/



























































































































Текст
Л.И.ТАРУТИНА
Ф. О. ПОЗДНЯКОВА
СПЕКТРАЛЬНЫЙ
АНАЛИЗ
ПОЛИМЕРОВ
Л.И.ТАРУТИНА
Ф. О. ПОЗДНЯКОВА
СПЕКТРАЛЬНЫЙ
АНАЛИЗ
ПОЛИМЕРОВ
ЛЕНИНГРАД
„ХИМИЯ”
ЛЕНИНГРАДСКОЕ ОТДЕЛЕНИЕ
1986
543
Т223
УДК 543.42:678
Тарутина Л. И., Позднякова Ф. О.
Спектральный анализ полимеров, —Л.: Химия, 1986,- 248 с., ил.
Посвящена анализу полимеризационных и поликонденсационных поли-
меров методами молекулярной и атомной спектроскопии. Рассмотрено
определение качества сырья, чистоты полимеров и их структурной неодно-
родности, содержания добавок в полимерных материалах, состава сополи-
меров. Описан спектроскопический контроль химических реакций в поли-
мерах. Представлены спектры наиболее распространенных полимеров, раст-
ворителей, стабилизаторов.
Предназначена инженерно-техническим и научным работникам, занимаю-
щимся изучением и исследованием полимерных материалов. Полезна препо-
давателям, аспирантам и студентам химико-технологических вузов.
Библиогр. 101 назв Ил. 234. Табл. 11
Рецензент: профессор докт. физ.-мат. наук М. А. Мартынов
Рекомендована физико-химической секцией Ученого совета ОНПО
„Пластполимера”
ПРОИЗВОДСТВЕННОЕ ИЗДАНИЕ
Людмила Ивановна Тарутина
Фатиха Олиевна Позднякова
СПЕКТРАЛЬНЫЙ
АНАЛИЗ
ПОЛИМЕРОВ
Редактор Э. Э. Ярцева
Техн, редактор 3. Е. Маркова
Переплет художника Б. Н. Осенчакова
Корректор М. 3. Басина
Операторы: 3. В. Васина, О. А. Семенова
Издание подготовлено к печати с использованием наборно-печатаюшей техники
в ордена „Знак Почета” издательстве „Химия”
ИБ № 1799
Подписано в печать 30.04.86. М-33804. Формат бумаги 60 z 901 /, 6. Бумага
типогр. N" 1. Офсетная печать. Усл.-печ. л. 15,5. Усл.кр.-отт. 15,5. Уч.-изд. л.
16,0. Тираж 4X00 экз. Зак. 129 Цена 1 р. 30 к. Изд. № 2702
Ордена „Знак Почета” издательство „Химия” Ленинградское отделение
191186, г. Ленинград, Д-186, Невский пр., 28
, Ленинградская типография №6 ордена Трудового Красного Знамени Ленин
градского объединения „Техническая книга” нм. Евгении Соколовой Союз-
полиграфпрома при Государственном комитете СССР по делам издательств,
полиграфии и книжной торговли. 193144, г. Ленинград, ул. Моисеенко, 10
1804000000-005
Т----------------5-86
050(011-86
© Издательство „Химия”. 198ь
ПРЕДИСЛОВИЕ
!i настоящее время нет необходимости пропагандировать целесообраз-
ность использования спектральных методов в химии. Они прочно вошли
I практику химических лабораторий, дополняя или заменяя традицион-
ные методы аналитической химии. В химии высокомолекулярных соеди-
нений применение спектральных методов по сравнению с другими часто
сказывается наиболее эффективно.
Основную роль в спектральном анализе полимеров играют методы
бсорбционной молекулярной спектроскопии. ПК- и УФ-спектры погло-
щения содержат информацию о строении макромолекул полимера, на-
личии функциональных групп и их природе, составе сополимеров, после-
ювательности присоединения мономерных звеньев и т. п.
Спектроскопические данные позволяют судить об изменении моле-
кулярной структуры конечного продукта при варьировании условий
полимеризации или поликонденсации. Это помогает выбрать технологи-
ческий режим, оптимальный для получения полимерного материала
с требуемыми свойствами. Спектры поглощения используют также для
шализа примесей в полимерах, дефектности макромолекул (разветвлен-
ности, ненасыщенности и т. п.), остатков реакционной смеси и. т. п.
Все это способствует повышению качества полимерных материалов и,
как указано в „Основных направлениях экономического и социального
развития СССР на 1986-1990 годы и на период до 2000 года”, дает воз-
можность при разработке новой техники и технологии более полно ис-
пользовать возможности материалов с заранее заданными свойствами,
особенно прогрессивных конструкционных, в том числе синтетических,
композиционных, сверхчистых и других, обуславливающих высокий
жономический эффект в народном хозяйстве.
Настоящая книга задумана как руководство по практическому
спектральному анализу полимеров и адресована сотрудникам завод-
ских лабораторий, лабораторий учебных и отраслевых институтов,
вязанных с синтезом, технологией и промышленным выпуском
3
полимеров. Она предназначена для тех работников, которые применяют
спектральные методы для решения стоящих перед нами задач, но не
являются спектроскопистами по своей основной специальности. Она
может быть использована также спектроскопистами, не имеющими
опыта работы с полимерными материалами. Имея связь с заводскими
и другими химическими лабораториями, мы убедились, что имеется
необходимость в книге такого плана.
В основу книги положен материал, накопленный за многие годы
в лаборатории спектроскопии ОНПО „Пластполимер”, организованной
проф. В. М. Чулановским, который много сил и внимания уделял вне-
дрению спектроскопии в практику химических исследований в на-
шей стране.
Кроме своего опыта и опыта коллег по лаборатории, мы использо-
вали также результаты других исследователей, хотя в наши цели не вхо-
дил обзор литературы по существующим методикам анализа полимеров.
В книге описаны методики, которые могут быть применены для
анализа как полимеризационных, так и поликонденсационных поли-
меров. В соответствии с интересами авторов большинство конкретных
примеров относится к первому типу полимеров. Спектроскопические
методики анализа наряду с другими физическими и химическими ме-
тодами исследования второго типа полимеров описаны в книге „Анализ
конденсационных полимеров”, выпущенной издательством „Химия”
в 1984 г.
Авторы благодарны проф. Г. С. Денисову за замечания, сделанные
им при подготовке рукописи, и проф. М. А. Мартынову за полезные
советы и обсуждение рукописи при ее рецензировании.
I. ОСНОВНЫЕ ПРЕДСТАВЛЕНИЯ ОБ АТОМНОЙ
И МОЛЕКУЛЯРНОЙ СПЕКТРОСКОПИИ
I. 1. МОЛЕКУЛЯРНЫЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ
Молекулярный спектральный анализ основан на свойстве вещества изби-
рательно поглощать отдельные участки электромагнитного излучения.
Имеется в виду оптический диапазон, который включает ультрафиоле-
товую, видимую и инфракрасную области. Ультрафиолетовая и видимая
области охватывают интервалы длин волн 0,01—0,38 и 0,38—0,76 мкм
соответственно. Инфракрасная область более протяженная. От видимой
области она простирается до радиоволнового диапазона (1000 мкм).
Границы областей условны. ИК-область делится на ближнюю (0,76—
2,5 мкм), среднюю (2,5—50 мкм) и дальнюю (50—1000 мкм). Длины
волн УФ и видимой областей принято выражать в нанометрах, ИК-облас-
ти — в микрометрах. Однако в ИК-спектроскопии обычно используют
волновые числа. Волновое число 7 измеряется в обратных сантиметрах
и определяет число длин волн Хо (в вакууме), укладывающихся в 1 см:
v = 1/А о
Частота волны v отличается от волнового числа множителем с рав-
ным скорости света в вакууме (с « 3 • 1010 см/с)
v= VC
В практике спектрального анализа волновое число принято для
краткости называть частотой и обозначают его v вместо гл Единица
измерения см-1 указывает на то, что в действительности имеют в виду
волновое число.
Проходя через слой вещества, излучение ослабляется. Ослабление
возникает из-за поглощения света * веществом, что является предметом
изучения молекулярной спектроскопии, а также в результате рассеяния
света на неоднородностях показателя преломления среды. Причиной
рассеяния света в полимерах могут быть посторонние включения, пу-
зырьки воздуха, микропустоты, флуктуации плотности, связанные с
надмолекулярными образованиями и т. п. Неоднородности можно рас-
сматривать, как частицы, вкрапленные в среду полимера и имеющие
оптические свойства, отличающиеся от свойств среды. Потери излучения
на рассеяние зависят от размеров частиц. Большие потери возникают,
когда размеры частиц сравнимы с длиной волны света. Если размеры
* Излучение в оптическом диапазоне длин волн обычно называют светом,
хотя первоначально этот термин относился к видимой области.
5
частиц гораздо меньше длины волны, то рассеяние обратно пропорцио-
нально четвертой степени длины волны (рассеяние Рэлея). Рассеянный
свет служит часто помехой спектральному анализу, особенно при работе
в ультрафиолетовом и видимом диапазонах длин волн.
Ослабление света, вызванное поглощением вещества, описывается
основным законом молекулярного спектрального анализа — законом
Б угера—Ламберта:
/ = /о<- К
где / и /„ - интенсивности света, прошедшего слой вещества и падающего на него
соответственно; d - толщина слоя вещества; к* ** - величина обратная расстоянию,
на котором интенсивность света ослабляется в е раз, называется натуральным по-
казателем поглощения*. Он определяет поглощающие свойства вещества и изме-
ряется в обратных сантиметрах (см"1) •
Поглощающая способность слоя вещества характеризуется оптичес-
кой плотностью (натуральной) D = 1п70/7. Показатель поглощения свя-
зан с оптической плотностью соотношением k'=D'jd. В последнее время
вместо натуральных логарифмов часто используют десятичные. Тогда
оптическую плотность определяют как D=]glo/I, а показатель поглоще-
ния к = D/d является величиной, обратной расстоянию, на котором
параллельный поток излучения ослабляется в 10 раз. Десятичные величи-
ны связаны с натуральными простыми соотношениями: к =0,434 к', D=
=0,434 D'.
Спектры поглощения записывают в виде зависимости пропускания
Т= 1ОО7/7о(в%) или оптической плотности 7)=lg( 100/ Г) от волново-
го числа.
Строго говоря, закон Бугера—Ламберта выполняется для монохро-
матического света. Однако немонохроматичность излучения не приводит
к существенным ошибкам при количественном анализе, когда полосы
поглощения значительно шире спектральной ширины щели спектро-
фотометров.
Если поглощающее вещество растворено в прозрачном растворителе,
то показатель поглощения пропорционален его концентрации в растворе
(закон Бера):
к = еС
Здесь С - молярная концентрация (число молей в единице объема); е - коэф-
фициент, который называют молярным показателем поглощения’1*. Если С выра-
жать в моль/л, а к - в см"1, то единица измерения е - л/ (моль • см).
Наряду с молярной концентрацией используется концентрация, вы-
ражаемая массой вещества, растворенного в единице объема растворите-
ля (г/л или г/см3). Будем обозначать ее также через С. Закон Бера
тогда записывается в виде
к = аС
где а - удельный показатель поглощения л/(г - см) или см2/г.
*В ГОСТ 7601-78 эта величина обозначена через а‘
**Используется также термин коэффициент экстинкции.
6
Объединяя законы Бугера—Ламберта и Бера можно написать для
оптической плотности выражение D = eCd uwD=aCd. Закон Бера приме-
ним и в том случае, когда поглощающее вещество распределено в твердой
прозрачной матрице. При этом содержание вещества часто характеризует-
ся массовой долей Ь. Тогда массовая концентрация С=Ьр, где р — плот-
ность пленки, и закон Бера принимает вид к=айр. Роль поглощающего ве-
щества могут играть отдельные группы полимера (например, концевые).
Закон Бера предполагает, что поглощающие молекулы не взаимо-
действуют друг с другом. При достаточно малых концентрациях это
справедливо. Для концентрированных растворов закон Бера может
нарушаться, т. е. е начинает зависеть от концентрации. Такие отклоне-
ния наблюдаются, если существенным становится взаимодействие между
молекулами растворенного вещества непосредственно или через окру-
жающие молекулы.
Кроме того, е может зависеть от природы растворителя, т. е. изме-
няться под влиянием взаимодействия между молекулами растворенного
вещества и растворителя. Если при этом концентрация раствора будет
мала настолько, что взаимодействие самих молекул растворенного ве-
щества еще не существенно, то пропорциональность между к и С будет
соблюдаться. Влияние растворителя скажется на угле наклона прямой,
выражающей зависимость к от С. Оптическая плотность смеси не взаимо-
действующих и не реагирующих друг с другом веществ аддитивно скла-
дывается из оптических плотностей отдельных компонент D =d (z?i С) +
+ а2С2 + Л3С3 +...). То же относится и к показателям поглощения.
1.1 Л. Электронные спектры поглощения
Под воздействием света УФ- и видимого диапазонов длин волн про-
исходит возбуждение электронных оболочек молекул вещества.
Молекулы переходят в возбужденное состояние с большей энергией.
Это сопровождается появлением полос поглощения в спектре при дли-
нах волн, соответствующих разности энергий возбужденного и невоз-
бужденного уровней. Каждому электронному уровню молекулы соответ-
ствует набор колебательно-вращательных уровней. Поскольку энергия
возбуждения электронных оболочек молекулы много больше энергии
возбуждения ее колебаний, то электронный переход обычно сопровож-
дается изменением колебательно-вращательного состояния молекулы.
Поэтому молекулярные электронные спектры жидкостей и твердых тел
состоят из широких полбе. Для насыщенных соединений энергия воз-
буждения электронов, как правило, значительно больше, чем для не-
насыщенных соединений. Поэтому большинство насыщенных соедине-
ний имеют поглощение в далекой, так называемой, вакуумной*
*В этой области составные части воздуха сильно поглощают, поэтому для по-
лучения спектров требуются специальные спектрометры с вакуумированием. Для
аналитических целей эта область спектра не используется. Поэтому, обсуждая в
дальнейшем поглощение того или иного соединения в УФ-области спектра, мы
имеем в виду только доступную для широкого использования аналитическую
область спектра, ограниченную прозрачностью воздуха.
7
УФ-области (до 200 нм). Возбужденные уровни расположены настолько
густо, что поглощение насыщенных соединений сплошное.
Избирательное поглощение в УФ- и видимой областях спектра харак-
терно для ненасыщенных соединений. Их поглощение определяется на-
личием в ненасыщенных связях легко возбудимых тг-электронов. Груп-
пы атомов, ответственные за избирательное поглощение, называют хро-
мофорами. Простейшими хромофорами являются группы с изолирован-
ными кратными связями С=С, С=С, С=О и т. п. Они также имеют погло-
щение в вакуумной УФ-области спектра или на границе рабочего интер-
вала (около 200 нм) обычных спектрометров. Положение полос погло-
щения хромофоров и их интенсивность может значительно изменяться в
зависимости от групп атомов, присоединенных к молекуле, содержащей
хромофор, и не имеющих собственного поглощения. Такие группы на-
зывают ауксохромами. Типичными ауксохромами являются ОН, ОСН3,
NH2, NCH3 и др. Ауксохромными свойствами обладают также атомы
галогенов. Под влиянием ауксохрома происходит сдвиг полос поглоще-
ния хромофора в сторону больших длин волн (батохромный эффект)
или к меньшим длинам волн (гипсохромный эффект). Смещение полос
поглощения и увеличение их интенсивности наблюдается также при вза-
имодействии хромофоров между собой. Так, соединения, содержащие
простую этиленовую связь, поглощают около 200 нм- Сопряжение этиле-
новых связей вызывает батохромное смещение поглощения к некоторой
предельной длине волны (рис. 1.1) и сопровождается появлением желтой
окраски. Интенсивность поглощения по мере увеличения длины сопря-
жения регулярно возрастает.
Сопряжение полиеновых систем с карбонильными группами сдвига-
ет поглощение в видимую область, вызывая углубление цвета (переход
окраски от желтой к фиолетовой по спектру видимого света). Взаимо-
действие с ауксохромами и эффект сопряжения приводит к тому, что
поглощение большинства хромофоров расположено в ближней УФ- и
и видимой областях спектра, удобных
для спектрального анализа. В табл. 1.1
представлены некоторые наиболее ра-
спространенные простые хромофоры
и положение максимумов их полос
поглощения.
Наиболее эффектные изменения на-
блюдаются в спектрах поглощения бен-
зола и его производных при присоеди-
нении ауксохромных групп или сопря-
жении. Эта группа соединений, благода-
ря своему химическому строению и
свойствам занимает особое положение
среди соединений, поглощающих в
Рис. 1.1. Зависимость положения максиму-
ма поглощения полиена Н(—СН=СН—)„Н
от числа сопряжений п.
8
УФ-области спектра. Бензол имеет три серии полос поглощения: две
очень интенсивные в вакуумной УФ-области и одну, так называемое
бензольное поглощение, меньшей интенсивности, в интервале длин волн
230—270 нм с условным максимумом при 255 нм. Замещение водорода
на ауксохром NH2 (в анилине) дает сдвиг бензольного поглощения до
280 нм. При этом интенсивность поглощения увеличивается на порядок.
Большое батохромное смещение наблюдается для полициклических аро-
матических соединений при увеличении числа конденсированных бензоль-
ных колец. Если бензол поглощает при 255 нм и является бесцветным
соединением, то нафтацен, поглощающий при 480 нм, имеет желтую
окраску, а пентацен, полоса которого сдвинута к 580 нм, окрашен в
голубой цвет [1, с.175]
нафтацен пентаием
При получении УФ-спектров поглощения веществ в разных раство-
рителях наблюдаются изменения контура полосы и ее интенсивности.
Полярность растворителя имеет большое значение. Это видно на
ТАБЛИЦА 1.1.
Некоторые хромофоры и их полосы поглощения
Хромофорная группа Соединение ^макс.нм
Этиленовая ^С=С^ Этилен (пар) 193
Ацетиленовая -С^С- Ацетилен (пар) 173
Карбонильная ^С = О Ацетон (в гексане) 279
„ (в спирте) 271
Циклогексанон (в гексане) 285
Ацетальдегид (в гексане) 291
„ (в спирте) 285
Карбоксильная —Cf Уксусная кислота (в воде) 204
ХОН Пальмитиновая кислота 210
Азо - N=N- Диазометан (в этаноле) 340
Нитрозо -N=0 Нитрозобутан (в эфире) 300; 665
Нитро -NO, Нитрометан (в спирте) 210; 271
Сульфоксидная ^3^*0 Циклогексилметил сульфоксид 210
Азометиновая /C=N— (в спирте) Ацетоксим 190
Нитрильная -С N Ацетонитрил < 160
Нитритная -O-N=O Октилнитрит (в гексане) 230
Примечания: 1. Вторая полоса карбонильного хромофора лежит ниже
200 нм. 2. Карбонильные группы кетона и альдегидов объединены в одну хромо-
форную группу. Это соответствует сходству свойств этих соединений и близости
их спектральных характеристик. 3; Присоединение гидроксила к карбонильной
группе резко меняет свойства карбонила, что отражается на его спектре поглоще-
ния и дает основание выделить карбоксильные группы в отдельный хромофор.
9
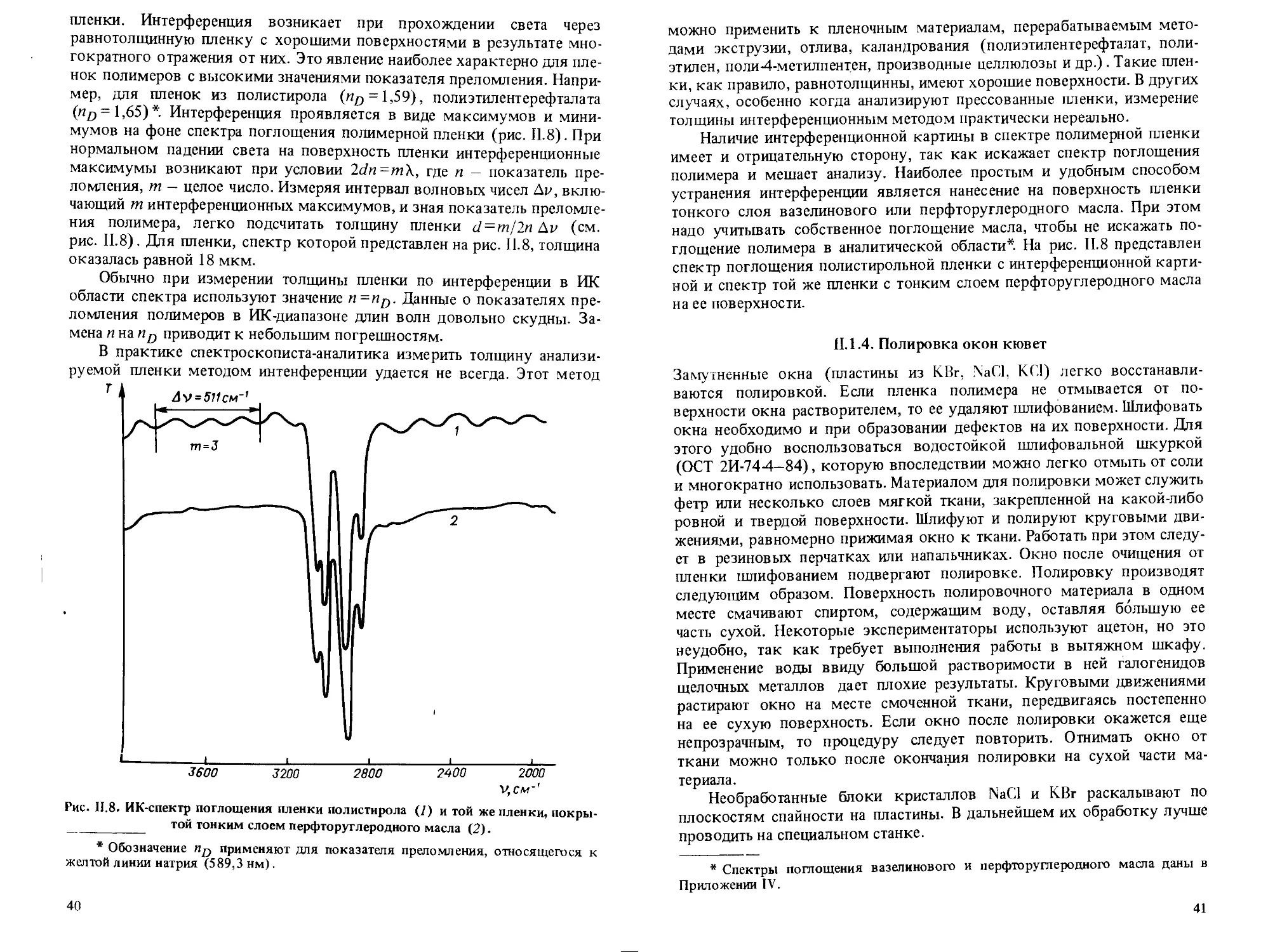
Рис. 1.2. Спектры поглощения раствора в гексане (/) и этиловом спирте (2).
Концентрация 0,1 г/л; толщина слоя 1 мм.
классическом примере фенола. Спектр раствора фенола в гексане имеет
тонкую структуру, которая отсутствует, когда фенол растворен в этило-
вом спирте (рис. 1.2).
Описанные выше общие закономерности относятся и к полимерам.
Так же как и большинство насыщенных соединений, полимеры, не со-
держащие в молекуле кратных связей, прозрачны в ближней УФ- и ви-
димой областях спектра (полиолефины, полимеры и сополимеры хлор-
и фторпроизводных этилена, поливиниловый спирт и т. п.). Полимеры
сложных эфиров акриловых кислот (полиакрилат, полиметакрилат
и т. п.), поливиниловые сложные эфиры (поливинилацетат, поливинил-
бутират и т. п.), а также другие полимерные эфиры карбоновых кислот,
содержащие карбонильный хромофор, поглощают на границе вакуумной
УФ-области (около 200 нм). Полимеры, содержащие карбоксильный
хромофор или бензольные кольца, поглощают в значительной части
УФ-области. Спектры полимеров в УФ-области, как правило, невырази-
тельные. Часто наблюдается лишь граница поглощения (рис. 1.3, спек-
тры 2-3). Положение границы определяется типом хромофора, интен-
сивностью его поглощения и зависит от толщины поглощающего слоя.
Например, бензольное поглощение полистирола при толщине пленки
6 мкм имеет сложный вид с максимумом при 262 нм (рис. 1.3, спектр У).
При толщине пленки 30 мкм структура поглощения уже не проявляется,
а наблюдается лишь резкая граница около 275—280 нм, которая по мере
увеличения толщины пленки полистирола сдвигается в сторону больших
длин волн. Сложная хромофорная группа полиэтилентерефталата (лав-
10
сана) —СО—С6Н4—СО— вызывает такое интенсивное поглощение УФ-
излучения, что в спектре полимера не обнаруживается структура даже
при толщине пленки 6 мкм и имеется только резкая граница выше
300 нм (рис. 1.3,спектр 5).
Образование в полимере полиеновых систем, так же как и в случае
низкомолекулярных соединений, вызывает сдвиг поглощения, связан-
ный с батохромным эффектом и сопровождается появлением окраски.
Примером этому может служить сополимер трифторхлорэтилена с ви-
нили денфторидом. При его термостарении в вакууме вследствие де-
гидрофторирования и дегидрохлорирования образуются длинные
цепи сопряженных связей С=С. Появляется желтая окраска (погло-
щение в синей области), и по мере увеличения продолжительности
старения наблюдается сдвиг границы поглощения к большим дли-
нам волн (рис.1.4) и изменение цвета до коричневого. Если такой обра-
зец подвергнуть окислению, которое вызывает образование кислород-
содержащих групп и разрушение полиеновых систем, то наблюдается
его „просветление”. Граница поглощения сдвигается к меньшим
длинам волн.
Вид УФ-спектров поглощения полимеров в большой степени зави-
сит от их рассеивающих свойств. Полимер может не иметь собственного
поглощения, но при этом задерживать УФ-излучение и даже видимую
и ближнюю ИК-области спектра. Примером служит политетрафторэтилен,
который вследствие высокой степени кристалличности является сильно
рассеивающим материалом. Из рис. 1.5 видно, что пленка политетрафтор-
этилена толщиной 0,2 мм задерживает УФ- и видимое излучение, а при
толщине 0,8 мм из-за больших потерь света на рассеяние становится
Рис. 1.3. Спектры поглощения образцов полистирола толщиной 6 мкм (7), 30 мкм
(2) и полиэтилентерефталата толщиной 6 мкм (3). (В спектре 3 проявляются
интерференционные полосы).
И
Рис. 1.4. Спектры поглощения образцов сополимера трифторхлорэтилеиа с вини-
лиденфторидом исходного (-------), после нагревания в вакууме при 300 °C
(-----------) и окисленного (--------).
Цифры у кривых — продолжительность нагревания.
непрозрачной также и в ближней ИК-области спектра. Модификация
политетрафторэтилена путем введения сомономера способствует
разрушению его мелкокристаллической структуры. Рассеянного све-
та становится меньше, светопропускание полимера увеличивается
(рис. 1.5, спектр 3).
Рис. 1.5. Спектры поглощения политетрафторэтилена (7, 2), модифицированного
политетрафторэтилена (5) и полиметилметакрилата (4).
Толщина пленки (в мм): 1,3 - 0,2; 2 - 0,8; 4 - 0,1.
12
Пологий склон границы пропускания характерен для рассеивающих
сред, поскольку интенсивность рассеяния зависит от длины волны. Для
сравнения на рис. 1.5 представлен спектр полиметилметакрилата, прак-
тически не рассеивающего излучения и имеющего поглощение ~ 200 нм.
Электронные молекулярные спектры для исследования структуры
полимера практического значения не имеют. Однако высокая интенсив-
ность полос поглощения хромофоров определяет применение УФ-спект-
ров для анализа примесей в полимерах, определения вводимых в поли-
меры добавок (антиоксидантов, пластификаторов, стабилизаторов и
т. п.). Задачи, которые могут быть решены при использовании видимого
диапазона длин волн, связаны с проблемами окраски полимеров: иссле-
дование комплексообразования в полимерах, выбор и анализ красите-
лей и др.
1.1.2. Инфракрасные спектры поглощения
Поглощение света веществом в ИК-области спектра связано с возбужде-
нием колебаний молекул. В ближней и средней ИК-областях (0,76 —
50 мкм; 13000 — 200 см-1) проявляются внутримолекулярные коле-
бания, при которых меняется относительное расположение ядер атомов,
составляющих молекулу. Такие колебания сопровождаются изменением
длин связей, соединяющих атомы (валентные колебания) и валентных
углов между связями (деформационные колебания) (рис. 1.6). Колеба-
ния тяжелых атомов, вращательные движения молекул имеют частоты,
соответствующие далекой ИК-области спектра (50—1000 мкм; 200—
10 см-1), примыкающей к субмиллиметровому диапазону радиоволн.
Частота колебаний молекулы v зависит от массы колеблющихся
атомов, а также от силовой постоянной f, которая характеризует квази-
упругие силы, возникающие в молекуле при отклонении атомов от
положения равновесия. Для двухатомной молекулы связь между этими
величинами выражается формулой:
где т=т1 тг / (mt +тг) - приведенная масса атомов.
Этой формулой можно воспользоваться для очень грубой оценки
частот колебаний многоатомной молекулы, рассматривая отдельные
Рис. 1.6. Колебания СХ5-групп;
Валентные: 1 — симметричные (vs) ; 2 — асимметричные (и^). Деформационные:
3 — ножничные (6); 4 — маятниковые (7Г); 5 — веерные (7W); 6 — крутильные
(7f)-
13
связи и группы атомов макромолекулы как независимые единицы.
Чем больше масса колеблющихся атомов, чем меньше силовая постоян-
ная, тем в более длинноволновой области спектра расположены полосы
поглощения. Изменение частоты колебания в зависимости от массы ко-
леблющихся атомов видно на примере валентных колебаний связей
С—Н и С—Hal. Если колебания С—Н поглощают вблизи 3000 см'1, то
колебания связей С— F, С—CI, С—Вг имеют частоты в интервалах 1400—
1000, 800—600 и 600—500 см-1 соответственно. В чистом виде зависи-
мость частоты колебания от массы атома видна на примере изотопичес-
кого замещения, когда сила взаимодействия между атомами не меняет-
ся. Например, валентное колебание связи С—Н имеет частоту около
3000 см 1, а связи С—D около 2200 см'1. Влияние силы взаимодействия
на частоту колебания хорошо проявляется при сравнении частот колеба-
ний простых, двойных и тройных связей одних и тех же атомов. Силовая
постоянная таких связей возрастает с увеличением их кратности. Так,
валентное колебание связи C-N имеет частоту около 1300 см'1, C=N
1640-1690 см'1, a C=N около 2250 см'1 [2, с.369, 378].
С квантовой точки зрения объяснить происхождение колебательных спектров
можно следующим образом. Энергия колебания не может меняться непрерывно,
но принимает лишь дискретный ряд значений-уровней энергии. Если колебание
гармоническое, то разность энергий двух соседних уровней ДДодна и та же. При
поглощении света происходит переход молекулы с исходного колебательного
уровня на соседний (верхний) и при этом, согласно правилу Бора, поглощается
фотон с волновым числом v:
и-ЛЕ/2 лЬс
где fi - постоянная Планка.
Ангармоничность (т. е. нелинейная зависимость квазиупругой силы от рас-
стояния между атомами) приводит к тому, что становится возможным переходы
между уровнями через один, два и т. д. При этом поглощаются фотоны с волновы-
ми числами 2v, 3v... К такому же эффекту приводит нелинейность связи дипольно-
го момента при колебаниях со смещениями атомов из положения равновесия.
Частота v соответствует в спектре поглощения основному тону колебания, 2v -
первому обертону, 3v - второму и т. п. Обычно ангармоничность мала и интенсив-
ность поглощения быстро убывает с увеличением номера обертона. Ангармоничес-
кое взаимодействие разных типов валентных и деформационных колебаний приво-
дит к появлению в спектре поглощения слабых полос, соответствующих сумме
или разности частот различных колебаний (составных частот). О применении обер-
тонов и составных частот для анализа полимеров и других веществ см. ниже в
этом разделе.
В каждом молекулярном колебании принимают участие в той или
иной мере вее атомы молекулы. Тем не менее можно выделить такие
колебания, в которых участвуют главным образом определенные атомы
или группы атомов. Роль остальных атомов молекулы оказывается
незначительной. Частоты этих колебаний сохраняются в спектрах различ-
ных соединений и называются характеристическими. Характеристичес-
кие колебания мало взаимодействуют с другими колебаниями молеку-
лы, и в этом смысле их можно рассматривать как независимые. Взаимо-
действие колебаний атомов, составляющих валентные связи в молеку-
ле, будет тем меньше, чем больше различаются массы этих атомов.
14
Например, вследствие большой разницы в массах углерода и водорода,
колебания связей С—Н и С—С в углеводородах можно рассматривать как
независимые. Колебания связи С—Н являются характеристическими.
Полосы поглощения, соответствующие валентным колебаниям этих
связей, независимо от соединения всегда наблюдаются вблизи 3000 см-1.
Напротив, взаимодействие колебаний связей С—F и С-С во фтороугле-
родных соединениях приводят к тому, что частоты колебаний связи
С—F варьируют в широком интервале в спектрах различных соединений
(1400—1000 см-1). Следует отметить, что в ИК-спектрах наблюдаются
полосы поглощения тех колебаний молекулы, которые связаны с изме-
нением дипольного момента. Чем сильнее это изменение при колебаниях,
тем интенсивнее соответствующая полоса поглощения.
Положение полос поглощения колебаний основных валентных свя-
зей и структурных групп представлены в табл. 1.2.
Из всех спектральных методов ИК-спектроскопия имеет наибольшее
применение в химии полимеров. В начале 50-х годов, когда метод ИК-
спектроскопии еще только начинал внедряться в практику химических
лабораторий, для решения аналитических задач использовалась ближняя
ИК-область спектра (0,76—2,5 мкм; 13 000-4 000 см-1), как наиболее
технически доступная. Поглощение в ней связано в основном, с оберто-
нами валентных колебаний связей С—Н, N—Н, О—Н, обертонами более
высокого порядка связей С—С, С=С, С—О, С=О и комбинационными
(составными) частотами (табл. 1.3).
В настоящее время большинство лабораторий оснащено современ-
ными ИК-спектрометрами и используется главным образом средний
диапазон длин волн (2,5 мкм - 50 мкм; 4000-200 см-1), в котором
поглощение обусловлено основными колебаниями молекул. Исследова-
ние спектров поглощения в этой области дает большую информацию о
строении молекул.
С развитием исследований в среднем диапазоне длин волн ближняя
ИК-Обласгь спектра стала редко использоваться. Между тем, примене-
ние ближней ИК-области для решения практических задач имеет ряд
преимуществ. Поскольку интенсивность полос поглощения первого
обертона обычно на два порядка меньше интенсивности основного тона
колебания и интенсивность уменьшается с увеличением номера оберто-
на, то для исследования поглощения обертонных колебаний можно ис-
пользовать большие слои исследуемого вещества. Это освобождает от
необходимости изготавливать тонкие пленки полимера, что связано час-
то с большими трудностями. Кроме того, ошибка при измерении толщи-
ны тонкой пленки уменьшает точность анализа. Например, валентные
колебания связи С—Н в спектре полистирола имеют интенсивные поло-
сы поглощения основного тона при 2923 и 3029 см-1. Если для получе-
ния спектра в этой области требуется пленка толщиной не более 20 мкм,
то чтобы получить спектр на первом обертоне этих колебаний (5846 и
6058 см-1) нужна пластина полистирола толщиной 2 мм (рис. 1.7).
Другое преимущество ближней ИК-области — использование в качестве
15
ТАБЛИЦА 1.2
Полосы поглощения основных колебаний связей и отдельных групп атомов
Группа Связь* V, см-1
3600 - 3100 см'1 -ОН (в спиртах, феноле) О-Н -СООН О-Н -nh2; ^NH—; —С—NH2; —с—nh-n-h z II II о о -С^СН С-Н 3650-3600 (несвязанные группы) 3400- 3200 (связанные группы) 3400-2500 (связанные группы) 35 00- 3 300 (несвязанные груп- пы) 3300-3250
3100-3000 см’1
-НС=СК2 С-Н 3090-3075 3040- 3010
sC-H (в ароматических соединениях) с-н 3030
— NH, N-H 3130-3030
3000 - 2800 см"1
-сн, С-Н 2960; 2870
-сн2- С-Н 2925; 2850
)сн С-Н 2890
-O-CHS (в алифатических соединениях) С-Н 2830-2815
-О-СНз (в ароматических соединениях) С-Н 2850
2800 - 2000 см'1
-NHj ,-NH+ N-H 2800-2000
-SH S-H 2600-2550
PH P-H 2440- 2350
-с=с- c=c 2260-2190
-C=N C^N 2260-2100
JSiH Si—H 2200-2100
1900- 1500 см'1
-</° -чс-0. с^° с=о
;о хон хо- хн
1880-1620**
% '
—C—NH2; C—NH—; —c—nc; C=O
II II 4
II о О о
1680-1630
-носи,, -НС=СН-
-HC=CFa, -FC=CF-
-FOCF,
-O-NOj
-О-NO
1680-1620
1755-1705
1800—1780
1650-1600
1650-161 0
16
Группа
ПРОДОЛЖЕНИЕ
v, см'
1610-1590)
1500-1490
—С—NH2;-С—NH;—NH2;/NH;~NH3 N-H4*
II II 7
О О
-C=N- ON
1690-1630
1690-1640
1500 - 1400 см'1
-СН3
- СН2 -
-СН=СН;
С-Н4*
С-Н4*
С-Н4*
1460; 1380
1470-1460
1420-1415
1350- 1000 см"1
\ /° //Q -УС—ОН; —С< ; ~ z хон хо— С-0
-с(° ; —СН2—О—СН2— -с<о -^CF; /CF2; —CF3 C-F
-Р-О-С (в ароматических соединениях) Р-О-С
-Р-О-С (в алифатических соединениях) Р-О-С
^Р=О Р=О
-O-NO2; >N-NO2
1000 - 700 см’1
1320-1050’*
1350-1200
1200
1030
1300-1280
1300-1250
-НС=СН2 С-Н6 *
/С=СН2 ; —НС=СН—
-(СН2)„- С-Н8*
Различные типы замещения ароматического С-Н6
кольца
1000-7407*
850-720’*
900-690
>СС1
^СВт
7CI
700 - 400 см -1
С-С1
С-Вг
С-1
900-600
600-500
~500
* Во всех случаях кроме отмеченных приведены данные для валентных коле-
баний. ** См. табл. III.2. 3 * Скелетные колебания. 4 * Деформационные колебания.
5 * См. табл. III.6. 6 * Внеплоскостные деформационные колебания.’* См. раздел
III.3.1. 8 * Маятник аиле колебания—’^См2;раздел IV.2.
-----------—--------------„-----. - . л—
„ ТАБЛИЦА 1.3
Положение полос поглощения обертонных колебаний
и составных частот различных групп атомов
Группа Обертон, составная частота Длина вол- ны, мкм Группа Обертон, составная частота Длина вол- ны, мкм
-СН,- 1 1,76и —ОН (в кис- 1 1,43-1,48
1,72 лотах) 2 0,96-1,00
2 1,22 3 0,71-0,76
3 1,05 -ОН (в спир- 1 1,42-1,55
4 0,93 тах) 2 0,95-0,97
5 0,76 3 0,74-0,80
^ + 6(СН2) 2,31 -ОН (вода) 1 1,39-1,45
-СНЭ 1 1,69 rs + « 1,93
2 1,19 С=О 1 2,90
3 1,02 2 1,92
4 0,90 3 1,45
5 0,74 4 1,16
=СН 1 1,77 5 0,97
=СН, 1 1,62 2р(С=О) + 2,18
СН(в арома- 1 1,69 + v (-О-С-)
тических 2 1,14 NH (в алифа- 1 1,49-1,50
соединениях) 3 0,87 тических сое- 2 1,02
динениях) 3 0,80
4 0,66
(NH2) 2,06
окон кювет негигроскопичных материалов (стекло, кварц). Это позво-
ляет проводить анализ растворителей, мономеров и других жидких
объектов на присутствие значительного количества влаги, что невозмож-
но сделать, используя средний диапазон ИК-излучения.
600 800 1000 1200 1400 1600 1800 2000 2200 МО 2600
Л, нм
Рис. 1.7. Спектры поглощения полистирола в ближней ИК-области.
Толщина поглощающего слоя (в мм) : 1 — 0,1; 2 — 2,0; 3 — 45.
18
Вследствие различной ангармоничности колебаний может оказаться,
что взаимное расположение аналитических полос и их интенсивность в
спектре анализируемого образца более благоприятны для анализа в
области обертонов и составных частот, чем в области основного тона.
Целесообразность использования той или иной области спектра
для решения конкретных практических задач будет обсуждена в соот-
ветствующих главах.
1.2 . АТОМНАЯ СПЕКТРОСКОПИЯ
Атомная спектроскопия, как метод анализа вещества, возникла и полу-
чила развитие значительно раньше, чем молекулярный спектральный ана-
лиз. Ее применение первоначально было связано с определением различ-
ных элементов, главным образом металлов, в виде примесей в сталях,
цветных металлах, рудах, минералах. В настоящее время сфера действия
атомной спектроскопии расширилась и ее используют в нефтехимичес-
кой, химической и других отраслях промышленности.
Метод атомной спектроскопии основан на использовании спектров
излучения или поглощения атомов в области УФ-, видимого и ИК-диапа-
зона длин волн. Кванты энергии такого излучения вызывают возбужде-
ние, так называемых, оптических или валентных электронов*.
Атомные спектры представляют собой набор отдельных линий.
Положение линий в спектре и их интенсивность обусловлены местом
атома в периодической системе элементов, т. е. числом валентных элек-
тронов и зарядом ядра.
Спектры атомов индивидуальны. Даже если некоторые элементы,
присутствующие в исследуемой пробе, имеют случайно совпадающие по
частоте линии, каждый элемент имеет характерные для него линии,
которые позволяют его идентифицировать. Это обстоятельство и лежит
в основе атомного спектрального анализа. В отличие от молекулярного
спектрального анализа, который сохраняет образец неизменным, атомная
спектроскопия является разрушающим методом. Анализируемый обра-
зец подвергается соответствующей обработке и переводится в атомарное
состояние. Это достигается, как правило, термическим воздействием в
пламени, угольной дуге, искре.
Кроме традиционного эмиссионного спектрального анализа, основан-
ного на исследовании спектров испускания вещества, успешно приме-
няется и развивается метод атомно-абсорбционной спектроскопии.
Если через атомарный пар пропустить излучение тех длин волн, которые
соответствуют переходу атома в возбужденное состояние, то излучение
будет поглощаться. Исследование этого поглощения и составляет содер-
жание метода атомно-абсорбционного анализа.
По сравнению с молекулярной спектроскопией, область приложения
которой при исследовании полимеров многообразна, метод атомной
* Для возбуждения внутренних электронов атома нужна энергия, соответствую-
щая рентгеновскому излучению.
19
спектроскопии имеет ограниченное применение. В основном использует-
ся эмиссионный анализ для определения металлов и других элементов,
остающихся в полимерах в виде примесей после полимеризации (остат-
ков катализаторов, инициаторов и т. п.).
Практически отсутствуют работы с привлечением метода атомной
абсорбции к анализу полимеров. Между тем, он заслуживает большего
внимания. Анализ неорганических примесей в полимерах несущественно
отличается от анализа других объектов этим методом, если полимер
подвергнут озолению. Кроме того, возможен анализ непосредственно
растворов полимеров.
Атомная спектроскопия может найти применение, например, при
исследовании химической модификации полимеров, связанной с введе-
нием в цепь макромолекул элементов, отсутствующих в исходном поли-
мере, и позволит при этом следить за процессом модификации.
Путем анализа промывных вод можно следить за чистотой промыв-
ки полимера от катализаторов, эмульгаторов и других компонентов
реакционной смеси, содержащих атомы металлов или неметаллов, а
также контролировать наличие этих примесей непосредственно в
полимере.
II. ЭКСПЕРИМЕНТАЛЬНАЯ ТЕХНИКА СПЕКТРАЛЬНОГО
АНАЛИЗА ПОЛИМЕРОВ
П.1. МОЛЕКУЛЯРНЫЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ
П.1.1. Выбор аналитической полосы поглощения
и измерение ее интенсивности
Необходимым условием молекулярного Спектрального анализа служат
правильно выбранные аналитическая полоса поглощения и толщина
поглощающего слоя, обеспечивающие необходимую чувствительность
анализа и его точность. Наиболее благоприятно наличие в спектре изо-
лированной аналитической полосы поглощения. Для определения ее
интенсивности проводят базисную линию — обычно это прямая, сливаю-
щаяся на краях полосы с фоном поглощения (рис. 11.1, а). В спектрах
полимеров и других соединений наличие изолированной полосы является
скорее исключением, чем правилом. В большей или меньшей степени
полосы поглощения налагаются друг на друга. При наличии двух или
более перекрывающихся полос можно провести общую базисную линию
(рис.11.1, в), но следует убедиться в том, что полосы поглощения, со-
седние с аналитической, не относятся к какому-либо переменному ком-
поненту, например, примеси, концевым группам и т. п. Различные ва-
рианты базисных линий даны на рис. II. 1. После выбора базисной линии
необходимо соблюдать однозначность ее проведения в спектрах анали-
зируемых образцов.
Измерение интенсивности полосы поглощения выполняют следую-
щим образом. Через точку В (см. рис. П.1), соответствующую минимуму
пропускания в полосе, проводят перпендикуляр к линии нулевого про-
пускания (точка С). За I принимают отрезок ВС. За 70 - отрезок А С,
равный расстоянию от нулевой линии (точка С) до пересечения пер-
пендикуляра с базисной линией (точка А). Отношение величин этих
отрезков (в процентах) дает значение коэффициента пропускания Т.
Базисная линия дает возможность учесть фон поглощения, связанный с
рассеянием света и его отражением от поверхностей образца. Кроме
того, при наложении перекрывающихся полос проведение базисной ли-
нии до некоторой степени учитывает вклад поглощения соседних полос.
Интенсивная аналитическая полоса не всегда оказывается пригод-
ной для анализа. Большая интенсивность поглощения обеспечивает вы-
сокую чувствительность определения какого-либо компонента или
структурного элемента полимера. Вместе с тем, она может.значительно
уменьшить точность анализа. Так, если исследуют полимер в виде плен-
21
Рис. ПЛ. Различные варианты базисной линии, совпадающей с фоном поглощения
(а), совпадающей с фоном поглощения с одной стороны группы полос (б) и каса-
тельной к кривой поглощения в минимумах (в, г).
ки, то чем выше интенсивность полосы поглощения, тем меньшая тол-
щина пленки требуется для проведения анализа. А это, как следствие,
увеличивает погрешность за счет точности измерения толщины пленки*.
С другой стороны, определение интенсивности проводится в этом случае
при малом пропускании, что также значительно увеличивает погреш-
ность. Поэтому интенсивность аналитической полосы должна быть
оптимальной, чтобы соответствовать условию наибольшей точности ана-
лиза. Таким условиям удовлетворяет интервал пропускания (Z/Zo) от
20 до 70%. Вне этого интервала погрешность возрастает. В связи с этим
иногда выгоднее использовать в качестве аналитической полосу мень-
шей интенсивности, даже если она не является изолированной.
Для уменьшения погрешности анализа, связанной с измерением
абсолютной интенсивности полосы поглощения, можно воспользоваться
отношением интенсивностей поглощения двух полос, выраженных в
оптических плотностях (Pi/D2). Одна из полос является аналитической.
Допустим, что ее интенсивность Di. В простейшем случае второй поло-
сой — полосой сравнения, может служить полоса спектра, принадлежа-
щая компоненту, концентрация которого в исследуемых образцах неиз-
менна („внутренний стандарт”). При определении состава сополимера
можно использовать отношение интенсивностей полос сомономеров. В
этом случае поглощение в максимуме полосы сравнения меняется в
зависимости от содержания анализируемого компонента.
Использование отношения интенсивностей полос поглощения исклю-
чает из расчетов значение толщины поглощающего слоя и, как правило,
увеличивает точность определений искомой величины.
Чтобы определить концентрацию анализируемого компонента,
строят градуировочный график - зависимость отношения интенсивностей
полос поглощения Di/D2 от его концентрации в эталонных образцах.
При использовании внутреннего стандарта, градуировочный график имеет
вид прямой линий (при соблюдении закона Бугера—Ламберта—Бера).
* Для измерения поглощения в области основных тонов обычно нужна пленка
полимера толщиной около 10 мкм и даже меньше. Часто равнотопшинную пленку
такой толщины изготовить трудно.
22
Рис. П.2. Наложившиеся полосы
индивидуальных веществ А и В (а), В
спектры индивидуальных веществ
А (б) и В (в).
Если интенсивность полосы
сравнения изменяется при изме-
нении О], то зависимость будет
нелинейная. Нелинейность графи-
ка не мешает его использовать
для анализа. Необходимо иметь
достаточное число эксперимен-
тальных точек для его досто-^
верности.
Выбор базисной линии влия-
ет на численное значение интен-
сивности аналитической поло-
сы, которое может быть или
частично завышено от вклада
поглощения соседних полос (см.
рис. II. 1. б), или занижено
а
(см. рис. II. 1, г). В этих случаях £
продолжение градуировочной в
кривой будет пересекать ось
ординат выше или ниже начала
координат. Такие градуировоч-
ные графики могут быть исполь-
зованы для анализа. Следует лишь учесть, что погрешность определения
малых концентраций при этом будет увеличена. Если интенсивность по-
лосы не искажена при ее измерении, то градуировочная кривая проходит
через начало координат.
Для определения интенсивности наложившихся полос поглощения с
близкими частотами можно пытаться проводить разделение спектра на
отдельные полосы. Разделение осуществляют графически, что является
трудоемким процессом, или используют ЭВМ*.
Для серийных анализов метод разделения полос поглощения не
всегда приемлем. В простых случаях, если известны слагаемые суммар-
ного контура и число наложившихся полос мало, можно обойтись без
полного разделения полос.
Рассмотрим в качестве примера случай, когда сложная полоса спектра пред-
ставляет собой наложение двух полос, относящихся к индивидуальным веществам
А и В (рис. П.2, а). Тогда оптическую плотность сложного контура D(y) можно
представить в виде:
D{v) = d [Сд6а(р) + Cb6b(p)J (II. 1)
*Вопросу разделения полос поглощения посвящен обзор [3].
23
где САи Св- молярные концентрации индивидуальных веществ; еА(г) и «в О') -
их молярные показатели поглощения.
Если еА(р) и ев (у) известны, то, написав уравнение (II.1) для двух значений
волновых чисел к, и v2, получим два уравнения для двух неизвестных СА и Св-
Решая эти уравнения сов местно, найдем:
с ев(^2) - Р(рг)ев(Р1)
(бд(Р1) бв(р2)~ев("1)еА(р2)] d
с Д(р2)^а(г>1) - Д(^1) еА(р2)
В [вд(У1)ев(ь’2)-бВ(р1)еА(р2^
В качестве vt и v2 удобно брать волновые числа, соответствующие максиму-
мам полос поглощения компонент А и В (см. рис. II.2), т. е. v
Отношение
1 = РА’
»2
"В-
са = р(ь’а) ев(^в) ~ Д(ь'в) ев(рА)
Св D(i>B')eA(vA)-D(vA) tA(pB)
(П.2)
можно переписать в виде:
СА D(i>a)-xD(vb)
CB D(vB)~ yD(uA) Z
_ e В ("a) _ 6A ("B) e В (рв)
где x------——У - -—-——; г - --—-——; - параметры, которые определяют
ев(|'в) 6 А ("а) са(^а)
из спектров индивидуальных веществ (см. рис. П.2, бив).
Разделив числитель и знаменатель формулы (П.З) на О(гв), получим:
Д(г’а) _
сА р(рв) •*
с в О(1*а)
у Г»(рв)
откуда можно определять отношение концентраций СА/СВ исследуемых соедине-
ний, не производя разделение наложившихся полос поглощения на Отдельные. Для
этого нужно измерить отношение оптических плотностей О (г’д)/О (гв) по экспери-
ментальному контуру (см. рис. II.2, а) и использовать предварительно определен-
ные параметры х, у иг.
Если форма и положение наложившихся полос неизвестны, то раз-
деление спектра, как правило, неоднозначно.
Рассмотрим пример. На рис. П.З, а представлены спектры поглощения двух
условных веществ (1 и 2) и их суммарный контур (3), полученный путем графи-
ческого сложения интенсивностей поглощения спектров (1 и 2). Предположим,
что надо решить обратную задачу - разделить спектр (5) на составляющие полосы,
форма и число которых неизвестны. Для выполнения этой работы проводим деле-
ние, варьируя параметры полос до достижения совпадения суммарного контура со
спектром (5) (рим. П.З, б). В качестве одного из вариантов получаем „индивиду-
альные” полосы (4, 5, 6). Сравнение с реальными спектрами (1 и 2) убеждает в
неправильно проведенном разделении. Если бы в процедуру разделения спектра
были бы заложены данные о форме и положении индивидуальных полос, то ре-
зультат разложения был бы ближе к действительности.
24
Рис. П.З. Разделение полос поглощения:
а — спектры индивидуальных веществ 1 и 2 и их графическое сложение 3\ б -
вариант разделения контура 3 на отдельные полосы.
т
-297J. , _з________
3000 2950 2900 2850
V, см~’
К сожалению, часто не удается получить такие данные для отдельных
перекрывающихся полос, составляющих сложный спектр. Особенно это
касается спектров полимеров. Тогда определение спектральных характе-
ристик перекрывающихся полос можно пытаться выполнить, например,
путем изменения состава исследуемого образца.
Так, на рис. II.4 представлены спектры поглощения сополимера
тетрафторэтилеиа с этиленом, содержащего разное количество звеньев
этилена. Полоса валентного колебания группы —СН2— под влиянием
атомов фтора смещается к большим частотам (2973 см-1) по сравнению
с полосой того же колебания по-
лиэтилена (2926 см"1). Полоса по-
глощения 2973 см-1 (рис. П.4, 7) в
спектре сополимера с молярной
долей этилена около 30%, когда
практически все этиленовые звенья
находятся в окружении звеньев
тетрафторэтилеиа, имеет практи-
чески „чистый” контур. При обо-
гащении сополимера этиленом по-
является вероятность присоедине-
ния нескольких звеньев этилена
подряд и в спектре наряду с поло-
сой 2973 см"1 появляется полоса
2950 см"1, значение волнового
Рис. П.4. ИК-спектры поглощения со-
полимера тетрафторэтилеиа с этиленом.
Молярное содержание звеньев этилена
(в %) : / - 30; 2 — 50; толщина пленки
(в мкм ) : I - 100; 2 - 40.
25
числа которой ближе к его значению в спектре полиэтилена (рис. П.4,2) .
Таким образом, чтобы разделить полосы 2973 и 2950, можно использо-
вать „чистый” контур полосы 2973 см"1.
Другую возможность в выявлении контура одной из перекрываю-
щихся полос может дать разная поляризация их колебаний. Тогда исполь-
зование поляризационных спектров облегчит задачу разделения полос
на отдельные.
Поясним принципы получения поляризационных спектров.
Молекулярные колебания, как правило, в той или иной мере поля-
ризованы, т. е. изменение дипольного момента при колебаниях происхо-
дит преимущественно либо вдоль оси молекулы, либо в плоскости
перпендикулярной оси. В исходном образце полимера оси макромоле-
кул обычно хаотически распределены по направлениям, поэтому в целом
нет преимущественной ориентации дипольного момента колебаний. Для
того чтобы такую ориентацию создать, полимерную пленку подвергают
одноосному растяжению. При этом макромолекулы, по крайней мере
частично, ориентируются в направлении растяжения.
Спектр поглощения такой пленки, снятый в поляризованном свете,
и называется поляризационным спектром. Для получения спектра ис-
пользуют свет, поляризованный в двух направлениях; когда вектор
электрического поля в световой волне колеблется по направлению рас-
тяжения и когда этот вектор колеблется в плоскости, перпендикулярной
направлению растяжения. В первом случае полосы, обусловленные ко-
лебаниями, поляризованными вдоль оси молекул, будут иметь увеличен-
ную интенсивность, а полосы, соответствующие колебаниям дипольного
момента в перпендикулярном направлении, - уменьшенную интенсив-
ность. Во втором случае повышается интенсивность полос поглощения,
связанных с теми колебаниями, при которых дипольный момент изме-
няется в направлении перпендикулярном оси молекулы. В зависимости
от степени ориентации полимерной пленки этот эффект будет наблю-
даться в спектре в большей или меньшей степени. Таким образом, поля-
ризационные спектры позволяют разделить полосы, отвечающие коле-
баниям, имеющим различные поляризации.
Примером влияния поляризации света на спектр ориентированной
полимерной пленки служат поляризационные спектры сополимера три-
фторхлорэтилена с этиленом (рис. II.5).
В тех случаях, когда перекрывающие полосы принадлежат разным
компонентам смеси, примеси в полимере и самому полимеру, раствори-
телю и растворенному веществу и т. п., для их разделения применяют
метод вычитания спектров. Например, при анализе примеси в полимере
в рабочий канал спектрофотометра помещают исследуемый образец, а в
канал сравнения — образец чистого полимера. Спектрофотометр ре-
гистрирует пропускание /2//х, где Ц и /2 — интенсивности света, про-
шедшего через испытуемый образец и образец сравнения соответственно.
Оптическая плотность lg/2//i представляет собой разность оптических
плотностей исследуемого образца Dr и образца сравнения Z)2,t. е. Dt-
D2. Зависимость этой разностной оптической плотности от частоты
26
т
1500 1400 1300 1200
v, см-'
Рис. 11.5- Поляризационный ИК-спектр поглощения сополимера трифторхлор-
зтилена с этиленом (d = 14 мкм).
Электрический вектор перпендикулярен (--------) и параллелен (-----)
направлению растяжения пленки.
и представляет собой разностный спектр. В этом спектре полосы, при-
надлежащие полимеру, скомпенсированы и остаются только полосы ана-
лизируемой примеси. Для контроля полноты компенсации используют
изолированную полосу, заведомо принадлежащую веществу, спектр ко-
торого требуется вычесть. Компенсирующий образец изготовляют в
виде клина, чтобы иметь возможность плавно менять толщину поглоща-
ющего слоя в канале сравнения.
Примеры применения метода компенсации см. в разделе III. 1.1.
В этом разделе описаны также применения метода компенсации для
выделения полос, связанных с кристаллическими структурами в
полимере.
Широкие возможности для получения разностных спектров пред-
ставляет соединение спектрального прибора с ЭВМ и, в частности, Фурье-
спектроскопия (см. раздел И. 1.7).
27
Остановимся на понятии „интенсивность полосы поглощения”.
Строго говоря, когда это понятие употребляется в количественном
смысле, то следует иметь в виду интегральную интенсивность, т. е.
площадь под кривой зависимости оптической плотности от волнового
числа. Однако в прикладной спектроскопии обычно используют, как ме-
ру интенсивности, оптическую плотность в максимуме полосы поглоще-
ния Омакс- Такая характеристика практически удобна и часто достаточ-
на для количественного анализа. Это так, если форма полосы в спектрах
образцов анализируемого полимера и эталонных веществ приближенно
одинакова. В качестве характеристики формы полосы обычно использу-
ют ее полуширину Ди,/, т. е. ширину на половине высоты (на уровне
D макс /2).
В некоторых случаях измерение ОМакс оказывается недостаточ-
ным. Например, если эталонами служат низкомолекулярные соединения
(обычно жидкие), то анализируемый полимер приходится плавить,
чтобы приблизить форму полосы поглощения к форме полосы эталона.
Чтобы этого избежать, в работе [4, с.821] (см. также раздел Ш.З) предла-
гается использовать в качестве меры интенсивности не 2)макс, а
-Омаке - величину, пропорциональную интегральной интенсивности.
Эта рекомендация основана на том, что для исследуемых в [4, с. 821]
полос поглощения интегральная интенсивность остается приблизительно
неизменной при плавлении полимера (хотя ОМакс при этом уменьшает-
ся, но Друвеличивается). Однако для полос, исследованных в [5],
оказывается постоянной при плавлении полуширина ДРу2- Чтобы избе-
жать плавления в серийных анализах, в этой работе рекомендуется
измерять D макс для твердых пленок, а при сравнении с эталоном
вводить заранее определенную поправку на изменение интенсивности
полосы при плавлении.
При измерении интенсивности полос поглощения полимера в спект-
ре расплава следует учитывать собственное излучение нагретого полиме-
ра. Этот эффект становится заметным при большой интенсивности по-
лос поглощения. Чтобы исключить его,следует записать спектр расплав-
ленного образца, перекрыв перед ним пучок света от источника спектро-
фотометра, и внести соответствующую поправку в поглощение расплава.
Интегральная интенсивность полос может зависеть от температуры.
В работе [6] исследована эта зависимость для различных типов колеба-
ний линейных полимеров. Показано, что изменение интегральной интен-
сивности может составлять от 1 до.30% при изменении температуры
на 100 °C.
II. 1.2. Приготовление образцов для анализа
Молекулярная спектроскопия позволяет исследовать полимеры в раз-
ных ростояниях: твердом (пленки, порошки, волокна), жидком (раст-
воры), вязкотекучем (расплавы), газообразном (низкокипящие про-
дукты пиролиза полимеров). Способ приготовления образцов опреде-
ляется поставленной задачей и свойствами полимера.
28
Пленки. Полимерные пленки готовят прессованием из расплава,
поливом из раствора, отверждением между окнами кювет, спеканием
из суспензии и т. п. Условия приготовления пленки определяют фор-
мирование различных надмолекулярных структур, влияют на конфор-
мационные переходы, кристаллизацию полимеров, ориентацию макро-
цепей. Поэтому спектры пленок, полученных разными методами, могут
иметь отличия. Следовательно, проводя подготовку образца к молеку-
лярному спектральному анализу, необходимо учитывать возможность
изменения спектра. В большой мере зто относится к кристаллизующимся
полимерам. Так, в зависимости от скорости охлаждения пленки поли-
этилентерефталата из расплава можно получить полимер в аморфном
или кристаллическом состояниях. Причем переход из аморфного состоя-
ния в кристаллическое сопровождается гош-граис-конформационным
переходом.
Пленки полимеров, полученные экструзией через плоскощелевую
головку, могут оказаться ориентированными в продольном направле-
нии. Рукавная пленка является двухосно ориентированной. Пленка то-
го же полимера, приготовленная горячим прессованием, как правило,
изотропна. При формовании полимеров из расплава, если их темпе-
ратура плавления близка к температуре разложения, возможны про-
цессы термо- или термоокислительной деструкции, которые также
находят отражение в спектре.
Если нет особых требований к качеству пленки, ее однородности по
толщине, что часто бывает при качественном анализе, то пленку из рас-
плава полимера можно быстро получить без специальных прессов и
пресс-форм. Для этой цели удобно использовать металлический держа-
тель образцов, прилагаемый к спектрофотометру. Образец полимера в
виде порошка или мелко нарезанных кусочков зажимается между дву-
мя стеклянными полированными пластинками толщиной 3—5 мм
(ГОСТ 7132—78 „Стекло листовое термически полированное”) - По-
скольку большинство полимеров имеют высокую адгезию к стеклу
и прессованные пленки плохо от него отделяются, поверхность стеклян-
ной пластинки покрывают пленкой(около 20 мкм толщиной) политетра-
фторэтилена (фторопласта-4), которая играет роль подложки.
Держатель с образцом в собранном виде нагревают. Во избежание
растрескивания стекла должны быть отделены от металла фторопласто-
вой или другой термически стойкой прокладкой. Постепенно по мере
просветления полимера при нагревании сжимают металлическую опра-
ву держателя, завинчивая гайки и раздавливая расплав полимера.
Стеклянные пластинки позволяют наблюдать за плавлением полиме-
ра и образованием пленки. После охлаждения держатель разбирают
и пленку образца отделяют от фторопластовой подложки. Для облег-
чения отделения пленки подложку иногда смачивают водой. Если прес-
сованная пленка не отделяется от подложки, то в большинстве случаев
можно получать спектр пленки вместе с подложкой, так как фторо-
плаСт-4 имеет большую область прозрачности в ИК-области. Вместо
стекла можно воспользоваться окнами из КВг или NaCl. Они менее
29
прочны, но при этом не требуется применения подложки и отделения
пленки, поскольку окна прозрачны для ИК-излучения. Условия для
получения пленки нужной толщины подбирают эмпирически, варьируя
количество образца и давление между окнами. Температуру нагрева
образца можно контролировать термопарой, просверлив отверстие в
окне или стеклянной пластинке.
Для получения полимерных пленок из термопластичных полимеров
в лабораторных условиях можно также использовать простое приспо-
собление, изготовленное из двух навинчивающихся друг на друга полых
металлических цилиндров. В специальный паз торцевой поверхности
наружного цилиндра вставляют окна из толстого стекла или галогенидов
щелочных металлов, между которыми помещают образец. Все устройст-
во нагревают в термостате до температуры плавления полимера. Ввинчи-
ванием внутреннего цилиндра, играющего роль пуансона, сдавливают
окна до образования между ними пленки полимера.
Для получения пленок путем спекания суспензии или прессования
полимеров часто используют в качестве подложки алюминиевую или
медную фольгу, которую затем растворяют. Для некоторых полимеров,
например химически стойких фторопластов, эта процедура не должна
повлиять на их химическую структуру. Однако в других случаях при
растворении фольги в концентрированных щелочах (алюминий) или
кислотах (медь) может измениться структура полимера, а следователь-
но, и его спектр. Мы предпочитаем при получении пленок для спектраль-
ного анализа как можно меньше подвергать полимер химической обра-
ботке, если это не является самим предметом исследования.
Из растворимых полимеров пленки получают поливом раствора на
разные подложки с последующим испарением растворителя. Удобно ис-
пользовать в качестве подложки, особенно для получения тонких (в
несколько микрометров) пленок, кюветные окна, прозрачные в анали-
тической области спектра. В качестве окон в зависимости от спектраль-
ного диапазона применяют стекло, кварц, фторид лития, фторид каль-
ция (флюорит), галогениды щелочных металлов (NaCl, КВг, КС1) и др.
Границы прозрачности этих металлов даны в Приложении I. Большая
гигроскопичность NaCl, КВг, КС1 не позволяет использовать окна из
этих кристаллов для работы с растворителями, содержащими воду.
Чтобы растворитель не стекал с окна, можно использовать в качест-
ве ограничителя стеклянные или металлические кольца. Очистить окно
от пленки можно ее растворением. Окна из КВг или NaCl часто дополни-
тельно шлифуют и полируют (см. разд. II. 1.4). В тех случаях, когда
требуется получить пленку, свободную от подложки, раствор отливают
на стекло* с ограничительным кольцом, например из металла с поли-
рованными поверхностями. Надо следить, чтобы раствор не подтекал
под кольцо.
Удобны для получения пленок из раствора чашки Петри биологи-
ческие. Они имеют полированную внутреннюю поверхность. Снять плен-
♦Можно воспользоваться отмытой от эмульсии фотопластинкой.
30
ку со стекла легче под слоем воды, слегка отделив края пленки лезвием
бритвы. Однако пленки толщиной 2—5 мкм даже при плохой адгезии по-
лимера к стеклу трудно от него отделяются. Полезно при этом использо-
вать способ приготовления пленок, применяемый авторами.
На стеклянную пластинку, смоченную водой, накладывают пленку
политетрафторэтилена толщиной 15—20 мкм. Пленку плотно притирают
к стеклу („приклеивают”), удаляя избыток воды безворсовой тканью.
Чтобы пленка не отставала от стекла, ее края по периметру смазывают
узкой полоской эмульсии поливинилацетата, которую высушивают.
Однако эта процедура требуется в редких случаях. Как правило, пленка
прочно удерживается на стекле. На поверхность фторопластовой пленки
наносят раствор полимера. Горизонтальность подложки, обеспечиваю-
щую равнотолщинность получаемой пленки, контролируют уровнем.
Такой способ использовали для приготовления пленок толщиной 2—5 мкм
из водных растворов поливинилового спирта, поливинилпирролидона и
сополимеров винилпирролидона, из раствора поливинилбутираля в сме-
си этилового спирта и бензола, а также из растворов некоторых фторо-
пластов в диметилформамиде. Массовая доля полимеров в растворе
составляла 0,5 —1,0%. Плохая смачиваемость политетрафторэтилена
водой способствует тому, что водный раствор полимера растекается на
определенную площадь и ограничителя не требуется. Хотя смачиваемость
политетрафторэтилена органическими растворителями выше, чем водой,
тем не менее растворы полимеров в них также удерживаются на пленке
фторопласта.
Обычно пленки, получаемые из растворов, равнотолщинные. Отли-
чия в толщине, обусловленные поверхностным натяжением раствора,
могут наблюдаться на краях пленки. Это в большей мере зависит от
концентрации раствора и особенно заметно для пленок малой площади.
При отливе пленок большой площади всегда можно выбрать равнотол-
щинный участок, необходимый для получения спектра.
Обычно пленку толщиной несколько микрометров можно получить
при концентрации порядка 1%. Однако условия растекания раствора,
а значит и толщина пленки, зависят также и от типа растворителя. Поэ-
тому в каждом случае условия приготовления пленки из раствора следует
отрабатывать эмпирически. Можно подобрать оптимальную для анализа
толщину, накладывая тонкие пленки друг на друга. Для количественно-
го анализа такой прием желательно не использовать, так как за счет
отражения излучения от поверхностей пленок возрастает общий фон
поглощения спектра, что может увеличить погрешность измерения.
Подбирая растворитель для приготовления пленки, необходимо
учитывать его температуру кипения, способность адсорбировать влагу,
его воздействие на полимер*.
Летучесть растворителя иногда определяет возможность его исполь-
зования при отливе пленок для спектрального анализа. Легко летучие
* Имеется в виду не химическое воздействие, которого не должно быть при
приготовлении пленки, а влияние растворителя на конформационные переходы,
условия кристаллизации и т. п.
31
растворители хорошо удаляются из пленки при комнатной температуре.
Однако их быстрое испарение может привести к получению пленки с не-
ровной поверхностью и появлению пузырей. Для устранения этих дефек-
тов следует уменьшить скорость испарения растворителя, покрыв, на-
пример, пленкообразующий раствор стеклянным колпаком. При этом
испарение растворителя замедляется, пленки получаются качественными.
Иногда высококипящие растворители плохо удаляются из пленки. Та-
ким растворителем, часто используемым в практике спектральных ис-
следований, является диметилформамид, который растворяет большин-
ство полимеров. Хорошие пленки из растворов в диметилформамиде
ряда фторопластов, ацеталей поливинилового спирта и других полиме-
ров можно приготовить по следующей методике. Растворение проводят
при температуре 50-60 °C. Раствор наносят на поверхность подложки
из стекла или политетрафторэтилена, находящихся при той же темпера-
туре. После образования пленки температура в термостате повышается
до 100 °C. Это обеспечивает полное удаление диметилформамида из
пленок толщиной 10—20 мкм в пределах 2 ч прогрева.
Следует иметь в виду, что увеличение температуры раствора полиме-
ров в диметилформамиде может привести к их деструкции, которая
сопровождается появлением окраски раствора. Так, поливинилиден-
фторид в растворе диметилформамида при 100 °C подвергается деги-
дрофторированию. Раствор окрашивается в красные тона. Спектр поли-
мерной пленки, полученный из такого раствора, указывает на химичес-
кие превращения в поливинилиденфториде [7].
Температура прогрева полимерной пленки для удаления растворите-
ля должна определяться стабильностью полимера. Полноту удаления
растворителя из пленки проверяют по отсутствию его полос поглощения
в спектре пленки*. Иногда пленку не удается освободить полностью от
растворителя. В этом случае следует учитывать наличие его полос в спек-
тре при выборе аналитической области.
Для получения пленок используют также смеси растворителей. Хо-
рошие пленки при этом образуются из смеси азеотропного состава. При
отклонении смеси от азеотропной и большой разнице ее компонентов в
скоростях испарения возможны такие условия пленкообразования, что
полимер высаждается и пленка может не образоваться. По-видимому,
этим можно объяснить образование мутных пленок поливинилбутира-
ля из смеси растворителей спирт-вода и прозрачных — из раствора в
диметилформамиде, содержащем во^у. Испарение растворителя прово-
дилось при комнатной температуре.
Ацетон, хорошо адсорбирующий влагу, обладает большой летучестью.
Пленки поливинилиденфторида, отлитые из раствора в ацетоне на откры-
той поверхности при комнатной температуре, но в вытяжном шкафу,
ускоряющим испарение растворителя, оказываются белыми, непрозрач-
ными и непрочными. Из того же раствора можно приготовить хорошую
прозрачную пленку поливинилиденфторида, если раствор нагреть до
* Спектры поглощения наиболее употребляемых растворителей даны
в Приложении IV и V.
32
40 °C и отливать пленку в термостате на подложку при этой же темпера-
туре. Таким образом, из одного и того же раствора можно отлить разные
по качеству пленки. Получение неударной пленки не дает еще основание
браковать растворитель, необходимо при этом поварьировать условия
пленкообразования, которые зависят не только от растворителя, но и от
свойств полимера.
Для исследования сшитых полимеров, например вулканизован-
ных каучуков, готовят пленки из раствора исходного каучука и за-
тем их подвергают вулканизации. ’ Аналогичным путем получают
пленки полимеров, сшивание которых достигается облучением, термо-
обработкой.
Сшитые полимеры, образующиеся путем взаимодействия низкомо-
лекулярных соединений с отвердителями, полимеризуют между окнами.
При этом по спектру можно судить о глубине конверсии. Толщина по-
глощающего слоя задается прокладкой. Окна после употребления исполь-
зовать вторично практически не удается. Для их предохранения можно
применить защитную пленку из политетрафторэтилена или эластичную
пленку ФУМ (ТУ 6-05-1383—80), которая легко натягивается на окно
и хорошо удерживается на нем.
Большая библиография, включающая работы по методам приготов-
ления пленок разных полимеров, содержится в книге [8, с.46] и обзор-
ной статье [9].
Порошки. Нерастворимые или неплавкие полимеры можно исследо-
вать в виде порошка, растертого с иммерсионной средой. В качестве им-
мерсионной среды используют вазелиновое или фторуглеродное (пер-
фторкеросин) масла, а также галогениды щелочных металлов (NaCl,
КС1, КВг). В первом случае суспензию полимера в масле помещают меж-
ду окнами, растирают, чтобы удалить пузырьки воздуха, и раздавливают
до нужной толщины. Во втором, порошок смешивают с галогенидами и
смесь таблетируют.
Поскольку полимерный порошок в иммерсионной среде представля-
ет собой оптически неоднородную систему, то большое значение приобре-
тает рассеяние света. Как известно, рассеяние света на неоднородностях
показателя преломления зависит как от размеров частиц, их формы,
так и от показателя преломления среды и порошка.
Комбинируя фторуглеродное и вазелиновое масла, имеющие поло-
сы поглощения в разных частях спектра, можно охватить большую
спектральную область при получении спектров порошков. Практически
выбор жидких иммерсионных сред ограничивается этими маслами. А
поскольку для многих полимеров показатели преломления в большей
или меньшей степени отличаются от показателей преломления масел, то
рассеяния света, связанного с различиями показателей преломления
порошка и иммерсионной среды, не избежать. Тем большее значение
приобретает размер частиц порошка. Используемый на практике диапа-
зон ИК-излучения (средняя ИК-область) охватывает длины волн от
2,5 до 50 мкм. Рассеяние света на одних и тех же оптических неоднород-
ностях в разных интервалах этого диапазона будет различаться, поскольку
33
Рис. П.6. Искаженный ИК-спектр поглощения порошка сополимера тетрафтор-
этилена с этиленом в таблетке КВт.
оно зависит от длины волны. Чтобы рассеяние света было мало, размер
частиц должен быть меньше X.
Существует еще одно ограничение на размер частиц. Порошок не
должен содержать частиц, размер которых превышал бы эффективную
толщину, необходимую для проявления наиболее интенсивных полос
поглощения спектра. Для большинства полимеров это слой толщиной от
одного до десятка микрометров. Если порошок неоднороден и содержит
частицы, не пропускающие излучение, спектр будет некачественным.
Это можно заметить по искаженной форме интенсивных полос погло-
щения, имеющих широкие тупые вершины (рис. II.6). Они могут сли-
ваться в линию сплошного поглощения, не соответствующую нулевому
пропусканию. Таким образом, размер частиц полимерного порошка для
получения хорошего спектра в широком диапазоне длин волн должен
быть порядка 1 мкм йли меньше.
Спектр может иметь искаженный вид и в том случае, если мелко-
дисперсный порошок плохо размешан с иммерсионной средой. Поэтому
вторым непременным условием получения хорошего спектра является
тщательное перемешивание образца с иммерсионной средой до получе-
ния однородной массы. Частицы порошка должны быть равномерно
распределены по толщине слоя образца. Если это условие не соблюдает-
ся, то на пути пучка света могут попадаться участки иммерсионной
среды без полимера и, наоборот, участки слипшихся частиц, дающих
сплошное поглощение.
Галогениды щелочных металлов (КВг, NaCl, К.С1) под давлением
сплавляются в прозрачную таблетку. Смешанный с ними измельченный
порошок полимера, распределенный по всей массе таблетки, как бы
находится в „твердом растворителе”, или матрице. Таблетирование
полимерного порошка предпочтительнее смешения их с жидкой иммер-
сионной средой, так как таблетка может быть сохранена для воспроиз-
34
ведения спектра и использована для количественного анализа. Однако
приготовить суспензию порошка в масле проще и быстрее, особенно
когда нужно дать качественную характеристику образца. Имея свои
достоинства и недостатки, оба метода исследования порошкообразных
материалов находят применение.
Из галогенидов щелочных металлов для таблетирования наиболее
употребителен КВг. Показатели преломления КВг (1,55) и NaC.l (1,53)
близки к показателям преломления большинства полимеров, так что
в этом отношении они взаимозаменяемы. Однако КВг по сравнению с
\аС1 обладает большей областью прозрачности для ИК-излучения и
требует меньших давлений при таблетировании. По показателю прелом-
ления КС1- (1,43) также подходит в качестве матрицы для ряда полиме-
ров, но он реже используется из-за сложных условий спекания (высокое
давление и повышенная температура).
В качестве основы для таблеток КВг используют кристаллы или
порошок особой степени очистки (ТУ 6-09-476 70). Бромид калия
должен быть измельчен. Оптимальный размер частиц порошка 150 мкм
[8, с.47]. Измельчение проводят ручным способом* или при помощи
вибромельницы. Размер частиц порошка влияет на качество таблетки.
Тонко размолотый порошок способствует увеличению адсорбции влаги
воздуха, в результате в его спектре появляются полосы поглощения
воды (3200—3600 см"1 и 1620 см-1). Удалить воду из порошка можно
длительным его прогреванием (около 20 ч) при температуре 150—200 °C
или в течение 2 ч при 500 °C. При систематической работе с таблетками
КВг желательно хранить его постоянно в термостате, отбирая нужное
количество для приготовления таблеток. Остужать порошок КВг до
комнатной температуры необходимо в эксикаторе над СаС12. Перед
приготовлением таблеток КВг с полимерным порошком необходимо
проверить КВг на присутствие влаги или других примесей, сняв спектр
его таблетки без полимера. Примеси из КВг удаляют прокаливанием
его в муфельной печи в течение нескольких часов при температуре 500 °C
и выше. Если прокаливанием не удается освободить КВг полностью от
примесей, то в канал сравнения спектрофотометра для компенсации их
поглощения можно ввести таблетку КВг без полимера.
Прессование таблеток осуществляется под давлением. Если спектро-
фотометр не снабжен прессом и приспособлениями для таблетирования,
то можно использовать любой гидравлический пресс, обеспечивающий
давление до 700 МПа. Для получения таблеток применяют специальные
пресс-формы [10]. Сведения о технике работы с КВг можно найти также
в работах [8. с.46; 11].
Прозрачность таблетки зависит от применяемого давления и време-
ни выдержки под ним. Хорошие результаты дает таблетирование при
давлении 700 МПа и выдержке от нескольких десятков секунд до 2 мин
* Растирание порошка в фарфоровой ступке может вызвать его загрязнение
частицами фарфора, что можно заметить по появлению в спектре КВг полосы
1100 см-1. Желательно использовать агатовую ступку.
2*
35
или 20-30 мин при давлении около 150 МПа. Для предотвращения
адсорбции влаги прессование проводят под вакуумом. Это усложняет
работу, особенно при проведении текущих анализов. Однако таблетиро-
вание КВг можно проводить и без вакуумирования пресс-формы. Боль-
шое значение при этом имеет влажность помещения, в котором произ-
водится работа.
Способ приготовления полимерного порошка, тщательность его
смешения с галогенидами влияет на качество получаемой таблетки и
спектр образца. Измельчение порошка полимера осуществляется теми
же средствами, что и измельчение КВг. При обработке в вибрационной
мельнице полимер подвергается более сильным механическим воз-
действиям, чем при растирании в ступке, поэтому измельчение может
сопровождаться механодеструкцией, нарушением надмолекулярных
структур и другими изменениями, хотя нет гарантии, что такие измене-
ния не произойдут и при ручном измельчении полимера. Если есть подо-
зрение на изменение структуры полимера, то следует сравнить его спектр
со спектром полимерного образца, приготовленного другим методом,
или поварьировать условия измельчения полимера и пытаться уловить
различия в спектре. Механодеструкция может быть обнаружена по по-
явлению полос поглощения кислородсодержащих групп, поскольку она
часто связана с процессами окисления полимеров.
Эластичные полимеры предварительно охлаждаются до хрупкого
состояния. Весь последующий процесс измельчения и смешения с иммер-
сионной средой должен проходить в присутствии инертного газа или
под вакуумом во избежание конденсации воды на порошке. Для серий-
ных анализов этот способ приготовления образцов мало пригоден. По-
рошок твердых поликонденсационных полимеров приготавливают пу-
тем истирания куска полимера мелким напильником с последующим
измельчением в шаровой мельнице или ступке.
Хорошее качество таблетки обеспечивается правильно выбранной
концентрацией исследуемого вещества. Оптимальная массовая доля
составляет 0,1—0,5%. Увеличивать содержание свыше 1% нельзя, так
как ухудшается качество таблеток. Если концентрация вещества не-
достаточна и спектр имеет малоинтенсивные полосы, то следует увели-
чить толщину таблетки.
В зависимости от размеров таблетки, которые определяются пло-
щадью пучка света спектрофотометра, на ее приготовление требуется от
300 мг до 2 г порошка КВг. Так, если 1 мг вещества смешать с 299 мг
КВг (концентрация 0,33%), то таблетка при диаметре 13 мм будет иметь
толщину 1 мм. Зная плотность КВг (2,75 г/см3), можно легко рассчи-
тать его количество, необходимое для получения таблетки заданного
диаметра и толщины. Когда исследуемого вещества меньше 1 мг (0,1-
0,05 мг), то при прессовании не следует распределять его по всей по-
верхности таблетки. Перемешав с КВг, образец распределяют с учетом
ширины пучка спектрофотометра так, чтобы он оказался на малом
участке в центре таблетки, и ограничивают этот участок диафрагмой или
прямоугольной рамкой.
36
При интерпретации спектра полимера, запрессованного с КВг, надо
иметь в виду, что истинный спектр образца может быть искажен. Это
связано с изменением структуры полимера в процессе измельчения,
возможным химическим взаимодействием между КВг и полимером,
адсорбцией влаги и др. Особенно важно это учитывать при исследовании
образца, спектр которого неизвестен. В большинстве случаев, когда
отработан метод таблетирования испытуемого полимера с КВг, спектры
образцов в виде таблеток и пленок одинаковы (см. рис. П.6 и П.7).
Растворы полимеров. Возможность использования спектров раство-
ров полимеров для аналитических целей зависит от собственного погло-
щения растворителей. В ИК-области спектра большинство растворителей,
как и других органических соединений, имеет интенсивные полосы по-
глощения при малой толщине поглощающего слоя. Увеличение толщины
слоя приводит к проявлению малоинтенсивных полос обертонных и
составных частот. Это усложняет спектр и ограничивает использование
растворов полимеров в аналитической работе. В ИК-спектроскопии
наиболее употребительным растворителем является тетрахлорметан.
Удобен для этой цели также сероуглерод. В ряде случаев можно подо-
брать растворитель с „окном прозрачности” в области аналитической
полосы. Для УФ-области спектра выбор растворителей богаче. Например,
предельные у гл еводороды .циклогексан .спирты, вода и другие со единения
1600 1500 1400 1500 1200 1100 1000 900 800 700
У,См'г
Рис. П.7. ИК-спектр поглощения образца сополимера тетрафторзтилеиа с этиленом:
/ — порошок сополимера, запрессованный с КВг (0,015 г полимера и 2 г КВг);
2 — прессованная пленка толщиной 12 мкм.
37
прозрачны для того диапазона УФ-излучения, который обычно исполь-
зуют в аналитических целях*.
При работе с растворителями важно знать, какие загрязнения наибо-
лее вероятны для данного растворителя и в какой области спектра они
поглощают. Особенно существенно это для УФ-спектроскопии. Так, если
неполярный растворитель загрязнен примесью, обладающей большим ди-
польным моментом, то такая примесь может повлиять на значение пока-
зателя поглощения анализируемого вещества. Прозрачность растворите-
лей в УФ-области спектра позволяет использовать большие толщины по-
глощающего слоя, а это существенно при определении малых количеств
примесей, добавок и т. п. Использование растворов выгодно, так как
исключает операцию отлива пленок и тем самым экономит время анализа.
Это особенно важно, когда велика интенсивность аналитической полосы
и для получения спектра требуются тонкие пленки.
Продукты пиролиза. В настоящее время для идентификации поли-
мерных материалов получил распространение метод пиролиза. Разлагая
полимер при высоких температурах и анализируя образующиеся низко-
молекулярные соединения, можно извлечь полезную информацию о его
составе и строении. Линейные полимеры при пиролизе, как правило,
разлагаются до мономера и низкомолекулярных осколков полимерной
цепи. Поэтому спектры полимера и продуктов его разложения могут
иметь много общего. Таким образом существует возможность исследо-
вать методом ИК-спектроскопии полимеры, из которых невозможно
приготовить образцы для прямого анализа на пропускание (сшитые
полимеры, наполненные композиции и т. п.). Если спектр окажется
слишком сложным и интерпретируется неоднозначно, то путем сочета-
ния ИК-спектроскопии с другими методами анализа, например газо-
жидкостной хроматографии, элементного анализа и других, можно до-
биться успеха в идентификации неизвестного образца.
Некоторые фирмы выпускают специальные пиролитические ячейки
[12, с. 89] для ИК-анализа. Например, удобная ячейка, позволяющая
проводить пиролиз и одновременно исследовать продукты разложения.
В специальную „лодочку”, нагреваемую до температуры разложения
полимера, помещают образец в количестве 10—20 мг. Для пиролиза по-
лимеров в зависимости от их природы используют температуру от
300 до 700 °C. Газообразные продукты пиролиза собирают в кюветном
отделении ячейки, позволяющем снимать спектры пропускания в про-
цессе разложения полимера. Более высокомолекулярные продукты
осаждаются на элементе нарушенного полного внутреннего отражения
(НПВО) (см. раздел II. 1.5), и, изменив положение ячейки относительно
пучка света спектрофотометра, получают спектры НПВО. Если в кон-
струкции пиролитической ячейки не предусмотрена съемка спектров, то
газообразные продукты можно перевести в вакуумированную газовую
кювету спектрофотометра, а жидкие или маслообразные продукты
* Спектры поглощения растворителей в ИК-и УФ-областях спектра представ-
лены в Приложениях IV и V.
38
разложения помещают между окнами кюветы, эмпирически подбирая
толщину слоя.
Спектры пиролизатов полиэтилентерефталата, политрифторхлор-
этилена, полигексаметиленадипамида, сополимера винилхлорида с ви-
нилиденхлоридом, эпоксидных смол, некоторых каучуков и других по-
лимеров даны в обзоре [13].
II. 1.3. Определение толщины поглощающего слоя образца
Для проведения количественного анализа необходимо знать толщину
поглощающего слоя образца. Иногда она не поддается прямому опреде-
лению измерительными инструментами (капля жидкости, раздавленная
между окнами кюветы, порошок полимера в иммерсионной среде и
т. п.). Прямое измерение толщины тонких и эластичных пленок из-за
механического усилия на них в индикаторе* дает неверные результаты.
В этих случаях толщину образца можно определить по оптической плот-
ности полосы поглощения, относящейся к основным структурным
группам исследуемого полимера с заранее измеренным показателем по-
глощения. Показатель поглощения выбранной полосы можно найти,
подобрав пленки того же полимера больших толщин, измерения кото-
рых надежны. Аналогично определяют толщину эффективного слоя по-
лимера в виде порошка, запрессованного с КВг , при условии, что поли-
мер известен или может быть однозначно идентифицирован по спектру.
Определив оптическую плотность этой полосы для образца полиме-
ра в виде таблетки и, зная значение показателя поглощения для нее
из спектров пленок, находят эффективную толщину** образца в таблет-
ке. Таким образом можно определить также эффективную толщину
порошка в вазелиновом масле или смешанного с другой иммерсионной
средой, толщину раздавленной капли жидкости и т. п. Интенсивность
выбранной полосы поглощения должна укладываться в интервал пропус-
кания 20—70%. Это условие должно соблюдаться как для испытуемых
образцов, так и для образцов, по которым определяют значение к.
Иногда для определения эффективной толщины порошкообразного
полимера в иммерсионной среде используют внутренний стандарт, т. е.
вводят вещество с известной концентрацией. Внутренний стандарт дол-
жен быть нейтральным по отношению к анализируемому порошку и
иммерсионной среде, иметь изолированную полосу поглощения, не по-
глощать в аналитической области полимерного порошка, хорошо с ним
смешиваться. На практике этот способ используется редко.
Тонкие пленки полимеров могут давать интерференционную карти-
ну в спектре поглощения, по которой также можно подсчитать толщину
* Индикатор многооборотный 1 МИГ, пределы измерения 0-1 мм, цена деле-
ния 1 мкм (ГОСТ 9696-82).
** Под эффективной толщиной образца в таблетке КВг понимают толщину
такой пленки полимера, которая имеет оптическую плотность, равную оптической
плотности полимера в таблетке. То же относится и к порошку в иммерсионной
среде.
39
пленки. Интерференция возникает при прохождении света через
равнотолщинную пленку с хорошими поверхностями в результате мно-
гократного отражения от них. Это явление наиболее характерно для пле-
нок полимеров с высокими значениями показателя преломления. Напри-
мер, для пленок из полистирола (нд = 1,59), полиэтилентерефталата
(ир = 1,65)*. Интерференция проявляется в виде максимумов и мини-
мумов на фоне спектра поглощения полимерной пленки (рис. П.8). При
нормальном падении света на поверхность пленки интерференционные
максимумы возникают при условии 2dn=mX, где п — показатель пре-
ломления, т — целое число. Измеряя интервал волновых чисел Дн, вклю-
чающий т интерференционных максимумов, и зная показатель преломле-
ния полимера, легко подсчитать толщину пленки d = m/2n Др (см.
рис. 11.8). Для пленки, спектр которой представлен на рис. 11.8, толщина
оказалась равной 18 мкм.
Обычно при измерении толщины пленки по интерференции в ИК
области спектра используют значение n=nD. Данные о показателях пре-
ломления полимеров в ИК-диапазоне длин волн довольно скудны. За-
мена п на nD приводит к небольшим погрешностям.
В практике спектроскописта-аналитика измерить толщину анализи-
руемой пленки методом интенференции удается не всегда. Этот метод
Рис. II.8. ИК-спектр поглощения пленки полистирола (7) и той же пленки, покры-
той тонким слоем перфторуглеродного масла (2).
* Обозначение «д применяют для показателя преломления, относящегося к
желтой линии натрия (589,3 нм).
40
можно применить к пленочным материалам, перерабатываемым мето-
дами экструзии, отлива, каландрования (полиэтилентерефталат, поли-
этилен, поли-4-метилпентен, производные целлюлозы и др.). Такие плен-
ки, как правило, равнотолщинны, имеют хорошие поверхности. В других
случаях, особенно когда анализируют прессованные пленки, измерение
толщины интерференционным методом практически нереально.
Наличие интерференционной картины в спектре полимерной пленки
имеет и отрицательную сторону, так как искажает спектр поглощения
полимера и мешает анализу. Наиболее простым и удобным способом
устранения интерференции является нанесение на поверхность пленки
тонкого слоя вазелинового или перфторуглеродного масла. При этом
надо учитывать собственное поглощение масла, чтобы не искажать по-
глощение полимера в аналитической области*. На рис. П.8 представлен
спектр поглощения полистирольной пленки с интерференционной карти-
ной и спектр той же пленки с тонким слоем перфторуглеродного масла
на ее поверхности.
П.1.4. Полировка окон кювет
Замутненные окна (пластины из KBr, NaCl, КС!) легко восстанавли-
ваются полировкой. Если пленка полимера не отмывается от по-
верхности окна растворителем, то ее удаляют шлифованием. Шлифовать
окна необходимо и при образовании дефектов на их поверхности. Для
этого удобно воспользоваться водостойкой шлифовальной шкуркой
(ОСТ 2И-74-4—84), которую впоследствии можно легко отмыть от соли
и многократно использовать. Материалом для полировки может служить
фетр или несколько слоев мягкой ткани, закрепленной на какой-либо
ровной и твердой поверхности. Шлифуют и полируют круговыми дви-
жениями, равномерно прижимая окно к ткани. Работать при этом следу-
ет в резиновых перчатках или напальчниках. Окно после очищения от
пленки шлифованием подвергают полировке. Полировку производят
следующим образом. Поверхность полировочного материала в одном
месте смачивают спиртом, содержащим воду, оставляя большую ее
часть сухой. Некоторые экспериментаторы используют ацетон, но это
неудобно, так как требует выполнения работы в вытяжном шкафу.
Применение воды ввиду большой растворимости в ней галогенидов
щелочных металлов дает плохие результаты. Круговыми движениями
растирают окно на месте смоченной ткани, передвигаясь постепенно
на ее сухую поверхность. Если окно после полировки окажется еще
непрозрачным, то процедуру следует повторить. Отнимать окно от
ткани можно только после окончания полировки на сухой части ма-
териала.
Необработанные блоки кристаллов NaCl и КВг раскалывают по
плоскостям спайности на пластины. В дальнейшем их обработку лучше
проводить на специальном станке.
* Спектры поглощения вазелинового и перфторугперодного масла даны в
Приложении IV.
41
С особой тщательностью следует готовить окна для газовых и жид-
костных кювет, к поверхности которых предъявляются высокие тре-
бования.
II.1.5. Исследование поверхностных слоев полимерных образцов
ИК-метод спектрального анализа в своем классическом варианте —
получение спектров путем пропускания излучения через образец, часто
не может быть использован при исследовании полимеров. Например, из
сшитых полимеров невозможно приготовить достаточно тонкую пленку
для съемки спектра, наполненные полимеры могут практически не
пропускать излучение даже в тонких слоях и т. п. В таких случаях может
быть полезен метод нарушенного полного внутреннего отражения
(НПВО), который за последние двадцать лет получил развитие как
самостоятельный метод и как дополнение к классическому методу
ИК-спектроскопии.
Метод НПВО основан на явлении полного внутреннего отражения
на границе раздела двух сред с различными показателями преломления
и позволяет исследовать тонкий поверхностный слой образца. Кратко
явление полного внутреннего отражения сводится к следующему. Луч
света (рис. П.9) из среды I с показателем преломления «j, падая под
углом i'i на границу раздела, частично зеркально отражается и частично
входит в среду II с показателем преломления п2. Если среда II оптически
менее плотная, т. е. пх >п2, то угол преломления i2 больше угла падения
и при некотором угле падения zKp преломленный луч скользит по грани-
це раздела сред, т. е. i2 становится равным 90 °C (рис. 11.9, б). При угле
падения z'i >zKp наблюдается только отраженный луч (рис. П.9, в), т. е.
происходит полное внутреннее отражение. Значение угла/кр определяет-
ся соотношением показателей преломления сред:
sin гкр = «г/г?!
Рис. II.9. Схема полного внутреннего отражения.
42
Рнс. 11.10. Схема нарушенного полного
внутреннего отражения: V
1 — элемент НПВО; 2 — образец. '
уЖЖКЛ’--------------'
Полное внутреннее отражение
действительности не является полным.
Свет проникает в среду II на глубину
порядка длины волны излучения. Если при данной X. среда II поглощает,
что в идеальном случае спектр НПВО должен повторить спектр пропуска-
ния в ослабленном виде.
Спектры поглощения НПВО получают на спектрофотометрах со
специальными приставками. Основой приставок является элемент НПВО.
Его назначение — создать условия, при которых луч света падал бы на
поверхность исследуемого образца из более плотной среды (рис. 11.10).
Элемент изготавливают в виде трапеции, ромба или призмы из кристал-
лических материалов с высокими показателями преломления. Такими
материалами являются: кристаллы бромид-иодид таллия (марка KRS-5),
кремния, германия, имеющих показатели преломления соответствен-
но 2,38; 2,0; 3,45; 4,05. К поверхности кристалла прижимается обра-
зец. Приставку устанавливают на пути луча спектрофотометра. Сис-
темой зеркал луч направляется на элемент НПВО и, отразившись на
границе с испытуемым образцом, падает на входную щель спектро-
фотометра.
Образцы должны иметь ровную гладкую поверхность, обеспечиваю-
щую хороший контакт с кристаллом. От жестких пленок с неровной
поверхностью можно вообще не получить спектр НПВО. В реальных
условиях спектры НПВО имеют ряд отличий от спектров пропускания.
Глубина проникновения света в образец зависит от его показателя
преломления, который в области полосы поглощения испытывает изме-
нения. Поэтому спектр может искажаться. Эти искажения становятся
существенными в области интенсивных полос поглощения и'при углах
падения близких к критическим.
В случае малой эффективной толщины поглощающего слоя
спектр имеет малоинтенсивные полосы. Для увеличения их интен-
сивности используют многократное отражение от исследуемого образца
(МНПВО).
Методы НПВО и МНПВО используются для решения разнообразных
задач полимерной химии, связанных с поверхностными явлениями.
Например, при исследовании процессов химической модификации по-
лимерных материалов, протекающих в поверхностных слоях, опреде-
лении стабилизаторов и других добавок, мигрирующих к поверхности
полимерного блока и т. п.
Подробно методы НПВО и МНПВО описаны в монографии [14] и
обзоре [15]. Описание приставок лабораторного исполнения и выпускае-
мых отечественной промышленностью к ИКспектрофотометрам дано в
специальном разделе книги [14, с. 303].
43
II. 1.6. Источники погрешностей в молекулярном спектральном анализе
Измеряемой величиной в абсорбционной спектроскопии является опти-
ческая плотность D, искомой — концентрация С. Согласно закону Буге-
ра—Ламберта—Бера D = Ced. Таким образом, погрешности в определении
С — складываются из погрешностей в измерении d,e nd.
При получении спектра следует свести к минимуму искажения, кото-
рые могут возникнуть из-за неправильного выбора условий записи спе-
ктра. Необходимо выбрать оптимальные значения спектральной ширины
щели спектрофотометра и скорости сканирования. Под спектральной
шириной щели подразумевают интервал волновых чисел, выделяемый
щелью монохроматора на выходе излучения. Спектральная ширина щели
связана с геометрической шириной щели спектрофотометра соотноше-
нием, зависящим от аппаратурных параметров прибора* *.
Изменение толщины щели возможно в определенных пределах, не
вызывающих существенного искажения контура полосы поглощения.
Верхний предел зависит от полуширины полос. Полушириной полосы
называют ее ширину на половине высоты, выраженной в единицах опти-
ческой плотности или показателя поглощения.
Для того чтобы не искажалась форма полосы, спектральная ширина
щели должна быть гораздо меньше полуширины полосы. Если ширина
щели сравнима с полушириной полосы или больше нее, то полосы в
спектре расплываются, детали структуры поглощения сглаживаются**.
Это видно на примере рис. II. 11 (спектры 1-3}, где представлен
участок спектра полистирола от 3200 до 2700 см”1, записанный на
спектрофотометре фирмы Бекман (модель 4250) со спектральной
шириной щели равной 4, 8 и 15 см-1 .С увеличением ширины щели узкие
полосы спектра сливаются, спектр искажается. Коэффициент пропуска-
ния Т в области полос увеличивается, а оптическая плотность D
уменьшается.
• Изменения в спектре наблюдаются и при увеличении скорости
сканирования (скорости изменения длин волн). Поскольку существует
инерционность приемно-регистрирующего устройства спектрофотомет-
ра, то необходимо выбирать такую скорость сканирования, чтобы избе-
жать временных искажений. Искажения происходят при больших ско-
ростях, когда перо самописца не успевает регистрировать все детали
спектра. Контуры полос искажаются несимметрично, их максимумы
смещаются в сторону сканирования.
Следовательно, условия записи спектра существенно сказываются
на измеренных значениях оптической плотности полос поглощения и
* Формула расчета спектральной ширины щели по значению ее геометричес-
кой ширины, а также определение погрешности спектрофотометра по шкале коэф-
фициента пропускания и шкале длин волн даны в ГОСТ 8.229—81 „Спектрофото-
метры инфракрасные, Методы и средства поверки”.
*С другой стороны, при слишком узкой щели происходят большие потери
интенсивности света, падающего от источника и возможны дифракционные
явления [16, с. 54].
44
Рис. 11.11. ИК-спектры полистирола, записанные при различных значениях спект-
ральной ширины щели.
данные о показателях поглощения, полученные при разных условиях,
могут отличаться. Поэтому, использовать литературные данные о значе-
ниях показателей поглощения в своей работе надо с большой осторож-
ностью. При воспроизведении какой-либо методики, взятой из литера-
турных источников, следует повторить измерение показателей поглоще-
ния для построения градуировочных кривых на спектрофотометре, ко-
торый будет использован для анализа. Необходимо также проверить
градуировку спектрофотометра по волновым числам и соответствие
пропускания 0 и 100% значениям на диаграммной бумаге. Погрешность
при измерении оптической плотности может быть связана и с неоднознач-
ностью выбора базисной линии, особенно когда на аналитическую полосу
накладываются соседние полосы.
Существенное различие данных о пропускании одного и того же
образца, полученных на разных приборах различными экспериментато-
рами, было продемонстрировано в специально предпринятом исследо-
вании, описанном в [17].
При оптимальных и постоянных условиях записи спектра погреш-
ность при определении оптической плотности в большой мере обусловле-
на качеством полимерной пленки, ее разнотолщинностью, поскольку
воспроизводимость значения Т для большинства спектрофотометров
обычно не превышает 0,5%.
В случае анализа какого-либо вещества, находящегося в растворе
при постоянной толщине кюветы,неточности в измерении будут главным
образом связаны с погрешностью в концентрации при составлении эта-
лонных растворов.
45
Как было уже отмечено в гл. I, наибольшая точность измерений
может быть достигнута в интервале значений коэффициента пропуска-
ния от 20 до 70%, что соответствует значениям оптической плотности
D от 0,7 до 0,15. Поэтому при выполнении количественного анализа
следует выбирать толщину пленки, концентрацию раствора такими,
чтобы определяемые значения D находились бы в указанном интервале.
Когда анализ проводится по градуировочному графику зависимости
оптической плотности от концентрации анализируемого вещества, то
наименьшей погрешности соответствует масштаб, при котором градуи-
ровочная прямая составляет угол 45° с осями координат. При благопри-
ятных условиях относительная погрешность молекулярного спектраль-
ного анализа обычно не превышает 5%.
Методы статистической обработки результатов измерений и вычисле-
ния погрешности в доступной форме изложены в книге [18].
II.1.7. Новые технические возможности инфракрасной спектроскопии
Развитие техники спектрального анализа полимеров связано с широким
применением электронно-вычислительных машин. Подключение спе-
циализированных ЭВМ непосредственно к спектральному прибору
позволяет программировать и автоматизировать процедуру анализа,
что особенно существенно при серийных испытаниях. Устройства запо-
минания и хранения спектральной информации на магнитных дисках
в сочетании с программами поиска, дают возможность создавать библио-
теки спектров, по объему и удобству пользования далеко превосходя-
щие обычные справочные пособия. ЭВМ позволяет представить однажды
записанный спектр в том виде (Т или D), который удобен для конкрет-
ного применения, с подробной разверткой заданного небольшого участ-
ка спектра или наоборот, в сжатом виде, позволяющем просмотреть
спектр в широкой области частот.
Большие преимущества представляет осуществление с помощью
ЭВМ операции со спектрами: их умножение на заданное число и вычи-
тание. Как уже упоминалось выше, в практике ИКспектроскопии часто
применяют метод компенсации, когда, например, для выявления спектра
примеси, записывают спектр образца с примесями относительно образца
сравнения (чистой полимерной матрицы). Для. точной компенсации
поглощения матрицы необходимо соответствующим образом подобрать
толщину образца сравнения. Применение ЭВМ избавляет от этой проце-
дуры. Компенсацию можно осуществлять, вычитая из спектра исследуе-
мого образца спектр образца сравнения, умноженный на число, которое
подбирает ЭВМ. При этом критерием компенсации, как и обычно,являет-
ся исчезновение какой-либо полосы поглощения заведомо принадлежа-
щей матрице.
Качественно новый уровень спектрального анализа связан с Фурье-
спектроскопией. В настоящее время этот метод широко используется
в научных исследованиях, однако еще не нашел повседневного примене-
ния в практике рутинных анализов. Фурье-спектрометры не содержат
46
призм или дифракционных решеток, пространственно разделяющих
излучение на спектральные составляющие. Принцип их работы основан
на интерференции двух лучей света. Луч от источника излучения, прохо-
дя через полупрозрачное зеркало (светоделитель), расщепляется на
два луча, которые, отразившись от двух разных зеркал, снова сводятся
и попадают на приемник. При этом лучи проходят разные пути. Разность
хода лучей можно менять. Для этого одно из отражающих зеркал сдела-
но перемещающимся*.
Вследствие интерференции результирующая интенсивность света,
регистрируемая приемником, зависит от разности хода лучей. Эта зави-
симость — интерферограмма — содержит информацию о спектральном
составе света. Для извлечения этой информации необходимо провести
над интерферограммой сложное математическое преобразование —
преобразование Фурье, которое выполняется с помощью встроенной
ЭВМ. Таким образом, Фурье-спектрометр содержит ЭВМ, как необходи-
мый элемент, преобразующий интерферограмму в спектр. Эта же ЭВМ
осуществляет функцию управления прибором и решения задач, которые
были перечислены выше.
Одним из главных преимуществ Фурье-спектроскопии для анализа
полимеров является существенное уменьшение потерь света. При полу-
чении спектра с одинаковым разрешением и при одной и той же спек-
тральной яркости источника излучения в Фурье-спектрометре на прием-
ник попадает примерно в 200 раз больше света, чем в обычном дифрак-
ционном спектрофотометре. Это обстоятельство, а также малое отноше-
ние сигнала к шуму позволяет получать информативные спектры от
слоев с оптической плотностью далеко за пределами интервала, доступ-
ного для дисперсионных или дифракционных спектрофотометров.
Возможно достаточно точное измерение пропускания, когда оно состав-
ляет доли процента, или когда, наоборот, очень мало поглощение. Фурье-
спектрометр дает информацию сразу обо всем спектральном диапазоне
(ограниченном лишь пропусканием светоделителя и чувствительностью
приемника), в то время как в обычном спектрофотометре спектр скани-
руется последовательно. Это вместе со способностью интерферометра
пропускать большие потоки энергии дает дополнительные преимущества
при изучении кинетики реакций, так как имеется возможность следить
за изменением во времени не одной полосы, а большого участка спектра.
Прмежутки времени между регистрациями спектра ограничены лишь
скоростью движения зеркала и составляют несколько секунд. Но воз-
можно и лучшее временное разрешение.
Возможности Фурье-спектроскопии для разделения перекрывающих-
ся полос поглощения методом компенсации хорошо иллюстрируется
проведенным в работе [19] исследованием спектров полимеров в поля-
ризованном свете. Как уже говорилось в разделе П.1.1., поляризация
после поглощения проявляется при одноосном растяжении полимерной
пленки, обеспечивающем ориентацию молекулярных цепей. Однако и в
* Для изменения разности хода лучей применяют также оптический клин.
47
пленке, специально не подвергнутой растяжению, имеется случайная
ориентация, обычно малая, но достаточная для разделения полос благо-
даря высокой точности записи спектров и операций над ними в Фурье-
спектрометрах. На рис. 11.12, а представлен спектр поглощения поли-
пропилена в интервале 1500—1400 см-1, полученный в плоскополяри-
зованном свете при некотором случайном расположении пленки (Do)
и спектр той же пленки, повернутой на 90° (£>д0). При этом плоскость
поляризации света сохранялась неизменной. На обоих спектрах видна
широкая полоса с максимумом около 1457 см’1 и более слабое плечо
около 1437 см”1. На первый взгляд, эти спектры не отличаются друг от
друга. Однако их вычитание явно показывает наличие ориентации в плен-
ке. На рис. П.12, б показаны разностные спектры (D0-qD90) при раз-
личных значениях масштабного
множителя q. Видно, что при
<7 = 1,10 плечо выделилось в
полосу 1436,5 см’1 и наблю-
дается симметричная отрица-
тельная полоса с максимумом
при 1463,5 см’1. При <7 = 1,00
на низкочастотном склоне этой
полосы появилась новая де-
таль — слабовыраженное плечо
вблизи 1450 см’1, а при q =
= 0,89 это плечо выдели-
лось в симметричную полосу
1452,5 см”1, в то время как
две предыдущие полосы исчезли
Dl а
-1___I. ।_____i__
7500 1460 1420
У,см~’
Рис. П.12. Поляризационные спектры поглощения полипропилена:
а - пленка в исходном положении (/) и повернута на 90 ° (2) ; б — разностные
спектры Т>0—д О90 при значениях q 1,10 (1), 1,00 (2) и 0,89 (?).
48
Таким образом оказалось, что широкая полоса в обоих спектрах на
рис. 11.12, а на самом деле состоит из двух полос. Спектр полипропи-
лена в рассматриваемой области состоит из трех полос 1436,5; 1463,5
и 1452,5 см-1, причем две первые выделяются в разностном спектре
при q = 1,10, когда полностью компенсируется последняя полоса, а при
<7 = 0,89, наоборот, в разностном спектре компенсируются две первые
полосы и выделяется последняя. Такой результат конечно обусловлен
противоположной поляризацией (относительно оси молекулы) колеба-
ний, ответственных за полосы 1436,5 и 1463,5 см-1 и полосу 1452,5 см 1
[8, с. 217].
Аналогичный анализ был проведен также для спектров полипропи-
лена в интервале 1350-1000 см-1 и поливинилиденфторида в области
650-400 см'1.
Фурье-спектроскопия значительно облегчает изучение дальней инфра-
красной области спектра. В этой области, в частности, расположены поло-
сы, принадлежащие колебаниям решетки кристаллических структур по-
лимера. Так, в спектре полиэтилена [20] в интервале 50—90 см'1 наблю-
дается широкий фон, который связывают с аморфными областями, и
относительно узкая полоса при 73 см'1, связанная с кристаллической
структурой. Этот спектр был использован для исследования изменений,
которые происходят при гамма-облучении отдельно в аморфных и кри-
сталлических областях полиэтилена.
Внедрение новых технических достижений в практику спектрально-
го анализа значительно расширяет возможности этого метода:
быстрое получение информации об исследуемом веществе;
исследование спектров отражения, позволяющих изучать процессы
в поверхностных слоях образца;
исследование водных растворов полимеров;
определение малых количеств примесей в тонких полимерных плен-
ках, путем накопления сигнала поглощения.
Современным достижениям инфракрасной спектроскопии посвя-
щен обзор [21].
П.2. АТОМНЫЙ СПЕКТРАЛЬНЫЙ
АНАЛИЗ
В полимерных материалах металлы и другие элементы могут присутство-
вать как в виде примесей, так и в составе минеральных добавок, спе-
циально введенных в полимер с целью модификации его свойств. Нали-
чие в полимерах примесей неорганического происхождения связано с
остатками веществ, участвующих в процессе полимеризации (инициато-
ров, катализаторов и т. п.). Они могут находиться в полимере в свобод-
ном состоянии и, следовательно, поддаваться вымыванию, или же в хи-
мически связанном с молекулами полимера состоянии, например, на
концах цепи макромолекулы.
49
Для обнаружения и количественного определения в веществе метал-
лов и других элементов используют методы атомной спектроскопии —
эмиссионный и атомно-абсорбционный анализы*. Поскольку доля приме-
сей в полимере по отношению к его массе мала, то анализируемые об-
разцы подвергаются концентрированию (минерализации). Для боль-
шинства полимеров - это метод озоления (ГОСТ 15973—82). Зола по-
лимера, состоящая из оксидов металлов, является объектом анализа.
При этом определяют массовую долю золы, значение которой необхо-
димо для окончательного расчета количества примесей.
При проведении озоления методом сжигания**следует иметь в виду,
что могут быть потеряны легколетучие элементы или уменьшена их кон-
центрация (например, ртути, мышьяка, свинца, сурьмы, висмута). Для
устранения потерь необходимо оптимизировать температурный режим и
условия озоления, исключающие открытое выгорание образца, или про-
вести его предварительную обработку. По указанному выше стандарту
рекомендуется обработка пробы полимера серной кислотой (метод
определения сульфатной золы). Образец, смоченный серной кислотой,
озоляют при постепенном повышении температуры с последующим про-
каливанием остатка до его постоянной массы (разность результатов
двух последовательных взвешиваний не должна быть более 0,0002 г).
Если полимер подвергается полной химической минерализации,
то нужно отдать ей предпочтение перед методом озоления при анализе
легколетучих элементов. Проведя такое концентрирование, необходимо
проверить чистоту реактивов и влияние вспомогательного инструмента
и посуды на результаты определения, сделав „холостой” опыт. Описание
методов концентрирования элементов можно найти в книге [27].
Эмиссионный спектральный анализ. В качестве источников возбуж-
дения спектров при эмиссионном спектральном анализе используют
пламя, дугу постоянного или переменного токов, высоковольтную
искру. Пламя — низкотемпературный источник света (около 3000 °C).
Температура плазмы дуги между угольными электродами достигает
7000 °C, между медными и железными около 5000-6000 °C, высоко-
вольтной искры до 10000 °C.
При возбуждении пламенем светятся атомы с низкими потенциалами
ионизации главным образом щелочных и щелочноземельных элементов.
При увеличении температуры плазмы свечение легковозбудимых эле-
ментов ослабевает за счет ионизации, наблюдается свечение элементов
с более высокими значениями энергий возбуждения/
* Методы атомной спектроскопии не являются основными при анализе поли-
мерных материалов и по широте применения уступают методам молекулярной
спектроскопии. Поэтому мы не будем подробно обсуждать обшие вопросы техни-
ки атомного спектрального анализа. Остановимся лишь на тех моментах, которые
касаются полимеров. Известен ряд монографий, подробно освещающих как теоре-
тические, таки практические аспекты этих методов [22-26].
** Кроме метода сжигания (сухое озоление) в практике атомного анализа
применяют метод химического концентрирования элементов, когда органическая
основа образца подвергается разрушению концентрированными минеральными
кислотами. Для подавляющего числа полимеров используют сухое озоление.
50
Таблица спектральных линий и значения их энергии возбуждения для
ряда элементов, встречающихся в полимерных материалах, представле-
ны в [2, 100, 101].
Концентрация элементов, которая может быть обнаружена методом
эмиссионной спектроскопии,зависит как от определяемого элемента, так
и от правильно выбранных условий метода: источника возбуждения,
стабильности его работы, условий регистрации спектра, добавок, влияю-
щих на скорость испарения пробы, и т. п. [22, 26]. Ту минимальную кон-
центрацию элемента, которая может быть обнаружена, принимают за
предел обнаружения данного элемента. Значение предела обнаружения
зависит от уровня фона (или шума). Отличают абсолютный предел
обнаружения — наименьшее количество определяемого элемента, вы-
раженное в граммах, и относительный — наименьшее количество опре-
деляемого элемента, выраженное в процентах. Абсолютный предел
обнаружения для некоторых элементов достигает КГ1 °-10"12г. При
использовании дуги переменного тока относительный предел обнаруже-
ния в зависимости от определяемых элементов составляет 10”6 — 10"2 %.
Эмиссионную спектроскопию применяют для качественного, полу-
количественного и количественного анализов. Этот метод позволяет
определять одновременно до 20 элементов в пробе.
Если объект исследования содержит примеси неизвестного состава,
то сначала проводят качественный анализ образца, идентифицируют
линии в спектре, определяют состав элементов в пробе. Для этого сни-
мают на фотопластинку* спектр железа, который хорошо известен, и
спектр анализируемого образца. В атласах наряду с линиями железа
даны положения наиболее чувствительных линий других элементов.
Отнесение линий пробы к длинам волн производят путем сравнения их
положения с линиями железа и данными таблиц. Полуколичественный
анализ дает сравнительную оценку количества элементов в определенных
интервалах концентраций. Во многих случаях сведений о приближенном
содержании элементов бывает достаточно для решения задачи. Полный
же количественный анализ требует большего времени.
Для выполнения количественного анализа полимера необходимо не
менее 10 мг золы. Золу разбавляют последовательно графитовым (уголь-
ным) порошком до необходимой концентрации (в 50, 100 и более раз),
которая определяется содержанием элемента в пробе и интенсивностью
аналитической линии в спектре.
При подготовке пробы необходимо исключить возможность ее за-
грязнения. На отсутствие определяемых элементов должны быть прове-
рены электроды и графитовый порошок. Для измельчения образца и
эталонов применяют агатовые или яшмовые ступку и пестик. Данные
о загрязнении пробы материалом ступки при ручном перемешивании
можно найти в книге [26, ч. I , с. 38].
Кроме анализа золы метод эмиссионной спектроскопии применим
для определения элементов в растворах полимеров, растворителях
* Несмотря на развитие фотоэлектрических методов регистрации спектров
распространенным методом еще остается фотографический.
51
и других жидких средах. При этом концентрирование пробы осущест-
вляется нанесением на графитовый электрод жидкости и выпариванием
ее под действием инфракрасной лампы. Угольный электрод, заточенный
на плоскость, предварительно покрывают тонкой пленкой полистирола,
окунув электрод в раствор полимера. Это дает возможность исключить
потери примеси за счет проникновения анализируемой жидкости в глубь
электрода.
Иногда возможен прямой анализ полимера без озоления, например
определение остатков катализатора в полиэтилене низкого давления
(см. раздел VI.2).
Непосредственное сжигание в угольных электродах органической
основы может сопровождаться выбросом пробы, образованием мешаю-
щего анализу фонового излучения. Устранения этих явлений можно
достичь введением в пробу угольного порошка.
Если полимер с минеральным наполнителем поддается растиранию
и перемешиванию, то количественное определение добавки может
быть выполнено также без озоления. Содержание наполнителя в по-
лимерных материалах составляет обычно единицы и десятки процентов.
Поэтому для проведения анализа необходимо разбавление анализируе-
мой полимерной пробы угольным порошком. Однако прямой анализ
полимерного порошка имеет много недостатков: увеличивается по-
грешность определения, сокращается число анализируемых совмест-
но элементов (особенно это касается анализа наполненных полиме-
ров) и т. п.
В основе количественного эмиссионного анализа лежит зависимость
интенсивности спектральной линии от концентрации излучающих ато-
мов анализируемого элемента. Эта зависимость выражается эмпиричес-
ким уравнением Ломакина—Шайбе: I =аСв. Значения параметров айв
для данного источника и выбранной спектральной линии остаются по-
стоянными в большом интервале концентраций. В практической работе
используют линейную зависимость между логарифмами интенсивности
и концентрации: lg 1=lgа + в 1g С.
При выборе аналитической линии следует руководствоваться ее
интенсивностью и содержанием анализируемого элемента в пробе.
При определении малых концентраций элементов особое значение
имеют, так называемые, последние линии — наиболее интенсивные линии,
исчезающие из спектра последними при уменьшении концентрации.
Однако для анализа в области больших концентраций эти линии часто
не пригодны из-за явления реабсорбции. Это явление связано с тем, что
при увеличении концентрации анализируемого элемента возрастает и
поглощение света парами этого элемента. Свет, испущенный атомами,
находящимися внутри объема, занятого парами, в значительной степени
поглощается, не успев выйти из этого объема. Степень этого эффекта
зависит от нижнего уровня, на который переходит возбужденный атом.
Если нижний уровень является основным (линия резонансная), то
реабсорбция минимальная. В этом случае для анализа используют менее
интенсивные линии.
52
При аналитических измерениях определяют не абсолютные интен-
сивности линий, а величины им пропорциональные. Для правильного
построения по этим измерениям градуировочного графика и проведения
анализов необходимо точное воспроизведение условий возбуждения и
регистрации спектра. Чтобы исключить влияние посторонних факторов
на интенсивность аналитической линии, используют внутренний стандарт,
т. е. линию спектра, интенсивность которой сохраняется постоянной при
изменении концентрации анализируемого элемента.
Линию внутреннего стандарта (линию сравнения) подбирают так,
чтобы влияние постороннего фона, светочувствительности фотоэмуль-
сии и других экспериментальных условий вносило бы одинаковый вклад
в оптическую плотность почернения этой пары линий. В качестве линии
внутреннего стандарта используют одну из линий основы пробы или
специального соединения, которое должно обладать такими свойствами,
чтобы определяемый элемент и элемент внутреннего стандарта поступали
бы в плазму разряда дуги одновременно. Выбор такого соединения
осуществляется эмпирическим путем. Иногда вместо линии сравнения
используют фон вблизи аналитической линии.
При фотографических методах регистрации за меру интенсивности
линии принимают почернение на фотопластинке, т. е. оптическую плот-
ность почернения: .S’=lg(/()/7), где 10 и I — интенсивности света, прошед-
шего через прозрачное место пластинки и через почернение фотоэмуль-
сии на пластинке, соответственно. Разность почернений AS между анали-
тической линией и линией внутреннего стандарта является мерой отно-
сительной интенсивности, которая используется в количественном
анализе.
Количественное определение элементов в образце наиболее часто
проводят при помощи градуировочных графиков зависимости разности
почернений аналитической пары линий от концентрации анализируемого
элемента в эталонных образцах. Эталоны должны быть идентичны пробе
и состоять из тех же оксидов металлов, что и испытуемый образец. Эта-
лоны, так же как и золу полимера, разбавляют угольным порошком. Он
служит не только для разбавления золы до необходимой концентрации,
но и играет роль „спектроскопического буфера”, который стабилизиру-
ет температуру плазмы, подавляет влияние других элементов на интен-
сивность аналитической линии.
Существуют разные методы количественного анализа, основанные
на фотографической регистрации спектра [22, 26]. Наибольшее примене-
ние при анализе полимерных материалов имеет метод трех эталонов.
В этом методе градуировочный график строят в координатах ДА и lgС.
В области нормальных почернений- график линеен. Для его построения
используют не менее трех эталонов. Эталоны следует выбирать так, что-
бы концентрации определяемых элементов в них образовали бы геоме-
трическую прогрессию. Тогда точки на графике будут равноотстоящи-
ми. Это обеспечивает наибольшую точность построения графика при
заданном числе эталонов. На каждую пластинку фотографируют
спектры эталонов и анализируемых проб. Этот метод длительный.
53
Его целесообразно применять для анализа большого количества одно-
типных проб, спектры которых можно снять на одну фотопластинку.
Когда необходимо анализировать пробу с примесью небольшой кон-
центрации, используют метод добавок. Он заключается в том, что на осно-
ве такой пробы готовят несколько проб, вводя известное содержание
добавок определяемой примеси. Добавки должны вводиться в виде та-
кого же химического соединения, как и примесь в исходной пробе.
Строится градуировочный график интенсивности линии от концентрации
вводимой добавки. Его пересечение с отрицательной частью оси абсцисс
даст значение концентрации примеси, содержащейся в анализируемой
пробе. Концентрация первой добавки должна соответствовать ожидае-
мой. Если она неизвестна, то концентрацию вводимых добавок подбира-
ют эмпирически.
Атомио-абсорбционный анализ. Для проведения атомно-абсорбцион-
ного анализа анализируемое вещество должно быть переведено в атомар-
ный пар. Для атомизации используют пламенный или электротермичес-
кий методы.
При пламенном методе образец в виде раствора подвергают фраг-
ментации — распылению до аэрозоля пневматическим распылителем и
вводят в пламя горелки, где и осуществляется атомизация. Обычно
применяют воздушно-ацетиленовое (максимальная температура 2250 °C)
пламя и пламя оксида азота (I) с ацетиленом (максимальная темпе-
ратура 2950 °C).
Атомарный пар освещают светом с длиной волны, соответствующей
резонансной линии анализируемого элемента, и регистрируют абсорбцию.
В качестве источника используют обычно лампу с полым катодом, в ко-
торой возбуждаются пары того же элемента, что и анализируемый. Та-
ким образом, в методе атомной абсорбции анализ на каждый элемент
проводится индивидуально, в отличие от эмиссионного спектрального
анализа, который позволяет анализировать пробу одновременно на
несколько элементов. Поэтому для обзорного качественного анализа
метод абсорбционной спектроскопии не применяется. В настоящее
время методом атомно-абсорбционного анализа может быть выполнено
количественное определение 70 элементов.
Электротермический метод атомизации основан на испарении пробы
в высокотемпературной кювете (печи) Львова, представляющей собой
графитовую трубку с системой электропитания, нагреваемую в течение
секунд до 3000 °C. Графитовая печь находится в атмосфере аргона.
Непламенной способ атомизации открыл новые возможности атомно-
абсорбционного анализа.
При испарении пробы в пламени возможно образование побочных
термостойких соединений (оксидов, гидроксидов), за счет которых
уменьшается степень атомизации и снижается абсорбционный сигнал.
Это ограничивает чувствительность* метода. Чувствительность, или
* В соответствии с рекомендациями ИЮПАК термин „чувствительность” за-
менен на термин „характеристическая концентрация”.
54
характеристическая концентрация, означает величину концентрации
определяемого элемента, которая вызывает поглощение, равное 1% от
интенсивности падающего излучения, т. е. поглощение в единицах опти-
ческой плотности, равное 0,0044.
Если для пламенных методов атомизации характеристическая кон-
центрация находится в пределах от 0,01 до 10 мкг/мл, то для непламен-
ных методов - 0,0001 -0,1 мкг/мл [24, с. 45].
Электротермическая атомизация, происходящая в среде инертного
газа, уменьшает влияние побочных реакций, свойственных пламени, и
увеличивает тем самым чувствительность определения*.
Высокая чувствительность электротермической атомизации обеспе-
чивается также тем, что при атомизации не происходит потерь анализи-
руемого образца. Весь образец, введенный в графитовую печь, подвер-
гается атомизации. При введении же в пламя аэрозоля лишь часть ве-
щества участвует в образовании сигнала абсорбции.
Применение кюветы Львова позволило понизить абсолютные пре-
делы обнаружения почти всех элементов до 10"12 - 10"15 г.
Определение малых количеств элементов ограничивается уровнем
шумов, на фоне которых регистрируется сигнал абсорбции. За предел
обнаружения принимают ту концентрацию элемента, которая дает сигнал
абсорбции, равный удвоенному стандартному отклонению. Стандартное
отклонение определяют по серии единичных измерений (не менее 10)
при содержании определяемого элемента, близком к уровню холостого
опыта [25, с. 159].
Сведения о технике атомно-абсорбционного анализа, способах уве-
личения чувствительности метода, устранении мешающего влияния дру-
гих элементов на результаты определения, влияние матрицы, чувстви-
тельности определения различных элементов в сравнении с другими ме-
тодами анализа, можно найти в книгах [23-25].
Для анализа полимерных материалов атомно-абсорбционная спектро-
скопия применяется еще редко.
При малой зольности полимера, когда ее невозможно определить
взвешиванием (разница масс тигля с золой и исходного тигля находит-
ся в пределах погрешности взвешивания) целесообразно применять
атомно-абсорбционный анализ. Следы золы в тигле смываются раствори-
телем, в качестве которого используют разбавленные минеральные кис-
лоты. Затем в полученный раствор солей добавляют воду и подвергают
его атомизации в пламени. При малом количестве пробы рационально
использовать электротермический атомизатор, поскольку в нем проба
практически вся превращается в атомный пар.
Полимеры могут быть проанализированы также в виде растворов
в воде или органических растворителях. Растворители влияют на свой-
* Помехи, возникающие при атомизации в графитовой печи (образование
карбидов, межслойных соединений) в результате взаимодействия металлов с мат-
рицей атомизатора, устраняются, так называемой, футеровкой печи танталовой
фольгой.
55
ства пламени. Ряд растворителей (ароматические соединения, галоген-
производные) непригодны для пламенного метода, поскольку из-за
неполного сгорания дают коптяшее пламя. Другие из-за большой скорос-
ти испарения в распылителе дают нестабильное пламя. Перечень полиме-
ров и их растворителей, используемых для анализа методом атомной
абсорбции,приведен ниже [25, с. 254].
Растворители
Метанол
Метилизобутил кетон
Ди метил ацетамид
Диметилформамид
Муравьиная кислота
Циклогексанон
Полимеры
Полиэфиры
Полистирол, ацетат целлюлозы,
ацетобутират целлюлозы
Поликарбонат, поливинилхлорид
Полиакрилонитрил, АБС-пластики
Полиамиды
Сополимеры винилхлорида и винилацетата
При анализе полимерных растворов могут возникнуть трудности,
связанные с матричным эффектом. Так, было исследовано влияние
полиакриламида в водном растворе на определение следовых количеств
Zi, К, Na, Са, Mg, Мп [28]. Наблюдали снижение абсорбционного сигнала
элементов в зависимости от концентрации и молекулярной массы поли-
мера, что связано с процессом распыления раствора. Введение в раствор
NaCl уменьшало влияние матричного эффекта.
III. ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНОЙ НЕОДНОРОДНОСТИ
ПОЛИМЕРОВ
В этой главе мы рассмотрим спектры полимеров, получаемых по реак-
ции полимеризации. Идеальная молекула таких полимеров представляет
собой бесконечно длинную цепочку повторяющихся звеньев. Реальные
молекулы имеют дефекты (нерегулярности). В зависимости от условий
и методов полимеризации они проявляются в образовании ответвлений,
ненасыщенных групп (для предельных полимеров), концевых групп, в
присоединении мономерных звеньев несимметричного строения по типу
„голова—голова” (—СН2СНКС11КСН2 — )„ и других отклонений от
„идеальной” молекулы.
Кроме того, при полимеризации, например, диеновых мономеров,
могут образовываться многообразные микроструктуры, которые ха-
рактеризуются не только разным типом присоединения мономерных
звеньев, но их расположением друг относительно друга, т. е. отличаются
как структурной, так и пространственной изомерией макромолекул*.
На процесс формирования полимерных молекул влияет множество
факторов: инициаторы и катализаторы полимеризации; температура и
давление; среда, в которой происходит реакция, и т. п.
Так, полиэтилен, получаемый при высоком давлении в присутствии
инициаторов (перекисей или кислорода) содержит боковые ответвле-
ния, среди которых преобладают этильные и бутильные группы. Коли-
чество ответвлений колеблется от 15 до 30 на 1000 атомов углерода.
Полимеризация этилена под действием металлорганических катали-
заторов при низком давлении приводит к образованию полимера со
значительно меньшим количеством групп —СН3 :1,5—5,0 на 1000 атомов
С. Практически отсутствуют ответвления (0,5—1 групп —СН3 на 1000
углеродных атомов) в линейном полиэтилене, полимеризация которого
осуществляется под влиянием окиснометаллических катализаторов
[29, с. 10]. Однако при таком способе полимеризации обрыв растущей
* Изомеры имеют одинаковый элементный состав, но отличаются порядком
расположения связей и атомов (структурная изомерия) или при одинаковом по-
рядке связей - пространственным расположением атомов (стереоизомерия).
цис-, граис-Изомерия характерна для полимеров, имеющих в цепи двойные связи:
57
полимерной цепи сопровождается образованием на конце макромолекул
полиэтилена двойной связи винильного типа (—СН=СН2). Йенасыщенные|
связи обнаружены также в полиэтилене, полученном другими методами’
полимеризации. Полиэтилен высокого давления содержит преимущест-
венно винилиденовые группы (^С=СН2).
На основе простейшего из диеновых мономеров — бутадиена
СН2=СН—СН=СН2 могут быть получены следующие изомеры
Н2С\ 2 э /СН2— сн2\ 2 3/СН2-СН2
/с=с\ /с=с\
W ХН н/ хн
1,4 -цис— полибутадиен
СН2 СН2х 2 3 /Н Нк 2 3 /СН
)С=С< 4 I /С—Cf
Н хсн2—сн2х хн
1,4 - транс—пол и бут ад иен
12 1 2
~сн2—сн—сн3—сн—сн2
ЗСН ЗСН
II II
4СН2 4СН2
1,2-полибутадиен
изотактический
сн2
II
с
I
~сн2—сн -сн2—сн—сн2~
сн
II
сн2
1,2-полибутадиен синдиотактический
В зависимости от условий полимеризации могут быть синтезированы
нерегулярные полибутадиены, молекулы которых содержат смешанные
структуры, или же практически „чистые” изомеры одной структуры.
Например, используя комплексные катализаторы кобальтового типа,
можно синтезировать полибутадиен, содержащий 93—98% звеньев струк-
туры 1,4-днс. Если же проводить полимеризацию в присутствии литий-
органического катализатора, то полученный полимер будет содержать
30—50% таких звеньев.
Применение методов стереоспецифической полимеризации приво-
дит к образованию стереорегулярных 1,4-транс-, 1,4-цис- полибутадие-
нов, а также синдиотактического и изотактического полибутадиена
1,2-конфигурации. Регулярный полибутадиен 1,4-транс-структуры может
существовать в двух кристаллических модификациях, отличающихся
58
__।_____। ।_____। 1 1______I—
1800 1400 1000 600
Рис. Ш.1. ИК-спектры погло- Т
щения полнбутадиена с преоб-
ладанием 1,4-цис- (1) и 1,4-
граис-изомера (2).
по температурам плавле-
ния (97 и 145 °C), плот-
ности и другим свойствам.
Таким образом, на-
правленное изменение ус-
ловий полимеризации по-
зволяет разнообразить мо-
мекулярное строение по-
лимеров, что, в свою
очередь, влияет на темпе-
ратуру плавления, плот-
ность, способность к крис-
таллизации, образование
надмолекулярных струк-
тур и т. п. А это, в ко-
нечном счете, дает воз-
можность варьировать физико-механические, диэлектрические и другие
свойства полимерных материалов. Поэтому при получении полимеров,
переработке, прогнозировании их работоспособности в изделиях необ-
ходим контроль за строением полимерных молекул, который в большин-
стве случаев может быть решен методами спектроскопии. Например, по
инфракрасным спектрам можно судить об относительном содержании
того или иного типа ненасыщенных групп в полиолефинах, полученных
разными методами полимеризации, о разветвленности их макромолекул,
о строении полимерной цепи. Так, различие в строении молекул 1,4-
транс- и 1,4-цпс-полибутадиенов находит отражение в ИК-спектре
(рис. П1.1), и позволяет получить информацию о структуре каучука.
П1.1. РАЗВЕТВЛЕННОСТЬ
В этом разделе мы рассмотрим методы определения разветвленности в
полимерах (полиэтилене, сополимерах этилена, поливинилхлориде),
имеющих наибольший удельный вес в промышленности полимерных
материалов.
Полиолефины. Если полиэтилен содержит метильные группы, то они
находятся не только на концах макромолекул, но и на концах ее боко-
вых ответвлений. Поэтому количество метильных групп в полимере при-
нято за меру его разветвленности.
Полоса поглощения метильных групп наблюдается при 1378 см’1.
Она относится к деформационным (ножничным) колебаниям и при-
сутствует в спектрах полиэтилена, сополимеров этилена и других
полимеров, содержащих метильные группы.
59
Рис. III.2. ИК-спектры поглоще-
ния образцов полиэтилена 1 при
25 °C и полиэтилена II при
25 (------) и 150 °C (--).
Полоса 1378 см-1 не
изолирована и перекрывает-
ся серией других полос,
связанных с веерными коле-
баниями метиленовых групп.
Эти полосы находятся при
1368, 1352, 1340 см’1.* К
веерным колебаниям отно-
сится также и сложная поло-
са при 1304 см’1, имеющая
асимметричный склон, ко-
торый маскирует малоинтен-
сивную полосу 1270 см’1.
На рис. III.2 показа-
на эта область спектра по-
глощения полиэтилена (1Г)
с концевыми метильными
группами и полиэтилена (/) , в котором они отсутствуют (концевыми
являются винильные группы). Рисунок сделан на основании спектров,
приведенных в работе [30], где подробно исследованы колебания моле-
кул парафинов при разных температурах, рассчитаны и интерпретирова-
ны их полосы поглощения, а также полосы в спектрах полиэтиленов.
При плавлении полиэтилена интенсивность полос веерных колебаний
увеличивается (см. рис. III.2). Это свидетельствует о том, что они свя-
заны с аморфным состоянием полиэтилена.
Чтобы измерить интенсивность полосы 1378 см’1 и определить
количество групп —СН3 в полимере необходимо учесть наложение
поглощения метиленовых групп (1368, 1352, 1340 см’1). Для этого
применяют разные способы: графическое разделение перекрывающих
полос на отдельные, запись разностного спектра с использованием ком-
пенсационного клина или отожженного образца (метод самокомпен-
сации) и др.
Определение —СН3 -групп путем измерения отношения оптических
плотностей неразделенных полос 1378 и 1368 см’1 в спектре пленки
полиэтилена, которое встречается в некоторых работах, дает лишь
грубую оценку разветвленности полимера.
* Макромолекулы полимеров обладают внутренним вращением вокруг оди-
нарных связей и могут принимать различные конформации. Показано [30], что
полосы 1368, 1352 и 1340 см связаны с различными поворотными изомерами
полиэтилена. Значение V полосы 1340 см 1 дано как среднее для двух полос 1338
и 1344 см’1. Полосы эти малоинтенсивные и расположены в виде плеча на склоне
полосы 1352 см’1.
60
Деление полос поглощения на отдельные производят при помощи
компьютерной техники. Такой метод анализа применен в работе [31]
для определения разветвленности полиэтилена. Положение выделенных
полос поглощения (рис. Ш.З) [31] сопоставлено и находится в хорошем
согласии с проведенным ранее отнесением [30]. Для градуировки, по ко-
торой проводится анализ коммерческих образцов полиэтилена разных
марок, использованы стандартные образцы с известным содержанием
ответвлений. Количество ответвлений в стандартных образцах находи-
лось в пределах от 0,5 до 30,0 —СН3- группы на 1000 атомов углерода.
Детальный анализ спектров поглощения полиэтилена показал, что ис-
следованные образцы имели, главным образом бутильные ответвления.
Другой путь учета метиленового поглощения состоит в его компен-
сации при помощи клина. Клин изготавливают из линейного полиэтиле-
на, не содержащего —СН3 -групп. Он представляет собой прямоугольной
формы пленку переменной толщины в одном направлении. Вставляют
клин в канал сравнения спектрофотометра и записывают разностный
спектр поглощения испытуемого образца полиэтилена относительно
клина. Подбирая толщину слоя клина, добиваются полной компенсации
поглощения полосы 1304 см-1. В разностном спектре остается только
полоса метильных групп.
Метод самокомпенсации состоит в том, что из одного и того же
полимера приготавливают „аморфный” и „кристаллический” образцы и
записывают их дифференциальные спектры. Такие образцы получают
отжигом („кристаллический”) или закалкой („аморфный”). Отожжен-
ный образец готовят медленным охлаждением расплавленного полиэти-
лена, запрессованного между двумя металлическими пластинами и
прокладками из алюминиевой фольги. При прессовании используют
плоские ограничительные кольца, обеспечивающие необходимую
толщину пленки. При охлаждении происходит кристаллизация по-
лимера.
Рис. Ш.З. Графическое разделение поглощения на отдельные полосы в области
деформационных колебаний полиэтилеиа.
61
—1 I____I_____I____I____I--
1400 1350 1300
V,CM-'
Рис. Ш.4. ИК-спектры поглоще-
ния полиэтилена до компенсации
метиленового поглощения (Z),
после частичной (2) и полной
(5) компенсации.
Для получения закален-
ного образца расплавлен-
ный полиэтилен, находящий-
ся между металлическими
пластинами, быстро охла-
ждают в смеси льда с водой.
От алюминиевой фольги по-
лимерная пленка хорошо от-
деляется.
При записи спектра по-
глощения отожженного по-
лиэтилена относительно за-
каленного, вычитается по-
глощение, связанное с „аморфными” полосами полиэтилена. Для кон-
троля степени компенсации спектра служит полоса 1304 см-1. Исчезно-
вение этой полосы в разностном спектре свидетельствует о полной ком-
пенсации. При этом остается поглощение —СН3 -групп кристаллической
части полимера (рис. III.4) [4, с.700]. Для достижения компенсации
спектра необходимо иметь несколько закаленных пленок полиэтилена
разной толщины.
Процедура определения ответвлений в полиэтилене (—СН3 -групп)
методом самокомпенсации описана в [4, с. 694]. Этот стандарт пред-
назначен для определения групп —СН3 в полиэтилене трех типов, отли-
чающихся значениями плотности: I - от 0,910 до 0,925 г/см3; II -
от 0,926 до 0,940 г/см3; III - от 0,941 до 0,965 г/см3. Длина определяе-
мых ответвлений больше трех углеродных атомов.
Процедура основана на предположении, что спектр образцов поли-
этилена можно представить как наложение спектров аморфных и кри-
сталлических структур. Поэтому для оптической плотности пленки
полиэтилена можно написать:
D = aaMb<ip + aKp(\-b)dp
где <?ам и <7Кр - удельные показателя поглощения в аморфной и кристаллической
структурах полимера, соответственно; р - плотность; Ь - доля аморфной струк-
туры в образце.
Учитывая, что в отожженной пленке (обозначенной индексом 1) и в
закаленной (индекс 2) присутствуют обе структуры (хотя конечно в
различных соотношениях), запишем разностную оптическую плотность
для полосы 1378 см-1 в виде:
47>1378 = [a’”8b,di/31 -I- a]?J9(l-6i)diyOi]- [aa3M7862d2jp2 + a^pS(\-b2)d2p2]
62
Это выражение может быть преобразовано к виду:
A^1378=(dlfl- Чгрг)а™ + (dljDtbl -d2Jo2b2)[a!”8-aL3p78] (П1.1)
Аналогичное выражение можно написать и для полосы поглощения
1304 см"1. Поскольку эта полоса принадлежит к колебаниям
-СН2 г- полиэтилена в аморфном состоянии, то a^p04 =0.
Тогда получим:
л г.1304 \ 1304
Введем обозначения
t>idi/>l-h2d2/>2 АР1378 Л£>1304
q -----, а = --------------------- з - ----------—
dipt-dzpi dipt-dxpz ' 1 dipi ^zp-,
тогда из формул (III. 1) и (III.2) найдем:
а== „1378 / 1378_ „1378ч. _ „„1304
Из этих двух уравнений можно исключить неизвестную величину
q и получить связь между а и 3:
Л“акР784-/ЗЛ (III.4)
1леЛ = (^’-^»)/^м04 '
групп
(III. 2)
(П1.3)
R определяет наклон прямой (Да/Д/3). В стандарте [4, с.694] указа-
но, что величина R близка к 0,5 для большинства образцов полиэтилена.
Уравнение (Ш.4) — основное для описываемой методики, которая
заключается в следующем: предварительно определяют pi и р2, dx и
d2 . .Затем измеряют разностные оптические плотности полос Д£)1 378 и
ДО1304, помещая в канал сравнения спектрофотометра закаленные
пленки разной толщины, в то время как в рабочем канале находится
отожженная пленка полимера. Поскольку трудно подобрать такую пару
пленок анализируемого образца, чтобы сразу достичь компенсации по-
лосы 1304 см"1, то для каждой пары закаленной и отожженной пленок
вычисляют значение а и (3 по формулам (III.3) и строят график зависи-
мости а от (3 (рис. III.5). Согласно уравнению (III.4) эта зависимость
должна изображаться прямой. Опыт показывает, что это действительно
так. Участок оси ординат, отсекаемый прямой, дает искомое значение
удельного показателя поглощения а’к378.
Точка (3=0 соответствует полной компен-
сации. Если плотность измерять в г/см3,
а толщину пленки в см, то а™78 получаем
2 / Н
В СМ /г.
Если предположить, что доля групп
-СН3 в аморфных и кристаллических
Рис. 111*5. График для определения удельного
показателя поглощения
кр
63
областях одинакова, то можно считать, что а'Кр78 пропорционален со-
держанию метильных групп в исследуемых образцах полимера.
Для определения содержания групп -СН3 необходимо сравнение
значения aiA78 с удельным показателем поглощения стандартного
образца Дет 8- В качестве стандарта можно использовать гексадекан.
Значение Дет78 стандарта определяют на основании измерения его опти-
ческой плотности 7 8, плотности рст и толщины слоя <7СТ:
р1378
„1378
«СТ —
^*СТ рст
Число групп —СН3 в полиэтилене на 100 атомов углерода (Л) опре-
деляют из формулы:
J1,CTuKp / «ст
где NCT = 12,5 - число групп -СН, на 100 атомов углерода для использованного
в качестве стандарта гексадекана.
Сравнительно простой метод определения метильных групп в сопо-
лимерах этилена с д-олефинами предложен в работе [5]. Метод разрабо-
тан для анализа сополимера этилена с бутеном-1, полиэтилена с этильны-
ми ответвлениями и может быть распространен на сополимеры этилена
с более длинными боковыми ответвлениями (сополимер этилена с пен-
теном, сополимер этилена с гексеном и т. п.)*. Эталонами служили
жидкие парафины C8Hi8—Ci 6Н34. В этом случае, как правило, полимер
анализируют также в расплавленном виде, так как интенсивность полос
меняется при плавлении. Установлено [5], что полуширина полосы
1378 см-1 в спектрах парафинов и исследуемых сополимеров этилена
практически одинакова, почти не меняется при плавлении полимера и не
зависит от степени его кристалличности. Поэтому предложено не прибе-
гать к плавлению анализируемых образцов, а вводить в их оптическую
плотность при 1378 см"1 поправочный множитель, учитывающий изме-
нение интенсивности при плавлении.
Правомочность использования парафинов в качестве эталонов до-
казана анализом образцов сополимера этилена с бутеном-1, состав ко-
торых определялся по данным расхода мономеров в процессе сополи-
меризации.
Метод основан на резделении перекрывающихся полос метильных
(1378 см"1) и метиленовых (1369 см"1) групп. При разделении пред-
полагается, что контур полос не зависит от степени кристалличности
образца. Тогда можно записать:
Р”78 = хР»69 ; D1™ = уD1378
*378 1369 \
где РСН2, Z>CH2 вклады ^-СН2-групп в оптические плотности при соответствую-
щих частотах; О1Л,78, - вклады -СН,-групп; хи у - коэффициенты мень-
K-rij С. г! з
ше единицы, которые определяются формой контура полос и предполагаются не
зависящими от степени кристалличности и разветвленности полимера.
* С увеличением длины ответвлений показатель поглощения при 1378 см-1
практически не меняется [5].
64
Обозначая оптические плотности полос для спектра исследуемого
образца через £)1378 ий1 369 и, учитывая, спектр представляет собой
наложение поглощения групп —СН2 — и —СН3, получим:
Z>i378 = pm8
СН3
4- D
1378 .
СН2 ’
р1369=п1369+р1369
П 2 С tig
Отсюда
п1378 ,-п1369
,,1378 D -XD
Снз 1-ху
(1П.5)
Значение х находят по спектру поглощения полиметилена или по-
лиэтилена с минимальным содержанием" метильных групп. Для опреде-
ления у методом компенсации выделяют полосу 1378 см 1 в спектре
сополимера (рис. Ш.6). Для компенсации был использован клин, отпрес-
сованный из полиметилена. Поскольку полной компенсации достичь
трудно, то коэффициент у рекомендуется определять как отношение
оптических плотностей выделенной полосы 1378 см-1 не при волновых
числах 1369 и 1378 см-1, а при 1387 и 1378 см"1, т. е. вместо низко-
частотного ее склона использовать высокочастотный, не перекрываю-
щийся поглощением метиленовых групп (предполагается, что контур
полосы симметричный).
Таким образом, по найденным поправочным коэффициентам а и у,
оптическим плотностям £>1 378 и О1 369, определенным из спектра образ-
ца, и, использовав формулу (III.5), можно получить значение оптичес-
кой плотности полосы 1378 см"1, обусловленное только поглощением
метильных групп Фен7 8) •
Число групп -СН3 на 100 атомов углерода Nнаходили по формуле:
N = (6D^/ed)(M/p)W0 (III.б)
где S - множитель, учитывающий уменьшение интенсивности полосы 1 378 см"1
при плавлении полимера; М=14 - молекулярная масса группы ^СН2; е - мо-
лярный показатель поглощения групп -СН3, определенный по спектрам парафи-
нов в л/ (моль • см).
Первый сомножитель в формуле (III.6) дает число молей групп
—СН3 в 1 дм3 расплавленного полимера, второй — обратное число молей
групп —СН2— в том же объеме. В условиях работы [5] 6=0,65, е =
= 13,4 л/ (моль • см).
Поливинилхлорид. На концах макромолекул поливинилхлорида
и боковых ответвлений могут образоваться в основном группы —СН3,
—СНС12, —СС1Н2, —СС1=СН2, —СН=СС12. Показано [32], что ничтожно
мало число концевых групп с ненасыщенными связями (менее 0,3 на
1000 атомов углерода). Поэтому общее количество ответвлений в по-
ливинилхлориде оценивают по концевым метильным и хлорсодержащим
группам. Определение ответвлений проводят после реакции восстанов-
ления поливинилхлорида обработкой ZiAlH4, в результате которой
происходит обмен хлора на водород. Таким образом, определение
разветвленности поливинилхлорида сводится к определению развет-
вленности полиэтилена. Анализ проводят [32] также по полосе 1378 см"1
65
V,CM''
Рис. III.6. ИК-спектр поглощения сополимера
этилеиа с бутеном-1 (/), полиметилена (2);
анализируемого образца после компенсации поли-
метиленом (J).
Толщина пленки 0,05 5 мм.
Для градуировки использовали углеводоро-
ды Cj8H38 - С58Н118. Строили зависи-
мость отношения оптических плотностей
при 1378 и 1368 см’1 (£>1 3 7 8 /£>1 368 )
от отношения количества групп —СН3 и
-СН2—. Поскольку поливинилхлорид со-
держит небольшое число ответвлений, то
для получения низких значений отношения
СН3/СН2 использовали смеси полиэтилена
с одним из углеводородов. Такие смеси
соответствовали углеводородам С80Н162,
С,з8Н286 > С2о0Н402
Найдено [32, 33], что боковыми ответвлениями в поливинилхлориде
являются группы, включающие один углеродный атом, например, ме-
тильные. По данным работы [33], которые согласуются с результатами,
полученными большинством авторов, среднее число ответвлений в про-
мышленных образцах поливинилхлорида равно 5—6 на 1000 атомов
углерода.
Ш.2. КОНЦЕВЫЕ ГРУППЫ. ОПРЕДЕЛЕНИЕ СРЕДНЕЧИСЛЕННОЙ
МОЛЕКУЛЯРНОЙ МАССЫ
Метод и условия полимеризации предопределяют те структурные груп-
пы, которые завершают макромолекулу полимера. Такими группами
могут быть группы, присущие химической структуре полимера или
„инородные” группы катализаторов, инициаторов и т. д. Иногда целе-
направленно проводят синтез с тем, чтобы на концах молекул находи-
лись функциональные группы, необходимые для последующей модифи-
кации полимера — сшивания его цепей, создания блоксополимеров.
При деструкции полимера, вызванной воздействием на него высоких
температур, облучения, механических напряжений и т. п., возникают
концевые ненасыщенные или кислородсодержащие группы.
Метод ИКспектроскопии в большинстве случаев позволяет иден-
тифицировать концевые группы и проводить количественный анализ
полимеров на их содержание (см. также разделы III.1, Ш.З, III.4).
Наличие концевых групп может служить основой для определения
среднечисленной молекулярной массы Мп. Определение Мп низкомоле-
кулярных полимеров методом ИК-спектроскопии по количеству конце-
вых групп часто оказывается более рациональным, чем использование
других методов, из-за меньших затрат времени, большей точности, про-
стоты. При этом должно выполняться условие, что полимер линеен, а кон-
цевые группы имеют интенсивную изолированную полосу поглощения.
66
Предположим, что в линейном полимере оба конца молекулы при-
надлежат одним и тем же группам. В этом случае, если Ск — концентра-
ция концевых групп в полимере (число концевых групп в единице объе-
ма), то концентрация молекул — Ск/2. Тогда число молей в единице
объема — CKI(2NА) (N^ — число Авогадро), а их суммарная масса,
т. е. плотность р, равнаМпСк12Ы^, отсюда следует, что
Mn = 2NAp/CK
Таким образом, если по спектру поглощения определить концен-
трацию концевых групп в полимере и знать плотность образца, то можно
подсчитать Мп. Для определения молекулярной массы полимера с раз-
ветвленными молекулами, а также содержащего разные концевые груп-
пы, использование спектрального метода нецелесообразно.
Удачным применением ИК-спектроскопии можно считать определе-
ние молекулярной массы деструктированных молекул полиэтилена [34].
Деструкция является следствием разрушения химических связей поли-
мера при длительном механическом нагружении. Исследовали образцы
полиэтилена низкого давления. ИК-спектры позволили обнаружить обра-
зование в полимере концевых кислотных групп по появлению полосы
1715 см-1. Количество кислотных групп в разрушенных образцах поли-
этилена использовано для определения их среднечисленной молекуляр-
ной массы по формуле:
Мп=М°
где М„ - молекулярная масса исходного полиэтилена; Ск - концентрация кар-
боксильных групп*
Эталонами для определения показателя поглощения полосы
1715 см-1 служили растворы предельных карбоновых кислот. Посколь-
ку наблюдалось различие в форме контура полос эталонов и полиэтиле-
на, то показатель поглощения рассчитывали с учетом поправки на изме-
нение полуширины полосы 1715 см-1.
Молекулярная масса деструктированного полимера, вычисленная
таким образои, хорошо коррелирует с данными, которые были рассчи-
таны на основании кривых молекулярно-массового распределения, по-
лученных методом скоростной седиментации. Исследованные образцы
имели Мп в пределах (10—15) • 103.
Формула (Ш.7) может быть использована для определения средне-
численной молекулярной массы других линейных полимеров, подверг-
* Молекула разрывается на несколько частей. Образование каждого нового
ее фрагмента сопровождается появлением двух концевых групп. Поскольку общая
масса полимера в результате разрушения молекул не меняется (масса образовав-
шихся карбоксильных групп мала), то можно написать равенство: =Mn(N0 +
+ N), где No - исходное число молекул_в единице объема, N+No - число молекул
после _разрушения; N=CK/2, тогда Мп=М„ 2N„/2NO + Ск, учитывая, что N„ =
-pNfJM^, получим формулу (III.7).
2pNA +М°(\
67
Рис. П1.7. Зависимость оптической плот-
ности полосы связи Si-H (2135 см"1)
стандартных (1,1' ) и исследуемых об-
разцов (2-5, 2* - 4* ) от массовой кон-
центрации раствора полимера в тетра-
хлорметаие.
Толщина слоя (в мм) : 0,1 (1—5) и 0,5
(Г-/)-
нутых деструкции, если образую-
щиеся при этом концевые группы
имеют в спектре полосу поглоще-
ния, пригодную для анализа.
Если полимер растворим, то для нахождения его молекулярной
массы можно применить простой метод, описанный в работе [35]. Опре-
деляли Мп полиметилсилоксанов по полосе 2135 см-1 валентного коле-
бания связи Si—Н в концевой группе —Si(CH3)2H.
Образцы исследуемых полимеров растворяли в тетрахлорметане и
строили зависимость оптической плотности полосы 2135 см-1 от массо-
вой концентрации раствора полимера (в г/л); стандартным образцом
служил 1,1,3,3,-тетраметилсилоксан, имеющий среднечисленную моле-
кулярную массу 134 (рис. III.7). Тангенс угла наклона S каждой из
прямых пропорционален удельному показателю поглощения для соот-
ветствующего образца. Произведение SMn дает значение оптической
плотности в расчете на один моль в литре раствора. Предполагается, что
для стандартного образца и исследуемых полимеров эти значения равны
друг другу (при одной и той же толщине поглощающего слоя). Поэтому
можно записать:
*УСТ 1^1 п ст
Таким образом, определив наклон прямых SCT и S,ji, зная молеку-
лярную массу стандартного образца, рассчитываем Мп испытуемого
полимера:
Мп ~ *5ст Л/п СТ/ S
Ниже приведены значения Мп полимеров,
полученные разными
методами:
О_бразец (см. рис. III. 7) Мп, определенная разными 2 3 4 5
методами
осмометрия 1150 1750 2900 —
титрометрия 1100 1795 2800 19000
ИК-спектроскопия 1100 1795 2725 20170
Результаты хорошо согласуются.
Данные ИК-спектроскопии получены на основании усреднения ре-
зультатов для двух толщин поглощающих слоев. Благодаря высокой
интенсивности полосы поглощения 2135 см"^спектроскопический ме-
тод может быть применен для определения Мп полисилоксанов с мас-
совым содержанием связей Si—Н в полимере 0,3%.
68
Идентификация концевых групп спектральным методом дает воз-
можность исследовать начальные стадии процесса полимеризации —
инициирование. Например, в работе [36] изучалась природа концевых
групп полистирола, полученного с бифункциональными диацильными
перекисями на основе фталевой и терефталевой кислот. В спектре по-
глощения перекиси диацетилфталоила
—О—О — с— СНз
— О—О—С — СНз
диацильная перекисная группировка имеет поглощение в виде дублета
при 1790 и 1815 см-1 (рис. III.8, 1). Распад перекиси приводит к обра-
зованию радикала
С—О—О — С—СНз
который инициирует полимеризацию стирола
С—о—О — С—СНз
С—О—СН2 —СН—СНо —сн
II 1’1
О С6Н5
С6н5
С6н5
В результате образуется полистирол с перекисными группами ради-
кала. Полученный полимер имеет спектр поглощения, где кроме полос
1790 и 1815 см-1, наблюдается полоса 1733 см-1 (рис. III.8, 3).Спектр
модельной перекиси
—О—о —с—СН3
аналогичен спектру синтезированного полимера (рис. III.8,2). Сопостав-
ление спектров показало, что полоса 1733 см"1 относится к поглощению
сложноэфирной группировки.
При полимеризации стирола в присутствии модельной перекиси
концевыми группами оказываются сложноэфирные. В спектре такого
полимера присутствует одна полоса 1733 см”1 (рис. III.8,4).
69
—j-----1_____।____।
1900 поо
V,C*r1
Рис. Ш.8. ИК-спектры поглощения диперекиси (Z),
модельной перекиси (2) и полистирола, полученного
в присутствии диперекиси (5) и модельной перекиси (4).
Полосы поглощения концевых групп (1815,
1790 и 1733 см-1) налагаются на собственное
поглощение полистирола в этой области спектра.
Для его компенсации в канал сравнения спектро-
фотометра помещали образец полистирола, полу-
ченный термической полимеризацией без участия
перекисей.
Авторы [36] использовали ИК-спектры по-
глощения для исследования полимеризации сти-
рола также с другими бифункциональными
перекисями на основе фталевой и терефталевой
кислот и показали, что все диперекиси образуют
концевые перекисные группы. Количественную
оценку содержания концевых групп проводили
по их аналитическим полосам поглощения. В
качестве модельных соединений использовали
диперекиси, применяемые в процессе полимери-
зации. Полученные результаты находятся в хо-
рошем согласии с данными йодометрического
титрования.
Ш.З. ГРУППЫ С НЕНАСЫЩЕННЫМИ СВЯЗЯМИ
П1.3.1. Спектральные характеристики полос поглощения связей С=С
В инфракрасном спектре существуют три основные области, в которых
проявляется поглощение колебаний связей С=С и которые могут быть
использованы для анализа. Это валентные колебания связейС=С, имею-
щие полосу поглощения в интервале волновых чисел 1680—1620 см-1;
валентные колебания связей С—Н при связях С =С,полосы которых рас-
положены около 3000 см-1 и внеплоскостные деформационные коле-
бания связей С=Сс участием атомов водорода, поглощающие от 1000
до 700 см”1.
Деформационные колебания групп “=СН, =СН2 около 1400 см"1, ана-
литического значения не имеют. Они обычно малоинтенсивны и их труд-
но идентифицировать на фоне интенсивного поглощения групп —СН2-
и- СН3.
По информативности данных о типе двойных связей, которые могут
быть получены из анализа спектров полимеров и других соединений,
указанные выше области не равнозначны.
Валентные колебания С—С (1680-1620 см”1). Интенсив-
ность поглощения этих колебаний мала, если двойная связь расположена
в середине молекулы (при колебании практически не происходит изме-
нения дипольного момента). Интенсивность полосы увеличивается при
70
замещении, нарушающем симметрию ненасыщенной группировки. На-
пример, наиболее интенсивной она становится, когда двойная связь
принадлежит концевой группе —СН=СН2 или —CR'R"=CH2. Наблюдается
также влияние заместителей и на частоту колебания С=С-связи. При
образовании сопряжения связей С=С или С=С и С=0 происходит сдвиг
частоты колебания в сторону меньших частот.
Большое влияние на частоту колебания связи С=С оказывает заме-
щение атомов водорода на фтор, сдвигающее ее к большим значениям.
Если полоса поглощения двойных связей в спектре полиэтилена нахо-
дится при 1643 см-1 и дает лишь информацию о суммарном содержании
ненасыщенных групп в полимере, то в спектрах фторсодержащих поли-
меров группа —CH=CF2 имеет полосу поглощения в интервале 1730—
1750 см-1, а группа —CF=CF2 поглощает уже при 1780—1800 см-1.
Таким образом, в отличие от спектров полиолефинов, спектры фтор-
полимеров могут дать некоторую дифференциальную характеристику
по типам замещения ненасыщенных групп. Однако этот факт следует
рассматривать как исключение из общей невыразительности данных,
получаемых на основании анализа спектров колебаний связей С=С.
Поэтому для количественных определений ненасыщенных групп в поли-
мерах эта область спектра практического значения не имеет. Частоты
колебаний разных групп, если и отличаются, то не настолько, чтобы мож-
но было идентифицировать тип связи в спектре неиндивидуальных сое-
динений, таких, как полимерные материалы. Это в равной мере относит-
ся как к предельным полимерам, для которых наличие связей С=С в
молекулярной цепи является „дефектом” ее строения и появление их
может быть также связано с процессами старения, так и к непредельным
соединениям, какими являются диеновые каучуки, несмотря на то, что
в спектрах каучуков полосы связей С=С в этой области имеют иногда
заметную интенсивность в зависимости от строения полимера.
Тем не менее исключать из рассмотрения область валентных коле-
баний связей С=С нельзя. В сочетании с данными, полученными из анали-
за полос поглощения других колебаний, она может дать полезные све-
дения о содержании в полимерах ненасыщенных групп.
Валентные кол ебания гру пп=СН,=СН2 (о кол о 3000см-1),
Полосы поглощения валентных колебаний связей С—Н присоединенных
к одиночным связям С—С(группы —СН2—, —СН3)расположены в интер-
вале от 2962 до 2853 см-1. Полосы, относящиеся к группам =СН и =СН2,
значительно смещены к большим значениям волновых чисел. Для груп-
пы =СН2 характерна достаточно изолированная полоса поглощения
3070 см-1, а для группы =СН — ЗОЮ см-1, причем ее положение практи-
чески не зависит от того, в какую ненасыщенную группу она входит:
—СН=СН- (цис, транс), —СН=СН2 [2, с.55]. Различия частот в спектрах
разных соединений небольшие и находятся в пределах около 20 см-1.
Значительный сдвиг полосы поглощения группы =СН2 от поглоще-
ния предельных групп (—СН2—, -СН3) позволяет использовать ее для
количественного определения групп —СН=СН2 и ^С=СН2 в диеновых
полимерах. При относительно небольшом содержании ненасыщенных
71
групп в образце полоса колебания =СН может быть замаскирована скло-
ном интенсивного поглощения групп -СН2- и -СН3. Так, в спектре по-
лиэтилена, содержащего ненасыщенные группы винильного типа, поло-
сы поглощения двойных связей (-СН=, =СН2) не проявляются. Их
нельзя обнаружить и при увеличении толщины пленки полиэтилена,
поскольку это приводит к расширению границ интенсивного погло-
щения основных групп, которое полностью перекрывает поглощение
связей =СН2.
Более удобна для определения количества групп =СН2 в каучуках
область первого обертона С-Н-колебаний. Различия в коэффициентах
ангармоничности колебаний валентных С—Н-связей, принадлежащих раз-
ным группам, приводят при переходе на первый обертон к таким изме-
нениям интенсивности и положения полосы поглощения группы
—СН=СН2, что она становится более изолированной, чем в спектре
основного тона колебаний. Кроме того, она становится и более интенсив-
ной относительно других полос спектра. Это обстоятельство было ис-
пользовано, например, для определения относительного количества
1,2*и 1,4-присоединений в полибутадиене (рис. Ш.9) [16, с. 289]. Ме-
тоды анализа микроструктуры каучуков подробно описаны в ра-
боте [37].
В работе [38] приведены спектры и дана интерпретация инфракрас-
ных полос поглощения технических каучуков разных марок.
Деформационные колебания групп =СН, =СН2. Наиболее
надежную информацию о типе ненасыщенных групп дают полосы погло-
щения, в области спектра 1000—700 см-1. В этой области поглощают
деформационные колебания, при которых атомы водорода выхо-
дят из плоскости двойной связи. Для разных групп они имеют отличаю-
щиеся по волновым числам полосы поглощения:
v, CM" '
Винильная RCH=CH2 ВСН = СНа 909; 995
Винилиденовая R\ уС=СН8 В 888
грвис-Виниленовая н\ /н 965
цис-Виниленовая нЧ=с/н R'-5' 740
Положение полос поглощения перечисленных групп достаточно
стабильно в спектрах соединений с разными заместителями. Исключение
составляют галогенозамещенные соединения. Для них нет надежных
корреляций. Известно, что замещение водорода на галоген сдвигает
частоту колебания к меньшим значениям. Так, полосу поглощения
830 см-1 хлоропренового каучука приписывают поглощению связи =СН
в группе R'C1C = CHR ”.
12-
el5 -CHj-
Рис. III.9. ИК-спектры поглощения раствора полибутадиена в тетрахлорметане
иа первом обертоне (л) и основном тоие (б) валентных колебаний связи С-Н.
Ш.3.2. Определение ненасыщенности полимеров
Для определения ненасыщенных связей С=С в полиолефинах и полиме-
рах диеновых мономеров используют главным образом область спект-
ра внеплоскостных деформационных колебаний. Полосы поглощения
этих колебаний достаточно изолированы, имеют отличающиеся частоты
(см. выше), что позволяет идентифицировать по спектру тип ненасыщен-
ной связи в полимере.
Полиолефины содержат двойные связи в небольшом количестве по
отношению к числу углеродных атомов (табл. III. 1).
Наличие двойных связей в полиолефинах обусловлено „дефектами”
макромолекул и в зависимости от метода полимеризации их относитель-
ное количество меняется. Число двойных связей в ненасыщенных поли-
мерах (каучуках), которые содержат их в основной цепи макромолекул,
на три порядка больше, чем в полиолефинах. Это отражается на методи-
ТАБЛИЦА III.1
Содержание ненасыщенных групп в различных типах полиэтилена
[29, с. 9], определенных методом ИКС
Содержание ненасыщенных групп от
общего количества, %
винильные винилиде- транс-ви-
новые ниленовые
Тип полиэтилена
Общее число
связей С=С на
1000 атомов
в углероде
Полиэтилен высокого 0,3-0,4 17 71 12
давления Полиэтилен низкого дав- 0,3-0,4 52 31 17
ления Полиэтилен среднего дав- 1,1-1,3 87 7 6
ления
73
ках определения ненасыщенности, хотя часто используют одни и те же
аналитические полосы и модельные соединения.
Анализ полиэтилена проводят на прессованных пленках толщиной
0,5—1,0 мм. Для количественного анализа полимеров, например поли-
бутадиенов, в которых ненасыщенность характеризует их микрострукту-
ру, требуются пленки толщиной от нескольких до 10—15 мкм. В этом
случае часто удобнее анализировать растворы полимеров.
Анализ полиолефинов. Как видно из табл. III.1, в полиэтилене низ-
кого и среднего давления содержится больше С=С винильных групп, а в
полиэтилене высокого давления — винилиденовых. транс-Виниленовые
группы составляют меньшую долю от общей ненасыщенности для всех
типов полиэтилена. В спектрах полиолефинов полосы поглощения этих
групп легко идентифицируют. О присутствии в полиэтилене цис -виниле-
новых групп с помощью инфракрасного спектра судить трудно. Полоса
поглощения 740 см"1 для такой цепи непригодна, так как она располо-
жена вблизи интенсивного дублета 720—730 см"1, который относится к
маятниковым колебаниям групп —СН2— и зависит от кристаллического
состояния полимера.
Определяя количество винилиденовых групп в образцах полиэтиле-
на по полосе 888 см"1, следует иметь в виду, что присутствие в полимере
боковых ответвлений, поглощение которых налагается на поглощение
винилиденовых групп, отразится на результатах анализа. Бутильные
группы имеют полосу при 895 см"1, приписанную маятниковым коле-
баниям метиленовых групп. Другие алкильные группы большей длины
поглощают при 889 см"1 [8, с. 205].
Чтобы учесть наложение алкильного поглощения на полосу 888 см"1,
используют компенсационный метод. Для этого полимерную пленку
подвергают бромированию, что приводит к насыщению двойных связей.
Записывают спектр поглощения исходной пленки полимера*относитель-
но бромированной (в канал сравнения спектрофотометра помещают
бромированную пленку) и получают разностный спектр, в котором при-
сутствуют только полосы поглощения ненасыщенных связей.
Используют два метода бромирования полимеров: обрабатывают
пленку парами брома или выдерживают ее в течение 3 чв 5% (по объему)
растворе брома в тетрахлорметане. Растворимые в тетрахлорметане поли-
меры обрабатывают парами брома, поместив в сосуд над жидким бромом
на 2—3 ч. Полимеры, нерастворимые в тетрахлорметане, например поли-
олефины, могут быть подвергнуты бромированию обоими методами.
Процесс обработки полимера следует проводить в сосуде, защищенном
от попадания света. Для проверки полноты бромирования необходимо
перед компенсационной записью спектра снять спектр поглощения обра-
ботанного образца. Отсутствие в его спектре полос поглощения нена-
сыщенных групп свидетельствует о прошедшей реакции замещения.
* Чтобы не вносить ошибки в измерения при компенсации поглощения за
счет различия в толщине пленок, желательно использовать равнотолщинную плен-
ку, разделив ее на две части; одну из них бромируют.
74
1________I_______ I ‘
WOO 950 900 850
VtCM'’
Рис. ШЛО. ИК-слектры поглощения различных образцов полиэтилена исходного
(/) и бронированного (2) и компенсационный спектр (3).
На рис. Ш.10 [39], приведены спектры поглощения образцов поли-
этилена до и после бромирования и компенсационный спектр. Такой
комбинированный метод позволяет определить винилиденовые группы
с содержанием связей С=С 0,05 на 1000 атомов углерода в цепи.
Рекомендуют [4, с. 821] использовать для количественного анализа
в качестве стандартного соединения 2,3-диметил-1,3-бутадиен, имеющий
две винилиденовые группы в молекуле. Это соединение растворяют в
сероуглероде. Концентрация раствора составляет 0,18 моль/л (14,8 г/л),
толщина слоя в кювете равна 0,1 мм.
Используют интегральную интенсивность полосы поглощения
888 см"1, вместо интенсивности полосы в максимуме (Омаке) • Инте-
гральная интенсивность приближенно сохраняется при плавлении поли-
мера, тогда как Омакс существенно изменяется при изменении агре-
гатного состояния. Хорошее приближение к интегральной интенсивности
достигается умножением D макс на полуширину полосы поглоще-
ния At'j/j.
Измерив полуширину полосы 888 см-1 стандартного образца, ин-
тенсивность ее в максимуме, и, зная концентрацию раствора, рассчиты-
вают интегральный молярный показатель поглощения (в л • моль/см2) :
Е = ( £>ст ивке/ *-ст д v 1/2 ст
где Сст - молярная концентрация винилиденовых групп, моль/л.
75
Вычисленное значение Е для стандартного образца используют при
определении молярной концентрации винилиденовых групп (С) в иссле-
дуемом в виде пленки образце полимера толщиной dj (для полиэтилена
0,5—1,0 мм) :
С ~ (^макс/Edij Д Piyi,
Число винилиденовых групп на 100 атомов углерода равно
ЛГ= (CMtp) • 100
где р - плотность образцов полимера перед бромированием: М - молекулярная
масса группы -СН2.
Определение в полиэтилене ненасыщенных связей других типов
можно выполнять по описанной выше схеме, используя соответствую-
щие модельные соединения. В бромировании образца нет необходимо-
сти, если проводить анализ, например, на содержание винильных групп
по полосе 909 см"1, поскольку она свободна от постороннего наложения.
Количество винильных групп в полиэтилене и сополимерах этилена
с пропиленом определяли в работе [40], используя в качестве эталонов
растворы гептена-1 в октане, изооктане, тетрахлорметане. Толщина слоя
раствора 0,1 мм. Молярные показатели поглощения для полосы 909 см"1
во всех растворах близки. Полимеры анализировали в виде пленок и
вводили поправку на изменение интенсивности полосы 909 см-1 при
плавлении аналогично описанной выше работе [5].
Как было указано выше, содержание двойных связей в полимерах
диеновых мономеров на три порядка больше, чем в полиэтилене. Поэ-
тому для анализа каучуков используют главным образом их растворы.
В ГОСТ 19920.2—74 (продлен до 1990 г.)* определение цис-1,4-полибу-
тадиена, транс-1,4-полибутадиена и 1,2-полибутадиена рекомендуется
проводить, соответственно по полосам поглощения 967, 910 см-1 и
740** используя растворы в сероуглероде. Для определения цис-1,4-
звеньев концентрация раствора должна составлять 2% (по массе) Г а
транс-1,4- и 1,2-звеньев —0,7% (по массе).
В качестве стандартных веществ следует применять регулярные по-
лимеры, содержащие максимальное количество соответствующих
звеньев, например транс- 1,4-полибутадиен со 100% числом транс- 1,4-
звеньев. Кроме того, можно использовать низкомолекулярные непре-
дельные углеводороды: гексен-1, децен-1, траис-2-гептен, гранс-2-децен,
цпс=4-октен.
Толщину слоя раствора подбирают так, чтобы достигнуть оптималь-
ного пропускания 20—70%. Если в полимере присутствуют добавки,
то перед анализом его необходимо переосадить.
* „Каучуки синтетические стереорегулярные бутадиеновые. Метод определе-
ния микроструктуры”.
** При определении содержания цис-1,4-звеньев необходимо учитывать, что
максимум полосы полибутадиена с высоким содержанием цис-1,4-звеньев (95%)
находится при 740 см“ С понижением содержания этих звеньев полоса смещает-
ся до 724 см~«.
76
Молярное содержание с цис-1,4-, транс-1,4- и 1,2-звеньев в образце
(в %) определяют по формуле:
Z^-lOO
с =------
dCe
где Dv - оптическая плотность полос 910, 967 или 740 см"' ; d - толщина слоя
раствора полимера, см; С - концентрация раствора, моль/л; е - молярный пока-
затель поглощения полос 910, 967 или 740 см’1, л/(моль-см).
Величину е вычисляют по данным для стандартного образца, исполь-
зуя формулу:
с Дст-100
d-CT ^ст С„
При нахождении е допускается использовать в качестве раствори-
теля тетрахлорметан для определения транс-1,4- и 1,2- звеньев и цикло-
гексанон для цис-1,4-звеньев. Иногда для анализа используют пленки
полимера, отлитые из раствора на пластинки NaCl или КВг. Толщину
пленки подбирают эмпирически в зависимости от содержания определя-
емых ненасыщенных групп. Чтобы исключить из расчетов толщину
пленки, следует использовать отношение интенсивностей аналитической
полосы и полосы внутреннего стандарта в оптических плотностях. Для
каучуков такой полосой может служить полоса поглощения деформа-
ционных колебаний связи С—Н при 1450 см-1. В качестве эталона может
быть использован каучук с известной микроструктурой Или непредель-
ный углеводород. В последнем случае должна быть внесена поправка на
различие контуров полос поглощения полимера и модельного соедине-
ния. Основой для определения ненасыщенных групп в каучуке служит
график зависимости отношения интенсивностей аналитической полосы
и полосы внутреннего стандарта в оптических плотностях от концентра-
ции ненасыщенных групп в модельных образцах.
Ш.4. КИСЛОРОДСОДЕРЖАЩИЕ ГРУППЫ
В этом разделе мы рассмотрим возможности обнаружения и количест-
венного определения с помощью спектроскопии кислородсодержащих
групп в тех полимерах, которые по своей химической природе их не со-
держат. Появление таких групп в полимерных материалах может быть
вызвано разными причинами: присоединением к концам макромолекул
фрагментов инициатора, как это было показано на примере полистирола
(см. рис. III.8); присутствием соединений, участвовавших в процессе
полимеризации и оставшихся в полимере в виде примесей, не вступив-
ших с ним в химическую связь и др.
Известно также, что полимеры под воздействием внешних условий
(повышенной температуры, облучения, механических нагрузок) в при-
сутствии кислорода воздуха могут подвергаться процессам окисления.
77
Окисление полимерного материала сопровождается образованием в его
макромолекулах кислотных, альдегидных, кетонных, сложноэфирных
и других групп. В большинстве случаев образовавшиеся группы являют-
ся концевыми, что свидетельствует об окислительной деструкции
полимера.
Причиной окисления полимера может служить несовершенство его
молекулярного строения (ответвления, двойные связи в середине цепи
или на ее концах, кислородсодержащие группы и др.). Так, полиэтилен
высокого давления имеет ответвления. Наличие третичного углеродного
атома в его макромолекуле является уязвимым местом, которое спо-
собствует возникновению процессов старения полимера. Нестойкими й
внешним влияниям оказываются полимеры с двойными связями, входя-
щими в основную цепь макромолекулы (например, диеновые каучуки),
так же как и полимеры, для которых ненасыщенные связи С=С являют-
ся результатом нерегулярности их строения.
Окисление полимера может происходить при его переработке или
эксплуатации. Обнаружение и количественное определение кислород-
содержащих групп диктуется необходимостью исследования процессов
старения с целью создания защиты полимеров от разрушения.
В области, где проявляется поглощение карбонильных групп (1600—
1900 см-1), большинство полимеров прозрачны. Благодаря высокой
интенсивности валентных колебаний связей С=О, карбонильные группы
могут быть легко обнаружены в ИК-спектре. Для полиэтилена предел
обнаружения групп ^С=О соответствует сотым долям процентов (по
массе). Зная количество карбонильных концевых групп, определенное
по спектрам поглощения, можно рассчитать среднечисленную молеку-
лярную массу деструктированных молекул Мп (см. раздел Ш.З).
Исследуя изменение количества связей С=О и С=С при старении
полимера в зависимости от времени при различных температурах опре-
деляют энергию активации того или иного процесса, изучают механизмы
окисления и стабилизации полимерных материалов. Для решения таких
задач методы спектроскопии дают полезную Информацию.
При исследовании окисления полимеров на основе ароматических
мономеров следует иметь в виду, что они имеют собственное поглощение
в области спектра 1650—2000 см-1, состоящее из серии малоинтенсив-
ных полос, в большей или меньшей степени перекрывающихся с полоса-
ми поглощения карбонильных групп. Эти полосы относятся к обертон-
ным и составным частотам внеплоскостных деформационных колебаний
бензольного кольца. Число полос и их интенсивность зависят от типа
замещения (рис. III. 11). Во избежание искажения спектра окисленного
полимера из-за наложения полос бензольного поглощения можно вос-
пользоваться компенсационной записью спектра, т. е. в канал сравнения
спектрофотометра поместить пленку исходного полимера той же толщи-
ны, что и окисленная.
Валентные колебания связи С= О. Частоты валентных коле-
баний карбонильных связей находятся в некотором интервале для
78
Рис. Ш.11. ИК-спектры поглощения полистирола
(/) и поли-2,4-деметилстирола (2).
Толщина пленки (в мм) : 0,02 (2) и 0,04 (2).
одного и того же типа групп и изменяются
в нем в зависимости от полярности сосед-
них связей, массы ближайших заместителей,
природы растворителя и т. п. Уменьшает
значение частоты карбонильного колебания
сопряжение связей С=О и С=С.
Основой для идентификации карбо-
нильных групп в спектрах окисленных
2000 1800 1600
V, см
полимеров служили полосы поглощения
соответствующих низкомолекулярных соединений (табл. Ш.2). Для
большинства полимеров положение полос поглощения колебаний кар-
бонильных групп в спектре коррелируют с данными, полученными для
низкомолекулярных соединений. Многие полимеры уже достаточно хо-
рошо изучены и отнесение полос поглощения колебаний С=О в их спек-
трах обычно не вызывает сомнения. Иногда возникает неоднозначность
в трактовке появившейся в спектре карбонильной полосы, поскольку
интервалы волновых чисел разных групп близки или перекрываются.
В этом случае интерпретацию полос следует подкреплять анализом воз-
можных химических реакций в полимере, приводящих к окислению.
Кроме того, может быть использован метод интерпретации полос, при-
мененный нами к спектрам фторированных полимеров и описанный ниже.
Значения волновых чисел полос карбонила галогенозамещенных
полимеров существенно отличаются от данных табл. Ш.2. Влияние ато-
мов галогенов приводит к сдвигу полосы в более высокочастотную
область спектра. Так, валентное колебание связи С=О кетона в спектре
полиэтилена поглощает при 1720 см-1, замещение на фтор у соседнего
с карбонилом углеродного атома (—CF2—СО—СН2—) сдвигает полосу
к 1770см”1, азамещениеу двух углеродных атомов (—CF2—СО—CF2—) —
к 1810 см”1.
Для интерпретации полос поглощения карбонильных групп в спект-
рах некоторых окисленных фторопластов были использованы характер-
ные реакции соединений с /С=О-группами. Например, в спектре поли-
трифторэтилена при его термостарении на воздухе наблюдалась полоса
1767 см”1, которая может быть приписана валентным колебаниям
связи С=О как кислотных, так и альдегидных групп. Было сделано
предположение, что полоса принадлежит поглощению альдегидных групп.
Чтобы доказать это, использовали реакцию взаимодействия альдегид-
ных групп с аммиаком:
R—СГ + NH3 —► R—CH=NH 4 Н2О
хн
в результате которой образуется альдегидимин, поглощающий в другой
области спектра.
79
ТАБЛИЦА 1П.2
Положение полос, поглощения валентных колебаний связи С=О
для различных групп
Группа
Волновое
число, см-1
Низкомолекулярные
соединения
Кето иная
Кислотная —С
1725—1700 Насыщенные карбоновые
кислоты
—сн2—С—СН2—
О
II
—сн=сн—с—
о
с6н5—С—
Альдегидная
-с(°
—сн=сн—с^°
хн
1725 -1705 Насыпанные кетоны с
открытой цепью
1685—1665 а, ^-Ненасыщенные
кетоны
1700-1680* Ароматические кетоны
1740-1720 Насыщенные алифати-
ческие альдегиды
1705-1680 а, (3-Ненасыщенные аль-
дегиды
Сложноэфирная
О
II
—CH—CH—С—О—
с6н5—с—о—
1750-1735 Насыщенные сложные
эфиры
1730-1720 а, ^-Ненасыщенные слож-
ные эфиры
,, Арилпроизводные слож-
ных эфиров
Ангидридная
1860-1800 Ангидриды кислот
1790-1740
*На положение полосы влияет замещение бензольного кольца.
Пленки полимера толщиной около 80—100 мкм помещали в аммиак
и выдерживали в нем несколько часов. Из спектра поглощения политри-
фторэтилена, подвергнутого обработке (рис. III. 12), видно, что полоса
1767 см-1 после выдержки образца в аммиаке в течение 5 ч исчезла, а
появилось поглощение с максимумом при 1645 см-1, относящееся к
валентному колебанию C=N, образовавшегося альдегидимина. Кроме
80
Рис. П1.12. ИК-спектры поглощения окис- D
леНвого политрифторэтилена до обработки
(7), после обработки аммиаком в течение
2 ч (2) и 5 ч С?). 1,0
Толщина пленки 0,08 мм.
того, при 3370 см-1 (рис. III. 13, а)
появилась полоса поглощения, ко- 0,5
торую можно приписать валентному
колебанию =N—Н. Изменения в спек-
трах свидетельствуют, что полоса по-
глощения 1767 см-1 принадлежит О
альдегидным группам.
Еще одним доказательством в
пользу альдегидных групп служит реакция взаимодействия альдегидов
с фенилгидразином:
✓°
В — + H2N—NHC6H5 —► В—CH^N—NHCAH5 + Н2О
\н
Для обработки пленку помещали в фенилгидразин и нагревали на
водяной бане. В спектре (рис. III. 14) наблюдали следующие изменения:
исчезала полоса 1767 см-1 и появлялись полосы поглощения, связан-
ные с колебаниями связи C=N (1650 см’1) и бензольного кольца (1600
и 1490 см-1), что свидетельствует об образовании фенилгидразона.
Исчезновение карбонильного поглощения после обработки полимера
реагентами еще не указывает на принадлежность к какому-либо типу
кислородсодержащих групп. Основным доказательством является
интерпретация конечных продуктов реакции по спектру поглощения.
Из спектров политрифторэтилена и перфторкислоты до и после обра-
ботки аммиаком видно, что появившиеся полоса 3370 см-1 (рис. III. 13, а)
на склоне валентного колебания С—Н основных групп полимера и по-
глощение связей N—Н в области 3300—2700 см-1 (рис. 111.13, б), при-
надлежат разным конечным продуктам реакции. .
Рис. 111.13. ИК-спектры поглощения полимеров:
а - окисленный политрифторэтилен до обработки (7), после обработки аммиа-
ком в течение 5 ч (2); б — перфторкислота до обработки (2) > после обработки
аммиаком в течение 1 ч (2). i .
1 ।
’ ’ » 4
ЯГ
1
В спектре окисленного политрифторхлорэтилена наблюдались по-
лосы поглощения 1875, 1805 и 1770 см-1 (рис. III. 15). На основании
сравнения со спектром перфторкислоты полоса 1770 см"1 была отнесе-
на к поглощению карбонила кислотной группы, полоса 1875 см"1 - к
фторангидридной группе —СО—F, поскольку колебания карбонила
этой группы имеют наиболее высокие частоты. Полоса 1805 см"1 пред-
положительно приписана хлорангидридным группам —СО—С1. Однако
при тех же волновых числах поглощает и кетонный карбонил. Для
доказательства предложенной интерпретации была использована харак-
терная для галогенангидридов реакция обмена атома галогена на другой
атом. Хлорангидридные (в нашем случае и фторангидридные) группы
хорошо реагируют с водой, образуя соответствующую карбоновую и
соляную кислоты:
/О
+ Н2О —► R— с(. + НС1
ХОН
Аналогично протекает реакция для фторангидридных групп. Исчез-
новение в спектре полос 1805 и 1875 см"1 (см. рис. III. 15,2) в результа-
те реакции гидролиза убедительно
их соответственно к хлорангидрид-
ным и фторангидридным группам.
Одновременно наблюдается увели-
чение интенсивности полосы кис-
лотных групп (1770 см"1).
Для доказательства отнесения
полосы поглощения карбонила к
Рис. Ш.14. ИК-спектры поглощения окисленного политрифторэтилена до обработ-
ки (7), после обработки фенилгидразином на водяной бане (2).
Толщина пленки 0,115 мм.
Рис. Ш.15. ИК-спектры поглощения окисленного политрифторхлорэтилена до
(7) и после реакции гидролиза (2).
Условия гидролиза — кипячение пленки в воде в течение 3 ч.
82
кислотным группам удобно также использовать их реакцию с хлористым
тионилом (SOC12), которая приводит к замещению карбоксильной груп-
пы на галоген с образованием хлорангидрида. Обратный перевод хлор-
ангидридных групп в кислотные возможен при кипячении полимерной
пленки в воде для осуществления реакции обмена.
Применение описанного метода спектрохимической интерпретации
полос возможно в том случае, если в реакцию вступают только анализи-
руемые группы, а остальная часть полимерной молекулы не взаимо-
действует с используемым реагентом. Наилучшие результаты достигают-
ся тогда, когда поглощение карбонильных групп проявляется в виде
изолированной полосы, а не сложного наложения перекрывающих-
ся полос.
Ниже приведены сравнительные данные о частотах колебаний кар-
бонильных групп (п, см'1) для полиолефинов и фторопластов (интер-
претация большинства полос фторопластов была выполнена спектрохи-
Полиэтилен Фторопласты
1715 1 790—1745
мическим методом):
— С — с — с—
—с—о—
1720 1 81 0-1790
1735 1790-1755
1745 1800-1780
1790
1820-1790
1890-1850
Введение атомов фтора в макромолекулу полимеров сдвигает к
большим волновым числам все частоты валентных колебаний связи
С=О. Для сополимеров фторированных мономеров с этиленом полосы
соответствующих колебаний находятся ближе к низкочастотной границе
указанного интервала.
Возможность количественного определения карбонильных групп
в полимерах определяется поставленной задачей, которую в каждом
83
Рис. 1П. 16. ИК-спектры поглощения образца фторированного сополимера исход-
ного (1) н после прогрева при 300 °C в течение 20 ч (2), 50 (5) н 70 (4).
Рис. III.17. ИК-спектры поглощения образцов полиэтилена исходного (7) , после
фотостарения в течение 175 ч (3) и после термостарения прн 110 ° в течение 250 ч
(2).
84
случае следует решать отдельно. Если требуется анализировать примеси,
содержащие кислородсодержащие группы, не связанные химически с
полимерной цепью, то при наличии аналитической полосы, их количест-
венный анализ проводится по обычной схеме с калибровкой показателя
поглощения на основании модельных соединений. Аналогично анализи-
руют концевые группы, если можно выбрать для них аналитическую по-
лосу поглощения (см., например, рис. III.8).
Вопрос о количественном определении карбонильных групп в поли-
мерах может возникнуть также в связи с работами по исследованию
процессов их окисления и стабилизации.
Окисление полимеров является в большинстве случаев многостадий-
ным процессом, приводящим к образованию кислородсодержащих групп
разного типа. Количество этих групп может меняться со временем по
разным законам, контур карбонильного поглощения бывает сложным
(как результат наложения перекрывшихся полос поглощения). В этом
случае нельзя рассчитывать на корректное количественное определение
карбонильных групп разного типа. Примером служат спектр поглощения
фторированного сополимера (рис. III. 16), изменение интенсивности по-
лос которого свидетельствует о многостадийности протекаемого процес-
са термоокисления, а также сложный контур карбонильного поглощения
полиэтилена,состоящий из многих компонент (рис. Ш.17).
Однако в простых спектрах окисленных полимеров, когда в процес-
се окисления происходит накопление одних и тех же кислородсодержа-
щих групп, возможно их количественное определение. В большинстве
случаев для исследования старения полимеров часто достаточно лишь
косвенной оценки количества этих групп, необходимой для построения
кинетических зависимостей.
Валентные колебания связи С—О. Кислоты, спирты, эфиры
и другие соединения имеют интенсивную полосу поглощения в области
спектра 1000—1350 см-1, относящуюся к валентным колебаниям связи
С—О. Это колебание чувствительно к природе ближайших структурных
групп или связей молекулы, что объясняет значительное различие в час-
тотах. В подавляющем большинстве соединений колебания С—О облада-
ют высокой интенсивностью, тем не менее они имеют ограниченное при-
менение для количественного анализа, особенно в рассматриваемом
случае, когда кислородсодержащие группы не являются основными для
макромолекул полимеров, а область спектра богата полосами поглоще-
ния других колебаний.
Ниже представлены интервалы волновых чисел*, соответствующие
колебаниям связей С—О, принадлежащим разным кислородсодержащим
группам:
Интервал волно-
вых чисел, см"1
О
II
— С—О—Н (карбоновые кислоты) 1320-1210
•Использованы данные монографии (2 ].
85
—с—о—с— (сложные эфиры) 1320-1000*
— СН2—О—снг— (простые эфиры) 1150-1050
=С—О— С (ароматические и простые ненасыщенные эфиры) 1270-1230
—С—О—Н О о II II — с—о—с— (спирты) 1150-1050
(ангидриды кислот) 1175-1050
*Сильное взаимодействие колебаний С— О и С— С.
Несмотря на широкий интервал значений частот колебаний С—О
и наличие постороннего поглощения, полосы связей С—О могут дать
полезную информацию при интерпретации, например, карбонилсодержа-
щих групп. Полиэтилен, в отличие от других полимеров, не имеет основ-
ных полос колебаний в обсуждаемой области спектра. На фоне сложного
карбонильного поглощения полиэтилена, в результате фотоокисления
последнего, трудно идентифицировать полосу сложноэфирных групп
(см. рис. III. 17). Однако появление полосы 1175 см-1 (рис. III. 18)
относящейся к валентным колебаниям С— О подтверждает образование
этих групп в полимере. Удобное расположение полос поглощения в
спектре сополимера этилена с винилацетатом (рис. III. 19) позволило
также наблюдать полосу колебаний СО при фотоокислительной де-
струкции сополимера. Положения полос сложноэфирных групп (1735
и 1175 см-1) в спектрах полиэтилена и сополимера этилена с винил-
ацетатом на рис. III.19 отмечены стрелками. Полоса С-0 колебания
ацетатных групп винилацетата расположена при 1245 см -1.
О-----------1---------1---------1-----
7400 1300 1200 1100
Чем"’
Валентные коле-
бания связ ей О—Н.По-
лосы поглощения, соот-
ветствующие колебаниям
гидроксильных групп
спиртов, кислот, гидро-
перекисей, проявляются в
ИК-спектрах полимеров,
как правило, в виде ши-
рокого сложного погло-
щения в области от
2700 до 3600 см-1. В
зависимости от природы
Рис. III.18. ИК-спектры погло-
щения образцов полиэтилена
исходного (/) и после фото-
старения в течение 100 ч (2).
86
т
Рис. III.19. ИК-спектры поглощения полиэтилена (Z), поливинилацетата (2), со-
полимера этилена с молярной долей винилацетата 87с (5) до (--------------) и
после (--------) фотостарения.
химических групп, наличия водородных связей форма полос поглощения
и их положение могут существенно изменяться. Поэтому интерпрета-
ция полос поглощения, появившихся в спектре в результате окислитель-
ной деструкции полимера, химической обработки или других превра-
щений сложна. Примером этому служат спектры, представленные на
рис. III. 19. Для окисленного полиэтилена и сополимера этилена с ви-
нилацетатом обнаружено поглощение с максимумом около 3350 см-1.
Окисление поливинилацетата приводит к появлению в спектре поглоще-
ния в широкой области от 2400 до 3600 см (рис. 111.19,2). Провести
отнесение полос к колебаниям связи О—Н в группах различного типа
на основании спектров трудно.
IV. АНАЛИЗ СОПОЛИМЕРОВ
Одним из наиболее результативных применений метода ИК-спектроско-
пии является исследование строения и состада линейных сополимеров,
хотя информация, получаемая из спектров, неравнозначна для разных
сополимеров.
Сополимеры - продукты совместной полимеризации разных мономеров. В
зависимости от реакционной способности мономеров и условий сополимеризации
возможно образование молекулярных цепей с регулярно чередующимися звеньями
АБАБАБ, ААББААББАА; присоединение мономеров по закону случая, что
приводит к созданию статистических сополимеров ААБББАБААБ; получение
полимеров, состоящих из блоков одинаковых мономерных звеньев разной длины
ААААБББББААА.
Говоря о сополимерах, имеющих блоки мономеров, мы имеем в виду со-
полимеры, полученные сополимеризацией, но не прививкой. Привитые или графт-
сополимеры имеют основную цепь из звеньев одного мономера, к которым при-
соединены боковые ветви из звеньев другого мономера:
ААААААААААА
Б Б
Зафиксировать спектроскопически факт Прививки, т.е. отличить привитой со-
полимер от смеси гомополимеров труднее, чем доказать факт сополимеризации,
в результате которой макромолекулы имеют блоки мономеров в основной цепи.
Информативность ИК-спектра сополимера в большой мере опреде-
ляется химической природой мономерных звеньев. В одних случаях,
когда сомономеры химически родственны, спектры их сополимеров соот-
ветствуют аддитивному наложению спектров гомополимеров. Это
видно на примере спектров сополимера бутилакрилата с винилацетатом
(рис. IV. 1).
Спектры сополимера и смеси гомополимеров практически иден-
тичны. Факт сополимеризации в данном случае из сравнения спектров
не очевиден и был доказан методом фракционирования. Если же со-
полимеризации подвергаются химически разнородные мономеры, то
спектр полученного сополимера существенно отличается от аддитивно
наложенных спектров гомополимеров. Хорошим примером этому
служит спектр сополимера тетрафторэтилена с этиленом (рис. IV .2
и IV.3). Спектры свидетельствуют о сильном влиянии групп —CF2- на
колебания этиленовых звеньев. Изменение содержания этилена приводит
к изменению числа полос поглощения групп —СН2 - и их интенсивности
(см. рис. IV.2), поскольку частоты валентных колебаний группы —СН2—,
88
Рис. IV. 1. ИК-спектры поглощения сополимера винилацетата с бутилакрилатом (7)
и смеси их гомополимеров (2).
Пленки отлиты на окнах NaCl из раствора в тетрахлорметане. Молярное соотноше
ние мономеров в сополимере и смеси 20 : 80.
изолированной от группы -CF2- другими группами СН2- и непо-
средственно присоединенной к группе —CF2— тетрафторэтилена, раз-
личны [41]. Сильное взаимодействие и малая разница в массах угле-
Рис. IV.2. ИК-спектры поглощения сополимеров этилена с тетрафторэтилеиом
с молярным соотношением мономеров 50 :50 (7) и 67 :33 (2).
Рис. ГУ.З. Ик-спектры поглощения полиэтилена (7) политетрафторэтилена (2)
и сополимера этилена с тетрафторэтилеиом эквимолярного состава (5).
Толщина пленки ( в мкм) : 1 - 20; 2 — 3; 3 — 15.
89
рода и фтора усложняет также спектр сополимера и в области погло-
щения скелетных колебаний цепи и валентных колебаний C-F (см.
рис. IV.3). Колебания эти не являются независимыми. Спектр сополи-
мера тетрафторэтилеиа с этиленом существенно отличается от простого
наложения спектров гомополимеров. Появление новых полос — струк-
турно-чувствительных — делает спектр сополимера информативным
для определения распределения звеньев сомономеров (см. раздел IV.2).
Мы рассмотрели два крайних случая влияния химического строения
мономеров на спектры их сополимеров. Спектры большинства сополи-
меров занимают промежуточное положение. При сохранении в общих
чертах индивидуальности каждого компонента в спектрах сополимеров
можно обнаружить новые полосы поглощения, отражающие чередование
мономерных звеньев, а также и другие особенности, связанные со строе-
нием макромолекулы, изменением ее геометрии.
Отличие спектров сополимеров от спектров смеси гомополимеров,
выражающееся в появлении новых полос поглощения или других изме-
нениях, может служить доказательством факта сополимеризации.
IV.1. ОПРЕДЕЛЕНИЕ СОСТАВА СОПОЛИМЕРОВ
Определению состава сополимеров разнообразных комбинаций моно-
меров методом ИКС посвящено большое число работ. Обсудить эти
работы или даже перечислить их — нереальная задача. Мы считаем не-
обходимым остановиться на общих вопросах разработки методик:
выборе аналитических полос и эталонных образцов, необходимых для
анализа состава сополимеров, и на нескольких примерах показать воз-
можные пути разработки методик.
IV. 1.1. Выбор аналитических полос
Для определения состава сополимеров выбирают такие полосы поглоще-
ния, интенсивность которых зависит только от концентрации соответ-
ствующих мономерных звеньев и не зависит от влияния окружающих
групп. Проверка осуществляется следующим образом. Измеряют ин-
тенсивность предполагаемых для анализа полос поглощения в оптичес-
ких плотностях. Вычисляют отношение интенсивностей поглощения
пары полос, относящихся к разным колебаниям одного из компонен-
тов сополимера. Если это отношение сохраняется постоянным при
изменении состава, то полосы являются структурно нечувствительными
и их можно использовать для определения состава сополимера. При этом
нет необходимости знать состав используемых для проверки образцов
(см. раздел IV. 1.4).
Одним из признаков правильного выбора аналитической полосы
для определения состава сополимера служит стабильность ее контура
и положения в спектрах сополимера и гомополимера. Так, удобная
90
для анализа изолированная полоса поглощения «валентного колебания по-
ливинилацетата 1736 см"1 изменяет полуширину в зависимости от соста-
ва сополимера. Если для поливинилацетата полуширина полосы со-
ставляет 24 см"1, то для сополимера этилена с винилацетатом с мо-
лярной долей винилацетата 2,5% (т. е. когда звенья оказываются изоли-
рованными друг от друга) ее значение равно 14 см"1*. В спектрах
растворов поливинилащетата или сополимера, обогащенного винилацета-
том, в тетрахлорметане полуширина полосы 1736 см"1 практически
не изменяется. Значит, уширение полосы определяется взаимодействи-
ем макромолекул поливинилацетата и отражает последовательность
присоединения мономерных звеньев сополимера.
Влияние распределения звеньев в сополимере этилена (Э) с винил-
ацетатом (ВА) на частоту колебания карбонила винилацетата обнару-
жили в работе [42] при скрупулезном исследовании спектров растворов
этого сополимера в хлороформе. Измерение волнового числа проводили
с точностью 0,5 см"1 и показали, что в зависимости от концентрации
триад BA—BA—BA, ВА—ВА—Э, Э—ВА—ВА, Э—ВА—Э полоса сдвигается
от 1737 до 1728 см"1. Сдвиг частоты наблюдается только в спектрах
растворов в трихлорметане и практически отсутствует в спектрах раство-
ров тетрахлорметане (в пределах 4 см “*). В спектрах пленок сополиме-
ра частота стабильна (1737 см"1). Изменение v авторы объясняют
образованием водородных связей между молекулами трихлорметана
и винилацетатными звеньями, на которое влияет расположение звеньев
в триадах. Если справедливость такой трактовки может быть спорной,
то факт влияния состава сополимера на положение полосы карбонила
в спектрах растворов сополимера не вызывает сомнений. Поэтому
к использованию полос поглощения таких функциональных групп,
как карбонильная группа сложного эфира, кетона и т. п., для анализа
состава сополимеров нужно подходить с осторожностью, особенно
тогда, когда анализируют растворы. Значительное изменение контура
полосы 1736 см "1 ** и волнового числа в спектрах растворов сополиме-
ра этилена с винилацетатом в трихлорметане относятся к крайним
составам. По-видимому, в узком интервале составов этими изменениями
можно пренебречь и полосу 1736 см"1 использовать как аналитическую.
Для выбора полос поглощения сополимера, пригодных для опре-
деления его состава, в работе [43] предложен графический метод. Он
позволяет отличить полосы в спектре сополимера, которые можно
использовать для определения состава, от полос, чувствительных к по-
* Изменение контура может быть учтено, если использовать интегральную
интенсивность полосы, вместо интенсивности полосы в максимуме. Однако при от-
сутствии интегрирующего устройства эта работа трудоемка и не оправдывает
затрат времени при массовом анализе образцов.
** Разные авторы приводят несколько различные значения частот колебаний.
Так, по нашим данным, волновое число ^.С-О-группы поливинилацетата 1736 см1,
авторы |42] дают значение 1 737 см"1, в некоторых работах 1735 и 1738 см"1.
91
Рис. IV.4. Графический метод определения показа-
телей поглощения.
следовательности распределения звеньев и
поэтому непригодных для определения сос-
тава. Для применения этого метода необхо-
дим широкий набор сополимеров раз-
ного состава (знание состава не требу-
ется) и присутствие в спектре изолирован-
ной полосы поглощения для каждого из
сомономеров. При выполнении этих условий метод позволяет также
определить удельные показатели поглощения аналитических полос.
Метод заключается в следующем. Измеряют оптические плотности
полос сомономеров для растворов сополимера с массовой концен-
трацией С = Сх + CY (Сх и CY - массовые концентрации сомономеров
X и Y в растворе сополимера, которые неизвестны). Оптические плот-
ности для аналитических полос какого-либо одного состава сополимера
можно записать в виде: Z>x = СхДдх, Z>Y = CYdaY, где ах и aY - удель-
ные показатели поглощения сомономеров X и Y в растворе сополимера.
Выразим Сх и CY из этих соотношений и подставим в формулу
C = CX+CY: п
х г £)х £>
---- ~ Q
dax day
или
ux \ dC ' aY\ dCI
Значения Z>x и Z>Y определяют из спектров, С - известна. Таким
образом, откладывая по осям координат величины D-%J(dC) и DY/ (dC)
для разных составов, получим линейный график, если на показатели
поглощения аналитических полос не оказывают влияние окружающие
группы сополимера (рис. IV .4). Отклонение этой зависимости от линей-
ной указывает на структурную чувствительность полос или одной из
полос. Из отрезков, отсекаемых прямой на осях координат, получаем
значения ах иву, а значит и состав каждого из сополимеров, поскольку
Сх _ ау
Су ах OY
Графический метод применим и тогда, когда, например, полоса
сомономера X изолирована, а вторая полоса представляет собой нало-
жение поглощений компонентов X и Y. Однако значения ах и aY полу-
чить невозможно, т. е. нельзя определить состав сополимера. Сходный,
но более сложный метод подбора полос предложен для анализа трой-
ных сополимеров и опробован при определении состава сополимера
окиси пропилена и тетрагидрофурана, а также тройного сополимера
на основе окиси пропилена, тетрагидрофурана и диэтиленгликоля [44].
92
IV.1.2. Эталонные образцы
Измерение показателя поглощения аналитических полос сополимера
осуществляется разными путями, выбор которых часто диктуется
возможностями экспериментатора в получении эталонных образцов.
За эталонные образцы могут быть приняты образцы сополимеров
с разным соотношением сомономеров. Определение их состава произ-
водят другими мегодамй (химическими, ЯМР и др.). Желательно анализ
эталонных образцов провести двумя независимыми методами, чтобы
исключить возможность систематической погрешности. К сожалению,
такая проверка не всегда возможна. Иногда метод ИК-спектроскопии
для определения состава оказывается единственным.
В качестве эталона часто используют гомополимер, если он имеет
изолированную полосу, или по крайней мере она может быть выделена
с достаточной определенностью. На основании измеренного показателя
поглощения этой полосы и ее интенсивности в спектре испытуемого
сополимера, определяют долю анализируемого компонента в бинарном
сополимере, т. е. его состав. При использовании гомо полимера как
эталона должны соблюдаться требования, обеспечивающие надежные
результаты измерения абсолютной интенсивности полосы и толщины
сополимерной пленки. Интенсивные полосы, для максимально точного
измерения интенсивности которых нужны пленки толщиной в несколько
микрометров, или даже несколько десятков микрометров, для указан-
ной цели мало пригодны. В случае растворимости гомополимера и со-
полимеров возможно использование растворов для определения показа-
теля поглощения аналитической полосы и последующего анализа. При
этом должна быть уверенность в том, что показатель поглощения анали-
тической полосы полимера сохраняется постоянным при переходе к со-
полимеру как при анализе пленок, так и растворов.
Искусственные смеси гомополимеров могут также служить основой
для количественного анализа сополимеров. Если из смеси гомополи-
меров готовят пленки, то должна быть обеспечена равномерность рас-
пределения в них гомополимеров. Неравномерность наиболее вероятна
при приготовлении пленок из раствора смеси гомополимеров, вследст-
вие различного взаимодействия последних с растворителем.
Для уменьшения погрешности анализа из-за допущенных ошибок
при составлении смеси Следует пользоваться градуировочными кривыми.
Построение градуировочных кривых необходимо и при использовании
растворов сополимеров и эталонных смесей гомополимеров, так как
из-за возможного влияния растворителя на показатель поглощения
аналитических полос зависимость их интенсивности от состава может
быть нелинейной. Градуировочные кривые применяют также при анализе
сополимера, когда в методике использовано отношение интенсивностей
пОлос поглощения его компонент.
При приготовлении эталонов хорошие результаты дает расчет состава
по анализу исходной смеси мономеров известного объема (или массы)
до полимеризации и смеси непрореагировавших мономеров, как это
93
Д
0,751-
•Q5
Рис.IV.5. Зависимость интенсивности поло-
сы поглощения трифторэтилена от его
содержания в смеси с тетрафторэтиленом.
Q25
О
показано на примере сополимера тет-
рафторэтилена с трифторэтиленом (см.
раздел IV.1.3). Этот метод становится
неэффективным, когда реакционные
способности мономеров сильно разли-
чаются, сополимер обогащается одним
компонентом и измерение состава
_1----1-----1----1----i-возвратной смеси мономеров оказы-
20 40- 60 60 ЭДОвается на грани точности анализа.
’ ° Анализ смеси мономеров до и после
полимеризации можно выполнить спектральным методом по интенсив-
ности полос поглощения мономеров (или одного мономера для двух-
компонентной смеси), построив соответствующий градуировочный
график. Поскольку зависимость интенсивности полосы поглощения
мономера от его концентрации в смеси может быть нелинейна (рис. IV.5),
то построение градуировочного графика для смеси мономеров разного
состава необходимо Для съемки спектров жидких мономеров с низ-
кими температурами кипения можно использовать газовые кюветы.
Перед заполнением кювету вакуумируют и заполняют парами исследу-
емой смеси мономеров. Такой метод получения эталонов для анализа
сополимеров трудоемкий, но дает надежные результаты.
IV. 1.3. Анализ по гомополимерам и по балансу мономеров
Наиболее удобно использовать гомополимер в качестве эталона при
наличии в его спектре изолированной полосы. Примером может служить
методика определения состава сополимера тетрафторэтилена с трифтор-
этиленом, которая основана на измерении интенсивности полосы погло-
щения 2987 см , относящейся к валентному колебанию С—Н трифтор-
этилена. Так как политетрафторэтилен в этой области прозрачен, то все
поглощение происходит за счет трифторэтилена (рис. IV.6). Проверено,
что контур полосы 2987 см-1 сохраняется постоянным в спектрах
политрифторэтилена и сополимеров разного состава. Интенсивность этой
полосы невелика, поэтому анализ можно осуществлять на прессованных
пленках толщиной 100—400 мкм.
Методика заключается в следующем. Записываем спектры пленок
политрифторэтилена разных толщин в области полосы 2987 см"1.
Определяем ее оптическую плотность для каждой пленки в максимуме
полосы. По графику зависимости оптической плотности от толщины
пленки находим показатель поглощения для полосы 2987 см"1 (& =
= 75 см"1). Получив из спектра сополимера значение оптической плот-
ности полосы 2987 см"1 D, находим эффективную толщину компо-
ненты трифторэтилена в пленке сополимера </] =D/k. Тогда величина
94
d\l(d-di) характеризует отношение эффективных толщин компо-
нент сополимера трифторэтилена с тетрафторэтиленом в образце.
Чтобы получить отношение числа молекул одной компоненты сополиме-
ра к числумолекулдруго^надо^/^—dj) умножить на х =PiAf2/p2Afi.
где Afj и М2 — молекулярные массы звеньев трифторэтилена и тетра-
фторэтилена, pi и р2 плотности соответствующих гомополимеров.
В нашем случае х близко к 1, поэтому отношение эффективных толщин
дает непосредственно отношение числа । молей мономеров, вступивших
в сополимеризацию. Молярное содержание одной из компонент со-
полимера, например трифторэтилена ct (в %), определяется так:
ci = dt -ioo/а
Применение этой методики основано на двух допущениях: во-пер-
вых, объем сополимера аддитивно составляется из объемов политетра-
фторэтилена и политрифторэтилена; во-вторых, поглощение на одну
группу ^СН— в спектрах политрифторэтилена и сополимера одинаково.
Справедливость этих допущений доказана хорошим совпадением значе-
ний для эталонных образцов с результатом их анализа по описанной
выше методике.
Эталонами для проверки методики служили образцы сополимеров,
состав которых определяли по балансу газообразных смесей мономеров
(тетрафторэтилена и трифторэтилена) до и после полимеризации. Для
анализа смесей мономеров использовали полосу поглощения трифтор-
этилена 750 см-1 (тетрафторэтилен прозрачен в этой области). Граду-
ировочная зависимость оптической плотности этой полосы от объемного
содержания трифторэтилена в смеси представлена на рис. IV .5. Она
позволила осуществить контроль за количеством вступившего в сополи-
мер трифторэтилена. Зная объемы смесей до и после сополимеризации,
можно рассчитать состав эталонных образцов. Результаты анализа эта-
лонных образцов, полученных по разработанной методике и по балансу
мономеров, хорошо согласуются:
Содержание трифторэтилена, %
по интенсивности полосы 44 44
2987 см1
по анализу смесей мономеров 43 39
Поскольку в методику заложено из-
мерение абсолютного значения интенсив-
ности полосы поглощения (а не отношения
интенсивностей полос), то для уменьше-
ния погрешности, связанной с определением
Рис. IV.6 ИК-спектры поглощения политрифтор-
этилена (7), сополимера тетрафторэтилена с
трифторэтиленом (2) и политетрафторэтилена (5).
Толщина пленки (в мкм): 1 - 90; 2- 100; 3 - 380.
57 74 26
59 74 23
95
толщины пленки, для каждого образца прессовали по 3 пленки сополи-
мера, отличающиеся по толщине.' Толщину каждой пленки измеряли
в 9 точках по всей ее площади, освещенной пучком спектрофотометра,
и вычисляли среднее значение. Разброс результатов анализа при парал-
лельных измерениях не превышал 3%.
IV .1.4. Анализ по эталонным образцам
Большинство сополимеров имеют сложные спектры с интенсивными
полосами, которые могут частично или Полностью перекрываться. Для
определения интенсивности таких полос нужны очень тонкие пленки
(несколько микрометров), изготовление которых связано с труднос-
тями, а измерение толщины вносит большую ошибку в значение ин-
тенсивности. Такие спектры не дают возможности использовать в качес-
тве эталонов гомополимеры или их механические смеси. В этом случае
за эталоны принимают образцы сополимеров, состав которых определен
другим методом.
Рассмотрим на примере сополимера этилена с винилацетатом методи-
ки определения его состава. Подход к решению этой задачи может
быть использован и при исследовании других сополимеров.
Спектры полиэтилена (рис. IV.7) имеют очень интенсивные полосы
поглощения валентных колебаний групп —СН2— при 2853 и 2925 см-1.
Менее интенсивное поглощение наблюдается при 1465 и 725 см-1 ,
* В спектре кристаллических парафинов и полиэтилена обе полосы поглоще-
ния расщепляются в дублет 1460-1470 и 720-730 см'1.
___ ।__।__1.11--------i__i--1 1 ।--1--1---1—1---1 —।
3600 2800 2000 1800 1600 1400 1200 1000 800
V, СМ’’
Рис. IV.7. ИК-спектры поглощения полиэтилена (1), сополимера этилена с моляр-
ным содержанием винилацетата 3 (2) и 32% (3) и полнвинилацетата (4).
Толщина пленки (в мкм) : 1 — 10; 2-25; 3, 4 — пленка на окне.
96
Рис. IV.8. Зависимость отношения
оптических плотностей полос погло-
щения DvjDv2 от состава сополимера
этилена с винилацетатом (с - моляр-
ное содержание этилена) :
1 - О2’25/О1465 ; 2 - D'333 /О1375 ; 3 -
р1125/£)1зЬ. 4 _ Д1020 ,р1375 . j _
О’45 /О1375 .
и относится к деформационным
колебаниям этих групп (ножнич-
ным и маятниковым, соответ-
ственно). Полоса 725 см"1 про-
является в спектре, если осу-
ществляется последовательность
из пяти и более групп -СН2 —
подряд, и может быть исполь-
зована для исследования рас-
пределения звеньев сополимера.
Интерпретация полос поглоще-
ния поливинилацетата приведена
в работе [45,46].
Слабое поглощение валент-
ных колебаний групп -СН2 —
и -СН3 винилацетатных звень-
ев маскируется интенсивным по-
глощением этиленовых звеньев
в спектре сополимера (см. рис.
IV.7). Интенсивная полоса v (С=О) при 1736 см'1 изменяет контур
в зависимости от состава сополимера. Нестабилен контур и полосы
1240 см'1, относящейся к колебанию v (С=О) .
Чтобы выбрать аналитические полосы поглощения, были построены
графики зависимости отношения оптических плотностей полос, при-
надлежащих к разным колебаниям каждого из сомономеров, от содер-
жания этилена в сополимере (рис. IV .8). Отношение интенсивностей
полос 2925 и 1465 см'1 этилена (рис. IV.8, 7), а также полос 1433
и 1375 см'1 винилацетата (рис. IV.8, 2) сохраняется постоянным при
изменении состава сополимера, следовательно, эти полосы могут быть
использованы для его определения. Изменение интенсивности полос
1 125, 1020 и 945 см'1 (рис. IV.8, 3-5) винилацетата от содержания
этилена свидетельствует о структурной чувствительности полос, и не
позволяет их использовать для определения состава. Таким образом,
установили, что определение состава сополимера этилена с винилаце-
татом можно проводить по полосам 2925, 1465, 1433 и 1375 см'1.
Для обеспечения необходимой чувствительности метода выбирают
ту или иную полосу в зависимости от ее интенсивности и состава поли'
мера.
97
В зависимости от состава сополимер может обладать свойствами
эластичных термопластов, каучукоподобных или твердых материалов.
Поэтому образцы для анализа могут быть получены в виде растворов
или прессованных пленок. Эталонами служили образцы сополимеров,
состав которых был определен химическими методами (омылением,
элементным анализом).
При определении состава сополимера этилена с винилацетатом
(с молярной долей этилена от 20 до 70%) использовали полосы погло-
щения 2926 и 1375 см -1. Методика основана на растворении сополимера
в тетрахлорметане. Концентрация раствора 10 г/л, толщина слоя раствора
0,25 мм (кювета постоянной толщины). Измерения проводились относи-
тельно растворителя, помещенного в кювету переменной толщины.
Строили градуировочную зависимость отношения оптических плотнос-
тей полос Z)2926 /7)1375 от отношения молярных концентраций этилена
и винилацетата в эталонных образцах сополимера. Зависимость линейная
(рис. IV.9). Поскольку в поглощение этилена при 2926 см-1 вносит
вклад поглощение винилацетата, то градуировочная прямая отсекает
по оси ординат отрезок, соответствующий этому поглощению.
Анализ состава сополимера в указанных пределах содержания
этилена можно выполнять, используя не только растворы, но и прес-
сованные пленки толщиной 100 мкм. Для этого в работе [47] была
использована малоинтенсивная полоса поглощения 797 см ~1, относя-
щаяся к маятниковым колебаниям групп —СН2— винилацетата. Строили
градуировочную зависимость интенсивности полосы 797 см -1 от состава
эталонных образцов, который оценивали также методами омыления
и элементного анализа.
от отношения молярных концентраций этилена
и винилацетата.
Рис. IV.10. Аналитические полосы поглощения сополимера этилена с винилаце-
татом (с молярным содержанием этилена от 70 до 95%).
98
Определение состава сополимера этилена с винилацетатом (с моляр-
ной долей этилена от 70 до 95% удобно проводить по полосам поглоще-
ния 1375 и 1465 см-1 (рис. IV.10). Градуировочный график аналогичен
рис. IV.9. Не все сополимеры этого интервала составов хорошо раствори-
мы. Поэтому образцы готовили в виде прессованных пленок толщиной
10-20 мкм. Чтобы исключить из расчетов значение толщины пленки ис-
пользовали отношение оптических плотностей полос.
Избежать приготовления тонких пленок можно применением метода
МНПВО. В работе [48] этот метод использован для анализа состава двух-
слойных пленок сополимера этилена с винилацетатом. Пленки получали
соэкструзией сополимера с молярной долей винилацетата (от 0 до 20%)
и полиэтилена. Аналитическими полосами служили полосы 1736 (винил-
ацетата) и 1465 см'1 (этилена). Содержание винилацетата определяли
по градуировочной зависимости отношения интенсивностей полос
D1,36/£)1 465 от состава эталонных образцов, для оценки которых ис-
пользован метод омыления. Градуировочный график линеен*. Примене-
ние метода МНПВО не только исключило из анализа процедуру приго-
товления тонких пленок, но дало возможность анализировать непосред-
ственно двухслойную пленку, а также определить в ней толщину слоя
сополимера dc по формуле:
CBAd/CBA
где еВА и еВА “ молярные доли винилацетата в сополимере, рассчитанные по
градуировочному графику при записи спектра пропускания и отражения соот-
ветственно; d - общая толщина двухслойной пленки.
Определение состава сополимера содержащего 85—90% этилена, мож-
но также проводить по малоинтенсивным полосам поглощения 607 см~1
(деформационные колебания ацетатных групп) и 3470 см-1 (первый
обертон С= О-колебаний). При этом использовали образцы в виде пле-
нок толщиной 0,1 мм (607 см'1) и пластин 1 мм (3470 см'1) [46].
Анализ проводили, используя значения показателя поглощения гомопо-
лимера, а для проверки результатов — эталонные образцы. Следует иметь
в виду, что при применении массивных образцов (толщиной 1 мм и бо-
лее) возможно проявление в спектре полос поглощения примесных
групп (остатков эмульгаторов, воды, а также таких, как концевые
и т. п.). Поглощение примесных групп может налагаться на поглощение
аналитических полос и вносить нерегулярные ошибки в их интенсивность.
Этот фактор следует учитывать при разработке методики: исследовать
спектры образцов сополимера разной степени чистоты, разного метода
получения и т. п., чтобы внести соответствующие коррективы в процеду-
ру измерения интенсивности полос, если удалить примеси окажется
невозможным.
Анализ состава тройных сополимеров принципиально не отличается
от анализа двойных. Возникают дополнительные трудности, связанные
* В ограниченных интервалах составов изменение полуширины полосы
1736 см"1 не сказывается.
99
с усложнением спектра, а следовательно, и с ограничением выбора ана-
литических полос поглощения. Однако для тройных сополимеров кроме
смеси гомополимеров в качестве эталонов целесообразно использовать
смеси двойных сополимеров, состав которых известен. Например,
для анализа тройного сополимера этилена с пропиленом и гексеном-1
использовали механические смеси сополимеров этилена с пропиленом
и пропилена с гексеном [49]. Такая смесь является лучшей моделью
анализируемого сополимера, чем механическая смесь отдельных го-
мополимеров.
Из тройных сополимеров большое промышленное значение имеют
АБС-пластики, получаемые прививкой сополимера стирола с акрилонит-
рилом на бутадиеновой, бутадиен-стирольный и другие каучуки. Сополи-
меры частично растворимы. Растворимая часть представляет собой в
основном сополимер стирола с акрилонитрилом, нерастворимая —
гель-фракцию (привитой сополимер). Несмотря на то, что раствор
АБС-пластика не является истинным, на окнах NaCI удалось получить
пленки после выпаривания растворителя [50, с. 54]*. Растворителем
служил бензол. Концентрация раствора 2-5% (по массе). Из-за негомо-
генности раствора пленки оказываются сильно рассеивающими. Компен-
сируя рассеянный свет путем ослабления пучка сравнения, получали
удовлетворительные для анализа спектры (рис. IV. 11). Отношение кон-
центраций САН/Спс акрилонитрила (АН) и полистирола (ПС) определя-
ли по отношению интенсивностей полос 2248 см-1 (валентное колеба-
ние связи С = N) и 1875 см-1 (составное колебание бензольного кольца).
В эталонных образцах сополимера содержание акрилонитрила определя-
ли по методу Кьельдаля [50, с. 7]. Для определения отношения концен-
траций каучука (К) и полистирола Ск/Спс использовали отношение
интенсивностей полос 967 см-1 (деформационное колебание связи С—Н
в группе —СН=СН—) и 1027 см-1 (деформационные колебания связи
С- Н бензольного кольца). Эталоны готовили на основе растворов сме-
сей каучука и полистирола, из которых отливали пленки. Определив
по градуировочным графикам САН/Спс и Ск/Спс и учитывая, что
* В брошюре [50] описаны методики определения состава двухкомпонентных
ударопрочных полистиролов и АБС-пластиков методами ИКС.
Т,7„
Ю0г
4000 3200 2400 1900 1700 1500 1300 1100 900 700
Рис. IV.11. ИК-спектр поглощения АБС-пластика.
Толщина пленки 32 мкм.
100
А, „мкм
Рис. IV.12. Спектры поглощения полистирола (1) и полиакрилонитрила (2) в
ближней ИК-области.
Толщина пленки (в мм) : 1 — I ; 2 - 0,4.
общее количество компонентов сополимера 100%, относительную долю
каждого компонента b рассчитывали по формулам:
=__________100____________
пс (Ск/Спс) + 1+(Сан/Спс) ;
t>ne{CK/Cnc)', Ьрм = Ьпс(Сдн/Спс)
Определенное преимущество для анализа сополимеров имеет ближ-
няя инфракрасная область спектра. В частности, это относится к анализу
сополимера стирола с акрилонитрилом. При этом можно использовать
удобные в работе прессованные образцы сополимера толщиной до 1 мм
и более. Аналитической полосой полистирола может быть полоса перво-
го обертона валентных колебаний С—Н 1,68 мкм (5952 см-1), полиак-
рилонитрила — полоса 1,95 мкм (5128 см-1), являющаяся полосой
составных валентных колебаний групп —C^N и —СН2— (рис. IV. 12).
Эталонами являются гомополимеры. По показателям поглощения,
рассчитанным из их спектров, находят содержание каждого ком-
понента сополимера. Поскольку количество стирола и акрилонитрила
в сополимере определяется независимо, то общий баланс (^пс+^АН =
= 100%) служит хорошей проверкой такой методики.
IV.1.5. Сочетание ИК-спектроскопии с пиролитической хроматографией
Иногда сополимеры из-за плохой растворимости, невозможности приго-
товления пленок необходимой толщины и т. п. оказываются трудными
объектами для анализа методом спектроскопии. Часто и химические
101
методы при этом недостаточно эффективны. Тогда прибегают к исполь-
зованию сочетания разных методов. Например, в настоящее время для
исследования строения полимерных материалов широко используют их
пиролитическое разложение. Комбинирование методов пиролиза и ИК-
спектроскопии позволяет обойти трудности препарирования образцов
и выполнить анализ состава сополимера. Кратко остановимся на некото-
рых работах по этому вопросу.
При помощи пиролитического разложения полиамида-6 и полиами-
да-6,6 проводили количественное определение его состава [51]. Пиролиз
осуществляли в атмосфере аргона при температуре 500 °C. Пиролизат,
растворенный в тетрахлорметане, исследовали методом ИК-спектроско-
пии. Для анализа использовали полосы 2920 см-1 (валентное колебание
групп —СН2— 1680 см-1 (валентное колебание группы С=О капролак-
тама) . Градуировочные кривые были построены на основе смесей гомо-
полимеров.
В работе [50, с. 9] рассмотрена методика определения содержания
стирола в сополимерах методом пиролиза со спектроскопическим окон-
чанием. Анализ стирола в пиролизате проводили по полосе 1492 см-1
(деформационное колебание бензольного кольца). Продукты пиролиза
и эталонные образцы стирола растворяли в тетрахлорметане. Аналогично
определяли состав сополимера стирола с акрилонитрилом [52]. Конден-
сат пиролизата состоял из мономеров и был исследован по полосам по-
глощения 2240 (акрилонитрил) и 3020 см"1 (стирол). Для градуиро-
вочного графика использованы соответствующие мономеры. При помо-
щи пиролитической газовой хроматографии был проанализирован состав
АБС-пластика [53]. Полученные результаты авторы сопоставили с данны-
ми ИК-спектроскопии. За аналитические полосы принимали полосы
бутадиенового каучука 967 см-1 (деформационное колебание группы
—СН=СН—) полистирола 1028 см"1 (деформационное колебание С— Н
бензольного кольца). Эталоны — смесь гомополимеров. Акрилонитрил
определяли обычным способом, описанным выше [50, 52].
Из небольшого перечня упомянутых выше работ по спектрально-пи-
ролитическому определению состава сополимеров видно, что они прово-
дятся по однотипной схеме: пиролитическое разложение сополимера и
анализ пиролизата методом ИКС. Анализ сополимера этилена с пропи-
леном рассмотрен в [54]. Газообразные, жидкие и смолообразные про-
дукты пиролиза сополимера исследованы в виде раствора в тетрахлор-
метане. В статье имеются сведения о полосах поглощения пиролизата
сополимера, которые мы считаем необходимым привести:
12, см"1 Отнесение
3090 р(СН) олефинов
2960-2940 (va (СН) в группе -СН3
ра(СН) в группе -СН,-
2870-2820 fvs(CH) в группе -СН3
|vs (СН) в группе -СН2 -
1630-1670 НС=С)
1470 (бдССН) в группе -СН3
(6 (СН) в группе -СН2—
102
1380
1150
990
970
910
890
6S(CH) в группе -СНЭ
(СН2)
(СН) в группе -СН= СН2
J(CH) в группе -СН=СН- (транс)
(СН) в группе -СН=СН2
(СН) в группе -RR'c=CH2
*Внеплоскостные деформационные колебания.
Нельзя считать, что к пиролитическому методу прибегают только в
случае, когда возможности других методов исчерпаны. Как видно из
приведенных данных (см. раздел IV. 1,4), состав сополимеров, например
АБС-пластиков, сополимеров стирола с акрилонитрилом, может быть
определен полностью одним методом ИК-спектроскопии. Однако при-
ходится преодолевать значительные трудности в приготовлении образ-
цов для получения ИК-спектра, и методические ухищрения для достиже-
ния результата не всегда оправдывают себя. Использование в помощь
ИКС пиролитического метода упрощает анализ сложных образцов. В
большинстве работ получение продуктов пиролиза и съемка их спектров
разделены методически. Удобнее, когда эти операции совмещены. Так,
например, для определения содержания акрилонитрила в хлоропрено-
вом каучуке [55], подвергаемый пиролизу образец, помещали в пироли-
тическую приставку к спектрофотометру. Она представляла собой ячей-
ку с окнами NaCl. После пиролиза (1000 °C) в течение 40 с и охлажде-
ния ячейки до 20 °C записывали спектры ИКС продуктов, сконденси-
рованных на окнах NaCl. Аналитической служила полоса акрилонитри-
ла 2250 см”1.
Если экспериментатор имеет в своем распоряжении спектрофото-
метр снабженный пиролитической ячейкой и одновременно систему
хроматографического разделения продуктов пиролиза и их анализа, то
без сомнения такой комбинированный метод обладает большими преи-
муществами перед традиционным методом ИК-спектроскопии при
исследовании сложных неразделенных объектов.
1V.2. ПОСЛЕДОВАТЕЛЬНОСТЬ РАСПРЕДЕЛЕНИЯ МОНОМЕРНЫХ
ЗВЕНЬЕВ В МАКРОМОЛЕКУЛАХ СОПОЛИМЕРОВ
Определение последовательности распределения мономерных звеньев
сополимеров базируется на исследовании структурно-чувствительных
полос спектров. ИК-спектры полимерных материалов, так же как и дру-
гих соединений, определяются их химической структурой и геометрией
молекулы. Рассмотрим сополимер с малым содержанием одного из
компонентов. Он представляет собой в основном цепи гомополимера
с включенными в них звеньями другого сомономера. Увеличение содер-
жания этого компонента изменяет геометрию макромолекулы, возника-
ют новые связи между звеньями. В зависимости от природы сополиме-
ризующихся мономеров все эти изменения могут в большей или
103
меньшей степени отражаться на спектре сополимера и быть основой для
исследования регулярности распределения мономерных звеньев. По-
скольку маятниковые колебания наиболее чувствительны к количеству
связанных между собой по цепи „маятников”, а также к влиянию окру-
жающих атомов, то исследование этих колебаний дает наибольшую
информацию о распределении звеньев мономеров. Большой фактический
материал накоплен по исследованию маятниковых колебаний групп
—СН2— сополимеров этилена с разной комбинацией мономеров.
Характер распределения звеньев влияет на внутримолекулярные
и межмолекулярные взаимодействия, что сказывается также и на других
колебаниях макромолекул, вызывая смещение полос поглощения,
изменение их интенсивности и контура. Однако по сравнению с полосами
маятниковых колебаний полосы других колебаний оказываются менее
информативными.
Кроме того, проявление распределения мономерных звеньев со-
мономеров может быть связано с нарушением стереорегулярного строе-
ния гомополимера при внедрении в его макромолекулу звеньев сомоно-
мера и, следовательно, затрагивает стереочувствительные полосы.
Исследование маятниковых колебаний сополимеров этилена с моно-
мерами типа (—СН2—CHR—)„, проведенное многими авторами*, позво-
лило выявить зависимость частоты маятниковых колебаний групп —СН2 —
от длины метиленовых последовательностей в сополимерах. Эта за-
висимость была установлена на модельных углеводородах и хорошо
коррелирует со значениями частот сополимеров этилена с пропиленом
[56, с. 143].
Ниже приведены значения волновых чисел маятниковых колебаний
для различных последовательностей метиленовых групп в сополиме-
рах этилена:
Значение и в последовательности (-СН2-)„ Волновое число v, см" 1 1 2 3 4 5
этилен с пропиленом [56, с. 143] 815 751 733 726 722
,, с трифторпропиленом 852 755 737 — 722
(57| ,, с винилхлоридом [58] 833 770 752 726 722
„ с винилацетатом [46] 797 — 738 — 720
Из представленных данных видно, что частоты колебаний, относя-
щиеся к одним и тем же последовательностям групп — СН2 - в различных
сополимерах различны. Такой эффект наблюдается, однако, лишь для
последовательностей малой длины, и максимальное различие имеется
для единичной группы — СН2 —. Это естественно, так как единичная груп-
па -СН2- с двух сторон испытывает непосредственное влияние сосед-
них, „чужих” звеньев.
Рассмотрим последовательность распределения звеньев наиболее
изученного сополимера этилена с пропиленом. Присутствие в спектре
сополимера полосы 815 см-1 свидетельствует о том, что звенья
* Литературу по этому вопросу см. в 18, 56].
104
пропилена изолированы от звеньев этилена и образуют блоки- Образо-
ванию последовательности
—сн3—сн—сн2—сн2—сн2—сн—
I I
СНз СН3
соответствует в спектре полоса 733 см"1, при этом единичное звено
этилена соединено с нормально расположенным звеном пропилена. Ано-
мальное присоединение пропилена
—сн2—сн—сн2—сн2—сн—сн2—
I I
СНз СНз
сопровождается появлением в спектре полосы при 751 см'1. Блокам
этилена всегда сопутствует в спектре полоса 722 см-1. Спектры сополи-
мера, содержание компонентов которых олизко к эквимолярному,
сложны в области маятниковых колебаний групп СН2 - из-за близких
частот и7следовательно, наложения полос поглощения. Если при этом
на основании индивидуальных соединений определить показатели по-
глощения полос в последовательностях (-СН2-)Л для разных п, то,
зная положение полос, при помощи ЭВМ можно получить количествен-
ные данные о распределении этих последовательностей в сополимере.
При малом содержании этилена в сополимере спектр проще, что
позволяет произвести графическое разделение полос и вычислить их
показатели поглощения. Так, например, в работе [59] исследована по-
следовательность присоединения мономерных звеньев при молярном
содержании этилена до 15%. На рис. IV.13 [59] представлен спектр погло-
щения сополимера пропилена с молярным содержанием этилена 9%, в
котором присутствуют полосы 733 и 722 см~1 *. Это означает, что кроме
одиночных звеньев этилена с последующим нормальным присоединени-
ем пропилена (733 см”1), цепи сополимера имеют блоки этилена, состо-
ящие в основном из двух звеньев. Авторы работы [59] рассчитали число
изолированных звеньев этилена и звеньев этилена, вошедших в блоки.
Молярные коэффициенты экстинции определяли по спектрам модель-
ных соединений.
Анализ полос поглощения маятниковых колебаний групп СН2- в
спектрах других сополимеров на основе этилена дает аналогичную инфор-
мацию о распределении звеньев. По полосам поглощения этих колебаний
можно выявить аномальное присоединение асимметричного звена и ха-
рактер чередования сомономеров. Наличие в снекзре сополимера этиле-
на с трифторпропиленом
—сн2—сн—сн2—сн2—сн--сн2—сн—сн2
CF3 CF3 CF.(
* Полоса 815 см-1, соответствующая блокам пропилена, наличие которой в
спектре не вызывает сомнений, в работе ]59] не обсуждается.
105
Рис. IV.13. ИК-спектры сополимера этилена с пропиленом (молярное содержание
этилена 9%>.
Толщина пленки 0,41 мм.
Рис. IV.14. ИК-спектры поглощения сополимеров этилена с трифторпропиленом.
Молярное содержание этилена (в %) ; 1 — 40; 2 — 77; 3 — 84.
слабой полосы 852 см"1, соответствующей единичной -СН2- группе,
указывает на образование блоков трифторпропилена даже в сополимере,
обогащенном этиленом (рис. IV.14) [57], а плеча полосы 755 см"1 -
на некоторое количество аномальных присоединений.
Последовательности, состоящие из четного числа групп (СН2)„
(л=2, 4), будут присутствовать в сополимере этилена, если асимметрич-
ное звено мономера (CH2=CHR) присоединено к звену этилена аномаль-
но, т. е. по типу „голова—голова”
—сн2—сн—сн2—сн2 —сн—сн2—
R R
Как видно из приведенных выше данных, такому присоединению припи-
саны полосы поглощения в спектрах сополимеров этилена с пропиленом,
этилена с трифторпропиленом и этилена с винилхлоридом. Аномаль-
ное присоединение может наблюдаться и в других сополимерах. Доказа-
тельством этому служат обычно спектры ИКС и ЯМР. Так, например,
присоединение „голова—голова” в молекулах сополимера этилена с
пропиленом подтверждено обоими методами. Однако было показано
[60], что наличие аномальных присоединений звеньев этого сополимера
наблюдалось не во всех случаях и зависело от применяемой каталити-
ческой системы, которая влияет на формирование структуры сополимер-
ной цепи. Аномальное присоединение звеньев сополимера этилена с ви-
нилацетатом по полосам поглощения маятниковых колебаний групп
—СН2— не доказано. На основании других методов исследования также
нет твердой уверенности в их существовании, однако не отрицается
и возможность таких присоединений [56, с. 137].
106
На рис. IV.15 представлен спектр поглощения этого сополимера
разного состава [47]. Установлено, что полоса 797 см-1 относится к
одиночной группе —СН2—. Уменьшение содержания винилацетата и уве-
личение доли звеньев этилена в сополимере вызывает следующие изме-
нения в спектре. Закономерно уменьшается интенсивность полосы
797 см-1, появляется сложное поглощение в низкочастотной области
спектра (рис. IV. 15, 2), в котором можно выделить полосу около
740 см-1, приписанную [46] последовательностям (-СН2—)3 - единич-
ное звено этилена и нормально присоединенное к нему звено винилаце-
тата. В спектре сополимера при молярном содержании этилена 68%
наиболее интенсивной в спектре оказывается полоса 720 см"1, относя-
щаяся к блокам этиленовых звеньев. Асимметричный склон ее возмож-
но скрывает полосу 740 см"1. Удивляет отсутствие полосы 720 см"1 в
спектре сополимера почти эквимолярного состава (43%) этилена (см.
рис. IV.15, 3) при достаточно интенсивной еще полосе 797 см-1 (блоки
звеньев винилацетата). Возможно, к последовательностям этиленовых
звеньев в этом случае относится полоса около 725 см-1.
Сведения о распределении звеньев в сополимерах этилена с пропиле-
ном или с винилацетатом, полученные из анализа спектров маятниковых
колебаний групп —СН2—, полезно дополнить информацией о поглощении
У, см"'
Рис. IV.15. ИК-спектры поглощения сополимеров этилена с винилацетатом.
Молярное содержание этилена (в %): 1 - 0; 2 - 25; 3 - 43; 4 - 68.
Рис. IV.16. ИК-спектры поглощения сополимеров этилена с винилацетатом.
Молярное содержание винилацетата (в %) : 1 — О; 2 — 18; 3 — 36; 4 - 100.
107
аналогичных колебаний групп -СН3. Полоса 935 см-1 в спектре сополи-
мера этилена с пропиленом приписана колебаниям изолированных
звеньев пропилена, а полоса 970 см”1 — блокам пропилена из четырех
и более звеньев. Отношение интенсивностей этих полос D935/£)970 было
использовано для оценки степени чередования звеньев этилена и пропи-
лена в сополимере [61]. Поскольку полосы поглощения в областях
700—800 см”1 и 900—970 см-1 характеризуют одно и то же распределе-
ние звеньев, но по разным мономерам сополимера этилена с пропиленом,
то одновременное исследование полос колебаний в этих областях даст
наиболее полное представление о строении молекулы сополимера.
Маятниковые колебания групп —СН3 проявляются в спектре сопо-
лимера этилена с винилацетатом при 945 см-1 (рис. IV. 16). Эта полоса
относится к единичной группе —СН3, т. е. к блокам винилацетата. По
мере увеличения содержания этилена в сополимере интенсивность поло-
сы уменьшается, на ее склоне появляется полоса 958 см-1, интенсив-
ность которой увеличивается. Интерпретация этой полосы не ясна. Со-
поставление спектров поглощения маятниковых колебаний групп —СН2~
и >СН-СН3 дает возможность предположить, что полоса 958 см-1 отно-
сится к маятниковым колебаниям групп —СН3 винилацетата при его
чередовании со звеньями этилена.
На примере маятниковых колебаний мы рассмотрели закономернос-
ти, наблюдаемые в спектре сополимеров в зависимости от состава, ко-
торые могут быть использованы для определения последовательности
присоединения мономерных звеньев.
Спектры сополимеров, мономеры которых склонны к стереоспеци-
фической полимеризации, часто обладают также пригодными для анали-
за структурно-чувствительными полосами. Например, полоса 1060 см-1
полиметилметакрилата характерна для его синдиотактической конфи-
гурации. При сополимеризации метилметакрилата с акрилонитрилом
полоса 1060 см-1 исчезает с увеличением доли акрилонитрила в сопо-
лимере. Это является следствием нарушения последовательности блоков
метилметакрилата синдиотактического строения звеньями акрилонитри-
ла, вошедшими в цепь. При эквимолярном составе сополимера блоков
метилметакрилата не обнаружено [62]. Полоса 1060 см-1 может служить
мерой длины блоков синдиотактического метилметакрилата. Она про-
является в спектре, когда число звеньев в блоке равно трем и больше.
Для определения количества блоков стирола в его сополимерах
с другими мономерами используют полосы поглощения в области 540-
560 см-1, относящиеся к колебаниям бензольного кольца и отражаю-
щие стереоспецифичность макромолекулы- Атактический полистирол
имеет интенсивную полосу поглощения 540 см-1, которая проявляется
в спектре при последовательности мономерных звеньев стирола более
5. В спектре изотактического полистирола изолированным звеньям со-
ответствует полоса 560 см-1, блокам из 2 и 3 звеньев — 550—555 см-1,
блокам из 4 и 5 звеньев - полоса при 545 см-1. Видно, что полосы по-
глощения сополимеров с большим содержанием стирола, когда возмо-
жен набор блоков разных последовательностей, сильно перекрываются.
108
Для статистических сополимеров вряд ли возможна однозначная интер-
претация этого спектра. Однако при исследовании структуры блоксопо-
лимеров стирола исследование указанных полос поглощения дает полез-
ную информацию [63 ].
Структурная чувствительность полос очень часто проявляется в
изменении их интенсивности, полуширины или положении в спектре.
Иногда эти изменения достаточно существенны, аргументированы и мо-
гут служить основой для разработки соответствующих методик анализа
сополимеров. Но значительно чаще — это предмет для исследования стро-
ения макромолекул, их внутримолекулярного и межмолекулярного
взаимодействий.
Как было уже упомянуто выше (см. раздел IV. 1.1), полоса погло-
щения валентных колебаний карбонильной группы кислородосодержащих
полимеров изменяет свою частоту, полуширину в зависимости от раст-
ворителя или типа соседних мономерных звеньев в сополимерах. Оста-
новимся подробнее на нескольких работах, посвященных исследованию
этих изменений. В разделе (IV.1.1) показано, что полоса группы
винилацетата 1736 см-1 изменяет полуширину в зависимости от состава
сополимера этилена с винилацетатом. Исследовали сополимерные плен-
ки. Уширение полосы составляло 10 см-1 [от 14 см-1 для сополимера
этилена с молярной долей 2,5% винилацетата до 24 см-1 для поливинил-
ацетата]. Растворение поливинилацетата или сополимера этилена с винил-
ацетатом в тетрахлорметане не влияло на полуширину полосы. Уширение
полосы связали с внутримолекулярным взаимодействием звеньев
сополимера.
Аналогичное явление наблюдали в работе [64], где исследовали изме-
нение полуширины полосы связи С=О в спектрах стирола с метилмета-
крилатом. Полуширина изменялась от 36 см-1 (полиметилметакрилат)
до 17,5 см- 1 (при молярном содержании метилметакрилата 22,5%) .Авто-
ры пытались связать эти данные с распределением звеньев, вычисляя ве-
роятности разных связок мономеров, и пришли к заключению, что основ-
ной причиной является внутримолекулярное возмущение колебаний
группы ^С=О.
Таким образом, видно, что факт изменения полуширины полосы
1736 см-1 (С=О) связан с распределением звеньев в сополимерах, но
этого недостаточно, чтобы делать какие-либо заключения о порядке их
распределения или величине блоков.
В работе [42] также исследовали поведение полосы группы ^СО=О
в спектрах сополимера этилена с винилацетатом. Сополимер растворяли
в трихлорметане. Наблюдали сдвиг полосы карбонила 1737 см-1, кото-
рый связывали с образованием триад в макромолекулах сополимера.
IV.3. ОПРЕДЕЛЕНИЕ ПРОИЗВЕДЕНИЯ КОНСТАНТ СОПОЛИМЕРИЗАЦИИ
Распределение звеньев с макромолекуле сополимера зависит от многих
факторов: типа и способа полимеризации, реакционной способности мо-
номеров, их концентрации в исходной смеси, применяемых катализато-
109
ров сополимеризации и т. п. Характер распределения звеньев (регуляр-
ное их чередование, образование блоков, статистическое присоединение
мономерных единиц) влияет на свойства сополимеров и поэтому подле-
жит изучению и анализу.
Поскольку ИК-спектры чувствительны к распределению мономер-
ных звеньев, то в ряде случаев можно определить долю звеньев, входя-
щих в блоки или являющихся изолированными. Наибольшую информа-
цию при этом из ИК-спектров поглощения можно получить для сополи-
меров этилена.
Порядок присоединения мономерных звеньев в процессе роста полимерной
цепи подчиняется определенным законам.
Напишем четыре основные типа элементарных реакций роста цепи:
-М1-+М1 -Mi*
~Мг + М2 —Мг*
~М2- + Mj -Mr-
—М2 • + Мг М2*
где М2 и М2 — мономеры; к - константы скорости реакции.
Присоединение звеньев М, и М2 определяется константами сополимеризации
г, и гг и концентрацией мономеров, при этом:
г 1~ ^11/^12 г2 = кгг/к21
Вероятности присоединения звеньев М, к М! обозначают через ft,, звеньев
М] к М2 - fi2> звеньев М2 кМ, - f 21 и звеньев М2 к М2 - /3 3.
В работе [65] даны формулы, позволяющие определять вероятность присое-
динения звеньев по известным константам сополимеризации. Они справедливы,
если состав исходной смеси остается постоянным.
Соотношение между fit и Д 2, т1 и т2 и произведением rtr2 имеет вид:
= + / (т2 / + Г'Ггт2. у
/12 2
=/ц Лг ’ =/22 "’"/’21 ’ /]2=/г1 (IV.2)
где mt и т2 - молярные доли мономера 1 и мономера 2 в сополимере соответ-
ственно.
Следует иметь в виду, что закономерности (IV.1) и (IV.2) справедливы толь-
ко при малых степенях конверсии. Если известно произведение констант сополи-
меризации rtr2, то,зная состав сополимера, можно установить последовательность
присоединения мономерных звеньев, т. е. рассчитать ftl,fl2, f2i На рис. IV.17
приведены зависимости f2l и/22 от содержания мономера т2 для разных значе-
ний произведения г,г2. Видно, что с уменьшением г2г2 вероятность присоединения
одинаковых звеньев друг к другу, т. е. образования блоков, уменьшается, а ве-
роятность чередования мономерных звеньев увеличивается.
Введем теперь величину п), которая показывает какая доля звеньев
мономера 1 (в %) содержится в блоках длиной в п мономерных звеньев. В работе
[66] выведена формула, выражающая эту величину через , и f\ 2:
(IV.3)
ПО
Рис. IV. 17. Зависимость доли связей
, (а) и f j 2 (6) от содержания т1 в со-
полимере для различных значений г,г2
(цифры у кривых).
Рассмотрим рис. IV.18, из которого можно определить долю звеньев мономе-
ра 1, входящих в блоки различной длины, в зависимости от содержания этого мо-
номера в сополимере для различных значений произведения г1г2. Например, при
эквимолярном составе сополимера (z«1 = 50%) и звенья мономера 1
распределяются по блокам разной длины следующим образом: единичных звеньев
82%, в блоках по две мономерные единицы 15%, в блоках из четырех единиц 0,2%.
Для того же состава сополимера но при г1г2=0,1, доля единичных звеньев умень-
шается до 56%, возрастает доля звеньев, входящих в блоки из двух и четырех мо-
номерных единиц. Когда г, г2 = 1, доля единичных звеньев близка к 25%, столько же
звеньев содержится в блоках из двух единиц и увеличивается доля звеньев, обра-
зующих блоки из четырех и более единиц. С уменьшением значения г2г2 растет
способность мономерных звеньев к чередованию.
Как было показано в работе [67], значение гхг2 можно определить
при помощи ИК-спектров. Для этого было получено следующее выра-
жение:
где/= m,/m2.
Авторы применили свой метод к определению произведения кон-
стант сополимеризации сополимера этилена с пропиленом, синтезирован-
ного на разных каталитических системах [59]. В качестве аналитических
полос поглощения были использованы полосы 733 и 722 см"1, относя-
щиеся соответственно к изолированным звеньям этилена и их блокам.
Показатели поглощения вычисляли на основании модельных парафино-
вых углеводородов. Рассчитанные значения гхг2 для этих сополимеров
были сопоставлены с данными, полученными по методу Файнемана-Рос-
са. Сравнение показало хорошее соответствие результатов.
Таким образом, было показано [59, 67], что для определения произ-
ведения констант сополимеризации достаточно знать состав сополимера
(/) и долю изолированных звеньев одного из сомономеров, которое
111
можно получить из ИК-спектров поглощения. Этим методом было опре-
делено произведение констант сополимеризации также для сополимера
этилена с винилхлоридом [58].
Спектроскопический метод определения произведения констант со-
полимеризации был также предложен в работе [61]. В отличие от выше-
изложенного этот метод основан на применении образцов сополимера
с известными значениями констант сополимеризации.
Использование ИК-спектров поглощения для определения произве-
дения констант сополимеризации позволяет проводить независимую
проверку этого параметра, а также получить дополнительные сведения
для других малоизученных сополимеров.
По формулам (IV.l) —(1V.3) легко построить зависимости у(тг,п)
от т1 для разных значений произведения констант сополимеризации.
Если исследовать изменение интенсивности структурно-чувствительных
полос поглощения сополимера от его состава (например, от тх), то
путем сопоставления полученных кривых с кривыми п) можно
выявить полосы, связанные с определенными последовательностями.
Хорошее совпадение экспериментальных данных с расчетными зависи-
мостями ¥> (^1, п) от т1 дает возможность произвести отнесение полос
112
поглощения к определенному типу последовательности мономерных
единиц сополимера при известном значении
Такое сопоставление было выполнено для сополимера этилена с
тетрафторэтиленом [68]. Получено удовлетворительное совпадение
теоретических кривых с экспериментальными при рассмотрении после-
довательностей звеньев этилена (Э) и тетрафторэтилена (Т) в триадах:
ЭЭЭ, ЭТЭ, ЭТТ, ТЭЭ, ТТТ, ТЭТ,
На основании полученных результатов (при известном значении
произведения констант сополимеризации г jr2=0,03) произведена интер-
претация полос поглощения, относящихся к разным последовательнос-
тям звеньев этилена и тетрафторэтилена и определены показатели погло-
щения этих полос.
В работе [63] была выполнена обратная задача: определено произве-
дение констант сополимеризации путем сопоставления эксперименталь-
ных и расчетных кривых. Исследовали сополимер стирола с пропиленом
блочной структуры. Полоса поглощения полистирола 565 см-1 приписа-
на к поглощению блоков изотактического строения. За полосу внутрен-
него стандарта принята полоса 1603 см-1. Измерены отношения интен-
сивности этих полос в оптических плотностях (D5 6 s/D160 3) для разных
составов сополимера. Из сопоставления этих данных с теоретическими
кривыми, построенными для разных значений произведения констант
сополимеризации для исследуемого сополимера, найдена величина гхг2= 5.
Аналогичный подход был применен и в работе [69] при исследовании
строения сополимеров 4-метилпентена-1 и гексена-1.
V. АНАЛИЗ МОДИФИЦИРУЮЩИХ ДОБАВОК
В ПОЛИМЕРНЫХ МАТЕРИАЛАХ
Практически во все полимеры на стадии переработки, а иногда и в процессе поли-
меризации вводят низкомолекулярные соединения (стабилизаторы, пластификато-
ры, наполнители, антистатики и др.). Они должны хорошо совмещаться с полиме-
ром, равномерно распределяясь в его массе.
В этой главе мы рассмотрим анализ добавок, которые не образуют с полиме-
ром прочных химических связей и могут быть выделены.
Назначение вводимых добавок различно: стабилизаторы предохраняют поли-
меры от процессов старения и удлиняют срок их службы, пластификаторы или
наполнители модифицируют исходные свойства полимеров, антистатики предотвра-
щают образование статического электричества при эксплуатации изделий из поли-
мерных материалов и т. п. Количество вводимых добавок регламентируется, а,
значит, подлежит контролю. Однако необходимость контроля содержания добавок
в полимере определяется не только этим. Еще на стадии разработки полимерных
материалов возникают вопросы выбора оптимального количества добавок, метода
введения, равномерности их распределения и. т. п.
В зависимости от назначения добавок их содержание в полимерном материале
может колебаться от десятых и сотых долей процента (стабилизаторы) до десятков
процентов (пластификаторы, наполнители) по отношению к массе полимера.
При содержании добавки,исчисляемой долями процента,возможно непосредствен-
ное ее определение по спектру полимерной пленки, если полимер не поглощает
в исследуемой области. В том случае, когда количество добавки сравнимо с массой
самого полимера (например, при пластификации), спектральные методы применя-
ются сравнительно редко. Это объясняется большой интенсивностью поглощения
пластифицированного полимера, для получения спектра которого требуются
тонкие (порядка 10 мкм) пленки, а их изготовление затруднено.
Чтобы не внести ошибок в результаты определения добавок, необходима
уверенность в том, что исследуемый полимер не содержит примесей, поглощаю-
щих в аналитической области спектра. Количественное определение добавок не-
органического происхождения таких как оксид магния, тальк, оксид кремния
(аэросил), можно осуществлять методами эмиссионного спектрального анализа
или атомно-абсорбционной спектроскопии. Для этого полимер подвергают озоле-
нию и полученную золу анализируют.
V. 1. ПЛАСТИФИКАТОРЫ
Назначение пластификаторов — модификация свойств полимеров. Введе-
нием пластифицирующих добавок достигается пластичность полимерно-
го материала, эластичность, увеличивается его морозостойкость или
огнестойкость. В качестве пластификаторов используют низкомолеку-
лярные органические соединения, главным образом сложные эфиры.
В табл. V.I. приведены основные пластификаторы, используемые для
полимерных материалов и наиболее характерные для данных соединений
полосы поглощения, которые могут быть использованы для аналити-
ческих целей.
114
ТАБЛИЦА V.l
Основные пластификаторы для полимерных материалов
Пластификатор
Структурная формула *
Наиболее
характерные
ИК-полосы
поглощения,
см'1
Эфиры о-фталевой кислоты и алифатических (ароматических)
спиртов
Фталаты
1725; дублет
1600-1580**;
1280; 1075
Эфиры алифатических карбоновых кислот и алифатических
спиртов
Адипинаты
Азелаинаты
Себацинаты
ROOC-(CH3)4 -COOR
ROOC-(CH2)7-COOR
ROOC-(CH,)S -COOR
j. 1725; 1175
Эфиры алифатических монокарбоновых кислот и гликолей
Эфиры триэтилен- СНг—сн2—о—(СНг)3—О—СН2—СН2 1735; 1175
гликоля и алифа- 1 о 1 о 1050
тических кислот 1 с=о 1 U 1
1 R 1 R
Триацетат глицерина сн2—сн—сн2 1 1 1 ООО 1 1 1 с=о с=о с=о । Ан Ан СНз СНз СНз 1735; 1120 1245;
Триоктил фосфат Эфиры фосфорной кислоты 1270; 1025
Трикрезилфосфат (СНЭС6Н4О)ЭР=О 1600; 1490;
Трифенилфосфат (С6Н5О)3Р=О 1300 1600; 1490;
Тритб-хлорэгил- (С1СН2СН2О)ЭР=О 1300 1280; 1080;
фосфат 1025
115
ПРОДОЛЖЕНИЕ
Пластификатор
Структурная формула *
Наиболее ха-
рактерные ИК-
полосы погло-
щения, СМ'1
Полиэфиры
Полипропилен- — ООС —(СН2)4—СООСН2—СН—0 — 1730; 1175;
гликольадипинат 1 1080
L СНЭ J п
Полиэтилен- — ООС — (СН2)4 — СООСН2—СН2о]„ То же
х'ликольадипинат Э п о ксидированные соединения СН2 СН —сн2 1735; 1180;
1 1 ООО 1 1 1 С=О С=О с=о 1 1 1 (СН2)„ (СНг)„ (СН2)п 1С НС НС | ро | '^О т /О 1С^ нс^ нс^ I I I R R R 830
*R — алкилы (С„Н2п + 1 )> К’ ~ бензил (СН2С6Н5) или фенил (С6Н5). **Аро-
матические соединения имеют большой набор полос поглощения, из которого
мы указали лишь полосы колебаний бензольного кольца 1600 и 15 80 см'1, не
отличающиеся больщой интенсивностью, но достаточно стабильные по частоте
и проявляющиеся в спектрах большинства фталатов в виде дублета.
Обычно анализ пластификаторов проводят методом экстрагирова-
ния с последующим расчетом содержания добавки по массе остатка
выпаренного раствора. Между тем, для определения пластификаторов
методы молекулярной спетроскопии также оказываются полезными.
Они могут служить дополнением к анализу экстракта, с их помощью
можно осуществлять контроль за полнотой экстракции или анализиро-
вать раствор пластифицированного полимера. Если же при пластифика-
ции полимеров применяют смеси разных пластификаторов, то возмож-
ности спектрального анализа ограничиваются. В этом случае может быть
полезным разделение смеси методом жидкостной хроматографии и
последующее определение ее компонентов при помощи УФ- и ИК-спек-
тров. Однако такое решение задачи в большей степени пригодно для
целей идентификации смеси. Другие добавки, вводимые в полимер
в малом количестве, могут не препятствовать анализу пластификаторов.
V.1.1. Фталаты
Фталаты используют для пластификации поливинилхлорида, нитро-
и этилцеллюЛозы, ацетобутирата целлюлозы, поливинилацетата, поли-
метилметакрилата и других полимеров. В ИК-спектрах фталатов наблю-
даются интенсивные полосы поглощения валентных колебаний связей
116
---J.-. ..1_ 1 i _ 1 _1 i I i 1 _ 1 1 _ k_ i --L 1 1 .1
3600 2300 2000 1800 1600 1400 1200 1000 800 600
V,CM’'
Рис. V.l. ИК-спектры поглощения дибутилфталатаG), ди-2-этилгексилфталата (2),
бензилбутилфталата (J).
' । i_____i. i___i _ i--1---1---i---1--1---i—
3600 2000 1600 1200 800
I'.CM'1
Рис. V.2. ИК-спектры поглощения дидецилфталата (2), поливинилхлорида, ие-
пластифицированиого (2) и пластифицированного дидецилфталатом (5).
I - капиллярный слой; толщина пленки (в мкм): 2 - 50; 3 - 30.
о
С=О и С-0 сложноэфирной группы -С-О-С- (см. раздел III), а в УФ-
спектрах — полосы бензольного поглощения.
На рис. V.1 представлены ИК-спектры поглощения трех пластифика-
торов, имеющих наибольшее применение: дибутилфталата, ди-2-этил-
гексилфталата и бензилбутилфталата. В общих чертах спектры очень
близки. Основные колебания, связанные со сложноэфирной группой,
имеют идентичные полосы поглощения (1725, 1280, 1120 см -1). Разли-
чия в алкильных и арильных радикалах обсуждаемых фталатов отража-
ются на числе и интенсивностях полос поглощения в областях спектра,
связанных с валентными (3100—2800см’1), деформационными (1500—
1400 см -1) колебаниями связи С-Н, а также в области 1000-700 см ’*,
в которой наблюдается поглощение скелетными колебаниями и погло-
щение, связанное с различным типом замещения в бензольном кольце.
Рассмотрим применимость ИК-спектроскопии для количественного
определения фталатов.
На рис. V.2 приведен спектр поглощения образцов неплнотифициро-
ванного поливинилхлорида и поливинилхлорида с добавкой 16% по
массе дидецилфталата (см. табл. V.l, R = Ci0H2i) в виде пленок толщи-
ной 30 мкм. Большинство полос поглощения фталата за исключением
малоинтенсивного дублета 1600, 1575 см’1 перекрывается полосами
поливинилхлорида или находится вблизи более интенсивных его полос.
В области поглощения аналитической полосы сложных эфиров 1725 см’1
поливинилхлорид прозрачен. По сравнению с другими полимерами
его спектр имеет менее интенсивные полосы поглощения. Так, для
основных полос полиэтилена сплошное поглощение достигается при
толщине 10-15 мкм, политетрафторэтилена около 5 мкм, эфиров
целлюлозы 5—10 мкм, а поливинил-
хлорида около 100 мкм. Сравнитель-
но небольшая интенсивность полос
поглощения поливинилхлорида поз-
воляет использовать для анализа плен-
ки толщиной до 200 мкм (рис. V.3).
Это дает возможность проводить опре-
деление фталатов в промышленных
пленках пластифицированного поли-
винилхлорида по одной из полос
дублета 1600 или 1575 см’1, ин-
тенсивность которого при указанной
толщине имеет оптимальное значение.
Если образцы поливинилхлорида
полностью растворяются в прозрачном
Рис. V.3. ИК-спектр поглощения пленки
поливинилхлорида, пластифицированного
дидецилфталатом.
Толщина пленки 200 мкм.
118
3600 2800 2000 1800 1600 1400 1200 1000 800 600
V,CM~’
Рис. V.4. ИК-спектры поглощения пропионата (I) и ацетобутирата целлюлозы (2).
Пленки на окне КВг,
растворителе (дихлорэтане, сероуглероде и др.), то можно анализиро-
вать также растворы. Концентрацию раствора подбирают с учетом интен-
сивности аналитической полосы, количества введенного пластификатора
и имеющегося в наличии набора кювет постоянной толщины. При этом
следует предварительно проверить постоянство полуширины полосы
поглощения фталата и ее положения при переходе к спектрам растворов.
Когда наблюдается влияние растворителя на контур аналитической поло-
сы (1725 см-1), определение фталата необходимо проводить по градуи-
ровочной зависимости интенсивности этой полосы от концентрации
эталонных растворов пластификатора, составленных на основе раствора
непластифицированного полимера. Нелинейный вид градуировочной
кривой не осложняет анализ.
Если для анализа поливинилхлорида может быть использована
ИК-область спектра, то для других полимеров, также пластифицирован-
ных фталатами, но содержащих карбонильные группы, непосредственное
определение пластификатора в растворе или пленке оказывается невоз-
можным. Это видно из сравнения ИК-спектров фталатов (см. рис. V.1)
со спектрами, например, производных целлюлозы (рис. V.4). То же
относится к поливинилацегагу и полимегилметакрилату. Можно восполь-
зоваться экстрагированием пластификатора растворителем, не вымываю-
щим низкомолекулярные фракции полимера. Обычно используют
в качестве экстрагента петролейный эфир, представляющий собой смесь
углеводородов. Полученный раствор можно проанализировать по полосе
1725 см-1 на основании эталонных растворов фталата в петролейном
эфире. Полноту экстракции можно контролировать методом, описанным
в разделе V.2.
119
ми: богаче выбор растворителей,
большая интенсивность полос по
ляет варьировать концентрацию и
Рис. V5. УФ-спектры поглощения раст-
вора дибутилфталата в этиловом спирте.
Концентрация раствора 0,2 г/л; толщина
слоя (в мм) : 1 — 5; 2 - 1.
Фталаты в отличие от других
сложноэфирных пластификаторов
поглощают в УФ-области спектра.
Это обусловлено присутствием в их
молекулах бензольного кольца. На-
пример, дибутилфталат имеет две
интенсивные полосы при 230 и
276 нм (рис. V.5) * Использование
УФ-спектров имеет ряд преиму-
ществ по сравнению с ИК-спектра-
прозрачных в аналитической области,
лощения. Сочетание факторов позво-
толщину поглощающего слоя и выби-
рать оптимальные для каждого частного случая.
Рассмотрим на примере пластифицированного поливинилацетата
методику определения дибутилфталата (см. табл. V.l, R=C4H9) по
УФ-спектрам поглощения. Поливинилацетат прозрачен в области погло-
щения фталатов. Из-за большой интенсивности полос поглощения ди-
бутилфталата трудно было анализировать непосредственно пленку
пластифицированного поливинилацетата. Поэтому для анализа использо-
вали раствор полимера. В качестве аналитической была выбрана полоса
276 нм, менее интенсивная, чем полоса 230 нм. Применение менее интен-
сивной полосы позволяет увеличить концентрацию пластификатора
в растворе или толщину поглощающего слоя раствора, что уменьшает
погрешность измерений. Кроме того, определение фталата по полосе
276 нм не требует компенсации „хвоста” полосы поглощения поливинил-
ацетата, максимум которой находится около 200 нм.
Чтобы избежать возможного влияния полимера на показатель по-
глощения аналитической полосы, дибутилфталат растворяли не в чистом
этаноле, а в растворе полимера в этаноле с концентрацией 1 г/л. Эталон-
ные растворы получали путем последовательного разбавления перво-
начального, концентрация пластификатора в котором составляла 2 г/л.
Для получения спектра использовали прямоугольные кварцевые кюветы
с толщиной слоя раствора 1 см. Измерение интенсивности полосы 276 нм
проводили от базисной линии, проведенной на уровне интенсивности
при 350 нм.
Массовую долю дибутил фталата ЬдБф в пленке пластифицирован-
ного поливинилацетата определяли по формуле:
* В действительности полоса 276 нм не является изолированной, а перекрыва-
ется полосой, находящейся около 283 нм, также принадлежащей поглощению дибу-
тилфталата. В дальнейшем мы используем полосу 276 нм, не разделяя эти полосы.
120
Ьдбф = <?(Z>276-P350)
где <7= l/aCd.
Концентрация полимера в растворе С всегда составляла 1 г/л. Удель-
ный показатель поглощения а определяли из спектров эталонных раство-
ров, усредняя значения, полученные для разных концентраций. При
массовой доле дибутилфталата 25% средняя абсолютная погрешность
его определения составляла 1%.
Подобным образом можно анализировать другие фталаты в поли-
мерах (выбрать аналитическую полосу, найти для нее показатель погло-
щения, подобрать концентрацию рабочего и эталонных растворов и т. д.).
V.I.2. Эфиры алифатических карбоновых кислот
и алифатических спиртов
Эфиры на основе алифатических кислот и спиртов (см. табл. V), так же
как и фталаты, имеют интенсивную полосу поглощения валентных
колебаний связи С=О сложноэфирной группы при 1725 см -1 (рис. V.6).
Однако полосы валентных колебаний связи С—О не являются такими
характерными и доминирующими по интенсивности, как это наблюдает-
ся в спектрах фталатов ниже 1400 см-1 (см. рис. V.1 и V.6). Взаимо-
действие колебаний связей С—О и С—С в адипинатах, азелаинатах, себаци-
натах и других пластификаторах этого класса приводит к сложному
спектру перекрывшихся полос поглощения в этой области, на фоне
которого выделяется полоса 1175 см -1, гак же как менее интенсивная
полоса 1245 см-1, отнесенная к колебаниям сложноэфирной группы
[70, с. 92].
В отличие от фталатов, рассматриваемые в этом разделе пластифика-
торы, не имеют полос поглощения в УФ-области спектра, поэтому для
их анализа может быть использована основная аналитическая полоса
сложных эфиров — 1725 см -1.
Поливинилхлорид, прозрачный в этой области спектра, может быть
проанализирован в виде раствора или тонкой пленки, толщина которой
зависит от количества введенного в полимер пластификатора. Если
массовое содержание пластификатора около 20%, то толщина пленки
должна быть не больше 30 мкм. Можно пытаться использовать полосу
1175 см-1, которая попадает в „окно прозрачности” поливинилхлорида
и имеет несколько меньшую интенсивность, чем полоса 1725 см-1. Это
дает возможность увеличить-толщину полимерной пленки.
Большинство полимеров (полиметакрилат, поливинилацетат, произ-
водные целлюлозы и т. п.), пластифицируемых адипинатами, себацина-
тами и другими представителями этого класса соединений, имеют, как
правило, спектры, перекрывающиеся полностью или частично со спектра-
ми пластификаторов. На рис. V.7 представлены спектры полиметил-
метакрилата и диоктилсебацината (см. табл. V.l, R=C8H17). Из сравне-
ния спектров видно, что практически все полосы диоутилсебацината
121
м,см
Рис. V.6. ИК-спектры поглощения диэтиладипииата (/), диоктилазелаииата (2)
и дибутил себацината (.?).
Капиллярные слои.
1 — пленка на окне КВг; 2 — капиллярный слой.
налагаются на полосы полимера, а если еще учесть, что толщина пленки
полиметилметакрилата около 5 мкм, то следует применять экстраги-
рование пластификатора с последующим анализом экстракта.
Иногда целесообразно экстрагировать пластификатор даже тогда,
когда возможен его прямой анализ в растворе. Например, когда требует-
ся определять не одну, а несколько добавок разного назначения, введен-
ных в полимер. В некоторых случаях это удается сделать, используя
один экстракт. Так, поливинилбутираль кроме пластификатора ди-
бутилсебацината содержит также термо- и светостабилизаторы. Послед-
ние удобно анализировать в виде экстрактов в тетрахлорметане по
УФ-спектрам поглощения (см. раздел V.2.1). При этом дибутилсеба-
цинат в поливинилбутирале также может быть определен по анализу
экстракта в тетрахлорметане по ПК-спектрам поглощения (полоса
1725 см-1). Спектральные характеристики стабилизаторов таковы,
что небольшая их концентрация в полимере не мешает определению
дибутилсебацината. Поливинилбутираль в тетрахлорметане не растворя-
ется. Слабое поглощение экстрагента в аналитической области пластифи-
катора было скомпенсировано. Высокая интенсивность полосы 1725 см-1
дает возможность использовать для анализа слой раствора толщиной
0,5 мм.
Для определения дибутилсебацината готовили эталонные растворы
в тетрахлорметане, концентрация которых составляла от 1 до 4 г/л;
они служили основой для определения показателя поглощения полосы
1725 см-1. Соотношение исследуемого полимера и растворителя равня-
лось 10 г/л. Экстрагирование пластификатора проводили при комнат-
ной температуре. Полное вымывание пластификатора достигалось за 4 ч.
Полноту экстрагирования оценивали по методу, описанному в разделе
V.2.I.
Если нет необходимости одновременного анализа стабилизаторов
и пластификатора в поливинилбутирале, то определение одного дибутил-
себацината можно проводить по полосе 1725 см -1, анализируя растворы
полимера, например в дихлорэтане *. Можно также использовать одну
из полос поглощения, относящихся к С—Н валентным колебаниям около
3000 см - l. Анализ в этой области спектра имеет некоторые преимущес-
тва, в частности вместо кювет с гигроскопичными окнами из КВт или
NaCl можно использовать кварцевые кюветы.
Часто адипинаты и себацинаты применяют для пластификации поли-
меров совместно с фталатами. Поскольку поглощение сложноэфирных
групп в их спектрах совпадает, то интенсивность полосы 1725 см-1
характеризует лишь суммарное содержание пластификаторов. Для анали-
за фталата можно использовать УФ-область спектра (если другие добав-
ки отсутствуют или не поглощают в ней). Определив содержание фталата,
* Поливинилбутираль имеет примесное поглощение в аналитической области
стабилизаторов (УФ-спектры), поэтому при одновременном определении добавок
использовали экстракцию в тетрахлорметане, а не растворение.
123
а также молярные показатели поглощения полосы 1725 см-1 для ком-
понентов смеси, можно по разности найти содержание адипината (се-
бацината и т. п.).
V. 1.3. Эфиры алифатических монокарбоновых кислот
и гликолей
Наибольшее применение из этого класса пластификаторов имеют эфиры
тризтиленгликоля и монокарбоновых кислот. Например, триэтилен-
гликоль-ди-(2-этилгексоат) и триэтиленгдиколь-ди-(2-этилбутират) (см.
табл. V.1)
R=—СН2—СН—(СН2)з—СНз
С2Н5
И —СН2—СН—СН2~СНз
С2н5
V, см-'
Рис. V.8. ИК-спектры поглощения поливинилбутираля (7), триэтилеигликоль-
ди-(2-этилбутирата) (2) и триэтиленгликоль-ди-(2-этилгексоата) (5).
Толщина слоя: 1 - 5мкм;2 и 3 - капиллярные слои.
124
применяются для пластификации поливинилбутираля, используемого
в виде пленок для производства триплексов. Их спектры (рис. V.8)
имеют много общего со спектрами сложных эфиров дикарбоновых
кислот и алифатических спиртов (рис. V.6 и V.7). Основной аналитичес-
кой полосой также служит полоса карбонильного колебания сложно-
эфирной группы (1735 см-1), волновое число которой несколько
больше, чем волновое число полосы пластификаторов (1725 см-1),
обсужденных выше. Определение этих пластификаторов может быть
основано на экстрагировании их из поливинилбутираля тетрахлормета-
ном и проведено по схеме, описанной для анализа дибутилсебацината
(см. в разделе V.1.2).
Наряду с полосой 1735 см-1 в качестве аналитической может быть
использована область спектра валентных колебаний С—Н. Показатели
поглощения полос колебаний как С=О, так и С—Н для триэтиленгли-
коль-ди-(2-этилгексоата), триэтиленгликоль-ди-(2-бутирата) и дибутил-
себацината близки • Концентрации растворов и процедура анализа
этих пластификаторов однотипны.
V.I.4. Фосфаты
Из соединений этого класса (см. табл. V.1) наибольшее применение
в качестве пластификаторов имеют трифенилфосфат (СбН5О)3Р=О
(совмещается с нитроцеллюлозой и этилцеллюлозой), трикрезилфосфат
(СН3С6 Н4О)3Р=О (совмещается с поливинилхлоридом, поливинил-
ацетатом, некоторыми эфирами целлюлозы), три-(3-хлорэтилфосфат
(С1СН2СН2О)3Р=О (совмещается с поливинилацетатом, поливинил-
бутиралем, эфирами целлюлозы, полиметилметакрилатом) и др.
Для фосфатов характерно наличие в спектре интенсивной полосы
поглощения, относящейся к валентным колебаниям связи Р=О. В спек-
трах трифенилфосфата и трикрезилфосфата она наблюдается около
1300 см-1 (рис. V.9). Триалкилфосфаты поглощают при несколько
меньших волновых числах: 1280—1270 см-1 (трихлорэтилфосфат,
триоктилфосфат и др.). К колебаниям группы Р— О—Сарил отнесена
полоса 1200 см-1 [2, с. 447]. Большинство соединений, содержащих
связи Р— О—Салкил, имеют полосу поглощения при 1030 см "1 [2, с. 448 ].
Спектры триарилфосфатов, являющихся ароматическими соедине-
ниями, харакеризуются полосами поглощения в области 1200—650 см"1,
относящихся к различным колебаниям ароматического кольца, на часто-
ты и интенсивности которых влияет тип заместителя. Эти полосы, нала-
гаясь на полосы поглощения связей Р—О—С, затрудняют точное отнесе-
ние последних, но не мешают использованию полос, присущих данному
фосфату, в качестве аналитических при благоприятном расположении
поглощения пластификатора, полимера, растворителя или экстрагента
Аналитическими полосами арилфосфатов могут . служить также
полосы характеристических колебаний бензольного кольца 1600 и
1500 см . Поэтому целесообразно для определения этих соединений
125
в полимерах использовать также УФ-спектры поглощения, как и при
анализе фталатов.
Методика определения трихлорэтилфосфата в поливинилацетате
основана на анализе растворов пластифицированного полимера в ди-
хлорметане. Удачное расположение аналитических полос поглощения
трихлорэтилфосфата и поливинилацетата позволяет проводить коли-
чественное определение этого пластификатора в полимере (рис. V.10).
Аналитической полосой пластификатора служила интенсивная полоса
1080 см "1. Она попадает в минимум поглощения поливинилацетата,
фон которого учитывается проведением базисной линии (см. рис. V.10).
Поскольку анализу подвергались растворы полимера, то в канал сравне-
ния спектрофометра помещали кювету с дихлорметаном для компен-
сации его слабого поглощения в аналитической области спектра. Поли-
винилацетат имеет полосу 1375 см"1, расположенную в „окне про-
зрачности” пластификатора и легко выделяемую базисной линией.
Наличие изолированной полосы полимера, сравнимой по интенсивности
с полосой 1080 см-1, позволило использовать для анализа отношение
интенсивностей полос поглощения 1080 и 1375 см"1 (в оптических
плотностях). Это исключает ошибки в концентрации при составлении
испытуемого раствора и дает возможность не придерживаться точного
значения выбранной для анализа концентрации.
Для построения градуировочного графика использовали растворы
эталонных смесей пластификатора и полимера в массовых соотношениях
т
3800 2800 2000 1800 1800 1400 1200 1000 800
V,cm-’
Рис. V.9. ИК-спектры поглощения трихлорэтилфосфата (7), трикрезилфосфата (2)
и трифенилфосфата (3).
Капиллярные слои.
126
Рис. V.10.HK спектры поглощения растворов в дихлорметане трихлорэтилфосфата
(/), поливинилацетата (2) и смеси поливинилацетат-трихлорэтилфосфат состава
80 : 20 (3).
(^пл/^пва) равных соответственно: 0:100, 20:80, 30:70, 40:60,
50 : 50. По оси абсцисс графика откладывали значения ^пл/^пва Д° оси
ординат — отношение оптических плотностей полос 1080 см-1 (£>1080)
и 1375 см"1 (Я1375 ). Полученная зависимость имеет линейный вид
и описывается уравнением:
Г11О8О/л1375_ , /.
D /и —аОпл/опвА
где а — постоянный коэффициент, определяемый по углу наклона градуировоч-
ной прямой к оси абсцисс.
Учитывая, что 6ПП+^ПВА = ^0%, получаем выражение для опре-
деления массовой доли трихлорэтилфосфата (в %) :
р1080
Ь ™ = р1375д +рЮ80 ’100
Абсолютная погрешность составляла 1% при йпл = 25%.
J27
V.l .5. Другие пластификаторы
Полиэфиры по химическому строению близки к другим сложным эфи-
рам на основе дикарбоновых кислот (адипиновой, себациновой и др.)
и спиртов, что обеспечивает сходство их спектров (рис. V.11 и V.6).
Наиболее интенсивной и изолированной полосой поглощения полиэфи-
ров, например полипропиленгликольадипината (см. рис. V.11), также
является полоса колебания связи С=О сложноэфирной группы при
1730 см -1.
Поскольку полиэфиры имеют молекулярную массу порядка 2000,
они не выделяются из полимера экстрагированием. Возможности спек-
трального анализа пластифицированных полиэфирами полимеров в
большой степени определяются прозрачностью полимера и раствори-
теля в аналитической области спектра главным образом при 1730 см -1.
Эпоксидированные соединения, используемые в качестве пластифи-
катора, представляют собой сложные эфиры, молекулы которых содер-
жат эпоксидные кольца. ИК-спектры эпоксидированных пластифика-
торов во многом не отличимы от спектров других сложных эфиров.
Однако наличие в структуре молекулы эпоксигруппы дает дополни-
тельные полосы поглощения, относящиеся к колебаниям этой группы.
Наиболее интенсивная ее полоса 1250 см-1 совпадает с поглощением
простой эфирной связи сложноэфирной группировки. Колебания эпок-
сидного кольца проявляются также в области 800—950 см-1 в виде
двух, а иногда одной полосы средней или малой интенсивности.
Рис. V.l 1. ИК-спектр поглощения полипропиленгликольадипината.
Капиллярный слой.
128
V.2. СТАБИЛИЗАТОРЫ
Стабилизаторы — низкомолекулярные органические и неорганические
соединения, которые замедляют процессы старения полимеров, протека-
ющие в них под влиянием повышенных температур, разного вида облу-
чения, механических напряжений как в присутствии кислорода воздуха,
так и в вакууме. Стабилизаторы вводят или на стадии полимеризации,
или в процессе переработки полимера. В настоящее время разработано
огромное число стабилизаторов, однако промышленное применение
находят лишь единицы.
Существуют разные типы классификации стабилизаторов. Посколь-
ку метод молекулярной спектроскопии служит для изучения хими-
ческого строения вещества, то целесообразно использовать классифи-
кацию по химической природе соединения. Для каждого стабилизатора
существует сокращенное название или торговая марка. При этом практи-
чески каждый стабилизатор имеет несколько наименований.
При рассмотрении возможностей спектроскопии для анализа стаби-
лизаторов мы взяли за основу химическую классификацию [71] и ис-
пользовали сокращенные названия, руководствуясь справочником [72].
Большинство стабилизаторов являются ароматическими соединени-
ями и, следовательно, имеют полосы поглощения в УФ-области спектра,
доступной для практического использования. Это имеет свои преиму-
щества для решения аналитических задач: полосы поглощения достаточ-
но интенсивные, имеется выбор прозрачных растворителей, поэтому
многие стабилизированные полимеры могут быть проанализированы
в виде растворов. Спектры некоторых стабилизаторов таких, как про-
изводные фенола, гидрохинона, сульфиды фенолов, эфиры фосфористой
кислоты, изменяются при добавлении в этанольный раствор стабилиза-
тора раствора щелочи, что проявляется в большем или меньшем бато-
хромном сдвиге поглощения. Это явление может быть использовано
при анализе стабилизированных полимеров.
Батохромный сдвиг может сопровождаться увеличением интенсив-
ности полосы. Примером служит три (л-нонилфенил) фосфит (фосфит
НФ, полигард), структурная формула которого [С9Н19—С6Н4—О]3Р.
В спектре его этанольного раствора имеется полоса при 272 нм (рис.
V.12) [73]. В спектре этого же соединения, растворенного в этаноле
с добавлением щелочи, наблюдается сдвиг полосы до 295 нм и значи-
тельное увеличение ее интенсивности.
Использование батохромного эффекта дает дополнительные воз-
можности для анализа. Например, требуется определить количество
стабилизатора фенольного типа в экстракте, содержащем другие добав-
ки, поглощение которых мешает анализу. Все ингредиенты раствора,
кроме фенольного стабилизатора, не изменяют спектр в щел очно-спирто-
вой среде. Тогда записывают спектр этанольного раствора смеси с до-
бавлением щелочи (КОН) относительно раствора без нее. Получают
дифференциальный спектр, в котором присутствует полоса стабилиэа-
129
Рис. V.12. УФ-спектры погло-
щения растворов три(и-ио-
нилфенил) фосфита в этаноле
(Z) и с добавлением 0,1 н.
КОН (2).
Концентрация 4 г/л; толщина
слоя 0,214 мм.
тора, смещенная к боль-
шим длинам волн, а по-
глощение других ингре-
диентов раствора ском-
пенсировано.
Однако при исполь-
зовании батохромного
смещения следует иметь
в виду, что поглощение спиртового раствора КОН постепенно возрастает
со временем при хранении. Возможно также иногда изменение погло-
щения анализируемого раствора. Это свойство щелочно-спиртовых
растворов необходимо учитывать при разработке методик.
В зависимости от спектральных характеристик стабилизирован-
ного полимера, стабилизатора, их растворимости в прозрачном раство-
рителе существуют различные варианты методик спектрального анализа.
Оптимальный путь — прямое определение стабилизатора по спектру
полимерной пленки или раствора. Может оказаться, что полимерная
пленка будет иметь малое светопропускание за счет рассеянного света
(см. раздел 1.1.1). Эти потери света могут быть скомпенсированы при
записи спектра пленки стабилизированного полимера относительно
пленки нестабилизированного образца.
Если полимер нерастворим, то для определения в нем стабилиза-
торов можно воспользоваться их экстрагированием. Однако одновре-
менно со стабилизаторами возможно вымывание также и примесей,
и низкомолекулярных фракций полимера, поглощающих в аналити-
ческой области спектра. Это приобретает существенное значение при
анализе именно стабилизаторов, поскольку в полимеры они вводятся
в небольших количествах. Если при этом наблюдается батохромный
сдвиг в спектре щелочно-спиртового раствора, то задача облегчается.
„Сдвинув” спектр стабилизатора от мешающего поглощения примеси,
можно проводить анализ, как описано выше. Контроль за полнотой
экстракции и обнаружение поглощающих примесей могут быть прове-
дены по методу,описанному в разделе V.2.1.
Иногда для определения содержания стабилизатора растворяют
стабилизированный полимер, затем его осаждают и анализируют полу-
ченный раствор.
В соответствии с химической классификацией [71 ] стабилизаторы раз-
деляют на классы, к которым относятся ароматические амины, гетероцик-
130
лические азотсодержащие соединения, производные фенола, эфиры фос-
фористой кислоты, а также соединения, содержащие серу и металлы.
Подробные сведения о классификации и номенклатуре стабилизато-
ров, химической структуре, свойствах, эффективности действия и дру-
гие содержатся в [71, 72]. Спектральные характеристики некоторых
широко применяемых стабилизаторов приведены в табл. V.2 (исполь-
зованы сведения из книг [71-73], также некоторые экспериментальные
данные авторов). Из данных табл. V.2 видно, что наиболее интенсивные
полосы поглощения характерны для производных аминов и других
азотсодержащих соединений.
Наряду с используемыми в промышленности стабилизаторами
идет поиск новых, обладающих более унифицированными свойствами,
т. е. стабилизаторов с несколькими функциональными группами в моле-
куле [74]. Например, в качестве бифункционального стабилизатора
полиэтилена предложен [75] дифениловый эфир о-гидроксифенил-
н-гидроксифениламинометилфосфоновой кислоты, обладающий свето-
и термостабилизирующими свойствами:
/°
NH —СН—р(-ОС6Н5
I ос6н5
сьн4он
Применение ИК-спектроскопии для серийных анализов в большой ме-
ре зависит от интенсивности аналитических полос стабилизаторов, проз-
рачности полимера или растворителя в аналитической области, поскольку
стабилизаторы вводят в полимеры в небольших количествах. Например,
для полиэтилена, одного из немногих полимеров, имеющих сравни-
тельно большие ..окна прозрачности” в инфракрасном спектре, оказалось
возможным проводить определение стабилизатора 4-алкокси-2-гидро-
ксибензофенона (бензон ОА) не-
посредственно по спектру поли-
мерной пленки [76]. Для этого
использовали интенсивную полосу
валентного колебания С=О связи
при 1622 см-1 (рис. V. 13).
Аналогичное решение зада-
чи возможно и для других ста-
билизированных полимеров.
Рис. V.13. ИК-спектры поглощения поли-
этилена, стабилизированного 4-алкокси-
2-гидроксибензофеноном.
Массовое содержание стабилизатора
0,5%. Толщина пленки 0,03 мм.
131
Химическое
СПЕКТРАЛЬНЫЕ ХАРАКТЕРИСТИКИ СТАБИЛИЗАТОРОВ В УФ-ОБЛАСТИ СПЕКТРА
Торговое
название название
Структурная формула
Темпера-
тура плав-
ления, °C
Раствори-
тели *
ТАБЛИЦА V.2.
Длина волны X, нм
0,1 н. рас-
этанол твор КОН
в этаноле
Производные ароматических аминов
К-Изопропил-М'-фенил-
фенилендиамин-1,4
Диафен ФП \
<^_р—ынсн(сн3)г
N, N'- Дифенил-
фенилендиамин-1,4
N.N'-Ди (нафтил-
2) фенилендиамин-1,4
4-гидроксиди-
фениламин
N-Фенилнафтил-
амин-1
Диафен ФФ
(диафен Ф)
Диафен НН
(диафен Н)
Дифенам О
(фенам О)
Неозон А
(нафтам-1)
80,5 Этанол Дихлорэтан Тетрахлор- метан 290(83) ** -
152 Этанол 304(105) —
Трихлорэтан - —
235 Трихлор- 246(74); —
этан + этанол 274 (63); 318(70); —
74 Этанол 283(85) —
Трихлорэтан — -
62 Этанол 253(79); 340 (40) -
Гептан — —
Трихлор- метан — —
Тетрахлор- метан - -
108 Этанол 272(122);
310(105);
348 (20)
Трихлор- -
метан
N-Фенилнафтил-
амин-2
Неозон Д
(нафтам-2)
* Указаны только растворители, прозрачные в УФ-области спектра. ** В скобках даны значения удел- лого показателя поглощения
полосы [в л/ (г • см) ].
ПРОДОЛЖЕНИЕ
Химическое Торговое
название
название
Структурная формула
Темпера-
тура плав-
ления, °C
Раствори-
тели *
Гетероциклические азотсодержащие соединения
2,2,4-Триметил-6-этокси- Хинол ЭД
1,2-дигидрохинолин
Жидкость Этанол
Длина волны X, нм
0,1 н. рас-
этанол твор КОН
в этаноле
229(100;
350(11)
1,2-дигидрохинолина (хинол Д)
Олигомер 2,2,4-триметил- Ацетонанил Р
2- (2-Гидрокси-5' Беназол П
метилфенил)-
бензотриазол
(мономер)
60 110 Этанол Трихлор- метан 236 (89); 296(13) -
132 Этанол Три хлор- метан - -
Тетрахлор- метан 300; 346(76) —
Производные фенола
2,6-Ди-грег-бутил-
4-метил фенол
Агидол-1
(алкофен Б,
алкофен БП,
ионол)
(СНз)зС
70 Этанол
278(8)
278(8);
307(2)
♦Указаны только растворители, прозрачные в УФ-области спектра.
ПРОДОЛЖЕНИЕ
Темпера- Длина волны X, нм
Химическое Торговое _ . нятаоиир Структурная формула HaJBdnHC naJBanMC г Раствори- тура плав- тели * 0,1 н. рас-
пения, С этанол твор КОН
в этаноле
2,4,6-Три-грег- Антиоксидант ОН 135 Этанол 274 (7) 274 (7);
бутилфенол П-23 (алкофен Б, алкофен Б,Б) (СН3)3С> 4JLZ,C(CH3)3 ту С(СН3)з 302(2)
2,2'-Метиленбис- (6-трет-бутил- 4-метилфенол) Антиоксидант НГ-2246, (бисалкофен, агидол 2) (СНз)зС- он ^-TzCHr ту он ^_Т/С(СН3)з ту 133 Этанол Трихлор- метан 281(12) 287(12) 308(17)
СНз СНз
2,2'-Метиленбис- [ 4 -метил-6- (1 -м етил- циклогексил-1) фенол ] Смесь а-метилбензил- Бисалкофен МЦП (бис- алкофен Ц) АО-20 (алко- _-,НзС он Х/снг'Хг- СНз 1 ъ УГГ-Т СНз 140 Жидкость Этанол Гексан Гептан Дихлорэтан Трихлор- метан Этанол 283(12) 280 (8) 288(12) 310(15) 302(15)
фенолов [2-, 4-, 2,4-да- фен МБ,
2,6-ди-, 2,4,6-триЧа-метил- агидол-20)
бензил) фенол]
4-Алкокси-2-гидрокси- Бензон ОА он Жидкость Этанол — -
бензофенон н /"А Дихлорэтан — -
R°—Т/“с—хУу 288; 330**
* Указаны только растворители, прозрачные в УФ-области спектра. ** Пленка полиэтилена.
ПРОДОЛЖЕНИЕ
Длина волны X, нм
Химическое Торговое
название
название
Структурная формула
Темпера-
тура плав-
ления, °C
Раствори-
тели *
0,1 н. рас-
зтанол твор КОН
в этаноле
Изоборнилкрезолы
Алкофен И
(алкофен ИП)
Жидкость Этанол -
Дихлорэтан -
Тетрахлор- 282(11)
метан
Другие стабилизаторы
Три (и-нонил-фенил)-
фосфит
Смесь а-метилбензил-
фенил фосфитов
Фосфит НФ
(полигард)
Фосфит П-24
(фосфит СФ)
Жидкость Этанол 272(4) 295(8)
Жидкость Этанол 269(6) 300(12)
279(7)
♦Указаны только растворители, прозрачные в УФ-области спектра.
Указаны только растворители, прозрачные в УФ-области спектра. ** Пленка полиэтилена
136
V.2.I. Анализ экстрактов полимеров
Наиболее широко при определении стабилизирующих добавок в поли-
мерах применяется анализ их экстрактов. Часто используют химические
методы, однако спектрофотометрическое определение стабилизатора
оказывается менее трудоемкой операцией.
При анализе экстрактов необходимо убедиться в полноте извле-
чения добавок, которая должна обеспечиваться правильным выбором
экстрагента, времени и температуры. Условия экстрагирования могут
быть подобраны спектроскопическим методом. Покажем это на примере
анализа поливинилбутиральной пленки, в состав которой входят термо-
стабилизатор алкофен И, представляющий собой смесь 2-изоборнил-
4-метилфенола и 2-изоборнил-5-метилфенола, и светостабилизатор
беназол П, 2-(2-гидрокси-5'-метилфенил) бензотриазол (см. табл. V.2).
Для анализа использованы УФ-спектры поглощения (рис. V.14).
Аналитическими полосами служили: полоса 282 нм термостабилизатора
и 346 нм светостабилизатора. В качестве экстрагентов были рассмотре-
ны этанол, трихлорметан и тетрахлорметан, прозрачные в аналитических
областях спектра. Для определения удельных показателей поглощения
указанных полос готовили эталонные растворы стабилизаторов
во всех использованных растворителях с концентрациями (в г/л):
для термостабилизатора — 0,016; 0,024; 0,032; 0,040 и для светоста-
билизатора 0,006; 0,008 ; 0,010 и 0,012. Значения показателей поглоще-
ния даны в табл. V.3.
Массовое содержание светостабилизатора Лс(в %) определяли
из формулы:
р346-д1г0
<v',)
где ZP46 и О420 - значения оптических плотностей в максимуме полосы и при
420 нм (фон поглощения); а346 - удельный показатель поглощения светостабили-
затора, л/ (г • см); С - содержание
исследуемого полимера в растворе,
г/л; d - толщина слоя, см.
Поскольку стабилизаторы
при введении в поливинилбути-
ральную пленку предварительно
растворяют в дибутилсебацинате,
Рис. V.14. УФ-спектры поглощения
термостабилизатора алкофена И (7)
и светостабилизатора бенаэолаП (2).
Концентрация исследуемого раствора
в тетрахлорметане (в г/л) : 1 — 0,04;
2 — 0,01. Толщина слоя 1 см.
137
ТАБЛИЦА V.3
Положения аналитических полос и значения удельных показателей
поглощения стабилизаторов
Растворитель Беназол П Алкофен И
X, нм а, л/(г • см) X, нм а, л/(г • см)
Тетрахлорметан 346 75 282 11
Трихлорметан 342 78 282 11
Этанол 338 70 282 11
то эталонные растворы готовили также в смеси с этим пластификатором.
В рассматриваемой области спектра он прозрачен и не влияет на резуль-
таты определения. Так как полоса поглощения термостабилизатора
282 нм налагается на полосу 300нм, принадлежащую светостабилизатору,
то при анализе учитывается поправка на это наложение. Поправка харак-
теризует долю вклада поглощения светостабилизатора при длине волны
282 нм и выражается через отношение показателей поглощения свето-
стабилизатора при длинах волн 282 и 346 нм с?*2 /а^6. Тогда массовое
содержание термостабилизатора в % определяется из формулы:
(D262—D420) — (a282/al46){D348—D420)
6Т = ~------------------------------------100 (V.2)
a282Cd
где D232, D34i, D*20 — значения оптических плотностей в максимумах полос 282
и 346 нм и фона при 420 нм соответственно; - удельный показатель поглоще-
ния термостабилизатора.
Выбор экстрагентов и условий экстракции проводили следующим
образом.
Для сокращения времени анализа и упрощения операции экстраги-
рования, последнюю проводили не в аппаратах Сокслета, а в колбах
с пришлифованными пробками, в которые помещали полимерную
пленку, нарезанную кусочками размером 2X5 мм (толщина пленки
1 мм). Экстракцию вели в стационарном режиме при комнатной тем-
пературе. Это позволяет одновременно проводить массовые анализы. Пе-
ред записью спектра полученный раствор перемешивали встряхиванием.
По формулам (V.1) и (V.2) находили содержание стабилизаторов
в экстрактах для разной продолжительности экстрагирования и строили
кинетические зависимости. для каждого растворителя (рис. V.15).
Количество введенных стабилизаторов отмечено на рисунке пунктир-
ными линиями. Время, при котором наблюдалось насыщение, соответ-
ствовало времени полного вымывания стабилизатора: экстрагирование
тетрахлорметаном термостабилизатора достигается за 6 ч, светостабилиза-
тора — за 2 ч. Использование в качестве экстрагента этанола и трихлор-
метана дает завышенное содержание термостабилизатора (кривые идут
значительно выше контрольной отметки). Это завышение объясняется
138
тем, что указанные растворители вымывают примеси, поглощение кото-
рых накладывается на поглощение термостабилизатора. А значит, этанол
и трихлорметан не могут быть использованы в качестве экстрагентов.
Если из измельченного образца, подвергнутого экстрагированию,
приготовить пленку, то можно еще раз удостоверить полноту извлече-
ния стабилизаторов по отсутствию в спектре пленки их полос поглоще-
ния. Исходя из кинетических зависимостей, были выбраны в качестве
экстрагента тетрахлорметан и продолжительность экстрагирования —
6 ч (см. рис. IV.15).
Для сокращения продолжительности анализа экстрагирование
проводили при повышенных температурах. При повышении температуры
до 40 °C длительность экстракции уменьшилась в 2 раза. Однако даль-
нейшее увеличение температуры нежелательно из-за возможного изме-
нения концентрации раствора вследствие испарения растворителя.
Легче осуществляется удаление стабилизаторов из пластифициро-
ванного полимера. В рассмотренном случае массовое содержание ди-
бутилсебацината в поливинилбутирале составляло 25%. Экстрагирование
из непластифицировэнного полимера затруднено. На полноту экстракции
влияет химическая природа поли-
мера и степень измельчения его
образца. Так, для бутадиен-метил-
стирольного каучука (СКМС-
ЗОПС) размер частиц не должен
превышать 1X1X1 мм [77].
Рис. V.15. Зависимость количества алкофеиа И (-----) ибеиазолаП (-----),
экстрагированных из полимера тетрахлорметаном (7), трихлорметаном (2) и
этанолом (3) от продолжительности экстракции.
Рис. V.16. Дифференциальные спектры поглощения щелочно-спиртовых растворов
антиоксиданта НГ-2246 (7) и антиоксиданта АО-6 (2) относительно их нейтраль-
ных растворов:
Концентрация (в г/л) : 1 - 0,0367; 2 - 0,0655; толщина слоя 1 см.
139
Таким образом, для каждого полимерного материала существуют
свои условия экстрагирования, которые могут быть подобраны с по-
мощью предложенного метода.
Рассмотрим анализ стабилизаторов с применением батохромного
сдвига. Такая методика использована, например, для одновременного
определения содержания антиоксидантов три (фенилэтилбензил) фосфи-
та — антиоксиданта АО-6 с 2,2'-метиленбис (б-трет-бутил-4-метиленфено-
лом) — антиоксидантом НГ-2246 в бутадиен-метилстирольном каучуке
СКМС-ЗОПС [77]. Стабилизаторы экстрагировали из каучука этанолом.
Анализ проводили по дифференциальным спектрам поглощения щелоч-
но-спиртового раствора стабилизаторов. Аналитическими полосами
служили полоса антиоксиданта АО-6 при 296 нм и антиоксиданта НГ-2246
при 310 нм (рис. V.16). Толщина слоя растворов равнялась 1 см.
Поскольку аналитические полосы поглощения перекрываются,
то для расчета содержания антиоксидантов составляли систему уравне-
ний с двумя неизвестными:
100Z>296= al^Cbjd + a226Cb2d
100Z)31o=afloCO]</ 4 (73210СЬ2<У
где D296 и D310 - значения оптических плотностей при 296 и 310 нм в спектре ана-
лизируемого раствора; а]96, а”6 и а]’°, а3™ - удельные показатели поглощения
антиоксиданта НГ-2246 и антиоксиданта АО-6 при длинах волн 296 и 310 нм,
соответственно, л/(г - см); С - масса навески каучука, рассчитанная на литр
анализируемого раствора; Ь, и Ьг - доли антиоксиданта НГ-2246 и антиоксидан-
та АО-6, % (по массе).
Методика [77] была опробована при анализе каучука с заданными
количествами антиоксидантов, введение которых в каучук осуществля-
лось путем вальцевания. Получено хорошее соответствие результатов.
V.2.2. Исследование полимерных пленок
Наиболее быстрым и удобным способом определения количества ста-
билизаторов в полимере является их прямое определение по спектру
стабилизированной пленки. Это возможно для полимеров, не имеющих
поглощения в области аналитических полос. Примером такого анализа
могут служить методики определения бисалкофена МЦП, антиоксидан-
та НГ-2246, бензона О А, неозона А с диафеном ФФ в полиэтилене [76].
Количесгво введенных стабилизаторов составляло 0,2—0,5% (по
массе). Аналитическими полосами поглощения (рис. V.17) служили:
полоса 285 нм (бисалкофена МЦП), 288 нм (антиоксидант НГ-2246),
252 нм (неозон А). Поскольку пленки полиэтилена сильно рассеивают
УФ-излучение, то для его компенсации в канал сравнения спетрофото-
метра помещали нестабилизированную пленку, относительно которой
записывали спектр полиэтилена со стабилизатором. Определение содер-
жания стабилизирующих добавок проводили по градуировочным графи-
140
кам. Зависимость оптической плотности полос от количества добавок
линейная.
Эталонами служили пленки полиэтилена с известным содержанием
стабилизаторов, которые вводили в полимер при перемешивании на
вальцах в течение 15 мин при 160 °C. В процессе приготовления эталон-
ных образцов существенное значение приобретает равномерность распре-
деления стабилизатора в полимерной пленке. При массовом содержании
стабилизирующей добавки от 0,01 до 0,1% эталоны готовили двухсту-
пенчатым перемешиванием, т. е. вводили в полимер добавку, предва-
рительно смешанную в отношении 1 : 10 с порошком КВг [76].
Для определения в полиэтилене 4,4'-тиобис(6-трег-бутил-З-мстил-
фенола) — тиоалкофена БМ (см. табл. V.1) была использована полоса
поглощения 250 нм (рис. V.18). Интенсивность этой полосы позволяет
определять массовое содержание добавки в пределах от 0,05 до 0,15%
при толщине пленки стабилизированного полимера 0,3 мм. Стабилиза-
D тор вводили также путем валь-
цевания при 160 °C в течение
10 мин. Представительность каж-
дого эталонного образца в данном
случае обеспечивалась измерением
оптической плотности аналитичес-
кой полосы для 10-15 образцов,
вырезанных из разных мест ста-
билизированной полимерной плен-
Рис. V.17. УФ-спектры поглощения полиэтилена, стабилизированного бисалкофе-
ном МЦП (7); антиоксидантом НГ-2246 (2); бензоном ОА (3); неозоном А +
+ диафеном фф (с соотношением 2 :1) (4).
Массовое содержание стабилизатора (в %) : 1 — 0,15; 2 — 0,2; 3 — 0,5; 4 — 0,1.
Толщина пленок 0,03 мм.
Рис. V.18. УФ-спектр поглощения (с компенсацией рассеинного света) полиэтилена,
стабилизированного тиоалкофеном БМ.
Толщина пленки 0,3 мм.
141
D 0,6
Рис. V.19. УФ-спектр поглощения пеитапласта с массовым содержанием диафеиа
НН 0,2%.
Толщина пленки 47 мкм.
Рис. V.20. ИК-спектры поглощения полиэтилена без стабилизатора (----)
с добавкой фенозана 23 (------).
Массовое содержание стабилизатора (в %) : 1 — 0,1; 2 - 0,3. Толщина пленки
0,9 мм.
ки. Хорошее расположение точек на градуировочной прямой (зависи-
мость поглощения полосы 250 нм от содержания тиоалкофена БМ
в полиэтиленё) свидетельствовало о равномерности распределения
стабилизатора в приготовленных эталонах.
Количественное определение М,М'-ди(нафтил-2)фенилендиамина-1,4
(диафена НН) в поли-3,3'-бис (хлорметил) оксациклобутане (пентаплас-
те) проводили по полосе 320 нм (рис. V.19). Методика позволяет опре-
делять диафен НН в интервале массовых концентраций 0,10—0,40%.
Эталонные образцы готовили для разных концентраций стабилизатора,
который вводили в полимер на скоростном смесителе с частотой враще-
ния 750 об/мин. Пленки прессовали при 190 °C в течение 5 мин. Спектры
эталонных и анализируемых образцов записывали относительно спект-
ров нестабилизированной пленки полимера, приготовленной из той же
его партии, в которую вводили стабилизатор.
Интенсивность аналитической полосы поглощения в максимуме
определяли с использованием базисной линии (см. рис. V.19). Зависи-
мость D/d от концентрации введенного стабилизатора описывается
прямой линией, проходящей через начало координат.
Мы рассмотрели возможности непосредственного определения
стабилизаторов в полимерах по УФ-спектрам поглощения. В зависимости
от интенсивности полосы вводимой добавки и прозрачности полимера
такие методики могут быть разработаны и с использованием ИК-об-
ласти спектра. Так, например, для определения стабилизатора эфира
3,5-ди-трет-бутил-4-гидроксифенилпропионовой кислоты и пентаэритри-
та (фенозана-23, тетраалкофена ПЭ)
142
СН2СН2СООСН2 — с
в полиэтилене можно использовать интенсивное поглощение валентного
колебания связи С=О сложноэфирной группы этого соединения. Про-
зрачность полиэтилена в аналитической области (полоса 1740 см-1)
позволяет использовать пленки толщиной около 0,9 мм (рис. V.20)
и определять содержание стабилизатора в интервале массовых концен-
траций 0,075—0,3%. Для равномерности распределения стабилизатора
в массе полимера сначала готовили его концентрат на скоростном смеси-
теле, затем на том же смесителе разбавляли концентрат полимерным
порошком до рабочих концентраций. Полученная смесь дополнительно
перемешивалась на вальцах при 160 С. Для каждой концентрации
прессовали по три пленки. Интенсивность полосы 1740 см "1 определяли
по методу базисной линии. Градуировочный график зависимости D/d
от концентрации введенной добавки линеен и проходит через начало
координат.
Погрешность определения концентрации добавки в полимере по
спектру пленки в большой мере зависит от неравномерности распреде-
ления стабилизатора по массе полимера эталонных образцов и разно-
толщинностью прессованных пленок. Поэтому отработка метода введе-
ния стабилизатора и приготовление эталонных пленок является основой
методики. Правильность выбранных условий можно контролировать
по разбросу точек градуировочного графика.
V.2.3. Исследование растворов полимеров
Определение стабилизатора по спектру раствора стабилизированного
полимера можно также рассматривать как прямой анализ. Для этой
цели наиболее пригодной является УФ-область спектра, поскольку
в ИК-спектрах трудно подобрать полосу поглощения стабилизатора,
необходимой интенсивности, в области прозрачности растворителя
и полимера.
Если на аналитическую полосу стабилизатора налагается поглощение
полимера, то можно использовать метод осаждения полимера из раство-
ра и анализировать полученный раствор. Так, например, раствор поли-
бутадиена, содержащего концевые хлорметилбензоатные группы
(—ОСОС6Н4СН2С1) не может быть проанализирован на содержание
антиоксиданта НГ-2246 из-за наложения их полос поглощения. Поэтому
была предложена [78] методика высаждения каучука из его раствора
в хлороформе пятикратным количеством спирта. При этом в растворе
остается более 95% антиоксиданта и менее 2% полимера. Последующий
анализ проводится по. обычной схеме.
143
V.2.4. Равномерность распределения стабилизатора в полимере
Введением стабилизирующих добавок в полимерные материалы про-
длевают срок службы, защищая от влияния внешних воздействий.
Большая эффективность стабилизирующих добавок обеспечивается
их равномерным распределением в массе полимера. Существуют разные
способы смешения стабилизаторов с полимерами. Для выбора наиболее
экономичного и оптимального метода введения добавок необходим
контроль за равномерностью их распределения. Это можно осуществить
при помощи спектральных методов анализа. Следует отметить, что
при исследовании равномерности распределения добавок методом
спектроскопии используется пленка небольшой площади примерно
10 Х20 мм (в зависимости от применяемого спектрофотометра), причем
в пучок света попадает еще меньший ее участок, по которому проис-
ходит усреднение определяемого количества стабилизатора. Чтобы
получить представление о распределении стабилизатора в большом
объеме гранулята или пленки по локальному участку, необходимо
установить то количество отобранных проб, которое будет характери-
зовать распределение стабилизатора в необходимом объеме полимера.
В работе [79] проводили сравнительное исследование эффектив-
ности введения стабилизатора бисалкофена МЦП разными способами.
Равномерность распределения стабилизатора оценивали с помощью УФ-
спектров поглощения [76] следующим образом. Из потока гранул отби-
рали „мгновенную” пробу массой в 3 кг, затем из разных мест пробы
брали 25—30 гранул, спрессовывали их в пленку и подвергали ее анали-
зу. Было показано, что характеристикой равномерности распределения
служат усредненные результаты, полученные для числа гранул равного
или большего 20.
Опыт приготовления эталонов для анализа стабилизаторов в пленке
свидетельствует, что наилучшее смешение достигается перемешиванием
в расплаве тех стабилизаторов, температура плавления которых ниже
температуры вальцевания или близка к ней.
V.2.5. Оценка эффективности действия стабилизаторов
Эффективность действия стабилизирующих добавок можно оценить
по продолжительности сохранения первоначальных свойств (например,
физико-механических, электрических) в процессе эксплуатации или
хранения полимера. Во многих случаях метод инфракрасной спектро-
скопии оказывается по сравнению с другими методами менее трудо-
емким, более информативным и требует небольшого количества поли-
мерного материала, что немаловажно, если работа проводится на стадии
лабораторных опытов. ИК-спектры поглощения позволяют следить
за увеличением числа появившихся или разрушившихся структурных
групп в полимерных молекулах в процессе старения в зависимости
от изменения условий. Кинетические кривые, полученные при разных
температурах старения, могут быть использованы для расчета энергии
активации того или иного процесса.
144
Рис. V.21. Кинетика накопления
групп С=О (а) и разрушение связей
С=С (б) ударопрочного полистирола
при его прогреве при 160 °C:
(-------) — нестабилизированный
образец; (---------) — образцы
стабилизированные 0,5% (по массе)
ФАУ-21 (/) иФАУ-13 (2).
Для выбора эффективных
стабилизаторов, оптимальных
доз вводимых добавок исполь-
зуют ускоренные мотоды ста-
рения полимеров, подвергая их
атмосферным воздействиям, на-
греванию, облучению и т. п.
В полимерах под влиянием ки-
слорода воздуха и повышен-
ных температур происходят
окислительные процессы, кото-
рые приводят к образованию
карбонильных групп разного
типа. Спектры поглощения поли-
мерной пленки фиксируют эти
изменения. т, ч
По мере увеличения продолжительности окисления возрастает ин-
тенсивность соответствующей полосы поглощения карбонильной
группы (кислотной, альдегидной, кетонной и др.). Зависимость ин-
тенсивности полосы группы С=О от продолжительности воздействия
на полимер внешней среды представляет собой кинетическую кривую,
которая дает представление о степени окисления полимера. Исполь-
зуя такие зависимости для полимеров, стабилизированных разными
добавками, получают данные об их защитных свойствах. Примером
определения сравнительной эффективности ряда стабилизаторов ударо-
прочного полистирола может служить рис. V.21 [80]. По оси ординат
(рис. V.21, а) отложено отношение оптических плотностей полосы
поглощения карбонильной группы 1720 и полосы 1875 см “1 полистиро-
ла, выбранной как „внутренний стандарт”. Увеличение этого отношения
отражает процесс накопления групп С=О в полимере. На рис. V.21, б
показано изменение содержания двойных связей полибутадиена, входя-
щего в состав ударопрочного полистирола (А, в процентах к исходному
количеству). Ход обеих кинетических кривых свидетельствует, что
количество связей С=С уменьшается по мере накопления групп С=О за
счет разрыва двойных связей.
Для стабилизированных образцов наблюдаются аналогичные из-
менения, но замедленные во много раз. Из сравнения кинетических
кривых видно, что наилучшим стабилизирующим действием обладает
стабилизатор бис (3,5 -ди-трет-бутил-4-гидроксифенилпропиогидразино)-
винилфосфин (ФАУ-21).
145
3600 2800 2000 1800 1600 1400 1200 1000 800 600 400
V,CM~'
Рис. V.22. ИК-спектры поглощения неорганических наполнителей в вазелиновом
масле.
Капиллярные слои между окнами КВг; 1 — тальк; 2 — слюда; 3 — диоксид титана;
4 — вазелиновое масло.
3600 2800 2000 1800 1600 1400 1200 1000 800
V, СМ'1
Рис. V.23. ИК-спектры поглощения исходного (I) полиэтилена и с добавкой SiO2
(2) и мела (5).
Толщина образцов 30 мкм.
V.3. НЕОРГАНИЧЕСКИЕ НАПОЛНИТЕЛИ
В полимерные материалы, кроме стабилизаторов и пластификаторов,
вводят также другие добавки разного значения, в том числе неоргани-
ческие наполнители. Содержание последних достигает 50% (по массе).
Среди минеральных наполнителей широко применяются диоксид
кремния (SiOj), мел (СаОО3), каолин (каолинит, Al4[Si4O10 ] (ОН) 8),
тальк (гидросиликат магния, Mg3[&4Oi0](OH)2), асбест (гидроси-
ликаты, например A^g3[S2O5 ](ОН)4), диоксид титана (11О2), слюда
(алюмосиликаты AB[AlSi3O10](OH, F)2, где А = К, Na; В = А1, Mg, Fe,
Zn) и др. Поскольку эти соединения имеют природное происхождение,
то они могут отличаться по составу элементов.
Большинство оксидов перечисленных минералов имеют интенсивное
поглощение около 1000 см-1, за исключением ТЮ2, а также полосы
поглощения в области 700—400 см -1 (рис. V.22).
Для полиэтилена, обладающего большой прозрачностью в ИК-об-
ласти спектра, можно проводить анализ какого-либо наполнителя по
спектру пленки. Например, на рис. V.23 приведен спектр полиэтилена с
добавкой 10% (по массе) мела при толщине пленки 30 мкм. В спектре
наблюдается интенсивное поглощение с максимумом в интервале
1500—1400 см-1, налагающееся на полосы деформационных колебаний
групп -СН2— полиэтилена, и узкая полоса при 880 см-1, обусловлен-
ная колебаниями иона СО3~ карбоната кальция [2, с. 489]. Наличие
узкой изолированной полосы 880 см-1 может служить основой для
количественного определения мела в полиэтилене. Для диоксида
кремния такой полосой может быть полоса около 1100 см-1 (см, рис.
V.23).
Однако наполнитель неорганического происхождения является не
единственной добавкой, которую вводят в полимер. Поглощающий до-
бавки органического происхождения приводят к наложению спектров и
осложняют анализ. Кроме того, большинство полимеров в области от
1400 см -1 и ниже имеют собственные полосы поглощения. Поэтому для
применения спектров поглощения необходимо выделение добавок из
полимера и их раздельное определение. Это трудоемкая работа. В связи с
этим наиболее целесообразно для определения количества минерального
наполнителя использовать метод эмиссионного спектрального анализа,
который, к сожалению, пользуется недостаточным вниманием экспери-
ментатора-аналитика. Метод требует озоления полимера, так же как и
химический элементарный анализ, но позволяет одновременно про-
водить количественное определение в полимере разных элементов.
V.4. ОПРЕДЕЛЕНИЕ ДОБАВОК НА ПОВЕРХНОСТИ
ПОЛИМЕРНОГО МАТЕРИАЛА
। Разная совместимость полимера и вводимой в него добавки определя-
ется главным образом их химической природой. Добавки, обладающие
ограниченной совместимостью с полимерным материалом, мигрируют
со временем к его поверхности. „Выпотевание” добавки наблюдается
147
1100
_____।--------1_______।—
1200 1100 1000
V, СМ-'
Рис. V.24. ИК-спектры поглощения НПВО
полиэтилена (а) и композиции полиэтиле-
на с полиизобутилеиом (б).
V,CM'1
a: 1 — исходная пленка; 2—4 — пленка с нанесением на ее поверхность антистатика
ЦС-17 из раствора с массовой концентрацией 2 (2), 5 (5) и 7,5% (4) ;
б: 1 — пленка с поверхностным слоем антистатика ЦС-17; 2 — пленка после снятия
слоя антистатика ЦС-17.
и в случае, когда ее концентрация превышает равновесный предел сов-
местимости с полимерной матрицей. На процесс миграции добавок
влияют также другие факторы (надмолекулярная структура полимера,
наличие других ингредиентов, температура и т. п.).
Задачи, связанные с изучением поверхностных явлений в полимерах,
могут быть решены методом нарушенного полного внутреннего отраже-
ния (НПВО) или МНПВО (см. раздел II.1.5)*.
Примером применения метода НПВО может служить исследование
миграции фталатных пластификаторов к поверхности поливинилхлорид-
ной пленки [81]. Это явление нежелательное, так как оно ухудшает
свойства пластифицированного полимера, придавая ему жесткость.
Изучение миграции проводили следующим образом. Сначала удаляли
пластификатор, находящийся на поверхности пленки, обработкой ее
в этаноле в течение 2-3 мин. Затем при помощи метода НПВО (элемент
из кристалла KRS-5, угол 45 °) записывали спектры поглощения пласти-
фикатора во времени. Содержание пластификатора на поверхности
пленки определяли по полосе 1725 см-1, относящейся к валентному
колебанию С=О сложноэфирной группы. Поскольку интенсивность
полосы поглощения пластификатора велика, то одного отражения (глу-
♦Метод НПВО (МНПВО) находит все большее применение в аналитических
работах, тем не менее его нельзя рассматривать как метод, пригодный для серий-
ных анализов в силу его специфики.
148
бина проникновения света в пленку толщиной порядка нескольких
микрометров) было достаточно для регистрации его спектра. Установ-
лено увеличение интенсивности полосы 1725 см-1 от времени наблюде-
ния, что позволило определить коэффициент диффузии фталатного
пластификатора в поливинилхлоридной пленке (полосой внутреннего
стандарта служила полоса 1425 см-1 поливинилхлорида).
Миграцию добавки можно исследовать и по спектру пропускания,
если следить за уменьшением интенсивности ее полосы поглощения,
периодически смывая с поверхности слоя полимера выделенную добавку.
На явлении миграции добавки к поверхности полимера основано
действие поверхностно-активных веществ как внутренних антистатиков.
Добавка вводится в массу полимера и мигрирует к его поверхности,
образуя слой, обладающий антистатическими свойствами. Скорость
миграции должна быть такой, чтобы обеспечить длительное действие
добавки. В отличие от внутренних наружные антистатики наносят на
поверхность полимерного материала его погружением в специальный
раствор или напылением.
Антистатическое действие поверхностно-активных веществ зависит
от толщины слоя вещества, скорости миграции и других факторов. Опре-
деление толщины поверхностного слоя добавки может быть выполнено
методом ИК-спектроскопии. Рассмотрим это на примере определения
количества антистатика ЦС-17 (продукта взаимодействия смеси цетило-
вого и стеарилового спиртов с молярным содержанием окиси этилена
17%) [82].
Антистатик вводили в расплав полимера на вальцах или наносили
на поверхность полимерной пленки погружением ее на 15 с в этанольный
раствор. Толщина прессованных образцов составляла 1,0; 0,1 и
0,05 мм.
Антистатик ЦС-17 имеет полосы поглощения 1060,1110и 1145 см -1,
в этой области полиэтилен прозрачен (рис. V.24). При комнатной темпе-
ратуре ЦС-17 находится в кристаллическом состоянии и его спектр
отличается от спектров расплава или раствора, что не позволяет послед-
ние использовать для определения показателя поглощения аналитичес-
кой полосы. Поэтому ЦС-17 вводили в полимер в виде смеси с объемной
долей диоктилсебацината 20%, который служил внутренним стандартом.
Пластификатор не влиял на форму полос антистатика. По известному
показателю поглощения полосы 1735 см-1 диоктилсебацината была
определена эффективная толщина слоя пластификатора и с учетом
его объемной доли рассчитан эффективный слой ЦС-17. Это дало воз-
можность определить показатель поглощения полос антистатика, нахо-
дящегося в кристаллическом состоянии.
Определение слоя ЦС-17, выпотевшего на поверхность, проводили
при помощи спектров НПВО, которые позволяли найти его толщину
при однократном отражении, начиная с 0,03 мкм. Кроме того, для
определения толщины слоя антистатика использовали спектры пропус-
кания, исследуя стопу тонких пленок.
149
VI. ОПРЕДЕЛЕНИЕ КАЧЕСТВА СЫРЬЯ И ЧИСТОТЫ
ПОЛИМЕРОВ
VL1. АНАЛИЗ ПРИМЕСЕЙ В НИЗКОМОЛЕКУЛЯРНЫХ
СОЕДИНЕНИЯХ
Основные свойства полимеров зависят от чистоты исходных продуктов —
мономеров, а также катализаторов, инициаторов, растворителей и дру-
гих вспомогательных веществ, участвующих в процессе полимеризации.
Необходимость контроля чистоты исходного сырья очевидна. Поэтому
при получении полимеров регламентируется допустимое количество
примесей.
Методы УФ- или ИК-спектроскопии часто оказываются неэффектив-
ными при определении примесей в низкомолекулярных смесях, особен-
но многокомпонентных. В настоящее время для этих целей успешно
используют методы газовой и газо-жидкостной хроматографии, которые
позволяют идентифицировать по хроматограмме основное соединение,
присутствующие в нем примеси и провести их количественное определе-
ние. Наложение же в спектре полос поглощения примесей и анализиру-
емого соединения затрудняет анализ. Такая задача может быть решена
при известных составе смеси и показателях поглощения аналитических
полос путем решения системы уравнений. В реальных условиях завод-
ских или отраслевых лабораторий это не всегда выполнимо, поэтому
использование хроматографических методов анализа наиболее рас-
пространено. Иногда удобно применять оба метода.
Рассмотрим, например, анализ винилацетата. Винилацетат может
содержать в виде примесей дивинилацетилен, бензол, ацетон, ацеталь-
дегид, кротоновый альдегид, гидрохинон, воду, уксусную кислоту. Боль-
шинство из перечисленных соединений имеют полосы поглощения в
УФ-области спектра (рис. VI.1). Склон интенсивного поглощения винил-
ацетата маскирует полностью поглощение бензола и одну из полос
дивинилацетилена (252 нм). Вторая его полоса при 266 нм на фоне
поглощения винилацетата может быть использована для анализа. Полосы
поглощения ацетона [275 нм; а = 0,27 л/(г ’см)], гидрохинона [294 нм;
а = 28 л/ (г • см) ], ацетальдегида [287 нм; а = 0,38 л/ (г • см) ] взаимно
перекрываются. Полоса поглощения кротонового альдегида [320 нм;
а = 0,3 8 л/(г *см)], присутствие которого в мономере не допускается,
практически не проявляется в спектре из-за слабого поглощения уже
при массовом содержании его 0,005%. Его вторая, интенсивная полоса
[219 нм; а =275 л/(г *см) ] недоступна для анализа.
Представленные на рис. VI.1 спектры поглощения соединений были
получены для растворов в винилацетате. Так как поглощение, винил-
150
ацетата в значительной мере перекрывает поглощение примесных сое-
динений в слое 1 см, который требуется для анализа в соответствии
с их реальным содержанием в мономере, то спектр каждого соединения
был получен с использованием поглощающего слоя в 1 мм с последую-
щим вычитанием спектра „чистого” винилацетата. Затем полученные
спектры пересчитывали на поглощение слоя 1 см.
Одновременное присутствие перечисленных примесей в винилацета-
те приводит к сложному и невыразительному спектру (см. рис. VI.1,
7), который не дает представления ни о составе примесей, ни об их
количестве.
Можно, например, установить критерий чистоты винилацетата,
определив суммарное количество примесей по интегральному поглоще-
нию в ограниченном интервале длин волн. Однако количественный
состав примесей нестабилен, а учитывая различия показателей поглоще-
ния полос каждого компонента смеси, такую оценку нельзя считать
корректной. В этом случае оказалось полезным сочетание методов
газовой хроматографии и спектроскопии.
Метод газовой хроматографии позволил идентифицировать и опреде-
лять количество большинства примесей винилацетата [83, с- 146[. Однако
дивинилацетилен, который является наиболее активной примесью,
ингибирующей процесс полимеризации винилацетата, определить не
удалось. Допустимая массовая доля его в мономере находится в преде-
лах 0,0003-0,0005%. Для его определения целесообразно использовать
методы спектроскопии так как благодаря высокому показателю погло-
щения дивинилацетилена
В КО г
[266 нм; а= 200 л/(г • см) ] чувствительность
Рис. VI. 1. УФ-спектры
поглощения примесей
в винилацетате:
1 — дивинилацетилен
(0,0003%); 2 - бензол
(0,01%); 3 — ацеталь-
дегид (0,05%); 4 —
ацетон (0,05%); 5 —
гидрохинон (0,0004%) ;
6 — очищенный и 7 —
загрязненный винилаце-
тат. В скобках — мас-
совое содержание при-
месей, толщина 10 мм.
его проявления в спектре
велика. Можно также опре-
делить показатели поглоще-
ния и форму полос приме-
сей винилацетата из спек-
тров их индивидуальных рас-
творов в винилацетате, а
затем при помощи ЭВМ рас-
считать поглощение, отно-
сящееся к дивинилацетиле-
ну, и найти количество по-
следнего.
0.8
0,6
0,4
0,2
290
ЗЮ
°250 270
330 350
Л, нм
151
Существует и более простой способ. Находят оптическую плотность
в одной точке спектра, соответствующей максимуму полосы поглоще-
ния 266 нм (D266 ). Это допустимо, так как оказалось, что суммарное
поглощение остальных примесей винилацетата соответствует оптической
плотности при Х= 266 нм на порядок меньшей, чем поглощение дивинил -
ацетилена. Определение дивинилацетилена можно проводить по формуле:
D^-D^-D^-iat.Ce + a^ + аацСац)
ад<7
где и Z>q66 - оптические плотности анализируемого и очищенного образцов
винилацетата; D350 — оптическая плотность фона при 350 нм; а — удельные показа-
тели поглощения каждой примеси при 266 нм; С - концентрация примесей по
данным газовой хроматографии. Индексы означают принадлежность а и С к ди-
винилацетилену (д), ацетону (ац), ацетальдегиду (а), бензолу (б).
Одной из примесей, присутствующей в растворителях, мономерах
и влиякпцей на ход процесса полимеризации, является вода.
В жидкой фазе молекулы воды находятся в ассоциированном со-
стоянии, что приводит в спектре к широкому поглощению с максиму-
мом при 3450 см-1, относящемуся к валентным колебаниям связи
О—Н.Деформационное колебание имеет полосу при 1640 см-1. В ближ-
ней ИК-области спектра наблюдаются полосы комбинированных коле-
баний 5 190 см -1 (1,92 мкм) и первого обертона валентных колебаний
связей О—Н 6 920 см “1 (1,45 мкм).
В зависимости от содержания влаги в образце можно использовать
полосы основных или обертонных колебаний при условии прозрачности
в аналитической области анализируемого соединения.
Для определения влажности растворителя с низким массовым
содержанием воды (до нескольких процентов) удобно использовать
полосы поглощения в ближней ИК-области. Примером может служить
спектр безводного (абсолютного) этанола в сравнении со спектром
технического спирта *, содержащего воду (рис. VI.2).
Влажность пластификатора триэтиленгликоль-ди (2-этилбутирата)
была оценена по полосе поглощения 1,91 мкм (рис. VI.3). Массовое
содержание воды в образце соответствовало 1,4%, что хорошо согласу-
ется с данными химического анализа (метод Фишера) — 1,3%. Спектр
был получен при толщине поглощающего слоя 1 мм.
С увеличением толщины слоя до 10 мм возрастает вклад соседних
интенсивных полос пластификатора в поглощение аналитической по-
лосы 1,91 мкм. Это приводит к искажению первоначально выбранной
(для d = 1 мм) базисной линии (существенно меняется угол ее наклона).
Кроме того, проявляются незаметные ранее полосы. Поэтому при пере-
ходе на новую толщину слоя при анализе необходимо измерять интен-
сивность поглощения как испытуемых образцов, так и эталонных с
* Этиловый спирт образует с водой азеотропную смесь, содержащую 4, 43%
воды (по массе).
152
Рис. VI.2. ИК-спектры поглощения технического (/) и абсолютного (2) этанола.
Толщина образца 1 мм.
Рис. VI.3. ИК-спектр поглощения пластификатора триэтиленгликольди (2-этил-
бутирата, содержащего воду (/) и осушенного (2).
Толщина образца 1 мм.
использованием вновь выбранной базисной линии. Большая интенсив-
ность поглощения валентных колебаний связей О—Н воды позволяет
с уверенностью определить влагу в образцах до 10—3% (по массе).
В некоторых случаях анализируемое вещество настолько прозрачно
в аналитической области, что можно увеличить толщину поглощения
слоя и тем самым уменьшить нижний предел определения влаги. Одна-
ко эта возможность чаще всего не реализуется, так как большинство
соединений, если и не имеют полос, непосредственно налагающихся
на полосы поглощения воды, то по крайней мере поглощают вблизи
них. Поэтому для каждого соединения предел обнаружения влаги мето-
дом ИК-спектроскопии будет зависеть от его спектральных характери-
стик.
Растворители часто загрязнены примесями, являющимися исходны-
ми или сопутствующими продуктами их синтеза, которые можно уста-
новить по спектру поглощения. Например, циклогексан, получаемый
гидрированием бензола, содержит его в качестве примеси. Бензол может
присутствовать также в гексане, абсолютном этаноле и некоторых
других растворителях. Серия узких интенсивных полос поглощения
бензола в УФ-области спектра (рис. VI.4) дает возможность контроли-
ровать чистоту абсолютного этанола.
153
Для определения малых количеств примесей в каком-либо соеди-
нении можно воспользоваться методом добавок, широко применяемом
в атомной спектроскопии. Его удобно использовать тогда, когда от при-
меси нельзя полностью очистить образец, а ее содержание подлежит опре-
делению. На основании испытуемого соединения с неизвестной концен-
трацией примеси Со составляют растворы с добавками примеси извест-
ных концентраций. Затем строят график зависимости D от С (концен-
трации добавленной примеси). Зависимость должна быть линейной*
(рис. VI.5). Прямая не проходит через начало координат, а отсекает на
оси ординат отрезок, величина которого соответствует оптической плот-
ности присутствующей в образце примеси. Экстраполяция прямой до
пересечения с осью абсцисс дает значение концентрации этой примеси.
Этот метод можно использовать, если есть уверенность, что анализи-
руемое соединение само не поглощает в данной области спектра или
если его поглощение удается скомпенсировать.
Чтобы не проводить каждый раз серию измерений,можно, убедив-
шись в линейности графика, ограничиться приготовлением растворов
двух каких-либо концентраций Са и Св. Оптические плотности полосы
поглощения для этих концентраций равны:
Z>a=((b+Са) ad ; 2>В=(С() + CB)orf
где а - удельный показатель по-
глощения аналитической полосы.
* Если наблюдается отклонение
от линейности, то методом добавок
пользоваться нельзя.
Рис. VI.4. УФ-спектр поглощения разных образцов абсолютного этанола, загряз-
ненного бензолом.
Толщина образца 10 мм.
Рис. VI.5. Градуировочный график для определения неизвестной концентрации
примеси Со по методу добавок.
154
Решая совместно эти уравнения, получаем выражение для опреде-
ления концентрации примеси в образце:
_ РаСв 7)вСа
С°~ Db-D&
Для определения микроколичеств примесей неорганического про-
исхождения полезны методы атомно-абсорбционной спектроскопии
и эмиссионного спектрального анализа.
В работе [84] предложен экспрессный спектрографический метод
определения микропримесей в растворах. По данным авторов, метод
отличается простотой, позволяет анализировать растворы различного
происхождения (сточные и промышленные воды, водно-спиртовые
смеси и др.) на элементы: Си, Ag, Zn, Cd, Al, Ti, Sn, Bi, V, Mo, Mn, Fe,
Ni, Co, а также ряд редкоземельных элементов. Источником возбуж-
дения служил разряд высоковольтной конденсированной искры от
генератора ИГ-3. Испытуемый раствор вводили непосредственно в зону
разряда, минуя операцию нанесения его на угольный порошок. Использо-
вание инертной среды, варьирование электрических параметров искро-
вого генератора позволило снизить пределы обнаружения примесей
практически для всех элементов до 10 -3 мг/мл.
VI .2. АНАЛИЗ ПРИМЕСЕЙ В ПОЛИМЕРАХ
Применение того или иного спектрального метода для определения
примеси зависит от ее природы — в основном от того, является ли при-
месь органическим соединением (остатки мономеров, инициаторов
органического происхождения, эмульгаторов, растворителей и т. п.)
или же неорганическими (главным образом, металлы — остатки ката-
литических систем).
Анализ органической примеси осуществляется методами молеку-
лярной спектроскопии. Эта задача принципиально не отличается от
любой другой, связанной с определением количества каких-либо функ-
циональных групп, добавок и т. п., рассмотренной выше, и сводится
к поиску аналитической полосы, выбору оптимальных концентраций,
необходимых для построения градуировочного графика, и вычисления
показателя поглощения. Например, по интенсивности полосы поглоще-
ния примеси можно следить за степенью отмывки от нее полимера. Та-
ким образом определяли чистоту сополимера этилена с винилацетатом
после удаления из него эмульгатора ОП-10. Содержание эмульгатора оце-
нивали по интенсивности его полосы поглощения при 277 нм. Для полу-
чения спектра со полимер растворяли в тетрахлорметане с концентрацией
10 г/л. После каждой промывки и удаления из полимера влаги опреде-
ляли оптическую плотность полосы 277 см-1 (О277) в спектре раствора
сополимера. Зависимость D211 от числа промывок (рис. VI.6) позволяет
следить за количеством оставшегося в полимере эмульгатора.
Рассмотрим анализ примесей неорганического происхождения
в полимерах на примере методики эмиссионного спектрального анализа,
155
Рис. VI.6. Зависимость интенсивности полосы эмульгатора (£)277 ) от числа
промывок полимера.
Рис. VL7. Градуировочный график для определения содержания А1 (266,04 нм)
и Ti (264,66 нм) в золе полиэтилена.
которая позволяет определять одновременно Mg, Fe, Mo, Si, Pb, Ca,
Cd, Zn, Mn, Cr, Al, Ti, Ni, V в золе полимеров и других органических
материалов. Состав элементов в эталонах должен соответствовать основ-
ному составу анализируемой золы. Все элементы вводили в эталоны
в виде оксидов. Сначала готовили головной эталон, который последо-
вательно разбавляли буферной смесью.
Буферная смесь состояла из угольного порошка и оксида меди в соотношении
1:1. Внутренним стандартом служила линия меди 302,26 нм. Навеску золы (не
менее 10 мг) разбавляли буферной смесью в 50, 150, 300 или 500 раз. Такое сту-
пенчатое разбавление позволяет выявить в спектре линии элементов, находя-
щихся в золе в разных концентрациях. Буферная смесь, эталоны и образцы для
анализа размешивали в яшмовой ступке в течение 30 мин. Готовые пробы и этало-
ны плотно набивали в канал нижнего электрода диаметром 3,3 мм, глубиной 2 мм.
Верхний электрод затачивали на усеченный конус.
Источником возбуждения служила дуга переменного тока. Использовали
кварцевый спектрограф ИСП-28.
Условия съемки: ширина щели спектрографа 0,015 мм, междуэлектродный
промежуток 3 мм, диафрагма на промежуточном конденсоре трехлинзовой систе-
мы 3,2 мм, сила тока 20 А, экспозиция 1 мин, фотопластинки „спектральные”,
тип 1.
Анализ проводили по методу трех эталонов. Градуировочные графики строили
в координатах Д5, IgC (например, для А1 и Ti, рис. VI.7).
Определив содержание искомых элементов в золе и зная зольность
полимера, рассчитывали содержание элементов по формуле: X - СВА/100,
где X — содержание анализируемого элемента в полимере, %; С — со-
держание анализируемого элемента в пробе, % В — коэффициент раз-
бавления, А — зольность полимера, %. Относительная погрешность
156
определения для большинства элементов составляла 10%. Ниже даны
аналитические линии элементов X и интервалы определяемых концен-
траций ДС:
Элемент А., нм ДС, % Элемент А, нм ДС, %
Mg 309,690; 277,669 0,03 -0,3 Zn 334,502 0,015-0,15
Ее 259,957 0,03-0,3 Мп 293,306 0,015-0,15
Мо 313,259 0,03-0,3 Сг 301,476 0,012-0,12
Si 251,612 0,03-0,3 А1 308,216 0,012-0,12
Pb 283,307 0,03-0,3 Ti 307,864 6,012-0,12
Са 317,933 0,015-0,15 Ni 305,082 0,012-0,12
Cd 326,106 0,015-0,15 V 318,540 0,006-0,06
Частным случаем рассмотренной методики служила методика опре-
деления А1 и Ti в золе полиэтилена низкого давления, полученного
в присутствии тетрахлорида титана и триэтилалюминия. Исходный
головной эталон содержал по 10% алюминия и титана в буферной смеси.
Если можно пожертвовать точностью анализа для сокращения его
продолжительности, то определение А1 и Ti в полиэтилене можно прово-
дить без предварительного озоления [85, с. 239]. В этом случае эталоны
готовили на основе смеси угольного порошка и порошка полиэтилена
высокого давления, не содержащего А1 и Ti. Сначала определяемые
элементы в виде оксидов вводили в угольный порошок и перемешивали
в ступке в течение 30 мин. Головной эталон готовили с содержанием
10% А1 и 107с Ti (в расчете на металл), который подвергался в дальней-
шем разбавлению угольным порошком до следующего содержания
металлов:0,01; 0,03; 0,05; 0,17. Затем каждый эталон разбавляли
порошком полиэтилена высокого давления в отношении 2 : 1 (по массе)
соответственно.
Для проведения анализа пробу полиэтилена низкого давления раз-
бавляли угольным порошком (1:2) путем перемешивания той же
продолжительности, что и эталоны. Условия съемки описаны выше,
глубина кратера нижнего электрода 5 мм. За внутренний стандарт при-
нимали фон около линий. Относительная погрешность определения
составляла 207.
Описанную выше методику, рассчитанную на определение 14 элемен-
тов в пробе, полезно использовать, когда нет никаких данных о составе
пробы и относительном содержании элементов. Если качественный
состав золы известен, как, например, при анализе полиэтилена низкого
давления, то нет необходимости использовать универсальные эталоны.
Эталоны при этом готовят с учетом анализируемых элементов и тех
интервалов концентраций, которые соответствуют их содержанию в про-
бе. Так, при анализе золы полиэтилена, полученного в присутствии
титан-магниевого катализатора на алюмосиликатном носителе [86 ].
аналитические линии и условия съемки спектров аналогичны указанным
выше, отличаются только интервалы концентраций определяемых эле-
ментов Ti. Mg. AT Si, Fe. Анализ золы полимера, т. е. оксидов металлов,
не отличается от анализа другого подобного объекта. Поэтому методики
157
мало отличаются по существу, схема их однотипна. При анализе полиме-
ра, непосредственно вводимого в зону разряда без озоления, возможно
влияние полимерной матрицы на интенсивность линий. В большей мере
это зависит от элементов, входящих в основу анализируемой пробы.
Чтобы обеспечить воспроизводимость всех физико-химических процес-
сов,протекающих в разряде, эталоны должны быть максимально иден-
тичны анализируемой пробе. Подготовку к анализу образцов порошко-
образных полимеров осуществляют по методике, описанной для поли-
этилена низкого давления (см. выше). Если же полимер не поддается
измельчению до порошкообразного состояния, например каучук, исполь-
зуют его раствор. В работе [87] методом эмиссионного анализа опреде-
ляли содержание кобальта в каучуке без озоления.
Эталоны готовили на основе растворов анализируемого каучука
в толуоле путем перемешивания их с растворами соли кобальта Co(NO3)2 •
•6Н2О в этаноле. Анализ проводили по методу добавок. Концентрация
первого эталона соответствовала предполагаемому содержанию кобальта
в каучуке. Последующие эталоны содержали кобальт в 2, 3 и 4 раза
больше. Смешанные растворы каучука и соли выдерживали до полного
испарения растворителей. Полученный таким образом каучук, с введен-
ными в него добавками кобальта, подвергали анализу, сжигая непосред-
ственно в кратере угольного электрода (диаметр 3 мм, глубина 6 мм).
Каучук измельчали до размеров 1—1,5 мм2. Относительная погрешность
определения составляла 13%. Наблюдали хорошее совпадение резуль-
татов анализа с данными фотокалориметрического метода при умень-
шении продолжительности определения в 2 раза.
Не многочисленные публикации по определению примесей металлов
и других элементов в полимерах методом атомно-абсорбционного
анализа посвящены исследованию растворов минерализованных хими-
ческими методами полимеров или растворов их золы. При анализе
полимерных растворов в пламени было обнаружено снижение интенсив-
ности аналитических линий при увеличении концентрации полимера
в растворе (для одной и той же концентрации определяемых элементов).
Интенсивность аналитических линий может понижаться из-за двух факто-
ров - снижения степени атомизации аэрозоля, уже поступившего в пла-
мя, и эффективности распыления (т. е. отношения количества образца,
достигшего рабочей зоны пламени, к полному его количеству, вступив-
шему в распылительное устройство [25, с. 46]). Последняя причина
носит название матричного эффекта.
Подробное исследование влияния полиакриламида на определение
следов элементов Li, К, Na, Са, Mg, Мп в его водных растворах прове-
дено в работах [28, 88]. Это исследование показайо, что полимер не
оказывает влияния на степень атомизации вещества в пламени и сниже-
ние интенсивности аналитических линий обусловлено главным образом
уменьшением эффективности распыления, т. е. матричным эффектом.
Среди многих факторов, от которых зависит эффективность распыления,
можно выделить скорость распыления и распределение капелек раствора
по их размерам. Крупные капли (размером более 10 мкм) задерживают-
158
ся в распылительной камере и не попадают в пламя. Из работы [88]
следует, что при увеличении концентрации полимера в растворе снижает-
ся качество распыления. Размеры капель зависят от физико-химических
свойств раствора, в том числе от вязкости и поверхностного натяжения.
При этом к растворам полимеров оказываются неприменимыми обыч-
ные закономерности, связывающие качество распыления со свойствами
раствора.
Хорошие результаты можно ожидать от применения непламенного
метода атомизации к анализу полимеров. В книге [24, с. 124] указано
на возможность быстрого определения следов элементов в полимерных
материалах без их предварительной подготовки, т. е. прямым анализом
в графитовой печи.
VII . ИЗУЧЕНИЕ ХИМИЧЕСКИХ РЕАКЦИЙ В ПОЛИМЕРАХ
МЕТОДАМИ МОЛЕКУЛЯРНОЙ СПЕКТРОСКОПИИ
Химические превращения в молекулах, сопровождающиеся разрывом
одних химических связей, образованием других, приводят к изменению
спектров полимеров, поэтому могут быть изученными методами мо-
лекулярной спектроскопии.
Рассмотрим возможности этих методов при исследовании хими-
ческих реакций, протекающих в полимерах.
VII. 1. ПОЛИМЕРИЗАЦИЯ И СОПОЛИМЕРИЗАЦИЯ
Реакция полимеризации, т. е. образования высокомолекулярных соеди-
нений за счет раскрытия двойных связей ненасыщенных молекул (моно-
меров) может быть исследована путем изучения интенсивности полос
поглощения связей С=С. Полимеризация, в отличие от поликонденсации,
не сопровождается выделением побочных продуктов и протекает без
изменения элементного состава. Если возможен отбор пробы из реакци-
онной смеси, то удается проследить за кинетикой процесса полимериза-
ции (или сополимеризации). В качестве иллюстрации приведем пример
спектроскопического исследования кинетики полимеризации N-винил-
пирролидона и его сополимеризации с диэтиламиноэтилметакрилатом
CH2=C(CH3)COOCH2CH2N(C2H5)2. В ультрафиолетовом спектре N-ви-
нилпирролидона наблюдается полоса при 235 нм, обусловленная погло-
щением связи С=С в его молекуле (рис. VH.l):
СН2 = СН
Поливинилпирролидон в этой области прозрачен. Прозрачны также
диэтиламиноэтилметакрилат (как и другие полимерные эфиры карбо-
новых кислот, поглощающих на границе вакуумной УФ-области (см.
раздел 1.1.1) и сополимер, полученный на основе указанных мономеров.
Процесс полимеризации N-винилпирролидона и его сополимеризации
с диэтиламиноэтилметакрилатом проводили в водной среде. Пробы
из реакционной смеси (по 0,1 г) разбавляли в 100 мл дистиллированной
воды и анализировали на содержание в них N-винилпирролидона. Кинети-
ческие зависимости расхода N-винилпирролидона при полимеризации
и сополимеризации для смесей мономеров разного состава приведены
на рис. УП.2.
Одновременно проводили анализ состава образующегося сополи-
мера в зависимости от состава исходной смеси мономеров по его спект-
160
Рис. VII.l. УФ-спектр водного раствора N-вннилпнрролндона:
Концентрация 0,5 г/л; толщина слоя 0,06 мм.
Рис. VII.2. Кинетические кривые полимеризации N-вннилпнрролндона в смеси
с диэтиламиноэтилметакрилатом.
Массовое содержание N-винилпирролидона(в %): 1 — 100; 2 — 70; 3 — 50; 4 — 30.
рам, используя полосы поглощения валентных колебаний связей С=0
звеньев N-винилпирролидона (1680 см"1) и дизтиламинозтилметакри-
лата (1725 см-1). Показатели поглощения определяли по спектрам
смесей гомополимеров. Анализу подвергали растворы сополимеров
в дихлорметане (концентрация 10 г/л). Таким образом спектры погло-
щения позволили получить данные об изменении состава исходной
смеси мономеров при сополимеризации, о составе образовавшегося со-
полимера в зависимости от условий полимеризации, определить констан-
ты сополимеризации *.
Применение спектральных методов особенно целесообразно для ис-
следования полимеризации или сополимеризации вновь синтезируемых
мономеров на стадии лабораторной разработки, поскольку количество
мономеров и полученных полимеров иногда недостаточно для выполне-
ния химических методов анализа. Если мономеры являются газообраз-
ными соединениями, то проведение анализа возвратной смеси мономе-
ров (после полимеризации до определенной конверсии) и сополимера
можно выполнить подобно тому, как это было описано в разделе IV. 1.3.
* Св едения о реакционной способности мономеров можно получить также
из спектров сополимеров, если на положение полос поглощения влияет последо-
вательность распределения мономерных звеньев (см. раздел IV.2.1).
161
VII.2. ПОЛИКОНДЕНСАЦИЯ
Поликонденсация — это процесс образования полимеров путем взаимо-
действия низкомолекулярных соединений, содержащих функциональные
группы. При этом происходит выделение побочных продуктов реакции:
воды, хлористого водорода, аммиака и т. п. Число функциональных
групп (ОН, СООН, NH2 и др.) должно быть не менее двух. Так, поли-
этилентерефталат получают в результате взаимодействия полизтилен-
гликоля и терефталевой кислоты:
пНО — СНг—СН2—ОН + пНООС—{ V—СООН
О—СН2 —СНг —О—СО
+ г?Н2О
При поликонденсации соединений, содержащих более двух функцио-
нальных групп (полифункциональные соединения), образуются часто
нерастворимые сетчатые или разветвленные полимеры. Однако в зави-
симости от различия в реакционной способности функциональных групп
мономеров, их взаимного расположения, условий проведения поли-
конденсации полифункциональные мономеры могут образовывать
как линейные, так и трехмерные полимеры [89 ].
Ряд задач, которые возникают при получении и исследовании поли-
конденсационных полимеров, могут быть решены средствами молеку-
лярного спектрального анализа*. Такими задачами являются: определе-
ние количества концевых групп, непрореагировавших исходных про-
дуктов, примесей металлов в полимерах, исследование степени хими-
ческих превращений в молекулах.
Например, на конце макромолекул полифениленоксидов, полу-
чаемых методом дегидрополиконденсации 2,6-дизамещенных фенолов,
находится гидроксильная группа:
В спектрах разбавленных растворов в тетрахлорметане полифенилен-
оксида (с массовой долей 0,43%) этой группе соответствует полоса
поглощения 3615 см-1, которая и была использована для определения
количества концевых гидроксильных групп [90, с. 142]. В качестве
эталонного образца для вычисления показателя поглощения был исполь-
зован 2,6-диметилфенол.
* Анализу поликонденсационных полимеров химическими, физико-химически-
ми и физическими методами посвящена книга |90]. В ней представлены также ме-
тодики спектрального анализа ряда поликонденсационных полимеров.
162
Методом спектрального анализа обнаружены гидроксильные группы
также в полисульфонах, полигликольтерефталатах, поликарбонатах
и других линейных поликонденсационных полимерах.
Положение .полос поглощения гидроксильных групп в зависимости
от химического строения полимера и используемого растворителя
несколько сличаются. Например, в спектрах поликарбонатов и поли-
сульфонов
эта полоса наблюдается при 3590 см-1 для растворов полимеров
в дихлорметане.
Кроме инфракрасной области спектра для анализа концевых групп
используют ультрафиолетовые спектры поглощения. Так, определение
концевых фенольных групп полифениленоксида проводили по полосе
318 нм [90, с. 139]. Метод основан на различии в поглощении фенольных
групп в спектрах нейтральных и основных растворов полимера в дихлор-
метане (после обработки раствором тетразтиламмония). Для большин-
ства полимеров указывают предел обнаружения концевых гидроксиль-
ных групп порядка 0,01% (по массе). Это дает возможность найти моле-
кулярную массу полифениленоксидов до 10s и полисульфонов до
2 • 10s. Для определения гидроксильных групп использовали различия
поглощения в УФ-спектрах нейтральных и основных растворах полиме-
ров в тетрагидрофуране при 302 нм [90, с. 145].
Спектры поглощения используют также для определения содержа-
ния побочных продуктов реакции поликонденсации, присутствие кото-
рых в полимере может влиять на его свойства. Например, промышлен-
ный метод получения полифениленоксидов основан на дегидрополи-
конденсации 2,6-диметилфенола в присутствии комплексов основных
солей меди (II) с аминами в среде органических растворителей. В резуль-
тате, кроме полимера, образуется низкомолекулярное соединение
3,3',5,5'-тетраметилдифенохинон (дифенохинон). Для его определения
используют интенсивную полосу при 420 нм. Высокий молярный пока-
затель поглощения порядка 80000 л/(моль*см) позволил определять
дифенохинон в интервале концентраций 10"4 -10-6 моль/л [90, с. 140].
163
Химические превращения в процессе поликонденсации могут быть
проконтролированы по изменению интенсивности полос поглощения
функциональных групп. Например, при образовании полиимида на пер-
вой стадии получают полиамидокислоту
—HN—ОС К-СО—NHH'
НООс/ ^соон
которая под воздействием термообработки при изготовлении пленки
превращается в полиамид
с выделением воды. Степень этого превращения, т. е. степень имидизации
(внутримолекулярной циклизации) может быть определена по уменьше-
нию интенсивности полосы 1645 см -1, которая относится к валентному
колебанию связи С=О в группе CONH полиамидокислоты [91 ].
Метод молекулярной спектроскопии полезен и широко применяет-
ся при исследовании кинетики отверждения рекционноспособных олиго-
меров. К таким соединениям относятся эпоксидные, фенолоформаль-
дегидные, резорциноальдегидные и другие смолы.
Большинство эпоксидных смол, структурная формула которых
имеет вид
СН2 —СНСН2О
ROCH2CHCH2O—
он
ROCHoHC— СН2
О
СНз
где и = •
СНз
получают конденсацией эпихлоргидрина и бисфенола А(4,4'-дигидроок-
сифенилпропана). Концевые эпоксидные и гидроксильные группы явля-
ются реакционноспособными и в присутствии отвердителей (полиамины,
фенолы, многоосновные кислоты и др.) смолы отверждаются, превра-
щаясь в твердые сшитые полимеры.
В инфракрасных спектрах эпоксидной смолы поглощение реакцион-
носпособных групп наблюдается в области 3450-3250 см(валентные
колебания связи ОН) и при 917 см-1 (деформационные'колебания
эпоксидного кольца). Эти полосы обычно используют для определения
количества эпоксидных и гидроксильных групп. В работе [92] анализ
164
эпоксидных групп в эпоксидированных полиоксипропиленполиолах
проводили по полосе 6050 см (1,65 мкм), соответствующей первому
обертону валентного колебания связи ОН при эпоксидном кольце.
Для определения гидроксильных групп служила полоса 4820 см-1.
Использование этих полос поглощения позволило авторам проводить
анализ одновременно на эпоксидные и гидроксильные группы по спек-
тру одной и той же пробы при толщине поглощающего слоя 5 мм. Гра-
дуировочные графики для определения эпоксидных групп строили на
основании спектров смесей метилглицидилового эфира с полиокси-
пропиленполиолом, имеющим молекулярную массу 3000, для опреде-
ления групп ОН — на основании полиоксипропиленполиолов с известным
содержанием этих групп, найденным химическим методом.
Процесс отверждения эпоксидных смол аминами протекает в две
стадии:
—NH2 + —СН—СН2 ——CHCHzNH —CHCHzNH---------h
О ОН ОН
ОН
4- —СН—СН2 —► —СН—СНг
>-
—СН—СН2
ОН
Для изучения кинетики отверждения удобно использовать полосы
поглощения реакционных групп как смолы, так и отвердителя. В работе
[93] первую стадию реакции контролировали по изменению интенсив-
ности полосы 4950 см-1 (составная частота валентного и деформацион-
ного колебаний групп -NH2), вторую - по полосе первого обертона
валентного колебания связей NH аминогрупп при 6500 см-1 (1,54 мкм).
Поскольку полоса 6500 см'1 представляет собой наложение поглоще-
ния связей NH первичных и вторичных аминов (группы -NH2, / NH),
то она была использована при изучении второй стадии процесса отверж-
дения, когда группы — NH2 полностью израсходовались в первой стадии
реакции, что легко контролируется по исчезновению полосы 4950 см “*.
Тогда все поглощение при 6500 см"1 можно отнести к колебаниям
вторичных аминогрупп.
VII.3. ПОЛ ИМ ЕР АНАЛОГИЧНЫЕ ПРЕВРАЩЕНИЯ
Многие полимеры получают из полимеров, содержащих в макромоле-
кулах реакционноспособные группы (СОН, COCI, CONH2, SO3H и т. п.)?
путем замещения этих групп на другие, т. е. проводя реакцию поли-
мераналогичных превращений. В результате такой реакции образуется
полимер практически с той же степенью полимеризации, что и исходный,
но обладающий комплексом новых свойств. Этот метод широко исполь-
165
зуют в промышленности. Замещая атомы водорода гидроксильной
группы на радикалы -СН3, -СН2СН3, -СН2СН2СН3, -СОСН3. -NO2
и другие, получают простые и сложные эфиры целлюлозы, нитроцел-
люлозу, а проводя реакцию омыления поливинилацетата, — поливинило-
вый спирт. Существует еще целый ряд полимеров, которые образуются
по реакции полимераналогичных превращений. Полнота превращения
влияет на свойства конечного полимера. Поэтому количественное опре-
деление остаточных реакционноспособных групп является предметом
анализа, для выполнения которого часто используют инфракрасные
спектры поглощения.
Иногда необходимо знать не только общее количество остаточных
групп, но и их распределение по полимерной цепи, поскольку оно часто
влияет на свойства полимера. Например, для получения поливинилового
спирта используют разные способы омыления поливинилацетата, в ре-
зультате чего поливиниловый спирт может содержать остаточные винил-
ацетатные звенья. Эти звенья в зависимости от способа омыления могут
быть по-разному распределены вдоль полимерной цепи:
~сн2—сн—сн2—сн—сн2—сн—сн2—сн—сн2—сн~
I I I I I
ОН ОН ОСОСНз он ОСОСНз
Показано, что одним из основных факторов, влияющих на физико-
химические и биологические свойства поливинилового спирта является
распределение остаточных звеньев винилацетата [94].
Метод ИК-спектроскопии позволяет изучать распределение моно-
мерных звеньев в полимерной цепи. Так, в спектрах поливинилацетата
и поливинилового спирта, содержащего блоки звеньев винилацетата,
валентные колебания групп С=О поглощают при 1736 см-1, а в спект-
рах образцов поливинилового спирта, содержащих изолированные
звенья винилацетата — при 1706 см-1. Измеряя интенсивности полос
поглощения при 1736 и 1706 см-1 в спектрах разных образцов поли-
винилового спирта, можно оценить в нем долю изолированных звеньев
и таким образом установить взаимосвязь
6,% свойств полимера с распределением мо-
7 номерных звеньев.
L * Спектральные методы удобно приме-
нятыакжедля изучения кинетических за-
\ \ кономерностей реакции полимеранапо-
I гичных превращений. При этом можно
50 \ 'Ча использовать полимерные образцы в ви-
X. у де пленок, поскольку они обычно сохра-
2 няют форму и размеры после обработки.
---А---А?
Рис. VII.3. Кинетика омыления сополимера
этилена с винилацетатом при разных тем-
—।—।—।—।—±—।—।—।—।—пературах:
Тч 1 - 20; 2 -40; 3- 60 °C.
166
Так,например,в сополимере этилена с винилацетатом при щелочном
гидролизе звенья винилацетата —СН2—СНОСОСН3— превращаются
в винилспиртовые — СН2 — СНОН— Измеряя изменение интенсивности
полосы 1025 см-1, относящейся к валентному колебанию связи С—О
групп —ОСОСН3, по отношению к интенсивности исходного образца,
можно найти количество оставшихся неомыленных винилацетатных
звеньев (в) и построить кинетические кривые для разных условий
гидролиза (рис. VII.3).
VII.4. ДЕСТРУКЦИЯ, СТРУКТУРИРОВАНИЕ
Под деструкцией понимают реакцию, сопровождающуюся разрывом
основной молекулярной цепи полимера. Деструкция активизируется
действием тепла, облучения, света, механических деформаций и может
сопровождаться окислением полимера, если протекает в присутствии
кислорода воздуха.
Изменение строения полимерных молекул под воздействием внеш-
них факторов и связанное с этим изменение первоначальных свойств
полимера называют старением. Методы молекулярной спектроскопии
находят применение при исследовании этих процессов, особенно процес-
сов окислительной термодеструкции полимеров, которые сопровожда-
ются образованием кислородсодержащих групп и появлением соответ-
ствующих полос поглощения в спектре.
Реакционная способность полимеров зависит от их химической
природы. Например, каучуки, содержащие в макромолекуле двойные
связи, могут разрушаться под действием кислорода воздуха уже при
комнатной температуре. Такие полимеры могут применяться лишь
с добавками антиоксидантов. К реакционноспособным полимерам
относятся также полимеры с ослабленной основной углеродной цепью,
когда в ней присутствуют единичные связи, или полимеры, содержащие
полярные заместители. Так, от молекул поливинилиденхлорида, поли-
винилхлорида под действием высоких температур, облучения, света
отщепляется НС1, при этом образуются двойные связи. При нагревании
поливинилхлорида в бескислородной среде можно наблюдать образо-
вание пространственных структур:
~сн2 —СНС1—СН2—СНС1- -сн2— СС1 — сн2 — СНС1~
+ * I
~СН2 —СНС1 —СН2—СНС1 — ~сн2 — СС1—сн2 — CHCI-
Если образование двойных связей, кислородсодержащих групп
фиксируется ИК-спектрами поглощения, то возникновение сшивок
между полимерными цепями методом молекулярной спектроскопии
наблюдать не удается, поскольку они не сопровождаются появлением
новых связей, отличных от основных связей в молекуле. В образовании
поперечных связей, или структурировании, при нагревании без кислоро-
да легко участвуют боковые винильные группы, например, бутадиено-
167
вых каучуков. Эти изменения можно обнаружить в спектре по умень-
шению интенсивности полос поглощения ненасыщенных групп. В присут-
ствии кислорода термическое структурирование развивается одновремен-
но с окислением и появлением в спектре полос поглощения кислород-
содержащих групп.
При нагревании полиакрилонитрила при температурах 150-200 °C
происходит разрыв связей С= N и циклизация:
/СН^СН^
сн сн
сн2 сн2 сн2/
сн сн сн
Возможно образование и других циклических структур, но они
также содержат связи C=N. В спектре полиакрилонитрила это проявляет-
ся в уменьшении интенсивности полосы 2240 <?м-1, относящейся к ва-
лентному колебанию связи C=N, и возрастанием интенсивности новой
полосы при 1550—1500 см-1, связанной с валентным колебанием
связи C=N, но мере увеличения продолжительности прогрева.
Для исследования реакций такого рода можно использовать гра-
фическую методику, аналогичную описанной в разделе IV.1.1 и применя-
емую для определения состава сополимера и показателей поглощения
аналитических полос. Предположим, что вещество А под действием
физических или химических факторов превращается в вещество В.
Если каждое из этих веществ имеет изолированную полосу в спектре,
то молярные доли веществ А и В в смеси (сА и св) выражаются через
оптические плотности соответствующих полос (Z)A и D#):
cA=DA/kAd; cB=DB/kBd (VII.1)
где А:а и А:в - показатели поглощения чистых веществ А и В.
Если реакция превращения веществ А и В мономолекулярная, то
сА + св = 1 и,следовательно
da + Ов
kAd kBd
(VII. 2)
Это уравнение показывает, что зависимость Лв от Z>A представ-
ляет собой прямую, отсекающую на координатных осях отрезки, со-
ответственно равные kAd и kQd (рис. VII.4, а). Обычно исходный обра-
зец содержит некоторое количество вещества В и уже первое измерение
показывает, что в спектре присутствует наряду с полосой поглощения
А и полоса В (рис. VII.4, б). На рис. VII.4, д оптические плотности полос
в начале измерений обозначены, как и£>в Откладывая их по соответ-
ствующим осям, получают первую точку графика D'. Повторяя эти
измерения через определенные промежутки времени и каждый раз
откладывая на осях значения оптических плотностей, получают последо-
вательно другие точки графика. При последнем измерении вещество А
168
может еще не превратиться в вещество В полностью. Поэтому в конце
измерений в спектре может все еще присутствовать полоса А. Через
Од и обозначены оптические плотности полос в конце измерений
и соответствующая точка графика D". Проводя через полученные точки
прямую и продолжая ее до пересечения с координатными осями, по-
лучают значения оптических плотностей для чистых веществ А и В,
т. е. значения fcAd и k#d, по которым можно определить показатели
поглощения этих веществ. Имея эти значения, можно с помощью уравне-
ния (VII.1) найти и св для каждого измерения и построить зависи-
мость концентраций от времени, т. е. кинетические кривые превращения
вещества А в вещество В.
Эта методика использована в работе [95] для изучения фоторазло-
жения светочувствительного соединения нафтохинон-1,2-диазида, кото-
рому, а также и эфирам на его основе принадлежит интенсивная полоса
поглощения 2130 см-1 (валентное колебание групп. CN2). При об-
лучении светом длин волн в интервале 300-420 нм нафтохинон-1,2-
диазид превращается в соединения (инденкарбоновые кислоты), имею-
щие поглощение при 1720 см -1 (валентное колебание С=О связи карбо-
ксильной группы).
Подобную методику можно применить и к изучению реакций в поли-
мерах, если в их спектрах наблюдаются полосы поглощения, характер-
ные для исходных групп и групп продуктов превращения, как, напри-
мер, в упомянутой выше реакции термостарения полиакрилонитрила.
При этом имеется то преимущество, что не требуется знание показа-
телей поглощения для чистых продуктов А и В. Они определяются
из графика (рис. VII.4), как описано выше.
Спектроскопическое изучение кинетических зависимостей при
различных температурах позволяет определять такие важные параметры,
как энергию активации процессов, протекающих в полимерах.
Рис. VII.4. Графический метод исследования мономолекулярной реакции:
а — зависимость Z>B от £>д в процессе превращения веществ А в В; б — спектры
поглощения исходного вещества (-----------) и обогащенного продуктом его
превращения (-----------) .
169
ПРИЛОЖЕНИЯ
I. ДЛИННОВОЛНОВАЯ ГРАНИЦА ПРОПУСКАНИЯ
НЕКОТОРЫХ МАТЕРИАЛОВ*
Материал Граница, мкм Толщина пластин- ки, мм Материал Граница, мкм Толщина пластин- ки, мм
Стекло 2,7 10 Бромид калия 25,0 10
Кварц (SiO2) 3,5 10 (КВг)
Сапфир (А12О3) 5,3 1 Бромид цезия 39,0 5
Фторид кальция 9,0 4 (CsBr)
(CaF2) Иодид-бромид 30,0 4
Хлорид натрия 16,0 10 таллия (KPS-5)
(NaCl) Иодид цезия (CsI) 50,0 5
* Приведенные данные соответствуют 60% пропускания пластинки. Граница
пропускания в зависимости от происхождения материала может несколько сдви-
гаться. Таблица составлена на основании данных книги [96, с. 159, 161-163].
Ц. КОРОТКОВОЛНОВАЯ ГРАНИЦА ПРОПУСКАНИЯ
РАСТВОРИТЕЛЕЙ В УЛЬТРАФИОЛЕТОВОЙ ОБЛАСТИ СПЕКТРА *
Растворители X, нм 20 nD Растворители X, нм 20 nD
Ацетон 326 1,3591 Пиридин 305 1,5102
Бензол 276 1,5011 1-Пропанол 210 1,3854
Бутанол 210 1,3993 2-Пропанол 210 1,3776
Вода 200 1,3330 Сероуглерод 376 1,6277
Гексан 210 1,3750 Тетрагидрофуран 235 1,4076
Гептан 210 1,3876 Тетрахлорметан 265 1,4603
цис-Декалин 215 1,4804 Тетрахлорэтилен 290 1,5055
граис-Декалин 215 1,4697 Трихлорметан 245 1,4456
М,М-Диметилформамид 270 1,4294 Толуол (d = 1 мм) 280 1,4969
Диоксан 213 1,4223 Циклогексац 210 1,4263
Дихлорметан 235 1.4237 Циклогексанол 220 1,461
1,2-Дихлорэтан 235 1,4443 (37 °C)
Изооктан 210 1,3916 Уксусная кислота 248 1,3715
Метанол 215 1,3286 ледяная
Метил формиат 260 1,3440 Этанол 210 1,3613
Me 1 илцикл огексан 210 1,4231 Этилацетат 251 1,3728
Октан 210 1,3977 Этиловый эфир 210 1,3528
Пентан 210 1,3577 Этил формиат 260 1,3597
* За коротковолновую границу пропускания условно принята та длина волны,
где пропускание слоя вещества толщиной 1 см составляет 1%. Данные таблицы
взяты из книги [97, с. 85[.
170
о
X
X
V
X
о.
о
и
о
е
3
х
X
X
S
5
о
ц
х С
и
о
О
сч
S
х
о
С
I
к"
U
I
X
X
X
<0
§
S
X
R
О
С
х
а-
о
5
§
X
X
§
ч
о
С
§
а-
о
•§
§
X
X
S
R
О
С
* Использованы данные книг [90, 98, 99]. ** В скобках указана температура измерения в
171
ПРОДОЛЖЕНИЕ
Полимер Строение звена Температура плавления, °C Показатель преломления "D* Растворители
Поливинилиден- фторид -СН2-CF2- 170-180 1,42 (20) Трихлорметан, тетрахлорметан, ацетон, диметилсульфоксид, диметилацетамид, диметил формамид
Политрифтор- хлорэтилен —cf2—cf— Cl 210-215 1,39-1,43 (20) Нерастворим
Полив инил ацетат —сн2—сн— ОСОСНз > 170 (разлагается) 1,47-1,49 Этанол, ацетон, дихлорэтан, трихлор- метан, тетрахлорметан, бензол, диоксан, циклогексанон, пиридин, уксусная кис- лота
Поливиниловый спирт —СН2—СИ- ОН >230 (разлагается) 1,49-1,53 При нагревании: вода, диметилформ- амид, диметил сульфоксид
Поливинил фор- маль* * —сн—сн,—СН—сн2— 1 1 о—снг—о > 150 (разлагается) 1,50 Дихлорэтан, уксусная кислота, пиридин, диоксан, смесь спирт-бензол (30:70)
Полив инилбути- раль* * —сн—СН2—СН—СНг— 1 1 о—сн—о 1 СН2СН2СН3 >160 (разлагается) 1,47-1,49 Этанол, ацетон, дихлорэтан, трихлорг метан, тетрагидрофуран, смесь спирт- бензол (30: 70)
Полиакриловая кислота — сн2—сн— 1 соон - 1,53 (25) Вода, метанол, диметилформамид, ди- оксан
* В скобках указана температура измерения в пы винилового спирта. °C. ** Как правило, в полимерах содержатся незамещенные гидроксильные труп-
ПРОДОЛЖЕНИЕ
Полимер Строение звена Температура плавления, °C Показатель преломления nD* Растворители
Полиметакрил о- вая кислота СНз 1 сн2—с— соон - Вода, этанол, диметилформамид, ди- оксан
Поли метил мет- акрилат СНз —СНг—С— СООСНз >200 (деполимеризу- ется) 1,49 (20) Ацетон, этилацетат, дихлорэтан, трихлор- метан, бензол, диоксан, тетрагидрофу- ран, пиридин, уксусная кислота
Полиакрило- нитрил Полиакриламид —сн2—сн— 1 C=N —сн2—сн— conh2 >200 (разлагается) > 100 (сшивается) 1,52 Диметилформамид, диметилсульфоксид Вода, диметилформамид, уксусная кис- лота
Поливинилпирро- лидон —сн2—сн— 1 N Н,С'''^'СО 1 1 н2с сн2 > 160 (разлагается) 1,53 Вода, ацетон, дихлорэтан, этанол, уксус- ная кислота
— СН2—СН=СН—СН2— 80-140
1,52 Сероуглерод, трихлорметан, бензол
Полибутадиен
* В скобках указана температура измерения в °C
ПРОДОЛЖЕНИЕ
Полимер Строение звена Температура плавления , °C Показатель преломления nD* Растворители
Полиизопрен — сн2—с=сн—сн2— СНз - 1,59 120) Сероуглерод, тетрахлорметан, трихлор- метан, циклогексан, бензол, толуол
Полихлоропрен —СН2—С = СН—СН2— CI - 1,55 (25) Дихлорэтан, тетрахлорметан, трихлор- метан
Полиметилен- оксид —сн2—о— 175-180 - Фенол (при t > 100 °C)
Полифенилен- оксид /СНз —^2/~°— 'СНз 260 — Диоксан, тетрагидрофуран, бензол, три- хлорметан, диметилформамид, диметил- сульфоксид
Целлюлоза СН2ОН \ \^\— °\J(OH .У ох он (разлагается при плавлении) 1,54 Водный раствор медноаммиачного комп- лекса
Триацетат цел- люлозы *В скобках СНгОАс । А—°\Г\ °\ ОАс Ас-—СОСНэ указана температура измерения в >210 (разлагается) °C. 1,54 Трихлорметан, дихлорэтан, уксусная кислота
ПРОДОЛЖЕНИЕ
Полимер Строение звена Температура плавления, °C Показатель преломления, "D* Растворители
Полиэтилентере- фталат —(СН2)2 — О—С—V /—С—о— II II о о 255-265 1,66 (25) При нагревании: фенол, анилин, пиридин
Полидифенилол- пропанкарбонат СНз о О 1 О II —о—(/ ч—с—(' ')—о—с— \=/ 1 \=/ СНз 220-270 - Трихлорметан, диоксан, диметилформ- амид
Поли-е-капро- амид (полиамид-6) —NH—(СН2)5—С — О 225 - Концентрированные серная, уксусная, му- равьиная кислоты, фенол
Полигексаметилен- адипинамид (поли- амид-6,6) —NH—(СН2)6—NH—С— О —(СН2)4— С— о 264 - То же
ПолиЧ-энанто- —NH—(СН2)6—С— 225
амид (полиамид-7) II
1,53-1,55
* В скобках указана температура измерения в °C.
ПРОДОЛЖЕНИЕ
Полимер Строение звена Температура 'плавления , °C Показатель преломления nD* Растворители
Поли-ш-ундекан- амид (полиамид-11) —NH—(СН2)10 —С О 185 1,53-1,55 Концентрированные серная, уксусная, му- равьиная кислоты, фенол
Полипиромеллит- имиды li ° II ^^11 о О - - В концентрированных серной, азотной кислотах растворяются при нагревании с деструкцией
Фенолоформаль- дегидная смола (новолак) он он - 1,5-1,7 Спирты, ацетон
Мочевиноформаль- дегидная смола —СН2—NH — С—NH — О - 1,54-1,56 Вода, ацетон
Меламиноформаль- —СН2—N—СХ^'ХС—NH— дегидная смола „ 1 J, ' НО — Н2С Nx С 1 nhoh2oh * В скобках указана температура измерения в °C. — То же
IV. ИНФРАКРАСНЫЕ СПЕКТРЫ ПОГЛОЩЕНИЯ ПОЛИМЕРОВ И ВСПОМОГАТЕЛЬНЫХ ВЕЩЕСТВ
Запись спектров осуществлялась на спектрофотометре UR-10 при следующих условиях: щелевая программа - 4; скорость сканиро-
вания v = 150 см'1 -мин'1; постоянная времени т = 4 с; масштаб записи - 12 мм на 100 см'1.
Полиэтилен высокого давления.
Прессованная пленка, d = 0,015 мм (7) и d = 0,3 мм (2).
Полиэтилен низкого давления.
Прессованная пленка, d = 0,015 мм.
177
178
Прессованная пленка, d = 0,01 мм.
Прессованная пленка, d = 0,107 мм.
т, %
100 г
0 La—1—।—।—1—1—।—।—।—l_i—।_।_।_।_।_।_।__।_।_।_।_।_।_।_।_।_।—।—।—।—।—l_J—।—।—।—।—।—J
4000 3600 3200 2800 2400 2000 1600 1200 800 700 600 500 400
Полистирол.
Пленка, отлитая из раствора в бензоле, d = 0,02 мм.
Пленка, отлитая из. раствора в бензоле, d = 0,01 мм.
Сополимер стирола с метилметакрилатом(массовое соотношение компонентов 40 :60).
Пленка, отлитая из раствора в трихлорметане, d = 0,025 мм.
100
°4000
80 -
60 -
40
20 -
3600
3200
2800
2400
2000
1600
1200
800 700 600 500 400
У,сиг'
Сополимер стирола с акрилонитрилом.
Пленка, отлитая из раствора в бензоле, d = 0,027 мм.
Сополимер стирола с метилметакрилатом и акрилонитрилом.
V, СМ'1
Пленка, отлитая из раствора в бензоле, d — 0,025 мм.
V, СМ"’
Привитой сополимер стирола с метилметакрилатом на бутадиеновый каучук.
Пленка, отлитая из раствора в дихлорэтане, d = 0,017 мм.
100r
4000 3600 3200 2800 2400 2000 1600 1200 800 700 600 500 400
V, СМ'1
Привитой сополимер стирола с акрилонитрилом на бутадиеновый каучук.
Прессованная пленка, d = 0,045 мм.
100,---
80
60 -
40 -
20
0 I-1—
4000
-L- 1 1 i ” 1 1 | _1 1 1 1 1 t 1 1 । 1 1 i 1 1 1 । 1 1 1 i t 1 а 1
3600 3200 2800 2400 2000 1600 1200 800 700 600 500 400
V, см-'
Поливинил толуол.
Пленка, отлитая из раствора в бензоле, d = 0,015 мм.
г, %
100 г
4000 3600 3200 2800 2400 2000 1600 1200 800 700 600 500 400
V, СМ-'
Поливинилхлорид.
Прессованная пленка, d = 0,026 мм.
т,%
100 г
V, см'’
Поливинилхлорид с массовым содержанием пластификатора дидецилфталата 16%.
Прессованная пленка , d = 0,03 мм.
Т °/
/о
100 Г
см~1
Политетрафторэтилен.
Прессованная пленка, d = 0,009 мм.
4000 3600 3200 2800 2400 2000 1600 1200 800 700 600 500 400
Поливинилиденфторид (а-форма).
Пленка, отлитая из раствора в ацетоне, d = 0,007 мм.
100
80
60 -
40
20 -
0 ь—1
4000
3600
3200
2800
2400
2000
1600
1200
800 700 600 500 400
Поливинилиденфторид (/3-форма).
Пленка, отлитая из раствора в ацетоне, d = 0,009 мм.
V, СМ~1
00
о\
т,%>
7001-------------,-----------------------------------------------------
j Uf W
п Ь—।—1—|—।—।—।—*—।—।—।—1—|—।—।—।—।—।_1—1_।_।—।—।—।—।—।—l -XVj_।_।_।—।—।—।—।—।—।—
4000 3600 3200 2800 2400 2000 1600 1200 800700 600 500 400
Политрифторхлорэтилен.
Прессованная пленка, d = 0,012 мм.
Прессованная пленка, d = 0,013 мм.
Прессованная пленка, d — 0,02 мм.
100
80
60
40
20
О Ь--L
4000
3600
3200
2800
2400
2000
1600
। । \ । «X—।_1__1_1_1__ 1 .
1200
800 700 600 500 400
Сополимер трифторэтилена с гексафторпропиленом.
Прессованная пленка, 4 = 0,022 мм.
V, СМ~1
T,%
100
80
60
40
20
О
4000
3600
3200
2800
240Q
2000
1600
1200
800 700 600 500 400
V, CM'1
Сополимер винилидеифторида с гексафторпропиленом.
Прессованная пленка, d = 0,011 мм.
100
80
60
40
20
О и—1
4000
3600
3200
2800
2400
2000'
1600
1200
800 700 600 500 401
V. СМ'1
Поливинилацетат.
Пленка, отлитая из раствора в трихлорметане, d = 0,005 мм.
Прессованная пленка, d = 0,015 мм.
Т *¥
100г
О L_।_ । _i__i—।—।—।—।—।—।—।—।—।—।—।—।—1—।—1—1—।—।—।—।—1—1__।_i । । 1 । , . ।
4000 3600 3200 2800 2400 2000 1600 1200 800 700 600 500 400
Сополимер винилацетата с этиленом (массовое соотношение компонентов 75 :25).
Пленка, отлитая на окне КВг из раствора в трихлорметане.
V, СМ-’
т
/, /о
100 г
V, СМ'1
Сополимер винилацетата с дибутилмалеинатом (массовое соотношение компонентов 75 : 25).
Пленка, отлитая из раствора в тетрахлорметане, d = 0.008 мм.
Пленка, отлитая из раствора в воде, d — 0,009 мм.
Сополимер винилового спирта с этиленом.
Пленка, отлитая из раствора в диметилформамиде, d = 0,005 мм.
192
Г, %
100 г
V, СМ'1
Поливииилбутираль (массовое содержание бутнральных групп 45%) •
Пленка, отлитая из раствора этанол - бензол (1:1), d = 0,01 мм.
Этилцеллюлоза.
Пленка, отлитая на окне КВг из раствора в трихлорметане.
Полиэтил ентерефталат.
Двуосноориентированная пленка, d = 0,01 мм.
100
SO
00
80
60
40
20
fh
О Lx.—।--1 1 1 l
4000 3600
3200
2800
2400
2000
1600
1200
800 700 600 500 400
V, СМ'1
Полидифен иленпропанкарбонат.
Пленка, отлитая из раствора в трихлорметане, d = 0,004 мм.
Т,°/.
100
80
60
40
20
О —4—। ।
4000 . 3600
3200
2800
2400
2000
1600
1200
800 700 600 500 400
V.CM-1
Эпоксидная смола ЭД-5-
Капиллярный слой.
100 г
17,СМ~1
Диглицидиловый эфир этиленгликоля.
Капиллярный слой.
so
SO
Полиэтиленполиамин.
Капиллярный слой.
ЦСМ~'
Полигексаметиленадипинамид (полиамид-6,6).
Пленка, отлитая из раствора в муравьиной кислоте, d = 0,007 мм.
Полигексаметилендодеканамид (полиамид-6,12) .
Пленка, отлитая из раствора в муравьиной кислоте, d = 0,005 мм.
Пленка, отлитая из раствора в муравьиной кислоте, d = 0,005 мм.
Пленка, d = 0,037 мм.
Ц СМ-'
Диоктилфталат.
Капиллярный слой.
Капиллярный слой.
Г,°/о
100 г
X ztf bJ ‘ * 1 1 1 1 1 1 1 1 1 1 1 ‘ * 1 1 1 I I 1 1 1 I 1 1 1 1 1 1 1 1 1 * I 1 1
^000 3600 3200 2800 2400 2000 1600 1200 800 700 600 500 400
Триэтилеигликоль-ди (2-этил гексоат).
Капиллярный слой.
V, СМ'1
100
80
60
40-
20
0 L—।—।—।—।—l
4000 3600
.3200
2800
2400
2000
1600
1200
J—I—I__I__I_I_‘ 1
800 700 600 500 400
v,cm-1
100
80
60 -
40 -
20
О Ь—L
4000
3600
3200
2800
Три (Д-хлорэтил) фосфат.
Капиллярный слой.
2400
2000
1600
1200
Вииил триметоксисилан.
Капиллярный слой.
800 700 600 500 400
V, СМ'1
d = 0,02 мм.
100
80
60
40
20
п U—।—।—।—1_1
4000 3600
3200
2800
2400
2000
1600
1200
Изооктан (эталонный).
d = 0,02 мм.
800 700 600 500 400
V СМ'1
d = 0,02 мм.
100
80
60
40
20
0 •-»—1
4000
3600
3200
W................................. ..
800 700 600 500 400
2800
2400
2000
1600
1200
Диоксаи (ч.).
</ = 0,02 мм.
Тетрагидрофуран (ч.).
V, см-1
</ = 0,02 ММ.
Т, %
4000 3600 3200 2800 2400 2000 1600 1200 800 700 600 500 400
Бензол (ч.).
</ = 0,02 мм.
V, СМ'1
100
208 209
80
60
40
20
0
4000
100
i__< i__।__i_i__i—i
3600 3200 2800
2400
2000
1600
1200
SOO 700 600 500 400
Толуол (ч.д.а.).
Капиллярный слой.
If, CM-1
Метилэтилкетон (ч.).
Капиллярный слой.
У,СМ'1
Циклогексанон (ч.).
d = 0,02 мм.
Диметилформамид (ч.).
Капиллярный слой (2); <2 = 0,02 мм (2).
цс/м'-’
210
Т,%
100 г
80
60
40
20
О Li—
4000
3600
3200
2800
2400
2000
1600
1200
800 700 600 500 400
V, см'1
Диметилацетамид (ч.).
Капиллярный слой.
Т,%
100
Диметилсулыррксид (х.ч.;.
d =0,02 мм.
1-Пропанол (х.ч.).
У, СМ'1
d = 0,02 мм.
212
d - 0,02 мм.
Полн-4- метилпентен:
d = 1,17 мм.
Сополимер стирола с метилметакрилатом н акрилонитрилом:
d = 2 мм.
Сополимер стирола с метилметакрилатом (массовое соотношение компонентов 40 :60) :
1 — d = 0,02 мм; 2 — d = 2 мм.
Привитой сополимер стирола с метилметакрилатом на бутадиеновый каучук:
d = 12 мм.
Т, %
Фторопласт-3 (/) и фторопласт-4 (2,5):
1 - d = 0,75 мм; 2 -d - 0,1 мм; 3 - d = 0,8 мм.
о
1 -d = 0,11 мм; 2 - d = 1,1 мм.
Т.%
Фторопласт-100:
d = 0,llMM; 2 - d = 1,15 мм.
Поливииилацетат:
1 - d = 0,06 мм; 2 —d = 1,08 мм.
Сополимер винилацетата с этиленом (массовое соотношение компонентов 8:92):
у % 1 —d =0,1 мм; 2 — d = 1 мм.
Поливиннлбутнраль с массовым содержанием пластификатора трнэтиленгликоль-ди (2-этилбутирата) 29% и стабилизатора
2-(2-гидроксн-5/-метилфенилбензотрназола) 0,1%:
d = 0,20 мм.
1 — d =0,1 мм; 2 — d — 3,5 мм.
</ = 0,36 мм.
224
К
Поли эт ил ентереф галат:
4 = 0,20 мм.
Полипиромеллитимид:
d ‘ 0,04 мм.
d = 0,16 мм.
г, %
Ди бу тил себацинат;
1 - d = 1 мм; 2 - d = 10 мм.
Диоктилфталат:
Трнэтиленгликоль-ди (2-этнлгексоат):
1 — d = 1 мм; 2 — d = 10 мм.
Т,%
Три (0-хлорэтил) фосфат:
1 — d = 1 мм; 2 — d = 10 мм.
Гептан (эталонный):
I — d = 1 мм; 2 — d = 10 мм.
Т,°/о
Изооктан (эталонный):
1 — d = 1 мм; 2 — d = 10 мм.
Циклогексан (ч.):
1 — d = 1 мм; 2 — d = 10 мм.
Диоксан (ч.) :
1 — d = 1 мм; 2 — d = 10 мм.
Бензол (ч.):
1 - d = 1 мм; 2 - d = 10 мм.
80
60
40
20
0
210 250 300 350 400 500 600 700 1000 1300 1800 1900 2200 2500 А,нм
Толуол (ч.дл.);
1 — d = 1 мм; 2 — d = 10 мм.
Ацетон (ч.);
1 — d = 1 мм; 2 — d = 10 мм.
Метилэтилкетон (ч.):
1 — d = 1 мм; 2 — d = 10 мм.
ю
Циклогексанон (я.):
1 — d = 1 мм; 2 — d = 10 мм.
Т,%
Диметилформамид <ч.):
1 — d = 1 мм; 2 — d = 10 мм.
ю
Этанол:
1 — d = 1 мм; 2 — d = 10 мм.
т,%
1-Пропанол (х.ч.):
1 — d = 1 мм; 2 — d = 10 мм.
Т, %
2-Пропанол (х.ч.):
1 _ d = 1 мм; 2 — d = 10 мм.
2-Метилпропанол (ч.д.а.) ;
1 — d = 1 мм; 2 — d = 10 мм.
bJ
u>
о
Изоамилацетат (ч.):
1 — d = 1 мм; 2 — d = 10 мм.
T,%
Этилацетат (я.):
1 — d = 1 мм; 2 — d = 10 мм.
1 — d = 1 mm; 2 — d = 10 мм.
Трихлорметан (ч.):
1 — d = 1 мм; 2 — d = 10 мм.
£
х
238
ЛИТЕРАТУРА
1. Штерн Э., Тиммонс К. Электронная абсорбционная спектроскопия в органичес-
кой химии. М.: Мир, 1957. 296 с.
2. Беллами Л. Инфракрасные спектры сложных молекул. М.: ИЛ, 1963. 590 с.
3. Денисов Г. С., Терушкин Б. С. - В кн.: Молекулярная спектроскопия. Л.:
Изд-во ЛГУ, 1981, вып. 5, с. 232.
4. Annual Book of ASTM Standards. Part 35. Philadelphia, 1980. 1114 p.
5. Гольденберг А. Л. - Ж. прикл. спектр., 1973, т. 19, № 3, с. 510.
6. Нельсон К. В., Австрийская Е. Е., Козлова Н. В., Березкина А. П. - Там же,
1971, т. 14, №2, с. 268.
7. Пирожная Л. Н., Максимов В. Л., Тарутина Л. И., Мадорская Л. Я. - Ж. прикл.
химии, 1982,т. 55,№ 12, с. 2758.
8. Дехант И., Данц Р., Киммер В., Шмольке Р. Инфракрасная спектроскопия
полимеров. М.: Химия, 1976. 472 с.
9. Мало Е. В., DuraoL. А. - J. Chem. Educ., 1973, v. 50, №3, р. 228.
10. Финкельштейн А. И., Малачевская Ф. Л., Фишер А- М., Рабовский Б. Г. —
Опт. и спектр., 1959, т. 8, № 6, с. 415; Аксельрод В. С. - Там же, I960, г. 8,
№ 3, с. 721; Макаревич Н. И., Борисевич Н. А. — Зав. лаб., 1963, № 8, с. 941.
11. Болдин А. А., Васильев Р. Ж. - Там же, 1961, № 7, с. 819.
12. Новейшие инструментальные методы исследования структуры полимеров/
Под ред. Дж. Кенига, М.: Мир, 1982. 264 с.
13. HarmsD.L. - Analyt. Chem., 1953, v. 25, №8, р. 1140.
14. Харрик Н Спектроскопия внутреннего отражения. М.: Мир, 1970. 336 с.
15. Золотарев В. М., Лыгин В. И., Тарасевич Б. Н. - Усп. химии, 1981, т. 50, № 1,
с. 24.
16. Чулановский В. М. Введение в молекулярный спектральный анализ. М.; Л.:
ГИТТЛ, 1951.462 с.
17. Никитин В. А., Воробьев В. Г. - Ж. прикл. спектр.. 1971, т. 24, № 6, с. 1050.
18. Зайдель А. И Погрешности измерений физических величин. Л.: Наука. 1985.
112 с.; Алексеев Р. И., Коровин Ю. И. Руководство по вычислению и обработ-
ке результатов количественного анализа. М.: Атомиздат, 1972, 72 с.
19. Krishnan К, - Appl. Spectroscopy, 1978, v. 32, № 6, р. 549.
20. Frank W. - J. Polymer Sci., Polymer Lett. Ed., 1977, v. 15, № 11, p. 679.
21. Локшин Б. В. - Ж. ВХО им. Менделеева, 1985, т. 30, № 2, с. 177.
22. Зайдель А. Н, Основы спектрального анализа. М.: Наука, 1965. 322 с.
23. Львов Б. В. Атомно-абсорбционный спектральный анализ. М.: Наука, 1966.
392 с.; Славин В. Атомно-абсорбцнонная спектроскопия. Л.: Химия, 1971.
296 с.; Спектроскопические методы определения следов элементов/Ред.
Дж. Вайнфорднер. М.: Мир, 1979. 494 с.
24. Хавезов И., Палев Д. Атомно-абсорбционный спектральный анализ. Л.: Химия,
1983. 144 с.
25. Прайс В. Аналитическая атомно-абсорбционная спектроскопия. М. Мир,
1976. 356 с.
26. Терек Т, Мика Й., Гегуш Э. Эмиссионный спектральный анализ. М.- Мир,
1982. Ч. 1,282 с.; Ч. II, 464 с.
27. Золотов Ю, А., Кузьмин И М. Концентрирование микроэлементов. М.- Химия,
1982. 278 с.
28. Lakatos L, Lakatos J. - Acta Chim. Acad. Sci. Hung., 1984, v. 116, № 4, p. 389.
29. Сирота A. T. Модификация структуры н свойств полиолефинов. Л.: Химия,
1984. 152 с.
30. Snyder R. G. - J. Chem. Phys., 1967, v. 47, №4, р. 1316.
31. RuedaD.R., Balta S. F.t Hidalgo A - Spectrochim. Acta, 1979, v. A35, № 7,
p. 847.
32. Riga A., Palma G„ Talamini G. - Makromol. Chem., 1972, Bd. 153, S.219.
239
33. Baker S., Maddams W. F„ Park G. 8. - Ibid., 1973, Bd. 165, S. 321.
34. Хайкин С. Я., Гольдман А. Я., Беляев В. M., Щербак В, В, — Высокомол
соед., 1979, т. 621, № И, с. 840.
35. Mddec Р. J., Marechai Е. - J. Polymer Sei.: Polymer Phys. Ed., 1980, v. 18, № 12,
p. 2417.
36. Севченко A. H, Зятьков И. П., Ольдекоп Ю. А. и др. Ж. прикл. спектр.,
1969, т. 11, №6, с. 1062.
37. Австрийская Е. Е„ Нельсон К. В. - Каучук и резина, 1968, № 9, с. 8.
38. Плиев Т. И., Чугай А. Д., Полетова В. Н., Чечик Л. Е. — Ж. прикл. спектр.,
1969,1. И, №6, с. 1068.
39. Bald G., Markert Н,-7. anal Chem., 1974, Bd. 268, №5, S. 360.
40. Гольденберг А. Л. - Пласт, массы, 1960, № 12, с. 59.
41. Тарутина Л. И. - Ж. прикл. спектр., 1968, т. 8, № 4, с. 654.
42. Ebdon J. R., Kandil 8. Н., Morgan К. J. - J. Polymer Sci.: Polymer Chem. Ed.,
1979, v. 17, №9, p. 2783.
43. Harwood H. J., Ang T. L., Bauer R. G. et al. - Am. Chem. Soc., Polymer Prep-
rints, 1966, v. 7, №2, p. 980.
44. Энтелис С. Г, Вайнштейн Э. Ф., Кумпаненко 3. Ф. - Высокомол. соед., 1969,
I- All, №12, с. 2615.
45. StokrJ., Schneider В. - Coll. Czech. Chem. Comm., 196 3, v. 28, №8, p. 1946.
46. Lesh W. - Kolloid-Z. u. Z. Polym., 1970, Bd. 240, № 1-2, S. 737.
47. Гольденберг А. Л., Зюзина Л. И. - Высокомол. соед., 1967, т. Б9, № 7, с. 542.
48. French A. R., Benham J. V., Pullukat Т. J. - ApplSpectr., 1974, v. 28, №5,р. 477.
49. Эффендиева Г. 3., Ибрагимов X. Д., Гусейнов Ф. О., Портя некий А. Е. -
Высокомол. соед., 1979, т. А21, № 10, с. 2392.
50. Методы исследования ударопрочных полистиролов/Под ред. В. М. Гальперина.
Л.: Химия, 1975. 76 с.
51. Shin Tsuge, Hajime Ito, Tsugi Takeuchi. - Бунсэки Кагаку, 1968, v. 17, № 11,
p. 1432.
52. GrossD. - Z. anal. Chem., 1969.Bd.248, S. 40.
53. Караснев С., Костов Г., Милина P„ Михайлов M. — Высокомол. соед., 1974,
т. А16,№9, с. 2162.
54. Tsugio Takeuchi, Shin Tsuge, HajimeIto. - Бунсэки кагаку, 1969, v. 18, № 3,
R. 383.
55. AgasheP. G., Sitaraman V., PatidarK. K. - Res. a. bid., 1981, v. 26, №4, p. 246.
56. Сополимеры этилена. Л.: Химия, 1983. 220 с.
57. Гольденберг А. Л., Литвинова М. А., Дунтов Ф. Н. - Высокомол. соед., 1967,
т. А9, №12, с. 2553.
58. Erussalimsky В., Tumarkun N., DuntoffF. etal. - Makromol Chem., 1967, Bd. 104,
S. 288.
59. Гольденберг А. Л., Пилиповский В. И. - Высокомол. соед., 1973, т. А15,
№11, с. 2610.
60. Van Schooten J., Duck E. W., Berkenbosch R. - Polymer, 1961, v. 2, № 3, p. 357.
61. Гоя C„ Valvassori A., Ciampelli F. - Europ. Polymer J.. 1968, v. 4, № 1, p. 107.
62. Schmolke R., Herma H., Grobe V. - Faserforsch. u.Textiltechn.,1965, v. 16, №12,
p. 589.
63. Данкович А., Киссин Ю. В. - Высокомол. соед., 1970, т. Al2, № 4, с. 802.
64. Kamiyama F., Matsuda H., Inagahi H. - J. Phys. Chem., 1967, v. 71, № 12, p. 4153.
65. Майо Ф. - Химия и технология полимеров, 1967, № 5, с. 3.
66. Гиндин Л. М., Абкин А. Д, Медведев С. С.-ДАН СССР, 1947, т. 56, № 2, с. 177.
67. Гольденберг А. Л. - ДАН СССР, 1968, т. 179, № 4, с. 900.
68. Пирожная Л. Н„ Тарутина Л. И. - Ж. прикл. спектр., 1981, т. 34, № 5, с. 862.
69. Бакаютов Н. Г, Киссин Ю. В., Вавилова И. И. и др. - Высокомол. соед., 1975,
т. А17,№ 10, С. 2163.
70. Хаслам Д, Виллис Г. А. Индентификация и анализ полимеров. М.: Химия,
1971.432 с.
240
71. Горбунов Б. И., Гуревич Я. А., Маслова И. П. Химия и технология стабили-
заторов полимерных материалов. М.: Химия, 1981. 368 с.
72. Химические добавки к полимерам: Справочник. 2-е изд. М.: Химия, 1984. 264 с.
73. Фихтенгольц В. С., Золотарева Р. В., Львов Ю. А. Атлас ультрафиолетовых
спектров поглощения веществ, применяющихся в производстве синтетических
каучуков. М.; Л.: Химия, 1965. 114 с.
74. Демидова В. М., Романцова О. Н. Стабилизация полипропилена: Обз. инф.
Серия „Полимеризационные пластмассы”, М.: НИИТЭХИМ, 1980, 24 с.
75. Ахмедзаде Д. А., Сахновская Е. Б-, Бабаева С. Г, Мустафаева С. К. - Пласт,
массы, 1984, № 10, с. 57.
76. Зюзина Л. И., Хайкин С. Я., Гольденберг А. Л. - Пласт, массы, 1970, № 7,
с. 66.
77. Москвин А. Ф., Докторова Л. И., Емельянова А. А, Боровкова 3. М. - Про-
мышленность синтетического каучука, М., 1979, № 7, с. 10.
78. Фихтенгольц В. С. Синтез и свойства жидких каучуков и эластомеров на их
основе. М.: ЦНИИТЭнефтехим, 1979, с. 66.
79. Василенко В. С., Недокунев В. В., Тумаркин И. Я. и др. - Пласт, массы, 1981,
№ 10, с. 55.
80. Кириллова Э. И., Малахова Г. П., Николаев А. Ф., Ленина Е. С. - Ж. прикл.
химии, 1979, т. 52, № 9, с. 2061.
81. Dutta Pradiz К., Gross К. R. - J. Appl. Polymer Sci., 1984, v. 29, № 6, p. 2247.
82. Василенок Ю. И., Гольденберг А. Л, Кононова Т. А. и др. - Пласт, массы,
1974, №5, с. 69.
83. Клещева М. С., Завьялов Ю. М., Коржова И. Т. Газохроматографический
анализ в производстве полимеризационных пластмасс. Л.: Химия, 1978.
222 с.
84. Карпенко Л. И., Фадеева Л. А., Гордеева А. Н., Ермакова Н. В. - Зав. лаб.,
1984, №12, с. 18.
85. Анализ полимеризационных пластмасс/Под ред. В. Д. Безуглого. М.; Л.:
Химия, 1965. 512 с.
86. Малахова Г. П, Попова Г. В., Яковлева М. Г - В кн.: Вопросы анализа полиме-
рнзационных пластмасс. Л.: ОНПО „Пластполимер”, 1986, с. 47.
87. Кириченко Э. А., Хурин В. Е. - Каучук и резина, 1969, № 2, с. 43.
88. Lakatos I. - Acta Chim. Acad. Sci. Hung., 1982, v. 109, № 3, p. 313.
89. Коршак В. В. - Высокомол. соед., 1982, т. А24, № 8, с. 1571.
90. Калинина Л. С., Моторина М. А., Никитина Н. И., Хачапуридзе Н. А. Анализ
конденсационных полимеров. М.: Химия, 1984. 296 с.
91. Власова К. Н., Суворова Л. Н., Строганов В. С., Попов В. А. - Пласт, массы,
1971, №8, с. 65.
92. Китухин Г. С., Жарков В. В. - Там же, 1981, № 6, с. 57.
93. Савельева А. Д, Жарков В. В. - Там же, 1969, № 6, с. 64.
94. Мнацаканов С. С., Сорокин А. Я. Еженкова Л. Л. и др. - Высокомол. соед.,
1972, т. А14, №4, с. 851.
95. Кольцов Ю. И. - Ж. аналит. химии, 1975, т. 30, № 9, с. 1822.
96. Козелкин В. В., Усольцев И. Ф. Основы инфракрасной техники. М.: Машино-
строение, 1974. 336 с.
97. Свердлова О. В. - Электронные спектры в органической химии. Л.: Химия,
1985.248 с.
98. Сперанская Т. А., Тарутина Л, И. Оптические свойства полимеров. Л.: Химия,
1976. 136 с.
99. Химический энциклопедический словарь. М.: Советская энциклопедия, 1983.
792 с.
100. Атлас спектральных линий для кварцевого спектрографа/Калинин С. К.,
Явнель А. А., Алексеева А. И. и др. М.: Госгеолтехиздат, 1959. 48 с.
101. Таблицы спектральных линий/Зайдель А. Н., Прокофьев В. К., Райский С. М.
и др. М.: Наука, 1977. 800 с.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Аддитивность оптической плотности 7
Адипиновой кислоты полиэфиры 128
Акриламид, сополимер с винилпирро-
лидоном и этилакрилатом, ИК-спектр 194
4-Алкокси-2-гидроксибензофенон см.
Бензон О А
Алкофен И
определение в поливинилбутирале
137-139
УФ-спектр 137
Антиоксидант АО-6
определениев бутадиен-метил старо ль-
ном каучуке 139,140
УФ-спектр 139
Антиоксидант НГ-2246
определениев бутадиен-метил стироль-
ном каучуке 139,140
УФ-спектр 139
Ауксохромы 8
Ацетобутарат целлюлозы 119
Ацетон, спектр в УФ-, видимой и ближ-
ней ИК-областях 231
Базисная линия 21-23
Батохромный эффект 8,9,11
использование для анализа 129,130
Беназол П
определение в поливинилбутарале
133-139
УФ-спектр 137
Бензилбутилфталат, ИК-спектр 117
Бензол
ИК-спектр 207
примесь
в винилацетате 150,151
в гексане, этаноле 153,154
спектр в УФ-, видимой и ближней
ИК-областях 230
Бензон ОА 131,141
УФ-спектр 140
Бера закон 6
Бисалкофен МЦП
определениев полиэтилене 140
УФ-спектр 141
Бугера-Ламберта закон 6
Бу г ера-Ламберта-Б ера закон 44
Бутилакрилат, сополимер с винилацета-
том, ИК-спектр 89,193
Винилацетат, определение примесей 150,
151
Винилацетат, сополимер
с буталакрилатом 89,193
Винилацетат, сополимер
с дибутилмалеинатом 190
с этиленом
ИК-спектр 87,96,189
определение эмульгатора ОП-Ю
155
распределение звеньев 104,107-
109
состав 98,99
спектр в УФ-, видимой и ближней
ИК-областях 222
степень гидролиза 167
Винилиденфторид, сополимер
с гексафторпропиленом, ИК-спектр
188
с трифторхлорэтиленом 11,12
Виниловый спирт, сополимер с этиленом,
ИК-спектр 191
N-Винилпирролидон
кинетика полимеризации 160,161
УФ-спектр 161
Винилтриметоксисилан, ИК-спектр 204
Внутренний стандарт 22,39,53
Вода
в триэтиленгликоль-ди (2-эталбутара-
те) 152,153
в этаноле 152,153
спектр в УФ-, видимой и ближней
ИК-областях 233
Гептан
ИК-спектр 205
спектр в УФ-, видимой и ближней
ИК-областях 227
2- (2гГидрокси-5-металфенил) бензотриа-
зол
Гипсохромный эффект 8
Диафен НН
определениев пентапласте 142
УФ-спектр 142 .
Дибуталсебацинат
ИК-спектр 122,202
определение в поливинилбутарале 123
спектр в УФ-, видимой и ближней
ИК-областях 225
Дибутил фталат
ИК-спектр 117
определение в поливинилацетате 120
УФ-спектр 120
Диглицидиловый эфир этиленгликоля,
ИК-спектр 199
Дидецилфталат 117
242
Диметилацетамид
ИК-спектр 210
спектр в УФ-, видимой и ближней
ИК-областях 233
Диметилсульфоксид, ИК-спектр 210
Диметилформамид
ИК-спектр 209
спектр в УФ-, видимой и ближней
ИК-областях 232
N,N'-Uh (нафтил-2) фенилендиамин-1,4,
см. Диафен НН
Диоксан
ИК-спектр 206
спектр в УФ-, видимой и ближней
ИК-областях 229
Диоксид кремния в полиэтилене, ИК-
спектр 146
Диоксид титана, ИК-спектр 146
Диоктилазелаинат, ИК-спектр 122
Диоктилсебацинат, ИК-спектр 122
Диоктил фталат
ИК-спектр 202
спектр в УФ-, видимой и ближней
ИК-областях 226
Дифенохинон, определение в полифени-
леноксиде 163
Дихлорметан
ИК-спектр 214
спектр в УФ-, видимой и ближней
ИК-областях 237
Диэтиладипинат, ИК-спектр 122
Ди-2-этилгексилфталат, ИК-спектр 117
Изоамилацетат
ИК-спектр 212
спектр в УФ-, видимой и ближней
ИК-областях 236
Изооктан
ИК-спектр 205
спектр в УФ-, видимой и ближней
ИК-областях 228
Иммерсионные среды 33,34
Интенсивность полосы поглощения 22,28
интегральная 28
Компенсационный клин 27,61
Композиция полистирол-поли-л-фениле-
ноксид, ИК-спектр 182
Концентрирование элементов 50,52
Ломакина-Шайбе уравнение 52
Масло вазелиновое,. ИК-спектр 146,215
фторированное (перфторкеросин),
ИК-спектр 215
Мел в полиэтилене, ИК-спектр 146
2,2'-Метиленбис (б-трет-бутил-4-метилфе-
нол) см. Антиоксидант НГ-2246
2,2’-Метиленбис [4-метил-6- (1 метилци-
клогексил-1) фенол] см. Бисалко-
фен МЦП.
2-Метилпропанол
ИК-спектр 212
спектр в УФ-, видимой и ближней
ИК-областях 235
Метил этилкетон
ИК-спектр 208
спектры в УФ-, видимой и ближней
ИК-областях 231
Метод
бромирования 74,75
добавок 54,154
компенсации 27,46,47,61,65
НПВО, МНПВО 38,42,43,99,148,149
озоления 50
определения элементов в полимерах
156,157
самокомпенсации 61,62
трех эталонов 53,156
Молекулярная масса, определение мето-
дами спектроскопии 66—68
Наполнители полимеров 146,147
Обертона 14,15
Оптическая плотность 6,44
влияние
скорости сканирования 44
ширины щели спектрофотометра
44
натуральная 6
Пиролиз полимеров 38,102,103
Пластификаторы, основные классы 115,
116
Пленка
измерение толщины
индикатором 39
по интерференционной картине 40,
41
приготовление
из расплава 29,30
из раствора 30-33
Показатель поглощения 6,44
натуральный 6
удельный 6
Показатель преломления полимеров 40,
171-176
Покровное стекло, спектр в УФ-, види-
мой и ИК-областях 225
Полиакрилонитрил," спектр в УФ-, види-
мой и ИК-областях 223
Полиамид-6, пиролиз 102
Полиамид-6,6
ИК-спектр 200
243
Полиамид-6,6
пиролиз 102
Полиамид-6,12, ИК-спектр 200
Полиамид-12, ИК-спектр 201
Полиамидо кислота 164
Полибутадиены
ИК-спектр 59,194
определение ненасыщенности 71,76
Поливинилацетат
ИК-спектр 87,96,188
определение
дибутилфталата 120,121
три (0-хлорэтилфосфата) 126,127
спектр в УФ-, видимой и ближней
ИК-областях 221
Поливинилбутираль
ИК-спектр 124,192
определение
алкофена И 137-139
беназолаП 137-139
дибутилсебацината 123
пластифицированный, спектр в УФ-,
видимой и ближней ИК-областях
222
Поливинилиденфторид, ИК-спектр 185
Полив инилкеталь, ИК-спектр 192
Поливиниловый спирт, ИК-спектр 190
Поливинилтолуол, ИК-спектр 183
Поливинилформаль, ИК-спектр 191
Поливинилхлорид
ИК-спектр 117,118,183,184
определение
разветвленности 65
фталатов 148,149
Полигард 129,130
Полигексаметиленадипинамид см. По-
лиамид-6,6
Полигексаметилендодеканамид см. По-
лиамид-6,12
Полидифеяиленпропанкарбонат, ИК-
спектр 198
Полидодеканамид, см. Полиамид-12
Полиметилеиоксид, ИК-спектр 195
По лим етилм егакрилат
ИК-спектр 122,193
спектр в УФ-, видимой и ближней
ИК-областях 12^223
Поли-4-метилпентен-1
ИК-спектр 178
спектр в УФ-, видимой и ближней
ИК-областях 217
Полиметилсилоксан, определение моле-
кулярной массы 68
Полипиромеллитимид
ИК-спектр 201
спектр в УФ, видимой и ближней
ИК-областях 224
Полипропилен
ИК-спектр 178
спектр в УФ-, видимой и ближней
ИК-областях 216
Полипропиленгликольадипинат, ИК-
спектр 128
Полировка окон 41
Полистирол
идентификация концевых групп 69
ИК-спектр 179
спектр в ближней ИК-области 18,
101
спектр в УФ, видимой и ближней
ИК-областях 217
стереорегулярность 108
УФ-спектр 11
Политетрафторэтилен
ИК-спектр 184
спектр в УФ-, видимой и ближней
ИК-областях 12
Политрифторхлорэтилен 186
Поли-п-фениленоксид
ИК-спектр 195
определение
дифенохинона 163
концевых групп 162
Полиэтилен
ИК-спектр 87,96,177
наполненный 146
определение
антиоксиданта НГ-2246 140,141
антистатиков 149
беизона ОА 131,140,141
бисалкофена МЦП 140
метильных групп 62-65
молекулярной массы 67
ненасыщенности 73-76
неозона А 140,141
тиоалкофена БМ 141
фенозана 23,142
элементов 157
Полиэтиленполиамин, ИК-спектр 199
Полиэтилентерефталат (лавсан)
ИК-спектр 197
спектр в УФ-, видимой и бпижней
ИК-областях 224
УФ-спектр 11
Полосы поглощения
интенсивность 28
полуширина 28,44
разделение 23-26,47,61
Поляризационные спектры 26,27,48
Порошки
галогенидов щелочных металлов 33-
36
полимеров, приготовление для ИК-
анализа 33-37
244
Предел обнаружения
абсолютный 51,55
относительный 51
Примеси металлов и других элементов
в полимерах 156-158
Произведение констант сополимериза-
ции, спектроскопическое определение
111,112
1-Пропанол
ИК-спектр 211
спектр в УФ-, видимой и ближней
ИК-областях 234
2-Пропанол
ИК-спектр 211
спектр в УФ-, видимой и ближней
ИК-областях 235
Пропускание 6,44,45
Растворители полимеров 56^8-63,171-
176
Растворы полимеров, использование для
спектрального анализа 37,38,56
Слой эффективный, определение толщи-
ны 39
Слюда, ИК-спектр 146
Составные частоты 15
Спектр
поляризационный 26,27,28
разностный 27,49,139
Спектральные диапазоны длин воли
5,13,15
Стабилизаторы
определение
в пленках 140-143
в растворе полимера 143
в экстрактах полимеров 137-139
основные классы 132—136
равномерность распределения в поли-
мере 144
эффективность действия 144
Степень разветвленности, определение
в поливинилхлориде 65
в полиэтилене 62-65
Стирол, анализ в пиролизатах сополи-
меров 102
Стирол, сополимер
с акрилонитрилом 101
ИК-спектр 180
с акрилонитрилом, привитой на бута-
диеновый каучук (АБС-пластик)
100,102
ИК-спектр 182
с метилметакрилатом
ИК-спектр 180
спектр в УФ-, видимой и ближней
ИК-областях 218
Стирол, сополимер
с метилметакрилатом и акрилони-
трилом
ИК-спектр 181
спектр в УФ-, видимой и ближней
ИК-областях 218
с метилметакрилатом, привитой на
бутадиеновый каучук
ИК-спектр 181
спектр в УФ-, видимой
и ближней ИК-областях 219
с пропиленом 113
Тальк, ИК-спектр 146
Тетрагидрофуран
ИК-спектр 207
спектр в УФ-, видимой и ближней
ИК-областях 229
Тетрафторэтилен, сополимер
с винилиденфторидом, ИК-спектр 187
с трифторэтиленом 94,95
с этиленом 25,37,88,89
Тетрахлорметан
ИК-спектр 214
спектр в УФ-, видимой и ближней
ИК-областях 238
Тиоалкофен БМ
определение в полиэтилене 141
УФ-спектр 141
4,4'-Тиобис (6-трет-бутил-З-метилфеиол)
см. Тиоалкофен БМ
Толуол
ИК-спектр 208
спектр в УФ-, видимой и ближней
ИК-областях 230
Триацетат целлюлозы, ИК-спектр 196
Трикрезилфосфат 125,126
Три (n-нонилфенил) фосфит см. Поли-
гард
Трипропионат целлюлозы 196
Трифенилфосфат 125,126
Три (фенилэтилбензил) фосфит см. Анти-
оксидант АО-6
Трифторхлорэтилен, сополимер
с винилиденфторидом 11,12
с этиленом 27,186
Трифторэтилен, сополимер с тетрафтор-
этиленом 94,95
Трихлорметан
ИК-спектр 213
спектр в УФ-, видимой и ближней
ИК-областях 237
Три (/3-хлорэтил) фосфат
ИК-спектр 125,204
определение в поливинилацетате 126,
127
->45
Три (р-хлорэтил) фосфат
спектр в УФ-, видимой и ближней
ИК-областях 227
Триэтиленглико л ь-ди (2-этилбутират)
ИК-спектр 124, 203
обнаружение влаги 152,153
Триэтиленгликоль-ди (2-этилгексоат)
ИК-спектр 124, 203
спектр в УФ-, видимой и ближней
ИК-областях 226
Фенозан-23, определение в полиэтиле-
не 142
Фенол, УФ-спектр 10
Фторопласт-3, спектр в УФ-, видимой'
и ближней ИК-областях 219
Фторопласт-4, спектр в УФ-, видимой
и ближней ИК-областях 219
ФгоропластЧМБ, спектр в УФ-, види-
мой и ближней ИК-областях 220
Фторопласт-10, спектр в УФ-, видимой
и ближней ИК-областях 220
Фторопласт-100, спектр в УФ-, види-
мой и ближней ИК-областях 221
Фурье-спектроскопия 46,47
Характеристическая концентрация 54,55
Характеристические колебания 14,15
Хромофоры 8,10,13
Циклогексан
ИК-спектр 206
спектр в УФ-, видимой и ближней
ИК-областях 228
Циклогексанон
ИК-спектр 209
спектр в УФ-, видимой и ближней
ИК-областях 232
Частоты колебаний
влияние изотопического замещения
14
зависимость от массы атомов 13,14
Частоты колебаний
обертонов 14,18
основные 16,17
составные 14,15,18
характеристические 14,15
Электронные спектры поглощения
влияние растворителей 9
полимеров 10;11
Элементы, определение методом атом-
ной спектроскопии 156,157
Эпоксидированные пластификаторы 128
Эпоксидная смола
ИК-спектр 198
определение реакционноспособных
групп 164,165
Этанол
обнаружение
влаги 152,153
бензола 153,154
спектр в УФ-, видимой и ближней
ИК- областях 234
Этил ацетат
ИК-спектр 213
спектр в УФ-, видимой и ближней
ИК-областях 2 36
Этилен, сополимер
с винилацетатом см. Винилацетат,
сополимер
с винилхлоридом 104
с пропиленом 102-106,108,111
с тетрафторэтиленом 25,37,88,89,113
с трифторпропиленом 104-106
Этилцеллюлоза, ИК-спектр 197
Эфир 3,5-ди-грет-бутил-4-гидроксифе-
нилпропионовой кислоты и пента-
эритрита см. Фенозан-23
Эффект
батохромный 8
гипсохромный 8
матричный 56
Эффективная толщина образца 39
СОДЕРЖАНИЕ
Предисловие .................................................... 3
I. ОСНОВНЫЕ ПРЕДСТАВЛЕНИЯ ОБ АТОМНОЙ И МОЛЕКУЛЯРНОЙ
СПЕКТРОСКОПИИ
1.1. Молекулярный спектральный анализ.......................... 5
1.1.1. Электронные спектры поглощения.....................
1.1.2. Инфракрасные спектры поглощения...................13
1.2, Атомная спектроскопия ....................................19
П. ЭКСПЕРИМЕНТАЛЬНАЯ ТЕХНИКА СПЕКТРАЛЬНОГО АНАЛИЗА
ПОЛИМЕРОВ
II.1. Молекулярный спектральный анализ......................... 21
II. 1.1. Выбор аналитической полосы поглощения и измерение ее ин-
тенсивности ........................................ 31
11.1.2. Приготовление образцов для анализа ............. 28
II.1.3. Определение толщины поглощающего слоя образца... 39
II. 1.4. Полировка окон кювет........................... 41
II.1.5. Исследование поверхностных слоев полимерных образцов ... 42
II. 1.6. Источники погрешностей в молекулярном спектральном анализе 44
II. 1.7. Новые технические возможности инфракрасной спектроскопии 46
II.2 . Атомный спектральный анализ............................. 49
Ш. ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНОЙ НЕОДНОРОДНОСТИ
ПОЛИМЕРОВ
III. 1. Разветвленность........................................ 59
III. 2. Концевые группы. Определение среднечисленной молекулярной массы 66
III. 3. Группы с ненасыщенными связями......................... 70
III.3. 1. Спектральные характеристики полос поглощения связей С=С 70
III.3. 2. Определение ненасыщенности полимеров ......... 73
Ш.4. Кислородсодержащие группы................................ 77
IV. АНАЛИЗ СОПОЛИМЕРОВ
IV.1. Определение состава сополимеров.......................... 90
IV. 1.1. Выбор аналитических полос...................... 90
IV.1.2. Эталонные образцы .............................. 93
IV. 1.3. Анализ по гомополимерам и балансу мономеров.... 94
IV. 1.4. Анализ по эталонным образцам .................. 96
IV. 1.5. Сочетание ИК-спектроскопии с пиролитической хроматогра-
фией ....................................................101
IV.2. Последовательность распределения мономерных звеньев в макро-
молекулах сополимеров 103
IV.3. Определение произведения констант сополимеризации .......109
247
V. АНАЛИЗ МОДИФИЦИРУЮЩИХ ДОБАВОК В ПОЛИМЕРНЫХ
МАТЕРИАЛАХ
V.I. Пластификаторы..............................................114
V . 1.1. Фталаты..........................................116
V .I.2. Эфиры алифатических карбоновых кислот и алифатических
спиртов....................................................121
V .1.3. Эфиры алифатических монокарбоновых кислот и гликолей . . . 124
V . 1.4. Фосфаты......................................... 125
V . 1.5. Другие пластификаторы ...........................128
V.2. Стабилизаторы ..........................................129
V .2.I. Анализ экстрактов полимеров.......................137
V .2.2. Исследование полимерных пленок....................140
V .2.3. Исследование растворов полимеров..................143
V . 2.4. Равномерность распределения стабилизатора в полимере.144
V .2.5. Оценка эффективности действия стабилизаторов......144
V .3. Неорганические наполнители..............................147
V. 4. Определение добавок на поверхности полимерного материала....147
VI. ОПРЕДЕЛЕНИЕ КАЧЕСТВА СЫРЬЯ И ЧИСТОТЫ ПОЛИМЕРОВ
VI .1. Анализ примесей в низкомолекулярных соединениях..........150
VI. 2. Анализ примесей в полимерах...............................155
VH. ИЗУЧЕНИЕ ХИМИЧЕСКИХ РЕАКЦИЙ В ПОЛИМЕРАХ
МЕТОДОМ МОЛЕКУЛЯРНОЙ СПЕКТРОСКОПИИ
VII. 1. Полимеризация и сополимеризация ........................160
VI I.2. Поликонденсация.........................................162
V II.3. Полимераналогичные превращения..........................165
VI I.4. Деструкция, структурирование............................167
ПРИЛОЖЕНИЯ
I. Длинноволновая граница пропускания некоторых материалов.....170
II. Коротковолновая граница пропускания растворителей в ультрафиоле-
товой области спектра......................................170
Ш. Полимеры, их структурные формулы, температуры плавления, пока-
затели преломления и растворимость ........................171
IV. Инфракрасные спектры поглощения полимеров и вспомо1ательных
веществ...................................................177
V. Спектры поглощения полимеров и вспомогательных веществ в ультра-
фиолетовой, видимой и ближней инфракрасной областях 216
Литература.................................................... 239
Предметный указатель ........................................... 242
Нет Сл?аи\л^
Ш-196,213-216
Copyleft ® Sanlcy incorporation
derevyaha + Q