц—2-
147
За систематичною номенклатурою дієнові вуглеводні називають так, як етиленові, але суфікс -єн в їх назві замінюють на суфікс -дієн. Положення подвійних зв’язків позначають цифрами, які вказують, після яких вуглецевих атомів розміщені ці зв’язки. Нумерують вуглецевий ланцюг так, щоб цифри, які вказують положення подвійних зв’язків, мали в номенклатурі найменше значення. Наприклад: Н2С=С—СН=СН2—2-метилбутадієн-1,3,
СН3
НаС—С=С=СН2 —З-метилбутадієн-1,2.
СН3
Дієни називають також за історичною номенклатурою. Наприклад, бутадієн-1,3 називають дивінілом, 2-метилбутадієн-1,3— ізопреном і т. д. (табл. 15).
Таблиця 15. Формули, назви і константи ряду спряжених дієнів
Формула Назва за номенклатурою т 1 кип* °С Густина 4° „20 о
систематичною історичною
Н8С=СН—сн=сн. Бутадієн-1,3 Дивініл —4,41 0,650 —
Н3С—сн=сн—сн=сн2 Пентадієн-1,3 Піперплен 43 0,683 1,4280
н2с=є—сн=сн2 сн3 2-Метилбута-дієн-1,3 Ізопрен 34 0,681 1,4216
н8с—сн=сн—сн=сн—сн8 Гексадієн-2,4 Дппропеніл 79 0,662 1,4540
Н2С=С—с=сн, 1 1 н3с сн8 2,3-Диметил-бутадієн-1,3 Діізопропе- НІЛ 69 0,745 1,4328
Залежно від взаємного розміщення подвійних зв’язків алка-дієни поділяють на три групи.
1. Кумулени, або алени, в молекулах яких два подвійних зв’язки розміщені біля одного атома вуглецю. Прикладом таких дієнів е пропадієн-1,2 Н2С=С=СН2 (ален).
2. Дієни з Ізольованими подвійними зв'язками. В молекулах цих дієнів подвійні зв’язки відділені однією або кількома групами -СН2—: Н2С=СН— (СН2)„^СН =СН2 (п = 1, 2,3,...). Наприклад, гексадієн-1,5 (діаліл) Н2С=СН—СН2—СН2—СН=СН2.
3. Спряжені (кон'юговані) дієни містять групу атомів >С=С—С=С<.
І І
У молекулах цих дієнів послідовно чергуються подвійний зв’язок С=С, одинарний зв’язок С—С і знойу п'бдвійнии зв’язок С=С (чергування я- і о-зв’язків). Першим представником гомо
118
логічного ряду спряжених дієпів є бутадієн-1,3, або дивініл, Н2С=СН—СН=СН2. У табл. 15 наведено константи і назви найважливіших представників спряжених дієнів.
Ізомерія. Ізомерія дієнових вуглеводнів може бути зумовлена положенням подвійних зв’язків у.молекулі і розгалуженням вуглецевого ланцюга. Наприклад: бутадієн-1,2 Н2С=С=СН—СН3 і бутадієн-1,3, пентадієн-1,3 Н2С=СН—СН=СН—-СН3 і 2-метил-бутадієн-1,3 Н2С=С—СН=СН2.
І сн3
Бутадієн-1,3 може існувати також у вигляді двох конформерів, які взаємно переходять один у другий в результаті обертання навколо простого зв’язку С—С:
Н\ /Н
/С=Сч Л
нх хн
«-трсис-Бутадієн-1,3
Н\ /Н
С=С „ и/ с/
II
нх хн в-^/С-Бутадкн-1,3
Ці конформери відрізняються за стійкістю. я-Трпнс-форма (від англ.8Іп§1еЬоп<1) на 10,9 кДж/моль стабільніша, ніж х-гр/с-форма.
Методи добування спряжених дієнів. Із спряжених дієнів найбільше значення мають бутадієн-1,3 і ізопрен. Для добування цих дієпів розроблені такі промислові методи:
1. З продуктів крекінгу нафти. Газоподібні продукти крекінгу, що містять бутан і бутилен, перетворюють на бутадієн термокаталі-тичною дегідрогенізацією їх над А12О3/Сг2О3. Вихід бутадієну-1,3 при цьому становить 20—25 % за одну конверсію. Хімізм цього процесу можна описати такою схемою:
Н2С=СН-СН2-СН3-, Н3С-СН2-СН2-СН8^~
Н8С—СИ=СН—сн3—І н2с=сн—сн=сн2.
550 °С
-н, ’
Аналогічно термокаталітичним дегідруванням пентан-пентено-вої крекінг-нафтової фракції добувають ізопрен:
Н3С—СН—СН2—СН3 Н2С=С—СН=СН2 н2с=с—сн2—сн3.
сн3 сн3 сн3
Метод добування бутадієну піролізом нафтової сировини розробив Б. В. Бизов.
2. З етилового спирту. С. В. Лебедєв (1927—1930 рр.) розробив пром.исловий метод добування бутадіену-1,3 з етилового спирту і на його основі в нашій країні уперше в свігі почали добувати синте
149
тичний бутадієновий каучук. Перетворення етилового спирту на бутадієн відбувається при температурі 400—450 °С при наявності дегідрогенізуючих та дегідратуючих каталізаторів (М§О+ 2пО):
2Н3С—СН„—ОН > Н,С=СН—СН=СН„ 4- 2Н„О -4- Н„.
Механізм цієї реакції включає кілька стадій (Ю. А. Горін);
5-й- ,о н3с-с + н—сн2—с -Чн / чн
н3с-сн2-он—н3с-сх 4II
Оцтовий альдегід
Н2;
+н
Н3С-СН=СН-С ------
н
Кротоновий альдегід
хо н3с—сн—сн2—сх
ОН ЧИ
□ -Оксимасляний альдегід
н3с-сн=сн-сн2-он
-н2о
Кротиловий спирт
——Н2С = сн - сн=сн,.
-н2о 2
Електронна будова спряжених дієнів. Вивчення будови бутадієну фізико-хімічними методами (рентгенографічний аналіз, дифракція електронів) дало можливість визначити між’ядерні віддалі, валентні кути в його молекулі і встановити, що вуглецевий ланцюг молекули бутадієну-1,3 має переважно енергетично вигіднішу транс-будову (гр./с-копфігурацію, енергія якої вища на 12,6 кДж/моль, мають всього ~4 % молекул). Встановлено також, що довжина зв’язку між атомами вуглецю С2—С3 дорівнює 0,148 нм, тобто вона менша, ніж довжина одинарного зв’язку С—С в молекулах алканів (для етану —0,154 нм). Отже, вуглецеві атоми С2 і С3 в молекулі бутадієну сполучені між собою крім одинарного зв’язку ще додатковими силами, внаслідок чого ядра цих атомів вуглецю зближуються, що приводить до зменшення довжини зв’язку між ними:
0,133 нм 0,148 нм 0,133 нм;
Н2С=СН-------СН====СН2
Отже, в молекулі бутадієну-1,3 спостерігається тенденція до вирівнювання віддалей між атомами вуглецю.
Застосування в органічній хімії методу МО дає можливість робити висновок про істинну будову органічних речовин. За цим методом для кожного хімічного зв’язку вираховується порядок зв’язку — ступінь відхилення даного зв’язку від одинарного. Порядок зв’язку С—Є в молекулі етану приймають таким, що дорівнює 1, етилену 2, ацетилену—3. Ці цифри прямо пропорційні значенням електронної густини посередині віддалі між атомами, які утворюють зв’язок. На основі уявлення про порядок зв’язку виво-
0,134 нм.
І Н2С==СН2
0,154 нм;
І Н8С--------сна
150
дять поняття ступінь зв' язаності атома. З цієї величини знаходять відносну ненасиченість атома вуглецю, або індекс вільної валентності, який відіграє важливу роль при встановленні характеру взаємодії органічних речовин, зокрема бутадієну, в реакціях приєднання. Індекси вільної валентності і порядок зв’язків у молекулі бутадієну-1,3 наведені на його молекулярній діаграмі:
Н3С—і—СН, Н,С—л-^СН, 0 1 О А £ С-
0,837
0,391 0,391
-Н > Порядок ЗВ’ЯЗКІВ
0,837 ч- Індекси вільної валентності
Як видно з молекулярної діаграми, в молекулі бутадієну-1,3 кратність зв’язку між атомами вуглецю Сх—С2 і С3—С4 однакова і дорівнює 1,894, а між атомами С2—С3 вона становить 1,45. Це означає, що між атомами С\ і С2, С3 і С4 порядок зв’язку менший, ніж в молекулі етилену. За о-зв’язкомвін дорівнює 1, аза л-зв’язком— 0,89. Між атомами С2 і С3 порядок зв’язку більший, ніж у етану, і відповідно дорівнює за о-зв’язком 1 і за л-зв’язком 0,45. Отже, в молекулі бутадієну-1,3 зв’язки Сг—С2 і С3—С4 не є повністю подвійними, а зв’язок С2—С3—частково подвійний.
Індекси вільної валентності в молекулі бутадієну-1,3 на кінцевих вуглецевих атомах С4 і С4 більші, ніж на середніх —С2 і С3. Вони мають відповідно значення 0,837 і 0,391. Це свідчить про більшу я-електронну густину на кінцевих атомах вуглецю в молекулі бутадієну-1,3, тобто про більшу їх ненасиченість порівняно з середніми С2 і С3 атомами вуглецю. Тому кінцеві атоми вуглецю в молекулі бутадієну-1,3 мають підвищену реакційну здатність у реакціях Л/? і Ае.
Електронну будову бутадієну-1,3 пояснюють за допомогою методу МО ЛКАО так. Усі чотири атоми вуглецю в молекулі бутадієну знаходяться в ‘.//-гібридизованому стані і лінійно сполучені трьома о-зв’язками, які напрямлені в просторі під кутами 120° і утворюють остов плоскої молекули. Атоми вуглецю утворюють також шість о-зв’язків з атомами водню. Отже, у молекулі бутадієну-1,3 є дев’ять о-зв’язків, на утворення яких ідуть 12 електронів атомів вуглецю з 16 валентних їх електронів. Після утворення о-зв’язків у молекулі бутадієну-1,3 залишаються ще чотири р-орбіталі (по одному негібридизованому р-електрону в кожного атома вуглецю), хмари яких перпендикулярні до площини молекули (рис. 37). При бічному (латеральному) перекриванні електронна густина цих чотирьох р-орбіталей зливається, утворюючи чотири молекулярні орбіталі, енергія яких поступово зростає.’Ці чотири МО описуються функціями ф1,'ф!.,ф3 іф4 (знаки «-Ц-» і«—» означають знаки функції) (рис. 37). З рис. 37 видно, що в молекулі бутадієну-1,3 з чотирьох л-МОдвіМО^іфа) зв’язуючі і дві МО*(ф3іф4) розпу
151
шуючі. Зв’язуюча МО з мінімальною енергією Е± відповідає бутадієну-1,3 з делокалізованими л-зв’язками, утвореними внаслідок взаємного перекривання (злиття) чотирьох р-АО, що приводить до утворення єдиної л-МО, яка охоплює всі чотири вуглецеві атоми і є спільною для всієї молекули (рис. 38). Утворення спільної л-МО в молекулі бутадієну-1,3 відбувається внаслідок того, що між собою перекриваються не тільки р-АО С4 і С2 та С3 і С4, а також р-АО С2 і С3 атомів вуглецю. Отже, електронні хмари двох л-зв’язків у молекулі бутадієну взаємодіють між собою. Така взаємодія називається л,л-спряженням.
Рис. 37. о-Зв’язки і л-молеку-' лярні орбіталі в молекулі бута-діену-1,3:
а — базисні АО спряженої системи; б — делокалізовані л-МО
Й
й
й
й
Рис. 38. Схематичне зображення будови молекули бутадієну-1,3 (а) і вигляд молекули зверху (б)
1 2 3 4 2 3
(перекривання електронних орбіталей між С—С і С—С більше, ніж між С—С)
Зв’язуюча МО з дещо більшою енергією відповідає бутадієну-1,3 з ізольованими л-МО подвійних зв’язків.
Розпушуючі МО* трз і МО* зр4 з високими енергіями в основному стані залишаються вакантними.
МОфг і МОя)і2 за звичайних умов найбільш стійкі і визначають
152
л-електронну будову бутадієну-1,3. Відповідно до концепції теорії резонансу, бутадієн-1,3 можна розглядати як резонансний гібрид структур І—IV. Резонанс зображують за допомогою двосторонньої стрілки між структурами:
Н2С=СН—СН=СН2оН2С—СН=СН—СН2 о н2с—сн=сн—сн2
І 2 2 II 2 2 III
Л .4 о н2с—сн=сн—сн2.
IV
Структури II і III вказують нате, що зв’язок між атомами Са іС3 частково подвійний. Формула IV зображує бірадикальнй характер молекули. Над крайніми атомами вуглецю зазначено неспарені електрони, а стрілками біля них вказаний напрямок їх спінів.
Спряжені дієни мають підвищену енергію утворення. На основі значень енергій зв’язків була теоретично обчислена енергія утворення бутадієну-1,3, яка повинна становити 3269,3 кДж/моль. Експериментально знайдена енергія утворення бутадієну-1,3 Дорівнює 3291,3 кДж/моль. Отже, експериментально знайдена енергія утворення молекули бутадієну-1,3 більша від теоретично обчисленого значення. Різниця, яка в даному випадку дорівнює 15 кДж, називається енергією спряження, або енергією резонансу Ео. Експериментально Еа можна визначити з порівняння значень теплот гідрування спряжених і неспряжених л-зв’язків. Наприклад:
І І2С=СН—-СН—СН2 + 2На ► Н3С—СН2СН2—СН3 + 239,66 кДж;
2Н2С=СН—СН2—СН, + 2На > 2Н3С—СН2—СН2—СН3 + 254,55 кДж; Н2С=СН—СН2—СН=СН2+2На > Н3С=СНа—СН2—СНа—СНд+254,14 кДж.
Отже, реальна молекула бутадієну-1,3 приблизно на 15 кДж стійкіша (маєменшийзапасенергії), ніж молекула, якій відповідає структура І.
Статичні І динамічні ефекти спряження. У молекулі бутадієну-1,3 та в інших спряжених системах внаслідок взаємодії сусідніх л-зв’язків і утворення єдиної л-електронної МО передача взаємного впливу може відбуватися через спряжені л-зв’язки. Така передача взаємного впливу через л-зв’язки називається мезомерним ефектом (Л4-ефектом), або ефектом спряження (іноді позначають С-ефект) (див. розд. 3). М-Ефекти викликають зміщення л-електронів подвійних зв’язків, а також р-електронів неподілених пар окремих атомів, і поляризацію цих зв’язків. М-Ефекти передаються без помітного затухання вздовж всього ланцюга спряження і можуть бути позитивними та негативними (див. розд. 3).
Розрізняють два види Л4-ефектів: статичні і динамічні ефекти спряження. Статичний М-ефект відповідає тій поляризації молекули, яка відбувається в ній ще до початку реакції. В симетрично побудованих спряжених системах, наприклад у бутадієну-1,3 в статичному стані, тобто до початку його взаємодії з яким-небудь реагентом, має місце зустрічне спряження, при якому відбувається рівномірне зміщення л-електронних орбіталей подвійних зв’язків
153
до одинарного зв’язку, який знаходиться між цими електронними орбіталями:
Тому інколи молекулу бутадієну-1,3 зображують ще так: Н2С—СН— СН--ілСН2. Пунктиром підкреслюють тенденцію до вирівнювання л-електронної густини і зв’язків у молекулі.
В несиметричних спряжених дієнах, наприклад у піперилену, виявляється електронна асиметрія молекули і виникає статичний Н-М-ефект, який полягає в постійній поляризації молекули і появі в неї дипольного моменту р:
Г І і і
Н3С —СН=^СН-^-СН=і=СН2 ф = 0,500).
Динамічним М-ефектом називають ефект, який спостерігається в молекулі зі спряженою системою подвійних зв’язків у процесі хімічної реакції. Як тільки молекула потрапляє в поле дії реагенту, в ній починають відбуватися електронні зміщення, які посилюються в міру наближення реагенту і супроводяться перенесенням л-електронів або неподіленої пари р-електронів атома ліганда по системі спряження в процесі хімічної реакції. Одночасно з цим відбувається зміна стану зв’язків і електричних зарядів на атомах. Прикладом реакцій, в яких виявляється динамічний ТИ-ефект, можуть бути Дс-реакції бутадієну-1,3 з галогенами або галогеноводнями, які розглядаються нижче.
Хімічні властивості. Для спряжених дієнів, як і для алкенів, характерними є реакції приєднання. Але спряжені алкадієни в реакціях іонного і радикального приєднання значно реакційноздат-ніші, ніж алкени і дієни з ізольованою системою подвійних зв’язків. Дивініл, ізопрен та інші спряжені дієни легко вступають в численні реакції приєднання і поступово приєднують спочатку один, а потім другий моль реагенту. Проте особливість у будові спряжених дієнів, яка полягає у взаємодії л-електронної густини одного л-зв’язку з електронною густиною другого л-зв’язку і про яку зазначалося вище, робить ці дієни здатними приєднувати різні реагенти як до першого і другого (в 1,2-положення), так і до першого і четвертого (в 1,4-положення) вуглецевих атомів. Температура, природа спряженого діену, природа реагенту і розчинника, тривалість реакції, наявність каталізаторів істотно впливають на співвідношення одержаних продуктів.
Гідрування. Атомарний водень приєднується до дивінілу переважно в 1,4-положення, утворюючи бутен-2, а водень при наявності каталізатора приєднується як в положення 1,4, так і в положення 1,2, утворюючи суміш ізомерних бутенів:
Н3С—СН=СН—СН3 -с;нвон+-№ Н2С=СН-СН=СН2
і-> Н3С—СН2—СН=СН2 (1,2-приєднання);
І-> Н3С—СН=СН—СН3 (1,4-приєднання).
154
Бутени, які утворюються на першій стадії реакції приєднання, можуть далі гідруватися і перетворюватися на бутан.
Г алогенування. Хлор і бром залежно від умов реакції можуть приєднуватися до спряжених дієнів як за іонним, так і за радикальним механізмом. Електрофільне приєднання (Л^-реакції) починається з утворення позитивно зарядженою частинкою, наприклад С1+, л-комплексу за місцем одного з подвійних зв’язків дієну. Потім катіон приєднується до одного з кінцевих атомів вуглецю дієну. Цьому сприяє підвищений індекс вільної валентності атомів вуглецю Сх і С4, а також зміщення л-електронів дієну в напрямку до частинки Х+ (динамічний М-ефект). В результаті утворюється відносно енергетично стійкий мезомерний алільний карбкатіон. Цей карбкатіон на наступній стадії взаємодіє з аніонною частинкою Х~, яка може приєднатися як до атома С2, так і до атома С4. Тому в процесі реакції бутадієну з хлором утворюється суміш 1,4-дихлорбутену-2 і 3,4-дихлорбутену-1, які здатні приєднувати другу молекулу хлору і перетворюватися на 1,2,3,4-тетра-хлорбутан:
сі :сі---►сП + :сГ ;
СІ++ Н2С = СН-СН = СН2 —[СІ-СН2-СН-СН=СН2»-СІ-СН2-СИ=СН-СН2] —
Мезомерний алільний катіон
СІ-СН2— СН СІ - СН = СН
СІ —СН2—СНСІ —СНСІ — СН2С1 .
Якщо в умовах реакції приєднання система близька до стану рівноваги, то кількість кожного ізомера в продуктах реакції залежить від положення рівноваги. Продукт 1,4-приєднання переважно енергетично вигідніший, і тому його утворюється більше. Коли ж система далека від стану рівноваги, то може утворюватися переважно продукт 1,2-приєднання, якщо енергія активації в реакції його утворення нижча, ніж продукту 1,4-приєднання. Так, при хлоруванні бутадієну-1,3 утворюється приблизно однакова кількість 1,2-і 1,4-дихлорбутенів. При бромуванні утворюється близько 66 % 1,4-дибромбутену-2, оскільки зв’язок С—Вг слабкіший від зв’язку С—СІ і рівновага для броміду досягається легше. Підвищення температури наближує систему до рівноважного стану.
Вивчення взаємодії дивінілу з бромом дає можливість уявити механізм електрофільного приєднання галогенів до спряжених дієнів і утворення продуктів 1,2- і 1,4-приєднання. Взаємодія брому, як і хлору, починається з приєднання до молекули дивінілу катіона Вг+ і утворення мезомерного карбкатіона, до якого далі може приєднатися аніон брому як в положення 2, так і в положення 4. Продуктами такого приєднання є відповідно 3,4-днбромбутен-1 (І) і 1,4-дибромбутен-2 (II):
155
Вг+ + * Вг”
Н2С=СН—СН=СН2 —> [ВгСН2—СН—СН=СН2 ВгСН2—СН=СН—СН2] -» А Б
Мезомериий карбкатіон
-> ВгСН2—СНВг—СН=СН2 (1,2-приєднання);
-> ЕгСН2—СН=СН—СН2Вг (1,4-приєднання);
Залежно від умов реакції, зокрема температурного режиму, співвідношення продуктів приєднання І і II може змінюватися.
ЙГ2+ СНг = СН- СН= СН2 вгСНі- сн=сн-сн2вг
Перетворення іонної гари 6 адВумп
Рис. 39. Зміна потенціальної енергії в реакції 1,2- і 1,4-приєднання Вг2 до бутадієну-1,3
Так, при —80 °С утворюється 80 % диброміду І і 20 % дибромі-ду II. Якщо ж реакцію здійснювати при 40 °С, то склад цих ізомерних продуктів зворотний. Пояснюють це тим, що утворення продуктів І і II визначається кінетичними (швидкісними) і термодинамічними факторами. При звичайних температурах швидше утворюється продукт І, частина якого при підвищеній температурі ізомеризується в дибромід II (алільне перегрупування). На рис. 39 зображено криву, яка показує зміну потенціальної енергії Е реакції 1,2- і 1,4-приєднання брому до дивінілу. З характеру ходу кривої випливає, що енергія активації Е реакції 1,2-приєднання менша, ніж 1,4-приєднання. Отже, кількість зіткнень аніона Вг - з карбка-тіоном А буде більшою, ніж кількість зіткнень Вг~ з карбкатіоном Б. Тому вихід продукту І буде вищим. Проте продукт II термодинамічно стійкіший, ніж продукт І. Оскільки потенціальна енергія продукту II нижча від потенціальної енергії продукту І, то при підвищених температурах енергетично менш стійкий продукті ізомеризується в енергетично стійкіший продукт II.
Реакції з г а логено во дня ми. Аналогічно галогенуванню спряжені дієни приєднують галогеноводні НХ (X — Р, СІ, Вг, І) і утворюють продукти 1,2-і 1,4-приєднання, які також мають здатність до алільного перегрупування:
Н3С—СНС1—СН=СН2 (1,2-приєднання):
Н2С=СН— СН=СН2 + НС1-
---->Н3С—СІ І==СН—СН2С1 (1,4-приєднання).
156
Дієновий синтез (реакція Дільса — А л ь-д е р а). Дієновим синтезом називають реакцію 1,4-приеднання до спряжених дієнів речовин, які мають подвійний або потрійний зв’язок, і утворення, таким чином, ненасичених шестичленних циклічних сполук. Реакцію дієнового синтезу можна зобразити такими загальними схемами:
Дієновий синтез відомий в органічній хімії як реакція Дільса— Альдера. О. Дільс і К- Альдер, починаючи з 1928 р., дослідили і узагальнили численні реакції спряжених дієнів з ненасиченими сполуками. У дієновому синтезі беруть участь спряжений дієн і неиасичена сполука (дієнофіл). Дієнофілами може бути велика кількість різноманітних органічних сполук. Сам етилен є слабким дієнофілом. Високореакційноздатними є такі дієнофіли, подвійний зв’язок яких активований електроноакцепторними групами. Дієнофіли, які найчастіше використовуються, за своєю активністю розміщуються в такий ряд:
КС.
’СМ -> Н?|//Ч?° > Н2С=СН- с^° > н2с=сн—см.
ХН Акрилонітрил
Акролеїн
С
Тетр ац і ан етил єн
О
Малеїновий ангідрид
Типовим прикладом дієнового синтезу є приєднання малеїнового ангідриду до дивінілу в 1,4-положення, в результаті якого утворюється циклічний продукт (аддукт)—ангідрид 1,2,3,6-тетрагід-рофталевої кислоти(Тпл = 103 °С):
Реакція спряжених алкадієнів з малеїновим ангідридом використовується для їх ідентифікації, оскільки ці аддукти мають чітку температуру плавлення, а також для кількісного визначення спряжених дієнів і в органічному синтезі.
157
Каучуки
Каучук має велике народногосподарське значення. З нього виготовляють гуму, а гумова промисловість виготовляє понад 70 тисяч назв різноманітних виробів. Каучук є також стратегічною сировиною. Щорічне виробництво каучуку в світі до 1990 р. досягло 32 млн т.
Розрізняють два види каучуків: натуральний і синтетичний.
Натуральний (природний) каучук був відомий ще індійцям до-колумбівської Америки, який вони добували з соку південноамериканського дерева гевеї. Від цього методу добування і походить назва — каучук (саа о-сЬи), що на стародавній мові майя означає «сльози дерева». В Європу каучук був завезений на початку XIX ст. Гевея в промислових масштабах культивується в Бразілії, Індонезії та в інших тропічних країнах. Одне дерево протягом року дає від 500 до 2000 кг каучуку у вигляді молочного соку, який називається латексом і складається на 35—40 % з каучуку, а на 55— 60 % —з води. Відомі також інші рослини-каучуконоси: звичайний фікус, деякі види кульбаби (кок-сагиз, тау-сагиз), гваюла, ваточник, який зустрічається на Україні. Проте вміст каучуку в них порівняно невеликий. З латексу каучук виділяють шляхом коагуляції. Для цього сирий латекс спочатку очищають, а потім коагулюють дією оцтової кислоти або нагріванням. Каучук, який при цьому виділяється у вигляді пружної аморфної маси, промивають, механічно обробляють, вносять наповнювачі, вулканізують добавлянням сірки і перетворюють таким чином на гуму.
Вивченням будови і хімічних властивостей природного каучуку було встановлено, що його склад можна зобразити загальною формулою (С5Н8)„, молекулярна маса близько 350 000 (п « 5000). А. Вільямсон у 1860 р. встановив, що під час сухої перегонки (пі-ролізу) природний каучук деполімеризуеться і перетворюється на ізопрен:
(СВН8)„ —>- ЯН2С=С-СН=СН2.
СН3
Каучук — ненасичена високомолекулярна сполука, яка каталітично гідрується, знебарвлює бромну воду і розчин КМпО4. Причому одна ланка його макромолекули приєднує два атоми водню, два атоми брому. К. Гаррієс (1905—1912 рр.), вивчаючи озоноліз натурального каучуку, виділив при цьому 4-оксопентаналь (леву ліновий альдегід), який може утворюватися тільки в тому випадку, коли природний каучук має будову 1,4-поліізопрену, тобто побудований з мономерних ланок ізопрену, сполучених між собою за принципом «голова до хвоста» (ізопренове правило): «голова»-* -+ Н2С =С—СН =СНа *- «хвіст»:
І
СН3
158
4-Н2О
-Н2°2
СНз
— -сн2-с=о
СН3
о3 — сн2- с=сн- сн2-сн2 -с= сн- сн2 ••• —*-
СН3 СН3
у—о у—у
--------СН2-Щ хСН-СН2-.СНа-С. ХСН -сн2 2 і о
сн3
о \\
+ с-сн2-сн2-с=о + о=сн-сн,2-------
Н7 СНз
Левуліновий альдегід
Фізико-хімічними методами (рентгеноструктурний аналіз) встановлено, що макромолекули природного каучуку мають структуру фіс-1,4-поліізопрену, в якому регулярно чергуються ланки ізопрену з ірю-розміщеними метиленовими групами біля подвійного С=С-зв’язку:
СН2, ,СН2-СН2Ч .СН2-СН2Х .СН2-СН„Ч сн2-...
2\с=с/ 2 У=с< 2 2
Н3С/ \н Н3С/ У * * * * ХН Н3СХ ХН Н3С/ хн або в скороченому вигляді:
\ Нзс/ \н
Ізомерною натуральному каучуку є нееластична гутаперча, яка міститься в латексі рослин родини сапотових (Раїацшшп §пНе та ін.), в нашому бересклеті бородавчастому і являє собою транс-1,4-поліізопрен:
...—сн2. ,н н8с. ,сн, сн2ч ,н н3с. ,СН2—...
ХС=С< хс=с< 2 У=с< ^С=С( 2 ,
Н8С/ ХСН2-СН/ ХН Нзс/ ХСН2-СНг/ ХН
або в скороченому вигляді:
—СН2х .Н \
2\с=с/ |
н3с/ хсн2-/„
У 1839 р. Ч. Гудьїр помітив, що липкий натуральний каучук при нагріванні з сіркою перетворюється на нелипкий еластичний
продукт, який отримав назву вулканізованого каучуку, а процес взаємодії каучуку з сіркою назвали вулканізацією. Процес вулканізації каучуку і перетворення його на еластичну гуму полягає в на-
гріванні і механічному перемішуванні каучуку з 2—-3 % сірки при
130—160 °С. При холодній вулканізації сирий каучук обробляють
хлоридом сірки (І) 82С12. В процесі вулканізації сірка приєдну-
ється за місцем подвійних зв’язків каучуку і утворює поперечні
159
(місткові) зв’язки між полімерними ланцюгами. Відбувається «зшивання» лінійних макромолекул каучуку в більші, сітчасті (тримірні) полімерні молекули:
СН3
.. —СН2—С=СН—СН2—СН2—С=СН—СН2-----
СН, + «8
• • —сн2—с=сн—сн2—сн2—с=сн—сн2----
СН8 сн3
СН,8,
І І
.. —сн,—с—сн—сн,—СН2—с=сн—сн,-----
І 1
—► 8 СН3
І
8
• • —СН„—С—СН—СН„—СН2—С=СН—сн2—. І І І
СН382 СН3
І
Поперечні зв’язки, які утворює при цьому сірка, надають такому каучуку еластичність, міцність і усувають липкість. Вулканізований каучук (гума) може розтягуватися до 900%. Якщо до каучуку добавляють більше сірки (20—-ЗО %), то утворюється твердий матеріал, відомий під назвою ебоніт.
Натуральний каучук використовують для виробництва шин, гумово-технічних виробів, клеїв, ебонітів, губчастих гум, побутових і медичних гумових виробів. Натуральний каучук використовують також у вигляді латексу.
Гутаперча, як і натуральний каучук, вулканізується (при наявності прискорювачів) з утворенням міцних вулканізатів. Вулканізована гутаперча —добрий діелектрик, стійка до дії води, кислот, може розтягуватися до 500—550 %. Її застосовують для ізоляції підводних електрокабелів, для гумування хімічної апаратури, у виробництві клеїв.
Синтетичні каучуки (СК). Перше в світі велике промислове виробництво СК було організоване в СРСР у 1930 р. на основі методу, розробленого С. В. Лебедєвим. Як вихідний дієн був використаний дивініл, з якого каталітичною полімеризацією добули синтетичний каучук бутадієновий (СКБ). У сучасній промисловості в усьому світі СК добувають переважно полімеризацією дивінілу, ізопрену, хлоропрену та деяких інших спряжених дієнів, а також їх співполімеризацією з такими мономерами, як стирол, акрилонітрил, ізобутилен тощо. СК, як і натуральний каучук, можуть вулканізуватися і перетворюватися при цьому на гуму.
160
З дивінілу, залежно від умов полімеризації, утворюється полібутадієн, який містить ланки як 1,2-, так і 1,4-приєднання:
(—СНа—СН—\ (—СНа— СН=СН—СНа—)п.
СН І ;
И /
СН2
Під час полімеризації бутадієну на каталізаторі Ціглера — Натта (див. розд. 14) добувають стереорегулярний цнс-1,4-полі-бутадієн1
якийвикористовуютьдлявиготовлення автомобільних шин, взуття, ізоляції для кабелів та інших гумово-технічних виробів.
У великих кількостях добувають співполімери дивінілу (3 частини) з стиролом (1 частина). Співполімеризацію ведуть під впливом радикальних ініціаторів:
п-
ЗпСНа =СН—СН=СН2+пСНа=СН—>
свнв
(—СН-—СН=СН—СН,—)3СН„—сн— Ін,
В результаті утворюється бутадієн-стирольний каучук (СКС— синтетичний каучук стирольний), який використовують для виготовлення автомобільних шин, підошв для взуття та інших гумових виробів.
Радикальною співполімеризацією дивінілу з акрилонітрилом добувають бутадієн-нітрильний каучук (СКН—синтетичний каучук нітрильний):
к-
пСНа= СН—СН=СНа+пСНа=СН—>- (—сн2—СН=СН—СНа—СН2—СН—)„. сн кі
В СКН бутадієн приєднує акрилонітрил в основному в положення 1,4. СКН —каучук спеціального призначення. Він має високу масло- і бензостійкість, стійкий проти нагрівання. Його використовують для виготовлення масло- і бензостійких шлангів, м’яких гум. У вигляді латексу СКН використовують при виробництві паперу ,і нетканих текстильних виробів.
Подібну до природного каучуку будову має синтетичний ізопре-новий каучук (СКІ — синтетичний каучук ізопреновий), який до
1 В СРСР цей каучук випускається під маркою СКД.
6 1-120
161
бувають стереорегулярною полімеризацією ізопрену. Технологію синтезу стереорегулярного ІЗОПреіІОЕОГО каучуку в СРСР розробив О. А. Коротков. Стереорегулярну полімеризацію ізопрену здійснюють при наявності каталізатора Ціглера—Натта і одержують при цьому цис-\,4-поліізопрен:
...—СН2Ч ,СН.,-СН,. ,сн2-сн2, ,сн2-сн2. _сн2—...
;С=С( )С=С< /С=С(
Нзс/ ХН Н// хн Н3СХ хн Н3СХ хн
Полімеризація ізопрену при наявності змішаного гідриду алюмінію і лужного металу та ТіС14 приводить до утворення транс-\,4-поліізопрену.
Ццс-1,4-поліізопрен за властивостями дуже подібний до натурального каучуку, а за деякими показниками, наприклад, за відносним видовженням, навіть перевершує його. СКІ використовують для заміни натурального каучуку у виробництві шин та інших гумово-технічних виробів.
Великого значення набув СК на основі хлоропрену (2-хлорбута-дієну-1,3), який полімеризується значно легше, ніж дивініл та ізопрен. Хлоропрен може полімеризуватися навіть без каталізаторів:
пСН2=СС1—СН=СН2—> (—СН2—СС1=СН—СН2—)
В результаті полімеризації хлоропрену утворюється хлоропреновий СК, який в СРСР називають совпреном, наїритом, авСША— неопреном, дюпреном. Хлоропреновий каучук має високу світлостійкість, маслостійкість, інертнийдо дії багатьох органічних розчинників і використовується як ізоляційний матеріал, а також для виготовлення бепзостійких гумових виробів, мікропористої гуми.
Контрольні запитання і вправи
1. Які речовини утворюються в процесі приєднання НС1 до бутадієну-1,3, 2-метилбутадієну-1,3, пентадієну-1,4, бутену-1, пентену-2? Яка з наведених речовин матиме найбільшу реакційну здатність до НС1? Чому?
2. Як за допомогою спектроскопічних методів та простих хімічних реакцій можна розрізнити такі речовини: а) пентадієн-1,3 і пентен-1; б) пентадієн-1,3 і пентин-1; в) пентадієн-1,3 і пентадієн-1,4; г) пентадієн-1,4 і пентен-2?
Розділ 17. АЛЬДЕГІДИ І КЕТОНИ
Альдегіди і кетони —це клас органічних сполук, молекули яких містять карбонільну групу >С=-0 (оксогрупу). Альдегіди мають
функціональну альдегідну групу—С^ і їх молекули описуються
162
О
н
загальною формулою Н—С
у якій 7? — атом водню або вуг
леводневий радикал. В молекулах альдегідів атом вуглецю карбонільної групи сполучений з атомом водню і вуглеводневим радикалом. Тільки в молекулі мурашиного альдегіду карбонільний атом вуглецю сполучений з двома атомами водню.
Загальна формула кетонів /?—-С—Ц'. В кетонах атом вуглецю
карбонільної групи сполучений з двома вуглеводневими радикалами, які можуть бути однаковими або різними.
Альдегіди і кетони утворюють гомологічні ряди, які можна легко записати (табл. 16, 17), виходячи з загальних формул цих речовин. Для цього необхідно підставити в них замість Е? відповідні вуглеводневі радикали.
Номенклатура. За систематичною номенклатурою назви альдегідів утворюють з назв відповідних вуглеводнів, додаючи до них суфікс -аль. Головний ланцюг в молекулі альдегіду вибирають так, щоб в нього обов’язково входив атом вуглецю карбонільної групи. Нумерацію вуглецевого ланцюга починають з атома вуглецю карбонільної групи, наприклад: метаналь, етаналь, 2-метилпропаналь і т. д. (табл. 16). Альдегіди часто називають також за історичними назвами, які походять від назв відповідних карбонових кислот. При цьому в їх назві слово «кислота» замінюється словом «альдегід»: мурашиний альдегід, оцтовий альдегід і т. д. (табл. 16).
Назву кетонів за систематичною номенклатурою також утворюють з назви відповідного вуглеводню, додаючи до неї суфікс -он. У назві зазначають також номер атома вуглецю, сполученого з карбонільним атомом кисню, наприклад: пропанон-пентанон-2 і т. д. (табл. 17). Кетони часто називають також за назвою радикалів, сполучених з карбонільною групою: диметилкетон, метилетилкетон і т. д. (табл. 17). Перший і найважливіший кетон—пропанон, або диметилкетон, має історичну назву ацетон.
Ізомерія. Ізомерія альдегідів пов’язана з розгалуженням вуглецевого ланцюга радикала і в гомологічному ряду цих сполук починається з четвертого члена ряду, який існує у вигляді масляного та ізомасляного альдегідів (табл. 16).
Ізомерія кетонів залежить як від будови- алкільних груп радикалів, так і від положення карбонільної групи в молекулі кетону. Кетон, для якого починається ізомерія, має в своєму складі п’ять атомів вуглецю. Серед ізомерів цього кетону пентанон-2 і 3-метил-бутанон-2.— ізомери, які відрізняються будовою алкільних радикалів, а пентанон-2 і пентанон-3 —• ізомери з різним положенням карбонільної групи (табл. 17).
Фізичні властивості. Мурашиний альдегід — газ, наступні гомологи альдегідів — рідини,вищі альдегіди—тверді речовини.
6*
Іба
Таблиця 16. Гомологічний ряд, номенклатура і фізичні властивості насичених альдегідів У?—С^
'Н
Радикал Ц Формула Назва за номенклатурою ^кип>
і сторичною раціональною систематичною
н— н—с^° \н Мурашиний (формальдегід) — Метаналь —21
Н3с- н,с—с^° 3 \н Оцтовий (ацеталь-дегід) Оцтовий Етаналь 21
н3с—сн2— н3с—сн2—с<^° хн Пропіоновий Метилоцтовий Пропаналь 49
Н3С-СН2— сн2— х0 н8с— сн,—сн2—с^ 2 \н Масляний Етилоцтовий Бутаналь 79
н3с—сн— (!н3 х0 Н,С—СН—С<* 1 ХН сн3 Ізомасляний Диметилоцтовий 2-Метилпропаналь 61
Н3С(СН2)2СН2— Н3С(СН2)3—с^н Валеріановий Пропілоцтовий Пентаналь 103
Н3С— сн—сн2— сн3 ^0 Н8С—СН—СН2—С< 8 І 2 \н сн3 Ізовалеріановий Ізопропілоцтовий З-Метнлбутаналь 92,5
Таблиця 17. Гомологічний ряд, номенклатура і фізичні властивості насичених кетонів 2?!—С—/?2 II О
Ні Я, Формула Назва за номенклатурою Т'кип-
раціональною систематичною
сн3- СН3— н3с—с—сн3 II о Диметилкетон* Пропанон 56
СН3— Н3С—СН2— Н3С—с—сн2—сн3 II 0 Лі етилетилкетон Бутанон 79,6
СНд— н3с—сн2—сн2— н3с—с—сн2—сн2—сн5 II о Метилпропілкетон Пентанон-2 101
Н3С—СН2— Н3С—СН2— н3с—сн2—с—сн2—сн, II 0 Діетилкетон Пентанон-3 102
СНз- Н-С—СН— (!н н3с—с—сн—сн3 0 СН3 Метилізопропілкетон З-Метилбутанон-2 93
о * Історична назва цього кетону—ацетон.
Ацетон і нижчі кетони —рідини, вищі кетони —тверді сполуки. Альдегіди і кетони киплять при значно нижчій температурі, ніж спирти з такою самою кількістю вуглецевих атомів. Це свідчить про те, що альдегіди і кетони, на відміну від спиртів, не є сильно асоційованими речовинами. В той же час, температури кипіння карбонільних сполук значно вищі за температури кипіння вуглеводнів з такою самою кількістю вуглецевих атомів, що пов’язано з високою полярністю молекул альдегідів і кетонів.
Мурашиний і оцтовий альдегіди, а також кетони з невеликою молекулярною масою розчинні у воді. При збільшенні молекулярної маси розчинність цих речовин у воді зменшується. Всі альдегіди і кетони добре розчиняються в органічних розчинниках (спирті, ефірі тощо).
Вважають, що карбонільна група —осмофор, тобто посій запаху. Мурашиний альдегід має досить різкий запах. Інші нижчі альдегіди мають задушливий запах, який при сильному розбавлянні стає приємним і нагадує запах овочів і фруктів. Кетони пахнуть досить приємно.
В ІЧ-спектрах валентне коливання карбонільної групи характеризується інтенсивною смугою поглинання в області 1740— 1720 см'1 (для альдегідів) і 1725—1705 см"1 (для кетонів). В аль-
дегідах група —Сб має ще характеристичну смугу .поглинання
ХН
валентних коливань зв’язку С—Н біля 2720 см-1.
В УФ-спектрах альдегіди і кетони поглинають в області 280 нм.
В спектрах ПМР сигнал протона альдегідної групи знаходиться в області 9—10 мли-1.
Методи добування. Альдегіди і кетони можна порівняно легко добути такими методами.
1. Дегідруванням спиртів. При пропусканні пари спирту над нагрітим каталізатором, наприклад міддю, від спиртів відщеплюється водень. З первинних спиртів при цьому утворюються альдегіди, з вторинних —кетони:
/°_Т^ хо
300-400°С ' + На:
Н н
Виходячи з цього методу добування, Ю. Лібіх у 1836 р. ввів назву «альдегід», що означає продукт дегідрування спирту, або де-
166
гідрований спирт (від лат. АІкоЬої <1ейус1го£епаіи8 — спирт без водню).
2. Окисленням спиртів (див. розд. 9).
3. Піролізом кальцієвих солей карбонових кислот. Р. Піріа в 1856 р. встановив, що при нагріванні кальцієвих солей карбонових кислот утворюються кетони. Наприклад, під час піролізу ацетату кальцію утворюється ацетон:
х°
Н3С-С _ НзС-С-СН3 + СаСОз.
•°ч І О
І СаІ
гУі
НзСТ< І
І о І
Піролізом суміші кальцієвих солей мурашиної і будь-якої іншої карбонової кислоти добувають відповідні альдегіди:
400 °С хО
(/?—СОО)2Са+(Н—СОО),Са ---> 2/?—+2СаСО3.
ХН
4. Оксосинтезом (див. розд. 14).
5. Гідролізом гемінальних дигалогеналканів. Атоми галогену в гемінальних дигалогеналканах під час гідролізу заміщуються за 5м -механізмом на гідроксильні групи. При цьому спочатку утворюються двохатомні спирти, в яких дві гідроксильні групи сполучені з одним атомом вуглецю. Такі спирти нестійкі (див. розд.9) і, відщеплюючи воду, перетворюються на альдегіди або кетони:
ИаОН /?-СНа-СНВг2+2НОН
нГ /ОН1
> /?—сн2—сн< —-?
!г '''ОН —12'">
.0
7?—СН,—С" ; ХН
/?—СВг2—К+2НОН
№1ОН
—2 НВг
'/?—С—7?’І о/Хо
І І
Ь Н Н -
-~нво
к-с-/?.
6. Гідратацією ацетиленових вуглеводнів (за реакцією Кучеро-ва, див. розд. 15).
7. Пінаколіновим перегрупуванням а-гліколів (див. розд. 9).
Електронна будова карбонільної групи. У карбонільній групі \с=О між атомом вуглецю і атомом кисню існує подвійний зв’язок, який за фізичною природою подібний до етиленового подвійного зв’язку С—С. Утворення подвійного зв’язку між атомами вуглецю і кисню, який також розглядають як поєднання о- і л-
167
X л
зв’язків ^>С=0, уявляють так. Вуглець має електронну конфігурацію І52252р3, а електронна конфігурація кисню —
С
І І І | І | І
8 Рх Ру Рг (5/72)3
и|<|и|Т 82 Рх Р^ Р?
О
о-Зв’язок в карбонільній групі утворюється 5р2-гібридизованою орбіталлю вуглецю і 2рх-атомпою орбіталлю кисню. Негібридизо-ваний р2-електрон вуглецю і р2-електрон кисню, перекриваючись, утворюють л-зв’язок. В основному стані р2-електрони вуглецю і кисню знаходяться на зв’язуючій л-МО, а у збудженому стані —
Рис. 40. Схематичне зображення будови молекули альдегідів і кетонів (а), їх карбонільної групи (б) та розміщення орбіталей в карбонільній групі (в)
переходять на розпушуючу л-МО*. Дві пари електронів карбонільного атома кисню залишаються вільними. Одна з цих пар знаходиться на 2ре-АО в площині, перпендикулярній до л-зв’язку, а друга — на 28-орбіталі. Вільна пара //-електронів атома кисню не чинить істотного впливу на хімічні властивості карбонільної групи. Ця пара електронів бере участь переважно в міжмолекулярних взаємодіях (утворення комплексів з солями металів, наприклад з 8пС14). На рис. 40 схематично зображено будову молекули альдегіду і розміщення орбіталей у карбонільній групі.
Подвійний зв’язок між атомами вуглецю і кисню в карбонільній групі має довжину 0,122 нм і енергію Ес=о= 750 кДж. Цей зв’язок утворений атомами, які значно відрізняються за електронегативністю. Тому електрони л-зв’язку в карбонільній групі зміщуються до електронегативнішого атома кисню, внаслідок чого електронна густина на ньому збільшується, а на атомі вуглецю — зменшується:
х б+! -[б-^>с=о.
Обчислене значення заряду становить 16 | «0,42. Отже, подвійний зв’язок /С=О полярний і молекули альдегідів і кетонів також полярні.
168
Карбонільна група виявляє негативні індукційний і мезомерний ефекти (—/ і —М-ефекти). У кетонів ці’ефекти деякою мірою менші, ніж у альдегідів, оскільки електронегативність кетопного кисню дещо менша, ніж кисню альдегідної групи.
Під впливом —І- і —М-ефектів карбонільної групи зменшується електронна густина на сусідньому, сполученому з нею «-вуглецевому атомі. Тому атоми водню, що знаходяться біля такого атома вуглецю (а-водневі атоми), набувають підвищеної активності. Підвищення активності атомів водню біля «-вуглецевих атомів викликане також взаємодією ст-орбіталей а-СН-зв’язків з л-орбіталями подвійного зв’язку (о, л-спряження), яке називають гіперкон’юга-цією і позначають зігнутою стрілкою:
Тому альдегіди і кетони можуть бути охарактеризовані також як СН-кислоти.
Підвищена активність атомів водню біля а-вуглецевих атомів у молекулах альдегідів і кетонів виявляється в їх порівняно легкій протонізації, у здатності брати участь у реакціях конденсації і гемолітичного заміщення.
Хімічні властивості. Альдегіди і кетони мають високу і різноманітну реакційну здатність. Вони вступають у численні реакції приєднання до карбонільної групи,в реакції заміщення атомів водню біля а-вуглецевих атомів, в реакції конденсації і полімеризації.
Реакції приєднання. Реакції приєднання альдегідів і кетонів зумовлені наявністю в їх молекулах карбонільної групи, зокрема подвійного зв’язку. Як уже зазначалося вище, подвійний \б+ в-
зв’язок між атомами вуглецю і кисню в групі /С=О полярний, тому його хімічна активність значно вища, ніж подвійного зв’язку С=С, наприклад, в етилені. У зв’язку з цим для альдегідів і кетонів можливі реакції приєднання нуклеофільного Ам- і електрофільного
АБ-механізму. В цілому ж група /С=О виявляє основні властивості, завдяки чому легко приєднує до атома кисню протон. При цьому заряд 6 + на атомі вуглецю зростає, що, в свою чергу, прискорює реакції з нуклеофільними реагентами:
\С=О+Н+ —С—О—н.
З цієї причини для альдегідів і кетонів перш за все характерні Адгреакції з реагентами Н+В", в яких першою починає процес приєднання нуклеофільна (аніонна) частинка В~. У загальному
169
вигляді механізм Л^-реакцій для карбонільних сполук можна зобразити у вигляді такої схеми:
ч I I
)с о г-в~ ; > —с—о- —с—о—н.
Повільно | Швидко І
в в
На першій, повільній, стадії нуклеофіл В~ приєднується до карбонільного атома вуглецю і утворює аніон тетраедричної структури з негативним зарядом на атомі кисню. На другій, швидкій, стадії протон приєднується до кисню і таким чином утворюється кінцевий продукт Лдгреакції. Радикали з негативним індукційним ефектом збільшують позитивний заряд на атомі вуглецю карбонільної групи і тим самим підвищують активність альдегідів і кетонів до реакцій нуклеофільного приєднання. Радикали з позитивним індукційним ефектом зменшують позитивний заряд на вуглецевому атомі карбонільної групи і тим самим зменшують активність групи /С=О до вищеназваних реакцій приєднання. В результаті такого впливу карбонільна група оцтового альдегіду менш активна до нуклеофільного приєднання, ніж трихлороцтового альдегіду:
с,х \ а+
СІ — С — С ; Н - С - С А* — С — /? с/ н НХ \
У випадку кетонів з карбонільною групою сполучені два вуглеводневих радикали, які мають позитивний індукційний ефект іще більше зменшують позитивний заряд на карбонільному вуглецевому атомі. Тому карбонільна група кетонів у хімічних реакціях значно менш активна порівняно з аналогічною групою альдегідів. Крім того, на реакційну здатність карбонільної групи в реакціях приєднання впливають також розміри сполучених з нею атомів і груп атомів. Збільшення розмірів/?, сполучених з групою/С=О, веде до зменшення реакційної здатності цієї групи у зв’язку з просторовими утрудненнями. За реакційною здатністю/С =О-групи карбонільні сполуки розміщуються в такий ряд:
,0 /О ,0 .о
н—С-С >Н,С—>Н3С—С" >(Н3С)3С-С<
Чі 3 \н Чн3 \С(СНз)з.
Приєднання синильної кислоти (ціангідринний синтез). Альдегіди і кетони при наявності основних каталізаторів (наприклад, №0Н) приєднують синильну кислоту і утворюють оксинітрили (ціангідрини).
170
Наприклад:
в-
8+ ,О 4-Н+С1ХГ —> ще—СН—СІМ;
Н3С-С^ |
хн он
Нітрил а-оксипропіо-нової кислоти й+ ?-
(Н3С)2С=О+Н+СМ- —> (н3с)2с—сьі.
он
Нітрил а-оксіїзо-масляної кислоти
Ціангідрини використовують як проміжні сполуки в синтезах окси- і амінокислот.
Приєднання гідросульфіту натрію. Альдегіди реагують з концентрованим розчином гідросульфіту натрію і утворюють кристалічні гідросульфіти! сполуки, важкорозчинні у воді. Ця Л^-реак-ція починається з атаки карбонільного атома вуглецю атомом сірки гідросульфіту натрію, який має вільну пару електронів:
„ « ОИа
8- і
Н3С-СН = О + :8 —ОН
О
О№
І
Н3С-СН-8-О: (її) — Н3С-СН-8О2О№ .
о~ о у ОН
----Гідросульфітиа похідна
З кетонів у цю реакцію вступають лише ті, що мають метильну групу, безпосередньо сполучену з карбонільним атомом вуглецю:
Н,С-С-/?.
II
О
Реакція з гідросульфітом натрію є якісною на карбонільну групу, використовується також для очистки та виділення альдегідів і кетонів з суміші з іншими речовинами. Гідросульфіти! похідні легко розкладаються кислотами і утворюють при цьому ті самі карбонільні сполуки:
/О
НЯС—СН—8О2ОМа+НС1 —> Н3С—+№С1+Н2О+8О2.
І ХН
ОН
Приєднання магнійорган-ічних сполук. Альдегіди і кетони легко приєднують реактиви Гріньяра.
В магнійорганічних сполуках зв’язок між атомами магнію і вуглецю полярний. На атомі вуглецю, який більш електронегативний, ніж магній, зосереджується надлишок електронної густини. Тому вуглеводневий залишок реактивів Гріньяра в реакціях поводить себе як нуклеофільна частинка і приєднується до атома вуглецю карбонільної групи, а залишок магнійгалогеніду —до атома кисню цієї групи. В результаті такого приєднання утворюються магнійгалогеналКоголяти, які швидко гідролізуються з утворенням відповідних спиртів і основної солі галогеніду магнію. При цьому
171
з мурашиного альдегіду утворюються первинні спирти, з інших альдегідів—вторинні, а з кетонів — третинні спирти:
8-
нон
н-с" + Н3С-СН2-МгВг-Н3С-СН2-СН2-О-Мв-Вг----------
Мурашиний альдегід
---- И3С—СН2—СН2 —ОН + МвВгіОН) ;
Пропанол-1
ЙХ"-5 ОМйВг он
Н3С—+ НзС-М^Вг-* Н3С-СН-СН3----------► Н3С-СН--СН3 + МдВг(ОН) •
'ЧН\—У Пропанол-2
Оцтовий альдегід
. ОН3 сн3
н3 сЛ^ГГн7с-Йвг-н3с-с-сн3-^ н3с—с—сн3 + МйВг(ОН)
13 II Я І •
0*4------ОМрВг ОН
Диметилкетон, 2-Метилпро-
ацетон панол-2
Отже, реакція магнійорганічних сполук з карбонільними є одним із загальних методів добування спиртів.
Приєднання води. На прикладі реакції гідратації чітко помітна відмінність у реакційній здатності подвійного зв’язку /С=Опорівняно з етиленовим зв’язком/С—С^. Як зазначалося вище, приєднання води до етиленових сполук відбувається при наявності сильних каталізаторів кислотного характеру, у відсутності яких реакція не йде (див. розд. 14). В той же час формальдегід швидко приєднує воду при кімнатній температурі без каталізатора:
8-
н-с. + но:ін=
н он чс
і/ хон
Ця реакція оборотна, оскільки утворений двохатомний спирт нестійкий (містить дві гідроксильні групи біля одного вуглецевого атома) і легко розкладається з відщепленням води за рахунок гідроксогруп. Гідратна форма мурашиного альдегіду може існувати тільки в розчині.
Набагато активніше порівняно з мурашиним альдегідом приєднує воду трихлоронтовий альдегід, який з водою реагує екзотермічно, утворюючи відповідний гідрат:
6-
.он
Хлораль
Хлоралгідрат
172
Воду здатні приєднувати найактивніші карбонільні сполуки, в яких на карбонільному атомі вуглецю досить великий заряд 6 *\ Формальдегід у водних розчинах знаходиться повністю (на 99,8 %) у відрахованій формі. Оцтовий альдегід, у якого завдяки 4-7-ефекту метильної групи зменшується величина заряду 6+ на карбонільному атомі вуглецю, гідратується тільки на 58 %:
б+ є- с+
Н8С—4-Н0:Н
.он н,с—сн<
4 он
Ацетон та інші кетони практично не гідратуються.
Приєднання спиртів. Альдегіди легко приєднують спирти. При еквімолярних співвідношеннях альдегіду і спирту утворюються ефіроспирти, які називають напівацеталями. При каталітичній дії сильних кислот (НС1, Н28О4) напівацеталі реагують ще з 1 моль спирту і утворюють прості ефіри 1,1-діолів, які називають ацета-лями:
8+ 3- 5-,8+
н3с-сн=о + с2н5о:)н-
х°н +нос2н5 нзС-снч _нп -ОС2Н3 2
хос2н3 н3с-снч
чос2н5
Ацеталь
Напівацеталь
Ацеталі — рідини з приємним ефірним запахом, при нагріванні з розбавленими кислотами гідролізуються і перетворюються на вихідні альдегіди і спирти.
Ацеталі кетонів (кеталі) прямою взаємодією з спиртами добути не вдається.
Приєднання аміаку і його похідних. Альдегіди енергійно реагують з аміаком і утворюють нестійкі продукти приєднання, які відразу ж дегідратуються і перетворюються на альдіміни:
Н3с-сн = о + ічн3
кн3
Н3С - сн - о~
-н2о
н3с-сн=ічн
Ацетальдімін '
Кетони з аміаком подібних сполук не утворюють.
Велике значення мають реакції взаємодії карбонільних сполук з такими похідними аміаку, як гідроксиламін, гідразин, арилгідра-зини. Внаслідок цих реакцій утворюються, як правило, кристалічні речовини з характерними для кожної карбонільної сполуки температурами плавлення. їх використовують для аналітичної характеристики альдегідів і кетонів.-В усіх цих випадках взаємодія відбувається через стадію нуклеофільного приєднання похідної аміаку до атома вуглецю карбонільної групи з подальшим відщепленням елементів води і утворенням кінцевого продукту реакції.
173
З гідроксиламіном альдегіди і кетони утворюють оксііміни, які називають оксимами. Механізм такої взаємодії можна зобразити 'ХЄМОЮ'
6- >.
н3с-сн = о + н2м —он
-------- Н3С - СН = N - ОН .
-й2о
Оксим оцтового альдегіду
Н3С —ЄН —\ —ОН
Аналогічно альдегіди і кетони реагують з гідразином і арилгід-разинами, утворюючи при цьому гідразони і арилгідразоніт.
Н3С—СН=О+Н2И—№і2 -> Н3С—СН=Ь1—МН2+Н2О;
Гідразин Гідразон оцтового альдегіду
Н3С—СН=О+Н2КТ—N11—С6Н6 -> Н3С—СН=К—кн—СвН8+Н2О.
Феніл гідразин Фенілгідразон оцтового альдегіду
Реакції з участю а-в однеє и х атомів. Як уже зазначалося, в молекулах альдегідів і кетонів під впливом електро-ноакцепторної карбонільної групи, яка виявляє —1-ї—Д4-ефекти, сс-водневі атоми набувають підвищеної активності, завдяки чому вони здатні протонізуватися і приєднуватися до карбонільного кисню. В результаті виникає ненасичений спирт—енол, гідроксильна група якого розміщена біля атома вуглецю з подвійним зв’язком. Енольна форма знаходиться в рухливій (динамічній) рівновазі з карбонільною формою (кето-енольна таутомерія, див. розд. 15):
а А)
СН2—Сф р—сн=сн—он.
\н
Ступінь енолізації насичених альдегідів і кетонів досить малий. Наприклад, за звичайних умов в ацетоні міститься всього 0,00025% енолвної форми:
Н8С—С—СН3 Н3С—С=СН2.
II І
О ОН
Кислоти, основи каталізують і значно прискорюють взаємоперетворення кетонної і енольної форм та досягнення рівноваги між ними.
Реакції конденсації. У зв’язку з наявністю в молекулах карбонільних сполук рухливих а-водневих атомів, альдегіди і кетони можуть сполучатися між собою, утворюючи при цьому нові вуглець-Вутлецеві зв’язки. Такого типу реакції сполучення називають
174
реакціями конденсації. Реакції конденсації відбуваються при наявності основних або кислотних каталізаторів. У ці реакції можуть вступати молекули як однакових, так і різних альдегідів і кетонів.
Альдольна конденсація. Конденсацію двох молекул альдегідів або кетонів, внаслідок якої утворюються альдегідо- або кетоно-спирти, називають альдольною конденсацією. Назва «альдоль» походить від назви одного з продуктів такої конденсації —альдегідо-спиртів (-аль —за систематичною номенклатурою суфікс для альдегідів, -ол—суфікс для спиртів). Сполучення (конденсація) двох молекул альдегідів або кетонів відбувається за рахунок розриву подвійного зв’язку карбонільної групи однієї молекули карбонільної сполуки і з участю «-водневого атома другої молекули такої сполуки. Наприклад, при нагріванні оцтового альдегіду з 10%-м водним розчином НаОН утворюється альдоль:
6+ 8- МаОН
Н3С-СН = О + н : сн2-с ------- н3с-сн-сн2-с
ЧН он
Альдоль, р-оксимасляний альдегід, 3-оксибутаналь
Альдольиу конденсацію відкрили в 1872 р. незалежно один від одного О. П. Бородін і Ш. Вюрц.
Під час конденсації інших альдегідів, наприклад пропіонового, у реакцію вступають тільки атоми водню, що знаходяться в «-положенні відносно карбонілу, які значною мірою активовані карбонілом:
/О /О . ,0
Н3С—СН,—Су Ч-Н—СН—->Н„С—СН,—СН—СН—.
ХН І \н І І хн
сн3 он сн3
З-Окси-2-метилпеитаналь
Якщо поряд з карбонілом знаходиться четвертинний атом вуглецю, то альдольна конденсація неможлива. Тому, наприклад, /°
триметилоцтовий альдегід (Н3С)3С—С<( в альдольну конденсацію
\н
не вступає.
Механізм альдольної конденсації, наприклад, оцтового альдегіду в лужному середовищі описується такими послідовними стадіями. На першій, повільній, стадії гідроксид-іон (каталізатор) гетеролітично відщеплює у вигляді протона рухливий « водневий атом молекули альдегіду. При цьому утворюється вода і карбаніон (а), який є сильним нуклеофілом. На другій стадії карбаніон (а) приєднується до карбонільного атома вуглецю другої молекули альдегіду і утворює новий аніон (б), який відщеплює від молекули води протон і перетворюється на кінцевий продукт конденсації —
175
альдоль. Одночасно на останній стадії вивільняється іон гідроксилу. Усі ці процеси можна зобразити такими схемами:
но~+ н):сн2-сн =о — Пов!-Л.Ї1?_2г :сн2-сн =о + н2о :
а
Н,с-с'° + : сн,—сн=о Н,с-сн- сн,- сі І=о:
ХО н3с-сн-сн,-сн=о + її): он—► н3с-сн-сн2-с' + но .
і- он н
Альдоль
Альдольна конденсація кетонів відбувається в жорсткіших умовах, при нагріванні кетонів з розчином Ва(ОН)2.
Кротонова конденсація. Якщо конденсацію альдегідів і кетонів при каталітичній дії основ проводити при нагріванні, то в цих умовах добувають ненасичені альдегіди або кетони. Такого типу конденсації називають кротоновими. Наприклад, оцтовий альдегід за таких умов конденсується в 2-бутеналь, або кротоновий альдегід, від назви якого дали назву даній конденсації:
н3с-сн = Го+ іСсн-с/-----►н3с-сн=сн-с + н2о.
!_---- Хн н
2 -Бутеналь, кротоновий альдегід
Кротонова конденсація, як встановлено, проходить через стадію утворення альдолю. Останній при нагріванні і каталітичній дії НО-дегідратуеться, перетворюючись на ненасичений альдегід:
нвс-сн<сн-сн=о—- н3с-сн=сн-сн=о + Н2О
Аналогічно, але з меншою швидкістю, конденсуються і кетони.
Окислювальн о-в ідновні реакції. Під дією відновлюючих реагентів карбонільна група відновлюється до спиртової. При цьому альдегіди відновлюються до первинних спиртів, кетони —до вторинних. Відновлення альдегідів і кетонів є одним з методів добування спиртів. Відновлення карбонільних сполук можна здійснювати воднем над нагрітими Ні, Рі, Ргі та іншими каталізаторами:
Я—СН=О+Н2 -> Р—СН2—ОН;
Р— С—А?+Н2 -> р—сн—р.
II І
о он
176
Електрохімічне відновлення кетонів, а також відновлення їх натрієм, амальгамами натрію або магнію приводить до утворення 1,2-діолів. Наприклад, ацетон при такому відновленні утворює пінакон:
СН3СН,
Н3СЧ .... II
3 \с—О:-»Н3С— С—С—СН
II ..СХ " 1-1-
сн3 сн3
-> Н3С—С—і-сн,
(Ж он Пінакон
Альдегіди легко окислюються і перетворюються на відповідні карбонові кислоти. У процесі реакції зберігається кількість вуглецевих атомів і структура вуглецевого ланцюга, а альдегідна група перетворюється на карбоксильну:
\н
\ж _
Окислювачами можуть бути кисень повітря, а також КМпО4, Н2СгО4, [А£(МН3)2]ОН, сполуки міді (II) та ін.
М’яким окислювачем альдегідів є аміачний розчин гідроксиду срібла (реактив Толленса), який готують перед проведенням реакції змішуванням розчину нітрату срібла з невеликим надлишком аміаку. При слабкому нагріванні реактиву Толленса з альдегідами останні окислюються до карбонових кислот, а срібло відновлюється до металічного і осідає на чистій поверхні скляного посуду у вигляді красивого срібного дзеркала. Тому ця реакція називається реакцією срібного дзеркала і є якісною на альдегіди, а також використовується у промисловості для сріблення дзеркал:
4 2ГЛЦОН
АЄМО3+№і4ОН-----------> А8ОН---------> [АЄ (МН3)2]+ОН“;
—МН4ЬІО, —н,о
Н-С< 4-2[АЄ(Ш3)2|+ОРГ-> Н-С< +2АбН-4М!3+Н2О.
ХН ХОН
Аналогічно альдегіди окислюються до карбонових кислот сполуками міді (II). З цією метою як окислювач використовують так звану рідину Фелінга, яка являє собою розчин комплексної солі гідроксиду міді (II) і натрієво-калієвої солі винної кислоти (сегнетової солі). Альдегіди при нагріванні з фелінговою рідиною окислюються до карбонових кислот, а мідь (II) відновлюється до міді (І) і кількісно випадає у вигляді червоного осаду Си2О:
СиЗО4+2МаОН -> Си(ОН)2-}Жа28О4;
177
СООК’а 1 СН—ОН 1 СН—он ( 1 ( + си(0Н)2—1 — Н2О ( ЗООИа /) 1 -н?-с< :н—о. чн /0 - /?-С^ + \он
1 , <•* ЗН—0х
соок 1 с + Си2О + •оок СОО№ 1 СН—ОН 1 +Н2О. сн—он соок
Реакція з фелінговою рідиною використовується для якісного і кількісного визначення альдегідів.
Кетони не дають реакції срібного дзеркала і не відновлюють фелінгову рідину, оскільки важко окислюються. Окислення кетонів відбувається в жорстких умовах при тривалому нагріванні їх з сильними окислювачами. При цьому відбувається розрив вуглецевого ланцюга з обох боків карбонільної групи відповідно до правил Попова, які були сформульовані в 1876 р., а потім розвинуті в роботах Є. Є. Вагнера. Згідно з цими правилами дія окислювача напрямлена на один з а-вуглецевих атомів. При цьому утворюється оксике-тон, який далі окислюється до а-дикетону. Подальша дія окислювача викликає розрив зв’язку між карбонільними групами в молекулі а-дикетону і приводить до утворення карбонових кислот. Оскільки окислення а-вуглецевих атомів може відбуватися по обидва боки кетонної групи, то в результаті утворюється суміш карбонових кислот. Схематично процес окислення, наприклад, етилбутилкетону можна зобразити так:
н3с—сн,—с—сн,—сн,—сн2—сн. «5 А А А А &
о 1ІО]
V? у
НЯС—СН—С—СН2—СН2—СН2—СН3 + Н3С—сн,—с—сн—сн,—сн,—сн.
І II II І
он о . о он
1[О1 І Ю!
Н3С—с—с—сн2—сн2—СН,—СН3 + н3с—сн2—с—с—сн2—сн,—сн.
II ! II
О| о
ЦО]
о о
1 Г03
Н3С—СООН+Н3С—СН2—сн2—СН2—СООН+Н3С— СН2—СООН+
Оцтова кислота Валеріанова Пропіонова кислота
кислота
+ Н3С—сн2~сн2—соон.
Масляна кислота
Реакція з фуксинсірчистою кислотою є якісною на альдегіди. Розчин фуксинсірчистої кислоти (реактив Шіффа) готують шляхом насичення оксидом сірки (IV) водного яскраво-рожевого розчину 178
барвника фуксину до його знебарвлення. Після добавляння альдегіду до такого безбарвного розчину фуксинсірчистої кислоти появляється фіолетово-пурпурове забарвлення, яке свідчить про наявність альдегідної групи в молекулі досліджуваної речовини.
Кетони, як правило, цієї реакції не дають. Проте кетони, в молекулах яких є метпльна група, безпосередньо сполучена з атомом вуглецю карбонільної групи Н3С—С—Р, зв’язують сірчисту кислоту
і розчин реактиву Шіффа рожевіє.
Реакція Канніццаро. Альдегіди, в молекулах яких немає водню /°
біля <7.-вуглецевого атома Р3С—, у лужному середовищі ХН
вступають в окислювально-відновну реакцію, відкриту С.Канніц-царо в 1853 р., в результаті якої одна молекула альдегіду відновлюється до спирту, а друга при цьому окислюється до карбонової кислоти:
Х° КОН хО
27?3С—С<^ + Н2О —> /?3С—СН2—ОН + ^зС—с\он •
Реакція Канніццаро відбувається з гідридним перенесенням водню альдегідної групи (див. розд. 35).
Кетони в цю реакцію не вступають.
' Реакція Тищенка. Альдегіди, на відміну від кетонів, при нагріванні з алкоголятами (краще за все з алкоголятами алюмінію) у неводному середовищі вступають в окислювально-відновну реакцію. відкриту В. Є. Тищенком у 1906 р. Продуктом цієї реакції є складний ефір:
,О
Н3С—
8 \н
+ фС-СН., н/
ІС-"-о,->1> Н<с_с^°
сн2—сн3.
Оцтовоетиловий ефїр
Реакції заміщення. Заміщення карбонільного кисню на галоген. При взаємодії альдегідів і кетонів з РС15 відбувається заміщення карбонільного кисню в їх молекулах на атоми хлору. В результаті утворюються геміпальні дихлоралкани. Наприклад:
Н3С—СН=О + РСЇе -> Н,С—СНС1» -4- РОСІ»;
Н3С—С—СН3 + РС16 -> Н3С—СС12—СН3 + РОС13.
Заміщення а-водневих атомів на галоген. Карбонільна група, як уже зазначалося, сприяє підвищенню активності а-водневих атомів. Тому карбонільні сполуки можуть вступати в реакції, в результаті яких відбувається заміщення а-водневих атомів. Так, альдегіди і кетони енергійно реагують з галогенами і утворюють
179
“-галогенозаміщені сполуки. Реакція каталізується кислотами • особливо лугами:
Н,С—С—СН4 + Вг2 -> Н3С—С—СН,—Вг 4- НВг. II о
О
Бромацетон
На галогени можуть заміщуватися послідовно всі «-водневі атоми альдегіду або кетону. Наприклад:
.Р +сі, +сц хО +сі2 хО
н3с—------------> сісн„-с^ --------> сі,сн—с<г --------> сі,с—С" ;
3 \н -неї 2 \н -неї 2 \н -неї 3 \н
Хлороцтовий Дихлороцтовий Трихлороцто вий
альдегід альдегід альдегід, або
хлораль
,О 4-С1, хО
Н,С-СН2-С^ —> Н3С-СНС1-С^ \н ~нс| Хн
о
4-С12
—н3с—ссі2—с
— неї ** *
\н
Полімеризація альдегідів. Альдегіди містять ви-сокореакційноздатний подвійний зв’язок С=О і завдяки його наявності можуть полімеризуватися. Для кетонів реакції полімеризації не характерні. Полімеризація альдегідів може бути циклічна і лінійна. Добре вивчена полімеризація мурашиного і оцтового альдегідів. Так, під час перегонки 6 %-го розчину формальдегіду при наявності каталітичних кількостей сірчаної кислоти утворюється кристалічний тример — 1,3,5-триоксан (Т„л =62 °С):
2*ХН?8О<
При тривалому зберіганні 40 %-го водного розчину формальде гіду (формаліну) поступово відбувається самовільна його полімеризація і утворюється білий осад не розчинного уводі полімеру по-ліформальдегіду, який називають параформом (ТІ]П = 120 °С):
пСН2=О + Н—ОН -> Н—(—О—СН2—)„—ОН.
ГІараформ
Ступінь полімеризації п у параформу коливається в широких межах—'Від 8 до 100. При нагріванні параформ деполімеризу-ється і перетворюється на мономер—формальдегід. Цим часто користуються в синтезах, у яких потрібен газоподібний формальдегід.
Оцтовий альдегід під дією кислот також полімеризується. Якщо до оцтового альдегіду добавити при 20°С кілька крапель концентрованої Н28О.і, то відбувається бурхлива екзотермічна реакція з
ПО
утворенням тримеру — 2,4,6-трпметил-1,3,5-триоксану, який називають паральдегідом (Гк,ш = 128 °С):
І
СІ І3
О
Н3С-СН 'Х'Н-СН3
І І
о О
'ЧС1І І СНз
Паральдегід
Окремі представники. Серед численних альдегідів і кетонів най важливіше промислове і народногосподарське значення мають формальдегід, оцтовий альдегід і ацетон.
Формальдегід. Крім загальних методів, за якими добувають альдегіди, для промислового виробництва формальдегіду розроблені такі:
1. З метанолу (термічним дегідруванням його над срібним каталізатором або окислювальним дегідруванням над мідним каталізатором):
Ар.600 °С О2, Сп, 550 °С
сн,=о---------н.с—он----------»сн,=о.
—н2О -н„о 2
2. З метану (неповним термічним окисленням його на срібному каталізаторі):
400—600 °С, Ар
СІІ4 + О2---------> СН,=О + Н..О.
Формальдегід — отруйний газ (ТКіт — —21 °С) з різким специфічним запахом, який подразнює слизові оболонки очей і дихальних шляхів. Добре розчиняється у воді; 40 %-й водний розчин формальдегіду у воді відомий під назвою формаліну і часто використовується для консервування анатомічних препаратів. У формаліні міститься 52 % води і 8 % метанолу. Формалін застосовують також для дезинфекції складів, зерпо- і овочесховищ, парників, теплиць, для протруювання насіння. Більша частина формальдегіду, який виробляється промисловістю, використовується для добування синтетичних полімерних смол, таких як фено-лоформальдегідні (див. розд.» 34), сечовіїноформальдегідні (карбаміди і) тощо.
При слабкому нагріванні і упарюванні формаліну з аміаком утворюється гексаметилентетрамін, відомий під назвою уротропіну, який вперше одержав О. М. Бутлеров у 1860 р.:
6СН2=О + 4№:3 е - > (СН2)6.М4 або
—о гіг*-)
161
Уротропін застосовують у медицині для дезинфекції сечовивідних шляхів, а також як протигрипозний препарат.
З уротропіну виготовляють вибухові речовини, наприклад гексоген. Спресований уротропін використовують як тверде пальне під назвою «сухий спирт».
Оцтовий альдегід (ацетальдегід) у промисловості, як уже зазначалося, добувають за реакцією Кучерова гідратацією ацетилену (див. розд. 15), дегідруванням або окисленням етилового спирту на срібному каталізаторі, каталітичним окисленням насичених вуглеводнів (див. розділи 7, 9).
Оцтовий альдегід використовують перш за все у виробництві оцтової кислоти, з нього також добувають оцтовий ангідрид, метил-ацетат, етиловий спирт та інші органічні речовини, його застосовують при виготовленні синтетичних смол і пластмас.
Ацетон у промисловості добувають кількома методами:
1. Сухою перегонкою деревини. Підсмольпа рідина, яка при цьому утворюється, МІСТИТЬ поряд з метанолом і оцтовою кислотою З—5 % ацетону.
2. Ацетопово-бутанольним бродінням сахаридів (див. розд. 26).
3. Дегідруванням або окисленням пропанолу-2.
4. Окисленням ізопропілбепзолу (кумолу) (див. розд. 34).
Ацетон у великих кількостях використовують як розчинник при виробництві лаків, фарб, ацетатного шовку, кіноплівки, бездимного пороху. Ацетон є також вихідним продуктом для синтезу ряду органічних сполук, наприклад органічного скла
Кетени
Кетенами називають сполуки з кумульованими подвійним С=О—і подвійним С=С-зв’язками. Найпростішим представником таких речовин єкетеи Н2С=С=О. Сам кетен і його монозаміщені
—СН=С=О називають альдокетенами, а двозаміщені кетени /?2С=С=О —кетокетенами. Кетен добувають піролізом ацетону:
050—700 °С н3с—со—сн,---------> Н2С=С=О+СН4.
Кетен не виявляє властивостей, пов’язаних з групою С=О. Він не дає фенілгідразону, оксиму. Проте кетен виявляє високу активність до реакцій приєднання, які відбуваються за С^С-зв’яз-ком. Так, він легко приєднує воду і утворює оцтову кислоту, з спиртами дає складні ефіри, а з карбоновими кислотами —ангідриди кислот, з аміаком — аміди кислот:
Завдяки високій хімічній активності кетену з нього добувають у великих кількостях оцтову кислоту, оптовий ангідрид, етилацетат та інші органічні речовини:
ІЬ2
,0
н3с-с^
МН3
—Н2С=С=|
нон о
------> н3с-с<г
\он
Оцтова кислота
Ацетамід
Ангідрид ОЦТОВОЇ кислоти
,С-С
ОН
с„нГ1—он О
--------> Н3С—С"
ХОС2Н5
Етилацетат
О
Контрольні запитання і вправи
1. Зобразіть графічні формули всіх можливих ізомерів альдегідів і кетонів складу С5Н10О, СсН, 2О і назвіть їх за міжнародною номенклатурою.
2. Які речовини утворюються під час дії на оцтовий альдегід і на ацетон синильної кислоти, гідроксиламіпу, етилмагнійброміду і етилового спирту при наявності іонів Н+?
3. У чому виявляється подібність і відмінність у властивостях альдегідів і кетонів? Якими якісними реакціями та спектральними даними можна відрізнити оцтовий альдегід від ацетону?
4. Як, використовуючи спектральні методи, можна розрізнити такі ізомерні сполуки: а) Н3С—СН=СН—С—СН3 і Н2С=СН—СН2—С—СН3;
II II
О О
б) Н..С—СН,—С—ОН і н„с—сн,—он?
II о
5. Напишіть стадії альдольпої конденсації пропапалю, яку каталізує гідроксид-іоп. Чому 2,2-диметплпропаналь не вступає в реакцію альдольпої конденсації під дією основ?
Розділ 18. МОНОКАРБОНОВІ КИСЛОТИ
Карбоновими кислотами називають такі органічні сполуки, у складі молекул яких міститься одна або кілька карбоксильних
,ОН груп —С<^ . Ки
слоти, в молекулах яких є одна
карбоксильна
група, називають монокарбоновими. Дикарбонові кислоти мають дві карбоксильні групи, трикарбонові —три і т. д. Кількість карбоксильних груп у молекулі карбонової кислоти визначає основність кислоти: кислоти з однією карбоксильною групою є однооспов-ними, здвома—двохосновними і т.д. Залежно від природи радикала, сполученого з карбоксильною групою, карбонові кислоти поділяють на насичені і ненасичені. За кількістю вуглецевих атомів у радикалі розрізняють кислоти нижчі (низькомолекулярні) і вищі (високомолекулярні).
Карбонові кислоти складають велику групу органічних сполук. В ивчення їх починають з монокарбонових кислот. Насичені моно-„О
карбонові кислоти мають загальну формулу СпН2п+і — ,
Х)Н
183
со Таблиця 18. Гомологічний ряд, номенклатура і фізичні властивості насичених монокарбонових кислот
х0 Формула Д—С" Ч0Н Назва за номенклатурою пл Гкип. °с рК“ (в Н,О)
систематичною історичною
н-соон Метанова Мурашина 8,4 100,7 3,75
н3с—соон Етанова Оцтова 16,7 118,1 4,73
Н3С—сн2—соон Пропанова Пропіонова —22,0 141,1 4,87
н3с—сн2—сн2—соон Бутан ова Масляна —7,9 169,1 4,82
н3с—сн—соон 2-Метилпроп анова Ізомасляна1 —47,0 154,4 4,86
СНз
н3с—сн2—сн2—сн2—соон Пентанова Валеріанова —34,5 187,0 4,86
н3с—сн—сн2—соон З-Метилбутанова Ізовалеріанова —17,6 176,7 4,77
СН3
Н3С—(СН2)4—соон Гексанова Капронова —4,0 205,0 4,88
Н3С— (СН2)В—соон Гептанова Енантова —10,0 223,5 4,86
Н3С-(СН2)14—соон Гексадеканова Пальмітинова 64,0 — —
Н3С—(СН2)15—соон Гептадеканова Маргаринова 60,6 — —
Н8С—(СН2)16—соон Октадеканова Стеаринова 70,0 — —
або/?—С\ , де /?—відповідний вуглеводневий радикал або
\он
атом водню, і утворюють гомологічний ряд (табл. 18), який можна записати, виходячи з загальної формули цих речовин. Для цього у формулі необхідно /? замінити на водень або інший відповідний вуглеводневий радикал. У табл. 18 наведені назви і константи найважливіших представників гомологічного ряду насичених монокарбонових кислот.
Номенклатура. Карбонові кислоти називають за систематичною та історичною номенклатурами. За систематичною номенклатурою назву кислот утворюють з назв відповідних насичених вуглеводнів, додаючи до них закінчення -ова і слово кислота. Головний ланцюг у молекулі кислоти вибирають так, щобу нього обов’язково входила карбоксильна група. Положення радикалів у вуглеводневому ланцюзі позначають цифрами. Нумерацію ланцюга починають з атома вуглецю карбоксильної групи. Наприклад, кислота будови Н3С—-СН—СН2—СН—має назву 2,4-диметилпентанова
І | кислота.
СН8 СН3
Для найпоширеніших карбонових кислот широко використовують історичні назви, такі як мурашина, оцтова, масляна, валеріанова кислота тощо (табл. 18). Ці назви пов’язані переважно з тими джерелами, з яких була виділена та чи інша кислота: мурашина — з мурашок, оцтова —з оцту, масляна —з масла і т. д.
У деяких випадках для назви кислот нескладної структури застосовують раціональну номенклатуру, в якій за основу назви беруть оцтову кислоту. Наприклад, 2-метилпропанову кислоту (ізомасляну) за цією номенклатурою називають диметилоцтовою.
Групу атомів 7?—С—О— в молекулах монокарбонових кислот І!
О
називають ацилатною групою (від лат. асібит —кислота). Назви ацилатних груп монокарбонових кислот такі: Н—С—О——форміат,
II
О
Н3С—-С—О------ацетат,. Н3С—СН2 —С—О----пропіонат і т. д.
II II
О О
Групу атомів /?—С—називають ацилом. Ацильні групи кис-II
О
лот мають такі назви: Н—С— — форміл, Н3С—С— — ацетил, II II
О О
Н3С—СН2—С-----пропіоніл і т. д.
185
Ізомерія. Монокарбоновим кислотам насиченого ряду властива ізомерія, пов’язана з розгалуженням вуглецевого ланцюга. Перші три представники гомологічного ряду цих кислот ізомерів не мають. Кислота з чотирма вуглецевими атомами — масляна — існує у вигляді двох ізомерів —н-масляної та ізомасляної (табл. 18), ваг леріанова— у вигляді чотирьох ізомерів і т. д. При збільшенні кількості вуглецевих атомів у молекулі карбонової кислоти кількість ізомерів різко збільшується.
Методи синтезу. Органічні кислоти досить поширені в рослинному і тваринному світі і можуть бути виділені з природних матеріалів. Разом з тим відомо досить багато загальних синтетичних методів добування монокарбонових кислот, які використовують як у промислових масштабах, так і в лабораторних умовах з препара-сивною метою. До найважливіших загальних методів добування монокарбонових кислот відносять такі:
1. Окислення спиртів і альдегідів (див. розділи 9, 17).
2. Окислення насичених вуглеводнів (див. розд. 7).
3. Карбонілування етиленових вуглеводнів (див. роздЛ 14).
4. Карбоксилування реактивів Гріньяра. Хлор-, бром- і йод-алканп порівняно легко можна перетворити на карбонові кислоти. Для цього з галогеналканів спочатку синтезують металоорганічні сполуки, зокрема магнійорганічні (реактиви Гріньяра), ефірний розчин яких потім насичують оксидом вуглецю (IV). При цьому утворюється змішана сіль карбонової і галогеноводневої кислот, яку потім розкладають розбавленою мінеральною кислотою. Так, з бромистого етилу цим методом можна добути пропіонову кислоту:
б+ б-*
Н-М&, ефір б- 6-і- 4-О=С—О
Н3С—СН2—Вг -------> Н3С—СН2—МйВг--------—>
ч-нон ЛО
-> Н3С-СНа-С-О-М8Вг — > Н3С-СН2-С^ + М8(ОН)Вг.
І н 4 ОН
о
Добування карбонових кислот з галогеналканів через магнійорганічні сполуки відбувається із збільшенням кількості вуглецевих атомів у молекулі синтезованої органічної кислоти. Наприклад, молекула бромистого етилу містить два вуглецевих атоми, а молекула добутої з нього кислоти •—три.
5. Гідроліз нітрилів. З галогеналканів карбонові кислоти можна синтезувати також через нітрили. У цьому випадку галогеналкан спочатку вводять у реакцію з сіллю синильної кислоти і одержують нітрил карбонової кислоти, який потім гідролізом перетворюють на відповідну кислоту. Гідроліз нітрилів здійснюють нагріванням їх з водою в кислому або лужному середовищі. Нітрил у цих умовах приєднує молекулу води і утворює спочатку нестійкий енол, який відразу ж ізомеризується (правило Ельтекова) в амід кислоти. Подальший гідроліз аміду приводить до утворення карбонової кислоти:
186
б+ б~
4-КСМ д+ С~ +Н ОН Ізомеризація
Р—Наї-----------> 7?-С^Н---------- Я—С=МН--------------
—Ї\гіа1 Нітрил | | |
ОН-1
Енол
+ 1ІО11
-> к—С-МН,---------> /?—с—он.
II “ II
о о
Амід
Добування карбонових кислот з галогеналканів через нітрили також супроводиться збільшенням кількості вуглецевих атомів у молекулі синтезованої кислоти.
Нітрильна група за електронною будовою аналогічна карбонільній. Оскільки азот більш електронегативний, ніж вуглець, то на атомі азоту в нітрильній групі зосереджується надлишок електронної густини (заряд б"), а на атомі вуглецю —її дефіцит (заряд 6+). Отже, атом вуглецю нітрильної групи є електрофільним ценсром, а атом азоту — нуклеофільним:
е+І |с--С=К.
Кислотний гідроліз нітрилів, який каталізується іонами Н+, відбувається за таким механізмом. Процес гідролізу починає іон Н+. Він приєднується до атома азоту нітрильної групи і утворює імінний карбкатіон, який потім взаємодіє з молекулою води і перетворюється на енол; При цьому одночасно регенерується протон. Енол є нестійким продуктом і відразу ж ізомеризується в амід:
—; ОН О
6+ 6~ + НО:|Н | II
С=Н4-Н+ р~С=НІ і Н++Я—с=кн->к—с— кн2.
При лужному гідролізі нітрилів каталізаторами реакції є іони ОН-, які і починають процес. Іон ОН “приєднується до вуглецевого атома нітрильної групи і утворює аніон з негативним зарядом на атомі азоту, який далі взаємодіє з водою і утворює енол. При цьому одночасно регенерується аніон ОН-. Енол відразу ж ізомеризується в амід:
<5+ Н!-ОН
/?_с=м-уон~ я—с=К“ -2ОЙ—> /?—с=ин -> т?—с—№іа. он он о
Аміди, які утворюються як проміжні продукти при кислотному і лужному гідролізі нітрилів, далі гідролізуються до карбонових кислот.
6. З малонового ефіру (див. розд. 22).
7. З ацетооцтового ефіру (див. розд. 24).
8. Гідроліз жирів (див. розд. 21). Цим методом добувають переважно вищі карбонові кислоти.
187
Фізичні властивості. Перші три члени гомологічного ряду кислот—мурашина, оцтова і пропіонова — безбарвні рідини з гострим подразливим запахом, які змішуються з водою в будь-яких співвідношеннях і розчиняються в деяких малополярних органічних розчинниках. Кислоти з кількістю вуглецевих атомів від С4 до Со — маслянисті рідини з неприємним запахом (запах згірклого масла), розчинність яких у воді зменшується в міру збільшення їх молекулярної маси. Вищі карбонові кислоти, починаючи з С;о, —тверді малолеткі парафіноподібні речовини, без запаху, не розчини і у воді, але розчинні в органічних розчинниках.
Карбоксильна група карбонових кислот —СООН є гідрофільною групою, а алкільний радикал /? — гідрофобною групою. Тому при збільшенні розміру алкільного радикала розчинність карбонових кислот у воді зменшується. З тієї ж причини густина тільки мурашиної кислоти більша за одиницю, а останніх карбонових кислот— менша від одиниці.
У кислот з нормальним вуглецевим ланцюгом в температурах плавлення виявляється так званий парно-непарний ефект. Кислоти з парною кількістю вуглецевих атомів плавляться при вищих температурах, ніж сусідні кислоти з непарною кількістю вуглецевих атомів (табл. 18). Таке чергування температур плавлення пояснюється різним просторовим розміщенням карбоксильної групи відносно кінцевої метильної групи. У кислотах парного ряду ці групи розміщені по різні боки, а непарного ряду—по один бік, що впливає на енергетичну міцність кристалічної структури речовини:
СНа .СН2 СНа
СНа СНз сна. оон.
Енантова кислота, (ТЬлг -1О°СЛ
СН'з; СН2 СН2 СН'2
Каприлова кислота (7?пл. =16°С,)
Температури кипіння монокарбонових кислот монотонно підвищуються. Як і в гомологічних рядах інших класів органічних речовин, кислоти з нормальним вуглецевим ланцюгом мають вищі температури кипіння, ніж кислоти з розгалуженим ланцюгом з такою самою кількістю атомів вуглецю.
Монокарбонові кислоти мають вищі температури кипіння, ніж спирти з такою самою кількістю вуглецевих атомів у молекулі, що зумовлено більшою полярністю зв’язку О—Н в карбоксилі, ніж у спиртів. Внаслідок цього кислоти утворюють міцніші міжмолекулярні водневі зв’язки за рахунок карбонільного кисню однієї молекули і водню карбоксилу другої молекули кислоти. Така асоціація може бути циклічною або лінійною. Фізико-хімічні дослідження, зокрема визначення молекулярної маси мурашиної, оцтової кислот, показало, що в рідкій фазі, а частково і в пароподібному стані переважають плоскі циклічні димерні молекули. На основі даних хвильових спектрів визначено розміри (у нанометрах) міжатомних віддалей в неасоційованій (І) і асоційованій (II) молекулах мурашиної кислоти:
188
2Н
0,1085
З цих даних випливає, що довжина зв’язку О—Н у молекулах карбонових кислот становить 0,1 нм, а довжина їх водневого зв’язку /С=О...Н—О 0,17 нм. Значення теплоти дисоціації димер-ної форми кислоти в мономерну становить близько 29 кДж/моль, а оскільки в димерній молекулі два водневих зв’язки, то енергія водневого зв’язку в кислотах у два рази менша і становить близько 14,5 кДж/моль. У кристалах мурашиної і оцтової кислот кільцевих димерів немає. Молекули цих кислот у даному випадку утворюють за рахунок водневих зв’язків —О—Н---О=С^ плоскі лінійні ланцюги:
0,1 0,17
—.0 О— Н-• • О О—Н'”ОЧ о-н-о о-н — с с с с
І III
А> /? А* V
Встановлено, що в 1 моль оцтової кислоти в рідкому стані при 20°С міститься 17 % мономерних молекул, 69 % кільцевих димерів і 14 % молекул, які являють собою ланцюгові асоціати. При температурах, вищих за температури кипіння, пара оцтової кислоти складається переважно з мономерних молекул Н3С—СООН.
В ІЧ-спектрах карбонових кислот є смуги поглинання як для карбонільної групи ^с=о = 1700—1725 см'1), так і для гідроксильної групи (їо-н =3670 см-1). При асоціації молекул карбонових кислот за рахунок міжмолекулярного водневого зв’язку смуга гідроксильної групи перетворюється на широку смугу і зміщується в область нижчих частот: Vо-н...о=с< =3000—2500 см-1.
У спектрах ПМР протон карбоксильної групи поглинає в інтервалі 9,0—14,6 млн-1.
Електронна будова карбоксильної групи. Карбоксильна група —С—О—Н являє собою поєднання карбонілу >С=О з гідрокси
лом —О—Н. Групи /С=О і —ОН в карбоксилі взаємно впливають одна на одну. Цей вплив виявляється в мезомерному ефекті р, л-спряження, в результаті якого р-електрони атома кисню гідроксилу зміщені до атома кисню карбонілу (4-Л4-ефект): Таке зміщен-0+^т-
-с - б - н.
189
ня електронів у карбоксильній групі приводить до підвищення полярності зв’язку О—Н і відповідно до його послаблення, а також до зменшення часткового заряду 6+ на карбонільному атомі вуглецю. В результаті атом водню в групі ОН карбоксилу стає значно активнішим, ніж в ОН-групі спиртів, і може відщеплюватися у вигляді протона Н+. Тому карбонові кислоти, на відміну від спиртів, дисоціюють у водних розчинах. При дисоціації карбонової кислоти крім протона виникає досить енергетично стійкий карбоксилат-аніон, який є резонансним гібридом двох граничних структур (ЇМ) і (ІІВ). У гібридному карбоксилатному аніоні відбувається виключно ефективна делокалізація негативного заряду і його рівномірний розподіл між двома ізовалентними атомами кисню карбоксилу. Така делокалізація веде до зменшення внутрішньої енергії цього аніона і до зменшення його тенденції зв’язувати протон, що, в свою чергу, приводить до зміщення рівноваги в бік дисоціації карбонової кислоти (тобто вправо). При сольватуючій дії розчинника (води) утворюється стабільний мезомерний карбокси-лат-аніон (III) і гідроксовій-катіон:
Ізовалентність атомів кисню в карбоксилат-аніоні підтверджується методами рентгеноструктурного аналізу та електронографії, з експериментальних даних яких випливає, що обидва зв’язки С—О в аніоні (III) однакові. Наприклад, у форміат-аніоні вони становлять0,127нм, тоді як у молекулі мурашиної кислоти прос-тий зв’язок С—О має довжину 0,136 нм, а подвійний зв’язок С=О— 0,123 нм:
Наявність в карбоксильній групі електронегативного карбонільного атома кисню зумовлює зміщення до нього електронної густини сусідніх о-зв’язків:
ХО—Н
Отже, карбоксильна група в молекулах карбонових кислот виявляє негативний індукційний ефект (—І-ефект).
Хімічні властивості. Властивості карбонових кислот зумовлені перш за все наявністю в їх молекулах карбоксильної групи.
190
Кислотні властивості. Карбонові кислоти у водних розчинах дисоціюють:
,0 .О
4?-С^ +Н2Оі=>/?-С^ +Н3О+.
4 о—н ХСГ
Проте порівняно з мінеральними кислотами, такими як НС1, НВг, НІ, Н28О4, НИО3, Н3РО4 та іншими, органічні кислоти набагато слабкіші, але сильніші від вугільної кислоти. Тому мінеральні кислоти витісняють карбонові кислоти з їх солей, а карбонові кислоти в свою чергу витісняють вугільну кислоту з її солей.
Згідно з теорією кислот і основ Бренстеда —Лоурі, такі сполуки, як вода, спирти, ацетилен, аміак, аміни, вуглеводні, можна розглядати також як кислоти. Однак вони порівняно з карбоновими кислотами набагато слабкіші. Відносна кислотність цих сполук змінюється в такому порядку:
неї > /?—СООН > Н—ОН > /?—ОН > НС=СН > N11, > 7?—Н.
Так, кислотні властивості атома водню гідроксильної групи карбоксилу карбонової кислоти за рахунок — І-ефекту і ефекту р, л-спряження •——С—--групи значно сильніші, ніж водню гідро
ксилу молекули води. Разом з тим, кислотні властивості водню групи ОН спирту завдяки -^/-ефекту алкільної групи виявляються склабкіше, ніж у гідроксилі води. Сказане підтверджують значення гл25
Дд для цих речовин:
Н3С-С Н-ОН Н3с—.СН2—,о-н.
0-Н
Яд* 1.76-10-5 І0-и ІО-18
Кислотні властивості карбонових кислот Ц—СООН залежать від природи і будови алкільного радикала 7?, наявності замісників у його вуглецевому ланцюгу. Групи з позитивним електронним ефектом зменшують величину часткового заряду 6+ на карбонільному С-атомі і тим самим послаблюють його електроноакцепторну дію на групу —ОН. Це приводить до послаблення сили карбонової кислоти4 Тому в гомологічному ряду монокарбонових кислот мурашина кислота, в молекулі якої біля карбонільної групи немає вуглеводневого радикала з позитивним електронним ефектом, є найсиль-нішою кислотою. Оцтова кислота порівняно з мурашиною в десять разів слабкіша, оскільки в її молекулі є метильний радикал, який виявляє -ф/-ефект. Атоми і групи атомів з негативним електронним ефектом збільшують величину заряду 6 + на карбонільному С-атомі У тим самим підсилюють електроноакцепторну дію карбонілу на
191
групу —ОН. Це зумовлює значне підвищення сили карбонової кислоти. Тому хлороцтова кислота приблизно в 20 разів сильніша, ніж оцтова. Отже, групи 7? з позитивним електронним ефектом зменшують, а з негативним електронним ефектом підвищують силу карбонових кислот. Це переконливо підтверджує порівняння значень рКк карбонових кислот, ЯКІ МІСТЯТЬ різної електронної природи (стрілками показано зміщення електронів у молекулі кислоти):
О о
Кислота: Н —С Н3С-»-С
о-н чо-н
рЛ'к 3,75 4,75
н3с
Н3С
Кислота:
О
сі — сн2 — с'
0—11
рАф 2,85
В міру віддалення замісника з —1 -ефектом від карбоксильної групи його вплив на цю групу послаблюється (затухає). Це видно з порівняння кислотних властивостей хлормасляних кислот:
“ Р Т
Кислота: Н,С—СНа—СН—. Н„С—СН—СНа—. НгС—СНа—СН,—С^
<51 он ’ А он ’ он ’
а-Хлормасляна Р-Хлормасляна у-Хлормасляна
рКк 2,86 4,05 4,42
Як видно із значень р/\к цих кислот, найбільший вплив на карбоксильну групу має атом галогену тоді, коли він знаходиться найближче до цієї групи, тобто в «-положенні відносно неї. Тому «-хлормасляна кислота значно сильніша від £- і у-хлормасляних кислот, а р-хлормасляна кислота з тієї ж причини сильніша від у-хлор-масляної.
Утворення солей. Карбонові кислоти при взаємодії з активними металами, основами, основними оксидами, солями вугільної кислоти утворюють солі:
2Н3С-СООН+М8 -+ (Н8С—СОО)яМе+На;
2Н3С—СООН+СаО (Н3С—СОО)2Са+Н2О;
Н3С—СООН+ШОН -> Н3С—СООМа-і-НаО;
Н3С—СООН + ИаНСОз -> Н3С— СОО№ + СО2+Н2О.
Солі карбонових кислот є кристалічними, іонно побудованими нелеткими речовинами. Амонійні солі і солі лужних металів органічних кислот добре розчинні у воді, але не розчиняються в неполярних розчинниках. Солі вищих карбонових кислот, наприклад пальмітинової, стеаринової, називають милами.
192
Основні властивості. У зв’язку в р, я-спряженням 2+^-.
-О - Н в групі основні властивості гідроксилу в карбоксилі СоЗ-
карбонових кислот дуже послаблені і даний гідроксил ці властивості виявляє тільки в реакціях з сильними мінеральними кислотами, такими як Н28О4, НС1О4.
При взаємодії карбонових кислот з Н28О4, НС1О4 протон цих сильних мінеральних кислот приєднується до карбонільного атома кисню і утворює таким чином спряжену кислоту (І)?
Р—С—О—Н + Н„8О4 =*=> /?—С—О—Н + Н80Г.
II II
О *ОН
і
Протон мінеральної кислоти може приєднатися також до атома кисню гідроксилу карбоксильної групи карбонової кислоти і утворити спряжену кислоту (II):
д—С—О—Н + Н„8О4 С—ОН„ + Н80Г.
II II
О О
п
Спряжена кислота (II) енергетично менш стабільна порівняно з її ізомером (І), у якого протон сполучений з киснем карбонілу.
Характерною реакцією карбонових кислот, яка відбувається з участю ОН-групи карбоксилу, є їх взаємодія при наявності мінеральних кислот із спиртами і утворення при цьому складних ефірів (див. розд. 19):
____ /О
Я—СГІ " + ні—О—Р Р—СС + Н.о.
|О—Н_____[ ''0—7?
Взаємодію карбонових кислот із спиртами називають реакцією етерифікації.
Реакції з участю а-водневого атома. Карбоксильна група, як зазначалося вище, є електрофільною і виявляє негативний електронний ефект (—/-ефект), під впливом якого електронна густина на а-вуглецевому атомі алкільного радикала зменшується:
Внаслідок цього даний атом вуглецю сильніше притягує до себе електронні пари від атомів водню і тим самим підвищує активність а-водневих атомів, які стають рухливими і можуть брати участь
7 мзо
193
в реакціях заміщення. Так, у молекулі оцтової кислоти «-водневі атоми можуть послідовно заміщуватись на атоми галогену:
н-0іЕ +сіг
Н.С—СООН----------► СІ—СІ 12—СООН------► С1„СІ-1—СООН —
-неї —неї
Монохлороцтова кислота Ди хлор оцтова кислота
-------* С13С—СООН.
—неї
Трихлороцтова кислота
Алкільні радикали інших карбонових кислот при взаємодії з галогенами можуть піддаватися безладному вільно-радикальному галогенуванню, характерному для алканів. Але якщо галогенування карбонових кислот проводити при наявності каталітичних кількостей фосфору, то в цьому випадку відбувається галогенування виключно в «-положення. Таке галогенування відоме як реакція Гель — Фольгард —Зелінського:
Вг2 Вг2
СН3—СН2—СООН-----------► СН3—СНВг—СООН---------*СН„—СВг,—СООН
—НВг —НВг
У молекулах карбонових кислот знаходиться карбонільна група, але її активність дещо відрізняється від активності такої самої групи в молекулах альдегідів і кетонів. Оскільки в карбонільній групі кислот має місце р,л-спряження між вільними р-електронами атома кисню гідроксилу і електронами л-зв’язку групи >С=О, то за рахунок цього величина заряду б+ на карбонільному атомі вуглецю карбонових кислот стає меншою, ніжу альдегідів і кетонів. Тому вуглець карбонілу кислот менш електрофільний, ніж у альдегідів і кетонів, і значно важче приєднує хімічні реагенти. Отже, реакційна здатність карбонільної групи в нижченаведеному ряду зменшується відповідно до зменшення величини заряду 6+ на карбонільному атомі вуглецю:
8+ 8- 84 8'- 8+ 8- 84" о
н-с=о > /?-с=о > /?-с = о > /?-с ; 8+>8+>8+>84".
х V \ О.
Н Н /? О-Н
Окремі представники. Мурашина кислота вперше була добута у XVII ст. перегонкою червоних лісових мурашок (Еогтіса гпіа, звідки пішла латинська та історична назви цієї кислоти Асідіші Іогтісиш). Солі мурашиної кислоти називають форміатами. Мурашина кислота досить поширена в природі. Вона трапляється у вільному стані в мурашках, у кропиві, виділяється також з тваринних організмів з потом та сечею.
У промисловості мурашину кислоту добувають взаємодією оксиду вуглецю (II) з розплавленим гідроксидом натрію. При цьому спочатку утворюється сіль мурашиної кислоти, яку потім дією мінеральної кислоти перетворюють на мурашину кислоту:
200 °С,6—10-102 Па неї
СО-І №ОН---------->-Н—СОО№----------► II—СООН.
— МаСІ
194
Для препаративних потреб мурашину кислоту можна добувати нагріванням щавлевої кислоти з гліцерином (див. розд. 22).
Мурашина кислота належить до кислот середньої сили і вступає в усі реакції, характерні для монокарбонових кислот. Проте ця кислота виявляє також ряд специфічних властивостей, які відрізняють її від інших членів гомологічного ряду карбонових кислот. У молекулі мурашиної кислоти, як першого члена гомологічного ряду кислот, карбоксильна група сполучена з атомом водню. Тому ця кислота містить у своїй молекулі одночасно як карбоксильну, так і альдегідну групи:
Карбоксильна група
но-
Альдегідна група
У зв’язку з цим мурашина кислота має крім кислотних властивостей також властивості альдегідів. Так, вона легко окислюється і виявляє сильні відновні властивості. Мурашина кислота відновлює аміачний розчин гідроксиду срібла (дає реакцію срібного дзеркала), окислюється КМпО4 та іншими окислювачами. При окисленні мурашина кислота перетворюється на нестійку вугільну кислоту, яка розкладається на оксид вуглецю (IV) і воду:
хон
4-І О]
х'1
н—о—
ХО—н
->СО2 + Н2О.
Мурашина кислота —сильний відновник і може відновлювати метали з їх солей:
Н-С^ +.нйсі2 -> + СО2 + 2НС1.
ХОН
Мурашина кислота має порівняно невисоку термічну стійкість. Так, при нагріванні з концентрованою Н28О4 вона розкладається на оксид вуглецю (II) і воду:
н—с<^° н“5°* , СО + Н2О.
хон
Цю реакцію використовують для добування чистого оксиду вуглецю (II).
Мурашина кислота застосовується в промисловому органічному синтезі, у текстильній промисловості (у вигляді алюмінієвої солі) для протравного фарбування тканини, при вичинці шкір, у харчовій промисловості для консервування соків і білково-вітамінних концентратів, для дезинфекції бродильних чанів при виробництві спирту, пива, для дезинфекції бджолиних вуликів, у медицині (1,25%-й спиртово-водний розчин мурашиної кислоти називають «мурашиним спиртом») і т. д.
1*
195
Оцтова кислота відома з глибокої давнини у вигляді оцту, який утворюється при окислювальному оцтовокислому бродінні спиртових напоїв:
+[О]
Н3С—СН,—ОН--------> Н.С—СООН + н,о.
Оцтова кислота — рідина з гострим запахом оцту, яка застигає (замерзає) при 16,7 °С у кристалічну масу, що нагадує лід. Тому чиста безводна оцтова кислота називається льодяною.
Технічну оцтову кислоту в промисловості добувають окисленням оцтового альдегіду, який у свою чергу синтезують гідратацією ацетилену (див. розд. 15), а також сухою перегонкою деревини. Широкого технічного застосування набув також метод добування оцтової кислоти окисленням бутану:
Н3С—СН,—СН,—СН3 —------► 2Н3С—СООН + нао.
Оцтову кислоту, яку використовують для харчових потреб і консервування харчових продуктів, добувають методом окислювального бродіння 6—10 %-го водного спирту під впливом особливих бактерій.
Оцтова кислота і її солі широко застосовуються в різних галузях народного господарства і в хімічній промисловості. У харчовій промисловості оцтову кислоту використовують як смакову і консервую іу речовину. У продаж надходять 5—8 %-й водний розчин оцтової кислоти під назвою харчовий оцет, а також оцтова есенція, яка являє собою 80 %-ну оцтову кислоту.
Практичне значення мають також добре розчинні солі оцтової кислоти, які називаюь ацетатами, наприклад, ацетати натрію,г алюмінію, заліза, хрому, міді, свинцю та інших металів.
Вищі монокарбонові кислоти. Пальмітинова С16Н81СООН і стеаринова С17Н8БСООН кислоти — тверді, не розчинні у воді речовини. У вигляді складних ефірів з гліцерином (гліцеридів) вони входять до складу більшості жирів (див. розд. 21). Пальмітинова кислота у вигляді складного ефіру є складовою частиною бджолиного воску (див. розд. 9).
Як самі ці кислоти, так і їх солі одержують каталітичним гідролізом жирів. Солі вищих карбонових кислот називають милами. Натрієві і калієві мила добре розчинні у воді. Суміш пальмітинової і стеаринової кислот називають стеарином і використовують для різних технічних потреб, наприклад для виготовлення свічок.
Значні кількості вищих карбонових кислот у техніці добувають каталітичним окисленням вищих насичених вуглеводнів (парафінів):
О,, Мпї+: 400—500 °С
Н8С-(СН2)„-СН3------------» Н8С-(СН2)СТ-СООН +
+ Н8С—(СН„)р—соон.
При цьому утворюється суміш кислот з т і р т 10—18, яку використовують для виробництва мила та миючих засобів.
196
Контроль ні запитання і вправи
1. Зобразіть графічні фомули всіх ізомерів карбонової кислоти складу СВНП—СООН і назвіть їх за міжнародною номенклатурою.
2. Які речовини утворюються при взаємодії масляної кислоти з етанолом, РС16, хлором, карбонатом кальцію?
3. Поясніть, до якого результату приведе спроба здійснити етерифікацію оцтової кислоти трет-бутиловим спиртом при наявності сухого НС1.
4. Поясніть, як за допомогою ІЧ-спектроскопії і методу ЯМР можна розрізнити: а) Н3С—СООН і Н3С—СН=О; б) Н3С—С—ОН і Н3С—С—-СН3;
II II
О О
в) Н3С—С—ОН і Н3С—сн2—он.
II о
б. Розмістіть речовини в порядку зростання їх кислотності: пропанова кислота, ацетилен, пропанол-1, пентан, 2-хлорпропанова кислота, 3-хлор-пропанова кислота.
Розділ 19. ПОХІДНІ КАРБОНОВИХ КИСЛОТ
Похідними карбонових кислот називають такі органічні речовини, які можна добути з органічних кислот в результаті хімічних змін у їх карбоксильній групі. Найважливішими похідними кислот є солі, галогенангідриди, ангідриди, складні ефіри, аміди і нітрили.
Галогенангідриди Г—С—Паї — це продукти заміщення гідро-II О
ксилу карбоксильної групи карбонових кислот на галоген. Розріз-няютьфтор-, хлор-, бром- і йодангідриди кислот. Серед галогенан-гідридів карбонових кислот найважливіше значення мають хлорангідриди. їх найчастіше добувають взаємодією карбонових кислот з хлоридом фосфору (V) або нагріванням кислот з тіонілхлоридом:
Я—С—ОН + РС15 Р—С—СІ + РОС13 + НС1;
II II
О О
Р—С— ОН + 8ОС12 Р—С—СІ + 8О2 + НС1.
II II
О О
Хлорангідриди називають переважно за назвою кислот. Наприклад, Н3С—С—СІ—хлорангідрид оцтової кислоти. Проте часто II О
їх називають також за назвою ацильного залишку /?—С—. Так,
II
О хлорангідрид оцтової кислоти називають хлористим ацетилом, хлорангідрид пропіонової кислоти — хлористим пропіонілом і Т. д.
Хлорангідрид мурашиної кислоти (хлористий форміл) нестійкий і в момент утворення розкладається на оксид вуглецю (II) і хлороводень:
ГН—С—С11
Ц ->СО + НС1.
І О І
197
Хлорангідриди, молекули яких містять від двох до шести С-ато-мів,—рідини з різким подразнюючим запахом і сльозоточивою дією. Наприклад, хлористий ацетил—легкокипляча рідина з Т™ = 52 °С.
Хімічні властивості хлорангідридів карбонових кислот зумовлені наявністю в їх молекулі атома хлору і карбонільної групи. У молекулах хлорангідридів біля вуглецевого атома, сполученого з атомом галогену, знаходиться карбонільний атом кисню, який відтягує електронну густину сусідніх зв’язків на себе:
?,+ 8-
В результаті Р - С - СІ
Со
на цьому вуглецевому атомі заряд 6+ більший, ніж на атомі сполученому з атомом хлору в молекулах хлоралканів - /?—СН2—СІ. Відповідно полярність зв’язку С—СІ у молекулах хлорангідридів значно вища, ніж полярність аналогічного зв’язку хлоралканів. Тому хлор у хлорангідридах набагато реакційноздат-ніший порівняно з хлоралканами і може надзвичайно легко заміщуватись різними нуклеофільними реагентами. Високу активність з тієї ж причини виявляють атоми галогену в молекулах інших галогенангідридів.
Заміщення галогену в галогенангідридах відбувається надзвичайно легко і, як і в галогеналканах, за нуклеофільним механізмом -реакції). Так, галогенангідриди гідролізуються навіть вологою повітря і утворюють при цьому відповідні карбонові кислоти- Екзотермічно реагують галогенангідриди з аміаком, спиртами і утворюють при цьому аміди і складні ефіри:
б-е+ б+е-
+с,н,он 6+ б- +нон
НС1 + Н,С—Є—ОС2ІІ5 ч-----------Н,С—С—СІ-----------» Н3С-—С—ОН 4-НС1.
II II II
О 0 0
Етилацетат | +К'Н3
Н3С—С—МН2 + НС1.
II
О
Ацетамід
У наведених реакціях відбувається заміщення окремих атомів у молекулах різних речовин (вода, спирт, аміак) на залишок карбонової кислоти —ацил. Тому взаємодію такого типу називають реакціями ацилування.
Галогенангідриди енергійно реагують також з первинними і вторинними амінами, утворюючи при цьому И-заміщені аміди карбонових кислот:
Н3С—С—СІ + С2Н.—ЙН, -> Н.С-С—N1-1—С2Н, + неї.
М-Етилацетамід
198
пероксиду водню:
Н3С—С—О—О—С—СН3.
II II
О о
Пероксид ацетилу
Заміщення галогену в молекулах галогенангідридів на групи МН2, —-МН7? або —Мї?2 називають реакціями амонолізу.
У практиці органічного синтезу хлорангідриди карбонових кис-$ лот використовують для добування пероксидів ацилів, наприклад пероксиду ацетилу, які є похідними
н—о—О—Н; Н3С—С—О—О—Н;
II
О
Пер оцтова (надоцтова) кислота
Пероксид ацетилу добувають взаємодією хлористого ацетилу з пероксидом натрію:
2Н3С—С—СІ + На—О—О—На — Н3С—С—О—О—С—СН3 + 2№С1.
II II II
О О О
Пероксиди ацилів при нагріванні легко розкладаються з утворенням вільних радикалів:
Н3С—С—О—О—С—СН32СН3 + 2СО2.
II II
О О
Тому їх застосовують як ініціатори вільно-радикальних реакцій, наприклад, при полімеризації ненасичених мономерів (див. розд. 14).
Ангідриди кислот Я—С—О—С—7? можна розглядати як про-
О О
дукти міжмолекулярного відщеплення води від двох молекул карбонових кислот за рахунок гідроксилів їх карбоксильних груп:
—Н2О
Оцтовий ангідрид
Найважливіше промислове і препаративне значення має оцтовий ангідрид. Його добувають нагріванням безводної оцтової кислоти, а також взаємодією хлористого ацетилу з ацетатом натрію та реакцією кетену з льодяною оцтовою кислотою (див. розд. 17):
.О /О
Н3С—С< + Н3С—-+ (Н3С—СО)2О + Масі.
___ чЗІ ОН’а
| Ангідриди кислот виявляють високу хімічну активність. Вони, як і хлорангідриди, є добрими ацилуючими реагентами.
Оцтовий ангідрид — рідина (7\Нп — 140°С) з різким неприємним запахом,важко розчиняється у воді і поступово гідролізується нею до оцтової кислоти. З спиртами він утворює складні ефіри, з аміаком — ацетамід:
(Н3С—СО) О2 + НОН - 2Н3С—СООН;
199
(Н8С—СО)2О -|- С2Н8ОН -> Н3С—С—О—С2Н8 -|~ Нзс—СООН;
О
(Н3С—С0)20 + 2МН3 -> Н3С—С—МІ2 + Н3С—СООК'П4.
о
Ацетамід
Оцтовий ангідрид широко застосовують у техніці для ацетилування.
Складні ефіри карбонових кислот Я—С—О—Я' можна розгля-
II
О
дати як продукти заміщення гідроксилу карбоксильної групи кислот на алкоксильну групу —ОТ?' або як продукти ацилування спиртів.
Назви складних ефірів походять від назв карбонової кислоти і спирту, з яких вони утворилися. Так, ефір Н3С—СООС2Н5 називають оцтовоетиловим ефіром, або етилацетатом, Н—СООСН3— мурашинометиловим ефіром, або метилформіатом і т. д.
Складні ефіри нижчих карбонових кислот являють собою рідини з приємним фруктовим запахом, легші за воду.
Складні ефіри можна добути взаємодією спиртів з хлорангідридами або ангідридами кислот. Проте^найважливішим методом добування складних ефірів є реакція взаємодії карбонових кислот із спиртами — реакція етерифікації. Реакції етерифікації каталізуються іонами водню Н + . Взаємодія карбонових кислот з спиртами — рівноважний процес:
- Р—С—ОН + /?'—ОН /?—С—О/?' + Н2О.
II II
О о
Утворення складного ефіру називають етерифікацією, а зворотний процес, тобто взаємодію складного ефіру з водою, —гідролізом складних ефірів.
Етерифікацію і гідроліз складних ефірів вивчали М. Бертло (1862 р.), М. О. Меншуткін, Я. Вант-Гофф(1877 р.) і багато інших.
Етерифікація-гідроліз —добре вивчений приклад застосування закону діючих мас до гомогенних рідких систем. У випадку взаємодії еквімолекулярних кількостей оцтової кислоти і етилового спирту при 20 °С для досягнення рівноваги потрібно 368 діб, а при нагріванні і каталітичній дії іонів водню рівновага встановлюється протягом кількох діб. При цьому вихід етилацетату досягає близько 70 %:
Н3С—С—ОН + С2Н8ОН Н3С—С—ОС2Н8 + Н2О.
II II
о о
Для повного перетворення оцтової кислоти на етилацетат, тобто щоб зсунути рівновагу даної реакції вправо, спирт необхідно брати у восьмикратному надлишку відносно кислоти.
200
Механізм реакції етерифікації. Цікавим е питання про утворення води в реакції етерифікації, а саме: за рахунок якого гідроксилу — кислоти чи спирту — утворюється вода. З застосуванням методу «мічених атомів» (у реакцію ввели спирт, який містив важкий ізотоп кисню 18О) було встановлено, що після етерифікації «мічений» атом кисню входив до складу складного ефіру, а вода містила звичайний атом кисню:
Я—С---ОН + Н | — «О—Я'
Я—С—18О—Я' + Н2О.
На основі цього дійшли висновку, що вода при етерифікації утворюється за рахунок атома водню ОН-групи спирту і гідроксилу — —СООН-групи кислоти. Отже, у реакції етерифікації відбувається нуклеофільне заміщення ОН-групи карбоксилу кислоти на алко-ксильну групу/?'О—спирту. Але у зв’язку з тим, щоспиртє слабким нуклеофілом, а карбонова кислота не сильним електрофілом, то реакція взаємодії карбонової кислоти і спирту відбувається досить повільно. Прискорюють етерифікацію кислотні каталізатори. З цією метою в реакційну суміш добавляють невеликі кількості концентрованої Н28О.і або вводять газоподібний НСІ. Каталізаторами можуть бути також і кислоти Льюїса, наприклад ВР3. Механізм реакції етерифікації при наявності каталітичних кількостей кислот можна подати у вигляді таких рівноважних реакцій:
&-
+ /х°-н . он
я-сч + н х=їЯ-С -----------Я-С
ОН хон { \)Н
+/он он
/?“С + Я'-О-Н^ХЯ-С-ОН
1 хі я-о-н
II
(1)
(2)
он
он
к-с-о-н
я — о\н
Я-С+
, І я -о
о
II
Я-Є-О-Я' • (3)
III IV V
На повільній стадії (1) до карбонільного атома кисню приєднується протон (каталізатор) і утворюється карбонієвий катіон (І), у якому значно збільшується позитивний заряд на карбонільному атомі вуглецю. На другій стадії (2) катіон (І) приєднує молекулу спирту і утворює зв’язок між карбонільним атомом вуглецю і гідроксилом спирту за рахунок вільної пари електронів його атома кисню. В результаті утворюється оксонієва сполука (II), яка потім
20»
перегруповується в гідроксонієву сполуку (III), здатну відщеплювати воду і перетворюватися на карбкатіон (IV). На останній стадії карбкатіон (IV) відщеплює протон, який починав каталітичний процес на першій стадії, і перетворюється на кінцевий продукт етерифікації—складний ефір (V) (реакція (3)).
Гідроліз складних ефірів. Гідроліз складних ефірів, наприклад етилацетату, в чистій воді (рН « 7) також належить до 5уу-реакцій і відбувається повільно, оскільки вода є дуже слабким нуклеофі-лом:
Н3С—С—О—СаН6 + Н—ОН Н3С—С—ОН + С2Н5—он.
Набагато швидше відбувається каталітичний гідроліз складних ефірів, який проводять у кислому або лужному середовищі.
Складні ефіри виявляють слабкі основні властивості, і механізм їх кислотного гідролізу можна зобразити, наприклад для етилацетату, у вигляді таких рівноважних реакцій:
+ /~н ?н
НзС-с + н нзіс-с =н3с-с-осгн5; (і)
ос,2н5 ЧОС,НЯ +]
он он
І І
Н'зС-С- ОС^На
о нх+хн II
о-н
о
НзС-С-О-С і!5Х=Г.Нз.С — С Ґ. О —С^Н;,^—ГН3С- + С2Н3ОН + Н +
За даними ІЧ-спектрів, складні ефіри протонізуються кислотами. Вони приєднують протон до карбонільного атома кисню і утворюють карбкатіон (І) (реакція (1)), який далі приєднує молекулу води і дає гідроксонієвий катіон (II) (реакція (2)). Катіон (II) потім перегруповується в катіон (III), розкладання якого приводить до утворення кінцевих продуктів гідролізу — карбонової кислоти (IV) і спирту (V). Одночасно на заклк! ній стадії відщеплюється протон, який почав каталітичний пропсс гідролізу складного ефіру (реакція (3)). Легко помітити, що наведена схема кислотного гідролізу складного ефіру обернена описаній вище схемі реакції етерифікації.
Лужний гідроліз протікає швидше, ніж кислотний, оскільки нуклеофільний аніон ОН-, який атакує карбонільний атом кисню, активніший і менший за об’ємом, ніж молекули води у ході кислот
202
ного гідролізу. Механізм лужного гідролізу складних ефірів можна зобразити такою схемою:
б- о-
С+ х?® Повільно І 18 Швидко хО
/?-с< 18 ----> /?—С—О—/?' Г -7? /?-С^ +
\Х о/ . «А- ШИ,ДК0 І Повільно \0Н
—+ ПІ) он
+ ^,180- Я'-О-Н + /?_с/° — .
\о- хОИа
За допомогою «мічених атомів» було доведено, що під час гідролізу в молекулі складного ефіру розривається зв’язок між ацилом і алкоксильною групою, оскільки мічений атом кисню 180 після реакції було виявлено у спирті, а не в карбоксилі кислоти.
Лужний гідроліз складних ефірів необоротний, оскільки на заключній стадії цього процесу утворюється карбоксилат-аніон, в якому відбувається повна делокалізація негативного заряду між обома атомами кисню, а такий аніон уже не чутливий до атаки спиртового гідроксилу.
Лужний гідроліз, який називають також омиленням складних ефірів, має широке практичне застосування. Так, омиленням багатьох природних складних ефірів водними розчинами лугів добувають солі вищих карбонових кислот (мило), гліцерин та інші спирти.
Реакція переетерифікації. Складні ефіри при нагріванні з спиртами (алкоголіз), кислотами (ацидоліз) або іншими складними ефірами вступають у реакції обміну, які називають реакціями переете-рифікації.
Алкоголіз:
/?—С—О—/?' + /?"—ОН 5* Р—С—О—Р” 4- /?'—ОН.
Іі II
О о
Ацидоліз:
Р—С—О—Р' + Р"—С—ОН ті Р"—С—О—Р' + Р—С—ОН.
II II II II
О О О О
Подвійний обмін:
Р—С—О—Р' + р"—С—О—Р'"
А>__С—О—/?"' + Р"— С—О—Р'.
Реакції переетерифікації притаманні фізіологічним процесам, що відбуваються в тваринних і рослинних організмах. Переетери-фікація каталітично прискорюється кислотами і основами.
Реакція переетерифікації оборотна. Для зміщення рівноваги в реакційну суміш вводять надлишок спирту при алкоголізі, надлишок карбонової кислоти при ацидолізі і надлишок іншого складного ефіру при подвійному обміні.
203
Амоноліз. Під час взаємодії складних ефірів карбонових кислот з аміаком, а також з первинними або вторинними амінами, утворюються відповідні аміди. Реакції відбуваються у відносно м’яких умовах, оскільки речовини з змінною групою виявляють вищу нуклеофільність, ніж спирти. Механізм цієї оборотної ^2-реакції аналогічний механізмові лужного гідролізу складних ефірів. Аміак спочатку приєднується до карбонільного атома вуглецю і утворює біполярний іон, який потім розкладається на амід і спирт. Механізм амонолізу етилацетату аміаком можна зобразити такими рівноважними реакціями:
г-
. О Оі ... о
8+4- .. Повільно і л-х Швидко //
УзС-С + МН3 ----------- Н3С - С - ОС2Н5^=Г НаС~ С + С2Н5ОН.
'оС2Нв НгІЧ.’О?) NN2
Складні ефіри досить поширені в природі. Вони входять до складу ефірних олій рослин, жирів, восків. Запах квітів і фруктів зумовлений наявністю в них суміші різних ефірів. Так, етиловий ефір мурашиної кислоти Н—СООС2Н5 має запах рому, етиловий ефір н-масляної кислоти С3Н-СООС2Н5 —запах ананаса, ізоамілацетат НЯС—СООС6Нц-гзо —запах грушевої есенції, н-аміловий ефір ізовалеріанової кислоти (Н3С)2СН—СН2—СООС5НП —запах яблук. Ці ефіри використовують для надання запаху різним напоям. Етилацетат та ізоамілацетат застосовують як розчинники синтетичних смол, для виготовлення лаків, фарб тощо.
Аміди карбонових кислот —МН2 — це похідні карбонових
II О
кислот, у молекулах яких гідроксильна група карбоксилу замінена на групу —МН2, яка у складі цих речовин називається амідною.
Аміди карбонових кислот можна добути, як уже зазначалося, взаємодією галогенангідридів і ангідридів кислот, а також складних ефірів з аміаком. Крім того, їх можна добути під час сухої перегонки амонійних солей карбонових кислот та неповним гідролізом нітрилів кислот:
Н3С—С/0 Перегонка^ н с_с^° + н о
\огш4 \гш2
С+ 6” б+ б~ ГТ+
Н3С—С=И + Н—ОН--------» ГН3С—С=ІЧН] -> Н3С—с—ин„.
І II
І он о
М-Заміщені аміди, як встановив у 1886 р. Е. Бекман, утворюються перегрупуванням оксимів альдегідів і кетонів, яке відбувається при нагріванні і наявності таких каталізаторів, як Н2ЗО4,
204
РСІБ, поліфосфорна кислота тощо. Таке перегрупування оксимів називають перегрупуванням Бекмана".
хМ—О—Н н т ,о
Р-С^ Н-С^ ;
ХН 'N11— Р
Альдоксим ЬІ-Заміщений формам ід
%с=ьі—ОН н*—* > р— с^°
р/ ЖН—р
Кетоксим К-Заміщений амід
Встановлено, що при перегрупуванні Бекмана гідроксильна група оксиму міняється місцем з радикалом, який знаходиться відносно неї в транс-положенні, за такою схемою:
/?-сч
"о
Хімічні властивості амідів кислот визначаються наявністю в їх молекулах груп —ІЧН2 і —С—. Проте завдяки взаємному впливу
II
О
атомів у молекулах властивості цих речовин дещо відрізняються від властивостей амінів і карбонільних сполук. Так, в амідній групі має місце р,л-спряження між вільними р-електронами атома азоту групи —МН2 і електронами л-зв’язку групи /С=О. При цьому вільні електрони азоту ПН2-групи виявляють ф-Л4-ефект, а група /С==О------ТИ-ефект. Таке спряження приводить до змі-
щення р електронів атома азоту до карбонільної групи:
8-
г°
іїна гін.
Внаслідок цього електронна густина на атомі азоту групи—ЛН2 зменшується і тому зменшується здатність цієї групи зв’язувати іони Н+. Отже, за рахунок р,л-спряження основні властивості —МН2-групи амідів послаблюються і вони стають практично нейтральними сполуками. У цьому можна переконатися, порівнявши константи основності, наприклад, ацетаміду і аміаку . Для аміаку при 25 °С вона дорівнює 1,8 • 10-6, а для ацетаміду при тій самій
205
температурі —10-1Б. Внаслідок послаблення основних властивостей аміди утворюють солі тільки з сильними мінеральними кислотами. Причому приєднання протона відбувається переважно до атома кисню карбонільної групи:
&-
&+£°
Н3С-С^ + неї
\н2
СІ. ,
Разом з тим аміди виявляють слабкі кислотні властивості, оскільки зменшення електронної густини на атомі азоту їх ГШ2-групи зумовлює сильніше зміщення до нього електронів зв’язку N ч- Н і, отже, підвищення активності атомів водню у цій групі:
Так, аміди в середовищі діетилового ефіру реагують з металічним натрієм, амідом натрію, оксидом ртуті (ТІ) і утворюють солі, які містять амбідентний аніон. Наприклад:
/О
Н3С—С<^ + На
\мн2
Н3С—С
'''МІГ
Отже, для амідів можлива внутрішньомолекулярна амідне імідна таутомерія:
/О
/?-С^ /?-С
хмн2
он ^ічн
проте амідна форма стійкіша від імідної на 94,2 Дж/моль.
Аміди крім групи —МН2 містять також групу —СО—. Однак внаслідок р,л-спряження, а саме за рахунок ф-ЛІ-ефекту МН2-групи, на карбонільному атомі вуглецю різко зменшується величина заряду 6 + . А це послаблює активність електрофільного центру молекул цих речовин. Тому реакції карбонільної групи в амідах виражені надзвичайно слабко.
Важливою властивістю амідів є їх здатність гідролізуватись і перетворюватись при цьому на карбонові кислоти:
.О /О
/?—СС + НОН Я— се + Ь!Н3.
ХНН2 4 ОН
Аміди вступають у реакцію з азотистою кислотою. При цьому вони, відщеплюючи азот, перетворюються на карбонові кислоти:
Я—+ НМО2 -> /?—с{ + К2 + Н2О.
хі\Н2 он
206
А. Гофманвстановив (1881 р.), що аміди, які не мають замісників біля атома азоту в групі —NН2, вступають у реакцію з гіпобро-мітом натрію (або бромом у розчині К’аОН) і через стадію утворення ізоціанатів перетворюються на первинні аміни, які містять на один атом вуглецю менше ніж вихідний амід:
К-С^° —Ла°Вг о /?—М=С=О Д?° Ь!На.
\мН “^аВг, —Н8О СО2 2
Таке перетворення амідів карбонових кислот на аміни називають перегрупуванням Гофмана.
Аміди при нагріванні з сильними водовідбирними реагентами дегідратуються і перетворюються при цьому на нітрили карбонових кислот. Наприклад:
/О
Н3С—Н,С—С=М.
—Н£О . .
2 Ацєтонітрнл
(нітрил оцтової кислоти)
Аміди нижчих карбонових кислот добре розчинні у воді, мають високі температури кипіння, що свідчить про високий ступінь лінійної асоціації їх молекул за рахунок утворення між ними водневих зв’язків.
Серед амідів монокарбонових кислот найважливіше значення мають формамід і М,М-диметилформамід.
Формамід Н——рідина з Ткип = 195 °С. Його добу-ХЙН2
вають взаємодією аміаку з метиловим ефіром мурашиної кислоти, який, у свою чергу, синтезують з метанолу і оксиду вуглецю (II): н3с—он-рсо НзС-ОІ,іа_> н—с<^° н—с^° -р н3с—он.
хосн3 хмна
Формамід використовують як розчинник, а також для добування синильної кислоти, яка легко утворюється при нагріванні цього аміду:
н—с<^° ^5-» нсм + н2о. хмн2
/°
N. ^-Димепшлформамід (ДМФА) Н—С<р —рідина
ХЙ(СН3)2
з Ткип = 152 °С. Застосовується як прекрасний біполярний апро-тонний розчинник у лабораторній практиці, а також у виробництві синтетичних волокон. ДМФА в промисловості добувають взаємодією метилформіату з диметиламіном:
,0
Н-С^ + НМ (СН3)2 -> Н-С< + н3с-он.
хосн3 ХМ(СН3)а
207
Аміди вугільної кислоти. Вугільна кислота двохосновна, тому утворює неповний амід (карбамінову кислоту) і повний амід (сечовину):
НО—С—ОН; II О
Вугільна кислота
Н2І\Т—С—ОН;
II о
Карбамінова кислота
Н2И—с—мн2. II о
Сечовина
Карбамінова кислота у вільному стані не виділена, оскільки вона не стійка і розкладається наСО2 і аміак. Стійкими є складні ефіри цієї кислоти —уретани В.—О—С—МН2. Окремі представ-
II
О
ники уретанів використовуються в медицині як снотворні і болезаспокійливі засоби.
Сечовина—тверда кристалічна речовина, добре розчинна у воді. Вона є кінцевим продуктом азотистого обміну в організмі людини та ссавців і виділяється з організму з сечею.
Синтетично сечовина була добута в 1828 р. Ф. Велером ізомеризацією ціанату амонію при його нагріванні (див. Вступ). У промисловості сечовину добувають за реакцією Базарова — нагріванням оксиду вуглецю (IV) з аміаком під тиском 2 • ІО’ГІа (200 атм):
900 °С
СО2 4- 2МН3 —> Н2М—С—ЬІН2.
Як амід, сечовина гідролізується при нагріванні в кислому або лужному середовищі:
Н2И—С—І\'Н, 4- НОН -> 2МН, -4- СО„. А А І а * а
о
Сечовина, як і аміди Інших кислот, виявляє склабкі основні властивості, оскільки в її молекулі відбувається спряження вільних р-електронів атомів азоту з я-електронами карбонільної групи:
Н2М-С-МН, . пл о/
Тому розчини сечовини мають нейтральну реакцію. З тієї ж причини сечовина утворює солі тільки з сильними кислотами. Так, при взаємодії з азотною кислотою вона утворює сіль з 1 моль цієї кислоти:
Н2Г4—С—НН24-Н8Ю, Н2М—С—МНа • НМО3.
Азотиста кислота розкладає сечовину з виділенням азоту:
НаМ—С—МНа 4- 2НО8ІО н2 + 2Н2О + НО—С—ОН СО2 + Н2О.
208
Аналогічне розкладання сечовини відбувається і при дії на неї гіпоброміту натрію (реакція Бородіна):
НаМ—С—І\ІНа + ЗК'аОВг СОа + 2НаО + 3№Вг + 2ІУа.
О
За допомогою цієї реакції, вимірявши об’єм азоту, що утворився, можна визначити вміст сечовини, наприклад, у сечі.
Сечовина має широке застосування в народному господарстві. Вона є вихідною речовиною при добуванні карбамідних смол, ціанатів калію і натрію, гідразину та ряду інших речовин. Серед похідних сечовини є високоактивні гербіциди. Сечовину використовують також як висококонцептроване добриво, яке легко засвоюється. Добавка сечовини до корму жуйних тварин замінює в їх кормових раціонах 25—ЗО % білків.
Сечовина є вихідною речовиною для виробництва ряду фармацевтичних препаратів, таких як веронал, люмінал, бромурал тощо.
Нагрівання сечовини при температурі 150—170 °С приводить до утворення речовини, яку називають біуретом".
н2к-с-!^Н9-+ н~|-мн-с-ічн, — н,іч-с-мн-с-ічн2 + хн3.
п '___2—-1 н 2 її її
о о о о
Біурет
Біурет — це кристалічна речовина (Тпл — 192,5—193 °С), слабко розчинна у холодній воді, але добре розчинна в лугах. Розчин біурету в лугах розчиняє Сп(ОН)2 з утворенням комплексної сполуки червоно-фіолетового кольору (біуретова реакція). Біуре-това реакція є характерною на наявність пептидного (—ИН—С—)
II О зв’язку в молекулах органічних речовин.
Розділ 20. ЕЛЕМЕНТООРГАНІЧНІ СПОЛУКИ
Елементоорганічними називають такі органічні сполуки, в молекулах яких крім елементів-органогенів, тобто крім вуглецю, водню, кисню, азоту, сірки і галогенів, містяться інші елементи періодичної системи Д. І. Менделєєва, атоми яких безпосередньо сполучені з атомом вуглецю. Цю область органічної хімії, яка знаходиться на межі між органічною і неорганічною хімією, О. М. Не-смеянов запропонував називати елементоорганічною хімією.
Елементоорганічні сполуки класифікують переважно за природою елемента. У зв’язку з цим розрізняють натрій-, магній-, алюміній-, кремній-, фосфорорганічні та інші сполуки.
Назва елементоорганічних сполук складається з назви елемента і назви сполученого з ним вуглеводневого радикала, наприклад: метилнатрій Н3С—№, триетилалюміній (С2Н5)3А1, феніллітій С6Н6Ьі, тетраметилсилан (Н3СЦ8І і т. д.
909
Металоорганічні сполуки. В цих сполуках атом металу безпосередньо сполучений з атомом вуглецю. Оскільки вуглець більш електронегативний, ніж метали, то електронна густина в молекулах металоорганічних сполук завжди зміщена до атома вуглецю. З металами І і II групи періодичної системи елементів, які мають порівняно низьку електронегативність, зв’язок С—Ме високополяр-ний і має значну частку іонності. Наприклад, у молекулі метилнат-
— )-
рію зв’язок між атомами вуглецю і натрію іонний: Н3С№.
Органічні сполуки металів І групи.З цих сполук найчастіше добувають літій-, натрій- і калійорганічні сполуки .
Загальним методом добування літій-, натрій- і калійалканів є взаємодія галогеналканів з відповідними металами:
/?— Х + 2Л/е -> Я— Ме-\-МеХ, де X—СІ, Вг, І; Ме— бі, К, №.
При цьому одночасно поряд з металоорганічною речовиною утворюється насичений вуглеводень — продукт реакції Вюрца (див. розд. 7), кількість якого тим більша, чим активніший лужний метал.
Літійалкіли і літійарили добувають (з високим виходом) взаємодією відповідних галогеповуглеводнів з металічним літієм у відсутності вологи в середовищі сухого ефіру, тетрагідрофурану або насиченого вуглеводню і в атмосфері інертного газу або азоту. Наприклад:
С4Н9С1 + 2Ьі —-сан' С4Н9—Ьі + ЬІС1;
С,.Н6Вг + 2Ьі Еф|р- ^~60°с_> С(]Н_Ьі + ЬіВг Бромбензол Фепіллітій
Фенілнатрій можна добути нагріванням бензолу з патрі калканами, наприклад з амілнатрієм:
С6НР + С6Н„-№ -> С„Н8№ + С5Н12.
Калійорганічні сполуки зручно добувати із ртутьорганічних сполук, наприклад:
(С2Нв)2Нб + 2К -> 2С2Н6К + НЄ.
Діетилртуть Етил калій
Реакційна здатність органічних сполук лужних металів зростає в ряду
Р— Еі < Я—№ < Р—К.
Натрій- і калійорганічні сполуки більш реакційноздатні, ніж літійорганічні. Проте оскільки зв’язки С—К’а, С—К мають більш іонний характер, ніж зв’язок С—Ьі, то натрій- і калійорганічні сполуки не розчиняються в органічних розчинниках, що обмежує їх застосування. Літійорганічні сполуки, зв’язок у молекулах яких має більш ковалентний характер, розчиняються в органічних розчинниках і тому найчастіше використовуються в органічному синтезі, зокрема, замість магнійорганічних сполук.
210
Літій-, натрій- і калійорганічні сполуки надзвичайно реакційно-здатні. Вони розкладаються під дією води, спиртів, кислот. При цьому атом металу в їх молекулах заміщується на водень:
С4Н9—М? + НОН С4Н10 + Ме— ОН.
Органічні сполуки металів І групи легко окислюються, часто з самозагорянням на повітрі, активно реагують з альдегідами, кетонами, оксидом вуглецю (IV) та іншими речовинами.
Органічні сполуки елементів II групи. З органічних сполук елементів II групи найкраще вивчені і мають найважливіше значення органічні сполуки магнію, цинку і ртуті.
'Магнійорганічні сполуки. Магнійорганічні сполуки є двох типів — повні /?2М§ і змішані /?—М§ — X, де /? — алкільний або арильний радикал, X — галоген. Повні магнійорганічні сполуки добувають взаємодією магнію з ртутьорганічними сполуками:
-* /?2М§+н8-
/
Змішані магнійорганічні сполуки добувають переважно взаємодією алкіл- або арилгалогенідів з магнієм (порошкоподібним або у вигляді стружки) в середовищі безводного (абсолютного) діетилового ефіру:
Н3С-І + М8 Н8С-Мб-І;
Етил ма гні й йодид
С6Н6Вг + М8 С6НВ—М§—Вг.
Бромбензол Фенілмагнійбромїд
Змішані магнійорганічні сполуки вперше добув у 1901 р
В. Гріньяр, вивчив їх і впровадив у практику органічного синтезу. Тому ефірні розчини /?М§Х називають реактивами Гріньяра.
Магнійорганічні сполуки мають високополярний, а тому високо-с- 6+
реакційноздатний зв’язок С—М§. Вони легко вступають у різноманітні реакції, які називають реакціями Гріньяра.
Особливо важливими є реакції приєднання магнійорганічних сполук до подвійного зв’язку карбонільної групи альдегідів, кетонів, складних ефірів карбонових.кислот, а також до оксиду вуглецю (IV) (див. розділи 17, 18).
Органічні сполуки елементів III групи. У металоорганічних сполуках III групи періодичної системи, будову яких можна виразити загальною формулою Г3Ме, атом металу має тільки три валентних електрони і незаповнену валентну орбі-таль. Тому такі металоорганічні сполуки є сильними акцепторами електронів (кислотами Льюїса).
Найкраще вивчені і найбільше практичне значення мають органічні сполуки бору і алюмінію.
Алюмінійорганічні сполуки. Органічні сполуки алюмінію стали доступними і набули промислового значення тільки в 60-х роках XX ст., коли К. Ціглер (1956 р.) відкрив реакцію прямого синтезу алюмінійтриалкілів з алкенів, водню і тонко подрібненого алюмі
211
нію. Так, триетилалюміній утворюється за такою сумарною реакцією:
2А1 + ЗН2 + 6Н2С=СН2 (-^-Н"),АІ-’ 1І5-°С’ 5’05-10а Па-> 2 (С2НБ)3А1.
Реакція ініціюється алюмінійорганічними сполуками.
Алюмінійтриалкіли можуть приєднуватися до алкенів і тому каталізують їх полімеризацію. Комплекс триетилалюмінію і чотири-хлористого титану (С2Н5)3А1 • ТіС14 (каталізатор Ціглера — Натта) каталізує полімеризацію алкенів і приводить до утворення полімерів з стереорегулярною структурою (див. розд. 14).
Органічні сполуки елементів IV групи. Свинецьорганічні сполуки. З цих сполук найбільшого значення набув тетраетилсвинець (скорочена назва ТЕС). У промислових масштабах ТЕС добувають взаємодією хлористого етилу з розплавленим сплавом свинцю і натрію:
4С2НБС1 + 4РЬ№ (СгНБ)4РЬ + 4МаС1 + ЗРЬ.
У 1920р. американські інженериТ. Бойд і Т. Мідглей помітили антидетонаційні властивості ТЕС. З тих пір він у великих кількостях використовується як антидетонатор, який підвищує октанове число моторних палив. В умовах згоряння палива в моторі ТЕС розкладається з утворенням вільних радикалів, які сповільнюють детонацію:
(С2НБ)4РЬ 4С2Нб + РЬ.
ТЕС та інші свинецьорганічні сполуки надзвичайно о т р у йн і . Допустима концентрація ТЕС у повітрі становить 3 • 10_® мг • л-1. Автомобільний парк світу щорічно викидає в атмосферу близько 250 кілотонн свинцю і його сполук і таким чином забруднює навколишнє середовище. Але поки що ТЕСє найефективнішим антидетонатором. У наш час інтенсивно ведуться пошуки неотруйних антидетонаторів, які б замінили ТЕС. Встановлено, що антидетонаційні властивості виявляють циклопентадієнільні похідні карбонілів марганцю, молібдену, такі як С5Н5Мп(СО)5, С5Н;,Мо(СО)3, метил-треш-бутиловий ефір Н3С—О—С(СН3)3. Ці сполуки, на відміну від ТЕС, не мають токсичної дії.
Розділ 21. СПОЛУКИ З ДВОМА АБО КІЛЬКОМА ФУНКЦІЯМИ
Галогеналкени (ненасичені алкілгалогеніди). Органічні речовини, в молекулах яких одночасно містяться подвійний зв’язок і атом галогену, називають галогеналкенами, або ненасиченими ал-кілгалогенідами. Галогеналкени, як і інші класи органічних речовин, утворюють гомологічний ряд, який можна легко записати виходячи з гомологічного ряду алкенів, заміщуючи в молекулі відповідного алкену один атом водню на галоген.
212
Галогеналкени називають так, як етиленові вуглеводні, а саме: спочатку називають галоген, вказуючи його місцеположення в молекулі, а потім називають відповідний алкен. Наприклад: Н3С—СН =СНС1 — 1-хлорпропен-1, С1СН2—СН=СН2 —3-хлор-пропен-1. Якщо в ненасиченій галогенопохідній містяться також вуглеводневі радикали, то назва галогену наводиться в алфавітному порядку разом з назвами вуглеводневих радикалів: Н2С=С—СН—СН2—СІ —2,3-диметил-4-хлорбутен-1. Галогенал-
СН3 СН3
кеші Н2С=СН—СІ і Н2С=СН—СН2—СІ часто називають за назвою радикала—хлористий вініл і хлористий аліл відповідно.
Залежно від взаємного розміщення атома галогену і подвійного зв’язку ненасичені галогенопохідні поділяють на три групи:
1) галогеналкени з атомом галогену біля атома вуглецю з подвійним зв’язком (Н2С=СН—СІ, Н2С =СС1—СН3);
2) галогеналкени з атомом галогену в а-положенні відносно подвійного зв’язку (Н2С=СН—СН2—СІ);
3) галогеналкени з атомом галогену ..віддаленим від подвійного зв’язку (Н2С=СН—СН2-СН2-С1).
Галогеналкени з атомом галогену біля атома вуглецю з подвійним зв’язком внаслідок взаємного впливу атома галогену і подвійного зв’язку характеризуються порівняно низькою реакційною здатністю як атома галогену, так і подвійного зв’язку .Галогеналкени з атомом галогену в а-положенні відносно подвійного зв’язку мають значно вищу реакційну здатність галогену, ніж галогенопохідні насичених вуглеводнів. Пояснюється це тим, що в процесі реакції з таких галогеналкенів утворюється алільний карбонієвий катіон, який за рахунок спряження має підвищену порівняно з алкільним катіоном стійкість:
~..І
н,с= сн—сн,—сі н.,сХсн-сн, + СГ.
Галогенопохідні третьої групи істотно не відрізняються за властивостями від галогеналканів і етиленових вуглеводнів.
Серед ненасичених галогенопохідних найбільше значення мають хлористий вініл і хлористий аліл.
Хлористий вініл (хлоретен) —безбарвний газ (7’кип = —13,8°С), виявляє наркотичну, нейротоксичну та канцерогенну дію, викликає так звану «вінілхлоридну хворобу». Добувають його каталітичним приєднанням хлороводню до ацетилену (див. розд. 15) або де-гідрохлоруванням 1,2-дихлоретану.
Хімічні властивості хлористого вінілу зумовлені наявністю в його молекулі подвійного зв’язку і атома галогену. В молекулі хлористого вінілу відбувається два види взаємного впливу атомів: індукційний вплив (/-ефект), який передається через о-зв’язки, і мезомерний ефект (А4-ефект), який передається черезр-електрони. Атом хлору, як більш електронегативний, ніж хр2-атом вуглецю, виявляє сильний —/-ефект і викликає зміщення електронів сусід
213
ніх зв’язків на себе: Н2С=СН —>С1. Разомз тим завдяки наявності на атомі хлору вільної пари р-електронів і близького розміщення атома хлору до подвійного зв’язку ця пара р-електронів вступає в спряження з л-електронами подвійного зв’язку (р,л-спряження), внаслідок якого електронна густина подвійного С=С-зв’язку і густина атома хлору в молекулі хлористого вінілу зливаються. Це спряження напрямлене від атома галогену до подвійного зв’язку і атом хлору виявляє при цьому Ч-М-ефект:
н2с=сн^й: .
Отже, атом хлору в молекулі хлористого вінілу виявляє двоїсту природу: сильний —/ -ефект і слабкий -|-Л4-ефект:
О
Н2С = СН * С): .
Результатом такого спряження є деяке зменшення заряду на атомі хлору, зменшення довжини зв’язку С—СІ (/) і дипольного моменту молекули (р) порівняно, наприклад, з хлористим етилом, в молекулі якого виявляється тільки —/-ефект атома хлору:
Сполука Н2С=СН—СІ Н3С—СНа—СІ
/С_С|, нм 0,169 0,177
ц, О 1,44 1,89
—/-Ефект атома хлору сильно зменшує електронну густину подвійного зв’язку. Внаслідок цього значно утруднюється електрофіль-не приєднання до хлористого вінілу. Проте все-таки за певних умов хлористий вініл приєднує галогеноводні, галогени та інші реагенти. Причому полярні реагенти (галогеноводні, вода тощо) приєднуються згідно з правилом Марковникова. Наприклад, приєднання НС1 приводить до утворення 1,1-дихлоретану:
Н2С=СНС1 + НС1 -* Н3С—СНС12.
Порядок такого приєднання галогеноводню пояснюється так. У молекулі хлористого вінілу в статичному стані, тобто до початку реакції, розподіл електронної густини такий, що заряд 6+ зосеред-
6+ о-
жений на р-вуглецевому атомі: Н2С=СН->СІ. Відповідно до такого розподілу електронної густини приєднання галогеноводню повинно відбуватися проти правила Марковникова. Однак у процесі реакції, коли до хлористого вінілу приєднується електрофільна частинка Н+ (стадія утворення л-комплексу), виявляється динамічний -ЦИ-ефект атома хлору, в результаті якого електронна густина від атома хлору зміщується до |3-атома вуглецю, на якому, таким чином, електронна густина збільшується:
Тому протон приєднується до р-атома вуглецю. При цьому виникає карбкатіон А з позитивним зарядом на а-атомі вуглецю. Карбкатіон А стабілізується вільними р-електронами сусіднього атома хлору, які зміщуються до атома вуглецю з позитивним зарядом і якоюсь мірою зменшують (компенсують) його позитивний заряд:
+ ГЛ Г7. , „ +СГ
Н,С-СН,-СІ—*-Н2С = СН-сі: + Н+СІ —НзС-СН-Сі:—-н3с—снсі2.
Б А
Якщо б протон приєднався до а-вуглецевого атома хлористого вінілу згідно з статичним розподілом електронної густини в молекулі, то виник би карбкатіон Б з позитивним зарядом на р-вугле-цевому атомі, який ізольований групою —СН2—від атома хлору, і р-електрони атома хлору в цьому випадку не мали б можливості зміщуватися і компенсувати позитивний заряд. Тому карбкатіон Б порівняно з катіоном А значно менш стійкий і відповідно менш енергетично вигідний. Внаслідок цього реакція відбувається у напрямку утворення енергетично стабільнішого карбкатіона А і закінчується швидкою стадією приєднання до нього аніона СІ-, тобто утворенням 1,1-дихлоретану.
Однією з найважливіших реакцій хлористого вінілу є його полімеризація, в результаті якої утворюється поліхлорвініл:
п СН2=СНСІ -> (—СН2—СНС1—)„.
Особливістю хлористого вінілу є надзвичайно мала хімічна активність атома хлору в його молекулі. Пов’язано це з тим, що внаслідок р,л-спряження атом хлору в молекулі хлористого вінілу значно міцніше, ніж у молекулі хлористого етилу, сполучений з атомом вуглецю і тому стає малореакційноздатним. Пасивність атома хлору хлористого вінілу в хімічних реакціях пояснюється також резонансною стабільністю молекули і частковою двоєз’вяза-ністю атома хлору (структура II), яка збільшує міцність зв’язку вуглець — хлор:
н2с = сн й: -----► н,с - сн = сі
і н
Тому атом хлору в молекулі хлористого вінілу, на відміну від хлору в хлористому етилі, майже не вступає в реакції нуклеофіль-ного заміщення. Так, хлористий вініл не реагує з ацетатом срібла, нітратом срібла, ціанідом натрію, аміаком. Хлор у молекулі хлористого вінілу не заміщується також на гідроксил.
Хлористий аліл (З-хлорпропен-1, Тккп ==44,6 °С) добувають високотемпературним хлоруванням пропілену в газовій фазі (див. розд. 14).
Хлористий аліл та інші галогеналіли, на відміну від хлористого вінілу, є високореакційноздатнимп сполуками. Галоген в їх моле
215
кулах має високу активність І легко вступає в різні реакції нук-леофільного заміщення:
н,с-с^
Н2С=СН—СН2—МО2 1чо-г- Н2С=СН—СН2—СІ ----
Нітроаліл |
| МаСИ
н2с=сн—сн2—см
Нітрил вінілоцтової кислоти
-> Н2С=СН—сн2—о—с—сн3.
II о Аліловий ефір оцтової кислоти
Реакції заміщення галогену в галогеналілах відбуваються із значно більшими швидкостями, ніж у випадку галогеналканів. Це пояснюється тим, що в процесі таких реакцій хлористий аліл, наприклад, порівняно легко утворює іонну пару — алільний ка-тіон-аніон хлору:
Н2С=СН—СН2—СІ Н2С=СН—СН8//С1“.
Енергія утворення алільного катіона за рахунок спряження р-електронів подвійного зв’язку з вакантною орбіталлю (тобто з позитивним зарядом) кінцевого атома вуглецю зменшується і одночасно значно підвищується його стабільність порівняно з первин-г
ним пропільним катіоном: алільний катіон — Н2С=СН—СН2, +
первинний пропільний катіон — Н3С—СН2—СН2. В результаті спряження в алільному катіоні виникають граничні (крайні) структури карбкатіона, в яких позитивний заряд розподілений фактично між усіма трьома вуглецевими атомами:
і + +1 : * л
Н2С=СНІСН2 Н.С-іСН=СН2, або Н..С—СН—СН„.
Тому в 5д4-реакціях, наприклад з ціанідом натрію, нуклеофіль-на групаСМ" може утворювати зв’язок як з першим, так і зтретім атомами вуглецю алільного катіона, що і доведено дослідом з використанням міченого атома 14С в хлористому алілі:
НаС=СН—14СН,—СІ Повільно Н2С=СН—«СЩ 4-Сі’}
2 3 2 І 2 3 *--- .* 2 з 2 1 2
Н2с=сн-«сн+ Н2С-СН=»СН2
-> Н2С=СН—14СН2—СМ + МС—СНа—СН=”СН2.
3 2 1 3 2 1
В результаті реакції утворюється суміш майже рівних кількостей ціанистого алілу з групою СМ як біля міченого атома вуглецю (14С), так і біля атома вуглецю 12С.
Ненасичені монокарбонові кислоти. Ненасичені кислоти містять у своєму складі подвійний або потрійний зв’язок, яких у молекулі може бути один або кілька. Ненасичені одноосновні кислоти ети-
216
левового ряду мають загальну формулу СпН2п_іСООН, а ацетиленового ряду-С„Н2„_3СОЗН. У табл. 19 наведено формули, назви та деякі константи найважливіших ненасичених монокарбонових кислот.
За систематичною номенклатурою назву ненасичених кислот утворюють з назви відповідного алкену (алкіну, дієну), додаючи до неї закінчення -ова, і цифрою вказують положення подвійного зв’язку в молекулі. Нумерацію вуглецевого ланцюга починають від атома вуглецю карбоксилу. Для ненасичених кислот з невеликою кількістю вуглецевих атомів використовують раціональну номенклатуру, беручи при цьому за основу акрилову або оцтову кислоту, наприклад: метакрилова, вінілоцтова кислота і т. д. Проте найчастіше користуються історичними назвами цих кислот (табл. 19).
Таблиця 19. Ненасичені монокарбонові кислоти
Формула Назва гпл. °С Т * кип» °С К^.10-6
н2с=сн—соон Акрилова, пропеиова 12,3 142 5,6
нас=с—соон с!н3 Метакрилова, 2-метил-2-пропенова 15 161 —
н3с—сн=сн—соон Кротонова, транс-2-бу-тонова 72 189 2,0
Ізокротонова, г{/іс-2-буте-нова 15,5 172 3,6
н2с=сн—сн2—соон Вінілоцтова, 3-бутенова —39 163 3,8
с17н33соон Олеїнова 14 286 —
с17н81соон Лінолева —5,2 — —
с17н28соон Ліноленова —11,3 — —
нс=с—соон Пропіолова, пропінова 9 144 8,6
Ізомерія ненасичених кислот етиленового ряду починається з кислоти з чотирма вуглецевими атомами в молекулі і залежить як від положення подвійного зв’язку, так і від структури вуглецевого ланцюга. Так, кротонова і вінілоцтова кислоти — ізомери з різним положенням подвійного зв’язку, а ізомерна цим кислотам метакрилова кислота відрізняється від них розгалуженням вуглецевого ланцюга (табл. 19). Деякі ненасичені кислоти можуть існувати також у вигляді цис- і тра«с-ізомерів:
Н3СЧ/Н
Нч хСНа с
н/хсоон
Кротонова кислота (тракс-І зо мер)
н/хсоон
Ізокротонова кислота (цис- ізомер)
217
Хімічні властивості ненасичених кислот зумовлені наявністю в їх молекулах карбоксильної групи і подвійного (потрійного) зв’язку. Ненасичені кислоти завдяки —/-ефекту етиленової (ацетиленової) групи атомів Н2С=СН ч- СООН та підвищенню стабільності аніона Н2С=СН—С=О за рахунок Л4-ефекту виявляють
По-
сильніші кислотні властивості і мають вищі константи дисоціації, ніж відповідні їм насичені кислоти. Вплив подвійного зв’язку на кислотні властивості ненасичених кислот тим сильніший, чим ближче цей зв’ язок розміщений до карбоксильної групи (табл. 19).
Ненасичені кислоти, подібно до насичених, утворюють похідні карбонових кислот: солі, галогенангідриди, ангідриди, складні ефіри, аміди, нітрили тощо. Разом з тим ці кислоти виявляють властивості ненасичених сполук і можуть приєднувати до подвійного зв’язку галогени, галогеноводні, воду та інші речовини. Ненасичені кислоти і їх похідні можуть також вступати в реакції полімеризації і співполімеризації.
Характерною особливістю акрилової, метакрилової, кротонової та інших кислот, у молекулах яких подвійний зв’язок знаходиться порядзСООН-групою(томуїх називають а, р-ненасиченими кислотами), е спряження л-електронів подвійного С=С-зв’язку з л-електронами подвійного С—О-зв’язку карбоксильної групи. Карбоксильна група виявляє при цьому досить сильний — М-ефект, внаслідок якого електронна густина подвійного С=С-зв’яз-ку в ненасичених кислотах зменшується і на Р-вуглецевому атомі виникає значний заряд б+:
н2с = сн -с
4 3 2 4
О-Н
Внаслідок такого —Лї-ефекту приєднання електрофільних реагентів до а, р-ненасичених кислот відбувається значно важче і повільніше, ніж, наприклад, до етилену, а приєднання полярних речовин, таких як галогеноводні, вода, аміак тощо, здійснюється до С=С-зв’язку проти правила Марковникова, відповідно до розподілу електронної густини в молекулах цих кислот. Вважають, що приєднання полярних реагентів до а, Р-ненасичених кислот відбувається в 1,4-положення спряженої, ристеми, як і у випадку спряжених дієнів (див. розд. 16). Так, НВг, взаємодіючи з акриловою кислотою, приєднує протон до карбонільного кисню, а аніон Вг- —до р-вуг-лецевого атома С=С-зв’язку. При цьому утворюється нестійкий проміжний ненасичений двохатомний спирт (енольпий діол),який відразу ж ізомеризується (правило Ельтекова) в стійку р-бромпро-піонову кислоту;
218
Енольнпй діод
За аналогічним механізмом акрилова кислота приєднує воду, аміак:
Н2С—СН2—СООН н2С=СН—соон —- н2с—сн2—соон.
ІЧН он
Р-Амїнопропіонова Р-Оксипропіонова кислота
кислота
Велике значення мають такі похідні акрилової кислоти, як її метиловий ефір (метилакрилат, 7,кип = 80 °С) і нітрил акрилової кислоти (акрилонітрил, Ткип =77°С), які є мономерами для промислового виробництва поліметилакри лату, поліакрилонітрилу та ряду співполімерів з іншими ненасиченими мономерами. Полімеризація метилакрилату і акрилонітрилу відбувається легко за такою схемою:
«Н2С==СН -> /—Н2С—СН— \ ;
СООСН3 \ СООСН3/„
пН2С=СН -» /—Н2С—СН—\ .
ск \ /„
Поліметилакрилат являє собою склоподібний полімер, з якого виготовляють органічне скло. Поліакрилонітрил використовують для виробництва синтетичного поліакрилового волокна, яке в СРСР має назву нітрон.
З похідних метакрилової кислоти велике значення має її метиловий ефір, який називають метилметакрилатом. Метилметакрилат легко полімеризується і утворює при цьому склоподібні полімери, які використовують для виробництва органічного скла:
п н2с=с—соосн3 сн3
(СН3 \
-Н2С-С— І.
СООСН3/ п
Пол і м етил мета кр и л ат (органічне скло, плексиглас)
Поліметилметакрилат у техніці має назву плексиглас.
Вищі ненасичені кислоти. До вищих ненасичених кислот належать олеїнова, лінолева і ліноленова кислоти, які містять відповідно один, два і три подвійних зв’язки. Молекули цих кислот мають такі загальні формули: олеїнова —С17Н33СООН, лінолева— СцНзіСООН, ліноленова —С17Н2УСООН.
Олеїнова (ццс-9-октадеценова) кислота
Нзс—(СН2)7—СН=СН-(СН2)7СООН досить поширена в природі. Ця кислота у вигляді гліцеридів (складних ефірів з гліцерином)
219
міститься майже в усіх природних жирах, особливо в рослинних оліях —соняшниковій, конопляній, льняній, оливковій,кукурудзяній та інших. З цих олій олеїнову кислоту можна добути гідролізом (омиленням).
Олеїнова кислота має г^цс-конфігурацію і при дії азотистої кислоти (точніше, оксидів азоту, які з неї утворюються) перетворюється на трпнс-ізомер —тверду елаїдинову кислоту (Тпл = 44 °С):
НХ/(СН2)7СН8 с
II
с н/\сн2)7соон
Олеїнова кислота
ньюв (КаК02+НС1)->
Н3С(СН2)7Ч ^Н
с
II с н/\сн2)7СООН. Елаїдинова кислота
До складу рослинних олій у вигляді гліцеридів входять також лінолева кислота, яка містить два ізольованих подвійних зв’язки
Н3С— (СН2)4СН=СН—СН2—СН=СН—(СН2)7СООН,
і ліноленова кислота, яка має три ізольованих подвійних зв’язки:
Н 3С—СН2—СН=СН—СН2—СН =СН—СН2—СН=СН—(СН2) 7СООН.
Жири. У природі дуже поширені складні ефіри гліцерину і вищих карбонових кислот, які називають жирами. Завдяки класичним роботам французького вченого М. Шевреля було встановлено (1811—1822 рр.), що при нагріванні жирів з водою утворюються гліцерин і різні карбонові кислоти. Пізніше, в 1854 р. М. Бертло безпосередньо взаємодією гліцерину з вищими карбоновими кислотами синтезував жири, а в 1859 р. А. Вюрц добув тристе-арин взаємодією 1,2,3-трибромпропану з срібною сіллю стеаринової кислоти. На основі цих досліджень було зроблено висновок, що жири —це повні складні ефіри трьохатомного спирту гліцерину і вищих насичених і ненасичених монокарбонових кислот:
Н2С—ОН
ні—ОН4-ЗС17Н35СООН-----
| 1 15 Зо — ЗН,О
Н2С—он
н2с—о—со—с17н38 ні-О-СО-Сі7Н36
Н2С—О—СО—С17Н35
^-ЗАеВг
ЗС17НзвСООА§ -}-
Н2С—Вг
НС—Вг.
Жири, у зв’язку з тим що до їх складу входить залишок гліцерину, називають ще гліцеридами.
220
Склад і будову жирів можна зобразити загальною формулою
НаС—О—С—Ц II ©
НС—О—С—Я'
Н2С—О—С—/?"
о
де 7? — радикали вищих насичених і ненасичених монокарбонових кислот. У природних жирах залишки кислот мають, як правило, нерозгалужений вуглецевий ланцюг і містять парну кількість вуглецевих атомів. Жири, що містять залишки однакових кислот (7? =/?' = /?"), називають простими. Якщо ж залишки /?, /?', Ц" різні, то такі тригліцериди називають змішаними.
За фізичними властивостями жири можуть бути твердими і рідкими. Тваринні жири (яловичий, баранячий, свинячий) —тверді і містять в основному залишки насичених кислот (пальмітинової і стеаринової). Рослинні жири —олії містять переважно залишки ненасичених кислот (олеїнової, лінолевої і ліноленової та деяких інших). Як приклад наведемо хімічний склад деяких жирів. Так, свинячий жир складається з тригліцеридів пальмітинової, стеаринової і олеїнової кислот, а коров’яче масло — з тригліцеридів пальмітинової, масляної і олеїнової кислот.
При довгому стоянні на повітрі жири частково окислюються киснем і гіркнуть.
Жири, які містять залишки ненасичених кислот, виявляють властивості ненасичених сполук: знебарвлюють бромну воду, розчин КМпО4> приєднують водень і т. д. Каталітичним гідруванням рідкі жири перетворюють на тверді:
НаС—О—СО—С17Н33 Н2С—О—СО—С17Н88
не—О—СО—С17Н33 не—о—со—с17нзв.
। X < ОО X • ЗО
н2с—о—со—с17н88 н2с—о—со—с17н88
Триолеїн Тристеарин
Вперше технічний метод гідрування жирів розробив у 1907— 1909 рр. російський хімік С. О. Фокін. Гідрування жирів широко використовується в харчовій промисловості і миловарінні.
Рідкі жири (олії) бувають висихаючими, напіввисихаючими і не-висихаючими. Висихаючі олії (лляна, бавовняна) мають здатність висихати, тобто ставати твердими і утворювати при цьому міцну прозору тонку плівку. Такі олії використовують для виготовлення оліфи, яку застосовують для приготування масляних фарб, клейонок, лінолеуму. Свіжа лляна або бавовняна олія висихає повільно.
Щоб прискорити цей процес, висихаючі олії варять, добавляючи до них каталізатори (сикативи). Як сикативи використовують сполуки свинцю (сурик, РЬ3О4), а в останні роки — деякі солі марганцю. Висихаюча олія, проварена при наявності сикативів, називається оліфою.
Найважливішою властивістю жирів є їх гідроліз, який відбувається при наявності води під впливом кислот, лугів або ферментів, наприклад ліпази. В результаті гідролізу жири розщеплюються на гліцерин і відповідні вищі карбонові кислоти:
Н2С—О—СО—/?
І н+
не—О—СО—+ ЗН2О--------»
Н2С—О—СО—
Н2С—ОН
-> не—ОН + /?—СООН + /?'—СООН + /?"—соон.
Н2С—он
Якщо гідроліз жирів проводять в лужному середовищі, то крім гліцерину утворюються солі вищих карбонових кислот — мило. Звідси і пішла назва реакцій взаємодії складних ефірів з водою — реакції омилення'.
Н2С—О—СО—7?
НС—О—СО—/?' + ЗНаОН ->
Н. С— О—СО— Р"
Н,С—ОН
І
НС—ОН + /?—СООїХа + /?'—СООК'а + /?"—СООКа.
І
н2с—он
Такий процес здійснюють у промисловості при виготовленні мила. Для цього жири нагрівають з надлишком розчину лугу .Натрієві солі вищих карбонових кислот (натрієве мило)—тверді речовини. Вони містять від 12 до 18 С-атомів. Калійні солі вищих карбонових кислот (калієве мило) —рідкі (зелене мило). Натрієве і калієве мила добре розчиняються у воді і утворюють у ній колоїдні розчини. Одночасно відбуваються і реакції гідролізу:
/?—СООі\а -р НОН /?СООН + МаОН.
Кальцієві і магнієві солі вищих карбонових кислот, на відміну від натрієвих і калієвих, у воді не розчинні. Тому в твердій воді, 'Ж>
яка містить багато іонів Са2+ і М§2+, мило втрачає миючу дію внаслідок утворення нерозчинних осадів магнієвих і кальцієвих солей:
27?—СОО№ + Мд8О4 -> (/?—СОО)2Мб| + №2804;
2/?—СОО№ + Са (НСО3)2 -> (/?—СОО)2Са| + 2КаНСО3;
Мило виготовляють також синтетичним методом із синтетичних вищих карбонових кислот, які, в свою чергу, добувають каталітичним окисленням рідких і твердих алканів нафти:
Н3С-(СН2)„-СН3 —- Н3С—(СН2)т—СООН + Н3С—(СН2)Р—СООН.
У процесі такого окислення утворюється суміш карбонових кислот, нейтралізацією якої добувають синтетичне мило.
У промисловості виготовляють також миючі засоби інших видів. Найпоширенішими є синтетичні миючі засоби—алкілсульфати, які належать до аніоноактивних поверхнево-активних речовин. Алкілсульфати — це солі кислих складних ефірів сірчаної кислоти і вищих спиртів Н3С—(СН2)„—О—8О3 На, які називають детергентами, наприклад:
я-С,1Н23СН2ОН м-С11Н23СН2-О-8О3Н
Додеканол-1 Лаурнлсульфат
(лауриловий спирт)
-> н-С11Н23СН2О8О3На.
Наїрієва сіль лаурилсульфату
Молекули детергентів містять від 12 до 18 С-атомів і мають неполярний вуглеводневий залишок, розчинний у жирах, і полярний залишок —О—8О3, розчинний у воді. Отже, молекули детергентів подібні за будовою до молекул мила.
Фосфатиди. Близькими за структурою до жирів є фосфатиди. Фосфатиди — це дигліцериди монокарбонових кислот, у яких третій гідроксил гліцерину етерифікований фосфорною кислотою, а остання своїм гідроксилом утворює складний ефір з аміноспиртом. Фосфатиди можна зобразити загальною формулою
Н2С—О—СО—/?'
НС—О—СО—7?"
І
Н2С—О—Р—о—сн2— сн2—н/?8.
Якщо 7? є Н (аміноспирт — етаноламін), то фосфатид називається кефаліном. Кефалін входить до складу клітин мозку. Якщо К~СН3 (аміноспирт—холін), то такий фосфатид називається лецитином. Лецитин міститься в яйцях, тканинах печінки, сої та в інших тваринних і рослинних об’єктах.
223
Розділ 22. ДИКАРБОНОВІ КИСЛОТИ
Дикарбоновими кислотами називають органічні сполуки, молекули яких містять дві карбоксильні групи. Розрізняють насичені і ненасичені дикарбонові кислоти. Склад насичених дикарбонових кислот можна зобразити загальною формулою С„Н2п(СООН)2. Найважливіші представники гомологічного ряду цих кислот з не-розгалуженою структурою, їх формули, назви і фізичні константи наведено в табл. 20.
Дикарбонові кислоти називають переважно за історичною номенклатурою, яка пов’язана,головним чином, з першим джерелом добування або виділення дикарбонової кислоти. Наприклад, щавлева кислота названа так тому, що вперше була виділена із щавлю. За систематичною номенклатурою пі кислоти називають так: до назви відповідного насиченого вуглеводню з такою самою кількістю вуглецевих атомів, як і в головному ланцюгу дикарбонової кислоти, додають закінчення -діова і слово кислота (табл. 20).
Всі дикарбонові кислоти, крім щавлевої і малонової, мають ізомери з розгалуженим вуглецевим ланцюгом. Наприклад, ізомером янтарної кислоти НООССН2—СН2СООН є метилмалонова кислота НООС-СН—СООН.
СН3
Методи добування дикарбонових кислот в принципі не відрізняються від методів добування однокарбонових. Однак при добуванні дикарбонових кислот як вихідні речовини застосовуються сполуки з двома функціональними групами, які можуть перетворюватися на карбоксильні. Так, дикарбонові кислоти можна добути окисленням двохатомних спиртів або діальдегідів, омиленням динітрилів тощо:
НО—СН2—СН2—ОН °Чс-С^0 ;
Етиленгліколь 8 НХ ХН НСИ ХОН
ГлІоксаль Щавлева кислота
СІ—СН2—СН2—СІ кс—сн2—сн2—сн
1, 2-Дихлоретан Динітрил
-> НООС—СН2—СН2—СООН.
Янтарна кислота
Дикарбонові кислоти можна також добути з малонового і ацетооцтового ефірів (див. розділи 22,24). Однак для добування окремих дикарбонових кислот застосовують спеціальні методи. Так, щавлеву кислоту в промисловості добувають швидким нагріванням форміату натрію. При цьому від нього відщеплюється водень і утворюється оксалат натрію, який під дією розбавленої сірчаної кислоти перетворюється на щавлеву кислоту:
1 Н-ІС00Ма СООИа ЄООН
І ; зео'с і +н2зо4 і
І {+ -Н,-* І —Ка88О4"* І
ІН—ІСООИа СООНа СООН
І—__!
224
1-120
Таблиця 20. Насичені дикарбонові кислоти
Формула Назва за номенклатурою тпл, °С Розчинність при 20 °С, г/ 100 г води Константа іонізації
систематичною історичною К,-10-4
ноос—соон Етандіова Щавлева 189* 9 5900 6,4
ноос—сн2—соон Пропандіова Малонова 136 74 177 0,47
НООС—(СН2)2СООН Бутандіова Янтарна 185 6,8 6,89 0,247
НООС—(СН2)3СООН Пентандіова Глутарова 98 64 4,6 0,534
НООС—(СН2)4СООН Гександіова Адипінова 153 2 3,7 0,39
НООС—(СН2)5СООН Г ептандіова Пімеліиова 103 5 3,3 0,487
НООС—(СН,)вСООН Октандіова Пробкова 140 0,2 3,07 0,47
НООС—(СН2),соон Нонандіова Азелаїнова 106,5 0,3 2,88 0,28
НООС-(СН2)8ЄООН Декандіова Себацинова 133 0,1 2,6 0,26
* Для безводної кислоти.
Малонову кислоту добувають в основному з натрієвої солі хлороцтової кислоти, взаємодією якої з НаСИ спочатку одержують натрієву сіль ціаноцтової кислоти, а потім нітрильну групу останньої гідролізують у карбоксильну:
СІ—СН2—СОО№ М=С-СН2-СОО№ >
-> НООС—СН2—СООН.
Янтарну кислоту добувають гідруванням малеїнової кислоти і на основі малопового ефіру (див. розд. 22).
Адипінову кислоту в промисловості добувають у великих кількостях окисленням циклогексанолу азотною кислотою (див. розд. 27).
Усі насичені дикарбонові кислоти —білі кристалічні речовини. Нижчі члени гомологічного ряду цих кислот добре розчинні у воді (табл. 20), але важко розчинні в неполярних розчинниках. Із збільшенням молекулярної маси дикарбонових кислот розчинність їх у воді зменшується. Дикарбонові кислоти мають порівняно високі температури плавлення, що зумовлено сильною асоціацією їх молекул за рахунок водневих зв’язків.
Хімічні властивості дикарбонових кислот визначаються перш за все наявністю в їх молекулах двох карбоксильних груп. Так, ці кислоти у водному середовищі ступінчасто дисоціюють і є сильнішими кислотами, ніж монокарбонові кислоти з такою самою кількістю вуглецевих атомів у молекулі:
НООС—СООН + Н2О НООС—СОО~ + Н3О+;
НООС—СОО - + Н2О -ООС—СОО- 4- Н3О+.
Посилення кислотних властивостей дикарбонових кислот пояснюється взаємним впливом карбоксилів, а саме сильною електро-ноакцепторною дією однієї карбоксильної групи на другу, яка полегшує дисоціацію:
го о
<11 II _ +
Н-О-С— С—О— НУ-ТГН-О-С-С-О +н (НІ II
1-0 о
Тому щавлева кислота (Дд НООС—СООН =5900 • 10~в) значно сильніша за оцтову (Л'д Н3С—СООН = 1,76 • 10-8), причому перша константа її дисоціації набагато більша, ніж друга Д2 (табл. 20) , оскільки карбоксилатний аніон —С—О- має нега-
II
О
тивний заряд і тому не виявляє електроноакцепторної дії. Із збільшенням кількості вуглецевих атомів у молекулі дикарбонової кислоти і в міру віддалення однієї карбоксильної групи від другої взаємний вплив їх послаблюється і сила дикарбонових кислот відповідно зменшується (табл. 20).
226
Дикарбонові кислоти виявляють такі ж властивості, як і моно-карбонові. Тільки завдяки наявності двох карбоксильних груп у молекулі вони можуть утворювати два ряди похідних: кислі і середні солі, повні і неповні похідні (складні ефіри, галогенангідриди, аміди тощо):
СООД СООН СООН соок соок
І я-он І я-он І КОН і кон І
| —що І *" —що —н2о~” | —що-*
СОО/? СООН СООН СООН СООК
Повний складний Неповний Кисла сіль Середня сіль
ефір складний ефір
Разом з тим наявність двох карбоксильних груп, їх взаємне положення в молекулі надає дикарбоновим кислотам особливих властивостей, які виявляються, наприклад, при нагріванні. Так, щавлева і малонова кислоти, а також одно- і двозаміщені мало нові кислоти при нагріванні легко декарбоксилуються, тобто розкладаються з відщепленням СО2 за рахунок однієї з карбоксильних груп. При цьому з щавлевої кислоти утворюється мурашина, а з мало-нової — оцтова кислота:
НООС—
НООС—СН2—
140—180сС
-Н
140—*160°С
—Н
Н—СООН + СО2;
Н8С—СООН + СО2.
Янтарна і глутарова кислоти, на відміну від попередніх, при нагріванні відщеплюють воду за рахунок обох карбоксильних груп і перетворюються на циклічні ангідриди. Особливо легко таке відщеплення води відбувається при наявності водовідбирних речовин, наприклад оцтового ангідриду:
Н2СХ хо -{ні
Янтарна кислота Янтарний ангідрид
Глутарова кислота
Глутаровий ангідрид
227
Адипінова і пімелінова кислоти при нагріванні декарбоксилу-ються і дегідратуються одночасно, перетворюючись при цьому на циклічні кетони:
СН2Го1
сн
^<4 ^сг сн2
Адипінова кислота
-о-ні о—
с=о + со2 + Н2О.
Н2С^ / сн2
Циклопентаном
Щавлева (оксалатна) кислота — біла кристалічна речовина, добре розчиняється у воді і кристалізується з неї у вигляді дигід-рату (СООН)2 • 2Н2О(Т„л = 101,5°С). У вільному стані, а також у вигляді солей щавлева кислота досить поширена в природі. Міститься в грибах, лишайниках, водоростях, папороті та багатьох інших рослинах. Вона входить до складу клітинного соку рослин. У значних кількостях щавлева кислота міститься в ревені, щавлі, кислиці (Охаїіз асеіозеїіа). Від назв останніх двох рослин ця кислота одержала свою історичну назву. У ссавців щавлева кислота виділяється з сечею у вигляді важко розчинної у воді кальцієвої солі —оксалату кальцію, який в паталогічних випадках утворює оксалатні камені в нирках або сечовому міхурі.
Щавлева кислота утворює середні і кислі солі, які називають оксалатами. Оксалат кальцію важко розчинний уводі. Не розчиняється він і в оцтовій кислоті. Цю його властивість використовують в аналітичній хімії.
Щавлева кислота при нагріванні з концентрованою Н2ЗО4 легко розкладається на оксиди вуглецю і воду:
НООС—СООН -90О-с-’-Нг—*-> со2 + СО + Н2О.
Різноманітні окислювачі, наприклад КМпО4, легко і кількісно окислюють щавлеву кислоту. У зв’язку з цим її використовують для встановлення титру перманганату калію в методі перманга-натометрії:
5НООС—СООН 4- 2КМпО4 + ЗНа8О4 = К28О4 + 2Мп8О4 + 10СО2 + 8Н2О.
Зовнішньою ознакою цієї реакції є знебарвлення розчину перманганату.
Щавлеву кислоту та и солі використовують у промисловості при дубленні шкіри, при фарбуванні тканин, для очистки урану, як каталізатор реакцій полімеризації і конденсації, для відбілювання соломи, при виготовленні чорнила, для обробки горіхового дерева тощо.
Малонова кислота. Свою назву ця кислота одержала від латинського слова шаіит — яблуко, оскільки була вперше добута окисленням яблучної кислоти (див. розд. 23). Малонова кислота міститься в соках цукрового буряка і ріпи. В промисловості мало-нову кислоту добувають переважно з натрієвої солі хлороцтової кислоти.
228
Для органічного синтезу особливо велике значення має діетиловий ефір малонової кислоти Н2С(СООС2Н5)2 (Ткнп = 199 °С), який називають просто малоновим ефіром. Характерною особливістю малонового ефіру є наявність в його молекулі метиленової групи з високореакційноздатними (рухливими)атомами водню. Атоми водню в групі —СН2— під впливом сусідніх етоксикарбонільних груп (—/-ефект і о,л-спряження) активуються настільки,що малоновий ефір являє собою СН-кислоту (Кл « 10“1Б), сила якої на три порядки вища, ніж кислотність етанолу (Л'Д С2Н,,ОН = 1018). У молекулі малонового ефіру спостерігається також о,л-спряження зв’язків С—Н і л-електронів двох карбонільних груп:
н Чос2н5
Внаслідок такого спряження порівняно легко виникають протон і резонансний енергетично стабільний карбаніон А, у якому негативний заряд метиленового атома вуглецю роззосереджений між двома карбонільними атомами кисню (В).
Активність атомів водню СН,-групи в малойовому ефірі настільки висока, що вони при дії натрію або етилату натрію легко заміщуються на натрій. В результаті утворюється моно- або динатрій-малоновий ефір. Ці натрійпохідні малонового ефіру являють собою іонно побудовані сполуки. Карбаніон у цих сполуках стабілізований спряженням негативного заряду з карбоксильними групами:
С2Н5о о
V- сн2—с' 2іа-аСо .
о"' чос2н5 сгн5он
д
С2Н5О о2
----------- с—сн—с"' 20- ос2н5
С2Н5°Ч _ />
с—сн—сх -—-° ОС2Н5
Малоновий ефір і його натрійзаміщені широко використовуються в органічному синтезі. Класичні дослідження в цій області провів М. Конрад (1881 р.).
Натріймалоновий ефір легко реагує з алкілгалогенідами і утворює при цьому алкілзаміщені малонового ефіру, які після гідролізу і декарбоксилування перетворюються на монокарбонові кис
229
лоти, наприклад:
Н3С— СН2—Вг + ЙаСН (СООС2Н6)2 _№в > Н3С-СН2-СН (СООС2Н5)2 ->
,,н п н+ /І СОО І Н
ЙйгаГ н,с-сн,-сн/Ь—1 н,с-сн,-сн,-соон.
ЧЛЛЛ"1 2 Масляна кислота
+2Н„О, Н+ Н3С\ /]522_ІН +2СІн,он- НаС/ \соон СО”
Виходячи з динатріймалонового ефіру, можна добути монокар-бонові кислоти з розгалуженим вуглецевим ланцюгом, наприклад;
Н2С (СООС2Н6)2 Иа2С (СООС2Н6)2 (Н3С)2С (СООС2НБ)2 -
Н8С—СН—СООН.
СН3
Ізомасляна кислота
Взаємодією натріймалонового ефіру з ефірами галогензаміщених органічних кислот можна синтезувати дикарбонові кислоти:
С2Н6О-С-СН2С1 + НаСН (СООС2НБ)2
ц
О
Етиловий ефір хлороцтової кислоти
-> С2НБО—С—СН2—СН (СООС2НБ)2 -►
О
^§с,н‘дй * но—С—СН2—СН^^^-И Гсо^* НООС—СН2—СН2—СООН.
|| 'СООН Янтарна кислота
О
Янтарна кислота була виявлена Г. Агріколою в продуктах перегонки бурштину (янтарю). Бурштин —це закам’яніла смола стародавніх хвойних дерев, яка плавиться в інтервалі 150—300 °С. У бурштині міститься близько 4,5 % янтарної кислоти. Цю кислоту і в наш час добувають перегонкою відходів бурштину. У невеликих кількостях янтарна кислота міститься в недозрілих ягодах агрусу. В промисловості янтарну кислоту добувають каталітичним гідруванням малеїнової кислоти:
Нч Ь \С=С' НООС/ \2ООН
+ Н2
НООС—СН2—СН2—СООН.
Янтарну кислоту застосовують як досить сильний стимулятор росту рослин. Поліконденсацією янтарної кислоти з гліколями добувають поліефіри, які використовують як пластифікатори синтетичних смол. В органічному синтезі використовують імід ЦІЄЇ кислоти — сукцинімід.
Адипінова кислота міститься в соку цукрового буряка. її інколи використовують у харчовій промисловості замість винної і молочної кислот. Проте переважно адипінову кислоту використовують для виробництва поліамідних смол і волокна найлон. При поліконденсації адипінової кислоти з 1,6-гексаметилендіаміном,
230
яку проводять при нагріванні в метанолі в атмосфері азоту, добувають поліамідну смолу з молекулярною масою 25 000 — 35 000 і Тпл =255 °С:
пНО—С—(СН2)4—С—ОН + пН2К—(СН2)в—НН2——->
II II
о о
Г—С— (СН2)4—С—МН— (СН2)в—МН—1 + иН2О.
II о Найлон
о
п
Поліаміди, подібно до білкових молекул (див. розд.25), містять амідну (пептидну) групу атомів —С—ГШ—. Вони легко перероб
ляються на хімічні волокна. Для цього розплавлений поліамід продавлюють крізь тоненькі отвори —фільєри.
Ненасичені дикарбонові кислоти. З ненасичених дикарбонових кислот найбільший інтерес являє симетрична етилендикарбонова кислота НООС—СН=СН—СООН, яка існує у вигляді двох геометричних ізомерів. і{«с-Ізомер цієї кислоти називається малеїновою кислотою, транс-ізомер — фумаровою кислотою'.
Н. /СООН
с :
н/ ^соон
цио Ізомер
(малеїнова кислота)
НООС^с/Н
II
Нх ХСООН
тран с-Ізомер
(фумарова кислота)
Відмінність у просторовій будові цих кислот зумовлює істотні відмінності у їх властивостях. У табл. 21 наведено для порівняння деякі властивості малеїнової і фумарової кислот.
Таблиця 21. Властивості малеїнової і фумарової кислот
Кислота Упл. ’С Розчинність у воді прн 25 °С, % Фізіологічна Дія Константа іонізації
К^5.10-‘
Малеїнова Фумарова 130 287 0,7 78,7 Отруйна Не отруйна 170 9,3 2,6 19
Хімічні властивості малеїнової і фумарової кислот зумовлені наявністю в їх молекулах подвійного зв’язку і двох карбоксильних груп. Фумарова кислота не здатна утворювати ангідрид навіть при нагріванні з водовідбирними речовинами.
Фумарова кислота міститься в багатьох рослинах, наприклад у рутці (Ришагіа оШсіпаїіз), звідки пішла назва цієї кислоти. Є вона також у лишайниках і грибах.
231
Малеїнова кислота, на відміну від фумарової, в природі не зустрічається. Назва її пішла від латинського таїши — яблуко, оскільки її можна добути дегідратацією яблучної кислоти (див. розд. 23). Малеїнова кислота добре розчиняється у воді; вона значно сильніша, ніж фумарова. При нагріванні малеїнової кислоти з водовідбирними речовинами, наприклад з оцтовим ангідридом, від неї легко відщеплюється вода за рахунок зближених у просторі карбоксильних груп і кислота перетворюється на ангідрид малеїнової кислоти:
Розділ 23. ОКСИКАРБОНОВАНІ КИСЛОТИ І ОПТИЧНА ІЗОМЕРІЯ
Оксикарбонові кислоти (оксикислоти), або спиртокислоти,— це органічні сполуки, молекули яких містять одночасно карбоксильну і гідроксильну групи. Отже, оксикислоти є біфункціональ-ними органічними сполуками. Кількість функціональних груп в оксикислотах може бути різна: одна гдіроксильна і одна карбоксильна, дві гідроксильні і одна карбоксильна, дві гідроксильні і дві карбоксильні і т. д.
Оксикислоти можна розглядати як гідроксильні похідні відповідних органічних кислот:
Органічна кислота Оксикислота
Н8С—СООН* * НО—СН,—СООН
Оцтова Оксіоцтова, або гліколева
Н8С—СН2—СООН Н8С—СН—СООН і Н2С—СН2—СООН
пропіонова он он
а-Оксипропіоио ва. 0-Оксипропіонова
або молочна
НООС—СНа—СНа—СООН НООС—сн2—СН—СООН
Янтарна І
он
Оксіяитариа, або яблучна
і НООС—СН—СН—СООН
І І он он
Діокс(янтарна, або винна
* Гомолог'чний ряд оксикислот починається з оксимурашиної кислоті. НО—СООН, яка називається вугільною.Ця кислота нестійка і легко розкладається на СО2 і Н2О.
282
Таблиця 22. Номенклатура і фізичні властивості деяких оксикислот
Формула Назва за номенклатурою ^пл -°С Питоме обертання 20 градуси Константа дисоціації у воді при 25 °С
систематичною історичною та за назвою карбонової кислоти
НО—СН2—СООН Оксіетанова Гліколева, оксіоцтова 63 Неактивна 1,48 —
н3с—сн—соон он 2-Оксипропанова (с±)-Молочна, а-оксипро-піонова (+)-Молочна (—)-Молочна 18* 26 26 Неактивна +3,82 —3,82 1,38 —-
но—сн2—сн2—соон З-Оксипроп анова Гідроакрилова, Р-оксипропіонова ** Неактивна 31,1 —
ноос—СН,—СН—соон 1 он 2-Оксибутандіова (±)-Яблучна, оксіянтарна (+)-Яблучна (—)-Яблучна 130— 131 100 100 Неактивна * ** «** 3,9 0,9
ноос—сн—сн—соон 2,3-Діоксибутандіова (с±)-Винна, виноградна 205 Неактивна 10,2 4,0
1 1 он он соон (+)-Винна (—)-Винна Мезовинна 170 170 140 + 12 -12 Неактивна 13,0 6,0 6,9 1.4
НООС-СН2-А-СН2-СООН он 2-Оксипропан-1,2,3-триова Лимонна 153 Неактивна 8,4 при 18 °С 1 у***» при 18 °С
* ТКИП = 122 °С при 206,15-10» Па.
** Сиропоподібна рідина, при нагріванні дегідратується.
*** Розчини, які містять менше, ніж 34% яблучної кислоти, при 20 °С обертають площину поляризації світла вліво, більш концентровані — вправо. В ацетоновому розчині ліве обертання постійне [а]^ = —5,7°.
**** Л3= 4-Ю-’.
І
Оксикислоти класифікують за основністю І за атомністю. Кількість карбоксильних груп вказує на основність кислоти, а кількість гідроксильних груп, враховуючи і гідроксил карбоксилу, — на її атомність. Наприклад,оксіоцтова кислота, а- і р-окси-пропіонові кислоти є одноосновними двохатомними оксикислотами, а яблучна кислота —двохосновною трьохатомною оксикислотою, винна —двохосновною чотирьохатомною оксикислотою і т.д.
Для оксикислот використовують переважно історичні назви, які походять від назв природних джерел знаходження цих оксикислот. Так, а-оксипропіонова кислота вперше була знайдена в кислому молоці і тому її називають молочною кислотою, оксіянтарна кислота вперше знайдена в яблуках і її називають яблучною, діок-сіянтарпа кислота міститься у винограді і тому її називають виноградною і т. д.
Часто оксикислоти називають за назвою відповідних карбонових кислот, позначаючи грецькими буквами положення спиртової групи в молекулі відносно карбоксилу, наприклад: а-оксипропіо-нова, р-оксипропіонова і т. д. За систематичною номенклатурою назву оксикислоти утворюють з назви відповідної карбонової кислоти, Перед якою ставлять префікс окси- (гідрокси-), що вказує на наявність ОН-групи в молекулах цих кислот. Місцеположення гідроксильної групи в ланцюгу вказують номером вуглецевого атома, з яким вона сполучена, наприклад: 2-оксипропанова кислота, 3-окси-пропанова кислота і т. д. У табл. 22 наведено назви найпоширеніших оксикислот.
Ізомерія в ряду оксикислот залежить від структури вуглецевого ланцюга і положення спиртового гідроксилу. Гліколева кислота ізомерів не має. Наступний гомолог ряду оксикислот існує у вигляді двох ізомерів — а-оксипропіонової і Р-оксипропіонової кислот, які відрізняються положенням спиртового гідроксилу. Одноосновна оксикислота з чотирма вуглецевими атомами може існувати у вигляді п’яти структурних ізомерів:
Н„С—СНа—СН—СООН;
Н3С—СН—СН2—СООН і
8-Окснбутановя, або сс-оксимасляна, кислота
З-Оксибутанова, або 0-оксимасляна, кислота
Н,С—СН,—СН,—СООНі
4 -Оксиб утановз, або у-окснмасляна, кислота
НаС—СН—СООН; І І
НО сн3
2-Метил-З-окси-пропанова кислота
сн3
Н3С—С—СООН.
он
2-Метил-2-окси-пропанова кислота
Перші три з наведених оксикислот є ізомерами, які відрізняються положенням спиртового гідроксилу, ізомерія останніх зумовлена ще й розгалуженням вуглецевого ланцюга. Крім того, оксикислоти, молекули яких містять асиметричні атоми вуглецю, існують ще у вигляді оптичних ізомерів.
ГМ
Одноосновнї оксикислоти
Методи добування. Ряд оксикислот, як, наприклад, молочна, яблучна, винна, лимонна та інші, є продуктами життєдіяльності організмів. Тому деякі оксикислоти і в промислових масштабах добувають шляхом біосинтезу— синтезу за допомогою живих організмів. Так, молочну кислоту добувають молочно-кислим бродінням вуглеводів. Крім того, існують також синтетичні методи добування оксикислот. З цих методів для добування а-оксикислот найчастіше використовують такі.
1. Кислотний гідроліз ціангідринів (оксинітрилів), які добувають з альдегідів або кетонів і синильної кислоти (див. розд. 17):
Н3С-СН=О Н3С—СН—СИ -1- Нзс-сн—СООН.
он он
2. Лужний гідроліз а-галогенокарбованих кислот:
Н3С—СН—СООН Н3С-СН—СООН.
Сі он
Р-Оксикислоти добувають одним з таких методів:
1. Кислотно-каталітичною гідратацією ненасичених кислот (див. розд. 21).
2. Реакцією Реформатського (1889 р.), який встановив, що складні ефіри сс-галогенокарбонових кислот при наявності цинку взаємодіють з карбонільними сполуками і утворюють складні ефіри р-оксикислот. Останні омиленням можна легко перетворити на р-оксикислоти. У реакції Реформатського як проміжні продукти спочатку утворюються цинкорганічні сполуки, які потім реагують з карбонільними сполуками. Так, Р-оксимасляну кислоту можна добути за допомогою цієї реакції з етилового ефіру бромоцтової кислоти і оцтового альдегіду:
в+ о— ®”
Вг—СН2—СООС2Н5 —- Вг—2п—СНа—СООСаНв ”зС~СН^-^
** Н3С—СН—СНа—СООС2НВ _2п (Он) вг, ~с,н,он **
О2пВг
Н3С—СН—СНа—СООН.
он
Фізичні властивості. Одноосновні оксикислоти з невеликою молекулярною масою — це густі сиропоподібні рідини або тверді речовини (табл. 22). Двохосновні оксикислоти — кристалічні речовини. Оксикислоти краще розчиняються в воді, ніж відповідні їм карбонові кислоти, але обмежено розчиняються в ефірі та інших органічних розчинниках. Оксикислоти мають значно вищітемпера-
235
тури плавлення і кипіння, ніж карбонові кислоти з такою самою кількістю вуглецевих атомів. Багато оксикислот виявляють оптичну активність.
Хімічні властивості. Оксикислоти містять дві функціональні групи і їх хімічні властивості визначаються наявністю як карбоксильної, так і гідроксильної груп. Тому оксикислоти одночасно виявляють властивості і карбонових кислот, і спиртів. Причому карбоксильна і гідроксильна групи можуть брати участь у хімічних реакціях окремо, незалежно одна від одної, або обидві разом. Так, оксикислоти, як і карбонові кислоти, у водних розчинах дисоціюють:
/?—СН—СООН + Н2О /?—СН—СОО- + н8о+.
он он
Однак оксикислоти, і в першу чергу а-оксикислоти, мають сильніші кислотні властивості, ніж відповідні їм карбонові кислоти. Наприклад, константи дисоціації гліколево'ї (/Сд = 1,48 • 10-4) і а-оксипропіонової (/<д = 1,38 • 10~4) кислот приблизно в 8—10 разів більші, ніж відповідно оцтової (/Сд = 1,75 • 10~6) і пропіо-нової (Яд =1,34 • 10-Б) кислот. Причиною посилення кислотних властивостей оксикислот є —/-ефект спиртової ОН-групи, яка зумовлює зміщення електронів по системі о-зв’язків НО СН2 ч- Сч- О+- Н. Тому атом водню карбоксильної групи
О
легше протонізується, ніж в карбонових кислотах, і сила оксикислоти зростає. Чим ближче спиртовий гідроксил розміщений до карбоксильної групи, тим сильнішою є оксикислота, оскільки в міру віддалення ОН-групи відСООН-групи вплив—/-ефекту поступово «затухає». Тому Р-, у-, 6-оксикислоти виявляють значно меншу кислотність, ніж відповідні їм а-оксикислоти.
Оксикислоти, подібно до карбонових кислот, утворюють солі, складні ефіри, аміди тощо. Наприклад:
Н3С—СН—СООС2Не «4^^ Н3С—СН—СООН Н3С—СН—СООИа.
с н он ОН
Етиловий ефір Лактат натрію
молочної кислоти
Оксикислоти, вступаючи в реакції як спирти, утворюють алкоголяти, прості ефіри, заміщують гідроксил на галоген. Так, при взаємодії оксикислот з лужними металами, лугами спочатку утворюються солі цих кислот, які далі реагують з цими ж речовинами і дають алкоголяти:
Е3С—СН—СООН Н3С—СН—СООК’а Н3С—СН—СООК'а.
ОН ОН О№
СІЛЬ молочно*
КИСЛОЇи
А ПКОГОЛЯТ молочної кислоти (і одночасно сіль)
236
При дії на оксикислоти галогеноводнів спиртовий гідроксил їх молекул заміщується на галоген і утворюються відповідні гало-генокислоти:
Н3С—СН—СООН + неї Н3С—СН—СООН + нао.
он сі
К-Оксипропіоно ва сс-Хлорпроп іонова
кислота кислота
П’яти хлористий фосфор заміщує на галоген одночасно як спиртовий гідроксил, так і гідроксил карбоксилу молекули оксикислоти і перетворює їх таким чином на хлорангідриди хлорорганічних кислот:
Н3С-СН—С^° + 2РС16 -> Н3С-С-С^ + 2РОСІ, 4- 2НС1.
| ХОН | ХС1
ОН СІ
а-Оксипропіонова Хлорангідрид
кислота а-хлорпропіоиової
кислоти
Оксикислоти, подібно до спиртів, можуть окислюватись. Ця властивість оксикислот відрізняє їх від карбонових кислот, які, як правило, стійкі до окислення. Оксикислоти, що містять первинну ОН-групу, при окисленні перетворюються на альдегідокислоти, а оксикислоти з вторинною ОН-групою—-на кетокислоти:
НО—СН2—СООН 2^- ^С—СООН; Гліколева кислота
Гліоксалева кислота
Н,С—СН—СООН -Дґо*Н„С—С—СООН.
І ~Н!° II
он о
Молочна кислота Піровиноградна
кислої а
Аналогічне окислення молочної кислоти до піровиноградної відбувається і в живих організмах.
Крім реакцій, які відбуваються з участю карбоксильної і спиртової груп, оксикислотам властиві також специфічні реакції, зумовлені взаємним впливом двох функціональних груп у їх молекулах. Так, а-, р-, у-, б- і є-оксикислоти по-різному перетворюються при нагріванні. Оксикислоти при підвищених температурах дегід-ратуються. Але залежно від взаємного розміщення спиртової і карбоксильної груп дегідратація їх проходить по-різному і з утворенням різних продуктів реакції.
сс-Оксикислоти при нагріванні відщеплюють воду міжмолекулярно за рахунок гідроксилу карбоксильної групи однієї молекули оксикислоти і атома водню спиртової групи другої молекули оксикислоти. Продуктами такої дегідратації є цикличні міжмолекулярні складні ефіри, які називають лактидами. Так, гліколева кис-
237
лота за таких умов дає кристалічний лактид 2,5-діоксо-1,4-діоксан, або гліколід (Тпл = 86 °С):
2Н2О +
Гліколід
Молочна кислота при дегідратації утворює лактид 3,6-диметил-2,5-діоксо-1,4-діоксан:
оч 4-0-н ^сн3
Л д__ і
Н3С ^О-_Н__Н^р^ о
2Н2О.
Лактид
р-Оксикислоти при нагріванні дегідратуються внутрішньомоле-кулярно. Вода при цьому відщеплюється за рахунок спиртового гідроксилу і а-водневого атома метиленової групи. В результаті утворюються а-, Р-ненасичені кислоти. Так, р-оксипропіонова кислота при цьому перетворюється на акрилову кислоту:
Н2С—СН—СООН дщт' Н2С=СН—СООН. Іно~ні
у- і б-Оксикислоти при нагріванні вступають у внутрішньомо-лекулярну етерифікацію між спиртовим гідроксилом і карбоксильною групою, утворюючи внутрішньомолекулярні циклічні складні ефіри, які називають лактонами. Так, у-оксимасляна кислота при дегідратації перетворюється на бутиролактон (Ткт, = = 204 °С):
Н2С —,СН2— СН.,- с = о --
І ---------4- -Н2о
о-н________он]
Реакцію лактонізації оксикислот відкрив у 1873 р. О. М. Зай-цев. Лактони є біологічно активними речовинами і використовуються для синтезу замінників крові. Деякі лактони мають приємний запах і їх застосовують у парфюмерії.
є-Оксикислоти і оксикислоти з ще більш віддаленою спиртовою групою при нагріванні вступають у реакції конденсації і утворюють при цьому лінійні полімери.
Найважливіші представники. Гліколова кислота (Тпл =79 °С) є найпростішою із стійких оксикислот. Вона міститься в соку недозрілого винограду, а також цукрової тростини. Гліколеву кисло
238
ту можна добути окисленням етиленгліколю або електролітичним відновленням щавлевої кислоти:
НО—СН„—СН,—ОН -±Гб-* но—СНа—СООН ноос-соон.
Гліколеву кислоту застосовують у ситцевибиванні і при дубленні шкіри.
Молочна кислота (Тпл = 18 °С) вперше була відкрита в 1780 р. К.Шеєле в кислому молоці, у зв’язку з чим і одержала свою назву. Молочна кислота є продуктом життєдіяльності молочнокислих бактерій Васіїіиз асісіі Іаеуоіасіісі, які перетворюють вуглеводи молока (переважно лактозу). Тому ця кислота крім кислого молока міститься також у таких кисло-молочних продуктах, як кефір, ряжанка тощо.
Рис. 41. Електромагнітні коливання в промені звичайного і поляризованого світла:
1 — звичайне світло; 2 — призма Ніколи;
З — поляризоване світло; 4 — площина поляризації
У промисловості молочну кислоту добувають молочнокислим бродінням вуглеводів (лактози, мальтози та інших) у підлужне-ному водному розчині. Цей процес описується такою загальною схемою:
С12НМОП + На0 БродІННЯ-> 4Н3С—СН—СООН. ін
Молочну кислоту добувають також ціангідринним методом і гідролізом а-галогенопропіонових кислот.
Солі молочної кислоти називають лактатами.
Молочна кислота вже досить давно використовується як консервуючий засіб, що запобігає гниттю при солінні капусти, огірків, помідорів та інших овочів і фруктів, при силосуванні кормів. Молочна кислота застосовується також для протравного фарбування тканин, у шкіряній промисловості, медицині. Так, лактати алюмінію і амонію запобігають кровотечі з ясен, лактат заліза приписують хворим на гіпохромну анемію, а саму молочну кислоту застосовують для припалювання.
Оптична ізомерія оксикислот. Відомо багато речовин, які здатні обертати (змінювати) площину поляризації світла, тобто відхиляти її вправо (за годинниковою стрілкою) або вліво (проти годинникової стрілки) (рис. 41, 42). Ця здатність речовин була названа оптичною активністю, а самі речовини, що мають таку здатність,—
239
оптично активними. Оптично активні речовини існують у вигляді оптичних ізомерів. Явище оптичної активності особливо поширене серед органічних речовин природного походження (оксикислоти, амінокислоти, вуглеводи, білки, нуклеїнові кислоти, алкалоїди тощо). Це явище має велике біологічне значення, оскільки воно пов’язане з асиметрією речовин, що входять до складу живих організмів, і з явищем життя.
Світло, згідно з електромагнітною теорією, являє собою поперечні електромагнітні хвилі, частинки яких роблять коливальні рухи, перпендикулярні до напрямку поширення хвилі. У зв’язку з цим коливання в промені звичайного світла відбувається в просторі у найрізноманітніших площинах, як це схематично показано
Рис. 42. Зміна площини поляризації при проходженні поляризованого світла крізь оптично активну речовину:
1 — площина поляризації до проходження світла крізь оптично активну речовину: 2 — оптично активна речовина; 3 — площина поляризації, повернута на кут а після проходження світла крізь оптично активну речовину
на рис. 41. Якщо таке світло пропустити крізь призму Ніколи, то ця призма пропустить тільки ті частинки світла, коливання яких відбувається в певній площині. Таке світло називають поляризованим. Площина, перпендикулярна до площини коливання частинок поляризованого світла, є площиною поляризації.
Якщо поляризоване світло направити на другу призму Ніколи, то світло пройде крізь неї тільки в тому випадку, коли площина, в якій відбувається коливання частинок світла, буде певним чином направлена відносно осі кристалів другої призми Ніколя. Якщо призму Ніколя встановити так, що крізь неї буде проходити поляризоване світло, а потім повернути цю призму на 90°, то поляризоване світло крізь неї проходити не буде (рис. 43, о). Призму Ніколя І поляризоване світло можна порівняти з книжкою і ножем. Ніж може пройти крізь книжку тільки в тому випадку, коли лезо ножа знаходиться в тій самій площині, що і сторінки книжки. Якщо книжку повернути на 90°, то ніж крізь неї пройти вже не може (рис. 43, б).
У 1848 р. французький вчений Л. Пастер встановив, що оптичну активність мають солі винної кислоти, які існують у вигляді «правого» і «лівого» оптичних ізомерів, а в 1860 р. цей вчений висловив
240
думку про те, що оптична активність е результатом просторової асиметричної будови цих речовин. Існування оптично активних ізомерів, що відхиляють площину поляризації світла вліво і вправо, по-яснилив 1874 р. Я-Вант-ГоффіЖ. Ле-Бель. Вчені майже одночасно незалежно один від одного опублікували роботи, в яких сформулювали стереохімічну гіпотезу, що стала потім теорією. Основні положення стереохімічної теорії оптичної ізомерії органічних речовин можна сформулювати так:
1. Оптично активні речовини, як правило, містять асиметричні атоми вуглецю, які позначають зірочкою: С*. Асиметричним називається атом вуглецю, сполучений з чотирма різними атомами або
Рис. 43. Схема проходження поляризованого світла крізь призму Ніколи при різних її положеннях
групами атомів (лігандами). Наприклад, гліцериновий альдегід» молочна кислота містять один асиметричний атом вуглецю:
Н
І НОСН2—с*—с
Існують органічні речовини, які мають не тільки один, а два, три і більшу кількість асиметричних вуглецевих атомів. Слід зазначити, що відомі такожоптично активні речовини, молекули яких не містять асиметричних атомів вуглецю. Оптична активність може бути зумовлена наявністю в молекулі і інших асиметричних атомів.
2. Чотири ліганди розміщені в просторі по вершинах тетраедра, в центрі якого знаходиться асиметричний атом С*. Просторова форма молекули, яка при цьому утворюється, називається конфігурацією. Конфігурація тетраедра асиметричного атома вуглецюзавжди неправильна, оскільки ліганди, які сполучені з цим атомом, різні.
3. Молекули, які містять асиметричні атоми вуглецю, несиметричні, тому що в них немає таких елементів, як центр і площина симетрії. Несиметричні молекули не можна сумістити з їх дзеркальним відображенням, як не можна сумістити праву і ліву руки. Тому такі молекули називають ще хіральними (від грецьк. «хірос» — рука). Останнім часом замість термінів «асиметричний агом», «аси
метрична молекула» використовують більш точні терміни «хіраль-ний атом», «хіральна молекула», оскільки термін «асиметричний» говорить про відсутність у молекулі взагалі будь-яких елементів симетрії, що є неточним. Хіральність —це властивість молекули (об’єкта) не суміщатися зі своїм дзеркальним відображенням.
4. Молекули з асиметричними атомами вуглецю існують у вигляді правих (ф-)-форм (конфігурацій) і лівих (—)-форм (конфігурацій), тобто у вигляді стереоізомерів, які відрізняються різним розміщенням лігандів у просторі навколо асиметричного атома С*. Правий і лівий стереоізомери дзеркально подібні між собою, але не сумісні (хіральні), або енантіоморфні (від грецьк. «енантіос» — протилежний). Отже, оптична (дзеркальна) ізомерія —це вид просторової ізомерії, при якій стереоізомери відрізняються розміщенням у просторі лігандів навколо асиметричного атома вуглецю, але подібні між собою як предмет і його дзеркальне відображення, тобто є енантіоморфними.
Право (+)- і ліво(—)-обертаючі ізомери мають однакову структуру (однакову послідовність хімічних зв’язків у молекулі), однакові температури кипіння і плавлення, розчинність, теплопровідність, електропровідність тощо, але розрізняються між собою такими властивостями:
І) у рідкому, газоподібному станах і в розчинах вони відхиляють площину поляризації світла на однаковий кут, але в протилежних напрямках;
2) обидва ізомери кристалізуються в енантіоморфних (дзеркально протилежних) формах один відносно другого, тобто є оптичними антиподами;
3) право (+)- і ліво (—)-обертаючі ізомери відрізняються реакційною здатністю відносно інших оптично активних реагентів;
4) ці ізомери виявляють різну фізіологічну дію на живі організми;
5) при змішуванні рівномолярних кількостей (-(-)- і (—^ізомерних форм утворюються оптично неактивні (недіяльні, абоінак-тивні) сполуки, які називають рацематами і позначають значком г(І) (від франц. асісіе гасешіяие —виноградна кислота, на прикладі якої Л. Пастер спостерігав це явище):
(+) -Ізомер 4- (—) -Ізомер -* г (і) (рацемат, і — інактисний).
Рацемати відрізняються від (-(-)- і (—)-ізомерів за фізичними та іншими властивостями.
Оптичну активність речовин визначають за допомогою поляриметра. Оптична активність речовини характеризується кутом питомого обертання [а]2д (£) —жовта лінія натрію) —стабільною величиною кута обертання площини поляризації світла розчином речовини при його концентрації С, що дорівнює 1 г речовини в 1 мл розчину, при товщині шару І, дм:
. ,зе а
[а]д = С1 •
242
При наявності в молекулі органічної речовини п асиметричних вуглецевих атомів кількість просторових ізомерів виражається формулою 2". У молекулі молочної кислоти міститься один асиметричний атом С* . Тому повинні існувати два (2і =2) просторових ізомери цієї кислоти. І дійсно, ще в 1847 р. Ю. Лібіх виділив із м’язів (м’ясного екстракту) молочну кислоту, яка відхиляє площину поляризації світла вправо. Тому (-|-)-молочну кислоту назвали м’ясо-молочною. Вона утворюється в м’язах у процесі анаеробного розщеплення вуглеводів. Лівообертаюча (—)-молочна кислота була добута підчас бродіннясахаристих речовин під впливом мікроорганіз
мів Васіїїиз асісіі Іаеуоіасіісі. Тетраедричні і кулестержневі моделі молекул цих кислот зображені на рис. 44. Молочна кислота, яка утворюється в процесі скисання молока, є оптично неактивною і являє собою рацемічну сполуку рівномолярних кількостей (-}-)-і (—)-молочних кислот: (+)- + (—)-молочна кислота =г-молоч-на кислота. Обидві (-}-)- і (—)-молочні кислоти мають однакову температуру плавлення (26°С), що відрізняється від температури плавлення рацемічної молочної кислоти, яка плавиться при 18 °С.
Е. Фішер у 1891 р. запропонував для зручності і простоти зображати стереоізомери не у вигляді рисунків їх молекул, а проекційними формулами, які являють собою проекції тетраедричних моделей відповідних молекул на площину (рис. 45). Для написання проекційної формули тетраедр розміщують відносно спостерігача так, щоб лінія горизонтального ребра зв’язку Н—С—О Н проходила через центр тетраедра (через ядро атома С*). При проектуванні зв’язків Н—С—ОН і Н3С—С—СООН на площину одержу
243
ють проекційну формулу у вигляді двох ліній, які перетинаються під прямим кутом. У точці перетину цих ліній знаходиться атом С*, а на кінцях ліній —ліганди:
СООН н-^-он
<ін3
(-) -Молочна кислота
СООН
(+)-Молочна кислота
Одне з найголовніших правил користування проекційними формулами полягає в тому, що ці формули можна повертати тільки в площині рисунка на 180°, але їх не можна повертати на 90° або виводити з площини рисунка. З рис, 45 видно, що як моделі, так
і проекційні формули правої форми молочної кислоти не суміщаються з формулами лівої її форми. В несумісності цих двох форм можна переконатися, переходячи від атома Н до групи ОН через карбоксил. Для (—)-форми такий перехід відбувається за годинниковою стрілкою, а для (-Ь)-форми — проти годинникової стрілки:
СООН
Н—ОН сн.3
СООН
но---н
СНз
(-(-Молочна кислота (Н'-Молочна кислота
Конфігурація і знак обертання. £>-і £-ряди. У стереохімії розрізняють поняття абсолютної конфігурації, тобто дійсного просторового розміщення лігандів у оптично активному стереоізомері, і відносної конфігурації, в якій розміщення лігандів порівнюють з стандартним або так званим опорним стереоізомером. У більшості випадків виходять з відносних, а не з абсолютних конфігурацій.
244
Для встановлення відносної конфігурації стереоізомера як опорну сполуку Е. Фішер (1891 р.) і М. А. Розанов (1906 р.) запропонували правообертаючий гліцериновий альдегід, якому довільно була приписана конфігурація Ь (від лат. Рехігацугиз — правообертаючий). Відповідно лівообертаючий гліцериновий альдегід (антипод) позначають буквою £ (від лат. Ееауо^угиз — лівообертаючий):
І н
Н'—с—он
сн.2он
СН.2ОН
(+)£>-Г.іІцериновий (")/- -Гліцериновий альдегід альдегід
Ж. Бійво в 1951 р. методом рентгеноструктурного аналізу встановив абсолютну конфігурацію (+)Р-гліцеринового альдегіду і підтвердив правильність приписаної йому конфігурації. (ф-)Р-Гліцериновий альдегід є найпростішою оксикарбонільною сполукою, яка містить один асиметричний атом С*, виявляє оптичну активність і існує у вигляді двох оптичних ізомерів. Оскільки цей •альдегід містить високореакційноздатні групи, то його можна, не порушуючи зв’язку з асиметричним атомом вуглецю, перетворити ні багато інших оптично активних сполук і тим самим встановити їх відносну конфігурацію. Оптично активні речовини, які були добуті з (Т-)Р-гліцеринового альдегіду, віднесли доР-ряду стереоізомерів незалежно від знаку обертання площини поляризації світла, а оптично активні речовини, добуті з (—)£-гліцеринового альдегіду, — до £-ряду стереоізомерів.Тому лівообертаюча(—)-молочна к іслота належить до Р-ряду, оскільки її відносна конфігурація може бути виведена з (Т-)Р-гліцеринового альдегіду такими послідовними перетвореннями:
О
Н—С*—ОН
Окислення
ґ^он
РІ_Відновлення
сн2—он (+) О-Гліцериновий альдегід
СН2ОН
(—) О-Глінери-нова кислота
-> н—с*—ОН.
І сн3 (—) И-Молочна кислота
Отже, (—)-молочна кислота належить до стереоізомерів Р-ряду. Позначення Р і Ь означають конфігурацію оптично активної речо
245
вини, а знаки (+) і (—) —напрямок обертання площини поляризації світла оптично активною речовиною.
Обертання площини поляризації світла залежить в основному від природи тих атомів і груп атомів, які входять до складу оптично активної речовини. Тому напрямок і величина обертання площини поляризації світла не залежить від конфігурації даної сполуки. Наприклад, солі, ефіри (-}-)-молочної кислоти відхиляють площину поляризації світла вліво, тоді як сама (ф-)-молочна кислота — вправо:
Н3С—СН—СООН; (Н3С—СН—СОО)22п;
он ОН
[а] р 4-3,3° —6,0°
Н3С—СН—СООСН3; Н3С—СН—СООН.
ОН ОСН3
[а]£> —8,2° —75,6°
Багатоосновні і багатоатомні оксикислоти
Багатоосновні оксикислоти можуть містити в молекулі дві, три і більше карбоксильних груп. Представниками дикарбонових оксикислот є оксі- і діоксіянтарні кислоти. Оксіянтарна, або яблучна, кислота вперше була виділена ще 1785 р. К. Шеєле з недостиглих яблук. Вона широко поширена в природі і міститься в кислих яблуках, горобині, агрусі, барбарисі, махорці тощо. Яблучна кислота має один асиметричний атом вуглецю С*, виявляє оптичну активність і існує у вигляді (-}-)£)-і (—)£-стереоізомерів (Тпл — = 100 °С):
СООН СООН
Н----ОН ; НО----Н
сн2соон СН2СООН
(+) £)-Яблучна (—) £-Яблучна
кислота кислота
У природі поширена (—^Ь-яблучна кислота. Рацемічну яблучну кислоту (Тил—130°С) можна добути гідратацією малеїнової кислоти або дією вологого оксиду срібла на рацемічну бром’ янтарну кислоту:
Н \с=с('[ 1 +н?” > НООС—СН—СН2—СООН
НООС/ хсоон | 8
Малеїнова кислота ОН
— НООС—СН—СН2—СООН.
Вг
Бром'янтарна кислота
Діоксіянтарні кислоти називають винними. Вони містять два * «
асиметричних атоми С*: НООС—СН—СН—СООН. Кількість оп-! І
ОН ОН
246
тичних ізомерів, як уже зазначалося, обчислюють за формулою 2", де п — число асиметричних атомів С*. Отже, стереоізомерних діок-сіянтарних кислот повинно бути чотири (два ізомери £)-ряду, два — £-ряду). Кожна пара цих оптичних ізомерів повинна давати по одному рацемату. В природі існують, а також добуті синтетично такі винні кислоти: (ф-)-винна (І), її антипод (—)-винна (II), мезо-винна (III) і рацемат — (±)-винна кислота, яка називається виноградною. У молекулі мезовинної кислоти обидва асиметричні атоми С* сполучені з однаковими лігандами і вона має внутрішню симетрію. Крім того, в мезовинній кислоті відбувається внутрішня компенсація обертання площини поляризації світла протилежно побудованими асиметричними центрами з однаковими лігандами. Тому мезовинна кислота є оптично неактивною і в неї немає антипода:
СООН
н——он
нох—н
СООН і
(+)-Винна кислота
II
і-)- Винна кислота
СООН но——н
Н— у-он
СООН
с :оон
у VII
( І :оон і
Мезовинна кислота
с :оон с :оон
но— ( п
Н п хюн с IV Огі хюн
Виноградна кислота
Причину оптичної неактивності мезовинної кислоти можна уяснити так. Припустимо, що розміщення Н, СООН і ОН біля асиметричного атома С* в такому порядку — від атома водню через карбоксил до гідроксилу за годинниковою стрілкою —зумовлює обертання площини поляризації світла вправо, а розміщення цих груп у протилежному порядку( проти годинникової стрілки) —обертання площини поляризації світла вліво. Після такого припущення легко помітити, що в мезовинній кислоті Н, СООН і ОН біля верхнього атома С* розміщені за годинниковою стрілкою і повинні зумовлювати праве обертання, а біля нижнього атома С* ці самі групи розміщені проти годинникової стрілки і повинні зумовлювати ліве обертання. Оскільки групи біля верхнього і нижнього атомів С* однакові, то праве обертання верхньої частини молекули повинно повністю компенсуватися лівим обертанням нижньої частини молекули. В кінцевому результаті така винна кислота, хоч і має асиметричні атоми С*, оптичної активності не виявляє.
(+)-» (—)-Винні кислоти є оптичними антиподами. Мезовинна кислота відносно цих винних кислот вже не є їх антиподом. Такі стереоізомери, які не є антиподами, називають діастереоізомерами. Отже, (ф-)-винна і мезовинна, (—)-винна і мезовинна кислоти є діастереоізомерами. Діастереоізомери різняться між собою не тільки оптичним обертанням, алей іншими фізико-хімічними властивостями, тоді як у антиподів фізико-хімічні властивості однакові, а відмінні тільки напрямки обертання площини поляризації світла (див. табл. 22). Діастереоізомери утворюють також відмінні за влас
•47
тивостями похідні. Так, солі діастереоізомерних винних кислот значною мірою відрізняються за розчинністю у воді. Ця властивість має важливе практичне значення і використовується для розділення даних діастереоізомерів.
З винних кислот найбільше значення має (-|-)-винна кислота, яка називається О-винною, або виннокам’яною, кислотою. Солі винної кислоти називають тартратами. Винна кислота і її солі містяться в багатьох плодах рослин, наприклад у винограді, горобині. Кисла калієва сіль винної кислоти важко розчиняється у воді. Вона міститься у виноградному соку і під час його переробки випадає в осад у вигляді «винного каменю». Діючи на «винний камінь» мінеральною кислотою, можна добути (-|-)-винну кислоту:
НООС—СНОН—СНОМ—СООК + НСІ (+)-Винна кислота.
Винну кислоту застосовують у харчовій промисловості, наприклад при виготовленні охолодних напоїв. Подвійна калій-натрієва сіль винної кислоти КООС—СНОН—СНОН—СООЬІа відома під назвою сегнетової солі і використовується для виготовлення реактиву Фелінга (див. розд. 17), а також у радіотехніці (п’єзокристали). Подвійна сіль калію і сурми 2С4Н4ОвК • (8ЬО)2 • Н2О під назвою «рвотпого каменю» застосовується в медицині, а також при протравному фарбуванні.
Антипод виннокам’яної кислоти —(—)-винна кислота в природі не зустрічається. її можна добути з рацемічної (ф-)-винної (виноградної) кислоти розділенням кристалів на ліво- і правообер-таючі ізомери.
Виноградна, або (±)-винна, кислота є рацематом. Вона являє собою хімічну сполуку рівномолярних кількостей (-(-)-винної і (—)-винної кислот і тому неє оптично активною. У водному розчині виноградна кислота має мономерну, а не подвоєну молекулярну масу. Це свідчить про те, що при розчиненні рацемат дисоціює на (-(-)- і (—)-винні кислоти. Виноградну кислоту можна добути при тривалому нагріванні (-(-)- або (—)-винної кислоти, а також при окисленні фумарової кислоти водним розчином КМпО4:
НООС. .Н н о КМпО
\с=с/ ——-----(±)-Винііа (виноградна)
Іг 'СООН кислота.
Під час кип’ятіння (4-)-, (—)-винних або виноградної кислот з лугами, а також при окисленні малеїнової кислоти водним розчином КМпО4 утворюється мезовиниа кислота:
\с=с/ Н,О, КМиО4 । ноос/ ~ /-СООН
Мезовинна кислота.
Представником трьохосновних оксикислот є лимонна кислота
ОН
НООС—СН2—С—СН2—СООН. соон
248
Вона не містить асиметричного атома С* і тому є оптично неактивною. Лимонну кислоту вперше добув у 1784 р. К. Шеєле з лимонного соку, в якому її міститься 6—10 %. Ця кислота є також у смородині, малині, агрусі, буряковому і виноградному соках, а у вигляді солей — в махорці, у невеликій кількості вона міститься в молоці і крові. Лимонна кислота відіграє важливу роль в процесах обміну речовин в живих організмах. У промисловості лимонну кислоту добувають лимонно-кислим бродінням глюкози і патоки, а також з махорки після виділення з неї нікотину. Лимонна кислота використовується в харчовій промисловості при виготовленні кондитерських виробів, у медицині, текстильній промисловості при фарбуванні тканин тощо.
Хлор'яблучні, або оксихлор' янтарні, кислоти належать До дикарбонових оксикислот з двома асиметричними атомами вуглецю: НООС— *СН—*СН—СООН. Тому у хлор’янтарної кислоти існує 4
І І
ОН СІ
(22 = 4) оптичних ізомери, які зображаються проекційними формулами І—IV:
2ООН СООН
Н —ОН но Н
н— —СІ СІ—н
( :оон СООН
І п
Назва —)-
Гпл- °С 173 173
СООН СООН
Н- -—ОН НО Н
СІ— н ’ н СІ
СООН СООН
ш IV
Назва Трео(+)- Трео(—)-хлор'-
яблучиа кислога
гпл- °с 167 167
Речовини, в молекулах яких однакові замісники розміщені біля асиметричних атомів С* з одного боку, називають еритро-ізомера-ми\ якщо ж ці замісники розміщені з різних боків, то їх називають трео-ізомерами. Крім стереоізомерів І—IV, існують дві рацемічні хлор’яблучні кислоти: рацемат (7’Ш1 = 146 °С), який складається 3 (+)• і (—)-еритро-форм і рацемат (7'пл= 153°С), який складається з (+)- і (—)-трео-форм.
Суміш стереоізомерних хлор’яблучних кислот можна добути приєднанням хлорнуватистої кислоти до малеїнової або фумарової кислоти:
НООС—СН=СН—СООН + НОСІ -> НООС—СН—СН—СООН.
і І
он сі
249
Методи розщеплення рацематів на антиподи. Класичні методи розділення рацематів на антиподи розробив Л. Пастер ще в середині минулого століття. Найважливішими з них є такі:
1. Метод кристалізації. При кристалізації рацематів у певному температурному режимі відбувається самовільне їх розщеплення на антиподи і утворення дзеркально подібних (енантіоморфних) кристалів право- і лівообертаючих оптичних ізомерів. Так Л. Пастер у 1848 р. вперше добув оптично активні ізомери з ранемату натрієво-амонієвої солі виноградної кислоти, яку він кристалізував з розчину при 27 °С, а потім механічно, за допомогою пінцета розділив їх на право- і лівообертаючі ізомери.
Самовільне розщеплення рацематів на антиподи методом кристалізації можна проводити ще й так. У пересичений розчин ранемату вносять «затравку» —кристал одного з антиподів. Після цього з розчину викристалізовується антипод, кристал якого був внесений у розчин, а другий антипод залишається в розчині.
2. Біохімічний метод. Суть цього методу полягає в тому, що деякі мікроорганізми в процесі життєдіяльності споживають тільки один з оптичних ізомерів. Так, чорнильна пліснява Репісіїїіиіп £Іаисит у розчині виноградної кислоти «поїдає» (ф-)-винну і залишає в розчині (—)-винну кислоту.
3. Хімічний метод базується на переведенні рацематів у діасте- -реоізомери. Для цього рацемат вводять у взаємодію з яким-небудь оптично активним реагентом. В результаті утворюються два діасте-реомерних продукти, які відрізняються розчинністю або іншими властивостями, що дозволяє виділити окремі антиподи. Наприклад, розчин рацемату, який містить (+)- і (—)-кислоту, за цим методом обробляють оптично активною (—)-основою. Такими основами є переважно складні гетероциклічні сполуки: бруцин, цинхонін та ін. В результаті утворюються діастереомери: добре розчинна сіль (-р)-кислоти з (—)-основою і важкорозчинна сіль (—)-кислоти з (—)-основою, яка випадає в осад. Діючи після цього мінеральною кислотою на окремо розділені солі, добувають (ф-)- і (—)-ізомери відповідної оптично активної кислоти:
(щ)-Кислота
(—)-Основа
Сіль (ф-)-кислоти в (—)-оєновою | НС1 (ф-)-Кислота
Сіль (—)-кислоти з (—)-основою
НС1
(—)-Кислота
4. Хроматографічний метод. За допомогою цього методу ра-цемати також переводять у діастереомери. З цією метою розчин рацемату піддають хроматографуванню на оптично активному адсорбенті. При цьому один з оптично активних ізомерів утворює
250
з таким адсорбентом діастереомер, який міцно утримується поверхнею адсорбента, а другий оптичний ізомер при цьому легко вимивається з адсорбційної колонки відповідним розчинником.
Стереохімія реакцій заміщення. Велике теоретичне і практичне значення мають реакції, які відбуваються з участю зв’язків асиметричного атома С*. зокрема реакції заміщення. Досить детально вивчені реакції нуклеофільного заміщення біля асиметричного атома С* (5дг-реакції). Такого типу реакції вперше спостерігав і вивчав П. Вальден у 1896 р. Він встановив, що реакції біля асиметричного атома С* у багатьох випадках супроводяться інверсією (обертанням) знака обертання площини поляризації світла. Це явище було назване вальденівським обертанням, тобто зміною конфігурації молекули в процесі реакції без рацемізації її продукту.
Зміна або збереження знака обертання, а відповідно і конфігурації молекул, істотно залежить від умов і механізму реакції заміщення. Так, (—)-хлор’янтарна кислота в процесі лужного гідролізу перетворюється на (-(-)-яблучну. А при дії на (—)-хлор’ янтарну кислоту зволоженого оксиду срібла, який важко розчиняється у воді і тому створює в розчині низьку концентрацію іонів ОН-, утворюється (—)-яблучна кислота. Отже, у першому випадку відбувається не тільки заміна атома хлору на гідроксильну групу, а й перегрупування (обертання) груп атомів, сполучених з асиметричним атомом вуглецю. Зміна конфігурації відбувається також при дії РСІ-, на оптичні ізомери яблучної кислоти, які при цьому перетворюються на хлор’янтарні кислоти з протилежними знаками обертання. Вищесказане можна зобразити такою схемою:
+Аеон, н.о
НООС—СН..—СІ І—СООН--------> НООС—СН„—СН—СОН;
2 І -АКС1 2 |
СІ он
(—)-Хлор’янтарна кислота
(—)-Яблучна кислота
кон
НООС—СН2—СН—СООН
он (4-)-Яблучна кислота
КОН ( |РС1в
4-АеОН, Н.0
-е--— -----НООС— СН2—СН—СООН.
—Авсі 2 |
СІ
(4-)-Хлор’янтарна кислота
Отже, заміна біля асиметричного атома С* галогену на гідроксильну групу під час гідролізу водним розчином лугу і заміна біля асиметричного С* ОН-групи на галоген під дією РС15 відбуваються із зміною конфігурації. Встановлено, що причиною такого обертання конфігурації є механізм реакції заміщення. Вальденівське обертання спостерігається у реакціях нуклеофільного заміщення, які відбуваються за бімолекулярним механізмом (5дг2-реакції) з утворенням перехідного стану (див. розд. 8). При такому механізмі заміщення аніон ОН" атакує асиметричний атом С* з протилежного відносно атома галогену боку —з «затилля». В результаті кінцевим
251
продуктом реакції є молекула з протилежною конфігурацією і відповідно з протилежним знаком обертання:
ноос сн2соон
НО---С-—;сі
НООС \н2соон
Н
----- НО—с
/\
НООС СН,СООІ І
(-)-Х.юр’яіпарна кислота Перехідний стан
(+)-Яблучна кислота
Отже, якщо в процесі хімічної взаємодії спостерігається обертання конфігурації (зміна знаку обертання), то можна робити висновок, що ця реакція напевне відбувається за 5уу2-механізмом. У тих випадках, коли нуклеофільні реакції заміщення біля асиметричного атома С* протікають за мономолекулярним механізмом (5/у1), відбувається рацемізація, яка супроводиться частковим вальденівським обертанням. Реакції цього типу проходять через стадію попередньої дисоціації вихідної речовини з утворенням карбонієвого іона, який має плоску конфігурацію, і аніон ОН” може його атакувати як справа, так і зліва. Часткове вальденівське обертання пояснюється тим, що карбонієвнй іон не встигає набути повністю плоскої конфігурації.
У тих випадках, коли молекула містить групу, наприклад карбоксильну, яка перешкоджає карбонієвому іону набути плоскої конфігурації, реакції 5дЧ відбуваються із збереженням конфігурації:
н
НООС—С+-------ОН'-1-”^0 .
СН2СООН
н
І + ноос—с + сі :
СН2СООН
СН2СООН
Контрольні запитання і вправи
І. Зобразіть фішерівські проекційні формули для всіх можливих оптичних ізомерів 1,2,3,4-тетрахлорбутану; 2,3-дибромпропанової кислоти; 3-бром-гександіолу-2,5; 2,3-дибромбутану.
2. Визначте відмінність у термінах структура, конформація, конфігурація. Який з цих термінів прийнятий для позначення взаємних переходів молекул без розриву або утворення зв’язків?
3. Порівняйте кислотні властивості молочної і пропіонової кислот; а-оксипропіонової і р-оксипропіонової кислот.
4. Які речовини утворюються при взаємодії молочної кислоти з оптовим ангідридом, хлороводнем, РС16, етанолом при наявності невеликої КІЛЬКОСТІ НдРОї, при окисленні молочної кислоти?
252
Розділ 24. АЛЬДЕПДО-І КЕТОКИСЛОТИ.
ТАУТОМЕРІЯ
Альдегіде- і кетокислотами називають органічні сполуки, у молекулах яких разом з карбоксильною групою одночасно міститься альдегідна або кетонна група. Отже, ці речовини належать до сполук з двома функціональними групами. У зв’язку з тим що карбонільна група називається ще оксогрупою, то альдегіде- і кетокислоти називають також оксокислотами.
Назви оксокислотпов’язані переважно з методами їх добування з природних джерел. Інколи ці речовини розглядають як похідні монокарбонових кислот. Наприклад, кислоту Н3С—С—СН2—СООН
називають ацетооцтовою. За систематичною номенклатурою назви оксокислот утворюють з назви відповідної монокарбонової кислоти, до якої додають префікс оксо-, і цифрою вказують положення карбонільної групи в молекулі. Так, вищезгадана ацетооцтова кислота за цією номенклатурою називається 3-оксобутановою кислотою.
Залежно від положення функціональних груп в молекулах оксокислот розрізняють а-, [3-, у- і т.д. альдегіде- і кетокислоти. Наприклад, ацетооцтова кислота є ^-кетокислотою.
Альдегідо- і кетокислоти, як і інші класи органічних речовин, утворюють гомологічні ряди. Гомологічний ряд альдегідокислот починається з гліоксилової кислоти, першою в гомологічному ряду кетокислот є піровиноградна кислота.
°\
Гліоксилова (рксоетанова) кислота ^С—СООН =
Ну
= 98 °С) міститься у недозрілих фруктах. Синтетично її добувають окисленням етиленгліколю або гліколевої кислоти, частковим відновленням щавлевої, а також гідролізом дихлороцтової кислоти.
Гліоксилова кислота дає всі реакції альдегідів і карбонових кислот.
У зв’язку з впливом —1- і М-ефектів карбонільної групи на карбоксильну
СП О
❖ ❖ с — с нх хо-н
гліоксилова кислота значно сильніша від оцтової, що підтверджують значення їх Ад (Ад О = СН—СООН =47 -10-6, Уд Н3С-СООН = 1,75 • 10-6).
Піровиноградна (ї-оксопропанова, або а-кетопропіонова) кислота Н;,С С СООН являє собою рідину (7’кип=165еС з розклада
253
нням). Цю кислоту вперше добув Й. Я. Берцеліус у 1835р. піролі-зом виноградної кислоти (звідки і пішла її історична назва):
НОООС—СН—СН—СООН
І І он он
------->
—со2, Н2О
Н2С=С—СООН1—> Н3С—С—СООН.
І II
он о
Піровиноградна кислота утворюється також при окисленні молочної кислоти, при гідролізі а, а-дихлорпропіонової кислоти або через нітрил.
Піровиноградна кислота дає всі характерні реакції кетонів і всі реакції карбонових кислот. Проте завдяки впливу «-карбонільної групи (—І- і —М-ефект)
О НзС-С-С4' г" V.
<о о — н
піровиноградна кислота значно сильніша від пропіонової (Кл Н3С — СО — СООН = 320 • 10-6, Н3С—СН2 — СООН =
= 1,34 • 10"Б).
Піровиноградна кислота є важливим проміжним продуктом в обміні вуглеводів, білків і жирів.
Ацетооцтова (3-оксобутанова, або $-кето масляна) кислота Н3С—СО—СН2—СООН (Тпл =37 °С) є найпростішою з 0-кето-кислот. Вона утворюється в організмах під час обміну жирних кислот, зокрема при їх Р-окисленні. У хворих на цукровий діабет ацетооцтова кислота разом з ацетоном виділяється з організму з сечею.
Ацетооцтову кислоту можна добути при обережному омиленні її ефірів або приєднанням води до дикетену:
+нон нон
НЯС—С—СН„—СООС.Н.-------► Н3С—С—СН,—СООН -<—- Н„С=С—СН,
8 ц 2 2 8 -с2н„он Ц II
О О О—с=о
Ацетооцтова кислота, як і інші Р-кислоти, нестійка і вже при слабкому нагріванні декарбоксилується і перетворюється на ацетон:
Н3С—С—СН2—СООН Н3С—С—снз + СО2.
II II
о о
Важливу роль у теоретичній органічній хімії і органічному синтезі відіграє етиловий ефір ацетооцтової кислоти, який називають переважно ацетооцтовим ефіром.
Ацетооцтовий ефіо Н8С—С—СН2—СООС2НВ — безбарвна рі-
II О дина з приємним запахом (ТКИп = 181 °С), яка змішується з органічними розчинниками і не розчиняється у воді. Ацетооцтовий ефір вперше добув А. Гейтер у 1863 р. нагріванням етилацетату з натрієм
254
і описав його як етиловий ефір р-оксикротонової кислоти Н3С—С=СН—СООС2Н6. Л. Клайзен (1877 р.) детально вивчив
ОН
цю реакцію і розробив метод складно-ефірної конденсації, а також встановив, що 2 моль етилацетату під впливом натрію або етилату натрію конденсуються і перетворюються на ацетооцтовий ефір, якому була приписана структура ефіру р-кетомасляної кислоти:
На або С2НБОМа
2Н3С—СООС2Н6 -----> Н3С—С—СН2—СООС2Н6 + С2НвОН.
II о
Ацетооцтовий ефір залежно від умов реакції реагує або як кетон (як похідна кетонокислоти) Н3С—С—СН2—СООС2Н5, або як енол
(як похідна пенасиченого спирту—еиолу) Н3С—С=СН—СООС2Н5.
І
ОН
Отже, ацетооцтовий ефір виявляє двоїсту реакційну здатність і дає реакції як кетонів, так і ненасичених спиртів (див. схему на с. 256).
У зв’язку з цим в органічній хімії протягом майже 50 років велася дискусія відносно будови ацетооцтового ефіру, яка формально закінчилася в 1911 р., коли Л. Кнорр розділив цей ефір і виділив з нього кетонну форму (І) у вигляді етилового ефіру ацетооцтової кислоти і енольну форму (II) у вигляді етилового ефіру [З-оксикро-тонової кислоти. Метод розділення цих форм полягав у охолодженні ацетооцтового ефіру до —78°С. Кетонна форма (І) при цьому кристалізувалася і виділялася у вигляді кристалів з 7’пл=—39 °С, а енольна форма (II) залишалася у вигляді маслянистої рідини. Енольна форма (II) з Тпл =—44 °С у чистому вигляді була добута дією сухого НС1 иа натрійацетооцтовий ефір.
Дещо пізніше К. Мейєр (1920 р.) розділив ацетооцтовий ефір на форми І і II перегонкою з кварцової посудини. Кварц, на відміну від скла, не викликає ізомеризації форми І у форму II і навпаки. Таку перегонку назвали «асептичною дистиляцією».
При підвищенні температури частина кетонної форми (І) поступово перетворюється на енольну форму (II), а чиста енольна форма (II), в свою чергу, частково перетворюється на кетонну форму (І). В результаті встановлюється рівновага між обома формами:
Н3С—С—СН2—СООС2Не Н3С—С=СН—СООС2Не.
II І
о он
І II
Після встановлення рівноваги при 20 °С у таутомерній суміші міститься 92,7 % кетонної форми (І) і 7,3 % енольної форми (II).
255
Реакції кетонної і енольноі форм ацетооцтового ефіру
Н3С-С-СН2-СООС2Н5 Н3С-С = СН-СООС2Н5
О ОН
Реакції кетонної форми ацетооцтового ефір. СN
НзС-С - СН2-СООС2Н5.
он
Оксинітрил 8О3№
н3с-с-сн2-соос2н3 н+-ОзМа ОН ------------------
Гідросульфітна похідна
н3с-с-сн2-соос2н5
Н-ннс6н3 Фенілгідразон Н3С - С- СН2 - СООС2Н 5
N - ОН Оксим
Н3С-С-СН3 О
Н3С — СН — СН2 —СОО2Н5 он
н2и-он
Розбавлений луг
Н,
[Н3С-СВг-СНВг-СООС2Н5]—-І “П ог
он
чЕтиловий ефір 2,3-дибром-3-оксимасляної кислоти
_________________РС1 5
Ацетооцтовий
ефір
СбН5-МН-МН2
Реакції енольноі форми ацетооцтового ефіру
На -----------» Н3С-С = СН-СООС2Н5
О№
Натрійацетооцтовий ефір Н3С-С = СН-СООС2Н5 о-ве^
Забарвлений комплекс Н3С-С-СНВг-СООС2Н, II О
СС-Бромацетооцтовий ефір
Н3С-С=СН-СООС2Н5
СІ
0 Етиловий ефір ^-хлоркротоново! кислоти НзС-С^
ЧСІ
Піридин
Етиловий ефір р -оксимасляної кислоти
НаС-С=СН-СООС2Н5 6 —СО —СН3
Етиловий ефір -ацетоксикротоново! кислоти
МН3
--------------- Н3С-С=СН-СООС2Н5
МН2
Етиловий ефір р -амінокротоновоі кислоти
Енольна форма являє собою г{цс-конфігурацію і утворює з іонами Ее3+ забарвлений комплекс (Па):
н3сч Ті
С=С
,Н3С —С = СН—СООС',11, 1 Ге3+ ---- б \-ОС,Н, .
ГІ ' ' \ II '
і ОН. Ее *- О
П Па
Наявність рівноваги між кетонною і енольною формами ацетооцтового ефіру досить легко довести експериментально. Так, найпростіший і легкодоступний такий дослід. При добавлянні до ацетооцтового ефіру розчину солі Ее3 + енольна форма дає забарвлену комплексну сполуку. Якщо до цього забарвленого розчину добавить бром (або бромну воду), то забарвлення зникає, оскільки енольна форма при цьому приєднує бром, а потім перегруповується в кетон— бромацетооцтовий ефір і таким чином виводиться з рівноважної суміші. Проте забарвлення в реакційній суміші досить швидко з’являється знову, оскільки частина кетонпої форми переходить (тауто-меризується) в енольну, яка дає з іонами Ее3+забарвлений комплекс:
РеЗ+
Н3С—С—СІІ2—СООСаІІ6 «=> Н3С—С=СН—СООС2Н5----->
II І
о он
Вг Вг
Вг2 | І
—Н..С—С=СН—С—ОС.Н5 —-> І ЦС—С—СН—соос,нБ-------->
І II І -нвг
о о он
—> 113С—С—СНВг—СООС2НВ.
II о
Таутомерія. Термін «таутомерія» (від грецьк. «таута» —той самий і «мерос» — частина) був введений в органічну хімію в 1885 р. Таутомерія —особливий вид ізомерії, який характеризується тим, що два ізомери зв’язані між собою рухомою рівновагою, тобто мають здатність переходити один в одного. Іншими словами, таутомерія — це оборотна, або динамічна, ізомерія двох структурних ізомерів. Ідею про динамічні перетворення органічних речовин вперше висловив О. М. Бутлеров у 1887 р.
Серед органічних сполук відомо багато вид в таутомерних перетворень. Назви таутомерним перетворенням Дають за назвою тих форм, у які ізомеризується даний таутомер. Так, вищерозглянутий ацетооцтовий ефір існує у вигляді кетонної і енольної форм (-єн і -ол —це за систематичною номенклатурою суфікси відповідно для етиленових вуглеводнів і спиртів). Тому така таутомерія називається кето-енольною, а ацетооцтовий ефір є класичним прикладом сполуки, здатної до кето-енольної таутомерії.
Механізм таутомерних перетворень кето-енольних систем. В ацетооцтовому ефірі атоми водню групи СН2 досить активні (рух
9 1*120
257
ливі), оскільки вони знаходяться під впливом двох карбонільних груп:
&+ 0 +
Н3С-С-ї-СН-С-ОС2Н5
С"
н3с-с = сн-с=о
Ої(Н9 ОС2Н5
Перетворення кетонної форми цього ефіру в енольну, а енольної в кетонну можна розглядати як молекулярне перегрупування, в якому рухливий атом водню метиленової групи кетоформи переміщується до кетонного атома кисню і утворює при цьому енольну форму. Активний атом водню енольного гідроксилу, в свою чергу, може приєднуватися до $/?2-гібридизованого вуглецевого атома етиленового зв’язку і утворювати кетонну форму ацетооцтового ефіру.
Кислі і основні каталізатори прискорюють процеси таутомери-зації і досягнення таутомерної рівноваги. Стан таутомерної рівноваги залежить від природи розчинника, температури і концентрації речовини. Полярні розчинники (вода, спирти) сприяють зміщенню рівноваги в бік кетонної форми, а в неполярних розчинниках (н-гексан) рівновага сильно зміщується в бік енольної форми.
Дані про вміст кетонної і енольної форм ацетооцтового ефіру в деяких розчинниках наведені в табл. 23. При підвищенні температури вміст енольної форми в розчинах переважно зменшується. Розбавляння розчинів приводить до збільшення в них кількості енольної форми.
Таблиця 23. Вміст кетонної і енольної форм ацетооцтового ефіру в розчинах (18 °С, концентрація 1—3 %)
Розчинник н2о н8с-он с2н„он свнв н-СвН„
Кетоформа, % Енольна форма, % 99,6 0,4 93,18 6,82 88,0 12,0 83,8 16,2 53,6 46,4
З даних термодинамічних розрахунків випливає, що внутрішня енергія нижча для кетонної форми. Різниця між значеннями внутрішньої енергії кетонної і енольної форм становить близько 75 кДж/моль. Тому ненасичені спирти з гідроксилом біля ^-гібридизованого атома вуглецю нестійкі і перегруповуються в енергетично стійкіші карбонільні сполуки (правило Ельтекова). Однак при утворенні енолу виникає спряжена система подвійних зв’язків і внутрішньомолекулярний водневий зв’язок, які стабілізують енольну форму:
.о.
хн
не /О
І
ОС2Н5
258
Виграш енергії за рахунок цих двох факторів компенсує деякою мірою затрату на утворення енолу, завдяки чому схильність до енолізації зростає. У тих випадках, коли група СН2 знаходиться між двома кетонними групами, як, наприклад, в молекулі ацетил-ацетону, енолізація досягає 80 %:
1І..С—С—СН,—С—СН3 НС3—С—СН=С—сн„. о & о о *>
(20 %)
(80 %)
С- і О-Алкілування ацетооцтового ефіру. Ацетооцтовий ефір виявляє слабко виражені кислотні властивості =7,1 • 10~ц, р/(к = 10,7). При взаємодії з етилатом натрію або з їдким натром він утворює кристалічний іонно побудований натрійацетооцтовий ефір, який містить амбідентний аніон:
Н3С—С—СН,—СООС,Н5 С^Н||О№ або КаОН ГН3С—С—СН—СООС,Н.
3 II 2 2 6-С2НвОНабо-Н2О 3 || 20
о о
Н3С—С=СН—СООСаНв"
о~
.Амбідентний аніон ацетооцтового ефіру має два сильних нуклео-фільних центри: карбаніонний атом вуглецю С~ і енолят-аніонний атом кисню О з кожним з них в ході реакції може утворюватися новий хімічний зв’язок. Обидва нуклеофільні центри зв’язані так, що реакція на одному з них зупиняє реакцію на другому. Залежно від природи реагенту і умов реакції амбідентний аніон ацетооцтового ефіру може утворювати два ряди похідних: С-похідні і О-похідні.
Отже, ацетооцтовий ефір виявляє двоїсту реакційну здатність, яка є однією з ознак таутомерії речовини.
Поняття про двоїсту реакційну здатність було розвинуте і сформульоване в роботах О. М. Несмеянова і його співробітників починаючи з 1950 р. Так, при взаємодії натрійацетооцтового ефіру з первинними або вторинними галогеналкілами реакція відбувається з участю С-нуклеофільного центру і утворенням С-алкіл-ацетооцтових ефірів. Наприклад, реакція з йодистим метилом дає метилацетооцтовий ефір. Метилацетооцтовий ефір містить активний атом водню в групі СН, який також може заміщуватися на натрій. Натрійметилацетооцтовий ефір при взаємодії з йодистим метилом дає диметилацетооцтовий ефір, у молекулі якого всі атоми водню метиленової групи заміщені на метальні радикали, тому для нього таутомерія не характерна і він вже не взаємодіє з етилатом натрію
9*
259
і їдким натром:
сн3і
-Н3С-С-СН - СООС2НБ І і\а+ —
О ]
Н3С—С-СН-СООСДК
II І
о сн3
<ДН,,ОІ\'а
-с2нвогГ
СН3 н3сі |
ГН3С-С-С-СООС2Н61№+ * Н3С-С-С-СООС2НБ. II І 'л II І
О СН8 о сн3
Ди метил ацетооцтовий еф і р
Якщо на натрійацетооцтовий ефір подіяти хлористим ацетилом, то реакція відбувається за 5Л-2-механізмом і в результаті утворюється С-ацетильна похідна ацетооцтового ефіру:
Н3С—С—СН—СООС2Н51К'а + Н3С—С—СІ -> Н3С—С—СН- СООС2Н6+ ИаСІ.
II II II І ‘ '
О ] О О СО—СН3
Етиловий ефір сі-ацетил-р-кетом аслякої кислоти
Якщо ж взаємодію натрійацетооцтового ефіру з хлористим ацетилом проводити в розчині піридину, то реакція відбувається за 5,у1 -механізмом з участю О“-нуклеофільного центру амбідентного аніона: піридин
ГН3С—С=СН—СООС2Н5' - -----
І О-
—-> Н3С—С=СН—СООС2НБ + Масі.
о—со—сн8
Етиловий ефір р-ацетоксикротонової кислоти
Кетонне і кислотне розщеплення С-алкільних заміщених ацетооцтового ефіру. Ацетооцтовий ефір і його С-алкільні заміщені при нагріванні з лугами або кислотами піддаються розщепленню. Залежно від концентрації лугу або кислоти можливе кетонне чи кислотне розщеплення цих речовин. Така властивість С-алкільних заміщених ацетооцтового ефіру використовується в органічному синтезі для добування кетонів і карбонових кислот.
Кетонне розщеплення відбувається при нагріванні ацетооцтового ефіру і його С-заміщених з розбавленими розчинами лугів або кислот (наприклад, Н2ЗО4). При такому гідролізі спочатку утворюється (3-кетокислота, яка легко декарбоксилується і перетворюється на кетон. З ацетооцтового ефіру в цих умовах утворюється ацетон, а із С-заміщених ацетооцтового ефіру — гомолога ацетону:
н3о+
—с,нвон
ЩО. КаОН
—С2Н,ОН
н3с—с—сн—СООС2НБ—
II І
о к
260
-> Н3С—С-СН—СООН —Н3с—С—СН,-К;
II І -СО* II
О Л О
н+
> Н3С—С—СН—СОО.Ма —-> Н3С—С—СН2—/?.
Аналогічно, виходячи з С-ацилзаміщених ацетооцтового ефіру, можна добути дикетони. Наприклад:
що+
Н3С—С—СН—СООС2НБ -- н3с—с—сн—соон —
ц । —с,щон ц । —со,
О СО—СІІ3 О СО—СН3
—> Н3С—С—СН„—С—СН3.
II II
о о
Ацетилацстон
Кислотне розщеплення відбувається при нагріванні ацетооцтового ефіру і його С-алкільних заміщених з концентрованими розчинами лугів. У таких умовах крім омилення складиоефірної групи відбувається відщеплення ацетильної групи. В результаті утворюються солі карбонових кислот, з яких при підкисленні добувають карбонові кислоти. Ацетооцтовий ефір за цих умов утворює оцтову кислоту, а його С-алкільні заміщені —-оцтову і іншу карбонові кислоти:
Н3С—С II О N<10
СН2—СООС2Н-ін І
2НС1
Н3С—С—ОК'а + Н3С—С—О№ -------
3 II II ~2№‘
О О
2КаОН
—С,Н,ОН
А ЗК'аОН 2І1С1
Н3С-С ;СН—СООС.Н, ------>1І3С—С-О№ + /?—СН,—С00№ —-»
|| і 26 -С,ЩОН 2 ц Г ’ -2КаС1
О О
—> Н3С—СООН -І- Я—СН2—СООН.
Кислотне розщеплення С-заміщених ацетооцтового ефіру є одним з методів добування моно- і дикарбонових кислот. Добираючи відповідні реагенти, з ацетооцтового ефіру можна добути різноманітні кетони і карбонові кислоти, в тому числі і дикетони та дикар-бонові кислоти. Наприклад, при дії на натрійацетооцтовий ефір йоду утворюється діетиловий ефір діацетил’янтарної кислоти, який при кислотному розщепленні дає янтарну кислоту, а при кетонно-
261
му — дикетон гександіон-2,5 (ацетонілацетоп):
2С,НаОКа —
2Н3С-С-СНа—СООСаНв > 2 ’Н3С-С-СН-СООСаН6-| і\'а+
ц —2с2гі8игі ц і
О І о ]
Н3С—С—СН—СООСаН8 II о
1І3С—С—СН—СООСаІ І3
Кислотне розщеплення
Кегониє роз1ЦЄ! 1Леиня
, 2Н3С—СООН 4- НООС—СН2-СІ І2—СООН;
г н3с—с—сна—сна—с—сн3.
II II
о о
Ацетон і л ацетон
Розділ 25, АМІНОКИСЛОТИ
Амінокислотами називають органічні сполуки, молекули яких одночасно містять карбоксильну і змінну групи. В молекулах цих речовин можуть знаходитися одна, дві або більше карбоксильних груп і одна, дві або більше аміногруп. У зв’язку з цим амінокислоти класифікують за кількістю карбоксильних груп (монокарбонові, дикарбонові і т. д.) і за кількістю аміногруп (моноаміно-, дізміно-і т. д.). Залежно від будови вуглецевого ланцюга амінокислоти поділяють на ациклічні і циклічні, а залежно від взаємного розміщення змінної і карбоксильної груп розрізняють а-, р-, у-, 6-і т. д.амінокислоти.
Амінокислоти, як і інші класи органічних речовин, утворюють гомологічний ряд, який можна записати виходячи з гомологічного ряду карбонових кислот. Для цього необхідно у вуглеводневому радикалі карбонової кислоти замінити один або більше водневих атомів на аміногрупу:
Карбонова кислота
Н—СООН — мурашина НаС—СООН — оцтова Н#С-«-СН4—СООН — пропіонова
Н8С—СНа—СНа—СООН - масляна
Амінокислота
Н21\’—СООН — аміномурашнна, карбамінова
Н2Н—СН2—СООН — амінооцтова, аміноетанова, гліцин
Н3С—СН—СООН — а-амінопропіонова, 2-амінопропано-| ва, а-аланін
ИН2
Н2И—СНа—СНИ—СООН — р-амінопропіонова, 3-аміно-пропанова, р-аланін
НаС—СН2—СН—СООН — сс-аміномасляна, 2-амінобута-| нова
ЬІН8
НаС—’СН—СН2—СООН р-аміномасляна, 3-амінобута-
| нова
КН2
Н а Н-СНа-»СНа-СНа-СООН =« у- ам І ном ас л я на, 4 - ам Ь нобутанова
262
Як видно з наведеного ряду сполук, гомологічний ряд монокар-боиових амінокислот слід було починати з аміномурашиної кислоти. Проте оскільки ця кислота одночасно є неповним амідом вугільної кислоти (карбамінова кислота), то вона вже розглядалася вище (див. розд. 19) при вивченні похідних вугільної кислоти.
Амінокислоти часто називають за назвою відповідної карбонової кислоти, до якої додають слово аміно. Положення аміногрупи відносно карбоксильної позначають грецькими буквами «, р, у і т. д., наприклад, «-, р-і у-аміномасляна кислота. За систематичною номенклатурою назву амінокислоти утворюють з назви відповідного вуглеводню, до якої додають префікс аміно-, закінчення -ова і слово кислота. Місцеположення аміногрупи позначають цифрою. Нумерацію вуглецевого ланцюга починають з атома вуглецю карбоксильної групи.
Амінокислоти досить поширені в природі. Так, «-амінокислоти є складовими частинами білків, беруть участь у найважливіших біологічних процесах і тому відіграють важливу роль у процесах життєдіяльності організмів. У наш час з гідролізатів білків виділено понад 20 «-амінокислот, будову деяких з них наведено в табл. 24. Для а-амінокислот, виділених з білків, у практику ввійшли їх історичні назви.
Амінокислотам, як і іншим органічним сполукам, властива ізомерія, яка для цих речовин може бути пов’язана з різним положенням аміногрупи відносно карбоксильної (наприклад, а-, р- і у-амі-номасляні кислоти), а також з розгалуженням вуглецевого ланцюга молекули амінокислоти. Таким ізомером а-, р- і у-аміномасля-них кислот є 2-аміно-2-метилпропанова кислота
сн3
Н3С—с—соон.
ЬІН2
Амінокислоти, молекули яких містять асиметричні вуглецеві атоми, виявляють оптичну активність і існують у вигляді дзеркальних ізомерів, наприклад «-амінопропіонова кислота:
СООН СООН
Н—С*—ИН2; Н2ЬІ—С*—Н.
І І
СН3 сн,
О (“-)-Аланін І- (+)-Аланін
Добування амінокислот. «-Амінокислоти А-ряду поширені в природі, Вони є складовими частинами білків. Амінокислоти П-ряду добувають шляхом синтезу. Із синтетичних методів добування амінокислот найважливіші такі:
1. Мікробіологічний синтез відбувається у клітинах мікроорганізмів або поза ними під впливом ферментів, які виділяють мікроорганізми . В наш час навчилися так спрямовувати життєдіяльність
263
Таблиця 24. Будова деяких амінокислот, виділених з білків
Формула Назва Формула Назва
Н2И—СН2—СООН Гліцин НООС— (СН2)2—СН—соон ин2 Глутамінова кислота
н3с—сн—соон іін2 Аланін НаИ—(СН2)4—сн—соон НН, Лізин
н2с—сн—соон 8Н ІШ2 Цистеїн Н2М-(СН.2)3-СН-С00Н \ н, Орні тин
Н2С—сн—соон он ^н2 Серин /ОУ<н2-сн _ соон \' ин2 Фенілаланін
н„с—сн,—сн—соон 1 1 8—СН3 НН2 Н3С—сн—сн2—сн—соон ін3 >?н2 НООС—сн2—сн—соон іін2 Метіонін Лейцин Аспарагінова кислота Н 0-\О7~СН 2 - СН " С00Н ' ' МН2 Тирозин Триптофан
Р її || '•-Л12 І ) ^Н2 NN
деяких мікроорганізмів, що вони не нагромаджують білок, а синтезують певну амінокислоту. Отже, з нехарчової сировини (вуглеводні нафти, газоподібні вуглеводні) за допомогою мікроорганізмів добувають у промислових масштабах лізин, глутамінову кислоту та деякі інші амінокислоти.
2. Гідроліз білків. Природним джерелом добування амінокислот є білкові речовини. Під час їх гідролізу утворюється суміш амінокислот, з якої відповідними методами виділяють окремі амінокислоти. Білки—це високомолекулярні речовини, макромолекули яких складаються із залишків а-амінокислот, сполучених між собою амідними (пептидними) зв’язками —ІЧН—С—. У загальному
вигляді частину молекули білка можна схематично зобразити так:
... —МН—СН—С—МИ—СН—С—МН—СН—С— .... І II І II І II /?х О /?2 О #3 О
де/?,,/?,, /?3 можуть бути Н,СН3,СН2—8Н та інші групи атомів. У процесі гідролізу білків амідний зв’язок розривається і при цьому утворюються відповідні амінокислоти:
і । і ।
... — ІМН—Сі 1—С — М’Н-СН—С !—ИН—сн—с —.
І І II І І II І II І
І /?! ОІ /?., О А’3 ОІ
І І ' І І
но|н но|н но|н но|н
Н2М—СН—СООН + Н2М—СН—СООН + Н„М—СН—СООН 4-..
І І І
/?1 /?2 /?з
У промислових масштабах гідролізу піддають відходи переважно м’ясної промисловості (кров, роги, копита).
Гідролізом білків добувають цистеїн, лейцин, ізолейцин.
3. Дія аміаку на солі а-галогенокарбонових кислот-.
Н2МІН + СІІ—СН2—СООМНа Н2М—СН2-СООМН4 + неї -* Н2М-СНа—СООН + МН4С1.
Спочатку в цій реакції утворюється амонійна сіль амінокислоти, яка далі взаємодіє з хлороводнем, що утворюється в процесі реакції і перетворює дану сіль на амінокислоту.
4. Дія аміаку і синильної кислоти на альдегіди або кетони (реакція Штрекера). В умовах цієї реакції карбонільна сполука спочатку взаємодіє з аміаком і утворює імін, який потім приєднує
265
ціановодень і перетворюється таким чином на нітрил «-амінокислоти. Під час гідролізу такого нітрилу добувають «-амінокислоту:
Н3С—С^° Н3С-СН=^Н ±н-.с> Н3С—СН— СН -н.г°-н* >
Ацетальдімін | ^Н3
ИН2
Нітрил аміно лропіонової кислої и
—*Н3С-~СН~СООН.
І
кно
М. Д. Зелінський вдосконалив цей метод добування а-амінокислот, застосувавши в реакції замість синильної кислоти суміш ціаніду калію і хлориду амонію.
5. Приєднання аміаку до ненасичених кислот (див. розд. 21).
Для синтетичного добування окремих амінокислот використовують спеціальні методи. Так, є-амінокапронову кислоту добувають, виходячи з фенолу за такою схемою:
СН, н,(г ^с=о н,с сн2
Циклогексаном
+ Н2К-ОН
^СНг
Н2СГ С=М-ОН Н28О4
Н2С СН, Бекманівське
\ перегрупування
'2
Оксим
циклогексанону
Капролактам (лактимла форма)
нон
сн2—сн2
Н2Ц - (сн2)5- соон.
6-Амінокапронова кислота
Капролактам (лактамна форма)
Фізичні властивості. Амінокислоти — безбарвні кристалічні речовини з високими температурами плавлення, які мало відрізняються для різних амінокислот. Тому температура плавлення не є характерною константою для цих речовин. Під час плавлення амінокислоти переважно розкладаються. Більшість амінокислот добре розчиняються у воді, гірше — в органічних розчинниках. а-Аміно-кислоти £-ряду, що входять до складу білків, гіркі на смак або не мають його, а їх £)-ізомери —солодкі на смак.
Хімічні властивості. Оскільки в молекулах амінокислот одночасно містяться карбоксильна і амінна групи, то ці речовини виявляють властивості як кислот, так і амінів, тобто є амфотерними сполуками. У вільному стані і в розчинах амінна та карбоксильна
групи в молекулах амінокислот взаємодіють між собою. Тому амінокислоти являють собою внутрішні солі (біполярні іони):
+. І +
Н2КІ—СН,—С— О~Н’ -> Н3М—СН2—С—О-.
II II
О о
Підтвердженням такої біполярної структури амінокислот є високі температури їх плавлення, відсутність у спектрах ліній, характерних для карбоксильної і змінної груп, а також деякі інші властивості амінокислот, зокрема нейтральність водних розчинів моноаміномонокарбонових амінокислот. Індикатори в таких розчинах не змінюють свого забарвлення.
Амінокислоти є типовими амфотерними сполуками. У кислому середовищі вони виявляють властивості основ, у лужному —кислот:
ІІ3ЬГ—СН2—С—О -|- Н+ Н3Г<—СН2—С—011 (кисле середовище);
II II
О О
-I-
ІІ3\'—СН2—С—0“-|-0П~-♦ Н2\’—СН,—С—О -|-Н20 (лужне середовище).
II II
О О
Якщо концентрації катіонів і аніонів у розчині амінокислоти однакові, то концентрація біполярного іона буде максимальною і в електричному полі він не переміщується. Такий стан амінокислоти називається її ізоелектричною точкою. Ізоелектрична точка амінокислоти характеризується певним значенням рН середовища.
Утворення солей. Оскільки амінокислоти мають амфотерний характер, вони можуть утворювати солі як з кислотами, так і з основами:
..* “Г
Н2Ї\Т—СН2—СООН + ТГС1 -> СГМН3—СН2—СООН;
І Хлористоводнева сіль
гліцину
Н.М—СН2—СООН -І- МаОН -» Н2И—СН,—СООИа + Н20.
Натрієва сіль гліцину
- х
Амінокислоти взаємодіють з гідроксидами важких металів, розчиняють їх і утворюють при цьому комплексні сполуки. Так, з гідроксидом міді амінокислоти утворюють забарвлені в темно-синій або синьо-фіолетовий колір комплекси:
2Н2К’-СН2-
С-ОН + Си(ОН),
Мідна сіль гліцину
У цій солі атом міді сполучений з атомами азоту координаційними (донорно-акцепторними) зв’язками.
267
Утворення складних ефірів. Амінокислоти, подібно до карбонових кислот, взаємодіють із спиртами і утворюють складні ефіри:
/°
Н Л— СН,—+ С2Н5ОН -> НЛ—СН,-С^ + Н,О.
' хон хос2нв
Етиловий ефір гліцину
Утворення хлорангідридів. Амінокислоти при взаємодії з пентахлоридом фосфору утворюють хлорангідриди (у вигляді гідрохлоридів):
О О
НЛ—СН2—+ РСІ5 -> НЛ—СН2—+ неї + Р0Сі3 ->
4 ОН ХС1
Хлорангідрид гліцину
СГНЛ—СН2—• \сі
Гідрохлорид хлорангідриду гліцину
Ацилування амінокислот. У молекулах амінокислот, як і амінів, атом водню аміногрупи може заміщуватися на залишки карбонових кислот (ацили):
,О
І !ас—с/ + НЛ-СН2—СООН --> Н3С—С-ИН—СН2-СООН + Н2О.
хсі II
Хлористий О
ацетил Ацетилгліцин
Дія азотистої кислоти. Під дією на амінокислоти азотистої кислоти виділяється азот аміногрупи і амінна група при цьому заміщується на гідроксогрупу. Продуктами цих реакцій є оксикислоти:
НЛ—СН2—СООН -І- НОНО -* НО—СН2—СООН + И2 + Н2О.
Гліколева кислота
Реакція амінокислот з азотистою кислотою використовується для кількісного аналізу вмісту амінокислот (за об’ємом азоту, який виділяється, методВан-Слайка).
Алкілування амінокислот. При алкілуванні амінокислот, залежно від кількості взятої алкілуючої речовини, утворюються вторинні, третинні алкіловані амінокислоти, а в кінцевому результаті —четвертинні амонійні основи, внутрішні солі яких називають бетаїнами:
СН3
+ +сня1 4- ч-енд +1 +сн31
Н,К—СН,—СОО~ Н2М—СН2СОО~---------> НН—СН2—СОО"------>
3 2 —ні 21 2 —ні । 2 —ні
СН3 СН,
К-Д’етилгліции N. ЬІ-Дим етил гліцин
---> (СН3)3М—СН2—СОО".
N. ЬІ, М-Тримегил гліцин,
бетаїн
268
Перетворення амінокислот при нагріванні. Залежно від взаємного розміщення аміногрупи і карбоксильної групи амінокислоти по-різному реагують на нагрівання, що використовують для встановлення їхньої будови.
а-Амінокислоти при нагріванні відщеплюють воду (міжмолекулярно) і утворюють циклічні аміди — дикетопіперазиніг.
НМІН нок н2с" с=о
о=с__________ ^сн2
!_ОН_
NN ^С=О
І + 2І12О.
о=с сн2
Дикетопіперазин
(і-Амінокислоти при нагріванні відщеплюють аміак (внутрішньо-молекулярно) за рахунок аміногрупи і атома водню СН2-групн, сусідньої з СООН-групою:
Н,К'—СН„—СН,—СООН -> Н,С=СН—СООН 4- N11...
у-, 6- і е-Аміпокислоти при нагріванні відщеплюють воду (внутрішньомолекулярно) за рахунок зближених у просторі ЬІН2-і СООН-груп. При цьому утворюються циклічні внутрішньомолеку-лярні аміди — лактами-.
.О
н,с—<_ __ не—с=о
н2с7' : ‘1 + н2о.
' \ |Н___І \
Н2С—Н2С ?ЛІ
Н Лактам 7 -аміномасляної
кислоти, бутиролактам
Атом водню ЬІН-групи лактамів виявляє підвищену рухливість. Внаслідок цього лактами можуть існувати як в лактамній, так і в лактимній (енольній) таутомерних формах, які зв’язані між собою рухомою рівновагою:
н2с—с=о^х
Н2С І
\ /
Н2С---К'-Н-
Бутиролактам, лактамна форма
н2с—с-о-н / І
Бутиролактам, лактимна форма
Тому лактами виявляють двоїсту властивість і можуть утворювати якМ-, атак і О-похідні, тобто похідні як лактамної, так і лактимної форм. Такого типу таутомерія називається лактам-лактимною.
Де кар б оксидування амінокислот. У живих організмах а-амінокислоти піддаються різноманітним перетворенням, зокрема декарбоксилуванню. При цьому утворюються аміни, які виявляють високу біологічну активність, тому їх назива
269
ють біогенними амінами.Так, при декарбоксилуванні глутамінової кислоти утворюється у’-аміномасляпа кислота:
НООС—СН—СН,СН,СООН —г> НЛ—СН,—СН,—СН,—СООН + СО».
| т-Аміномасляна кислота
ин2
у-Аміномасляна кислота міститься в мозгсвій тканині і виконує в організмі функції нейрогуморального інгібітори.
Біогенні аміни утворюються також при декарбоксилуванні таких амінокислот білків, як аспарагінова кислота, тирозин, триптофан, гістидин та ряду інших.
Пептиди. Пептиди—це аміди, що утворюються внаслідок взаємодії аміногруп і карбоксильних груп амінокислот. Амідна група —МН—С— у таких сполуках називається пептидним зв'яз
ком. Залежно від кількості амінокислотних залишків у молекулі пептиди поділяють на дипептиди (два залишки амінокислот), три-пептиди (три залишки) і т.д., аж до поліпептидів (багато залишків амінокислот). Поліпептидами домовилися вважати пептиди з молекулярною масою до 10 000, а пептиди з більшою молекулярною масою вважають білками.
Утворення пептидів можна уявити як результат взаємодії карбоксильної групи однієї амінокислоти з амінною групою іншої:
нл—сн2—с—!он + н| N н—сн—с—он -* нл-сн2-с- №1 - сн - с - он; II ----------------- III II і II
О СН3 о о СН3 о
Гліцин Аланін- Дипептид гліцил-аланін
Н2№ СН2-С- МН - сн - с £он + н]мн - сн -с - ОН -
о СН3 О С»2 О
Дипептид1 54
Цистеїн-
----.н2м-сн2-с-мн-сн-с-М'н-сн-с-он + Н2О •
О СН. О сн2 о 8Н
Трипептид гліцил-аланіл-цистеїн
Назву пептидів утворюють з назв амінокислот, що входять до їх складу. При цьому всі амінокислоти в пептиді за винятком останньої амінокислоти, яка має вільну карбоксильну групу, змінюють у закінченні назви букву «н» на букву «л». Наприклад, вище-наведений дипептид має назву гліцил-аланін, а трипептид — гліцил-аланіл-цистеїн. Синтетичні методи добування поліпептидів розробив німецький вчений Е. Фішер із співробітниками. Одним з таких методів є взаємодія хлорангідридів а-галогенокарбоно-вих кислот з «-амінокислотами:
270
Н3С—СН—С—!С1“+ НІN11—СН2—С—ОН 357Г
І II '-------' II
Вг О О
2МІЯ
---> Н3С—СН—С—N11—СН2—С—ОН ———>
І II II -М1‘ВГ
Вг О О
—> Н3С—СН—С—НН—СН2—С—ОН.
І II II
МН2 о о
Дипептид аланіл-гліцин
За цим методом Е. Фішер і Е. Абдергальден синтезували поліпептиди, до складу яких входило 18—19 залишків амінокислот. Синтетичні поліпептиди за деякими властивостями подібні до природних речовин —білків.
Поліамідні полімери. Поліамідні високомолекулярні сполуки, як і білки, містять амідну групу атомів —N14—С—, яка сполучає
окремі ланкиполімерноголанцюга.Однимзтаких поліамідів єнап-лон, розглянутий вище (див. розд. 22). До синтетичних поліамідних полімерів належать також капрон і енант.
Капрон добувають шляхом полімеризації капролактаму, яку здійснюють нагріванням його при наявності невеликої кількості води:
сн2-сн2-с=о
п сн2 | -------► МН —(СН2)5—с-]д
сн-2— СН2—ИН о
Капролактам Капрон
Капрон використовують для виготовлення хімічного волокна з такою самою назвою, а також для виготовлення різних деталей машин і механізмів—шестерень, гайок, втулок, стійких до дії мастил і проти корозії.
При поліконденсації аміноенантової кислоти добувають полімер, з якого виготовляють хімічне волокно енант. Поліконденсація аміноенантової кислоти відбувається за схемою:
4 Н2\'—(СИ2)в—С—ІОН + 4 Н|МН—(СН2)„-С—он ->
2 II І_____2__| II
о о
МН—(СН2)в—С—1 + лН2О. II о
Енант
п
Волокно енант порівняно з капроновим волокном має значно вищу термо-, світло- і хімічну стійкість, а також приблизно в два
271
рази стійкіше від капронового волокна до багаторазових деформацій і стирання.
Енант використовують для виробництва різних трикотажних виробів і корду.
Розділ 26. ВУГЛЕВОДИ
Органічні речовини, які відносять до класу вуглеводів, широко розповсюджені в живій природі. Представниками вуглеводів є виноградний цукор (глюкоза), фруктовий цукор (фруктоза), буряковий і тростинний цукор (сахароза), крохмаль, целюлоза та ряд інших. У результаті процесу фотосинтезу рослинами щорічно на нашій планеті синтезується велика маса вуглеводів, яка оцінюється вмістом вуглецю, що становить 4 • 1010 т. Близько 80 % сухої речовини рослин припадає на вуглеводи. В зерні, картоплі, овочах, плодах вуглеводи є резервною поживною речовиною.
Вуглеводи входять до складу їжі і є одними з найважливіших харчових продуктів людини. Потреба людини в енергії покривається при харчуванні значною мірою якраз за рахунок вуглеводів. Поряд з білками, нуклеїновими кислотами і ліпідами вуглеводи є необхідною складовою частиною організму людини і тварин, де вони виконують важливі біологічні функції. Деякі складні білки, нуклеїнові кислоти, тобто речовини, які тісно пов’язані з процесами життєдіяльності, також містять вуглеводи.
Назву «вуглеводи» запропонував у 1844 р. К. Шмідт на тій основі, що ці речовини за складом можна формально розглядати як сполуки вуглецю з водою, наприклад, С6Н12ОВ, або 6С • 6Н2О. Тепер добре відомо, що вуглеводи —це не гідрати вуглецю, але стара назва залишилася. Крім того, існують вуглеводи з іншим співвідношенням С, Н і О.
Вуглеводи поділяють на дві великі групи: прості вуглеводи, або моносахариди (монози), і складні вуглеводи, або полісахариди (поліози). Простими називають такі вуглеводи, які не піддаються гідролізу з утворенням простіших сахаридів. Хімічний склад простих вуглеводів можна подати загальною формулою С„Н2„О,г, де п = 4, 5, 6, 7 і т. д. Складні вуглеводи здатні гідролізуватися і утворювати при цьому прості сахариди. Полісахариди, в свою чергу, поділяють на низькомолекулярні, або олігосахариди, і високо-молекулярні, або полісахариди. Олігосахариди (від грецьк.волі-гос» — небагато) утворені з невеликої кількості залишків молекул моносахаридів — від двох до десяти. Найпростішими оліго-сахаридами є дисахариди, молекули яких складаються з двох залишків моносахаридів. Полісахариди складаються з великої кількості залишків молекул моносахаридів.
Моносахариди
Хімічний склад моносахаридів описується загальною формулою С„Н2пО„. Назву моносахаридів утворюють з грецької назви числівника, який вказує кількість вуглецевих атомів у молекулі моноса-
272
хариду, і закінчення -оза. Так, моносахариди, в молекулі яких є п’ять С-атомів, називають пентозами, моносахариди, молекули яких мають шість вуглецевих атомів, називають гексозами і т. д.
Молекули моносахаридів містять від 4 до 10 атомів вуглецю, проте найбільше значення мають моносахариди з 5 і 6 атомами вуглецю, тобто пентози С5Н10О5 і гексози С6Н)2О6, а з гексоз —глюкоза (виноградний цукор) і фруктоза (фруктовий цукор). Структурна формула глюкози виведена на основі її хімічних властивостей:
Висновки
Експериментальні факти
Глюкоза — багатоатомний спирт;
Глюкоза містить:
п’ять ОІІ-груп;
альдегідну групу;
нерозгалуженип ланцюг з шести вуглецевих атомів
Враховуючи всі ці експериментальні факти, а також те, що дві гідроксильні групи не можуть заходитися біля одного атома вуглецю (правило Ерленмейєра), дійшли висновку, що глюкоза — п’ятиатомний альдегідоспирт, і приписали їй таку формулу (А. Байєр, Р. Фіттіг, 1870—1871 р.):
6 5 4 3 2 1 хО
СН2—СН— СІ І—СІ і—сн—с-с
І І І І І хн.
ОН ОН ОН 011 оп
Аналогічно було встановлено, що і в молекулі фруктози міститься п’ять ОН-груп, а шість атомів вуглецю її молекули також утворюють иерозгалужений вуглецевий ланцюг.Проте фруктоза, на відміну від глюкози, окислюється в жорстких умовах. При такому окисленні вуглецевий ланцюг її молекули розривається після другого вуглецевого атома і утворюються щавлева НО—С—С—-ОН
II II
О О
і винна НО—С—СН—СН—С—ОН кислоти. Таке окислення ха-II І І II О ОН ОН О
рактерпе для кетонів (див. розд. 17). Оскільки при окисленні фруктози утворилося дві кислоти — щавлева, яка містить два С-атоми, і винна, яка містить чотири С-атоми, то було зроблено висновок,
273
що кетонна група в молекулі фруктози знаходиться біля другого вуглецевого атома. На основі цього фруктозі приписали будову п’ятиатомного кетоноспирту:
6 5 4 3 2 1
СН,—СН— сн-сн—с—сн,.
І І І І II І
он он он он о он
Класифікація. Моносахариди поділяють на групи залежно від: 1) кількості атомів вуглецю в молекулі; 2) наявності в них альдегідної або кетонної групи; 3) конфігурації їх молекул, тобто від просторового розміщення груп атомів біля найвіддаленішого від карбонільної групи асиметричного атома вуглецю. Так, залежно від кількості вуглецевих атомів моносахариди поділяють на тетро-зи, пентози, гексози і т. д. Разом з тим моносахариди, які містять альдегідну групу, відносять до групи альдоз (альдегід-}- оза), а моносахариди з кетонною групою—до групи кетоз (кетон-\-оза). Глюкоза є альдогексозою, оскільки в її молекулі є альдегідна група і шість вуглецевих атомів. Фруктоза є кетогексозою.
Ізомерія. Для моносахаридів характерні кілька видів ізомерії.
1. Ізомерія, зумовлена наявністю альдегідної або кетонної групи. Прикладом такого виду ізомерії є глюкоза і фруктоза. Вони мають однакову молекулярну формулу —СсН]2О6, але відрізняються тим, що глюкоза містить альдегідну групу, а фруктоза — кетонну.
2. Ізомерія, зумовлена наявністю асиметричних атомів вуглецю. У молекулі альдогексози (наприклад, глюкози) і кетогексози (наприклад, фруктози) є асиметричні (сполучені з чотирма різними атомами або групами атомів) вуглецеві атоми. Вони в хімічних формулах позначені зірочками:
СН..-СН—СН—СН—СН-С^°; СН,-СН-СН—СН-С—СН2.
І І ! І І ХН І ' І І І II І
ОН ОН ОН ОН ОН ОН он он ОН о он
Як видно з наведених формул, альдогексоза має чотири асиметричних вуглецевих атоми, кетогексоза — три. Отже, глюкоза і фруктоза є оптично активними речовинами, вони обертають площину поляризації світла. Тому вищенаведені хімічні формули належать не тільки глюкозі і фруктозі, а і їх оптичним ізомерам.
Між кількістю асиметричних атомів вуглецю і числом просторових оптичних ізомерів існує математична залежність N = 2" (де N — кількість оптичних ізомерів, п — число асиметричних вуглецевих атомів). На основі цієї формули легко обчислити, що для альдогексози, яка має чотири асиметричних атоми вуглецю, повинно існувати 16 просторових оптичних ізомерів (24 — 16). Усі 16 просторових ізомерів альдогексози відомі. Одним з цих ізомерів є глюкоза. Вісім з цих 16 ізомерів мають £)-, тобто праву конфігурацію, а вісім —£-, тобто ліву. Для кетогексози, яка містить три асиметричних атоми вуглецю, повинні існувати 8 просторових ізомерів (23 =8). Чотири з них мають О-конфігурацію, а чотири — £-конфігурацію. Одним з цих ізомерів є фруктоза.
274
У молекулі природної глюкози гідроксильні групи біля асиметричних вуглецевих атомів розміщуються в просторі так: біля другого, четвертого і п’ятого вуглецевих атомів —справа, а біля третього —зліва:
/О /О
рн рн
Н—С*—ОН НО—с*—н
І І
НО—С*—Н Н—с*—он
І І
н—с*—он но—с*—н
Н—с*—он но—с*—н
І і
сн2—он СН2ОН
Д (Ч-)-Глюкоза 1 (—-)-Глюкоза
Отже, природну глюкозу можна було б назвати, враховуючи конфігурацію (розміщення ОН-груп біля всіх асиметричних атомів вуглецю), Р, Ь, О, Р-глюкозою, а її дзеркальне відображення (антипод), добутий синтетичним способом, — Ь, Р, Ь, Ь-глюкозою. Проте в назвах моносахаридів не зазначають конфігурації всіх асиметричних атомів вуглецю, а беруть до уваги лише конфігурацію найвіддаленішого від карбонільної групи асиметричного атома вуглецю (в молекулах альдо- і кетогексоз —п’ятого) і порівнюють її за пропозицією М. А. Розанова з конфігурацією гліцеринового альдегіду (див. розд. 23):
Н—С*—ОН; І СН2ОН
Р(4-)-ГліцериновнГі альдегід
но—с*—н.
І
СН2ОН
£(—)-Гліцериновий альдегід
Якщо моносахарид можна добути з Р-гліцеринового альдегіду шляхом надбудови вуглецевого ланцюга зверху над асиметричним вуглецевим атомом, тобто з того боку, з якого знаходиться альдегідна група, то такий моносахарид відносять до Р-ізомерів (до речовин Р-ряду). Нас. 277 наведено схему генетичного взаємозв’язку О-гліцеринового альдегіду, тетроз, пентоз і гексоз Р-ряду. Легко помітити, що при будь-якій надбудові вуглецевого ланцюга в молекулі гліцеринового альдегіду залишаться без зміни групи атомів, сполучені з двома нижніми його вуглецевими атомами:
І Н—С*—ОН;
І СНаОН Група атомів й -гліцеринового альдегіду
но—с*—-н.
І
СН„ОН
Група атомів £-гліцеринового альдегід}
275
Тому якщо в молекулі моносахариду гідроксильна група біля найвіддаленішого від карбонільної групи асиметричного вуглецевого атома розміщена в просторі справа, як у Д-гліцеринового альдегіду, то такому моносахариду приписують О-конфігурацію. Якщо конфігурація біля цього асиметричного С-атома така, як у Ь-гліцеринового альдегіду, то такому моносахариду приписують Ь-конфігурацію.
Молекула фруктози має таку конфігурацію:
СН2ОН
С=О
І
НО—є*—н
І ;
н—с*—он
Н—С*—он
сн2он £>(—.)-фруктоза
сн2он
І с=о
І н—с*—он
І но—с*—н
І но—с*—н
І сн2он £(-|-)-Фруктоза
Великий внесок у вивчення конфігурації моносахаридів внесли класичні дослідження Е. Фішера.
Перед назвою моносахариду в дужках стоїть знак (ф) або (—). Цей знак вказує, в який бік обертає площину поляризації світла розчин даного моносахариду. Якщо розчин моносахариду обертає площину поляризації світла за годинниковою стрілкою, тобто вправо, то перед назвою такого моносахариду ставиться знак «плюс» (+), у протилежному напрямку —знак «мінус» (—). Знак обертання площини поляризації світла визначають експериментально за допомогою поляриметра.
Слід зазначити, що не існує безпосереднього зв’язку між знаком обертання площини поляризації світла і конфігурацією/)- або £-. Трапляються оптично активні речовини, які мають конфігурацію Р- і знак обертання (+), конфігурацію Ь- і знак обертання (—), тобто конфігурація і знак обертання яких збігаються. Прикладом такої речовини є глюкоза. Проте є і такі речовини, наприклад фруктоза, які мають О-конфігурацію і знак обертання (—), а при /.-конфігурації —знак обертання (ф).
Р- і /.-ізомери є дзеркальними ізомерами, антиподами. Тому вони мають однакову назву, перед якою ставиться символ Р або /., наприклад, Р-глюкоза і /.-глюкоза. Альдогексози, які відрізняються від глюкози конфігурацією хоча б біля одного асиметричного С-атома, вже не є дзеркальними відображеннями глюкози, тобто не є її антиподами. Вони є оптичними ізомерами глюкози, але відрізняються від неї за властивостями. Такі ізомери називають діа-стереоізом ерами (від грецьк. «діа»—окремо, нарізно). Діастерео-ізомери глюкози мають різні назви, що підкреслює відмінність у їхніх властивостях. Просторовим ізомером глюкози, тобто її діастереоізомером, є широко поширена в природі галактоза (див. схему на с. 277). Діастереоізомером глюкози і галактози є маноза.
276
Генетичний взаємозв’язок гліцеринового альдегіду, тетроз, пентоз і гексоз Д-ряду
Гексози Пентози Тетрозн
/-І
І '-н н—с—он
сн2—он
Л* Гліцериновий альдегід
сн=о СН=О
н—С—он но—С—н
н-с-он Н-С-ОН 1
сн,—он Д-Еритроза сн,—он Д-Треоза
сн=о сн=о сн=о 1 сн=о |
н-с-он НО-С-Н Н-С-ОН но-с-н
н-с-он н-с-он но-с—н но-с-н |
н-с-он н-с-он н-с-он н-с—он 1
сн,—он Д-Рибоза 1 сн,-он Д-Арабіноза сн,—он Д-Ксилоза сн,-он Д-Ликеоза
сн=о СН=О СН=О сн=о сн=о сн=о і сн=о СН=О 1
1 н-с—он 1 НО-С-Н 1 1 но-с-он НО-С-Н н— с— он І но—с—н 1 Н-С2-ОН | но—с—н
н—с—он н-с-он 1 1 НО-С-Н НО-С-Н . н-с-он і Н-С-ОН 1 но-с-н 1 но-і-н 1
н-с—он Н-с-он Н—С—ОН Н—С—ОН но-с-н но-с-н 1 но—С—н 1 НО—С—н 1
н-с-он н-с-он 1 1 Н-С-ОН н-с-он н-с-он 1 н—с—он 1 н-с-он 1 Н-С-ОН 1
1 СН,—ОН 0-А лоза СН,—ОН О-Альтроза 1 1 сн2-он сн,-он Д-Глюкоза Д-Маноза сн2-он Д-Гулоза сн,—он Д-Ідоза СН2-ОН Д-Галактоза сн,-н Д-Талоза
Моносахариди, які відрізняються конфігурацією тільки біля одного, переважно біля другого вуглецевого атома, називають епімерами. Так, епімерами є глюкоза і маноза (див. схему на с. 277).
3. Ізомерія, зумовлена існуванням таутомерії. Згідно з вище-наведеними формулами, глюкоза і фруктоза повинні виявляти реакції, характерні відповідно для альдегідів і кетонів, а також реакції багатоатомних спиртів. Проте не всі властивості цих моносахаридів можна пояснити виходячи з такої будови глюкози і фруктози. Глюкоза, наприклад, не забарвлює розчину фуксинсір-чистої кислоти, не вступає в реакцію з гідросульфітом натрію. А ці реакції є характерними для альдегідів. Було встановлено також, що один з п’яти гідроксилів глюкози і фруктози має вищу реакційну здатність, ніж чотири інших, тому він легко вступає в реакцію із спиртами, утворюючи кристалічні речовини типу простих ефірів —глікозиди. Однак глікозиди, на відміну від простих ефірів, легко гідролізуються розбавленими мінеральними кислотами, утворюючи при цьому одну молекулу моносахариду і одну молекулу спирту.
Якщо виходити з наведених вище формул глюкози, фруктози, то не можна пояснити утворення глікозидів, оскільки кожний з п’яти спиртових гідроксилів цих моносахаридів міг би дати із спиртом сполуку типу простого ефіру. Крім того, з цих формул не зрозуміло також, чому тільки один гідроксил бере участь в утворенні глікозиду і чому глікозиди —речовини типу простого ефіру —так легко гідролізуються, тоді як прості ефіри дуже стійкі до гідролізу (див. розд. 10).
При дослідженні оптичної активності свіжоприготовлених розчинів глюкози та інших моносахаридів виявилося, що при стоянні цих розчинів їх оптична активність протягом деякого часу змінюється, а потім набуває певної для кожного моносахариду величини. Явище зміни оптичної активності розчину моносахариду після його приготування називають мутаротацією. Мутаротацію також не можна пояснити виходячи з наведеної вище формули глюкози або іншого моносахариду.
Всі ці та інші особливості моносахаридів стали зрозумілими після того, як було доведено, що вони можуть існувати ще в іншій ізомерній (таутомерній) циклічній формі. Уявлення про циклічну структуру глюкози вперше висловив у 1870 р. О. А. Коллі, а пізніше, в 1895 р., така її структура була доведена роботами ряду хіміків, і в першу чергу Е. Фішера, Б. Толленса і Ш. Танре. Утворення цієї форми молекули глюкози можна уявити так. Вуглецевий ланцюг молекули глюкози в просторі може мати велику кількість конформацій. Одна з них така, що перший і п’ятий С-атоми розміщуються близько. У цьому випадку близько розмістяться і ті групи атомів, які знаходяться біля цих атомів вуглецю. В результаті карбонільна і гідроксильна групи молекули моносахариду зближуються. А оскільки ці групи полярні, то вони притягуються і взаємодіють між собою відповідно до полярності їх зв’язків. Внаслідок такої
278
взаємодії розривається л-зв’язок карбонільної групи і атом водню гідроксилу п’ятого вуглецевого атома приєднується до карбонільного атома кисню. Таким чином біля першого С-атома утворюється новий гідроксил, який називається глікозидним, а у випадку глюкози — глюкозидним. Атом кисню гідроксильної групи п’ятого С-атома при такій взаємодії сполучається з вуглецевим атомом карбонільної групи. Перший і п’ятий С-атоми молекули глюкози при цьому сполучаються через атом кисню і молекула глюкози набуває циклічної будови:
сн2он
У такій взаємодії цих функціональних груп у молекулі глюкози або іншого моносахариду нічого дивного немає, оскільки спирти легко реагують з альдегідами і утворюють при цьому напівацеталі (див. розд. 17). Циклізація молекули глюкози, як видно з вищена-веденої схеми, приводить до утворення шестичленного кільця, в якому крім вуглецевих атомів є атом кисню:
Таке циклічне угруповання називається піранозним, а глюкоза в такій циклічній формі називається глюкопіранозою.
Глюкоза може мати і таку конформацію, при якій у просторі близько розмістяться перший і четвертий вуглецеві атоми. У цьому випадку буде відбуватися взаємодія карбонільної групи з ОН-групою, яка знаходиться біля четвертого С-атома:
Глюкозидний ;ОН. гідроксил
6 сн2-он-
ІВ результаті такої взаємодії також утворюється циклічна форма глюкози, але в ній атом кисню сполучає перший і четвертий С-ато-
279
ми. У цьому випадку циклізація приводить до утворення п’ятичлен-ного кільця, в якому крім вуглецевих атомів є атом кисню:
Таке циклічне угруповання атомів називається фуранозним, а глюкоза в такій циклічній формі називається глюкофуранозою.
Циклічні форми моносахаридів називають ще напівацетальніши формами, оскільки вони утворюються внаслідок взаємодії карбонільної і спиртової груп. Така взаємодія відбувається у моносахаридів в межах однієї молекули. Отже, циклічні форми моносахаридів являють собою внутрішні напівацеталі. З вищесказаного випливає, що моносахариди можуть існувати як у нециклічній формі, яка має карбонільну групу і називається оксоформою, так і в ізомерній їй циклічній формі. Така ізомерія моносахаридів називається цикло-оксотаутомерією.
В результаті циклізації перший вуглецевий атом а ль до гексози стає асиметричним. Тому пірапозні та фуранозні форми моносахаридів існують у вигляді стереоізомерів, які відрізняються розміщенням глікозидного гідроксилу в просторі. Такий тип стереоізомерії називається аномерією, а такі стереоізомери — аномерами (від грецьк. «ано»—вгорі). При циклізації, наприклад, глюкози одночасно виникають два аномери глюкопіранози і два — глюко-фуранози, які відрізняються між собою розміщенням глюкозидно-го гідроксилу:
НО.
н * .он ______
н—с—он І
но—н 9
н-<р—он І
Н—Є—ОН-сн,2-он
сн2—он
Й-Д'-Глкжопіраноза! ргЛ-Глюкопіраноза а-О-Глюкофураноза.р-Дг&юііофураноза
Щоб розрізнити аномери, їх позначають буквами «альфа» (а) і «бета» (Р). Циклічна форма моносахариду, в якій глікозидний гідроксил розміщений у просторі з того боку, що й гідроксил, який визначає конфігурацію цього моносахариду, називається а-аноме-ром. Якщо ці гідроксили розміщені по різні боки, то це р-апомер. У молекулі вищерозглянутої глюкопіранози гідроксил, за яким визначають конфігурацію, пішов на утворення циклічної форми. У зв’язку з цим у а-аномера глюкопіранози глюкозидний гідроксил розміщений у просторі з того самого боку (справа), що і кисневий місток, а у р-аномера —з протилежного боку.
2й0
Для фруктози також існують піранозні і фуранозні циклічні форми. У фуранозній формі фруктоза входить до складу бурякового і тростинного цукру:
сн2-он с=о
но-с—н н-с—он н-с—он
СН2-ОІІ £> -Фруктоза
СН2-ОН
НО—С—н""'] н—с—он 0 н—с—он \
І
сн2
СС-7)-Фрукто-піраноза
сн2-он А *
но—С—Н І н-с-ОН О н-с—он| сн/'"^
В-О -Фрукто-’
1 ліраноза
сн2-он І».
С—ОН
но-с— н-с—он 9 н-с-^
ін2-он
СС-£>-Фрукто-фураноза
сн2-он но-с* ___
НО-С—н А
-О -Фрукто -фураноза
Наведені вище структурні формули лінійних та циклічних форм молекул глюкози, фруктози та інших моносахаридів називають проекційними. Вони були запропоновані Е. Фішером. Для більш реального зображення циклічних молекул моносахаридів використовують так звані перспективні формули, які запропонував у 1925 р. англійський вчений Н. Хеуорс. У перспективних формулах фуранозні і піранозні цикли молекул моносахаридів зображають у вигляді правильних п’яти-і шестикутників, розміщених перпендикулярно до площини рисунка. Атом вуглецю, з яким сполучений глікозидний гідроксил, як правило, розміщують на рисунку справа, кисневий атом у циклі —за площиною рисунка. Групи атомів, які розміщені в просторі біля асиметричних атомів вуглецю справа, у перспективних формулах записують знизу, під циклом. Зверху, над циклом, записують групи атомів, розміщені в просторі зліва. Атоми вуглецю в циклі, як правило, не записують:
а-О-Глюко-піраноза
8_£>-Глюко- а-О-Фрукто-
“ піраноза фураноза
носн2 он
он н Р~£>-Фрукто-фураноза
Легко помітити, що піранозні і фуранозні форми окремих моносахаридів ізомерні між собою. Наприклад, ізомерами є глюкопіра-ноза і глюкофураноза. Ізомерія в цьому випадку викликана розміром циклу в молекулі моносахариду.
Циклічні піранозні форми моносахаридів стійкіші, ніж фуранозні. Іншими словами, шестичленні цикли молекул моносахаридів міцніші, ніж п’ятичленні. Тому моносахариди в твердому стані, як правило, перебувають у формі шестичленних піранозних циклів. У водних розчинах більша частина розчинених молекул моносахаридів також існує у вигляді циклічних піранозних форм, оскільки фуранозні (п’ятичленні) циклічні форми моносахаридів нестійкі.
281
Причому, якщо моносахарид у фуранозній формі входить до складу складного сахариду, то такі складні сахариди легко гідролізуються.
Вивчення хімічних і фізичних властивостей індивідуальних моносахаридів дало можливість дійти висновку, що в кристалічному стані моносахариди існують виключно у формі циклічних напів-ацеталів. Під час розчинення однієї з піранозних форм моносаха-риду у воді або іншому розчиннику відбувається часткове перетворення (ізомеризація) його в другий аномер, у фуранозні форми і в карбонільну форму. Така ізомеризація відбувається до тих пір, доки не встановиться рівновага між піранозними і фура-нозними формами та між а- і 0-аномерами даного моносаха-риду.
У наш час методом спектроскопії ядерного магнітного резонансу встановлено склад таутомерпої суміші деяких моносахаридів. Хід таутомерного перетворення моносахариду можна простежити за зміною оптичної активності його розчину при його стоянні протягом деякого часу. Це явище, як уже зазначалося вище, називається мутаротацією. Так, кут питомого обертання щойно приготовленого розчину а-О-глюкопіранози дорівнює 4-112,2°. При стоянні цього розчину кут питомого обертання його змінюється (зменшується) і через деякий час набуває постійного значення 4-52,7°. Питоме обертання свіжоприготовленого розчину р-О-глюкопірано-зи становить 4-18,7°, а при стоянні розчину воно теж змінюється (збільшується) і набуває постійного значення 4-52,7°.
Явище мутаротації пов’язане з утворенням рівноважної системи між аномерними формами моносахариду:
4-! 12,2°------------=» 4-52,7° <------------1-18,7°
а-О-Глкжопіраноза Суміш таутомерів: Р-Р-Глюкопіраноза
а-Р-глюкопіранози — 36 %
Р-Р-глюкопіранози — 64 %
Процес мутаротації можна прискорити, добавивши до розчину моносахариду невелику кількість (кілька крапель) розчину аміаку.
У розчинах деяких моносахаридів виявлені і фуранозні форми. Наприклад, у водному розчині О-фруктози при 20 °С встановлюється рівноважна суміш, яка складається з 76,4 % р-О-фруктопі-ранози, 19,5 % р-£)-фруктофуранози і 4,1 % а-Л-фруктофуранози. Карбонільні форми моносахаридів містяться в розчині у дуже малій кількості. Так, полярографічним методом дослідження встановлено, що розчин глюкози містить тільки 0,0026 % альдегідної форми, а остання кількість цього моносахариду перебуває в циклічній формі.
З вищесказаного можна зробити висновок, що в розчині моносахариди перебувають у вигляді кількох таутомерних форм, які 28$
зв’язані між собою рухомою рівновагою. Таутомерні перетворення О-глюкози можна зобразити такою схемою:
Отже, згідно з сучасними уявленнями, моносахариди — це внутрішньомолекулярні напівацеталі багатоатомних оксиальдегі-дів і оксикетонів, які у водних розчинах зв’язані рухомою рівновагою з своїми таутомерними карбонільними формами —•багатоатомними альдегіде- і кетоноспиртами.
4. Конформаційна ізомерія. Для сахаридів у циклічній формі можливий ще один вид ізомерії —поворотна, абокопформаційна, ізомерія, яка пов’язана з просторовим розміщенням вуглецевих атомів шестичленного циклу. Пірапозний цикл, подібно до цикло-гексанового (див. розд. 27), може існувати у вигляді кількох кон-формерів з різною стійкістю. Від циклогексанового піранозний цикл відрізняється несиметричністю, яка викликана наявністю в циклі атома кисню. У зв’язку з цим збільшується кількість можливих конформаційних ізомерів. У той час як для циклогексанового циклу можливі конформери у вигляді «крісла», «ванни» і «твісту», піранозне кільце може існувати у вигляді восьми ненапружених конформацій, дві з яких мають вигляд «крісла», а шість —«ванни». Ванноподібні конформації піранозного циклу енергетично менш стійкі і їх існування можна не враховувати. Стійкішими є дві кріслоподібні конформації піранозного циклу, оскільки вони мають мінімальну торсійну і вандерваальсівську напругу. Ці конформації мають такий вигляд:
. Для моносахаридів найстійкішими є такі кріслоподібні конформації, у яких об’ємні групи —СНаОН і —ОН мають незатруднене
283
екваторіальне положення. Іак, а- і р-аномери Д-глюкопіранози існують у вигляді таких конформацій:
СС 0-1 люкопіраноза В~-О -4'люкопіраноза
Легко помітити, що в молекулі р-Р-глюкопіранози СН2ОН-і всі ОН-групи розміщені в просторі в екваторіальному положенні, в молекулі сс-О-глюкопіранози глюкозидний гідроксил має аксіальне розміщення, а всі інші ОН-групи і СН2ОН-група — екваторіальне. Тому р-Д-глюкопіраноза має стійкішу конфігурацію і найпоширеніша в природі.
к-Д-Глюкопіраноза з аксіальним розміщенням глюкозидного гідроксилу менш стійка внаслідок взаємодії цієї групи з двома аксіальними атомами водню. У випадку інших альдогексоз най-стійкішою, як правило, є та конформація, при якій найбільш об’ємна група —СН2ОН займає екваторіальне положення.
У метильованих і ацетильованих піранозах об’ємні групи також намагаються зайняти екваторіальне положення. Однак існує один загальний виняток, який називається аномернин, або Д2-ефектом: метокси- або ацетоксигрупа біля першого вуглецевого атома намагається розміститись у просторі аксіально. Аномерний ефект пояснюють відштовхуванням диполів у атома кисню біля першого вуглецевого атома і атома кисню піранозного циклу:
Стійкіша Аномерний ефект, менш
конформація стійка конформація
Для вільних сахаридів, розчинених у воді, аномерний ефект урівноважується іншими факторами. Так, Д-глюкоза, наприклад, існує переважно як р-аномер з ОН-групою біля першого вуглецевого атома в екваторіальному положенні.
Методи добування моносахаридів. У природі вуглеводи утворюються в зелених рослинах з вуглекислого газу і води в процесі фотосинтезу і їх можна легко виділити з природної сировини. Для синтетичного добування моносахаридів найбільше значення мають такі методи:
1. Гідроліз дп- і полісахаридів. Цей метод є одним з основних для добування моносахаридів.
2. Конденсація альдегідів і кетонів. При дії вапняної або баритової води на мурашиний альдегід утворюється суміш оксіальде-гідів або оксикетонів, у якій містяться також гексози. Таким мето-
284
домО. М. Бутлеровщев 1861 р.добув густий сироп (метиленітан), у якому, як він довів, міститься цукриста речовина. Це був перший синтез цукристої речовини в лабораторії. Пізніше Е. Фішер виділив з такого сиропу гексозу С6Н12О6 і назвав її акрозою. Він встановив, що акроза — це рацемічна суміш антиподів: кетози складу СН,ОН—(СНОН)3—С—СН2ОН, а саме О-фруктози, яка трапля-
ІІ О
ється в природі, і її антиподу—/.-фруктози, яка в природі не зустрічається.
Реакцію конденсації формальдегіду в акрозу можна подати у вигляді такого сумарного рівняння:
,0
6Н—С< >- сн2—сн—сн—сн—с—сн2.
ХН І І 2 І І І II | 2 он он он он о он
Ця реакція відбувається, напевне, послідовно через стадії утворення проміжних сполук — продуктів альдольпої конденсації (див. розд. 17) деякої кількості молекул формальдегіду.
Синтез моносахаридів шляхом альдольної конденсації альдегідів і кетонів відіграє важливу роль у біологічних процесах. •
3. Оксинітрильний синтез, за допомогою якого добувають вищі моносахариди з нижчих (див. далі).
Хімічні властивості. Вище зазначалося, що кожний моносаха-рид існує в розчині у вигляді кількох таутомерних форм: карбонільної і циклічних, які зв’язані між собою динамічною рівновагою. У зв’язку з цим моносахаридам властиві реакції як багатоатомного альдегідо- або кетоноспирту, так і циклічної напівацеталь-ної форми.
Реакції моносахаридів розглянемо на прикладі глюкози. У рівняннях реакцій будемо записувати тільки ту форму моносахариду, яка безпосередньо вступає в реакцію.
Реакції карбонільних форм моносах ари-д і в. Відновлення. При відновленні О-глюкози воднем при наявності каталізатора утворюється шестиатомний спирт—£)-сорбіт:
І І
н—с—ОН н—с—он
НО—С—Н —±!Е.-_И---„ НО—С—н
І І
н—с—он н—с—он
І І
н—с—он н—с—он
І І
сн2—он сн2—он
С-Глюкоза О-Сорбіт
Сорбіт вперше був виділений з плодів горобини. Він приблизно в два рази менш солодкий, ніж сахароза. Сорбіт не підвищує вмісту
285
глюкози в крові, тому його використовують як замінник сахарози в харчуванні людей, хворих на цукровий діабет. Крім того, сорбіт використовують для виробництва аскорбінової кислоти(вітаміну С).
Окислення. Усі моносахариди легко окислюються. При цьому залежно від характеру окислювача можуть утворюватися одноос-новні (альдонові) або двохосновні (сахарні) кисдоти. М’які окислювачі, наприклад бромна вода, при кімнатній температурі окислюють тільки альдегідну групу моносахариду. Так, £)-глюкоза за таких умов окислюється до £>-глюконової кислоти, кальцієва сіль якої — глюконат кальцію —• використовується в медицині: /О Л)
| ХН |4ОН
Н—С—ОН Н—с—он
НО—С—Н ________________> но—с—н
І Вг.'ІСО і
н—с—он н—с—он
І І
н—с—он н—с—он
І І
СН2ОІІ СН2ОН
©-Глюкоза Р-Глюконова кислота
Окислення альдегідної групи моносахариду можна здійснити тільки в нейтральному або кислому середовищі. У лужному середовищі окислення моносахаридів відбувається з розщепленням їх вуглецевого ланцюга. При цьому утворюється ряд продуктів, які виявляють сильні відновні властивості. Тому навіть такі слабкі окислювачі, як оксид срібла А§20 і гідроксид міді Сн(ОН)2 при легкому нагріванні з альдозами відновлюються до металічного срібла і оксиду міді (І). Отже, розчини моносахаридів мають відновні властивості.
Сильні окислювачі, наприклад концентрована НМО3, крім альдегідної групи окислюють також первинну спиртову групу моносахаридів і перетворюють їх таким чином на двохосновні сахарні кислоти:
,0 /О
ІХН І\он
н—с—ОН н—с—он
но—с—н г°1- нко" > НО—С—н
Н—С—он н—С—он
н—С—он н—с—он
сн2—он © -Глюкоза V хон Сахарна кислота
Окислення пероксидом водню. Зменшення довжини вуглецевого ланцюга альдоз. Моносахариди можна також піддавати вибірко-
286
вому окисленню. Наприклад, якщо глюкозу спочатку окислити бромною водою до глюкопової кислоти, а далі проводити її окислення пероксидом водню при наявності ацетату заліза (III), то в процесі реакції відщеплюється вуглекислий газ і утворюється альдоза (арабіноза), яка містить на один атом вуглецю менше, ніж вихідний моносахарид:
о к У_ о о 'Ч/ С •) ,О С\ 1 Н
н—с—он Н—( 2—ОН НО—с—н
но—с—н [0] НО—( 2— II Н.О2, Ге8+ , н—с—он
—со2 —н,о
н-с-он Н—( 2—ОН н—с—он
Н-с-он Н—< "—ОН сн2-он
1 сн2—он 1 СН2—ОН Р-Арабіноза
Р-Глюкоза Р-Гліокоиова кислота
Приєднання синильної кислоти (оксинітрильний синтез). До
карбонільної групи моносахаридів легко приєднується синильна кислота. При цьому утворюються відповідні оксинітрили, які використовують для синтезу вищих моносахаридів з нижчих. Так,
глюкоза, приєднуючи синильну кислоту, утворює оксинітрил, який омиленням перетворюють на оксикислоту. Дегідратація ос-
танньої дає гептози: с-
|\н н—с—он
но—с—н
н—с—он
лактон, відновлення якого приводить до утворення /О
си рон
н—с—он н—с—он
І І
+Н+СК- Н—С—ОН +211,0 н—С—ОН
-------> І -------> І —я»
НО—С—Н НО—С—Н "Н’°
Н—(2—ОН Н—І—ОН
н—с—он
СН2—ОН Р‘Глюкоза
И—(2—ОН
І
СН2—ОН Оксішітрил
Н—С—ОН
СН2—он Оксикислота
Н—с—он
І н—с—он о
НО—Є— Н
І Н—С------
Н—(2—ОН
СН2—ОН
Лактон
н-с—он
Н—с— он но—с—н
Н—С—ОН
Н—с—он
сн2—он
Гептода
287
Отже, за допомогою оксинітрильного синтезу з гексози можна синтезувати гептозу.
Взаємодія з фенілгідразином. Реакція моносахаридів з фенілгід-разином мала велике значення для встановлення конфігурації моносахаридів і детально вивчена Е. Фішером (1884 р.). Спочатку моносахарид реагує з фенілгідразином з участю карбонільної групи і утворює фенілгідразон, який далі перегруповується через стадію утворення таутомерного енгідразину (в процесі внутрішньо-молекулярної окислювально-відновної реакції)в моноімін кеталь-дегіду. Останній, взаємодіючи з фенілгідразином, утворює озазои. Озазони — не розчинні у воді кристалічні речовини жовтого кольору, добре очищаються кристалізацією. Тому їх використовують для характеристики моносахаридів. Під дією водних розчинів кислот від озазонів легко відщеплюються обидві молекули фені лгід-разину і утворюється так званий озон (кетальдегід), у молекулі якого поряд знаходяться дві карбонільні групи. При відновленні озону утворюється кетоза. Отже, реакцією моносахаридів з фенілгідразином можна перейти від альдоз до кетоз:
СН=О + Н2И-МН—С6Н5
Н—С—ОН
но—с—н
Н—с— он
І
н—с—он
І
сн.2—он
Р-Глюкоза
сн=и—мн—свн5 н—с—он
—Н2О
но—с—н
І н—с—он
І н—с—он
сн2—он
Фенілгідразон
СН—МН— МН-СвНв
сн=ин
С— О—І-Н І но—с—н
н—с—он
І н—с—он
СН2—он Енгідразин
-с„н6-кні,>
НО—с—н
н—С—ОН
2СГ,НЛ—N31—N13, -Н2О. -N13; *
н—с—он
І
СН2—ОН
Моноімін кетальдегіду
СН=М—МН—С(ІНБ
С=М—ИН—свн8
но-с-н 2НгО •
н—С—он -2С,НВ-КН-ИН,'
н—с—ОН
І
сн2—он Озазон
288
,0 с-^ І хн с=о
НО—<1—Н 2Н
І
н—с—он
н—с—он
сн2—он Озон (кетальдегід)
сн2—он І
с=о
но—с—н .
І
н—с—он
н—с—он
І
сн2—он
Л-Фруктоза
Перетворення моносахаридів під впливом лугів. Луги і слабкі основи при кімнатній температурі дуже легко викликають ізомерні перетворення біля першого і другого С-атомів у молекулах моносахаридів. Це приводить до утворення рівноважної суміші альдоз і кетоз. Так, О-глюкоза за таких умов перетворюється на 2,5 % вО-манозу і на 31 % —вО-фруктозу. Аналогічно ізомеризуються і кетози, наприклад фруктоза. Можливо, що ця ізомеризація проходить через стадію утворення нестійкого енолу (ненаси-ченого двохатомного спирту):
н—с—он
І но—с—н н—с—он
І н—с—он
І сна—он Л-Глюкоза
- сн—он — II с—он
но—с—н
1 н—с—он
н—С—он
СН2—он_ Енол
сн2он с=о
но—с—н
Н—с—он
Н—с—он
І
СН2ОН Л-Фруктоза
но—С—н
но—с—н
І н—с—он
н— с— он
І сн2—он Л-Маноза
Такого типу ізомерні перетворення моносахаридів під впливом лугів називають епімеризацією, оскільки вони приводять до утворення суміші епімерів, наприклад, глюкози і манози. Подібні
Ю 1420
289
перетворення моносахаридів відбуваються і в живих організмах під дією спеціальних ферментів.
Отже, під впливом лугів з глюкози, манози і фруктози утворюється рівноважна суміш цих трьох моносахаридів. Тому розчини фруктози відновлюють аміачний розчин оксиду срібла, фелінго-ву рідину, хоча у фруктози немає альдегідної групи. Насправді ці реактиви відновлює не фруктоза, а глюкоза і маноза, які під впливом лугу утворюються з фруктози.
Концентровані розчини лугів осмоляють моносахариди. При цьому утворюється велика кількість різноманітних продуктів, оскільки осмолення моносахаридів супроводиться їх конденсацією, полімеризацією, розщепленням, частковим окисленням тощо.
Реакції циклічних форм моносахаридів. Взаємодія з лугами і гідроксидами важких металів. Наявність гідроксильних груп у молекулах моносахаридів підтверджується відповідними характерними реакціями спиртів. Так, при дії лугів і гідроксидів важких металів легко утворюються похідні моносахаридів типу алкоголятів, які називають сахаратами. Моносахариди розчиняють гідроксид міді і утворюють при цьому забарвлений у синій колір розчин. Ця реакція аналогічна взаємодії гідроксиду міді з гліцерином і свідчить про те, що моносахариди є багатоатомними спиртами.
Утворення складних ефірів. Моносахариди, подібно до спиртів, взаємодіють з ангідридами і галогенангідридами карбонових кислот, з утворенням складних ефірів. Залежно від кількості взятого ангідриду або галогенангідриду з моносахаридів можна добути ряд складних ефірів, аж до повного ефіру. При цьому поряд з спиртовими гідроксилами реагує і глікозидний гідроксил:
н он
+ 5(СН,СО)2О
-5Н3С~ СООН
СН2ОСОСН3
НІ—----он
|/н \
КОСОСНз Н/І
Н3С-С-о\ У ОСОСНз
II 1 І
О Н ОСОСНз
1.‘2,3,4,6-Пентаацетпл-а-£>-г.іюкоп1раноза (пентаацетнл глюкоза)
Велике значення мають деякі моно- 1 дифосфорнокислі ефіри моносахаридів, такі як, наприклад, 6-фосфат глюкози, 1,6-дифос-фат фруктози та інші:
6-Фосфат глюкози
ноч хон
О = Р-О-СІЬ.О СІ12-О-Р = О
НС)/ кн нои Х°Н
он н
І.С-Дпфосфат фруктози
290
Фосфати глюкози і фруктози є проміжними продуктами біосинтезу вуглеводів, процесів бродіння та інших важливих біологічних процесів. Фосфати таких пентоз, якО-рибоза і 2-дезокси-О-ри-боза, є структурними фрагментами нуклеїнових кислот.
Взаємодія із спиртами. Ця реакція характеризує моносахариди як циклічні напівацеталі.
Хімічні властивості циклічної напівацетальної форми моносахариду в першу чергу визначаються наявністю глікозидного гідроксилу. Цей гідроксил виявляє набагато більшу реакційну здатність, ніж інші, неглікозидні, гідроксили. Підвищення активності глікозидного гідроксилу пов’язане з впливом ефірного атома кисню циклу, який викликає зміщення електронної густини сусідніх зв’язків на себе і тим самим збільшує полярність зв’язку між вуглецевим атомом і глікозидним гідроксилом:
Н он
Тому із спиртами при наявності хлороводню реагує тільки глікозидний гідроксил моносахаридів, неглікозидні гідроксили за цих умов у реакцію не вступають. В результаті реакції утворюються ацеталі (прості ефіри) моносахаридів, які називаються глікозидами. Ацеталі глюкози називають глюкозидами'.
сн.,он
а-Д-Глюкопіраноза
Н ОН
СС-О-Метил-Д-гліокопіранозид
Глікозиди — похідні моносахаридів, у молекулах яких атом водню глікозидного гідроксилу заміщений алкільним радикалом або більш складним замісником В. Будову глікозидів для а-£>-глю-копіранози можна зобразити такою загальною формулою:
сн2он
В/—о.н
Ун \ Крн н/ н°г“Го~/? • н он
Замісник 7? у глікозидах називається агліконом. Глікозиди можуть мати у своєму складі залишки як глюкози, так і інших моносахаридів.
Глікозиди широко розповсюджені в природі і мають велике біологічне значення. У рослинному світі вони містяться в листі,
10*
291
насінні, корі дерев. Глікозиди є в багатьох лікарських рослинах. Так, вони виявлені в наперстянці і використовуються в медицині для стимулювання діяльності серця (серцеві глікозиди). У медицині застосовують також глікозиди-антибіотики, такі як стрептоміцин, який інгібірує синтез білків у рибосомах. По типу глікозидів побудовані складні сахариди.
Глікозидний гідроксил моносахаридів вступає також у реакцію з амінами. При цьому він заміщується на аміногрупу. Продуктами такої взаємодії є так звані N-глікозиди, або глікозиламіни:
N -Етил-8-0-глюкопіранозиламін
ААГлікозидами є нуклеозиди — складові частини нуклеїнових кислот.
Утворення простих ефірів. Метилюючі реагенти —диметил-сульфат (СН3О)28О2 або йодистий метил при наявності А§20 — заміщують у моносахаридах атоми водню всіх гідроксильних груп на метальні радикали. Так, а-П-глюкопіраноза в цих умовах утворює 1,2,3,4,6-пентаметил-а-П-глікопіранозид (пентаметил-глюкозу):
-|-5(СН3О)28О2
-5 Н3С —О—8О2 —ОН
Бродіння. Бродінням називають складний процес розщеплення моносахаридів під впливом різних мікроорганізмів. У більшості випадків цей процес супроводиться виділенням газоподібних продуктів (СО2, Н2тощо). Продуктами бродіння можуть бути різні речовини. Залежно від кінцевого продукту розрізняють кілька видів бродіння: а) спиртове; б) молочнокисле; в) маслянокисле; г) лимоннокисле і ряд інших. Бродіння з утворенням етилового спирту можна зобразити такою схемою:
С6Н12ов Др-!ждж1^ 2С2Н8ОН + 2СО2.
Глюкоза
В організмі людини постійно відбуваються процеси біохімічного розщеплення і синтезу моносахаридів. Так, при скороченні м’язів у результаті розщеплення сахаридів утворюється молочна кислота.
Процеси бродіння широко використовуються в промисловості.
Дія кислот на моносахариди (дегідратація з циклізацією). Пентози при нагріванні з розбавленою сірчаною кислотою утворюють фурфурол, а гексози — оксиметилфурфурол (див. розд. 40).
292
Окремі представники. Пентози. Пентози — це моносахариди з загальною формулою С5Н10О8. У природі вони зустрічаються переважно в складних сахаридах (С5Н8О4)П, які називаються пентозанами. Пентозани в значній кількості (10—25 %) містяться в деревині, соломі тощо. Під час гідролізу пентозанів при наявності мінеральних кислот утворюються пентози:
(С8Н8О4)П -р «НаО-► лС6Н10Ов.
Пентози, як 1 гексози, існують у таутомерних карбонільних і циклічних формах. Вони дають усі реакції моносахаридів, які були описані вище. Найважливішими представниками пентоз є арабіноза, ксилоза, рибоза і дезоксирибоза:
Арабіноза:
|хн н—є—он
но—с—н
но—н
сн2-он А (+)-Арабіноза.
а-іД+)-АрабопІраноз»
Арабінозу можна добути кип’ятінням з кислотами вишневого клею або подрібнених буряків:.
Ксилоза:
о
£>(+) -Ксилоза
Ксилоза входить до складу полісахариду ксилану, який міститься в соломі, деревині тощо. Ксиланпри гідролізі дає ксилозу.
Рибоза:
О -Рибоза
СС-£> -Рибофураноза
293
Рибоза має важливе біологічне значення. Вона входить до складу рибонуклеїнових кислот (РНК) ядер клітин.
Дезоксирибоза'.
ХО
Сх ї'н
н—с—н
І *
н—С—ОІ!
н—он
сн2—он
О-Дезоксирибоза
К-О-Дезоксирибофураноза
Дезоксирибоза, як і рибоза, також має велике біологічне значення. Вона входить до складу дезоксирибонуклеїнових кислот (ДНК) ядер клітин.
Гексози. Глюкоза, або виноградний цукор. О (-\-)-Глюкоза широко поширена в природі. Вона міститься у винограді та інших солодких плодах. Глюкоза входить до складу організму людини. Так, укрові людини завжди міститься від 0,08 до 0,11 % глюкози. При деяких захворюваннях (цукровий діабет) кількість глюкози в крові зростає і вона з’являється в сечі.
У промисловості глюкозу добувають гідролізом крохмалю і використовують як дешевий замінник бурякового або тростинного цукру при виготовленні варення, солодких напоїв, а також у медицині. Крім того, глюкозу застосовують як відновник і як загусник для фарб.
В-Галактоза. Ця альдогексоза входить до складу молочного цукру лактози, гідролізом якого її добувають, а також до складу деяких глікозидів та тканин мозку.
Фруктоза, або фруктовий цукор. Ця кето гексоза входить до складу соку багатьох фруктів, а також меду. Разом з глюкозою фруктоза є складовою частиною бурякового і тростинного цукру (сахарози), з якого її добувають шляхом гідролізу. Фруктоза солодша за глюкозу. Смак меду зумовлює головним чином фруктоза. Слід зазначити, що фруктофуранози практично не мають солодкого смаку, а носієм його є Р-Д-фруктопіраноза.
Дисахариди
Дисахаридами називають складні сахариди, молекули яких під час гідролізу утворюють дві молекули моносахаридів. Хімічний склад дисахаридів, молекули яких складаються із залишків гексоз, можна подати загальною формулою С12Н22О11:
^-12^22^11 “Г н2о * Ссн12ой + С„н12ов.
Дисахарид Моносахариди
Дисахариди побудовані як глікозиди, тому що їх утворення відбувається в результаті відщеплення молекули води від двох
294
молекул моносахаридів за рахунок двох ОН-груп, одна з яких обов’язково є глікозидним гідроксилом.
Для встановлення будови і характеристики властивостей дисахаридів необхідно знати: 1) які моносахариди утворюються при гідролізі даного дисахариду; 2) у якій формі (фуранозній чи піра-нозній) перебувають залишки моносахаридів у дисахариді; 3) у якій формі (ос- чи Р-) моносахариди входять до складу дисахариду; 4) які гідроксильні групи моносахаридів беруть участь в утворенні зв’язку (кисневого містка) між залишками моносахаридів.
Утворення дисахаридів можна уявити як процес, зворотний гідролізу дисахаридів:
С6Н11ОЬ-ІОН+ЦО-С6Н11ОВ —> СеНпОз-О-^НцО, + Н2О.
(СвНі2Ов) (СвН]2О6) (С12Н22О11)
Оскільки в молекулах моносахаридів активність гідроксилів різна (глікозидні гідроксили більш реакційноздатні), може бути два випадки утворення зв’язків між залишками моносахаридів у молекулі дисахариду; тому за будовою та хімічними властивостями дисахариди поділяються на два типи:
1. Якщо відщеплення води відбувається за рахунок глікозидних гідроксилів обох молекул моносахаридів, то в молекулі такого дисахариду вже не буде глікозидного гідроксилу. Дисахариди, молекули яких не містять глікозидного гідроксилу, існують тільки в циклічній формі і не можуть ізомеризуватись в альдегідну форму. Тому вони не відновлюють метали з їхніх оксидів. У зв’язку з цим такі дисахариди називають невідновлюючими. Водні розчини невідновлюючих дисахаридів при стоянні не піддаються мутаротації, що підтверджує існування в них тільки однієї (циклічної) форми дисахариду.
2. У випадку відщеплення води за рахунок глікозидного гідроксилу однієї молекули моносахариду і одного з неглікозидних гідроксилів другої в молекулі такого дисахариду залишається один глікозидний гідроксил, який може легко ізомеризуватися в альдегідну групу. Дисахариди, молекули яких містять глікозидний гідроксил, існують у двох таутомерних формах — циклічній і карбонільній, які зв’язані між собою рухомою рівновагою, і за властивостями подібні до моносахаридів. Так, вони легко вступають у реакції, характерні для альдегідів, зокрема, відновлюють метали з їхніх оксидів. Тому такі дисахариди називають відновлюючими. Водні розчини відновлюючих дисахаридів при стоянні піддаються мутаротації, що підтверджує існування в них кількох таутомерних форм дисахариду.
Прикладом невідновлюючих дисахаридів є сахароза і трега-лоза, відновлюючих — мальтоза, лактоза і целобіоза.
Сахароза — найбільш відомий і широко поширений у природі дисахарид. Добувають сахарозу з цукрового буряка та цукрової тростини. Тому її називають іце буряковим, або тростинним, цукром. У цукрових буряках сахарози міститься від 16 до 20 %, а в
295
цукровій тростині — 14—26%. Підчас гідролізу сахарози утворюються Д-глюкопіраноза і О-фруктофураноза. Сахароза не виявляє властивостей ні альдегідів, ні кетонів. Це свідчить про те, що залишки глюкози і фруктози сполучені між собою в її молекулі за рахунок своїх глікозидних гідроксилів:
Отже, сахароза є а-Д-глюкопіранозил-р-Д-фруктофуранози-дом (глюкозидо-фуранозидом).
Як уже зазначалося, сахароза під час кислотного або ферментативного гідролізу розщеплюється на глюкозу і фруктозу:
СіаН220ц -р Н2О -> СвН12О„ + СвНІ2Ов.
£)(+)-Сахароза £)(+) Глюкоза О(—-)-Фруктоза
[а]£)=4-66,5° +52.5 ° [а]£>=—92°
Як видно з даної схеми, в результаті гідролізу сахароза перетворюється на суміш рівних кількостей глюкози і фруктози. Реакційна суміш обертає площину поляризації світла до гідролізу і після нього. Проте оскільки фруктоза має більший кут лівого обертання (—92°), ніж глюкоза —правого (4-52,5°), то суміш глюкози і фруктози, яка утворилася в результаті гідролізу сахарози, має ліве обертання (—39,5°), тоді як вихідний продукт — сахароза — обертає площину поляризації світла вправо (4-66,5°). Така зміна внаслідок гідролізу правого обертання розчину на ліве називається інверсією (від лат. іпуегзіа — обертання), а суміш рівних кількостей глюкози і фруктози, що утворилася при цьому, — інвертним цукром, або штучним медом. Інвертний цукор широко використовується в кондитерській промисловості.
За фізичними властивостями сахароза являє собою білу кристалічну речовину з температурою плавлення 184—185 °С, яка добре розчиняється у воді. Сахароза широко використовується в харчовій промисловості і є важливим харчовим продуктом.
Трегалоза, або мікоза, вперше була знайдена в ріжках (Сіауі-серз рпгригеа Тиі.), а потім — у деяких бактеріях і в багатьох грибах (звідси пішла її друга назва — грибний цукор).
Трегалоза являє собою 1-О-(а-Д-глюкопіранозил)-а-Д-глюкопі-ранозид, тобто глюкозидо-глюкозид, в якому ефірний зв’язок
2,96
між двома залишками глюкози утворений з участю обох глюкозид-них гідроксилів:.
Таку структуру трегалози підтверджують її властивості. Так, під часи гідролізу утворюється О-глюкопіраноза, а водні розчини трегалози не піддаються мутаротації, що свідчить про відсутність у даного дисахариду таутомерії і про відсутність в його молекулі глікозидного гідроксилу.
Мальтоза, або солодовий цукор (від лат. таїіит —солод). Залишки глюкози в молекулі мальтози сполучені між собою а-1,4-глкжозидним зв’язком. Отже, у молекулі мальтози є один глюкозидний гідроксил. Тому вона здатна до мутаротації, існує в а- і р-формах, у розчині перебуває в таутомерних формах — циклічній і альдегідній, які зв’язані між собою рухомою рівновагою:
Отже, мальтоза є 4-0-(а-£)-глюкопіранозил)-а-£)-глюкопірано-зою (глюкозидо-глюкозою), тому вона виявляє відновні властивості: дає реакцію «срібного дзеркала», відновлює фелінгову рідину. Альдегідна група в її молекулі при цьому окислюється в карбоксильну і мальтоза перетворюється на мальтобіонову
297
[О]
Мальтобіонова кислота
У техніці мальтозу добувають при неповному гідролізі крохмалю ферментами, які містяться в солоді (у пророслому зерні злаків), або при нагріванні крохмалю з водними розчинами кислот, наприклад Н28О4. Ліальтозу часто можна виявити у тваринних і особливо рослинних організмах як проміжний продукт ферментативного розщеплення крохмалю.
Мальтоза кристалізується з однією молекулою води у вигляді білих голчастих кристалів, які плавляться при 102—103°С. Кристалічна мальтоза являє собою |3-форму. Розчини мальтози обертають площину поляризації світла вправо і при стоянні піддаються мутаротації: [До = -фі 11,7° -> ф-130,4°.
Мальтоза приблизно на 40 % менш солодка, ніж сахароза.
Лактоза, або молочний цукор (від лат. Іасішп —молоко), міститься в молоці. Так, у коров’ячому молоці вміст лактози становить 4—5 %. Добувають лактозу з молочної сироватки.
Лактоза являє собою 4-0-(Р-Д-галактопіранозил)-а-£)-глюкопі-ранозу і при гідролізі утворює Д-галактопіранозу і£)-глюкопірано-зу. В молекулі лактози є один глікозидний гідроксил. Тому вона здатна до мутаротації, існує в а- і р-формах, у розчинах перебуває в таутомерних формах — циклічній і альдегідній, які зв’язані між собою рухомою рівновагою. У зв’язку з цим лактоза виявляє відновні властивості. Альдегідна група в її молекулі легко окислюється в карбоксильну і лактоза при цьому перетворюється на лакто-біонову кислоту:
Лактоза
(циклічна а-форма)
Лактоза (альдегідна форма)
Лактоза існує переважно в а-формі, кристалізується з однією молекулою води. Кристалогідрат лактози плавиться при 202 °С. Кут питомого обертання для рівноважного розчину лактози дорів
298
нює [а]д = +52,6°. Лактозу використовують для харчування немовлят, при виготовленні фармацевтичних препаратів.Лактоза в 4—5 разів менш солодка, ніж сахароза.
Целобіоза е 4-0-(Р-0-глюкопіранозил)-|3-£)-глюкопіранозою і залишки глюкози вії молекулі з’єднані 0-1,4-глюкозиднимзв’яз-ком. Під час гідролізу целобіоза утворює Д-глюкопіранозу. В молекулі целобіози є один глікозидний гідроксил. Тому целобіоза здатна до мутаротації, існує в а- і Р-формах, у розчинах перебуває в таутомерних формах. У зв’язку з цим целобіоза виявляє відновні властивості і при окисленні перетворюється на целобіонову кислоту:
Н он
Целобіоза (циклічна й-форма>
Целобіоза (альдегідна форма)
Целобіонова кислота
Целобіоза є проміжним продуктом ферментативного гідролізу целюлози. Кристалічна целобіоза являє собою р-ізомер з Тпл = = 225 °С. Розчини целобіози обертають площину поляризації , ,20 світла вправо і при стоянні піддаються мутаротації: [сс]о = = 4-14,2° — 4-34,6°.
Целобіоза добре розчиняється у воді, майже не солодка.
Вищі полісахариди
Вищими полісахаридами називають природні високомолекуляр-ні сполуки (біополімери), молекули яких складаються з великої кількості залишків молекул моносахаридів. Утворення полісахаридів відбувається шляхом поліконденсації молекул моносахаридів:
ц-о-цні0о4ІШ+н;-о-дн10о4бн+ні--о—с6н10о4-^5н4-----------
—* (я — 1) Н2О 4- • • • —О СсН10О4- О—С,1 ІІ(,О. —О С0Н10О4- —• • , або в скороченому вигляді:
пС6Н12Ос_- (С6Н10О5)„4- (п- 1) Н2О.
Полісахариди побудовані подібно до дисахаридів: залишки циклічних форм моносахаридів у молекулах полісахаридів сполучені міжсобою кисневими містками, в утворенні яких беруть участь глікозидний гідроксил однієї молекули і гідроксил четвертого вуглецевого атома другої молекули моносахариду.
299
Основними представниками вищих полісахаридів є крохмаль, глікоген і целюлоза (клітковина).
Крохмаль —дуже поширений в природі полісахарид. Він є одним з найважливіших продуктів фотосинтезу і відіграє роль резервної поживної речовини рослин. Крохмаль міститься в насінні, коренях, бульбах, листках. Наприклад, у бульбах картоплі його 12—24 %, в зернах рису — 62—82 %, кукурудзи — 65—72 %, пшениці —57—75 %. Крохмаль має вигляд білого аморфного порошку. Висушений при 100—110°Свіндуже гігроскопічний. Крохмаль не розчиняється в холодній воді, а в гарячій утворює колоїдний розчин, який називається клейстером. Розчини крохмалю обертають площину поляризації світла вправо: [сс]о =4-201,5... ...205°.
ооососоозоосоооооооо-—————<юоооооооосоооосоооо
Рис. 46. Схематичне зображення будови полісахаридів: а — амілоза; б — амілопектин; в — глікоген
З розчином йоду в йодиді калію крохмаль дає характерне синє забарвлення. Ця реакція досить чутлива і дає змогу виявити за допомогою розчину йоду навіть невеликі кількості крохмалю і, навпаки, за допомогою крохмалю — незначні кількості йоду.
Крохмаль — це суміш двох полісахаридів —амілози і амілопектину, які відрізняються будовою ланцюга молекули. В більшості рослин крохмаль складається з ~25 % амілози і ~75 % амілопектину. Амілоза має в основному ниткоподібну (лінійну) будову (рис. 46, а), середня молекулярна маса її знаходиться в межах ЗО 000 — 160 000. Амілоза розчинна в гарячій воді, але ці розчини нестійкі і при стоянні з них випадає осад амілози. Розчин амілози дає синє забарвлення з йодом. Молекули амілози побудовані із залишків а-О-глюкопіранози, сполучених між собою в положенні 1 ->4, так, як і в мальтозі:
0С-І,4-Гліокозидний зв’язок
300
або скорочено
Амілопектин — полісахарид розгалуженої будови, який містить від 600 до 60 000 залишків глюкози. Його молекулярна маса становить від 100 000 до 1 000 000. Амілопектин, як і амілоза, складається із залишків а-Д-глюкопіранози. Проте частина їх сполучена в положенні 1 4, а частина (у місцях розгалуження)—
1 -> 6-зв’язками (рис. 46, б):
Амілопектин, на відміну від амілози, при нагріванні з водою утворює клейстер.
Крохмаль практично не виявляє відновних властивостей. Він не відновлює ні оксид срібла, ні гідроксид міді, ні фелінговий розчин.
У молекулі крохмалю містяться гідроксильні (спиртові) групи, але відкрити їх за допомогою Сн (ОН)2, як у випадку моно- і дисахаридів, не вдається, оскільки крохмаль утворює колоїдні розчини і тому взаємодія його молекул з Сн(ОН).2 відбутися не може. Однак іншими методами було доведено, що кожний залишок глюкози в молекулі крохмалю містить в середньому по три гідроксильні групи.
При швидкому нагріванні крохмалю з невеликою кількістю води (10—20%) відбувається його частковий гідроліз, в результаті якого макромолекули крохмалю розщеплюються на менші молекули полісахаридів, які називаються декстринами. Декстрини мають таку' саму загальну формулу, як і крохмаль, — (СвН^ОзД, але меншу молекулярну масу, тому х у формулі декстринів має менше значення, ніж п у формулі крохмалю.
Декстрини в міру поступового зменшення молекулярної маси дають з йодом відповідно синьо-фіолетове, фіолетове, червоно-фіолетове, оранжеве і жовте забарвлення. Утворення декстринів відбувається під час випікання хліба, варіння харчових продуктів.
ЗОЇ
Декстрини краще розчинні у воді, ніж крохмаль, і тому краще засвоюються організмом.
Повний гідроліз крохмалю (амілози і амілопектину) приводить до утворення Й-глюкопіранози. У процесі такого гідролізу можна виділити і проміжні продукти —декстрини, олігосахариди, в тому числі і дисахарид мальтозу:
п
(СсН10Об)п-> Розчинний крохмаль -> (С6Н10О5)х -> 7 С] 2 Н22ОП -> лСвН12Ов.
(де х<гі) Мальтоза Глюкоза
Декстрини
Каталізаторами гідролітичного розщеплення крохмалю можуть бути як мінеральні кислоти, так і ферменти, наприклад амілаза слини.
Крохмаль має широке застосування. Він входить до складу таких важливих харчових продуктів, як хліб, картопля, різноманітні крупи, які є основним джерелом вуглеводів у харчуванні людей і тварин. У промисловості значну кількість крохмалю добувають з картоплі, кукурудзи і використовують для харчових потреб. Шляхом гідролізу з нього добувають декстрини, патоку, глюкозу. Крохмаль застосовують також у текстильній промисловості (для приготування шліхти, апретури, загусників для фарб), у поліграфічній, паперовій, сірниковій, косметичній промисловості, у палітурній справі.
Глікоген, або тваринний крохмаль, (СвН10О.,)п відіграє в організмі людей і тварин виключно важливу роль як запасний полісахарид. Він міститься в усіх тканинах людського організму. Особливо багато глікогену в печінці (до 20 %) і м’язах (4%).Усі процеси життєдіяльності, і в першу чергу робота м’язів, відбуваються одночасно з розщепленням глікогену. В тканинах організму з глікогену в результаті ряду складних перетворень утворюється молочна кислота. Цей процес називається гліколізом.
Глікоген — білий аморфний порошок, добре розчинний навіть у холодній воді. При розчиненні він утворює колоїдні розчини. Молекули глікогену побудовані аналогічно молекулам амілопектину. Від амілопектину глікоген відрізняється тільки більшою розгалуженістю молекули (рис. 46, в), у зв’язку з чим молекулярна маса глікогену значно вища, ніж амілопектину, і досягає кількох мільйонів. З йодом розчини глікогену дають забарвлення від винно-червоного до червоно-бурого, залежно від походження глікогену, тобто залежно від виду тварин, з організму яких був виділений глікоген.
В організмах людей і тварин глікоген синтезується з глюкози і запасається в тканинах, а в період між прийманням їжі він розщеплюється і постачає організм глюкозою. Проте кількість глікогену, яка може відкластися про запас у тканинах, обмежена. Після запасання 50—60 г глікогену на кілограм тканини з глюкози починає синтезуватися жир, а не глікоген. З цього часу починається ожиріння організму.
302
Резервним полісахаридом є також інулін, який міститься в бульбах деяких складноцвітих рослин і в деяких водоростях. Інулін — природний полісахарид, що має формулу (С8Н10О5)п, під час гідролізу утворює Р-фруктофуранозу і невелику кількість (2—3 %) Р-глюкопіранози. Макромолекули інуліну лінійні і складаються із залишків р-Р-фруктофуранози, сполучених між собою [3-2 ->1--глюкозидними зв’язками:
НОСН2 6 о
Закінчуються макромолекули інуліну залишком а-Р-глюко-піранози, як молекули сахарози. Молекулярна маса інуліну не перевищує 6 000, кут питомого обертання його розчинів становить від —34° до —40°.
Інулін міститься у бульбах жоржин (до 12 %), цикорію (до 10 %), звідки його добувають екстрагуванням гарячою водою. Використовують інулін для добування Р-фруктозн.
Целюлоза (від лат. сеіініа — клітина), або клітковина, є головною складовою частиною оболонок клітин рослин. Волокна бавовни-ка (очищена вата) і фільтрувальний папір —це зразки майже чистої целюлози. Целюлоза складається із залишків Р-Р-глюкопіра-нози, сполучених між собою, як у молекулі целобіози, Р-1.4-ГЛЮ-козидними зв’язками. Середня молекулярна маса целюлози різного походження становить від 100 000 до 1 000 000 і більше. Молекули целюлози містять від 600 до 6 000 залишків глюкози:
303
Порівнюючи будову целюлози з будовою крохмалю, легко помітити, що макромолекули целюлози складаються із залишків 0-£)-глюкопіранози, а крохмалю — із залишків а-£)-глюкопі-ранози.
Целюлоза є складовою частиною стінок клітин рослин. Вона надає тканинам рослин механічної міцності, еластичності і утворює немовби їхній скелет. У рослинах целюлоза утворюється в результаті складних біохімічних перетворень, які починаються з фотосинтезу найпростіших вуглеводів. Тому в природі целюлоза зустрічається не в чистому вигляді.. Нитки бавовнику містять 92— 95 % целюлози, у різних видах деревини вміст целюлози коливається від 40 до 60 %. Найважливішими супутниками целюлози є лігнін, геміцелюлози, пентозани, пектинові речовини, які відділяють від неї, діючи на рослинні матеріали різними хімічними реагентами (лугом, гідросульфітом кальцію тощо). Чиста целюлоза являє собою білу аморфну на вигляд речовину без запаху і смаку. Однак при рентгенівському дослідженні вона дає характерні рентгеногра -ми, які свідчать про значну впорядкованість її структури. Макромолекули целюлози мають лінійну волокнисту будову. Довжина їх становить 1570 нм і більше, поперечні розміри — 0,4 нм X X 0,8 нм.
Целюлоза не розчиняється у воді, спирті, ацетоні та інших органічних розчинниках, але добре розчиняється в концентрованому розчині хлориду цинку і аміачному розчині гідроксиду міді (реактив Швейцера). Розчин целюлози в реактиві Швейцера використовують для добування мідно-аміачного шовку. З цією метою целюлозу розчиняють в аміачному розчині гідроксиду міді, а потім добутий розчин продавлюють крізь фільєри в кислу ванну, яка містить Н28О4 і гідросульфати. Внаслідок нейтралізації мідно-аміачного комплексу целюлоза виділяється з розчину у вигляді нитки.
Целюлоза не виявляє відновних властивостей і важче, ніж крохмаль, піддається гідролізу. При нагріванні целюлози з мінеральними кислотами, наприклад з Н28О4, можна добути проміжні продукти гідролізу — амілоїд і целобіозу:
(СеН10О5)п —» (СсН10О5)х -5* "2" С12Н22О1Х -> пС6Н12Ое.
Целюлоза (де х<п) Целобіоза Д-Глюкоза Амілоїд
Хімічні властивості целюлози визначаються в основному наявністю в її молекулах гідроксильних груп. У кожному залишку глюкози є три таких групи і для зручності формулу целюлози можна зображати так: [С6Н7О2(ОН)3]„. Тому за властивостями целюлоза подібна до спиртів. Так, вона легко реагує з концентрованим розчином лугу і утворює при цьому лужну целюлозу (алкаліцелюлозу):
[СсН,О2(ОН)3]„ + пМаОН-*- [СеН7О2(ОН)2—О—Ма]п + Н2О.
У промисловості цей процес називають мерсеризацією (Д.Мер-сер, 1848 р.). Лужна целюлоза, взаємодіючи з сірковуглецем 804
С82, утворює натрієву сіль ефіру ксантогенової кислоти — ксантогенат целюлози:
8- &+/ &А8-
[С6Н7О2(ОН)2-О-На]„+ «С =8 — [С6Н7О2(ОН)2-О-С-8-№] п ------------------"^8 8
Цей ксантогенат має здатність розчинятися в лугах. Такий розчин називають віскозою (від. лат. уізсозпз —в’язкий, липкий, клейкий). При продавлюванні віскози крізь фільєри у водний розчин Н28О4 виділяється целюлоза, але вже у вигляді нитки. Таким методом добувають волокно, яке називають віскозним шовком-. [СвН7О2(ОН)2—О—С—8—№]п + «Н28О4 -*[СвН7О2(ОН)3]„+ лС52+ «№Н8О4-
8
Якщо віскозу, пластифіковану гліцерином, продавлювати в розчин кислоти крізь вузькі щілини, то утворюється тонка прозора пластина целюлози —целофан.
Целюлоза легко взаємодіє з азотною кислотою і утворює її ефіри —нітроцелюлозу (нітроклітковину). Залежно від кількості взятої азотної кислоти та від умов реакції можна добути нітроефіри з участю однієї, двох або трьох гідроксильних груп у кожному залишку глюкози:
Г /ОІП
С0Н7О2<-ОН 4-«НО—МО2пН2О + С6Н7О2(-ОН Хон „
/О-т/
- «Н20 + С6Н7О2г—он 2 [ 2\он
Мононітрат клітковини Г ,О—N0/ 2/гН„О + С6Н7О2^-О—ІУО2 2 [ 2\ОН ]
Динітрат клітковини
г УО—N021
І \О-К'О2|„
Тринітрат клітковини
Суміш моно- і динітроклітковини називають колодійною ватою, або колоксиліном. Розчин колоксиліну в суміші спирту й ефіру називають колодієм і застосовують у медицині для заклеювання невеликих травм та для закріплення пов’язок. Колоксилін використовують також для виготовлення нітролаків. Із суміші колоксиліну з камфорою виготовляють целулоїд. Повністю нітрована целюлоза (тринітрат клітковини) називається піроксиліном. Піроксилін є вибуховою речовиною і використовується для виготовлення бездимного пороху.
При взаємодії клітковини з сумішшю оцтового ангідриду і оцтової кислоти (при наявності каталізатора Н28О4) утворюються ефіри клітковини і оцтової кислоти. Залежно від кількості введеного в реакцію оцтового ангідриду можна добути моно-, ді- і три-ацетилклітковину:
Г ,ОСОСН31
сбн7оХон
І “ХОН
Моноацетилклітковина
\он
0Н1
С«Н7О,^-ОН + 2«Н0—N0» . 6 \он]„
' .. _ /Он-Се. 7'-,2 у,. .
\0Н
\он
п
СвН,О»<-ОН + 3«НО—ИЮ2 -> 3«Н2О + С6Н7О2^-О—N0.
\0Н„ [ О -Г.’с:
/ОСОСНді С6Н7О.^-ОСОСН3
Хон ]„
Д і аце ти л кл і тковин а
,ОСОСН3і с„н7о,—ососн, ХОСОСНЦ, Т риацетилклітковин'а
п
305
Найширше застосування мають ді- і триацетилклітковина, з яких виготовляють ацетатний шовк (ацетатне волокно), негорючу рентгенівську і кіноплівку, лаки тощо.
Лігнін. Постійним супутником целюлози в рослинних тканинах є природний полімер лігнін. У середньому на кожний кілограм целюлози припадає близько 400 г лігніну. Неоднорідність деревини вперше помітив у 1838 р. французький хімік Г. Пайєи. Целюлоза, як виявилося, насичена «інкрустуючим матеріалом», який назвали лігніном (від лат. Іі^пит —деревина). Якщо порівняти, наприклад, структуру деревини із структурою залізобетону, то в цій аналогії целюлозі відводять роль арматури, а лігніну — роль бетону. Спроби виділити лігнін у тому стані, в якому він знаходиться в деревині, до цих пір не увінчались успіхом. Тому точну будову лігніну поки що не встановлено.
Хітин (від грецьк. «хітон»—покриття) — це полісахарид, з якого побудовані міцні покриття комах, ракоподібних та деяких грибів. Він не розчиняється у воді, органічних розчинниках, реактиві Швейцера, дуже стійкий до дії лугів. Хітин побудований із залишків М-ацетилглюкозаміну (хітозаміну), які сполучені між собою р-1,4-глікозидними зв’язками:
Хітин
Молекулярна маса хітину становить 150 000 —200 000.
Поняття про фотосинтез. Фотосинтез — це процес синтезу в зелених частинах рослин вуглеводів, а також білків і жирів з вуглекислого газу і води за рахунок енергії сонячного світла і з участю зеленого пігменту рослин —хлорофілу. Утворення вуглеводів з СО2 і води в процесі фотосинтезу можна зобразити загальною схемою:
Мц хлорофіл
ЛїСО2 *-{- ЛН2О "4“ я02«
Фотосинтез має надзвичайно велике значення в житті нашої планети, оскільки завдяки йому атмосфера збагачується киснем, а з атмосфери одночасно видаляється надлишковий вуглекислий газ. Крім того, зелені рослини щорічно в процесі фотосинтезу утворюють 100 млрд т біомаси, 2 млрд т якої використовується для харчування людей і тварин.
306
Контрольні запитання і вправи
1. У ПМР-спектрах альдопіраноз і їх похідних сигнал одного з протонів зсунутий в область більш слабкого поля порівняно з іншими протонами. Який не протон і чому відбувається цей зсув?
2. Поясніть, чому в рівноважній суміші метил-а- і метил-Р-П-глюкозидів переважає а-аномер.
3. Целюлозу можна окислити за допомогою М2О4 до [(С5Н7О4)СООН]П. Яка структура цього продукту? На яку речовину він перетвориться при гідролізі? Як називається продукт гідролізу? Декарбоксилування [(С6Н7О4)СООН]П дає (С5Н8О4)П. Що утворюється під час гідролізу цієї речовини і як називається продукт гідролізу?
4. Із шкаралупи арахісу виділений полісахарид арабан, гідроліз якого дає тільки £-арабінозу. Метплювання цього полісахариду з наступним гідролізом приводить до утворення еквімолярннх кількостей 2,3,5-три-,2-3-ди-і З-О-метил-Ь-арабіноз. Запропонуйте структурну формулу для арабану.
б. Пектинова кислота є основним компонентом пектину, який зумовлює утворення желе з фруктів і ягід. Метплювання пектинової кислоти з наступним гідролізом дає тільки 2,3-ди-О-метнл-£)-галактуронову кислоту. Припускають, що в молекулі пектинової кислоти є а-глікозидні зв’язки. Запропонуйте структурну формулу для пектинової кислоти.
6. Агар, який виділяють з морських водоростей, використовують як поживне середовище для вирощування мікроорганізмів. Під час гідролізу агару утворюються П-галактоза, /.-галактоза і сірчана кислота у співвідношенні 9:1:1. Метилювання агару з його подальшим гідролізом дає 2,4,6-три-О-метил-£>-галактозу, 2,3-ди-0-метил-І.-галактозу і сірчану кислоту в тому ж співвідношенні (9:1 ; 1). Запропонуйте структурну формулу для агару.
ЧАСТИНА II
Аліциклічні сполуки
Органічні сполуки, залежно від того, як сполучені між собою атоми в їх молекулах, поділяють на нециклічні і циклічні. В молекулах нециклічних вуглеводнів атоми вуглецю сполучені між собою у вигляді відкритих вуглець-вуглецевих ланцюгів, а в циклічних — у замкнені цикли (кільця). Якщо цикли молекул побудовані тільки з вуглецевих атомів, то такі речовини називають циклічними вуглеводнями. За своїми властивостями вони подібні до відповідних сполук аліфатичного ряду, тому їх назвали аліфатичними циклічними, або скорочено аліциклічними, вуглеводнями.
Аліциклічні сполуки класифікують за кількістю циклів, наявних у молекулі, за величиною і будовою циклу, а також залежно від наявності в циклі кратних зв’ язків і функціональних груп. За кількістю циклів у молекулі аліциклічні сполуки поділяють на моно-, ди-, трициклічні і т. д. За величиною і будовою циклу розрізняють сполуки циклопропанового, циклобутанового, циклопептанового, циклогексанового та інших рядів. Існують також ненасичені аліциклічні сполуки: циклоолефіни, циклодіолефіни, циклоацетилени і т. д. У молекулах цих сполук між вуглецевими атомами в циклі крім одинарних зв’язків існують кратні (подвійний, потрійний) зв’язки, яких може бути як один, так і декілька. Відома також велика кількість функціональних і нефункціональних похідних аліциклічних сполук.
Циклічні структури є основою молекул багатьох природних сполук. Особливо поширеними є сполуки з п’яти- і шестичленними циклами. Найкраще вивчені і найбільше значення мають моно-циклічні аліфатичні сполуки.
Розділ 27. МОНОЦИКЛИ
Основу моноциклів становлять насичені циклічні вуглеводні, склад молекул яких можна подати загальною формулою СпН2п, або (—СН2)„. Оскільки за властивостями такі сполуки дуже подібні до алканів (парафінів) з відкритими вуглець-вуглецевими зв’язками, то їх назвали циклоалканами, або циклопарафінами. Циклоларафіни називають також поліметиленовими вуглеводнями, або нафтенами. Останню назву для цих сполук запропонував
308
В. В. Марковников, який встановив, що кавказька нафта складається в основному з циклопарафінів, цикли молекул яких містять 5—7 вуглецевих атомів.
Нафта широко поширена в природі і відома людині з глибокої давнини. Слово «нафта» походить від персидського «нефт», що означає — витікати. За хімічним складом нафта — це природна суміш великої кількості органічних речовин, близько 90 % якої складають різні вуглеводні — алкани, циклоалкани, ароматичні тощо. Середній вміст вуглецю в нафті становить 82—88 %, водню — 12—14 %. У нафті в невеликій кількості містяться також кисне-, сірко-, азотовмісні та інші органічні сполуки. Для кожного нафтового родовища характерний певний склад нафти (табл. 25).
Таблиця 25. Вміст вуглеводнів у нафті (у % від загальної маси)
Родовище Алкани Нафтени Ароматичні вуглеводні
Бакинське зо 61 9
Північнокавказьке 61 24 15
Сибірське 52 36 12
Поволзьке (Татарстан) 58 25. 19
Провідна роль у вивченні циклопарафінів і розвитку хімії цих сполук належить вітчизняним ученим, таким як В. В. Марковников, Г. Г. Густавсон, М. Я. Дем’янов, М. Д. Зелінський, С. С. Намьот-кін, М. М. Кіжнер, Б. О. Казанський та ін. Великий вклад у хімію аліфатичних циклічних сполук внесли також зарубіжні вчені А. Байєр, Л. Ружичка, К. Неніцеску та ін.
Моноцикли утворюють гомологічний ряд (табл. 26), у якому фізичні властивості змінюються так, як і в ряду алканів і алкенів. Температура кипіння і густина циклопарафінів дещо вища, ніж
Таблиця 26. Гомологічний ряд, номенклатура і фізичні властивості циклопарафінів
Формула Назва Т'пл» °С А<ИП> О<- № 4 „20 по
с3нв Циклопропан — 126,9 —33,3
С4Н8 Циклобутан —90,7 12,5 0,708 1,3752 (при 0 °С)
Свн1о Циклопентан —93,8 49,3 0,751 1,4065
СвН1а Циклогексан 6,5 80,7 0,779 1,4262
с,н14 Циклогептан — 12,0 117,0 0.810 1,4449
СЯН1В Циклооктан 14,2 146,0 0,839 1,4585
Циклононан 10,8 170,0 0,850 1,4666
с10н20 Циклодекан 9,6 201,0 — —
Циклотриконтан 56,0 — 0,854 —
С-зіНвв Циклотетратрикон-тан 66,0 — 0,856 —,
309
у відповідних алканів і алкенів з нормальним вуглецевим ланцюгом. ІЧ-спектрп циклопарафінів дуже подібні до спектрів алканів. Циклопарафіни, подібно до алканів, поглинають УФ-світло з довжиною хвилі, меншою за 200нм. У зв’язку з цим їх використовують як розчинники при вивченні УФ-спектрів інших сполук.
Номенклатура. Назву аліциклічних сполук з одним циклом утворюють додаванням префіксу цикло- до назви відповідного насиченого вуглеводню:
СН2
Н2С----сн2
,сн2
сн2
Н2с-----сі і.
ІКС—СІ І.
Циклопропан
Циклобутан
Циклопентан
сн2
'ч Л"2; с сн, с<
Циклогексан
н2с.
Н2СХ
"СН2 сн2.
.сн,
Циклопентан
Існує багато аліциклічних сполук, молекули яких містять кілька конденсованих циклів. У назві таких сполук вказують кількість циклів і кількість всіх атомів вуглецю, які входять у цикли, а в дужках зазначають число атомів вуглецю, розміщених між спільними вуглецевими атомами. Нумерацію атомів вуглецю у циклі починають від розгалуження і продовжують по найдовшому вуглецевому ланцюгу. Наприклад:
Біцикло-[3,1,0]-гексан
Біцикло-[2,2,0]-гексан
'І ри ції кл о -[3,3,1,1]-декан, адамантап
При наявності замісників у циклі в назві вказують номери атомів вуглецю, біля яких розміщені ці замісники. Нумерацію в циклі здійснюють так, щоб атоми вуглецю, біля яких знаходяться замісники, отримали найменше цифрове значення:
1,3-Диметилцнклогексан
2-Етил-1-метилцнклопептан
Для зручності аліциклічні сполуки часто зображують у вигляді простих геометричних фігур: циклопропан—трикутником, циклобутан — квадратом, циклопентан—п’ятикутником, циклогексан — шестикутником і т. д.:
Циклобутан
310
Ізомерія. Для моноциклів можливі кілька видів ізомерії.
Структурна ізомерія. Структурна ізомерія циклоалканів може бути зумовлена різною величиною циклу або замісників у циклі, положенням і будовою замісників у циклі. Наприклад, циклогексан, метилциклопентан, етилциклобутан і пропілциклопропан мають однакову загальну формулу СвН12 і є структурними ізомерами. Ізомерія їх пов’язана з різною величиною циклу:
Циклогексан
-сн?-сн3-
СІ12-СІ І-.СН3 Г
Етилциклобуїаі! Пропілциклопропан
Ізомерами є 1-метил-2-пропілциклогексан і 1,2-діетилцикло-гексан:
Ізомерія цих сполук зумовлена різною величиною замісників у циклі. Прикладом ізомерів з різним положенням замісників у циклі є 1,2-, 1,3- і 1,4-диметилциклогексани:
Ізомерами є також пропілциклогексан та ізопропілциклогексан:
сн2-сн3
Для циклопарафінів, у циклі яких міститься два замісники, можлива також геометрична і оптична ізомерія.
Стереоізомерія моноциклів. Геометрична {іще-, транс-ізомерія властива двозаміщеним циклопарафінам, у молекулах яких замісники розміщені біля різних вуглецевих атомів циклу. Причиною геометричної ізомерії в цьому випадку є різне розміщення замісників (лігандів) у просторі відносно площини циклу. Геометричну ізомерію моноциклів називають також цис-/пряяс-ізомерією, оскільки вона подібна до цис-транс-ізомерії етиленових сполук (див. розд. 14). Проте геометрична ізомерія циклів відрізняється від геометричної ізомерії етиленових сполук. Для
311
етиленових сполук цей вид ізомерії викликаний відсутністю ВІЛЬНОГО обертання однієї частини молекули відносно другої навколо С=С-зв’язку, а цнс-/пранс-ізомерія циклопарафінів зумовлена неможливістю переміщення лігандів з одного боку циклу на другий (без розриву одного із зв’язків у циклі).
Отже, жорсткість циклу відіграє ту саму роль, що і подвійний зв’язок у молекулах етиленових сполук. Ліганди в молекулах циклопарафінів можуть розміщуватися у просторі як з одного боку циклу, так і з різних його боків. Наприклад, 1,2-циклопропанди-карбонова кислота, 1,3-диметилциклобутан, 1,2-і 1,3-диметилцик-логексани та інші двозаміщені циклопарафіни існують у вигляді двох геометричних ізомерів, які відрізняються за фізичними і хімічними властивостями:
н/н\соон
СООН Н
Ч«с-1,2-Цнклопропандикарбонова кислота (Тпл=139°С)
тдояс-1,2-Циклопропа„дикарбонова кислота (7'ПЛ = 175°С)
Оптична ізомерія у циклопарафінів з’являється в тих випадках, коли в їх циклічній молекулі немає площини симетрії. Однозамі-щені циклоалкани завжди мають площину симетрії, яка проходить через замісник. Наприклад:
і
і
і
н—с—н
І
Тому молекули однозаміщених циклопарафінів симетричні і стереоізомерів не мають. Молекули двозаміщених циклоалканів, у молекулах яких замісники в циклі знаходяться біля різних вуглецевих атомів, площини симетрії не мають. Тому для таких циклоалканів існують оптичні ізомери. Так, у молекулі циклопропанди-карбонової кислоти атоми вуглецю, сполучені з карбоксильними групами, є асиметричними. Тому 1,2-циклопропандикарбонова кислота існує у вигляді стереоізомерів. Молекула цнс-1,2-цикло-пропандикарбонової кислоти симетрична (має площину симетрії) і внаслідок внутрішньої компенсації, подібно до мезовинної кислоти (див. розд. 23), оптичну активність не виявляє. Молекула транс-1,2-циклопропандикарбонової кислоти не має площини симетрії і тому ця речовина існує у вигляді двох оптичних ізомерів (антиподів І
812
і II), які обертають площину поляризації а другий вліво) на однаковий кут ±84,5°:
світла (один вправо,
і
ноос /н
н соон
п
н
н
Відома також рацемічна трснс-І,2-циклопропандикарбонова кислота (7’ПіП = 162 °С), яка складається з рівномолекулярних кількостей антиподів І і II. У циклогексановому ряду оптична ізомерія спостерігається при 1,2- і 1,3-транс-положенні замісників:
Напруження в циклах. Моноциклопарафіни залежно від розміру циклу (від кількості атомів у циклі) поділяють на циклоалка-ни з малим циклом (3- і 4-членні), нормальним циклом (5-, 6- і 7-членні), середнім циклом (8—11-членні) і великим (макро-) циклом (12-членні і з більшою кількістю атомів вуглецю в циклі). Різні цикли відрізняються за міцністю. Тому циклічні сполуки, залежно від величини циклу, мають різну стійкість. Малі цикли (циклопропан, циклобутан і їх похідні) нестійкі і в хімічних реакціях, а також при нагріванні порівняно легко розриваються. Нормальні, середні і великі цикли мають високу міцність. Для пояснення такої відмінності у міцності циклів А. Байєр запропонував в 1885 р. теорію напруження, згідно з якою мірою стійкості циклу є відхилення валентних кутів у циклі від тетраедричного (нормального) — 109°28'. Чим більше таке відхилення, тим меншу міцність має цикл. Графічне зображення величини відхилення (А) валентних кутів перших членів ряду моноциклопарафінів подано на рис. 47. Внутрішній кут а правильного багатокутника (внутрішній кут циклу) обчислюють за формулою
2а (п — 2) а =----п---’
деп —кількість атомів у циклі; гі=90°. Для визначення величини відхилення валентних кутів А від кута 109°28' віднімають значення кута а і одержану величину ділять на два, оскільки у відхиленні беруть участь два валентних зв’язки: д = ~а,
313
Отже, мірою напруженості циклів, за А. Баіієром, є половина різниці між тетраедричним кутом і валентним кутом атома вуглецю в циклі.
Теорія А. Байєра змогла пояснити, чому три- і чотиричленні цикли нестійкі і розриваються і чому цикл у молекулі циклопентану стійкий. Проте з цієї теорії виходило, що вже циклогексан і інші багаточленні цикли повинні мати напруження (негативне напруження), оскільки внутрішні кути в їхніх циклах повинні бути більшими від кута 109°28'. Наприклад, п’ятнадцятнчлеиний циклічний кетон мускон С]ЬН2ВО, відхилення валентних кутів у якого становить—23с16', повинен бути таким же нестійким, як і циклопропан. Насправді ж циклогексан та інші циклоалкани, в тому числі і мускон, мають таку саму стійкість, як і циклопентан.
Рис. 47. Кутове відхилення напрямку валентності вуглецю в циклах
Рис. 48. Конформації молекули циклогексану: а — форма «крісла»; б — форма «ванни»; в — форма «твіст»
Дальшим розвитком теорії напруження була гіпотеза Г. Саксе (1890 р.) і Е. Мора (1919 р.), яка пізніше стала теорією про непла-нарність молекул циклоалканів. Згідно з цією теорією, атоми вуглецю в циклі розміщуються в просторі не в одній площині. Зокрема, для циклогексану були запропоновані форми «крісла» і «ванни» (рис. 48), в яких атоми вуглецю розміщені у двох площинах, що наближає величину кутів між атомами вуглецю в циклі до 109°28'.
Згідно з сучасними уявленнями про напруження в циклах, важливого значення набула теорія конформаційного аналізу, у розвиток якої великий вклад вніс К. Пітпер. Теорія конформаційного аналізу при характеристиці енергетичного стану молекул органічної сполуки, а отже, і стійкості циклічних сполук, враховує не тільки стан валентних кутів, але й можливості обертання навколо зв’язків та коливання і взаємне відштовхування атомів у молекулах. Це дало можливість сформулювати загальну теорію напруження в циклах. Основні положення цієї теорії такі:
1. У циклах існує кутове і конформаційне напруження.
314
2. Кутове («байєрівське») напруження виявляється внаслідок відхилення валентних кутів від кута 109°28'.
3. Конформаційне («пітцерівське») напруження виникає в результаті вимушеного повороту зв’язків і відхилення відзатормо-жених конформацій, які є енергетично найвигіднішими. При цьому виникають крутильні (торсійні) сили, які приводять до інверсії циклу.
4. Напруження може виникати внаслідок взаємного відштовхування зближених у просторі атомів, переважно атомів водню. В цьому випадку напруження виникає в результаті дії внутріш-ньомолекулярних ван-дер-ваальсівських сил.
5. Напруження в циклі може виникати як результат зміни міжатомних віддалей (зменшення або збільшення довжини зв’язків).
З урахуванням вищеназваних факторів молекули намагаються набути такої конформації (просторової форми), в якій сума напружень була б мінімальною.
Конформаційна ізомерія циклоалканів. У молекулі циклогексану при плоскому розміщенні атомів вуглецю в циклі внутрішні кути правильного шестикутника повинні були б дорівнювати 120°, а всі шість пар атомів водню мусили б знаходитися в енергетично невигідній заслоненій конформації, в якій атоми водню однієї СН2-групи протистояли б атомам водню другої СНа-групи:
Все це створювало б як кутове, так і конформаційне напруження в циклі. Щоб набути мінімального напруження в циклі, крутильні (торсійні) сили виводять три СН2-групи з площини. При цьому молекула циклогексану набуває конформаційної форми «крісла» або «ванни» (див. рис. 48). Тому реальні молекули циклогексану мають майже ненапружені цикли. Найстійкішою є конформація у вигляді «крісла», при якій кути між зв’язками практично дорівнюють тетраедричним (109°30'), а крім того, немає заслонених конформацій. У конформації «крісла» атоми вуглецю розміщуються вдвох площинах: атоми Сх, С3 і С5 —у нижній, а Са, С4 і С6 —у паралельній верхній площині. Молекула циклогексану може набувати також конформації у вигляді викривленого човна, або «твіст»-форми (рис. 48, в).
У кріслоподібній формі молекула циклогексану має 12 зв’язків С—Н. Шість з них орієнтовані в просторі паралельно один одному і паралельно осі симетрії. Такі С—Н-зв’язки називають аксіальними (а-зв’язки). Почергово три «-зв’язки напрямлені вгору, а інші три «-зв’язки —-вниз. Останні шість С—Н-зв’язків називають екваторіальними (е-зв’язки). е-Зв’язки напрямлені радіально від
315
Рис. 49. Аксіальне (а) і екваторіальне (е) розміщення С — Н-зв’язків у молекулі циклогексану (форма «крісла»)
циклу і орієнтовані в просторі паралель, но площині, в якій знаходяться атоми циклу (рис. 49).
Форма «крісла» (І) у молекулі циклогексану зазнає безперервних коиформа-ційних змін і швидко перетворюється на форму Іа (рис. 50). Таке взаємне перетворення конформаційних форм називається інверсією конформацій. У процесі перетворення форм ІіІа одночасно відбувається перехід аксіальних зв’язків в екваторіальні і навпаки.
У монозаміщених циклогексану замісник Я може займати в просторі як аксіальне, так і екваторіальне поло-
ження. Екваторіальна та аксіальна форми таких монозаміщених є конформерами і знаходяться в кон-формаційній рівновазі. Ці конформери при кімнатній температурі зазнають швидкого взаємного перетворення. Тому розділити
Рис. 50. Взаємний перехід аксіальних і екваторіальних зв'язків у молекулі циклогексану
їх не можна, можливо тільки виявити методами спектроскопії:
Р
Аксіальний конформер Екваторіальний конформер
де /?~СН3, СІ, СООН та Інші групи •
У зв’язку з конформаційною напругою екваторіальні конформери стійкіші, ніж аксіальні. Тому в рівноважній суміші значно більше екваторіального конформера. Так, при 25°С 95 % молекул метилциклогексану (/?—СН3) перебувають в екваторіальній конформації, і тільки 5 % —мають аксіальну конформацію.
Конформери відрізняються за фізичними і хімічними властивостями.
Методи добування. Основним джерелом циклопарафінових вуглеводнів е нафта, з якої методом розгонки (або - іншими методами) можна виділити індивідуальні циклопарафіни. Разом з тим відомо
316
багато реакцій, за допомогою яких циклопарафіни можна добути синтетично.
Загальні методи. 1. Дегалогенування а, ш-дигало-генопохідпих алканів натрієм (Фрейнд, 1882 р.) або цинком (Г. Г. Густавсон, 1887 р.):
н2<
СНо-С1 /
'сНд-СІ
/пСІ2 .
За таким методом можна добувати циклоалкани з кількістю атомів вуглецю у циклі від 3 до 6. Проте найлегше так добути циклопропан, його гомологи і похідні.
2. Взаємодія дигалогенопохідпих алканів з натріймалоновим ефіром. Залежно від того, яку дигалогенопохідну (1,2-, 1,3-, 1,4-або іншу) вводять у реакцію, за цим методом можна добути циклічні сполуки, цикли яких містять від 3 до 6 вуглецевих атомів:
СНаВг СООС2Н8
СН2 + Н2С\ 2Сгн,О№
\_____ хс.оос.„н.
Н2С сн2
2№Вг+ /СООС^І,
Н2С-----С.
ХСООС2Н5
Діетиловий ефір ї ,1-циклобутзн-дикарбонової кислоти
2НЯО
—2С2НвОН
1,1 -Циклобутанди- Ц икл обутан карбонова
карбонова кислота кислота
3. Піроліз солей дикарбонових кислот. Цей метод зручний для синтезу циклічних сполук, які містять 5,6 і більшу кількість атомів вуглецю в циклі:
Н2с—сн2—с
І ?>ва
Н2С—сн2—
о
Адипінат барію
Н2Є--СН2
| ^С = О + ВаСО3
н2с—сн2
Циклопентаном
Добутий кетон можна потім відновити у відповідний цикло-алкан.
Спеціальні методи. Моноциклічні сполуки з шестичленними циклами добувають гідруванням відповідних ароматичних сполук (див. розд. 28), а також реакцією дієнового синтезу (див. розд. 16).
Циклоалкани, цикли яких містять ЗО і більше атомів вуглецю, добувають сухою перегонкою торієвих со-лей дикарбонових кислот. Під час добування таких макроциклів з процесами внутрішньомоле-кулярної циклізації конкурують реакції міжмолекулярної взаємо-
дії
дії, яких відбувається особливо багато, якщо віддаль між реагуючими групами (наприклад, групамиСООН) велика. Перебіг таких небажаних побічних реакцій можна зменшити, використовуючи метод надрозбавляння і швидкого перемішування, оскільки при дуже низьких концентраціях реагуючих речовин переважають процеси циклізації. Наприклад, використовуючи розбавлений ефірний розчин а,ш-динітрилів і А-метиланілін як основу, за допомогою цього методу були добуті макроциклічні кетони:
(СН2)„ с=к
4Н2О г
—2МН3
І х° 'і '
/’С^-ОНІ ___
(сн2)л і [ —— (сн2)я с=о;
’ ГАЇ < “СС') X /
С.~І°И | -Н2б --------
хо—
л-15—34.;
Електроциклічні реакції спряжених дієнів. Правила Вудвор-да—Хофмана. Органічні сполуки із спряженою системою подвійних зв’язків мають здатність до циклізації, яка може відбуватися під впливом світла (фотохімічно) або при нагріванні (термічно). В результаті такої циклізації виникає новий простий зв’язок, який сполучає два реакційноздатних кінці спряженої системи, а інші подвійні зв’язки цієї системи переміщуються. Продуктами циклізації спряжених дієнів є циклоолефіни. Останні під дією тих самих факторів здатні рециклізуватися і перетворюватися на ті самі спряжені дієни з відкритим вуглецевим ланцюгом:
Бутадієн-1,3 Циклобутен
Гексагрієн -1,3,5 ЦиклогексадІєн-1,3
Реакції такого типу і оборотні їм називають електроцикліч-ними. Електроциклічні реакції відбуваються за механізмом узгоджених реакцій. Узгодженими називають такі реакції, в ході яких перерозподіл електронів реагуючих речовин здійснюється синхронно (одночасно), без виникнення проміжних іонів або радикалів. Узгоджені реакції, які проходять через циклічний перехідний стан, називають пер [циклічними реакціями. Узгоджені реакції стереоспецифічні, мало чутливі до дії каталізаторів і до впливу електронних ефектів лігандів.-
Стереоспецифічність електроциклічних реакцій можна уяснити на такому прикладі. Експериментально встановлено, що при термічній циклізації 1,4-дизаміщеної бутадієну-1,3 утворюється цис-дизаміщений циклобутен, а при фотохімічній циклізації цієї ж 1,4-дизаміщеної бутадієну-1,3 утворюється траяс-дизаміщений циклобутен. Аналіз елементарних процесів показує, що при термічній активації атоми вуглецю Сг і С, повертаються в один і той самий бік, внаслідок чого радикали в продукті реакції виявляються розміщеними в ч«с-положенні. У такому випадку говорять, що реакція
318
відбувається конротаторно. При фотохімічній активації спостерігається дисротаторний процес, який приводить до утворення продукту з трпнс-будовою:
Симетрія ВІДНОСНО площини
Симетрія ВІДНОСНО ОСІ /
/
Конротаторне обертання
Дисротаторне обертання
Р. Вудворд і Р. Хофман (1965 р.) сформулювали правила, згідно з якими напрямок перебігу узгоджених реакцій визначається симетрією вищих зайнятих орбіталей (ВЗО). Наприклад, длябута-дієну-1,3 і його гомологів ВЗОєф2МО (рис. 51). Ці правила дають
Розпушуючі ’ ордіта/іі
> Зв'язуючі орбіталі
Рис. 51. Схема молекулярних л-орбіталей бутадієну-1,3
можливість передбачити, яку енергію активації — низьку чи високу — матимуть узгоджені реакції при підвищенні температури і дії УФ-світла. Узгоджені реакції, для яких передбачена низька енергія активації, називаються дозволеними за симетрією, тоді як реакції з високою енергією активації називають недозволеними за симетрією.
Правила Вудворда — Хофмана. 1. Узгоджені реакції під дією теплоти і світла можуть відбуватися б тому випадку, якщо зв’язуючі
319
МО вихідних молекул переходять у зв’язуючі МО кінцевих продуктів з такою самою симетрією (принцип збереження орбітальної симетрії).
2. Для спряжених систем, які містять 4п р-електронів (и = = 1,2,3,4 іт.д.), наприкладдлядієнів К—СН—СН—СН=СН— дозволені (можливі) термічні узгоджені реакції циклізації при конротаторному процесі, фотохімічні —при дисротаторному процесі. Наприклад, у бутадієну-1,3 при конротаторній циклізації відбувається узгоджене обертання орбіталей атомів вуглецю С4 і С4 л-зв’язку ВЗО в один бік. Елементом симетрії цих орбіталей є вісь молекули. Одночасно з таким обертанням відбувається регібридизація цих двох атомів вуглецю з хр2-в зр3-стан і перекривання позитивних частин орбіталей атомів Сі і С4. В результаті один л-зв’язок перетворюється на о-зв’язок, що в цілому приводить до зменшення енергії системи. Повороти орбіталей атомів Сг і С4 спричинюють повороти зв’язків^—С2іС3—С, і одночасно повороти
Рис. 52. Конротаторна циклізація
лігандів /?. Тому в процесі такої циклізації утворюється цнс-диза-міщений циклобутен. При цьому зберігається симетрія р-орбіталей відносно осі симетрії (рис. 52). Отже, основним елементом симетрії при конротаторній циклізації є вісь симетрії молекули.
У випадку фотохімічної циклізації відбувається збудження молекули дієну, яке супроводиться переходом електрона на ф3 МО і ця МО перетворюється на ВЗО. ф3МО має площину симетрії. Для того, щоб у цьому випадку при циклізації утворився о-зв’язок, орбіталі атомів вуглецю С, і С4 повинні обертатися дисротаторно, тобто в протилежних напрямках. В результаті такої циклізації виникає транс-дизаміщений циклобутен. При цьому зберігається симетрія р-орбіталей відносно площини симетрії (рис. 53). Отже, при дисротаторній циклізації основним елементом симетрії є площина симетрії молекули.
Електронна будова циклопропану. Остов молекули циклопропану являє собою жорстку систему з С—С-зв’язків, сполучених у вигляді правильного трикутника з внутрішніми валентними кутами 60°. Величина цих кутів досить сильно відрізняється від тетраедричного кута 109°28'. В результаті осі валентних ^-гібридизованих орбіталей, які утворюють С— С-зв’язки, не можуть розміщуватися так, щоб бути напрямленими під кутом 109° 28'. Тому хр3-орбіталі вуглецевих атомів у молекулі циклопропану змушені ви
320
гинатись, що приводить до меншого їх перекриття і утворення більш слабких зв’язків, які розміщуються поза тричленним циклом. Такі хімічні зв’язки називають «банановими» (рис. 54). «Бананові» зв’язки можна розглядати як проміжні між о- і л-зв’язками, оскільки у них відбувається деяка делокалізація електронів за типом кільцевої л-електронної хмари, яка оточ1 є цикл і розміщена в одній з ним площині. Отже, зв’язки в молекулі циклопропану можна вва-
Рис. 54. Схема утворення о-зв'язків (а) і «бананові» зв’язки (б) в молекулі циклопропану
жати близькими до етиленового. Підтвердженням цьому є схильність циклопропану до реакцій приєднання з розривом С—С-зв’яз-ків у циклі.
Крім С—С-зв’язків у молекулі циклопропану є ще о-зв’язки С—Н. Валентні кути зв’язків Н—С—Н у циклопропані, як видно з електронографічних даних
11 1-120
321
близькі до 118°.' Атоми водню в циклопропані розміщені в просторі у найбільш енергетично невигідному стані, один навпроти другого, тобто в заслонених конформаціях:
н
Це створює додаткове конформаційне напруження в циклопропа-новому циклі.
Хімічні властивості. Хімічні властивості циклоалканів в основному визначаються розміром циклу. Три- і чотиричленні цикли нестійкі і легко руйнуються. Тому для них характерні реакції приєднання, які супроводяться розривом циклу. При цьому циклопропан і циклобутан та їхні похідні в хімічних реакціях виявляють властивості, подібні до властивостей етиленових сполук. П’яти-, шестичленні та вищі цикли стійкі і тому хімічні властивості циклопентану, циклогексану і вищих циклопарафінів подібні до властивостей насичених вуглеводнів. Для таких циклоалканів характерні реакції заміщення, які відбуваються без розриву циклу молекули;-
Реакції з воднем. Циклопропан при наявності каталізаторів (Рі, Г\ті) і температурі 80°С легко приєднує водень. При цьому тричленне кільце його молекули розривається і утворюється пропан:
80°С / \ + н,----------- н,с-сн,~сн3.
. - /__\ 2 Р(, Мі 3 - 3
Циклобутан при каталітичному гідруванні також приєднує водень і перетворюється на н-бутан. Проте для розриву циклобу-танового кільця необхідна вища температура:
20 ох
н2------► н3с — сн2 — сн2 — сн3
ГҐятичленні никли стійкі і розриваються важко. Тому циклопентан і його гомологи взаємодіють з воднем тільки при високих температурах. Продуктами таких реакцій також є алкани:
О, ЗОО’С
+ н2-------- Н3С-СН?-СН2-СН2-СН3 .
Циклогексан і його гомологи при нагріванні з каталізаторами (Рі, Реї тощо) піддаються дегідруванню і перетворюються на бензол або його гомологи. Наприклад:
300°С
Рі
322
Ця реакція стала основою методу ароматизації циклопарафіно-вих сортів нафти.
Реакція з галогенами. Циклопропан приєднує бром і йод При цьому циклопропанове кільце розривається і утворюються відповідно 1,3-дибром- і 1,3-дийодпропани:
Х-СН2-СН2-СН2-А' (деХ-Вг,І)
1
З хлором циклопропан вступає переважно в реакцію заміщення з утворенням 1,1-дихлорциклопропапу:
+ 2НС1 .
Циклобутан приєднує бром при нагріванні. Реакція супроводиться розривом циклобутанового кільця:
+ Вг,----Вг-си2- СН,-СІІ?-СН_,- Вг .
П’яти- і шестичленні циклоалкани вступають з галогенами в реакції заміщення, в результаті яких п’яти- і шестичленні цикли зберігаються і утворюються відповідні галогенопохідні циклопентану і циклогексану:
Відомі також полігалогеноциклоалкани. Так, при освітленні суміші бензолу з хлором УФ- або сонячним світлом відбувається гемолітичне приєднання хлору до бензолу, яке приводить до утворення гексахлорциклогексану (гексахлорану):
80°С
УФ-свІтло
СНСІ
Сігу '"'СНСІ СІНС^ СНСІ •
СНСІ
Гексахлоран — безбарвний кристалічний порошок (Тпл = = 112°С) із специфічним запахом (запах дусту). Він є сильним інсектицидом. Гексахлоран являє собою суміш геометричних стереоізомерів гексахлорциклогексанів. Інсектицидну властивість вияв-11*
323
ляє тільки один з них, так званий у-ізомер, який має а, а,а,е,е,е-конформацію. Вміст-у-ізомера в суміші становить ~ 13 %:
Гексахлоран,'(-Ізомер (аааеее)
Реакції з галогеноводнями. Циклопарафіни з малими циклами приєднують галогеноводні, а також інші сильні кислоти. Цикли їх молекул при цьому розриваються і утворюються відповідні галогенопохідні насичених вуглеводнів:
+ НВг ----- Н(С — СН2—СН2—Вг'
+ НІ
н3с—сн2—сн2—сн2—і.
Циклопентан, циклогексан і циклопарафіни з більшою кіль-кістю.атомів вуглецю в циклі з галогеноводнями не взаємодіють.
Дія окислювачів. Циклопарафіни не реагують з озоном і з розбавленим розчином КМпО4. Під дією сильніших окислювачів (НМО3, кисень при наявності каталізаторів тощо) відбувається поступове окислення одного з атомів вуглецю циклу, внаслідок якого спочатку утворюються циклічні спирти, які потім окислюються в циклічні кетони, а останні далі окислюються з розривом циклу і перетворюються на дикарбонові кислоти з тією самою кількістю атомів вуглецю, що і в циклі циклопарафіну. Практичне значення має окислення циклогексану, яке в кінцевому результаті приводить до утворення адипінової кислоти — вихідного продукту для виробництва найлону (див. розд. 22):
НООЮ—(СНз^-СООН,.
Циклогексан, Циклогексанол. Циклогексаном. Адипінова кислота
Терпени і терпеноїди
Терпени — це природні вуглеводні з загальною формулою (С5Н8)п (п = 2,3,4 і т. д.). Особливо велике значення мають терпенові вуглеводні складу С10Н16 (п =2). Більшість відомих терпенів побудована з ізопренових фрагментів, сполучених між собою за типом «голова до хвоста» (ізопренове правило Ружички; 1921 р.).
524
У зв’язку з цим терпени називають ще ізопреноїдами. Назва «терпени» походить від назви терпентинного дерева, із смоли якого був виділений перший представник цього класу природних речовин. Відомі також численні кисневмісні похідні терпенів (спирти, альдегіди, кетони), які називають терпеноїдами.
Моноциклічні терпени. В основі будови моноциклічних терпенів лежить вуглеводень циклопарафінового ряду С1оН2о — ментан (1-метил-4-ізопропілциклогексан), який існує у вигляді цис- і транс-стереоізомерів. Найважливішою похідною мептану є ментол (1-метил-4-ізопропілциклогексанол-3):
Молекула ментолу містить два асиметричних атоми вуглецю. Природний ментол обертає площину поляризації світла вліво [а]д = —49°. Він є основною складовою частиною олії перцевої м’яти, в якій ментолу міститься до 80 %. Ментол має характерний Смак і запах м’яти. Він виявляє властивості слабкого антисептика і тому використовується в медицині як заспокійливий, знеболюючий і дезинфікуючий засіб. Ментол використовують також у харчовій і парфюмерній промисловості.
Біциклічні терпени. Біциклічні терпени, подібно до моноциклічних, можна розглядати як похідні ментану. У молекулах цих терпенів в утворенні другого циклу (три-, чотири- чи п’ятичлен-ного) найчастіше бере участь восьмий атом вуглецю ізопропільної групи ментану. Біциклічні терпени поділяють на групи карану, пі-нану і камфану:
Каран, пінан і камфан не є природними речовинами. У природі зустрічаються похідні цих біциклічних вуглеводнів.
325
Найважливішими представниками біциклічних терпенів є «-пінен, борнеол і камфора:
а-Пінен є основною складовою частиною скипидару, в якому його міститься до 60 %. Скипидаром називають олію, яка утворюється під час перегонки соснової смоли живиці з водяною парою. За хімічним складом скипидар-—це суміш моно-і біциклічних терпенів.
Борнеол (борнійська камфора) є основною складовою частиною ефірної олії дерева ПгуоЬаІапорз сашрЬога, яке росте на островах Борнео і Суматра. Від назви першого з цих островів одержав назву даний терпеноїд.(—)-Борнеол знаходиться в природі у вигляді ефіру оцтової кислоти —борнілацетату, який міститься у великих кількостях (до 40 %) в олії з хвої сибірської ялиці. Борніл-ацетат виявляє властивості складного ефіру і легко омилюється, утворюючи при цьому (—)-борнеол:
Камфора є найважливішим похідним камфану. Вона відома на Сході з давніх часів. Її добувають перегонкою з водяною парою листя або деревини камфорного дерева Баигиз сашрЬога, яке поширене у В’єтнамі, Південному Китаї, на о. Тайвань. Камфорний лавр культивується і в СРСР. На Україні росте однорічна рослина—камфорний бази лик, з якого також можна добути камфору.
Камфора має важливе практичне застосування. Її з давніх-давен застосовують у медицині для збудження серцевої діяльності, а також як дезинфікуючий препарат. У техніці камфору використовують для виготовлення целулоїду, ацетилцелюлози, бездимного пороху, як пластифікатор високомолекулярних сполук тощо.
ЧАСТИНА III
Сполуки ароматичного ряду
Розділ 28. ВУГЛЕВОДНІ РЯДУ БЕНЗОЛУ І ЇХ ХАРАКТЕРИСТИКА
і До ароматичних відносять сполуки, молекули яких містять цйкЛічне угруповання з атомів вуглецю, яке називається бензольним ядром:
І
і
Це угруповання атомів одержало таку назву від назви першого члена гомологічного ряду цих речовин—бензолу. Ароматичні сполуки можуть містити одне, два або більшу кількість бензольних ядер. Термін «ароматичні сполуки» виник на початку розвитку органічної хімії, коли органічні речовини поділяли на аліфатичні (жирні) і ароматичні. До ароматичних сполук у той час відносили різноманітні речовини, які мали приємний запах.
, "Найпростішою ароматичною сполукою є бензол. Крім бензолу до ароматичних сполук належать численні його гомологи і похідні, а також сполуки, молекули яких містять кілька бензольних ядер (дифеніл, трифеніл, нафталін, антрацен та ін.). )
Бензол. Бензол вперше виділив у 1825 р. М. Фарадей з рідини, яка утворювалась із світильного газу. Тоді ж виникла проблема відносно будови бензолу, яка була вирішена тільки після встановлення основних положень теорії будови органічних сполук, одним з основоположників якої був О. М. Бутлеров. Властивості бензолу, які були незрозумілими на основі існуючих на той час уявлень, одержали пояснення в 1865 р., коли А. Кекуле запропонував зображати структурну формулу бензолу у вигляді шестичленного циклу з трьома подвійними зв’язками:
сн нс^сн
II І нс\ ^сн "'сн
і.
Бензол.
(формула Кекуле)
327
Подвійні зв’язки в циклі молекули бензолу були введені переважно для збереження чотиривалентності атомів вуглецю. Незважаючи на те, що формула бензолу (І) пояснювала багато експериментальних даних і створила основу для теорії ароматичних сп олук, вона не відображала ряду істотних фізичних і хімічних властивостей бензолу, а в деяких випадках перебувала з ними в протиріччі. Так, формула І не пояснювала причин особливої міцності бензольного ядра, зокрема, його стійкість до нагрівання і до дії окислювачів. Незрозумілим було також, чому бензолу не властиві реакції етиленових сполук, тобто реакції приєднання, а характерні реакції заміщення атомів водню в циклі на галогени та інші групи атомів. Формула І не пояснювала також, чому немає ізомерів у 1,2-і 1,3-дизаміщених бензолу (в останньому випадку —з різними атомами або групами атомів у бензольному ядрі). Якщо ж виходити з принципів хімії ненасичених сполук, то повинні були б існувати два ізомерні, наприклад, 1,2-диметилбензоли (II і Па), а також два ізомерні 1-нітро-З-хлорбензоли (III і І На) з різним положенням подвійних зв’язків у бензольному ядрі:
Однак у гомологів і похідних бензолу такого виду ізомерія ніколи не спостерігалася, а пізніше було точно встановлено, що такі ізомери добути неможливо.
У 1872 р. А. Кекуле для пояснення причини відсутності ізомерів у 1,2-дизаміщених бензолу запропонував осциляційну гіпотезу, згідно з якою подвійні вуглець-вуглецеві зв’язки в бензольному ядрі не закріплені, а безперервно переміщуються (осцилюють) у кільці молекули, і зображав це формулами І і Іа (для зручності групи СН у молекулі бензолу та інших ароматичних сполук не записують):
Іа
Для підтвердження осци.яяційної гіпотези використовували експериментальні факти, наприклад окислення 1,2-дизаміщених гомологів .бензолу. Так, 1,2-диметилбензол, подібно до бензолу, легко окислюється озоном. При цьому утворюється суміш, яка складається з метилгліоксалю, гліоксалю і діацетилу у співвідношенні 3:2: 1. Утворення цих речовин можна пояснити такою схемою:
328
НдС-С-С-СН
II II о о
Виходячи із схеми, легко помітити, що метилгліоксаль має бути
одним з продуктів окислення осциляційного ізомеру І, а діацетил — ізомеру І а.
Сучасні дані про структуру бензолу не підтверджують гіпотезу осциляції подвійних зв’язків у молекулах бензолу та інших арома-
тичних сполук і наявності в них одинарних і подвійних зв’язків, що чергуються між собою. А результати озонолізу 1,2-ди-метилбензолу свідчать тільки про те, що шість вуглець-вуглецевих зв’язків у бензольному ядрі є рівноцінними.
Велике значення для встановлення структури бензолу мали фізичні методи дослідження, які в поєднанні з хімічними
дали змогу створити уявлення про струк- рис 55 структура туру бензолу і геометричну будову ЙОГО молекули бензолу молекули, що зображена на рис. 55. На
основі даних рентгеноструктурного аналізу та інших фізичних методів (рефрактометрія, визначення дипольних моментів, УФ-, ІЧ- та ЯМР-спектроскопія, спектри комбінаційного розсіювання, електронографія тощо) встановлено:
1. Молекула бензолу являє собою плоский правильний шестичленний цикл, у якому всі шість атомів вуглецю і шість атомів водню розміщені в одній площині і утворюють правильний шестикутник з кутом 120°. Довжини всіх С—С-зв’язків однакові —
0,14 нм і мають приблизно середнє значення між довжинами простого (0,154 нм) і подвійного (0,134 нм) вуглець-вуглецевих зв’язків. Довжини всіх зв’язків Н—С також однакові —0,108 нм
(рис. 55).
2. Молекула бензолу симетрична і має вісь шостого порядку, а не третього, як це випливає з формули А. Кекуле.
3. Ядра атомів і валентні електрони в молекулі бензолу розподілені симетрично. Бензол є неполярною сполукою. Його дипольний момент дорівнює нулю (р. =0).
4. Бензол —діамагнітна сполука і має анізотропію магнітної сприйнятливості. Його молекула є симетричною в магнітному відношенні (рис. 68). Це свідчить про наявність у його молекулі делока-лізованої системи л-електронів в ароматичному ядрі, якає причиною кільцевого струму, що виникає при накладанні магнітного поля,
329
і відповідно причиною магнітної анізотропії. Наявність у молекулі такої л-електронної системи і діамагнітна анізотропія молекули є основною ознакою ароматичності органічних сполук.
5. Загальною властивістю бензолу та інших ароматичних сполук є здатність їх п-електронної орбіталі до деформації під впливом замісників і полярних реагентів. Під впливом замісників електронна густина в бензольному ядрі може збільшуватись або зменшуватись.
6. Бензол і бензольні ядра в молекулах алкілбензолів характеризуються двома основними смугами поглинання в УФ-області: біля 200 нм і біля 260 нм. Перша смуга для бензолу з Хмакс == 198 нм (ємакс = 800) відповідає збудженню л-електронів (л л*-перехід). Друга смуга з Хмакс = 255 нм має невелику інтенсивність (єМакс = 23), називається бензольною смугою і також відповідає л ->л*-переходу низької енергії молекули бензолу. Резонансні сигнали протонів, сполучених з атомами вуглецю ароматичного ядра, у спектрах ЯМР розміщені в області 6,5—8 млн-1, тобто в більш слабкому магнітному полі, ніж резонансні сигнали протонів, які знаходяться біля атомів вуглецю етиленових зв’язків (4,6—6,9 _млн-1).
Квантово-хімічне описання молекули бензолу. Для описання е лектронної будови бензолу квантова хімія використовує два методи: метод валентних зв’язків (ВЗ) і метод молекулярних орбіталей (МО). Згідно з методом ВЗ, молекулу бензолу уявляють як проміжну між граничними структурами І—¥,дві перші з яких (І, II) є структурами Кекуле^а три інші (III—V) — структурами Дьюара:
У граничних структурах Дьюара (III—V) на протилежно розміщених С-атомах крапками зображають р-електрони з протилежними спінами, які здатні брати участь в утворенні ароматичного секстету л-електронів. Граничні структури (І—V) зберігають високу симетрію рівностороннього шестикутника і мовби накладаються одна на одну (резонують), що приводить до делокалізації шести л-електронів у циклі. Отже, молекула бензолу за методом ВЗ характеризується хвильовою функцією фе, яка являє собою лінійну комбінацію хвильових функцій фх —ф3 п’яти граничних структур (І—V). Розрахунки варіаційним методом дають змогу зробити висновок, що найбільший внесок у резонанс дають структу-ри Кекуле — І і II (по 39 % кожна). На кожну із структур Дьюара (III—-V) припадає по 7,3 % . Крім того, можливі полярні граничні структури, на частку яких припадає менш як 1 % і які в розрахунках не враховуються. Структури Кекуле (І і II), які дають найбільший внесок у резонанс і мають найменш)7 енергію, називають основними структурами.
Велика заслуга у становленні сучасних уявлень про будову ароматичних сполук належить Е. Хюккелю, який вперше застосу-
330
вав один з розрахункових методів квантової механіки — метод молекулярних орбіталей в л-електронному наближенні (метод МО ЛКАО) —до ароматичних сполук. Згідно з цими уявленнями, у молекулі бензолу кожний атом вуглецю за рахунок 2з- і двох 2р-орбі-талей утворює о-зв’язки (з двома сусідніми атомами вуглецю і одним атомом водню, зр2-гібридизація, рис. 56). Отже, шість атомів вуглецю в молекулі бензолу сполучені о-зв’язками в рівносторон-ній шестичленний цикл. Кожна орбіталь о-зв’язку має циліндричну симетрію вздовж лінії, яка з’єднує ядра атомів. Крім того, у молекулі бензолу біля кожного з атомів вуглецю є ще по одному 2р-електрону, які мають здатність утворювати л-зв*язки (рис: 57). Отже, у молекулі бензолу є шість 2р-електронів, кожному з яких відповідає свій енергетичний рівень і власна МО. МО бензолу фе
Рис. 56. Схема розміщення а-зв’язків у молекулі бензолу
Рис. 57. 2р-Орбіталі в молекулі бензолу
являє собою лінійну комбінацію хвильових функцій атомних орбіталей ф з коефіцієнтами с, які характеризують внесок кожної МО:
’Ц = С1 Ті + С2Т2 + СзТз + ««Ті + С5ф» + С6(рс.
Шляхом наближеного розв’язування рівняння Шредінгера визначають енергію і вигляд одноелектронних хвильових функцій.
Шість 2р-АО дозволяють побудувати шість л-МО (<рг—<рв). Причому в основному стані шість л-електронів займають орбіталі з найнижчою енергією, якій відповідають зв’язуючі МО —ср3. Останні три орбіталі (<р4—фс) — розпушуючі МО і є вакантними (рис. 58). Оскільки молекула бензолу високоспметрична, то вона має однакові за енергією (вироджені) рівні (Е2=£3і£4==£5). Тому замість шести існує всього чотири енергетичних рівні, на кожному з яких можуть перебувати тільки два електрони. З цих орбіталей найстійкішою є зв’язуюча МО з найнижчою енергією Ех. Ця МО утворюється шляхом взаємного перекриття всіх шести 2р-АО бензольного ядра. л-Електронна орбіталь такої зв’ язуючої МОохоплює все бензольне ядро, є делокалізованою (роззосередженою) поза площиною молекули (рис. 59). Така замкнена шести-л-електронна
331
система (л-електронний секстет) утворює у бензольному циклі ароматичний зв’язок, зумовлює всі найважливіші фізичні та хімічні властивості сполук, які містять бензольне ядро, і є основною ознакою ароматичності органічних речовин. Електронна густина такої л-МО в молекулі бензолу рівномірно розподілена по всій молекулі. Тому дипольний момент бензолу дорівнює нулю.
Отже, в молекулі бензолу, в бензольних ядрах інших ароматичних сполук немає дійсних подвійних зв’язків, таких, як у молекулах етиленових сполук, тому що р-електрони в бензолі не групуються парами (не локалізуються), а електронна орбіталь кожного р-елек-
Рис. 58. Графічне зображення л-молекулярннх орбіталей бензолу
трона перекривається з р-електронною густиною сусідніх атомів. В результаті р-електрони розподіляються симетрично так, що кожний атом вуглецю бензольного ядра сполучений з двома сусідніми атомами не тільки ст-зв’язком, але й р-електронами. При цьому на кожний о-зв’ язок припадає електронна густина одного р-електро-на. Такий зв’язок називають ароматичним і часто зображають у вигляді комбінації риски і пунктирної лінії —С—С—-. Виходячи з такої електронної будови, формулу бензолу найправильніше слід зображати у вигляді рівностороннього шестикутника з вписаною в нього циклічною л-орбіталлю, яка символізує секстет л-електронів:
332
Інколи молекулу бензолу зображають також у вигляді шестикутника з трьома спряженими подвійними зв’язками:
Слід зазначити відмінність між ароматичним і звичайним подвійним етиленовим зв’язком. У молекулі етилену на один о-зв’язок С—С припадає два р-електрони, а бензолу — один р-електрон.
Методом МО для бензолу обчислені порядок С—С-зв’язків та індекси вільної валентності С-атомів, значення яких наведені на молекулярній діаграмі (рис. 60). Як видно з цієї діаграми, поря
Рис. 59. Розподіл л-електронної густини в молекулі бензолу
Рис. 60. Молекулярна діаграма молекули бензолу
док всіх С—С-зв’язків у бензолі однаковий — 1,67. Значення його середнє між порядком одинарного і подвійного зв’язків. Індекс вільної валентності атомів вуглецю у молекулі бензолу також однаковий — 0,4, що характеризує здатність його молекули до реакцій приєднання.
Масштабна модель молекули бензолу Стюарта —Бріглеба показана на рис. 61, а схематичне зображення цієї молекули — на рис. 62.
Ароматичні властивості ароматичних сполук. Своєрідна електронна будова бензолу надає йому і сполукам, які містять бензольне ядро, такі специфічні хімічні властивості:
1. Ароматичні вуглеводні ряду бензолу мають загальну формулу СпН2,і_6 і порівняно з циклопарафінами, загальна формула яких С„Н2п, є сполуками, ненасиченими атомами водню. Виходячи з цього, слід було чекати, що бензол вступатиме в реакції, типові для ненасичених сполук, тобто в реакції приєднання. Проте бензол не знебарвлює бромної води, не приєднує бром, розчинений у СС1}. І навіть тоді, коли бензол реагує з бромом, бром не приєднується до бензолу, а витісняє водень з бензольного ядра.
Отже, бензол і його гомологи більш схильні до реакцій заміщення, ніж до реакцій приєднання. Для бензолу і його гомологів характерні реакції електрофільного заміщення (галогенування,
333
нітрування, сульфування, алкілування, ацилування тощо), у процесі яких зберігається ароматичне ядро. Така стійкість бензолу і його гомологів, незважаючи на високий ступінь ненасиченості, до реакцій приєднання брому та інших електрофільних реагентів є одним із способів визначення ароматичності сполук.
2. Сам бензол і бензольне ядро в молекулах його гомологів стійкі до дії окислювачів. Так, бензол не знебарвлює (не окислюється) водний розчин КМпО4. У гомологів бензолу окислюються
Рис. 61. Масштабна модель молекули бензолу за Стюартом — Бріглебом
Рис. 62. Взаємне перекривання 2р-орбіталей у молекулі бензолу: а — вигляд збоку; б —• вигляд зверху
алкільні радикали, а ароматичне ядро залишається без зміни (див. розд. 29). Отже, бензольне ядро стійкіше до дії окислювачів, ніж насичені вуглеводні.
3. Атоми і групи атомів, сполучені з бензольним ядром, набувають особливих властивостей. Так, ОН-група фенолів набуває кислотних властивостей (див. розд. 34), ИН2-група ароматичних амінів знижує свої основні властивості (див. розд. 36) і т. д.
4. У різноманітних реакціях утворення ароматичних ядер відбувається досить легко, часто з виділенням теплоти. Наприклад, екзотермічно відбувається перетворення циклогексадієну-1,3 в бензол:
+
Д/У—23,4 кДж-моль ' .
334
М. Д. Зелінський у 1911 р. встановив, що з виділенням теплоти відбувається також перетворення циклогексену на бензол:
В результаті реакції з циклогексену з кількісним виходом утворюється суміш бензолу і циклогексану у молярному співвідношенні 1 : 2. Цю реакцію називають реакцією необоротного каталізу. У процесі її відбувається перерозподіл (диспропорціонування) одинарних і подвійних зв’язків та атомів водню. Причому загальна кількість усіх зв’язків С—С, С=С, С—Н після реакції залишається такою самою, як і до реакції. Однак три ізольованих до реакції подвійних (етиленових) зв’ язки в процесі її переходять в одну молекулу —у бензол, в циклі якого зв’язки ароматичні. Особливо цікавими те, іцо реакція супроводиться виділенням енергії,яка становить 150 кДж • моль'1. Це є свідченням того, що молекула бензолу за рахунок спряження трьох подвійних зв’язків стабільніша за молекулу, яка б містила три неспряжених подвійних зв’язки етиленового типу, тобто стабільніша за молекулу циклогексатрієну-1,3,5 (структура молекули бензолу, запропонована Кекуле). Отже, бензол є термодинамічно стійкою сполукою і утворення його відбувається енергетично вигідно. Енергія, яка виділяється під час цієї реакції, являє собою різницю між енергією бензолу і теоретично обчисленою енергією для молекули циклогексатрієну-1,3,5. Ця енергія називається енергією спряження, або енергією резонансу.
Слід зазначити, що бензол, як термодинамічно стійка система, одночасно є термостійкою сполукою і витримує нагрівання до 900 °С без розриву циклу.
Розглянуті вище своєрідні властивості бензолу, ЙОГО ГОМОЛОГІВ і похідних називають ароматичними. Сам бензол прийнятий за еталон ароматичності. Наявність подібних ознак у інших органічних сполук є критерієм їх ароматичності.
Німецький вчений Е. Хюккель у 1931 р. на основі квантово-механічних розрахунків і методу МО узагальнив поняття ароматичності і сформулював правило, яке стало критерієм ароматичності органічних сполук. Згідно з цим правилом, ароматичні властивості матиме будь-яка плоска циклічна сполука із спряженою системою подвійних зв’язків, якщо її цикл містить 4п -|- 2 л- або я ф-р-елек-тронів (и — натуральний ряд чисел). При п = 0,1,2,3 і т. д. число л-електронів у циклі дорівнюватиме відповідно 2,6,10,14 ... .
Бензол є типовим прикладом ароматичної системи з секстетом л-електронів (п — 1). До ароматичних систем належать також вуглеводні з конденсованими бензольними ядрами, такі як нафталін, антрацен, фен а нтр єн тощо, які також відповідають вимогам правила Хюккеля. В той же час циклооктатетраєн-1,3,5,7 ароматичних
335
властивостей не має, а є типовою ненасиченою сполукою:
Циклооктатетраен
Дослідження показали, що причина цього полягає в його електронній будові. Циклооктатетраєн не задовольняє вимогам правила Хюккеля. Кількість л-електронів у циклі цього вуглеводню — 8 (чотири спряжених ПОДВІЙНИХ зв’язки) І не відповідає формулі 4п 2. Довжини С—С-зв’язків у його молекулі такі, як у спряженого дієну —дивінілу. Крім того, молекула циклооктатетраєну непланарна і має конформації «крісла» (І) або «ванни» (II), що не дає можливості електронним орбіталям восьми р-електронів злитися в одну молекулярну орбіталь.
Методи утворення-бензольного ядра. Термодинамічна стійкість бензольного ядра сприяє тому, що в багатьох реакціях і піролітич-них процесах органічних речовин легко утворюються енергетично стабільні бензольні цикли. Так, бензол утворюється при дегідруванні циклоалканів над нагрітою платиною:
Бензол утворюється також у процесі дегідроциклізації алканів» яку проводять над нагрітими оксидами металів. Наприклад:
ССН3-
сн3
АІ2О,ч 450°С
+ 4Н2 .
Утворення бензолу відбувається і при циклічній трпмеризації ацетилену (див. розд. 15).
Хімічні властивості бензолу. Бензол може вступати в реакції приєднання, заміщення, фотохімічної ізомеризації, а також у реакції, які супроводяться розривом бензольного ядра.
. Реакції приєднання. Структурні особливості бензолу зумовлюють міцність і стійкість бензольного ядра до реакцій приєднання. Для того щоб відбулося приєднання реагенту до бензольного ядра, необхідно подолати енергетичний бар’єр спряженої системи л-електронів, що дорівнює 150 кДж • моль-1. Тому бензольне ядро приєднує тільки багаті на енергію реагенти. Такими реагентами є частинки з неспареними електронами, тобто атоми або вільні радикали. У зв’язку з цим приєднання до бензолу відбувається при атомарно-радикальних процесах. До таких реакцій належить приєднання водню при наявності каталізатора, приєднання деяких галогенів на світлі та ін. Так, приєднання водню до бензолу відбувається при нагріванні і наявності каталізатора (Рі), у відсутності £36
якого реакція не йде. Продуктом такого гідрування бензолу є циклогексан:
(з н .2, Р1) 80-1504: ’
При каталітичному гідруванні бензолу не вдається виділити частково гідровані продукти, такі як циклогексен або циклогекса-дієн. Якщо ж відновлення бензолу проводити натрієм у рідкому аміаку при наявності етилового спирту (відновлення за методом Бйорча), то можна добути циклогексадієн-1,4:
(На, КН3, С2Н6ОН)
Хлор і бром приєднуються до бензолу на сонячному світлі (УФ-опромінення) у відсутності каталізатора. Продуктом такого хлорування бензолу є гексахлорциклогексан:
+ 6СІ-
(ЗСІ2І
УФ-свІтло
СНСІ
СІНС^ '''СНС1
1 і
СІНС ^.СНСІ .
>чснсі
При опроміненні УФ-світлом бензол вступає також у реакцію Дільса — Альдера і приєднує 2 моль малеїнового ангідриду, утворюючи при цьому кристалічний аддукт (Тп.„ = 350 °С)-
Реакції, що супроводяться розривом бензольного ядра. Розрив вуглець-вуглецевих зв’язків бензольного ядра відбувається перш за все під час реакцій окислення. Як уже зазначалося, бензол за звичайних умов стійкий до дії окислювачів. Так, він не знебарвлює водного розчину КМпО4 навіть при тривалому кип’ятінні. Однак бензол приєднує 3 моль озону і таким чином окислюється. При цьому утворюється надзвичайно нестійкий триозонід, який під дією води розкладається на гліоксаль і пероксид водню:
Триозсгнід бензолу
+ЗН2О
-ЗН2О2
СН = О
З І
СН=О
Гліоксаль
337,
Кисень повітря при звичайній температурі практично не діє на бензол. Якщо ж бензол підпалити, то він горить на повітрі кіптявим полум’ям. При достатній кількості кисню (кисневе дуття) він згоряє до оксиду вуглецю (IV) і води:
СвНв + 7,5О2 -> 6СО2 + ЗН2О 4* 5350 кДж • моль-1.
Якщо суміш пари бензолу з киснем пропустити над нагрітим каталізатором (У2О5), то відбувається часткове окислення бензолу до малеїнового ангідриду:
+ А, 50,
У2О5
450"С
Ь 2СО2 + 2Н.2О .
Фотохімічна ізомеризація бензолу. Бензол під дією світла може вступати в реакцію фотохімічної ізомеризації, в результаті якої в 60-х рр. нашого століття були добуті валентні ізомери бензолу — бензвален, біциклогексадієн, призман та його структурний ізомер — фульвен, які відрізняються від бензолу кількістю о- і л-зв’язків, а у випадку фульвену —положенням л-зв’язків. Так, при тривалому опроміненні бензолу світлом з довжиною хвилі 254 нмутворюєтьсябензвален (вихідстановить ~ 1 %). Реакція відбувається через стадію утворення бірадикала. Бензвален при дальшому освітленні перетворюється на фульвен:
При швидкому опроміненні бензолу світлом з довжиною хвилі 206 нм утворюється біциклогексадієн (бензол Дьюара), який при дальшому освітленні перетворюється на призман (бензол Ладен-бурга):
Біциклогексадієн Призман
(бензол Дьюара) (бензол Ладенбурга)
Усі валентні ізомери бензолу при нагріванні або освітленні взаємно перетворюються один на другий і легко ізомеризуються в бензол.
'Електрофільне заміщення* в молекулі' бензолу. Для бензолу та інших ароматичних сполук особливо
338
характерні реакції електрофільного заміщення (<$£-реакції) атомів водню бензольного ядра на позитивно заряджені електрофільні частинки Х + . 5£-Реакції для бензолу можуть бути описані такою загальною схемою:
Оскільки в бензолі всі аюми вуглецю і всі атоми водню рівноцінні, то в процесі 5£-реакцій імовірність заміщення кожного з атомів водню однакова. Найважливішими 5д-реакціями ароматичних сполук є галогенування, нітрування, сульфування, алкілування, ацилування та ін. Електрофільні реагенти в цих реакціях або самі виявляють електроноакцепторні властивості, або в процесі 5£-реакцій генерують електрофільні частинки Х+, найважливішії-ми з яких є іони МО2, 8О3Н, /?—С=О, СІ+, Вг+ та деякі інші. Іони цього ряду більш електрофільні, ніж протон, а тому витісняють і таким чином заміщують його у бензольному ядрі. Засвоїм значенням 5£-реакції посідають важливе місце в хімії ароматичних сполук і використовуються для добування різноманітних похідних бензолу і інших ароматичних речовин.
Механізм електрофільного заміщення в бензольному ядрі.У більшості випадків реакції електрофільного заміщення: в бензолі є бімолекулярними, тобто 5£2-реак-ціями.
Бензольне ядро з його секстетом я-електронів є нуклеофільним реагентом і донором електронів, тобто л-основою. Кінетичними і хімічними методами встановлено, що електрофільне заміщення в бензольному ядрі є полярним постадійним процесом, який охоплює утворення кількох проміжних комплексів і частинок. Цей процес можна зобразити у вигляді таких послідовних реакцій:
Першим елементарним актом при електрофільному заміщенні в бензольному ядрі є повільне зближення електрофільної частинки з л-електронною орбіталлюбензольного ядра. Дія частинки напрям
339
лена перпендикулярно до площини бензольного кільця і спрямована на л-орбіталі. В результаті виникає донор но-акцепторний л-комплекс (І), у якому електрофільна частинка фіксована всіма л-елек-тронами бензольного ядра (рис. 63). В окремих випадках л-комплек-си (І) були виділені в індивідуальному стані або помічені спектрофотометрично.
Наступна елементарна реакція — повільне перетворення л-комплексу (І) в нестійкий о-комплекс (II), який являє собою карбо-нієвип катіон. о-Комплекс (II) утворюється внаслідок приєднання електрофілу X + до одного з атомів вуглецю бензольного ядра. В результаті між атомом вуглецю і електрофілом виникає о-зв’язок за рахунок пари л-електронів ароматичного секстету електронів. У бензольному ядрі внаслідок цього залишається тільки чотири л-електрони і воно набуває позитивного заряду.
о-Комплекс — це не перехідний стан, а проміжна сполука, існування якої в ряді випадків було помічено спектрофотометрично. В окремих випадках о-комплекси були виділені у вигляді хімічних сполук. У о-комплексі атом водню залишається ще сполученим з атомом вуглецю ароматичного ядра, до якого приєднався електрофіл X+. У процесі утворення о-комплексу (II) атом вуглецю, який приєднав електрофіл, переходить із тригонального $/?-стану в тетраедричний $/Авалентний стан. У о-комплексі (II) як частинка X, так і атом водню розміщені у вершинах тетраедра і не знаходяться в одній площині з бензольним кільцем. За своєю природою о-комплекс (II) являє собою циклогексадієновий іон карбонію, який вже не має ароматичних властивостей. Чотири л-електрони, які залишилися в циклі о-комплексу (II), роззосереджені між п’ятьма атомами вуглецю бензольного ядра.Користуючись теорією резонансу, о-комплекс (II) можна уявити у вигляді гібрида трьох структур (а—в), які відрізняються положенням подвійних зв’язків і позитивного заряду, або у вигляді мезомерної формули (г):
5£2-Реакція закінчується останнім швидким актом—відщепленням протона від 5р3-гібридизованого атома вуглецю. При цьому пара електронів, яка сполучала цей атом вуглецю з атомом водню, повертається в бензольний цикл і регенерує таким чином ароматичний секстет електронів. Протон Н+, який при цьому утворився, на деякий чає утворює л-комплекє (III). Розпад останнього приводить до утворення кінцевого продукту 5£2-реакції (IV). Утворення проміжних л- і о-комплексів (І і II) не потребує великої затрати енергії, оскільки енергія спряження о-комплексу (109 кДж • моль-1) близька до енергії спряження бензолу (150 кДж - моль-1).
340
Зміна вільної енергії Е системи реакції електрофільного заміщення зображена на енергетичному профілі кривої (рис. 64). Перший невисокий пік і наступний мінімум на цій: кривій характеризують перехідний стан л-комплексу (І). Більш високий пік і наступний мінімум відповідають о-комплексу (II). Потім іде пік і неглибокий мінімум, які відповідають л-комплексу (III).Останній пік і неглибокий мінімум відповідають продукту реакції (IV).
Нуклеофільні частинки, на відміну від електрофільних, не мають вакантних р-орбіталей, тому вони не можуть утворювати л-комплекс з л-орбіталлю бензольного ядра. Крім того, нуклео-
Рис. 63. Схема утворення зт-ком-лексу з бензольним ядром
Координата реакції
Рис. 64. Зміна потенціальної енергії в процесі реакції електрофільного заміщення в молекулі бензолу
фільні частинки не можуть наблизитися до бензольного ядра, оскільки його л-МО ці частинки відштовхує. В результаті для бензолу реакції нуклеофільного заміщення не характерні.
Розділ 29. БЕНЗОЛ ТА ЙОГО АЛКІЛЗАМІЩЕНІ
Бензол є першим членом гомологічного ряду ароматичних вуглеводнів, склад яких відповідає загальній формулі С„Н2,г_е (табл. 27). Як і в інших гомологічних рядах, в ряду ароматичних вугле-
Таблиця 27. Ароматичні вуглеводні ряду бензолу
Формула Назва 7пл» °С ГКнп. °С 20 Густина
с6н6 Бензол 5,5 80,1 0,879
свн„сн3 Толуол (метилбензол) —95 110,6 0,867
С6Н4(СН3)а К силол (диметилбензол)
орто- (1,2-) —28 144,5 0,880
меіїїа- (1,3-) —53 139 0,864
пара- (1,4-) 13 138,4 0,861
Етилбензол —94 136 0,867
С„Н6СН2СН2СН3 Пропілбензол —99,5 159 0,863
С0Н5СН-СН3 Ізопропілбензол (кумол) —97 152,4 0,862
1,2,3-Т риметилбензол —25,4 176,1 0,894
сн3 1,2,4-Трпметилбензол —43,8 169,4 0,875
С6Н3(СН3)3 1,3,5-Трпметилбензол —44,7 164,7 0,865
341
воднів кожний наступний член відрізняється від попереднього на одну або кілька груп —СН2.
Номенклатура. Ароматичні вуглеводні називають як за історичною (толуол, ксилол, кумол і т. д.), так і за систематичною номенклатурою. Історичні назви походять переважно від назви джерела, з якого вперше був виділений даний вуглеводень.
За систематичною номенклатурою всі сполуки ароматичного ряду розглядають як заміщені бензолу, метилбензол, етилбензол, пропілбензоліт.д. При наявності в бензольному ядрі двох або більшої кількості груп атомів (замісників) положення їх зазначають цифрами. Нумерацію вуглецевих атомів бензольного ядра ведуть за годинниковою стрілкою, пам’ятаючи, щоб величина цифр у назві була найменшою. Наприклад, двозаміщений гомолог бензолу будови
сн3
&
СН3
за систематичною номенклатурою має назву 1,3-диметилбензол, а не 1,5-диметилбензол. Для багатозаміщених бензолу нумерацію атомів вуглецю бензольного ядра починають від «старшого» замісника. За «старшинством» замісники розміщуються в такий ряд:
СН3— > С2Не— > С3Н7— > —8О3Н > /?—С— >
>—ОН > —>—МО2 > -Р > —СІ > —Вг >—І Зменшення старшинства
Наприклад:
1-Метил-2-ети.т4-хлорбензол
.При відщепленні одного або кількох атомів водню від ароматичного вуглеводню утворюється радикал цього вуглеводню. Будова і назви деяких радикалів наведені в табл. 28.
Ізомерія. Ізомерія гомологів і похідних бензолу зумовлена ізомерією радикалів (замісників), їх кількістю та положенням у бензольному ядрі. Сам бензол ізомерів не має, оскільки всі шість атомів водню в його молекулі рівноцінні. При введенні в бензольне ядро замість атома водню алкільного радикала або будь-якого іншого атома чи групи атомів можливе існування тільки одного продукту заміщення, тому що всі шість положень у бензольному ядрі рівноцінні. У зв’язку з цим для однозаміщених бензолу немає
342
Таблиця 28. Радикал ароматичних вуглеводнів
таких ізомерів, ізомерія яких була б викликана положенням радикала, атома або групи атомів у бензольному ядрі. Отже, толуол, етилбензол ізомерів не мають. Однозаміщені бензолу можуть мати тільки такі ізомери, ізомерія яких зумовлена ізомерією самого радикала, на який замінили атом водню бензольного ядра. Наприклад:
Пропілбензол
Ізопропілбензол
Ізомерія двозаміщених бензолу пов’язана з положенням замісників (атомів або груп атомів, які заміщують у бензольному ядрі атоми водню), а також ізомерією самих радикалів і їх величиною. Двозаміщені бензолу можуть мати замісники в 1,‘2-(орто-), 1,3-(мета) і 1,4 (пора-) положеннях. Наприклад, диметилбензол (ксилол) існує у вигляді таких ізомерів:
1,2-Диметилбензол, с-ксилол
і,3-Диметилбензол, . х-ксилол
1,4-Диметилбензол, я-ксилол
Ізомери орто-, мета- і пара- позначають скорочено літерами о-, м-, п-.
Починаючи з метилпропілбензолу крім о-, м-, и-ізомерів існують ізомери з ізомерними радикалами (о-, м-, п-метилпропілбензоли
343
і о-, м-, п-метилізопропілбензоли) та ізомери, ізомерія яких викликана різною величиною радикалів (о-, м-, и-метилпропілбензо-ли І О-, м-, п-діетилбензоли).
Заміщені бензолу з трьома однаковими замісниками існують у вигляді трьох ізомерів. Наприклад, для триметилбензолу вони мають таку будову:
1,2,3-Триметилбензол
1,3,5-Трпметилбензол
Якщо три замісники різні, то для такої тризаміїценої бензолу можливі 10 ізомерів, які відрізняються положенням замісників у бензольному ядрі.
£, г
230 260 250 260
Довжина лвиД, нм
Рис. 65. УФ-спектр поглинання бензолу (в циклогексані), на якому видно бензольні смуги
Фізичні властивості.Гсензол і його гомологи, як правило, — безбарвні рідини, які сильно заломлюють світло, іноді це тверді речовини. Вони мають специфічний запах. Температури їх кипіння дещо вищі,ніж температури кипіння насичених вуглеводнів з такою самою кількістю вуглецевих атомів. Наприклад, 7’ІШП бензолу становить 80,1 °С, а н-гексану— 68,8°С. Температури кипіння ізомерних ароматичних сполук відрізняються незначною мірою. Орто-ізомери киплять при вищій температурі, ніж пара-ізомери. Кожна нова група —СН2 підвищує температуру кипіння ароматичного вуглеводню приблизно на ЗО °С. Ізомери з симетричною будовою мають вищу температуру плавлення. З дизаміщених бензолу пара-ізомер плавиться при найвищій температурі (див. табл. 27). Густина ароматичних вуглеводнів значно вища, ніж насичених вуглеводнів нециклічної і циклічної будови.
Для ароматичного ядра характерне поглинання УФ-світла в області 180—300 нм., Інтенсивність цієї смуги в УФ-спектрі неве
344
лика. При високій роздільній здатності приладу вона розпадається на кілька вузьких піків (рис. 65). Такий вигляд спектра досить характерний для ароматичних вуглеводнів, у зв’язку з чим його часто називають бензольною смугою поглинання.
В ІЧ-спектрах для ароматичного ядра характерні дві смуги поглинання в областях 1600 і 1500 см-1, які відрізняються за інтенсивністю і пов’язані з валентними коливаннями С—С-зв’язків ароматичного ядра, і чіткі піки в області 3030 см-1, характерні для ароматичних зв’язків С—Н. Надзвичайно важливим є те, що за ІЧ-спектрами можна визначити кількість і положення замісників у бензольному ядрі. Так, смуги поглинання в областях 1650—2000, 1225—950,900—700см-1 дають змогу встановити характер заміщення в бензольному ядрі. На рис. 66 наведені ІЧ-спектри толуолу, о-, м- і п-ксилолів, на яких чітко виділяються смуги поглинання валентних коливань С^С- і С—Н-зв’язків, а за характером смуг в областях 2000—1650 і 800—690 см-1 легко встановити кількість і положення замісників у ядрі.
У спектрах ПМР резонансні сигнали ароматичних протонів (6,5—8 млн-1) розміщені в слабшому магнітному полі, ніж резонансні сигнали протонів, які знаходяться біля звичайних подвійних зв’язків (4,6—6,9 млн-1). Ця різниця переважно має величину близько 2 млн-1.Так, бензол дає у ПМР-спектрі один вузький сигнал при 7,37 млн-1 (рис. 67). Наявність різниці в хімічних зсувах ароматичних протонів і протонів у алкенах, незважаючи нате, що вони як в ароматичному ядрі, так і в алкенах пов’язані з 5р2-вуглецевими атомами, зумовлена особливими властивостями л-електронної системи ароматичних сполук, електрони якої можуть вільно циркулювати над і під площиною бензольного ядра, як це зображено на рис. 68. Коли л-електронна система молекули бензолу піддається дії зовнішнього магнітного поля Но, в ній збуджується потік л-елек-тронів вздовж ядра, що п ри водить до появи так званого кільцевого струму. Цей кільцевий струм, у свою чергу, збуджує аксіальне магнітне поле, яке підсилює поле Но ззовні ядра, в районі атомів водню і напрямлене проти нього всередині кільця( рис. 68). В результаті протони молекули бензолу дезекрануються і вступають у резонанс при нижчих значеннях Но, тобто в області слабшого поля. Виявляється, що кільцевий струм такого напрямку і такої сили виникає тільки в молекулах ароматичних сполук. Тому наявність його, про що свідчить зміщення сигналів хімічних протонів в область слабшого поля,є найпоширенішим експериментальним критерієм ароматичності. У цьому легко переконатися, порівнявши спектри ПМР бензолу(ароматичного вуглеводню), фурану (ароматичної гетероциклічної сполуки) і циклооктатетраєну (неароматичної циклічної ненасиченої сполуки; див. рис. 67).
Добування гомологів бензолу. У наш час основними джерелами добування ароматичних вуглеводнів є кам’яне вугілля і нафта. При коксуванні кам’яного вугілля добувають кокс, газоподібні продукти і кам’яновугільний дьоготь. Газоподібні продукти сухої перегонки кам’яного вугілля називають коксовим газом. Йо складу
345
Довжина хвилі
З 0 5 5 Є 7 8 9 10 12 10
3600 '28ОО ' 2000 2000 1800 /600 1000 1200 1000 800
Частота, см-і
Довжина хвилі
цього газу входить водень (приблизно 50 %), метан (приблизно ЗО %), етиленові, ароматичні та інші вуглеводні. Коксовий газ пропускають крізь ряд поглиначів і таким чином з нього виділяють легке масло, яке містить до 60 % бензолу, толуолу та інших ароматичних вуглеводнів. У наш час більшу частину бензолу в промисловості добувають цим методом.
З кам’яновугільного дьогтю виділено більш як 120 різних речовин, серед яких є ароматичні вуглеводні, ароматичні кисневмісні речовини (феноли), азотовмісні речовини (піридин, хінолін), сірковмісні речовини (тіофен)та ін. Під час фракційної перегонки кам’яновугільного дьогтю добувають кілька фракцій:
1) легке масло, яке переганяється при температурі до 160 °С. Ця фракція містить бензол, толуол, ксилоли та інші ароматичні вуглеводні;
2) середнє, або карболове, масло, яке переганяється при 160—230 °С і складається в основному з ряду фенолів;
3) важке, або креозотове, масло, яке містить нафталін і переганяється при 230—270 °С;
346
Довжина хвилі
3600 2800 2000 2000 1800 1600 1400 1200 1000 800
Частота, сиг1
Довжина хвилі
3 4 5 5 6 7 8 9 10 12 74
толуолу, О-, ЛІ- і п- ксилолів
4) зелене, або антраценове, масло, яке переганяється при 270— 360°С. До складу цього масла входить антрацен.
Після перегонки кам’яновугільного дьогтю залишається твердий залишок —• пек.
Важливим джерелом ароматичних вуглеводнів є нафта. Більшість видів нафти містять ароматичні вуглеводні у невеликій кількості Однак є нафти, багаті на ароматичні вуглеводні (див. розд. 27). З метою підвищення масової частки ароматичних вуглеводнів у нафті її піддають ароматизації, в результаті чого з нафти вдається добути 15—18 % ароматичних вуглеводнів.
Відомо також багато синтетичних методів добування ароматичних вуглеводнів. З них найчастіше використовують такі:
1. Дія металічного натрію на суміш галогенобензолу з галоге-налкілом (використовують переважно броміди), тобто реакція Вюрца —Фіттіга (1864 р.), яка є окремим випадком реакції Вюрца (див. розд. 7). Наприклад, етилбензол за цим методом добувають з бромбензолу і бромистого етилу за схемою:
С6Н6—Вг + 2Ка 4- Вг—С2Нв-> С6НВ—С2НБ + 2№Вг.
347
2,0 ЗР 4,0. 5,0 Є,0 7,0 8,0 -9,0 10 Чг,млн4
|і7Г.ігі,Ігііі1іГііііі?.ііІн.іІ,<ііІііііі!<ГнІІі|іПндЦіпи/Іі|Ііпііі^ДптіЦііііІіі>ііІпз
500 400 300 200 100 0 Гц
8,0 7,0 6,0 5,0 4,0 3,0 2,0 1,0 8, млн'
2,0 3,0 4,0 5,0 6,0 а 7,0 8,0 9,0 ^мт'1
ГПігіі|іппі|і!Гі іинін і;лтп гіі ггіімііііііі1цііііі|і.шпіґніпт7тгпгілігп 500400300?ОП 100 0 Гц
8,0 7,0 6,0 5,0 4,0 х 3,0 2,0 1,0 0 8,млн-'
2,0 3,0 4,0 50 6,0 й 70 8,0 9,0 ІО^млн''
500 400 300 200 100 0 Гц
8Р 7,0 6,0 5,0 4,0 3,0 2р 1р 0 &,мл8'
6
Рис. 67. Спектри ПМР бензолу (а), фурану (б) і цикло-октатетраєну (в)
Механізм цієї реакції аналогічний механізмові реакції Вюрца.
У процесі реакції Вюрца —Фіттіга утворюються також побічні продукти, такі як дифеніл і алкани:
2СвН6—Вг + 2№ > С6Н5—СеН5 + 2ИаВг;
Дифеніл
2С2Н5—Вг + 2№ —> С4Н10 + 2№Вг.
Бутан
2. Алкілування ароматичних вуглеводнів (метод Фріделя — Крафтса, 1877 р.). Ароматичні сполуки, наприклад бензол, при нагріванні з галогеналкілами або алкенами при наявності каталіза-
348
торів алкілуються і утворюють гомологи бензолу. Наприклад, з бензолу і хлористого етилу або етилену за цим методом добувають етилбензол:
СН2-СН3
н2с = сн 2
АІС13
Н3С —СН2—СІ ,.АІСІ3
-НСІ
сн2-сн3
Каталізаторами таких реакцій є безводний А1С13 та інші кислоти Льюїса (РеС13, 8пС14, ВР3, 7пС12 , НР, Н28О4, Н3РО4). Алкілуючими-речовинами у цій реакції можуть бути також спирти та їх ефіри. Алкілування за методом Фріделя — Краф-тса є бімолекулярною реакцією електро-фільного заміщення 5е2, швидкість якої відповідає рівнянню:
V = к [Ароматична сполука] [Алкілуючий реагент].
Рис. 68. Електронна густина, кільцевий струм і силові лінії магнітного поля бензольного ядра
Початковою стадією цієї реакції є утворення між А1С13 і алкіл-галогенідом комплексу з високополярним зв’язком вуглець—• галоген:
. С+ 6+
Н3С—Сі: + А1С13 Н,С---СІ---А1С13.
Комплекси А1С13 з первинними алкілгалогенідами без попередньої дисоціації утворюють з бензолом о-комплекси, які потім розкладаються з утворенням гомолога бензолу, ПСІ і А1С13:
С-Комп.'іекс
Третинні алкілгалогеніди дають з А1С13 комплекси, які дисоціюють на третинний катіон і А1С1“. Катіони, що при цьом-' утворилися, взаємодіють з бензолом за схемою:
(Н3С)3С— СІ + АІСІ3
ЇНзОаС + А1СІ
-Комплекс
349
Прикладом промислово важливої реакції алкілування є синтез етилбензолу та ізопропілбензолу (кумолу) з бензолу та відповідно етилену і пропілену:
сн2— сн3
н2с=сн2
АІСІз
Ізопропілбензол, кумол
У цьому випадку алкілування також є реакцією електрофільного заміщення, в якій карбкатіон виникає в процесі каталітичної дії кислот Льюїса:
АІСІз + Н2о (сліди) неї 4 АІ(ОН) СІ2 ; АІ СІ з + НСІ Н+ [АІСЦГ і
сн2=сн2 + н+—- н3с - сн2+;
8~ + + .
Н3С-СН=СН2 4 Н -—► Н3С-СН -СН3;
СН-СНз [^ҐсНз+іГ.
3. Промислове значення для добування бензолу і його гомологів має ароматизація циклопарафінів (див. розд. 27) і алканів нафти. Принципи ароматизації нафти розробили радянські вчені М. Д. Зелінський, Б'. О. Казанський, А. Ф. Плате, роботами яких було встановлено, що насичені вуглеводні, молекули яких містять шість і більше С-атомів, при пропусканні над нагрітими каталізаторами (Рі або^рксид хрому, нанесений на А13О3) перетворюються на ароматичні вуглеводні. Так, з н-гексану за таких умов утворюється бензол, а з н-гептану —-толуол:
• - Рі
н3с - сн2 - сн2 - сн2- сн2 - сн3--
Н3С—СН2—СН2~СН2 —СН2 —СН2 —СН3
4 4Н2 :
4 4 Н2 .
Після такої дегідроциклізації вміст ароматичних вуглеводнів у нафті підвищується до 50 %.
, Електронна будова гомологів бензолу. Якщо в бензольне ядро замість атома водню ввести який-небудь вуглеводневий радикал, то цей радикал, виявляючи позитивний індукційний ефект, збільшує електронну густину бензольного ядра і поляризує його, тобто порушує в ньому рівномірний розподіл електронної густини. Це впливає на реакційну здатність гомологів бензолу. Наприклад, метальна група в молекулі толуолу збільшує електронну густину 350
бензольного ядра і поляризує його так, що на вуглецевих атомах 2, 4, 6 виникає надлишок електронної густини:
Н
Н—с— Им
Якщо електронну густину на атомах^'вухлейю в молекулі бензолу взяти за одиницю, то розподіл -електронної густини в молекулі
толуолу порівняно з бензолом буде таким:
Тому заміщення в молекулі толуолу підчас електрофільних реакцій відбуватиметься в основному біля вуглецевих атомів з найбільшою електронною густиною, тобто в положеннях 2,4,6. Якщо в реакцію вводити на 1 моль толуолу 1 моль електрофільного реагенту, то утворюється суміш орто- і иара-ізомерів монозаміщеної толуолу, а при достатній кількості реагенту—-тризаміщена толуолу:
351
Усі алкільні радикали збільшують електронну густину бензольного ядра, тому швидкість електрофільного заміщення в бензольному ядрі гомологів бензолу набагато більша, ніж у молекулі бензолу. Д. Бекер і його учень В. Натан помітили, що різні алкільні радикали мають різний вплив на активність бензольного ядра. Так, швидкість реакцій хлорування і бромування алкілбейзолів змінюється таким чином:
[ Алкілбензоли
Швидкість хлорування
Швидкість бромування
З наведених даних видно, що найбільше підвищує активність бензольного ядра До електрофільного заміщення група —СН3, а найменше —група —(СН3)3С. Такий вплив алкільних радикалів на активність бензольного ядра пояснюють так. Завдяки безпосередньому зв’язку між бензольним зр2-атомом вуглецю і яр8-атомом вуглецю метильної або іншої алкільної групи електрони о-СН-зв’язків збуджені і перебувають на енергетично вищих орбі-талях, які наближаються до р-орбіталей. Внаслідок цього відбувається спряження між електронами о-СН-зв’язків алкільних радикалів і я -електронами бензольного ядра (о, л-спряження). Таку взаємодію електронів о-зв’язків з л-електронами Д. Бекер і В. Натан назвали надспряженням, або гіперкон’югаціею (ефект Натана — Бекера).
Чим більше о-СН-зв’язків спряжені з л-електронамибензольного ядра, тим сильніше виявляється ефект надспряження і тим більшою буде електронна густина ароматичного ядра, а відповідно тим вищою буде його здатність до реакцій електрофільного заміщення. Тому толуол, метальна група якого має три о-СН-зв’язки, виявляє більшу активність до реакцій електрофільного заміщення, ніж етилбензол, ізопропілбензол та тре/п-ізобутилбензол, у молекулах яких кількість спряжених з бензольним ядром о-СН-зв’язків менша. З тієї ж причини здатність бензольного ядра до електрофільного заміщення в ряду етилбензол, ізопропілбензол, ізобутил-бензол зменшується відповідно до зменшення в молекулах цих речовин кількості спряжених з бензольним ядром о-СН-зв’язків.
Отже, чим більше в алкільному радикалі гомолога бензолу о-СН-зв’язків у а-положенні відносно бензольного ядра, тим більший ефект спряження такого радикала, тим вищу електронодонор-ність він виявляє. Тому заелектронодонорністю алкільні радикали, сполучені з бензольним ядром, розміщуються в такий ряд:
СН3— > СН,—СН,— > (СН,), СН— > (СН,),С— ,
<---2-—----2----'_і—1---______1—_--_______Надспр яження
-)- /-Ефекі-------------------------------------
352
У цьому ряду надспряження (ефект Натана — Бекера) зменшується в послідовності, зворотній до зменшення -(-/-ефекту алкільних радикалів. Отже, група СН3—та інші алкільні радикали виявляють ф-/- і 4-Л4-ефекти, які напрямлені в бік бензольного ядра і збільшують його електронну густину, чим підвищують здатність ароматичного ядра до реакцій електрофільного заміщення. Крім того, ці радикали поляризують бензольне ядро так, що на вуглецевих атомах в орто- і пора-положеннях збільшується електронна густина і тому електрофільне заміщення відбувається переважно в цих положеннях:
Наслідком надспряження в молекулі толуолу є деяке послаблення зв’язків атомів водню з атомом вуглецю метильної групи, тобто підвищення активності атомів водню цієї групи. Надспряження в молекулі толуолу зображають у вигляді граничних формул І—III:
Формули II і НІ відображають певну рухливість і підвищену реакційну здатність атомів водню метильної групи, а також пояснюють, чому підчас електрофільних реакцій заміщення в молекулі толуолу відбувається в орто- і пора-положеннях.
Підвищення активності атомів водню метильної групи в толуолі виявляється в деякій їх кислотності (рКтол =40,9), внаслідок чого атом водню метильної групи може заміщуватись атомами лужних металів:
С„Н3-СН3 + С2Н6-Ма С6Н6—СН2—Ма + С2Нв (рКетану = 42). Етил натрій Бензилнатрій
. Окислення бічних вуглецевих ланцюгів а л к і лбе нзо л і в. Алкілбензоли, на відміну від бензолу, порівняно легко окислюються різними окислювачами (КМпО4, К2Сг2О7, О2 та іншими) до ароматичних кислот. При цьому незалежно від довжини бічного вуглецевого ланцюга окислення відбувається так, що карбоксильна група ароматичної кислоти, яка утворюється, завжди сполучена з бензольним ядром. При окисленні 12 1-120 353
моноалкілбензолів утворюється бензойна кислота. Окислення діалкілбензолів дає бензолдикарбонові кислоти:
Бензойна кислота
+ Н3С-СООН+Н2О
Бензойна кислота
ЗО2,СоВг 130'160-С
Терефталева кислота
Ароматичні вуглеводні з подвійними зв’язками в бічному ланцюгу. Останнім часом важливого значення набули ароматичні вуглеводні з подвійними С=С-зв’язками в бічному ланцюгу. Найпростішим представником таких вуглеводнів є стирол (вінілбензол, або фені лети лен).
Стирол (7КИП = 146 °С) у промисловості добувають
СН=сн2
Стирол
дегідрогенізацією етилбензолу над оксидом цинку при температурі ' 600 °С:
Стирол виявляє всі властивості ненасичених сполук етиленового ряду. Так, він легко приєднує за місцем подвійного зв’язку галогени, галогеноводні, воду та інші реагенти. Причому приєднання полярних реагентів до стиролу відбувається за правилом Марковникова:
1-Хлор-1 -фей Іл ета н (а-хлоретилбензол)
1,2-Дибром-1-фенІлетаи (диброметилбензол)
354
Найважливішою властивістю стиролу є його здатність до легкої полімеризації, яка відбувається за схемою:
Полістирол
Полістирол має високий електричний опір, високу вологостійкість і використовується як електроізоляційний матеріал, а також для виготовлення іграшок, упаковочного і термоізоляційного матеріалу. Полістирол, фенільні залишки якого містять різні функціональні групи (—8О3Н, —ЬІ(СН3)2 тощо), використовують як іонообмінні смоли (іоніти).
Стирол використовують також для співполімеризації з бутадієном. При цьому добувають синтетичний бутадієн-стирольний каучук СКС (див. розд. 16).
Розділ ЗО. ПРАВИЛА ОРІЄНТАЦІЇ ЕЛЕКТРОФІЛЬНОГО ЗАМІЩЕННЯ В БЕНЗОЛЬНОМУ ЯДРІ
І У молекулі бензолу, як уже зазначалося, всі атоми водню рівноцінні. То»му будь-який з них з однаковою імовірністю може бути заміщений при Зе-реакціях. Отже, вхід першого замісника в бензольне ядро є рівноімовірним процесом. Якщо ж в бензольному ядрі замість атома водню стоїть яка-небудь група атомів або атом галогену, то ці замісники порушують рівномірність розподілу електронної густини в ароматичному ядрі і поляризують його. Внаслідок цього електронна симетрія бензольного ядра порушується. Підтвердженням цього є наявність у монозаміщених бензолу дицрльних моментів р, тоді як сам бензол дипольного моменту не має) (табл. 29).
Таблиця 29. Дипольні моменти монозаміщених бензолу
Формула И. о Формула ц. о
с6н6 0,00 С6Н5-МО2 4,23
СвН6—СН3 0,34 СеН5—СК! 4,39
свн6-сі 1,56 /О с„н5-с^ хн 2,77
СеН6—он 1,40 СсН6-МН2 1,48
12*
355
У монозаміщених бензолу С6Н3—7 є два орто-, два мета- і одне ла/?а-положення:
ї І
\ орто орто
мета IX—У4 мета
пара
; У зв'язку з впливом замісника 7 на бензольне ядро атоми водню в молекулах таких монозаміщених стають нерівноцінними для наступного електрофільного заміщення^;
У молекулах ароматичних вуглеводнів існує кілька видів взаємного впливу. їх називають ефектами. Передача впливу від замісника на ароматичне ядро через о-зв’язки називається сигма-ін-дукційним ефектом (/0). У хімічних формулах він схематично зображається прямою стрілкою.
Наявність біля вуглецевого атома ароматичного ядра замісника з вільною електронною парою або вакантною р-орбіталлю приводить до змішування (спряження) вільних, або вакантних р-орбіталей замісника з л-орбіталлю ароматичного ядра і перерозподілу електронної густини в цілому ядрі. Такий ефект називається мезо-мерним ефектом (М-ефект). Залежно від напрямку зміщення електронної густини ЛІ-ефект може бути позитивним (4-М) і негативним (—М). М-Ефект, в результаті якого електронна густина л-молекулярної орбіталі бензольного ядра збільшується, називають позитивним. Протилежний ефект називають негативним. Зфект спряження в хімічних формулах зображають зігнутою стрілкою:
4-М-Ефект виявляють замісники:
—Р, —СІ, —Вг, —І, —ЬЇН2, —Ш2.
—М-Ефект виявляють замісники:
-с-к, -с\ .
II 4 он о
М-Ефект зменшується при збільшенні електронегативності атома, який несе електронну пару, внаслідок пониження тенденції такого атома віддавати цю пару:
а також при збільшенні об’єму атома, який несе електронну пару:
> :ер- > : вГ^ > „
Тому бензольне ядро фторбензолу більш здатне до реакцій електрофільного заміщення, ніж хлор- і бромбензолу. Так, встановлено, що швидкості нітрування фтор-, хлор- і бромбензолів відносяться як 5 : 1, 1 : 1. Ефект спряження зменшується в тому випадку, якщо атом, який несе вільну електронну пару, сполучений з групою — акцептором електронів. У зв’язку з цим -фУИ-ефект ацета-міногрупи менший, ніж аміногрупи:
Нок' - > Н3С — С — гії-П- .
У спряженні електронної густини беруть участь р-електрони вільних електронних пар і л-електрони бензольного ядра. На відміну від індукційного ефекту, ефект спряження поширюється тільки по ланцюгу спряження. Ефекти спряження можуть бути протилежні за знаком індукційним ефектам груп або атомів. Наприклад, у молекулах фенолу, аніліну індукційний ефект приводить до зміщення електронної густини бензольного ядра до атомів кисню, азоту, які більш електронегативні, ніж вуглець. Схематично у формулах це показують прямою стрілкою. Ефект спряження в молекулах цих речовин діє в протилежному напрямку. Викликано це тим, що електронна густина р-електронів кисню, азоту і л-молекуляр-ної орбіталі бензольного ядра за рахунок спряження стає спільною. В результаті р-електронна густина цих гетероатомів змішується з л-електронною густиною бензольного ядра і зміщується з гідроксильної групи, з аміногрупи на бензольне ядро, що схематично зображають зігнутими стрілками:
На основі кількісних і кінетичних досліджень встановлено, що подальше електрофільне заміщення у молекулах монозаміщених бензолу відбувається з участю будь-якого з п’яти водневих атомів бензольного ядра, тобто порто-, мета- і пара-положеннях. Проте оскільки замісник 2 впливає на електронну густину ароматичного ядра (збільшує або зменшує її) і відповідним чином поляризує його, то вхід другого замісника в ароматичне ядро є конкурентним процесом і місце входження його в ароматичне ядро визначається електронною природою першого замісника 2. Залежно від природи 2 і умов реакції заміщення водню в орто-, мета- і пара-положеннях монозаміщених бензолу відбувається з різними швид
357
костями. Внаслідок цього орто-, мета- і пара-дизаміщені бензолу утворюються з різними виходами.
Отже, природа 7 визначає місце входження в ароматичне ядро наступного замісника. Тому замісник 7, введений першим в ароматичне ядро, називають орієнтантом. Замісники (орієнтанти) за своєю орієнтуючою дією в реакціях електрофільного заміщення (5£-реакціях) поділяють на дві групи: замісники першого роду, або орто-, пара-орієнтанти, і замісники другого роду, або мета-орієнтанти. До замісників першого роду належать такі групи атомів, як —ОН, —ОД, ДСОО—, —8Н, —8Д, —ЬІН„ —МНД, —МД2, —МНСОД, —И=М—, —СН3, —СН.2Д, —СД3‘а також атоми —Р, —СІ, —Вг, —І (де Д —вуглеводневий радикал). Замісники першого роду (за винятком галогенів) збільшують електронну густину бензольного ядра і поляризують його так, що на атомах вуглецю в орто- і пара-положеннях виникають часткові заряди 6": '
Тому при наявності в ароматичному ядрі замісника першого роду електрофіл £+ у 5£-реакціях атакуватиме переважно орто- і пара-положення монозаміщених бензолу і заміщення відбуватиметься переважно в цих положеннях^Заміщення в орто- і пара-положен-нтх у молекулах таких ароматичних сполук, крім того, є енергетично вигіднішим, ніж заміщення в жта-положенні, оскільки при цьому виникають о-комплекси (карбкатіони І—IV), в яких замісник 7 бере участь у делокалізації позитивного заряду катіона, що стабілізує такі катіони. Якщозамісник7, наприклад, алкільний радикал, не має вільної електронної пари, то він стабілізує о-комплекси (І і II) за рахунок -(-/-ефекту, в результаті якого відбувається зміщення пари електронів від цього радикала до бензольного карбонієвого іона і зменшення таким чином позитивного заряду в о-комплексі:
н
, Якщо замісник 7 має вільну пару електронів, наприклад —ГЯН2, —ОН, —ОСН3 та інші групи, то в цьому випадку він передає цю пару електронів у спільне користування сполученому з ним атому вуглецю бензольного ядра. Це приводить до утворення позитивно
368
зарядженого амонієвого, оксонієвого або іншого катіона, що також стабілізує а-катіон (III, IV):
пі
Якщо ж електрофіл £ + атакує в таких сполуках мета-поло-ження, то утворюються енергетично менш стійкі о-катіони (V. VI), оскільки у них не відбувається компенсації позитивного заряду за рахунок замісника 7:
г
Том^за->ііщеиня в ліета-ноложення в таких сполуках є енергетично несприятливим.
Швидкість реакції електрофільного заміщення в бензольному ядрі залежить від швидкості уворення л-комплексу (див. розд. 28). А оскільки замісники першого роду збільшують електронну густину ароматичного ядра, то з таким ядром електрофіл утворюватиме л-комплекс швидше, ніж з бензолом і відповідно електрофіль-не заміщення відбуватиметься легше і активніше, ніж з бензолом. Отже, замісники першого роду підвищують здатність бензольного ядра до реакцій електрофільного заміщення (виняток складають галогени, які дезактивують бензольне ядро до Зд-реакцій), тому їх називають електронодонорними, або позитивними, замісниками.
Замісники першого роду за характером орієнтуючої дії поділяють на такі групи:
1. Слабкіорієнтанти. Ці орієнтанти виявляють -(-/-ефект, мезо-мерний ефект у них виявляється слабко, інколи практично дорівнює нулю. До слабких орієнтантів відносять алкільні групи, фенільну групу С6Н5—, групу —СН2СООН. Алкільні групи за силою -(-/-ефекту розміщуються в ряд
(СН3)3С— > (СН3)2СН- > СН3—СН2— > СН3—.
Незважаючи на те що алкільні радикали мають слабку орієнтуючу дію, вони,виявляючи позитивний індукційний ефект (-(-/-ефект), збільшують електронну густину ароматичного ядра і поляризують його так, що на вуглецевих атомах 2,4,6 (тобто в орто- і пара-поло-женнях) зосереджується більше електронної густини, тобто виникають часткові заряди 6". На л/е/ио-положення електронний ефект замісника в цьому випадку майже не поширюється. Відносні
359
значення зарядів 6“ у молекулі толуолу, розраховані за методом МО, мають такі значення:
2. Орієнтанти сильної [середньої сили. Сильними орієнтантами є —О~, —N7^2. —№НД, —Т4Н2, —ОН, —ОСН3. До орієнтантів середньої сили відносять —N14—С—Д, —б—С—/?. Орієнтанти
II " II О О
сильної і середньої сили містять атоми кисню, азоту (гетероатоми), сполучені з атомом вуглецю бензольного ядра, які мають вільні пари електронів і виявляють позитивний ефект р, л-спряження (ф-Л1-ефект). Разом з тим ці замісники мають негативний індукційний ефект (—/-ефект). Однак у цьому випадку ф-ЛЇ-ефект значно перевищує —/-ефект (ф-ЛІ-ефект >—/-ефекту).Тому такі замісники підвищують електронну густину бензольного ядра і активують- його до Зй-реакцій, а також поляризують це ядро так, що часткові заряди б- зосереджуються в орто- і наро-положеннях. Відносні величини зарядів 6~, наприклад, у молекулі аніліну мають такі значення:
- 0,02
Передачу р-електронної густини гетероатома 7. по системі р,п-спряження у випадку таких орієнтантів можна зобразити граничними структурами І—IV (мезоформулами):
(де 2 —ЙН2, —ОН, — ИН—Аік, —б—АІк, — ЙН—С— та ін )
II
О
В результаті спряження вільних пар р-електронів кисню,азоту 8 л-електронною густиною бензольного ядра їх електронна густина зливається, стає спільною і, отже, утворюється єдина спряжена л-молекулярна орбіталь (див. рис. 70, 71, розд. 34, 36).
360
3. Орієнтанти, що дезактивують ароматичне ядро. До таких орієнтантів належать атоми галогенів у молекулах арилгалогені-дів. Атоми галогену, на відміну від інших замісників першого роду, не прискорюють, а сповільнюють реакції електрофільного заміщення. Пояснюється це тим, що атом галогену, виявляючи значний негативний індукційний ефект (—7-ефект), відтягує електрони з ароматичного ядра. В результаті електронна густина бензольного ядра, наприклад, у молекулі хлорбензолу стає меншою, ніж у бензолі . В статичному стані (доки молекула не вступає в реакцію) переважає негативний індукційний ефект хлору, що видно із схеми розподілу зарядів у молекулі хлорбензолу:
Отже, у молекулах галогенобензолів до початку реакції в орто-і пара-положеннях утворюються часткові заряди б + . Однак у зв’язку з тим, що на атомі галогену є вільні пари р-електронів, які перебувають у спряженні з л-орбіталлю бензольного ядра (р, л-спряжен-ня), під час реакції у бензольному ядрі молекули галогенобензолу відбувається зміна полярності зв’язків, тобто виявляється динамічний ефект. У момент утворення з молекулою галогенобензолу я- і о-комплексів посилюється ефект спряженняр-електронів атома галогену з л-орбіталлю бензольного ядра. Атом галогену при цьому виявляє динамічний 4-Л1-ефект і тому має сильно виражену орто- і иара-орієнтуючу дію:
При цьому вільна пара електронів галогену бере також участь у делокалізації позитивного заряду о-комплексу. Отже, атом галогену в ароматичному ядрі виявляє статичний -/-ефект і динамічний -}-Л1-ефект. Така неузгодженість динамічних і статичних факторів значно знижує швидкість З^-реакцій для галогенобензолів, і для їх перебігу з такими ароматичними ядрами необхідно створювати жорсткіші умови.
Співвідношення орто- і пара-ізомерів. У молекулах монозаміщених бензолу є одне пара- і два рівноцінних орто-положення, тобто співвідношення між пара- і орто-положеннями становить 1:2. Слід було чекати, що заміщення в оршо-положенні відбуватиметься більшою мірою, ніж у мара-положенні. Проте в більшості Зе-реакцій переважає заміщення в пара-положенні і виходи пара-ізомерів, як правило, вищі, ніж орто-ізомерів.Утворення переважної кількості пара-ізомеру пояснюють впливом таких електронних і стеричних факторів:
361
1. У процесі Зд-реакцій легше утворюється енергетично більш стійка пнрп-хіноїдна форма (І) карбонієвого о-комплексу, ніж ізомерна їй орто-форма (II):
2. Співвідношення орто- і пара-ізомерів визначається силою -/-ефекту замісника 7. Чим більший —/-ефект виявляє замісник 7, тим сильніше він дезактивує орто-положення бензольного ядра.
3. Замісник 7. в ароматичному ядрі екранує (закриває) орто-положення і тим самим утруднює взаємодію електрофілу з атомами водню в цих положеннях.
Замісники другого роду, або мета-оріентинти. До замісників другого роду належать
-5 -ОН
Со
ОН
0 + ^ 0+//
- с • - с
о/? ГІН2
8+ г, 8+/-ч 8+^. + +
-С55Й. -ССІ3 , - СР3, - (МН3, - М/?3
(де /? —алкільний радикал).
Замісники другого роду направляють (орієнтують) вхід наступного замісника Е + переважно в ме/ш.ьположення відносно замісника
2, який вже знаходиться в бензольному ядрі. Орієнтанти —НА//?8, —СГ3 і —СС13 виявляють сильний -/-ефект, а мезомерний ефект їх дорівнює нулю (М = О). Для всіх інших замісників другого роду характерним є дефіцит електронів (заряд б+) або наявність повного позитивного заряду на атомах, які сполучені з атомами вуглецю бензольного ядра.
Отже, лщща-орієнтанти є електроноакцепторними групами. Вони за рахунок -/-ефектів і —ЛІ-ефектів типу р, зт-спряження зменшують електронну густину бензольного ядра і поляризують його так, що на атомах вуглецю в орто- і пара-положеннях буде найбільший дефіцит електронів (заряд б+), а менше зниження електронної густини буде на атомах вуглецю в л/ета-положеннях:
О^+о,77^-° °-42
+0,039
4 0,025
+0,039
+0,025
+0,036
При наявності в ароматичному ядрі замісника другого роду електрофіл Е+ в -^-реакціях атакуватиме переважно лщ/па-положеннямо-нозаміщеної бензолу, оскільки в цьому положенні під впливом замісника меншою мірою знижуватиметься електронна густина і тому заміщення буде відбуватись в основному в мета положення. Крім того, в цьому випадку виникає найбільш енергетично стійкий ц-комплекс (карбкатіон), в якому позитивний заряд у будь-якій з трьох граничних структур (І—III) знаходиться не на атомі вуглецю, сполученому з атомом азоту нітрогрупи, і делокалізується з участю спряженої системи кільця:
При наявності в бензольному ядрі замісника другого роду електрофільне заміщення в орто- і парп-положеннях є енергетично невигідним процесом, оскільки при заміщенні в цих положеннях повинні утворюватися а-комплекси (карбкатіони IV і V), в яких позитивні заряди зосереджені на розміщених поряд атомах —на атомі вуглецю і сполученому з ним атомі азоту нітрогрупи, що надзвичайно енергетично невигідно:
Отже, електронні ефекти, змінюючи стійкість проміжного о-комплексу, полегшують або утруднюють взаємодію з певними атомами бензольного ядра.
Швидкість реакції електрофільного заміщення в бензольному ядрі, як уже зазначалось, залежить від швидкості утворення п-комплексу (див. розд. 28). А оскільки замісники другого роду зменшують електронну густину ароматичного ядра, то з таким ядром електрофіл утворюватиме л-комплекс повільніше,ніж з бензолом, і відповідно електрофільне заміщення відбуватиметься повільніше і важче, ніж з бензолом. Отже, замісники другого роду зменшують здатність бензольного ядра до реакцій електрофільного заміщення. Тому ці замісники називають електроноакцепторнИми, або негативними.
Лїетп-орієнтанти, зменшуючи електронну густину бензольного ядра, роблять можливим підхід до нього нуклеофільних частинок.
заз
Тому замісники другого роду підвищують здатність ароматичного ядра до реакцій нуклеофільного заміщення (див. розд. 33).
Крім замісників першого роду (орто-, пара-орієнтантів) і другого роду (же/па-орієнтантів) існує невелика кількість замісників проміжного характеру. Такі замісники виявляють змішану орієнтацію, яка приводить до утворення суміші приблизно однакових кількостей орто- , мета- і пара-ізомерів. До орієнтантів проміжного характеру належать групи —СНС12, —СН2ОН, —СН2—МО2 і деякі інші.
Орієнтація в дизаміщених бензолу. При наявності в ароматичному ядрі двох замісників проблема орієнтації є більш складною, але і в цих випадках часто можна передбачити, в якому переважно напрямку буде входити новий, третій замісник.
Два замісники в бензольному ядрі можуть бути розміщені так, що орієнтуюча дія одного з них підсилюватиме орієнтуючу дію другого. Така орієнтація двох замісників називається узгодженою. Наприклад, у молекулах речовин І,II,III орієнтація є узгодженою, оскільки обидва замісники орієнтують вхід наступного електрофі-лу в одне і те саме положення (вхід третього замісника в бензольне ядро показаний стрілками):
Якщо орієнтуюча дія одного замісника не співпадає з орієнтуючою дією другого, то в цьому випадку орієнтація цих двох замісників є неузгодженою. Наприклад, в речовинах IV, V, VI орієнтація двох замісників є неузгодженою:
Для того щоб передбачити напрямок входження в бензольне ядро третього замісника, користуються такими емпіричними правилами:
1. Місце входження третього замісника визначається переважно орієнтантом першого роду. Наприклад:
364
Вхід третього замісника полегшується узгодженою орієнтацією обох замісників.
Якщо електронодонорний і електроноакцепторний замісники перебувають у ларл-положенні, то має місце узгоджена орієнтація в орто-положення відносно електронодонорного замісника. Прикладом такої узгодженої орієнтації може бути нітрування толуолу в 2,4,6-тринітротолуол:
2. Якщообидва замісники є електронодонорними і розміщуються в орто - або ллрл-положенні, то орієнтація є неузгодженою і вхід третього замісника визначається сильнішим орієнтантом. За силою орієнтуючої дії замісники першого роду розміщуються в такий ряд:
—N112 > —ОН > —О/? > —СІ > —І > -Вг > —СН3.
Наприклад, при нітруванні 2-хлортолуолу утворюється переважно І-метил-З-нітро-5-хлорбензол, оскільки хлор є сильнішим орієнтантом, ніж метильна група:
Якщо в бензольному ядрі два електронодонорні замісники перебувають у лщ/лл-положенні, то їх орієнтація є узгодженою. Наприклад, при нітруванні 3-хлортолуолу або 1-хлор-З-бромбензолу утворюються відповідні нітросполуки з такими виходами:
З наведених даних про вихід продуктів 5£-реакцій випливає, що заміщення на атомі вуглецю між двома замісниками відбувається незначною мірою. Можливо, це пов’язано із стерйПніТХш факторами двох замісників, які утруднюють підхід електрофілу до цього вуглецевого атома. ‘
365
3. Якщо обидва замісники електроноакцепторні, вони сильно зменшують електронну густину бензольного ядра і тому дуже дезактивують його до 5£-реакцій. У зв’язку з цим наступне електро-фільне заміщення в ароматичному ядрі відбувається дуже важко.
Перебування в бензольному ядрі двох електроноакцепторних замісників в орто- або пара-положенні робить наступне електро-фільне заміщення неузгодженим і вхід третього замісника в цьому випадку визначається сильнішим орієнтантом. За силою орієнтуючої дії в 5е-реакціях замісники другого роду розміщуються в такий ряд:
—СООН > —8О3Н > —МО2.
Наприклад, при нітруванні о-нітробензойної кислоти електрофіль-не заміщення відбуватиметься переважно в мета-положення відносно СООН-групи, яка є сильнішим люта-орієнтантом, ніж МО.2-група:
СООН
Якщо дві електроноакцепторні групи знаходяться в мета-положенні, то в цьому випадку наступне електрофільне заміщення хоч і проходить дуже важко, але є узгодженим. Так, при нітруванні .«-динітробензолу можна добути симетричний тринітробензол:
Контрольні запитання і вправи
1. Поясніть, чому під час взаємодії бензолу з хлором при наявності АІВгз не утворюється бромбензол.
2. Під час радикального хлорування етилбензолу утворюється 91 % 1-хлор-1-фенілетану і 9 % 2-хлор-1-фенілетану. Чому основним продуктом реакції є 1-хлс.р-І-фенілетан?
3. Чому при галогенуванні бензолу за допомогою ІС1 утворюється йод-бензол, а не хлорбензол?
4. Розмістіть такі речовини в порядку зменшення їх здатності до реакцій 8£2: а) бензол, 1,3,5-триметилбензол, толуол, л-ксилол, п-ксилол; б) бензол, бромбензол, нітробензол, толуол; в) бензойна кислота,толуол, толуїлова кислота, п-ксилол; г) хлорбензол, п-метихлорбензол, п-нітрохлорбензол, п-хлорфенол.
5. За допомогою яких реакцій можна розрізнити: бензол і циклогексан, беизол і гексен-1, бензол Етолуол, толуол і н-гептан, бензол і стирол, толуол і стирол?
36в
Розділ 31. ГАЛОГЕНОПОХІДНІ БЕНЗОЛЬНОГО РЯДУ
Галогенопохідні бензольного ряду поділяють на арилгалоге-нід ч і арилалкілгалогеніди.
Арилгалогенідами називають сполуки, які містять атом галогену, безпосередньо сполучений з атомом вуглецю бензольного ядра. Прикладом таких галогенопохідних є фторбензол, хлорбензол, бромбензол, йодбензол тощо.
Методи добування. Найпоширенішим методом добування арил-ралогенідівє пряма дія галогенів на ароматичні вуглеводні. В ряду Г >СІ >Вг >1 реакційна здатність галогену зменшується. Найактивнішим з галогенів є фтор, який руйнує ароматичні вуглеводні, найменш активний —йод. У промисловості та лабораторній практиці найчастіше використовують хлорування і бромування ароматичних вуглеводнів.
Хлор на світлі у відсутності каталізаторів, як уже зазначалося (див. розд. 28), приєднується до бензолу і утворює гексахлорцнкло-гексан. При наявності каталізаторів (А1Сі3, ЕеС13, 8ЬС13 та інших) хлор заміщує атом водню в бензольному ядрі:
Каталітичне хлорування бензолу є реакцією електрофільного заміщення, яка відбувається за 8£2-механізмом. Швидкість цієї реакції прямо пропорційна добутку концентрації бензолу і хлору:
V = к [СвНв] [С12].
Електрофілом у цій реакції є катіон С1+, який утворюється в процесі поляризації, а потім іонізації молекули хлору під дією електронодефіцитного атома заліза каталізатора ЕеС13:
8+ В- СІ
.•СІ: сі:-► :С1: -* сі:.Ее:сі—► :сі+[Еесц].
сі
Катіон С1+ далі реагує з бензолом за 5£2-механізмом:
367
Аналогічно за 5с2-механізмом відбувається і бромування бензолу, продуктом якого є бромбензо":
РєВг, + Вг2-----2.
НВг .
Толуол та інші алкільні гомологи бензолу можуть реагувати з галогенами як з участю атомів водню бензольного ядра, так і з участю атомів водню бічної алкільної групи. Хлорування і бромування толуолу при наявності каталізаторів РеС13, А1С13, РеВг3 та інших відбувається з участю атомів водню бензольного ядра за 5£-2-механізмом. Оскільки метальна група є орієнтантом першого роду, то в процесі галогенування утворюється суміш орто- і пара-галогенотолуолів:
При нагріванні і опроміненні УФ-світлом (/IV) або при наявності радикальних реагентів галогенування гомологів бензолу відбувається за радикальним механізмом у бічному вуглеводневому радикалі. Так, при хлоруванні толуолу за цих умов відбувається поступове заміщення атомів водню метальної групи на хлор з утворенням спочатку хлористого бензилу (І), який потім хлорується в хлористий бензиліден (II), а він у подальшому —в трихлормети л-бензол, або бензолтрихлорид (III):
Пряме йодування бензолу, яке також належить до 5^-реакцій, проводять йодом при наявності окислювачів, наприклад концентрованої азотної кислоти. Окислювачі у цьому процесі необхідні для зв’язування НІ, який утворюється в процесі йодування:
При дії вільного фтору на ароматичний вуглеводень відбувається руйнування (деструкція) органічної речовини. Перспективним фторуючим реагентом є фторид ксенону ХеР2, який реагує з над-
368
лишком бензолу (в середовищі ССІ4 і при наявності невеликої кількості НЕ) і утворює фторбензол з виходом 68 %:
Радянські хіміки під керівництвом М. М. Ворожцова (мол.) встановили, що при нагріванні гексахлорбензолу з безводним КЕ утворюється гексафторбензол:
+ 6КР
500°С
+ 6КСІ .
Гексафторбензол —рідина з температурою кипіння 80,3 °С, яка дуже близька до температури кипіння бензолу (Т^СвНв = — 80,1 °С). Така порівняно низька температура кипіння гексафтор-бензолу пояснюється малими дисперсійними силами взаємодії між його молекулами (слабкі сили Ван дер Ваальса).
Арилгалогеніди можна також добувати розкладанням солей арилдіазонію (див. розд. 37). Арилйодиди, крім того, можна добути взаємодією арилбромідів з магнієм і йодом (див. нижче).
Фізичні властивості. Моно- і дигалогенарили є рідкими або кристалічними речовинами, які розчиняються в більшості органічних розчинників, але практично не розчинні у воді. Густина їх більша за одиницю. При збільшенні атомної маси галогену густина арилгалогенідів зростає (табл. ЗО).
Таблиця ЗО. Моногалогенобензоли
Формула Т °С 1 кип» Довжина зв’язку вуглець—галоген, им л20 64
Свн5р 84,7 0,134 1,024
С8Н6С1 132 0,139 1,106
СвН6Вг 156 0,184 1,495
С6НеІ 188 0,206 1,831
Ізомерні дигалогенобензоли мають близькі температури кипіння, але лета-ізомери, як правило, киплять при нижчій температурі, ніж відповідні орто- і пара-ізомери (табл. 31).
Будова зв’язку вуглець — галоген у молекулах арилгалогенідів. Зв’язок вуглець —галоген в арилгалогенідах ковалентний полярний. Однак на йоговластивості впливає бензольне ядро. Так, довжини зв’язків вуглець —галоген у молекулах арилгалогенідів,
369
Таблиця 31. Дигалогенобензоли
Формула Ї-КІ1П( °С 7ПЛ. °С
орто - мета- пара- орто- мста- пара-
С6Н4Ея 92 83 89
СвН*С1, 180 173 175 — — 52
С,.П4Вг5 221 217 219 — — 87
СвН4І2 287 285 285 27 35 129
менші, ніж у галогеналканів. Наприклад:
як і вінілгалогенідів.
Сполука
Довжина зв’язку
' вуглець-галоген, нм
[і, О
0,169
1,73
Н2С = СН-С1
Н3С—СН2 —Сі
0,169
0,177
1,44
2,05
Це вказує на додаткову взаємодію атома галогену з атомом вуглецю ароматичного ядра. У галогенобензолів, наприклад у молекулі хлорбензолу, як і хлористого вінілу, атом хлору сполучений з 5/?2-атомом вуглецю, який більш електронегативний, ніж хр3-атом у молекулі хлористого етилу. Крім того, в хлорбензолі та інших арилгалогенідах відбувається спряження між вільними р-елект-ронами галогену з л-орбіталлю бензольного ядра. Тому р-електро-ни в галогенобензолах зміщені від атома галогену до 5р2-атома вуглецю. Внаслідок цього збільшується електронна густина і міцність зв’язку вуглець —галоген, а дипольні моменти рарилгало-генідів, які напрямлені від ядра до атома галогену, стають меншими, ніж ц галогеналканів:
Отже, атоми галогенів у молекулах арилгалогенідів, які сполучені з ароматичним ядром, мають двоїсту природу. Як більш електронегативні, ніж вуглець, вони притягують електрони до себе і тим самим зменшують загальну електронну густину бензольного ядра. Разом з тим вони можуть подавати електрони на зв’язок вуглець — галоген, а потім у бензольне ядро.
Електронна будова хлорбензолу і орієнтуюча дія хлору. Як уже зазначалося (див. розд. ЗО), атоми галогенів виявляють статичний —/-ефект і динамічний +Л1-ефект і належать до дезактивуючих замісників першого роду. У статичному стані розподіл електронної густини в хлорбензолі, як видно з молекулярної діаграми його молекули (див. розд. ЗО), не сприяє орто- і пора-електро-фільному заміщенню. Проте в момент реакції домінує динамічний
370
+Л4-ефект(спряження парир-електронів атома хлору з ^-електронами бензольного ядра). Крімтого,парар-електронів хлору бере участь у делокалізації (ф-)-заряду о-комплексу, який виникає в процесі 5£-реакції (див. розд. ЗО). Наприклад, при хлоруванні хлорбензолу при наявності А1С13 електрофіл —катіон С1+ приєднується до орто- або /шра-атомів вуглецю бензольного ядра і утворює о-катіон, позитивний заряд якого делокалізується ^-електронами атома хлору по системі р,л-спряження. В результаті утворюється суміш орто • і иаря-ізомерів дихлорбензолу:
Хімічні властивості. Галогенобензолам властиві реакції 8 участю атома галогену і з участю бензольного ядра.
Реакції з участю атома галогену. Атоми галогену в молекулах арилгалогенідів безпосередньо сполучені з атомами вуглецю бензольного ядра і їх вільні р-електрони перебувають у спряженні з л-електронами бензольного ядра. У зв’язку з цим дані атоми, як і атоми галогену у вінілгалогенідах (див. розд. 21), виявляють малу реакційну здатність. Так, вони не взаємодіють з такими нуклеофільними реагентами, як їдкі луги, солі срібла, ціаніди лужних металів, алкоголяти, аміак за тих умов, за яких ці реагенти заміщували атом галогену в галогеналканах. Наприклад, при нагріванні бромбензолу з спиртовим розчином А&МО3 протягом кількох днів не вдається помітити утворення навіть незначних кількостей А«Вг, що свідчить про відсутність нуклеофільного заміщення брому в бромбензолі. Для нуклеофільного заміщення атома галогену в арилгалогенідах необхідно створювати жорсткі умови.
Рухливість атома галогену в арилгалогенідах, тобто його здатність до нуклеофільного заміщення, найбільша у фтору: Р^>С1, Вг, І. Пояснюється це переважно високою полярністю зв’язку 6+ с-
С->-Р, зумовленою великим індукційним ефектом атома фтору. Проте здатність атома галогену в арилгалогенідах до обміну на метал, наприклад, при дії на них алкіллітію зменшується в ряду І >Вг >С1 »Р.
Хлор- і бромбензоли реагують з їдкими лугами тільки при 800 °С, а з аміаком — при наявності каталізаторів (солі міді або мідний порошок) при 180—200°С. За них умов галоген заміщується
371
на гідроксил або аміногрупу. Продуктами таких 5дг-реакцій є відповідно фенол і анілін:
Активність атомів галогенів у арилгалогенідах підвищується під впливом електроноакцепторних груп, таких як —МО2, —8О3Н та інших, якщо вони перебувають у бензольному ядрі в орто- або пара-положенні відносно атома галогену. Так, ипра-нітрохлорбен-зол легко реагує з лугом і обмінює атом хлору на гідроксил, перетворюючись при цьому на пара-нітрофенол. У даному випадку нітрогрупа виявляє узгоджені —/- і —Л4-ефекти, в результаті яких на вуглецевому атомі зв’язку С—СІ виникає значний за величиною заряд 6+, що сприяє взаємодії нуклеофілу (ОН-) з цим атомом вуглецю і утворенню карбаніона, який далі перетворюється на /шра-нітрофенол.
Реакції заміщення активованого атома галогену біля атома вуглецю ароматичного ядра відбуваються за механізмом 5^2. Наприклад, утворення и-нітрофенолу з п-нітрохлорбензолу і гідроксиду натрію описується такими елементарними реакціями. На першій, повільній стадії до позитивно зарядженого атома вуглецю, сполученого з атомом хлору, приєднується нуклеофільна група ОН". В результаті утворюється негативно заряджений о-комплекс (карбаніон), який є відносно стійким внаслідок делокалізації ніт-рогрупою негативного заряду (структури І—III). У деяких 5д.-2-реакціях такий карбаніон може бути помічений і індентпфікова-ний. На другій, швидкій стадії відбувається відщеплення атома хлору з парою електронів у вигляді аніона СІ", який, сполучаючись з катіоном На+, дає хлорид натрію. Одночасно регенерується секстет л-електронів, а замість хлору стає гідроксил, тобто утворюється нітрофенол:
№ОН^=±\та+ + он ;
372
Атака нуклеофілом атома вуглецю в молекулах арилгалогенідів відбувається не «з затилля», а «збоку», і при цьому утворюється не л-комплекс, а відразу карбаніон. Крім того, розрив зв’язку вуглець —'Галоген і утворення нового зв’язку вуглець —нуклео-філ відбуваються асинхронно (у різний час). Отже, механізм 8^2 ароматичного типу відрізняється від механізму 8^2 для аліфатичних сполук (див. розд. 8).
У випадку лг-нітрохлорбензолу нітрогрупа не бере участі в де-локалізації негативного заряду карбаніона (о-комплексу) і тому хлор у цій сполуці неактивний.
Заміщення в арилгалогенідах неактивованих атомів галогену відбувається під дією сильних основ. У цьому випадку реакція відбувається не за таким механізмом, який ми щойно розглянули. Так, якщо хлор- або бромбензол обробляти в рідкому аміаку такою сильною основою, як амід натрію або водний розчин лугу, то відбувається нуклеофільне заміщення атома галогену на КН2- або ОН-групу з утворенням відповідно аніліну і фенолу. Проте під дією цих сильних нуклеофільних реагентів від арилгалогенідів спочатку відщеплюється галогеноводень. При цьому утворюється надзвичайно реакційноздатний проміжний продукт, який називають дегідробензолом.Дегідробензол далі швидко приєднує нуклеофільний реагент, наприклад воду або аміак, і утворює фенол або анілін:
МаКНг
-НВг
Дегідробензол
Як випливає із схеми, заміщення в цьому випадку включає дві стадії —'спочатку елімінування (відщеплення), а потім приєднання. Отже, неактивовані атоми галогену в арилгалогенідах заміщуються за механізмом елімінування—приєднання.
Арилгалогеніди порівня но легко реагують з активними металами. Так, з магнієм вони утворюють магнійорганічні сполуки — магнійарилгалогеніди, які широко використовуються в органічному синтезі. Бромбензол, наприклад, у середовищі абсолютного ефіру утворює фенілмагнійбромід, з якого можна добути йодбензол, бензойну кислоту та інші органічні речовини:
Д—Свн5-~І + МеВгі ;
СЛ-ВГ + - С6Н5-Мц-ВГ-~^ м _нс^ н.
6 5 -М^ВгСІ
Під дією металічного натрію на суміш арилгалогенідів з алкіл-галогенідами утворюються гомологи бензолу (реакція Вюрца — Фіттіга; див. розд. 29).
373
Реакції арилгалогенідів з участю ароматичного ядра. Арилгалогеніди вступають у реакції електрофільного заміщення атомів водню бензольного ядра, а саме в реакції галогенування, нітрування, сульфування та ін. Атоми галогену, як уже зазначалось, є дезактивуючими замісниками першого роду і орієнтують вхід наступного електрофіла в орто- і иа/хг-поло-ження.
Арилалкілгалогеніди. Органічні сполуки цього класу містять атом галогену в бічному ланцюгу. Прикладом таких речовин є хлористий бензил С6Н5—СН2—СІ, бромистий бензпл С6Н5—СН2—Вг та інші.
Хімічні властивості арилалкілгалогенідів визначаються наявністю ароматичного ядра і атома галогену в бічному вуглеводневому ланцюгу. Так, ароматичне ядро арилалкілгалогенідів вступає у всі Хн-реакції, характерні для ароматичних сполук ряду бензолу (галогенування, нітрування та ін.). Атоми галогену в арилалкіл-галогенідах надзвичайно реакцій нездатні. За активністю вони подібні до галогенів у галогеналілах (див. розд. 21) і вступають у різноманітні реакції нуклеофільного заміщення. Так, хлористий бензил при нетривалому нагріванні з водним розчином карбонату натрію легко гідролізується і перетворюється на бензиловий спирт:
Ця нуклеофільна реакція відбувається за механізмом Хууі. На першій, повільній стадії хлористий бензил дисоціює на іонну пару з утворенням енергетично стійкого бензильного катіона, стійкість якого зумовлена спряженням л-електронів бензольного ядра з (+)-зарядом (вакантною орбіталлю) на вуглецевому атомі в бічному ланцюгу. На другій стадії бензильний катіон швидко реагує з нук-леофілом — іоном ОН' —і утворює продукт реакції —бензиловий спирт:
374
Розділ 32. АРОМАТИЧНІ СУЛЬФОКИСЛОТИ
Ароматичними сульфокислотами називають сполуки, які містять сульфогрупу —8О3Н,^атом сірки якої сполучений з атомом вуглецю ароматичного ядра/ В ароматичному ядрі може бути одна, дві або три сульфогрупи. Залежно від цього ароматичні сульфокислоти поділяють на моно-, ди- і трисульфокислоти. (Прикладом ароматичної моносульфокислоти є бензолсульфокислота
<()>-8О3Н .
Методи добування. Ароматичні сульфокислоти добувають переважно реакцією сульфування, тобто взаємодією ароматичної сполуки з сульфуючим реагентом. Як сульфуючі реагенти застосовують концентровану Н28О, (96 %-у, густина 1,84), моногідрат (100 %-на сірчана кислота), олеум [розчин оксиду сірки (IV) в Н28О4], оксид сірки (VI), хлорсульфонову кислоту НО8О2С1, хлористий сульфурил 8О2СІ2, комплексні сполуки оксиду сірки (VI) з піридином або діоксаном. Наприклад:
СвНв + НО8О3Н <=± СвН5—8О3Н ф- Н2О.
Сульфування — це реакція електрофільного заміщення (5/;-реакція). Сірчана кислота, оксид сірки (VI) та інші сульфуючі речовини є електрофільними реагентами з дефіцитом електронів, тобто з позитивним зарядом на атомі сірки. При сульфуванні сірчаною кислотою сульфуючою частинкою є активний електрофіл —•
катіон гідросульфонію8О2ОН, який утворюється в концентрованій сірчаній кислоті за схемою:
Н0-8О2Ибн~4^4+80чНг^2ГН!2.О + НО-8О.2 + Н8О; *
Реакція сульфування, як електрофільний процес, відбувається за 5г2-механізмом (див. розд. 28):
Взаємодія ароматичних сполук із сірчаною кислотою є оборотною реакцією. Вода, що утворюється в процесі реакції, зміщує рівновагу вліво. При сульфуванні концентрація сірчаної кислоти в міру перебігу реакції зменшується і ч^рез деякий час досягає 65 %. При такій концентрації сірчаної кислоти реакція сульфування зупиняється.
Алкілбензоли сульфуються значно легше, ніж бензол, і утворюють суміш переважноорто- і пяря-алкілсульфобензолів. Толуол,
375
наприклад, при нагріванні з концентрованою Н28О4 утворює толуолсульфокислотп:
Активнішим сульфуючим електрофільним реагентом, ніж сірчана кислота, є оксид сірки (VI). Оскільки в молекулі цього оксиду три атоми кисню, як більш електронегативні, ніж сірка, сильно зменшують електронну густину на атомі сірки, то на цьому атомі виникає частковий позитивний заряд, який і надає 8О3 властивостей активного електрофілу:
ОС-
II Л_1_
О О
Механізм сульфування бензолу оксидом сірки (VI) можна описати такою схемою електрофільного заміщення:
7Г-Комплекс а-Комплекс
Характерною особливістю реакції сульфування ароматичних сполук є її ізотопний ефект: звичайний водень (протій) заміщується в ароматичному ядрі приблизно в два рази швидше, ніж дейтерій у молекулі дейтерованого бензолу. Причиною цього є оборотність реакції сульфування. Якщо сульфувати дейтерований бензол, то швидкість розриву зв’язку С—О в карбкатіоні І (И) менша, ніж швидкість розриву зв’язку С—Н у протієвому карбкатіоні І (Н):
КН) 1(0)
Тому сульфування дейтерованого бензолу відбуватиметься повільніше, ніж звичайного (протієвого). Різниця у швидкостях сульфу
376
вання дейтерованого і звичайного бензолу являє собою величину ізотопного ефекту.
Сульфокислоти з реакційної суміші виділяють переважно у вигляді натрієвих солей. Для цього використовують метод висолювання, який базується на обмеженій розчинності натрієвих солей сульфокислот у концентрованих розчинах хлориду або сульфату натрію. Тому після проведення реакції сульфування реакційну суміш виливають у насичений розчин кухонної солі. При цьому в осад випадає малорозчинна натрієва сіль ароматичної сульфокислоти, яку потім відділяють:
СвНБ8О3Н + МаСІ -> СвН58О3Ма | + НС1.
Для того щоб виділити вільні сульфокислоти, реакційну суміш після закінчення сульфування обробляють гідроксидами або карбонатами кальцію або барію. Ароматичні сульфокислоти за таких умов утворюють добре розчинні у воді сульфонати кальцію або барію, які фільтруванням відділяють від важкорозчинного сульфату кальцію або барію. При добавлянні до цих сульфонатів еквівалентної кількості Н28О4 добувають вільні ароматичні сульфокислоти:
2СвНБ8О3Н + Н28О4 + 2Са (ОН)2 _2НгО > Са8О4 | +
+ (СвНБ8О3)2Са > 2С„Н58О3Н.
Ароматичні сульфокислоти є кристалічними гігроскопічними речовинами, добре розчинними у воді і спирті.
Хімічні властивостцРеакції ароматичних сульфокислот визначаються наявністю в їх молекулах сульфогрупи і бензольного ядра. Ці реакції можна розділити на три типи: 1) реакції сульфогрупи; 2) реакції заміщення сульфогрупи; 3) реакції з участю бензольного ядра.
Реакції сульфогрупи. У сульфогрупі атом сірки сполучений з гідроксильною групою і двома атомами кисню. Атоми кисню, як більш електронегативного, ніж сірка, відтягують електронну густину на себе:
- 5 —О-Н .
6
В результаті атом водню ОН-групи може протонізуватися. Отже, сульфокислотщкюдібно до сірчаної кислоти, є сильними кислотами і'у водному розчині повністю дисоціюють. Проте вони дещо слабкіші, ніж сірчана кислота:
СвН6 - 8О3Н + 1 ДО Н3О+ + СяН5-8О^.
377
Дослідження показали, що в сульфонатному аніоні негативний заряд розподілений між трьома атомами кисню і тому ці атоми в сульфогрупі рівноцінні:
хОТ
СеН-—8^-0 .
^'О
Виходячи з цього, електронну будову бензолсульфокислота можна зобразити у вигляді граничних структур І—III:
Отже, у сульфогрупі немає подвійних зв’язків, а два атоми кисню сполучені з сіркою семіполярними зв’язками:
:бі
—+5+—О—Н.
:ОЇ
Ароматичні сульфокислота вступають у реакцію нейтралізації з лугами, гідроксидами інших металів і утворюють солі, які називають сульфонатами:
С6Н6 — 8О3Н + ИаОН С«Н6 — 8О3і\та ф Н2О.
Сульфонати більшості металів добре розчинні у воді. Сульфо-нати магнію, кальцію, барію і свинцю (II), на відміну від сульфатів цих металів, також добре розчинні у воді.
З ароматичних сульфокислот можна добута їх похідні, такі як хлорангідриди, складні ефіри, аміди тощо. Так, при взаємодії бензолсульфокислота або її натрієвої солі з РС13 або з хлористим тіонілом утворюється хлорангідрид бензолсульфокислота (бензол-сульфохлорид):
X’
СвН6—8О2Оі\та С«Н® - 8~° С6Н6—8О2—ОН.
ХС1 —5О2
Арилсульфохлориди утворюються також при взаємодії ароматичних сполук з хлорсульфоновою кислотою:
Сенс ф 2НО8ОаС1 — СвН8—8О2С1 ф Н28О4 ф НС1.
Цей метод добування арилсульфохлоридів простіший від попереднього, тому користуються переважно цим методом.
Хлор у хлорангідридах сульфокислот надзвичайно реакційно-здатний. Тому хлорангідриди використовують для синтезу різно
878
манітних похідних сульфокислот. Сульфохлориди, подібно до хлорангідридів карбонових кислот, легко взаємодіють із спиртами і утворюють при цьому складні ефіри сульфокислот, а з аміаком — аміди сульфокислот. Наприклад:
СвН5—8О2—і\Н2 с—кн4Сі ^вН6 8О2 Сі * С6Н3—8О2—ОС2Н5.
Бензолсульф- Етиловий ефір
амід бензолсульфокислоти
Ефіри сульфокислот застосовують як алкілуючі засоби, а аміди сульфокислот, які називають переважно сульфамідами, знайшли застосування в медицині, оскільки деякі з них виявляють антимікробну дію.
Під дією гіпохлоритів атоми водню сульфамідної групи заміщуються на хлор. В результаті утворюються так звані хлораміни:
9 НОСІ НОСІ
С6Н6-8О2-іУС12 -ггщо СвНв-8О2ГШ2 СвН6-8О2-КНС1;
Дихлорамін Б Хлорамін Б
л-СН3С6Н4—8О2—ГШ2 + НОСІ -> л-СН3С3Н4—8О2—1УНС1 + Н2О.
Хлорамін Т
Хлор- і дихлораміни виявляють окислювальні властивості:
С6Н5— 8О2-ИС12 + 2НОН->- С„Н5—8О2—і\Н2 + 2НС1 + 2 [О].
Тому водні розчини хлорамінів використовують як дезинфікуючі засоби, а також для дегазації отруйних речовин.
Реакції електрофільного і нуклеофіль-н о г о заміщення су льфогру п и.~ Як уже зазначалося, сульфування ароматичних сполук є оборотною реакцією. Отже, ароматичні сульфокислоти можуть гідролізуватись (десульфува-тись) і перетворюватись на ароматичні вуглеводні. Так, бензолсуль-фокислота при нагріванні з розбавленою соляною кислотою гідролізується і перетворюється на бензол і сірчану кислоту:
8О3Н + Н3О* 150—200°с”> + Нї$О4.
Гідроліз сульфокислот є реакцією електрофільного заміщення сульфогрупи на протон) Сульфогрупа може заміщуватись і іншими електрофільними реагентами. Наприклад, електрофільне заміщення сульфогрупи на нітрогрупу відбувається при добуванні пікринової кислоти (див. розд. 34).
Більш характерними для сульфокислот є реакції нуклеофіль-ногозаміщення сульфогрупи. Так, натрієві солі ароматичних сульфокислот при нагріванні з нуклеофільними реагентами (Г » 200— 300 °С) перетворюються на феноли, тіофеноли, ароматичні кислоти та їх нітрили:
СеН5СООН
СвН6-Сі\
НСООПа
МаСМ
—КагЗО8 СвН5-8О3№ _№г5Оз
—№28О3 і №Г<Н2 СвНБ-МН2
і^иєвН6-ОН;
~-->СйН,-5Н,
379
Такі реакції широко використовуються в органічному синтезі. Зокрема, сплавляння натрієвих солей з твердим лугом називається реакцією лужного плаву і використовується для добування різноманітних фенолів (див. розд. 34).
Реакції з участю бензольного ядра/ Відсутність у сульфогрупі подвійних зв’язків і неплоска, тетраедрична її будова перешкоджають р,я-спряженню цієї групи з л-електронами бензольного ядра. Тому 8О3Н-група виявляє тільки —/-ефект. За рахунок цього ефекту сульфогрупи виявляє значну електронегатнвність і сильно притягує л-електрони бензольного ядра. Тим самим вона зменшує його електронну густину, особливо в орто-і пара-положен-нях. Отже, сульфогрупа є орієнтантом другого роду (лгелш-орієн-тантом) і спрямовує вхід електрофільних частинок у реакціях галогенування, нітрування, сульфування та інших в л/ешп-поло-ження бензольного ядра:
Сульфогрупа, як досить сильний електроноакцептор, пасивує бензольне ядро до 5е-реакцій. Тому такі реакції для ароматичних сульфокислот, наприклад для сульфобензолу, відбуваються набагато важче, ніж для бензолу, і в порівняно жорстких умовах.
Застосування сульфокислот та їх похідних. Ароматичні сульфокислоти мають різноманітне застосування. Вони є основою для добування поверхнево-активних речовин, миючих засобів, їх використовують для виробництва розчинних у воді барвників, дубильних речовин, іонообмінних смол тощо.
Розділ 33. НІТРОСПОЛУКИ АРОМАТИЧНОГО РЯДУ
Ароматичними нітросполуками називають речовини, що містять нітрогрупу —N02, атом азоту якої сполучений з атомом вуглецю ароматичного ядра. Ароматичні нітросполуки можуть містити одну, дві і три нітрогрупи. Загальна формула ароматичних мононітро-сполук Аг—N02- Прикладом найпростішої ароматичної нітросполуки є нітробензол С6Н3—КТО2.
. Методи добування. Ароматичні нітросполуки добувають переважно реакцією нітрування, тобто взаємодією ароматичної сполу
380"? -
ки з нітруючим реагентом: Як нітруючі реагенти найчастіше застосовують суміш концентрованих НМО3 і Н28О4 (нітруюча суміш), суміш селітри (К’аІ\’О3 або ККО3) з концентрованою Н28О4, азотну кислоту різної концентрації, оксиди азоту.
Класичним прикладом такої реакції є нітрування бензолу. Бензол при перемішуванні з нітруючою сумішшю за порівняно м’яких умов (40—50 °С) утворює нітробензол з виходом ~95 %:
С6Н6 + НОі\О2 !^2-4-» СвН8—ЬІО2 + Н2О.
Реакція нітрування за механізмом є 5в2-реакцією. Нітруючим реагентом при електрофільному нітруванні є нітроній-катіон
а
,’р ----- N = о: $рг
б
Рис. 69. Будова катіона нітронію ІЧО2:
а — схема будови електронної оболонки атома азоту в катіоні нітронію; б — схема просторової будови катіона нітронію: два о-зв’язки розміщені лінійно; два Л -зв’язки розміщені у взаємно перпендикулярних площинах
МО2.У самій концентрованій азотній кислоті міститься близько 4 % катіонів ЕЮ2, які утворюються в результаті дисоціації цієї кислоти за схемою:
21 Юі\()2 N0^ + N07 + Н2°-
В нітруючій суміші нітроній-катіон генерується в процесі рівноважної реакції:
НКО3 + 2Н25О4 N0+ + Н30+ + 2Н5О^.
Існування катіона М02 підтверджується тим, що електропровідність нітруючої суміші значно вища, ніж електропровідність окремо взятих НМ03 і Н28О4.
В нітроній-катіоні є два розміщених лінійно о-зв’язки кисень — азот і два л-зв’язки, які знаходяться у взаємно перпендикулярних площинах (рис. 69).
Відносна енергетична стабільність нітроній-катіона пояснюється взаємодією вільних пар р-електронів «/Аатомів кисню з вакантною р-орбіталлю атома азоту, який знаходиться в хр-гібриди-зованому стані (рис. 69).
Механізм нітрування. Електрофільне нітрування ароматичних сполук відбувається за описаним вище (див. розд. 28) 5е2-механіз-
381
мом. Згідно з цим механізмом нітрування бензолу можна зобразити такою схемою-
^-Комплекс о-Комплекс
(карбкатіон)
Реакція нітрування є необоротною, тому в цій реакції не спостерігається ізотопного ефекту (див. розд. 32), тобто ЯК ПрОТІЄВИЙ І (Н), так і дейтерієвий 1(0) карбкатіони перетворюються на кінцевий продукт (нітробензол), а не на вихідну речовину (бензол):
І (Н)
1(0)
При нітруванні бензолу нітруючою сумішшю сульфування бензолу не відбувається і бензолсульфокислота практично не утворюється, тому що швидкість нітрування в 10® рази більша від швидкості сульфування.
Гомологи і похідні бензолу з електронодонорними замісниками в ароматичному ядрі нітруються легше, ніж сам бензол, а похідні бензолу з електроноакцепторними замісниками нітруються важче і в жорсткіших умовах, ніж бензол. Так, толуол нітрується в 24 рази швидше, ніж бензол, а для нітрування нітробензолу необхідно значно підвищити температуру реакційної суміші.
Під час нітрування толуолу нітруючою сумішшю при кімнатній температурі утворюється переважно орто- і /шра-нітротолуол і тільки невелика кількість ле/па-нітротолуолу:
НОКО2+Н25О4 20°С
Для дальшого нітрування орто - або па,ра-нітротолуолу температуру реакційної суміші необхідно підвищити до 70 °С. При цьому утворюється 2,6-динітротолуол. Нітрування 2,4-дииітротолуолу вимагає ще жорсткіших умов — нагріванняпри ПО °С. За цих умов утворюється 2,4,6,-тринітротолуол, який є вибуховою речовиною і під назвою тротил, або тол, широко використовується в промисловості і воєнній справі:
382
Якщо нітрування толуолу здійснювати розбавленою азотною кислотою при температурі 120—150 °С, то нітрується бічний ланцюг (з участю атома водню метальної групи). В результаті атом водню метальної групи толуолу заміщується на нітрогрупу (реакція Коновалова, див. розд. 7) і утворюється фенілнітрометан:
СвН6—СН3 + НО—N0,120~1Б0°с-> СаНв—СН2—N0, + Н2О.
Нітрування гомологів бензолу розбавленою азотною кислотою при температурі 120—150 °С відбувається за радикальним механізмом:
НМО3 + НЬЮ2 2ЬЮ2 + Н2О;
с0н6-сн3 + йо2 - сен5-сн2 + НК’О2:
СеН6-СН2 + НСЖО2 - СеН5-СН2-і\О2 + НО.;
С6Н5—СН3 + НО- Н2О + С„Н5—СН2 і т. д.
Легко нітрується фенол, оскільки його молекула містить гідроксильну групу, яка є замісником першого роду. При нітруванні фенолу можна добути орто- і паро-нітрофеноли, а також ди-або тринітрофеноли (див. розд. 34).
У реакцію нітрування вступають і галогенобензоли. Так, хлорбензол при нітруванні утворює суміш орто- і поро-нітрохлорбензо-лів. Проте нітрування хлорбензолу відбувається важче, ніж бензолу, оскільки атом хлору виявляє значний статичний — /-ефект і є дезактивуючим орієнтантом першого роду.
Хімічні властивості. Реакції ароматичних нітросполук визначаються наявністю в їх молекулах нітрогрупц і бензольного ядра. Найважливішою реакцією нітросполук, яка відбувається з участю нітрогрупи, є їх відновлення до ароматичних амінів (див. розд. 36):
С6Н5—ІЧО2 + 6Н -> СвН5—N1-1, + 2Н2О.
Ця реакція була відкрита М. М. Зініним у 1842 р.
Реакції ароматичних нітросполук з участю бензольного ядра подібні до реакцій самого бензолу. Однак на активність ароматичного ядра нітросполук впливає нітрогрупа. Електронна будова нітрогрупи така, що вона, як замісник другого роду, відтягує
383
електрони бензольного ядра на себе, зменшує його загальну електронну густину і тим самим знижує його здатність до реакцій електрофільного заміщення:
"О. +Со
N х
8+
Найменша електронна густина в молекулі нітробензолу залишається на атомах вуглецю в орто- і псрл-положеннях, на яких виникають часткові позитивні заряди. Тому нітрогрупа орієнтує вхід електрофільних замісників у лщ/па-положення, в яких ця густина вища, ніж в орто- і лара-положеннях:
Такі реакції, як бромування, сульфування, нітрування нітробензолу, відбуваються у значно жорсткіших умовах, ніж аналогічні реакції з бензолом. Так, бензол легко нітрується нітруючою сумішшю при 40—50 °С, а нітробензол— уже при температурі 90— ПО °С. Нітрування динітробензолу проводять при тривалому нагріванні його (протягомп’яти днів) з великим надлишком димучих азотної і сірчаної кислот, тобто введення третьої нітрогрупи в бензольне ядро здійснюється зі значними труднощами:
Нітрогрупа, зменшуючи здатність ароматичного ядра до реакцій електрофільного заміщення, разом з тим підвищує його активність до реакцій нуклеофільного заміщення, яке для нітробензолу відбувається у положеннях з найменшою електронною густиною, тобто в орто- і ллрл-положеннях. Так, при нагріванні нітробензолу з порошкоподібним КОН утворюється суміш орто- і пара-нітрофенолятів:
+ Н2 .
384
Ще легше, ніж нітробензол, у реакції нуклеофільного заміщення вступає динітробензол, який при нагріванні з гідроксилам!ном (Н2\—ОН) і з ціанідом каліюутворює відповідно 2,6-динітроанілін і 2,6-динітробензонітрил:
Розділ 34. ФЕНОЛИ ТА АРОМАТИЧНІ СПИРТИ
Фенолами називають органічні сполуки, які містять гідроксильну групу, безпосередньо сполучену з вуглецевим атомом бензольного ядра. Найпростішим з них є оксибензол. Його переважно називають фенолом. Термін «фенол» походить від стародавньої назви бензолу — «фено» (від грецьк. «файно» — той, що несе світло). Якщо гідроксильна група ароматичного вуглеводню сполучена з вуглецевим атомом бічного ланцюга, то такі оксисполуки називають ароматичними спиртами, наприклад бензиловий спирт:
Феноли залежно від кількості гідроксильних груп в їх молеку-. лах поділяють на одноатомні, двохатомні, трьохатомні і т.д. Атомність фенолу вказує на кількість гідроксильних груп в його молекулі.
Одноатомні феноли
Одноатомні феноли містять одну гідроксильну групу. Вони утворюють гомологічний ряд, який можна легко записати виходячи з гомологічного ряду ароматичних вуглеводнів. Для цього потрібно замінити в їх молекулах один атом водню ароматичного ядра на гідроксильну групу. Першим представником гомологічного ряду цих речовин є фенол. Від толуолу можна утворити орто-, мета-і /шра-окситолуоли, які називають крезолами (табл.32).
Методи добування. Фенол вперше виділив Ф. Рунге у 1834 р. з кам’яновугільної смоли і назвав його карболовою кислотою оскільки ця речовина взаємодіє з лугом. Цим методом1фенол добувають і в наш час. Крім фенолу під час сухої перегонки (коксування) кам’яного вугілля, торфу та інших органічних матеріалів добувають також крезоли. Для виділення фенолів з кам’яновугільної смоли її обробляють лугом. Феноли при цьому реагують з лугом і
13 [-120
385
Таблиця 32. Одноатомні феноли
Назва
Фенол
орто-Крезол
ліето-Крезол
пара-Крезол
утворюють добре розчинні у воді феноляте, які легко відділити від нерозчинної у воді кам’яновугільної смоли. Наприклад:
СБНБ—ОН + ИаОН -ч- СвН5—О№ + Н2О.
Фенолят натрію
Добутий водний розчин феноляту обробляють вуглекислим газом або розбавленою мінеральною кислотою, які розкладають феноляте з утворенням фенолів:
СвНБ—ОКа + НаО + СО2 -> СвНБОН + К’аНСО3.
У наш час таким шляхом добувають близько 5 % фенолу від загального його виробництва. Враховуючи велике значення фенолу і продуктів, які добувають на його основі, розроблено ряд синтетичних методів його добування. З них найважливіше значення мають такі:
1. Окислення ізопропілбензолу (кумолу). Метод розроблений радянськими хіміками під керівництвом П.Г.Сергєєва. Кумольний метод добування фенолу, за яким крім фенолу одночасно утворюється й ацетон, має найбільше промислове значення. Цим методом добувають більш як 3 млн т фенолу в рік, що становить майже 90% його світового виробництва. За цим методом кумол при наявності основних каталізаторів спочатку окислюють киснем повітря до гідропероксиду кумолу (І), який потім розкладають розбавленим розчином сірчаної кислоти. Розкладання пероксиду відбувається
386
за механізмом так званого секстетного перегрупування. А саме, гідропероксид (І) спочатку перетворюється на іон оксонію (II) з секстетом електронів на атомі кисню. Іон (II) після відщеплення молекули води перетворюється на катіон (III), в якому атом кисню має шість електронів і заряджений позитивно (несе (+)-заряд). У катіоні (III) відбувається 1,2-зсув фенільної групи від атома вуглецю до електронодефіцитного атома кисню.В результаті такого зсуву катіон (III) перетворюється на карбкатіон (IV), який при взаємодії з водою утворює оксосполуку (V). Оксосполука (V) є напівацеталем і легко розпадається на фенол і ацетон. Цей процес можна зобразити такими схемами реакцій:.
с6н5
Н3С-С-п
СНз Кумол
Ог(ОН) 130’С
((-бЧв н.(с-с^о-р-н
СНз
?н.5.о +
н3с—с—о : о - н —-
ІНз Н
-н2о
н3с—с—о—свн5
СН3 IV
• н):о- н
______~ н,с-сго-с6н5---------н3с—с—сн3 + С6Н5-ОН + н+ .
І II
сн3 О
у 1 Ацетон Фенол
2. Лужний плав сульфокислот (див. розд. 34). Цим методом добувають двохатомні феноли; сам фенол у промисловості так не добувають.
3. Гідроліз арилгалогенідів (див. розд. 31). Цей метод є одним із сучасних промислових способів добування фенолів.
4. Гідроліз солей арилдіазонію (див. розд. 37).
Фізичні властивості. Фенол являє собою білу кристалічну речовину з характерним запахом, добре розчинну в більшості органічних розчинників, але обмежено розчинну у воді. З водою він утворює кристалогідрати, а з парою води легко переганяється. Фенол — отруйна речовина, при попаданні на шкіру утворює опіки. Він вбиває багато мікроорганізмів, тобто має дезинфікуючі і антисептичні властивості.
Феноли мають високі температури кипіння, що пов’язано із значною асоціацією їх молекул за рахунок міжмолекулярних водневих зв’язків.
Хімічні властивості. Хімічні властивості фенолів визначаються гідроксильною групою і сполученим з нею бензольним ядром. Гідроксил, яквідомо, виявляє—/-ефект(див.розд. ЗО). Разомзтим вільні пари р-електронів атома кисню взаємодіють з л-електронами бензольного ядра (явище р, л-спряження). В результаті такої
13*
387
взаємодії р-електронна густина кисню змішується з л-електронною густиною бензольного ядра і утворюється єдина, спільна, спряжена електронна орбіталь, яка охоплює як бензольне ядро, так і атом кисню (рис. 70). Отже, електронна густина ароматичного ядра збільшується і гідроксильна група в молекулі фенолу виявляє
п „ 4-/И-ефект, який є значно сильнішим від її — I-
Рис. 70. Схема р, п-, и
спряження В молекулі ефекту. Наслідком р, л-спряженняє зменшення фенолу довжини зв’ язку С—О (/) і величини дипольного
моменту (р) у фенолу порівняно, наприклад, з етиловим спиртом. Від спирту фенол відрізняється також і положенням смуг поглинання коливань зв’язків С—О в ІЧ-спектрах:
Речовина
0,140 1,53 1230
І, нм р, С
V, СМ-1
Н3С —> СН 2-> 6-<— ]і
0,144
1,66 1050—1200
Внаслідок спряження електронна густина на атомі кисню в молекулі фенолу зменшується. Тому кисень, як більш електронегативний елемент, ніж водень, притягує до себе електронну густину від атома водню, але значно сильніше, ніж у випадку аліфатичних спиртів, у яких немає р, л-спряження. Це приводить до збільшення полярності Н—О-зв’язку в молекулах фенолів і підвищення активності атома водню цієї групи. В результаті кислотні властивості фенолів посилюються і виражені значно сильніше, ніж у спиртів. Проте фенол є досить слабкою кислотою, слабкішою навіть від вугільної, синильної і карбонових кислот. Це наочно демонструє порівняння констант іонізації
Речовина Н,С—СООН Н,СО3 НСН СВН8—ОН Н,0 С.,Н,ОН о 4 И О О а А О
Кд 1,76 • 10-& 4,9 • 10-’ 7,2 • 10-1° 1,3 • 10"м 1,8 • Ю*16 10'1»
Виходячи з величини константи іонізації, легко помітити, що кислотні властивості фенолу приблизно в 3770 разів слабкіші, ніж вугільної кислоти. Тому вугільна та інші кислоти'витісняють фенол з розчину феноляту:
С6Н6— ОНа -ф СО2 + Н2О С6Н5—ОН + КаНСОя
На кислотні властивості фенолу впливають введення в ароматичне ядро замісника і його положення в цьому ядрі відносно ОН-групи (табл. 33).
Електронодонорні замісники, такі якСН3-група (орієнтанти першого роду), збільшуючи електронну густину ароматичного ядра, одночасно підвищують її на атомі кисню ОН-групи, тому щозавдяки
388
Таблиця 33. Константи кислотності Л’д фенолів
Фенол
Фенол
р,л-спряженню електронна хмара ароматичного ядра і кисню є спільною. Тим самим вони послаблюють (зменшують) кислотні властивості фенолів (табл. 33):
о —н .
Електроноакцепторні замісники, такі як ЬЮ2-група (орієнтанти другого роду) і атоми галогенів, зменшуючи електронну густину ароматичного ядра, одночасно зменшують її і на атомі кисню ОН-групи. Тим самим вони підсилюють (підвищують) кислотні властивості фенолу.
Особливо значний вплив на зміну кислотності фенолів мають замісники в орто- і иара-положеннях, які в даному випадку знаходяться в р, л-спряженні з електронами ОН-групи. Замісники в Лівта-положенні, не спряжені з фенольним гідроксилом, мало впливають на зміну кислотності фенолу (табл. 33). Узгоджене
389
^-спряження, наприклад, вяара-нітрофенолі сприяє підвищенню кислотної іонізації цієї сполуки:
Нагромадження в ароматичному ядрі фенолу електроноакцеп-торних груп значно підсилює кислотні властивості фенольного гідроксилу. Тому 2,4,6-тринітрофенол (пікринова кислота) є досить сильною кислотою. Кислотні властивості фенольного гідроксилу є наслідком -рЛЇ-ефекту, в результаті якого в молекулах фенолів виникає відносно стабільний резонансний аніон, який можна зобразити мезомерними формулами (І—IV):
Негативний заряд у таких аніонах роззосередженпй по всій спряженій системі бензольного ядра, що знижує електронегативний потенціал атома кисню порівняно з таким же потенціалом алко-голят-іонів А1к-О~, в яких негативний заряд локалізований на атомі кисню. Внаслідок цього у фенолят-іона С6Н8О~ зменшується здатність до сполучення з протоном. Тому фенолят-іон є слабкішою основою, ніж, наприклад, етокси-аніон С2Н5О“.
Кислотно-основні властивості фенольного гідроксилу. Як уже зазначалося, в результаті р,л-спряження атом водню фенольного гідроксилу легше, ніж у спиртах, протонізується, а валентні електрони атома кисню роззосереджуються в орто- і мара-положеннях бензольного ядра:
Посилення кислотних властивостей фенолів приводить до того, що вони, на відміну від спиртів, утворюють феноляти не тільки при взаємодії з лужними металами х, але й з лугами:
СвН5—ОН + ИаОН ^=±г СвН5—О\'а + Н2О.
В результаті реакції утворюються феноляти (солі фенолів), які можуть бути виділені випарюванням реакційної суміші. Феноляти
* Взаємодія фенолів з лужними металами відбувається дуже бурхливо і може супроводжуватись вибухом,
390
лужних металів легко гідролізуються і тому їх водні розчини мають сильнолужну реакцію:
С(;Н —СЖа + НОН С6НВ—ОН + Иа+ ОН.
Фенол легко утворює з хлоридом заліза (III) забарвлений у фіолетовий колір комплексний фенолят заліза-
ОС6Н6
н
І
.О— СВН3
6С6Н,ОН + Ге СІ,-----
ЗНСІ
Ця реакці я є характерною якісною пробою на фенольний (енольнпй) гідроксил і на феноли взагалі.
Феноли, подібно до спиртів, можуть утворювати прості ефіри, які добувають переважно взаємодією фенолів з діалкілсульфатами або взаємодією фенолятів лужних металів з галогеналкілами. Наприклад:
СвН5—ОН + (СН3)28О4 ->С(ІНВ—О—СН3 + СН3—О8О3Н;
Фенілметиловий ефір (анізол)
СеН6—ОіМа + С2Н5—Вг-> СвНв—О—С,Н5 + ИаВг.
Фенілети ловий ефір (фенетол)
Внаслідок спряження р-електронів атома кисню з л-молекуляр-ною орбіталлю бензольного ядра атом кисню гідроксильної групи фенолу додатково сполучається з вуглецевим атомом бензольного ядра (див. рис. 70), і активність фенольного гідроксилу різко зменшується порівняно з активністю ОН-групи спиртів. Тому нуклео-фільне заміщення гідроксильної групи фенолів відбувається надзвичайно важко. Так, галогеноводні зовсім не заміщують ОН-групу в молекулі фенолу, а при взаємодії фенолу з РС1- утворюється тільки невелика кількість хлорбензолу, основним же продуктом є трпфенілфосфат:
(С6Н;О)5Р +
Нуклеофільне заміщення ОН-групи в молекулі фенолу відбувається за механізмом відщеплення — приєднання (див. розд. 31). Електроноакцепторні замісники в орто- і па/?с-положениях бен-
391
зольного ядра фенолу, зменшуючи електронну густину цього ядра, підвищують здатність фенольного гідроксилу до реакцій нуклеофільного заміщення. Так, 2,4,6-тринітрофенол(пікринова кислота) при взаємодії з РСІ- легко перетворюється на 2,4,6-тринітрохлор-бензол (пікрилхлорид):
Карбаніон
+ ОН .
Внаслідок р,л-спряження, як уже зазначалося, електронна густина на атомі кисню фенольного гідроксилу зменшується. Тим самим послаблюються основні властивості фенолів. Тому складні ефіри фенолів добувають взаємодією фенолу з хлорангідридами карбонових кислот у водному розчині їдкого лугу (реакція Шот-тен — Баумана, 1884 р., 1886 р.):
с6н5—он + сі—с—сн3МаОН' НаО^ сен6—о—с—сн3 + на.
Феніл ацетат
Реакції з участю бензольного ядра. Реакції фенолів, які відбуваються з участю ароматичного ядра, подібні до відповідних реакцій бензолу (див. розд. 28).
Реакції приєднання. Фенол при наявності каталізаторів легко гідрується і перетворюється на циклогексанол:
Реакції електрофільного з а м і щ е н н я. Найважливішими реакціями електрофільного заміщення фенолів є їх нітрування, галогенування, сульфування, алкілування, азоспо-лучення та ін. Оскільки в ароматичному ядрі фенолу міститься ОН-група, яка є сильним орієнтантом першого роду і збільшує електронну густину бензольного ядра:
і,053
1,000
1,053
то 5г-реакції для фенолу відбуваються надзвичайно легко в орто-і ппра-положеннях ароматичного ядра. Так, фенол нітрується роз-
392
бавленою (20 %-ю) азотною кислотою при 10—15 °С. При цьому утворюється суміш орто і ио/ш-нітрофенолів, яку розділяють перегонкою з водяною парою, оскільки орто-нітрофенол з нею легко відганяється. Леткість орто-нітрофенолу зумовлена внутрішньо-молекулярним водневим зв’язком між ОН- і ЬЮ2-групами:
Мононітрофеноли можуть далі нітруватись, але вже за допомогою концентрованої азотної кислоти. При цьому утворюються 2,4- і 2,6-динітрофеноли, які нітруються в ще більш жорстких умовах з утворенням 2,4,6-трйнітрофенолу (пікринової кислоти):
Пряме нітрування нітрофенолів концентрованою азотною кислотою супроводиться значним окисленням і осмоленням реакційної суміші. Тому технічно важливий 2,4-динітрофенол у промисловості добувають лужним гідролізом 1-хлор-2,4-динітробензолу:
і\аОН, Н2О
-неї
Концентрована азотна кислота окислює і осмолює фенол ще гильніше, ніж нітрофеноли. Тому 2,4,6-тринітрофенол (пікринову кислоту) у промисловості добувають так. Фенол спочатку сульфу-ють і добуті дисульфофеноли перетворюють потім нітруванням у пікринову кислоту:
393
На останній стадії цього процесу сульфогрупи заміщуються на нітрогрупи.
Фенол енергійно вступає в реакцію з галогенами. Залежно від кількості введеного в реакцію галогену можна добути орто- і пара-галогенофеноли, 2,4- і 2,6-дигалогенофеноли або 2,4,6-тригалогеио-фенол. Важливою є реакція фенолу з бромом (бромною водою), в результаті якої утворюється важкорозчинний у воді 2,4,6,-трибромфенол (ТІІЛ — 96°С). Тому реакція з бромом є якісною на фенол. За її допомогою можна виявити фенол у розчині навіть при його концентрації 1 : 100 000. При дії надлишку брому 2,4,6-три-бромфенол бромується далі і’перетворюється'иа2,4,4,6-тетрабром-циклогексадієн-2,5-он-1 •
Аналогічно фенол реагує і з хлором.
Галогенофеиоли виявляють дезинфікуючу дію, яка значно сильніша, ніж у фенолу.
Фенол і його похідні активно вступають у реакцію азоеполучен-ня, яка широко використовується в синтезі азобарвників (див. розд. 37).
Окислення фенолів. Наявність гідроксильної групи в ароматичному ядрі робить його нестійким до дії окислювачів. Залежно-від природи окислювача і умов проведення окислення добувають різні продукти. Так, при окисленні фенолу пероксидом водню при наявності залізного каталізатора утворюється двохатомний фенол — пірокатехін з невеликим виходом:
ОН
н,о .
При окисленні хромовою сумішшю фенол перетворюється на ипрс-хінон. Можливо, що при такому окисленні фенол спочатку
394
окислюється до гідрохінону, при дальшому окисленні якого і утворюється паро-хіион:
парй-Хінон уже не має характерної для бензольного ядра системи спряжених трьох подвійних зв’язків, тому він не виявляє ароматичних властивостей, а має властивості ненасичених кетонів.
Феноли легко окислюються навіть киснем повітря. При цьому вони змінюють свій колір і забарвлюються в рожевий- червоно-рожевий або темно-червоний колір. Домішки прискорюють окислення і тому неочищені феноли дуже швидко темніють. Утворення при окисленні фенолів забарвлених речовин пов’язане з проміжним утворенням хінонів, що містять хіноїдну групу атомів, яка є носієм забарвлення (хромофором) молекул органічних речовин.
Взаємодія фенолів з .альдегідами. Феноли можуть вступати в реакції конденсації з багатьма речовинами, зокрема з альдегідами. Особливого значення набула відкрита в 1872 р. А. Байєром конденсація фенолу з формальдегідом, в результаті якої утворюється фенолоформальдегідна смола.Якщо поліконденсацію фенолу з формальдегідом, яка відбувається при наявності кислот або лугів, проводити при надлишку фенолу, то утворюються низькомолекулярні полімерні сполуки, які називають поволоками'
Новолачна смола
При нагріванні фенолу з надлишком формальдегіду поліконденсація відбувається з утворенням полімеру з розгалуженою просторовою сітчастою структурою — резиту.
У промислових масштабах конденсацію фенолу з формальдегідом здійснив у 1908 р. А. X. Бакеланд. Тому добуті таким методом смоли були названі бакелітами. У конденсацію цього типу вступають багато фенолів і альдегідів. Синтетичні смоли, які при цьому утворюються, називають фенопластами.
Багатоатомні феноли
Серед багатоатомних фенолів найважливішими є пірокатехін, резорцин, гідрохінон, пірогалол і флороглюцин. Формули і температури плавлення їх наведені в табл, 34,
395
Таблиця 34. Багатоатомні феноли
Формула
Назва
Пірокатехін, 1, 2-діокснбензол
Резорцин, 1, 3-діоксибензол
Гідрохінон, 1, 4-діоксибензол
Пірогалол, 1,2, 3-триоксибензол
Флороглюцин, 1, 3, 5-триокснбензол
105
110
173
133
219
У промисловості пірокатехін добувають з кам’яновугільної смоли або синтетично — з орто-хлорфенолу за схемою:
Пірокатехін використовують в органічному синтезі для добування, наприклад, адреналіну, а також як проявник у фотографії.
Резорцин добувають лужним плавом бензол-1,3-дисульфокис-лоти:
+ г.Ч'ааЗОз + 2Н2О .
У резорцину ароматичний характер виражений менше, ніж у фенолу. Тому вів має здатність до кето-енольної тауто-
396
мерії, рівновага якої зміщена в бік енольної (фенольної) форми:
он
Внаслідок цього з деякими реагентами, наприклад з оцтовим ангідридом, резорцин реагує в енольній формі, тобто оксигрупами, і утворює діацетат, а з гідроксилам! ном він взаємодіє в кетонній формі, тобто кетонними групами, і утворює цпклогексен-4-діок-сим-1,3:
Резорцинта його похідні використовують у медицині як антисептичні, дезинфікуючі, антигельмінтні препарати, а також у промисловому органічному синтезі для добування барвників.
Гідрохінон у промисловості добувають відновленням сірчистою кислотоюпара-уЛнону, який, у свою чергу, добувають окисленням аніліну або анодним окисленням бензолу:
Анодне окислення
Гідрохінон є прекрасним відновником. Тому його широко використовують як проявник у фотографії, а також як стабілізатор (інгібітор полімеризації) ненасичених сполук. •
Хінон. Хінон вперше добув О. А. Воскресенський у 1838 р. окисленням хінної кислоти, виділеної з кори хінного дерева. Синтетично хінон добувають окисленням аніліну або анодним окисленням бензолу.
Хінонмістить спряженусистему подвійних зв’язківС=С—С=О. Тому за структурою і хімічними властивостями він є ненасиченим дпкетоном і виявляє властивості як кетону, так і ненасичених сполук.
При зміщуванні рівномоляриих кількостей хінону і гідрохінону утворюється молекулярна сполука — хінгідрон, який являє собою темно-зелені кристали з температурою плавлення 171 °С.
337
В хінгідроні молекули хінону і гідрохінону сполучені водневими зв’язками. Крім того, в хінгідроні відбувається перенесення заряду від гідрохінону до хінону. Гідрохінон при цьому є донором електронів, а хінон — їх акцептором:
Хінгідрон
Хінгідрон використовують як електрод (хінгідронний електрод) для визначення концентрації іонів Н+ у розчині.
Серед трьохатомних фенолів найважливіше значення мають пірогалол і флороглюцин. Пірогалол добувають піролітичним де-карбоксилуванням галової кислоти (див. розд. 35):
Пірогалол є сильним відновником. Лужний розчин пірогалолу швидко взаємодіє з киснем повітря (поглинає кисень). Тому його використовують у газовому аналізі для кількісного визначення кисню в газових сумішах.
Флороглюцин і його похідні поширені в природі, зокрема у вигляді глікозиду вони містяться в корені яблунь. Синтетично флороглюцин добувають нагріванням водного розчину хлоргідрату 1,3,5-триамінобензолу:
НСІ-Н^
змн4сі.
У флороглюцину найсильніше з усіх фенолів виявляється кето-енольна таутомерія і пов’язана з нею двоїста реакційна здатність. Залежно від природи реагенту в реакцію може вступати або фенольна (І), або кетонна (II) форма флороглюцину. Так, при взаємодії флороглюцину з діазометаном у реакцію вступає енольна форма (І) і утворюється 1,3,5-триметоксибензол (Іа), а при взаємодії флороглюцину з гідроксилам!ном у реакцію вступає кетонна форма і утворюється трикетоксим флороглюцину (Па):
398
11а
Флороглюцин використовують для відкриття ваніліну, лігніну, а також в органічному синтезі.
Пестициди. Пестицидами називають хімічні речовини, які використовують для боротьби з мікроорганізмами, рослинами і тваринами, шкідливими чи небажаними з точки зору економіки або охорони здоров’я.
За характером дії пестициди поділяють на Інсектициди — засоби боротьби з шкідливими комахами, акарициди — засоби боротьби з кліщами, гербіциди — засоби боротьби з бур’янами, водоростями у водоймищах і зрошувальних системах, бактерициди — засоби боротьби з бактеріями, фунгіциди — засоби боротьби з нижчими рослинами, зокрема з грибами, зооциди — засоби боротьби з шкідливими хребетними тваринами, нематоциди (антигельмінти) — засоби боротьби з круглими червами і глистами та деякі інші.
Пестицидну дію виявляють багато органічних речовин. Так, активними гербіцидами є солі і ефіри хлорфеноксіоцтової кислоти. Широко застосовується гербіцид 2,4-ДУ (2,4-дихлорфеноксіоцтова кислота), який добувають з 2,4-дихлорфеноляту натрію і натрієвої солі хлороцтової кислоти:
ОКа + СІ - СН2-СОО№—-СІ
сі
О-СН2— СООК'а + Касі .
Гербіцид 2,4-ДУ
Бактерицидами є фенол, крезоли, резорцин, хлорфеноли, хлор-крезоли. Пентахлорфенолят натрію СвС15Ойа використовують як фунгіцид. Його добавляють у промислові води для запобігання росту в них слизевих грибів і водоростей. Розчином пентахлорфено-ляту просочують деревину для запобігання руйнуванню її грибами і термітами.
З метою охорони навколишнього середовища кожний пестицид може бути використаний тільки з дозволу компетентних організацій. В СРСР такий дозвіл дає Державна комісія по боротьбі з шкідниками, хворобами рослин і бур’янами.
399
Контрольні запитання і вправи
1. Розмістіть нижченаведені речовини в порядку посилення їх кислотності: а) о-нітрофенол, .«-нітрофенол, «-нітрофенол; б) бензолсульфокислота, бензойна кислота, бензиловий спирт, фенол; в) лебромфенол, лі-крезол, л«-нітрофенол, фенол; г) «-хлорфенол, 2,4-дихлорфенол, 2,4,6-трихлорфенол.
2. За допомогою яких хімічних реакцій можна розрізнити: а) фенол і о-ксилол; б) «-етилфенол, «-метиланізол, «-метилбензиловий спирт; в) ацетилсаліцилову кислоту, етиловий ефір ацетилсаліцилової кислоти, саліцилову кислоту; г) л-динітробензол, лі-нітробепзойну кислоту, лі-нітрофенол?
3. Коніфериловий спирт С10Н12О3 добувають із соку хвойних дерев. Він розчиняється у водному розчині ЙаОН, але не розчиняється в розчині МаНСО3. Взаємодія коніферилового спирту з: а) хлористим бензоїлом і піридином дає речовину А (С24Н20О5); б) холодною НВг дає С10НиО2Вг; в) гарячою йодистоводневою кислотою дає йодистий метил; г) йодистим метилом і основою дає речовину Б (СПН14О3). Речовини А і Б не розчинні в розчині ИаОН і швидко знебарвлюють розчини КМпО4 і Вг2/ССІ4. При озонолізі коніферилового спирту утворюється ванілін. Яка структура коніферилового спирту? Напишіть рівняння для всіх вищеописаних реакцій.
Розділ 35. АЛЬДЕГІДИ, КЕТОНИ І КАРБОНОВІ КИСЛОТИ РЯДУ БЕНЗОЛУ
Альдегіди. Ароматичні альдегіди містять функціональну карбо-
нільну групу —С"
яка безпосередньо сполучена з атомом вуг-
лецю бензольного ядра. Найпростішим представником ароматичних альдегідів є бензальдегід (бензойний альдегід):
Якщо карбонільна група знаходиться в бічному ланцюгу, то такі альдегіди називають жирно-ароматичними, наприклад фенілоцто-вий альдегід:
Альдегіди бензольного ряду утворюють гомологічний ряд, найважливішим представником якого є бензальдегід.
Методи добування бензальдегіду. У промисловості бензальдегід добувають з толуолу одним з таких методів:
1. Каталітичним окисленням толуолу в паровій фазі киснем повітря при наявності каталізатора У265:
4 С6Н5-СН3^->С6Н5-С^° + Н2О. і
Окислення толуолу можна проводити і в рідкій фазі. Для цього окислювачем необхідно брати хромову суміш або МпО2 в 65 %-й сірчаній кислоті.
400
2. Каталітичним гідролізом (каталізатор Ре) хлористого бензи-лідену, який добувають хлоруванням толуолу (див. розд. 31):
с6н6-сн3 _2нсі~* СсН6 СНС12 95 _ 100оС
С0Н8—+2НСІ. хн
Аналогічно можна добути й інші ароматичні альдегіди.
Бензальдегід — безбарвна оліїста рідина (Ткап = 179 °С) з сильним запахом гіркого мигдалю, важко розчиняється у воді, але добре розчиняється в органічних розчинниках. Бензальдегід використовують для виробництва барвників, пахучих речовин та інших сполук.
Хімічні властивості. Бензальдегіду властиві реакції з участю альдегідної групи і з участю бензольного ядра.
Реакції з участю альдегідної г р у п и. Ароматичні альдегіди, в тому числі і бензальдегід, вступають у більшість реакцій, характерних для альдегідів аліфатичного ряду. Так, бензальдегід реагує з синильною кислотою, гідроксиламіном, феніл-гідразином, гідросульфітом натрію і утворює відповідно оксиніт-рил, оксим, фенілгідразон, гідросульфітну похідну:
СсН5—СН = И—N11—С(ІН6 Фенілгідразон
с.н.-ічн-кн,
б* /О
ХН
СЬІІЬ—СН—8О3К’а-
он
Гідросульфїтиа похідна
-> С6НБ—СН—СМ;
Оксинітрил
с„н5—СН = и—он.
Оксим
Разом з тим ароматичні альдегіди за деякими властивостями відрізняються від аліфатичних. Так, бензальдегід окислюється значно легше, ніж аліфатичні альдегіди. При цьому він перетворюється на бензойну кислоту:
/О п Ю
2СсН6—С< —2С6НБ—С^ .
ХН ХОН
Окислення бензальдегіду відбувається за ланцюговим атомарно-радикальним механізмом, прискорюється світлом (Иу>) і солями заліза, марганцю тощо.
Характерною для ароматичних альдегідів є реакція Канніц-царо (І853 р.), яка є окислювально-відновним диспропорціонуван-ням двох молекул альдегіду в розчині концентрованого лугу (50%-го
401
\гаОН або КОН). В результаті реакції утворюється ароматичний спирт і карбонова кислота (її сіль). Наприклад:
2С СДІа-С^-ОН + СЙИ5-С^°
У > Н Бензібючиш Спирт. * ^СЖа.
Натріегіа $1ль, бен^Йнр/ к иСлсуі и
Реакція підпорядковується кінетичному рівнянню третьогопо-рядку: V ~ к [СвН5—ОНОРЖаОН]. Механізм реакції Канніццаро складається з ряду елементарних реакцій, першою з яких є швидке і оборотне приєднання нуклеофільної групи ОН “лугу до карбонільного атома вуглецю однієї з молекул альдегіду. На другій,' повільній стадії відбувається перенесення гідрид-аніона Н~ на карбонільну групу другої молекули альдегіду. При цьому утворюється карбонова кислота і алкоголят-аніон, які обмінюються протоном і перетворюються на стійку пару аніон кислоти — спирт:
8-
6+хР Швидко
с6ц,-с + он с^щ-сн-о* :
\ і "З І
- II он
(" О
V' Повільно
СвНв-С-О“ + -------- СвНу-С=.О + С^-СНї-О -
ОН ' Н * о:(н
і\а+
С4,н5 - (/ + с,н^- сн, - он.
\'У\'ат
Реакція Канніццаро легко відбувається також між ароматичними і аліфатичними альдегідами. Так, при нагріванні в лужному середовищі надлишку формальдегіду з бензальдегідом останній повністю перетворюється на бензиловий спирт:
Са[ 1.-0^° -р Н—с/° С-Нн-СН,—ОН + Н—с/°
' _ ЧІ НІ 5™ ЧО№.
Бензальдегід дуже легко вступає в реакції конденсації з альдегідами та іншими органічними речовинами. Однією з таких реакцій є бензоїнова конденсація, в результаті якої 2 моль бензальдегіду конденсуються в оксикетон — бензоїн:
СвНв-С-СН-С6Н5.
II І о он
Бензоїн
Бензоїнова конденсація відбувається під впливом водного розчину КСМ, який створює лужне середовище. Цю конденсацію вперше здійснив М. М. Зінін у 1839 р.
402
Реакції з участю бензольного ядра. Ароматичним альдегідам, як і бензолу, властиві реакції електрофільного заміщення в бензольному ядрі: галогенування, нітрування, сульфування та ін. Альдегідна група електроноакцепторна і в зв’язку з цим є, наприклад у бензальдегіді, литпа-орієнтантом (замісником другого роду). Вона за рахунок -7- і —М-ефектів зменшує електронну густину бензольного ядра. Причому найбільше цієї густини зменшиться в орто- і пора-положеннях. Тому бензальдегід значно важче, ніж бензол, вступає в реакції електрофільного заміщення атомів водню ароматичного ядра і заміщення відбувається переважно в летла-положенні відносно альдегідної групи. Наприклад, нітрування бензальдегіду відбувається в досить жорстких умовах і дає лгелш-нітробензальдегід:
Н2О
Кетони. Кетони ряду бензолу є двох типів: чисто ароматичні, у молекулах яких кетонна група сполучена з атомами вуглецю двох ароматичних ядер, і жирно-ароматичні, в яких кетонна група сполучена як з ароматичним ядром, так і з алкільним радикалом. Прикладом ароматичного кетону є бензофеноп (дифенілкетон), жирно-ароматичного — ацетофенон (мети лфепіл кетон):
Бензофеноп
Ацетофенон
Ацетофенон добувають переважно ацилуванням бензолу хлористим ацетилом або оцтовим ангідридом при наявності каталізатора— безводного хлориду алюмінію (реакція Фріделя — Крафтса, див. розд. 29):
Ацилування за Фріделем — Крафтсом є реакцією електрофільного заміщення (5£2-реакція). Механізм, наприклад., ацетилування, бензолу хлористим ацетилом складається я таких послідовних еле-
ментарних реакцій:
Н3С-С-СІ + АІСІз
О
Н3С-С + Л1С14 ;
II О
Аналогічно за реакцією Фріделя — Крафтса добувають також і ароматичні кетони. Наприклад, при взаємодії бензолу з хлористим бензоїлом утворюється бензофенон:
Хлористий Бензофенон
бензоїл
Ароматичні кетони вступають у реакції, які відбуваються як з участю кетонної групи, так і з участю ароматичного ядра. Проте ароматичні кетони менш реакційноздатні, ніж аліфатичні кетони.
Карбонові кислоти ароматичного ряду. Ароматичними карбоновими кислотами називають похідні ароматичних вуглеводнів
що містять функціональну карбоксильну групу —С-^ ,яка без-
ХОН
посередньо сполучена з вуглецевим атомом бензольного ядра. Залежно від кількості карбоксильних груп ароматичні кислоти поділяють на одно-, двох-,трьох- і більше основні. Найпростішим представником одноосновних ароматичних кислот є бензойна кислота СвН5—СООН. Ароматичні кислоти утворюють гомологічний ряд. Найважливіші представники цього ряду, їх формули, назви, константи наведені в табл. 35.
Якщо карбоксильна група сполучена з атомом вуглецю бічного ланцюга, то такі кислоти належать до жирно-ароматичного ряду. Прикладомтакої кислоти єфенілоцтова кислотаС6НГч—СН2—СООН (7™ = 76,7 °С).
Одноосновні ароматичні кислоти — кристалічні речовини, важко розчиняються в холодній воді, краще—-в гарячій, добре розчиняються в полярних розчинниках. Температури кипіння і плавлення цих кислот вищі, ніж кислот аліфатичного ряду з тією самою кількістю вуглецевих атомів
Методи добування. Деякі ароматичні кислоти і їх похідні поширені в природі. Вони входять до складу рослинних смол, бальзамів або виконують певні функції в живих організмах. Разом з тим для найважливіших ароматичних кислот розроблені спеціальні методи синтезу. Наприклад, бензойну кислоту можна добути всіма методами, відомими для добування кислот жирного ряду, зокрема:
404
Таблиця 35. Кислоти ароматичного ряду
Формула Назва гпл> °с „25 кл Розчинність , г/100 г води
СН,—СООН Н3С—С6Н4—СООН Бензойна Т олуїлова 122 6,5-10-5 0,34
орто- 106 1,35-10-5 0,12
мета- 112 5,32-10-5 0,10
пара- 180 4,33-10-5 0,03
П-С1—С(Н4—СООН О2М—СвН4— СООН пара-Хлорбензойна Нітробензойна 242 1,04-10~4 0,009
орто- 147 6,95-10-3 0,75
мета- 141 .3,4-10"4 0,34
пара- 242 3,93-10-4 0,03
о-НО—С0Н4—СООН Саліцилова (орто-оксибепзойна) 159 1,07-Ю-з 0,22
П-Н2М—СсН4—СООН /шрп-Амінобен-зойна 187 1,2-10-5 0,30
1. Окисленням бензойного альдегіду:
/О ГП1 ,()
СРН6—с,Нг—С-^ ХН ХОН.
2. Гідролізом бензонітрилу:
С(;Нг, СМ _мн3
О
3. З бромбензолу через магній- або літійорганічні сполуки, діючи на останні вуглекислим газом:
С6Н8-Вг + Мй ->С6Нб-М8-Вг С,.Н6—СООМрВг _м"°гН(онГ СвН5СООН
СО нон
С6НВ—Вг + 2ЕІ С6Н5-Ьі —- С„Н5—СООІл С6НВСООН + Ьі+.
У промисловості бензойну кислоту добувають термічним окисленням толуолу киснем повітря під тиском:
Інший промисловий метод добування бензойної кислоти базується на гідролізі бензолтрихлориду, який, у свою чергу, добувають хлоруванням толуолу (див. розд. 31):
їй
Хімічні властивості. Хімічні властивості ароматичних кислот визначаються наявністю в них карбоксильної групи і ароматичного ядра.
Реакції з участю карбоксильної групи. Ці реакції ароматичних кислот аналогічні реакціям для аліфатичних кислот. Перш за все, наявність карбоксильної групи надає цим речовинам кислотних властивостей. У водних розчинах ароматичні кислоти дисоціюють з утворенням протонів:
Однак одноосновні ароматичні кислоти сильніші, ніж аліфатичні. Так, бензойна кислота, Лд якої становить 6,5 • 10-Б, більш як у два рази сильніша від оцтової кислоти, Кд якої дорівнює 1,76 X X 10-6. Пояснюється це впливом бензольного ядра на карбоксильну групу, яке відтягує електронну густину від карбоксилу і тим самим підсилює її кислотні властивості1:
н.
Сила ароматичних кислот залежить від природи і положення замісника в бензольному ядрі. Електронодонорні замісники, наприклад мети льна група СНЯ, зменшують силу цих кислот, а електроно-акцепторні замісники, такі як КО2-група, збільшують силу ароматичних кислот (табл. 35):
Замісники в /га/щ-положеннях мають значно більший вплив на силу ароматичної кислоти, ніж у лге/тш-положенні. Пов’язано це з тим, що в першому випадку зміщення електронної густини відбувається за спряженою системою (М-ефект), а вплив замісника в лщ/иа-положенні здійснюється переважно його слабким /-ефектом. Електронегативні замісники в орто-положенні особливо сильно збільшують силу ароматичної кислоти, оскільки у них крім —/-і —Лї-ефектів утворюється внутрішньомолекулярний водневий зв’язок, який ще більшою мірою послаблює зв’язок О—Н і тим самим сприяє протонізації атома водню карбоксилу:
1 Залежно від електронної природи груп, сполучених з бензольним ядром, воно може виявляти електронодонорні або електроноакцеторні властивості .
Тому оряю-нітробензойна кислота значно сильніша, ніж пара-і .иета-ізомери цієї кислоти.
Ароматичні кислоти, як і аліфатичні, вступають у реакції з лугами, карбонатами і утворюють солі:
С6Н6—СООН + ІХ'аОН -> СвН5—СОО№ + Н2О.
Бензоат натрію
Бензоат натрію виявляє дивну властивість. Різні люди по-різному сприймають його на смак. Одним він здається гірким, іншим — кислим, солодким або зовсім без смаку.
Із спиртами ароматичні кислоти утворюють складні ефіри, а з РСї5 або 8ОС12 — хлорангідриди:
г н . рс|‘ г н г^° с*н»он- н* г н с^°
С6НВ-С^ СвН5-С*_ _І)?ОСсН5-С<
ХС1 —-РОС!а ХОН 'О—С2На.
Хлористий Бензоііиоетиловий
бензоїл ефір
Важливе значення має хлорангідрид бензойної кислоти — хло-
ристий бензоїл СвН5—С/
ХСІ
' Хлористий бензоїл (Ткип — 197 °С) — безбарвна рідина з різким запахом, повільно гідролізується і перетворюється на бензойну кислоту:
,0 /£>
С.Н5—С" -І- НОІІ -> С.НС" +НС1.
6 6 \сі чон
Хлористий бензоїл використовується як ефективний бензоїлую-чий реагент, за допомогою якого можна вводити в молекули інших речовин бензоїльну груп)' СсН5—С—. Наприклад, бензоїлуванням
II О
фенолу за реакцією Щоттен — Баумана (див. розд. 34) добувають феніловий ефір бензойної кислоти:
С6Н5—С—СІ + с„н5-он с6нв-с-0-с6н5.
о о
Бензоїлуванням солей карбонових кислот добувають ангідриди органічних кислот:
С„Н6—С-С1 -ф На—О—С—С6Н8-> СГ)Н8—С—О—С—СвНв + ІХ’аСІ.
Ангідрид бензойної кислоти
407
При взаємодії хлористого бензоїлу з пероксидом водню в розчині їдкого лугу добувають пероксид бензоїлу (перекис бензоїлу, 7ПЛ = 104 °С):
н г СІ Т4 о 2№ОН_»с6Н5—С—о—О-С—С„Н6. 2С6Н6—С—СІ -р Н2О2~2иаС1-* 8 ц II
« -н*° О О
Пероксид бензоїлу
Пероксид бензоїлу використовують як ініціатор радикальної полімеризації ненасичених сполук, а також для відбілювання харчових жирів, олій, борошна та інших продуктів.!
Бензойна кислота порівняно легко декарбоксйлу'ється, особливо при нагріванні з гідроксидами лужних, лужно-земельних металів, і перетворюється на бензол:
СвН5—СООН -а (°-); -> СвН6 + СО2.
Реакції з участю ароматичного ядра. Ароматичним кислотам, як і бензолу, властиві реакції електрофільного заміщення в бензольному ядрі: галогенування, нітрування, сульфування та ін. Карбоксильна група — електрофільна і є замісником другого роду (лета-орієнтантом, див. розд. ЗО). Вона за рахунок —І- і —А1-ефектів зменшує електронну густину бензольного ядра і тим самим знижує його активність до реакцій електрофільного заміщення. Тому реакції електрофільного заміщення атомів водню бензольного ядра ароматичних кислот відбуваються значно важче, ніж бензолу. Крім того, карбоксильна група поляризує бензольне ядро так, що на атомах вуглецю в орто- і яарп-положен-нях дефіцит електронів (заряд 6+) найбільший, а в мета -положеннях електронна густина знижується менше:
Тому електрофільне заміщення атомів водню, наприклад, у молекулі бензойної кислоти відбувається біля атомів вуглецю 3 і 5 (в мета-положенні). Наприклад, при нітруванні бензойної кислоти нітруючою сумішшю можна добути 3-нітробензойну, а потім 3,5-динітро-бензойну кислоту:
Бензойну кислоту використовують для синтезу барвників, пахучих речовин, фармацевтичних препаратів, а також для консерву-
408
вання харчових продуктів, оскільки вона має сильну антисептичну дію.
Ароматичні оксикислоти (фенолокислоти). З оксибензойних кислот найважливіше значення має саліцилова (2-оксибензойна) кислота, яка зустрічається в деяких рослинах у вигляді глікозидів, наприклад в корі верби (від лат. заііх — «верба» походить назва кислоти), в ефірних оліях квітів:
Саліцилова кислота .
У промисловості саліцилову кислоту добувають термічним кар-бокснлуванням феноляту натрію оксидом вуглецю (IV):
Ця реакція відкрита Г. Кольбев 1859 р., вдосконалена в 1885р. Р. ІІІміттом і називається реакцією Кольбе—Шмітта. Реакція Кольбе — Шмітта відбувається за механізмом електрофільного заміщення:
Саліцилова кислота (Л'д = 1,07 • 10~3) у 17 разів сильніша, ніж бензойна (/(д СеНБСООН = 6,5 • 10-6). Пояснюється це утворенням внутрішньомолекулярного водневого зв’язку між функціональними групами, що приводить до збільшення величини 6+-заряду на карбоксильному атомі вуглецю і сильнішого зміщення до нього електронних пар ОН-групи карбоксилу, що, в свою чергу, полегшує протонізацію атома водню карбоксилу (орто-ефект):
+ ЬҐ.
Хімічні властивості саліцилової кислоти визначаються наявністю в її молекулі фенольного гідроксилу, карбоксильної групи і бензольного ядра. Як фенол, саліцилова кислота утворює з РеС13 комплексну сполуку з характерним фіолетовим забарвленням.
409
З карбонатами лужних металів у реакцію вступає тільки карбоксильна група молекули саліцилової кислоти. При цьому утворюються солі — саліцилати'
+ Н,о -І- со, .
З лугом, при наявності достатньої його кількості, в реакцію вступає як карбоксильна, так і гідроксильна групи саліцилової кислоти:
2№ОН
Саліцилова кислота утворює два види похідних: похідні з участю фенольного гідроксилу і з участю карбоксильної групи. Так, при дії на саліцилову кислоту галогенангідридів або ангідридів кислот в реакцію вступає фенольний гідроксил. У випадку її взаємодії з ангідридом або хлорангідридом оцтової кислоти утворюється аспірин (ацетилсаліцилова кислота, Тш — 137 °С), який широко використовується як жарознижувальний медичний препарат:
н3с - соон-
З хлороксидом фосфору саліцилова кислота утворює хлорангідрид:
Під час взаємодії цього хлорангідриду з фенолом утворюється феніловий ефір саліцилової кислоти (фенілсаліцилат, Тпл= 43°С), який під назвою салол використовується в медицині як дезинфікуючий препарат при деяких кишкових захворюваннях:
хО чо-с6н6 + неї, он
Фен іл саліцилат Ісало.і і
410
У промисловості салол добувають нагріванням саліцилової кислоти і фенолу в середовищі хлороксиду фосфору.
Бензольне ядро саліцилової кислоти може вступати в реакції електрофільного заміщення з галогенами, азотною, сірчаною кислотами та іншими електрофільними реагентами. За рахунок впливу ОН-групи ароматичне ядро саліцилової кислоти значно активніше до цих реакцій, ніж бензольне ядро молекули бензойної кислоти. Так саліцилова кислота легко взаємодіє з бромною водою, тоді як бензойна кислота за цих умов бромну воду не знебарвлює:
Ві
3,5-Дибромсаліцилова кислота
2НВг.
Ефективним протитуберкульозним препаратом є /ш/щ-аміноса-ліцилова1(4-аміно-2-оксибензойна) кислота, яку скорочено називають ПАСК. Вихідною речовиною для добування цієї кислоти служить резорцин, який спочатку взаємодією з аміаком перетворюють на л/еща-амінофенол, а його потім карбоксилуваниям за реакцією Кольбе — Шмітта — на ПАСК-
NN9
ПАСК
ПАСК — кристалічна речовина (Тпл = 147 °С), розчиняється в більшості органічних розчинників, важко розчиняється у воді, легко декарбоксилується і перетворюється при цьому на мета-амінофенол.
Ароматичні триоксикарбонові кислоти. З цієї групи кислот найважливіше значення має 3,4,5-триоксибензойна, або галова, кислота (Т!Ш = 253°С). Галова кислота і її похідні входять до складу дубильних речовин (чорнильні горішки дуба, дубова кора, листя чаю тощо). У промисловості галову кислоту добувають гідролізом природної дубильної речовини таніну або лужним плавом 5-бромпротокатехової кислоти:
5-Б|>о.м п ро гокатехова кисло га
СООН
Галова кислота
Галова кислота при нагріванні декарбоксилується і перетворюється на трьохатомний фенол пірогалол (див. розд. 34).
411
Галова кислота виявляє сильні відновні властивості. її використовують як барвник для приготування чорнила, похідні галової кислоти застосовують при дубленні шкіри.
Дикарбонові ароматичні кислоти. Беизолдикарбонова кислота існує у вигляді трьох ізомерів:
Фталева кислота
.СООН
'СООН
Ізофталева кислота
СООН
Т ерефталева кислота
З цих дикарбонових кислот найбільше значення мають фталева і терефталева кислоти. Фталеву (бензол-1,2-дикарбонову) кислоту в промисловості добувають каталітичним окисленням (каталізатор У2О5) о/япо-ксилолу або нафталіну.При цьому спочатку утворюється ангідрид фталевої кислоти, який потім гідролізують у фталеву кислоту:
Фталева кислота (Тпл = 206°С) при нагріванні до температури плавлення легко дегідратується і перетворюється на фталевий ангідрид (7ПЛ = 130 °С):
+ Н2о.
Фталевий ангідрид є важливою вихідною речовиною в органічному синтезі і виробництві синтетичних смол. При взаємодії фталевого ангідриду зі спиртами 7?—ОН утворюються ефіри фталевої кислоти, а при взаємодії його з аміаком — фталімід:
-Н,О
ОТ0"
соон
412
Ефіри фталевої кислоти, наприклад дибутилфталат (7? — С4Н9; Ткип == 340 °С), застосовують як висококиплячі розчинники і пластифікатори синтетичних полімерів. Диметил-, діетил-і дибу-тилфталати (7? — СН3, С2Н5, С4НЙ) використовують як репеленти — завоби проти комарів і гнусу.
Фталімід (Тпл = 238 °С) у вигляді N—Вг- і М—К-похідних використовується в органічному синтезі, зокрема для добування первинних амінів і амінокислот. У молекулі фталіміду вільна пара р-електронів атома азоту спряжена з л-зв’язками двох карбонільних груп (р,л-спряження). Внаслідок цього електронна густина на імідному атомі азоту зменшується і він досить сильно притягує до себе електронну пару від атома водню, що приводить до послаблення зв’язку Г\—Н і підвищення його кислотних властивостей. Тому фталімід виявляє слабкі кислотні властивості і водень імідної групи може заміщуватись на атоми металів. При цьому утворюються солеподібні продукти. Так, при взаємодії фталіміду зі спиртовим розчином КОН утворюється іонно побудований фталімід калію, який легко вступає в реакцію з галогеналкілами і утворює М-алкілпохідні фталіміду. М-Алкілпохідні фталіміду при гідролізі утворюють первинні аміни (реакція Габріеля, 1888 р.):
Гліфталеві смоли. Значні кількості фталевого ангідриду використовують для добування високомолекулярних поліефірів, які називають гліфталями, або гліфталевими смолами. Гліфталі добувають термічною конденсацією фталевого ангідриду з багатоатомними спиртами, перш за все з гліцерином:
Гліфталь
413
Макромолекули гліфталевих смол мають просторову сітчасту структуру.
Гліфталі являють собою тверду смолисту масу, яку використовують як основу для лаків і плівок. При добавлянні до гліфталевої смоли висихаючих олій добувають алкідні смоли, які використовують для виробництва стійких лаків і фарб.
Терефталева (бензол-1,4-дикарбонова) кислота. У промисловості цю кислоту добувають каталітичним окисленням /шрп-ксило-лу або пйра-метилізопропілбензолу (цимолу):
02,СоВгг,130-1600С
Терефталеву кислоту добувають також каталітичним термічним карбоксилуванням бензойної кислоти оксидом вуглецю (IV) під тиском при наявності каталізатора (КНСО8):
<^-С-ОН.+ СО,
КНСО3,340“С
За фізичними властивостями терефталева кислота — кристалічна речовина, яка сублімується при температурі понад 300°С.
Надзвичайно великого значення набули ефіри і поліефіри те-рефталевої кислоти. З метанолом терефталева кислота утворює диметиловий ефір (диметилтерефталат, Ткип = 140 °С), який далі шляхом переетерифікації перетворюють на етиленглікольтерефта-лат. При поліконденсації етиленглікольтерефталату утворюється поліетилентерефталат з молекулярною масою 15 000—20 000:
-2Н2О
-2Н,С-О!І
О - СНз +2НО - СН., - СН, -оі І
414
О—СН,~СН
— СН,—
он +
Пбліетилентерефталат
+ п НО~СН2 —СН2—ОН • п
Поліетилентерефталат використовують переважно для виготовлення поліефірного волокна (лавсану1, або терилену), яке прядуть з розплаву. Волокно лавсан не мнеться, стійке до різних погодних умов.
Розділ 36. АРОМАТИЧНІ АМІНИ
Ароматичними амінами називають сполуки, в молекулах яких міститься аміногрупа (—ЬІН2, —МНУ? або —М/?2), безпосередньо сполучена з вуглецевим атомом ароматичного ядра. Ароматичні аміни, як і аміни аліфатичного ряду, є похідними аміаку. Вони можуть бути первинними (Аг—МН2), вторинними (Аг—ІЧН—Аг) і третинними (Ага1Ч, де Аг — ароматичний радикал, наприклад С6НБ-----феніл, о-, м-, п-СНа—СвН4 о-, м-, п-толіл і т. д.).
Найпростішим первинним ароматичним аміном є анілін С6Н6—1ЧН2, вторинним — дифеніламін (С0Н5)2ГЧН, третинним — трифеніламін (С0Н5)аІЧ. Існують також жирно-ароматичні аміни, до складу яких входять не тільки ароматичні, а й аліфатичні радикали: метилані-лін СвН5—1ЧН—СНа, диметиланілін С«НВ—М(СНа)2. Крім того, відомі аміни з аміногрупою в бічному ланцюгу, наприклад бензи-
Таблиця 35. Ароматичні аміни
Формула Назва 7ПЛ, °С ЛсИП» °С «о
СдН6—НН2 Анілін —6,2 184,4 3,82-10"10
Н8С—С6Н4—мн. Толуїдин
орто- —28 199,7 2,47-10~10
мета- —43 203 4,92-10-1°
пара- 45 200 1,18.10-»
О2М—С(,Н4—мн2 Нітроанілін
орто- 71 284 6- ю-»4
мета- 112 306 3,2-10~12
пара- 148 332 0,1-10-12
п-СІ—СсН4—мн2 ппрп-Хлоранілін 70 232 1,5-10-ю
п-ІЮ83—СвН4— ИН. Сульфапілова кислота 288 —- —
(СвНЕ)2МН Дифеніламін 52 302 7,6-10-и
(С«Н5)3М Трифеніламін 127 365 —
сен8—ічн—сна Метиланілін —57 196 7,1-10-ю
С,Н6-М(СН3), Диметиланілін 3 194 1,15-Ю-о
1 Назва «лавсан» утворена з початкових букв таких слів: лабораторія високомолекулярних сполук Академії наук.
415
ламін С0Н5—СН2—МН2. Формули, назви, температури плавлення і кипіння, константи основності (Л’о) найважливіших представників ароматичних і жирно-ароматичних амінів наведені в табл. 36.
Номенклатура. Ароматичні аміни називають за систематичною або тривіальною номенклатурою За систематичною номенклатурою назву ароматичного аміну утворюють з назви відповідного ароматичного вуглеводню і префікса аміно-, наприклад амінобензол, амінотолуол і т. д. Для багатьох ароматичних амінів використовують тривіальні назви, які виникли історично. Так, амінобензол називають аніліном, амінотолуоли — толуїдинами тощо.
Ізомерія. Ізомерія ароматичних амінів залежить від різного розміщення в бензольному ядрі аміногрупи відносно іншого замісника (орто-, мета- і пара-ізомери), від положення аміногрупи в ядрі або в бічному ланцюгу, а також від положення алкіль-ного радикала в ядрі або біля атома азоту. Наприклад, нижченаве-дені аміни є ізомерами:
ІЧН2 МН2 №І2 СН2-МН2 ІЧН-СН3
о-Толуїдин
.«-Толуїдин
СН3
л-Толуїдин Бензиламін И-Метиланілін
Методи добування. З сучасних промислових методів добування ароматичних амінів найважливіше значення мають такі:
1. Відновлення ароматичних нітросполук. Так,анілін добувають відновленням нітробензолу. Реакцію добування аніліну відновленням нітробензолу відкрив у 1842 р. видатний російський хімік М. М. Зініи, використовуючи як відновник сульфід амонію:
СвН6-К'О2 + З (ИН4)28 -> СвН5—ЬІН2 + 2Н,0 + 6МН3 + 38.
Відновленням інших ароматичних нітросполук М. М. Зінін добув ряд інших ароматичних амінів. Тому реакцію відновлення ароматичних нітросполук в ароматичні аміни називають реакцією Зініна. Економічно найвигідиішимє відновлення ароматичних нітросполук залізом (стружками або ошурками) в соляній кислоті. Відновлення в цьому випадку відбувається за такою схемою:
С6Н5— Г\О2 + ЗЕе + 4Н2О —СГ)Н6-КН2 + ЗЕе (ОН)2;
С6Н5—МО2 + 6Ее (ОН)2 + 4Н2О С6Нв-МН2 + 6Ее (ОН)3;
або сумарно:
неї
С6Н5-ЛГО2 + 2Ее + 4Н2О-----> С6Н5—№Н2 + Ее (ОН)3.
На початковому етапі реакції відбувається утворення хлориду заліза (II), який каталізує процес відновлення і зв’язує кисень нітрогрупи.
41 6
2. З галогенопохідних ароматичних вуглеводнів і аміаку. Реак-ціюдобуваиня аніліну амонолізом хлорбензолу відкрили в 1870 р. російські вчені О. М. Енгельгардт і П. О. Лачинов. У сучасних промислових установках реакцію ведуть з надлишком аміаку, під тиском і при наявності мідних каталізаторів. Вихід, наприклад, аніліну з хлорбензолу за таких умов досягає 90 % теоретичного'-
СвН5—СІ + 2МНа 200°С’ М-Па-* СеНБ—ИН2.
Орто- і наря-нітрохлорбензоли взаємодіють з аміаком значно легше, ніж хлорбензол, оскільки нітрогрупа підвищує реакційну здатність атома хлору (див. розд. 31).
Механізм відновлення нітросполук. Відновлення ароматичних нітросполук відбувається через ряд стадій і проміжних сполук. Утворення кінцевого продукту відновлення залежить від рН середовища і природи відновника. Якщо нітробензол відновлювати в кислому середовищі залізом або оловом, то за цих умов спочатку утворюється иітрозобензол, який потім перетворюється на И-фе-нілгідроксиламін, а останній відразу ж відновлюється до аніліну:
С6НБ-МО2 2НЕ^-7-> С„НБ-М = О -- С6НБ—ИН—ОН —5- СБНБ-МН2.
Нітрозо- Гч'-Фенілгід-
бензол роксиламіи
У нейтральному середовищі, коли застосовують як відновник . цинк і хлорид амонію, кінцевим продуктом відновлення нітробензолу є ІЧ-фенілгідроксиламін (7ПЛ — 81 °С).
У лужному середовищі, наприклад, коли застосовують як відновник цинк і гідроксид натрію, нітробензол відновлюється до гідразобензолу, утворення якого відбувається через ряд проміжних продуктів. Спочатку з нітробензолу утворюється нітрозобен-зол і і'і-фенілгідроксиламін, які далі взаємодіють між собою і утворюють азоксибензол. Наступне відновлення азоксибензолу приводить до утворення азобензолу, відновлення якого дає гідразобензол:
СвН5—ИО2 - » СвНБ—N = О СвНБ—ИН—ОН;
СвНБ—N=0 + СеНБ—ИН—ОН СеНБ—М=М—СвНБ
О-
Азоксибензол
-СБНБ-М=М-С6НБ СеНБ—ИН—ИН—СвНБ.
Азобензол Гідразобензол
М. М.Зінін (1842 р.) встановив, що при нагріванні гідразобен-волу з кислотами (НС1 або Н28О4) відбувається так зване бензиди-нове перегрупування, в результаті якого утворюється технічно -важливий діамін — бензидин (4,4'-діамінодифеніл, Тпл = 128 °С). »1/2 14 ьі20 417
Бензидин широко застосовується для добування азобарвників!
Рис. 71. Схема р,л-сгіря-жения в молекулі аніліну
Фізичні властивості. Ароматичні аміни — рідини або тверді речовини (табл. 36) з характерним неприємним запахом. Дуже о т р у й н НУ воді мало розчинні. Анілін, як найпростіший представник ароматичних амінів, являє собою оліїсту рідину, важко розчинну у воді. Сам анілін частково розчиняє воду (5 %), від якої його осушують твердими лугами (№ОН або КОН). Анілін є полярною речовиною. Його дипольний момент становить р — 1,52 Б.
В ІЧ-спектрах аніліну є дві інтенсивні смуги поглинання в області 1180 — 1360 см-1, які відповідають валентним коливанням зв’язків С—N. і сильні смуги деформаційних коливань в області 650 — 900 см-1, характерні для зв’язків №—Н.
Електронна будова молекули аніліну.
У молекулі аніліну та інших ароматичних амінів, на відміну від аліфатичних амінів (див. розд. 13), середнє значення
валентного кута атома азоту збільшується і досягає близько 120°. Внаслідок цього атом азоту аміногрупи цих амінів перебуває в і/?2-гібридизованому стані. Тому аміногрупа в молекулі ароматичних амінів набуває плоскої конфігурації.
Атом азоту в молекулі аніліну дві свої $р2-орбіталі використовує на утворення двох о-зв’язків з 8-орбіталями двох атомів водню, а третю 8/г-орбіталь — на утворення <т-зв’язку з ^-гібридизованим атомом вуглецю бензольного ядра. Четверта 5/г-орбіталь змінного азоту зайнята вільною парою електронів.
У молекулі аніліну атоми і групи атомів взаємно впливають одні на одних: змінна група впливає на бензольне ядро, а бензольне ядро, в свою чергу, — на амінну групу. Цей вплив виявляється у вигляді двох електронних ефектів: індукційного і мезомерного. Амінна група виявляє — 7-ефект (див. розд. ЗО). Разом з тим вільні пари /7-електронів атома азоту аміногрупи взаємодіють з л-електро-нами бензольного ядра (явище р, л-спряження). В результаті р-електронна густина азоту змішується з л-електронною густиною бензольного ядра і утворюється єдина спільна спряжена електронна орбіталь, яка охоплює як бензольне ядро, так і атом азоту (рис. 71).
Отже, електронна густина ароматичного ядра збільшується і амінна група виявляє в аніліні -ф/И-ефект, який значно сильніший від її — /-ефекту. Наслідком р.л-спряження є зменшення в молекулі аніліну довжини зв’язку С—N (І) і дипольного моменту р порівняно з тими ж величинами для аліфатичних амінів, наприклад
418
для етиламіну, етильна група якого виявляє -(-/-ефект і викликає зміщення електронів до атома азоту аміногрупи:
р.,р
/с-и 'нм
1.53
0,127
н3с-сн2—к’н2
-і------
3.19
0,147
У зв’язку із спряженням електронна густина на атомі азоту в молекулі ароматичних амінів зменшується. Внаслідок цього зменшується здатність змінного азоту приєднувати протони. Отже, основні властивості ароматичних амінів значно послаблюються порівняно з основними властивостями аміаку. Разом з тим за рахунок сильного -р/И-ефекту аміногрупи підвищується електронна густина бензольного ядра ароматичних амінів, переважно аорто-і иара-положеннях. В результаті підвищується його активність до реакцій електрофільного заміщення (див. розд. ЗО). З молекулярної діаграми аніліну також випливає, що л-електронна густина в його бензольному ядрі на орто- і пора-атомах вуглецю більша, ніж на мета-атомах. Індекси вільної валентності дещо вищі на ор/по-атомах вуглецю:
Порядок ЗВ'ЯЗКІВ
Розподіл електронної густини в молекулі аніліну
Хімічні властивості. Хімічні властивості ароматичних амінів визначаються аміногрупою і сполученим з нею ароматичним ядром.
Реакції з участю змінної групи. \. Кислотно-основні властивості. Ароматичні аміни, подібно до аміаку, виявляють основні властивості, оскільки в них на атомі азоту аміногрупи є вільна пара електронів. Проте, як уже зазначалося, у ароматичних амінів за рахунок р,л-спряження основні властивості виражені значно слабкіше, ніжуаміаку і амінів аліфатичного ряду. Так, константа основності (/\о) аніліну становить 3,8 • 10-10, тоді як Ко аміаку і метиламіну — відповідно 1,8 • 10~Б і 4,4 10 ®. З тієї ж причини водний розчин аніліну і його емульсія у воді не змінюють забарвлення таких індикаторів, як фенолфталеїн, лакмус. Наявність в ароматичному ядрі аніліну груп атомів, які відтягують електрони на себе, приводить до зменшення основності аміногрупи. Наприклад, значення Ко нітроанілінів значно менші, ніж аніліну (табл. 36).
14*
419
Введення в молекулу аніліну біля атома азоту аміногрупи алкільних груп підвищує основність таких амінів, тому що алкільні радикали виявляють позитивний індукційний ефект і збільшують електронну густину на атомі азоту. Так, М-метиланіліну С6Н5—МН-<-СН3 дорівнює 7,1 • 10-10, Ко N. М-диметиланіліну
,.^сн3
СбН5-К
сн3
—1,1 • 10~е, тобто більші, ніж /Со аніліну.
Аналогічно впливають на основність аніліну алкільні радикали, введені в ароматичне ядро (табл. 36).
Введення біля атома азоту аміногрупи другого ароматичного ядра ще більшою мірою зменшує її основність. /Со дифеніламіну становить 7,6 • 10-1в. Таке значне пониження основних властивостей аміногрупи в молекулі дифеніламіну пов’язане з тим, що в спряженні вільної пари електронів атома азоту бере участь два бензольних ядра і тому електронна густина на атомі азоту в молекулі дифеніламіну значно менша, ніж, наприклад, аніліну. Три-феніламін з тієї ж причини основних властивостей не має:
Дифеніламін
Трифеніламін
За основністю ароматичні аміни можна розмістити в такий ряді
ГШ8 > СвНБ—N (Л/й)2 > С6Н8—N114Ік > С„Н8—ИН8 > (СвН6)я МН > > (СвН8)8М.
Отже, чим більше водневих атомів у молекулі аміаку заміщено на бензольні ядра, тим нижча основність такого ароматичного аміну.
Основні властивості ароматичних амінів виявляються в реакціях з сильними кислотами, з якими вони утворюють солі. Так, анілін з соляною кислотою утворює легко розчинну у воді сіль—солянокислий анілін:
.1 І
СвН8—МН2 + Н+СГ->[СсНБ—МН8]+СГ, або С8НБ—НН2 . НС1.
Аналогічно анілін реагує з сірчаною кислотою і утворює важкорозчинний сірчанокислий анілін СвН5—МН2 • Н2ЗО4.Солі ароматичних амінів легко гідролізуються водою. Тому їх водні розчини мають кислу реакцію:
[С6Н8-КН8ГСГ + Н2О <=> [С6Н8-МН2]ЮН- + Н+ + сг.
420
З слабкими кислотами, наприклад оцтовою, вугільною та іншими, ароматичні аміни солей не утворюють. Це також свідчить про послаблення основних властивостей даних амінів порівняно з аміаком.
2. Реакції алкілування. Ароматичні аміни, подібно до аліфатичних, вступають у реакцію з галогеналкілами і алкілуються. За допомогою цієї реакції з ароматичних амінів і галогеналкілів можна добути М-алкілароматичні аміни. Так, при взаємодії аніліну з йодистим метилом можна добути П-метиланілін, а з нього — М,І\[-димети лані лін:
І |о+ б- +
С6Н5—МН2 + СН3—І->С6Н5—МН2Г,абоСвН5—NN НІ;
сн3 сн3
Йодид Ь]«метилфенІламонію,
модистокисл ц.1 N - метнла и і л ін
СвН5—NN - НІ СсН5—НН-СН3;
СН3
р їб+ іг~ 4-
С6НЙ-№1-СН3 + СН3-І-*С6Н5-МН-СН3Г, або С„Н5—N (СН8)2 - НІ;
СН3
Йодид N. К-диметилфепіламоиііо, модистокислиіі N. К-днмсіпланілін
СвН6—N (СНЯ)2 . НІ С0І15—N (СН3)2.
При подальшій взаємодії М,П-диметиланіліну з йодистим метилом утворюється четвертинна сіль — йодид триметилфеніла-монію:
б* є- +
С6Н5—N (СН3)2 + СН3-І-> [СвН5-Н (СН3)3]Г.
3. Реакції арилування. При нагріванні аніліну з його сіллю відбувається заміщення водню аміногрупи на феиільний (ароматичний) радикал. В результаті утворюється вторинний ароматичний амін дифеніламін:
СвН5-МН2+СвН6-НН2 • НС1-> (СвН6)8ИН + ИН4С1.
Трпфеніламін добувають за реакцією Ульмана (1903 р.)— нагріванням дифеніламіну з бромбензолом при наявності поташу і мідного каталізатора:
(СДф^Н + С6Н3-ВгКг2.нв-г?--~> (СсН.,)8Н.
4. Реакції ацилування. Під дією ацилуючих реагентів на ароматичні аміни атом водню аміногрупи заміщується на ацильну групу. Такого типу реакції називають реакціями ацилування. Як ацилуючі реагенти використовують кислоти (мурашину, оцтову, бензойну), ангідриди і хлорангідриди кислот. Ацилуючі групи часто вводять у молекулу аміну тимчасово для захисту аміногрупи
14 1.120
421
від окислення або для розділення первинних, вторинних і третинних амінів. Так, анілін при взаємодії з оцтовим ангідридом утворює ацетанілід, який можна розглядати як амід оцтової кислоти, в молекулі якого атом водню амідної групи заміщений на ароматичний радикал:
С6НБ—МН2 + (СО3—С)2О -> С6НБ—№—С-СН8 + СН8—СООН.
II II
о о
Ацетанілід
Аніліди основних властивостей не мають.
5. Реакція з альдегідами. Анілін та інші первинні ароматичні аміни при слабкому нагріванні легко вступають у реакцію з альдегідами і утворюють кристалічні сполуки, які містять азоме-тинову групу атомів •—СН=И—. У зв’язку з цим такі речовини назвали азометанами, або основами Шіффа (1864 р.):
СБНБ—МН2 + С6НБ-СН = О -> С6Н5—N = СН—СеНв + Н2О.
Дифені лазометин
Азометани легко гідролізуються з утворенням ароматичних амінів. Тому їх добувають для тимчасового захисту аміногрупи від окислення. Деякі азометини виявляють властивості рідких кристалів.
6. Взаємодія ароматичних амінів з азотистою кислотою. Ця реакція має особливо велике значення. Первинні ароматичні аміни при взаємодії з азотистою кислотою утворюють високореакційнездатні солі діазонію, механізм утворення і хімічні властивості яких розглядаються в наступному розділі (див. розд. 37):
С6НБ—Ш2 + НМО2 + неї -> [С6НБ—N = М]*СГ + 2Н2О.
Хлористий фенілдіазоній
Вторинні ароматичні і жирно-ароматичні аміни з азотистою кислотою утворюють М-нітрозаміни:
С6НБ—М—СН3 С8НБ—М—СН3 + Н2О.
І_____ І
|НН-НО|НО N=0
ГЧ-Нітрозо-М-метилані лін
При взаємодії з азотистою кислотою третинних ароматичних амінів відбувається електрофільне заміщення атома водню в бензольному ядрі на нітрозогрупу і утворення ароматичних нітрозо-похідних. Нітрозогрупа заміщує водень переважно идра-положен-ня ароматичного ядра третинного аміну, а якщо иард-положення зайняте — то водень орто-положення. Наприклад:
(Сн3)2іч—N°(СН3)2ІЧ—^^-N = 0 .
л-Нітрозо-М,Н-диметиланілін
Реакції з участю бензольного ядра. Оскільки МН2-група в молекулах ароматичних амінів виявляє сильний 422
4-Л1-ефект і таким чином збільшує електронну густину бензольного ядра, вона підвищує його активність до реакцій електрофільного заміщення. В результаті електрофільне заміщення у ароматич-нихамініввідбуваєтьсянабагатолегше, ніжу бензолу, іпроходить в орто- і паро-положеннях відносно аміногрупи. В більшості таких реакцій утворюються продукти заміщення в поро-положенні. Найважливішими 5Е-реакціями для аніліну, його похідних та гомологів є галогенування, нітрування і сульфування.
1. Галогенування. Анілін дуже легко вступає в реакцію з бромною водою і з кількісним виходом утворює важко розчинний у воді 2,4,6-триброманілін (Т..л — 119 °С). Реакція може бути якісною пробою на анілін:
Для того щоб добути моноброманілін, амінну групу попередньо ацетилують, а потім бромують ацетанілід. Ацетамідна група внаслідок свого електронного і стеричного ефекту орієнтує наступне електрофільне заміщення переважно в ппра-положення. Після гідролізу ацетамідної групи в /шрс-бромацетаніліді добувають па-рп-броманілін:
(СНзСОкО "Н3С-СООН
МН-СО-СН, N9 —СО—СН3
Ацетанілід
Вг
НОН
-Н3С-СООН
Хлорування ароматичних амінів супроводиться значним їх окисленням, оскільки хлор виявляє сильні окислювальні властивості. У зв’язку з цим аміногрупу ароматичного аміну спочатку захищають і хлорують не самі аміни, а їх ацильні похідні, які значно стійкіші до окислення. Добуті хлорацетаніліди потім гідролізом перетворюють на орто- і псрд-хлораніліни:
нон
-н3с - соон
14*
423
2. Нітрування. Анілін та інші ароматичні аміни при дії азотної кислоти легко окислюються і осмоляються. Тому пряме нітрування азотною кислотою здійснити неможливо. Якщо нітрування аніліну проводити в концентрованій сірчаній кислоті (нітруюча суміш), то крім окислення аніліну відбувається перетворення його на іон анілінію з позитивно зарядженим атомом азоту. МН3-Група є замісником другого роду (лгелш-орієнтантом) і орієнтує вхід нітроніш-катіона в лїета-положення. В результаті з невеликим виходом утворюється лешо-нітроанілін:
У промисловості леша-нітроанілін добувають відновленням мета-динітробензолу сульфідом натрію,під дією якого відновлюється тільки одна нітрогрупа:
Для добування иара-нітроаніліну в молекулі аніліну спочатку захищають аміногрупу ацетилуванням і нітрують ацетанілід. При цьому добувають лшра-нітроацетанілід, який гідролізом перетворюють на пара-нітроанілін:
8. Сульфування. Сульфування аніліну розбавленою сірчаною кислотою є 5е-реакцією і приводить до утворення суміші орто-і лара-анілінсульфокислот:
ч2-і
Анілінсульфокнслоти застосовують для синтезу барвників, а також для добування різних похідних бензолу (див. розд. 32) та лікарських препаратів. Особливо велике значення має пара-анілінсульфокислота (4-амінобензолсульфокислота), яку переважно називають сульфаніловою кислотою. Сульфанілову кислоту добувають методом «запікання» — короткотривалим нагріванням аніліну з концентрованою Н28О4. При цьому спочатку утворюється сірчанокислий анілін, який потім при нагріванні дегідрату-ється і перетворюється на сульфамінову кислоту. Сульфамінова кислота далі ізомеризується (перегруповується) в сульфанілову кислоту (І). Кислота (І) містить МН2-групу, яка надає молекулі основних властивостей, і 8О3Н-групу, яка надає цій же молекулі кислотних властивостей. При взаємодії цих груп утворюється внутрішня сіль (II):
Сульфанілова кислота (Тпл = 228 СС) виявляє сильні кислотні властивості. З основами вона легко утворює солі. Так, з ИаОН вона утворює добре розчинну у воді натрієву сіль:
л-НаМ—С6Н4—8ОаН + іХ'аОН -> л-Н2М—СвН4-8О8№ + Н2О.
Ця кислота має надзвичайно велике значення як складова частина хіміко-терапевтичних лікарських засобів, які називають сульфамідами. Сульфаміди є амідами сульфанілової кислоти. Г. Домагк у 1935 р. встановив, що сульфаміди виявляють сильну антибактеріальну (протистрептококову і протипневмококову) дію і разом з тим мають низьку токсичність. Найпростішим сульфамідним препаратом є амід сульфанілової кислоти, який назвали білим стрептоцидом. У промисловості білий стрептоцид добувають з ацетаніліду:
Білий стрептоцид
Пізніше були синтезовані більш ефективні сульфамідні препарати — альбуцид, норсульфазол, етазол тощо.
425
Контрольні запитання і вправи
1. Розмістіть такі сполуки в порядку підвищення їх основності:а) аміак, анілін,циклогексиламін; б) анілін, п-метоксіанілін, га-нітроанілін;в) бензила-мін, .и-хлорбензиламін, .м-етилбензиламін; г) п-хлор-М-метилачілін, л-фтор-І\І-метиланілін, п-метил-К’-метиланілін, /і-нітро-М-метиланілін.
2. Як за допомогою простих хімічних реакцій можна розрізнити: а) И-метиланілін і о-толуїдин; б) анілін і циклогексиламін; в) анілін і ацетанілід; г) 2,4,6-тринітроанілін і анілін?
3. Запропонуйте прості хімічні методи для розділення таких сумішей: а) анілін і н-гептан; б) анілін і анізол; в) о-нітроанілін і сульфанілова кислота; г) ї\т-метиланілін і N. И-диметиланілін; д) о-нітротолуол і о-толуїдин. Кожну з цих речовин необхідно виділити в практично чистому вигляді.
4. Поясніть, чи однакову активність виявляють анілін і бензол у реакціях бромування, нітрування і сульфування.
5. Які речовини утворюються під час взаємодії п-метиланіліну («-толуїдину) з бромистоводневою кислотою, хлористим метил ом, хлористим ацетилом, азотистою кислотою при наявності соляної, з сірчаною кислотою, бромною водою?
Розділ 37. АРОМАТИЧНІ ДІАЗО- І АЗОСПОЛУКИ
Ароматичними діазосполуками називають органічні речовини, молекули яких містять діазогрупу —М.2—, яка складається з двох атомів азоту, сполучених між собою подвійним або потрійним зв’язком. У молекулах ароматичних діазосполук один з атомів азоту діазогрупи сполучений з атомом вуглецю ароматичного ядра. Склад ароматичних діазосполук можна виразити загальною формулою Аг——X, де Аг — ароматичний радикал, X— СІ, Вг, 8О4Н, і\Ю3, ВР4, ОН, СН3СОО та інші залишки. Найважливішими з ароматичних діазосполук є солі діазонію.
Солі діазонію. Солі діазонію мають загальну формулу іАг—Нн=Й]+Х_, де Х~—-будь-який аніон — СІ-, Вг-, N0', Н8О7, ВР7 тощо. Вперше солі діазонію добув у 1857 р. І. П. Гріс, який і ввів у хімію термін «діазо». У дослідженні будови, механізму утворення, реакційної здатності ароматичних діазосполук брали участь хіміки багатьох країн. Проте найбільший внесок у розвиток хімії діазосполук внесли німецький хімік А. Ганч і радянський хімік Б. О. Порай-Кошиць.
Назву солей діазонію утворюють так, як і назви солей мінеральних кислот. Спочатку називають аніон, наприклад хлористий, сірчано-кислий тощо, а потім — ароматичний радикал діазосолі (феніл-, ннра-толіл-, орто-нітрофеніл- і т. д.). Закінчується назва словом «діазоній», яке і підкреслює, що ці речовини є солями. Наприклад:
Хлористий феніл-діазоиїй, фсніл-діазонійхлорид
Борфторид л-нітро-фенілдіазонію, /г-нітро-фенілдіазонійборфторид
Бромистий /г-толілдіазоній, п-толілдіазонійбромід
Ароматичні солі діазонію добувають взаємодією первинних аро» матичних амінів з азотистою кислотою в розчині мінеральної кис-
426
лоти. Реакцію утворення діазосполук з амінів називають діазоту-ванням. Оскільки азотиста кислота — сполука нестійка, то її генерують при наявності аміну взаємодією нітриту натрію з мінеральною кислотою, переважно соляноюабосірчаною. У загальному вигляді реакція діазотування описується рівнянням:
Аг—МН2 + І\’аІ\’О2 + 2НХ °~ 5---> [Аг—К’2|+Х' + + 2Н2О.
Діазотування, наприклад, аніліну в соляній кислоті відбувається за такою схемою:
СеН8—МН2 + 2НС1 + №ЬЮа [С„Н5-Н2]+СГ 4- №С1 ф-
Хлористий фенілдіазоиій
+ 2Н2О + 126 кДж.
Оскільки діазотування — екзотермічна реакція, а азотиста кислота і сіль діазонію, які утворюються в процесі реакції, при підвищених температурах розкладаються, то діазотування ведуть при охолодженні. Суміш аміну і кислоти охолоджують до температури —5...—10 °С, а потім до неї добавляють розчин нітриту натрію з такою швидкістю, щоб температура реакційної суміші не піднімалась понад 4-5 °С.
Солі діазонію — кристалічні речовини, більшість з-яких добре розчинні у воді. В сухому стані солі діазонію вибухають. У зв’ язку з цим їх рідко виділяють у кристалічному стані, а використовують переважно у вигляді розчинів. Солі діазонію повільно розкладаються навіть при температурах льодяної бані. Тому їх розчини використовують відразу ж після добування.
Механізм реакції діазотування. У процесі реакції діазотування з діазотуючих реагентів утворюється надзвичайно активна електро-фільна частинка — нітрозоній-катіон N==0 з позитивним зарядом на атомі азоту. Нітрозоній-катіон виникає з азотистої кислоти в сильнокислому середовищі. Так, у соляній і сірчаній кислотах азотиста кислота утворює відповідно хлористий нітрозил і нітро-зонійсульфат, які є джерелом нітрозоній-катіонів:
НО—N0 4- НС1 <=> Н2О 4- СІ—N=0 *=? СГ 4~ И=О;
НО—N0 4- НО8О3Н <=& Н20 4- О=ІУ-8О4І І 0=Н Н80~.
У реакцію діазотування вступає тільки вільна основа первинного ароматичного аміну, яка завжди наявна в реакційній суміші і в кислому середовищі перебуває в рівновазі зі своєю сіллю:
С.Н,—НН2. НС1 С„Н,—І\’Н2 4- неї.
Механізм утворення солі арилдіазоніюна прикладі діазотування аніліну можна описати такими послідовними елементарними реакціями. Першою стадією при діазотуванні в розбавлених кислотах є взаємодія нітрозуючого реагенту X—N=0 (X—СІ, 8О4Н, О—N=0, ОН) з ароматичним аміном і утворення іона нітрозамо-
427
нію [реакція (1)], Ця стадія є повільною і визначає швидкість реакції діазотування. Іон нітрозамонію, як сильна кислота, на наступній стадії легко депротонізується до нітрозаміну (реакція (2)]. Можливо, ці обидві стадії відбуваються одночасно. На третій стадії здійснюється таутомерне перегрупування нітрозаміну в поки що не виділений у вільному стані діазогідрат [реакція (3)1. Діазогід-рат у кислому середовищі миттєво (швидка стадія реакції діазотування) утворює катіон діазонію [реакція (4)]:
СвН5—ЙН2 + Х-И=О С6НБ—МН2—N0 + Х~і (1)
Іон фенілнітроз-амонію
С6Н5—Йн2—N0 Н+ + СсН5—ГШ—N == О; (2)
Фені лн ітрозам ін
С6НБ—ІУН—N=0 СвН5—N=14—ОН;
Фенілдіазогідрат
СвН8— І\=К—ОН + неї «=>
С6Н5—ГЧТ СГ+ Н20.
N
(3)
(4)
Хлористий фені лді а зоні й
Електронна будова катіона солей арилдіазонію. Діазосолі складаються з діазокатіона і аніона. На основі даних рентгено-структурного аналізу і інших фізико-хімічних методів встановлено,
Рис. 72. Структура молекули хлористого фенілдіазонію: а — бензол: б — хлористий фенілдіазоній
що катіони арилдіазонію, наприклад фенілдіазонію, побудовані лінійно (рис. 72). Обидва атоми азоту діазогрупи копланарні і розміщені на одній лінії, яка є продовженням осі бензольного ядра. В катіоні фенілдіазонію під впливом діазогрупи порушується симетрія бензольного ядра, яке неначе «стискається», про що свідчить зменшення віддалі між першим і четвертим вуглецевими атомами (рис. 72, б) порівняно з аналогічною віддаллю в молекулі бензолу (рис. 72, а).
Розрахунки показали, що в діазогрупі крайній атом азоту має дещо більший частковий позитивний заряд, ніж атом азоту, сполучений з атомом вуглецю бензольного ядра. Довжина зв’язку між атомами азоту і вуглецю бензольного ядра приблизно дорівнює 428
довжині одинарного зв’язку. Аніон солі діазоніюС1~з електростатичних і стеричних причин розміщується від діазокатіона на віддалі 0,32 нм і по один бік площини діазокатіона, ближче до крайнього атома азоту діазогрупи. Позитивний заряд у діазокатіоні розподілений між обома атомами азоту діазогрупи, але частково він компенсується за рахунок л-електронів бензольного ядра. Це можна зобразити у вигляді граничних (резонансних) структур (І—V) і мезомерних формул (VI і VII), які відображають стійкість катіонів арилдіазонію:
Величина заряду на атомах азоту, стійкість катіона арилдіазо-нію змінюються при введенні в бензольне ядро замісника і залежать також від природи цього замісника та його положення в бензольному ядрі.
Форми діазосполук. Ароматичні діазосполуки відомі у вигляді кількох форм, які у розчинах залежно від рН середовища можуть перетворюватися одна в одну:
Кисле середовище Нейтральне середовище Лужне середовищ-?
+ МаОН +_ №ОІІ №ОН
д г— мсг Аг—НОН Лг—М=М—ОН «==* Аг—Н=Н—ОНа+.
ці неї ці неї неї
N N
І П ПІ IV
Арилдіазоніпхло- Аршідіазоній- Арилдіазоній* Арилдіазотат
рид (сіль діазонію) гідроксид гідрат натрію
У кислому середовищі діазосполуки перебувають у формі солей діазонію (І), стійких на холоді 0°С). Солі діазонію є кристалічними речовинами, більшість з яких добре розчинні уводі. У водних розчинах солі арилдіазонію повністю дисоціюють на іони:
Аг—Н2—СІ ч=і Аг—4- СГ.
Катіон арилдіазонію Аг—М* є сильною апротонною кислотою, яка з аніонами мінеральних кислот утворює нейтральні (на лакмус) солі.
429
При обережній дії на солі арилдіазонію (І) зволоженого оксиду срібла або лугу утворюються арилдіазонійгідроксиди (II), які є сильними основами і в розбавлених водних розчинах повністю дисоціюють на іони:
Аг—Ма—ОН Аг—Ьі+ +ОН-.
Це свідчить про наявність у арилдіазонійгідроксиду іонного зв’язку між гідроксильною групою і катіоном арилдіазонію. При підкисленні арилдіазонійгідроксид (II) перетворюється на сіль арплдіа-зонію (І). Арилдіазонійгідроксид (II) у нейтральному середовищі повільно ізомеризується в арилдіазоній гідрат(ІП), який відрізняється від солі діазонію (І) і діазонійгідроксиду (II) тим, що в нього атоми азоту перебувають у $р2-гібридизованому стані і гідроксильна група сполучена з атомом азоту діазогрупи ковалентним зв’язком. Утворення цього зв’язку пояснюють тим, що при взаємодії гідроксильного іона з діазокатіоном позитивний заряд останнього повністю зосереджується на крайньому атомі азоту діазогрупи і таким чином між ним і ОН-групою виникає ковалентний зв’язок:
[Лг -к = м]+ + ОН------- Аг — № ІЧ-О-Н;.
Діазогідрати (III) є проміжними сполуками і мають велику лабільність. У водних розчинах діазогідрати виявляють амфотерні властивості: під дією кислот діазогідрати перетворюються на діазо-солі (І), а під дією лугів— швидко утворюють діазотати (IV).
Хімічні властивості. Ароматичні діазосполуки легко розкладаються і тому є надзвичайно реакційноздатними. Для них характерні реакції, що відбуваються з виділенням азоту діазогрупи або без виділення азоту діазогрупи. Обидва типи реакцій знайшли широке застосування в промисловому і препаративному органічному синтезі.
Реакції солей діазонію з виділенням а з о т у. У процесі реакцій ароматичних солей діазонію, які супроводяться виділенням азоту, відбувається заміщення діазогрупи на інші атоми або групи атомів,- зокрема на Р, Сі, Вг, І, СМ, ОН, Н, Аг, МО2, ОР тощо. За допомогою цих реакцій можна вводити в ароматичне ядро найрізноманітніші замісники.
Розрив зв’ язку між атомом вуглецю ароматичного ядра і атомом азоту діазогрупи може відбуватись гетеролітично або гемолітично. Відповідно заміщення діазогрупи може здійснюватися за нуилєо-фільним або за радикальним механізмом.
1. Гетеролітичні реакції солей діазонію. Гетеролітичні реакції ароматичних діазосолей відбуваються за двостадійним мономолекулярним нуклеофільним механізмом (5д? І-реакції):
Повільно
[Аг:К21+Х ------> Ат* + X +КІ2;
Дрил-катіон
430
Повільною стадією, яка визначає швидкість усієї реакції, є розкладання солі арилдіазонію на арил-катіон, азот і аніон Х~. На другій, швидкій стадії, арил-катіон Аг + взаємодіє з нуклео-філом Х~ і утворює продукт реакції Аг—X. Найважливішими типовими 8м 1-реакціями розкладання ароматичних діазосолей з виділенням азоту є такі:
с6нв—ьі8—х НОН, 50-80-С, Н25О4
——нх ОН, (1) щс-он ф
С6ІІ5 —О СІ 13, (2) С»НП-ОН ,гтг/° /чч
^6^6 (СН2)3Сх (3) НВР< ->СН Р <Л\
С6Нб 1 . (4)
Розкладання водних розчинів солей арилдіазонію в кислому середовищі [реакція (1)], яке проводять при слабкому нагріванні, використовують для препаративного добування фенолів, крезолів, нітрофенолів тощо. Утворення, наприклад, фенолу з сульфату фе-нілдіазонію описується такими рівняннями:
с6н5(Тмр нзо;----- + ,\2 + н§о; •,
СбН* + но?)н-----►С6Н5-ОН + н + :
Н+ 4- Н8О7---*-Н28О4 .
Взаємодія солей діазонію із спиртами [реакції (2) і (3)] відбу-вається удвох напрямках і може приводити до утворення простих ефірів фенолів [реакція (2)] та ароматичних вуглеводнів [реакція (3)]. Наприклад, взаємодія хлористого фенілдіазонію з метиловим спиртом дає з виходом понад 90 % метилфені.товий ефір (анізол). Реакція солей діазонію з більш високомолекулярними спиртами, наприклад з аміловим [реакція (3)], є окислювально-відновною. В ній окислювач — діазоній-катіон, а відновник — спирт, від якого а-водневий атом мігрує у вигляді гідрид-аніона до арильного катіона. Одночасно від гідроксильної групи спирту відщеплюється протон. В результаті утворюються ароматичний вуглеводень і альдегід. Наприклад, при взаємодії хлористого фенілдіазонію з аміловим спиртом утворюються бензол і валеріановий альдегід:
С6Н5(Гу2 СІ + Н3с (СН2)з-СН-О: (н— С6Н6 + Н3С-(СН2)3-с'
(н) чн *
З етиловим спиртом солі діазонію реагують з утворенням як ароматичного вуглеводню, так і простого ефіру фенолу:
,0
Лґ-Н + 4- неї + Н3С-С^ ;
Аг— И2—СІ + Н3С-СН2-ОН— Н
и Дг-О-СН2-СН3+ 4-неї.
431
Термічний розклад борфторидів арилдіазонію [реакція (4)] називають реакцією Шімана (1927 р.). Борфториди арилдіазонію, які добувають взаємодією хлориду арилдіазонію з борфтористовод-невою кислотою, при нагріванні перетворюються на фторпохідні ароматичні вуглеводні. При розкладанні, наприклад, борфториду фенілдіазонію утворюється фторбензол:
СвН5-М2—СІ + І ІВІ7„ тднсГ- счн5—ьі2—вг, С„НБ—Р + N3 + ВР8.
2. Гемолітичні реакції солей арилдіазонію. Механізм радикального розкладання солей арилдіазонію поки що недостатньо вивчений. Гемолітичні реакції діазосолей відбуваються переважно з участю каталізаторів, таких, як солі міді або металічна мідь. Мідь, якперехідний метал, виявляє в сполуках змінну валентність. Каталітична дія галогенідів одновалентної міді полягає, напевне, в тому, що галогеніди арилдіазонію утворюють з галогенідами міді подвійні солі, в яких мідь переносить електрон на зв’язок N—С діазогрупи. Як проміжні частинки при цьому виникають іон-ра-дикали, які гемолітично розпадаються на вільний азот, арильний радикал і галогенід двовалентної міді. Галогенід двовалентної міді потім передає атом галогену артільному радикалу. В результаті утворюється арилгалогенід і регенерується галогенід одновалентної міді. Так, утворення хлорбензолу з хлориду фенілдіазонію при каталітичній дії хлориду міді (І) можна описати такими реакціями:
С6Н5^СГ+ СиС1^[С6Н5Кй]+СиС1^[СвН5: ЙгмҐСиСІ? ——
Іон-радикал
-------- С6Н5 + СТ).СиСГ--► С6Н5-СІ + СиСІ.
Найважливішими 5^-реакціями розкладання ароматичних солей діазонію з виділенням азоту є такі:
С6НЛвХ’
неї, СиСІ (Єн)
НВг, СиВг (Си)
К1
СиСК’
С6н5-С1;
С6Н5—Вг;
СеН5-І;
С„.Н6-СМ.
(6)
(7)
(8)
(9)
Розкладання солей діазонію при нагріванні з галогенідами одновалентної міді приводить до утворення арилгалогенідів. Добування арилхлоридів і арилбромідів за реакціями (6) і (7) було відкрите Т. Зандмейєром (1884 р.) і називається реакціями Занд-мейєра.
Арилхлориди і арилброміди утворюються також при нагріванні відповідно хлоридів і бромідів арилдіазонію при наявності каталі
432
затора — мідного порошку (реакція Гаттермана, 1890 р.).У цьому випадку мідь передає на розрив зв’язку С—N два електрони.
Арилйодиди добувають добавлянням йодиду калію або натрію до розчину солі діазонію [реакція (8)]. У цьому випадку спочатку утворюється нестійкий йодид арилдіазонію, в якому аніон І ~ передає один електрон атому азоту діазоній-катіона. В результаті утворюється іон-радикал, який потім розпадається на молекулярний азот і арил-радикал. Арил-радикал і атомарний йод далі сполучаються і утворюють арилйодид. Утворення, наприклад, йодбензолу з діазотованого аніліну описується такими послідовними реакціями:
•’НСі №МО2,НСІ + - К+1
С6Н5НН2---►С6Н5МН2-НС1---------* С6Н5№С1 _КС1 -
-----[с6н5-ьіСьі?^г [с9н;(ічзм]+і-^*с6нв + і—-свн5-і
Ця реакція є одним з найзручніших методів введення йоду в ароматичні сполуки.
Виходячи з діазосолей в ароматичне ядро можна ввести і ніт-рильиу групу [реакція (9)]. Наприклад, нітрил бензойної кислоти утворюється при взаємодії хлористого фенілдіазонію з ціанідом міді:
2С,.НВ—И2—СІ + Си2 (СИ)2 -> 2СсН5—СМ + 2М2 + СиС12.
Діазогрупа всоляхдіазоиіюможезаміщуватись і на атоми металів. О. 'А. Несмеянову 1929 р.. встановив, іцо при дії металів на подвійні солі діазонію, які добуваюсь взаємодією цих солей з галогенідами перехідних металів, виділяється азот і утворюються металоорганічні сполуки. Наприклад:
С6Н6М+С1- • НЄС12 + 2Си -г СсН6НЄС1 + М2 + 2СиС1;
Подвійна сіль
(СвН5м+СГ)2 • 8пС14 + 2п - (С6ІІ6)28пС12 + N. + ХпС12.
Подвійна сіль
Дотування металоорганічних сполук цим методом було назване реакцією Несмеянова.
Реакції солей діазонію без виділення азоту діазогрупи. Без виділення азоту діазогрупи ароматичні солі діазонію вступають у реакції відновлення і азосполу-чення.
Відновлення солей діазонію хлоридом олова (II) в соляній кислоті приводить до утворення арилгідразинів:
[Дг-М^М+С1- + 28пС12 + 4НСІ -> С„Нв—Г4Н-ИН2 . НС1 + 28пС14.
У промислових масштабах арилгідразини добувають відновленням солей діазонію гідросульфітом натрію:
[СвН5-К’^МГСі- + 2МаН8О3 + 2Н2О СвН4—МН— ІМН2 + 2МаН8О4-
433
При повному відновленні солі діазонію перетворюються на ароматичні аміни і аміак:
С.Н6-К+ СІ- + ЗН2-^ С6Н6-ЛНа + NN. +НС1.
Реа кція азо сполучення. Азосполуки. Солі діазонію за певних умов реагують з деякими ароматичними сполуками, утворюючи речовини з загальною формулою Аг—И=П—Аг', які називають азосполуками. Групу атомів —П=Г<— називають азогрупою, а реакцію утворення азосполук — азосполученням. При азосполученні атоми азоту діазогрупи зберігаються в продукті реакції. Вони сполучені з атомами вуглецю ароматичних ядер:
Аг—+Аг— Н -> Аг— К=К-Аг’ + Н+.
Отже, азосполучення — це реакція заміщення атома водню ароматичного ядра під дією іона діазонію на групу Аг——.У реакцію азосполучення вступають тільки такі ароматичні сполуки, молекули яких містять сильну електронодонорну групу, переважно ОН, МТ?2, №Н/?, НН2. Атоми водню бензольного ядра в таких сполуках виявляють високу активність до реакцій електрофільного заміщення (див. розд. ЗО).
У реакції азосполучення беруть участь два реагенти — сіль діазонію (діазоскладова реакції) і ароматична сполука з електро-нодонорнпм замісником 7? (азоскладова реакції). Ці реагенти вводять у реакцію в еквімолярних кількостях:
Діазоскладова Азоскладова
Аі— N = N
/? + нх; Х-СІ, 8О4Н.
Азосполука
Азосполуки Аг—N=14—Аг' називають як похідні азобензолу. Положення замісників у ароматичних ядрах позначають цифрами. Причому для позначення положень у різних ядрах використовують цифри з штрихами. Наприклад:
Азобензол
4-Нітроазобензол
4-Н|тро-4'-окс І азобензол
434
Інколи азосполуки називають також як похідні ароматичних вуглеводнів, у яких атом водню ароматичного ядра заміщений на арилазогрупу Аг—КТ=М—. Наприклад:
л-(и'-Нітрофенілазо) -М,М-диметиланіл1н
Механізм реакції азосполучення. Азосполучення є типовою реакцією електрофільного заміщення в ароматичному ядрі (8Е 2-реак-ція). Діазосіль вступає в цю реакцію тільки у вигляді діазокатіо-на, який є електрофільним реагентом. Інші форми діазосполук у реакції азосполучення участі не беруть. Електрофільні властивості діазокатіона досить слабкі, оскільки-у ЛІ-ефект л-електронів ароматичного ядра значною мірою зменшує позитивний заряд діазогрупи, для якої можливі діазо-і азо-мезомерні структури (І і II):
і п
На електрофільну активність діазокатіона істотний вплив має замісник /? в ароматичному ядрі, особливо якщо він розміщений в пара- або оршо-положеннях відносно діазогрупи. Електронодо-норні замісники (групи СН3, СН3О та інші) збільшують електронну густину бензольного ядра і внаслідок А-М-ефекту зменшують позитивний заряд діазогрупи, зменшуючи тим самим електрофільну активність діазосполук. Електроноакцепторні замісники (НО2, 8О3Н, СООН та інші), навпаки, зменшують електронну густину бензольного ядра і тим самим збільшують позитивний заряд діазогрупи, що приводить до підвищення електрофільної активності таких діазосполук. Тому як діазоскладові в реакції азосполучення дуже часто використовують діазосолі, добуті діазотуванням первинних ароматичних амінів, таких як нітроаніліни, сульфанілова кислота, антранілова кислота тощо, в бензольному ядрі яких містяться електроноакцепторні замісники. Азоскладовими в реакції азосполучення можуть бути тільки аміно- і оксипохідні ароматичного ряду, ароматичне ядро яких має підвищену електронну густину. Бензол, гомологи бензолу, галогене-, сульфо-, нітропохідні ароматичного ряду, ароматичні карбонові кислоти в реакцію азосполучення не вступають.
За своїм механізмом реакція азосполучення, як і будь-яка інша реакція електрофільного заміщення в ароматичному ядрі, прохо
435
дить через стадії утворення л- і о-комплексів і закінчується витісненням азоскладовою атома водню з ароматичного ядра:
а-Комплекс
При цьому азоскладова завжди атакується крайнім атомом азоту діазогрупи (форми II), а не амонійним азотом форми І, оскільки в останньому випадку повинна була б утворюватись енергетично нестійка проміжна структура такої будови:
Лг-И = М
Реакція азосполучення відбувається з великою швидкістю. У зв’язку з цим та враховуючи термічну нестійкість діазосолей, цю реакцію проводять при охолодженні. Азосполучення відбувається переважно в псра-положення, а якщо це положення зайняте, — то в орто-положення азоскладової. Пояснюється це тим, що замісник У? азоскладової екранує орто-положення бензольного ядра і створює стеричні утруднення для перебігу реакції.
Умови проведення реакції азосполучення. Реакцію азосполучення проводять при певній кислотності середовища — в інтервалі рН від 5 до 9. Оптимальне значення рН залежить від властивостей азоскладової. Азосполучення фенолів найкраще здійснюється при 0 °С у слабколужному середовищі, оскільки електронна густина в ароматичному ядрі феиолят-аніона набагато вища, ніж у неіонізованого фенолу, що полегшує атаку його молекули електрофільним діазо-ній-кагіоном. У сильнолужному середовищі, при рН > 10, азосполучення не відбувається, оскільки катіон арилдіазонію перетворюється на діазотат-аніон, який не має здатності вступати в реакцію азосполучення. Практично реакцію азосполучення здійснюють так, що кислий розчин діазоскладової добавляють до лужного розчину азоскладової. Наприклад, при взаємодії кислого розчину хлористого фенілдіазонію з лужним розчином феноляту утворюється азосполука ппрс-оксіазобепзол:
436
У випадку ароматичних амінів в азосполучення з катіоном арилдіазонію вступають тільки вільні аміни (основи), а не їх солі. У більшості випадків азосполучення з ароматичними амінами проводять при 15—20 °С в нейтральному або слабкокислому (оцтовокислому) середовищі. У такому середовищі солі ароматичних амінів гідролізуються і перебувають у рівновазі з вільним аміном. У більш кислому середовищі, при рН < 5, утворюється сіль аміну з пози-4* тивним зарядом на змінному атомі азоту. А амонійна група є акцептором л-електронів ароматичного ядра і інактивує його до реакцій азосполучення.
У процесі азосполучення вільні р-електрони амінного азоту беруть участь у стабілізації ст-комплексу, делокалізують його позитивний заряд. У цьому випадку о-комплекс азоскладової набуває плоскої хіноїдної структури. Взаємодією хлористого фенілдіазонію з аніліном, яка приводить до утворення псро-аміноазобензолу — анілінового жовтого азобарвника (7\ІЛ = 126 °С), можна зобразити такою схемою:
п -Аміноазобензол
Азобарвники. Азосполуки мають забарвлення. Забарвлення речовини пов’язане з її здатністю поглинати у видимій частині спектра (в інтервалі довжин хвиль від 800 до 400 нм) тільки деякі промені з певними довжинами хвиль, тобто з певною енергією. При цьому промені спектра, які не поглинаються, є додатковими до поглинутих і сприймаються як видимі. Тому речовина стає забарвленою. Наприклад, якщо речовина поглинає всі промені, за
437
Рис. 73. Схема поглинання світла речовиною
винятком червоних (620 — 400 нм), то вони відбиваються і забарвлюють речовину в червоний колір (800 — 620 нм, рис. 73).
Поглинання світла речовиною визначається станом електронів у її молекулах. Оскільки енергія поглинутих променів іде на збудження зовнішніх електронів, стан яких може бути різним (а- або л-електрони), то змінюючи хімічну будову молекули барвника, можна в широкому інтервалі змінювати величину і характер поглинання світла. Для збудження електронів, які утворюють прості о-зв’язки (о-електрони), необхідна більша енергія, тобто потрібні промені світла з більшою енергією. Тому насичені сполуки поглинають тільки в далекій УФ-області, промені якої мають достатню для цього енергію. В той же
час сполуки, що мають подвійні зв’язки, будуть поглинати у видимій області або на її межі, оскільки л-електрони потребують для свого збудження меншої енергії. Отже, для появи забарвлення
речовини необхідна наявність в її молекулі досить довгої спряженої системи подвійних зв’язків або таких груп, як ——, л-елек-
трони яких легко збуджуються променями видимого світла і поглинають деякі з них.
Згідно з сучасною теорією колірності, для того щоб речовина була забавлена, необхідна наявність в її молекулі хромофорної групи (від грецьк. «хромос» колір і «форос» — носій). Хромофорними групами є довга система спряжених подвійних зв’язків, хіноїдна
система подвійних зв’язків —^=3* азогрупа —ХТ=ІЧ—- та інші.
Введення в молекулу забарвлених речовин таких полярних груп, як —ОН, —ЬІН2, —СООН, —8О3Н та інших, сприяє поглибленню забарвлення, підвищенню його інтенсивності, надає молекулам забарвлених речовин здатності зв’язуватися з волокнами тканини. Ці групи атомів називають ауксохромами (від грецьк. «ауксео» — збільшую, «хромос» — колір). Ауксохромні групи, маючи електро-нодонорні або електроноакцепторні властивості, викликають зміщення л-електронної густини по системі спряжених подвійних зв’язків під впливом світла.
Азосполуки, молекули яких можуть міцно сполучатися з волокном або іншими матеріалами і фарбувати їх, називають азобарвниками. Азобарвники є найчисленнішим класом барвників. У сучасній анілінобарвниковій промисловості їх нараховується близько 2000. Азобарвники застосовують для фарбування текстильних,
438
вовняних і синтетичних тканин, паперу, шкіри, гуми тощо. Деякі азобарвники використовують як індикатори дня визначення кислотності середовища.
Розглянуті вище пара-окси- і лара-аміноазобензоли, хоч і е азобарвниками, проте на практиці майже не використовуються, оскільки вони не дають міцного забарвлення. Типовим представником азобарвиків є лшра-диметиламіноазобензол (диметиловий жовтий), який добувають азосполученням сірчанокислого фенілдіазонію з диметиланіліном:
.\'^8О4Н + Н
N = N
^'(СЬІ3)2.
-н2$о4
Диметиловий жовтий
Азобарвники-індикатори. Деякі азобарвники при зміні кислотності середовища у певних межах рН різко змінюють своє забарвлення. Такі барвники використовують як кислотно-основні індикатори. В основі індикаторних властивостей азобарвників лежить азогідразонна таутомерія, при якій хромофорна азогрупа, знаходячись у ланцюгу спряжених подвійних зв’язків, переходить в інший хромофор — азиногрупу:
—К=М—; =ІУ—К=.
Азогрупа Азиногрупа
У процесі таутомерних перетворень виникають також хіноїдні угруповання. Азо- і хінонгідразонні форми азобарвників мають різне забарвлення, яке залежить від характеру замісника в бензольному ядрі і рН середовища. Типовим азобарвником-індикатором є ппра-диметпламіноазобензолсульфокислий натрій, відомий під назвою метиловий оранжевий (метилоранж), або геліантин, який добувають азосполученням діазотованої сульфанілової кислоти з диметиланіліном:
Метилоранж, жовто-оранжевого кольору: дма =472нм, нейтральне або лужне середовище (рН^7)
Метилоранж, червоного кольору; Амакс =506нм» кисле середовище (рН < 7)
439
У нейтральному і лужному середовищі метилоранж має жовто-оранжеве забарвлення, яке при підкисленні переходить у червоне. Забарвлення метилоранжу в нейтральному і лужному середовищі надають хромофорні групи — спряжена система подвійних зв’язків, у яку входить і азогрупа —, та ауксохромні групи (СН3)2Н—, —8О3На. Вільні р-електрони диметиламіногрупи за рахунок спряження збільшують електронну густину сполученого з нею бензольного ядра, що приводить до збільшення електронної густини всієї хромофорної групи. Група —8О3Кга викликає зміщення електронної густини хромофора на себе.
При підкисленні, наприклад, соляною кислотою іон водню приєднується до атома азоту азогрупи. В результаті відбувається перенесення реакційного центру по ланцюгу спряження з димети-ламінового атома азоту до атома азоту азогрупи. Одночасно відбувається електронна перебудова спряжених подвійних зв’язків бензольного ядра у хіноїдну систему подвійних зв’язків. При цьому одна хромофорна група (азогрупа—N=N—) зникає, а з’являється інша хромофорна група (хіноїдне ядро), що приводить до зміни забарвлення речовини.
Спрощеною характеристикою забарвлення речовини є довжина хвилі світла Хмакс, при якій спостерігається найбільша інтенсивність його поглинання і яка вимірюється спектрофотометрами. У нейтральному або лужному середовищі, при рН > 7, розчини метилоранжу мають Амак0 — 472 нм; у кислому середовищі, при рН < 7, спостерігається поглиблення забарвлення, відбувається так званий батохромний.зсув і Хмакс досягає 506 нм. Інтервал зміни забарвлення метилоранжу від жовтого до червоного знаходиться в межах рН від 3,1 до 4,4.
Розділ 38. БАГАТОЯДЕРНІ АРОМАТИЧНІ СПОЛУКИ З НЕКОНДЕНСОВАНИМИ БЕНЗОЛЬНИМИ ЯДРАМИ
Органічні сполуки з двома або більшою кількістю бензольних ядер, сполучених між собою простим зв’язком, називають багатоядерними ароматичними сполуками з неконденсованими бензольними ядрами. Відповідно до кількості бензольних ядер їх називають дифенілами, трифенілами і т. д. Наприклад:
Цифен іл
Трифеніл
Відома велика група жирно-ароматичних багатоядерних сполук з неконденсованими бензольними ядрами, які можна розглядати як похідні алканів, алкенів і алкінів. Наприклад:
440
1,2-ДифенІлетилен, стильбен Дифенілацетилен, толан
Дифеніл у невеликих кількостях міститься в кам’яновугільній смолі. Крім того, його можна синтезувати за реакцією Вюрца — Фіттіга:
2СвНг,—Вг + 2№ СвН6—СсНБ + 2>\аВг.
З високим виходом дифеніл утворюється також при нагріванні йодбензолу з мідним порошком (реакція Ульмана, 1903 р.):
2СсНг-1 + 2Си СвН6—СвН8 + 2Си1.
Дифеніл — безбарвна кристалічна речовина (Тт = 70 °С), має високу термічну стійкість, радіаційну стійкість, низьку залишкову радіоактивність, незначне газовиділення, корозійну інертність. Тому його використовують як теплоносій при охолодженні ядерних реакторів.
Положення замісників у молекулі дифенілу позначають цифрами або префіксами орто-, мета-, пара- (формула І):
0,148 нм
5' 6' 6 5
м'— о'— । о— м—
Бензольні ядра в молекулі дифенілу сполучені між собою о-зв’язком (І), довжина якого (0,148 нм) менша, ніж довжина аналогічного зв’язку, наприклад, у молекулі етану. Причиною зменшення довжини цього зв’язку є спряження між я-електронами бензольних ядер, яке в молекулі дифенілу відбувається якраз за місцем о-зв’язку. При нагріванні дифенілу до пароподібного стану в його молекулі можливе обертання фенільних груп навколо п-зв’язку. Якщо хоча б в одному з положень 2,2',6,6' наявний замісник, то таке обертання бензольних ядер неможливе і ці ядра розміщуються в просторі під кутом (формула II):
При дослідженні просторового розміщення бензольних ядер у похідних дифенілу було встановлено, що при заміщенні атомів
15 1-120
441
водню в орто-положсшіях обох бензольних ядер дифенілу на об’ємні групи атомів у таких похідних виникають два оптичних ізомери. Наприклад, 6,6'-динітродифенова кислота існує в таких двох енан-тіоморфних формах:
Такий вид ізомерії називають поворотною оптичною Ізомерією, або атропоізомерією, а самі ізомери — атропоізомерами.
Дифеніл — типовий ароматичний вуглеводень, хімічні властивості якого аналогічні властивостям одноядерних сполук бензольного ряду. Фенільні групи в молекулі дифенілу одна відносно другої є орто- і пара-орієнтантами. Тому реакції електрофільного заміщення відбуваються переважно в положення 4 і 4'.
Трифенілметани. Трифенілметан і його похідні є одними з найважливіших багатоядерних сполук з неконденсованими бензольними ядрами. Його добувають алкілуванням бензолу хлороформом або хлористим бензиліденом за Фріделем — Крафтсом:
А1С1 А1С1
ЗС6Н6 + СНС13=знй- (С8Н8)8СН 2С6Н0 + С6Н5СНС1а.
Трифенілметан — безбарвна кристалічна речовина з ТІ!Л = = 93 °С . У молекулі трифенілметану атом водню біля третинного атома вуглецю під впливом трьох фенільпих груп стає рухливим і набуває кислотних властивостей. Константа кислотності цього атома водню становить Л'д = 10 ~33:
Метановий атом водню в молекулі трифенілметану заміщується на калій, хлор, бром, легко окислюється:
(СсН5)3С—СІ (С6НБ)8С-Н^-> (СГ.Н,)3С-К.
ТрифенІл- [О] Трифеніл-
хлормстап метилкал їй
(СвН6)3С-ОН
Трифенілкарбінол
Трифенілметильні катіони і трифенілметнльні аніони виявляють високу стійкість, оскільки в них відбувається спряження трьох фенільпих груп з зарядом па третинному атомі вуглецю:
442
Трифеїплметильний аніон Трифенілметильний катіон
В результаті такого спряження в трифенілметильному аніоні відбувається роззосередження негативного заряду переважно в орто і шрс-положеннях бензольних ядер, а в трифенілметильному катіоні, навпаки, позитивний заряд «погашається» зміщенням л-електронів бензольних ядер, що стабілізує цей аніон і катіон.
Трифенілметильні іони мають забарвлення, оскільки заряд на третинному атомі вуглецю приводить до спряження л-електрон-них хмар трьох бензольних ядер і до виникнення хромофору — довгої системи спряжених подвійних зв’язків.
Окислювачі перетворюють трифенілметан на третинний спирт — трифенілкарбінол (Тпл = 162 °С). Цей же спирт легко утворюється при гідролізі трифенілхлорметану:
(СвН5)3С-С1 (С6НБ)3С—ОН.
Гідроксильна група в молекулі трнфенілкарбінолу під впливом трьох фенільних радикалів виявляє високу активність (рухливість). Так, з НС1 вона легко заміщується на хлор, а з спиртами при наявності мінеральних кислот трифенілкарбінол надзвичайно легко утворює прості ефіри:
(С0Нв)3С-О-С2Н6<-^^ (С6НБ)3С-ОН±й§-> (С6Н3)3С-С1.
При розчиненні трнфенілкарбінолу в концентрованій Н28О4 розчин забарвлюється в інтенсивно жовтий колір (явище галохро-мії). Поява забарвлення в цьому випадку зумовлена виникненням забарвлених катіонів (С6Н5)3С+. Під час розбавляння розчину водою забарвлення зникає, оскільки трифенілметильні катіони реагують з водою і утворюють трифенілкарбінол:
(С6Н5)3с(Х)Н + Н28О4^=^(С6Н5)3С++ + НОН .
Такого ж жовтого кольору розчини були добуті при дії на три-фенілхлорметан порошкоподібного срібла в бензолі у відсутності кисню повітря. М. Гомберг (1900 р.) довів, що в цьому випадку утворюються забарвлені в жовтий колір вільні трифенілметильні радикали, які в розчині перебувають у рівновазі зі своїми димерами:
2 (СЙП6)3С Ц СІ + 2ЛЄ 2 (С„Н8)3С - Димер.
15*
443
Трифенілметил був першим виділеним стійким вільним радикалом. Причиною стійкості трифенілметильного радикала є спряження неспареного електрона з л-молекулярпими орбіталями трьох бензольних ядер і роззосередження його в орто- і /гара-положення кожного з цих ядер:
Отже, для цього радикала можлива мезомерна стабілізація:
В алкільних радикалах, наприклад в метильному, етпльному та інших, такого роззосередження неспареного електрона немає, тому алкільні радикали надзвичайно нестійкі і виділити їх у вільному стані неможливо. Стійкість вільних радикалів у нижченаве-деному ряду зменшується:
(СсН5)3С • > СвНв—СН, > Н2С=СН—СН2 > Н«С-СН2.
У такій же послідовності зменшується і час існування цих радикалів.
Барвники трифенілметанового ряду. З похідних трифенілмета-нових вуглеводнів найбільше значення мають їх аміно- і оксипо-хідні, які є барвниками.
Найпростішим барвником трифенілметанового ряду є малахітовий зелений. Його добувають конденсацією диметиланіліну з бензальдегідом, яку здійснюють нагріванням цих речовин при наявності безводного хлориду цинку або концентрованої Н28О4. При цьому спочатку утворюється безбарвний 4,4-бїс(диметиламіно)трифе-нілметан (І), який називають лейкоосновою (від грецьк. «лейкос» — білий, безбарвний). Лейкосполука (1) при окисленні оксидом свинцю (IV) перетворюється на безбарвний 4,4-бїс-(диметиламіно)трифеніл-карбінол (II), який називають карбінольною основою барвника.
444
Карбінольна основа (II) при взаємодії з надлишком кислоти, наприклад соляної, перетворюється на забарвлену сіль (III) — барвник малахітовий зелений:
Бензальдегід
Лейкоснолука
Носієм забарвлення (хромофором) цього барвника є довга система спряжених подвійних зв’язків, в яку входить і хіноїдне угруповання атомів. Диметиламінна і диметиламонійна групи є ауксо-хромами. Перша з них за рахунок р, л-спряження вільної пари електронів збільшує електронну густину хромофору, а друга — викликає зміщення цієї густини на себе.
Малахітовий зелений фарбує бавовну, шовк, вовну у красивий зелений колір, однак світлостійкість цього барвника невисока.
Представником амінопохідних барвників трифенілметанового ряду є також кристалічний фіолетовий. Цей барвник добувають конденсацією, /г,п'-диметиламінобензофенону (кетону Міхлера) з диметиланіліном при наявності хлороксиду фосфору. Спочатку в результаті конденсації утворюється безбарвна карбінольна основа, яка потім з кислотою утворює барвник кристалічний фіолетовий:
445
Кристалічний фіолетовий фарбує матеріали у яскраво-фіолетовий колір. Його використовують також для виготовлення фіолетового чорнила і пасти для кулькових авторучок.
Типовим представником оксипохідних трифенілметанового ряду є фенолфталеїн, який добувають конденсацією фенолу з фталевим ангідридо?л при наявності водовідбирних засобів, наприклад концентрованої Н.28О,. При конденсації утворюється безбарвна (лак-тонна) форма фенолфталеїну:
Фенолфталеїн безбарвний (лактонна форма)
Фенолфталеїн — кристалічна речовина (ТПл — 261 °С), не розчинна у воді, але добре розчинна в спирті. Його використовують V медицині як слабке проносне під назвою пурген. Широкого застосування фенолфталеїн набув як індикатор (інтервал переходу забарвлення рН = 8—10). У кислому і нейтральному середовищі
фенолфталеїн безбарвний, а з розбавленими розчинами лугів утворює діаніон червоно-фіолетового кольору, в якому хромофором є довга система спряжених подвійних зв’язків разом з хіноїдним уг-
рупованням, групи —О , —СОО , >С=О є ауксохромами: О*На'
он
Фенолфталеїн безбарвний
№ОН
2ИаОН
-н2о
О
О На
ОН
СОО“Иа +
б* Фенолфталеїн забарвлений (динатрієва сіль)
Фенолфіалеїн безбарвний (тринатрієва сіль)
ОИа
446
Збільшення концентрації лугу приводить до перетворення забарвленого фенолфталеїну в безбарвну тризаміщену карбінольну основу (тринатрієва сіль), яка не має хромофору — довгої системи спряжених подвійних зв’язків і хіноїдного угруповання. Тому в концентрованих лугах забарвлення фенолфталеїну зникає.
Розділ 39. АРОМАТИЧНІ ВУГЛЕВОДНІ З КОНДЕНСОВАНИМИ БЕНЗОЛЬНИМИ ЯДРАМИ
Ароматичні вуглеводні, в молекулах яких два або кілька бензольних ядер мають спільні атоми вуглецю, називають вуглеводнями з конденсованими бензольними ядрами. До найважливіших з них належать нафталін, антрацен і фенаитрен:
сн сн
Нафталін
Антрацен
Нафталін
Фенаитрен
Нафталін був відкритий у кам’яновугільній смолі раніше від бензолу, в 1819 р. Елементний склад його — С10Н8— встановив О. А. Воскресенський у 1838 р., а структурну формулу запропонували Е. Ерлеимейєр і К. Гребе (1866—1868 рр.).
У промисловості нафталін добувають з кам’яновугільної смоли, в якій його міститься близько Ю %. Знаходиться нафталін також і в продуктах піролізу бензину і гасу. Синтетично нафталін можна добути, наприклад, з ацетилену такими послідовними реакціями:
знс==сн
НС~СН
Ацетилен Бензол
НС==СН
-н2
Нафталін
Цей синтез одночасно є доказом структури нафталіну.
Вуглецеві атоми у молекулі нафталіну нумерують. Нумерацію ведуть за годинниковою стрілкою і так, щоб спільні атоми вуглецю нумерувались останніми:
447
Рис. 74. Розподіл л-елек-тронної густини в молекулі нафталіну
Положення 1,4,5,8 позначають буквою а і називають а-положеннями, 2,3,6,7— буквою р і називають (3-положеннями.
Електронна будова нафталіну. Молекула нафталіну має десять вуглецевих атомів, які лежать в одній площині і розміщені по кутах двох сполучених між собою (конденсованих) бензольних ядер. Атоми вуглецю в молекулі нафталіну перебувають у 5р2-гібридизованомустані. В цьому валентному стані кожний вуглецевий атом має ще по одному р-електрону. Отже, у молекулі нафталіну є десять р-електро-нів, які мають форму об’ємних вісімок,
взаємно протилежні спіни і паралельні осі. Кожний з цих р-елек-тронів взаємодіє з р-електронами сусідніх вуглецевих атомів. У результаті всі десять р-електронів взаємно перекриваються між собою, утворюючи при цьому спільну електронну орбіталь, яка охоплює всі десять атомів вуглецю і розміщується поза площиною молекули (наді під нею, рис. 74). Цю електронну орбіталь можна розглядати як сукупність двох секстетів л-електронів, які частково перекриваються і мають два спільних л-електрони.
Рентгеноструктурний аналіз показує, що в молекулі нафталіну, на відміну від бензолу, віддалі між вуглецевими атомами не однакові:
Як видно з молекулярної діаграми нафталіну, зв’язки між атомами вуглецю 1,2 за довжиною більш подібні до подвійних, ніж зв’язки між атомами 2,3, а зв’язки між атомами 1,2 і 3,4 деякою мірою аналогічні спряженій системі зв’язків 1,3-бутадієну. Внаслідок неоднакової віддалі між вуглецевими атомами л-електронна хмара в молекулі нафталіну розподілена хоч і симетрично, але нерівномірно. А це приводить до нерівноцінності положень в його молекулі. На а-вуглецевих атомах електронна густина більша, ніж на Р-атомах. Менша симетрія кожного з конденсованих бензольних ядер у молекулі нафталіну позначається на індексах вільної валентності і на кратності зв’язків:
448
Отже, нафталін являє собою циклічну спряжену систему, яка містить десять л-електронів, і його молекулу можна описати такими граничними структурами:
Враховуючи, що в нафталіну десять л-електронів делокалізова-ні і утворюють спільну електронну орбіталь із збереженням енергетично вигідної ароматичності обох ядер, його молекулу можна зобразити без подвійних зв’язків:
Внаслідок нерівноцінності положень у молекулі нафталіну його однозаміїцені існують у вигляді двох ізомерів—а - і р-. Наприклад:
а -Метилнафталін р -Метилнафталін
<Х-Хлорнафталін р-ХлорнафталІп
Двозаміщені нафталіну з однаковими замісниками існують у вигляді десяти ізомерів. Наприклад:
1,2- або орто~ Диметил нафталін
1,3- або мета -Диметилнафталін
1,8- або пері -Диметилнафталін
2.,3-ДиметнлнафталІн 2,6-або амфі-Диметилнафталін 2,7-ДиметилнафталІн
449
При наявності в нафталіновому ядрі двох різних замісників кількість ізомерів таких двозаміщених збільшується і досягає 14.
Наявність у молекулі нафталіну двох бензольних ядер і їх рівноцінність доводять такими реакціями. При окисленні нафталіну утворюється фталева кислота (див. розд. 35). А це свідчить про те, іцо одне з ядер у його молекулі є бензольним.
Нафталін легко нітрується і утворює ос-нітронафталін. Окислення сс-нітронафталіну дає сс-нітрофталеву кислоту. Так доводиться, що ядро І, яке містить нітрогрупу, є бензольним. При відновленні а-нітронафталіну утворюється сс-нафтиламін, який при окисленні дає фталеву кислоту. Приокисленніа-нафтиламінуруйнується ядро І, оскільки його електронна густина за рахунок позитивного мезо-мерного ефекту аміногрупи більша, ніж ядра II. Отже, при окисленні залишається ядро 11. А оскільки продуктом такого окислення є похідна бензолу — фталева кислота, то це свідчить, що і ядро II є бензольним:
Фізичні і хімічні властивості. Нафталін — біла кристалічна речовина (7ПЛ = 80,3°С), дуже летка і легко сублімується. Максимум найбільш довгохвильової смуги поглинання нафталіну в УФ-області становить А = 275 нм.
Нафталін задовольняє вимогам правила ароматичності Хюкке-ля (див. розд. 28) і виявляє ароматичні властивості, подібні до властивостей бензолу. Тому нафталін, які бензол, вступає в реакції приєднання і заміщення.
Реакції приєднання. Десять я-електронів у молекулі нафталіну розподілені між двома ядрами. Оскільки з цих електронів можна скласти тільки один л-електронний секстет, то енергія спряження в молекулі нафталіну (255,2 кДж • моль-1) значно менша, ніж£с двох молекул бензолу (2 • 150,5 = ЗОЇ кДж • моль-1). А це свідчить про меншу термодинамічну стійкість нафталіну порівняно з бензолом і відповідно про більш «ненасичений» характер хімічних властивостей нафталіну. Так, нафталін, на відміну від бензолу, гідрується воднем у момент виділення і перетворюється при цьому на 1,4-дигідронафталін, який легко ізомеризується в стійкіший 1,2-дигідронафталін:
450
При каталітичному гідруванні нафталіну спочатку гідрується одне бензольне ядро і утворюється тетрагідронафталін (тетралін), який при дещо вищій температурі гідрується далі з утворенням декагідронафталіну (лекалі ну):
Тетралін і декалін використовують у техніці як розчинники, а також як добавки до моторних палив для підвищення їх октанового числа.
Реакції за м і щ е н н я. Нафталін, як і бензол, вступає в реакції електрофільного заміщення. Проте ці реакції відбуваються для нього легше, ніж для бензолу. При цьому замісник, як правило, заміщує атом водню «-положення, вуглецевий атом якого має більшу електронну густину. Крім того, в цьому випадку виникає енергетично стійкіший (приблизно на 40 кДж • моль'1) о-ком-плекс (карбкатіон), ніж толі, коли б заміщення здійснювалося в Р-положепні. Пов’язано це з тим, що при заміщенні в «-положенні порушується ароматичність тільки одного бензольного ядра, а при заміщенні в р-положенні — обох бензольних ядер:
Менш стійкі карбкатіони
' Більш стійкі карбкатіони
де А'+ = С1+, Вг+, N0^, 8О3Н+ та ін.
1. Галогенування. У реакціях нафталіну з хлором і бромом чітко виявляється його «ненасичений» характер. Першою стадією в цих реакціях є приєднання молекули галогену в «-положення одного з бензольних ядер і утворення нестійких продуктів 1,4-приєднання, які легко дегідрогалогенуються і перетворюються на а-галогено-нафталіни. Так, нафталін за звичайних умов приєднує хлор і послідовно утворює 1,4-дихлордигідронафталін і 1,2,3,4-тетрахлортетра-
451
гідронафталін, які при нагріванні дегідрохлоруються і перетворюються відповідно на 1-хлор- і 1,4-дихлориафталін:
Аналогічно відбувається і бромування нафталіну:
Взаємодію нафталіну з хлором і бромом каталізують солі заліза.
2. Нітрування нафталіну нітруючою сумішшю легко відбувається уже при 50—60°С. При цьому з виходом понад 95 % утворюється а-нітронафталін (Т,,ГІ = 01 °С) і невелика кількість (~4,5 %) (3-нітронафталіну:
3. Сульфування — найважливіша 5ц -реакція нафталіну, оскільки нафталінсульфокислоти є вихідними продуктами для добування багатьох барвників та інших похідних нафталіну. Залежно від температури сульфування нафталіну може здійснюватися в а-або |3-положенні. У м’яких умовах при температурі 80 °С нафталін взаємодіє з концентрованою Н2ЗО4 з утворенням а-нафталінсульфо-кислоти, а в жорсткіших умовах (нагрівання з концентрованою Н2ЗО4 до 160—165 °С) — |3-нафталі нсульфокислоти:
452
Сульфування нафталіну, як і сульфування бензолу, є оборотною реакцією.
Окислення нафталіну. Нафталін значно легше, ніж бензол, окислюється, в чому також виявляються його «ненасичені» властивості. Енергійне окислення нафталіну, зокрема киснем повітря при наявності каталізатора і 400°С, приводить до утворення фталевого ангідриду, який має важливе промислове значення (див. розд. 35):
-2СО2, -Н2О
При обережному окисленні нафталіну хромовим ангідридом в оцтовій кислоті або хромовою сумішшю утворюються нафтохінони:
1,2-НафтохІнон '1,4-НафтохІнон 2,6-НафтохІнон
Властивості нафтохінонів подібні до властивостей бензохінонів (див. розд. 34). З нафтохінонів найважливіше значення має 1,4-нафтохінон, який сублімується при 100 °С і являє собою кристалічну речовину жовтого кольору. Похідними 1,4-нафтохінону є деякі природні барвники і вітаміни групи К. Синтетичним аналогом вітамінів групи К є 2-метилнафтохінон-1,4, який під назвою метінон (вітамін К3) широко застосовується в медицині. Вітамін К3 добувають окисленням 2-метилнафталіну:
о
о
Похідні нафталіну. Нафталін, як і бензол, утворює галогено-, нітро-, сульфо-, гідрокси-, амінопохідні, які за своїми властивостями подібні до аналогічних похідних бензолу. Найбільше значення з похідних нафталіну мають гідрокси-, аміно- і сульфопохідні. Гідроксильні похідні нафталіну називають нафтолами, а амінопохідні — нафтиламінами:
а-Нафтол
Р-Нафтол
СС-НафтиламІн ^-Нафтиламін
453
За хімічними властивостями нафтоли подібні до фенолів, а наф-тиламіни — до ароматичних амінів.
Нафтоли і нафтиламіни використовують для добування азобарвників.
Антрацен
Антрацен вперше виділили Ж. Дюма і О. Лоран 5 1832 р.з кам’яновугільної смоли, звідки пішла назва цього вуглеводню (греньк. «антракс» — вугілля). У молекулі антрацену містяться три бензольних ядра, які лінійно конденсовані між собою. Нумерацію вуглецевих атомів у молекулі антрацену проводять так:
Положення 1,4,5,8 називають а-положеннями, 2,3,6,7 — |3-по-ложеннями і 9,10 — у- або ц- («мезо» — середній) положеннями.
У промисловості антрацен добувають з кам’яновугільної смоли, зокрема з її фракції, яка переганяється при 300—350 °С і називається антраценовим, або зеленим, маслом. У кам’яновугільній смолі міститься 0,1—1 % антрацену. Синтетично антрацен добувають за реакцією Фріделя — Крафтса (нагріванням бензолу з тетрабром-етаном при наявності безводного броміду алюмінію):
АІ Вг з
-4НВг
Антрацен можна добути також конденсацією фталевого ангідриду з бензолом. У цьому випадку спочатку утворюється антрахінон, який потім відновленням перетворюють на антрацен:
АІСІз
—н2о
6Н
-2Н,О
Антрацен являє собою кристалічну речовину (Тпл = 217 °С), яка флуоресціює в УФ-світлі. Максимум найбільш довгохвильового поглинання його розчинів становить ХМ0КС= 370 нм.
454
Молекула антрацену має чотирнадцять вуглецевих атомів, які лежать в одній площині. Рентгеноструктурний аналіз показує, що віддалі між вуглецевими атомами в його молекулі неоднакові:
Молекулярна діаграма вказує, що відносна ненасиченість атомів вуглецю в положеннях 9 і 10 (у-положення) більша, ніж в а(1,4,5,8)- і Р(2,3,6,7)-положеннях, а порядок зв’язку між С\- і С2-атомами більший, ніж між С,- і С3-атомами:
Отже, антрацен являє собою циклічну спряжену систему з 14 л-електронами і його будову можна зобразити граничними структурами І—IV:
IV
З вищенаведених даних випливає, що у молекулі антрацену розподіл електронної густини між атомами вуглецю ще більш нерівномірний, ніж у нафталіну. Значення енергії спряження для антрацену становить Ес = 351,5 кДж • моль-1, що набагато менше, ніж потроєна енергія спряження в молекулі бензолу (3 • 150,5 = = 451,5 кДж • моль"1) .Аце свідчить про меншу термодинамічну стійкість антрацену порівняно з бензолом і, відповідно, про більш «ненасичений» характер його хімічних властивостей.
Антрацен задовольняє вимогам правила ароматичності Хюккеля (див. розд. 28) і виявляє ароматичні властивості, подібні до властивостей бензолу. Проте внаслідок нерівномірності розподілу електронної густини ароматичні властивості антрацену виражені значно меншою мірою, ніж нафталіну, і набагато слабше, ніж бензолу. Антрацен виявляє квазіароматичні властивості. Разом з тим два
455
крайніх бензольних ядра, намагаючись зберегти свою ароматичність, частково локалізують в положеннях 9 і 10 по одному неспа-реному електрону. Тому вуглецеві атоми 9 і 10 мають підвищену реакційну здатність, що виявляється в сильно вираженій «ненаси-ченості» і високій активності цих атомів вуглецю до реакцій приєднання.
Антрацен, як і бензол, може вступати в реакції приєднання і заміщення. і
Реакції приєднання. Приєднання до антрацену різних реагентів відбувається переважно до 9 і 10 атомів вуглецю реакції (1—6)]і
Так, антрацен легко гідрується воднем в момент виділення і перетворюється при цьому на 9,10-дигідроантрацен [реакція (1)]. Приєднання галогенів (С12, Вг2) відбувається з утворенням 9,10-дигалогенодигідроантраценів, які при нагріванні легкодегідрога-логенуються і перетворюються на 9-галогеноантрацен [реакція(2)[. Антрацен, подібно до спряжених дієнів, приєднує в положення 9, 10 лужні метали [Па, реакція (3)], а з таким дієнофі лом, як малеїновий ангідрид, дає аддукт[реакція (4)]. При опроміненні антрацену УФ-світлом він фотохімічно циклізується і перетворюється на ді-антрацен [димер, реакція (5)], а при наявності повітря за цих умов антрацен приєднує кисень [реакція (6)] і утворює пероксид.
Реакції заміщенн я. Антрацен може вступати в реакції електрофільного заміщення: нітрування, галогенування, сульфування та ін., які відбуваються переважно з заміщенням атома водню положення 9. Проте ці реакції поки що не мають великої синтетичної цінності.
Окислення антрацену. Антрахінон. Антрацен значно легше, ніж бензол, окислюється такими окислювачами, як О2, НЬЮ3, СгО3
456
тощо. Окислення антрацену відбувається за місцем вуглецевих атомів 9 і 10 і приводить до утворення дикетону — антрахінону:
Найважливішою реакцією антрахінону є його сульфування, яке широко використовують у промисловості. При нагріванні антрахінону з концентрованою Н28О, утворюється р-антрахінон-сульфокислота. Якщо сульфування антрахінону проводити при наявності солей ртуті (II), то, як вперше помітив М. О. Ільїнський (1903 р.), утворюється а-аитрахінонсульфокислота:
Антрахінонсульфокислоти використовують для синтезу барвників
Алізарин. Найважливішим похідним антрахінону є барвник алізарин (1,2-діоксіантрахінон). Алізарин у вигляді глікозиду зустрічається в природі в коренях марени (крапу), звідки .його добували до 1875 р. У 1868 р. К. Гребе і Т. Ліберман встановили структуру алізарину і вперше здійснили його синтез (1869 р.). Результати цієї роботи стали основою дешевого промислового методу виробництва алізарину. У наш час алізарин у промисловості добувають лужним плавом антрахінон-Р-сульфокислоти при наявності окислювачів (О2 повітря, КС1О3, №МО3 тощо.) При цьому одночасно відбувається нуклеофільне заміщення сульфогрупи на гідроксил і окислення атома водню «-положення відносно гідроксильної групи:
№ОН,[0},200°С
-№28£)3>
Синтетичний алізарин повністю витіснив природний.
457
Алізарин являє собою кристалічну речовину червоного кольору (7’,,л — 290 °С), добре розчинну у водних розчинах лугів. Алізарин фарбує попередньо змочені (протравлені) солями дво- і тривалентних металів (алюмінію, хрому, заліза) тканинні утворює на них надзвичайно міцні забарвлені сполуки (лаки) комплексної будови. Наприклад:
Тканини, протравлені солями заліза, алізарин фарбує у фіолетовий колір, після хромової протрави — в червоно-коричневий, після алюмінієвої — в червоний колір. Такий метод фарбування називають протравним.
Фенаитрен
Фенантрен є ізомером антрацену. В молекулі феиантрену одне з трьох бензольних ядер конденсоване ангулярно (під кутом). Для вуглецевих атомів феиантрену прийнята така нумерація:
Фенантрен
Фенантрен, як і антрацен, міститься в кам’яновугільній смолі, з якої він вперше був виділений і описаний у 1872 р. К. Гребе.
У молекулі феиантрену формально можна виділити два л-електронних секстети, які зумовлюють його ароматичність. Фенантрен задовольняє вимогам правила ароматичності Хюккеля (див. розд. 28) і виявляє ароматичні властивості. Ароматичний характер у феиантрену виражений дещо сильніше, ніж в антрацену, і тому енергія спряження його (Ас = 387 кДж/моль -1) дещо вища, ніж у антрацену.
Для феиантрену можливі реакції електрофільного заміщення, які відбуваються переважно біля вуглецевих атомів 9 і 10. Разом з тим у молекулі феиантрену зв’язок між атомами С9 і С1() має характер етиленового подвійного С=С-зв’язку. Тому для феиантрену характерні реакції приєднання за місцем цього зв’язку, які поки що не мають синтетичної цінності.
458
Повністю гідроване ядро фенантрену називається пергідрофен-антреновим. Воно входить до складу багатьох природних продуктів —стероїдів, гормонів, алкалоїдів тощо. Так, до складу стероїдів (див. розд. 39) входить угруповання атомів, яке складається з конденсованих між собою пергідрофенантренового і пиклопентанового ядер. Таке угруповання атомів називається циклопентанпергідро-фенантреновим:
Пергідрофенантрен
Циклопентан пергідрофенантрен
Канцерогенні багатоядерні ароматичні вуглеводні
Ряд вуглеводнів з конденсованими бензольними ядрами мають здатність викликати утворення ракових пухлин. Такі речовини називають канцерогенними (від лат. сапсег — рак). Одними з найсиль-ніших канцерогенних ароматичних сполук є заміщені в мезополо-женні 1,2-бензантрацену, наприклад 10-метилбензантрацен, а також дибензантрацен, бензпірен та ін.:
1,2-Бензантр.ацен
Бензпірен
Канцерогенні багатоядерні ароматичні вуглеводні містяться в кам’яновугільній смолі, сажі, продуктах піролізу органічних речовин. Бензпірен утворюється при неповному згорянні різних органічних речовин. Забруднення атмосфери бензпіреном різко збільшилось у зв’язку з розвитком автомобільного транспорту. Щоб викликати рак легенів або шкіри людини, достатньо кількох міліграмів цієї речовини. Потрапляючи з повітря в грунт, воду, рослини, бензпірен може забруднювати і продукти харчування. Бензпірен міститься також у тютюновому димі і є причиною захворювання курців на рак легенів.
Канцерогенні вуглеводні мають сильно виражені ароматичні властивості. Ці вуглеводні особливо легко вступають у реакції електрофільного заміщення, навіть у такі, як азосполучення. •
459
Стероїди
Стероїди — це природні сполуки переважно тваринного походження, які мають велике значення в життєдіяльності організму. Назва «стероїди» пішла від грецького «стерос»— твердий. Відомо більш як 15 000 стероїдів, з яких понад 100 застосовують у медицині.
Стероїди у своїй структурі містять циклопентанпергідрофенант-ренове угруповання атомів,'в якому прийнято таку нумерацію вуглецевих атомів у циклах:
де Ц — переважно вуглеводневий залишок, а X — гідроксильна або карбоксильна група.
Природні стероїди містять асиметричні атоми вуглецю і є оптично активними речовинами.
До стероїдів належать стерини, жовчні кислоти, статеві гормони, гормони надниркових залоз, серцеві аглікони, сапоніни, отрути жаб і стероїдні алкалоїди.
ЧАСТИНА IV
Гетероциклічні сполуки
Гетероциклічними називають такі сполуки циклічної будови, у циклах яких поряд з атомами вуглецю знаходяться атоми інших елементів. Ці інші атоми називають гетероатомами (від грецьк. «гетерос» — інший). Найчастіше такими гетероатомами є атоми кисню, сірки, азоту В гетероциклі може знаходитись один, два, три і більше гетероатомів.
Гетероциклічні сполуки для систематики і зручності вивчення класифікують. Класифікацію гетероциклів здійснюють залежно відвеличини циклу. Відповідно до цього розрізняють три-, чотири-, п’яти-, шестичленні гетероцикли та гетероцикли з більшою кількістю атомів. Серед них найважливіше значення мають п’яти- і шестичленні гетероциклічні сполуки.
Гетероциклічні сполуки називають за тривіальною, раціональною і систематичною номенклатурою. Для давно відомих гетероциклічних сполук часто використовують тривіальні назви. Наприклад, пірол, піридин,фуран, індол, пуринтаін. В раціональній номенклатурі за основу беруть назву певного гетероциклу — фура-ну, тіофену, піролу, піридину чи іншого, а положення замісників у них позначають цифрами або буквами грецького алфавіту. В гете-роциклах з одним гетероатомом нумерацію починають з цього гетероатома:
Фуран
Піридин
Уп’ятичленних гетероциклах з одним гетероатомом положення 2 і 5 позначають відповідно буквами а і а', положення 3 і 4 — Р, Р'. У шестичленних гетероциклах з одним гетероатомом положення 2,3 і 4 позначають відповідно буквами а, р, у, а положення 5 і 6 — а' і Р'.
Сучасна наукова номенклатура гетероциклічної системи включає величину циклу, його ненасиченість, кількість гетероатомів, їх вид і положення. Назва гетероциклу за цією номенклатурою
461
складається з трьох частин: кореня, який вказує розмір циклу, суфікса (часто суміщеного з коренем), який вказує ступінь пена-сиченості гетероциклічної системи, і префікса, який вказує вид гетероатомів і їх кількість. Тричленний цикл має корінь -ір, чотиричленний ----єт, п’ятичленний-----о/і, шестичленний-----ін.
Насичені гетероцикли затомом азоту мають суфікс-ідін-, насичені гетероцикли без атома азоту мають суфікс -ан-, ненасичені гетероциклічні системи мають суфікс -ін-.
Природа гетероатома вказується префіксами окса-, тіа- і аза-відповіднодля кисню, сірки і азоту; префікси діокса-, ді-'тіа-, діаза-означають відповідно два атоми кисню, сірки і азоту. Якщо в гете-роциклі два або більше різних гетероатомів, то вони перелічуються за старшинством: кисень раніше від сірки, а сірка раніше від азоту, і їх нумерують у такому порядку: 0,8, N. При наявності в гетеро-циклі одного атома кисню і одного атома азоту використовують префікс оксаза-, а при наявності одного атома сірки і одного атома азоту — тіаза-. При одночасному перебуванні в циклі третинного атома азоту і групи ИН цифрою 1 позначають атом азоту групи ЦН. У цьому випадку нумерацію проводять у такому порядку: О, 8, ИН, N.
Ці правила ілюструють наведені нижче приклади (у дужках дано тривіальну назву):
Оксол (ФУ ран)
Тіол (тіофен)
Азол Азин ’.З-Оксазол ЬЗ-Тіазол
(пірол) (піридин)
Розділ 40. П'ЯТИЧЛЕННІ ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ
З ОДНИМ ГЕТЕРОАТОМОМ
З п’ятичленних гетероциклічних сполук з одним гетероатомом найважливіше значення мають фуран, тіофен і пірол. Для фурану, тіофену, піролу і їх похідних типовими є реакції електрофільного заміщення: нітрування, сульфування, галогенування, ацилування та ін. Така особливість властивостей цих гетероциклічних сполук пов’язана з їх електронною будовою. У циклах цих речовин містяться як атоми вуглецю, так і гетероатоми. Вуглецеві атоми і гетероатоми сполучені з сусідніми атомами вуглецю о-зв’язками. Для утворення цих зв’язків кожний атом циклу використовує .«^-гібридизовані орбіталі. Крім цього, у кожного вуглецевого атома циклу залишається ще по одному р-електрону, а в гетероатома — два р-електрони. р-Електрони атомів вуглецю і вільні р-електрони гетероатома перекриваються між собою і утворюють л-електрон-ний ароматичний секстет, густина якого розміщена над і під площиною циклу (рис. 75). Отже, у молекулах фурану, тіофену, піролу
462
відбувається спряження між вільними р-електронами гетероатома і електронами л-зв’язків (на схемі це показано зігнутими стрілками):
Фуран
Таке спряження л-електронів подвійних зв’язків циклу і р-електронів гетероатома, в результаті якого утворюється стійкий
Рис. 75. Розподіл п-електронної густини (а) і схема перекривання р-орбіталей (б) в молекулі піролу
л-електронний секстет, є енергетично вигідним. Наприклад, енергія спряження для фурану становить 67,8 кДж • моль"1, для піролу— 90,4 кДж • мольД, для тіофену—121,8 кДж - моль-1. Тому фуран можна розглядати як гібрид структур І—-Іб, тіофен — як гібрид структур 1І-—П6, а пірол — як гібрид структур III— ІІІб:
Делокалізацію електронів за рахунок їх р, л-спряження підтверджують значення дипольних моментів і довжин зв’язків, дані УФ- і мікрохвильових спектрів, теплоти згоряння п’ятичленних гетероциклів. Так, дипольні моменти фурану і тіофену значно менші
463
від дипольних моментів відповідно тетрагідрофурану і тетрагідро-тіофену:
0,55
1,87
Це свідчить про те, що за рахунок спряження у фурану і тіофену відбувається деяке зміщення електронної густини в напрямку, протилежному дипольному моменту, зумовленому різницею елек-троноакцепторних властивостей кисню і вуглецю, сірки і вуглецю у о-зв’язках С—О і С—8. За різницею дипольних моментів фурану і тетрагідрофурану, тіофену і тетрагідротіофену можна оцінити внесок мезомерного ефекту .гетероатомів у загальний дипольний момент молекули.
Оскільки у фурану, тіофену, піролу шість л-електронів(чотири л-електрони двох подвійних зв’язків і два р-електрони гетероатома) утворюють, як і в бензолу, спільну електронну орбіталь (див. розд. 75), то їх молекули, подібно до молекули бензолу, можна зобразити такими формулами:
В результаті спряження цикли цих речовин стають плоскими, зв’язки від атомів вуглецю до гетероатома коротшають порівняно з аналогічними зв’язками в інших органічних сполуках, а між’ядерні віддалі атомів вуглецю стають подібними до міжатомних віддалей у молекулі бутадієну. Рентгенографічні вимірювання дали такі значення міжатомних віддалей в молекулах фурану, тіофену і піролу, нм:
Утворення в циклах фурану, тіофену, піролу спільної молекулярної орбіталі з шести л-електронів (число л-електронів відповідає правилу ароматичності Хюккеля:4н Ц-2, л = 1) приводить до того, що ці речовини виявляють ароматичні властивості (див.
464
розд. '28). Проте наявність у гетероциклі гетероатома впливає на властивості цих речовин. Гетероатом виявляє індукційний і мезо-мерний вплив (ефект). Оскільки електронегативність гетероатома більша, ніж вуглецю, то індукційний ефект гетероатома негативний (-/-ефект). М-Ефект гетероатома виявляється за рахунок вільної електронної пари, яка зумовлює р,л-спряження в циклі. В результаті такого спряження електронна густина в п’ятичленних гетероциклах стає більшою, ніж у молекулі бензолу. Отже, гетероатом у цих сполуках виявляє -фМ-ефект і —/-ефект:
Реакційна здатність гетероциклів визначається перш за все густиною л-електронного секстету. Тому основний вплив на властивості фурану, тіофену, піролу має М-ефект гетероатома.
У фурану, тіофену, піролу, на відміну від бензолу, спостерігається нерівномірний розподіл л-електронної густини в молекулі. Електронна густина в цих п’ятичленних гетероциклічних сполуках за рахунок р, л-спряження зміщена від гетероатома в бік вуглецевих атомів циклу і стає найбільшою в а,«'-положеннях:
Це приводить до того, що в а-положеннях молекул фурану, тіофену і піролу найлегше і перш за все відбуваються реакції електрофільного заміщення. А оскільки в циклах цих п’ятичленних гетероциклів електронна густина більша, ніж у бензолу, то фуран, тіофен, піролактивніші до реакцій з електрофільними частинками, ніж бензол. Наприклад, реакції електрофільного заміщення для тіофену відбуваються у 103 разів швидше, ніж для бензолу. За здатністю до реакцій електрофільного заміщення фуран, пірол і тіофен можна розмістити в такий ряд:
Пірол Фуран Тіофен Бензол
Природа гетероатома, його електронегативність впливають на хімічну активність п’ятичленних гетероциклічних сполук. Від електронегативності гетероатома залежить здатність його пари електронів до взаємодії (спряження) з-л-електронамиподвійних зв’язків циклу. Оскільки електронегативність атома сірки менша, ніж ато
465
мів азоту і кисню, то р,л-спряження у молекулі тіофену виявляється більшою мірою, ніж у піролу і фурану. Внаслідок цього тіо-фен найбільш подібний за ароматичними властивостями до бензолу і з п’ятичленних гетероциклів він найбільш «ароматичний». Підтверджує цей висновок і порівняння значень енергій спряження Ес п’ятичленних гетероциклів з Ес бензолу.
Сполука Фуран Пірол Тіофен Бензол
Ес, кДж • моль-1 67,8 90,4 121,8 150,5
Як видно з наведених даних, £сДЛя тіофену менше всього відрізняється від Ес для бензолу. А це свідчить про більшу термодинамічну стійкість тіофену і сильнішу «ароматичність» його властивостей порівняно з піролом і фураном. Найменша термодинамічна стійкість порівняно з бензолом у фурану, що свідчить про «нена-сичений» характер його властивостей і найслабкішу «ароматичність». Отже, за ароматичністю порівняно з бензолом п’ятичленні гетероцикли розміщуються в такий ряд'
Бензол
Тіофен
Пірол Фуран
Фуран і його похідні. Фуран, як уже зазначалося, виявляє ароматичні властивості, але вони в нього виражені меншою мірою, ніж у піролу і тіофену. Разом з тим реакції електрофільного заміщення у фурану відбуваються легше, ніж у бензолу, оскільки в а- і «'-положеннях фуранового ядра значно підвищена електронна густина за рахунок /цл-спряження вільних ц-електронів атома кисню і л-електронів «-вуглецевих атомів.
Ароматичність фурану, як і бензолу, виявляється в реакціях електрофільного заміщення, які відбуваються досить легко і переважно в положення 2. Атака електрофілу в положення 3 приводить до утворення карбонієвого іона, який являє собою гібрид структур Іг і Ід, тоді як атака електрофілу в положення 2 дає карбонієвий іон, який є гібридом не тільки структур Іа і Іб (аналогічних структурам Іг і Ід), але й структури Ів. Структура Ів надає додаткової стабілізації карбоиієвому іону, що робить цей іон стійкішим:
Менш СТІЙКІ Іони
тче
Реакції галогенування, сульфування, нітрування і ацилування фурану, які відбуваються за 5в-механізмом, можна зобразити схемами (1)—(4):
Як видно із схеми, взаємодія фурану з галогенами [реакція (1)1 проходить через стадію утворення нестійкого проміжного продукту приєднання (наприклад, 2,5-дибромдигідрофурану), який легко втрачає галогеноводень і перетворюється на а-галогенофуран.
Фурановий цикл нестійкий до дії мінеральних кислот. Так, під впливом сірчаної кислоти фуран осмолюється, а азотна кислота руйнує цикл і окислює фуран у малеїнову кислоту. Тому для сульфування і нітрування цього гетероциклу підбирають «м’які» умови проведення реакцій. Радянські вчені О. П. Терентьєв і Л. О. Японська запропонували сульфувати фуран комплексом піридину або діоксану з 8О3 [реакція (2)]. Нітрують фуран [реакція (3)] ацетил-нітратом Н3С—С—О—Г\Ю2 у піридині. Реакція в цьому випадку
II О також проходить через стадію утворення проміжного продукту приєднання, від якого потім відщеплюється оцтова кислота і утворюється а-нітрофуран. Оцтовий ангідрид [реакція (4)] ацилує фуран з утворенням а-ацетилфурану.
Фуранове ядро має досить реакцій нездатні подвійні зв’язки. Зниження ароматичності фуранового ядра приводить до того, що фуран легше, ніжтіофен і пірол, вступає в реакції приєднання. Так, він легко приєднує водень і перетворюється при ньому на тетра-гідрофуран (ТГФ, ТКІШ = 65 °С):
не—сн
// V
не сн
2Нг
К'і
Фуран
Н,С--СН,
/ \2
Н2С СН2
хк
Тетраї ідрофуран
467
Фуран, подібно до бутадієну, виявляє властивості спряжених дієнів і може приєднувати в положення 1,4-дієнової системи фу-ранового циклу малеїновий ангідрид:
Ангідрид ендоксотетрагідро-фталевої кислоти
Отже, фуран має властивості як ароматичних сполук, так і спряженого дієну.
Для п’ятичленних гетероциклічних сполук з одним гетероатомом характерні реакції взаємного обміну гетероатомів. Радянський вчений Ю. К. Юр’єв встановив, що такий обмін гетероатомів відбувається під час пропускання пари відповідної гетероциклічної сполуки з Н,8, ИН3 або Н2О над А12О3 при температурі 400 °С (реакція Юр’єва):
Так фуран можна перетворити на тіофен і пірол, а тіофен і пірол — у фуран, тіофен — у пірол, а пірол — у тіофен.
Важливою похідною фурану є альдегід фурфурол (від лат. їигїнг — висівки). Фурфурол легко утворюється при нагріванні з розбавленими кислотами пентозовмісних речовин, таких, як висівки, солома, соняшникове лушпиння, кукурудзяні качани, деревна тирса тощо:
ч Н2О
(С5Н8О4)Л-------
Пентозани
СНОН-СНОН _
І І г>°
сн, сн- с
І * І \
он он н
Н28О!
-ЗН2О
фурфурол
Альдопсіітози
ФуРФУР0Л (^кип = 162 °С) використовують як селективний розчинник при очищенні нафтових фракцій, у виробництві пластмас, деяких лікарських препаратів (фурациліну, фурадоніну, фуразолі-
468
дону та ін.). При термокаталітичному декарбоксилуванні фурфуролу в промислових масштабах добувають фуран (Ткиа — 31,8 °С):
400°С, оксиди металів
+СО
Тіофен. Тіофен (7,КИП=84,ГС) утворюється при коксуванні кам’яного вугілля і міститься в кам’яновугільному дьогті, з якого його відганяють разом з бензолом, оскільки він дуже подібний до бензолу за своїми фізичними і хімічними властивостями. У промисловості тіофен добувають термічною реакцією ацетилену з сірководнем або взаємодією бутану з сіркою в газовій фазі:
400°С
21ІС = С1І+Н.,8------'
" -н2
-ЗН..8
сн2-сн2
/ \
СН3 СНз
+
48
Тіофен виявляє ароматичні властивості, подібні до властивостей бензолу. Реакції електрофільного заміщення для тіофену відбуваються набагато легше і швидше, ніж для бензолу, але повільніше, ніж для піролу і фурану. Вища реакційна здатність тіофену порівняно з бензолом пов’язана з нерівномірністю розподілу л-електронної густини в тіофеновому циклі і збільшенням її в а-і а'-положеннях за рахунок р,л-спряження вільної пари електронів атома сірки. Ароматичні властивості тіофену, як уже зазначалося, виражені значно сильніше, ніж фурану і піролу.
З п’ятичленних гетероциклічних сполук тіофен є найстійкішою сполукою. Він не руйнується кислотами. Тому його нітрують, сульфують, як і бензол, дією відповідно концентрованих азотної і сірчаної кислот, а галогенують безпосередньою взаємодією з галогенами. Електрофільне заміщення (Хв-реакції) в тіофеновому циклі здійснюється переважно в а-положення:
2-АцетилтІофен
2-Сульфотіофен
Тіофен досить стійкий до дії окислювачів і відновників. Він важче, ніж фуран, гідрується, але при наявності каталізаторів
469
тіофен все-таки приєднує водень і перетворюється натетрагідротіо-фен (тіофан):
+2Н2
Р<1/С
Тіофан
Тіофан при окисленні Н2О2 перетворюється на тіофансульфоксид, а при окисленні КМпО4— на сульфон тіофандисульфоксид (суль-фолан):
о
Тіофансульфоксид
Сульфолаїї
Пірол. Пірол виділяють з кам’яновугільної смоли та з кісткового жиру. Назва сполуки — пірол — походить від грецько* го слова «пір» — вогонь і латинського «олеум» — олія, масло-
За звичайних умов пірол є нестійкою безбарвною, не розчин* мою у воді рідиною (Ткцп = 129,7 °С), яка легко окислюється кис* нем повітря. Хімічні властивості піролу подібні до властивостей бензолу. Пірол, як і бензол, виявляє ароматичні властивості і тому схильний до реакцій електрофільного заміщення. Проте в молекулі піролу, як і інших гетероциклів, завдяки наявності гетероатома не досягається тієї рівномірності в розподілі електронної густини, як у молекулі бензолу." Розподіл електронної густини в молекулі піролу, обчислений методом МО, вказує, що ефективні заряди на а-вуглецевих атомах більші, ніж на (З-вуглецевих:
-0,06.__-0,06
-о,іо^ \ -о,ю
N40,31 І н
Тому реакції електрофільного заміщення для піролу здійснюються переважно в а-положенні, де електронна густина більша, ніж у [3-положенні. Оскільки в циклі піролу електронна густина більша,ніж у бензолу, то електрофільне заміщення в нього відбувається значно — легше, ніж у бензоЛіМрапрпкладГгалогенування піролу відбувається під дією~хлористого сульфурилу (ЗО2С12) в ефірному розчині при 0 °С. Продуктом такого галогенування є а-хлорпірол:
+ $о2сі2
і н
І II
СІ ь неї + ЗО2
470
Пірол надзвичайно чутливий до дії кислот. Азотна і сірчана кислоти повністю руйнують його молекулу. Тому нітрування і сульфування пі ролу здійснюють у «м’яких» умовах, а саме нітрують розчином азотної кислоти в оцтовому ангідриді, а сульфують за методом О. П. Терентьєва і Л. О. Японської комплексом піридину або діоксану з 8О3:
2-Сульфопірол
НОГЮ2.(Н3С-СО)2О
-н2о
При відновленні піролу воднем у момент виділення утворюється піролін (Ткип = 90 °С), а при каталітичному гідруванні — піролі-дин (Ткип = 88 °С):
НгС—сн2
Н2С сн2
2Н______
(7п +Н3С-СООН)
2Н,, Рі
Гііролідин
Наявність у молекулі піролу групи >І\ІІ дає підставу чекати, що він має основні властивості. Проте ці властивості в піролу виражені дуже незначною мірою, оскільки р-електрони атома азоту за рахунок р,л-спряження втягнуті в ароматичний секстет і тільки в сильнокислому середовищі він приєднує протон. Приєднання протона до піролу відбувається за рахунок пари електронів атома азоту, яка при цьому вилучається з шести-я-електронної системи. Після такого приєднання протона спряження в пірольному ядрі порушується і ароматичність піролу зникає. В результаті виникає дуже нестійка дієнова система, яка швидко полімеризується з утворенням смолистого продукту, і замість солеутворення відбувається осмолення.
Як уже зазначалося, вільна пара електронів атома азоту в молекулі піролу бере участь в утворенні ароматичного секстету л-електронів. У зв’язку з цим електронна густина на атомі азоту зменшується і тому азот, як високоелектронегативний елемент, сильно притягує електронну пару зв’язку Н. А це приводить до посилення полярності цього зв’язку, підвищення активності (рухливості) атома водню і до появи в піролу кислотних властивостей. Кислотні властивості у піролу дуже слабкі, слабкіші навіть, ніж у фенолу: /<д піролу становить 1,3 • 10~16, Кд фенолу —10~10. Пірол виявляє кислотні властивості в реакціях з лужними металами, з амідами цих металів і з магнійорганічними сполуками. При взаємодії піролу з цими речовинами атом водню зв’язку >К’Н у його молекулі заміщується на метал і утворюються солеподібні
471
М-металічні похідні піролу, які використовують для синтезу заміщених піролу:
Природні сполуки піролу. Пірол і його заміщені досить поширені в тваринному та рослинному світі і мають велике біохімічне значення. Особливо важливими є сполуки, побудовані з чотирьох пірольних ядер, сполучених між собою в а-положеннях метановими (=СН—) групами. Плоский 16-членний цикл, що виникає при цьому, називають порфіном. Ядро порфіну входить до складу гемоглобіну крові, хлорофілу, вітаміну В12, пігментів жовчі та інших речовин.
Порфін. Порфін вперше синтезував у 1935 р. Г. Фішер, нагріваючи 2-піролальдегід з мурашиною кислотою, яка у цій реакції є дегідратуючим реагентом і донором атомів водню:
Порфін
Порфін — темно-червона кристалічна речовина, досить стійка до нагрівання, витримує без розкладання температуру до 300 °С, важко розчиняється в органічних розчинниках, не розчиняється в лугах, але добре розчиняється в кислотах.
Порфін є вищим небензоїдним гетероциклом, який задовольняє вимогам правила ароматичності Хюккеля (див. розд. 28). 16-Член-ний цикл порфіну містить 11 спряжених подвійних зв’язків (22 л-електрони) і чотири пари р-електронів (8р-електронів) атомів азоту. У спряженні порфінбвого ядра беруть участь ЗО л- і р-електронів. Порфін має дуже високу енергію спряження (£с = = 840 кДж • моль-1) і йому, як і іншим ароматичним системам, властиві реакції електрофільного заміщення атомів водню гетгчо-циклу. Так, порфін вступає в реакції сульфування, нітрування, ацилування та інші.
472
У молекулі порфіну атоми водню біля атомів азоту двох п’ятичленних гетероциклів можуть заміщуватись на атоми Ге, М§, Си та ін., які додатково утворюють донорно-акпепторні зв’язки з вільними парами р-електронів атомів азоту двох інших п’ятичленних гетероциклів. Такі заміщені порфіну називають порфіринами. Особливо велике значення мають комплекси порфіринів, зокрема гемін і хлорофіл.
Гемін (гем) — майже чорні голчасті кристали складу С34Н32С1ГеП4О4 — є дихальним пігментом крові. Будову гемоглобіну встановлювали протягом більш як 40 років. Підсумком цих структурних досліджень був його синтез у 1930 р. Г. Фішером. Молекула геміну являє собою комплекс порфіну з Ре2+. Вільний гемін легко окислюється на повітрі до гематину, який має таку ж будову, але містить Ре3+. При дії на гемін хлориду натрію в його склад входить хлорид-іон:
Гемін входить до складу складного білка крові — гемоглобіну, є його небілковою частиною, міститься в червоних кров’яних тільцях і зумовлює забарвлення крові. Гемоглобін — це комплекс геміну з розчинним у воді білком глобіном. На кожну молекулу гемоглобіну припадає 4 атоми заліза. Молекулярна маса гемоглобіну становить 67 000. З цього випливає, що гемоглобін містить близько 4 % геміну і 96 % глобіну (за масою).
Гемоглобін у живих організмах переносить (транспортує) кисень від легенів до тканин.
З залізом гемоглобіну міцніше, ніж кисень, зв’язується оксид вуглецю (II) (чадний газ), молекули якого блокують гемоглобін. Внаслідок цього при наявності в повітрі чадного газу у людей наступає ядуха (чадіння).
Хлорофіл. Зелені частини вищих рослин містять забарвлену речовину хлорофіл, який входить до складу хлорофілових зерен листків і становить від 0,6 до 1,2 % маси сухої речовини. В листках хлорофіл сполучений з білком пластеїном, від якого він легко відділяється при висушуванні. Хлорофіл вперше виділив Я. Берце-ліус у 1837 р. у вигляді темно-зеленої воскоподібної речовини. Хлорофіл складається з двох дещо відмінних за складом і забарвленням речовин —• хлорофілу а 75 %), який має склад С53Н72О5М4М§, і хлорофілу Ь (~25 %), який має склад
\6 М2й - 473
Сб5Н7(А№М£. Хлорофіли а і Ь виділив і розділив у 1906 р. М. С. Цвєт за допомогою розробленого ним хроматографічного методу. Велика заслуга в дослідженні і встановленні структури хлорофілу належить Г. Фішеру і Р. Вудворду, який у 1960 р. здійснив повний синтез хлорофілу.
За структурою хлорофіл являє собою плоский порфіновий цикл, подібний до циклу геміну крові, що свідчить про єдність походження рослинного і тваринного світу:
Н;(С
/?- >СНСН2СН2-
Н3С
СНз СН3
—сн2снсн2сн— -сн2-с=сн-сн2-
— -12.
Залишок фітолу
Хлорофіли а і Ь відрізняються між собою тим, що метальна група першого окислена в другому до альдегідної групи. Від геміну хлорофіл а відрізняється не тільки наявністю в порфіриновому ядрі магнію замість заліза, але і наявністю залишку ненасиченого спирту фітолу, сполученого складноефірним зв’язком з залишком пропіонової кислоти, наявністю двох атомів водню в пірольному ядрі, а також додатковим карбоциклічним ядром.
Хлорофіл бере участь у фотосинтезі (див. розд. 26). Він поглинає квант світла і використовує поглинуту енергію для фотолітач-ного розкладання води.
У молекулі хлорофілу є три асиметричних С-атоми, тому він має сильно виражене ліве обертання площини поляризації світла.
Індол. Молекула індолу складається з двох конденсованих ядер — бензольного і пірольного:
і Н
474
Тому індол можна назвати бензо(о)піролом. Свою назву індол одержав від назви синього барвника індиго, з продуктів розкладання якого його вперше добув А. Байєр.
Індол і його похідні досить поширені в природі. Так, ядро індолу міститься в барвнику індиго, в амінокислотах білків, алкалоїдах, ефірних оліях квітів тощо.
Сполуки індолу містяться в кам’яновугільній смолі, зокрема в її фракції з Ткип = 220—260 °С, в якій їх 3—5 %. Синтетично індол можна добути термічною конденсацією аніліну з ацетиленом (О. Є. Чичибабін, 1915 р.) або конденсацією форміл-ор/ио-толуїдину при наявності щрет-бутилату калію:
Індол — безбарвна кристалічна речовина (Тпл = 52 °С), розчиняється в гарячій воді, має неприємний запах, але в малих концентраціях пахне приємно (запах жасмину), завдяки чому його використовують у парфюмерії.
Індол можна виявити якісно — він забарвлює змочену соляною кислотою соснову скалочку в червоний колір.
Оскільки за структурою індол являє собою конденсовану біциклічну систему бензольного і пірольного ядер, він є біциклічною ароматичною сполукою, в якій поєднуються електронні структури бензолу і піролу. Як видно з молекулярної діаграми індолу, пірольне ядро має більшу електронну густину, а відповідно і більшу реакційну здатність, ніж бензольне. Спряження електронної густини бензольного і пірольного ядер створює підвищену електронну густину в Р-положенні:
-0,023
-о,оз9|^Х-_^-о,і7о -0,0374^^.^+0,001
-0,037 7 +0,379 н
Ефективні заряди на атомах в молекулі індолу
Індол за властивостями подібний до піролу, але внаслідок спряження між л-електронними орбіталями пірольного і бензольного ядер електронна густина в пірольному ядрі індолу стає меншою, ніж у піролу. Тому пірольне ядро в молекулі індолу має меншу реакційну здатність, ніж пірол.
Для індолу характерні реакції електрофільного заміщення (нітрування, галогенування, азосполучення та ін.). Першим у ці реакції вступає багатше електронною густиною пірольне ядро. Причому Х^-реакції для індолу відбуваються у р-положенні пірольного ядра, електронна густина якого вища, ніж у а-положенні.
475
Індол, подібно до піролу, виявляє також слабкі основні властивості. Л'іінеральні кислоти полімеризують і осмоляють індол, ЯК і пірол. Лужні метали заміщують атом водню в ІМН-групі індолу на атом металу (На, К) і утворюють М-металічні похідні індолу.
Велике значення мають кисневмісні похідні індолу:
2-Оксіндол
З-Оксіндол, ^-Індоксил
Ізатин
Найважливішим з цих кисневмісних похідних індолу є ^-індоксил, який є проміжним продуктом промислового синтезу барвника індиго. Індоксил добувають такими методами.
1. Конденсацією аніліну з хлороцтовою кислотою:
+ С1-СН2-СХ
І\’Н2 Ч°н
2. Конденсацією аніліну з оксидом етилену:
Індоксил (Тпл = 85°С) у вигляді глікозиду (індикану) міститься в рослинах виду Іпсіі§оїегае, а також у сечі у вигляді сульфату. Індоксил існує в двох таутомерних формах — кетонній (І) і еноль-ній (II):
Енольна форма індоксилу легко розчиняється в лугах.
Індиго. Синій барвник індиго був відомий ще в стародавньому Єгипті та Індії. Його добували з рослин Іпс1і£оГегае, які ростуть в Індії і містять до2 % глікозиду індикану. При екстрагуванні подрібнених частин цих рослин водою індикан під впливом ферменту і ндоксилази гідролізується з утворенням глюкози та індоксилу, який відразу ж окислюється киснем повітря до індиго:
476
Індиго
У кінці XIX ст. були розроблені промислові методи синтезу індиго, які повністю витіснили його добування з рослин. У наш час відомо понад тридцять методів синтезу індиго, з яких найважливіше практичне значення мають методи Г. Геймана (1897 р.). Ці методи зводяться до добування індоксилу, який потім окислюють в індиго.
Дослідження будови і властивостей індиго показали, що цей барвник як у твердому стані, так і в розчинах має стійку транс-конфігурацію, в якій між атомами водню пірольних атомів азоту і атомами кисню карбонільних груп існують водневі зв’язки:
Індиго являє собою темно-синій порошок (7ПЛ — 390—392 °С), який при 300 °С сублімується з утворенням червоної пари. Цей барвник не розчиняється у воді, лугах, розбавлених кислотах і звичайних органічних розчинниках. При відновленні синього індиго гідросульфітом натрію або цинком у лужному середовищі утворюється безбарвна сполука — біле індиго (лейкоіндиго), якемаєенольну будову і тому розчиняється в лугах. У техніці лужний розчин білого індиго називають «кубом», оскільки відновлення індиго здійснюють у дерев’яних апаратах, які мають форму куба. Тканину, яку потрібно пофарбувати, опускають у куб, де вона змочується і сполучається з білим індиго. Потім цю тканину вміщують в окислювальну ванну або вивішують на повітрі, де відбува-єься окислення лейкоіндиго у барвник. Такий метод фарбування називають кубовим. Процес фарбування барвником індиго можна зоб-
477
разити такою схемою послідовних реакцій:
[О]
-н2’о
2Н
(2п + №ОН)
Біле індиго (куб на тканині)
о
Синє індиго (на тканині)
Носієм забарвлення(хромофором) індиго є спряжена система подвійних зв’язків, в яку входять також подвійні зв’язки бензольних ядер і карбонільних груп.
Біологічне значення похідних індолу. Ядро індолу входить до складу багатьох біологічно активних речовин, зокрема 0-індоліл-оцтової кислоти, триптофану, серотоніну та інших:
^-Індолілоцтова кислота, або гетероауксин, міститься в рослинах у дуже малих кількостях, але вона має надзвичайно великий вплив на їх ріст, оскільки є гормоном росту. У великих концентраціях гетероауксин діє на рослини як сильна отрута.
Триптофан (Р-індоліл-а-амінопропіонова кислота) є однією з найважливіших незамінних гетероциклічних амінокислот, необхідних для життєдіяльності організму людини і тварин. Входить до складу білків.
Серотонін, або 5-окситриптамін, — порівняно недавно відкритий гормон, який підвищує тиск крові. Серотонін, очевидно, відіграє певну роль у розвитку гіпертонії, а разом з тим регулює і психічну діяльність.
Розділ 41. ШЕСТИЧЛЕННІ ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ З ОДНИМ ГЕТЕРОАТОМОМ
Найважливішим представником шестичленних гетероциклічних сполук з одним гетероатомом є піридин. Структурну формулу піридину запропонував у 1869 р. В. Кернер по аналогії з структу
478
рою бензолу, замінивши у формулі останнього одну =СН-групу на атом тривалентного азоту:
не.
Наявність гетероатома азоту приводить до нерівномірного розподілу л-електронної орбіталі в піридиновому циклі і до нерівноцінності положень в його молекулі. Положення 2,5 називають відповідно а і а', положення 3,5 — (3, |3', положення 4 — у. Тому монозаміщені піридину, на відміну від монозаміщених бензолу, існують у вигляді трьох ізомерів — а-, р- і у-. Наприклад:
2-Метшшіридин, ОС.-метилпіридин, СХ-ПІКОЛІН
З-Метилпіридин, й-метилпіридин, ґ Й-ПІКОЛІН
4-Метилпіридин, у-метилпіридин, у-піколін
Піридин і його гомологи називають піридиновими основами. Піридинові основи містяться в продуктах сухої прегонки кам’яного вугілля, торфу, деревини, кісток тварин та інших органічних матеріалів. Піридин вперше був виділений з кісткової олії (Т. Андер-сон, 1849 р.), яка утворюється при піролізі кісток. У 1854 р. його виділили з кам’яновугільної смоли. В наш час піридин і його гомологи добувають з кам’яновугільної смоли і синтетично, оскільки вміст піридинових основ у кам’яновугільній смолі незначний (менше 0,1 %). Піридин, наприклад, добувають піролізом суміші ацетилену з синильною кислотою:
Гомологи піридину синтезують конденсацією карбонільних сполук з аміаком. Так, при нагріванні акролеїну з аміаком утворюється [3-піколін, а з оцтового альдегіду і аміаку — 2-метпл-5-етил-піридин:
ХО
2Н2С = СН -СХ+МН3-------
чн -2Н2О
Акролеїн
хо
4 Н3С -СХ + МНз чн
479
Будова піридину. Структур а піридину дуже подібна доструктури бензолу. Встановлено, що молекула піридину, як і молекула бензолу, плоска. Проте піридинове ядро, на відміну від бензольного, не є рівностороннім шестикутником, тому що довжина зв’язків С—С (0,139 нм) і С—N (0,134 нм) у молекулі піридину не однакова. Крім того, азот більш електронегативний, ніж вуглець, і молекула піридину, на відміну від молекули бензолу, полярна (р = 2,260). Вектор дипольного моменту напрямлений у молекулі піридину ВІД ядра до атома азоту:
Електронна будова піридину також подібна до електронної будови бензолу. В молекулі піридину, як і в молекулі бензолу, існує єдина делокалізована шести-л-електронна система, яку утворюють
Рис. 76. Розподіл я-елсктронної густини (а) і схема перекривання р-орбіталей (б) в молекулі піридину
п’ять р-електронів п’яти вуглецевих атомів і один р-електрон атома азоту. Електронна густина нього секстету р-електронів розміщена над і під площиною циклу молекули піридину (рис. 76). Підтвердженням такої електронної будови піридину є енергія спряження його молекули: Ес -- 116,7кДж - моль-1. Отже, за будовою молекула піридину відповідає вимогам правила ароматичності Хюккеля (див. розд. 29).
Проте у зв’язку з більшою електронегативністю азоту відносно вуглецю в молекулі піридину, на відміну від бензолу, електронна густина шестп-л-електронної системи розміщена в циклі нерівномірно і зміщена до атома азоту. Крім того, на атомі азоту в піриди-новомущщслі міститься вільна пара електронів (рис. 76). Виходячи з вищесказаного, молекулу піридину можна розглядати як гібрид структур Іа—Ід і зобразити структурною формулою І, подібною до структурної формули бензолу, в якій коло всередині циклу означає я-електронний секстет:
480
Фізичні і хімічні властивості. Піридин і його найближчі гомологи— безбарвні рідини з неприємним запахом, розчинні у воді, досить отруйні. Температура кипіння піридину дорівнює 115,6°С, тобто дещо вища, ніж бензолу.
УФ-спектри піридину і бензолу дуже подібні. Так, бензол має смуги поглинання при 179 нм (інтенсивна), 200 нм (середня) і 255 нм (слабка), а піридин має максимуми поглинання при 170 нм (інтенсивний), 200 і 250 нм.
Піридин, як і бензол, має замкнену електронну систему із секстету л-електронів. Завдяки ньому він має ароматичні властивості і вступає, як і бензол, у реакції заміщення і приєднання.
Атом азоту в молекулі піридину, на відміну від піролу, виявляє негативні як індукційний, так і мезомерний ефекти (—1-і —М-ефекти). Підтвердженням цього є більший дипольний момент піридину (2,26 П) порівняно з дипольним моментом піперидину (1,170):
Внаслідок —І- і —/И-ефектів електронна густина в піридиновому ядрі стає меншою, ніж у бензольному. Тому піридин значно важче, ніж бензол, вступає в реакції електрофільного заміщення і, на відміну від бензолу, вступає також у реакції нуклеофільного заміщення.
Реакції заміщення. Атоми водню піридинового ядра можуть заміщуватись як електрофільними, так і нуклеофільними частинками.
Електрофільне заміщення.В піридиновому ядрі розподіл електронної густини під впливом гетероатома азоту стає нерівномірним, і в а-, а'- і у-положеннях електронна густина знижується. Це відповідає значенням ефективних зарядів на атомах в молекулі піридину, обчислених методом МО:
481
Тому реакції електрофільного заміщення для піридину відбуваються переважно в Р-положення.
Піридин досить важко і в жорстких умовах вступає в Хг-реакції, а в реакцію ацилування за методом Фріделя — Крафтса зовсім не вступає (подібно до нітробензолу). Низька активність піридинового ядра до Хг-реакцій зумовлена не тільки —І- і —7И-сфектами атома азоту, а також і тим, що при Х^-реакціях електрофіл £+ спочатку утворює з азотом гетероциклу М-комплекс. В результаті на атомі азоту виникає позитивний заряд. А це приводить до ще сильнішого зміщення л-електронної густини ароматичного секстету піридинового ядра до атома азоту і до ще більшого пониження активності цього ядра до Хг-реакцій:
Аналогічна інактивація піридинового циклу відбувається і при наявності сильних кислот, кислот Льюїса, які також атакують атом азоту піридину і утворюють на ньому електропозитивний центр.
Піридин бромується тільки при сильному нагріванні і утворює при цьому р-бромпіридин. Сульфування піридину також здійснюється під час тривалого нагрівання його з 20 %-м олеумом при наявності каталізатора (солі Н§2+). При цьому утворюється Р-піридин-сульфокислота (вихід ~70%). Нітрування піридину відбувається під час його нагрівання з сумішшю селітри з концентрованою Н28О4 і приводить до утворення з невисоким виходом (~20 %) Р-нітропіридину, який при відновленні може перетворюватись у Р-амінопіридин:
Реакція не відбувається
З-Нітро- З-Аміно-
піридин піридин
4Ь>
Атака електрофільного реагенту Е+ в положення 3 піридинового ядра приводить до утворення іона, який являє собою гібрид структур І, II і III:
Атака в положення 4 піридинового ядра приводила б до утворення нестійкого іона, який був би гібридом структур IV, V і VI:
VI
IV
Особливо нестійкою є структура VI, оскільки у неї електронегативний атом азоту має тільки шість електронів. Тому атака в положення 4 відбувається надзвичайно повільно. Внаслідок цього 5н-реакції піридину відбуваються переважно в положення 3. Оскільки атом азоту відтягує електрони на себе, то структури І—III менш стійкі, ніж аналогічні структури, які виникають при реакціях бензольного ядра. Внаслідок цього піридин набагато повільніше, ніж бензол, вступає в 8е -реакції.
Нуклеофільне заміщення. Зменшення електронної густини піридинового ядра полегшує нуклеофільне заміщення в ньому .Тому піридин, на відміну від бензолу, досить легко реагує з нук-леофільними реагентами. Нуклеофільне заміщення (Зуу-реакіїії) здійснюється в положення 2,4 і 6 піридинового ядра і ^-положення), в яких електронна густина найменша. Реакційна здатність піридину в ^-реакціях настільки висока, що під час їх перебігу в піридиновому ядрі заміщується гідрид-аніон :Н_. Найважли-вішими8дг-реакціямипіридинуєйоговзаємодіязамідом натрію і з порошкоподібним КОН.
Піридин при нагріванні з амідом натрію утворює сс-амінопіри-дин (7пл — 57 °С, реакція Чичибабіна):
МаМНг
-н2
Реакція з амідом натрію найлегше відбувається у рідкому аміаку. Механізм цієї реакції можна зобразити такою схемою:
+ :іїн
+ : н~.
МИ 2
Нуклеофільпа атака в положення 2 приводить до утворення
карбаніона, який являє собою гібрид структур І, II і III:
Особливо енергетично вигідною є структура III, оскільки у неї негативний заряд локалізований на найбільш електронегативному атомі — атомі азоту.
При нагріванні піридину з порошкоподібним КОН при 250— 300 °С утворюється а-оксипіридин (О. Є. Чичибабін):
Отже, висока електронегативність атома азоту зумовлює малу реакційну здатність піридину в реакціях електрофільного заміщення і високу активність у реакціях нуклеофільного заміщення.
Реакції приєднання. Піридин, подібно до бензолу, може приєднувати водень. Проте, на відміну від бензолу, він гідрується воднем у момент виділення (гідрування за методом О. М. Вишнеград-ського) і перетворюється при цьому на гексагідропіридин (піпери-дин, Ткип = 106 °С):
І н
Піперидин — рідина з характерним запахом, виявляє сильні основні властивості (/\сси = 1,58 • 10~3).
Реакції окислення. Піридинове ядро, подібно до бензольного, стійке до окислення. Піридин, як і бензол, не знебарвлює розчин КМпО4, а (3-піколін, подібно до толуолу (див. розд. 29), окислюється КМпО4 до 0-піридинкарбонової, або нікотинової,, кислоти:
При окисленні піридину надоцтовою кислотою (суміш Н2О2 + + СН3СООН) відбувається приєднання кисню до атома азоту за рахунок вільної пари електронів останнього. В результаті утворюється кристалічний КГ-оксппіридин (7ПЛ = 67 °С):
+ НзС-С-о-о-н-------- НзС-С-О-Н .
О о
М-Оксипіридин
484
ЬІ-Оксипіридин, на відміну від піридину, значно активніше вступає в реакції електрофільного заміщення, що використовують при добуванні, наприклад, у-нітропіридину. З цією метою М-оксипіри-дин нітрують сумішшю концентрованих азотної і сірчаної кислот і добувають у-нітро-М-оксипіридин, який подальшою взаємодією з РС13 перетворюють на у-нітропіридин:
НМО.„ Н28О<
-н2о
До М-оксипіридину подібний за будовою піридинсульфотриок-сид, який утворюється при взаємодії піридину з оксидом сірки (VI):
Піридинсульфотриоксйд-
О. П. Терентьєв (1946—1949 рр.) запропонував використовувати пірпдинсульфотриоксид як ефективний м’який сульфуючий реагент для сульфування органічних речовин (див. розд. 40).
Основні властивості. Піридин завдяки вільній парі електронів на атомі азоту виявляє слабкі основні властивості. Константа основності піридину становить 1,9 • 10~и. Основні властивості піридину сильніші, ніж аніліну (Ко аніліну дорівнює 4,0 • 10“10), але набагато слабкіші, ніж піперидину (гексагідропіридину) і аліфатичних амінів (Аф піперидину становить 1,33 • 10"3 , Ко аліфатичних амінів — ~10~4).
Послаблення основних властивостей піридину порівняно з пі-перидином і аліфатичними амінами пов’язане з тим, що у піридину пара електронів атома азоту, яка зумовлює ці властивості, знаходиться на 5/Аорбіталі і утримується азотом сильніше, ніж пара електронів атома азоту у піперидину і аліфатичних амінів, яка в цих сполуках перебуває на 8р3-орбіталі.
Послаблення основних властивостей аніліну викликане р,п-спряженням вільної пари електронів атома азоту з я-електронною орбіталлю бензольного ядра (див. розд. 36). У піридину вільна пара електронів атома азоту в такого виду спряження не вступає. Тому основні властивості піридину сильніші, ніж аніліну. Разом з тим основні властивості піридину значно сильніші, ніж піролу (Ко піролу дорівнює 2,5 • І0~и), оскільки -у піролу пара електронів атома азоту спряжена з я-електронами С=С-зв’язків циклу і бере участь в утворенні секстету л-’електронів пірольного ядра. Внаслідок цього в молекулі піролу на атомі азоту немає такої електронної пари, як у піридину, і пірол, приєднуючи протон кислоти, одночасно втрачає ароматичність ядра (див. розд. 40).
485
Основні властивості піридин виявляє в реакції з водою, при взаємодії з кислотами, такими, як соляна, бромистоводнева, сірчана та ін., з якими він легко утворює солі. З водою піридин утворює гідроксид піридинію. У зв’язку з цим піридин, на відміну від бензолу, легко розчиняється у воді. Водний розчин піридину спричинює посиніння червоного лакмусового папірця, а при добавлянні до цього розчину солей заліза випадає осад гідроксиду заліза, що також підтверджує основний характер водного розчину піридину:
+ н+он
Взаємодія піридину з кислотами, наприклад з соляною, відбувається за такою схемою:
Хлорид піридинію, солянокислий піридин
У процесі такої взаємодії протон кислоти, як і протон води, утворює з атомом азоту за рахунок його вільної пари електронів координаційний зв’язок, не порушуючи ароматичного секстету л-електронів піридинового ядра. Аналогічні сполуки з водою і кислотами утворюють і гомологи піридину. Піридин і його гомологи утворюють солі також з пікриновою кислотою, які називають пікратами. Пікрати використовують для ідентифікації піридину і сполук, що містять піридиновий цикл.
Піридин, як третинний амін, легко приєднує галогеналкіли і утворює солі алкілпіридинію, які при нагріванні перегруповуються в солі а- або у-алкілпіридинію. Наприклад:
Йодид Йодид
-етил піри ди н і ю 2-етил піридинію
Реакцію піридину з галоГеналкілами використовують для добування деяких гомологів піридину.
Біологічне і фармакологічне значення похідних піридину. Багато похідних піридину є фізіологічно активними речовинами. Наприклад, вітамін РР (Вв), або нікотинамід, є амідом Р-піридин-карбоїювої (нікотинової) кислоти. Цей вітамін запобігає розвитку
486
пелагри і є складовою частиною небілкових компонентів ряду ферментів, що каталізують окислювально-відновні процеси в організмах. Похідну цієї ж кислоти — її діетиламід під назвою кордіамін використовують як засіб, що збуджує серцеву діяльність:
Нікотинова Амід нікотинової Діетиламід
кислота кислоти нікотинової кислоти
До похідних піридину належать також вітаміни групи Вв:
Піридоксол Піридоксаль ГІІридоксамін
З них біологічну активність мають піридоксаль та піридоксамін. Ефір піридоксалю з фосфорною кислотою входить до складу ряду ферментів, які каталізують перебіг окремих груп реакцій перетворення амінокислот в організмі, а також біосинтез пуринових основ та гемоглобіну.
Ядро гідрованого піридину (піперидину) входить до складу деяких алкалоїдів, зокрема алкалоїду перцю — піп ери ну (від лат. рурег — перець), алкалоїду рослини болиголова — коніїну та інших.
Розділ 42. ГЕТЕРОЦИКЛИ З КІЛЬКОМА ГЕТЕРОАТОМАМИ
П’ятичленні гетероцикли з двома гетероатомами. З п’ятичленних гетероциклів з двома гетероатомами розглянемо такі, в яких обидва або хоч один гетероатом є атомом азоту. Ці гетероцикли називають азолами. Найважливішими з них є тіазол, піразол, імідазол і оксазол:
Тіазол Піразол Імідазол Оксазол
Всі азоли виявляють ароматичні і слабколужні властивості. Вони входять до складу багатьох лікарських речовин, вітамінів, антибіотиків. Так, ядро тіазолу входить до складу вітаміну Вь антибіотика пеніциліну, сульфамідного препарату норсульфазолу:
487
Найбільш відомі пеніциліни відрізняються тільки природою замісника 7?: —СН2—СеН5 — пеніцилін 6 . (бензилпеніцилін), —СН2—О—СвН5 — пеніцилін V (фенокснметилпеніцилін). Пеніцилін виділений з культури плісеневих грибів Репісіїїіиш поіаіит і використовується при лікуванні бактеріальних інфекційних захворювань.
Імідазольне ядро входить до складу пуринових основ нуклеїнових кислот, деяких алкалоїдів, наприклад пілокарпіну, вітаміну Н (біотину), а також ряду синтетичних фармацевтичних препаратів (дибазолу), які застосовуються при лікуванні гіпертонії, нервових захворювань:
О п
С н\х Х.\'Н
не----сн
І І
Н2СХ ^СН -СНв- СН2- СН2 - СІ і2 - соон
Вітамін Н(біотин)
Н 1+ _ КСІ
уС-СН2-С6Н5 .
N
І н Дибазол, хлоргідрат 2-бензилбенз1м1дазолу
Шестичленні гетероцикли з двома гетероатомами. З цієї групи сполук винятково важливе значення має піримідин, який являє собою безбарвну речовину (7ПЛ = 22 °С, Ткип = 124 °С), добре розчинну у воді. Піримідин у складі своєї молекули містить два атоми азоту, які розміщені один відносно другого в положеннях 1 і 3:
СН
^сн
НС^З^СН дг *
Піримідин — ароматична гетероциклічна сполука з секстетом л-електронів, два з яких є р-електронами атомів азоту.
Піримідин має велике біологічне значення, оскільки бере участь у побудові небілкових компонентів ряду ферментів, вітаміну Вг, а також нуклеїнових кислот — речовин, які відповідають за збереження спадкових ознак і передачу їх за потомством. У складі нуклеїнових кислот піримідин знаходиться у вигляді оксн- та аміно-
488
похідних — піримідинових основ: цитозину, урацилу і тпміну, які можуть існувати як у лактамній, так і в лактимній формі (лак-тим-лактамна таутомерія):
.Лактамна форма Лактнмна форма
Цитозин
Лактамна форма
Лактнмна форма
Н^^С-СН, ХС. .СН
С)' N
і
Н
Лактамна форма
Урацил
Лактнмна форма
Тимін
Конденсовані системи гетероциклів. До конденсованих гетероциклів належать такі гетероциклічні системи, які складаються щонайменше з двох гетероциклічних ядер, сполучених між собою кількома спільними для обох ядер атомами. Серед таких сполук особливе місце посідає пурин, відкритий у 1848 р. німецьким хіміком Е. Фішером:
сн
8СН
І н
Пурин — ароматична гетероциклічна сполука, молекула якої складається з двох гетероциклів — піримідипового та імідазоль-ного, в яких два атоми вуглецю (С4 і С8) є спільними. Делокалізо-вана л-електронна система у його молекулі містить десять спряжених р-електронів (4л + 2, п = 2) два з яких є електронами атома азоту в положенні 9. Пурин (Т„л = 217 °С), як і піримідин, має основні властивості і утворює ряд похідних, серед яких слід зазначити аденін (6-амінопурин) і гуанін (2-аміно-6-оксипурин). Аденін і гуанін називають пуриновими основами. Гуанін може існувати в лактамній і лактимній формах (лактам-лактимна таутомерія):
Аденін Лактамна форма Лактнмна форма
Гуанін
489
Аденін і гуанін разом з піримідиновими основами цитозином, урацилом, тиміном є обов’язковими складовими частинами нуклеїнових кислот. Поряд з цим аденін бере участь у побудові небіл-кових компонентів ряду ферментів, а також аденозинтрифосфорної кислоти (АТФ) — переносника енергії в біохімічних реакціях і важливого фосфор плюючого реагенту.
Деякі похідні пурину, наприклад 6-меркаптопурнн, який затримує і зменшує поділ клітин, застосовують у медицині для лікування гострих і підгострих лейкозів.
Окснпохідні пурину, такі як ксантин (2,6-діоксипурин), сечова кислота (2,6,8-триоксипурин), можуть існувати у двох таутомер-них формах:
Лактамна форма Лактимна формі
Ксантин
Лактамна форма Лактимна форма
— Сечова кислота
Ксантин трапляється в рослинах. Так, він разом з кофеїном міститься в чаї. У тваринних організмах ксантин знаходиться в крові, печінці, сечі.
Сечова кислота виділяється з сечею людей і деяких тварин.
Велике значення в медичній практиці мають метильовані ксан-тини — теофілін, теобромін і кофеїн:
Теофілін Теобромін Кофеїн
Кофеїн (Тип — 236 °С) збуджує різні відділи центральної нервової системи, підвищує працездатність поперечно-смугастих м’язів, є сильним сечогінним засобом, підвищує збудливість і силу серцевих м’язів. Кофеїн міститься в листках чаю (~5 %), в зернах кофе 1 %).
Теобромін відкрив О. А. Воскресенський у 1842 р. Він міститься в бобах какао (~1,8 %) і в невеликій кількості — в листках чаю.
49 ц
Його застосовують при спазмах судин серця, набряках серцевого і ниркового походження.
Теофілін міститься в листках чаю. Його застосовують як сечогінний препарат.
Контрольні запитання і вправи
1. Які органічні сполуки називаються гетероциклічними? Як їх класифікують?
2. Порівняйте електронну будову та хімічні властивості фурану, піролу, тіофену. Поясніть, у чому полягає подібність і відмінність у властивостях цих речовин.
3. Поясніть, у чому полягає подібність і відмінність в електронній будові та у властивостях піридину і бензолу.
4. Які речовини утворюються під час взаємодії піридину при нагріванні з бромом, азотною і сірчаною кислотами?
5. Як якісно можна відрізнити^піридин від бензолу?
Розділ 43. НЕБЕНЗОЇДН9 АРОМАТИЧНІ СПОЛУКИ
До небензоїдних ароматичних сполук відносять плоскі циклічні сполуки, які не містять бензольних ядер, але задовольняють квантово-механічним умовам ароматичності (правилу Хюккеля) — мають 4п 4- 2 зт-електронів у циклі — і виявляють ароматичні властивості (див. розд. 28).
Діамагнітні властивості ароматичних систем. Речовини за відношенням до зовнішнього магнітного поля можуть бути діамагнітними і парамагнітними. У діамагнітних речовин атоми при відсутності магнітного поля не мають магнітного моменту, а під дією магнітного поля ці речовини намагнічуються і діамагнітна намагніченість з'авжди напрямлена назустріч зовнішньому полю (див. розд. 28, рис. 68). Тому діамагнітна намагніченість таких речовин і їх діамагнітна сприйнятливість (у) завжди мають від’ємний знак.
Парамагнітні речовини намагнічуються під дією зовнішнього магнітного поля так, що власні магнітні моменти їх атомів орієнтуються переважно в напрямку поля. В результаті такі речовини набувають сумарного магнітного моменту, який пропорційний напруженості поля. Магнітна сприйнятливість таких речовин має додатний знак.
Бензол та інші ароматичні системи діамагнітні, вони не мають власного магнітного моменту (див. розд. 28).
Для ароматичних сполук спостерігається значна різниця Д (екзальтація) між експериментально встановленою і розрахованою магнітною сприйнятливістю 7. Екзальтація діамагнітної сприйнятливості може бути ознакою ароматичності сполуки (табл. 37).
Небензоїдні ароматичні системи можна класифікувати за кількістю «ароматичних» зт-електронів.
Ароматичні системи з двома л-електронами. Найпростішою не-бензоїдною ароматичною карбоциклічною системою з двома зт-елек-тронами (4« + 2, п 0) е циклопропенілій-катіон, у якого обидва
•йи
Таблиця 37. Екзальтація діамагнітної сприйнятливості як характеристика ароматичності
Сполука Магнітна сприйнятливість Екзальтація Д Екзальтація Д
експериментальна розрахована Кількість бензольних циклів
Бензол —55,6 -36,9 18,7 18,7
Нафталін —91,9 —55,7 36,2 18,1
Антрацен — 129,4 —74,5 54,9 18,3
Фенантрен — 127,9 —74,5 53,4 17,8
Дифеніл — 102,9 —68,0 34,9 17,5
Азулен —91 —55,7 35,3 —
Трополон -61 —45,8 15,2 —
Тропон —54 —41,2 12,8 —
л-електрони знаходяться на єдиній зв’язуючій молекулярній орбіталі в полі трьох копланарно розміщених атомів вуглецю. Будову циклопропенілій-катіона краще описувати структурою І, а не формулою II з л-зв’язком:
1 п пі
Солі незаміщеного катіона циклопропенілію внаслідок його нестійкості поки що не добуті. Стійкими є похідні цієї ароматичної системи, наприклад, добутий бромід трифенілциклопропенілію (III).
Ароматичні системи з шістьма л-електронами. До таких систем належать сполуки, які містять циклопентадієніл-аніон, тропілій-катіон, а також п’ятичленні гетероцикли (див. розд. 40).
Циклопентадієніл-аніон. Небензоїдні ароматичні сполуки з п’ятичленним циклопентадієніл-аніоном С5ІЦ у вигляді солі С5Н5К+ були відомі хімікам ще в 1901 р. Сполуки з таким аніоном можна добути прямою дією металів або реактивів Гріньяра на циклопентадієн:
Негативний заряд у цій сполуці, іншими словами пара електродів, утворює з електронами двох подвійних зв’язків секстет спряжених л-електронів. які зумовлюють ароматичність п’ятичленного ядра:
492
В циклопентадієніл-аніоні, як і в бензолі, шість л-електронів розміщені на трьох зв’язуючих молекулярних орбіталях, а копланарно розміщені атоми вуглецю рівноцінні, довжини зв’язків між ними однакові (0,143 нм). Цей аніон енергетично стабільний. Його енергія спряження становить Ес = 167 кДж • моль'1 і близька до Ес бензолу (Ес — 150 кДж • моль'1). Ароматичність циклопептадіє-ніл-аніона виявляється в реакціях електрофільного заміщення, в які вступають сполуки з таким аніоном.
Ф е р о ц е н. Найважливішим представником небензоїдних ароматичних сполук, які містять ядра циклопеитадієніл-аніона, є синтезоване вперше в 1951 р. Т. Кілі і П. Паусон дициклопентадієніл-залізо, яке за пропозицією Р. Вудворда було назване фероценом. Фероцен добувають з циклопентадієну і заліза при 300 °С або з цик-лопентадієн-1,3-іл-5-магнійгалогенідів і солей заліза (II):
Фероцен
Фероцен, як встановив Р. Вудворд (1952 р.), має структуру «сендвіча». У молекулі фероцену рівні між собою всі Ре—С- (0,203нм), С—С- (0,143 нм) і С—Н- (0,1 нм) зв’язки. Ця сполука утворюється за рахунок перекривання зв’язуючих л-МО циклопентадієніл-аніо-на з вільними Згі-АО іона Ре2+. Атом заліза знаходиться на однакових віддалях від усіх атомів вуглецю обох п’ятичленних ядер.
Для фероцену характерна чітко виражена ароматичність, яка в нього вища, ніж у бензолу. Фероцен і його похідні вступають у типові реакції електрофільного заміщення атомів водню циклопента-дієнільних кілець: сульфування, алкілування, ацилування тощо. Слід зазначити, що молекулафероцену руйнується при гідруванні і дії галогенів, а окислювачі перетворюють його на ферициній-катіон з Ре3+.
Семичленні небензоїдні карбоцикли. Тропілій-катіон. Семичлен-ний вуглеводневий цикл з секстетом л-електронів і позитивним зарядом С7Щ називається тропілій-катіоном (циклогептатрієнілій-катіоном). Формально тропілій-катіон можна розглядати як тро-піліден (циклогептатрієп-1,3,5), від якого відщеплений атом водню в парою електронів:
Тропіліден
493
Солі тропілію, наприклад бромистий тропілій, відомі ще з 1891 р. Хлористий тропілій, який утворюється особливо легко і з високими виходами, добувають взаємодією тропілідену з РС15:
||^СН2 + РСІ5
СІ + РСІ3 + неї
Тропілій-катіои — один з рідкісних випадків стійкості карб-катіонів. На відміну від циклопентадієніл-аніона, тропілій-ка-тіон не вступає в реакції електрофільного заміщення, але легко вступає в реакції з нуклеофільними реагентами і утворює при цьому похідні тропілідену, найважливішими з яких є тропон і тро-полон. Тропон (ТККП — 113 °С при 15 мм) можна добути гідролізом бромтропілій-катіона:
Н2О
-2НВг
Тропон належить до класу кетонів. Проте він не реагує з гідро-ксиламіном, семікарбазидом і відрізняється від інших кетонів значно більшим дипольним моментом (р = 4,3 В). Ці особливі властивості тропону пов’язані з можливістю утворення в ньому секстету л-електронів. Тому структуру тропону краще всього відображає мезомерна форма II.
Трополон (2-оксициклогептатрієн-2,4,6-он-1) утворюється під час окислення тропілідену:
0И2
КМпО4
Трополон — кристалічна речовина (ТШІ = 49 °С), добре розчинна у воді. Цикл молекули трополону має плоску структуру з однаковою довжиною всіх зв’язків С—С (0,139 нм). Енергія спряження трополону становить 163 кДж/моль. Все це узгоджується з ароматичними властивостями цієї речовини.
За хімічними властивостями трополон подібний до фенолу. Він, як і фенол, дає характерне забарвлення з ЕеС13, вступає в реакції електрофільного заміщення. Однак кислотні властивості трополону виражені сильніше, ніж фенолу (рЛ'д трополону становить 6,7; р/(д фенолу — 10). Ароматичність трополону, як і тропону, здійснюється за рахунок поляризації карбонільної групи і утворення шестиелектронної тропілієвої системи:
Ароматичні системи з кількістю л-електронів більше шести.
А з у л е н. Азулен, або біцикло-[5,3,0]-декапентаєн — кристалічна речовина синього кольору (Тт = 99 °С). Молекула азулену складається з конденсованих п’яти- і семичленного циклів, які містять п’ять спряжених подвійних зв’язків, 10л-електронів (по одному р-електрону від кожного з 10 атомів вуглецю). У зв’язку з намаганням молекули азулену утворити секстет електронів у кожному кільці, один електрон семичленного кільця має здатність переходити до п’ятичленного. В результаті виникає біполярна структура, в якій частково поділені ароматичні секстети є в кожному кільці (два електрони — спільні для двох кілець). Квантово-механічні розрахунки свідчать, що п’ятичленне кільце має підвищену, а семичленне кільце — понижену електронну густину:
аоо
1,173 0,855
ОҐЙА0-986
1 027/ 0,870 • 0,986
1,173 0,855
Семичленне кільце азулену стабілізоване л-електронним секстетом, тоді як у п’ятичленному кільці електронна густина на першому і третьому атомах вуглецю підвищена. Такий розподіл електронної густини в молекулі приводить до появи в ній дипольного моменту (р азулену дорівнює 1,0б).
Азулен задовольняє вимогам правила ароматичності Хюккеля і має ароматичні властивості. Енергія спряження азулену становить 188,4 кДж • моль-1. Ароматичний характер азулену виявляється в схильності вступати в реакції електрофільного заміщення в п’ятичленному кільці. Азулен у таких реакціях значно активніший, ніж його ізомер нафталін. Перший замісник в 5е-реакціях завжди вступає в положення 1, а наступний — в положення 3. Крім того, азулен вступає також у реакції нуклеофільногозаміщен-ня, які відбуваються з участю семичленного кільця.
Аннулени. Циклічні системи з великою кількістю спряжених подвійних зв’язків називають аннуленами. Аннулени, які задовольняють вимогам правила ароматичності Хюккеля, виявляють ароматичні властивості. Прикладом такого аннулену є цикло-октадеканоєн (18-аннулен), молекула якого майже плоска і має 18 л-електронів:
495
До макроциклічних небензоїдннх ароматичних систем належать також порфірини (див. розд. 40).
Антиароматичні системи. Циклічні ненасичені сполуки з кількістю 4/2 л-електронів, які не задовольняють вимогам правила ароматичності Хюккеля, називають антиароматичними. Антиароматичні сполуки мають властивості, протилежні властивостям ароматичних сполук. Прикладом сполук з антиароматичними властивостями є циклобутадієн, циклооктатетраєн та ін. Антиароматичні системи, в яких відбувається делокалізація л-електронів у циклі, нестійкі. У зв’язку з цим циклобутадієн не добутий у вільному стані. В момент утворення він димеризується, виявляючи властивості спряженого дієну і дієнофілу:
За розрахунками молекула циклобутадієну прямокутна і має ізольовані подвійні зв’язки, довжина яких відповідає довжині
0,151
зв’язків у молекулах етиленових вуглеводнів:,
Антиароматичні властивості має також і циклооктатетраєн-1,3,5,7 (див. розд. 28).
СПИСОК РЕКОМЕНДОВАНОЇ ЛІТЕРАТУРИ
1. Артеменко А. И. Органическая химия.— М. : Вьісш. шк., 1980.— 44С с.
2. Беккер Г. Введение в злектоонную теорию органических реакций.— М. : Мир, 1977,— 658 с.
3. Гауптман 3., Грефе Ю., Ремане X. Оргаиііческая химия.— М. : Химия, 1979.— 832 с.
4. Днепровский А. С., Темникова Т. И. Теоретические основи органиче-ской химии.— Л. : Химия, 1979.— 520 с.
5. КазицинаЛ. А., КуплетскаяН. Б. Применение УФ-, ИК- и ЯМР-спект-роскопии в органической химии,— М. : Внсш. шк., 1971.— 264 с.
6. Дери Ф., Сандберг Р. Углубленньїй курс органической химии : В 2 кн.— М. : Химия, 1981,— Кн.1 — 2.
7. Моррисон Р., Бойд Р. Органическая химия.— М.: Мир, 1974,— 1132 с.
8. Несмеяное А. Н., Несмеянов Н. А. Начала органической химии : В 2 кн.— М. ; Л. : Химия, 1969— 1970.— Кн. 1—2.
9. Перекалин В. В., Зонис С. А. Органическая химия.— М. : Просвещение, 1982.— 576 с.
10. Петров А. А., Бальян X. В., Трощенко А. Т. Органическая химия.— М. : Вьісш. шк., 1981.— 592 с.
11. Робертс Дж., Кассерио М. Основи органической химии : В 2 ч.— М. : Мир, 1968,— Ч. 1—2.
12. Сайкс П. Механизмн реакций в органической химии,— М. : Химия, 1977,— 319 с.
13. Степаненко Б. Н. Курс органической химии: В 2 ч.— М. : Вьісш. шк., 1981,— Ч. 1—2.
14. Терней А, Современная органическая химия : В 2 т.— М. : Мир, 1981.— Т.1—2.
496
ПРЕДМЕТНИЙ ПОКАЖЧИК
Аденди 118
Аддукт 337, 456
Аденін 489, 490
Азобарвники 394, 418, 437—440, 454
Азоли 487
Азометинп 422
Азосполуки 434—440
Акрилонітрил 132, 143, 157, 160, 161, 219
Акролеїн 157, 479
Алізарин 457, 459
Алкадієни 147—162
Алкалоїди 487, 488
Алкани 34—56, 309, 310, 348
Алканоли див. Спирти
Алкени 111—134
Алкіли 36
Алкілування 84, 124, 137, 259, 268, 348—350, 421
Алкіни 135—147
Алкоголяти 79, 94, 236
Альдегід (и) 162—182, 395, 422
ароматичні 400—403 валеріановий 430 гліцериновий 245, 275—277 жирио-ароматичні 400 мурашиний 163, 166, 284 оцтовий 166, 180, 182 фенілоцтовий 400
Альдегідокислоти 253
^Альдози 274, 288 ___.
'< Аміди 204-209, 579 .?
Амілоза 300
Ашни 104—109, 270, 383, 413, 415—
Амінокислоти .262—272, 413, 487 ізоелектрична точка 267
Аміноспирти 110 /
Ангідрид (и]_ 1^8, 407, 410_„ малеїновий 337, 338, 456, 468 оцтовий 467, 471 фталевий 412, 413, 454 хромовий 453
Анілін 360, 372, 373, 397, 415, 416,
“ 418 45ГГ47"—~ ‘ ......
Аннулени 495"—
Аномери 280, 284
Аномерія 280
Антиподи 245, 248, 250, 312, 313
Антрахінон 454, 457
Антрацен 335, 347, 454—456
Арабіпоза 287, 293
Арилалкілгалогеніди 374
Арилгалогеніди 367—374, 432
Арилгідразини 433
Арилгідразони 174
Арилсульфохлориди 378
Арилування 421
Атропоізомери 442
Атропоізомерія 442
Ауксохроми 438, 445
Ацеталі 173
Ацетати 196
Ацетилен 135, 137, 138, 336, 447, 469, 479
Ацетиленіди 144, 145
Ацетилхолін ПО, 111
Ацетон 9, 85, 143, 163, 167, 177, 182, 254, 260, 386
Ацил (и) 185, 268
Ацилування 268, 403, 421, 422
Бензальдегід 400—403, 404
Бензидин 417, 418
Бензин (и) 43, 44, 52, 53, 447
Бензол 145, 322, 327—333, 341—351, 376, 379, 382, 408, 450, 451, 469
Білки 265, 270
Біополімери 299
Бромбензол 368, 371, 373, 405
Вальденівське обертання 251, 252
Віск бджолиний 88, 196
Вуглеводні
аліциклічні 308—316
ацетиленові див. Алкіни
етиленові див. Алкени
насичені 46, див. також Алкани
Галактоза 276, 294
Галогеналкени 212
Галогенангідриди 197, 198
Галогенопохідні 56
алканів 56—70, 324
бензольного ряду 367—374, 417
Галогенування 123, 140, 155, 156, 367—369, 423, 451, 452
Галохромія 443
Гексахлоран 323, 324
497
Гексози 273, 294
Геліантин див. Метилоранж
Гемін (гем) 473
Гемоглобін 472, 473
Гербіциди 209, 399
Гетероауксин див. Кислота індоліл-оцтова
Гетероатоми 461
Гібридизація атомних орбіталей 11—
16
Гідразони 174
Гідратація 123, 141, 167, 172, 235
Гідрогалогенування 119, 120, 141
Гідроліз 66, 167, 187, 200, 202, 203,
222, 235, 284, 387, 405
білків 265
Гідроксиламін 398, 401
Гідроформілування див. Оксосинтез
Гідрохінон 395—397
Гідрування 118, 119, 140, 154, 322,
337, 450, 451
Гіперкон’югація 169, див. також Ефект
Натана — Бекера
Глікозиди 278, 291, 409, 457
Гліколі 131, 167, див. також Спирти двохатомні
Гліколіз 302
Глікозиламіни 292
Гліфталі 413, 414
Гліцериди 220
Гліцерин 94—96, 220, 413
Глюкоза 272, 273, 285 289 294
Глюкопіраноза 279—281, 300, 303
Глюкофураноза 280, 281
Гомологи бензолу 34, 350—352, 373
Гомологічна різниця 34
Гуанін 489, 290
Гутаперча 159, 160
Дегідратація 83, 114, 238, 287, 292
Дегідрування 85, 115, 166, 322, 336
Декарбоксилування 269, 398
Декстрини ЗОЇ
Детергенти 223
Детонація 53
Дивініл 148, 160, 161
Дикетопіперазини 269
Дипольний момент 18, 41, 60, 329,
355, 418, 464
Дисахариди 272, 294—299
Дифеніл 348, 441,,442
Діазосполуки 426—437
Діазотування 427, 428
Діаміни 109, 110
Діастереоізомери 247, 250, 276
Екзальтація 491, 492
Електронегатпвність 17
Електрофіли ЗО
Енергія
активації 48, 319
спряження 153, 335, 340, 450, 455,
458, 466, 493
Еноли 142, 174
Епімери 278, 289
Епімерпзація 289
Етан 39, 116, 144
Етанол 5, див. також Спирт етиловий Етаноламін 110
Етилацетат 182
^Етилен 84, 115—118, 123, 127, 129 133, 144, 350
Етиленгліколь 89, 93, 131
Ефект
аномерний 284 ізотопний 376, 382 індукційний 21, 22, 60, 79, 169, 190, 213, 356, 362, 371, 418, 465
Натана — Бекера 26, 352 сигма-індукційний 356 спряження, або мезомерний, 23— 26, 153, 154, 169, 213, 356, 362, 418, 465
Ефір (и) ацетооцтовий 187, 224, 254—256 259—261 вінілові 143 диметиловий 5 діетиловий 66, 98, 211 малоновий 187, 224, 229 метилетиловий 84 оцтовоетиловий 66, 83, 179 прості 83, 91, 96—99, 236, 292, 391 443
складні 66, 83, 91, 95, 179г 200— 204, 236Г268; 290,' 379
Жири 95, 220—223
Зооциди 399
Ізомер (и) 5, 26, 38, 39, 328, 343, 349, 449 геометричні 114
оптичні 234, 240, 274, 312
поворотні 39
Ізомеризація 54 фотохімічна 338
Ізомерія 26, 27, 76, 89, 97, 137, 149, 163, 217
геометрична або цис-, транс- 112, 113, 311
динамічна 257
оптична 239—246, 312, 313
поворотна, або конформаційпа, 39, 58, 283, 315
поворотна оптична див. Атропоізо-мерія
Ізопрен 148, 149, 158, 160, 324
Ізопреноїди див. Терпени
Ізосполуки 27, 35
конформацій 316
Індиго 476—478
Індол 474—476, 478
Інверсія 296
Інсектицид (и) 323, 399
49 8
Карбінол див. Спирт метиловий Каталізатор Ціглера— Натта 129, 161, 162, 212
Каучуки 158—162 бутадієн-нітрильний 161 бутадієн-стирольний 161, 355 натуральний 158—160 синтетичні 160—162 хлоропреновий 162
Кетени 182, 183
Кетози 274, 288, 289
Кетон (и) 162—182, 254, 255 ароматичні 403, 404 Міхлера 445
Кислота (и)
адипінова 226, 228, 230, 324 азотиста 108, 208, 268, 422, 427 азотна 305, 368, 381, 383, 384, 393 424, 467, 471 ароматичні 404—409 аскорбінова 286 ацетооцтова 254
бензойна 401, 404, 408, 421 виноградна 234, 247, 248 вінілоцтова 217 вугільна 388, 421 галова 398, 411 гліколева 238, 239 гліоксилова 253 глутарова 227 гримуча 26 дикарбонові 224—232, індолілоцтова 478 карбамінова 208 карбонові 178, 26! -кротонова 217 лимонна 248, 249 ліполева.219, 220 ліноленова 219, 220 Льюїса 76, 211, 349 малеїнова 231, 232, 246, 248, 467 малонова 224, 226, 228 масляна 185, 186, 221 молочна 237—239, 243, 244, 254, 292 302
монокарбонові 183—196, 216—220 мурашина 185, 188, 194, 195, 227, 421, 472
нікотинова 484, 486 нуклеїнові 488, 490 окспкарбонові, або оксикислоти 232—252
оксіоцтова 234 олеїнова 219, 220 оцтова 22, 95, 111, 132, 182, 185 188, 196, 227, 232, 261, 305,. 406, 467 пальмітинова 196, 221 пікринова 390, 392, 393 піровиноградна 237, 254 пропіонова 186, 188 саліцилова 409 сечова 490
синильна 490
сірчана 180, 292, 375, 377, 379, 381, 384, 420
соляна 420, 427, 433
стеаринова 196, 221
сульфанілова 425
терефталева 412, 414
фосфорна 223, 487
фталева 412, 450
фуксинсірчиста 178
фумарова 231, 248
хлороцтова 22, 226, 399, 476
хлор’яблучні 249
ціанова 26
щавлева 4, 91, 224, 227, 228, 239, 273
яблучна 232, 234, 246, 251
янтарна 224, 226, 230, 232, 261
Коламін, див. Етанолсшін
Колодій 305
Конденсація 284, 395, 402, 413, 444, 445, 476
Конформація 39—41, 315, 316
Конформери 39—41, 149, 316
Крезоли 385, 399
Крекінг 5Т, 52,115, 149
Крохмаль 272, 294, 300, ЗОЇ
Ксилол 342, 412, 414
Кумол 342, 350, 386
Кумулени 148
Лавсан 415
Лактами 269
Лактати 239
Лактиди 237, 238
Лактоза 294, 295, 298
Лактони 238, 287
Латекс 158
Лейцин 265
Лецитин 223
Лігнін 304, 306
Лізин 265
Магнійарилгалогеніди 373
Мальтоза 295, 297, 298
Меркаптани, або тіоспирти, 99, 100
Мерсеризація 304
Метакрилат 219
Метамерія 27, 97
Метанол 181, див. також Спирт метиловий
Метплетилкетон 9
Метилоранж 439, 440
Моносахариди, або монози, 272—294
Моноцикли 308—311
Надспряження див. Ефект Натана —
Бекера
Напівацеталі 173, 280
Наркотик (и) 59, 88
Нафталін 335, 346, 412, 447, 495
Нафтиламіни 453, 454 , *
Нафтохінони 453
499
Нематодиди 399
Нікотннамід 486
Нітрили 66, 186, 187, 207, 379
Нітробензол 380, 382, 384, 416
Нітрозаміни 109
Нітросполуки ароматичні 380—385,
416
Нітрофенол 372, 383, 393
Новолаки 395
Озон 288, 337
Озоніди 133
Окислення 286, 324, 337, 394, 395, 153,
456, 457, 484
Оксалати 228
Оксими 174, 205, 401
Оксибензол див. Фенол
Оксинітрпли 170, 287, 401
Оксокислоти 253—261
Оксосіінтєз 124, 167
Олеум 375, 482
Олефіни див. Алкени
Олігомери 125
Олігосахарпди 272, 302
Олії 221, 222, 414
Парафіни 45, див. також Алкани
Параформ 180
Пеніциліни 488
Пентозани 293
Пентози 273, 293, 294
Пептиди 270, 271
Пікрати 486
Пінаколін 93
Пінакон 177
Пінан 325
Пінен 326
Піридин 346, 478—487
Піроіалол 395, 398, 411
Пірокатехін 395, 396
Пірол 462—472
Піроліз 52, 53, 166, 167, 182, 317
Поліетилен 127—129
Поліізопрен 158, 159, 162
Полімеризація 125—130, 145, 146, 180,
181, 219, 271, 355
іонна 128
каталітична 160—162
ланцюгова, або лінійна, 126, 180
радикальна 126, 127
стереорегулярна 162
ступінчаста 125
Полісахариди, або поліози, 272
вищі 299—306
Порфірини 473, 496
Правило (а)
Вудворда —Хофмана 319, 320
Ельтекова 186, 218, 258
Ерленмейєра 273
Зайцева 67, 84, 126
Зайцева — Вагнера !?1
ізопренове 158, 324
Корнблюма 103
Попова 178
Хюккеля 335, 336, 450, 455 464 480, 491, 495
Проба Лукаса 86
Пропілен 120, 121, 129, 133, 350
Рацемат (и) 242, 247, 248, 250
Рацемізація 64, 252
Реактив (н)
Гріньяра 69, 73, 98, 145, 171 186 211,492
Лукаса 86
Толленса 177
Швейцера 304
Шіффа 178
Реакція (ї) азосполучення 394, 434—437 алкілування 108 амонолізу 199, 204 ацилування 108, 198 бімолекулярні 31 біуретова 209
Вагнера 131 відщеплення 28, 52, 67—69 Вюрца 42, 210, 347, 348
Вюрца — Фіттіга 347, 348, 379, 441
Габріеля 413
Гаттермана 433
гетеролітичні 29, ЗО, 431, 432 гемолітичні 29, 432, 433
Гофмана 106, 108
Гріньяра 211 дегідратації 92 дегідрогалогенування (елімінував ця) 67—69 дегідрогенізації 52
Дільса—Альдера 157, 337 електроциклічпі 318—320 електрофільні 49 етерифікації 83, 193, 200—209 заміщення 28, 31, 46—52, 133, 143— 145, 179, 180, 323, 333, 451
Зіиіна 383, 416 і
, Канніццяря 179, 401,-402_Д
Кольбе 42
Кольбе— Шмітта 409, 411 конденсації 174—176, 238 Коновалова 50, 383
Кучерова 141, 167 необоротного каталізу 335 Несмеянова 433 окислення 131—133, 146, 186 окислювально-відновні 176—179 пергетерифікації 203 приєднання 28, 140—143, 154—156, 168—174, 322—324, 336, 337 392, 450, 456, 484
Прилежаева 132
срібного дзеркала 177, 195, 297
Тищенка 179
Ульмана 421, 441
Фіпкельштейна 58
5о0
Чичибабіпа 483
Чугаєва — Церевітинова 79
Шімана 432
Шоттен — Баумана 392, 407
Штрекера 265
Резорцин 395—397, 399, 411
Рибоза 293, 294
Сахароза 272, 295, 296
Синтез
Вільямсона 84
дієновий 157, 317
Дюма 43
оксинітрпльний 285, 287
ціангідринний 170
Смоли фенолоформальдегідні 395
Спектр (и)
14-74, 97, 115, 166, 344, 345 молекулярні 7
ПМР 10, 11, 60, 74, 75, 115, 166, 345 поглинання 6
ЯМР 44, 330
Спирт (и) 70—96, 172, 173, 291 алкілування 84 ароматичні 385 багатоатомні 290 бензиловий 374, 385 вищі 88 двохатомні 89—93 дегідратація 83 етиловий, або винний, 66, 71, 87, 88, 109, 149, 292, 388, 430 метиловий, або деревний 71, 87, 430 мірициловий 88 одноатомні 71—88 'Трьохатомні 93—96
цетиловий 88
Сульфокислоти ароматичні 375—380
Таутомерія 143, 257, 258
кето-енольна 174, 257, 397, 398 лактам-лактимна 269, 489
Теорія
електронних зміщень 20—26
ЖМКО 77
кислот і основ Бренстеда — Лоурі 76, 191
конформаційного аналізу 314, 315 напруження А. Байєра 313, 314 напруження в циклах 314, 315 напрямлених валентностей 11—20 оптичної ізомерії органічних речовин 241
хімічної будови органічних сполук О. М. Бутлерова 4, 5, 26, 327
Теофілін 490, 491
Терпени 324—326
Тетраетилсвинець 69, 70, 212
Тіофен 346, 462—470
Тіоефіри 100—102
' Толуол 342, 346, 351, 368, 382, 400, , . 40Т7 '403-----------------
Трегалоза 295—297
Тринітротолуол 365, 382
Тринітрофенол 390, 392
Триптофан 270, 478
Тропілій-катіон 493, 494
Урацил 489, 490
Уретани 208
Уротропін 181
Фенантрен 335, 458, 459 Фенілгідразин 288, 401 Фенол (и) 266, 346, 372, 373, 379, 383, 385—399, 431, 471, 494
Феноляти 386, 390, 391
Флороглюцин 395, 396
Формальдегід 164, 172, 173, 180, 181, 285, 395, 402
Формула (и) Кекуле 327 Ньюмена 40 Фосфатиди 111, 223 Фотосинтез 306, 474 Фруктоза 272, 273, 285, 289, 294 Фруктопіраноза 281, 282 Фруктофураноза 281, 282, 303 Фуран 462—469 Фурфурол 292, 468, 469
Хімічний зв’язок ароматичний 332 «бананові» 321 довжина 16 енергія 16 ковалентний 16, 369, 431 пептидний 265, 270 поляризованість 19 полярність 19 порядок 150, 151
Хінгідрон 397, 398
Хінон 394—398
Хлорангідриди 378, 379, 407, 410 Хлорбензол 367, 371, 383, 391, 432 Хлоропрен 146, 160, 162 Хлорофіл 473, 474 Хлороформ 442
Целобіоза 295, 299, 304
Целулоїд 305, 326
Целюлоза'272, 303, 304
Циклізація 292, 318—320 Циклоалкани 308—324, 336 Циклобутан 322, 323
Циклогексан 315, 31§^322» ЗД4, 337 Цикло-оксотаутомерїя 280 ~ "
Циклопарафінп див. Циклоалкани Циклопропан 310, 317, 320—322
501
ЗМІСТ
Вступ........................................................ З
Розділ 1. Теорія хімічної будови.............................4
Розділ 2. Теорія напрямлених валентностей....................11
Розділ 3. Теорія електронних зміщень .......................20
Розділ 4. Ізомерія..........................................26
Розділ 5. Класифікація органічних реакцій....................27
Розділ 6. Класифікація органічних сполук.....................32
Частина І. Ациклічні сполуки аліфатичного ряду .... 34
Розділ 7. Алкани (насичені вуглеводні)........................34
Розділ 8. Галогенопохідні алканів............................54
Розділ 9. Алканоли (спирти) ... ...................70
Одноатомні спирти . . ............. .71
Двохатомні спирти . . '. . . . . . 89
Трьохатомні спирти ... ..... 93
Розділ 10. Прості ефіри ......................................96
Розділ 11. Тіоспирти, тіоефірп та інші сполуки сірки .... 99
Розділ 12. Нітросполуки аліфатичного ряду....................102
Розділ ІЗ.-Аійіш аліфатичного ряду ..........................104
Розділ 14. Алкени (етиленові вуглеводні, олефіни)...........111
Розділ 15. Алкіни (ацетиленові вуглеводні)..................135
\РозоГд~Т(ії Алкадієни (дієнові вуглеводні)..................148
Ка'учуки . .......................................158
Розділ 17. Альдегіди і кетони...............................162„
Кетени............................................182
Розділ 18. Монокарбон ові кислоти.........................-18Х
Розділ 19. Похідні кар бонових .кислот.................... 197
Розділ 20. Елементоорганічні сполуки......................209
Розділ 21. Сполуки з двома або кількома функціями.........212
Розділ 22. Дикарбонові кислоти . . . ..................Ж.
Розділ 23, Оксикарбонові кислоти і оптична ізомерія .... 232
Одноосновні оксикислоти........................ 235
Багатоосновні і багатоатомні оксикислоти.........246
Розділ 24. Альдегідо- і кетокислоти. Таутомерія............253
Розділ 25. Амінокислоти....................................£62
Розділ 26. Вуглеводи .....................................272
Моносахариди . ............................ .... 272
Дисахариди . •.................•.................294
Вищі полісахариди ..................... .... 299
Частин а II. Ациклічні сполуки ............................308
Розділ 27. Моноцикли . , , „ ,...........................308
Терпени і терпеноїди.............................324
502
Части наПІ. Сполуки ароматичного ряду ............327
Розділ 28. Вуглеводні ряду бензолу і їх характеристика . . . 327
Розділ 29. Бензол та його алкілзаміщені......................341
Розділ ЗО. Правила орієнтації електрофільного заміщення в бензольному ядрі ....... і ... .
Розділ 3/. Галогенопохідні бензольного ряду...............
Розділ 32/\Ароіцдтичні сульфокислоти .... . . .
Розділ ЗЗСШЧтросполуки ароматичного ряду...............
Розділ 34. Феноли та ароматичні спирти....................
Одноатомні феноли...............................
Багатоатомні феноли , . . . ...................
Розділ 35. Альдегіди, кетони і карбонові кислоти ряду бензолу . 400
Розділ 36. Ароматичні аміни.................................4Т5
Розділ 37. Ароматичні діазо- і азосполуки...................426
Розділ 38. Багатоядерні ароматичні сполуки з неконденсованими бензольними ядрами..........................................440
Розділ 39. Ароматичні вуглеводні з конденсованими бензольними
ядрами ...................................... . . 447
Нафталін . .................................... , 447
Антрацен...............-........................ 454
Фенантрен .................................... . . 458
Канцерогенні багатоядерні ароматичні вуглеводні . . 459
Стероїди , .......................................460
. 367
. 385
. 395
Частина IV. Гетероциклічні сполуки ....... 461
Розділ 40. П’ятичленні гетероциклічні сполуки з одним гетероатомом ........................................... 462
Розділ 41. Шестичленні гетероциклічні сполуки з одним гетероатомом ............................................ 478
Розділ 42. Гетероцикли з кількома гетероатомами , , . . . 487
Розділ 43. Небензоїдні ароматичні сполуки .... . . 491
Список р е к о м е н д о в а н о ї л і т е р а т і/ р и . . , 496
Предметний покажчик...................... , . . • . 497
Домбровський Л. В., Найдан В. М.
Д66 Органічна хімія: Навч. посібник.— К.: Вища шк., 1992.—503 с.: ід.
І8ВК! 5-11-002504-5
д 1705000000—180 74_91
Д М211(04)-92
ББК24.2я73
Учебное ііздание
Домбровский Андрей Владимирович Найдан Владимир Матвеевич
ОРГЛНИЧЕСКЛЯ химия
Допущено Министерстеом народного образования Украиньї в качестве учебного пособия для студентов ертесіпвенньїх и естественно-географических факультетов педагогичєских институтов
Киев
' «Вища школа»
На украинском язьіке
Оправа художника Л. О. Дикарєва Художній редактор О. Д- Бондаренко Технічний редактор С. Л. Свєтлова
Коректор С. Г. Чиркіна
ИВ № 14286
Здано до набору 29.12.90. Підписано до друку 31.10.91, Формат 60х90*/1в. Папір ДРук. № 2. Гарнітура літературна. Високий друк. Умов.-друк. арк. 31,Б. Умов, фарбовідб. ЗІ ,79. Обл.-вид. арк. 31.61. Тираж 8000 пр. Вид. № 8826. Замовлення 1-120. Ціна 2 крб. 90 к.
Видавництво «Вища школа», 282084. КнТв-84,вул. Гоголівська, 7
Книжкова фабрика їм. М. В. Фрунзе, 310057, Харк1в-57, вул. Доиець-Захаржевського, 6/8