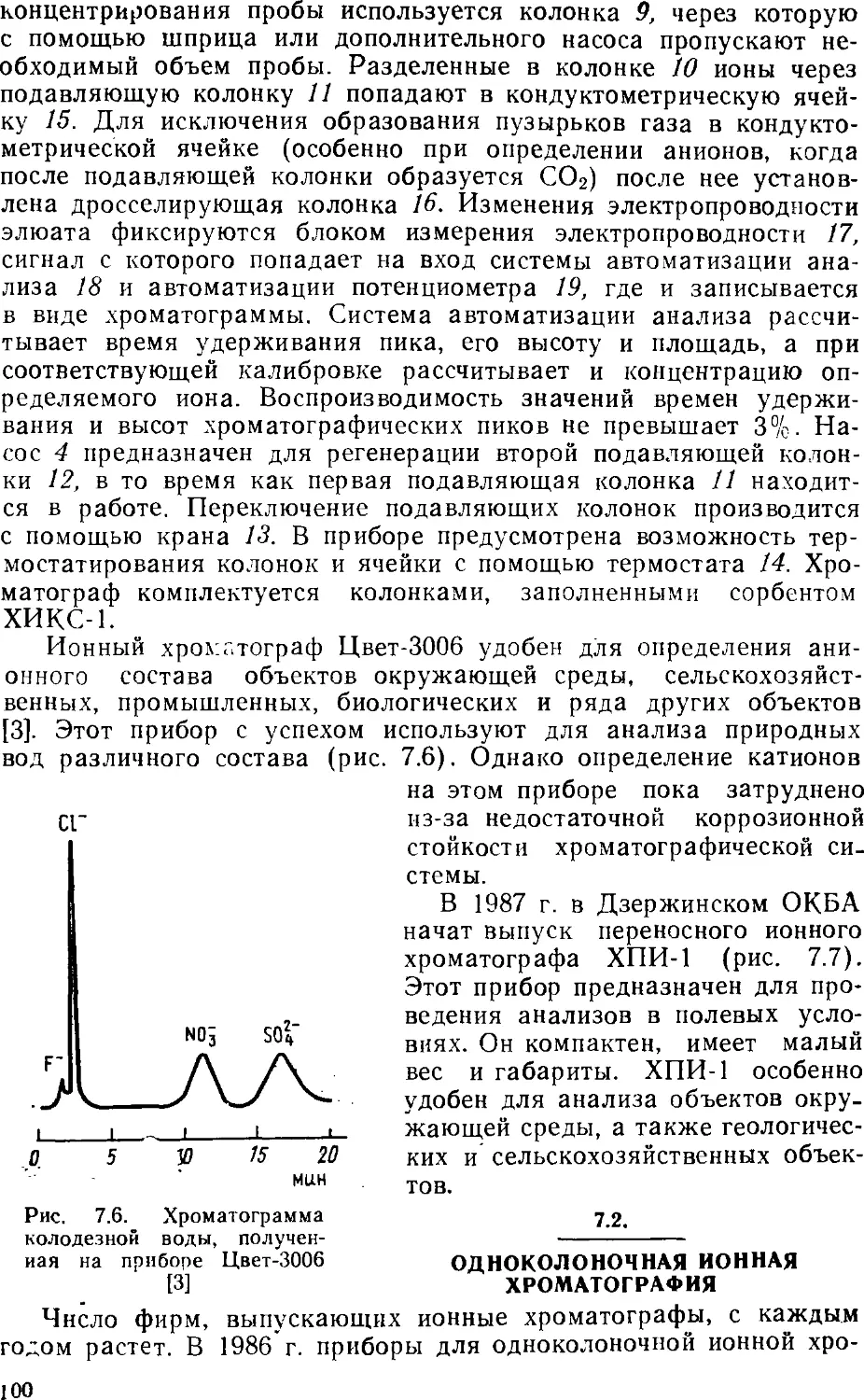
/































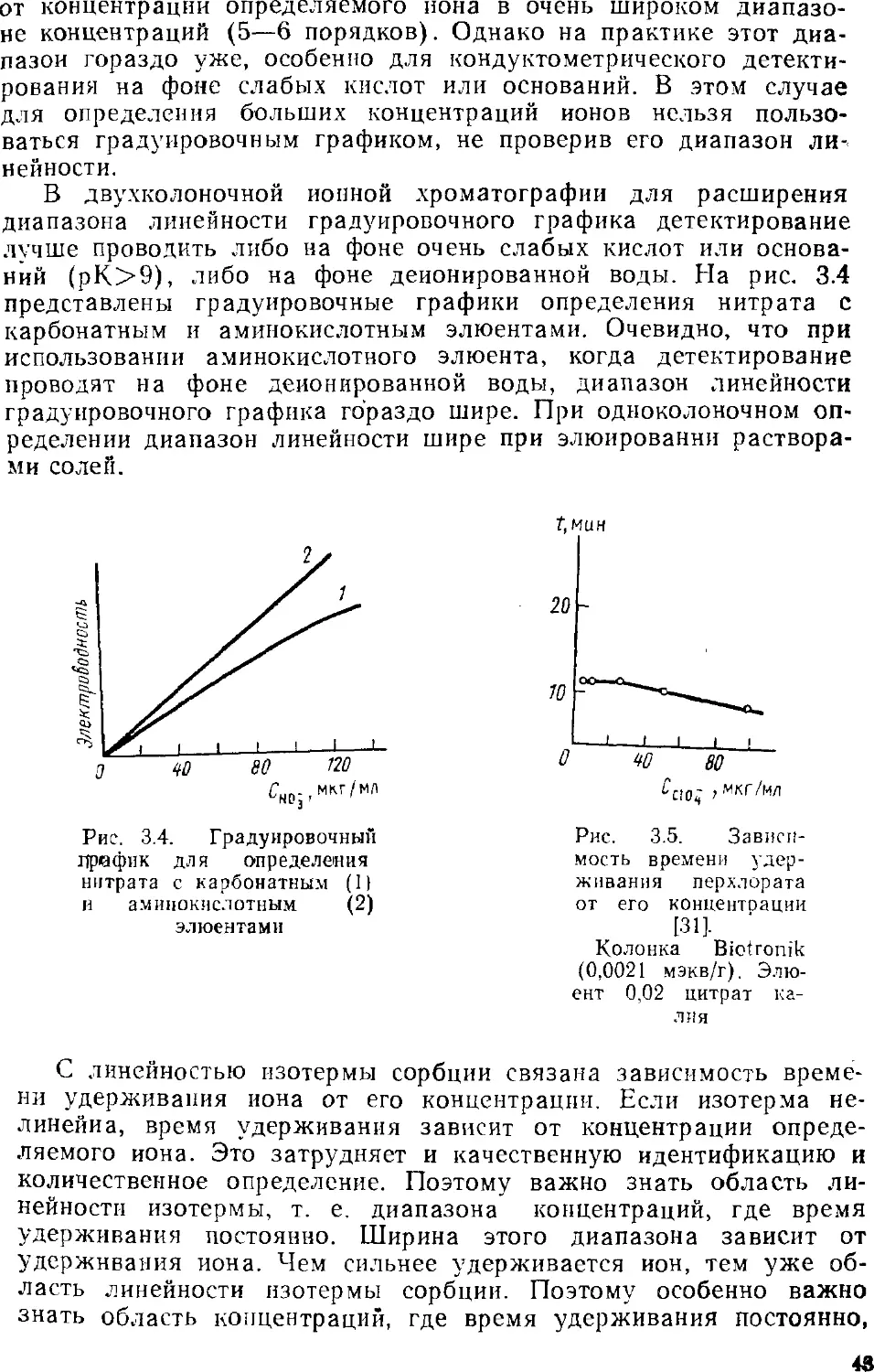

















































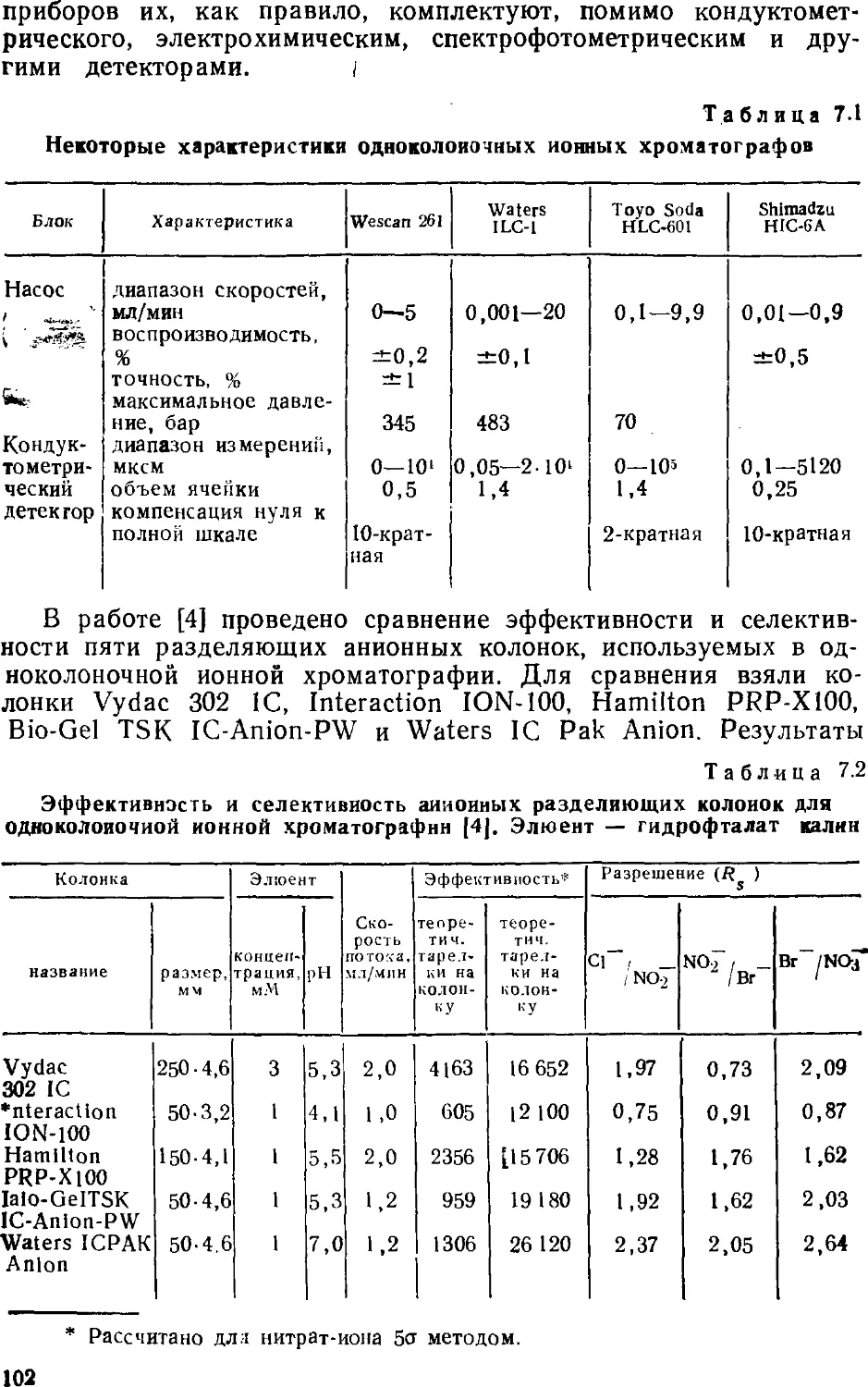



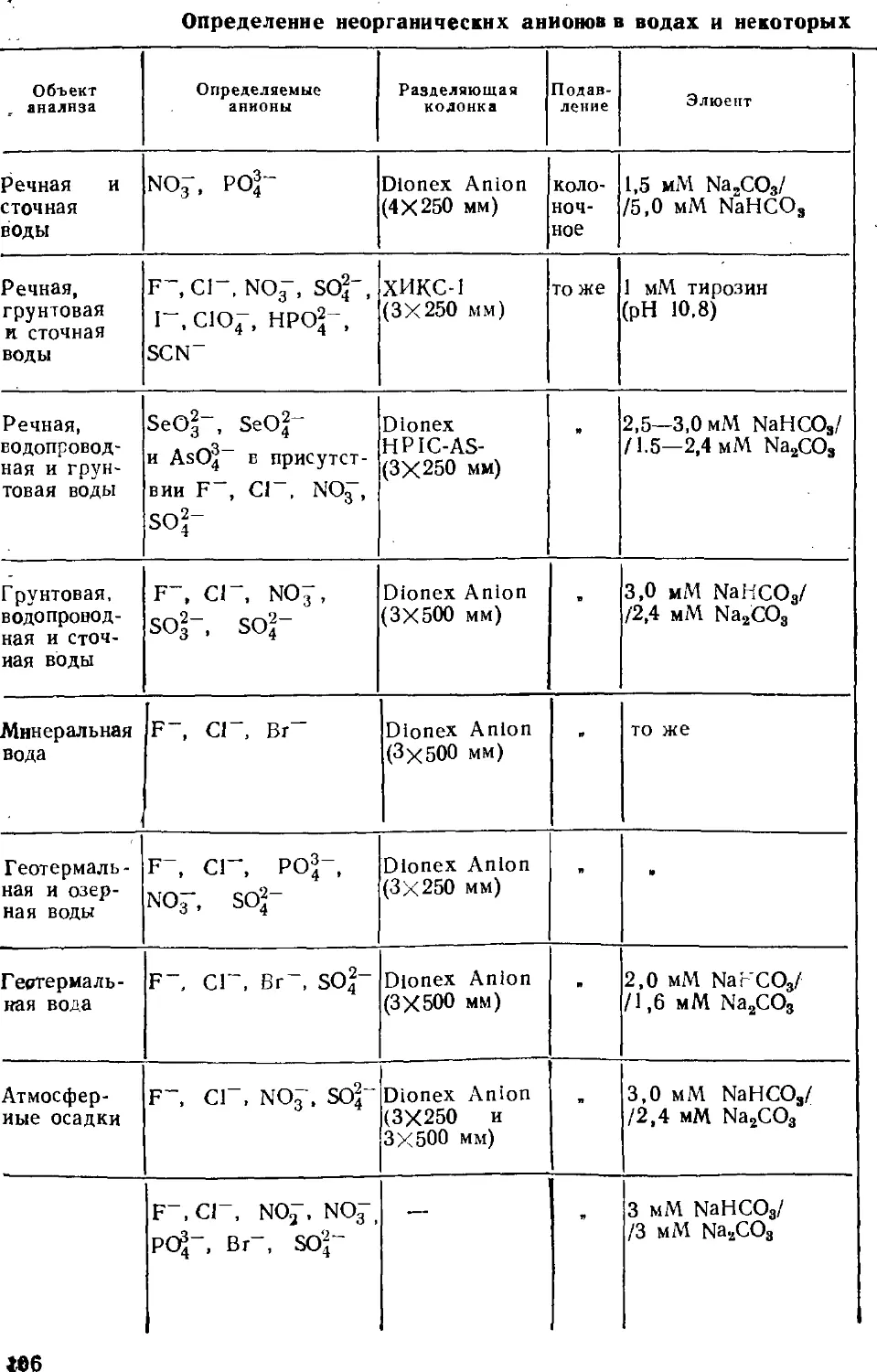
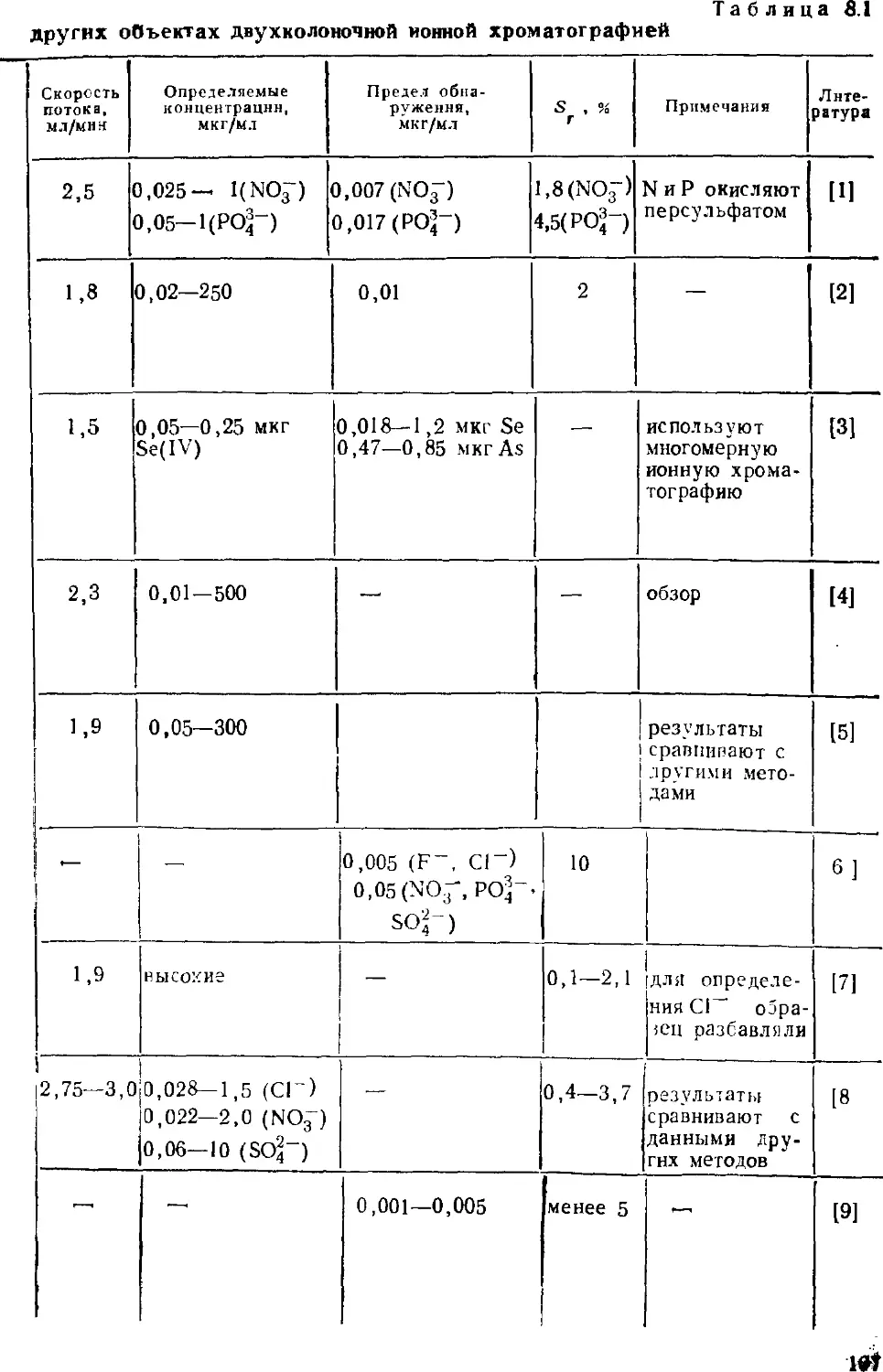
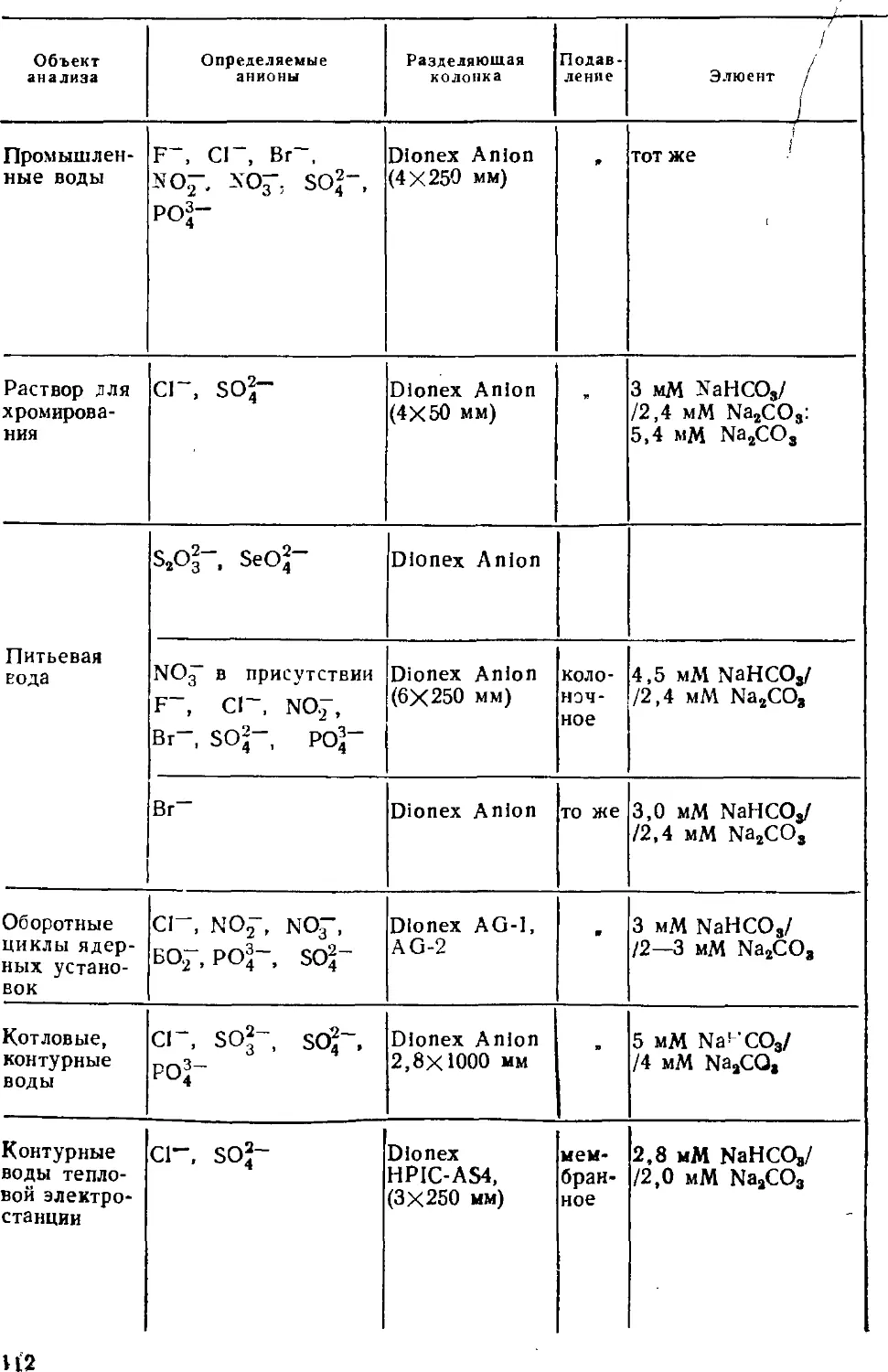
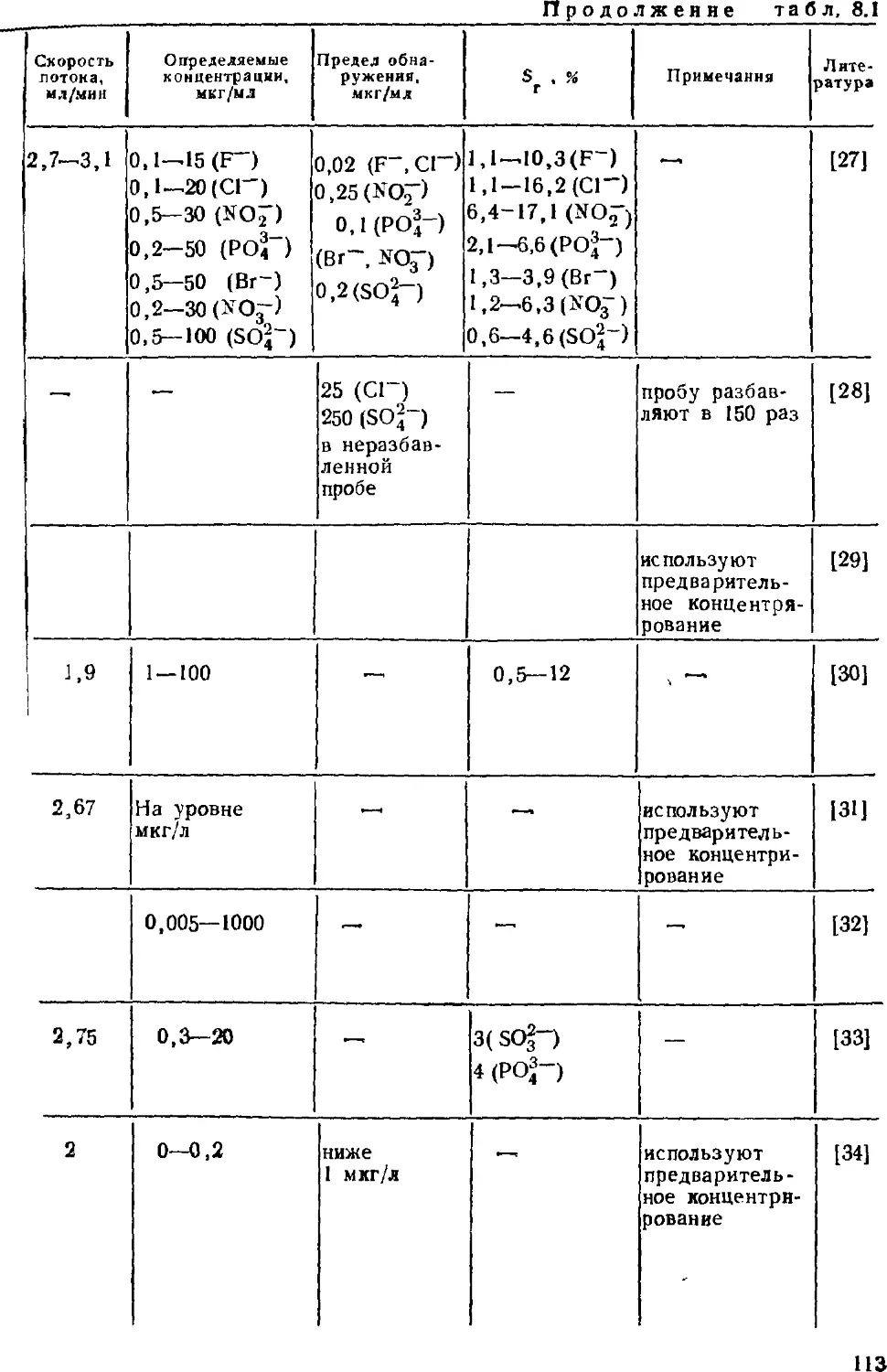
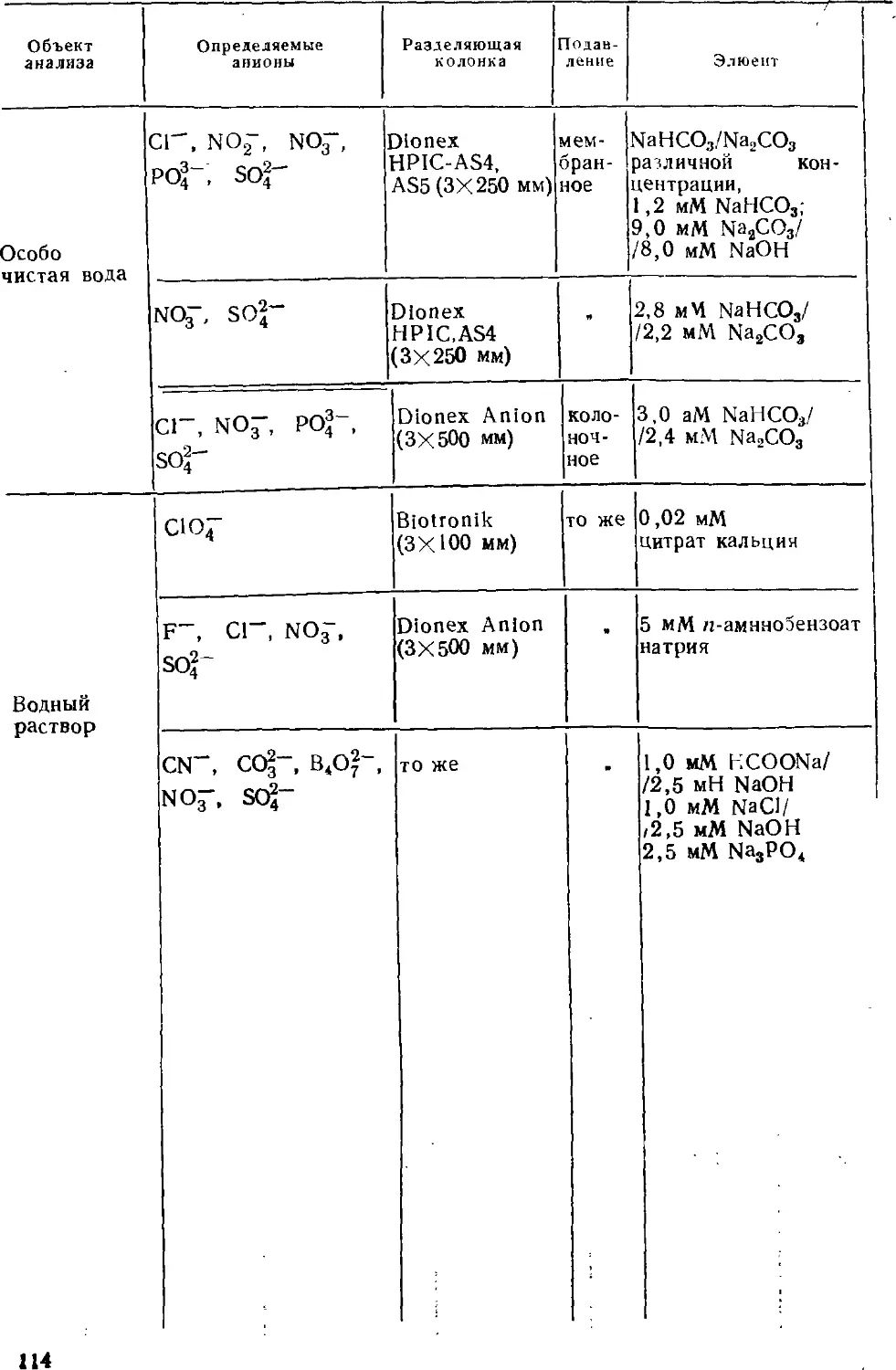


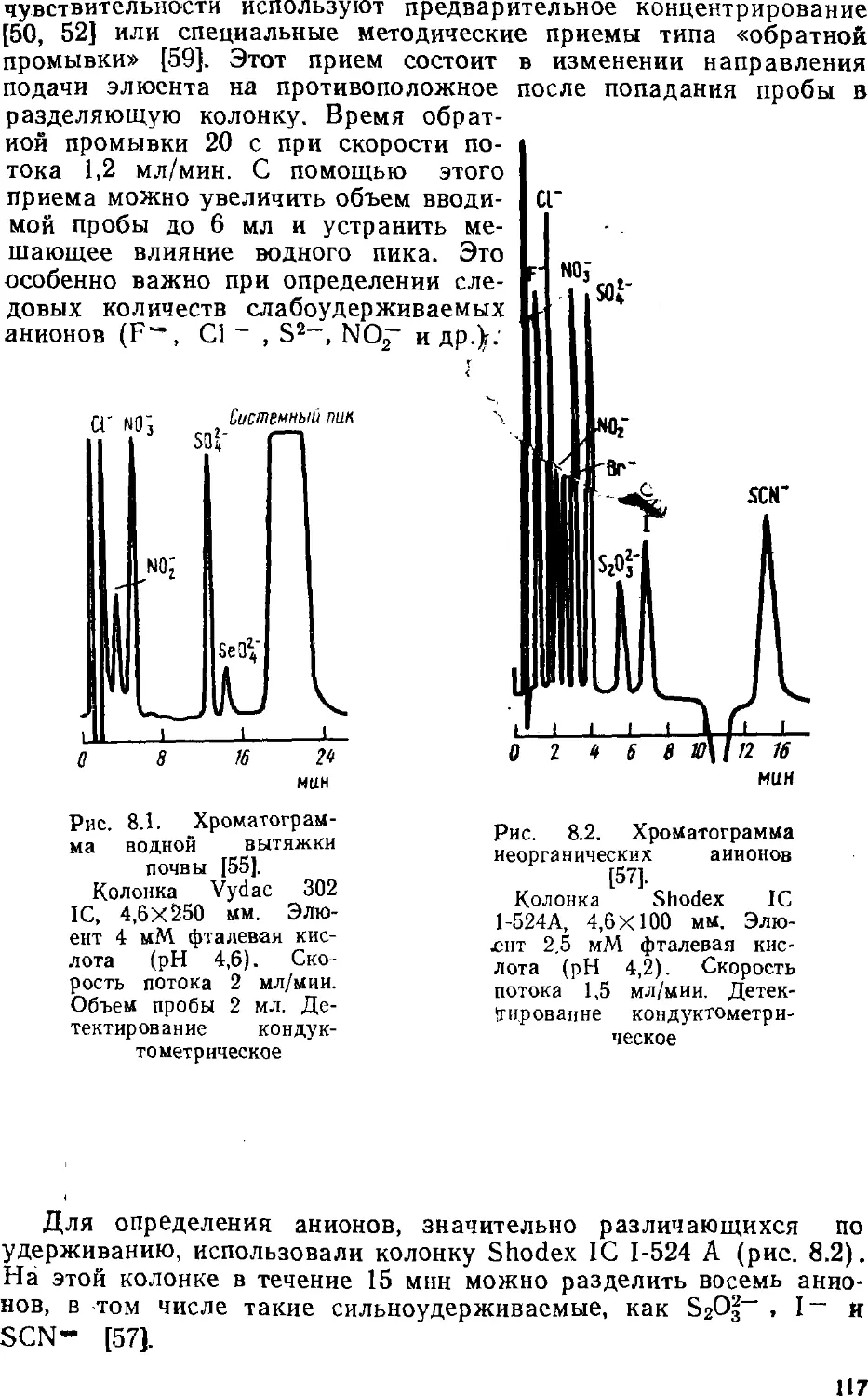







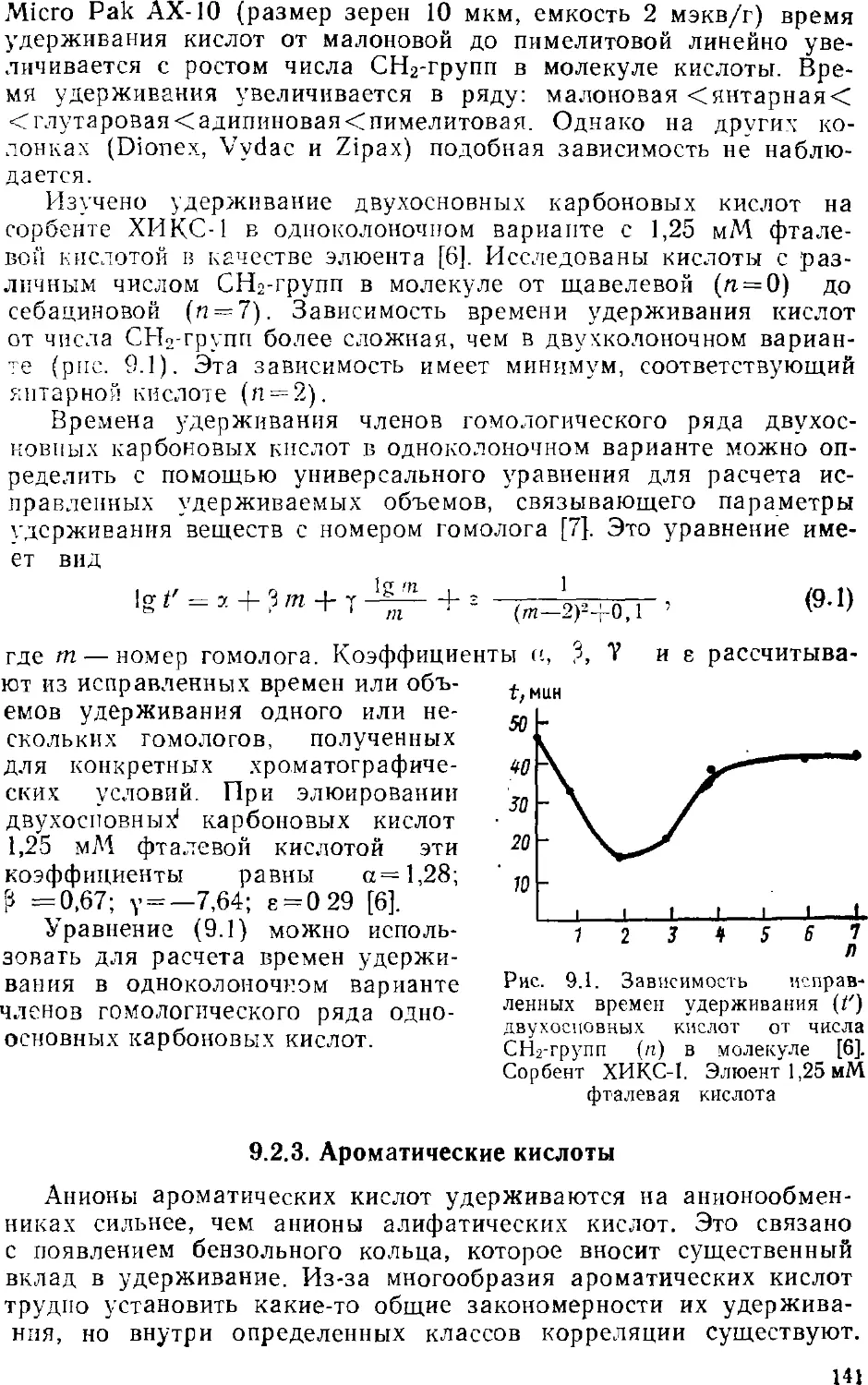















































Автор: Золотов Ю.А. Шпигун О.А.
Теги: другие физико-химические методы анализа (кроме оптических) анализ воды химия водоснабжение хроматография водоподготовка
ISBN: 5—211—00903—7
Год: 1990




Текст
>ДК 543.544.6:543.8
Шпигун О. А., Золотов Ю. А. Ионная хроматография и ее применение в
анализе вод. — М.: Изд-во МГУ, 1990. — 199 с: ил. ISBN 5—211—00903—7.
В монографии впервые в отечественной литературе рассмотрены основы
иоиохроматографического анализа вод — лучшего современного метода
определения анионов в растворах. Описаны последние достижения в развитии
ионной хроматографии, существенно расширяющие ее возможности, такие как
новые системы подавления фонового сигнала, детекторы и сорбенты. Особое
внимание уделено определению неорганических анионов. Обсуждаются способы
определения органических веществ, главным образом кислотного характера.
Приводятся методы определения металлов, в частности описан разработанный
авторами метод определения металлов в виде оксоанионов. Отдельно рассмотрен
анализ вод различных типов — поверхностных пресных, сточных, морских, а
также атмосферных осадков.
Для химиков-аиалитиков, хроматографистов, специалистов по охране
окружающей среды, работников промышленности и сельского хозяйства, а также
преподавателей и студентов вузов химического профиля.
Рецензенты:
доцтор химических наук Г. В. Кудрявцев,
доктор химических наук И. А. Ревельский
Печатается по постановлению
Редакционно-издательского совета
Московского университета
1707000000— 145
Ш 077@2)-90
__ © Издательство Московского
ISBN 5—211—00903—7 ^ университета, 1990
ПРЕДИСЛOB ИЕ
В анализе объектов окружающей среды анализу вод
принадлежит важнейшее место, причем воды каждого типа имеют свои
особенности: морские и многие подземные воды сильно
минерализованы, сточные могут иметь нестабильный разнообразный
состав, питьевые воды содержат определяемые компоненты в
очень низких концентрациях и т. д. Число веществ, которые
приходится определять в водах, весьма велико и имеет тенденцию
к увеличению. Особенно важно определять загрязняющие воду
компоненты, присутствие которых нежелательно или
недопустимо. Среди таких веществ, например, тяжелые металлы и
токсичные анионы. Наибольшие трудности связаны с анализом вод
на органические вещества из-за многообразия последних, поэтому
разработка и совершенствование методов, позволяющих решать
задачи анализа вод, — важная проблема аналитической химии.
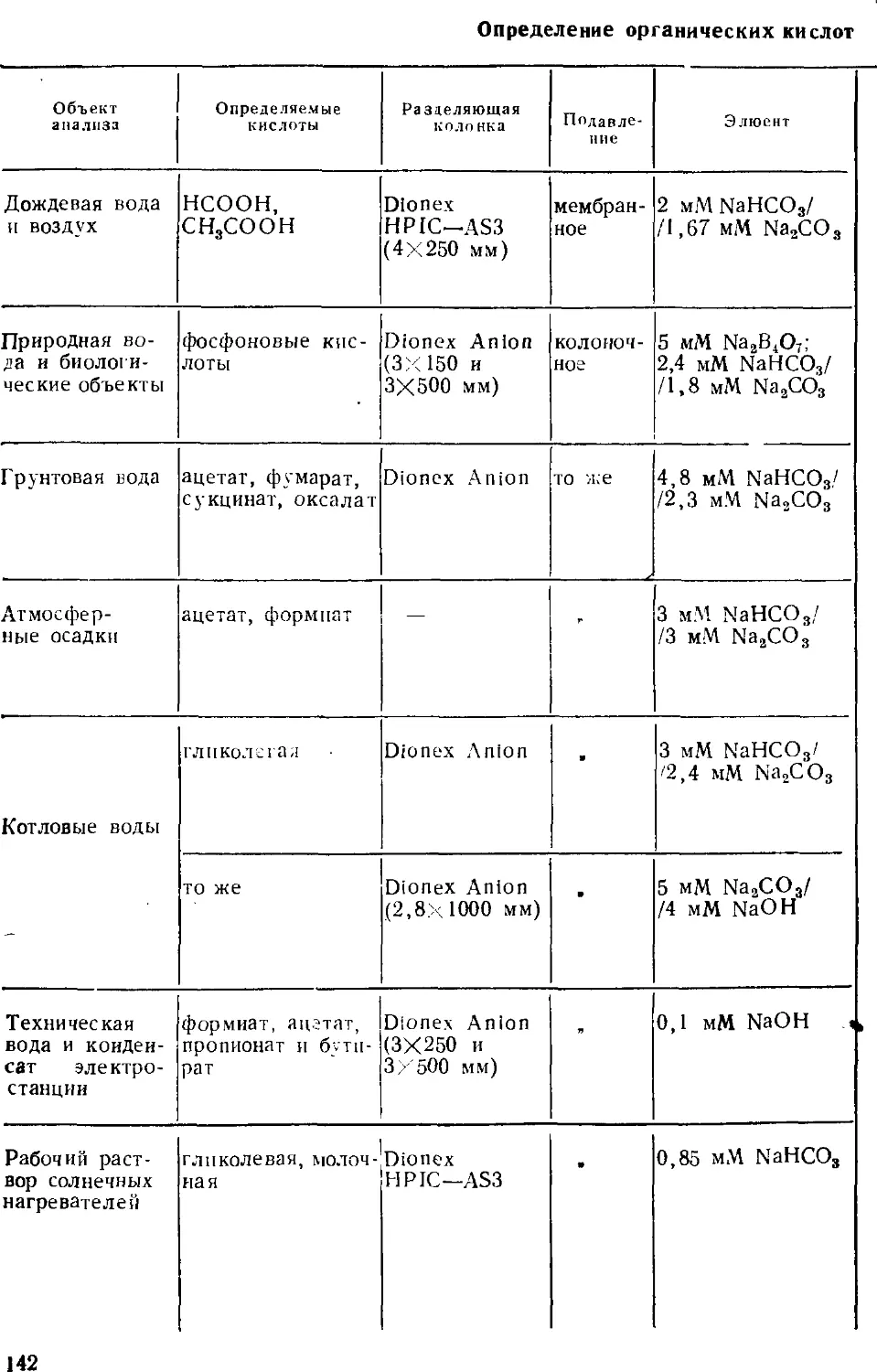
Среди таких методов значительное место заняла ионная
хроматография — относительно молодой, но очень эффективный
«гибридный» метод анализа. Гибридный, потому что он
позволяет и разделять сложные смеси веществ, находящихся в
ионной форме, и определять их содержание; другими словами,
ионная хроматография, как и прочие современные хроматографиче-
ские методы, одновременно является и методом разделения, и
методом определения. Кроме того, этот метод позволяет
определять неорганические анионы (это вообще лучший метод
определения анионов), органические кислоты и основания, катионы
щелочных, щелочноземельных и переходных металлов;
разработаны приемы определения ряда тяжелых токсичных металлов.
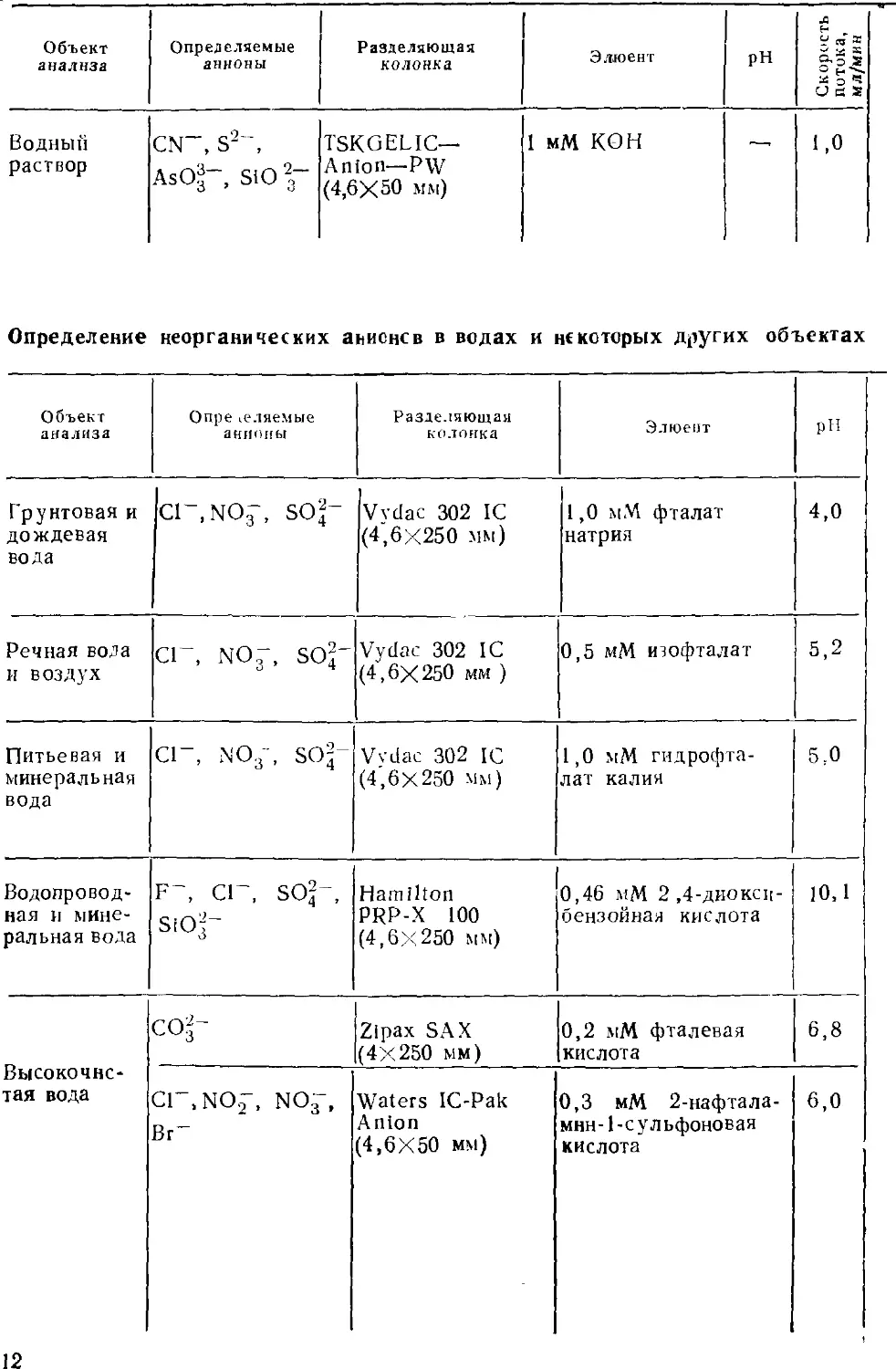
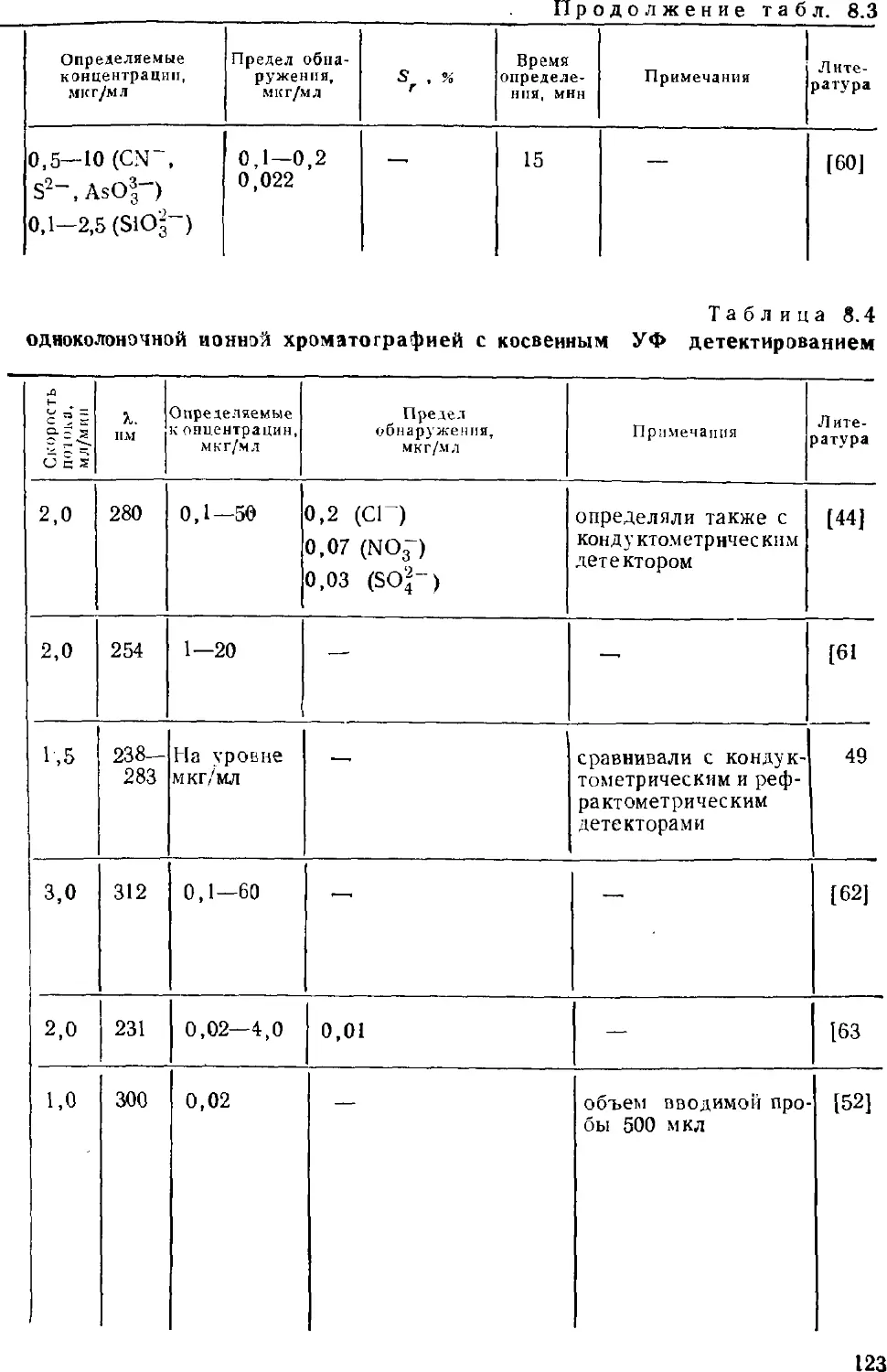
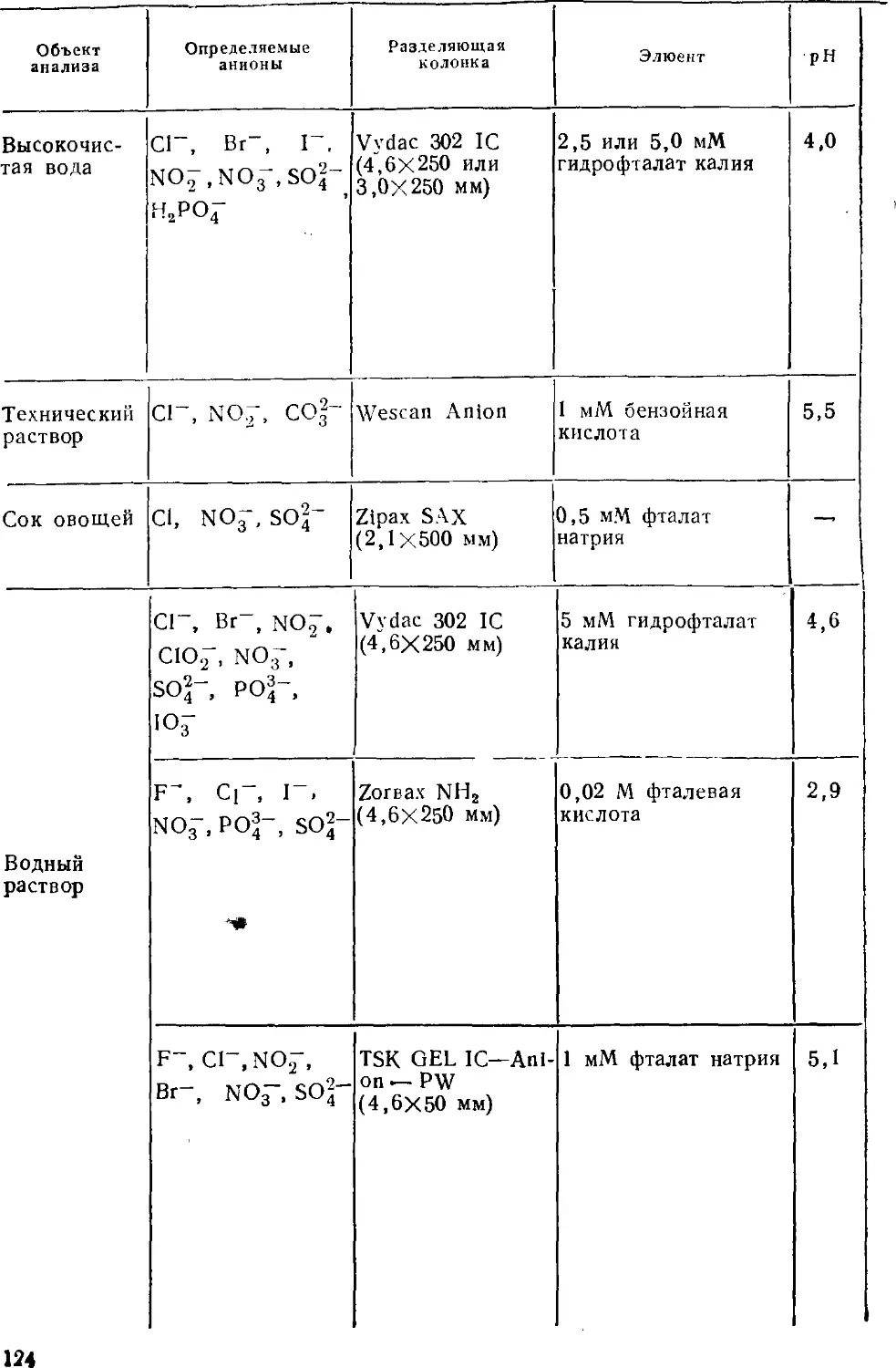
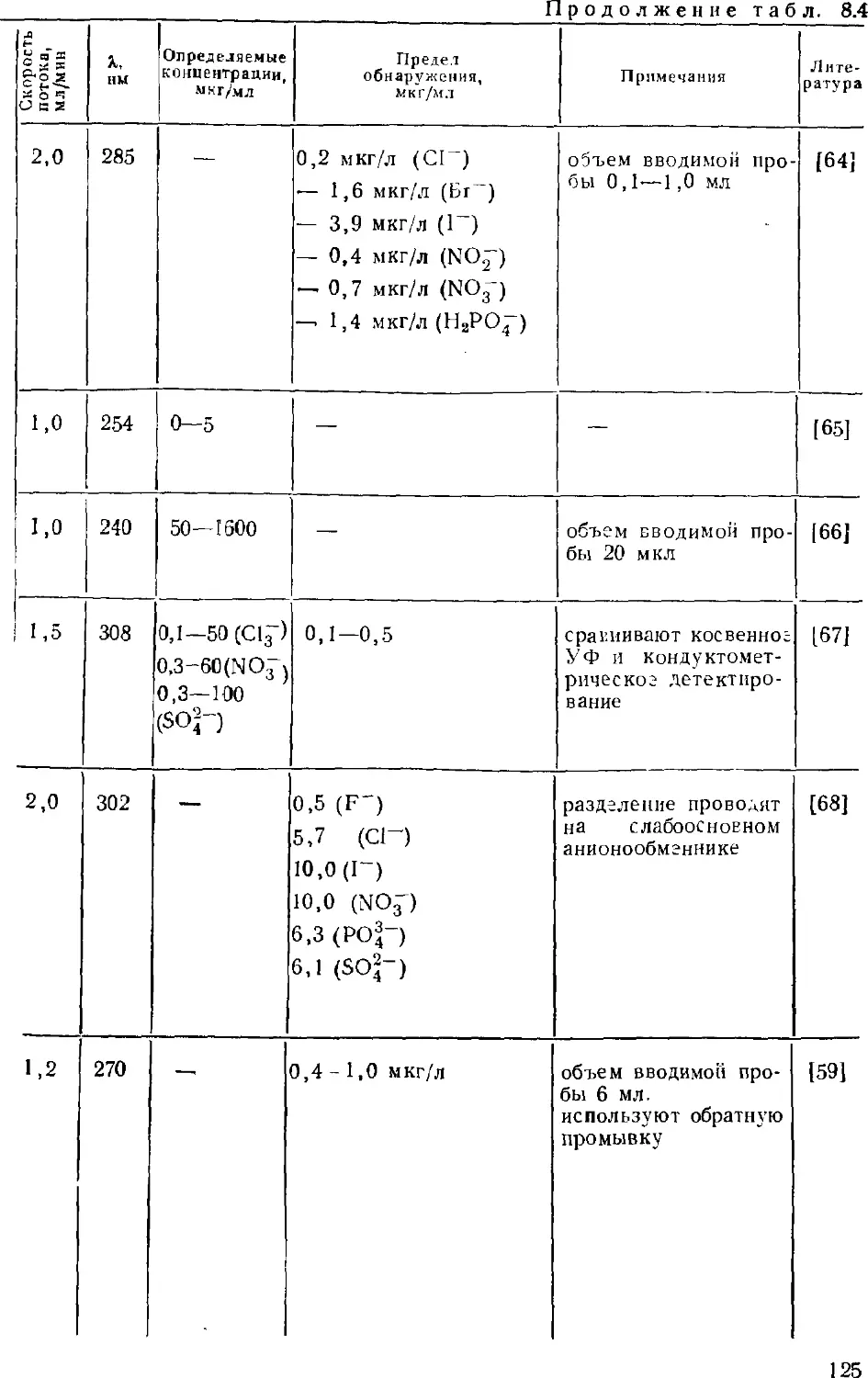
Интенсивно развиваются примыкающие к ионной хроматографии
методы ион-парной и ион-эксклюзионной хроматографии.
Ионной хроматографии посвящены сотни публикаций, в том числе
несколько книг, ряд фирм изготавливает ионные хроматографы.
Однако применение этого метода в анализе вод ранее не было
обстоятельно рассмотрено. Было стремление осветить и оощпе
основы метода, и его приложения как средства анализа вод
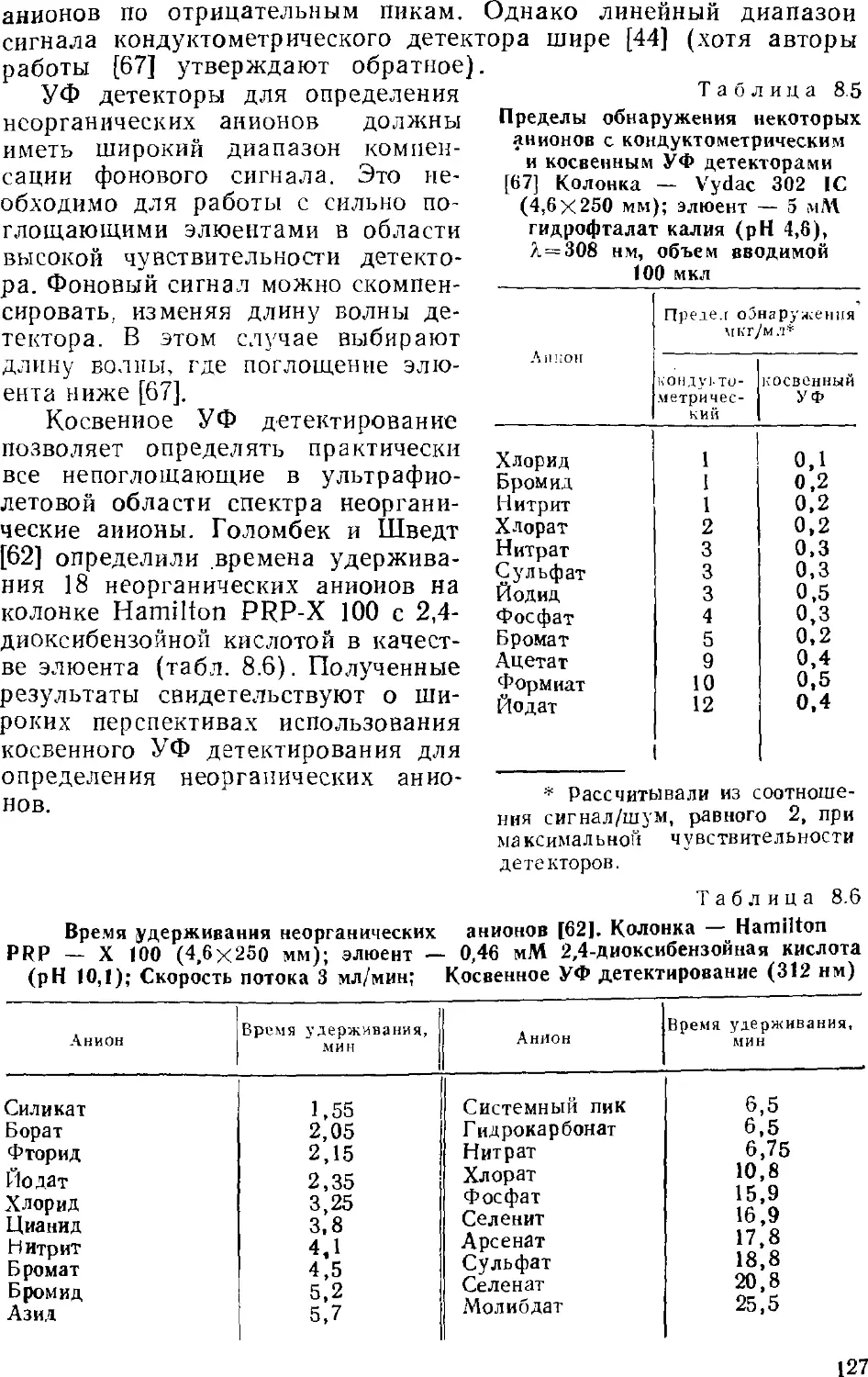
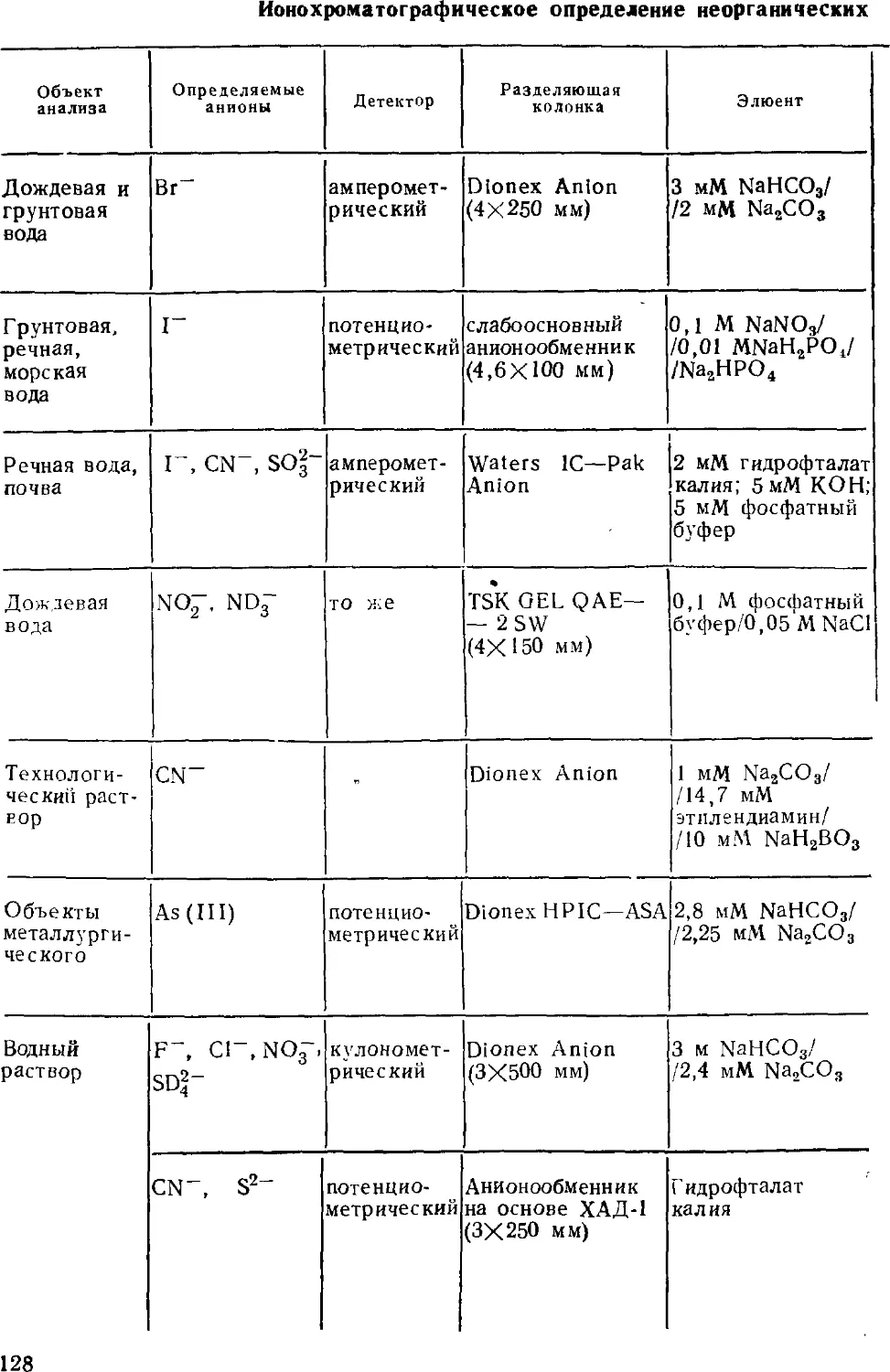
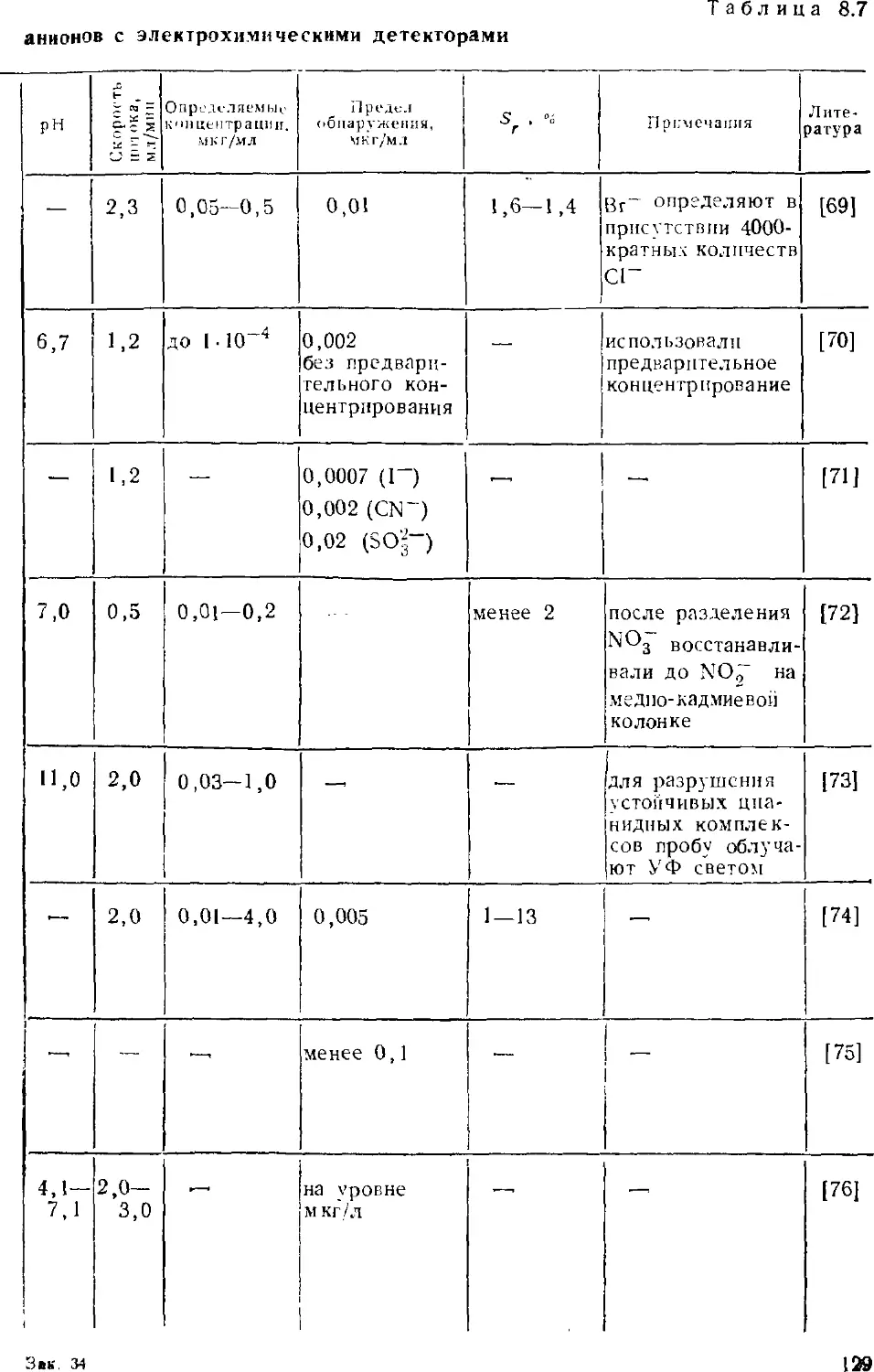
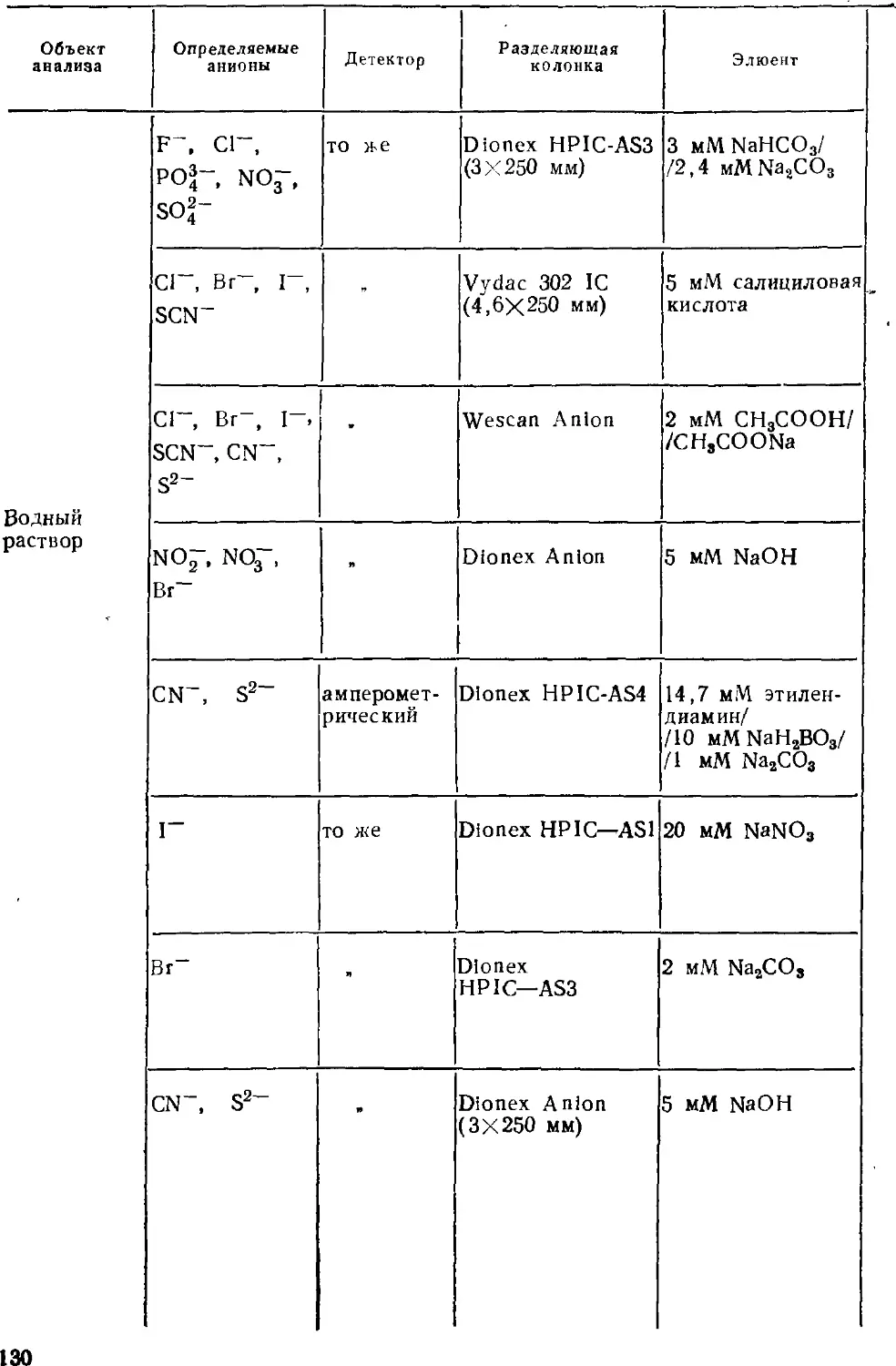
различного типа. При этом авторы опирались на публикации по ионной
хроматографии, появившиеся в основном до конца 1986 г. В
какой-то степени в книге нашли отражение и результаты
собственных исследований авторов, проводившихся на химическом
факультете в Московском государственном университете с 1980 г.
Много внимания уделено недавним успехам в развитии
метода, тем более что они не могли быть освещены в
монографиях Гьерде и Фритца, Смита и Ченга. К последним достижениям
относятся, например, развитие градиентного варианта, исполь-
зование новых систем подавления фонового сигнала,
применение оригинальных элюентов, разработка теоретических основ
метода.
Книга построена следующим образом. Первая глава
посвящена основам ионообменной хроматографии. Здесь рассмотрены
ионообменные равновесия, общие сведения об ионообменниках,
термодинамические и кинетические параметры,
характеризующие элюентное хроматографическое разделение. Во второй
главе обсуждаются основные принципы двухколопочной и одноко-
лоночной ионной хроматографии, закономерности удерживания
анионов и катионов на ионообменниках. Требования,
предъявляемые в ионной хроматографии к сорбентам, элюентам,
детекторам и системам подавления фонового сигнала, а также вопросы
их оптимального использования изложены в главах с третьей
по шестую. В седьмой главе обсуждаются характеристики
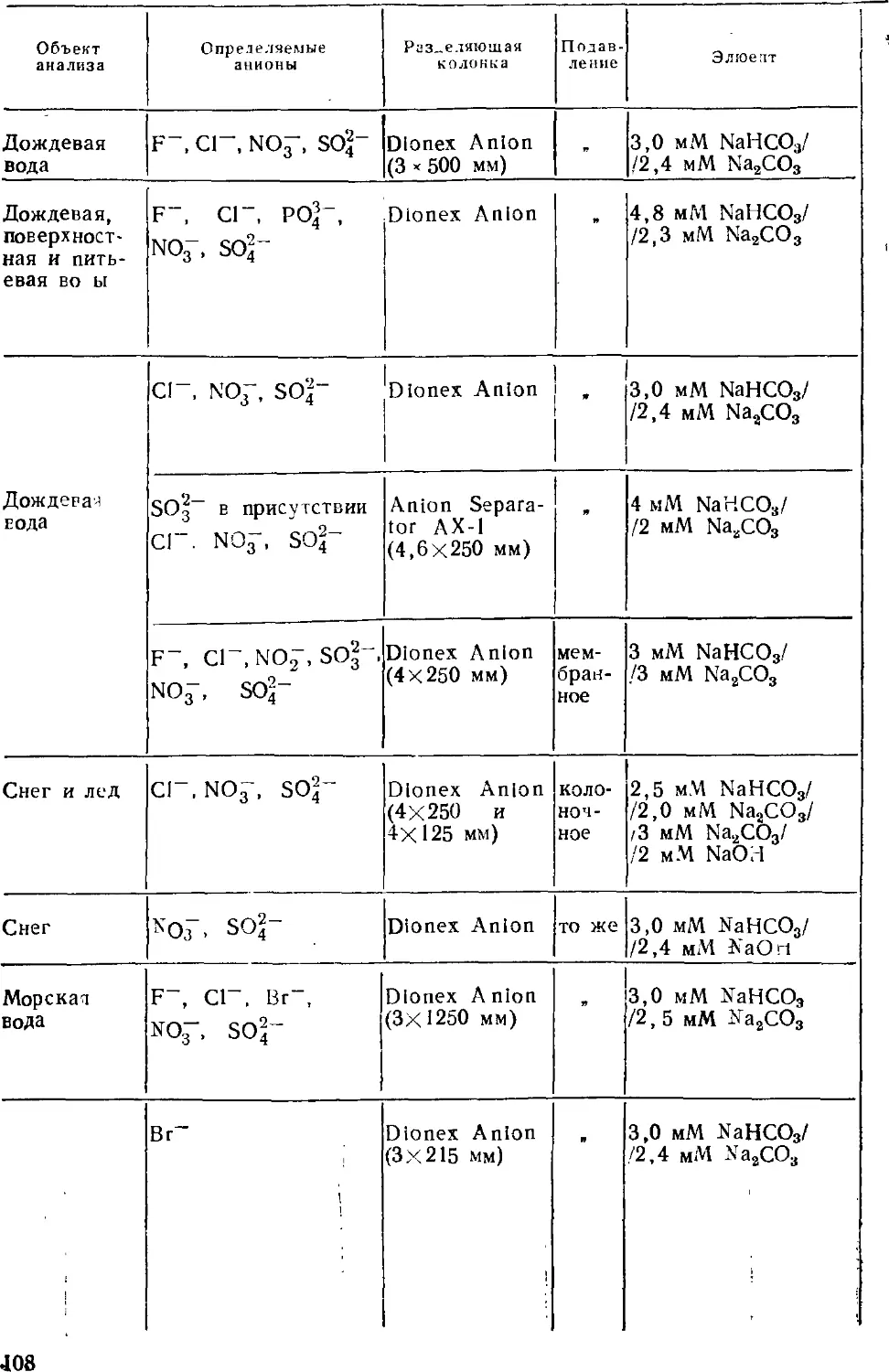
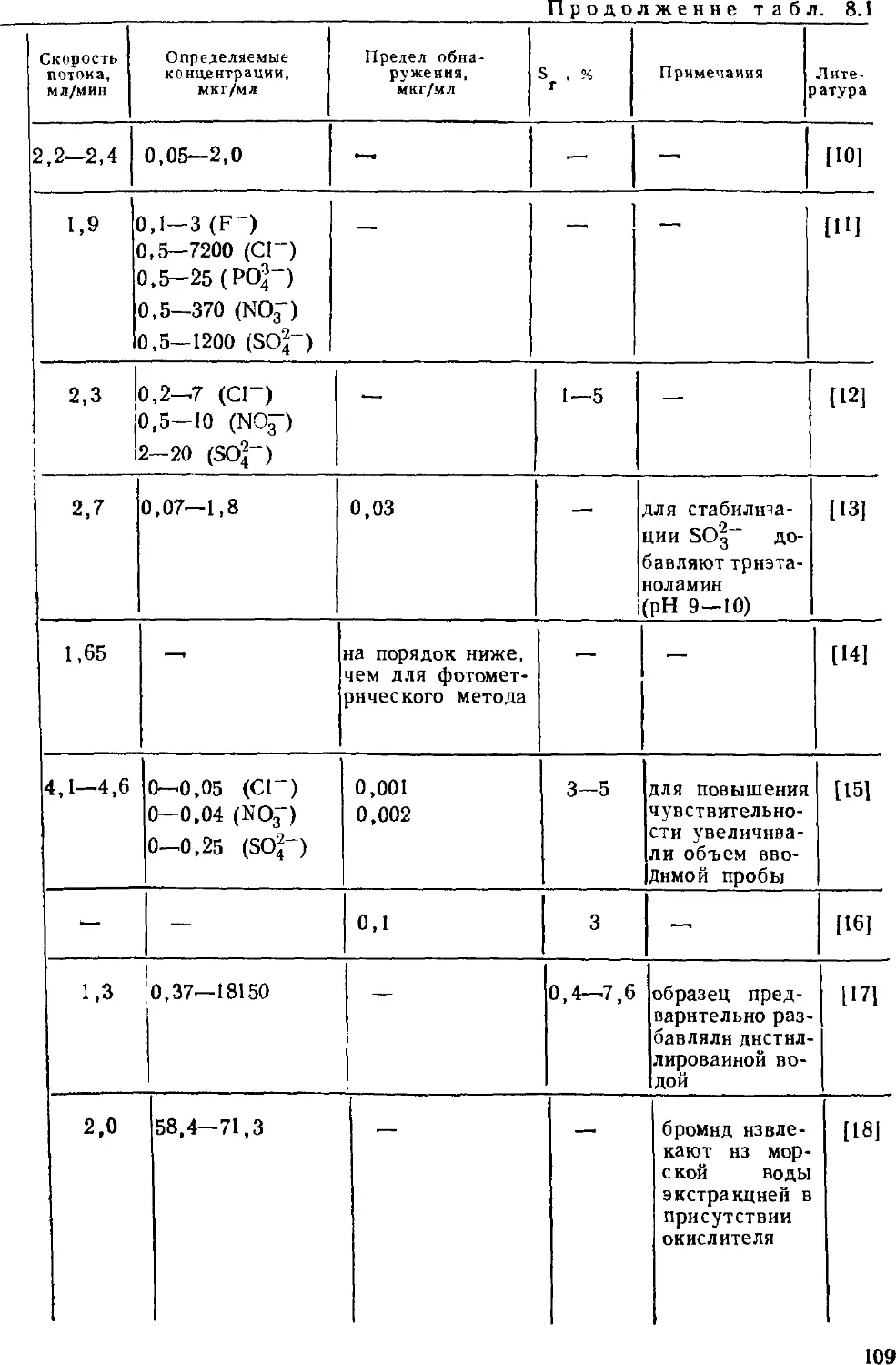
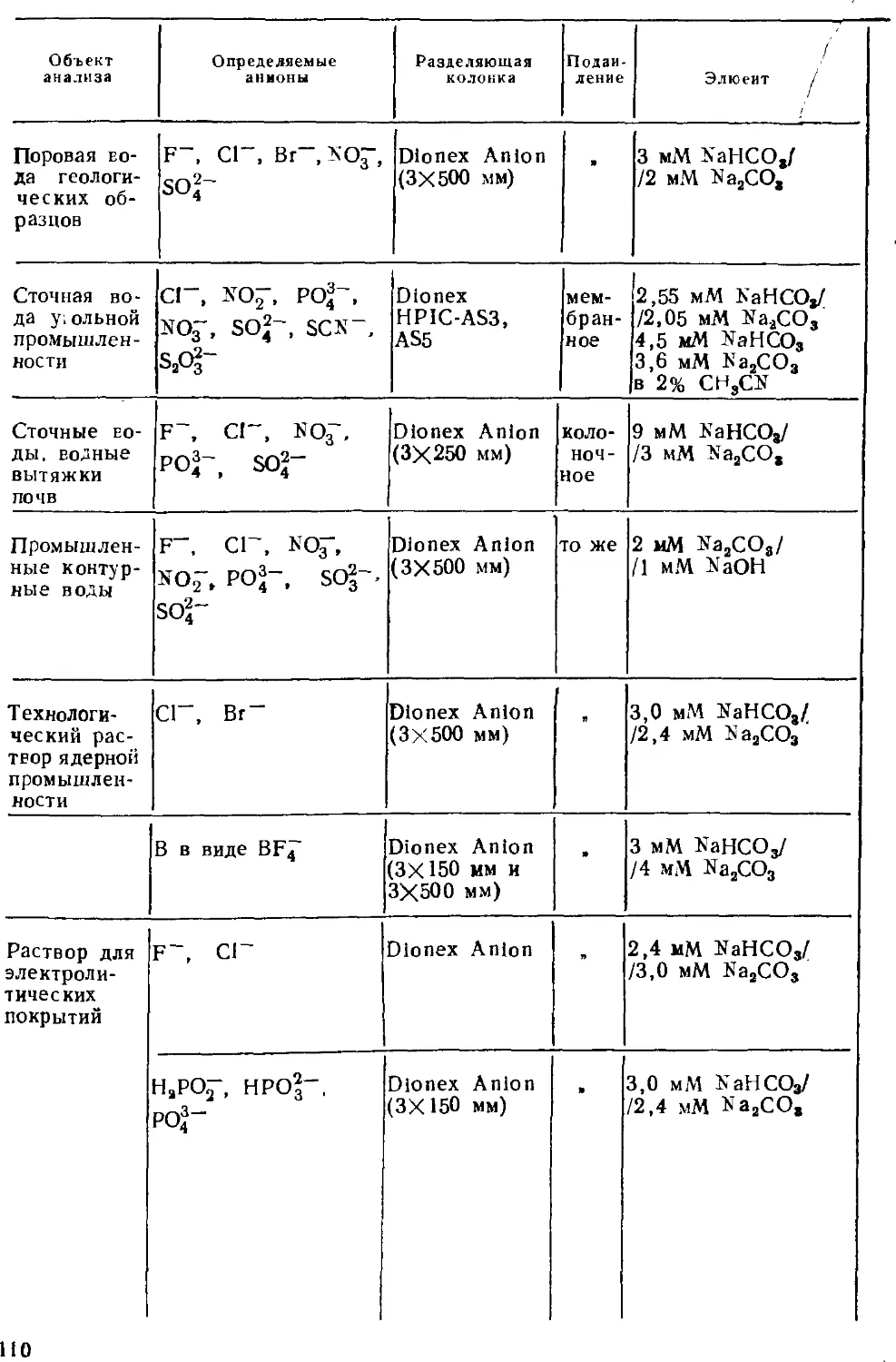
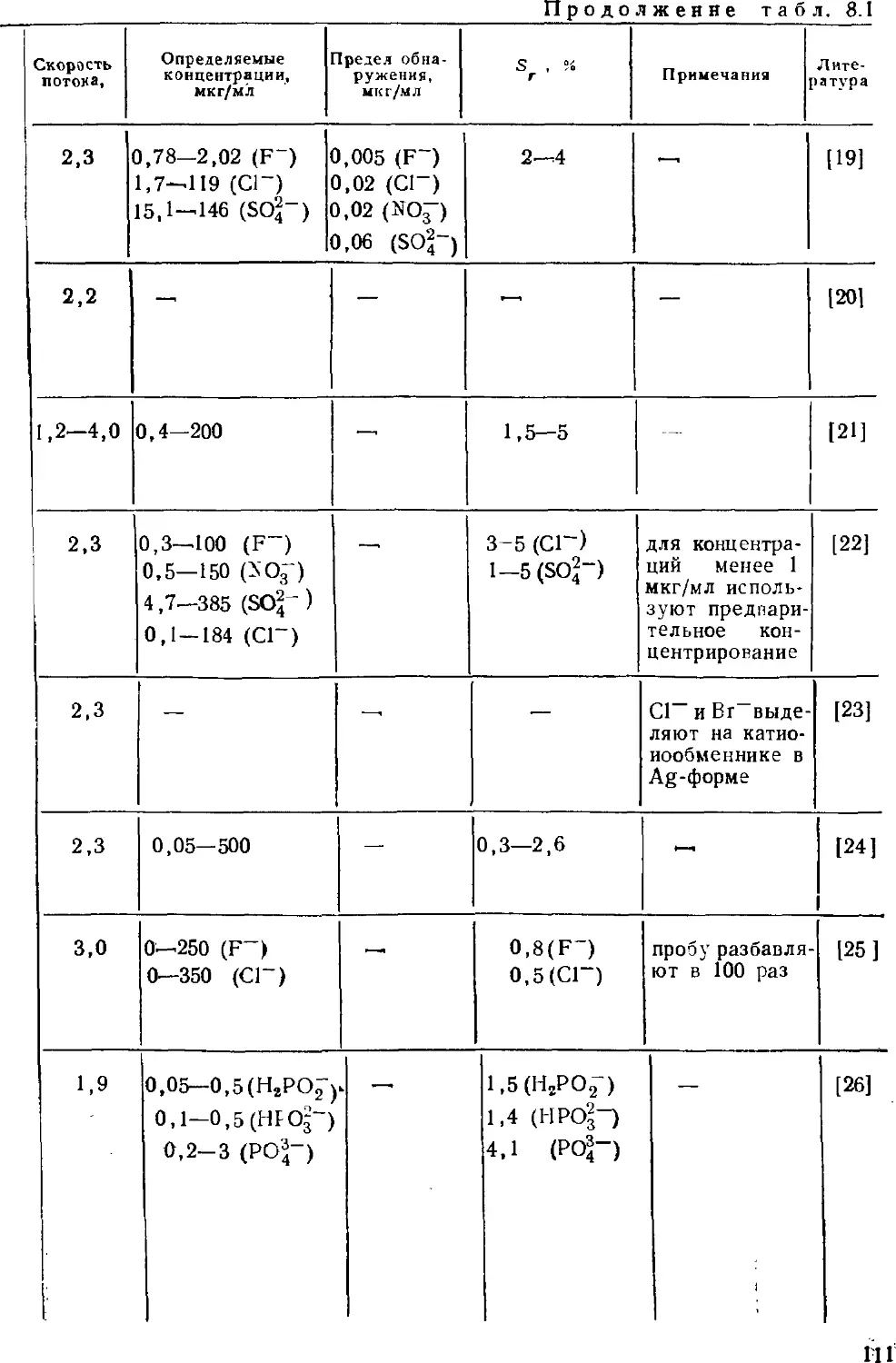
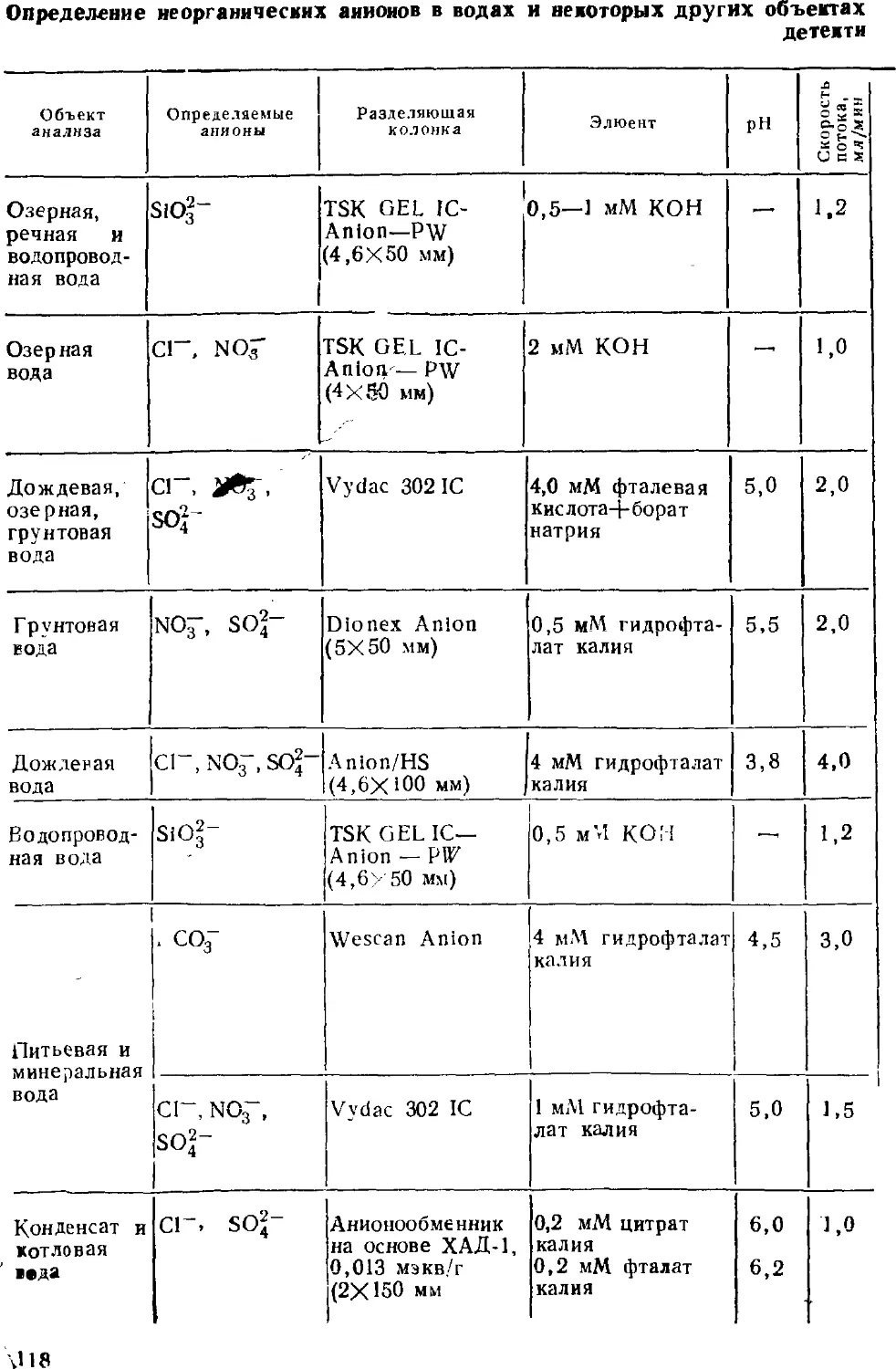
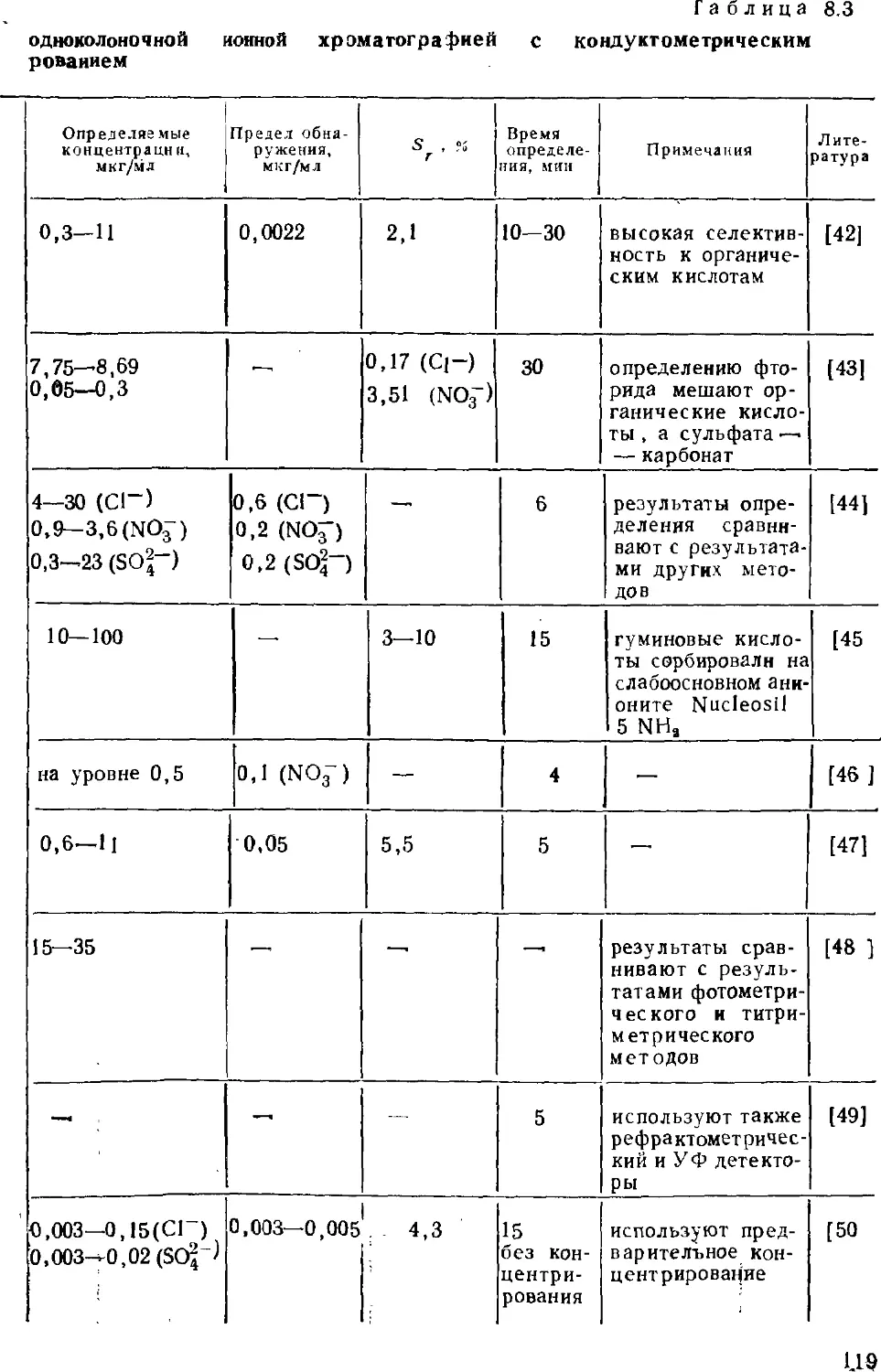
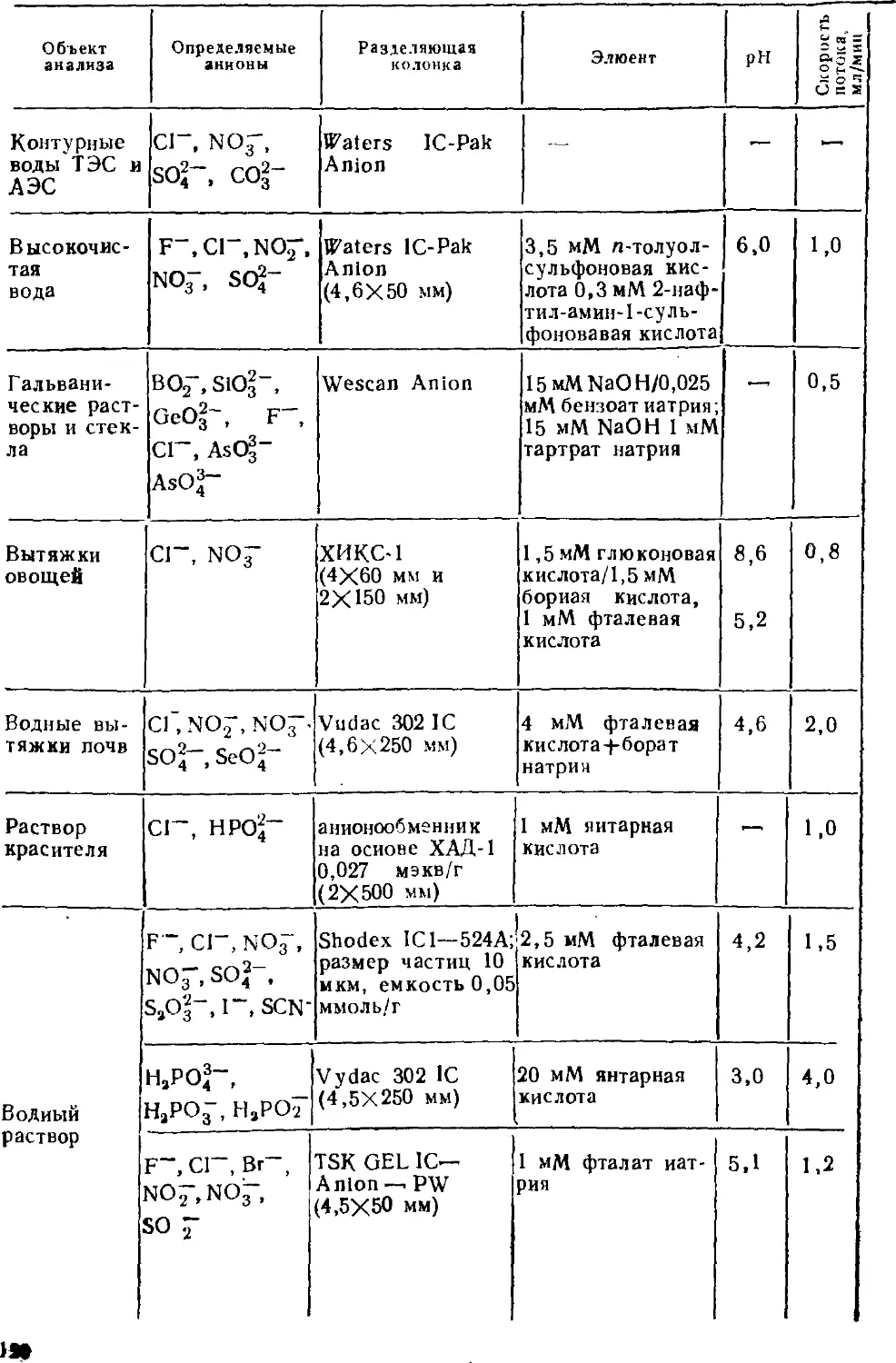
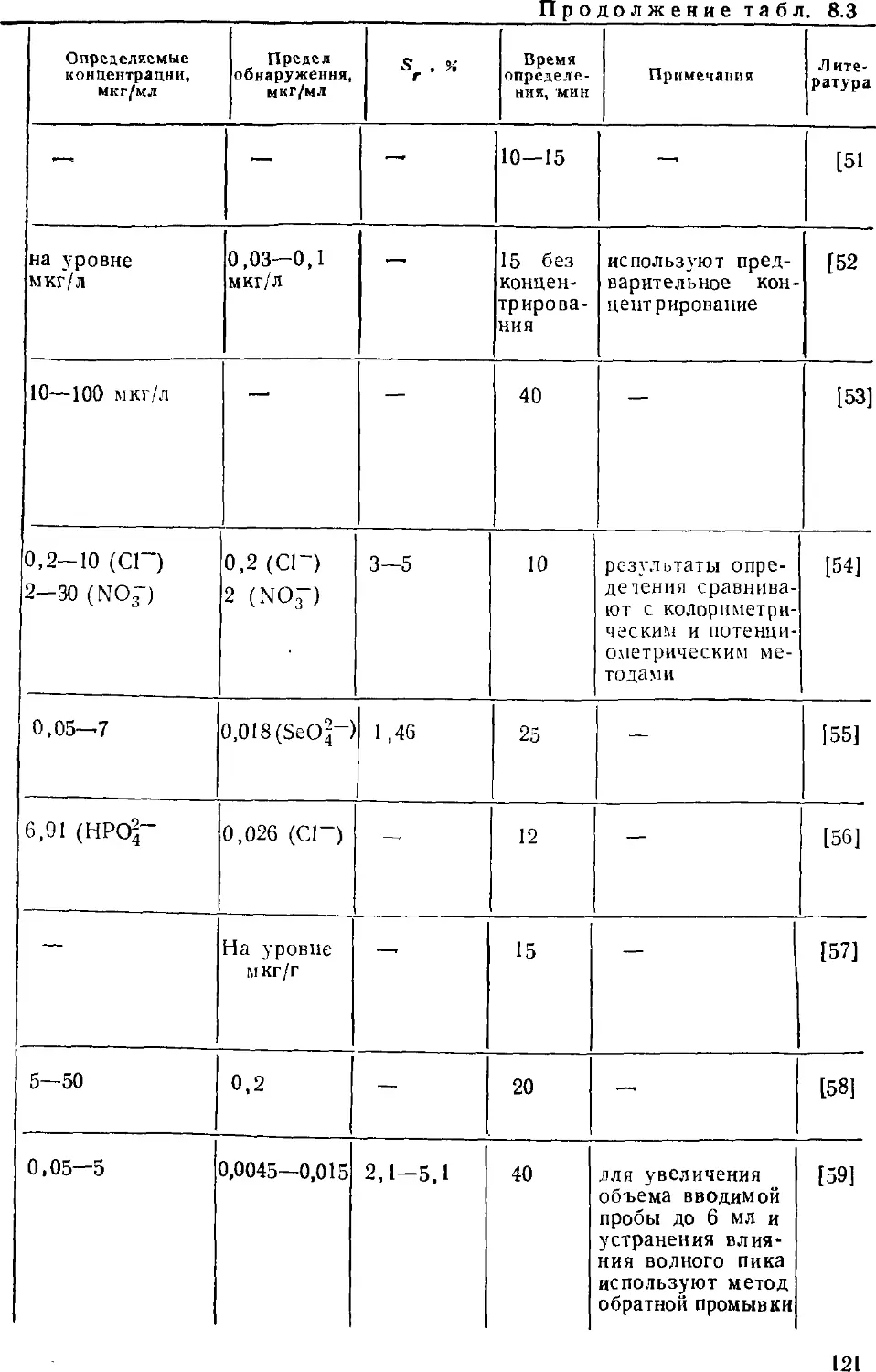
современных ионных хроматографов. Определению неорганических
анионов в водах посвящена восьмая глава; имеются в виду не
только обычные анионы, но и анионы слабых кислот. В
следующей главе собран материал об определении органических
соединений — алифатических и ароматических карбоновых кислот,
поверхностно-активных веществ. Тема десятой главы —
определение металлов; помимо щелочных и щелочноземельных
рассмотрены тяжелые металлы, существующие в катионной и анионной
формах. Одиннадцатая глава посвящена определению аминов.
Отдельная глава характеризует основные принципы совместного
определения анионов и катионов. Тринадцатая глава посвящена
особенностям ионохроматографического анализа вод различного
состава — поверхностных, пресных, атмосферных осадков,
сточных и сильно минерализованных вод, включая морские.
Авторы выражают благодарность своим сотрудникам и
коллегам за активное участие в исследованиях, результаты которых
нашли отражение в книге, за помощь в подготовке рукописи.
Все замечания о книге будут приняты с искренней
благодарностью.
ВВЕДЕНИЕ
История трех видов хроматографии — газовой, жидкостной
адсорбционной и ионообменной — оказалась очень похожей. Эти
методы проходят одни и те же этапы развития, как бы
заимствуя опыт друг у друга. Поэтому полезно вспомнить ход
становления первых двух, более сформировавшихся, направлений
хроматографического анализа, а затем и историю ионообменной и
ионной хроматографии. В сущности ионная хроматография
является современным автоматизированным вариантом
ионообменной хроматографии, но с принципиальным отличием: это уже
не только метод разделения, но и метод определения. Точно так
же, как и современная газовая и жидкостная адсорбционная
хроматография. ,
Начало хроматографии положила работа М. С. Цвета [1],
опубликованная в 1906 г. После этого почти тридцать лет
хроматографию практически не использовали. Возрождение
метода относится к 1931 г., когда Р. Кун, А. Винтерштейн и Е. Леде-
рер [2] выделили а- и ? -каротин из сырого каротина, используя
для этого метод Цвета. Широкое применение хроматография
получила в начале 40-х годов, после работы Мартина и Синга [3],
предложивших представлять хроматографическую систему как
некоторое число теоретических тарелок и создавших
распределительный вариант хроматографии. Ими впервые были даны
количественные характеристики хроматографического процесса, что
позволило определить пути повышения эффективности
хроматографического разделения.
Вначале эти пути были использованы в газовой
хроматографии. В 50-fe годы появились первые газовые хроматографы, в
которых для повышения эффективности разделения газообразных
веществ применяли длинные колонки, заполненные
мелкозернистым сорбентом. Для повышения скорости перемещения
подвижной фазы в этих приборах использовали мощные компрессоры.
Были созданы универсальные чувствительные детекторы для
газовой хроматографии, такие как детектор по теплопроводности
(катарометр), пламенно-ионизационный и др. В 1957 г. М. Го-
лей [4] предложил наносить сорбент на внутренние стенки
капиллярной трубки, что позволило еще больше повысить
эффективность разделения многокомпонентных смесей. Благодаря
многочисленным достижениям газовая хроматография уже более
тридцати лет является высокоэффективным методом
разделения и определения компонентов в сложных смесях газообразных
веществ.
В отличие от газовой жидкостная хроматография долгое вре-
мя не находила широкого применения и развивалась медленно.
Одной из основных причин этого было отсутствие
высокочувствительных универсальных детекторов для жидкостей. Это
вызвало необходимость применять химические методы анализа
растворов, вымываемых из хроматографической колонки, что в свою
очередь требовало относительно больших объемов исследуемых
веществ и времени анализа. Имевшийся в распоряжении
адсорбционной жидкостной хроматографии органических веществ
набор сорбентов не обеспечивал эффективного разделения смесей.
В конце 60-х годов интерес к жидкостной хроматографии
резко возрос. Родилась высокоэффективная жидкостная
хроматография. Этому способствовало создание высокочувствительных
детекторов (ультрафиолетовый, рефрактометрический), новых
селективных полимерных сорбентов, новой аппаратуры,
позволяющей работать при высоких давлениях. Все это привело к
значительному увеличению скорости хроматографического
процесса, повышению эффективности разделения смеси веществ и
возможности определять малые концентрации. Если в
классической жидкостной хроматографии разделение смеси обычно
проводилось в довольно длинных колонках диаметром 10—12 мм,
заполненных сорбентом с диаметром зерен 150—250 мкм, то в
современной высокоэффективной жидкостной хроматографии
(ВЭЖХ) применяют колонки диаметром 1—3 мм и сорбенты с
размером частиц менее 50 мкм. Благодаря этому по
эффективности разделения веществ жидкостная хроматография
практически не уступает газовой. Таким образом, современная
жидкостная (не ионообменная) хроматография, во многом благодаря
использованию опыта газовой хроматографии, стала
высокочувствительным, селективным и экспрессным методом разделения
и определения многокомпонентных смесей в растворах и методом
определения компонентов, главным образом органических [5].
Однако все это относилось не к жидкостной хроматографии
вообще, а лишь к ее вариантам, основанным на адсорбции, а
также на распределении между двумя жидкостями.
Особое место занимала ионообменная хроматография.
Явление ионного обмена было открыто более ста лет назад,
но интерес к нему не ослабевал, а наоборот возрастал. Большое
значение ионный обмен имеет в агрохимии, в процессах
жизнедеятельности, в химическом анализе и других областях.
Применение ионного обмена в химическом анализе заключалось в
концентрировании следов определяемых компонентов, удалении
мешающих ионов, в разделении близких по свойствам ионов. В
1935 г. Адаме и Холмс [6] разработали методику синтеза
высокомолекулярных органических сорбентов — ионообменных смол,
значение которых для развития ионного обмена трудно
переоценить.
Долгое время ионообменная хроматография являлась лишь
методом разделения смесей ионов. На эту тему написано
несколько монографий [7—9]. Многие методики отделения мешающих
ионов и разделения определяемых катионов или анионов с
помощью ионообменной хроматографии, несомненно, полезны и
широко используются в аналитических лабораториях. Однако эти
методики имеют ряд недостатков, обусловленных использованием
для разделения ионитов высокой емкости B—5 мэкв/г).
Использование таких ионитов требовало больших объемов элюентов
высокой концентрации. Разделение проводят в колонках
диаметром 10—20 мм, заполненных ионитом с размером зерен 100—
200 мкм. Элюент через такую колонку обычно двигался под
действием собственной массы, что не позволяло осуществлять
разделение быстро и эффективно. Отсутствовали универсальные
проточные детекторы для непрерывного анализа элюента.
Поэтому элюат собирали фракциями определенного объема,
каждую из которых анализировали обычными методами.
Для перевода ионообменной хроматографии в ранг
высокоэффективного метода определения ионов необходимо было решить
те же проблемы, какие стояли перед жидкостной адсорбционной
хроматографией 5—10 годами ранее. Надо было найти
чувствительный универсальный проточный детектор и создать
аппаратуру, позволяющую проводить высокоэффективное разделение,
ионов на мелкодисперсных ионитах.
Эти проблемы во многом были решены в 1975 г., когда Смолл,
Стивене и Бауман опубликовали работу [10], посвященную
новому аналитическому методу, названному ионной
хроматографией. С появлением этой работы начался новый этап в
развитии ионообменной хроматографии. В предложенном авторами
[10] варианте ионная хроматография представляет собой
высокоэффективную ионообменную хроматографию с кондуктометри-
ческим детектированием. Поскольку в ионообменной
хроматографии в качестве элюентов используют растворы сильных
электролитов, для снижения их фоновой электропроводности было
предложено после разделяющей колонки устанавливать вторую
колонку, названную подавляющей. Еще одной особенностью
ионной хроматографии является использование в разделяющей
колонке мелкозернистого ионита (диаметр зерен менее 40 мкм) с
очень низкой обменной емкостью @,01—0,1 мэкв/г). Это
позволило использовать сильно разбавленные растворы элюентов.
Специфическое строение ионита и высокое давление в системе
(до 10 МПа) обеспечивают высокую эффективность разделения
определяемых ионов. |
Удачно сочетание особенностей (кондуктометрическое
детектирование, подавляющая колонка, специфический
низкоемкостный ионит для разделения) делают ионную хроматографию
одним из наиболее эффективных методов определения ионов в
растворе.
Первоначальный вариант ионной хроматографии постоянно
совершенствуется. Появились разновидности метода. В 1979 г.
Гьерде, Фритц и Шмуклер [11] предложили вариант без
подавляющей колонки (одноколоночную ионную хроматографию). Вме-
сто кондуктометрического все чаще используют другие
детекторы, особенно прямой и косвенный УФ-детектор [12]. Ион-парный
вариант определения неорганических ионов на обращенной фазе
тоже часто относят к ионной хроматографии [13]. Поэтому
современную ионную хроматографию можно охарактеризовать как
метод высокоэффективного хроматографического разделения
ионов с последующим определением в проточном детекторе.
Прогресс ионной хроматографии поразителен. По темпам
развития (рост числа публикаций, выпуск приборов) этот метод
опережает ВЭЖХ. За немногие годы существования метода
опубликовано более 600 статей, несколько книг [14—17], большое
число обзоров [18—40].
Широкое распространение ионной хроматографии обусловлено
рядом ее достоинств:
а) возможность определять, очень большое число
неорганических и органических ионов, а также одновременно определять
катионы и анионы;
б) высокая чувствительность определения (до 1 нг/мл без
предварительного концентрирования) ;
в) высокая селективность и экспрессность (можно
определять 10 ионов за 15—20 мин, а при градиентном элюировании —
22 иона за 25 мин [41]);
г) малый объем анализируемой пробы (требуется не более
2 мл образца);
д) широкий диапазон определяемых концентраций (от
1 нг/мл до 1000 мг/л без разбавления);
е) возможность использования различных детекторов и их
комбинаций, что позволяет обеспечить селективность и малое
время определения;
ж) возможность полной автоматизации определения;
з) во многих случаях полное отсутствие предварительной про-
боподготовки.
Вместе с тем, как и любой аналитический метод, ионная
хроматография не лишена недостатков. На данном этапе развития
к недостаткам ионной хроматографии следует отнести:
а) сложность синтеза ионообменников, что существенно
затрудняет развитие метода;
б) низкую эффективность разделения по сравнению с ВЭЖХ
(максимальная эффективность была достигнута на анионообмен-
нике TSK-GelIEX-52O и составляет 25 000 теоретических
тарелок на метр [42]) ;
в) необходимость высокой коррозионной стойкости хроматог-
рафической системы, особенно при определении катионов.
Но эти недостатки несущественны и не могут повлиять на
развитие ионной хроматографии и на ее использование в
различных областях. Наиболее широко этот метод распространен в
анализе объектов окружающей среды [14, 15], медицине [43—
47], пищевой промышленности [48], геологии [49—511,
энергетике [52—55].
Одно из важнейших направлений использования ионной
хроматографии — анализ вод. Известно, насколько важно
определить компоненты вод разного типа — поверхностных пресных,
атмосферных осадков, морских, подземных, а также сточных.
Среди этих компонентов существенное место занимают
неорганические аиионы, ионы металлов, ионогенные органические
вещества. Ионная хроматография быстро заняла значительное место
в ряду аналитических методов, пригодных для определения
указанных компонентов.
В СССР метод развивается с 1980 г., первоначально в
лаборатории авторов в Московском университете. Первая научная
публикация, посвященная ионной хроматографии, опубликована в
Докладах АН СССР в 1982 г. [56]. Работы ведутся в Москве
(МГУ, ГЕОХИ АН СССР), Дзержинске (ОКБА НПО
«Химавтоматика»), Таллинне (Институт химии АН ЭССР). Исследования
направлены на создание новых, эффективных сорбентов, поиск
новых элюентов, систем подавления и детектирования,
разработку методик ионохроматографического анализа различных
объектов. В 1986 г. начат выпуск отечественного ионного
хроматографа Цвет-3006. Большую заинтересованность во
внедрении ионной хроматографии проявляют аналитические службы
Госкомгидромета, Госагропрома, Минхимнефтепрома, Минэнерго,
Минцветмета и ряда других министерств и ведомств.
ЛИТЕРАТУРА
1. T s w e 11 M. // Ber. Deut. Botan. Ges. 1906. В. 24. S. 384—387.
2. Kuhn R., Winter st ein A., Leder e г Е. 33 Hoppe Seyler's Z. Phi-
siol. Chem. 1931. B. 197. S. 141—145.
3. Martin A. J. P., Synge R. L. M. // Biochem. J. 1941. V. 35, № 8.
P. 1358—1365.
4. G о 1 a y M. J. // Anal. Chem. 1957. V. 29. № 7. P. 928—937.
5. Современное состояние жидкостной хроматографии. Пер. с англ. / Под
ред. Дж. Киркленда. М.: Мир, 1974. 325 с: нл.
6. Adams В. A., Holmes E. L.//J. Soc. Chem. Ind. London. 1935. V. 54,
№ 1. P. 1—10.
7. Самуэльсон О. Ионообменные разделения в аналитической химии.
Пер. с англ. М.: Мир, 1966. 416 с: ил.
8. Мархол М. Ионообменнвши в аналитической химии. Свойства и
применение в неорганической химии. Пер. с англ. — М.: Мир, 1985. — 545 с: ил.
9. Сенявин М. М. Ионный обмен в технологии и анализе неорганических
веществ. М.: Химия, 1S80. 272 с.
10. S m а 1 1 H., S t e v e n s T. S. В a u m a n W. S. // Anal. Chem. 1975. V. 47,
№ 10. P. 1801—1809.
11. G j erde D. T., Fritz J. S., Schmuck 1er G. // J. Chromatogr. 1979.
V. 186. P. 509—519.
12. Ha ddad P. R., Hecken ber g A L. // J. Chromatogr. 1984. V. 300,
№ 3. P. 357—394.
13. Weiss J., G ob 1 M // Fresenius Z Anal. Chem 1985. B. 320. S. 439—
444.
14. Ion Chromatographie Analysis oi Environmental Pollutants/Ed, by
E. Sawicki, J. D. Mulik and E. Wittgenstein Ann Arbor: Ann Arbor Sei., 1978.
210 p.
15. Ion Cvormifographic Analysis of Environmental Pollutants / Ed. by J. D.
Mulik and E. Sawicki. Ann Arbor: Ann Arbor Sei., 1979, 435 p.
16. Фритц Дж., Гьерде Д., Полаид К. Ионная хроматография. Пер.
с англ. / Под ред. В. Г. Березкина. М..: Мир, 1984. 221 с.
17. S m і t h F. С, С h a n g R. C. Practice of Ion Chromatography. New York:
John Wiley and Sons, 1983. 218 p.
18. V u k a m о H. // Kem.-Kerni. 1979. V. 6, № 4. P. 190—192.
19. M a с D о n a 1 d J. // Am. Lab. 1979. V. 11, № 1. P. 45—55.
20. R і с h W. // Instrum. Technol. 1977. V. 24, № 8. P. 47—51.
21. R і с h W. // Anal. Instrum. 1977. V. 15. P. 113—117.
22. J a n s e n K. // Labor. Prax. 1978. B. 2, № 3. S. 30—34.
23. N о m u r a T. // Kazaku to Kogio. 1978. V. 52. P. 448—454.
24. T s u с h і t a n і J. // Bunseki Kagaku. 1979. V. 29, № 8. P. 603—608.
25. Smith F., С h a n g R. // Crit. Rew. Anal. Chem. 1980. V. 9 № 3.
P. 197—217.
26. Bogoczek R., M і e m u s G. // Przem. Chem. 1980. V. 59, №9.
P. 471—474.
27. Pohl С. A., Johnson E. L. // J Chromatogr. Sei 1980. V. 18 № 9.
P. 442-452.
28. S m a 1 1 H. // Trace Anal. 1981. V 1. P. 267—273.
29. S t r a y H // Kiemi. 1981. V. 4, № 1. P. 30—35.
30. Q і D, Q u C, Z h о u T. // Huaxue Tongbao. 1982. V. 8. P. 492.
31. Rokushika S. // Kagaku. 1982. V. 37, № 3. P. 557—560.
32. Шпигун O. A, 3 о л о т о в Ю. А. // Зав. лаб. 1982. Т. 48, № 9.
С. 4—14.
33. M о s e s С. // GIT Fachs. Lab. 1982. V. 26, № 3. P. 241—245.
34. W e і s s J. // Chem. Labor. Betz. 1983. B. 34. S. 293—342.
35. В u с h e r P. // Schweiz. Lab. Z. 1983. B. 40. S. 91 —157.
36. S m a 11 H. // Anal. Chem. 1983. V. 55, № 2. P. 235 A—242 A.
37. R u s e v a E. // Khim. Ind. 1983. V. 55, № 2. P. 126—133.
38. S с h w e d t G. // Labor. Prax. 1984. B. 8, № 1. S. 30—34.
39. P о h 1 a n d t C. // South. Afr. J. Sei. 1984. V. 80. P. 208—213.
40. F r і t z J. S. // HPLC Mag. 1984. V. Z. P. 446—449.
41. J о h n s о n L. // Analitica. 1986. № 1. P. 43—47.
42. M a t s u s h і t a S. et al. // J. Chromatogr. 1983. V. 259, № 3. P. 459—464.
43. Anderson С.//Clin. Chem. 1976. V. 22, № 9. P. 1424—1426.
44. L і n d g r e n M., CedergrenA. Lindberg J. // Anal. Chim Acta.
1982. V. 141. P. 279—286.
45. Whit taker J., Lemke P. // J. Pharm. Sei. 1982. V. 71. P. 334—337.
46. De Jong P., Berggraaf M. // Clin. Chim. Acta. 1983. V. 132.
P. 63—67.
47. H і e f t j e G. // Clin. Chem. 1983. V. 29, № \Q. p. 1659—1662.
48. E d w a r d s P. // Food Techno]. 1983. V. 37, № 1. P. 53—56.
49. Smee В., Hall G., Koop D. // J. Geochem. Explor. 1978. V. 10.
P. 245—249.
50. L a s h R., H і 1 1 C. // Anal. Chim. Acta. 1979. V. 108. P. 405—409.
51. Evans K., Tarter J., Moore С. // Anal. Chem. 1981. V. 53, № 6.
P. 925—930.
52. Rich W., Wetze 1 R. // Actual. Chim. 1980. V. 6, № 1. P. 51—54.
53. Borman S. // Anal. Chem. 1980 V. 52, № 12. P. 1409A—1412A.
54. L о w J. // Power Eng. 1981. V. 85. P. 94—98.
55 В r a n d t F. Trost R. // VGB Kraftwerktech. 1984. В. 64. S. 74—80.
56. Зо лотов Ю. А и др. окл. АН СССР. 1982. Т. 263, № 4, С. 889—892.
ГЛАВА 1
ОСНОВЫ
ИОНООБМЕННОЙ ХРОМАТОГРАФИИ
Поскольку разделение веществ в ионной хроматографии
базируется на ионном обмене, полезно рассмотреть основные
положения ионообменной хроматографии.
Этот метод основан на эквивалентном обмене ионов раствора
на ионы твердой фазы. В отличие от адсорбции ионный обмен
описывается стехиометрическим химическим уравнением, что
важно и для ионной хроматографии. Однако четкую грань
между адсорбцией и ионным обменом провести трудно, так как на
ионообменниках часто наблюдается и физическая адсорбция.
Ею нельзя пренебрегать особенно при ионохроматографическом
определении органических веществ или их использовании в
качестве элюентов.. И все-таки основную роль при ионообменном,
а следовательно, и ионохроматографическом разделении
веществ играет ионообменное равновесие.
1.1.
ИОНООБМЕННОЕ РАВНОВЕСИЕ
Есть три точки зрения на ионообменный процесс: его
рассматривают как мембранное равновесие [1], осмотический процесс
[2] и гетерогенную химическую реакцию двойного обмена [3].
Существуют соответствующие теории. Наиболее распространенной
является последняя.
Согласно этой теории реакцию обмена двух однозарядных
катионов А+ и В+ можно записать следующим образом (R —
матрица ионообменника) :
Константа равновесия этой реакции (Кв), называемая
константой ионного обмена, имеет вид
[ВЪ _ к л [В+]т
Здесь [A]s, [В] s — равновесные концентрации в фазе ионита
ионов А+ и В - соответственно, [А +]„,, [В+]т— равновесные
концентрации ионов А+ и В + в жидкой фазе.
И
Физический смысл константы ионного обмена заключается в том,
что она позволяет дать количественную характеристику способности
ионообменника к обмену с теми или иными ионами из растра,
т. е. выражает преимущественную сорбцию одного из двух
обменивающихся ионов. Поэтому константу ионного обмена часто
называют коэффициентом селективности.
Коэффициент селективности может быть больше, меньше или
д
равен единице. Если/Гв >1, то ион В+, находящийся в растзоре,
имеет большее сродство к ионообменнику, чем ^ион А+ , и пре-
имущественно сорбируется ион В+. Если Кв < 1, то направление
процесса меняется, и большее сродство к иониту имеет ион А+ .
ПриКв = 1 сродство ионов А+ и В+ к твердой фазе одинаково.
Если обмениваются ионы, имеющие разные заряды,
коэффициент селективности равен
л\
[Bis
[А-Ч„
1/2,
A.2)
Сродство к ионообменнику в значительной степени зависит
от заряда ионов. Многозарядные ионы сорбируются, как
правило, сильнее, что может быть объяснено возрастанием энергии
взаимодействия многозарядных противоионов с фиксированными
ионами ионитов. Из уравнения A.2) можно сделать вывод, что
уменьшение концентрации ионов в растворе при сохранении
постоянства отношения их концентраций приводит к увеличению
сорбции попов с большим зарядом.
Наряду с коэффициентом селективности для количественной
характеристики ионообменников пользуются концентрационным
коэффициентом распределения D: дд __ (АЬ л з)
["¦lin
Он представляет собой
отношение равновесной концентрации
иона в сорбенте к его
равновесной концентрации в растворе.
Важное значение при изучении
ионообменного равновесия имеет
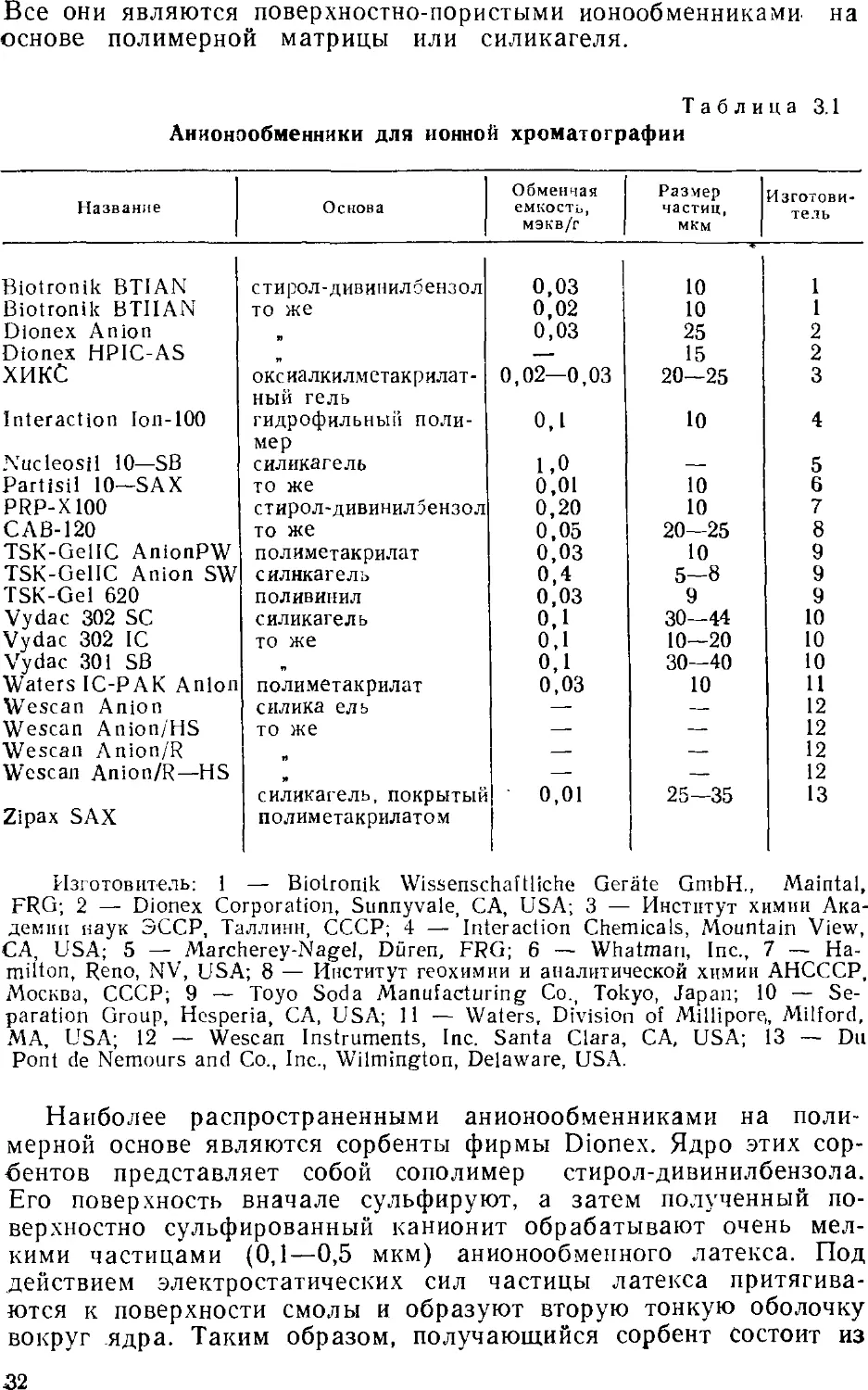
изотерма ионного обмена. Как
видно из рис. 1.1, изотерму
обмена можно
характеризовать графиком зависимости
равновесной концентрации иона в
ионнообменнике от его
равновесной концентрации в растворе.
Рис. 1.1. Изотермы ионного Существует три основных типа
обмена. изотерм ионного обмена—ли-
/ - пып>™^огн2т~ линейная> нейная, выпуклая и вогнутая. Ча-
12
ще всего встречаются выпуклые изотермы, характерные для
случая, когда поглощаемый ион имеет большее сродство к иониту,
чем ион, первоначально в нем находившийся. Если поглощаемый
ион сорбируется слабее, чем ион, находившийся в ионообменни-
ке, то получается вогнутая изотерма. Если сродство обоих
ионов к смоле одинаково, изотерма ионного обмена линейна.
На практике изотермы обмена, линейные в широком
интервале концентраций, наблюдаются лишь в исключительных
случаях. Если обмену подвергаются очень малые количества ионов,
то изотерму обмена можно считать линейной. Иными словами,
отношение [А] ДА]m в узком интервале концентраций можно
считать постоянным (но не равным единице). Этот случай важен
для аналитических разделений методом элюентной
хроматографии, это наблюдается и в ионной хроматографии.
1.2.
ЭЛЮЕНТНОЕ РАЗДЕЛЕНИЕ
Ионная хроматография представляет собой вариант
колоночной элюентной ионообменной хроматографии, поэтому
целесообразно рассматривать основные понятия элюентного
ионообменного хроматографического разделения. В элюентной колоночной
хроматографии подвижная фаза (элюент) служит, как правило,
только для перемещения вещества через хроматографическую
систему. Разделение в этом случае происходит благодаря
разному сродству компонентов определяемой смеси к неподвижной
фазе и, следовательно, разным скоростям перемещения по
колонке. Неподвижной фазой в ионообменндй хроматографии является
ионообменник.
1.2.1. Объем и время удерживания
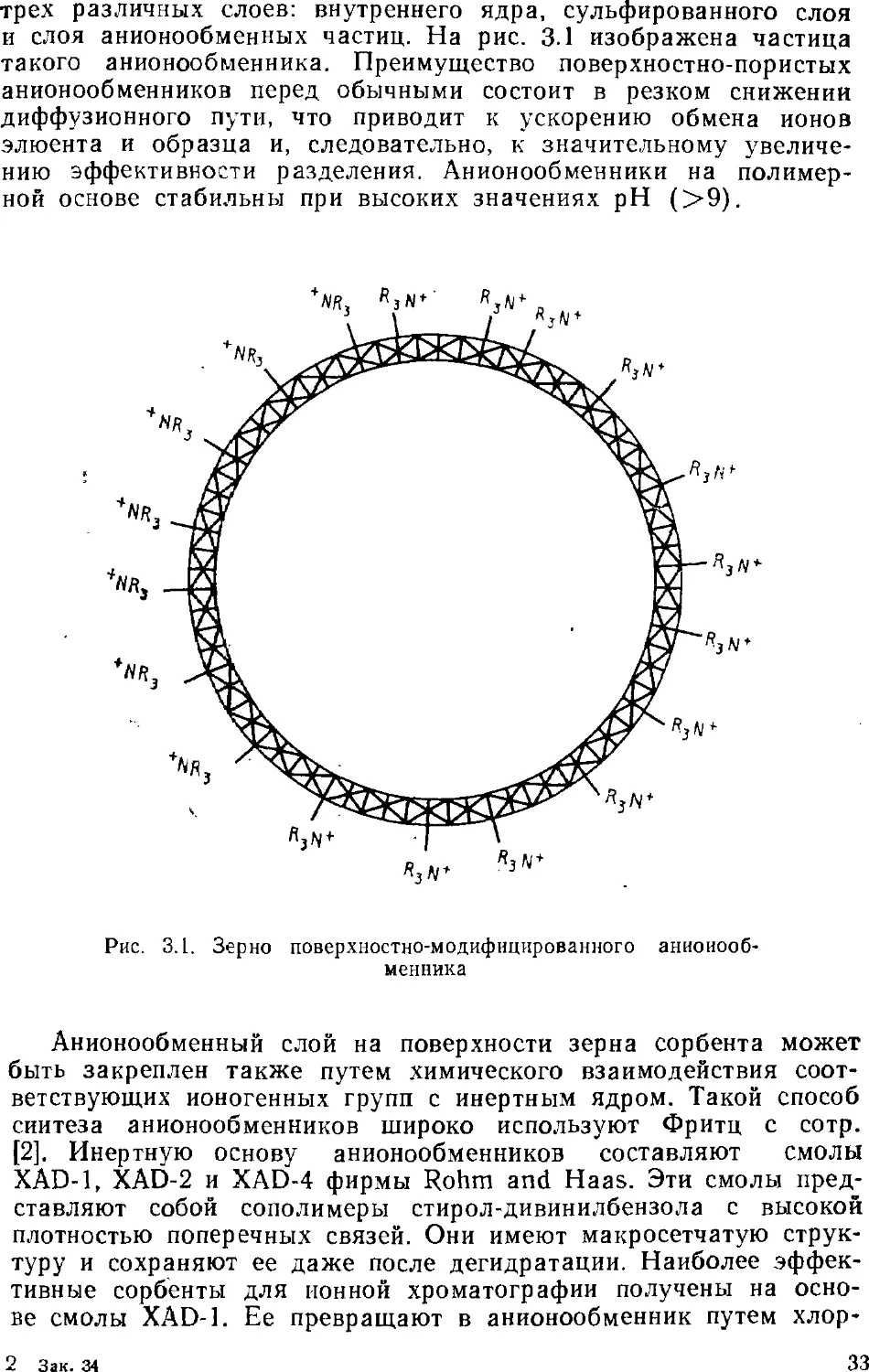
Величины, характеризующие хроматограмму, полученную
элюентным методом, приведены на рис. 1.2.
Рнс. 1.2. Хроматограмма, полученная элюентным методом
13
Объем элюента, необходимый для извлечения из хроматогра-
фической колонки максимальной концентрации вещества,
называют общим объемом удерживания Kr . «Мертвый» объем хрома-
тографической системы Vo равен общему свободному объему
системы, включающему систему ввода, колонки, трубопроводы и
детектор. На практике Vo определяют как время удерживания
иесорбируемого компонента. Вычитая из V R величину Vo,
получают исправленный объем удерживания Vі :
VR = VR-V0. A.4)
При автоматической регистрации хроматограмм часто
используют время удерживания t^. Это время, необходимое для элюи-
роваиия вещества до его максимальной концентрации. Скорость
движения диаграммной ленты постоянна, поэтому время
удерживания можно измерить непосредственно по хроматограмме. По
величине tu и объемной скорости F протекания элюеита через
колонку находят объем удерживания:
VR=^RF. A.5)
Общий объем удерживания иона связан с его коэффициентом
распределения уравнением
Vk=DVs+V0, A.6)
где Vs—объем ионита.
Учитывая уравнение A.4), получаем
VR = DVS. A.7)
Уравнения A.6) и A.7) являются основными уравнениями
хроматографии. Они показывают, что объем или время
удерживания иона пропорциональны его коэффициенту
распределения и объему ионита. Чем выше коэффициент распределения
иона, тем ниже скорость его движения по колонке.
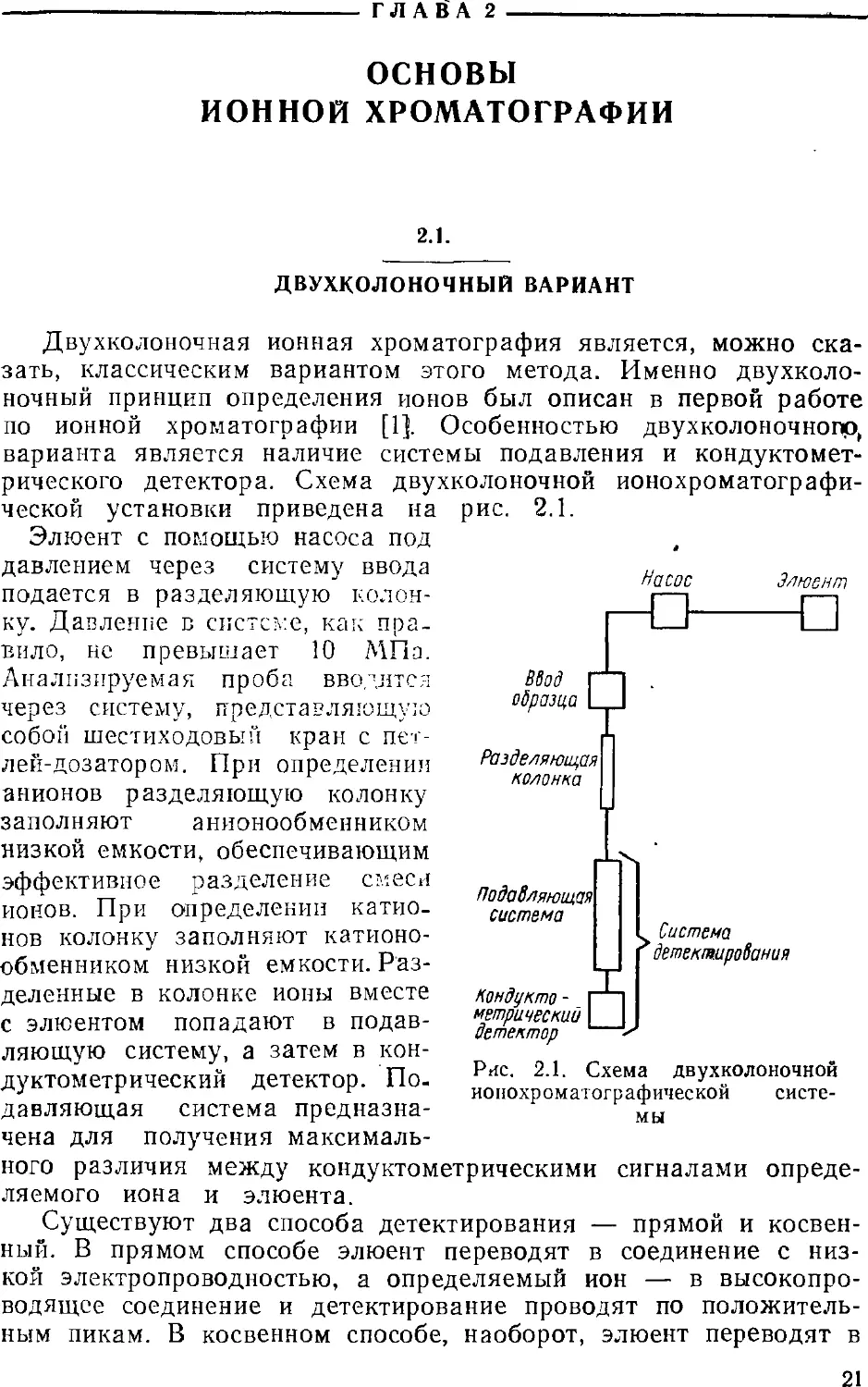
Форма хроматографнческого пика и время (объем)
удерживания зависят от типа изотермы ионного обмена (рис. 1.3). В
случае линейной изотермы пик симметричен, а объем
удерживания не зависит от концентрации определяемого иона. Для
выпуклой изотермы хроматографический пик имеет асимметричную
форму с различным задним фронтом, а объем удерживания
уменьшается с увеличением концентрации определяемого иона.
Обратная картина наблюдается для вогнутой изотермы. В этом
случае различен передний фронт хроматографического пика, а
объем удерживания увеличивается. Очевиден вывод, что
концентрации определяемых ионов должны находиться в области
линейной изотермы ионного обмена.
1.2.2. Селективность
Селективность является мерой взаимного распределения двух
или более определяемых веществ в ходе хроматографического
процесса. Хроматографическое разделение основывается на
селективности сорбента и различиях в термодинамических свой-
М
ствах определяемых веществ по отношению к
хроматографической системе. Таким образом, селективность является мерой
относительного удерживания или относительной подвижности двух
веществ. На рис. 1.2 разность в удерживаниях представлена как
расстояние между центрами зон AV, где центры зон
соответствуют максимумам хроматографических пиков.
Согласно уравнению A.6) AV равна
AV = К,- V1 = (D, Vs+V0) - (Dy Vs+V0) = (Dt-DJ Vs. A.8)
Из уравнения A.8) видно, что селективность зависит от
различия коэффициентов распределения определяемых веществ и
количества неподвижной фазы. Если Di=D2, то хроматої рафиче-
ское разделение невозможно.
Наиболее распространенным способом повышения
селективности является увеличение V ,. за счет увеличения длины
хроматографической колонки. Однако увеличение длины колонки
ведет к возрастанию размывания хроматографических пиков.
На практике селективность чаще характеризуют величиной
относительного удерживания а:
я у* -У»YI A~?* /19)
Если а=1, то это означает, что в данной системе отсутствует
термодинамическое различие между обоими компонентами и их
нельзя разделить. Но даже в том случае, когда коэффициенты
распределения двух веществ различаются достаточно сильно,
нельзя заранее сказать, что разделение будет хорошим.
Необходимо знать, какова эффективность хроматографической системы.
1.2.3. Эффективность
Под эффективностью в хроматографии понимают способность
системы препятствовать размыванию хроматографических зон
(пиков) определяемых веществ. Эффективность характеризует
кинетические свойства хроматографической системы.
Мерой размывания хроматографической зоны является высота,
эквивалентная теоретической тарелке (ВЭТТ). Чем меньше
ВЭТТ, тем меньше размывание хроматографической зоны и тем
выше эффективность разделения. ВЭТТ для одного из
компонентов можно определить непосредственно из хроматограммы
(см. рис. 1.2), пользуясь уравнением
где h — ВЭТТ, L — длина колонки, W — ширина пика у
основания, / — время удерживания данного компонента.
15
Для характеристики размывания используют понятие числа
теоретических тарелок N:
N = L/h = 16(//W)*. A.11)
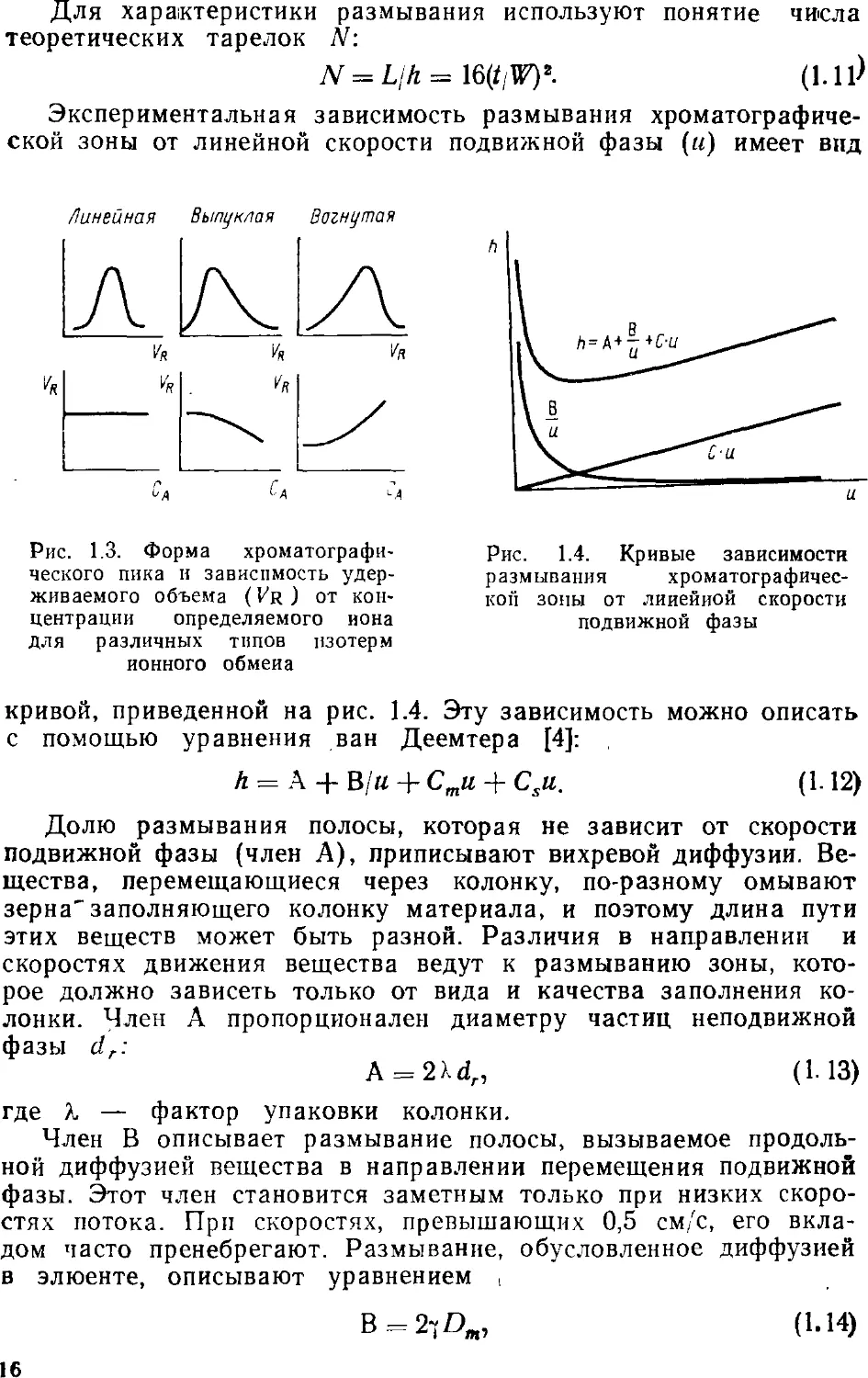
Экспериментальная зависимость размывания хроматографиче-
ской зоны от линейной скорости подвижной фазы (и) имеет вид
/Іинеиная Выпуклая Вогнутая
A /V/Y
Рис. 1.3. Форма хроматографи-
ческого пика и зависимость
удерживаемого объема (Vr ) от
концентрации определяемого иона
для различных типов изотерм
ионного обмена
Рис. 1.4. Кривые зависимости
размывания хроматографичес-
кон зоны от лииейиой скорости
подвижной фазы
кривой, приведенной на рис. 1.4. Эту зависимость можно описать
с помощью уравнения ван Деемтера [4]: ,
А = А + В/и + Сти + Csu.
A.12)
Долю размывания полосы, которая не зависит от скорости
подвижной фазы (член А), приписывают вихревой диффузии.
Вещества, перемещающиеся через колонку, по-разному омывают
зерна'заполняющего колонку материала, и поэтому длина пути
этих веществ может быть разной. Различия в направлении и
скоростях движения вещества ведут к размыванию зоны,
которое должно зависеть только от вида и качества заполнения
колонки. Член А пропорционален диаметру частиц неподвижной
фазы dr:
где к — фактор упаковки колонки.
Член В описывает размывание полосы, вызываемое
продольной диффузией вещества в направлении перемещения подвижной
фазы. Этот член становится заметным только при низких
скоростях потока. При скоростях, превышающих 0,5 см/с, его
вкладом часто пренебрегают. Размывание, обусловленное диффузией
в элюенте, описывают уравнением ,
В = 2-(o , A.14)
16
где Dm—коэффициент диффузии вещества в подвижной фазе,
Y учитывает ограничение пути диффузии в заполненной колонке.
Эффективная диффузия в направлении оси колонки в
жидкостной хроматографии при высоких скоростях больше, чем
продольная диффузия. Эту долю размывания зоны, вызванную
диффузией в движущемся элюенте, связывают с членом Ст. Он
зависит от диаметра частиц неподвижной фазы и обратно
пропорционален Dm:
DJ. A.15)
Величина Ф является функцией только коэффициента емко-
сти К', равного -f •
При движении через колонку молекулы или ионы
анализируемой пробы постоянно переходят из подвижной фазы в
неподвижную или обратно. Если молекула сорбируется, то она
отстает от центра зоны, которая продолжает двигаться вдоль
колонки. Когда эта молекула возвращается из неподвижной фазы в
подвижную, она движется быстрее, чем точка центра
удерживаемой зоны, так как скорость элюента всегда выше, чем средняя
скорость продвижения зоны вещества. Этот процесс ведет к
размыванию зоны вещества, которое описывается так называемым
членом массопередачи Cs в неподвижной фазе:
С, = const/ (K'X^'DJ- A-16)
Хіробь df/Ds служит мерой длительности задерживания в
неподвижной фазе.
Ценность уравнения ваи Деемтера A.12) в том, что с его
помощью можно определить условия, позволяющие свести к
минимуму размывание зон и достичь максимального разрешения. В
соответствии с уравнением A.13) вклад вихревой диффузии
можно свести к минимуму, добиваясь однородного заполнения
колонки и используя частицы неподвижной фазы малого диаметра.
Применительно к колоночной хроматографии было найдено, что
величина X уменьшается с уменьшением диаметра колонки. Для
ограничения диффузии в подвижной фазе (уравнение (L.15))
следует использовать мелкодисперсные частицы и стремиться к
однородному заполнению колонки. Кроме того, частицы малого
диаметра необходимо применять для более плотного заполнения
колонки и уменьшения объема подвижной фазы, через которую
перемещается вещество перед переходом в неподвижную фазу.
Анализ уравнения ван Деемтера показывает, что наиболее
эффективным способом уменьшения размывания является
использование мелкодисперсной неподвижной фазы. Поэтому в
современных высокоэффективных жидкостных хроматографах
используют неподвижные фазы с диаметром частиц 10 мкм и менее.
17
1.2.4. Разрешение
Разрешение хроматографической системы является мерой
полноты разделения двух веществ. В то время как селективность
характеризует разделение центров зон, разрешение является
характеристикой разделения самих зон. Разрешение R является
функцией селективности и эффективности системы и
определяется расстоянием между максимумами пиков (выраженным как
разность времен удерживания) и средней арифметической
шириной обоих пиков у основания:
В хроматографии стремятся не к наибольшему, а к
оптимальному разрешению, т. е. пики должны отстоять друг от друга
только на требуемое расстояние. Если пики имеют форму гауссовых
кривых, то для количественного анализа достаточно разделение
с R=l,5 (называемое также бст-разрешением), так как для
гауссовой кривой R = 4a. В этом случе пики отделены друг от друга
практически до нулевой линии. Большего разделения
добиваются, увеличивая длительность определения. Если # = 1,0,
расстояние между двумя пиками точно равно 4сг (в этом случае
говорят о 4ст-разрешении). Для количественного анализа такое
разрешение еще достаточно, так как только 2% площади пиков
перекрывается.
1.3.
ОБЩИЕ СВЕДЕНИЯ ОБ ИОНООБМЕННИКАХ
Свойствами ионообменников обладает довольно большое
число различных природных и синтетических соединений.
Важнейшими из них являются синтетические полимерные смолы и
некоторые минеральные ионообменники. Любой ионообменник
органической или неорганической природы представляет собой
матрицу, содержащую способные к обмену ионогенные группы.
Природные минеральные ионообменники являются, как
правило, кристаллическими силикатами, жесткая решетка которых
несет избыточный заряд. Наиболее важными представителями
этой группы ионообменников считаются цеолиты, способные к
обмену катионами (катионообменник). Они обладают
правильной пространственной сетчатой структурой со сравнительно
большими расстояниями между узлами решетки. Роль противоионов
играют ионы щелочных и щелочноземельных металлов.
Наибольшее практическое применение нашли синтетические
органические иониты. Большинство этих ионообменников имеет
матрицу из сополимера стирола с дивинилбензолом (ДВБ). Этот
сополимер легко образуется и обладает достаточно высокой
физической и химической устойчивостью в различных условиях.
Требуемые ионогенные группы могут быть относительно легко при-
18
соединены к этой матрице при соответствующих химических
реакциях.
Свойства матрицы определяются в значительной степени
соотношением количеств индивидуальных мономеров. Ионообмен-
ники с низким содержанием ДВБ (менее 4%) сильно набухают
в водных растворах. Механическая прочность матрицы
уменьшается с уменьшением доли ДВБ. Ионообменники с матрицами,
доля поперечносшивающего вещества в которых велика (более
15% ДВБ), набухают в водных растворах в незначительной
степени. Их механическая устойчивость высока. Для матриц с
высокой степенью сшивки число ионогенных групп, которое может
быть присоединено к каркасу, ограничено и уменьшается с
увеличением процента ДВБ. Обычно для синтеза ионообменников
используют матрицы с 5—8% ДВБ.
Наиболее распространены два типа структур матриц. Если
матрицу синтезируют без добавления других веществ
(особенно растворителей), то в результате сополимеризации стирола и
ДВБ образуется каркас гелевого типа. Этот тип матрицы
состоит из взаимопроникающих сеток, образованных
индивидуальными цепями. Размер пор, который определяется расстоянием
между индивидуальными полимерными цепями, очень мал.
Ионообменники, имеющие каркас с подобными свойствами, называются
гелевыми, или микросетчатыми смолами. Другой тип структуры
матрицы образуется при введении в полимеризационную
систему растворителя, растворяющего мономер. Полученная таким
образом матрица имеет макропористую или макросетчатую
структуру. Такие матрицы имеют губчатую структуру, состоящую из
агрегатов сфер нормальной гелевой пористости, пронизанных
порами большого диаметра. Размер пор можно регулировать в
процессе получения матрицы. Ионообменники, имеющие такую
матрицу, называют макропористыми.
Полимер может быть использован в качестве ионообменника
только после введения в матрицу ионогенных групп. Ионогенная
группа состоит из двух ионов. Один из них прочно фиксируется
на матрице за счет ковалентной связи и называется
функциональной группой (фиксированным ионом). Ионы
противоположного заряда связываются с фиксированным ионом за счет
электростатического взаимодействия. Они называются противоионами.
Эти ноны могут обмениваться на эквивалентное количество
ионов того же заряда из раствора.
В соответствии с характером фиксированных ионов
ионообменники делятся на следующие: ,
катионообменники — содержат анионы (фиксированные
группы — SO-, —COO-, — РО|-, AsO^-);
анионообменники — содержат катионы (фиксированные
группы —NR:!, —NHR2, -NH2R, др.);
амфотерные ионообменники — содержат анионы и катионы.
1»
Реакция обмена ионов на катионообменнике записывается в
виде
RH+Na+*«=R—Na+H+,
а на анионообменнике
RC1
Символом R принято обозначать матрицу с фиксированным
ионом.
В зависимости от силы сопряженой кислоты (или
основания) фиксированного иона ионообменники делятся на
сильнокислотные, среднекислотные и слабокислотные (или основные).
Классификация ионообменников дана в табл. 1.1.
Таблица 1.1
Классификация ионообменников
Ионооэменник
Катионообменник
Анионообменник
Тип
сильнокислотный
сред некие лотньііі
слаэокислотный
сильноосновный
среднеосновный
слабоосновный
Фиксированные ноны
-РО§-, -Аз0|-
—СО О H
NR3
—NR3/-N!!Ra
—NHSR
К важнейшим свойствам ионообменников относится их
обменная емкость. Каждый ионообменник содержит определенное
число фиксированных ионов. Число таких ионов в единице
массы (объема) ионообменника характеризует его обменную
емкость. Обменную емкость принято выражать в миллиэквивалентах
обменивающегося иона на грамм сухого ионообменника. Для
наиболее распространенных в ионообменной хроматографии смол
обменная емкость составляет 3—5 мэкв/г.
ЛИТЕРАТУРА
1. D о п п а п F. // Elekrochem. 1911. V. 17. Р. 572—585.
2. G reg or H. // J. Am. Chem. Soc. 1951. V. 73, № 4. P. 642—650.
3. Никольский Б. П., Парамонова В. И. // Успехи химии. 1939.
Т. 8, № 10. С. 1535—1567.
4. VanDeemterJ. J., ZuiderwegF. J., Klinkenberg А. // Chem.
Eng. Sei. 1956. V. 5. P. 271—280.
ГЛАВА 2
ОСНОВЫ
ИОННОЙ ХРОМАТОГРАФИИ
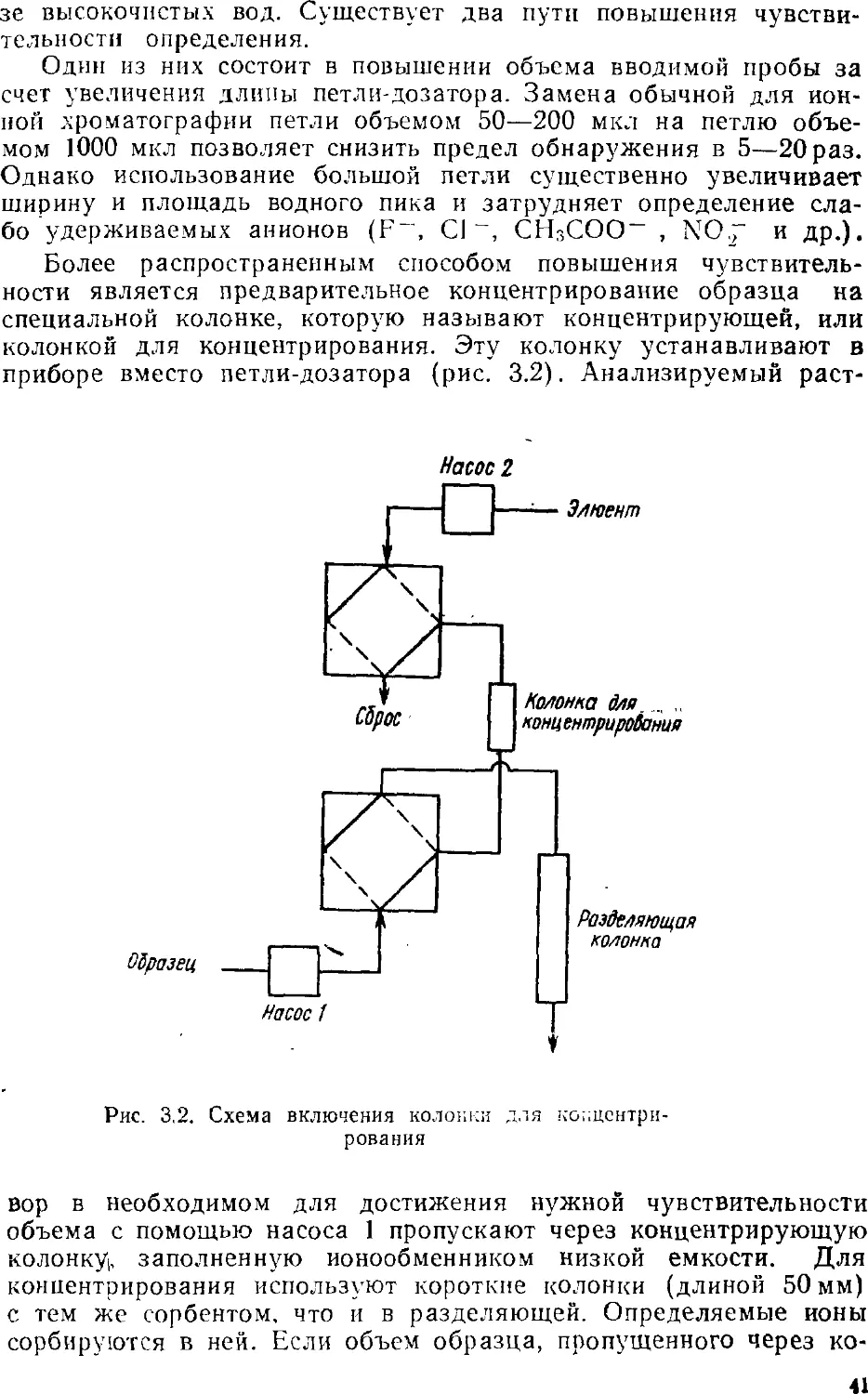
2.1.
Насос
Элюент
-а
образца
а
Разделяющая
колонка
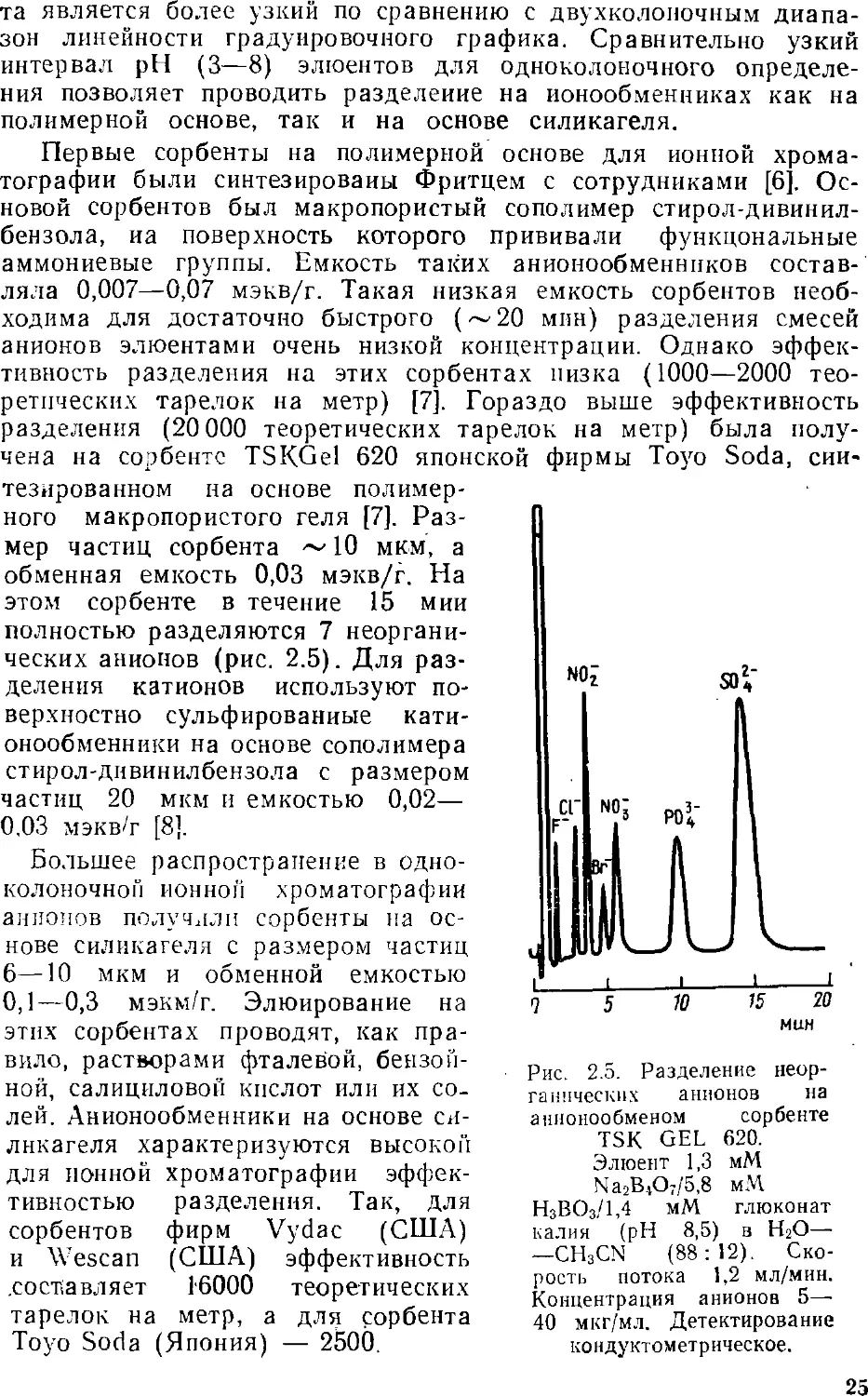
ДВУХКОЛОНОЧНЫЙ ВАРИАНТ
Двухколоночная ионная хроматография является, можно
сказать, классическим вариантом этого метода. Именно двухколо-
ночный принцип определения ионов был описан в первой работе
по ионной хроматографии [1]. Особенностью двухколоночнопр,
варианта является наличие системы подавления и кондуктомет-
рического детектора. Схема двухколоночной ионохроматографи-
ческой установки приведена на рис. 2.1.
Элюент с помощью насоса под
давлением через систему ввода
подается в разделяющую
колонку. Давление в системе, как
правило, не превышает 10 МПа.
Анализируемая проба вводлтс.-і
через систему, представляющую
собой шестиходовый кран с
петлей-дозатором. При определении
анионов разделяющую колонку
заполняют анионообменником
низкой емкости, обеспечивающим
эффективное разделение смеси
ионов. При определении
катионов колонку заполняют катионо-
обменником низкой емкости.
Разделенные в колонке ионы вместе
с элюентом попадают в
подавляющую систему, а затем в кон-
іїуктометпический летектоо По
дуктометрическии детектор, но.
давляющая система предназна-
чена для получения
максимального различия между кондуктометрическими сигналами
определяемого иона и элюента.
Существуют два способа детектирования — прямой и
косвенный. В прямом способе элюент переводят в соединение с
низкой электропроводностью, а определяемый ион — в высокопро-
водящее соединение и детектирование проводят по
положительным пикам. В косвенном способе, наоборот, элюент переводят в
Подавляющая
система
"
^ Система
детектироВания
Кондукто -
метрический
детектор *
Prfc. 2.1. Схема двухколоночной
ионохроматографической
системы
21
высокопроводящее соединение, а определяемый ион — в низко-
проводящее и детектируют по отрицательным пикам.
Принцип двухколоночного определения ионов можно
рассмотреть на примерах определения неорганических анионов (С1~ ?
NO3~~) и катионов щелочных металлов (Na+ , К+).
Наиболее распространенными элюентами для определения
анионов являются разбавленные растворы солей угольной
кислоты, например NaHCO3. Анионит в разделяющей колонке
находится в НСОз-форме. Подавляющей системой служит колонка,
заполненная катионообменником высокой емкости в Н+ -форме.
В этом случае в разделяющей и подавляющей колонках имеют
место следующие равновесия.
Разделяющая колонка:
R—HCO,+Na++(Cl-, NO3-) **R-
Подавляющая колонка:
R—H+Na++HCO-^R— Na+H2CO3,
R—H+Na++(C1-, NO-)*^R—Na+H++(C1-, NO3-).
Определяемые анионы попадают в кондуктометрическии
детектор в виде сильных кислот (НС1, HNO3), имеющих высокую
электропроводность на фоне слабой угольной кислоты Н2СОз.
Эквивалентная электропроводность Н+ выше электропроводности
катионов, поэтому такая система детектирования обеспечивает
максимальную чувствительность определения. .
При определении катионов щелочных металлов в качестве
элюента используют разбавленный раствор азотной кислоты.
Разделяющая колонка заполнена катионообменником в Н-фор-
ме. Подавляющая колонка заполнена анионообменником
высокой емкости в ОН-форме. На колонках устанавливаются
следующие равновесия.
Разделяющая колонка:
R-H+NO3-+(Na+, K+)**R—(Na: K)+NO3-+H+.
Подавляющая колонка: :
R—OH+H++NO3-^R—NOj+HjO,
R—OH+(Na+, K+)+NO3-^R—NOs+OH-+(Na+, K
Катионы щелочных металлов детектируют в виде
гидрооксидов, имеющих высокую электропроводность на фоне деионизо-
ванной воды, что обеспечивает максимальную чувствительность
определения. Типичные хроматограммы смесей неорганических
анионов и катионов щелочных металлов приведены на рис. 2.2
и 2.3.
22
Емкость ионообменников, которыми заполняют разделяющие
колонки для двухколоночной ионной хроматографии, колеблется
от 0,02 до 0,1 мэкв/г. Для определения катионов могут быть
использованы поверхностно-модифицированные катионообменники
как на полимерной основе, так и на основе силикагеля с
функциональными сульфогруппами. Однако для определения анионов
CI72
мин
Рис. 2.2. Хрома-
тограмма
неорганических
анионов с
карбонатным элюентом
t В 12 16 20
мин
Рис. 2.3. Хроматограм-
ма катионов с
азотнокислым элюентом
сорбенты на основе силикагеля непригодны. Это связано с тем,
что растворы солей слабых кислот, которые используют как элю-
енты в двухколоночной ионной хроматографии анионов, имеют
pH выше 8. Силикагель в этих условиях разрушается, поэтому
предпочтение отдается сорбентам иа основе стирол-дивииил-беи-
зола или полиметакрилата.
2.2.
ОДНОКОЛОНОЧНЫЙ ВАРИАНТ
В 1979 г. Гьерде, Фритц и Шнуклер [2] предложили для
определения анионов ионохроматографическую систему без
подавляющей колонки. Впоследствии этот принцип получил название
23
Насос
Элюент
ВШ
образца
Разде/іяю -
щая полент
одноколоночнои ионной хроматографии. В предложенном
варианте (рис. 2.4) кондуктометрическии детектор был непосредственно
соединен с разделяющей колонкой. Для сохранения высокой
чувствительности определения,
которая в двухколоночном варианте
достигается благодаря
использованию системы подавления, в одно-
колоночном варианте используют
элюенты с очень низкой
электропроводностью. Это чаще всего
анионы ароматических кислот,
имеющих низкую эквивалентную
электропроводность, но в то же время
обладающих высоким сродством к
анионообменнику, что позволяет
достичь быстрого и селективного
разделения определяемых анионов. В
качестве элюентов применяют либо
растворы солей ароматических
кислот с концентрацией A—5)X10 ~
М, либо растворы самих кислот с
концентрацией ( 1—5) • 10 ~~3 М. Ве-
Детектор І I
Рис. 2.4. Схема
одноколоночнои нонохроматогра-
фнческой системы
личина pH элюентов изменяется от 3 до 8. Первоначально одно-
колоночный вариант был предложен для кондуктометрического
детектирования, позднее его стали широко использовать с
косвенным УФ [3, 4] и электрохимическим [5] детекторами.
К преимуществам одноколоночнои ионной хроматографии по
сравнению с двухколоночной следует отнести:
а) более высокую эффективность разделения, которая
достигается за счет уменьшения мертвого объема системы, а также
благодаря возможности использовать сорбенты на основе сили-
кагеля;
б) простоту аппаратуры;
в) отсутствие необходимости периодически регенерировать
подавляющую систему;
г) возможность использовать различные детекторы;
д) менее жесткие требования к химической устойчивости
сорбентов и коррозионной стойкости хроматографической системы;
е) более широкий выбор элюеитов.
Однако пределы обнаружения ионов в одноколоночнои
ионной хроматографии обычно выше, чем в двухколоночной. Поэтому
при определении очень малых концентраций (менее 0.1 мг/л)
предпочтение в большинстве случаев отдается двухколоночному
варианту. Использование электрохимического детектора
позволяет снизить пределы обнаружения некоторых ионов по
сравнению с двухколоночным, но при этом теряется универсальность
определения. Кроме того, недостатком одноколоночного вариан-
24
та является более узкий по сравнению с двухколоночным
диапазон линейности градуировочного графика. Сравнительно узкий
интервал pH C—8) элюентов для одноколоночного
определения позволяет проводить разделение на ионообменниках как на
полимерной основе, так и на основе силикагеля.
Первые сорбенты на полимерной основе для ионной
хроматографии были синтезированы Фритцем с сотрудниками [6].
Основой сорбентов был макропористый сополимер стирол-дивинил-
бензола, иа поверхность которого прививали функцональные
аммониевые группы. Емкость таких анионообменннков
составляла 0,007—0,07 мэкв/г. Такая низкая емкость сорбентов
необходима для достаточно быстрого (~20 мин) разделения смесей
анионов элюентами очень низкой концентрации. Однако
эффективность разделения на этих сорбентах низка A000—2000
теоретических тарелок на метр) [7]. Гораздо выше эффективность
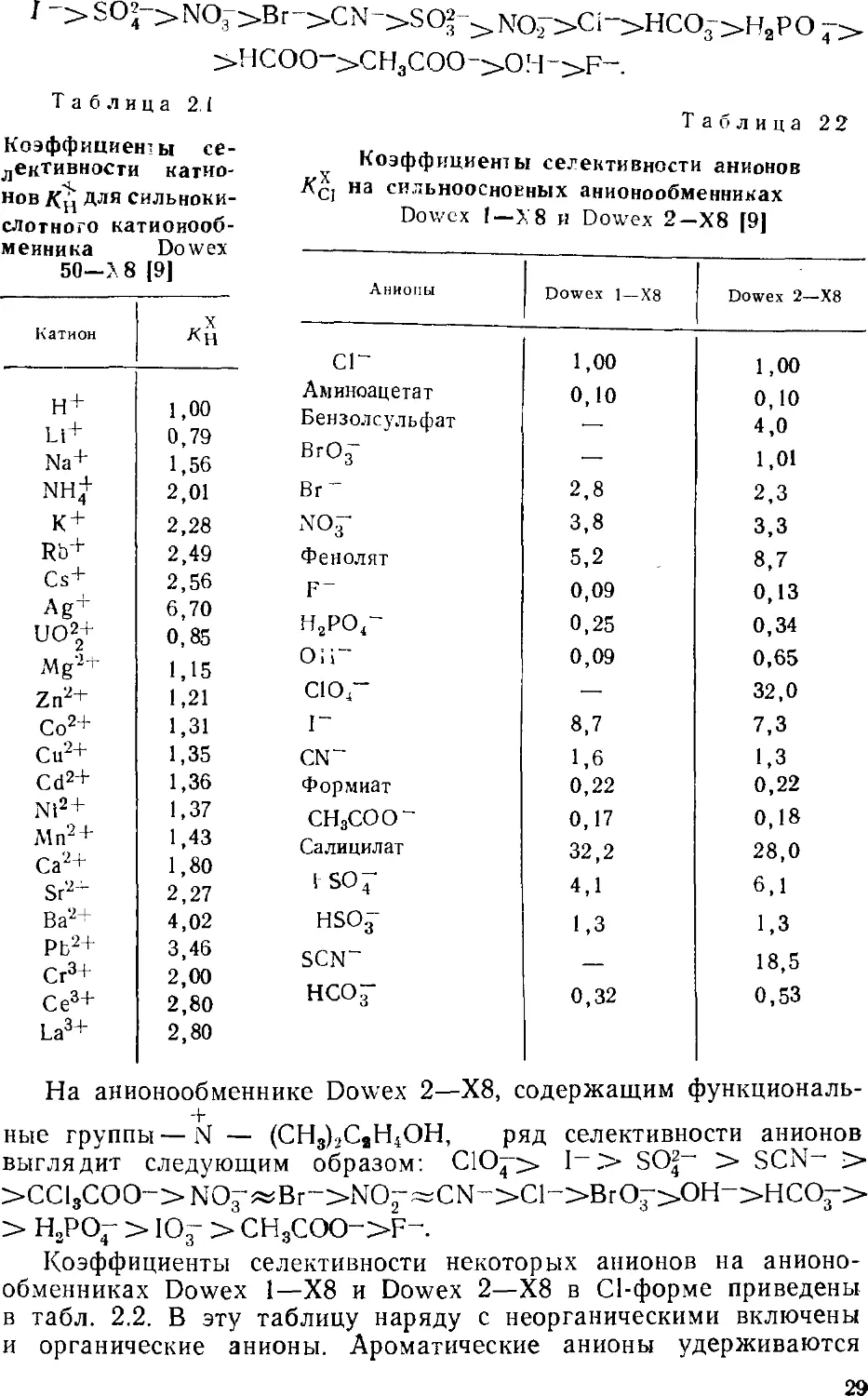
разделения B0 000 теоретических тарелок на метр) была
получена на сорбенте TSKGel 620 японской фирмы Toyo Soda,
синтезированном на основе
полимерного макропористого геля [7].
Размер частиц сорбента ~ 10 мкм, а
обменная емкость 0,03 мэкв/г. На
этом сорбенте в течение 15 мии
полностью разделяются 7
неорганических анионов (рис. 2.5). Для
разделения катионов используют
поверхностно сульфированные кати-
онообменники на основе сополимера
стирол-дивинилбензола с размером
частиц 20 мкм и емкостью 0,02—
0,03 мэкв/г [8].
Большее распространение в одно-
колоночной ионной хроматографии
анионов получлли сорбенты на
основе силикагеля с размером частиц
6—10 мкм и обменной емкостью
0,1—0,3 мэкм/г. Элюирование на
этих сорбентах проводят, как
правило, растворами фталевой,
бензойной, салициловой кислот или их
солей. Анионообменники на основе сл-
лнкагеля характеризуются высокой
для ионной хроматографии
эффективностью разделения. Так, для
сорбентов фирм Vydac (США)
и Wescan (США) эффективность
.составляет 16000 теоретических
тарелок на метр, а для сорбента
Toyo Soda (Япония) — 2500.
25
Рис. 2.5. Разделение
неорганических анионов на
анионообменом сорбенте
TSK GEL 620.
Элюент 1,3 мМ
Na2B4O7/5,8 мМ.
НзВОз/1,4 мМ глюконат
калия (pH 8,5) а Н2О—
—CH3CN (88:12).
Скорость потока 1,2 мл/мин.
Концентрация анионов 5—•
40 мкг/мл. Детектирование
кондуктометрическое.
2.3
РАВНОВЕСИЕ ИОННОГО ОБМЕНА
В УСЛОВИЯХ ИОННОЙ ХРОМАТОГРАФИИ
Оптимизация условий ионохроматографического определения
заключается в поиске наиболее подходящего сочетания
быстрого, селективного и эффективного разделения ионов и их
чувствительного, а в ряде случаев и селективного детектирования.
Оптимальными являются условия разделения, при которых время
выхода последнего хроматографического пика не превышает
20 мин, а разрешение соседних пиков равно 1,5. Условия
детектирования должны быть такими, чтобы чувствительность и
воспроизводимость определения отвечали требованиям,
предъявляемым к анализу данного объекта.
В настоящее время условия определения выбирают, как
правило, полуэмпирически, путем подбора подходящего сорбента,
элюента и детектора. При выборе сорбента и элюента
руководствуются закономерностями, ионообменной и ВЭЖХ, а также
практическим опытом ионной хроматографии. Выбор детектора
зависит от условий разделения, характера анализируемого
образца и задач анализа.
Равновесие ионного обмена между определяемым и элюи-
рующим ионами является основой оптимизации условий
разделения. При элюировании иона X ионом Е равного заряда
концентрации С в системе устанавливается ионообменное равновесие
R—E + X^RX+E. B.1)
Оно характеризуется константой ионного обмена, или
коэффициентом селективности Кх' который равен
КІ = '*! 1Ц1 B.2)
[X] [Е] '
где [X] и [Е]— равновесные концентрации определяемого и элюи-
рующего ионов в фазе ионообменника, а [X] и [Е] — равновесные
концентрации этих ионов в подвижной фазе, ;
Отношение [Х]ДХ] является коэффициентом распределения
(Dx ) определяемого иона X и характеризует способность этого
иона удерживаться сорбентом. Тогда
*х-О*Щ- B-3)
Если удельная обменная емкость сорбента Q, то при малых
заполнениях колонки [E] = Q—[X]»Q, a [51-сСе . Заполнение колон-
26
ки не должно превышать 10%. Иными словами, количество
определяемого иона должно быть как минимум в 10 раз меньше
обменной емкости сорбента. В этом случае
B.4)
Согласно основному уравнению хроматографии
исправленный удерживаемый объем V^ равен
B.5)
где Vs — объем сорбента.
Отсюда, учитывая уравнение 2.4,
В ионной .хроматографии удерживание иона на сорбенте
чаще характеризуют величиной исправленного времени
удерживания t'R которое равно
где F — объемная скорость элюента. Объем сорбента обычно
определяют по его пористости (б) и общему объему колонки V:
Vt = (l-*)V. B.8)
Тогда уравнение 2.7 принимает вид (
Ы ~лх СЕ F ' \ >
Таким образом, время удерживания иона X при элюировании
ионом Е прямо пропорционально коэффициенту селективности
Кх, обменной емкости и объему сорбента. В то же время
удерживание иона обратно пропорционально концентрации элюирую-
щего иона и объемной скорости элюента. Уравнения 2.7 и 2.9
используют при выборе условий ионохроматографического
определения. Эти уравнения позволяют теоретически оценить
возможность использования того или иного элюента или сорбента
для разделения анализируемой смеси.
2.4.
УДЕРЖИВАНИЕ КАТИОНОВ
Для разделения катионов используют поверхностно
сульфированные катионообменники низкой емкости. Из катионообмен-
ников высокой емкости ближе всего к этим сорбентам по
свойствам сильнокислотный сульфокатионит Dowex 50—Х8. Поэтому
можно предположить, что закономерности удерживания катио-
27
нов на Dowex 50—Х8 и на катионитах для ионной
хроматографии во многом сходны.
Способность катионов удерживаться сульфокатионитами
зависит от заряда и радиуса гидратированного иона. Катион
удерживается тем сильнее, чем больше его заряд и чем меньше
радиус гидратированного иона. При этом иногда радиус
гидратированного иона оказывает большее влияние на удерживание, чем
заряд. Способность катионов удерживаться на катионите
Dowex 50—Х8 изменяется в рядах
Ag+>Cs+>Rb+>K+>NHf >H+>
Ba2+>Sr-'+>Ca2+>Mg2+>Be2+;
Ba2+>Pb2+> Sr2+>Ca2+>Ni2+ « Cu2+> Cd2+> Co2+>Zn2+ > Mn2+
La3+>Ce3+>Cr3+.
Количественной характеристикой способности катионов
удерживаться на катионообменнике является коэффициент
селективности. Чем он больше, тем сильнее удерживание. Коэффициент
селективности некоторых катионов для катионообменника
Dowex 50—Х8 в Н-форме приведены в табл. 2.1.
Зная величины коэффициентов селективности, .можно рассчитать
коэффициенты селективности любой пары ионов. Например, для
обмена между катионом рубидия и катионообменником Dowex
50—Х8 в Li +-форме
#Rb= /CR-Y/Су = 2,49/0,79=3,15.
Аналогично для обмена Na+ на Ва2+
(К*$»= №I/2Лна= 4,02 1,56=2,58.
Значения коэффициентов селективности, приведенные в табл. 2.1,
можно использовать для оценки времен удерживания катионов
по уравнению 2.9.
2.5.
УДЕРЖИВАНИЕ АНИОНОВ
Способность анионов удерживаться сильноосновными анио-
нообменниками, как и катионов, зависит от заряда и радиуса
гидратированного иона. Чем больше отрицательный заряд и
меньше радиус гидратированного иона, тем сильнее этот анион
удерживается на анионообменнике.
По способности удерживания на анионообменнике Dowex
I—Х8, содержащем функциональные группы (—СН3)з—N,
анионы располагаются в следующий ряд:
28
>HCOO->CF^COO->OH->F-.
Таблица 2. і
Коэффициенты се-
лективности
катионов к^ для
сильнокислотного катиоиооб-
меиника Dowex
50—> 8 (9]
Катион
н+
Li+
Na+
NH +
К +
Rb+
Cs+
Ag+
UO2+
Mg-
Zn2+
Co2+
Cu2+
Cd2+
Ni2+
Mn2 +
Са2+
Sr2i
Ba2+
РЬ2+
Cr3+
Ce3+
La3+
1,00
0,79
1,56
2,01
2,28
2,49
2,56
6,70
0,85
1,15
1,21
1,31
1,35
1,36
1,37
1,43
1,80
2,27
4,02
3,46
2,00
2,80
2,80
Таблица 22
х Коэффициенты селективности анионов
KCJ на сильноосновных анионообменниках
Dovvcx 1-Х 8 и Dowex 2—Х8 [9]
Анионы
с\~
Аминоацетат
Бензолсульфат
ВгО3-
Вг~
NOJ-
Фенолят
р-
н2ро4-
Оіі~
сюг
I-
CN~
Формиат
СН3СОО~
Салицилат
I-SO7
hso^-
SCN~
HCO3-
Dowex 1—Х8
1,00
0,10
.—
—
2,8
3,8
5,2
0,09
0,25
0,09
—
8,7
1,6
0,22
0,17
32,2
4,1
1,3
0,32
Dowex 2—Х8
1,00
0,10
4,0
1,01
2,3
3,3
8,7
0,13
0,34
0,65
32,0
7,3
1,3
0,22
0,18
28,0
6,1
1,3
18,5
0,53
На анионообменнике Dowex 2—X8, содержащим
функциональные группы — N — (CH3JCaH4OH, ряд селективности анионов
выглядит следующим образом: С1О;р> I~> SO^~ > SCN~ >
CONOCNClBO
> Н2РО- > Юз~ > CH3COO->F-.
Коэффициенты селективности некоторых анионов на анионо-
обменниках Dowex 1—Х8 и Dowex 2—Х8 в С1-форме приведены
в табл. 2.2. В эту таблицу наряду с неорганическими включены
и органические анионы. Ароматические анионы удерживаются
29
на сильноосновных анионообменниках гораздо сильнее
алифатических. Коэффициенты селективности бензолсульфоната,
фенолята и салицилата могут быть использованы для оценки элюи-
рующей силы этих анионов.
ЛИТЕРАТУРА
1. Small H., S te v e n s T. S., В a u m a n W. S. // Anal Chem 1975
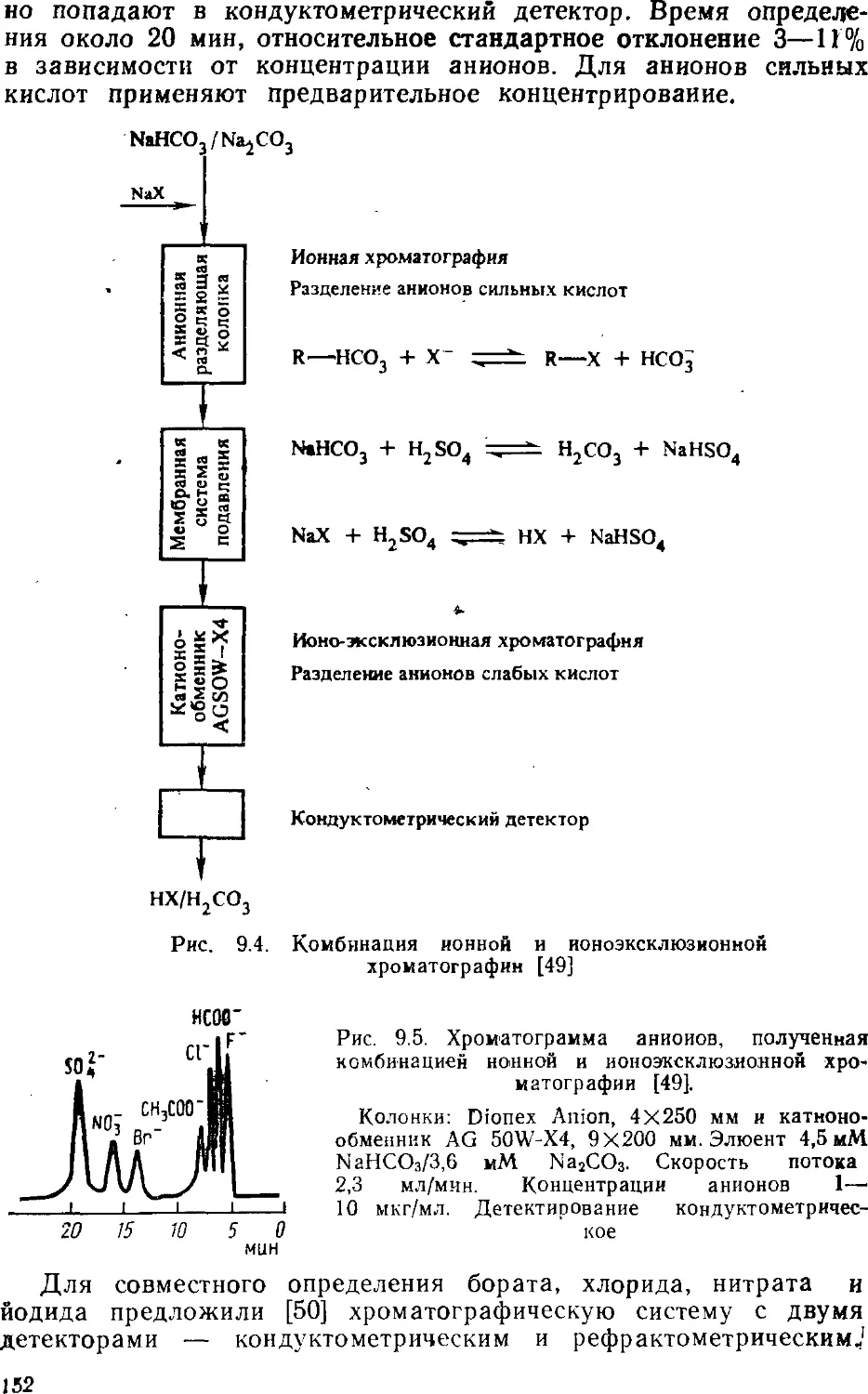
V. 47, № 10. P. 1801—1809.
2. GjerdeD. T., F r і t z J. S., Schmuckler G.//J. Chromatogr 1979.
V. 186. P. 509—519.
3. S m a 11 H., M і 1 1 e r T. E. // Anal. Chem. 1982. V. 54, № 2 P. 462—466
4. Cochrane R A., H і 1 1 m a n D. E. // J Chromatogr 1982 V. 241.
P. 392—394.
5. В о n d A. M. et al. // Anal. Chem. 1982. V. 54, № 3. P. 582—585.
6. Фритц Дж., Гьерде Д., Поланд К. Ионная хроматография. Пер.
с англ. (Под ред. В. Г. Березкнна). М.: Мир, 1984. 221 с.
7. HaddadP. R., Heckenberg A L Ці. Chromatogr. 1984 V. 300,
№ 3. P. 357—394.
8. F r і t z J. S., G j e r d e D. T., В e с k e r R. M. // Anal. Chem. 1980.
V. 52, №9. P. 1519—1522.
9. Map хол M. Ионообменники в аналитической химии. Свойства и
применение в неорганической химии. Пер. с англ. М.: Мир, 1985. С. 46—47.
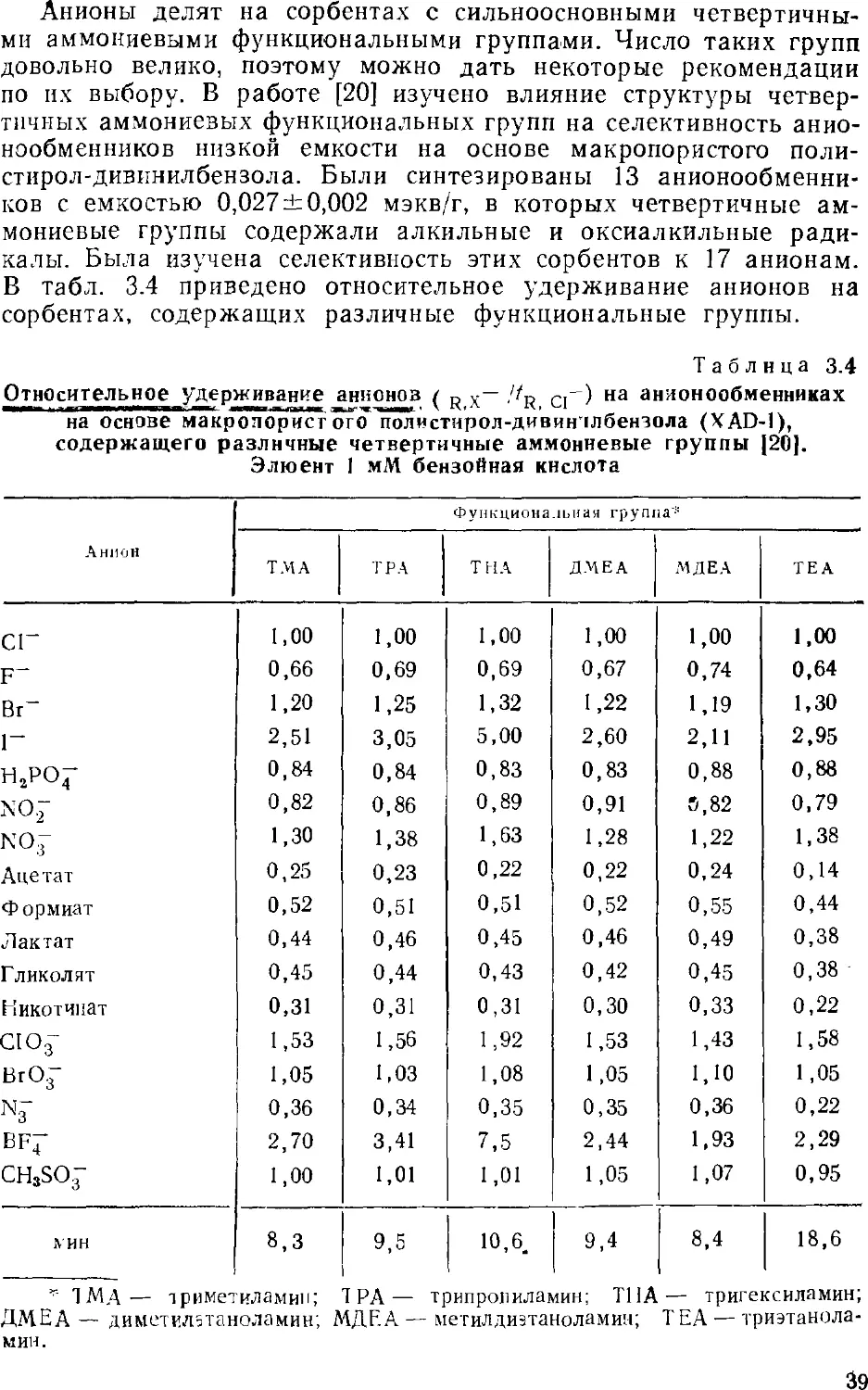
ГЛАВА З
СОРБЕНТЫ
3.1
ТРЕБОВАНИЯ К СОРБЕНТАМ
Выбор неподвижной фазы имеет большое значение при
проведении любого хроматографического разделения. Синтез
сорбентов для ионной хроматографии затруднен, поскольку к ним
предъявляется довольно много требований.
1. Сорбент должен иметь очень низкую ионообменную емкость
@,001—0,1 мэкв/г). Это связано с использованием кондуктомет-
рического детектирования, при котором необходимы элюенты
концентрации менее 0,01 М. Для эффективного
хроматографического разделения такими разбавленными элюентами
требуются иизкоемкостные ионообменные сорбенты.
2. Диаметр зерен сорбента не должен превышать 50 мкм.
Только в этом случае можно достичь высокой эффективности
разделения.
3. Зерна сорбента должны обладать высокой механической
прочностью и устойчивостью к давлению, которое возникает при
работе с мелкодисперсной неподвижной фазой.
4. Сорбент должен обладать высокой химической
устойчивостью по отношению к элюирующему раствору. Он должен
сохранять стабильность в широком интервале pH.
Этим требованиям удовлетворяют поверхностно-пористые (пел-
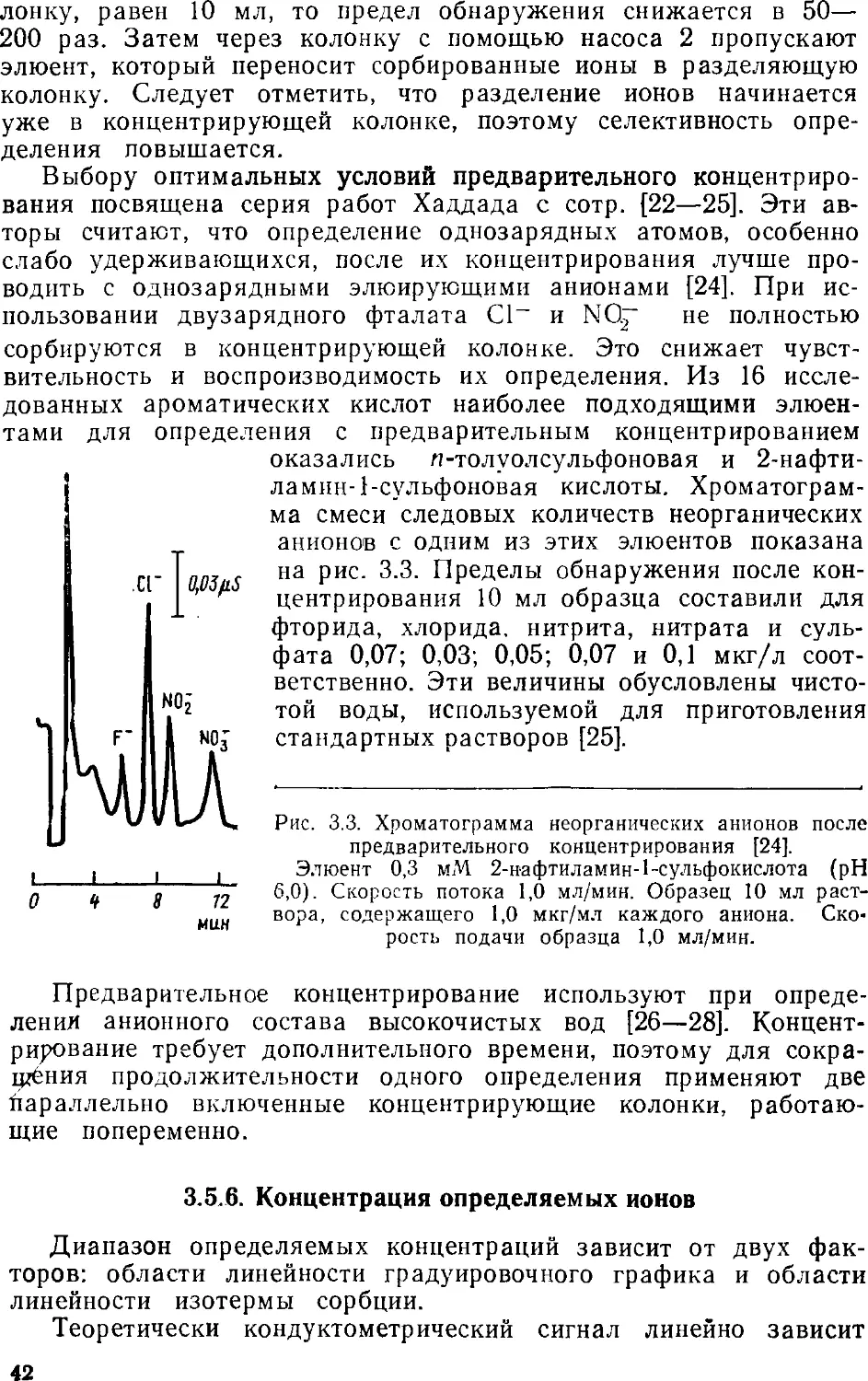
ликулярные) ионообменники, которые состоят из твердого
инертного ядра, покрытого тонким слоем ионита. На таких сорбентах
быстро устанавливается равновесие, поскольку диффузия в
тонкую ионообменную пленку занимает мало времени. В
результате ускоряется хроматографический процесс. Кроме того, эти
сорбенты обладают очень низкой ионообменной емкостью.
Работы по синтезу и использованию поверхностно-пористых ионооб-
менников появились за несколько лет до опубликования первой
статьи по ионной хроматографии [1]. Но именно в ионной
хроматографии эти сорбенты нашли самое широкое применение.
3.2
АНИОНООБМЕННИКИ
Продажные анионообменники, используемые в качестве
неподвижной фазы в ионной хроматографии, приведены в табл. 3.1.
31
Все они являются поверхностно-пористыми ионообменниками на
основе полимерной матрицы или силикагеля.
Т а б л и
ца 3.1
Анионообменники для ионной хроматографии
Название
Biotronik BTIAN
Biotronik BTIIAN
Dionex Anion
Dionex HPIC-AS
ХИКС
Interaction Ion-100
.\ucleosil 10—SB
Partisil 10—SAX
PRP-X100
CAB-120
TSK-GellC AnionPW
TSK-OellC Anion SW
TSK-Oel 620
Vydac 302 SC
Vydac 302 1С
Vydac 301 SB
Waters IC-РАК Anion
Wescan Anion
Wescan Anion/HS
Wescan Anion/R
Wescan Anion/R—HS
Zipax SAX
Основа
стирол-дивинилбензол
то же
оксиалкилметакрилат-
ный гель
гидрофильный поли-
мер
силикагель
то же
стирол-дивинилэензол
то же
полиметакрилат
силнкагель
поливинил
силикагель
то же
полиметакрилат
силика ель
то же
„
силикагель, покрытый
полиметакрилатом
Обменная
емкость,
мэкв/г
0,03
0,02
0,03
0,02—0,03
0,1
1.0
0,01
0,20
0,05
0,03
0,4
0,03
0,1
0,1
0,1
0,03
—
—
—
0,01
Размер
частиц,
МКм
10
10
25
15
20—25
10
_
10
10
20—25
10
5-8
9
30-44
10—20
30—40
10
25-35
Изготовитель
1
1
2
2
3
4
5
6
7
8
9
9
9
10
10
10
11
12
12
12
12
13
Изготовитель: 1 — Bioironik Wissenschaftliche Gerate GmbH., Maintal,
FRG; 2 — Dionex Corporation, Sunnyvale, CA, USA; 3 — Институт химии
Академии наук ЗССР, Таллинн, СССР; 4 — Interaction Chemicals. Mountain View,
CA, USA; 5 — Marcherey-Nagel, Duren, FRG; 6 — Whatman, Inc., 7 —
Hamilton, Reno, NV, USA; 8 — Институт геохимии и аналитической химии АНСССР,
Москва, СССР; 9 — Toyo Soda Manufacturing Co., Tokyo, Japan; 10 —
Separation Group, Hesperia, CA, USA; 11 — Waters, Division of Millipore,, Milford,
MA, USA; 12 — Wescan Instruments, Inc. Santa Clara, CA, USA; 13 — Du
Pont de Nemours and Co., Inc., Wilmington, Delaware, USA.
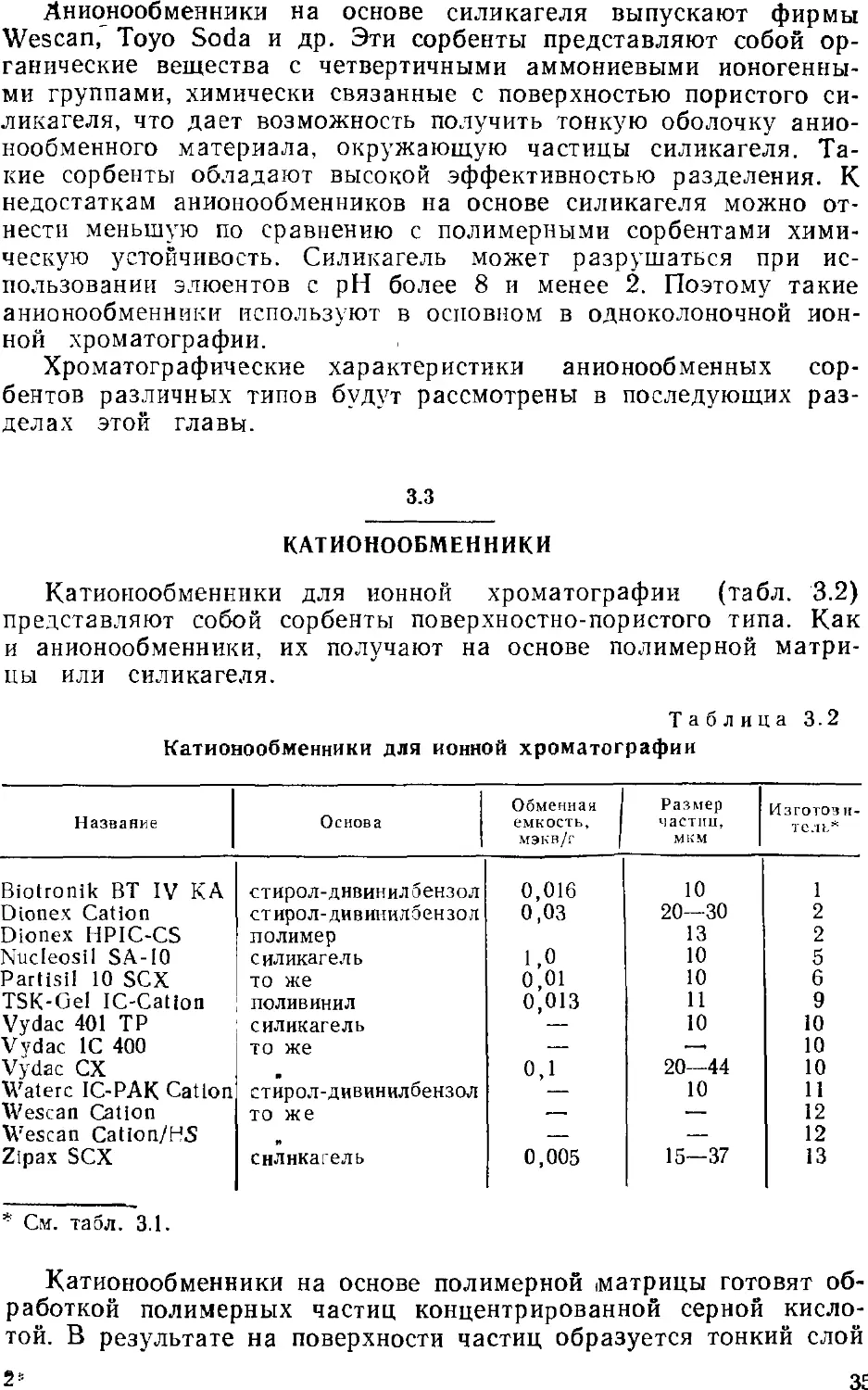
Наиболее распространенными анионообменниками на
полимерной основе являются сорбенты фирмы Dionex. Ядро этих
сорбентов представляет собой сополимер стирол-дивинилбензола.
Его поверхность вначале сульфируют, а затем полученный
поверхностно сульфированный канионит обрабатывают очень
мелкими частицами @,1—0,5 мкм) анионообменного латекса. Под
действием электростатических сил частицы латекса
притягиваются к поверхности смолы и образуют вторую тонкую оболочку
вокруг ядра. Таким образом, получающийся сорбент состоит из
32
трех различных слоев: внутреннего ядра, сульфированного слоя
и слоя анионообменных частиц. На рис. 3.1 изображена частица
такого анионообменника. Преимущество поверхностно-пористых
анионообменников перед обычными состоит в резком снижении
диффузионного пути, что приводит к ускорению обмена ионов
элюента и образца и, следовательно, к значительному
увеличению эффективности разделения. Анионообменники на
полимерной основе стабильны при высоких значениях pH (>9).
Рис. 3.L. Зерно поверхностно-модифицированного аниоиооб-
менника
Анионообменный слой на поверхности зерна сорбента может
быть закреплен также путем химического взаимодействия
соответствующих ионогенных групп с инертным ядром. Такой способ
синтеза анионообменников широко используют Фритц с сотр.
[2]. Инертную основу анионообменников составляют смолы
XAD-1, XAD-2 и XAD-4 фирмы Rohm and Haas. Эти смолы
представляют собой сополимеры стирол-дивинилбензола с высокой
плотностью поперечных связей. Они имеют макросетчатую
структуру и сохраняют ее даже после дегидратации. Наиболее
эффективные сорбенты для ионной хроматографии получены на
основе смолы XAD-1. Ее превращают в анионообменник путем хлор-
Зак. 34
33
метилирования и последующего аминирования третичным
амином: !
осп-, осп
ZnCl,
ЇЦСІ
(CIKKN
CH,N'(CH3K CI
В мягких условиях при небольшой продолжительности хлорме-
тилирования получают анионообменники с различной, но всегда
малой обменной емкостью. Синтез проводят по следующей
методике.
Частицы смолы XAD-1 размалывают и просеивают, пыль
удаляют, а фракцию 40—60 мкм сушат при 60°С в течение ночи. В
закрывающийся сосуд помещают 2,1 г безводного хлорида
цинка, 12,0 мл метиленхлорида, 9,6 мл хлорангидрида уксусной
кислоты и 6,0 мл метилаля и встряхивают сосуд до полного
растворения хлорида цинка. При проведении реакции хлорметилирова-
ния 1,5 мл этого раствора шприцем переносят в колбу, в
которую помещены 5 г смолы XAD-1 и 60 мл метиленхлорида.
Содержимое колбы выдерживают при комнатной температуре в
атмосфере азота в течение 15—120 мин (степень хлорметплиро-
вания зависит от времени). Останавливают реакцию
добавлением воды. Затем частицы отфильтровывают, промывают водой
и метанолом и сушат на воздухе. Для проведения аминирования
к хлорметилированным частицам добавляют большой избыток
25%-го раствора триметиламина в метаноле или воде и смесь
оставляют на ночь. Полученный сорбент промывают 1 M HC1,
пропанолом-2 и водой, затем сушат в течение ночи при 60°С.
Установлено, что при проведении хлорметилирования в
течение 15; 30; 60 и 120 мин получаются анионообменники с емкостью
0,009, 0,012, 0,017 и Q,021" мэкв/г соответственно.
Приведенные в табл. 3.1 анионообменники на полимерной
основе (Biotronik, Toyo Soda, ХИКС-1) также получают путем
обработки поверхности соответствующей инертной матрицы анионо-
обменным материалом. Так, сорбент ХИКС-1 получают
приклеиванием анионообменного слоя на поверхности зерен
полимерного геля [3].
Другой подход к синтезу анионообменников для ионной
хроматографии предложен в Институте геохимии и аналитической
химии Академии наук СССР. Предлагается использовать
центрально-привитой анионообменник, названный CAB-120. Сорбент
получают [4] путем удаления большей части ионогенных
аминогрупп из зерна высокоосновного полистирольного анионообмен-
ника AB-17, имеющего высокую обменную емкость C,5 мэкв/г).
Снижение емкости сорбента до 0,05 мэкв/г достигается в
результате его обработки раствором концентрированной серной
кислоты в течение 2 ч при температуре 150°С. і
34
Анионообменники на основе силикагеля выпускают фирмы
Wescan, Toyo Soda и др. Эти сорбенты представляют собой
органические вещества с четвертичными аммониевыми ионогенны-
ми группами, химически связанные с поверхностью пористого
силикагеля, что дает возможность получить тонкую оболочку анио-
нообменного материала, окружающую частицы силикагеля.
Такие сорбенты обладают высокой эффективностью разделения. К
недостаткам анионообменников на основе силикагеля можно
отнести меньшую по сравнению с полимерными сорбентами
химическую устойчивость. Силикагель может разрушаться при
использовании элюентов с pH более 8 и менее 2. Поэтому такие
анионообменники используют в основном в одноколоночной
ионной хроматографии.
Хроматографические характеристики анионообменных
сорбентов различных типов будут рассмотрены в последующих
разделах этой главы.
3.3
КАТИОНООБМЕННИКИ
Катионообменники для ионной хроматографии (табл. 3.2)
представляют собой сорбенты поверхностно-пористого типа. Как
и анионообменники, их получают на основе полимерной
матрицы или силикагеля.
Таблица 3.2
Катионообменники для ионной хроматографии
Название
Biotronik ВТ IV КА
Dionex Cation
Dionex HPIC-CS
Nucleosll SA-10
Partisil 10 SCX
TSK-Gel IC-Cation
Vydac 401 TP
Vvdac 1С 400
Vydac CX
Waterc IC-РАК Cation
Wescan Cation
Wescan Cation/HS
Zipax SCX
Основа
стирол-днвинил бензол
стирол-дивинилбензол
полимер
силикагель
то же
поливинил
силикагель
то же
стирол-дивинилбензол
то же
енлнкагель
Обменная
емкость,
мэкв/г
0,016
0,03
1,0
0 01
0,013
—
0,1
—
—
0,005
Размер
частиц,
м к м
10
20—30
13
10
10
11
10
,—,
20—44
10
—
—
15—37
Изготоз и-
Т С Л1. *
1
2
2
5
6
9
10
10
10
11
12
12
13
См. табл. 3.1.
Катионообменники на основе полимерной матрицы готовят
обработкой полимерных частиц концентрированной серной
кислотой. В результате на поверхности частиц образуется тонкий слой
сульфогрупп. Обменная емкость катионообменника связана с
толщиной этого слоя и зависит от типа смолы, диаметра частиц,
температуры и времени нагревания с серной кислотой.
Методика получения таких сорбентов описана в первой статье по
ионной хроматографии [5].
Длина диффузионного пути в поверхностно-пористом катионо-
обменнике низкой емкости меньше, чем в обычной катионообмен-
ной смоле, поскольку инертное гидрофобное ядро сорбента
ограничивает доступ катионов в его объем. Это приводит к ускорению мас-
сообмена катионов и, следовательно, к повышению
эффективности разделения. Кроме того, благодаря жесткости ядра
частицы сорбента подвергаются меньшему сжатию. Поэтому с
такими катионообменниками можно работать при больших
давлениях, а соответственно и скоростях потока, чем с обычными
смолами. Катионообменники низкой емкости на основе полимерной
матрицы устойчивы при pH 1—14 и практически не набухают.
Катионообменники на основе силикагеля получают
сульфированием в імягких условиях поверхностно-пористых силикагелей
Эти сорбенты обладают всеми достоинствами, свойственными
катионообменникам на основе полимерной матрицы. Однако они
устойчивы только при pH 2—8, что ограничивает область их
применения.
3.4.
КОМПЛЕКСООБРАЗУЮЩИЕ СОРБЕНТЫ
Селективность разделения ионов многих металлов на катио-
нообменниках невысока вследствие малого различия их
коэффициентов распределения. Этот недостаток можно устранить
введением в сорбент подходящих комплексообразующих групп. В
этом случае селективность определяется не различием констант
ионного обмена, как для катионообменников, а различной
прочностью связи определяемых ионов с комплексообразующими
группами. Наиболее широко комплексообразующие сорбенты
используют для разделения ионов переходных и тяжелых металлов.
Используемые в ионной хроматографии хелатообразующие
сорбенты указаны в табл. 3.3.
Разделение на комплексообразующих сорбентах может
осуществляться и по ионообменному механизму. Таким образом
разделяются анионные хлоридные комплексы платиновых металлов
на сорбентах с амидными или дитизоновыми функциональными
группами, которые благодаря наличию протонированного азота
обладают анионообменными свойствами [8, 9].
На сорбентах с привитыми краун-эфирами катионы
щелочных металлов разделяются по механизму комплексообразования.
А поскольку комплексы щелочных металлов с краун-эфирами
заряжены положительно, то сорбент может разделять
неорганические анионы по ионообменному механизму [11, 12].
36
Пока в ионной хроматографии предпочтение отдается
ионообменным сорбентам. Однако высокая селективность и
эффективность разделения тяжелых и переходных металлов на хела-
тообразующих сорбентах позволяют надеяться на их широкое
использование, особенно для определения катионов.
Таблица 3.3
Комплексообразующие сорбенты длч ионной хроматографии
Хе.іатообра-
зуюшие
группы
Иминодиуксусиан кисло-*
та
Тиогликолевая кислота
Амиды
Дитизон и дегидротизон
8-Оксихинолин
Краун-эфнры
4-(Пиридил-2-азо)резор-
д ин
5ис (салиинлальдегид)-
этилендиамин
Основы
стирол-дивинил-
бензол
то же
г/
„
силикагель
то же
целлюлоза
то же
3.5.
Разделяемые ионы
Cu(II),
Zr (IV)
Au(III)
Bl(III),
Au(III)
U (VI), Th (IV),
Ag(I), Hg(II),
Cd(II). Cu(II)
платиновые
металлы
Bi(III),
Sn(II),
Fe(III).
Au(III)
Cd(II), St(III),
Zn(II), Cr(VI),
Pb(II). Cu(II),
. Ag(I), платино-
вые металлы
Mn (II)
Co(II),
, Cd(II), Zn(II),
Pt(II). Ni(il)
катионы щелочных
металлов.
авионы
Fe(III),
Fe(III),
неорганические
Cu(II), U(VI)
Cu(II)
Литература
[6]
[7]
[8]
[9]
[10]
111.121
[13,14]
[13,14]
ПРАКТИКА ВЫБОРА СОРБЕНТА
Число сорбентов для ионной хроматографии невелико. В
значительной степени это связано со сложностями их синтеза. Как
правило, аналитик имеет в своем распоряжении колонки,
которыми комплектуются ионные хроматографы. В связи с этим
оптимизация условий ионохроматографического определения за
счет изменения свойств неподвижной фазы весьма затруднена.
Однако некоторые рекомендации по выбору сорбента,
основанные на опыте ионной и высокоэффективной жидкостной
хроматографии, можно сформулировать.
3.5.1. Размер частиц и параметры колонки
Согласно теории высокоэффективной хроматографии
эффективность разделения зависит от размера частиц сорбента. Чем
меньше размер частиц, тем выше эффективность. С другой
стороны, эффективность разделения зависит от однородности
фракции сорбента. Чем меньше различие в размере частиц, тем мень-
37
ше «вихревая» диффузия и выше эффективность. Добиться
однородности фракции при очень малом размере частиц (~5 мкм)
очень сложно. Кроме того, при использовании таких малых
частиц для обеспечения нужной скорости подачи элюента
необходимо очень высокое давление. А это требует специальной
конструкции прибора. Поэтому оптимальный размер частиц
сорбентов для ионной хроматографии от 10 до 20 мкм с дисперсией
по фракции не более ±20%. Использование таких сорбентов
позволяет добиться высокой эффективности разделения при
относительно невысоком давлении.
В качестве разделяющих, как правило, используют колонки
длиной от 50 до 250 мм и диаметром 3—5 мм. Увеличивая длину
колонки, можно повысить эффективность разделения, поскольку
увеличится число теоретических тарелок на колонку. Однако
следует помнить, что при этом увеличатся времена удерживания
ионов вследствие увеличения объема сорбента (см. уравнение
B.9)). Иначе говоря, при увеличении длины колонки надо
пропорционально увеличивать концентрацию элюирующего иона или
использовать более сильный элюент.
3.5.2. Матрица
Сорбенты для ионной хроматографии получают
модифицированием кремнезема, сополимера стирол-дивинилбензола и поли-
метакрилата. Свойства сорбентов во многом определяются их
матрицей. Сорбенты на основе кремнезема устойчивы лишь
при pH 2—7, поэтому их применяют только в одноколоночном
варианте с нейтральными элюентами. Сорбенты на полимерной
основе устойчивы при pH 1 —13, поэтому их можно использовать
как в одноколоночном, так и в двухколоночном варианте.
Большое значение имеет матрица сорбента при определении
гидрофобных сильноудерживаемых ионов. Для их определения
следует использовать сорбенты с менее гидрофобными
матрицами. В работах [15—17] показано, что при определении сильно
удерживаемых катионов щелочноземельных металлов, а также
неорганических анионов (I"" , БгОд2"" . SCN~ , С1О4~) и хлорид-
ных комплексов платиновых металлов лучше использовать
сорбенты на основе кремнезема или полиметакрилата. На этих
сорбентах ионы удерживаются слабее, чем на стирол-дивинилбен-
зольных. Это объясняется различной гидрофоблостью матрицы,
которая возрастает в ряду [18]: кремнезем <полиметакрилат<
<стирол-дивинилбензол. Поэтому для определения гидрофобных
ионов надо выбирать менее гидрофобные матрицы.
Подробное изучение и сравнение хроматографических
характеристик сорбентов Vydac, Interaction, PRP-XIOO, TSK-GellC и
Waters, имеющих различную матрицу, проведено в работе [19].
3.5.3. Функциональные группы
Для разделения катионов используют сильнокислотные катио-
нообменники низкой емкости с сульфогруппами.
38
Анионы делят на сорбентах с сильноосновными
четвертичными аммониевыми функциональными группами. Число таких групп
довольно велико, поэтому можно дать некоторые рекомендации
по их выбору. В работе [20] изучено влияние структуры
четвертичных аммониевых функциональных групп на селективность анио-
нообменников низкой емкости на основе макропористого поли-
стирол-дивинилбензола. Были синтезированы 13 анионообменни-
ков с емкостью 0,027±0,002 мэкв/г, в которых четвертичные
аммониевые группы содержали алкильные и оксиалкильные
радикалы. Была изучена селективность этих сорбентов к 17 анионам.
В табл. 3.4 приведено относительное удерживание анионов на
сорбентах, содержащих различные функциональные группы.
Таблица 3.4
Относительное удерживание анионов ( R x— //R С]-) на анионообменниках
на основе макроїГорїїсг (ГгсГ полистирол-дивинчлбензола (XAD-1),
содержащего различные четвертичные аммониевые группы [20).
Элюент 1 мМ бензойная кислота
А ни он
er
F~
Вг-
1~
Н2РОГ
N07
N07
Ацетат
Формиат
Лактат
Гликолят
Никотчиат
ClO-f
ВгО3-
МГ
BF7
CH3SO3"
НИН
Функциональная группа-4
TM А
1,00
0,66
1,20
2,51
0,84
0,82
1,30
0,25
0,52
0,44
0,45
0,31
1,53
1,05
0,36
2,70
1,00
8,3
"ГРА
1,00
0,69
1,25
3,05
0,84
0,86
1,38
0,23
0,51
0,46
0,44
0,31
1,56
1,03
0,34
3,41
1,01
9,5
TUA
1,00
0,69
1,32
5,00
0,83
0,89
1,63
0,22
0,51
0,45
0,43
0,31
1,92
1,08
0,35
7,5
1,01
10,6.
Д.МЕА
1,00
0,67
1,22
2,60
0,83
0,91
1,28
0,22
0,52
0,46
0,42
0,30
1,53
1,05
0,35
2,44
1,05
9,4
МДЕА
1,00
0,74
1,19
2,11
0,88
5,82
1,22
0,24
0,55
0,49
0,45
0,33
1,43
1,10
0,36
1,93
1,07
8,4
TEA
1,00
0,64
1,30
2,95
0,88
0,79
1,38
0,14
0,44
0,38
0,38
0,22
1,58
1,05
0,22
2,29
0,95
18,6
'- IMA— іриметкламин; Т PA— трипрониламин; TUA— тригексиламин;
ДМЕА — диметилїтаноламин; МДЕА — метилдизтаноламин; TEA — триэтанола-
мин.
39
Видно, что с увеличением длины радикала алкильных
аммониевых групп увеличивается селективность сорбента к более
поляризуемым, сильнее удерживаемым анионам (NO^~, BF4~ , СІО^"
и І~). В то же время удерживание анионов слабых кислот не
зависит от длины алкильного радикала аммониевой группы.
Таким образом, изменяя длину алкильного радикала, можно
повысить селективность разделения сильно удерживаемых анионов.
Что касается оксиалкильных аммониевых групп, то для них
трудно выявить какие-либо закономерности. В работе [20]
установлено, что лучшая селективность разделения НСОО~, Cl ~ , Вг ~
NO3~ , С1О3~ достигается на анионообменнике, содержащем три-
гексиламмониевые функциональные группы.
В ряде случаев для повышения селективности определения
анионов их разделение можно проводить на сорбентах со
слабоосновными первичными аминогруппами. Так, неорганические
анионы разделяли [21] на колонке Zorbax NH2 с фталатным элю-
ентом (pH 2,9). Времена удерживания анионов приведены в табл.
3.5. Видно, что ряд селективности анионов на слабоосновном
анионообменнике отличается от аналогичного ряда на
сильноосновном (см. табл. 3.4).
3.5.4. Обменная емкость
Основным критерием выбора обменной емкости сорбента
является относительное удерживание определяемых ионов. Для
Таблица 3.5 определения слабо удержива-
Время удерживания анионов на слабо- емых ионов нужны сорбенты
основном ан-юнэобменнчке [211. _ J г
Колонка ZorbaK NH2 D,6x250 мм); с большей емкостью, а для
элюент 0,02 M фталевая кислота определения сильно удержи-
(рН 2,9), скорость элюента 2 мл/мин ваемых — с меньшей.
Количественно оценить емкость
сорбента, необходимую для
быстрого и селективного
разделения, можно по уравнению B.9).
На практике изменение
обменной емкости сорбента для
оптимизации используют редко.
Чаще оптимальные условия
находят изменением состава
и свойств элюента.
3.5.5. Колонки для концентрирования
В некоторых случаях даже при оптимизации всех условий
ионохроматографического определения его чувствительность
бывает недостаточной. Чаще всего с этим сталкиваются при анали-
40
Аи ион
Сульфат
Фосфат
Хлорид
Йодид
Нитрат
Фторид
Время удерживания,
мин.
3,4
4,0
5.2
2,0
2,3
1,2
зе высокочистых вод. Существует два пути повышения
чувствительности определения.
Один из них состоит в повышении объема вводимой пробы за
счет увеличения длины петли-дозатора. Замена обычной для
ионной хроматографии петли объемом 50—200 мкл на петлю
объемом 1000 мкл позволяет снизить предел обнаружения в 5—20 раз.
Однако использование большой петли существенно увеличивает
ширину и площадь водного пика и затрудняет определение
слабо удерживаемых анионов (F~, CI ~, СНзСОО~ , NO.r и др.).
Более распространенным способом повышения
чувствительности является предварительное концентрирование образца на
специальной колонке, которую называют концентрирующей, или
колонкой для концентрирования. Эту колонку устанавливают в
приборе вместо петли-дозатора (рис. 3.2). Анализируемый раст-
Насос 2
Ооразец
Насос 1
Элюент
(I Колонка д/ія _
конц ентриробания
Разделяющая
колонка
Рис. 3.2. Схема включения колонки для
концентрирования
вор в необходимом для достижения нужной чувствительности
объема с помощью насоса 1 пропускают через концентрирующую
колонкуі, заполненную ионообменником низкой емкости. Для
концентрирования используют короткие колонки (длиной 50мм)
с тем же сорбентом, что и в разделяющей. Определяемые ионы
сорбируются в ней. Если объем образца, пропущенного через ко-
лонку, равен 10 мл, то предел обнаружения снижается в 50—
200 раз. Затем через колонку с помощью насоса 2 пропускают
элюент, который переносит сорбированные ионы в разделяющую
колонку. Следует отметить, что разделение ионов начинается
уже в концентрирующей колонке, поэтому селективность
определения повышается.
Выбору оптимальных условий предварительного
концентрирования посвящена серия работ Хаддада с сотр. [22—25]. Эти
авторы считают, что определение однозарядных атомов, особенно
слабо удерживающихся, после их концентрирования лучше
проводить с однозарядными элюирующими анионами [24]. При
использовании двузарядного фталата С1~ и NQj" не полностью
сорбируются в концентрирующей колонке. Это снижает
чувствительность и воспроизводимость их определения. Из 16
исследованных ароматических кислот наиболее подходящими элюен-
тами для определения с предварительным концентрированием
оказались л-толуолсульфоновая и 2-нафти-
ламин-1-сульфоновая кислоты. Хроматограм-
ма смеси следовых количеств неорганических
анионов с одним из этих элюентов показана
СІ' 1005 S на РИС- ^" Пределы обнаружения после кон-
I ' центрирования 10 мл образца составили для
фторида, хлорида, нитрита, нитрата и
сульфата 0,07; 0,03; 0,05; 0,07 и 0,1 мкг/л
соответственно. Эти величины обусловлены
чистотой воды, используемой для приготовления
стандартных растворов [25].
Рис. 3.3. Хроматограмма неорганических анионов после
предварительного концентрирования [24].
, , , , Элюент 0,3 мМ 2-нафтиламин-і-сульфокислота (pH
п и я 12 6.0). Скорость потока 1,0 мл/мин. Образец 10 мл раст-
мин вора, содержащего 1,0 мкг/мл каждого аниона.
Скорость подачи образца 1,0 мл/мин.
Предварительное концентрирование используют при
определения анионного состава высокочистых вод [26—28].
Концентрирование требует дополнительного времени, поэтому для сокра-
ц^ения продолжительности одного определения применяют две
параллельно включенные концентрирующие колонки,
работающие попеременно.
3.5.6. Концентрация определяемых ионов
Диапазон определяемых концентраций зависит от двух
факторов: области линейности градуировочного графика и области
линейности изотермы сорбции.
Теоретически кондуктометрический сигнал линейно зависит
42
от концентрации определяемого иона в очень широком
диапазоне концентраций E—6 порядков). Однако на практике этот
диапазон гораздо уже, особенно для кондуктометричєского
детектирования на фоне слабых кислот или оснований. В этом случае
для определения больших концентраций ионов нельзя
пользоваться градуировочным графиком, не проверив его диапазон
линейности.
В двухколоночной ионной хроматографии для расширения
диапазона линейности градуировочного графика детектирование
лучше проводить либо на фоне очень слабых кислот или
оснований (рК>9), либо на фоне депонированной воды. На рис. 3.4
представлены градуировочные графики определения нитрата с
карбонатным и аминокислотным элюентами. Очевидно, что при
использовании аминокислотного элюента, когда детектирование
проводят на фоне депонированной воды, диапазон линейности
градуировочного графика гораздо шире. При одноколоночном
определении диапазон линейности шире при элюированни
растворами солей.
Т, мин
20
70
-J 1 1 і
W
80
Рис. 3.4. Градуировочный
график для определения
нитрата с карбонатным (I)
и аминокислотным B)
элюентами
Рис. 3.5.
Зависимость времени
удерживания перхлората
от его концентрации
[31].
Колонка Biotronik
@,0021 мэкв/г). Элю-
ент 0,02 цитрат
калия
С линейностью изотермы сорбции связана зависимость
времени удерживания иона от его концентрации. Если изотерма
нелинейна, время удерживания зависит от концентрации
определяемого иона. Это затрудняет и качественную идентификацию и
количественное определение. Поэтому важно знать область
линейности изотермы, т. е. диапазона концентраций, где время
удерживания постоянно. Ширина этого диапазона зависит от
Удерживания иона. Чем сильнее удерживается ион, тем уже
область линейности изотермы сорбции. Поэтому особенно важно
знать область концентраций, где время удерживания постоянно,
43
для сильно удерживаемых ионов. Было установлено [30], что
область линейности изотермы сорбции перхлората зависит от гид-
рофобности матрицы сорбента (рис. 3.5). Для сорбента ХИКС-1
с менее гидрофобной полиакрилатной матрицей область
линейности изотермы шире, чем для сорбента Biotronik со стирол-ди-
винилбензольной матрицей. Следовательно, для определения
.Сильно удерживаемых ионов лучше использовать сорбенты с
менее гидрофобными матрицами (полиметакрилат или кремнезем).
ЛИТЕРАТУРА
1. H о г v a t h С, Р г є і s s В., L і р s k y S. R. // Anal. Chem. 1967. V. 39,
№ 12. P. 1422—1428.
2. Фритц Дж., Гьерде Д., Полаид К. Иоииая хроматография. Пер. с
-англ. / Под ред. В. Г. Береэкииа. М.: Мир, 1984. С. 119.
3. H а 1 d п a U. et. al. // J. Chromatogr. 1985. V. 350. P. 296—298.
4. Д ол roHocoB A. M. // Ж. физ. химии. 1984. T. 48, № 8. С. 1989—1991.
5. Small H., Stevens T. S., В a u m a n W. S. // Anal. Chem. 1975.
V. 47, № 10. P. 1801 — 1809.
6 Mo y ers E. M, Fritz J. S // Anal. Chem. 1977. V. 49, № 3. P. 418—
423.
7. Phillips R. J., Fritz J. S. // Anal. Chem. 1978. V. 50, № 11.
P. 1504—1508.
8. Pohl a n dt G, Fritz J. S // J. Chromatogr. 1979. V. 176, № 2.
P 189—197.
9. G r о t e M, К e 11 r u p A. // Anal. Chim. Acta. 1985. V. 172. P. 223—239.
10. Jez orek J. R., F reiser H. // Anal. Chem. 1979. V. 51, № 3.
P. 366—373.
11. Blasius E. et al. // Fresenius Z. Anal. Chem. 1977. B. 284, № 5.
g 337 3gg
12. В 1 a s і u s E. et al. // Fresenius Z. Anal. Chem. 1985. B. 320. S. 435—438.
13. Burba P., G ! e і t s m a n n В., L і e s e r K. H. // Fresenius Z. Anal.
Chem. 1978. B. 289, № 1—2. S. 28—34.
14. Burba P., Lies er К. H. // Fresenius Z. Anal. Chem. 1978. B. 291.
№ 3 S. 205—209.
15. Zol oto v Yu. A. et al. // J. Env. Anal. Chem. 1987. V. 31, № 2—4.
P. 99—106.
16. Miyazaki M., H a y a k a w a K-, S e u n g-G i-Ch о і // J.
Chromatogr. 1985. V. 323, Ni 2. P. 443—446.
17. Шпигун О. А., П азу хии а Ю. Э. // Ж. аналнт. хнмни. 1987. Т. 42,
№ П. С. 2003—2006.
18. D u v а 1 D. L, F г і t z J. S. // J. Chromatogr. 1984. V. 295. P. 89—101.
19. H a d d a d P. R., J a с k s о n P. E., H e с k e n b e r g A L // J.
Chromatogr. 1985. V. 346. P. 139—148.
20. В a r r о n R. E., F r і t z J. S. // J. Chromatogr. 1984. V. 284. P. 13—25.
21. Cortes H. J., Stevens T. S. // J. Chromatorg. 1984. V. 295.
P. 269—275.
22. H a d d a d P. R., H e с k e n b e r g A. L. // J. Chromatogr 1984. V. 318,
№ 2. P. 279—288.
23. H e с k e n b e r g A. L., H a d d a d P R. // J. Chromatogr 1985. V. 330,
№ 1. P. 95—111.
24. J a с k e s о n P. E., H a d d a d P. R. // J. Chromatogr 1986 V 355, № 1.
P. 87—97.
25. H a d d a d P. R., J a с k s о n P. E. // J. Chromatorg 1986. V. 367
P. 301—309.
26. Wetzel R. A. et al. // Anal. Chem. 1979 V 51 № 9. P. 1532—1535
27. T rette r H. et al. // Fresenius Z. Anal. Chem. 1985. B. 321 № 7.
S. 650—652.
44
28. В а I к о n і L., P a s с а 1 і R., S і g о n F. // Anal. Chim. Acta. 1986.
V 179. P. 419—425.
29. M о s к о J. A. // Anal. Chem. 1984. V. 56, № 4. P. 629—633.
30. Пазу хина Ю. Э. Ионохроматографическое определение сильно
удерживаемых неорганических анионов, в том числе хлоридных комплексов
платиновых металлов: Дне. ...канд. хим. наук: М., 1987. 141 с.
31. Шпигун О. А., Обрезков О. Н., Зол ото в Ю. А. // Ж. аналит.
химии. 1986. Т. 41, № 4. С. 692—695.
ГЛАВА 4-
ЭЛЮЕНТЫ
4.1.
ДВУХКОЛОНОЧНАЯ ИОННАЯ ХРОМАТОГРАФИЯ
Элюенты, используемые в двухколоночной ионной
хроматографии, должны отвечать двум основным требованиям.
Во-первых, они должны быстро и селективно разделять определяемые
ионы в разделяющей колонке. Во-вторых, после прохождения
подавляющей системы элюент должен превращаться в
соединение, электропроводность которого максимально отличается от
электропроводности определяемого иона. Как правило, элюент
подбирают такой, чтобы время удерживания последнего
определяемого иона на хроматограмме не превышало 20—25 мин.
Выбор элюента зависит от числа и характера определяемых
ионов. Концентрация элюентов для двухколоночной ионной
хроматографии обычно колеблется от 1 до 10 мМ. Низкая
концентрация элюента связана, во-первых, с низкой емкостью
разделяющих ионообменников, поэтому для селективного разделения
необходимы элюенты низкой концентрации, а, во-вторых, с
временем работы подавляющей системы: чем меньше концентрация
элюента, тем больше время работы подавляющей системы между
регенерациями.
4.1.1. Определение анионов
Наиболее распространенными элюентами в двухколоночной
ионной хроматографии анионов являются разбавленные
растворы солей слабых кислот. Эти элюенты используют для определения
анионов сильных кислот, рК которых ниже 5. При этом в
подавляющей системе элюент переводят в малодиссоциированную
кислоту, имеющую низкую электропроводность, а определяемые
анионы — в сильную кислоту, имеющую высокую
электропроводность.
Для определения анионов слабых кислот, рК которых выше
6, в качестве элюентов используют разбавленные растворы
солей сильных кислот. В этом случае в подавляющей системе оп-
ределямый анион переводят в малодиссоциированную кислоту,
а элюент — в сильную кислоту с высокой электропроводностью
[1, 2]. Детектирование проводят по уменьшению кондуктометри-
ческого сигнала.
46
В табл. 4.1 приведены элюенты, используемые в двухколоноч-
ной ионной хроматографии анионов. Элюенты, обладающие
низкой элюирующей способностью (NaOH, Na2B4O7), используют
Таблица 4.1
Элюенты, используемые в двухколоночной ионной хроматографии
авионов
Элюент
NaOH
Na2B4O,
Na2CO3
NaHCO3/Na2CO3
Na2CO3/NaO. і
NaNOa/NaOH
NaOPh
Аминокислота
Элюирующий
анион
o:.-
B07
СОз^
i.co~, co-~
COf-, 0 ;-
NOf, OH~
PhO~
HaNRCOO~
Элюирующая
способность
низкая
низкая
средняя
средняя
высокая
средняя
средняя
зависит от
свойств
кислоты
Реакция подав ления
R—H+Na~-+OH-=^R—Na+H2O
R— H+Na" -1-BO.^R— Na+HBO2
2R~H+2N'a +CO3~±f2R—Na+
+H2COS
3R_H+ 3Na""+CO|"+HCO-=b>=
^ЗН—Ыа+г 1!2CO3
3R_-l-|_3Xa'f+ СОз~+ОН~±р
*5:3R—Na+H2CO3+H2O
2R—H +2Na+ + NO^+OH""^
R—H+Na++ PhO --ье
^R—Na+PhOH
2R—H+Na++ H^RCOO-^R - Na+
-f-
+R—NH.RCOOH
для определения слабоудерживаемых анионов, таких как F~
CI~, NO~ 1 и некоторых алифатических одноосновных кислот.
Низкая элюирующая сила не позволяет использовать эти
элюенты для определения среднеудерживаемых анионов. Для этого
используют наиболее распространенную в двухколоночной
хроматографии элюирующую смесь карбоната и гидрокарбоната
натрия. Однако использование этого элюента вызывает ряд
сложностей, связанных с детектированием на фоне угольной кислоты.
К таким сложностям относится наличие на хроматограмме
отрицательных водного и карбонатного пиков, затрудняющих
определение слабоудерживаемых анионов, а также узкий диапазон
линейности градуировочного графика, связанный с подавлением
диссоциации угольной кислоты в зоне определяемого аниона, и
ряд других. Многие эти сложности можно устранить, используя
в качестве элюентов растворы аминокислот. В этом случае
детектирование осуществляется на фоне деионизованной воды, что
повышает чувствительность определения.
4.1.2. Определение катионов
В двухколоночной ионной хроматографии катионов, как и при
определении анионов, используют два варианта детектирования.
47
В первом из иих, наиболее распространенном, элюент в
подавляющей системе переводят в соединение с низкой
электропроводностью, а определяемые катионы — в соединения с высокой элек^
ропроводностью. Этот вариант детектирования "используют дЛя
определения катионов щелочных, щелочноземельных и
некоторых переходных металлов. В качестве элюентов
используют разбавленные растворы сильных кислот, а также солей
слабых оснований, серебра, бария, свинца, цинка. Для
определения слабо удерживаемых катионов щелочных металлов
пригодны элюенты с низкой элюирующей силой, а для определения
сильно удерживаемых катионов щелочноземельных и
переходных металлов нужны элюенты с высокой элюирующей силой.
Во втором варианте детектирования элюент переводят в
соединение с высокой электропроводностью, а определяемый
катион — в соединение с низкой электропроводностью. Этот вариант
применяют для определения катионов слабых оснований, в
частности аминов [1].
Таблица 4.2
Элюенты, используемые в двухколоночной ионной хроматографии
катионов
Элюент
Элюируюший
Элюируюшап
способность
Реакция подавления
HNO3
AgNO3
Фенилендиамин Х
X2HCI
ВаС12
P1(NO3J
Zn(NO3yp.NQs
KC1/KC1
Аминокислота
-fhCI
H+
Ag+
Ba2+
Pb2+
Zn2-, H+
K+, H+
H3NRC00H
низкая
средняя
высокая
высокая
высокая
высокая
средняя
зависит от
свойств
кислоты
R-O ;
-NOa+
+AgCl
2R—О!
2R—S0i-^-Ba2-^-2Cr±s2R--Cl+
+BaSO4 1
2R—Ю3+РЬ2++
5R—
+
2R—OH+H3NRCOOH+CI
-Cl+2H2O+R-0OCRN Ha
Элюенты, используемые в двухколоночной ионной
хроматографии катионов, приведены в табл. 4.2. Следует отметить, что
двухколоночный вариант используют в основном для
определения "катионов щелочных и щелочноземельных металлов с
растворами сильных кислот или солей слабых оснований в качестве
элюентов.
48
4.2.
ОДНОКОЛОНОЧНЫЙ ВАРИАНТ
Элюенты, используемые в одноколоночной ионной
хроматографии, должны быстро и селективно разделять определяемые
ионы, иметь низкую электропроводность- и максимальное различие
величин эквивалентной электропроводности элюирующего и
определяемого ионов. Элюенты, отвечающие этим требованиям,
могут быть использованы и в системах с косвенным УФ-детек-
тором.
4.2.1. Определение анионов
Для определения анионов в качестве элюентов обычно
выбирают растворы органических кислот или их солей. Анионы этих
кислот имеют высокое сродство к разделяющему сорбенту,
поэтому быстрое и селективное разделение достигается при низких
концентрациях элюирующего иона. Большинство органических^
кислот, используемых в качестве элюентов в одноколоночной
ионной хроматографии анионов, приведено в табл. 4.3.
Таблица 4.3
Органические кислоты, используемые в качестве элюентов
в одиоколоиочной ионной хроматографии анионов, и нх сорбция
иа сорбенте XAD-I [3|
Кислота
Сульфаниловая
Никотиновая
Пиколнновая
Лимонная
Янтарная
Фумаровая
Сульфосалициловая
п -Аминобензойная
Бензолсульфоновая
3,5-Диоксибензойная
Дипиколиновая
2,4-Диоксибензойная
1,3,5-Бензолтрикарбокснльная
Фталевая
Бензойная
Салициловая
л- г, итробензойная
Время удерживания,
pH элюента 2,1
0,90
0,98
1,00
1,04
1,06
1,10
1,20
1,44
1,60
1,96
2,40
4,46
4,60
7,15
21
30
30
мин
pH элюента 6,7
1,09
0,98
1,00
0,86
0,98
0,98
1,05
1,00
1,60
0,98
0,98
1,40
0,98
1,04
1,45
2,75
2.С5
Для элюирования можно использовать растворы как солей,
так її самих кислот, которые частично диссоциированы в водном
49
растворе. Однако при использовании растворов некоторых
ароматических кислот (фталевой, бонзойной, салициловой) на хро-
матограмме появляется дополнительный, системный пик [3].
Изучение природы этого пика показало [4, 5], что его появление
связано с неионообменной сорбцией недиссоциированных молекул
слабой органической кислоты на матрице сорбента, а также
нарушением сорбционного равновесия в колонке в результате
изменения pH в зоне определяемого аниона. Положение пика на
хроматограмме зависит от способности недиссоциированных
молекул кислоты, используемой для элюирования, сорбироваться
на матрице, и от концентрации этих молекул. Из данных,
приведенных в табл. 4.3, видно, что полимерной матрицей XAD-1
сильнее всего сорбируются гидрофобные молекулы ароматических
Ni.Zn
Э
1
ю
20
30
мин
Рис. 4.1. Хроматограмма
неорганических анионов [38].
Колонка TSK GE1 1С —
Апіоп — PW. Элюент 2 мМ
КОН. Скорость потока
1,0 мл/мин. Концентрация
анионов 10 мкг/мл.
Детектирование косвенное
кондуктометр ическое
Со
6 8 Ю
мин
Рис. 4.2. Хроматограмма
двузарядных катионов [26].
Колонка Dionex CS2,
4X250 мм. Элюент 2 мМ
лимонная кислота/2 мМ
этилендиамин (pH 4,0).
Скорость потока 1,5 мл/мин.
Концентрация катионов 5—
20 мкг/мл. Детектирование
кондуктометрическое
кислот, в то же время анионы этих кислот практически не
сорбируются. Поэтому при использовании в качестве элюентов солей
органических кислот отрицательные пики отсутствуют.
60
Помимо кислот, указанных в табл. 4.3, и их солей в качестве
элюентов использовали растворы сульфобензоата аммония,
перхлората и малоната натрия [6, 7], а также п-толуол-сульфоновую
и 2-нафтиламин-1-сульфоновую кислоту [8].
Однако все названные элюенты эффективны при определении
анионов сильных кислот и кислот средней силы и не годятся
для определения анионов слабых кислот, таких как цианид,
борат, арсенит и силикат, существующих в растворе только при
высоких pH. Для определения таких анионов в качестве элюен-
та используют гидроксиды натрия или калия [9]. Поскольку
гндроксил имеет большую эквивалентную электропроводность,
Таблица 4.4
Константы диссоциации и время удерживания ор'анических кислот.
Элюент — г и дроке ид калия различной концентрации (указана в таблице),
сорбент — TSK GelIC-620-SA [9|
Кислота
Бензойная
о- Толуоловая
.«-Толуоловая
л-Толуоловая
о-Хлорбензойная
.м-Хлорбензойная
л-Хлорбензойная
п-Аминобензойная
о-^итробензоіїная
.м-Нитробензойная
/г-Нитробензойная
о-Анисовая
л-Аннсовая
Салициловая
Бензолсульфоновал
л-Толуолсульфоновая
«-Хлорбензолсульфоновая
Фенол
о-Крезол
л-Крезол
гс-Крезол
3.5-Диметилфенол
и-Этилфенол
Муравьиная
Уксусная
Гликолевая
Щавелевая
Янтарная
Малеиновая
Фумаровая
Винная
рК
а
4,21
3,91
4,73
4,36
2,94
3,82
3,99
4,85
2,17
3,45
3,44
4,09
4,49
3,00
0,07
—
—
10,00
10,29
10,09
10,26
—
—
3,76
4,76
3,83
1,27; 4,27
4,21; 5,64
1,92; 6,23
3,02; 4,38
3,04; 4,37
Время
2 мЛ1
11,33
8,60
17,38
17,71
10,49
27,44
28,60
8,85
8,85
26,83
28,20
6,48
15,20
а*
12,28
17,96
29,48
16,50
19,47
18,53
20,03
28,00
37,30
4,10
3,68
3,60
а
а
а
а
а
удерживания, *
4 мМ
8,23
6,24
11,77
12,07
7,55
17,88
18,03
6,44
6,57
17,48
17,90,
5,25
10,44
25,9
8,60
11,99
18,0
9,66
13,67
12,60
13,80
16,18
22,30
2,79
2,59
2,54
а
а
а
а
а
6 мМ
5,76
4,46
8,01
8,37
5,34
9,75
10,10
4,63
4,81
9,60
9,64
4,18
7,18
22,0
5,97
8,14
15,34
5,98
9,46
8,78
9,51
10 80
15,40
2,23
2,27
2,09
13,13
11,03
11,53
16,17
11,10
>а — ис злк ируется.
чем другие анионы, детектирование проводят по отрицательным
пикам. Иными словами, при прохождении зоны образца через
детектор наблюдается снижение электропроводности, равное
разности между проводимостью определяемого аниона и
эквивалентно вытесненного гидроксила. Разделение необходимо
проводить на полимерных сорбентах, поскольку силикагель в
щелочной среде разрушается. На рис. 4.1 приведена хроматограм-
ма смеси неорганических анионов, полученная на полимерном
сорбенте TSK GelIC-Anion-PW с элюентом 1 мМ КОН.
В работе [9] изучено удерживание некоторых органических
кислот на полимерном сорбенте при элюировании растворами
гидроксида калия различной концентрации (табл. 4.4). Эти
данные могут быть полезными при выборе элюента для одноколо-
ночного определения. Поскольку для элюирования, как правило,
используют сильноудерживаемые ионы, то из приведенных в
табл. 4.4 в качестве элюентов могут быть использованы все
кислоты, удерживающиеся сильнее бензойной.
4.2.2. Определение катионов
При определении катионов щелочных металлов и аммония
для элюирования, как правило, используют разбавленные
(~1 мМ) растворы азотной или соляной кислоты [10, 11].
Поскольку эквивалентная электропроводность ионов водорода
наибольшая, детектирование проводили по уменьшению
электропроводности элюента при прохождении через детектор зоны
определяемого катиона.
Для определения катионов щелочноземельных и переходных
металлов в качестве элюентов используют соли этилен- или фе-
нилендиамина [10]. В этом случае, как и при определении
анионов, на хроматограмме появляется дополнительный пик.
Связано это, по-видимому, с сорбцией и десорбцией диамина
матрицей катионообменника при перемещении зоны определяемого
катиона по колонке. Для повышения селективности разделения
и сокращения времени определения двузарядных катионов в
качестве элюентов используют диаммониевые соли с комплексо-
образующим анионом винной, а-оксиизомасляной и других
кислот [12, 13]. Комплексообразующии анион смещает
ионообменное равновесие, частично образуя либо незаряженный, либо
меньшего заряда комплекс. Это приводит к уменьшению
коэффициента распределения металла и соответственно его времени
удерживания. Соответствующий анион, а также концентрацию и
pH элюента следует выбирать таким образом, чтобы связь ионов
в комплексе была достаточно слабой. Если устойчивость
комплекса высока, то время удерживания катионов будет слишком
мало и разделение не произойдет. Хроматограмма смеси
двузарядных катионов с использованием цитрата этилендиаммония
приведена на рис. 4.2.
52
4.3.
ПРАКТИКА ВЫБОРА ЭЛЮЕНТА
Оптимизация условий ионохроматографического определения
чаще всего достигается за счет изменения состава и свойств
подвижной фазы. Это более простой и доступный вариант
оптимизации, поскольку изменение свойств неподвижной фазы
сопряжено с большими сложностями. Оптимальным считается элю-
ент, позволяющий быстро, селективно и чувствительно
определять нужные ионы. Поиск такого элюента, как правило,
заключается в изменении состава и концентрации элюирующих ионов.
Часто оказывается эффективным введение в состав элюента комп-
лексообразующих агентов или органических добавок.
4.3.1. Состав
Первая задача, которая стоит перед аналитиком,
разрабатывающим метод ионохроматографического анализа определенного
образца, заключается в выборе подходящего элюирующего иона.
В этом случае, как и при решении любой другой аналитической
задачи, необходимо учитывать характер и особенности
анализируемого образца, а также свойства определяемых ионов.
Предположим, что стоит задача количественного
определения анионного состава образца воды, в котором наиболее
удерживаемым анионом является сульфат, а концентрация
определяемых анионов в диапазоне 1—10 мг/л. В этом случае можно
использовать как одноколоночный, так и двухколоночный
вариант ионной хроматографии. Поскольку определяемые анионы,
а это могут быть фторид, ацетат, фосфат, хлорид, нитрит,
бромид, нитрат, относятся к среднеудерживаемым, элюент должен
обладать средней элюирующей способностью. Такими элюента-
ми могут быть растворы бензойной, фталевой кислот или их
солей, смеси карбоната и гидрокарбоната.
Эмпирический принцип выбора элюента заключается в том,
что способность элюирующего и определяемых ионов
удерживаться на ионообменнике должны быть близки. Другими
словами для определения слабоудерживаемых ионов используют
слабые элюенты, для определения среднеудерживаемых — элюен-
ты средней силы и т. д. Некоторые элюенты, используемые для
определения анионов и катионов, и их элюирующая сила
приведены в табл. 4.5. и 4.6.
Одной из самых сложных задач является одновременное
определение слабо-, средне- и сильноудерживаемых ионов. Как
правило, изокрэтическим элюированием разделить такую смесь
за короткое время (до 30 мин) очень сложно. Чаще всего для
разделения смеси ионов, сильно различающихся по своему
удерживанию, используют градиентное элюирование. Однако
частично проблему можно решить и с помощью одного элюента.
Так, в работе [14] для разделения восьми различающихся по
удерживанию анионов предложено использовать в качестве
элюента щелочной раствор тирозина. Но обычно одним элюентом
53
можно разделить смесь либо слабо- и среднеудерживаемых
ионов, либо средне- и сильноудерживаемых. і
При выборе элюента следует помнить, что состав элюирую-
щих ионов влияет не только на селективность и быстроту
определения, но и на его чувствительность. Элюент надо выбирать
такой, чтобы различие фонового и аналитического сигналов было
Таблица 4.5
Элюенты для определения анионов и их элюнрующая способность
Элюирующая
способность
Слабая
Средняя
Сильная
Элю1!р\'ющии анион
ОА~, борат, HCO.f ,
аминоацетат, глицинат
HCO-^-f- СО3~, глутами-
нат, бензоат, аминобен-
зоат, гидрофталат, бен-
золсульфонат, тартрат,
оксалат
со|~, со'^~+ о .~ ,
салицилат, аминосалици-
лат, тирозинат, фталат.
цитрат, сульфосалицилат
Определяемые анионы
СНзСОО", С1СН3СССГ
п\'риват, гликолят, F~,
СГ, ВгО3~, Ю3~. H3POj
", HCOO-,
CN-. S2--
" HSiO7
адипат. бутират, бутилфосфат,
СігСНСОО". фумарат,
малеат, малоиат,
сукцинат, AsO^~, Nj". i
глутарат.
пропионат
ЧОГ. NOT
SOg", SO'4-", ClOJ", Вг~ , BFr,
HP05-. S-O^-, FeO|-
аскорэат, С13ССОО~,
CrO2-, ReO^". WO24-,
A&O]-.
MoO^j~ ,
с oa~ SCN" S->O-~ Cl^>~ T —
RuCIg", SeO|"
1-:- RhCl'l-
максимальным. При использовании прямого детектирования
фоновый сигнал элюента должен быть минимальным, а при
проведении косвенного детектирования — максимальным.
Таблица 4,6
Элюенты для определения катионов и их элюирующая способность
Элюирующая
способность
Слабая
Высокая
Элюирующий катион
Н+ . Li +
Zn2+, PJL, Ag+, См'2+
зтилендиаммониіі, фени-
лендиаіимоний, лизин
Определяемые катионы
катионы щелочных металлов. NH^
этаполамин. изопроііаноламин,
метиламин, диметиіамин, триметиламин,
гуанидиіі
катионы щелочноземельных и
переходных металлов, Н-йутиламин, тет-
раэтиламмоний
54
4.3.2. Концентрация
Если состав элюента выбирают эмпирически на основании
закономерностей удерживания определяемых и элюирующего
иона, то концентрацию элюента выбирают такую, чтобы время
определения не превышало 20 мин, а селективность разделения
соответствовала предъявленным требованиям.
Для определения оптимальной концентрации элюирующего
иона можно использовать уравнение B.9). Предположим, что
для определения фторида, хлорида, нитрата и сульфата на
колонке 3X250 мм, заполненной анионообменником с удельной
емкостью 0,03 мэкв/мл и пористостью 0,5, в качестве элюента
выбрали раствор бензолсульфоната (BSO~). Необходимо
определить, при какой концентрации элюирующего иона время
удерживания SO]~ (при скорости элюента 2 мл/мин) будет равно
15 мин. Поскольку анионообменники, используемые в ионной
хроматографии, по свойствам близки к Dowex 2—Х8, то,
пользуясь табл. 2.2, можно оценить коэффициенты селективности
бензолсульфоната и определяемых анионов. Эти величины будут
равны
HSO HSO BSO,
RSO, CI ' Cl ' ' '
КН0* =3,3/4 0 = 0,82; КС[ - 1,0/4 0 = 0,25;
BSO3 ' ' ' ' ESO, ' ' ' '
KF =0,13 4,0=0/32-
BSO;, '
Подставляя в уравнение B.9) Q = 0,03, є = 0,5, F = 2, V=l,87 и
SO
К * =1,52, находим что для V = 15 концентрация бензолсульфо-
BSOj
ната равна
п 1,52X0,03X0,5X1,87 , . 1П ,
Cbso, = — ~[ъх2 — = 1,4-10-3м.
Учитывая, что время удерживания несорбируемого компонента
не превышает 2 мин, а ширина пика су.льфата при V = 15 мин
равна приблизительно 3 мин, общее время анализа смеси фторида,
хлорида, нитрата и сульфата будет не более 20 мин.
Исправленные времена удерживания первых трех анионов при концентрации
бензолсульфоната 1,4-10-3М, рассчитанные по уравнению B.9),
составляют 0,32 мин (F~), 2,5 мин (С1~) и 8,2 мин (NO3~ ), что
свидетельствует о хорошем разделении. Время определения
можно уменьшить, повысив концентрацию элюирующих ионов, но
при этом ухудшится селективность разделения и снизится
чувствительность определения [15].
Изменяя концентрацию элюирующего иона, можно повысить
селективность разделения ионов с различными зарядами. В слу-
55
чае, когда заряды элюирующего и определяемого иона
различны, равновесие ионного обмена имеет вид
у R2—E-*+zX-y^zRy—X-y+yE-*, D.1)
где гну — заряды элюирующего и определяемого ионов
соответственно. Тогда в соответствии с уравнением B.4) и B.7)
Dx=(K*)»-(Q,CEy". D.2)
tR = (Kly-(QCE)y*.(Vs;F). D.3)
Логарифмируя обе части уравнения D.3), получим
ig tR = (-y(z)lgCE )+(y(z)lgQ)+const. D.4)
Аналогичная зависимость может быть получена
логарифмированием уравнения D.2). Из уравнения D.4) следует, что если
в разделяющей колонке содержится смола с заданной емкостью
Q, а концентрация элюента меняется, то график зависимости
логарифма исправленного времени удерживания (или
коэффициента распределения) от логарифма концентрации элюирующего
иона представляет собой прямую с наклоном (—у/г). На рис. 4.3
представлена подобная зависимость для хлорида, нитрата и
сульфата при их элюировании карбонатом [16]. Наклон прямых
для хлорида и нитрата равен (—1/2), а для сульфата (—1).
Благодаря этому удается повысить селективность разделения
нитрата и сульфата.
Для быстрого и селективного разделения ионов, сильно
различающихся по своему удерживанию, используют градиентное
элюирование с изменением концентрации элюента. Этот способ
применяли для определения серосодержащих анионов (SOj ,
SO24-, S2O32-) [17], а также Cl", Вг-, NO-f, SO2" и S2O2- [18].
4.3.3. Величина pH
Концентрация анионов слабых кислот и катионов слабых
оснований в растворе зависит от его pH. Поэтому, если такие
ионы используют как элюирующие, их концентрацию можно менять,
изменяя pH элюента. Зависимость равновесной концентрации
аниона слабой кислоты Н„А от pH выражается уравнением
Р''Н+l-' СнА, D.5)
л п
п
о
где Си а—общая концентрация слабой кислоты в растворе, а
п
?,- — константа диссоциации кислоты.
Сн а = [Н„ А1+[Нп_,А-| + [Н,,_.,А2-1-[-----МА«-1, D.6)
56
fr =*!•*,•....*,, D.7)
здесь Кі — ступенчатые константы диссоциации кислоты.
По уравнению D.5) можно рассчитать равновесную
концентрацию любой формы слабой кислоты при заданном значении pH
раствора. При использовании Нп-с А'~в качестве элюирующего
аниона уравнение B.9) принимает вид
Р Q(i—s) v zpjH-f]-'
При элюировании анионами слабых кислот увеличение pH
раствора элюента приводит к уменьшению времени удерживания
определяемых анионов. Это вызвано двумя причинами. При
незначительном увеличении pH увеличивается концентрация элюи-
рующих ионов, а при значительном увеличении возможно
изменение состава элюента. Другими словами, кислотно-основное
равновесие в растворе сдвигается в сторону образования аниона
с большим отрицательным зарядом и более высокой
элюирующей силой. Например, при использовании раствора о-фталевой
кислоты (pKi = 2,93, рКг = 5,41) с рН = 3,5 элюирующим является
гидрофталат, обладающий средней силой. Увеличение pH
элюента до 4,0 приводит к увеличению концентрации гндрофталата и
незначительному увеличению элюирующей силы. Но при
увеличении pH до 6,5 основной анионной формой элюента
становится двузарядный фталат с высокой элюирующей силой.
ig К1:
-5,0
-2,0
lg[HC|-]
Рис. 4.3. Зависимость
коэффициентов
распределения
неорганических анионов (D) от
равновесной
концентрации гидрокарбоната в
элюенте [15]
Рис. 4.4. Зависимость
удерживания неорганических
анионов (k') от pH 5,0 мМ
фталатиого элюенга [18]
Зависимость удерживания некоторых неорганических
анионов от pH 5мМ фталатного элюента приведена на рис. 4.4.
Видно, что изменение pH элюента изменяет не только время опре-
57
деления, но и его селективность. Наклон зависимостей для одно-
и двузарядных анионов различен, поэтому можно найти такое
значение pH элюепта, при котором селективность определения
таких пар анионов, как йодид—тиосульфат, бромид—сульфат
будет максимальной. Кроме того, изменяя pH элюента, можно
повысить селективность определения анионов слабых
многоосновных кислот, например фосфата. В зависимости от pH этот
анион может находиться в растворе в виде однозарядного дигид-
рофосфата, двузарядного гидрофосфата или трехзарядного
фосфата, удерживание которых различно. Поэтому, изменяя pH
элюента, можно перевести фосфат в форму, удерживание
которой будет оптимальным. В работе [19] установлено, что при
работе на колонке Vydac 302 1С D,6x250 мм) с элюентом 5 мМ
гидрофталат калия при скорости элюента 2 мл/мин оптимальное
Cl В НР2 I | О^
|-
р /
разделение ионов Cl-, Вг~, Н2РО4-, SO42~. I~, S2O|
достигается при pH элюента 5,3.
При выборе pH элюента для одноколоночного определения
следует помнить, что с увеличением pH увеличивается
концентрация элюирующих ионов. Это приводит к повышению
фонового сигнала и снижению чувствительности определения, особенно
при кондуктометрическом детектировании [15].
При определении анионов слабых кислот pH элюента должен
быть выше рК самой слабой из определяемых кислот. Тольки в
этом случае кислоты будут находиться в растворе в виде
анионов и возможно их ионообменное разделение [20].
э
I
4.3.4. Устранение посторонних пиков
При определении анионов на хромато-
грамме могут появляться два
посторонних пика (рис. 4.5). Первый из них (с
малым временем удерживания)
называют водным, пли инжекционным, а второй
(с большим временем удерживания) —
системным.
Появление водного пика связано с
прохождением через детектор неудержи-
вающейся зоны воды, содержащей элю-
ирующий анион в количестве,
эквивалентном анионному составу пробы.
Наличие водного пика мешает определению
анионов, удерживающихся слабее хло-
Рис. 4.5. Хроматограмма неорганических
анионов с инжекционным и системным пиками [21].
Колонка Vydac 302 1С, 4,6X250 мм. Элюент
2,5 мМ гвдрофталат калия (pH 4,0). Скорость
потока 2,0 мл/мин. Детектирование косвенное
УФ B85 им)
12 16
MUH
рида. В двухколоночной ионной хроматография водный пик
проявляется в виде неудерживаемого отрицательного пика. По
времени выхода этого пика можно судить о «мертвом»
объеме хроматографической системы. Для устранения
водного пика двухколоночное определение лучше проводить
на фоне депонированной воды или использовать элюенты,
дающие очень низкий фоновый сигнал. В одноколоночном
варианте водный пик может быть как положительным, так и
отрицательным в зависимости от концентрации и объема вводимой
пробы. В этом варианте устранить водный пик без усложнения
хроматографической системы очень трудно [21].
Появление системного пика характерно для одноколоночно-
го варианта. Подробно механизм возникновения пика изучен в
работах [22—24]. Появление системного пика вызывается
десорбцией с разделяющей колонки молекулярной формы элюента (не-
диссоциированной кислоты) при введении пробы. Эта форма
удерживается гидрофобной частью сорбента по обращенно-фаз-
ному механизму. Системный пик имеет большое время
удерживания и .мешает идентификации и определению сильноудержива-
емых анионов, особенно сульфата [21]. Кроме того, наличие
системного пика увеличивает время определения.
Высота и площадь системного пика, а также время его
появления зависят от pH, концентрации и объема вводимой пробы,
свойств определяемых ионов, pH и концентрации элюента, гид-
рофобности матрицы сорбента. Эти факторы влияют на кислотно-
основное равновесие в системе и на удерживание по обращенно-
фазному механизму.
Наиболее эффективным способом устранения системного
пика является использование элюентов с высоким значением pH,
при котором молекулярная форма отсутствует. Установлено, что
системный пик исчезает при элюировании раствором винной
кислоты (pKi = 3,04, рК2 = 4,37) с pH более 5 [23] и фталевой
кислоты (pKi = 2,95, pK2 = 5 41) с pH более 7 [21].
4.3.5. Использование комплексообразования
Для повышения селективности и сокращения времени
определения можно использовать комплексообразование. Для этого
в элюент вводят соединения, образующие с определяемыми
ионами комплексы различной устойчивости. В этом случае
коэффициент распределения определяемого иона будет равен
2
о
Здесь Dx — коэффициент распределения нона в отсутствие ком-
плексообразующего агента, [Ц — равновесная концентрация
этого агента и ?i — константа устойчивости комплекса. Таким
образом, коэффициент распределения, а соответственно и время
59
удерживания иона уменьшаются с увеличением концентрации
комплексообразующего лиганда и повышением устойчивости
комплекса. Это связано с образованием в подвижной фазе
комплексов, которые либо не удерживаются в разделяющей
колонке, либо удерживаются значительно слабее свободных ионов.
Время удерживания иона в присутствии комплексообразующего
агента согласно уравнению B.9) будет равно
1-?) V
CE 2 ?і [Ц! F
D.10)
Комплексообразование используют в основном для повышения
селективности определения катионов щелочноземельных и
переходных металлов. В качестве комплексообразующих добавок
применяют винную, лимонную, щавелевую, оксиизомасляную,
фталевую и салициловую кислоты или их соли с этилендиамином
[25—29]. Хроматограмма смеси семи катионов с цитратом этилен-
диаммония в качестве элюента приведена на рис. 4.2. Введение
в элюент комплексообразующего вещества позволяет добиться
быстрого и селективного разделения катионов.
Селективность разделения можно повысить, изменяя
концентрацию комплексообразующего агента. В табл. 4.7 приведены
исправленные времена удерживания катионов при различной
концентрации тартрата. Изменение времен удерживания зависит от
устойчивости тартратных комплексов. Чем выше устойчивость
комплекса, тем сильнее изменяется время. Поэтому время
удерживания РЬ2+ изменяется сильнее, чем у других катионов. С
другой стороны, Mg-+ при рН = 4,5 не образует тартратных
комплексов, и его время удерживания не изменяется.
Таблица 4.7
Исправленное время удерживания (мин) иоиов металлов
при постоянной концентрации этилендиамина B,0-10~3 М), pH
D,50) и различной концентрации тяртрата |25]
Ион
мє галла
Mg2+
Zn2+
Ni2+
Mn2+
Cd2+
Sr2^
Pb2+
0,0
2,8
3,2
3,4
4,3
5,2
11,2
41,2
Концентрация
1,0
2,9
2,6
3,0
4,0
5,0
11,0
12,7
2,0
2,8
2,0
2,4
3,8
4,5
10,3
7,8
тартрата, M>c
3,0
2,8
1,6
2,0
3,7
4,0
9,7
5,7
4,0
2,9
1,5
1,8
3,6
4,0
9,6
4,9
5,0
2,
1,
1,
3,
3,
9,
4,
8
4
6
5
7
1
1
Комплексообразование можно использовать для повышения
селективности определения анионов. Предложен способ опреде-
60
ления неорганических анионов (F~, PO^-j, образующих
устойчивые комплексы с Fe (III) [30]. Анионы разделяют на катионо-
обменнике низкой емкости с элюентом РеСіз/НСІ. Определяемые
анионы разделяются в соответствии с константами устойчивости
комплексов с Fe (III). Анионы, не образующие комплексов, на
катионообменнике не удерживаются. Фоновый сигнал подавляют
на анионообменнике высокой емкости в ОН-форме. Определяемые
анионы проходят через подавляющую колонку в виде
комплексов с железом, остальные анионы сорбируются и не дают
сигнала. Для определения применяют кондуктометрический
детектор. В этом случае использование комплексообразования
позволило существенно повысить селективность определения фторида
и фосфата.
Комплексообразование можно использовать для переведения
иона в более удобную для определения форму. Например,
определение бора в виде бората в двухколоночном варианте
затруднено, а в виде комплекса BF4 не представляет сложности
[31]. По-видимому, этот подход может быть применен для
определения и других элементов.
4.3.6. Введение органических добавок
Хроматографические пики сильноудерживаемых ионов С1О4~ ,
SCN~, SSO|~, Ba2+, РЬ2+ и ряда других часто бывают
различны, что затрудняет количественное определение таких ионов.
Размывание связано с вкладом необменных сорбционных процессов,
которые особенно заметны для ионов с большими временами
удерживания [32]. Для предотвращения размывания пиков в элю-
ент вводят органические добавки. При определении сильно
удерживаемых катионов в элюент добавляют метанол, ацетонитрил,
2-пропанол [29], а при определении анионов — ацетонитрил [33}.
В табл. 4.8 и 4.9 приведены зависимости удерживания и
эффекта б лица 4.8
Влияние органической добавки на удерживание катионов щелочноземельных
металлов [29]. Сорбент — Zorbax SIL элюент 3,0- 10 ~3 M Na3Cit в смеси Н2О:
органическая добавка = 9:1, рН = 7,1, F=l мл/мин
Органическая
доЗавка
Метанол
Ацетонитрил
2-Пропанол
Sr +'
тарелки/м
12 000
11 000
12 000
Фактор емкости, k'
1,30
1,17
0,95
2-1-
Са
1,47
1,48
1.07
Sr2+
4,22
3,56
2,35
Ва"^
6,95
6,18
3,92
тивности разделения двузарядных катионов от свойств
добавки и ее концентрации. В работе [29} влияние органических
добавок на удерживание объясняют изменением устойчивости цитрат-
61
ных комплексов металлов в водно-органической среде. При
увеличении концентрации добавки устойчивость комплекса
возрастает, а время удерживания уменьшается.
Таблица 4.9
Влияние концентрации метанола в элюенте на удерживание катионов
щелочноземельных металлов [29]. Условия такие же, как в табл. 4.8
Метанол, %
0
10
20
Sr2+
тарелки/м
20 000
12 000
8 300
Фактор емкости, k'
2,71
1,30
1,01
Са" '
2,71
1,47
1,23
Sr '
6,27
4,22
2,90
Ва2+
10,2
6,95
5,17
Введение метанола или ацетонитрила в элюент использовали
для повышения селективности определения калия и метиламина
[34]. Для разделения этих катионов в качестве элюента
предложили 0,01 M HC1 в 40%-ном метаноле. Селективность
повышается благодаря существенному увеличению времени удерживания
К + при введении органической добавки. Влияние метанола на
удерживание калия, по мнению авторов, связано с увеличением
набухания полимерного сорбента и изменением свободной
энергии сольватации катионов.
4.3.7. Аминокислоты как элюенты
В двухколоночной ионной хроматографии для определения
анионов и катионов обычно применяют четыре типа элюентов:
гидроксильный, или карбонатный, для определения анионов, а
сильнокислотный, или диаминовый, для определения катионов
щелочных и щелочноземельных металлов. Но каждый из этих
элюентов наряду с достоинствами имеет и недостатки. Так,
использование гидроксильных и сильнокислотных элюентов
позволяет детектировать ионы на фоне деионизированной воды. При
таком детектировании высока чувствительность, широк
линейный диапазон градуировочного графика. Однако из-за низкой
элюирующей способности эти элюенты можно использовать
только для определения слабоудерживаемых ионов. Средне- и силь-
ноудерживаемые ионы определяют с карбонатными или диами-
новыми элюентами. Но эти элюенты имеют ряд недостатков,
обусловленных детектированием ионов на фоне слабой кислоты или
оснований.
Перспективными элюентами для двухколоночной ионной
хроматографии являются растворы аминокислот или их солей [35—
37]. Принципиальная возможность использования этих
соединений в ионной хроматографии связана с их амфотерными
свойствами. Известно, что в зависимости от pH аминокислоты нахо-
62
дятся в растворе в трех формах: анионной, катионнои и цвит-
тер-ионной. Кислотно-основное равновесие для простейшей ами-
нокарбоновой кислоты, глицина, выглядит следующим образом:
Н+ + h++
NH2CH2COO-^[NHSCH2COOH^NH3CH2COO-] ^ NH3CH2COOH.
В кислой среде аминокислота находится в растворе в виде
катиона и может элюировать катионы, а в щелочной среде — в
виде аниона, способного элюировать анионы. В нейтральной
среде аминокислота преимущественно находится в виде цвиттер-
иона, способного элюировать и анионы, и катионы. При
определении анионов на колонках имеют место следующие равновесия:
разделяющая
R—An+NHaCH2COO-=^R—OOCCH1NH,+An-,
подавляющая
2 R—H -HNH2CH2COO-+Na+^:R—Na+R—NH8CH2COOH.
Для определения катионов используют катионную форму
аминокислоты. При этом на колонках имеют место следующие
равновесия:
разделяющая
R-Kt+NH3CH2COOH^R-NH3CH2COOH+Kt+,
подавляющая
2 R—OH+NH8CH8COOH+Cte=3-Cl +R—ООССНг\Нг+2Н»О.
Помимо катионнои и анионной форм в качестве элюента
можно использовать и цвиттер-ионную форму аминокислоты.
Сравнение некоторых характеристик аминокислотного и других элю-
ентов для двухколоночной ионной хроматографии приведено в
табл. 4.10. Из данных, приведенных в этой таблице, видно, что
аминокислотный элюент имеет все преимущества, свойственные
другим элюентам. Вместе с тем у него нет недостатков,
присущих другим элюентам.
Основным достоинством аминокислотного элюента является
возможность определения ионов на фоне деионизованной воды,
благодаря сорбции аминокислоты на подавляющей колонке. Это
происходит при определении и анионов, и катионов.
Детектирование на фоне деионизованной воды позволяет повысить
чувствительность определения и расширить линейный диапазон гра-
дуировочного графика по сравнению с карбонатным и диамино-
вым элюентами. В отличие от гидроксидного и сильнокислотного
элюентов можно использовать аминокислоты с различной элюи-
рующей силой и определять ионы независимо от их удерживания.
Так, с помощью элюента на основе я-аминосалициловой кислоты
63
Таблица 4.10
Сравнение характеристик аминокислотного и традиционных элюентов
для двухколоночной ионной хроматографии
Характеристики
Фоновый раствор
Элюирующая
способность
Предел
обнаружения, мкг/мл
Линейный
диапазон градуировоч-
ного графика
Время работы
подавляющей
колонки, ч
Водный пик
Системный пик
Градиент pH
Градиент
концентрации без
изменения фонового
сигнала
Элюент
Аминокислотный
Н2О
низкая,
средняя,
высокая
0,001
5—6
порядков
30
нет
нет
да
да
Гидро-
ксидиый
Н2О
низкая
0,001
5—6
порядков
10
нет
нет
да
,г,а
Карбонатный
Н3СО3
средняя,
высокая
0,01
3—4
порядка
15
есть
есть
да
нет
Сильнокислотный
н2о
низкая
0,001
5—6
порядков
15
нет
нет
да
да
Диаминовый
диамин
средняя,
высокая
0,01
3—4
порядка
15
есть
есть
да
нет
[38] можно определять слабо-,
анионы (рис. 4.6). А поскольку
N0",
сю:
0 9 18 27 36 45
мин
Рис. 4.6. Хроматограмма
неорганических анионов [37].
Колонка ХИКС— І, ЗХІ50 мм.
Элюент 1 мМ га-аминосалицнло-
вая кислота/1,5 мМ КОН (pH
9,8). Скорость потока 1,9 мл/мин.
Детектирование: коидуктомет-
рическое.
средне- и сильноудерживаемые
элюирующая способность л-ами-
носалицилата выше, чем
карбоната, то концентрация
катионов в аминокислотном элю-
енте ниже. Благодаря этому
время работы подавляющей
колонки с аминокислотным
элюентом больше, чем с
карбонатным.
Детектирование на фоне
деионизованной воды
устраняет водный пик, мешающий
определению слабоудерживае-
мых анионов. Поэтому
определение фторида, хлорида,
нитрита, формиата, ацетата и
других слабоудерживаемых
анионов лучше проводить с
аминокислотным элюентом.
Наличие на хроматограмме
водного пика свидетельствует
о недостаточной чистоте воды
и реактивов, использованных
64
для приготовления аминокислотного элюента. Благодаря полной
сорбции аминокислоты на подавляющей колонке устраняется и
системный пик.
Аминокислотный элюент удобен для градиентного элюирова-
ния. Независимо от того, какой градиент создается (pH или
концентрации), фоновый сигнал в процессе элюирования не
изменяется, что облегчает аналитическое определение.
ЛИТЕРАТУРА
1. Шпигун О. А.., ВолощикИ. H., 3 о л о т о в Ю . А. // Ж. аналит.
химии. 1987. Т. 42, № 6. С. 998—1003.
2. Р і n s с h m і d t R. K- // Ion Chromatographie Analysis of Environmental
Pollutants / Ed. by J. D. Mulik and E. Sawicki Ann Arbor- Ann Arbor Sei., 1979
P. 41—50.
3. G j e r d e D. T., F r і t z J. S. // Anal. Chem. 1981. V. 53, № 11. P. 2324—
2327.
4. F r і t z J. S., D u v a 1 D. L., В a r r о п R E. // Anal. Chem. 1984. V. 56,
№7. P. 1177-1182.
5. P a p p E. // J. Chromatogr. 1987. V. 402. P. 211—220.
6. G j er de D. T., Fritz J. S., Schmuckler G. // J. Chromatogr. 1979.
V. 186. P. 509—519.
7. G j e r d e D. T., S с h m u с k 1 e r G., F r і t z J. S. // J. Chromatogr. 1979.
V. 187. P. 35—45.
8. J a с k s о n P. E., H a d d a d P. R. // J. Chromatogr. 1986. V. 355, № 1.
P. 87—97.
9. Oka da T., К u w am о t о T. // Anal. Chem. 1983. V. 55, № 7. P. 1001 —
1004.
10. V e e V. T., К a n n e d y J. M. // Anal. Chem. 1982. V. 54, № 9. P. 1631 —
1633.
11. J u p і 1 1 e T. H., G j e r d e D. T. // J. Chromatogr. Sei. 1986. V. 24, № 10.
P. 427-433.
12. Seve nie h G. E., Fritz J. S. // J. Chromatogr. 1986. V. 371, № 2.
P. 361—372.
13. Z о 1 о t о v У u. A. et al. // J. Env. Anal. Chem. 1987. V. 31, № 2—4.
P. 99—106.
14. Glatz J. A, Girar d J. E. // J. Chromatogr. Sei. 1982. V. 20, № 2.
P. 266—271.
15. Шпигун О. А. и др. // Ж. аналит. химии. 1985. Т. 40, № 10. С. 1925—
1929.
16. Suden T. LindgrenM., С e d e r g r e n А. // Anal. Chem. 1983.
V. 55, № 1. P. 2—4.
17. Jenke D., Mitchell Р. К-, Pagenkopf G. К. // Anal. Chim.
Acta. 1983. V. 155. P. 279—285.
18. H a d d a d P. R., С о w і e С L. // J. Chromatogr." 1984. V. 303. P. 321 —
330.
19. Oka da T., К u w a m о t о T. // Anal. Chem. 1985. V. 57. № 4.
P. 829—833.
20. О k a d a T., К u w a m о t о T. // J. Chromatogr 1985. V. 350. P. 317—
323.
21. Jackson P. E., Ha d d a d P. R. // J. Chromatogr. 1985. V. 346.
P. 125-137.
22. Hershcovitz H., Yarnitzky Gh., Schmupkler G. // J.
Chromatogr. 1982. V. 244, № 1. P. 217—224.
2'3. Okada T., Kuwamoto T. // Anal. Chem. 1984. V. 56. № 12.
P. 2073—2078.
3 Зак. 34 G5
24. S even ick G. J., Fritz J. C. // Anal. Chem. 1983. V. 55, № 1.
P. 12—18.
25. S h p і g u n О. А., С h о p о г о v a 0. D., Z о 1 о t о v Y u. A. // Anal.
Chim. Acta. 1985. V. 172. P. 341—346.
26. YanD, SchwedtG. // Fresenius Z. Anal. Chem. 1985. B. 320, № 2.
S. 121 — 124.
27. S e v є п і с h G. J., F г і t z J. S. // J. Chromatogr. 1985. V. 347. P. 147—
154.
28. S m і t h R. L., P і e t r z y к D. J. // Anal. Chem. 1984. V. 56, № 4.
P. 610—614.
29. Золотов Ю. А., Шпигун О. А., Обрезков О. Н. Способ
определения фосфора в растворах, содержащих нитрат-ионы. / Авт. заявка СССР
№ 3740800/31—26 от 14.05.84 г.
30. Willshire J. P., Brown W. A. // Anal. Chem. 1982. V. 54, № 9.
P. 1647—1650.
31. Них R. A., Cantwell F. F. // Anal. Chem. 1984. V. 56, № 8.
P. 1258—1263.
32. Пазухина Ю. Э. ert al // Ж. аналит. химии. 1988. T. 43, № 1.
С. 117—119.
33. Bue с he le R. С, Re ut ter D. J. // J. Chromatogr. 1982. V. 240.
P. 502—507.
34. I v e y J. P. // J. Chromatogr. 1984. V. 287. P. 128—132.
35. S h p і g u n O. A., V о 1 о s h с h і k I. N., Z о 1 о t о v Yu. A. // Anal.
Sei. 1985. V. 1, № 2. P. 335—339.
36. Волощик И. H. Аминокислоты как элюенты в ионной хроматографии:
Дне. ... канд. хим. наук: М., 1987. 257 с.
37. Шпигун О. А., Волощик И. Н., Золотое Ю. А. // Ж. аналит.
химии. 1987. Т. 42, № 7. С. 1209—1216.
38. Ok a da Т., Ku warn о to Т. // Bunseki Kagaku. 1983. V. 36, № 1.
P. 20—25.
ГЛАВА 5
ПОДАВЛЯЮЩИЕ СИСТЕМЫ
Подавляющая система является составной частью системы
детектирования и предназначена для обеспечения максимального
различия кондуктометрических сигналов элюента и
определяемого нона. Широко используют две системы подавления:
колоночную и мембранную. На раннем этапе развития двухколоночной
ионной хроматографии использовали колоночное подавление.
Однако ряд недостатков этой системы вызвал необходимость
совершенствовать подавляющую систему, что привело к созданию
мембранной системы подавления. Эта система имеет ряд
существенных преимуществ перед колоночной. По-видимому, работы
по совершенствованию систем подавления будут продолжены, и
хочется надеяться, что в ближайшем будущем будут предложены
новые оригинальные системы.
5.1.
КОЛОНОЧНОЕ ПОДАВЛЕНИЕ
Колоночная система подавления была предложена в первой
работе по ионной хроматографии [1]. Реакция подавления, т. е.
перевод элюента и определяемого иона в соединения,
сильно различающиеся по своей электропроводности,
осуществляется в колонке, заполненной ионообменником высокой емкости. Эта
колонка устанавливается после разделяющей перед кондукто-
метрическим детектором. При определении анионов (см. табл.
4.1) подавляющую колонку заполняют катионообменником
высокой емкости в Н-форме. В большинстве случаев в
подавляющей колонке элюент переводят в малодиссоциированную кислоту,
а определяемые анионы в сильные кислоты. При определении
анионов слабых кислот наблюдается обратная картина: элюент
переводят в сильную кислоту, а анионы — в слабые кислоты.
При определении катионов подавляющую колонку заполняют
анионообменником высокой емкости в ОН-форме. При этом
элюент переводят в воду или малодиссоциирующее основание, а
определяемые катионы — в сильно проводящие основания.
В качестве сорбентов для подавляющей колонки могут быть
использованы катионообменник Dowex 50—Х8 и анионообменник
Dowex 1—Х8 или другие ионообменники, близкие к ним по свой-
3* 67
ствам. Размер зерен сорбентов 20—40 мкм, обменная емкость
3,5—5,0 мэкв/г. Мелкое зернение сорбента в подавляющей
колонке необходимо для уменьшения «мертвого» объема хромато-
графической системы и повышения эффективности разделения.
Для подавления используют сорбенты на полимерной основе
(чаще всего стирол-дивинилбензол), что позволяет работать как с
кислыми, так и со щелочными элюентами (pH 1—13). Это
особенно важно для определения анионов, где используют
щелочные элюенты с pH более 8.
В процессе работы подавляющая колонка постепенно
отрабатывается, т. е. сорбент переходит из протонной или гидрок-
сильной в солевую форму. Полностью отработанная колонка
перестает выполнять функции подавляющей системы и ее
необходимо регенерировать. Это один из недостатков колоночной
системы подавления, поскольку надо прерывать выполнение анализов
либо для проведения регенерации, либо для установки другой
подавляющей колонки. Поэтому в современных приборах
устанавливают две подавляющие колонки, одна из которых
находится в работе, а другую в это время регенерируют. Регенерацию
проводят пропусканием через колонку 2М H2SO4 или 0.5 M
NaOH и последующим промыванием колонки водой. Время
работы между регенерациями подавляющих колонок размером
6X250 мм фирм Dionex и Biotronik составляет 6—10 ч, в
зависимости от концентрации элюента и емкости подавляющей
смолы. Число образцов, которые можно проанализировать до полной
отработки подавляющей колонки, определяется по уравнению [1]
N=VbCb:(VaCaKX), E.1)
где V\ — объем слоя разделяющего сорбента, Vb — объем слоя
подавляющего сорбента, Сд — емкость разделяющего сорбента,
Св — емкость подавляющего сорбента, /С/— константа обмена
определяемого иона х, наиболее сильно сорбирующегося на ио-
нообменнике, на элюирующий ион у. Из уравнения E.1)
следует, что для увеличения времени работы подавляющей колонки
наряду с повышением емкости подавляющего сорбента можно
увеличивать объем подавляющей колонки. Однако это приведет к
увеличению «мертвого» объема системы и снижению
эффективности разделения. Поэтому отношение Vb /Va должно быть не
очень большим. С этой целью в современных приборах
устанавливают две подавляющие колонки малого объема CX150 мм),
работающие, как уже сказано, попеременно.
Определяемые анионы и катионы не должны удерживаться
на подавляющей колонке. Для анионов сильных кислот и
катионов сильных оснований это требование соблюдается, и
подавляющая колонка не вносит дополнительного вклада в
удерживание. По-иному ведут себя анионы слабых кислот, а точнее
молекулы малодиссоциированных слабых кислот, которые
образуются в результате реакции обмена в подавляющей колонке. Вслед-
63
ствие мембранного эффекта Доннана [2] эти молекулы могут
проникать в глубь зерен подавляющего катионообменника и
удерживаться на подавляющей колонке. Степень удерживания
слабых кислот зависит от величин их рКй, размера и степени
отработанности подавляющей колонки [3]. Этот эффект
наблюдается для анионов многих органических кислот [4, 5]i, нитрита [6J,
фосфата, цианида, карбоната, бората и фторида [7].
Удерживание на подавляющей колонке сказывается на высотах
хроматографических пиков анионов слабых кислот [3]. Чем больше время
работы подавляющей колонки, тем больше высота пиков.
Поэтому количественное определение анионов слабых кислот в двух-
колоночном варианте с подавляющей колонкой лучше проводить
по площадям, а не по высотам пиков.
Эксклюзионное удерживание угольной кислоты в
неотработанной части подавляющей колонки приводит к появлению так
называемого «карбонатного» отрицательного пика на хромато-
граммах, полученных с карбонатным элюентом [3, 8]. Наличие
этого пика и особенно зависимость его положения на хромато-
грамме от степени отработанности подавляющей колонки
снижает воспроизводимость определения таких анионов, как F~, Cl,
NO.j" и некоторых органических анионов. Несмотря на ряд
недостатков, колоночное подавление широко используют. В то же
время знание недостатков этой системы подавления необходимо
для правильного ее использования.
5.2.
NqCI,
МЕМБРАННОЕ ПОДАВЛЕНИЕ
Недостатки колоночной системы подавления, связанные с
размыванием хроматографических зон, необходимостью
регенерации подавляющей колонки а
эксклюзионны(м удерживанием
некоторых анионов слабых кислот
на подавляющей колонке, можно
устранить, используя
мембранную систему подавления. Эту
систему начали использовать с
1981 г. [31.
Принцип действия
мембранной системы подавления показан
на рис. 5.1. Элюент и
определяемые анионы в в.іде солей
направляют после разделяющей
колонки внутрь тонкой трубки,
стенки которой изготовлены из
катионообменного материала.
N<f
I
Рис. 5.1. Принцип мембранного
подавления
69
Снаружи мембранной трубки противотоком непрерывно подается
регенерирующий раствор серной кислоты. Через стенки трубки
происходит обмен ионов Na+ на Н+, и на выходе элюент
превращается в слабую кислоту, а определяемые анионы — в
сильные кислоты. При этом анионы регенеранта, элюента и
анализируемого образца в силу доннановской эксклюзии не могут
проникать через стенку такой трубки.
Процесс переноса частицы через мембрану включает три
стадии: 1) конвективный перенос частицы внутри трубки, 2)
диффузия частицы в неподвижном двойном электрическом слое на
поверхности мембраны, 3) диффузия частицы непосредственно
через мембрану. В случае ламинарного потока через
незаполненную трубку скоростьопределяющим будет диффузионный
процесс, и обмен между катионами элюента и регенеранта будет
происходить медленно. Для достижения полного обмена нужно
уменьшать скорость подачи элюента.
Описанная в работе [3] мембранная система подавления
состояла из собранных в пучок восьми полых катионообменных
мембранных трубок длиной 6 футов и внутренним диаметром
0,3 мм, которые снаружи регенерировали 0,01 M H2SO4. Такая
система подавления действительно позволяет устранить
недостатки, связанные с необходимостью регенерации и эксклюзионным
удерживанием на подавляющей колонке. Однако из-за большого
«мертвого» объема размывание зон в такой системе даже
больше, чем в подавляющей колонке. Кроме того, полный обмен Na+
на Н+ достигается только при скорости подачи элюента не
более 1 мл/ыин.
Для повышения скорости ионного обмена и уменьшения
мертвого объема, а следовательно, и повышения эффективности
разделения предложено использовать заполненные мембранные*
трубки, свернутые в спираль [9—12]. Схема одной из таких
систем подавления приведена на рис. 5.2.
выход
элюента
Вход
реггмеранта
Щель
Меморанная трудна
Выход
регенеранта
Вход
элюента
1 ,25 хц дюйма
Цилиндр
1,51* Ч дюйма
Рис. 5.2. Спиралевидная мембранная система
подавления [9]
Существует два способа заполнения трубки мембранных
подавляющих систем. В первом из них [9] катионную мембранную
трубку Nafion 811-Х (внутр. диаметр 0,8 мм) заполняют
инертными стирол-дивинилбензольными сферическими частицами диа-
70
метром 0,5 мм. Авторы [9] считают, что такое соотношение
диаметров трубки и частиц является оптимальным. Система позволяет
повысить скорость подачи элюента до 2,7 мл/мин (при
давлении 3 бар), а эффективность разделения ионов при ее
использовании выше, чем для колоночного и мембранного подавления
с незаполненной трубкой (рис. 5.3).
?
\
Л Мембрана
Рис. 5.3. Хроматограмма,
полученная с заполненной
мембранной системой подавления
[9]
Регенерант
Рис. 5.4. Схема микромем-
бранной системы подавления
[15]
Другой способ заполнения мембранной трубки предложен
Дезгупта с сотр. [10—12]. Для заполнения трубки Nafion 811-Х
(внутр. диаметр 0,7 мм) предлагается помещать в нее
нейлоновую нить диаметром 0,66 мм. Эта система была названа filament
filled helical (FFH). Мембранная трубка помещается в систему
в виде спирали, что позволяет повысить эффективность обмена
катионов за счет увеличения скорости переноса частиц из
раствора к стенкам трубки [10, 11]. При длине трубки 100 см FFH
позволяет . полностью перевести в угольную кислоту элюент
B,4 мМ Na2CO3/3MM NaHCO3) при скорости его подачи до
4 мл/мин. Однако давление в системе при этом составляет 25—
30 бар. Чтобы повысить эффективность обмена, предложено [12]
использовать двойную мембранную систему подавления (ДМАН).
Система состоит из FFH мембраны, которую помещают внутрь
мембранной трубки большего диаметра и сворачивают в спираль.
Элюент проходит через ДМАН между внутренней и внешней
мембранными трубками, а раствор регенеранта подается двумя
путями: внутри внутренней трубки и снаружи системы. Такая
конструкция позволяет существенно увеличить площадь контакта
элюеита и регенеранта.
71
В 1985 г. фирма Dionex создала новую подавляющую
систему, названную микромембранным подавителем (MMS) [13].
Принцип действия MMS изображен на рис. 5.4. В отличие от
трубчатой системы подавления в микромембранной используют тонкие
ионообменные пластины, закрывающие с двух сторон пластину,
иа которую по тонкому каналу подается элюент. Снаружи
ионообменные пластины омываются раствором регенеранта.
Преимуществом микромембранной системы является малый объем,
высокая эффективность ионного обмена и возможность
использования для градиентного элюирования. Сравнительные
характеристики различных систем подавления приведены в табл. 5.1.
Микромембранная система подавления была разработана для
определения как анионов, так и катионов, і
Таблица 5.1
Сравнительная характеристика систем подавления [13]
Характеристика
Возможность
непрерывной регенерации
Чувствительность
Емкость
Свободный объем
Возможность
градиентного элюирования
Колоночная
(]97j г.)
нет
высокая
высокая
2000
нет
Мембранная
A983 г.)
Да
высокая
низкая
200
Нет ,
Мнкромембранная
A985 г.)
да
высокая
высокая (в 10—'15
раз выше, чем для
мембранной)
45
да
Для удаления СО2, образующегося в элюенте после
подавляющей системы, предложено [14} использовать
газопроницаемую тефлоновую трубку. Такая трубка позволяет снизить
содержание СО2 в элюенте на 90%, что повышает чувствительность
определения и расширяет возможности градиентного
элюирования. Однако установка подобной конструкции после
подавляющей системы увеличивает «мертвый» объем и снижает
эффективность разделения, і
ЛИТЕРАТУРА
1. Small H., Stevens T. S., В a u m a n W. S. // Anal. Chem. 1975.
V. 47, № 10. P. 1801—1809.
2. D on n a n F. // Electrochem. 1911. V. 17. P. 572.
3. S t e v e n s T. S., D a v і s J. S., S m a 1 1 H. // Anal Chem. 1981. V 53,
№ 9. P. 1488—1492.
4. И в a h о в Д. А., Ш п и г у н О. А., 3 о л о т о в Ю. А. // Ж. аналит. химии.
1986. Т. 4I, № 1. С. 134—139.
5. T u r k e 1 s о n V. T., Richards M. // Anal. Chem. 1978. V. 50, № И.
P. 1420—1423.
6. К о с h W. F. // Anal. Chem. 1979. V. 51, № 9. P. 1571—1573.
72
7. Т а п а к а К., I s h і z u к a T., S u n a h a r a H. // J. Chromatogr. 1979.
V. 174. P. 153—160.
8. 1 s h і b a s h і W., К і к u с h і R., Y a m a m о t о К // Bunseki Kadaku.
1982. V. 31, №2. P. 207—211.
9. Stevens T. S., Jewett G. L., В r e d e w e g R. A. // Anal. Chem.
1982. V. 54, № 7. P. 1206—1207.
10. Da s gup ta P. K. // Anal. Chem 1984. V. 56, № 1 P 96—103.
11. D a s g u p t a P. K. // Anal. Chem. 1984. V. 56, № 1. P. 103—105.
12. D a s g u p t a P. К., В 1 і g h R. Q., M e r с u r і о M. A. // Anal Chem.
1985 V. 57, № 2. P. 484—489.
13. J о hn s о n L. // Analitica. 1986. № 1. P. 43—47.
14. Sunden T., С e d e r g r e n A., S і e m e r D. D. // Anal. Chem. 1984.
V. 56, № 7. P. 1085—1089.
15 F г a n к 1 і n G. O. / Amer. Lab. 1985. № 6. P. 65—72.
ГЛАВА 6.
ДЕТЕКТОРЫ
ел.
ТРЕБОВАНИЯ К ДЕТЕКТОРАМ
Для автоматического определения ионов после их разделения
в колонке используют проточные детекторы. В ионной
хроматографии наиболее распространены детекторы трех типов: кондук-
тометрический, спектрофотометрический и электрохимический.
Кондуктометрический детектор универсален (неселективен),
поскольку все ионы, находящиеся в растворе, проводят
электрический ток. Спектрометрический и электрохимический детекторы
являются селективными, поскольку они реагируют только на
определенные ионы. Конструкция всех трех детекторов, принципы
их работы и примеры использования подробно описаны в обзорах
[1-3].
К детекторам предъявляют следующие основные требования.
1. Высокая чувствительность. Детектор должен чувствовать
одну часть (или меньше) определяемого вещества в 106 частях
элюента.
2. Малый объем ячейки и такая ее конструкция, чтобы зона
определяемого вещества не размывалась при прохождении через
детектор.
3. Линейная зависимость величины сигнала детектора от
концентрации определяемого вещества в широком диапазоне
концентраций.
4. Высокая стабильность фонового сигнала.
5. Малое время отклика, что позволяет быстро и точно реги
стрировать определяемое вещество.
6. Стабильность сигнала детектора при изменении скорості
подачи элюента и температуры.
6.2.
КОНДУКТОМЕТРИЧЕСКИЙ ДЕТЕКТОР
6.2.1. Принцип работы
Действие кондуктометрического детектора основано на не
прерывном измерении электропроводности проходящего через не
го раствора. Электропроводность раствора G есть величина, of
ратная его сопротивлению R, и измеряется в обратных ома
(Ом^1) или сименсах (S):
G=l,/?. F.
74
Удельная электропроводность k определяется как
k =-9L-, F.2)
где А — площадь электродов (см2) и / — расстояние между
электродами (см). Таким образом, k измеряется в единицах
Ом-1 -см-1.
Постоянная кондуктометрическои ячейки К описывается
соотношением
Л' = і; А F.3)
и выражается в единицах см-1 . Из двух последних уравнений
следует, что
k = G К. F.4)
Эквивалентная электропроводность ). зависит от концентрации
вещества в растворе и выражается как .
1000 k ,с сч
'• = -с—, F-5)
где С — концентрация вещества в эквивалентах на 1000 см3.
Эквивалентная электропроводность измеряется в единицах
Ом -см^экв.
Сочетание уравнений F.4) и F.5) дает выражение,
связывающее эквивалентную электропроводность с измеряемой
электропроводностью G:
° <66>
Электропроводность раствора соли ЩАп с концентрацией С
выражается уравнением ,
C
KtAn
В ионной хроматографии соблюдаются принцип
электронейтральности и эквивалентности ионного обмена: общее число
эквивалентов катионов и анионов, участвующих в обмене, равно.
Поэтому при элюированни однозарядного аниона элюентом
электропроводность элюента равна
іс >+(C-c)>-;-cs /.s -]
, F.8)
где ?ье-~ и '-к ' — зквивалелтпые электропроводности элюента
(катиона и аниона соответственно), As~ — эквивалентная
электропроводность элюируемого аниона, Се и Cs — концентрации
элюента и определяемого аниона.
При элюировании катиона s+ элюентом Е+
электропроводность элюента равна
[ОЕ >.Р._+(СЕ-
G
п
где Ks+ — эквивалентная электропроводность элюируемого
катиона.
Таким образом, электропроводность элюента зависит от
эквивалентной электропроводности элюирующих и определяемых
ионов и от их концентрации. Величины эквивалентных электро-
проводностей некоторых катионов и анионов приведены в табл. 6.1.
Таблица 6.1
Предельные значения эквивалентной электропроводности некоторых
ионов в водном растворе при 25°С
Анионы
он-
Чг СЮ1~
V, SO4-
Вг~
1~
С1-
»/• с2о42-
Чг СО,2-
NO3-
ч, ро43-
SCNT
НСОСГ
F~
НСО3-
СН3СОО-
С2Н5СОО~
СвНбСОО"
Я., Он^гсн'.экв
198
85
80
78
77
76
74
72
71
69
66
55
54
45
41
36
32
Катионы
н+
к+
NH+
V, Рь2+
• V, La3+
1/3 Fe3+
V, Ва2+
V. Са?+
V2 Sr2+
CH3NH3+
V, Cu2+
V2 Mg2+
V. Co2+
V. Zn2+
Na+
Li +
N(C,H5)+
Ju Ом .см'-экв
350
74
73
71
70
68
64
60
59
58
55
53
53
53
50
39
33
Постоянную кондуктометрической ячейки вычисляют из
величины измеренной электропроводности раствора с известной
удельной электропроводностью (например, 0,001 M KC1) по
уравнению F.4). Если постоянная ячейки известна, то, измерив
G, можно рассчитать удельную электропроводность других
ионов.
6.2.2. Типы детекторов и их конструкция
Если к двум электродам, находящимся в растворе
электролита, приложено напряжение, то в растворе возникает
электрический ток, а проводящая среда имеет электрическое
сопротивление. На измерении этого сопротивления основан принцип
действия большинства кондуктометрических детекторов.
«76
Детектор состоит из проточной ячейки, в которую подается
анализируемый раствор, индикатора и системы регистрации кон-
дуктометрического сигнала. Индикатор градуируется в единицах
Ом -1 или мкОм —1 .
Кондуктометрическая ячейка представляет собой камеру
объемом менее 10 мкл, соединенную с двумя электродами,
изготовленными из пластины, золота, нержавеющей стали или другого
инертного проводящего материала. Сопротивление ячейки, как
правило, измеряют с помощью моста сопротивлений Уитстона.
Конструкция кондуктометрической ячейки, используемой в
ионных хроматографах фирмы Биотроник, приведена на рис. 6.1.
Г
г
Рис. 6.1. Схема кондуктометрической ячейки фирмы
Biotronik.
/ — вход злюеита, 2 — корпус ячейки, 3 — электрод,
4 — выход элюента, 5 — позолоченный электрод, 6 —
контакт с измерительным устройством, 7 — болт для
изменения постоянной ячейки, 8 — контрогайка, 9 — уп-
лотнительное кольцо
Электропроводность большинства растворов возрастает
примерно на 2% при увеличении температуры на 1°С. Поэтому в
кондуктометр ических детекторах предусмотрена температурная
компенсация. Осуществляют ее с помощью термистора и
компенсационной схемы, в которой сопротивление линейно меняется
с изменением температуры раствора. Полезно изолировать
ячейку детектора, чтобы предотвратить случайные колебания ее
температуры.
Кондуктометрические детекторы включают средства
компенсации фонового сигнала, который может на три порядка
превышать сигнал от образца. Компенсацию можно осуществить с
помощью дифференциальной схемы измерения, включающей
ячейку сравнения. Однако в большинстве современных кондуктомет-
рических детекторов фоновый сигнал компенсируют с помощью
электронной схемы, что позволяет использовать только одну
ячейку.
77
При измерении сопротивления ячейки могут возникать
нежелательные явления — образование на электродах двойного
электрического слоя, имеющего собственное емкостное
сопротивление, или электролиз. Эти явлення можно устранить, подав/я на
электроды переменное напряжение. При изменении знак/
приложенного напряжения меняются направление перемещения
ионов, характер электролиза и образования емкостного
сопротивления. По мере увеличения частоты влияние электролиза
снижается или совсем устраняется, и ток в ячейке определяется
емкостным сопротивлением. Влияние емкостного сопротивления!
можно устранить, измеряя мгновенный ток, т. е. ток,
возникающий в момент подачн напряжения, когда двойной слой еще не
образовался.
На электроды ячеек некоторых детекторов подают
напряжение синусоидальной формы частотой — 10—10 000 Гц. На этом
принципе работает детектор фнрмы Wescan модель 213. На
электроды этого детектора подается переменное напряжение с
частотой 10 кГц при напряжении 20 В. При таком режиме работы
электролиз отсутствует.
В других детекторах для устранения электролиза используют
метод биполярной импульсной проводимости [4, 5]. На ячейки
последовательно подают два импульса напряжения малой
длительности A00 мкс). Импульсы имеют противоположную
полярность, но равны по амплитуде и длительности. Сразу по
окончании второго импульса измеряют ток в ячейке и по закону Ома
определяют ее сопротивление. Поскольку при биполярном
импульсном питании в ячейке измеряют величину мгновенного тока,
емкостное сопротивление не влияет на измерения, и можно точно
определить сопротивление ячейки.
Для уменьшения поляризации, коррозии и других
нежелательных процессов, способствующих изменению поверхности
электродов и постоянной ячейки, используют ячейку, в которой
электроды не находятся в гальваническом контакте с анализируемым
раствором [6]. Для этого электроды покрывают тонким слоем
силиконового лака или тефлона.
6.2.3. Прямое детектирование
Этот принцип детектирования наиболее распространен в двух-
колоночной ионной хроматографии. В подавляющей системе элюент
переводят в низкопроводящее соединение, а определяемый ион —
в соединение с высокой электропроводностью. Детектирование
осуществляется по увеличению кондуктометрического сигнала
при прохождении зоны определяемого иона через детектор.
При прямом определении анионов сильных кислот в качестве
элюента используют соли слабых кислот, чаще всего угольной.
Фоновый сигнал элюента подавляют либо на катионообменнике
в Н-форме, либо в мембранной системе подавления. В этом
случае определяемые анионы детектируют в виде сильных кислот
на фоне слабой угольной кислоты. Равновесная концентрация
78
протонов и гидрокарбонат-ионов в растворе угольной кислоты
равна
[Н+] = [НСО3-] = VkaCHico3 ,
где ка — первая константа диссоциации угольной кислоты
(второй константой пренебрегаем), Сн2со, — общая концентрация
угольной кислоты.
Величина фонового кондуктометрического сигнала согласно
уравнению F.7) будет определяться выражением ,
GH!CO, = ^Н+}А/г"СНгСО,, +1НСО,~У feaCH2CO3 . F.10)
В соответствии с принципом эквивалентности ионного обмена
общая концентрация угольной кислоты в зоне определяемого
аниона уменьшается на концентрацию аниона (Сап)- Согласно
принципу электронейтральности [Н+]=[НСО3~] + САп . Тогда
электропроводность раствора в зоне определяемого аниона равна
Г 'з
An ~ 1000 К
F.11)
Изменение кондуктометрического сигнала (AG) при
прохождении зоны определяемого аниона через детектор равно
Д G =GAn - GH.2co, =
1000 /Г
t У к (СН2соз -CAn)
TooT? ¦ F-12)
Поскольку величина эквивалентной электропроводности
протона в несколько раз выше, чем других катионов (см. табл. 6.1),
кондуктометрическое детектирование анионов в виде кислот
наиболее чувствительно.
Однако прямое кондуктометрическое детектирование на фоне
слабой кислоты имеет ряд недостатков. Самым существенным
из них является сравнительно узкий диапазон линейной
зависимости аналитического сигнала от концентрации определяемого
аниона. Из уравнения F.12) видно, что только при САп <С Сн2со,
AG = (^+AAn)-CAn F.13)
— сигнал линейно зависит от концентрации аниона, а при
САп^кСн2со, зависимость имеет нелинейный характер. Кроме того,
при увеличении концентраций определяемого аниона в его зоне
уменьшается pH, что приводит к подавлению диссоциации слабой
79
кислоты и уменьшению величины фонового сигнала. Поэтому
происходит как бы «провал» аналитического сигнала, и область
его линейной зависимости заметно сужается. /
Для устранения этих недостатков в качестве элюентов/сле-
дует выбирать соли кислот, рК которых больше 9 или, что еще
лучше, проводить детектирование на фоне деионизованной/воды,
используя для элюирования растворы гидроксидов или
Аминокислот [7]. В этом случае величина фоновой электропроводности
практически равна нулю, и изменение сигнала описывается
уравнением F.13) в широком диапазоне концентраций определяемого
аниона. Более подробно теория сигнала при кондуктометриче-
ском детектировании на фоне слабых кислот изложена в рабо-
те [8].
Для бензоата натрия, одного нз наиболее распространенных
элюентов, электропроводность его раствора (соль диссоциирует
полностью) будет равна (см. уравнение F.7))
I^+^) CBzl /с 1Л
— • FЛ4)
Электропроводность в зоне определяемого аниона равна
G An = ———Bz iwo к —"—~ ' F.15)
тогда
п \ \г
Л /1 П /1 ' 'An Bz^ An _ in im
Из уравнения F.16) следует, что сигнал детектора линейно
зависит от концентрации определяемого аниона в широком
диапазоне. Уравнение зависимости сигнала от концентрации
определяемых катионов при использовании солей органических оснований
в качестве элюентов будет иметь такой же вид, как уравнение
F.16).
При элюировании растворами органических кислот
зависимость изменения кондуктометрического сигнала от концентрации
определяемого аниона будет описываться уравнением F.12),
поскольку, как и в двухколоночном варианте, речь идет о
детектировании анионов на фоне слабой кислоты. По-видимому, элюи-
рование растворами органических кислот менее удобно, чем элюи-
рование их солями. При использовании солей шире линейный
диапазон градуировочного графика и отсутствует системный пик.
Использование прямого кондуктометрического
детектирования в двухколоночной ионной хроматографии катионов
ограничивается определением щелочных и щелочноземельных
металлов. В случае щелочных металлов используют наиболее удобное
детектирование на фоне деионизованной воды с растворами НС1
или HNO3 в качестве элюентов и подавлением фонового
сигнала на анионообменнике в ОН-форме. Катионы щелочноземель-
ве
ных металлов их детектируют на фоне слабых оснований (эти-
лендиамины, фенилендиамин). В связи с этим возникают такие
же проблемы, как и при определении анионов на фоне слабых
кислот. Прямое кондуктометрическое детектирование можно
использовать только для определения анионов кислот, рК которых
меньше 6.
6.2.4. Косвенное детектирование
Для определения анионов слабых кислот (рК>7) в двухко-
лоночной ионной хроматографии используют косвенное
кондуктометрическое детектирование [9, 10]. В этом варианте элюиро-
вание проводят растворами солей сильных кислот (хлорид,
сульфат, фосфат) с добавлением гидроксида для создания щелочной
среды. В подавляющей колонке, заполненной катионообменником
в Н-форме, элюент переводят в сильную кислоту, а определяемые
анионы — в слабую. Детектирование проводят по уменьшению
фоновой электропроводности при прохождении через детектор
зоны определяемого аниона. ,
При использовании раствора хлорида натрия в качестве элю-
ента после подавляющей колонки образуется соляная кислота и
фоновая электропроводность определяется уравнением
Г? , (>н + Xci)Cci F.17)
UHC1 — iooo к
При определении аниона слабой кислоты, константа диссоциации
которой k, электропроводность раствора в зоне определяемого
аниона (Ап) равна
.
1000 К
F 1
Тогда изменение кондуктометрического сигнала при
прохождении через детектор зоны определяемого аниона будет равно
AG = G д „ - GhC. = "^ ALHAn - -HAn^W^HAn -*С1^НАп . F. Щ
1000 К
Если степень диссоциации кислоты мала и]^*СНАп<с;Снап
уравнение F.16) принимает вид
'— F.20)
В этом случае величина изменения сигнала линейно зависит от
концентрации определяемого аниона в широком диапазоне.
Косвенное кондуктометрическое детектирование, по-видимому,
может быть использовано и для определения катионов слабых
оснований. Элюентом может служить раствор хлорида натрия с
добавлением соляной кислоты для создания кислой среды, а по-
81
давляющая колонка должна быть заполнена анионообменником
в ОН-форме. Детектирование будет осуществляться по
уменьшению фонового сигнала гидроксида натрия при прохождении
через детектор слабого основания. Однако пока сведения о
подобном способе определения катионов слабых оснований в
литературе отсутствуют. 1
При проведении косвенного детектирования необходимо
помнить о возможности удерживания слабых кислот на катионо-
обменннке в Н-форме по эксклюзионному механизму. Для
устранения этого нежелательного явления следует использовать либо
короткие подавляющие колонки, либо мембранную систему
подавления.
6.2.5. Заместительная ионная хроматография
Это сравнительно новый вариант детектирования. Его
особенностью является наличие в подавляющей системе двух
последовательно соединенных колонок, что позволяет заменить кондук-
тометрический детектор на другой, например атомно-эмиссион-
ный или атомно-абсорбционный [11, 12]. Таким образом, этот
вариант представляет собой трехколоночную ионную
хроматографию.
Способ использовали для определения щелочных
металлов [11]. Элюентом служил раствор соляной кислоты. После
разделения на катионообменнике низкой емкости и прохождения
подавляющей колонки с анионообменником высокой емкости в
ОН-форме определяемые катионы в виде гидроксидов на фоне
деионизованной воды попадают в третью колонку
(заместительную), заполненную катионообменником высокой емкости в Li-
форме. В этой колонке разделенные катионы полностью
обмениваются на Li+, который затем определяют
высокочувствительным пламенным атомно-эмиссионным литиевым детектором.
Достоинство этого варианта — возможность использовать
простой, доступный и в ряде случаев чувствительный пламенный
атомно-змиссионный детектор. Недостатком является снижение
эффективности хроматографического разделения из-за
увеличения «мертвого» объема системы. Аналогичный вариант
использовали и для определения некоторых неорганических анионов [11].
6.3.
ДРУГИЕ ЭЛЕКТРОХИМИЧЕСКИЕ ДЕТЕКТОРЫ
Помимо кондуктометрического к электрохимическим
относятся потенциометрический, амперометрический и кулонометриче-
ский детекторы. Эти детекторы, как правило, реагируют на один
или несколько ионов и поэтому относятся к селективным.
Принцип действия и аналитические возможности электрохимических
детекторов описаны в обзорах [2, 13].
82
6.3.1. Потенциометрические детекторы
Определение ионов потенциометрическим детектором
осуществляется с помощью ионоселективных электродов. Принцип
действия этих электродов основан на измерении потенциала,
возникающего между электродом и анализируемым раствором.
Величина потенциала зависит от концентрации определяемых ионов
в растворе. Теория измерения, типы ионоселективных
электродов и их конструкции описаны в ряде книг и обзоров [14—19].
Основная проблема, возникающая при создании ячейки
проточного потенцнометрического детектора, заключается в
миниатюризации ионоселективного электрода. Объем ячейки должен
быть не более 10 мкм, но в то же время должен быть обеспечен
хороший контакт электрода с элюатом и быстрое время отклика
детектора на изменение концентрации определяемого нона.
В настоящее время в потенциометрических детекторах,
используемых для высокоэффективного хроматографического
определения ионов, используют ионоселективные электроды
нескольких типов. Для определения изомеров фталевой кислоты, а также
нитрат- и нитрит-ионов использовали водородный электрод [20].
Для определения неорганических анионов использовали
серебряные электроды [21, 22) и на основе триоктилметиламмония
[23]. Медь-селективный электрод использовали в детекторе при
определении переходных и редкоземельных металлов [25]. В этой
работе элюент после прохождения через разделяющую колонку
смешивали с раствором комплекса меди с ЭДТА, а определение
проводили по увеличению концентрации свободной меди в элюен-
те вследствие вытеснения этого элюента из комплекса металлами.
Потенциометрические детекторы характеризуются высокой
чувствительностью и воспроизводимостью. Но специфичность и
сложность конструкции этих детекторов ограничивают
возможности их применения.
6.3.2. Амперометрические детекторы
Амперометрические детекторы применяют для определения
электроактивных веществ, способных окисляться или
восстанавливаться на электродах. Возникающий ток электролиза
фиксируется в детекторе.
Зависимость величины предельного тока электролиза (і) от
концентрации электроактивных частиц (С) и скорости потока
(Fr ) выражается уравнением
t=k.n.F.cF?. ¦ F.21)
Здесь п — число электронов, участвующих в
окислительно-восстановительной реакции, F — число Фарадея, /г — константа,
зависящая от вязкости жидкости, коэффициента диффузии
электроактивных частиц и геометрии электрода. Величина а для
стационарного электрода в потоке колеблется от 1/3 до 1/2.
83
Поскольку величина тока зависит от концентрации
электроактивных частиц в элюенте, амперометрические детекторы с
успехом используют для определения различных веществ, в
частности ионов. Способность вещества окисляться или
восстанавливаться зависит от его окислительно-восстановительного
потенциала, поэтому амперометрические детекторы являются
селективными. Селективность можно изменить, подбирая оптимальный
потенциал электродов ячейки.
Амперометрические детекторы характеризуются высокой
чувствительностью, они позволяют определять нанограммовые
количества ионов. Достоинствами этих детекторов является
широкий интервал определяемых концентраций D—5 порядков
величины) и малый объем ячейки A мкл). Они просты, дешевы и
надежны. К недостаткам этих детекторов следует отнести
специфичность, что существенно ограничивает их использование.
Кроме того, амперометрические детекторы чувствительны к
изменению скорости потока элюента и значениям pH, что снижает
воспроизводимость результатов [25].
В проточных амперометрических ячейках обычно используют
три электрода: рабочий, сравнения и вспомогательный.
Электродом сравнения служит насыщенный каломельный элемент или
хлорид-серебряный электрод. Вспомогательный электрод
изготавливают из платины или нержавеющей стали. Окисление или
восстановление определяемых веществ проходит на рабочем
электроде, потенциал которого измеряют с помощью электрода
сравнения. Выбор материала рабочего электрода очень важен,
поскольку он во многом определяет возможности детектора. В
настоящее время используют рабочие электроды двух типов:
ртутный капающий и твердые
Аа -электрод
сравнения
Элюат из
колонки
электрод
Элюат
Hg - пробка
¦*- Copoe
V
Рис. 6.2. Схема проточной амперометричес-
кой ячейки с капающим ртутным электро-
дом
(углеродный,
платиновый, золотой или сереб-
РЬ - Вспомогательные р я н ы й ).
Капающий Hg -
электрод
Схема проточной
ячейки с капающим ртутным
электродом представлена
на рис. 6.2. Детектор с
такой ячейкой иногда
называют
полярографическим. К его достоинствам
следует отнести
возможность работы в широком
диапазоне отрицательных
потенциалов в катодной
области (менее — 2 В).
Кроме того, поверхность
постоянно
ляется, что устраняет
84
проблемы, связанные с загрязнением поверхности рабочего
электрода. Недостатками полярографического детектора является
узкий рабочий диапазон в анодной области (менее +0,2 В),
ограниченный собственным окислением ртути, и относительная
сложность конструкции ячейки. Этот детектор использовали для
определения сульфид- и цианид-ионов [26].
Более распространены амперометрические детекторы с
твердыми электродами. Материалами электродов служат стеклоуг-
лерод [27], серебро [27], золото [26] и другие [2]. Конструкция
ячейки детектора с твердым рабочим электродом представлена
на рис. 6.3.
Электрод
сравнения
КонтактггчО стержень
РаЪочий зттрод
Рис. 6.3. Проточная амперометрическая ячейка с твердым рабочим
электродом [27]
По сравнению с полярографическим детекторы с твердым
электродом имеют ряд преимуществ — более высокую
чувствительность, простоту конструкции ячейки, широкий рабочий
диапазон потенциалов (для стеклоуглеродного электрода от —1,3 до
+ 1,5 В). Это значительно расширяет возможности детекторов.
Недостатком таких детекторов является невысокая
воспроизводимость результатов, связанная с изменением поверхности
рабочего электрода в результате адсорбции загрязняющих веществ
или окисления.
Для повышения воспроизводимости и селективности
детекторов, а также устранения нежелательной зависимости величины
тока от скорости подачи элюента применяют детекторы,
работающие в импульсном или дифференциальном импульсном ре-
85
жиме [28]. Для повышения чувствительности и селективности
детектора в ячейке устанавливают два или несколько рабочих
электродов [29].
Амперометрические детекторы с твердым рабочим
электродом использовали для определения сульфид- и цианид- [26], а
также йодид-, цианид- и сульфит-ионов [27]. Для определения
катионов использовали косвенное амперометрическое
детектирование [30].
6.3.3. Кулонометрические детекторы
Наряду с амперометрией для детектирования
электроактивных частиц в хроматографии используют кулонометрию при
постоянном потенциале. Ячейка потенциостатического кулонометри-
ческого детектора сходна с ячейкой амперометрияеского
детектора с твердым электродом. Она включает три электрода:
рабочий электрод, электрод сравнения и вспомогательный. В
качестве рабочих используют графитовый, серебряный или платиновый
электроды. Принцип работы кулонометрического детектора при
постоянном потенциале для определения неорганических ионов
подробно описан в работах [25, 31].
В отличие от амперометрического детектора, где в
соответствующий продукт превращается лишь 1 —10% электроактивных
частиц, в кулонометрических детекторах конверсия
электроактивных частиц осуществляется полностью. Это достигается за
счет применения электродов с большой поверхностью и ячеек с
эффективным массопереносом частиц к электроду. Кулонометрн-
ческий детектор менее чувствителен к изменениям скорости
потока и температуры элюента, чем амперометрический.
Количественное определение ионов кулонометрическим детектором можно
проводить, исходя из площади хроматографического пика, не
используя градуировочного графика. Однако кулонометрические
детекторы сложны в изготовлении, обслуживании и
эксплуатации. Их электроды легко загрязняются и трудно очищаются.
Существует два способа использования кулонометрических
детекторов при постоянном потенциале. Один из них —
первичная кулонометрия, когда элюент из разделяющей колонки
подается непосредственно в ячейку детектора. При этом
определяются все электроактивные частицы, находящиеся в элюенте. Этот
подход использован для определения большого числа
неорганических анионов [31]. Другой способ — это вторичная
кулонометрия с использованием послеколоночной реакции между
определяемыми ионами и селективным реагентом для перевода этих
ионов в электроактивные частицы. При определении катионов
металлов этим способом использовали их реакцию со щелочным
раствором комплекса >лсдн (И) с диэтилентрнаминпентаук-
сусной кислотой [31], а для аиио'юв сильных неорганических
кислот и алифатических аминов — реакцию с хинолином [31—33].
ас.
в.4.
СПЕКТРОФОТОМЕТРИЧЕСКИЕ ДЕТЕКТОРЫ
6.4.1. Принцип работы и конструкция
Спектрофотометрические детекторы давно и успешно
используют в ВЭЖХ. Спектрофотометрическое детектирование можно
использовать и для определения ионов, поэтому эти детекторы
находят все более широкое применение в ионной хроматографии.
Используется основной закон светопоглощения Ламберта—
Бера, согласно которому количество поглощенного раствором1,
монохроматического излучения прямо пропорционально
концентрации поглощающих частиц в этом растворе С и длине
поглощающего слоя /:
Л*-=г*-С/, F.22)
где Ак — поглощение раствора при длине волны 'к, ех —
молярный коэффициент поглощения.
При прохождении раствора элюента через спектрофотометри-
ческий детектор его сигнал будет определяться уравнением
А\ = *\СЪ1, F.23)
при прохождении зоны определяемого вещества сигнал будет равен
А)=*\ИрЕ-С)+г\1Сг F.24)
Изменение сигнала выражается соотношением
А$ -А\= ($—4) 1СГ F.25)
Таким образом, изменение сигнала детектора при
прохождении определяемого вещества пропорционально концентрации
этого вещества, что позволяет использовать спектрофотометриче-
ский детектор в количественном хроматографическом анализе.
Оптическая схема спектрофотометрического детектора
представлена на рис. 6.4. Световой поток от источника излучения
Лампа
Дифракционная -- -" л
решетка __ - - " V J
Делатель Yl~\ о )Детектор
потока
проточная"
ячейка
Рис. 6.4. Оптическая схема спектрофотометрического
детектора
87
проходит через монохроматор, выделяющий излучение с
определенной длиной волны, и разделяется зеркалами на два потока.
Один из них проходит через* измерительную ячейку, другой —
через сравнительную.
Затем потоки излучения попадают на фотоэлементы.
Возникающий фототок усиливается дифференциальным
логарифмическим усилителем. При прохождении через измерительную ячейку
вещества, поглощение которого отличается от поглощения элю-
ента, возникает разбаланс фотоумножителя, что и фиксируется
на регистрирующем приборе в виде хроматограммы.
Большое значение для нормальной работы
спектрометрического детектора имеет конструкция проточной ячейки. Она
должна обеспечивать быстрое прохождение всей массы жидкости
через ячейку. Используют ячейки двух типов. В ячейке Z-формы
анализируемый раствор поступает с одного конца, омывает
кварцевые окошки и выводится с другого. В ячейке Н-формы
раствор, попадая через отверстие снизу в центр ячейки, разделяется
на два потока и выводится сверху. Объем ячеек обоих типов
должен быть минимальным, обычно не более 10 мкл.
Современные спектрофотометрические детекторы работают
как в УФ B00—400 мм), так и в видимой области спектра D0Q—
700 мм). Существуют детекторы с фиксированной длиной волны
излучения (фотометрические) и переменной
(спектрофотометрические) . В фотометрических детекторах в качестве источника
излучения., как правило, применяют ртутные лампы низкого
B54 мм) или среднего'давления. Мопохроматизация излучения
в этих детекторах осуществляется с помощью светофильтров. В
спектрофотометрических детекторах в качестве монохроматора
служит дифракционная решетка. При работе в УФ области в
качестве источника излучения используют дейтериевую лампу,
а в видимой области — вольфрамовую.
Существует три способа спекърофотометрического
детектирования: прямой, косвенный и с послеколоночной спектрофотомет-
рической реакцией.
6.4.2. Прямое детектирование
Прямое спектрофотометрическое детектирование проводят по
увеличению сигнала детектора при прохождении через него
зоны определяемого вещества. При этом способе поглощение
определяемого иона должно быть выше, чем элюирующего.
Поэтому длину волны детектора выбирают таким образом, чтобы
разность поглощений определяемого и элюирующего ионов была
максимальной.
Наиболее эффективным этот способ детектирования может
быть при определении органических ионов, особенно
ароматических, которые имеют сильное поглощение в УФ области. В
качестве элюентов в этом случае можно использовать растворы
неорганических солей, как правило, слабо поглощающих в этой
88
области спектра. Прямое спектрофотометрическое
детектирование также используют для определения неорганических анионов,
поглощающих в УФ области (табл. 6.2). По сравнению с кон-
Таблица 6.2
Пределы обнаружения некоторых неорганических анионов в УФ области
с различными элюентами [1]. Элюеиты: А — 10 мМ HCI; В — 3 мМ NaHCO3 /
/2,4 мМ Na2CO3; С — 6 мМ Na2CO3
Анион
AsO33-
AsC^-
Br~
BrO3-
ci-
CIO3-
1~
ІОГ
Длина
волны, HM
200
200
195
195
192
195
195
195
Э люент
А
А
В
в
в
15
С
В
Предел 1]
обнаружения,
м\т/мл |1
1,2
1.5
0.1
0,16
2.0
4,0
0.15
0,08
AHLOH
NO3-
NOJ-
с1' —
SCN~
S:CN~
SeO'^
Длина
волны,
HM
195
195
195
200
195
195
195
195
Элюент
в
в
в
А
с
с
в
в
Предел
обнаружения,
мкг/мл
0,3
0,1
0,1
0,4
0,2
0.4
0,5
15,0
дуктометрическим прямое спектрофотометрическое
детектирование позволяет повысить селективность и чувствительность
определения нитрита, нитрата и бромида [34, 35]. Возможности
использования этого способа детектирования для определения
неорганических анионов рассмотрены в работах [1, 34, 36].
6.4.3. Косвенное детектирование
При косвенном спектрофотометрическом детектировании
определение проводят по уменьшению сигнала детектора при
прохождении через него зоны определяемого вещества. Этот способ
используют для определения ионов, поглощение которых ниже
поглощения элюирующего иона.
Способ предложен Смоллом и Миллером [37]. В их работе
излагаются теоретические аспекты метода и приводятся примеры
его использования для определения катионов и анионов. Важное
значение для косвенного детектирования имеет правильный
выбор элюента. Элюент должен эффективно разделять
определяемые ионы и в то же время его оптическая плотность должна
находиться в интервале 0,2—0,8, где точность спектрофотометриче-
ского измерения максимальна.
Для определения неорганических анионов косвенным
методом в качестве элюентов используют 1-10~3 M растворы фта-
лата натрия, 1-10—4 M сульфобензоата натрия и 1-10 2 M три-
мезиата натрия [37, 38]. При определении щелочных и
щелочноземельных металлов элюеитом служил 1,25-10 ~3 M раствор
сульфата меди (II), а при совместном определении катионов и
анионов — 5-Ю-3 M раствор нитрата меди (II) [37]. Чувстви-
89
тельность определения неорганических анионов в варианте
косвенного спектрофотометрического детектирования
приблизительно на порядок выше, чем при использовании кондуктометричес-
кого детектирования без подавляющей колонки [38].
6.4.4. Послеколоночная реакция
Такую реакцию проводят для повышения чувствительности и
селективности определения. Чаще всего прием используют при
определении катионов металлов. Послеколоночная спектрофото-
метрическая реакция — это реакция, с помощью которой
разделенные на колонке катионы металлов переводят в сильно
поглощающие, чаще всего окрашенные соединения. Для этого в элю-
ат, прошедший через колонку, вводят спектрофотометрический
реагент. Реагент и элюент перемешиваются в смесительной
камере, которая устанавливается между колонкой и детектором.
Схема камеры, приведенная в работе [39], показана на рис. 6.5.
Капиллярная | Реагент
труока
К детектору Е^у. v^ Этет
Рис. 6.5. Разрез смесительной камеры для
проведения послеколоночной реакции [39]
Элюент после колонки поступает в камеру с одной стороны, а
спектрофотометрический реагент — с другой. Мертвый объем
такой камеры не превышает 2 мкл, что обеспечивает
эффективное перемешивание и препятствует размыванию хроматографи-
ческих зон.
Послеколоночные реакции должны быть быстрыми,
собственное поглощение спектрофотометрического реагента — слабым
или вовсе отсутствовать, а поглощение соединения металла с
реагентом — максимальным. Как правило, реакцию проводят
при большом избытке реагента. Послеколоночные реакции с кси-
леновым оранжевым и арсеназо III использовали для
определения редкоземельных металлов [40, 41], а с пиридилазорезорци-
ном — для определения переходных металлов [42].
6.5.
ПРАКТИКА ВЫБОРА ДЕТЕКТОРОВ
Выбор детектора зависит от числа определяемых ионов, их
концентрации и желаемого времени анализа. Для определения
большого числа ионов в одном образце необходим универсаль-
90
ный детектор. Если нужно определять несколько ионов, близких
по своим свойствам, задачу можно решить с помощью
селективного детектора. В ряде случаев для повышения селективности и
уменьшения времени анализа используют комбинации
универсальных и селективных детекторов.
6.5.1. Универсальные детекторы
Хроматография является многокомпонентным методом
анализа, поэтому использование универсальных детекторов в этом
методе предпочтительно. В ионной хроматографии
распространенным универсальным детектором является кондуктометрический.
Для него разработаны различные варианты метода (двухколо-
ночный, одноколоночный, косвенный). Как уже говорилось,
кондуктометрический детектор позволяет определить за 20 мин до
10 ионов, а с градиентным элюированием — более 20 ионов [43].
Чувствительность определения с кондуктометрическим
детектором максимальная, особенно в двухколоночном варианте (до
1 нг/мл без предварительного концентрирования). Правда, при
работе с этим детектором необходимо строго контролировать
температуру, поскольку температурный коэффициент кондукто-
метрнческого сигнала довольно высок.
Помимо кондуктометрического как универсальные можно
использовать косвенные УФ [37] и атомно-абсорбционный [44]
детекторы, а также послеколоночную СФ реакцию [39]. Косвенный
УФ детектор чаще используют для определения непоглощающих
в этой области спектра анионов, а косвенный
атомно-абсорбционный и послеколоночную реакцию — для определения
катионов. При работе с этими детекторами нет необходимости строго
контролировать изменения температуры, но чувствительность
определения ниже, чем в случае кондуктометрического детектора.
6.5.2. Селективные детекторы
Для быстрого и селективного определения небольшого числа
ионов можно использовать селективные детекторы. Самыми
распространенными являются электрохимические детекторы. Их
применение наиболее эффективно в тех случаях, когда
определению электроактивных ионов с универсальным детектором
мешают неэлектроактивные ионы.
Потенциометрический детектор используют для
чувствительного и селективного определения С1~, Вг~~ , 1~ , SCN~, S~'2 ,
CN- , N0^ и NOg- [45, 46]. Амперометрический детектор можно
использовать для определения бромида, йодида, цианида,
сульфида [47—49]. Как правило, при использовании
электрохимических детекторов в одном образце можно определить не больше
пяти ионов. Укажем на использование атомно-абсорбционного
детектора [50—52].
91
6.5.3. Комбинации детекторов
Для повышения селективности и чувствительности
определения и сокращения его времени можно использовать комбинации
двух или трех селективных и неселективных детекторов.
Для определения Cl~, NO^", Br-, SO«" и S2O|~
предложено [53] использовать пары из трех детекторов — кондуктомет-
рического, спектрофотометрического и рефрактометрического.
Помимо повышения селективности и быстроты определения
сочетания двух детекторов позволяют расширить диапазон линейности
градуировочного графика до 4 порядков величины при расчете
по площадям пиков. Для определения NO2~- , NO3~ , С1~ и SO^"
использовали [34] комбинацию из кондуктометрического и
электрохимического детекторов. Нитрит определяли
электрохимически, а остальные анионы — кондуктометрически. Удалось
повысить селективность совместного определения нитрита в
присутствии больших количеств хлорида и фосфата.
Можно использовать также комбинации из трех детекторов.
Сланина [55] предложил определять F~ , Cl~, NO2~, РО4'~ , Br ~
NO~ и SOj- тремя детекторами — кондуктометрическим, УФ и
потенциометрическим. Это позволило существенно сократить время
определения и повысить селективность. Описан [56] трифункцио-
нальный (УФ, кондуктометрический и флуоресцентный)
детектор, который может быть использован в ионной хроматографии.
ЛИТЕРАТУРА
1. H a d d a d P. R., H e с k e n b e r g A. L. // J. Chromatogr. 1984. V. 300,
№ 3. P. 357—394.
2. W h і t e R. С // Analyst. 1984. V. 109. P. 677—697.
3. M a j о r s R. E., В a r t h H. G., L о h m u 11 e r С H. // Anal. Chem.'
1984. V. 56, № 5. P. 300R—349R.
4. Johnson D E., Enke С G. // Anal. Chem. 1970. V. 42, № 3.
P. 329—335.
5. К e 1 1 e r J. M // Anal. Chem. 1981. V. 53, № 2. P. 344—345.
6. PungorE. Pal F. TohtK // Anal. Chem. 1983. V. 55, № 11.
P. 1728—1731.
7. Shpigun O. A, Voloshchik I N., Z о 1 о і о v Y u. A. // Anal.
Sei. 1985. V. 1, № 2. P. 335—339.
8. Va n О s M. J. et al. // Anal. Chim. Asia. 1984. V. 156. P. 169—180.
9. Шпигун О. А., Во лощи к И. А., Золотое Ю. А. // Ж. аналит.
химии. 1987. Т. 42, № 6. С. 998—1006.
10. Р і n s с h m і d t R. K. // Ion Chromatographie Analysis of Environmental
Pollutants / Ed. by J D. Mulik and E Sawicki. — Ann Arbor: Ann Arbor Sei.,
1979. P. 41—50.
11. Downey S W., Hieftje G M. // Anal. Chim. Acta. 1983. V. 153.
P. 1—13.
12. Galante L. J., Hieftje G. M. // Abstracts Pittsburgh Conference
on Analytical Chemistry and Applie Spectrascopy. — Atlantic City. 1986. № 981.
13. S t u 1 ik K., P а с a k о v a V. // J. Electroanal. Chem. Interfacial Electro-
chem. 1981. V. 129, № 1. P. 1—10.
14. Морф В. Принципы работы ионоселективных электродов и мембранный
транспорт. Пер. с англ. М.: Мир, 1985. 280 с.
92
15. Лакшминараянайах Н. Мембранные электроды. Л,: Химия, 1979.
360 с.
16. Байулеску Г., Кошофрец В. Применение ионоселективных
мембранных электродов в органическом анализе. М.-. Мир, 1980. 232 с.
17 Д а р с т Р. А. Ионоселективные электроды. Пер. с англ. М.: Мир, 1972.
432 с.
18. Никольский Б, П., Мат ер о ва Е. А. Ионоселективные электроды.
М.: Химия, 1983.
19. Петру хин О. М. Ионоселективные электроды. М.: Знание, 1986 32 с
20. Schultz F. A, Ma this D. E. // Anal. Cliem. 1974. V. 46, '№ 14.
P 2253 2255
21. H e r s h с о v і t z H., J a r n і t z k y C, S с h m u с k 1 e r G. // J.
Chroma togr. 1982. V. 252. P. 113—119.
22. Butler E. С V., G e rs hey R. M. // Anal. Chim. Acta. 1984. V. 164.
P. 153—161.
23. Ar ug a H. et al. // Buhseki Kagaku. 1983. V. 32, № 6. P. 585—591.
24. Dore y R. С // Diss. Abstr. Int. B. 1980. V. 40. P. 1672—1685.
25.1 R ti с k і R. J. // Talanta. 1980. V. 27, № 2. P. 147—156.
26. Bond A. M. et al. // Anal. Chem. 1982. V. 54, № 3. P. 582—585.
27. J a n di k P., Со x D., W on g D. // Amer. Lab. 1986. V. 18. № 3.
P. 114—125.
28. S w a r t z f a g e r D. G. // Anal. Chem. 1976. V. 48, № 14. P. 2189—2192.
29. MoClintockS. A., PurdyW. С // Int. Lab. 1984, № 9. P. 70—76.
30. Wallace G. G. et al. // Abstracts Pittsburgh Conference on
Analytical Chemistry and Applied Spectroscopy. — Atlantic City 1986 № 977.
31. G і r a r d J. E. // Anal. Chem. 1979. V. 51, № 7. P. 836—839.
32. TanakaJ, M u t о G. // Anal. Chem. 1973. V. 45, № 11. P. 1864—1868.
33. Tanaka K., Ishizuka T., S u n a h a r a H. // Chromatogr. 1979.
V. 172. P. 484—489.
34. W і 1 1 і a m s R. J. // Anal. Chem. 1983. V. 55, № 6. P. 851—854.
35. Tanaka K. // Bunscki Kagaku. 1982. V. 31, № 6. P. T106—T111.
36. R e e v e R. N. // J. Chromatogr. 1979. V. 177. P. 393—397.
37. S m a 11 H., M і 11 e r T. E. // Anal. Chem. 1982. V. 54, № 2. P. 462—466.
38. N a і s h P. J. // Analyst. 1984. V. 109. P. 809—812.
39. Elch uk S., Cassidy R. M. // Anal. Chem. 1979. V. 51, № 3.
P. 1434—1438.
40. Hayakawa T., Moriyasu M., Hashimoto J. /'/ Bunseki
Kagaku. 1983. V. 32, № 2. P. 136—140.
41. H і г о s e A. et al. // J. High Resolut. Chromatogr., Chromatogr. Commun.
1981. V. 4, № 3. P. 531—537.
42. H w a n g J. M., С h a n g F. C, Y e h Y. С // Clin Chem. Soc. 1983. V. 30,
№2. P. 167—172.
43. J о h n s о n L. // Analitica. 1986. № 1. P. 43—47.
44. Tarter J. G., Make ton S. // Abstracts Pittsburgh Conference on
Analytical Chemistry and Applied Spectroscopy. — Atlantic City. 1986. № 632.
45. S u z u k і К. et al. // Bunseki Kagaku. 1983. V. 32, № 8. P 585—590.
46. Lockridge J. E. et al. // Anal. Chim. Acta. 1987. V. 192. P. 41—48.
47. P y en G. S., E r dmann D. E. // Anal. Chim. Acta. 1984. V. 164.
P. 153—160.
48. Whe a 1 s B. B. // J. Chromatogr. 1987. V. 402, № 1. P. 115—126.
49. R о с k 1 і n R D. Johnson E. L // Anal Chem. 1983. V. 55, № 1.
P. 4—7.
50. R icci G. R. et al. // Anal. Chem. 1981. V. 53, № 4. P. 610—613.
51. P e t t e r s e n J. M. // Anal. Chim. Acta. 1984. V. 160. P. 263—266.
52. M a k e t о n S., О t t e r s о n E. S, T a r t e r J. G. // J. Chromatogr.
1986. V. 368. P. 395—400.
53. J e n k e D. R., Mitchell P. K., Pagenkopf G. K. // Anal. Chim.
Acta. 1983. V. 55. P. 279—285.
54. К о r d о г о u b a V. et al. // Anal. Chim. Acta. 1982. V. 143. P. 173—178.
55. Slanina J. et al. // Anal. Chim. Acta. 1981. V. 130. P. 1—13.
56. G о n z a 1 о F. P. // Quim. e ind. 1985. V. 31, № 2. P. 265—269.
93
- ГЛАВА 7 —
ПРИБОРЫ
В настоящее время ионные хроматографы серийно выпускают
около десяти фирм и предприятий США, Японии, ФРГ, СССР и
Швеции. Приборы обеспечивают высокую селективность,
чувствительность и воспроизводимость определения ионов. Все
ионные хроматографы снабжены высокоэффективными беспульса-
ционными насосами, благодаря чему стабильность сигнала
детектора очень высока. Такие насосы обеспечивают давление до
20 МПа. Использование петли-дозатора для ввода пробы
повышает воспроизводимость результатов определения. Поскольку
для элюирования часто используют кислые или щелочные
растворы, особое внимание уделяется коррозионной стойкости
приборов. Низкая коррозионная стойкость хроматографической
системы оказывает негативное влияние на определение ионов,
особенно катионов [1]. Тяжелые и переходные металлы, которые
попадают в элюент в результате его взаимодействия с
металлическими частями хроматографической системы, могут
сорбироваться в разделяющей колонке и снижать ее селективность и
эффективность. Для повышения коррозионной стойкости ионных
хроматографов, особенно двухколоночных, всю систему, через
которую проходит элюент, выполняют из полимерных материалов.
7.1.
ДВУХКОЛОНОЧНАЯ ИОННАЯ ХРОМАТОГРАФИЯ
Пионером в развитии двухколоночной ионной хроматографии
является Dionex Corporation (Sunnyvale, СА, USA). Первые
ионные хроматографы были выпущены этой фирмой в 1976 г.
Первые патенты по ионной хроматографии также принадлежат этой
фирме. Dionex специализируется только в области ионной
хроматографии. Это позволило фирме накопить богатый опыт по
синтезу сорбентов, созданию новых систем подавления,
усовершенствованию приборов и практическому применению метода.
Ионные хроматографы серии Dionex 2000i сконструированы
по модульному принципу. Прибор комплектуется пятью
детекторами: кондуктометрическим с микромембранной системой
подавления для анионов и катионов, спектрофотометрическим
(видимая область), спектрофотометрическим для видимой и УФ обла-
94
сти, флуоресцентным с послеколоиочным реактором, а также ам-
перометрическим. В зависимости от задачи можно устанавливать
любой из этих детекторов или их комбинацию. Кроме того, Dio-
пех производит высокоэффективные колонки для ионообменных,
ионоэксклюзионных, ион-парных и обращеино-фазных разделений.
Широкий выбор детекторов и разделяющих колонок, наличие
микромембранной системы подавления, высокая коррозионная,
стойкость хроматографической системы позволяют использовать
приборы серии Dionex 200CH для быстрого, селективного и
чувствительного определения многих неорганических и органических
веществ.
В 1986 г. на Питсбургской конференции фирма Dionex
продемонстрировала новый ионный хроматограф серии 4000І (рис. 7.1).
Этот прибор снабжен высокоэффективным, коррозионно-стойким
насосом, позволяющим проводить элюирование с градиентом по
четырем растворам. Использование градиентного элюирования
существенно повышает возможности ионной хроматографии. На
приборе Dionex 4000І за 25 мин разделяют с хорошим
разрешением 22 неорганических и органических аниона [2].
Рп;. 7 1. Ионный хроматограф Dionex 4'JOUi
С 1981 г. начала выпускать двухколоночные ионные
хроматографы фирмы Biotronik Wissenschaftliche Gerate GmbH (Main-
95
5 (M
Рнс. 7.2. Блок-схема ионного хроматографа Biotronik IC-5000.
1, 2, 3 — система подачи инертного газа; 4 — емкости для элюента; 5 — кран выбора элюента- 6 — насос подачи
ента; 7 — демпфер; 8 — манометр; 9 — кран ввода пробы; 10 — кран выбора разделяющей колонки; // — разделяющая
элюента
колонка; U — мембранная система подавления; 13— кондуктометрическая ячейка; 14, 15 — емкости для регенеранта
и воды: 16 — электромагнитные клапаны; 17 — кран выбора регенеранта; 18 — воздухоуловнтель; 19 — насос для подачи
tal. FRG). Фирма производит ионные хроматографы 1С 1000 и
1С 5000. Приборы этой фирмы снабжены высокоэффективными,
беспульсационными, коррозионно-стойкими насосами, все
трубопроводы и колонки выполнены из полимерных материалов, что
обеспечивает высокую коррозионную стойкость всей хроматогра-
фической системы. Эти приборы снабжены колоночной и
мембранной системами подавления. Они комплектуются пятью
детекторами: кондуктометрическим, спектрофотометрическим для
видимой и УФ областей, рефрактометрическим, флуоресцентным,
электрохимическим. Фирма производит колонки для
ионообменного, ионоэксклюзионного и ион-парного разделения. Ионный
хроматографіє 1000 предназначен для выполнения ручных
анализов, он надежен и прост в эксплуатации. Для
автоматизированных определений используют прибор 1С 5000. Этот хроматограф
имеет программатор с семью программами и автоматический
пробоотборник. Блок-схема хроматографа 1С 5000 приведена на
рис. 7.2, внешний вид — на рис. 7.3.
Рис. 7.3. Ионный хроматограф Biotronik IC-5000
Элюент, находящийся в одной из шести емкостей 4, под
давлением инертного газа через кран выбора элюента 5 подается с
помощью двухпоршневого беспульсационного насоса 6 через
кран для ввода пробы 9 в разделяющую колонку //, выбор
которой осуществляется краном 10. Разделяющая колонка может
быть соединена с мембранной системой подавления 12 или через
4 Зік. 34
97
8-ходовой кран 20 с одной из двух подавляющих колонок 21.
После прохождения системы подавления поток поступает в кондук-
тометрическую ячейку 13, а затем направляется на слив. На этой
схеме можно выделить отдельный контур регенерации.
Регенерирующие растворы находятся в емкости 14, 15, вода для
промывки отрегенерированнои подавляющей колонки в емкости 15а.
Выбор регенерирующего раствора осуществляют краном 17. При
работе с подавляющей колонкой 21 требуемый раствор выбирают
с помощью электромагнитных клапанов 16. Регенерирующие
растворы подают насосом 19.
Такая схема позволяет проводить одновременно определение
и регенерацию отработанной подавляющей колонки. Например,
при работе с левой разделяющей и правой подавляющей
колонками левую подавляющую колонку можно регенерировать. Это
дает возможность работать с короткими подавляющими
колонками и уменьшить влияние эксклюзионных эффектов. Приборы
фирмы Biotronik успешно используют для определения многих
неорганических и органических катионов и анионов.
Рис. 7.4. Ионный хроматограф Цвет-3006
В 1986 г. в Дзержинском ОК.БА НПО «Химавтоматика»
начат выпуск ионного хроматографа Цвет-3006 (рис. 7.4). Прибор
работает по двухколоночной схеме (рис. 7.5). Элюент подается
в систему беспульсационным насосом 3. Скорость подачи элюен-
та от 0,5 до 5,0 мл/мин при максимальном давлении 16 МПа.
Анализируемую пробу вводят в элюент с помощью устройства
для ввода пробы 7 и дозирующего крана 8. При необходимости
98
j 5
1
y
S 4
2
fi
Рис. 7.5. Блок-схема ионного хроматографа Цвет-3006.
1 — элюент; 2 — регенерант; 3 — насос для подачи элюента; 4 — насос для
подачи регенерата; 5, 6 — блок ограничения давления; 7 —.ввод пробы; 8 —
шестнходовон дозируюшпй кран; 9 — колонка для концентрирования; 10 —
разделяющая колонка; //, 12 — подавляющая колонка; 13 — сдвоенный четы-
рехходовой переключающий кран; 14 — термостат; 15 — кодуктометрическая
ячейка; 16 — дросселирующая колонка; 17 — блок измерения
электропроводности; 18 — система автоматизации анализа; 19 — автоматический потенциометр
концентрирования пробы используется колонка 9, через которую
с помощью шприца или дополнительного насоса пропускают
необходимый объем пробы. Разделенные в колонке 10 ионы через
подавляющую колонку // попадают в кондуктометрическую
ячейку 15. Для исключения образования пузырьков газа в кондукто-
метрической ячейке (особенно при определении анионов, когда
после подавляющей колонки образуется СО2) после нее
установлена дросселирующая колонка 16. Изменения электропроводности
элюата фиксируются блоком измерения электропроводности 17,
сигнал с которого попадает на вход системы автоматизации
анализа 18 и автоматизации потенциометра 19, где и записывается
в виде хроматограммы. Система автоматизации анализа
рассчитывает время удерживания пика, его высоту и площадь, а при
соответствующей калибровке рассчитывает и концентрацию
определяемого иона. Воспроизводимость значений времен
удерживания и высот хроматографических пиков не превышает 3%.
Насос 4 предназначен для регенерации второй подавляющей
колонки 12, в то время как первая подавляющая колонка //
находится в работе. Переключение подавляющих колонок производится
с помощью крана 13. В приборе предусмотрена возможность тер-
мостатирования колонок и ячейки с помощью термостата 14.
Хроматограф комплектуется колонками, заполненными сорбентом
ХИКС-1.
Ионный хроматограф Цвет-3006 удобен для определения
анионного состава объектов окружающей среды,
сельскохозяйственных, промышленных, биологических и ряда других объектов
[3]. Этот прибор с успехом используют для анализа природных
вод различного состава (рис. 7.6). Однако определение катионов
на этом приборе пока затруднено
Q- из-за недостаточной коррозионной
стойкости хроматографической
системы.
В 1987 г. в Дзержинском ОКБА
начат выпуск переносного ионного
хроматографа ХПИ-1 (рис. 7.7).
Этот прибор предназначен для
проведения анализов в полевых
условиях. Он компактен, имеет малый
вес и габариты. ХПИ-1 особенно
удобен для анализа объектов
окружающей среды, а также
геологических и сельскохозяйственных
объектов.
Рис. 7.6. Хроматограмма 7.2.
колодезной воды,
полученная на приборе Цвет-3006 ОДНОКОЛОНОЧНАЯ ИОННАЯ
[3] ХРОМАТОГРАФИЯ
Число фирм, выпускающих ионные хроматографы, с каждым
годом растет. В 1986'г. приборы для одноколоночной ионной хро-
100
матографии производили около десяти фирм. Среди них такие
известные фирмы, как Millipore (США), Wescan (США), Тоуо
Soda (Япония), Shimadzu (Япония), Varian (США), Tecator
(Швеция).
Рис. 7.7. Переносной ионный хроматограф ХПИ-І
Одноколоночные ионные хроматографы, как правило, имеют
эффективные беспульсацнонные насосы для подачи элюента,
коррозионностоикую хроматографическую систему. Для
уменьшения влияния температуры флуктуации в приборах
предусмотрено термостатирование или электронная термокомплексация
сигнала. Поскольку в одноколоночном варианте величина
фонового сигнала сравнительно высока, то все одноколоночные
ионные хроматографы имеют широкий диапазон компенсации
фонового сигнала. Характеристики насосов и кондуктометрических
детекторов некоторых приборов для одноколоночной ионной
хроматографии приведены в табл. 7.1. Для расширения возможностей
101
приборов их, как правило, комплектуют, помимо кондуктомет-
рического, электрохимическим, спектрофотометрическим и
другими детекторами. і
Таблица 7.1
Некоторые характеристики одноколоиочных ионных хроматографов
Блок
Насос
Кондук-
то
метрический
детек гор
Характеристика
диапазон скоростей,
мл/мин
воспроизводимость,
%
точность, %
максимальное
давление, бар
диапазон измерений,
мксм
объем ячейки
компенсация нуля к
полной шкале
Wescan 261
0-5
±0,2
rt 1
345
0—10і
0,5
10-кратная
Waters
ILC-l
0,001—20
:t0,l
483
0,05—2-10'
1,4
Toyo Soda
HLC-60I
0,1—9,9
70
0-103
1,4
2-кратная
Shimadzu
HIC-6A
0,01—0,9
=?0,5
0,1—5120
0,25
10-кратная
В работе [4] проведено сравнение эффективности и
селективности пяти разделяющих анионных колонок, используемых в од-
ноколоночной ионной хроматографии. Для сравнения взяли
колонки Vydac 302 1С, Interaction ION-100, Hamilton PRP-XIOO,
Bio-Gel TSK IC-Anion-PW и Waters 1С Pak Anion. Результаты
Таблица 7.2
Эффективность и селективность анионных разделяющих колонок для
одноколоиочиой ионной хроматографии [4]. Элюент — гидрофталат калин
Колонка
название
Vydac
Ova IVj
*nteraction
ION-100
Hamilton
PRP-X100
Ialo-GelTSK
IC-Anion-PW
Waters ICPAK
Anlon
размер.
250 ¦ 4,6
50-3,2
150-4,1
50-4,6
50-4.6
Элюент
концентрация,
м M
3
1
1
1
1
pH
5,3
4,1
5,5
5,3
7,0
Скорость
потока,
мл/мин
2,0
1,0
2,0
1,2
1.2
Эффективность*
теоре-
тич.
тарелки на
колонку
4163
605
2356
959
1306
теоре-
тич.
тарелки на
колонку
16 652
12 100
І15 706
19 180
26 120
Разрешение (R )
/noJ"
1.97
0,75
1,28
1,92
2,37
N°2 /вг~
0,73
0,91
1,76
1,62
2,05
Вг /NO7
2,09
0,87
1,62
2,03
2,64
* Рассчитано для нитрат-иона 5сх методом.
102
сравнительных испытании в оптимальных условиях с гидрофта-
латным элюентом приведены в табл. 7.2. Было установлено, что
из пяти проверенных колонок, четыре (Vydac, Hamilton, Bio-
Gel TSK и Waters) имеют примерно одинаковую эффективность и
селективность. Однако при работе с колонкой Vydac pH элюента
не может быть больше 6,0, что мешает устранению системного
пика и затрудняет определение анионов слабых кислот. Кроме
того, при работе с колонками Vydac и Interaction выше фоновый
сигнал элюента, что снижает чувствительность определения.
ЛИТЕРАТУРА
1. Bogen D. G., Na go urne y S. J. // Ion Chromatographie Analysis
of Environmental Pollutants / Ed. by J. D. Mulik and E. Sawicki. — Ann Arbor:
Ann Arbor Sei., 1979. P. 319—328.
2. Anal. Chem. 1986. V. 58, № 2. P. 148A—152A.
3. A p a T с к о в а А. А., Орлов В. И., Яшин Я. И. //Ж. аналит. химии.
1987. Т. 42, № 2. С. 3O5--369.
4. H a d d a d P. R., Jackson P. E., H e с k e n b e r g A. L. // J. Chro-
matogr. 1985. V 346. P. 139—148.
ГЛАВА 8-
ОПРЕДЕЛЕНИЕ
НЕОРГАНИЧЕСКИХ АНИОНОВ
8.1.
ОПРЕДЕЛЕНИЕ АНИОНОВ В ВОДАХ
КАК АНАЛИТИЧЕСКАЯ ЗАДАЧА
Анализ вод разного типа чаще всего включает определение
анионов. Например, в случае подземных
высокоминерализованных вод, употребляемых как лечебные или столовые напитки,
обычная аналитическая задача — определение сульфата,
хлорида, карбоната и ряда других анионов. В питьевых водах
водопроводов определяют хлорид, нитрат, сульфат, а также фторид и
другие неорганические компоненты анионной природы. Более
сложная задача — контроль анионного состава сточных вод; состав
этот может быть самым разнообразным.
Диапазоны концентраций неорганических анионов в водах
могут быть исключительно широкими — от ничтожных
содержаний в особо чистой воде до макроконцентраций хлоридов в
морской воде. До появления ионной хроматоїрафии не было
эффективного метода определения неорганических анионов. Спектро-
фотометрическиїї, титриметрический, гравиметрический методы
и даже потенциометрический с ионоселективными электродами
не всегда удовлетворяют современным требованиям. По
аналитическим характеристикам — чувствительности, - селективности,
скорости и воспроизводимости — эти методы определения
неорганических анионов заметно уступают ионной хроматографии.
По этой причине ионную хроматографию чаще всего
используют именно для определения анионов, особенно неорганических.
Метод позволяет существенно сократить время анализа, повысить
его чувствительность и селективность. Природные и сточные
воды, атмосферные осадки, многие технологические растворы
являются наиболее удобными объектами для ионной
хроматографии. Их анализ в большинстве случаев не требует пробоподго-
товки и занимает не более 20 мин.
Настоящая глава посвящена этой наиболее важной области
использования ионной хроматографии. Будут рассмотрены
примеры использования метода в различных его вариантах для
анализа вод и некоторых других объектов.
8.2.
ДВУХКОЛОНОЧНЫЙ ВАРИАНТ
В большинстве случаев анионы определяют, используя
карбонатные элюенты. Чаще всего разделение анионов проводят на
колонке Dionex Anion с элюентом 3 мМ NaHCO3/2,4 мМ Na2CO3
104
при скорости потока 2,3 мл/мин. Этот элюент иногда называют
стандартным. Примеры использования двухколоночиой ионной
хроматографии для определения неорганических анионов
приведены в табл. 8.1. .
Вильяме [41} определил, что время удерживания І9
неорганических анионов при использовании стандартного элюента не
превышает 30 мин, а сильноудерживаемые анноны элюнруются
6 мМ раствором Na2CO3 менее чем за 20 мин (табл. 8.2).
Следовательно, все анноны, перечисленные в табл. 8.2, можно
определять двухколоночным вариантом. Правда, селективность
определения некоторых из них со стандартным элюентом невысока.
Так, стандартные условия определения непригодны для
совместного определения фторнда, сульфида и йодата; хлорита и дигид-
рофосфата; бромата и хлорида; гидрофосфата и селенита;
бромида и сульфита; нитрата н хлорида. В этом случае требуется
дополнительная оптимизация условий.
Двухколоночная ионная хроматография больше всего
подходит для анализа вод с содержанием анионов 1—500 мкг/мл. В
этом случае не требуется никакой пробоподготовки, кроме
фильтрования образца. Результаты определения характеризуются
высокой воспроизводимостью E не более 5%). К объектам с
таким анионным составом относятся речные, озерные,
поверхностные, дождевые и грунтовые воды. Поэтому ионную
хроматографию в основном н используют как раз для их анализа.
Определение анионов в высокочистых водах с содержанием
менее 1 мкг/мл требует предварительного концентрирования.
Следует отметить, что продолжительность одного анализа с
концентрированием на 5—10 мнн дольше, а воспроизводимость
результатов хуже (Sr не более 10%). Однако эти недостатки
компенсируются повышением чувствительности в 100—200 раз.
Анализ объектов с высоким содержанием анионов (более
500 мкг/мл) требует предварительного разбавления пробы. В
противном случае наблюдается переполнение разделяющей
колонки и нсвоспрокзводимость результатов по временам
удерживания и площадям пиков. Предварительное разбавление
необходимо при анализе морской воды н некоторых технологических
растворов. ,
Определение неорганических анионов в водах и некоторых
Объект
анализа
Определяемые
анионы
Разделяющая
Подавление
Элюент
Речная
сточная
воды
, PO|
Dlonex Anion
DX250 мм)
ночное
1,5 мМ Na2CO3/
/5,0 мМ NaHCO3
Речная,
грунтовая
и сточная
воды
F", Cl", NO3
SCN
ХИКС-1
CX250 мм)
тоже
1 мМ тирозин
(pH 10,8)
Речная,
водопроводная и
грунтовая воды
и AsO|-
вии F~
SO
в
присутстCl", NO^,
Dionex
HPIC-AS-
CX250 мм)
.2-
2,5—3,0 мМ NaHCO3/
/1.5—2,4 мМ Na2CO3
Грунтовая,
водопроводная и сточ-
иая воды
F", СГ, N07
'4 "
so?-
Dionex Anion
3X500 мм)
3,0 мМ NaHCO3/
/2,4 мМ Na2CO3
Минеральная
вода
F", Cl", Br"
Dionex Anion
Cx500 mm)
то же
Геотермальная и
озерная воды
F", Cl-, РОЗ",
Dlonex Anion
Cx250 мм)
Геотермальная вода
F", СГ, Br~, SO|
2-
Dlonex Anion
CX500 мм)
2,0 мМ NarCO3/
/1,6 мМ Na2COs
Атмосферные осадки
, СГ
SOi~
Dionex Anion
CX250 и
3X500 мм)
3,0 m M NaHCO3/
/2,4 мМ Na2CO3
F~,Cr
З мМ NaHCO3/
/3 m M Na„CO„
других оОъектах двухколоночной ионной хроматографией
Таблица 8.1
Скорость
потока,
мл/мни
2,5
1,8
1,5
2,3
1,9
1
1,9
2,75—3,0
Определяемые
концентрации,
мкг/мл
3,025— ЦМОзГ) (
Э,05— 1(РО^-) (
0,02—250
0,05—0,25 мкг
Se(IV)
0,01 — 500
0,05—300
—
нысохие
0,028—1,5 (СГ)
0,022—2,0 (NO-p
0,06—10 (SO4~)
Предел
обнаружения,
M КГ/МЛ
),007(NO.p
3,017 (РО^)
0,01
0,018—1,2 мкг Se
0,47—0,85 мкг As
.
0,005 (F~, Cl~)
0,05(NOj~, PO^.
—
—
0,001—0,005
s , %
г
,8(NOJ-)
*,5(РОЗ-)
2
10
0,1—2,
0,4—3,7
менее 5
Примечания
N и Р окисляют
персульфатом
—
используют
многомерную
ионную
хроматографию
обзор
результаты
сравнивают с
лругими
методами
і
[для
определения С1— оэра-
ієц разбавляли
результаты
сравнивают с
данными
других методов
Литература
[1]
[2]
[3]
[4]
[5]
6]
[7]
[8
[9]
l»i
Объект
анализа
Дождевая
вода
Дождевая,
поверхностная и
питьевая во ы
Дождерая
Еода
Снег и лед
Снег
Морскач
вода
I
!
Определяемые
анионы
F~, СГ, NOjf, SO^~
F", СГ, PO|-,
NO3-, SO^
СГ, NO]", SO^-
SOj~ в присутствии
СГ. NOjf, SO^~
F~, cr.NO^", SO^.
NO3-, S0^~
er, NO3-, so^-
F~, СГ, Br~,
m-, so24-
Br~
і
1
Раз_еляюшая
колонка
Dionex Anlon
C « 500 mm)
Dionex Anlon
Dionex Anlon
Anion
Separator AX-1
D,6x250 mm)
Dionex Anlon
Dx250 mm)
Dionex Anlon
Dx250 и
4x125 мм)
Dionex Anlon
Dionex Anion
(ЗХІ250 мм)
Dionex Anlon
Cx215 mm)
Подавление
-
бранное
ночное
то же
•
Элгоелт
3,0 мМ NaHCCy
/2,4 мМ Na2CO3
4,8 мМ NaHCCy
/2,3 мМ Na2CO3
3,0 мМ NaHCCy
/2,4 мМ NaaCO3
4 мМ NaHCOs/
/2 мМ Na2CO3
3 мМ NaHCO3/
/3 мМ Na2CO3
2,5 мМ NaHCO3/
/2,0 мM NaaCO3/
/3 мМ Na2CO3/
/2 м.M NaOH
3,0 мМ NaHCCy
/2,4 мМ HaOn
3,0 мМ tfaHCO3
/2, 5 мМ Na2CO3
3,0 мМ NaHCO3/
/2,4 мМ xVa2CO3
і
¦108
Продолжение табл. 8.1
Скорость
потока,
мл/мин
Определяемые
концентрации,
мкг/мл
Предел
обнаружения,
мкг/мл
s , %
Примечания
Литература
2,2—2,4
0,05—2,0
[10]
1,9
0,1—3 (F")
0,5-7200 (СГ)
0,5— 25 ^
0,5—370
0,5—1200
[И]
2,3
0,2—7 (С
0,5—10
2-20 (
[12]
2,7
0,07—1,8
0,03
для
стабилизации SO g""
добавляют трнэта-
ноламин
(pH 9-Ю)
[13]
1,65
на порядок ниже,
чем для
фотометрического метода
[14]
4,1—4,6
0—0,05 (Cl"")
0—0.04 (NO3-)
0-0,25 (SOj-)
0,001
0,002
3-5
для повышения
чувствительности
увеличивали объем вво-
Днмой пробы
1151
0,1
[16]
1,3 '0,37—18150
0,4—7,6
образец
предварительно
разбавляли
дистиллированной
водой
2.0
58,4—71,3
бромид
извлекают из
морской ВОДЫ
экстракцией в
присутствии
окислителя
Объект
анализа
Определяемые
анионы
Разделяющая
Подаи-
Элюеит
Поровая Ео-
да
геологических
образцов
F", С1~, ВГ
so2.-
Dionex Anion
CX500 мм)
З мМ КаНСО,/
/2 мМ Na2CO,
Сточная
вода уюльной
промышленности
СІ",
Dionex
HPIC-AS3,
AS5
мем- 2,55 мМ КаНССУ
бран- /2,05 мМ NaaCOs
4.5 мМ NaHCOs
3.6 мМ NajCOj
2% CH.CN
Сточные Ео-
ды. водные
вытяжки
почв
CI", NO3
Dionex Anion
CX250 мм)
ночное
9 м M Na HCC)»/
/З мМ iiajCO,
Промышленные
контурные воды
F~, СГ, NQf,
NO^", PO4-, SO:
soi-
Dionex Anion
CX500 mm)
то же
2 иМ Na2eO3/
/1 мМ NOH
Технологический
раствор ядерной
промышленности
СГ, Вг"
Dionex Anion
CX500 мм)
3,0 мМ NaHCOg/
/2,4 мМ SaaC(V
В в виде BF4
Dionex Anion
CX150 им и
3X500 мм)
3 мМ y
/4 мМ Na2CO3
Раствор для
электролитических
покрытий
F"", С|-
Dionex Anion
2,4 мМ NaHCOj/
/3,0 мМ KajCOg
-, hpo^-
Dionex Anion
CX150 mm)
3,0 мМ KaHCO3/
2,4 мМ NaaCO,
110
Продолжение табл. 8.1
Скорость
потока.
Определяемые
концентрации,
мкг/мл
Предел
обнаружения,
ик г/мл
S , К
Примечания
Литература
2,3
0,78—2,02 (F~)
1,7—119 (Cl")
,1 — 146 (SO|-)
0,005 (F~)
0,02 (СГ)
0,02
0,06
2—4
[19]
2,2
[201
,2—4,0
0,4—200
1,5—5
[21]
2,3
0,3—100 (F~)
0,5—150 (SO3)
4,7—385 (SO^
0,1 — 184 (СГ)
3-5 (Cl-)
1-5 ^
для
концентраций менее 1
мкг/мл
используют предпари-
тельное
концентрирование
[22]
2,3
Cl и Вг
выделяют на катио-
иообменнике в
Ag-форме
[23]
2,3
0,05—500
0,3—2,6
[24]
3,0
0—250 (F~)
0—350 (СГ)
0,8(F-)
0,5(С1~)
проб}'
разбавляют в 100 раз
125 ]
1,9
0,05— 0,
0,1—0,
0,2-3
1,4 (НРО|-)
4.1
[26]
111
Объект
анализа
Определяемые
анноны
Разделяющая
колонка
Подав
ление
Элюент
Промышленные воды
КО"
Br~,
C, SO,2",
Dionex Anion
DX250 мм)
тот же
Раствор лля
хромирования
er, soj-
Dlonex Anion
DX50 мм)
З мМ КаНССУ
/2,4 мМ Na2COs:
5,4 мМ Na2COs
-, SeO2-
Dionex Anion
Питьевая
Еода
NO3 в присутствии
F~, СГ
Br~, SO?
no;
2.
РОІ
Dionex Anion
FX250 мм)
ночное
4,5 мМ NaHCOs/
/2,4 мМ Na2COs
Br"
Dionex Anion
то же
3,0 мМ NaHCOj/
/2,4 мМ Na2COs
Оборотные
циклы
ядерных
установок
CI-, NO2-, NO",
Dlonex AG-1,
AG-2
З мМ NaHCO,/
/2—3 мМ Na2COs
Котловые,
контурные
воды
er, so
*-
Dionex Anion
2,8x1000 мм
5 мМ Na^CO3/
/4 мМ NajCO,
Контурные
воды
тепловой
электростанции
ci-, soj-
Dionex
HPIC-AS4,
CX250 мм)
бранное
2,8 мМ NaHCOj/
/2,0 мМ NajCO3
H2
Продолжение табл. 8.1
Скорость
лотока,
мл /ми H
2,7—3,1
J,9
2,67
2,75
2
Определяемые
концентрации,
икг/ыл
0,l-15(F-)
0,1—20 (Cl')
0,5—30 (SOJ)
0,2—50 (PU4~)
0,5—50 (Вг~)
0,2—30 (NO^-)
0,5—100 (SO^~)
1 — 100
На уровне
мкг/л
0,005—1000
0,3—20
0—0,2
Предел
обнаружения,
АІКГ/МЛ
0,02 (F~,C1~)
0,25 (Noj")
0,1 (PO^-)
(Br-, NOJ")
0,2 (SO^-)
25 (Cl")
250 (SO^-)
в
неразбавленной
пробе
¦—'
—
—
ниже
1 мкг/л
1,1—10,3(F~)
1,1 —16,2 (Cl—)
6,4-17,1 (NO7)
2,1—6,6 (PO^-)
1,3—3,9(Br-)
1,2—6,3 (XO^-)
0,6—4,6 (SO^~)
0,5—12
—
3( SO23-)
4 (PQ3-)
Примечания
пробу
разбавляют в 150 раз
используют
предварительное концентря-
рование
используют
предварительное
концентрирование
—
—
используют
предварительное
концентрирование
Лите-
>атура
[27]
[28]
[29]
[30]
131]
[32]
[33]
[34]
113
Объект
анализа
Определяемые
анноны
Разделяющая
Подавление
Элюент
Особо
чистая вода
NO,f,
Dlonex
HPIC-AS4,
AS5 CX250 мм)
NOf
мем-
бран-
ное
NaHCO3/Na2CO3
различной
концентрации,
1,2 мМ NaHCO3;
9,0 мМ NaaCO3/
/8,0 мМ NaOH
Dionex
HPICAS4
CX250 мм)
2,8 мМ NaHCO3/
/2,2 мМ Na2CO3
Cl", NO- PO3;
sol"
Dionex Anion
CX500 мм)
ночное
3,0 aM NaHOy
/2,4 мМ Na2CO3
cio:
Biotronik
CX100 mm)
о же
0,02 мМ
цитрат кальция
F~, Cl~, NO-f
set
Dionex Anion
CX500 мм)
5 мМ /1-амнноэензоат
натрия
CN~, CO|-, B4
то же
1,0 мМ KCOONa/
/2,5 мН NaOH
1,0 мМ NaCl/
2,5 мМ NaOH
2,5 мМ Na3PO4
чувствительности используют предварительное концентрирование
[50, 52] или специальные методические приемы типа «обратной
промывки» [59]. Этот прием состоит в изменении направления
подачи элюента на противоположное после попадания пробы в
разделяющую колонку. Время
обратной промывки 20 с при скорости по- |
тока 1,2 мл/мин. С помощью этого
приема можно увеличить объем
вводимой пробы до 6 мл и устранить
мешающее влияние водного пика. Это
особенно важно при определении
следовых количеств слабоудерживаемых
анионов (F-, Cl - , S2~, N0^ и др.):
Системный пин
SeOV
ч.
О 8 16 2"
мин
Рис. 8.1. Хроматограм-
ма водной вытяжки
почвы [55].
Колонка Vydac 302
1С, 4,6x250 мм. Элю-
ент 4 мМ фталевая
кислота (pH 4,6).
Скорость потока 2 мл/мии.
Объем пробы 2 мл.
Детектирование кондук-
тометрическое
SCN"
О 2
Рис. 8.2. Хроматограмма
неорганических анионов
[57].
Колонка Shodex 1С
1-524А, 4,6x100 мм. Элю-
?нт 2.5 мМ фталевая
кислота (pH 4,2). Скорость
потока 1,5 мл/мии.
Детектирование кондуктометри-
ческое
Для определения анионов, значительно различающихся по
удерживанию, использовали колонку Shodex 1С 1-524 А (рис. 8.2).
На этой колонке в течение 15 мнн можно разделить восемь
анионов, в том числе такие сильноудерживаемые, как 5гО|~ , 1~ и
SCN- [57].
117
Определение неорганических анионов в водах и некоторых других объектах
детеяти
Объект
анализа
Озерная,
речная и
водопроводная вода
Озерная
вода
Дождевая,
озерная,
грунтовая
вода
Грч'нтовая
вода
Дожлерая
вода
Водопроводная вода
Питьевая и
минеральная
вода
Конденсат и
котловая
¦•да
Определяемые
анионы
sio^-
CI-, ЙОГ
ci-, v6i,
soi-
NO3-, SO*~
Cr.NO^.SOf-
SiC.2-
'CO3-
CI-, NOf,
so42-
cr. so^~
Разделяюшая
колонка
TSK GEL IC-
Anion—PW
D,6X50 mm)
TSK GEL IC-
Anion — PW
DX50 mm)
Vydac 302 1С
Dionex Anlon
EX50 mm)
Anion/HS
D,6Xi00 mm)
TSK GEL 1С—
Anion — РГ
D,6>50 mm)
Wescan Anion
Vydac 302 1С
Анионообменник
на основе ХАД-1,
0,013 мэкв/г
BX150 мм
Элюент
0,5—1 мМ КОН
2 мМ КОН
4,0 мМ фталевая
кислота+борат
натрия
0,5 мМ гидрофта-
лат калия
4 мМ гидрофталат
калия
0,5 мM КОН
4 мM гидрофталат
калия
1 МіМ
гидрофталат калия
0,2 мМ цитрат
калия
0,2 мМ фталат
калия
pH
1—'
,
5,0
5,5
3,8
4,5
5,0
6,0
6,2
Скорость
потока,
мл/мин
1.2
1,0
2,0
2,0
4,0
1,2
3,0
1,5
1,0
ОДНОКОЛОНОЧНОЙ
роваиием
ионной хроматографией с
Таблица 8.3
кондуктометрическим
Определяй мые
концентрации,
мкг/мд
Предел обна
ружения,
мкг/мл
Время
определения, ИНН
Примечания
Литература
0,3-11
0,0022
2,1
10-30
высокая
селективность к
органическим кислотам
7,75—8,69
0,65—0,3
0.17 (Q-)
3,51 (NO3~)
30
определению
фторида мешают
органические кисло
ты , а сульфата •—
— карбонат
4—30 (CI")
0,9— 3,6 (N0^)
0,3—23 (SO|~)
0,6 (Cl")
0,2 (N0^)
0,2
результаты
определения
сравнивают с
результатами других
методов
10—100
3—10
15
гуминовые
кислоты сорбировали на
слабоосновном ани-
оните Nucleosil
5 NHa
на уровне 0,5
0,1 (NO.")
0,6—11
0,05
5,5
15—35
результаты
сравнивают с
результатами
фотометрического и титри-
метрического
методов
используют также
рефрактометрический и УФ
детекторы
0,003—0,15(СГ)
0,003-^0,02 ^
0,003—0,005 . . 4,3
15
без
центрирования
используют пред-
вар ителъное
концентрирование
Объект
анализа
Определяемые
анионы
Разделяющая
колонка
Элюент
pH
Контурные
воды ТЭС и
АЭС
IC-Pak
Anion
Высокочистая
вода
F~, С
NO3-,
Waters IC-Pak
Anlon
D,6X50 mm)
3,5 мМ я-толуол-
сульфоновая
кислота 0,3 мМ 2-наф-
тил-амин-1-сулъ-
фоновавая кислота
6,0
Гальванические
растворы и
стекла
, SIO3
-
Wescan Anion
С\
15мМЫаОН/0,025
мМ бензоат натрия;
15 мМ NaOH І мМ
тартрат натрия
Вытяжки
овощей
CI
ХИКС-І
DX60 мы и
2X150 мм)
1,5мМ глю коновая
кислота/1,5 мМ
борная кислота,
І мМ фталевая
кислота
8,6
5,2
Водные
вытяжки почв
Cl , NOjf, N(
SOf~, SeO^
Vudac 302 1С
D,6X250 mm)
4 мМ фталевая
кислота+борат
натрии
4,6
Раствор
красителя
анионообмэнник
на основе ХАД-1
0,027 мэкв/г
BX500 мм)
I мМ янтарная
кислота
Водный
раствор
F~, СГ, NOJ",
NO", SO42".
ijO^-.r, SCN-
Shodex ICI—524A;
размер частиц 10
мкм, емкость 0,05
ммоль/г
2,5 мМ фталевая
Н,РО
з—
Vydac 302 1С
D,5X250 мм)
4.2
кислота
20 мМ янтарная
кислота
3,0
F-.Cr, Br-,
SO
TSK GEL 1С—
Anlon — PW
D,5X50 мм)
І мМ фталат иат-
рия
5,1
Продолжение табл. 8.3
Определяемые
концентрации,
мкг/мл
Предел
обнаружения,
мкг/мл
Время
определения, мин
Примечания
Литература
10—15
[51
на уровне
м кг/л
0,03—0,1
мкг/л
15 без
трирования
используют
предварительное
концентрирование
[52
10—100 мкг/л
40
[53]
0,2-10 (С1~)
2—30 (NO~)
0,2 (СГ)
2 (NO3-)
3—5
10
результаты опре
деіения
сравнивают с
колориметрическим и потенци-
о.иетрическим
методами
[54]
0,05—7
1,46
25
[55]
6,91 (НРО5-
0,026 (Cl")
12
[56]
На уровне
м кг/г
15
[57]
5-50
0,2
20
[58j
0.05—5
0,0045-0,015
2,1—5,1
40
для увеличения
объема вводимой
пробы до 6 мл и
устранения
влияния водного пика
используют метод
обратной промывки
[59]
121
Объект
анализа
Определяемые
анноны
Разделяющая
колонка
Элюент
pH
У я *
m
Водный
раствор
CN~, S2~,
|~, SiO g
TSKGELIC—
Anion— P\V
D,6X50 мм)
1 мМ КОН
1,0
Определение неорганических аьиснев в водах и некоторых других объектах
Объект
Опре .еляемые
Разделяющая
ко.торгка
Элюеит
pH
Грунтовая и
дождевая
вода
Речная вола
и воздух
Vydac 302 1С
D,6X250 мм)
, no
Vydac 302 1С
D,6X250 мм )
1,0 m M фталат
натрия
4,0
0,5 мМ иэофталат 5,2
Питьевая и
минеральная
вода
Cl~, NO3', SO5- Vydac 302 1С
D,6x250 мм)
1,0 мМ гидрофта-
лат калия
5,0
Водопроводная и
минеральная вода
F~, СГ
Hamilton
PRP-X 100
D,6X250 мм)
0,46 мМ 2 ,4-диоксц-
бензойная кислота
10,1
со-
Высокочнс-
тая вода
Zipax SAX
Dx250 мм)
0,2 мМ фталевая
кислота
6,8
:r,NO;r, no-
Waters IC-Pak
Anion
D,6X50 мм)
0,3 мМ 2-нафтала-
мнн-1-сульфоновая
кислота
6,0
12
Продолжение табл. 8.3
Определяемые
концентрации,
мкг/мл
0,5— 10(CN-,
S2-,AsO33-)
0,1—2,5(S1O5~)
Предел
обнаружения,
мкг/мл
0 1—0,2
0,022
sr.%
—'
Время
определения, мнн
15
Примечания
—
Литература
[60]
Таблица 8.4
одноколоночной ионной хроматографией с косвенным УФ детектированием
п I а
Определяемые
< онцентрацин,
мкг/мл
Предел
обнаружения,
мкг/мл
Примечания
Литература
2,0
280
0,1—50
0,2 (СГ)
0,07 (NO3~)
0,03 (SO^-
определяли также с
КОНД у КТОМеТрнЧеС КИіМ
детектором
[44]
2,0
254
1—20
[61
1,5
238—
283
На уровне
л кг/мл
сравнивали с кондук-
тометрическим и
рефрактометрическим
детекторами
49
3.0
312
0,1—60
[62]
2,0
231
0,02—4,0
0,01
[63
1,0
300
0,02
объем вводимой
пробы 500 мкл
[52]
123
Объект
анализа
Высокочистая вода
Технический
раствор
Сок овощей
Водный
раствор
Определяемые
анионы
СГ, Br-, I-.
NO^.NO^.SO^-
н2ро4-
С1-, NCXf, COf-
С1, NO3-, SO^
Cl-, Br-, NO^,
C1O.J, NO3-,
so*-, роз-,
IO3-
F~. СГ, I",
NO^, PO^-, SO^-
F", Cr.NO^,
Br-, NOJ-, SQ2-
Разделяющая
колонка
Vvdac 302 1С
D,6X250 или
3,0X250 мм)
Wescan Anton
Zipax SAX
B,1x500 мм)
Vvdac 302 1С
D,6X250 мм)
Zorisax NH2
D,6X250 mm)
TSK GEL 1С—Anl-
on— PW
D,6X50 mm)
Элюент
2,5 или 5,0 мМ
гидрофталат калия
1 мМ бензойная
кислота
),5 мМ фталат
натрия
5 мМ гидрофталат
калия
0,02 M фталевая
кислота
1 мМ фталат натрия
pH
4,0
5,5
—
4,6
2,9
5,1
124
Продолжение табл. 8.4
ИМ
Определяемые
концентрации,
м к г Діл
Предел
обнаружения,
мкг/мл
Примечания
Литература
2,0
285
0,2 мкг/л (CI")
¦— 1,6 мкг/л (Бг~)
— 3,9 мкг/л A~)
— 0,4 мкг/л (NO-)
— 0,7 мкг/л (NOg")
— 1,4 мкг/л (Н2РО~)
объем вводимой про
бы 0,1—1,0 мл
[64]
1,0
254
0-5
[65]
1,0
240
50—1600
объем вводимой
пробы 20 мкл
[661
1,5
308
0,1— 50(С1з~
0,3—100
(SOJ-)
0,1-0,5
сравнивают косвенно;
УФ и кондуктомет-
рическоз
детектирование
[67]
2,0
302
0,5 (F")
5,7 (Cl")
10,0A-)
10,0
6,3
6,1
разделение проводят
на слабоосновном
анионообмгннике
[68]
270
0,4-1,0 мкг/л
объем вводимой
пробы 6 мл.
используют обратную
промывку
[591
125
8,3.2. Косвенное УФ детектирование
Условия разделения анионов с косвенным
спектрометрическим детектированием в УФ области спектра идентичны
условиям разделения с кондуктометрическим детектором. Разделение
чаще всего проводят на колонке Vydac 302 1С размером 4,6х
Х250 мм фталатным элюентом с концентрацией 0,5—5,0 мМ. Для
обеспечения нужной элюирующей силы и устранения
мешающего влияния системного пика устанавливают pH от 4 до 6.
Скорость подачи элюента, как правило, составляет 1,0—2,0 мл/мин.
Длина волны детектора 230—310 нм в зависимости от типа
детектора и условий разделения. Определение в этих условиях
характеризуется высокой чувствительностью, селективностью и эк-
спрессностью (рис. 8.3).
сг
погл
ЮООмкг/ш сг
N0,
Вр"
12
Рис. 8.3. Хроматограмма
неорганических анионов [61].
Колонка Vydac 302 1С, 4,6x250 им.
Элюент 1 мМ фталевая кислота (pH
4,0). Скорость , потока 2 мл/мин.
Концентрация анионов 4,50—6,75 мкг/ил.
Детектирование косвенное УФ B73 нм)
Рис. 8.4. Определение бромида в
хлориде натрия с амперометрнческим
детектором [80]
Примеры практического использования одноколоночной
ионной хроматографии с косвенным УФ детектированием для
определения неорганических анионов представлены в табл. 8.4.
Косвенное УФ детектирование имеет ряд преимуществ перед
кондуктометрическим. При сравнении чувствительности этих
вариантов детектирования [44, 59, 67] было показано, что
косвенное УФ детектирование приблизительно на порядок
чувствительнее кондуктометрического (табл. 8.5). К преимуществам
косвенного УФ детектирования можно также отнести возможность
использовать аппаратуру для ВЭЖХ, а также детектирование всех
126
Таблица 85
Пределы обнаружения некоторых
анионов с кондуктометрическим
и косвенным УФ детекторами
[67] Колонка — Vydac 302 1С
D,6x250 мм); элюент — 5 мМ
гидрофталат калия (pH 4,8),
Я = 308 нм, объем вводимой
100 мкл
анионов по отрицательным пикам. Однако линейный диапазон
сигнала кондуктометр ического детектора шире [44] (хотя авторы
работы [67] утверждают обратное).
УФ детекторы для определения
неорганических анионов должны
иметь широкий диапазон
компенсации фонового сигнала. Это
необходимо для работы с сильно
поглощающими элюентами в области
высокой чувствительности
детектора. Фоновый сигнал можно
скомпенсировать, изменяя длину волны
детектора. В этом случае выбирают
длину волны, где поглощение элю-
ента ниже [67].
Косвенное УФ детектирование
позволяет определять практически
все непоглощающие в
ультрафиолетовой области спектра
неорганические анионы. Голомбек и Шведт
[62] определили времена
удерживания 18 неорганических анионов на
колонке Hamilton PRP-X 100 с 2,4-
диоксибензойной кислотой в
качестве элюента (табл. 8.6). Полученные
результаты свидетельствуют о
широких перспективах использования
косвенного УФ детектирования для
определения неорганических
анионов.
Ли пои
Хлорид
Бромид
Нитрит
Хлорат
Нитрат
Сульфат
Йодид
Фосфат
Бромат
Ацетат
Формиат
Йодат
Предел оЗнар
м кг/мл
;онду]-то-
четричес-
кий
1
I
1
2
3
3
3
4
5
9
10
12
ужения
косвенный
УФ
0,1
0,2
0,2
0,2
0,3
0,3
0.5
0,3
0,2
0,4
0,5
0,4
* Рассчитывали из
соотношения сигнал/шум, равного 2, при
максимальной чувствительности
детекторов.
Таблица 8.6
Время удерживания неорганических анионов [62]. Колонка — Hamilton
PRP — X 100 D,6x250 мм); элюент — 0,46 мМ 2,4-диоксибензойная кислота
(pH 10,1); Скорость потока 3 мл/мин; Косвенное УФ детектирование C12 нм)
Анион
Силикат
Борат
Фторид
Йодат
Хлорид
Цианид
Нитрит
Бромат
Бромид
Азид
Время удерживания,
мин
1,55
2,05
2,15
2,35
3,25
3.8
4,1
4,5
5.2
5,7
Анион
Системный пик
Гидрокарбонат
Нитрат
Хлорат
Фосфат
Селенит
Арсенат
Сульфат
Селенат
Молибдат
Время удерживания,
мин
6,5
6,5
6,75
10,8
15,9
16,9
17,8
18,8
20,8
25,5
127
Ионохроматографическое определение неорганических
Объект
анализа
Дождевая и
грунтовая
вода
Грунтовая,
речная,
морская
вода
Речная вода,
почва
Дождевая
вода
Технологический раст-
ЕОр
Объекты
металлургического
Водный
эаствор
Определяемые
анионы
Вг~
1~
I", CN", SO^~
CN~
As (III)
F^_C!-,NO3-
Детектор
амперомет-
рический
потенциометр ический
амперомет-
рический
то же
-
потенцио-
метрический
кулономет-
рический
потенцио-
метрический
Разделяющая
колонка
Dionex Anion
DX250 мм)
слабоосновный
анионообменник
D,6X100 мм)
Waters 1С—Pak
Anion
TSK GEL QAE—
- 2 SW
DX150 mm)
Dionex Anion
Dionex H PIC—ASA
Dionex Anion
CX500 мм)
Анионообменник
на основе ХАД-1
3X250 мм)
Элюент
3 мМ NaHCO3/
/2 мМ Na2CO3
0,1 M NaNO3/
/0,01 MNaH2PO4/
/№2HPO4
2 мМ гидрофталат
калия; 5 мМ КОН;
5 мМ фосфатный
буфер
0,1 M фосфатный
буфер/0,05 M NaCl
1 мМ Na2CO3/
/14,7 мМ
этнлендиамин/
/10 м M NaH2BO3
2,8 мМ NaHCO3/
/2,25 мМ Na2CO3
3 м NaHCO3/
/2,4 мМ Na,CO3
"идрофталат
калия
128
Таблица 8.7
анионов с электрохимическими детекторами
pH
—
6,7
7,0
11,0
1
¦ .
4,1—
7,1
Л
H
Pi
2,3
1,2
1,2
0,5
2,0
2,0
2,0—
3,0
Определяемы^
¦С'їнцентрацин.
мкг/мл
0,05—0,5
до I-IO
0,01—0,2
0,03—1,0
0,01—4,0
—-•
Предал
обнаружения,
мкг/мл
0,01
0,002
без
предварительного
концентрирования
0,0007 (Г)
0,002 (CN~)
0,02 (SO*-)
—'
0,005
менее 0,1
на уровне
м кг/л
С* о'
г ' °
1,6—1,4
¦—¦
менее 2
—
1 — 13
—
Примечания
цг- определяют в
присутствии 4000-
кратныл количеств
er
использовали
предварительное
концентрирование
¦—¦
после разделения
NO3
восстанавливали до NO^" на
медио-кадмиевоіі
колонке
для разрушения
устойчивых цпа-
нидных
комплексов пробу
облучают УФ светом
¦—'
¦
Литература
[69]
[70]
[71]
[72]
[73]
[74]
[75]
[76]
3»к. 34
129
Определяемые
Детектор
Разделяющая
колонка
Элюент
то >ье
soi-
, NO",
Dionex HPIC-AS3
CX250 мм)
3 MMNaHCO3/
/2,4 мМ№2СО3
СІ~, В г",
SCN-
Vydac 302 1С
D,6X250 мм)
CI", Bf", Г
SCN-, CN~,
N0-, NO3~,
Br~
=2-
Br"
-, s2
амперомет-
рический
то же
Wescan Anlon
Dionex Anion
Dionex HPIC—ASl
Dionex HPIC-AS4
Dionex
HPIC—AS3
Dionex Anion
CX250 мм)
5 мМ салициловая
кислота
2 мМ СНзСООН/
/CH,COONa
5 мМ NaOH
14,7 м M этилен-
диамин/
/10 MMNaH2BO3/
/1 мМ Na2CO3
20 мМ NaNO3
2 мМ NaaCOs
5 мМ NaOH
Продолжен и е т а б л. 8.7
4.12
6,4
~-
11,0
Скорость
-потока,
мл/мип
—
2.0-
—2,5
2.0
I3.9
2,5
2,5
2.5
0,9
Определяемые
концентрации,
мкг/мл
—
—
0,05—1,0
0,1-10
1—'
Предел
обнаружения,
мкг/мл
ниже 5
менее 0,1
менее 0,05
0,002 (CN~)
0,03 (S2")
0,01
0,01
0,25 (S2-)
1,5 (CN-)
s , %
—'
¦—¦
—•
—
—
менее 5
Примечания
детектор
регистрирует изменения
pH элюента
объем вводимой
пробы 100 мкл
—
1—
—'
Вг~ определяют в
чистом хлориде
натрия
Литература
[77]
[78]
[79]
[79
[80]
[80]
[80]
[81]
131
8.4
ЭЛЕКТРОХИМИЧЕСКОЕ ДЕТЕКТИРОВАНИЕ
Электрохимические детекторы пока не нашли широкого
практического применения для определения неорганических анионов
(табл. 8.7). В большинстве публикаций предлагаются детекторы
различных типов и конструкций и изучаются аналитические
возможности на водных растворах солей. Чаще всего
электрохимические детекторы используют для определения галогенидов, а
также цианида и сульфида. Определение галогенидов с
электрохимическим детектором характеризуется более высокой
селективностью по сравнению с кондуктометрическим
детектированием [69, 80]. Селективность настолько высока, что можно
определять бромид в высокочистотном хлориде натрия (рис. 8.4).
Выбор электрохимических детекторов для определения галогенидов
широк, поскольку многие из них селективны к этим анионам.
Определение цианида и сульфида с кондуктометрическим
детектором сопряжено с рядом трудностей. Эти анионы
удерживаются на анионите слабо, поэтому многие неорганические (F~,
Cl-, NOj- ) и органические (формиат, ацетат, пропионат) ионы
мешают их определению. Кроме того, чувствительность
кондуктометр ического детектирования цианида и сульфида невысока
@,1—0:2 мкг/мл) [60]. Использование потенциометр ического [76]
или амперометрического [73, 80] детекторов позволяет снизить в
50—100 раз пределы обнаружения этих анионов и повысить
селективность определения.
Потенциометрический детектор предложено использовать как
альтернативу кондуктометрическому для измерения pH элюата
после подавляющей системы. Однако чувствительность такого
детектирования существенно ниже, чем кондуктометр ического.,
Перспективно, видимо, использовать электрохимический
детектор в комбинации с неселективным, например с
кондуктометрическим [80]. Это позволяет повысить чувствительность и
селективность определения некоторых анионов, а также сократить
время анализа. В ряде случаев могут быть полезными и другие
комбинации детекторов [82]. ,
8.5
СРАВНЕНИЕ ИОННОЙ ХРОМАТОГРАФИИ
С ДРУГИМИ МЕТОДАМИ ОПРЕДЕЛЕНИЯ АНИОНОВ
Определение анионов, особенно неорганических, всегда было
более трудоемким, чем определение катионов. Для определения
неорганических анионов в водах, как правило, применяют
колориметрический, потенциометрический и титриметрический методы
(табл. 8.8). Однако эти методы могут быть использованы лишь
для определения одного аниона; другие ионы могут в ряде слу-
132
чаев оказывать мешающее влияние, поэтому необходимо их
предварительное отделение. Кроме того, эти методы требуют
значительного объема анализируемой пробы и большого числа
реагентов.
Таблица 8.8
Обычные методы определения неорганических анионов в водах
Анионы
Метод
Фторид
Хлорид
Нитрит
Фосфат
Нитрат
Сульфат
Потенциометрический с нон-селективным электродом
Колориметрический с Hg (SCN)o
Титриметрический с KMnO, или колориметрически:! с
образованием азосоединения
Колориметрический с аскорбиновой кислотой
Колориметрическим с Си—Cd-восстановлением и
последующим определением в виде азосоединения
Колориметрический С Ва + МЄТИЛТИМ0ЛОВЬПІ СПНИ:'[ ИЛИ Ііе-
фелометрическии в виде BaSOt
В отличие от традиционных методов ионная хроматография
не требует длительной пробоподготовки и большого числа
реагентов. Определение можно проводить из малого объема пробы,
при этом чувствительность выше, чем у обычных методов.
Поскольку ионная хроматография является гибридным методом и
определению анионов предшествует их разделение, селективность
этого метода существенно выше.
Результаты ионохроматографического определения
неорганических анионов в водах сравнивали с результатами, полученными
другими методами. Это позволяет оценить правильность ионохро-
матографических определений. Кроузер и Мак Брид [8]
использовали двухколопочную ионную хроматографию для
определения фторида, хлорида, нитрата и сульфата в атмосферных
осадках. Результаты определения сульфата сравнивали с данными,
полученными колориметрическим методом с метилтимоловым
синим: содержания весьма близки. Чувствительность
ионохроматографического определения приблизительно в 30 раз выше.
Хорошо совпадают результаты при определении хлорида и нитрата
ионохроматографическим и колориметрическим методами. В то
же время для фторида получены завышенные значения по
сравнению с потенциометрическим и колориметрическим методами.
Поэтому ионную хроматографию не рекомендовали для
определения фторида в атмосферных осадках.
Моско [27] сравнивала результаты определения
неорганических анионов в различных водах ионной хроматографией и
другими методами (табл. 8.9). Все результаты, кроме данных для
сульфата, хорошо согласуются.
Онкава и Саито [83] провели тестовое определение нитрата и
сульфата в атмосферных осадках и дождевой воде. Для концент-
133
Таблица 8.9
Сравнение результатов определения неорганических авионов в водах (мкг/мл), полученных двухколоночиой ионной
хроматографией н традиционными методами [27]
Вода
Питательная котлоиая
Котлоьам
Охлаждающая
Городская водопроводная
Артезианская
Фторид
ИХ
0,1
1,2
0,4
1.0
0,6
А
—
—
—
1.0
0,6
Хлорид
ИХ
16
140
23
16
2,2
в
16
125
28
16
3,0
Фосфат
ИХ
0.2
14
4,2
0,2
0,2
С
0,05
14,4
4,2
0,05
0,05
Нитрат
ИХ
2,0
1,0
6,0
9,0
0.2
D
1.9
1,0
—
8,3
0,2
Сульфат
ИХ
42
70
46
23
10
Е
40
60
45
25
5
Методы: А — потенциометрический с ионоселективным электродом; В — колориметрический с Hg(SCN)a; С —
колориметрически;'] с аскорбиновой кислотой; D—колориметрический с Си—Cd-восстановлением н образованием азосоединенин;
Е колориметрический с метилтимоловым синим.
раций 10—100 мкг/мл получено хорошее совпадение как
межлабораторных данных, так и результатов, полученных ионохромато-
графическим, колориметрическим и нефелометрическим методами.
В области более низких концентраций совпадение результатов
хуже. Близость результатов определения неорганических анионов
двухколоночной ионной хроматографией и гравиметрическим, тит-
риметрическим, спектрофотометрическим, потенциометрическим
и нефелометрическим методами получена в работах [1, 4, 5, 22,
33, 84}.
Для оценки правильности результатов, полученных одноко-
лоночной ионной хроматографией, их также сравнивали с
данными других методов. Херн и др. [44] сопоставляли результаты
определения нитрата и сульфата в водах с данными турбидиметри-
ческого и УФ спектрофотометрического методов (табл. 8.10).
Результаты определения, полученные различными методами,
хорошо согласуются. і
Таблица 8.10
Сравнение результатов опрэделенил сульфата и нитрата (мкг/мл) в водах,
полученных одноколоночной ионной хроматографией и другими методами
J44.] В скобках указан 95%-ный доверительный интервал
Вода
Грунтовая
Поверхностная
Дождевая
Образец
G1
G2
G3
SI
S2
R2
Сульфат, как S
ИХ
22,30@,48)
9,35@,26)
12,59@,34)
7,63@,26)
3,91@,30)
1,60@,30)
Турбидимет-
рический
23,02@,85)
9,16@,42)
11,91@,60)
7,42@,58)
3,91@,27)
1,75@,36)
Нитрат, как N
ИХ
2,89@,17)
1,59@,18)
2,58@,17)
3,57@,16)
3,21@,18)
0,86@,20)
УФ
2,91@,09)
1,57@,09)
2,74@,30)
4,04@,08)
3,49@,08)
0,91@,09)
Правильность результатов ионохроматографического
определения силиката была проверена спектрофотометрическим
методом [42], хлорида и нитрата — потенциометрическим и
спектрофотометрическим [54], селената — атомно-эмиссионной
спектроскопией с ИСП [55]. Окада и Кувамото [43] получили хорошее
совпадение результатов определения хлорида и нитрата с
данными спектрофотометрического метода. Но результаты
ионохроматографического определения фторида оказались сильно
завышенными, что связано с мешающим влиянием органических кислот.
Хорошее совпадение результатов ионохроматографического
определения неорганических анионов с данными, полученными
другими методами, свидетельствует о правильности ионной
хроматографии.
ЛИТЕРАТУРА
1. Т a k a m і К- et al. // Bunseki Kagaku. 1982. V. ЗІ, № 7. P. 362—367.
2. Z о 1 о t о v Y u. A. et al. // Int. Env. Anal. Chem. 1987. V. 31, № 2—4.
P. 99—106.
135
3. Hoover Т. В., Yager G. D. // Anal. Chem. 1984. V. 56, № 2.
P. 221—225.
4. R і с h W. E. // Instrum. Techn. 1977. V. 24, № 8. P. 47—51.
5 N a к a о к a H et al. // Bunseki Kagaku. 1981. V. 30 № 10. P. T97—T101.
6 L і p s к і A. J , V a і г о С. J. // Canad. Res. 1980. № 2. P. 45—48.
7. L a s h R. P., H і 11 С. J. // Anal. Chim. Acta. 1979. V. 108. P. 405—409.
8 Grow t her J Me Bride J. // Analyst. 1981. V. 106. P. 702—209.
9. Muller К. Р. // Fresenius Z. Anal. Chem. 1984. B. 317, № 3—4.
S 345—346.
10. T y r e e S. Y., S t о u f f e r J. M., В о І І і n g e r M. // Ion
Chromatographie Analysis of Enveironmental Pollutants / Ed. by J. D. Milik and E. Sa-
wicki. — Ann Arbor: Ann Arbor Sei. 1979. P. 295—304.
11. Schwabe R. et al. // J. Env. Anal. Chem. 1983. V. 14, № 3.
P 169—179.
12 Wagner F. Valenta P., Nurnberg H. W. // Fresenius Z. Anal.
Chem. 1985. B. 320, № 5. S. 470—476.
13. T a n a к a S et al. // Bunseki Kagaku. 1986. V. 35, № 5. P. 465—470.
14 Mat s um о to M., I tan о Т. // J. Jap. Air Pollut. 1984. V. 19, № 3.
P. 247—254.
15. L e g r a n M., De A n g e 1 і s M., D e I m a s R. J. // Anal. Chim. Acta.
1984. V. 156. P. 181 — 192.
16. Galvin P. J, С line J. A. // Atoms. Environ. 1978. V. 12, № 11.
P. 1163—1167.
17 It oh H., S h і n b о r і Y. // Bunseki Kagaku. 1980. V. 29, № 4.
P. 239—242.
18. К a to h K. // Bunseki Kagaku. 1983. V. 32, № 7. P. 567—570.
19. P y e n G. S., F і s h m a n M. J. // Ion Chromatographie Analysis of
Environmental Pollutants / Ed. by J. D. Mulik and E. Sawicki. — Ann Arbor:
Ann Arbor Sei. 1979. P 235—244.
20. Potts M. E. P о t a s T. A. // J Chromatogr. Sei. 1985. V. 23, № 9.
P. 411—414.
21 Шпигун О. А. и др. // Ж. аналит. химии. 1985. Т 40 № 11 С 1925—
19-29.
22. R a w a J. А. // Ion Chromatographie Analysis of Environmental
Pollutants / Ed. by J. D. Mulik and E. Sawicki. — Ann Arbor: Ann Arbor Sei 1979
P. 245—270.
23. S ie m e r D. D. // Anal. Chem. 1980. V. 52, № 12 P 1874—I877
24. Hill C. J.. Lash R. P. // Anal. Chem. 1980. V 52 № 1 P 24—27
25. Ko r t h \V„ Ellis J. // Talanta. 1984. V. 31, № 6. P. 467—468
26. Та пака T. et a!. // Bunseki Kagaku. 1M3. V. 32 N> 12 P 771—773
27. M о s к о J. // Anal. Chem. 1984. V. 56. № 4. P. 629—633.
28. Smith R. E., Davis W. R. // Plat, and Surface Finish 1984. V. 71,
№ 11. P. 60—63.
29. Зо лотов Ю. А. и др. Докл. АЛ СССР. 1982. Т. 263, № 4. С. 889—892.
30. Darimont T., Schulze G., Sonne born M. // Fresenius Z. Anal
Chem. 1983. В. 314, № 4. S. 383—385.
31. Morrow С M, Min ear R. A. // Water Res. 1984 V 18 № 9
P. 1165—1168.
32. Brandt F., Trost R. // VGB-Konf. Chem. Kraftwerk 1983. № 1.
S. 24—27.
33. S t e v e n s T. S., T u r k e 1 s о n V T., A I b e W. R // Anal Chem. 1977
V. 49, №8. P. 1176—1178.
34. В a I с о n і L., P a s с a 1 і R., S і g о n F. // Anal. Chim Acta 1986 V. 179
P. 419—425.
35. T r e t t e r H et al. // Fresenius Z. Anal. Chem. 1985 В 321 № 7.
S. 650—652.
36. Weiss J., Gobi M. // Fresenius Z Anal. Chem 1985 В 320 № 5
S. 439—444.
37. Wetzel R. A. et a/. // Ana,'. Chem. 1979. V. 51, № 9. P. 1532—1535.
38. Шпигуй О. А., Обрезков О. H., Золотое Ю А. ,// Ж. аналит
химии. 1986. Т. 41, № 4. С. 692—695.
136
39. S h p i g u n О. A., V о 1 о s h с h і к І. N.. Z о 1 о t о v Y u. A. // Anal.
Sei. 1985. V. 1, № 2. P. 335—339.
40. P і n s с h m і d t R. K. // Ion Chromatographie Analysis of
Environmental Pollutants / Ed. by J. D. Mulik and E. Sawicki. — Ann Arbor: Ann Arbor Sei.
1979. P. 41—50.
41. Williams R. J. // Anal. Chem. 1983. V. 55, № 6. P. 851—854.
42. Oka da T., Kuwamoto T. // Anal. Chem. 1985. V. 57, № 1
P. 258—262.
43. О к a d a T., Kuwamoto T. // Bunseki Kagaku. 1983. V. 32 № 7.
P 595—599.
44. Hern J. A., Rutherford G. K., Van Loon G. W. // Talanta.
1983. V. 30, № 9. P. 677—682.
45. Mark o-V a r g a G., С s і к у I., J o n s s о n J. A. // Anal. Chem. 1984.
V. 56, № 12. P. 2066—2069.
46. In focus. The Tecator Journal of technology for chemical analysis 1986.
V. 9, № 12. P. 12—13.
47. О к a d а Т., К u w a m о t о T. // Anal. Lett. 1984. V. 17 (A15). P. 1743—
1751.
48. Yan D. R., Rossner В., Sch we dt G. // Anal. Chim. Acta. 1984.
V. 162. P. 451—455.
49. Lu с к as В. // Fresenius Z. Anal Chem. 1985. B. 320, № 5. S. 519—524.
50. Roberts К. M., G j er de D. T., Fritz J. S. // Anal. Chem. 1981.
V. 53, № 11. P. 1691—1695.
51. Pec e vick P. M., Jones W. R. // Power. Eng. 1986. V. 90, № 1.
n Qg Q7
52. J a с к s о n P. E., H a d d a d P. R. // J. Chromatogr. 1986. V. 355, № 1.
P. 87—97.
53. M с С г о r y-Y о у С // Anal. Chim. Acta. 1986. V. 181. P. 277—282.
54. Pentchuk J., Haldna Y u., Ilmoia K. // J. Chromatogr. 1986.
V. 364. P. 189—192.
55. KarlsonU., Frankenberger W. T Ці. Chromatogr. 1986.
V. 368. P. 153—161.
56 F r і t z J. S., D u v a 1 D L., В a r r о n R E. // Anal. Chem. 1984. V. 56,
№7. P. 1177—1182.
57. T о к u d a T. et al. // Abstracts Pittsburgh Conference on Analytical
Chemistry and Applied Spectroscopy. — Atlantic City. 1986. № 630.
58. Ryder D. S. // J. Chromatogr. 1986. V. 354. P. 438—441.
59. О к a d a T., K u w a m о t о T. // J. Chromatogr. 1985. V. 350. P. 317—323.
60 OkadaT., KuwamotoT // Anal Chem. 1985. V. 57, № 4.
P. 829—833.
61. N a і s h P. J. // Analyst. 1984. V. 109. P. 809—812.
62 GolombekP. SchwedtG //J. Chromatogr. 1986 V. 367, № 1.
P. 69—76.
63. Brandt G, Matuschek G. К e 11 r u p A. // Fresenius Z. Anal.
Chem. 1985. B. 321, № 6. S. 653—654.
64. H e с к e n b e r g A. L. H a d d a d P. R. // J. Chromatogr. 1984. V. 299,
№ 1. P. 301—305.
65. Andrew B. E. // High. Resolut. Chromatogr. and Chromatogr.
Commun. 1984. V. 7, № 10. P. 580—581.
66. H a u a к a w a K. et al. // Bunseki Kagaku. 1984. V 33, № 7. P. 390—392.
67. Cochrane R A. H і 11 man D E. // J. Chromatogr. 1982. V. 241.
P. 392—394.
68. Cortes H. J., Stevens S // J. Chromatogr. 1984. V. 295.
P. 269—275.
69. P yen G. S., Erdman n D. E. // Anal. Chim. Acta. 1984. V. 164.
p 355 358
70. В u't 1 e r E. C. U., G e r s h e y R M. // Anal. Chim. Acta. 1984. V. 164.
P. 153—161.
71. Jandik P., Сох D., W о n g D. // Amer. Lab. 1986. V. 18, № 3.
P. 114—125.
72. I s h і g am і A. et al. // Bunseki Kagaku. 1986, № 10. P. 944—946.
6 Зак. 34 137
73. Р о h 1 а п d t С. // S. Afr. J. Chem. 1984. V. 37, № 3. P. 133—137.
74. T a n L. K., D u t r і z a с J. E. // Anal. Chem. 1986. V. 58, № 7. P 1383—
1389.
75. Girard J. E. // Anal. Chem. 1979. V. 51, № 7. P. 836—839.
76. Wang W., Chen Y., W u M. // Analyst. 1984. V. 109. P. 281—289.
77. T a r t e r J. G. // Lig. Chromatogr. 1984. V. 7. № 8. P. 1559—1566.
78. HerschcovitzH., YarnitzkyCh., Schmuckler G. //
J. Chromatogr. 1982. V. 252. P. 113—119.
79. Suzuki K. et al. // Bunseki Kagaku. 1983. V. 32, № 8. P. 585—590.
80. Rocklin R. D., Johnson E. L. // Anal. Chem. 1983. V. 55, № 1.
P. 4—7.
81. Bond A. M. et al. // Anal. Chem. 1982. V. 54, № 3. P. 582—585.
82. Jenke D. R., Mitchell P. K., Pagenkopf G. K. // Anal. Chim.
Acta. 1983. V. 155. P. 279—285.
83. О і к a w a K., S a і t о h H. // Bunseki Kagaku. 1983. V. 32, № 6. P. T105—
T109.
84. T a b a t a b a і M. A., Dick W. A. // J. Environ Quai. 1983. V. 12,
№ 3. P. 209—213.
ГЛАВА 9
ОПРЕДЕЛЕНИЕ
ОРГАНИЧЕСКИХ КИСЛОТ
9.1
ОРГАНИЧЕСКИЕ КИСЛОТЫ В ВОДАХ
Многие компоненты вод имеют органическую природу.
Прежде всего это естественные компоненты — гуминовые и фульво-
кислоты. Ряд примесей антропогенного происхождения также
представляют собой органические соединения, диссоциирующие
в воде по кислотному типу. К ним относятся, например,
анионные поверхностно-активные вещества.
Ионная хроматография оказалась удобным методом
обнаружения и количественного определения таких кислот.
9.2
УДЕРЖИВАНИЕ ОРГАНИЧЕСКИХ АНИОНОВ
9.2.1. Одноосновные алифатические кислоты
Способность анионов одноосновных алифатических кислот
удерживаться на анионообменниках возрастает с увеличением
длины углеводородной цепи. В табл. 9.1 приведены времена удер-
Таблица 9.1
Время удерживания одноосновных алифатических кислот на
анионообменнике Dionex Anion Cx500 мм). Элюент — 0,1 мМ NaOH;
скорость элюента — 2,3 мл/мин
Кислота
Глнколевая
Молочная
Муравьиная
Уксусная
Пропноновая
Масляная
Изомасляная
Валериановая
рК
3,83
3,83
3,76
4,76
4,86
4.82
4,86
4 86
Рремя
удерживания, мин
V«
6,0
7,5
8,3
9,0
11,1
10,5
13,6
Кислота
Изовалериановая
Пивалиновая
Капроновая
Каприловая
Каприновая
Фтористоводороіная
Хлористоводородная
рК
4,78
4,88
4,89
3,21
—
Время
удерживания, мин
10.7
12,2
21,4
25,2
30,1
6,5
16,1
живания одноосновных алифатических кислот и некоторых
неорганических анионов [1]. Времена удерживания кислот не корре-
лируются с их константами ионизации. Это свидетельствует о
вкладе неионообменных процессов в механизм сорбции кислот
6* 139
анионообменниками. Вклад этот возрастает с увеличением
длины алкильного радикала. Возможно, необменные процессы
связаны с гидрофобным взаимодействием алкильного радикала и
матрицы анионообменника [2].
Существует закономерность и в удерживании изомеров. Изо-
кислоты (изомасляная, изовалериановая) удерживаются слабее,
чем соответствующие нормальные кислоты. Это, по-видимому,
связано с влиянием стерических факторов на удерживание.
Оксикарбоновые кислоты (гликолевая, молочная)
удерживаются слабее, чем соответствующие карбоновые кислоты
(уксусная, пропионовая). Вероятно, введение в молекулу кислоты окси-
группы уменьшает ее гидрофобность и способность
удерживаться на анионообменнике.
Вайс [3] исследовал закономерности ионохроматографического
удерживания анионных поверхностно-активных веществ. -'Были
изучены алкилсульфонаты R—СН2—SQp, Na+, алкилсульфа-
ты R—СН2О—SO- Na+. оксиалкилсульфонаты R
SO3'"Na+ и олефинсульфонаты R—СН2—СН = СН—
СН2- SO3-Na+.
Закономерности удерживания анионных ПЛВ и карбоноБЫХ
кислот во многом сходны. Так. времена удерживания алкилсуль-
фонатов увеличиваются с ростом длины алкильного радикала.
При его одинаковой длине удерживание уменьшается в ряду ал-
килсульфат > алкилсульфонат > оксиалкилсульфонат.
Олефинсульфонаты удерживаются слабее, чем алкилсульфонаты с такой
же длиной радикала.
Если сравнивать анионы одноосновных алифатических
кислот с другими органическими и неорганическими анионами, то
их можно охарактеризовать как очень слабоудерживаемые.
Большинство из них удерживаются слабее хлорида. Поэтому для
определения таких анионов нужны слабые элюенты с
концентрацией менее 1 мМ [!, 4].
9.2.2. Двухосновные алифатические кислоты
Время удерживания анионов двухосновных кислот зависит от
pH элюеита. При использовании нейтральных и кислых элюентов
эти кислоты будут разделяться в виде однозарядных анионов, а
при использовании щелочных элюентов — в виде двузарядных.
Кроме того, молекулярная форма кислот может удерживаться
на подавляющей колонке по ионоэксклюзионному механизму.
Поэтому порядок удерживания двухосновных кислот в одноколоноч-
ной и двухколоночной системах может различаться.
Баг [5] исследовал удерживание двухосновных карбоновых
кислот на четырех различных сорбентах в двухколоночном
варианте. В качестве элюента использован 3 мМ NaHCO3/2,4 мМ Na2SO3
с pH более 9. При этом pH кислоты находятся в растворе в
виде двухзарядных анионов. Было установлено, что на колонке
но
Micro Рак АХ-10 (размер зерен 10 мкм, емкость 2 мэкв/г) время
удерживания кислот от малоновой до пимелитовой линейно
увеличивается с ростом числа СН2-групп в молекуле кислоты.
Время удерживания увеличивается в ряду: малоновая <янтарная<
<глутаровая<адипиновая<пимелитовая. Однако на других
колонках (Dionex, Vydac и Zipax) подобная зависимость не
наблюдается.
Изучено удерживание двухосновных карбоновых кислот на
сорбенте ХИКС-1 в одноколоночном варианте с 1,25 мМ
фталево ii кислотой в качестве элюента [6]. Исследованы кислоты с
различным числом CHs-групп в молекуле от щавелевой (п = 0) до
себациновой (п = 7). Зависимость времени удерживания кислот
от числа СНо-групп более сложная, чем в двухколоночном
варианте (рис. 9.1). Эта зависимость имеет минимум, соответствующий
янтарной кислоте (л = 2).
Времена удерживания членов гомологического ряда
двухосновных карбоновых кислот в одноколоиочном варианте можно
определить с помощью универсального уравнения для расчета
исправленных удерживаемых объемов, связывающего параметры
удерживания веществ с номером гомолога [7}. Это уравнение
имеет вид
где m — номер гомолога. Коэффициенты с, 3, V и е
рассчитывают из исправленных времен или
объемов удерживания одного или
нескольких гомологов, полученных
для конкретных хроматографиче- но
ских условий. При элюировании
двухосновных1 карбоновых кислот
1,25 мМ фталевой кислотой эти
коэффициенты равны а=1,28;
? =0,67; у = —7,64; є = 0 29 [6].
Уравнение (9.1) можно
использовать для расчета времен
удерживания в одноколоночном варианте
членов гомологического ряда
одноосновных карбоновых кислот.
Рис. 9.1. Зависимость
«справленных времен удерживания (Ґ)
двухосновных кислот от числа
СН2-групп (п) в молекуле [6].
Сорбент ХИКС-1. Элюент1,25мМ
фталевая кислота
9.2.3. Ароматические кислоты
Анионы ароматических кислот удерживаются на анионообмен-
никах сильнее, чем анионы алифатических кислот. Это связано
с появлением бензольного кольца, которое вносит существенный
вклад в удерживание. Из-за многообразия ароматических кислот
трудно установить какие-то общие закономерности их
удерживания, но внутри определенных классов корреляции существуют.
14»
Определение органических кислот
Объект
анализа
Определяемые
кислоты
Разаеляющая
колонка
Подавление
Элгоент
Дождевая вода
и воздух
нсоон,
СН3СООН
Dionex
H PIC—AS3
Dx250 мм)
мембранное
2 міМ NaHCCy
/1,67 мМ Na2CO3
Природная
вола и
биологические объекты
фосфоновые
кислоты
Dionex Anlon
CX150 и
3X500 мм)
колоночное
5 мМ Na2B4O7;
2,4 мМ NaHCO3/
/1,8 мМ Na2CO3
Грунтовая вода
ацетат, фумарат,
сукцинат, оксалат
Dionex Anion
то л; є
4,8 мМ NaHCOg/
/2,3 мМ Na2CO3
Атмосферные осадки
ацетат, формпат
3 мM NaHCOg/
/З мМ Na2CO3
глпколЕга;:
Dionex Anion
Котловые воды
то же
Dionex Anion
B,8X1000 мм)
З мМ NaHCOg/
'2,4 мМ Na2COs
5 мМ Na2CO3/
/4 мМ NaOH
Техническая
вода и коидеи-
сат
электростанции
формиат, ацгтат,
пропионат и бути-
рат
Dionex Anion
CX250 и
3X500 мм)
0,1 мМ NaOH
Рабочий
раствор солнечных
нагревателен
глчколевая, молоч-Dionex
пая HPIC-AS3
0,85 мМ NaHCOg
142
двухколоночной ионной хроматографией
Таблица 9.2
Скорость
потока,
мл/мин
Предел
обнаружения,
МІСГ/МЛ
Примечания
Литература
0,5
в воздухе
0,05 мкг/'м:> (НСООН)
0,15 мкг/м1 (СНзСООН)
[Ю]
2,3
0,1—0,25
[И]
1,9
необходимо
предварительное концентриро
вание
[12]
Менее 5
[13]
2,3
[HI
1,75
3-4
гликолят образуется
при разложении ЭДТА
[151
2,3
A,5—3,1I0
—3
1,1—1,5
[1]
2,0
кислоты образуются
при разложении
этилен- и
пропилен-гликоля
[16]
143
Объект
анализа
Сточная вода
Водные
вытяжки грибов и
огурцов
Гемигидрат
кальция
Воздух
Водный раствор
Определяемые
кислоты
анионные ПАВ
бензойная
лимонная
муравьиная
пировиноградная,
метан-с\ льфоно-
вая
кислоты цикла
кребса
Разделяющая
колонка
Dionex Anion
CX500 мм)
Dionex Anion
CX250 мм)
Dionex
HPIC—AS4
DX50 мм)
Dionex Anion
CX500 мм)
Dionex Anion
Dionex
HP 1С AS4
'4X250 mm)
Подавление
•
-
мембранное
колоночное
то же
мембранное
Элюент
0,1 мМ NaHCO3/
/0,1 мМ Na,CO,
3 мМ Na2COs/
/1,5 мМ*КОН
10 мМ Na.2CO3
2,5 мМ Na2B4Or
0,8 мМ Na2B4Or;
0.17 мМ №гСО3/
/0,22 мМ NaHCO3
20 мМ Na2BA;
20 мМ Na2CO3/
/2 мМ NaOH
Было изучено удерживание некоторых ароматических кислот на
анионообменниках низкой емкости в двухколоночном и одноко-
лоночном вариантах [8, 9]. Для метил-, хлор- и нитрозамещенных
бензойной кислоты в зависимости от положения заместителя
удерживание изменяется в ряду: орто<мета<пара. Причем времена
удерживания мета- и пара-изомеров близки, a орго-изомера от
них отличается сильно. Эта зависимость коррелируется с
коэффициентом Гаммета для этих кислот. Для орго-галогензамещенных
бензойной кислоты время удерживания зависит от
поляризуемости заместителя и увеличивается в ряду F<Cl<Br. Аналогичная
зависимость наблюдается для замещенных салициловой кислоты,
для которых время удерживания увеличивается в ряду
CH3<OH<SO3<CH3O. Для арилжирных кислот время
удерживания растет с увеличением длины боковой цепи и с
появлением двойной связи в заместителе. При замене карбоксильной на
другие кислотные группы (арсоновую, селеноновую, сульфоно-
вую) время удерживания кислот возрастает с уменьшением
поляризуемости анионов кислот. і
144
Продолжение табл. 9,2
Скорость
потока,
мл/мин
2,3
2,3
2,0
1,9
—
1,5
Предел
обнаружения.
мхг/мл
0,005
1,0
1,0
80 мкг/м3
0,05—0,08
—
1,5
1,0—2,2
—
7,3
—
Примечания
—
—
—
—
—
—
Литература
[4]
[8]
[17J
[181
[19]
[20]
9.3.
ИОНОХРОМАТОГРАФИЧЕСКОЕ ОПРЕДЕЛЕНИЕ
9.3.1. Двухколоночный вариант
В большинстве случаев пределы обнаружения органических
анионов выше, чем неорганических. Это связано со степенью
диссоциации кислот. Поскольку органические кислоты слабее,
неорганических, чувствительность их кондуктометрического де-
тектироЕания ниже. Воспроизводимость результатов определения
органических и неорганических анионов примерно одинаковая.
Примеры использования двухколоночной ионной хроматографии
для определения органических кислот в водах и других
объектах приведены в табл. 9.2.
Результаты ионохроматографического определения
органических кислот сравнивали с данными, полученными другими
методами [1, 8, 21]. Во всех случаях получено хорошее совпадение ре-
145
зультатов. Кроме того, установлено [21], что воспроизводимость
определения щавелевой кислоты двухколоночной ионной
хроматографией выше, чем ферментным методом.
Однако при двухколоночном определении органических
кислот следует помнить о возможности их эксклюзионного
удерживания на подавляющей колонке. Поэтому определение лучше
проводить либо с короткими подавляющими колонками, либо- с
мембранной системой подавления.
9.3.2. Одноколоночный вариант
с кондуктометрическим детектором
Этот вариант для определения органических кислот пока
используют очень редко. В большинстве публикаций показана
сама возможность такого определения без оценки аналитических
характеристик методик.
В одной из первых работ по одноколоночной ионной
хроматографии Гьерде и Фритц [22] установили, что 1 мМ бензойную
кислоту можно использовать как элюент для совместного
определения 9,2 мкг/мл ацетата, 30 мкг/мл пропионата и 9,4 мкг/мл
формиата. Анионы разделяли на поверхностно-модифицированном
анионообменнике емкостью 0,023 мэкв/г. В более поздней работе
Фритц и др. [23] для определения муравьиной, уксусной,
молочной и гликолевой кислот использовали 1 мМ раствор янтарной
или никотиновой кислоты.
Для определения уксусной и некоторых аминокислот было
применено [24] косвенное кондуктометрическое детектирование с
1 мМ КОН в качестве элюента. Этот же прием использовали [9]
для изучения ионохроматографического поведения большого
числа алифатических и ароматических кислот на полимерном
анионообменнике TSK-Gel IC-620 SA.
Некоторые аналитические характеристики одноколоночного
определения муравьиной, уксусной, молочной, янтарной и
бензойной кис/ют приведены в работе [25]. Кислоты разделяли на
колонке TSK-Gci IC-Anion-SW D,6X50 мм) в потоке 1,0 мМ
раствора фталевой кислоты (pH 3,4). Пределы обнаружения
кислот при соотношении S/N = 2 составляют 0,005—0,02 мкг/мл.
Относительное стандартное отклонение при определении 3—Юмкг/мл
изменяется от 0,1% для бензойной кислоты до 6,9% для
янтарной. Метод был использован для определения кислот в
алкогольных напитках и фармацевтических препаратах.
Для ионохроматографического определения органических
кислот в винах [26] в качестве элюента был взят раствор смеси
3 мМ сульфамовой кислоты и 0,7 мМ сульфамата аммония. Было
отмечено, что для одного анализа методом ионной
хроматографии требуется 15 мин, а методом обращенно-фазной
хроматографии — 1ч.
. Представляет интерес использование ионной хроматографии
для определения нейтральных органических соединений. Опре-
146
деляемые вещества вначале переводят в соединения ионного
характера. Низкомолекулярные альдегиды и ацетон переводили в
оксиалкилсульфонаты по реакции с гидросульфитом натрия [27].
Затем полученные сульфонаты разделяли 20 мМ лимонной
кислотой (элгоент) и определяли кондуктометрически. Для
определения Сахаров [28] использовали в качестве элюента борную
кислоту. Сахара образуют с борной кислотой комплексы, константа
диссоциации которых существенно выше, чем борной кислоты.
Это позволяет определять сахара с кондуктометрическим
детектором.
9.3.3. Одноколоночный вариант с УФ детектором
Для определения органических кислот используют как прямое,
так и косвенное УФ детектирование.
При прямом детектировании измеряют собственное
поглощение кислот в УФ области спектра. Р. Боуман [29] определял
замещенные бензойной кислоты в вытяжках почв при 205 нм.
Кислоты разделялись на колонке Partisil lOSAX элюирующей смесью
(90:10) 5 мМ фосфатного буфера (pH 4,0) и CH3CN. Предел
обнаружения на уровне 10 мкг/л. Для определения муравьиной,
уксусной, малеиновой, бензойной и лимонной кислот также
использовали [30] в качестве элюента фосфатный буфер. Кислоты
детектировали при 210 нм.
Описан ионохроматографический метод определения
бензойной кислоты в атмосфере [31]. Воздух пропускали через тефлоно-
вый фильтр с объемной скоростью 1—30 л/мин. После
прибавления 1 мкг и-толуиловой кислоты (внутренний стандарт) и 1 мл
0,1 M HC1 пробу экстрагировали 5 мл эфира в течение 15 мин,
а затем центрифугировали для разделения слоев. Эфирный слой
выпаривали, а остаток растворяли в 2 мл карбонатного буферного
раствора B,6 мМ Na2CO5/3,3 мМ NaHCO3), используемого в
качестве члюентя. Раствор (-100 мкл) вводили в хроматограф.
Разделение проводили на колонке Dionex HPIC, а детектирование при
224 нм. Растворы подчиняются закону Бера при содержании
бензойной кислоты от 20 нг до 20 мкг. Относительное
стандартное отклонение 10% при содержании кислоты 20—50 нг и 1 %
при содержании 5—20 мкг.
УФ детектирование B10 нм) использовали [26] для
определения органических кислот в вимах с элгоентом: 3 мМ сульфамо-
вая кислота / 0,7 мМ сульфамат аммония.
Косвенное УФ детектирование используют для определения
органических аипомоз, поглощение которых ниже, чем элюирую-
щих. Этот способ детектирования был применен [32] для
определения муразьинон и уксусной кислот. Кислоты определяли с
5 мМ гидрофталатсм калия (pH 4,6) при 308 нм.
Чувствительность определения кислот е УФ детектором в 20 раз выше, чем
с кондуктометрнчеекпм. Показана также возможность исполь-
147
зования косвенного УФ детектирования для определения
уксусной, молочной, глюконовой и щавелевой кислот [33].
Ларсон [34] предложил метод определения алкилсульфонатов
при использовании л-сульфобензойной кислоты в качестве
поглощающего в УФ области спектра элюента. Определение
проводят на колонке Partisil 10SAX с элюентом F0:40) CH3N—Н2О,
содержащим 5 мМ лг-сульфобензойной кислоты (pH 3,6) при
298 нм. Сигнал детектора линеен в области концентраций
алкилсульфонатов 10—1000 мкг/мл.
9.4.
ИОНОЭКСКЛЮЗИОННАЯ ХРОМАТОГРАФИЯ
9.4.1. Принцип метода
Ионоэксклюзионная хроматография — довольно
распространенный метод разделения слабых кислот, особенно органических.
Разделение проводят на сильнокислотных катионообменниках в
Н-форме. Метод был описан Уитоном и Бауманом [35], а затем
в работах [36, 37].
В основе ионоэксклюзионного разделения лежит доннановская
эксклюзия. Твердая матрица сульфокатионита является как бы
полупроницаемой мембраной между жидкостью, находящейся в
порах смолы, и подвижной фазой (рис. 9.2). Анионы сильных
кислот из-за отталкивания суль-
фогруппами катионообменника не
проникают в его поры и не
удерживаются. В то же время
слабые кислоты, находящиеся в
растворе в неионизированной,
молекулярной форме проходят в
поры смолы и удерживаются ею.
Способность слабых кислот
удерживаться сильнокислотными ка-
тионробменниками в Н-форме
определяется рядом факторов
[38] (табл. 9.3).
Основным фактором является
способность кислоты диссоци-
ировать в водном растворе [38].
,0-ССН,
ник
ііо/іупроницае -
мая мемірана
Т
Жидкая
Рис. 9.2. Принцип ионоэксклюзи-
онного разделения цем выше p^fl кислоть1) теМ
сильнее она удерживается на катионите. Для кислот, имеющих
примерно равные рКа (уксусная, пропионовая, масляная,
валериановая), удерживание растет с увеличением длины
углеводородного радикала, т. е. с ростом гидрофобное™ молекулы.
Харлоу и Мормап [361 установили некоторые общие
закономерности удерживания органических кислот.
148
1. Кислоты гомологического ряда элюируются в порядке
увеличения кислотности и уменьшения растворимости в воде.
2. Двухосновные кислоты удерживаются слабее одноосновных.
3. Кислоты изомерного строения удерживаются слабее
соответствующих кислот нормального строения.
4. Наличие двойной связи в молекуле кислоты способствует
увеличению удерживания.
Таблица 9.3
Удерживание карбоновых кислот на катионообменнике в Н-форме
и пределы обнаружения с кондуктометрическим детектором и 0,5 мМ
бензойной кислотой в качестве элюента [38]
Кислота
Серная
Муравьиная
Уксусная
Пропионовая
Масляная
Валериановая
Щавелевая
—2
3,8
4,8
4,9
4,8
4,8
1,3
VR , мл
3,2
5,4
7,4
10,4
16,3
34,3
3,3
Предел
обнаружения
(S/N=2),
мкм |
0,16
1,28
5,19
11,7
15,6
46,7
0,30
Кислота
Малеиновая
Малоновая
Винная
Лимонная
Молочная
Янтарная
Угольная
1,9
2,9
3,0
3,2
3,9
4,2
6,4
VR , мл
4,2
3,6
3,6
3,7
5,2
5І9
9,2
Предел
обнаружения
(S/N=2),
мкм
0,71
0,86
0,76
0,70
2,30
2,26
2,33
5. Кислоты с бензольным кольцом удерживаются сильнее.
Закономерности удерживания некоторых кислот изучены
также в работе [39].
Поскольку в ионоэксклюзионной хроматографии кислоты
разделяют в виде недиссоциированных молекул, то для элюирования
чаще используют кислые водные растворы. В этом случае
достигается высокая селективность разделения слабых и сильных
кислот. В качестве элюентов используют разбавленные растворы
A—10 мМ) серной [40—42], соляной [37, 43, 44], угольной [45],
бензойной и янтарной [38] кислот. Можно использовать для
элюирования и деионированную воду [38, 46, 47].
При использовании сильнокислотных элюентов (соляная,
серная кислоты) наиболее подходящим является УФ детектор с
длиной волны 210 нм [36, 37, 40]. Поскольку поглощение
минеральных кислот в этой области спектра существенно ниже, чем
органических, то чувствительность определения с этим детектором
сравнительно высокая.
Кондуктометрический детектор чаще используют со слабо
проводящими элюентами (угольная, бензойная, янтарная кислоты и
деионизованная вода) [38, 42, 45, 46]. Хроматограмма смеси
органических кислот, полученная с кондуктометрическим
детектором, приведена на рис. 9.3. Для проведения кондуктометрического
детектирования с солянокислым элюентом фирма Дайонекс
предлагает устанавливать между разделяющей колонкой и детекто-
149
ром вторую колонку, заполненную катионитом высокой емкости
в Ag-форме. Эта колонка предназначена для снижения фоновой
электропроводности элюента. Соляная кислота удаляется из элю-
ента в результате образования осадка
хлорида серебра. Органические кислоты не
взаимодействуют с серебром и проходят
через колонку без изменения.
Помимо УФ и кондуктометрического
детекторов для иоиоэксклюзионного
определения органических кислот используют
кулонометрический [48],
рефрактометрический [41], электрокинетический [47] и даже
масс-спектрометрический [44] детекторы.
Разделение проводят на сильнокислотных
катионообменниках высокой емкости в Н-
форме с размером частиц 5—10 мкм. На
таких сорбентах можно достичь высокой
эффективности разделения.
20 3$
MUH
Рис. 9.3. Ионоэксклюзионное разделение
щавелевой A), малоновой B), малеиновой C),
муравьиной D), янтарной E), уксусной (б) кислот [38].
Колонки: катионообменник TSK, 8X200 мм и
7,5x100 мм, соединенные последовательно. Элюент
0,5 мМ бензойная кислота. Скорость потока
1,0 мл/мин. Концентрация кислот 1 мМ
Детектирование кондуктометрическое
9.4.2. Определение органических кислот
Ионоэксклюзионную хроматографию использовали для
определения органических кислот в различных объектах. Ито и Шин-
бори [45] предложили использовать метод для определения одно-
и двухосновных кислот в фармацевтических препаратах. Элюен-
том служил раствор угольной кислоты, который получили
пропусканием смеси NaHCO3 и Na2CO3 через колонку, заполненную
катионообменником в Н-форме. Разделенные кислоты
детектировали кондуктометрически. Предел обнаружения органических
кислот больше или равен 0,04 мкг/мл, а относительное
стандартное отклонение 1,3%.
Для определения молочной кислоты в пищевых продуктах
предложен [40] метод с 9 мМ H2SO4 в качестве элюента и УФ
детектированием при 210 нм. Сигнал детектора линеен в области
концентрации молочной кислоты 0,2—2,5 мг/мл, относительное
стандартное отклонение 2,0—4,9%.
Мешающее влияние катионов металлов, которые снижают
150
эффективность разделяющей колонки, устраняли добавлением к
образцу 0,05% ЭДТА.
Танака и Фритц [38J изучили условия ионоэксклюзионного
определения двенадцати алифатических карбоновых кислот с кон-
дуктометрическим детектором. Была показана перспективность
использования растворов слабых кислот (бензойной и янтарной)
в качестве элюентов. Для 0,5 мМ бензойной кислоты пределы
обнаружения органических кислот колеблются от 3-10~7
до 510~7 M (табл. 9.3). Добавка 5% ацетонитрила к элюенту
заметно уменьшает времена удерживания сильно сорбирующихся
кислот.
Глод и Кемуля [17] использовали ионоэксклюзионную
хроматографию с электрокинетическим детектированием для
определения двухосновных алифатических и ароматических кислот.
Элюирование проводили деионированной водой. Эффективно
разделяются кислоты с рКа от 2,5 до 6,5. Пределы обнаружения
кислот зависят от рКа и находятся в интервале 2-Ю'2—3- Ю-7 М.
Метод использовали для определения бензойной кислоты в
кормах.
9.5
СОЧЕТАНИЕ ИОННОЙ И ИОНОЭКСКЛЮЗИОННОИ ХРОМАТОГРАФИИ
Для совместного определения анионов слабых и сильных
кислот перспективны методы, сочетающие ионную и
ионоэксклюзионную хроматографию. Для определения F^ , НСОО~ , СНзСОО~
Cl" ,. NO2-, HPOf- , Br- , NO- и SO42- предложена [49] хрома-
тографическая система, состоящая из анионной разделяющей
колонки, мембранной системы подавления, ионоэксклюзионной
разделяющей колонки и кондуктометрического детектора (рис. 9.4).
В качестве элюента использовали смесь NaHCO3/Na2CO3. Все
анионы разделяли на первой разделяющей колонке. А для
полного разделения фторида, хлорида, формиата и ацетата
использовали ионоэксклюзионную колонку. Элюентом является
угольная кислота, которая образуется в мембранной системе
подавления. Хроматограмма смеси семи анионов, полученная этим
методом, представлена на рис. 9.5. При объеме пробы 100 мкл
предел обнаружения формиата равен 0,03 мкг/мл, а ацетата —
0,15 мкг/мл. Метод использовали для определения анионов в
атмосферных осадках. 1 ,
Рич и др. [46] использовали сочетание ионной
ионоэксклюзионной хроматографии для определения НСОО~, СН3СОО~
СО| - , Cl", ClOj- и SO\~ в рассоле и каустике. Вначале на
ионоэксклюзионной колонке разделяют анионы слабых кислот,
элюируя их деионированной водой. Анионы сильных кислот при
этом не удерживаются и направляются на анионообменную
колонку, где их разделяют карбонатным элюентом. Разделенные
таким образом анионы слабых и сильных кислот последователь-
151
но попадают в кондуктометрический детектор. Время
определения около 20 мин, относительное стандартное отклонение 3—П%
в зависимости от концентрации анионов. Для анионов сильных
кислот применяют предварительное концентрирование.
NaHCOj/NajCOj
Ионная хроматография
Разделение анионов сильных кислот
Na*
Анионная
разделяющая
колонка
Мембранная
система
подавления
і
Катионо-
обменник
AGSOW-X4
R—HCO3 +
R—X +
N»HCO3 + H2SO4 '^=±: Н2СО3 + NaHSO4
Ионо-эксклюэионная хроматографня
Разделение анионов слабых кислот
Кондуктометрический детектор
20 15 10
нх/н2со3
Рис. 9.4. Комбинация ионной и ионоэксклюзионнон
хроматографии [49]
Рис. 9.5. Хроматограмма анионов, полученная
комбинацией ионной и ионоэксклюзионнон
хроматографии [49].
Колонки: Dionex Anion, 4X250 мм и катноно-
обменник AG 50W-X4, 9X200 мм. Элюент 4,5 мМ
NaHCO3/3,6 мМ Na2CO3. Скорость потока
2,3 мл/мин. Концентрации анионов 1—
10 ыкг/мл. Детектирование кондуктометричес-
5 О
мин
Для совместного определения бората, хлорида, нитрата и
йодида предложили [50] хроматографическую систему с двумя
детекторами — кондуктометрическим и рефрактометрическим«1
152
Борную кислоту отделяют на ионоэксклюзионнои колонке и
определяют с рефрактометрическим детектором. Остальные
анионы определяют обычным ионохроматографическим методом с
кондуктометрическим детектором. Метод характеризуется
высокой воспроизводимостью (S, менее 1%). Он может быть
использован для анализа контурных вод атомных электростанций.
ЛИТЕРАТУРА
I. Иванов А. А., Шпигун О. А., 3 о л о т о в Ю. А. // Ж. аналит. химия.
1986. Т. 41, № 1. С. 134-139.
2 С а п t w е 1 1 F. F, Р и о п S. // Anal. Chem. 1979. V. 51, № 6. P. 623—632.
3. W e і s s J. H J. Chromatogr. 1986. V. 353. P. 303—310.
4 И в а но в A A. et al. // Ж. аналит. химии. 1985. T. 40, № 9. С. 1699—1702.
5. В a g S. P. // Talanta. 1985. V. 32, M! 8B. P. 779—784.
6. Иванов А. А. Определение органических карбонових кислот иоиной
хроматографией: Дис. ...канд. хим. наук: M., 1989.
7 Г с л о в н я Р. В., Г р и г о ,р ь є в а Д. H. // Ж. аиалит. химии. 1985. T. 40,
Ne 2. С. 316—329.
8 Золотое Ю. А., Иванов А. А, Шпигун О. А. // Ж. аналит. химии.
1983. Т. 38, № 8. С. 1479—1483.
9. О k a d а Т., К u w a m о t о Т. Ц Anal. Chem. 1983. V. 55, № 7. P. 1001—
1004.
10 Brocco D., Tappa R. Ц J. Chromatogr. 1986. V. 376, № 1.
P. 240—246.
II. Schiff L. J , P 1 e v a S. G., S a г v e r E. W. // Ion Chromatographie
Analysis of Environmental Pollutants / Ed. by J. D. Mulik and E. Sawicki. —
Ann Arbor: Ann Arbor Sei. 1979. P. 329—344.
12. S с h w a b e R. et al. // J. Env. Anal. Chem. 1983. V. 14, № 3. P. 169—179.
13. Muller К Р. // Freseniuz Z. Anal. Chem. 1984. B. 317, № 3—4.
S. 345—346.
14. R і с h W. E. // Instrum. Techn. 1977, № 8. P. 47—51.
15. Stevens T. S., T и r к e 1 s о n V. T., A Ы e W. R. // Anal. Chem.
1977. V. 49, № 8. P. 1176—1178.
16. К о s s і t e r W. J, Brown P. W., G о dette M. // Solar Energy
Mater. 1983 V. 9, № 3 P. 267—279.
17. M a t s и і H. et al. // Gyps, and Lime. 1984. V. 189. P. 69—75.
18. H a y n e s D. L. // Ion Chromatographie Analysis of Environmental
Pollutants / Ed. by J. D. Mulik and E. Sawicki — Ann Arbor: Ann Arbor Sei 1979.
P. 157—170.
19. G r о s j e a n D, N i e s D. J. //Anal Lett. 1984. V. A17, № 2. P. 89—96.
20. R о с к 1 і n R. D, S 1 і n g s b y R. W., P о h 1 С A. // J. Liq. Chromatogr.
1986. V. 9, № i. P. 757—775.
21. С I a s s e n A, H e s s e A. // Fresenius Z Anal. Chem. 1986 B. 324, № 3—
4. S. 303—305.
22. G je r d e D. T., F r і t z J. S. // Anal. Chem. 1981. V. 53, № 14. P. 2324—
2327.
23. FritzJ. S., DuvalD. L, BarronR. EJ/ Anal. Chem 1984. V. 56,
№7. P. 1177—1182.
24. В ол ощ h к И. H.. Ш п игу н О. А., 3 о л о т о в Ю. А //Ж. аналит.
химии. 1986. Т. 41, № 6. С. 1089—1092.
25. Hoshin о Y., S ai ton H., О і к a w a K. jj Bunseki Kagaku. 1983.
V. 32, № 4. P. 273—276.
26. Fujimura K., T s u с h і y a M. // Bunseki Kagaku. 1986. V. 35, № 1.
P. T50-T56
27. D u v a 1 D. L., R о g e r s M. FritzJ. S. // Anal Chem. 1985. V. 57,
№8. P. 1583—1586.
28. Oka da T., Ku warn о to T. // Anal. Chem. 1986. V. 58, № 7.
P. 1375—1379.
153
29. В о w m a n R. S. // J. Chromatogr. ] 984. V. 285. P. 467—477.
30. H a r r і s о n К. et al. // Int. Lab. 1986. № 4. P. 90—94.
31. F u n g K., Grosjean D. // Anal. Lett. 1984. V. A17, № 4. P. 475—480.
32. Cochrane R. A., Hi lim an D. E. // J. Chromatogr. 1982. V. 24t.
P. 392—394.
33. Jenke D. R. // Anal. Chem. 1984. V. 56, № 14. P. 2674—2681.
34. L a r s о n J. R. // J. Chromatogr. 1986. V. 356, № 2. P. 379--381.
35. W he a ton R. M., Bauman W. С // Ind. Eng. Chem. 1953. Y. 45,
№ 2. P. 228—236.
36. H a r 1 о w G. A., M о r m a n D. H. // Anal. Chem. 1964. V. 36, № 13.
P. 2438—2442.
37. T u r ke 1 s о n V. T., Richards M. // Anal. Chem. 1978. V. 50, № 11.
P. 1420—1423.
38. T a n a к a K., F r і t z J. S. // J. Chromatogr. 1986. V. 36t. P. 151—160.
39. Tanaka K-, Ishinzuka T. // J. Chromatogr. 1979. V. 174.
P. 153—160.
40. A s h о о r S. H., W e 11 y J. // J. Chromatogr. 1984. V. 287. P. 452—456.
41. P e ci n a R. et al. // J. Chromatogr. 1984. V. 287. P. 245—248.
42. J u p і 1 1 e T. // Int. Lab. 1985, № 11—12. P. 82—89.
43. Lebe 1 A., F u Y. T. // Anal. Chem. 1984. V. 56, № 4. P. 807—808.
44. P a с h о 1 e с F., E a t о n D. R., R о s s і D T // Anal. Chem. 1986. V. 58,
№ 12. P. 2581—2583.
45. Iton H., Shinbori Y. // Bull. Chem. Soc. Jap. 1985. V. 58, № 11.
P. 3244—3247.
46. Rich W. et al. // Ion Chromatographie Analysis of Environmental
Pollutants / Ed. by J. D Mulik and E Sawicki. — Ann Arbor: Ann Arbor Sei. 1979.
P. 17-30.
47. G 1 о d В. К., К e m u 1 a W. // J. Chromatogr. 1986. V. 366. P. 39—50.
48. T a к a t a Y., M u t о G. // Anal. Chem. 1973. V. 45, № 11. P. 1864—1868.
49. P і m min ge r M. et al. // Fresenius Z. Anal. Chem. 1985. B. 320, № 4.
S. 445—450.
50. J о n e s W. R., H e с к e n b e r g A L. J a n d і к P. // J. Chromatogr.
1986. V. 366. P. 225—233.
ГЛАВА 10
ОПРЕДЕЛЕНИЕ МЕТАЛЛОВ
Методом ионной хроматографии металлы определяют
главным образом в виде катионов; реже используют анионные
комплексы. Можно определять большинство металлов. Разработаны
методики ионохроматографического определения щелочных,
щелочноземельных, d-переходных, редкоземельных, платиновых и
других элементов. Методики обычно характеризуются высокой
чувствительностью, селективностью и экспрессностьго. Однако
публикаций о практическом использовании ионной
хроматографии для определения металлов значительно меньше, чем работ по
определению анионов, особенно неорганических. Это по крайней
мере частично объясняется тем. что другие современные методы
определения металлов могут успешно конкурировать с ионной
хроматографией в отличие от методов определения
неорганических анионов.
10.!.
ЩЕЛОЧНЫЕ МЕТАЛЛЫ И АММОНИЙ
Аммоний, конечно, не относится к металлам, но в ионной
хроматографии его чаще всего определяют вместе с катионами
щелочных элементов. Поэтому целесообразно рассматривать
аммоний вместе со щелочными металлами. Поскольку эти катионы
удерживаются на катионообменннках слабо, то для их
определения используют злюенты с низкой элюирующей способностью.
10.1.1. Двухколоночный вариант
В двухколоночном варианте щелочные металлы и аммоний
определяют, как правило, с использованием сильнокислотных элю-
ентов — растворов соляной или азотной кислот. Затем на
подавляющей колонке, заполненной анионообменником в ОН-форме,
элюенты превращаются в воду. Определяемые катионы
детектируют кондуктометрически в виде гидроксидов.
Оценена возможность определения лития, натрия, аммония и
калия в геотермальных водах [1]. Катионы элюировали 3 мМ.
HNO3. Относительное стандартное отклонение результатов
определения составило 0,4—1,8%, что удовлетворяет требованиям,
155
предъявляемым к анализу вод указанного типа. Полученные
результаты хорошо согласуются с данными других методов. Хро-
матограмма образца геотермальной воды приведена на рис. 10.1.
Li
Na+
П 16 20
MUH
Рис. 10.1. Определение лития и ам-
моиия в геотермальной воде [1J.
Колонка Dionex Cation, 6x250 мм.
Элюент 3 мМ HNO3. Скорость
потока 3,0 мл/мин. Детектирование кон-
дуктометрическое
6 0
мин
Рис. 10.2. Определение натрия,
аммония и калия в водопроводной
воде [9].
Колонка 2X340 мм с катионооб-
менником емкостью 0,017 мэкв/г.
Элюент 1,25 мМ HNO3. Скорость
потока 1,0 мл/мин. Детектирование
косвенное коидуктометрическое
Двухколоночная ионная хроматография с 5 мМ НС1 в
качестве элюента использована [2] для определения ничтожных
количеств натрия, калия и аммония в антарктическом снеге и льде.
Для повышения чувствительности определения использовали
петлю-дозатор объемом 5 мл. Градуировочный график линеен в
интервале концентраций 0—30 нг/г для натрия и 0—10 нг/г для
аммония и калия. Предел обнаружения натрия и аммония
0,5 нг/г, а калия 1,0 нг/г. Результаты определения натрия
хорошо согласуются с данными неитроноактивационного метода.
Для повышения чувствительности определения натрия и
калия в особо чистой воде использовали [3] предварительное
концентрирование. Это позволило определять катионы на уровне
1 мкг/л при объеме пробы 10 мл. Элюирование проводили 7,5 мМ
соляной кислотой. Относительное стандартное отклонение
156
зультатов при определении 1 мкг/л составило 13,5% для натрия
и 8,7% для калия.
Известна методика определения натрия, калия и аммония в
дождевой воде [4]. В качестве элюента использовали 6 мМ
азотную кислоту. При объеме вводимой пробы 100 мкл предел
обнаружения натрия и аммония 5 мкг/л, а калия 20 мкг/л. При
концентрации определяемых катионов выше 0,1 мкг/мл результаты
определения хорошо согласуются с данными атомно-абсорбцион-
ной и атомно-эмисснонной спектрометрии, а также
колориметрии. Трудность определения более низких концентраций связана
с малой коррозионной стойкостью хроматографической системы.
Двухколоночная ионная хроматография с кондуктометриче-
ским детектированием использована для определения натрия,
калия и аммония в растворах электролитических ванн [5],
биологических объектах [6], воздухе [7]. Методика определения
щелочных металлов и аммония в этих объектах сходна с методикой
определения в водах.
Помимо кондуктометрического можно использовать и другие
детекторы. Для определения катионов щелочных металлов и
аммония в водах после биологической очистки предложен куло-
нометрический детектор [8]. Смесь Li + , Na+, К+, Rb+, Cs+
п NH^~ элюировали 3 мМ азотной кислотой. Катионы
переводили в гидроксиды в мембранной подавляющей системе. Пределы
обнаружения катионов с кулонометрическим детектором ниже,
чем с кондуктометрическим для Li+ и Na+ в 10 раз, а для К +
Rb+ , Cs+ и NH+ в 2—3 раза. Однако линейный диапазон гра-
дуировочного графика для некоторых катионов уже.
10.1.2. Одноколоночный вариант
10.1.2.1. Кондуктометрическое детектирование
При одноколоночном кондуктометрическом определении
щелочных металлов элюирование в основном проводят, как и в
двухколоночном варианте, растворами сильных кислот. Однако
эквивалентная электропроводность протона гораздо выше, чем у
других катионов, поэтому используют косвенное детектирование.
Катионы разделяют на сильнокислотных катионообменниках
низкой емкости.
В одной из первых работ по одноколоночной хроматографии
катионов Фритц с^сотр. [9] предложили методику определения
натрия, калия и аммония в водопроводной воде. Разделение
проводили на низкоемкостном катионообменнике @,017 мэкв/г) с
1,25 мM азотной кислотой в качестве элюента. Нижняя граница
определяемых содержаний для натрия составила 0,1 мкг/мл,
время определения не превышает 10 мин. Для удаления кальция и
магния, которые в этих условиях сорбируются на колонке, после
157
элюирования щелочных металлов в колонку вводят 0,2 M
азотную кислоту. Хроматограмма водопроводной воды приведена на
рис. 10.2.
Для определения аммония в атмосферных осадках в
качестве элюента предложено [10] использовать 3 мМ азотную кислоту.
При объеме вводимой пробы 0,1 мл предел обнаружения
аммония равен 0,02 мкг/мл. Определению 0,52 мкг/мл аммония не
мешают до 400 мкг/мл натрия и калия. На результаты
определения практически не влияет величина pH пробы в интервале pH
3—7.
Описан [11J способ определения натрия и калия в питьевой
воде с использованием раствора азотной кислоты с pH 2,5 в
качестве элюента. Мизобучи и др. [12] сравнивали результаты ио-
нохроматографического определения натрия, калия и аммония в
сточных водах с данными, полученными атомно-абсорбционным
и атомно-эмиссионным спектральными методами. Отмечено их
.хорошее совпадение.
Катионы щелочных металлов (Na~, K+. Rb+, Cs + )
разделяли на силнкагеле [13]. Поскольку силикагель проявляет ка-
тионообменные свойства только в нейтральной среде, то в
качестве элюента предложено использовать 2,5 мМ хлорид лития.
Катион лития имеет более низкую эквивалентную
электропроводность, чем катионы других щелочных металлов, поэтому
определяемые катионы детектируют по положительным пикам.
10.1.2.2. Спектрофотометрическое детектирование
Катионы щелочных металлов не поглощают излучение УФ
области спектра, поэтому для их определения применяют
косвенное детектирование. Предложен [14] метод определения
щелочных металлов с использованием
разбавленного раствора сульфата меди (II) в качестве
->люентя. Разделение проводили на колонке
Zipax SCX с последующим детектированием
гри 220 нм, гце катион меди имеет высокое
гоглощение. Хроматограмма смеси катионов
щелочных металлов приведена на рис. 10.3.
Этот метод использован [15] для определения
щелочных металлов в питьевой воде. Преде-
лі-і обнаружения натрия, калия и аммония
•~гв,чы 0.02; 0 07 и 0,3 мкг/мл соответственно.
Результаты хорошо согласуются с данными
атомно-абсорбциоыного метода. Такой же
Рис. 10.3. Хроматограмма катионов щелочных
металлов [14].
Колонка Zipax SCX, 4,6x120 мм. Элюент
0,25 мМ сул:.фат меди. Скорость потока 0,5 мл/мин.
Детектирование косвеннее УФ B20 нм)
158
О
мин
10
принцип применяли для определения щелочных металлов в соке
овощей [16J, водных настоях [17].
Для элюирования катионов щелочных металлов и аммония
можно использовать растворы ароматических оснований [18].
Высокая чувствительность и селективность определения достигнута
на колонке, заполненной катионообменником Waters IC-Pak С,
[три элюировании 0,5—1,0 мМ растворами пиколиновой кислоты,
анилина, бензнламина с последующим косвенным УФ
детектированием. Пределы обнаружения, полученные для бензиламина как
элюента при объеме вводимой пробы 0,01 мл, составили 1,1 мкг/л
для Li+, 90 мкг/л для Na+ , 16,0 мкг/л для К+ и 19,5 мкг/л
для NH+.
10.2.
ЩЕЛОЧНОЗЕМЕЛЬНЫЕ МЕТАЛЛЫ И МАГНИЙ
Двузарядные катионы щелочноземельных металлов
удерживаются на катионообменниках значительно сильнее однозарядных.
Их удерживание зависят от радиуса гидратированного иона и
увеличивается в ряду Mg2+<Ca2+ <Sr2+ <Ba2+.
Для быстрого и селективного разделения катионов
щелочноземельных металлов и магния необходимы элюенты с высокой
элюирующей способностью. Поэтому для элюирования
используют двухзарядные катионы некоторых переходных металлов
или органических оснований. Для уменьшения времени
удерживания катионов и повышения эффективности разделения в элю-
ент иногда вводят комплексообразующие добавки. Разделение
проводят на катионообменниках низкой емкости.
10.2.1. Двухколоночный вариант
Для определения магния и кальция в природных водах
предложено [19] элюировать эти катионы растворами солей бария или
свинца с последующим снижением фонового сигнала на
высокоемкостном аннопообмеинике в ЗО^-форме. Минимально
определяемые концентрации магния и кальция находятся на уровне
0,04 мкг/мл. Результаты сравнивали с данными атомно-абсорб-
ционного определения (табл. 10.1), установлено достаточно
хорошее согласие. Для повышения чувствительности определения
после подавляющей колонки перед кондуктометрическим
детектором устанавливали колонку, заполненную катионообменником
высокой емкости в Н-форме. Благодаря высокой эквивалентной
электропроводности протона кондуктометрическое детектировзг-;
ниє в виде кислот оказывается более чувствительным.
Недостаток этого метода — сложность регенерации подавляющей
колонки. Образующиеся здесь сульфаты бария или свинца трудно
перевести в растворимые соли, поэтому рекомендовано не регене-
159
рировать колонку, а заменять ее на новую. Кроме того, этим
методом нельзя определить барнй.
Таблица 10,1
Результаты определения магния и кальция
в природных водах методом ионной хроматографии
(ИХ) и атомно-абсорбциовной спектрометрии (АЛС)
[19]. Элюент — 1,0 мМ витрат бария при pH 4
Образец*
PW 1***
PW 2'**
PW 3***
PW 4s**
ЕРА 1І74В****
их
389
149
98
150
7,40
мкг/мл
А А О*
399
145
97
146
7,29
Ca'-'1,
ИХ
572
381
214
340
24,3
миг/мл
А А С*
561
370
210
334
23,8
* Указаны условные номера проб.
** Найдено атомно-абсорбционным методом.
*+* Образец разбавлен в 25 раз.
•*** Стандартный образец. ЕРА 7,20 мкг/мл Mg
22,2 мкг/мл Са'2+.
2+
Для определения трех щелочноземельных металлов и магния
в природных водах предложено [20] использовать в качестве элю-
ента смесь 1 мМ Pb(NO3J/0,1 мМ HNO3 (pH 4) с последующим
подавлением фонового кондуктометрического сигнала на
высокоемкостном анионообменнике в Ю3-форме. Хроматограмма
образца воды представлена на рис. 10.4. Для повышения
чувствительности определения между подавляющей колонкой и кондуктомет-
рическим детектором устанавливали колонку с анионообменни-
ком в ОН-форме. Минимально определяемые концентрации
щелочноземельных металлов и магния составляют 0,05—1,1 мкг/мл.
При определении щелочноземельных металлов в качестве элю-
ента использована также смесь Zn(NO3J/HNO3 с подавлением
фонового сигнала на анионообменнике в ОН-форме и кондукто-
метрическнм детектированием [21]. Подавление сигнала
достигается за счет сорбции аниона ZnO2" на подавляющей колонке.
Отработанную колонку легко регенерировать, пропуская через
нее 0,5 M NaOH. Хроматограмма смеси катионов приведена на
рис. 10.5.
Для определения кальция, стронция и бария предложен [22]
двухколоночпый вариант с использованием хлорида или тартра-
та этилендиаммония в качестве элюентов и
кондуктометрическим детектированием. Для снижения фонового сигнала
использовали колонку с анионообменником в ОН-форме. При
определении 20 мкг/мл относительное стандартное отклонение
составляет 1,5—5,0%, предел обнаружения на уровне мкг/л. В качест-
160
ве элюента можно также использовать смесь ж-фенилендиамина
с азотной кислотой [23].
Перспективны аминокислотные элюенты. Такой элюент
использован [24] для определения следовых количеств магния и
Мдг+ Саг+
О 5 Ю
мин
Рис. 10.4. Хромато-
грамма природной
воды [20].
Элюент 1,0 мМ
Pb(NO3h (pH 4,0).
Скорость потока 2,3
мл/мин.
Детектирование кондуктомет-
рическое
W
10
0
мин
Рис. 10.5. Хроматограм-
ма катионов
щелочноземельных металлов и
магния [21].
Колонка Dionex
Cation, 6x250 мм.
Элюент 3 мМ HNO3/5 мМ
Zn(NO3h. Скорость
потока 3 мл/мин.
Концентрация катионов 5—
20 мкг/мл.
Детектирование кондуктометрическое
кальция в концентрированных растворах хлоридов натрия и
калия. При анализе 30%раствора хлорида натрия пределы
обнаружения катионов составляют 0,025 мкг/мл, относительное
стандартное отклонение менее 6%. Катионы щелочных и
щелочноземельных металлов определяли одновременно в морской воде [25],
используя элюенты на основе лизина и аргинина. Применяли
метод ступенчатого элюировання: изменяли pH элюента в
колонке, заполненной анионообменником в СН3СОО-форме.
Двухколоночную ионную хроматографию с кулонометрическим
детектором использовали [8] для определения кальция в
очищенных водах. Элюирование проводили смесью 2,5 мМ ж-фенилендиа-
мина с 2,5 мМ HNO3. Пределы обнаружения (мкг/мл)
составляют 0,177 для Mg 2+ , 0,329 для Са2+,'0,489 для Sr2+ и 1,169 для
Ва2+ .
161
10.2.2. Одноколоночный вариант
10.2.2.1. Кондуктометрическое детектирование
В качестве элюентов при определении щелочноземельных
металлов, как правило, используют растворы солей органических
оснований—фенилендиамина и этилендиамина. Для повышения
селективности и уменьшения времени определения в элюент вводят
комплексообразующие анионы, например цитрат, оксалат или тарт-
рат. Принципы выбора условий одноколоночного определения
щелочноземельных металлов и магния изложены в работах [26, 27].
Фритц и сотр. [9] предложили метод определения магния,
кальция, стронция и бария с 1 мМ раствором нитрата этиленди-
аммония (pH 6,1) в качестве элюента. Катионы разделяли на
поверхностно-сульфированном катионообменнике емкостью
0,017 мэкв/г. Время определения четырех катионов не
превышает 15 мин. Метод использован для определения магния и кальция
в водопроводной воде. При определении 2,4 мкг/мл кальция
относительное стандартное отклонение составило 2,6%. Хромато-
грамма водопроводной воды представлена на рис. 10.6.
Описана [28] методика определения магния и кальция в
питьевой, минеральной и речной воде с элюентом 2 мМ этилендиа-
мин/2 мМ цитрат (pH 5,0). Пределы обнаружения магния и
кальция составляют 10 мкг/л и 20 мкг/л соответственно.
Результаты определения сравнивали с данными метода комплексоно-
метрического титрования. Примеры одноколоночного определения
щелочноземельных металлов в различных водах приведены в
работах [11, 29—31].
10.2.2.2. Спектрофотометрическое детектирование
Для определения щелочноземельных металлов и магния
используют прямой и косвенный варианты спектрометрического
детектирования. Косвенное определение проводят в УФ области
спектра, а прямое — в видимой. Для прямого детектирования
используют послеколоночную спектрометрическую реакцию.
Примером использования косвенного детектирования может
служить метод определения магния и кальция в питьевой воде
[15]. Катионы разделяли на катионообменнике Zipax SCX
(матрица его — силикагель) с элюентом 2,5 мМ сульфатом меди (II).
Длина волны детектирования 220 нм. Пределы обнаружения
магния и кальция составили 0,09 мкг/мл и 0,3 мкг/мл
соответственно. Получено хорошее совпадение результатов ионохроматогра-
фического определения с данными комплексонометрического
титрования. Этот же метод использовали для определения магния и
кальция в соке овощей [16] и водных настоях [17].
Прямое детектирование использовали [32] для определения
магния и кальция в биологических жидкостях. Катионы элюиро-
вали смесью 2 мМ этилендиамин/4 мМ винная кислота.
Выходящий из колонки элюат смешивали с потоком раствора реагента
162
неоторина A50 мкг/мл в 20 мМ аммонийном буферном растворе
с pH 10) и детектировали при 570 нм. Пределы обнаружения
металлов на уровне 0,03 мгк/мл, относительное стандартное
отклонение не превышает 5%. Сделан вывод, что спектрофотомет-
рическое детектирование чувствительнее и свободнее от помех,
чем кондуктометрическое.
Для определения магния и кальция в винах и соках [33] в
качестве элюента использовали смесь 2мМ этилендиамин / 1,5 мМ
щавелевая кислота (pH 4,0) и послеколоночную реакцию с
ПАР—Zn—ЭДТА. Длина волны детектирования 490 нм.
10.2.2.3. Электрохимическое детектирование
Хаддад и др. [34] использовали для детектирования катионов
щелочноземельных металлов металлический медный электрод.
Са
8 П
MUH
Рис. 10.6. Определение
магния и кальция в
водопроводной воде
[9].
Колонка 2x340 мм с
катионообменником
емкостью 0,017 макв/г.
Элюент 1 мМ нитрат
этилендиаммония (pH
6,1). Скорость потока
1,0 мл/мин.
Детектирование
кондуктометрическое
Рис. 10.7. Хромато-
грамма катионов
щелочноземельных
металлов и магния [34].
Колонка Wescan
Cation. Элюент 1 мМ тар-
трат/1 мМ этилендна-
мип (pH 4,6). Скорость
потока 0,3 мл/мин.
Концентрация катионов
20 мкг/мл.
Детектирование потенцнометри-
ческое
Этот электрод чувствителен к ионам и молекулам, образующим
устойчивые комплексы с медью, к тартрату, цитрату и некоторым
другим. Если один из этих ионов входит в состав элюента,
детектор фиксирует сравнительно высокий фоновый сигнал. При
прохождении через детектор катиона, образующего комплекс с этим
ионом, сигнал детектора уменьшается пропорционально
концентрации катиона. При элюировании тартратом такими катионами
могут быть катионы щелочноземельных металлов. Поэтому для
определения этих катионов в качестве элюента была взята смесь
1 мM тартрат/1 мМ диэтилентриамин (pH 4,6). Катионы
разделяли на катиокообменнике фирмы Wescan. Хроматограмма
смеси катионов приведена на рис. 10.7; видно, что чувствительность
и селективность довольно высоки. Градуировочный график
линеен в диапазоне 0—40 мкг/мл.
10.3.
ПЕРЕХОДНЫЕ МЕТАЛЛЫ
10.3.1. Двухколоночный вариант
Катионы переходных и тяжелых металлов двухколоночным
вариантом определяют крайне редко: предпочтение отдают од-
ноколоночному варианту. Сложность двухколоночного
определения связана с возможностью образования малорастворимых гид-
роксидов металлов в подавляющей колонке, заполненной анио-
нообменником в ОН-форме.
При определении марганца (II), железа (II), кобальта (II),
никеля (II), меди (II), цинка (II) и кадмия (II) с кондуктомет-
рическим детектором [!9] для элюирования использовали соли
бария и свинца, а подавляющую колонку заполняли анионообмен-
ником в сульфатной форме. Селективность разделения катионов
переходных металлов невысока. Однако использование
подавляющей колонки позволяет достичь пределов обнаружения на
уровне 0,05 мкг/мл.
Выпадение осадков гидроксидов на анионообменнике в ОН-
форме можно избежать, добавляя в элюент комплексообразующие
вещества. Использование раствора тартрата этилендиаммония
[22] позволяет быстро и селективно определить медь (II), никель
(II), кадмий (II), цинк (II), марганец (II) и свинец (II).
Первые три катиона детектируют кондуктометрически в виде
положительных пиков, а остальные — в виде отрицательных. Предел
обнаружения катионов — микрограммы на литр, а относительное
стандартное отклонение не превышает 5%.
10.3.2. Одноколоночный вариант
10.3.2.1. Кондуктометрическое детектирование
Одноколоночный вариант с кондуктометрическим
детектированием, как и двухколоночный, для определения катионов пере-
164
ходных и тяжелых металлов используют очень редко. Пока этот
вариант не нашел широкого применения в анализе вод. В
качестве элюентов используют соли диаминов с комплексообразующи-
ми анионами. Было изучено влияние природы комплексообра-
зующих анионов на селективность разделения катионов
многовалентных металлов [26]. Исследование показало, что введение
тартрата или оксиизобутирата в 1,5 мМ раствор этилендиамина
повышает селективность и экспрессность определения двух- и
трехзарядных катионов d-переходных и редкоземельных
металлов. Селективность определения переходных металлов можно
повысить, добавляя комплексообразующий реагент в образец. Так,
для повышения селективности определения марганца (II) в
образец добавляли нитрилотриуксусную кислоту (pH 3,0), а в
случае цинка, железа (II) и железа (III) — сульфосалициловую
кислоту (pH 4,5) [35]. Эти рекомендации могут быть полезными
при разработке методик определения катионов металлов в водах.
Одноколоночный вариант использовали [36] для
определения алюминия в стандартном образце
воды с 8,23 мМ раствором n-фенилендиамина (рНЗ,0)
в качестве элюента. Хроматограмма стандартного
образца представлена на рис. 10.8. Определению
алюминия не мешают более чем 10-кратные
количества цинка, урана, кальция, магния, стронция,
кобальта и марганца. Относительное стандартное
отклонение не превышает 6,5%.
10.3.2.2. С пектрофото метрическое детектирование
Одноколоночный вариант определения
катионов переходных, в том числе редкоземельных, а
также тяжелых металлов со спектрофотометриче-
ским детектором наиболее распространен.
Разделенные в катионообменной колонке катионы с
помощью послеколоночной реакции переводят в
интенсивно окрашенные соединения, которые
детектируют спектрофотометрически в видимой об- ¦
ласти спектра.
Рис. 10.8. Определение алюминия в стандартном образце [36].
Колонка Wescan Cation, 2x250 мм. Элюент 8,23 мМ
n-фенилендиамин (pH 3,0). Скорость потока 0,97 мл/мин.
Концентрация алюминия 0,383 мМ. Детектирование кон-
дуктометрическое
5 3 0.
мин
Для определения микроколичеств марганца, железа, кобальта,
никеля и цинка в охлаждающем агенте первичного контура
ядерного реактора [37] катионы разделяют на колонке, заполненной
катионообменником Aminex A9, раствором винной кислоты и пе-
эеводят в окрашенные соединения с эриохромом черным Т. Для
)пределения кобальта, содержание которого ниже 0,1 мкг/л, не-
165
обходимо предварительное концентрирование. Медь, цинк,
железо, никель и марганец в соках и винах определяли f33] на
колонке Nucleosil SA 10 DX250 мм) с раствором 1,0 мМ щавелевая
кислота/2,5 мМ этилепдиамин (pH 3,5). Послеколоночным спект-
Fe
10
20
мин
Рис. 10.9. Определение
металлов в яблочном соке
[33].
Колонка Nucleosil SA=
= 10,4X250 мм. Элюент
1 мМ щавелевая
кислота/2,5 мМ этнлендиамин
(pH 3,5). Скорость потока
2,0 мл/мин.
Детектирование спектрофотометричес-
кос D90 нм) с послеко-
лоночнон реакцией с
ПАР — Zn — ЭДТА.
ть
Gd
11
18
24
MUH
Рис. 10.10. Хроматограм
ма редкоземельных
элементов [40].
Колонка Nucleosil SA=
10,4X300 мм. Элюирова-
нпе градиентное от 0,01 до
0,04 M 2-метплмолочной
кислотой (рН4,6). Скорость
потока 1,0 мл/мин.
Концентрация катионов 10 мкг/мл.
Детектирование спектро-
фотометрическое F00 мм)
с послеколоночиой
реакцией с Арсеназо-1
рофотометрическим реагентом служила смесь ПАР—Zn—ЭДТА.
Катионы детектировали при 490 нм (рис. 10.9). Результаты ио-
нохроматографического определения переходных металлов
сравнивали с данными, полученными атомно-абсорбционным методом
(табл. 10.2). Результаты удовлетворительно согласуются.
Можно упомянуть метод [38] определения железа и алюминия
в питьевой воде и пищевых продуктах. Катионы разделяли на ка-
тионообменнике Dionex CS-2, элюируя смесью 20 мМ сульфоса-
лициловая кислота/2 мМ этилендиамин (pH 5,0). Детектирова-
166
ли при 610 нм после реакции со смесью 0,01% хромазурол S/
0,4 M цетилтриметиламмоний/ 0,05% тритон Х-100 (pH 5,9).
Достигнуто хорошее разделение AI и Fe (II), но времена
удерживания Fe (III) и Al совпадают. Предел обнаружения AI и Fe (II)
1 и 2 мкг/л соответственно при объеме вводимой пробы 1 мл.
Таблица 10.2
Результаты определения (мкг/мл) переходных металлов в винах
н соках [33J методом ионной хроматографии (ИХ)
и атомно-абсорбционной спектрометрии (ААС)
Определяемый ион
Cu(II)
Zn(II)
Fe(Il)
Mn(II)
Рейнское красное
вино
ИХ
0,29
1,2
5,4
1,3
ААС
0,27
1,2
5,5
1,3
Белое вино
ИХ
0,48
0,57
4,6
7,5
ААС
0,44
0,67
3,9
8,4
Яблочный сок
ИХ
0,40
0,38
5,8
5,3
ААС
0,32
0,32
5,2
4,7
Для определения урана в технологических растворах
использовали C9] послеколоночную реакцию с 4-
B-пиридилазо)резорцином и детектирование при 520 нм. Уран отделяли на катионо-
обменнике Dionex CS-2 с элюентом 0,02, M (NH4hSO.,/0,2 M
H2SO4. Градуировочный график линеен до 15 мкг/мл, а предел
обнаружения составляет 0,04 мкг/мл. ,
Описаны методики определения d-переходных и
редкоземельных металлов иа уровне мкг/мл [40]. Катионы разделяли на
колонке Nucleosil SA градиентным элюированием. Переходные и
тяжелые металлы (Fe, Си, Zn, Ni, Со, Pb, Cd, Mn) элюировали
раствором лактата натрия (от 0,04 до 0,12 М) с послеколоночной
реакцией с ПАР—Zn—ЭДТА и детектированием при 510 нм.
Редкоземельные металлы элюировали 0,01—0,04 M раствором и 2-
метилмолочной кислоты с послеколоночной реакцией с арсеназо
I и детектированием при 600 нм. Время разделения 14 катионов
редкоземельных металлов не превышает 30 мин (рис. 10.10).
Методики характеризуются высокой воспроизводимостью
результатов — относительное стандартное отклонение не превышает
2%. Методы определения редкоземельных металлов в природных
и технологических образцах описаны в [41, 42]. Методы основаны
на градиентном элюировании с 2-оксиизомасляной кислотой,
послеколоночной реакции с ПАР и детектировании при 520 нм.
Пределы обнаружения металлов на уровне 0,1 мкг/мл, а
относительное стандартное отклонение не превышает 10%.
10.3.2.3. Электрохимическое детектирование
Для определения некоторых переходных металлов в морской
воде использовали [43, 44] косвенное амперометрическое детекти-
167
рование после разделения на колонке Dionex CS-2c элюентом
0,04 M винная кислота / 0,012 M лимонная кислота (pH 4,3).
Селективность разделения кадмия, свинца, меди (II) и железа (II)
высокая, но медь и никель в этих условиях не разделяются. Хро-
матограмма морской воды аналогична представленной на рис. 1Q.11,
Ni'
0 2 4 6 8
мин"
Рис. 10.11. Хроматограм-
ма катионов переходных
металлов [44].
Колонка Dionex CS = 2.
Элюент 0,04 M винная
кислота/0,012 M лимонная
кислота (pH 4,3). Скорость
потока 1,0 мл/мин.
Концентрация катионов 1 —
8 мкг/мл. Детектирование
косвенное амперометриче-
ское
П 18
24
мин
Рис. 10.12. Хромато-
грамма оксоанионов
[51].
Колонка Dionex Anion,
3x500 мм. Элюент
5 мМ Na2CO3. Скорость
потока 3,8 мл/мин.
Детектирование кондукто-
метрическое
но с пиком неуде ржи вающегося на колонке хлорида. Метод
позволяет определять в морской воде до 0,5 мкг/мл кадмия,
свинца и никеля, до 0,2 мкг/мл меди и 2,0 мкг/мл железа. Косвенное
амперометрическое детектирование основано на уменьшении тока
окисления пирролидиндитиокарбамата .в результате образования
устойчивого комплекса с определяемым катионом. ,
Для определения цинка, меди, никеля, кобальта и кадмия на
уровне 0,5 мкг/мл был применен [45] кулонометрический детектор.
Катионы разделяли на колонке Aminex-A4 с элюентом 0,18 M
тартрат натрия/0,04 M винная кислота/0,04 M хлорид натрия.
После разделяющей колонки в элюент вводят раствор 0|,01 M
диэтилентриаминопентаацетата меди и определяемые катионы
детектируют кулонометрически по вытесненной из комплекса
меди. Для определения катионов переходных и тяжелых металлов
можно использовать косвенное потенциометрическое
детектирование с металлическим медным электродом [46, 47].
168
10.3.2.4. Другие детекторы.
Для определения металлов помимо спектрометрического и
электрохимического можно использовать и другие детекторы. Для
определения 15 редкоземельных металлов использовали [48] атом-
но-эмиссионное детектирование с индуктивно связанной плазмой.
Пределы обнаружения катионов при объеме вводимой пробы
100 мкл составляют 0,001—0,3 мкг/мл. Катионы разделяли гради-
нетным элюированием лактатом аммония (от 0,4 до 1,0 М) при
pH 4,22. Представляет интерес метод определения марганца (II),
кобальта (II), никеля (II) и цинка (II) с косвенным атомно-аб-
сорбционным детектированием [49]. Катионы разделяли на ка-
тионообменнике Dionex CS-1 1 мМ раствором меди (II) в качестве
элюента и детектировали атомно-абсорбционным методом с
медной лампо« по уменьшению сигнала.
В работе [50] для определения кобальта (II) и марганца (II)
использовали электрокаталитнческий эффект, возникающий при
взаимодействии определяемых катионов с винной кислотой.
Разделение проводили с элюентом 0,155 M тартрат натрия/ 0,04 M
винная кислота/0,50 M нитрат натрия. Пределы обнаружения
кобальта н марганца составляют 0,003 и 0,03 мкг/мл
соответственно. Относительное стандартное отклонение не превышает 5%.
10.4.
АНИОННЫЕ КОМПЛЕКСЫ МЕТАЛЛОВ
10.4.1. Оксоанионы
Такие металлы, как молибден, вольфрам и хром, можно
определять в водах в виде оксоанионов. Молибдат, вольфрамат и
хромат относятся к сильноудерживаемым анионам, поэтому для
их разделения необходимы сильные элюенты
Для определения молибдена, вольфрама и хрома в природных
и сточных водах оксоанионы элюировали 5 мМ карбонатом
натрия [51, 52). Время определения не превышает 30 мин.
Оксоанионы определяли двухколоночным вариантом с кондуктометриче-
ским детектированием. Хромато-
грамма смеси оксоанионов
приведена на рис. 10.12.
Минимально определяемые концентрации
Таблица 10.3
Минимально определяемые
концентрации и ПДК (мкг/л)
без использования колонки для металлов, образующих оксоанионы
концентрирования при прямом
вводе пробы объемом 1,0 мл не
превышают предельно
допустимые концентрации (ПДК) метал-
лов в природных водах (табл.
10.3). Для перевода определя-
емых металлов в оксоанионы
пробу предварительно обріаба-
тывали 5%-м пероксидом водо-
7 Зак. 34
[51]
Опредетяемый
металл
Вольфрам
Хром
мин
35
25
50
ПДК
50
250
50
169
рода в щелочной среде. Результаты ионохроматографического
определения вольфрама и хрома в сточной воде хорошо
согласуются с данными химико-спектрального и спектрофотомстриче-
ского методов.
Двухколоночное определение молибдата и вольфрамата [53]
проводили на колонке Dionex Апіоп CX250 мм), используя в
качестве элюента 6 мМ раствор карбоната натрия. Метод
применили для определения молибдена и вольфрама на уровне
микрограммов в литре в природных водах. Результаты определения
хорошо согласуются с данными атомно-абсорбционного и атом-
но-эмиссионного методов. Молибден и вольфрам предварительно
концентрировали на колонке с комплексообразующим сорбентом
Chelex 100. Хромат в лигносульфонате определяли [54] двухко-
лоночным вариантом с атомно-абсорбционным детектором.
Анионы разделяли на короткой колонке DX40 мм) Dionex AS-1 с
элюентом 8 мМ Na2CC>3. Градуировочный график линеен в
интервале концентраций хромата от 2 до 10 мкг/мл. Для определения
хромата вместе с другими неорганическими анионами в речной
и водопроводной воде использовали [55J одноколоночный
вариант с кондуктометрическим детектированием. Анионы разделяли
на колонке Vydac 302 1С, применяя в качестве элюента 0.1 —
1,0 мМ раствор орто- и изофталевой кислот.
10.4.2. Комплексы с неорганическими и органическими лигандами
Ионную хроматографию использовали [56] для определения
золота, палладия и платины в виде анионных хлоридных
комплексов. Метод включает разделение анионов на колонке Dionex
AS-5 Dx250 мм) с элюентом 0,3 M NaClO4/0,05 M HC1 и спект-
рофотометрическое детектирование при 215 нм. Хроматограмма
хлоридных комплексов металлов приведена на рис. 10.13.
Пределы обнаружения металлов без предварительного
концентрирования составляют 0,03 мкг/мл для Pt 0,05 мкг/мл для Pd и
0,1 мкг/мл для Аи. Кроме благородных металлов возможно
определение железа (III) и свинца (II).
Для определения хлоридных комплексов платины, палладия
и иридия [57] использовали одноколоночный вариант с УФ
детектированием при 2Є0 нм. Анионы разделяли на колонке,
заполненной полиміетакрилатньїм анионообменником ХИКС
обменной емкостью 0,003 мэкв/г с элюентом 1 мМ сульфосалици-
ловая кислота (pH 4,2). Такой элюент имеет меньшую по
сравнению с кислотным коррозионную активность. ,
Танака [58] предложил определять кадмий, кобальт, никель,
медь и цинк в виде их анионных комплексов с ЭДТА.
Разделение проводили на колонке Dionex Anion CX500 мм) с элюентом
3 мМ NaHCO3/2,4 мМ Na2CO3 и кондуктометрическим
детектором. К пробе предварительно добавляют раствор ЭДТА. Метод
был использован для определения никеля в сточных водах и галь-
170
ванических растворах (рис. 10.14). Полученные
результаты хорошо согласуются с данными метода АЭС—ИСП.
Относительное стандартное отклонение не превышает 5%.
Рис. 10.14. Хроматограм-
ма сточной воды из
гальваническом ванны
никелирования [58].
Колонка Dionex Anion,
3x500 мм. Элюент 3,0 мМ
ЫаНСОз/2,4 мМ Na2CO3.
Скорость потока 1,9 мл/мин.
Детектирование кондукто-
метрическое
0 Ю 20 30
мин
Рис. 10.13. Хромато-
грамма хлоридных
комплексов
металлов [56].
Колонка Dionex
НР1С = AS5,4 X
Х250 мм. Элюент
0,3 M NaClO4/0,05 M
НС1. Концентрации
металлов 0,2—
5,0 мкг/мл.
Детектирование УФ B15
нм)
ЛИТЕРАТУРА
1. Lash R. Р., Hill С. J. // Anal. Chim. Asta. 1979.
2. L е g r a n M., De Angelis M., Delmas R. J.
1984. V. 156. P. 181—192.
3. F u 1 m e r M., P e n k n о t J., N a d 1 і n R. // Ion Chromatographie
Analysis of Environmental Pollutants / Ed. by J. D. Mulik and E. Sawicki. — Ann
Arbor: Ann Arbor Sei. 1979. P. 381—400.
4. Bogen D. C, N a g о u r n e y S. J. // Ion Chromatographie Analysis of
Environmental Pollutants / Ed. by J. D. Mulik and E. Sawicki. — Ann Arbor:
Ann. Arbor Sei 1979. P. 319—328.
. 5. Dionex Application Note. 1983. June, № 50.
6. Anderson С // Clin. Clietn. 1976. V. 22, № 9. P. 1424—1426.
7. M і z u о с h і M. et al. // Bunseki Kagaku. 1983. V. 32, № 6. P. 620—625.
8. T a n a k a K., I s h і h a r a Y., N a k a j і m a K- // Bunseki Kagaku. 1983.
V. 32, № 6. P. 626—631.
9. F r і t z J. S., G j e r d e D. T., В e с k e r R. M. // Anal. Chem. 1980. V. 52,
№ 9. P. 1519—1522
10. Dokij a Y., Essh о S // Anal Sei. 1986. V. 2, № 2. P 187—190.
11. Tecator Application Note. 1986. № 37.
12. Mizobuchi M. et al. // Zenkoku Kogaiken Kaishi. 1982. V. 7, № 2.
P. 67—71. (Chem. Abst. 1983. V. 98, № 185225).
V. 108. P. 405—409.
// Anal. Chim. Acta.
171
13. Smith R. L., Pietrzyk D. J. // Anal. Chem. 1984. V. 56, № 4.
P. 610—614.
14. Hayakawa K-, H ira к і H., Miyazaki M. /'/ Bunseki Kagaku.
1983. V. 32, № 8. P. 504-505.
15. Miyazaki M. Hayakawa K., Choi S. G. // I. Chromatogr 1985.
V. 323. P. 443—446.
16. H a y a к a w a K. et al. // Bunseki Kagaku. 1984 V. 33 № 7. P. 390—392
17. I s h і к a w a M. et ai. // Bunseki Kagaku. 1986. V. 35 № 3. P. 309—311.
18. Foiey R. C. L, Haddad P. R. // J. Chromatogr. 1986. V. 366.
P. 13—26.
19. Nordmeyer F. R. et ai. //'Anal. Chem. 1980 V. 52, № 6 P. 852—856.
20. L a m b J. D. // Anal. Chem. 1981. V. 53, № 4. P. 749—750.
21. Wim be r ley J. W. // Anal Chem. 1981 V. 53 № 13 P 2137—2138.
22. Slip і gun О. A., et ai. // Anal. Cliim. Acta 1985. V. 172 P 341—346.
23. Wimberley J. W. // Anal. Chem. 1981. V. 53, № 11. P. 1709—1710.
24. Не n r y L. E. // Abstracts Pittsburgh Conference on Analytical Chemistry
and Applied Spectroscopy. — Atlantic City. 1986. Nb 983.
25. Шпигун О. А., Вол ощик И. Н., Золото в Ю. А. // Ж- аналит.
химии. 1988.
26. Se ven ich G J., Fritz J. S. // Anal. Chem. 1983. V. 55 № 1.
P. 12—16.
27. Y a n D. R, S с h w e d t G. // Anal. Chim. Acta. 1985. V. 178. P. 347—353.
28. Yan D. R., Schwedt G. // Fresenius Z. Anal. Cliem. 1985. B. 320, № 2.
S. 121 — 124.
29. S с h o 1 1 e r F., О 1 1 r a m F. // osterr. Wasserwirt. 1983. В. 35, № 3—4.
§ 73 77
30. В' e h n e r t J., В e h r e n d P. // Lab. Prax. 1984. V. 8, № 11. P. 1204—1208.
31. J u p і 1 I e T.. В u r g e D.. T о g a m і D. // Res. and Develop. 1984. № 3.
P. 135—139.
32. Nagashima H. // Bunseki Kagaku. 1986. V. 35, № 1. P. 7—11.
33. Y a n D R., S t u m p p E., S с h w e d t G. // Fresenius Z. Anal. Chem.
1985. B. 322, № 4. S. 474—479.
34. Haddad P. R., A 1 e h a n d e r P. W., T r о j a n о w і с z M. // J.
Chromatogr. 1984. V. 294. P. 397—402.
35 Seve nie h G J., Fritz J S. // J. Chromatogr. 1985. V. 347.
P. 147—154.
36. Fort і er N. E., Fr it z J. S. // Talanta. 1985. V. 32, № 11. P. 1047—1050.
37. Jones P., В a r r о n K-, E b d о n L. // Anal. Proc. 1985. V. 22, № 12.
P. 373—375.
38 Yan D R S с h w e d t G. // Fresenius Z. Anal. Chem. 1985. B. 320,
№ 2. S. 252—257.
39. В y e r I e y J. J., S с h a r e r J. M., Atkinson G. F. // Analyst. 1987.
V. 112, № 1. P. 41—47.
40. Wang W., С h e n Y., W u M. // Analyst. 1984. V. 109. № 3. P. 281—286.
41. M a z z u с о t e 1 І і A. et al. // J. Chromatogr. 1985. V. 349. P. 137—142.
42. T і e 1 r о о y J. A., К r a a к J. C, M a e s s e n F. J. // Anal. Chim. Acta.
1985. V. 176. P. 161—174.
43. Hojabri H., La vin A. G, W a 11 a с e G. G. // Anal. Proc. 1986.
V. 23, № 1. P. 26—27.
44. H oj a b ri H. et. al. // Anal. Chem. 1987. V. 59 № 1. P. 54—57.
45. G і r a r d J. E. // Anal. Chem. 1979. V. 51, № 7. P. 836—839.
46. Alexander P. W., Haddad P. R., T r о j a n о w і с z M. // Anal.
Chim. Acta. 1985. V. 177. P. 183—195.
47. Haddad P. R., Alexander P. W., T r о j a n о w і с z M. // J.
Chromatogr. 1985. V. 324, № 2. P. 319—332.
48 YoshidaK- HaraguchiH. // Anal. Chem. 1984. V. 56, № 13,
P. 2580—2585.
49 M a к e t о n S., О t t e r s о n E. S., T a r t e r J G. // J. Chromatogr.
1986. V. 368. P. 395—400.
\-,2
50. Heuser J. R, Girard J. E. // Anal. Chem. 1985 V. 57, № 14.
P. 2847—2850.
51. Зо лотов Ю. А., Шпигун О. А., Бубчнкова Л. А. // Двкл. АН
СССР. 1982. Т. 266, Я? 5. С. 1144—1147.
52. Z о 1 о t о v Y и. A., S h р і g u п О. A., Bubchikova L. A. //
Fresenius Z. Ana]. Chem. 1983. B. 316, № 1. S. 8—12.
53. Ficklin W. H. // Anal. Lett. 1982. V. AI5, № 10. P. 865—871.
54. Pet ter sen J. M. // Anal. Chim. Acta. 1984. V. 160. P. 263—266.
55 G I a t z J. A., Girard J. E. // J. Chromatogr. 1982. V. 20, № 6.
P. 266—273.
56. Rockli n R, D. // Anal. Chem. 1984. V. 56. № 11. P. 1059—1062.
57. Шпигун О А., Па зу хина Ю. Э. // Ж. аналит. химии. 1987. Т. 42,
№ 7. С. 1285—1289.
58 Т a n a k а Т /' Fresenius Z. Ana]. Chem. 1985. В. 320, № 2. S. 125—127.
ГЛАВА 11.
ОПРЕДЕЛЕНИЕ
АЛИФАТИЧЕСКИХ АМИНОВ
Алифатические амины являются токсичными веществами, что
обусловливает необходимость контроля их содержания в водах.
Ионную хроматографию для этой цели пока не использовали.
Поэтому в данной главе обсуждаются аналитические
характеристики методик ионохроматографического определения
алифатических аминов в других объектах. Эти сведения могут быть
полезными при создании методик определения аминов в водах.
11.1.
УДЕРЖИВАНИЕ АМИНОВ НА КАТИОНООБМЕННИКАХ
НИЗКОЙ ЕМКОСТИ
Алифатические амины — слабые основания, поэтому для их
определения используют сильнокислотные (рН<3) элюенты. В
таких элюентах амины протонированы и находятся в виде ка-
Таблица 11.1
Время удерживания (мин) некоторых алифатических аминов.
Колонка — Dionex Cation Fx250 мм); элюент — 10 мМ
и различные количества метанола; F=l,53 мл/мин [1]
К.ітиоі
Калия
Этаноламипа
Днэтанодямина
Метиламина
Этиламина
Триэтаноламина
Тримьтиламииа
Днэтиламнна
Триэтиламина
(-одер
0
16,16
15,26
16,50
16.84
18,66
19,29
23,13
26,88
39,66
i:aii!ie метабола
10
16,61
15,21
16,11
16,53
17,92
18,18
21,61
24,07
31,68
20
18,23
15,61
16,51
16,86
17,91
17,93
21,55
23,19
28,15
174
тионов, которые разделяют на катионообменниках низкой
емкости.
Катионы алифатических аминов относятся к средне- и сильно-
удерживаемым. В одинаковых условиях их времена
удерживания, как правило, больше, чем время удерживания катиона
калия [1]. Удерживание аминов на катионообменниках низкой
емкости зависит от структуры и числа алифатических радикалов
(табл. 11.1). Чем больше число радикалов и чем они гидрофоб-
нее, тем сильнее амин удерживается на катионообменнике.
Поэтому при использовании кислотных элюентов третичные амины
удерживаются сильнее вторичных и первичных. С увеличением
числа углеродных атомов в радикале растет его гидрофобность,
поэтому этиламин
удерживается сильнее метиламина.
Введение в радикал гидроксогруппы
уменьшает его гидрофобность,
йрэт,ому время удерживания
этанол-амина меньше, чем эти-
ламина. Зависимость
удерживания аминов от гидрофобностп
алифатических радикалов
свидетельствует о вкладе неионо- ,
обменного сорбционного взаи- [/\
модействия в механизм
удерживания. Введение метанола
в элюент уменьшает
неионообменную сорбцию аминов и
их времена удерживания.
Селективность разделения
аминов изменяется при
введении в элюент гидрофобных
органических катионов [2]. Так,
при использовании в качестве
элюента раствора хлорида о 8 Ш
бензилтриметиламмония (рис.
11.1) наблюдается обращение
порядка элюирования
первичных, вторичных и третичных
аминов по сравнению с
сильнокислотным элюентом. Сле-
П
мин
Рис. 11.1. Хроматограмма алифатичес-
ких аминов [2].
/ — триметиламин; 2 — диметиламин;
3 — метиламин. А — колонка Dionex
довательно, селективность on- HP1C-CS2, 4,6X250 мм. Элюент 7,5 мМ
ределения аминов можно ре- бензилтриметиламмоний хлорид.
Скорость потока 0,5 мл/мин. Детектирова-
гулировать введением в элю-
ние косвенное УФ B75 нм)
ент гидрофобных добавок. Это ? _колонка Оіойех HPIC-CS2, 4,бх
особенно важно при
определении вторичных и третичных
аминов.
Х250 мм. Элюент 10 мМ НС1. Скорость
потока 1,0 мл/мин. Детектирование
кондуктометрическое
175
11.2.
ИОНОХРОМАТОГРАФИЧЕСКОЕ ОПРЕДЕЛЕНИЕ
Метиламин, диметиламин и триметиламин определяли [3] в
атмосфере двухколоночным методом с кондуктометрическим
детектором. Амины разделяли на колонке Dionex Cation FX250мм),
используя в качестве элюента 2,5 мМ раствор азотной кислоты.
Градуировочный график линеен в интервале концентраций 5—
25 мкг/мл, относительное стандартное отклонение не
превышает 5%. Амины сорбировали на фильтре, импрегнированном
щавелевой кислотой и глицерином, а затем десорбировали водой.
Тетраметиламмоний определяли в моллюсках [4] двухколоночным
методом на двух последовательно соединенных колонках Dionex
CS-2 Dx50 и 4X250 мм) с элюентом 10 мМ НС1. Предел
обнаружения амина составляет 5 мкг/мл, относительное
стандартное отклонение 4,4%. Двухколоночный вариант использовали [5]
для определения этилендиамина в медицинских препаратах. Амин
определяли на колонке Dionex Cation DX50 мм) с элюентом
4 мМ НС1/2,5 мМ ZnCl2. Подавляющую колонку заполняли
анионообменником в ОН-форме. При этом этилендиамин
детектировали кондуктометрически в виде комплекса с цинком.
Минимально определяемая концентрация амина менее 1 мкг/мл, а
относительное стандартное отклонение составляет 2,8%.
Предложен одноколоночный вариант определения
алифатических аминов с косвенным УФ детектированием при 275 нм [2].
Амины разделяли на колонке Dionex CS-2 D,6x250 мм) с 7,5 мМ
раствором хлорида бензилтриметиламмония в качестве элюента.
Пределы обнаружения восьми изученных аминов колеблются от
0,5 до 4,9 мкг/мл. Градуировочный график линеен в диапазоне
концентраций 10—3000 мкг/мл, относительное стандартное
отклонение равно 0,6%'¦
Для определения аминов с
кулонометрическим детектором
использовали [6] реакцию
окисления гидрохинона в щелочной
среде. Пределы обнаружения
аминов составляют 0,06V—
0,32 мкг/мл (табл. 11.2).
Градуировочный график линеен в
интервале концентраций 1 —
200 мкг/мл. Амины разделяли
на анионообменнике высокой
емкости Hitachi 2632 в ОН-форме, а
элюентом служила вода или смесь
воды—40%-й ацетон. Разделение
(Основано на адсорбции аминов
матрицей анионообменника.
Таблица 11.2
Пределы обнаружения
алифатических аминов
с кулонометрическнм детектором
F1
Амин
Метиламин
Этиламин
н-Пропиламин
Изопропиламин '
н-Бутиламин
Диэтиламин
н-Амиламин
G-Гексиламин
н-Гептиламин
Триэтиламин
н-Октиламин
Предел
обнаружения.
мкг/мл
0,06
0,П
0,16
0,16
0,19
0,23
0,24
0,26
0,29
0,30
0,32
176
ЛИТЕРАТУРА
1. Buechele R. С, Re utter D. J. // J. Chromatogr. 1982. V. 240.
P. 502—507.
2. Me Aleese D. L. // Anal. Chem. 1987. V. 59, № 3. P. 541—543.
3. К і і u n е J., О і к a w а К. // Bunseki Kagaku. 1979. V. 28, № 7. P. 587—
590.
4. S а і t oh H. et al. // J. Chromatogr. 1983. V. 281. P. 397—402.
5. Buechele R. C, Reut ter D. J. // Anal. Chem. 1982. V. 54, № 11.
P. 2113—2114.
6. T a n a к a K., Ishizuka T., S u n a h a r a H. // J. Chromatogr. 1979.
V. 172. P. 484—489.
ГЛАВА 12
СОВМЕСТНОЕ ОПРЕДЕЛЕНИЕ
КАТИОНОВ И АНИОНОВ
Ионная хроматография позволяет в ряде случаев совместно
определять катионы и анионы. Предложены некоторые методики
такого определения. Использование этих методик может
существенно сократить время определения катионов и анионов, что
особенно важно при выполнении рутинных анализов вод. Однако
это направление, несмотря на его перспективность, развивается
медленно, ему посвящено лишь несколько работ.
12.1.
ИСПОЛЬЗОВАНИЕ ПОСЛЕДОВАТЕЛЬНО СОЕДИНЕННЫХ КОЛОНОК
Такое определение обычно проводят на последовательно
соединенных катионной и анионной разделяющих колонках с элю-
ент^м, способным разделять как катионы, так и анионы. Для
совместного определения Na + , К+ , Mg2+ , Са 2+, F~ , С\~
Рг ~ , NO^~ и SOI"" использовали [1] хроматографическую
систему, состоящую из катионной разделяющей колонки,
кондуктометрического детектора, анионной разделяющей колонки,
анионной подавляющей колонки и кондуктометрического или амперо-
метрического детектора. Однозарядные катионы и анионы элюи-
ровали смесью 1,6 мМ Li2CO3/2,4 мМ CH3COOLi (pH 10,4), а
двухзарядные — 3,3 мМ фталатом меди. Два детектора
позволяют одновременно фиксировать катионы и анионы (рис. 12.1).
Определение однозарядных ионов длится менее 15 мин, а двух-
зарядных — около 30 мин. Предел обнаружения большинства
ионов не превышает 2 мкг/мл. Метод использовали для анализа
водопроводной и дождевой воды. Результаты определения
кальция и магния хорошо согласуются с данными комплексонометри-
ческого титрования.
Для совместного определения большого числа одно- и двух-
зарядных катионов и анионов использовали [2] колонку,
заполненную смесью катионообменника и анионообменника, и кондук-
тометрический детектор. В зависимости от удерживания
определяемых ионов элюировали разбавленными растворами ацетата
и фталата лития, а также фталатом этилендиаммония.
178
Использование двух последовательно соединенных колонок,
дифференциального кондуктометрического детектора и 4 мМ гид-
рофталата лития в качестве элюента позволило [3] совместно
!
2,00
1,50
1,00'r
0,50
О
9,0
6,0
ПА
3,0
О
Вг
J L
0,5
0
' 6,0
3,0
\ 1,5
-NO,
8 16 24
О в
16 24 32
мин
Рис. 12.1. Хроматограмма водопроводной воды [1].
Скорость потока 1,5 мл/мин.
а — двухэарядиые катионы и анионы: колонки —
Vydac 302 1С, 4,6x250 мм и Dionex HPIC-CSI,
4x200 им. Элюент 3,3 v.M. фталат меди.
Детектирование кондуктометрпческое.
б — однозарядные катионы и анионы- колонки —
Dionex HPIC-AS3, 4ХІ50 мм и Dionex HP1C-CSI,
4X200 мм. Элюент 1,6 мМ Li2CO3/2,4 мМ CH3COOLi
Детектирование кондуктометрпческое и амперометрн-
ческое.
Концентрации ионов 0,7—44 мкг/мл
определить Na+,
K+ , Cl- , NO,-
и SO|-
.
Чувствительность определения менее 1 мкг/мл, а относительное стандартное
отклонение не превышает 2%. Время определения шести ионов
не более 15 мин.
Для совместного определения Na+, NH+, К+, Cl-, ~NO-, NO-,
Вг - и SO|- использовали [4] косвенное УФ детектирование
при 240 и 270 им. Ионы разделяли на двух
последовательно соединенных анионной и катионной разделяющих колонках,
17«
а элюентом служил 0,5 мМ раствор о-сульфобензоата меди. При
объеме пробы 100 мкл пределы обнаружения (мкг/мл)
составляют 1,32 для Cl-, 0,69 для NH + , 0,90 для К+ и 1,10 для SO<~.
12.2.
ДРУГИЕ ВАРИАНТЫ
В работе [5] Cl- , NO2-, NO3~, H2PO4-, SO2- , Са2+ и Mg2+
определяли на анионной колонке, заполненной сорбентом TSK
GEL IC-Anion-SW с кондуктометрическим детектором. Ионы элюи-
ровали раствором 1 мМ ЭДТА (pH 6,0). Анионы определяли по
положительным пикам, а кальций и магний в виде анионных
комплексов с ЭДТА — по отрицательным пикам (рис. 12.2).
Пределы обнаружения (мкг/мл) для С1~ 0,04,
для NO2-. NO3-, Mg2+ и Са2+ 0,05, для
НгРО;^ и SO2 0,1. Метод использовали
для анализа водопроводной, дождевой и
морской воды (табл. 12.1). Морскую воду
предварительно разбавляли в 100 раз.
10 15 20
мин
Рис. 12.2. Хроматограмма водопроводной воды
[5].
Колонка TSK GEL IC-Anion-SW, 4,6X50 мм.
Элюент 1 мМ ЭДТА (pH 6,0). Скорость потока
1,0 мл/мин. Детектирование кондуктометрическое
Таблица 12.1
Результаты совместного определения катионов н анионов
в водах (мкг/мл) [5]
Вода
Дождевая
Водопроводная
Cl
2,
4,
5
6
NC
1
0
з-
,1
,9
so-
0,38
0,43
Ca
0
7
,9
,7
0,5
0,9
ЛИТЕРАТУРА
1. J о п е s V. К., Т а г t е г J. G. // Int. Lab. 1985. № 9. P. 36—45.
2. Pietrzyk D. J., Brown D. M. // Anal. Chem. 1986. V. 58, № 12.
P. 2554—2557.
3. Tarter J. G // J. Chromatogr. 1986. V. 367, № 1. P. 191—194.
4. Iskandarani Z., Miller T. E. // Anal. Chem 1985. V. 57, № 8.
P. 1591—1594.
5. Yamamoto M. et al. // Anal. Chem. 1984. V. 56, № 4. P. 832—834.
180
ГЛАВА ІЗ
ИОНОХРОМАТОГРАФИЧЕСКИЙ АНАЛИЗ
ВОД РАЗЛИЧНОГО ТИПА
13.1.
ВОДЫ КАК ОБЪЕКТ АНАЛИЗА
По увеличению концентрации ионных компонентов можно
выделить следующие типы вод: высокочистые, атмосферные осадки,
водопроводные и питьевые, поверхностные, грунтовые,
высокоминерализованные подземные, морские. Особое место занимают
сточные воды, состав которых может быть самым разнообразным.
Воды разные типов отличаются и содержанием других
компонентов, часто очень сильно. Так, особенностью поверхностных
пресных вод является наличие в них относительно высоких
концентраций органических веществ, прежде всего гуминовых и
фульвокислот. Эти вещества обладают комплексообразующей
способностью по отношению к ионам металлов и иногда
выступают в качестве восстановителей.
Для определения ионного состава вод ионная
хроматография — прекрасный метод; особенно это относится к анионному
составу. С другой стороны, воды служат идеальным
анализируемым объектом для ионной хроматографии. Из-за различия
качественного и количественного состава вод универсальной ионо-
хроматографической методики их анализа не существует.
Условия определения ионных компонентов в водах зависят от
концентрации ионов, их соотношения, наличия мешающих веществ
и ряда других факторов.
13.2
ОТБОР И ПОДГОТОВКА ПРОБЫ
Правила отбора проб для ионохроматографического. анализа
не отличаются от общих правил отбора проб воды. Это
относится, видимо, и к условиям консервации проб, хотя большинство
компонентов, определяемых ионной хроматографией,
достаточно стабильно в условиях хранения и транспортировки.
При ионохроматографическом определении ионов в водах
требуется минимальная подготовка пробы, что обеспечивает
быстроту анализа, его высокую производительность. Обычно
подготовка образца воды для ионной хроматографии заключается в его
очистке от взвешенных частиц, способных засорить тонкие
трубопроводы хроматографа, и удалении гидрофобных органических
181
веществ, отравляющих разделяющий сорбент. Для этого
образец пропускают через сорбент, содержащий щетки Ci8, a затем
через пористый фильтр @,45 мкм). Однако эта процедура
может привести к загрязнению образца [1] ионами свинца,
хлорида, нитрата и сульфата. После пропускания деионизованной
воды через щеточный сорбент Waters Assoc (Milford, MA, USA)
Сів Sep-Pak содержание ионов в воде составляло 30—70 мкг/л.
После прохождения воды через фильтры Millipore (Bedford, MA,
USA) 0,45 мкм Millex НА и HV в ней найдено 13,2 мкг/л хлорида
и 8,7 мкг/л сульфата. Это надо учитывать при анализе
высокочистых вод.
13.3.
ВЫСОКОЧИСТЫЕ ВОДЫ
К высокочистым относят воды, содержание ионов в которых
ниже 1 мкг/л. Анализ таких вод требует очень высокой
чувствительности, которую не может обеспечить обычная ионная
хроматография. Поэтому для повышения чувствительности либо
увеличивают объем вводимой пробы [2, 3], либо используют
колонку для концентрирования [4—7].
Обычно объем петли-дозатора равен 0,1 мл, но при анализе
высокочистых вод его увеличивают до 6 мл, при этом
пропорционально увеличивается чувствительность определения. Но
следует помнить, что увеличение объема вводимой пробы приводит
к увеличению «водного пика», мешающего определению слабо-
удерживаемых анионов (F~, ацетат). Для устранения «водного
пика» можно использовать метод обратной промывки [2].
В качестве колонки для концентрирования используют
короткую E0 мм) разделяющую колонку. Эта колонка через шести-
ходовой кран соединена с разделяющей. Анализируемую воду с
помощью насоса пропускают через колонку для концентрирования.
Объем пропущенной воды зависит от чувствительности
определения. Если необходимо повысить чувствительность в 100 раз,
то пропускают 10 мл во,ды, обычно со скоростью 2—4 мл/мин.
Ионы, сорбированные в колонке для концентрирования, затем
десорбируют элюентом и направляют в разделяющую колонку.
При анализе высокочистых вод используют как двухколоноч-
ный, так и одноколоночный вариант. При двухколоночном
определении анионов элюентом служит карбонатно-бикарбонатный
раствор, концентрация которого зависит от природы
определяемых анионов. При одноколоночном определении анионы элюи-
руют растворами фталевой или других ароматических кислот.
Элюентом для двухколоночного определения однозарядных
катионов служит раствор минеральной кислоты (соляной или азотной).
Другие условия анализа высокочистых вод не отличаются от
условий анализа других объектов (см. гл. 8—10).
Примеры использования ионной хроматографии для анализа
высокочистых вод приведены в табл. 13.1.
182
В качестве примера ионохроматографического анализа особо
чистой воды можно рассмотреть методику определения Cl- и
SO?— в конденсате пара одноколоночной ионной
хроматографией [6]. Поскольку речь идет об анализе очень чистого образца,
Таблица 13.1
Примеры ионохроматографического анализа высокочистых вод
Определяемые
ионы
СГ, SO;j-, Na+, K+
В виде BF7
СГ, POJj", NOJ",
4
NOr. SO?"
О 4
er, SO4-
CO2"
3
F", СГ, NO-,
NO-, Br-, S02-
Na+, K+, NH+ ,
СГ, NO3~, SO|~
Na+, NH+, СГ,
NO3-, SO2-
Fe(III). Cti(II), Z
(II), Со A1), Fe(ll)
Fe(III), Cu(ll
N1A1), Zn(ll), Fe(ll)
Вариант
вухколо-
ОЧНЫЙ
то же
щ
одноколо-
ночный
то же
двухколо-
ночный
то же
одкоколо-
ночиый
одноколо-
ночный
Элюент
7,5 мМ НС1;
3 мМ NaHCO3/
2,4 мМ Na2CO3
3 мМ NaHCO3/
4 мМ Na2CO3
3 мМ NaHCO3/
/2,4 мМ Na2CO3
,8 мМ NaHCO3/
2,2 мМ Na3CO3
0,2 мМ фталат
калия, pH 6,2
2 мМ фталевая
кислота, pH 6,8
1 мМ фталат
натрия, pH 5,1
5 мМ НСІ;
2,5MMNaHCO3
/2 мМ Na,CO3
4 мМ HNO3;
2 м M Na3CO3
0,1 "Л тартра і-,
pH 3,05
пиридин-дикар
боновая кислот
(
Детектор
ондукто-
етрический
То же
УФB31нм)
кондукто-
меіричес-
кий, УФ
B70 им)
кондукто-
метрический
то же
СФ с ПАР
СФ с ПАР
Способ
повышения
чувствительности
А
А
А
А
А
—
В
в
в
А
А
Лите-
затура
[8]
[91
[41
[51
161
[10]
[3]
[И]
[2]
V 1
1121
А—использование колоржи для концентрирования;
В —. увеличение объема вводимой пробы.
то необходима предельная осторожность при выполнении всех
операций. Любая неосторожность может привести к загрязнению
образца и искажению результатов анализа. Важно, чтобы
образец как можно меньше времени находился в контакте с
воздухом. Поэтому образец быстро помещают в полистирольную или
пропиленовую бутылку, которую плотно закрывают. В таком
контейнере конденсат не загрязняется. Нужный объем конденсата
(около 20 мл) со скоростью 2,5 мл/мин пропускают через
колонку для концентрирования B,5X30 мм), заполненную анионооб-
менником XAD-4 емкостью 0,5 мэкв/г. Образец пропускают с
183
помощью насоса для ввода пробы. Использование шприца
нежелательно из-за возможности загрязнения образца. Для удаления
примесей перед колонкой для концентрирования устанавливают
предколонку, заполненную катионообменником Dowex 50X8 в
Н-форме. Затем через колонку для концентрирования
пропускают элюент @,2 мМ фталат калия, pH 6,2). Скорость подачи элю-
ента 1 мл/мин. Элюент направляют в разделяющую колонку
BX150 мм), заполненную анионообменником ХАД-1 емкостью
0,013 мэкв/г. Для устранения необратимой сорбции ионов
образца в колонке для концентрирования элюент пропускают через
нее в направлении, обратном пропусканию образца. Для этого
перед пропусканием элюента соединения колонки для
концентрирования с 6-ходовым краном меняют местами. Кондуктометри-
ческое детектирование проводят на шкале 0,1 mkS. Эта методика
позволяет за 30 мин определять в конденсате менее 0,02 мг/л
хлорида и сульфата. Элюент и стандартные растворы готовят
растворением соответствующих высокочистых солей или кислот
в воде с сопротивлением 18 МОм.
13.4.
ПИТЬЕВАЯ И ВОДОПРОВОДНАЯ ВОДЫ
Концентрация большинства ионов в питьевой и
водопроводной водах равна 1—10 мг/л. Концентрации хлорида и сульфата в
некоторых образцах достигают 100 мг/л. Для определения таких
концентраций ионов можно использовать как двухколоночную,
так и одноколоночную ионную хроматографию с кондуктометри-
ческим, УФ и электрохимическим детектированием. Объем
вводимой пробы 0,1 мл достаточен для определения большинства
ионов. Условия определения анионов и катионов в этих
образцах зависят от природы определяемых ионов и рассматриваются
в гл. 8—10. Как и в случае анализа других вод, обязательным
является фильтрование образца и элюента через пористый
0,45-мкм фильтр. Предварительное концентрирование или
повышение чувствительности и селективности другими способами
требуются только для определения ионов, концентрация которых
ниже 0,1 мет/мл [13].
Примеры использования ионной хроматографии для анализа
питьевой и водопроводной вод приведены в табл. 13.2.
Для анализа питьевой и водопроводной воды используют
стандартные ионохроматографические методики. Примером
такой методики может служить определение кальция и магния в
питьевой воде одноколоночной ионной хроматографией с кон-
дуктометрическим детектированием [25]. Анализируемые катионы
разделяют на колонке Dionex CS-2 DX250 мм) с элюентом 2 <мМ
этилендиамин / 2 мМ лимонная кислота (pH 4,0). Скорость
подачи элюента 1,5 мл/мин. В этих условиях время удерживания
более удерживаемого кальция около 6 мин. Объем вводимой про-
184
ды [51] его окисляли с помощью NaCIO до брома, который затем
экстрагировали ССЦ. Выделенный бром восстанавливают
кислым раствором Н2О2 до бромида и определяют ионной
хроматографией.
Для определения состава морской воды используют как одно-
колоночный, так и двухколоночный вариант ионной
хроматографии. Образцы перед анализом фильтруют через пористый 0,45-мкм
фильтр.
Примеры использования ионной хроматографии для анализа
морской воды и близких ей по составу образцов представлены
в табл. 13.6.
Таблица 13.6
Примеры ионохроматографического анализа морской воды и близких ей
по составу образцов
Определяемые
ионы
Вт-, NO3-
Вг-
F~
Mg2+, Ca2+
50^ , MgZT,
Са2+
F~, СП, Вг~,
SO?,", L1+, Na+,
Вариант
ОДИОКОЛОНОЧНЫЙ
двухколоночный
оді:околоноч-
кый
одноколокоч-
ный
двухколоночный
одноколоноч-
ный
двухколоноч-
ный
Элюент
5 мМ ортофталат.
pH 3,9
ЗмМ NaHCO3/2,4 мМ.
Na2CO3
2,5 мМ фталевая
кислота, pH 4,0
0,1 M NaNCyO.OlM
NaHaPOa/0,01M
Na2hPO4
Аминокислота
1мМ ЭДТА
3 мМ HNO3; 2 мМ
NaHCOs/1.6mM
NaXO,
Детектор
косвенный УФ
B98 нм)
кондуктомет-
зический
то же
потенциометри-
ческий
кондуктомет-
рический
то же
я
Литература
[42]
151]
[21]
[16]
152]
[28]
[49]
13.8.
СТОЧНЫЕ ВОДЫ
Ионный состав сточных вод зависит от профиля предприятия,
выбрасывающего эти воды. Поэтому задача
ионохроматографического анализа сточной воды должна решаться для каждого
образца отдельно. Обычно содержание ионных компонентов в
сточных водах довольно высоко, поэтому надо помнить о возможности
перегрузки разделяющей колонки.
Имеются сведения об использовании ионной хроматографии
для определения в сточных водах хлорида, бромида, нитрита и
нитрата [46], азота и фосфора [53], хрома и вольфрама [54], нит-
191
рата, фторида, хлорида, фосфата и сульфата [55], никеля,
хлорида, нитрата и сульфата [561.
ЛИТЕРАТУРА
1. В a geh і R. H a dd a d P. R. // J. Chromatogr. 1986. V. 351. P. 541 —
547.
2. IveyJ. P., DaviesD. M. // Ana]. Chim. Acta. 1987. V. 194. P. 281—
286.
3. Oka da Т. К u w a m о t о Т. // J. Chromatogr. 1985. V. 350. P. 317—323:
4. Wetzel R. A. et al. // Anal. Chem. 1979. V. 51, X« 9. P. 1532—1535.
5. Weiss J., G о Ы M. // Fresenius Z. Anal. Chem. 1985. В 320, №4.
S. 439—444.
6. Roberts К. M., Gjerde D. T. Fritz J. S. // Anal. Chem. 1981.
V. 53, Ks 11. P. 1691—1695.
7 Y a n D. R., S с h we d t G. // Fresenius Z. Anal. Chem. 1987. B. 327, № 5.
S. 503—508.
8. F u 1 m e r M., Penknot J., N a d 1 і n R. // Ion Chromatographie
Analysis of Environmental Pollutants / Ed. by J. D. Mulik and E. Sawicki. — Ann
Arbor: Ann arbor Sei. 1979. P. 381—400.
9. Hill С J., Lash R. P. // Anal. Chem. 1980. V. 52, № 1. P. 24—27.
10. Brandt G. M a t u s с h e k G., Ke 11 r u p A. // Fresenius Z. Anal.
Chem. 1985. B. 321, № 5. S. 653—654.
U Leg r and M., De Ange lis M., Del ma s R. J. // Anal. Chim.
Acta. 1984. V. 156. P. 181—192.
12. F r a n k 1 і n G. O. // Amer. Lab. 1985. № 5. R. 65—79.
13. H oo ver T. В., Yager G. D. // Anal. Chem. 1984. V. 56, № 2.
P. 221—225.
14. Oka da T., Kuwamoto T. // Anal. Chem. 1985. V. 57, № 1. P. 258—
262.
15. Yan D. R., Rossner В., S eh we dt G. // Anal. Chim. Acta. 1984.
V. 162. P. 451—455.
16. Butler E. C. V, G e r s h e y R. M. // Anal. Chim. Acta. 1984. V. 164.
P. 153—161.
17. Fie kl in W. H. // Anal. Let. 1982. V. A15, № 10. P. 865—871.
18. Brandt G., К e t t r u p A. // Fresenius Z. Anal. Chem. 1985. B. 320,
№ 4. S. 485—489.
19. Tecator Application Note 1986 ASTN 4D/86. 861015.
20. Golombek R., S с h w e d t G. // J. Chromatogr. 1986. V. 367.
P. 69—76.
21. Oku t a n і T., Tanaka M // Bunseki Kagaku. 1987. V. 36, № 2.
P. 169—173.
22. M u 1 1 і n s F. C. P. // Analyst. 1987. V. 112. P. 665—671.
23. H о s h і k a Y., M u r a y a m a N., M u t о G. // Bunseki Kagaku. 1987.
V. 36, № 3. P. 174—178.
24. Fritz J. S., Gjerde D. T., Becker R. M. // Anal. Chem. 1980.
V. 52, № 9. P. 1519—1522.
25. Yan D. R., Schwedt G. // Fresenius Z. Anal. Chem. 1985. B. 320,
№ 3. S. 252—257.
26. Yan D. R., Schwedt G. // Fresenius. Z. Anal. Chem. 1985. B. 320,
№ 2. S. 121 — 124.
27. J о n e s V. K, T a r t e r J. G. // Int. Lab. 1985. № 9. P. 36—39.
28. Yam am ot о M et al. // Anal. Chem. 1984. V. 56 № 4 P. 832—834.
29. H e r n J. A. Rutherford G. К., V a n L о о n С W. // Talanta.
1983. V. 30, № 9. P. 677—682.
30. Wagner F., V a 1 e n t a P., Nurnberg H. W. // Fresenius Z. Anal.
Chem. 1985. B. 320, № 4. S. 470—476.
31. Crow t her J., Me В ri de J. // Analyst. 1981. V. 106. P. 702—709.
32 In focus. The Tecator Journal of technology for chemical analysis 1986.
V. 9, № 12. P. 12—13.
92
33. О і к a w а К., Saitoh H. // Bunseki Kagaku. 1983. V. 32, № 6.
P. T105—T109.
34. I s h і g a m і A. et al. // Bunseki Kagaku. — 1986. V. 35, № 10. P. 944—
946.
35. P yen G. S., Erdmann D. E. // Anal. Chim. Acta. 1983. V. 149.
P. 355—358.
36. T a n a к a S. et al. // Bunseki Kagaku. 1986. V. 35, № 7. P. 465—470.,
37. Da vies D. M., Ivey J. P. // Anal. Chim. Acta. 1987. V. 194.
P. 275—279.
38. Fuchs G. R., В ach mann К. //Fresenius Z. Anal Chem. 1987.
B. 327, №2. S. 205—212.
39. Bogen D. C, Na go urne y S. J. // Ion Chromatographie Analysis
of Environmental Pollutants / Ed. by J. D. Mulik and E. Sawicki. — Ann Arbor:
Ann Arbor Sei. 1979. P. 319—328.
40. H і 1 1 R., Lie se г К. H. // Fresenius Z. Anal. Chem. 1987. B. 327, № 2.
S. 165—169.
41. M a г к o-V a r g a G., С s і к y I., J o n s s о n J. A. // Anal. Chem. 1984.
V. 56, № 12. P. 2066—2069.
42. N a і s h P. J. Ц Analyst. 1984. V. 109. P. 809—812.
43. Motomizu S. et al. // Bunseki Kagaku. 1987. V. 36, № 1. P. 77—82.
44. Oka da T., Kuwamoto T. // Bunseki Kagaku. 1983. V. 32, № 6.
P. 595—599.
45. L am b J. D. et al. // Anal. Chem. 1981. V. 53, № 4. P. 749—750.
46. R o s s n e r В., В e h n e r t J., К і p p 1 і n g e r A. // Fresenius Z. Anal.
Chem. 1987. B. 327, № 6. S. 698—700.
47. N a к a о к a H. et al. // Bunseki Kagaku. 1981. V. 30. P. T97—T101.
48. Попов H. И., Федоров К. H., Орлов У. M. Морская вода. / Под
ред. А. С. Мониад. М.: Наука, 1979. С. 145
49. L a s h R. P., H і 1 1 С. J. // Anal. Chim. Acta. 1979 V. 108. P. 405—409.
50. S і e m e r D. D. // Anal. Chem. 1980. V. 52, № 12. P. 1874—1877.
51. К a t oh K. // Bunseki Kagaku. 1983. V. 32, № 8. P. 567—570.
52. H e n r y L. E. // Abstracts Pittsburgh Conference on Analytical Chemistry
and Applied Spectroscopy. — Atlantic City. 1986. № 983.
53. T a k a m і К., Kazunobu О., N a k a m о t о M. // Bunseki Kagaku.
1982. V. 31, № 4. P. 362—367.
54. Золотое Ю. A., Шпигун О. А., Бубчикова Л. А. // Докл. АН
СССР. — 1982. Т. 266, № 5. С. 1144—1147.
55. Шп игу н О. А. и др. // Ж. аналит. химии. 1985. Т. 40, № П. С. 1925—
1929.
56. Та naka T. / Fresenius Z. Anal Chem. 1985. В. 320, № 1. S. 125—127.
ЗАКЛЮЧЕНИЕ
Попытаемся оценить место ионной хроматографии как метода
анализа.
Нет сомнения, и об этом уже говорилось, что ионная
хроматография вне конкуренции, когда речь идет об определении
анионов, особенно сразу нескольких или многих. Действительно, нет
нн одного другого метода, позволяющего определять, например,
десять обычных неорганических анионов за 20 мин. А с помощью
градиентного элюирования, используя ионный хроматограф Дай-
онекс-4000, можно за полчаса определить даже 25—30 анионов —
практически все известные неорганические или большой набор
органических.
Но даже при определении 3—5 анионов ионная хроматография,
конечно, лучший метод. Альтернативой по некоторым показателям
(скорость, автоматизация) мог бы служить проточно-инжекци-
онный анализ, но это в основном однокомпонентный метод, как,
впрочем, и практически все другие известные методы
определения анионов. Тем более что проточно-инжекционный анализ —
это в сущности не метод, а способ осуществления ряда
аналитических методов, как например, фотометрического или потенцио-
метрического. Обычные же широко применяемые химические
методы обнаружения и определения анионов не идут ни в какое
сравнение с ионной хроматографией. Разве что иногда они могут
иметь большую точность.
Все это относится, конечно, и к анализу вод на анионы. Как
раз задача многокомпонентного анионного анализа весьма
типична в анализе вод.
Для определения катионов, например катионов металлов,
аналитическая химия имеет много различных методов. Среди них
есть многоэлементные, отличающиеся к тому же хорошими
метрологическими характеристиками. В числе наиболее интересных
и перспективных с этой точки зрения — метод, сочетающий
плазменный источник возбуждения (индуктивно-связанная плазма) и
квадрупольный масс-спектрометр. Но даже обычный атомно-эмис-
сионный анализ, особенно с индуктивно-связанной плазмой,
успешно решает многочисленные задачи многоэлементного
анализа.
При определении небольшого числа элементов или тем более
одного, широко применяют атомно-абсорбционную
спектрометрию, а также спектрофотометрию в видимой и ультрафиолетовой
области, вольтамперометрию и многие другие методы.
Ионная хроматография, как показано в гл. 10, позволяет
определять и металлы, однако ей нелегко конкурировать в этой
сфере с указанными методами. И тем не менее разработка ио-
194
нохроматографических методов определения металлов вполне
оправдана, как и само практическое использование метода для
обсуждаемых целей. Определение катионов металлов становится
рациональным, если лаборатория уже имеет ионный хроматограф
для определения анионов, а анализ на металлы в данной
лаборатории не является массовым и регулярным. В этом случае на
одном приборе можно решать обе задачи. ,
Кроме того, ионохроматографический метод имеет то
преимущество, что позволяет в ряде случаев осуществлять
вещественный анализ, т. е. определять отдельные ионные формы, а не
валовое содержание элемента. Спектроскопические методы как
правило такую возможность не обеспечивают. При наличии
анионных форм металлов (разд. 10.4) их определение относительно
легко вписывается в обычный анионный анализ.
Когда речь идет об определении органических веществ,
ионную хроматографию следует сопоставлять прежде всего с
другими хроматографическими методами: высокоэффективной
жидкостной; газовой (в том числе реакционной); тонкослойной,
хроматографией. Место ионохроматографнчсского метода
определяется прежде всего его физико-химической сущностью: он
позволяет определять ионные формы, т. е. относительно легко
ионизуемые соединения. Именно такие (обычно водорастворимые)
формы не всегда можно определять другими хроматографическими
методами. В этом может быть отличие от ВЭЖХ. Достоинство
по сравнению с газовой хроматографией в том же, чем ВЭЖХ
превосходит газовую: можно работать с нелетучими и термически
нестойкими соединениями. Тонкослойная хроматография проще
и доступнее, но редко обеспечивает возможность определения
сразу многих веществ.
Другой вопрос, который здесь следует кратко рассмотреть,—
вопрос об общих перспективах развития ионной хроматографии
как таковой. Несомненно метод имеет большое будущее. Его
использование расширяется, чему способствуют разработка и
серийный выпуск приборов и аксессуаров к ним, сорбентов и т. д.
С точки зрения развития основ самого метода большое значение
имела бы дальнейшая проработка его теории. Есть перспективы
совершенствования, возможно радикального, стадии
детектирования. Пример с введением аминокислот в качестве элюентов
показывает, что задача выбора эффективных элюентов остается
важной и шаги в этом направлении — многообещающими. Это
относится и к сорбентам. Использование градиентного элюирова-
ния значительно расширит — и уже расширяет — возможности
ионной хроматографии.
Дискуссионен вопрос о перспективности основных вариантов
метода: двух- и одноколоночного. По-видимому, сохранятся оба,
хотя в последнее время больший акцент, особенно в прикладном
отношении, делался на одноколоночный способ. Перспективы
двухколоночного варианта связаны с поиском и отработкой
новых систем подавления фонового сигнала.
195
ОГЛАВЛЕНИЕ
Предисловие . . 3
Введение 5
Глава 1. Основы ионообменной хроматографии 11
1.1. Ионообменное равновесие 11
1.2. Элтоентное разделение 13
1.2.1. Объем и время удерживания 13
1.2.2. Селективность 14
1.2.3. Эффективность 15
1.2.4. Разрешение 18
1.3. Общие сведения об ионообмеиниках 18
Глава 2. Основы ионной хроматографии 21
2.1. Двухколоиочный вариант 21
2.2. Одноколоночный вариант 23
2.3. Равновесие ионного обмена в условиях ионной хроматографии . . 26
2.4. Удерживание катионов 27
2.5. Удерживание анионов 28
Глава 3. Сорбенты 31
3.1. Требования к сорбентам 31
3.2. Анионообменники 31
3.3. Катионообмениики 35
3.4. Комплексообразующие сорбенты 36
3.5. Практика выбора сорбента 37
3.5.1. Размер частиц и параметры колонки 37
3.5.2. Матрица 38
3.5.3. Функциональные группы ' . . 38
3.5.4. Обменная емкость 40
3.5.5. Колонки для концентрирования 40
3.5.6. Концентрация определяемых ионов 42
Глава 4. Элюенты 46
4.1. Двухколоночная ионная хроматография 46
4.1.1. Определение анионов 46
4.1.2. Определение катионов 47
4.2. Одноколоночный вариант 49
4.2.1. Определение анионов 49
4.2.-2. Определение катионов 52
4.3. Практика выбора элюента 52
4.3.1. Состав : 53
4.3.2. Концентрация 55
4.3.3. Величина pH 56
4.3.4. Устранение посторонних пиков 58
4.3v5. Использование комплексообразования 59
4.3.6. Введение органических добавок 61
4.3.7. Аминокислоты как элюекты 62
196
Глава 5. Подавляющие системы 67
5.1. Колоночное подавление 67
5.2. Мембранное подавление 69
Глава 6. Детекторы 74
6.1. Требования к детекторам 74
6.2. Кондуктомегрический детектор 74
6.2.1. Принцип работы 74
6.2.2. Типы детекторов и их конструкция 7»
6.2.3. Прямое детектирование 7°
6.2.4. Косвенное детектирование 81
6.2.5. Заместительная ионная хроматография 82
6.3. Другие электрохимические детекторы 82
6.3.1. Потенциометрическіие детекторы 83
6.3.2. Амперомет.рнческие детекторы 83
6.3.3. Кулоиометрические детекторы 86
6.4. Спектрофотометрические детекторы 87
6.4.1. Принцип работы и конструкция .87
6.4.2. Прямое детектирование 88
6.4.3. Косвенное детектирование 89
6.41.4. ПослеколЪночвая реакция 90
6.5. Практика выбора детекторов 90
6.5.1. Универсальные детекторы 91
6.5.2. Селективные детекторы 91
6.5.3. Комбинации детекторов 92
Глава 7. Приборы 94
7.1. Двухколоночная ионная хроматография 94
7.2. Одиоколоиочная ионная хроматография 100
Глава 8. Определение неорганических анионов 104
8.1. Определение анионов в водах как аналитическая задача .... 104
5.2. Двухколоночный вариант 104
8.3. Одноколоночный вариант 116
8.3.1. Кондуктометрическое детектирование 116
8.3.2. Косвенное УФ детектирование 126
8.4. Электрохимическое детектирование 132
8.5. Сравнение ионной хроматографии с другими методами определения
анионов : : 132
Глава 9. Определение органических кислот 139
9.1. Органические кислоты в водах 139
9.2. Удерживание органических анионов 139
9.2.1. Одноосновные алифатические кислоты 139
9.2.2. Двухосновные алифатические кислоты 140
9.2.3. Ароматические кислоты 141
9.3. Иоиохроматографическое определение 145
9.3.1. Двухколоиочный вариант 145
9.3.2. Одноколоночный вариант с кондуктометрическим детектором . 146
9.3.3. Одноколоночный вариант с УФ детектором '47
3.4. ИоноэкокЛ'Юзионная хроматография 148
9.4.1. Принцип метода 148
9.4.2. Определение органических кислот 150
Э.э. Сочетание ионной и ноноэкеклюзиоиной хроматографии .... 151
197
Глава 10. Определение металлов 155
10.1. Щелочные металлы и аммоний 155
10.1.1. Двухколоночный вариант 155
1Ю.1.&. Одноколоночный вариант і . 157
10.1.2.1. Кондуктометрическое детектирование 157
10.1.2^2. Спектрофотометрическое детектирование .... 158
10.2. Щелочноземельные металлы и магний 159
10.2.1. Двухколоночный вариант 159
10.2.2. Одноколоночный вариант 162
10.2.2.1. Кондуктометрическое детектирование 162
,10.2.2.2. Спектрофотометрнческое детектирование 162
10.2.2.3. Электрохимическое детектирование 163
10.3. Переходные металлы 16*
10.3.1. Двухколоночный вариант 164
10.3.2. Одноколоночный вариант 164
10.3.2.1. Кондуктометрическое детектирование '^4
10.3.2.2. Спектрофотометрическое детектирование 165
10.3.2.3. Электрохимическое детектирование 167
10.3.2.4. Другие детекторы 169
10.4. Анионные комплексы металлов 169
10.4.1. Оксоанионы : : . 169
10.4.2. Комплексы с неорганическими и органическими лигандами . 170
Глава 11. Определение алифатических аминов 174
11.1. Удерживание аминов на катионообменниках низкой емкости . . 174
11.2. Ионохроматографическое определение ...... і 176
Глава 12. Совместное определение катионов и анионов 178
12.1. Использование последовательно соединенных колонок .... 178
12.2. Другие варианты : '80
Глава 13. Ионохроматографический анализ вод различного тнпа ... 181
13.1. Воды как объект анализа '81
13.2. Отбор н подготовка пробы 181
13.3. Высокочистые воды '82
13.4. Питьевая и водопроводная воды '84
13.5. Дождевая вода : 185
13.6. Поверхностные и грунтовые воды '87
13.7. Морская и подземные высокоммнерализованные воды .... 189
13.8. Сточные воды '9'
Заключение '94
Научное издание
Шпигун Олег Алексеевич, Золотое Юрий Александрович
ИОННАЯ ХРОМАТОГРАФИЯ И ЕЕ ПРИМЕНЕНИЕ В АНАЛИЗЕ ВОД
Зав. редакцией Н. М. Глазкова
Редактор О. В. Апентьева
Обложка художника Н. В. Андриевича
Художественный редактор А. Л. Прокошев
Технический редактор М. Б. Терентьева
Корректоры М. И. Элычус, Т. И. Алейникова
И Б № 3704
Сдано в набор 31.01.90. Подписано в печать 14.02.91. Формат
— еч.
р. 70 к
Сдано в набор 31.01.90. Подписано в печать 14.02.91. Формат б
Бумага офс. № 2. Гарнитура литературная. Печать высокая. Усл. печ. л.
У'ч-изд. л. 12.Є6. Тираж 2800 экз. Заказ. 34. Изд. № 881. Цена 3
Ордена «Знак Почета» издательство Московского университета.
103009, Москва, ул. Герцена, 5/7.
Калужское ПО «Полиграфист», 509281, г. Калуга, пл. Ленина, 5
О.А.Шпигун, Ю.А.Золотов
ИОННАЯ ХРОМАТОГРАФИЯ
и её применение в анализе вод
Издательстро Московского университета
1990