/



















































































































































































































































































































































Текст
АКАДЕМИЯ НАУК СССР
СИБИРСКОЕ ОТДЕЛЕНИЕ
К. К. АНДРЕЕВ
Термическое разложение
и горение
взрывчатых веществ
ИЗДАНИЕ ВТОРОЕ
(ПЕРЕРАБОТАННОЕ И ДОПОЛНЕННОЕ)
ИЗДАТЕЛЬСТВО «НАУКА»
МОСКВА • 19 66
541.427.6;541.126+552.92
Ответственный редактор
профессор, доктор физико-математических иаугг
Р. И. СОЛОУХИН
127
569 6
*
•> ШУГ
. SlSJi/k f 1-
2-5-4
538-66
ОТ РЕДАКТОРА
Автор этой книги — один пз крупнейших специалистов в области тео-
рии взрывчатых веществ — профессор Константин Константинович Анд-
реев начал свою научную деятельность в конце 20-х годов, еще будучи
студентом Московского высшего технического училища.
Одной цз первых его работ было исследование термического распада
азида бария, выполненное им в 1928—1929 гг. в Берлине, где он около
года стажировался в Физпко-химическом институте Берлинского универ-
ситета, Несколько позже, в начале 30-х годов, в Институте химической
физики в Ленинграде, работая под руководством Ю. Б. Харитона,
К. К. Андреев впервые предпринял исследование горения вторичных
взрывчатых веществ. С тех пор, главным образом в Московском химико-
технологическом институте нм, Д. И. Менделеева (в котором К. К. Андреев
работал с 1935 г. и до кончины), им было выполнено более 150 работ по
различным вопросам теории взрывчатых веществ. Однако термический
распад и теория горения ВВ всегда оставались направлениями, наиболее
близкими ему.
В 1957 г. вышла в свет монография профессора К. К. Андреева «Тер-
мическое разложение и горение взрывчатых веществ». Эта книга быстро
разошлась и была переведена на ряд европейских языков.
Бурное развитие исследований в области горения ВВ в последние годы
потребовало значительного пополнения книги новым материалом и пере-
смотра некоторых се разделов. Поэтому с 1963 г. К. К. Андреев начал
подготовку книги к переизданию. Многое в ней было изменено, а боль-
шая часть материала написана заново.
В феврале 1964 г. К. К. Андреев заболел. В течение последних меся-
цев жизни, несмотря на несколько перенесенных им операций, он мно-
го и интенсивно работал над книгой, написав более половины вновь вве-
денного в нее материала, и в конце апреля в общих чертах завершил эту
работу. К. К. Андреев скончался 9 мая 1964 г. па 60-м году жизни в пол-
ном расцвете творческих сил.
Несмотря па то, что большая часть книги в машинописном варианте
была прочитана и проверена Константином Константиновичем, многое он
закончить пе успел. Около 300 страниц пришлось напечатать по рукопис-
ным материалам, изложенным хотя и в достаточно закопченном виде
(Константин Константинович, как правило, писал свои работы сразу на-
бело и впоследствии коренных изменений в них обычно не вносил), но тем
не менее не выверенных и нс правлеиных автором. Ближайшими ученика-
ми и последователями К. К. Андреева был подготовлен к печати обширный
иллюстрационный материал, составлены таблицы, библиография.
В научных трудах К. К. Андреева мы видим пример умелого сочетания
ясности и последовательности научной мысли с .правильным выбором
метода исследования и с непосредственным практическим применением по-
лученных результатов. Проблемы химического превращения взрывчатых
веществ решаются им с учетом совокупности многих факторов, способных
3
оказывать влияние на ход процесса в тех пли иных конкретных условиях,
А если учесть при этом необычайно широкий выбор наиболее интересных
в практическом отношении взрывчатых веществ, становится ясной уни-
кальность и эпциклопедичность данной монографии. Описание физико-
химической структуры и различных видов превращений основных типов
ВВ и насыщенность фактическим материалом делают книгу незаменимым
пособием для широкого круга научных работников, инженеров и студентов
физико-химических и химических специальностей.
Научный кругозор К. К. Андреева необычайно широк. По существу,
в каждом разделе книги ему лично п его ученикам принадлежат основ-
ные результаты к основополагающие факты. Это относится прежде всего
к термическому распаду и теории горения вторичных ВВ, к анализу кри-
тических явлений и физической сущности различия в свойствах ряда
взрывчатых веществ.
После описания основных видов взрывчатых веществ и их способно-
сти к химическому превращению того или иного типа (устойчивое горе-
ние, нестационарное горение и детонация), которому посвящена первая
глава, автор рассматривает основные вопросы химической кинетики воз-
никновения и развития взрыва под влиянием изменения температурных
условий. Несмотря па законченность и широкую известность кинетической
схемы самовоспламенения газов, разработанной Н. Н. Семеновым,
Д. Л, Франк-Каменецким и другими советскими учеными, описание дина-
мики термического разложения конденсированных ВВ еще далеко от сво-
его заверпюния. Здесь, пожалуй, следует дополнительно отметить серию
недавних работ сотрудников Института химической физики АВ СССР,
где выполнены расчеты теплового взрыва для ряда веществ, которые мо-
гут быть полезны при анализе опытных данных.
Последующие главы посвящены описанию различных видов «медлен-
ного» горения главным образом конденсированных веществ. Актуальное ть
систематизации научных результатов в этой области, многие из которых
принадлежат автору книги, связана с быстро растущим кругом задач но-
вой техники. В последовательном виде рассматриваются особенности
сложной физико-химической модели процесса горепия, включающей в себя,
в форме неразрывной связи, как индивидуальные особенности химического
превращения выбранного вещества, так и влияние физических условий
(температуры, давления, плотности, гидродинамических факторов ит.п.)
на процесс химического превращения. Здесь особенно интересны вопросы
устойчивости горения и перехода в детонацию в применении к горению
порохов и других сложных конденсированных систем.
Разумеется, не следует считать недостатком большого труда автора
отсутствие столь же последовательного н обстоятельного анализа детона-
ционного вида химического превращения взрывчатых веществ- Эта рабо-
та по своему объему выходпт далеко за рамки замысла данной книги.
Однако нельзя не отметить интенсивно продолжающегося поиска ясной
физической схемы детонационного горения, В особенности это относится
к разделу о конденсированных взрывчатых веществах, где описаны пока
лить самые первые шаги в построении такой схемы.
К. К. Андреев чрезвычайно тщательно работал пад всем, что им пуб-
ликовалось, и в том случае, если бы ему удалось самому подготовить руко-
пись к печати, он, несомненно, внес бы в псе немало изменений и допол-
нений. Однако работавшие над книгой не сочли возможным что-нибудь
изменить в написанном К. К. Андреевым тексте.
Основная работа по подготовке рукописи к печати была выполнена
Б. С. Светловым (главы I и II), Л. П. Глазковой (глава III) и Б, Ц. Конд-
риковым (главы IV, V и VI). В обсуждении вопросов, связанных с под-
готовкой книги к печати, принимал участие [,Д. И. Гольбиидщ!
Р. Я . СОЛОУХИН
4
ПРЕДИСЛОВИЕ
Процессы горения конденсированных веществ представляют значи-
те л ьцын практический интерес. Эти процессы широко применяются в тех-
нике, главным образом в огнестрельном оружии разных типов, в пиротех-
нических изделиях, дистанционных трубках, замедлителях, бикфордовом
шнуре, электродетоваторах замедленного действия и др.
Горение взрывчатых веществ (ВВ) может возникать также по разным
принимай па заводах ц складах при производстве и хранении ВВ, равно
как и при их применении.
Предупреждение возникновения горения, способы борьбы с начавшим-
ся пожаром, предупреждение перехода горения в детонацию представляют
одну из важнейших задач обеспечения безопасности производства, хране-
ния и применения ВВ.
Вопросы технического использования горения ВВ освещаются в соот-
ветствующих специальных руководствах [1], и их рассмотрение выходит за
рамки данной книги.
Наряду с практическим значением вопросы горспия ВВ представляют
большой теоретический интерес. При горении химическая реакция про-
текает при очень высоких температурах, труднодоступных при иных усло-
виях проведения реакции, за исключением детонации. Кроме того, можно
осуществлять процесс также при очень больших и регулируемых давлени-
ях. Таким образом, в принципе возможно, исследуя процессы горения,
изучать особенности течения химических реакций в указанных необычных
условиях.
Но меньшее теоретическое значение представляют вопросы устойчиво-
сти горения ВВ и условия его перехода в детонацию. Современная взрыв-
ная техника базируется на существовании трех основных классов ВВ.1
инициирующих, вторичных и метательных (порохов). Основное различие
между этими классами ВВ состопт в различной степени устойчивости их
горения.
Несмотря на большой теоретический и технический интерес к проблеме
в целом, она оставалась до недавнего времени почти неизученной. За по-
следние 20 лет этот пробел в значительной мере восполнен главным обра-
зом работами советских ученых по исследованию процессов горения ВВ.
Эти работы позволили дать общую картину процесса и построить количе-
ственную теорию горения, знание которой, несомненно, полезно и для ис-
следователей, работающих над вопросами технического применения горе-
ния ВВ.
Большинство работ по горению опубликовано в различных физико-
химических и физических журналах и других, непериодических, изданиях
разного профиля, следить за которыми затруднительно. Кроме того, все
работы посвящены отдельным частным вопросам проблемы; работ обзор-
ного характера нет. По этим причинам было целесообразным дать сводку
п обобщение экспериментального и теоретического материала по исследо-
ванию горения ВВ, что и составляет задачу данной книги.
5
Наибольшее внимание уделено в книге экспериментальным работам
последи его периода, проведенным в условиях, позволяющих использовать
результаты эксперимента как материал для построения теории и ее про-
верки. Многочисленные исследования, проведенные главным образом над
порохами в конце прошлого и в текущем столетии для решения техниче-
ских задач ствольной артиллерии, затронуты в малой степени, тем более,
что эти работы, равно как ц выводы пз них, более или менее подробно
освещены в ряде монографий по внутренней баллистике.
Книга включает шесть глав. Первая глава посвящена общей характе-
ристике основных форм химического превращения ВВ и главным типам
ВВ, по составу, химической структуре и способности к устойчивому горе-
нию, а также к детонации.
Во второй главе рассматривается медленное химическое превращение
ВВ под влиянием повышенной температуры.
Часть этой главы, которая в первом издании была посвящена общим
вопросам химической кинетики, в настоящем издании отсутствует, по-
скольку эти вопросы были недавно подробно рассмотрены в целом ряде
монографий [2—5], В главе излагаются экспериментальные данные по тер-
мическому распаду основных представителей различных классов ВВ —
нитроэфиров, питросоединений ароматического и алифатического рядов,
твердых ВВ (пикраты, стифпаты, азиды, перхлораты). Этот раздел главы
существенно пополнен if модернизирован па основе большого числа выпол-
ненных и опубликованных за последние годы в советской н зарубежной
литературе работ, В заключение дастся обобщение рассмотренных данных
и таблицы основных кинетических характеристик различных ВВ.
В третьей главе излагаются экспериментальные данные по устойчиво-
му горению ВВ. Она начинается с описания различных методов исследо-
вания (бомбы переменного п постоянного давления). Далее рассматри-
ваются критические условия горения: критический диаметр, температура
и давление. Эти вопросы в последнее время приобрели большое значение
особенно в связи с выгоранием промышленных ВВ, поэтому раздел су-
щественно пополнен фактическими и теоретическими данными. Далее
последовательно описаны для основных ВВ зависимости скорости горения
от давления, начальной температуры и кубической плотности. Данные для
отдельных ВВ сильно сокращены, и читатель отсылается к первому из-
данию, где они подробно рассмотрены. Кратко рассматривается состав
газообразных продуктов горения и экспериментальные данные по опреде-
лению его температуры.
В четвертой главе рассматриваются основные теории горения — Ми-
хельсона и Малляра — Ле-Шателье, Зельдовича — Беляева и производит-
ся сопоставление экспериментальных данных с выводами теории примени-
тельно к различным тинам ВВ (летучие, нелетучие, быстрогорящие).
В пятой главе излагается теория устойчивости горения, эксперимен-
тальные данные по этому вопросу и сопоставление этих данных с теоре-
тическими заключениями применительно к горению твердых сплошных,
порошкообразных и жидких ВВ.
В шестой главе рассматриваются особые случаи — вспышка и воспла-
менение ВВ в различных условиях (интенсивное нагревание, действие
света и др.).
Книга предназначена для исследователей, работающих в различных
областях технического использования горения ВВ, для работников заво-
дов, производящих ВВ и пороха, и для специалистов ио горному и взрыв-
ному делу. Она может быть использована также в качестве учебного посо-
бия для студентов старших курсов соответствующих специальностей.
|К. К. Андреев^
ГЛАВА ПЕРВАЯ
ОБЩАЯ ХАРАКТЕРИСТИКА ВЗРЫВЧАТЫХ ^ВЕЩЕСТВ
И ОСНОВНЫХ ФОРМ
ИХ ХИМИЧЕСКОГО ПРЕВРАЩЕНИЯ
Взрывчатые вещества, так я;е как и вещества певзрывчатые, способны
к медленному химическому превращению. Эта форма протекания химиче-
ской реакций не показывает в основном принципиальных особенностей,
которые были бы свойственны только ВВ.
Характерным же отличием ВВ является их способность к химическому
превращению в двух других формах—горению и детонации. В противо-
положность медленному химическому превращению, протекающему более
пли менее равномерно во всем объеме ВВ, при горении и детонации возни-
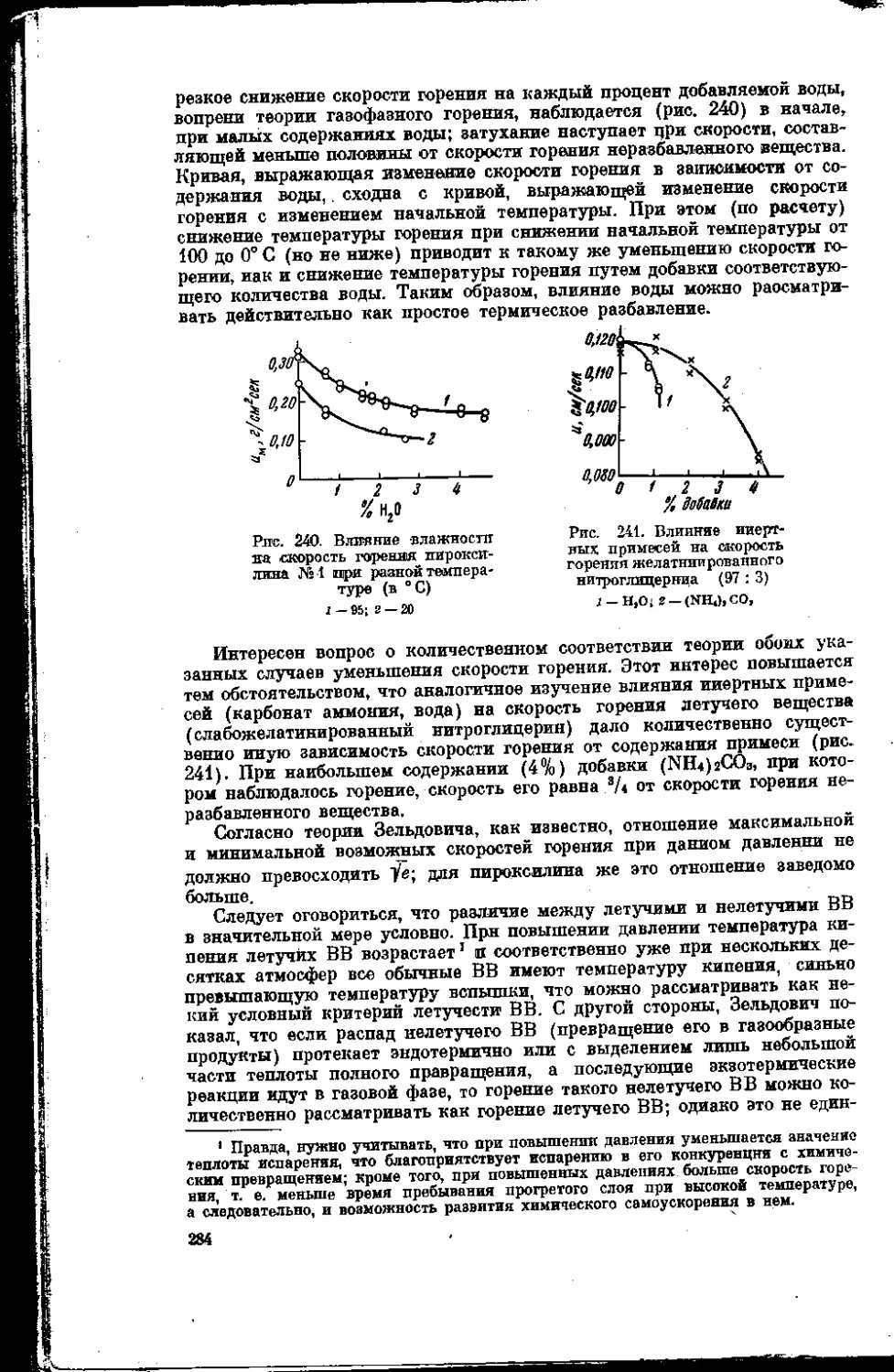
кает фронт превращения — зона интенсивной химической реакции, отде-
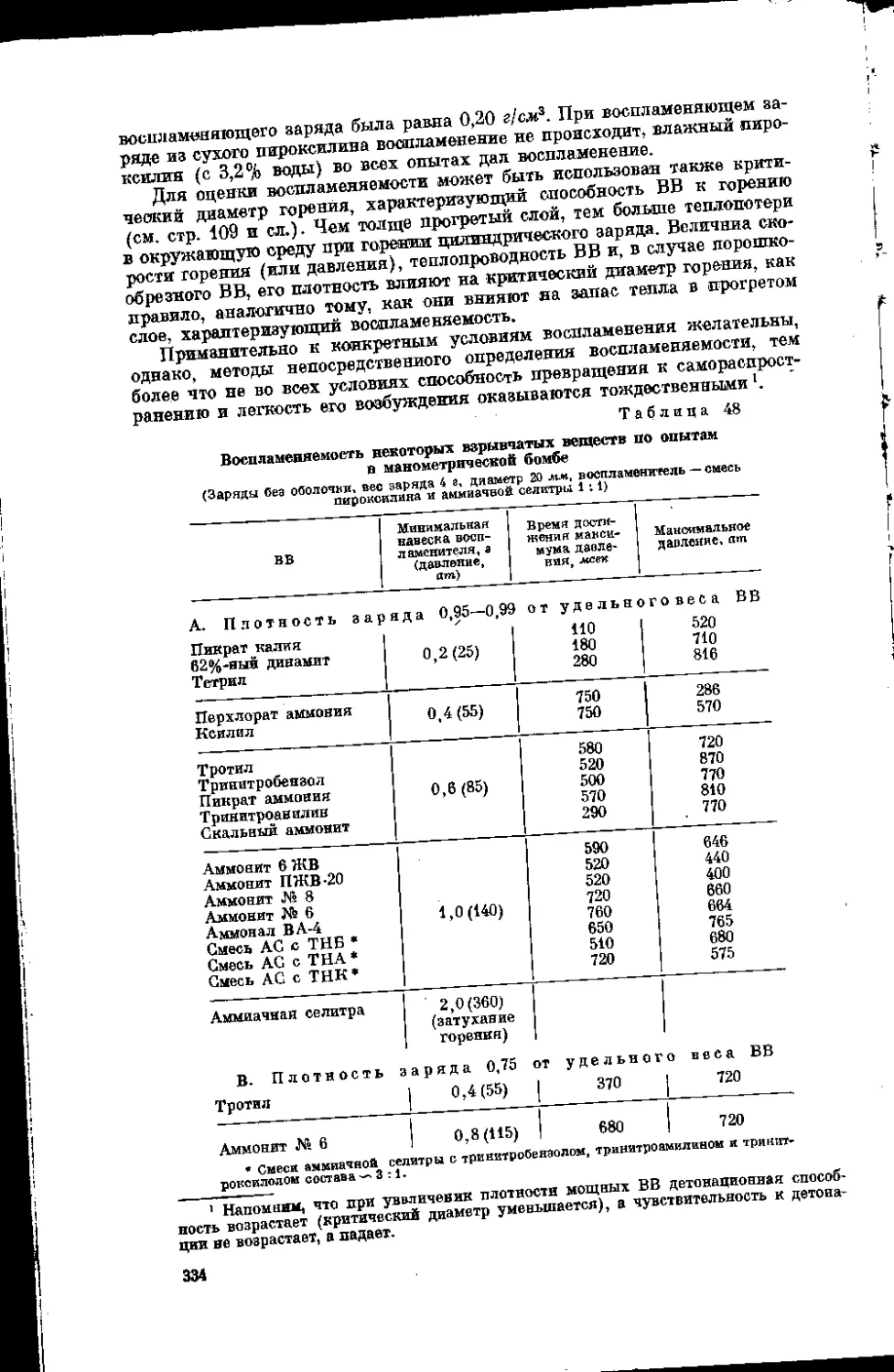
ляющая нспрореагировавшее вещество от продуктов реакции и переме-
щающаяся по ВВ.
Основное формальное различие между горением и детонацией заклю-
чается в величине линейной скорости распространения фронта превраще-
ния, измеряемой для типичных ВВ миллиметрами в секунду при горении
н километрами в секунду при детонации.
Одним из основных следствий большой скорости распространения реак-
ции при детонации является возникновение чрезвычайно большого и бы-
стро возрастающего давления, производимого газообразными продуктами
превращения на окружающую среду. В результате возникновения разности
давлений между продуктами детонации и окружающей средой потен-
циальная энергия продуктов детонации в обычпых условиях протекания
последней превращается в механическую работу. При этом, так как вели-
чина давления очень велика (порядка сотен тысяч атмосфер), то и работа,
совершаемая газами при детонации, отнесенная к единице времени, иначе
говоря мощность, крайне велика, больше, чем у всех иных обычных источ-
ников работы. Исключительно большая мощность, легко достигаемая при
детонации, и составляет основу ее технического использования.
При горении В В (в отсутствие оболочки) вследствие медленности об-
разования газов повышение давления весьма незначительно и механиче-
ское действие практически отсутствует.
Способность к детонации является наиболее специфичной для ВВ не
только ио существу механизма процесса, но и по внешней картине его
течения и действия. Горение же, как известно, наблюдается и в системах,
нс способных к детонации (горение любого горючего на воздухе), и внеш-
не его течение в этих системах очень сходно с горением ВВ. Существенное
различие состоит в том, что горение невзрывчатых веществ, как правило,
требует доступа воздуха или иного газа, участвующего в реакции, поступ-
ление которого в значительной степени определяет течение горения. Горе-
ние же ВВ протекает без участия внешней среды. По существу примене-
ние одного и того же термина «горение» к обоим процессам определяется
только их внешним сходством, и правильнее было бы горение ВВ обозна-
чить иначе, например, как «самостоятельное» горение.
Чтобы химическое превращение могло протекать в форме горения и де-
тонации, оно должно быть экзотермичным и идти с большой скоростью,
7
продукты превращения должны.быть полностью иля частично газообраз-
ными и, наконец, превращение должно быть сдмораспрострапягощимся.
Остановимся несколько подробнее на значении этих условий. Химиче-
ское превращение в форме горения или детонации по самому определению
этих процессов является самораспространяющимся. При этом в послед-
нем случае самораспространенне протекает с чрезвычайно большими ско-
ростями. Чем же обусловливается способность химической реакции к са-
мораспространению и скорость его?
Известно, что скорость химической реакции (число молекул, реаги-
рующих в единицу времени в единице объема) зависит от концентрации
активных частиц. Поскольку эта концентрация возрастает с температурой
по известному закону е_, EiRT. где Е~ энергия активации), то скорость
реакции растет с температурой. Скорость реакции растет также с увели-
чением концентрации вещества, а следовательно, в случае газов — с дав-
лением, которому пропорциональна концентрация. При реакциях первого
порядка, скорость которых пропорциональна первой степени концентра-
ции, влияние концентрации объясняется просто увеличением числа моле-
кул в единице объема. При реакциях второго порядка, где скорость реак-
ции пропорциональна произведению или квадрату концентрации, влияние
последней объясняется тем, что число соударений между способными к
реакции молекулами пропорционально квадрату давления.
При гомогенном течении процесса молекулы, вступающие в химиче-
скую реакцию, получают энергию активации при обмене энергией друг
с другом за Счет того общего запаса энергии, который содержится в веще-
стве при данной температуре. Этим и определяется зависимость скорости
реакции от температуры.
Чтобы вызвать самораспространяющееся превращение в форме горе-
ния, необходимо поджечь заряд ВБ в каком-либо месте. Поджигание озна-
чает сильный и локальный разогрев; в нагретой зоне с большой скоростью
(соответственно высокой температуре) идет экзотермическая химическая
реакция; тепло, при ней выделяющееся, передается соседним слоям ВВ и,
разогревая их, вызывает быструю химическую реакцию, которая затем
аналогично возбуждается в дальнейших слоях. Во-первых, скорость рас-
пространения превращения в этих условиях зависит от его теплового эф-
фекта, за счет которого происходит разогрев вещества, и от константы
скорости реакции. Во-вторых, она зависит от условий передачи тепла от
зоны реакции в непрореагировавшее вещество, которые определяют ско-
рость распространения зоны высокой температуры.
Наиболее быстро превращение распространялось бы в том случае, если
бы продукты распада одной молекулы были в состоянии непосредственно
активировать одну или тем более несколько близлежащих частиц.
При распаде типичных ВВ, например гремучей ртути или тротила, вы-
деляется 120—230 ккал/моль. Энергия активации медленного термиче-
ского разложения этих ВВ составляет 30—40 ккал/молъ. Если бы распад
происходил в одну стадию, то каждая распавшаяся молекула могла бы
активировать 4—6 других, распад каждой из которых вызвал бы в свою
очередь распад такого же числа соседних молекул.
Распад отдельных молекул происходит в любом веществе даже при
комнатной, а тем более при повышенных температурах. Он, несомненно,
происходит также под влиянием проникающего излучения, а в радиоактив-
ных ВВ, вроде азида радия,— и под действием собственного излучения.
Тем не менее быстрого ускоренного разложения ВВ не наступает. Его не
наблюдается даже при действии осколков деления урана, энергия кото-
рых па несколько порядков превосходит энергию активации ВВ.
Поскольку таким образом распад отдельной молекулы ВВ не приводит
к быстрому развитию превращения, следует заключить, что не выполняет-
ся вторая предпосылка такого развития — распад молекулы в одну стадию
8
до продуктов полного превращения. Действительно, опыт показывает, что
при всех формах превращения этот распад идет ступенчато, тепловой
эффект отдельных ступеней гораздо меньше полного теплового эффекта
превращения; помимо этого, энергия активации некоторых промежуточ-
ных стадий может быть значительно больше, чем энергия активации пер-
вичной стадии или суммарного процесса распада ВВ. В итоге цепного раз-
вития превращения не может произойти — энергия выделяется ступенчато
н передается соседним молекулам порциями, меньшими, чем энергия акти-
вации, распределяясь в них в виде тепла. Все это приводит к тому, что
зона максимальной температуры располагается далеко от поверхности ВВ,
i-радиент температуры мал, и скорость распространения тепла, если оно
происходит путем теплопроводности, также мала.
Следует добавить, что эта особенность для ВВ используемых в технике,
неслучайна. ВВ, которые взрывались бы от распада отдельной молекулы
или небольшого числа пх, настолько опасны при производстве и примене-
нии, что их использование было бы практически нереальным. Поэтому
технический отбор из большого числа химических соединений, способных
к взрыву, выделил такие, вызвать взрыв которых можно лишь достаточно
интенсивным воздействием, случайное возникновение которого было ис-
ключено- Собственно говоря, открытие современных — вторичных ВВ, со-
вершившееся около ста лет назад, в том и заключалось, что были найде-
ны ВВ, неспособные к взрыву от действия пламени, слабых ударов, тре-
ния и других возможных в практике случайных воздействий.
Если распад отдельной молекулы ВВ не Приводит к самораспростра-
неггию превращения, то локальный распад значительного числа молекул,
образование очага распада, может к нему привести как при цепной, так
и при тепловой передаче реакции. При цепной реакции число богатых
энергией частиц продуктов превращения, приходящихся на одну молекулу
в соседнем с очагом слое вещества будет больше. В случае теплового
ускорения реакции экзотермичность превращения приводит к повышению
температуры и, если размеры очага велики, то при распространении теп-
ла в окружающее вещество будет поддерживаться высокая температура
и соответственно большая скорость тепловыделения.
Рассмотренная схема распространения химического превращения, объ-
ясняя горение, не включает распространения в форме детонации; для этого
необходимо учесть, что скорость горения зависит также от давления.
Кроме того, если горение идет па поверхности, значительно большей, чем
сечение заряда, то количество реагирующего на единицу поверхности этого
сечения ВВ будет соответственно больше. Поэтому, если создать одно-
временно с разогревом высокое давление и развитую поверхность горения,
то массовая скорость распространения превращения (в а/сл2сек) будет
иметь тот порядок, который характерен для детонации. Очевидно, что дав-
ление при химическом превращении может возникнуть лишь в том случае,
если продукты его при достигаемой температуре хотя бы частично яв-
ляются газами.
Другая особенность детонационной формы превращения состоит в ха-
рактере передачи энергии: при быстром протекании превращения возни-
кает резкое локальное повышение давления, которое распространяется в
виде ударной волны со сверхзвуковой скоростью; это распространение при-
водит к возбуждению химического превращения в форме горения с пара-
метрами (давление н удельная поверхность), характерными для детонации.
Образование ударной волны также возможно лшпь, если превращение
образует газы. Таким образом, возникновение давления, создаваемого
газами, является не только одним из двух основных условий быстрого про-
текания реакций горения (другим условием является высокая температу-
ра), но н необходимым фактором быстрого распространения процесса, не
осуществляющегося для реакций, которые не образуют газов.
!>
Все формы химического превращения ВВ — медленное термическое
превращение, горение и детонация — связаны между собою как по сущ-
ности происходящих при них процессов, так и генетически. Медленное
химическое превращение может в определенных условиях приводить к
вози икн о ней ию горения, горение может переходить в детонацию; возмо-
жен также и переход детонации в горение.
Как стационарные процессы каждого из трех типов, так и их взаим-
ные переходы представляют значительный практический интерес. При не-
которых процессах производства, снаряжения и применения технологиче-
ски желательна возможность нагрева ВВ до относительно высокой тем-
пературы. В то же время такой разогрев может привести к загоранию ВВ
пли значительному разложению, делающему В В не пригодным для упо-
требления. Чтобы предупредить эту возможность, нужно знать условия
перехода медленного превращения в горение.
Медленное химическое превращение происходят и при обычных тем-
пературах храпения ВВ, особенно в жарком климате, определяя их хими-
ческую стойкость. Помимо этого, при длительном хранении больших масс
ВВ медленное химическое превращение может привести даже к самовос-
пламенению. Это превращение является, как правило, сам оу окоряющимся
процессом, и если хранение длится Очень долго, а масса хранимого ВВ
очень велика, то возможно накопление тепла, приводящее к самовоспла-
менению, Задача предотвращения такого явления аналогична предыдущей,
по зачастую сложнее, так как требует знания закономерностей распада
nptt столь низких температурах, при которых их не посредственное опре-
деленно обычным методом требует чрезвычайно большого времени.
Характеристики стационарного горения (скорость горения, завис и мог те.
скорости горения и его теплового эффекта от давления, зависимость ско-
рости горения от начальной температуры) представляют интерес но всех
условиях технического использования горения, особенно же для гореппл
в полузамкнутом объеме.
Переход горения в детонацию интересен в ряде аспектов. Во всех при-
меняемых в технике взрывчатых веществах (кроме азида свинца) удар,
трение, пламя, если они достаточно интенсивны, вызывают первоначально
горение, которое может затем перейти во взрыв. Ущерб, создаваемый го-
рением ВВ, то 1’0 же характера и масштаба, как и при горении (пожаре) без
участия ВВ. Иначе обстоит дело в том случае, если горение заменяется
взрывом, вызывающим как в его очаге, так и на более или менее значи-
тельных расстояниях в окрестности сильные механические разрушения.
Одной из важных особенностей современных вторичных ВВ, существенно
ограничивающей их опасность, является именно значительная устойчи-
вость их горения в различных возможных его условиях. Горение этих ВВ,
вызванное поджиганием или случайно возникшее, например в результате
перегрева и самовоспламенения при производстве, за исключением особых
условий, которые мы рассмотрим ниже, протекает без возникновения де-
тонации.
Еще большее значение имеет предотвращение горения от перехода во
взрыв при использовании ВВ для целей метания. Емкости, в которых про
походит горение в метательных аппаратах (ствол огнестрельного оружья,
камера ракетного двигателя) рассчитаны на относительно небольшие дав-
ления, во много раз меньшпе, чем те, которые возникают при детонации.
Возникновение последнегг означает не только неуспех выстрела, ио п раз-
рыв аппарата и часто гибель обслуживающего его персонала.
Поэтому обязательным требованием к ВВ, используемым для метания
(порохам) и к условиям их применения является надежная устойчивость
их горения. Собственно, говоря, и важнейший этап развития порохов я
прошлом — переход от черного пороха к современным бездымным поро-
хам— н значительной мере диктовался соображениями устойчивости го-
10
рения. Черный порох перестал в середине прошлого века удовлетворять
возросшим требованиям в отношении устойчивости горения, и хотя при
нарушении этой устойчивости процесс горения не развивался до детона-
ции, все же неустойчивость нормального горения черного пороха выну-
дила к отказу от его применения в пользу новых порохов — порохов пласт-
массового типа па основе высокополимера — нитроклетчатки. Физические
свойства этих порохов так отличаются от свойств вторичных ВВ, которые
являются их основными компонентами, что горение нс переходит во
взрыв даже при тех особых условиях, при которых этот переход возможен
в случае вторичных ВВ.
Устойчивость горения бездымных порохов была столь велика, что во-
прос о ней долгое время практически не стоял, Оп возник вновь в связи
с новым применением порохов — для реактивного метанпя, при котором
нарушения устойчивости нормального горения еще оцаспсе, чем при
ствольг<ом мотанин. С другой стороны, этот вид мстацпя потребовал раз-
вития новых — смесовых порохов, по многим своим свойствам промежу-
точных между чисто пластмассовыми порохами и черным порохом. Одним
из этих свойств является повышенная ио сравнению со своими предше-
ственниками склонность к нарушению нормального режима горения осо
бепцо в новых условиях применения. Устранение этой склонности поэтому
вновь приобрело актуальность,
В рассмотренных примерах неустойчивость горения является серьез-
ным недостатком. Сто лет назад эта неустойчивость сыграла решающую
положительную роль и изыскании способа возбуждения взрыла новых —
вторичных взрывчатых веществ, и этот способ без существенных измене-
ний сохраняет свое значение и в настоящее время.
Черный порох взрывается от подяспгапмя пламенем, вторичные ВВ,
как известно, этой способностью не обладают п, чтобы обеспечить возмож-
ность их технического применения, потребовался простой п надежный
способ возбуждения их взрыва. Такой способ был найден. Его основой
явилась главная особенность инициирующих ВВ — крайняя неустойчи-
вость их горения1, обусловливающая способность давать при поджигании
практически мгновенный переход горев ня во взрыв. Маленький заряд ини-
циирующего ВВ, запресовапный в металлическую гильзу, при поджигании
иа расстоянии тем же способом, который рацее применялся для черного
пороха, пли иным безотказно детонирует и вызывает детонацию сколь
угодно большого заряда вторичного ВВ. Из приведенных примеров видно,
что основой тех свойств, которые обусловливают способы современного
технического применения ВВ, являются характеристики их горения и в
первую очередь степень его устойчивости.
Выше отмечалось, что возможно и явление перехода детонации в горе-
ние. Для конденсированных ВВ это явление практически не изучено. Од-
нако такая возможность вызвала в последние годы значительный интерес
в связи с взрывными работами в угольных шахтах, в которых может об-
разоваться метано- или пылевоздушпая взрывчатая смесь. При взрыве
заряда ВВ в шпуре в тех условиях, в которых этот взрыв сам по себе
безопасен в отношении воспламенения горючей атмосферы, прекращение
детонации и возникновение взамен нее горения резко увеличивает опас-
ность указанного воспламенения. В этом плане возможность прекращения
детонации заряда ВВ и последующего возникновения горения остатка за-
ряда привлекает внимание многих исследователей, видящих в нем основ*
ной источник опасности взрывных работ применительно к возникновению
взрыва метано- или пыл с воздушных смесей,
1 Понятно, что подразделение ВВ на вторичные, инициирующие и метательные
цо признаку устойчивости горения в известной гире ус job ио, поскольку эта устойчи-
вость зависит не только от .мшц'цч:ь’ой природы ВВ, ио и от физической структуры
заряда, а также от условий горе пи я; и амоис и нем атцх факторов можно существенно
и;пчеиит|, степень устойчивости горении бо;1ьши|гстка В В.
ГЛАВА ВТОРАЯ
МЕДЛЕННОЕ ХИМИЧЕСКОЕ ПРЕВРАЩЕНИЕ
ВЗРЫВЧАТЫХ ВЕЩЕСТВ
Горение ВВ представляет собой самора сп рос трапяющуюся экзотерми-
ческую реакцию. Это самораспространенке реакции осуществляется в ре-
зультате передачи тепла, выделяющегося в реагирующем слое, прилегаю*
щему к нему, еще не вступившему в реакцию слою ВВ. Под влиянием
разогрева слоя ВВ в нем начинает идти химическая реакция со все уве-
личивающейся соответственно возрастающей температуре скоростью. Так
как реакция экзотврмична, то она ускоряет разогрев от предыдущего слоя
и на известном этапе своего развития становится главным поставщиком
тепла.
Таким образом, одним из .элементов процесса горения является хими-
ческое превращение при повышенных и возрастающих температурах. Та-
кое превращение происходит также при воспламенении ВВ в поверхност-
ном слое его. с которого начинается горение.
Знание закономерностей медленного химического превращения ВВ яв-
ляется необходимой предпосылкой для построения теории их горения и
возможности ее приложения к конкретным ВВ. Наибольший интерес при
этом представляют быстрые процессы, идущие при относительно высоких
температурах: в условиях горения прогрев слоя, вступающего в реакцию,
осуществляется сравнительно очень быстро, и химическое превращение
в основном протекает в области высоких температур, где его характери-
стики и закономерности течения могут быть существенно иными, чем при
НП31Д1Х температурах. Однако большая часть соответствующих исследо-
ваний выполнена именно в области относительно низких температур, с од-
ной стороны, потому, что количественное изучение кинетики химического
превращения ВВ прд высоких температурах труднее, и, с другой стороны,
потому, что в большинстве случаев исследования преследовали чисто прак-
тическую цель — установление химической стойкости изучаемого ВВ при
хранении. Поэтому не всегда можно непосредственно переносить резуль-
таты исследования низкотемпературного распада ВВ на условия течения
их химического превращения при горении.
При относительно высоких температурах исследования производились
только для вспышки ВВ по установлению температуры ее возникновения
в различных условиях и зависимости времени задержки вспышки от тем-
пературы, Эти исследования дали большой и интересный материал,
но теоретическая его интерпретация ввиду большой сложности яв-
ления имеется лишь для немногих случаев, главным образом для газовых
систем.
Приложение закопомерпостей течения химических реакции, рассмат-
риваемых классической теорией химической кинетики, к медленному тер-
мическому превращению жкдкпх и твердых ВВ, происходящему при тем-
пературах ниже температуры их вспышки, затрудняется сложностью
реакций, протекающих в этом случае,
12
Как правило, термическое превращение ВВ включает не одну, а не-
сколько параллельно или последовательно идущих реакций, причем в за-
висимости от условий преобладают различные реакции.
Так, например, аммиачная селитра при температурах около 100° С
обратимо разлагается на аммиак и азотную кислоту по уравнению
КН4КО3ггЛ’ПзЧ-HNOa.
При температуре около 200°С основной реакцией является образова-
ние закиси азота и воды, согласно уравнению
Nir4NO3= N-.0 + 2ILO.
При еще более высоких температурах разложение аммиачной, селитры
можзт быть выращено уравнением
NH4NO3 3/4 1/2NO, + 21GO.
Подобное же изменение состава продуктов разложения с изменением
температуры установлено для термического распада нитроглицерина и
нитроклетчатки [7]. Состав газообразных Продуктов распада (без воды),
полученных при различных температурах опыта, приведен в табл, 1,
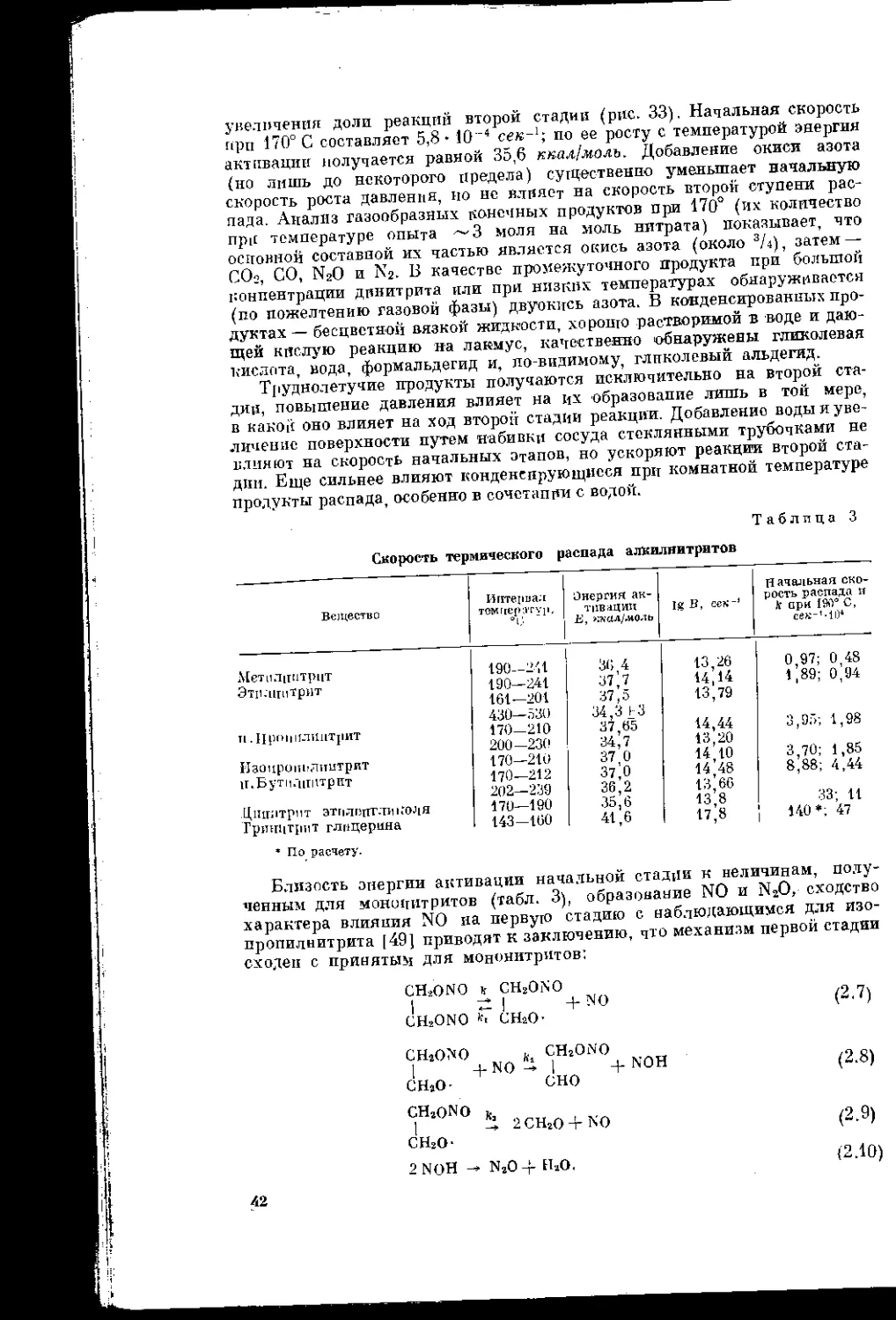
Т а б л п к а 1
Зависимость состава газообразных продуктов распада нитроглицерина
и нитроклетчатки от температуры
Сиет,|п ородунтов распада. %
Продукты распада 1 иггрсни ' ii е [ища 1к:троклстчагкн
Ofj—98J G lik/ C 12')Q C 14d° C
(.ill; 34 30 30,2 35,7
СО (j и 17,4 16,8
1XO 37 21 4 23,G
X.. .\д> 8 11 31 J) 18,5
Объем газов, см3/г , , . . 156,3 ’ 280
Общая потеря а аесе, % — — 48,6 6.5
Даже такие простые соединения, как азиды, например азид кальция
(Са]\б), дают при разных температурах различные продукты распада: при
низких температурах (100—150“ С) [(Случается вещество, соответствующее
эмпирической формуле (CaN)n, в то время как при вспышке образуются
нитрид и металл [8].
* Более того, дли многих ВВ не только при разных температурах полу-
чаются разные продукты распада, но и во время одного н того же опыта,
проводимого при постоянной температуре, состав продуктов также может
меняться.
Из данных Сапожникова [7] видно, что при разложении нитроклетчат-
ки при температуре, близкой к температуре вспышки, в начале опыта со-
держание соединений азота в газообразных продуктах, выраженное в
грамм-атомах, и (>,5 раза превышает содержание соединений углерода;
в конце же опыта содержание азота в газообразных продуктах распада
только в 1,5 раза больше содержания углерода.
При разложении жидких питроэфпров (нитроглицерин, нитро глико лг.,
цетнлинтрат) о эвакуированной и запаянной ампуле при различных тем-
15
перату pax от 70 до 240° С и начальной стадии разложения наблюдалось
сильное побурение газовой фазы в результат!' образования двуокиси азота.
Затем интенсивность окраски газов уменьшалась, и в конце концов они
становились бесцветными. Поскольку анализ показывал е, конечных газо-
образных продуктах распада значительное содержание окиси азота, то
естественно предположить, что обссцвечпвание газов происходило в ре-
зультате окисления двуокисью азота органических соединений, образую-
щихся па начальных стадиях распада, а может быть и исходных пптро-
эфнров.
Как бы то пи было, описанные осложнения течения термического рас-
пада ВВ существенно уменьшают надежность тех выводов, которые были
сделаны в ряде исследований без учета Ьго особенностей.
Рассмотрим исследования по медленному термическому распаду ВВ
для основных пх представителен и некоторые обобщения, которые позво-
ляют сделать имеющиеся данные,
I. НИТРАТЫ И НИТРИТЫ МНОГОАТОМНЫХ СПИРТОВ
Техническое значение имеют преимущественно полные эфиры азотной
кислот]»! многоатомных спиртов — глицерина, этиленгликоля, ди этиленгли-
коли. дпэтаполнитрамина, пептаэритрита, клетчатки ц поэтому термический
их распад изучался бодее подробно. При умеренно повышенных темпера-
турах первые четыре нитроэфира находится в жидком состоянии, поита-
эрптриттетранптрат плавится при 140° С, по может быть «ожижеш» и
ври более низких температурах путем растворения в пилколлавком
растворителе; нитроклетчатка до температуры вспышки не переходит
в жидкое состояние, по как высокополимер с сильно развитой поверх-
ностью ведет себя в некоторых отношениях сходно с жидкими штгроэфи-
рамн. Низкомолекулярные ннтроэфнры могут быть получены и в виде
паров.
Изменение агрегатного состояния существенно отражается на величине
скорости распада, на ее изменении во времени я влиянии на распад неко-
торых примесей. Поэтому рассмотрение и анализ экс пер и ментальных дан-
ных по распаду пнтроэфпров мы проведем с учетом их агрегатного состоя-
ния.
1. НИТРОГЛИЦЕРИН И НИТРОГЛИКОЛЬ
Кинетика термического разложения жидкого нитроглпиерпна впервые
была изучена Робертсоном [9]. Уже в то время по опытам Внлля [10] было
известно, что разложение нитроклетчатки в токе инертного газа идет с по-
стоянной скоростью, а в присутствии продуктов распада самоускоряется
по мерс пх накопления. Поэтому Робертсон, чтобы устранить предположи-
тельное ускоряющее действие газообразных продуктов распада, проводил
свои опыты с нитроглицерином, так же цац и Билль, в токе инертного газа
(СО2), Он установил, что в этих условиях практически весь азот отщеп-
ляется в виде NO2; определение двуокиси азота производилось спектро-
скопически н контролировалось восстановлением окпелов азота над рас-
каленной медью до азота; скорость отщепления азота остается па протя-
жении всего временл опыта (4 часа при 120° С общее количество
отщепленного азота 3%) постоянной п сильно зависит от температуры.
По данным, иолучепцым при различных температурах (90—120° С), укла-
. AN. 1 ,
дывающимся в координатах 1g —дт‘----па прямую, могут быть рас-
считаны энергия активации Е — 43 700 кал/молъ и иредэкспопенциальиып.
множитель В = Ю16’64 сек-1 в уравнении Аррспиуса k = Ве~ЕРт.
14
Обстоятельное исследование термического распада нитроглицерина
было проведено Рогинским и сотр. [11]. Разложение изучалось нри постоян-
ном объеме, и о его скорости судили по повышению давления газообраз-
ных продуктов распада.
Было установлено, что разложение протекает по-разному в зависимо-
сти от температуры ц от степени заполнения сосуда S — отношения объема
нитроглицерина к объему сосуда, в котором он заключен1.
При высоких температурах (150° С) п малых б скорость газообразо-
вания пропорциональна количеству неразложцвгпсгося нитроглицерина.
При моггыних температурах и больших 6 газообразование вначале идет
с малой скоростью, а затем резко ускоряется.
Рогинский объяснил эти закономерности тем, что распад нитроглицери-
на имеет автокаталитический характер, причем катализирующие продук-
ты распада нрп температуре опыта находятся в газообразном состоянии,
и концентрация их в жидком нитроглицерине соответственно зависит от
температуры и давления, которое на определенной стадии распада в свою
очередь пропорционально 6.
Так же как и Робертсон, Рогинский получил большой температурный
коэффициент скорости при малых степенях распада, соответствующий
значительной энергии активации. Поскольку константа скорости также
довольно велика (2,2 • 1СН сек~1 при 120° С), это приводит к аномально
высоким значениям предэкспоненциального множителя (102°—1023,5), не-
сколько изменяющимся при изменении температуры.
Последующие исследовании [12—20] проводились также манометриче-
ским методом, но с использованием стеклянного манометра типа Бурдона,
исключающего недостатки обычного манометриче-
ского метода (контакт паров манометрической или
запорной жидкости с ВВ и продуктами его распа-
да, возможность нх разделения вследствие пере-
гонки в холодные части прибора, особенно при
опытах в вакууме и др.).
Манометр (рис. 1)' представляет собой тонко-
стенную стеклянную мембрану 1 серповидного се-
чения, заканчивающуюся стрелкой 2. Реакцион-
ный сосуд в который помещается навеска иссле-
дуемого ВВ, сообщается с внутренним пространст-
вом мембраны. Образующиеся при разложении ВВ
газы давят па стенки мембраны, вследствие чего
она выгибается, и стрелка отклоняется от перво-
начального положения. В трубку 4. окружающую
мембрану и соединенную с жидкостным маномот-
Рис. 1. Схема стг,вляппо-
го компенсационного ма-
нометра типа Б у рдО | । а
ром, впускают воздух до возвращения стрелки в
нулевое положение. Давление впускаемого возду-
ха отсчитывают (с точностью до 0,5 лея) но жидко-
стному манометру.
Отвод 5 предназначен для введения ВВ, после
чего он запаивается, трубочка 6 с перетяжкой соединяет реакционный со
суд 3 с вакуум-насосом и после откачивания воздуха запаивается.
Опыты показали, что разложенце жидкого нитроглицерина протекает
отчетливо двухстадпппо. При этом наступление и развитие второй стадии
связано с накоплением в жидкости легколетучих продуктов распада, кото-
рое зависит от 6, При малых значениях- 6 вторая стадия может не насту-
пить до конца разложения, напротив при больших- 6 Опа начинается очень
> Часто степень эаполлелия сосуда характеризуется отношением веса ВВ к сво-
бодному объему (m/о). Поскольку т/е = , где j—удельный вес ВВ, то при
1—0
небольших степенях заполнения сосуда б пропорциональна т/г.
(5
о
Рис. 2. Распад Ill’ll, лрн lit)’С ц различных уме-
ренных отношениях mjv (в г/лн3> 1О-4):
Т—5,0; 2 — 9.8; 3—18,9; 4 — 18,0; о — 47
рано. Наступление второй
стадии может быть суще-
1 ственпо ускорено искус-
ственным введением в нит-
роглицерин примесей не-
которых из веществ, кото-
рые образуются при рас-
паде, в частности воды.
Наконец, если вести раз-
ложение при очень малых
значениях б, когда весь
нит.роглицсфин находится
в парообразном состоянии,
то ход распада отличается
от распада жидкого веще-
ства.
В соответствии со ска-
занным мы рассмотрим
разложение нит рог: i яце рл-
па: а) при умеренных б;
б) при больших б; в) в при-
сутствии воды или других
примесей, ускоряющих по-
добно воде или задержи-
вающих развитие разложе-
ния и г) при малых б, т, о,
в парах.
Одновременно с нитро-
глицерином будут рас-
смотрены и данные для
нигроглоколя, сходного с
ним по многим характе-
ристикам распада, хотя
этот нитроэфир изучен го-
раздо меньше.
Разложенце жидкого нитроглицерина при умеренных" степенях запол-
нения сосуда, изучалось между 80 и 11)5° С. Во всем этом интервале газо-
образование идет со скоростью, лишь слабо (по абсолютному значению)
возрастающей но времени. Ускорение выражено тем меньше, чем выше
при прочих равных условиях температура. При 125° С (б = 9,7 ДО-4) мак-
1 ецмальная скорость газообразования превышала начальную лишь при-
мерно на 1/з- Возможно, что даже это незначительное ускорение не яв-
ляется особенностью первой стадия реакции, а обусловлено тем, что одно-
временно хотя и в небольшой степени (соответственно малому давлению
продуктов распада) протекали реакции второй стадия- Более того, по-ви-
димому, па первую стадию б оказывает влияние, обратное тому, которое
она производит на вторую стадию. Так, при разложении нитроглицерина
и нптрогликоля при увеличении 5 в области малых се значений начальная
скорость газообразования (рис. 2 и 3) отчетливо уменьшается, особенно
при повышенных температурах. Для нитроглицерина уменьшается также
максимум скорости, что можно видеть на рис, 2; эти влияния б наблю-
даются и для расплава тэнд.
Разложение жидкого нитроглицерина при больших степенях заполне-
ния сосуда на начальных этапах, пока давление газообразных продуктов
распада мало, идет практически с такой же низкой скоростью, пак и прп
малых б (рис- 4), слабо (~ рр,/з) возрастающей в .ходе распада.
После того, однако, как давление' достигнет некоторого Критического
16
значения скорость начина-
ет расти гораздо быстрее
(~р2) и может даже про
низких температурах до-
стигать весьма больших
значений, в сотни и тыся-
чи раз превышающих на-
чальное. При Этом меняет-
ся и состав газов, о чем
можно судить по резкому
усилению их бурой окрас-
ки (накопление NO2) ц по
сильному увеличению доли
газов, конденсирующихся
при комнатной температу-
ре. Светлов [21] определил
растворимость в нитрогли-
церине двуокиси азота в
интервале 20—80u С и при
изменении ее давления от-
100 до ООО мм рт. ст. Рас-
творимость пропорцио-
нальна давлению в степе-
ни 1,5—2,3, что следует,
по-видимому, объяснить
преимущественным рас-
(_ творением димера N2O4.
^Действительно, расчет по-
J называет, что если отно-
\сить концентрацию N2O4 в
^растворе к концентрации
\ее в газовой фазе, то меж-
ду ними соблюдается пря-
мая пропорциональность.
Константа растворимости
меняется от 0,00170 .мм-1
при 20° до 0,00021 мл»-1
при 80° С. При критиче-
ском давлении (80°) кон-
центрация N2O4 в нитро-
глицерине колеблется в
пределах 0,001—0,004%.
Сопоставляя эти данные с
зависимостью скорости
второй стадии распада от
давления, Светлов заклю-
чил, что она оцределяет-
Рцс. 3. Распад НI'Jj [грп 140° С и различных уме
ренггых отношениях m/v (в г/см3-IO-1):
7 — 10,9; г — 13,5; 3 — Г8,3; 4 — 45,9; 5 — Г17.2
Рис. 4. Распад НГЦ при 10(Г (1 и повышенных от-
ношениях m/v. Цифры пад кривыми — отноше-
ние m/v в г/слГ.Щ4
ся окислением, осуществ-
ляемым четырехокисью
азота. При 100° С в зна-
чительном интервале
S(14-10"4 - 440-Ю-4)
резкое измонопне темпа роста скорости газообразования наступает практи-
чески при одинаковом давлении (180 — 200 мм), хотя времена его достиже-
ния различаются в десятки раз. При этом, если до достижения критического
давления скорость газообразования несколько зависят ыт S, уменьшаясь
в общем при ее увеличении, то при давлениях выше критического зпаче-
2 К. К. Андреев
17
Рис. 5. Влияние давления продуктов
распада на скорость разложения
НГЦ при 80, 100 и 120° С и умерен-
ных отношениях miv (в г/см$)
Г — 543'10-'; 2 — 507,2-10-'; 3 — 537'10-'
ния скорости при данном давлении продуктов распада практически оди-
наковы, независимо от значения 5. Лишь при очень больших 5, т. е. при
быстром росте давления во времени, иначе говоря при малых индукцион-
ных периодах, величина критического давления растет; при 6, близкой к1,
оно составляет 800—900 мж.
Таким образом, критическое давление в некоторой мере зависит от
степени распада; на более поздних его стадиях газы несколько более эф-
фективны, чем в самом начале распада,
По-видггмому это связано с изменением
состава газов \ в частности с обогаще-
нием их водой. Время наступления рез-
кого ускорения, естественно, зависит от
5, уменьшаясь с ее ростам, и при 6, близ-
кой к 1, составляет около 9 час.; количе-
ство образовавшихся к этому времен!
газов порядка 1 см3!г.
Критическое давление зависит так
же от температуры опыта, возраста!
с ее увеличением (рис. 5). При 80° он<
составляет 60 —80 ж, при 120°С-
400—500 зсм. Эти данные относятся ।
Ю-2; при 6, близкой к 1, притиче
ское давление при 80° возрастает Д1
500 .иле; при 60° С оно составляет 250 .нм
Эта зависимость, по-видимому, опре-
деляется в основном изменением раство
римостн газообразных продуктов распада в жидком нитроглицерине, а так
же изменением констант скорости первой и второй стадий. На скорость вто
рой стадии температура практически не влияет. Точнее говоря, это влия
иие, почв ид и мом у, настолько слабо, что увеличение скорости при повышены]
температуры компенсируется уменьшением концентрации газообразны:
продуктов. При низких температурах и больших д влияние температур!
на скорость второй стадии становится более заметным (рис. 6), но он,
по-прежнему остается гораздо более слабым (Е = 15 ккал/моль), чем дои
первой стадии.
Зависимость скорости распада на ранних его стадиях от температурь
была рассчитана для опытов как при малых, так и при умеренных б- В обо
их случаях она выражается прямой в аррениусовских координатах, но :
первом зависимость сильнее (Е = 43,7 ккал/моль и IgB = 18,4 сек-1), чез
во втором (Е = 39,3 ккал/моль и 1g В — 15,4 сек-1), Интересно, что пер
вая пара величин тождественна той, которая была определена Робертсон»!
при удалении газообразных продуктов распада. Это подтверждает, что и;
первую стадию последние действительно не оказывают ускоряющее
влияния.
Вопрос о реальности критического давления является спорным, Воз
можно [16, 17], что наблюдаемый характер кривой Иг(р) обусловлен тем
что газообразование определяется двумя одновременно протекающим:
реакциями, скорость одной из которых не зависит от давления, а дру
гой — пропорциональна квадрату давления. Аргументом в пользу этог
предположения является выполнение зависимости, характерной для втс
рой стадии на протяжении значительной части индукционного период,
если по оси ординат откладывать текущую скорость газообразования з
вычетом начального ее значения (рис. 7).
Разложение нитроглицерина в присутствии ускоряющих или задержь
вающих развитие процесса примесей (вода, кислоты, окислы азота, code
1 Другое объяснение зависимости критического давления от 5 см. в работе [1S
18
мел). Горбунов [22] определил растворимость воды в нитроглицерине'
в интервале давлений 16—120 мм и температур 30—90° С. Константа рас-
творимости (отношение концентрации воды в нитроглицерине к равно-
весному давлению ее паров) уменьшается с температурой по уравнению
1g Л'— 9,114 -4-1550/Г.
Ниже приводится содержание воды в насыщенном ее растворе (в %)
при различных температурах:
20° С 40° С 60° С 80°С 100° С 120° С
0,26 0,38 0,51 0,66 0,81 0,97
Таким образом, вода плохо растворима в нитроглицерине; поэтому се
содержание в нем можно изменять только в относительно узких пределах,
что достигается изменением давления паров воды над нитроэфиром.
Характер кривых p(t) при разложении нитроглицерина в присутствии
воды зависит от ее относительного содержания; при малых количествах
воды он в общем такой же, как для безводного нитроглицерина (рис. 8).
Давление растет сначала медленно, но быстрее, чем для безводного нитро-
глицерина, и обычное ускорение газообразования наступает раньше. Оба
эти отличия выражены тем сильнее, чем больше содержание воды.
Рис. 7. Влияние давления продуктов
распада на скорость разложения НГЦ
I — опыты при 80, 100 и 120° С (из теку-
щей скорости вычтена начальная скорость)'
г — исходные данные для опыта при 100° С
Рис. 6. Влияние давления продуктов
распада па скорость разложения НГЦ
при 60, 80 л 100° С и степени заполне-
ния, равной ~ 1
При умеренных количествах воды (выше 30—50 мм) изменение дав-
ления во времени своеобразно (рис. 9). После установления равновесного
давления оно некоторое время остается постоянным, затем быстро и силь-
но падает, после чего начинает более или менее ускоренно расти. Индук-
ционный период (время до наступления падения давления) сильно колеб-
лется, но в общем имеет тенденцию к росту при увеличении количества
воды; повышение температуры сокращает индукционный период.
1 Горбунов определил также растворимость воды при 120° С в дигликольдинитрате
и динитроглицерине; в первом из них она меньше, чем в нитроглицерине (констан-
та растворимости 6,6 10-6 мм-1 для нитроглицерина и 5,2 10-в льи-1 (для дигликоль-
динитрата), во втором — значительно больше: 36 10-6 мм-1.
2*
19
Интересно изменение конденсирующихся при комнатной температуре
газов в ходе разложения (рис. 10). Оно уменьшается па стадии падения
давления и медленно начинает расти при последующем его подъеме. Ха-
рактер изменения давления после временного перерыва разложения раз-
личен до минимума давления и после него. Газы, не конденсирующиеся
при температуре жидкого азота, появляются только на стадии резкого
ускорения газообразования.
Рис. 8. Распад НГЦ при 100° С и ма-
лых количествах добавленной воды
Числа у кривых — равновесное давле-
ние паров воды, в скобках — 6 Ю*1
Рис. 9. Распад НГЦ при 100° С и умерен-
ных количествах воды при б = 300-10-4
Числа пад кривыми соответствуют начальному
равновесному давлению паре а воды
При больших количествах воды характер кривой p(t) вновь меняется.
В начале опыта наблюдается медленное и небольшое падение давления
и затем столь же медленный его рост. Резкое ускорение наступает значи-
тельно позже, чем даже при разложении сухого нитроглицерина. Двой-
ственный характер влияния воды (ускорение при малых содержаниях,
замедление при больших) особенно отчетливо проявляется в опытах при
малых 6 (рис, 11).
Рис. 10. Изменение дав-
ления и количества кон-
денсирующихся гдзов
при 20 и (Г С в ходе раз-
ложения ПГЦ при 80° G
в присутствии воды
PliiO = I35 л,-ч Рт' ст.; 8 “
—
Увеличение 6 при данном давлении паров нодьг, означающее умень-
шение общего ее количества при постоянном начальном содержании в
нитроглицерине, ведет к сокращению индукционного периода.
2»
Опыты по влиянию воды были поставлены также пр» низких темпера-
турах и больших 6 [23]. В этих условиях время до резкого ускорения со-
кращается водой относительно слабее, например при 100° С и 0,2% воды
(PiijO — ЮО л/л/) только в 2,5 раза, а при 60 и 40° С — возможно даже не-
сколько меньше; при дальнейшем увеличении содержания воды (до 0,4%)
индукционный период продолжает сокращаться, но медленнее. На стадии
резкого ускорения наблюдается та ще зависимость w р2, но коэффи-
циент пропорциональности несколько больше, нем для сухого нитроглице-
рина.
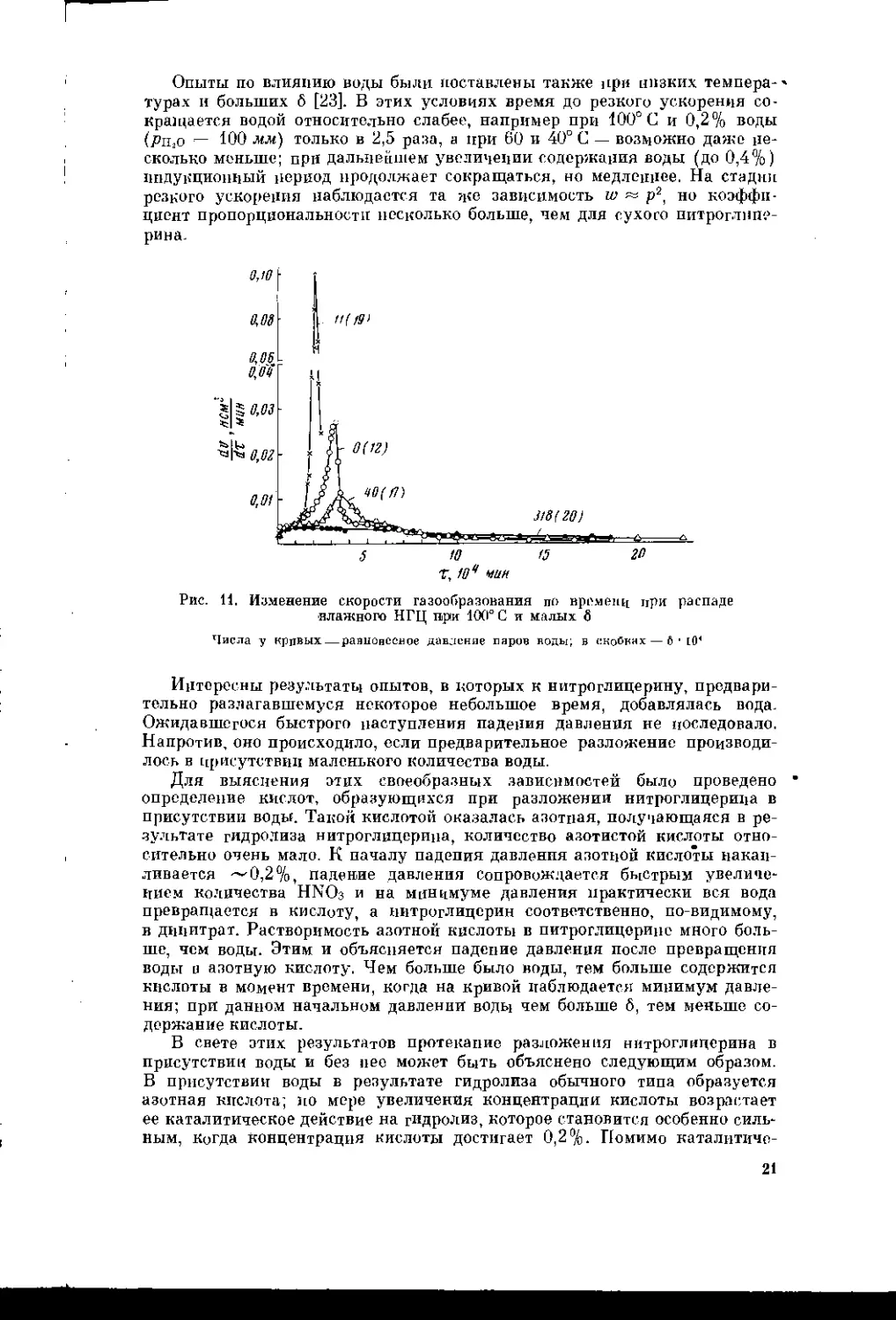
Рис. 11. Изменение скорости газообразования по времени при распаде
влажного НГЦ при 100° С я малых 5
Числа у кривых — равновесное давление паров поды; в скобках — б1 [0<
Интересны результаты опытов, в которых к нитроглицерину, предвари-
тельно разлагавшемуся некоторое небольшое время, добавлялась вода.
Ожидавшегося быстрого наступления падения давления не последовало.
Напротив, оно происходило,если предварительное разложение производи-
лось в присутствии маленького количества воды.
Для выяснения этих своеобразных зависимостей было проведено
определение кислот, образующихся при разложении нитроглицерина в
присутствии воды. Такой кислотой оказалась азотная, получающаяся в ре-
зультате гидролиза нитроглицерина, количество азотистой кислоты отно-
сительно очень мало. К началу падения давления азотной кислоты накап-
ливается -—-'0,2%, падение давления сопровождается быстрым увеличе-
нием количества HNO3 и на минимуме давления практически вся вода
превращается в кислоту, а нитроглицерин соответственно, по-видимому,
в дииитрат. Растворимость азотной кислоты в нитроглицерине много боль-
ше, чем воды. Этим и объясняется падение давления после превращения
воды в азотную кислоту. Чем больше было воды, тем больше содержится
кислоты в момент времени, когда на кривой наблюдается минимум давле-
ния; при данном начальном давлении воды чем больше 6, тем меньше со-
держание кислоты.
В свете этих результатов протекание разложения нитроглицерина в
присутствии воды и без нее может быть объяснено следующим образом.
В присутствии воды в результате гидролиза обычного типа образуется
азотная кислота; ио мере увеличения концентрации кислоты возрастает
ее каталитическое действие на гидролиз, которое становится особенно силь-
ным, Когда концентрация кислоты достигает 0,2%. Помимо каталитиче-
21
ско г о действия, накопление кислоты, несомненно, благоприятствует гидро-
лизу вследствие увеличения растворимости воды в нитроглицерине —
увеличивается эффективная концентрация этого участника гидролиза. На-
ряду с этим следует учитывать, что скорость катализированного азотной
кислотой гидролиза, очевидно, проходит через максимум при некотором
содержании кислоты. По-видимому, сочетание всех этих влияний и при-
водит к резкому ускорению гидролиза, наблюдающемуся на стадии паде-
ния давления.
* Наряду с каталитическим действием па гидролиз образующаяся азот-
ная кислота оказывает и окисляющее действие. Однако при тех концен-
трациях, которые уже сильно влияют на гидролиз, окисляющее действие
азотной кислоты особенно в отсутствии окислов азота еще относительно
слабо. По-видимому, когда гидролиз закончился, концентрация азотной
кислоты оказывается уже достаточной для значительного убыстрения окис-
ления тем более, что одновременно возросла и концентрация динитроглн-
церипа, окисляющегося легче, чем нитроглицерин.
Разложение смесей нитроглицерина с динитроглицерином и азотной
кислотой протекало очень сходно с тем, которое наблюдалось после мини-
мума давления при разложении нитроглицерина в присутствии воды. Оки-
сление азотной кислотой, которое развивается тем быстрее, чем больше
было воды и чем больше 6, является самоускоряющимся процессом — обра-
зующаяся двуокись азота является гораздо более энергичным окислите-
лем, чем одна азотная кислота, а при окислении динитрата, очевидно, так-
же образуются высшие окислы азота. Наряду с этим получается вода, ко-
торая п кислой среде быстро гидролизует нитроглицерин, поставляя таким
образом дополнительную пищу для окисления, а возможно ускоряя также
и само окисление, как об этом можно судить по резкому усилению уско-
ряющего действия смеси NO и NCb на газообразование в присутствии
воды [24].
Эти данные и соображения позволяют объяснить и различный характер
кривой p(t) в присутствии различных количеств воды и отсутствие влия-
ния предварительного разложения сухого нитроглицерина на последующее
его разложение в присутствии воды. Если воды мало, то роль гидролитиче-
ской реакции по сравнению с «безводным» распадом мала и быстро дости-
гается такая концентрация кислоты, при которой преобладает ее окисли-
тельное действие; скорость окисления, однако, невелика из-за малой кон-
центрации кислоты. Восстановление превращает высшие окислы азота
в NO, влияние которой на распад очень мало. В итоге разложение разви-
вается по типу безводного распада, но в целом быстрее за счет того, что
дополнительно к безводному распаду окисление производится образующей-
ся при гидролизе азотной кислотой.
При умеренных количествах воды гидролиз превращает ее в азотную
кислоту, которая, окисляя преимущественно динитрат, восстанавливается,
образуя NO2 и воду, ускоряющие как гидролиз, так и окисление органи-
ческих промежуточных продуктов распада.
При большом количестве воды она разбавляет.образующуюся при гид-
ролизе кислоту и этим не дает возможности развиваться как гидролизу,
так и окислительным реакциям. Поэтому наступление ускорения может за-
держиваться даже по сравнению с сухим нитроглицерином.
* При разложении сухого нитроглицерина образующаяся двуокись азота,
по-видимому, в основном восстанавливается и после добавления воды кис-
лоты, во всяком случае азотной, не образуется 1. Если же разложение на-
чинается в присутствии воды, то окисление в начале из-за разбавления
1 Если нитроглицерин разлагается в присутствии кислорода, то ускорение насту-
пает быстрее, очевидно, благодаря превращению образующейся при восстановлении
окцен азота обратно в двуокиси.
22
не идет и кислота накапливается в количестве, достаточном для ускорения
гидролиза дополнительно введенной водой.
При разложении сухого нитроглицерина без последующего введения
воды резкое ускорение начинается, по-видимому, тогда, когда в резуль-
тате окисления накапливается достаточно воды и окислитель образуется
не только в процессе «сухого» распада, но и гидролиза, идущего быстро,
так как концентрация кислоты велика.
Развитие окислительных процессов зависит не только от содержания
летучих окислителей, но и от количеств и характеристик окисляемых ве-
ществ. Это заключение было подтверждено как при «водном», так и при
«безводном» разложении нитроглицерина. Из нитроглицерина, разлагавше-
гося в присутствии воды, после достижения минимума давления откачи-
вали летучие продукты и затем продолжали опыт. Газообразование в на-
чале было очень близко к тому, которое давал неразлагавшийся нитрогли-
церин, но ускорение наступало несколько раньше.
Если подготовленный таким же образом нитроглицерин разлагать в
присутствии паров воды, то ход Кривой p(t) и индукционный период при-
мерно такие же, но последующее ускорение развивается быстрее.
Наконец, если сухой нитроглицерин разлагать, добавив к нему лету-
чие продукты распада, то разложение развивается медленнее, чем на со-
ответствующей стадии разложения влажного нитроглицерина; это пока-
зывает, что перазлагавшикся нитроглицерин менее чувствителен к дей-
ствию продуктов распада, чем частично разложенный.
Заключения, вытекающие из этих опытов, подтверждаются также дан-
ными по разложению динитроглицерина в присутствии воды — окисление
наступало раньше и происходило более интенсивно, чем при разложении
нитроглицерина.
Образование промежуточных продуктов установлено и при «безводном»
разложении нитроглицерина па стадии резкого ускорения [25]. Нитрогли-
церин разлагался на ~10% при 100°С, после длительной откачки летучих
продуктов разложение продолжалось при этой и более низких температу-
рах (рис. 12). Газообразование идет со скоростью, слабо зависящей от тем-
пературы (£ = 20 ккал/молъ), по насыщающейся кривой во много
( ~ 1000) раз быстрее, чем при разложении свежего нитроглицерина. Лишь
значительно позже, при р > 600—700 мм наблюдается резкое ускорение
распада. Авторы отмечают сходство полученных кривых p(i) с кривой
разложения нитроглицерина, к которому добавлено немного щавелевой
кислоты.
Если добавление воды как агента гидролиза, образующего азотную
кислоту, ускоряющую гидролиз и окисляющую органическую часть моле-
кулы, ускоряет разложение нитроглицерина, то естественно ожидать того
же влияния при добавлении непосредственно азотной кислоты а также и
других кислот {26]. Действительно, введение небольшого (0,3 %) количе-
ства азотной кислоты увеличивает при 100° С начальную скорость газо-
образования и сокращает время до резкого ускорения, хотя это действие
при равных весовых количествах значительно слабее, чем воды; по-види-
мому, устойчивость нитроглицерина к окислению значительно больше, чем
к гидролизу.
Принимая во ввимание ускоряющее влияние азотной кислоты па гидро-
лиз, можно было ожидать, что она будет еще сильнее увеличивать скорость
разложения влажного нитроглицерина. Действительно такое ускорение
наблюдается, если содержание азотной кислоты составляет по отношению
к нитроглицерину 0,1% и более, но отсутствует при 0,02% кислоты. Это
влияние проявляется в значительном сокращении индукционного периода,
но не отражается на последующем ускорении. Последнее возрастает лишь
при значительно больших содержаниях кислоты, например 1,5%. Ана-
логичное влияние на индукционный период оказывает трихлоруксусная
23
кислота. Влияние щавелевой кислоты более сложно, поскольку она не
только кислота, но и восстановитель. При малых содержаниях ее наблю-
дается увеличение индукционного периода, при больших ускорение газо-
образования наступает быстрее, чем для чистого нитроглицерина. Однако
форма кривой p(t) существенно иная.
Наступление убыстряющегося разложения нитроглицерина ускоряется
также окислами азота — слабо окисью, сильнее двуокисью и еще сильнее
при их совместном присутствии
[24]. Оцпсь азота не влияет на
величину индукцЕЮнцого перио-
да п присутствии воды, но устра-
няет цаде.нце давления, вероят-
но, в результате восстановления
ааотцой кислоты до азотистой,
которая предположительно ме-
нее растворима в нитроглицери-
не, так же как и окислы азота.
нгц too"
ЮО 200 300 000 500
г, MU ft
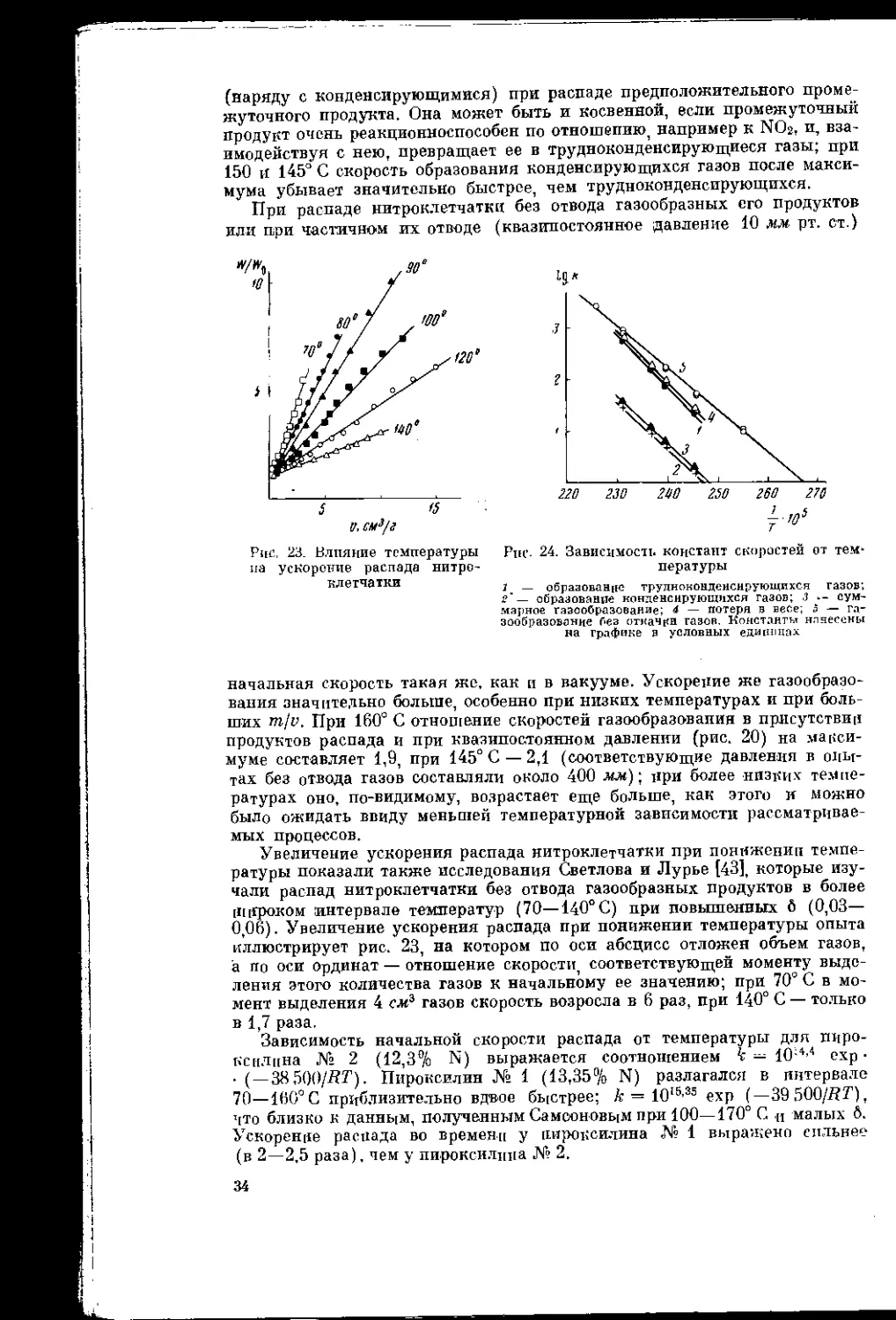
Рис. 12. Распад предварительно разлагавшегося Рис. 13, Влияние состава до-
и эвакуированного НГЦ ври о = 0,03—0,04 ба вл с лисп кислоты на время
индукционного периода при
распаде НГЦ
по энергичнее его окисляет. Особенно сильно ускоряется газообразование
при совместном присутствии воды, окиси азота и двуокиси азота; индукци-
онный период в этом случае исчезает и время достижения определенного
давления сокращается более чем в 100 раз.
Описанные опыты цо влиянию кислот проводились при умеренных б
и 100° С. При больших б [23] влияние азотной кислоты относительно сла-
бее. Так, при 100° С и б, равной —'l, добавление 0,2% HNOj сокращает
время до быстрого ускорения в 1,5 раза слабее, чем то же количество воды-
При более цизких температурах действие азотной кислоты больше и силь-
нее, чем воды. Так, при 40° С ускорение разложения яри 0,17% HNCb
наступило в 4 раза раньше, чем в присутствии воды. По-видимому, ско-
рость окисления слабее зависит от температуры, чем скорость гидролиза;
кроме того, при низких температурах скорость «безводного» распада сни-
жается столь сильно, что он практически перестает играть роль источника
кислоты.
Было изучено также влияние концентрации водной азотной кислоты,
добавляемой в количестве 0,2% на индукционный период при 40 ц 60J С
(рис. 13). Сильнее всего влияет 25%-ная кислота.
Если в развитии разложения нитроглицерина в присутствии воды зна-
чительную роль играет азотная кислота как катализатор гидролиза и окис-
литель, то нейтрализация ее должна замедлять процесс. Сода, введенная
в малых количествах (0,07%) в безводный нитроглицерин, практически
не изменяет течения его распада при 100° С. Это следует объяснить тем,
что сода не влияет на первичные процессы распада, а на стадии резкого
24
ускорения количество кислоты несравненно больше, нем указанное выше
количество соды. Напротив, в присутствии воды сода значительно увели-
чивает индукционный период (количество кислоты к началу падения дав-
ления соизмеримо с количеством соды) При применении больших коли-
честв соды (введенной не в жидкость, а н боковой отросток реакционного
сосуда) ускорения разложения не наступило в течение 30 суток при 100° С,
в то время как без соды оно начинается через несколько часов. Аналогич-
ное действие оказывает мел. Если разбавленной азотной кислоты добав-
лено так много, что часть ее не растворяется в нитроглицерине, то индук-
ционный период может сильно возрастать.
По полученным данным в опытах с большим б был рассчитан [18] эк-
страполяцией индукционный период при 20° С и 5, близкой к 1. Для без-
водного и нейтрального нитроглицерина он составляет 17 лет, для нитро-
глицерина, содержащего 0,2% примеси воды— 10 лет, безводной азотпои
кислоты — 1,4 года, разбавленной азотной кислоты — 3 месяца.
2. ПЕНТАЭРИТРИТТЕТРАНИТРЛТ (ТЭН) fC(CH2ONO2)4]
Медленный распад тзца привлекал внимание исследователей по трем
причинам. С одной стороны, тэн как твердое ВВ с большой теплотой взры-
ва применялся в боеприпасах, предназначенных для длительного хране-
ния. С другой стороны, стойкость тэпа гораздо больше, чем других, близ-
ких к нему по энергии взрыва нитроэфиров. В-третьих, добавление к тэну
таких стойких ВВ, как тротил, или даже некоторых певзрывчатых веществ,
снижало при испытании по обычпым пробам его стойкость [27—30].
Исследования показали [31, 32], что две последние особенности связаны
с тем, что тэн испытывался при тех температурах, при которых он нахо-
дится в твердом состоянии. Будучи переведен расплавлением или раство-
рением в жидкое состояние, тэн распадается в десятки раз быстрее 1 и по
скорости и характеру распада становится близким в нитроглицерину и
питрогликолю. В ходе распада твердого тэна образуются конденсирован-
ные промежуточные продукты, растворимые в тэде п снижающие соответ-
ственно температуру его плавления. Это приводит к тому, что все боль-
шая часть твердого тэна переходит в ходе распада в жидкое состояние,
благодаря чему общая скорость распада возрастает дополнительно к тому
увеличению, которое наблюдается при распаде твердого тэна. Этим же
объясняется и увеличение скорости распада тэна при добавлении к нему
химически стойких, но растворяющих его примесей. При температурах
ниже эвтектической точки такое влияиие не должно проявляться.
Зависимость скорости распада от температуры не укладывается для
твердого тэна в уравнение Аррениуса, возможно, вследствие изменения
доли тэна, находящегося в жидком состоянии при различных температу-
рах, из-за примесей или конденсированных продуктов распада, снижаю-
щих температуру плавления. По точкам для низких температур Е —
= 51,5 KKitjiMOAb. Бода или кислород ускоряют распад твердого тэна; осо-
бенно сильно проявляется это ускорение мри их совместном присутствии.
Термический распад тэна в жидком состоянии изучал Л. Робертсон [33]
при относительно высоких температурах (160—225° С) с малыми навес-
ками (1 мг и несколько выше), чтобы обеспечить быстрый прогрев и све-
сти до минимума саморапогрев. Тэп помещали в стеклянной ложечке в
верхней части пробиркн, нижний конец которой был погружен в баню.
При повороте ложечки тэн падал в пробирку. Течение распада характери-
зовалось по повышению давления. В пробирке находился воздух или азот
(под давлением 5; 10 и 76 cat), так как в вакууме тэн быстро испарялся
1 Отношение скоростей в жидком н твердим состоянии увеличивается при пони-
жении температуры; при 130° оно равно 40, при 110° С — 100.
25-
и перегонялся в холодные части прибора. Присутствие указанных газов
И величина их давления не влияли на скорость распада.
Кривые давление — время в интервале 161—233° С показывают почти
постоянную скорость на протяжении приблизительно всей первой поло-
вины распада, после чего скорость падает в соответствии с уравнением
реакции первого порядка.
Константа скорости для первых этапов распада определялась по на-
клону начального прямого участка кривой и общему повышению давле-
ния при опыте
к = exp (—47 000/7??1).
В 5%-ных растворах тэпа в дициклогексилфталате при 171 —238° С ход
кривых давление — время в общем тот ясе, что и для расплава. Значения
константы скорости при умеренных температурах (171° С) очень близки
для расплава и для раствора (4,8-10"4 и 5,0-IO"4 еек~1 соответственно);
при повышенных температурах (папример при 238° С) они существенно
отличаются, составляя 0,49 сек"1 для расплава и 0,17 сек-1 для раствора.
Для раствора 1016-1 ехр (—39500/Й71).
Таким образом, по Робертсону кинетические коэффициенты (£ и В)
распада тэна в расплаве значительно больше, чем в растворе. Вероятнее
всего это объясняется тем, что в случае расплава при повышении темпе-
ратуры опыта все большая часть тэна находится в парообразном состоя-
нии, в котором скорость распада выше чем в жидкости. Соответственно
увеличивается температурный коэффициент скорости, а также Е и В.
В другом исследовании [31, 32] распад жидкого тэна изучали при более
низких температурах посредством измерения давления газообразных про-
дуктов при помощи манометра типа Бурдона. В расплаве (145—171° С)
и в растворе в тротиле (100—145° С) при малых б тэн распадается сходно
с нитроглицерином (рис. 14), но заметно (вдвое при 145° С) медленнее.
Абсолютная скорость растет до ~30% распада и на максимуме в 2,5 раза
превышает начальное значение. Кривые давление — время хорошо совме-
щаются путем изменения масштаба времени, что дает Е = 39 ккал/моль
и 1g В — 15,6 сек"1; константа скорости при 160° С равна 6,86-10-5 сек-1.
Опыты в растворах показали, что растворитель не увеличивает скоро-
сти распада, а несколько снижает ее, особенно в разбавленных растворах;
скорость не зависит также от концентрации тэна, т. е. распад является
реакцией первого порядка.
При больших б, так же как и для нитроглицерина, зависимость W — р
в логарифмических координатах может быть представлена в виде двух
прямых, причем на участке резкого ускорения скорость (при 100° С) про-
порциональна квадрату давления; при очень больших б скорость на обоих
участках несколько меньше чем при умеренных 6. По начальным скоро-
стям газообразования Е — 40,1 ккал/моль и IgB = 15,8 сек-1, что очень
близко к величинам, полученным для расплава.
Как и в случае нитроглицерина, вода сильно сокращает время до на-
ступления быстрого ускорения при распаде тэна (в тротиловом растворе).
Кислород не влияет на скорость газообразования на начальном этапе рас-
пада тэна при 145° С, но сокращает время до резкого ускорения.
В работе Робертсона [33] был определен также состав газообразных
продуктов распада тэна. После двухминутного нагревания тэна при 210°
продукты распада наряду с формальдегидом и водой содержали следующие
газы (в молях на моль тэна): NO2 — 0,51; NO — 2,11; N2O — 0,42; N2—
0,07; На — 0,09; CO — 0,93; CO2 — 0,28. При продолжении нагревания
реакционного сосуда двуокись азота в результате вторичных реакций ис-
чезает, а количества окиси и двуокиси углерода увеличиваются.
26
Рис. 14, Влияние температуры на разложение тана при малых fi
Чтобы определить количество первично образующейся двуокиси азота,
распад проводился в специальном приборе в токе азота. Этим методом было
получено 1,1 моля NO2 на моль тэна при полном разложении при 190° С.
Остальной азот получался главным образом в виде окиси и закиси, как п
при статических опытах. Проточным методом подтверждено также образо-
вание формальдегида при распаде. Робертсон предполагает, что первич-
ной ступенью при распаде тэна является прямое отщепление молекулы
NOa, за которым следует или отщепление остатка — СНгО — в виде фор-
мальдегида или окисление Этого остатка соседней группой NO2. В послед-
нем случае образуется остаток ONO, от которого может непосредственно
отщепиться окись азота.
3. ДИЭТИЛЕНГЛИКОЛЬДННИТРАТ (OaNOCHstCHsOCHjCHaONOa)
Наиболее существенной особенностью термического распада рассмот-
ренных выше нитроэфиров (нитроглицерин, нитрогликоль, тэн) является
двустадпйность распада, причем вторая стадия развивается под влиянием
кислот, образующихся при распаде и ускоряющих гидролиз нитроэфира
2;
водой, являющейся продуктом распада или примесью. Для того чтобы
гидролиз протекал быстро, необходимо накопление значительного количе-
ства азотной кислоты. Однако азотная кислота и высшие окисли азота,
из которых она может образоваться, яцляются энергичными окислителями.
Поэтому, если сам нптроэфир пли промежуточные продукты его превра-
щения достаточно полно восстанавливают азотную кислоту, то ускорения
гидролиза кислотой, а следовательно, и второй стадии распада не насту-
пает. Примерами таких нитроэфиров являются диэтплеи гликоль динитрат
и дипа, изучавшиеся Светловым и сотрудницами [34—36], а также нитро-
клетчатка.
Распад дпэтилснгликольдипнтрата исследовался в интервале 60—150° С
в значительном диапазоне 6 (0,0013—0,95). Начальная скорость умень-
шается, как н для других нитроэфиров, при увеличении 6 и возрастает с
температурой; к = 10t6"5 exp (—42500//?/'). По абсолютной величине ско-
рость при 100° приблизительно в 5 раз меньше, чем для нитроглицерина, и
близка к скорости распада нитрогликоля и нитроклетчатки. В отличие от
нитроглицерина зависимость скорости от температуры не меняется во всем
изученном ее интервале. Вследствие этого при ппзких температурах, на-
пример цри 6О°С, скорость распада диэтиленглцкольдпнитрата оказывает-
ся значительно (в 20 —Зо раз) меньше, чем нитроглицерина.
Газообразование идет с малым и практически постоянным ускоре-
нием — скорость на максимуме лишь в 3—4 раза превышает начальную.
Известно, что наступлению резкого самоускорепия благоприятствуют
низкая температура и большая степень заполнения сосуда. Однако для
дпэтиленгликольдиннтрата при увеличении 5 до 0,95 и снижении темпе-
ратуры опыта до 80—60° С перехода распада во вторую стадию не наблю-
дается. С этим согласуется и отсутствие влияния воды на ход и скорость
разложения. По-видимому, скорость гидролиза мала по сравнению со ско-
ростью безводной реакции.
Наблюдающийся небольшой рост скорости, вероятно, обусловлен про-
теканием послодо1вательпых реакций, включающих связывание, а затем и
восстановление NO2; за это говорит, в частности, изменение состава гадов,
происходящее уже ла ранних стадиях распада,— доля конденсирующихся
при компатпой температуре газов заметно уменьшается. В продуктах рас-
пада при различных ого условиях двуокиси азота в измеримых (>1 м.и)
количествах не обнаруживается. Это не означает, что двуокиси азота не
образуется при первичном акте распада, но она быстро исчезает, образуя
конденсированные соединения или восстанавливаясь путем взаимодей-
ствия с остатком молекулы; опыты показали, что двуокись азота, добав-
ленная к диэтилепгликольдинитрату, исчезает.
Связывание двуокиси азота происходит и независимо от разложения
нитрата, поскольку, с одной стороны, оно наблюдается и для самого ди-
этиленгликоля. а с другой стороны, для питрата происходит со скоростью,
значительно, превосходящей скорость разложения особей по прн низких
(например 60° С) температурах. В связи с этим в начале наблюдается не-
большое падение давления, в дальнейшем же оно растет гораздо быстрее,
чем в отсутствие примеси. Чем ниже температура, тем больше увеличение
скорости распада, вызываемое двуокисью азота.
Снижение давления наблюдается и при распаде диэтилевглнкольдипп-
трата в присутствии окиси азота; последующий рост скорости происходит
в этом случае несколько быстрее, чем в ее отсутствие. Кислород влияет на
ход распада аналогично двуокиси азота. В начале наблюдается падение
давления, а затем его рост со скоростью, которая при больших количе-
ствах кислорода может в десятки раз превышать скорость газообразова-
ния при распаде нитрата без добавок. Одпако ускорения газообразования
во времени не наблюдается. Прп распаде в присутствии кислорода наблю-
дается образование значительных количеств NOa- Роль кислорода по ис-
28
черпывается образованием NOa, на что указывает связывание кисло рю да
в начале опыта, которое наблюдается не только для нитрата, но и для
самого диэтнленгликоля.
Концентрированная азотная кислота приводит после короткого индук-
ционного периода к быстрому разложению, идущему, однако, с умень-
шающейся во времени скоростью. Разбавленная азотная кислота вызывает
падение давления в начале опыта и более резкое ускорение его роста в
дальнейшем, но-видимому, в результате гидролиза и последующего окис-
ления. Ускорение распада днэтилепгликольдипитрата наблюдается такясо
в присутствии щавелевой кислоты.
Существенным отличием распада диэтплепглпкольдипнтрата с изучав-
шимися примесями и без них является то, что значительного роста ско-
рости во времени не наблюдается, хотя под влиянием примесей скорость
и может в десятки раз превышать скорость распада чистого вещества. Ос-
новным фактором антостабцлизации распада диэтиленгликольдпнитрата
является большая скорость восстановления высших окислон азота, образу-
ющих с водой кислоты, до низших, не способных к этому. В результате
при распаде не осуществляются условия для быстрого протекания гидро-
лиза, создающего материал, способный к быстрым самоускоряюшимся
окислительно-восстановительным реакциям.
4. ДИНА [ДИНИТРОКСИЭТИЛ НИТРАМИН, ЫКОЙ(СНЙСНЙОКО2)21
Светлов и Лурье [36] установили, что при относительно высоких тем-
пературах и малых значениях б дина разлагается качественно и количе-
ственно сходно по характеру кривых p(t) с диэтилепгликольдиннтратом.
Рис. 15. Разложение диры при 160° С п
малых 6;
а: 1 — 0,0005; 2 — 0,002; б — проверка подчине-
ния разложения дины первому порядку при
6 - о.оооо
Рис. 16. Изменение скорости газо-
образования при распаде дипы в за-
висимости от количества выделив-
шихся продуктов разложения при
б = 0,3—0,5 и различных температу-
рах
Относительная скорость газообразования при малых S уменьшается во вре-
мени (рис. 15), при больших — может значительно возрастать. При пони-
жен ин температуры опыта п увеличении 6 усиливается рост скорости на
начальном этапе распада (при 60° С она возрастает в 250 раз по сравнению
с начальным значением) и уменьшается степень распада, при которой до-
стигается максимум скорости (рнс. 16). При этом после максимума ско-
рость особенно при низких температурах значительное время остается по-
стоянной, при повышенных — слабо растет. Другим отличием является
отсутствие двуокиси азота в продуктах распада обеих стадий, а также от-
29
и-, сн3/г см*/г
Рис. 17. Термическое
разложение дины при
разных температурах
и различных степе-
нях заполнения сосу-
да (числа у кривых).
При 120° С время
в мин.
т, часы
Т, мин
Рис. 18. Влияние воды на термическое раз-
.тожепие дины при 100° С и Ъ = 0,03
носительно малое содержание газов, конденсирующихся нри комнатной
температуре, особенно при низких температурах распада (3—6%). По-ви-
димому, нри распаде не накапливается значительных количеств воды.
Общая картина распада (рис. 17) указывает на его сложный двухста-
дийный характер, особенно отчетливо проявляющийся при низких темпе-
ратурах, когда различие между скоростями обеих стадий становится боль-
ше и наблюдается значи-
тельный рост скорости газо-
образования, обусловленный,
по-видимому, накоплением
промежуточного продукта.
Влияние б на ход процесса
(рис. 17) указывает на учас-
тие в нем газообразных про-
дуктов. Энергия активации
при повышенных температу-
рах составляет около
42 ккал1моль (]g 2? =17).
Близость ее к энергии актива-
ции распада нитратов спиртов
говорит за то, что и при распа-
де дины первичным процес-
сом является отщепление ни-
троксильной нитрогрунпы'.
Распад дины изучался
также в присутствии паров
воды. Начальный участок
Числа у кривых — содержание воды по отношению
к количеству дины (в %)
1 Следует, однако, отметить, что и энергия активации распада N-нитраминов при-
близительно та же.
кривой p(t) в присутствии
НгО сходен с наблюдающим-
30
ся для нитроглицерина (рис. 18). Время до наступления ускорения распада
в присутствии воды сильно (в 6—8 раз) сокращается. Однако ускорение,
наблюдающееся после падения давления, идет, особенно при небольших
содержаниях воды, замедляясь, что, но-видимому, связало с уменьшением
концентрации двуокиси азота, установленным специальными опытами; это
уменьшение обусловлено быстрым вступлением NOs в реакцию, подобно
тому, как это наблюдается при разложении дины и в отсутствие воды.
Лзотпая кислота ускоряет распад, если исключить начальный участок,
сходно с водой.
твердом состоянии.
Рис. 19. Ход разложе-
ния нитроклетчатки в
токе инертного газа
при 135° С
б. НИТРОКЛЕТЧАТКА
Нитроклетчатка — высокомолекулярное соединение — существенно от-
личается от низкомолекулярных нитроэфиров своей неплавкостью, вслед-
ствие чего оца разлагается при всех температурах в
Наряду с этим нитроклетчатка имеет физически
сложную структуру, обычно очень развитую поверх-
ность и не представляет собой, как правило, химиче-
ски однородного вещества. Наконец, при получения
нитроклетчатки образуются различные, часто нестой-
кие, трудно удаляемые полностью, примеси. Все эти
обстоятельства существенно усложняют течение рас -
пада.
Исследования показали (10,37—39], что скорость
газообразования и ее изменение во времени сильно
зависят от того, остаются ли газообразные продукты
распада в контакте с нитроклетчаткой или удаляют-
ся. В первом случае распад идет со значительным
ускорением, во втором оно существенно меньше —
абсолютная скорость газообразования не растет во
времени, а остается на значительном участке пре-
вращения постоянной. Билль удалял газообразные
продукты распада пропусканием через разлагающуюся нитроклетчатку
тока инертного газа (СО2) и определял в нем соединения азота в виде N2,
объем которого и служил мерой скорости распада (рис. 19). Отсутствие
существенного роста скорости распада нитроклетчатки наблюдалось и при
распаде ее в растворах. Термическое разложение нитроцеллюлозы
(11,85% азота) изучалось для ее разбавленного (1%) раствора в инерт-
ных растворителях (1-хлор-2,4-динитробензол, бензофенол, 1-нитронаф-
талин) при 165—200° С[40]. Процесс идет с образованием 5 молей газов на
1 моль нитроклетчатки и представляет собой реакцию первого порядка,
хотя и наблюдаются некоторые отклонения от обычного ее течения. Так на
протяжении первой трети распада скорость газообразования в хлордини-
тробензольном растворе уменьшается относительно мало и константа ско-
рости возрастает приблизительно на 25%; на протяжении последней чет-
верти распада, напротив, константа уменьшается почти на 60% по сравне-
нию с ее значением на промежуточной стадии, это последнее значение
приближенно одинаково в разных растворителях. Константа скорости
к = 1018-0 ехр (—43 000/ЙГ). Среднее отклонение экспериментальных зна-
чений константы от рассчитанных по этому уравнению не превышает 7%.
Робертсон и Нэпнер [41] определили спектроскопически содержание
двуокиси азота в продуктах распада в токе инертного газа при 135° С и
показали, что 40—50% азота содержатся в виде NOs; при разложении в те-
чение 4 час. без отвода газов доля азота в виде NOs составляла лишь 25 %;
содержание двуокиси азота уменьшается также при замедлении тока
инертного газе. Все это указывает на сравнительно большую скорость окис-
лительно-восстановительных реакций с ее участием. По мере хода распада
31
отношение NOj/N в его продуктах уменьшается в одном из опытов с 54 до
8 % к шестидесятому часу, хотя абсолютная скорость выделения азота
оставалась постоянной. Это говорит об изменении удельного веса отдель-
ных реакций в ходе распада и, возможно, об увеличении роли вторичных
процессов,
Если предположить, что распад нитроклетчатки включает первичную
реакцию, скорость которой не зависит от наличия продуктов распада,
и вторичные реакции, которые от них зависят, то постоянство абсолютной
скорости, т. с, значительный рост относительной скорости при опытах
в токе инертного газа, могло быть обусловлено неполнотой удалении газо-
образных продуктов распада. В этом случае при более полном их удало-
Рис. 20, Кривые изменения скоростей во време-
ни при распаде паткроклегчатки (температура
160° С)
1 — труцноконденсирующиеея газы; 2 — конденсиру-
ющиеся газы; 3 — сумма труднонопденсирующихся и
конденсирующихся газов; 4 — убыль я весе’ 5 — га-
аосбрэзование без откачки газон (при 1GO° С, m.'v «
= i&,8*i0-4 г/сма); fi—газообразование при квэзипо-
ctchjhhom давлении
ыии, осуществляемым, на-
прттмер, пткачиианием,
моя: в о было бы получить
характерное для реакции,
идущей без самоускорения,
уменьшен не абсолютной
скорости во времени.
Для этой цели разло-
жение велось [42, 44] при
откачивании газообразных
его продуктов диффузион-
ным насосом, так что дав-
ление их не поднималось
выше 10-3 мм. Откачивае-
мые газы проходили через
одну из двух параллель-
ных попеременно включае-
мых ловушек, охлаждае-
мых жидким азотом. Изме-
рение давления после ра-
зогрева выключенной ло-
вушки до комнатной тем-
пературы позволяло опре-
делять объем конденсиру-
ющихся газов, выделен-
ных за определенный ин-
тервал времени, Кратко-
временное отключение насоса и измерение подъема давления в системе
(он нс превышал Ю-3 мм) использовалось для расчета скорости образова-
ния трудноконденсирующихся газов. При помощи кварцевых пружинных
весов одновременно измерялась потеря в весе нитроклетчатки. Кривые
трех упомянутых скоростей представлены для трех температур (160, 150
и 145° С) на рис. 20 — 22. Выделение конденсирующихся газов при 160° G
замедляется во времени. Средний молекулярный вес газов, рассчитанный
по потере веса нитроклетчатки, в начале распада близок к 46, т- е. к мо-
лекулярному весу NOa, Поскольку в начале распада доля конденсирую-
щихся газов составляет около 96%, эти данные говорят за то, что первич-
ным актом распада нитроклетчатки является отщепление NOa, Скорость
образования трудноконденсирующихся газов существенно растет во време-
ни; общее их количество, если экстраполировать на т = к, составляет
120 с.м?[г. Доля трудноконденсирующихся газов к копцу распада состав-
ляет около 1/з от общего объема газов. Суммарная скорость гл зовы деления
некоторое время остается приближенно постоянной, а затем падает. Сход-
ный характер имеет кривая скорости убыли в весе, хотя падение ее проис-
ходит более резко, что, по-видимому, связано с уменьшением среднего мо-
лекулярного веса газов.
32
С понижением температуры характер кривых несколько изменяется.
Скорость выделения конденсирующихся газов падает во времени вначале
значительно медленнее, и это падение не компенсирует рост скорости вы-
деления трудпоь'онденсирующпхся газов; в итоге общее газовыделецие от-
четливо ускоряется во времени. Еще значительнее ускорение на кроной
скорости потери веса. Объем газов несколько уменьшается, так же как
и доля труднокоидепсирующихся газов.
Ряс. 21. Кривые изменения скоростей во вро-
mgeeii при распаде нитроклетчатки (темпера-
тура 150° С)
Обозначения те же, нто и на рис.. 20
Рис. 22. Кривые изменении скоро-
стей во времени при распаде нитро-
клетчатки (ври М5°С, mjv = 13,9'
10“4 г/гл3)
Обозначения те ясе, что ее на отес. 20
При 135° С разложение проводилось только 13 час., т- е. па начальном
участке (конечная потеря веса 4%); отчетливый рост показывает лини,
кривая тРУДноконденсирующнхся газов; для конденсирующихся газов и
потери веса скорости практически постоянны.
Различие в кривых Иг(£) указывает, что при распаде нитроклетчатки
протекают до крайней мере две последовательные реакции.
По характеру кривых образование конденсирующихся газов, скорость ко-
торого наибольшая в начальный момент, следует считать результатом пер-
вичных процессов, а трудиоцонденсирующихся — результатом вторичных.
Образование конденсирующихся газов нельзя рассматривать как про-
стую мономолекулярную реакцию. Если при 160° С скорость распада не-
прерывно падает во времени приближенно по закону реакции первого
порядна, то при более низких (150 и 145°С) температурах падение ско-
рости происходит в начале очень медленно: на протяжении значительно-
го интервала времени она остается практически постоянной. Очевидно,
этот процесс является сложным и возможно протекает через образование
промежуточного соединения, также дающего при разложении копденси1-
рующиеся газы. Накопление этого соединения до некоторой квазистацпо-
нарной концентрации могло бы дать ту форму кривой Hz(t), которая на-
блюдалась при опыте.
Образование трудноконденсирующрхся газов, по-видимому, также свя-
зано с накоплением некоторого промежуточного продукта, тем более, что
максимумы на кривых скоростей потери в весе и образования труднокон-
денсирующихся продуктов близки по времени наступления. Эта связь
может быть прямая, т. е. трудиокопдецсцрующиеся продукты образуются
3 К. К. Андреев
33
(наряду с конденсирующимися) при распаде предположительного проме-
жуточного продукта. Она может быть и косвенной, если промежуточный
продукт очень реакционноспособен по отношению, например к NOa, и, вза-
имодействуя с нею, превращает ее в труди ©конденсирующиеся газы; при
150 и 145° С скорость образования конденсирующихся газов после макси-
мума убывает значительно быстрее, чем трудноконденсирующихся.
При распаде нитроклетчатки без отвода газообразных его продуктов
или при частичном их отводе (квазипостоянное давление 10 мм рт. ст.)
Рис, 23. Влияние температуры
на ускорение распада нитро-
клетчатки
Рпс. 24. Зависимость констант скоростей от тем-
пературы
1. — образование трулнокондеисирующихся газов;
2' — образование конденсирующихся газов; 3 — сум-
марное газообразование; 4 — потеря в весе; 5 — га-
зообразование без oTXaHita газов. Константы нанесены
на графике в условных единицах
начальная скорость такая же, как и в вакууме. Ускорение же газообразо-
вания значительно больше, особенно при низких температурах и при боль-
ших m/f. При 160° С отношение скоростей газообразования в присутствии
продуктов распада и при квазипостоянном давлении (рис. 20) на макси-
муме составляет 1,9, при 145° С — 2,1 (соответствующие давления в опы-
тах без отвода газов составляли около 400 мм); при более низких темпе-
ратурах оно, по-видимому, возрастает еще больше, как этого и можно
было ожидать ввиду меньшей температурной зависимости рассматривае-
мых процессов.
Увеличение ускорения распада нитроклетчатки при понижении темпе-
ратуры показали также исследования Светлова и Лурье [43], которые изу-
чали распад нитроклетчатки без отвода газообразных продуктов в более
пигроком интервале температур (70—140°С) при повышенных б (0,03—
0,06). Увеличение ускорения распада при понижении температуры опыта
иллюстрирует рис. 23, на котором по оси абсцисс отложен объем газов,
а по оси ординат — отношение скорости соответствующей моменту выде-
ления этого количества газов к начальному ее значению; при 70° С в мо-
мент выделения 4 см3 газов скорость возросла в 6 раз, при 140° С — только
в 1,7 раза.
Зависимость начальной скорости распада от температуры для пиро-
ксилина № 2 (12,3% N) выражается соотношением t — 10;4,4 ехр
• (— 38 5(W/RT). Пироксилин № 1 (13,35% N) разлагался в интервале
70—160° С приблизительно вдвое быстрее; к — 1015’35 ехр (—39 500//??1),
что близко к данным, полученным Самсоновым при 100—170° С .и малых б.
Ускорение распада во времени у пироксилина № 1 выражено сильнее
(в 2—2,5 раза), чем у пироксилина № 2.
34
Несмотря на то, что ускорение распада нитроклетчатки растет при по-
нижении температуры, рост скорости, если исключены побочные влия-
ния вроде саморазогрева, не бывает столь большим, как это наблюдалось
для нитроэфиров тина нитроглицерина, тэна и др. По-видимому возмож-
ности самоускорения распада нитроклетчатки более ограничены. Это под-
тверждается также сопоставлением скоростей газообразования, полу-
ченных без отвода газообразных продуктов, при квазилостоянном давле-
нии и в вакууме, где максимальные давления различались на много по-
рядков, скорости же газообразования лишь в несколько раз.
Рост скорости во времени вплоть до максимума [44] может быть вы-
ражен соотношением
— - Ае*!
где А — постоянная, зависящая от температуры; <р зависит от температу-
ры и от 6. При 120° С и б = 8 • 10“4, если скорость выражать в кубических
сантиметрах на грамм в секунду, постоянные А и <р имеют значения
1,49-10~4 и 1,34-10-6 соответственно.
В свете полученных данных вероятно, что влияние газообразных про-
дуктов распада на его течение заключается главным образом во взаимо-
действии их реакционноспособных компонентов друг с другом и с твердым
веществом, ведущим к ускорению газообразования и к образованию допол-
нительных количеств газов.
Сопоставление различных скоростей при разных температурах позво-
ляет рассчитать кинетические коэффициенты соответствующих процессов.
При вычислении энергии активации для реакций, приводящих к убыли
в весе, использовались коэффициенты совмещения интегральных кривых,
полученных при разных температурах, По начальной скорости убыли в
весе п энергии активации вычислялся предэкспоненциальный множитель,
логарифм которого равен 17,8. Энергии активации для общего газообразо-
вания и образования конденсирующихся газов рассчитывались по ходу
прямых, полученных в координатах IgA — 1/Т. Энергия активации для
Таблица 2
Кинетические коэффициенты скоростей реакций при термическом распаде
нитроклетчатки в вакууме при 135—160° С
Характеристика реакции, по которой рассчитывалась константа скорости Начальные значения констант скоростей (К), сея-МО’ Я, кал/лсоль [g Вт сек -1 Метод расчета Е
135° С 145° С 150’ С 160’ с
Убыль в весе * 0,090 0,3.3 0,65 2,4 44 600 17,8 Совмещением кри- вых убыли в ве- се во времени
Образование газов 0,18 0,60 1,1 3,3 43 400 17,5 По начальным скоростям газо- образования
Образование конденси- рующихся газов Образование труднокон* денсирующихся газов 0,24 0,77 1,5 4,9 42 100 16,9 То же
0,019 0,084 0,16 0,50 45 700 17,8 Совмещением кри- вых в коордвЕ1а- тах Др/Дт = /(т)
Газовыделение без от- качки газов ** 0,10 0,40 — 1,9 39 200 15,0 Совмещением кри- вых р ~ / (т)
* Ковставта скорости убыли в весе рассчитывалась относительно
для га сообразовав ин относительно объемов газов, выделившихся при Тд,.
** Опыты Проводились при 100—170° С.
убыли в весе ори тот
3* 35
трудно конденсирующихся газов рассчитывалась по прямой 1g /гсовмсщ — 1/7
на основе совмещения дифференциальных кривых Лк/Лт — /(т).
Результаты расчетов приведены в табл. 2 и представлены графически на
рис. 24.
Значения энергии активации и предзкспоненциального множителя для
всех улавливаемых видов газов и убыли в весе близки друг к другу. Дан-
ные для скорости образования газов в вакууме практически совпадают
с теми, которые даст расчет по данным Билля для скорости отщепления
соединений азота в токе инертного газа. Величина энергии активации
Рис. 25. Влияние воды на термический распад нитроклетчатки
при 120° С (т/и ~ 50—70 10-1 г/сл:1) и различН[)М давлении
паров воды (в .км рт. ст.);
1 — 0; г — 138, .1 — СОЗ
несколько больше, чем для простейших алкилнитратов и соответствует тако-
вой нитроглицерина. Это обстоятельство и завышенное значение предэкс-
поненцнального множителя в сочетания с рассмотренным выше характе-
ром изменения скорости во времени говорят за то, что образованно
различных определявшихся видов газов является прд распаде нитроклет-
чатки результатом совокупности нескольких реакций и не может быть
непосредственно использовано для расчета кинетических параметров от-
дельных реакций.
Распад нитроклетчатки изучался также в присутствии некоторых ве-
ществ, которые образуются при распаде (вода, двуокись и окись азота,
азотная кислота). Определялось также влияние на распад кислорода н
серной кислоты.
Гужон и другие исследователи [45, 46], изучавшие распад влажной ни-
троклетчатки, наблюдали значительное падение давления после некото-
рого индукционного периода с последующим резким ростом скорости газо-
образования. Однако в этих опытах нитроклетчатка находилась в контакте
также с воздухом, что могло усложнять картину влияния воды. Действи-
тельно, опыты, проведенные в присутствии одной воды [47], показали, что
характер распада не меняется, а ускорение его гораздо меньше, чем в
опытах Гужона (рис. 25).
36
Иначе обстоит дело, если, помимо воды, присутствует кислород
(рис. 26). Давление длительное время почти не меняется, а затем быстро
падает ц столь же резко начинает расти; скорость возрастает в 100—150 раз
7, MUH.
Рис. 26. Совместное влияние воды и кис-
лорода па термический распад (нитроклет-
чатки при 120с С и различных от[гоше(ги-
ях m/p - 104 {числа у кривых)
В скобках у кривых — давление плрьв волы
в ли рт. ст.
Давление кислорода (в мм рт. ст.): J — 0;
2 — 63,5; J — 2В4; 4 — 87; j — 274
по сравнению с гой, которая наб-
людается в отсутствие добавок;
бурное газообразование сопровож-
дается побурением газовой фазы.
Ускорение распада нитроклетчат-
ки наблюдается и в присутствии
одного кислорода, без воды, но ход
Рис. 27. Влияние кислорода на тер-
мический распад нитроклетчатки
при 120°С (m)v = 60—80-10-4 г/см3)
и различном давлении кислорода (в
мм рт. ст.):
1 — 0; 2 — 267; 5— 265; 4 — 168,5,
Рис. 28. Влияние содержания двуокиси
азота на скорость разложения нитро-
клетчатки при 120° С
изменения давления в этом случае
иной (рис. 27) и рост его не столь
резок. По-виднмому, в присутствии
воды я кислорода в начале проте-
кает медленный распад и накапли-
вается небольшое количество не-
конденспрующихся газов. Одно-
временно пары воды частично свя.
1 — без добажж, mlv - [3-[□-• г/с.ч!; г —
с 2% NOi при Руо2 ” 65 «л рт. Ст., пг,'о **
= 56,110-* г/сжэ; 3 — с 2% NO; в присут-
ствии P;Os, m/n 46-iO-4 г!см\ P;\'OS^22 J*”
рт. Ст.; 4— mlv = <3.7-[0-* г/елр, рц,Ог=
= 275 мм рт. Ст.; J — m/о - [7,5-10-* е/ем\
р NO, = 218 мм рт. ст.
потен конденсированной фазой, ибо
давление над нитроклетчаткой во время индукционного периода остается
почти постоянным, т. е. количество конденсирующихся газов соответст-
венно уменьшается. После достижения определенной концентрации про-
межуточных продуктов происходит сильное падение давления, очевидно,
за счет взаимодействия воды с нитроклетчаткой и поглощения ею образую-
щейся азотной кислоты и воды; продукты гидролиза вступают в окисли-
тельно-восстановительные реакции, сопровождающиеся быстро ускоряю-
щимся газообразованием.
37
Рис, 29. Влияние азотной кислоты
низкой концентрации на термиче-
ский распад нитроклетчатки при
120° С
J — 2,38% кислоты 16,5%-иой концентра-
ции; г — 1,31% кислоты 11,3%-ной кон-
центрации; а —1,89% кислоты 10,3%-цой
концентрации
Рис. 30, Влияние азот-
[10 й кислоты высокий
концентрации на терми-
ческий распад нитро-
клетчатки при 120° С
1 — 1,03% кислоты 73.2%-
ной концентрации: г —
2,76% кислоты 43,7%-ной
концентрации; 3 — 2,26%
кислоты 42,1%-ной кон-
центрации
Сухая двуокись азота нри малых концентрациях (?no , = 55 мм,
т!и = 56 • 104 г/сле3> слабо ускоряет распад нитроклетчатки; при увеличе-
нии концентрации ускорение возрастает (рис. 28). На кривой скорости
газовыделения нри больших содержаниях NO2 наблюдаются два макси-
мума, Первый из них больше второго и сопровождается полным обесцве-
чиванием газовой фазы. Повышение начальной скорости в присутствии
NO? можно объяснить ее окисляющим действием. Однако ускорение газо-
образования развивается медленно. Одной из возможных причин его яв-
ляется накопление NO: ее присутствие в случае распада нитроглицерина
существенно усиливало действие двуокиси азота. Падение скорости после
максимума естественно связать с исчерпанием NCh. Второе относительно
слабое ускорение газообразования, по-видимому, соответствует тому, кото-
рое наблюдается без добавок; возможно, оно усилено гидролизом за счет
воды, образовавшейся при окислении. Общий объем газообразных продук-
тов в присутствии двуокиси азота возрастает на 30—40 см?1г.
На распад нитроклетчатки влияет также окись азота, хотя и, естествен-
но, гораздо слабее, чем двуокись. Начальная скорость несколько меньше,
но ускорение значительно больше, чем для нитроклетчатки без добавок,
При изучении влияния азотной кислоты на распад нитроклетчатки ее
вводили в виде водного раствора, количество которого составляло около 2%
по отношению к нитроклетчатке а концентрацию кислоты в нем изменяли
от 0 до 100%. Кривые рис. 29 30 показывают, что азотная нислота уско-
ряет распад нитроклетчатки. Наибольшее ускорение дает 10%-ная кисло-
та; при меньших и больших концентрациях влияние ее меньше, Кривые
скорости газовыделения имеют два максимума; их величина связана с
концентрацией азотной кислоты, На первом максимуме кривой бурая
окраска газов наиболее интенсивна, на втором максимуме наблюдается
полное обесцвечивание газовой фазы.
38
Еще сильнее влияет на распад концентрированная (95%-ная) серная
кислота, причем это влияние возрастает при увеличении б. На рис. 31 по-
казана зависимость характера и скорости распада от количества серной
кислоты По-видимому, нелетучая серная кислота вытесняет из нитро-
клетчатки азотную кислоту, последующее разложение которой приводит
к развитию окислительпо-восстановнтельпых процессов, возможно уско-
ряемых серной кислотой.
Сопоставление опытов по распаду нитроклетчатки и нитроглицерина
показывает известное различие. Прн распаде нитроклетчатки в отсутствие
воды ускорение по порядку величины существенно меньше, чем при рас-
паде нитроглицерина, Это различие могло быть приписано одной из двух
причин (или их совокупности) — недостатку воды или недостатку кислоты
для развития гидролиза. Опыты по распаду нитроклетчатки в присутствии
Рис. 31. Влияние концентрированной
серной кислоты на термический рас-
пад нитроклетчатки при 100° G
Содержание кислоты (в %};
J— 3,55; 2 — 1,57; 3—1,28; 4 — 0,51
воды показывают, что дело не в первой причине. Присутствие воды не
меняет характера кривой и лишь слабо увеличивает скорость газообразо-
вания, Эти опыты показывают также, что скорость восстановления выс-
ших окислов азота, получающихся при распаде и в присутствии воды, об-
разующих азотную кислоту, в опытах с нитроклетчаткой достаточно вели-
ка, чтобы предотвратить накопление кислоты. В этом отношении нитро-
клетчатка сходна с дин тиле нтликольдини трат ом, для которого Светлов так-
же установил отсутствие ускоряющего влияния воды па распад. Однако
восстановительные свойства нитроклетчатки в условиях распада выраже-
ны не столь сильно и в присутствии кислорода, особенно же при совмест-
ном присутствии воды и кислорода распад принимает характер, свойствен-
ный нитроглицерину. Точно так же добавление совместно с водой азотной
кислоты приводит к сильному увеличению скорости распада; если добавить
2% кислоты (10%-ной концентрации), например, скорость возрастает при
120°в 32 раза.
В заключение следует отметить, что, поскольку нитроклетчатка при
температуре распада находится в твердом состоянии, в пей возможен топо-
химический распад, связанный с локальным образованием и развитием его
зародышей. Некоторые наблюдения говорят в пользу этого предположения.
Так, Гужон [45] установил, что скорость распада нитроклетчатки возрас-
тает при уменьшении размеров частиц, т. е. при увеличении удельной по-
верхности, При разложении нитроклетчатки под слоем инертной жидкости
скорость распада сильно уменьшается, что также указывает на влияние
1 Сходный характер имеют, по исследованиям Виляя [10], кривые разложения не-
полностью стабилизированной нитроклетчатки.
39
состояния поверхности на развитее реакднп. Наконец, Митташ [37]
наблюдал своеобразное влияние некоторых твердых инертных примесей па
распад, которое трудно объяснить, не допуская влияния твердой поверх-
ности па развитие реакции.
6. ГАЗООБРАЗНЫЕ НИТРИТЫ И НИТРАТЫ
Термический распад нитратов многоатомных спиртов является слож-
ным процессом. Одним из факторов сложности этого процесса является
образование в качестве промежуточного продукта двуокиси азота, кото-
рая, с одной стороны, является энергичным окислителем, взаимодействую-
щим с промежуточными продуктами, а также и с самим нитратом, а, с дру-
гой, с водой образует кислоты. В процессе окисления образуется вода,
которая особенно в присутствии кислоты быстро гидролизует нитроэфпр;
при распаде в жидкой фазе влияние гидролиза в большей или меньшей
мере отражается па течении первичных стадий распада. Если устранить
условия, благоприятствующие развитию гидролиза (например, разлагая
пптроэфиры в парах при низких давлениях), то распад протекает Кггнетп-
чесКп значительно проще.
Усложнение распада, вызываемое двуокисью азота, отсутствует также,
как правило, при распаде нитритов, первичным продуктом которого вме-
сто двуокисд азота является окись азота (окислитель несравненно более
слабый); в остальном же следует ожидать сходства по характеру распада
с нитратами на его первых стадиях.
Исходя из этих соображений и было изучено термическое разложение
моно- и полиннтрггтов, а также нитратов некоторых спиртов в газовой
фазе.
Полиндтриты спиртов
Термический распад алкрлнитритов (метил-, этцл- н.прогтил-, изопро-
пил-, н.бутил-) в парах в основном гомогенен и рост давления подчи-
няется закону реакции первого порядка [48]. Энергия активации для всех
членов ряда почти одинакова и близка к 37 ккал]молъ.
Объяснение этих зависимостей, а также состава продуктов распада п
влияния различных примесей может быть для этцлнитрита дано на основе
следующего механизма распада, включающего как первую стадию обрати-
мый отрыв NO-гуппы [49]
Тс
CH3CHs0N0^GH3CH20‘ 4-N0 (2.1)
Кд
СН3СНгО. + NO - CHsCHO + NOH (2.2)
CH3CH2O СН3 4- СН2О (2.3)
2NOH-»NSO+HSO (2.4)
СН»СЩО • + СНзСНО CHsCHsOII 4- (СНЭСО-^ С1Г9 4- СО). (2.5)
Скорость исчезновения исходного вещества
rflafCsHgONO] _ -гл д,
~= Лч 4- А;, А>
Для динитритов [50] при сравнительно высоких температурах
( — 300сС) первой ступенью распада также является образование NO
И окспрадикала. Дальнейшая судьба оксирадикала (распад или перегруп-
пировка с отщеплением ТКОД зависит от его структуры.
Исследование термического распада зтиленгликольдииитрита в парах
(т. кип. 96° С) было проведено Кондриковым манометрическим методом
при 120—190° С и начальных давлениях 50—1000 л.к [51—53].
40
Рис. 32. Кинетика газообразования при распаде гликольди-
нитрита при 170’С; начальное давление (в мм рт. ст.);
1— 248; 2— 248; 3 — 251; кривая с максимумом — в координатах
1/pcdp/dt—т; эй а пени я р/р0 на оси ординат справа
Основная особенность распада этого вещества заключается в ярко вы-
раженной двустадийпости (рис. 32). проявляющейся тем сильнее, чем
выше давление и чем ниже температура. При очень больших давлениях
п низких температурах вторая стадия наступает столь рано, что кривая
p(t) принимает обычную для автокаталитической реакции S-образную
форму. Начальная скорость (при 170’С) от концентрации не зависит1.
Рис. 33. Влияние начальной концентрации на разложение гли-
кольдииитрита при 170°С в различном давлении (в мм рт. ст.):
1 — 76; г —221; 3 — 320; 4 — 405 и 5 — 059
и при малых давлениях уменьшение скорости во времени происходит при-
ближенно по закону реакции первого порядка. Общее время распада при
повышенных начальных давлениях существенно сокращается за счет
1 Поаже наблюдается некоторое самоторможение первой стадии, тем большее, чем
выше начальное давление (рис. 33).
41
увеличения доли реакций второй стадии (рис. 33). Начальная скорость
при 170° С составляет 5,8 * 10 "4 сек-1; по ее росту с температурой энергия
активации получается равной 35,6 ккал/моль. Добавление окиси азота
(но лишь до некоторого предела) существенно уменьшает начальную
скорость роста давления, но нс влияет на скорость второй ступени рас-
пада. Анализ газообразных конечных продуктов при 170° (их количество
прц температуре опыта ~3 моля на моль нитрата) показывает, что
основной составной их частью является окись азота (около 3Л), затем —
СО;, СО, N2O и N2. В качестве промежуточного продукта при большой
концентрации двнитрита пли при низких температурах обнаруживается
(по пожелтению газовой фазы) двуокись азота. В конденсированных про-
дуктах — бесцветной вязкой жидкости, хорошо растворимой в воде и даю-
щей кислую реакцию на лакмус, качественно обнаружены гликолевая
кислота, вода, формальдегид и, по-видимому, гликолевый альдегид.
Труднолетучие продукты получаются исключительно на второй ста-
дии, повышение давления влияет на их образование лишь в той мере,
в какой оно влияет на ход второй стадии реакции. Добавление воды и уве-
личение поверхности путем набивки сосуда стеклянными трубочками не
влияют на скорость начальных этапов, но ускоряют реакции второй ста-
дии. Еще сильнее влияют конденсирующиеся при комнатной температуре
продукты распада, особенно в сочетании с водой.
Таблица 3
Скорость термического распада алкилнитритов
Вещество Интервал томперзтур, %' Энергия ак- тив ации >Ж<1ЛДИ0ЛЬ 1g В, сек-' Начальная ско- рость распада и )с оои 190е С.
сек- 101
Метилцптрпт 190—241 36 4 13,26 0,97; 0 48
Этплпитрит cf о F5 77'? 0-^-0 Cs О ТО 37,7 37,5 34,3 1-3 14,14 13,79 1,89; 0,94
п. Црепилиптрит 170—210 200-230 37,65 34,7 14,44 13,20 3,95; 1,98
Нзоирош’Лпцтрлт 170—210 37 0 14 10 3,70; 1,85 4,44
П.Бути.-цтПТрПТ 170—212 202—239 37^0 36,2 14’48 13,'66 8,88;
Цтгятрпт этпдапт.тнкодя 170—190 35,6 13,8 3 3; 11
Гринцтрит глицерина 143—160 41,6 17,8 140 ♦; 47
* По расчету.
Близость энергии активации начальной стадии к величинам, полу-
ченным для моноинтритов (табл. 3), образование NO и NaO, сходство
характера влияния NO на первую стадию с наблюдающимся для изо-
пропилнитрита [49] приводят к заключению, что механизм первой стадии
сходен с принятым для мононитритов:
CH2ONO s CH2ONO
I I + NO
CHsONO СНгО-
CHiONO ъ CHiONO
| + NO A | 4- NOH
CHaO- OHO
CH2ONO й
I X 2CH2O + NO
CH2O-
2 NOH N2O A HaO.
(2.7)
(2.8)
(2.9)
(2.10)
42
8 присутствии NOs возможна также реакция
CHiONO CH2ONO
I A NOa - |
CHaO- CHaONOa.
Предполагая, что концентрация радикала CHaONOCHsO стационар-
на, для скорости изменения давления получаем
ь гvпт । 'Ji. ГCHaONO
fra lAUj Ц- । /9 1 9\
(^Т + A) [NO] + A CHaONO.
Для начальной спорости газообразования при распаде имеем
к = |ЖО. (2.13)
концентрации и позволяет
dp [ ft 0 J
₽ к\
(2.11)
(2.14)
ИС, —- — ") = Зк или
° A Wr Л
Это согласуется с независимостью ее от
определить константы скорости по начальным скоростям газообразова-
ния, а также постоянные В и Е уравнения Аррениуса для к
к — 10ls'8exp( — 35 600/2?Г)сек_|.
Для опытов с добавлением окиси азота начальная скорость газо-
образования определяется выражением
гуьго = _?_/Ё£\ ь I^Q]q +
° Ро \dr (МА /с2) [NO]o-|- fcg
Из этого выражения следует, как и показал опыт, что начальная ско-
рость зависит только от концентрации NO и при ее увеличении стре-
ккя
мится к пределу .
Анализ экспериментальных данных позволяет рассчитать отношения
констант скоростей вторичных реакций первой стадии: k2;kt — б;
кз/ki ~ 120 (концентрации — в мм рт. ст.). Объяснимы также самотормо-
жение реакции на первой стадии и зависимость его от начальной кон-
центрации динитрита.
Вторую стадию распада Кондриков рассматривает как взаимодействие
гликольдинитрита и, возможно, промежуточных продуктов его распада
с водой, первоначально образующейся, напрпмер, по реакции 2NOH-*-
—- N2O + Н2О. Это приводит к замене нитритной группы на окенгруппу
и образованию азотистой кислоты, которая разлагается на опись и дву-
окись азота п воду. Двуокись азота окисляет органические продукты, об-
разуя СО, СОй ц воду. Общие соображения, а также влияние поверхности
и труднолетучих продуктов указывают на то, что эти реакции идут на
стенке сосуда в тонкой пленке покрывающей ее жидкости.
Распад глицеринтринцтрита изучали при 100—160° С. Сходство кине-
тических Кривых (рис. 34) и закономерностей с получающимися для ди-
нптрита гликоля велико: независимость начальной скорости от концентра-
ции, увеличение при повышения концентрации максимальной скорости и
уменьшение времени ее достижения, увеличение роли реакций второй
стадии при понижении температуры, уменьшение начальной скорости при
добавлении NO и отсутствие влияния NO на вторую Стадию, ускоряющее
влияние конденсированных продуктов распада и воды, влияние поверхно-
сти на вторую стадию и отсутствие этого влияния на первую.
Имеются ц некоторые отличия. При близких временах полного рас-
пада роль реакции второй стадии при распаде трииитрита значительно
больше. Скорость на первой стадии падает менее резко и угнетающего
влияния начальной концентрации на скорость первых этапов распада
не проявляется. Желтая окраска газовой фазы выражена сильнее, осо-
бенно при низких температурах.
При значительном снижении температуры (до 100—120° С) характер
кривой p(t) несколько меняется; скорость на начальной стадии не умень-
43
пгается, а медленно растет, как это обычно бывает при автокаталитиче-
ской реакции и как это наблюдалось .и для гликольдинитрита в тех же ус-
ловиях.
Кинетические характеристики распада различных нитритов приведены
в табл, 3. Из табл. 3 видно, что увеличение молекулы в ряду алкилмоио-
питритов приводит к увеличению скорости распада. При переходе от
одного гомолога к следующему скорость реакции возрастает приблизи-
тельно вдвое. Увеличивается при усложнении молекулы и скорость рас-
пада оксирадикала, не играющего почти никакой роли при распаде этпл-
нитрита и весьма существенного при распаде н.пропилпитрита.
Ряс. 34. Кинетика гаппобразопапин прп распада глицеринтри-
нитрпта (температура 150° С) и различием начальном давлении
(в мм рт. ст):
1 — 545; 3 — ЗЮ; 3--215; 4—142; 5 — 69
Если молекулу этилнитрита усложнить, заменив водород р-углеродпо-
го атома па О — NO, т- е, превратить этилнцтрпт в динитрит этиленгли-
коля, то на начальной стадии распада это приведет к увеличению скоро-
сти разрыва связи О — NO в 5—6 раз большему, чем при введении СНз-
группы, а также к некоторому увеличению скорости распада оксирадикала
без принципиального изменения механизма этой стадии.
То же самое произойдет при переходе от пропилиитрита к триннтриту
глицерина: основные реакции начальной стадии распада сохранятся, но
скорость разрыва связи О — NO и скорость распада оксирадикалов суще-
ственно увеличатся.
При повышении температуры соотношения между Отдельными скоро-
стями, очевидно, изменяются. Скорость распада оксирадикала, вероятно,
растет быстрее, чем скорость реакций взаимодействия его с окисью азота.
Таким образом, если рассматривать только начальные этапы распада,
переход от мононитритов к полинитритам принципиальных изменений
в механизме разложения не вызывает. Естественно, скорости отдельных
основных реакций и соотношения между константами этих скоростей ме-
няются, прежде всего при распаде оксирадикала и в реакциях его с окисью
азота (но в пользу последних). О;щако, если перейти к рассмотрению всего
процесса распада в целом, то обнаруживается существенное принципиаль-
ное отличие механизма газофазного пиролиза полинитритов от мопонит-
ритов. Оно состоит в наличии у полинитритов в определенных условиях
второй самоускоряющейся и существенно полимолскуляриой стадии рас-
44
пада, а также в появлении в промежуточных продуктах двуокиси азота,
тесно связанном со степенью развития самоускоряющсйся реакции.
Наиболее вероятное объяснение механизма второй стадии заключается
в том, что нолинитриты с их большой склонностью к гидролизу распа-
даются на стенке сосуда в пленке покрывающих ее труднолетучих про-
дуктов распада. Образующаяся при этом азотистая кислота быстро рас-
падается с образованием двуокиси азота, которая и обеспечивает ггро-
хождепие реакций окисления, дающих обнаруженные в конечных про-
дуктах органические кислоты и углекислоту.
Рис. 35, Изменение но времени
давления газообразных про-
дуктов (7) и скорости распа-
да (2) нитроглицерина в парах
при 140“ С
Рис. 36. Изменение скорости газо-
образования во времени при рас-
паде тана (температура 171° С)
:Ц малых В-104;
1 — 0,43; 2 — 0,83; 3 — 11,4
Переход от алкилмщюнитритов к дицитрпту этиленгликоля и тринит-
риту глицерина увеличивает не только п не столько скорость термического
распада, сколько скорость гидролиза нитрита. По-видимому, существен-
ную роль здесь играет расположение нитритных групп рядом друг с дру-
гом- Для динитрита 1.4-бут плен гликоля, который по скорости термиче-
скою распада почти не отличается от динитрита 1,2-этиленгликоля,
удельный вес второй стадии значительно меныпе.
Распад нитроглицерина в парах [13] изучали при 140, 150 и отчасти
105° G при значениях б, рассчитанных цз зависимости упругости пара от
температуры. Абсолютная скорость газообразования пропорциональна
концентрации паров, в несколько раз больше, чем у жидкого нитрогли-
церина, и непрерывно падает во времени (рис. 35); это падение отклоня-
ется по своему характеру от того, которое свойственно реакции первого
порядка. При заполнении реакционного сосуда стеклянными капиллярами
(поверхность стала больше в 7 раз) скорость увеличилась в 2—3 раза,
что указывает на протекание одновременно с гомогенной гетерогенной ре-
акции на стекле. Газообразные продукты распада, впачале бесцветные,
буреют, а затем вновь утрачивают окраску; одновременно уменьшается
доля конденсирующихся при комнатной температуре газов.
Распад паров нитрогликоля при 140—170° С протекает в общем сход-
но с распадом нитроглицерина; относительная скорость не зависит от
начальной концентрации, но в начале несколько возрастает (в отличие
от нитроглицерина) во времени. Конечный объем газов растет ври по-
вышении температуры опыта, Кривые р(/) хорошо совмещаются на
участке распада 20—60% (от конечного давления) путем изменения
масштаба времени, и по зависимости коэффициента трансформации от
температуры энергия активации получается равной 34,2 ккал^моль-^ она
меньше, чем для жидкости; для паров нитроглицерина энергия активации
45
значительно больше, чем для паров нитрогликоля, Поскольку скорости
распада близки (при 140° С нитроглицерин разлагается лишь в 5 раз
быстрее, чем нитрогликоль), то соответственно различаются и значения
предэкспоненциального множителя.
Распад тэна при 171° С изучали (31, 32] при малых б (11 * 10~4—
"0,4‘10-4), когда значительная часть вещества находится в парообразном
состоянии (рис, 36), Чем меньше б, тем меньше, как и в случае нитро-
глицерина, ускорение распада и тем больше его скорость. Ориентировоч-
ный расчет показывает, что тэн в парообразном состоянии разлагается в
6—8 раз быстрее, чем в жидком.
Увеличение поверхности при разложении тэна и нитрогликоля не из-
меняет скорости газообразования, что указывает на отсутствие заметной
гетерогенной реакции на стенках сосуда. Результаты исследования рас-
пада полинитритов гликоля и глицерина, а также алкилмононитратов по-
зволяют сделать некоторые заключения и в отношенгги распада поли-
нитратов.
Общая схема этого распада может быть (в применении к простей-
шему динитрату) представлена [53] в виде совокупности следующих
реакций;
GHs0NO2 ->• СН2О* | | -f-NOj CH2ONCh «- CH2ONO2 (2,15)
СН2О* I — 2 СНгО + КО2 CH2ONO2 (2,16)
СН2О 4- NO; — NO, CO, CO-2, ЩО (2,17)
CH2O- CH-jONO | 4-NO -» | CH2ONOs CHjONO-j (2.18)
CH2O- CHO | 4- NO -» | -p NOH CHjONO, CH2ONO2 2 NOH -> N2O-|-H2O (2,19) (2.20)
и т. Д.
По сравнению с этилнитратом скорость распада оксирадикала бу-
дет больше, а соотношение констант скоростей образования нитритнит-
рата и нитрата оксиальдегида изменится в пользу последнего, В итоге
та особенность распада этилнптрата, которая заключается в образова-
нии большого количества нитрита как промежуточного продукта, по-ви-
димому, не присуща распаду тринитрата глицерина и динитрата гли-
коля по крайней мере в изучавшемся интервале температур.
Отдельные данные по распаду полинитратов говорят в пользу при-
веденного механизма. Скорость распада этиденгликольдияитрата и
нитроглицерина не зависит от начальной концентрации.
Энергия активации распада этиленгликольдинитрата близка к энер-
гии активации этилнитрата; при добавлении окиси азота начальная
скорость газообразования уменьшается (при 170°С), причем характер
ее зависимости от конпентрации такой же, как для динитрита. На после-
дующие стадии NO не влияет* В продуктах распада в присутствии NO
обнаруживается закись азота.
Наличие максимума скорости для глмкольдинитрата возможно свя-
зано с накоплением промежуточного продукта в значительной кояпент-
раипп, который, распадаясь, образует газы. Отсутствие такого макси-
мума при распаде нитроглицерина может быть связано с тем, что ква-
зцравновеснаи концентрация промежуточного продукта невелика и до-
стигается рано. Обращает на себя внимание в этой связи повышенные
46
энергия активации и значение предэкспоненциальвого множителя нитрата
и нитрита глицерина в сочетании с повышенной константой скорости
распада. Нет оснований ожидать повышения энергии активации роакпгш
отщепления NO или NO2 от молекулы у производных глицерина по
сравнению с производными гликоля, скорее наоборот. Поэтому указанная
особенность производных глицерина, вероятнее, связана с тем, что ве-
дущая реакция сложнее, чем в случае распада нитратов двухатомных
спиртов и, возможно, представляет собой сочетание обратимого эндотерми-
ческого распада молекулы с образованием промежуточного продукта, не-
обратимо распадающегося с образованием газов. При указанных условиях
могут получиться именно то зависимости, которые установлены для три-
питрата и тринитрита глицерина.
В целом распад нитритов имеет значительные черты сходства с рас-
падом нитратов, особенно в начальной стадии, которая качественно тож-
дественна, представляя собою соответственно отрыв NO- и КОг-группы.
Различия обусловлены возможностью образования воды и кислоты как
участника и катализатора гидролиза, который является причиной наступ-
ления второй стадии. Эта возможность больше у нитратов. С другой сто-
роны, большая относительная легкость гидролиза нитритов, а также об-
разование нелетучих жидких продуктов (в случае распада нитратов они
окисляются в газы) способствуют наступлению второй стадии даже в тех
условиях, в которых она при распаде нитратов не наблюдается (разложе-
ние паров). В то же время эта стадия у нитратов носит гораздо более
бурный и самоускоряющийся характер, так как образует основной окис-
литель и источник кислоты (NO2) в гораздо больших количествах, чем
могут дать нитриты. Соответственно достаточно много получается и воды
для продолжения гидролиза.
То, что именно гидролиз является решающим фактором сильного уско-
рения распада, наглядно показывает распад нитроглицерина и метнлнпт-
рата в парах: окислитель (NO2) здесь образуется в больших количествах,
но самоускорения процесса тем не менее не наблюдается. Поэтому'имен-
но возможность развития гидролиза, которая зависит от наличия воды,
константы скорости ги.чролиза и влияния на нес кислоты, а также от
накопления этой кислоты, определяет степень самоускорения распада.
В свою очередь накопление кислоты возможно, если образующаяся дву-
окись азота не восстанавливается слишком быстро до окиси. Этот послед-
ний фактор и обусловливает сильное различие в склонности к самоус-
корению распада пидроэфпров типа нитроглицерина и типа диэтиленглп-
кольдинитрата.
II. НИТРОМЕТАН
Кипетику термического разложения нитрометана изучали [54] стати-
ческим и проточным методами при различных температурах при низких
(4—400 мм) и повышенных (12—40 ат) давлениях. Разложение про-
текало по закону реакции первого порядка в обоих случаях. При низкпх
давлениях (4 — 400 леи) порядок реакции несколько возрастал при по-
вышении температуры.
Изучение влияния различных газов на разложение при низких дав-
лениях показало, что в большей части они не влияют па разложение (ге-
лий, азот, окись азота, углекислота, вода); кислород ускоряет разложе-
ние, водород замедляет его; добавление окиси или двуокиси азота прп
повышенных Давлениях не оказывает влияния на скорость разложения,
если количество их невелико,- при больших количествах опи замедляют
разложение. Кислород при больших давлениях не влияет на скорость
реакции, но изменяет состав ее продуктов. Свободные радикалы снижают
температуру разложения нитрометана.
47
Состав продуктов разложения существенно различается в зависимости
от давления, при котором оно проводится.
При низких давлениях (200—400 .о) и в интервале температур 280—
300° С продукты разложения содержат окись азота, закись азота, воду,
окись углерода, метан^ углекислоту, малые колпчества этилена и этана
и следы двуокиси азота. Главным азотсодержащим соединением является
окись азота, се содержание уменьшается по мерс хода распада. Отноше-
ние СО СЩ ; СОг (106,2 : 1,4) после разложения нитрометана на 5%
остается постоянным.
При повышенных давлениях и 355° С 'основными продуктами разло-
жения являются углекислота, окись углерода, метай, синильная кислота,
окись азота, азот, вода и в меньших количествах метилциапид, этилциа-
нид, формальдегид и закись азота. При 5% разложения отношение
СО : СН$: СОг было равным 10 ; 5 : 10, Наиболее существенным отличием
состава продуктов разложения при повышенных давлениях является
образование па ранних стадиях большого количества синильной кислоты
как основного углеродсодержащего продукта,
В качестве начального этапа разложения при низких давлениях пред-
лагались две реакции: распад нитрометана на нитрозометап и атом кис-
лорода, связанный с разрывом свизи между азотом и кислородом
CHsNO2 -+ CH3NO 4- О (2.21)
и распад на метил-раддкал к двуокись азота, связанный с разрывом связи
между азотом и углеродом
CH3NO2 -> СН’з -|- NO., (2.22)
Механизм (2,21) неприменим к разложению при низких давлениях,
поскольку при пом не образуется синильной кислоты, Которая легко
образуется при распаде питрозометана. С другой стороны, образование
свободных: радикалов и двуокиси азота говорит в пользу механизма (2,22).
Значения энергия активации, найденные в большинстве исследований,
лежат в пределах 50—53 ккал]моль. Энергия связи С — N, равная
57 ккал, больше энергии активация. Это указывает па возможность ра-
дикального механизма распада, Поскольку, однако, реакция тормозится
только большими количествами окиси азота, можно допустить существо-
вание лишь очень коротких цепей.
Для начальных стадия при низких давлениях принимается следующая
схема реакций:
CII3NO^aH3-| NO2, ' (2.23)
СН'й- CII9N0.2 - CHH-CHA'Oj, (2,24)
(!H2NO3 + NO. -* CH2O 4- NO 4- NO., (2.25)
(’П2О 4- NOo^ CO — NO 4. H2O, (2.26)
C,H8 4-CHa-» G2HC. (2.27)
Образование больших количеств HCN при повышенных давлениях мо-
жет быть объяснено образованием питрозометана по реакции (2.21),
Однако он может образоваться к при взаимодействии метил-радикал а
с окисью азота. Этой реакции благоприятствуют тройные соударении:
поэтому ее роль мала при низких давлениях и становится большой при
высоких. В пользу этого объяснения говорит также то, что количество
сиппльной кислоты увеличивается даже при повышении давления до-
бавлением инертной примеси (СО2); так же влияет и добавление NO.
Изучалось также влияние различных окислов па воспламенение нитро-
метана; основные окислы катализируют реакцию в отличие от кислотных,
которые не оказывают на нес влияния, По-видимому, при каталитическом
воспламенении играет существенную роль ациформа нитрометана, веро-
ятно, взаимодействующая с основными окислами.
4S
Разложение питроэтана л нитро пропанон изучалось н меньшей степе-
ни, чем нитрометана. По Котреллу, Грэхэму и Рейду [55] в интервале тем-
ператур 355—405°С питроэтац разлагается в основном гомогенно при
данлоппях 50 — 300 мм но закону реакции первого порядка. Доля гете-
рогенной реакции ц указанных условиях, возрастающая при понижении
температуры, составляет около 10%, Добавление окиси азота не влияет
па скорость реакции. Основными газообразными продуктами реакции
являются этилен и окись азота. По энергетическим соображениям, а так-
же учитывая образован нс олефина, авторы исключают отщепление дву-
окиси азота и допускают внутримолекулярную перегруппировку с зго-
следующим мономолекулярпым распадом на олефин и азотистую кисло-
ту, быстро распадающуюся затем на воду, окись и двуокись азота, окис-
ляющую олефин.
Грэй, Иоффе п Розелаар [56] установили образование свободных ра-
дикалов при распаде нитроэтана, а также значительное ускорение его раз-
ложения при введении метильных радикалов. Помимо этого, состав про-
дуктов распада более сложен, чем этого можно было бы ожидать, если
бы первичная реакция исчерпывалась внутримолекулярным отщеплением
HNO->. При 477° С распад может быть выражен уравнением
lOOCiHjNOj -> 40С2НЦ + 3CsHe 4- ЗН,7СН4 + 22СНЙО +
4- 48г3 СО + 73’0; + Й9,7НйО + 29N2 + 34NO + 8NO2 (2.27)
Все это говорит за то, что первичным этапом является распад нитроал-
кана па радикал и NOj. Эта реакция может объяснить последующее обра-
зование всех остальных экспериментально обнаруженных продуктов —
этилена, окиси азота, СО, формальдегида и метана,
Продукты первичного распада взаимодействуют также с самим пптро-
этаном, вследствие чего наблюдаемая скорость распада оказывается боль-
ше, чем скорость разрыва связи С—N. Энергия активации составляет
39 ккал/мольг что значительно меньше энергии указанной связи. Для
1-нитропроиана получена энергия активации, равная 48,5 ккал/молъ, хотя
этот результат не может считаться точным, для 2-витроцропана — Е *=
= 39 ккал/моль. В остальном разложение пцтропропанов сходно с раз-
ложением нитроэтапа.
Применение нитрометана в качестве ракетного топлива осложняется
псобходгимостью применения более длинной камеры, чем у других топлив.
Это обстоятельство, по-видимому, связано с медленностью сгорания нит-
рометана, которое может быть ускорено применением катализаторов Сг^Оз
пли ЕедОз. Изучение распада нитрометана в присутствии катализаторов
проводилось в интервале температур 220—245°С при начальном давле-
нии 36 ат [57]|. Окись железа (в отличие от окиси хрома) теряет свою
каталитическую активность после того как распад пройдет на */з.
R присутствии окиси хрома распад идет по уравнению реакции пер-
вого порядка:
k = 10псхр (—36 000//?7').
Отличием катализированного распада является большая скорость
распила соответственно различным значениям меньшей энергии актива-
ции (36 и 49.2 к кал /моль). Кроме того, в продуктах распада не содер-
жится синильной кислоты, которая содержится в больших или меньших
(в зависимости от давлении) количествах в продуктах некэтэлизированного
распада. Другим отличием является содержание больших количеств бикар-
боната аммония (особенно при повышенных давлениях) и отсутствие чер-
ного твердого остатка, которого много без катализатора.
Эти отличия могут быть объяснены двумя различными механизмами
распада. Один из них в качестве первого этапа предполагает ту же ре-
акцию, что н при пскатализированпом распаде — разрыв связи С—N.
4 К. К. Андреев
49
Чтобы объяснить образование бикарбоната аммония и отсутствие
HCN, следует допустить, что последняя в присутствии окисла полностью
гидролизуется с образованием окиси углерода и аммиака, который за-
тем с водой и углекислотой образует бикарбонат.
Это объяснение было подтверждено нагреванием HCN с водой в от-
сутствие и в присутствии катализатора при 230е С. В первом случае гид-
ролиза не происходило, во втором образовывался аммиак. Второй меха-
низм в качестве первого этапа предполагает разрыв свяли N—О с образо-
вал ном метилоксима и гидроксила.
Влияние катализаторов может быть сопоставлено с влиянием кисло-
рода, который также приводит к исчезновению HGN из продуктов рас-
пада и, подобно им, требует минимально допустимой длины камеры сго-
рания, По-видимому, большое значение этой длины при горении нитроме-
тана обусловливается медленностью распада HCN.
III. НИТРОСОЕДИНЕНИЯ АРОМАТИЧЕСКОГО РЯДА
Первые попытки сравнительной оценки скорости термического рас-
пада н нт росоедп нений ароматического ряда были сделаны Фармером
п Робертсоном, Фармер [58] определил температуры, при которых 1 а ВВ
выделяет за 100 час. нагревания 1 см3 газов (табл. 4). Робертсон [59] для
топ же цели устанавливал скорость газообразования при некоторой по-
стоянной температуре (140 и отчасти 120° С) (табл. 5).
Таблица 4
Температура образования 1 см3 газа
при распаде некоторых ВВ (1 г) за 100 час.
ВВ Температура плавления, °C Темпера- тура, “С
ТривнтробеНЗОЛ 123 190—195
Тринитрофенол 122,5 150—155
2,4,6-Тринитротолуол 80,6 135—140
2,3,4-Трияитротол уол 110,3 135—140
2,4,5 -Т р ив итр ото лу о л 102,3 130—135
Тетрил Нитрат целлюлозы 129 115—120
(13% N) —-* 100
Данные этих работ показывают, что изучавшиеся нитросоединоняя го-
раздо более стойки, чем питроэфиры п, как правило, также нитрамины.
Из тршгитросоединений во много раз медленнее других разлагается три-
нитробензол, близко к нему примыкают трмнитромезитилен и тринитро-
ксилол. По-видимому, малая скорость распада последних двух нитросо-
единений обусловлена в значительной мере тем, что они при температуре
опыта находятся в твердом состоянии. Несимметричные изомеры трини-
тротолуола и тринитробензола разлагаются в общем заметно быстрее,
чем симметричные.
Систематическое изучение кинетики .термического распада многих ни-
тросоединений (моно-, ди- и трицитробепзол и ряд производных послед-
него — метил-, окси-, диокси-, нитро-, хлор-, дихлор-, трихлор- и ами-
но-) было недавно проведено Максимовым- [60] и отчасти (тринитрофенол
и тринитро резорцин) Лю Бао-фен [61, 62].
Для оолыпинства соединений разложение проводилось как в жидкой
фазе, так и в парах, для соединений с большой упругостью пара и ма-
лой константой скорости распада (моно- и динитробензол) только в па-
рах, для соединений с очень малой упругостью пара и высокой темпера-
турой плавления — в расплаве (или в растворе) и в твердом состоянии.
50
Таблица 5
Скорость газообразования W (в слеа/кг час) при распаде некоторых ВВ
вв Темпера- тура плявле- ния. °C Скорость газообразования после 40 час. нагревании при
SO’ 120° 140“ ISO’ | 180°
1,3-Динитробензоп 90 , 0*
2,4-Динитроянилин 127 — — 0* -— —
1,3,5-Тринитробензол 123 — — 0* — 0,05
1,4,6-Тринитромезитилеп 235 — 0,003 — 8,4
2,4,6-Тринитро-л<-ксилол 182 —<• 0,5 8,0 — 1
2,4,6-Тринитрофенол 122,5 -— — 0,6 — - —
2,4,6-Тринитроянилип 198 — — 4 — —-
1.2,4-Тринитробензол 61 — 8 — —
2,4,6-Тринитротолуол 80,6 — 9 — —
2,4,6-Три ни тро-л«- кр езол 109,5 — — 10 —
2,3,4-Тринитротолуол 110,3 — — 13 —
2,3,5-Тр и нит р ото лу о л 92,5 — 1,5 24** — —
2,4,5-Трикитротолуоп 102,3 — — 32 — —
2,4,6-Тринитр о ан из о л 68,4 — —— 32 -— “—
Тетрил 129 — 3 128** — —
2,3,4,6-Тетранитроанплин 217 — 18 288** —г
3,4,5-Тринитротолуол 132 — 22 352** — —
Окситетрил 182 .— 23 370** — —
Нитроцеллюлоза (13% N) — 2500 (135°) 5000** — —
Нитроглицерин 13,3 — 1660 (115°) 86000** — —
Азид свинца —1 — .— 40
Гремучая ртуть 2,5 — — — —
* Скорость неизмерима при 140° С.
‘* Скорость газовыделении на ранней стадии разложения до ускоренна.
1. РАЗЛОЖЕНИЕ НИТРОСОЕДИИЕНИЙ В ПАРАХ
Моно-, ди- и тринитробензол
Разложение в парах мононитробеизола изучалось в интервале началь-
ных давлений от 80 до 1150 мм при 416°, динитробензола — от 190 до
740 мм (395°С), а трииитробензола — от 90 до 250 -мле- Эти данные ис-
пользовались для определения влияния концентрации на скорость распа-
да и ее изменение во времени. Зависимость скорости распада от темпера-
туры изучалась в интервалах температур’ 390— 415° (мононитробензол),
345 — 410° (динитробензол) и 270 — 355°С (тринитробензол).
Разложение протекает с образованием газов, конечное давление
в 2 (мононитробенэол), в 3 (динитробензол) и в 4,5 (тринцтробензол) раза
превосходит начальное. Одновременно образуются конденсированные про-
дукты в наибольшем количестве для мононитросоединендя.
Во всем изученном интервале температур относительная скорость рос-
та давления для динитробензола больше, чем для мононитробензола, а для
трииитробензола больше, чем для динитробензола. При 350° С и ро —
= 500 .о динитробензол разлагается в 1,6 раза быстрее, чем мононитро-
бензол при 1000 мм; при той же температуре тринитробензол (р0 =
— 200 мм) разлагается в 20 раз быстрее, чем динитробензол (при р0 =
== 500 мм).
Начальная скорость и характер кривой роста давления во времени
зависят от начального давления паров, У мононитробеизола с увеличени-
ем начального давления начальная скорость растет, цо-видимому, стре-
1 Интервал температур брали таким, чтобы было удобно измерить скорость га-
зообразования применявшимся методом.
51
4*
мясь к некоторому пределу (рис. 37); во времени скорость падает,
причем при малых давлениях (около 100 мм) по закону реакции первого
порядка, при больших начальных давлениях скорость в начале опыта
уменьшается медленнее (рис. 38).
У дипитробензола начальная скорость такящ увеличивается нри уве-
личении начального давления, но раньше (при меиыпем давлении) чем
для монопитробепзола достигает максимального значения; качествен-
ным отличием от мопонитробензода является то, что даже при наимень-
шем из изучавшихся давлений скорость растет во времени, причем тем
сильнее, чем больше начальное давление (рпс. 39),
Рис, 37. В.тцяггие давления паров мо-
нонитробевзола на начальную отно-
сите л гягую скорость распада при
415° С
Р[1С. 38. Изменение скорости газообразова-
ния при расраде мононитробеизола (тем-
пература 415° С) и падальном давлении
(в мм рт. ст,) '
/ — 80; 2 — 2(5; 3 — 57:,; 4 — 1000; 5-IW
У тршгптробензола при давлении от 92 до 250 мм (рис. 40) начальная
скорость остается постоянной; по-видимому, ее рост с давлением, если
он имеет место, происходит в области еще меньших давлений, чем для
динитробензола и тем более мопонитр'обеизола. Ускорение газообразо-
вания во времени выражено еще сильнее, чем для ди нитробензола.
Возможно, что описанные различия между моно-, ди- и трннитробеп-
зо,ламп в некоторой мере связаны с тем, что опыты проводились при раз-
личных Относительных давлениях (Роти = po/p.s, где р., — давление насы-
щенного пара). Так, например, для монондтробензола при 416° С замед-
ление падения скорости, отсутствующее при /то = 80 мм (ро/рв — 0,004),
становится отчетливым при 1150 мм (ро/р.с = 0,05); вероятно, при еще
больших давлениях наблюдалось бы ускорение газообразования, подобное
тому, которое происходит при распаде дицитробензола при 395° С ир0 ~
= 185 мм (ро/р, = 0,03), Значительное ускорение, наблюдавшееся для
трипитробензола уже при ро = 92 .w.w (po/ps = 0,29), возможно, в извест-
ной мере связано с том, что опыты ставились при относительно низкой
температуре, когда процессы ускорения выражены гораздо сильнее, чем
при более высоких температурах (при 312° С, например, б; — 4,5,
а при 355° О и том же начальном давлении — только 1,5).
Увеличение поверхности сосуда (помещение в него капилляров) су-
щественно влияет па характер кривой р(1) и на величину начальной ско-
рости: уменьшается также конечный объем газов. У мопонитробензола
исчезает замедление падения скорости, а у трипнтробонзола — ее макси-
мум; начальная скорость резко возрастает, а во времени скорость непре-
рывно падает по закону, близкому к закону реакции первого порядка.
Кривая р(1) для дипитробензола при увеличенной поверхности сосуда по
52
своему виду отличается от кривых для мононитробензола и тринитробен-
зола, но это отличие заключается в первую очередь в том, что для нее
заметен участок роста начальной скорости, который не был уловлен
в услониях опытов с мононитробензолом и трииитробензолом.
Полученные закономерности качественно можно объяснить, допуская,
что в условиях производившихся опытов при распаде паров нитробензо-
лов с соизмеримыми скоростями идет, с одной стороны, гомогенная
реакция и, с другой,—гетерогенная реакция на стекле; удельный вес
последней возрастает при переходе от мононитробензола к динитробензолу
Рис. 39. Изменение скорости газо<о-
разоваппя во времени ара распада
1,3-ди нитробензола в парах (темпе-
ратура 395° С) и различном да пле-
нив (в рт. ст.):
J — 185; — ЛОТ', а — 450; 4 — 740;
.5 — 00В (поверхность стекли увеличена
в [0 раз)
Рис. 40. Изменение скорости газообразования
во времени при распаде три нитробензола в
парах (температура 312° С) и различном дав-
лении (в мм рт. ст.):
1 — 92; 2 — [37; .7 — 230; 4 — 247; .5 — 135
(поверхность стоила увеличена в г0 раа)
И трицитробензолу. Гомогенная реакция происходит с ускорением, слабо
выраженным у моиопнтробенаола, сильнее у динитробензола п еще силь-
нее у тринцтробензола; скорость же гетерогенной реакции, идущей, воз-
можно, без ускорения, падает по закону, близкому к закону реакции пер-
вого порядка; при увеличении давления возрастает удельный вес гомо-
генной реакции, максимум скорости становится больше и наступает
раньше.
Зависимость скорости реакции от температуры была для мононитробен-
зола определена путем построения аррепиусовской прямой, причем кон-
станта скорости рассчитывалась для начального участка кривых
dp 1____t
dt ро
(величина ро изменялась для сравнивавшихся опытов в относительно уз-
ких пределах — от 300 до 500 мм).
Из построенных графиков значения Е — 53,4 ккал/молъ и 1ц В = 12,65.
Аналогичным образом для динитробензола Е — 52,6 ккал/молъ и 1ц В —
= 12,70; щ[я тринцтробензола Е — 51,9 ккал/молъ, 1g В — 13,6.
53
Понятно, что при одновременном протекании с соизмеримыми скоро-
стями гетерогенной и гомогенной реакций, удельный нес которых меняется
с температурой, использованный метод расчета по суммарной скорости
носит в известной мере формальный характер тем более, что при равен-
стве абсолютных начальных давлений относительные давления были дли
всех трех веществ различны; различной может быта п их способность
адсорбироваться на стекле, С этой оговоркой рз полученных данных сле-
дует, что при одинаковых температурах температурный коэффициент ско-
рости приблизительно одинаков для всех трех веществ, что и выражается
в примерно одинаковом значении Е.
Температура влияет и на характер кривых РИ (£) динитробензола
и трииитробензола; чем выше температура, тем меньше относительный
максимум скорости И^макс/И^ и тем раньше он наступает. По-видимому,
как это обычно наблюдается, энергия активации самоускоряющейся ре-
акции меньше, чем первичной, и соответственно при повышении темпера-
туры удельный вес последней возрастает. Подобная картина может, впро-
чем, наблюдаться и при последовательных реакциях, если скорость вто-
ричной стадии сильнее зависит от температуры, чем первичной,
Моцо-, ди- и трихлортри нитробензол
Разложение моиохлортринитробензола в парах идет при сравнимых
условиях в 4 раза быстрее, чем тринитробензола; ускорение газообразова-
ния несколько меньше и также, хотя и слабее, чем для трииитробензола,
возрастает при увеличении отношения т/v. Введение стеклянных капил-
ляров, аналогично тому, как это наблюдалось для всех изучавшихся нит-
росоединений в парах, сильно увеличивает скорость газообразования и из-
меняет характер кривой p(t).
Зависимость скорости распада от температуры значительно слабее,
чем для тринитробензола; к — 1О®-45 ехр (— 37 000 RT}. Ди- и трпхлор-
тринитробензолы по скорости распада (при 330° С, ро = 150 льм) близки
к монохлорсоединению.
Тринитротолуол
Разложение метилтринитробензола — тринитротолуола, изучавшееся в
интервале температур 280—320° С при начальном давлении 70—200
идет гораздо (в 1000 раз) быстрее, чем трипитробепзола; наблюдается
ускорение, хотя и меньшее (в 2—3 раза по величине Пмакс/ТРо), нем
у трпнитробензола. Начальная скорость, а также степень ускорения воз-
растают (рис. 41) как и для тринитробензола при увеличении отношении
min (начального давления паров ро); при ро = 40 мм скорость (абсолют-
ная) почти постоянна во времени; при ро = 160 мм начальная скорость
в 3, а максимальная в 6 раз больше, чем при 40 мм. При увеличении
отношения т!и сокращается также время достижения максимума.
Увеличение поверхности сосуда (в 5 раз) путем введения стеклянных
капилляров резко (в 60 раз) увеличило начальную скорость распада по-
добно тому, как это наблюдалось для трипитробепзола; скорость в этом
случае пе росла, а падала во времени; это падение скорости (для гетеро-
генной компоненты) хорошо укладывается на прямую первого порядка.
Построение аррениусовской зависимости для начальной скорости при
постоянном начальном давлении (80 льв) не дает прямой липки. Прямая
получается, если при различных температурах брать скорость при оди-
наковых приведенных давлениях (po/ps, где ps — упругость пара тринитро-
толуола при данной температуре). Для отношений ро/ра = 0,19—0,25
энергия активации составляет 34,5 ккал[молъ, а предэкопонепт 108,15; при
большем отношении ро/рз (0,41—0,46) эти величины соответственно рав-
ны 31,5 и 107’4,
54
Тринитрофенол
I рипитрофенол в парах, так же кап трииитробензол и тринитротолуол,
разлагается при низких давлениях (30 мм) с малым ускорением и (при
290е С) вдвое быстрее тринитротолуола. Увеличение начального давления
г, ад
Рис. 41, Влияние начального давления на из-
менение давления (Л) и скорости газообразо-
вания (В) во времени при распаде тринитро-
толуола
Числа у кривых — давление <Ро, в лив г>т. ст.);
I8i> леч рт. ст.— давление насыщенного пара;
о опмте с индексом (m/и в г/елг1' 10”*)—смесь
пара и ^нидиости
увеличивает как начальную
скорость, тар и максималь-
ную, особенно сильно, когда
давление паров приближает-
ся к давлению насыщенного
пара. Время достижения .мак-
симума сокращается. Увели-
чение поверхности в 10 раз
резко (в 100 раз) увеличивает
Рис. 42. Разложение тринитрофе-
но^а в парах ггри 290э С
Д;1я верхней кривой относительная
величина поверхности стекла рас-
[ta 10, дли нижней—1
начальную скорость распада (рис. 42); достигнув максимума, давление
начинает медленно падать, по-видпмому, в результате образования кон-
денсированных продуктов,
Трцнитроанилив
Тринитроанилпи разлагается в парах при 300° С значительно медлен-
нее тринитротолуола (в 20 раз) и всего в 2 раза быстрее, чем тринитро-
бензол. Из зависимости начальной скорости от температуры Е =
— 38,5 ккал/молъ и ]g В = 8,8. Начальная скорость и максимум скорости
возрастают с давлением подобно тому, как это наблюдается для других
описанных выше нитросоеДинсппй. Сходное влияние (сильное увеличение
начальной скорости, исчезновение ятапа ускорения) оказывает и набивка
сосуда капиллярами. Гетерогенная реакция идет по закону реакции пер-
вого порядка,
2. РАЗЛОЖЕНИЕ НИТРОСОЕДИНЕНИЯ В ЖИДКОМ СОСТОЯНИИ
Три нитробензол
Разложение жидкого трнпитробензола в интервале температур 250 —
310° С идет приблизительно в 10 раз быстрее, с тем же удельным газо-
образованием, но с большим ускорением, чем в парах.
55
Рис. -53. Разложение жидкого три-
нитробензо.та при 271° С
Числа у кривых — отношение m:v
Рис. 44. Разложение жидкого тришггро-
беизоиа нри 312° С
Числа у кривых—отношение m:'v
(г/с.ч3-1О-4)
Вид кривых изменения скорости газообразования но времени при низ-
ких температурах (рис. 43) указывает на сложный характер превраще-
ния; скорость вначале надает, а затем начинает расти. Увеличение отно-
шения иг/г, т. с. давления газообразных продуктов на определенной ста-
дии распада, слабо увеличивает скорость газообразования и сильнее ее
рост во времени, а также максимум скорости. Она растет также и неза-
висимо от роста давления по мере развития процесса, как это показывает
сопоставление значений скорости при одном ц том же давлении, но на
разных стадиях разложения. Влияние отношения mlt> проявляется н при
более высоких температурах (рис. 44). Самоускорение газообразования пе
является, ио-видимому, проявлением автокатализа, так как добавление
частично разложившегося продукта к свежему не приводит к увеличению
скорости сверх аддитивной. Введение стеклянного порошка в жидкость
пе ускоряет газообразования, стеклянные капилляры над жидкостью силь-
но увеличивают скорость газообразования. Зависимость скорости от тем-
пературы определена на начальном (2 — 3% распада) участке при т/и =
— 100- Ю-4 г/с-м3 и следует уравнению к = 1О10-9 ехр ( —43 000/Л?’).
Тетранитробензол
Разложение жидкого тетрапитробеизола идет не только значительно
ГйАМроб l,Ppn 22tf С и mJv = Ш) > г|сл£ приблизительно в 300 раз),
чем тринптробензола, но и существенно отлично от последнего в отноше-
нии характера кривой и влияния mfv на скорость распада.
Так, при 22(Г С, а также и более низких температурах (рис. 45) рас-
пад идёт с большой начальной скоростью, однако роста ее во времени
пе наблюдается- помимо этого, газообразование почти прекращается после
выделения некоторого объема газов (300 ext3 при 220 С), значительно
меньшего, чем при полном распаде трипитробензола. По-видимому при
распаде тетранитробепзола образуется стойкий сравнительно с ним самим
промежуточный продукт, способный, однако, к дальнейшему разложению
с образованием газов.
Другой особенностью распада тетранитробензояа является то, что
скорость его как при 220, так и при 180 я 140° С не возрастает, подобно
скорости распада всех остальных изучавшихся нптросоедипенци бензоль-
ного ряда, но существенно уменьшается при увел [[‘[они и отношения
(рис. 46).’ Причины этого отличия не установлены, можно предположить
образование промежуточного продукта, обратимо разлагающегося с об-
5U
Рис. 45. Разложение жидкого тетра-
[(птробелзола при различных темпе-
ратурах
Параллельный опыт при 220° С был
продолжен при 300° С
Рис. 46. Влияние отношения m/п на
разложенце тетраяцтробензола при
220° С
Числа у кривых— т'ъ (г-'см1-10-1)
разованием газов; та же картина получилась бы, если бы разложение шло
практически только в парах, относительное количество которых обратно
пропорционально отношению ml о.
Моно-, ди- и трихлортрр нитробензо л
Разложение моно-, ди- и трихлортринитробензолов в жидком состоя-
нии идет со скоростью, в 5 —10 раз превышающей скорость в парах и зна-
чительно быстрее, чем тринитробензола. При переходе от моно- к трч-
хдорзамещенному бензолу скорость распада уменьшается. При сопоста-
вимых условиях (2лО°С, mjv = 100 • 10“4 г/сз*3) скорости для тринитро-
бензола, монохлортриннтробензола, дпхлортринцтробензола ц трвхлортри-
гцттробензола относятся как 1:80:50:5, В ходе распада скорость растет
в 5 — 10 раз.
Тринитротолуол
Рогинским и сотр. [63] было установлено, что термический распад
тринитротолуола в интервале температур 220—270° С идет с самоускоре-
цием, которому при низких температурах иногда предшествует значи-
тельный по длительности индукционный период. Ускорение, по-видимо-
му, обусловлено накоплением конденсированного авто катализатора, по-
скольку, как показали опыты Ю. П, Захарова, откачивание газов во время
идущего разложения не приводит к заметному изменению скорости газо-
образования. Чем выше температура, тем больше максимум скорости
и тем быстрее он достигается.
А. Робертсон [64] подтвердил самоускоряющийся характер распада
тринитротолуола — давление возрастало во времени по показательному
закону р = A.eki, Константа ускорения росла с температурой k =
= 10’14 exp ( — 34 400/2??'), где к выражено в обратных секундах.
На участке замедления газообразования скорость его уменьшается по
закону реакции первого порядка, причем Е = 34 ккал/молъ и 1g В = 11.
Энергия активации, рассчитанная ио линейной зависимости логарифма
задержки вспышки от обратной температуры, составляет 32 ккал/молъ,
Робертсон изучал также влияние различных- добавок на распад rpii-
нцтрс>толуола. Многие из добавок в небольших количествах увеличивают
k и соответственно уменьшают задержку вспышки. Ни одна из изучен-
ных добавок не оказывала замедляющего действия.
57
По Максимову, разложение жидкого тротила при 290° С идет, как
и в случае распада тринитробензола, приблизительно в 10 раз быстрее,
чем паров, с ускорением, которое слабо возрастает при увеличении отно-
шения m/о. Из разложившегося при 230° С наполовину вещества может
быть выделено растворяющееся в ацетоне и тротиле вязкое бурое веще-
ство. разлагающееся при той же температуре по закону первого порядка
Рис. 47. Разложение тротила при 230° С и выде-
ленного из него промежуточного продукта.
Проверка течения распада последнего па первому
V — V
оо
порядку, 1g -------------------(т)
Рис. 48. Влияние добавления 3%
промежуточного продукта разло-
жения на распад тротила при
230“ С
со скоростью, в десятки раз превышающей скорость распада тротила
(рис. 47). Добавление к тротилу этого вещества в количестве всего 3%
втрое сокращает время достижения максимума скорости (рис. 48). По-
скольку экспериментальная кривая идет значительно выше аддитивной,
следует заключить, что промежуточный продукт оказывает ускоряющее,
возможно, каталитическое действие на распад тротила, что согласуется
и с наблюдениями Робертсона [64].
Для определения коэффициентов температурной зависимости распад
был прослежен при четырех температурах (193, 210, 230 и 250° С) при
таких отношениях тп/п, при которых доля конденсированной фазы была
одинаковой (0,9). Полученные кривые (до 25% Ркоч) хорошо совмеща-
ются при изменении масштаба времени; по аррениусовской прямой Е —
— 26,2 ккал/молъ.
Тринитрофенол
Основные опыты но разложению жидкого трннитрофенола проводи-
лись при малых значениях m/v (10* 10~4 з/елг3), позволяющих просле-
дить весь ход распада. Распад в жидком соМоянип идет гораздо быстрее,
чем в парах, скорость распада жидкого трипптрофепола, экстраполирован-
ная от 183—270° на 290°, в 10 раз больше, чем паров. Ход кривых рас-
пада (рис. 49) своеобразен и указывает на сложное протекание разложе-
ния, в котором можно различить по крайней мере 5 этапов, отличающих-
ся по характеру зависимости скорости газообразования от времени. В целом
ускорение при распаде тринитрофенола выражено Очень слабо, го-
раздо меньше, чем при распаде тротила и других замещенных тринитро-
бензОлов. Первым этапом, отчетливо наблюдающимся при низких тем-
пературах (и больших значениях m/к), является индукционный период,
затем следует второй Этап (II) —- ускорение газообразования; третьим
58
этаном (777) является быстрая реакция, скорость которой падает во
времени, количество выделившихся газов к концу этого этапа растет
с температурой и при 180—270° С составляет 5—20% от конечного их
количества. Четвертый этап (Л') характеризуется значительно (в 3—
9 раз) меньшей скоростью газообразования, чем максимальная скорость
па третьем этапе; при этом скорость не надает, а растет, хотя и слабо,
Рис. 49. Изменение скорости распада во
времени (lg W — 1g т) при распаде трпнпт-
рофенола при различных температурах
Рис. 50. Разложение жидкого тринит-
рорезорцина при различных темпе-
ратурах
Римскими цифрами указаны отдельные этапы
распада
но времени. Рост этот выражен тем сильнее, чем ниже температура. Ско-
рость возрастает при увеличении отношения m/к. На пятом этапе (И)
скорость падает, по-видимому, в результате уменьшения количества ис-
ходного вещества. По значенпям средней скорости на IV этапе при
разных температурах можно рассчитать энергию активации (Е —
= 38,5 ккал/молъ), и принимая первый порядок реакции, предэкспонент
(IgS = 11,7).
Трппитрорезорцин
Изучение термического разложения трипитрорезорцияа затрудняется
тем, что он относительно крепко удерживает воду. Скорость разложения
значительно больше (приблизительно в 10 раз), чем у тринитрофенола.
Общая картина распада (рис. 50) анат^ична наблюдавшейся для трипи-
трофенола. Распад сопровождался небольшим начальным ускорением га-
зообразования (первый этап), разложением со значительной, но быстро
уменьшающейся скоростью (второй этап), участком слабо растущей ско-
рости (третий этап) и, паконец, ее падением. Первый этап обнаружива-
ется лишь в опытах, проведенных при наиболее низких из изученных
температур (180 — 200° С); это отчасти относится и ко второму этапу,
длительность которого составляла при этой температуре около 10 мин.,
а количество образовавшихся газов— 20 — 35 нсм2/г. Третий этап — слабо
ускоряющееся газообразование — охватывает основную часть распада;
скорость его гораздо (в 6 — 8 раз) меньше, чем на предыдущем этапе,
мал и рост скорости; при 200° С он составляет всего 1,56, при более вы-
соких температурах — еще меныпе. Температурная зависимость, оцепои-
пая так же, как для трпнитрофепола, приводит к Е = 34,6 ккал/молъ,
1д/7 = 11,2.
Т р 11 п п т р о а и и л и н
Жидкий тринитроанплин разлагается значительно быстрее газообраз-
ного; при экстраполировании данных по распаду паров на температуру
275° С, при которой изучалась жидкость, скорости различались в 15 —
20 раз (m/у — 30’ 10-4 г/см2).
59
Однако скорость газообразования сильно возрастает при увеличении
значений т/и, так что при больших значениях т/и разница между жид-
костью и парами еще больше. Скорость распада растет и во времени
и па максимуме превышает начальную приблизительно в 10 раз; при
больших значениях m/г ускорение распада во времени уменьшается и
даже прекращается (рис. 51).
Рис. 51. Разложение жидкого трипитроаиилипа
при 275° С и различных значениях отношении т/и
Числа у кривых — mlv (г/с*1-11}-1)
Зависимость скорости газообразования от температуры была определе-
на путем совмещения кривых v{t), полученных при разных температурах
и приблизительно одинаковом отношении т/и (100 — 150- Ю’"1 г/ем3).
Кривые удовлетворительно совмещаются лишь па начальном участке (до
у = 0,025 Ркол),‘ расчет по Аррениусу дает Е = 31 ккал/моль и 1g В = 7,1.
Соединения с двумя бензольными ядрами
Жидкий гексанитро дифенил разлагается с ускорением (при 245“ С
К макс/Жо = 8) (рис. 52), приблизительно таким же, как и у тринитробен-
зола, но начальная скорость (при 290° С) в 10 —15 раз больше, чем у три-
ннтробспзола. Ускорение газообразования возрастает при увеличении от-
ношения m/v и тем сильнее, чем ниже температура. Определение зависи-
мости скорости от температуры было произведено совмещением кривых
v(t) на участке от начала распада до достижения и — 0,131(ОН, оно дает
Е = 49,5 ккал/моль и 1g В = 16,1.
Жидкий гексанитродифенилсульфид разлагается при 245° С в 10 раз
быстрее гексанитродифенила, т. е. в 100 раз быстрее трииитробензола.
Разложение идет со значительным ускорением. При 235“ С скорость на
максимуме в 10 раз превышает начальную. Повышение температуры (при
постоянном т/и) увеличивает отношение П'макс/Жо. Изменение отноше-
ния m/v лишь слабо влияет на течение распада. Добавление частично
разложившегося вещества приводит к некоторому ускорению распада.
При относительно низких температурах (235—255° С) и значительном
м/г (!()()• 1()—J| г/ем3) распад имеет на начальных этапах отчетливый
двухстадинный характер (рис. 53), Совмещением кривых для разных
температур (на участке до 6% распада) можно определить
зависимость скорости газообразования от температуры, что дает Е =
— 43 ккал/моль.
Рис. 52. Разложение
жидкого гексанитроди-
фенила при различных
температурах (а) и со-
вмещение кривых и — т
с изменением масштаба
времени (6) (m/у = 10-
ТО-4 г/с-м3, кТц — коэф-
фициент трансформа-
ции)
Рис. 53. Разложение
жидкого гоксанитроди-
фе ни л сульфида при раз-
личных температурах
а — изменение давания
во времени; б — измене-
ние скорости газообразо-
вания во времени
в
Жидкий гексил (т. пл. 243° С) при относительно высоких температу-
рах разлагается со скоростью, несколько большей, нем трииитрофеиол,
и уменьшающейся во времени. Увеличение отношения m/ь’ уменьшает
общую скорость газообразования особенно на начальной стадии. При са-
мой низкой из изучавшихся температур (250° С) па начальной стадии рас-
пада наблюдается небольшое ускорение,
сходное с тем, которое установлено и
для тринптрофенола в близких усло-
виях.
Аналогичный ход кривой к(0 и у
гексанптрооксанилида (т. пл. 332°) при
338°С (рис. 54). «Время полураспада»
в 3 раза больше, чем для тринитрофено-
ла, но в 200 раз меньше, чем для три-
нитробензола. ж
Рис. 34. Разложение жидкого гекса
нитроокса нил и да при 338° С
Разложение при больших'
значениях m/v
Все рассмотренные выше данные по
распаду н итросоединений н жидкой
фазе были получены при m/к, не пре-
вышающих 500-Ю-4 г/с№; это позволя-
ло проследить процесс распада на заметном его протяжении. В условиях
применения распад может идти при значительно больших степенях запол-
нения емкости. В связи с этим распад некоторых ВВ был изучен также
нри больших значениях m/v (2000—6000 • 10-4 г/см3). Для тринп-
тробензола начальная скорость распада (250°) при увеличении значения
m/v от 330 Ю-4 до 6500 10~4 г/см3 продолжает возрастать, ио относитель-
но слабо (в указанном интервале m/u всего в 1,3 раза); несколько сильнее
возрастает ускорение распада.
Сходным образом влияет увеличение отношения m/u на разложение
монохлортринитробензола: при его увеличении от 40-10"4 до 40 000-
10~4 г/с.ч3 начальная скорость возрастает лишь в 3 раза. Относительное
ускорение (ТУмакс/И^) не зависит от от/гл Для тетранитробензола (220° С)
при переходе от ш/п = 3‘10-4 к 1750-10~4 г/см3 скорость распада
уменьшается в 50 раз. Ускорения распада во времени для тетранггтро-
бензола не наблюдается как при малых, так и при больших значе-
ниях ш/о.
Более сложное (увеличение скорости газообразования на ранних ста-
диях при увеличении в области малых его значений и прекращение этого
увеличения при больших m/о) влияние оказывает m/v па распад дихлор-
тринитробензола и трихлортрииитробензола. Речь идет, однако, о выде-
лении очень малых количеств газов, составляющих несколько кубических
сантиметров на грамм, которое может быть связано также с разложением
возможных примесей.
При сопоставлении с тринитробензолом расположение ВВ в ряду стой-
кости сохраняется тем же, что и при малых m/v, но количественные раз-
личия значительно меньше; так монохлортринитробензол оказывается уже
пе в 80, а лишь в 5 раз менее стойким, чем тринитробензол.
3. РАСПАД ПОЛИНИТРОСОЕДИНЕИИЙ В ТВЕРДОМ СОСТОЯНИИ
Введение некоторых заместителей в молекулу тринитробензола суще-
ственно повышает температуру плавления; то же наблюдается и для со-
единений, содержащих два или более тринитробензольных кольца. Повы-
шение температуры плавления может быть столь значительным, что ско-
62
рость термического разложения становится доступной измерению уже
ниже температурь! планления.
Скорость распада ВВ в твердом состоянии, как и всех других исследо-
ваппых в этом отношении веществ, значительно меньше, чем в жидком
состоянии. Благодаря этому казалось возможным, вводя в тринитробепзол
заместители, повышающие температуру плавления, пли получай много-
кольцевые полинитросоединения, повышать термическую стойкость.
Исследования показали, однако, что введение любого заместителя в трини-
тробензол повышает скорость распада, и это повышение при всех темпе-
ратурах преобладает над понижением ее вследствие изменения агрегат-
ного состояния. Таким образом, этот путь повышен пн термической стой-
кости может иметь практическое значение лишь в том случае, если обя-
зательным требованием является твердое агрегатное состояние вещества
(напомним^ что трииитро бензол является относительно низкоплавиим - -
т. пл. 122d С — соединением); одним из обоснований этого требования
может быть большая плотность твердого вещества по 'сравнению с рас-
плавом.
Распад полинитросоедппенпй в твердом состоянии, таких как гексани-
тродпфеннл, гексанитродпфенилсульфид, гексанптродифенилоксид, гекса-
нптродифепиламин, гексацитрооксанилид, диамипотрипптробензод и тря-
амппотрицитробензо.т, изучался Максимовым.
Гексанитродифенпл плавится при относительно низкой температуре
(240’С), при которой скорость его распада еще очень мала — 1 г веще-
ства за 15 дней выделил лишь 15 см3 газа. При 216 и 231° С начальные
скорости газообразования составили соответственно 2,7 10-5 и 9,75 -
• 10'6 нсл«3/гсек, что приблизительно в 10 раз мепыпе, чем дает экстра-
поляция на эти температуры скорости газообразования жидкого гексапя-
тродифенила.
Летучие примеси, содержащиеся в неочищенном гексанитродифеппле,
значительно ускоряют его распад как в жидком, так и особенно в твердом
состоянии.
Распад твердого гексанцтродифенилсулъфида (т. пл. 233° С) изучали
яри 200—220° С. Ускорение распада И’макс/И’о велико и равно 100 для
200° и 70 для 220°, что почти в 10 раз больше, чем при распаде расплава
Н, по-видимому, связано с прогрессирующим расплавлением вещества
в ходе распада. Скорость газообразования слабо возрастает при увеличе-
нии значений m/с. Наступление быстрого ускорения распада происходит
гораздо раньше при меньших кристаллах, но скорость на этом последнем
этапе больше для крупных кристаллов. Скорость распада твердого веще-
ства в 3—5 раз меньше, чем расплава. Для твердого вещества расчет по
температурной зависимости коэффициента трансформации интегральных
кривых дает Е = 42 700 ккал/молъ.
Распад гексанитродифенцлоксида (т. пл. 266° С) в твердом состоянии
протекает с ускорением равным 10. Скорость на этапе ускорения растет
по показательному закону. Чем больше чистота продукта (оцениваемая по
т. пл.), тем меньше скорость распада.
Гексанитродифенпламнн (т. пл. 242° С) и в твердом состоянии разла-
гается относительно быстро, ио со скоростью, падающей во времени. Она
растет с температурой относительно медленно — на участке приблизи-
тельного постоянства скорости Апо5 = 1,8, в то время как для расплава
при более высоких температурах к ~ 3. Содержание конденсирующихся
при комнатной температуре соединений в газообразных продуктах распада
по мере его течения значительно возрастает.
Распад гексапитрооксанилида (т. ил. 332° С) в жидком состоянии
(338° С), идущий с уменьшением скорости во времени, при температурах
ниже температуры плавления, идет по-разному в зависимости от т/г-.
Увеличение отношения m/v оказывает двоякое действие — скорость
63
г/р язи ‘л г/р из и V?
а: 1 — 10; 2 — 75; 3 ~ 303.
б: J--2; 2—10; 3--И2; 4 — 264.
в: t — 10; й— 10; 3 - 60; 4 ~
11)1); .5 — '270. г: 1 — 10; 2 — 18;
3 — 80
Рис, 55, Разложение твердого
гекса шпроо пеан или да при
различных температурах и
отношениях m/v( г/с.и3 Ю-4);
Т, МИ
Рис. 56, Изменение скорости распа-
да твердого дяаминотринитробс'нзо
да пн времени npti т-/н = - 10- 10 4
г/с,иа tt различных температурах
(в “С):
I — 259; 2 — 209; 3 — 279
газообразования уменьшается, ускорение увеличивается. Количествен о
оба эти влияния зависят также от температуры опыта (рис, 55), Из совме-
щения начальных участков интегральных кривых для твердого вещества
= 51,5 ккял/моль, 1g В = 16,0. Скорость газообразования несколько
уменьшается при увеличении размеров кристаллов.
Рис. 57. Изменение газо-
образования (Л) и ско-
рости газообразования
при распаде три-
амин о три нитро бе и з о ла
при т/» =10-10“11 ?!см3
и различных температу-
рах (°C):
1 — 284; В — 2ВЗ- 3 — 305;
4 — 311: 5 — 320
Распад диаминотринитробензола (т. пл. 289° С) сопровождается час-
тичным ожижением, тем большим, чем выше температура распада. На
кривых скорость — время наблюдаются два максимума скорости, тем более
выраженных, чем выше температура опыта (рис, 56). Первый из них на-
ступает при выделении 2—5% от общего количества газов, второй — около
50%. При более низких температурах характер кривых существенно ме-
няется, Изменение размеров кристаллов в 10 раз не оказывает значитель-
ного влияния на характер и скорость газообразования.
Распад триаминотрпнитробепзола (при определении температуры плав-
ления разлагается не плавясь) слабо зависит от Jn/v и от размеров кри-
сталлов. В интервале 284—320° С распад идет (рис. 57) с относительно не-
большим (2,5 —2,9) ускорением, величина которого не зависит От темпера-
туры; максимум скорости наступает сравнительно поздно, при 50—80%
разложения. Интегральные кривые хорошо совмещаются до конца распада,
что дает Ё = 41,8 ккал!молъ, 1g В = 11,56.
IV. N-НИТР АМИНЫ
Распад нитраминов алифатического ряда исследовался для метилен-
динитрампна и этилендинитрамина.
Метилендинитрамин (медина, NOjHN — CHs—NHNO2)
Распад медины (т. пл. 105—106° с разложением) изучался [65] мето-
дом непрерывного (посредством весов Вестфаля) или периодического оп-
ределения потери в весе при нагревании в открытых пробирках, чтобы
предотвратить накопление ускоряющих процесс летучих продуктов. Ко-
нечная потеря в весе составляла около 45%; остаток представлял собой
белый порошок, нерастворимый в воде, бензоле, ацетоне и спирте, раз-
лагающийся при ~ 208° С.
5 К. К, Андреев
65
Распад расплава в интервале температур ПО —130° С идет без уско-
рения со скоростью, пропорциональной количеству перазложпвпюгося ве-
щества. Ее константа к = 1015’6 ехр ( — 35 400/7?/).
Распад твердого вещества при 70 —100° С начинается индукционным
периодом, во время которого разлагается около 1 % вещества и скорость
падает. За пим следует период ускорения. Добавление пикриновой кислоты
в раствор с последующим удаленном растворителя це влияет па длитель-
ность индукционного периода и развитие последующего ускорения; одна-
ко количество вещества, разложившегося в индукционном периоде, су-
щественно уменьшается, в чем и проявляется ингибирующее действие
пикриновой кислоты. Это действие по предположению авторов исследова-
ния связано с затруднением поверхностной реакции образования зароды-
шей, поскольку пикриновая кислота была введена так, что она находи-
лась только па поверхности частиц вещества. Сходство характера распада
с тем, который наблюдался Для твердых неплавящихся солей, позволяет
предположить ого топохимическое течение, связанное с образованием
зародышей реакции на поверхности кристаллов и последующим их рос-
том. Одпако близость температуры опытов к температуре плавления ис-
ходного вещества и наблюдавшееся в других работах полное расплавление
мсдины при распаде вблизи температуры плавления указывает па воз-
можность другого механизма ускорения — прогрессивного перехода твер-
дого вещества в расплав в результате накопления растворимых в нем
продуктов распада. Скорость же распада расплава Медины, как и следо-
вало ожидать, гораздо больше, чем твердого вещества.
Этплендинитрамцн (эдна, NOjHN — СН2СН2—NHNO2)
Термический распад этплендиндтрамина (т. пл. 176,5° С, высушен ггад
серпов кислотой, степень чистоты 99.95%) изучался при температурах
ниже температуры плавления (60—120° С) и выше нее (180—254° С),
а также в водных растворах {66, 64].
Рпс. 58. Влияние различных при-
месей (й) и тротила (ТНТ) (б)
на разлоя;еиие эдны
При разложении сухой эдиы в вакууме
(начальное давление 2—5 мм, фракпия
порошка между ситами с ячейками 0,147 —
0,295 льи) кривая р — т при 100" С имеет,
по крайней мере вначале, насыщающийся
характер; при 120°С скорость в точение
50 час. приближенно постоянна, а затем
растет по показательному закону на участ-
ке до 2—3% распада.
При давлении 1 ат [фи 120° С наблю-
дается индукционный период, длящийся
100—150 час., за которым следует экспо-
ненциальный рост скорости, продолжаю-
щийся до 25% распада; в 50%-ной смеси
с тротилом ускорение по показательному
закону наблюдается па протяжении всего
распада.
Примеси воды (0,1%) или гликоля (0,1
и особенно 50%) ускоряют распад; смеси
с тротилом (50 '. 50) также разлагаются
значительно быстрее, чем сама эдна
(рис. 58). Газообразные продукты распада
по ускоряют его; упоминается без конкрет-
пых данных, что увеличение размера на-
вески к удельпон ее поверхности ускоря-
ют распад. В отличие от того, что должно
6G
быть по формуле Аррениуса, температурный коэффициент скорости
реакции пе уменьшается, а возрастает при повышении температуры:
эффективное значение энергии активации возрастает от 36—37 ккал/моль
при 100—120° до 51,5 ккал/моль при 135— 1451* С,
При высоких температурах [64] в вакууме (Ю-3 мм) вещество испа-
рялось и перегонялось в холодные части прибора, прежде чем происходи-
ло заметное разложенце. Для предотвращения улетучивания применяли
инертный газ, обычно азот, под давлением 10 см. При этих условиях жид-
кое ВВ разлагалось в интервале температур 184—254° С Со скоростью,
пропорционально ii количеству не разложившегося вещества. Период полу-
распада составлял 43 сек. при 184° и ‘/г сек. ври 254° С. Константа ско-
рости при низких температурах не зависела от количества вещества; это
указывает на гомогенность реакции распада. Для кинетических опытов
при более высоких температурах, чтобы избежать вспышки и обеспечить
быстрый прогрев, приходилось применять очень малые навески.
Константа скорости li — 1012,s ехр (— 30 500/Д77).
Газообразные конечные продукты распада при 195° С содержали в мо-
лях на моль вещества: NO — 0,10; NaO — 1,4; Na — 0,39; СО — 0,028 и СО2
— 0,065. Образовывались также вода и ацетальдегид. *
В водных растворах одна разлагается, гидролизуясь, с образованием
закиси азота, этиленгликоля и ацетальдегида. Скорость гидролиза доволь-
но велика и возрастает при увеличении кислотности (H2SO1) раствора
к температуры; во времени она уменьшается по закону реакции первого
порядка; таким образом, автокаталитического ускорении гидролиза пе
наблюдается. Переход от нейтрального раствора к 0,009 Аг увеличивает
константу скорости почти в 2,5 раза, в 0,5 А кислоте она в 127 раз боль-
ше, чем в нейтральном растворе.
Причиной ускорения распада твердой эдны во времени следует счи-
тать накопление конденсированных продуктов распада. Однако отнюдь
не обязательно их действие является каталитическим в обычном смысле
этого слова. Против этого говорит отсутствие ускорения при распаде рас-
плава здпы н опытах Робертсона [64]; однако эти опыты проводились при
относительно высоких' температурах*, когда роль автокатализа может ста-
новиться относительно малой; поэтому более убедительной была бы
констатация отсутствия самоускорения при разложении растворов эдны
в инертном растворителе, например, тротиле при относительно низких
(100—120° С) температурах. Наиболее вероятной причиной ускорения яв-
ляется перевод вследствие накопления конденсированных продуктов рас-
пада возрастающей во времени доли эдны в жидкое состояние благодаря
взаимной растворимости исходного вещества и продуктов его распада.
Скорость же распада жидкой эдны гораздо больше, чем твердо if. Так же
следует объяснить ускоряющее влияние тротила, в котором эдпа заметно
растворима. Это объяснение ускорения распада представляется более ве-
роятным, чем связанное с развитием топохимического ускорения распада
в твердой фазе. Показательный закон роста скорости во времени, наблю-
дающийся при распаде твердого продукта, нельзя считать достаточным
основанием, чтобы относить распад, как это делает Томлинсон [66], к цеп-
ным процессам. Выполнение этой зависимости в присутствии тротила
является дополнительным свидетельством в пользу объяснения ускорения
распада ожиженном исходного нещества.
Т р н н и т р о ф е п и л м е т и л п и т р а м н и
[тетрил, CeHa^AO^gNCHaNOs]
Тетрил разлагается с заметной скоростью уже при температуре плав-
ления (131,5° С); поэтому его разложение могло быть изучено при темпе-
ратурах, лежащих как выше, так п ниже температуры плавления [67],
5*
67
Для установления порядка реакции была определена скорость распада
расплава тетрила и его растворов в тротиле (табл. 6). Последний был вы-
бран в качестве растворителя, поскольку в данном интервале температур
он практически не разлагается и в то же время устойчив по отношению
г: тем химически активным продуктам, которые могут образоваться при
распаде тетрила.
Таблица 6
Зависимость скорости распада тетрила
от его концентрации
Число молей газа на t моль тетрила
Время, часы ЧИСТЫЙ тетрил 33%-ный раствор тетрпла 16,6%-иый раствор тетрила
1 0,18 0,23 0,19
2 0,49 0,63 0,54
3 0,86 1,00 0,84
4 1,19 1,28 1,09
5 1,40 1,50 1,30
в 1,64 1,65 1,43
Результаты опытов показывают, что скорость пе зависит от концентра-
ции тетрила, откуда следует, что основная реакция при распаде является
моно молекулярной.
Однако на начальных стадиях распад идет с ускорением. Это показы-
вает. что наряду с мономолекулярной реакцией протекает реакция авто-
каталитическая. Предположительным автокатализатором является обра-
зующаяся при реакции пикриновая кислота.
При простейшем допущении, что ускорение реакции катализатором
пропорционально первой степени его концентрации, для суммарной ско-
рости применимо выражение
4- Ь + 41п (! + £> = const = <2-28)
где /с = — отношение констант мономолекулярной и автокаталити-
ческой реакций.
По данным для двух замеров при 140° С было вычислено значение
константы к ~ 0,187. Зная эту величину, можно было проверить постоян-
ство константы S на протяжении всего опыта, т. с. справедливость для
разложения тетрила уравнения автокаталитической реакции. Расчеты
показывают хорошее постоянство константы, которая изменяется в пре-
делах, пе превышающих ± 4%.
Опыты, в которых к тетрилу была прибавлена пикриновая кислота,
также количественно подтвердили предположение о ее катализирующем
действии, связанном, по-видцмому, с действием иона водорода.
Катализирующее действие пикргшовой кислоты на термическое раз-
ложение тетрила подтверждено также в работе Рогинского и Лукина
[68], методом задержки вспышки.
В стеклянную ампулу емкостью 2,5 с .и 3 вводили навеску тетрила
(0,3 г); ампулу запаивали, помещали в термостат и определяли время
до возникновения вспышки. При 156° С время задержки от добавления
5% пикриновой кислоты сократилось с 29 мин. до 21 мин,, при 20% —
до 13,5 мин. Дальнейшее увеличение добавки пикриновой кислоты не
уменьшало время задержки; по-видимому, сказывалось ее разбавляющее
действие.
68
В этой же работе было показано, что, помимо образования пикрино-
вой кислоты, па возникновение вспышки тетрила влияют также условия
опыта. Было установлено, что в открытых ампулах в определенных усло-
виях опытов вспышки при 150—170° С не происходило. В запаянных же
ампулах вспышка происходила, начиная от 150° С, при этом чем больше
было количество вещества па единицу объема ампулы, иными словами,
чем больше давление газов, развиваемое при разложении, тем ниже пре-
дельная температура, при которой еще происходит вспышка1. Это влияние
«плотности заряжания», наблюдавшееся для многих ВВ, по Рогинскому
наиболее естественно объяснить, допустив, что газообразные, промежуточ-
ные продукты разложения, но-видимому, оказывают сильное влияние на
скорость реакции и возникновение вспышки. Концентрация этих продук-
тов в жидкой фазе, очевидно, пропорциональна их давлению над жидко-
стью. Это объяснение, однако, в случае тетрила опровергается прямыми
опытами Р. Ю, Сусловой, установившей, что откачка газообразных про-
дуктов разложения или замена их на азот практически не влияет па ско-
рость медленного термического распада тетрила при 140° С.
Возможны иные объяснения влияния отношения т'с- на возникнове-
ние вспышки.
Так, при больших значениях m/v разрыв ампулы может быть вызван
ле вспышкой ВВ, а просто возникновением в результате медленного рас-
пада давления, достаточного для разрыва ампулы. Понятно без пояснений,
что в этом случае чем больше -n/iy тем быстрее будет достигнуто давле-
ние. необходимое для разрыва ампулы.
Если опыт ставить при таких значениях m/r, прп которых максималь-
ное давление изотермического распада недостаточно для разрыва ампулы,
то повышенные значения m/v и соответственно давления газов при рас-
паде могут все же благоприятствовать возникновению взрыва вследствие
того, что меньше будет объем пузырьков газов, образующихся при разло-
жении, размешивающих жидкость и увеличивающих, таким образом, теп-
лоотдачу2. Далее, чем больше значение m/v, тем меньше объем свобод-
ного пространства над жидкостью и тем меньшая часть вещества может
превратиться в пары; соответственно большая часть вещества останется
в жидком виде, ц условия для возникновения теплового взрыва будут
более благоприятны. Наконец, возможно, что в замкнутой ампуле вспыш-
ка может возникнуть в парах ВВ по тепловому механизму, т. е. если
давление (плотность) паров превышает критическое; максимальная же
плотность паров зависит от отношения m/v.
Выяснить, какое из этих возможных влиянии давления играет основ-
ную роль, можно было бы, изучив воспламенение паров ВВ в отсутствие
жидкости, а также возникновение вспышки в условиях, исключающих
образование значительных концентраций паров ВВ.
Скорость распада твердого тетрила в 50—100 раз меньше, чем скорость
распада жидкого, рассчитанная для той же температуры, но быстро рас-
тет по мере хода распада. Рост скорости количественно объясняется про-
грессивным переходом твердого тетрила в ходе разложения в жидкое со-
стояние, вследствие понижения температуры его плавления конденсиро-
ванными продуктами распада. Это явление имеет, по-видимому, общий
характер и наблюдалось для всех ВВ, изучавшихся в этом отношении',
скорость разложения в твердом состоянии значительно меньше, чем в рас-
плаве или в растворе, и сильно растет во времени по указанной причине.
1 Опытами IO. П. Захарова, проводившимися при по стоя пн ой навеске (0.5 г) и
при гп/а от 0,15 до 0,44 г/смг (температура 180QC). показано, что задержка вспышки
в этих условиях практически не зависит от гп/1.. Возможно поэтому, что в данном
случае определяющим фактором является величина яавески ВВ, а не величина объе-
ма ампулы,
г Предварительные опыты ire обнаружили существенного влияния этого фактора
на возникновение вспышки.
Добавки азотной и уксусной кислот не ускоряют разложения, вероят-
но, вследствие летучести этих кислот; серная кислота вызывает быстрое
выделение газов.
Температурный коэффициент скорости велик —около 4 иа 10° С при
температурах 120—150° С. Это значение коэффициента приводит к боль-
шой величине энергии активации и предзкепоиенциалыгого множителя
в выражении константы скорости реакции распада.
По расчетам Рогинского Е = 00 ккал/моль, а В = 1027’5. Правда, по
данным позднейшей работы [64], проведенной при более высоких темпера-
турах (211—26(FC). k = 1015’4 ехр (—38 ООО/ЯТ1), т. е. значение В лишь
немного превышает обычную для мономолекулярпых реакций его величи-
ну. Этот вопрос требует дальнейшего уточнения.
Рис. 59. Влияние степени заполнения сосуда иа разло-
жение тетрила при 150° и различных отношениях
m/u (г/сл3 10“’):
j— 353,5; 2 — 49,4; Л— 555; 4 — 8,0; 5 — 0 (с откачкой);
— глубина превращения
Мержанов с сотр. [69], изучали разложение тетрила в значительно
большем (в 44 раза) интервале значений m/v. Опыты проводили в сосуде
из нержавеющей стали, конечное давление при наибольшем значения т / v
достигало 6000 ми. Изменение павески (в 7 раз при 150° С) не повлияло
иа ход разложения.
Полученные кривые степень разложения — время (рис. 59) показы-
вают, что степень заполнения относительно слабо влияет на скорость.
При увеличении значения m/г от 8-Ю"4 до 353,5 Ю*4 время полураспада
уменьшилось незначительно. Разница в скорости газообразования была
еще слабее, поскольку при бблыних давлениях образуется меньше газов.
Помимо определения кривых давление — время, было определено из-
менение состава газообразных продуктов в ходе некоторых опытов нрн
150° С. Оно показало, что вначале в газах содержатся значительные коли-
чества NOs и Ns1, причем в дальнейшем ходе разложения количество
1\О2 проходит через максимум и уменьшается значительно быстрее, чем
количество NO. При меньших значениях т/и максимум для обоих газов
меньше и наступает позже.
Изменение скоростей во времени при удалении газообразных продук-
тов реакции описывается выражением для автокатализа первого порядка.
Попытка учесть аналогичным образом влияние газообразных продуктов,
допустив что они катализируют распад и что это действие пропорцио-
нально нх давлению над жидкостью, т. е. концентрации в ней, успеха
не имела. Это неудивительно: наличие в продуктах разложения таких
г Возможно, что и при распаде нитраминов первичным процессом является, как
и при распаде питро&фиров, отщепление NCB.
70
реакционноспособных газов, как окислы азота, и изменение их концентра-
ции н ходе процесса ясно указывают, что он включает последовательные
реакции с участием газообразных продуктов, идущие с образованием га-
зов. Уменьшение скорости ирп откачке газов и постепенное ее нараста-
ние при продолжении опыта одновременно с накоплением КОг в продук-
тах распада подтверждают эти соображения. Нет оснований поэтому рас-
сматривать влияние газообразных продуктов распада как каталитическое1
действие* конечных продуктов подобно тому, как Это делал для конденси-
рованных продуктов распада - пикрцповоц кислоты — Хцгансльвуд. До-
бавим, что соотношение между газообразными соединениями азота Н
углерода, полученные в данной работе, существенно отличаются от того,
Которое следовало ожидать при превращении трнпитрофепилметилпитра-
мина в трпннтрофенол: число грамматомов азота в 3 раза больше, чем
углерода.
Г е к с о г е п и о к т о г е п
Разложение расплава гексогена, циклотриметил епт рииитрамина
NO2
СН2—
OaN—N СН2 ,
\lli-
^NOi
изучал Робертсон [70] при 213—299° С в навесках, достаточно малых, что-
бы обеспечить прохождение разложения без вспышки. Во всем этом ин-
тервале температур разложение протекает по закону реакции первого
порядка 1 с периодом полураспада 410 сек. при 213 и 0,25 сек- при 299° С.
Скорость реакции нс изменяется в присутствии воздуха, азота или водо-
рода при различных давлениях вплоть до атмосферного или от контакта
вещества с поверхностью слюды или меди.
Зависимость константы скорости от температуры выражается аррениу-
совским соотношением при Е = 47,5 ккал/молъ и 1g В = 18,5.
Опыты с разбавленными (2 н 5%) растворами гексогена в дицикло гек-
силфталате п тротиле выше 250° С показывают мопомолекуляряое тече-
ние реакции. Ниже 250° С наблюдается самоускореппе реакции, судя ко
линейному начальному участку кривой. Это самоускорение выражено тем
сильнее, чем ниже температура, и при 200° С лилейная зависимость соблю-
дается до достижения давления, составляющего половину от конечного,
после чего выполняется уравнение реакции первого поря;ща. На наличие
автокатализа указывают также результаты опыта, в котором был приме-
нен растворитель от предыдущего опыта; скорость при этом была значи-
тельно повышенной. Это свидетельствует, в частности, и о том, что ката-
лизатор является довольно стойким соединением по сравнению с самим
гексогеном.
Для 2%- и 5%-ных растворов гексогена копставты скорости рассчиты-
вались по начальному участку прямой с учетом конечного давления. Для
циклогексилфталатных растворов Е — 41,0 ккал/молъ t 1g В — 15,46. Ско-
рость распада в тротиловых растворах немного меньше.
’ Этот вывод расходится с результатами позднейшей работы Максимова [71], ко-
торый для растворов гексогена в ди нитробензоле паблюдал при 150—200° С заметное
увеличение (в 2—5 раз) скорости газообразования на начальном его участке; ирп
этом ускорение возрастало при повышении температуры опыта. Ускорение, однако,
зависит от отношения ™/,!, в чем, возможно, и следует искать причину расхождения;
кроме того, Робертсон в опытах с растворами исключал из рассмотрения начальный
участок, па котором происходил прогрев и испарение растворителя.
71
Сопоставление скоростей для расплава и растворов показывает, что
в последнем случае скорость распада меньше; разница очень малая при
200° С, увеличивается с температурой.
Разложение октогена, циклотетраметилентетраиитрамина,
OjN NOa
'n - GH2-N
i I
Gila C.H2
- CH,-J
O.N^ N'Oa,
идет медленнее, чем гексогена; оно изучалось в среде водорода или возду-
ха под давлением 5 см. При опытах выше 280° С вещество быстро плави-
лось и в жидком виде разлагалось мономолекулярно. Период полураспада
составлял 16 сек. при 271 и 0,45 сек. при 314° С. Для гексогена по расчету
он составляет при этой температуре 0,015 сек, т. е. в 30 раз меньше.
Анализ газообразных продуктов разложения гексогена (при начальном
давлении азота 6 см), а также октогена дал результаты, приведенные
в табл. 7.
Таблица 7
Газообразные продукты разложения гексогена и октогена (в молях на 1 моль ВВ)
при различных условиях опыта
Темпера- тура, °C Продукты разложения
NO N,0 N, н, СО СО!
Гек сосен
267 ] 0.75 | 0,76 I 1,03 I 0,06 I 0.29 I 0,44
225 | 0,54 1 0,98 | 1,16 [ 0,09 I 0,40 | 0,48
Гексоген в виде 5%-ного раствора е дицихлогексил^талате
225 | 0,15 | 1,13 | 0,89 | 0,05 ] 0,16 | 0,35
Гексоген е виде 5%-ного раствора в тротиле
264 I 1,02 I 0,47 I 1,33 I 0,30 I 0,32 I 0,85
220 I 0,70 | 0,63 | 1,65 | 0,20 | 0,57 | 0,83
Охтоген (после разложения в течение 2 мин)
280 | 0,95 I 1,51 | 1,16 [ — | 0,57 | 0,64
Кроме газов, указанных в таблице, образуются значительные количе-
ства воды и формальдегида, а также твердый остаток.
Из таблицы видно, что состав продуктов распада меняется с темпера-
турой; это говорит о сложном характере реакций. Состав газов заметно
изменяется также при переходе от расплава к растворам гексогена и за-
висит от примененного растворителя.
Максимов изучал манометрическим методом термический распад гек-
согена, очищенного перекристаллизацией из ацетона (т. пл. 202,8 —
203,0° С) в твердом виде (150—197° С) при m/v от 3,6-10"4 до
1870 10-4 г/см2. Скорость газообразования растет во времени, увеличи-
ваясь на максимуме в 5—20 раз по сравнению с начальным значением ’.
1 При разложении визуально обнаруживается частичное, а при повышенных тем-
пературах (197° С) и полное ожижение вещества, рано приводящее к спеканию на-
вески; вероятно, это оплавление является одной из причин наблюдающегося ускоре-
ния газообразования.
Скорость сильно зависит от отношения m/г, уменьшаясь в отличие от
многих других ВВ при его увеличении (рис. 60). Максимум скорости на-
ступает тем позже, чем больше значение m/г. Увеличение m/г увеличивает
отношение максимальной скорости к начальной.
Изменение размеров кристаллов в 10—20 раз практически не отрази-
лось па скорости распада; когда кристаллы были помещены под слой
инертной (силиконовой) жидкости, скорость несколько снизилась.
При значительных m/v (100-10-4 г/смI * 3 * * * * В) рост скорости но времени
может быть выражен соотношением W = &;еы, причем А-ч и к2 растут с
температурой. Таким образом, скорость газообразования растет пропор-
ционально количеству образовавшихся продуктов распада.
Рис. 60. Термическое разложение гексогс- Ряс, 61. Влияние небольших количести
. на при 190° и различной степени запол- тротила на разложение гексогена при
| нения сосуда веществом 150°С и т/и=10-10-4 г/смг
, Значения отношения m/v (г/см3-10-'): 1 — 3,6;
г —11; 3 — 101; 4 — 540; 5 — 1870
I
I На начальных участках приблизительно до 1% распада кривые v(t)
1 могут быть совмещены и это совмещение приводит к значениям Е —
= 51,0 ккал/моль и ]g В = 18,6.
Двойственное влияние на распад ^оказывает добавление небольших ко-
личеств тротила (рис. 61). Вначале газообразование больше, чем в отсут-
; ствие тротила, но замедляется во времени; позже наблюдается его ускоре-
ние. Начальное увеличение скорости может быть связано с ожижением
। части гексогена в результате его растворения в жидком тротиле. В пользу
этого объяснения говорит также то, что начальная скорость в присутствии
тротила быстрее растет с температурой.
В том же интервале температур изучалось разложение гексогена, раст-
воренного в динитробензоле. Разложение (рис. 62) идет быстрее, чем
твердою вещества (при 180°С в 16 раз), но с меньшим ускорением, кото-
рое возрастает при повышении температуры (рис. 63). Скорость на мак-
симуме превышает начальную в 2—5 раз. Это наблюдение противоречит
с5 приведенным выше данным Робертсона, который наблюдал уменьшение
ускорения при повышении температуры, правда, в области более высоких
температур (выше 250° С).
73
Гис. 63. Разложение гексогена, растворенного
в дпнитробспзолс, при 190° С
Числа у кривых—весовая концентрация гексогена
в процентах
Увеличение концентра-
ции гексогена с 4 до 15%
практически не влияет на
распад; на стадии падения
скорости опа изменяется в
соответствии с уравнением
реакции первого порядка.
В ходе распада окраска
раствора меняется от сла-
бо зеленой до темпо-внш-
певой.
Температурная зависи-
мость, определенная сонме-*
щепием кривых на участке
до выделения 30% газов,
дает значения энергии ак-
тивации 41,3 ккал/молъ и
Ig В — 15,3.
Константа скорости
прц 200° С составляет
1,5 10'4 сек-1, в 2 раза
меньше, чем получил Робертсон в дициклогексилфталатиом растворе. Ско-
рость распада растворенного гексогена слабее зависит от температуры, чем
твердого; поэтому при низких температурах различие между ними в отно-
шении скорости, распада увеличивается.
Разложение октогена (т. пл, 272,5° С) в твердом состоянии (176 —
230° С) при m/к, равном 5 10~4—500 10—4 г/см3 идет с ускорением во
времени большим, чем для гексогена. При 150° С начальная скорость рас-
пада октогена такая же, как и гексогена; при более высоких температу-
рах октоген разлагается медленнее. Как и для гексогена, скорость газооб-
разования при малых значениях m/v больше, чем при больших, по уже
начиная с m/v = 10- Ю4 г/см3, при дальнейшем увеличении m/v пе меня-
ется. Отношение скорости на максимуме к начальной при m/v = 5-
• 10~4 г/см? составляет 8; при увеличении m/v оно возрастает так, что, на-
чиная с некоторой степени разложения, скорость газообразования яри
больших значениях т/и становится больше, чем при малых.
Крупнозернистый октогеп разлагается несколько быстрое, чем мелкозер-
нистый, Начиная с 1 °/о разложения ц до 20%, изменение скорости в
ходе распада может быть выражено степенной зависимостью И7 = кхп,
где показатель степени п
пе зависит от температуры
it равен 1,3—1,4, а констан-
та /с растет с температурой.
Совмещение кривых v(t)
па участке до образования
10 см3 газов па 1 г (полное
разложение дает 600 см3/г)
для интервала температур
180—230°С приводит к
Е = 3(1,5 ккал/молъ и
1g В = 10,7-
Распад октогена изу-
чался также для его раст-
вора в дпнитробензоле
(171 —215° С, содержание
октогена от 0,5 до 2%);
скорость его в 8 раз боль-
Рпс. 63. Влияние температуры иа характер изме-
нения скорости во времени при распаде 4—5%-яого
раствора гексогена в динигробензо.те при различ-
ных температурах
ше, чем твердого октогена. Изменение концентрации почти не влияет
на скорость газообразования И ее изменение во времени. Вначале распад
идет подобно тому, как это наблюдается для гексогена, со слабым ускоре-
нием, возрастающим как ц степень распада на максимуме при повышении
температуры.
Кривые к(/) хорошо совмещаются до ?/з распада. Рассчитанные по
коэффициенту совмещения Е — 42,6 ккал!моль, Е = 14,0. Сопоставле-
ние результатов, полученных для растворов октогона л гексогена, пока-
зывает, что при 150 -200’ октоген разлагается медленнее, чем гексоген.
V. АЗОТИСТОВОДОРОДНАЯ КИСЛОТА (НА,)
Азотистоводородная кислота очень чувствительная и опасная взрывча-
тая жидкость, кипящая при 37° С, Поэтому распад ее был изучен только
в газообразном состоянии [72]. Опыты проводились в кварцевых и стек-
лянных сосудах в интервале температур 306—330° С при давлениях 30 —
200 мм. Распад HNj почти количественно ведет к образованию аммиака,
согласно уравнению
3HNs^=NHs+.4N2 (2.29)
Скорость реакции становится измеримой прп температурах выше 250° С.
Реакция протекает в основном па стенках сосуда. Течение ее подчиняется
мономолекулярпому закону; период полураспада при 330° в стеклянном
сосуде составляет около 12 мин. Незначительные прямее и сильно катали-
зируют распад и легко приводят к взрыву. Скорость реакции сильно зави-
сит от величины поверхности. Увеличение отношения величины поверх-
ности к объему в 8 раз увеличивает скорость реакции приблизительно
в 5 раз, В кварцевых сосудах реакция протекала приблизительно в 3 раза
медленнее, чем в стеклянных. Значения константы скорости сильно коле-
бались, что не позволило рассчитать энергию активации. Повышение тем-
пературы на 10° приводило в стеклянном сосуде к увеличению скорости
в 1,5—2 раза; добавление инертных газов (Не, Н2 и N) не оказывает влия-
ния. Необходимо отметить, что такое чувствительное ВВ, как азотистово-
дородная Кислота, в то же время обладает относительно очень высокой
термической стойкостью.
VI- ВЗРЫВЧАТЫЕ ВЕЩЕСТВА, РАЗЛАГАЮЩИЕСЯ
В ТВЕРДОМ СОСТОЯНИИ
В предыдущих разделах мы рассмотрели законы распада тех ВВ, кото-
рые (за исключением нитроклетчатки) при температурах опыта могли
находиться в жидком состоянии. Для многих В В (преимущественно солей
тяжелых металлов) температура плавления лежит выше температуры
вспышки. Для других температура плавления достаточно высока, и они
разлагаются со значительными скоростями ниже згой температуры в
твердом состоянии.
Как известно, автокаталитический распад твердых веществ моя;ет быть
сведен к двум случаям распада; топохимическому ц цепному. Топохимиче-
ский распад в свою очередь протекает по-разному в отношении зависи-
мости скорости реакции и количества прореагировавшего вещества От
времени при малом и постоянном числе зародышей, с одпой стороны, и
при большом их числе,— с другой.
Примером топохимического распада (1-й слушай) может служить рас-
пад азида кальция (CaNe).
75
При манометрическом исследовании распада этого вещества [8] при
температурах 100—140° С были получены S-образные кривые, показываю-
щие изменение давления азота, образующегося при распаде, во времени
(рис. 64), Процесс разложения можно разбить на 3 этапа:
1) |Индукционлый период с очень малой и практически постоянной ско-
ростью (АВ) ;
2) резкое увеличение скорости распада (ВС);
3) падение скорости (CD),
Индукционный период при разложении азида кальция, по-видимому,
обусловлен в значительной мере действием примесей, уничтожающих
зародыши реакции и препятствующих, таким образом, ее развитию. Исчер-
пание этих примесей делает возможным рост зародышей, скорость реакции
начинает быстро расти.
р, мы
Т, мин.
Рис. 64. Термический распад
азида кальция в вакууме
.1 — под слоем парафина;
2 — без парафина
Iff?
Л 5 IQ I. 1- »_____L. —I
0гё КО £4 U 23 2,0
Рис. 65. Зависимость скорости
распада ааида кальция от
количества разложившегося
вещества
В пользу такого объяснения говорит, с одной стороны, большое непо-
стоянство продолжительности индукционного периода и, с другой,— опы-
ты по разложению в атмосфере водяных паров или кислорода. В присутст-
вии обоих газов скорость реакции оказывается почти равной нулю, откачи-
вание газов вызывает резкий рост скорости в отличие от медленного раз-
вития процесса, наблюдающегося в начале разложения свежего образчика.
В периоде ускорения скорость реакции довольно хорошо следует топо-
химическому закону для случая, когда скорость образования зародышей
равна нулю: dx/dt — кх'/г. Последнее условие достигалось путем предвари-
тельного нагрева азида при более высокой температуре, пока не прибли-
жался конец индукционного периода. Затем температура понижалась, и
разложение прозодилось при этой пониженной температуре, при которой
новых зародышей практически не образовывалось, а шел лишь рост заро-
дышей !„ возникающих при предварительном нагреве. Особенно наглядно
можно установить выполнение указанного уравнения, строя график
1gИ’ —Igp, причем должна получиться прямая с тангенсом утла наклона,
равным 2/3. Опыт действительно дает прямую, причем tg а — 0,65 (рис. 65).
На последнем этапе падение скорости математически подчиняется мо-
номолекулярному закону подобно тому, как это наблюдалось для ряда дру-
гих твердых взрывчатых веществ.
Вычисление энергии активации показало, что в интервале 80 —100° С
она равна 20 ккал/молъ. Интересно, что при повышении температуры энер-
гия активации не остается постоянной, но растет, достигая при 140° С
34 000 кал/моль.
1 Образование зародышей черного цвета, хорошо видимых на фоне белых кристал-
лических агрегатов, можно наблюдать визуально даже при небольшом увеличен и и.
76
При исследовании разложения азида кальция было установлено, что
введение в пробирку с азидом парафина так, чтобы анид находился под
слоем парафина, приводит к резкому торможению распада (см. рис. 64,
кривая 1). Индукционный период длится в 3 раза дольше, переход к уско-
рению не так резок, и величина скорости при дальнейшем течении реак-
ции значительно меньше. Тормозящее действие парафин оказывает и в
том случае, когда его вводят во время уже идущей реакции: скорость
тотчас же становится в 40 раз меньше. Аналогичное влияние парафина
было позже установлено при разложении оксалата серебра [73] и при тер-
мическом распаде формиата никеля [74].
Механизм действия парафина не вполне ясен. Наиболее вероятной при-
чиной торможения реакции является дезактивация парафином какого-
то промежуточного продукта реакции, ускоряющего процесс. Это влияние,
почвидимому, имеет общий характер и может быть использовано как кри-
терий для установления топохимического характера реакции. В этой связи
уместно напомнить также о флегматизирующем действии парафина и по-
добных ему жидкостей в отношении чувствительности ВВ к удару. Воз-
можно, чго аналогия между действием парафина в обоих отношениях не
случайна.
Гарнер и Ривз [75] изучали влияние ультрафиолетового облучения па
последующее термическое разложение азнда кальция. Облучение сильно
сокращает индукционный период и увеличивает скорость распада. Сте-
пенной закон роста скорости сохраняется.
Другим примером топохимического распада является распад оксалата
серебра [76]. Ход разложения изображается кривыми, аналогичными кри-
вым для азида кальция. Количество прореагировавшего вещества возраста-
ет пропорционально tm, где т = 3,5'. Предварительная сушка, изменение
условий изготовления и измельчение отражаются на скорости распада.
Однако различия в скорости при этом меньше, чем те, которые иногда
наблюдаются между одинаково изготовленными и обработанными образ-
цами оксалата. Облучение оксалата серебра перед разложением светом с
длиной волны менее 520 ммк сильно увеличивает скорость последующей
термической реакции. Контакт с кислородом во время облучения заметно
снижает влияние облучения по сравнению с облучением в атмосфере азо-
та, углекислоты или в вакууме. Возрастание количества прореагировав-
шего вещества для малых периодов облучения приблизительно пропор-
ционально числу поглощенных квантов, для больших времен облучения
оно меньше.
По такому же типу распада, как распад азида кальция и оксалата
серебра, разлагаются многие другие изученные взрывчатые и невзрывча-
тыо вещества — гремучая ртуть [77], окись серебра [78], азид бария [75],
азид стронция [75]. Наряду с исследованием распада кристаллического по-
рошка эти вещества исследовали (чтобы избежать осложняющих влияний,
обусловленных неодинаковостью размеров и свойств отдельных кристал-
лов) в виде отдельных больших кристаллов. Такой кристалл весом 0,5—
5 лег, помещенный в платиновой коробочке, подвешенной на тонкой про-
волоке, опускали при помощи особого приспособления в сосуд, тщательно
эвакуированный при температуре опыта. Изменение давления, составляв-
шее несколько десятых миллиметра, измеряли манометром Мак-Леода или
Пирани.
Внешняя картина разложения для некоторых изученных ВВ следую-
щая. Для гремучей ртути [IlgfOXCis] при температуре 100 —115° С сначала
наблюдается очень быстрое незначительное выделение газов, которое Гар-
1 Следует добавить, что, согласно другим работам [73], разложение оксалата се-
ребра подчиняется на этапе ускорения цепному закону. Не исключено, что причиной
расхождения являс'1 ся различие в условиях приготовления соли-
77
Г.
Рис. 66. Термический
распад азпда свинца
(навески ОДООО а)
нер первоначально приписывал выделению газов, адсорбированных на вне-
шней и внутренних поверхностях кристалла и па поверхности плати-
новой коробочки. Исследования Апина [79] по разложению азида свинца
показали, однако, что в действительности начальный подъем давления
связан с прохождением первичной поверхностной реакции. Это ясно вид-
но из кривой, изображающей скорость как функцию времени (рис. 86);
после максимума скорость резко падает, и лишь спустя некоторое время
начинается новый подъем скорости, обусловленный реакцией, протекаю-
щей в случае распада азида во всей массе крис-
талла. Реальность обоих максимумов подтверж-
дается также измерениями саморазогрева при
реакция. Подобные же двойные максимумы па
кривой скорости наблюдались и при разложении
других- твердых взрывчатых и невзрывчатых ве-
ществ.
После начального ускорения реакции распа-
да гремучей ртути наступает индукционный
период, в течение которого скорость выделения
газов очень мала, причем давление возрастает
линейно сп временем. Одновременно с зтнм кри-
сталл становится коричневым, В конце индукционного периода кристалл
разламывается, причем большая часть осколков окрашена в желтый цвет.
Окраска эта в основном поверхностная.
По окончании индукционного периода реакция ускоряется. Это ускоре-
ние продолжается до тех пор, пока давление газа над кристаллом не дохо-
дит до 7s—2/з конечного его значения, после чего в течение короткого
периода времени скорость реакции остается постоянной. В конце периода
ускорения весь кристалл становится коричневым, хотя большая часть гре-
мучей ртути при этом еще не разложилась. На последнем этапе скорость
реакции пропорциональна количеству яеразложившегося вещества, т. е.
7 1 , а
выражается уравнением реакции первого порядка А = -у 1п —вы-
полняющимся до конца разложения. При раздавливании кристалла индук-
ционный период спльпо сокращается; при измельчении он практически
отсутствует, в остальном общий ход разложения остается пеизмепным.
Такой же характер распада и у а-азида свинца при условии примене-
ния мелких кристаллов. Ускорение наблюдается до тех пор, пока не рас-
падется около половины вещества.
Кристаллы азида бария (BaNe-I^O) [80] теряют кристаллизационную
воду даже ори комнаткой температуре, образуя белую нсевдоаморфную
массу. Дегидратированные кристаллы после индукционного периода раз-
лагаются с измеримой скоростью при температурах выше 95° С, причем
зародыши, число которых растет со временем, видимы под микроскопом,
возникают как иа внешних, так и на внутренних поверхностях кристалла,
имеют неправильную форму и сгруппированы на поверхности очагами.
Общий ход разложения качественно тот же, что и для гремучей ртути, но
давление на участке ускорения растет пропорционально шестой степени
времени.
Случай, когда число зародышей велико [81] и продукты реакции быстро
покрывают поверхность кристалла, по Гарперу, наблюдается при разложе-
нии рзцдад свинца 1 и серебра [82]. По-впдпмпму, ой имеет место также
1 Следует указать, что данные о типе распада азида свинца противоречивы. Пара-
ду с наблюдениями Гарпера [82], указывающими па быстрое наступление зтапа паде-
ния скоросги, имеются данные того а;е автора, показывающие палиппо аплчптслвного
участка роста < ко росит, если применять кристаллы меньших размеров н бо.тее низкие
температуры. ЛпнП [79] с несомненностью установил наличие длительных периодов
индукции и рост газообразовании по показательному закону при расца.че азида сини-
ца в широком интервале температур.
78
при разложении трицитротриазидобензола- на гексанитрозобензол и азот
[83] при низких температурах (100° и ниже), идущем, согласно уравнению
Ce(NO-,)3(N3)a=Cc(NO)e4-3N2. (2.30
Зависимость между количеством прореагировавшего вещества и време-
нем изображается кривой па рис. 67. Анализ этой кривой показал [84], что
разложение очень близко следует закону
]
а'о — (и — я)’1'» = -у kt.
Сопоставление значений констант скорости распада при разных темпе-
ратурах (20 —100° С) приводит к энергии активации распада твердого
тринитротриазидобензола, ранной 32 300 кал/молъ.
Турен [85] изучал по выделению азота разложение трииитромоноазидо-
бензола, тринитро диазидобензола и тринитротрпазпдобепзола в твердом
виде при 50° н в виде 1%-ных растворов в
в четыреххлористом углероде при 61 и 76,5° С.
По данным для растворов, в которых распад
идет без заметного ускорения, энергии акти-
вации близки и составляют соответственно
22 400, 25 300 и 23 800 кал/молъ. Интересно
отметить, что скорость распада больше всего
у трпнитромоноазидобензола и меньше всего
у трипитротриазидобензола. Эти скорости
различаются в 7 раз в растворе при 76,5° и в
5 раз в твердом состоянии при 50° С. Все три
вещества разлагаются в растворе значитель-
но быстрее, чем в твердом состоянии,
Распад трипитродиазидобензола при 50° С
так же идет, по опытам Иоффе [96], распад
Рис. 67. Термический распад
триня тротрпазидобс1ыо.1а
идет с ускорением; точно
твердого тринитротриазидо-
бензола нри повышенных температурах. Это ускорение следует объяснить
постепенным переходом твердого вещества в расплав вследствие снижения
температуры плавления продуктами распада. Добавим, что на скорость
распада влияет, по опытам Иоффе, также давление, под которым идет
распад; скорость распада тем больше, чем выше давление.
Характерным для топохимического процесса является наличие участ-
ка ускорения, на котором скорость возрастает со временем по степенному
закону, обусловленному ростом трехмерных зародышей реакции.
Для некоторых твердых веществ (взрывчатых и певзрывчатых) также
наблюдается значительный период ускорения, по скорость при нем растет
со временем но но степенному, и ио показательному закону 1
4г- =
at
В качестве примера можно привести разложение триннтрорезорцнпата
свинца [87] [СбН(КО2)3О2РЪ • Н2О] (рис. 68). При нагревании этого веще-
ства в вакууме до 120° С прежде всего теряется кристаллизационная вода,
причем дегидратация не вызывает изменения формы и прозрачности кри-
сталла. При температурах выше 200° С кристаллизационная вода теряется
очень быстро и кристаллы раскалываются на кусочки. За дегидратацией
следует термическое разложение, и если температура выше 225 ° С, то оно
заканчивается взрывом. Аналогичный характер кривых для участка уско-
1 Нужно указать, что не всегда можно однозначно сделать выбор между показа-
тельным и степенным законом. Так, Гарнер, первоначально относившей гремучую
ртуть и азпд бария к числу веществ, характеризующихся экспоненциальным ростом
скорости со временем, в настоящее время считает правильным выразить этот рост
степенном зависимостью.
7»
Рис. 68. Термический распад трвиитро-
резорцината свпнца 1 — р=/(т); 22 —
Igdp/dt = /(т)
рения наблюдался при разложении оксалатов некоторых тяжелых метал-
лов и перманганатов.
Термическое разложение пикратов и стифнатов аммония, калия и
свпнца в интервале температур 190—300° изучала Лю Бао-фен [88]. По
изменению давления газообразных продуктов распада устанавливался ход
разложения во времени при разных температурах, влияние на него давле-
ния газообразных продуктов путем
изменения степени заполнения сосу-
да и зависимость скорости от темпе-
ратуры.
Для всех солей абсолютная ско-
рость газообразования растет со вре-
менем на значительной доле распада.
Падение скорости, обусловленное
уменьшением концентрации (а в дан-
ном случае количества) реагентов на-
ступает относительно поздно.
Начальная скорость распада всех
изучавшихся солей меньше, чем со-
ответствующих кислот. Так как, од-
нако, скорость распада солей быстрее
растет с температурой и со временем,
то максимальные скорости в ряде
случаев оказываются выше у солей, чем у кислот.
Третьей особенностью является стадийность распада для многих со-
лей, отчетливо видимая даже на интегральных кривых. Основным типом
этой стадийности является сочетание двух этапов ускорения, подобного на-
блюдавшемуся при распаде стифната аммония (табл. 8), но с большими
вариациями рак в величине ускорения и в доле газообразования на каж-
дом из этих этапов (в резкости их перехода друг в друга), так и в отно-
сительном времени их протекания. Как правило, ускорение сильнее на
первом этапе, чем на втором. В некоторых случаях в начале разложения
наблюдается индукционный период, более (пикрат и двузамещенный
стифнат калия) или менее (однозамещенный стифнат калия) выра-
женный.
В табл. 8 приводится общее представление о характере кривых v — т
для изучавшихся солей, а также исходных кислот, и некоторые количест-
венные характеристики скоростей распада, выраженные через период до-
стижения давления, равного '/г от максимального.
Ввиду сложного характера кривых v — т описать их простым матема-
тическим выражением не удается; однако на более или менее значитель-
ных участках кривые могут быть представлены различными полуэмпири-
ческими соотношениями, в частности степеиной или показательной форму-
лой (табл. 9). Иногда, чтобы получить прямую в этих координатах, при-
ходится вычитать начальные значения скорости или объема газообразных
продуктов.
Как правило, ускорение газообразования относительно больше при
низких температурах Так, для пикрата аммония при 230° С максималь-
ная скорость превышает начальную в 18 раз, при 270° С — только в
5.5 раза. Во всем температурном интервале, ускорение значительно боль-
ше, чем для соответствующих кислот. Различие в величине ускорения
в ряде случаев приводит к тому, что кривые газообразование — время для
солей и кислот пересекаются.
Зависимость скорости распада от температуры определялась различны-
ми приемами: по начальной скорости, путем совмещения кривых v — т
1 Исключение составляет двузамещенный стифнат калия, для которого ускорение
возрастает при увеличении температуры опыта.
80
Таблица 8
Основные характеристики термического распада пикриновой и стифниновой
кислот и их солей
Вещество Интервал темпера- тур (в °C) и агрегат- ное сос- тояние Характер распада Время полу- распада при 230° С, мнн. *1 4’
Пикриновая кис- лота Стпфниповая кис- лота Пикрат аммония Стифпат аммония Пикрат калия Однозамещенный стифнат калия 183—270 (жидкая) 180—250 (жидкая) 200—270 (твер- дый) 190—230 (твер- дый) 250—300 (твер- дый) 170—200 (твер- дый) V,« 30 го >0 0 50 0 «00 200 Ш 200 300 200 >00 300 200 too сиЗ/г tS3e | 1 f 1 500 ' i000 5000т, пик Ю0* 'г 1 / 1 > , too ' 300 9'00 /250 " /230° SOO /000 50 ;0B i I cz! , . too toss г ooo 470 45 720 65 8600 262 *** (вспышка при 200°) 1 1,53 18,3 1 1,44 5,83
• к, — отношение периодов полураспада пикратов и пикриновой кислоты.
• * X, — отношение периодов полураспада етифнатов и стифниновой (кислоты.
* ** Определено расчетом.
6 К. К. Андреев ' 81
Таблица 8 (окончание)
Вещество Интервал темпера- тур (в °C) и агрегат- ное сос- тояние Характер распада время полу- распада при 2W" С, мни. к2
Д в у а вмещенный
стифнат калия
Пикрат свинцч
Стифнат свинца
200—240
(твер-
дый)
230-260
(твер-
дый)
200—230
(твер- :
дый)
210
(вспышка
при 250°)
920
(вспышка
при 200°)
143
(взрыв при
240°)
1,96
4,6?
Таблица 9
Энергия активации (Е) термического распада пикриновой и стифниновой
кислот и их солей
Вещество 1 интервал температур l °C Е, ккал/моль Метод расчета Е
Пикриновая кислота 183—270 38.6 По средним значениям скорости при 5—50% распада
Пикрат кадия 250—300 41,2 Совмещением кривых v = /(т)
Пикрат аммония 200—270 43,0 40,5 По средней скорости при распаде от 3—И до 45% (первое значение) и по тангенсам угла наклона пря- мых 1g (v — рй) -= А + fcr
Пикрат свпнца 230—260 60,2 58,7 По тангенсам угла наклона прямых 1g И7 = А -)- к т (первое значение) и по средней скорости прп 10— 50% распада
Стифнинопая кислота 180—250 34,6 По аначенпям средних скоростей при 22—45% распада
Стифнат калия одноза- мещенный 170—200 47,6 По значениям скорости при 5—35% распада
Стифнат калия двуза- мещепный 200—240 51,7 52,9 По тангенсам угла наклона прямых 1g v = А — kt прц 5—27 % распада (первое значение) и по средней скорости па этапе ускорения
Стифнат аммония 190—230 45,5 45,5 По средней скорости при распаде от 2—9 до 45% (первое значение) и по углу наклона прямых 1g W = А 4- —|- к т
Стифнат свинца 200-230 34,8 36,4 По значениям скорости при -—30% распада (первое значении) и по углу наклони прямых 1gи =А kt
82
(особенно хорошо совмещаются кривые для пикрата калия), сопоставле-
нием средних значений скорости по константам уравнений, определяющих
зависимость скорости от времени, и т. п, (см. табл. 9). Скорость второго
этапа распада слабее зависит от температуры, чем первого. Как правило,
скорость распада солей сильнее зависит от температуры, чем кислот (ис-
ключение составляет стифнат свинца, для которого энергия активации
практически совпадает с энергией активации кислоты). Особенно велики
энергии активации двузамещенного стифната калия и пикрата свинца
(~ 60 ккал/моль),
Так как по абсолютным значениям в изучавшемся интервале темпера-
тур скорости отличаются не сильно, то соответственно предэкспоненциаль-
ный множитель велик для веществ с сильной температурной зависимо-
стью скорости и мал, приближаясь к нормальному, для веществ, скорость
распада которых слабее изменяется с температурой.
Степень заполнения сосуда веществом (т/и), определяющая давление
газообразных продуктов распада, оказывает некоторое, но, как правило,
не сильное влияние на ход разложения; скорость и рост ее во времени
возрастают1; при увеличении значения т/а повышается также и скорость
газообразования на втором этапе.
Сабо и Сава [89] изучали термический распад пикрата натрия в эва-
куированном сосуде, определяя задержку вспышки т (или время дости-
жения максимума скорости) в интервале температур 300 —310° С, которая
лежит в пределах 1 — 13 мин. и была достаточно хорошо воспроизводимой.
(Навеска и ее размер и расположение не указаны в работе). Но зависи-
мости т (Т) была рассчитана энергия активации (Е-т = 73 ккал/моль) и
предэкслоненциальный множитель (1^5 — 5). Кривые W(t) показывают
максимум скорости в течение первой 0,5 мин,, который не устраняется
сушкой или нагреванием до 300° при атмосферном давлении, но падает по-
сле обработки в вакууме при 270° С. Давление растет во времени по пока-
зательному закону W — Aevt, причем по температурной зависимости
ср энергия активации составляет 65 ккал/моль.
Для тонко измельченного пикрата индукционный период на 4—5 мин.
короче, Et значительно больше (93 ккал/моль) г — 68 ккал/моль,
Пикрат, полученный кристаллизацией при избытке карбопата калия,
разлагается и взрывается гораздо быстрее, чем в отсутствие примеси; кро-
ме того, сильнее становится зависимость т от температуры, частичное
Предварительное разложение при немного более низкой температуре за-
трудняет возникновение вспышки при последующем повышении темпера-
туры; избыток пикрата калия при кристаллизации такого влияния не ока-
зывает.
Изучалось влияние различных газов на индукционный период при
310° С,
Под влиянием давления кислорода (до 300 .<ч) индукционный период
сокращается, при ро? = 600 мм он возрастает до прежнего значения; при
добавлении С Оз индукционный период, напротив, возрастает при
300 .им и возвращается к прежнему значению при J600 мм, азот увеличи-
вает t во всем изученном интервале давлений (до 600 .и.и), водород еще
сильнее задерживает и затрудняет вспышку. Откачка газообразных про-
дуктов распада не изменяет индукционного периода.
Старение пинрата (до 1,5 лет) при обычной температуре не влияет ню
кинетику его распада; при повышенных (120, 150, 170 и 210° С) тем-
пературах 15-часовой нагрев сокращает индукционный период и умень-
шает энергию активации; чем выше температура старения, тем более1
стоек его результат; при 120° последствия старения исчезают уже че-
рез 1 день.
1 При распаде пикрата в стифната аммония увеличение значения m/v уменьшает
начальную скормят,.
83.
6*
Анализ результатов исследования распада твердых ВВ, ускоряющегося
по показательному закону, был проведен Семеновым [90]; он показал хо-
рошее согласие полученных результатов с цепной схемой развития реак-
ции. Эта схема позволяет количественно объяснить наличие и длитель-
ность индукционного периода, образование видимых зародышей в одних-
ВВ и отсутствие их в других, влияние измельчения, зависимость задерж-
ки взрыва от температуры и т. д.
Следует остановиться здесь лишь на индукционном периоде, посколь-
ку механизм его в этом случае, по-видимому, отличен от механизма, при-
нятого нами для топохимического распада азида кальция и автокаталити-
ческого распада тротила [6]. В последних двух случаях индукционный
период обусловливается присутствием примесей, тормозящих автокатали-
тическую реакцию. При цепном же распаде Семенов считает индукцион-
ный период кажущимся, получающимся в результате медленного разви-
тия цепи. При этом вначале, когда давление очень мал» и не может быть
измерено применяющимся прибором, создается впечатление отсутствия
реакции. На самом деле реакция идет, ускоряясь по одному и тому же за-
кону, с самого начала и до «юнца периода ускорения, только мы начина-
ем замечать ее лишь с того времени, когда давление образовавшихся га-
зов достигает некоторого значения. Подтверждением этой точки зрения
служит тот факт, что произведение величины индукционного периода на
Ф всегда сохраняет постоянное значение, как этого требует цепная тео-
рия.
VII. ПЕРХЛОРАТ АММОНИЯ
Мы ие останавливаемся здесь на ранцих исследованиях по термическо-
му распаду перхлората аммония, к тому же освещенных в обзорных рабо-
тах [82, 91, 92], и ограничиваемся рассмотрением ряда новейших работ,
опубликованных преимущественно зарубежными исследователями.
Термический распад перхлората аммония, изучавшийся Биркдмшоу л
Ньюмэком [93, 94], протекает Очень своеобразно, Следовало ожидать, что
он будет идти по схеме; твердое вещество -> газ. В действительности же
в большинстве изученных условий распад шел по схеме: твердое вещест-
во А—>твердое вещество В + газ. В высоком вакууме или в токе инерт-
ного газа при температурах ниже 290° С распад останавливался после того,
как потеря в весе 'достигала 30%. Остаток после распада, который охва-
тывает весь кристалл, изоморфен с исходной солью, но порист 1 ц представ-
ляет собой полностью перхлорат аммония.
Состав продуктов распада ниже 300° С может быть выражен уравне-
нием
4NH,C10j 2CIH-302+8H,0+2(VL.0. (2,31)
Выше 350° С составу продуктов больше соответствует следующее урав-
нение, отличающееся от предыдущего образованием окиси азота взамен
закиси:
4NH4CL01^ 2Ch+2NO2+81[2O+2NO. (2.32)
Кроме того, образуются хлорная и соляная кислоты и хлористый нитро-
зил,
Характер распространения реакции по кристаллу перхлората, наблю-
давшийся под микроскопом лад отдельными маленькими кристалликами
на разных стадиях распада, отчетливо указывает на его топохимическое
> Удельная поверхность остатка [95] была равна 1,5 л2/г, что соответствует разме-
ру составляющих его частиц 3 лк,
84
течение — возникают дискретные непрозрачные зародыши, которые рас-
тут, сливаются и образуют оболочку продукта, окружающую неразложив-
шееся вещество.
В большей части кинетических опытов распад вели в эвакуированном
сосуде, и скорость газообразования при 250—300° С определяли по росту
давления постоянных газов (Мак-Леод) за время непродолжительного вы-
ключения насосов; остальные газы конденсировались в жидком кислороде;
при 400°С и выше применялось небольшое давление азота (1—4 см), что-
бы подавить сублимацию, причем о ходе распада судили по росту давления
постоянных газов V
Кривые p(t) имели S-образцый характер; наблюдался такя;е неболь-
шой индукционный период, длительность которого зависела от темпера-
туры и истории образчика. Участок падения скорости был всегда больше,
чем участок ускорения; время достижения максимума скорости умень-
шалось с повышением температуры; при 380—450° С. кривые почти цели-
ком соответствовали падению скорости. Дейтерированный перхлорат раз-
лагался несколько быстрее, чем обычный.
Предположение о химическом отравлении реагирующей поверхности
в результате адсорбции газообразных продуктов распада как причине оста-
новки превращения не подтвердилась. Возможно, что необходима некото-
рая определенная конфигурация решетки, чтобы реакция разложения
могла конкурировать с сублимацией.
Остаток яе-разложившегося перхлората полностью разлагался при на-
гревании выше 400° С, полностью разлагался при этой температуре и ис-
ходный перхлорат. Это говорит о том, что механизмы разложения различ-
ны в обоих температурных интервалах.
Гальви и Джэкобс [95], считая, что размеры частицг составляющих оста-
ток, такие же, как и у элементов мозаичной структуры кристалла, пола-
гают, что 30%-ный низкотемпературный распад представляет собой рас-
пад напряженного вещества, связывающего элементы мозаики; высокотем-
пературный распад охватывает и сами эти элементы.
Пары воды и других растворителей оказывали «омолаживающее» дей-
ствие на остаток от разложения, и при последующем нагреве распад прю-
должался. Степень «омоложения» оказалась пропорциональной раствори-
мости перхлората в растворителе и времени экспозиции.
При 240° С происходит переход перхлората аммония из орторомбиче-
ской в кубическую форму, это оказывает заметное влияние на величину
максимума скорости. Из рис. 69 видно, что максимум растет до 238°, за-
тем падает до 250° С и выше этой температуры вновь растет. Индукцион-
ный период монотонно убывает с температурой,
Опыты проводились также под давлением азота (3 ем) при 260—300° С
(при помощи кварцевых весов); они дали результаты, аналогичные полу-
ченным в эвакуируемом сосуде. Повышение давления за счет образовав-
шихся неконденсирующихся газов составляло около 2 мм, Дальнейшее по-
вышение давления инертного газа изменяет характер кривой (потеря
веса — время); влияние это наиболее сильно проявляется в увеличении
скорости превращения на той стадия, где опа в отсутствие инертного газа
становится близкой к нулю. Почти полного прекращения распада после
превращения 30% при повышенных давлениях инертного газа не наблю-
дается. '
Изучалось влияние перерыва 1 2 разложения (10 мин.). Ниже 240°С он
не оказывал влияния на скорость последующего распада. Выше 240° С
1 Состав постоянных газов был практически одинаков в широком интервале тем-
ператур (240—330° С) и по ходу отдельного опыта, что позволяло использовать их ко-
личество как меру течения распада.
2 Перхлорат аммония во время перерыва выдерживали в вакууме.
85
220 200 2S0(-
Температура, ’С “
Рис. 69. Влияние температуры
на величину максимальной
елорости при разложении пер-
хлората аммония
скорость всегда стало вилась больше, чем до перерыва, предположительно
вследствие растрескивания кристаллов при модификационном переходе,
происходившем при охлаждении. Па участке ускорения скорость после пе-
рерыва возрастала в 2—3 раза, но затем быстро падала. При температурах
недалеких or переходной — до 255ПС — кривая скорости после перерыва
имела два максимума и минимум. Это обь-
ясняется близостью к температуре перехо-
да и тем, что проба пе сразу разогревалась
до нужной температуры после возобновле-
ния опыта; растрескивание при охлажде-
нии обусловливало первый максимум;
новый переход в кубическую форму вновь
приводил к образованию свежей поверх-
ности, что и давало второй максимум. Сте-
пень распада и в Этих опытах не превосхо-
дила 28—30%.
Изучалось также влияние на распад не-
которых примесей, в частности нитрата
аммония, который мог быть промежуточ-
ным продуктом. Присутствие нитрата не-
сколько сокращало индукционный период
(с 30 до 15 мин. при 230° С). Хлорная кислота, которая, так же как и
Н-ионы, обнаруживается среди продуктов распада, тоже приводит к сок-
ращению индукционного периода, хотя максимальная скорость была не
столь высока, как в присутствии нитрата аммония.
Влияние аммиака изучалось по следующим соображениям. Если бы
первой стадией был распад перхлората на хлорную кислоту и аммиак, то
последний должен бы был увеличивать индукционный период; это и на-
блюдалось при опытах. Индукционный период перхлората аммония, пере-
кристаллизованного из водного раствора аммиака, увеличился до 50 мин.,
гораздо меньше была и скорость последующего распада, хотя обычный его
предел — 28% — не был превышен. Замедляюще влияют такясе пары ам-
миака. При нагревании пробы в течение 2 час. при давлении NH3 3,5 см
(230° С) степень распада уменьшилась с ~30% до 3%.
Известно, что хлористый аммоний сильно ускоряет термический рас-
пад нитрата аммония. Добавление его к перхлорату оказало, однако, об-
ратное действие; индукционный период при 230s С увеличился на 10 мин.
Кривые p(t) анализировались ца основе уравнения Проута — Томпкин-
са для участка ускорения
+ (2.34)
^КОВ С
с различными значениями константы для периодов ускорения и падения
скорости; период ускорения (до а < 0,25) может быть описан также сте-
пенным выражением р = Kt” при п ~ 6.
Для участка падения скорости использовалось уравнение реакции пер-
вого порядка
— Ig(Piw — р) =/rat(2.35)
Для роста скорости цо формуле Проута — Томпкинса в аррепиусов-
ских координатах получаются как для орторомбической, так и для куби-
ческой формы прямые линии. Линии для участка падения скорости пока-
зывают отчетливый перелом при температуре перехода, по не являются
прямыми. Расчет энергии активации был произведен также по уравнению
хе-Е;нт ~ С0П5ц где т время разложения определенной части вещества
(0.1 п л и 0,5) и дал результаты, близкие к тем, которые были получены по
степенному выражению.
86
Полученные данные для энергии актинации приведены в табл. 11,
Ввиду сложности процесса распада физический смысл этой величины в
данном случае не вполне ясен,
Таблица И
Величины энергии активации
Применяемые формулы Энергия активации, ккал/люль <240’0 | > 240’0
Участок ускорения на кривой р (() Е т=т0„ру т = т01!ру 28,4 18,4
28,8 17,1
/ р \ 24,9 23,0
Участок падения скорости на кривой 1g (Р/ ~~Р) = fat -1-0(1 -го порядка) 26,8 20,5
/ р \ 27,7 17,5
1 ff 1 1 — гСо Е —*— О в \Pf-p! ’
Средние величины 27,8 18,9
Влияние размеров частиц на скорость распада изучали при 230° [100].
Соль измельчали и рассеивали на фракции, по кривым скорости и раз-
личным ее константам определяли влияние размеров частиц. Максимум
скорости быстро растет при уменьшении размера частиц до определенной
величины, а затем падает. Воспроизводимость результатов при резко
фракционированном составе была меньше; кроме того, влияло распределе-
ние навески в сосуде; если навеска была распределена па значительной дли-
не трубки, то скорость была меньше, чем в том случае, когда навеска была
сосредоточена в конце трубки, При равных размерах частиц перхлорат,
полученный измельчением крупных кристаллов, разлагается быстрее, чем,
если мелкие частицы получены быстрой кристаллизацией. При получении
частиц различных размеров рассеиванием кристаллического перхлората
без его предварительного измельчения скорости распада различных фрак-
ций почти не различаются, предположительно потому, что крупные ча-
стицы представляют собой агломераты мелких кристаллов.
Гальии и Джэкобс [101] изучали распад перхлората аммония в виде
целых кристаллов, измельченного порошка и прессованных (775 ат) таб-
леток.
Методика этих опытов заключалась в нагревании образчика в вакууме
с измерением объема образующихся газов двумя манометрами Мак-Лсода
для разных интервалов давления. В опытах с таблетками выше 240° С при-
менялось измерение общего давления всех газов при помощи манометра из
стеклянной спирали.
Изучалось, в частности, влияние различной обработки на поведение
остатка после разложения при последующем его использовании для опы-
та, Встряхивание для разрушения больших комков, выдержка на воздухе
в течение 20 мин., двухдневная выдержка в вакууме не дали существенно-
го омоложения; напротив, после измельчения с последующей семидневной
выдержкой перхлорат омолаживался практически полностью.
87
Сравнение Проута — Томпкинса, которым пользовались Биркамшоу и
сотрудники, обычно применяется для описания квазицепного процесса
ускорения; в данном же случае наблюдается образование и рост трехмер-
ных зародышей, Кроме того, это уравнение не очень хорошо .изображает
течение распада 1 * особенно для кристаллов и порошка, Поэтому Гальви и
Джзкобс применили уравнение Аврами — Ерофеева, представив его в виде
Jn(1 — а) ~ I4 для начальной и — 1п(1 — а) ~ i3 для конечной стадии рас-
пада. Эти уравнения хорошо описывают распад на большом его протяже-
нии. Так, для целых кристаллов н = 4 в интервале а 0,02 4- 0,20 и п — 3
для а 0,20 -г 0,90; соответствующие значения Е составляют 20,6 и
16,9 ккал/моль- для кубической формы п = 2 и Е = 25,3 ккал/моль; для
порошка, как и следовало ожидать, п — 4 в большем интервале а — до 0,70,
Е = 24,6 (орторомбическая форма) и 24,8 ккал/моль (кубическая
форма); значения Е для таблеток практически тождественны для
обеих форм, но значительно больше (30,1 и 29,9 ккал/моль), причем п = 3
(для орторомбической формы) в интервале а от 0,05 до 0,75,
Для малых а указанная зависимость может быть приближенно выра-
жена как степенная, но при показателе степени п = 4, который и получи-
ли Гальви и Джзкобс в отличие от Биркамшоу, в опытах которого он был
равен 6.
Из полученных интегральных энергий активации могут быть рассчита-
ны Е} для возникновения зародышей и Ег для их роста. Для кристаллов
Е( = 31,7 и Ег = 16,9 ккал/моль, для порошка и таблетки Е соответствен-
но равны 22 и 30,1 ккал/моль. Такие большие различия авторы приписы-
вают различиям в физической структуре связующего элементы мозаики ве-
щества.
Выше 240° Сп т. е. в кубической форме, перхлорат аммония разлагает-
ся одинаково в кристаллах, порошке и таблетках (п = 2 и значения Е со-
ставляют соответственно 25,3; 24,6 и 29,9 ккал/моль). Это связано с тем,
что модификационный переход снимает те различия, которые определя-
лись подготовкой образчика,
В одной из работ (102] распад перхлората аммония проводили в замк-
нутом сосуде, по без вымораживания конденсирующихся продуктов с
измерением давления стеклянным манометром типа Бурдона. О ходе рас-
пада судики но кривым p(t), пересчитывая давление на объем при нор-
мальных условиях. В интервале 160—280° С общий ход кривых S-образ-
ный (рис. 70, 71) и включает индукционный период 0А, период быстрого
роста скорости АВ, медленное ее падение ВС, участок постоянства нлп
очень медленного падения скорости CD и период обычного, т. е. относи-
тельно быстрого ее уменьшения DE.
Первые три периода охватывают небольшую долю распада и отчетли-
во наблюдаются лишь при относительно низких температурах (160—
200° С). Так, индукционный период, составляющий при 160° около
1000 мин., при 190° уменьшается до 100 мин.; период ускорения при 160°
заканчивается через 2600 мин., при а = 0,026, при 230° С — через 50 мин.
при степени распада, равной ~ 0,09,
Таким образом, чем ниже температура, тем короче (по степени рас-
пада) этап ускорения. Основную часть распада представляет особенно при
повышенных температурах период постоянства скорости, заканчивающий-
ся при а = 0,91—0,98. Изменение степени заполнения сосуда перхлора-
том (m/v) не оказывает существенного влияния на ход распада, так же как
1 То, что уравнение Проута — Томпкинса все же применимо в определенном ин-
тервале а в известных условиях распада, Гальви и Джзкобс объясняют возможностью
разветвлении распространяющейся по связующему веществу реакции в местах стыка
нескольких кристаллитов.
88
Рис. 70. Разложение перхлората аммония при m/v = 10 • I0-4 г/см3
(температура 190° С)
J и 2 — чистый продукт; 3 — под слоем парафина
присутствие инертного газа (400 мм Nj). Течение и скорость распада
слабо изменялись и при изменении (в 70 раз) размеров частиц пер-
хлората.
На участке ускорения скорость растет пропорционально давлению в
степени, увеличивающейся с повышением температуры и заключающейся
между 3,2 (при 180°) и 3,8 (при 210°). Падение скорости может быть опи-
сано выражением для сжимающейся сферы. Последующее постоянство ско-
рости является, по-видимому, итогом двух влияний — падения скорости за
счет уменьшения количества реагирующего вещества и ее увеличения
за счет «омолаживающего» действия конечных продуктов распада. Это
действие слабо проявляется на начальных этапах распада, когда мало
Рис. 71. Разложение перхлората аммония при т/у = 10‘10-’ г/см3
(температура 250° С)
J и 2 — чистый продукт; 3 и 4 — под слоем парафина
89
3
Рис. 72. Разложение перхлора-
та аммония в присутствии нор-
мального гексана щри 220° С и
т / г = 10’ К-1 а/сл3
1 — без гексана; г — давление гексана
40 мм рт. ст.; а — давление гексана
450 мм рт. ст.
Рис. 73. Разложение перхлората аммония (7),
его смеси с парафином (2) и с веществом В
(3 и 4) при 220Р С и m/v = 10 • 10—4 г/см?
содержание конденсирующихся компонентов 1 и мало количество перхло-
рата, требующею «омоложения». «Омоложение» может быть предотвра-
щено вымораживанием конденсирующихся продуктов распада подобно
тому, как это наблюдалось в опытах Биркамшоу, а также присутствием в
сосуде парафина или паров гексана в достаточной концентрации. Началь-
ные этапы распада в присутствии этих веществ ускоряются, но затем на-
ступает остановка процесса или, точнее, очень сильное его замедление —
скорость газообразования уменьшается в 20 раз (рис. 70 и 72). Перерывы
опыта, связанные с временным охлаждением реакционного сосуда, ведут в
результате «омоложения» к преходящему ускорению распада; в присутст-
вии парафина этого ускорения не наблюдается.
Другие более химически активные органические примеси ускоряют рас-
пад в первой его трели, но затем, подобно парафину, задерживают его. Не-
которые из них, однако, не оказывают такого влияния в температурной
области устойчивости орторомбической модификации, но сильно его прояв-
ляют при 240° С и выше. Тогда повышение температуры в области моди-
фикационного перехода сопровождается более резким уменьшением ско-
рости, чем его дает перхлорат без добавок (рис. 73 и 74).
Температурная зависимость, определенная для разных участков распа-
да, выражается прямыми Аррениуса; значения Е по разным формулам ч
для разных участков близки и заключаются в пределах 26—31 нкйл/люль;
при разложении под слоем парафина кривые р(7) в отличие от самого пер-
хлората хорошо совмещаются на всем протяжении (рис. 75) и по коэффи-
циенту совмещении Е — 25,1 ккал/моль.
1 Содержащее конденсирующихся продуктов возрастает в ходе распада, по-види-
мому, в результате газофазных реакции; по достижении половины распада оно со-
ставляет ~ 50% (при 20°С) и ~ 80% (при — 180°С) и в дальнейшем почти не ме-
в вется.
90
Абсолютная величина скорости также невелика: при 270° С период по-
лураспада составляет 225 мин., который гораздо больше, чем, например,
для распада нитрата аммония. Известную роль здесь может играть разли-
чие агрегатного состояния.
Распад перхлората аммония изучали [100] также при повышенных
температурах (380—450° С). Кривые изменения давления во времени при
этих температурах значительно отличаются от тех, которые наблюдались
при более низких температурах; нет участка ускорения и скорость пада-
ет на всем протяжении распада, который не останавливается па 30%,
а идет до конца. Эти кривые можно выразить формулой р = ktn при п
меньшем 1 и растущим при повышении температуры от 0,5 при 380—
400° до 0,8 при 450° С.
В дальнейшем высокотемпературный распад был изучен более деталь-
но ,и по несколько ипой методике [104]. Опыты проводились в стеклянном
сосуде, в который после эвакуации впускали некоторое количество азота,
чтобы замедлить сублимацию; течение процесса регистрировали при помо-
щи весов из кварцевой спирали.
При 300—380° С опыты не давали воспроизводимых результатов. Выше
340° реакция на всем ее протяжении была замедляющейся и не показы-
вала признаков остановки до самого конца. При 400—440° С получались
воспроизводимые результаты, остатка почти не получалось. Константа ско-
рости пропорциональна давлению азота в степени 0,6. Кривые выражают-
ся уравнением т2/3— kt + const, где т —масса неразложившейся соли.
По температурной зависимости константы скорости 'расчет дает энергию
активации, равную 73,4 ккал/молъ.
В области относительно высоких температур распад перхлората изу-
чали в присутствии инертного газа (азота) также Гальви и Джэкобс [103].
Давление продуктов 'распада измерялось спиральным стеклянным маномет-
ром. Они подтвердили, что распад идет по степенному закону р = kt71, при-
чем п < 1. При увеличении навески п несколько уменьшается (применял-
ся кусочек прессованной таблетки), иногда кривая p(t) состоит из двух
участков с разным га, меньшим в начале. Анализ экспериментальных дан-
ных по формуле сжимающейся оболочки 1 —- (1 — а)'/э = kt показал по-
стоянство к в значительном интервале а для кусочков таблетки от 0,2 до
0,8, для целых кристаллов от 0 до 0,7. Энергия активации для таблетки,
рассчитанная по этой формуле, составляет 38.8 ккал/молъ.
Рис. 74. Разложение перхлората
аммония (7), его смеси с парафином (2)
и с веществом Б {3) при 250° С
И m/v = 10 • Ю-4 г/см?
/00
~7оо
Т, мин.
о - 1
• -2
&-3
Л.-1;
□ - 5
Рис. 75. Сопмещение кривых v = /(т) по
оси времени для парафиновой смеси на
этапе быстрого роста давления. Коэффи-
циент трансформации при различной
температуре:
2 — 180" С, 18,5; Т —100°С, B.G; 3 — 200’ С, j,4;
<1 — 220’0. 1,7; 3 — 230= С. 1
1)1
Анализируя полученные им результаты, Биркамшоу рассматривает
три конкурирующие реакции, происходящие при нагревании перхлората:
низкотемпературную реакцию, высокотемпературную реакцию и сублима-
цию. Первую из них следует считать реакцией, идущей в твердой фазе с
типическими чертами такой реакции. Высокотемпературную реакцию
(380—440° С) следует считать разложением паров перхлората, на что ука-
зывает и влияние давления инертного таза. Ведущей реакцией при этом
является, вероятно, распад С1О4-иона. В пользу этого говорят опыты с пер-
хлоратом калия, который прн 500—550° С дает энергию активации, рав-
ную 69,3 ккал/моль и близкую к энергии активации перхлората аммония.
Кроме того, для перхлората калия трудно представить себе иную реакцию,
нежели распад СЮгИОна.
Гальви и Джзкобс определяющим этапом высокотемпературного распа-
да считают переход протона на поверхности твердой фазы с последующим
окислением аммиака радикалами, образовавшимися при распаде хлорпой
кислоты,
Свои заключения о трех возможных механизмах превращения перхлора-
та аммония Биркамшоу мотивирует следующими общими соображениями.
1. Протонный переход — обратимый процесс, контролирующий субли-
мацию. Если кислота неустойчива, разложение может идти этим путем,
как это можно увидеть, сравнивая случаи, где анионом являются NO2,
CIOs, СЮз, М11О4, N3, с темп, где анионом являются NOa, SO4, СЮ4.
В первой группе как кислота, так и аммониевая соль неустойчивы.
2. Реакция перехода электрона между анионом и межузловым положи-
тельным ионом является существенным этапом при разложении щелочно-
земельных и щелочных азидов, а также азида свинца,
3. Термический распад аниона, по-видимому, происходит в случае
СЮгЛОна в перхлоратах лития и калия, где образуются хлориды.
Можно ожидать, что ион NH4 будет устойчивым, так как возможная
реакция его распада сильно эндотермична
N1L-+NHS + И — 200-7- 220 ккал. (2.36)
При сублимации перхлората аммония основным процессом должен быть
переход прогона. Маловероятно, однако, чтобы он был начальным этапом
при разложении.
Перхлорат-ион мог бы распадаться термически. Перхлораты лития и
калия разлагаются при 400° С и выше, перхлорат же аммония распадается
с измеримой скоростью уже при 200° С, Поэтому прогонным переходом,
ведущим к сублимации, можно объяснить лишь высокотемпературное раз-
ложение перхлората — выше 380° С, Для разложения между 200 и 300° С
остается механизм электронного перехода.
Межувловыми ионами в перхлорате аммония являются ГчНд-цоны, так
как они гораздо меньше, чем СКД-ионы. Начальная реакция перехода
электрона выражается соответственно уравнениями
СЮ: - + NHt+ СЮ, + NH,, (2.37)
NHt — NH3+ Н. (2,38)
Радикал С1О4, образовавшийся внутри кристалла, либо захватывает
электрон от соседнего С1О4-лона, диффундируя таким образом к поверх-
ности, где он может разложиться, либо захватывает атом водорода, обра-
зуя ИСЮ4. В первом случае то место, где СЮ4 появится на поверхности, те-
ряет электрон и становится положительной дыркой. Этот заряд может быть
нейтрализован либо поступлением электрона изнутри кристалла, либо миг-
рацией КН4-иона из соседнего места. Этот NH4-hoh становится межузло-
вым ионом, который может принять участие в реакдци перехода электро-
па по соседству От точки разложения, Продолжение этого процесса приво-
92
дит к образованию зародыша. Решетка внутри зародыша должна быть на-
рушенной, и эта нарушенность увеличивается с его ростом. Два эффекта
становятся существенными в этой области.
1. Электроны поступают внутрь кристалла на границе раздела между
упорядоченным и неупорядоченным веществом. Это будет происходить
из-за наличия вакансий вблизи этих СЮд-ионов; С1О4-радикал, образую-
щийся при миграции электрона, может разлагаться. Разложение на внут-
ренней поверхности зародыша будет затруднено тем, что при переходе
электрона от иона к радикалу (С1О4_->С1О4 + е) захват электрона дру-
гим радикалом внутри кристалла будет труднее из-за нарушенное™ ре-
шетки и будет не так много межузловых NH4-hohob, которые могут иници-
ировать переход электрона из-за возросшего числа вакансий в местах на-
хождения NH4-hohob в решетке.
2. Протонный переход в нарушенной решетке будет легче, так как
увеличится поверхность, т. е. условия для сублимации будут благоприят-
нее.
Итак, два процесса всегда соревнуются за С1О4-ионы на поверхности —
сублимации (путем перехода протона) и разложение (путем перехода
электрона). Реакция разложения образует нарушенную решетку и это
предположительно благоприятствует сублимации за счет разложения. Воз-
можно поэтому, что на некоторой стадии разложения сублимация стано-
вится гораздо более вероятной, чем разложение, так что последнего не про-
исходит. Этим объясняется остановка разложения.
Что касается судьбы Н-атома, образовавшегося при распаде NH4-pa-
дикала (NH4->NH3 + Н + 26 ± 10 ккал), то он, так же как и NH3, мо-
жет проДиффундировать к поверхности и перейти в объем. Другая возмож-
ность состоит во встрече Н-атома с С[О4-радикалом внутри или на поверх-
ности; в этом случае образуется НСЮ4 и разложения не происходит
Н + С]О4 (внутри) —»-]]C]Oi(внутри) + 106 ккал, (2.39)
С11О4(внутри)-рСЮ4(на пов.)->-С1О1(на лов.)-|-С]О4 (внутри), (2.40)
0 +СЮ4(па пов.)-+НС]О4 (па пов.), (2.41)
]]C10i(na пов,)-+НС1О4(пар), (2.42)
NH.,+ -j- СЮ; NHs + ЛС1О4(внутри или на поверхности). (2-43)
Поскольку НС1О4 внутри решетки не так быстро будет диффундиро-
вать из кристалла, как NH3, следует ожидать, что разложившийся перхло-
рат будет слегка кислым, что и наблюдалось. Известно, что следы НС1О4
уменьшают .индукционный период, в то время как NH3 увеличивает его и
ведет к замедленному разложению.
Маловероятно, чтобы НС1О4 в решетке образовывала Н- и С1О4-ионы ио
сильно эндотермичной реакции
НСГО4 — СЮ4 + Н - 287 ккал,
(2.44)
нереальной поэтому при 200—300° С.
Другой возможный механизм с участием Н-атомов состоит в следую-
щем:
И + НС1О4 —НгО -|- С[Оз + 67 ккал. (245)
Таким образом, Н-атомы могут реагировать или с С104-радикалом, об-
разуя НС1О4, или с НС1О4, образуя СЮз-радикалы. Такие С1О3-радикалы
также будут действовать как ловушка электронов и соответственно уско-
рять разложение. Этим можно объяснить каталитическое действие НС1О4.
Добавление аммиака может иметь двоякое действие.
93
а) Если в решетке уже имеется НСЮз, то могут образоваться NH; и
СЮ4-ИОНЫ. Поскольку между ГШг-иопами в узлах решетки и в между-
у алиях существует равновесие, число последних не может возрасти очень
сильно.
б) Реакция
СЮ; - + NH;+ -> СЮ; + NH(2.37)
N.H; -* NII3 + Н (2.38)
может быть обратимо?! в кристалле. Добавление аммиака будет коатому
уменьшать скорость образования С1О«-радпг<алов и вследствие этого —
скорость распада. Кроме того, огг будет уводить молекулы HCIO4 и воз-
можно Н-атомы. Итогом этого будет увеличение индукционного периода
и уменьшение скорости распада в присутствии аммиака.
Гальви и Джэкобс предлагают другой механизм распада. Как первую
ступень, учитывая состав продуктов распада, они принимают образование
и распад молекулы NH4CIO4, причем этапом, определяющим скорость реак-
ции, является образование положительной дырки. Распад молекулы проис-
ходит с отщеплением воды, образованием атома азота и СЮ2; последую-
щее взаимодействие молекулы азота и атома кислорода (образовавшегося
при распаде СЮа) дает закись азота.
Влияние добавок на разложение перхлората аммония.
Выло изучено [94] влияние окпелов ряда металлов (АЕОз, CaO, FejOa,
МиОа) на распад перхлората мри 230° С. Перекись марганца оказывает
наибольший каталитический эффект; через 2 часа распад проходит на
100%; по характеру кривая распада сходна с кривой, наблюдавшейся
прп 400° о тем отличием, что образуется большое количество СЮг. а не
нптрозилхлорида. Окись железа также оказывает каталитическое дейст-
вие за 2 часа разложилось 44%, за 4 чара — 60% вещества. Кривая раз-
ложения имеет своеобразный вид с двумя максимумами. Окись кальция за-
медляет разложение, по-видимому, в результате образования аммиака,
окись алюминия влияния не оказывает.
Гальви и Джэкобс [96] изучили (при 137—212° С) разложение смесей
перхлората аммония и МнОг, Распад идет в два этапа, первый из которых
представляет собой каталитическое разложение перхлората аммония. Две
константы для разных интервалов распада дают ту же £ = 32 000 ккал^моль.
Она предопределяет механизм реакции, в которой начальным этаном яв-
ляется образование положительных дырок, а каталитическое действие
окисла приводит к увеличению среднего времени их жизни. Второй этап
реакции предполагает разложение чистого перхлората аммония и де зави-
сит от присутствия катализатора.
Действие катализаторов (МпОг, NisOs, MgO и смесь окислов кобальта
СогОз + СО3О4) на термический распад перхлората аммония изучали так-
же в связи с их влиянием на горение Хермопп и Сал мои [97]. При 170—
200° С разложение прослеживалось гравиметрически, выше 200° С приведе-
ны давления невыморажцвающихся газов (Ch + Ns). Все перечисленные
окислы ускоряют распад перхлората аммония при 170° С, особенно сильно
действуют окислы кобальта. Действие окиси магния уменьшается при уве-
личения ее содержания, очевидно, в результате ее взаимодействия с пер-
хлоратом аммония с образованием перхлората магния.
Выше 200° С увеличение количества МнОа (сверх 5 %) почти пе увели-
чивало скорость распада, но конечное давление несколько возрастало. Ско-
рость газообразования па этапе ее падения описывается уравнением сжи-
мающейся оболочки. Расчетное Е изменяется для разных катализаторов в
пределах 27—48 ккал/моль и больше При повышенных температурах
(табл. 10), что, возможно, связано с изменением механизма распада. При-
сутствие катализаторов влияет предположительно как на реакции в кон-
44
Таблица 10
Лррениусовские параметры Л и Е, полученные в различных
температурных интервалах
(100 а# перхлората аммония -{-15 .иг катализатора)
Катализатор Интервал темпера- тур. ’С В К, ккал
МпО3 170—200 4-10s 28
NiaO3 170—200 3-1OI'2 33
СоаОэ 4" СО3О4 170—200 9КР3 33
N12O3 210—235 3-1013 43
МпО2 200—240 1Q12 34
Со^Оз GoaOd 210—230 1,5-101" 42
Ст203 210-230 110’ 27
МпО2 249—272 1()м 48
ИйОз 245-265 8.101* 40
СтаОэ 250—275 Ш5 38
денсированной, так и в газовой фазах; они изменяют и состав газообразных
продуктов распада, который зависит также от температуры.
Распад шел до конца во всем изучавшемся интервале температур. Ка-
тализатор сокращал задержку вспышки тем сильнее, чем больше его (до
некоторого предела) добавлено; прн крупнопористом перхлорате необхо-
димое для максимального снижения задержки количество катализатора
меньше; задержка вспышки уменьшалась также при увеличении внешне-
го давления.
Гальви и Джзкобс [98] изучали также влияние древесного угля на рас-
пад перхлората аммония. Ниже 240° С уголь не влияет па распад и не реа-
гирует с его продуктами. При 240 — 260° С он сокращает индукционный пе-
риод; выше 260° распад быстро ускоряется и приводит к мягкому беспла-
менному взрыву, сопровождающемуся разрушением таблетки. Давление
при распаде растет по степенному закону при показателе степени больше
1, зависящем от массы таблетки и растущем с температурой, по-вндимому,
вследствие саморазогрсва. Авторы Отмечают, что в присутствии утля оста-
новки распада на 30% не наблюдается, уже начиная с 240° против 350°
для чистого перхлората
Изучалось влияние закиси меди на термический распад перхлората ам-
мония [99]. Осколки прессованных таблеток разлагались при 265° С и дав-
лении азота 250 дьм; начиная с краткого индукционного периода, констан-
та скорости и степень разложения уже не зависят от давления. Ход рас-
пада описывается выражением
lg = (2.33)
Смесь с Сп2О (4,56 молярных %) при 260° С показывала экспоненци-
альный вначале и затем более быстрый рост давления, заканчинающпйся
вспышкой через 540 сек. При меньших навесках и более низких темпера-
турах кривая p(t) S-образна, после максимума наблюдается некоторое па-
дение давления. £, рассчитанная по скорости газообразования при разных
температурах, составляла 29,0, по задержке вспышки — 28,1 ккал/моль.
За де ряжа вспышки сложно зависит от содержания С112О, по-впдпмому,
в результате многостадийности процесса в присутствии СщО, о которой
можно судить по кривым p(t) до возникновения вспышки при разных со-
держаниях закиси меди.
1 Возможно, однако, что эти разногласия связаны с различиями в методике в
большей стеиецп, чем с нлияписм угля.
95
VIII. ОБЩИЕ ЗАКОНОМЕРНОСТИ
ТЕРМИЧЕСКОГО РАСПАДА ВВ
Рассмотренные исследования позволяют сделать по вопросу о меха-
низме и особенностях течения медленного термического распада ВВ неко-
торые общие выводы.
Установлено, что термический распад многих ВВ представляет собой,
как правило, совокупность по меньшей мере двух реакций. Одна из них —
самопроизвольная, для изученных в этом отношении веществ — первого
порядка, скорость ее зависит только от температуры вещества и соответ-
ствует минимальной возможной скорости его распада.
Вторая реакция — самоускоряющаяся по автокаталитическому или
иному механизму, причем вещества, вызывающие ускорение, могут быть
как конечными продуктами распада (например, вода в случае нитроэфи-
ров), так и промежуточными его продуктами (например, двуокись азота
при распаде нитроэфиров).
Если ускоряющие реакцию продукты ее являются веществами нелету-
чими, то обе реакции — первичная и сам ©ускоряющаяся — протекают не-
избежно .совместно. Если эти продукты при температуре распада летучи,
то в известных условиях (высокая температура, большой относительный
объем сосуда, пропускание тока инертного газа и т. д.) они могут быть уда-
лены из конденсированной фазы, и тогда можно наблюдать мономолекуляр-
ную реакцию в чистом виде. Подавления самоускоряющейся реакции мож-
но в принципе достигнуть и при нелетучих катализаторах, добавляя к
взрывчатому веществу такие примеси (стабилизаторы), которые быстро
вступают в реакцию с автокаталиэаторами и сводят к минимуму их уско-
ряющее действие. Наконец, для изучения первичной реакции могут быть
использованы начальные ступени распада, когда концентрация и действие
продуктов распада малы.
Изучая температурную зависимость скорости мономолекулярной реак-
ции, можно установить кинетические константы этой реакции: энергию ак-
тивации £', предэкспоненциальный множитель В и период полураспада,
дающий наглядное представление о максимально возможной сравнитель-
ной стойкости того или иного ВВ. Впервые такой анализ кинетики мономо-
лекулярного распада ВВ был проведен Рогинским на основании собствен-
ных экспериментальных данных и материалов предшествовавших работ.
Этот анализ обнаружил некоторые своеобразные кинетические особенно-
сти распада ВВ.
В В распадаются с заметной измеримой скоростью при умеренно повы-
шенных температурах 100—300° С, т. е. константы скорости их распада до-
вольно велики. В то же время этот распад характеризуется большим тем-
пературным коэффициентом скорости; скорость распада при повышении
температуры на 10° С увеличивается для многих изученных ВВ приблизи-
тельно в 4 раза. Такой большой температурный коэффициент скорости не-
избежно означает большую энергию активации.
В самом деле
кг =
ki = В-е~Е!КТ‘.
Температурный коэффициент скорости
ftj e~E/RT,
(2.46)
(2-47)
(2.48)
М “ е-ь’/кт, '
Логарифмируя, получаем
In*? = - Е/ВТ^Е/ВТ^
LU j. ““ D ТГ -TI 1
(2-49)
A‘i Г1Т а 1
(2.50)
96
откуда
E = {2.51)
“I -t 2““< 1 ' ?
Таким образом, если при данном значении произведения Т\Т% отноше-
ние /i2/A\ велико, то Е и должна быть велика. С другой стороны, поскольку
! величина k значительна, то нри большой Е значение В = keB:RT должно
быть также большим. Действительно, как видно из табл. 12, для многих
ВВ величина Е велика и находится между 40 000 и 60 000 кал/.моль; то
»же относится и к величине В, лежащей для ряда ВВ между 10тб и 1028.
То, что величина энергии активации ВВ велика, само по себе не явля-
ется удивительным. Способность к взрыву необязательно предполагает пе-
1 устойчивость молекулы, но определяется соотношением энергии реакции,
i вызывающей возбуждение реакций соседних молекул, и энергии актива-
4 ции, измеряющей устойчивость молекулы по отношению к энергетическим
воздействиям-
Иначе обстоит дело с большими значениями постоянной В. Согласно
общепринятым представлениям, В не может быть существенно больше час-
тоты внутримолекулярных колебаний 101/2—1014. Поэтому установление
•’ Рогинским для всех изученных тогда ВВ гораздо больших значений В вы-
звало значительный интерес. Можно было допустить, что эта закономер-
ность является принципиальной характеристикой ВВ.
Естественное предположение о наличии длинных цепей с v — 10s—
107, объяснило бы большие значения В. В связи с этим для нитроглице-
рина были поставлены опыты по разложению в растворах, которые не
подтвердили этого предполо/кеипя.
Дальнейшие исследования [33, 106] показали, что большие значения В
не являются неизбежной особенностью ВВ. Для ряда ВВ, в том числе и
для некоторых нитратов спиртов, были установлены умеренные значения £
и нормальные значения В. Более того, для одного из нитроэфпров (тэн)
и одного нитрамина (гексоген), дающих при разложении в чистом виде
бадьппго В, при разложении в виде раствора В имеют нормальное значение.
Все это делает вероятным заключение о том, что большие В пе представля-
ют собой характеристики моиомолекулярной реакции, по обусловливаются
некоторыми особенностями течения реакции распада отдельных ВВ.
В работе [109] указывалось па увеличение количества газообразных про-
дуктов распада при повышении температуры распада, как па одну из таких
особенностей. Если при этом характеризовать отношение скоростей при
двух температурах отношением абсолютных количеств газов, выделенных
в единицу времени единицей веса ВВ без учета указанного изменения ко-
личества газов, то получится завышенное значение температурного коэф-
фициента скорости, а следовательно, и завышенные значения Е и В. Изме-
нение характера кривой р ~ /(£) при изменении температуры также мо-
жет быть источником ошибок при сравнении объемов.
Другая особенность [110] распада заключается в том, что он состоит из
двух ступеней: эндотермического и обратимого перехода исходного вещест-
ва Л в промежуточный продукт Б и необратимой реакции распада послед-
него А В —> С. Тогда при повышения температуры суммарная скорость
i реакции возрастает но двум причинам;- вследствие повышения концснтра-
1 цпп промежуточного продукта В и вследствие увеличения скорости пере-
хода ого в конечный продукт С. В итоге мы получаем завышенное значе-
; пце температурного коэффициента, а следовательно, и большие значения
. Е и В L Наконец, возможны и другие механизмы, приводящие к завышен-
ным температурным коэффициентам. Так, если при разложении есть ко-
поткие и поэтому трудно обнаруживаемые цепи, длина которых возра-
1 Следует указать, что вовейшпе работы во термическому распаду алкилвитратоп
и нитритов (стр, 40). показывающие, что первый этап реакции является эндотерми-
ческим и обратимым, говорят в пользу этого предположения;
7 К. К. Андреев 97
Таблица 12
Значения анергии активации, предэкспоиенцнального множителя и периода
полураспада для различных В В
вв Интервал темпера- тур, ° С Е, ICOA/lWlb IR В Период полу- распада при 120’ С по рас- чету •. часы Лите- ратура
Мотил^итрат ...... в»». ~ ~ 212—239 39 500 14,4
Этилнцтрат 161—181 41 230 16,25
Нитроглнколь .......... —- 35 000 14
» .......... 85—105 39 000 15,9
Нитроглицерин 150—190 50 000 23,5
» . в . . . . в . . . 125—150 45 000 19,2
1» .......... 90—125 42 600 18,0
» . 75—105 40 300 17,1
а 90-120 43 700 18,64
Нитроглицерин при малых 5 . . . 80-140 43 700 18,4
Нитроглицерин при больших 6 . - 80—140 39 300 15 4
Пропилецгликольдипитрат 80—100 37400 15 2
Триметпленглинольдипитрат .... 85—110 38 100 15,2
Три.метилолннтрометаятринитрат . . 75—95 36 400 15,3
Тэн 160—225 47 ООО 19,8
Т:>и при малых 8 . . . 145-171 39 000 15 6
Тли в растворе 171-238 39 500 16,1
ДиэтилеягликОльдинитрат 60—150 42 500 16,5
Дина 100—160 42 000 16 5
Лтилепдиампндинитрат 230-357 40 000 13Д
Нитрат аммония 243-361 40 500 13,8
Пироксилин JVs 1 (13% N) • . . . , 90—135 49 000 21,0
Пироксилин № 1 (13% N) 140—155 48 000 20,0
То же 1г>г>—175 56 000 24,0
Пироксилин Хз 1 (13,35% N) ... 135-160 39 200 15,0
То же 70-160 39 500 15 .35
Ппрог-гсилин М 2 (12,3% N.) . . . . 70-160 38 500 14,4
Кол roi.c-’лпн (11,85% N), раствор . 165-200 43 000 18,0
коллодионный хлопок — 50 000 21,0
.)т г-тендицитрамнг 184—254 30 500 12,8
Moiioi[ijTpo6o;[30.T (пары) ..... 390—415 53 400 12,65
Дипзтробепзол (пары) 346—355 52 600 12 70
Тр«нитробензол (пары) 270—355 51 900 13,60
Трипитробензол (жидкость) .... 250-310 43 000 10,9
Тротил (поры) 280—320 34 500 8,45
Тротп-Л (жидкость) 193-250 26 200
'1 ротпл 223—270 ,53 500 19,0
1ринатроанпЛпн (пары) 2.50—320 38 500 8,8
1 рппптроапжигн (жидкость) .... 2.50—3(10 31 000 7 1
Пикриновая кислота — 57 500 23,0
РСтГСатгТрОД'гфспнЛ Гексдвптродифеиилсульф !д | 240—300 49 500 16,1
Яч’ДНИЙ 235—2->5 43 000 15,5
твердый 200—220 42 700 12,0
Медина 110—130 35 400 15,6
Гексоген 213—299 47.500 18,5
Гексоген в растворе . 41 000 15,46
]’окс<>гец 150-197 51 001) 18,6
Гексоген в растворе 150—197 41 300 15 3
Оптоген — 52 71)0 19 7
t 176—230 36 500 10 7
Оотопчг в растворе Тетрил жидкий 176-230 129,9- 42 600 60 000 14,9 27,5
138,6
* Расчет периода полураспада ирнияаедсн по данным, полученным
состояния, в котором вещество находится при температурах, приведс
всех случаях, соответствующих агрегатному его состоянию при 120" С.
•• ТИЛЛаЯ ПАПнЦиМО пРпнЛТТо гтг.ттггг.л^,. — - —------- - - •» -
для
п уведен tn .ix
•• Малал величина периода полураспада этнлеильпитрамнна объясняется тем, что для рас-
чета испольиованм данные [64] расплааа, скорость ьасцада которого гораздо больше чем твер-
дого вещества.
7120 1 1Ю5]
922 1106
56 [107
119 [108
3,91 И]
129 И
95 11
40 [108]
80 [9
140 [16
540 [16
77 [ 108
188 [108
17 [108
421 [33
230 32
142 33
2400 35
1400 [36
27 ЛО1 [331
1№ [331
343 [11
954 [Н[
2680 [11
1200 144
770 43
1900 W
150 40
1230 11
2 8** 64
17 -1012 60
6 Л 01а [60
3,4 Л 0” 60
1,9-10» 60]
10’ 60
— 60
5,8-10® 11
7,5-10» 60
2,6-10» 60
18-10* 111
4,9-10’ 60
4,9-Ю4 [60
9,7-10’ |60
2 3 [65
16-103 [70
4 2-10» [70
1 1-10’ (71
8’240» [71
7,8-103 70
7 2-105 71
1 1-105 [71
142 11
того агрегатного
в графе 2, т, е. во
98
Таблица 12 (окончание)
ВВ Интервал темпера- тур, “С Е, кал/миль 1g в Период полу- распада при 120° С по рас- чету*, часы Лите- ратура
Тетрил жидкий То же Тетрил твердый То же Т рин п трот риа з и до бен з ол а-Лзид свинца То же (З-Азид свинца То же Трпнитрорезорцинат свинца .... Гремучая ртуть . , , , Азид натрия ..... Азид калия А.чвд кальция . То же .... Азид стронция Азид бария Азид серебра То же . 211—260 20—100 245—275 220—260 237—252 200—270 225—255 100—115 240—275 222—255 60—130 80-100 100—135 1 Ор—150 210—270 55 500 38 400 52 000 36 600 32 300 55 000 38 000 40000 37 000 46 700 25 31 И) 34 400 36100 18 000— 19 000 20 000 20 200 23 500 29000 40 000 1 1 1 1 II И 1 1 1 1 1 | 1 1 1 | 1 1 ] | | i 1 Mill [Ilf [641 (111 (84 [79 [82 (79 [82 84 77 82 82 82 82 [ 82) 82) [82) 82)
стает с температурой, если имеет место саморазогрев, растущий с повы-
шением температуры, если при повышении температуры возрастает кон-
центрация летучего автокатализатора в жидкой фазе вследствие того, что
скорость диффузии растет с температурой медленнее, чем скорость реак-
ции, то все эти факторы могут привести к ионы пи: иным значениям Е и И.
Уточнение этих вопросов должно быть предметом дальнейших экспери-
ментальных исследований. Во всяком случае многие типичные ВВ не по-
казывают в отношении характеристик мономолекулярной реакции каких-
либо существенных отличий от невзрывчатых веществ.
Песравнопио более сложный характер имеет самоускоряющсеся разло-
жение ВВ, Обычно это самоускорнющееся разложение рассматривается как
автокаталитическое. Однако лишь в немногих случаях (тротил, тетрил,
гексоген) образование автокатализаторов подтверждено непосредственны-
ми опытами добавления продуктов реакции к свежему веществу, которое
приводило к увеличению скорости газообразования. Даже и в этом случае
только для тетрила ход разложения согласуется с простой схемой автока-
талитической реакции, скорость которой пропорциональна первой степени
концентрации автокатализатора.
Возможно, что в ряде случаев ускорение газообразования при распаде
является следствием того, что он представляет собой совокупность после-
довательных реакций, включающих образование малостойких промежуточ-
ных продуктов, разлагающихся с большей скоростью, чем исходное вещест-
во. Скорость образования газов (или тепла) при распаде будет возрастать
во времени также и в том случае, если превращение промежуточного про-
дукта дает газон (или тепла) значительно больше, чем первичное превра-
щение исходного вещества.
Протекание распада, как ряда последовательных реакций особенно ве-
роятно в случае распада ВВ, содержащих насколько одинаковых групп,
с которых начинается распад молекулы — полиннтратов многоатомных
спиртов, по ли нит росе единений ,и т. п, Естественно, что первичным процес-
сом и этом случае является реакция одной группы, которая необязательно
должна сразу привести к полному разрушению молекулы. Сказанное не
исключает возможности автокаталитического влияния продуктов распада
как конечных, так и промежуточных. Это влияние усложняется том об-
7*
99
стоите лье твои, что некоторые катализаторы летучи и их концентрация в
жидкой фазе зависит от условий опыта, определяющих давление газов
над жидкостью.
Если учесть, что и само направление реакции, поскольку о нем иожнл
судить по конечному составу продуктов распада, меняется с изменение и
температуры, то стагговится ясной сложность точения распада в условиях
наличия самоускоренпя. Добавим, что состав продуктов распада во всех
изученных случаях itc соответствует равновесному, так что указанное его из-
менение не может быть объяснено влиянием температуры на равновесие.
Термический распад ВВ в твердом состоянии при принципиальном его
сходстве с распадом жидких ВВ имеет две особенности. Во-первых, реак-
ция начинается с появления зародышей преимущественно на поверхности
кристаллов ВВ. Зародыши эти врастают в вещество неравномерно. Во-вто-
рых, реакция зачастую имеет не одни, а два максимума скорости. По-вп-
димому, оба зтп обстоятельства существенно влияют и на механизм горе-
ния таких ВВ. Для тех твердых- ВВ, которые могут быть расплавлены, ус-
тановлено, что скорость распада в жидком состоянии значительно больше,
чем в твердом, Если при распаде образуются конденсированные продукты
и твердое ВВ, растворяясь в цих, частично переходит в жидкое состояние,
то это приводит к прогрессивному ускорению реакции независимо от дру-
гих возможностей ее ускорения.
В заключение отмстим, что почти все исследования термического рас-
пада ВВ проводились манометрическим методом. Этот метод был принят не
потому, что он наиболее пригоден для изучения хода распада, а лишь в
силу его простоты и доступности. В то же время этот метод дает при слож-
ных реакциях лишь суммарный их итог и то лишь постольку, поскольку
каждая из реакций сопровождается выделением не конденсирующихся при
температуре опыта газов. Судить же по общему газовыделению о течении
отдельных реакций можно лишь в исключительных случаях. Поэтому и
в методическом отношении экспериментальное исследование термического
распада ВВ нуждается в существенном усовершенствовании.
Рассмотренные механизмы химического превращения ВВ — мономоле-
кулярныд и самоу окоряющийся — изучены для медленного гомогенного
термического распада ВВ. Возникает естественный вопрос о том, в какой
мере они будут иметь место при горении ВВ, Несомненно, что при возник-
новении горения — яри восплнмепенли ВВ и при вспышке — процессы са-
моускорения реакция играют важнейшую роль. Именно благодаря посте-
пенному развитию самоускоренпя реакции достигается нарушение тепло-
вого равновесия — теплоприход становится больше теплоотвода, после чего
и разложение быстро ускоряется по тепловому механизму7 до возникнове-
ния горения. Однако при автокаталитическом самоускорснии роль автока-
тализаторов при возникновении вспышки или горения определяется также
тем, как высока температура ВВ ц как быстро она достигается.
При быстром нагревании критическая для теплового механизма ско-
рость реакции может быть достигнута за счет мономолекулярпой реакции
ня столь ранних ее стадиях, когда концентрация и действие катализаторов
заметно ire сказываются. Помимо этого, с повышением температуры вели-
чина константы мономолекулярного распада растет быстрее, чем величи-
на константы автокаталитической реакции соответственно различию энер-
гий активации, и при высоких температурах поэтому соотношение скоро-
стей менее благоприятно для автокатализа, чем при низких.
В обычных условиях стационарного горения, по-видимому, это условие
выполняется, и при горении многих ВВ, особенно летучих и при низких
давлениях, автокатализ в жидкой фазе не играет существенной роли,
Мепее ясен вопрос для нелетучих ВВ, в том числе и для нитроклетчат-
ки. В Этом случае поверхностные и автокаталитические реакции Могут,
по-видимому, играть большую роль, определяющую существенное отличие
механизма горения нелетучих- ВВ от механизма горения летучих ВВ,
ГЛАВА ТРЕТЬЯ
УСТОЙЧИВОЕ ГОРЕНИЕ 1ЫРЫ1ВЧАТЫХ ВЕЩЕСТВ
I. МЕТОДИКА ИССЛЕДОВАНИЯ
1. МАНОМЕТРИЧЕСКАЯ БОМБА
Основным и до недавнего времени практически единственным лабора-
торным прибором для определения давления при горении, количества и
состава газов, а также зависимости скорости горения от давления была ма-
нометрическая бомба Сарро и Вьеля [111]. Этот прибор позволял изучать
горение порохов при давлениях до нескольких тьесяч атмосфер, при кото-
рь[х они применяются в ствольном огнестрельном оружии. Манометриче-
ская бомба в свое время дала возможность установить некоторые праи[ци-
пиальвые закономерности горения черного пороха, явившиеся теоретиче-
ской основой для разработки современньЕХ порохов коллоидального типа,
баллистическое исследование которых и поныне в основном проводится
в манометрической бомбе.
МаЕЕОметрЕЕческая бомба, схематически изображенная на рпс. 76, пред-
ставляет собой толстостенную прочную стальную трубку 2 внутренним
объемом От 22 cat3 л выше, в которую с обеих сторон ввинчиваются проб-
ки 1 и 4.
Пробка 1 имеет центральный электрически
и служит для воспламенения порохового заряда
изолированный стержень
при помощи воспламени-
теля, поджигаемого в свою очередь накалом
электрическим током тонкой проволочки.
Пробка 4 имеет центральный канал, в кото-
ром движется хорошо пригнанный поршень 3.
Между головкой этого поршня 5 и пробкой 8,
ввинчиваемой в пробку 4, помещается изме-
ритель давления, в качестве которого был
применен и широт;о используется и в настоя-
щее время медный крешерный цилиндрик 7
определенных размеров, сжатие которого ха-
рактеризует 'величину давления. Чтобы полу-
2 3 5 о
Рис. 76. Схема устройства ма-
нометрической бомбы
чить характеристику изменения давления во времени, движение поршня
записывается обычно непосредственно укрепленным на eio головке сталь-
ным пером 6 на заЕсопчеипой бумаге, натянутой на нращагощЕЕЙся барабан.
Эта запись дает изменение давления от момента начала обжатгЕЯ модееого
цилиндрика до момента достижения максимального давления — пониже-
ние давления при применсЕЕии модного цнлнпдртПЕа, естествсЕЕНо, записано
быть пе может. Перевод обжатия модееого цилиндрика в сдеееенцы давления
ПрОИЗВОдЕЕТСЯ С учетОМ ЕЕОперечЕЕОГО сечеЕЕЕЕЯ ЦОрШЕЕЯ ЕЕЭ ОСЕЕОВаНЕЕЕЕ опытов
обжатия серии цилиндриков на прессах при определенных статических
давлениях.
101
Определение давления при горении в бомбе осложняется рядом факто-
ров. Так, обжатие медного цилиндрика зависит не только от величины
давления, но и от скорости, с которой достигнуто это давление. При мгно-
венном нарастании давления обжатие получается за счет инерции порш-
ня вдвое больше, чем при статическом или медленном обжатии. Расчет
давления в случаях, промежуточных между этими предельными, сложен.
Поскольку величина обжатия зависит от массы поршня, то, изменяя по-
следнюю, можно приблизиться к одному из крайних случаев.
Далее, применяя обычные цилиндрические медные крешеры, нельзя
детально фиксировать изменение давления в области малых давлений.
Этот недостаток устраняется применением медных цилиндриков, сточен-
ных на конус, предложенных и введенных в применение Серебряко-
вым [112]. Наконец, применение в качестве датчиков необратимо деформи-
рующихся металлических цилиндров позволяет регистрировать только по-
вышение давления, но не его падение.
Разработан ряд приборов с малой инерционностью для регистрации
быстро изменяющихся как в сторону повышения, так и в сторону пони-
жения давлений [ИЗ]. В большинстве их в качестве чувствительного эле-
мента используется круглая диафрагма или пластинка, жестко закреплен-
ная по периферии. Прогиб диафрагмы может быть определен различными
способами — применением оптического указателя, оптической интерферен-
ции, электрического конденсатора, переменного магнитного сопротивле-
ния, тензометра сопротивления. Особенно пригоден для измерения очень
быстро изменяющихся давлений пьезоэлектрический датчик; возникающий
при приложении силы заряд записывается после усиления шлейфовым или
катодно-лучевым осциллографом.
Фактором, искажающим результат измерения в манометрической бом-
бе, является охлаждение газов горения вследствие теплоотдачи стенкам
бомбы. Правда, при больших средних давлениях, т. е. при малых временах
горения, теплоотдача сравнительно невелика и может быть приближенно
учтена; однако при малых средних давлениях ее влияние значительно.
Опытами в манометрической бомбе в принципе можно определить так-
же скорость горения и ее зависимость от давления. Однако такое опреде-
ление значительно менее надежно, чем измерение давления.
О величине скорости горения опыт в бомбе позволяет судить лишь кос-
венно — по величине давления газообразных продуктов горения, предпо-
лагая определенную зависимость между этим давлением и количеством
сгоревшего вещества.
Это суждение, однако, является вполне надежным лишь ври выполне-
нии ряда предпосылок.
I. Если воспламенение происходит мгновенно и одновременно по всей
поверхности заряда. Это условие невыполнимо. Особенно при малых дав-
лениях, развиваемых воспламенительным зарядом, имеет место относи -
тсльно большая задержка воспламенения, а также и неодповремеппость
его. При больших давлениях воспламенителя начальный участок кривой
давления отражает одновременное горение и заряда и воспламенителя.
2. Если состав газообразных продуктов горения и их температура не-
изменны при всех давлениях. Это условие не выполняется как при низких
давлениях, когда состав газов не соответствует равновесному (азот полу-
чается при этом главным образом в виде NO), так и при повышенных дав-
лениях, когда равновесие между отдельными компонентами продуктов го-
рения может смещаться в зависимости от давления.
3. Если выполняется так называемый геометрический закон горения,
т. е. если горение идет по всей поверхности нормально к ней и с постоян-
ной скоростью. Этот закон также не выполняется, особенно в тех случаях,
когда при горении газы со значительной скоростью движутся вдоль горя-
щей поверхности.
102
4. Если зависимость скорости горения от давления во всем исследуемом
интервале одна и та ясе. Опыт показывает, что для широких интервалов
давления это условие такясе не выполняется.
Кроме того, поскольку процесс идет не при постоянном, а при возра-
стающем давлении, сгорание некоторого (прогретого) слоя в начале горе-
ния моясет протекать в зависимости от условий воспламенения с большей
пли .меньшей скоростью, чем это соответствует стационарному горению при
данном давлении. Далее, поскольку давление при горении возрастает, тол-
щина прогретого слоя меняется, что такясе отражается в принципе на ве-
личине скорости горения. Наконец, сгорание в конце по той же причине
неизбежно является нестационарным.
Все эти обстоятельства влияют на скорость горения, причем в тем
большей степени, чем меньше толщина пороховых элементов. Особенно
большое значение описанные факторы получают при изучении горения под
умеренным давлением. Сам Вьель, описывая свой метод, отмечает несовер-
шенство сведений о горении, получаемых из опытов в манометрической
бомбе, а также отсутствие методов прямого определения скорости горения
нри высоких давлениях.
2. ПРИБОРЫ ПОСТОЯННОГО ДАВЛЕНИЯ
Учитывая рассмотренные выше соображения, целесообразно было бы
разработать способы исследования горения при постоянном давлении тем
более, что в реактивном огнестрельном оружии, сменяющем ствольное,
обычным режимом является именно горение при постоянном давлении.
Методически, однако, изучать процесс в этих условиях более сложно, так
как нельзя использовать, как в манометрической бомбе, изменение давле-
ния в качестве критерия хода горения; при этом необходимо было преду-
смотреть возможность проведения опытов при различных температурах;
желательной также была возможность непосредственного наблюдения про-
цесса.
Скорость горения при постоянном давлении можно определить, осу-
ществляя трение заряда в камере с соплом. Если поверхность заряда по-
стоянна, то давление на протяжении горения почти не меняется. В этом
случае линейная скорость горения может быть рассчитана как отношение
полутолщины стенки (толщины свода) пороховой трубки ко времени го-
рения. Преимуществом метода определения является близость условий
горения к условиям реального применения, недостатком — необходимость
готовить сравнительно большие образчики цороха. Кроме того, при опыте
трудно исключить влияние некоторых побочных факторов (веодновремен-
ность воспламенения заряда ио всей поверхности, подъем давления, обус-
ловленный сгоранием воспламенителя, движение газов вдоль горящей по-
верхности и др.). Более простым в лабораторном выполнении и не требу-
ющим больших количеств пороха является определение скорости горения
при постоянном давления цилиндрического бронированного с боровой по-
верхности заряда, поджигаемого с торца, с регистрацией времени горении
участка определенной длины пли перемещения зоны горения во времени.
Первый прибор, разработанный дчя этих целен Варга [114], представлял
собой стеклянную трубку диаметром около 30 мм, запаянную снизу. Труб-
ка имеет в верхней части два боковых отвода, Один из них соединяет труб-
ку с .манометром, другой —с емкостью большого объема, в которую при го-
рении поступают газы, благодаря чему в трубке сохраняется практически
постоянное давление. Сверху трубка закрывается резиновой пробкой, че-
рез которую проходит тонкая, запаянная снизу стеклянная трубочка для
термопары и вторая трубочка для проводников тока, заканчивающаяся вос-
пламенительной спиралью из тонкой проволоки. На этой второй трубочке
укрепляют столбик пороха пли ВВ обычно так, чтобы он не касался боко-
103
вой своей поверхностью трубки-удержа теля во избежание дополнительных
теплопотерь за счет теплоотдачи трубке-держателю.
Если исследуемое вещество жидкое или порошкообразное, то оно по-
мещается в стеклянную трубочку малого диаметра; если вещество способ-
но сохранять свою форму, то
его применяют в виде цилин-
дрической шашкц, защищен-
ной с боковой поверхности
какой-либо негорючей или
трудногорючей изоляцией,
чтобы воспрепятствовать пе-
реходу горения с торца на
боковую поверхность; особен-
но существенно это при го-
рении в атмосфере воздуха.
Температуру ВВ можно
регулировать, погружая труб-
ку па достаточном ее протя-
жении в термостат обычного
типа плц в сосуд Дьюара.
Давление, при котором
должен быть проведен опыт,
устанавливают, либо откачи-
вая частично воздух из сис-
темы, либо впуская в нее воз-
дух или лучше инертный газ.
Недостатком этого вари-
анта прибора является то,
что резиновая пробка, зак-
рывающая трубку, пе дер-
жится в пей уже при давле-
нии 2—2,5 ат. В то же время
сама стеклянная трубка вы-
держивает значительно боль-
шее давление и разрывается
только при 70—80 ат. По
этим причинам был принят
другой вариант (рис. 77).
Металлическую трубку с
внутренним диаметром, не-
сколько превышающим на-
ружный диаметр стеклянной
трубки, соединяли с ней при
Рис. 77, Схема прибора для изучения горения
при умерснно-повышеппых давлениях
1 -- стальная трубка; 2 — навиптяая крышка; 3 — ма-
нометр; < — стеклянная трубка; 5 — емкость; й —
Стеревпи-алектропровстдккки; Г — держатель шашки;
J — внутренняя стеклянная трубка; 5—пороховая
шашка; J0— игла; 11— спираль для воспламенения;
12 — менделеевская замазка; 13 — распределительная
коробка; 14 л 15 — вентили для соединения с вану-
ум-яасосом и газобаллоном; 16—штуцер для соеди-
нения системы с атмосферой
помощи менделеевской за-
мазки. Сверху металлическую трубку закрывали навинтной крышкой с
проходящим через нее изолированным стержнем, на котором и закреп-
ляли столбик испытуемого ВВ. Металлическая трубка имела два боковых
отвода — для соединения с манометром и с емкостью. В такой системе
было проведено большое число опытов при давлениях около 20 ат, причем
разрывы стеклянной трубки были нечастым явлением, особенно если при-
менять промежуточную стеклянную трубку для смягчения разогрева
газообразными продуктами горения наружной стеклянной трубки.
При медленно горящих веществах время горения можно измерять се-
кундомером. При малых временах горения и если оно не образует непро-
зрачных продуктов, определяют общее время свечения, фотографируя го-
рящий столбик с торца. При прозрачной оболочке столбика можно фото-
графировать перемещение светящегося фронта горения. В обоих случаях
104
фотографирование производится па движущейся с определенной скоростью
фотопленке. Второй способ предпочтительнее, так как дает возможность
не только установить общее время горения, но и количественно показать
изменение но времени скорости горения, если таковое имеет место.
В некоторых случаях, если свечение фронта горения слишком слабое
или образуются непрозрачные продукты, фотографирование свечения мо-
жет не дать ясной картины распространения горепия. Уменьшить погло-
щение свечения непрозрачными продуктами горения можно, если поме-
стить между зарядом и стенками прозрачной трубки прозрачную призму с
плоскопараллельными стенками, которая предотвращает попадание непро-
зрачных продуктов в пространство между столбиком ВВ и объективом фо-
тоустановкп. В таких случаях применим теневой способ фотографирования
Рпс. 78. Схема устаповки для теневого фо-
то гр а фиче ско го способа определения ско-
рости горения
Рис. 79. Фотография горепия
пикрата калия
1 источник света; s — цилиндрик; J — 1цсль;
4 -- объектна; 5 — барабан с пленкой
горения [115] (рис. 78). Столбик ВВ освещается узкой интенсивной поло-
сой света от источника 1 и на движущейся фотопленке регистрируется
изображение этой полосы, ширина которой растет соответственно сгоранию
столбика и уменьшению высоты образуемой им тени (см. фото дли пикра-
та калия, рис. 79). Еще более целесообразно, по сложнее в выполнении и
поэтому мало использовалось, фотографирование пере'Мещенпя фронта го-
рения в отраженном свете.
При всех этих вариантах применима также кадровая киносъемка горя-
щего столбика с определенным интервалом между кадрами. Уменьшение
высоты столбика позволяет рассчитать скорость горения. Наконец, воз-
можна фиксация времени горения при помощи двух тонких проволок из
соответствующего металла, проходящих через столбик ВВ на определен-
ном расстоянии друг от друга и расплавляющихся при горении [116]. Мо-
менты разрыва проволочек фиксируются по прекращен ию тока, проходяще-
го через каждую из них, каким-либо из многих существующих для этой
цели способов. Этот метод дает, естественно, лишь среднюю скорость горе-
ния; кроме того, при быстром горении, особенно в тех случаях, когда
температура на поверхности раздела конденсированной фазы и первичных
продуктов горения низка, не исключено заметное и неравномерное запаз-
дывание переплавления проволочек. Преимуществом способа, особенно су-
щественным если опыт проводят при повышенной или пониженной темпе-
ратурах, является возможность работать в непрозрачном сосуде.
Поскольку, однако, в большинстве случаев фотографический способ ре-
гистрации распространения горения заслуживает предпочтения, то наряду
с прибором, в котором горепне протекает в стеклянной трубке, т. е. при
давлениях, не превосходящих нескольких десятков атмосфер, был разра-
ботан ряд конструкций бомбы постоянного давления в виде стального
сосуда, снабженного одним или несколькими окошечками из органическо-
го дли неорганического стекла. Бомба имеет достаточно большой объем
или же соединяется с емкостью достаточных размеров для того, чтобы по-
вышение давления при опыте не превышало заданной величины; соответ-
ственно выбирается также сечение трубки, соединяющей бомбу с емко-
стью. Первая из советских моделей бомбы была сконструирована Баке-
евым [117].
105
фотографирование производится на движущейся с определенной скоростью
фотопленке. Второй способ предпочтительнее, так как дает возможность
не только установить общее времн горения, но и количественно показать
изменение во времени скорости горения, если таковое имеет место.
В некоторых случаях, если свечение фронта горении слишком слабое
или образуются непрозрачные продукты, фотографирование свечопия мо-
жет ио дать ясной картины распространения горения. Уменьшить погло-
щение свечения непрозрачными продуктами горения моцщо, если поме-
стить между зарядом и стенками прозрачной трубки прозрачную призму с
плоскопараллельными стенками, которая предотвращает попадание непро-
зрачных продуктов в пространство между столбиком ВВ и объективом фо-
тоустаповкн. В таких случаях применим теневой способ фотографирования
Рис. 78. Схема установки для теневого фо- Рис, 79. Фотография горения
тографйческото способа определения ско- пикрата калия
рости горения
I — источник света; 2 — цилиндрпн; 5 — щель;
< — объектив; 5 — Оарабаи с пленкой
горепия [115] (рис. 78). Столбик ВВ освещается узкой интенсивной поло-
сой света от источника 1 и на движущейся фотопленке регистрируется
изображение этой полосы, ширина которой растет соответственно сгоранию
столбика и уменьшению высоты образуемой нм тени (см. фото для пикра-
та калия, рис. 79). Еще более целесообразно, но сложнее в выполнении я
поэтому мало использовалось, фотографирование перемещения фронта го-
рения в отраженном свете.
При всех этих вариантах применима также кадровая киносъемка горя-
щего столбика с определенным интервалом между кадрами. Уменьшение
высоты столбика позволяет рассчитать скорость горения. Наконец, воз-
можна фиксация времени горения при помощи двух тонких проволок из
соответствующего металла, проходящих через столбик ВВ на определен-
ном расстоянии друг от друга и расплавляющихся при горения [116]. Мо-
менты разрыва проволочек фиксируются по прекращен ию тока, проходяще-
го через каждую из них, каким-либо .из многих .существующих для этой
цели способов. Этот метод дает, естественно, лишь среднюю скорость горе-
ния; кроме того, при быстром горении, особенно в тех случаях, когда
температура на поверхности раздела конденсированной фазы и первичных
продуктов горения низка, не исключено заметное и неравномерное запаз-
дывание псреплавления проволочек. Преимуществом способа, особенно су-
щественным если опыт проводят при повышенной или пониженной темпе-
ратурах, является возможность работать в непрозрачном сосуде.
Поскольку, однако, в большинстве случаев фотографттческий способ ре-
гистрации распространения горения заслуживает предпочтения, то наряду
с прибором, в котором горение протекает в стеклянной трубке, т. с. при
давлениях, нс превосходящих нескольких десятков атмосфер, был разра-
ботан ряд конструкций бомбы постоянного давления в виде стального
сосуда, снабженного одним или несколькими окошечками из органическо-
го или неорганического стекла. Бомба имеет достаточно большой объем
или же соединяется с емкостью достаточных размеров для того, чтобы по-
вышение давления при опыте пе превышало заданной величины; соответ-
ственно выбирается также сечение трубки, соединяющей бомбу с емко-
стью. Первая из советских моделей бомбы была сконструирована Баке-
евым [117].
105
Бомба, сконструированная в Институте химической физики АН СССР,
представляет собой толстостенный стальной стакан внутренним диамет-
ром 10 см и объемом 1 л, прочность которого позволяет проводить опыты
прй давлениях до 150 ат. В цилиндрической части бомба имеет два плек-
сигласовых окна, расположенных друг против друга. Сверху бомба закры-
вается герметически при помощи резиновой прокладки стальной крышкой
(рис. 80), имеющей 10 отверстий, которыми крышка надевается на болты,
закрепленные в корпусе бомбы; болты имеют нарезку, на нее навинчива-
ются гайки, прижимающие крышку к торцу бомбы. На крышке бомбы
смонтировано несколько электровводов, используемых для поджигания за-
ряда, измерения температуры и т. д. На ней имеется также вентиль, сое-
диненный с манометром и служащий для наполнения бомбы газом и вы-
пуска газов горения после опыта. На нижней стороне крышки укреплены
дна стержня; к ним присоединена подставка, в центре которой устанавли-
вается образчик ВВ. Поджигающая спираль закреплена на пружинке, рас-
положенной в верхней частя бомбы, и при помощи нитки подтягивается к
торцу ааряда. При воспламенении ВВ нитка перегорает и спираль отво-
дится пружинкой от горящей поверхности. Еслп этого не делать, спираль
Рис. 80. Схема бомбы с приспособле-
ниями для поджигания пороха
.4 - корпус бомбы; Б — крышка
оомбы; Й — подставка для испы-
туемого абразив; Г — образлиц
ВВ; Ь’ — изолированные электро-
вводы
Рис. 81. Схема бомбы постоянного давле-
ния на 350 ат
I — корпус; 2 — крышка с уплотняющим коль-
цом; з—прижимное кольцо; 4— обна для ре-
гистрации процесса горения; 5 — окна для Под-
светки; s — Столик для образца; 7 — зарнд
ВВ; J — изолированные электровводы; 9 —
Спираль из нихромовой проволоки для воспла-
менения; ц) и И — вводы для соединении с
компрессором и нейтилеи для сброса газа па
боМоЫ после опыта
10G
может сильно накаливаться в результате каталитического догорания на
ней промежуточных продуктов и это может влиять на процесс горения и
особенно на состав его продуктов.
Бомба постоянного давления другой конструкции [118], позволяющая
проводить опыты при давлениях до 350 ат, изображена на рис. 81. Корпус
бомбы представляет собою толстостенный стальной сосуд емкостью 3 л
с окнами из органического стекла. Через одно из окон регистрируется
процесс горения; остальные служат для подсветки при фотографировании
горения или при наводке фоторегистра на фокус.
На крышке бомбы смонтирован столик. В крышке
закреплены токопроводящие свечи, изолированные '
от корпуса бомбы. Цилиндрический заряд испы-
туемого ВВ в прозрачной трубочке помещается в
специальном держателе (рис. 82), установленном
в центре столика. Держатель имеет тонкие гори-
эортальные полоски, приходящиеся против сред-
ней части заряда, расстояние между которыми
можно точно измерить. Эти полоски дают тень на
снимке и позволяют определить высоту той части
заряда, которая находится между ними.
Для опытов при повышенных температурах в
бомбе может быть установлен термостат. Этот тер-
мостат (рис. 83) представляет собой массивный
цилиндр из красной меди, нагреваемый током и
хорошо термоизолированный. Температуру цилин-
дра намеряют и контролируют двумя термопарами;
ее поддерживают постоянной при помощи автома- Ряс. 82. Трафарет для
тпческого электронного регулятора с регистрирую- испытуемого образчика
щим устройством. Термостат имеет глубокий ка-
нал по оси для помещения заряда и две узкие прорези, которые при за-
крытой крышке бомбы располагаются против ее окон. Прорези плотно за-
крыты стеклами для уменьшения теплообмена за счет конвекция. Высота
термостата, а также расположение нагревательных элементов обеспечи-
вают практически постоянную температуру заряда по всей его длине.
Определение скорости горения при постоянном давлении производи-
лось и при значительно более высоких давлениях — до 10 000 ат [119].
Следует указать, что различие монаду бомбой возрастающего давления
и бомбой постоянного давления больше относится к методике работы, чем
к устройству бомбы. По существу это различие заключается в величине
плотности заряжания, при которой ставится опыт. Если она велика, то
давление растет и этот рост может быть использован для измерения вре-
мени горения, по которому может быть рассчитана скорость горения.
Именно так определялась средняя скорость горения в опытах А. П. Глаз-
ковой но горению аммиачной селитры п ее смесей с различными катализа-
торами. Аналогичный прием применили Беляев и сотр. [120], которые из-
меряли среднюю скорость горения некоторых ВВ, используя комбиниро-
ванные заряды из последовательно расположенных столбиков разных
веществ. Кривая записи давление — время имела при Этом форму лестни-
цы (рис. 84), по ширине ступенек которой можно было определить время
горения каждого столбика при некотором среднем давлении, приближенно
соответствующем середине высоты ступеньки. ГО. А. Корпин, изменяя по-
верхтшеть охлаждения в манометрической бомбе в соответствии со скоро-
стью тепловыделения, осуществлял в ней горение при практически по-
стоянном давлении; однако в этом случае необходимо обеспечить фикси-
рование момента окончания горения, что связано с известными трудно-
стями.
107
При исследовании горения под низкими давлениями иногда необходи-
мо определение объема и состава образующихся газов. Для этой цели
Сарро и Вьель [121] применяли следующий метод. Взрывчатое вещество
помещали в стеклянную трубочку (d — 4 мм), соединенную с резиновым
баллоном, в который при горении поступали образующиеся газы. Соеди-
нптельную стеклянную трубку перед опытом наполняли углекислотой,
объем которой учитывали
Рнс.
ведения
давлении и
при последующем анализе газов.
Во многих работах определение объема
газов производилось следующим образом, На-
веску ВВ помещали в круглодонную колбу,
которая наполнялась инертным газом для
предотвращения возможного участия возду-
ха в реакции горения и последующего взаи-
Рпс. 81. Кривая р (т) при горении ком-
эиниропа иного заряда, включающего
три цилиндрика тротила (7, 2, 3) из-
вестной длпцьг и четыре цилиндрика
вспомогательного вещества с большим
значением dp/dx
83. Схема термостата для
опытен ври постоянном
повышенных темпера-
турах,
J ™ блок ьз красной меди; 2 — цилинд-
рический канал, r который помещается
держатель с нарядом; 5 — щелеыщные
прорези; 4 — рамка; 5 — стекло; — на-
гревающая обмотка с изоляцией; 7 — ко-
жух
про-
модействия кислорода с окисью азота. Перед опытом в колбе создавалось
некоторое разрежение с таким расчетом, чтобы среднее давление при го-
рении соответствовало желаемому. Взрывчатое вещество поджигали спи-
ралью. После охлаждения газов горения до температуры окружающего
воздуха измеряли их давление и по обычным формулам рассчитывали
объем неконденсирующнхся газов.
Для определения состава газы собирались в приборе, показанном на
рис. 85. В пижнюю часть стеклянной трубки 1 диаметром около 15 лш
помещали трубочку 2 с ВВ, Трубка 1 соединена троЙшиком 5 с сосудом 3,
наполненным ртутью и соединенным резиновой трубкой с уравнительной
склянкой 4. При зажигании ВВ газы выходят на воздух, промывая от
108
пего систему. После того как сгорит некоторое количество ВВ, колено 6
тройника закрывается, и газы поступают в сосуд при одновременном опу-
скании уравнительной склянки. Недостатком метода является то, что его
трудно применить для собирания газов при горении под повышенными
постоянными давлениями. Другой вариант схемы прибора, примененного
для зтон дели, описан в работе [122].
Для определения состава и количества газообразных продуктов горе-
ния, в первую очередь соотношения между отдельными их компонентами,
можно применять и бомбу постоянного давления, особеппо если ипертггый
газ, который используется для создания давления иной, чем то газы, ко-
торые образуются при горении.
Определение состава газов обычно производится с некоторыми допол-
нениями тем методом, который имеет широкое применение для анализа
печных газов. Газы последовательно пропускают через поглотительные
склянки с соответствующими реагентами: 1) для поглощения углекислоты
концентрированный (60%-ный) раствор едкого калия; 2) для поглощения
окиси азота раствор сернокислого железа (закисного), подкисленный сер-
ной кислотой; 3) для поглощения кислорода щелочной раствор пирогалло-
ла; 4) для поглощения окиси углерода аммиачный раствор полухлор истой
меди. После поглощения паров аммиака остаточные газы смеогнваются с
кислородом пли воздухом и поступают па дожигание над платиновой спи-
ралью. Количество метана определяли ко количеству образовавшейся
углекислоты, количество водорода — по уменьшению объема. После дожи-
гания вновь определяли количество кислорода, чтобы убедиться в отсут-
ствии закиси азота, а также других углеводородов, кроме метана. Азот
рассчитывали по разности.
Двуокиси азота обычно образуется очень мало, и она не определяется;
в случае необходимости двуокись азота можно определить колориметриче-
ски или другим способом в жидкости первой склянки, которая поглощает
NOa вместе с углекислотой.
Наиболее надежным методом определения закиси азота, поскольку уп-
ругость се паров при низких температурах сильно отличается от упругости
паров других (мжелов азота, олеме псарного азота и СО, являете в дробное
вымораживание. После отделения NaO от других газов можно произвести
сожжепие ее в атмосфере водорода или поглощение в Этиловом спирте.
II. ПРЕДЕЛЬНЫЕ УСЛОВИЯ ГОРЕПИЯ
Простейшим случаем горения является одномерное его распростране-
ние в удлиненном, например, цилиндрическом заряде, зажженном с торца.
Опыт показывает, что такое горение возможно лишь, если диаметр заряда
превышает некоторую величину — критический диаметр.
Критический диаметр обычно определяют как диаметр затухания горе-
ния слегка суживающегося заряда, подожженного с широкого конца.
Можно ставить опыт также в телескопических трубках с переходными
участками в виде усеченного конуса. Критический диаметр может быть
определен также последовательными опытами с цилиндрическими уши-
ряющимися с поджигаемого конца зарядами различных диаметров. При
проведении опытов при различных давлениях целесообразно помещать
заряды в жидкость (например в воду), чтобы обеспечить постоянство
температуропроводности окружающей среды.
Критический диаметр зависит пе только от природы вещества, по и от
условий трения, в первую очередь от давления и начальной температуры,
а для порошкообразных ВВ также от кубической плотности. В табл. 13
приведены скорости горения ряда ВВ при низких давлениях п критиче-
ские условия горения для тех ВВ, для которых они были определены.
109
Таблица 13
Значения скоростей и критических диаметров горения * для ряда ВВ
при атмосферном давлении (на воздухе)
Вещество ILiot- новть, Критический диаметр го- рения, М.И Скорость ГОрСН (IH , Примечание
Тротил...............
Тетрил...............
Гексоген v ..........
Тан..................
Дина...............
При несколько повышенных
давлениях тэн при этом
диаметре не горел
0,051
0,060
0,14
0,030
0,012
Порох Ц ...........
Ннтрог.’пптрир . ,
Нитрог.ишоль , . .
ДпглИ|-;ольД[[1[1[Трат
Перхлорат аммония
Приведенное значение сеп-
тического диаметра отно-
сится к токкодиеперсному
продукту заводского из-
готовления; измельчен-
ной в ступке уссстый пер-
хлорат не горел даже при
диаметре заряда 40 -и,ч
* В той случае, когда в графе дли критического диаметра приведенье два числа, мстц-шсе и<
htlx попадает наибольший диаметр цилиндрического заряда, при котором горспне не распростра-
няется, большее — наименьший диаметр, црц котором око распространяется. Есин приведено
<>дпо число, оно означает диаметр затухания конического заряда.
Существование критических условий горения обусловлено следующи-
ми причинами. Распространение горения происходит за счет передачи
тепла, выделяемого экзотермическими реакциями, происходящими и реа-
гирующем слое, прилегающему слою ВВ, Это тепло нагревает слой ВВ,
вступающий в реакцию до такой температуры, начиная с которой скорость
экзотермической реакции достаточно велика, чтобы теплоприход компен-
сировал теплопотери и скорость реакции самостоятельно возрастала. Од-
нако имеют место теплопотери от прогретого слоя как на нагрев следую-
щего слоя ВВ, так и в окружающую среду. Если температура, до которой
нагревается вступающий в реакцию слой недостаточна, то скорость реак-
ции,. а соответственно и теплоприход меньше, и последний не может
компенсировать теплоотвода. Тогда температура слоя не может возрасти
за счет развивающейся вследствие саморазогрева реакции до максималь-
ной температуры горения, а, пройдя через некоторый менявши максимум,
падает. Следующий слой получает еще меньше тепла, максимум темпера-
туры в нем оказывается еще ниже и т, д. Таким образом, в результате
тенлопотерь горение быстро прекращается.
Все факторы, которые затрудняют теплоотвод в окружающую среду,
а также, как правило, и в глубь ВВ, благоприятствуют горению. Точно
так же способствуют ему те факторы, которые увеличивают скорость хими-
ческих реакций, выделяющих терло,
В свете этих соображений становится понятным влияние на возмож-
ность горения диаметра заряда, скорости горения, давления, начальной
температуры ц кубической плотности, а также характеристик его обо-
лочки,
110
Влияние диаметра и других параметров на возможность горения объя-
сняется в принципе так же, как их влияние на тепловое самовоспламене-
ние, возникающее при равномерном нагреве ВВ. В последнем случае теп-
лоприход пропорционален кубу диаметра, теплоотвод при конвективном
теплообмене — его квадрату; при кондуктивном теплоотводе его величина
растет с диаметром медленнее; в критерий Франк — Каменецкого диаметр
входит во второй степени — при увеличении диаметра возрастает путь
тепла от места его выделения до стенки,
Причины существования критического диаметра горения в принципе
сходны также и с теми, которые определяют существование критического
диаметра детонации, Возможность распространения последней обусловли-
вается соотношением скорости выделения энергии и скорости ее отвода
в окружающую среду. Это сотношение определяет величину достигаемого
давления, от которого зависит скорость детонационного превращения.
Опять-так н количество выделяемой энергии пропорционально объему
заряда, количество энергии, теряемой в окружающую среду, зависит от
относительной поверхности заряда. Основное различие состоит в том, что
при детонации скорость превращения весьма сильно возрастает с увеличе-
нием давления я соответственно снижается «потерями» (падением) давле-
ния. Теплоотвод же из-за большой скорости процесса не играет реальной
роли. В этом смысле понятие критического диаметра детонации более
сложно; оно появилось в науке о взрывчатых веществах позже (1940 г.),
чем понятие критического диаметра горения (1934 г.). Естественно, что
критический диаметр детонации в отличие от критического диаметра горе-
ния не зависит от внешнего давления — давление, достигаемое при дето-
национном превращении, иа несколько порядков выше тех давлений, кото-
рые можно реализовать при эксперименте.
В свете роли диаметра, как фактора, определяющего величину тепло-
нотерь, естественно, что величина критического диаметра, вообще говоря,
тем меньше, чем больше скорость горения. При большей скорости горения
доля теплоты, Отдаваемая окружающей среде, меньше (при прочих рав-
ных условия^). ’
Помимо величины скорости горения, необходимо, однако, учитывать
характер горения. Если речь идет о летучем веществе вроде нптрогликоля,
у которого конденсированная фаза прогревается только до температуры
кипения, то потери тепла за Счет теплопроводности жидкости малы, и кри-
тический диаметр мал (1 м.ч), несмотря на малую скорость горения
(0,03 см/сек). Если горение сопровождается значительным прогревом кон-
денсированной фазы, как при горении бездымного пороха, то из-за боль-
ших тепловых потерь критический диаметр гораздо больше, несмотря на
большую скорость тореиия.
Большой критический диаметр горения обусловлен в данном случае
потерями тепла и это можно показать на примере нитроглицеринового по-
роха, который в асбестовой обмотке горит лишь при диаметре не меньше
20 .ил, а, будучи измельчен в порошок, притом относительно грубый, го-
рит в стеклянной трубке даже при вдвое меньшем диаметре. Но той же
причине пироксилин горит в стеклянных трубках диаметром около 5 мм,
а пироксилиновый порох даже при очень малом содержании летучпх
не горит при значительно больших диаметрах.
Влияние давления на возможность горения объясняется следующим
образом.
Повышение давления, увеличивая скорость горения, уменьшает отно-
сительные теплопотери и соответственно критический диаметр. Аналогич-
но влияет начальная температура, ее повышение увеличивает скорость
горения и, кроме того, уменьшает перепад температуры между ВВ и окру-
жающей средой; по этим причинам относительные теплопотери при горе-
Н[1ц уменьшаются.
nt
Для порошкообразных ВВ величина критического диаметра зависит
также от кубической плотности. Повышение кубической плотности может
как облегчать горение, т. е. уменьшать критический диаметр (плавящиеся
ВВ, например тетрил), так и затруднять его, т. е. увеличивать критиче-
ский диаметр (пеплавящиеся ВВ, например нитроклетчатка). С первого
взгляда противоположность влияния плотности в обоих случаях парадок-
сальна. Мы увидим, однако, ниже (стр. 190), что в данном случае оно мо-
жет быть объяснено простыми соображениями относительно влияния плот-
ности иа теплоотвод.
На возможность горения влияет также оболочка заряда — теплопро-
водность ее материала и толщниа стенок. Для материалов со значитель-
ной теплопроводностью (стекло, металлы и т. п.) при большей толщине
стенки трубки минимальный диаметр больше. Объясняется ото тем, что
при горении на воздухе величина теплопотерь определяется в основном
коэффициентами перехода тепла от ВВ и продуктов горения к трубке,
теплопроводностью и толщиной стенок ее. Теплоотдача от трубки к возду-
ху относительно незначительна так же, как и теплопроводность и теплоем-
кость воздуха.
Проще всего влияние толщины стенки может быть иллюстрировано
следующим опытом. Узкая трубочка с нитрогликолем или гексогеном по-
гружается нижней своей половиной в воду. Горение, нормально распро-
страняющееся в верхней части трубочки, прекращается, дойдя до уровня,
соответствующего уровню воды вне трубочки. Если взять вещество, горя-
щее более быстро, например метиллитрат, то вода но успевает принимать
участие в теплообмене и горение не затухает, а продолжается, причем
скорость его лишь незначительно меньше скорости горения верхнего
участка.
Более детально влияние толщины стенки изучалось на примере горе-
ния гексогена в гипсовых трубках (табл. 14). Было установлено, что при
слишком большой толщине стенки трубки (внутренний диаметр ее был
постоянным — 7,8 льм) горение затухает. Если толщина стедки меньше
4 мм, оно распространяется до конца заряда. При этом, в случае бо-
лее толстой стенки, скорость горения несколько меньше. Если вещество
предварительно подогрето, то горение возможно при большей толщине
стенки.
Т а б л и я а 14
Влияние толщины стенки гипсовой трубки на горение гексогена
Толщина степь'11, I,8 2,2 4,0 3,9 5,7 5,8 3,9 10,3 20,8 20,8 + 8 10,3 | 20,8 1
Л писиная скорость горения, си/сек 0,066 0,067 0,055 Горение затухло 0,052 Горение аатухло
Примечание. Кубическая плотность ijo.Te6a.incb в продолах 0,97-1,04 ?]см\
температура вещество в первых десяти опытах составляла -|-20,5’С, в [ioc.ie.iiiах
трех опытах +85° С; внешнее давление 7л8 ч,и, внутренний диаметр трубки во всех
Опытах 7,8 мм.
Аналогично ведет себя тротил. В стеклянных трубках горение распро-
страняется при диаметре 27 мм и выше; обмотка трубки асбестовым шну-
ром несколько увеличивает скорость горения.
В бронзовых трубках диаметром 30 мм при толщине стопки 1; 5; 8 и
10 мм горение не распространяется. Если в трубке с толщиной стопки
1 мм уменьшить теплоотвод в воздух, обмотав ее асбестовым шнуром, то
горщгие становится возможным, причем скорость его (0,015 с.м/сек) за-
112
метно больше, чем в стеклянных трубках (0,011 см/сек). При этом горение
к концу своему ускоряется и становится неравномерным, очевидно, вслед-
ствие прогрева ВВ в нижней части трубки за счет передачи тепла
по ее стенкам. В более толстостенных (5, 8 и 10 мм) бронзовых трубках
горение не распространяется также и при наличии обмотки асбестовым
шнуром.
Приведенные соображения и примеры поясняют главным образом за-
трудняющее влияние оболочки заряда на горение. Оболочка, однако, может
иметь и обратное влияние. При не слитком большой относительной тол-
щине стенок оболочки, большой теплопроводности ее материала и малой
скорости горения тепло от продуктов горения может передаваться В В по
стенкам трубки быстрее, чем через фронт горения, и это влияние может
преобладать над влиянием потерь тепла на нагрев стенок и окружающей
трубку среда. Наиболее наглядно это влияние проявляется, если в стек-
лянную трубку с медленно горящим ВВ — ннтросликолем — ввести стер-
жень небольшого диаметра из металла с большей теплопроводностью
(красная медь), скорость горения жидкости при этом сильно возрастает.
Несомиеиио, что причиной этого увеличения скорости горения является
доиолпительиый подвод тепла к жидкости по медному стержню. Ту же
роль — дополнительного подвода тепла — могут играть и тонкие стенки
трубки, в которой идет горение, если они сделаны из материала большой
теплопроводности. Действительно, в тонкостенных металлических трубках
скорость горения оказывается выше, чем, например, в стеклянных. Так,
гремучий студень в латулиых трубках (d = 28 ли, s — 1 лик, I — 5 см)
горел со средней скоростью 0,195 см1сек, в то время как в стеклянных
трубках скорость горения составляла только 0,142 с.и/сек. Более того,
в латунных трубках горение имело ускоряющийся характер — средняя
скорость при длине трубки 24 см увеличивалась до 0,340 см/сек.
При опытах с гремучей ртутью, запрессованной до большой плотности
в краспомедную гильзу кач сю ля-детонатора, наблюдался переход горения
в детонацию, которого не происходило, если В В запрессовывалось непо-
средственно и толстостенную стальную матрицу. Очевидной причиной
возникновения детонации в первом случае был разогрев тепловой волной,
быстро распространяющейся по стопкам гильзы.
Для тэна, горящего относительно медленно, ускорение горелия вслед-
ствие распространения тепла по стенкам трубки наблюдалось даже в стек-
лянных трубках: в трубках меньшего диаметра горение шло быстрее
[123].
Если вести горение питрогликоля в одном из колея сообщающихся
стеклянных сосудов, поддерживая уровень горящей жидкости постоян-
ным, то из-за усиленного подвода тепла по стенкам скорость горения
сильно увеличивается.
То же наблюдали Адамс и Стокс [124] при горении гидразина.
96,7%-ный пидразип горел iipir атмосферном давлении в азоте (диаметр
трубки 3 л<л() со скоростью 0,031, а при диаметре 5 и 10 мм —со скоро-
стью 0,027 CMjce.K. Авторы связывают это влияние диаметра трубки с
зависимостью от него площадки поверхности газофазного пламени, кото-
рая была относительно больше в узких трубках. С этим объяснением
можно согласиться, уточняя, что увеличение относительной поверхности
пламени является не причиной увеличения скорости горения, а следствием
того, что за счет усиления теплоподвода по стенкам увеличивается ско-
рость парообразования. Чтобы большее количество паров успело сгорать,
естественно, поверхность пламени должна стать больше, подобно тому,
как это происходит в горелке Бунзена при увеличении скорости поступ-
ления газа,
Попятно, что описанные эффекты могут иметь место только при малой
скорости горепия, когда скорость распространения тепловой волны по
8 К. К. Андреев
113
материалу оболочки соизмерима со скоростью горения. Поэтому, напри-
мер, при повышении давлении, увеличивающего скорость горения, но нс
изменяющего теплопроводности оболочки, ускоряющее влияние послед-
т же относится и к отрицательному влиянию
оболочки на возможность горения.
Влияние давления на критический диа-
метр горения нитрогликоля изучалось А. Ф.
Гущиной. При давлениях 0,57; 1,0 и 2,0 аг
горение в конических трубках затухало при
достижении диаметра соответственно 1,7; 1,0
и 0,5 мм. Интересно, что произведение давле-
ния на критический диаметр является по-
стоянной величиной. В. Э. Анников опреде-
лил критический диаметр горения литого
тротила в интервале 10—350 ат. ВВ находи-
лось в сужающихся стеклянных трубках, по-
груженных в основной части .в воду при 10° С
и поджигалось с широкого конца трубки в
бомбе постоянного давления. Как видно из
рис. 86, критический диаметр горения составляет при 10 ат ~ 10,5 мм и
уменьшается при 350 ат до 0,2 лш. Н. А. Афонина определяла изменение
критического диаметра горения порошкообразного тетрила при относитець-
ной плотности ~1в зависимости от давления. Критический диаметр силь-
но уменьшается при повышении давления, падая от 21 мм при 1 ат до
~ 1 мм при 30 ат. По И. В. Бабанцеву, в конических трубках из кварце-
вого стекла, погруженных в воду, горение тона (при ₽ = 25 ат) затухает
при 3,8, тротила при 4,6 и тринитробензола при 6,2 мм.
Если учесть, что скорость горения приближенно пропорциональна
давлению, а поверхность теплоотвода диаметру трубки, то полученные для
нитрогликоля и тротила результаты можно истолковать как постоянство
относительных теплопотерь при критическом диаметре. Такого простого
соотношения можно, однако, ожидать лишь при одинаковом типе горения
и постоянстве его тепловых характеристик.
ней будет уменьшаться. То
й
ДО
200
р, кг/см3
Рис. 86. Зависимость критиче-
ского диаметра горении литого
тротила от давления
Таблица 15
Значения критических диаметров и скоростей горения
ряда порохов при атмосферном давлении
Условное обозна-
чение пороха
Критический диаметр
горения *, мм
Скорость горения,
см!сек
1
2
3
4
5
6
Порох Н
1,8;
2,2-
3,5;
4,1;
6,5;
7,3;
9,0;
2,7
3,4
4,5
5,1
7,5
8,1
11 2
0,077
0,120
0,066—0,069
0 103—0 120
0,05,3
0 051
0,060
О
• См. сноску п табл. (3 йа стр. 110.
Г. Н. Беспалов определил критический диаметр горения при атмосфер-
ном давлении ряда образчиков пороха ла нелетучем растворителе, отли-
чавшихся по составу (табл. 15). Значения диаметра различались более
чем в 4 раза, скорости горения (при диаметрах значительно больше кри-
тического) приблизительно в 2 раза. Обнаруживается некоторое соответ-
ствие между скоростью горения и критическим диаметром, одиако
имеется много исключений, показывающих, что по одной скорости горения
114
пороха судить об его способности к горению еще нельзя. Беспалов опре-
делил способность порохов к горению также и при повышенном (20 ат)
давлении. Так как готовить конические заряды очень малых диаметров
трудно, он использовал клиновидные заряды, бронированные перхлорви-
ниловым лаком, горевшие в окружении воды. Скорость горения (табл. 16)
Таблица 16
Значения критических диаметров и скоростей горения
для клиновидных зарядов некоторых порохов при 20 ат
Условное обозна- чение пороха Критически ft диаметр горения, ллс. Скорость горения. C3i/C6K
1 0,18; 0,20 0,64
2 0,43; 0,50 0,43
3 0,39; 0,48 0,44
4 0,32; 0,38 0,73
5 0,63; 0,72 0,47
6 0,20; 0,34 0,22
Порох Н 0,72; 0,80 0,30—0,37
была больше, чем при атмосферном давлении, хотя и в разное (3,6—8,9)
число раз, критическая толщина клина меньше, чем критический диаметр
при атмосферном давлении. Опять-таки наблюдается известное соответ-
ствие между критическим размером и скоростью горения, хотя наблюда-
ются и исключения, особенно в тех случаях, когда состав пороха суще-
ственно отличается от остальных (порох 6, например, горел с наименьшей
скоростью при наименьшем критическом диаметре).
Более отчетливо зависимость критического Диаметра от скорости горе-
ния проявляется в его зависимости от давления. На рис. 87 эта зависи-
мость изображена для двух порохов, Для одного из них рост скорости с
давлением сильно ослабевает в интервале 30—80 аг; “Соответственно почти
постоянным остается в этой области давления и критический диаметр
горения; для другого же пороха скорость растет и dur уменьшается. Кста-
ти, для обоих порохов в области малых давлений (до 30 ат) скорости
горения близки, а значения критического диаметра различаются в не-
сколько раз.
Отметим также, что для пороха 2, определяя занисимость по кривой
(р), можно быстрее и проще оцепить характер зависимости и(р), чем
непосредственно определяя последнюю.
Из сказанного о сущности влияния давления на критический диаметр
горения следует, что, увеличивая диаметр трубки, можно получить горе-
ние при любом сколь угодно малом давлении1. Практически это, по-види-
мому, не так по той причине, что химическое превращение ВВ при горении
на начальных стадиях обычно идет с поглощением тепла и тогда не может
быть самораспростраияющимся. Последующие стадии превращения экзо-
термнчны; одиако, если дааление очень мало, то они практически не
имеют места, и горение становится невозможным. Если повышать давле-
ние, то, начиная с некоторого предела (зависящего также от внешних
условий), процесс становится суммарно экэотермичным и способен к само-
распространению.
Аналогично предельному критическому давлению может существовать
и предельная критическая температура (от которой зависит конечная тем-
1 Если нс учитывать потерь тепла излучением, которые, согласно Я. Б. Зельдови-
чу, определяют существование предела способности к горению в случае газовых
смесей.
8*
f 15
нература начального этапа превращения), особенно, если скорость экзо-
термических реакций сильно зависит от температуры. При более высоких
температурах горение возможно, по лишь при диаметрах, превышающих
критический.
Б. Н. Кукиб изучал влияние температуры на способность к горению
уплотненного гексогена (с 3% дигликольди нитрата для облегчения прес-
сования). Критический диаметр, уменьшающийся, хотя и не очепь сильно,
при повышении температуры, составлял 5,5 мм при +90°С п 6,5 мм при
+15° С. Критический диаметр тетри-
ла, по опытам Н. А. Афониной, так-
же уменьшается при повышении тем-
пературы (рис. 88) — вдвое при
изменении температуры от —20
до 90° С. В этих опытах применялся
порошкообразный тетрил при плотно-
Рис. 87. Зависимость скорости
и критического диаметра горе-
ния от давлении для двух по-
рохов
1 — и(р), I — dnr(p) для пороха I;
II — и(р), 2 — d«r(p) дли поро-
ха II
-20-юз югозоto®zasoзот
TtMfiepfimtjpu. 'С
Рис. 88. Зависимость критическо-
го диаметра горения тетрила
от температуры
1 — тетрил сгорел; 2 — горение за-
тухло
сти, равной —'1, при которой на возможность горения сильно влияет про-
никновение расплава в глубь порошка, роль расплава в свою очередь мо-
жет зависеть от температуры.
При замораживавши жидкого ВВ увеличивается критический диаметр.
Например, желатинированный нитроглицерин горел в трубке диаметром
5,4 .ад; для получения его устойчивого горения и замороженном виде
потребовалось увеличить диаметр трубки до 11 мм. Влияние заморажива-
ния вполне понятно, так как оно, аналогично охлаждению, приводит к
уменьшению скорости горения и его теплоты (а соответственно и темпе-
ратуры газов) па величину скрытой теплоты плавления.
Помимо давления н температуры, на возможность горения влияет еще
ряд факторов. Иногда эти факторы тривиальны. Например, при горении
метилнитрата в стеклянной трубочке, опущенной в воду на ее стенках
конденсируются пары воды, образующейся при горении. Капельки воды,
стекая вниз, попадают на поверхность горящей жидкости п тушат го-
рение.
Менее тривиальным является затухание горения жидкости, например
нитро гликоля при встряхивании трубочки, в которой идет горение. Это
встряхивание нарушает прогретый слой жидкости, примыкающий к ее
поверхности при стационарном горении. Теплоотвод в глубь жидкости
возрастает настолько, что не компенсируется теплочриходом от газооб-
разных продуктов горения, ц это приводит к затуханию горения.
Подобным же образом следует объяснить затухание горения, наблю-
давшееся в известных условиях при пропускании через жидкость пузырь-
116
Рис. 89. Область горения нитроглицерина
при малых давлениях (заштриховано)
ков воздуха. При перемешивании прогретого слоя с холодной жидкостью
(при прохождении пузырька через поверхность) горение прекращается.
Сходные явления могут наблюдаться при горении жидких (или пла-
вящихся) ВВ, если горение переходит с нормального режима па турбу-
лентный, при котором отвод тепла в глубь вещества и в окружающую
среду существенно возрастает. В этом случае может наблюдаться пара-
доксальное явление: вещество при определенном диаметре заряда горит
при низкпх давлениях, по горение затухает, если давление превышает
некоторый предел. Такое явление впервые было установлено для жидкого
нитроглицерина [125] и для
твердого нитромандита (126].
Более подробно оно изучалось
для нитроглицерина и некото-
рых других ннтроэфпров Бес-
паловым (127] и дли тана Ионо-
вой (123].
Опыты с нитроглицерином
показали, что при турбулентном
горении критический диаметр 1
даже при относительно высоких
(13—100 ат) давлениях ве-
лнк — значительно больше, чем
при нормальном горении, и
сильно возрастает при уменьше-
нии давления. При 100 ат он со-
ставляет 0,35 -и-м; при экстрапо-
ляции на 1 ат = 14,5 см, в то
время как при нормальном режиме нитроглицерин может гореть при ат-
мосферном давлении уже при диаметре трубки 0,5 мм.
Поскольку давление влияет на характер горения жидкости, зависи-
мость критического диаметра горения нитроглицерина от давлении более
сложная (рис. 89), чем в отсутствие такого влияния. Б области малых
давлений нитроглицерин способен гореть при относительно малых диамет-
рах. например при 200 мм рт. ст. в трубке диаметром ~1 мм. Увеличение
давления, как обычно уменьшает критический диаметр, в этих условиях
горение нитроглицерина идет на нормальном режиме — из-за малого диа-
метра турбулентность нс м'Х+>т развиваться.
Если, одпако, при давлениях выше 300 мм увеличить диаметр выше
3—4 мм. то горение затухает. Увеличение диаметра, с одной стороны, бла-
гоприятствует распространению горения вследствие уменьшения относи-
тельных теплопотерь; однако, с другой стороны, при этом становится
возможным развитие турбулентности. Это последнее влияние преобладает
и горепие прекращается. При дальнейшем увеличении диаметра влияние
уменьшения теплопотерь становится сильнее, чем их увеличения за счет
турбулентности, и горение вновь становится возможным
В пользу этого объяснения говорит влияние желатинизации па воз-
можность горения. Увеличение вязкости путем желатинизации, затруд-
няющее турбулизацию горения и снижающее область ее проявления в
сторону больших давлений, сильно уменьшает критический диаметр.
Кривая <7iT (р) принимает при этом своеобразный вид (рис. 90). В области
! Критический диаметр определялся обычно как диаметр затухания в конических
трубках, сужающихся (или расширяющихся, см. ниже) книзу.
2 Для обеспечения горепия питр о глицерида при тех значениях диаметра и давле-
ния. при которых оно возможно, важную роль играет метод поджигания. Наиболее,
а в некоторых условиях и единственно эффективным оказалось поджигание при по-
ниженном давлении с последующим ростом последнего.
117
Рис. 90. Зависимость критического диамет-
ра горения (dHr) нитроглицерина и его же-
латин от давления (р) в интервале выше
атмосферного
I — нитроглицерин <1 — и окружении воздуха,
S — в окружении воды); II—3,2%-ная жела-
тина; III — 5%-ная желатина; IV — 8,5%-ная
желатина
малых давлений, где горение идет на невоз-
мущенном режиме, критический диаметр
уменьшается (участок кривой 1 —10 аг); по-
явление турбулентности при давлениях выше
10 ат приводит к росту критического диамет-
ра (участок кривой 10—30 аг); при более
высоких давлениях преобладает влияние
уменьшения теплопотерь и dl(r вновь падает.
Чем больше вязкость желатины, тем слабее
выражено увеличение критического диамет-
ра, обусловленное турбулизацией.
Опыты по определению d1(r при разных
давлениях были поставлены также для ни-
трогликоля. Ожидалось, что при переходе на
турбулентный режим обнаружится интервал
давлений, где горение будет затухать. Этого, однако, не наблюдалось, хотя
вблпзи соответствующего давлении, если сразу начиналось турбулентное
горение, затухание часто происходило при гораздо больших диаметрах,
чем в его отсутствие (рис. 91).
Не наблюдалось затухания горения или заметного роста критического
диаметра и при переходе на турбулентный режим горения динитроглнце-
рипа (рис. 92).
Затухание горения при повышении давления наблюдалось также у
твердого нитроэфира — тэна. Было известно, что при диаметре трубки
6 мл тэн способен гореть при атмосферном давлении и умеренно повы-
шенной температуре (100° С). При комнатной температуре, однако, горе-
ние не распространялось. Вероятной причиной этого был большой крити-
ческий диаметр. Действительно, при увеличении диаметра до 30 м.и
горение (р = 1,2 г} см?) стало возможным; скорость горения мала
(0,023 г/см2сегс). Однако при несколько повышенных давлениях тэн при
этом диаметре трубки не горел. При поджигании в полузамкнутом сосуде
(бомба с открытым вентилем) утрата способности к горению при повы-
шенных давлениях проявлялась -своеобразно. Тэн зажигался при атмо
сферлом давлении, когда давление за счет газообразования поднималось
до 2,5 ат, горение затухало. Вследствие истечения газов через вентиль
давление падало и тэн вновь (воспламеняющая спираль не была выклю-
чена) загорался, затухал и это повторялась несколько раз.
Затухание горения твердого, но плавящегося при горении, тзна следует
объяснить турбулизацией расплавленной части прогретого слоя, ведущей
к увеличению теплопотерь. При малой скорости горения этого увеличения
оказывается достаточно для затухания. Как и у нитроглицерина, отсут-
ствие способности к горению проявляется у тэна лишь в некотором интер-
вале давления в данных условиях опыта —до 16 ат. При больших давле-
ниях тэн горит, ио скорость горения в отличие от нитроглицерина
соответствует нормальному горению: толщина расплавленной части про-
гретого слоя при значительной скорости торения, соответствующей повы-
шенному давлению, мала и это ограничивает развитие в нем турбулент-
НЬ
носги. С другой стороны, ввиду достижения полноты горения (до азота
вместо NO) увеличивается тепло приход превращения.
Затухание горения тэна при повышении давления (до — 2 ат) наблю-
далось и при повышенной начальной температуре (100°С, d = 7 мм).
Затухание горения происходило и в том случае, если тзн поджигался при
повышенном давлении (20 ат) и затем давлению давали медленно падать;
оно наступало при ~ 10 ат (опыт ставился при 20°С).
Рис. 91. Зависимость критического
диаметра горения нитрогликоля
от давления при 40 и 20° С
Рис. 92. Зависимость критического
диаметра и скорости горения ди нит-
роглицерина от давления
1 — <(нг(р); 2 — u(p)
Опыты с расплавом тэна при 140° С показали, что горение при 8—9 ат
переходит па пульсирующий режим; затухания в этом случае не наблю-
дается, как и при горении других жидких нитроэфиров при повышенных
давлениях и температурах. Скорость пульсирующего горения, естественно, °
гораздо больше, чем получающаяся при экстраполяции экспериментальных
данных при невозмущенном горении. При желатинированном (полиметил-
метакрилатом) расплаве тэна горение переходит на пульсирующий режим
при несколько большем давлении (9—13 ат).
Затухание горения при повышении давления наблюдалось также при
горении жидкого гидразина {124]. Скорость горения гидразина растет при-
близительно пропорционально корню квадратному из давления. Выше
10 ат данные воспроизводятся хуже и средние значения стремятся к неко-
торой постоянной величине, не зависящей от давления. Выше некоторого
давления жидкость не воспламеняется от накаленной проволоки. Затуха-
ние наблюдалось и в том Случае, если жидкость зажигалась при малых
давлениях и затем давлению давали подниматься. Критическое давление
зависит от диаметра трубки, увеличиваясь с 22 до 34,5 ат при его увели-
чении с 3 до 5 мм п уменьшается при разбавлении гидразина водой.
Уменьшения скорости горения перед затуханием не наблюдалось.
Исследователи объясняют затухание горения следующими соображе-
ниями. Давление затухания в трубках диаметром 5 мм прямо пропорцио-
нально скорости горения при атмосферном давлении. Соответственно
можно думать, что давление затухания зависит от скорости течения, плот-
ности газа, а также диаметра трубки и что эффект этот имеет гидродина-
мическую природу. Поскольку массовая скорость горения пропорциональна
корню квадратному цз давления, скорость течения несторевших паров
вблизи поверхности жидкости уменьшается при повышении давления.
119
Вблизи предела затухания она сравнима по величине с конвекционными
скоростями, которые могут, возникая вследствие того, что плотность про-
дуктов горения мала по сравнению с окружающем атмосферой, удалять
пламя от поверхности жидкости. В результате этого увеличиваются боко-
вые тсплопотери и распространение горения становится невозможным.
Если эта причина реальна, то уменьшение плотности окружающей атмо-
сферы должно было .затруднить затухание, Что и наблюдалось ври замене
азота па гелий. В азоте давление затухания составляло 8—9 ат, в гелии
горение продолжалось при 13 ат.
Затухание горения гидразина очень напоминает аналогичные явления
при горении нитроглицерина. К сожалению, опыты с гидразином не были
проведены при больших давлениях, когда (если сходство с нитроглицери-
ном не случайно) можно было бы ожидать возобновления горения, но уже
па пульсирующем режиме.
Остановимся еще на влиянии дробной газификации при горении, зна-
чение которой поясним на примере горения стехиометрического раствора
жидких самостоятельно не горящих компонентов, сильно различающихся
по летучести. При ноджпгапии1 такого раствора сначала, естественно, бу-
дет испаряться преимущественно более летучий компонент и над поверх-
ностью ВВ образуется малокалорийная смесь паров. Лишь после того как
поверхностный слой жидкости обогатится труднолетучим компонентом,
в газовую фазу станет поступать смесь состава, соответствующего макси1-
мальной калорийности, сгорание которой в состоянии поддерживать горе-
ние. Воспламенение вещества будет затруднено из-за затраты тепла на
удаление легколетучего компонента. Затруднено будет и последующее
горение, поскольку летучий компонент должен будет поступать из глубин-
ных слоев, диффундируя, или иным путем. Таким образом, различие лету-
чести взаимодействующих компонентов смеси будет снижать способность
ее к горению *.
Аналогичные явления могут наблюдаться и при горении смеси, со-
стоящей из нелетучих компонентов, разлагающихся независимо друг от
друга ,и с сильно различающимися скоростями. Тогда один из пих (напри-
мер горючее) разложится раньше другого (окислителя), и когда темпера-
тура на поверхности станет достаточной для разложения окислителя, горю-
чее должно будет поступать для участия в реакции из глубинных слоев.
Больше того, отклонение состава газовой фазы ври воспламенении от
состава ВВ возможно даже при горении индивидуальных ВВ. Известно,
что для органических нитратов, а, по-видимому, и для питросоедипеппн
ароматического ряда начальным этапом распада является отщепление
двуокиси азота, для большинства ВВ более летучей, чем остальная часть
молекулы. Таким образом, синхронность распада и взаимодействия ком-
понентов смеси млн определяющих тепловыделение групп в молекуле ин-
дивидуальных ВВ является важным фактором способности к горению л,
в частности, воспламеняемости ВВ.
Кроме описанных, имеется еще ряд наблюдений по затуханию горения,
которые не получили пока надежного теоретического объяснения. Так,
пикрат калпя горит при атмосферном давления с большой скоростью, но
утрачивает зту способность уже при 500 лш, хотя скорость горения еще
велика и объяснить затухание горения теплопотерями трудно. Это отно-
сится и к пикратам ряда других металлов, например натрия, которые пе
горят и при атмосферном давлении. В то же время другие быстро горящие
ВВ горят устойчиво при гораздо мепыпих давлениях; например гремучая
ртуть горит при 20—10 .« и ниже, перекись трнппцлоацетона — при
40 мм, трипнтротриазидобензол — даже при 10 мм.
’ Впервые такое явление было открыто и изучено А. И. Гол (.биндером на примере
смеси бензола с тетр aim троне та но и [128].
120
Смеси стифната свинца в порошкообразном виде при большом содер-
жании талька не горят, при малом его содержании дают вспышку; полу-
чить устойчивое горение с умеренными скоростями не удается. Смеси же
стифната свипца с нитроклетчаткой (также в порошкообразном виде)
горят устойчиво, если содержание стифната не слишком велико; в про-
тивном случае горение переходит во взрыв. Смеси трннитротриавидобеп-
зола со стцфнатом свинца в прессованном виде устойчиво горят при атмо-
сферном давлении при содержании до 20% последнего. При 30—40%
горение пе распространяется; 60—70%-ные смеси вновь оказываются
способными к горению.
Вероятная причина затухания в описанных примерах заключается в
чрезмерной интенсивности диспергирования конденсированной фазы: ко-
личество твердого вещества, поступающего в зону горячих газон и охлаж-
дающего их, становится столь болы дим, что распространение горения ока-
зывается невозможным.
Для определения критического диаметра торепця перхлората аммония
был применен топкодисперсный продукт заводского измельчен ня, поме-
щавшийся в стеклянные трубки, погруженные в воду. При малой плот-
ности (0,6 г/с.и3) затухание происходило1, если диаметр трубки был равен
26 .и„м, при большей (1,0 г/с.ч3) — 33 мм2.
Следует добавить, что попытки воспроизвести эти результаты с чистым
перхлоратом, измельченным в ступке, успеха не имели: перхлорат не
горел даже при диаметре заряда 40 мм.
Возможно поэтому, что упомянутый вытцс образчик содержал какие-то
примеси, которые существенно увеличивали способность к горению. Опы-
ты, произведенные с лабораторным образчиком при 30 ат, показали, что
критический диаметр его горения составляет 10 мм. Добавление топко-
измольчениого алюминия сильно уменьшает критический диаметр.
Органические примеси повышают способность перхлората к горению.
Влияние плотности наблюдается и для этих смесей. На массовую скорость
горения уплотнение, как в случае перхлората, так и его смесей, не оказы-
вало значительного влияния.
Таким образом, в отличие от некоторых составов на основе аммиачной
селитры уплотнение уменьшает способность перхлората аммония к горе-
пню. Возможно, что это различие связано с тем, что перхлорат аммония в
отлупе от аммиачной селитры при горении нс плавится. В целом эти дан-
ные показывают, что перхлорат аммония характеризуется относительно
низкой способностью к горению, уступая в этом отношении тротилу, по
существенно превосходя аммпачпую селптру-
Для перхлората аммония при малом поперечном размере заряда наблю-
дается нс только пцжний предел способности к горению по давлению,
но п верхний предел {129].
Прн увеличении диаметра заряда, по опытам Глазковой [130], нижнпй
предел давления снижается3, а верхний предел повышается; при 250 ат
1 При горении перхлората ла внутренних стенках трубки остается слон несго-
ревшего вещества, особенно толстый, если трубка окружена водой. В ионических
трубках этот слой по мере уменьшения диаметра сечения трубки становится толще
и в момент затухания близок к радиусу сечения. Само горение идет на торце заряда
неравномерно — в виде очага (или нескольких очагов) диаметром 3—й переме-
щающегося по поверхности. Поверхность затухшего заряда — неровная, испещренная
раковинами.
2 В этих же условиях dKr гексогена составляет 8 мм.
1 По Леви и Фридману, напротив, нияеггий предел, равный но пх опытам для
столбиков квадратного сечения (4 X 4 мм) при интенсивном воспламенении 22 ат, по
зависит от размеров образчика,- он пе зависит также от того, является ли окружаю-
щей атмосферой азот пли телий; они связывают зто с тем, что предположительно оп-
ределяющую роль играют тепдояотерн за счет излучении и в качестве дополнитель-
ного аргумента указывают, что охлажденный до — 18° С образчик перхлората не горел
даже при 270 ат.
121
<iltr составляет (для голых шашек) около 5 льи. Существование верхнего
предела для перхлората аммо<ния связано, по-видимому, с тем, что при
повышении давления в определенном его интервале скорость горения
вместо того, чтобы продолжать расти, начинает падать.
Такое объяснение требовало, однако, подтверждения того, что умень-
шение скорости горения при повышении давления является причиной
увеличения теплопотерь, а не следствием этого увеличения, которое может
быть связано, например, с увеличением плотности окружающего газа,
Рис. 93. Зависимость скорости горения от
давления для перхлората аммония, запрес-
сованного в плексигласовые трубочки раз-
личного диаметра
1 — d = 7 леи* 2 — <1 = 5 лен; 5 — d = It) мм
Рис. 94. Скорость горения перхлората
аммония при различном (числа у
кривых) содержании хромита меди
усилением конвекции и т. п. Для этого скорость горения была определена
для зарядов, находившихся в пластмассовой (плексигласовой) трубке,
достаточно хорошо термоизолирующей их от внешней среды Уменьше-
ние скорости горения в области 160—200 ат наблюдалось и в этом случае
(рис. 93), подтверждая таким образом независимость падения скорости
от теплопотерь в окружающую среду. Дополнительно этот вывод под-
тверждают также опыты по горению при различных диаметрах трубки
(5, 7 и 10 мм), которые пе показали различия в скорости горения.
Несколько иначе по методике выполнения были поставлены аналогич-
ные опыты Леви п Фридманом [131]. Они жгли столбики перхлората
в асбестовой обмотке, и не обнаружили до изученного предела давления
1 Толкование результатов этого опыта требует одной оговорки; материал оболоч-
ки (плексиглас) и горящий перхлорат аммония могут взаимодействовать друг с дру-
гом; зто возможное взаимодействие вносит новый фактор в явление, влияние которо-
го оцепить априори затруднительно. Однако судя по тому, что смеси порошкообраз-
ного плексигласа с перхлоратом в данной области давлении горит медленнее, чем сам
перхлорат, вряд ди их взаимодействие при несравненно менее Тесном контакте — за-
ряда со стенкой — может играть значительную роль.
122
(340 ат) прекращения горения или уменьшения скорости. Авторы приш-
ли к заключению, по-видимому, без достаточных оснований, что верхний
предел способности к горению по давлению голых шашек обусловлен кон-
вективными теплопотерями.
Своеобразное влияние на нижний предел оказывает добавка катали-
затора, например хромита меди (рис. 94). Большая добавка понижает
его, что, естественно, объясняется увеличением скорости экзотермической
газофазной реакции; напротив, малая добавка, даже всего 1 : 20000, силь-
но (в 4 раза) повышает нижний предел, что авторы объясняют повыше-
нием излучающей способности эоны горения и, следовательно, тепло-
потерь.
ВЫВОДЫ
Горение столбика ВВ возможно лишь в том случае, если теплопотери
горящего слоя не превосходят некоторого предела, при котором нару-
шается устойчивое равновесие между теплоприходом и теплоотдачей в
этом слое. В соответствии с этим изменение условий горения в сторону
увеличения теплопотерь или уменьшения теплоприхода может привести
к тому, что горение станет невозможным.
Основными из этих условий являются диаметр заряда (при горении
столбика ВВ с торца), давление, температура и кубическая плотность
(для порошкообразных ВВ). Соответственно существуют критический
минимальный диаметр, минимальное давление и минимальная темпера-
тура, ниже которых горение не распространяется.
Значения этих параметров не являются абсолютными характеристика-
ми ВВ, но взаимно связаны. На возможность горения влияют также теп-
лопроводность и толщина оболочки. Прекращение горения может насту-
пать, далее, при всех условиях, которые ведут к нарушению прогретого
прифронтового слоя ВВ.
III. ВЛИЯНИЕ ДАВЛЕНИЯ НА СКОРОСТЬ ГОРЕНИЯ ВВ
Зависимость скорости горения ВВ от внешнего давления является
одной из двух основных теоретически важных характеристик горения.
Одновременно она представляет значительный технический интерес.
Устойчивое горение заряда в полузамкнутом объеме возможно лишь в
том случае, если скорость горения растет с давлением медленнее, чем
скорость истечения газов через сопло, пропорциональная давлению. При
сильной зависимости скорости горения от давления склонность его к пе-
реходу во взрыв обычно выше.
Скорость горения, как правило, возрастает при повышении давления.
Это вполне естественно в том случае, когда экзотермические реакции при
горении протекают в газовой фазе. Повышение давления, увеличивая
абсолютную скорость этих реакций, приближает зону их протекания к
поверхности конденсированной фазы, увеличивает градиент температуры
«близи этой поверхности и соответственно передачу тепла последней.
Это основное влияние давления на скорость горения согласуется с тем
фактом, что для ряда ВВ, характеризующихся малой летучестью, где
предполагается, что экзотермические реакции идут в конденсированной
фазе, скорость горения не зависит от давления.
Отсутствие такой зависимости наблюдалось и у бездымных порохов
при сильном снижении давления и объяснялось тем. что в этих условиях
скорость газофазных реакций уменьшалась настолько, что вклад этих реак-
ций в тепло, получаемое поверхностью конденсированной фазы, стано-
вился пренебрежимо малым по сравнению с теплом, выделяемым реак-
циями, протекающими в конденсированной фазе. В известных условиях,
123
однако, ускорение горения при повышении давления может происходить
и независимо от его влияния на скорость газофазной реакции: вследствие
увеличения плотности газовой фазы увеличивается количество тепла, пе-
редаваемого поверхности конденсированной фазы.
Менее попятно наблюдавшееся у некоторых ВВ значительное умень-
шение скорости горения при повышении давления. Возможно, это падение
связано с затруднением диспергирования вещества при горении или с
влиянием звуковых колебаний, возникающих при горении, па его течение.
Все эти разнообразные формы зависимости скорости от давления имеют
одну существенную общую черту: скорость горения обычно растет мед-
леннее, чем давление (если исключить короткие по интервалу давления
участки перехода от постоянства или падения скорости к ее росту). Если
в опыте dii/dp растет с давлением, то это почти всегда является признаком
того, что горение перешло с нормального па возмущенный решим.
Основными формами возмущенного горения являются турбулентное
горепие жидкостей п конвективное горение пористых твердых ВВ.
Для всех изучавшихся жидкостей нормальное горение по достижении
определенного давления переходит на пульсирующий режим. Если Этот
переход происходит при ояе.иь низких давлениях, то он мо?кет приводить
К затуханию горения; при высоких давлениях наблюдается сильное уве-
личение скорости и более быстрый ее рост с давлением. Давление перехода
в сильной степени зависит от скорости горения, присущей данному веще-
ству, уменьшаясь с ее увеличением. Соответственно у быстрогорящего
нитроглицерина смена режимов горения происходит при 0,5 аг, у медлен-
но горящего дигликольдицитрата — 55 аг. Повышением вязкости жидко-
сти, например, растворением в пей высокополимера можно сильно расши-
рить интервал устойчивости нормального горения по давлению.
У порошкообразных (точнее газопроницаемых) ВВ точпо так же
наблюдается пе который предел давления, выше которого нормальное го-
рение переходит на режим, связанный с проник и овен нем газообразных
продуктов горения в глубь заряда. Оба эти вида возмущенного горения
будут рассмотрены далее.
Математически выразить единой формулой различные виды зависимо-
сти п (р) затруднительно потому, что даже для одного и того же вещества
в разных интервалах давлений характер зависимости нередко бывает
различен. Соответствующие выражения н(р) носят эмпирический харак-
тер и приложимы поэтому лишь для того интервала давлений, в котором
они определены. Некоторые из них мы рассмотрим применительно к кон-
кретным ВВ-
Наиболее простым и распространенным в теории трения является
выражение и = А + 2?р, причем для многих веществ член А относительно
мал, так что зависимость н(р) приближается к прямой пропорциональ-
постп- Линейная зависимость была установлена [132] в области относитель-
но низких давлений для метилнитрата. нитрогликоля (жидкого и же-
латинированного), желатинированного нитроглицерина, дигликольдинит-
рата. .нитроклетчатки, гексогена, тетрила и гремучей ртутп. При расши-
рении интервала давления вверх (для отдельных ВВ до 1000 ат) линейная
зависимость для ряда ВВ сохраняется, хотя и с несколько иными коэф-
фициентами. К числу этих ВВ Относятся тэн, тротил и пикрпиовая кис-
лота.
Для некоторых ВВ (тетрил, гексоген, пороха па трудполетучем рас-
творителе) кривая п(р) в интервале давлений от нескольких до 100—
250 ат явно отклоняется от прямой; скорость растет .медленнее, чем
пропорционально давлению и приближенно может быть выражена соот-
ношением и — Ai + Bip\ где Ai л Bi — постоянные, a v меньше единицы.
Выше некоторого предела давления скорость начинает расти линейно:
и = Ла + В^р, причем Ai>At.
124
Для веществ третьей группы (гремучая ртуть, черный порох) насы-
щающийся рост скорости горения с давлением сохраняется во всем изучен-
ном интервале последнего.
Для ряда быстрого рящих ВВ скорость горения в области низких дав-
лений быстро растет, затем этот рост сильно замедляется или даже совсем
прекращается. Примерами таких веществ могут служить пикрат и стиф-
нат свинца.
Наконец, для некоторых ВВ быстрый подъем скорости в области ма-
лых давлений затем переходит в ео более или менее значительное падение,
при дальнейшем повышении давления вновь сменяющееся относительно
медленным ростом. Так недут себя, например, пикрат калия и перхлорат
аммония.
Повышение давления обычно сопровождается изменением состава
продуктов горения, который при больших давлениях соответствует хими-
ческому равгговесшо; при низких же давлениях продукты горения содер-
жат много компонентов, указывающих па неполноту превращения, типич-
ным из которых для цитратов и нитросоединспий является окись азота.
В продуктах горения пптрогликоля при атмосферном давлении отношение
содерясания окиси азота к сумме содержаний углеродсодержащих газов
(СОг + СО + СН$) равпо 0,9, т. е. почти весь азот выделяется в виде NO;
при 9 ат это отношение составляет 0,7, а при 14 ат окись азота в продук-
тах горения отсутствует-
Для некоторых ВВ (нитрогликоль, нитроглицерин и др.) изменение
состава продуктов горения при повышении давления является резким и
сопровождается появлением вторичного пламени, в котором, очевидно,
и происходит взаимодействие окиси азота с горючими продуктами на-
чальных стадий превращения. Возможно, что для других 13В, папример
нитроклетчатки [133], переход от горения до NO к горению до элементар-
ного азота несколько растянут по давлению.
Давление, при котором появляется вторичное пламя, не одинаково
для изучавшихся нитроэфиров и, Кроме того, зависит от диаметра заряда,
уменьшаясь с его увеличением. У пптрогликоля при диаметре 5 .w.w оно
появляется при 9,5 аг, при диаметре 9 мм — уже при 5,6 ат. У дигли-
ко ль динитрата (яшдкого или слабожплатинированного) вторичное пламя
возникает трудное; при диаметре 5,2 лш оно появляется при 16 ат, при
диаметре 10,5 мм — при 7,5 ат. С нитроглицериновой желатиной
(3—5% коллоксилина) опыты проводились в трубках меньшего диаметра
('''З лиг), вторичное пламя появлялось при более высоком давлении
(15 ат)-, однако граница его при низких давлениях очень неровная вслед-
ствие частого подхода к первичному пламени, ц лишь начиная с 30 ат,
приобретает четкие очертания. При еще большем давлении (35—40 ат)
появляется вторичное пламя при горении этилпнтрата.
Изменение начальной температуры желатинированного пптрогликоля
на 40° (с 20 до 60° С) практически не отразилось на давлении, при кото-
ром появляется вторичное пламя. Ilo-видцмому, процессы, ведущие к его
возникновению, протекают при сравнительно высоких температурах, по
Отношению к которым указанное ее изменение является малым.
Некоторые минеральные добавки в малых количествах (1%) сильно
(вдвое) снижают давление, при котором появляется вторичное пламя,
другие, напротив, несколько повышают его. Скорость горения при этом
давлении в обоих случаях была ниже, чем в отсутствие добавок. Соот-
ветственно выше в присутствии добавок был и предел давления, начиная
с которого становилось возможным горение.
Второй важной характеристикой двухпламенного горения является
расстояние первичного пламени от вторичного и изменение этого рас-
стояния с давлением. Эти данные для жидкого и желатинированного иит-
рогликоля [271] представлены па рис. 95; для нитроглицериновой желатины
125
Рис. 95. Изменение расстояния
между первичным и вторичным
пламенами с давлением
1 — жидкий и ит р о гликоль; 2 — же*
патинированный нитрогликоль. Пунк-
тиром обозначена область пульсиру-
ющего горения
расстояние значительно больше, чем для нитрогликоля, что может быть
связано с большей скоростью горения последнего; при сравнении следует,
однако, учитывать, что диаметры заряда в обоих случаях были различ-
ными.
У твердого тэна вторичное пламя отсутствует при атмосферном дав-
лении и продукты горения содержат около 40% окиси азота. При 16 ат
окиси азота в продуктах горении не содержится, но вторичное пламя на-
ходится так ближе от первичного, что ин-
тервал между иимиперазличим; ото опять-
таки, в известной мере, может быть связа-
но с относительно малой скоростью горе-
ния тэна в сочетании с высокой темпера-
турой, достигаемой во вторичном пламени.
Расстояние между вторичным и пер-
вичным пламенем зависит также от на-
чальной температуры ВВ: возрастает при
ее повышении. Это, по-видимому, связано
с тем, что температура процессов, приво-
дящих к возникновению вторичного пла-
мени, гораздо выше, чем ведущая темпе-
ратура первичного пламени; поэтому по-
вышение температуры на одну и ту же ве-
личину оказывается для вторичного пла-
мени относительно меньше, чем для пер-
вичного.
Поскольку конечную температуру пер-
вичного .пламени в отсутствие теплопотерь
можно рассчитать, определение влияния
начальной температуры на индукционный период появления вторичного
пламени может быть использовано для расчета кинетических характерис-
тик этого процесса.
Двухпламенное горение было обнаружено Зельдовичем и Шауловым
и при горении паров метилнитрата [134]. Второе пламя возникает лишь
при значительном сжатии продуктов первичного распада, подобно тому
как это имеет место при горенки жидкого нитрогликоля при повышенных
давлениях. К сожалению, первое пламя не удалось сфотографировать
ввиду' слабого его свечения, и поэтому нельзя было точно установить
скорость горения паров.
1. НИТРОЭФНРЫ
Изучение зависимости и(р) для нитроглицерина затрудняется тем, что
горение этого жидкого ВВ уже при низких (< 1 ат) давлениях переходит
на турбулентный режим, что обыч но вызывает его затухание *. Поэтому
указанная зависимость была непосредственно определена лишь в интер-
вале давлений от 20 до 300 мм, и = 0,0067 + 0,216 р0,831. По этой формуле
было легко, в частности, рассчитать скорость горепия при давлении зату-
хании, равную 0,072 см! сек-, непосредственно измеренная вблизи давления
затухания скорость составляла 0,077 см)сек.
Опыты с нитроглицерином были проведены также при повышенной
температуре (98°С). Давление, начиная с которого горепия при зажига-
нии пе возникает, приблизительно то же: 320—380 мм. Однако при повы-
шенной температуре скорость горения, естественно, больше; поэтому if
предельная скорость вблизи затухания больше и составляет около
0,13 см!сек. Кроме того, если давление при горении возрастает до кри-
тического, то горение не всегда или во всяком случае не сразу заканчи-
। Критическое давление зависит от диаметра трубки, см. стр. И8>
126
г
Рис. 96. Зависимость скорости
горения нитроглицерина
от дэндеции
вается затуханием. Первым этапом при повышении давления является
пульсирующее горение, в некоторых опытах очень своеобразное: пламя
как бы проникает вдоль одной стороны стенки трубки па большую глуби-
ну, и фронт горения жидкости становится почти вертикальным. Возможно,
что это явление представляет собою пульсацию очень большого периода.
Экстраполируя кривую и(р) на большие давления, можно рассчитать
скорость горения при атмосферном давлении, равную 0,23 г/смгсек. Эта
величина соответствует результатам непосредственного измерения скоро-
сти горения при атмосферном давлении в
трубках диаметром 0,5—1,5 .иле. Были сде-
ланы попытки определить скорость горе-
ния также следующим путем: если доба-
вить к нитроглицерину 2% пироксилина
№ 1, то он сравнительно легко поджига ст-
оя и горит при атмосферном давлении
со скоростью 0,28 г[см?сек.
Опыты при пониженных давлениях по-
казали, что примесь 2% пироксилина не
оказывает заметного влияния на скорость
горения нитроглицерина. Если это было
бы справедливо и для атмосферного давле-
ния, то можно было бы заключить, что ско-
рость горения нитроглицерина равна
0,28 г!см1сек. Однако, сопоставляя эту
величину с полученной экстраполяцией л
непосредственным определением, следу-
ет заключить, что 2% пироксилина неполностью подавляют турбулиза-
цию при горении под атмосферным давлением, и что указанная скорость,
является несколько завышенной.
Попытка определить скорость горения твердого нитроглицерина за-
трудняется тем, что критический диаметр для него гораздо больше, чем
для жидкости. Поэтому определение было проведено при 27 ат; скорость
горения разнялась 2,68 г]см2сек, т. е. гораздо меньше, чем для жидкости
из-за различия режимов горения и отчасти из-за более низкой температу-
ры горения. В желатинированном (2% коллоксилина) виде нитрогллпевин
(d = 11 мм) при + 9°С и атмосферном давлении горел в незамерзшем
состоянии со скоростью 0,113 см]сек, в замерзшем — 0,0838 см1сек.
Повышение вязкости путем растворения высокополимера увеличивает
способность нитроглицерина к горению. Скорость горения (при атмосфер-
ном давлении) уменьшается сначала вяачительно, при дальнейшем ясе
увеличении содержания высокополимера — гораздо слабее. Так, при рас-
творении 1% коллоксилина она уменьшалась на 0,02, при переходе от-
1 к 2% уже только на 0,006 и от 2 к 3% —на 0,001 см! сек. Различие в
скорости не проявляется заметно при низких давлениях, когда благодаря
малой скорости горения и сам нитроглицерин горит на нормальном ре-
жиме; при повышении давления горение нитроглицерина затухает
(рис. 96), а маловязкие растворы горят с повышенной скоростью вслед-
ствие появления турбулизации; при еще большем давлении затухают
и они.
Применением коллоксилинов различной вязкости, сравнением способ-
ности к горению до и после растворения высокополимера, а также раство-
ров мономера и полимера было показано [135], что влияние последнего
имеет не химическую, а физическую природу. Повышение вязкости за-
трудняет возникновение турбулентности и повышает давление перехода
от нормвльного к возмущенному горению. Благодаря этому становится
возможным определение зависимости скорости горения от давления в
большем, чем для жидкости, интерввле последнего. От 0,3 до 2,2 ат эта
127
зависимость для 3%-ной коллоксилиновой желатины может быть выра-
жена линейной формулой = 0,0315 + 0,146 р. В более широком (до
150 ат) интервале давлений зависимость а(р) была определена Г. Н. Бес-
паловым для желатин, содержащих 3,2; 5 и 8,5% коллоксилина (рис. 97).
В этом диалазоне содержания высокополимера скорость горения заметно
Рис. 97. Зависимость скорости горении нит роглиц ери копыт
желатин от давления
Содержание коллоксилина (%): Т—3,2; г—5; з — 8.5
не изменяется. Рост ее с давлением особенно в области умеренно повы-
шенных давлений (до 30 ат) отчетливо замедляется.
Другие изучавшиеся жидкие питроэфиры (метилпитрат, нитрогликоль,
дигликольдинитрат и этилнитрат) горят медленнее, чем нитроглицерин.
Соответственно интервал давления, в котором горение идет на нормальном
режиме, для них больше: для металнитрата до 1,75, для интрогликоли
Рис. 98. Фотография пульсирующего го ре пи я жидкого нитро-
гликоля в трубке диаметром 3,5 .«.и
1 — первичное пламя; 2 — вторичное пламя
до 20, для дигликольдинитрата до 55 и для этилгпгтрата до 80 ат. Скорост!
нормального горения при давлениях перехода для последних двух В£
составляет соответственно 0,6—0,7 и 0,5 см! сек. В небольших интервала:
давления .вблизи атмосферного скорость горения может быть выражен;
линейной формулой и — А + Вр (табл. 17), причем член А мал по срав
нению с В и соответственно зависимость близка к прямой пропорцио
нальности.
128
Таблица 17
Значения коэффициентов Л и В в уравнении горения и = А ”!>
для некоторых нитроэфиров
Вещество Интервал давле- ния, в котором справедлива фор- мула, лмерт. ст. А В
Нитрогликоль 300—770 0,005 0,0375
Нитроглицерин * Желатинированный 3%-ный нитро- 20—300 0,0067 0,216
глицерин 200—1600 0,0315 0,146
Метилнитрат 130—1520 0,010 0,133
* Для ж [Тро гл-. [Церина: u — А -|- Bpv , v = 0,83b
ь
3
х
а
Рис. 99. Зависимость скорости
горения жидкого нитрогликоля
от давления (271)
Так, например, для питрогликоля между 0,4 и 1 ат им —0,005 + 0,0375 р. , ,
В более широком интервале давлений, особенно вблизи перехода па воз- 17
мущенный режим, скорость горения растет несколько быстрее; при дав-
лении перехода характер горения существенно меняется. Поверхность
жидкости явно перестает быть спокойной и горизонтальной, все горение
представляет собой как бы ряд быстро следующих друг за другом вспы-
шек, скорость горения резко увеличивается и колеблется в больших
пределах. Склонность горения к переходу во взрыв в этой области давле-
ния существенно повышается.
Фоторегистрация пульсирующего горения отличается тем (рис. 98),
что расстояние между первичным и вторичным пламенами не остается
постоянным. Вторичное пламя через одинаковые шли неодинаковые про-
межутки времени подходит к поверхно-
сти жидкости и затем удаляется от нее.
По-вцдимому, турбулизация на поверх-
ности горящей жидкости приводит к ох-
лаждению прогретого слоя и испарение
замедляется. Тогда зона вторичного
пламени приближается к поверхности.
Тен временем прогретый слой успевает
восстановиться и, возможно, даже стать
толще нормального; испарение л пер-
вичное горение усиливаются; скорость
отхода пх продуктов возрастает и вто-
ричное пламя отбрасывается от поверх-
ности жидкости.
При еще больших давлениях горение
вновь приобретает видимую равномер-
ность, но идет уже со скоростью, на по-
рядок большей, чем при нормальном
горении1 (рис. 99).
Более детальное научение С. В. Чуйко переходной области показало, что
переход с прямой н(р) для нормального горения на прямую2 турбулентного
горения происходит по сложной кривой. Для нитрогликоля скорость в этой
области больше, чем это соответствовало бы прямой турбулентного горения
1 По опытам С. В. Чуйко, для яитрогликоля предельное давление нормального
горения несколько уменьшается при увеличении диаметра трубки; наряду с этим от
диаметра трубки зависит также du! dp при горении нд быстром режиме, возрастая
при его увеличении от 4 до 7 -««; для днгликчльдинитрата и этилпитрата этого влия-
ния диаметра не наблюдается.
2 Для дигликольдивитрата линия а(р) на участке турбулентного горения имеет
некоторую выпуклость, обращенную к оси ординат.
9 К. К. Андреев 129
! (рис. 100); для дигликольдинитрата и этилнитрата в узких трубках
(4 — 5 льи) она оказывается несколько ниже. Эти особенности, по-видимому,
; связаны с двойственным влиянием турбулизации на горение — нарушая
. прогретый слой, она затрудняет горение и может приводить даже к его
затуханию, как это наблюдается для нитроглицерина при низких давле-
ниях. С другой стороны, увеличивая поверхность контакта между газооб-
разными продуктами горения и жидкостью, турбулизация может ускорять
горение. В зависимости от тсрмокинетических характеристик вещества,
Ряс. 100. Зависимость скорости горения некоторых нитроэфнрев
от давления
— н&трогликоль; 2 — дцгликольлинитрат; J — ншлцитрат
а также области давления, где наступает турбулизация, может преобладать
то или иное влияние. Для загущенной путем желатинизации жидкости
давление изменения режима горения сильно повышается н вплоть до
150 ат питрогликолевая желатина горит с нормальной скоростью, липедно
растущей с давлением (рис. 101), как и для нитроглицерина, несколько
меньшей, чем нормальная скорость горения незагущенной жидкости.
Твердый тэн, не горящий при умеренно повышенных (2—16 ат) дав-
лениях в трубках небольшого диаметра, приобретает эту способность при
больших давлениях. Скорость горения от 16 до 100(1 ат растет пропор-
ционально давлению, причем коэффициент пропорциональности пря
1000 ат приближенно тот же. что и при 1 —2 ат.
Зависимость скорости горения высокомолекулярного нитроэфнра —
нитроклетчатки — от давления в относительно небольшом интервале низ-
ких давлений может быть выражена линейной формулой [136], по со зна-
чительно большим, чем для низкомолекулярных нитроэфпров членом А.
Это проявляется, в частности, в существенно большей скорости горения
нитроклетчатки при атмосферном давлении в сравнении с летучими нит-
роэфирамл. близкими к пен по теплоте горения. В более широком интер-
вале давлений (до 150 ат) скорость горения нитроклетчатки растет не
пропорционально давлению, а медленнее (рис. 102).
130
Сопоставление зависимостей скорости горения разных нитроэфиров от
давления обнаруживает некоторые различия между ними. Так, если для
тэна и цитроглюколя1 эта зависимость выражается прямой линией, про-
ходящей, особенно для тэна, практически через начало координат, то для
.нитроглицерина (желатинированного) кривая и(р) имеет вначале отчет-
ливо насыщающийся характер и лишь выше 30 ат может быть прибли-
женно выражена прямой при значительном члене Л в выражении
и — А + Вр. Сжорость горепия нелетучей и неплавящейся нитроклетчат-
ки во всем изученном интервале давления — до 150 ат — растет по насы-
щающейся кривой. Скорость горения нитроклетчатки тем больше, чем
выше содержание азота.
Определение .зависимости и(р) при нормальном горении было прове-
дено также для о0%-пых желатин па основе нитроглицерина .п пптрог.чи-.
коля, исходя из следующих соображений. Скорость горения тэна растет
Рис. 102, Зависимость скорости
горения нитроклетчатки от дав-
ления
с давлением ио одному и тому же простому закону как при низких, так
н при высоких даплеп.иях. Это позволяет предположить, что ведущим во
всем диапазоне давлений является один и тот же Комплекс реакций,
а нмонпо тот, который ведет процесс при низких давлениях. Сопостав-
ление скорости горщшн нитроглицерина п нитро гликоля дает возмож-
ность проверить ото предположен и с. Действительно, оба нптроифира. очень
близкие но составу и термохимически, горят при атмосферном давлении
с существенно (в 4 раза) различающимися скоростями, очевидно, вслед-
ствие различия кинетики ведущих горение реакций2. Если эти реакции
сохраняют свою ведущую роль и при высоких давлениях, то разница в
скоростях должна сохраниться. Если же при каком-то давлении ведущая
роль переходит к заключительным реакциям (NO + СО + Hs-»-N2 +
+ (Х)г + НгО), то скорости должны стать практически одинаковыми.
Сравнить по скоростям горения непосредственно жидкости не представля-
ется возможным, так как уже при небольшом давлении горение перехо-
дит па возмущенный режим. Поэтому пришлось применить желатины с
большой виз костью, хотя в этом случае мощно было опасаться, что инди-
видуальность жидких шыроэфпров будет нивелироваться большим содер-
жанием нитроклетчатки.
Опыты показали (рис, 103), что даже при 1000 ат нитроглицериновая
желатина горит значительно быстрее иитрогликолевой, отсюда можно
1 Интересно, что для желатинированного яитро гликоля линия «(р) при относи-
тельно низких давлениях состоит как бы из двух прямых с близким углом накло-
на, причем участок для р < 2 ат идет чняю участка, соответствующего давлениям
выше 4 ат [137].
2 При умеренно повышенных давленных скорости горения различаются меньше,
но все же значительно.
9*
13t
заключить, что реакции, ведущие горение при низких давлениях, играют
свою роль вплоть до указанного давления.
Беляев и Комкова [138] изучали горение трех жидких нитрозфиров и
раствора нитробензола в тетранитрометане при повышенных (10—150 ат)
давлениях инертного газа (азот). Опыты проводили в стеклянных стакан-
чиках диаметром 5 и высотой 15 леи при поджигании нихромовой спи-
ралью.
Все изученные вещества способны к устойчивому горению \ проте-
кающему, однако, со значительно большей скоростью, чем моишо было бы
ожидать, экстраполируя зависимости, полученные для нормального горе-
\1 ния при низких давлениях. Это подтверждается сопоставлением скорости
b горения жидкого и твердого2 нитроглицерина (рис. 104). Последний горел
в 10—20 рал медленнее прежде всего потому, что из-за большой вязкости
горение пе переходило на ускоренный режим, так же как в опытах других
исследователей при горении желатинированного нитроглицерина,
Зависимость скорости горения от давления может быть выражена соот-
ношением и = А + Вр\ причем для тетранптрометаяа с нитробензолом и
метилинтрата v ss 1, для нитроглицерина и нитрогликоля несколько боль-
ше 1.
Обращает на себя внимание значительно большая скорость горения
смеси тетранитрометана с нитробензолом, хотя температура се горения
(по расчету) лишь немногим выше, чем .нитроглицерина. В свою очередь
нитроглицерин горит лишь немного быстрее, чем питрогликоль, в то время
как при нормальном режиме оц горит в 4 раза быстрее, чем нитроглмколь.
Известно, что при горении нитроэфиров многоатомных спиртов при
атмосферном и близких к нему умеренно повышенных давлениях большая
часть азот.а содержится в продуктах горения в виде окдси.
Окись азота содержится как основное соединение азота в продуктах
горения эт.нлнптрата, а в более холодных продуктах горения пропилннтра-
та наряду с окисью азота содержатся большие количества нитритов и
питросоединений. Вместе с тем в продуктах горения всегда содержится
также небольшое количество элементарного азота, причем его образо-
вание идет гораздо быстрее во фронте пламени, чем в более горячей и
восстановительной атмосфере позадп фронта,
В свете этих фактов представляет интерес способность к горению нит-
ритов, реакция распада которых с образованием окиси азота является
практически термонейтральной, тан что тепло может выделяться только
за счет восстановления окиси азота. Это относится, в частности, к метил-
нитриту. который в парах (т. кип. —18° С) способен гореть даже при
атмосферном давлении, очевидно, благодаря образованию в пламени про-
дуктов, легко восстанавливающих окись азота; при горении метилиитрата
эта реакция протекает менее полно.
Горение паров метилнитрита осуществлялось в двух вариантах [139].
Один из них был основан на использовании горелки плоского пламени
диаметром 3,8 см. Пламя было почин невидимым и обнаруживалось тене-
вым методом. На различных расстояниях от поверхности горелки опреде-
лялся химичсскигй состав и температура пламени. Газы горепия медленно
отсасывались через кварцевую трубочку ц замораживались в ловушке с
жидким азотом. Газовая фракция анализировалась химическим П инфра-
красным методом, франция паров и жидкости — только последним, вода
определялась по методу Фишера. Результаты анализов, применительно к
! В частя опытов, особенно для нптроглияерипа и метилнвтрата, прп повышен-
ных давлеппнх наблюдался переход тореняя во взрыв.
2 Отверждение нитроглицерина достигалось охлаждением его в жидком азоте,
По-видимому, исследователи подучали при этом но кристаллический, а переохлажден-
ный питрорлнцорцн, который ври последующем проведении опыта в бомбе при ком-
натной температуре не успевал ее принят!,.
132
основным компонентам продуктов реакции, представлены на рис. 105.
Температура пламени измерялась платино-платинородиевой термопарой,
покрытой тонким слоем пирекса и натянутой горизонтально в пламени.
Кривая Ц1) изображена на верхнем рисунке. Точка перегиба является
реальной, судя по наличию двух
участков па теневой фотографии.
Скорость гореиия по отношению к
парам при 200° С составляла
7,5 см/сек.
Ниже описан процесс горения
на основе имеющихся данных.
Пары мотилнитрита, нагретые до
160—200° С излучением матери-
ала горелки, поступают в зону
пламени и распад, ио-видимому,
200 000 600 600 ЮОО
р, кг/см2
Рис. 103. Зависимость скорости горения нит-
рогликолевой (7) и нитроглицериновой (2)
желатин от давлении
g ЮОО
| 800
§ 600
| 000
| 200
Рис. 104. Зависимость скорости горении
от давления
I — жидкий нитроглицерин; г — иитрогли-
ноль; з — метилнптрат; 4— твердый ни-
троглицериц; 5 — смесь тстраццтрометапа
с нитробензолом
Рис. 105, Распределение температу-
ры и продуктов в пламени метил-
нитрита
завершается до точки излома кривой температуры, т- е. до 700" С. Суммарное
уравнение первой Стадии CHjONO—> 7гСН5ОН + ’/зСНгО + МО — 1 ккал
показывает практическую ее термонейтральность. Затем протекает экзо-
термическая реакция с образованием воды, СО и азота между ХО и други-
ми продуктами указанной реакции или продуктами их распада. Эта реак-
ция пе продолжается после того как метанол и формальдегид распадутся
на СО я Нг, что происходит па расстоянии щигблизительно 9 .и.и от фронта
пламени; при этом около половины ХО остается невосстановленной в ко-
нечных продуктах при температуре ~1100° С.
Реакциями, потребляющими нитрит, могут быть следующие
CH3ONO-+CILO' + NO—~38 ккал.
(3.1)
СН3О' + CII3ONO-> СЩОН + С1ЬО + NO— -37 ккал. (3.2)
133
Реакция (3.1) рассматривается как первый итак при термическом рас-
паде метилннтрита, однако последующие реакции между ее продуктами
окончательно не установлены. В частности, применительно к термиче-
скому распаду Леви отвергает реакцию (3.2).
Стиси и Шоу допускают последующий распад метоксила на формаль-
дегид п атом водорода, последний с метоксилом образует метиловый спирт.
Леви ключевую роль при горении приписывает реакции
СПзО’ + NO — NOII + СПгО + ккал (3.31
при медленном распаде NOII образует закись азота в воду, а также с мирт
СН3О’ + NOII -> СНзОН + NO. (ЗЛ)
При высоких температурах горения может стать существенной
реакция
NOH -+ NO + Н.
Во втором этапе реакции, начинающемся на расстоянии 0,5 мм от
фргщта, участвуют следующие основные соединения: NO (42% но объему),
С.НзОН (19%) и СНаО (20%). Экзотермические реакции, которые поддер-
живают распространение горения, должны происходить между этими-ин-
гредиентами, тем более что восстановление NO прекращается, когда
СНзОН и СНэО израсходовались.
Любопытна, что при горении изопропцлиитрата, где все эти три ингре-
диента присутствуют примерно и том Же соотношении, .значительного
восстановления NO не происходит. Различие между обеими системам и
заключается, во-первых, в том, что в пламени нзопропнлнптрата содер-
жатся многочисленные другие ингредиенты и, но-вторых, что максималь-
ная температура составляет всего около 450° G, так что СНзОН п С‘Н;()
нс разлагаются слишком быстро. Наиболее вероятное объяснение состоит
в том, что активный продукт, образующийся и системе СИ3ОН/СП2О и
реагирующий с NO, легче взаимодействует с другими веществами, содер-
жащимися н и зов ропмл нитратном пламени, пли же этот продукт не обра-
зуется достаточно быстро нри 450° С. Возможно, что этим продуктом
является атомарный водород, образующийся при высокотемпературном
распаде СНзОН или С,Н2О.
При взрыве паров мет пл нитрита при атмосферном давлении Добавле-
ние NO к нитриту увеличивает сил;- взрыва. Этого трудно было ожидать,
если учесть, что в конечных продуктах горения нитрата около 50% N()
остается невосстановленной.
Точно так же нпжпий концентрационный предел смеси по содержанию
метилннтрита очень мал — 17,6% (по объему), в то время как в смеси с
азотом он составляет 42,8%.
Разбавление метилннтрита четырехкратным объемом NO не приводит
к продуктам полного окисления. Большая часть NO остается неизменен-
ной. образуется меньше СО и СОо, но СО остается главным ингредиентом
продуктов горения; несмотря на избыток ок делите.!] я количество N’sO
сипыш возрастает, метоксида, что особенно существенно, если и образует-
ся, то очень мало; количество СН2О изменяется мала.
Возможным объяснением влияния NO является реакция (3.3).
В условиях, в которых метилнитрит легко воспламеняется н горит,
оказалось невозможным воспламенить смесь двух молей NO, моля СН3ОП
и моля СН<?(); это тем более интересно, что метилнитрит, на первой Стадии
распадается именно на эти части и притом термонейтралыю. Из резуль-
татов опытов с парами метилннтрита, разбавленными окисью азота, сле-
дует заключить, что NO реагирует с рано образующимися промежуточ-
134
ныли продуктами распада метилиитрита. которые в противном случае
быстро реагировали бы с самим метил ни тритом. Наиболее вероятными из
них являются СНзО или Н особенно потому, что добавление NO ингиби-
рует образование метанола.
В другой работе [140] горение метилннтрита проводилось па воздухе
в вертикальной стеклянной трубке диаметром несколько сантиметров. При
этом образуется серовато-желтое диффузионное
пламя. Па внутренней поверхности диффузионно- ?
го пламени образуется в виде конуса на конце £
трубки красно-оранжевая зона. Если уменьшить
скорость истечения паров, то это слабое оранжево- \ 3
красное пламя отрывается от внешнего дпффузи- /
онного пламени и медленно спускается в трубке в
виде плоского диска с расположенным над ним сла-
бым коническим послесвечением того же цвета,
легко водимым в темноте. Выше этого пламени на
стенках трубки отлагается белый налет, сильно
пахнущий формальдегидом, Пламя распада метил-
нитрита распространяется в пирексовой трубке
диаметром всего 0,7 с.м. По-видимому, оно способ-
но распространяться даже и при меньших диамет-
рах (0,65 и 0,53 с.и), хотя в этих условиях оно не
60 !20 <30 200
Температура, “С
Рис. 106. Изменение ско-
рости горения метя.тни-
трига с температурой
в трубке диаметром
2,56 мм
Скорость горения измеря-
лась ijo отношению к го-
рячему нагоревшему газу
видимо даже в темноте.
Скорость горения очень слабо возрастает при
увеличении диаметра трубки от 1 до 6 св и при 18° С сое гавднот около
3,2 см!еек. Несмотря на такую малую скорость, критический диаметр мал,
Произведение ud составляет всего ~ 3 см2[сек, в то время как для смесей
углеводородов с окисью азота оно равно ~30 см^сек, а для смесей угле-
водород— кислород—инертный газ ~ 12 см2/сех, возможно, потому, что
при горении метилннтрита осуществляется необычная для других смесей
реакция, протекающая с значительной скоростью при относительно низ-
ких температурах.
Личей пая скорость горения метилиитрита растет при повышении на-
чальной температуры (рис, 106), массовая скорость горения при этом
остается приблизительно постоянной; при 240°С ц выше горения не на-
блюдается. При приближенном измерении голой термопарой температура
пламени оказалась равной 1100° С; расчетная температура (по составу
продуктов горения) — lf)80° С. Состав продуктов горения приведен в
табл. 18; в третьей графе таблицы прпиедоп состав, скорректированный
для приведения в соответствие со стехиометрией.
Таблица 18
Состав продуктов горения метилннтрита
(в молях па 100 молей сгоревшего мет ид нитрита)
ПРОДУКТ Moan, най- дештые ^ке.[Тс- р[Тм<щтальт[О Состав, скоррек- тированной для приведении в соответствие сп стехиометрией П[1ОДУт<Т Моли, най- денные экспе- риментально Состав, екоррек- твроеанннй для проведения в соответствие со стехиометрией
со 84 77 ( Lfil 112^4 1,5 1,5
1Ь 59 64 СН2О 1 1
HiO 51 51
NO 35 34 Бала нс по элементам
N-.O 26 24 с; 107 100
С IEOH 11 И И 289 300
Na 5 5 О 210 200
СИЛОН 2 2 N 105 100
135
Этому составу соответствует уравнение горения
CH30N0-> 0,77СО + 0,64Н2 + 0,42НЕО +
+0.34NO + 0,24Л’гО + 0,05Ni + 0,11СН30Н +
+ 0,10611,0 + 0,06NH3 + 0,Q2CH3XOII + 26,4 ккал/моль. (3.6)
Спектроскопический анализ пламени (с добавлением небольшого коли-
чества кислорода для его усиления) тгоказывает, что пиролиз проходит в
основном до образования пламени, причем образуется окись азота, которая
лишь частично реагирует в этой зоне.
Весьма интересно отсутствие горения при предварительном подогреве
до 240° С и выше. По-видимому, причиной этого является образование
продуктов, которые не в состоянии участвовать в самоподдерживающихся
реакциях в пламени или могут даже затруднять их. Время пребывания
метилнитрита в нагревательной трубке составляет около 7 сек., разложе-
ние при 240° может пройти при этом на 2—3%, что оказывается доста-
точным, чтобы сделать невозможным горение.
Не исключено, что с этим связало ц отсутствие влияния начальной
температуры (при меньших ее значениях) на массовую скорость горения.
Естественное увеличение скорости при повышении температуры компен-
сируется ее уменьшением, которое выше 240° С делает горение невоз-
можным.
Попытка рассчитать скорость по формуле Зельдовича — Франк-Каме-
нецкого, исходя из допущения, что ведущей является та же реакция
первого порядка, которая определяет скорость медленного распада
(Е = 36,4 ккаЛ/моль, В = 1,84-1013 сек—1), дала пл = 120 см/сек, вместо
3,2 см/сек, полученной при опыте. Это обстоятельство наряду с различием
состава продукт он превращения в обоих случаях приводят и заключению,
что реакция начального этана термического распада не является опреде-
ляющей при процессе горения.
Холл и Вольфхард [141] изучили спектроскопически многозональ-
ные пламена, образующиеся при горении некоторых нитритов или
смесей нитратов, нитритов и китросоедипенпй с кислородом или воз-
духом.
Известно, что ряд пламен, поддерживаемых окисламп азота при атмо-
сферном давлении, состопт из двух зон, различающихся но цвету и спектру
и иногда разделенных несветящимся промежутком. Такие пламена наблю-
даются при горении углеводородов с двуокисью азота, водорода с.о смесью
кислорода и окиси азота. Было установлено, что первые зоны получаются
от реакции между горючим и NOs и горючим и О? соответственно, а вто-
рые зоны связаны с реакцией между горючим и NO, поскольку последи но
реакции требуют более высокой температуры ц идут медленнее, чем пер-
вые. Было также установлено, что окись азота реагирует по одному из
двух путей в зависимости от химической природы горючего; с углеводо-
родами и особенно с аммиаком реакция проходит при умеренных темпе-
ратурах по радикальному механизму, но в Отсутствие соответствующих
радикалов, как это имеет место в пламенах с На или СО, окись азота под-
держивает горение смеси, если конечная температура пламени достаточно
высока для быстрого выделения кислорода в результате разложения
•окиси азота.
Учитывая эти данные и результаты спектроскопического исследования
различных вон при горении нитратных, нитритных и других смесей, авто-
ры приходят п следующим заключениям относительно механизма хими-
ческих процессов в этих случаях, протекающего трехзона.чьно, причем
зоны пламени отличаются по цвету и спектру.
136
Для пламени метилнитрата без примесей следует принять, что в на-
чальной первой зоне молекула эфира распадается, возможно, по урав-
нению
CH3ONOs -> СНзО + NOS -* СО + Нг (и (или) СЩО) +
+ NO + О2 4- NOa. (3.7)
Во второй зоне
На + СО + Оз + NOs — СО + СО2 + Нг + НгО + NO, (3,8)
В третьей зоне На, СО и СНгО легко реагируют с NO лишь при темпе-
ратурах выше, чем те, которые получаются в метилпитратпых пламенах
при горении под низкими постоянными давлениями, поэтому третьей зоны
lie наблюдается. Она получается, однако, при взрыве в замкнутом сосуде,
когда достигается значительно более высокая температура.
Для стехиометрической смеси метилпитрнта с кислородом в первой
(голубой) зоне большая часть эфира распадается на малые осколки,,
формальдегид и NO; одпако образуются и относительно большие молекулы
предположительно СНаОН и, возможно, немного NOa. Во второй (оран-
жевой) выраженной слабо зоне не происходит существенных реакций.
В третьей (фиолетовой) зоне
Н.СО + Ог -> СО + Нг + НаО, (3.9)
Органическое горючее сгорает за счет имеющегося кислорода и закиси
азота, если она есть, и затем с частью NO, образуя полосы CN и NH. Реак-
ции с О2 предшествует реакция с N2O и NO.
В пламени стехиометрической смеси нитрометана и кислорода первая
зона отсутствует, во второй зоне происходит распад
CH;tNO3 -+ СПз + КОг (3.10)
п дальнейшее окисление кислородом и N'O2. В третьей зоне органические
молекулы, предположительно образовавшиеся из начальных осколков
(СНз, СГ12 и т. п-), реагируют с О2 и затем немного позже в пла-
мени с NO.
Для аналогичной смеси этнлнитрата в первой зоне эфир распадается
на NO2, формальдегид, ацетальдегид п другие органические молекулы
близких размеров. Во второй зоне происходит частичное сгорание молекул
горючего за счет NO2, в третьей — кислород реагирует с органическим го-
рючим п затем последнее реагирует с NO.
Термический распад метилннтрита при умеренно повышенной темпе-
ратуре образует окись азота, метиловый спирт и формальдегид. Эта реак-
ция приближенно термонептральна, и поэтому была неожиданной способ-
ность метилннтрита к горению. Определение состава продуктов последнего
показало, однако, что при горении часть азота восстанавливается до за-
киси азота и даже до элементарного азота. В итоге реакция является
экзотермической, что делает понятной ее способность к самораспростра-
неиию.
Первым этапом химического превращения при горении, как и при
медленном термическом распаде, является отщепление окиси азота. Оста-
валось неясным, как и Почему происходит при горении переход от термо-
не и тральной к экзотермической реакции и какие из происходящих реак-
ций являются ведущими и этом случае. Для выяснения этих вопросов
изучалось горение и медленный термический распад паров метилнитри-
та [142].
137
Аналитическое определение состава продуктов горения приводит к сле-
дующему уравнению превращения:
CILONO = 0.72СО + 0,36Н2 + 0,54NO + 0,4211=0 + ().14N2 + ОЛЗСПзОН +
+ 0,10CII20 + 0,06NH3 + 0,05N20 + 0,02CJI2NOH + 0,02CHt +
+ 0,01 CO2 4- 27,1 ккал, (3.11)
что соответствует температуре горения 1110° С.
При повышении начальной температуры скорость растет (от 3,2 сл/сек
при 16° до 6,08 см!сек при 180° С). Расчет энергии активации по этим дан-
ным дает 28,5 ккал/моль.
Инертные разбавители, понижая температуру горснця п изменяя теп-
лопроводность и коэффициент диффузии, уменьшают скорость горения;
сильнее всего влияет гелий, за ним следует аргон и азот. Расчетные тем-
пературы горения хорошо согласуются с экспериментально измеренными.
Расчет энергии активации по этим последним температурам дает
32 ккал/моль.
Изучалось также влияние на характер, скорость, температуру и
устойчивость пламени добавок закиси азота, окиси азота и кислорода
(рис. 107). Добавление до 30% (молярных) окиси азота делает пламя
бо.тш! ярким, но не меняет его структуры. Температура горения при этом
повышаемся от 1100 до 1200° С, а скорость возрастает от 3,2 до 4,3 см/сек.
При 30% характер пламени резко меняется. Из оранжево-красного мгаго-
зональпого оно превращается в двузональное с ярким сине-фиолетовым
внутренним конусом, окруженным тонкой красной второй зоной. Присут-
ствие закиси азота даже в небольших количествах, не меняя вида и харак-
тера пламени, приводит к сильному увеличению скорости горения. Воз-
можной причиной этого увеличения является образование при разложении
N2O кислорода. При добавлении последнего скорость горения сильно воз-
растает.
Термический распад метплнмтрита изучался проточным методом и в
отличие от предшествующих исследований также и при более высоких
температурах (от 250 до 700°С). Уж? при 250° ('. в отличие от «теорети-
ческого» уравнения, предусматривающего образование только метанола,
формальдегида m окиси азота, получались заметные количества воды и
закиси азота. При 700° С восстановление азота было еще больше:
CH3ONO = 0,11СН3ОН + 0,10СН2О + 0,42 Н2О +
+ 0,46N2O + 0,53NO + 0,15N2 + 0,72СО + 0,5ЭН2 +
+ 0,02CH2NOH + 0,06NH3 + 0,01СН4 + 0,01С02, (3.12)
соответственно этому экспериментально определенный саморазогрев при
распаде увеличивался: при 250° он составлял 50°, при 700—300°С.
Вероятной причиной образования закиси азота является взаимодей-
ствие метокс пра дикая о в с окисью азота с образованном формальдегида и
нитроксила, дающего затем закцсь азота и воду.
Менее вероятно образование закиси азота по реакции
NO + HNO — ОН + ]Ч2О. (3.13)
Закись азота в сною очередь может восстанавливаться до азота атома-
ми водорода, образующимися при пиролизе метоксила млн формальде-
гида.
Вопрос о ведущей при горении реакции остается неясным. Расчет
скорости горения при допущении, что ведущим является отщепление NO,
дает скорость горения, равную 23 см/сек, против экспериментально наблю-
133
дающейся 3.2 см/сек. Возможно, поэтому, что первичная реакция проте-
кает при горении при более низкой температуре, чем максимальная.
Изучение горения при низких давлениях [143] имеет то существенное
преимущество, что зона протекания реакции сильно растягивается в про-
Рис. 107. Влияние добапол-
на скорость горения мотил-
питрита
Гис, 108. Зависимость скорости
пламети в парах этилнитрита
от рдавленчя
странстве. Это облегчает как измерение
температурного профиля, так и особенно
отбор проб из разных частей зоны.
Температура
р, .И.«- р/li. Ггуг.
Рие, 109. Профили температуры и состав
продуктов горения этилнжтрата
Для этил нитрата, кацрпмер, при 30 мм рт. ст. толщина зоны горении
составляет 8 лмт, и то время как при атмосферном давлепПн она равна
всего 0,5 лом.
Кроме того, при низких давлениях отсутствуют различные вторичные
реакции — восстановление окиси алота1 пиролиз продуктов реакции -
it конечная температура соответственно ниже.
1 Годача паров этплпитрата осуществлялась путем испарения жидкости
и они поджигались в горелке с плоским пламенем. Ввиду слишком слабого
свече’гпя фронта горения поверхность его фотографировалась гплиреп-
методом. Одновременно измерялся профиль температуры при помощи пе-
ремещающейся в вертикальном направлении тонкой термопары и ироиз-
водилсл отбор газов посредством тонких (наружный диаметр d — 0,2 мм}
кварцевых капилляров.
Толщина зоны горения уменьшается при повыше пин давления и ско-
рости горения (рос. 108). В том интервале давлений (до 150 льм), где ли-
нейная скорость го ре пи я постоянна, толщина зоны горении обратно про-
порциональна давлению. Скорость горения рассчитывалась делением объе-
ма сгоревшего газа па поверхность фронта горения, Прц начальной тем-
пературе 60° С скорость горения составляет 12,8 см/сек ц конечная темпе-
ратура равна 786° К.
Но зависимости скорости горения от конечной температуры, изменяе-
мой разбавлением аргоном, рассчитывалась энергия активации ведущей
реакции Е ~ 38 ккал/молъ, что соответствует энергии разрыва связи
ВО -NO,.
139
Изменение состава и температуры газообразных продуктов горении
в его фронте изображено на рис. 109 (р = 35 ,и.и, и = 15 см!сект
Т = 800° К). По полученным зависимостям и тепловым характеристикам
реагирующей смеси могут быть рассчитаны скорости кондуктивного и
конвективного теплообмена и тепловыделения и. в частности, установ-
лено, что более 50% этилнитрата реагирует в зоне толщиной всего 2 мм
л в узкой области температур 750—800° К.
Одним из промежуточных продуктов горения является этилнитрит.
Следует отметить также образование -около 80 молей форм альдегида на
100 молей этилнитрата, несмотря па то, что ’/s ого распалась на Са-соеди-
ленпя — ацетальдегид н этанол. Помимо 5 молей метана и 2 модем
нитрометана, остальной углерод получается в виде соединении, содер-
жащих С — О-связи. Отсюда следует, что связь С — С рвется сравнительно
легко с последующим окислением. Другим существенным обстоятельством
является наличие в продуктах горения этанола, эт-илн-итрита, метанола н
метилннтрита, что говорит за участие в процессе горения как этоксила,
так и метоксила. Поскольку эти.тнитрнт является одним -из основных
продуктов медленного термического распада, хщжпо заключить, что в
низкотемпературной зоне пламени химические реакции сходны с проте-
кающими при медленном распаде.
Распад этоксила, незначительный ниже 200'С, когда при медленном
распаде образуется преимущественно этанол и ацетальдегид, становится
быстрым выше 200° С, когда среди продуктов преобладает формальдегид
и продукты реакций метильных радикалов. Допуская, что мгтнл-радикал,
образующийся в высокотемпературной зцпо, реагирует с NOs быстрее,
можно Припять следующую общую схему горения этилнитрата ирп низких
давлениях;
СгН5ОКО2(+М) ->C2HSO + NOa(+M), (3.14}
С2Н5О Х-> Сг-соедивения, (3.15)
СгН5О(+М) ->СН., + СН20(+М), (3.16)
CH., + NO, -> СН2О + NO, (3.17)
СНз, СТРО 4- X —> С 1-соединения, (3.18}
В этой схеме М обозначает любую молекулу, которая, сталкиваясь с мо-
лекулой или радикалом, активирует его, а X — соединение, способное
реагировать со свободным радикалом; им может быть как молекула, так
и сам свободный радикал.
В низкотемпературной области соединения X для второго типа реак-
ций могут быть подразделены на такие, как NO2, которые отбирают
Н-атом у этоксила, и такие, как формальдегид, которые отдают Н-атом,-
соединения типа окиси азота реагируют с этоксилом, возможно, при
тройном соударении.
При повышении температуры в зоне пламени отношение С2: С.;
уменьшается в результате -изменения механизма распада; по-видимому,
происходит термический распад этоксирадикала па формальдегид и мс-
тид-радикал; энергия активации этих реакций (предположитолэю в
пределах 12—34 ккал/молъ) ffonnie, чем для большинства реакций отби-
рания водорода.
Авторы полагают, что эндотермическое расщепление этцлнитратд на
этоксил н NO2 является ведущей реакцией, протекающей при р < 150 .н.«
псевдомолекулярио. Последующие быстрые экзотермические реакции а,т-
коксильных и алкильных радикалов с двуокисью азота поддерживают
пламя.
140
2. ГИДРАЗИН
Горение, основанное на экзотермическом распаде одного соединения,
является наиболее простым из возможных его типов. Его примером мо-
ще г быть горение гидразина [144], образующего при распаде только азот,
водород и аммиак. Это упрощает анализ и термохимические расчеты.
Кипетика начального этапа при гомогенном распаде (разрыв связи N—N)
также известна. Основными являются зависимость скорости горения от
давления м(р), по которой может быть установлен суммарный порядок
реакции, и зависимость скорости реакции от температуры a(i), по кото-
рой может быть рассчитана энергия активации.
Горение изучалось как для газообразного гидразина в закрытом сосуде
шлирен-методом при разных давлениях — до 10 ем рт. ст. (62° С), так и
для жидкого в узких трубках при давлениях от 10 см до атмосферного.
Рис. 110. Давление зату-
хания для пламени раз-
ложения жидкого гид-
разина
ГЫп-пснтрацив ЬКЩ (в %):
J — 37,7; 2 — 95,9'. .3 — 92,7,
4 — 9t ,4
Рис. 111. Пределы стабиль-
ности и скорости горения
жидкого гидразина в труб-
ке диаметром 5 мм. Резуль-
таты выше 1 ат взяты из
работы [147]
Рис. 112. Влияние раз-
бавления на зависимость
скорости горения гидра-
зина от давления
Концентрация N.H, (в %):
1 — 100; 3 — 99; 3 — 97.1
Скорость горения (в см'сгх-}:
1 — 0.0С,; а — 0,05; 3 — 0,04;
4 — 0,03; 5 — 0,02
Состав продуктов торопил жидкого и парообразного гидразина не за-
висит от давления и близко соответствует уравнению
N,H4->-NHs-i-l/2N2-rl/2H2. (3.19)
Изменение давления в 4 раза (от 2 до 8 см рт. ст.) слабо увеличивает
линейную скорость горения паров гидразина, причем это увеличение, не-
видимому, обусловлено особенностями методики. Поэтому следует заклю-
чить, что ведущей является реакция второго порядка. При пересчете на
скорость горепця относительно несгоревшего газа пн получается равной
117 см! сек. Разбавители (Пе, Аг, N2 и NH3) уменьшают скорость горе-
ния. Водород и азот, имеющие большую теплоемкость, чем аргон и гелий,
сильнее снижают скорость. Влияние инертного газа определяется прежде
всего снижением температуры горения, а также изменением теплопровод-
ности. Скорость больше в смесях с большей теплопроводностью; для сопо-
ставления с теорией теплопроводность рассчитывалась применительно к
температурам, достигаемым при горении.
В опытах по горению жидкого пидразина измерялось также критиче-
ское давление (давление затухания). Оно возрастало при уменьшении
диаметра трубки, а также при разбавлении гидразина водой. Величина
141
диаметра затухания пропорциональна обратной величине давления
(рис. 110).
При постоянном давлении скорость горения меньше в трубках боль-
шего диаметра. Область горения, в зависимости от содержания воды,
показана на рис. 111, ограниченная нижним и верхним пределами но
давлению. Влияние давления и концентрации на этом рисуй не предетав -
лены в виде линий равных скоростей.
Скорость горения, если ее определять в трубках одинакового диаметра,
растет с увеличением давления, пе пропорпионально ему, а медленнее.
Предполагается, что это связано с теплообменом со Степками трубки.
Если скорость горения при каждом давлении измерять, применяя диаметр
трубки, равный пятикратному значению критического диаметра, то полу-
ченные данные показывают (для 97,7% гидразина) и интервале давлений
0,5 — 1 ат прямую пропорциональность скорости горения давлению. Сопо-
ставляя зависимость скорости горения от температуры горения, изменяе-
мой разбавлением инертными газами (с учетом влияния этого разбавле-
ния па теплопроводность), получаем энергию активации, равную
96 ккал/молъ. Эта величина, так же как и полученная предыдущими
исследователями в опытах по горению (30—45 ккал/молъ), значительно
меньше величины энергии активации реакции
NJE-^NIb (3.20)
(А — G0 ккал/молъ).
Кроме того, при расчете скорости, ос нова и пом на этой послед пой
величине, w = 30 см/сек, вместо 200 см/сек, полученных в опыте.
Таким образом, первичная реакция распада не является ведущей при
горении.
Антуан [145] изучал горение Жидкого гидразина в толстостенных
пирексовых трубочках различного внутреннего диаметра (5,6; 9.5 и
12,7 мм) при давлениях от 1 до18аг.
На графике (рис. 112) показана зависимость и(р) для гидразинов
концентрация 100; 99 и 97,1%. В некотором интервале давлений, тем
большем, чем меньше Диаметр трубки, скорость горения растет с давле-
нием по насыщающейся кривой, затем скорость начинает расти линейно
И гораздо быстрее. Для гидразина, содержащего воду, скорость горения,
естестве оно, меньше и переход горения да быстрый режим наступает
при большем давлении.
Изучалось также влияние некоторых других разбавителей, добавлен-
ных в количестве 1% по объему, которые оказывали действие, аналогич-
ное воде и возрастающее при переходе от метилгидразпна [кривая и(р)
для которого почти не отличается от кривой для чистого гидразина] г; эти-
ловому спирту, этилен дна мину, н. ами.чамппу, третичному бутиловому
спирту, пиридину п н. бутиловому спирту.
Обращает па себя внимание большое сходство указан вых зависимостей
с установленными ДлЧ яитрогликоля, который близок ц гидра зилу и но
абсолютной величине скорос.ти горения; по-видимому, малые скорости
соответствуют режиму нормального горения, большие — турбулентного:
последнее наступает тем позже, при больших .давлениях, чем меньше ско-
рость горения и диаметр трубки. Отличие от рптрогликоля состоит в том,
что скорость нормального горения пропорциональна це первой, а значи-
тельно меньшей степени давления. Возмояшо, что это связано с тем, что
нормальное горение ведет, как и медленный термический распад, реакция
первого порядка; заключительная л;о реакция, которая становится веду-
щей при горении на турбулентном режиме, является реакцией второго
порядка, в соответствии с чем скорость растет пропорционально дав-
лению.
142
Мюрре и Холл [146] определяли, измеряя размеры конуса пламени,
Нормальную скорость горения гадразила распадающегося, согласно экс-
псрименталыю подтвержденному суммарному уравнению
2КНэ 4" Na 4" (3,21)
При начальной температуре 150° распад по этому уравнению дает по
расчету температуру 1660° С; распад до элементов дает значительно более
низкую температуру ~1100°С. Е, по Шварцу, равпо 60 ккал/молъ. Опре-
делялась также скорость горения смесей парен с кислородом, которое
предположительно осуществляется в дна этапа, т, е. первоначально обра-
зуется смесь аммиака с азотом и водородом, а затем водород п аммиак
реагируют с кислородом. Сопоставляя скорость горения этой смеси со
скоростью горения искусственной смеси (2NHj 4- [Ь + Оа), температура
горения которой по расчету на 1100° С выше, можно установить влияние
температуры горения на его скорость. Это можно сделать также, разбав-
ляя гидразин водой.
Горение гидразина привлекало в последние годы внимание исследо-
вателей [148] по трем основным причинам.
С одной стороны, гидразин имеет значительную отрицательную теплоту
образования, Поэтому распад его на элементы1 2 достаточно экзотермичен.
чтобы можно было ожидать протекания его в форме саморарнространяю-
тцегося процесса.
Во-вторых, молекула гидразина содержит атомы всего двух элементов,
it поэтому естественно ожидать простоты состава как конечных, так я
промежуточных продуктов распада по сравнению, например, с нитритами
или цитратами многоатомных спиртов. Это делает его более удобным
объектом для сопоставления экспериментальных закономерностей горения
с закономерностями, вытекающими из различных теоретических пред-
ставлений об Этом процессе. Далее, гидразин и некоторсче его производ-
ные в сочетании с окислителями получили в последнее время значитель-
ное применение в ракетной технике. Наконец, изучение гирения самого
гидразина и систем гидразин 4- окислитель может помочь выяснению роли
пиролиза горючего при горении таких систем.
При исследовании горения гидразина, поддерживаемого кислородом,
закисью азота н окисью азота, был установлен распад избытка реагсптов
в песгехпометричесццх смесях и исследованы условия перехода при обога-
щении смеси горючим от относительно горячего пламени окислщгпя к
пламени разложения.
Горение паров гидразина изучалось при 62° С шлиреп-методом. В этих
условиях скорость горения мало меняется при изменении давления от
20 до 80 мм рт. Ст. Эта независимость скорости от давления соответствует
второму порядку ведущей его реакции.
Состав продуктов горения хорошо соответствует уравнению
ГУН; —>- l.OONHj 4- 0,!50N2 4- 0,50Нг 4- 33,74 ккал[молъ. (3.22)
Как показал расчет, температура горения составляет 1904° К, Линей-
ная скорость горения при 40 льм — И2 см/сек.
Разбавление гидразида замедляет горение (рис. 11,3). Основную роль
ирд этом нтрает теплоемкость разбавителя, определяющая снижение тем-
1 Ь’эмфорд [147] показал, что пары гидразина могут взрываться от искры или на-
гретого тела. Имеются ппжнпй п верхней пределы давления; ипжпин предел воспла-
менения от искры составляет 38 мм в трубке диаметром 3 ел.
2 Любопытно, что при торении гидразина в продуктах содержится довольно
много аммпрка, вследствие чего тепловой эффект горепря оказывается заметно
больше, чем од был бы при распаде па элементы.
143
пературы горения; азот и недород, имеющие большую теплоемкость, чем
аргон и гелий, оказывают более сильное влияние.
Существенна также теплопроводность, влияние которой проявляется
особенно наглядно, когда разбавитель (например гелий и аргон) имеют
одинаковую теплоемкость — горение распространяется быстрее в системах
с большей теплопроводностью. Расчет энергии активации ведущей реак-
ции дает 35 000 ккал/молъ, по температурной зависимости скорости горе-
ния [146] Е = 36 500 ккал/моль.
Скорость пламени в гидразин-кислородных смесях (4020 см!сек для
стехиометрической смеси
Парциальное ваеление разбави-
теля, еж рт, ст.
Рис. 113, Влияние разбавите-
лей на скорость распростране-
ния пламени (и) в парах
гидразина. Давление КгЩ —
5,02 см рт. ст.
при 40 мм) значительно больше, чем самого
гидразина (1270 см/сек), В смесях, содержа-
щих гидразина больше, чем соответствует
стехиометрии, величина повышения давления
показывает, что стехиометрическое количе-
ство гидразина сгорает, а избыток разлагает-
ся, причем до содержания гидразина 75%
продуктами разложения являются водород и
азот, при большем содержании гидрази-
на продукты горения содержат также ам-
миак.
Для стехиометрической смеси с закисью
азота скорость пламени при 40 .м.и составляет
2400 см/сек (скорость горения 164 см/сек).
В смесях с избытком гидразина он разлагает-
ся, при недостатке гидразина избыток закиси
азота разлагается па азот и кислород,
В смесях с окисью азота наибольшая ско-
рость распространения наблюдается при не-
котором избытке гидразина. В смесях, содер-
жащих избыток NO, при химическом анализе
обнаружено ее разложение.
Теплота сгорания изучавшихся смесей приблизительно в 5 раз больше,
чем теплота горения самого гидразина; однако скорость и температура
пламени различаются гораздо меньше. Главной причиной является увели-
чение диссоциации продуктов горения при повышении его температуры,
а также различие теплоемкости продуктов горения.
Интересно, что при почти тождественных по температуре и составу
продуктов горения смесях гидразина с закисью азота и с окисью азота
(разбавленных азотом) скорость первой смеси 164, а второй только
115 см!сек, что соответствует относительно низкой реакционной способ-
ности окиси азота. Однако в сочетании с гидразином окись азота все же
оказывается очень реакционноспособной по сравнению со смесями, где
горючим является углерод или водород; реакционная способность NO по
отношению к гидразину даже выше, чем по отношению к аммиаку, воз-
можно, потому, что окись азота легко взаимодействует с радикалом Nib.
образующимся в аммиачных пламенах, и, вероятно, также в смесях на
основе гидразина,
При толковании горепия смесей гндразипа с окислителями в принципе
возможно предположение, что ведущим является распад гидразина, а роль
последующего окисления ограничивается повышением температуры, Одна-
ко постепенное увеличение скорости горения при добавлении кислорода
пли окиси азота говорит против этого предположения и в пользу того, что
окисление в обоих случаях протекает быстрее разложения, хотя пельзя
считать исключенной возможность одновременного протекания обоих про-
цессов, в пользу чего говорят также спектроскопические наблюдения.
В смесях с закисью азота соотношение обоих процессов зависит от содер-
жания горючего.
144
3. НИТРОСОЕДИНЕНИЯ И НИТРАМИНЫ
Вблизи атмосферного давления скорость горения тетрила и гексогена
[149] линейно растет с давлением.
В интервале давлений от 1 до 50 ат по опытам М. С. Плясунова для
прессованных в плексигласовые трубочки тетрила и гексогена, скорость
растет не пропорционально давлению, а медленнее (рис. 114), для тетри-
ла она может быть выражена соотношением нм — 0,065 р9"69, для гексоге-
па — и-и — 0,054 p0|So.
В более широком диапазоне давления — до 1000 ат — зависимость ско-
рости горения от давления для тротила, пикриновой кислоты, тетрила и
гексогена, по опытам Глазковой и Терешкина [118], имеет вид, изобра-
женный на рис. 115. Для пер-
вых двух веществ эта зави-
симость во всем интервале дав-
лений линейная. Для тетрила
кривая н(р) до 250 ат имеет
Рис. 115. Зависимость скорости горения
тротила (7), пикриновой кислоты (2),
тетрила (3), тэна (4). гексогена (5) и же-
латинированного нитроглицерина (б) от
давления
Рис. 114, Зависимость скоро-
сти горения тетрила (2)
и гексогена (2) от давления
насыщающийся характер, а при больших давлениях скорость растет с дав-
лением линейно; линейной формулой может быть приближенно выражена
в интервале 100—ЮОО ат и скорость горения гексогена. Численные значе-
ния коэффициентов формул приведены в табл. 19.
Таблица 19
Значения коэффициентов Л и £ в формуле и = А 4- Вр
для ряда ВВ
ВВ Интервал давле- ний, в котором еораведлива формула, ат А В
Тротил 20—950 0,04 0,00716
Пикриновая кислота . 25—950 0 14 0,00805
Тетрил * 250—950 0,04 0,0114
Гексоген 100—1000 0,90 0 0216
• В интервале давлений 10—250 am им =0,063 рО,6Э5_
10 К. К, Андреев
145
4. ИНИЦИИРУЮЩИЕ ВЗРЫВЧАТЫЕ ВЕЩЕСТВА
Приведенные выше данные относятся ко вторичным, т. о. относительно
устойчиво горящим ВВ. Большой интерес, естественно, вызывало также
определение зависимости и(р) для инициирующих ВВ тем более, что
неустойчивость их горепия предположительно связывалась с большой
величиной скорости горения и влиянием на нее давления. Однако именно
неустойчивость горения осложняла указанное определение — можно было
опасаться, что инициирующие ВВ вообще неспособны стационарно гореть
и что такими методами, как, например, повышением плотности иди по-
нижением внешнего давления, удастся лишь уменьшит!, неустойчивость,
увеличить участок горения до возникновения взрыва. Поэтому первые
исследования [150] в указанном направлении были проведены с ини-
циирующими ВВ, разбавленными инертными примесями или вторичными
ВВ, уменьшающими скорость горения. Предполагалось, что, опреде-
лив скорость горения как функцию содержания разбавителя, можно
будет экстраполяцией установить скорость горения чистого инициирую-
щего ВВ.
Опыты были проведены с гремучей ртутью и трпиитрорезорцинатом
синица с тальком, тетрилом и нитроклетчаткой в качестве разбавителей.
В зарядах малой плотности смеси гремучей ртути с тальком горели нерав-
номерно — с большой и сильно колеблющейся скоростью, смеси тринигро-
резорцината свипца с тальком или тетрилом при малых содержаниях
разбавителя дают вспышку или взрыв, при больших — зге торят. Смеси
трпиитрорезорцината с нитроклетчаткой в определенном интервале со-
става способны к горению и по скорости, быстро растущей при увеличении
содержания тринитрэрезорципата или давления, в несколько раз превос-
ходят нитроклетчатку.
Впервые Беляев показал, что многие цз инициирующих или быстро-
горящих ВВ (гремучая ртуть, тринитротриазцдобепзол, перекись тряцик-
.чоацетона, диазодинитрофенол, пикрат калия) в сильно уплотненном
состоянии способны к стаппонарному горению не только в вакууме, но
и при повышенных (до 150 ат) давлениях [151]. Была измерена их ско-
рость горения, в десять и бодее раз превосходящая скорость горения
наиболее быстро горящих вторичных ВВ. Беляев открыл также суще-
ственную особенность горения инициирующих ВВ — протекание рто с
интенсивным диспергированием. Этот режим горения типичен для ипп-
циирующих ВВ, но осуществляется также и для вторичных ВВ при неко-
торых особых условиях горения.
Внешне горение изученных инициирующих ВВ выглядит следующим
образом. При горении гремучей ртути под пониженным давлением
(150 мм) к поверхности заряда непосредственно прилегает темная зона
значительного протяжения и яркое пламя возникает лишь на некотором
расстоянии от поверхности. При давлении ниже 40—50 мм пламя исче-
зает полностью, таблетка «тает» без видимого света, а на стенках сосуда
образуется значительный налет загрязненной гремучей ртути, количество
которой составляет пе меньше 2/з первоначального заряда. При горении
спрессованной гремучей ртути в вакууме иногда наблюдается разламы-
вание таблетки па кусочки малой величины, которые или догорают или
остаются несгоревшими.
В опытах И. Ф. Похила горение гремучей ртути в вакууме идет устой-
чиво при давлении 10—15 мм и температурах выше —20°; температура
на повер-хности конденсированной фазы составляет 270—320° С. С ростом
давления наблюдается холоднопламеяное горение, а при 25—30 .«.и на
значительном расстоянии над поверхностью образчика появляется пламя,
видимое при дневном свете. Тепловой эффект реакции и конденсирован-
ной фазе составляет, по расчетам Похила, 48—60 кал!?-,
Рис. 116. Фотография горения гремучей ртути
I__таблетка гремучей ртути непосредственно перед началом горенья; г — начало газифика-
ция, времц J • 10-3 сек; ь— появление слабого свечения, времл 4- 1П-3 сек; 4—появление
пламени на Спирали, греми 6 1(М сек; S — проскок пламени в спираль, время 14 10J сек;
6 — спираль сдвинулась в сторону, сгорело ','з таблетки, время 130- 10—3 сек
р, кгfa*2
% азида свинца
Рис. 117. Зависимость скорости горний® Рис, 118. Скорость горения смесей азида
гремучей ртутя (7) я пикрата палия (2) свинца с гремучей ртутью при темп?
от давления ратурс жидкого азота
10*
Другие изученные Беляевым ВВ ведут себя при пониженных давле-
ниях несколько иначе. При горении перекиси трициклоацетона под дав-
лением 100 мл на стенках колокола появляется белый налет. Если собрать
и поджечь этот налет, то он легко вспыхивает. Однако это не перекись,
а вещество с более высокой температурой плавления и менее летучее.
При горении тринитротриазидобензола по мере понижения давления
пламя вначале делается более бледным и вытягивается, затем оно умень-
шается в размерах. При 10 лл пламя очень слабое и маленькое.
Для определения зависимости н(р) гремучей ртути при повышенных
давлениях (152] таблетки ее (<? = 4 мм, h = 5 — 6 л.и, относительная плот-
ность 0,96), покрытые с боковой поверхности слоем шеллака или вазе-
лина, поджигались нихромовой проволочкой в бомбе, наполненной азотом.
Регистрация горения производилась при помощи лупы времени в слабом
проходящем свете (рпс. 116). В этих условиях опыта при атмосферном
давлении видимого свечения в начале горения не обнаруживается; если
пламя позже появляется, то оно находится ла значительном расстоянии
от поверхности таблетки и существенного влияния на скорость и харак-
тер горения не оказывает. Не наблюдается этого влияния и при повышен-
ных давлениях, когда пламя приближается г; поверхности заряда. Зави-
симость скорости горения от давления изображена на рис. 117. С повы-
шением давления рост скорости замедляется. Если ранее [153] на
основании результатов опытов при пониженных давлениях он был выра-
жен соотношением = 0,4 + 1,1 р, то при повышенных давлениях
п.ч = 1 + 1,04 p0,s.
Вплоть до еще больших давлений (1000 ат) скорость горения гремучей
ртути (содержащей 5% парафина) определил С. В. Чуйко. При относи-
тельной плотности 0,95—0,99 горение в плексигласовых трубочках- про-
ходило в большинстве опытов равномерно и без перехода во взрыв. Ско-
рость горения велика; она росла с давлением по насыщающейся кривой
и достигала при 400 ат 18 г/сл»2се«.
Беляев [154] определил также скорость горения прессованных смесей
некоторых инициирующих или быстрогорящих веществ в зависимости от
соотношения компонентов. Добавление к гремучей ртути всего 2% азида
свинца приводит к детонации при поджигании. При температуре жид-
кого азота смеси горят вплоть до содержания 15% азида, причем скорость
горения значительно возрастает при увеличении содержания последнего
(рис. 118). Скорость и устойчивость горения сильно зависят от характера
бронировки боковой поверхности заряда. Если вместо вазелина приме-
нить тонкий слой коллодия, то скорость горения становится значительно
больше и оно уже при меньшем содержании азида переходит во взрыв.
Скорость горения смеси относительно медленно горящего (0,70 см/сек)
трпнитротриазтщобе изола с более быстро горящей гремучей ртутью
(1,6 см/сек) растет при увеличении содержания последней сначала мед-
ленно, а затем, начиная с 70% гремучей ртути, быстрее (рис. 119).
Скорость горения тринитротриазпдобензола с тринитрорезорципатом
свинца (рис. 120) также возрастает при увеличении содержания быстрее
горящего компонента; как и в предыдущем случае, рост идет сначала
медленно, затем гораздо быстрее. Особенностью этой смеси является
неспособность ее к устойчивому горению в некотором (30—40%) интер-
вале содержания тринитрорезорцината — при поджигании горение воз-
никает, по происходит оно неравномерно, как бы отдельными толчками,
и ио прекращении действия поджигателя затухает.
Беляев и Кондрашков изучали горение пикрата калия при повышен-
ных (до 125 ат) постоянных давлениях [155]. Светлов и Фогельзанг опре-
делили зависимость скорости горения от давления (до 400 ат) пикратов
ряда металлов — лития, патрия, калия, рубидия, бериллия, магния, бария,
ципка, серебра, свинца и железа (двухвалентного и трехвалентного),
148
тринитробензоата калия, а также некоторых стпфнатов, в частности,
стифната свинца [156]. Опыты производились с прессованными до отно-
сительной плотности 0,9—0,95 лакированными с боковой поверхности или
запрессованными в плексигласовые трубочки цилиндриками вещества в
бомбе постоянного давления и атмосфере азота.
Скорость горения пикрата калия (как н большинства других пикра-
тов) при атмосферном давлении невелика (0,40 см/сек), но очень быст-
ро растет с давлением, проходит при 8 ат через максимум (7 см/сек),
уменьшается при 80 ат до минимума (2,50 см!сек) и затем вновь, но уже
медленно растет, достигая при 350 ат 5 см}сек (рис. 121),
Кривая зависимости скорости горения от давления для некоторых из
изученных пикратов п стпфпатов того же типа, что п для пикрата калпя;
для других она существенно отличается, приближаясь к типу кривых, ха-
рактерных для гремучей ртути или пикрата свинца. Сильно отличаются
различные пикраты п по величине скорости при повышенных давлениях,
которая изменяется от 3 до 30 см!сек.
Удалось получить устойчивое горецле наиболее быстро горящего нз
изучавшихся веществ—стифната свинца. В толстостенных (2 .и.и) плек-
сигласовых трубочках малого диаметра стифнат свпцца горел устойчиво
во всем изучавшемся диапазоне внешнего давления —от 15 мм рт, ст. до
400 ат. Скорость горе идя очень велика (25—26 сж/сгк) уже в вакууме,
слабо растет при повышении давления до 50 ат, а затем — до 400 ат —
остается почти постоянной (рис. 122). По характеру кривой и(р) стифнат
свинца сходен с пикратом свинца. Последний отличается, однако, неспо-
собностью гореть в данных условиях опыта при давлениях шгже 20 ат.
При том же давлении (18 пт), начиная с которого пикрат свинца приоб-
ретает эту способность, скорость его горения почти в 30 раз меньше, чем
стифната, но быстро растет с давлением и выше 200 ат лишь па 10—1,т%
меньше, чем скорость горения стифната,
Б, С. Светлов и А. Е. Фогельзанг в своих опытах обнаружили своеоб-
разное влияние оболочки заряда быстро горящего ВВ па его горнице. Так,
с пикратом свинца в трубках внутренним диаметром 7 мм и толщиной
стенки 1 мм при давлениях выше 30 ат происходили взрывы, возможно,
из-за недостаточной прочности трубки, расширявшейся при горении; при
Этом возможно образование щели, куда могли проникать газы. Утолщение
стеики до 2 м.м с одновременным уменьшением внутреннего диаметра до
4 льм устраняло возникновение взрывов.
Далее было обнаружено, что, если торец находится у среза трубки
или заряд не имеет прочной оболочки, но лишь лакирован сбоку, то ско-
рость горения значительно (приблизительно в 4 раза) больше, чем в том
случае, если он углублен в трубку. Соответственно при заряде, целиком
заполняющем трубку, скорость горения уменьшается по мере того, как
оно уходит в глубь трубки, достигая при глубине свободного участка
2—4 мм постоянной величины.
Этим объясняется, в частности, различие абсолютных значений ско-
рости горения пикрата калия, полученных Б. С Светловым и А. Ф, Бе-
ляевым; последний ставил опыты с шашечками, бронированными путем
лакировки боковой поверхности, которые соответственно горели быстрее,
чем шашки, углубленные в плексигласовые трубки в опытах Светлова и
Фогельзанга. Добавим, что общий характер зависимости п(р) сохраняет-
ся, хотя и наблюдаются некоторые количественные различия (рис. 123),
Любопытное влияние кубической плотности на устойчивость нормаль-
ного горения пикрата калия обнаружил В. В. Горбунов. При атмосферном
давлении пикрат насыпной плотности (~ 1 г/смъ) горел с обычной ско-
ростью, пикрат же повышенной плотности (~1,2 г/с.и3). т, е, меньшой
газопроницаемости, горел уже в условиях ускоренного режима со ско-
ростью, в 3 раза большей.
149
о is го за so so so то зо зо да
% гремучей ртути ..
Рис. 120. Скорость горения смесей
трииитроре:чорци||пта свинца с три-
шпрот риал и добрнзолоо
Гнг, 111). Скорость горения смесей
гремучей ртути с трипитротрпззидо-
бснЗРЛОМ
Ряс. 121. Зависим осп. скорости горе
пня пикрата калия от давления
Рис, 1 22. Зависимость скорости горо-
|ши стифцатв свинца (?) и пикрата
сзцпца (5) от давления
Рис. 123. Зависимость ско-
рости горения пикрата ка-
лия (цилиндрики диамет-
ром 4 мм, покрытые с боко-
вой поверхности лаком) от
давления ио данным
Беляева [[35]
Рис. 124. Влияние кислого серн ок целого калия
на скорость горения калия
1 — пикаат калия; г — 10'i'i сернокислого ка-
лии; 3 — ?(>-[< Т —30%
Изучалось также влияние некоторых примесей на горение пикрата
калин, пикриновая кислота при содержании 20% в 2—3 раза замедляет
горение при атмосферном давлении, значительно меньше — при 5—50 ат;
при 125 ат скорости горения практически одинаковы, Смесь пикрата ка-
лия с перхлоратом (1'1) не показывает максимума скорости. Кислый
сернокислый калий, по Светлову и Фогельзангу, снижает скорость горе-
ния в интервале давлении от 1 до 30 ат и увеличивает ее при больших
давлениях (рис. 124); максимум скорости несколько смещается в сторону
меньщих давлений. Присутст-
вие углекислого аммония влия-
ет аналогичным образом (рис.
125), по равномерность и устой-
чивость горения сильно умень-
шаются.
А. Е. Фогельзанг изучал
также горение солей динцтрофе-
нола (лития, калия, свинца),
прто- и парамононитрофенола
(калия и свинца) и монопптро-
резорцина (калия и свинца).
Большинство изученных солей
обладает гораздо большей спо-
собностью 1 и скоростью горе-
ния, чем можно было бы ожи-
1 — пикрат нллия; 2 — 10%; 3 — 20% (N НО Д’О а
дать, судя по возможной не.ти-
Рис. 126- Влияние углекислого аммония на
скорость горения пикрата калия
чине тецлоты горения, которая
из-за большого недостатка кис-
лорода гораздо меньше, чем, например, у трннцтрогоедниеиий тех
же фенолов. По-видпмому, это связано с тем, что реакции при горении
солен (в отличие от иолннитрофецолов) проходят до конца, с полным теп-
Рис. 126. Зависимость скорости горения
дняитрофенолята свинца от давления
р, кг/см2
Рис. 127. Зависимость скорости
горения азида барии от давления
ловым эффектом; возможно, что здесь играет роль и каталитическое дейст-
вие металла. Наиболее распространенным тип горения — быстрый рост
скорости при малых давлениях и сильно замедленный при умеренно повы-
шенных; в качестве примера на рис. 12G приведена кривая для динитрофе-
нолята свинца. Для калиевых солей наблюдается (аналогично пикрату
калия) зависимость с максимумом. Налпсвыр соли орто- и нараиитрофсно-
ла сильно различаются по способности к горению.
А. Е. Фогельзангом было изучено также горение азида бария (рис. 127),
протекающее в отношении зависимости н(р) сходно с гремучей ртутью,
Иц со значительно меньшими скоростями.
1 В частности, ди- и моноиптрофеполят свинца начитают гореть при гораздо
меньших давлениях, чем трплитрофенолят.
151
Малой способностью к горению характеризуется также азид кальция
[157], несмотря на то, что скорость его горения в тех немногих опытах-,
в которых оно имело место, была относительно велика (0,5—0,7 а/сле2сек
при атмосферном давлении). Горение сопровождается интенсивным дис-
пергированием несгоревшего вещества. При повышенных давлениях азид
кальция способен к устойчивому горению; при 21 ат скорость горения
составляла 4 г/см2сек.
5, АММОНИЙНЫЕ СОЛИ ХЛОРНОЙ И АЗОТНОЙ КИСЛОТ
И СМЕСИ НА ИХ ОСНОВЕ
Хлорная кислота является более активным окислителем, чем азотная.
Это отличие сохраняется и в аммониевых солях. Способность к горению
хлорнокислых солей, способных к внутримолекулярному окислению, зна-
чительно больше, чем азотнокислых. Это позволило изучить зависимость
н(р) для перхлората аммония без органических добавок.
В бомбе постоянного давления, наполненной азотом при давлениях
до 1000 ат, горение перхлората аммония изучала Глазкова [130]. Цилинд-
рики соли, прессованной до плотности, близкой к удельному несу, в обо-
лочке или без нее поджигались с верхнего торца накаленной проволочкой;
скорость измерялась футореглстром.
Зависимость а(р) своеобразна (см. рис. 93) и отчасти сходна с наблю-
давшейся для пикрата калия; она включает участок падения скорости
при повышении давления; в области давлений 500—1000 ат кривая »(р)
обращена выпуклостью к оси абсцисс.
Чтобы выяснить, в какой степе<Н|Г указанная зависимость характерна
для перхлората и в какой она зависит от побочных влияний, опыты при
разных давлениях были проведены в различных условиях. Опыты при
различных диаметрах заряда (5; 7 и 10 леи), представленные на рис. 93,
показывают, что в интер-
вале давлений до 400 ат
скорость горения нс зави-
сит от диаметра. Это гово-
рит о том, что значения
скорости не искажены вли-
янием теплопотерь. По-
скольку продукты горения
перхлората обладают окис-
лительными свойствами, а
материал оболочки, приме-
нявшийся в опытах (плек-
сиглас), горюч, их взаимо-
действие в принципе могло
влиять на скорость распро-
странения процесса. Чтобы
д, кг/смг
Рис. 128. Зависимость скоро-
сти горения образчиков пер-
хлората аммония в различных
оболочках от давления
1 — без оболочки; 2— в перхлор-
виниловом лаке (Од лии); з—в
трубке иа перхлорвинилового ла-
ка <1 лыи); 4 — в плексигласовых
трубках; 5 — во фторированной
смазке. На кривой 1 пунктиром
обозначена область пульсирующе-
го горения
152
установить реальность этого влияния, применялись разные оболочки —
плексигласовая трубка и тонная (0,1 леи) трубка из перхлорвинилового
лака; в последнем случае при давлениях выше 150 ат (рис. 128) скорость
заметно больше; однако она больше п в случае стехиометрической смеел
перхлората с тонкоизмельченным плексигласом, где предполагать химиче-
ское взаимодействие с оболочкой трудно. Причина различия в скоростях и
разных оболочках в обоих случаях остается неясной.
Изменение толщины слоя перхлорвиниленого лака от 0,1 до 1 мм не
изменило общего характера зависимости н(р), а в области давлений до
200 ат — даже и величины скорости.
Если при давлениях до 400 ат влияние оболочки па скорость горения
оказывается слабым, то ырп 500—1000 ат оно становится значительным
(см, рис. 128). Наибольшую скорость горения при 10(10 ат (14 г/см2сек)
Показывает перхлорат н плексигласовой оболочке толщиной 1 .илг, значи-
тельно медленнее он горит в тонких (0,1 льч) трубках пз перхлорвинила
и еще медленнее в виде голых цилиндрiukob дли щггпндриков, покрытых
тонким слоем фторированной смазкп.
Чтобы выяснить, не связаны ли эти различия с влиянием теплопотерь,
опыты в тонкой перхлор виниловой оболочке был и проведены (прд
1000 ат) при разливных диаметрах; при увеличении диаметра от 3 до
10 мм скорость горения возрастала от 8,3 До Ю,0 г/см2 сек слишком мало,
чтобы объяснить все наблюдавшиеся различия. Более того, при горении
голых цилиндриков перхлората скорость горения при малых диаметрах
(3 и 7 мм) была заметно больше (7,5 г/см2сек), чем при диаметре 15 мм
(6,0 г/смРсек), в противоположность тому, что можно было бы ожидать,
если бы различие Определялось только относительными теплойотерями.
Кроме того, при больших диаметрах (15 .м.м) скорость горения голых
зарядов почти вдвое меньше, чем при зарядах в топкой перхлорвиниловой
оболочке, По-видимому, при горении перхлората аммония сказываются
какие-то факторы, не играющие существенной роли при горении других,
ранее изучавшихся ВВ, может быть подобные тем, которые проявляются
(и частности в отноигенгш влияния оболочки) для пикрата калия. По-ви-
димому, при больших давлениях горение в плексигласовых трубках опре-
деляется взаимодействием перхлората со стенками трубки, слабее выра-
женным при горении перхлората в перхлорвиниленой оболочке. Смеси
перхлората с парафином в обоих оболочках (а также без оболочки) дают
близкие скорости.
Скорость горения и характер кривой и(р) перхлората аммония сильно
изменяются в присутствии катализаторов (рис. 129) (оговоримся, что
опыты проводились в плексигласовых трубочках). Впхромат калия уско-
ряет также горение смеси перхлората с коксом (рис. 130).
Были изучены (рис. 131) также стехиометрические смеси перхлората
с некоторыми горючими (парафин, кокс, сахароза) [/58]. Смеси с коксом
п сахарозой в области низких давлений, как и плексигласовые смеси,
горят медленнее, чем сам перхлорат, хотя способность их к горению, ха-
рактеризуемая критическим давлением, значительно больше. Это указы-
вает на то, что ведущую роль при горении в этих условиях играет хими-
ческое превращение перхлората; взаимодействие я,е его продуктов с го-
ркнгим происходит слишком далеко от поверхности горения, чтобы влиять
на скорость ее перемещения. В целом зависимость и(р) имеет сложный
характер. Теплота сгорания смеси сама по себе не оказывает определяю-
щего влияния на величину скорости горения и на ее зависимость от дав-
ления. *
Леви и Фридман [131] изучали горение перхлората при постоянном
подводе к горящему торцу лучистой энергии. Если этот подвод состав-
ляет не менее 10 кал/см2сек, перхлорат воспламеняется через несколь-
ко секунд и продолжает гореть до конца. На рнс. 132 показана давцси-
163
Рис. 1^9. Влиянии каталитически
добавок (5%) па скорость го pi
ин я перхлората аммония
J — с бцхроматом нллищ 2 — ч;
стый перхлорат; ,9 — с окисью jp
ма (CraOj)
Рис. 1,30. Влияние бихромата кал,
(5%) на скорость горения сгехл
метрической смеси перхлората а
мония с коксом
1 — чистая смес,,; S — смесь с би*’[
матом калия
Рис. 131. Зависимость скорости горения некото-
рых стехиометрических смесей перхлората ам-
мония с горючими от давления
1 — перхлорат аммония; г — смесь с парафином;
.1 — с коксом; 4—е сахарозой
Рис. 132. Влияние интенсивно,
иалучення нэ скорость ropei
чистого ц катализиронанного в
хлората аммония
2 _ 3,0% хромита меди: г — С
хромата меди; 3 — NH.CICH без
банок
MiH’Tb скорости такого горения от интенсивности излучения. Присутствие
'.аталлзатора сильно увеличивает скорость горения и уменьшает мини-
мальную интенсивность светового потока, необходимую для получения го-
рения. На наклон прямых оказывает, по-виднмому, влияние изменение
коэффициента поглощения поверхности перхлората в присутствии черного
катализатора.
Исследователи определяли также состав газообразных продуктов горе-
пня перхлората при различных давлениях, чтобы получить представление
о последовательности происходящих при этом реакций. При повышенных
давлениях газообразные продукты состояли пз азота, закиси азота и
кислорода. Вода, которая находилась в сосуде для сжигания, содержала
Пптрат-ион, а также попы аммония и перхлората в небольших эквива-
лентных количествах; последние, очевидно, получались вследствие того,
что часть перхлората избегала сгорания, возможно, в результате диспер-
гирования. Отсутствие питрит-иона объясняется тем, что образовавшаяся
при горении окись азота реагировала с кислородом (которого к тому же
было больше) быстрее, чем с хлором. Образующаяся при взаимодействии
?>О2 с полон азотистая кислота хлором в присутствии воды окислялась
до азотной. При атмосферном давлении состав продуктов был несколько
ни он, по это отчасти объяснялась тем, что условия отбора проб были
другими, чем при повышенных давлениях.
Влияние давления на соотношение между отдельными составными
частями продуктов горения показано в табл. 20.
Таблица 20
Влияние давления на химический состав продуктов горения перхлората аммония
/[явление, m Количество молей тала на моль сгоревшего перхлората аммония • Валаяс
NO- N+O N, о, □о N по О *•
Атмосфер- ное 35 70 140 (J 55+0,02 (.)) 0,31 + 0.01 (5) 0.23+0.01 (12) 0,23± [) .01 (3) 0,10+0,01 (2) 0.11 (1) 0.12+0,01 (2) 0,05(2) 0,11+0.01 (2) 0,05 + 0,02 (2) 0,75(1) 0,80(2) 0 80+0,0-1 0.97 3.95 4,23 4,18 4 И
* Цифры в скобках — число экспериментов, результаты которых усреднены.
♦’ В ед л о сделано цредцоллжетще о том, что и а моль сгоревшего перхлората аммонии ofjpa’
зуегся 2 моля воды. При расчете баланса ио кислороду я опытах при атмосферном даЕменш!
<’дпн ид двух атомов кислорода в NO2 получается цз реакций с водой, так что лишь ня
кислородов рассчитываете и в балансе. При бо-тее высоких давлениях второй кислород п двуоки-
си заота берется ии продуктов горецинт соответствендо принимаются и расчет газиобраяпьЕЦ кисло-
род и оба кислорода в двуокиси азота.
Диля азота, окисленного до окиси, зависит от давления, уменьшаясь
от 0.5;> при атмосферном давлении до 0,23 при 70 лг и затем, по-виднмому,
оставалась постоянной: выход закиси азота не зависит от давления до
70 ат, но падает при его увеличении до 140 ат. Присутствие катализатора
(при 70 иг) сильно уменьшает содержание закиси азота.
То же влияние хотя и в меньшей степени оказывает увеличение попе-
речного сечения образчиков.
При расчете температуры горения (для 70 ат), исходя ,цз экспери-
ментально установленного соотношения соединений азота (N2 : ХО : N2O ~
= 1 :0.87 0,45) и допущения, что хлористый водород, хлор, вода и кис-
лород находятся н термодинамическом равновесии
NH4GIO4 - О,265Хг ф 0,12N20 +0.23NO -|-1,0150. + 1,62НгО +
-|-О,76ЯС1-]-0,12С1г (3.23)
ее значение составляет 987°С. что хорошо согласуется с экспериментально
установленной величиной, заключенной в пределах 930—970° С.
155
Для атмосферного давления, когда весь хлор получается в элементар-
ном виде, уравнение горения имеет следующий вид:
NH4G1O4 O,125N2 + 0,10ХгО. ф 0.55NO + 0,5Ck 2Hs0 + 0,6750s (3.24)
и соответствующая расчетная температура равна 967° С.
Уменьшение количества окиси от 0,55 до 0,23 моля ври переходе от
атмосферного давления к 70 ат свидетельствует о том, что окись азота
образуется в результате окисления аммиака, а азот —из окиси азота.
При этом разложение окиси азота при 1000° С идет слитком медленно,
чтобы за время горения оно могло образовать азот в установленных опы-
том количествах. Наиболее вероятно, что азот образуется и результате
взаимодействия между окисью азота в аммиаком. Уменьшение количе-
ства окиси азота при увеличении давления следует связать с тем, что
давление сильнее влияет на взаимодействие аммиака с окисью азота,
чем па образование ее путем окисления аммиака.
Уменьшение количества закиси азота при увеличении поперечных
размеров образчиков или в присутствии катализатора, возможно, обуслов-
лено повышением эффективной температуры горепия, в результате чего
преимущественно выгорает более реакционноспособная (по сравнению
с NO) закись азота.
Адамс, Ньюмэн и Робинс [159] изучали зависимость скорости горения
перхлората аммония и смесеных порохов на его основе от ряда факторов
(размера частиц окислителя и горючего, давления, соотношения окисли-
тель : горючее, химического состава горючего).
Образчики сечением 0,5 X 0,5 см и длиной 8 см готовились попереч-
ным прессованием под давлением 500 кг/см- ц сжигались в атмосфере
азота в небронированном виде в бомбе постоянного давления с окнами.
Скорость горения измерялась теневым способом п была постоянной в пре-
делах ±5%. Наибольший разброс наблюдался в области независимости
скорости горения от давления.
Скорость горения самого перхлората аммония и в меньшей степени
нижний предел способности к горению зависят от метода подготовки
перхлората, главной причиной этого являются, по-видимому, следы
Примесей. Одна из таких примесей — хлористый свинец, который в не-
больших количествах сильно уменьшает скорость горения. Однако медлен-
ное горевие наблюдалось и в образчике, не содержащем свинца. Отсюда
следует, что добавление свипна, по-впдямому, подавляло действие какого-
то положительного катализатора горения, содержавшегося в быстро горев
шем перхлорате. В смесях с горючим эти различия сглаживаются.
Основной причиной существования нижнего предела способности i
горению под давлением авторы считают уменьшение теплоты горения пр,
снижении давления, ведущем к образованию значительного количеств
окиси азота.
Нижний предел может быть снижен такими изменениями, благодар
которым пламя может поддерживаться при более низкой температур
например, повышением начальной температуры, добавлением неболыш
количеств горючего или помещением в атмосферу горючего газа. Та
при атмосферном давлении горение распространяется, если повыси’
начальную температуру до 250°С; в этих условиях измеренная темпер
тура горепия составляла 1200° К, что соответствует полному превраи
нню до элементарного азота.
Зависимость п(р) может быть выражена степенным законом при г
казателе степени 0,5; вблизи нижнего предела скорость может падать п
уменьшении давления быстрее; размеры частиц оказывают лишь ела?
влияние па ее величину.
Горючие могут быть разделены на две основные группы: 1) раз,
гающиеся при нагревании с образованием только газообразных продукт
156
Рис. 133. Влияние весового отношения го-
pvooero к окислителю (числе у кривых)
на скорость горения смесей перхлората
аммония с полистиролом
Размер частиц 1ЧН,СЮ4— 74—105 ж поли-
стирола <74 лк
Pile. 134. Влияние изменения отноше-
нии окислителя к горючему (числа у
кривых) на скорость горения смесей
перхлората аммония с параформальде-
гидои
например, параформальдегид, углеводороды; 2) плавящиеся и обугли-
вающиеся пли только обугливающиеся с образованием твердой и газо-
образной фаз, например углеводы и ацетилированные углеводороды. К их
числу относится и сажа. Так как размер частиц горючего оказывает
пренебрежимо малое влияние па скорость горения, размеры частиц го-
рючего были взяты постоянные.
На рис. 133 приведена зависимость Щр) для смесей перхлората аммо-
ния с полистиролом при различном содержании последнего от 0 до
избытка, дающего ту же расчетную температуру горения, что и чистый
щ-рхлорат.
Рис. 136. Влияние изменения отношения
окислителя к горючему (числа у кри-
вых) ца скорость горения смесей пер-
хлората аммония с ацетилцеллюлозой
Рис. 135. Влияние сажа на скорость
горения смесей перхлората аммония
с параформадьдсгидом.
Содержание перхлората — Ta.S’i. содер-
жание сажи: 1 — 7 5; 7 — 2,4;';!—[2;
Г — 0,5 и J —(КЬ.
Показатель Степени для стехиометрической смеси ниже б() ат заклю-
чается между 0,5 и 0,6; между 60 л 100 ат он уменьшается до 0,25 и затем
вновь возрастает. Из графика видно также, что при увеличении содер-
ясапия полистирола скорость горения быстро возрастает, проходат через
максимум при ~8% и затем медленно падает. Смесь, содержащая 28%
полистирола, горит вдвое быстрее, чем перхлорат, хотя расчетные темпе-
157
рату.ры горения их одинаковы. Кривая для смеси с нафталином почти
тождественна полистирольной кривой. Опыты по влиянию размера частиц
окислителя показали, что, начиная от 100 мк и ниже, уменьшение раз-
мера частиц до 60 ат пе влияет на скорость горения; при больших давле-
ниях смесь с мелкими частицами горит быстрее.
Скорость горения смесей с параформальдегидом (рис. 134) растет
пропорционально pG'5 до 55 ат; при дальнейшем повышении давления по-
казатель степени снижается н становится равным нулю при 90 ат. Наи-
большая скорость горения наблюдается при избытке окислителя. С умень-
шением размера частиц (85—30 мк) скорость горения смеси с 20% пара-
формальдегида в интервале 4—20 ат увеличивается.
Рис. 138. Влияние размера частиц окис-
лителя па скорость горения стехиомет-
рических смесей перхлората аммонит
с онтаацстагом сахарозы
1 — размер частап «; 2 — 1k—105 л;
дл LJ---1-1—1—1------1--L
<5 20 30 t0 50607000/00 /50 200
и. кг/смг
Рис. 137. Влняппе изменения отноше-
ния окислителя к горючему (числа у
кривых) на скорость горения смесей
перхлората аммония с октаацетатом са-
харозы
В смесях с сажей при давлении до 50 ат наблюдается тот же рост
скорости с давлением, что и для рассмотренных выше смесей, но npi
дальнейшем повышении давления показатель степени непрерывно растет
п выше 100 ат скорость не могла быть определена; тот же рост v обнару
живается для смесей с сахарозой, образующей при термическом разло
яши-ии твердый углеродистый остаток.
Интересно поведение стехиометрических параформальдегидпых сме
сей, в которых часть параформальдегида заменена на сажу (рис. 135)
Присутствие уже 1% сажи снимает независимость скорости горения о
давления; 2,4% сажи несколько увеличивает скорость при низких flai
лениях; выше 70 ат зависимость п(р) медленно возрастает с давлением
при 7,3% сажи кривая п(р) почти тождественна кривой смеси, соде!
жащей одну сажу.
Кривые смесей различного состава на основе ацетилцеллюлоз:
(рис. 136) показывают большую скорость при избытке окислителя. Нез;
вцеимо от состава наблюдается интервал давления, где скорость пе зав;
сит от давления. Влияние размера частиц окислителя иное, чем в случ:
смесей с полистиролом. Уменьшение размера частиц увеличивает скорое'
горения; участок независимости скорости горения от давления па крив<
п(р) при этом сохраняется.
Влияние ацетилированных углеводородов на зависимость скорое:
горения от давления было изучено на смесях на основе октаацетата сах
розы (рис. 137). До 60 ат соотношение компонентов слабо влияло j
скорость горения, хотя максимум лежал при избытке горючего. При бол
высоких давлениях скорость уменьшалась при повышении давления
увеличении содержания горючего. Выше ~100 ат она меньше, чем сь
рость горения чистого перхлората.
Влияние' размера частиц невелико: оно проявляется в том направо
нии, что уменьшение размеров ведет к увеличению скорости при низк
давлениях и уменьшает ее при высоких (рис. 138).
Описанные результаты, полученные при торцевом горении, особен
влияние размера частиц и тягла горючего на зависимость скорости горен
158
от давления и(р), были сопоставлены с опытами по горению в маленькой
ракетной камере (d = 5 см, I — 15 см, вес заряда ~ 300 г). Влияние аце-
тилированного горючего было изучено на составе, содержащем в качестве
связующего ацетат целлюлозы, растворенный в триаретине; показатель
степени был почти равен пулю во всем изученном интервале давлений
от 35 до 150 ат, скорость при этом изменялась от 0,5 до 0,533 см!сек. При
торцевом горевши скорость имела Отрицательное v выше 55 ат и заряд
прекращал гореть при 130 ат.
Аналогичным образом изучалось влияние размеров частиц на зависи-
мость скорости горепия от давления для состава, содержащего 89% пер-
хлората, 10% полиизобутилена и 1% смачивающего вещества.
Опыты ставились в двух вариантах. В первом порох готовили па двух
образчиках перхлората — крупнозернистом, пе подвергавшемся измельче-
нию, и на измельченном до ущельной поверхности, равной 2000 елг/с.м3.
Уровень энергии пороха меняли заменой части перхлората на пикрат
аммония. Результаты, приведенные в табл. 21, показывают, что при дав-
лениях ниже 70 ат, независимо от содержания пикрата, изменение размера
частиц окислители оказывает лишь слабое влияние как на скорость горе-
ния, так и на се зависимость от давления. При более высоких давлениях
крупнозернистый окислитель оказывает значительное влияние особей по в
случае составов, содержащих много горючего: при 20—50% пикрата
выше 70 пт зависимость скорости горения от давления отсутствовала.
Ниже 2о и выше 50% размер частил окислителя тоже влиял, во нс так
Сильно.
Таблица 21
Влияние размера частиц окислителя на скорость горения
ври различной калорийности топлива
Содержание б смеси пер- хлората н пикрата аммо ! Скорость горения при 70 iP'rt, Показатель степей я при павлеции
ння, NH*C1O4 %
1 пикрат 1 аммония
89 (м) 1,88 0,67
89 (г:) — 1,62 0.60
79 (м) 10 1,30 0,60
79 (к) 10 1,04 0,60 (до 70 ат) 0,375 (выше 70 ат)
69 (м) 20 0,94 0.54
69 (к) 20 0,87 0,685 Нуль (выше 77 ат)
59 (м) 30 0,4»
59 (к) 30 0,685 Нуль (выше 77 ami
49 (м) 40 0,485 О.‘4б
49 (к) 40 0,485 Нуль (юнце 70 ат)
39 (м) 50 0,356 0.52
39 (к) 50 0,34 Пуль (выше 70 ат)
29 (м) 60 0,27 0,52
29 (к) 60 0.25 0,66 до 70 ат 0,28 (выше 70 ат)
Примечание- м — мелкодисперсный; к — крупнодис-
псрспый-
Во втором варианте опыта содержание пикрата было иостоянпым
(30%). Окпслитель извлекали из пороха и определяли размеры его частиц.
Различные размеры частиц окислителя получали изменением доли круп-
нозернистого перхлората. Результаты опытов (табл. 22) показывают силь-
ное влияние размеров частиц на зависимость скорости горения от дав-
ления. Чем они больше, тем меньше растет скорость особенно при повы-
шенных давлениях; при наиболее крупных из изучавшихся частиц
скорость даже уменьшалась при повышении давления. При давлениях
70 ат и ниже увеличение размеров частиц уменьшает скорость горения,
15S
хотя при смесях, содержащих меньше 10% (по весу) горючего, это
уменьшение слабо.
При более высоких давлениях зависимость скорости горения от дав-
ления изменяется и становится более чувствительной к размерам частиц
окислителя, к содержанию и химической природе горючего.
Таблица 22
Влияние размера частиц окислителя на зависимость
скорости горения от калорийности топлива
% партии Удельная поверхность NHxClO,. С.,1*. C.U4 Скорость горения при 70 сна, ел, сек Показатель степени при давлении
1 4500 0,685 0,48
2 3050 0,710 0,25
3 2500 0,66 Нуль (выше 77 am)
4 2620 0,710 Нуль (выше 70 ат)
5 2300 0,66 Нуль (70—98 ат) —0,5 (выше 98 ат)
6 1920 0,635 —0,33 (выше 77 ат)
В общем для летучих топлив зависимость скорости горения от давле-
ния в интервале 70—200 ат уменьшается при увеличении размеров частиц,
содержания кислорода в горючем и, возможно, содержания горючего.
Показатель степени при р может быть отрицательным, а скорость горения
меньше, чем самого перхлората аммония даже для смесей со стехиомет-
рическим содержанием горючего.
В случае, изображенном на рис. 137 и 138, знак влияния изменения
размеров частиц окислителя и отношения горючее: окислитель меняется,
переходя от области давлений с положительным значением v к области
высоких давлений с отрицательным значением v.
Скорость горения смеси перхлората аммония с полистиролом [160]
может быть больше плд меньше, чем самого перхлората, в зависимости от
давления, состава смеси и гранулометрии (рис. 139).
Для крупных зерен окислителя при малом его содержании скорость
горения смеси выше при низких давлениях и меньше при высоких дав-
лениях, чем таковая чистого перхлората.
При большом содержании в смеси тонкозернистого перхлората ско-
рость горения последней в изученном интервале давлений выше, чем
чистого окислителя. Допуская, что скорость сгорания частиц перхлората
в смеси во всяком случае не меньше, чем чистого перхлората, авторы
заключают, что в смесях горящих медленнее самого перхлората, выступы
па горящей поверхности представляют собой горючее.
С этим заключением следует сопоставить данные по эрозионному го-
рению некоторых перхлоратных смесей, которые показывают, что при
повышенных давлениях (100 ат) и .значительной скорости течения газов
(> 100 м[еек) различие в величине скорости горения исчезает.
При средних (50 ат) и низких (20 ат) давлениях дело обстоит ипаче
и влияние прироста температуры продолжает сказываться, хотя влияние
состава смеси (при 50 ат) исчезает. При этом, если при больших давле-
ниях скорость безэрозцоцного горения меньше скорости горения самого
перхлората, то эрозия снимает влияние состава; если же скорость горения
больше, тем перхлората, то влияние состава сохраняется и ири эрозии.
Причины уменьшения скорости горения некоторых быстрогорящих
ВВ. а также перхлората аммония при увеличении давления нс выяснены.
Повышение давления обычно увеличивает скорость горения. Eg уменьше-
ние может быть истолковано двояко: во-первых, давление может также
/60
происходит, например, на верхнем
2,0-
0,2
0,f
i,0 -
6 0,8 -
nh4cib4
• - !
o-2
II 1 I______________I___|_|------1
S fO 20 80 60 SO 100 200
80-28} Тонкое
j измельчение
80-20\ Was
? измемченм
/J- j£J )
Рис. 139. Зависимость скорости горения
от давления для смеси перхлората аммо-
ния с полистиролом (числа у кривых —
соотношение компонентов) и другими
горючими
I — полисульфилная смола 75:25; 2 — поли-
су льфидиая смола 65 : 35; 3 — полиэфир 65 : 35
уменьшать скорость горения, причем это влияние в пикрате калия и др.
преобладает в определенном интервале давлений, во-вторых, какие-то фак-
торы увеличивают скорость горения указанных веществ в области умерен-
но повышенных давлений, а при дальнейшем повышении давления, где
скорость падает, их влияние не проявляется и скорость горения, а также
ее рост с давлением имеют нормальные величину и характер.
Известны некоторые случаи, когда повышение давления уменьшает
скорость химических реакций. ""
пределе цепных реакций, когда
обрыв цени определяется трой-
ными соударениями, а продол-
жение ее двойными. Если в по-
следовательность реакций вхо-
дит обратимая реакция диссо-
циации с последующим моно-
молекулярным превращением
одного из ее продуктов, то повы-
шение давления, уменьшая кон-
центрацию последнего, также
может уменьшать скорость об-
разовании конечных продуктов.
Нет, однако, конкретных осно-
ваний допустить подобные ме-
ханизмы в данном .случае.
Можно себе представить так-
же и физические причины ука-
занной закономерности. Если
экзотермическая реакция раз-
вивается через отдельные заро-
дыши, то наличие в контакте с ними более плотного газа моя:ет смягчать
локальные разогревы и уменьшать вследствие этого скорость роста заро-
дышей.
Далее, при горении твердых веществ большое значение может иметь
диспергирование вещества; повышение же давления, уменьшая объем
образующихся пузырьков газообразных продуктов распада, должно за-
труднять диспергирование.
В связи с тем, что ВВ, для которых обнаруживается максимум ско-
рости на кривой п(р), при высоких температурах ожижаются вследствие
плавления и разложения, Светлов и Фогельзанг предполадают, что умень-
шение скорости горения обусловлено именно этим ожижением, насту-
пающим при горении под повышенными давлениями и уменьшающим
скорость химического превращения. Можно представить себе и косвенное
влияние ожижения. Если диспергирование при горении осуществляется
в результате кипения жидкого промежуточного продукта, то увеличение
давления, повышая его температуру кипения, будет уменьшать диспер-
гирование, а следовательно, и скорость горения.
Наконец, пе исключено, что в данном случае влияние давления связано
с существенно новым для теории горения ВВ фактором. Известно, что в
определенных условиях горение многих ВВ, в частности быстрогорящих,
идет неравномерно, пульсируя. Между колебаниями давления и других
параметров горения и зарядом или его частями при определенных соот-
ношениях между периодами колебаний может иметь место резонанс,
ведущий к взаимному усилению колебаний. Усиление колебаний в зоне
горения может, услливая диспергирование, ускорять горение. Возможно
также, что оно может переводдть горение каждой из частиц, образующих-
ся в результате диспергирования, с диффузиоппо-кондуктивного на кон-
вективный режим; соответственно уменьшается время сгорания частиц, и
зона максимальной температуры приближается к горящей поверхности —
11 K. li. Андреев
161
скорость горепия возрастает. В той области условий горения, где резонанс
отсутствует, скорость горения имеет обычное низкое значение; после па-
дения скорость горения пикрата калия близка к скорости горения пикри-
новой кислоты и так же медленно растет с давлением.
Шидловский [161] изучал горение аммониевых солей азотной, хлорной,
йодноватой и хромовой кислот в манометрической бомбе. Прессованные
таблетки воспламенялись черным порохом в навесках, создававших дав-
ление от 75 ат (в опытах с нитратом) до 120 ат (в опытах с остальными
солями). Первые три соли сгорали полностью, хромат — частично. Дли-
тельность горения в параллельных опытах для зщтрата сильно колебалась
Рис. 141. Влияние бихромата калия
(5%) на скорость горения амматола
80:20
? — амматол 80 : 20; 2 — аяматол 80 : 20 +
+ КгСг^О?
Рис. 140. Зависимость скорости горения
амматола 80 : 20 от давлений
(0,57 и 0,93 сек); соответственно различалось (из-за теплопотерь) и макси-
мальное давление (580 и 410 ат). Перхлорат сгорал быстрее и равномер-
нее цо давлению и по времени. Иодат занимал промежуточное положение.
В условиях бомбы постоянного давления, по опытам Глазковой [162],
аммиачная селитра, залитая в стеклянный стаканчик, не горит даже при
диаметре 35 мм и давлении 1000 ат- Водоустойчивая селитра, содержащая
небольшие количества железных солей жирных кислот, способна гореть
в плексигласовых трубках диаметром 7 лш уже начиная с 210 ат. Скорость
горения очень слабо растет с давлением л ври 1000 ат составляет всего
1,5 з/с№сек. С другими каталитическими добавками (КгСгОг, PbCrCU,
(NH^aCraO?) и в больших количествах (5%) спорость больше и при
1000 ат заключается между 1,8 и 2,7 г/сл(2сек. Изучалось также влияние
хлоридов (NaCl, BaCh и СгС1з). Селитра с 7% NaCl начинает гореть при
12 ат. Скорость растет линейно и относительно быстро до 200 ат и мед-
ленно от 200 до 1000 ат. Селитра с 10% хлористого бария начинает гореть
с 27 ат; ио своему влиянию иа скорость горения селитры этот хлорид
близок к хлористому натрию, несколько отличаясь от него по характеру
Кривой н(р).
Тротил относительно слабо увеличивает способность аммиачной селит-
ры к горению. Для смеси селитры с тротилом (80 :20) в трубках диамет-
ром 7 леи критическое давление горения составляет 145 ат. Другим отли-
чием смеси является малая скорость гореппя и слабый ее рост с давлением
(рис. 140); при 1000 ат скорость горения составляет всего 5 з/с,м2сек, для
тротила, близкого по теплоте горения,— 6,5 г/с.м?еек.
В интервале давлений 150 — 350 ат скорость горения растет с увели-
чением давления не линейно, а несколько медленнее, латаная с 400 ат.
зависимость скорости от давления может быть выражена соотношением
= — 0,58 + 0,00554 р.
162
В отличие от тротиловой смесь аммиачной селитры с древесным углем
(91,4 :8,6) способна к горению в трубках диаметром 7 мм даже при атмо-
сферном давлении, хотя скорость ее горения при высоких давлениях мала
п медленно растет с давлением, В интервале 25—1000 ат она может быть
выражена соотношением им = 0,0156 р0,778.
Введение в состав тротиловой смеси 5% бихромата калия сильно
увеличивает как способность к горению, так и скорость его (рис, 141);
так же влияет хромат калия у__%
па горение угольной смеси
(рис. 142)'. Значительное ч
влияние катализаторов ука- з г
зывает на то, что малая око- "9"'
рость в их отсутствие опреде- > % ' (з
ляется медленностью проте-
кающих экзотермических ре-
. , ** /) I L t _ 1 J 1-1. 1.1___ J
Ц?“‘ 200 Й0 600 800 <000
-Катализаторы влияют и кг/см?
на горение йодата аммония; ’ '
Рис. 142. Влияние хромата калия на скорость
горения аммиачной селитры с древесным углем
в присутствии бихромата ка-
лия он способен гореть и при
атмосферном давлении (диа-
J — динаммоя 91,4:8,5; 2 — смесь, содержащая 82%
NHiNOj, 8% утпя и 10% К,СгО4
метр заряда 17,5 мм). Спо-
собствует горению также повышение начальной температуры. При 100,
80 и 60° С скорость горения составляла соответственно 0,138; 0,132 и
0,090 г/см2 сек.
Аналогично ведет себя и гидроксиламинсульфат (NHsOH^SOi; эффек-
тивными катализаторами для этого соединения являются полухлористая
медь, особенно безводная, и двуокись марганца. При 100° С и атмосфер-
ном давлении гидроксил амипсульфат также способен гореть (диаметр
наряда 20 мм), причем скорость горения составляет 0,07 см/сек.
Опыты с прессованным бихроматом аммония показали, что в зарядах
диаметром 20 зеч он способен гореть, начиная с 1 ат, причем скорость
горения в интернале давлений 1—20 ат растет почти пропорционально
давлению.
При горении небронированных цилиндрических шашек в манометри-
ческой бомбе средняя скорость горения составляла 4 г!см2сек при среднем
80 + 640 9СП
давлении —= 460 ат. Скорость горения при атмосферном давлении
определялась также при разных температурах от комнатной до 150° С
(при —70° бихромат в данных условиях опыта не горел). Она сильно
возрастает с температурой, составляя при 18° — 0,035, при 95° — 0,11 и
при 150° — 0,16 г1см2сек. Была определена также скорость горения три-
хромата аммония (ГШфСгзОю; при 0,5 ат она составляла 0,038, при 5 ат —
0,5 cMjceK. Повышение температуры с 20 до 98° увеличивало скорость
горения трихромата при атмосферном давлении вдвое.
6. ГОРЕНИЕ ВВ ПРИ СВЕРХВЫСОКИХ ДАВЛЕНИЯХ
Все рассмотренные данные по зависимости и(р) относятся к давле-
ниям, не превосходящим 1000 ат. При несколько более высоких давлениях
(до 4000 ат) скорость горения некоторых ВВ была определена Беляевым
и сотр. [120]. Опыты проводились в манометрической бомбе с брониро-
ванными цилиндрическими зарядами, состоящими из последовательно
расположенных слоев прессованных (под давлением 4000— 5000 ат) ВВ
с различной скоростью горения, сгорание которых па кривой p(t) изобра-
жалось ступеньками с различным наклоном (см. рис. 84), Кроме того,
применялся метод ионизационных датчиков. Результаты опытов изобра-
11* 163
Рис. 143. Зависимость скорости
горения от давления
1 — тротил; г — тэн; 3 — дымный
порох (ДРЙ-З); 4 — смесь 79% ам-
миачной селитры и 21%
3 — смесь 86,6% КСЮ,
битума; S — смесь 61.5%
38,5% тротила
тротила;
и 13,4%
КСЮ, и
обладающее значение,
жены на рис. 143. Скорость горения тротила и тэна растет с давлением
линейно и почти ему пропорциональна. Также зависит от давления и
скорость горения смеси аммиачной селитры с тротилом. При 3000 ат
точка для тэна лежит несколько выше прямой, что предположительно
связано с нарушением послойного горения. Для смесей перхлората калия
с битумом и с тротилом скорость горения растет не пропорционально
давлению, а медленнее. Особенно это от-
клонение скорости от пропорциональности
давлению заметно у черного пороха. До
2000 ат скорость его горения медленно воз-
растает; при 2000 ат достигается предель-
ное значение скорости —6 см/сек, кото-
рое при дальнейшем росте давления боль-
ше не увеличивается. Авторы полагают,
что более слабая (чем прямая пропорцио-
нальность) зависимость и(р) для смесей
есть общее их свойство, причем для раз-
личных смесей оно проявляется в разных
интервалах давления — дли смеси битума
с перхлоратом калия раньше, для тро-
тиловой смеси позже, для смеси тротила
с аммиачной селитрой этого проявле-
ния следует ожидать при еще более вы-
соких давлениях, лежащих выше интер
вала давления, изученного в данной ра
боте.
Причиной замедления роста скорости i
области высоких давлений является пре
в этих условиях получает диффузия, ли
митирующая скорость реакции во фронте горения-
Очень интересные данные по горению ряда ВВ, в том числе и пни
цнирующих при сверхвысоких давлениях до 12 000 аг, были получен!
Мюрауром и Вассе [163]. Опыты с азидом свинца были поставлены еле
дующим образом. Взрывчатое вещество в виде комочков или таблетог
спрессованных иод давлением 3000 аг, помещали на подкладке из листик
слюды или латунной пластинки в камере высокого давления емкости
25 ел3. Впуская соответствующий газ, создавали нужное давление, поел
чего ВВ воспламеняли нагреваемой током платиновой проволокой.
При атмосферном давлении детонация азида пробивает в патуя
отверстие диаметром 10 льм; в среде азота, сжатого до 1000 кг/см2, диамет
отверстия уменьшается до 4 мм. Механическое действие взрыва при да1
лении 3000 аг становится еще меньше, а начиная с давления 5000 а
практически отсутствует. При замене азота на аргон, сжатый до 9000 а
аенда свинца на нитро диазобензолперхлорат, а также при замене быстро!
нагревания проволоки медленным при давлении 10 000 ат результат
опыта не изменились. Таким образом, при сверхвысоких давлениях азт
свинца и нитродиазобензолперхлорат сгорают, не производя механич
ского действия. Возможной причиной этого влияния является разбавлен!
ВВ газом значительной плотности, заполняющим пространство меж;
частицами, а также растворяющимся в ВВ. Помимо этого, при сверхвыс
ких давлениях различие в плотности между исходным ВВ и газообра
нымй продуктами горения сильно снижается. Это уменьшает дисперг
рующее действие последних, которое является основным фактором, опр
деляющим большую скорость горения и переход его я детонацию.
Аналогичные опыты при сверхвысоких давлениях с гремучим студш
и прессованной пикриновой кислотой показали, что они в этих условие
также горят не давая детонации.
164
7. БЕЗДЫМНЫЕ ПОРОХА
Горение бездымных порохов изучалось в различных интервалах дав-
лений. В течение длительного периода времени, когда единственной об-
ластью применения порохов было огнестрельное ствольное оружие, прак-
тический интерес представляли давления от нескольких сот до 3000 ат.
Исследования в этом диапазоне давлений производились в основном при
помощи манометрической бомбы и привели, особенно в многочисленных
Рис. 145. Зависимость скорости горения
пироксилинового пороха от давления
I — в атмосфере азота; г — в атмосфере про-
дуктов горения; a—для нитроглицеринового
пороха (в атмосфере азота)
h, ММ
Рис. 144. Зависимость температу-
ры от расстояния при горении
пироксилинового пороха
1 — беспламенное горение (рпол <
^2 мм); г — холоднопламенпое горе-
ние (р — 3—4 -ил*): a — двухпламен-
ное горение (ряои — 10 *и)
работах Мюраура и его сотрудников, к утверждению линейной зависи-
мости скорости горения от давления и — А + 5р. При этом член А неве-
лик, поэтому при больших давлениях .линейная зависимость может быть
заменена выражением прямой пропорциональности, которое в силу своей
простоты широко использовалось во внутренней баллистике ствольного
оружия.
В дальнейшем в связи с усилившимся интересом к относительно низ-
ким давлениям, при которых порох горит в реактивных двигателях, а так-
же для выяснения механизма горения стали исследовать область низких
давлений — от атмосферного до нескольких сот атмосфер, а также горение
в вакууме — от долей миллиметра до атмосферы. В двух работах изуча-
лось также горение порохов при сверхвысоких давлениях —до 10000 и
даже 60000 ат.
В порядке возрастания интервала давления и будут рассмотрены
соответствующие исследования.
Похил [164] изучал горение пироксилинового пороха при нагреве его
в вакууме до самовоспламенения и при искусственном поджигании.
Горение, возникающее в результате самовоспламенения, протекает по
внешним своим признакам по-разному в зависимости от давления. При
давлении (конечном) ниже 2 мм горение идет беспламенпо и с относи-
тельно большой скоростью. Выше 2 мм наблюдается холоднопламениое
горение, сопровождающееся свечением сине-голубого цвета. При увели-
чении давления выше 2 леле высота холоднопламениой зоны возрастает,
начиная с ~10 мм на расстоянии 7 леи над поверхностью пороха появ-
ляется второе сине-желтое пламя, которое в отличие от холодного пла-
мепи видимо и при горении пороха при дневном свете. Если поджечь
порох при давлениях выше 10 ат, то на значительном расстоянии над его
поверхностью появляется язычок пламени желтого цвета, усиливающего-
ся с давлением по яркости и приближающегося к поверхности пороха.
165
Давление, при котором появляется это пламя, понижается при повышении
начальной температуры пороха и при увеличении диаметра заряда.
Горение пороха в вакууме сопровождается образованием наряду с га-
зами большого количества дыма —до 70% при беспламенном горении
При повышении давления количество дыма уменьшается — до 50% npt
20 мм и до 30% при 100 лш; однако он наблюдается и при значительна
больших давлениях: даже при 100 ат образуется около 0,6% дыма. Колп
чество дыма зависит также от начальной тем
пературы, уменьшаясь с се повышением. П<
цвету дым, образующийся при низких давле
ниях, не отличается от мелко растертого по
роха и при поджигании сгорает с желты}
пламенем. При повышении давления дым ста
ловится более тоикодисперсным и темным.
При помощи топких термопар была изме
рена температура на поверхности горящег
пороха и ее изменение с расстоянием. И
рис. 144 видно, что при беопламенном ropf
нпп эта температура составляет 280 — 300° (
максимум температуры (~330°) достигаете
[[а расстоянии ~2 лкм. При давлении 3—4 м,
максимум температуры выше (~400°С
При 5—7 м.и — с появлением второго план.
Ряс. 146, Зависимость темпе-
ратуры от расстояния н зоне
дымогазовой смеси
J _ пироксилиновый порох (d —
«5 м»«); 2 — нитроглицериновый
порох JWJWI); з—НИтрОГЛИ-*
цериновый порох (d - 10 ж)
этом интервал давления, в
nil -- максимум температуры значятельБ
больше (600—700° С) и находится гораз;
дальше от поверхности пороха.
Те же типы пламен наблюдаются при и
кусственном поджигании пороха, нагрето:
до температуры, мепыпей, чем та, при кот
рой происходит самовоспламенение. П]
котором наблюдается беспламенное или сое
ветственпо холоднопламеннос горепце, шире; этот интервал тем больп
чем нцже температура. Так, при 90° С порох горел беспламенно до давл
ния 20 — 30 мм вместо 2 лкм при самовоспламенении.
Сопоставляя температуру па поверхности пороха при беелламешл
горении (280—300°) с начальной температурой (85—90°С), Похил, по.т
гая, что горение идет без теллопотерь в окружающую среду, заключ!
что тепловой эффект реакции в конденсированной фазе составля
Q = 0,4 ( 280— 85) — 80 кал[г, где 0,4 — теплоемкость образующейся дьп
газовой смеси.
Зависимость скорости горения от давления своеобразна (рис. 14;
В области беспламенного горения, т. е. малых давлений, скорость оста,
ся постоянной; при больших давлениях ока линейно растет и = А + Q,'
(Р — 0,026); для нитроглицеринового пороха и = 0,05 + 0,315 (р —0,0
Температура вблизи поверхности была измерена и при больших д
лениях, когда порох в столбиках применявшегося диаметра горит р и
комнатной температуре. Из рис, 146 видно, что увеличение давлен
приводит к повышению максимума температурь! и приближает его
поверхности пороха. Увеличение диаметра образчика повышает тем
ратуру горения. Повышение давления приводит к увеличению, хотя
сравнительно слабому, также и температурь! поверхностного слоя го
щего пороха (табл. 23).
Был определен состав газообразных и конденсированных продук
горения при разных давлениях (табл. 24). При беспламенном горет
образуется особенно много окислов азота, причем половина из нт
двуокись азота. Содержание горючих газов и продуктов окисления неп
порционально мало. По-видимому, на первой стадии горения происхо,
166
Таблица 23
Приближенные значения температур в зоне (в °C) над поверхностью пороха
в зависимости от расстояния при p — i>ama
(диаметр образца пороха 4—5 л*л*)
Зона пад по- верхностью пороха (h), ж Пироксили- новый порох Н|1тРОГЯ11ЦВ- РГШОВ1.1Й порох Зона над по- верхностью пороха (h), хч Пирок сила- новый порох Нитроглице- риновый порох
1 5,30 5,30 7 800 820
2 620 650 10 750 750
3 670 720 15 600 600
5 750 780 20 400 400
Таблица 24
Состав продуктов при горении в разных условиях
Давление, мм рт. Ст- Процент от пйъема газовой фазы *Дым>, % К весу пороха Элементар- ный состав •дыма», % нао, % К весу пороха
NO, NO соа СО сн. НЕ непре- дельные углево- дороды к.
<2 (беспламенное горение) 32 32 12 20 4 — — — 70 С 32,2 О 54,7 Н 2,1 N 11,0 7,2
~12 (двухпламен- ное горение) 15 44 13 25 3 — о,3 1,6 ^42-50 1 С 50-55 О 38—42 Н 2—1 5 N 6-4 13,6
~ 100 4
(двухпламен-
яое горение)
преимущественно Отщепление кислорода, связанного с азотом, а окисли-
тельные реакции протекают в гораздо меньшей степени.
При повышении давления образование NO? быстро падает, содержа-
ние NO сначала растет, а затем слабо уменьшается; значительно возра-
стают количества СО и Н2О. Сопоставляя эти данные с уменьшением
количества дыма и обогащением его углеродом, следует заключить, что
изменение состава газовой фазы происходит преимущественно за счет
газификации азота и кислорода дыма в виде окиси азота и взаимодей-
ствия с дымом двуокиси азота с переходом ее в окись азота.
На основании дапдых таблицы уравнение беспламенного горения по-
роха имеет следующий вид:
C^HatOteNn = 2,350, -|- 2,3 К О + 1.45СО +O,85CQ2 + 0,ЗСН4 + 4,5Н2О +
~Г С21,4Н18,Ер27,Л,38 + 119 ККв/1. (3.25)
Похц.т полагает, что и при относительно высоких давлениях горение
порохов идет через стадию образования дыма. Чтобы его обнаружить,
шашку пороха плотно вставляли в металлический цилиндр; в пего
до поверхности пороха был введен дырчатый поршень, через отверстия
167
которого выходили при горении газообразные продукты с дымом. Б этих
условиях опыта количество дыма было гораздо больше (например, при
100 ат в 10 раз), чем в обычных условиях гореция. Поскольку количество
улавливаемого дыма зависит от интенсивности процесса торможения,
а также поскольку температура поверхности лишь очень слабо завист
от давления, Похил считает экспериментально доказанным, что механизм
горения порохов коллоидального типа в области повышенных давленш
тот же, что и: в вакууме, т. е. в обоих случаях первоначально образуете}
большое количество дыма.
Образование порошкообразного остатка («белого вещества») при го
рении нитроклетчатки при низких (ниже 5 мм рт. ст.) давлениях наблю
дали также американские исследователи [165]. Большая часть этого остат
ка состоит из частично деиитроваяной и окисленной нитроклетчатК!
со средним молекулярным весом около 1500. Образуются также ниэ
комолекулярные продукты — муравьиная и щавелевая кислоты, фур
фурол, глиоксаль и формальдегид, окислы азота и углерода, водоро
и вода.
При больших давлениях разложение становится более полным. Пр
100 мм около 40% навески получалось в виде сложной смеси жидки
продуктов («красное вещество»), состоящей главным образом из глии
саля, муравьиной кислоты, формальдегида, воды и небольших количест
глицеринового альдегида, мезоксалевого альдегида и других более слоя
ных соединений.
Для начальных реакций, ведущих к образованию «красного вещества
авторы предлагают следующую схему:
I
HCONOa
I
OaNOCH
H<io----
1
HGO-----
1
H2CONOu
I----
---CH
I
HC=O
O=CH
HCO----
H(io---
+3NOa.
(3.2
HaC=O
Первым этапом является разрыв связей RO — NO2 с последующ
перегруппировкой образующихся свободных радикалов и разрывом угл
род-углеродных связей 2—3 и 5—6. По этой схеме глиоксаль получает
из углеродных атомов 1 н 2 путем гидролиза ацетальных связей. Форма;
дегид образуется из углеродного атома 6; углеродные атомы 3, 4 и 5 <
разуют альдегиды с трехчленной цепью, обнаруживаемые в продукт
горения. Дальнейшие реакции между NO2 и этими первичными ирод;
тами приводят к более глубоко окисленным соединениям, обнаруя
ваемым в «красном веществе». Дальнейшее повышение давления уме1
тает количество образующегося «красного вещества», но альдеги
содержатся в продуктах горения вплоть до давлений в несколько .
мосфер.
Опыты с нитроглицериновым и динитродиэтиленгликолевыми поре:
ми [164] показали те же три типа горения при пониженных давлени;
несколько выше была необходимая температура предварительного по;
грева (110—115°) и температура на поверхности (300—310°С); бе;
отчетливо наблюдалось образование на горящей поверхности ожшкеиш
кипящего реакционного слоя.
Хис и Херст [166] изучали при умеренно-повышенных давлениях i
140 ат) горение с торца бронированных цилиндриков кордита [нит
целлюлозы (12,2% N) — 48%, НГЦ — 41 %, карбамита — 9%’, криолита :
168
лия — 2%] в бомбе постоянного давления в токе азота. Помимо измерения
скорости горения, толщины темной зоны и зоны пламени при разных
давлениях производились спектрографические измерения зоны пламени,
которые показали, что спектр его излучения непрерывен, а цветная и
яркостная температуры одинаковы (< 2340—2370°К), т. е. излучение
имеет характер излучения абсолютно черного тела. Вероятным излуча-
телем являются частицы углерода, образующиеся в ходе горения. Изме-
ренная температура ниже расчетной (2470°К при 70 ат) на 80—100°С
из-за потерь тепла излучением. Ниже 7 ат кордит (d = 6 мм) не горел,
яркое пламя появлялось выше 18 ат. Скорость горения росла с давле-
нием, согласно выражению и = Вр\ где v = 0,56. Зависимость толщины
темной зоны от давления может быть выражена соотношением S =
причем а = 1,9 в отличие от данных (а = 3) Крауфорда, полученных для
пороха другого состава.
Спимки горящей поверхности показывают наличие на ней ярких
пузырьков и меньших по размерам черных шариков, общий вид поверхно-
сти напоминает поверхность жидкости, на которой горящие шарики регу-
лярно появляются и исчезают так, что общее число их остается постоян-
ным. Время жизни шарика (миллисекунды) уменьшается при увеличении
давления, их концентрация (на единицу поверхности), напротив, возрас-
тает; размер шарика составляет 0,01—0,03 мм. По-видимому, эти шарики
представляют собой пузырьки, образующиеся на ожиженной поверхности
пороха, в которой идут реакции, заканчивающиеся образованием пламени,
которое, когда пузырьки лопаются, поднимается к основной пламенной
зоне. Черные шарики являются гораздо более долгоживущими и предпо-
ложительно не являются пузырьками, а образуются неорганическими
компонентами пороха.
По-видимому, лишь очень тонкий слой пороха, прилегающий к поверх-
ности зерна, принимает участие в горении. При уменьшении скорости
горения толщина прогретого слоя быстро возрастает и тепловой эффект
экзотермических реакций, в нем происходящих, составляет все большую
н большую долю общей энергии, необходимой для поддержания горения.
Количество тепла, выделяющееся за счет реакции в конденсированной
фазе, может возрастать при уменьшении давления и скорости горения.
Экспериментально установлено, что для большинства порохов на трудно-
летучем растворителе зависимость п(р) становится меньше при пони-
женных давлениях. Точно так же медленно горящие пороха обнаружи-
вают меньшую зависимость и(р) в области низких давлений, чем быстро-
го рящие.
Поскольку ракетные двигатели по большей части работают при срав-
нительно низких давлениях — ниже 140 ат, то реакции в конденсирован-
ной фазе могут иметь большое практическое значение и заслуживают
тщательного изучения.
Оценийая температуру поверхности горящего пороха по методу рас-
плавления и разложения маленьких частиц щелочноземельных карбона-
тов, введенных в порох, Даниэльс, Вильфонг и Пеннер [167] полагают, что
она превосходит 1000° С. Эта оценка, по-видимому, является завышенной,
поскольку поверхность пороха не светится, и обусловлена каталитически-
ми реакциями на поверхности твердых частиц, как это установлено прп
попытках измерения температуры пламени голыми термопарами. Другой
возможный источник ошибки состоит в том, что твердые частицы могут
выдаваться над поверхностью пороха и попадать таким образом в область
пламени с высокой температурой.
Лейпунский и Аристова [168] намерили калориметрически запас тепла
быстро затушенного зерна пороха и, исходя из Михельсонова распреде-
ления температуры, рассчитали температуру поверхности пороха. Для
нитроглицеринового пороха она составила 330 ± 45°, для пироксилинового
169
— 252 ± 48°. Поскольку опыты проводились при атмосферном давлении,
когда роль реакции в конденсированной фазе значительна, а расчет
производился без учета этого обстоятельства, ошибка в определении 7\
может быть значительной.
Температура поверхности может быть определена также непосред-
ственным измерением температурного профиля при помощи термопар.
При этом должна соблюдаться линейпая зависимость 1g {Т — То) от %-
В области низких температур это соотношение выполняется, при повы-
шенных — наблюдается значительная кривизна, очевидно, обусловленная
тепловыделением в конденсированной фазе. Если плоскость, где начи-
нается отклонение принять за поверхность горения, то температура ее
получается для пироксилинового пороха — 250°, что возможно несколько
занижено, поскольку не учитывается тепловой эффект реакции.
Хотя оба рассмотренные метода нельзя считать падежными, следует
полагать, что температура на поверхности горящего нитроглицеринового
пороха 300°С.
Для типичного нитроглицеринового пороха толщина темной зоны в ее
зависимости от давления выражается соотношением
i = J-£.
J Тепловой эффект реакций, заканчивающихся в темной зоне, составляет
' около 500 кал/г, т. е. меньше половины полного теплового эффекта. Соот-
ветственно изменяется и состав продуктов реакции (рис. 1-47). В области
темной зоны образуется значительное количество окиси азота, восстанав-
ливающейся в зоне пламени. В этой зоне количество окиси углерода
уменьшается, количество СОг возрастает, так же как и количество водо-
рода, образующегося по реакции водяного газа.
Таким образом, пламя горения может быть разделено на две обла-
сти — темную и светящуюся с существенно различными свойствами,
в которых происходят различные’ химические процессы. Часть темной
области, прилегающей к поверхности пороха, американские исследова-
тели называют зоной шппепия, поскольку газообразные продукты отте-
кают нормально к поверхности горения со значительной скоростью, вызы-
вающей шипящий звук. Характер оттока продуктов горения указывает
на то, что реакция в основном происходит очень близко к горящей поверх-
ности. Как и следовало ожидать, температурный градиент вблизи горящей
поверхности возрастает с давлением, поскольку оно ускоряет газофазные
реакции. Температурный профиль зоны горения был измерен [169] при
помощи термопар (рис. 148). Эта зона очень узка (несколько сотых сан-
тиметра), значительно меньше, чем темная зона при том же давлении.
За пределами этой узкой реакционной зоны температура поднимается до
уровня 1400° С, па котором к остается до конца темной зоны. Эта тем-
пература подтверждается результатами спектроскопического измерения
интенсивности полос поглощения NO. Этот метод, хотя и менее точный,
чем метод термопар, имеет то преимущество, что в пламя пе вводится
никакого постороннего тела. Расчеты, основанные на измерения теплоты
реакций темной зоны и анализе образующихся в ней продуктов, приво-
дят к близким значениям ее температуры. Характер изменения темпера-
туры в темной области показывает, что наружные ее области сравни-
тельно мало активны. Причина этой задержки в наступлении конечной
серии реакций неизвестна. Ее можно сопоставить с индукционным пе-
риодом, часто наблюдающимся при газовых реакциях. Исчерпание веще-
ства, обрывающего цепи, образование критической концентрации проме-
жуточного продукта, являющегося автокатализатором, пли достижение
критической температуры для теплового взрыва могут быть причиной
170
внезапного и резкого усиления реакции, приводящей к продуктам, соот-
ветствующим химическому равновесию.
Нормальная скорость горения пороха и характер ее зависимости от
свойств пороха п условий горения имеют чрезвычайно большое значение
для 'Определения пригодности пороха в качестве реактивного горючего.
Выявление факторов, которые влияют на скорость горения, является
основной задачей и целью изучения механизма процесса горения. В свою
Рис. 147. Изменение продуктов
горения топлива HES4016 с рос-
том давления инертного таза
Рис. 148. Экспериментальные профили темпе-
ратуры при горении пороха, содержащего
нитроцеллюлозу с 13,5% азота +1 % лти.т-
цеигралпта
Предполагается, что температура па поверхности
равна 250° С; числа у кривых — давления (в
очередь изучение влияния какого-либо фактора на скорость горецня может
дать существенные сведения о механизме горения. Целесообразно исполь-
зовать оба пути: во-первых, рассмотреть зависимость скорости горения от
характеристик пороха и внешних условий и там, где возможно, связать
эту зависимость каузально с механизмом горения; во-вторых, использо-
вать данные эмпирического изучения скорости горения для развития
качественной картины процесса горения.
Зависимость скорости горения от давления выражают часто формулой
более общего вида:
и = А + Вр'1. (3,27)
Однако для большей части смесевых порохов, для многих баллиститпых
порохов, содержащих различные катализаторы горения, и для большин-
ства порохов при очень низких давлениях даже это выражение не доста-
точно для описания реальной зависимости скорости горения от давления.
По-видимому, эту зависимость нельзя выразить ин одним простым за-
коном.
Имеющиеся экспериментальные данные позволяют сделать следующие
обобщения.
1. При относительно высоких давлениях (выше предела, лежащего
между несколькими десятками и 140 ат) в зависимости от состава закон
горения является монотонной функцией давления и может быть выражен
формулами и = Вр\ и = А + Вр или более того и = Л + Bpv.
2. При промежуточных давлениях зависимость скорости горения от
давления может быть более сложной и сильно обусловленной составом
пороха. (Иногда наблюдаются интервалы давлений, в которых скорость
не зависит от давления или даже уменьшается при его повышении.)
3. При очень низких давлениях экстраполяция скорости па давление,
равное нулю, дает конечное се значение, хотя порох перестает гореть при
171
давлениях, лежащих где-то между долями атмосферы и десятками ат-
мосфер,
С точки зрения механизма горения, по-нидимому, при высоких давле-
ниях большая часть энергии, необходимой для вызова поверхностного
разложения (которое обусловливает перемещение поверхности, т, е, ско*
рость горения), поступает из высокотемпературного пламени. Скорость
передачи энергии определяется теплопроводностью пламепи и его рас-
стоянием от горящей поверхности. Поскольку скорости реакций в пламени
сильно зависят от давления, увеличение последнего приводит к увеличе-
нию передачи энергии и, следовательно, к плавному возрастанию скорости
горения.
При промежуточных давлениях зона пламени удалена от горящей
поверхности или совсем отсутствует. Значительная часть энергии, необ-
ходимой для поддержания горения, поступает из зоны шипения. Реакции
в этой зоне вялы, ио могут быть ускорены катализаторами. Они сильно
зависят от состава пороха в противоположность пламенным реакциям,
которые практически одинаковы для всех баллистных порохов. Как след-
ствие зависимость н(р) сильнее различается в этой области.
При очень низких давлениях очень мало энергии возвращается к го-
рящей поверхности из газовой фазы и скорость становятся почти незави-
симой от давления. Источником энергии, независимо^рт давления, стано-
вится при Очень низких давлениях (атмосфера или* ниже) реакция i
конденсированной фазе.
Зависимость скорости горения от начальной
температуры
Изменение*скорости горения пороха при изменении его начальной тем
пературы сравнительно мал б — обычно меньше 0,5% на градус, а часн
и еще меньше. Попытка связать этот малый температурный коэффициент
с энергией активации ведущей реакции приводит или к неправдоподобие
малым значениям энергии активации, или к слишком высоким значениям
температуры поверхности. Однако, по-видимому, такое простое рассмот-
рение температурной зависимости неприменимо в данном случае. Реакция
идет не изотермически, а в области очень большого градиента температу-
ры, где точное приложение концепции энергии активации становится за-
труднительным. Определяющей скорость горения является скорость по-
ступления энергии к пороху; температура в реакционной зоне подстраи-
вается так, чтобы поддерживать соответствующую скорость.
Повышение начальной температуры пороха вызывает близкое по ве-
личине повышение конечной температуры пламени, точное значение ко-
торого может быть рассчитано с учетом теплоемкости пороха п продуктов
реакции.
Можно допустить, что температурный профиль повышается приблизи-
тельно на одинаковую величину во всех точках. Однако температура го-
рящей поверхности не может повыситься на эту величину, поскольку
вследствие увеличения скорости горения поверхностные слои в течение
меньшего времени получают энергию от пламени и реакций в конденси-
рованной фазе. Поэтому температура на горящей поверхности повысится,
но на меньшую величину, чем То и Г1(0Н.
Влияние начальной температуры на скорость горения можно рассмат-
ривать: а) учитывая повышение температуры и соответственно теплопе-
редачи от пламени к пороху и б) учитывая, что потребуется меньше
энергии для нагревания пороха до температуры Ти.
КоРяеР рассчитал, что при повышении начальной температуры
на 10 С скорость горения кордита SC должна возрасти на 2,2%; опыт дает
значения, лежащие между 3 и 4%.
172
Уравнение
<3-28)
выражает зависимость скорости горения от начальной температуры. Т' —
константа, зависящая от состава пороха и имеющая размерность темпе-
ратуры; она соответствует той температуре, при которой скорость стано-
вится бесконечной, т. е. при которой горение произошло бы во всем зерне.
Значения Т, полученные экстраполяцией, заключаются между 200 и 350° С,
что находится в разумном согласии с оценкой температуры поверхности
и температуры вспышки.
В соответствии с этими соображениями порох с большим значением
Tf должен иметь низкий температурный коэффициент. Экспериментально
было установлено, что пороха, которые содержат относительно стойкие
компоненты, например динитротолуол, и которые соответственно имеют
более высокую температуру разложения, чем «горячие» составы, обычно
показывают меньшую зависимость скорости горения от температуры. Мно-
гтщ смесевые пороха, у которых вследствие исключительно высокой стой-
кости их компонентов следует ожидать высокую температуру поверхности,
также известны своими малыми температурными коэффициентами.
При высоких давлениях скорость горения предположительно опреде-
ляется конечной температурой горения, и влиянием реакций в конденси-
рованной фазе можно пренебречь. При низких давлениях пренебрегать
ролью реакций в конденсированной фазе нельзя. Поэтому не удивительно,
что температурная зависимость при низких и промежуточных давлениях
является более сложной.
Существуют пороха с нулевой или слабо отрицательной зависимостью
скорости горения от температуры в этой области давлений. Изменение
состава часто влияет очень специфично, но экспериментальных данных
недостаточно, чтобы сделать обобщения. Поэтому было бы очень полез-
ным тщательное изучение температурной зависимости скорости горения
при низких и промежуточных давлениях.
Влияние состава пороха на скорость горения
Скорость горепия пороха при постоянной температуре и давлении за-
висит в первую очередь от его состава.
Изменения скорости горения, в зависимости от изменения его состава,
могут быть вызваны как итоговым влиянием, которое может быть связано
с изменением температуры горения, так и специфическим влиянием, ко-
торое зависит от конкретного физико-химического действия на опреде-
ленном промежуточном этапе горения.
Мюраур [171] на основании своих многочисленных исследований, кото-
рые привели его к формуле, выражающей зависимость скорости горения
от давления
и — а/2+Ь/2р
установил, что коэффициент b (скорость горения выражена в мм/сек)
связан с температурой горения соотношением
Ig(1000fe) = 1,214+0,308Гг/1000 (3.29)
или
lgb/2 = —2,087+0,308Тг/1000. (3.30)
Таким образом, предполагается, что скорость горения зависит только
От теплоты, выделяющейся при горении, или температуры, при нем до-
стигаемой, но не от кинетики химического превращения.
173
Эти эмпирические соображения были выведены па основании данных
по скорости горения, полученных при высоких давлениях. В этих усло-
виях скорость горения в значительной мере определяется передачей энер-
гии из зоны пламени и соответственно должна зависеть от температуры
горения.
Нет оснований, однако, ожидать, что указанные зависимости окажут-
ся справедливыми для низких и промежуточных (ниже 150 ат) давлений,
где пламени либо нет, либо оно находится на значительном расстоянии
от горящей поверхности. В таких условиях скорость горения порохов
с близкими тепл стами горения может различаться больше чем вдвое.
В этой области низких давлений влияние различных компонентов может
быть значительным.
Поскольку при очень низких давлениях скорость горения в значитель-
ной мере определяется экзотермическими реакциями в конденсированной
фазе, можно ожидать, что составы, в которых могут развиваться такие
реакции, будут гореть в этих условиях с относительно большой скоростью.
Пороха, содержащие значительные количества стабилизаторов, например
этилцентралита или дифениламина, относятся к этому классу п устойчиво
горят вплоть до очень пизких давлений. Близкая к ним добавка — п-фе-
нилепдиамип особенно активен в этом отношении.
Более устойчивые охлаждающие добавки, например дибутнлфталат пли
триацетин, не так охотно вступают в низкотемпературные реакции; по-
этому пороха, содержащие эти добавки, характеризуются ограниченной
способностью к горению при пизких давлениях. Вещества, способные к
эндотермической реакции вблизи горящей поверхности, замедляют горе-
ние еще сильнее. Примером такой добавки может служить параформаль-
дегид. Вероятно, пара формальдегид диссоциирует на формальдегид, а за-
тем п на окись углерода и водород в зонах поверхности и дает реакции
с поглощением большого количества энергии. Порох, содержащий 5% па-
раформальдегнда не горел при пизких давлениях, но при бодее высоких
давлениях при приближении пламени к горящей поверхности скорость
горения быстро росла с давлением и приближалась к величине, соответ-
ствующей теплоте горения.
В области давлений, близких к атмосферному [172], зависимость ско-
рости горения нитроглицеринового пороха Н от давления изучалась при
повышенных температурах (рис. 149). Вблизи критического давления ско-
рость быстро растет с давлением, а, начиная с некоторого значения по-
следнего, рост скорости становится более медленным и дйнейным. Чем
выше температура, тем раньше (при меньшем давлении) начинается ли-
нейный участок зависимости н(р). На этом участке при 85° нл = 0,142 +
+ 0,0142 р; при 115° С ггл — 0,184 + 0,0180 р. Интересно, что А и В уве-
личиваются при повышении температуры в одинаковое число раз.
Порох с большим содержанием нитроглицерина был способен к горе-
нию в этих условиях, начиная с более низкой температуры; член А был
для него несколько меньше, а В значительно больше, больше соответст-
венно и скорость горения.
Способность порохов к горению увеличивается также при содержании
в них стабилизаторов химической стойкости. При низких температурах
эти вещества, представляющие собой обычно слабо основные ароматиче-
ские соединения, связывают окислы азота, образующиеся при самопро-
извольном распаде иитроэфиров. При более высоких температурах, име-
ющих место в прогретом слое при горении, стабилизаторы могут реаги-
ровать непосредственно с нитроэфирами с выделением тепла. Благодаря
этому способность к горению возрастает. В определенных условиях опыта
порох с большим содержанием стабилизатора горел при 35° С при атмос-
ферном давлении, в то время как без стабилизатора горение наблюдалось
лишь, начиная с 14 пт.
174
Горение пороха Н изучалось в широком интервале давлений и при
обычной температуре. При низких давлениях (до 4 ат) наблюдается так-
же относительно быстрый рост скорости который при больших давле-
ниях (4—8 ат) замедляется; однако затем скорость вновь начинает расти
быстрее. Между 8 л 100 ат зависимость и(р) может быть выражена:
600 800 100012001400 ЮОО
р, мм рт. ст.
Рис, 149. Зависимость скорости горения
нитроглицеринового пороха от давления
при повышенных температурах
р, кг/смг
Рис, 150. Зависимость скорости
горения нитроглицеринового
пороха от давления
(рис. 150) соотношением и = А 4- причем v для пороха Н равно 0,84..
Сходную зависимость даст и порох с большим содержанием нитроглицери-
на (43%), хотя, по-видимому, для пего v остается немного меньше I и
мри давлениях, превышаютдих 100 ат [116].
Глазковой [118] скорость горения пороха I] была определена при по-
стоянном давлении в атмосфере азота в интервале давлений от 12 до
1000 ат. Для опытов применялись цилиндрики пороха диаметром 5 или
9 мм, покрытые с боковой поверхности перхлорвиниловым лаком2. До
50 ат зависимость скорости горения от давления может быть выражена со-
отношением
= 0,12+0,0158 /Л9®; (3-31)
в интервале 50—1000 ат скорость растет медленнее я лпнейпо;
ик = 0,62+0,00926 р. (3-32)
Жак и Джзймс Бассз определили скорость горения ряда порохов в
атмосфере инертного газа в еще болео широком интервале давлений: от
Тйа блица 25
Состав порохов (в %), применявшихся в работе Жака и Джеймса Бассо
Номер пороха Азот Нитроклет- чатка Нитрогли- церин Централит Дифенил- уретан
1 13,04 59,2 31,2 3 6,6
2 12,30 69,7 22 8,55 .
3 14,21 58,7 36,5 4,8 .—
4 14,77 58 41 0,99 .
5 11,73 66 21 13,1 .
6 Перхлоратный порох
1 Чтобы охватить область давлений ниже о ат, помимо шашек диаметром 5 мм»
которые применялись для основной части опытов, были использованы также шашки
диаметром 10 и 20 мм.
2 Различия в скорости горения при этих диаметрах не наблюдалось.
175
50 до 10 000 ат. Скорость рассчитывалась по интервалу времени, которое
требуется для разрушения двух свинцовых проволочек диаметром 0,1 мм,
пропущенных через бронированный шеллаком столбик (d — 3,5 мм) по-
роха на расстоянии 40 мм друг от друга. Составы изученных порохов при-
ведены в табл. 25, зависимость скорости горения от давления представле-
на на рис. 151. Выше 1000 ат скорость горения всех порохов линейно
Рис. 151. Зависимость скорости горения порохов, изученных
Жаком и Джеймсом Бассэ, от давления
। Цифры у кривых — вомер пороха
растет с повышением давления; ниже 1000 ат кривые имеют выпуклость,
очень слабую для пороха № 5, более заметную для № 1, 2, 3 и 4 и очень
сильно выраженную для смесевото пороха, скорость горения которого в
значительном интервале давлений почти не меняется [173}.
Анализ результатов опытов Бассэ с точки зрения их соответствия фор-
муле Мюраура и Они, связывающей скорость горения пороха при 1000 ат
с расчетной температурой горения, а также некоторые экспериментальные
данные по горению ряда порохов в бомбе постоянного давления в интер-
вале 40—600 ат приведены в статье Мюраура и Фово [174].
8. ЧЕРНЫЙ ПОРОХ
Опуская старые работы по установлению зависимости скорости го-
рения черного пороха от давления (175], исследования по горению трубоч-
ных порохов в дистанционных кольцах [176} и результаты опытов
по определению скорости горения смесей черного пороха с нитроглице-
рином [126], остановимся на ряде последних исследований по данному
вопросу.
Зависимость скорости горения черного пороха (KNOs — 78%, S — 10%,
уголь —12%) в виде бикфордова шнура от давления (0,17—30 ат} была
определена в бомбе постоянного давления [177]. Эта зависимость может
быть выражена соотношением и = Вр\ но значения коэффициентов фор-
мулы в разных интервалах давлений различны. Для белого шиура прп
давлении до 2 ат В = 0.848 и v = 0,528; выше 4 ат В — 1,21, v = 2,238.
Беляев и Мазнев [178] в опытах с прессованными (р = 1,75 г/е.и3)
столбиками пороха (KNOg— 75%, 3 — 10%, уголь —15%) с тонким
покрытием боковой поверхности для обеспечения торцевого горения, так-
же получили более быстрый рост скорости горения (а = 0,88 р0,5) в об-
ласти давлений до 5 ат, чем при больших давлениях; от 5 до 125 ат они
выражают этот рост формулой и = А 4- Bpv, причем А = 1,5; В = 0,2
и v ~ 0,47.
Одновременно, но в большем диапазоне давлений (от 1 до 1000 ат},
скорость горения черного пороха, запрессованного до большой плотности
176
в плексигласовые трубочки внутренним диаметром 7 .и.м, определила Глаз-
кова [118]. В интервале 10—1000 ат скорость может быть выражена фор-
мулой = 2,30 р°>216. В изученной Беляевым и Мазневым области дав-
лений формулы Беляева и Глазковой дают близкие результаты; однако
при больших давлениях расхождение между формулой Беляева и экспе-
риментальными значениями скорости становится значительным, что ука-
зывает на ограничеинреть области ее применения в широком интервале
да злений.
Рис. 152. Зависимость скорости горения первого пороха
от давления во данным разных исследователей:
7— данные Андреева (для бикфордова шнура); Д — данные Беляе-
ва (в манометрической бомбе при плотности образчиков 1.76 г/слГ);
,7—данные Глазковой (в бомбе постоянного давлении, образчики
запрессованы в плексигласовые трубки до плотности 1,8 —1.9 г/см3)
Интересно отметить, что еще Вьель [111] на основании опытов в ма-
нометрической бомбе заключил, что средняя скорость горения предельно
уплотненного черного пороха в опытах, при которых максимальное дав-
ление изменялось от 1000 до 3000 ат, пропорциональна давлению в степе-
ни 1/4.
Беляев изучил также зависимость и(р) для «бсссерного» пороха
(KNO3— 85%, уголь —15%). Во всем диапазоне давлений до 125 ат
скорость горения значительно меньше и растет, согласно выражению
и = 0,195 \
В еще большем интервале давлений (до 4000 ат) методом слоеных
зарядов в манометрической бомбе зависимость и(р) для черного пороха
определена Беляевым и сотр. [120]. В области давлений от 5 до 2000 ат
скорость растет по закону а = Вр'" в согласии с результатами работ [118,
177], от 2000 до 4000 ат скорость остается постоянной (~ 6 см/сек).
На рос. 152 сопоставлены результаты всех упомянутых работ.
Резкое отличие черного пороха от вторичных ВВ в отношении зави-
симости и(р) не является удивительным. Горение черного пороха ио сво-
ему механизму, несомненно, отличается от горец ня вторичных ВВ. Это
можно заключить хотя бы уже из того, что скорость его горения при
атмосферном давлении приблизительно в G0 раз больше скорости горения,
например, тротила, близкого к черному пороху по теплоте горения. Воз-
можно, что слабая зависимость скорости горения от давления обуслов-
ливается тем, что экзотермические реакции при горении черного пороха
идут в значительной мере в конденсированной фазе, их скорость пе за-
висит от давления, передача тепла горения цепрОреаги реванше му веще-
ству также пдет цс только через слой паров или газов, как при горении
вторичных ВВ; а непосредственно от твердых пли жидких продуктов
реакции. Кроме того, превращение каждого из компонентов черного по-
роха При нагреве идет с поглощением тепла; тепло выделяется лишь при
взаимодействии компонентов или продуктов их первичного превращения,
смешение которых затрудняется при повышении давления.
12 К. К. Андреев
177
По данным Баума [176], величина V в выражении для скорости горения
трубочных порохов при прочих равных условиях тем меньше, чем меньше
объем газообразных продуктов горения на единицу веса пороха. Так, для
трех порохов, для которых объем газон составляет 515, 330 и 250 л1кг.,
v имеет значения соответственно 0,607, 0,242 и 0,117. Для составов, горя
1цих без выделения газов, скорость горения не зависит от давления.
Относительно слабый рост скорости горения с ростом давления п его
уменьшение при высоких давлениях, возможно, являются [170] одной из
причин низкой детонационной способности черного пороха.
Я. ТЕРМИТЫ
Как исходные, так п конечные продукты горения термитов являются
труднолетуч ими я относительно химически устойчивыми веществами,
реакция должна протекать поэтому полностью или во всяком случае пре-
имущественно в конденсированной фазе. Если это так, то скорость реак-
Рцс, 153. Зависимость скорости горения
термитов от давлении
1 — маргонскллюмиппевый термит; 2 -- и;е-
ЛЕ>яоа.тюм1|Циевый; >v — хромпМтииевый;
4 — XIHIMIKUIKIM и пневый терме г
ции, а также, как правило, и ско-
рость горения не должны зависеть
от давления.
Беляев [180] изучал при раз-
личных давлениях до 150 ат го-
рение ряда термитов: хромоалю-
миииопого термита (Сг^Оз + Д1)
со сравнительно низкой темпера-
турой горения (2000" по расче-
ту), желез оалюмнн ново го термита
(Ке20з + А1) с более высокой рас-
четной температурой горения, хро-
мом агниевого термита (Сл'аОз +
+ 3Mg) и маргапецалюмипиевого
термита , (1,5 МиО2-~2А1)
(рис. 152). Термиты прессовали в
виде цилиндрических столбиков
диаметром ~ 13 .м.м до относитель-
но низкой (0,5—0,6) плотности.
Скорость горения хромоалюмпнно-
вого термита (кривая 4) практически не зависит от давления; скорость
горения л.'рлезоалюмлиневого термита (кривая 2} растет с дав лепи ем сна-
чала — до 40 ат — быстро, потом медленно. Своеобразную зависимость дает
хромомаги новый термит (кривая 3\. При малых давлениях скорость горе-
ния растет лаже несколько быстрее, чем у железоалюмипневого термита,
при давлении 40—50 ат достигается 1максимум скорости, а при дальнейшем
увеличении давления наблюдается явное падение скорости. Прн самых,
больших изучавшихся давлениях горепве становится пульсирующим, что,
возможно, связано с малой плотностью применявшихся образчиков. Мар-
гансцалюмип новый термит (кривая 1) ведет себя аналогично железо алю-
миниевому термиту.
Независимость скорости горения хромоалюмппиевого термита от дав-
ления предположительно обусловливается тем, что при его низкой тем-
пературе горения не образуется паров компонентов и реакция идет только
в конденсированной фазе. Если это объяснение справедливо, то следует
ожидать, что у других термитов при снижении их температуры горения,
например, путем разбавления термита продуктами горения, скорость дол-
жна также стать не зависящей от давления,
Газовой фазой при горении жслезоалюминисвого термита следует счи-
тать парообразный алюминий- Ввиду незначительного количества испаря-
ющегося алюминия роль реакции в газовой фазе ограниченна.
178
IV. ВЛИЯНИЕ НАЧАЛЬНОЙ ТЕМПЕРАТУРЫ
НА СКОРОСТЬ ГОРЕНИЯ
Естественно ожидать, что повышение начальной температуры ВВ при-
ведет к увеличению скорости горения, поскольку весь интервал темпе-
ратур. который проходит вещество от начала превращения до его конца,
перемещается в область более высоких температур.
Теория горсиггя Зельдовича - Фраи к-Камо пен кого, исходящая из пред-
положения, что ведущая реакция происходит прп температуре, равной
максимальной температуре горечщя, именно так п рассматривает в.тпяппе
начальной температуры на скорость горения — полы ш с1 н г го начальной тем -
пературы приводит к соответственному повышению конечной темпера-
туры, вследствие чего возрастает вираже и гц> е~ЕРТ, определяющее в ос-
новном скорость химического превращения при горения, а слщщвателгяго,
и влияние температуры на его скорость.
Известно, что для большинства реакции, рассматриваемых' в хпмпие-
ской кинетике, повышении температуры па 10° в 2—4 раза увеличивает
скорость реакции. Н случае горении повышение начальной температуры
па КГ увеличивает скорость горыпгя всего па несколько процептов. ,г)то
различие определяется прежде всего тем, что при горении реакция про-
текает обычно при очень высокой температуре. Повышение начальной
температуры па 10° повышает температуру горепия приблизительно на
ту же величину; относительное повышение температуры горения гораздо
меньше, чем начальной температуры; соответственно меньше увеличниа-
1'гся п скорость реакции при температуре горения.
Однако опыт показывает, что реакция, приводящая к конечным про-
дуктам, не обязательно является ведущей. Зто наглядно рндпо па при-
мере тех ВВ, при горении которых наблюдается отчетливо выраженная
зональность реакций, как что имеет место для нитро гликоля п некоторых
других пптродфиров. Когда при достаточном повьгпгепип давления при
горении начинает протекать завершающая его стадия, приводящая к ко-
вочным продуктам превращения, никакого увеличения скорости ['прения
пе наблюдается. Зщ обстоятельство не удивительно, так как зона завер-
шающей реакции находится при со появлении далеко от поверхпости
жидкости и влиять на происходящие в ней процессы не может. В атом
случае ведущей может быть одна цз промежуточных реакции [гревраще-
ния вещества, проходящая спответстлонщ) при более низкой темпоратуре.
Тогда nprt прочих равных- условицх скорость гороппп будет сильнее за-
висеть ут начальной температуры, поскольку при некотором ео иопышо-
ппн увеличение температуры ведущей реакции будет относительно боль-
ше, чем повышен не коночной болею высокой температуры.
Наконец, возможен п такой случай, когда температура, а следователь-
но, и скорость недушей реакции по зависят от начальной температуры.
Как пример можно указать горение жидкого вещества, прп котором ве-
дущая экзотермическая реакция идет в копдечгворованной фазе, я тем-
пература последней ограничивается температурой кипения. Несмотря па
постоянство температуры ведущей реакции и независимость этой темпера-
туры от начальной, скорость горепия будет зависеть от начальной тем-
пературы, поскольку при ее повышении будет уменьшаться количество
тепла, которое пу/Ыю сообщить жидкости для пагрева со до температуры
кипения. Критической температурой в указанном смысле может быть не
только температура кипения, но ц температура питонепнпого газообразо-
вания, ведущего к диспергированию вещества.
Представление о разогреве вещества при горекпп до некоторой кри-
тической температуры как основно-Ц факторе, определяющем скорость
горения, содержится уже в гипотезе Малляра п Д[е-П.1ате.тьо, особенно
в ее применении к горению конденсированных ВВ. Ошибочным в этой
12*
179
гипотезе является в основном отождествление критической температуря
с температурой вспышки, физический смысл которой в то время пе был
еще достаточно выяснен. Разумеется, зту гипотезу нс следует противо-
поставлять теории Зельдовича — Фр анк-Камен едкого. Реализация того
или другого механизма горения зависит от конкретных свойств вещества.
Такое противопоставление тем более было бы не обоснованным, поскольку
Зельдович по существу разработал и представление о горении нрн условии
протекания ведущей реакции в конденсированной фазе, температура ко-
торой не зависит от начальной температуры, с учетом кинетики тепловы-
деляющей реакции, чего не было сделано Малляром и Ле-Шателье.
Такая теория приводит к закономерностям процесса, Отличным от тех,
которые получаются при горении, когда ведущая реакция протекает при
максимальной температуре. Обе эти гипотезы приводят, в частности, к
различным зависимостям скорости горения от начальной температуры,
а также от давления. Опыт показывает, что многие, в том числе и прак-
тически наиболее интересные, случаи находятся в лучшем согласии с вы-
водами, вытекающими из предположения о горении при постоянной тем-
пературе ведущей реакции, чем с гипотезой газофазного процесса,
Зависимость скорости горения от начальной температуры представляет
большой технический интерес. Желательно, особенно для порохов, пред-
назначенных для горения в полузамкнутом объеме, чтобы эта зависи-
мость была минимальной, что обеспечивает большее постоянство равно-
весного давления при разных температурах двигателя.
Определение скорости горения при температурах, отличных от тем-
пературы окружающей среды при повышенных давлениях, сложно по-
тому, что требует термостатировать те довольно громоздкие приборы (бом-
ба постоянного давления, манометрическая бомба), которые применяются
для изучения горения при повышенных давлениях, или же помещать
термостат в эти приборы. По этой причине большая часть исследований
проведена при атмосферном пли близких к нему давлениях.
Измерение зависимости скорости горения от температуры ограничи-
вается ипжиим и верхним пределами последней. Нижним пределом яв-
ляется та минимальная температура, ниже которой при данных условиях
(диаметр заряда, давление) горение не распространяется, Верхний пре-
дел температуры во всяком случае ограничен температурами, лежащими
вблизи температуры вспышки данного ВВ. Однако для некоторых ВВ,
особенно жидких или пористых, нормальное течение горения нарушается
при температурах, еще далеких от температуры вспышки; при этом горе-
ние может становиться пульсирующим, конвективным, идущим с большой
скоростью пли даже переходить во взрыв.
Рассматриваемые ниже зависимости скорости горения от температуры
различных ВВ относятся к тем интервалам температуры и других пара-
метров, влияющих на устойчивость горения, в которых нормальное то-
чение горения пе нарушается.
ЗАВИСИМОСТЬ СКОРОСТИ ГОРЕНИЯ
ОТ НАЧАЛЬНОЙ ТЕМПЕРАТУРЫ
ПРИ АТМОСФЕРНОМ И БЛИЗКИХ К НЕМУ ДАВЛЕНИЯХ
1, НИТРОЭФПРЫ И НИТРОСОЕДИНЕНИЯ [81, 114, 1811
Зависимость скорости горения метплнитрата от температуры изуча-
лась в пределах от 0 до 53° С (рис. 154); при 60°C после короткого участ-
ка горения возникает взрыв.
Нитроглпколь в трубках малого диаметра горит даже при — 20°С.
Скорость его горения значительно меньше, чем метплнитрата. В ряде ра-
180
бот скорость горения при атмосферном давлении измерялась в интервале
температур от 0° до почти температуры кипения (~ 200°С) (рис. 155).
Вблизи температуры кипения рост скорости усиливается и опа достигнет
1—5 см. что связано с образованием пузырьков вследствие разложен ян,
выделения растворенного воздуха и парообразования. Измерялась также
скорость горения нитрогликоля, желатинированного 3% коллоксилина;
при комнатной температуре она меньше, чем нежелапгиировяппой жид-
кости, но растет с температурой быстрее и при 100° С скорости почти
одинаковы.
Диэтилен гликольди нитрат горит еще медленное, чем пптрогллколб,
и лишь при больших диаметрах заряда. Зависимость скорости горении
жидкого и желатинированного вещества От температуры сходня с уста-
новленной для нитрогликоля (рис. 156).
Для жидкого нитроглицерина систематических опытов по определе-
нию зависимости скорости горения от температуры проведено не было.
Он горит при 20° и 300 мм со скоростью 0,068 см/сек, при 98° она воз-
растает до 0,117 см/сек, т- е. (в 1,7 раза) заметно сильнее, чем для жидкого
нитрогликоля (при атмосферном давлении — в 1,4 раза). Желатиниро-
ванный (97 ; 3) нитроглицерин горит со скоростью, приблизительно
в 4 раза большей, чем нитроглнколь, и быстрее растущей с температурой
(рис. 157).
Изучалось также горение растворов пнтроэфпров в зависимости от
соотношения Компонентов. Скорость горения раствора нитроглрколя в ни-
троглицерине при комнатной температуре растет с увеличением содер-
жания нитроглицерина, не аддитивно, а медленнее (рис. 158). Ирл 56—
60% нитроглицерина после короткого участка горения (5 — 7 .мл) насту-
пает затухание, а при 75% нитроглицерина раствор не загорается, а дает
при зажигании вспышку с расплсскивапиом, подобную той, которая на-
блюдается в опытах с чистым нитроглицерином. Скорость горения при
затухании по экстраполяции составляет около 0,09 г/см^сек, т. е. только
в 2 раза превышает скорость горения нитрогликоля.
При болео высокой температуре (95° С) скорость горения раствора ра-
стет с увеличением содержания нитроглицерина быстрее, чем при ком-
натной температуре, т. е. температурный коэффициент скорости горения
также возрастает с увеличением содержания нитроглицерина. При этом
способными к горению оказываются дая;е смеси со значительно большим
содержанием нитроглицерина (до 70%). Скорость горения 70%-ного ра-
створа сравнительно велика ц составляет 0,29 г/см2сек. Ирл больших со-
держаниях нитроглицерина горение идет с сильной пульсацией (иногда
затухая), и средняя скорость резко увеличивается.
Дигликолъдинитрат + нитроглицерин. При комнатной температуре го-
рение растворов Этих нцтроэфпров в трубках с внутренним диаметрам
6 лыг затухает, не дойдя до конца столбика жидкости. При повышенной
температуре (92—97° С) чистый дпглпкольлинптрат в трубках того же
диаметра горит со скоростью 0,022 см/сек. По мере увеличения содержа-
ния нитроглицерина скорость горения возрастает; прц содержании 60 %
нитроглицерина она составляет 0,09 елг/сек, при 70%—0,14 см/сек. а
при 80% нитроглицерина в смеси при зажигании происходит типична}!
для нитроглицерина вспышка с расплескиванием жидкости и горение не
распространяется.
Скорость горения нелетучего иитроэфира — нитроклетчатки — изуча-
лась в зависимости от температуры при разных плотностях. При 0° скоро-
сти горения нитроклетчатки (пироксилин № 1) при разных плотностях
были одинако-вы, при 95° более плотная нитроклетчатка (р = 0,6—ОД г/см3)
горела почти в 1,5 раза быстрее, чем мало плотная (0,19—0,26 г Дм3).
Сопоставление скорости горения при разных плотностях ц двух темпера-
турах (18 и 95° С) приведено на рис. 159. При низкой температуре
181
Рис. 154. Зн виги мог ti, скорости горении
митцлиптрита от Fin чалbiioii темпера-
туры
Рис. 155. Злнпснмость скорости горения
пптрогликоля от пачп.тьлоп темпера-
туры
Рис. 15G. Зависимость скорости горения
жидкого п желатинированного диэтп-
,чс11 тли кол ьдинптрата от начальной тем-
пературы
1 — жидкость; 2 — желатина
Рис. 157. Зависимость скорости горения
желатинированного нитроглицерина
(97 : 3) от начальной температуры
О 20 00 60 80 !00
Содержание ишпраглицерина,
8ее. %
Рис. 159. Зависимость скорости горения
пироксилина № 1 от плотности при раз-
Jin'iwx температурах-
J — rjiii С; 2 — при Э5° С
Рис. 158. ЗивцснмосТ|, скорости горения
растворов Пцтрогли|(о;гя в нитроглице-
рине от начальной температуры
1 I’oppHifr?; 2 — пульсирующее
горение
скорость горения но зависит от плотности почти во всем изученном ее
интервале. Напротив, при 95° С наблюдается отчетлнпое увеличение скоро-
сти как при низких, так и при высоких плотностях. Увеличение скорости
при малых плотностях обусловливается тривиальной причиной — проник-
новением газов в глубь сгорошка, имеющим место для данного образчика
пироксилина уже при р = 0,2 Дел/3. При повышенной температуре это
проникновение происходит легче, чем при низкой. Увеличение скорости
при больших плотностях, вероятно, связано с увеличением теплопровод-
ности нитроклетчатки при повышении плотности. Для горизонтальной
части кривой температурный коэффициент скорости горения составляет
1,97. При увеличении плотности выше 0,6 <щ становится еще больше.
Рис, 160, Зависимость
скорости горения гексо-
гена от начальной тем-
пературы
Рис. 161, Зависимость
скорости горения тетри-
ла от начальной темпе-
ратуры
Рпс. 162, Зависимость
скорости горения троти-
ла от начальной темпе-
ратуры
Из иптросоедпнепий изучались гексоген, тетрил п тротил (рос. 160,
161 и 162). Скорость горения тротила очень медленно растет с темпера-
турой и лнп1ь пЦп температурах выше 250° С (в стеклянных трубках) V
наблюдается значительное ускорение горения. Однако это ускорение на-
ступает не сразу после начала горения, а лишь после того как оно проб- -5
дет некоторый путь и сопровождается пульсацией и выбрасыванием брызг
горящей жидкости. Ио-впдимому, это связано с разогревом ц разложе-
нием жидкости в результате передачи тепла по стенкам трубки. В медных
трубках соответственно цх большей теплопроводности пульсация наступа-
ет при более низких температурах и тем раньше, чем выше температура
опыта.
2, ИНИЦИИРУЮЩИЕ ВВ
Зависимость скорости горения прессованной гремучей ртути от тем-
пературы определялась при разных давлениях ниже атмосферного [182].
Полученные данные с поправками па динамическое повышение Давления
приведены на рис, 163. Скорость горепин растет с температурой не ли-
нейно, а Несколько быстрее. При повышении температуры на 100° С
скорость горения возрастает приблизительно в 1,7 рапа, близко к тому, что
наблюдается для многих вторичных ВВ, На рис. 164 показапа также
зависимость скорости горения от давления при разных начальных тем-
пературах. При всех температурах Эта зависимость является линейной.
При этом, если продолжить прямые влево, то все они пересекутся с осью
183
абсцисс в одной точке. При 20° С и — 0,47 + 1,05 р; при 90° С и — 0,65 +
+ 1,44 р и при 105° С и — 0,71 + 1,60 р. Отношение Л : В при всех темпе-
ратурах постоянно, т. е. А и В одинаково меняются с изменением темпе-
ратуры.
Были поставлены также опыты, в которых было увеличено время пре-
дварительного прогрева таблетки гремучей ртути. На рис. 164 сплошным
квадратом показано среднее значение скорости, соответствующее предва-
рительному прогреву при 90° С в течение 1 часа вместо 15 мин. Более
длительный прогрев привел к некоторому снижению скорости горения,
которое сохраняется и по охлаждении при комнатной температуре.
Рис. 163, Зависимость скорости горе-
ния прессованной гремучей ртути
от начальной температуры
р, мм рт. ст
Рис. 164. Зависимость скорости горения
гремучей ртути от Д.твлегтия нри рапных
•начальных температурах (в ЕС):
i — 20; 2 — ЙО; з — 105
Беляева несколько расходятся с дан-
Описанные результаты опытов Беляева несколько расходятся с дан-
ными Мюраура и Вольгемута [183], которые при зажигании перепрессо-
ванной гремучей ртути уже при 108° С, а тем более при 120° С наблюдали
заметное механическое действие на свинцовую подкладку; нагревание
при 130 С приводило к самовоспламенению, носившему характер взрыва
с очень сильным действием на подкладку.
Рве, 165. Зависимость скорости горения
пороха от температуры
* 0 -
~°'S ~1S0 -00 О 00 00 ‘26
Температура, ’С
Рис. 166- Зависимость скорости горе-
ния пороха от температуры в коор-
динатах lg и — Т
В опытах Беляева [184] при этих температурах лишь иногда возникал
взрыв, обычно же имело место неустойчивое горение с проскоком пламе-
ни по поверхности со скоростью 2,5—3,5 см!сек, но без механического
действия. Возможно, что ото различие связано с различием в размерах
184
таблеток или режимов прогрева, которые применились в обеих работах.
Во всяком случае значительные колебания скорости от опыта к опыту
показывают, что горение находится на пределе устойчивости и весьма
склонно к ускорению и переходу во взрыв.
3. ПОРОХА
Ряд работ, проведенных с бронированными шашками пороха Н [114,
185, 186] в интервале температур от —18 до 125°С в несколько разли-
чающихся условиях, дали зависимость скорости горения от температуры,
приведенную на рис, 165 и 166. Обращает на себя внимание силгяюе и
резкое увеличение температурного коэффициента скорости горения воро-
ха, происходящее при температуре около 20° С.
В несколько иных условиях [187] и в более узком интервале темпера-
тур (75 —130°С) скорость горения различных нитроглицериновых поро-
хов определялась Мюрауром и Шумахером. Особенно резкое ускорение
горения наблюдается вблизи 130° С, вероятно, в результате протекающей
при этой температуре экзотермической реакции разложения пороха, по-
вышающей температуру и изменяющей структуру пороха.
Все эти данные получены при атмосферном давлении; они показывают
сильный рост скорости горения с повышением температуры. При повы-
шенных давлениях температурный коэффициент скорости горения пада-
ет, стремясь к некоторой постоянной величине.
Так, если на 1 ат скорость горения нитроглицеринового пороха Н при
повышении температуры от 15 до 95° возрастает в 2,45 раза, то ирп
2 ат она увеличивается только в 2,07 раза, а при 20 ат — только в 1,7 раза
при изменении температуры от —60 до +60°. Дальнейшее повышение
давления не изменяет температурный коэффициент в области положи-
тельных температур.
По данным Баума [176], скорость горения трубочного пороха Слабо
зависит от температуры, при ее изменении от —30 до + 44° скорость
возросла только на 5%.
По опытам А. Л. Шидловского скорость горения прессованного чер-
ного пороха (р = 1,7 г/см3) при 13 и 95° С составляла соответственно
0,95 и 1,04 сл/сек, т. е. увеличилась только на 15%. Такой же темпера-
турный коэффициент получили Беляев и Лукашеня [188] в интервале
температур 20—250° С. Таким образом, скорость горения черного пороха
по сравнению с другими изучавшимися ВВ очепь слабо зависит от тем-
пературы. Интересно, однако, что при повышении давления до 10 ат
температурный коэффициент скорости горения черного пороха, по дан-
ным Беляева, не уменьшается, как это наблюдается для других ВВ. а
сильно (вдвое) возрастает[. Температурный коэффициент для «бессер-
ного» пороха (KNOg — 85%, уголь — 15%) больше, чем обычного
(5 • 10-3 f/градус); при повышении давления (до 10 ат) ои снижается до
3 • 10'3 1/градус.
ЗАВИСИМОСТЬ СКОРОСТИ ГОРЕНИЯ ОТ ТЕМПЕРАТУРЫ
ПРИ ПОВЫШЕННЫХ ДАВЛЕНИЯХ
Значительная трудность при фотографическом определении скорости
горения при повышенных температурах и давлениях заключается в том,
что из-за усиленной конвекции при маленьких размерах термостата труд-
но поддерживать постоянство температуры в образчике по всей его
( Это, кстати, означает, что зависимость скорости горения черного пороха от дав-
ления при высоких температурах становится сильнее.
185
Рис. 167. Зависимость скорости горения
вторичных взрывчатых веществ от тем-
пературы при 25 ат
Числа у кривых—Jf, (см. тайл. 26): m —
твердый THE; ж — жидкий
Рис. 168. Зависимость скорости горения
вторичных взрывчатых веществ от тем-
пературы при 50 ат
Числа у кривых имеют тв иге значении, что
и на рис. 167
К
',5
f,3
1,Z
_J__________I_________L_
50 tOO 150
Температура,aC
Рцс. 169. Зависимость температурного
коэффициента скорости горения коллок-
силина и гексогена от температуры при
1 ат (к = щ-ио / Щ-jo)
Рцс. 170. Зависимость температурного
коэффициента скорости горения вто-
ричных взрывчатых веществ от темпе-
ратуры при 25 ат (к = иг + то / at_ю)
Рпс. 171. Зависимость температурного
коэффициента скорости горении вто-
ричных взрывчатых веществ от темпе-
ратуры при 50 ат (к = «ц-ю I iit-jo)
а)
-О----о--о—о—о
______I_____-I_______L—
50 f00 t50
Температураt 'С
Рис. 172. Зависимость спорости горения
тетрила от температуры в интервале
20-160= С при 25 ат
m — твердый тетрил; ж — жидкий
1
4W-
высоте п н течение всего цремспп горения, Эти трудности были преодоле-
ны; был сконструирован термостат в виде термоизо.лнроваиного красномед-
пого блока квадратного сечения (35 X 35 мм) высотой 120 .м.м с централь-
ным каналом и окнами, закрытыми стеклами, ВВ помещалось в плекси-
гласовой трубке, что исключало возможность опережающего прогрева
заряда при горении тепловой волной, распространяющейся по стенкам
трубки, а также в результате возможного повышения температуры термо-
стата аа счет нагрева его теплом, выделяю-
щимся при горении. Прозрачность трубки
цозно.чяла регистрировать скорость и харак-
тер горения фотографически, применение
же плексигласовой трубки давало возмож-
ность прессовать НЕ до высокой плот-
ности. Суммарная ошибка измерения ш>
превышала ±4%. Подробности устрой-
ства прибора ц методики опыта см. в ра-
ботах [122, 189].
Зависимость скорости горения от тем-
пературы в пптервале от 20° идо верхнего
предела (ПО—200° для разных ВВ) опре-
делилась [фн давлениях от 1 до 50 пт. Ско-
рость горенпн всех изучавшихся В В (ни-
троклетчатка, таи, дигликольдппитрат,
тетрил, гексогеп, три нитробензол, диами-
нотрипитробепзол, гексапитродифопиламии) растет с повышением темпе-
ратуры, Темпы этого роста существенно различны для разных В В
(рис. 167) и зависят также от давления, уменьшаясь с его ростом
(рис. 168).
В табл. 2G приведены температурные коэффициенты скорости горения
изучавшихся В В при различных давлениях. Сильнее всего возрастает
<' температурой скорость горения пптрозфнров — нитроклетчатки, дигли-
кольдинитрата и тэна; наименьший рост наблюдается у гексогена, трдни-
тробопзоЛа и тетрила; остальные ВВ занимают промежуточное поло-
жение.
Температурный коэффициент для большинства изучавшихся В В воз-
растает при повыгнецпн температуры (рис, 169). Эта зависимость сильнее
пыражепа у нитроклетчатки, слабее — у гексогена; она уменьшается при
по вышей и и давления. если исключить для некоторых В В область вы-
соких температур (рис. 170 и 171).
/ 00'------1-------1------1----
' В 50 100 150
р, кг/смг
Рис. 173. Зависимость темпера-
турного коэффициента скорости
горения нитроглицеринового по-
роха от давления
1 — WWi 2 — Utf/ilj."
Таблица 26
Температурные коэффициенты = н(Г10Ди50о скорости горения вторичных
нарывчатых веществ при различных давлениях
Давление, am
ВБ 1 5 W 12 20 25 30 50
Гексоген Так Тетрил ТИБ ДАТПБ Гсксцл ДГДН Пироксилин 5» 1 . . . 1Со.ТЛ(,К( 11.111(1 1,25 1,08 1,19 1,17 1,35 1,51 1,37 1 16 1.45 1 23 1 21 1,32 1 29 1J13 1,60 1,2(1 1,40 1,33 1,11 1 3(1 1 17 1,17 1 ,3(1 1,26 1,40
187
Для двух ВВ — тринцтробензола и тетрила — темпера!ура плавления
лежит в изучавшемся интервале температур. При переходе от твердого
состояния к жидкому скорость горения возрастает; этот переход, увели-
чивающий внутреннюю энергию ВВ на теплоту плавления, равноценен
повышению температуры тринитробензола на 70 и тетрила на 90° С,
Несмотря на примерно одинаковое повышение теплосодержания (и рас-
четной температуры), скорость горепия тетрила особенно сильно (при-
близительно в 10 раз) возрастает при 25 и 50 ат (ряс, 172) и гораздо
слабее (на 20—30%) и тех же условиях возрастает скорость горепия три-
питробепзола. В последнем случае горение идет па пульсирующем режи-
ме, не приводящем еще, однако, к большой скорости горения; тетрил же,
очевидно, горит па устойчивом быстром микротурбулентном (поскольку
неравномерности горения не наблюдается) режиме, сопровождающемся,
вероятно, и диспергированием вещества.
Возможно, что с неустойчивостью нормального режима горепия рас-
плава связано и упоминавшееся выше резкое ускорение горения тэна и
тетрила при высоких начальных температурах и давлениях. При повы-
шении начальной температуры увеличивается толщина прогретого слоя,
в частности расплавленной его части. Если слой расплава тонок, то тур-
булизация в нем не развивается. Это следует из того, что при обычной
температуре твердые, но плавящиеся ВВ горят во много раз медленнее,
чем в жидком состоянии в условиях, когда осуществляется турбулентный
режим горения. Если же расплавленный слой становится достаточно тол-
стым, то турбулизация получает возможность развиться и горение ус-
коряется 1 * * *. Развитию этих процессов может способствовать также сниже-
ние температуры плавления вследствие накопления конденсированных
продуктов распада в прогретом слое.
Наконец, при горении под повышенными давлениями становится боль-
шой и концентрация в конденсированной фазе газообразных продуктов
разложения, как известно, сильно ускоряющих, например термический
распад тэна.
В опытах при повышенных температурах горение отдельных ВВ от-
личается некоторыми особенностями. Так, для гексогепа и тэна наблюда-
ется раздвоение пламени на первичное и вторичное, При определении «(f)
для дигликольдинитрата при 25 ат и 150°, а также при 5 аг и 120s про-
исходили сильные взрывы; при 100° в нескольких опытах наблюдалось
затухание горения после прогорания некоторого участка. По-видимому,
эти явления связаны с переходом горения жидкости па турбулентный
режим.
Уменьшение температурной зависимости скорости горения при повы-
шении давления наблюдалось не только у индивидуальных ВВ, по и у
баллистцтяых порохов. На рис. 173 приведена зависимость температур-
ного коэффициента скорости горения нитроглицеринового пороха е 43%
нитроглицерина от давления [116). В интервале температур 25—50° С этот
коэффициент падает при повышении давления до 35 ат; при дальнейшем
увеличении давления вплоть до изученного предела (250 ат) температур-
ный коэффициент остается постоянным.
Аналогичное изменение претерпевает температурный коэффициент
скорости горения в интервале температур от 25 до 0°; отлцчце состоит
лишь в том, что постоянное значение температурного коэффициента до-
стигается при более высоком давлении (50—60 ат).
1 Этим объясняется, вероятно, и то, что, начиная с некоторой температуры, под-
жигание тэна приводит не к стадионарному горению, а к вспышке; при поджигании
вспышка возникает при тем меньшей температуре, чем выше давление (начиная со
130° при 25 аг и со 11<ГС при 50 аг).
188
Скорость горения нитроглицеринового пороха при разных температу-
рах (—80°, -[-18° и -(-97° С) была определена также при еще более вы-
соких давлениях в манометрической бомбе, т. е. при горении под возра-
стающим давлением [190], В качестве се характеристики была принята
обратная величина J pdt. Опыты показали, что 1/Jpdi возрастает с увели-
чением температуры от —80 до +97°С в 1,7 раза.
ВЫВОДЫ
Скорость горения всех изучавшихся ВВ возрастает с температурой1.
Этот рост невелик, В табл. 26 приведены для ряда ВВ значения темпера-
турного коэффициента — относительного увеличения скорости горения
яри повышении температуры на 1°С,
При повышении начальной температуры на 100° С скорость горения
при атмосферном давлении увеличивается, как правило, на 30—100%.
Вообще говоря, наблюдается известное соответствие между температур-
ным коэффициентом скорости горения при атмосферном давлении и тем-
пературой вспышки. Чем выше температура вспышки (тротил, черный
порох), тем слабее зависит скорость горения от температуры.
Для многих ВВ температурный коэффициент скорости горения не по-
стоянен, ио возрастает с температурой, для некоторых ВВ (гексоген) сла-
бее, для других (нитроклетчатка, тэн) значительно сильнее.
Зависимость скорости горения от начальной температуры может быть
1 т п
выражена в значительном ее интервале прямыми в координатах — —т в
1
соответствии с полуэмпприческим соотношением — — —В\То, Из
этого выражения следует, в частности, что температурный коэффициент
А = увеличивается с температурой. Количественно это увели-
чение зависит от соотношения между констаптами А\ ц Вц а также от Гц;
в области низких температур оно мало, при повышении температуры воз-
растает.
Температурный коэффициент, как правило, уменьшается при повыше-
нии давления. В области малых давлений это уменьшение идет быстрее,
чем при больших. Возможно, поэтому, что при достаточном повышении
давления температурный коэффициент снижается до некоторого посто-
янного значения, как это наблюдалось для изучавшегося в относительно
большом интервале давлении нитроглицеринового пороха с 43% нитрогли-
церина.
Наблюдается также некоторый параллелизм между изменением с дав-
лением температурного коэффициента и относительной теплоты горения
(отношения фактической теплоты к максимально возможной). Если от-
носительная теплота горения велика уже при атмосферном давлении (гек-
соген), то температурный коэффициент мал и слабо уменьшается с дав-
лением; если она мала (питроклетчатка), то температурный коэффициент
велик и сильнее уменьшается при повышении давления.
Температурный коэффициент скорости горения жидких ВВ зависит,
но-вндимому, также от их физических свойств и от изменения этих свойств
с изменением температуры.
Температурный коэффициент скорости горения нитроклетчатки зави-
сят от относительной плотности, при больших плотностях он возрастает,
вероятно, пз-.яа реакции в конденсированной фазе, ведущей к увеличению
скорости горения при повышенной температуре.
1 В литературе встречаются нек о икре газированные упоминания о том, что для
никоторых порохов в определенном интервале температур скорость горения при по-
вышении температуры уменьшается.
189
V. ВЛИЯНИЕ ОТНОСИТЕЛЬНОЙ плотности,
ДИАМЕТРА ЗАРЯДА И ИНЕРТНЫХ ПРИМЕСЕЙ
НА ВОЗМОЖНОСТЬ И СКОРОСТЬ ГОРЕНИЯ
Влияний относительной плотности порошкообразного ВВ на возмож-
ность if скорость горения представляет значительный интерес по следу-
ющим соображениям [191].
Изменение относительной плотности, очевидно, цо может илнять на
скорость .химических реакций, протекающих в га зоной фазе- Не может
оно также оказывать прямого влияния и па течение реакции в конденси-
рованной фазе. В то же время условия распространения тепла в конден-
сированной фазе могут существенно меняться как в результате измене-
ния теплопроводности порошка, так п в результате изменения его про-
ницаемости для материальных носителей тепла — жпдипх (расплав веще-
ства) или газообразных (газообразные продукты испарения и распада).
Таким образом, изучение влияния относительной плотности па горение
позволяет выявить роль распространения тепла в конденсированной фазе
в процессах горения. Изменением условия распространения тепла в но-
рописс it объясняется своеобразное влияние относительной плотности ла
возможность и спорость горения различных твердых ВВ.
Утл влияние изучалось иа двух плавящихся порошкообразных ВВ --
тетриле н гексогене п двух печяавящихся — нитроклетчатке и измель-
ченном в порошок нитроглицериновом порохе (.
1. ВЛИЯНИЕ ОТНОСИТЕЛЬНОЙ плотности вв
ПА ВОЗМОЖНОСТЬ ГОРЕНИЯ
Опыты показали, что для плавящихся ВВ существует минимальный
предел относительной плотности, вблизи которого горение, равномерное
при более высоких плотностях, становится пульсирующим, а ниже зату-
хает. Предельная плотность зависит от диаметра трубки: при большем ди-
аметре она меньше. Она зависит также от размеров кристаллов ВВ. Опы-
ты ио выясненцн) влияния размеров кристаллов вещества [192] были по-
ставлены с тремя образчиками тетрила; первый образчик представлял
относительно крупные кристаллы, преходившие через сито № 49* 2 и оста-
вавшиеся па ('цте № 73, второй образчик — кристаллы средних размеров,
проходившие через сите № 73, и третий образчик самые мелкие кристал-
лы, полученные растиранием тетрила в ступке. Сжигание производили и
стеклянных трубках с внутренним диаметром 19 -ч.м.
Результаты опытов, приведенные иа диаграмме рис. 174, показывают,
что чом крупнее кристаллы, тем больше нижний предел плотности. При
атом массиваи скорость горения вблизи предела зависит от размеров кри-
сталлов: при крупных кристаллах опа Отчетливо больше.
Причина перехода горения на пульсирующий решим и затухания его
при малых плотностях при горении плавящихся веществ заключаются в
слишком большой скорости отвода тепла в конденсированную фазу. Нри
малых плотностях :»тот отвод происходит в значительной части путем про-
никновения расплава вещества в промежутки между частицами. Если ото
проникновенно невелико, то вещество горит, причем скорость его может
быть даже несколько выше вследствие увеличения тепло up их ода от цро
дуктон горения, что мы и наблюдаем в опытах с крупными кристаллами.
Если, однако, вследствие увеличения размеров промежутков .между чн-
! Пптроглицсрнвовый порох значительно размягчается при нагреве др высоких
температур, но сохраняет достаточно большую вязкость в ведет себя в условиях го-
рении как исплавнщееся вещество,
2 Размер B'ii‘rif,'n сита М 49—(J.123 .«•«, сита № 73- 0,G8i л-.it.
190
стнцами проникновение расплава облегчится чрезмерно, то теплоотвод в
глубь заряда может стать столь большим, что он пе будет компенсиро-
ваться теплопрпходом от газов, и горение затухает. Понятно, без пояс-
нении, значение при описанном механизме влияния размеров кристаллов
и относительной плотности; увеличение диаметра заряда уменьшает по-
терн тепла в окружающую среду п сдвигает соответственно предельные
значения двух других параметров. Добавим, что для образчиков тетрила
с разной формой кристаллов наблюдались ^злачные значения предельной
плотности, что естественно связать с различиями в проницаемости порош
На для расплава, зависящей нс толь-
ко от размеров, цо и от формы час-
тиц-
Аналогичные наблюдения в отно-
шении влияния плотности и разме-
ров кристаллов на возможность горе-
ния были сделаны и для второго
изученного плавящегося вещества —
гексогена.
Влияние относительной плотности
на горение пеплавящихся порошко-
образных ВВ несколько отличается
от описанного выше. I [рп малых плот-
ностях также может наблюдаться ми-
нимум плотности', ниже которого
равномерное горение уступает место
пульсирующему; однако причина
пульсации в данном случае другая:
при уменьшении сопротивления по-
Рис- 174. Влияние на возможность
и скорость горения тетрила отло(-птсл|,-
iroii плот пости и размеров частиц:
7 — равномерное горение (цифры Cytpapu по-
кидывают массовую скорость гсц»е? «тп; г в
генсек); 2 — неравномерное горение; J —
затухание
роп[ка вследствие увеличения проме-
жуткой между частицами начинает нграц, роль проникновение газообраз-
ных продуктов горепия в глубь порошка. Эффективная поверхность горе-
ния возрастает, увеличивается количество вещества, сгорающего в единицу
времени. Обычно ускорение горения в условиях описанных опытов было
преходящим и сменялось замедлением, за которым вновь следовало уско-
рение п т. д.— горение шло пульсируя.
Причину пульсации следует видеть в том, что при проникновении га-
зов в глубь порошка сильно увеличивается поверхность контакта газон
с частицами порошка; газы при этом сильно охлаждаются, частицы же
порошка нагреваются недостаточно, чтобы сразу загореться. Однако после
некоторого индукционного периода в прогревшемся слое ВВ происходит
вспышка в форме быстрого сгорания, Опа приводит к повышению давле-
ния, которое заставляет горячие газы проникнуть вновь на значительную
глубину в В В, После выгорания вспыхнзшшего слоя давление падает, ско-
рость Горения также временно уменьшается; затем вновь происходит
вспышка прогретого газами слоя и т. д.
Падение давления, отсутствие прогретого слоя, который выгорел, по-
нижение температуры газов, приводящие при пульсирующем горении к
временному периодическому замедлению процесса, могут привести и к
затуханию горения, которое наблюдалось п эксиернменталг.ио при из
вестпых соотношениях между размерами частиц, восиламспяемостп их и
условий опыта. Если же интенсивность вспышки слоя, прогретого про-
никшими газами, достаточно велика, то опа может привести к лавинооб-
разному ускорению проникновения газон и горения в глубь заряда, за-
к?пчивеющемуся взрывом.
1 Этот мпвиммум не всегда можно обнаружить — он может быть меньше насып-
ной ПЛОТНОСТИ,
191
Таким образом, поведение при низких плотностях неплавящихся ВВ
с точки зрения возможности проникновения в порошок носителей тепла
(с соответствующими его последствиями) сходно с плавящимися1.
Существенное различие между плавящимися и неплавящимися ВВ
наблюдается яри повышенных плотностях. Для неплавящихся ВВ суще-
ствует некоторый верхний предел относительной плотности, выше кото-
рого горение при прочих равных условиях опыта не распространяется.
Так, например, в опытах с пироксилином № 1 в трубках с внутренним
диаметром 5—10 мм горение затухало, если плотность порошка превосхо-
дила 0,5—0,6 г!см?. Измельченный в порошок нитроглицериновый порох
устойчиво горел в стеклянных трубках с внутренним диаметром 9 да при
относительной плотности 0,4, при плотности около 0,6 горение затухало,
не доходя до конца заряда. Шашка пороха (р = 1) не горит даже при
диаметре 15 да.
Причина существования верхнего предела относительной плотности по
существу та же, что и нижнего ее предела,— чрезмерное увеличение ско-
рости распространения тепла в конденсированной фазе. При больших
плотностях порошка теплопроводность его возрастает настолько, ито тепло
отводится в глубь вещества быстрее, чем оно подводится из газовой фазы,
вследствие этого горение затухает.
Казалось бы, что такой эффект должен проявляться и при горении
плавящихся веществ. Однако состояние зоны повышенной температу-
ры — зоны расплава — и толщипа ее при больших плотностях здесь прак-
тически не зависят от плотности; не зависят поэтому от последней и теп-
лопотери прогретого слоя конденсированной фазы. Поэтому верхнего пре-
дела относительной плотности для плавящихся ВВ не наблюдается.
2. ВЛИЯНИЕ ОТНОСИТЕЛЬНОЙ ПЛОТНОСТИ ВВ
НА СКОРОСТЬ ГОРЕНИЯ
Изменение относительной плотности не отражается прямо на ходе ре-
акций в газовой фазе и теплопередаче от газов конденсированной фазе.
Однако, так как теплоемкость единицы объема вещества прямо пропорци-
ональна относительной плотности, то объем вещества, разогреваемого га-
зами до некоторой определенной температуры в единицу времени, прп
уменьшении плотности соответственно увеличивается. Поэтому линейная
скорость горения должна возрастать с уменьшением относительной плот-
ности. Опыты подтвердили, что с изменением относительной плотности
порошка линейная скорость горения меняется.
Для ряда изученных порошкообразных ВВ (тетрил, гоксогеп, нитро-
клетчатка, порошок нитроглицерппового пороха) величина линейной ско-
рости горения приблизительно обратно пропорциональна относительной
плотности, и произведение этих величин, т. е. массовая скорость горения,
приближенно постоянна [192]. Для тетрила это было установлено в ин-
тервале относительной плотности от 0,4 до 0,6 а/см3, для гексогена — от
0,38 до 0,64, для нитроклетчатки — от 0,12 до 0,36, для пороха — от
0,37 до 1.
На рис. 175 приведены экспериментальные результаты для индивиду-
альных ВВ. Из графика видно, что постоянство скорости не является стро-
гим- В случае тетрила, гексогена и порохового порошка массовая ско-
рость несколько возрастает с увеличением плотности.
В конце предыдущего раздела уже отмечалось, что у плавящихся ВВ
поверхностная зона конденсированной фазы представляет собой расплав,
г При горении плавящихся ВВ проникновение газообразвых продуктов горения
в глубь порошка при малых п.чотпостдх ве происходит, так как газы отделены от по-
рошка слоем расплава.
192
12
• -1
ь-г
О-J
□ -i
£р дд
о (Р оо со
0,5
° 0,2 0,1;
-1—I—I—I—I—I—I___I t I [_
0,0 0,8 1,0 1,2 1,0 1,6
fi, г/см3
1,0 1,5
/. г/см3
Рис. 175. Зависимость скорости горении
некоторых ВВ от кубической плотности
? — пироксилин X? 1; 2 — тетрил; 5 — гексо^
геи; 4 — порох
Рис. 176. Влияние относительной плот-
ности па массовую скорость горения
тэна при 52 ат
1 —размер частиц —5 лгк; 2 — 100—400 №
[[ се состояние поэтому не зависит от относительной плотности порошка
(при высоких плотностях). С этим обстоятельством Тэйлор [193] связыва-
ет я независимость массовой скорости горения от относительной плотно-
сти, наблюдавшуюся яри горении тэна с разными размерами частиц при
давлении 52 ат (рис. 176). Массовая скорость горения приближенно по-
стоянна; однако прп малых и больших плотностях наблюдается некоторое
ее увеличение, аналогичное тому, которое было установлено Г. В. Оран-
ской для нитроклетчатки н, вероятно, определяемое теми же причинами.
Добавим, что при малой плотности тэна и значительной плотности инерт-
ного газа можно бььчо бы ожидать из-за разбавления некоторого уменьше-
ния скорости, которое, очевидно, преодолевается усилением конвективной
теплопередачи, приводящим к увеличению скорости. Для крупнозериисто-
i‘o тэна при малой плотности (при большой плотности кристаллы дробятся
во время прессования) скорость Отчетливо больше, чем для мелкозерни-
стого. по-вндпмому, по той же причине.
Несколько меньше н скорость горения порошкообразного октогеиа прп
малой плотности (рис. 177), причем разница становится более заметной
при больших давлениях, когда сильнее разбавление. При 205 ат эта раз-
ница, по-видимому, уменьшается, возможно, потому, что уже сказывается
проникновение газообразных продуктов горения в глубь заряда.
Опыты И. И. Полякова с нитро-
клетчаткой (коллоксилин), проведен-
ные в широком интервале плотности
(0,15—0,93), показали, Ито при плот-
ностях больше 0,6 массовая скорость
горения существенно (примерно в
1,м раза) увеличивается с плот-
Рис. 178. Зависимость скорости горения
коллоксилина от плотности при различных
давлениях (цифры у кривых)
Пунктиром обозначена области пулт/енрующе*
го горения
Рис. 177. Зависимость скорости горения
октогева разной дисперсности от дав-
ления
I — размер частип кристаллов 5 мк; 2 —
124—132 .ик
13 Н h‘. Анлрееэ
193
ностыо. Ю. А. Рассудив, работавший с другим образчиком коллоксилина,
который горел значительно медленнее чем изучавшийся И. И. Поляко-
вым, не обнаружил при 1 и 3 аг влияния плотности на скорость горения,
которая оставалась постоянной. Однако при 10 ат и более высоких давле-
ниях такая зависимость наблюдалась — скорость значительно возрастала
при увеличении плотности (рис. 178). Правда, в этих условиях влияние
Рис. 179. Зависимость скоро-
сти горения некоторых предо-
хранительных ВВ от кубиче-
ской плотности при 100—110
плотности может быть частично связано с
разбавлением ВВ инертным газом, которое
уменьшается при увеличении плотности.
Подобное Hie влияние относительной плот-
ности на скорость горения наблюдалось в
опытах Ю. А. Рассулина при горении тэна,
скорость которого не зависит от плотности
при 20 ат и заметно возрастает с плотно-
стью при 150 ат\ однако для тетрила уве-
личения скорости горения при повышении
плотности не наблюдалось ни при 20, ни
при 100 ат.
Влияние плотности на скорость горе-
ния может быть связано с разными причи-
нами: изменением теплопроводности ВБ,
1 — победит ВП-3; г — побелит
ВП-i; 3—водоустойчивый аммо-
нит ПЖВ-20; 4 — аммонит Л1 8;
3— победит ПУ-2
которое может влиять и на скорость горе-
ния (особенно если в конденсированной
фазе идет экзотермическая реакция), и на
теплопотери в окружающую среду, а так-
же с изменением теплового эффекта на
единицу объема ВВ, в то время как теплопотери на нагрев оболочки оста-
ются ПОСТОЯННЫМИ И др.
Для всех изучавшихся ВВ при малых плотностях наблюдался переход
горения на пульсирующий ускоренный режим; относительная плотность,
при которой это наступало, была тем больше, чем выше давление
(рис. 178).
При большой проницаемости порошка скорость горения может возра-
стать вследствие проникновения газообразных продуктов горения в глубь
порошка. В опытах с измельченным в сравнительно крупный порошок
нитроглицериновым порохом наблюдалось отчетливое влияние размеров
частиц порошка на скорость горения [81]. Мелкий порошок прп малой
плотности (0,4) горел с пониженной скоростью (0,071 г/сле2сек), очевидно,
потому, что влияние проникновения газов в узкие поры практически не
сказывалось. При горении крупного порошка это влияние имело место
и скорость горения была заметно больше (0,092 г/см2сек). При большей
плотности обеих фракций (около 0,69) проникновение газов не играло
роли ни при горении крупного, ни при горении мелкого порошка, и ско-
рости горения были одинаковы (0,084 г/ем2сек).
Влияние проницаемости порошка может сказываться и при горении
плавящихся ВВ. В предыдущем разделе уже отмечалось, что в боль-
ших кристаллах тетрил (малой плотности) горел быстрее, чем в мелких,
предположительно из-за проникновения расплава в глубь порошка.
Аналогичные наблюдения были сделаны Тэйлором при горении тэна
и октогена при умеренно повышенных постоянных давлениях (табл. 27
и 28) а.
1 Различные образчики нитроклетчатки при одинаковом содержании азота не-
редко значительно различались по скорости горения при атмосферном давлении; воз-
можно, это связано с различным содержанием минеральных веществ, катализирую-
щих горение и перешедших в нитроклетчатку из сырья (клетчатка, кислоты, про-
мывные воды).
г Тэйлор большую скорость горения крупнозернистого порошка объясняет обра-
зованием в этом случае некоторого микрорельефа поверхности жидкого слоя.
194
Рассмотренные примеры иллюстрируют простейший случай, когда эк-
зотермические реакции идут преимущественно в газовой фазе и роль плот-
ности ограничивается ее влиянием на проницаемость вещества для рас-
плава или газов, а также на теплопроводность порошка.
В общем случае зависимость скорости горения от относительной плот-
ности может быть более сложной. Так, например, по опытам М. А. Раби-
нович, для смеси бариевой селитры с идитолом (89 :11) при увеличении
плотности в области малых плотностей наблюдается слабое уменьшение
скорости горения; при дальнейшем увеличении плотности скорость горе-
ния быстро растет; при больших плотностях наблюдается резкое падение
скорости.
Т а б л и ца 27
Влияние размера частиц на скорость
горения тэна при 27,2 ат
Диаметр частиц, лк Плотность заряда! г/см9 Массовая скорость горения, г/см9 сек
5 0,87 0,461 0,480
353—500 0,96 0,516 0,535
500—853 0,92 0,544 0,531
Таблица 28
Влияние размера частиц на скорость
горения октогена при 12,6 ат
Диаметр частиц, лсх Плотность заряда, а/сле Массовая скорость горения, г/с-к'-сек
~5 1 02 0,48
67—76 1,05 0,52
104—124 1,04 0,56 '
200—600 1,19 0,69
Необычное влияние плотности на скорость горения наблюдалось у ряда
аммонитов [194], преимущественно предохранительных (рис. 179). При
увеличении плотности от 1 до 1,7 г [см? массовая скорость горения (под
давлением азота 100—110 аг) существенно уменьшается, хотя скорее,
учитывая уменьшение разбавления порошка инертным газом при повы-
шении плотности, следовало ожидать обратного ее влияния.
3. ЗАВИСИМОСТЬ критической плотности вв от давления
Мы видели, что нарушение равномерности горения и переход его на
пульсирующий режим с увеличенной скоростью наблюдается при атмос-
ферном давлении в том случае, если уменьшить относительную плотность,
иначе говоря, увеличить размеры пор в порошке до некоторого предела.
Естественно ожидать, что при данной относительной плотности можно
получить переход горения с нормального на ускоренный режим, если уве-
личить давление, под которым идет горение. Это заключение было прове-
рено опытами по горению ряда порошкообразных ВВ при различных по-
стоянных давлениях и при возрастающем давлении.
Горение при постоянном давлении
Опыты производились в различных приборах постоянного давления.
Данные для пироксилина № 1 представлены на рис. 180. При некоторой
определенной плотности порошка массовая скорость горения при малых
давлениях линейно растет с увеличением давления. Однако эта зависи-
мость выполняется только до некоторого давления, выше которого ско-
рость горения начинает расти быстрее и становится менее однообразной,
хотя нарушения равномерности горения визуально обнаружить еще не
удается. При дальнейшем повышении давления меняется характер горе- (
ния, оно становится отчетливо неравномерным, пульсирующим, Скорость
13* 195
горения (точнее количество вещества, сгорающего в 1 сек на 1 см1 сече-
ния заряда) резко возрастает и становится еще более колеблющейся.
Величина критического давления зависит от относительной плотности,
возрастая с ее увеличением (рис. 181). То, что основной причиной зави-
симости критического давления от плотности являлось в условиях этих
опытов влияние последней на проникновение газов, показывает кривая
рис. 181, изображающая зависимость удельного сопротивления пирокси-
лина прохождению воздуха при небольших перепадах давления от куби-
ческой плотности [г = Ар/у, где Др — перепад давления, мм вод. ст. на
1 см длины трубки сечением 1 см2, a и — объем прошедшего воздуха
(в см3/мпн)].
Рис. 181. Зависимость предельного
давления для пироксилина № 1 от
кубической плотности
I — горение равномерное; 2— горение с
проскоком; з — уд. сопротивление
Рис. 180. Зависимость скорости горе-
ния пироксилина № 1 от давления
(р = 0,37 — 0,45 г/см3, d — 10 мм,
t = 8—КУС)
В более широком интервале давлений (до 1000 ат) переход горения
на ускоренный режим и особенности горения в этом режиме изучал Чуй-
ко [195] на примере тэна, гексогена, тетрила и Тэйлор — на примере окто-
гена, тэна и гексогена при давлениях до 200 ат. В опытах Чуйко ВВ в виде
фракций с определенным размером частиц прессовалось порционно в плек-
сигласовые трубочки диаметром 5 мм (толщина стенок 1 мм, высота
35 мм). Горение осуществлялось в бомбе постоянного давления, наполнен-
ной азотом, и фотографировалось на вращающемся барабане.
Зависимость скорости горения от давления для двух образчиков тэна
с разными размерами частиц и при различных относительных плотностях
представлена на рис. 182.
В области малых давлений массовая скорость горения порошкообраз-
ного ВВ малой плотности та же, что и спрессованного, и также растет
с давлением. Однако по достижении некоторого критического давлении,
величина которого тем больше, чем больше относительная плотность и
чем меньше размер частиц, горение переходит на ускоренный режим, при-
чем Аим/Ар становится гораздо (в 10—120 раз) больше. Характер кривой
ц(р) различен в зависимости от плотности, а, возможно, и от давления,
поскольку при различных плотностях смена режимов горения происходит
при разных давлениях.
На зависимость скорости горения от давления на ускоренном режиме
влияют два частично взаимосвязанных фактора — проникновение газооб-
разных продуктов горения в глубь порошка, которое собственно и явля-
ется причиной повышенной скорости горения, и разбавление ВВ инертным
газом, тем большее, чем меньше плотность порошка и чем больше давле-
ние. Проникновение газообразных продуктов приводит к тому, что горе-
ние идет не только на торце заряда, но и в слое порошка некоторой тол-
щины, на поверхности его частиц. Соответственно большей эффективной
196
поверхности горения на ускоренном режиме больше и удельная (па еди-
ницу поверхности сечения заряда) скорость газообразования. О том, что
частицы горят в слое значительной толщины, говорит и наблюдавшийся
особенно при крупнозернистых порошках выброс несгоревшего вещества,
оседавшего после опыта на дне бомбы. Диспергирование вещества, интен-
сивно развивающееся при горении порошков малой плотности, является
одним из Стабилизирующих факторов, определяющих возможность горе-
р, кг/см?
ния с большими скоростями без пе-
рехода его во взрыв. Унос вещества
препятствует утолщению слоя горя-
щей взвеси и ограничивает связан-
ный с ним рост давления в зоне го-
рения.
Разбавление порошка ВВ малой
плотности снижает температуру газо-
образных продуктов, проникающих в
порошок, содержащимся в послед-
нем инертным газом. Особенно на-
глядно влияние разбавления показано
от
ООО
юо
°0,г 0,0 0,0 0,0 1,0
Рас. 182. Зависимость скорости горения
тэна (различных размеров частиц и отно-
сительных плотностей) от давления
В — зависимость для высокоялотиого вещества.
Равмеры частиц (в ж к) и плотности (в г/еж’):
1 — 200 и 0,57; Э — 200 и 0,66; 3 — 5 и 0,28;
4 — 5 я 0,40; 5 —5 и 0,66
Рис. 183. Зависимость скорости горения
при 1000 ат мелкокристаллического
5 лк) тэна от относительной
плотности
на рис, 183, изображающем изменение скорости горения мелкокристалли-
ческого тэна в зависимости от плотности. Падение массовой скорости горе-
ния при увеличении плотности (ветвь ВС кривой) вполне естественно
и является следствием уменьшения зазоров между частицами, а следова-
тельно, и газопроницаемости порошка. Восходящая же ветвь кривой АВ
может быть, очевидно, объяснена лишь уменьшением разбавления, которое
особенно сильно проявляется при большом давлении (плотности) инерт-
ного газа и малой относительной плотности, когда доля объема, приходя-
щаяся на поры, велика. При плотности 0,27 и 1000 аг содержание азота
в порошкообразном тэне составляет 44,5%, при плотности 0,4— 28%.
Разбавление в условиях опытов при постоянном давлении оказывает
на устойчивость и особенно на характер ускоренного горения своеобразное
стабилизирующее влияние. Чем выше давление, тем больше скорость го-
рения и динамическое повышение давления во фронте, заставляющее газы
горения проникать в глубь порошка, и тем выше эффективная темпера-
тура последних. Однако одновременно повышение давления, усиливая
разбавление, снижает температуру газов, увеличивает теплоемкость
смеси ВВ + инертный газ, понижает температуру горения, т. е. ско-
рость его.
В области умеренно повышенных давлений (75—450 яг) для мелко-
кристаллического тэна при увеличении плотности, приводящем к увели-
197
чению давления срыва, разбавление остается приближенно одинаковым;
отношение же Ардт. ц сильно уменьшается. Это указывает на то, что пре-
обладающее влияние имеет повышение температуры газов (вследствие
увеличения полноты превращения), проникающих в порошок. При боль-
ших давлениях (1000 ат) роль этого фактора, естественно, уменьшается
н преобладающее значение получает разбавление.
В делом разбавление в большей или меньшей степени нейтрализует
влияние проникновения газов. При этом именно в тех условиях, где газо-
проницаемость велика (малая плотность), или велико газодинамическое
повышение давления (большие давления, т. е. большие скорости горе-
ния), велико также разбавление. Поэтому благоприятные условия для
проникновения горения могут реализоваться лишь в том случае, если это
проникновение происходит при относительно низких давлениях, т. с.
либо в крупнозернистом порошке, либо для ВВ с большой скоростью го-
рения. Действительно, наибольшее увеличение скорости горения наблю-
далось у крупнозернистого тэна и гексогена при сравнительно умеренно
Рис. 184. Фотозапись горения тетрила в ускоренном режиме
повышенных давлениях. Обращает на себя внимание и то, что непрерыв-
но ускоряющееся горение мелкокристаллического тзна наблюдалось при
некоторой средней плотности и давлении; при большой плотности проник-
новение начиналось при большом давлении, когда разбавление было уже
велико; при малой плотности оно начиналось рано, но при этом давлении
скорость горения и динамическое повышение давления были Ьще малы.
При средних давлениях могут сочетаться все условия, благоприятствую-
щие проявлению проникновения — достаточно большая скорость и темпе-
ратура газов горения и не слишком большое разбавление ВВ.
Естественно и то, что при умеренно уплотненной гремучей ртути про-
никновение газов горении проявляется столь сильно, что приводит к пе-
реходу горения в детонацию уже при атмосферном давлении. Одной из
причин этого является большая скорость горения гремучей ртути. При
горении вторичных В В, имеющих при атмосферном давлении значительно
меныпке скорости и эффективные температуры горения, динамическое по-
вышение давления недостаточно, чтобы обеспечить интенсивное проник-
новение горения.
Фоторегистрация горения ВВ в ускоренном режиме показывает неко-
торые его особенности. По сравнению с нормальным горенпем фронт его
размыт, особенно при горении крупнокристаллического порошка; переме-
щение фронта менее равномерно — иногда эта неравномерность имеет ха-
рактер проскоков, иногда она меньшего масштаба и имеет относительно
«равномерный» характер. Свечение обычно имеет неравномерную поло-
сатую структуру. Частым ее типом является тот, который наблюдался при
горении тетрила (рис. 184). У окоренный режим обладает известной устой-
чивостью и обычно, раз возникнув, уже не переходит в нормальный; сред-
няя величина скорости приближенно постоянна — в отдельных опытах,
однако, наблюдался ее непрерывный рост. Особенностью ускоренного ре-
жима является также характер свечения в конце горения: оно продолжа-
198
ется (в отличие от нормального горения) более или менее длительное
время.
При неравномерном горении гексогена и особенно тетрила на снимках
заметны отдельные почти горизонтальные
пеньки. Их возникновение, по-видимому,
с вяз а но с проникновением горячих газов в
глубь порошка отдельными струями.
О струйчатом проникновении газов в ре-
жиме ускоренного горения говорят также
опыты, в которых дном заряда служила
тонкая бумага. Следы действия высокой
температуры’ были на ней в виде отдель-
ных пятен, в то время как при нормальном
горении обжиг был равномерен.
Условия и результаты опытов Тэйлора
сходны с описанными выше. Влияние раз-
меров частиц октогена на переход горения
на ускоренный режим показано на рис. 185.
Мелкий (5 мк) порошок даже при малой
плотности во всем изучавшемся интервале
давлений горел в нормальном режиме со
скоростью, линенио растущей с давлени-
ем; более крупнозернистые образчики да-
вали переход на ускоренный режим,
интенсивно светящиеся сту-
который наступал тем раньше, чем круп-
нее были частицы. Зависимость скорости
горения от давления при горении в быст-
ром режиме сильнее для крупнозернистого
порошка, аналогично горели в опытах Тэй-
лора тэн и гексоген.
Рис. 185. Скорости горения спрес-
сованного порошка октогена при
различных давлениях
Размер частиц (в лк) и плотность
(в е1см‘): 1 — 200—600 зос и Р - 1,20
е/см3; 2 — 104—124 и 1.05; 3 — 64—76
и 1.07; 4 — 53—64 и 1,08; 5 —5 и 1.02
1
Для подтверждения механизма горения
в быстром режиме Тэйлором были постав-
лены опыты по горению в плексигласовых трубках длиной около 8 см в
трех вариантах: 1 — с закрытым нижним концом; 2 — с нижним концом,
закрытым сеткой; 3 — с закрытым сеткой нижним концом и суженным
Рис. 186. Данные Тэйлора по горе-
нию крупнозернистого октогена в
различных условиях (давление азота
27,2 аг, размер частиц 200—600 зек,
плотность 1,08 г/сле3)
Рис. 187. Зависимость массовой скорости
горения етружия пороха, Н от относитель-
ной плотности заряда
приблизительно вдвое верхним отверстием. Результаты опытов, произво-
дившихся с крупнозернистым октогеном (200—600 лк)т представлены на
рис. 186. Начальная скорость горения во всех трех вариантах одинакова
В дальнейшем, однако, горение ускоряется — больше всего в третьем
199
варианте и меньше во втором. По мере уменьшения высоты оставшегося
столбика В В сопротивление его уменьшается и соответственно усиливает-
ся проникновение газов из фронта. В первом варианте, напротив, на по-
следних сантиметрах горение замедляется, очевидно, вследствие того, что
проникновение газов затрудняется возникающим противодавлением, кото-
рого не возникает при втором варианте. В третьем варианте сужение
выходного сечения трубки создает дополнительное повышение давлении,
добавляющееся к динамическому его повышению, вследствие чего усили-
вается проникновение газов в порошок. Само по себе увеличение давления
слишком мало, чтобы объяснить наблю-
дающееся увеличение скорости горения.
Кондриков [196] изучал зависимость
скорости горения некоторых порохов
(в виде стружки) от относительной
плотности (рис. 187). В области малых
плотностей скорость быстро возрастает
с плотностью, проходит через максимум,
на котором линейная скорость в 30—70
и больше раз превышает нормальную
скорость горения, и затем падает, при-
ближаясь к скорости, характерной для
сплошного вещества. Ари дальнейшем
увеличении плотности вплоть до пре-
Рис. 188. Влияние давлении на ско-
рость горевия нороховой стружни
1 — смесевой порох; г — порох Н
дельного ее значения скорость горения
слабо возрастает. Максимум скорости
достигается для разных порохов при
различных значениях относительной
плотности, не показывающих прямой
зависимости от величины нормальной скорости. Сильно различаются Так-
же значения отношения «макс/ия-
Горение в быстром режиме идет неравномерно, скорость его то резко
возрастает, то падает; значительная часть горящих частиц уносится газа-
ми от поверхности заряда и догорает вдали от нее. Непосредственным на-
блюдением можно обнаружить, что газообразные продукты превращения
со взвешенными в них частицами дыма распространяются, опережая пла-
мя на сантиметр и более. Реакционная зона становится широкой и в про-
тивоположность нормальному горению, по-видимому, тем шире, чем боль-
ше скорость горения. Одной из особенностей быстрого режима горения яв-
ляется сильная зависимость его скорости от диаметра; так, при увеличе-
нии последнего в 5 раз скорость возрастает более чем в 5 раз. Она сильно
возрастает также с давлением (рис. 188).
Режим быстрого квазистационарного горения сохраняется при условии,
что длина трубки не слишком велика. В противном случае после значи-
тельного участка горения с небольшой и почти постоянной скоростью она
внезапно возрастает в сотни раз и достигает значений порядка сотен мет-
ров в секунду. Большая скорость достигалась не скачкообразно, а в ре-
зультате прогрессирующего ускорения горения на протяжении нескольких
десятков сантиметров и в течение приблизительно 10 миллисекунд
(рис. 189).
При переходе горения на ускоренный режим обращает на себя вни-
мание резкость этого перехода, хотя если считать, что он обусловлен только
динамическим повышением давления, следовало бы скорее ожидать
постепенного увеличения скорости горения соответственно постепенному
увеличению динамического давления. Тэйлор (и независимо от него Бе-
ляев) высказал предположение, что проникновению горения в глубь
порошка препятствует слой расплава, образующийся па горящей поверх-
ности. При повышении давления, а следовательно, и скорости горения тол-
200
щипа расплавленного слоя уменьшается и при переходном давления он
становится слишком тонким, чтобы предотвратить проникновение газов
в порошок. Реальность расплавленного слоя Тэйлор, помимо логических
заключений, подтверждает фотографиями пламени (рис. 190), на которых
видна на границе ВВ и пламени узкая полоска большей ширины, толщи-
на которой тем меньше, чем больше давление и скорость горения. Мень-
шую ширину изображения пламени он объясняет отклонением лучей из-за
различной плотности продуктов горения и стекла; малая ширина изобра-
жения столбика порошка объясняется тем, что из-за плохого оптического
контакта частиц со стеклом на периферии заряда происходит отклонение
лучей, не попадающих поэтому в объектив.
Рис, 189. Горение пороховой стружки в заряде мзлой
плотности, помещенном в длинную стеклянную трубку
Съемка камерой СКС-2М с частотой 1000 кадров/сек (Л — рас-
стояние от верхнего конца заряда, т — время; а — весь за-
ряд; б — часть заряда, и которой произошел взрыв)
Зависимость критического давления от размера пор, большую легкость
перехода горения на ускоренный режим для высокоплавкого (278°С)
и быстрее горящего октогена по сравнению с плавящимся значительно
ниже (140° С) тэном 1 Тэйлор рассматривает как подтверждение отмечен-
ной выше роли расплавленного слоя.
Известную трудность для трактовки Тэйлора представляет то обстоя-
тельство, что для нитроклетчатки наблюдаются, как мы видели выше, за-
кономерности, аналогичные плавящимся ВВ. Он обходит эту трудность,
считая, что при горении нитроклетчатки тоже образуется расплавленный
слой, препятствующий при большой его толщине проникновению горения
в глубь порошка.
Хотя наличие расплавленного слоя при горении низкомолекулярных
ВВ бесспорно и неоднократно отмечалось и использовалось раньше для
объяснения некоторых явлений, имеющих место, при горении плавящихся
ВВ [197], в частности как препятствие для проникновения газов горения
в глубь пористого порошка, наблюдения, сделанные при изучении перехо-
да горения порошков на ускоренный режим, могут быть объяснены ина-
че, чем это делает Тэйлор.
Фактором, непосредственно вызывающим проникновение газов в глубь
порошка, является динамическое повышение давления, возникающее у го-
рящей Поверхности вследствие оттока газов. Это повышение давления, как
мы увидим ниже, пропорционально (при малых его значениях) скорости
1 Тэйлор не учитывает, что н температура кипения тэна (270® С) много ниже,
чем неизвестная (но во всяком случае большая, чем у гексогена (34(ГС)] температура
кияевия оятогена. Поэтому вопрос о том, для какого ВВ слой расплава будет тоньше,
нельзя оценить только по температуре плавления и скорости горения.
201
горения, т. е. быстро возрастает с увеличением давления над горящим за-
рядом. Так как, кроме того, при повышенных давлениях больше будет
плотность газов, то их масса и внутренняя энергия, проникающие в глубь
порошка, будут расти еще быстрее. Поэтому, повышая давление (при го-
рении под постоянным давлением), можно получить эффект опережения
Рис. 190. Фронт пламени тэна со светящейся линией, соответст-
вующей расплавленному слою
Л — светящаяся линия» видимая по всему диаметру щелк» В — предел
видимости порошка
газами фронта горения не только при малых, но и при повышенных плот-
ностях порошкообразного ВВ. При этом, чем выше плотность, тем больше
должно быть при прочих равных условиях (в частности равных размерах
частиц) давление, начиная с которого проявляется указанный эффект.
Точно так же при равной плотности возможность проникновения газов
должна зависеть от размера частиц, определяющего размеры пор, ниаче
говоря, сопротивления порошка проникновению газов. Следует ожидать
также, что при прочих равных условиях ВВ с большей скоростью горения
будет при большей плотности или при меньшем давлении давать наруше-
ние нормального хода горения.
Виляние скорости горения ясно видно из результатов опытов, приве-
денных А. Ф. Беляевым, по горению гремучей ртути при атмосферном
давлении и различных относительных плотностях (рис. 191). Заметный
рост скорости с уменьшением плотности наблюдается уже при плотности
около 0,68, при плотности 0,64 он резко возрастает и иногда наступает
даже детонация.
Таким образом, влияние пронннновения газов в глубь порошка при
горении быстро горящей гремучей ртути сказывается при атмосферном
давлении уже при уменьшении плотности до 0,7 от удельного веса *, в то
время как для относительно медленно горящей нитроклетчатки это наблю-
1 Правильнее было бы вести сравнение не по объему пор, а по их поперечному
сечению. Однако величина этого сечения зависит от величины частиц, а также их
формы, в отношении которых данных нет. Кроме того, уменьшение размеров частиц
увеличивает их поверхность; в итоге, хотя проникновение газов затрудняется, интен-
сивность возникшего горения возрастает. Суммарное влияние всех этих факторов
в общем случае оценить затруднительно.
202
дается лишь при плотности около 0,12 от удельного веса. Для нитроклет-
чатки же с относительной плотностью 0,7 критическое давление составляет
40—50 ат.
Если бы проникновение горения в глубь заряда определялось только
величиной динамического повышения давления, то следовало бы ожидать
А-Рдин .
постоянства отношения-----(где г — удельное сопротивление порошка
проникновению газов). Более того,
при данном размере частиц, посколь-
ку увеличение сопротивления путем
увеличения плотности заряда сопро-
вождается увеличением количества
вещества в единице объема, следо-
вало ожидать, что указанное отноше-
ние будет возрастать при повышен-
ных давлениях срыва нормального
Рис. 191. Зависимость скорости горе-
ния гремучей ртути от кубической
плотности
горения, которые характерны для
сильно уплотненных порошков.
В действительности же, чем выше
давление срыва, тем меньше соответ-
ствующее значение Особенно резко это уменьшение проявляется
в области умеренно повышенных давлений — до 100 аг.
Уже отсюда следует, что влияние давления, под которым идет го-
рение, на возможность его пронннновення в глубь порошкообразного
заряда не исчерпывается изменением динамического повышения дав-
ления.
Известно, что горение ВВ идет, кап правило, ступенчато, причем при
низких давлениях оно может не доходить до конца и останавливаться на
некоторой промежуточной стадии. В этом случае выделение тепла непол-
ное и температура газообразных продуктов горения относительно низка.
В то же время ясно, что поджигающая способность газообравных продук-
тов горения, естественно, зависит от их температуры. Прохождение сту-
пенчатого превращения ВВ при горения в сильной мере зависит от дав-
ления; повышенно давления способствует его прохождению. Для разных
ВВ предел давления, пачнная с которого превращение в определенных
условиях горения доходнт до конца, различен. Поэтому реальная темпе-
ратура, достигаемая при горении при определенном давлении, зависит
от величины последнего.
Эта конечная температура достигается не непосредственно у поверх-
ности ВВ, а на некотором от нее расстоянии, которое тем меньше, чем
выше давление. Поэтому даже при давлениях, лежащих выше того, при
котором достигается полнота превращения, профиль температуры вблизи
поверхности ВВ различен: при низких давлениях средняя температу-
ра слоя определенной толщины меньше, чем при высоких. Поскольку,
как уже отмечалось, при повышении давления увеличивается и плот-
ность газов, то увеличение внутренней энергии прилегающего к горя-
щей поверхности слоя газов становится на единицу их объема еще
больше.
По этим трем причинам поджигающее действие газообразных продук-
тов горения при повышении давления может сильно возрастать.
Очевидно, что для тех ВВ, температура горения которых выше и при
данном давлении зона с максимальной температурой находится ближе к
горящей поверхности, проникновение горения в глубь заряда, переход его
с нормального на ускоренный режим будут происходить легче, при мень-
шей газопроницаемости заряда. В свою очередь, при данной проницаемо-
сти порошка давление, начикая с которого будет осуществляться режим
проникновения, для таких ВВ будет меньше.
203
1
Однако проникновение газообразных продуктов горения в поры по-
рошкообразного заряда не тождественно еще проникновению в них самого
горения. Недостаточно поджечь ВВ по внутренней поверхности лор, необ-
ходимо, чтобы горение могло в них развиваться. Это зависит, помимо при-
роды ВВ, от диаметра пор, от их длины * 1 и давления. Известно, что при
поджигании с торца заряда неплавятцегося порошкообразного ВВ горение
идет лишь в том случае, если плотность и соответственно теплопровод-
ность порошка, определяющая теплоотвод от поверхности, не слишком ве-
лики.
Аналогично в канале (поре) малого диаметра теплоотвод в радиаль-
ном направлении может быть столь велик, что не будет полностью ком-
пенсироваться теплоприходом.
Однако, помимо этого физико-геометрического аспекта, есть и другая
существенная сторона явления. Известно, что первичные реакции превра-
щения ВВ, как правило, выделяют сравнительно мало тепла, а иногда
являются даже эндотермичяыми. Вторичные реакции происходят, когда
ВВ горит по свободной поверхности, на некотором расстоянии от нее. Если
близко к этой поверхности находится охлаждающее тело, например по-
верхность плиты из хорошо проводящего тепло металла, то вторичные
реакции не осуществляются. Сходное явление имеет место и при близком
расположении пороховых поверхностей, например в канале малого диамет-
ра пороховой трубки. При увеличении диаметра канала горение становит-
ся возможным.
Увеличение длины канала затрудняет горение по той причине, что ско-
рость течения газов вдоль горящей поверхности возрастает. Это, с одной
стороны, может приводить к увеличению скорости горения в канале и к
повышению давления в нем, ведущего к выгоранию прогретого слоя; по-
следующее падение давления может приводить к затуханию. С другой
стороны, большая скорость движения газообразных продуктов горения
в узком канале может приводить к перемешиванию холодных внутренних
слоев газообразных продуктов с горячими наружными слоями и тормо-*
зить, таким образом, завершение реакций в последних2.
По этим причинам горение в узких порах при низких давлениях не-
устойчиво. Если горение проникло в пору малого диаметра, то оно будет
из нее выброшено. Этот выброс может быть столь энергичен, что горение
затухает. Повышение давления ускоряет газофазные реакции при горе-
нии и когда толщина зоны превращения уменьшается до размеров порядка
радиуса поры, горение в порах становится возможным. Тогда для проник-
новения горения в поры становится достаточно даже того небольшого ди-
намического повышения давления, которое возникает при нормальном
горения.
Проникновение горения в поры соответственно увеличению фактиче-
ской поверхности горения сильно увеличивает динамическое повышение
давления, т. е. движущую силу проникновения газов. Тогда это проникно-
вение должно неограниченно развиваться, если только нет противодейст-
вующих этому развитию факторов.
Таким образом, динамическое повышение давления нормального го-
рения при указанных выше условиях выполняет роль только своеобраз-
ного спускового механизма, вызывающего проникновение горения в поры,
1 Замкнутые поры, не соединенные друг с другом, например в виде пузырьков,
могут привести лишь к некоторому увеличению средней скорости горения без нару-
шения его равномерности. Напротив, глубоко проникающие поры, как оия существу-
ют в обычном спрессованном порошке, при соответствующих условиях могут приво-
дить к нарушению равномерности горения.
1 Это явление сходно с тем, которое наблюдается при наступлении турбулиза-
ции горения нитроглицерина при низких давлениях, приводящей к затуханию го-
реяия.
204
после этого проникшее горение становится самостоятельно развивающим-
ся процессом
Одним из факторов, препятствующих ускорению конвективного горе-
ния, может быть сгорание частиц на поверхности заряда, перемещающее
эту поверхность в направлении горения в глубь порошка. Им может
быть унос частиц, увлекаемых быстротекущими газами, благодаря чему
эффективная толщина слоя горящей взвеси остается постоянной.
Однако само по себе постоянство толщины слоя горящей взвеси ча-
стиц ВВ не является еще достаточным условием стационарности процес-
са. Существенно также, чтобы эта толщина (точнее величина газообра-
зования) не превосходила определенного предела, который при данном
давлении соответствует количеству оттекающих газов. Если газообразо-
вание больше газоотвода и растет с давлением не пропорционально ему,
а быстрее, то равновесного давления при горении не существует — оно
растет и соответственно ускоряется конвективное горение.
Там, где отсутствуют предпосылки для ускорения горения, конвектив-
ное горение распространяется квазистационарно со скоростями в десят-
ки и сотии сантиметров в секунду, как это и наблюдалось в опытах Чуйко,
Тэйлора и Кондрикова.
Если ограничения для ускорения горения отсутствуют, то возникают *
процессы, распространяющиеся со скоростями в несколько сот метров в се-
кунду, легко приводящие к возникновению ударных волн и детонации.
Роль уноса частиц при развитии конвективного гореник Б. Н. Кондриков
иллюстрировал следующим опытом. При определенных условиях заряд,
состоящий из крупнозернистого пороха, горит быстро, но устойчиво с по-
стоянной скоростью; если порох слегка смочить ацетоном так, чтобы
зерна его, не меняя своего размера, слиплись, и затем удалить ацетон, то
горение такого заряда быстро ускоряется, так как унос частиц становится
невозможным и толщина горящего слоя непрерывно растет.
Конвективное горение отличается некоторыми особенностями. Чувстви-
тельность его скорости к внешнему давлению гораздо больше, чем нор-
мального горения. Скорость последнего определяется суммарным давле-
нием (внешнее давление + относительно небольшое динамическое повы-
шение давления). Скорость же конвективного горения определяется
прежде всего скоростью проникновения газов, зависящей от динамиче-
ского повышения давления. Относительно малое изменение внешнего
давления будет по отношению к динамическому повышению давле-
ния большим и может сильно повлять на скорость конвективного го-
рения.
Это влияние настолько сильно, что горение переходит с устойчивого
конвективного режима на ускоренный даже если взять большую длину
заряда в трубке. По мере углубления горения от верхнего среза трубки
сопротивление освободившегося ее участка истечению газов становится
все больше и больше и, наконец, вызванное этим повышение давления
оказывается достаточным, чтобы обеспечить утолщение горящего слоя и
соответственно ускорение горения. Естественным является также непо-
стоянство скорости в переходной области. Когда внешнее давление увели-
чено до такого значения, при котором уже может происходить для данного
ВВ проникновение газов, то оно происходит сначала лишь в немногие
наиболее крупные поры, число которых мало и непостоянно. Лишь при
дальнейшем повышении давления начинают «работать» все или большая
часть пор и влияние крупных пор сходит на нет.
1 В опытах по горению под возрастающим давлением необходимое дня перехода
на быстрый режим давление во много раз больше, чем динамическое повышение дав-
ления. Это также указывает на то, что для повышенного давления нормальное горе-
ние служит только начальным толчком в возникновении режима быстрого горения.
205
На скорость конвективного горения сильно влияет, как показал
Б. Н. Кондрннов, диаметр заряда, причем это влияние не определяется
его обычным влиянием на соотношение теплопотерь и теилоприхода.
Одно из возможных объяснений этого влияния заключается в том, что
при большом диаметре на общую поверхность заряда приходится больше
крупных пор, по которым легче распространяется горение-
Следует указать, что если горение проникло в пору, То дальнейший
процесс является саморазвивающимся, так как разгорание поры делает
горение в ней, как правило, более устойчивым1.
Определенную роль играет глубина поры. Если она слишком велика,
то скорость горения стенок поры может усилиться из-за большой скорой
сти течения газов вдоль нее. Это же в свою очередь приводит к пульси-
рующему горению, а в пределе и к затуханию. С другой стороны, рас-
пространение горения в поре лишь при небольшой ее глубине определяет-
ся внешним давлением. Если глубина велнна и сопротивление выходу
газов стало значительным, то ведущая роль может перейти от внешнего
давления к внутреннему и горение начинает ускоряться независимо от
внешнего давления.
Этим объясняется тот парадоксальный факт, что при данной величине
порошкообразного заряда в определенных условиях горения увеличение
высоты заряда (при соответственном уменьшении диаметра) приводит
к увеличению интенсивности горения, хотя условия для проникновения
газов в порошок (при данном внешнем давлении) затрудняются.
Горение при возрастающем давлении
В предыдущем разделе мы рассмотрели переход нормального горения
на ускоренный режим в результате прониниовеяия в пористый заряд га-
зообразных продуктов горения, начинающегося под влиянием того ло-
кального повышения давления, которое возникает вблизи горящей по-
верхности заряда.
Гораздо более благоприятные условии для проникновения горения в
глубь пористого заряда осуществляются, если оно идет под возрастающим
давлением, как это, в частности, имеет место в огнестрельном оружии
ствольного типа. В этом случае проникновение газов в поры происходит не
только из-за местного повышения давлении у фронта горения, но и вслед-
ствие возрастания давления во всем объеме газов. Поэтому даже неболь-
шая проникающая пористость ведет к нарушению нормального горения.
Именно это обстоятельство явилось в свое время одним из основных мо-
тивов отказа от черного пороха и перехода на пороха коллоидального
типа. Даже при значительных технологически реальных плотностях Вьель
наблюдал при высоких возрастающих давлениях нарушение нормального
горения черного пороха, что не давало возможности регулировать в долж-
ной мере длительность горении. Он показал также, что такая регулировка
возможна, если прессовать черный порох под очень большими давления-
ми, дающими плотность, близкую к удельному весу.
Бездымные пороха благодаря сплошности и прочности своих частиц
обеспечивают нормальный режим горения во всем интересующем технику
диапазоне давлений.
Исследования по горению ВВ под возрастающим давлением производи-
лись в основном по двум методам: в так называемых трубках Андреева п
в манометрической бомбе Вьеля.
Первый из методов заключается в поджигании с торца значительного
(50 и более граммов) заряда ВВ в стальном стакане; стакан имеет крыш-
1 Это обстоятельство, возможно, играет роль при развитии прогаров в пороховых
шашках.
206
i Рис. 192. Три типа разрыва трубки
а — при горении; 6 — при взрыве; в — при детонации
ку с отверстием большого сечения, закрытым металлическим диском, вы-
рывающимся по достижении определенного давления. При малой прочно-
сти диска горение приводит к его вырыванию без нарушения целостности
стакана. При большой прочности, несмотря на вырывание диска, стакан
разрывается на большее или меньшее число кусков (рис. 192) в пределе
соответствующее тому, которое дает детонация, вызванная капсюлем-де-
тонатором. Минимальная прочность диска, начиная с которой происходит
разрушение стакана, служит мерой склонности горения ВВ к переходу на
взрывной режим.
Обычно применялся стакан (рис. 193) диаметром 40 леи, с толщиною
стенки 4—6 мм и высотой 200 леи. Диаметр отверстия крышки — 30 лек;
оно закрывается диском из свинца (или иного металла), прочность
207
3 4 г 8 1 < 9 6 5
Рис. 193. Трубка для опытов по горению
под возрастающим давлением
1 — железный корпус; г —железная крыш-
ка; 3 — свинцовая пластинка; 4 — асбестовая
пластинка; 5-—железное дно; 6 — гипс;
з — ВВ; & — зажигательная шашка;
S — провода для воспламенения
которого определяется опытами по горению в стакане крупнозернистого
бездымного пороха. Такая тарировка, естественно, определяет прочность
диска применительно к относительно медленно нарастающему давлению;
при быстром росте давления приобретает значение также инерционное со-
противление диска. Границей прочности является сопротивление разрыву
самого стакана, которое при указанных размерах составляло около 1000 ат.
Воспламенение производится обычно
при помощи двухграммовой шашки
пиротехнического состава (KNOa —
68%, Mg —20%, идитол—12%),
воспламеняемой при помощи стопина
и накаливаемой током проволочкп.
Для увеличения плотности заряжа-
ния стакан на некоторую высоту мо-
жет быть залит гипсом.
В одном из вариантов метода при-
меняют крышки для стакана с ма-
леньким открытым очком определен-
ного сечения. Диаметр очка, разгра-
ничивающий отсутствие и наличие
разрыва стакана, принимается за меру склонности горения испытуемого
ВВ к переходу во взрыв. Этот вариант менее надежен, чем описанный
выше, так как возможность истечения газов через очко затрудняет вос-
пламенение ВВ и может даже привести к затуханию начавшегося горения
вследствие быстрого ;спада давления. Поэтому отрицательный результат
опыта может быть следствием не устойчивости горения ВВ, а плохой его
воспламеняемости; металлический же диск обеспечивает падежное воспла-
менение. Применение в качестве воспламенителя пиротехнического соста-
ва, развивающего высокую температуру при относительно низком давле-
нии, не способном вырвать диск обычной прочности, также обеспечивало
надежность воспламенения.
Опыты показали, что для каждого ВВ есть некоторый предел прочно-
сти диска, выше которого поджигание дает переход горения во взрыв, со-
провождающийся разрывом стакана. Одиако величина этого предела зави-
сит не только от природы ВВ. Одно и то же ВВ ведет себя совершенно
различно в зависимости от относительной плотности, т. е. от степени по-
ристости.
Так, в одной из работ горение порошкообразного гексогена (относи-
тельная плотность от 0,4 до 0,6) переходило в детонацию (заряд 50 г)
уже при давлении 45 ат; гексоген же, спрессованный до плотности, близ-
кой к 0,9, горел, не детонируя, даже при давлении 700 аг; одиако уже
при давлениях 200 ат и выше наблюдалось некоторое дробление стакана,
свидетельствующее об ускоренном по сравнению с нормальным горении.
Горение порошкообразного тэна малой плотности (около 0,6) также
переходило в детонацию уже при применении диска, вырывающегося при
45 ат; при этом горение прессованного тэна (р ~ 0,96) приводит только
к срыванию диска без нарушения целостности стакана, и лишь при давле-
нии 500—700 аг в двух из трех опытов наблюдалась детонация. При мень-
шей плотности (0,92) разрушение стакана наблюдалось уже при диске,
выдерживающем давление 210 аг.
На характер горения влияет также величина заряда. Так, при диске,
вырывающемся при 40 аг, заряд тэна весом 10 з (размеры частиц 0,25—
0,5 мм) не вызывал разрыва стакана ', такой разрыв наблюдался при уве-
1 Под зарядом порошкообразного тзна помещался литой тротил. Отсутствие его
взрыва служило дополнительным свидетельством того, что горение тэна не перешло
в детонацию.
208
личении веса заряда до 13—20 г. Влияние величины заряда установлено
и для гексогена; однако при прочих равных условиях переход горения во
взрыв происходил, начиная с величины заряда 22 а, почти вдвое большей,
чем для тэна.
Минимальная величина заряда, дающая переход горения во взрыв,
уменьшается при увеличении прочности диска. При диске, выдерживаю-
щем давление 260 и 400 ат, десятиграммовый заряд тзна давал переход
горения во взрыв, которого не наблюдалось при диске прочностью 40 ат.
Такое же влниние прочности диска на минимальную величину заряда на-
блюдалось и для флегматизированного гексогена, но, естественно, в об-
ласти больших величин заряда.
Играет роль также высота заряда, увеличение которой в определенных
пределах (при постоянной его величине) благоприятствует переходу го-
рения во взрыв. Так увеличение высоты десятиграммового заряда тзна
с 9 при которой взрыва не было, до 14 жж приводило к переходу
горения во взрыв.
Значительное влияние на устойчивость горения оказывают также
размеры частиц ВВ. Если размер частиц тэна составлял 0,01 мм, то пере-
ход горения во взрыв наблюдался, начиная с величины заряда, равно-
го 10 г, при 0,25—0,5 мм — начиная с 12 г и при 1—2 мм — начиная с 40 г.
Заряды весом 50 г при прочности диска 20 ат детонировали, если размер
частиц составлял 0,01 и 0,25—0,5 жж; если размеры частиц были равны
2—3 жж, наблюдалось только горение.
При горении мелкого гексогена переход горения в детонацию наблю-
дался, если размер частиц составлял 0,1—0,25 мм, вес заряда 50 г, проч-
ность диска 22 ат и плотность 0,5—0,6; при 17 ат происходило горение
без разрыва стакана. Для зарядов гексогена, состоявших из кристаллов
размером 0,5 мм и 1—2 мм, при прочности диска 38 ат наблюдался пере-
ход горения во взрыв, но дробление стакана было гораздо меньше. При
увеличении размера кристаллов до 2—3 жж и более прочность диска,необ-
ходимая для взрыва, резко увеличивается и перехода горения во взрыв не
наблюдается даже при прочности диска, превышающей прочность стака-
на (1000 ат).
Естественно ожидать, что и чрезмерное уменьшение размеров частиц
затруднит переход горения во взрыв из-за уменьшения газопроницаемо-
сти. Для тэна и гексогена при применившихся размерах частиц и относи-
тельно невысокой плотности это влияние не обнаруживалось. Однако оно
отчетливо наблюдалось для смесей перхлората аммония с алюминиевой
пудрой; при размере частиц перхлората 10 жк (размер частиц алюминия
1 мк) и относительной плотности 0,56 перехода горения во взрыв не
наблюдалось во всем интервале прочности диска (22—1500 ат); если раз-
мер частиц был равен 40 и 120—150 мк, взрыв происходил уже при проч-
ности диска 20—25 ат-, при применении крупных частиц (50—1000 и
1000—2000 ж«) взрыва при этой прочности диска не наблюдалось.
Флегматизация тэна и гексогена смесью стеарина и церезина значи-
тельно повышает давление, при котором горение переходит во взрыв. Если
добавить инертную примесь в порошкообразном виде (крахмал), то флер
матизирующее ее действие в отношении перехода горения во взрыв про-
является значительно слабее.
Относительно легко (начиная с прочности диска 100 ат) взрываются
при поджигании также смеси (82 : 18) аммиачной селитры (преобладаю-
щий размер частиц 0,1—0,25 жж) с алюминиевой пудрой (1 жк); взрыв
получается даже несколько легче, чем для тротиловой смеси (82 : 18).
Еще активнее ведут себя смеси перхлората аммония с алюминиевой
пудрой. Сам перхлорат аммония достаточно инертен и не давал взрыва во
всем диапазоне прочности диска (50—1500 аг). При содержании в нем
5 % пудры дробление стакана наблюдается уже при прочности диска 22 аг;
14 К. К. Андреев
209
разрыв стакана, хотя и на меньшее число кусков, наблюдался также при
добавлении к перхлорату 9 и 18% алюминия; при 28% алюминия (сте-
хиометрическая смесь) взрыва не наблюдалось даже при 1000 ат. Смесь
перхлората аммония с тротилом (84; 16) ведет себя значительно пассив-
нее, чем алюминиевая; она взрывается при 400 ат и взрыва не происхо-
дит при 260 ат.
Другие изучавшиеся порошкообразные ВВ дают сходную картину
разрушительного действия в зависимости от относительной плотности.
Однако стакан дробится слабее чем в опытах с тэном или гексогеном.
Аммониты на основе тетрила (79 : 21; 88:12) при насыпной плотности
только при давлении менее 200 ат не разрывают стакан; при давлении
500— 700 ат наблюдается значительное его дробление, близкое по интен-
сивности к детонационному; при давлении 1200 ат все аммониты детони-
руют.
Порошкообразный тетрил (р ~ 0,6) только при прочности диска 45 ат
не производит дробления стакана; при давлении 65 ат стакан разрывает-
ся на небольшое число (3 и 4) кусков; увеличение прочности диска
усиливает дробление, но даже при давлении 500—700 ат детонации не
происходит как при заряде, равном 50 г, так и нри заряде 150 г.
Пикриновая кислота (р ~ 0,6) при давлении 500—700 ат вызывает
еще более слабое дробление, интенсивность ноторого несколько возраста-
ет при увеличении веса заряда. В оболочке, разрывающейся при 1200 ат
и заряде весом 425 а, той же плотности пикриновая кислота дала детона-
цию. Ксилил и тринитробензойная инслота показывают сходство в по-
ведении, уступая по интенсивности дроблении стакана пикриновой ки-
слоте.
Горение тротила1 вызывает наиболее слабое разрушение Оболочки.
Третил при прочном диске, как правило, горит и разрывает стакан не
дробя его. Даже в оболочках, разрывающихся при давлениях 1200 и
1600 ат, целиком заполненных кристаллическим порошком (р ~ 0,55),
горение не переходи? в детонацию и наблюдается лишь умеренное дроб-
ление оболочки. Влизок к тротилу по действию гранулированный трини-
тронафталин.
Динитросоединения, естественно, более пассивны в описанных усло-
виях опыта, чем даже тротил — горение их часто затухает, не приводя к
разрыву стакана.
В прессованном или литом виде тетрил, пикриновая кислота, аммо-
ниты даже при давлениях 500—700 ат только горят. Опыты с ВВ, поме-
щенными в более прочные оболочки (давление разрыва 1200 и 1600 ат),
показали, что и в этих условиях при заряде тротила 570 а дробление обо-
лочки гораздо более слабое, чем при детонация; литая пикриновая кисло-
та в оболочке, разрушающейся при давлении 1200 ат, производит слабое ее
дробление г.
Изучение горении порошкообразных ВВ в стальных стаканах позво-
лило сопоставить различные ВВ по склонности горения к переходу во
взрыв и установить влияние ряда факторов (плотность, размеры частиц,
примеси) на этот переход. Основным достоинством метода использования
1 По-видимому, в случае горения тротила существенное влияние оказывает его
способность плавиться без разложения, которая в условиях данного опыта способ-
ствует уплотнению заряда, исключающему проникновение горения в глубь его.
1 Были проведены также опыты с зарядами тана, гексогена, тетрила, тротила и
амматола (80:20) весом SO г при повышенных температурах, приводивших к само-
воспламенению (давление разрыва оболочки 500—700 ат). Очевидна, что в момент
воспламенения указанные ВВ находились в жидком состоянии. Тэн и гексоген дали
детонацию; степень дробления оболочки при горении тетрила была близка к детона-
ционной, амматол вызвал только разрыв стакана. Танин образом, несомненно, что по-
вышение температуры, в данном случае с расплавлением ВВ, увеличивает интенсив-
ность горении и склонность его к переходу во взрыв.
210
стальных стаканов является простота и возможность быстро получить ха-
рактеристику склонности того или иного ВВ к переходу горении во взрыв.
По существу возможность получения такой характзристики по столь гру-
бому методу была несколько неожиданной. По-видимому, различия между
изучавшимися ВВ и влияние их физической структуры столь велики, что
обнаруживаются даже при этом методе.
Недостатком метода является необходимость использования бронека-
меры, рассчитанной на довольно сильный взрыв и обеспечивающей улав-
ливание металлических осколков* и использование для опытов значи-
тельного количества ВВ, что также иногда является затруднительным.
Кроме того, метод дает только конечный результат опыта и притом
лишь в полуколкчественной форме (число и размеры осколков) и не
характеризует развития процесса во времени.
Указанные недостатки отсутствуют в другом методе изучения пере-
хода горения во взрыв, который состоит в поджигании небольшого (1—
10 г) заряда ВВ в манометрической бомбе я регистрации изменения давле-
нии во времени. Этот метод по существу представляет собой вариант ме-
тода Вьеля, развитый и модернизированный применительно к заряду,
состоящему, как правило, из одного крупного «зерна». В несколько ином
варианте этот метод был использован в работе Горбунова [198]; он заклю-
чался в определении относительной плотности порошкообразного ВВ, ко-
торая отделяет область его нормального горения при возрастающем дав-
лении от области ускоренного горения, обусловленного проникновением
горения в глубь заряда. В манометрической бомбе с датчиком давлении,
позволяющим регистрировать не только повышение, но и понижение дав-
ления1, поджигается наряд исследуемого порошкообразного ВВ с опреде-
ленным размером частиц в виде равномерно уплотненной шашки опре-
деленного диаметра, бронированной со всех сторон, кроме одного торца.
При постоянном весе заряда, т. е. при постоянной плотности заряжания,
определялась та относительная плотность рк ВВ, ниже которой горение
переходило с нормального на ускоренный режим. Этот переход проявлял-
ся в том, что кривая р(т), в начале горения совпадающая с кривой для
предельно уплотненного заряда, откланялась от иее вверх, т. е. в сторону
больших Др/Дт (рнс. 194), Это означало, что горение переходило с нор-
мального режима на режим, связанный с его проникновением в глубь за-
ряда. Соответственно увеличению поверхности горения быстрее возраста-
ла и скорость нарастания давления во времени. Чем больше критическая
плотность, тем больше устойчивость горения данного ВВ.
Собственно говоря, правильнее характеризовать устойчивость горения
не критической плотностью, а критической газопроницаемостью, которая
в большей мере, чем плотность, определяет возможность проникновения
продуктов горения в глубь заряда. Это тем более целесообразно, что,
как показывает опыт, для разных ВВ при одинаковой относительной плот-
ности (и, естественно, одинаковых начальных размерах кристаллов) газо-
проницаемость может существенно различаться.
Сопоставляя при указанных одинаковых условиях разные ВВ, можно
установить сравнительную склонность гореини каждого из них к пере-
ходу на ускоренный режим.
Можно решать этим методом н другую задачу, работая с одним ВВ,
определить влияние на устойчивость гореиня различных характеристик
заряда, напримзр, размера частиц, соотношения размеров заряда, харак-
теристик воспламенения и др. В работе Горбунова [198] метод использо-
вался для получения указанных выше характеристик.
1 В указанной работе был применен тензометрический датчик; возможно н даже
более целесообразно применение пьезозлектрического датчика, позволяющего запи-
сывать более быстрые изменении давления.
14*
2»
Не нужно, применяя данный метод, забывать об известной условности
постоянства плотности заряжания при сравнении разных ВВ. Непосредст-
венной причиной проникновения газообразных продуктов горения в глубь
заряда является достижение при горении определенного давления. Вели-
чина же давления при данной плотности заряжания может скльно разли-
чаться для разных ВВ. Кроме того, в данных условиях опыта величина
давления очень сильно зависит от длительности горения вследствие отно-
сительно очень большой теплоотдачи газов стенкам бомбы. Наконец, кри-
вые р(т) в опытах Горбунова не показывали однозначной зависимости
ускорения горения от до-
стигнутого давления, поче-
му в качестве характери-
стики явления и использо-
Рис. 194. Вид кривой р(т) для
предельно уплотненного ВВ
(I) и для ВВ пониженной
плотности (2).
Носил.— кривая при горении од-
ного воспламенителя
Рис. 195. Влияние плотности заряда на устойчи-
вость нормального горения тротила
Относительная плотность заряда: 1 — 0.87; 2 — 0,73;
з — 0,06; 4 — кривая р(т) при горении одного вос-
пламенители (1,0 в)
Удельное сопротивление в условных единицах: 1 —
2,5; 2 — 5,8
вался указанный выше критерий — критическая плотность при постоян-
ной плотности заряжания.
Были изучены восемь ВВ. Заряд из частиц определенного интервала
размеров, выделенных отсеиванием на ситах, порционно уплотнялся в
плексигласовом стаканчике диаметром 10 мм и толщиной стенок 2 мм.
Вес заряда составлял обычно 1 или 2 г, при этом плотность заряжания
была равна 0,02 и 0,04 г/см3. Для воспламенения применялся черный по-
рох № 3.
Критерием режима нормального горения служила независимость кри-
вой р(Т) от длины заряда — начальный участок этой кривой для большо-
го заряда повторяет тот, который дает опыт с малым зарядом; в отличие
от этого при ускоренном горении кривая большого заряда сравнительно
рано начинает идти выше кривой нормального горения.
Общий характер кривых р(т) рассмотрим на нескольнях типичных
примерах. Для тротила (рис. 195) при плотном заряде (кривая 3) после
быстрого небольшого подъема давления, создаваемого воспламенителем,
следует довольно длительный участок слабого падения давления, обуслов-
ленный, очевидно, тем, что повышение давления за счет нормального го-
рения превосходит падение давления из-за охлаждения газов стенками
бомбы. В дальнейшем, по-видимому, из-за того, что поверхность сте-
нок несколько прогревается и теплоотвод в них замедляется, давление
начинает расти, ускоряясь, поскольку скорость горения растет с дав-
лением.
Если плотность заряда несколько меньше (кривая 2), то в начале
кривая идет так же, как и для предельно уплотненного заряда; в даль-
нейшем она идет несколько выше, вероятно, потому, что расплав вдавли-
212
вается возрастающим давлением в глубь порошка, и эффективный коэф-
фициент теплопередачи соответственно возрастает.
При значительном уменьшении плотности (кривая 1) относительно
рано наступает быстрый рост давления, который и служит показателем
перехода горения на ускоренный режим. Для некоторых ВВ (тетрил, тэн,
гексоген, гремучая ртуть) ускорение горения происходило настолько рез-
ко, что не могло количественно быть записано применявшимся прибором
обычно в этих случаях плексигласовый стаканчик оказывался раздроблен-
ным.
Однако гексоген отличается некоторыми особенностями (рис. 196);
для плотного заряда кривая р(т) идет вначале выше, чем для менее плот-
ного. В последнем случае, по-видимому, проникновение газов воспламени-
Рис. 196. Влияние плотности заряда на
устойчивость нормального горения гексо-
гена
Относительная плотность заряда: 1,1' — 0.98;
SX — 0,93. Удельное сопротивление в услов-
ных единицах: 2,2' — НО. Вес заряда: I'— 1,0 г;
1—2,0 г; 2.2’—1,50 г: 3— кривая р(т) при
горении одного воспламенителя (1,0 г). Пунк-
тиром обозначен переход горения во взрыв, со-
провождавшийся дроблением стаяанчика
Рис. 197. Влияние плотности заряда на
устойчивость нормального горения пи-
крата калия с размером частиц 50—
60 мк
Числа у кривых означают относительную
плотность ааряда и (в скобках) удельное
сопротивление Вес заряда 2,0 г; Воспл.—
кривая р(т) при горении одного воспламе-
нителя (1 г)
теля в глубь заряда, недостаточное, чтобы сразу его поджечь по внутрен-
ней поверхности, приводит к их сильному охлаждению, что и проявляется
в более низком уровне давления. По истечении некоторого времени насту-
пает резкий подъем давления. Возможно, что период очень медленного
роста давления, предшествующий этому подъему, представляет собой
торцевор горение слоя ВВ, оплавленного и подпресованного газообразными
продуктами горения воспламенителя; после выгорания этого слоя проис-
ходит проникновение горения в глубь остальной части заряда. В пользу
этого объяснения говорит характер кривых для неплавящегося пикрата
калия (рис, 197), где участок медленного роста давления практически
отсутствует.
Сопоставление изучавшихся ВВ в отношении склонности их горения к
переходу на ускоренный режим приводится в табл. 29, в которой ВВ рас-
положены в порядке возрастания указанной склонности. Меньше всего
она у тротила и ксилила , затем следует пикрат калия, пикриновая лис-
лота, тетрил, гексоген, тэн и гремучая ртуть.
1 Тензометрический датчик имел частоту собственных колебаний 18—20 кгц и
в сочетании с тензостанцией УТС-12к и вибратором осциллографа МП02 позволял
регистрировать с достаточной точностью сигналы с длительностью нарастания дав-
ления не менее 1 мсек.
2 Переходная область изучалась недостаточно подробно, чтобы установить, какое
из этих двух ВВ следует поставить на первое место; ксилил горит вдвое дольше, чем
тротил; правда, и величина максимального дапления при горении ксилила значитель-
но меньше (150—200 ат), чем тротила (190—300 аг).
213
Таблица 29
Исходные данные в результаты опытов по сравнению устойчивости горев ни ВВ
(Вее воспламенителя 1,0 а, диаметр 10 л*)
ВВ । Относительная плотность Размер частиц, мк 1 —-у- 1 Высота, мм, вес । заряда, а 1 Время горения, сек Максимальное дав* левде, ат Время до ускоре- ния» «к Удельное сопро- тивление, лл рт. от. 1см1 Результат опыта Степень дробления стаканчика (число осколков)
1 2 а 4 В в 7 8 9 10
Тротил 0,9В 50—60 15,2/2,00 4,40 192 — Нормальное горение Нет
0,73 20,8/2,06 4,00 190 — 5,8 То же »
0,67 23,0/2,00 1,46 300 0,60 2,5 Ускоренное горение *
Кснлил 0,96 50—60 15,4/2,00 8,10 162 , — Нормальное горение Нет
0,78 19,3/2,00 8,20 150 —— —— То же »
0,73 20,8/2,00 2,40 175 0,70 3,0 Ускоренное горение »
0,63 23,5/2,00 1,60 217 0,06 — То же
Пнират 0,95 50-60 14,1/2,00 0,47 224 Нормальное горение Нет
калия 0,89 15,1/2,00 0,45 224 —— 26 То же >
0,86 15,7/2,00 0,14 276 0,03 14 Ускоренное горение »
0,77 17,6/2,00 0,11 272 0,03 То же »
Пикрат 0,87 20 15,5/2.00 0,47 210 - —— Нормальное горение Нет
калия 0,84 16,1/2,00 0,46 214 — 39 То же *
0,81 16,7/2,00 0,16 270 0,03 17 Ускоренное горение
0,81 16,7/2,00 0,16 270 — —— То же »
Пнири- 0,97 20 14,1/2,00 3,96 148 — — Нормальное горение Нет
новав 0,94 15,0/2,00 4,00 160 —— —— То же >
кислота 0,87 16,2/2,00 4,00 160 —— — » > >
0,81 17,4/2,00 0,94 230 0,30 21 Ускоренное горение >
0,67 15,7/1,50 0,13 334 0,06 — То же »
Тетрил 0,96 50—100 15,1/2,00 1,84 230 Нормальное горение Нет
0,89 16,2/2,00 1,81 230 —— То же »
0,86 16,9/2,00 0^16 327 0,06 37 Ускоренное горение »
0,83 17,3/2,00 0,07 380 0,05 —• Взрыв До 20
Гексоген 0,98 50—60 13,6/2,00 0,44 295 Нормальное горение Нет
0,98 6,8/1,00 0,29 168 — — То же
0,93 10,8/1,50 0,39 270 0,37 110 Взрыв 10-15
0,96 10,8/1,50 0,36 306 0,33 но » 10—15
Тэн 0,90 10 15,1/2,00 2,25 205 0,20 Нормальное горение Нет
0,90 7.6/1,00 1,43 96 0,06 — То же »
0 82 12,6/1,50 0,74 308 — 400 Взрыв 10—12
0,75 13,9/1,50 0,08 410 — — » До 50
Грену- 0,99 50—100 13,1/0,96 0,11 87 Нормальное горение Нет
чаи 0,95 13,8/0,96 0,11 83 — То же »
ртуть* 0,89 14,0/0,95 0,03 115 0,03 1900 Взрыв 10-15
* Диаметр заряда гремучей ртути 4,8 мм, толщина стенок стакана 2,5 .ши, длина 20 лип.
Влияние размеров частиц (рис. 198) заключается в том, что при при-
менении очень мелких частиц проникновения горения в глубь заряда не
происходит, несмотря на относительно малую плотность (0,57); горение
прошло практически за то ясе время, что и для предельно уплотненного
порошка. При использовании крупных частиц быстро начинается горение
на ускоренном режиме; однако интенсивность его меньше, чем при при-
менения некоторого промежуточного размера частиц. Аналогичное влия-
ние на характер горения оказывает изменение плотности при постоянном
размере частиц; для мелких частиц (10 лек) даже при малой плотности
Рис. 198. Влияние размеров частиц на характер горения тэна
Интервал размеров частиц в жя: I — 400—500; 2 — 300—400; з —
100—160- 4 — 63—100; 5 — 10; в — кривая р(т) при горении одного
воспламенителя (0,5 а). Дли тана с размером частиц 63—100 лек
наблюдался взрыв с дроблением стаканчика, во всех остальных
опытах — гореиее без разрушедня стаканчика
(0,71) и при значительном весе заряда (1,5 г) проникновения горении в
глубь заряда не происходило; при меньших плотностях оно происходило
и доводило ускорение горения др взрыва; ускорение наступало тем рань-
ше, чем меньше плотность.
Своеобразное влияние на переход горения на ускоренный режим и во
взрыв оказывает высота заряда. Увеличение высоты заряда тонкозерни-
стого тэна малой плотности (0,25) ускоряло переход горения зо взрыв.
При больших плотностях (0,40 и 0,58) малые заряды не давали взрыва;
при большей высоте заряда взрыв наступал, но значительно позже, чем
при малой плотности. Аналогичные опыты в том же интервале веса заря-
дов, но при вдвое большем диаметре, т. е. при меньшей высоте заряда, по-
казали такое же влияние увеличения заряда, но все времена были гораз-
до меньше.
Однако во всех этих опытах по существу сочеталось влияние двух
факторов — высоты заряда и плотности заряжания. Чтобы разделить
влияние этих факторов, были поставлены опыты при постоянной плот-
ности заряжания (весе заряда). Из рис. 199 видно, что заряд малой высо-
ты горит, большой — взрывается, хотя этот взрыв наступает тем позже,
чем больше высота, очевидно, в связи с тем, что становится больше время
проникновения. Еще более наглядно значение высоты заряда иллюстри-
руется опытом, в котором заряд определенного веса был размещен в двух
стаканчиках того же диаметра; взрыва не произошло, хотя время до на-
ступления ускорения, естественно, несколько сократилось.
Если условия для проникновения горения в глубь заряда трудны
(мелкие частицы, значительная плотность), то на возможность проникно-
вения горения может оказывать влияние давление воспламенителя. При
малой навеске воспламенителя горение до конца идет без проникновения;
215
ряло (для тэна) развитие процесса:
Рис. 199.
ние тэна
пости 0,57 и постояплом весе заряда 1,0 з
Числа у кривых означают диаметр и (в скоб-
ках) высоту заряда в лик; Воспл,—кривая р(т)
при горении одного воспламенителя (1 г).
Пунктиром о В означен переход горения во взрыв
Влплпле высоты заряда на горе-
(2 -мк) при относительной плот-
при больших — ускорение наступает тем раньше, чем больше навеска вос-
пламенители. Играет роль, однако, не только максимальное давление
воспламенителя, но и быстрота нарастания давления; быстрогорящий вос-
пламенитель труднее вызывал взрыв тэна умеренной плотности, возможно,
это было связано с тем, что он наряду с воспламеняющим действием про-
изводил некоторое уплотнение заряда.
Ускорение горения и взрыв наступают раньше, если под зарядом в
стаканчиие поместить инертный пористый порошок (песок), что умень-
шает противодавление, возникающее в глубине заряда и тормозящее про-
никновение газов.
Предварительное введение инертного газа в бомбу (50—85 ат) уско-
увеличение скорости горения и по-
вышение эффективной его тем-
пературы, по-видимому, преоб-
ладает над разбавлением ВВ
инертным газом, заполняющим
поры.
Как и при горении под воз
р а стающим давлением больших
зарядов, на проникновение го-
рения в глубь заряда сильное
влияние могут оказывать неко-
торые примеси.
Одним из типов Этих приме-
сей являются катализаторы го-
рения. Так, при добавлении к
измельченному в порошок ни-
троглицериновому пороху тон-
кодисперсного бихромата аммо-
ния горение в манометрической
бомбе при прочих равных усло-
виях гораздо раньше перехо-
дило на ускоренный режим, чем в отсутствие катализатора (рис. 200)-.
То же наблюдалось, если горение начиналось под повышенным давлением
(100 ат) предварительно введенного в бомбу инертного газа. Опыты по
горению под постоянным давлением показали, что и в этих условиях в при-
сутствии добавки переход на режим проникновения наблюдается при го-
раздо меньшем давлении и соответственно меньшей скорости горения, чем
для порошка без добавок.
Примером второго типа добавок может служить алюминий в сочета-
нии с перхлоратом аммония. По опытам В. М. Рогожникова, добавка
тонкодисперсного алюминия к перхлорату аммония повышает его способ-
ность к горению. Так, если при 30 ат критический диаметр горения пер-
хлората (р = 1,17 г/см3) составляет ~ 10 льи, то в присутствии 5% алю-
миния он снижается до 5 льм. При увеличении содержания алюминия
наблюдается дальнейшее хотя и не столь сильное уменьшение критиче-
ского диаметра; при 25% алюминия он составляет ~3 мд. Интересно,
что при добавлении 5% алюминия скорость горения перхлората аммония
(при 50 ат) не только не увеличивается, но даже заметно уменьшается,
что, впрочем, наблюдается также и при введении некоторых органических
горючих добавок в перхлорат аммония.
В манометрической бомбе перхлорат аммония в стеклянном стакан-
чике (d = 17 льм) воспламеняется и горит с трудом; требуется значитель-
ный по весу воспламенитель; особенно трудно идет горение при повышен-
ной плотности порошка.
Добавление тонкодисперсного (1 jkk) алюминия сильно повышает вос-
пламеняемость, способность к горению и склонность его к переходу на
216
режим проникновения (рис. 201) Сам перхлорат в условиях этих опы-
тов воспламеняется лишь при вдвое большей навеске воспламенителя и
давление достигает максимума лишь через 400 л»се«; при содержании
27 % алюминия максимум давления наступает через 20 мсек, а при мень-
ших содержаниях алюминия горение переходит во взрыв, который раньше
всего (через 12 мсек) происходит при добавления 5% алюминия.
Как и для органических ВВ, переходу горения на режим проникнове-
ния горения в глубь заряда благоприятствует и ускоряет его увеличение
высота заряда (при постоянном весе его). Сильное уменьшение размеров
частиц перхлората (до 10 мк) задерживает наступление ускорения и за-
трудняет переход горения во взрыв. Аналогичное влияние оказывает уве-
личение относительной плотности смеси.
Рис. 200. Влияние бихромата аммония (5%)
на горение измельченного в порошок нит-
роглицеринового пороха в манометриче-
ской бомбе
В ос пл.— 0.5 г черного пороха; Н — порох;
Н + К — порох с бихроматом. Числа у кривых
означают относительную плотность заряда
Рис. 201. Влияние содержания алюми-
ния на характер горения перхлората
аммония
Числа у кривых показывают количество до-
бавленного алюминии в процентах
При поджигании смеси воспламенителем, помещенным в донной частя
стаканчика (с тем, чтобы исключить влияние проникновения горения),
ускорение газообразования шло быстрее, но скорость его пе достигала
взрывных значений. При давлении до 300 ат скорости горения перхлората
с алюминием и без него близки; выше 300 ат смесь горит быстрее. При
содержании 25% алюминия скорость горения значительно больше, чем
при 5%; при «торцевом» горении соотношение времен горения н склон-
ностей к переходу его во взрыв было обратным.
При горении по всей поверхности цилиндрических зарядов большой
плотности под давлением до 350 ат перхлорат горел быстрее, чем его
смесь с 5% алюминия.
Опыты В. В. Горбунова по добавке алюминия к нитроклетчатке
(пироксилин № 1) показали влияние, обратное тому, которое наблю-
далось в случае перхлората аммония — заметное замедление роста газо-
образования, особенно выраженное при повышенной плотности смеси.
Из совокупности полученных данных следует, что алюминий при ма-
лых содержаниях сильно способствует переходу горения перхлората ам-
мония на конвективный режим. Наиболее вероятную причину этого влия-
ния следует видеть во взаимодействии алюминия с первичными продук-
тами распада перхлората аммония; при этом вследствие большого
1 Опыты ставили в стеклянном стаканчпле (d = 15 мл. и = 3 г, р= 1.1 г/слг,
воспламенитель—0,5 г дымного пороха, рвоспл =30 ат).
217
теплового эффекта окисления алюминия образуются газы с высокой тем-
пературой, которые, проникая в порошок, гораздо легче поджигают его
частицы, чем относительно холодные продукты распада самого перхлора-
та. Судя по тому, что наиболее эффективным оказывается небольшое по
сравнению со стехиометрическим содержание алюминия, речь идет об его
взаимодействии с небольшим количеством продуктов распада перхлората,
выделяемым новерхяостными слоями частиц. Избыток алюминия не толь-
ко не' помогает проникновению горения в глубь заряда, но даже 'затруд-
няет его, так как ка нагрев этого избытка расходуется часть тепла.
Решающая роль проникновения горения подчеркивается тем обстоятель-
ством, что алюминий не увеличивает скорость нормального горения
перхлората аммония. Не удивительно н то, что алюминий не интенсифици-
рует горение пироксилина; горение последнего образует продукты, срав-
нительно бедные кислородом и менее реакционноспособные, чем в случае
перхлората аммония. Даже при горении аммиачной селитры влияние алю-
миния на этот переход гораздо слабее.
VI. ГОРЕНИЕ ГЕТЕРОГЕННЫХ СИСТЕМ
1. МЕХАНИЗМ ГОРЕНИЯ
Лейпунский [199] рассмотрел механизм горения гетерогенной системы,
в которой компоненты (горючее и окислитель) содержатся в виде макро-
скопических частиц, причем один из них имеет низкую упругость пара.
Эти условия выполняются, капример, при горении черного пороха осо-
бенно в беосерком его варианте. В этом случае реакция не может проис-
ходить в газовой фазе к должна протекать на поверхности частиц нелету-
чего компонента (угля).
Принимается следующая последовательность процессов при горении
черного пороха: селитра разлагается, выделяя кислород, который диффу-
зией в газовой фазе поступает к частичке угля, реагирует на ее поверхно-
сти, а продукты реакции также диффузией в газе удаляются от угольной
частицы. Существенной стороной механизма горения является зависи-
мость скорости элементарного акта горения — сгорания угольной части-
цы — от давления.
В кинетической области скорость горения угля пропорциональна дав-
лению кислорода. В диффузионной области, когда реакция углерода с
кислородом идет настолько быстро, что скорость диффузии лимитирует
скорость сгорания, время сгорания угольной частицы будет зависеть от
условий диффузии. Пусть ?о — количество кислорода, потребное для сго-
рания частицы угля рассматриваемых размеров, D — коэффициент диф-
фузия (~--), Со — концентрация кислорода на бесконечности (•"-'₽),
Гэфф — эффективный путь диффузии (градиент концентрации равен
Со/г»фф), т — время сгорания частицы и о — ее поверхность. Тогда
q^D^-xS (3.33)
Гвфф
и
Знаменатель в выражении (3.34) не зависит от давления, до также не
зависит от нега Зависимость времени сгорания частицы от давления оп-
ределится зависимостью от давления эффективного путн диффузии; При
сгорании угольного шарина в атмосфере кидлорода эффективный путь
218
диффузии равен радиусу шарика, т. е. не зависит от давления; соответст-
венно время сгорания угольного шарика не зависит от давления кисло-
’' рода. То же может происходить, если источикк кислорода (частица
селитры) и угольная частица находятся в твердой фазе на неизменном
расстоянии друг от друга. Прк сгорании плоской пластины путь диффузии
равен толщине слоя кислорода, нужного для сгорания пластины. Толщи-
на этого слоя '-'1/р; поэтому время сгорания будет обратно пропорцио-
нально давлению кислорода. Таким образом, время сгорания частицы угля
'f может с ростом давления уменьшаться или оставаться неизменным в за-
висимости от внешних условий.
j • Поскольку селитра разлагается при относительно низкой 1 температуре,
при которой, как предполагал^Тейпунский, горения угля еще не проис-
ходит, то газообразные продукты распада селитры должны уносить не-
сгоревшие частицы угля, которые реагируют в газоугольном факеле над
поверхностью пороха. Таким образом осуществляется случай сгорания
угольного шарика в атмосфере кислорода, когда время сгорания не зави-
сит от давления.
Скорость распространения пламени в углевоздушной смеси составляет
от 10 до 50 см/сек, что соответствует скорости потребления утля 3,3—
11 мг/сек • см2, а в пересчете на скорость горения пороха 0,015—
0,05 см/сек. Эта величина значительно меньше, чем скорость горения чер-
ного пороха (1 CMjeeK), однако различие все же не превосходит 1—2 по-
рядков.
Независимость времени сгорания частицы утля от давления не означа-
ет отсутствия зависимости скорости горения от давления. По теории Зель-
довича, линейная скорость газификации пороха пропорциональна корню
квадратному из величины скорости объемного выделения тепла в зоне
реакции. При горении черного пороха с ростом давления пропорционально
ему возрастает количество угля и кислорода в единице объема и соответст-
венно объемная скорость тепловыделения. Таким образом, рассматривае-
мая схема горения черного пороха формально аналогична схеме горения
бездымного пороха. Сгорающие частицы угля аналогичны большим моле-
кулам, реагирующим по мопомолекулириому закону; унос угля в потоке
4 соответствует газификации, тепло, необходимое для диспергирования по-
| роха и подготовки горючей смеси подается из газовой фазы. Скорость
горения должна в таком случае возрастать пропорционально корню квад-
1 ратному из величины давления в соответствии с кааэимономолекулярным
законом реакции.
Другой ход рассуждения относительно зависимости скорости горения
от давления при независимости от него скорости сгорания отдельной ча-
стицы основан на рассмотрении слоя горящей газоугольной вмеси, харак-
] теризуемой определенной глубиной выгорания, т. е. определенным вре-
менем горения. При увеличении давления линейная скорость потока га-
зов пропорционально уменьшается, а тан как время сгорания частицы не
зависит от давления, то расстояние от поверхности, на которой окажутся
сгорающие пылинки и горячие продукты горения рассматриваемого слоя,
уменьшится и градиент температуры, определяющий поток тепла к по-
верхности пороха, соответственно увеличится пропорционально давле-
нию.
Увеличение градиента температуры в рассматриваемом слое горящей
газоугольной смеск не приведет к его' охлаждению потому, что пропор-
ционально давлению увеличится концентрация газоугольной смеси,
т. е. объемная скорость выделения тепла. Поэтому температура рассмат-
риваемого газового слоя окажется неизменной, он лишь пододвинется к
поверхности. Увеличение градиента температуры приведет к увеличению
потока тепла к поверхности пороха, т. е. к увеличению скорости газифи-
кации.
219
Из равенства количеств тепла, подводимого из области реакции и
потребляемого при газификации пороха:
Гкрк(Тп-Т0)Ск = т1^^- = т1^^Н, (3-35)
^эфф *гтэфф
Т^ГрГ ~ V КрЮ
где Рк, рк, — скорость газификации, плотность, теплоемкость конденси-
рованной фазы; Го, Гц, ГЭфф — начальная температура пороха, темпера-
тура поверхности пороха, эффективная температура области реакции,
прилегающей к поверхности пороха и определяющей теплопередачу к по-
верхности; Хэфф — расстояние от поверхности до области в газе с темпе-
ратурой Гафф; Тэфф — время сгорания того количества угля, которое необ-
ходимо для достижения температуры ГЭфф; ц — коэффициент теплопро-
водности
^иРк)2 - Л • (3.36)
ср 1 и 7 о тэфф
Так как с увеличением давления пропорционально возрастут не толь-
ко градиенты температуры, но и объемные скорости выделения тепла,
то Гафф не должно изменяться с увеличением давления. Так как Гп мало
зависит от давления, а т) и Г5фф не зависят от давления, то согласно урав-
нению (3.36)
(3.37)
т. е. скорость гонения пропорциональна корню квадратному из величины
давления. В случае, если горение частиц угля происходит в кинетической
области, то скорость реакции на поверхности угля пропорциональна р‘\
где 0 < и 1. Соответственно скорость сгорания частиц т г/р"; тогда,
согласно формуле (3.36)
т. е. скорость гореикя пропорциональна давлению в степени (14- и) /2
и обратно пропорциональна корню из радиуса частицы.
Таким образом, при сгорании угля в кинетической области скорость
горения пороха сильнее зависит от давления, чем при сгорании в диффу-
зионной области, и может быть пропорциональна давлению (при п— 1).
Зависимость скорости горения от размера частиц в кинетической обла-
сти слабее, чем в диффузионной.
Более точный расчет зависимостей и(р, г) для рассмотренной схемы .
горения был проведен Новожиловым [200] с учетом, в частности, того,
что скорость движения частиц угля может быть меньше, чем скорость
движения газа. При ряде допущений он приходит к заключению, что
граница диффузионного и кинетического режимов должна лежать около
1 ат; ниже этого давления, т. е. при горении в кинетическом режиме,
скорость должна расти пропорционально первой степени давления, а при
больших давлеикях на диффузионном режиме пропорционально корню
кубичному из давления. Абсолютная величина скорости по расчету долж-
на составлять ~ 1 мм/сен.
При дальнейшем уточнении теории [201], помимо более нревильного
учета диффузик окислителя и сил, действующих на частицу, движущуюся
в газе, было принято во внимание, что воспламенение частицы угля про-
исходит не сразу после того как она оторвется от поверхности пороха,
а лишь тогда, когда температура окружающего газа достигнет некоторой
температуры воспламенения. Расчетная оценка этой температуры приме-
220
нительно к черному пороху показывает, что она значительно выше тем-
пературы поверхности, принимаемой равной 300° С. Расчет дает для ско-
10-s
рости горения выражение п ж р , При d = 10~ см получаем для
атмосферного давления и = 10-2 см/сек, т. е. на три порядка меньше
экспериментального значения скорости горения черного пороха.
Это несоответствие приводит автора к заключению, что истинная схе-
ма горения должна включать воспламенение частицы значительно раньше,
чем в газовой фазе будет достигнута температура ее воспламенения, воз-
можно, на поверхности твердой фазы, причем
температура частицы 'будет выше темпера- N р
туры окружающего окислителя; в газовой же
фазе будет происходить догорание горючего.
Реакция; • определяющая скорость горения,
будет происходить не в газовой, а в конден-
сированной фазе.
Новожилов [202] рассмотрел также горе-
ние смесевого пороха, оба компонента кото-
рого способны газифицироваться в диффу-
зионной и кинетической областях. На
рис. 202 изображена схема горения для диф-
фузионного случая. Порох состоит из слоев
веществ А и В одинаковой толщины 2h; если
Рис.
202. Схема горения
смесевого пороха
и В — слои вещества одина-
ковой толщины
компоненты одинаковы по своим физическим
свойствам и скорость химического распада
одинаково зависит от концентраци А и В,
картина горения будет периодической по про-
странству ;с периодом 2h. Чтобы найти ско-
рость горения такого пороха, необходимо рассмотреть элементарную ячей-
ку, ограниченную плоскостями MN и PS. Очевидно, максимум тепловыде-
ления будет находиться по линии Ог; в связи с этим поток тепла из газо-
вой фазы на поверхность твердой фазы не будет ревномерно распределен
на поверхности последней. Это обусловливает неревномерное выгорание
различных-частей поверхности, и горящий inopox будет иметь поверхность,
подобную изображенной на рис. 202.
Для упрощения задачи принимается, что поверхность пороха плоская
и совпадает с плоскостью х = 0. Далее допускается как предельный слу-
чай, что скорость химического распада бесконечна и соответственно ско-
рость горения лимитируется скоростью диффузии. При этом зона химиче-
ской реакций будет бесконечно тонка: она будет совпадать с плоскостью
у = 0. Кроме, того, концентрация реагирующих веществ в зоне реакции
будет равна нулю. Плоскость у ведет себя как абсолютно поглощающая
поверхность, причем она отделяет область у < 0, где есть вещество В и
продукты реакции С и D и нет вещества Л, от области, где есть А, С и D
и нет В.
Математическое рассмотрение этого случая показывает, что для него
скорость горения не зависит от давления и обратно пропорциональна дис-
персности. В том случае, когда отношение скоростей диффузии к скорости
химической реакции велико и лимитирующим процессом является хими-
ческая реакция, скорость горения пропорциональна давлению и (если
реакция бимолекулярна) не зависит от дисперсности.
Теория горения смесевых порохов Саммерфилда
Саммерфилд, Сазерленд, Уэбб, Табак и Холл [203] предлагают физи-
ко-химическую модель зоны горения, описывающую изменение темпера-
туры, скорость реакции, диффузионные процессы и условия течения газов,
22i
которая позволяла бы предсказывать влияние давления, размеров частиц
и соотношения горючего и окислители.
Объектом исследования был смесевой порох на основе тонко измель-
ченного перхлората аммония и кополимера эфир — стирола. Опыты долж-
ны были дать ответ ка вопрос о том, имеется ли на поверхности жидкий
слой окислителя или горючего. В положительном случае газы, образуе-
мые горючим к окислителем, могут поступать в зону тазофазной реакции,
Расстояние от мйерйнмти герсРия
Рис. 203. Теоретическая модель уста-
новившейся воны пламени со средним
по времени профилем температуры
будучи частично смешаны я на-
оборот. Второй, связанный с пар-
ным вопрос состоит в том, проис-
ходит ли смешение путем молеку-
лярной или турбулентной диффу-
зии.
Фотографии горения в собст-
венном его свечении, а также шли-
раи-фотографии показывают, что в
тонком (1 мм) слое, прилегающем
и поверхности, турбулентности
нет; ту неравномерность, которая
обнаруживалась при цветной вы-
сокоскоростной киносъемке и прк
измерении температуры, ълеяу&г
приписать колебаниям в степени
смешения в реакционном слое
Микрокинофотографии в проходу
щрм свете, показывающие зубча-
тый профиль поверхности горе-
ния, указывают ка то, что плавле-
ния на ней пе происходит. Это лодпверщдается сравнением с горением со-
става на основе перхлората калия, продукт разложения которого (КС1)
плавится перед испарением, что приводит к ровной поверхности с проры-
вающимися через нее пузырьками. *
Измерение профиля температуры тонкими платино-родневыми термо-
парами, а также измерение излучения’чхламеик доказало, что,при дав-
лениях выше 14 ат толщина реакционной эоны меньше 100 жж, если при-
нять, что температура поверхности равна 700—800° С, Последний метод
показывает также, что зона пламени иикроиеоднородва,. причем масштаб
неоднородности по крайней мере равен 10 мк. Сведеикя о химической
природе частиц, образующихся в пламени, были получены спектроскопи-
ческим методом.
Рассмотрение экспериментальных данных приводит к следующему
представлению о механизме горения. Квазистационарное газообразное
пламя прилегает к горящей поверхности, как показано ка рис. 203. По-
верхность сухая, т. е. газы, образуемые окислителем и горючим, выделя-
ются непосредственно твердой фазой в результате сублимации или пиро-
лиза. Смешение горючего и окислителя происходит только в газовой фазе.
В твердой фазе не происходит никаких реакций, которые влияли бы на
пламя, не выделяется и тепло. Реакции сублимации или пиролиза на по-
верхности могут быть экзотермическими или эндотермическими. В ре-
зультате окислительных реакций тепло выделяется в тонкой реакционной
зоне в газовой фазе.
Пары горючего или окислителя выделяются в виде отдельных кузырь-
ков, которые затем горят в атмосфере другого компонента. Каждый пу-
зырек в среднем меньше по массе отдельного кристаллика окислителя,1 но
тем не менее их массы находятся в некоторой зависимости — чем больше
кристалл, тем больше и пузырек. Допускается, что масса пузырька не за-
висят от давления. Пузырьки, проходя через зону пламени, постепенно
222
реагируют со скоростью, определяемой диффузионным смешением и ки-
нетикой, выделяя тепло. Эта структура зоны пламени называется пузырь-
ково-диффузионной.
Физическая основа образования таких пузырьков и, в частности, неза-
висимость их величины от давления неизвестна; наиболее вероятно, что
они представляют собой пузырьки газифицированного горючего, которые
увлекаются потоком продуктов газов, образовавшихся из окислителя. Рас-
пространение горанкя происходит в результате передачи энергии от эоны
пламени к поверхности пороха, которая осуществляется преимуществен-
но теплопроводностью.
Ниже приводится количественная формулировка рассмотренных пред-
ставлений.
Количество тепла, необходимое для разогрева пороха от Та до Тщ сооб-
щается экзотермической реакцией газификации его компонентов и
теплопроводностью в газовой фазе при среднем градиенте температуры.
Г> —
-L~~, (3.39)
(Тп Тй) — ---
(3.40)
где ии — массовая скорость горедая; са — теплоемкость пороха; Тп —
I ф некоторая средняя температура его поверхности; То — начальная темпе-
ратура; Q# — положительный тепловой эффект газификации компонентов
пороха; — коэффициент теплопроводности газов у поверхности поро-
ха; Т] —конечная температура горения; L — толщина зоны горения.
Линейная скорость пиролиза обоих компонентов может быть выражена
соотношением
ця = В ехр (— Е / ЙТП). (3.41)
При стационарном горении обе скорости равны друг другу (и скорости го-
рения пороха).
Поскольку энергии активации пиролиза окислителя и горючего раз-
личны, то и температуры на поверхности их частиц должны быть разны-
ми. Поэтому понятие средней температуры является некоторым условным
приближением. Эта температура принимается независящей от давления,
размеров чаотиц и соотношения окислителя и горючего, но следует ожи-
дать, что она зависит от содержания хромита меди, являющегося катали-
затором разложения перхлората.
.Для определения толщины зоны реакции L рассматриваются два пре-
дельных случая. При очень низких давлениях скорость^ молекулярной
диффузии гораздо больше скорости реакции окисления, так что послед-
няя происходит в смеси газов. При очень высоких давлениях скорость
1 химической реакции столь велика, что скорость горения полностью кон-
тролируется взаимной диффузией. Реально возможен и промежуточный
случай.
При малых давлениях пламя можно рассматривать как поток со
скоростью Им/рг, в котором происходят реакции второго порядка. Тогда
г__ м 1 __ и _______1_____/____/о доч
Pg (&\ Pg U — е)’рЛЛехр ЯТе) , < ‘ '
Wt/cp.
причем под е, d&jdt, pg и т. д. подразумеваются их средние значения.
Подставляя уравнение (3.42) в (3.40), можно найти
2 - . Уг ~ ги) ЛехР (" Е/дгг)
( «и—
223
(3.44)
Таким образом, скорость горения пропорциональна давлению в пер-
вой степени. Толщина реакционной зоны обратно пропорциональна
давлению.
1
Pg К(Гп~ Го) ~ ^Г'’' Иехр(-В/ЯТ#)]Ч,
Для второго предельного случая при допущении, что на поверх-
ности образуются пузырьки горючего с массой р и размером d,
= ц = (3.45)
Длительность жизни пузырька определяется скоростью газовой диф-
фузии внутрь его и наружу.
Обозначим буквой D коэффициент диффузии, одинаковой для обоих
газов и усредненной по реакционной зоне. Тогда толщину зоны и ско-
рость горения
можно рассчитать следующим образом:
_ <Р г _ «иН*4
^Dg ’
П)- :
У/у‘4(Л-Тд)‘А
(3.46)
(3-47)
массовая скорость горения пропорциональна,давлению
что согласуется с наблюдениями, сделанными для по-
Видно j что
в степени */з»
рохов на основе перхлората аммония.
Для промежуточного случая можно допустить, что толщина зоны
с давлением меняется» отчасти так, как если бы она контролировалась
кинетикой, и отчасти — как если бы она контролировалась диффузией
Г — Г MT1 —тп) TZ* ( г1 I 1
[МГв-П)-<28] Vgp«P(-^Tg)p
(3.48)
Это выражение для толщины зоны может быть введено в выражение
баланса энергии и для скорости горения получим:
1 _ (Ре1й8(Гп~Го)~~ |V* f_______«1________। zap‘4 ] 3 4g.
“и t %(T1-Tn) J (р/И«Р(-В’/ДГв)]'Г ^'*pH ‘ j
>V*
Отсюда зависимость скорости горения от давления получается в виде:
— = -+4 • (3-5°)
им Р ' /4 ' *
Эта зависимость дает возможность проверить теорию путем сопостав-
ления с нею экспериментальной зависимости скорости горения от давле-
ния в широком интервале последнего. Наряду с этим параметр скорости
реакции а должен быть очень чувствителен к температуре пламени,
а параметр диффузии b должен быть чувствителен к среднему размеру
частиц. Катализаторы разложения повышают скорость разложения, сни-
жая Та и соответственно уменьшая в одинаковой степени а и Ь.
Для проверки теории в бомбе постоянного давления методом расплав-
ления проволочек, пропущенных через заряд, определялась скорость го-
рения при давлениях от 1 до 106 ат. Точность определения составляла
±3% ив основном зависела от неравномерности горения пороха, обус-
ловленной его неоднородностью и неодиообразием бронировки.
224
Полученные данные в координатах р/а — р’1'1 на графике дают прямые
(рис. 204), причем для крупнозернистого перхлората (средний размер
120 jkk), мелкозернистого (16 мк), «холодной смеси (75% окислителя) и
«горячей» смеси (80% окислителя) получаются следующие значения
а и Ъ:
а Ь
1 — «холодная» смесь (крупнозернистый окислитель) 365
2 — «холодная» смесь (тонкозернистый окислитель) 400
3 — «горячая» смесь (крупнозернистый окислитель) 245
4 — «горячая» смесь (тонкозернистый окислитель) 160
39,0
19,8
27,0
17,3
Таким образом, оба параметра изменяются в зависимости от размера
частиц в соответствии с теорией.
Повторные определения скорости горения при разных давлениях,
произведенные другим исследователем, дали результаты, согласующиеся
с приводимыми выше. Однако влияние со- ц/ц
отношения окислителя и горючего на а,
которое было получено ранее и предусмат-
ривается также теорией, не обнаруживает-;
ся; при этом значении а существенно
меньше. Введение в порох хромита меди
уменьшает а и b в одинаковое число раз,
как и следовало ожидать по теории.
Таким образом, простая концепция, за-
ключающаяся в том, что в области низких
давлений скорость горения определяется
скоростью химической реакции, а в обла-
сти высоких --скоростью диффузии, под-
тверждается тем фактом, что размеры час-
тиц окислителя не сказываются на скоро-
сти в области низких давлений и получа-
ют решающее значение в области высоких
давлений.
Позднейшие опыты, произведенные,
кроме сополимера эфир-стирола, с поли-
сульфидными и эпоксисмолами (в ограни-
ченном интервале размера частиц), пока-
зали, что при использовании 1полисульфид-
норо горючего и при давлениях выше 70 ат
частицы перхлората пиролизуются гораздо
быстрее, чем горючее, и оставляют на по-
верхности маленькие углубления, которые
можно обнаружить под микроскопом на
Рис. 204. Сравнение данных Са-
терленда по скоростям горения с
теорией Саммерфилда (прямые
линии)
I — в — в,25; а — 9,9; У — в — 4.5;
о — 4.35; 3 — « — 3.2; а " 10.fi; 4 — в“
— 2,7; а — 6,6, Соотношение ме:кду
окислителем и горючим и размер их
частиц показывают цифры у кривых
затушенных шашках. Таким
образом, ведущей в этих условиях является газификация перхлората. Ско-
рость горепия близка к скорости'чтения перхлорета. При низких
(<35 ат) давлениях состояние поверхности соответствует описанной выше
теории; это относится также и к величине скорости горения. Отклонения
от теории исчезают, когда применяются смеси двух фракций окислителя
или когда применяются смеси с большим содержанием окислителя.
Последующие исследования имели целью уточнить роль передачи теп-
ла в реакционной зоне излучением, связь между размерами частиц окис-
лителя и параметром Ь, а также соответствие между значением скорости
при горении цилиндрического заряда с торца и ее значением при горении
заряда в ракетной камере по каналу.
Скорость торцезого горения определялась в бомбе постоянного давле-
ния при пропускании тока азота, выходившего в «трубу», расположенную
над зарядом. При скоростях азота в трубе от 18 до 61 см/сек при 35 ат
влияния на скорость горения не обнаруживалось; примесь до 20% кисло-
15 К. К, Андреев
225
W 1,2 M f,S f,S 2,0 2,2
js-№J(4)-i 1
Рис. 205. Примеры линейных ско-
ростей пиролиза для некоторых
окислителей и связок
А — нитрат аммония; В — полисти-
рол; С — перхлорат аммонал: D —
каучук ORS-32; Е — пластик GS-2
рода также ие изменяет скорости горения. При 7 ат не оказывало влияния
также наличие бронирован. Шаппа диаметром 11,2 мм горели при 35 аг
на 3% быстрее, чем при диаметре 6,4 мм.
Измерения скорости горения пташки пороха с центральным каналом
по этому ианалу в направлении к периферии шашки дали значения ско-
рости на 7%. меньшие, чем при торцевом горении (d = 6,4 лж), в проти-
воположность тому, что можно было ожидать; причины этого различия
не выяснены.
Измерение излучения пламени и учет его влияния показали, что оно
увеличивает скорость незначительно — на 1 % при горении заряда с тор-
ца и на 6 % при горении пташки по канаку.
Интенсивность излучения становится боль-
ше при увеличении размера частиц; это
влияние проявляется при низких давле-
ниях и отсутствует при высоких.
Более конкретную картину процесса
горения рассматривают Чайкин и Андер-
се® [204]. Они исходят иа двухтемпера-
турпого постулата Шулице и Деккера, со-
гласно которому разложения твердого
окислителя и связки являются в микро-
скопическом масштабе независимыми про-
цессами. Линейные скорости пиролиза
окислителя и горючего равны друг другу
и скорости горения пороха.
Линейные скорости пиролиза могут
быть определены методом горячей пла-
стинки; их значения при равных темпера-
турах для ряда веществ приведены на
рис. 205. Эти скорости для обоих основных
компонентов пороха при данной темпера-
туре могут быть не одинаковыми. Отсюда
делается заключение, что соответственно
должны быть неравными и температуры на поверхности частиц.
Для смесевых порохов иа остове нитрата аммония но этой гипотезе
принимаются следующие этапы процесса:
а) газификация окислителя, сиорость которой определяется процессом,
включающим эндотермическую реакцию десорбция с поверхности;
б) газофазная окислительно-восстановительная реакция продуктов пи-
ролиза окислителя, образующая пламя с температурой около 1250° К;
в) эндотермический пиролиз связки в пламени;
г) газофазные экзотермические реакции между продуктами пиролиза
связки и окислителя.
Пламя от стадии б заставляет газифицироваться частицы связки, и на
некотором расстоянии от поверхности конденскровакиой фазы между
газообразными продуктами превращения окислителя и связки возникает
диффузионное илами. Однако лишь небольшая часть тепла, выделяюще-
гося в этом пламени, передается конденсированиой фазе.
Устеновлено. например, что большие изменения, в 10—100 раз, ско-
рости пиролиза полистироЛьной связки не влияют на скорость горения
соответствующего пороха на аммяачной селитре. Это говорит за то, что
поверхность частиц окислителя получает тепло преимущественно из тон-
кой зоны газофазной реакции между NH3 и HNO3, а диффузионное пла-
мя возникает слишком далеко, чтобы влиять иа скорость горения. Такой
вывод подтверждается и тем фактом, что сама аммиачная селитра (с ка-
тализатором) горит с той же скоростью, с какой горят и многие пороха
на ее основе.
226
Применение простой схемы процесса, основанной на двухтемператур-
ном постулате, к порохам на основе перхлората аммония осложняется не-
которыми обстоятельствами.
При горении пороха на поверхности окислителя первоначально проис-
ходит диссоциация на NHg и НСЮч (сублимация). Эти продукты реагиру-
ют вблизи поверхности, образуя окислительно-восстановительное пламя,
как в случае нитрата аммония. Однано харантеристики поверхностного
разложения перхлората и нитрата различаются, и полной аналогии в про-
цессах горения порохов на их основе нет. Во-первых, при горении пер-
хлората аммония возможна, помимо сублимации, на поверхности экзо-
термическая реакция в твердой фазе, за счет теплового эффекта которой
уменьшается эндотермичность сублимации.
Удельный вес твердофазной реакции зависит, в частности, от скорости
горения. Во-вторых, температура поверхности окислителя при горении
перхлоратных порохов гораздо выше, чем аммиачно-селитренных, и близ-
ка к температуре пиролиза связок. Это обстоятельство влияет на степень
взаимодействия окислителя и горючего вблизи поверхности пороха.
При горении аммиачно-селитренных порохов, если передача тепла в
реакционной зоне около частицы осуществляется теплопроводностью и
имеется линейное изменение температуры в ней, толщину зоны можно
приближенно выразить соотношением
б = го (1 + Го /V яКт у*1, (3.51)
где Го — средний радкус частицы; К — средняя теплопроводность газовой
фазы; т — период полупревращения окислительно-восстановительной
реакции в газовой фазе.
Значение б связано со скоростью, с которой расширяется газ и со ско-
ростью происходящих в нем реакций.
К = Mo/Cppg, __, (3.52)
где М — средний молекулярный вес газов: о — средняя их теплопровод-
ность; pg — плотность; ср—средняя молярная теплоемпость. Отсюда сле-
дует, что толщина реакционной зоны зависит от давления.
Для окислительно-восстановительной реакции второго порядка в
газах
т — 1/cgkg — 2М fpgltg, (3.53)
где cg — концентрация одного из реагентов; kg— константа скорости.
Если выполняется закон идеальных газов, то, используя уравне-
ния (3.52) и (3.53), получаем
й _________________________£»_______=
1+ * 1+V
(3-54)
где 7?g — газовая постоянная; Тй — температура, при которой реагирует
основная масса газа; р — давление. При постоянном размере частиц
Ф можно считать постоянной.
Приравнивая скорость теплоотвода в глубь частицы скорости пере-
дачи тепла, необходимой для обеспечения стационарности пиролиза на
ее поверхности, получаем
4лпго<з Tf 8Гз'° = 4лг<гор,ДЯЙо, (3.55)
где ДЯ— эндотермичность реакции пиролиза ла поверхности окислите-
ля (кал/г-моль); р,-—плотность окислителя (г/см3); п — доля открытой
поверхности частицы; Во — линейная скорость пиролиза окислителя.
227
15*
Ив уравнений (3.40), (3.54) и (3.55) получаем скорость горения
<3-56>
Вр~АЯ'аехр(-Ег10/ЯТ1'0). (3.57)
Скорость газофазного взаимодействия между NH3 и HNO3 предпо-
ложительно определяется реакцией
NH»+ NOa-s. NHa+HNOa. (3.58)
Участвующая в ней NO> образуется в результате относительно быстро-
го разложения HNO3.
Влияние связки на реакционную зону
Представления о двухстадийном процессе превращения частицы ве-
щества приложимы к однокомпонентному пороху. Однако они справедли-
вы и при горении смесевых порохов на основе аммиачной селитры. Тем-
пература на поверхности частиц окислителя низка (600° К) по сравне-
нию с температурой"на поверхности частиц горючего (^ ПОСТ К). Вслед-
ствие этого газификация горючего происходит относительно далеко от по-
верхности частиц окислителя, а взаимодействие продуктов газификации —
еще дальше и теплота его не влияет на общее течение процесса.
Рис. 206. Влияние связи на тепловой слой
Рис. 207. Изменение е с давлением
При горении порохов на основе перхлората аммония температура на
поверхности частиц окислителя выше (^ 1100° К), возможность взаимо-
действия продуктов его газификации с горючим и влияния на процесс в
реакционной зоне окислняия больше. Это влияние может быть различ-
ным (рис. 206).
1. Газифицированное горючее может диффундировать в реакционную
зону, но не реагировать в ней. Оно будет в этом случае оказывать раз-
бавляющее действие, увеличивая длительность окислительно-восстанови-
тельной реакции 6 и скижая температуру превращения окислителя.
2. При диффузии происходит относительно медленное взаимодействие
и окислительно-восстановительная реакция будет замедлена, 5 толще, но
конечная температура Tf выше.
3. При диффузкй горючего извне реакция происходит быстро. Тогда
толщина слоя остается неизменной, но температура повышается.
4. Наконец, при диффузии горючего извне в реакционную зону не
происходит взаимодействия — толщина зоны не будет меняться, одиако
может иметь место разбавление пламенной зоны, т. е. снижение Т/.
Наконец, при диффузии горючего в реакционную зону и взаимодейст-
228
вии его со скоростью, большей, чем скорость окислительно-восстанови-
тельной реакции, толщина реакционной зоны будет зависеть от скорости
диффузионной реакции между горючим и окислителем.
Количественное рассмотрение всех перечисленных возможностей пред-
ставляет собой очень слоящую задачу. Особенностями второго случая, ио-
видимому, наиболее распространенного, являются: а) появление нереаги-
рующего горючего в реакционной зоне, что призодит к уменьшению
концентрации реагентов и увеличивает длительность реакции; б) диффун-
дирующее горючее быстро реагирует в высокотемпературной зоне первич-
ного пламени, повышая его максимальную температуру по сравнению с
той температурой, которую дает чистый окислитель.
Иа уравнения (3.53) видно, что разбавление можно учесть, умень-
шая cg. Тогда общее давление газа в реакционной зоне может быть
выражено из закона идеальных газов как
Р = Ро + Рь = (2c# ,о + с^.ь) RgTg, (3.59)
где Ро и рь — парциальные давления газов окислителя (т. е. NHs и HNOa)
и горючего; се,о — молекулярная концентрация одного иа продуктов диссо-
циации окислителя; cgib — горючего.
Решая уравнение (3.59) относительно Cgp и подставляя полученное
решение в уравнение (3.53), получаем
¥ = (3-6°)
где L = pb]p.
Используя уравнения (3.51) и (3.52) и закон идеальных газов, по-
лучаем
<зм)
и
д =---------. (3.62)
Сравнение уравнений (3.54) и (3.62) показывает, что влияние раз-
бавления уменьшает значение ф для окислителя в (1—L)tlt раз.
Реакцию горючего в первичном пламени можно учесть, принимая
повышенное значение температуры пламени на наружной границе ре-
акционной зоны.
Т t = т !,Q + 8. (3.63)
Используя уравнения (3.62) и (3.63), можно переписать уравнение
(3.56) для скорости горения в виде
В ~ + е)(1+<рр ]/~1—£)
Из уравнения (3.64) видно, что, за исключением е и L, параметры
в выражении для скорости горения имеют те же значения, что и дли
чистого окислителя.
8, как и L, зависит от скорости диффузии горючего в реакционную
зону. Если допустить, что максимум L соответствует массовому отно-
шению окислителя к горючему, то может быть показано, что (1—L)'!* ~
для стехиометрического состава. Это вытекает из следующих сооб-
ражений.
Пусть массовое отношение окислителя и горючего, а Мг соот-
ветствующее отношение молекулярных весов продуктов газификации.
Тогда из закона идеального газа следует
₽> = £₽ (3-65)
229
о
l = (3-66)
mr p 7
Оцениа значзний mr и Mr дает 4 и 0,4 соответственно. Тогда
Д — 0,1^^0,1 и (1 —
Зависимость в от скорости диффузии довольно сложна, и точная
оценка ее затруднительна. Однако для тех смесевых порохов, для ко-
торых взаимодействие окислителя с горючим сильно влияет на скорость
горения, е должна быть прямо пропорциональна коэффициенту диффу-
зии и обратно пропорциональна размерам частиц. Поскольку коэффи-
циент диффузии обратно пропорционален давлению, следует ожидать,
что и е будет уменьшаться при уменьшении давления.
На основании экспериментальных данных по горению (табл. 30) и
уравнения (3.64) можно установить изменение е в зависимости' от давле-
ния, размеров частиц и массового отношения компонентов. В градусах
Кельвина для аммиачно-селитренных порохов
7 30.10..^ _
1 + 22I-0J5
(3.67)
Дли перхлоратных порохов
е— ЖЛ (3.68)
1 + 56#7гор ' '
При выводе этих соотношений предполагалось, что (1 — L)'11 ж 1 и пре-
небрегалось изменением Та в зависимости от скорости горения.
На рис. 207 приводится зависимость е от давления для следующих со-
ставов;
Кривые Окислитель Отношение окислителя к связке Средний радиус частицы окис- лителя * мк
А NH*G1O* 80 :20 8
В NH*C1O* 75:25 8
С NH4CIO4 80:20 60
В NH4CIO* 75:25 60
Е N 11*010» 85 :15 10
Физическая роль связки при горении порохов
Смесевые пороха готовятся различными способами — полимеризаци-
ей или литьем, включая формование под вакуумом или под давлением,
прессованием, выдавливанием и вальцеванием.
От способа изготовления обычно зависит конечная плотность пороха,
й, как правило, чем она больше, тем медленнее горит порох. Отсюда сле-
дует, что основное физическое влияние связки на характеристики горе-
ния определяется ее влиянием на эффективную поверхность окислителя,
по которой может идти горение. Это следует и из рассмотренных выше
теоретических соображений. Связки, которые разлагаются при меньшей
поверхностной температуре, чем окислитель, дают возможность его горе-
ния на большей поверхности, горенке в иавестной степени переходит в
«объем». Способ изготовления пороха, летучесть связки, сцепление ее с
окислителем могут влиять на горение частично также физически.
То же относится и к способности связки плавиться. Связки, которые
плавятся при низких температурах, но быстро испаряются лишь при вы-
соких температурах, могут «заливать» горящую поверхность, приводя к
временному затуханию горения окислителя.
230
Данные по скорости горения
Таблица 30
Топливо Отаошеаие масс окисли- теля и связки Эмпатическое уравнение скорости горения (Р, от; В, см/сек)
Полистирол нитрат аммония Л-3 вес. % (NH4)2Cr3O7 85:15 10 В = = 0,092 4-0,002/»
80:20 60 £ В 3,62 4,39 " Р +
Смола на основе стиро- ла 4- перхлорат аммония 80:20 75:25 8 60 Д В £ В 4,99 2,64 - р + pV, 6,30 6,28 - р + р’/.
75:25 8 £ В 7,19 , 3,39 - , + А
Структура и природа поверхности пороха могут зависеть от способа
изготовления. Например, литые и выдавленные пороха представляют со-
бою тесно расположенные частицы окислителя, расположенные в сплош-
ной массе горючего.
Прессованные заряды, вероятно, содержат дисперсные частицы горю-
чего, распределенные в сплошной среде окислителя. Такие пороха обычно
пескольно различаются по характеристикам горения.
2. ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ ПО ГОРЕНИЮ
ГЕТЕРОГЕННЫХ СИСТЕМ
При горении гетерогенной смеси, тепловой эффект которого опреде-
ляется взаимодействием составляющих ее компонентов, например смеси
окислителя и горючего, скорость горения, естественно, может зависеть от
размеров их частиц, поскольку предпосылкой реакции является смеше-
ние реагирующих веществ.
Предельными случаями такой смеси являются, с одной стороны, моле-
кулярное смешение компонентов, как оно осуществляется, например,
в растворах или газовых смесях, и, с другой — система, в которой полу-
пространство, заполненное горючим, граничит с полупространством, за-
полненным окислителем. Возможность горения в этих последних услови-
ях до недавнего времени была известна лишь по отдельным наблюдениям.
Так, Дотриш установил, что горение аммонитов существенно облег-
чается, если заряд контактирует с углем; в опытах А. П. Глазковой по
горению перхлората аммония в плексигласовых трубках распространение
горения по поверхности контакта окислителя и оболочки заставило искать
другие материалы для последней. Контактное горение, по-видимому, мо-
жет быть вызвано в определенных условиях в результате взаимодействия
концентрированной азотной кислоты со стенками алюминиевого аппарата.
Потенциальное техническое значение контактного горения заключается
в том, что оно указывает на возможность получить составы, которые мо-
гут гореть, но не способны детонировать.
Систематически контактное горение было изучено в работах Бахмана,
Беляева и их corp. [205, 206]. Горение осуществлялось в зарядах, состоя-
щих из твердого неорганического окислителя, запрессованного в ви-
де стержня или пластины в толстостенные цилиндричеслие или плоские
231
оболочки из плексигласа. Е тех опытах, где в качестве горючего приме-
нялся металл, окислитель запрессовывался в латунную оболочку, в нем
сверлилось отверстие, в которое прессовался порошок металла. В опытах при
насыпной плотности порошок металла насыпался в стакан из кальки
(с? от 4 до 12 лип), который устанавливался по центру в латунной оболоч-
ке, зазор между калькой и латунной оболоч-
6,- o_f кой засыпался порошкообразным окислите-
Й • -% лем. Заряд поджигался на верхнем торце
V д —<? вдоль границы горючее — окислитель, после
* /„ короткого участка разгона устанавливалась
1 -______,_______, постоянная скорость горения, фронт которо-
5 i0 а, мм го перемещался в виде конической выемки
Рис. 208. Зависимость скорости
пламеия от характерного раз-
мера слоя окислителя (КС10<;
рк, = 10 аги)
1 — цилиндрическая оВолочка; 2—
плоская кювета; 3 — стальная
кювета
ния исходных компонентов
ВНИЗ.
Распространение пламени вдоль поверх-
ности контакта твердого горючего и окисли-
теля в этих условиях включает в себя гази-
фикацию компонентов под влиянием тепло-
вого потока от пламени, смешение (за счет
столкновения потоков, направленных под уг-
лом друг к другу, а также за счет молекуляр-
ной диффузия), прогрев продуктов разложе-
и последующее сгорание. При этом вблизи но-
сика, по-видимому, происходит горение гомогенной смеси (смешение успе-
вает произойти в пределах зоны прогрева), а выше устанавливается диф-
фузионное пламя.
Измерялась зависимость скорости распространения от характерного
размера d слоя окислителя, давления, под которым шло горение, и отно-
сительной плотности окислителя, а также формы выемки. При увеличе-
ния d скорость распространения
Рис. 209. Зависимость ui(p) для различных
окислителей в цилиндрической оболочке
(d = 6 мм) из плексигласа
7 —КСЮ* (р - 0,88); II — KClOj (Р - 0.88);
III — ВаО, (р - 0.35); IV — КМпО, (р - 0,55);
V — стехиометрическая смесь плексигласа о
КСЮ* (размер частиц ~10 жк)
и в случае перхлората калия
уменьшается, стремясь к неко-
торому пределу. Это изменение
скорости связано с тем, что при
малых расстояниях между пла-
стинами пламена, распростра-
Рис. 210. Асимптотический
пни зависимости скорости пла-
мени от размера частиц
1 — упорядоченная смесь; 8— не-
упорядочеилая смесь
няющиеся вдоль правой и левой границ слоя с?, влияют друг на друга пу-
тем теплообмена. При увеличении толщины слоя окислителя, т. е. рас-
стояния между пламенами, это взаимодействие уменьшается. Если
232
I - - I _ t _ 1 L
25 50 75 fOO f25
/, кг/см*
Рис. 211. Зависимость скорости горения от давления при различном размере
частиц окислителя для стехиометрической смеси КС1О< с битумом:
1 — d — 0,01 жж; г — d — 0,07; з— 0,2; 4— 1,7 жж
применять в качестве окислителя перекись бария, то образуется твердый
и пористый черный шлак, препятствующий тепловому взаимодействию
пламен, и скорость распространения при увеличении d возрастает, также
стремясь к постоянной величине. Независимость и от d указывает на то,
что скорость определяется в основном условиями вблизи носика выемки.
За это говорят и результаты опытов по влиянию толщины пленок ряда
пластмасс, наносившихся на пластину плексигласа, горевшую в контакте
с перхлоратом калия. Пленка полиэтилена (толщиной 0,1 лим) на 15—
20% увеличивает скорость, а пленка полифтортетраэтилена (фторо-
пласт 4), толщиной 0,06 льм, почти вдвое уменьшает ее. При увеличе-
нии толщины пленки скорость плавно растет (для полиэтилена) или
уменьшается (для фторопласта), выходя на плато тем раньше, чем выше
давление. В последнем случае по достижении некоторой толщины пленки
(тем большей, чем выше давление) горение прекращается.
Распространение горения зависит от химической природы окислите-
ля и горючего. Не все окислители способны к распространению горения.
Так, не поддерживает горения (в контакте с плексигласом) ни один из
изучавшихся нитратов [РЪ(Ж)з)2, Ba(NOs)2, Sr(NOg)2, LiNOal хотя тепло-
вой эффект, рассчитанный на полное превращение, дли них больше, чем
для ВаС3, Не наблюдалось распространения горения в контакте с KCIOi
(р 60 ат) для тех горючих (полиэтилентерефталат, перфоль, галалит,,
фенолформальдегидная смола ФКПМ-15), при термическом разложении
которых в присутствии кислорода образуется большой конденсированный
остаток.
При изучении систем, в которых горючим является металл, были уста-
новлены некоторые особенности горения таких систем. Способность к го-
рению, в отличие от того, что наблюдается для органических горючих,
монотонно убывает по мере уменьшения теплового эффекта взаимодейст-
вия, определяемого теплотой (на моль кислорода) разложения окислителя,
В опытах при насыпной плотности для алюминия была установлена1
233
следующая последовательность (в порядке убывания <?) окислителей:
КС1О4, AhO, PbO2, КМ11О4, ВаОг, Ba (NO3) 2, М11О2, CuO, С02О3, РЬО, КегО3,
5п0, ZnO, Сг2О3. Последние 4 системы не горели при атмосферном дав-
лении, но приобретали эту способность при 20 ат, за исключением систе-
мы с СГ2О3, которая не горела при давлении меньше 50 аг.
Способность к горению уменьшается при увеличении относительной
плотности порошка металла из-за слишком большого теплоотвода из
зоны реакции. Продельная плотность
Рис. 212. Зависимость скорости горе-
ния от размера частиц онислителя
для стехиометрической смеси КСЮ4
с битумом при различных давлениях
(в кг/см1): .
1 — р — 1; 2 — р — з; J — р-5;
4 - р - 10
растет при увеличении давления и за-
висит от диаметра стержня металли-
ческого порошка. При увеличении
диаметра стержня, так же как и дия
органических горючих, наблюдается
уменьшение скорости распростране-
ния до некоторой постоянной вели-
чины. Скорость уменьшается также
при увеличении плотности металли-
ческого порошка, причем ир, также
уменьшаясь, стремится к некоторому
постоянному пределу; она не зависит
(при малых скоростях горении) от
размера частиц. Обе эти особенности
наблюдались для относительно легко-
плавкого металла (AI) и объясняют-
ся образованием на поверхности
стержня пленни расплава.
Зависимость скорости распространения от давления для систем А1 —
КМ11О4 и Al — Ba(NOs)2 была линейной; для системы W — КМпО4 ско-
рость горения до давления ~10 аг быстро растет, от 10 до 60 ат она
остается постоянной.
Величина скорости и вид эакисимости и(р) для разных окислителей
различны (рис. 209); она различается также для разных горючих (при
данном окислителе — KCIO4).
При уменьшении относительной плотности для перхлората калия при
больших давлениях п возрастает по-видимому, потому, что начинает иг-
рать роль конвективная теплопередача от продуктов горения, проникаю-
щих в глубь окислителя. Для перакиси бария и перманганата калия опы-
ты были проведены только при малых плотностях окислителя, так как
при больших р горение не распространялось.
При уменьшении размеров частиц скорость горения растет; однако,
если оба компонента летучи, этот рост невелнн и при повышении давле-
ния, отношение скоростей ик/ан уменьшается. Для смеси KCIO4 — плекси-
глас это соотношение показано на рис. 209 (кривая V); ин при размере
частиц перхлората 10 лк всего- ~ 3 раза больше ик.
Увеличение скорости горения по мере уменьшения размера частиц бу-
дет происходить лишь до тех пор, пока шкрина зоны смешения паров реа-
гентов (~ unap as/D) не станет меньше ширины зоны прогрева
(1пр ~ к/сри ~ ‘/Рп), соответствующей гомогенной смеси паров. Отсюда
следует, что минимальный размер частиц, ниже которого скорость го-
рения перестает от него зависеть, уменьшается с увеличением дав-
ления.
Таким образом, кривая зависимости скорости горения от размера
частиц (рис. *210) для систем с непрерывной границей раздела реагентов
должна иметь S-образную форму, а для обычной неупорядоченной смеси
(при конечном значении диаметра) обрываться при некотором достаточно
большом размере частиц.
234
Это заключение было сделано при изучении горения стехиометриче-
ской смеси перхлората кания с битумом 1 [207]. Определяющим парамет-
ром здесь является размер частиц перхлората, труднее газифицирующего-
ся, чем битум. На рис. 211 показана зависимость и(р) для смесей с раз-
ным размером частиц перхлората. Эта зависимость имеет вид пальмовой
ветви, т. е. относительный рост скорости горения с давлением больше
для тонкодисперсиого перхлората.
В координатах и — d результаты опытов приводятся на рис. 212.
Каждая кривая схематически может быть представлена как совокупность
трех участков: участок 1 от 0 до d, на котором скорость не зависит от
d; участок 2 от d] до d2, на котором скорость уменьшается при увеличе-
нии d, и участок 3 при d > di, на котором скорость вновь не зависит от d.
Согласно теоретическим соображениям, протяженность участка 1
уменьшается при увеличении давления (точнее, скорости горения). Уча-
сток 1 при уменьшении размеров частиц растягивается. На участке 3
скорость горения слабее зависит от давления, чем на участке 2. Поэтому
при малых давлениях отношение скоростей и1/н3 меньше, чем при боль-
ших.
Физический смысл указанных различий состоит в том, что при малых
скоростях горения (малых давлениях), когда спорость газофазных реак-
ций мала, смешение продуктов газификации отдельных компонентов ус-
певает произойти (даже при наличии крупных частиц) раньше, чем
смесь веществ разогреется до температуры быстрой реакции. Поэтому,
вступая в реакцию, газы уже не помнят, каковы были размеры частиц
тех твзрдых компонентов, из которых они образовались. При больших же
скоростях горения скорость химического превращения ограничивается
необходимостью смешения первичных продуктов газификации, длитель-
ность которого тем больше, чем больше были частицы (участок 2).
Представляет интерес возможность количественной оценки того раз-
мера частиц dunn, ниже которого горение смеси идет в кинетическом ре-
жиме [208]. Одно из условий осуществления этого режима заилючается
в том, чтобы взаимная растворимость компонентов при существующих в
подготовительной зоне температурах была достаточно велика, вторым ус-
ловием является достаточно большая скорость взаимной диффузии ком-
понентов в том состоянии, в которое они переходят в подготовительной
зоне.
Каждый из двух твердых компонентов в подготовительной зоне может
либо остаться в твердом состоянии, либо расплавиться, либо газифициро-
ваться. Из возможных шести вариантов полное смешение заведомо воз-
можно в случае газификации обоих компонентов и практически исключе-
но, если одни из компонентов газифицируется, другой же остается в твер-
дом или переходит в жидкое состояние.
Сопоставляя скорость перемещения границы зоны смешения и зоны
прогрева, можно показать, что <2мил по порядку величнны^для того случая,
когда оба компонента превращаются в газы, равен
+ (3.69)
где «м — массовая скорость горения при d <7МИЛ (в г/сж2сек); X — тепло-
проводность (в кал)см • сек • град); с — теплоемкость в кол/з • град); для
обеих этих величии принимаются средние по подготовительной зоне зна-
чения; D — коэффициент диффузии (в сж2/сек); То и Тт— начальная и
1 Сравнение смеси типа бессерного пороха (15% угла, 85% KNOa) повязало, что
она сходна с перхлорат-битумиой смесью в отношении слабого влияния размеров ча-
стиц на скорость горения. При переходе от частиц размером 400 лк к частицам в 10—
20 мк скорость гореплн при 1—51 ат уменьшалась только вдвое.
235
конечная температуры горения; 7'1 — ниясняя температурная граница
зоны смешения.
— для всех газов которые могут явить-
С
ся продуктами газификации различных конденсированных смесей, ме-
няется в сравнительно узких пределах. Кроме того, если не рассматри-
вать зависимость izM(p, Т), которая задается из опыта, то правая часть
уравнения (3.69) не зависит от давления и сравнительно слабо зависит
от выбора в разумных пределах Тт, Л и То. Приняв, например, что темпе-
ратуры Тт и Т{ могут независимо меняться в интервале 1500—3000я К и
600—1000° К, из выражения (3.69) получим следующий интервал значе-
ний:
(3.70)
“и
Данные для смеси перхлората калия с битумом показывают, что при
— 0,2 г1см2сек составляет около 70 мк, что согласуется с уравне-
нием (3.70).
Вариантом осуществления кинетического режима является также тот
случай, когда реакция идет на поверхности частиц одного компонента,
а скорость подвода второго не лимитирует скорость суммарного процесса.
Для этого необходимо, чтобы степень гетерогенности и Тт не были слиш-
ком велики и чтобы Di ^D?, где D[ и Ds — соответственно коэффициенты
диффузии первого (образующего частицы) и второго компонента в про-
дукты реакции. В этом случае для системы твердое вещество — газ ско-
рость горения зависит от размера частиц (и ~ d~'ls) и, следовательно,
гетерогенная система не эквивалентна гомогенной.
В известной степени этим предпосылкам удовлетворяют смеси перхло-
рата калия с вольфрамом и графитом, горение которых изучалось Беляе-
вым и Цыгановым [209]. В этих смесях только окислитель может разла-
гаться с образованием газовой фазы (кислорода), горючие же при темпе-
ратуре горения имеют ничтожную упругость пара. Процесс горения в
этих условиях естественно представить себе как раакцию расплавившего-
ся окислителя или газообразного кислорода на поверхности горючего.
Определялась скорость горения уплотненных зарядов в зависимости
от давления, соотношения компонентов и величины частиц. Смеси с гра-
фитом стехиометрического состава (15% графита) горят с трудом вотли-
чие от смесей с вольфрамом, которые горят на порядок быстрее, легко под-
жигаются и как при избытке, так и при недостатие горючего горят устой-
чиво.
Зависимость ы(р) при изменении давления от 5 до 100 ат у большин-
ства изучавшихся смесей сходная и значительная; для смесей обоих горю-
чих, не слишком далеких от стехиометрических, она выражается степен-
ным законом и = 6pv, причем значения постоянных b и v (изменяющихся
в пределах 0,3—0,7) сложно зависят от соотношения между горючим и
окислителем, размера частиц горючего и др.
Влияние размера частиц горючего позволяют оценить результаты опы-
тов со смесями перхлората и графита (70 :30), в которых размер частиц
графита различался в 50 — 100 раз; скорость горения при мелких части-
цах была больше в 10 раз. Таким образом, в рассматриваемом случае
зависимость u(d) сильнее, чем в системах, в ноторых не только окисли-
тель, но и горючее способны газифицироваться: в смеси КС1О4 — плекси-
глас скорость горения при уменьшения размера частиц в 100 раз увели-
чивалась только в 1,5—2 раза.
Сильное и своеобразное плииние на скорость горения оказывает соот-
ношение компонентов. На рис. 213 по оси абсцисс отложена а — кисло-
236
родный коэффициент смеси, а по оси ординат — отношение скорости го-
рения при данной а к скорости горения при а = 1. Кривая 1 показывает
влияние соотношения компонентов на скорость горении смеси с газифи-
цирующимся горючим — декстрином, кривая 2 — к смеси с графитом (при
малом размера частиц) и кривая 3 — к смеси с вольфрамом. Для обеих
последних смесей при увеличении содержания горючего скорость горения
растет вплоть до очень больших его содержаний — для вольфрама, на-
пример, до 97%. Смесь 95% W и 5% KC1CU (а = 0,14; Tv ~ 2000° К)
Рис. 213. Зависимость us/ua = 1
от а
Рис. 214. Влияние размера частиц нелетучего
компонента на скорость горения смеси газифи-
цирующихся окислителя и горючего (Z — отно-
шение скорости горения тройной смеси к ско-
рости бинарной)
горит со скоростью 20 см/сек. Для смеси с крупными частицами графита
(не приведенными на графике) зависимость гг(а) близка к таковой для
вольфрамовой смеси.
Полученные результаты авторы объясняют следующим образом. Ре-
акция идет на поверхности частиц горючего, и скорость ее w пропорцио-
нальна удельной поверхности S. Эта поверхность а следовательно и ш,
пропорциональна 1/d. Поскольку то и~й-°15 в согласии с резуль-
татами опытов.
Зависимость скорости горения от давления можно объяснить тем, что
скорость реакции связана с концентрацией кислорода у поверхности час-
тиц, которая пропорциональна давлению, или же с количеством кислоро-
да, адсорбированным на этой поверхности. При этих предположениях
w ~ р (или при адсорбции несколько слабее, чем пропорционально); так
как и ~ и?0"5, то и ~ р0,5 или несколько слабее.
Сильное увеличение скорости при большом избытке вольфрама может
быть обусловлено большим увеличением теплопроводности конденсиро-
ваяной фазы; скорость же горения w Л0-5. Кроме того, при разбавлении
горючим возрастает удельная поверхность.
Бахман и Кондрашков {210] рассмотрели также случай горения трой-
ной смеси, состоящей ив двух газифицирующихся компонентов (напри-
мер, КС1О« + битум) и третьего высококалорийного нелетучего компо-
нента, например вольфрама. Устанавливалось влияние размера частиц
этого компонента, которое характеризовалось отношением п/м0 скорости
горения в присутствии третьего компонента к скорости горения в его от-
сутствие.
При достаточно больших размерах частиц u/uq 1 потому, что круп-
ные частицы могут воспламеняться и сгорать только далеко за фронтом
горения продуктов газификации КСЮд и битума и не будут передавать
исходному веществу сколько-нибудь значительную часть энергии сгора-
237
бия. Из-за больших размеров частиц затрата тепла на их прогрев и фа-
зовые превращения до окончания взаимодействия «летучих» компонентов
также будет невелика. В итоге скорость горения будет соответствовать,
скорости объемного тепловыделения при взаимодействии продуктов га-
зификации КСЮ« и битума (Ф| кал/см3сек) при несколько пониженной
температуре по сравнению с бинарной смесью.
Опыт подтвердил это заключение: для смеси КСЮ< (d ~ 10 жк) с би-
тумом (ао = 0,75) при добавке 13,1 % алюминия в виде крупных (190 лея)
частиц u/ио при 1 ат равнялось 0,99, при 10 ат — 0,94 и при 100 ат —
0,96. Если частицы добавки очень мелки, настолько, что они будут вос-
пламеняться и сгорать между поверхностью конденсированной фазы
и фронтом горения бинарной смеси, то взаимодействие компонентов будет
идти в две пространственно разделенные стадии: сначала частицы реа-
гируют с продуктами газификации КСЮч, а затем происходит реакция
между продуктами газификации битума и оставшейся частью продуктов
газификации КСЮ4. Эта реакция по-прежнему остается ведущей, но про-
исходит она теперь уже при иной, более высокой (за счет сгорания час-
тиц третьего компонента) температуре и при ниом составе газа (обед-
ненного окислителем).
При среднем размере частиц, когда зона горения продуктов газифи-
кации и зона горения частиц пространственно не разделяются, скорость
горения будет зависеть как от Ф|, так и от Фг, где Фа — скорость объем-
ного тепловыделения за счет горения частиц. Если снижение температу-
ры газов за счет теплоотдачи частицами будет преобладать над повыше-
нием температуры за счет сгорания частиц ' и суммарная скорость тепло-
выделения будет меньше, чем в отсутствие частиц, то и/«о может стать-
меньше единицы. Кривая и/но — d будет иметь вид, изображенный на
рис. 214. Наличие минимума на кривой h/uq — d было проверено косвен-
ным путем: при постоянном размере частиц повышалось давление опыта;
u/uo при этом уменьшалось, по-видимому, по той причине, что частицы
становились «крупнее» по отношению к толщине зоны горения бинарной
смеси.
Рассматривая зависимость скорости горения бинарной смеси газифици-
рующихся компонентов от размера частиц, мы видели (см. рис. 211), что
в некотором интервале малых размеров скорость постоянна, при даль-
нейшем увеличении размеров частиц она уменьшается, а затем вновь
становится постоянной. Это постоянство, нонвидимому, обусловлено тем,
что ведущей стадией процесса становится контактное распространение-
горения. Однако независимость и от d может быть связана и с другой
стороной явления, а именно с тем, что при достаточно больших размерах
частиц (несколько сот микрон и выше), помимо молекулярной диффу-
зии, начинает играть существенную роль конвективное перемешивание;
в этом случае коэффициенты массо- и теплообмена растут с увеличе-
нием^ [211].
Возникновение турбулентности связано с тем, что исходная конденси-
рованная смесь с беспорядочным расположением и формой частиц пред-
ставляет собой своего рода «замороженную» турбулентность, которая
«оживает» при газификации компонентов. Уровень такой турбулентности
наиболее высок вблизи поверхности конденсированной фазы, т. е. в тон
зоне, которая обычно оказывает наибольшее влияние на -скорость горе-
ния. Основной масштаб турбулентности в данном случае должен быть
пропорционален d. Причинами, которые могут приводить к взаимному
внедрению объемов, заполненных продуктами газификации компонентов,
1 Бинарные смеси окислителя (КСЮ,) и металлов горят значительно медленнее,,
чей соответствующие двойные (окислитель + битум) или тройные (окислитель +
+ битум + металл) смеси; это говорит о том, что Фа мало по сравнению с Ф|.
238
являются прежде всего непараллельность потоков продуктов газифика-
ции и неравенство абсолютных их скоростей. Поверхность конденсиро-
ванной фазы при горении не является плоской — вдоль границ контакта
компонентов образуются клинообразные выемки, которые растут с уве-
личением d. В непосредственной близости к поверхности конденсирован-
ной фазы продукты гафизикации движутся перпендикулярно к ней; по-
этому направление движения газа на поверхности меняется от точки
к точке и происходит столкновение газовых потоков, ведущее к их пе-
ремешиванию.
Из условия сохранения потока массы для каждого из компонентов
v = ри / рг, где р и рг — плотность конденсированного компонента и про-
дуктов его газификации, и — скорость газификации. Средине по времени
значения щ и И2 пропорциональны, но текущие их значения в любой мо-
мент времени могут находиться в произвольном отношении; плотности
до и после газификации также, вообще говоря, не равны. Поэтому v2^
Неравенство абсолютных величии скоростей продуктов газификации
способствует их перемешиванию в тангенциальном (± пи) направлении
и делает возможным конвективное перемешивание в аксиальном (]]иЕ) на-
правлении. В момент времени, когда в данной точке поверхности один
компонент при горении сменяется другим, по потоку, поскольку Vj v2t
должна пойти либо элементарная ударная волна (если скорость стала
больше), либо элементарная волна разрежения (если скорость стала
меньше), которые, отражаясь от неоднородностей плотности, существую-
щих в потоке, и от поверхности конденсированной фазы, породят посте-
пенно затухающие вторичные, третичные и т. д. волны. В результате
поток более быстро текущего газа будет внедряться в объем другого газа,,
причем граница между ними будет реамываться пульсациями давления
и скорости газа, возникающими при прохождении элементарных волн.
Перемешивание газа в аксиальном направлении играет незначительную
роль по сравнению с перемешиванием в тангенциальном направлении,
так как при газификации кубика твердого компонента мы получаем пря-
моугольный объем газа, у которого размеры основания не изменились,
а высота (и путь смешения) в аксиальном направлении увеличилась
в р/рг раз, т. е. на один — три порядка.
Для рассматриваемой турбулентности из параметра с размерностью
длины (d) и параметра с размерностью скорости (v 1 — v2) можно соста-
вить комбинацию, имеющую размерность коэффициента турбулентного'
обмена (в см?/сек):
-^турб^ d(i>i — Уг), (3.71)
при этом для перемешивания в тангенциальном направлении следует
брать (vjt — v2f)t а для перемешивания в аксиальном направлении (у и —
— ^2«) • Учитывая, что tij ~ ~ получаем
-^турб ~ duH • (3.72)
Коэффициент молекулярной диффузии Рмол ~ 1/р не зависит от d, а
обычно пропорциональна р0,Б °'7. Отсюда следует, что роль конвек-
тивного перемешивания возрастает по мере увеличения d и р. Она долж-
на возрастать также и по мере искривления поверхности конденсированной
фазы, поскольку при этом увеличивается угол столкновения струй и по-
скольку путь смешения в тангенциальном направлении много меньше,
чем в аксиальном.
Для качественной проверите роли конвективного перемешивания опре-
делялась высота h ядра факела при горении (р = 1 erj = 20°) цилин-
дрических зарядов смеси перхлората калия с разными размерами частиц
239
(10, 80 и 700 жк) с битумом. Эта высота не только не возрастала, но,
напротив, несколько убывала при переходе от частиц размером 10 .мк
к 700 мк, чтс говорит в пользу возрастания коэффициента массообмена
при увеличении d.
Определение высоты факела при контактном горении порошкообраз- .
ного перхлората калия, запрессованного в толстые цилиндрические обо-
лочки из плексигласа с различным внутренним диаметром (от 0,4 до
1,4 еле), показало, что ~ d, что и должно быть при турбулентном сме-
шении, в то время как для молекулярного смешения должно было бы
выполняться соотношение ~ d2.
Изучалось [212] также горение двойных смесей перхлората кдлия с
алюминием (60:40). Смесь помещали в канал (d = 9,5 лии) с полиро-
ванными стенками, высверленный в плексигласовом стержне. Иницииро-
вание осуществлялось зарядом тетрила (0,36 г), подрывавшегося при по-
мощи электродетонатора, закрепленного в отверстии канала, Профили
зоны реакция на фотоснимках могут быть разделены на три типа; одним
из них является облако со светящейся центральной областью; два дру-
гих отличаются тем, что имеют удлиненную сильно светящуюся зону,
распространяющуюся в глубь заряда; в случае третьего типа ей пред-
шествует предволна — относительно слабо светящаяся зона, сохраняю-
щая постоянную протяженность во время всего процесса вероятно, она
имеется и при втором типе, но свечение слишком слабо, чтобы быть ре-
гистрированным, Фотография на движущемся фильме показывает, что
за зоной свечения следует темная зоца, после которой наблюдается дли-
тельное «размазанное» догорание.
Скорости распространения постоянны, однако при соотношении ком-
понентов в смеси 60:40 иногда наблюдалось резкое увеличение ско-
рости — приблизительно вдвое (от 480—590 до 810—910 м/сек). Малая
скорость соответствует процессу первого типа, большая — одному из двух
остальных; соответственно переход к большой скорости, по-видимому,
связан с образованием предволны.
Размеры частиц перхлората слабо влияют на явление, размеры частиц
алюминия, особенно при переходе от мелких к крупным, приводят к
уменьшению скорости горения; это указывает на то, что сгорание про-
исходит на поверхности частиц алюминия. Наличие темной зоны предпо-
ложительно связано с образованием газообразного AhO и А1, послевол-
на — с образованием твердого AI2O3, когда’ температура цадает ниже кри-
тической.
VII. ВЫГОРАНИЕ ВЗРЫВЧАТЫХ ВЕЩЕСТВ
ПРИ ВЗРЫВНЫХ РАБОТАХ В ШАХТАХ
При взрывных работах в шахтах иногда вместо взрыва заряда наблю-
дается его выгорание. Это очень нежелательное явление по многим при-
чинам. При выгорании ВВ не совершает той работы, которую оно долж-
но выполнить, образуются в повышенных количествах ядовитые газы
(NO и СО), отравляющие воздух шахты. Горение после значительного
промежутка времени может перейти во взрыв, который поражает взрыв-
ника, вернувшегося к забою. Однако к наиболее тяжелым последствиям
1 Допущение, что эта зона является предволной, подтверждается большим уси-
лением свечения при встрече зон, получаемых при инициировании заряда с обоих
торцов, Кроме того, рентгенограммы также показали образование и распространение
зоны повышенной плотности. Свечение в этой зоне может быть связано с разогревом
газовых включений или с локальной химической реакцией.
Z40
может привести выгорание в шахтах, опасных по газу или пыли, вызы-
вая воспламенение метано* или пылевоздушных смесей.
Возможные причины выгорания и факторы, ему благоприятствующие,
многообразны. Существенную роль при возникновении выгорании играет
способность ВВ к горению, которая изучалась в ряде работ путем опреде-
ления различных ее показателей — критического диаметра, критического
давления и критической температуры.
Был определен [194] критический диаметр горения и его зависимость
от плотности для ряда порошкообразных промышленных ВВ на основе
аммиачной селитры. ВВ находилось в,
конической целлофановой или стеклян-
ной гильзе, помещенной в воду, и под-
жигалось с широкого конца в бомбе по-
стоянного давления, Горение проводи-
лось при начальном давлении 100 ат,
поднимавшемся к концу опыта до
120 ат. Результаты опытов (рис. 215)
показывают, что критический диаметр
изучавшихся веществ относительно ве-
лик и лежит между 6 и 13 льм, т. е. зна-
чительно больше, чем для относительно
мало способного к горению троткла
(йиг при 1 ат 21 хи, при 100 ат — 1 .ил);
Содержание в аммоните жидких ни-
троэфиров повышает способность к го-
рению. Критический диаметр всех изу-
чавшихся ВВ (за исключением победи-
та ПУ2) заметно уменьшается при уве-
личении кубической плотности. Такое
влияние является несколько неожидан-
Рис. 215. Зависимость критического
диаметра горения некоторых предо-
хранительных В В от кубической
плотности при 100—110 ат
1 — породный аммонит АП-1; 3—побе-
дит ВП-1 в целлофановой оболочке;
3 — победит ВП-1 в стеклянной оболоч-
ке’ 4 — водоустойчивый аммонит
ПЖВ-20; 5 — аммонит М 8; в — побе-
дит ПУ-2
ным, поскольку скорость горения при
повышении плотности для всех без исключения изучавшихся аммонитов
уменьшается, и скорее следовало бы ожидать соответственного роста кри-
тического 'диаметра.
Малая способность аммонитов к горению, по-видимому, определяется
большим содержанием в них аммиачной селитры, .способность которой к
горению очень низка. Катализаторы, однако, сильно повышают эту спо-
собность как у самой селитры, тдк и у ВВ на ее основе. Так, селитра,
содержащая 7% хлористого натрия, горит уже при 12 ат и диаметре за-
ряда, равном всего 7 л.и, в то время как сама селитра в аналогичных
условиях не горит даже при диаметре 35 лмг и давлении 1000 ат.
Каталитическое действие различных минеральных веществ на горе-
ние аммиачной селитры изучалось Глазковой [213]. В манометрической
бомбе исследовалось горение прессованных шашек, содержавших 10%
примеси, при плотности заряжания 0,2 г/см?. Недостатком метода яв-
ляется то, что горение идет дегрессивно и конец его трудно точно опре-
делить, тем более что давление измерялось медным крешерным цилинд-
риком и падения давления при остывании нельзя было наблюдать. В ка-
честве воспламенителя применялся черный порох в количестве, дававшем
при сгорании максимальное давление 135 ат. Показателем каталитиче-
ской эффективности примеси служило отношение средней массовой ско-
рости горения в ее присутствии к скорости горения селитры без примеси.
Соответствующие данные приведены в табл. 31.
Непосредственное сравнение горения селитры с примесями и без них
затрудняется тем обстоятельством, что из-за разного времени горения
сильно различаются также и давления, которые, естественно, значитель-
но ниже, чем рассчитанные для горения без теплопотерь. Тем не менее
16 К. К, Андреев
241
Таблица 31
Влияние минеральных добавок на горение аммиачной селитры
в манометрической бомбе
Примесь ₽ср» ОЯ1 Ицг % Примесь ₽ср* “Ч ми, в/см’-свк «м> %
Без примеси 187 1 192 J 190 0,521 0,55 J 0,53 100 KiGf^Oi 366 1 360 362 1,36) 1,66 1,47 270
NaCl КС1 355 1 360 J 378 1 291 / 358 335 1,451 1,41 J 1,251 1,30 J -1,43 1,27 270 240 К,СгО4 БаС13 361 298 ) 285 J 320 ) 324 J 292 322 1,38 1,241 1.09J 1,60) 1,55 J 1,17 1,57 220 290
СаСОв * SiOt** Горение затухло
* Лштух. =154 ат-
** Ata-тух. в 145 апг-
из таблицы видно, что, за исключением мела и двуонлси кремния, все
изучавшиеся примеси существенно ускоряют горение аммиачной селит-
ры; сильнее всего влияет хлористый барий, за ним следуют бихромат
калия, хлористый натрий, хлористый калий и хромат калия,
В предохранительных аммонитах содержание щелочных хлоридов
значительно больше, и в этом случае проявляется не только их каталити-
% Nad
Рис, 216, Влияние содержания NaCl на
скорость горения амматола 80 : 20 в мано-
метрической бомбе
1 — воспламенитель 1,5 г черного пороха;
Я — 2,5 а черного пороха
ческое, но и разбавляющее
влияние как инертной примеси.
Влияние содержания хлори-
стого натрия иа горение изуча-
лось Глазковой [214] на приме-
ре амматола (80 : 20) в мано-
метрической бомбе. В одной се-
рии опытов амматол применял-
ся в виде двух шашек (d =
= 11 мм, h = 13—14 мм, об-
щий вес 4 г). Воспламенение
осуществлялось черным поро-
хом (1,5 г), развивавшим давле-
ние 60 ат\ в другой серии при-
менялась одна шашка весом 4 г
(d =: 13 ж, h = 17—18 мм) и
воспламенитель весом 2,5 г
(давление по сгорании 90 ат).
Результаты опытов (рис. 216) показывают, что 1% соли вдвое уве-
личивает скорость горения; при увеличении содержания катализатору до
6,5%' скорость продолжает возрастать; при большем содержании соли
скорость уменьшается и при 33% амматол не загорается даже при уве-
личении веса воспламенителя до 5 г. При оценке этих результатов сле-
дует учитывать, что, поскольку общий вес заряда был постоянным, ко-
личество собственно ВВ по мере разбавления солью уменьшалось; соот-
ветственно уменьшалось и дииленпе, развивающееся при горении. Один
опыт был проведен при 9 %-ном содержания соли, но с соответственно
увеличенным зарядом так, чтобы количество самого амматола составляло
4 г; давление при этом опыте было заметно больше, но скорость горения
не увеличилась.
Бы да поставлена серия опытов для сравнения влияния различных
примесей (9,1%) на горение амматола (80:20) в виде четырехграммо-
вых шашек при воспламенителе, дававшем давление 90 ат.
242
Таблица 32
Влияние минеральных добавок яа горение амматола (80 : 20)
Примесь Гор , am г/сл&ек UM' % Примесь Гер , am u^cp), Um. %
Без 251 2,19 100 2381 3,35 1
примеси 341 (NHibCr.O» 227 248,6 3,88 3,69 169
4,15) 281 , 3,85
LiCl 194 245 • 259,5 2,46 3,30 3,46 158 KjCtQi 207) 214 ‘210,5 3,361 3 31J 3,34 153
NaCl 258 243 3,94 3,69 169 (NH^CaOa 146 1 198 ‘172,0 1,06) 0,91 j 0,98 45
КС1 301 300 294,3 3,601 3,86 • 3,58 164 KNOa 2491 255 ‘ 252,0 4 18) 3,34) 3,76 172
282 3,29 217 2,94
Ва,С1ц 264) 245 J 254,5 3,98) 4,48 i 4,23 196 КСЮ4 256 263 -250,9 3 45 3,07 -3,34 153
247 2,161 267 3,84
NH4G1 267 > 259,0 2,58 h,32 106 258) 2,081
263 2,24 CaFa 272 267,6 2,30 2,07 105
HgCi 213 3,66 273. 1,83
224 222 224,2 2,28 1,88 2,52 115 Уголь 2161 213 J 214,5 2,181 2,24 j 2,22 102
238 2,23 250 1 3,04
KjCrOi 264 11,89 Графит 248 253,0 2,24 2,78 127
261 I 264,3 12,66 112,19 560 261 3,05 .
KgCrgO? 268 2351 237) 236,0 12,01 5,701 5,40 j ‘5,55 254 GaGOa SiOa 2501 215 J 269 1 294 -232,5 281,5 2,691 2,43 j 2,111 2,25 „ 2,56 2,18 118 100
Результаты опытов (табл. 32) показывают, что сильнее всего горение
дмматола ускоряется соединениями шестивалентного хрома, особенно
хроматом калия; значительное и приблизительно одинаковое ускорение
дают хлориды щелочных металлов и хлористый барий; щавелевокислый
аммоний сильно замедляет горение; меньшее по сравнению с хлори-
стым калием влияние перхлората калин может быть связано с тем, что
действующим началом является именно КС1, которого при разложении
перхлората образуется вдвое меньше, чем было добавлено в опыте
с хлоридом.
Сопоставление данных для аммиачной селитры и для амматола пока-
зывает, что тротил ускоряет горение селитры; одиако действие катали-
тических примесей в последнем случае сказывается слабее. Тан, хлори-
стый натрий горение селитры ускоряет в 2,7 разд, амматола — лишь
в 1,7 раз.
Обращает на себя внимание значительный разброс данных параллель-
ных опытов как в отношении рМакс, так и ц, причем отклонении этих ха-
рактеристик не всегда соответствуют друт другу по величине и даже по
знаку; в известной мере разброс может быть связдн с задержкой воспла-
менении и с тем, что оно происходит не одновременно во всех точках
поверхности, тем более что был применен относительно слабый (90 ат)
воспламенитель, Возможно, что это обстоятельство является причиной и
значительного расхождения давлений при горении амматола с КС1 и
NaCl. Вопреки ожиданию в последнем случае максимальное давление на
100 ат меньше.
Каталитическое действие на горение селитры и аммонитов оказывает
и та добавка железных солей органических кислот, которая вводится для
придания им водоустойчивости. Критический диаметр горения аммонитов
на-о желе зне нн ой селитре как содержащих хлористый натрий, так и не
U*
243
содержащих его, вдвое меньше, чем в отсутствие солей железа. По-види-
мому, соли железа повышают способность к горению лишь при относи-
тельно повышенных давлениях - при сравнении двух победитов сильное
различие наблюдалось при 100 ат и отсутствовало при 18 ат. Добавим,
что различие в скоростях горения было весьма небольшим.
Опыты при низких давлениях показали, что некоторые катализа-
торы, например бихромат аммония, проявляют свое действие уже при
атмосферном давлении, другие, например хлориды калия, натрия и бария,
как было установлено А. П. Глазковой,— лишь начиная с умеренно повы-
шенных давлений.
При изучении влияния некоторых комбинированных каталитических добавок,
включающих бихромат и другой катализатор или вещество, самостоятельно не про-
являющее каталитического действия, наблюдалось как усиление, так и ослабление
эффекта. Если принять скорость горения смеси аммиачной селитры с бихроматом ка-
лия (р ~ 1 г!см\ р — 2 ат, dTp = 16 о), равную 0,13 г/см1 сек, за 1W, то замена
1/5 бихромата на хлорид уменьшает скорость до 13, СаСО3, SiO2 н К2СО5, напротив,
увеличивают ее до 4il2, 126 и 173 соответственно. Сами по себе эти три вещества не
сообщают аммиачной селитре способности к торению при низких давлениях, а два
первых даже при высоких давлениях уменьшают эту способность.
Интересно, что горючие вещества, как правило, сильно снижают ка-
талитическое действие бихроматов, вероятно, в результате восстановления
последних [213].
Рис. 217. Влияние состава аммонита на критиче-
ский диаметр горения при атмосферном давле-
нии и насыпной плотности
I — заряды в оболочке ив бумаги, пропитанной рас-
твором антипиренов; 2— в оболочке ив стекла.
Рис. 218. Влияние плотности на
критический диаметр горения
тротила (7) и аммонитов 50: 50
(2) и 60; 40 (3) в стеклянной
трубке
Своеобразно влияние каталитических добавок, не обладающих окислительной
функцией, на горение аммиачной селитры с древесным углем. Как известно, добав-
ка угля сильно повышает способность аммиачной селитры к горению. Смесь
(84,2:15,8) горит уже при атмосферном давлении при диаметре 8,5 лл (и ~
~ 4,5 г/смР-сек); при добавления хлористого натрия (1—9%) способность к горению
утрачивается; при 3 ат н добавлении 1—3% NaCl горение идет, но в отдельных опы-
тах затухает; при добавлении 5 и 6,5% NaCl оно идет устойчиво со скоростью, зна-
чительно превышающей скорость горения смеси без добавок; при содержании
9% NaCl скорость горения состава становится меньше и вновь наблюдаются затуха-
ния; по-видимому, здесь уже сказывается разбавляющее действие NaCl.
В. Г. Хотин определял критический диаметр горения смесей аммиач-
ной селитры с тротилом при атмооферном давлении в стеклянных трубках
и в трубках из бумаги, пропитанной антипиреном (рис. 217). При увели-
чении содержания селитры в смеси критический диаметр растет и при
50% селитры достигает (в стеклянных трубках) 30 лии. Амматол (70 : 30)
не горел даже при диаметре стеклянного стакана 74 мм и при обмотке
его асбестовым шнуром.
w
При увеличении кубической плотности критический диаметр горения
уменьшается как для тротила, так и для аммдтолов (рис. 218). Опыты
при повышенных давлениях были проведены только с амматолом
(70 : 30), чтобы установить влияние на критический диаметр некоторых
примесей, которые вводятся или могут быть введены в аммонит, в пер-
вую очередь древесной муки. Эта добавка вводится дня уменьшения уплог-
няемости ВВ и, как следовало ожидать по аналогии с относительно легко.
горючими дицаммонами, может повышать способность к горению.
Рис. 219. Влияние некоторых добавок
на критический диаметр горения амма-
тола 70: 30 при умереино-повышенных
давлениях (плотность насыпная, обо-
лочка стеклянная)
1 — амматол 70 : 30: г — амматоп 70 : 30 +
+ 10% NaCl; а —5% тротила в составе ам-
матола 70/30 замерены на алюминиевую
пудру; 4 — 4% тротила заменены на древес-
ную муку; s — 4% тротила заменены на ни-
троглицерин
Рис. 220. Влияние некоторых добавок
на зависимость скорости горения амма-
тола 70:30 от давления в стеклянной
оболочке при насыпной плотности
1 — амматол 70 : 30; 2 — амматол 70 : 30 +
+ 10% NaCl; 3 — амматол 70 : 30. в котором
4% тротила заменены на алюминиевую пуд-
ру; 4 — амматол 70 : 30. в котором 4% тро-
тила ваменены на древесную муку; з — ам-
матол 70 : 30, в котором 4% тротила заме-
нены на нитроглицерин
Результаты опытов (рис. 219) подтвердили, что древесная мука (4%)
очень сильно снижает критический диаметр при низких давлениях, так
что смесь оказывается способной к горению даже при 1 ат и не слиш-
ком большом диаметре (19 лл). Однако зависимость ^кр(р) для этой
смеси своеобразна. При ~20 ат критический диаметр проходит через ми-
нимум (8 лии), а затем вплоть до 30 ат растет и при 30 ат лишь немногим
меньше, чем для смеси без муки. Хлористый натрий снижает критиче-
ский диаметр амматола, хотя, по-видимому, не так сильно, как самой
селитры. Его влияние становится значительным лишь при давлениях
выше 30 ат. При 100 ат введение 20% NaCl в состав аммонита, сенсиби-
лизированного тэном, вдвое снизило критический диаметр горения. До-
бавка нитроглицерина (4%) для данного бинарного аммонита не умень-
шает критического диаметра; при 50 ат смесь дает при поджигании
вспышку.
Интересно, что изменение скорости горения от присутствия добавок
(рис. 220) не находится в соответствии с их влиянием на критический
диаметр: меньше всего скорость в присутствии древесной муки и алюми-
ния, которые сильнее всего уменьшают критический диаметр. Не обна-
руживается связи и при сопоставлении с возможной максимальной
теплотой горения, как показывает пример того же алюминия или энер-
гетически инертного хлористого натрия, введенного к тому же в значи-
тельном количестве (10 %).
Изучалось также сравнительное влияние добавок тэна и гексогена
(6—10%) к амматолу (90 : 10) на способность к горению при 60—100 ат.
245
При добавке тэна критический диаметр несколько больше при насыпной
плотности, при увеличении последней способность к горению возрастает,
а различие уменьшается.
Для смесей аммиачной селитры с каменным углем (90 : 10), который,
как известно, сильно способствует горению, было изучено влияние дис-
персности компонентов на способность к горению при атмосферном дав-
лении. Смесь, в которой оба компонента были измельчены тонко, горит
при диаметре (10 лии) приблизительно вдвое меньшем, чем три другие
смеси (оба компонента или одни из них крупночастичные). По-видимому,
при горении данной смеси существенную роль играют реакции, происхо-
дящие уже при температурах ниже температуры плавления аммиачной
селитры.
Одибер и Дельма [215] определили критическую начальную темпера-
туру горения при атмооферном давлении ряда предохранительных ВВ, при-
менявшихся во Франции. Результаты этих опытов показывают, что ско-
рость горения была мала и составляла несколько миллиметров в минуту.
Наличие в составе ВВ нитроглицерина благоприятствует горению. В еще
большей степени ему способствует одновременное присутствие древесной
муки и хлористого натрия в хлористом гризудинамите.
VIII. СОСТАВ ГАЗООБРАЗНЫХ ПРОДУКТОВ ГОРЕНИЯ
Как уже указывалось, состдв газообразных продуктов горения при низ-
ких давлениях для изученных в этом отношении ВВ (преимущественно
нитроэфиры и бездымные пороха) не соответствует равновесному. В част-
ности, большая часть азота получается в виде окиси азота, в то время как
при установлении равновесия практически весь азот должен получаться
в элементдрном виде.
В опытах Сарро и Вьеля [216] для нитроглицерина (в виде гурдинами-
та) и пироксилина при горении под давлением, близким к атмосферному,
получен состав газов, приведенный в табл. 33. Результаты новейших ис-
следований по описанной выше методике над метилнитратом, нитрогли-
колем, .слабо желатинированным нитроглицерином, дигликольдимМ тр а то м,
пироксилином, нитроглицериновым порохом и дндаммоном также приве-
дены в табл. 33. От опыта к опыту данные несколько колеблются; здесь,
в частности, может оказывать влияние догорание газов на воспламени-
тельной проволочке, пОлнота которого может меняться.
В целом, однако, различии между результатами опытов разных авто-
ров, производившихся при не строго одинаковых условиях, показывают
относительно мдлые колебания. Это свидетельствует о том, что реакции
завершающей фазы горения, включающей превращение NO в Na, в этих
условиях опыта протекают со сравнительно малой скоростью и не успе-
вают заметно повлиять на состав продуктов начальных этапов горения.
Об устойчизости данного состава продуктов горения говорит и слабое
влияние ня этот состав начальной температуры, установленное для некото-
рых ВВ.
Из данных табл. 33 видно, что при горении нитроэфиров большая часть
азота, а в некоторых случаях и практически весь ааот, содержится в про-
дуктах горения в виде окиси.
Большая часть углерода содержится в виде окиси углерода. Водород
в основной его части окисляется до воды. Наряду с этим для некоторых
ВВ (метилнитрат, дигликольдннитрат и др.) относительное количество уг-
лерода в гдзах в отличие от азота значительно меньше, чем в ВВ. Это по-
казывает, что наряду с углеродсодержащими газами получается некоторое
количество конденсирующихся при обычной температуре углеродистых
246
Таблица 33
Состав гавообразных продуктов горения (без воды) некоторых ВВ
Вещество Содержание, % Отношение чнола атомов углерода к числу атомов азота в Прняечание
СО, СО СП, NO О, н» Ni
вв про- дуктах горе- ния
Метиниятрат 11,5 33,3 0,4 39,6 0,4 5,5 9,4 1,00 0,77 Среднее иа пя- ти опытов
Нитрогликоль 8,4 39,6 0,4 45,2 — 4,1 2,4 1,00 0,97 Среднее из трех опытов
Д игликольдинит - рат при 18° С 9,4 45,3 2,7 30,1 0,2 10,9 1,5 2,00 1,74 Среднее из де- сяти опытов
То же при 95° G 9,3 45,7 3,1 29,7 о,1 11,3 0,9 2,00 1,84
Дигликольдинят- рат желатины- 9,7 45,5 3,0 29,4 0,1 10,2 2,0 2,02 1,74 Среднее из трех опытов
ревенный (97:3) при 18° С 0,1 9,7 1,6
То же, при 95° G 9,9 44,7 3,0 31,1 2,02 1,68
Нитроглицерин жел атиниров ан - 14,0 36,8 0,15 45,4 0,3 1,5 I,8 1,04 1,04 Среднее из двух опытов
ный(97:3) 1,59
Нитроглицерин в виде гурдинамя* 12,72 35,86 0,27 48,18 — 1,29 По опытам Gap- ро и Вьеля
та
Пироксилин К* 1 18,3 44,7 1,6 23,9 — 5,3 6,4 1,76 Среднее из трех опытов
19,6 18,39 42,2 41,94 1,4 1,28 24,1 24,68 о,2 6,3 7,90 6,3 5,81 2,18 1,72 Среднее из восьми опы- тов по дан- ным другой работы По Gappo и Вьелю
Нитроглицер ино- 16,7 42,3 1,6 28,0 °,4 5,2 6,2 2,35 1,50
вый порох с 28% нитрогли-
Смесь аммиачной селитры с углем (84:16) 20,6 5,8 1,4 30,4 0,3 0,2 • 41,3 0,515 0,246
соединений; из этих соединений для метилнитрата, дигликольдннитрата,
а при пониженных давлениях также и для нитроглицерина обнаружен
фор ма ль дегид.
При более высоких давлениях происходит догордние продуктов непол-
ного сгорания, в первую очередь СО за счет кислорода NO. У нитрогли-
коля это догорание наступает при давлениях между 10 и 14 ат и сопро-
вождается появлением второго пламени. Если при давлении 1 ат содержа-
ние окислов азота в продуктах горении соответствует 0,9 содержания
азота в ВВ, а при 9 ат — 0,7, то при давлении 14—14,5 ат содержание
окиси азота в продуктах горения падает до нуля.
Для других ВВ также установлен факт догорания, но минимальные
значения соответствующих давлений не определялись.
247
Анализ газообразных продуктов горения гремучей ртути при давлении
10 Л4-и, по данным Беляева, приводит к следующему уравнению реакции:
Hg (ONC)2 ж 0,61 NOa + 0,37 СО2 + 0.19 C3N3 +
+ 0,51 N8 + 0.05 CO 1,2 C +1,0 Hg 4- 71,5 ккал/молъ. (3.73)
Если учесть, что из одного моля исходной гремучей ртути разлагают-
ся только 0,35 моля, а остальная дасть проходит зону реакции без раз-
ложения и дает при низких давлениях осадок, то суммарное уравнение
принимает следующий вид:
Hg (ONC)3 Ж 0,65Hg (ONC)a 4- 0,21NOa + 0,13 COa 4-
4- 0,07 CaNa 4-0,18Na 4- 0,02 CO -f-0,42C 4-
4- 0,35 Hg 4- 24 ккал/молъ. (3.74)
Таким образом, гремучая ртуть сгорает (при низких давлениях) осо-
бенно неполно. Неполнота эта проявляется не только в составе газов, но и
в том, что большая часть вещества проходит зону гореиня, не разложив-
шись, в виде пыли. Соответственно расчетная температура горения по-
лучается очень низкой — 750—800° С [217].
Катализирующие добавки сильно влияют на состав газообразных про-
дуктов горения (при атмосферном давлении) смесей на основе аммиач-
Та блица 34
Влияние каталитических добавок (6,5%) на состав продуктов
горения смеси аммиачная селитра—тротил—уголь *
(Теоретическое отношеаие XN : ХС равно 2.4; р =1.4 —1,6 г/с-ч'; температура 100’С)
Добавка Состав газов, объемн. %" X N: X С (экспервиен- тальное}
СО, NO О, СО сн, Н, N,
- 14,9 25,8 0,3 12,3 1,3 0,14 48,8 4.5
NaCl 20 4 12,3 0,5 12,6 1,5 2,6 50,1 3,3
КС] 19,1 13,1 0,3 11,6 3,4 6 1 46,2 3,1
* К амматолу (80 : 20) добавлялось 5% угла.
*• Среднее из двух опытов.
Таблица 35
Влияние каталитических добавок (6,5%) на состав продуктов горения
динамона (84: 16)
(Теоретическое отношение SN : SC равно 1.9)
Добавка Состав газов, объема.% Х№: SC (эксперимен- тальное)
СО, NO О, СО СН. н, N,
20,6 30,4 0,3 5,8 1,4 0,2 41,3 4,1
к3сг04 22,4 0,0 1,2 10,3 1,8 7,8 56,5 3,3
ВаС1в 17,7 0,1 1,2 8,1 1,7 2,3 68,9 5,0
KsCr-jOv 22,2 3,9 0,5 13 6 2,3 10,8 46,8 2,6
NaCl 23,5 8,3 0,6 7,9 1,6 3,2 54,8 3,6
КС1 22,4 24,5 0,5 6,1 1,4 2,8 42,3 3.6
NH4GI 22,7 26,2 0,6 6,7 0,4 0,1 43,4 3,8
Примечание. В таблице приведены средние значения из 2—3опытов. Сжига-
ние производилось в трубках диаметром 15 мм.
248
Таблица 36
Влияние каталитических добавок (6,6%) на состав продуктов горения
динамона (84,2:15,8)
(Теоретическое отношение SN: SC равно 1,94; ₽ 1,3—1,4 d— 25,4 «л)
Добавка Состав газов, объемн.% SN : ВС (эксперим ви- тав ьное)
СО, NO 01 СО СН! н, N,
22,4 14,4 0,3 3,9 0,7 1,2 57,1 4,8
Без примеси 18,5 27,3 0,3 3,5 1,0 1,4 48,0 5,4
27,6 0,1 1,0 2,4 1,4 2,5 65,1 4,2
BaCla 20,0 0,0 6,0» 1,3 1,3 1,0 70,4 —
23,2 0,6 2,6 1,6 1,8 1,4 68,9 5 2
КС1 29,0 1,5 0,4 1,5 4,5 2,6 60,5 3,5
27 5 7,2 0,5 6,3 0,7 1,0 56,9 3,5
NaCl 27,4 3,9 0,1 6,5 1,4 3,2 57,5 3,4
* Повышенное количество кислорода обусловлено, по-видимому, непоглотнвшейся СО,-
ной селитры; в первую очередь это влияние проявляется в уменьшении
содержания окиси азота и в уменьшении отношения SN : SC, приближаю-
щем его к расчетному. Это последнее обстоятельство указывает на то,
что окисление углерода, очень неполное в отсутствие добавок, увеличи-
вается сильнее, чем распад аммиачной селитры (исключение составляет
хлористый барий).
В табл. 34—36 приведены соответствующие данные для ряда смесей.
IX. ТЕМПЕРАТУРА ГОРЕНИЯ
Любые спектроскопические явления, зависящие от температуры газа,
могут быть использованы для ее измерения. Спектроскопическими мето-
дами для измерения этой температуры являются: метод резонансного об-
ращения спектральных линий, принятый как для видимой, так и для ин-
фракрасной областей спектра, метод распределения интенсивности во
вращательных линиях полосатого спектра, двухцветный пирометрический,
абсолютно радиационный метод и метод допплеровского расширения
спектральных линий. Первый из этих методов заключается в следующем.
Если излучение пламени является тепловым, т. е. если различные
участвующие в процессе испускания состояния находятся в термодинами-
ческом равновесии, то температура пламени может быть измерена мето-
дом обращения спектральных линий. При введении в пламя, например,
хлористого натрия при его испарении и диссоциации могут образоваться
атомы натрия, которые цогут возбуждаться и испускать желтый D-дублет
натрия с длиной волны 5890—5896 А. Если поместить позади пламени
черное тело и направить на него через плдмя щель спектроскопа, то при
некоторой температуре черного тела, так называемой температуре обра-
щения, яркость его в спектральной области D-линий будет равна яркости
света, проходящего в этой области через пламя, плюс яркость D-линий
от самого пламени. Только при этой температуре в спектроскоп будет ви-
ден сплошной спектр, в то время кдк при любой другой температуре
линии натрия будут выделяться как яркие или темные на фоне сплош-
ного спектра черного тела в зависимости от того, будет ли температура
этого тела выше или ниже температуры обращения. Если определить
температуру обращения и поглощательную способность пламени, то мож-
но рассчитать его температуру.
249
Температура плдмени может быть изучена путем сопоставления ко-
личества ввергни, излучаемой тонкой металлической проволочкой, нагре-
ваемой электрическим током и находящейся в одном опыте в высоком
вакууме, а в другом — в изучаемом пламени. Очевидно, что проволочка
в пламени излучает при данной силе нагревающего тока столько же энер-
гии, сколько и в вакууме, лишь при условии, если температура проволоч-
ки равна температуре пламени и она не теряет тепла за счет теплопро-
водности, а отдает его, как и вакууме, только за счет излучения. Темпе-
ратура же нагретой проволочки в вакууме может быть измерена опти-
ческим пирометром. \
В принципе описанные и подобные им методы могут быть использо-
ваны и для определения температуры горения взрывчатых веществ, од-
нако соответствующих работ, особенно при низких давлениях, не опуб-
ликовано.
Оптический метод сине-красного отношения был использован Мальце-
вым и др. {218] для получения распределения температуры в газовой
фазе (от 800—1000° С и выше) при горении баллиститного пороха под
давлением 20 — 70 ат. Распределение температуры на осциллограмме име-
ет вид ступеньки, которая по мере увеличения давления приближается
к поверхности.
В ряде работ для измерения температурного профиля горения были
использованы термопары.
Распределение температуры в зоне горения является результатом вы-
деления или поглощения тепла химическими реакциями и передачи его
от горячих слоев более холодным. Поэтому знание профиля температуры
необходимо для количественной характеристики физико-химических про-
цессов при горении.
Одной из возможностей определения указанного профиля является
измерение изменения температуры в ходе горения при помощи тонких
малоииерционных термопар. Детальная разработка и теоретическое обо-
снование этого метода как для газовой, так и для конденсированной ча-
стей зоны горения приводятся в работе Зенина {219].
Термопары применялись для данной цели и раньше, однако методика
их применения содержала существенные ошибки, которые и были обна-
ружены в указанной работе. Непременным условием правильности термо-
парных измерений является применение термопар специальной П-образ-
ной формы. -
Зенин рассмотрел теплообмен термопары с газовой средой, экспери-
ментально определил коэффициенты теплоотдачи применяемых термопар
и рассчитал занижение намеряемой температуры термопарой наиболее
общей П-образ ной формы за счет тепловой инерционности термопары и
теплопотерь в концы термопары посредством теплопроводности и излуче-
ния. Относительцая ошибка вследствие теплопотерь в концы термопары
уменьшается с увеличением скорости горения, она резко возрастает при
уменьшении величины плеча; ошибка за счет тепловой инерции быстро
растет с увеличением толщины термопары; термопары толщиной 7 мк
дают достаточно хорошую запись температуры до скорости горения
0,5—0,8 см/сек.
Экспериментально и расчетно показано, что тепловая инерционность
тонких термопар, покрытых для предотвращения каталитического эффек-
та тонким слоем расплавленной буры, равна тепловой инерционности го-
лых термопар той же толщины.
Это определяется тем, что в данных условиях скорость прогрева тер-
мопар настолько сильно лимитируется теплоподводом из газовой фазы,
что даже у плохих проводников тепла отсутствует изменение темпера-
туры по толщине термопары, т. е. их тепловая инерционность не зависит
от теплопроводности матерцдла.
250
Аналогично был рассмотрен теплообмен термопары с конденсирован-
ной средой. На этой основе наряду с оценкой жесткости П-образной тер-
мопары были выработаны критерии, которым Должны удовлетворять па-
раметры термопар, чтобы обеспечить надежную запись температурного
профиля. Разработанные методы были применены для изучения горения
нитроглицеринового порохд Н при различных давлениях и температурах.
Полученные результаты позволяют рассчитать количества тепла, выде-
ляющегося вследствие химических реакций в конденсированной и газо-
вых фазах, подвод тепла из газовой фазы в конденсированную и эффек-
тивную энергию активации разложения конденсированной фазы.
Возможны два способд использования термопары для измерения в га-
зовой фазе распределения температуры образующегося при устойчивом
горении конденсированного ВВ. При одном из них положение термопары
относительно зоны горения неизменно. Это условие может быть осущест-
влено двумя путями — перемещением термопары, помещенной в некото-
рую точку температурного распределения, со скоростью движения пла-
мени или перемещением горящей поверхности относительно термопары
со скоростью горения.
В этих условиях тепловая инерционность термопары не имеет значе-
ния, следует только принять меры против теплоотдачи в концы термо-
пары (или теплоприхода из концов на термоспай) и выбрдть размеры
термопары значительно меньше ширины зоны переменной температуры.
Этот способ трудно применить при малой толщине области перемен-
ной температуры и большой скорости распространения горения. В этом
случае больше подходит второй способ — термопара покоится относитель-
но исходного конденсированного ВВ, а пламя проходит через нее со ско-
ростью горения. Кроме учета теплоотдачи в концы термоцары и необхо-
димой достаточно малой толщины термопары, должна быть учтена и сде-
лана малой тепловая инерционность термопары.
Распределение температуры при горении в твердом веществе можно
получить только вторым способом. Для правильного измерения распреде-
ления температуры важна форма термоцары. Расположение углом ведет
к большим тепловым потерям в концы, при расположении термопары
вдоль изотермы теряется жесткость ее закрепления; оба эти недостатка
устраняются в П-образной термопаре.
Расчеты, проведенные для конденсированной фазы, показали, что от-
носительная ошибка за счет тепловой инерционности термопары растет
пропорционально толщине термопары и скорости горения. Для термопар,
толщиной 3,5—7 мк она достаточно мала. Ошибки за счет теплоотвода в
концы могут быть уменьшены увеличением величины плеча термопары.
В общем условия измерения являются более жесткими в газовой фазе,
и если они выполнены, то измерения в конденсированной фазе будут так-
же безошибочными 1.
Зенин измерял распределение температуры при горении порохд Н.
Применялись две группы ленточных П-образных термопар: 1) медь —
константан, манганин—константан (толщина 5 и 2 мк), имеющие вы-
сокую термоэлектродвижущую силу и различную теплопроводность, но
не позволяющие измерять температуру выше 1000° С и 2) вольфрам +5%
рения — вольфрам + 20 % рення, вольфрам — молибден + 5 % алюминия
(толщина 7—3,5 лек), термозлектродвижущая сила этих последних тер-
мопар в 4 раза меньше, чем у термопар медь — константан, но, будучи
тугоплавкими, они позволяют измерить температурный профиль пол-
ностью вплоть до максимальной температуры в пламени.
Термопары завлеивались попарно при помощи ацетона в продольно
или поперечно разрезанные шашки пороха; это давало возможность
1 Оценки даются применительно к баллиститному пороху Н.
251
одновременно с определением профиля температуры измерять скорость
горения в данном опыте.
Кривые t(s) для разных давлений ниже атмосферного при начальной
температуре пороха 125° С приведены на рис. 221. В вакууме темпера-
тура газовой фазы не растет, а пульсирует с амплитудой 10—20° С. При
повышении давления максимальная температура увеличивается, и зона
ее приближается к поверхности пороха. Начиная с 200 мм рт. ст. газо-
Рис. 221. Профили температуры при
горении пороха Н в вакууме при
120° С и различном давлении (в мм
1 —р ’ 0;
рт. ст.):
Z — р » 100;
4 — р _ goo
3 — р - 300;
вая фаза начинает влиять на
скорость горении посредством
теплоподвода в конденсирован-
ную фазу; начиная с 300 льм
подвод тепла возможен из зоны
максимальной температуры.
Температура поверхности
увеличивается при повышении
начальной температуры пороха
и слабо растет также с давле-
нием.
Тепловыделение в конденси-
рованной фазе от давления поч-
ти не зависит, тепловыделение
в газовой фазе быстро растет
при повышении давления до
200 мм, когда оно становится
вдвое больше тепловыделения в
конденсированной фазе; при
дальнейшем повышении давле-
ния до 600 мм тепловыделение
растет о тень медленно. Доля
200—400 мм около 5% от общего
теплоприхода от газа составляет при
количества тепла конденсированной фазы. Скорость горения от темпе-
ратуры конденсированной фазы зависит очень слабо.
I, мм
Рис. 222. Профиль температуры (J) и скорость тепловыделения
(2) при горении пороха Я (р = 5 лт-, и - 1,38 мм/сек)
Температурный профиль был определен вольфрдмо-рениевыми термо-
парами и при повышенных давлениях (до 150 ат). На рис. 222, 223, 224
показаны профили температуры и тепловыделение при разных давле-
ниях. За температуру поверхности условно принята та, при которой тем-
пературная кривая меняет свой вид. Тепловыделение рассчитывалось из
уравнения теплопроводности в стационарно распространяющейся тепло-
вой волне с тепловыделением
х£'-срцй'+ф^ = 0’ <3-75)
252
Рис. 223. Профиль температуры (2, 2’) и скорость тепловыделения
(2, для пороха Н при 30 ат
Рис. 224. Профиль температуры (2) и скорость тепловыделения (2) для
пороха Н При 75 ат
первые два члена которого рассчитывались дифференцированием темпе-
ратурной кривой. Первый максимум тепловыделения представляет, оче-
видно (он наблюдался и при горении в вакууме), тепловыделение в кон-
денсированной фазе, т. е. соответствует той ее точке, где температура наи-
большая. Соответственно эта температура и принимается за температуру
поверхности конденсированной фазы. Значения этой температуры при
разных давлениях представлены на рис. 225. Эти температуры выше
(на —40°), чем измеренные [220] термопарой, прижатой к поверхности
пороха.
Кривые распределения температуры в конденсированной фазе не-
сколько отличаются от Михельсоновских, ширина зоны прогрева больше;
это обстоятельство следует приписать в первую очередь увеличению тем-
пературопроводности при повышепки температуры, которое было под-
тверждено и измерено независимыми опытами. Это позволило, в част-
ности, рассчитать запас тепла в прогретом слое при стационарном горе-
нии (табл. 37).
253
Таблица 37
Запас тепла в конденсированной фазе при стационарном горении
пороха Н при различных давлениях
р, am Б 10 20 Б0 75 100
Q, кал/ем? 1.0 0,9 0,72 0,50 0,45 0,40
Для распределения температуры в газовой фазе характерно, что она
не сразу растет до максимального значения, соответствующего полному
сгоранию, а имеет ступеньку в области 1000° С; при 20 ат появляется
пламя с максимальной температурой, которое с ростом давления при-
ближается к поверхности (рис. 226), а температура его растет до неко-
торой постоянной величины (2050°С). Рассчитанные кривые тепловы-
деления в зависимости от температуры при 10 и 20 ат приведены на
рис. 227. По-видимому, они являются следствием нескольких параллельно
и последовательно идущих реакций, что особенно существенно допустить
для дымогазовой золы, где реакции могут протекать и в газе и в дисперги-
рованных частицах.
А. А. Зенин произвел также расчеты порядка реакции и ее энергии
активации для разных зон. Эти расчеты основаны на формальном допу-
щении, что скорость тепловыделения пропорциональна концентрации реа-
гентов в степени, равной порядку реакции
(J7’ Ф \ v
7г~——ж—) (3-76)
Если взять значение Ф при фиксированной температуре, но при различ-
ных давлениях, то по наклону прямой
Ig®KOH = В + vlgp (3.77)
можно получить v. Для дымогазовой фазы при 600 и 700° С v получается
равным 1, а энергия активации составляет 5 ккал/моль. Для начала зоны
пламени по зависимости
1пФ(7)=/(1) (3.78)
Е — 50 ккал/молъ, порядок реакции в ней равен 2.
Полученные данные позволяют рассчитать тепловыделение в различ-
ных зонах в зависимости от давления (рис. 228). Тепловыделение в кон-
денсированной фазе растет при 5—50 ат, насыщаясь при 50—100 ат, воз-
можно, в связи с уменьшением диспергирования. Рост тепловыделения в
дымогазовой и пламенной зонах происходит, по-вмдимому, вследствие уве-
личения полноты реакций: общая полнота горения достигается около
60 аг.
Видно, что зависимость и(р) имеет сложный характер: при давлениях
1—4 ат наблюдается быстрый рост скорости, затем он замедляется, и при
4—8 ат наблюдается почти плато на кривой зависимости скорости горе-
ния от давления. При 8—10 ат наблюдается опять рост скорости, но уже
значительно меньший. Из рис. 229 видно, что дальнейший ход скорости
горения от давления не имеет особенностей.
Своеобразную зависимость и(р) в области даалений до 10 ат Зенин
связывает с тем, что £кф, рассчитанная по зависимости и(Тп), при малых
давлениях мала (6,5 ккал/молъ), а при р > 10 ат значительно больше
26 ккал/молъ. На этом основании можно предположить, что механизм
распада конденсированной фазы различен в обеих областях.
254
Рис. 225. Температура поверхности горя-
щего пороха Н в зависимости от давле
ния
Рис. 226. Изменение расстояния
пламени от поверхности в зависимо-
сти от давления для пороха Н
Рис. 227. Скорость тепловыделения в
пародымогазовой зоне как функция
температуры при горении пороха Н
7 — р ” 10 кг/см2- 2 — р = 20 кг/см*
Рис. 229. Зависимость скорости горе-
ния пороха Н от давления
Рис. 228. Количество тепла, выделяющееся
вследствие химических реакций (порох И):
I — в конденсированной фаве: г— в пароды-
могавовой воне; я — в зоне пламени; t—об-
щее тепнсвыделение при горенки
О V V и и и
Рис. 230. Доля тепла, подводимого из газовой
фазы в конденсированную, по сравнению с теп-
лом, содержащимся в конденсированной фазе
в зависимости от давления
Рис. 231. Типичная осциллограм-
ма горения перхлората аммония
в стабильной области
Поток тепла из газовой фазы в конденсированную увеличивается с
ростом давления вследствие увеличения градиента температуры у поверх-
ности. Доля тепла, подводимого из газовой фазы в конденсированную, по
сравнению с теплом, содержащимся в последней, уменьшается при по-
вышении давления (рис. 230). Одновременно она существенно влияет на
скорость горения, и в этом смысле во всем изученном интервале давлений
ведущими являются как реакции в конденсированной фазе, так и в ды-
могазовой зоне.
Профиль температуры при горении сильно спрессованного (р = 1,93—
— 1,94 с/см3) перхлората аммония в шашках диаметром 7 мм изучался
[221] при помощи П-образных термопар вольфрам — рений 5/20, круглых
(d = 30 и 15 мк) и ленточных толщиной 7 и 3,5 мк.
В интервале давлений 30—150 ат записи имеют обычный характер
(рис. 231). Совершенно иначе выглядят они в интервале давлений 160—
350 ат (рис. 232). В большинстве случаев наблюдается плато температу-
ры в начале температурного профиля (250—300° С) и затем резкие пуль-
сации температуры, достигающие по амплитуде 500° (1000—500° С) с
периодом ~50 мсек.
Фотографии горения, полученные на фоторегистре, также имеют не-
обычный вид — наблюдаются нерегулярные остановки горения и пра-
вильные чередования потемнений и посветлений на пленке, средний пе-
риод которых близок к периоду колебаний температуры.
Распределение скорости выделения тепла при двух давлениях пока-
зано на рис. 233 и 234.
Температура поверхности в области нестабильного горения, измерен-
ная термопарой с грузиками, составила 270° С в интервале давлений
160—350 ат. Максимальная температура горения проходит через макси-
мум при 150 ат (1050°) и уменьшается до 900° С при 300 ат.
Тепловой баланс горения перхлората аммония в устойчивой его об-
ласти представлен на рис. 235. Особенностью его является падение тем-
пературы поверхности с ростом давления, особенно низкой в области не-
стабильного горения, и сильное выделение тепла в конденсированной
фазе.
Обращает на себя внимание также диспергироваине реакционного
слоя, которое можно было наблюдать на стеиле, поставленном рядом с
шашкой, на высоте, в 2—3 раза превышающей высоту шашки. В интер-
вале 40—150 ат наблюдается рост суммарного тепловыделения, что свя-
зано с сильным его ростом в дымогазовой фазе. Однако подвод тепла из
дымогазовой фазы в конденсированную мал и слабо изменяется с давле-
нием, д тепловыделение в конденсированной фазе, составляющее около
95% общего тепла в прогретом слое, с ростом давления падает.
Таким образом, наблюдающийся рост скорости с давлением нельзя
объяснить с помощью обычного теплового механизма газофазного го-
рения.
По кривым скорости выделения тепла в газовой фазе энергия акти-
вации, рассчитанная по формуле
u~p*%TE/RTn, (3.79)
при 30—150 ат составляет 10—12 ккал/молъ.
Полинг и Смит [222] определили путем измерения излучения ппи оп-
ределенных длинах волн в инфракрасной части спектра (3,7 и 7,1 мк)
температуру поверхности при горении смесей перхлората аммония с легко
газифицирующимся горючим (параформальдегид) при низких давлениях
(25—760 мм). При атмосферном давлении температура поверхности не
зависит от скорости горения (последняя изменялась, в частности, путем
256
Рис. 232. Осциллограмма горения перхлората аммония в нестабильной области
(р = 250 ат)
Рис. 233. Профиль температуры (1) и
распределения скорости выделения теп-
ла (2) при горения перхлората аммо-
ния (р = 50 ат)
Рис. 234. Профиль температуры (7) и
распределение скорости выделения теп-
ла (2) при горения перхлората аммония
(р = 100 ат)
Рис. 235. Зависимость суммарного тепло-
выделения (7), тепловыделения в газовой
(2) и конденсированной (2^ фазах, а так-
же теплоприхода ив газовой фазы (4) при
говения перхлората аммония от давления
Рис. 236. Зависимость температуры по-
верхности от давления при горении
смесей перхлората аммония с парафор-
мальдегидом (в %) :
/ — 10; г —12,5; 3 —24.3. Длина волны
ппи определении температуры составляет
7,1 мк
17 К. к. Андреев
изменения содержания горючего). Отсюда было сделано заключение, что
на горящей поверхности осуществляется термодинамическое равновесие
между твердым перхлоратом аммония и продуктами его диссоциации - -
газообразным аммиаком и хлорной кислотой. При справедливости этого
Заключения температура поверхности должна была зависеть от давления,
что и было подтверждено экспериментально (рис. 236).
Температура поверхности определялась для смесей, содержащих 10—
24,3% горючего в интервале давлений 760 — 25 мм. При данном давлении
содержание горючего слабо влияет на температуру поверхности, хотя ско-
рость горения при изменении содержания горючего меняется сильно. За-
висимость 1g р — яг- выражается прямой линией, причем из тангенса
* д
угла цаклона тепловой эффект процесса получается равным 57 ккал/моль,
что практически совпадает с теплотой диссоциации твердого перхлората
аммония на аммиак и хлорную кислоту (56 ккал/молъ). Применение го-
рючих, газифицирующихся труднее, чем параформальдегид (например,
полистирола), дает более высокие температуры поверхности, но наклон
1
прямой 1g р — у не меняется. Возможно, что это повышение температуры
обусловлено тем, что на поверхности наряду с перхлоратом находятся
также частицы горючего или продукты его распада, имеющие более вы-
сокую температуру. Впрочем, высокие температуры поверхности наблю-
дались лишь в том случае, если горючее трудно газифицируется или со-
держание его велико. Чтобы обеспечить возможность горения смеси при
низких давлениях, приходится добавлять относительно много горючего,
например при 25 лсм около 50% от стехиометрически необходимого.
Независимость скорости горения от температуры поверхности пока-
зывает, что реакция, идущая на поверхности, является равновесной и не
ведущей, а ведомой экзотермическими газофазными реакциями.
Было также при помощи тонких термопар измерено распределение
температуры в твердой и газовой фазах, показавшее изменение темпера-
турного градиента на поверхности и медленные вторичные реакции в га-
зовой фазе. Эти определения были сделаны также для смесей, содержа-
щих катализатор (3% хромита меди) и горящих вдвое быстрее, чем в
отсутствие катализатора. Температуры поверхности были при этом прак-
тически одинаковы, градиент же температуры в газовой фазе в присут-
ствии катализатора больше, что опять-таки подтверждает отсутствие ве-
дущей роли процессов, идущих на поверхности. С этим согласуется также
тот факт, что предварительный подогрев перхлората аммония до 350° С
не влияет на температуру поверхности.
Однако описанная ситуация реализуется лишь при низких давлениях;
при повышенных давлениях зона высокой температуры приближается к
поверхности и концентрация NH3 и HCIO4 стремится к нулю.
Возможно, что с этим связано резкое уменьшение зависимости ско-
рости горения от давления, наблюдающееся около 70 ат для слабых сме-
сей горючих с перхлоратом аммония.
Была сделана попытка [125] измерить температуру горения нитрогли-
коля путем введения в жидкость через дно трубки серебряной и медной
проволочек толщиной 0,25 мм с заостренными концами. В двух опытах
серебряная проволочка оплавилась, а медная нет. Таким образом, кдза-
лось возможным заключить, что температура горения нитрогликоля ле-
жит выше 960° и ниже 1080° С. Однако не было исключено, что разогрев
и оплавление серебряной проволочки обусловливаются в известной мере
идущей на ее поверхности каталитической реакцией догорания промежу-
точных продуктов горения. Ца это указывал, в частности, тот факт, что
при зажигании платиновой или нихромовой проволочкой последняя часто
продолжала светиться долгое время после выключения тока.
258
Для проверки этого предположения опыт с нитрогликолем был повто-
рен, причем, во-первых, температура иитрогликоля была выше (75° С) и,
во-вторых, кроме серебриной и медной проволочек, в нитрогликоль была
введена платиновая проволочка. Можно было ожидать (волн температура
горения нитрогликоля при 20° С лежит ближе к 1080°, чем к 960б), что
при повышении начальной температуры нитрогликоля на 55° соответст-
венно повысится температура горения и оплавится также медная прово-
лочка. Однако по-прежнему оплавления медной проволочки не произошло.
В то же время оплавилась (и значительно сильнее, чем серебряная) пла-
тиновая проволочка. Таким образом, при этом методе ориентировочной
оценки температуры горения (а таиже при измерении температуры горе-
ния термопарами из соответствующих металлов) надежным является
лишь отрицательный результат, т. е. отсутствие оплавления. Кроме того,
нужно иметь в виду, что при описанной постановке опыта температура
проволочки вследствие хорошей теплопроводности металла не достигает
температуры газов. Чтобы уменьшить влияние теплопроводности в одном
из опытов с нитрогликолем была применена медная проволочка в виде
горизонтальной спирали. В этом случае проволочка оплавилась, т. е. тем-
пература была не ниже 1100° С.
Опыты с желатинированным нитроглицерином (97: 3) при прямых
(серебряной и медной) проволочках дали оплавление обеих проволочек.
Таким образом, температура горения желатинировдияого нитроглицерина
выше температуры горения нитрогликоля. При горении тротила обе проц-
вел очки остались неон л явленными.
Измерение температуры горения гремучей ртути в непосредственной
близости от конденсированной фазы было проведено Беляевым следую-
щим образом. Опыты производились при иязком давлении, когда видимое
пламя отсутствует; температура измерялась тонкой (0,05 мм) медно-коя-
стантдновой термопарой, спай которой перед началом горения касался по-
верхности ВВ. Измеренная таким образом температура очень низка и со-
ставляла около 500° С.
По ориентировочной оценке А. Ф. Беляева температуры горения не-
которых других изученных им инициирующих ВВ также относительно
низки, хотя и выше температуры горения гремучей ртути.
Температуры, измеренные как по оплавлению проволочек, так и тер-
мопарами, нельзя считать достаточно надежными, поскольку они могут
быть заниженными вследствие теплопотерь и завышенными вследствие
каталитического действия металла. Если теплопотери можно в основной
их части устранить, например применением большего диаметра трубки и
неподвижной по отношению к проволочке зоны горения, то надежно
исключкть возможность катализа труднее. В условиях опытов с нитро-
эфирами преобладающее влияние имеют, по-виднмому, теплопотери, так
как измеренные температуры нитрогликоля и нитроклетчатки значитель-
но ниже рассчитанных по уравнению горения.
Таким образом, температуры горения нитроэфиров при атмосферном
давлении, измеренные по оплавлеияю проволочек, гораздо ниже расчет-
ных температур детонации в соответствии с тем, что горение в указанных
условиях является менее полным, чем при детонации.
Температуры горения изученных инициирующих ВВ значительно
ниже температур горения нитроэфиров.
Вследствие теплопотерь и возможного катализа при горении измерен-
ные температуры следует считать менее надежными, чем расчетные.
Следует, впрочем, учитывать также возможность искажения и при рас-
четном определении температуры горения, если имеют место догорание
или вторичные реакции во время охлаждения продуктов горения.
17»
ГЛАВА ЧЕТВЕРТАЯ
ТЕОРИЯ СТАЦИОНАРНОГО ГОРЕНИЯ В В
I. МЕХАНИЗМ ГОРЕНИЯ
Химическое превращение любого ВВ до продуктов, соответствующих
термодинамическому равновесию, является экзотермическим. Если такое
экзотермическое превращение протекает без потерь энергии в окружаю-
щую среду, то температура системы должна повышаться. Поскольку же
скорость реакции быстро возрастает с температурой, то химическое пре-
вращение ВВ в указанных условиях является самоускоряющимся. В ре-
альных условиях, когда имеют место теплопотери в окружающую среду,
условием самоускорения является нагрев ВВ до некоторой критической
температуры, начиная с которой теплоприход за счет теплоты идущей
реакции превышает теплопотери. Превосходство теплоприхода над тепло-
потерями сохраняется и при всех более высоких температурах, которые
достигаются во ВВ в результате саморазогрева. В этом и заключается
сущность теории теплового взрыва [2, 223].
Критическая температура представляет собой ту минимальную тем-
пературу, при разогреве до которой реакция самопроизвольно развивает-
ся до самовоспламенения. Это развитие, поскольку оно начинается при
указанной относительно низкой температуре, протекает сравнительно
медленно, и самовоспламенение наступает с заметной задержкой;
В теории теплового взрыва рассматривается тот случай, когда неко-
торый объем ВВ равномерно цагрет внешним источником тепла и проис-
ходит теплообмен между этим объемом и окружающей средой. Тепло при-
ход и теплоотвод находятся в устойчивом равновесии, если теплоотвод
быстрее растет с температурой, чем теплоприход, и, наоборот, равновесие
является неустойчивым, если теплоприход растет быстрее, чем теплоот-
вод. В этом последнем случае скорость реакции возрастает до очень боль-
ших значений, соответствующих максимальной достигаемой при реакции
температуре.
Аналогичное явление можно представить себе и для того случая, когда
нагревается не все ВВ, а лишь некоторый поверхностный слой его.
Допустим Для простоты, что ВВ имеет форму цилиндра, что потерь
тепла в радиальном направлении нет и нагреву подвергается поверхност-
ный слой ВВ с торца цилиндра. В этом слое под влиянием нагрева идет
реакция и выделяется тепло. С другой стороны, тепло от слоя отдается
в глубь вещества, соседнему «холодному» слою. Очевидно, что есть и в
этих условиях некоторая температура, при которой теплоприход компен-
сирует теплоотвод. Начиная с этой температуры, в слое происходит локаль-
ный «тепловой взрыв» — быстрая химическая реакция, подобная гомоген-
ному тепловому варыву. ____
При стационарном горении мы имеем внешне сходное явление:
ВВ, нагреваясь, также достигает критической температуры. СуществеМИ
260
разница заключается, однако, в том, что нагрев производится не внешним
источником тепла определенной температуры, значительно более низкой,
чем температура горения, который прекращает поэтому свое действие пе
достижении критической температуры, но, как правило, непрерывно про-
должается и после достижения этой температуры. Таким образом, понятие
критической температуры в этих условиях почти полностью утрачивает
свой обычный смысл, ибо и после ее достижения разогрев слоя продолжа-
ется в основном извне продуктами реакции предыдущего слоя.
Химическая реакция при быстром внешнем разогреве пе успевает в
заметной мере пройти при критической температуре и в главной своей
части протекает вблизи максимальной температуры, равной — (теплоте
, р
реакции, деленной на среднюю теплоемкость продуктов ее).
। По взрывчатому веществу при его горении распространяется тепловая
| волна, прохождение которой вызывает химическую реакцию. Тепловыде-
i леияе этой реакции, в свою очередь, поддерживает постоянство распреде-
ления температуры в пространстве и постоянную скорость распростране-
ния тепловой волны. Холодный слой, вступающий в эону реакции, полу-
чает тепло от предыдущего слоя, отдает последующему и разогревается
за счет тепла идущей реакции. Температура слоя при этом растет от
начальной до максимальной температуры горения.
Основными факторами, определяющими скорость горения, являются,
с одной стороны, температура, достигаемая при реакции, и скорость реак-
ция при этой температуре, а с другой стороны,— скорость теплообмена
между продуктами реакции и непрореагировавшим веществом.
Скорость теплообмена определяется также помимо механизма передачи
тепла (теплопроводность, конвекция, излучение) и расстояния эоны мак-
симальной температуры от слоя, вступающего в реакцию, величиной по-
верхности контакта продуктов горения и непрореагировавшего вещества.
При горении цилиндрического заряда по торцу минимальная площадь
этой поверхности равна поперечному сечению цилиндра, однако она мо-
жет становиться значительно больше, например, если газообразные про-
дукты горения проникают в глубь порошкообразного вещества, если по-
верхность перестает быть плоской и приобретает некоторый макро- или
микрорельеф, если частицы твердого ими капли жидкого ВВ попадают в
струю газов и уносятся ею и т. д.
Помимо этого, если газообразные продукты горения движутся относи-
тельно несгоревшего вещества, то это движение также ускоряет горение,
особенно если оно сопровождается турбулизацией на поверхности их кон-
такта, резко увеличивающей разогрев несгоревшего вещества продуктами
горения.
Во всех Этих случаях скорость горения, если оно идет, становится боль-
ше, чем при плоском фронте, так как теплота горения сгоревшей части
вещества полнее используется для возбуждения химической реакции в
последующих слоях ВВ. Очевидно, одиако, что скорость теплообмена не
должна быть и чрезмерно большой, ибо в этом случае температура, до
которой нагревается вещество, вступающее в реакцию, станет столь низ-
кой, что скорость реакции резко снизится и не сможет компенсировать
неизбежных теплопотерь в окружающую среду. Если встряхнуть трубку
с горящим нитрогликолем, то горение затухает именно по этой причине.
Критерием скорости реакции при ее равномерном протекании во всем
объеме заряда ВВ может служить температура вспышки. Однако темпе-
ратура «вспышки» — температура равенства теплоприхода и теплоотво-
да — применительно к условиям горения, очевидно, должна быть выше,
чем при гомогенном тепловом взрыве по двум причинам. Вещество здесь
подвергается нагреву только с одной стороны и одновремейно идет те-
плоотдача в глубь холодного вещества, в то время как при возбуждении
26t
гомогенного теплового взрыва весь объем ВВ окружен средой повышенной
температуры, G другой стороны, при гомогенном процессе разогрев обыч-
но идет медленно, и скорость реакции возрастает не только вследствие повы-
шения температуры, но и за счет изотермического самоускорения реакции.
То, что температура вспышки непостоянна по самому своему опреде-
лению, ясно без пояснений. Условность температуры вспышки как крите-
рия скорости реакции при горении определяется также различием меха-
низма вспышки и горения, рассмотренным выше. Однако в известных
условиях можно, по-видимому, рассматривать процесс горения как непре-
рывный ряд вспышек последовательных слоев ВВ, прогреваемых продук-
тами горения до некоторой критической температуры, при которой про-
исходит скачкообразное изменение состояния ВВ, в результате чего дан-
ный слой перестает передавать тепло нижележащим или по крайней мере
эта передача сильно уменьшается. Так, например, обстоит дело, если ВВ
способно превращаться в пар, теплопередача от которого гораздо слабее,
чем передача тепла по жидкости или если ВВ диспергируется, превраща-
ясь во взвесь частиц в газообразных продуктах превращения. Можно пред-
ставить себе указанное изменение состояния ВВ и как следствие быстрого
развития реакции, подобного вспышке, в результате химического самоуско-
рения, которому может сопутствовать и повышение тепловыделения. В этих
условиях скорость горения можно рассматривать как скорость прогрева
вещества от его начальной температуры до критической температуры.
Именно так подходили к выводу выражении для скорости горения
Михельсон и Малляр и Ле-1Пателье, рассматривавшие этот случай как
общий механизм процесса горения [224].
П. ГИПОТЕЗА МИХЕЛЬСОНА
И МАЛЛЯРА - ЛЕ-ШАТЕЛЬЕ
Если продукты горении, имеющие температуру Гг, нагревают слой газа
{или взрывчатого вещества1), имеющего температуру То, до температуры
вспышки Тк, то, приравнивая количество тепла
= (4.1)
передаваемое продуктами горении несгоревшему газу (ВВ), количеству
тепла, получаемому им в 1 сек.
'92~исй(Тк — То), (4.2)
<х(Тг-Т0) = «сб(Тк-Т0), (4.3)
приходим к соотношению
_ а(Тг-Т„)
Ц~с5(Тк-Г0) ’
(4.4)
где а — коэффициент теплопередачи; с — объемная теплоемкость; б —
плотность газа (вещества),
Так как
^-ro = |.
где Cj—удельная теплоемкость продуктов горения, то
ctQ
(4.5)
и сС15(Тк-Т0) *
(4.6)
1 Гипотеза Михельсона и Малллра — Ле-Ш ателье была развита для газов, но она
по идее своей приложима без существенных изменений и к горению конденсирован-
ных взрывчатых систем.
Применительно к конденсированным ВВ эта формула содержит вависи-
мость скорости горения от начальной температуры, которая имеет вид
“ = т ~т~
ИЛИ
1 = ^(ТК-ТО). (4.8)
Она дает также зависимость линейной скорости горения от кубиче-
ской плотности:
w6 = const, (4.9)
Михельсон и Малляр и Ле-Шателье не рассматривали зависимости
скорости горения от давления. Увеличение скорости горения с давлением,
исходя из позиций их гипотезы, очевидно, следует истолковать как след-
ствие увеличения с давлением количества тепла, передаваемого продук-
тами горения взрывчатому веществу. По-видимому, под влиянием экспе-
риментальной зависимости
и—Л-гВр,
установленной для бездымных порохов, Мюраур [225] принимал, что
теплопередача при горении осуществляется бомбардировкой поверхности
ВВ молекулами прилегающего к ней газового слоя, имеющего некоторую
достоянную, не зависящую от давления температуру. Количество пере-
данного тепла при этом пропорционально числу ударов молекул газа, т. е.
его давлению. Допускалось также, что передача тепла осуществляется не
только ударами молекул, но иными путями, не зависящими прямо от дав-
ления, например излучением от газов, теплопроводностью по конденсиро-
ванной фазе. Этим объяснялось наличие в выражении и(р) члена Л, не
зависящего от давления.
Однако в настоящее время сама зависимость
и = Л+Др
утратила тот универсальный характер, который ей в свое время приписы-
вали; допущение о независимости от давления температуры газового слоя,
прилегающего к поверхности конденсированной фазы, также нельзя счи-
тать обоснованным. Поэтому эта сторона явления - влияние давлении на
ско1рость горения — количественно не объясняется гипотезой Михельсона
и Ле-1Пателье, хотя увеличение теплопередачи при повышении плотности
газа, контактирующего с поверхностью конденсированной фазы, качест-
венно является довольно естественным.
Зависимость и(Т), вытекающая из рассматриваемой гипотезы, во мно-
гих случаях согласуется с экспериментальной: обратная величина скоро-
сти горения линейно уменьшается с начальной температурой, как это ус-
тановлено для бездымных порохов и ряда ВВ. При этом значения Тк
близки к температурам вспышки, но выше их, как это и должно быть.
При повышении давления Тщ возрастает, что также можно объяснить тем,
что при большей скорости горения в меньшей степени успевает развиться
химическое самоускорение реакции.
Далее, во многих случаях массовая скорость горения порошкообраз-
ных ВВ не зависит, в согласии с гипотезой, от относительной плотности
порошка; там же, где наблюдаются отклонения от этой закономерности,
они, как правило, могут б_ыть объяснены наличием экзотермической реак-
ции в конденсированной фазе, тепловыми потерями, передачей тепла по
стенкам трубки и т. п.
Гипотеза Михельсона и Малляра — Ле-Шателье была первой количест-
венной гипотезой, предложенной для объяснения явления горания. Она
263
сыграла большую роль в познании горения и не утратила в известной
мере своего значения и в настоящее времи,
Одиако согласие выводов гипотезы с опытом не является полным да и
не может быть таковым. Так, хотя -в целом для однотипных по свойствам
(например летучих) ВВ наблюдается известное соответствие между ско-
ростью горения и температурой вспышки (нитросоединения ароматическо-
го ряда, например, горят медленнее, чем нитроэфиры) и между темпера-
турой горения и его скоростью (нитроэфиры с большой теплотой горения
вроде нитроглицерина горят быстрее, чем нитроэфиры с малой теплотой
горения вроде дигликольдинитрата), даже в пределах этих классов соеди-
нений обнаруживается слишком много отклонений от этого соответствия.
В частности, нитроглицерин горит в 4 раза быстрее, чем нитрогликоль,
хотя температуры вспышки и теплоты их горения близки; тэн горит в
6 раз медленнее твердого нитроглицерина. Еще больше отклонений наблю-
дается для нелетучих быстрогорящих ВВ, например, тринитрорезорцинат
свинца горит во много раз быстрее гремучей ртути, хотя температура его
вспышки выше, чем гремучей ртути, а расчетная теплота горения (на
грамм-атом) значительно меньше. Отсюда следует, что температура
вспышки не является надежным критерием скорости химического пре-
вращения при горении. Это и не удивительно: вспышка конденсированных
ВВ при всей схематической простоте представляет собой сложный процесс
взаимодействия явлений превращения, протекающих как в конденсирован-
ной, так и в газовой фазе, сильно зависящий от относительно низкотемпе-
ратурных и медленно развивающихся реакций химического превращения
ВВ. Поэтому она мало пригодна для характеристики кинетики превраще-
ния, протекающего при горении, и приходится удивляться скорее тому, что
соответствие между скоростью горения и температурой вспышки все же
в ряде скучаев наблюдается.
Наряду с этим даже, если бы температура вспышки была определена
для тех малых времен задержки, которые соответствуют условиям горе-
ния, представление о мгновенности реакции по достижении некоторой
критической температуры, лежащее в основе гклотезы Малляра — Ле-Ша-
телье, неправильно в принципе. Сзюрость реакции п при горении зависит
от температуры и концентрации и эти зависимости должны учитываться
теорией горения.
* Мы опускаем рассмотрение ряда последующих работ [226—228] по го-
рению бездымных порохов, исходивших из неправильного допущения, что
конечные продукты превращения при горении непосредственно граничат
с поверхностью пороха. При горения порохов конечная температура газов
на 1000—3000 °C выше температуры конденсированной фазы, и если бы
даже имел место непосредственный контакт этих газов с поверхностью
твердого пороха, то наряду с небольшим числом активирующих ударов
подавляющая часть энергии газов передавалась бы пороху в виде тепла
порциями, меньшими энергии активации. Соответствующий разогрев по-
верхностных слоев пороха в большей мере определял бы реакцию, чем
непосредственная активация его частиц. Кроме того, при указанной рез-
вости температур тепловой поток от газа к пороху был бы нереально велик
п превосходил скорость выделения тепла реакцией горения.
Следует полагать, далее, что (как вытекает из многих наблюдений)
при любом давлении, в частности и при давлениях, измеряемых тысячами
атмосфер, реакция химического превращения пороха, или, точнее, основ-
ных составляющих его нитроэфиров — нитроклетчатки и нитроглицери-
на,— в условиях горения протекает не в одну ступень, а по крайней мере
в две, а еще более вероятно в большее число Ступеней,
Этот вывод вытекает из результатов экспериментального изучения про-
цесса горения, особенно при низких давлениях. Теоретически ступенчатое
протекание реакции не является неизбежным; по-влдимому, оно является
264
кинетически более выгодным, т. е. каждая из ступеней требует меньшей
энергии активации, чем реакция полного распада, Поэтому при медленном
нагреве протекает практически только та реакция, энергия активации
которой наименьшая; затем продукты первичного распада подобным же
образом, г, е. по пути, связанному с наименьшей затратой энергии на акти-
вацию, реагируют между собой и т. д. до предписываемого термодинами-
кой конца. Для некоторых нитроэфиров идентифицированы даже вероят-
ные схемы отдельных ступеней.
Следует иметь в виду при Этом, что первая ступень распада является
либо эндотермической, либо слабо экзотермической. Поэтому допущение
при горении непосредственного контакта продуктов реакции полного пре-
вращения, имеющих максимальную температуру горения, с поверхностью
конденсированной фазы совершенно не соответствует действительности.
Зона максимальной температуры отделена от поверхности конденсирован-
ной фазы более или менее толстым слоем газов, в котором происходят
прогрев и химическое превращение. Передача энергии конденсированной
фазе происходит в простейшем случае путем теплопроводности черее ука-
занный слой газов. При этом температура поверхностного слоя газа, при-
мыкающего к поверхности конденсированной фазы, лишь немного (на
несколько градусов) может превышать температуру поверхности, так что
непосредственная активация ударами горячих частиц никакой реальной
роли играть не может. Поэтому все расчеты гипотез, основанных на допу-
щении непосредственного контакта ВВ с конечными продуктами реакции,
ве имеют реальной физической основы, не соответствуют физической кар-
тине горения реальных ВВ. Согласие выводов из этих гипотее с фактами
(тогда, когда оно имеет место), не может поэтому служить свидетельством
справедливости исходной гипотезы, которая опровергается прямым путем.
Все эти обстоятельства делали необходимым развитие теории горения
для общего случая на основе современных представлений о механизме хи-
мических реакций и других физических процессов, в совокупности своей
составляющих процесс горения. Теория должна дать физически правиль-
ную картину горения, т. е. установить последовательность химических ре-
акций при горении, причин, вызывающих эти реакции, пространственное
распределение реакций, распределение веществ (исходного ВВ и продук-
тов его превращения) и температуры по обе стороны фронта горения.
При этом желательной была возможность расчета абсолютной величи-
ны скорости горения, исходя из кинетических констант реакции и других
свойств ВВ и продуктов его превращения, и зависимостей скорости горе-
ния от давления и от температуры. Естественное требование к теории со-
стоит также в том, чтобы предсказания, сделанные на ее основе, согласо-
вались с результатами опыта и позволяли предвидеть последние.
III. ТЕОРИЯ ГОРЕНИЯ ЗЕЛЬДОВИЧА - БЕЛЯЕВА
Развитие теории горения конденсированных ВВ было осуществлено со-
ветскими исследователями, в первую очередь Я. Б, Зельдовичем и
А. Ф. Беляевым.
В 1938 г, Зельдовичем и Франк-Каменецким [229] была разработана
теория стационарного горения газов. Были сформулированы дифференци-
альные уравнения процесса и количественно рассмотрено течение хими-
ческой реакции в условиях переменной температуры, меняющейся как
вследствие выделения тепла реакцией, так и за счет притока тепла от ра-
нее прореагировавших слоев и отдачи его последующим слоим; одновре-
менно необходимо было учитывать перемещение вещества путем диффу-
зии продуктов реакции в направлении распространения горения и исход-
ного вещества в противоположном налравлелии.
265
Л ш
При выводе выражений для скорости горении и ее зависимостей ar
давления и от начальной температуры было принято допущение, что Ят-
мическая реакция при горении практически целиком протекает при тем-
пературе, близкой к максимальной температуре горения. Это допущение
справедливо для наиболее важного, типичного для химической кинетики
случая сильной зависимости скорости реакции от темперетуры, что имеет
место при Е RT.
$ ч Тогда же в 1938 г. Беляев обратим внимание на возможность приложе-
ния теории горения газов к горению конденсированных ВВ. Многие ВВ
обладают значительной летучестью, и
при умеренных темперетурах, когда
скорость реакции во ВВ еще мала, ско-
рость испарения может быть значитель-
ной. Это определяется тем, что теплота
испарения гораздо меньше, чем энергия
активации, я соответственно вероят-
ность испарения молекулы ВВ иа по-
верхности гораздо больше, чем вероят-
ность ее распада ^В .результате этого
при быстром прогреве очередного тон-
кого слоя ВВ в процессе горения снача-
ла происходит испарение ВВ, а химиче-
ские реакции протекают целиком в па-
рах. Для тех условий, в которых это
утверждение справедливо (малое внеш-
нее давление, высокая летучесть ВВ в
сочетании с малой константой скорости
реакции в кондеисироваииой фазе), го-
рение ВВ можно рассматривать просто
как горение газа (шара) с той лишь
разницей, что этот газ образуется в про-
цессе самого горении путем испарения
конденсированной фазы.
Таким образом, летучесть оказалась тем свойством ВВ, которое истори-
чески сыграло весьма важную роль в развитии теории горения.
Рассмотрим стационарное горение летучего ВВ в трубке постоянного
сечения. Распространяющийся фронт горения представляет собой тонкую
зону ЕЕСС (рис. 237), отделяющую холодное исходное ВВ от горячих
продуктов реакции, в которых вся химическая энергия перешла в тепло-
вую. Ведущей стадией процесса в этом случае является распространение
экзотермической химнческой^доакции в парах^ЦГепло, передаваемое от
зоны реакции к поверхности ВВ, заставляет его испаряться со скоростью,
определяемой скоростью подвода тепла.\В конденсированной фазе хими-
ческих реакций не происходит, а имеет место лишь нагрев вещества'oi
его начальной температуры до температуры кипения^
Сопоставление уравнений теплопроводности и диффузии показывает,
что температура и состав меняются одновременно в результате химиче-
ской реакции. Но они меняются одновременно и в той зоне, где химиче-
ская реакция не происходит: температура изменяется за счет теплопро-
водности, состав — за счет диффузии. В том частном случае, когда физиче-
ские свойства (молекулярный вес) участников реакции и продуктов ее
близки между собой, кинетическая теория газов предсказывает, что вели-
чины коэффициентов диффузии и теплопроводности будут близки; тогда
i |
j с
Рис. 237. Схема распределения тем-
пературы и протекания реакции при
горении летучих ВВ по Беляеву
I — исходное ВВ; II — прогретый слой
конденсированного ВВ; III — эона про-
грева паров ни; IV — зона химической
реакции в парах; V — эона продуктов
.горения
1 При сопоставлении указанных вероятностей нужно, впрочем, учитывать, что
испаряться могут лишь молекулы, находящиеся ян поверхности жидкости, а реаги-
ровать способны и молекулы, находящиеся и в прилежащих слоях, прогретых до до-
статочно высокой температуры.
266
во фронте связь между температурой и составом будет особенно простой,
такой, как если бы реакция шла адиабатически без обмена теплом и без
обмена веществом с соседними слоями.
Распределение температуры в пространстве показано на рис, 237:
Го — начальная температура ВВ, Гк — температура кипения, Гг — маиж-
мальная температура, достигаемая при горелни. Горение распространяет-
ся слева направо. Поверхность ВВ в рассматриваемый момент времежи
находится в плоскости АА. Плоскость АА, отделяющая конденсированное
ЁВ от его паров (а поскольку мы рассматриваем стационарный процесс,
то а плоскости ЕЕ, ДД и СЁ), перемещается со скоростью и слева на-
право.
Будем рассматривать процесс в системе координат, движущейся вместе
с фронтом горения. В такой системе плоскость АА неподвижна, а взрывча-
тое вещество будет втекать в нее справа налево со скоростью и, равной
скорости горения. Превратившись в пары, ВВ будет течь со скоростью во
столько раз большей, во сколько раз плотность паров меньше плотности
конденсированного вещества. По мере разогрева паров вследствие переда-
чи тепла из соседних ^лоев и выделении тепла при химической реакции,
протекающей в них, скорость движения будет расти и после завершения
реакции достигнет своего максимального значения. При этом поскольку
процесс стационарен, массовая скорость (up), т. е. масса вещества, проте-
кающая через каждую зону в единицу времени, одна и та же.
К плоскости АА прилегает прогретый слой взрывчатого вещества
ААСС. За толщину его АС, математически удобно принимать участок, на
котором разогрев В В уменьшается в е раз, отсюда
ГС-70
Пары,* текущие справа налево, нагреваются вследствие поступления
тепла путем теплопроводности из зоны высокой температуры, а также
вследствие выделения тепла химической реакцией. При этом до точки пе-
региба кривой Г = f(x) градиент температуры растет (по абсолютному
значению), в точке перегиба градиент проходит через максимум, а затем
падает, достигая нуля при Т = Тг. Соответственно изменяется и баланс
потока тепла; справа от точки перегиба элемент объема получает больше
тепла от горячих слоев, чем отдает его холодным; слева от точки d элемент
объема движущихся газов отдает тепла холодным слоям больше, чем по-
лучает его от горячих. Это, конечно, не означает, что запас тепла и темпе-
ратура елемента. объема в этой зоне понижаются, наоборот, они растут
вследствие того, что по мере повышения температуры резко возрастает
скорость выделения тепла химической реакцией.
Уравнение теплового баланса для елемеята объема реагирующего газа
(пара), расположенного на некотором расстоянии от плоскости АА, мож-
но написать в виде
ЭТ йГ , W 4Л.
где Ср, р,ц—соответственно теплоемкость при постоянном давлении, плот-
ность и теплопроводность газа (предполагается, что теплоемкость и теп-
лопроводность не зависят от температуры); F — количество тепла, выде-
ляемого реакцией в единице объема в единицу времени (объемная ско-
рость тепловыделения).
Это уравнение показывает, что изменение тепловой энергии элемента
объема происходит: 1) в результате движения газа со скоростью и; 2) за
счет «чистой» теплопроводности; 3) за счет выделения тепла прн протека-
нии химической реакции.
267
Так как свежая смесь всегда поступает из более холодной части объе-
ма, то первое слагаемое правой части уравнения всегда отрицательно и
уменьшает количество тепла в данной точке.
Второй член уравнения (4.10), равный т]д2Т /б^г2, представляет собой
изменение тепловой энергии элемента объема за счет теплопроводности и
может быть как больше, так и меньше нуля. 'При низких температурах
(рис. 237) (участок cd — выпуклость кривой обращена вниз) он положи-
телен, а при высоких температурах (участок de — выпуклость кривой
обращена вверх) отрицателен.
Третий член уравнения (4.10) выражает тепловыделение за счет иду-
шей реакции, скорость которой зависит от концентраций реагентов и от
температуры.
Поскольку речь идет о стационарном процессе и принимается, что зона
горения неподвижна, то распределение температуры, изображенное на
рис. 237, не меняется во времени, поэтому в выражении (4.10) можно при-
нять отношение дТ / dt = 0 и заменить частные производные полными.
Тоцца это уравнение принимает вид
+ +F = 0- <4Л1>
Если рассматривать ту зону, где температура и скорость химиче-
ской реакции, а следовательно и тепловыделения так малы, что тепло-
выделением можно пренебречь, то, полагая F = 0, выражение (4.11)
можно написать в виде
+ П ---°- (4-12)
’ dT
Введя переменную ф — , получаем
Срриф -j- n § = о
или
rf<p __________________________ __ СРРИ ,
откуда после интегрирования находим
с„р их
1п<р =---+
Так как
V 1
Л X ’
где % — коэффициент температуропроводности, то
(4.13)
откуда
Г=ес* е-™'*+Сг. (4.14)
Постоянные интегрирования и С2 определяем из условия, что,
при х — оо Т = 70, а при х = О' Т*= Тк; тогда = Т й и
ес* = ~(Та-Т0).
Подставив эти постоянные в выражение (4.14), получаем зависимость
температуры Т от расстояния х, измеряемого от плоскости с темпера-
турой Тк, в виде
Т = Та + (Т^Т0)е~7Х. (4.15)
268
Это соотношение в теории горения было впервые выведено Михель-
•соном, оно справедливо для зоны, в которой тепловыделением химиче-
ской реакции можно пренебречь.
В той области температур, где тепловыделение за счет реакции
становится существенным, член F в выражении (4.11) сохраняется. Это
уравнение можно преобразовать следующим образом.
Скорость тепловыделения F зависит от концентрации реагирующих
веществ и от температуры. Связав концентрацию с температурой, мож-
но выразить скорость реакции и скорость тепловыделения как функ-
цию одной температуры.^Лри этом получается уравнение второго поряд-
ка, нелинейное ввиду нелинейной зависимости F(T’).
Для решения этого уравнения Зельдович использовал свойство за-
кона Аррениуса, заключающееся в том, что скорость реакции сильно
зависит от температуры, если, как обычно, выполняется условие
(4.16)
Зависимость скорости тепловыделения реакций нулевого, первого
и второго порядков от температуры для типичных условий горения
(70 = ЗОО°К; Тг = 1900°К; Е = 50000 кял/.иогь) приведена на рис. 238.
Когда выполняется условие уравнения ’(4.15), можно считать, что
практически вся реакция протекает в узком температурном интервале 0,
т. е. между температурами Тг — 0 и Тт, причем
е <(/’,—т0).
Поскольку этот интервал узок, то для зоны реакции расходом теп-
ла на нагрев реагирующего вещества можно пренебречь, т. е. положить
исрр^г = °-
Тогда уравнение
Уравнение (4.17)
проводыостью равен
теплового баланса примет вид
+ = (4-17)
означает, что в зоне реакции отвод тепла тепло-
теплу, выделяющемуся при химической реакции.
При помощи уравнения (4.17) можно определить величину потока
тепла
dT
Ч^-
Снова вводим переменную-ф = dT I dx. Очевидно, что
d2T rf<p
dx2 dT
Подставляя это значение в уравнение (4.17), получаем
или
церйф - — FdT.
Интегрируя по зоне реакции, получаем
Тг-е т, ТР
--FdT^S — W = 5 FdT'
тг Тр Тц
откуда градиент температуры на границе зоны реакции приближенно
равен
ср =
и F'IT
269
или
1 г
2ц С FdT.
(4.18)
В выражении (4.18) интеграл скорости выделения тепла по темпе-
ратуре берется по всей области, в которой скорость реакции отлична от
нуля. Вследствие быстрого уменьшения скорости реакции^рри падении
температуры основное слагаемое дает сравнительно узкий интервал
температуры, примыкающий к температуре горения.
Поскольку расходом тепла на нагрев реагирующего гааа мы пренеб-
регли, допустив, что оно целиком отводится теплопроводностью, то
поток тепла, очевидно, можно приравнять общему количеству тепла,
выделяющемуся в зоне горения в единицу времени, т. е. произведению
количества сгоревшего вещества
на его теплотворную способ-
ность Q:
</т Л
Подставляя это выражение в
формулу (4.18), получаем
(4-19)
Так как F = wQ, где w — объем-
ная скорость реакции, то
Рис. 238. Зависимость объемной скорости
тепловыделения при горении от темпера-
туры
По оси ординат отложена скорость в услов-
ный единицах, максимальное значение ско-
рости дин каждой из кривых принято за еди-
ницу.
О — реакция нулевого порядка, Г — реакция
первого порядка; в — реакция второго порядка
uM = j/^$wdT (4.20)
Это выражение показывает, что скорость горения возрастает с увеличе-
нием теплопроводности и скорости реакции при температуре горения. При
этом скорость горения пропорциональна не первой степени, а корню
квадратному из скорости реакции.
Подставляя в формулу (4.20) вместо F его выражение через концент-
рации реагентов, пропорциональные давлению в степени, соответствую-
щей порядку реакции, и учитывая, что к моменту достижения максималь-
ной температуры часть вещества уже прореагировала и оно несколько раз-
бавлено продуктами реакции, Зельдович получил для массовой скорости
горения следующее выражение:
' Им = т/ QWTr<
У х X а /
(4.21)
где им — массовая скорость горения; Е — энергия активации реакции го-
рения; п — порядок реакции; uvc —скорость реакции при температуре
горения.
Формула (4.21) позволяет найти зависимость скорости горения от дав-
ления и начальной температуры. В эту формулу давление входит в скры-
том виде, поскольку от давления зависит скорость реакции wr в газовой
физе.
270
Обозначив через В произведение всех входящих в уравнение (4.21)
величин, не зависящих от давления, можно написать
Ми = Bw%aF .
Как известно, зависимость скорости газовой реакции n-го порядка от
давления имеет вид
и>тг = крп,
где к — коэффициент пропорциональности.
Отсюда
и = Bip”/® , (4.22)
где
Вх = В к12.
Таким образом, при маномолекулярной реакции горения скорость его
должна быть пропорциональна корню квадратному из давлении; при би-
молекулярной реакции скорость горения должна
быть пропорциональна первой степени давления.
< Физический смысл влияния давления заклю-
чается в том, что при большем давлении скорость
\ реакции выше и зона, в которой она происходит
\ (зона высокой температуры), придвигается ближе
] к поверхности ВВ. Количества передаваемого теп-
/ ла, а следовательно, и испаряющегося ВВ, возра-
\ стают соответственно увеличению скорости хими-
\ческой реакции.
Зависимость скорости горения от начальной
температуры ВВ определяется в первую очередь
зависимостью скорости реакции от температуры
горения:
wTr^Ae'E!RT-.
скорости горения лету-
чих ВВ от температуры
горения по теории Беля-
ева
Температура горения Тг = То + Qh'v, здесь ср — средняя теплоем-
кость продуктов горения при постоянном давлении. Таким образом, чем
выше начальная температура, тем выше температура горения, тем больше
скорость реакции и соответственно скорость горения.
В формуле (4.21) температура горения в явном или скрытом виде
входкт еще в ряд множителей: Q ~ Гг, ц растет с температурой, концент-
рация в выражении для скорости реакции обратно пропорциональна тем-
пературе. Зависимость от температуры всех множителей, входящих в вы-
ражение (4.21) (кроме е-я?втг), можно представить в виде
/(Тг) = т7’Л
где у включает все множители, не зависящие от температуры.
• Суммарную зависимость скорости горения от температуры можно вы-
размть приближенным соотношением
и = fTvFe~EI2RTr (4.23)
или
— = (
логарифмируя последнее, получаем
1п^ = -£/2ЯТг-|-1пт. (4.24)
* г
271
Таким образом, в координатах т зависимость скорости горе-
г г
ния летучих ВВ от максимальной температуры горения должна изобра-
жаться прямой линией, причем тангенс угла ее наклона равен E/2R
(рис. 239).
От плотности порошка скорость реакции в газовой фазе, естественно,
не должна зависеть, поэтому массовая скорость горения не должна изме-
няться при изменении плотности. Отсюда следует, что линейная скорость
горения должна изменяться обратно пропорционально изменению плот-
ности.
IV. СОПОСТАВЛЕНИЕ ЭКСПЕРИМЕНТАЛЬНЫХ
ДАННЫХ И ЗАКОНОМЕРНОСТЕЙ
С ВЫВОДАМИ ТЕОРИИ ЗЕЛЬДОВИЧА — БЕЛЯЕВА
Первым взрывчатым веществом, на котором было проведено подробное
сопоставление данных теории и опыта, был нитрогликоль [230]. Оно вклю-
чало расчет абсолютного значения скорости горения и ее зависимостей от
начальной температуры и давления.
При расчете скорости Беляев принимал приближенное уравнение реак-
ции горения нитрогликоля, рассчитанное по составу продуктов горения в
атмосфере азота
C2H4(ONO2)2 =2NO+1,7CP 4- 1,7Н30 .4- 0,ЗСО2 + 0,ЗН2 + 70 кмл (4-25)
Средняя теплоёмкость продуктов горения при постоянном давления
ср = 0,34 кал!г > град. Отсюда повышение температуры при горении ни-
трогликоля ДГг = Гг — У’о = Q / Ср = 460/0,34 = 1350°, где 460 — теплота
горения на грамм вещества. Максимальная температура Тт = Тъ + &Тт =
=3004-1350=1650° С. Коэффициент теплопроводности продуктов горения
при его максимальной температуре ц = 2 • 1СН »йл/сл • сек • град.
Поскольку реакция горения нитрогликоля есть прежде всего реакция
распада, то Беляев считает эту реакцию мономолекулярной. Однако, если
расчет скорости вести по выражению для реакции первого ,порядка й ис-
пользовать значения энергии активации и предэкспоненциального множи-
теля, полученные для медленного термического распада [й = 10 м’3 •
• ехр (—35 700/Я71)], то результат расчета примерно в 200 раз превышает
экспериментальное значение скорости горения Поэтому Беляев, учиты-
вая экспериментально установленную пропорциональность скорости горе-
ния первой степени давления (что соответствует второму порядку ведущей
реакции), предположил, что скорость реалции распада нитрогликоля при
давлениях, близких к атмосферному, пропорциональна числу соударений.
При этом предположении он в формуле (4.21) взял п = 1, но вместо га!
принял (п + 1)1, т. е. 2.
Число столкновений z = У"2 лз2«т2, где а — диаметр молекулы, и —
ее скорость, m — число молекул в кубическом сантиметре; число реаги-а
рующих молекул вдвое больше числа столкновений
» = /6 Лб2]/, (4.26)
где М — молекулярный вес нитрогликоля и Л’ - число Авогадро.
1 Чтобы при этом расчете получить значение скорости, соответствующее экспери-
ментальному, надо принять температуру реакции равной ~87О°С. Поскольку при
стационарном горевии скорости распространения всех зон должны быть одинаковы;
то, возможно, именно при этой температуре и протекает при горении нитрогликолп
первичная стадия процесса, не являющаяся в этих условиях ведущей.
272
самом же деле у нитрогликоля скорость горения пропорциональна давле-
нию в первой степени, в то время как медленный термический распад
представляет собой типичную мономолекулярную реакцию.
Чтобы разрешить это противоречие, Беляев предполагает, что моно-
молекулярная реакция в тех условиях ее течения, которые имеют место
во фронте горения, превращается в бимолекулярную в том смысле, в ка-
ком это происходит со всеми мономолокулярными реакциями при очень
низких давлениях. Как известно, кажущаяся независимость образования
активированных молекул при мономолекулярных реакциях от числа со-
ударений обусловливается тем, что при столкновении наряду с активаци-
ей идет также дезактивация активных молекул. Если время жизни актив-
ной молекулы велико по сравнению с временем между соударениями, то
устанавливается некоторая определенная концентрация активных моле-
кул, которой (а черев нее и общей концентрация вещества) пропорцио-
нальна скорость реакции. Беляев допускает, что при тех высоких темпе-
ратурах, при которых идет реакция во фронте горения, число дезактиви-
рующих столкновений относительно мало, и моиомолекулярная реакция
идет поэтому со скоростью, пропорциональной числу столкновений, т. е.
пропорциональной квадрату давления. Это же дает пропорциональность
скорости горения давлению.
В дальнейшем попытки расчета абсолютного значения скорости горе-
ния были произведены применительно к другим ВВ. Так, Арден и Паулинг
[139] рассчитали скорость горения паров метилннтрита, приняв темпера-
туру горения, соответствующую экспериментально определенному составу
его продуктов, и кинетические константы, установленные для медленного
распада, дающего, правда, иные конечные продукты. Этот расчет дал
и = 120 см/сек вместо 3,2 см/сек.. определенных экспериментально.
Сопоставление теоретического и экспериментального влияния началь-
ной температуры на скорость горения не могло быть произведено, так как
выяснилось, что массовая скорость горения не увеличивается при повы-
шении начальной температуры. Формально отсюда следовал бы вывод, что
либо температура горения очень высока (это опровергается результатами
ее измерения), либо энергия активации ведущей реакции очень мала. Ве-
роятнее, однако, что эта аномалия связана с некоторыми особенностями
течения превращения, которые приводят, в частности, к тому, что при по-
вышении начальной температуры выше 240° С горение перестает распро-
страняться.
Грэй [144] рассчитал скорость горения гидразина, используя константу
скорости медленного распада (первого порядка) (Е = 60 ккал/моль), и по-
лучил и = 30 см/сек вместо 200 см/сек, определенных экспериментально
(скорость рассчитана для паров). Опыты по разбавлению паров различ-
ными инертными газами также показали относительно невысокую энер-
гию активации (36 ккал/моль). Наконец, скорость горения растет пропор-
ционально первой степени давления, что указывает на бимолекулярность
ведущей реакции, в то время как медленный распад идет по закону перво-
го порядка.
Все эти данные говорят за то, что ведущей при горении нельзя считать
ту первичную реакцию (или совокупность их), которая определяет тече-
ние медленного распада.
К этому же выводу приводит более углубленное сопоставление экспе-
риментальной и расчетной температурной зависимостей скорости горения
и скорости медленного термического распада для нитро гликоля и ряда
других ВВ.
Согласно теории для реакции первого порядка (для реакции второ-
го порядка различие невелико),
w = [J2’;!'VE'sRTr. (4.28)
274
Отсюда температурный коэффициент скорости горения—отношение
скоростей горения при двух температурах и Тг
^E,/RT I \ 3/2 В(Тг-Т()
* — р а а - I 2k 1 /!нт;т;'
л — рГз/2 -в*/нт, \ Т1 /
(4.31)
Чем выше температура горения, тем при данной Е меньше должен
быть к. Или, чем больше к, тем больше при данной температуре горе-
ния должна быть Е.
Логарифмируя выражение (4.31), получаем
1п * ~ А + i111 ? (4.32)
ИЛИ
In А = Л In jr-. (4.33)
Таким образом, приближенно величина In А определяется отношени-
ем у , так как Т2П\ близко к 1, а логарифм единицы равен нулю.
Таким образом,
или
In к = А = const. (4.34)
Для ряда ВВ на основании экспериментально установленного состава
газообразных продуктов горения, теплоемкости ВВ и рассчитанной на этой
основе температуры горения для разных начальных температур ВВ и тем-
пературного коэффициента скорости горения вычислены по уравнению
(4.34) значения энергии активации, которые сопоставлены с эксперимен-
тальными значениями Е для медленного термического распада. Эти дан-
ные сведены в табл. 38. При расчетах для метилнитрата принята теплоем-
кость, равная 0,4 кал/г по аналогии с нитрогликолем и нитроглицерином.
Температура горения взята для нитрогликоля выше, чем принимал Беля-
ев. Как указывалось, он при расчете этой температуры исходил из прибли-
женного уравнения горения, полагая, в частности, что весь азот выделяется
в виде NO. В действительности же некоторая небольшая часть азота полу-
чается в элементарном виде. Учет образования азота значительно отра-
жается на результатах расчета. Теплота горения нитрогликоля, по урав-
нению Беляева, равна 460 кал/г, при полном превращении NO в Na эта
теплота составляет около 1600 кал/г. Таким образом, естественно, что пре-
вращение даже небольшой части NO в Na значительно повышает темпера-
туру горения.
Таким образом, для большинства изученных в этом отношении ВВ,
в том числе и для иитрогликоля, на котором Беляев впервые проверил
теорию, не получается даже удовлетворительного соответствия между зна-
чениями энергии активации, вычисленными по температурной зависимо-
сти скорости горения и полученной для медленного термического распада.
Следует подчеркнуть, что значения Е, которые дает расчет для горения,
иногда столь велики, что крайне сомнительно, чтобы они могли представ-
лять энергию активации какой-либо из реакций, происходящих при
горении.
Причины такого несоответствия между теорией и опытом могут быть
двоякого рода; допустим, что ускорение горения с повышением начальной
температуры ВВ происходит не только по тому механизму, из которого
исходит теория, но и по другим, возможно, даже не связанным с кинети-
кой реакции. Тогда температурный коэффициент скорости оказывается
18* 275
Таблица 38
Характеристика горения некоторых ВВ
ВВ Расчетная температура горевия (в “К) яри начальной температуре Отношение ско- ростей горения яри 100 и 0“ С Принятая теплоем- кость В В, кал/г Ерасч. Еэнея-
О’С 100“ с
Метилнитрат . . 2003 2050 (при 50° С) 1,47 ( “50“ \ 0,4 121000 39 500
Нитрогликоль. . 1894 1992 1,52 0,379 52000 35 700
Нитроглицерин .. 2030 2129 1,82 0,356 91000 50000
завышенным, а это, в свою очередь, как ясно из формулы (4.34), приводит
к завышенному значению энергии активации.
Вторая возможность заключается в ошибочности предположения о том,
что течение процесса горепия определяется одной единственной реакци-
ей — той же, которая происходит при относительно низкотемпературном
термическом распаде и характеризуется соответствующими кинетически-
ми константами.
Имеющиеся экспериментальные данные не дают исчерпывающего от-
вета на вопрос о том, какие именно из возможных причин определяют рас-
хождение теории и опыта По-видимому, реальны причины обеих групп.
Предположение о том, что ведущая при горении реакция та же, что и
при медленном термическом распаде, не является органическим элементом
теории Зельдовича. Можно допустить, что течение горения определяется
какой-то другой реакцией с иной энергией активации, и это допущение
также может быть проверено сопоставлением с экспериментом следующи-
ми путями (122].
Из теории следует определенная зависимости температурного коэф-
фициента скорости горения от температуры горения (4.34), а следователь-
но, и от начальной температуры ВВ. При повышении То возрастает и Тг,
следовательно, должен уменьшаться, хотя и незначительно (так как воз-
можное увеличение Го невелико, а Тг высока), температурный коэффи-
циент скорости горения. Опыт, однако, показывает, что для всех изучав-
шихся в этом отношении ВВ (рис, 169—171) температурный коэффициент
скорости горения не только не уменьшается, но отчетливо возрастает при
повышении начальной температуры ВВ. Это увеличение выражено, вооб-
ще говоря, слабее при повышенных давлениях*, например при 50 ат по
сравнению с 25 или 1 ат.
Другую возможность проверни теории дает установление влияния дав-
ления на температурную зависимость скорости горения. Теория не преду-
сматривает такого влияния. Опыт показывает, однако, что оно проявляет
ся (см. табл. 26). Для всех изучавшихся ВВ температурный коэффициен:
1 Недавно Беляевым была рассмотрена еще одна возможная причина такого pat
хождения. Он предположил. что ведущая реакция в газовой фазе, даже при относа
Тельио низких давлениях, идет не при максимальной температуре (рассчитанной в
составу продуктов горения), а ври некоторой более низкой «эффективной» темпер;
туре, которую в принципе можно было бы определить, например, намеряя при пом;
щи термопары температурный профиль волны горения {270] (Прим. ред.).
г Для некоторых ВБ при повышенных давлениях в области высоких температу
Недалеких от температуры самовоспламенения, наблюдается реакий рост темпер:
турного коэффициента, но этот рост имеет уже другую природу, будучи связан
экзотермическим распадом в конденсированной фазе и, возможно, с возмущение
расплавленного слоя.
276
уменьшается при повышении давления; для некоторых ВВ, например гек-
согена,— относительно слабо: при переходе от 1 к 50 ат А уменьшается от
1,25 до 1,14; для других веществ — значительно сильнее, для тетрила, на-
пример, от 1,35 при 42 ат до 1,17 при 50 ат.
Эта последняя зависимость противоречит концепции, согласно которой
ведущей является одна, не зависящая от других реакция, протекающая
при постоянной, не зависящей от давления температуре, равной То + -у-,
где Q — ее тепловой эффект, ас — средняя теплоемкость продуктов. Од-
нако, .если учесть бесспорно установленную многостадийность газофазного
превращения, то объяснить эту зависимость можно было бы, допустив,
что прн повышении давления зона протекания следующей экзотермиче-
ской ступени приближается к зоне ведущей реакции и несколько повыша-
ет температуру последней, что делает ее менее «чувствительной» к на-
чальной температуре. Если подобным образом влияет не только повыше-
ние давления, но и начальной температуры, то это могло бы объяснить и
рост температурного коэффициента с повышением последней. Объясни-
мым было бы и уменьшение роста температурного коэффициента с началь-
ной температурой при повышении давления. Есни давление повышает эф-
фективную температуру ведущей реакции, то она становится менее чув-
ствительной к другим влияниям, дающим тот же эффект. Понятным было
бы и относительно слабое проявление обоих указанных влияний для бо-
лее «горячего» вещества — гексогена по сравнению, например, с тетрилом.
Рассмотрим зависимость скорости горения от давления. Выше уже от-
мечалось постоянство этой зависимости, установленное экспериментом.
Для простейшего случая летучих ВВ скорость горения определяется вы-
ражением и = А Ц- Вр, причем член А очень мал и, по-видимому, связан с
побочными причинами, так что скорость горения можно считать пропор-
циональной давлению в первой степени. Согласно теории это означает, что
ведущая реакция бимолекулярна. Предположение Беляева о том, что при
повышении давления будет наблюдаться переход от зависимости и(р), со-
ответствующей бимолекулярному течению ведущей реакции (и ~ р),
к зависимости, соответствующей ее мономолекуляриому течению
(и ~ р'/г), не подтвердилось. Для всех изучавшихся твердых относительно
летучих органических нитратов и нитросоединений скорость горения во
всем широком исследованном интервале давлений (до 1000 аг) растет
приближенно пропорционально давлению. В то же время для всех этих ВВ
медленный термический распад является реакцией первого порядка.
Простейшее объяснение экспериментальной зависимости скорости го-
рения от давления заключается в том, что ведущая реакция является
реакцией бимолекулярной, точнее протекающей через двойные соударе-
ния. Отсюда следует также вывод, что этой ведущей реакцией ие является
реакция, определяющая скорость мономоленулярного распада.
Рассмотрим более подробно возможные объяснения эксперименталь-
ных зависимостей скорости горения от температуры и вероятную схему
течения процесса горения.
Прежде всего остановимся на одном тривиальном возможном объясне-
нии расхождения теории и опыта. Горение всех изученных ВВ при низ-
ких давлениях вблизи атмосферного приводит, как указывалось выше,
к продуктам незавершенного превращения. При повышенных давлениях
полнота сгорания возрастает и, наконец, начиная с некоторого давления,
мы получаем состав газообразных продуктов горения, соответствующий
термодинамическому равновесию. Можно было допустить, что аналогич-
ное влияние оказывает повышение начальной температуры ВВ и что при
повышенных начальных температурах состав продуктов горевня меняется
в сторону увеличения полноты горения и температуры газообразных про-
дуктов его. Это обстоятельство, если бы оно имело место, могло бы дать
277
дополнительное увеличение скорости горения, без учета которого получи-
лось бы завышенное значение энергии активации. Для выяснения реаль-
ности этой возможности Г. В. Оранской были проведены определевия со-
става газообразных продуктов горения нитрогликоля и пироксилина № 1
при комнатной температуре и при 95° С. Было установлено, что слабое из-
менение состава газов при повышении начальной температуры, по-види-
мому, имеет место, но оно очень мало и недостаточно, чтобы объяснить
чрезмерно высокие значения энергии активации1.
Опыты показали, что скорость горения жидких ВВ суфествеино зави-
сит от вязкости жидкости. Значительное увеличение последней (путем
желатинизации небольшим количеством высокополимера) уменьшает ско-
рость горения нитрогликоля на l/s, нитроглицерина на 1/з- При этом
уменьшение скорости горения с увеличением вязкости ВВ наблюдается
при низких (комнатных) температурах, а при повышенных температурах
(около 100° С) уменьшения скорости горения нет, вероятно, потому, что
вязкость желатины при повышенных температурах резко снижается.
Влияние вязкости на скорость горения ВВ может быть связано с тем,
что на поверхности, граничащей с газами, жидкость при горении кипит в
обычном значении этого слова или же получается явление, внешне подоб-
ное кипению, вследствие выделения пузырьков растворенных газов. Это
бурление жидкости, возможно влекущее поступление брызг ее в газы,
создает некоторый микрорельеф поверхности и усиливает теплообмен га-
зов с жидкостью, в результате чего увеличивается скорость горения. Оче-
видно, что интенсивность явления должна зависеть от вязкости, чем и
обусловливается влияние последней. Однако при таком механизме влия-
ния вязкости на скорость горения ясно, что температурный коэффициент
скорости горевия может быть, по крайней мере для жидкостей с большой
и сильно зависящей от температуры вязкостью, значительно завышен за
счет указанного эффекта.
Следует добавить, что для относительно быстрогорящих жидкостей
возможны искажение поверхности и увеличение скорости также вслед-
ствие появления эффекта Ландау (см. ниже стр. 303). Не исключено, на-
пример, что большое ускорение горения метилнитрата с температурой свя-
зано именно с этой причиной.
Помимо указанных физических причлн, возможно влияние вязкости,
ближе связанное с кинетикой химического процесса горения. При горении
таких ВВ, как органические нитраты и нитросоединення, распад молекулы
происходит не сразу, сначала отщепляются преимущественно газообраз-
ные соединения азота с кислородом, в первую очередь NOj, относительно
быстро восстанавливающаяся до NO. В результате этого поверхностный
слой жидкости обедняется окислителем, который в него поступает затем
путем диффузии из глубжележащих слоев. Скорость, диффузии тем мень-
ше, чем больше вязкость жидкости. Повышение визкости затрудняет так-
же конвективное перемешивание, которое может играть роль при малых
скоростях горения. То обстоятельство, что повышение вязкости сильнее
влияет при относительно быстрогорящих жидкостях, говорит в пользу
указанного объяснения.
При рассмотрении возможных причин нарушения «нормальной» зави-
симости скорости горения от начальной температуры в сторону получения
повышенного температурного коэффициента предполагалась правильность
допущения Беляева о том, что скорость горения определяется именно ско-
ростью моно молекулярной реакции, тождественной с реакцией, происхо-
дящей при медленном термическом распаде.
1 Отсюда следует заключить, что для восстановления окиси азота требуется го-
раздо большее повышение температуры горения, чем то, которое достигается при по-
вышении начальной температуры ВВ на 100° С.
278
Это допущение, однако, не имеет серьезных обоснований; основным
аргументом в его пользу является совпадение энергий активации, которое,
казалось, было получено Беляевым. Если бы химическое превращение при
горении шло в одну ступень, то, естественно, эта реакция и была бы оп-
ределяющей, как это следует из теории горения.
Однако почти несомненно, что для всех рассмотренных ВВ это не так
и что даже те продукты неполного сгорания, которые получаются при ат-
мосферном давлении, являются результатом двух или более реакций. Есть
серьезные, хотя и косвенные, основания полагать, что, например, при го-
рении нитроэфиров и нитросоединений первичной реакцией является эн-
дотермическое или слабо экзотермическое отщепление двуокиси азота, ко-
торая затем реагирует с другими промежуточными продуктами распада,
возможно также и с исходным веществом.
Вполне возможно и вероятно, что именно эта первичная реакция оп-
ределяет скорость медленного гомогенного термического распада ВВ. Хотя
при этом распаде также идут вторичные реакции и в общем, по-виднмому,
в том же направлении, что и при горении (судя по сходству продуктов
реакции), но общая скорость распада может определяться одной, самой
медленной из последовательных реакций. Эндотермична или экзотермична
эта реакция в условиях медленного распада, не существенно, так как реак-,
кия идет при постоянной температуре, поддерживаемой теплообменом с
внешней средой. Судя по тому, что двуокиси азота в конечных продуктах
реакции в обычных условиях течения последней не наблюдается, следует
заключить, что именно первичная реакция (ведущая к ее образованию)
является наиболее медненной.
Совершенно другое значение имеет тепловой эффект реакции при го-
рении. В этом случае температура течения реакции и скорость ее опреде-
ляются именно положительным тепловым эффектом реакции. Поэтому *
первичная эндотермическая реакция также мало может определять ско-
рость горения 1, как и эндотермический физический процесс испарения,
скрытая теплота которого не входит в выражение скорости горения по
Зельдовичу.
Скорость горения определяется течением той реакции (или тех реак-
ций), при которой выделяется основная часть теплового эффекта горения.
Скорость же эндотермической первичной реакции автоматически устанав-
ливается соответственно скорости ведущей реакции. Если константа ско-
рости первичной реакции велика, то зона максимальной температуры на-
ходится далеко от зоны первичной реакции, и температура в этой по-
следней зоне относительно низка, а именно такова, чтобы скорости обеих
реакций были одинаковы, что является необходимым условием при ста-
ционарном процессе.
Если представить себе, что скорость первичной реакции уменьшилась
и она стала распространяться в глубь вещества медленнее, чем ведущая ,
реакция, то фронт последней будет приближаться к зоне первичной реак-
ции, и температура в этой зоне будет возрастать до тех пор, пока скорость
ее распространения не станет равной скорости распространения ведущей
реакции.
Таким образом, если химическое превращение при горении представ-
ляет собой совокупность двух последовательных реакций — эндотермиче-
ской и экзотермической (идущей «самостоятельно» за счет выделяемого
1 Исключение составляет лишь тот (в принципе возможный) случай, когда кон-
станта скорости вторичной экзотермической реакции очень велика по сравнению со
скоростью первичной реакции, и продукты последней сразу реагируют друг с другом
так, что рассматриваемого ниже распределения реакций по зонам не наблюдается.
В этом случае скорость превращения лимитируется кинетическими характеристика-
ми первичной реакции, а температура ее течения — тепловым эффектом экзотерми-
ческой реакции,
279
ею тепла), течение эндотермической первичной реакции, так же как и эн-
дотермического испарения, является вынужденным и скорость этих про-
цессов автоматически регулируется ведущей реакцией.
Сложнее обстоит дело в том случае, когда при горении происходит
несколько последовательных экзотермических реакций. Здесь, по-видимо-
му, ведущей является та реакция, которая при данных условиях ее
течения (температура и давление) распространяется с наибольшей
скоростью. Если таковой является, например, вторая из двух последова-
тельных вкзотермических реакций, то она будет «поджимать» первую,
идущую медленнее реакцию; за счет передачи тепла скорость первой реак-
ции должна будет возрасти до скорости более быстро распространяющейся
реакции. Если же вторая экзотермическая реакция идет медленнее, то она
будет отставать от фронта и на течение горения прямого влияния может
не оказывать.
Следует, однако, учитывать, что при изменении температуры горения
(путем изменении начальной температуры ВВ) или давлении соотноше-
ние между скоростями реакций изменяется я роли их могут перемениться.
Необходимо, кроме того, иметь в виду возможное влияние теплопотерь,
особенно существенное при медленно идущих реакциях. В качестве при-
мера укажем на горение того же нитрогликоля при атмосферном давлении.
Основным соединением азота является NO, углерода — СО, т. е. газы, спо-
собные к взаимодействию; однако это взаимодействие в обычных условиях
опыта практически не идет, очевидно, потому, что понижение температу-
ры в результате теплопотерь превосходит теплоприход, малый по причине
малой скорости реакции между СО и NO. При повышении давления вслед-
ствие уменьшения теплопотерь1 появляется второе пламя, в котором окись
азота восстанавливается до азота с большим тепловым эффектом. Это пла-
мя находится на большом (1—2 см) расстоянии от поверхности жидкости
п за счет теплопроводности на скорость горения влиять не может и в
действительности не влияет, о чем свидетельствует отсутствие заметного
изменения скорости при появлении указанного пламени. Однако при даль-
нейшем повышении давления следует ожидать приближения второго пла-
мени к поверхности жидкости, и это давало основания полагать, что имен-
но эта заключительная реакция и является для иитроэфиров ведущей при
повышенных давлениях.
У При низких же давлениях ведущей является другая реакция, возможно
та, которая приводит к продуктам, являющимся конечными при этих
давлениях. Аргументом в пользу этого допущения является близость по
порядку величины экспериментальных значений скорости горения и рас-
считанных теоретически, исходя из температуры горения, соответствующей
конечному составу газов при атмосферном давлении. То, что при этом та-
кое согласие получается при применении разумной величины энергии ак-
тивации медленного распада в тех случах, когда эта близость реальна2,
само по себе ничего не говорит, тем более, что расчетная величина скоро-
сти горения довольно мало чувствительна даже к значительным измене-
ниям -значений ЕяТ, используемых для расчета 3 *.
1 Теплопроводность газа не зависит от давления, количество энергии в единице
объема газов пропорциональна давлению, время горения меньше.
2 При расчете возможны различные его варианты: можно принять порядок реак-
ции равным 1 или 2, предзкспоненциальный множитель равным числу соударений
или меньшим, тепловой эффект рассчитывать по экспериментально установленному
составу газов или корректировать этот состав некоторыми допущениями. При некото-
рых из этих вариантов энергия активации, рассчитанная по величине скорости горе-
ния, получается близкой к анергии активации медленного термического распада.
2 Эта величина определяется в основном значением Если Е Припять
равной 40 000 кал и Т = 1800*, то при увеличении температуры на 100° величина
e-E!zRTv возрастает только в 1,35 раза; если при неизменной температуре энергия
активации будет не 40 000, а 50 000 кал, то e~E''s®Tr уменьшится лишь в 4 раза.
280
Если согласие между расчетной и экспериментальной величинами ско-
рости горения получено для какого-либо одного из нитроэфиров, то оно
будет наблюдаться для всех других нитроэфиров, поскольку скорость го-
4 рения их (при атмосферном давлении) отличается, вообще говоря, мало
(примерно от 0,015 см! сек у дигликольдинитрата до 6,11 см)сек у метил-
нитрата), а температуры горения тех нитроэфиров, у которых на каждый
углеродный атом приходится одна группа ONO2, также близки. Возмож-
но, что ведущая реакция однотипна, подобно тому, как, по-виднмому, од-
нотипны и первичные реакции. Даже для летучих ВВ других классов,
горящих по рассмотренному механизму, нельзя ожидать скоростей горе-
| ния, во много раз больших, чем, например, для метилнитрата, так как это
означало бы или нереально большую теплоту горения, или очень малую
энергию активации, также нереальную, особенно в сочетании с первым ус-
ловием. Таким образом, скорость горения летучих ВВ ограничена, вообще
говоря, сравнительно невысоким верхним пределом.
i Однако (независимо от этих соображений) согласле в пределах немно-
|гих порядков между теоретическим расчетом абсолютной величины ско-
рости горения для ряда ВВ с расчетом, не включающим никаких эмпи-
рических констант, и опытом можно считать достаточным показателем
правильности теории в ее основе. Делом дальнейших исследований яв-
ляется установление реакций, которые ведут процесс, их кинетических
f характеристик и температур, при которых они протекают.
Интересным является вопрос, в какой области давлений ведущая роль
। переходит от реакции, образующей NO, к реакции, приводящей к ее вос-
становлению. Обычно принимается, что этот переход происходит при дав-
лениях -около 100 ат; основным соображением при этом, по-видимому,
является то, что около этого давления вторичное пламя настолько прибли-
жается к первичному, что темная эона между ними становится незамет-
ной. Есть, однако, основания как экспериментальные, так и логические,
полагать, что в действительности указанный переход происходит при го-
; раздо более высоких давлениях. Так, нитрогликоль и нитроглицерин энер-
гетически очень близки; в то же время нитрогликоль (при атмосферном
давлении) горит в 4 раза медленнее, чем нитроглицерин, что можно при-
писать различиям как в эффективных температура к i прения, так и в кине-
тике ведущих при низких давлениях реакций. Очевидно, однако, что на
заключительной стадии горения оба эти соображения отпадают, посколь-
ку речь идет в основном о взаимодействии СО и Hj с NO, а тепловой эф-
фект реакции одинаков. Поэтому там, где ведущая роль переходит к за-
ключительной стадии, скорости горения должны стать одинаковыми. Опыт
; показывает, однако, что даже при 1000 ат иитроглииоль горит в 1,4 раза
! медленнее нитроглицерина (оба нитроэфира желатинизировались нитро-
, клетчаткой, чтобы предотвратить переход нормального горения на возму-
щенный режим). Отсюда можно заключить, что даже при 1000 ат ведущей
является в основном та же реакция, которая выполняет эту роль при низ-
ких давлениях. В то же время при горении обоих нитроэфиров в их обыч-
ном жидком состоянии при турбулентном режиме уже при 100 ат скорости
। горении близки; в этих условиях зональное распределение температуры
устраняется турбулентным перемешиванием и скорость испарения опре-
деляется (конечной температурой горения.
' Для многих ВВ (например тэна, тротила, пикриновой кислоты) спо-
рость горения во всем изученном (до 1000 ат) интервале растет про-
* порциональпо давлению, причем коэффициент пропорциональности не
К меняется. Простота этой зависимости и одинаковость ее для различных
▼ ВВ позволяет думать, что она не случайна и не представляет собой итога
f компенсации разных влияний, а является следствием бимолекулярности
| ведущей реакции, характеристики которой не меняются в указанном диа-
пазоне давлений. Поскольку же при низких давлениях, когда заключп-
281
тельная стадия реакции вообще отсутствует, ведущей может быть только
начальная или одна иа промежуточных стадий, следует заключить, что
эта стадия сохраняет ведущую роль и при 1000 ат.
Эти экспериментальные результаты не являются неожиданными. При
низких давлениях заключительная стадия вдет медленнее, чем ведущая,
и соответственно зона ее протекания находится вдали от горящей поверх-
ности. Повышение давления ускоряет обе стадии. Ведущая стадия, судя по
зависимости и(р), является бимолекулярной; трудно допустить, чтобы
заключительная стадия представляла собой реакцию более высокого по-
рядка. Это означает, что влияние давления на обе стадии должно быть
одинаковым. Если все же при повышении давления зона заключительной
реакции приближается к зоне ведущей реакции, то это происходит, в част-
ности, потому, что при повышении плотности газа уменьшаются теплопо-
тери 1 и самовоспламенение продуктов заключительной реакции насту-
пает раньше.
Другой причиной ускорения обеих стадий, может быть повышение тем-
пературы их протекания для первой за счет получении тепла от воны
второй, а для последней за счет уменьшения теплопотерь. Однако умень-
шение теплопотерь вряд ли может сильно повысить конечную температу-
ру тем более, что она и так высока. В то же время повышение температу-
ры ведущей реакции, относительно низкой, должно сильно увеличивать ее
скорость2 и, как следствие этого, вызывать отбрасывание зоны заключи-
тельной реакции. Таким образом, исходя из соотношений, осуществляю-
щихся при горении под низкими давлениями, нет оснований ожидать бы-
строго перехода при повышении давления ведущей роли к заключитель-
ной реакции.
То, что температура конечных продуктов не обязательно является той
температурой, при которой идет ведущая реакция, показывает простой
опыт. Если рассчитать температуру горения иитрогликоля по анализу га-
зов, то можно было бы получить резкий скачок скорости горения около
15 ат, которого в действительности не наблюдается. Кроме того, примене-
ние при расчете температуры порядка 4000°, соответствующей конечному
(в данном случае полному) распаду при давлении 15 ат, дало бы сильно
завышенное значение скорости горения.
Невыполнение соответствующей момомолекулярному течению реакции
зависимости скорости горения от давления можно также объяснить, если
допустить, что ведущей является одна из вторичных реакций. Эта реакция
между осколками молекулы, например между NO2 и формальдегидом или
радикалами, естественно, должна протекать при соударениях соответст-
вующих частиц; число же соударений пропорционально произведению
концентраций, т. е. квадрату давления- Хотя это обстоятельство не исклю-
чает возможности следования химического превращения закону реакции
первого Порядка, но при учете простоты осколков эта возможность являет-
ся маловероятной. Замена при расчете реакции распада молекул нитро-
эфира реакцией между продуктами его первичного распада может почтя
не отразиться на численном результате расчета.
Таким образом, теория Зельдовича — Беляева в рассмотренном ее тол-
ковании, кстати, вполне естественном и ничуть не колеблющем основной
идеи теория, может быть приведена в согласие с фактами, установленны-
ми при горении летучих ВВ.
1 Давление, при котором появляется вторичное пламя, снижается при увеличении
диаметра трубки, в которой идет горение.
2 Если только энергия активации ведущей реакции не слишком мала; судя по
значительной температурной зависимости а(Т), это условие выполняется.
282
V. МЕХАНИЗМ ГОРЕНИЯ НЕЛЕТУЧИХ ВВ
^Основным отличием горения нелетучих В В является то, что при горе-
нии в конденсированной фазе могут быть достигнуты относительно высо-
кие температуры, прн которых скоростью химического превращения уже
нельзя пренебрегать, как это было допустимо при рассмотрении горения
летучих ВВ, когда температура конденсированной фазы на горящей по-
верхности не могла быть выше температуры кипения.
Опыт показывает, что горение нелетучих органических вторичных (вы-
с окопе л и мерные нитраты) ВВ не показывает принципиальных отличий от
горения летучих ВВ в отношении зависимости скорости горения от давле-
ния. Точно так же не наблюдается изменения зависимости н(р) при зна-
чительном повышении давления, когда летучие ВВ «утрачивают» свою
летучесть. Наиболее ощутимым отличием этой зависимости для нелету-
чих ВВ является наличие в распространенной линейной зависимости
и = А + Вр значительного члена А, не зависящего от давления и в 2—
4 раза (при обычной единице для выражения массовой скорости и давле-
ния) превосходящего В, Это означает, что скорость горения уже при ат-
мосферном давлении велика по сравнению с летучими веществами анало-
гичного химического строения и теплоты горения. Наиболее вероятное
объяснение этой особенности, как предполагал еще Мюраур, заключается
в том, что этот член отражает протекание экзотермической реакции
в конденсированной фазе и приток тепла от нее к вступающему в горе-
ние слою порохаг. Эта теплота передается по конденсированной фазе
путем теплопроводности, которая велика (по сравнению с теплопровод-
ностью газа) и поэтому более полно используется для распространения
процесса горения, чем теплопроводность газофазных реакций, зону кото-
рых от поверхности ВВ отделяет более (при низких давлениях) или менее
(при высоких давлениях) толстый слой газообразных продуктов первич-
ных стадий химического превращения.
Иное объяснение наличия члена А, считая его кажущимся, предложил
Беляев [231]. Он предполагал, что при горении нелетучих ВВ основная
реакция проникает в поры и идет в них под несколько повышенным дав-
лением рс, что и дает в уравнении член А. Аргументом в пользу этого
объяснения является постоянство рассчитанного повышения давления Др
в опытах с гремучей ртутью при различных температурах. Абсолютная
величина предполагаемого повышения давления для гремучей ртути неве-
лика — 300 jmc, для трниитротриазидобензола — 150 мл. Однако при го-
рении других ВВ, например пироксилина № 1, коллоксилина, пришлось
бы допустить гораздо большее Ар (2,2 и 4,5 ат соответственно), для нит-
роглицеринового же пороха еще больше — 100—150 ат, что явно неправ-
доподобно.
Другим отличием нелетучих ВВ (конкретно нитроклетчатки) от лету-
чих низкомолекулярных иитроэфиров является относительно большой тем-
пературный коэффициент скорости горения, который существенно воз-
растает с повышением начальной температуры. К тому же температурный
коэффициент зависит от плотности, возрастая с ее увеличением. Это по-
следнее обстоятельство необъяснимо с точки зрения ведущей роли газо-
фазного химического превращения и явно указывает на существенную
роль процессов в конденсированной фазе.
Своеобразно также влияние инертных примесей на горение исследо-
ванного в этом отношении нелетучего ВВ — пироксилина № 1. Особенно
1 В некотором противоречии с этим объяснением, исходя из автономности
члена А, является то, что при увеличении начальной температуры он увеличи-
вается в то же число раз, что и В, как это было установлено для нитроглицери-
нового пороха Н.
резкое снижение скорости горения на каждый процент добавляемой воды,
вопреки теории газофазного горения, наблюдается (рис. 240) в начале,
при малых содержаниях воды; затухание наступает при скорости, состав-
ляющей меньше половины от скорости горения неразбавленного вещества.
Кривая, выражающая изменение скорости горения в записимости от со-
держания воды,. сходна с кривой, выражающей изменение скорости
горения с изменением начальной температуры. При этом (по расчету)
снижение температуры горения при снижении начальной температуры от
100 до 0° С (но не ниже) приводит к такому же уменьшению скорости го-
рении, иак и снижение температуры горения путем добавки соответствую-
щего количества воды. Таким образом, влияние воды можно рассматри-
вать действительно как простое термическое разбавление.
5£нго
Рис. 240. Влияние влажности
на скорость горения пирокси-
лина №1 при разной темпера-
туре (в °C)
1 —85; 0 — 20
Рис. 241. Влияние инерт-
ных примесей на скорость
горения желатинированного
нитроглицерина (97 : 3)
1 -Н,О; 2 — (NHJ.CO,
Интересен вопрос о количественном соответствии теории обоих ука-
занных случаев уменьшения скорости горения. Этот интерес повышается
тем обстоятельством, что аналогичное изучение влияния инертных приме-
сей (карбонат аммония, вода) на скорость горения летучего веществе
(слабожелатинированный нитроглицерин) дало количественно сущест-
венно иную зависимость скорости горения от содержания примеси (рис.
241). При наибольшем содержании (4%) добавки (NH^JsCOa, при кото-
ром наблюдалось горение, скорость его равна аА от скорости горения не-
разбавленного вещества.
Согласно теории Зельдовича, как известно, отношение максимальной
и минимальной возможных скоростей горения при данном давлении не
должно превосходить Уе; для пироксилина же это отношение заведомо
больше.
Следует оговориться, что различие между летучими и нелетучими ВВ
в значительной мере условно. При повышении давлении температура ки-
пения летучих ВВ возрастает1 п соответственно уже при нескольких де-
сятках атмосфер все обычные ВВ имеют температуру кипения, сильно
превышающую температуру вспышки, что можно рассматривать как не-
кий условный критерий летучести ВВ. С другой стороны, Зельдович по-
казал, что если распад нелетучего ВВ (превращение его в газообразные
продукты) протекает эндотермично или с выделением лишь небольшой
части теплоты полного правращения, а последующие экзотермические
реакции идут в газовой фазе, то горение такого нелетучего ВВ можно ко-
личественно рассматривать как горение летучего ВВ; одиако это не един-
1 Правда, нужно учитывать, что при повышении давления уменьшается значение
теплоты испарения, что благоприятствует испарению в его конкуренция с химиче-
ским превращением; кроме того, при повышенных давлениях больше скорость горе-
ния, т. е. меньше время пребывания прогретого слоя при высокой температуре,
а следовательно, и возможность развития химического самоускорения в нем.
284
ственная возможность при горении нелетучих ВВ. Если реакции газифи-
кации конденсированной фазы достаточно экзотермична, то она может
стать ведущей. В частности, если повышать давление, под которым идет
горение летучих ВВ, то температура кипения будет возрастать и соот-
ветственно этому увеличиваться скорость превращения и тепловыделения
в конденсированной фазе. При некотором давления, особенно учитывая,
что плотность (концентрация) конденсированной фазы на три порядка
больше, чем газовой, скорость тепловой волны в конденсированной фазе
может стать соизмеримой и даже больше скорости волны, создаваемой
газофазной реакцией; тогда скорость горения будет определяться ско-
ростью перемещения той зоны жидкости, в которой за счет идущей в ней
реакции жидкость нагревается до температуры кипения.
Для этого случая Зельдович рассмотрел (пренебрегая изменением кон-
центрации реагирующих молекул при нагреве до температуры, близкой
к Т>{) скорость распространения*
<435)
где р, Ср, г|—плотность, теплоемкость и коэффициент теплопроводности
жидкости; Тк — температура кипения ее; Q — теплота реакции в конден-
сированной фазе (мл/см®); В — предэкспонент; Е— энергия активации.
Как и в случае горения, при котором ведущими являются газофазные
превращения, теория может быть сопоставлена с опытом расчетом зави-
симости скорости горения от начальной температуры и ее абсолютного
значения. Возможна и оценка по выражению зависимости скорости горе-
ния от давления, хотя эта оценка ввиду зависимости скрытой теплоты
испарения от давления, не учитываемой выражением (4.35), может быть
распространена лишь на ту область давления, где изменение X мало.
Многие черты горения бездымных порохов при низких давлениях гораздо лучше
согласуются с выводами, вытекающими из предположения о том, что ведущей
в этих условиях является реакция в конденсированной фазе. Беляев [232] рассчитал
скорость горения пироксилинового пороха нри тех весьма низких давлениях, при ко-
торых она не зависит от давления и соответственно определяется лишь реакциями
в конденсированной фазе. Этот расчет он провел по формуле Зельдовича для скоро-
сти тепловой волны, обусловленной реакцией в конденсированной фазе и нагреваю-
щей вещество до некоторой предельной температуры (в случае летучих веществ рав-
ной температуре их книения).
“= рСр(Тп —Го) j/"2ц^рВе“£/ДТо (4.36)
Приняв, по данным других исследователей, р = 1,6 г/сл<’, ср = 0,29 ккал/арад,
п = 5,5-10_* кял/см сеж • гроЗ,ф === 80 квл/е, В = 10” сек-1, Е = 44600 кал/малъ,
ГП = 573°К (300°С), Та = 363°К, Беляев получил и=3,4-10~1 см/сек, всего лишь
вдвое меньшую, чем было установлено ранее экспериментом. Учитывая возможные
ошибни в значениях ряда величин, входящих в формулу (4.36), согласие результата
расчета с опытом следует считать удовлетворительным.
Зависимость скорости горения от начальной температуры непосредст-
венно вытекает из формулы (4.35). Обозначив через <р члены выражения,
1 При горении твердых плавящихся ВВ уравнение (4.35) может быть написано
в виде; ___________________
ии~ 1 f 2т)рВе-^тк_ ЯГ® (4.35а)
cp(TK-To) + I. “Г
где L — скрытая теплота плавления БВ (в жал/г); р — плотность жидкой фазы
(в г/сж’). (Прим, рей.).
285
не зависящие от То, получаем
— То (4-37)
и для температурного коэффициента
где Ту — Т2 начальные температуры ВВ.
Из формулы (4.37) следует, что
1) зависимость скорости горения от начальной температуры должна
определяться изменением при изменении То количества тепла, необходимо-
го для нагревания конденсированной фазы от начальной температуры до
температуры кипения. В общем случае чем выше Tttj тем эта зависимость
должна быть слабее;
2) при повышении давления температурная зависимость скорости го-
рения должна уменьшаться в соответствии с ростом Тк;
3) с повышением начальной температуры скорость горения должна
увеличиваться тем сильнее, чем больше То, т. е. температурный коэффи-
циент скорости горения, рассчитанный для последовательных равных ин-
тервалов изменения То, с повышением То должен расти.
В координатах 1/и — То зависимость скорости горения от начальной
температуры должна изображаться прямой.
Эти следствия, за исключением второго, формально почти пе отлича-
ются от тех, которые следуют из гипотезы Михельсона и Малляра —
Ле-Шателье, но в противоположность им следуют из концепции, учиты-
вающей физико-химические и кинетические характеристики вещества.
До недавнего времени зависимость и (То) была определена для боль-
шинства изучавшихся летучих и нелетучих ВВ лишь при атмосферном
и близких к нему давлениях. Лишь для нитроглицеринового пороха это
определение было распространено на умеренно повышенные давления,
причем установлено, что зависимость и (То) с ростом давления ослабева-
ет. Для атмосферного давления скорость растет с Го не линейно, а быст-
рее. Этот рост в координатах 1/и — То выражается на графике прямой
линией. При этом точка пересечения этой прямой с осью абсцисс дает
разумное значение критической температуры, умеренно превышающее
значения, получаемые при стандартном определении температуры
вспышки.
Однако зависимость и (То) для нитрогликоля, как это показал Беляев
[230], меняет быть описана и выражением
Г'-е-^яГг, (4.38)
вытекающим из газофазного горения, и выяснить этим сопоставлением
реальность существования того или иного механизма нельзя.
Такую возможность дает выяснение влияния начальной температуры
и давления на температурный коэффициент скорости горения.
По газофазной теории, как отмечалось, этот коэффициент должен
слабо уменьшаться при повышении температуры; при горении, когда ве-
дущая реакция протекает в конденсированной фазе,— возрастать. В дей-
ствительности температурный коэффициент для всех изучавшихся ВВ
растет, при этом особенно сильно для заведомо нелетучего ВВ — нитро-
клетчатки; на эту зависимость, уменьшая ее, влияет также повышение
давления. «Горячие» (например гексотен) или относительно1 летучие (на-
пример тринитробензол) ВВ показывают меньшую зависимость темпера-
турного коэффициента от температуры.
’ Подразумевается соотношение летучести я скорости термического распада.
286
Таким образом, совокупность всех зависимостей, установленных при
изучении влияния начальной температуры на скорость горения, качест-
венно согласуется с предположением о ведущей роли экзотермической
реакции в газовой фазе.
Это заключение согласуется и с результатами попыток обнаружения
химического превращения в конденсированной фазе путем прерывания
горения.
Прекращение горения достигалось или проведением горения (при ат-
мосферном давлении) в суживающихся конических трубках или путем
заливания горящего в трубке заряда охлаждающей жидкостью (жидким
азотом). Наличие термического разложения в прилегающем к поверхности
слое определялось по снижению температуры плавления ВВ. Во всех слу-
чаях обнаружено более или менее значительное понижение температуры
плавления, наибольшее для тетрииа (25° С) и наименьшее для тротила
(1°С).
Следует, однако, еще раз подчеркнуть, что не наблюдается качествен-
ного различия в зависимости и(Т0) между летучими и нелетучими ВВ.
Больше того мы видели, что установленное влияние давления и началь-
ной температуры можно объяснить и с точки зрения газофазного горения.
В этом случае приходится, однако, делать некоторые допущения, непо-
средственно экспериментом не проверенные, в чем нет необходимости при
допущении ведения процесса в конденсированной фазе.
До сих пор мы рассматривали только зависимость скорости горения
от начальной температуры, которую дает реакция в конденсированной
фазе. Однако она позволяет оценить и зависимость м(р). Температура
кипения ВВ зависит от давления 1пр = — Используя это со-
*?Тп
отношение, получаем
и„ = = ррЕ/ах, (4.39)
где
В табл. 39 сопоставлены значения V, полученные при опыте (Vi) и при
расчете по формуле (4.39) и (Уз) и с учетом изменения Тк / (Тн — То)
с давлением (уз).
Таблица 39
Экспериментальные и расчетные показатели степени при
давлении в выражении ы = Вр* в интервале 1—50 ат
ВВ Vj
Гексогея 0,85 0,81 0,81
Тэн 1,00 0,87 0,845
Тетрил 0,69 0,74 0,73
Согласие получается вполне удовлетворительным, особенно если учесть
недостаточную надежность значения X для высококипящих ВВ. Помимо
этого, при повышении давления X уменьшается и это должно приводить
к увеличению V. Возможно, впрочем, что Это влияние ослабляется вслед-
ствие уменьшении времени пребывания ВВ в прогретом состоянии и умень-
шения химического самоускорения реакции.
Формула (4-35а) позволяет рассчитать и абсолютные значения скоро-
сти горения названных В В; результаты расчета, приведенные в табл. 40,
показывают, что экспериментальные значения отличаются от расчетных
не больше чем в 4 раза. Учитывая весьма приближенный характер многих
287
величин, входящих в выражение (4.35а), следует признать такое согласие
вполне удовлетворительным.
Таким образом, и в этом применении выражение для ведущей реакции
в конденсированной фазе дает результаты, еще более близкие к экспери-
ментальным, чем формула газофазовой реакции, особенно при использо-
вании для расчета в последнем случае константы скорости термичеслого
распада. Следует добавить, что расчет для конденсированной фазы ближе
к экспериментальным данным, так как, во-первых, протекание реакции
в жидкости больше соответствует условиям медленного распада и, во-вто-
рых, различие в температурах гораздо меньше, чем если данные медлен-
ного распада экстраполировать на максимальную температуру горения.
Поэтому в целом теории ведущей реакции в конденсированной фазе не
хуже, а скорее даже лучше описывает экспериментальные данные, полу-
ченные для плавящихся вторичных ВВ, в отношении абсолютной величи-
ны скорости, ее зависимости от начальной температуры, а также от дав-
лении.
Возможно, что нет оснований противопоставлять обе концепции; можно
представить себе протекание и взаимодействие при горении экзотерми-
ческих реакций как в газовой, так и в конденсированной фазе. При изме-
нении условий горении удельный вес каждой из этих реакций меняется,
соответственно горение в большей или меньшей степени приобретает чер-
ты, характерные для одного из режимов. Количественные заключения
о соотношении обеих реакций можно будет, по-видимому, получить лишь
путем определения температурного профиля при горении и расчета на
этой основе удельного веса каждой из реакций.
Есть и другая возможность, ограничивающая развитие реакции в кон-
денсированной фазе для нелетучих ВВ, в известной мере аналогичная
испарению.
Эта возможность заключается в диспергировании значительной части
твердого ВВ при горении. В общем виде диспергирование связано с со-
четанием трех особенностей химического превращения ВВ при горении -
переходом конденсированного ВВ в газы, имеющие гораздо меньшую
плотность, с протеканием при горении химического превращения не толь-
ко на поверхности, но и на заметной глубине в прогретом слое конденси-
рованной фазы и с неравномерностью химического превращении в про-
странстве.
Первые две особенности присущи также и летучим плавящимся ВВ,
но ограничение температуры поверхностного и прогретого слоев эндотер-
мическим процессом испарения, возможность выравнивания локальных
разогревов и концентрации продуктов превращения путем диффузии
и конвекции затрудняет возможность диспергирования и оно наблюдает-
ся только при некоторых специфических условиях. Некоторые исследова-
тели, впрочем, допускают возможность диспергирования жидкости путем
отрыва от поверхности и увлечения ее капель быстро оттекающим газом
[233]. Особенно характерным диспергирование является для твердых
веплавящихся ВВ или же ВВ, переходящих при нагреванни в состояние
очень вязкой жидкости.
Значение диспергирования для осуществления протекания химических
реакций в глубине заряда легко может быть иллюстрировано па примере
горения порошкообразных ВВ. В этих условиях газообразные продукты
горения (а вместе с ними и химическое превращение) могут проникать
в заряд и воспламенять его на значительной гдубние.
Возникновение взвеси частиц ВВ здесь обусловлено тем, что распро-
странение реакции идет неравномерно: в глубь заряда по поверхности
частиц ВВ скорость его больше, чем скорость распространения ее от по-
верхности частицы к ее центру. Такое явление наблюдается, например,
при горении крупнодисперсяого пороха малой гравиметрической плотности..
288
Таблица 40
Расчетная скорость горения гексогена, тана и тетрила
[по уравнению (4.35а)]
А. ГЕКСОГЕН
Давление, кг/см1 Примечание
i 5 10 25 50
Темиература кипе- ния, “К 613 667 690 725 760 Ср=0,24 кал/г-град
ер(Тк-Го) + Ь • • 106,1 119,1 124,6 133,0 141,4 tj=4-10-4 кал/см- • сек -г рад
ии (расч.), г/смг-сек 0,045 0,18 0,30 0,63 1,22 Е = 41 000 кал/молъ
ии (эксп.), г/см*-сек 0,054 0,21 0,33 0,94 1,51 1g А = 15,5
врасч/ывксп • • • • • 0,83 0,86 0,91 0,67 0,81 р = 1.5 г/сл^ К = 26 000 кал/молъ L = 29,3 кал/е Та = 293° К.
В. ТЭН
Давление, ке/сл" Примечание
20 25 40 50
Температура кипении, °К 631 640 656 665 ср = 0,4 кал/г-град
ер(Тк-Т0)-£ 152,9 156,5 162,9 166,5 tj = 4-10-4 кал/см-сек-
град
ми (расч.), г/см*сек .... 0,10 0,13 0,18 0,22 Е = 40000 кал/молъ
1g А = 15,6
вм (эксп.), .... 0,38 0,47 0,76 0,97 р = 1,5 г/см3 7. = 23 000 кал/молъ
мрасч'ивксп 0,26 0,28 0,24 0,23 L = 17,7 кал/г Го- 293° К.
В. ТЕТРИД
Давление, Примечание
1 12 25 30 50
Температура кипе- ния, °К ср(Тк-Го) + £ • • им (расч.), г/см*сек им (эксп.), г/см-сек “расч/^аксн • • • • 583 86,2 0,045 0,07 0,64 653 102,3 0,25 0,36 0,70 680 106,5 0,45 0,58 0,78 687 110,1 0,56 0,66 0,85 710 115,4 0,81 0,89 0,91 ср = 0,23 кал/г-град ?] - 2,5 -10"4 кал/см- •сек-г рад Е — 38 400 кал/молъ 1g А = 15,4 р = 1,5 г/см* X, = 26 000 кал/молъ i = 19,5 кал/г Го = 293° К
|9 К. К. Андреев
289
При этом газообразные продукты горения легко проникают в зазоры меж-
ду частицами пороха, горение идет в слое большой толщины; отходящие
газы увлекают горящие частицы пороха.
В случае применения сильно уплотненного индивидуального ВВ 1, сво-
бодного от пор, такой механизм проникновения, очевидно, не осуществим
при условии, разумеется, если поры не образуются при горения, напри-
мер, путем растрескивания. Однако даже гомогенная реакция в твердом
веществе, идущая с образованием газов, неизбежно приводит уже на ран-
них своих стадиях к нарушению целостности кристаллической решетки,
к образованию взвеси ВВ в газообразных продуктах его распада. Если же
ВВ кинетически неоднородно, то условии образования взвеси еще более
благоприятны. При прогреве прифронтового слоя в нем идет химическое
превращение. Это превращение в случае твердых ВВ имеет локальный са-
моуСкОряющийся по топохимическому или цепному закону характер и
начинается с появления отдельных, сравнительно немногих зародышей
реакции. Возникновение этих очагов распада происходит с трудом, с боль-
шой анергией активации; рост зародышей—автокаталитический или цеп-
ной — идет гораздо легче, с меньшей энергией активации*.'
Существенную роль может играть также наблюдавшаяся в ряде слу-
чаев ступенчатость течения реакции, проявляющаяся в наличии двух мак-
симумов на кривой скорости. При разогреве возникает первичная локаль-
ная реакция, идущая с выделением газов, которые и уносят большую часть-
вещества в неразложившемся виде; после некоторого времени полета, со-
ответствующего периоду индукции, предшествующему развитию второй
реакции, происходит сгорание частиц в струе, имеющее место на значи-
тельном расстоянии от поверхности конденсированной фазы3. Косвенным
свидетельством в пользу такого механизма является наблюдение Беляева,
заключающееся в том, что гремучая ртуть, нагревавшаяся некоторое время
при повышенной температуре, горит медленнее. Так оно и должно быть
в свете рассматриваемой гипотезы, поскольку предварительный нагрев вы-
зывает частичное медленное прохождение первичной реакции, играющей
важную роль в процессе диспергирования. Гарнер [234] при термическом
разложения гремучей ртути наблюдал распадение кристалла на началь-
ных ступенях разложения на ряд осколков. При горении этот распад дол-
жен происходить более бурно и может сопровождаться разлетом осколков,
как это наблюдалось Боуденом и сотр. [235] при горении ’ отдельных кри-
сталлов некоторых ВВ.
Если нагревать азид кальция в пробирке, то при вспышке совершен-
но отчетливо видны летящие горящие частицы; отдельные из них летят
даже независимо от общего потока газов по реактивному принципу. Пос-
тавщиком газов может служить не только одна химическая реакция. Если
ВВ содержит много растворенного воздуха, выделяющегося при нагрева-
ния, или легколётучие вещества, например воду, переходящую при этом
1 В случае смесей, экзотермическое превращение которых возможно в результате
взаимодействия компонентов фаз, неравномерное распространение горения может, не-
видимому, иметь место в виде «контактного» гогреиия (стр. 231) и при отсутствии лор.
Для сильно уплотненных индивидуальных ВВ нет оснований допускать преимуще-
ственное раскространеияе химической реакции по поверхности частиц по сравнению
с распространением в глубь частицы.
а Негомогенному началу и течению химического процесса способствует кинети-
ческая неоднородность вещества в случае твердого порошка. Энергия активации на
поверхности и внутри частицы может быть здесь существенно различной. При быст-
рых процессах может играть существенную роль также саморазогрев за счет экзо-
термической реакции.
’ Более наглядно можно представить себе такой процесс горения, если допу-
стить, что мы имеем порошкообразную смесь частиц двух ВВ — с низкой и высокой
температурами воспламенения. Возможен случай, когда гореть будет одно вещество,
а частицы другого будут уноситься струей газов, а при малой скорости его горения
и при сильном охлаждении газов даже выбрасываться иа трубки несгоревшнми.
290
в пар, то они, выделяясь в виде пузырьков при разогреве поверхностного
слоя, вступающего в горение, также будут способствовать отрыву частиц
конденсированной фазы при горении.
11ри описанном механизме горения влияние различных факторов, на-
пример давления, температуры и др., на горение может, очевидно, быть
существенно иным, чем при нормальном «теплопроводностиом» процессе,
так как эти факторы будут влиять не только обычным образом на ско-
рость газофазной реакции, но и на течение газифицирующих процессов.
Так, повышение давления, замедляя отток газов, будет, несомненно*
приводить к повышению эффективной температуры в диспергирующемся
слое. Помимо этого, давление будет влиять и на длительность горения
диспергированных частиц, на среднее расстояние их сгорания от места
образования и т. д.
Диспергирование не может играть существенной роля при горении ле-
тучих ВВ в обычных условиях. Однако и для них можно создать тайне
условия, при которых будет происходить диспергирование. Поясним это
на примере следующего опыта: будем медленно нагревать значительную
навеску пикриновой кислоты; при достижении определенной температуры
наступит бурное разложение, сопровождающееся выбросом снопа горя-
щих капель ВВ. Аналогичный характер процесса наблюдается при горе-
нии нагретого до возможно высокой температуры тротила: горение ста-
новится пульсирующим и преходящее его ускорение сопровождается вы-
бросом с газами капель жидкости.
При длительном нагреве указанных ВВ. в них проходит процессы раз-
ложения, сопровождающиеся накоплением автокатализаторов. В резуль-
тате этого скорость химической реакции сильно возрастает и вероятность
ее может стать соизмеримой с вероятностью испарения, особенно если
учесть большую толщину слоя, где может пройти реакция, по сравнению
с поверхностным слоем, в котором идет испарение. Вследствие этого пи-
криновая кислота, нагретая до 300° С, прн большой навеске не успевает
заметно испариться до того, как в ней возникнут очаги интенсивного раз-
ложения; это и приводит к описанной картине вспышки. То же происхо-
дит и при горении сильно нагретого тротила; испарение происходит
только на поверхности жидкости, а разложение — в горячем слое значи-
тельной толщины и с большим содержанием автокатализатора. Возникно-
вение внутри слоя очагов распада ведет к их быстрому росту как вслед-
ствие саморазогрева, так и вследствие автокатализа. Очевидно, что число
очагов невелико; если бы их было очень много, то частицы (в данном слу-
чае капли) ВВ были бы очень малы и диспергирование было бы трудно на-
блюдать, так как частицы сгорали бы очень быстро.
Возможно, что эти соотношения выпонияются при горения некоторых
твердых ВВ, например нитроклетчатки, в тех обычных его условиях, в ко-
торых не наблюдается видимого диспергирования. Если для той же нит-
роклетчатки изменить условии горения в сторону уменьшения скорости
вознияновения зародышей и увеличения скорости их роста, то можно на-
блюдать, как и для тротила, интенсивное диспергирование.
Таким образом, горение с диспергированием явниется общим меха-
низмом горения, и разница в этом отношении между вторичными и быст-
рогорящими ВВ заключается лишь в том, что для последних этот меха-
низм является нормальным, для первых наблюдается лишь в некоторых
(необычных) условиях горения *.
1 Есть основания полагать, что этот механиам проявляется и при детонации. При
сжатии взрывчатого вещества ударной волной (даже в случае гомогенного вещества)
разложение может возникать в отдельных точках и развиваться в виде очагов по
теиновому или даже по цепному механизму, что будет приводить к диспергированию
ВВ. Такой путь образования во фронте детонационной волны смеси частиц ВВ и про-
дуктов его распада, которое допускают современные теории детонации, представляет-
ся иам для гомогенных ВВ наиболее вероятным.
19* 391
Нет достаточного материала для того, чтобы, не ограничиваясь кон-
статацией различия, ответить на вопрос о его причинах; по-видимому, оно
связано с различием в удельной поверхности, а также в скорости (энер-
гии активации) появлении зародышей и скорости (энергии активации)
их роста в различных условиях: например, чрезмерно большая, так же
как и чрезмерно малая, скорость появления зародышей не благоприят-
ствует проявлению диспергирования; изменение температуры ВВ, меняя
соотношение между скоростями обоих процессов, должно отражаться и на
проявлении диспергирования.
Существенны также условия роста зародышей в связи с величиной
скорости горения: при неблагоприятных соотношениях очаги газообра-
зования могут не успевать развиться.
Во всяком случае общая теория горения должна рассматривать не
только разобранный Зельдовичем и Беляевым случай горения жидких ВВ
при низких давлениях в условиях равномерного испарения, но и тот слу-
чай, когда значительная, а иногда даже бблыпая часть неразложившегося
ВВ увлекается потоком газообразных продуктов горения и продолжает
реэлагаться и реагировать с ними более ини менее далеко от границы не-
разложившегося ВВ, а иногда и совсем избегает химического превре-
щения.
Одна из сторон этого процесса — превращение конденсированной фазы
во взвесь частиц ВВ в газообразных продуктах его превращения — может
быть описана, исходя из рассмотренной выше теории ведущей экзотерми-
ческой реакции в конденсированной фазе, причем максимальная темпе-
ратура конденсированной фазы ограничивается некоторым пределом. При-
менительно к кипящим ВВ этот предел принимается равным температуре
нияения; применительно к диспергирующимся ВВ можно представить себе
некоторый аналог этой температуры, когда скорость диспергирующей
реакции становится столь большой, что конденсированная фаза переходит
из своего первоначального состояния в состояние взвеси.
Приложимость этой теории и к случаям диспергирования является,
по-видимому, причиной сходства зависимостей, наблюдаемых при горении
диспергирующихся и летучих ВВ при повышенном давлении или диспер-
гирующихся и ограниченно летучих ВВ, где реакции в конденсированной
фазе происходят и при низких давлениях.
Одним из следствий диспергирования, отличающем горение на этом
режиме от того случая, когда ведущая экзотермическая реакция в конден-
сированной фазе не образует взвеси, а только газы, является наличие
большой поверхности коятанта между газообразными продуктами реакции
и частицами взвеси. Поэтому теплообмен сильно возрастает, а соответст-
венно увеличивается и количество реагирующего вещества на единицу
площади сечения заряда. Это напоминает процесс, который осуществляет-
ся при проникании горящих газообразных продуктов горения в глубь по-
рошкообразного пористого заряда. Разница лишь в том, что в одном слу-
чае газы проникают в порошок, а в другом случае порошок (образующиеся
при диспергировании частицы) проникает в газ.
До сих пор речь шла о диспергирующей реакции, протекающей с вы-
делением тепла и ведущей благодаря этому процесс горения.
Такое диспергирование назовем условно «активным». Можно предста-
вить себе и другой случай — «пассивного» диспергирования, когда это дис-
пергирование не является ведущим, а протекает вынужденно, т. е. под
влиянием теплоприхода от слоя реагирующей в газообразных продуктах
взвеси, прилегающего к поверхности конденспроваяного В В.
Если температура взвеси достаточно высока, то скорость теплопереда-
чи будет большой; велика будет и скорость перемещения зоны дисперги-
рования. Кстати, способность к горению здесь может быть и не слишком
большой; при слишком интенсивном диспергировании газовая часть
292
взвеси может оказаться не в состоянии «переварить» слишком обильно
поступающие в нее частицы, которые, чрезмерно охладив горячую зону,
могут привести к затуханию горения. Соответствующая минимальная
скорость горения может быть (сравнительно с летучими ВВ) относитель-
но велика, так как все зависит от скорости «переваривания» и скорости
поступления «пищи», последняя не должна быть чрезмерно большой.
Точно так же влияние различных факторов, например, давления, на
скорость горения будет двойственным, складываясь из влияния на ско-
рость тепловыделения в горящей зоне и скорость диспергирования.
В принципе возможен и такой случай, что повышение, например, давле-
ния, увеличивает скорость в горячей зоне, но сильное уменьшение дис-
пергирования приведет к уменьшению скорости горения.
Вопрос о том, для каких веществ и условий реализуется «активное»
или «пассивное» диспергирование, может быть непосредственно выяснен
лишь установлением профиля температуры и тепловыделения в ходе го-
рения. Косвенные заключения в этом отношенни позволяют сделать не-
которые данные по исследованию термического распада и горения ряда
быстрогорящих ВВ.
ГЛАВА ПЯТАЯ
НЕУСТОЙЧИВОЕ ГОРЕНИЕ ВВ
В предыдущих главах мы рассмотрели устойчивое стационарное го-
рение ВВ, т. е. гореиие, идущее на всем своем протяжении равномерно
с постоянной скоростью, определяемой свойствами ВВ и постоянными
внешними условиями (давление, температура, условия теплоотдачи и др.).
Опыт показывает, однако, что при известных условиях горение пере-
стает быть устойчивым, становится неравномерным. Изменение скорости
горения может быть периодическим, причем средняя скорость остается
постоянной (пульсирующее горение). Может иметь место также непре-
рывное, равномерное или пульсирующее, возрастание скорости.
При этом впоние естественны те случаи, когда изменение скорости го-
рения происходят вследствие изменения внешних условий, например
из-за возрастания давления при горении ВВ в замкнутом постоянном объе-
ме, вследствие прогрессирующего внешнего разогрева ВВ. Однако уско-
рение горения может иметь место и при неизменных внешних условиях.
Нарушение стационарности горения может заканчиваться возникновени-
ем детонации.
Вопрос об условиях устойчивости горения представляет большой тео-
ретический и практический интерес. Как уже указывалось, современное
техническое применение ВВ основано на существовании трех их классов,
главное различие между которыми заключается в степени устойчивости
горения: минимальной у инициирующих ВВ, максимальной у порохов;
вторичные ВВ занимают в этом отношении промежуточное положение.
Для того чтобы обеспечить наибольшую степень устойчивости горения ВВ,
необходимую при их применении в качестве порохов, нужно знать те фак-
торы, которые обусловливают устойчивость горения. То же самое следует
сказать и о предотвращении перехода горения в детонацию при пожарах
с участием ВВ на заводах и складах.
Нестационарное горение обычно наблюдается при воспламенении ВВ
на начальном участке горения.
Наконец, так называемая вспышка ВВ, возникающая при относитель-
но медленном равномерном нагреве некоторого количества ВВ до высоких
температур, также может протекать как нестационарное горение. Подоб-
ного рода явление может происходить, и притом в больших масштабах,
при самовоспламенении ВВ в производственных аппаратах.
I. ОБЩАЯ ТЕОРИЯ УСТОЙЧИВОСТИ ГОРЕНИЯ [236]
При горении ВВ полностью или частично превращается в газы. Вслед-
ствие малой плотности, особенно при высоких температурах, достигаемых
при горении, эти газы имеют объем, во много раз превосходящий объем ВВ.
Если бы горение происходило в собственном объеме ВВ, то продукты его
294
имели бы очень высокое давление, измеряемое сотнями тысяч атмосфер.
Если же поверхность горящего заряда соединяется с атмосферой, то газы
расширяются и давление у поверхности ВВ получается гораздо меньше.
Однако оно все же имеет некоторое конечное значение, а именно такое,
какое нужно, чтобы сообщить газам то количество движения, которое они
приобретают при расширеши. Это количество движения тем больше, чем
больше скорость образования газов, т. е. скорость горения, и чем меньше
давление окружающей среды, от которого зависит конечная плотность
и скорость движения расширившихся газов. Одиако динамическое повы-
шение давления в свою очередь влияет на горение, поскольку скорость
горения растет с увеличением давления. Увеличение же скорости горении,
очевидно, означает увеличение количества образующихся в единицу вре-
мени газов и, следовательно, скорости их движения, т. е. увеличение ди-
намического повышения давления. Обе эти характеристики процесса, та-
ким образом, взаимно связаны. Одиако характер связи различен. Горение,
т. е. образование газов может происходить и независимо от оттока газов,
отток же газов возможен лишь в том случае, если они образуются, т. е.
если идет горение.
< И образоваяие газов и отток их зависят от давления. Возможность
устойчивого равновесия между газоприходом и газоотводом, очевидно, оп-
ределяется соотношением скоростей роста их с давлением.
Если газопряход растет с давлением быстрее газоотвода, то устойчи-
вого равновесия между ними быть не может — увеличение газоприхода
не будет компенсироваться газоотводом. Если газоотвод растет быстрее
газоприхода, то такая компенсация осуществляется. Эти соотношения ста-
новятся особенно наглядными при графическом их представлении.
В качестве простейшего случая рассмотрим горение цилиндрического
заряда, находящегося в прочной трубке сечением 1 см2 и горящего с одно-
го, открытого торца при внешнем давлении ро. Образующиеся газы горе-
ния оттекают в направлении, противоположном направлению распрост-
ранения горения со скоростью, зависящей от скорости их образования
(рис. 242).
Происходящие при горевди процессы образования газов и истечения
их из трубки в зависимости от давления внутри трубки представлены на
рис. 243. По осн абсцисс отложено давление у поверхности фронта горе-
ния. По оси ординат отложены значения — количества газов, образу-
ющихся при данном давлении в 1 сек с 1 см2 поверхности фронта горе-
ния (массовая скорость горения) и та — количества газов, вытекающих за
единицу времени при давлении внутри трубки р и внешнем давлении ро
через каждый квадратный сантиметр ее сечения (расход газа).
Линия, изображающая расход газа в зависимости от давления, имеет
как известно из газодинамики, следующий внд. При р = ро, т. е. если дав-
ление у фронта равно внешнему давлению, расход газа равен нулю. При
увеличения давления расход газа возрастает, причем вначане этот рост
идет по кривой, а затем, начниая с давления, приблизительно в 2 раза
превышающего внешнее давление, расход газа возрастает прямо пропор-
ционально давлению:
та = А'р, (5.1)
где К — постоянная, зависящая от температуры, плотности и теплоемко-
сти газа.
Линия газоприхода mi = есть по существу линия, изображающая
массовую скорость горения как функцию давления, так как 5 — площадь
поверхности горения мы приняли равной единице. Применительно к вы-
полняющейся для многих ВВ зависимости
ия = А + Вр
295
построим на графике а, рис. 243 для две прямые / и II соответственно
двум значениям В', В < К и В > К.
Лег ио показать, что при В < К горение будет устойчивым. В самом
деле, если мы зажжем ВВ при давлении ро, то при этом давлении гаэоотвод
равен нулю, газоприход изображается ординатой m'i- Из-за разности га-
зоприхода и газоотвода давление у фронта горения будет расти до тех
пор, пока не будет достигнуто давление Pi, при котором газоотвод стано-
вится равным газоприходу. Дальше при В < Л, давление расти не может,
Рис. 242. Схематическое изоб-
ражение движения газов яри
горении в трубке
Рис. 243. Зависимость скорости образования га-
зов при горении и их оттока от давления
так как при больших давлениях расход газа больше газоприхода. Поэтому,
если бы давление по какой-либо случайной причине и выросло, напри-
мер до значения Ра, оно неизбежно затем должно снизиться до значе-
ния рь
Так как К при данном составе газов величина постоянная, то газодина-
мическая устойчивость горения зависит от значения В (точнее du / dp) при
равновесном давлении. Чем меньше В, тем больше устойчивость. Таким
образом, независимость скорости горения от давления (В = 0), а тем
более ее уменьшение при повышении давления (В <_ 0} дают наиболь-
шую устойчивость горения. Напомним, что первая зависимость (В = 0)
наблюдалась в определенной области давлений для ряда быстрогорящих
ВВ, вторая (В < 6) — для пикрата калия н некоторых других, сходных
с ним солей.
Легко показать, что при В > К давление в трубке будет непрерывно
возрастать, и устойчивого равновесия между газообразованием и газоотво-
дом ни при каком давлении установиться не может.
Построение соответствующих графиков зависимости скорости горения
от давления нм = А + Вру при v < 1 и при v > 1 (рис. 243, графики б
и в) показывает, что в первом случае горение может быть либо неустой-
чивым при всех давлениях, когда кривая газоприхода II расположена це-
ликом над линией газоотвода, либо в некотором интервале давлений (до
р ~ pz, кривая I графика в) горение может быть устойчивым, при боль-
ших же давлениях — ускоряющимся.
Подчеркнем, что равенство газорасхода и газоприхода не является до-
статочным для устойчивости горения. Это наглядно видно на графике
рис. 243.
В точке А газоприход и газорасход равны, и равновесие их устойчи-
вое, так как если бы давление повысилось, то газорасход стал бы больше
газоприхода, и давление упано бы вновь до pj.
Иначе обстоит дело в точке С, где газорасход и газоприход также
равны. Бели давление опустится ниже ра, то оно будет продолжать падать,
так как в этом интервале давлений газоприход меньше газорасхода.
Напротив, если давление повысится несколько выше ра, то оно будет про-
должать расти, так как газоприход будет больше газорасхода. Таким об-
разом, отклонение от равновесия в обе стороны будет вызывать не возвра-
296
и
д
£
300
щеиие к нему, а дальнейшее усиление первоначального отклонения. Это
и означает неустойчивость равновесия.
Таким образом, возможность устойчивого горения при данном давле-
нии определяется соотношением между скоростью горения (точнее скоро-
стью газообразования) и скоростью ухода газов при этом давления, или,
правильнее говоря, соотношением между ускорением газоприхода и уско-
рением газорасхода с давлением.
Если при равенстве газоприхода и газорасхода газоприход растет
с давлением быстрее, чем газорасход, то горение неустойчиво и, наоборот,
если газоприход растет медленнее, чем газорасход, то мы имеем устойчи-
вое горение,
Следовательно, при рассмотренном газодинамическом подходе к вопро-
су об устойчивости горения эта устойчивость должна определяться значе-
ниями величины К — тангенса угла наклона прямой газорасхода и вели-
чины В = dnM /dp — производной скорости горения (точнее, скорости га-
зообразования) по давлению. Поскольку те инн иные параметры (напри-
мер давление, температура, относительная плотность) внияют на В или
К, то они будут влиять и на устойчивость горения.
На основе этих общих соображении об условии устойчивости горения
рассмотрим имеющиеся данные применительно к конкретным ВВ и кон-
кретным условиям горения.
Газорасход при повышенных давлениях, как указывалось, прямо про-
порционален давлению. Коэффициент пропорциональности
к / 2 m-i , Г 2Т0 _ ..Г к / 2 m-i у
k-hlU + 1/ И Po^Tf~a^ + + J Л
, Г к / 2 т-1 ,/' 2 273 Л7
Ро 224107'1 ~ ° V fc + iU + 1/ V 1,013.10»-22410Т1 ’
(5-2)
где о — сечение отверстия для выхода газов (с№), а к = ср / с„; М —
молекулярный вес газов (в г); ро — атмосферное давление — 1,013-
10е дин/'см2; vo — удельный объем газов (в см3/г); 7’о -273° К; h —
температура газов (в °К).
* Для двухатомных газов
> /41(гпГ‘,=0'485
И
j К = а 0,485 0,0001555 ~ = 0,0000752 з |/ ^
’> Из формулы видно, что К не является строго постоянной величиной,
но может меняться соответственно различным возможным значениям
температуры газообразных продуктов горения и их молекулярного веса.
Для оценки величины К, необходимой для сопоставления теории с опытом,
рассчитаем ее значение, приняв для простоты, что о = 1, температура горе-
t. ния равна 2730° К, а средний молекулярный вес газов равен 28 (окись угле-
рода и азот). Тогда численное значение К равно -'7,4 (г/сек см2}-. (кг/см2).
При повышенных температурах значение К несколько уменьшается
f соответственно уменьшению cp/cv; если ВВ (например гремучая ртуть)
образует тяжелые тазы, величина К становится больше. Если горение идет
с диспергированием, так что в газы превращается лишь часть конденси-
’ рованного ВВ, то величина К уменьшается, так как уменьшается скорость
движения газов.
Для оценки устойчивости горения нужно сравнить значение К со зна-
чением duM/dp из выражения зависимости скорости горения от давления
(табл. 41). Если эта зависимость при нормальном горения является
297
Таблица 41
Значение производной массовой скорости горения по давлению (du/dp)
вв du/dp, г/свксл’/хг/сл* ВВ йиЦр,
Коллоксилин 0,0162 Тетрил ........ 0,0513
Тан 0,0180 Метилнитрат жидкий 0,1330
Нитрогликоль желати- Нитроглицерин тела- 0,1460
нированный 0,0290 тинированный . . . .
Нитрогликоль жидкий 0,0390 Гремучая ртуть .... ' 4,18
Пироксилин 1 . . . 0,0405 Т рин итротри аз идо бензол 0,85
Гексоген 0,0505
линейной, duM/dp есть постоянная величина, которую мы обозначили че-
рез В. Если зависимость и(р) иная, то duK/dp рассчитывалось графически
для определенного интервала давления.
Для всех изученных вторичных ВВ значения difa/dp гораздо (в 50—
21*0 рез) меньше, чем предельное значение, начиная с которого газоди-
намически невозможно устойчивое горение. Таким образом, способность
вторичных ВВ к устойчивому горению является теоретически вполне
естественной.
Те из инициирующих ВВ, которые в прессованном до большой отно-
сительной плотности состоянии способны к устойчивому горению, харак-
теризуются при низких давлениях более сильной зависимостью скорости
горения от давления, чем вторичные ВВ; значение dii^/dp для них при-
ближается к предельному.
При повышенных давлениях для изучавшихся в этом отношении ини-
циирующих и быстрогорящих ВВ отношение dii^/dp уменьшается; одна-
ко скорость гореиня по абсолютной величине велика, а следовательно,
велико и динамическое повышение давления.
Возможно, что те инициирующие ВВ, для которых не удалось экспе-
риментально наблюдать устойчивого горения, характеризуются еще боль-
шнми значениями duH/dpt что и должно приводить к невозможности
устойчивого равновесия между газоприходом и газоотводом, иначе гово-
ря, к невозможности устойчивого горения.
Однако сильная зависимость скорости нормального горения от давле-
ния не единственная возможная причина неустойчивости горения. Мас-
совая скорость горении равна нормальной его скорости, умноженной на
эффективную поверхность горения. До сих пор предполагалось, что по-
верхность горения равна площади торца горящего заряда. В действитель-
ности это не обязательно так — поверхность горения может становиться
значительно больше. Это осуществляется, например, если горение идет
с интенсивным диспергированием, фактическая поверхность горения тог-
да во столько раз больше сечения заряда, во сколько раз суммарная по-
верхность частиц больше площадя торца заряда. Диспергирование может
иметь не только химическую природу (газообразующая реакция в кон-
денсированной фазе), но и физическую — частицы ВВ могут образовы-
ваться в результате сильного растрескивания его кристаллов под вния-
инем теплового удара, вследствие выделения паров легиопетучих приме-
сей или газовых включений и т. д.
Увеличение поверхности при горении особенно естественно, если ВВ
характеризуется значительной газопроницаемостью и горение происхо-
дит в слое некоторой конечной толщины. Такое, конвективное, горение
отличается не только значительно большей скоростью, чем нормальное,
т. е. гораздо большим Др, но и гораздо большим du^/dp, поскольку это от-
ношение в данном случае определяется не только и не столько отиоситель-
298
но слабой зависимостью нормальной скорости горения от давления, сколь-
ко гораздо более сильной зависимостью толщины горящего слоя (поверх-
ности горения) от давления. Эта толщина определяется Др, которое мало
по сравнению с р; поэтому небольшое изменение давления, мало меняю-
щее скорость нормального горения, будет большим по отношению к Др,
сильно изменит толщину горящего слоя и duM/dp.
Имеющиеся немногочисленные данные говорят скорее в пользу того,
что ускорение горения инициирующих ВВ, приводящее к переходу его
в детонацию, обусловлено не чревмерно сильной зависимостью нормаль-
ной скорости горения от давления \ а возрастанием поверхности горе-
ния по одному из указанных выше путей. Ряд особенностей инициирую-
щих ВВ, в частности высокая температура и большая скорость горения
при низких давлениях, обусловливающие повышенную склонность их
горения к переходу на конвективный режим, способствуют переходу го-
рения в детонацию.
Таким образом, один из фундаментальных фактов, характеризующих
ВВ,— устойчивость горения вторичных ВВ и неустойчивость горения
инициирующих ВВ — естественно объясняется теорией.
Однако известно, что горение вторичных ВВ, в большинстве случаев
устойчивое, в некоторых условиях переходит в детонацию. Точно так же
и те инициирующие ВВ, которые по газодинамическому критерию долж-
ны гореть устойчиво и действительно горят так при определенных усло-
виях, например в сильно спрессованном состоянии, в вакууме, в других
условиях, например при меньших давлениях прессования, дают быстрый
переход горения в детонацию.
С точки зрения теории возникновение перехода горения в детонацию
в обоих случаях должно быть истолковано как следствие изменения зави-
симости скорости горения от давления в сторону увеличения duMjdp. Что-
бы выяснить, как осуществляется это изменение, рассмотрим последова-
тельно экспериментальные данные по горению жидких ВВ и порошкооб-
разных ВВ в тех условиях, когда нарушается стационарное течение про-
цесса, и теоретическое истолкование этого нарушения й.
II. ВЛИЯНИЕ ДАВЛЕНИЯ, ТЕМПЕРАТУРЫ И ФИЗИЧЕСКИХ
СВОЙСТВ НА УСТОЙЧИВОСТЬ ГОРЕНИЯ ЖИДКИХ ВВ
ТЕОРИЯ УСТОЙЧИВОСТИ ГОРЕНИЯ ВЗРЫВЧАТЫХ ЖИДКОСТЕЙ
В главе третьей мы рассмотрели экспериментальные данные по стаци-
онарному горению ряда жидких ВВ — метилнитрата, нитрогликоля, нит-
роглицерина и др. Однако горение всех этих веществ является стационар-
ным и равномерным лишь при определенных условиях. Основными из
этих условий являются давление и температура, при которых идет горе-
ние. Существенную роль играет также вязкость жидкости.
Экспериментальные исследования [237] дали следующую общую кар-
тину влияния различных факторов на характер горения.
1 Так, например, для смесей азида свпнца с жидкими вторичными ВВ в условиях
нормального горения скорость его лишь немного больше, чем вторичных ВВ. Для гре-
мучей ртути в свиьно уплотненном состоянии при больших давлениях скорость нор-
мального горении растет даже медленнее, чем при низких давлениях.
8 Ниже рассматриваются экспериментальные данные только по неустойчивому
горению жидких ВВ. Раздел о неустойчивости горения порошкообразных ВВ (кроме
той части экспериментальных данных по горению порошков, которые имеются
в гл. 3, стр. 195 и далее) К. К. Андреев написать не успел (Прим. ред).
299
Рис. 244. Зависимость сиорости
горения иитрогликоля от давле-
ния при различных температурах
Если давление лежит ниже некоторого предела, то горение не распрост-
раняется. Этот нижний предел обусловливается тепловыми потерями реа-
гирующего слоя как в окружающую среду, так и в глубь ВВ и зависит
поэтому от диаметра заряда, свойств материала, стенок оболочки и т. д.
При давлениях выше нижнего предела горение является устойчивым,
и скорость его растет с увеличением давления. Наблюдение за горением
показывает, что поверхность жидкости, оставаясь горизонтальной и ви-
димо спокойной, перемещается сверху
вниз. При некотором давлении, различном
для разных ВВ, характер горения меняет-
ся — оно становится пульсирующим. Пуль-
сация эта различна по своему характеру
для разных ВВ и в различных условиях
горения. При горении нитрогликоля по-
верхность жидкости начинает как бы дро-
жать, период пульсации, амплитуда коле-
баний поверхности малы.
У метилнитрата характер пульсации
тот же, но амплитуда и период колебаний
значительно больше. У нитроглицерина
при обычной температуре пульсация про-
исходит в форме, напоминающей вспыш-
ку, сопровождается расплескиванием жид-
кости и обычно приводят к затуханию го-
рения. На характер пульсации жидких ВВ
влияет также начальная температура.
Величина давления, при котором горе-
ние переходит на пульсирующий режим,
и значения скорости горения различны
для разных ВВ. Так, для метилнитрата это
происходит при 1,75 ат, причем скорость го-
рения составляет около 0,26 г/смг сек, для
нитрогликоля пульсирующее горение наступает при 17 ат и скорости го-
рения 0,79 г/см2сек, для нитроглицерина (t = 98° С) — при 0,4 ат и ско-
рости горения 0,12 г/см2сек.
Повышение температуры облегчает возникновение пульсации, снижая
давление, при котором она начинается.
На рис. 244 приведены результаты опытов И. А. Терешкина по влия-
нию температуры на значение давления перехода горения нитрогликоля
на турбулентный режим и зависимость скорости горения в этом режиме
от давления.
В тех случаях, когда пульсация приводит к затуханию горения, повы-
шение температуры затрудняет это затухание, приводя к повышению
давления затухания или даже (полностью предотвращая его. Так, для
жидкого и слабо желатинированного нитроглицерина при обычной темпе-
ратуре происходит затухание, при умеренно повышенной — горение идет
с пульсацией, при сильно повышенной — возникает взрыв. Для метилни-
трата повышение температуры приводит к тому, что вместо равномерной
пульсации возникает взрыв.
Повышение вязкости жидкости затрудняет возникновение пульсации.
Так, если жидкий нитроглицерин не горит при атмосферном давлении, ни-
троглицерин, желатинированный растворением в нем высокополимерного
вещества, нормально горит как при атмосферном, так и дри умеренно по-
вышенных давлениях. При этом чем выше вязкость желатины, тем выше
давление, при котором горение переходит на пульсирующий режим или
затухает. То, что стабилизирующее влияние желатинирующей добавки
связано именно с повышением вязкости, показывает отсутствие этого
300
влияния при добавке того же вещества не в виде полимера, а в виде моно-
мера.
Аналогично поведение нитрогликоля: горение жидкого нитрогликоля
переходит на пульсирующий режим при давлении около 20 ат, слабоже-
латинированное (97 •. 3) вещество равномерно горит даже при 450 ат.
Такова в основных чертах общая картина нарушения равномерности
горения и влияния на возникновение этого нарушения различных фак-
торов.
Теоретический анализ нарушений нормального характера горения
жидкостей был дан Зельдовичем {238] на основе взглядов Беляева и (не-
зависимо от экспериментальных исследований) Ландау [239].
Зельдович исходит из следующих соображений. Горение летучих жид-
ких ВВ идет при ниаких давлениях таким образом, что жидкость1 пре-
вращается в пары в результате нагрева путем теплопередачи от газооб-
разных продуктов реакции. Пары, нагреваясь дальше, реагируют, причем
выделяется тепло реакции. Однако наряду с реакцией в газовой фазе про-
исходит реакция в жидкости, при которой также выделяется тепло, вслед-
ствие чего в глубь жидкости распространяется тепловая волна. При
повышении давления, под которым происходит горение, увеличиваются
скорость реакции в газовой фазе и соответственно скорость горения. Од-
нако одновременно возрастает скорость реакции в жидкой фазе, так как
при повышении давления увеличивается температура на поверхности
жидкости, равная температуре ее кипения.
При определенных соотношениях между величинами энергии актива-
ция и теплоты испарения жидкого ВВ скорость волны нагрева, распро-
страняющейся в жидкости при горении, возрастает с давлением быстрее,
чем скорость горения. Поэтому, начиная с некоторого предельного давле-
ния, скорость волны нагрева может стать больше скорости горения. Тогда
вместо испареиня жидкости на поверхности горения происходит вскипа-
ние ее в слое некоторой конечной толщины, что ведет, с одной стороны,
к резкому увеличению поверхности горящей жидкости и, с другой —
к поступлению пузырьков пара из жидкости в зону горячих газообраз-
ных продуктов горения и в итоге к сильному увеличению количества сго-
рающего в единицу времени вещества, что приводит к детонации.
Зельдович дал выражение для предельного давления рпр, которое поз-
волило приближенно рассчитать его значения для нитрогликоля и ме-
тилнитрата [240]. Однако эти расчеты дают значения предельного давле-
ния, на несколько порядков превышающие экспериментальные его зна-
чения. Кроме того, как это указывает и сам Зельдович, выведенные им
соотношения применимы лишь в том случае, если рп₽ значительно ниже
критического давления, которое для нитрогликоля хотя и не установлено,
но по аналогии с другими веществами не превышает значительно 50 ат.
Поэтому полученные значения предельного давления не имеют физиче-
ского смысла.
Следует, однако, указать, что использованные при расчете данные по
теплоте реакции рассчитаны по составу газообразных продуктов горе-
ния, а не непосредственно определены для реакции в жидкой фазе; кро-
ме того, при расчете величина предельного давления в сильной степени
зависит от отношения скорости горения к скорости тепловой волны. Ско-
рость же горения, как показывает опыт, зависит от вязкости жидкости,
что теорией не учитывается. Далее реакция в жидкой фазе сопровожда-
ется выделением пузырьков газа, перемешивающих ее, что может влиять
на коэффициент передачи тепла. Из-за этих возможных источников неточ-
1 По существу эти соображения приложимы и к твердым ВВ, плавящимся при
горении перед превращением в пары.
301
ности количественные расхождения результатов расчета и опыта сами по
себе не имеют решающего значения.
Известную дополнительную возможность проверки гипотезы пред-
ставляет сопоставление температурной зависимости предельного давле-
ния, которая из нее вытекает, с экспериментальной. По формулам Зель-
довича, изменение предельного давления с температурой зависит от изме-
нения с температурой скорости горения и скорости тепловой волны. По-
1
следняя пропорциональна отношению i т. е. относительно слабо
возрастает в области низких температур и сильно — при приближении
начальной температуры к температуре кипения. Поскольку скорость го-
рения нитрогликоля растет с давлением более равномерно и медленно,
то предельное давление уменьшается с повышением начальной темпера-
туры сначала медлеыио, а затем все быстрее и быстрее. При переходе от
20 к 100° С предельное давление уменьшается по расчету в 3,5 раза, что
согласуется с опытом. При более высоких температурах расчет дает бо-
лее сильное уменьшение предельного давления, чем это, по-видимому,
имеет место в действительности.
В рассматриваемом механизме возникновения взрыва есть, однако,
один принципиальный спорный момент. Он заключается в том, что ре-
акция в газовой фазе рассматривается независимо от реакции в жидко-
сти. В то же время несомненно, что ускорение экзотермической реакции
в конденсированной фазе приведет также к ускорению горения в газовой
фазе, и разрыва между обеими скоростями не получится.
По-видимому, наличие заметной реакции в жидкости будет приводить
только к некоторому ускорению горения по сравнению с тем случаем,
когда реакция в конденсированной фазе отсутствует и сама по себе
к взрыву привести не может. В пользу этого говорит тот эксперимен-
тальный факт, что нелетучие вещества, например пироксилин, у которых
реакция заведомо идет также в конденсированной фазе, горят спокойно
(без перехода во взрыв) в очень большом интервале давлений. Это отно-
сится также ко многим летучим ВВ, горящим прп повышенных давле-
ниях, т. е. при высоких температурах кипения.
Гипотеза Зельдовича рассматривает повышение давления как фактор,
стимулирующий переход горения во взрыв. Имеются, однако, наблюдения,
указывающие на возможность возникновения взрыва при быстром пони-
жении давления над горящей жидкостью, приводящем к ее вскипанию.
Так, Беляев наблюдал в этих условиях переход горения метилнитрата во
взрыв. При толиовании этих результатов следует учитывать большую
склонность паров метилнитрата и их смесей с воздухом к взрыву при вос-
пламенении [241], а также малую устойчивость горения самого метнннит-
рата, уже при давлении около 2 ат переходящего на возмущенный режим.
Взрыв при вскипании горячей жидкости может возникнуть также по
следующей причине: при горении трубка прогревается, эвакуация приводит
к бурному вскипанию применяющегося малого количества жидкости, па-
ры оказываются в нагретой трубке и, разогреваясь, взрываются. При боль-
ших количествах жидкости можно было бы ожидать затухания горения
вследствие чрезмерного поступления холодной жидкости в зону горения
и падения температуры газов.
Возникновение взрыва в результате вскипания в общем случае мало-
вероятно и потому, что ускорение горения вследствие киления вызовет по-
вышение давления, повышение же давления будет подавлять кипение. Та-
ким образом, ускорение горения в результате вскипания должно быть, как
правило, самотормозящимся процессом и только в особых случаях, когда
ускорение горения происходит очень резко’, может привести к детонации.
1 При этой существенную роль могут играть, как указывает Беляев [243], разо-
грев и воспламенение пузырьков пара вследствие адиабатического их сжатия.
302
То, что состояние кипения само по себе не является достаточным усло-
вием для перехода горения в детонацию, видно из того факта, что многие
ВВ, температура кипения которых лежит ниже температуры вспышки, не
дают детонации при вспышке [242]. Очевидно и доказано прямым наблю-
нием, что самовоспламенение при атмосферном давлении наступает для
указанных веществ в состоянии кипения.
Другой механизм ускорения горения жидких ВВ был выдвинут Лан-
дау [239]. Он показал, что нормальный «теплопроводностный» режим го-
рения, заключающийся в испарении жидкости, прогреве и химической ре-
акции в парах, является, вообще говоря, неустойчивым. Возмущение, воз-
никающее во фронте такого горения, стремится возрастать по амплитуде
во времени. Чем больше скорость горения, тем меньше устойчивость пло-
ского фронта горения.
Факторами, препятствующими искажению фронта, являются сила тя-
жести, поверхностное натяжение и вязкость жидиости. Стабилизация го-
рения может обеспечиваться также малой величиной диаметра трубки.
Если нарушения нормального режима горения имеют место, то они вы-
ражаются в турбулизации фронта горения, ведущей к увеличению коли-
чества сгорающего вещества на единицу площади сечения столбика
жидкости *,
Учет стабилизирующего влияния силы тяжести и поверхностного на-
тяжения приводит к следующему выражению для максимальной скорости
горения, допускающей нормальное невозмущенное горение:
= (4otKgpr6ffl)^4 , (5-3)
где им — массовая скорость горения (г/см2сек); ак — поверхностное
натяжение между жидкостью и ее насыщенным паром при температуре ки-
пения (дин/см); рг — плотность газообразных продуктов горения (с/см3);
6ж — плотность жидкости (г/см3).
Если скорость горения превышает значение, рассчитанное по формуле
(5.3), то горение все же может быть устойчивым, если жидкость находит-
ся в сосуде достаточно малого диаметра (d). Условие устойчивости в этом
случае имеет вид
(5-4)
Расчет показывает обычно, что условие (5.4) приводит даже при горе-
нии под атмосферным давлением, а тем более при повышенных дав-
лениях к очень малым значениям диаметра. В обычных условиях опыта го-
рение в таиих узких трубках не распространяется ив-за тепловых потерь.
Выражение (5.3) можно преобразовать [244], воспользовавшись при-
ближенным соотношением для парахора
где М — молекулярный вес.
Для критической массовой скорости горения получаем выражение
и* = 7,91 . (5-6)
Для критической лянейной скорости
и = 7,91 . (5.7)
1 В принципе этот механизм увеличения скорости мог бы иметь место и при го-
рении твердых плавящихся ВВ. Однако малая толщина расплавленного слоя (особен-
но при больших давлениях) предотвращает в этом случае возможность ускорения го-
рения вследствие турбулизации.
303
Использование выражения (5,6), хотя и несколько менее точного, для
характеристики устойчивости горения особенно удобно потому, что вели-
чина парахора может быть рассчитана по структурной формуле вещества
и не изменяется с температурой в широком интервале изменения послед-
ней. Таким образом, можно избежать экспериментального определения
поверхностного натяжения ВВ при темперетуре кипения, в большинстве
случаев практически неосуществимого; для многих ВВ неизвестны и труд-
ноопределимы даже и сами температуры кипения, особенно при повышен-
ных давлениях.
Помимо этого, выражение (5.8) позволяет оценить влияние химиче-
ской структуры жидкого ВВ на устойчивость нормального горения и по-
казать, что значение предельной скорости для различных ВВ изменяется
в очень узких пределах.
В самом деле, множитель Р/М физически соответствует удельному
объему вещества при температуре, при которой а = 1. Плотности жидких
органических ВВ заключаются при обычной температуре в интервале
1,2—1,8 г/см?. Соответственно мало отличаются и значения Р/М, посколь-
ку состав различных органических ВВ (соотношение С, Н, N и О) меняет-
ся мало *. Этот вывод может быть иллюстрирован данными табл. 42.
Таблица 42
Значения парахора я отношения Р/Д£ для различных ВВ
ВВ Пара- хор Р • МОЛ©- куляр* ный вес Р/М вв Пара* хор Р* Моле* куляр- ний вес Р/М
Метилнитрат . - -
Нитрогликоль . .
Нитроглицерин . .
Пентаэритриттетра-
нитрат ...........
149,1 77
264,0 152
378,9 227
532,8 316
1,94
1,74
1,67
1,68
Тринитробензол . .
Тринитротолуол . ,
Пикрлновая кислота
Тринитрофенилме-
тклннтрамин (тет-
рил) .............
374,8 213 1,76
413,8 227 1,82
394,8 229 1,72
499,3 287 1,74
* Значения парахора и молекулярного веса во веет случаях расчетные.
Величина 6 ж соответственно небольшому интервалу изменения
меняется мало, тем более бж, если говорить о линейной скорости го-
рения.
Плотность газообразных продуктов горения
рг = 273/^77 , (5.8)
где 1’о — объем газообразных продуктов горения 1 г вещества при нор-
мальных условиях; 77 — температура горения.
Некоторое изменение рг возможно за счет изменения уравнения реак-
ции горения с ивменением давления. Однако это изменение невелико, как
легко показать на примере ВВ с большой калорийностью, при которой
влияние изменения уравнения горения должно быть особенно значитель-
ным. При сгорании нитрогликоля до продуктов полного превращения без
учета диссоциации рг было бы равно около 8,8 • 10~5 г/см3. При сгорании
нитрогликоля при атмосферном давлении, по данным эксперимента, рг =
= 1,87 Ю '4 г/см3. Таким образом, для наибольшего изменения уравнения
1 Значительное увеличение Р/М наблюдается лишь для богатых водородом
(Р/М = 17,1) веществ кии смесей, например гидразина, раствора метанола в переки-
си водорода, метана в кислороде и т. п. Однако для талях веществ влияние большой
величины отношении Р/М на величину критической скорости будет в известной мере
компенсироваться понижением плотности продуктов горения.
304
горения, которое превосходит возможное в действительности, рг изменяет-
ся только в 2,2 раза, а рг'^ в 1,5 раза. Нет оснований также ожидать суще-
ственной разницы между рг для различных органических взрывчатых
жидкостей. Для метилнитрата, нитрогликоля, нитроглицерина значения,
рассчитанные по экспериментально определяемому составу продуктов го-
рения, составляли соответственно 1,93 • 10“\ 1,87 • Ю~4 и 1,72 • 10-4 г/см3.
Из сказанного вытекает, что правую часть равенства действитель-
но можно рассматривать как приближенно постоянную величину
(~ 0,25 г/см2 сек при атмосферном давлении) *.
Отсюда следует, что устойчивость нормального горения может быть ха-
рактеризована величиной скорости горения при данных условиях давления
и температуры.
Скорость горения- должна быть определена экспериментально. Однако
в принципе она может быть также рассчитана теоретически, на основе тео-
рии Зельдовича — Беляева, если известны термохимические и кинетические
характеристики ведущей реакции горения,в определенных условиях послед-
него.
Формула (5. 6) показывает зависимость предельной скорости от давле-
ния, при котором идет горение. При изменении давления в правой части
формулы меняется в основном только плотность газов, пропорциональная
давлению. Учитывая эту зависимость, получаем
Пн(кр} = , (5-9)
где р = 0, 25, если р выражать в атмосферах, а пМ(кр) в граммах на квадрат-
ный сантиметр в секунду.
Таким образом, условие устойчивости нормального горения жидкости
может быть сформулировано следующим образом: если скорость горения
при данном давлении больше, то горение неустойчиво, и наоборот. Из вы-
ражения (5.9) следует также, что при повышении давления устойчивость
горения уменьшается.
В самом деле, скорость горения можно положить приближенно пропор-
циональной давлению. Таким образом, при повышении давления скорость
горения растет быстрее, чем расчетное значение предельной скорости, и при
каком-то давлении скорость горения становится равпой предельному значе-
нию. Это давление можно определить из следующих соотношений. Полагая
Uf=Bp, получаем
Вркр~Рр1/г
или
7>КР~(|)2. . (5.10)
Вычисленное значение ркр обычно будет завышенным, так как опыт по-
казывает, что ужо при давлениях, значительно меныпих, скорость горения
начинает расти с давлением не пропорционально последнему, а быстрее.
Кроме того, рассмотренные соотношения и, в частности, постоянство пре-
дельной скорости сохраняют свою справедливое ть^только до тех пор, пока
давление, под которым происходит горение, не слишком близко к критиче-
скому давлению. При этих последних условиях поверхностное натяжение
жидкости становится равным нулю fc соответственно надает до нуля и зна-
чение предельной скорости, определяемое выражением (5.6).
Поскольку критическое давление органических взрывчатых жидко-
стей равно, как показывает опыт, ~50 кг/сл*2, следует заключить,
1 Рассчитанные на основе экспериментальных данных для аи. рг и значения
вя (кр) для метилнитрата, нитрогликоля и нитроглицерина составляли соответствен-
но 0,26; 0,26 и 0,25 г/с№сек.
20 К. К, Андреев
305
что устойчивое горение жидкостей выше 50 кг/см2 должно быть невозмож-
ным. Поэтому при повышении давления (особенно при приближении
к критическому) скорость горения должна расти быстрее за счет эффекта
Ландау; кроме того, должна появляться пульсация. Эти обстоятельства не
исключают, впрочем, возможности устойчивого течения горения, а при
малом периоде пульсации даже видимой его равномерности.
Формула (5.6) показывает влияние начальной температуры ВВ
на устойчивость его горения. Правая ее часть почти не зависит от темпе-
ратуры (точнее, очень слабо уменьшается с температурой соответственно
уменьшению плотности жидкости). Левая же часть формулы (скорость
горения) возрастает с температурой; иначе говоря, устойчивость горения
должна уменьшаться при повышении начальной температуры.
Есть, одиако, разница во влиянии давления и температуры. Давление
мы можем повышать неограниченно, вплоть до его значений, соответствую-
щих неустойчивому горению для любой жидкости; возможность же повы-
шения температуры ограничивается температурой самовоспламенения ве-
щества. Поэтому не для всякого жидкого ВВ можно этим последним путем
прийти к неустойчивому горению.
Выше мы установили, что достижение критической скорости приводит
к прекращению режима нормального («теплоироводиостного») гбрения.
Вопроса о том, какие последствия будет иметь искривление плоской по-
верхности фронта, теория детально не касается.
Искривление, иначе говоря увеличение поверхности контакта газов и
жидкости, должно влечь за собой усиление теплообмена между продуктами
горения и жидкостью и вследствие этого ускорение парообразовании и го-
рения. Однако это ускорение лимитируется тем обстоятельством, что коли-
чество выделяемого при горении тепла ограничено скоростью химической
реакция в газовом слое, где идет реакция, и площадью его поверхности (по-
верхностью первичного пламени). Если кинетические и термохимические
константы реакции в газовой фазе таковы, что горячие газы в состоянии
«переварить» усиленно поступающие пары, иначе говоря, если ускорение
парообразования ведет за собой ускорение химической реакции в газах
(на единицу сечения трубки) вследствие увеличения поверхности горения
паров или турбулизации зоны реакции, то горение может ускориться.
В противном случае горение сначала несколько ускоряется, но вследствие
усиления парообразования слой паров становится толще, удаляет горячую
зону от поверхности жидкости и уменьшает поэтому количество тепла,
передаваемого ей газами в единицу времени. В то же время при искрив-
лении поверхности жидкости утончается в большей или меньшей степени
прифронтовой прогретый ее слой, что ведет к усиленной теплоотдаче в
глубь жидкости. В результате всего этого ускорение парообразования
является в этом случае самого рмозящимся (в этом заключается, в частно-
сти, возможная причина возникновения пульсации, сопровождающейся
приближением вторичного пламени к поверхности жидкости).
Торможение горения после его временного ускорения может быть столь
значительным, что горение затухает, как зто и наблюдалось для некоторых
жидкостей. Особенно благоприятны условия для затухания, если турбули-
зация возникает при низких давлениях, когда отсутствует вторичное пламя
и тепловыделение горения мало, как это имеет место, например, при горе-
нии нитроглицерина. При более высоких давлениях, когда имеется вторич-
ное пламя, турбулизация приводит к сильному увеличению скорости горе-
ния, так как образующееся повышенное количество паров сжигается во
вторичном пламени.
Существенную роль при этом режиме горения жидкостей может играть
переход на турбулентный режим вторичного пламени, поскольку при тур-
булизации жидиости скорость движения паров и продуктов первичного го-
рения становится столь значительной, что даже в применявшихся узких
306
трубках число Рейнольдса превосходит критическое значение. Для нитро-
гляколя по приближенному расчету при 25 ат Re = 3200.
Таким образом, ускорение газообразования за счет турбулизации по-
верхностного слоя жидкости одновременно приводит к переходу движения
газообразных продуктов с ламинарного на турбулентный режим. Это об-
стоятельство резко увеличивает скорость вторичного газофазного горения
и приближает зону его к поверхности жидкости. Тепло, выделяемое во
вторичном пламени, передается теперь поверхности жидкости уже не теп-
лопроводностью через относительно толстый слой тазов и паров, а конвек-
цией, скорость горения жидкости соответствует не температуре первич-
ного пламени, а гораздо более высокой температуре вторичного пламени.
Ускорение горения вследствие турбулизации вторичного пламени мо-
жет быть существенным фактором не только ускорения горения и перехо-
да его во взрыв, но и стабилизации горения в отношении возникновения
взрыва. Опыт показывает, что склонность к возникновению взрыва при го-
рении больше в области промежуточных давлений между нормальным и
турбулентным горением. Вероятной ее причиной является накопление в
газовой фазе в результате турбулизации жидкости большого количества
продуктов неполного сгорания со взвешенными в них капельками жидко-
сти, последующий взрыв которых создает скачок давления, вызывающий
взрыв несгоревшей части жидкости. Ускорение сгорания промежуточных
продуктов в результате турбулизации вторичного пламени предотвращает
их накопление и делает горение более равномерным. Действительно,
в переходной области наблюдаются относительно большие колебания ско-
рости горения; при больших давлениях горение становится гораздо более
равномерным.
Мы рассмотрели основные выводы теории устойчивости горения жид-
костей. В какой мере согласуются с ними экспериментальные данные?
Такие данные были получены при изучении горения четырех жидкос-
тей: метилнитрата, нитрогликоля, дигликольдииитрата и нитроглицерина.
Все эти вещества горят при комнатной температуре и атмосферном давле-
нии. Скорость горения нитроглицерина — 0,23, метилнитрата — 0,14, нит-
рогликоля — 0,044 и дигликольдииитрата — 0,021 г/см2сек, т. е. меньше
критического значении (0,25 г/см2сек), Следовательно, согласно теории,
горение этих веществ может быть устойчивым. Скорость горения нитро-
глицерина очень близка к критической. Ограниченная способность нитро-
глицерина к нормальному горению является естественным следствием
относительно большой скорости горения.
При повышенной температуре (60° С) горение метилнитрата переходит
во взрыв; предельная расчетная скорость горения для него составляет
0,24 г/см? сек, скорость, определенная экстраполяцией экспериментальных
результатов при этой температуре, равна 0,20 г/см2сек. Для нитрогликоля
скорость горения вблязи температуры самовоспламенения равна
0,09 г/слс2сек, при этом он не взрывается; предельная скорость по расчету
составляет 0,25 г/см? сек. Таким образом, влияние температуры на устой-
чивость горения обоих нитрозфиров также согласуется с теорией.
При горении под повышенными давлениями для метилнитрата (начиная
с 1,75 ат)' для нитрогликоля (с 19 ат) и для дигликольдииитрата (с 55 ат)
наблюдается переход от равномерного горения к ускоренному пульсирую-
щему горению. Значения расчетных максимальных скоростей устойчиво-
го горения составляют 0,32 г/сж2сек для метилнитрата и 0,88 г/см2сек
для нитрогликоля; экспериментальные скорости находятся между 0,26 и
0,46 г/см2сек в первом и около 0,90 г/слс2сек во втором случае. Для дигли-
кольдинитрата критическая скорость составляет около 0,7 г/с№сек, что
значительно меньше расчетного значения. Это обстоятельство неудиви-
тельно, так как для дигликольдииитрата переход горения с нормального
на возмущенный режим происходит вблизи критического давления (впри-
20*
307
нятом в физической химии значении этого термина), когда поверхностное
натяжение (а следовательно, и критическое значение скорости горения)
Стремится к нулю.
Для нитроглицерина при атмосферном давлении скорость горения, как
упоминалось, близка к допустимой для устойчивого горения. Согласно тео-
рии, следует заключить, что при пониженном давлении нитроглицерин дол-
жен гореть. Действительно, как мы видели, при давлениях 100 — 200 мм ни-
троглицерин горит; если горение идет в замкнутом-сосуде и давление газов
возрастает, то по достижении давления несколько более 300 ми горение за-
тухает. Расчетное значение предельной скорости при 310 мм составляет
0,16 г/сл2 *сек, фактическая величина скорости вблизи затухания меньше и
составляет 0,12 zjcMpcen. При повышенных температурах вместо затуха-
ния горение при повышении давления переходит на пульсирующий ре-
жим, иногда приводящий к затуханию. Максимальная скорость горения
без видимой пульсации составляет 0,21 г/сж2сек, т. е. больше теоретиче-
ски допустимой.
Таким образом, теоретические выводы и экспериментальные данные
в отношении возможности устойчивого горения ряда жидких ВВ при атмо-
сферном давлении и влияния температуры и давления на устойчивость го-
рения в целом хорошо согласуются друг с другом.
Теория Ландау рассматривает стабилизацию горения за счет сил по-
верхностного натяжения и тяжести *. Аналогичное влияние должна ока-
зывать вязкость жидкости. Желатинированный нитроглицерин устойчиво
горит в большом интервале давлений [245]. В опытах под возрастающим
давлением, при которых жидкий нитроглицерин детонировал при поджи-
гании его в сосуде, разрывающемся при давлений около 200 ат, желати-
нированный нитроглицерин (93:7) не давал детонации даже при поджи-
гании его в сосуде, разрывающемся при 1200 ат.
В опытах по горению под постоянным давлением нитрогликоль, желати-
нированный 3% коллоксилина, равномерно и устойчиво горел, как мы ви-
дели, во всем изученном интервале давлений — до 150 ат, в то время как
жидкий нитроглпколь показывал нарушение равномерности горения уже
при давлении, в 10 раз меньшем.
Теоретический анализ стабилизирующего влияния вязкости на основе
тех же соображений, какими руководствовался Ландау, привел Левича
[246], к следующему выражению для предельной скорости нормального
горения жидкости в зависимости от вязкости:
им = (3 уз ?т]р№)ш, (5.11)
где — массовая скорость горения;,
ц — вязкость;
рг — плотность газообразных продуктов горения;
бж — плотность жидиости.
Из четырех подробно изученных жидких иитроэфиров наиболее вяз-
кий — нитроглицерин; вязкость его 0,36 пуаз. Расчет дает при рг = 1,72 X
Х10“4 г!см? и дж = 1,6 г/см2 предельное значение скорости — 0,17 г/см2сек;
фактическое значение скорости равно 0,23 г/см2сек. Таким образом, вяз-
кость нитроглицерина недостаточна для стабилизации его горения.
Если принять те же значения рг и то для возможности устойчивого
горения жидкости за счет вязкости последняя должна была иметь значение,
равное или превосходящее один пуаз. Были поставлены опыты с нитрогли-
1 Если трубка с жидким ВВ находится в состоянии невесомости, то в соответ-
ствии с формулой Ландау нормальное горение должно быть неустойчивым при лю-
бом давлении. Напротив, если трубке с горящей жидкостью сообщить ускоренно
(в осевом направлении), значительно превышающее ускорение силы тяжести, то,по
замечанию Ю, Б. Харитона, устойчивость горения возрастет. При ускорении, равном
100 g, критическая скорость будет больше в 3,2 раза.
308
церином, вязкость которого была увеличена растворением коллоксилина.
То, что способность желатины к устойчивому горению определяется имен-
но повышенной вязкостью, показывает сравнение смеси нитроглицерина
с коллоксилином (1%) до желатинизации, когда ее вязкость практически
равна вязкости нитроглицерина, и после желатинизации (вязкость
3,5 пуаз). Первая смесь не горела, вторая горела устойчиво со скоростью
0,24 zjcM^ceK. При такой вязкости допустимая максимальная скорость по
расчету составляет 0,37 з/с.и2сек. Нитроглицерин с добавкой 1% мономе-
ра метилметакрилата не горит, добавка такого же количества полимера
метилметакрилата, резко повышающего вязкость нитроглицерина, при-
водит, в согласии с теорией, к устойчивому горению.
По формуле (5.11) предельная скорость горения зависит и притом так
же, как это следует из формулы Ландау (5.3), от давления, под которым
идет горение. Скорость горения возрастает пропорционально первой степе-
ни давления, правая же часть равенства (через рг) пропорциональна дав-
лению в степени '/г- Таким образом, левая часть растет быстрее, чем пра-
вая, и при некотором давлении скорость горения начинает превосходить
допускаемое теорией значение.
Подобно давлению, уменьшает скорость горения повышение температу-
ры. Скорость горения с повышением температуры растет, предельное же
значение ее уменьшается вследствие уменьшения вязкости.
Для экспериментальной проверки влияния давления опыты были поста-
влены с желатиной (99 : 1) при различных давлениях* При 800 лсч горение
шло равномерно со скоростью 0,27 г/см2сек; допустимая при данной вяз-
кости студня скорость горения равна 0,38 г! см2 сек. При 1000 мм горение
шло уже на пульсирующем режиме со скоростью около 0,59 г/см2сек\ эта
скорость была бы стабильной при вязкости в 9,4 пуаз, т. е. в 2,5 раза
большей, чем имела желатниа.
Таким образом, влияние давления таково, как его предусматривает тео-
рия. Влияние температуры на желатинах не могло быть проверено потому,
что, как показали опыты, при нагревании вязкость желатины не остается
постоянной, а меняется в зависимости от длительности нагрева.
Нарушение нормального режима горения и стабилизация его повыше-
нием вязкости наблюдались не только для индивидуальных жидких ве-
ществ, в частности нитратов спиртов. Уиттэкер и др. [233] описывают
это явление для многих двух- и поликомпонентных растворов горючих
(2-нитропропан, нитрил себациновой кислоты) в окислителях (азотная
кислота, четырехокись азота) и связывают его с увлечением при испаре^
нни горящей жидкости ее капелек оттекающим паром. Этими исследовате-
лями изучалось влияние вязкости, упругости паров горючего, диаметра и
формы трубки. Увеличение вязкости, достигавшееся растворением полиме-
тилметакрнлата, уменьшало скорость горения, которая стремилась к неко-
торому пределу, и увеличивало давление перехода на турбулентный ре-
жим. Это давление несколько уменьшалось при увеличении диаметра
трубки. При 14,6 ат смесь 2-нитропропана с 97%-ной азотной кислотой
горела со скоростью 0, 114 см!сек\ при содержании в этой смеси 0,1, 0,5 и
0,75% полиметилметакрилата скорость горения оказалась равной 0,079,
0,076 и 0,074 см!сек, а критическое давление перехода горения на турбу-
лентный режим жидкой и желатинированных смесей нитропропана и
азотной кислоты составило 76, 83, 117 и 130 ат соответственно.
Опыты с желатинированными жидкостями позволили выяснить также
вопрос о том, не обусловлено ли появление пульсации горения вторичным
пламенем. При горении желатины это пламя появляется при том же давле-
нии и расположено даже несколько ближе к поверхности, чем при горении
жидкости; в то же время пульсирующее горение наблюдается в первом и
отсутствует во втором случае даже и тогда, когда вторичное пламя рас-
полагается совсем близко у поверхности горящего вещества. Отсутствие
309
прямой связи между появлением вторичного пламени и переходом горения
на турбулентный режим подтверждается также тем фактом, что для неко-
торых жидкостей, например для нитроглицерина, этот переход происходит
при таких низких давлениях, когда вторичного пламени еще нет.
Как указывалось выше, для обеспечения стабильности горения даже
при атмосферном давлении требуется значительная вязкость, порядка
1 пуаз; при повышении давления необходимая для стабилизации величи-
на вязкости быстро растет; например при давлении в 100 ат она в 1000 раз
больше, чем при атмосферном давлении.
Все описанные опыты были проведены при постоянных давлениях,
были проведены также опыты при возрастающем давлении преимущест-
венно в стальных стаканах (см. стр. 200).
Изучались нитрогликоль, нитроглицерин, тротил и его растворы в
азотной кислоте.
Нитрогликоль легко дает переход горения в детонацию. Лишь в опытах
с диском, выдерживающим давление 65 ат, в одном из двух опытов диск
был вырван без повреждения трубки. В другом опыте с диском, вырывае-
мом давлением 200 ат и более, наблюдалась детонация (заряд 50 г). Ин-
тересно, что в опытах с наиболее прочными дисками дробление трубки
в двух из трех опытов было заметно меньше, чем в опытах с дисками
обычной прочности. *
Нитроглицерин при этих же условиях (200 аг) показывает умерелиое
дробление, при 65 ат в обоих опытах диск был вырван без повреждений
трубки. Увеличение навески нитроглицерина до 200 г не изменило степени
дроблеикя трубки, несмотря на применение более прочного диска (500—
700 ат).
Для жидкого тротила (200 г) при 100° С наблюдается только горение
при максимальной прочности трубки. Совершенно иную картину показы-
вает тротил в растворенном в азотной кислоте виде. Раствор с содержа-
нием азотной кислоты 40% детонирует при диске, вырывающемся под
давлением 500—700 ат; даже при 200 яг наблюдается взрыв со значитель-
ным дроблением трубки.
Были изучены также жидкости в желатинированном состоянии. Же-
латинированный нитроглицерин (93 : 7) в количестве 50—200 г зажигался
в трубке, закрытой дисками, выдерживающими 200, 500—700 ат 1(плотяость
заряжания 0,4—1,2 г/сл»3), и даже в корпусе снаряда, выдерживающем
1200 аг. Ни в одном из этих опытов детонация не возникала.
Аналогичное, но несколько более активное поведение показал 62%-ный
желатни-дкиамит на основе натриевой селитры. При 200-граммовых заря-
дах некоторому дроблению подвергалась лишь донная часть трубки.
Были поставлены опыты также с нитроглицерином, иммобилизирован-
ным не растворением в нем нитроклетчатки, а путем добавки инертного
порошка (тонкоизмельчениый кизельгур) в количестве */з от нитроглице-
рина; при этом получается смесь мазеобразной консистенции. Такая смесь
ведет себя подобно желатине, не давая перехода горения в детонацию,
если даже диск вырывался под давлением 500—700 аг. Испытание анало-
гичной по физической структуре смеси нитроглицерина с мелким порош-
ком гексогена не дало того эффекта, который наблюдался при смеси с
кизельгуром.
Суммируя результаты опытной проверки теории устойчивости горения
жидких ВВ, следует констатировать, что все полученные результаты в
отношении влияния на эту устойчивость физических характеристик жид-
кости и условий горения хорошо согласуются с предсказаниями теории.
ГЛАВА ШЕСТАЯ
ВСПЫШКА И ВОСПЛАМЕНЯЕМОСТЬ ВВ
I. ВСПЫШКА ВВ
1. МЕХАНИЗМ ВОЗНИКНОВЕНИЯ ВСПЫШКИ
Если ВВ Нагревается внешним источником тепла, начальная темпера-
тура которого относительно низка и постепенно возрастает, то по дости-
жении некоторого ее значения наблюдается самовоспламенение (вспыш-
ка) ВВ. Значения температуры вспышки для ряда ВВ при некоторых
стандартных условиях определения приведены в табл. 43 и 44.
Несколько более трудоемким, но физически более простым является
определение температуры вспышки путем нагревания ВВ при постоянной
температуре источника тепла. Устанавливают при прочих равных услови-
ях ту минимальную температуру этого источника, начиная с которой дан-
ное вещество дает вспышку. Другую характеристику явления представ-
ляет задержка вспышки — время от начала нагревания до ее возникнове-
ния. Иногда определяют температуру, при которой вспышка происходит
с определенной задержкой, например не более 5 сек.
Сущность явления с точки зрения теплового эффекта заключается
схематично в следующем. При низких температурах скорость экзотерми-
Таблица 43
Температура вспышки различных В В по Касту
вв Температура вспышки. °C ВВ Температура вспышки. “С
Нитроглицерин .... Нитрогликоль ..... Пироксилин Коллодионный хлопок Пентаэритригтетраниг- . рат ' Гремучий студень , . . Желатлндинамит . . . Динитробеизол .... J Тринитробензол .... Тринитротолуол .... Трицитро хлорбензол . . Пикриновая кислота Тринитрокрезол .... Тринитроакизол .... 200—205 195-200 195—200 204—205 205—215 202—208 180—190 Нет вспышки То же 295—300 395—397 3Q0—310 270—276 290—296 Гексанитр о дифениламин Тетрил Гексоген Шеддит (тип 60) .... Аммониты Черный порох .... Гремучая ртуть .... Азид свинца Тринитрорезорцинат свинца Пикрат свинца .... Тринитрокрез ил ат свии- Да . 248—252 190—194 230 258—265 Не дают вспышки или вспыхивают выше 300° 310—315 169-175 305—320 275 285—287 255—257
Примечание, Температура вспышки определялась при нагревании пробир-
ки с навеской ВВ в жидкостной бане (с определенной — 20 град/лши— скоростью).
311
Таблица 44
Температура вспышки различных ВВ
вв Температура ВСПЫШКИ, ’С " вв Температура вспышки, °C
Гликольдинитрат 257 Тринитротолуол 475
Диэтиленгликольдинитр ат 237 Пикриновая кислота .... 322
Нитроглицерин 222 Пикрат аммония 318
Эритриттетранитрат . . . 225 Тетрил 257
Тэн 225 Гексил 325
Дипентаэритритгексанит- Гремучая ртуть 210
рат 255 Гремучее серебро 170
Маянитгексанитрат .... 205 Азид свинца кристаллический 345
Нитроклетчатка (13,3% N) 230 Диазо динитрофено л 180
Этилендинитрамин .... 190 Тринитрореэ орцин ат свинца 265
Нитрогуанидин 275 Динитрорезорцинат свинца 265
Гексоген 260 Тетразен 154
Октоген 335 Гексаметилентрипероксид-
Три нитробензол 550 диамин <149
Примечание. 0,02 г ВВ в медной гильзе помещается в баню со сплавом
Вуда. Устанавливаетсн температура, при которой вспышка происходит через 5 сек.
ческого распада ВВ, а следовательно, и теплоприхода мала и растет с тем-
пературой медленнее^чем скорость теплоотвода; все выделяющееся тепло
отводится в окружающую среду — между теплоприходом и теплоотводом
существует устойчивое равновесие.
При более высоких температурах скорость реакции п теплоприход
растут с температурой быстрее, чем теплоотвод. Температура самовоспла-
менения представляет собой ту минимальную температуру, начиная с ко-
торой равновесие между теплоприходом и теплоотводом становится не-
устойчивым: теплоприход начинает расти быстрее теплоотвода, скорость
реакции быстро возрастает и наступает самовоспламенение.
Так как количество выделяющегося тепла пропорционально объему
ВВ, а теплоотвод (при конвективном теплообмене) — его поверхности, то
температура самовоспламенения зависит от количества ВВ. Чем больше
зто количество, тем меньше (при данной форме заряда) отношение по-
верхности к объему, тем ниже температура самовоспламенения. Отсюда
ясно, что температура вспышки не является абсолютной константой ВВ;
при больших размерах заряда вспышка данного ВВ может происходить
при гораздо' более низких температурах, чем указано в таблицах. За-
держка ее может быть в этом случае также весьма большой.
Условия теплоотвода относительно мало изменяются при переходе от
одного ВВ к другому; то же относится и к теплоте, выделяющейся прп
распаде единицы веса ВВ. Поэтому главным параметром, определяющим
температуру вспышки, является скорость термического распада ВВ. По-
этому по температуре вспышки можно приближенно судить о величине
этой скорости при высоких температурах. Следует напомнить, что скорость
распада ВВ может сильно зависеть от условий, в которых он протекает
и часто более сложно зависит от температуры, чем это вытекает из форму-
лы Аррениуса. Поэтому оценка по температуре вспышки скорости распада
ВВ при низких температурах может быть лишь сугубо приближенной и
условной.
Вспышка является очень сложным процессом. В ряде работ [247—
249] показано, что при равных условиях теплоотвода температура само-
воспламенения может меняться, например, в зависимости от скорости ра-
зогрева в результате химического (автокаталитического или иного) са-
моускорения реакции. Интервал времени между моментом прогрева (при
постоянной температуре окружающей среды) и моментом самовоспламе-
нения может быть очень велик, несравненно больше возможного времени
развития чисто теплового процесса, что с несомненностью указывает на
большую роль химического развития процесса при возникновении само-
воспламенения. Именно вследствие развития этого процесса скорость ре-
акции 1растет, несмотря на то, что температура остается постоянной. Когда
скорость реакции достигает значения, при котором теплоприход становит-
ся больше и растет с температурой быстрее, чем теплоотвод, возникает
самовоспламенение.
Интересные исследования по выяснению механизма вспышки и роли
отдельных факторов в ее 'возникновении провел Гольбиндер [250], Опыты
производились со смесями тетранитрометана или азотной кислоты с раз-
личными горючими. Смеси тетранитрометана с анилином [воспламеняются
при комнатной температуре со значительной задержкой1 (десятки се-
кунд), что позволяет получить при смешении гомогенные смеси. Вспышка
возникает лишь, если количество смеси превосходит определенный предел.
Однако это влияние не исчерпывается в данном случае известным из тео-
рии теплового самовоспламенения изменением соотношения теплоприхода
и теплоотвода. Если приготовить смесь в стеклянном сосуде, находящемся
на воздухе, и в стальном тех же размеров, помещенном в воду, то, несмот-
ря на различные условия теплоотвода, минимальные количества смеси и
индукционные периоды оказываются одинаковыми. В опытах с сосудами
разной формы (рис. 245) при прочих равных условиях в сосуде а мини-
мальное количество смеси, необходимое для самовоспламенения, оказы-
вается в 2—4 раза меньше, чем в сосуде 6, хотя условия теплоотвода весь-
ма близки.
Измерение температуры в жидкости и над нею показало, что в течение
всего индукционного периода температура жидкости остается постоянной
и равной начальной температуре компонентов смеси. Это также показыва-
ет, что причиной самовоспламенения нельзя считать тепловое самоускоре-
ние процесса в конденсированной фазе. Воспламеняется и затем горит
практически холодная смесь. Над жидкостью же перед воспламенением
происходит быстрый (сотни градусов в секунду) подъем температуры. Са-
мовоспламенению предшествует «кипение», при котором выделяющиеся
газы увлекают мельчайшие капельки жкдкостн, образуя своего рода
«дым». Следует допустить, что основным процессом в индукционном пе-
риоде является термонептральная или слабоокзотермическая реакция
амина с окислителем, ведущая к образованию конденсированного относи-
тельно стойкого промежуточного продукта2. Дальнейшие стадии процес-
са идут со скоростью, достаточной для самовоспламенения, лишь тогда,
когда концентрация этого продукта достигает некоторого предела.
Эти процессы заключаются в образовании при взаимодействии проме-
жуточного продукта с тетранитрометаном газов высокой реакционной
способности, которые, накопившись <в достаточной концентрации над жид-
костью, самовоспламеняются. Одновременно с выделением газов происходит
их разбавление окружающим воздухом. Чем толще слой жидкости, тем
большее количество газов выделяется на единицу ее поверхности. Этим
объясняется влияние количества жидкости на возникновение самовоспла-
менения. Более легкое возникновение вспышки в сосуде а объясняется
большим количеством газов, выделяющихся в нем, на единицу объема
воздуха в сосуде. Независимость индукционного периода От условий теп-
1 Другие амины (толуидин, ксилидин и трпэтиламин) дают значительно мень-
шую задержку.
s В соответствии с этим предположением при получении смеси в два приема за-
держка воспламенения уменьшается при увеличении времени выдержки первой пор-
дии смеси.
313;
лоотвода также согласуется с заключением, что основную часть этого
периода составляет изотермическое развитие реакции в конденсированной
фазе; помимо этого, коэффициент теплопередачи от газа в окружающую
среду меняется при переходе от стеклянного сосуда к стальному гораздо
меньше, чем от жидкости.
Изучались также смеси тетранитрометана с горючими, воспламеняющие-
ся лишь при нагреве до повышенных температур. Температура начала ки-
Рис. 245. Стеклянные сосуды
для опытов по самовоспламе-
нению смеси тетранитромета-
на и анилина
пения этих смесей была ниже температуры
их вспышки. По этой причине в малых на-
весках они давали вспышку лишь при отно-
сительно высокой (>• 160°) температуре. При
увеличении навески температура вспышки
заметно снижается. Так, смесь тетранитро-
метана и бензола в количестве 0,1 мл вспы-
хивает только при температуре термостата
~ 500° С с малой задержиой. Если же взять
25 мл смеси то после нагревания при 130°
в течение 3—5 мин, возникает вспышка,
в большинстве опытов заканчивающаяся
а — полузамкнутый; б - открытый взрывом. Смесь
зольной головкой»
нефти), взятая в
пламеняется при 190°, при 1 мл воспламенение
при 20 мл оно наблюдается уже при 15—18° С.
тетранитрометана с *бен-
(легкая фракция пиролиза
количестве 0,05 мл, вос-
происходит при 40—45° С,
Весьма интересные данные были получены с бензольными смесями,
к которым добавлялось небольшое количество стирола. Присутствие всего
0,7—1,0% стирола снижало температуру вспышки примерно на 200° С.
Смеси на основе азотной кислоты ведут себя немного иначе, нежели те-
траншрометановые. Реакции в конденсированной фазе в этом случае экзо-
термичны и температура жидкости быстро поднимается до некоторого по-
стоянного для данной смеси значения, соответствующего температуре
кипения и им ограничиваемого (80—130° для разных смесей). Сильный
подъем температуры, приводящий к самовоспламенению, происходит в га-
зовой фазе, содержащей не только реакционноспособные газы, но и пары
или продукты их термического распада; кроме того, в отличие от предыду-
щего случая горит не холодная, а нагретая жидкость.
Отличным от большинства ВВ, для которых роль химического самоуско-
рения превращения при возникновении вспышки велика, является, по
опытам Апина [251], жидкий хлористый азот. В запаянной ампуле при на-
веске 0,1 г ов. при 55° вспышки не дает; при 60° всегда и притом почти
мгновенно возникает вспышка в форме взрыва. По-видимому, такой резкий
переход от медленного разложения и взрывному при повышении темпера-
туры следует объяснить чисто тепловым характером самовоспламенения.
Другие изучавшиеся ВВ можно нагреть до сравнительно высокой тем-
пературы, при которой начальная сиорость разложения относительно мала
и тепловое равновесие не нарушается; лишь при последующем развитии
ускорения реакции достигается та критическая скорость, при которой теп-
лоприход превышает теплоотвод. Если же при разложении вещества уско-
рения реакции не происходит (для хлористого азота это было показано
Вант-Гоффом), то, очевидно, температурная граница нарушения теплового
равновесия должна быть очень резкой; возникновение вспышки может
лишь в очень малой мере зависеть от времени, что и наблюдается для хло-
ристого азота.
Напротив, чем более выражено самоускорение, тем шире интервал
температуры — от минимальной температуры вспышки до той температу-
ры, при которой величина задержки определяется лишь временем прогре-
ва ВВ. Соответственно в гораздо больших пределах изменяется и величина
314
задержки вспышки. Эти характеристики явления вспышки могут быть ис-
пользованы для быстрого установлении кинетического типа термического
распада ВВ.
Задержка вспышки т уменьшается при повышении температуры. Во
многих случаях зависимость между ними изображается в координатах
1g т — уг прямой линией. Такая зависимость получается и теоретически для
теплового, цепного, топохимического и автокаталитического ускорений
реакции [252]. Однако тангенс угла наклона прямой, выражающей связь
задержки вспышки и обратной абсолютной температуры, зависит от вели-
чины и формы заряда, условий теплообмена и других характеристик опыта.
Поэтому зависимость между кинетическими характеристиками реакций,
ведущих к вспышке, и ее задержкой более сложна, чем ее дают указанные
теории, и нельзя отождествлять тангенс утла наклона прямой 1g т — с
энергией активации химического превращения, приводящего к вспышке.
2. ХАРАКТЕР ВСПЫШКИ
Температура самовоспламенения является лишь одной из характери-
стик явления вспышки. Существенную роль играет характер вспышки,
т. е. характер явлений, следующих за самовоспламенением. Опыт показы-
вает, что инициирующие ВВ, а также некоторые вторичные ВВ, например
нитроглицерин и нитроманиит, дают вспышку в форме взрыва, большин-
ство других дает мягкую вспышку в виде более' или менее бурного горе-
ния.
Этим различиям давались разные объяснения. Так, Беляев предпола-
гал [253], что характер вспышки определяется соотношением между тем-
пературой кипения ВВ и температурой его вспышки. Если температура
кипения много выше, чем температура вспышки, то вспышка имеет ха-
рактер взрыва и наоборот. Однако при сопоставлении температур кипения,
температур вспышки и характера последней для большого числа ВВ,
а также влияния на характер вспышки давления это предположение не
подтвердилось [254].
Действительную причину различий в интенсивности вспышки можно
установить на основе следующих соображений [255].
Вспышку можно рассматривать просто как горение ВВ. Отличие от
обычных условий горения заключается в следующем. Во-первых, при
вспышке ВВ горит при предельно высокой температуре, до которой оно
может быть нагрето. Во-вторых, горит не ВВ в первоначальном виде,
а расплавившееся (если температура плавления ниже температуры вспыш-
ки), частично разложившееся вещество, содержащее конечные и проме-
жуточные продукты распада, и с изменившимися физическими свойствами
(уменьшение вязкости, переход в пенообразное состояние, в состоянии ки-
пения и т. п.). В-третьих, горение возникает не от внешнего поджигания,
а в результате самовоспламенения.
Характер явлений, следующих за самовоспламенением, должен опре-
деляться тем, устойчиво ли горение в этих условиях. В положительном
случае мы получим мягкую вспышку, в отрицательном может наблюдать-
ся переход начавшегося горения во взрыв.
Следует указать, что суждение о характере вспышки, в частности об
ее интенсивности, до недавнего времени имело весьма субъективный ха-
рактер; она оценивалась на слух по звуку, по наличию дробления стеклян-
ной пробирки и т. п. Желательно было иметь более объективную оценку
этой стороны вспышки. Этой задаче и была посвящена работа Кондрико-
ва [256], который изучал интенсивность вспышки различных ВВ. Мерой
последней служила высота или энергия подскока шарика, лежащего на
315
отверстии пробирки, помещенной в термостат постоянной температуры,
в которой вспыхивала небольшая навеска ВВ.
Основные черты явления следующие. При умеренно повышенных тем-
пературах ВВ разлагается спокойно без появления пламени, и шарик ос-
тается неподвижным. По достижении некоторой минимальной для данных
условий температуры происходит вспышка, обычно сопровождающаяся
<7,<?ГМ
1000-
600-
400
200 -
too
Д1—id-1---------1-----i------•-
130 150 170 190
Збмператррч, “С
Рис. 246. Зависимость интенсивности
вспышки гремучей ртути от темпе-
ратуры
Навески: I — 0,02 г; г — 0.05 г. Л — от-
сутствие воспламенения (J — произведе-
ние веса шарика на высоту его цтскоиа)
пламенем и подбрасывающая вверх ша-
рик на некоторую высоту. По характе-
ру изменения этой высоты при измене-
нии температуры и других условий опы-
та, а также по максимальной высоте
подскока шарика при данной навеске
разные ВВ отличаются друг от друга
весьма существенно.
На рис. 246 изображена зависимость
интенсивности вспышки от температу-
ры для гремучей ртути, типичная и для
других изучавшихся быстрогорящих ВВ
(пикраты свинца, натрия и калия, тет-
разен, стифнаты свинца и калия, гекси-
лат калия). После более или менее ко-
роткого участка начального роста ин-
тенсивность вспышки при всех темпера-
турах вплоть до весьма высоких остает-
ся практически постоянной. Изменение
размеров частиц гремучей ртути не
отражается на интенсивности вспышки. Если применить грамучую ртуть
не в виде порошка, а в виде спрессованной таблетки, то кривая интенсив-
ность вспышки — температура имеет максимум, величина которого в 3—
4 раза меньше, чем для порошка. При поджигании холодной таблетки в
пробирке шарик остается неподвижным; при поджигании в аналогичных
условиях порошка шарик подскакивает, но на высоту в 2—3 раза мень-
шую, чем прп вспышке.
Основное отличие вторичных и метательных ВВ от быстрогорящих
заключается в том, что, начиная с некоторого значения температуры, ин-
тенсивность вспышки падает — высота подскока шарика постепенно
уменьшается в большинстве случаев практически до нуля.
Особенностью тротила является то, что, начиная с некоторой достаточ-
но высокой температуры (в данных опытах 365° С), самовоспламенение
внезапно прекращается, и шарик уже более не подбрасывается (верхний
температурный предел самовоспламенения).
Между отдельными ВВ наблюдаются известные различия в отношения
роста интенсивности вспышки вблизи минимальной ее температуры. Для
некоторых из них (нитроклетчатка, тетрил, дина, высокопроцентные рас-
творы нитроклетчатки в нитроглицерине, тротил) увеличение интенсивно-
сти вспышки до максимума происходит в значительном (иногда десятки
градусов) интервале температур (рис. 247). Другие ВВ (дигликольдини-
трат, гремучий студень, отчасти тэн) по достижении минимальной
температуры самовоспламенения или в интервале 1—2° С дают вспышку
максимальной интенсивности (рис. 248). Нитроглицерин (1 канля =
=0,02 г) сразу же подбрасывает шарик на высоту, близкую к максималь-
ной для гремучего студня. Однако при дальнейшем повышении температу-
ры интенсивность вспышки не падает, а возрастает в несколько раз, при-
чем в части опытов пробирка дробилась на куски; если продолжать по-
вышать температуру, интенсивность вспышки резко падает до нуля.
Сходная картина зависимости интенсивности вспышка (однако без Дроб-
ления пробирки) от температуры наблюдается и для нитрогликоля,
316
Максимальные интенсивности вспышки и некоторые другие характе-
ристики опыта приведены в табл. 45.
Наибольшую из изученных ВВ интенсивность вспышки показыва-
ют нитроэфиры — нитроглицерин, метилнитрат, нитроклетчатка, нитро-
гликоль, наименьшую — нитрамины и ароматические нитросоединения,
Рис. 247. Зависимость интенсивности вспышки от темпе-
ратуры
1 — пироксилин Л1 1 (навеска 0.05 г); г — раствор (1:1) пи-
троклетчатки в нитроглицерине (0 02- г) — ось ординат слева;
з — тетрил (ОД г); 4 — тротил (ОД г)—ось ординат справа»
— отсутствие (верхний предел) вспышки тротила
особенно гексил, гексан птродифенилсульфпд, гексоген, тротил, энергия
нодброса шарика для которых не превышает нескольких процентов от той,
которую дает нитроглицерин. Из
ароматических нитросоединений
выделяется стифниновая кислота,
в навесках ~0,04 г подбрасываю-
щая, хотя и весьма нерегулярно,
шарик почти так же высоко, как и
нитроглнколь.
Бы стр о горящие ВВ по интен-
сивности вспышки отличаются
друг от друга меньше, чем вторич-
ные, причем ни для одного из них
она не достигает максимального
значения, наблюдаемого для нит-
роглицерина.
При увеличении навески ве-
щества максимальная интенсив-
ность вспышки большинства изу-
чавшихся ВВ может сильно возра-
Рис. 248. Зависимость интенсивности
вспышки от температуры
Г — гремучий студень (10% коллоксилина),
навеска 0,02 г (ось абсцисс — верхний рнд чи-
сел); г — тэн (0,05 г); 3 — дигликольдинитрат
(0 05 г) (ось абсцисс — нижний ряд чисел)
стать, в пределе — до возникнове-
ния взрыва. Однако характер за-
висимости интенсивности вспыш-
ки от величины навески для раз-
ных ВВ различен. Для инициирую-
щих и бы строго рящ их ВВ, нитро-
глицерина, нитроклетчатки, грему-
чего студня интенсивность вспышки при увеличении навески прогрессив-
но растет. Уже при зарядах в десятые доли грамма измерение ее по при-
мятому методу становится невозможным — происходит взрыв, дробящий
пробирку на куски. Для дигликольдинктрата, тэна и некоторых порошко-
образных взрывчатых смесей (например смеси 95% перхлората аммония
317
Таблица 45
Максимальные интенсивности вспышки некоторых инициирующих и вторичных ВВ*
вв Навеска, г Интервал темпе- ратур, в котором изучалась к была получена вспыш- ка"*, °C Максималь- ная интен- сивность вспышки (J, а>см) 10“* Температура, при которой наблюдалась вспышка макси- мальной интен- сивности, ’С
Гремучая ртуть Тетразен Пикрат натрии Пикрат калия Пикрат свинца Гексилат калия Нитроглицерин Нитрогликоль * Метилнитрат * Дигликольдинитрзт * . . . . Тэн Дина * Гремучий студень (10% кол- локсилина) ........ Пирок силки порошкообраз- ный Пироксилиновый порох . . . Тетрил Тротил Гексоген Октоген Гексил Г вис анптродифен илсульфид Пикриновая кислота •* . . . Пикрат аммония4* Стифниковая кислота .... /0,02 1.0,05 /0,01 10,02 0,02 0,02 0,02 0,02 —0,02 —0,05 -0,02 0,05 /0,05 10,3 /0,05 10,2 0,02 0,05 0,1 0,1 0,1 0,1 о,1 0,1 0,1 0,1 0,02 —0,04 147—200 136—200 136—200 142-180 290—335 310-370 260—300 325—375 199 Л-320 202,5—308 267—270 200.5—242 204—276 799-248 209-243 202—320 195—235 176-225 170-2051 202—265 309-365 230—305 265-330 ' 250—365 280-410 300—325 260—325) 230-320 2,5 10 0,8 2,4 3,1 2,9 5,6 8 И 8 8,1 0,7 1 1,6 1 0,5 2,8 8 0—0,2 До 4 155—200 145—200 140 165 305-335 320 270 345 225—290 215—270 200 206 202 250 230 195 195 225 365
* Опыты с веществами, отмеченными звездочкой, проводились в пробирках диаметром 2 и вы-
сотой 20 сх, остальные — в пробирках диаметром 1,7 к высотой 15 см (яижякя часть пробирки
на глубину 5—7 с.н погружена в термостат со сплавом Вуда).
’ Критические температуры самовоспламенения набраны курсивом.
"* Из 2В опытов в трех наблюдались слабые вспышки, в остальных —бесплкненное разложе-
ние.
•• Иа 15 опытов в пяти наблюдались очень олабые вспышки, в остальных — беспламенное раз»
пожевав.
и 5% алюминиевой пудры) изменение интенсивности вспышки происхо-
дит иначе: вначале при увеличении навески (для тэна, например, от 0,05
до 0,3 г) интенсивность почти не меняется, однако, когда величина заря-
да достигает некоторого критического значения, вспышка принимает, без
какого-либо заметного участка роста интенсивности, характер сильного
взрыва. Такой взрыв вызывают, например, 2 г диглмкольдинитрата. При
этом вначале наблюдается спокойное горение умеренно кипящей жидко-
сти, скорость горения и интенсивность кипения возрастают, затем вся
жидкость вспенивается, и тут же следует сильный взрыв. В случае неко-
торых ВВ, например дины, при увеличении количества от 0,05 до 0,2 г
(опыт ставился в атмосфере воздуха) наблюдалось даже уменьшение ин-
тенсивности вспышки. Возможно, однако, что при значительно больших
зарядах, которые не были применены в данных опытах, вспышка может
принять характер взрыва и для дины и подобных ей ВВ *.
' Несколько позже было установлено, что при увеличении навески до 5 г вспыш-
ка дины действительно может принимать характер взрыва [Прим. реЗ.),
318
Описанные опыты в основной их части были проведены по обычной
для определения температуры вспышки методике: пробирка находится в
термостате и в нее вводится ВВ *. При изучении вспышки нитроглице-
рина и других ВВ была использована также другая методика [257]. В стек-
лянную плоскодонную пробирочку диаметром около 4 мм и высотой 15 мм
вводилось ВВ. Пробирочка в нижней части на некоторую высоту была об-
мотана нагревающей проволокой. Опыты с нитроглицерином были прове-
дены при разных высотах заряда: 5, 3, 2 и 1 ли. При 5 и 3 мм через корот-
кое время при бурном «кипении», идущем с выделением бурых окислов
азота, регулярно происходил взрыв, дробивший пробирочку на мелкие ку-
сочки, разрушавший спираль и оставлявший отпечаток на свинцовой пла-
стинке, на которую была поставлена пробирочка. Этот взрыв достаточно
интенсивен, чтобы возбудить взрыв холодного столбика нитроглицерина,
находящегося в трубочке ниже зоны нагрева. При уменьшении высоты
столбика нитроглицерина до 2 мм пробирочка дробится, но слабее, при
1 мм она остается целой.
Если нагревающую спираль поместить на пробирочке со значитель-
ным количеством нитроглицерина так, чтобы нижняя ее часть была на
уровне поверхности жидкости, то нагрев спирали приводит первоначально
лишь к вспышке с частичным выгоранием нитроглицерина.
Возбудить взрыв нитроглицерина можно не только внешним нагревом,
но и ваодя внутрь жидкости нагревающую спираль; она должна, одна-
ко. находиться на некоторой глубине (1,5—2 ли). Взрыв при нагревании
извне можно было получить и для нитрогликоля; для этого, однако, тре-
бовалось вести опыт при повышенных давлениях; при 10 ат взрывы по-
лучались регулярно. Напротив, если при опытах с нитроглицерином пони-
зить давление до 40—50 мм рт. ст., то вспышка его принимает мягкий ха-
рактер. Это, однако, относится лишь к опытам в короткой (15 мм)
пробирочке. Увеличение ее высоты до 40 мм приводит к взрыву. То же
наблюдается и для нитрогликоля: при атмосферном давлении в 15-милли-
метровой пробирочке получается мягкая вспышка, в 40-миллиметровой —
взрыв.
Увеличение длины пробирочки, по-видимому, приводит к увеличению
давления, возникающего при вспышке. Если образующейся при воспламе-
нении взвеси ВВ приходится совершать более длинный путь в пробироч-
ке, то горение каиелек продолжается и давление растет до величины, до-
статочной для возникновения взрыва; в короткой пробирочке взвесь быст-
ро попадает в воздух, и повышения давления не происходит 1 2.
На характер явления влияет также интенсивность нагрева. Если он
осуществляется очень быстро то большая часть нитрогликоля просто вы-
брасывается из пробирочки. Если прогрев вести медленнее, и жидкость
успевает прогреться, то возникает взрыв; если нагрев еще более слаб, то
протекает разложение со слабой вспышкой или даже без нее. Это влияние
быстроты нагрева, по-видимому, обусловливается, с одной стороны,. влия-
нием начальной температуры на скорость горения (а через нее и на на-
ступление турбулизации), с другой стороны, она определяет величину
объема жидкости, охваченного термическим разложением, а также разви-
тие последнего.
Изучалось влияние газовой фазы над ВВ в пробирке на характер и
интенсивность вспышки [256]. Замена воздуха на углекислоту (опыты ста-
вились с дигликолъдяиитратом, нитрогликолем, метилнитратом и нитро-
клетчаткой) существенно уменьшает интенсивность вспышки, а также
увеличивает критическую температуру и задержку вспышки. В случае
1 В некоторых опытах в термостат вводилась холодная пробирка с ВВ.
2 При горении нитроглицерина в трубочке, длина -которой была равна вы-
соте столбика жидкости (5 лм»), взрыва при наружном нагреве (при атмосферном
даваенин), как правило, не происходило.
319
нитроглицерина при переходе от воздуха к углекислоте интенсивность
вспышки при низких температурах существенно уменьшается, при высо-
ких — почти не меняется. Для большинства жидких и плавящихся ВВ
(особенно для тэна, диглмкольдинитрата, дины, тетрила, гексогена, окто-
гена и гексила) перед вспышкой образуется, иногда в значительных ко-
личествах, пена. Повышение температуры уменьшает образование пены.
Влияние вспенивания на скорость сгорания ВВ при вспышке нагляднее
проявляется в опытах с большими (1—2 г) навесками тэна, тетрила и ди-
мы. При высокой температуре расплавы этих ВВ, воспламенившись с по-
верхности, сгорают спокойно ровлым фронтом и почти беззвучно. При
более низкой температуре все жидкости за время предпламенного разло-
жения превращаются в объемистую рыхлую пану, сгорающую очень быст-
ро, иногда с сильным звуком,
и в форме взрыва.
а в случае тэна в определенных условиях
О, г-см
300
200
/да
0,8- CM
&M0
I 300
£U
200
(00
о 20 но да so too % НГЦ
।___।_।__।_।_।
уда SO SO 40 20 0
% msetmsM
Рис. 250. Влияние температуры и вели-
чины навески на возможность возник-
новения взрыва при самовоспламенении
нитроглицериновой желатины
J — взрыв; 2 — мягкая вспышка
Рис. 249. Влияние состава
растворов коллоксилина н
нитроглицерине на макси-
мальную интенсивность их
вспышки (навеска 0,02
И. В. Бабайцевым п Б. Н. Коидриковым изучалось также поведение
при вспышке растворов коллоксилина в нитроглицерине. Введение 10%
коллоксилина очень сильно снижает интенсивность вспышки нитроглице-
рина (рис. 249). При увеличении содержания коллоксилина до 60% она
остается приближенно постоянной, а затем снова резко падает, при 75%
практически до нуля.
Интенсивность вспышки зависит также от величины на вески и от тем-
пературы. Результаты изучения совместного влияния этих двух факторов
приведены на рис. 250. Показателем интенсивности вспышки служило
дробление пробирки. При увеличении навески температурный интервал,
в котором вспышка дробит пробирку, сначала расширяется, составляя для
1 г почти 50°, а затем сужается; правая его граница — в области высоких
температур — сильнее, чем низкотемпературная, смещается влево; поэто-
му при весе желатины 10—50 г температурный интервал интенсивной
вспышки сокращается до 10—15°С. Однако при соответствующих условиях
этот взрыв достаточно интенсивен и может при опытах в стальной трубке
давать дробление ее, близкое к детонационному.
Измерение температуры в центре сферических или цилиндрических
зарядов желатин показало (рис. 251), что при невысоких температурах
термостата прогрев идет с уменьшающейся скоростью до достижения тем-
пературы термостата, после чего начинается ускоряющееся повышение
температуры, заканчивающееся вспышкой. При высоких температурах,
напротив, центр заряда к моменту возникновения вспышки не успевает
прогреться до температуры термостата и горит более холодная желатина.
.320
При определенной промежуточной температуре в момент возникновения
горения температура в центре заряда равна температуре термостата. Для
некоторых желатин в этих условиях наблюдается максимальная интен-
сивность вспышки.
Были поставлены также опыты, в которых осуществлялась интенсифи-
кация вспышки желатин с относительно малым содержанием нитроглице-
рина. Желатина нагревалась при температуре ниже критической и затем
переносилась в термостат, нагретый до более высокой температуры. Ин-
тенсивность вспышки в этих условиях может быть существенно выше
(рис. 252).
Рис. 251. И вменение температуры в центре сфериче-
ского заряда нитроглицериновой желатины при нагре-
вании (навеска — 1г)
Числа у кривых — температура термостата
Наблюдения, сделанные при изучении вспышки нитроглицериновых
желатин, согласуются с результатами, полученными по методике, описан-
ной на стр. 319. Желатины, содержащие 90, 80 и 70% нитроглицерина,
при определенной интенсивности нагревания давали взрыв, сопровождав-
шийся дроблением пробирочки, иногда разрывом нагревающей спирали и
образованием отпечатка на свинцовой пластинке. При слабом нагревании
происходит медленное разложение, внешне напоминающее кипение. При
более сильном нагреве желатина до самовоспламенения успевает про-
греться, разжижиться из-за повышения температуры и, вероятно, депо-
лимеризации нитроклетчатки и «закипеть». В этих условиях вслышка
имеет характер взрыва. При слишком сильном нагреве желатина воспла-
меняется на периферии заряда раньше, чем он весь успеет прогреться,
тогда вспышка имеет мягкий характер и сопровождается выбросом из про-
бирочки большей части заряда в горящем состоянии. Взрыв можно полу-
чить также, хотя и труднее, при меньших содержаниях нитроглицерина —
60, 50 и даже 40%. В этих случаях, однако, необходимо увеличить длину
пробирки и высоту столбика ВВ. Разложение желатин с большим содер-
жанием нитроклетчатки сопровождается, помимо описанных явлений, зна-
чительным вспениванием.
Наблюдения, сделанные при изучении вспышки различных ВВ, могут
быть объяснены на основе следующих соображений.
Отсутствие вспышки ниже некоторой критической температуры тер-
мостата, естественно, объясняется с точки зрения теплового самовоспла-
менения с учетом химического самоускорения реакции во времени. Влия-
ние газа, наполняющего пробирку, говорит за то, что вспышка непосред-
ственно возникает в газовой фазе, представляющей собой в данном случае
смесь паров или летучих продуктов распада с атмосферой пробирки.
21 К. К. Андреев
321
। । । <__i_____
/60 /80 200 220
Температура, ’С
Рис. 252. Зависимость интенсивности
вспышки желатины с относительно
малым содержанием нитроглицерина
при 200° С от времени предварительно-
го прогрева ее при 145° С (кривая 2);
интенсивность вспышки той же жела-
тины в зависимости от температуры
при однократном нагревании (кривая 2)
Интенсивность вспышки определяется скоростью газообразования при
ней, которая в свою очередь зависит от скорости горения ВВ. Эта послед-
няя зависит не только от природы ВВ, но и от режима — нормального или
возмущенного,— в котором проходит горение.
При вспышке порошкообразных ВВ одной из форм возмущения являет-
ся горение с проникновением процесса в глубь заряда (конвективное горе-
ние), а при вспышке жидкостей —
турбулентное горение. Переход на
возмущенный режим сопровождается
сильным увеличением скорости газо-
образования. Возможность перехода
зависит от ряда условий, в частности
от удельной поверхности ВВ, от ве-
личины нормальной скорости горения,
а следовательно; и от температуры,
при которой оно идет.
Влияние удельной поверхности
наглядно видно при сопоставлении
результатов опытов с пироксилино-
вым порохом, который вообще не под-
брасывает шарика, и порошкообраз-
ным пироксилином, который подбра-
сывает его почти так же высоко, как
гремучая ртуть. Между гремучей
ртутью и ее аналогами, с одной сто-
роны, и пироксилином, с другой, есть
существенное рааличие, состоящее в
том, что для нее интенсивность
вспышки не убывает при повышении
температуры опыта. По-видимому,
способность горения к проникнове-
нию в поры порошка быстрогорящего
ВВ столь велика, что проникновение
происходит в данных условиях опыта
независимо от температуры навески.
В случае же пироксилина проникно-
вение происходит лишь, если он перед самовоспламенением успел про-
греться. В случае жидких (при температуре вспышки) ВВ существенную
роль играет возможность возникновения турбулизации горения. Она обус-
ловливается прежде вседо соотношением их скорости горения в условиях
вспышки и предельного значения скорости нормального горения по Ландау
(«0,25 г1см'-сек при атмосферном давлении).
В табл. 46 приведены приближенные значения скоростей горения ряда
ВВ при температуре вспышки, полученные экстраполяцией иа основании
данных для более низких температур, и характеристяка поведения ВВ
при вспышке. Те вещества, которые имеют скорость горения, превышаю-
щую предельное значеяне, дают взрыв при самовоспламененян, в то время
как для веществ с мягкой вспышкой скорость горения много меньше.
При вспышке возможны некоторые дополнительные явления, влияю-
щие на ход гореиня.
В принципе возможен тот случай, когда при нагреве сохраняется пол-
ная термическая и химическая гомогенность нагреваемого вещества. Тогда
вещество будет к моменту воспламенения нагрето до предельно высокой
температуры, центры воспламенения возникнут сразу в очень большом
числе и время от момента самовоспламенения до момента завершения
реакции будет очень мало. Это наиболее быстрый гомогенный взрыв ве-
щества.
322
Таблица 46
Зависимость характера вспышки от величины скорости горения ВВ
(Навеока ВВ 0,3 г)
вв Температура вспышки, "С Скорость горения при температуре вспышки, е/см’сек Характер вспышки
Нитроглицерин .... 200 —0,5 Y Резкий звук, пробирка дро-
бится на мелкие куски;
сплав Вуда выплескивается
Нитроманнит 180—190 ~0,7 d из бани
Нитрогликоль 200 0,096
Тан 205 0,120 Слабый звук, пробирка во
Тетрил 190 0,210 всех случаях без поврежде-
Гексоген 230 0,084 ний мягкая вспышка
Тротил 300 0,040
Очевидно, однако, что в реальных условиях возникновения вспышки
при экзотермическом и самоускоряющемся характере распада ВВ нельзя
ожидать одновременного возникновения центров воспламенения во всей
его массе. Реакция, возникшая в какой-либо точке, создает условия для
реакции соседних молекул, более благоприятные, чем в массе вещества
в целом. Кроме того, ВВ в разных точках находится в различных услови-
ях в отношении теплоотдачи (и, следовательно, температуры), концент-
рации ускоряющих реакцию продуктов этой реакции, контакта с воздухом
и т. д. Поэтому следует полагать, что началом вспышки может служить
появление одного (первого) центра устойчивого воспламенения
Дальнейшее течение горения зависит от ряда факторов, в первую оче-
редь от возможности или невозможности устойчивого горения нагретого
до дайной температуры и частично разложившегося вещества. Если даже
в момент самовоспламенения состояние вещества допускает устойчивое
горение, то это условие необязательно будет выполняться на всем протя-
жении горения.
Существенную роль при этом играет масса ВВ. Если состояние вещест-
ва в последующее после возникновения центра воспламенения время не
меняется, то при малой массе все вещество может сгореть от первого и
единственного центра воспламенения. При большей массе вещества за
время распространения горения, начавшегося от первого центра воспла-
менения, возможно возникновение новых центров, что приведет к увели-
чению в той и ин иной степени интенсивности явления.
Это осложнение первичного процесса тем более вероятно, что обычно
состояние вещества не остается неизменным за время после воспламене-
ния, а продолжает изменяться в сторону увеличения вероятности появле-
ния центров воспламенения (повышение температуры вследствие внеш-
него разогрева и саморазогрева, увеличение концентрации промежуточ-
ных или конечных продуктов, ускоряющих превращение).
Развитие химической реакции распада может отразиться на изменении
хода горения даже независимо от появления новых центров воспламене-
ния. Эта реакция идет, как правило, с выделением газов; интенсивное вы-
деление пузырьков газа, поднимающихся кверху и прорывающихся черев
фронт горения, нарушает этот фронт и приводит к ускорению горения,
сопровождающемуся увлечением капель жидкости в поток оттекающих
газов. Аналогичные последствия имеет образование пены в разлагающем-
ся жидком ВВ.
1 Под центром устойчивого воспламенения мы разумеем такой очаг воспламене-
ния, который способен к самораспространению. Судя по некоторым работам, его воз-
никновению может предшествовать образование центров меньших размеров, не даю-
щих неограниченного самораспространения пламени.
21* 323
Именно этими факторами следует объяснить и своеобразный характер
горения, наблюдавшегося при самовоспламенении в результате внешнего
нагрева навески (100 а) пикриновой кислоты в колбе Эрленмейера емко-
стью 250 ел»3. Сначала вещество расплавилось, затем наблюдалось образо-
вание белых паров и их слабая вспышка с загоранием жидкости. Через
30 сек произошло вспенивание с выбросом снопа горящих брызг, закон-
чившееся глухим хлопком с разрывом колбы.
Та же картина наблюдается при горенки сильно нагретого жидкого
тротила, возникающем от внешнего поджигания или в результате само-
воспламенения. Равномерное при меньших температурах, оно становится
Пульсирующим и сопровождается, как и при горении пикриновой кислоты,
выбросом горящих брызг жидкости.
Помимо разложения вследствие самопроизвольного развития реакции,
в этих условиях известную роль при малой величине скорости горения
тротила может играть таиже прогрев жидкости за счет передачи тепла
по стенкам сосуда: при более низких температурах горение вначале было
равномерным, но, подходя к концу трубки, становилось пульсирующим,
так как трубка стояла на подставке иа плохо проводящего тепло материа-
ла, теплу было некуда уходить, и происходил сильный разогрев стенок.
Интересны в атом отношении также опыты Гольбиндера [258] по пере-
ходу горения во взрыв смеси виилина с тетранитрометаном, механизм
самовоспламенения которой был описан выше.
Сильно экзотермическая реакция, приводящая к самовоспламенению,
возиняает в газовой фазе или точнее в газопарокапельной взвеси над по-
верхностью жидкости. Воспламенение газов или паров приводит к горе-
нию жидкой смеси.
Это горение распространяется со значительной скоростью, превосходя-
щей то ее значение, при котором обычно возникает автотурбулизация
поверхности горящей жидкости, неравномерно и с сильной пульсацией.
Так же как и для некоторых других взрывчатых жидкостей, при перехо-
де горения на турбулентный режим в определенных условиях наблюдается
затухание горения, в данном случае временное; реакции, приводящие к
самовоспламенению, продолжаются и после затухания и вновь вызывают
самовоспламенение. В результате этого горение может протекать в форме
чередующихся вспышек с сильно различающейся для разных ВВ частотой.
Изучение условий перехода горения во взрыв показало, что ему сильно
благоприятствует увеличение высоты (а не общая масса) столбика жид-
кости; критическая высота для смесей на основе анилина и о-толуидииа
составляет около 10 сж. Длина сгоревшего до перехода во взрыв участка
не показывает закономерной зависимости от общей высоты столбика жид-
кости. Повышение начального давления увеличивает скорость горения и
облегчает переход его во взрыв.
Переход горения во взрыв и влияние на него высоты заряда объясняет-
ся следующим образом. Горение само по себе идет со скоростью, доста-
точной дия его ускорения за счет возникновения турбулизации. Это уско-
рение дополнительно усиливается прохождением через жидкость пувырь-
ков газов, создающих рельеф поверхности жидкости и уносящих капли
жидкости в зону горячих продуктов горения. Это дополнительное ускоре-
ние, как показали опыты, в данном случае является решающим. При вы-
соте заряда 10 см в трубках диаметром 5 и 18 леи горение проходило без
взрыва. В комбинированной трубке, нижний участок которой (4 см) имел
диаметр 18 леи а верхний (6 си) — 5 леи, горение регулярно переходило
во взрыв. Активный характер промежуточных газообразных продуктов,
содержащихся в пузырьках, образующихся в жидкости, также как и на
массе пены, ячейки которой наполнены теми же продуктами, способству-
ет возникновению взрыва. Известие^ по опытам Боудена, что ему благо-
приятствует наличие в жидкости даже пузырьков инертного газа; несом-
32«
неино, что при горячем газе, способном к быстрой экзотермической реак-
ции, это влияние пузырьков будет еще более выражено.
Газовый или пенный взрыв, естественно, вызывает взрыв нагретой
жадности.
Подобное горение больших количеств ВВ, особенно при продолжаю-
щемся внешнем разогреве сосуда, сопровождающееся выбросом брызг
жидкости в пламя, может закончиться детонацией, которая нередко на-
блюдалась при пожарах в производстве.
Помимо указанного, количество вещества может иметь еще следую-
щее влияние. Если горение неустойчиво и способно переходить в детона-
цию, то при слишком малом количестве вещества ВВ может выгореть,
прежде чем будет достигнут детонационный режим. В опытах по неустой-
чивому горению порошкообразных, а также жидких ВВ переход горении
во взрыв наблюдался лишь в том случае, если высота заряда превосходила
некоторый минимум.
В некоторых случаях, как мы видели, в возникновении вспышки при-
нимают участие пары ВВ или смесь их с окружающим газом (обычно воз-
духом). Вспышка паров может отразиться на всем явлении по двум при-
чинам. Воспламенение паров или их смеси с воздухом может приводить к
их детонации, которая в свою очередь может вызвать детонацию нагретой
конденсированной фазы.
Известно, что детонация газа при достаточной его плотности способна
вызывать детонацию конденсироввиного ВВ [259]. Помимо непосредствен-
ного возбуждения детонации нагретой жидкости газовым взрывом, послед-
ний может способствовать возникновению детонации следующим косвен-
ным образом. Степень устойчивости горения, как мы видели, сильно зави-
сит от давления. Повышение давления, создаваемое газовым взрывом,
может быть в некоторых случаях достаточным, чтобы перевести горение
в неустойчивую область. При нагреве метилннтрата в пробирке до темпе-
ратуры, близкой к температуре кипения, и поджигании паров возникала
детонация их, приводившая к детонации жидкости.
Очень своеобразно, не при нагревании, а при охлаждении, возникает
в результате самовоспламенения паров вспышка жидкого хлористого азота
[251]. Верхний конец запаянной трубочки с хлористым азотом, находился
при комнатной температуре, инжиий был погружен в охлаждающую
смесь. Когда температура жидкости снижалась до —25°, происходил
взрыв. Механизм возникновения этого взрыва, согласно Апиву, следую-
щий. Для паров хлористого азота вследствие цепного механизма его рас-
пада существует верхний предел давления, ниже которого даже при ком-
натной температуре происходит их самовоспламенение. При охлаждении
жидкого хлористого азота давление паров над инм понижается и, когда
достигается верхний предел, пары воспламеняются, вызывая воспламене-
ние и обычно взрыв жидкости.
3. ВЕРХНИЙ ТЕМПЕРАТУРНЫЙ предел вспышки
При изучении вспышки было обнаружено [260] своеобразное явление,
названное верхним пределом температуры вспышки. Оно заключается в
том, что для некоторых летучих ВВ (тротил, пикриновая кислота, ксилил)
вспышки, наблюдающейся при введении небольшой навески ВВ в нагре-
тую пробирку, не происходит, если температура пробирки выше опреде-
ленного предела; вместо вспышки наблюдается беспламенное разложение,
У нитроглицерина детонация, имеющая место при вспышке в интервале
температур 200—220°С, сменяется при температуре выше 250° быстрым
сгоранием без заметного звука и механических эффектов. У тетрила верх-
него предела температуры вспышки не наблюдается.
Существование верхнего предела объясняется тем, что возникновение
вспышки зависит не только от температуры, но и от концентрации продук-
тов распада, промежуточных или конечных. Так как развитие термиче-
ского распада, включающего ряд реакций, зависит, в частности, от темпе-
ратуры, можно предположить, что при высоких температурах опыта и со-
ответственно быстром прогреве ВВ до температуры кипения не достигает-
ся той высокой концентрации промежуточных продуктов, ускоряющих
превращение, которая необходима для самовоспламенения. Это объясне-
ние косвенно подтверждается опытами, при которых применялся частично
разложенный при низкой температуре тротил и
которые давали вспышку даже при температурах
выше верхнего предела.
Процессы, происходящие при нагревании в опи-
санных выше условиях опыта надо представлять
себе следующим образом [254]. Если температура
бани невысока и нагрев идет медленно, то к момен-
ту достижения температуры кипения концентра-
ция ускорителей достаточна для возникновения са-
мовоспламенения и последнее происходит.
Если же температура бени высока и нагрев идет
быстро, то к моменту достижения 'температуры
кипения концентрация продуктов реакции будет
недостаточной для возникновения самовоспламене-
Рис. 253. Изменение во
времени температуры
тротила при быстром
иагреваили
I — при температуре Пани
340° С; 2 — при температуре
Вани 380“ С
меньшей плотности), а
ния, и вещество превращается в пар. Разложение
будет проходить без вспышки, так как в парообраз-
ном состоянии при одинаковых кинетических ха-
рактеристиках для возникновения самовоспламене-
ния нужна более высокая температура: количест-
во тепла, выделяющегося в единице объема, значи-
тельно (в 200—1000 раз) меньше (соответственно
теплопроводность паров меньше теплопроводности
жидкости только приблизительно в 10 раз. Кроме того, самоускорение
распада большинства изучавшихся ВВ в парах меньше, чем в жидком
состоянии.
Наглядное подтверждение этого объяснения дают результаты опытов
[26Ц в которых измерялось изменение температуры небольшой (0,1 г)
навески тротила, сброшенной в пробирку, погруженную на х/з в нагретый
сплав Вуда. Кривая 1 (рис. 253) показывает изменение температуры тро-
тила при температуре сплава 340° С; через 19 сек. была достигнута темпе-
ратура 337° С, близкая к температуре кипения, и произошла вспышка.
При температуре сплава 380° С (кривая 2) эта температура была достиг-
нута через 8 сек.; после некоторой остановки подъема температуры, оче-
видно, вызванной вскипанием тротила, она продолжала подниматься и
была прослежена до 360°С, причем вспышки так и не произошло.
Верхний предел температуры вспышки дают не все ВВ, но лишь те,
температура кипения которых лежит сравнительно низко по отношению
к температуре вспышки; ВВ, температура кипения которых лежит выше
температуры вспышки (тетрил, гекоотен и др.) или которые вообще не
способны к кипению (пироксилин), не имеют верхнего предела темпера-
туры вспышки.
Гольбиндер [250] обратил внимание на аналогию явлений, наблюдав-
шихся <им при вспышке жидких смесей, происходящей на верхнем пре-
деле температуры вспышки индивидуальных ВВ. Многие летучие смеси,
взятые в небольших количествах, показывают явление верхнего предела
во всем интервале температур от комнатной до 500° С, когда уже правиль-
нее говорить не о вспышке ВВ, а о поджигании его паров нагретой по-
верхностью. Если увеличивать толщину слоя смеси, то время испарения
растет и самоускорение распада успевает произойти раньше чем испарит-
ся все ВВ, поэтому верхний предел снижается.
Очень наглядно иллюстрируют роль химического самоускорения и
летучести опыты по добавке небольших количеств стирола к смеси бензо-
ла с тетранитрометаном, резко (на 200° С) снижающего температуру
вспышки. Сама по себе смесь бензола с тетранитрометаном химически
сравнительно инертна; поэтому самовоспламенение ее возникает только
при такой температуре термостата, при которой вспыхивает смесь паров,
т. е. около 500° С. При добавлении 0,5 % стирола условия испарения смеси
существенно измениться не могли. Сильное снижение температуры вспыш-
ки объясняется тем, что наличие в смеси реакционноспособного стирола
приводит при более низкой температуре к образованию с достаточной
, скоростью активных продуктов. Необходимая для самовоспламенения их
концентрация создается за время, меньшее, чем время полного испарения.
В заключение следует отметить, что верхний предел температуры
' вспышки не является абсолютным пределом, но лишь ограничивает снизу
интервал температуры, в котором вспышка отсутствует. При более высо-
ких температурах вспышка появляется вновь, но это уже, очевидно,
вспышка или даже воспламенение паров,
ВЫВОДЫ
Вспышка ВВ представляет собой горение нагретого вещества, возни-
1 кающее путем самовоспламенения. Это самовоспламенение происходит
вследствие саморазогрева ВВ за счет экзотермической и, как правило,
самоускоряющейся реакции его термического распада. Минимальная тем-
пература вспышки зависит от размера заряда и длительности нагрева,
( определяющей химическое ускорение превращения.
Характер вспышки определяется устойчивостью горения при темпера-
туре самовоспламенения. Если горение устойчиво, вспышка имеет мягкий
характер, напоминающий нормальное горение; если горение резко не-
устойчиво, то вспышка принимает характер взрыва, особенно, если масса
ВВ велика.
Интенсивность вспышки инициирующих и быстрогорящих ВВ в по-
рошкообразном виде возрастает при увеличении температуры термостата
, до максимума и при дальнейшем повышении температуры остается посто-
янной. Для вторичных ВВ после максимума наблюдается понижение ин-
тенсивности вспышки, связанное с тем, что горение заряда, не успевшего
прогреться, является более устойчивым.
Если условия нагрева и летучесть ВВ таковы, что оно быстро превра-
. щается в пар, прежде чем разложение пройдет в значительной степени
и накопится достаточное количество промежуточных продуктов, ускоря-
ющих превращение, то вспышка с пламенем отсутствует. По этой причине
при повышении температуры нагрева для летучих ВВ наблюдается верх-
ний предел температуры, выше которого вспышка прекращается и появ-
ляется вновь уже в виде вспышки или воспламенения паров при значи-
тельно более высоких температурах.
II. ВОСПЛАМЕНЯЕМОСТЬ ВВ
Воспламеняемость является важным свойством ВВ, особенно сущест-
венным в некоторых конкретных условиях его применения. Это прежде
всего выстрел ствольный и реактивный. Задачей воспламенителя является
обеспечение (за возможно короткое время и без чрезмерного повышения
давления) возникновения горения на всей поверхности заряда. Дефекты
327
воспламенения могут иметь, особенно в реактивных системах, и более
тяжелые последствия — увеличение горящей поверхности вследствие про-
никновения горении в образующиеся при воспламенении трещины в заря-
де, итогом которого является разрыв двигателя.
Воспламенение ВВ может происходить при взрывных работах в шах-
тах; при откаф детонации или при прекращении ее по той или другой при-
чине оставшаяся часть заряда может загореться, если воспламеняемость
ВВ велика и осуществляются условия для воспламенения: достаточно
интенсивное по длительности и величине давления действие газообразных
продуктов детонации.
Наконец, и во многих условиях возбуждения взрыва механическим
импульсом первой стадией процесса является горение, а его возникнове-
ние определяется, в частности, воспламеняемостью ВВ.
Между вспышкой, происходящей при медленном разогреве ВВ, и вос-
пламенением, возникающим при очень быстром его нагреве, есть сущест-
венная разница.
Если заряд ВВ нагреть до некоторой умеренно повышенной темпера-
туры, то оно, если эта температура не слишком низка, а размеры заряда
не слишком малы, даст вспышку. Физический смысл этого явления был
рассмотрен в предыдущем разделе; ВВ целиком прогревается до темпера-
туры термостата, при которой идет развивающаяся экзотермическая реак-
ция. Когда теплоприход за счет этой реакции начинает расти быстрее
теплоотдачи, возникает вспышка, представляющая собой самовоспламене-
ние и последующее гореине вещества.
Можно рассчитать минимальное количество тепла, подводимого веще-
ству извне, необходимое для получения вспышки. Оно равно произведе-
нию массы ВВ на его теплоемкость и на ДГ — разность температуры термо-
стата и начальной температуры вещества. Таким образом, это количество
пропорционально массе вещества: при большей массе оно будет большим.
Если на тот же заряд действовать источником тепла высокой тем-
пературы, например пламенем, то также наступит воспламенение, но
количество необходимого для воспламенения тепла будет значительно
меньше, чем в первом случае, и практически от массы зависеть не будет *.
Таким образом, хотя оба явления представляют собой воспламенение
ВВ, вызываемое внешинм нагревом, но вследствие различия температур
чагрева они существенно отличаются друг от друга.
Каков же механизм процесса во втором случае?
При действии высокой температуры, например, нагретого газа на по-
верхность АВ взрывчатого вещества (рис. 254) температура на этой по-
верхности не сразу достигает своего максимального значения. Величина
температуры поверхностного слоя определяется двумя процессами: сооб-
щением тепла нагретым газом поверхности ВВ и отводом тепла от поверх-
ности и в глубь вещества1 2.
Величина теплоотвода в вещество определяется разностью температуры
поверхности и температуры прилегающих слоев. Вначале (рлс. 254, а)
1 Иногда, пытаясь определить задержку вспышки при очень высоких температу-
рах, вводят ВВ в сильно нагретую пробирку. Фактически в условиях такого опыта
реализуется не вспышка, а воспламенеиле ВВ и, естественно, его результаты анало-
гичны тем, которые получаются при поджигании В В прикосновением к нему сильно
нагретого стержня или при поджигании сильно нагретым сжатым газом и т. п., т. е.
в большей или меньшей мере характеризуют воспламеняемость ВВ.
2 Здесь, как и во всем дальнейшем рассмотреиля, предполагается, что в конден-
сированной фазе химической реакции не происходит. Это полностью справедливо
лишь для горения летучих ВВ при илзилх давлениях. Если при горении имеет место
существенное выделение тепла в конденсированной фазе (или нарушение ее физиче-
ской структуры, например, диспергирование при торёнил некоторых ВВ), излагаемые
соображения и выводы утрачивают в большей или меньшей степени свою прило-
жимость.
32S
эти слои имеют температуру, равную начальной температуре ВВ (Го)- По
мере прогрева температура этих слоев возрастает, и в результате этого
теплоотвод в глубь вещества падает (рис. 254, б). Если считать скорость
сообщения тепла постоянной, то уменьшение теплоотвода означает быстрое
повышение температуры поверхностного слоя. Когда эта температура до-
стигает некоторого предала Тк — температуры кипения (рис. 254, в) для
летучих ВВ или температуры быстрой газификации для нелетучих ВВ, то
образуются пары или газообразные продукты первичной реакции разло-
жения. Эти пары быстро — соответственно их малой объемной теплоем-
кости — нагреваются до такой температуры, при которой наступает весь-
ма быстрая экзотермическая реакция, приводящая к конечным при дан-
Рис. 254. Изменение температуры на поверхности ВВ
(пороха) при воспламенении
а—в начале нагревания температура поверхностного слоя равва
То; б — в ходе нагревания температура поверхностного слоя Воль-
те То. но меньше Тк; в — в момент воспламенения температура
поверхностного слоя равна Ти
Если в момент воспламенения прекращается действие внешнего источ-
ника тепла, то судьба возникшего горения может быть различной в зави-
симости от толщины образовавшегося при нем прогретого слоя конденси-
рованной фазы.
Действительно, при стационарном горении при данном давлении и на-
чальной температуре, глубина зоны прогрева, толщина прогретого слоя
имеет вполне определенную величину. Эта толщина определяется ско-
ростью тепловой волны, убегающей вглубь вещества, и скоростью движе-
ния фронта горения, догоняющего и подталкивающего эту волну.
Теория [238] показывает, что вблизи поверхности имеется определен-
ное распределение температуры в конденсированной фазе.
Пусть интенсивность нагрева при воспламенении будет танова, что в
момент достижения поверхностью АВ температуры Тк распределение тем-
ггературы в прогретом слое конденсированной фавы будет соответствовать
тому, которое имеет место при стационарном горении. В этом случае вос-
пламенение приведет к возникновению устойчивого горения (рис. 255,а).
Если интенсивность нагрева будет меньше, то в момент достижения
поверхностью предельной температуры Тк толщина прогретого слоя будет
больше, чем это соответствует стационарному горению, другими словами,
кривая падения температуры от поверхности в глубь конденсированной
фазы будет более пологой, чем в нервом случае (рис. 255, б). Тогда при
неизменном теплоприходе от газов горения теплоотвод в глубь вещества
будет меньше. За счет увеличенной разности теплоприхода и теплоотвода
газообразные продукты горения будут иметь больший запас тепла, т. е.
большую конечную температуру. Поэтому скорость горения вначале боль-
ше, чем при стационарном горении. Картина будет такая же, как если бы
вначале горело предварительно подогретое вещество.
Когда фронт горения, догоняя ушедшую вперед тепловую волну, по-
дойдет к холодному веществу, градиент температуры вблизи поверхности
увеличится, скорость же горения уменьшится до значения, соответствую-
щего стационарному режиму, и горение будет продолжаться с этой ско-
ростью.
329
Если горение идет в условиях, при которых повышение давления за
счет газов горения не Происходит, то единственное отличие второго случая
от первого будет заключаться в более быстром горении начального
участка.
Если интенсивность нагрева будет больше, чем в первом рассмотрен-
ном случае, то к моменту, когда на поверхности АВ будет достигнута
предельная температура, толщина прогретого слоя будет меньше, чем со-
ответствующая стационарному горению (рис. 255, в).
Рис. 255. Изменение толщины прогретого слоя при воспла-
менении в зависимости от интенсивности нагревания
о — прогретый слой нормальной для стационарного горения толщи-
ны; о — прогретый слой толще нормального; в — прогретый слой
тоньше нормального
Если источник тепла в этот момент прекратит свое действие, то гра-
диент температуры у поверхности будет больше стационарного, тепло-
приход не будет компенсировать теплоотдачу и возникшее горение может
затухнуть. Понятно, что если действие источника тепла не прекратится,
то оно будет компенсировать теплопотери, и горение, медленное вначале,
ускоряясь, достигнет нормальной скорости, а прогретый слой, утолщаясь
за счет медленности горения, достигнет своей нормальной толщины.
Так протекает воспламенение при разных температурах теплоносителя
и в условиях, в которых заметное повышение давления при воспламене-
нии отсутствует.
Для этих условий можно рассчитать на основании законов теплопере-
дачи время воспламенения, например, при действии струей горячего газа.
Это — время, необходимое для достижения на поверхности определенной
температуры, температуры кипения Тк или температуры разложения Тр.
Точно так же можно рассчитать и время нормального воспламенения,
приводящего к образованию прогретого слоя с толщиной, соответствую-
щей стационарному горению; для этого при подсчете надо температуру
теплоносителя принять равной температуре горения.
Перейдем к вопросу о связи воспламеняемости с характеристиками
ВВ и процесса горения.
Основным вопросом нормального воспламенения является образование
прогретого слоя соответствующей данным условиям (ро и То) толщины.
Запас тепла в этом слое может быть мерой воспламеняемости вещества.
Если в конденсированной фазе, прилегающей к фронту (поверхности
раздела конденсированной фазы и паров или газов), существует теплопро-
водиостное михельсоново распределение температуры \ то температура
Т на расстоянии х от фронта определяется выражением (4.15) (стр. 268).
Г = 7’0 + (7’к-7’0)/1', (6.1)
1 Некоторыми исследователями указывалось, что применительно к практически
наиболее интересному случаю — воспламенению порохов — указанное условие непри-
менимо вследствие того, что при нагревании в конденсированной фазе идет экзотер-
мическая реакция разложения, что нарушает михельсоново распределение темпера-
туры; в принципе это замечание, весьма вероятно, правильно. Возможно [262], одна-
ко, что влияние экзотермической реакции будет столь незначительно, что оно не от-
разится существенно на возможности применения к данному случаю теории прогре-
того слоя в простейшем ее виде.
330
где Тк— температура на поверхности конденсированной фазы, в едучае
жидких летучих ВВ — температура кипения, в случае нелетучих ВВ— не-
которая температура, предполагаемая Зельдовичем приближенно посФбянл
ной, при которой конденсированная фаза быстро переходит в газы.
Количество тепла Q в прогретом слое может быть рассчитано по выра-
жению
СО
Q—^срд(Т ~T0)dx = ^(Тк-—Т0). (6.2)
О
Это выражение показывает, что нельзя, как это иногда делается, при-
нимать в качестве характеристики воспламеняемости температуру само-
воспламенения ВВ. Во-первых, эта температура не является постоянной,
но зависит, как мы видели, от длительности нагрева и других факторов
и, вообще говоря, ниже Тк. Только при очень малых временах задержки
температура самовоспламенения приближается к 7'к. Далее, Q зависит не
только от Гв, но и от теплопроводности ВВ и скорости его горения. В то же
время как в отношении теплопроводности, так и особенно в отношении
скорости горения различные ВВ существенно отличаются друг от друга.
Поэтому два вещества, имеющие близкую температуру самовоспламене-
ния, например тротил и черный порох, могут иметь резко различную вос-
пламеняемость соответственно различию в 60 раз их скоростей горения.
Беляев [263] сопоставлял воспламеняемость с летучестью, полагая, что
чем больше летучесть, тем больше и воспламеняемость. Этот вывод спра-
ведлив лишь постольку, поскольку при прочих равных условиях необхо-
димый запас тепла для летучих веществ тем больше, чем выше темпера-
тура кипения. Однако прочие условия, как правило, не бывают равными;
кроме того, понятие летучести условно, и для нелетучих ВВ 7р может
быть меньше, чем температура кипения для некоторых летучих веществ.
Нельзя считать теоретически обоснованным также экспериментальный
метод, которым пользовался Беляев для определения воспламеняемости;
накаливаемая в течение 5 сек. проволочка касалась поверхности вещества;
устанавливалась та температура, до которой должно было быть нагрето ве-
щество, чтобы произошло воспламенение. При этих условиях происходит
воспламенение не ВВ, как такового, а смеси его паров или продуктов
разложения с воздухом, воспламеняемость которой может не иметь ничего
общего с воспламеняемостью ВВ. Таким способом может быть определена
и воспламеняемость горючих, не способных к самостоятельному горению.
Формула для запаса тепла дает и зависимость воспламеняемости от
температуры вещества. Необходимый запас тепла уменьшается с возраста-
нием температуры, так как уменьшается разность Тк—Т0 и, кроме того,
увеличивается скорость горения.
Зависимость воспламеняемости от давления мы можем получить, заме-
няя и выражением ее зависимости от давления. Принимая для этой зави-
симости формулу прямой пропорциональности и = В-р имеем:
^^.(П-П), (6.3)
т. е. необходимый запас тепла приближенно обратно пропорционален дав-
лению.
Известно, что при вылете недогоревших пороховых зерен из ствола ору-
дия при выстреле наблюдается их затухание. Запас тепла в прогретом
слое при горении под большим давлением в канале ствола орудия мал и
он оказывается недостаточным для горения под атмосферным давлением,
хотя порох, зажженный при этом давлении, способен гореть.
При уменьшении плотности порошкообразного вещества его воспламе-
няемость возрастает, так как увеличивается линейная скорость горения и
одновременно уменьшается теплопроводность порошка.
331
ЭКСПЕРИМЕНТАЛЬНОЕ ОПРЕДЕЛЕНИЕ ВОСПЛАМЕНЯЕМОСТИ ВВ
Наиболее распространенными являются два метода экспериментально-
го определения воспламеняемости.
Над ВВ помещают иа некоторой высоте вертикально отрезок бикфор-
дова шнура или электровоспламенитель. Устанавливают то максимальное
расстояние, на котором воспламеняется ВВ. Это расстояние и принимают
за меру воспламеняемости. Такой метод, естественно, позволяет дать толь-
ко сравнительную оценку воспламеняемости.
Второй метод заключается в том, что поверхность ВВ подвергают не-
прерывно или периодически действию высокой температуры, например га-
зового пламени. Минимальное время действия источника тепла, приводя-
щее к воспламенению, служит мерой воспламеняемости. В отношении это-
го метода следует заметить, что, во-первых, воспламеняются первично не
пары ВВ, а смесь их с продуктами горения газового пламени, и, возмож-
но, с воздухом. Во-вторых, температура нагрева может не соответствовать
той, при которой в момент воспламенения толщина прогретого слоя равна
толщине его при стационарном горении. Если температура горения газо-
вого пламени ниже температуры горения ВВ, то результаты (количество
тепла) получатся завышенными; если температура будет выше, то явле-
ние устойчивого воспламенения будет иметь более сложный характер, и
результаты опыта трудно будет оценить.
Этим методом определял воспламеняемость различных порохов Крон-
квист [264]. Пороховое зерно, закрепленное в нижнем конце маятника,
проходило при его движении через пламя. Продолжительность прохожде-
ния зерна в пламени до воспламенения служила мерой воспламеняемости.
Полученные времена выражаются сотнями миллисекунд и для бездымных
порохов в несколько раз меньше, чем для разных видов черного пороха.
Если судить по этим данным, то следовало бы заключить, что воспла-
меняемость черного пороха гораздо меньше, чем бездымного, что не соот-
ветствует ни опыту практики, ни результатам методически правильно по-
ставленных лабораторных опытов (см. ниже). Ошибочность результатов,
полученных Кроиквистом для черного пороха, объясняется тем, что интен-
сивность нагрева (температура пламени) в его опытах была слишком
мала. Поэтому для черного пороха необходимое время нагрева было так
велико, что по сути дела определялась не воспламеняемость его, а темпе-
ратура вспышки. Чтобы получить правильное представление о воспламе-
няемости черного пороха, необходимо применить при испытания более
высокую температуру нагрева.
Принципиально правильным был бы такой метод определения воспла-
меняемости, при котором поверхность вещества подвергалась бы действию
химически инертного теплоносителя с температурой, равной температуре
горения вещества. Это может быть осуществлено, например, в виде паде-
ния тонкой пластинки вещества (пороха) в сосуде с нагретым инертным
газом или продуктами горения пороха. Фиксируется время до воспламе-
нения и время горения. При низких температурах газа при этом опреде-
ляется просто температура вспышки. Чем выше будет температура, тем
меньше будет время до воспламенения, и тем больше время сгорания. Та
температура инертного газа, при которой время горения равно времени
стационарного горения пластинки данной толщины, и есть температура
горения пороха, а время до воспламенения при этой температуре являет-
ся мерой воспламеняемости. Экспериментально этот метод не реализован.
Один из возможных, теоретически обоснованных и экспериментально
осуществленных методов [265] состоит в том, что на торец цилиндриче-
ского заряда вещества, воспламеняемость которого надо определить, поме-
щается вплотную столбик воспламеняющего вещества, в качестве кото-
рого применяется неплавящееся порошкообразное ВВ, например пирокси-
332
лин. Устанавливается минимальная плотность воспламеняющего вещест-
ва, при которой происходит воспламенение испытуемого вещества. Чем
меньше эта плотность, тем больше воспламеняемость.
В основе метода лежит зависимость запаса тепла в прифронтовом слое,
с которым идет горение, от плотности. Из соотношения Q = Х/м (Тк—То)
следует, что запас тепла уменьшается при уменьшенин плотности, так как
линейная скорость горения возрастает приблизительно обратно пропорци-
онально плотности (массовая скорость постоянна), а теплопроводность
уменьшается. Поэтому залас тепла, который передает воспламеняющее
вещество поверхности воспламеняемого, зависит от плотности первого и
даже может быть количественно определен, если известны все величины,
входящие в выражение для Q. Если этот запас больше, чем необходимый
для стационарного горения испытуемого вещества, то воспламенение на-
ступает, и наоборот. При неполном знании величин, входящих в выра-
жение для Q, минимальная плотность воспламеняющего,вещества може/
служить относительной мерой воспламеняемости.
Недостатком метода является необходимость большой точности при вы-
полнении опыта в отношении равномерности плотности тем более, что ре-
зультат опыта определяется не средним значением плотности воспламеня-
ющего заряда, а плотностью тонкого слоя, прилегающего к поверхности
воспламеняемого заряда. Для более точного определения предельной плот-
ности целесообразно производить ие один, а несколько опытов при каж-
дой плотности. В табл. 47 приведены результаты опытов по определению
воспламеняемости ряда ВВ по данному методу.
Таблица 47
Опыты по определению воспламеняемости ВВ
Испытуемое вещество Кубическая плотность испытуемого ВВ, е/ем1 Кубическая плотность воспламеняющего вещества, е/см* Результат опыта
Нитроглицериновый порох 1,58 1,08: 1,19; 1,20; 1,21 1.20,1,21; 1,21; 1,21; 1,22; 1,28 Не воспла- менился Воспламе- нился
Порох ВТ 0,91 0,38; 0,51; 0,54; 0,61; 0,67; 0,67; 0,70 0.62; 0,65; 0,67; 0,69; 0,70; 0,70; 0,73; 0,73; 0,74; 0,75 Не воспла- менился Воспламе- пился
Черный порох (мякоть) 1,25 0,20; 0,21; 0,21 То же
Гексоген 0,80 1,48; 1.48; 1,49; 1,52; 1,53; 1,57- 1,53; 1,55 Не воспла- менился Воспламе- нился
Тетрил 0,77 1.45; 1,50; 1,62 Не воспла- менился
Примечание. В качестве воспламеняющего вещества во всех опытах
применялась смесь пироксилина с гексогеном 80 : 20,
Любопытны результаты опытов по сопоставлению воспламеняющего
действия влажного и сухого пироксилина. Скорость горения влажного
пироксилина значительно меньше и поэтому запас тепла в прифронтовом
слое больше. Следствием этого являются, с одной стороны, меньшая
воспламеняемость влажного пироксилина и, с другой,— его более высокое
воспламеняющее действие, чем сухого, несмотря на то, что температура
газообразных продуктов гореиня ниже. При опытах плотность воспламе-
няемого заряда (пироксилина) была одинаковой (0,60 г/см3); плотность
22 К. К. Андреев 333
воспламеняющего заряда была равна 0,20 г/см3. При воспламеняющем за-
ряде из сухого пироксилина воспламенение не происходит, влажный пиро-
ксилин (с 3,2% воды) во всех опытах дал воспламенение.
Для оценки воспламеняемости может быть использован также крити-
ческий диаметр горения, характеризующий способность ВВ к горению
(см. стр. 109 и сл.). Чем толще прогретый слой, тем больше теплопотери
в окружающую среду при горении цилиндрического заряда. Величина ско-
рости горения (или давления), теплопроводность ВВ и, в случае порошко-
обрезного ВВ, его плотность влияют на критический диаметр горения, как
правило, аналогично тому, как они внияют на запас тепла в прогретом
слое, характеризующий воспламеняемость.
Применительно к конкретным условиям воспламенения желательны,
однако, методы непосредственного определения воспламеняемости, тем
более что не во всех условиях способность превращения к самораспрост-
раяеяию и легкость его возбуждения оказываются тождественными *.
Таблица 48
Воспламеняемость некоторых взрывчатых веществ по опытам
в манометрическое бомбе
(Заряды без оболочки, вес заряда 4 г, диаметр 20 л», воспламенитель—смесь
пироксилина и аммиачной селитры 1 :1)
вв Минимальная навеска восп- ламенителя, а (давление, пт) Время дости- жения макси- мума даоле- ВЯЯф МСВК Мансимяльяое дапленме, ат
А. Плотность заряда 0,95—0,99 от удельноговеса ВВ
Пикрат калия 110 520
62%-ный динамит 0,2 (25) 180 710
Тетрил 280 816
Перхлорат аммония 0,4(55) 750 286
Ксилил 750 570
Тротил 580 720
Тринитробензол 520 870
Пикрат аммония 0,6(85) 500 770
Тринитроанилин 570 810
Скальный аммонит 290 770
Аммонит 6 ЖВ 590 646
Аммонит ПЖВ-20 520 440
Аммонит 8 520 400
Аммонит № 6 1,0(140) 720 660
Аммонал ВА-4 760 664
Смесь АС с ТНБ * 650 765
Смесь АС с TH А * 510 680
Смесь АС с ТНК* 720 575
Аммиачная селитра 2,0(360) (затухание
горения)
В. Плотность заряда 0,75 от удельного веса ВВ
Тротил | 0,4(55) | 370 720
Аммонит № 6 | 0,8(115) | 680 720
» Смеси аммиачной селитры с триивтробеяаолом, тринитроамилиаом я трятп-
роксилолом состава — 3 :1.
' Напомним, что при увеличении плотности мощных ВВ детонационная способ-
ность возрастает (критический диаметр уменьшается), а чувствительность к детона-
ции не возрастает, а падает.
334
Таблица 49
Воспламеняемость порохов лучистой энергией в зависимости от типа пороха,
состояния поверхности и формы образчиков
Порох Формй пороха и состояние поверхности Тепловой импульс Поглощеяная часть тепло- вого импульса — а) Коэффи- циент отраже- нии, а (прнбли- женно)
1СС 1Д/СЛ41
Нитроглицериновый (Н) Цилиндр, поверхность 8,2 6,56 0,2
гладкая Цилиндр, торцевая поверх- ность порезана на квад- раты со стороной — 2 мм и глубиной — 1 эм»; по- верхность обеспылена Цилиндр, торцевая поверх- ность порезана на квад- раты со стороной ~ 2 мм и глубиной — 1 эм»; по- верхность не обеспыле-
г? 5,0 4,5 0,1
* на . - . Цилиндр, поверхность, об- разовавшаяся при пере* 3,5 3,15 0,1
ломе пороха 5,3 —- —-
Нитр or л и церииовып Шар, поверхность гладкая Цилиндр, поверхность 9,1 7,28 °,2
(Н) -I 0,5% сажи То же гладкая ........ Цилиндр, торцевая поверх- ность нарезана на квад- раты со стороной — 2 эм» и глубиной — 1 эм»; по- 3,2 2,72 0,15
Нитр огли це ринов ый верхность обеспылена Цилиндр. поверхность 2,5 2,37 0,05
(Н) + 3% сажи П ироксилиновый гладкая Цилиндр, поверхность 3,1 2,63 0,15
гладкая ... Цилиндр, на торцевой по- верхности сделаны мел- 5,0 3,0 0,4
кие насечки з,о 2,4 0,2
Шар, поверхность гладкая 5,6 3,36 0,4
Пироксилиновый Н 0,5% сажи Шар, поверхность гладкая 4,0 3,2 °,2
Спрессованная таблет- ка (95% нитроглице- ринового пороха -|-5% сажи) Цилиндр 0,89 0,85 0,05
Одним из таких методов является определеяие минимального давле-
ния (или веса) воспламенителя, дающего в определенных условиях вос-
пламенение заряда испытуемого ВВ. В опытах В. М. Рогожникова 1 прес-
сованный заряд ВВ помещался в манометрической бомбе, и определялся
минимальный заряд воспламенителя (смесь пироксилина и аммиачной
селитры 1 : 1), при котором происходило воспламенение. В табл. 48 при-
ведены результаты этих опытов. В графе второй в скобках указаны соот-
ветствующие давления сгорания воспламенителя.
Данные таблицы показывают, что из числа изучавшихся ВВ наиболь-
шей воспламеняемостью характеризуются 62%-ный желатин-динамит и
пикрат калия, наименьшей — аммиачная селитра. Они показывают тан-
1 Можно осуществлять поджигание и при постоянном повышенном давлении,
как это делал Л. В, Дубнов, и за меру воспламеняемости принимать то значение ука-
занного давления, начиная с которого поджигание приводит к горению,
22*
335
же, что в условиях этих опытов воспламеняемость обычно возрастает при
уменьшении плотности. Необходимое для воспламенения давление вос-
пламенителя уменьшается при увеличении поверхности воспламеняемого
заряда — горение последнего по большей поверхности предотвращает
падение давления за счет отвода тепла в стенки бомбы.
Каталитические добавки к аммиачной селитре повышают ее воспла-
меняемость.
Применялись также другие методы оценки воспламеняемости. Так,
Г. Н. Беспалов изучал воспламеняемость порохов при атмосферном дав-
лении, помещая на торец шашки большого диаметра пороха столбик ВВ,
имеющего малый критический диаметр горения.
Определялся минимальный диаметр воспламеняющего заряда, ина-
че говоря диаметр горячего «иятиа», создаваемого на торце воспламеняе-
мого заряда, при котором происходит воспламенение последнего. Между
диаметром «пятна» и критическим диаметром горения для разных поро-
хов наблюдалось определенное соответствие. При повышении давления
скорость горения разных порохов увеличивается неодинаково; соответ-
ственно и соотношение критических диаметров может быть при повышен-
ных давлениях иным, чем при атмосферном давлении.
Похил [266] изучал воспламенение порохов лучистой энергией. Источ-
ником света служила дуговая лампа, излучение которой фокусировалось
при помощи двух параболоидных отражателей. Плотность лучистой энер-
гии в фокальном теле определялась калориметрическй. Показателем вос-
пламеняемостн пороха служило минимальное количество тепла
(в кал/см2), которое ему нужно было сообщить для воспламенения. Рас-
чет этого количества производился с учетом коэффициента отражении
пороха. Некоторые результаты опытов приведены в табл. 49. Из табли-
цы видно, что пироксилиновый порох воспламеняется значительно легче,
чем нитроглицериновый, что связано со значительно большей прозрач-
ностью последнего. Вследствие этого поглощенная лучистая энергия рас-
пределяется в более толстом слое и достигаемая температура оказывает-
ся ниже. Это объяснение подтверждается сильным повышением воспла-
меняемости при добавлении к пороху сажи (0,5%), в десятки раз умень-
шающей его прозрачность. Воспламеняемость такого пороха становятся
больше, чем пироксилинового. С другой стороны, добавление сажи к пи-
роксилиновому пороху практически не уменьшает его малой прозрачно-
сти и слабо отражается и на воспламеняемости.
При шероховатой поверхности, воспринимающей излучение, порох
воспламеняется легче отчасти в связи с тем, что при этом уменьшается
коэффициент отражения, отчасти ив-за того, что выступы и ребра вос-
пламеняются вообще легче: соотношение теплоприхода и теплоотвода
для них более благоприятно, чем для гладкой поверхности. Спрессован-
ная таблетка нитроглицеринового пороха с 5% сажи требует двя воспла-
менения в несколько раз меньше лучистой энергии, чем сплошной заряд.
Воспламеняемость зависит также от формы заряда — шар воспламеняет-
ся труднее, чем цилиндр с торца.
ВЫВОДЫ
При интенсивном нагреве ВВ с поверхности по достижении на послед-
ней некоторой критической температуры, представляющей двя летучих
ВВ температуру кипения, а двя нелетучих ВВ — температуру газифика-
ции, образуются -пары или газообразные продукты первичного распада.
Эти пары быстро нагреваются до температуры самовоспламенения и на-
чинается горение ВВ. Если в этот момент прогретый слой конденсирован-
ной фазы соответствует слою, который имеется при стационарном горе-
нии, то горение начинается и идет с постоянной скоростью. Если указая-
336
ный слой тоньше или толще, чем «стационарный», то горение вначале
идет соответственно медленнее или быстрее, чем стационарное. Время
нормального воспламенения соответствует времени нагрева, необходимо-
го для образования «стационарного» прогретого слоя.
Мерой воспламеняемости ВВ может служить запас тепла в прогретом
слое при стационарном горении. Чем больше необходимое количество теп-
ла, тем меньше воспламеняемость. Теория показывает, что запас тепла
в прогретом слое при стационарном горении прямо пропорционален теп-
лопроводности, разности температур — критической и начальной,— об-
ратно пропорционален скорости горения и, следовательно, давлению.
Экспериментальные методы определения воспламеняемости должны
быть основаны на определении запаса тепла, необходимого двя возбужде-
ния нормального горения. Сравнительная оценка воспламеняемости мо-
жет быть произведена так же по величине критического диаметра горе-
ния, минимальному давлению продуктов горения воспламенителя, необ-
ходимому для (воспламенения ВВ в замкнутом объеме, а также по разме-
ру очага высокой температуры, достаточному двя возбуждения горения.
Ш. РАСПРОСТРАНЕНИЕ ГОРЕНИЯ
ПО ПОВЕРХНОСТИ ЗАРЯДА
При зажигании заряда ВВ на некоторой малой части его открытой по-
верхности горение во многих случаях быстрее распространяется по по-
верхности заряда, чем в глубь вещества. Это может приводить к тому, что
заряд, зажженный в одной точке его поверхности, быстро охватывается
пламенем и горит по всей поверхности от периферии в глубь заряда. Та-
кая картина процесса наблюдается, например, при зажигания шашки тет-
рила и некоторых других ВВ на воздухе. Внешке аналогично протекает
горение и при воспламенении порохового заряда в замкнутом или полу-
замкнутом объеме, например в огнестрельном оружии.
Большая скорость поверхностного распространения пламени при го-
рении на воздухе обычно связана с тем обстоятельством, что у всех изу-
чавшихся ВВ реакция при горении под атмосферным давлением не идет
до конца, причем образуются горючие газы даже у веществ с нулевым
кислородным балансом, например нитрогликоля. При горении на воздухе
эти газы, смешиваясь с ним, догорают до конца, что ведет к значительно-
му повышению температуры. Таким образом, поверхностные слои заряда
по контуру фронта горения нагреваются газами более высокой темпера-
туры, чем поверхностные слои центральной части сеченин фронта го-
рения. Естественно поэтому, что первые горят быстрее.
То, что поверхностное распространение пламени с большой скоростью
связано в случае бездымных порохов именно со вторичными реакцними,
идущими при контакте с воздухом, следует из опытов [267], показавших,
что в инертном газе обе скорости — поверхностного горения и горения
в глубь заряда — равны. Больше того, даже при горении на воздухе в ус-
ловиях, когда отсутствует вторичное пламя, поверхностное распростра-
нение горения не имеет места [268]. Этот вывод подтверждается также
тем фактом, что величина скорости поверхностного распространения
пламени гораздо больше у веществ с большим недостатком кислорода,
образующих при горении много горючих газов, могущих затем догорать
при соприкосновении с воздухом.
Так, при горении прессованного тетрила, скорость которого равна
0,047 см!сек, скорость поверхностного распространения горения дости-
гает 1,70 см/сек:, точно так же у нитроглицеринового пороха скорость
горения по поверхности составляет 2,78 см}сек, при скорости горения
337
вглубь — 0,075 см/сек; у 62%-ного динамита — вещества с избытком кис-
лорода, имеющего скорость горения, приблизительно вдвое большую, чем
тетрил, скорость поверхностного распространения пламени гораздо мень-
ше, чем у тетрила.
Известную роль в процессе поверхностного распространения пламени
при значительных скоростях горения может играть также некоторое по-
вышение давления, образующееся во фронте горения. Под влиянием это-
го повышения давления газы могут распространяться во все стороны,
в том числе и по поверхности заряда, вызывая его воспламенение. Выска-
зывалось предположение, что при поверхностном распространении горения
на воздухе может иметь значение приход энергии за счет лучеиспуска-
ния. Если бы это было так, то и скорость горения в глубь зерна должна
была бы возрастать. Прямые опыты указывают, что влияние излуче-
ния вторичного пламени нитроглицеринового пороха на скорость его
горения в заметной мере не имеет места [269]. При горении цилиндров
пороха на воздухе скорость горения составляла 0,0797 сл/сек; при горе-
нии в токе углекислоты, когда вторичного пламени не образуется —
0,0765 сж/сек. При горении цилиндра пороха на воздухе, но без асбесто-
вой обмотки, скорость горения, рассчитанная по поверхностной скорости
и углу конуса, составляла 0,0747 см/сек.
Большая скорость распространения горения по поверхности при го-
рении в замкнутом объеме имеет другую причину. Давление при горении
растет, образующиеся при горении газы быстро заполняют свободный
объем камеры сгорания, вытесняя и сжимая тот воздух, который в нем
находился, при одновременном смешении с этим воздухом. Газы имеют
высокую температуру и нагревают поверхность пороха, что и приводит
к его воспламенению.
ЛИТЕРАТУРА
1. И. П. Г р а в е. Внутренняя баллистика. Л., 1938; М. Е. Серебряков, К. К. Гро-
те н, Г. В. О п п о к о в. Внутренняя баллистика. М,— Л., Оборонена, 1939;
Е. В. Антулаев. Подрывное дело, ч. 1. М,—Л., ОНТИ, 1934, М. Е. Серебря-
ков. Внутренняя баллистика ствольных систем и пороховых ракет. Обороигиа,
1962.
2. Н. Н. Семенов. О некоторых проблемах химической кинетики и реакционной
способности. М., Изд-во АН СССР, 1958; Цепные реакции. Л., ГХТИ, 1934.
3. В. Н. Кондратьев. Кинетика химических газовых реакций. М., Изд-во АН
СССР, 1968.
4. Н. М. Эмануэль, Д. Г. Кнорре. Курс химической кинетики. М., Изд-во
«Высшая школа», 1962.
5. С. Б е и с о и. Основы химической кинетики. М., изд-во «Мир», 1964.
6. К. К. Андреев. Термическое разложение и горение взрывчатых веществ. 1-е
издание. М., Госэнергоиздат., 1957.
6а. К. К. Андреев и А. Ф. Беляев. Теория взрывчатых веществ. М., Оборои-
гиз, 1960.
7. А. Я. Лукин. ЖФХ, 3, 406 (1932) ;А. В. Сапожников и др. ЖРфХО, 36, 836
(1904); 37,822 (1905); 38, 1186 (1906).
8. К. К. А н д р е е в. Сборник статей по теории взрывчатых веществ (под редакцией
проф. К. К. Андреева и Ю. Б. Харитона). М., Оборонена, 1940, стр. 84.
9. R. R о Ь е г t s о n. J. Chem. Soc., 95, 1241 (1909).
10. W. Will. Z. angew. Chem., 14, 743 (1901); Mitteilungen Centralstelle f. Wissensch-
techn. Untersuch. Abt. 2 (1900); Abt. 3 (1902).
11. С. 3. P о г и н с к и й, A. M. Сапожников. ЖФХ, 2, 80 (1931).
12. К. К. Андреев, А, П. Глазкова. ДАН СССР, 105, 286 (1955).
13. К, К. Андреев, А. П. Гл а з ко в а, Н. Д. М аурина, Б. С. Светлов. ЖФХ,
32, 1727 (1958).
14. К. К. Андреев. ЖПХ, 31, 484 (1958).
15. Б. С. Светлов. Научные доклады высшей школы СССР. Химия и хим. технбл.,
№3,422 (1958).
16. Б. С. Светлов. Кинетика и катализ, 2, 38 (1961).
17. Сб. «Теория взрывчатых веществ» (под редакцией К. К. Андреева, А. Ф. Беляе-
ва, А. И. Гольбиндера, А. Г. Горста). М., Обороигиа, 1963, стр. 184.
18. В. В. Г о р б у н о в. Б. С. С в е т л о в. См. ссылку J17, стр. 190].
19. К. К. Андреев, Г. Н. Беспалов. ЖФХ, 35, 2437 (1961).
20. К. К. А н д р е е в, Г. Н. Б е с п а л о в. См. ссылку [17, стр. 131].
21. Б. С. Светлов. Кинетика и катализ, 2, 179 (1961); См. ссылку [17, стр. 208].
22. В. В. Г о р б у н о в. См:, ссылку [17, стр. 219].
23. В. В. Г о р б у н о в, В. С. С в е т л о в. См. ссылку f 17, стр. 197].
24. К. К. Андреев, В. П. Шелапутииа. ЖПХ, 34, 1371 (1961).
25. В. В. Г о р бу н о в, Б. С. С в е т л^Гв. См. ссылку [17, стр. 214]
26. К. К. А н д р е е в, Г. Н. Б е с п а л о в. См. ссылку [17, стр, 172].
27. М. То n е g u t ti. La chimica e I’lndustria, 17, 517 (1935).
28. T. Urbanski B. Kwiatkowski, M. Milaaowski. Z. Shiess- und Spreng-
s toff we sen, 32, 1, 29, 57 85 (1937).
29. T. Urbanski, M. Miladowski. Z. Schiess- und Sprengstoffwesen, 33, 247
(1938).
30. G. Bonrjol. Mem. des poudres, 35, 83 (1953).
31. К. К. А и д p e e в, Б. И. К а й ды м о в. ЖФХ, 35. 2676 (1961).
32. К. К. А н д p e e в, Б. И. К а й д ы м о в. См. ссылку [17, стр. 241].
33. А. I. В. R о Ъ е г t s о n. J. Soc. Chem, Ind., 67, 221 (1948).
339
34. В. G. С в е т л о в. См. ссылку [17, стр. 274].
35. Б. А. Л у р ь е, В. С. С в е т л о в. См. ссылку [17, стр. 281].
36. В. С. С в е т л о в. В. А. Л у р ь е. ЖФХ, 37, 1979 (1963).
37. А. М i 1t а s с h. Z. angew. Chem., 16, 29 (1903).
38. А. В. Сапожников. ЖРФХО, 36, 836 (1904); 37, 822 (1906); 38, 1186 (1906).
39. J. G о u j on. Mem. des poudres, 34, 2 (1930—1931).
40. B. D. Smith. Nature, 170, 844 (1952).
41. S.S. Napper B. Bobortson. J. Chem. Soc., 91, 761 (1907).
42. К. К. А н д p e e в, В. С. С а м с о я о в. ДАН СССР, 114, 815 (1957).
43. Труды МХтИ. Сборник статей по теории нарывчатых веществ. М., изд-во «Выс-
шая школа», 1966.
44. К К. Андреев, В. С. Самсонов. См. ссылку [43].
45. J. G о u i он. Мёт. art. franf., 8, 807 (4929).
46. Е. В е г 1, G. Ru f f, Ch. Carpenter. Ind. Eng. Chem., 30, 219 (1938).
47. К. К Андреев, В. С. Самсонов. См. ссылку [43].
48. W. R. Steacie a. coll. Proc. Roy. Soc., A146, 388 (1934); A151, 685 (1935);
J. Chem. Phys., 2, 345 (1934); 3, 344 (1935); 4, 323 (1936); 4, 504 (1936); 5, 125
(1937); J. Chem. Soc., 4, 05 (1936).
49. J. B. Levy. J. Am. Chem. Soc., 78, 762 (1956); Ind. Eng. Chem., 48, 762 (1956).
50. L. P. Kuhn, R. Wright, L. De AngeliB. J. Am. Chem. Soc., 76, 328 (1954);
78, 2719 (1956); L. 0. Kuhn, L. de Angelis. J. Am. Chem. Soc., 52, 59 (1956).
51. Б. H. К о н д p ико в. ЖФХ, 32,1175 (1958).
52. Б. H. Кендриков. Научные доклады высшей школы. Химин и хим. технол..
Ml, 19 (1959).
53. Б. Н. К о н д р и к о в. См. ссылку [17, стр. 296].
54. Т. L. Cottrell, Т. Е. Graham, Т. J. Reid. Trans. Faraday Soc., 47, 564 (1951).
55. T. L. Gottrell, T. E. Graham T. J. Reid. Trans. Faraday Soc., 47, 1089
(1951).
56. P. Gray, A. D. Yoff e, L. Roselaar. Trans. Faraday Soc., 51, 1489 (1965).
57. A. He r moni, T. B. Gr un wal d. 8--th Symposium on Combustion. N. Y., Reinhold
Publishing Corporation, 1962, p. 1084.
58. R. C. Fa r m в r. J. Chem. Soc., 117, 1432 (1920).
59. R. Robertson. J. Chem. Soq., 119,1 (1921).
60. Ю. Я. Максимов. См. ссылку [17, стр. 338]; Кандидатская диссертация, МХТИ
им. Д. И. Менделеева, 1963.
61. К-К. Андреев, ЛюБао-фен. См. ссылку [17, стр. 349].
62. Лю Ба о-фен. Кандидатская диссертация. МХТИ им. Д. И. Менделеева, 1961.
63. С. 3. Рогинский, А. М. Матид. ЖФХ, 2 263 (1931).
64. A. J. В. R о b е г t s о n. Trans. Faraday Soc., 44, 677 (1948).
65. М. Tobin, J. Fowler, H. Hoffman, C. Sauer. J. Am. Chem. Soc., 76, 3249
05. V. R? T о m I i n s о a. J. Org. Chenv 17, 648 (1952).
67. R. G. Farmer. J. Chem. Soc., 117, 1432 (1920); C. N. Hi nah el wood. J. Chem.
Soc., 119-129, 721 (1921).
68. S. Z. R о g i n s k y, A. Y. L u k i n. Acta physico-chimica, 2, 385 (1935).
69. Ф. И. Дубовицкий, В. H. Струнин, Г. В. М ан ели с, А. Г. Мержанов.
ЖФХ, 35,306 (1961).
70. A. J. В. R о b е г t з о n. Trans. Faraday Soc., 45, 85 (1949).
71. Ю. Я. М а к с и м о в. См. ссылку [43].
72. R. М е у е г, Н. J. S с h u m а с h е г. Z. phys. Chem., А170, 33 (1934).
73. J. J. М а с d о n а 1 d. J. Chem. Soc., 1936, 832, 839; 1937, 273.
74. C. 3. Рогинский, Д. П. Добычжн Г. Ф. Зелинская. ЖФХ, 13, 1367
(1935Н
75. W. Е. G а г в е г, L. Е. R е е v е s. Trans. Faraday Soc., 51 694 (1955).
76. A. F. Benton, G. H. Gunningham. J. Am. Chem. Soc., 57, 2227 (1935);
F. С. T о m p k i n s. Trans. Faraday Soc., 44, 206 (1948).
77. W. E. G а г n e г, E. W. H а у с о c k. Proc. Roy. Soc., A211, ЗЭ5 (1902).
78. W. E. G a r n e r, L. E. Reeves. Trans. Faraday Soc., 50, 2a4 (4954).
79. А. Я. А пив. См. осылку [8, стр. 106].
80. F. E. Harvey. Trans. Faraday Soc., 29, 633 (1933).
81. См. ссылку [6, стр. 56].
82. W. E. Gamer. Chemistry of the Solid State, Butterworth Scientific Publications,
1965, p. 2321
83. O. Turn. China, et ind., 28, 781 (1931).
84. S. Z. Roginsky, К. К Andreev. J. Chem. phys., 30, 467 (1933).
85. О. T u r e k. China, et ind., 29, 507 (1933).
86. A. D. Y о f f a. Proc. Roy. Soc., A208, 188 (1951).
87. H. R. H a i 1 e s. Trans. Faraday Soc., 29, 544 (1933).
83. K.K. Андреев, ЛюВао-фен. См. ссылку [17, стр. 363].
89. Z. G. S za bб, Y. S z a va. 7-th Symposium on Combustion. N. Y., 1959.
340
90. Н. Н. Семенов. Сб.« Инициирующие взрывчатые вещества», выл. 2. М., ОНТИ,
1905, стр. 47.
91. J. С. Schumacher. Perchlorates, their Properties, Manufacture and Uses. N. Y.,
Reinhold Publishing Corporation, I960.
92. Сун Цюань-цай. Кандидатская диссертация, МХТИ им. Д. И. Менделеева,
1961.
93. L. Bircumshaw, В. Newman. Proc. Roy. Soc., A227, 115 (1954); A227, 228
<1955Ъ
94. L. L. В i r c u m s h a w, В. N e w m a n. J. Chem. Soc., 1957, 4741.
95. A. K. G a 1 w e у, P. W. M. J а с о b s. Trans Faraday Soc., 55, 837—844 (1959).
96. A. K. Galwey, P. W. M. Jacobs. Trans. Faraday Soc., 55, 1165 (1959).
97. A. Hermoni, A. Salmon. 8-th Symposium on Combustion, N. Y., 1932, p. 656.
98. A. K. Galwey, P. W. M. Jacobs. Trans. Faraday Soc., 56, 581 (I960).
99. P. W. M. Jacobs, A. R. T. Kuzeishy. 7-th Symposium on Combustion. N. Y..
1959, p. 672.
100. L. L. Bircumshaw, В. H. Newman. Proc. Roy. Soc. A227, 228 (1955); A227,
115 (1954).
101. A. K. G a 1 w e у, P. W. M. J а с о b s. Proc. Roy. Soc., A254, 455 (1960).
102. К- К. Андреев, Сун Цюань-цай. См. ссылку [431.
103. А. К. G а 1 w е у, Р. W. М. J а с о b s. J. Chem. Soc., 1959, 837.
104. L. L.Bircumshaw, Т. R. Phillips. J. Chem. Soc., 1957, 4741.
105. А. Я. Адин, О. M. Тодес, Ю. Б. Харитон. ЖФХ, 8, 866 (1936).
106. J. В. Levy. J. Am. Chem. Soc., 76, 3254, 3790 (1954); F. Н. Pollard, А. Е. Ped-
le г, С. I. Н ard у. Nature, 20, 979 (1954),
107. А. Ф. Б е л я е в. ЖФХ, 14, 1009 (1940).
108. L. Phillips. Nature 160, 753 (1947); 165, 564 (1950); F. Pollard R. Wyatt,
H. M. Marshall. Nature, 165, 564 (1950).
109. H. Mura our. Chim. et inti’29. 507 (1933).
140. К. K. Andreev. J. Chim. phys.. 31, 14'1 (1904).
111. P. Vieille. Мёт. des poudres 6, 256 (1893).
112. M. E. Серебряков, К К Грете н, Г. В. Оппоков. Внутренняя баллисти-
ка. М.— Л., Оборонгиз, 1939, стр. 117.
1'13 . Т. В. Захарова и Ю. Б. Харитон. Сб. «Физика взрыва», № 1. М., Изд-во
АН СССР, 1952, стр. 117; А. И. Коротков. То же, № 4, 1955, стр. 4.
114. К. К Андреев, А. Е. Варга. Дополнительная статья к русскому переводу
книги М. Патри «Горение и детонация взрывчатых веществ». М.—Л., Оборонгиз,
1938, стр. '161.
115. А. Ф. Беляев, А. Е. Беляева. ЖФХ, 20 1381 (1941).
116. В. L. Crauford С. Hugget, F. Daniels. В. Е. Wilfong. Analyt. Chem..
19,630 (1947).
117. К К. Андреев, А. П. Бакеев. ДАН СССР, 48, 595 (1945).
118. А. П. Глазков а, И. А. Терешкин. ЖФХ, 86, 1622 (1961).
119. J. Basset. С. г., 185 349 (1927); 191, 1925 (1930); 165, 1242 (1932).
120. А. Ф. Б е л я е в, А. И. К о р о т к о в, А. К. Парфенов А. А. Сулимов. ЖФХ,
37,150 (1963).
121. В. S a r г a u, Р. Vi е i 11 е. Мёт. des poudres, 2, 453 (1884—1889).
122. М. С. Плясунов. Кандидатская диссертация, МХТИ им. Д. И. Менделеева.
1960.
123. К. К. Андреев, П. П. Попова. ЖФХ, 86, 1979 (1961).
124. G. К. A d a m s, G. W. S t о c k s. 4-th Symposium on Combustion. N. Y. 1953, p. 239.
125. К К Андре ев. См. ссылку [8, стр. Эй]; ЖФХ, 20, 467 (1916).
126. К К. Андреев. См. ссылку [6, стр. 139].
127. См. ссылку (17, стр. 430].
128. А. И. Гольбиндер. См. ссылку [17 стр. 4571.
129. R. FriedmanR. С. N u g е n t, К. Е. Rumhel, A. S с и г 1 о с k. 6-th Sumposium
on Combustion. N. Y., 1957, p. 612.
130. А. П. Глазкова. Ж. нрнкл. механики и технич. фианни, № 5, 421 (1963).
131. J. В. Levy, R. Friedman. 8-th Symposium on Combustion. N. Y., 1962, p. 663.
132. К. К. Андреев. См. ссылку [8, стр, 39 и 53]; Ф. Наоум. Нитроглицерин и нит-
роглицериновые взрывчатые вещества. М.—Л. ГХТИ, 1934, стр. '149; К. К. Анд-
реев. ЖФХ, 20, 467 (1Ш); К. К. Андреев, М. М. Пуркалн. ДАИ СССР, 56,
281 (1945); А. Ф. Беляев и А. Е. Беляева. ДАН СССР, 33, 41 (1941); 52, 507
(1946) ;_ЖФХ. 20, 1381 (1916).
183. М. L-Wolfrom, Е. Е. D i с к е у, Н. С. Prosser a. coll. С. F е n i m о г е. Gom-
bustion Processes vol. II. High Speed Aerodinamics and Jet Propulsion. Princeton
University Press, 1956, p. 520.
134. Я. Б. 3 e л ь д о в н ч, Ю. X. Ш а у л о в. ЖФХ, 20 1869 (1946).
135. См. ссылку |6, стр. 130—133].
136. К. К. Андреев. ДАН СССР, 51, 29 (1946); 53, 237 (1946).
137. См. ссылку [6, стр. 125—126].
341
136. А. Ф. Беляев и Л. Д. Комкова. Сб. «Физика взрыва», № 2. М. Изд-во АН
СССР, 1953, стр. 175.
139. Е. A. Arden, S. Fowling. 6~tb Symposium on Combustion. N. Y., 1957, p. 177.
140. Peter Gray, M. W. T. Pratt. 6-th Symposium on Combustion. N. Y., 1957, p. 183.
141. A. R. Hall, G. W. W о 1 f h a r d. 6-th Symposium on Combustion, N, Y., 1957,
p. 190.
142. Peter Gray, A. Williams. 8-th Sympeeium on Combustion. N. Y., 4962, p. 496.
143. J. A. H i c k s. 8-th Symposium on Combustion. N. Y., 1962, p. 487.
144. Peter Gray, J. C. Lee (in past), D. G. Taylor. 6-th Symposium on Combustion.
N. Y., 1957, p. 255.
145. A. C. Antoine. Combustion and Flame, 6, 363 (1'962).
146. R. С. M u r r a y, A. R. H a 11. Trans. Faraday Soc., 47, 743 (1951).
147. С. H. В a m f о r d. Trans. Faraday Soc., 35,1239 (1939).
148. P. G r a y, J. C, Lee. 7-<th Symposium on Combustion. N. Y., 1959, p. 61.
149. К. К. Андреев. ДАН СССР, 1 220 (1935)1 см. ссылку [114, стр. 149]; см. ссылку
[132р см. ссылку [8, стр. 39]; ЖФХ, 20, 467 (1946).
150. К К. Андреев. ДАН СССР, 51, 29 (1946).
151. А. ф. Беляев, А. Е. Беляева. ДАН СССР, 33, 41 (1941); 52, 507 (1946).
152. А. Ф. Беляев и А. И. Коротков. Сб. «Физика взрыва», № 1-. М. Изд-во АН
СССР, 1052, стр. 177.
153. А. ф. Беляев и А. Е. Беляева. ЖФХ, 20 1381 (1946).
1&4. А. Ф. Беляев. Сб. «Физика варыва», № 1. М., Изд-во АН СССР 1953, стр. 185.
155. А. Ф. Беляев и Ю. А. Кондрашков. ДАН СССР, 131, 364 (‘I960).
156. Б. С. Светлов, А. Е. Фогельзанг. ДАН СССР, 137, 654 (1961).
157. См. ссылку [6, стр. 143].
158. А. П. Глазкова. ЖФХ, 37, 1119 (1963).
159. G, К Adams, В. Н. N е w m а п, А. В. Robins. 8-th Symposium on Combustion.
N. Y., 1962, p. 693.
160. J. Vandenkerckhove A. Jaumotte. 8-th Symposium on Combustion. N. Y.,
1962, p. 689.
161. А. А. Шидловский, С. А. Оранжерее в. ЖФХ, 26, 27 (1953); А. А. Ши д-
ловский. ЖПХ, 35, 511 (1962)- Известия высших* учебных заведений СССР.
Химия и хим. технол., №3, 405 (1960).
162. К. К Андреев, А. П. Глазкова. См. ссылку [4В].
163. Н. М и г а о и г, J. В a s s е t. С. г., 208, 809 (1939).
164. П. ф. Похил. Сб. «Физика взрыва», Xs 2. М., Изд-во АН СССР, 1953, стр. 181;
№ 3, 1955, стр. 93.
165. М. L. Wolf г от, Е. Е. Dickey, G. G. Maher. The thermal decomposition of
cellulose nitrpte under reduced pressures. Ohio State Univ. Research Foundation,
1950; M. L. W о 1 f г о m, E. E. Dickey, H. C. Prosser. A possible free radical
mechanism for the initial reaction in the thermal decomposition at reduced pressure
of cellulose nitrate (and other organic nitrates). Ohio State Univ. Research Founda-
j'] од
166. G. A. Heath, R. Hirst. 8-th Symposium on Combustion. N. Y., 1962, p. 711.
167. R. E. Wilfong, S. S. Penner, F. Daniels. J. Phys. Chem., 54, 863 (1956).
168. О. И. Л ейпунский, 3. И. Аристова. Об. «Фнзика взрыва», № 2. М., Изд-во
АН СССР, 1953, стр. 225.
169. R. Klein, М. М е n t sе г, G. von Е 1 b е, В. Lewis. J. Phys. Chem., 54, 877 (1i950).
170. J. Corner. Theory of the Interior Ballistiks of Guns, Wiley, (1950); Trans. Fara-
day Soc., 43, 635 (1947).
171. H. M u г a о u r- Cahiers de physique, premiere serie, 1942, p. 29.
172. К. К- Андреев, A. E. Варга. См. ссылку [114, стр. 138 и 161].
173. Jaques et James В a s s e t. С. г., 231, 649 (1950).
174. H. Muraour, J. Fauveau. Ghim. et Ind., 65, 53 (1951).
175, И. П. Г p а в e. Пиростатика. Л., Иад-во Артакадемии, 1988, стр. 145.
176. Ф. А. Баум. Трубочные пороха и дистанционные составы. М., Обороигиа, 1940,
стр. '10.
177. К К. Андреев. ДАН СССР, 49, 437 (1945).
178. А. Ф. Б е л я е в, С. Ф. М а з н е в. ДАН СССР, 13f, 887 (1960).
179. А. Ф. Беляев, Р. X. Курбангалина. ЖФХ, 33, 579 (1964).
180, А. Ф. В е л я е в, Л. Д. Комкова. ЖФХ, 24, 1302 (i960).
181. А. Ф. Беляев. ЖФХ, 14, 1009 (1940); см. ссылку [6 стр, 156].
182. А. Ф. Беляев А. Е. Беляева. ДАН СССР, 33, 41 (1944); 52 507 (1946);
ЖФХ, 20, 1381 (1946).
183. Н. Muraour, J. Wolgemuth. Chem. et ind., 38, 472 (1936).
184. А. Ф. Беляев. Докторская диссертация, НХФ АН СССР, 1944.
185. К. К- Андреев. ЖФХ, 20, 467 (1946).
186. А, И, Коротков О. И. Лейпунский. Сб. «Физика взрыва», Хе 2. М., Изд-ва
АН СССР, 1953, стр. 213.
187. Н.Muraour, W. Schumacher. Chim. et ind., 33 84 (1935).
188. А. Ф. Беляев, Г. В. Лукашеня, ЖФХ, 85, 1950 (1962).
342
189. К. К. А и д,р е е в, М. С. Пл я с у и о в. См. ссылку [43].
190. Н. Ми га cur. С. г., 220, 198 (1946).
191. К. К. А н д р е е в. ДАН СССР, 53, 237 (1946).
192. К. К. Андреев. ЖФХ, 20, 467 (1946)' см. ссылку [8].
193. J. W. Taylor. Trans. Faraday Soc., 2o, 561 (1962); Combustion and Flame, 6,
103 (1962).
194. К. К. А н д p e e в, П. П. Попова. ДАН СССР, 134 1142 (I960).
195. К. К. Андрее в, С. В. Ч у Й ко. ЖФХ, 37, 1304 (1963).
196. Б. Н. Кендриков. См. ссылку [43].
197. См. ссылку [6, стр. 168].
196. В. В. Горбунов. Кандидатская диссертация, МХТИ им. Д. И. Менделеева,
1963.
199. О. И. Лейпунский. ЖФХ 34 177 (i960).
200. Б. В. Новожилов. ДАН СССР, 131, 1400 (1960).
201. В. В. Н о в о ж и л о в. ЖФХ, 36, 1803' (1962).
202. Б. В. Новожилов. ЖФХ, 36, 3508 (1962).
203. М. Summerfild a coll. Solid Propellant Rocket Research Progress in Astranna-
tics and Rocketry, vol. I. N. Y.— London, Academic Press, 1960, p. 141.
204. R. F. Chai ken, W. H. Andersen. Solid Propellant Rocket Research, edited
by Martin Summerfild, Princeton University. New-Yersy. N. Y.— London, Academic
Press, *1660, p. 229.
205. H. H. Б a x м а н. ДАН СССР, 129, 1079 (1959).
206. H- H. БахманД. П. Поликарпов. Изв. АН СССР, ОТН. Энергия и автома-
тика, № 4, 37 (1961).
207. Н. Н. Бахман и А. Ф. Беляев. ДАН СССР, 133, 866 (1960).
208. Н. Н. Б а х м а н. ДАН СССР, 140, 141 (1961).
209. А. Ф. В е л я е в С. А. Ц ы г а и о в. ДАН СССР, 146, 383 (1962).
210. Н. Н. В а х м а н и Ю. А. К о в д р a m к о в. ЖФХ, 1, 216 (1963).
211. Н. Н. Бахман. ДАН СССР, 137, 1141 (1961).
212. J. Hershkowitz, F. Schwartz, J. V. R. Kaufman. 8-th Symposium on
Combustion. N. Y., 1962, p. 720.
213. К. К. Андреев, А. П. Глазкова. ДАН СССР, 86, 801 (1952); см. также ссылку
[43].
214. К. К. Ан др ее в, А. П. Глазкова. См. ссылку [43].
215. Е. Audibert, L. Delmas. Annaies des Mines, [12] 20, 443 (1931).
216. В. S a г г a u, P. V i e i 11 e, Mem. des poudres, 2, 153 (1884—1889).
217. А Ф. Беляев, A E. Беляева. ДАН СССР, 33, 41 (1941); 52, 507 (1946); ЖФХ,
20, 1381 (1946).
218. Л. Н. Гальперин В. М. Мальцев, П. Ф. По хи л. ДАН СССР 127, 131
(1959); П. Ф. Похил, В. М. Мальцев Г. В. Лукаше ня. ДАН СССР, 135,
913 (1960).
219. А. А. Зе пин. Кандидатская диссертация, ИХФ АН СССР, 1962.
220. П. Ф. Похил Л. Д. Ром од а н ов а и М. М. Белов. Сб. «Физика варыва»,
№ 3. М., Изд-во 'АН СССР 1956, стр. 98.
221. В. К. Боболев, А. П. Глазкова, А. А. Зенин, О. И. Лейпунский. ДАН
СССР, 151 604 (1963); Ж. прикл, механики и техн, физики, АЙ 3 153 (1964).
222. J. Fowling, W. A. W. Smith. Combustion and Flame, 7, 269 (1963),
223. См. ссылку [6а’ стр. 49].
224. В. А. Михельсон. О нормальной скорости воспламенения гремучего газа. М.,
1890; Е. Mallard Н. Le Chatelier. Annaies des Mines, 4 274 (1883).
225. II. Mur a our. Z. phys. chem. A. Hater —Band 163 (1928); C. r., 196 478 (1933).
226. A. D. Crow, W. E. Grimshaw. Phil. Trans. Roy. Soc., A230, 387 (1932).
227. G. Letang. Mem. art. franQ., 1, 955 (1922); G. Schweikert. Innere Ballistik.
Leipzig, 1923.
228. N. Y am a ga. Z. f. das gesamte Schiens- und Sprengstoffwesen, 25, 60 (1930).
229. Я. В. Зельдович, Д. А. Франк-Каменецкий. ЖФХ, 12, 100 (1938).
230. А. Ф. Be ляев. ЖФХ, 12, 930 (1938); ЖФХ, 14, 1009'(1940).
231. А. Ф. Б е л я е в, А. Е. Б е л я е в а. См. ссылку [182].
232. А. Ф. Б е л я е в. ДАН СССР, 129, 635 (1959).
233. A. G. Whittaker, Т. М. Donovan, Н- Williams. J. Phys. Chem., 62, 908
(1952).
234. W. Е. G а г и е г, A. S. G о m ш, Н. R. Н a i 1 е s. J. Chem. Soc., 1933, 1393.
2S6. Ф. Боуден, А. Иоффе. Быстрые реакции в твердых веществах. М., ИЛ, 1963,
стр. 216.
236. К. К. Андреев. ДАН СССР 29, 469 (1940); 51, 29 (1946); А. Ф. Беляев. ДАН
СССР, 28, 715 (1940).
237. К- К. Андреев и corp. ДАН СССР, 51, 119 (1946); 50, 281 (1945); 50, 277 (1945);
53,237 (1946).
238. Я. Б. Зельдович. ЖЭТФ, 12, 496 (1942).
239. Л. Д. Л а н д а у. ЖЭТФ, 14, 240 (1944).
240. См. ссылку (6, стр. 245].
343
241. К. К. Андреев. ЖПХ, 21, 462 (1948).
242. А. Ф. В е л я е в. ДАН СССР, 27, 131 (1940).
243. А. Ф. В е л я е в. ЖПХ, 23 &2 (1950).
244. К. К. Андреев. ДАН СССР. 54, 39 (1946).
245. К. К. А я д р е е в. См. ссылку (17, стр. 404].
246. В. Г. Л е в и ч. ДАН СССР, 109, 975 (1956).
247. S. Z. R о g 1 a sky. Sow. Phys., 1, 603 (1932).
248. А. Л. Лукин. ЖФХ, 3 406 (1932); А. В. Сапожников и др., ЖФХО, 36, 836
(1904); 37, 822 (1906); 33,’ 1186 (1906).
249. К. К. А н д р е е в. См. ссылку [8, стр. 134].
250. А. И. Гольбиндер. См. ссылку [17, стр. 499].
251. А. Я. А п и н. ЖФХ, 14, 494 (1940).
252. См. ссылку [6, стр. 2791.
253. А. Ф. В е л я е в. ДАН СССР, 27,131 (1940).
254. К. К. Андреев. ЖПХ, 21, 462 (1948).
255. К. К. А н д р е е в. ДАН СССР, 54, 39 (1946).
256. В. Н. Кендриков. См. ссылку [17, стр. 515].
257. К. К. Андреев, В. С. Самсонов. Научные доклады высшей школы. Химия
и хим. технол., № 2, 220 (1958).
258. А. И. Гольбиндер. См. ссылку (17, стр. 4661.
259. К. К. Андреев, В. П. Маслов. ДАН СССР, 25, 195 (1969); см. ссылку [8,
стр. 150].
260. К. К. Андреев. См. ссылку [8, стр. 137].
261. А. Ф. В е л я е в. ЖФХ 20, 613 (1946).
262. О. И. Л ейпунскив, 3. И. Аристова. ЖФХ, 20, 1391 (1946).
263. А. Ф. Б е л я е в, Е. Н. С а м бу р с к а я. ДАН СССР, 30, 627 (1940).
264. W. Cronquist. Z. f. das gesamte Schiess- and Sprengstoffwesen, 1, 106 (1906).
265. К. К. Андреев, И. Д. Костин. ДАН СССР, 54, 231 (1946).
266. П. Ф. Похял М. М. Белов. Сб. «Физика взрыва», № 5. М. Изд-во АН СССР,
1956, стр. 104.
267. Н. Muraou г, W. Schumacher. Chim. et ind. 33, 64 (1936).
268. К. К. А н д р е е в. ДАН СССР,. 50, 275 (1945).
369. К. К. А н д р е е в. ЖФХ, 20, 467 (1946).
270. А. Ф. В е л я ев, Г. В. Л у к» ш е и я. Ж. прикл. механики и техник, физики,
№ 6, 114 (4963).
271. К. К. Андреев, А. П. Глазкова, И. А. Терешкин. ЖФХ, 35, 426 (1961).
ОГЛАВЛЕНИЕ
От редактора .........................................................
Предисловие ..................................................... •
Глава первая. Общая характеристика взрывчатых веществ и основных
форм их химического превращения......................................
Глава вторая. Медленное химическое превращение взрывчатых веществ
I. Нитраты и нитриты многоатомных спиртов.................... . . .
1. Нитроглицерин и нитрогликоль.................................
2. Пентаэритриттетранитрат (тэн)................................
3. Диэтиленгликольдинитрат......................................
4. Дина.........................................................
5. Нитроклетчатка...............................................
6. Газообразные нитриты и нитраты...............................
II. Нитрометан...........*.........................................
III. Нитросоединения ароматического ряда............................
1. Разложение нитросоединений в парах...........................
2. Разложение нитросоединений в жидком состоянии................
3. Распад полинитросоединений в твердом состоянии...............
IV. N-Нитрамины...............................*.....................
Метилендинитрамин (медина)......................................
Этилондинитрамии (эдна).........................................
Тринитрофенилметилнитрамин (тетрил)..............’..............
Гексоген и октоген..............................................
V. Азотистою дородная кислота......................................
VI. Взрывчатые вещества, разлагающиеся в твердом состоянии..........
VII. Перхлорат аммония..............................................
VIII. Общие закономерности термического распада ВВ....................
Глава третья. Устойчивое горение взрывчатых веществ...................
I. Методика исследования...........................................
1. Манометрическая бомба .,.....................................
2. Приборы постоянного давления.................................
II. Предельные условия горения.....................................
III. Влияние давления на скорость горения В В.......................
1. Нитроэфиры............................................. . . .
2. Гидразин ....................................................
3. Нитросоединения и нитрамины .................................
4. Инициирующие взрывчатые вещества.............................
5. Аммонийные соли хлорной и азотной кислот и смеси на их основе
6. Горение ВВ при сверхвысоких давлениях.................... . .
7. Бездымные пороха ............................................
8. Черный порох.................................................
9. Термиты......................................................
IV. Влияние начальной температуры на скорость горения.................
7
12
14
14
25
27
29
31
40
47
50
51
55
62
65
65
66
67
71
75
75
84
96
101
101
101
103
109
123
126
141
145
146
152
163
165
177
178
179
345
Зависимость скорости горения от начальной температуры при атмосферном
и близких к нему давлениях....................................... 180
1. Нитроэфиры и нитросоединения............................... 180
2. Инициирующие ВВ ............................................ 183
3. Пороха...................................................... 185
Зависимость скорости горения от температуры при повышенных давлениях 185
V. Влияние относительной плотности, диаметра заряда и инертных примесей
на возможность и скорость горения.................................... 190
1. Влияние относительной плотности ВВ на возможность горения . . . 190
2. Влияние относительной плотности ВВ на скорость горения .... 192
3. Зависимость критической плотности ВВ от давления................ 195
Горение при постоянном давлении..............................195
Горение при возрастающем давлении............................206
\ VL Горение гетерогенных систем.....................................218
* 1. Механизм горения ................................................218
2. Экспериментальные данные по горению гетерогенных систем . . . 231
VII. Выгорание взрывчатых веществ при взрывных работах в шахтах . . . 240
VIII. Состав газообразных продуктов горения . . . .......... 246
IX. Температура горения............................................249
х Глава четвертая. Теория стационарного горения ВВ .
V I. Механизм горения.............................................ТТ. . .
II. Гипотеза Михельсона и Малляра — Ле-Шателье.......................
III. Теория горения Зельдовича — Беляева........................
IV. Сопоставление экспериментальных данных и закономерностей с выводки
теории Зельдовича — Беляева......................................< Г .
V. Механизм горения нелетучих ВВ...........................
Глава пятая. Неустойчивое горение ВВ.............................
I. Общая теория устойчивости горения..................................
II. Влияние давления, температуры и физических свойств на устойчи$$>еть
горения жидких ВВ..............................................₽<< •
Теория устойчивости горения взрывчатых жидкостей.......................
Глава шестая. Вспышка и воспламеняемость ВВ.............................
пи
1. Механизм возникновения вспышки .
2. Характер вспышки ................
3. Верхний температурный предел вспышки . . . .
II. Воспламеняемость ВВ...........................
Экспериментальное определение воспламеняемости ВВ
III. Распространение горения по поверхности заряда . . .
262
265
272
283
294
294
299
299
311
311
311
315
325
327
332
337
Литература........................................339
Константин Константинович Андреев
Термическое разложение
и горение взрывчатых веществ
ОПЕЧАТКИ И ИСПРАВЛЕНИЯ
j Страница С ока Напечатано Должно быта
8 16 63 181 105 238 309 11 СВ. ?. ся. If сн. : св. . • св. > СИ. . ) СВ. Г, E/RT РР1,‘) ккал/моль СМ на см. рис. 211 . скорость 1 1966 г. -Я/RT 80,4. ,, зак. 261 (~/*) кал /моль см/сек при см. рис. 212 устойчивость
К. К. Авдеев