




































































































































































































































































































































































































































































































































































































Автор: Анчита X.
Теги: вода для напитков и других промышленных целей минеральные и лечебные воды пищевой лед технология топлива нефть теплоэнергетика нефтяная промышленность переводная литература
ISBN: 978-5-91884-068-9
Год: 2015




X. Анчита
ПЕРЕРАБОТКА
ТЯЖЕЛОЙ НЕФТИ
Реакторы и моделирование процессов
- >10 Т • Л в с т t о
ПР » ФЕССИЯ
Modeling ol Processes
and Reactors for
Upgrading of Heavy
Petroleum
Jorge Ancheyta
1^)
CRC Press
Taylor &. Francis Group
Boca Raton London New York
CRC Press Is an imprint of the
Taylor & Francis Croup, an Informa business
Хорхе Анчита
ПЕРЕРАБОТКА
ТЯЖЕЛОЙ НЕФТИ
РЕАКТОРЫ И МОДЕЛИРОВАНИЕ ПРОЦЕССОВ
Перевод с английского языка
под редакцией О. Ф. Глаголевой, В. А. Винокурова
Санкт-Петербург
2015
ЦЕНТР
ОБРАЗОВАТЕЛЬНЫХ
ПРОГРАММ
ПРОФЕССИЯ
УДК 663.63
ББК 35.514
Хорхе Анчита
Ан64 Переработка тяжелой нефти. Реакторы и моделирование процессов :
пер. с англ. яз. под ред. О. Ф. Глаголевой, В. А. Винокурова. — СПб. :
ЦОП «Профессия», 2015. — 592 с., ил.
ISBN 978-5-91884-068-9
ISBN 978-1-4398-8046-3 (англ.)
Приведены классификация процессов и сопоставление различных техно-
логий переработки тяжелой нефти: от термических до термокаталитических
и гидрогенизационных процессов. Рассмотрены важные вопросы совмести-
мости и стабильности тяжелых видов нефтяного сырья, особое внимание уде-
лено асфальтенам и их роли в образовании кокса.
Выполнен анализ основных факторов, влияющих на сложную цепь реак-
ций, равновесие фаз, массоперенос, диффузию в частицы катализатора.
Отдельные главы посвящены моделированию процесса газификации и тер-
мических процессов висбрекинга и коксования.
Для каждого процесса приведены экспериментальные данные, даны прак-
тические рекомендации по реализации моделей, определению ключевых па-
раметров и их влиянию на выбор технологии и типа реактора. Результаты
моделирования процессов и реакторов подтверждены сравнительными экс-
периментальными и фактическими данными.
УДК 663.63
ББК 35.514
All Rights Reserved. Authorized translation from English language edition published by CRC Press,
an imprint of Taylor & Francis Group LLC.
Все права защищены. Никакая часть данной книги не может быть воспроизведена
в какой бы то ни было форме без письменного разрешения владельцев авторских прав.
Информация, содержащаяся в данной книге, получена из источников, рассматриваемых из-
дательством как надежные. Тем не менее, имея в виду возможные человеческие или технические
ошибки, издательство не может гарантировать абсолютную точность и полноту приводимых све-
дений и не несет ответственности за возможные ошибки, связанные с использованием книги.
ISBN 978-1-4398-8046-3 (англ.) © 2013 by Taylor & Francis Group, LLC
ISBN 978-5-91884-068-9 © ЦОП «Профессия», 2015
© Перевод, оформление: ЦОП «Профессия», 2015
Оглавление
Предисловие к русскому изданию........................................17
Предисловие...........................................................19
Благодарности.........................................................23
Птава 1. Тяжелая нефть.............................................. 27
1.1. Определение................;.................................27
1.2. Классификация нефтей.........................................28
1.3. Свойства.....................................................29
1.3.1. Физические и химические свойства.........................29
1.3.2. Асфальтены...............................................31
1.3.3. Оценка химического состава асфальтенов...................34
1.3.3.1. Экспериментальная часть..............................36
1.3.3.2. Результаты......................................... 38
1.3.4. Склонность к коксообразованию............................42
1.3.5. Вязкость.................................................43
1.3.5.1. Вязкость нефтей......................................43
1.3.5.2. Вязкость смеси нефтей................................45
1.3.5.3. Другие свойства смесей...............................52
1.3.6. Стабильность и совместимость нефтей......................53
1.3.6.1. Определения..........................................53
1.3.6.2. Методы оценки стабильности и совместимости...........54
1.4. Анализ тяжелой нефти.........................................57
1.4.1. Определение..............................................57
1.4.2. Цели анализа.............................................58
1.4.3. Виды анализа.............................................59
1.4.4. Свойства некоторых тяжелых нефтей........................59
1.5. Проблемы при переработке тяжелой нефти.......................66
Литература........................................................68
Птава 2. Технологии переработки тяжелой нефти.........................71
2.1. Общая классификация процессов................................71
2.2. Текущее положение в области переработки тяжелого сырья.......75
6
Оглавление
2.3. Технологии обогащения водородом............................76
2.3.1. Гидровисбрекинг........................................77
2.3.2. Гидропереработка в неподвижном слое....................77
2.3.3. Гидропереработка в движущемся слое.....................78
2.3.4. Гидропереработка в кипящем слое........................79
2.3.5. Гидропереработка в фазе суспензии......................80
2.4. Технологии обеднения углеродом.............................81
2.4.1. Экстрактивная деасфальтизация..........................82
2.4.2. Газификация............................................82
2.4.3. Коксование.............................................83
2.4.4. Висбрекинг.............................................83
2.4.5. Каталитический крекинг остаточного сырья в псевдоожиженном слое ....83
2.5. Перспективные технологии................................. 84
2.6. Комбинированные процессы облагораживания...................85
2.6.1. Комбинированные технологии обеднения углеродом.........85
2.6.2. Комбинированные технологии обогащения водородом........92
2.6.3. Комбинированные технологии обогащения водородом
и обеднения углеродом..........................................93
Литература......................................................100
Етава 3. Моделирование висбрекинга.................................105
3.1. Введение...................................................105
3.2. Описание процесса..........................................106
3.3. Способы висбрекинга........................................106
3.3.1. Висбрекинг в змеевике...................................108
3.3.2. Висбрекинг с выносной реакционной камерой...............108
3.3.3. Различия между процессами...............................109
3.4. Параметры процесса.........................................109
3.4.1. Свойства сырья...........................................НО
3.4.2. Температура.............................................111
3.4.3. Давление..................................................Ш
3.4.4. Время пребывания в реакционной зоне.....................111
3.4.5. Закачка пара............................................112
3.4.6. Главные параметры процесса..............................112
3.5. Химия процесса..............................................112
3.5.1. Разрыв связи С-С........................................114
3.5.2. Дегидрирование..........................................114
3.5.3. Изомеризация............................................114
3.5.4. Полимеризация и конденсация.............................114
3.5.5. Реакции с участием гетероатомов.........................114
3.6. Кинетика реакций..............................................115
3.7. Моделирование реакторов.....................................118
Оглавление
7
3.7.1. Эмпирические зависимости.............................118
3.7.2. Моделирование реакторов..............................119
3.7.2.1. Общие характеристики реакторов висбрекинга........119
3.7.2.2. Моделирование процессов в змеевике и в реакционной камере ....120
3.7.3. Моделирование процесса висбрекинга...................125
3.7.3.1. Характеристики реакторов и рабочие условия........125
3.7.3.2. Свойства сырья и продуктов........................126
3.7.3.3. Результаты моделирования..........................127
3.7.3.4. Заключительные замечания и рекомендации...........131
Обозначения....................................................133
Литература.....................................................135
Глава 4. Моделирование процесса газификации.......................137
4.1. Введение..................................................137
4.2. Типы процессов газификации................................138
4.2.1. Газификация в стационарном слое........................138
4.2.1.1. Противоточные газогенераторы стационарного слоя ;..138
4.2.1.2. Параллельноточные газогенераторы стационарного слоя.139
4.2.2. Газогенераторы с кипящим (псевдоожиженным) слоем.......139
4.2.3. Факельные газогенераторы...............................140
4.2.4. Другие способы газификации.............................140
4.3. Параметры процесса.........................................141
4.3.1. Температура............................................141
4.3.2. Давление...............................................141
4.3.3. Скорость начала псевдоожижения.........................142
4.3.4. Соотношение воздух/пар.................................142
4.3.5. Коэффициент избытка окислителя.........................142
4.3.6. Размер частиц..........................................142
4.4. Описание процесса..........................................142
4.5. Химия и термодинамика процесса.............................144
4.6. Моделирование реактора газификации.........................145
4.6.1. Уравнения модели........................................146
4.6.1.1. Баланс масс..........................................146
4.6.1.2. Термодинамическое равновесие.........................148
4.6.1.3. Баланс энергий.......................................154
4.6,1.4. Теплота сгорания синтез-газа и КПД газификации.......156
4.6.2. Решение уравнений модели................................157
4.7. Моделирование процесса газификации...........................158
4.7.1. Проверка модели.........................................158
4.7.2. Влияние условий реакций.................................160
4.7.2.1. Влияние давления......................................160
8
Оглавление
4.7.2.2. Влияние температуры.....................................162
4.7.2.3. Влияние кратности кислорода к сырью.....................162
4.7.2,4. Влияние кратности воды к сырью..........................164
4.7.3. Применение модели..........................................165
4.7.3.1. Применение модели к вакуумным остаткам с различными
свойствами................................................165
4.7.3.2. Оптимизация выхода водорода.............................167
Литература...........................................................168
Глава 5. Моделирование процесса коксования..............................170
5.1. Введение........................................................170
5.2. Процессы коксования.............................................171
5.2.1. Замедленное коксование.......................................172
5.2.2. Непрерывное коксование (флюидкокинг).........................173
5.2.3. Флексикокинг.................................................174
5.3. Описание процесса................................................175
5.4. Параметры процесса...............................................177
5.4.1. Температура на выходе из печи и на входе в камеры коксования.177
5.4.2. Давление в камере коксования.................................178
5.4.3. Коэффициент рециркуляции сырья...............................179
5.4.4. Виды сырья...................................................179
5.5. Теоретические основы процесса коксования.........................180
5.5.1. Химия процесса................................................180
5.5.2. Кинетика реакций..............................................181
5.5.3. Термическое разложение асфальтенов............................182
5.6. Кинетика реакций коксования........................................184
5.6.1. Разделение атмосферного остатка...............................184
5.6.2. Неизотермическая кинетика......................................185
5.6.3. Термическое разложение.........................................186
5.6.4. Кинетические параметры.........................................189
5.6.5. Примечания.....................................................193
5.7. Зависимости для прогнозирования выхода кокса.......................193
5.7.1. Зависимости....................................................193
5.7.1.1. Зависимости Гэри и Хэндверка................................194
5.7.1.2. Зависимости Мейплза..........................................195
5.7.1.3. Зависимости Шаброна и Спайта.................................197
5.7.1.4. Зависимости Кастильони.......................................198
5.7.1.5. Зависимости Смита и соавторов................................201
5.7.1.6. Зависимости Фолька и соавторов...............................202
5.7.2. Применение зависимостей..........................................203
5.7.2.1. Влияние свойств сырья........................................203
Оглавление
9
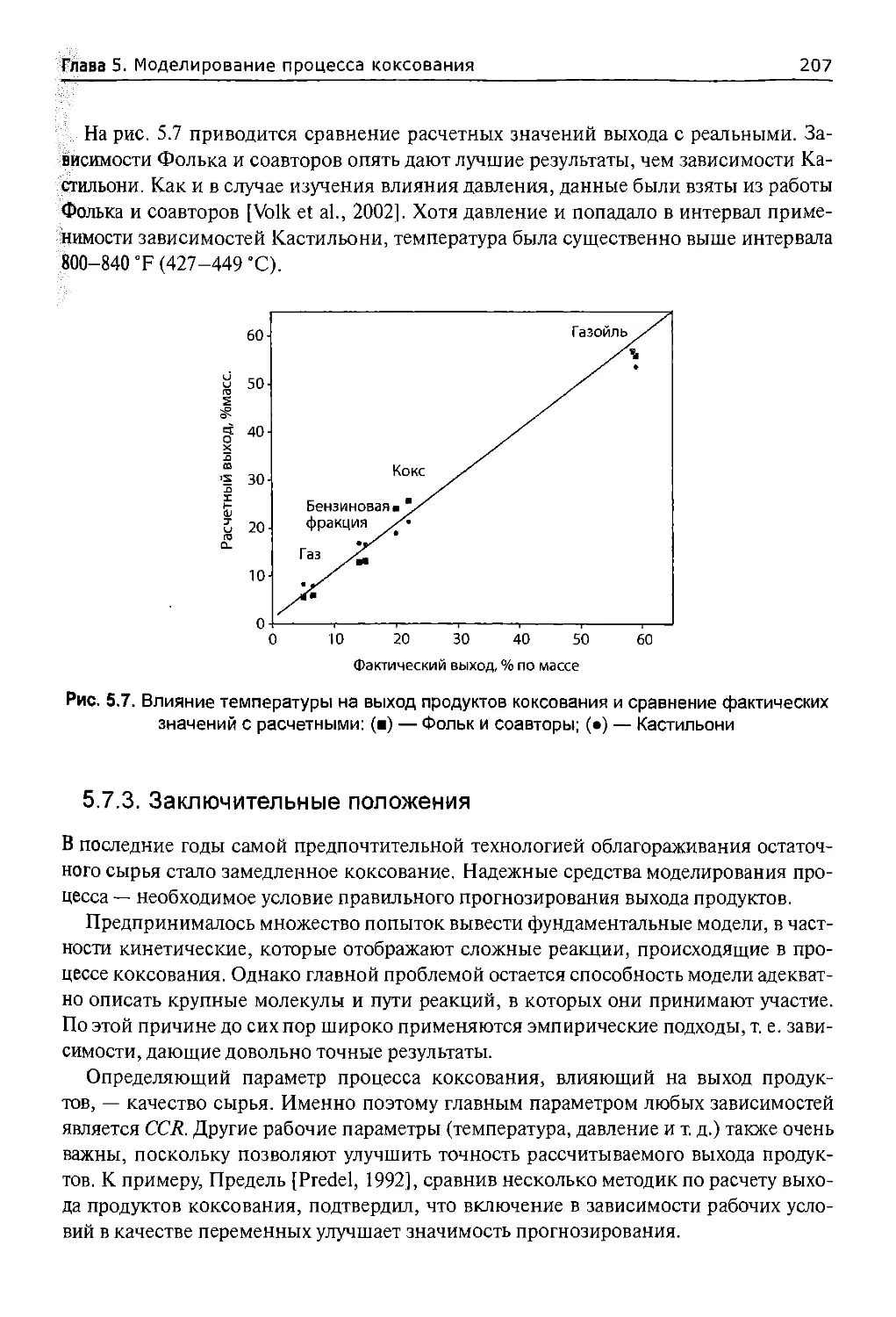
5.7.2.2. Влияние давления.....................................205
5.7.2.3. Влияние температуры..................................206
5.7.3. Заключительные положения................................207
Обозначения......................................................208
Литература...................................................... 209
Гпава 6. Некаталитическая (термическая) гидропереработка............212
6.1. Введение....................................................212
6.2. Экспериментальная часть.....................................214
6.2.1. Сырье...................................................214
6.2.2. Оборудование............................................215
6.2.3. Условия реакций.........................................217
6.2.4. Методы анализа..........................................217
6.3. Результаты..................................................218
6.3.1. Двухреакторная установка................................218
6.3.1.1. Некаталитическое гидрообессеривание..................218
6.3.1.2. Избирательность к некаталитическим процессам
гидрообессеривания и гидродеметаллизации......................221
6.3.1.3. Влияние температуры и соотношения FmT/ KSiC на плотность
продукта......................................................222
6.3.1.4. Влияние температуры и соотношения FmT! KSiC на кривую
разгонки продукта............................................223
6.3.1.5. Влияние на состав суммарного жидкого продукта........223
6.3.1.6. Распределение температур по оси реакторов............227
6.3.2. Однореакторная установка................................228
6.3.2.1. Кинетика некаталитической гидроочистки...............228
6.3.2.2. Кинетика превращения вакуумного остатка..............230
6.3.2.3. Кинетика некаталитического гидрокрекинга.............232
Обозначения......................................................237
Латинские......................................................237
Нижние индексы.................................................238
Литература.......................................................238
Гпава 7. Моделирование каталитической гидроочистки..................243
7.1. Введение....................................................243
7.1.1. Значение гидроочистки в нефтепереработке................244
7.1.2. Текущая ситуация в нефтеперерабатывающей отрасли........247
7.2. Описание процесса...........................................249
7.3. Типы реакторов..............................................251
7.3.1. Реакторы с неподвижным слоем............................251
10
Оглавление
7.3.1.1. Охлаждение в реакторах с неподвижным слоем катализатора.254
7.3.1.2. Внутреннее оборудование реакторов..................256
7.3.2. Реакторы с движущимся слоем катализатора..............258
7.3.3. Реакторы с кипящим слоем катализатора.................259
7.3.4. Реакторы с суспензионным слоем........................261
7.4. Теоретические основы......................................262
7.4.1. Реакции гидроочистки..................................262
7.4.1.1. Гидрообессеривание......................:..........263
7.4.1.2. Гидродеазотирование................................265
7.4.1.3. Гидродеоксигенация.................................265
7.4.1.4. Гидродеметаллизация................................266
7.4.1.5. Реакции гидрирования................................266
7.4.1.6. Гидрокрекинг........................................266
7.4.1.7. Гидродеасфальтизация................................267
7.4.2. Кинетика реакций........................................267
7.4.3. Термодинамика...........................................272
7.4.4. Катализаторы............................................275
7.5. Параметры процесса.........................................277
7.5.1. Температура реакций.....................................278
7.5.2. Парциальное давление водорода...........................280
7.5.3. Скорость подачи сырья...................................281
7.5.4. Кратность водорода к сырью и скорость циркуляции........281
7.5.5. Активация катализатора..................................283
7.6. Моделирование гидроочистки газойля, полученного из тяжелой нефти.285
7.6.1. Экспериментальная часть.................................285
7.6.1.1. Материалы и оборудование..............................285
7.6.1.2. Эксперименты..........................................286
7.6.1.3. Методы анализа........................................287
7.6.2. Построение модели реактора..............................288
7.6.2.1. Допущения, принимаемые в рамках модели................288
7.6.2.2. Баланс масс в нестационарном состоянии................289
7.6.2.3. Баланс теплоты в нестационарном состоянии.............290
7.6.2.4. Граничные условия.....................................291
7.6.2.5. Метод интегрирования..................................292
7.6.3. Модели кинетики реакций..................................292
7.6.3.1. Гидрообессеривание....................................292
7.6.3.2. Гидродеазотирование...................................292
7.6.3.3. Гидродеароматизация...................................293
7.6.3.4. Гидрирование олефинов.................................295
7.6.3.5. Мягкий гидрокрекинг...................................295
7.6.4. Расчет параметров........................................ 295
7.6.4.1. Кинетические параметры................................295
Оглавление
11
7.6.4.2. Коэффициент эффективности катализатора............298
7.6.4.3. Гидродинамические параметры.......................299
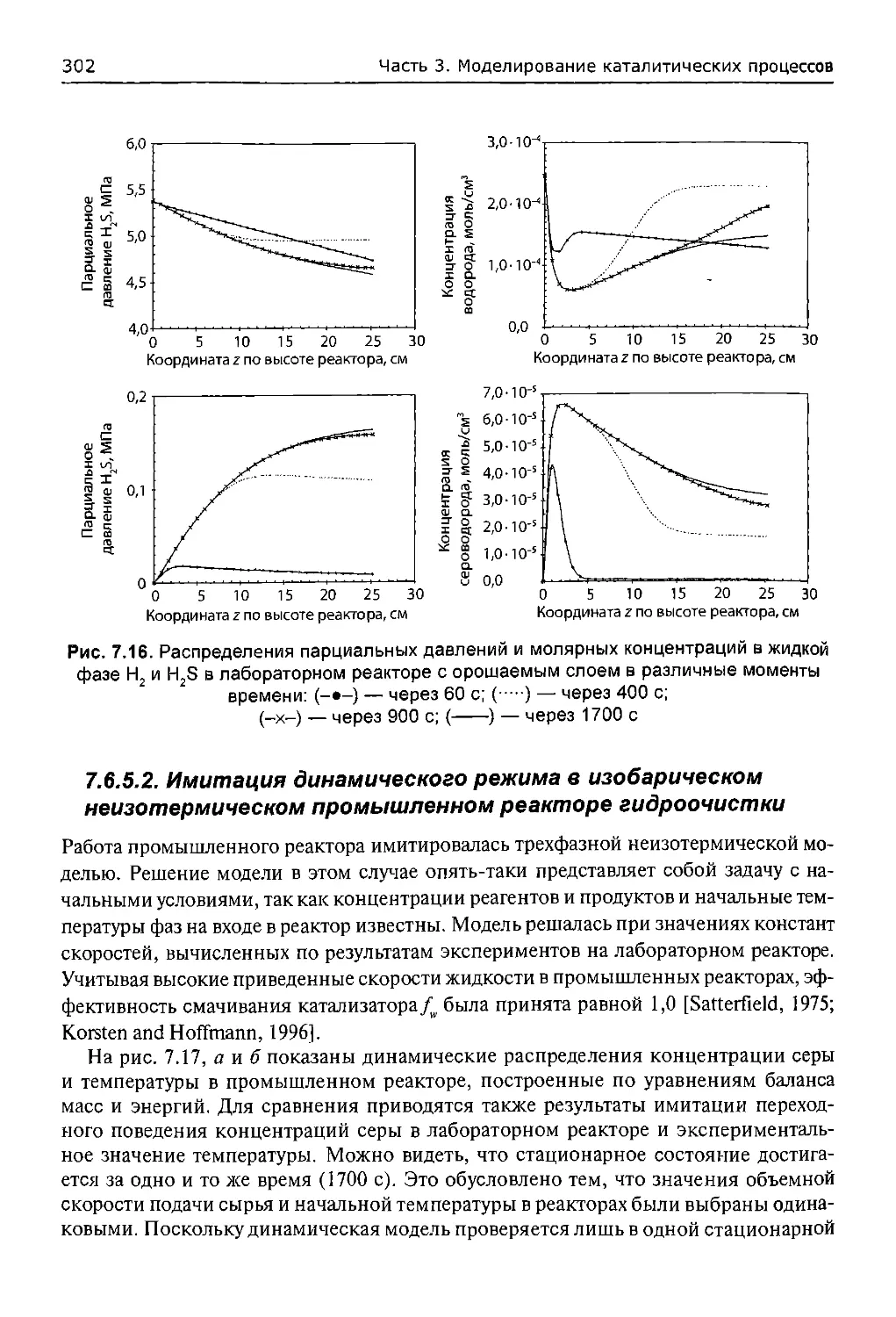
7.6.5. Анализ результатов...................................300
7.6.5.1. Имитация динамического режима в изотермическом
лабораторном реакторе гидроочистки.........................300
7.6.5.2. Имитация динамического режима в изобарическом
неизотермическом промышленном реакторе гидроочистки.........302
Обозначения...................................................309
Греческие....................................................309
Латинские....................................................309
Нижние индексы...............................................311
Верхние индексы..............................................312
Литература.....................................................312
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти...316
8.1. Введение..................................................316
8.2. Описание процесса гидрооблагораживания Мексиканского
нефтяного института............................................317
8.3. Экспериментальные исследования............................319
8.3.1. Получение кинетических данных.........................319
8.3.2. Исследование влияния тяжелого сырья на деактивацию
катализатора..................................................323
8.3.3. Испытание катализатора на долговременную стабильность..327
8.4. Моделирование..............................................328
8.4.1. Стационарные уравнения баланса масс и теплоты..........329
8.4.2. Динамические уравнения баланса масс и тепла............331
8.4.3. Кинетика реакций.......................................333
8.4.4. Масштабирование кинетических данных....................334
8.4.5. Деактивация катализатора...............................335
8.4.6. Метод решения..........................................337
8.4.6.1. Моделирование установившегося процесса..............337
8.4.6.2. Моделирование динамического процесса................337
8.5. Согласование данных..........................................338
8.5.1. Кинетические параметры..................................338
8.5.2. Параметры деактивации...................................340
8.6. Моделирование процесса на лабораторной установке.............341
8.6.1. Моделирование реактора при стационарной активности
катализатора...................................................341
8.6.2. Моделирование реактора при переменной активности
катализатора...................................................345
8.6.2.1. Влияние типа сырья и температуры реакций на деактивацию
катализатора...................................................345
12
Оглавление
8.6.2.2. Результативность процесса при испытании катализатора
на долговременную стабильность..............................347
8.7. Масштабирование кинетических данных, полученных
на лабораторной установке......................................349
8.8. Моделирование процесса в промышленном реакторе............352
8.8.1. Выбор компоновки и моделирование работы реактора
при постоянной активности катализатора.......................352
8.8.2. Моделирование и анализ работы реакторов в течение рабочего
цикла........................................................355
8.8.3. Переходное поведение реактора при пуске...............358
8.8.3.1. Охлаждение.........................................359
8.8.3.2. Температура сырья..................................362
8.8.3.2. Тактика пуска......................................364
Обозначения....................................................365
Латинские и русские..........................................365
Греческие....................................................367
Нижние индексы...............................................368
Литература......................................................368
Гпава 9. Моделирование лабораторного реактора для гидроочистки
мексиканской тяжелой нефти «Майя»..................................370
9.1. Введение...................................................370
9.2. Модель.....................................................371
9.2.1. Допущения при разработке модели........................371
9.2.2. Описание модели........................................372
9.2.2.1. Стехиометрические коэффициенты реакций ГДО..........373
9.2.2.2. Коэффициенты скоростей реакций......................374
9.2.2.3. Определение кинетических параметров.................376
9.2.2.4. Оценка транспортных и термодинамических свойств.....377
9.2.3. Решение модели.........................................383
9.3. Экспериментальная часть....................................384
9.3.1. Характеристики сырья....................................384
9.3.2. Экспериментальный реактор...............................385
9.3.3. Изотермическая работа реактора..........................385
9.3.4. Свойства катализатора...................................385
9.3.5. Загрузка катализатора...................................386
9.3.6. Активация катализатора..................................386
9.3.7. Сопротивление массопереносу.............................386
9.3.8. Влияние условий реакций.................................387
9.4. Результаты..................................................387
9.4.1. Стехиометрические коэффициенты..........................387
Оглавление
13
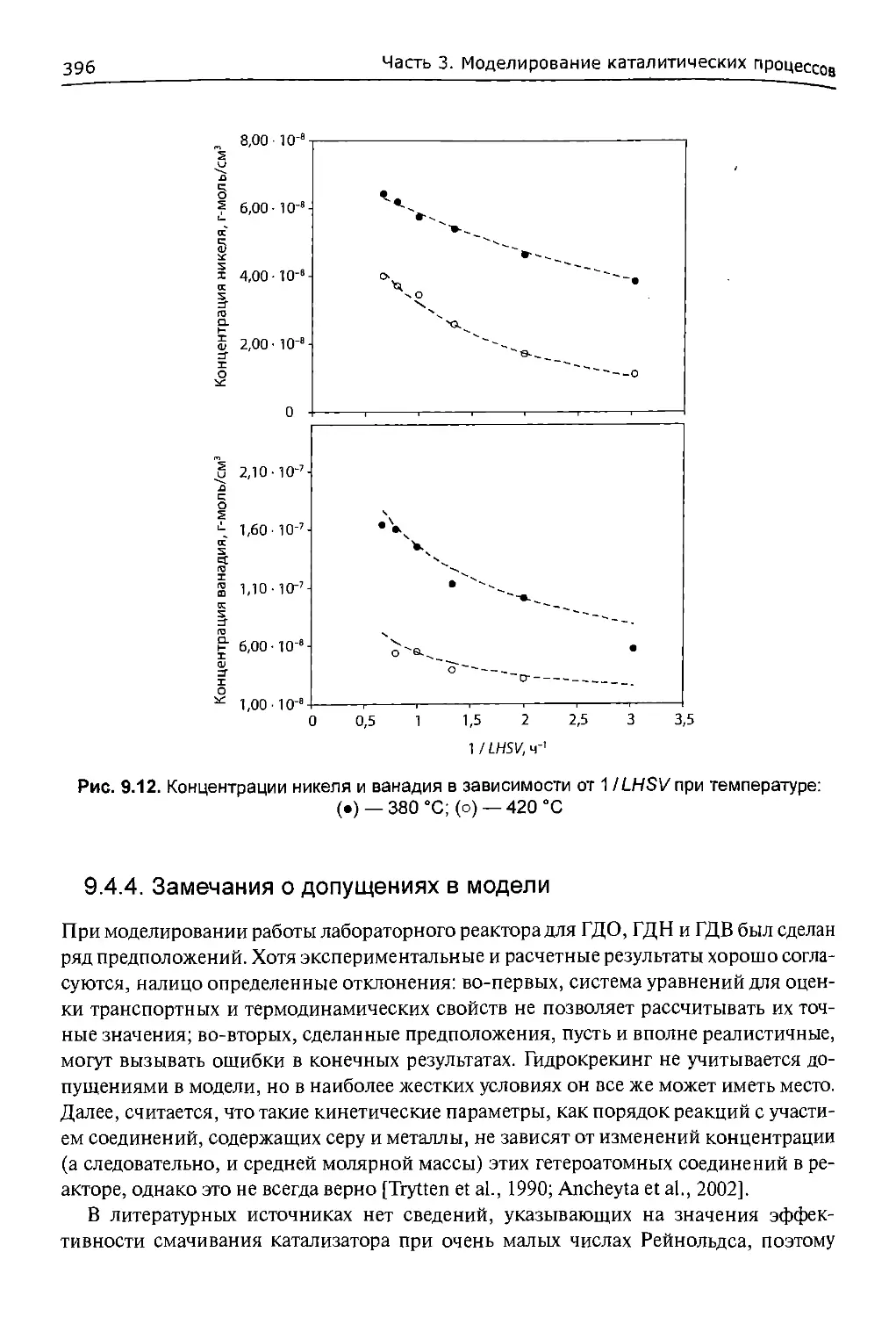
9.4.2. Кинетические параметры реакций гидрообессеривания
и гидродеметаллизации........................................388
9.4.3. Моделирование лабораторного реактора.................391
9.4.4. Замечания о допущениях в модели......................396
Обозначения....................................................398
Греческие...................................................398
Латинские...................................................398
Нижние индексы..............................................400
Литература.....................................................400
Гнава 10. Моделирование реакторов с кипящим или суспензионным
слоем.............................................................403
10.1. Введение.................................................403
10.2. Реакторы с кипящим слоем.................................405
10.2.1. Элементы реактора с кипящим слоем....................407
10.2.1.1. Рециркуляционный поддон...........................407
10.2.1.2. Система распределения потока......................407
10.2.1.3. Распределительная тарелка.........................408
10.2.1.4. Стояк.............................................408
10.2.1.5. Разрыхляющий насос................................408
10.2.2. Достоинства и недостатки реакторов с кипящим слоем.....409
10.2.3. Рабочий заряд катализатора.............................410
10.2.4. Образование осадка.....................................411
10.2.5. Износ частиц катализатора..............................412
10.2.6. Деактивация катализатора...............................413
10.2.7. Экономические аспекты процесса.........................417
10.3. Промышленные технологии кипящего слоя......................418
10.3.1. Процесс H-OU...........................................418
10.3.2. Процесс T-Star.........................................419
10.3.3. Процесс LC-Fining......................................420
10.4. Моделирование реактора с кипящим слоем.....................422
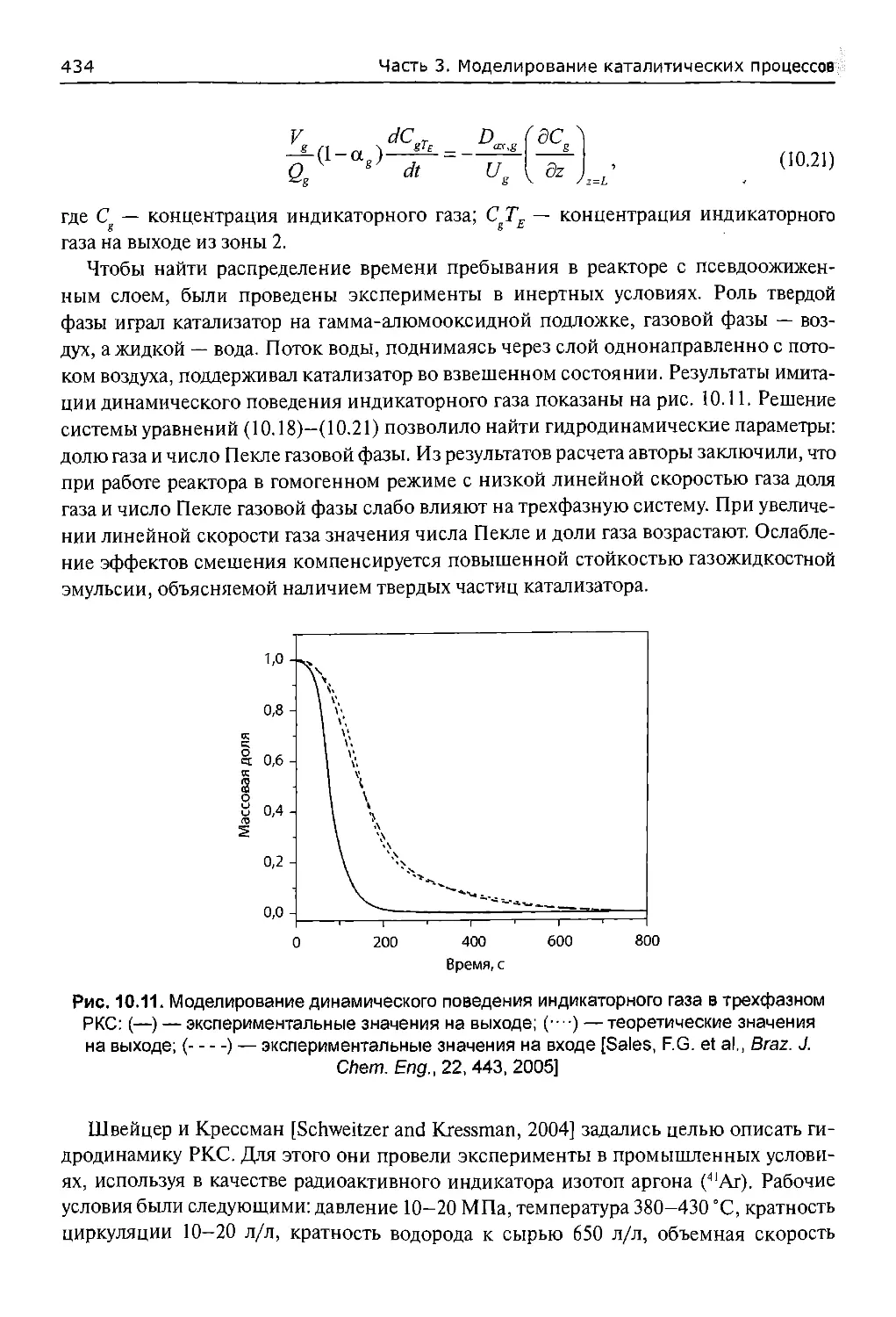
10.4.1. Исследования гидродинамики.............................423
10.4.2. Исследования обратного масштабирования.................425
10.4.3. Моделирование реактора.................................431
10.5. Моделирование реакторов с суспензионным слоем...............439
10.6. Исследование кинетики гидрокрекинга тяжелой нефти в проточном
реакторе смешения.................................................442
10.6.1. Экспериментальная часть................................444
10.6.1.1. Оборудование.........................................444
10.6.1.2. Загрузка и активация катализатора....................445
10.6.1.3. Эксперименты и анализ продуктов......................445
14
Оглавление
10.6.2. Анализ результатов...................................450
10.6.2.1. Ограничения массопереноса.........................450
10.6.2.2. Моделирование кинетики............................451
10.6.3. Выводы...............................................454
10.7. Заключительные замечания.................................454
Обозначения....................................................455
Латинские....................................................455
Греческие....................................................458
Нижние индексы...............................................459
Литература.................................................... 459
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием
кинетики......................................................463
11.1. Введение..................................................463
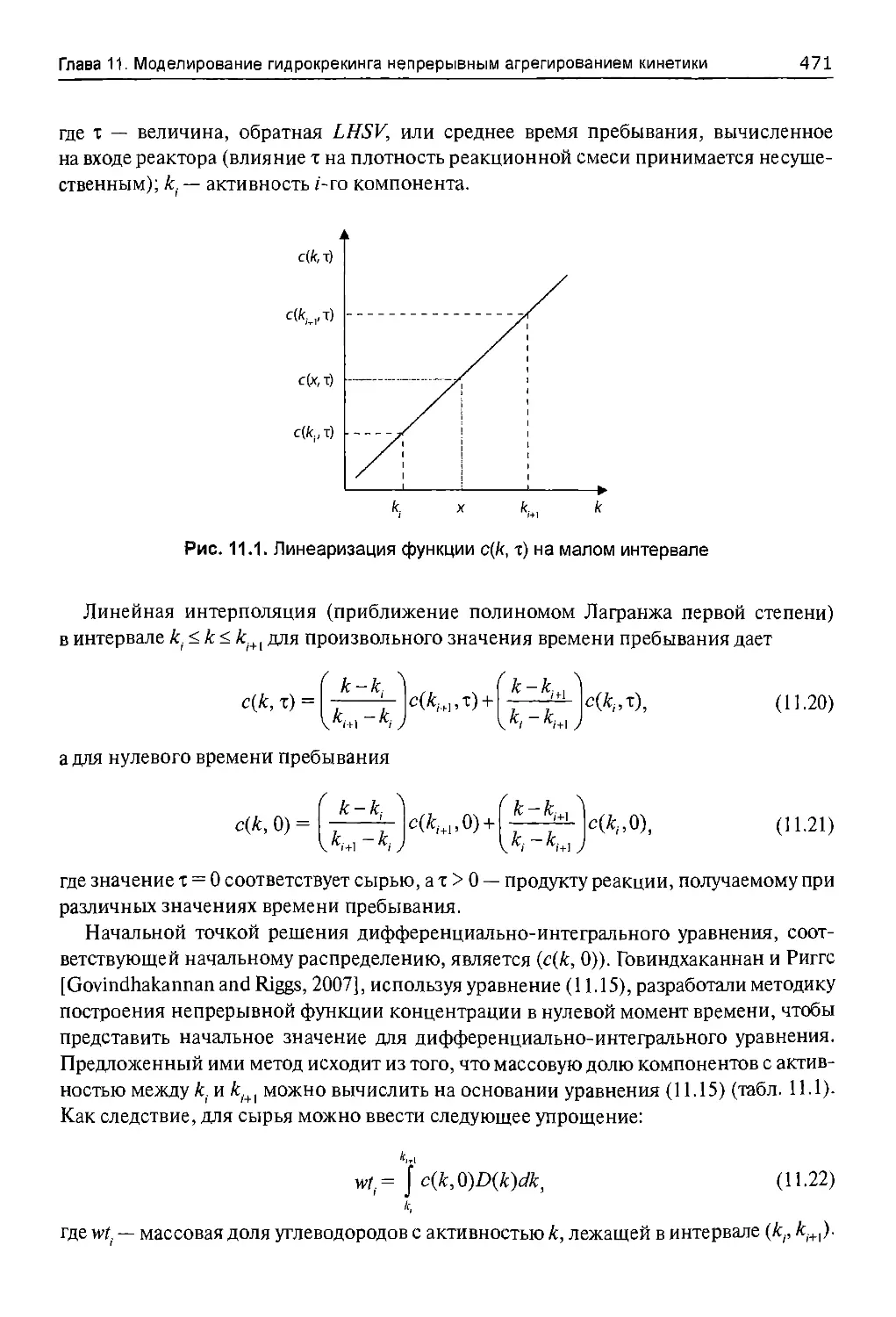
11.2. Непрерывное агрегатное моделирование кинетики.............467
11.2.1. Описание модели........................................467
11.2.2. Допущения модели для реактора с неподвижным слоем......470
11.2.3. Решение модели.........................................470
11.3. Экспериментальная часть...................................477
11.3.1. Гидрокрекинг нефти «Майя»..............................477
11.3.2. Влияние давления на гидрокрекинг нефти «Майя»..........478
11.3.3. Одновременные гидрообессеривание и гидрокрекинг тяжелой
нефти.........................................................478
11.4. Пошаговый пример применения модели.........................479
11.4.1. Исходные данные........................................479
11.4.2. Учет температуры кипения...............................479
11.4.3. Численное решение......................................480
11.4.4. Результаты.............................................480
11,4.4.1. Максимальная температура кипения.....................481
11.4.4.2. Разбиение области изменения и линейное приближение
функции выхода.................................................482
11.4.4.3. Шаг приращения времени пребывания....................482
11.4.4.4. Значения параметров модели...........................482
11.4.4.5. Результаты применения модели.........................483
11.5. Моделирование гидрокрекинга нефти «Майя».....................483
11.5.1. Экспериментальные результаты............................483
11.5.2. Расчет параметров.......................................485
11.5.3. Проверка модели.........................................486
11.5.4. Применение модели.......................................488
11.6. Моделирование влияния давления и температуры на гидрокрекинг
нефти «Майя».......................................................490
Оглавление
15
11.6.1. История исследований...................................490
11.6.1.1. Обзор литературы...................................490
11.6.1.2. Влияние давления...................................491
11.6.1.3. Значимость влияния давления........................492
11.6.2. Учет влияния давления..................................492
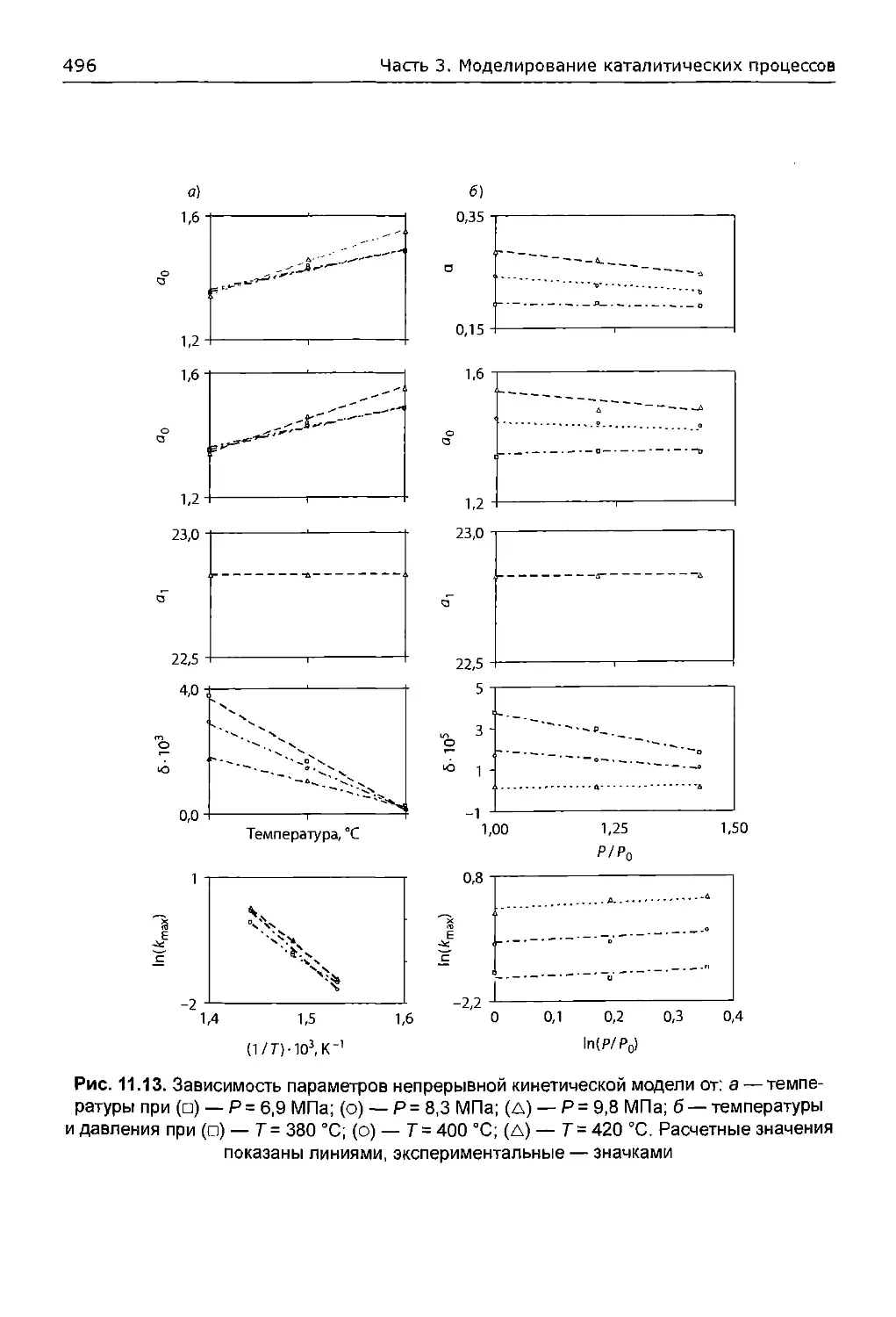
11.6.3. Анализ результатов.....................................494
11.6.3.1. Экспериментальные данные...........................494
11.6.3.2. Зависимость параметров модели от давления и температуры.494
11.6.3.3. Значения параметров модели как функции давления....497
11.6.3.4. Расчетные кривые разгонки..........................498
11.7. Совместное моделирование гидрообессеривания тяжелой нефти..501
11.7.1. Описание модели........................................501
11.7.1.1. Модель гидрокрекинга...............................501
11.7.1.2. Модель гидрообессеривания..........................504
11.7.2. Решение модели.........................................506
11.7.3. Анализ результатов.....................................506
11.7.3.1. Гидрокрекинг.......................................506
11.7.3.2. Гидрообессеривание.................................506
11.7.3.3. Заключительные замечания...........................508
11.8. Значимость параметров непрерывной модели...................509
11.8.1. О параметрах модели....................................509
11.8.2. Другие факторы, влияющие на параметры модели...........510
11.8.3. Открытые вопросы.......................................510
Обозначения......................................................511
Латинские и русские............................................511
Нижние индексы.................................................512
Верхние индексы................................................513
Греческие......................................................513
Литература.......................................................513
Гпава 12. Зависимости и прочие вопросы..............................516
12.1. Зависимости для прогнозирования свойств продуктов
гидроочистки тяжелой нефти......................................517
12.1.1. Описание зависимостей..................................519
12.1.2. Анализ результатов.....................................523
12.1.2.1. Экспериментальные данные...........................523
12.1.2.2. Расчет по оригинальным значениям параметров........524
12.1.2.3. Расчет по оптимизированным значениям параметров....528
12.1.2.4. Корреляция значений параметров со свойствами сырья.530
12.2. Расход водорода при каталитической гидроочистке............533
12.2.1. Расход водорода........................................535
16
Оглавление
12.2.1.1. Расчет по балансу водорода в газовых потоках.........537
12.2.1.2. Расчет по общему балансу водорода....................537
12.2.1.3. Расчет по потреблению водорода в химических реакциях...538
12.2.1.4. Расчет по среднему вкладу отдельных реакций...........540
12.2.1.5. Расчет по кинетической модели.........................541
12.2.2. Растворимость водорода...................................542
12.2.3. Анализ результатов.......................................543
12.2.3.1. Экспериментальные данные..............................543
12.2.3.2. Расчет по общему балансу водорода.....................545
12.2.3.3. Расчет по балансу водорода в газовых потоках..........548
12.2.3.4. Расчет по потреблению водорода в химических реакциях...548
12.2.3.5. Расчет по среднему вкладу отдельных реакций...........549
12.3. Истинные значения степени превращения и выхода продукта
при гидропереработке тяжелой нефти.................................549
12.3.1. Экспериментальные данные..................................551
12.3.2. Методика..................................................552
12.3.3. Результаты................................................554
12.4. Пересчет содержания металлов на свежий катализатор............556
12.4.1. Постановка задачи.........................................557
12.4.2. Пробы катализатора........................................559
12.4.3. Результаты................................................559
12.5. Применение функций распределения вероятностей
для приближения кривых кипения нефти..................................562
12.5.1. Функции распределения вероятности..........................564
12.5.2. Методика...................................................566
12.5.2.1. Данные..................................................566
12.5.2.2. Иллюстрация расчета параметров на примере...............567
12.5.2.3. Расчет параметров всех функций распределения............571
12.5.3. Результаты.................................................573
12.5.3.1. Ранжирование функций....................................573
12.5.3.2. Проверка наилучших функций..............................579
Обозначения...........................................................583
Латинские...........................................................583
Нижние индексы......................................................585
Верхние индексы.....................................................585
Греческие...........................................................586
Литература............................................................586
Предисловие к русскому изданию
Предлагаемая вашему вниманию книга содержит обширный материал по термиче-
ским и каталитическим процессам переработки тяжелого сырья. Многие сведения
почерпнуты из открытых литературных источников, в том числе патентов, научных
трудов и материалов конференций.
В последние годы проблема переработки тяжелых нефтей и нефтяных остатков
привлекает все большее внимание ученых, практиков и проектировщиков. Перера-
ботка тяжелых видов сырья представляет собой тяжелую задачу вследствие наличия
в них серы, азота, тяжелых металлов и смолисто-асфальтеновых веществ. Такую зада-
чу не решить без новых технологий и катализаторов, а также без высокого расхода во-
дорода, позволяющего обеспечить надлежащее качество получаемых продуктов. Все
эти вопросы получают здесь достаточное освещение.
Каждую главу предваряет краткое изложение ее содержания. Это дает читателю
возможность быстрее ориентироваться в материале как отдельных глав, так и всей
книги в целом.
В первой главе приводится характеристика тяжелых нефтей, добываемых на уже
разработанных месторождениях США, Канады и Венесуэлы. Особое внимание уде-
лено асфальтенам — как наиболее высокомолекулярной части нефти — и их роли
в образовании кокса. Затрагиваются важные вопросы, связанные со стабильностью
и совместимостью тяжелых видов нефтяного сырья.
Далее рассматриваются классификация процессов и сопоставление различных
технологий переработки остаточного сырья — от термических процессов до термока-
талитических и гидрогенизационных.
При освещении каждого процесса автор подробно описывает моделирование
на основе анализа кинетики реакций как некаталитических, так и каталитических
процессов: гидрообессеривания, гидродеазотирования, гидродеметаллизации, ги-
дродеароматизации. В ряде случаев рассмотрены результаты опытов, технологиче-
ские параметры и их влияние на выход и качество продуктов. Анализируются основ-
ные факторы, влияющие на сложную цепь реакций, равновесие фаз, массоперенос,
диффузию в частицы катализатора.
Отдельные главы посвящены моделированию термических процессов: висбрекин-
га (печного или с выносной реакционной камерой) и коксования. Особо рассмотрен
процесс газификации, а также процессы некаталитического гидрооблагораживания
с использованием инертных материалов. Наряду с описанием опытов, приводятся
технологические параметры и оценка их влияния на результаты процесса.
18
Предисловие к русскому изданию
Описан процесс гидрогенизационной переработки тяжелого сырья примени-
тельно к реакторам со стационарным, движущимся, кипящим слоем, а также в фазе
суспензии.
В книге немало конкретных примеров, связанных с переработкой тяжелой неф-
ти «Майя».
Подробно рассмотрены моделирование реакторов, кинетика реакций, стехиоме-
трические коэффициенты, даются уравнения и методы их решения.
Книга может быть рекомендована специалистам отрасли, стремящимся к углу-
бленному изучению сущности процессов (как термических, так и термокаталити-
ческих), протекающих в реакторах разного типа. Вместе с тем она будет полезна
руководителям производства, людям, занятым в нефтепереработке, а также исследо-
вателям механизма и кинетики реакций, протекающих в тяжелом нефтяном сырье.
Несомненный интерес книга представляет для учащихся вузов нефтегазового профи-
ля и молодых специалистов — всех тех, кто стремится повысить свою квалификацию,
пополнить и углубить знания об особенностях переработки тяжелого сырья.
О. Ф. Глаголева,
д-р техн, наук, профессор
В. А. Винокуров,
д-р хим. наук, профессор
Предисловие
Серьезной проблемой современной нефтепереработки является то, что добыча тя-
желой нефти обгоняет добычу легкой. Тяжелая нефть содержит повышенное ко-
личество примесей (серы, азота, металлов и асфальтенов); ее перегонка дает боль-
ше остатка и меньше дистиллятов (бензина и дизельного топлива). Эти особенности
снижают ценность тяжелой нефти. Кроме того, действующие нефтеперерабатываю-
щие заводы рассчитаны на переработку легкой нефти; перерабатывать одну лишь тя-
желую нефть они не могут. Поэтому тяжелую нефть разбавляют легкой, хотя это воз-
можно лишь до определенной степени.
Переработка тяжелой нефти требует или существенных изменений в кон-
струкции действующих установок, или сооружения новых. В обоих случаях вви-
ду технологического разнообразия установок не обойтись без значительных
капиталовложений.
Одно из возможных решений проблемы — пропустить тяжелую нефть через уста-
новку облагораживания, прежде чем отправить на нефтеперерабатывающий завод.
Такие установки будут преобразовывать тяжелую нефть в промежуточную и легкую
с малым содержанием примесей и повышенным содержанием дистиллятов. Описан-
ные в литературе процессы облагораживания опираются на один из двух принципов:
1) обеднение углеродом и 2) обогащение водородом. Основной технологией среди
первой категории процессов является замедленное коксование, которое в нефтепе-
рерабатывающей промышленности применяется шире остальных. На втором месте
по объемам применения стоит каталитическая гидроочистка, относящаяся ко вто-
рой категории.
Для расчета процессов облагораживания тяжелой нефти нужны инструменты
имитационного моделирования, опирающиеся на лабораторные эксперименты. Эти
инструменты представляют собой математические модели, описывающие явления
в реакторах превращения тяжелой нефти.
Есть публикации, посвященные имитационным моделям для реакторов различ-
ного типа и назначения, но информация о моделировании реакторов для перера-
ботки тяжелой нефти крайне скудна. Это обусловлено в основном трудностью полу-
чения экспериментальных данных, на основании которых можно было бы вывести
значения параметров моделей.
Кроме того, моделирование и расчет реакторов усложняет то обстоятельство, что
они должны перерабатывать тяжелую нефть. Состав и свойства последней таковы,
20
Предисловие
что в одной и той же реакционной системе фазы реакционной смеси, типы катали-
заторов, конструкции реакторов, условия реакций, скорость деактивации катали-
заторов и другие факторы могут быть столь разнообразны, что разработка модели
становится крайне трудной задачей. Тяжелая нефть содержит сотни различных ком-
понентов, которые вступают в реакции по разнообразным маршрутам и конкуриру-
ют за активные центры катализаторов, что вносит дополнительную сложность в фор-
мулировку кинетики реакций и моделей реакторов.
Существует множество литературных источников, рассматривающих различные
аспекты реакторов нефтепереработки. Но ни один из них не раскрывает разработки
и применения моделей кинетики, а также реакторов облагораживания тяжелой нефти.
Эта книга содержит новейшие сведения о моделировании реакторов, применя-
емых в основных процессах облагораживания тяжелой нефти. Подробно рассма-
триваются фундаментальные аспекты каждого процесса: термодинамика, химия
и кинетика реакций, технологические схемы и рабочие параметры. Обстоятель-
но описываются вывод зависимостей и разработка моделей кинетики и реакторов
с использованием экспериментальных данных, полученных на реакторах различ-
ных масштабов. Благодаря строгому изложению различных вопросов и пошаговому
описанию формулировки и применения моделей, эта книга станет незаменимым
источником не только для профессионалов, разрабатывающих модели реакторов
в нефтяной промышленности, но и для студентов, получающих химико-техноло-
гическое образование.
Книга состоит из трех частей. Первая часть имеет дело с общими аспектами
свойств тяжелой нефти и ее переработки. Вторая и третья части описывают модели-
рование соответственно некаталитических и каталитических процессов.
Часть 1 содержит две главы.
• Глава 1 дает углубленное представление о таких вопросах, как определение тя-
желой нефти, ее свойства и анализ. Там же рассматриваются проблемы, часто
встречающиеся при ее облагораживании и переработке.
• Глава 2 сообщает о некоторых аспектах промышленного облагораживания тя-
желой нефти. Описываются распространенные варианты облагораживания
методами обеднения углеродом и обогащения водородом, а также комбиниро-
ванными методами. Освещаются и новые технологии, пока не нашедшие про-
мышленного применения.
Часть II состоит из четырех глав.
• Глава 3 посвящена моделированию процесса висбрекинга. В ней описаны наи-
более важные и фундаментальные особенности этого процесса. Заключитель-
ный раздел охватывает моделирование висбрекинга в змеевике и в реакцион-
ной камере.
Глава 4 имеет дело с моделированием процесса газификации. В начале главы
описываются основы процесса, его типы и рабочие параметры, затем рассма-
тривается моделирование реакторов.
Предисловие
21
• Глава 5 описывает моделирование и имитацию процесса коксования. В первых
разделах рассматриваются химия процесса и другие теоретические вопросы.
Остальные разделы имеют дело с моделированием реакторов коксования.
• Глава 6 посвящена термической гидропереработке. Подробно исследуется вли-
яние условий реакций на различные аспекты некаталитических процессов ги-
дрообессеривания, гидродеметаллизации и гидрокрекинга.
Часть III содержит шесть глав.
• Глава 7 служит введением в вопросы, требующие рассмотрения при моделиро-
вании гидрокаталитических реакторов. Она содержит всесторонний обзор важ-
нейших особенностей реакторов, предназначенных для переработки тяжело-
го сырья, в том числе их классификации, характеристик, параметров процесса
и теоретических основ. Моделирование иллюстрируется на примере реактора
гидроочистки газойля, полученного из тяжелой нефти.
• Глава 8 посвящена моделированию облагораживания тяжелой нефти путем ка-
талитической гидропереработки. Описываются получение кинетических дан-
ных, исследование влияния сырья на деактивацию катализатора и испытание
последнего на долговременную стабильность. Приводятся уравнения баланса
масс и теплоты для моделирования поведения реактора в установившемся и пе-
реходном режимах. Описывается также имитация процессов в лабораторных
и промышленных реакторах в различных ситуациях.
• Глава 9 рассматривает моделирование лабораторного реактора, в котором про-
водятся гидродеметаллизация и гидрообессеривание нефти «Майя». Опи-
сываются все аспекты, которые необходимо принимать во внимание при
моделировании реакторов этого типа, в том числе стехиометрические коэффи-
циенты, коэффициенты скоростей реакций, транспортные и термодинамиче-
ские свойства.
• Глава 10 охватывает моделирование реакторов каталитической переработки
в кипящем слое и в суспензионной фазе. Описываются характеристики и про-
мышленные варианты технологий кипящего слоя. Глава завершается деталь-
ным описанием моделирования реакторов.
• Глава 11 описывает моделирование гидрокрекинга непрерывным агреги-
рованием кинетики. Модель иллюстрируется пошаговым описанием ее
применения.
• Глава 12 дает представление о других важных аспектах гидрокаталитической
переработки. Описываются зависимости для прогнозирования свойств про-
дуктов, расчеты расхода водорода, степени превращения сырья и содержания
металлов в пересчете на свежий катализатор, а также применение функции рас-
пределения вероятностей для подгонки кривых кипения.
Каждая глава сопровождается детальными экспериментальными данными, пояс-
нениями определения параметров модели и сравнениями результатов расчета по мо-
делям с экспериментальными данными в различных ситуациях.
22
Предисловие
Можно надеяться, что эта книга станет хорошим руководством для читателя, так
как уделяет особое внимание моделированию реакторов, проверке моделей с исполь-
зованием экспериментальных данных различного уровня и практическим вопросам,
касающимся моделирования реакторов для облагораживания тяжелой нефти.
MATLAB^ является зарегистрированным торговым знаком компании The Math-
works, Inc. Контактная информация для запроса сведений о продукте:
The Math Works, Inc.
3 Apple Hill Drive
Natick, MA, 01760-2098 USA
Тел.: 508-647-7000
Факс: 508-647-7001
Электронная почта; info@mathworks.com
Веб-страница: www.mathworks.com
Благодарности
Благодарю всех магистров, докторов наук и докторантов, внесших свой вклад в под-
готовку этой книги. Отдельное спасибо фонду Маркоса Мошинского, предоставив-
шего мне исследовательский грант (Catedra de Investigation).
Хорхе Анчита
Об авторе
Хорхе Анчита — доктор наук. Получил в 1989 г. степень бакалавра нефтехимии,
в 1993 г. — степень магистра химико-технологических наук и в 1997 г. — степень ма-
гистра администрирования, планирования и экономики переработки углеводоро-
дов в Национальном политехническом институте Мексики. Прошел аспирантуру
в Независимом университете Мехико и в Имперском колледже в Лондоне. В 1999 г.
стал научным сотрудником в лаборатории каталитических процессов Национально-
го центра научных исследований Франции в Лионе (CPE Lyon). Читал лекции в лабо-
ратории катализа и спектральной химии Канского университета (2008—2010) и в Им-
перском колледже в Лондоне (2009).
С 1989 г. работает в Мексиканском нефтяном институте (IMP). В настоящее время
руководит научно-исследовательскими проектами. С 1992 г. читает лекции для сту-
дентов старших курсов и аспирантов школы химико-технологических и добывающих
отраслей Национального политехнического института (ESIQIE-IPN), с 2003 г. — для
аспирантов Мексиканского нефтяного института. Являлся научным руководителем
более чем ста бакалавров, магистров и докторов наук, а также ряда профессоров.
Доктор Анчита работает над разработкой и применением катализаторов, кине-
тических моделей, моделей реакторов и процессов каталитического крекинга, ка-
талитического риформинга, гидроочистки средних дистиллятов и облагораживания
тяжелой нефти. Является автором или соавтором ряда патентов, книг и более 200 на-
учных публикаций. За свою исследовательскую работу доктор Анчита был удостоен
правительством Мексики высшей (третьей) степени отличия. Член Академии наук
Мексики. Выполняет обязанности внештатного редактора ряда международных жур-
налов, в том числе Catalysis Today, Petroleum Science and Technology, Industrial Engineer-
ing Chemistry Research, Chemical Engineering Communications и Fuel. Председательствовал
на многих международных конференциях.
Часть I
Свойства тяжелой нефти
и технологии ее переработки
Глава 1. Тяжелая нефть
В настоящей главе, помимо определения тяжелой нефти, указываются способы ее
классификации и ее основные свойства (плотность, вязкость, содержание асфальте-
нов и т. д.). Описываются критерии анализа, подкрепляемые экспериментальными
данными. Освещаются проблемы, сопутствующие добыче, транспортировке, подго-
товке и переработке тяжелой нефти.
1.1. ОПРЕДЕЛЕНИЕ
Тяжелой принято называть нефть с большим сопротивлением течению. Свойство
нефти быть тяжелой или легкой чаще всего характеризует ее плотность по шкале
APT, или просто плотностью API. Относительная плотность при 60 °F (15,56 °C), или
удельный вес (иначе удельная масса, англ, sg — specific gravity) и плотность в градусах
API нефти связаны следующими формулами:
141 5
°АР1 = 131 5;
__60 °F
Sg(,0'< F
(1.1)
(1.2)
__6o°f _ 141,5
g60°F “°ЧР/+131,5
Запись Jgsoop означает отношение плотности жидкости при температуре 60 °F
к плотности воды при той же температуре. Чем тяжелее нефть, тем меньше числен-
ное значение ее плотности по API, т. е. плотность в градусах API обратно пропор-
циональна физической плотности. Тяжелой обычно считают нефть с плотностью
меньше 20 °АР1 (0,9340), очень тяжелой — с плотностью менее 10 °АР1 (1,000). Абсо-
лютная (физическая) плотность, т. е. масса нефти в единице объема, и вязкость тя-
желой нефти заметно выше, чем у обычной [Schmidt, 2010]: дегазированная тяжелая
нефть имеет вязкость от 100 до 10 000 сП при пластовой температуре.
Основные проблемы при добыче и переработке тяжелой нефти создаются ее
низкой подвижностью как в пластовых условиях, так и на поверхности. Ее добы-
ча и транспортировка к нефтеперерабатывающим заводам связаны с большими
1 В англо-американской технической литературе. У нас относительная плотность обозначается
как dfi. — Примеч. науч. ред.
28
Часть 1. Свойства тяжелой нефти и технологии ее переработки
трудностями и материальными затратами. Максимальная вязкость нефти, допуска-
ющая ее трубопроводную транспортировку, составляет около 100 сП. По этой причи-
не тяжелую нефть с плотностью менее 15 ° API, вязкость которой при пластовой тем-
пературе достигает нескольких тысяч и даже миллионов сантипуаз, часто не удается
перекачивать без предварительного снижения вязкости.
На рис. 1.1 показаны значения вязкости четырех проб нефти, плотность и кине-
матическая вязкость которой при 100 °F (38 °C) варьируются от 10 до 27 ° API. Из ри-
сунка видно, что чем тяжелее нефть (меньше значение плотности в градусах APT), тем
сильнее она сопротивляется течению под действием силы тяжести.
Рис. 1.1. Сравнительные различия в текучести разных сортов нефти
1.2. КЛАССИФИКАЦИЯ НЕФТЕЙ
Способов классификации нефти довольно много. Они определяются ее свойствами,
химической структурой, происхождением, свойствами дистиллятов и т. д. Все эти
способы проверены на практике, так что нет никакой необходимости в их пересмотре.
К примеру, по содержанию серы сорта нефти подразделяют на высоко- и малосер-
нистые. По удельной массе нефть может быть тяжелой или легкой (соответственно
низкие или высокие значения плотности в градусах APT).
Ниже представлены некоторые критерии классификации нефти, напрямую опре-
деляющие ее пригодность для переработки.
• Цена. Целесообразность поставок или закупок нефти определяется не толь-
ко ее плотностью, но и другими свойствами. Например, цена на легкую нефть
снижается, если она содержит много серы. Кроме того, при классификации
Глава 1. Тяжелая нефть
29
по географическому происхождению транспортировка нефти из одного реги-
она в другой может обойтись слишком дорого даже при высоком ее качестве.
• Полезность. Легкая нефть предпочтительнее для переработки в ценные продук-
ты (например, в бензин и дизельное топливо), так как дает больший выход этих
продуктов, чем тяжелая нефть.
• Экологичность. При сжигании продуктов переработки малосернистой нефти
выбросов меньше, чем в случае с высокосернистой нефтью.
По физическим свойствам нефть классифицируется следующим образом:
• Легкая, или обычная нефть’, плотность не менее 20 ° API, вязкость меньше 100 сП.
• Тяжелая нефть’, плотность не выше 20 ° API, вязкость не менее 100 сП. Это гу-
стая и вязкая нефть, характеризующаяся высоким содержанием асфальтенов
(соединения, состоящие из сложных и крупных молекул).
• Очень тяжелая нефть’, плотность меньше 10 ° API.
• Битум, или битумная нефть. Схожа по свойствам с тяжелой нефтью, но более
густая и вязкая. В отличие от тяжелой нефти вообще не течет. Природный би-
тум представляет собой нефть с вязкостью выше 10 000 сП.
1.3. СВОЙСТВА
Опубликован ряд книг, подробно рассматривающих физические и химические ха-
рактеристики нефти [Speight, 1999, 2001], а также методы прогнозирования этих ха-
рактеристик с помощью определенных зависимостей [Riazi, 2004]. Поэтому здесь
описываются не все свойства тяжелой нефти, а лишь те из них, которые крайне важ-
ны для ее переработки. Сначала дается краткий обзор основных физических и хими-
ческих свойств нефти. Затем подробнее рассматриваются ключевые свойства тяже-
лых нефтей и методики их расчета.
1.3.1. Физические и химические свойства
Тяжелая нефть — густая, темная липкая жидкость, транспортировка и переработ-
ка которой в ценные продукты требуют значительных затрат. Тяжелая нефть име-
ет большую вязкость, т. е. оказывает сильное сопротивление течению. Характерные
свойства тяжелой нефти таковы:
• высокая физическая плотность (малая плотность по шкале API)’,
• низкое соотношение водорода и углерода;
• большой коксовый остаток (высокая коксуемость);
• высокое содержание асфальтенов, тяжелых металлов (главным образом вана-
дия и никеля), серы и азота.
Ввиду дефицита водорода по сравнению с обычной нефтью, для переработки тя-
желой нефти в полезные продукты необходимо либо повысить содержание в ней во-
дорода, либо снизить долю углерода.
30
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Тяжелая нефть образовалась из обычной, которая мигрировала из глубоко залега-
ющих пластов в верхние зоны залежей, где подверглась биологическому разложению
и воздействию воды, а также сложным микробиологическим процессам и (иногда)
улетучиванию легких углеводородов из неглубоко залегающих пластов, не перекры-
тых сверху непроницаемыми породами.
Физические и химические свойства, а также точный химический состав тяжелой
нефти зависят от ее происхождения. Нефть, в особенности тяжелая, содержит боль-
шие количества асфальтенов (высокомолекулярных полярных соединений). Кроме
них, в состав нефти входят смолы, жирные кислоты (в том числе нафтеновые), пор-
фирины, парафин и другие компоненты, которые способны связываться асфальте-
нами и влиять на стабильность нефти. Нефть может содержать также механические
примеси, состоящие из оксидов кремния и железа, из глин и т. д.
В табл. 1.1 представлены отдельные свойства нефтей некоторых сортов. Как
видно, некоторые физические и химические свойства тяжелых нефтей (такие как
вязкость, плотность, интервалы кипения, цвет) подвержены заметным колеба-
ниям. В то же время данные элементного анализа значительного числа проб со-
средоточены в узком интервале. Содержание углерода довольно постоянно.
Основные различия обусловлены переменным содержанием водорода и гетеро-
атомных соединений. В некоторых сортах тяжелой нефти, состоящих преиму-
щественно из углеводородов, азот, кислород и сера содержатся лишь в следовых
количествах.
Существенное влияние на структуру выхода продуктов нефтепереработки ока-
зывают именно гетеросоединения. Компоненты сырья претерпевают изменения
во время нефтезаводских операций, поэтому переработка тяжелого сырья пред-
ставляется трудной задачей. Отсюда вытекает безусловная необходимость про-
верки физических свойств сырья, позволяющей делать выводы о наиболее це-
лесообразных способах переработки. Среди всех процедур анализа исходных
материалов, предназначенных для использования в качестве сырья нефтеперера-
батывающего завода, главенствующее место занимает оценка нефти по данным
о физических свойствах, на основании которой принимаются решения о методах
ее переработки.
Намного более точный индикатор поведения нефти — ее химический состав. Све-
дения о типах или классах присутствующих в ней соединений позволяют точнее
определить характер реакций, протекающих в процессах переработки. Поэтому та-
кие сведения играют важную роль в определении свойств получаемых продуктов,
а также выборе методов, которыми следует перерабатывать сырье.
Таким образом, изучение свойств исходного сырья позволяет судить о наибо-
лее целесообразных способах переработки либо соотнести различные свойства
с представленными структурными типами и выбранной классификацией сырья.
Без сомнения, тщательная оценка на основании данных о физических свойствах
играет ведущую роль в первичных исследованиях сырья нефтепереработки. Пра-
вильная интерпретация результатов анализа нефти немыслима без понимания
их смысла.
Глава 1. Тяжелая нефть
31
1.3.2. Асфальтены
Как правило, тяжелая нефть характеризуется низким соотношением водород/угле-
род, высокими показателями относительной плотности (низкой плотностью в граду-
сах ЛАТ), коксуемости и концентрации асфальтенов, серы, азота и металлов (главным
образом ванадия и никеля), небольшим выходом дистиллятов и высоким выходом
остатка перегонки. Это хорошо видно из табл. 1.2, где сравниваются основные свой-
ства и состав тяжелой и легкой нефтей.
Из тяжелой части нефти можно выделить компоненты двух основных типов: ас-
фальтены и мальтены. К тяжелым относятся такие фракции с высокими температура-
ми кипения и молекулами, содержащими более 25 атомов углерода, как асфальтены
и смолы [Merdrignac and Espinat, 2007]. Асфальтены являются главными компонен-
тами тяжелых нефтей, а их структура и строение прямо влияют на состав нефти. По-
этому они заслуживают отдельного упоминания.
Согласно наиболее признанному определению, основанному на растворимости ас-
фальтенов, последние нерастворимы в таких алканах, как «-пентан, н-гексан и «-геп-
тан, но растворимы в ароматических углеводородах (в бензоле, толуоле). Содержа-
ние асфальтенов чаще всего рассматривают как содержание веществ, нерастворимых
в «-гептане. Строго говоря, эту величину следует указывать как разность содержаний
веществ, нерастворимых в «-гептане и толуоле.
Асфальтены принято выделять из нефти осаждением «-гептаном, который (как
и высшие алканы) не оказывает существенного влияния на их свойства [Andersen, 1994].
Асфальтены — главные предшественники шлама и осадков. Асфальтены пред-
ставляют собой сложные полиароматические соединения с температурой кипения
выше 500 °C и содержат следующие компоненты [Demirba?, 2002]:
• ароматические кольца с присоединенными алкильными цепями, содержащи-
ми до 30 атомов углерода;
• серу в бензотиофеновых кольцах, а также пиррольный и пиридиновый азот;
• кетоны, фенолы, карбоновые кислоты;
• никель и ванадий в комплексах с пиррольными атомами азота в порфириновых
кольцах.
Мальтены, другая составляющая нефти, содержат следующие компоненты:
• смолы, структурой схожие с асфальтенами, но имеющие меньшую молярную
массу;
• кислород, азот и серу, не всегда содержащиеся в ароматических структурах;
• нафтены и другие предельные углеводороды с прямыми и разветвленными
цепями.
Сложность тяжелых нефтей сильно затрудняет компонентный анализ, поэтому раз-
деление осуществляется не по отдельным соединениям, а по типам соединений. При
отделении нерастворимых компонентов (асфальтенов) соотношение водород/углерод
в растворимых фракциях (мальтенах) возрастает, а содержание серы, азота и кислоро-
да снижается. Как правило, нерастворимая фракция содержит от 7 до 21 гетероатомов
в молекуле, а растворимая — в среднем от 0,8 до 1,7 [Sharma et al., 2007].
W
Таблица 1.1
Свойства тяжелых нефтей различного происхождения
Свойства Сорт нефти
Кали- фор- ния 11 (США) Бос- канская (США) Кали- фор- ния 15 (США) Белри- джская тяжелая (США) Бета (США) Бача- керо (Вене- суэла) Аджо (Ка- нада) Эо- цен (Ку- вейт) Блок 65 Грин Каньон (США) Хондо (США) Альба (США) Хеброн (Кана- да) Эме- ральд Ин- гларетта (Велнко- бнтания) Кар- пин- терия (США) Энди- котт (США) Ат- кинсон (Кана- да)
Плот- ность, °АР1 10,3 10,9 13,2 13,6 13,7 16,64 16,8 18,6 19,5 19,6 20 20,1 22 22,9 23 23,7
Общая сера, % масс. 3,3 5,5 5,5 1,03 3,78 2,4 0,19 4,55 1,87 4,3 1,33 0,75 1,88 1,34 0,86
Вязкость, сП (сСт), при: О’С 15 °C 20 °C 25 °C 30 °C 40 °C 50 °C 220 000 34 000 8 826000 485 500 31 000 6400 92 600 12610 90 210 13 380 (294) 165 62 514 177 102 3507 735 (259) (62) 680 185 (170) (47) (28) 790 164 501 84 65
Содер- жание, %масс.: пре- дельных углево- дородов 25 19 28 80 38 33 44 47
Часть 1. Свойства тяжелой нефти и технологии ее переработки
арома- тических углево- дородов смол асфаль- тенов 16 35 22 18 35 23 23 39 30 3 19 1 0 40 14 8 31 24 12 30 17 9 4 зИ 14 3
Метал- лы, мг/кг (РР™): Ni V 106 245 117 1320 111 266 70 86 112 146 1 0,6 75 196 6 1 1 13 49 112 5 17 2 9
Вода, %масс. 8,6 0,1 2,4 1,7 7,1 1,5 0,9 0,9 0,1
Давле- ние паров по Рейду, кПа 11 8 < 1 4 25 6
Темпера- тура теку- чести, °C 0 21 -9 2 3 -23 -29 -28 — 15 -30 —4 -29 -21 —2 -46
* Содержание предельных и ароматических углеводородов, а также смолисто-асфальтеновых веществ (анализ 54Л4).
Глава 1. Тяжелая нефть
ио
ио
34
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Таблица 1.2
Сопоставление свойств тяжелой и легкой нефтей
Свойства Тяжелая нефть Легкая нефть
Сорт нефти «Майя» «Истмус»
Плотность, "API 21,3 33,3
Сера, %масс. 3,52 1,80
Азот, %масс. 0,32 0,14
Ni + V, мг/кг 322,5 99,7
Асфальтены, %масс. 12,7 3,06
Коксуемость по Рамсботтому, %масс. 10,8 4,13
Состав, %об.:
газы С,—См 0,5 0,7
фракция бензина С,-170 °C 15,0 23,0
фракция реактивного топлива 170-230 °C 8,4 И,7
фракция керосина 230—290 °C 8,0 10,5
легкий газойль 290—360 °C 9,1 11,4
тяжелый газойль 360—538 °C 21,4 25,3
вакуумный остаток 538 °С+ 37,6 17,4
Существуют две модели асфальтеновых молекул.
1. Модель континентального типа. Характеризуется одним периконденсирован-
ным ядром, включающим в себя не менее семи ароматических колец.
2. Модель типа «архипелаг». Молекула представлена множеством небольших
островков-ядер, соединенных между собой алкановыми мостиками с алкиль-
ными и сульфидными связями.
На рис. 1.2 [Zhao et al., 2001; Sheremata et al., 2004] представлены предполагаемые
структуры асфальтеновых молекул этих двух типов. Молекулы разных типов агреги-
руют разными способами. Асфальтены типа «архипелаг» образуют в растворах пло-
ские агрегаты, тогда как континентальные молекулы способны наслаиваться, фор-
мируя вертикально организованные пачки [Murgich et al., 1996].
1.3.3. Оценка химического состава асфальтенов
В зависимости от происхождения нефти, из которой осаждены асфальтены, они мо-
гут значительно различаться по составу и структуре. Общеизвестен разброс молярных
масс и ароматичности асфальтенов, обусловленный широкими колебаниями числа
Асфальтены континентального типа
Асфальтены типа «архипелаг»
ь
Q)
CD
Q)
1. Тяжелая нефть
Рис. 1.2. Усредненная структура асфальтенов континентального и архипелагового типов. Заимствовано из [Zhao, S. et al., Fuel,
80(8), 1155, 2001; Sheremata, J-M. et al., Energy Fuels, 18(5), 1377, 2004]
OJ
cn
36
Часть 1. Свойства тяжелой нефти и технологии ее переработки
и длины боковых алкильных цепей, а также числа ароматических колец [Ancheyta and
Speight, 2007; Ancheyta et al., 2010].
Распределение молярных масс асфальтенов и содержание в них гетероатомов
во многом определяют выбор катализаторов, пригодных для переработки этих тяже-
лых веществ: распределение размеров их пор должно быть оптимальным в том смыс-
ле, что, наряду с высокой металлоемкостью ограничения, диффузии были бы ми-
нимальны и крупные молекулы имели бы возможность беспрепятственно достигать
каталитических центров. Ниже сравниваются свойства асфальтенов, осажденных
из различных нефтей с плотностью от 10 до 33 ° API [Leyva et al., 2012].
1.3.3.1. Экспериментальная часть
1.3.3.1.1. Осаждение асфальтенов
Асфальтены осаждали н-гептаном при объемно-массовой кратности растворите-
ля, равной 5:1. Смесь 250 г нефти и 1250 мл н-гептана перемешивали в реакторе, ос-
нащенном мешалкой, с азотной подушкой при температуре 60 °C и давлении азота
25 кг/см2 в течение 30 мин при скорости 700 об/мин [Centeno et al., 2004]. Затем смесь
осаждали в течение 30 мин и под вакуумом пропускали через фильтр Ватмана 943 АН
с диаметром пор 1,5 мкм, чтобы отделить асфальтены от фракции, растворимой
в н-гептане. Асфальтены промывали н-гептаном до тех пор, пока отфильтрованная
жидкость не становилась бесцветной.
Затем асфальтеновую фракцию промывали в течение 8 ч в аппарате Сокслета сме-
сью толуола и гептана в соотношении 2:1 при температуре 96 °C. Вещества, нераство-
римые в н-гептане, в течение 12 ч высушивали при температуре 100 °C. Пробам по-
лученных асфальтенов присваивались наименования в формате А-Х, где X означала
плотность нефти, из которой пробы были выделены. Таким способом были получе-
ны пробы Л-10, Л-13, Л-16, Л-21 иЛ-33. Свойства соответствующих нефтей представ-
лены в табл. 1.3. Они изменялись в широких пределах: например, плотность образцов
нефти колебалась от 10 до 33 ° API, а содержание асфальтенов — от 1,7 до 25,1 %масс.
Более подробное описание свойств нефтей приводится в дальнейших подразделах.
Пробы нефти одного и того же сорта могут иметь разные свойства, если отбираются
в разные моменты времени.
Таблица 1.3
Свойства исследованных проб нефти и выделенных из них асфальтенов
Свойства нефтей и асфальтенов При максимальной плотности образцов, °АР1
10 13 16 21 33
Свойства нефтей
Удельный вес 1,0008 0,9801 0,9598 0,9262 0,8584
Плотность, API 9,9 12,9 15,9 21,3 33,3
Никель, мг/кг 94,2 83,4 69,2 51,1 8,9
Глава 1. Тяжелая нефть
37
Таблица 1.3, окончание
Свойства нефтей и асфальтенов При максимальной плотности образцов, °АР1
10 13 16 21 33
Ванадий, мг/кг 494 445 361 247,4 37,1
Сера, %масс. 5,7 5,4 4,6 3,5 1,5
Компоненты, нерастворимые в н-С7, %масс. 25,1 18,0 15,7 11,4 1,7
Выход вакуумного остатка при температурах ниже 538 °C, %об. 54 48,4 45,4 36,7 17,1
Свойства асфальтенов
Общий никель, мг/кг 322 217 196 187 181
Общий ванадий, мг/кг 1520 1391 1286 1467 1658
Отношение V/Ni 4,7 6,4 6,6 7,9 9,2
Асфальтеновый никель, % 85,7 46,9 44,4 41,8 33,7
Асфальтеновый ванадий, % 77,1 56,3 55,8 67,8 73,7
Углерод, %масс. 77,5 81,2 82,1 82,6 82,9
Водород, % масс. 7,9 8,31 8,5 7,7 8,7
Азот, %масс. 1,0 1,14 1,1 1,3 1,1
Общая сера, %масс. 13,4 9,2 8,2 8,3 7,2
Асфальтеновая сера, %масс. 58,8 31,0 28,0 26,7 8,1
Атомное отношение Н/С 1,215 1,220 1,226 1,106 1,250
Атомное отношение S/C 0,065 0,043 0,037 0,038 0,033
Атомное отношение N/C 0,011 0,012 0,011 0,013 0,011
1.3.3.1.2. Методы, использованные для оценки асфальтенов
• Атомно-абсорбционная спектрометрия. Содержание никеля и ванадия из-
мерялось атомно-абсорбционным спектрометром SOLAAR АА. Для удале-
ния возможных органических включений пробы твердого вещества нагревали
до 550 °C, затем помещали в смесь хлористоводородной и азотной кислот и по-
догревали до полного растворения. Раствор отфильтровывали и исследовали
на спектрометре.
• Элементный анализ. Содержание углерода, водорода, серы и азота в пробах ас-
фальтенов определяли элементным анализатором Perkin-Elmer 240. Содержа-
ние кислорода вычисляли по разности. Пробу массой 1 мг размалывали и про-
сеивали через сито с размером ячеек менее 0,2 мм. За окончательный результат
принималось среднее значение по четырем измерениям. Во всех случаях по-
грешность эксперимента составляла менее 0,5 % от абсолютной величины.
• Эксклюзионная хроматография. Этим методом определяли распределение мо-
лярных масс асфальтенов. Анализ проводили на колонках типа Mixed-D длиной
300 мм и внутренним диаметром 7,5 мм с полистирол-полидивинилбензольным
38
Часть 1. Свойства тяжелой нефти и технологии ее переработки
сорбентом, предоставленных компанией Polymer Laboratories, Черч-Стреттон,
Великобритания. Элюентом служила смесь 1-метил-2-пирролидона и хлоро-
форма (СНС13) в объемном соотношении 6:1, подаваемая дозатором со ско-
ростью 0,5 мл/мин. Колонка работала при температуре 80 °C. Детектирование
осуществлялось по ультрафиолетовым (УФ) спектрам поглощения на длинах
волн 280, 300, 350 и 370 нм с помощью спектрофотометрического детектора
Perkin-Elmer £С290, соединенного последовательно с испарительным детекто-
ром светорассеяния (нефелометром) модели £151000 компании Polymer Labo-
ratories. Образцы вводились в растворах концентрацией от 0,4 до 2,5 мг/мл.
• Ультрафиолетовая флуоресцентная спектроскопия. Спектры УФ-люминесцен-
ции снимались люминесцентным спектрометром Perkin-Elmer LS50 при скоро-
сти сканирования 240 нм/мин и оптической ширине щели 5 нм. Применялась
кварцевая кювета с длиной оптического пути 1 см. Спектрометр имел систе-
му автоматической поправки на изменение интенсивности источника в зави-
симости от длины волны. Для растворов всех образцов в хлороформе получа-
ли спектры излучения и возбуждения и спектры синхронной люминесценции.
В синхронном режиме спектры получали при постоянной разности длин волн,
равной 20 нм. Во избежание эффектов самопоглощения растворы разбавляли
хлороформом; степень разбавления увеличивали до тех пор, пока интенсив-
ность сигнала люминесценции не начинала снижаться, а относительные интен-
сивности спектральных максимумов не возрастали, становясь различимыми.
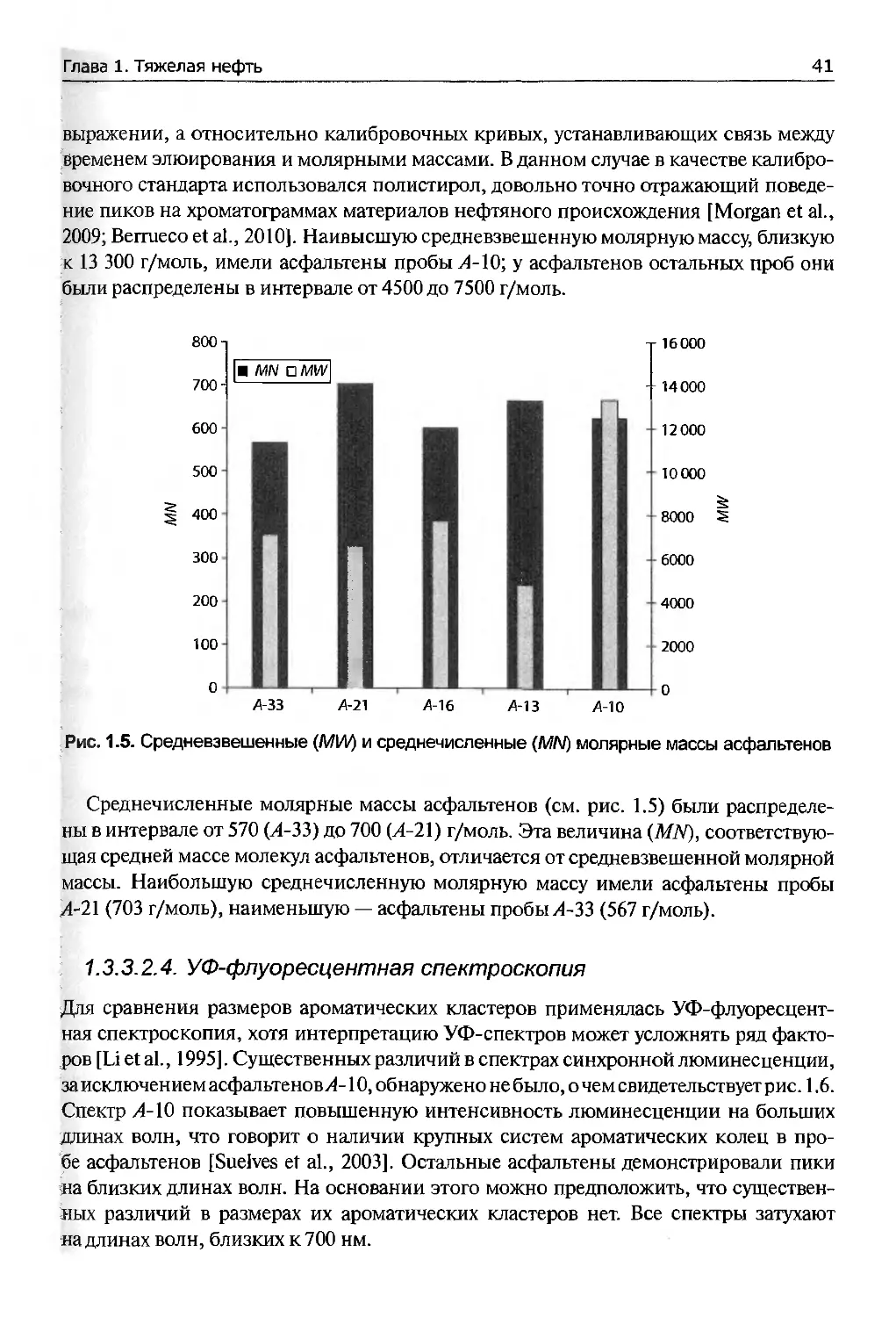
1.3.3.2. Результаты
1.3.3.2.1. Атомно-абсорбционная спектрометрия
Содержание никеля и ванадия в образцах нефти и выделенных из них асфальтенах
показано в табл. 1.3. Наблюдается общая тенденция увеличения концентрации ме-
талла по мере того, как нефть становится тяжелее. Но для ванадия эта тенденция
не соблюдается: асфальтены легких нефтей (21 и 33 ° АРР) содержат больше ванадия,
чем асфальтены тяжелых нефтей (13 и 16 °APL) [Reynolds, 1990].
В табл. 1.3 показан также расчетный процент содержания металлов в асфальте-
нах, определенный по балансу масс исходя из свойств тяжелых нефтей и асфальте-
нов. Тяжелая нефть демонстрирует повышенное содержание никеля в асфальтенах.
Что касается ванадия, повышенные его количества содержатся и в асфальтенах лег-
ких нефтей. Этот факт можно объяснить тем, что, хотя общее содержание асфальте-
нов в нефти с плотностью 33 °APLневелико, ванадий в ней сконцентрирован именно
в асфальтенах. Видно также, что с уменьшением удельного веса нефти соотношение
ванадия и никеля растет. Эти выводы отличаются от других результатов, полученных
на аналогичных нефтях [Ancheyta et al., 2002]. Различия могут быть объяснены раз-
ными источниками и разным временем отбора проб. Проверка свойств нефтей под-
тверждает их изменение со временем, даже если пробы отбирались из одного источ-
ника. В нефтях с промежуточной плотностью ванадий не всегда концентрируется
в асфальтеновой фракции.
Глава 1. Тяжелая нефть
39
1.3.3.2.2. Элементный анализ
Результаты элементного анализа асфальтенов представлены в той же табл. 1.3. Там же
указаны расчетный процент асфальтеновой серы и содержание атомов различных
элементов по отношению к углероду. Количество асфальтеновой серы следует той же
тенденции, что и содержание никеля: чем тяжелее нефть, тем больше доля серы и ни-
келя в асфальтенах. Это свидетельствует о том, что количество серы в нефти в целом
соответствует ее количеству в асфальтеновой фракции нефти.
Соотношение Н/С может служить индикатором ароматичности асфальтенов: чем
оно выше, тем меньше в последних ароматических колец [Acevedo et al., 2009; Marca-
no et al., 2011]. Высокое соотношение Н/С говорит о том, что асфальтены нефти Л-33
менее ароматичны, чем асфальтены, выделенные из других проб; наибольшей аро-
матичностью обладает проба нефти Л-21. Образцы асфальтенов других нефтей (Л-16,
Л-13 иЛ-10) показывают схожие уровни ароматичности. Высокое значение соотно-
шения Н /С в асфальтенах нефти Л-33 можно объяснить повышенным содержанием
длинных алифатических цепей. Этим же обусловлено и низкое содержание гетеро-
атомов (N и S), которые обычно концентрируются в ароматических кольцах [Mill-
er et al., 1998].
С увеличением удельной массы атомное соотношение S/С обычно возрастает, что
согласуется с увеличением содержания этого гетероэлемента в асфальтенах. Соотно-
шение N /С демонстрирует более или менее постоянные значения.
1.3.3.2.3. Эксклюзионная хроматография
На рис. 1.3 сравниваются хроматограммы асфальтенов, выделенных из различных
нефтей. Для всех образцов на хроматограммах наблюдается два максимума. Пик,
соответствующий большему времени удерживания (приблизительно 15-25 мин),
дает материал, разрешаемый колонкой. Более ранний пик (10-14 мин) обуслов-
лен заполнением мертвого объема колонки несорбируемым веществом. Согласно
[Trejo et al., 2007], впадина между пиками свидетельствует о перестройке конформа-
ции молекул в жесткую трехмерную структуру.
Асфальтены из всех проб показали схожее распределение молярных масс.
На рис. 1.4, a-в показаны пики, соответствующие сорбируемому материалу. Ранние
(нарастающие) части пиков представлены на рис. 1.4, б, поздние (убывающие) —
на рис. 1.4, в. На рис. 1.4, б видно, что асфальтены проб Л-13 иЛ-21 несколько сме-
щены в сторону большего времени элюирования. Максимумы интенсивности
на хроматограммах достигаются за разное время элюирования, составляющее
от 18,4 мин для Л-21 до 20 мин для Л-33. Из рис. 1.4, в видно, что асфальтены про-
бы Л-33 смещены в сторону большего времени элюирования. Это говорит о том,
что они состоят из молекул меньших размеров, чем асфальтены из остальных проб.
Эти особенности элюирования свидетельствуют о различиях в распределении мо-
лярных масс асфальтенов, пусть даже незначительных, причем их среднечислен-
ные молярные массы располагаются в следующем порядке: Л-33 < Л-16 < Л-10 <
<Л-13<Л-21.
40
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Рис. 1.3. Эксклюзионные хроматограммы асфальтенов, выделенных из различных нефтей
Рис. 1.4, Эксклюзионные хроматограммы асфальтенов: а — пик несорбируемого мате-
риала (10-15 мин); б — нарастающая часть пика сорбируемого материала (15-20 мин);
в — убывающая часть пика сорбируемого материала (20-25 мин)
По полученным хроматограммам асфальтенов были вычислены их средневзве-
шенные (MW) и среднечисленные (MN) молярные массы; результаты представле-
ны на рис. 1.5. Молярные массы по хроматограммам определяют не в абсолютном
Главе 1. Тяжелая нефть
41
выражении, а относительно калибровочных кривых, устанавливающих связь между
временем элюирования и молярными массами. В данном случае в качестве калибро-
вочного стандарта использовался полистирол, довольно точно отражающий поведе-
ние пиков на хроматограммах материалов нефтяного происхождения [Morgan et al.,
2009; Berrueco et al., 2010]. Наивысшую средневзвешенную молярную массу, близкую
К 13 300 г/моль, имели асфальтены пробы >1-10; у асфальтенов остальных проб они
были распределены в интервале от 4500 до 7500 г/моль.
Рис. 1.5. Средневзвешенные (MW) и среднечисленные (MN) молярные массы асфальтенов
Среднечисленные молярные массы асфальтенов (см. рис. 1.5) были распределе-
ны в интервале от 570 (Л-33) до 700 (Л-21) г/моль. Эта величина (MN), соответствую-
щая средней массе молекул асфальтенов, отличается от средневзвешенной молярной
массы. Наибольшую среднечисленную молярную массу имели асфальтены пробы
Л-21 (703 г/моль), наименьшую — асфальтены пробы Л-33 (567 г/моль).
1.3.3.2.4. УФ-флуоресцентная спектроскопия
Для сравнения размеров ароматических кластеров применялась УФ-флуоресцент-
ная спектроскопия, хотя интерпретацию УФ-спектров может усложнять ряд факто-
ров [Li et al., 1995]. Существенных различий в спектрах синхронной люминесценции,
за исключением асфальтенов Л-10, обнаружено не было, о чем свидетельствует рис. 1.6.
Спектр Л-10 показывает повышенную интенсивность люминесценции на больших
длинах волн, что говорит о наличии крупных систем ароматических колец в про-
се асфальтенов [Suelves et al., 2003]. Остальные асфальтены демонстрировали пики
на близких длинах волн. На основании этого можно предположить, что существен-
ных различий в размерах их ароматических кластеров нет. Все спектры затухают
надлинах волн, близких к 700 нм.
42
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Рис. 1.6. Спектры синхронной люминесценции асфальтенов
Полученные результаты свидетельствуют о следующем:
• содержание асфальтенов хорошо согласуется с плотностью исследованных
проб нефти;
• тяжелые нефти показывают также возрастающие концентрации серы, до-
стигающие уровней выше 5 %масс., и следовых количеств металлов (никеля
и ванадия);
• структура асфальтенов никак не связана с распределением их молярных масс,
причем все пробы показывают схожие интервалы молярных масс;
• для выявления зависимостей в размерах ароматических кластеров химическую
структуру асфальтенов исследовали методом УФ-флуоресцентной спектроско-
пии. Каки в случае эксклюзионной хроматографии, все асфальтены обнаружи-
вают схожие спектры, за исключением асфальтенов нефти с наименьшей плот-
ностью (10 ° API), которые показывают наличие крупных полиароматических
кольцевых систем.
1.3.4. Склонность к коксообразованию
Мацусита и соавторы [Matsushita et al., 2004] ввели следующий параметр, который
связывает атомные соотношения Н / С асфальтенов и мальтенов и позволяет судить
о растворимости асфальтенов и их влиянии на образование кокса в процессах пере-
работки нефти:
Н/С асфальтенов
RSI =---------------, (1.3)
Н/С мальтенов
где RSI — относительный коэффициент растворимости (англ, relative solubility index).
Глава 1. Тяжелая нефть
43
Сообщалось, что чем выше относительный коэффициент растворимости, тем
ниже склонность к коксообразованию; кроме того, при низких значениях RSI отла-
галось больше кокса, который имел меньшее отношение Н/С (более ароматический
кокс). Из трех проб нефти были выделены асфальтены и мальтены, которые под-
вергли элементному анализу, в том числе для вычисления их значений RSI. Результа-
ты представлены на рис. 1.7. Можно ожидать, что чем «тяжелее» нефть, тем выше ее
склонность к коксообразованию при переработке.
0,625 т
0,620 -
0,615 -
0,610-
сс
0,605 -
0,600 -
0,595
0,590 -
5
10 15 20
Плотность нефти, °АР1
Рис. 1,7. Относительная растворимость тяжелых нефтей
1.3.5. Вязкость
1.3.5.1. Вязкость нефтей
Как говорилось выше, вязкость — еще одно свойство, определяющее пригодность
нефти для транспортировки. Как правило, тяжелые нефти (с низкой плотностью
по шкале API) имеют высокую вязкость, что можно видеть из рис. 1.8, На рисун-
ке отдельно показаны плотность в градусах API и вязкость мексиканской нефти
и нефтей из других стран. Наблюдается практически одна и та же тенденция: если
для транспортировки допускается вязкость не более 100 сСт при 100 °F (37,8 °C),
минимально допустимая плотность близка к 20° API. Некоторые источники [Hed-
rick et al., 2006] допускают возможность транспортировки при вязкости до 250 сСт
(при 100 °F) и плотности до 16 ° API. Эти значения тоже хорошо согласуются с кри-
выми на рис. 1.8,
Предложен ряд методов расчета вязкости жидкостей (табл. 1.4), однако боль-
шинство из них применимы лишь для чистых компонентов в ограниченном интер-
вале температур. Для предсказания вязкости тяжелых (плотность от 10 до 20 ° API)
и очень тяжелых (плотность менее 10 ° API) нефтей из всех опубликованных фор-
мул пригодны лишь модифицированные уравнения Эгбога—Джека [De Ghet-
to et al., 1995].
44
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Рис. 1.8. Зависимость между плотностью по шкале API и вязкостью различных нефтей:
° — мексиканская нефть; • — нефть из других стран
Таблица 1.4
Зависимости для расчета вязкости жидкостей
Источник Зависимости
[Beal, 1946] ( 1,8-10’Y 360 'I l^= 0,32 + - — a i API Дт-260 J a _ j QO.43+8,ЗЗ/’ЛР/.
[Beggs and Robinson, 1975] ци/= 10х- 1; x°=y(T- 460)-1163; y— IQ3.0324—0,02023 "API где ц — вязкость дегазированной нефти; а, х, у — эмпирические коэф- фициенты
[Glaso, 1980] (1^= (3,141 • 1010)(Т-460)-3’444 [log (’ЛИ)]»! а°= 10,313 [log (Т- 460)] - 36,447
[Labedi, 1992] Ю’.™ . РГ4.7О13ТО.6739 > где т,- температура, F /111 If
[Kartoatmodjo and Schmidt, 1994] х — 5,7526log Tf- 26,9718
[Elsharkawy and Alikhan, 1999] 1^= antilgX- 1,0; Х= antilg у; у = 2,16924 - 0,02525 ° API-0,68875 1g Tf
[De Ghetto et al., 1995] (зависимости Эгбога—Джека) Тяжелая нефть (10 < ° АРК 20): 1g (Р0(/ + 1) = 2,06492 - 0,0179 ° API-0,702261g Tf. Очень тяжелая нефть (< 10 "АРГу. lg(po<y + 1)= 1,90296 -0,012619 "API— 0,61748 1g Tf
Глава 1. Тяжелая нефть
45
1.3.5.2. Вязкость смеси нефтей
Определение вязкости нефти не представляет особых проблем, так как в большин-
стве случаев под рукой всегда есть лабораторный вискозиметр. Сложности возника-
ют при необходимости расчета вязкости смеси нефтей. (Чтобы получить пригодную
к переработке смешанную нефть по минимальной себестоимости, т. е. при мини-
мальном расходе высокосортной нефти, низкосортную нефть для повышения про-
дажной цены или улучшения технологичности часто смешивают с высокосортной.)
Для получения смеси с заданной вязкостью обычно необходимо заранее знать, ка-
кие сорта и в каких объемных соотношениях следует смешивать. Так что задача опре-
деления вязкости смеси становится скорее математической, чем эксперименталь-
ной. Вязкость смесей вычисляют с помощью формул смешивания. Известно в общей
сложности 26 опубликованных формул смешивания (см. [Centeno et al., 201 Г] и упо-
мянутые в этой работе источники), которые можно распределить по категориям в со-
ответствии с количеством и типами используемых параметров и необходимых экспе-
риментальных данных (табл. 1.5и 1.6).
Таблица 1.5
Отвлеченные формулы смешивания и формулы смешивания,
использующие индексы вязкости в смеси
Формула/закон/метод Запись
Отвлеченные формулы смешивания
Аррениуса Ц = ИМ”; log р = xjog р4 + xjog цд, где х — мольная доля компонента; ц — динамическая вязкость; А, В — компоненты
Бингама 1=2х+Дг_ и Pj
Кендалла-Монро и1'3 = w.^ + ИЖ'3
Линейный закон
Краго с <1000In20^1 ц - 5 • 10 4expl 1, где Z -
Рейда v_ xAvA + xBvs где w — массовая доля компонента; v — кинематическая вязкость
Чириноса log log (v + 0,7) = w(log log (vA + 0,7) + w^log log (vs + 0,7)
Формулы смешивания, использующие индексы вязкости в смеси
Метод REFUTAS В форме десятичного логарифма: FBI. = 23,097 + 33,4691g 1g (v. + 0,8). В форме натурального логарифма: FBI. = 10,975 + 14,534 In In (v, + 0,8);
46
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Таблица 1.5, окончание
Формула/закон /метод Запись
|'l-Wp-2?.(>973 V=1OJ ’>* J-0,8; v = IO10 — 0,8, где e — основание натурального логарифма
Метод Chevron logv,. ' 3 + logv,. И57 = Т^уВ!,- " |’=| р = £х,Ж
Источник: [Centeno, G. et al., Fuel, 90, 3561, 2011].
Таблица 1.6
Формулы смешивания, использующие дополнительные параметры, параметр бинарного
взаимодействия и функцию отклонения от идеальности
Формула/закон/ метод Запись
Формулы смешивания, использующие дополнительные параметры
Уолтера log log (v + Q = xjog log (ул + Q + x^log log (yg + Q
Латура _ (ep(l-wg)]+ln -l) VR n = 0,9029^+0,1351 a = ln(ln vA - In ve + 1)
Ледерера In p = x'A . a X,1 = OXA i-l= Pi где x'A, x метр, уч 1пцл+х^1пцг, X. , 1 , , xD =1 хА ; + хя aln — х,, IpJJ ' — мольные доли компонентов Л и В; a — эмпирический пара- итывающий разность энергий когезии между компонентами Ан В
Шу 17,О4Дро,5237рл2745р'/1"’
Hk+X) где рл, ps — плотность компонентов; Др — разность плотностей
Ишикавы А-1 = ра где .К — константа
Глава 1. Тяжелая нефть
47
Таблица 1.6, окончание
Формула/закон/ метод Запись
Лобе V = + фвгае*'|“'; ал = -1,7^; Va aa = 0,27^- + fl,31n^| ; VB 1 ) , fi.V, , m„V„ Фл = — , Фа = — ’ maPa+maPa mtiVA+mBVB где ф — молярный объем жидкости
Степенной закон Р = (»Х+^)'"
Барруфет и Се- тиадармы а= 0,35422695m,1'7"54
Тву и Буллса In In (v + 0,7) = m In T + b, где T — температура, К; b — константа
Панченкова v = Xp^7’^[exp(e/AT)- 1], где At — эмпирическая константа; e — энергия связи между молекулами в смеси; Я — универсальная газовая постоянная
Рейка V =^> 4 7,1/3
Лима 1-2,9 УтлМл+твМв\
Формулы смешивания, использующие параметр бинарного взаимодействия
Ван дер Вайка In v — ml In | I + 2m. In | I + In v_ A 2 Al IS \VAB ) I VB )
Грюнберга и Ниссана In p = m,ln рд + m^n pa + mAmBGAB, где G — параметр взаимодействия
Тамуры и Кураты log v = mAv^A + таУЛ +
Формулы смешивания, использующие функцию отклонения
Ратклиффа и Хана 0nvpej = X^lnv-:t^nv)£; (lnvi№J = I>,lnv,.; (lnv)/;=o’/1BwA, где E — избыточная функция
Уэдлейка и Рат- клиффа In ц = In ц + Вг; 1 *пдеал ~ J ln = р£=рл + рг;
Источник: [Centeno, G. et al., Fuel, 90, 3561, 2011].
48
Часть 1. Свойства тяжелой нефти и технологии ее переработки
• Отвлеченные формулы смешивания. Удобство их применения связано с тем, что
достаточно знать величины вязкости компонентов и состав смеси, выражен-
ный в массовых или объемных долях. К подобным формулам относятся лога-
рифмический и линейный законы, а также формулы Аррениуса, Бингама, Кен-
далла-Монро, Краго, Рейда и Чириноса.
• Формулы смешивания, использующие индексы вязкости в смеси. Эти правила раз-
работаны для расчета вязкости смесей всех нефтепродуктов — от бензина до ва-
куумного остатка. Речь, в частности, идет о методе REFUTAS. Для определения
вязкости смеси в рамках данного метода необходимо знать индексы вязкости
в смеси VBI. для каждого компонента. По ним последовательно вычисляются
индекс VBI^ и вязкость смеси (v). Метод Chevron — еще один способ расчета
вязкости смеси по индексам вязкости компонентов в смеси.
• Формулы смешивания, использующие дополнительные параметры. Для расчетов
по этим формулам необходимы дополнительные параметры, вычисляемые ма-
тематическими методами. К ним относятся формулы Уолтера, Латура, Ледере-
ра, Шу, Ишикавы, Лобе; степенная формула; формула Барруфет—Сетиадармы;
метод ASTM D341, оптимизированный Тву и Буллсом; формулы Панченкова,
Рейка и Лима.
• Формулы смешивания, использующие параметр бинарного взаимодействия. Такие
формулы предложили ван дер Вайк, Грюнберг и Ниссан, Тамура и Курата. Ма-
каллистер предложил метод с двумя параметрами бинарного взаимодействия,
которые вычисляются по известным значениям вязкости смеси. Согласно это-
му методу, параметры взаимодействия линейно зависят от величины, обрат-
ной температуре. Если известны их величины при двух значениях темпера-
туры, величины при других температурах можно найти по соответствующим
номограммам.
• Формулы смешивания, использующие функцию отклонения. Ратклифф и Хан вы-
числяют вязкость смеси, исходя из абсолютных величин вязкости компонен-
тов. Их метод предусматривает функцию для учета отклонения от идеального
поведения смеси. Уэдлейк и Ратклифф сообщили о модели с функцией откло-
нения, которая вычисляется по структурной постоянной, числу групп в соеди-
нении и весу отдельных групп в смеси.
Недавно была проверена точность расчетов вязкости смесей тяжелых нефтей по не-
которым из этих методов. Основные выводы представлены ниже [Centeno et al., 2011].
Для расчета вязкости смесей используется ряд методов — от чисто эмпириче-
ских, для которых нужны экспериментальные данные, вплоть до тех, которые тре-
буют вычисления сравнительно сложных математических функций. Простейшими
из них являются отвлеченные формулы расчета, которые получили широкое рас-
пространение благодаря своей простоте и небольшому объему необходимых экс-
периментальных данных (нужны лишь данные о вязкости компонентов и их кон-
центрациях в смеси). Эти формулы имеют простую математическую формулировку
(линейная, логарифмическая, степенная и обратная зависимости). Вместе с тем
такая простота расчета приводит к тому, что его успешность можно считать чуть
Глава 1. Тяжелая нефть
49
ли не делом случая. Излишнюю простоту подобных формул компенсирует другая
группа методов — формулы смешивания с дополнительными параметрами. Для
этого в расчет вводятся константы, которые, являясь, по сути, коэффициентами
корреляции, вычисляемыми по экспериментальным данным, действительны лишь
для того интервала условий, для которого они были вычислены. Авторы этих мето-
дов утверждают, что они точнее, чем отвлеченные формулы. В этом есть своя прав-
да, поскольку с увеличением числа параметров расчетные величины лучше согласу-
ются с экспериментальными данными.
Хорошо известно, что при смешивании линейно ведет себя не вязкость, а зави-
сящая от нее функция — индекс вязкости в смеси. Линейное поведение вязкости
означает, что вязкость смеси равна просто арифметическому среднему вязкостей
компонентов, взвешенных по их концентрации в смеси. В принципе, прави-
ла смешивания с использованием индексов вязкости в смеси [REFUTAS and Chev-
ron] должны показывать хорошее согласие с экспериментальными результатами.
Но практика показывает, что это справедливо лишь для компонентов с низкой вяз-
костью. Причина в том, что вязкость является неаддитивным свойством и ведет
себя иначе, чем другие свойства, основанные на массе (такие как содержание при-
месей или плотность).
Правила смешивания с параметром бинарного взаимодействия или функцией от-
клонения опираются на термодинамические принципы и способны давать лучшее
согласие, чем прочие методы. Во всяком случае, параметры должны вычисляться для
широкого диапазона экспериментальных данных и смесей, так что эти правила мо-
гут быть распространены на пробы, отличные от тех, которые применялись при вы-
воде величин параметров.
И наконец, следует упомянуть, что реологическое поведение нефти, как прави-
ло, близко к ньютоновскому, однако отклонение сильно зависит от состава нефти.
При низких температурах нефть вполне может оказаться настолько вязкой, что чис-
ло Рейнольдса резко снижается (по логарифмическому закону). Изменчивость вяз-
кости больше связана с составом, чем с плотностью или температурой. Ввиду этого
обстоятельства вязкость относится к свойствам, определение которых расчетным пу-
тем наименее надежно. Коммерческие модели технологических процессов использу-
ют несколько зависимостей, но применительно к тяжелым нефтям они обычно дают
неверные результаты.
Сентено и соавторы [Centeno et al., 2011] пришли к следующим выводам. Рас-
чет вязкости нефти по отвлеченным формулам дает высокую среднеквадратическую
погрешность. Расчеты по наиболее известным и широко применяемым формулам
[REFUTAS and Chevron] приводят, как правило, к оценкам вязкости, завышенным
по сравнению с экспериментальными данными. Сравнительно низкую погрешность
дают лишь формулы, использующие дополнительный параметр: например, форму-
лы Уолтера и Эйнштейна и степенной закон. Ни один отдельно взятый метод не спо-
собен достаточно точно предсказывать вязкость всех нефтей: с уменьшением плот-
ности (по шкале АРГ) расчетные и экспериментальные результаты расходятся все
больше. Тот факт, что точность предсказания вязкости смеси всеми этими методами
50
Часть 1. Свойства тяжелой нефти и технологии ее переработки
снижается с увеличением удельного веса компонентов, подтверждается исследова-
нием смесей легких нефтяных дистиллятов. Результаты свидетельствуют о том, что
предсказание вязкости тяжелых нефтей и их смесей остается трудной задачей, так что
потребность в адекватных правилах смешивания по-прежнему существует.
Сентено и соавторы [Centeno et al., 2011] не исследовали правила смешивания,
использующие параметр бинарного взаимодействия или функцию отклонения. Для
проверки их способности предсказывать вязкость смесей в качестве примера были
взяты данные, опубликованные в литературных источниках (табл. 1.7). Согласно ме-
тоду Ратклиффа и Хана, отклонение от идеальности учитывается введением функции
отклонения в уравнение
(1П Vpe J = X- 1 <1П V)£’ (1 -4)
где функция отклонения определяется выражением
(1пуГ=ял^Л. (1.5)
Вязкость смесей различных нефтей
Таблица 1,7
Температура, °C Кинематическая вязкость нефтей, сСт
14,8 24,7 36,0
10 2193,10 163,40 14,22
20 887,90 81,46 9,51
30 426,40 51,81 7,14
40 223,50 34,63 5,96
60 78,71 17,76 4,01
Плотность, °АР1 Состав смеси, массовых долей, для сорта нефти
1 2 3 4
14,8 о,з 0,1 0,7 0,2
24,7 0,3 0,7 0,1 0,3
36,0 0,4 0,2 0,2 0,5
Температура, °C Кинематическая вязкость смесей, сСт
10 122,1 171,78 27,09 207,16
20 70,75 83,38 16,01 110,30
30 37,25 50,51 11,59 62,71
40 25,56 31,98 8,43 39,43
60 13,88 16,27 5,61 19,70
Глава 1. Тяжелая нефть
51
В соответствии с рис. 1.9, для трехкомпонентных смесей возможны три различных
бинарных сочетания, т. е. необходимо вычислить три параметра: а12, а[} и а2}. Приме-
няя к ним уравнение (1.5), получаем следующую систему зависимостей:
(In v)/ = Oj/jX1 + + a23x2lx3l; (1.6)
(In v)/ = aI7x(2x22 + ajcft + a23x22x32; (1.7)
(In v)3£ = fl12Xj3x23 + aI3x,3x33 + a23x23x33; (1.8)
Для решения этой системы уравнений необходимо знать концентрацию нефти
каждого сорта в смеси х/ и функцию отклонения (In v)£. Величины х/ уже приведены
в табл. 1.7, а величину (In v)£ можно вычислить исходя из экспериментальных данных
и уравнения (1.4). Решение системы после подстановки данных смесей 1—3 дает зна-
чения параметров, которые приведены в табл. 1.8.
Таблица 1.8
Значения параметров л12, а13 и аи в зависимости от температуры
Температура,°C Я12 «13
10 0,2174 -4,2731 -1,0310
20 -0,3685 -3,8685 -0,9811
30 -0,7992 -3,4638 -0,9312
40 -1,0748 -3,0592 -0,8814
60 -1,1605 -2,2500 -0,7816
На рис. 1.10 показаны расчетные и измеренные значения вязкости смеси 3 в зави-
симости от температуры. Для сравнения приведены результаты, вычисленные по ши-
роко распространенному методу REFUTAS. Кроме того, с использованием параметров
52
Часть 1. Свойства тяжелой нефти и технологии ее переработки
бинарного взаимодействия была вычислена вязкость смеси 4, которая в расчетах этих
параметров не участвовала. Хорошо видно, что во всех случаях наилучшие результа-
ты получаются с использованием параметров бинарного взаимодействия. В осталь-
ном значения параметров, указанные в табл. 1.8, показывают обратную зависимость
от температуры. Исходя из этого, можно построить зависимость каждого параметра
от температуры, что позволит без труда вычислять вязкости смесей при различных
значениях температур.
Рис. 1.10. Сравнение расчетных и экспериментальных значений вязкости смесей нефтей:
(») — измеренные значения; (—) — расчетные значения по методу REFUTAS;
(—) — расчетные значения с использованием параметра бинарного взаимодействия
1.3.5.3. Другие свойства смесей
Для расчета других свойств смесей можно использовать подход, уже применявшийся
к определению вязкости смесей.
Глава 1. Тяжелая нефть
53
1. Аддитивные свойства. Это свойства, зависящие от массы (например, плотность
шкале API, содержание примесей (сера, металлы, азот) и т. д.) и подчиняющие-
ся при смешивании линейному закону
= (1-9)
/=1
где Р4 — значение свойства смеси; у. — объемная или массовая доля /-го компонента
в смеси; р. — значение свойства /-го компонента в отдельности.
2. Неаддитивные свойства. Это свойства, не зависящие от массы: вязкость, дав-
ление паров по Рейду и т. д. Для расчета неаддитивных свойств необходимо
знать индексы свойств компонентов в смеси (индексы вязкости, давления па-
ров и т. д.), что позволяет привести поведение определенного свойства смеси
к линейному виду, отвечающему следующему уравнению:
N
В1=^уЫ, (1.10)
1=1
где BI — индекс свойства /-го компонента в смеси.
Зависимости для индексов некоторых свойств смеси приводятся в литературе
и представлены ниже.
Давление паров по Рейду (RVP):
BIRyp = (RVP)^. (1.11)
Температура вспышки (FP):
2414
16Д/„--6,1188+гр_42|6. (1.12)
Температура текучести (РР):
BIpf = РР1'0№. (1.13)
Анилиновая точка (АТ):
BIAT= l,124e°'0065W. (1.14)
1.3.6. Стабильность и совместимость нефтей
1.3.6.1. Определения
Стабильность нефти можно определить как ее способность оставаться в неизменном
состоянии несмотря на обстоятельства, способные вызывать изменения. И наоборот,
нестабильность нефти — это ее склонность образовывать отложения асфальтеновых
осадков под воздействием факторов времени и/или температуры.
Совместимость двух или более сортов нефти разного происхождения выражает
меру их смешиваемости без флокуляции (осаждения) асфальтенов или выпадения
54
Часть 1. Свойства тяжелой нефти и технологии ее переработки
парафина. Нефти одного и того же типа, как правило, совместимы. Тем не менее
даже в пределах одного и того же типа или группы нефтей может наблюдаться несо-
вместимость. В таких случаях предсказание проблем несовместимости требует зна-
чительной экспериментальной работы.
Осажденные асфальтены быстро растворить трудно. Осаждение асфальтенов мо-
жет вызывать образование стабильных эмульсий воды в нефти, закупорку линий пе-
рекачки, загрязнение теплообменников и закоксовывание змеевиков печей перегон-
ных установок.
Стабильными и совместимыми можно считать лишь следующие смеси разных нефтей:
• сохраняющие однородность непосредственно после смешивания;
• остающиеся однородными в нормальных условиях хранения;
♦ не образующие существенного осадка (и не проявляющие склонность к такому
образованию).
Асфальтены в значительной мере определяют реологическое поведение, совме-
стимость и стабильность нефтей при хранении и транспортировке. Этот факт под-
тверждается результатами обширных реологических исследований нефтей раз-
личного происхождения. Хотя нефть в целом принято относить к ньютоновским
жидкостям, некоторые ее сорта с высоким содержанием сложных асфальтенов про-
являют выраженное псевдопластичное, неныотоновское поведение.
Принято считать, что асфальтены содержатся в нефти в мицеллярной форме. Смо-
лы, иначе называемые мальтенами, можно рассматривать как низкомолекулярные
асфальтены; они играют роль растворителя для остальных компонентов. По обще-
принятому представлению, дисперсная фаза тяжелой нефти состоит из асфальтенов,
образующих с высокомолекулярными компонентами мальтенов (смол) и жидкими
углеводородами комплексы в виде мицелл. В этих условиях имеет место равновесие
и мицеллы находятся в пептизированном состоянии, т. е. образуют коллоидную дис-
персию. Но если соотношение С / Н в молекулах мальтенов снижается, абсорбиро-
ванные асфальтенами смолы частично десорбируются. Вследствие этого состояние
равновесия изменяется. Частицы асфальтенов оказываются окруженными молеку-
лами смол не полностью и в результате начинают испытывать взаимное притяжение.
Все это заканчивается осаждением асфальтенов.
1.3.6.2. Методы оценки стабильности и совместимости
Общепринятые методы оценки стабильности и совместимости нефтей и их смесей
можно подразделить на прямые и косвенные [Speight, 2001].
Прямые методы
• Метод пятна. Стабильность нефти определяется визуально, поэтому результа-
ты в некоторой степени субъективны.
• Испытание на фильтруемость в горячем состоянии. Метод позволяет определить
общий осадок нефти, но не дает никаких сведений о ее стабильности.
• Метод ксилольного эквивалента. Определяется наименьшая концентрация кси-
лола в такой смеси с изооктаном, которая при смешивании с пробой нефти
Глава 1. Тяжелая нефть
55
такого же объема оставляет на хроматографической бумаге однородное пятно
без кольца в центре. Чем меньше необходимая концентрация ксилола, тем луч-
ше совместимость пробы.
• Испытание в склянке. Гравиметрический по сути и легко осуществляемый ме-
тод. Общепринят при определении совместимости нефти и жидкости для за-
канчивания скважин.
• Определение стойкости к смолообразованию. Метод дает приближенную оценку
склонности нефти к образованию смол при хранении.
• Определение содержания воды, осадка и солей. Чем выше содержание воды и дон-
ного осадка, тем больше ожидаемая скорость образования шлама и отложений.
Косвенные методы
• Элементный анализ (определение содержания С, Н, О, N, S, металлов и золы).
Чем больше азота и серы содержит нефть, тем сильнее ее склонность к образо-
ванию шлама.
• Определение удельного веса и плотности по шкале API. Нефти с низкой плотно-
стью по шкале API (тяжелые нефти) вследствие высокого содержания поляр-
ных асфальтеновых компонентов более склонны к образованию шлама.
Определение вязкости. Как правило, продукты процессов, ухудшающих ста-
бильность или совместимость нефтей, увеличивают вязкость.
• Определение температуры текучести. Нефть с высокой температурой текуче-
сти содержит больше тугоплавких парафинов, способствующих образованию
шлама.
• Определение содержания асфальтенов. Чем выше содержание асфальтенов, тем
сильнее склонность нефти к образованию шлама, особенно в смеси с несовме-
стимыми сортами.
• Определение общей кислотности (кислотного числа). Нефти с высокой кислотно-
стью могут быть склонными к нестабильности.
• Определение характеризующего фактора нефти. Если он выше 12 (парафиновая
нефть), вполне вероятно образование отложений парафина.
Методы, основанные на анализе SARA
• Показатель коллоидальной неустойчивости (colloidal instability index — СП). Он вы-
ражает устойчивость асфальтенов через содержание предельных и ароматиче-
ских углеводородов, смол и асфальтенов (SARA) и определяется как отношение
суммы массовых долей асфальтенов и компонентов, способствующих их неста-
бильности (т. е. флокулянтов, которыми являются предельные углеводороды),
к сумме массовых долей пептизирующих агентов (смол и ароматических углево-
дородов), способствующих стабильности асфальтенов в нефти [Asomaning, 2003]:
Процент асфальтенов + Процент предельных углеводородов
Процент смол + Процент ароматических углеводородов
СП можно также представить в виде графика, по осям которого откладывают-
ся сумма содержания смол и ароматических углеводородов и сумма содержания
56
Часть 1. Свойства тяжелой нефти и технологии ее переработки
асфальтенов и предельных углеводородов. Чем меньше величина СП, тем устойчивее
асфальтены в нефти. СП служит удобным показателем сравнительной устойчивости
нефтей. Асфальтены неустойчивы, если СН> 0,9, и устойчивы, если СП < 0,7. Если
величина С//находится между 0,7 и 0,9, ничего определенного об устойчивости ас-
фальтенов сказать нельзя.
• Диаграмма Станкевича [Stankiewicz et al., 2002]. По ее осям откладываются со-
отношение предельных и ароматических углеводородов и соотношение ас-
фальтенов и смол. Диаграмма состоит из областей стабильности и нестабиль-
ности и позволяет быстро определить риск осаждения асфальтенов.
♦ Сводная диаграмма стабильности [Sepulveda et al., 2010]. Она состоит из четырех
графиков, позволяющих определить стабильность асфальтенов нефти любого
типа по следующим соотношениям содержаний компонентов:
1. (Смолы/асфальтены)/(предельныеуглеводороды/ароматическиеуглеводо-
роды) либо Ароматические углеводороды/асфальтены.
2. (Смолы/асфальтены)/(предельные углеводороды/ароматическиеуглеводо-
роды) либо Смолы/асфальтены.
3. Предельные углеводороды /ароматические углеводороды либо Смолы /ас-
фальтены.
4. (Смолы/асфальтены)/ (предельные углеводороды/ароматические углеводо-
роды) и, как вариант, Ароматические углеводороды/предельные углеводоро-
ды/асфальтены.
По утверждениям, надежность этого метода выше (0,92), чем у других (0,72 для СП
и 0,86 для диаграммы Станкевича).
Другие методы
• Диаграмма де Бура [de Boer et al., 1995]. По ее осям откладываются плотность
пластовой нефти и разность между пластовым давлением и давлением насыще-
ния в пластовых условиях. Повышенные риски наблюдаются в случае легкой
нефти при большом превышении давления насыщения.
• Титрование по Хайтхаусу (Р- Value). Определяется общее количество «-гептана,
которое можно добавить к пробе нефти, прежде чем она станет нестабильной.
Титрование по Хайтхаусу применимо лишь к компонентам нефти, раствори-
мым в толуоле.
Приготавливают три раствора нефти в толуоле с различными концентрациями
и титруют их слабым растворителем, например изооктаном. Для каждого рас-
твора в момент, когда начинается осаждение асфальтенов, регистрируют мас-
су пробы нефти (BQ, объем толуола (Vs) и объем добавленного титрующего изо-
октана (кр. Вычисляют по следующим формулам коэффициент флокуляции
(flocculation ratio — FR) и концентрацию разбавления (С) [Heithaus, 1962]:
Глава 1. Тяжелая нефть
57
Строят график зависимости FR от С и находят координаты точки пересечения
(FAmax и СУ). Вычисляют параметры Хайтхауса:
• пептизируемость асфальтенов
р =1 — FR . (1.18)
• растворяющая способность мальтенов
+ 1) (1Л9)
• общая совместимость остатка
Большие значения р соответствуют пептизируемым асфальтенам, а большие зна-
чения Р свидетельствуют об общей совместимости системы. Большие значениямо-
гут толковаться по-разному. Нефть с Р < 1 рассматривается как нестабильная.
• Собственная стабильность. С помощью оптического прибора измеряется соб-
ственная стабильность асфальтенов в нефтяном носителе. Как и в предыдущем
методе, определяют три параметра:
• У. Параметр состояния пептизации асфальтенов в нефти, характеризую-
щий общую стабильность пробы. Асфальтены нефти с низким значением 5"
склонны к флокуляции. В нефтях с высоким значением 5" они удерживаются
в пептизированном состоянии и при смешивании не флокулируют.
• Sa. Параметр потребности асфальтенов в пептизирующих агентах, характе-
ризующий ароматичность асфальтенов. Иначе говоря, он показывает спо-
собность асфальтенов оставаться в коллоидной дисперсии. Чем меньше ве-
личина Sa, тем выше ароматичность.
• So. Параметр пептизирующей способности нефти, характеризующий арома-
тичность смол и их способность удерживать асфальтены в растворе. Чем боль-
ше величина So, тем выше ароматичность.
1.4. АНАЛИЗ ТЯЖЕЛОЙ НЕФТИ
1.4.1. Определение
Анализ нефти — это совокупность лабораторных исследований для определения фи-
зических и химических свойств и испытаний на опытных установках, результаты ко-
торых характеризуют данную пробу нефти. Аналитическая разгонка осуществляется
на атмосферной и вакуумной установках, которые в совокупности обеспечивают опре-
деление истинных температур кипения (ИТК). Типичная установка для эксперимен-
тального определения ИТК (модель Minidist компании GECIL Process) емкостью 40 л
показана на рис. 1.11. Цикл испытаний занимает от 3 до 5 суток и позволяет собрать
достаточное количество дистиллятных фракций, которые могут быть использованы
в дальнейших испытаниях. Установки атмосферной и вакуумной разгонки дают те же
58
Часть 1. Свойства тяжелой нефти и технологии ее переработки
дистилляты и остатки, что и полномасштабные перегонные установки. Интервалы от-
бора фракций обычно определяют в зависимости от номенклатуры продуктов нефте-
перерабатывающего завода. Наиболее распространенные и общепризнанные интерва-
лы выкипания фракций при анализе нефти определяются следующим образом:
• легкий прямогонный бензин (ЛПрБ): НК—71 °C;
• средний прямогонный бензин (СПрБ): 71—177 °C;
• тяжелый прямогонный бензин (ТПрБ): 177—204 °C;
• реактивное топливо (РТ): 204—274 °C;
• керосин: 274—316 °C;
• прямогонный газойль (ПГ): 316—343 °C;
• легкий вакуумный газойль (ЛВГ): 343—454 °C;
• тяжелый вакуумный газойль (ТВГ): 454-538 °C;
• вакуумный остаток (ВО): 538 °С+.
Рис. 1.11. Установка для экспериментального определения НТК
Иногда описывают как отдельную фракцию атмосферный остаток (интервал ки-
пения 343 °С+, смесь легкого и тяжелого вакуумных газойлей и вакуумного остатка).
Проводят также детальный анализ углеводородов отходящих газов.
1.4.2. Цели анализа
В нефтепереработке анализ нефти проводят чаще всего:
• для получения экспериментальных данных, позволяющих судить о возможно-
сти переработки нефти на данном заводе;
Глава 1. Тяжелая нефть
59
• выявления соответствия нефти требованиям к выходу, качеству и объему продукции;
• выявления соответствия нефти, ее продуктов и процессов переработки эколо-
гическим и иным требованиям;
• при принятии решений о внесении изменений в производственные процессы,
при разработке планов выпуска продукции, оптимизации процессов перера-
ботки и проверке намечаемых производственных мероприятий;
• для обеспечения проектно-конструкторских организаций сведениями, необхо-
димыми при технологическом и техническом проектировании нефтеперераба-
тывающих заводов;
• решения вопросов, связанных с ценами, и согласования штрафных санкций, зави-
сящих от уровня содержания примесей и других нежелательных свойств нефти,
1.4.3. Виды анализа
Глубина и сложность анализа нефти зависят от ее типа и предназначения. Анализ мо-
жет быть приемочным и полным (всесторонним). Существует несколько видов ана-
лиза, значительно различающихся объемом получаемых экспериментальных данных:
• исследования выхода и свойств тех продуктов, которые используются в каче-
стве сырья каталитического риформинга (бензиновая фракция) и каталитиче-
ского крекинга (газойли);
• исследования, дающие более подробные сведения о потенциале получения
масляных фракций и/или асфальта (гудрона);
• исследования, дающие минимум необходимой информации: например, кри-
вую разгонки (НТК) или кривую плотности;
• полный и всесторонний анализ (характеризуемый экспериментальными ме-
тодами) как нефти в целом, так и различных ее фракций, дающий сведения
об ИТК, плотности и содержании серы.
1.4.4. Свойства некоторых тяжелых нефтей
В табл. 1.9 представлены физические и химические свойства различных сортов тя-
желой нефти с плотностями от 10 до 21 ° API. Хорошо видно, что чем тяжелее нефть
(т. е. чем меньше плотность по шкале API), тем выше содержание примесей (серы,
азота, коксуемых веществ, асфальтенов, металлов). Вязкость, значения которой
обычно приводят при как минимум трех значениях температуры, с увеличением
удельной массы нефти измеряют при все более высоких температурах. По графи-
ку зависимости двойного логарифма вязкости от температуры можно легко найти
значения вязкости при других температурах. Чтобы быть уверенным в правильном
экспериментальном определении значений вязкости, точки на графике должны до-
статочно точно ложиться на прямую линию. Как видно из таблицы, нефти с плотно-
стями 10 и 13 ° API имеют высокую вязкость и не отвечают требованиям транспорти-
ровки; такие нефти нуждаются в улучшении свойств.
60
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Таблица 1.9
Физические и химические свойства тяжелых нефтей
Свойства Метод ASTM Плотность, ’API
10 13 21
Относительная плотность, 60/60 °F £11298 1,0008 0,9801 0,9260
Плотность, ’API £1287 9,89 12,87 21,31
Кинематическая вязкость, сСт, при температуре, °C: 15,5 21,1 25,0 37,8 54,4 60,0 70,0 0445 7081 4426 2068 19646 5102 1235 299,2 221,6 181,4
Характеризующий фактор (Кио],) 6Л9Р375 11,50 11,60 11,71
Температура текучести, °C £>97 + 12 0 —
Коксуемость по Рамсботтому, %масс. £>524 20,67 16,06 10,87
Коксуемость по Конрадсону, %масс. £>189 20,42 17,94 11,42
Вода и осадок, %об. 04007 1,40 0,10 0,20
Общая сера, %масс. 04294 5,72 5,35 3,57
Содержание солей, фунтов на 1000 баррелей 03230 744,0 17,7 15,0
Общее кислотное число, мг- КОН/г 0664 0,48 0,34 0,30
Общий азот, мг/кг 04629 5650 4761 3200
Основный азот, мг/кг t/ОРЗ 13 1275 1779 748
Компоненты, нерастворимые в«-С7, %масс. 03279 25,06 18,03 11,32
Компоненты, нерастворимые в толуоле, %масс. 04055 0,41 0,20 0,11
Металлы, мг/кг: никель ванадий Атомно-аб- сорбционный То же 94,2 494,0 83,4 445,0 53,4 298,1
Хлориды, мг/кг 0808 86 10 4
В табл. 1.10 приведены данные о свойствах различных фракций, полученных ат-
мосферной и вакуумной перегонкой из нефти плотностью 13 ° API. На рис. 1.12 пред-
ставлены кривые ИТК, плотности по шкале API и содержания серы, построенные
для трех проб нефти, перечисленных в табл. 1.9. Более тяжелые нефти дают меньший
выход дистиллятов, а остатки их разгонки имеют меньшую плотность по шкале API
и содержат больше серы.
Таблица 1.10
Свойства дистиллятов, полученных разгонкой нефти с плотностью 13 'API
Свойства Метод Тяжелая нефть ЛПрБ СПрБ ТПрБ РТ Керосин пг лвг твг ВО
Интервал кипения, °F 45ГМР2892 НК-160 160-350 350-400 400-525 525-600 600-650 650-850 850-1000 > 1000
Интервал кипения, °C НК-71 71-177 177-204 204-274 274-316 316-343 343-454 454-538 >538
Отогнанный объем, % 100 1,45 7,20 2,25 6,50 4,73 3,39 13,98 12,14 48,36
Средний отогнанный объем,% 0,73 5,05 9,78 14,15 19,77 23,83 32,51 45,57 75,82
Отогнанная масса, % 100 0,99 5,54 1,84 5,55 4,24 3,09 13,20 11,98 53,57
Интервал отгона, %об. 0-1,45 1,45- 8,65 8,65— 10,9 10,9- 17,4 17,4— 22,13 22,13— 25,52 25,52— 39,50 39,50- 51,64 51,64-100
Средняя температура кипения, °C 129 185 237 287 316 400 484
Относительная плот- ность, 60/60 °F ASTMD1298 0,9801 0,6659 0,7542 0,8001 0,8371 0,8780 0,8945 0,9255 0,9669 1,0856
Плотность, °АР1 ASTMD1K1 12,87 80,99 56,12 45,35 37,54 29,66 26,69 21,39 14,84 -1,16
Кинематическая вяз- кость, сСт, при темпе- ратуре, °C: 25,0 37,8 54,4 98,9 121,1 135,0 150,0 ASTMDAA5 19 646 5102 1235 1,41 1,14 0,73 2,32 1,80 1,05 4,57 3,21 1,61 6,80 4,51 2,09 34,21 17,47 5,13 418,10 136,00 18,89 10 186 934 360 872 68 833 15 365
Коксуемость по Ко- нрадсону, %масс. ASTMDW) 17,94 35,61
Общая сера, %масс. ASTM £4294 5,35 0,0620 0,4115 1,5110 2,4430 3,6140 3,7840 4,4512 4,9622 6,7741
Глава 1. Тяжелая нефть
Таблица 1.10, окончание
Свойства Метод Тяжелая нефть ЛПрБ СПрБ ТПрБ РТ Керосин пг лвг твг ВО
Давление паров по Рейду, psi ASTMD323 4,9
Температура текуче- сти, °C ASTMD97 0 -66 -42 -18 -6 + 18 +39
Анилиновая точка, °C ASTMD6W 53,8 55 53,8 57,8 60,4 61,2
Общий азот, мг/кг ASTMD4629 4761 0,2 о,3 2 14 249 650 1145 2600 6848
Основный азот, мг/кг UOP3Y3 1779 0,1 1 8 186 292 496 745 246
Компоненты, нераство- римые в л-С5, %масс. ASTMD2077 20,93 40,21
Компоненты, нера- створимые в л-С7, %масс. ASTMD3279 18,03 34,37
Металлы, мг/кг: никель ванадий Ni +V ААС 83,4 445 528,4 149 792 941
Структурно-групповой анализ, %об.: ароматические угле- водороды олефиновые углево- дороды предельные углево- дороды А?7МЛ1319 Тоже » 8,40 0,50 91,10 15,50 0,60 83,90 21,90 1,00 77,10 34,10 1,20 64,70
Бензол, %об. 0,89 0,55
Анализ PIONA, %об.: парафины изопарафины ASTMD6730 То же 39,46 36,20 36,54 31,83
Часть 1. Свойства тяжелой нефти и технологии ее переработки
олефины нафтены ароматические угле- водороды неопределенные сое- динения всего, %об. » » » » 0,56 18,14 5,14 0,50 100,00 1,27 17,03 11,18 2,15 100,00
Показатель преломле- ния при 20 °C ASTM D1218 1,5153 1,5368
Дизельный индекс 60,2 49,4 38,7 37,3
Цетановый индекс 39,3 43,5 42,9 43,0
Высота некоптящего пламени, мм ASTMD1322 22 20,0 19,0 18,0
Фактор ароматичности 0,2427 0,3180 0,3988
Индекс корреляции Горного бюро 49,45 53,87 65,48
Вязкостно-весовая константа 0,8743 0,8860 0,9141
Разгонка, °C НК/5 %об. 10/20 %об. 30/40 %об. 50/60 %об. 70/80 %об. 90/95 %об. кк ИТК 29/132 192/297 382/458 529/- D86 £>86 61/87 94/105 113/122 130/137 145/152 161/168 180 £>86 158/175 177/179 180/182 183/185 187/190 194/201 206 £>86 214/222 224/227 229/232 235/238 242/247 254/258 267 £>86 273/280 281/281 282/284 285/287 288/291 295/298 304 £>86 305/309 311/312 313/315 316/317 319/320 323/325 340 />1160 340/360 367/373 380/386 394/403 414/426 442/456 481 />1160 262/393 433/456 468/478 488/498 508/520 534/547 567 />1160 435/-
Отгон, %об. 51,64 при 538 °C 98,3 98,0 98,6 98,9 98,6 99,0 99,0 1,5 при 522 °C
Глава 1. Тяжелая нефть
Сокращения'. ЛПрБ — легкий прямогонный бензин; СПрБ — средний прямогонный бензин; ТПрБ — тяжелый прямогонный бен-
зин; РТ — реактивное топливо; ПГ — прямогонный газойль; ЛВГ — легкий вакуумный газойль; ТВГ — тяжелый вакуумный газойль;
ВО — вакуумный остаток; ААС — атомно-абсорбционная спектроскопия.
СП
w
64
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Средний отогнанный объем, %
Рис. 1.12. Кривые ИТК, содержания серы и плотности по шкале API тяжелых нефтей
со следующей плотностью: (—) — 10 ° API; (—) — 13 ° API', (- -) — 21 ° API
Чтобы быть уверенным в правильности данных экспериментальной разгонки, а также
в характеристике нефти и ее фракций, необходимо проверить баланс. Масса пробы
нефти должна быть равна сумме масс всех дистиллятов, а масса серы в нефти — сумме
масс серы во всех дистиллятах; это же требование должно выполняться для остальных
составляющих. На рис. 1.13 показан общий баланс масс и баланс серы при разгонке
пробы нефти с плотностью 13 ° API. Полученные расхождения невелики и вполне допу-
стимы; они объясняются погрешностью измерений при разгонке и анализе.
лава 1. Тяжелая нефть
Общий баланс Баланс серы
Фракция Начальный отогнанный объем, % Конечный отогнанный объем, % %об. Средний отогнанный объем, % Удельная масса (плотность (абс.)), г/мл Масса, г %масс. %масс. Масса, г
Проба нефти плотностью 13 °ЛР/
Разгонка —* ЛПрБ —” СПрБ —•• ТПрБ —‘ РТ *Керосин —” ПГ —’ ЛВГ —” ТВГ - ВО 0,00 1,45 1,45 0,73 0,6659 0,966 0,99 0,062 0,001
Масса, г 1,45 8,65 7,20 5,05 0,7542 5,430 5,54 0,412 0,022
Объем, мл Относительная плотность при 60 "F/60 °F Масса, г Общая сера, %масс. 100 8,65 10,90 2,25 9,78 0,8001 1,800 1,84 1,511 0,027
0,9801 10,90 17,40 6,50 14,15 0,8371 5,441 5,55 2,443 0,133
17,40 22,13 4,73 19,77 0,8780 4,153 4,24 3,614 0,150
98,01
22,13 25,52 3,39 23,83 0,8945 3,032 3,09 3,784 0,115
5,35 5,244 25,52 39,50 13,98 32,51 0,9255 12,938 13,20 4,451 0,576
39,50 51,64 12,14 45,57 0,9669 11,738 11,98 4,962 0,582
51,64 100,00 48,36 75,82 1,0856 52,500 53,57 6,774 3,556
Всего: 100,00 0,9800 97,999 100,00 5,163
По нефти: Расхождение, %: 98,010 5,244
0,011 1,542
Рис. 1.13. Общий баланс масс и баланс серы при разгонке нефти с плотностью 13 ° API
(сокращенные обозначения фракций см. в табл. 1.10)
66
Часть 1. Свойства тяжелой нефти и технологии ее переработки
1.5. ПРОБЛЕМЫ ПРИ ПЕРЕРАБОТКЕ ТЯЖЕЛОЙ НЕФТИ
Как уже говорилось выше, в тяжелых нефтях высока концентрация гетероатомных
соединений, что влияет на все аспекты переработки таких нефтей. Как правило,
в наибольших концентрациях содержится сера, которую довольно легко удалить; су-
ществует множество катализаторов, позволяющих удалять до 90 % и более всей серы.
Азот удалить труднее, чем серу, а номенклатура катализаторов, предназначенных для
его удаления, не столь широка. Если азот и серу не удалять, есть вероятность выбро-
сов вредных оксидов азота (NOx) и серы (SO*) при переработке нефти и потреблении
нефтепродуктов.
Нефти большинства сортов содержат металлы (больше всего никеля и ванадия).
Тяжелые нефти содержат их в повышенных концентрациях в виде солей или металло-
органических соединений (металлопорфиринов), которые чрезвычайно трудно уда-
лить из сырья. Практически весь металл исходной нефти концентрируется в остатке
перегонки. Исключение составляют соединения металлов, которые в условиях пере-
гонки могут оказаться летучими и перейти в высококипящие дистилляты. Металлы
вызывают особые затруднения, поскольку они отравляют катализаторы процессов
удаления серы и азота, а также других процессов (например, каталитического кре-
кинга). В связи с этим ведется активная работа по разработке катализаторов, способ-
ных выдерживать высокие концентрации металлов без существенного ухудшения ак-
тивности или сокращения срока службы.
Большинство высокомолекулярных компонентов тяжелой нефти в пластовых ус-
ловиях остаются растворенными в жидкой фазе, но на определенных стадиях перера-
ботки могут выпадать в виде твердой фазы, в том числе в виде асфальтенов.
В каталитических процессах эти твердые вещества отлагаются на поверхности ка-
тализатора и снижают его активность, в конечном счете закупоривая поры. В тяже-
лых и очень тяжелых нефтях высокомолекулярные компоненты содержатся в значи-
тельных количествах, что увеличивает вязкость нефти. Общеизвестно, что тяжелые
компоненты — основная причина проблем, возникающих при хранении и транспор-
тировке нефти. Поэтому важно понять, как тяжелые компоненты нефти влияют на ее
свойства, а также на образование и отложение твердых веществ.
Признано, что проблемы переработки тяжелого сырья можно связать с химиче-
скими свойствами, а также количеством сложных и высококипящих компонентов
сырья. Переработка тяжелой нефти — задача, требующая досконального знания хи-
мической структуры и химического поведения этого сырья, куда более сложного,
чем обычная нефть. Асфальтены, будучи наиболее сложными соединениями нефти,
представляют в то же время и наиболее трудную для переработки ее часть.
Из проблем, встречающихся в процессах гидропереработки тяжелых нефтей
с большим содержанием асфальтенов, можно особо выделить следующие:
• осаждение на поверхности катализатора;
• закупорка устьев пор катализатора;
• действие в качестве предшественников кокса, что завершается потерей актив-
ности катализатора;
• ограничение глубины превращения вследствие образования осадка.
Глава 1. Тяжелая нефть
67
Так как основная часть металлов (главным образом это ванадий и никель) тяжелых
нефтей концентрируется в асфальтенах, удаление этой высокомолекулярной состав-
ляющей приведет к уменьшению количества металлов в сырье других процессов, на-
пример гидрокаталитических.
В гидрокаталитических процессах кокс образуется очень быстро — в течение не-
скольких часов. Затем потеря активности катализатора быстро достигает псевдорав-
новесного состояния, так как металлы сырья переходят в сульфиды, отлагающиеся
в порах и вызывающие необратимую потерю активности. Деактивация катализатора
металлами протекает дольше. По данным литературных источников, масса образую-
щегося кокса достигает 25 % от исходной массы катализатора, из-за чего происхо-
дит значительное уменьшение (50-60 %масс.) активной поверхности катализатора.
Нарастание отложений металлов, главным образом ванадия, происходит до полной
закупорки устьев пор. При переработке сырья с низким содержанием металлов по-
сле сравнительно быстрой начальной потери активности из-за отложения кокса на-
ступает период более медленной деактивации, обусловленной отложением металлов.
Но в случае сырья с высоким содержанием металлов деактивация после начального
отложения кокса происходит быстрее. С началом закупорки пор падение активности
ускоряется, и в конечном счете катализатор достигает состояния, требующего прекра-
щения цикла процесса переработки. По этой причине рабочую температуру процесса
в течение цикла постоянно повышают. Кроме того, повышение температуры компен-
сирует потери активности катализатора. Конечно, рано или поздно температура до-
стигает такого уровня, когда ее повышением уже становится невозможно компенси-
ровать падение активности, и цикл приходится прерывать. Это хорошо иллюстрирует
рис. 1.14 на примере различных видов сырья. Видно, что срок службы катализатора
при переработке тяжелой нефти короче, а температуру приходится повышать быстрее.
Минусы:
• повышенное содержание металлов;
• пониженная активность катализатора;
• сокращенный срок службы катализатора;
• повышенная необходимая температура
в реакторе
Срок службы катализатора, годы
Рис. 1.14. Повышение необходимой температуры в реакторе и срок службы
катализатора при переработке нефтяного сырья:
(о) — температура в начале цикла; (•) — температура в конце цикла
68
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Основная причина потери активности катализатора — уменьшение числа актив-
ных центров, вызываемое главным образом их отравлением сильно адсорбируемыми
веществами, перекрытием центров отложениями кокса и металлов, сужением и за-
купоркой устьев пор и спеканием активной фазы. В процессах гидрокаталитической
переработки нефтяных фракций выявлены три стадии потери активности:
• деактивация коксом, который образуется при переработке практически любого
сырья (в случае тяжелой нефти главный источник кокса — асфальтены);
• необратимая деактивация металлами, скорость которой зависит от содержания
металлов в сырье;
• сужение и закупорка пор, характеризующиеся очень сильной потерей активно-
сти, после чего цикл работы установки приходится прерывать.
Очевидно, что наиболее важны две первые стадии, так как их длительность опре-
деляет срок службы катализатора.
Характер образования отложений кокса и металлов, которые обычно распределе-
ны между структурами порфириновых и других типов, зависит от происхождения сы-
рья. Таким образом, потеря активности катализатора при переработке разного сырья
характеризуется своими особенностями.
ЛИТЕРАТУРА
Acevedo, S., Cordero, J.M., Carrier, Н., Bouyssuere, В., Lobinski, R. 2009. Trapping of paraffin and other
compounds by asphaltenes detected by laser desorption ionization-time of flight mass spectrometry (LDI-
TOF MS): Role ofAl and A2 asphaltene fractions in this trapping. Energy Fuels 23:842—848.
Ancheyta, J., Centeno, G., Trejo, F, Marroquin, G., Garcia, J.A. 2002. Extraction and characterization
of asphaltenes from different crude oils and solvents. Energy Fuels 16:1121-1127.
Ancheyta, J., Speight, J.G. 2007. Hydroprocessing of Heavy Oil and Residua, CRC Press, Taylor & Francis
Group, NewYrrk.
Ancheyta, J., Trejo, E, Rana, M.S. 2010. Asphaltenes: Chemical Transformations during Hydroprocessing
of Heavy Oils, CRC Press, Taylor & Francis Group, New York.
Andersen, S.I. 1994. Dissolution of solid boscan asphaltenes in mixed solvents. Fuel Sci. Technol. Int.
12(11—12):1551—1577.
Asomaning, S. 2003. Test method for determining asphaltene stability in crude oils. Petrol. Sci. Technol.
21:581-590.
Beal, C. 1946. Viscosity of air, water, natural gases, crude oil and its associated gases at oil field
temperature and pressures. Trans. AIME (Am. Inst. Min. Metall. Petrol. Eng.) 165:114—127.
Beggs, H.D., Robinson, J.R. 1975. Estimating the viscosity of crude oil systems. J. Petrol. Technol.
27:1140-1141.
Berrueco, C., Afenditti, S., Morgan, TJ., Alvarez, P, Millan, M., Herod, A.A., Kandiyoti, R. 2008.
Calibration of size-exclusion chromatography columns with 1 -methyl-2-pyrrolidinone (NMP)/chloroform
mixtures as eluent: Applications to petroleum-derived samples. Energy Fuels 22:3265—3274.
de Boer, R.B., Leeriooyer, K.., Eigner, M.R.P, van Bergen, A.R.D. 1995. Screening of crude oils for
asphalt precipitation: Theory, practice, and the selection of inhibitors. SPE Prod. Facil. 10(l):55—61.
Centeno, G., Sanchez-Reyna, G., Ancheyta, J., Munoz, J.A.D., Cardona, N. 2011. Testing various
mixing rules for calculation of viscosity of petroleum blends. Fuel 90:3561-3570. Centeno, G., Trejo, E,
Ancheyta, J., Carlos, A. 2004. Precipitacion de asfaltenos del crudo maya en un sistema a presion. Rev. Soc.
Quim. Mex. 48:186-195.
Глава 1. Тяжелая нефть
69
De Ghetto, G., Paone, E, Villa, M. 1995. Pressure—volume—temperature correlations for heavy and
extra heavy oils. In: 1995 SPE International Heavy Oil Symposium, Calgary, Alberta, Canada, June 19—21,
SPE 30316.
Demirbas, A. 2002. Physical and chemical characterizations of asphaltenes from different sources. Petrol.
Sci. Technol. 20(5—6):485—495.
Elsharkawy, A.M., Alikhan, A. A. 1999. Models for predicting the viscosity of middle east crude oils. Fuel
78:891-903.
Glaso, O. 1980. Generalized pressure—volume—temperature correlation for crude oil system. J. Petrol.
Technol. 2:785-795.
Hedrick, B.W, Seibert, K.D., Crewe, C. 2006. A New Approach to Heavy Oil and Bitumen Upgrading,
UOP LLC, Meta Petroleum, Bogota, Colombia.
Heithaus, J.J. 1962. Measurement and significance of asphaltene peptization. J. Inst. Petrol. 48:45-53.
Kartoatmodjo, E, Schmidt, Z. 1994. Large data bank improves crude physical property correlation. Oil
Gas J. 4:51-55.
Labedi, R. 1992. Improved correlations for predicting the viscosity of light crudes. J. Petrol. Sci. Eng.
8:221-234.
Leyva, C., Ancheyta, J., Berrueco, C., Milkin, M. 2012. Chemical characterization of asphaltenes from
various crude oils. Fuel Proc. lech, (submitted).
Li, C.-Z., Wu, E, Xu, B., Kandiyoti, R. 1995. Characterization of successive time/temperature-resolved
liquefaction extract fractions released from coal in a flowing-solvent reactor. Fuel 74:37—45.
Marcano, E, Flores, R., Chirinos, J., Ranaudo, M.A. 2011. Distribution of Ni and V in Al andA2
asphaltene fractions in stable and unstable Venezuelan crude oils. Energy Fuels 25:2137-2141.
Matsushita, K., Marafi, A., Hauser, A., Stanislaus, A. 2004. Relation between relative solubi-
lity of asphaltenes in the product oil and coke deposition in residue hydroprocessing. Fuel 83(11 — 12):
1669-1674.
Merdrignac, L, Espinat, D. 2007. Physicochemical characterization of petroleum fractions: The state of
the art. OilGasSci. Technol. Rev. /F7>62(1):7—32.
Miller, J.T, Fisher, R.B., Thiyagarajan, P, Winans, R.E., Hunt, J.E. 1998. Subfractionation and
characterization of mayan asphaltenes. Energy Fuels 12:1290-1298.
Morgan, T, George, A., Alvarez, R, Herod, A.A., Millan, M., Kandiyoti, R. 2009. Isolation of size
exclusion chromatography elution-fractions of coal and petroleum-derived samples and analysis by laser
desorption mass spectrometry. Energy Fuels 23:6003—6014.
Murgich, J., Rodriguez, J., Aray, Y 1996. Molecular recognition and molecular mechanics of micelles of
some model asphaltenes and resins. Energy Fuels 10( 1):68—76.
Reynolds, J.G. 1990. Trace metals in heavy crude oil and tar and bitumens. In: Eastern Oils Shale
Symposium, Lexington, K\ November 6-8.
Riazi, M.R. 2004. Characterization and Properties of Petroleum Fractions, ASTM stock book number
MNL50, Philadelphia, PA.
Schmidt, V 2010. Defining heavy oil. World oil. FindArticles.com, October 04, 2010. http://findarticles.
com/p/articles/mi_m3159/is_l 2_226/ai_nl 5979981 /
Sepulveda, J.A., Bonilla, J.R, Medina, Y 2010. Stability prediction for asphaltenes using SARA
analysis for pure petroleum. Rev. Ing. Reg. 7:103-110.
Sharma, B.K., Sharma, C.D., Bhagat, S.D., Erhan, S.Z. 2007. Maltenes and asphaltenes of petroleum
vacuum residues: Physico-chemical characterization. Petrol. Sci. Technol. 25(1—2):93—104.
Sheremata, J.M., Gray, M.R., Dettman, H.D., McCaffrey, WC. 2004. Quantitative molecular
representation and sequential optimization of athabasca asphaltenes. Energy Fuels 18(5): 1377-1384.
Speight, J.G. 1999. The Chemistry and Technology of Petroleum, 3rdedn., Marcel Dekker, Inc., New
York.
Speight, J.G. 2001. Handbook of Petroleum Analysis, John Wiley & Sons, New Yark. Stankiewicz, A.B.,
Flannery M.D., Fuex, N.Q., Broze, G., Couch, J.L., Dubey, S.T, Iyer, S.D. 2002. Prediction of asphaltene
70 Часть 1. Свойства тяжелой нефти и технологии ее переработки
deposition risk in Е&Р operations. In: Proceedings of 3rd International Symposium on Mechanisms and
Mitigation of fouling in Petroleum and Natural Gas Production, AIChE 2002 Spring National Meeting, New
Orleans, LA, March 10—14, paper 47C, pp. 410—416.
Suelves, 1., Islas, C.A., Millan, M., Galmes, C., Carter, J.E, Herod, A.A., Kandiyoti, R. 2003.
Chromatographic separations enabling the structural characterisation of heavy petroleum residues.
Fuel 2:1-14.
Trejo, E, Ancheyta, J., Morgan, TJ., Herod, A.A., Kandiyoti, R. 2007. Characterization of asphaltenes
from hydrotreated products by SEC, LDMS, MALDI, NMR, and XRD. Energy Fuels 21:2121—2128.
Zhao, S., Kotlyar, L.S., Wtods, J.R., Sparks, B.D., Hardacre, K., Chung, K.H. 2001. Molecular
transformation of athabasca bitumen end-cuts during coking and hydrocracking. 80(8): 1155— 1163.
Глава 2. Технологии переработки
тяжелой нефти
Здесь рассматриваются основные процессы переработки тяжелых нефтей, описан-
ные в литературных источниках, т. е. процессы, основанные на обеднении углеродом
или обогащении водородом. Затрагиваются также характеристики новых техноло-
гий, имеющие отношение к тяжелой нефти. Рассматриваются достоинства и недо-
статки комбинированных процессов.
2.1. ОБЩАЯ КЛАССИФИКАЦИЯ ПРОЦЕССОВ
Низкая плотность по шкале API тяжелых нефтей объясняется меньшим соотно-
шением водорода и углерода (Н/С), что явствует из рис. 2.1. Таким образом, для
облагораживания тяжелой нефти следует увеличить соотношение Н/С, что мож-
но сделать двумя способами: увеличением числа атомов водорода (обогащение во-
дородом) либо уменьшением числа атомов углерода (обеднение углеродом). Оба
этих способа могут быть как каталитическими, так и некаталитическими. В чис-
ло некаталитических процессов обеднения углеродом входят экстрактивная де-
асфальтизация, газификация, коксование и висбрекинг. К каталитическим про-
цессам относится каталитический крекинг остатков. Из числа некаталитических
процессов обогащения водородом можно упомянуть гидровисбрекинг; к каталити-
ческим принадлежат гидроочистка и гидрокрекинг. Эту классификацию иллюстри-
рует рис. 2.2. Применительно к переработке тяжелых нефтей как процессы обога-
щения водородом, так и процессы обеднения углеродом имеют свои недостатки
[Rana et al., 2007].
Общепризнано, что сырье с низким содержанием металлов лучше всего перераба-
тывать посредством процессов каталитического крекинга остатков в псевдоожижен-
ном слое (RFCC). Для сырья с высоким содержанием металлов рекомендуются тех-
нологии обогащения водородом и обеднения углеродом. В случае тяжелой и очень
тяжелой нефти выбор между коксованием и гидропереработкой определяется тех-
нико-экономическим обоснованием и обуславливается не только производитель-
ностью процесса, но и влиянием качества полученных дистиллятов на последую-
щие процессы очистки. К примеру, дистилляты, получаемые в процессах коксования
72
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Плотность, °АР1
Рис. 2.1. Зависимость между атомным соотношением Н/С
и плотностью по шкале API тяжелых нефтей
(бензин и газойль коксования), требуют дальнейшей гидроочистки, тогда как дис-
тилляты гидрокаталитических процессов, в зависимости от требований специфика-
ций на конечные продукты, в такой доочистке могут не нуждаться. В любом случае
выбор технологии, пригодной для облагораживания нефти конкретного сорта, не яв-
ляется простой задачей и должен делаться с учетом нескольких факторов. К важней-
шим из них относятся [Ancheyta and Speight, 2007]:
• цена нефти;
’ уровень содержания примесей в сырье;
• цель облагораживания нефти;
• поточная схема завода, на который будет направляться облагороженное
сырье.
Существует несколько общих критериев, помогающих выбрать технологию, наи-
более подходящую для переработки конкретной тяжелой нефти; некоторые из них
представлены на рис. 2.3. Чаще всего ориентируются по двум критериям: количеству
металлов и коксуемости атмосферного остатка (выкипающего выше 343 °C), получа-
емого из тяжелой нефти [Houde and McGrath, 2006].
Как правило, тяжелые и очень тяжелые нефти характеризуются следующими
особенностями:
• высоким содержанием серы, металлов и прекурсоров кокса;
• низкой ценой;
• сложностью и высокой себестоимостью переработки.
Глава 2. Технологии переработки тяжелой нефти
73
Технологии
обогащения
углеродом
Технологии
обеднения
углеродом
Гидровисбрекинг
(некаталитический)
Экстрактивная
деасфальтизация
(ЭДА)
RFCC
(обеднение
углеродом)
Газификация
Каталитические технологии
Гидроочистка
в неподвижном
слое
Гидроочистка
в подвижном
слое
Висбрекинг
Замедленное
коксование
Гидрокрекинг
в кипящем слое
Флюидкокинг
Гидрокрекинг
в суспензионном
реакторе
Флексикокинг
Рис. 2.2. Технологии обогащения водородом и обеднения углеродом,
применяемые для облагораживания тяжелого нефтяного сырья
74
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Общее содержание Ni и V, мг/кг
Рис. 2.3. Общие критерии выбора технологий облагораживания тяжелых нефтей исходя
из свойств атмосферного остатка: (□) — содержание серы;
(•) — коксуемость; (Д) — плотность, °АР1
Выбрать технологию, лучше остальных подходящую для облагораживания тяже-
лых и очень тяжелых нефтей, нелегко. В прошлом сырье с высоким содержанием ме-
таллов традиционно облагораживали технологиями обеднения углеродом, чаще всего
путем замедленного коксования. Однако было установлено [Ancheyta et al., 2010; At-
kins et al., 2010], что переработка очень тяжелой нефти методом коксования влечет
за собой следующие негативные последствия;
• низкий выход облагороженного сырья;
• высокий выход кокса;
• высокое содержание ароматических углеводородов в продуктах облагора-
живания;
• снижение выхода и качества бензина и дизельного топлива при последующей
переработке;
• низкая ценность облагороженного сырья.
В целях охвата таких «проблемных» нефтей область процессов гидропереработки
расширили, но не так, как при традиционной гидропереработке с жесткими условия-
ми реакций и большой глубиной превращения. В результате возник вариант процес-
сов гидроочистки с умеренной глубиной.
Из-за интенсивного образования осадка глубина превращения в современ-
ных процессах гидропереработки тяжелых и очень тяжелых нефтей при высоком
Глава 2, Технологии переработки тяжелой нефти
75
парциальном давлении водорода ограничена величиной 50 % (рис. 2.4). Такое огра-
ничение и высокие капитальные и эксплуатационные затраты делают эти процес-
сы непривлекательными для переработки тяжелых и очень тяжелых нефтей. Они
целесообразны при большой глубине превращения, но для этого склонность сы-
рья к образованию осадка должна быть низкой. Такому требованию отвечают лишь
обычные нефти (с низким содержанием асфальтенов и других примесей). Для пере-
работки проблемных нефтей можно применять процессы гидропереработки с уме-
ренной глубиной превращения. Переработка с умеренной глубиной превращения
(менее 50 %) означает, что нефть частично облагораживается — так, что становится
возможным транспортировать и перерабатывать ее на обычных нефтеперерабатыва-
ющих заводах, а также сбывать по более высокой цене.
Глубина превращения остатка выше 1000 °F (538 °C), %об.
Рис. 2.4. Ограничения глубины превращения при гидропереработке различного
сырья, вызываемые образованием осадка
2.2. ТЕКУЩЕЕ ПОЛОЖЕНИЕ В ОБЛАСТИ ПЕРЕРАБОТКИ
ТЯЖЕЛОГО СЫРЬЯ
В табл. 2.1 представлены итоговые данные по объемам облагораживания тяжелых
нефтей. Преобладают некаталитические технологии, особенно коксование, на ко-
торые приходится в общей сложности 59,34 % мирового объема переработки. Не-
фтеперерабатывающие заводы продолжают предпочитать технологии коксования,
главным образом из-за сравнительно низких капитальных затрат и несложности осу-
ществления. Лишь японские заводы уделяют особое внимание технологиям гидро-
очистки в неподвижном слое, но они перерабатывают сырье с относительно невысо-
кой концентрацией примесей [Liu et al., 2009].
76
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Таблица 2. ]
Мировые мощности по облагораживанию остаточного сырья, тыс. баррель/сут
Технология США Евро- па Канада, Мексика и Венесуэла Япония Осталь- ные страны Всего в мире Доля в общем объеме, %
Некаталитические процессы
Висбрекинг 44 2260 331 24 1635 4293 25,85
Коксование 2245 673 951 66 1169 5104 30,73
Деасфальтизация 283 46 39 16 75 458 2,76
Всего 2572 2979 1321 106 2879 9855 59,34
Каталитические процессы
Гидроочистка в неподвиж- ном слое 499 149 30 591 1042 2312 13,92
Гидрокрекинг в кипящем слое 102 79 244 23 49 497 2,99
Гидрокрекинг в фазе суспен- зии — — 4 — — 4 0,02
RFCC 831 681 281 318 1832 3942 23,73
Всего 1432 909 559 932 2923 6755 40,66
Всего 4004 3888 1880 1038 5802 16 610 100
Давая продукты сравнительно высокого качества и ценности, технологии обога-
щения водородом с ростом цен на нефть станут еще прибыльнее, что приведет к уве-
личению соответствующих мировых производственных мощностей. Кроме того,
технологии облагораживания тяжелых и очень тяжелых нефтей методом обеднения
углеродом теряют привлекательность. В результате хорошей альтернативой им станут
процессы гидроочистки с умеренной глубиной превращения.
2.3. ТЕХНОЛОГИИ ОБОГАЩЕНИЯ ВОДОРОДОМ
Технологии обогащения водородом характеризуются тем, что [Rana et al., 2007]:
• посредством механизма гидрокрекинга и гидрирования увеличивают выход
жидких продуктов за счет снижения выхода кокса;
• увеличивают отношение Н/С в перерабатываемом сырье;
• при переработке тяжелых и очень тяжелых нефтей привлекательнее других
технологий;
• нуждаются в водороде под высоким давлением и с высокой температурой, так
как в тяжелых нефтях высока концентрация углерода;
• проводятся, за исключением гидровисбрекинга, в присутствии катализаторов;
• снижают содержание серы, азота, металлов и асфальтенов и за счет гидрокре-
кинга увеличивают выход жидких топлив;
Глава 2. Технологии переработки тяжелой нефти
77
• сталкиваются с определенными трудностями, вызванными ускоренным отло-
жением металлов и кокса на катализаторе;
• различаются главным образом типами катализаторов и реакторов, а также ра-
бочими условиями;
• проводятся в реакторах с неподвижным, подвижным, кипящим и суспендиро-
ванным слоем катализатора;
• потребляют больше водорода, зато дают больше жидких продуктов;
• рассчитаны большей частью на получение сырья для последующих процессов;
• нуждаются в катализаторах, способных выдерживать высокие концентрации
металлов и асфальтенов в тяжелом сырье.
Каталитическая система, выбираемая из соображений активности, избирательно-
сти и срока службы, состоит, как правило, из кобальт-молибденовых и никель-мо-
либденовых катализаторов на алюмооксидном носителе, рассчитанных на решение
определенных задач. В число последних входят гидрообессеривание, гидродеметал-
лизация, гидродеазотирование, гидродеасфальтизация, гидрокрекинг и снижение
коксуемости по Конрадсону.
2.3.1. Гидровисбрекинг
1. Процесс HYCAR) Некаталитический процесс, основанный на висбрекинге
и предусматривающий обработку водородом в нежестких условиях. Проводится по-
следовательно в трех реакторах: висбрекинга, гидродеметаллизации и гидрокрекинга.
• Реактор висбрекинга. Умеренный термический крекинг в присутствии водоро-
да, дающий более стабильные продукты, чем обычный висбрекинг. Прежде чем
начнется закоксовывание змеевика, можно достигнуть большей глубины пре-
вращения и получить менее вязкие продукты.
• Реактор гидродеметаллизации. Металлы удаляются в присутствии катализатора
с поровым объемом, достаточным для диффузии и адсорбции высокомолеку-
лярных соединений.
• Реактор гидрокрекинга. В нем удаляются сера и азот и одновременно осущест-
вляется гидрокрекинг.
2. Процесс Aqua-conversion. Каталитический процесс с переносом водорода от воды
в фазе суспензии. Гомогенный катализатор в присутствии водяного пара контактиру-
ет с тяжелым сырьем, способствуя переносу к его молекулам водорода воды. Про-
цесс проводится в реакционной системе обычной установки висбрекинга, состоящей
из змеевика и реакционной камеры. Реакции образования кокса подавляются, ас-
фальтеновые вещества не отделяются [Houde et al., 1998].
2.3.2. Гидропереработка в неподвижном слое
1. Процессы RDS и VRDS. Процесс RDS применяется для гидроочистки остатка ат-
мосферной перегонки, процесс VRDS — для гидрообессеривания вакуумного остат-
ка. Сырье контактирует с катализатором и водородом при умеренных температурах
78
Часть 1. Свойства тяжелой нефти и технологии ее переработки
и давлениях, расход водорода составляет около 700-1300 ст. куб. футов на баррель
сырья [Lars et al., 1984; Otterstedt et al., 1986].
2. Процесс Hyvahl-F. Применяется для гидроочистки атмосферных и вакуумных
остатков и превращения их в более ценные продукты (бензиновую фракцию и сред-
ние дистилляты). Проводится в реакторе с неподвижным слоем катализатора. Пер-
вый (входной) катализатор устойчив к загрязнению, закоксовыванию и закупорке
асфальтеновыми компонентами, имеет высокую металлоемкость и выполняет ги-
дродеметаллизацию и большую часть превращения. Второй, высокоактивный ката-
лизатор, защищенный таким образом от отравления металлами и закоксовывания,
осуществляет глубокое гидрообессеривание и очистку и завершает гидрокрекинг
[Kressmannetal., 1998, 2000].
3. Процесс Hyvahl-S. Используется два переключаемых защитных реакторов с не-
подвижным слоем и простым внутренним устройством. Когда активность катали-
затора в защитном реакторе становится ниже приемлемой, процесс быстро пере-
ключается на второй реактор со свежим катализатором. Главная особенность этого
процесса — переключаемые реакторы с малым временем контакта с неподвижным
слоем катализатора, работающие при высокой температуре и высоком парциальном
давлении водорода [Morrison et al., 1994].
4. Процессы гидрокрекинга остатков (HCR). Применяются катализаторы с двойной
функциональностью — крекинга и гидрирования. Технологическая схема типичной
установки гидрокрекинга остатков в неподвижном слое включает в себя два реактора:
• реактор первой ступени содержит высокоактивный катализатор гидроочистки
(никель-молибденовый) для удаления металлов и гетеросоединений;
• реактор второй ступени содержит катализатор крекинга на кислой подложке
(цеолиты, смешанные оксиды).
Примеры — одно- и двухступенчатый процессы гидрокрекинга Института неф-
ти Франции (IFP), процесс Isocracking, процесс мягкого гидрокрекинга (MRH), про-
цессы APCU и HyCycle Unicracking из семейства процессов Unicracking компании UOP,
2.3.3. Гидропереработка в движущемся слое
1. Процесс HYCON. Применяется для улучшения качества нефтяных остатков пу-
тем удаления серы, металлов и асфальтенов. Фактически проводится в режиме не-
подвижного слоя, но по мере увеличения содержания металлов в сырье подключа-
ются в качестве входных один или несколько «бункерных» реакторов с движущемся
слоем, осуществляющих гидродеметаллизацию. Есть возможность быстрой замены
(частичного удаления или добавления) катализатора посредством шлюзовых бунке-
ров без прерывания работы установки. Достоинства реакторов с неподвижным сло-
ем, работающих в пробковом режиме, сочетаются с удобством замены катализатора.
Это обеспечивает высокую гибкость процесса при подготовке сырья для реакто-
ров обессеривания, в особенности с высоким содержанием металлов. Рабочие ус-
ловия и скорость добавления и отбора катализатора можно регулировать так, что-
бы отбирался полностью отработанный катализатор, а в реакторе поддерживался
Глава 2. Технологии переработки тяжелой нефти
79
приемлемый средний уровень каталитической активности [Scheffer et al., 1998; Yu-
andong et al., 2009].
2. Процесс OCR. Представляет собой гидропереработку тяжелых нефтей и остат-
ков с высоким содержанием металлов в противоточном режиме в реакторе с подвиж-
ным слоем при высоких температурах и давлениях. Катализатор движется в реакторе
сверху вниз, сырье — снизу вверх. В противоточном режиме часть сырья с наивыс-
шим содержанием примесей контактирует сначала с наименее активной частью ка-
тализатора. Добавление свежего и отбор отработанного катализаторов осуществля-
ются периодически без прерывания работы установки [Chevron Lummus, 2010, 2011].
3. Процесс Hyvahl-M. Проводится в противоточных реакторах с подвижным сло-
ем и рекомендуется для переработки сырья с высоким содержанием металлов и ас-
фальтенов. Как и процесс OCR, нуждается в специальном оборудовании и техноло-
гиях для осуществления безопасной и эффективной подачи и отбора катализатора
из реакторов высокого давления. Катализатор, находящийся под атмосферным дав-
лением, подается в реактор с высоким парциальным давлением водорода. Затем от-
работанный катализатор отбирается из сосуда с высоким давлением и переносится
в среду с атмосферным давлением [Kressmann et al., 1998, 2000].
2.3.4. Гидропереработка в кипящем слое
1. Процесс П-Oil. Проводится в одно-, двух- или трехступенчатых реакторах
с кипящим слоем в широком интервале глубин превращения. Особенно хорошо при-
способлен для переработки тяжелых вакуумных остатков с высоким содержанием
металлов и коксовым остатком (коксуемостью) в дистиллятные продукты, а также
для обессеривания и деметаллизации сырья установок коксования и каталитическо-
го крекинга в псевдоожиженном слое — для получения нефтяного топлива с низким
содержанием серы и производства компонентов битума. Свойства продуктов под-
держиваются постоянными в течение всего цикла. Уникальный реактор смешения
с псевдоожиженным слоем катализатора, позволяющий проводить экзотермические
реакции и перерабатывать сырье с механическими примесями, гибко приспосабли-
вается к изменениям свойств сырья или целей переработки [Dickenson et al., 1998;
Kressmann et al., 1998, 2000].
2. Процесс Т-Star. Расширение процесса H-Oil. Способен обеспечивать общую
глубину превращения от 20 до 60 % и глубину гидрообессеривания 93-99 %. Может
использоваться для гидроочистки сырья каталитического крекинга в псевдоожижен-
ном слое или гидрокрекинга вакуумного газойля. Процесс можно проводить на ката-
лизаторе H-Oil. Реактор T-Star можно установить последовательно с реактором H-OU
для улучшения качества дистиллятных продуктов последнего. Процесс T-Star, про-
водимый в режиме мягкого гидрокрекинга на катализаторе, нечувствительном к со-
держанию серы и азота, обеспечивает постоянство выхода и качества продуктов при
неизменной глубине превращения до 60 % [Gragnani, 2011].
3. Процесс LC-Fining. Процесс гидрирования, используемый для гидрообессери-
вания, гидродеметаллизации и гидрокрекинга остатков атмосферной и вакуумной
80
Часть 1. Свойства тяжелой нефти и технологии ее переработки
перегонки. Хорошо подходит для гидроочистки очень тяжелого остаточного сырья,
асфальтов и вакуумных остатков; демонстрирует длительный цикл работы. Общие
достоинства процесса LC-Fining— малые капиталовложения, больший выход легких
фракций, низкие эксплуатационные затраты и меньшие потери водорода. Процесс
дает полный спектр высококачественных дистиллятов. Тяжелый остаток можно ис-
пользовать как нефтяное топливо или сырье для процессов каталитического крекин-
га в псевдоожиженном слое, коксования, висбрекинга или экстрактивной деасфаль-
тизации (ЭДА) [Daniel et al., 1998].
2.3.5. Гидропереработка в фазе суспензии
1. Процесс CANMET. Процесс гидрокрекинга тяжелых нефтей и остатков атмо-
сферной и вакуумной перегонки, первоначально разработанный для облагоражива-
ния тяжелой и битумной нефти, а также остатков перегонки. Вместо катализатора
используется добавка, подавляющая коксообразование и способствующая глубоко-
му превращению в низкокипящие продукты. Процесс проводится в трехфазном ре-
жиме в вертикальном одноступенчатом реакторе без внутренних устройств. Частицы
твердой добавки и жидкая углеводородная фаза образуют суспензию, через которую
в виде пузырьков всплывают водород и газообразные продукты. Отработанная до-
бавка остается в непревращенном вакуумном остатке. Типичные рабочие температу-
ры в реакторе составляют 440—460 °C, давление — 10—15 МПа [Silva et al., 1984; Lu-
nin et al., 1985; Pruden et al., 1993].
2. Процесс SOC (Super Oil Cracking). Реакции протекают в горизонтальном змее-
вике печи. Суспензия тонко размолотого катализатора промотирует крекинг тяже-
лого сырья. Процесс проводится при сравнительно коротком времени пребывания
в жестких условиях реакции — парциальном давлении водорода выше 20 МПа и тем-
пературе около 480 °C. Известная до настоящего времени максимальная производи-
тельность процесса составляет 3500 баррелей вакуумных остатков в сутки [Dicken-
son et al., 1996].
3. Процесс Microcat-RC. Гидрокаталитический процесс превращения в кипящем
слое, проводимый при сравнительно невысоких давлениях и температурах. Рассто-
яние между частицами катализатора, образующими однородную суспензию в сы-
рье, уменьшено до минимума, и молекулы реагентов быстрее находят активные ка-
талитические центры. В реактор подаются углеводородное сырье, микрокатализатор
и водород. Поток реактора поступает в испарительный сепаратор для извлечения во-
дорода, углеводородных газов и жидких продуктов. Остаток направляется в колон-
ну вакуумной перегонки, где разделяется на продукт с верхней границей кипения
565 °C и остаток выше 565 °C, содержащий непревращенное сырье, микрокатализа-
тор и практически все металлы исходного сырья [Bearden, 1997; Mart et al., 2005].
4. Процесс MRH. Процесс мягкого гидрокрекинга, предназначенный для перера-
ботки такого сырья с большим содержанием металлов и асфальтенов, как вакуумные
остатки и битумы, преимущественно в средние дистилляты. В реакторе поддерживает-
ся смешанная трехфазная суспензия сырья, тонкого порошка катализатора и водорода,
Глава 2. Технологии переработки тяжелой нефти
81
способствующая эффективному контакту. Суспензия катализатора в тяжелом сырье
подогревается в печи и подается в реактор. Из нижней секции реактора отбирается от-
работанная суспензия, состоящая из катализатора, некрекированного сырья и неболь-
шого количества вакуумного газойля. Газойль извлекают в суспензионном сепараторе,
а оставшиеся катализатор и кокс направляют в регенератор [Zhang et al., 2007].
5. Процессы VCC и HDH plus. Процесс гидрокрекинга и гидрирования для пре-
вращения остатков перегонки и другого тяжелого сырья. Технология VCC переросла
в HDH {Hydrocracking Distillation Hydrotreating), которая параллельно разрабатывалась
компанией INTEVEP с 1984 г. В настоящее время лицензируется лишь процесс HDH.
Недавно INTEVEP объявила о внедрении технологии HDHplus на двух нефтеперера-
батывающих заводах в Венесуэле. Тяжелое сырье, суспендированное небольшим ко-
личеством тонкого порошка добавки и смешанное с водородом и циркулирующим
газом, подвергается гидрированию (гидрокрекингу) на катализаторе в жидкофаз-
ном реакторе гидрирования, работающим при температуре 440-485 °C и давлении
15-30 МПа [Wenzel and Kretschmar, 1993].
6. Процесс EST (ENI Slurry Technology). Гидроочистка тяжелого сырья в фазе су-
спензии с диспергированным катализатором в присутствии водорода. Поток из ре-
актора подается в колонну фракционирования. Катализатор в составе нижнего про-
дукта колонны проходит через установку экстрактивной деасфальтизации и вместе
с асфальтенами возвращается в суспензионный реактор. На опытной установке про-
цесс продемонстрировал глубину превращения остатков 98-99 %, гидрообессерива-
ния — выше 80 %, гидродеметаллизации — выше 90 % и показал удаление более 96 %
коксового остатка. Часть сырья сразу превращается в легкие и средние дистилляты,
остальные продукты можно перерабатывать на установке гидрокрекинга или катали-
тического крекинга в псевдоожиженном слое. Сообщается о таких достоинствах про-
цесса EST, как гибкость по сырью, оптимальные показатели использования и потре-
бления водорода, гибкость номенклатуры продуктов [Rispoli, 2009; Sanfilippo, 2009].
7. Процесс НСАТ. Технология гидрокаталитического облагораживания тяжелых
нефтей, разработанная компанией HTIG {Headwaters Technology Innovations Group).
Процесс НСАТ перерабатывает низкокачественное сырье — тяжелую и битумную
нефть и остатки перегонки — в высококачественную синтетическую нефть. Процесс
протекает в присутствии жидкофазного нанокатализатора, частицы которого пред-
ставляют собой отдельные молекулы. Катализатор химическим путем вырабатывает-
ся в самом реакторе из прекурсора, вводимого вместе с сырьем, и обеспечивает высо-
кую степень превращения. Процесс существенно увеличивает глубину превращения
тяжелой нефти и уменьшает образование шлама. Недавно HTIG объявила о первом
в мире успешном промышленном внедрении технологии НСАТна заводе компании
Neste Oil Corporation в Порвоо, Финляндия [HTI, 2011].
2.4. ТЕХНОЛОГИИ ОБЕДНЕНИЯ УГЛЕРОДОМ
Ниже указаны некоторые главные особенности технологий обеднения углеродом
[Solari et al., 1997; Vartivarian and Andrawis, 2006].
82
Часть 1. Свойства тяжелой нефти и технологии ее переработки
• Суть этих процессов — удаление углерода в виде кокса с низким отношением
Н / С или в виде асфальта (в случае деасфальтизации) и получение среднего вы-
хода жидких продуктов.
• Технологии представляют собой важный и наиболее широко применяемый
в промышленных масштабах способ превращения остатков.
• Термический крекинг остаточного сырья, часто называемый коксованием,
проводится под сравнительно невысоким давлением.
• Процессы проводятся при температурах от 480 до 550 °C при времени пребыва-
ния в паровой фазе 20 с и более и сопровождаются существенным крекингом
и дегидрированием сырья, что затрудняет последующую переработку и дает
значительный выход малоценных продуктов в виде легких газов и кокса.
• Происходит перенос водорода от тяжелых молекул к более легким, в результате
чего образуется кокс (углерод). Остаточное сырье становится донором водоро-
да при высоких температурах.
• Процессы дают сравнительно большое количество таких газов, как метан, этан,
пропан, бутан, а также вторичных продуктов в виде СНГ, тощего газа и др.
• Существенным побочным продуктом является кокс, механизм образования
которого отличается от механизма образования других продуктов.
• Большинство проверенных временем процессов превращения тяжелого сырья
проводится при невысоком давлении в отсутствие катализатора.
• Процессы обеднения углеродом способны перерабатывать наиболее тяжелые
фракции нефти, давая кокс, в котором концентрируется большая часть серы,
азота и металлов исходного сырья.
2.4.1. Экстрактивная деасфальтизация
Процесс деасфальтизации растворителями — наиболее распространенный способ
осаждения асфальтенов. Остаточное сырье с помощью растворителя (легкие пара-
финовые углеводороды С3, С4, С5 и С7) разделяют на деасфальтизат (DAO) и битум
(асфальт) деасфальтизации, в котором концентрируется большая часть примесей ис-
ходного сырья. Деасфальтизат обычно служит сырьем установки гидрокрекинга или
каталитического крекинга в псевдоожиженном слое. На нефтеперерабатывающих
заводах этот процесс применяют для облагораживания тяжелых остатков перегонки
до деасфальтизата, который можно переработать в моторные топлива. Процесс мож-
но использовать также на нефтепромыслах для увеличения ценности нефти до от-
правки на нефтеперерабатывающие заводы [Billon et al., 1997; McGrath, 2008].
2.4.2. Газификация
Предусматривает полный крекинг остатков, в том числе асфальтенов, до газообразных
продуктов, проводимый при высоких температурах (выше 1000 °C) и дающий в каче-
стве основных продуктов синтез-газ (состоящий преимущественно из водорода, оки-
си углерода, углекислого газа и воды), техническую сажу и золу. Синтез-газ можно
Глава 2. Технологии переработки тяжелой нефти
83
переработать в водород или сжигать на теплоэлектростанциях в целях выработки де-
шевого электричества и пара для нефтеперерабатывающих заводов [Marano, 2003; Stie-
gel, 2005].
2.4.3. Коксование
1. Замедленное коксование. Полунепрерывный процесс термического крекинга, при-
меняемый на нефтеперерабатывающих заводах для облагораживания и превращения
остатков атмосферной и вакуумной перегонки в жидкие и газообразные продукты. В ре-
зультате остается твердый материал, обогащенный углеродом, — нефтяной кокс, цен-
ность которого зависит от таких свойств, как содержание серы, металлов и т. д. Про-
дуктами замедленного коксования являются жирный газ, бензиновая фракция, легкий
и тяжелый газойли и кокс. Кокс, который дает замедленное коксование, — почти чи-
стый углерод и используется как топливо или, в зависимости от качества, для производ-
ства электродов [Hamilton, 2002; Elliott, 2003; Haniford, 2003; Elliott and Wedlake, 2007].
2. Коксование в псевдоожиженном слое (флюидкокинг). Непрерывный процесс,
в котором концепция псевдоожижения материала, состоящего из твердых частиц,
используется для превращения сырья в более ценные продукты. Псевдоожижение
слоя позволяет проводить реакции коксования при более высоких температурах и за
более короткое время, чем при замедленном коксовании. Такие условия способству-
ют снижению выхода кокса и повышению выхода жидких продуктов [Parrish et al.,
1996; Furimsky, 2000; Kamienski et al., 2007].
3. Флексикокинг. Расширение процесса коксования в псевдоожиженном слое,
предусматривающее газификацию производимого кокса с получением синтез-газа.
Однако температура, при которой проводится процесс (1000 °C), недостаточна для
сжигания всего кокса [Furimsky, 2000; Kamienski et al., 2007].
2.4.4. Висбрекинг
Развитая технология, применимая к остаткам атмосферной и вакуумной перегонки
и даже к битуму деасфальтизации, снижающая вязкость путем термического разло-
жения. Термическое превращение остаточного сырья осуществляется нагреванием
его до высоких температур в печи специальной конструкции. Основные цели висб-
рекинга — снизить вязкость сырья, уменьшить количество остаточного нефтяного
топлива и увеличить долю средних дистиллятов в продукции, вырабатываемой заво-
дом [Phillips and McGrath, 1998; Shell, 2011].
2.4.5. Каталитический крекинг остаточного сырья
в псевдоожиженном слое
Процесс каталитического крекинга остаточного сырья в псевдоожиженном слое
(RFCC) — расширение традиционной технологии каталитического крекинга
84
Часть 1. Свойства тяжелой нефти и технологии ее переработки
в псевдоожиженном слое (FCC). Процесс был разработан в начале 1980-х гг. с це-
лью увеличить избирательность к бензину и снизить выход легких газов по сравне-
нию с процессами гидропереработки и термическими процессами. Он проводится
при температурах от 480 до 540 °C в реакторе, аналогичном используемому в процес-
се FCC (т. е. в реакторе, где слой катализатора приводится в псевдоожиженное со-
стояние), и рассчитан на переработку остаточного сырья с коксуемостью (коксовым
остатком) по Конрадсону, превышающим 4 %масс. Процесс требует сырья лучшего
качества, чем гидрокаталитические процессы (необходимы более высокое соотноше-
ние Н/С и низкое содержание металлов и асфальтенов), что делает его менее пред-
почтительным. (Высокое качество сырья позволяет избежать высокого выхода кок-
са и расхода катализатора и улучшить работоспособность установки.) Но такое сырье
стоит дороже, а доступные его объемы на нефтеперерабатывающих заводах ограни-
чены [Letzsch, 1997; Meyers, 2004].
2.5. ПЕРСПЕКТИВНЫЕ ТЕХНОЛОГИИ
1. Процесс HTL (Heavy-То-Light). Технология превращения тяжелого сырья в жид-
кие продукты, разработанная компанией Ivanhoe Energy. Представляет собой бы-
стрый термический крекинг, не нуждающийся в катализаторе и водороде. Весь кокс
и газы идут на выработку энергии. Эффективно использует концепцию тонких пле-
нок, дает высокий выход синтетической нефти; проводится в менее жестких услови-
ях, чем коксование.
2. Процесс GHU. Разработан компанией Genoil Inc. Проводится в реакторах с не-
подвижным слоем разных катализаторов и использует концепцию переключаемых
защитных реакторов. Входной реактор содержит катализатор гидродеметаллизации
для предварительного удаления металлов, последующий — слой высокоактивного
катализатора гидрообессеривания или комбинированный слой катализаторов гидро-
обессеривания и гидродеазотирования для удаления серы и азота и заключительного
облагораживания остаточного сырья или превращения в легкую нефть.
3. Процесс VISCOSITOR. Разработан компанией Wescorp Energy, Inc. Сырье рас-
пыляется водяным паром и расщепляется, сталкиваясь на высокой скорости с подо-
гретым песком. Процесс проводится при низких температурах и давлениях и не ну-
ждается в катализаторе. Потребность во внешнем источнике энергии невелика, так
как энергию дает топливо, производимое самим процессом. Технология рассчитана
в первую очередь на облагораживание тяжелой нефти через превращение ее в более
легкую и ценную синтетическую нефть.
4. Процесс IMP. Разработан Мексиканским институтом нефти. Проводится по-
следовательно в реакторах с неподвижными слоями разных катализаторов, рассчи-
танных на удаление металлов и серы и гидрокрекинг асфальтенов. Реакторы работа-
ют в умеренных условиях, поэтому количество образующихся осадков минимально.
Используются собственные катализаторы, разработанные с учетом типа и каче-
ства облагораживаемой тяжелой нефти. Непрерывность процесса обеспечивается
Глава 2. Технологии переработки тяжелой нефти
85
надлежащей подготовкой сырья, а также правильным выбором рабочих условий, рас-
положения реакторов и свойств катализаторов [Ancheyta et al., 2010].
2.6. КОМБИНИРОВАННЫЕ ПРОЦЕССЫ
ОБЛАГОРАЖИВАНИЯ
Наблюдаемая во всем мире тенденция к добыче все более тяжелой нефти ставит пе-
ред нефтеперерабатывающей отраслью задачу «синтезировать» существующие тех-
нологии облагораживания такой нефти. Даже взятые в отдельности, процессы обо-
гащения водородом или обеднения углеродом и сами по себе способны снизить
содержание примесей и уменьшить плотность сырья. Выбор наиболее подходящего
способа определяется главным образом свойствами нефти, целевым качеством обла-
гороженной нефти, ценами на нефть и спросом на продукты.
До сих пор предпочтение оказывалось технологиям коксования, в особенности за-
медленного, поскольку это сложившийся процесс с многолетним опытом промыш-
ленной эксплуатации, хоть и характеризующийся невысоким выходом дистиллятных
продуктов и необходимостью их дальнейшей очистки. Но сегодня, по мере того как
нефть дает все больше кокса и меньше жидких продуктов, подход к вопросу начина-
ет меняться. Постепенно возвращается имевший место в прошлом интерес к другому
направлению облагораживания — обогащению водородом.
Неплохим вариантом представляется сочетание нескольких процессов облаго-
раживания тяжелой нефти. Есть публикации о нескольких успешных попытках,
но в каждом конкретном случае нужны немалые научно-исследовательские и опыт-
но-конструкторские усилия. Так или иначе, сопряжение технологических установок
требует особого внимания, поскольку различия в рабочих условиях и необходимость
обеспечения нужных температур и давлений технологических потоков могут вызвать
потребность в дополнительном оборудовании (компрессоры, насосы, теплообмен-
ное оборудование и т. д.), что приведет к увеличению капитальных и эксплуатацион-
ных затрат. Поэтому решение об интеграции различных процессов должно прини-
маться исходя из технико-экономических исследований.
Публиковались также предложения о совместном применении нескольких техно-
логий облагораживания тяжелых нефтей с тем, чтобы получить максимальную вы-
году. Главное преимущество такого совместного применения заключается в том, что
оно дает большую выгоду, чем может дать каждая из технологий по отдельности. Наи-
более перспективные комбинации можно получить соединением технологий деас-
фальтизации, висбрекинга, замедленного коксования, газификации, гидрокрекинга
и гидроочистки [Castaceda et al., 2012].
2.6.1. Комбинированные технологии обеднения углеродом
1. Деасфальтизация плюс газификация. Сырье для газификатора поступает не-
посредственно из колонны отпарки асфальта. До подачи в отпарную колонну
86
Часть 1. Свойства тяжелой нефти и технологии ее переработки
и отделения растворителя, когда теплопроводность асфальта выше, его подогре-
вают до температуры, необходимой для оптимальной перекачки в газификатор.
В результате устраняются ограничения вязкости асфальтенов. Что важнее, до-
стигаемое увеличение глубины деасфальтизации означает больший выход деас-
фальтизата и, соответственно, больший объем производства дизельного топлива.
Совместная работа установок деасфальтизации и газификации дает ряд преиму-
ществ, в том числе следующие [Wallace et al., 1998; Biasca et al., 2003; McGehee,
2006; Shell, 2011]:
• выгодное использование асфальтенов, получение низкокалорийного топлива
дня внутреннего потребления;
• производство водорода для гидроочистки деасфальтизата и извлечение водоро-
да из отходящего газа гидроочистки;
• водород, необходимый для гидроочистки деасфальтизата, выступает в качестве
первичного продукта газификатора и может быть произведен из асфальтенов,
что исключает потребность во внешнем его источнике;
• увеличивается гибкость переработки нефтяного сырья, а значит, и пропускная
способность по нефти без нежелательного увеличения выхода тяжелого нефтя-
ного топлива;
• возрастают объемы производства заводом дизельного топлива;
• возрастает ценность нефти, так как деасфальтизация удаляет тяжелые компо-
ненты, снижает содержание металлов и коксовый остаток, а также снижает аб-
солютную плотность;
• для отделения растворителя от деасфальтизата и асфальта в соответствующих
отпарных колоннах и возвращения его в процесс нужны значительные количе-
ства тепла (это тепло может давать процесс газификации);
• баланс энергии между установками исключает потребность во внешнем источ-
нике тепла для отделения растворителя от деасфальтизата;
• снижается тепловая нагрузка на отпарные колонны и печи подогрева
асфальта;
• обеспечивается выгодная интеграция продуктов деасфальтизации и гази-
фикации;
• отходящий газ гидроочистки можно направить на установку газификации;
• появляется возможность улавливать серу из деасфальтизата на месте без увели-
чения нагрузки на заводские мощности по извлечению серы.
2. Замедленное коксование плюс газификация. Нефтяной кокс можно газифициро-
вать в целях производства электроэнергии для внутреннего потребления и поставки
во внешние сети. Такая комбинированная схема дает целый ряд преимуществ [Bres-
san and McGrath, 2007]:
• номенклатура тяжелой продукции полностью исчерпывается высококаче-
ственной серой и шлаком;
• так как газификация способна полностью перерабатывать сырье, возникает
удобная возможность производить синтетическую нефть;
Глава 2, Технологии переработки тяжелой нефти
87
• можно, начав с сырой нефти, разделять ее на тяжелую и легкую части; далее
из тяжелой нефти можно производить электроэнергию, а легкую (синтетиче-
скую) нефть сбывать на нефтяном рынке).
3. Деасфальтизация плюс замедленное коксование. Этот комбинированный про-
цесс получил название ASCOT. Сырье подогревается до необходимой температуры
экстракции, после чего поступает в экстрактор. Восходящий поток растворителя
в экстракционной колонне способствует извлечению из сырья большего количе-
ства парафиновых углеводородов. Асфальтены не растворяются и вместе с неболь-
шим количеством увлекаемого растворителя отбираются с низа экстрактора. Сы-
рой деасфальтизат выходит сверху и поступает в систему извлечения растворителя.
Растворитель возвращается в экстрактор. Деасфальтизат вместе с тяжелым газой-
лем коксования отпаривается в колонне фракционирования секции коксования,
давая сырье для крекинга. Нижняя фаза, содержащая асфальт и немного раствори-
теля, с контролируемой скоростью отбирается с низа экстрактора и поступает не-
посредственно в секцию коксования. Асфальт с некоторым количеством рециркуля-
та колонны и растворителя проходит через печь коксования, где быстро нагревается
до температуры, необходимой для камер коксования. Преимущества такой комби-
нированной схемы заключаются в следующем [Phillips and McGrath, 1998; Elliott and
McGrath, 2009]:
• отделение деасфальтизата до коксования увеличивает выход жидких продуктов
и снижает выход кокса;
• тепловая интеграция секций коксования и деасфальтизации снижает потребле-
ние энергоносителей;
• растворитель, оставшийся в неотпаренном асфальте и деасфальтизате, извлека-
ется в колонне фракционирования;
• растворитель, отбираемый с верха колонны фракционирования, можно исполь-
зовать как абсорбент для извлечения углеводородов С3/С4, что исключает необ-
ходимость доставки абсорбента из колонны стабилизации бензиновой фракции;
• растворитель, попадающий вместе с асфальтом в печь и камеры коксования,
сильно снижает парциальное давление асфальтового сырья по сравнению
с обычной установкой замедленного коксования;
• низкое парциальное давление асфальта ведет к снижению выхода кокса и уве-
личению выхода жидких продуктов в реакциях коксования.
4. Деасфальтизация плюс замедленное коксование плюс газификация. Дополнение
предыдущей схемы процессом газификации дает, кроме уже упомянутых, следующие
выгоды [Haniford, 2003; Elliott and McGrath, 2009]:
• электроэнергию для внутреннего потребления и поставки во внешние сети;
• водород для последующих процессов переработки;
• продукты для химической промышленности (метанол, синтез-газ и т. д.).
Комбинации этих технологий обеднения углеродом показаны на рис. 2.5, 2.6. Ко-
нечные продукты отдельных процессов в совокупности образуют облагороженную
нефть.
88
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Дистилляты
Атмосферная I Вакуумная
перегонка перегонка
Дистилляты
Тяжелая
нефть
Деасфальтизация
Деасфальтизат
Колонна
отпарки
асфальта
Вакуумный
остаток
Асфальт
Сепаратор
; деасфальти-
зата
Атмосферный
остаток
Колонна
отпарки
деасфальтизата
Газификация
Кислород
Синтез-газ
В колонну
------>
отпарки
кислой воды
Никель-вана-
диевая зола
Деасфальтизация + газификация
Облагоро-
женная
нефть
Рис. 2.5. Комбинированные процессы: деасфальтизации;
Сокращения: БФ — бензиновая фракция; КК — камера
отфильтрованного осадка; ТГ — тяжелый газойль
Глава 2. Технологии переработки тяжелой нефти
89
Дистилляты
Атмосферная
Печь
—;>
Газификация
Колонна
фракцио-
нирования
Отпарная
колонна
Вакуумная
перегонка
Дистилляты
Синтез-газ
Фильтрование
Никель-вана-
диевая зола
Тяжелая
нефть
В колонну
отпарки
кислой воды
Атмосферный
остаток
Замедленное коксование
Вакуумный
остаток
Кокс
ЯГ
ТГ
-с.
БФ
Облагоро-
женная
нефть
Кислород
Пар
Скруббер
Котловая
вода
Реак- котел
тор Охлаж-
дение
шлака
Замедленное коксование + газификация
газификации и замедленного коксования; газификации,
коксования; ЛГ — легкий газойль; ООО — отделение
90
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Дистилляты
Атмосферная
перегонка
Вакуумная
перегонка
Дистилляты
Тяжелая
нефть
Атмосферный
остаток
Экстракци-
Деасфальтизация
Колонна
отпарки
асфальта
отпарки деас-
фальтизата
Вакуумный
остаток
Деасфальтизат
Деасфальтизация + замедленное коксование
Облагоро-
женная
нефть
------»
Рис. 2.6. Комбинированные процессы: деасфальтизации; замед-
газификации. Сокращения: БФ — бензиновая фракция; КВ —
ООФ — отделение отфильтрованного осадка; СД — сепаратор
Глава 2. Технологии переработки тяжелой нефти
91
Атмосферная
перегонка
Дистилляты
Вакуумная перегонка
Дистилляты
Тяжелая
нефть
Атмосферный
остаток
ЛГ
Кокс
Кокс
Облагоро-
женная
нефть
Экстракци-
онная
Вакуумный
остаток
Деасфальти-
зация
деасфальг
тизата-~
Замедленное коксование
Колонна
фракциони-
отпарки
асфальта
Асфальтч
Асфальт Деасфальтизат
[ Синтез-газ
БФ
Газификация
Кислород
4
Реак- Котел
тор Охлаж-
дение
шлака
Пар
Скруббер
КВ
। В колонну
! отпарки
I КИСЛОЙ ВОД|;|
1 Никель-
] ванадиевая
1 зола
Фильтрование ।
Деасфальтизация + замедленное коксование + газификация
оксования и деасфальтизации; замедленного коксования;
ленного к _________легкий газойль; ОК — отпарная колонна;
котловая вода J । __ газойль
деасфальтизата, п
92
Часть 1. Свойства тяжелой нефти и технологии ее переработки
На рис. 2.7 сравниваются комбинированные схемы обеднения углеродом, вклю-
чающие процесс газификации. Наибольший выход жидких продуктов дает схе-
ма ЭДА-ЗК-Гф, поскольку его увеличивает предварительная деасфальтизация. Как
и в двух других схемах, асфальт коксуется, а кокс направляется в газификатор. Плот-
ность по шкале API облагороженной нефти прямо зависит от количества тяжелых
компонентов, поступающих в газификатор.
100
ЗК-Гф ЭДА-Гф ЭДА-ЗК-Гф
Рис. 2.7. Сравнение комбинированных схем, включающих газификацию:
ЗК-Гф — замедленное коксование + газификация; ЭДА-Гф — экстрактивная деасфаль-
тизация + газификация; ЭДА-ЗК-Гф — экстрактивная деасфальтизация + замедленное
коксование + газификация
Влияние изменений, вносимых в процесс каталитического крекинга в псевдоожи-
женном слое (FCQ, на октановое число и выход бензина часто выражают, используя
понятие октано-барреля (это просто произведение октанового числа на выход бен-
зина в баррелях). По аналогии с этим, при облагораживании тяжелой нефти можно
ввести понятие «АР/-барреля», учитывающее увеличение плотности в градусах API
и изменение объемного выхода. При данных, представленных на рис. 2.7, величина
этого индекса составляет 2253 А/7-барреля для комбинации ЗК-Гф, 1662Л/7-барреля
для комбинации ЭДА-Гф и 1953 А/7-барреля для комбинации ЭДА-ЗК-Гф. Из трех
схем наибольший индекс в А/V-баррелях показывает комбинация ЗК-Гф. Это под-
тверждается более высоким выходом и большей плотностью по API по сравнению
с другими комбинациями. Комбинация ЭДА-Гф показывает наименьший индекс
в Я/7-баррелях в основном из-за того, что дает наименьший выход, хотя по показате-
лю плотности в градусах API она сравнима с комбинацией ЭДА-ЗК-Гф.
2.6.2. Комбинированные технологии обогащения водородом
1. Комбинации процессов в кипящем слое. Типичный представитель этой груп-
пы процессов — двухступенчатая установка Н-Oil с последовательно соединенными
Глава 2. Технологии переработки тяжелой нефти
93
реакторами. Такая схема увеличивает глубину превращения вакуумного остатка, по-
вышает качество жидких продуктов и улучшает обессеривание [Kressmann et al.,
2000]. В этой конфигурации поток реактора первой ступени (смешанная фаза) раз-
деляется на пары и жидкие продукты. Жидкость направляется в реактор второй сту-
пени, а пары соединяются с верхним продуктом горячего сепаратора высокого дав-
ления, расположенного после реактора второй ступени. Промежуточное разделение
дает следующие преимущества:
• улучшается кинетика реакций в реакторе второй ступени;
• увеличивается пропускная способность по сравнению с одноступенчатой
установкой;
• улучшаются показатели процесса и качество продуктов;
• вместо повышения производительности установки можно уменьшить пропуск-
ную способность второго реактора и тем самым снизить капиталовложения;
• оптимизируется подача водорода в отдельные реакторы.
2. Гидрокрекинг в фазе суспензии плюс гидроочистка в неподвижном слое. Гидроочист-
ку остатков можно комбинировать с процессом Uniflex. Реактор Uniflex можно устано-
вить перед реакторами с неподвижным слоем катализатора уже существующих устано-
вок гидроочистки. Получается комплексный процесс, сочетающий в себе достоинства
обеих технологий. Кроме реактора Uniflex, может потребоваться дополнительное обору-
дование для отделения асфальта от других продуктов процесса Uniflex перед их перера-
боткой в главных реакторах гидроочистки. Такая комбинированная установка потребует
меньших капиталовложений, чем установка замедленного коксования [Gillis et al., 2009].
3. Гидрокрекинг в фазе суспензии плюс гидропереработка в кипящем слое. Как сооб-
щает Neste Oil [HTI, 2011], существующие установки облагораживания в кипящем
слое для увеличения производительности можно сочетать с технологией НСАТ (ги-
дрокрекинг в фазе суспензии).
2.6.3. Комбинированные технологии обогащения водородом
и обеднения углеродом
1. Система переключаемых защитных реакторов (Permutable reactor system — PRS)
плюс RFCC. Такой подход сочетает в себе технологии гидроочистки Hyvahl и гидро-
крекинга R2R. Установка обессеривания, использующая катализаторы нескольких
типов, перерабатывает вакуумный остаток либо в нефтяное топливо с очень низким
(0,3-0,5 %масс.) содержанием серы, либо в сырье для процесса R2R, в случае чего
производство нефтяного топлива можно вообще исключить [Plain et al., 2011]. Ос-
новные преимущества такой интеграции представлены ниже.
• В течение срока службы катализатора глубина превращения в дистилляты
с температурой кипения до 565 °C меняется в широких пределах: от 25 % в на-
чале цикла до 55 % в конце.
• Вакуумный газойль с интервалом кипения 370—565 °C не отделяют от ва-
куумного остатка 565 °С+; на установку крекинга направляется целиком вся
94
Часть 1. Свойства тяжелой нефти и технологии ее переработки
фракция 370 °С+. Таким образом, установка RFCC перерабатывает как гидро-
очищенный вакуумный газойль установки мягкого крекинга, так и гидроочи-
щенный вакуумный остаток.
• Единственный побочный продукт, используемый в качестве технологическо-
го топлива, получается фильтрованием некондиционного газойля установки
RFCC. Он полностью потребляется как нефтезаводское топливо; его выход со-
ставляет менее 3,5 % от нефтяного сырья.
• Высокая гибкость и система переключаемых защитных реакторов позволяет
производить из сырья, на 100 % состоящего из вакуумного остатка, три разных
сорта нефтяного топлива и ежегодно поставлять его на рынок по меньшей мере
в течение И мес. непрерывной работы.
2. Гидрокрекинг в кипящем слое плюс замедленное коксование. Непревращенный
остаток процесса H-Oil направляется в процесс замедленного коксования. Смесь ди-
стиллятов коксования и H-Oil проходит гидроочистку в реакторах с неподвижным
слоем и дает на выходе синтетическую нефть [Kressmann et al., 1998]. Преимущества
этой схемы таковы:
• налицо экономическая целесообразность в регионах, где есть рынок сбыта кок-
са (однако необходимость дальнейшей гидроочистки все равно остается);
• даже при том, что конечный остаток представляет собой кокс, его выход со-
ставляет лишь около 10 % от массы сырья процесса H-Oil.
3. Гидрокрекинг в кипящем слое плюс деасфальтизация. Непревращенный остаток
процесса H-Oil направляется на установку деасфальтизации и разделяется на деас-
фальтизат и остаточный асфальтовый продукт. Деасфальтизат можно возвращать
в реактор H-Oil для дальнейшего превращения или вместе с дистиллятами H-Oil на-
правлять на гидроочистку. Основные преимущества схемы приведены ниже [Kress-
mann et al., 1998].
• Асфальт, производимый в сравнительно небольших количествах, можно сбы-
вать или направлять в газификатор для производства водорода.
• Конечный остаток — асфальт, выход которого сравним с выходом кокса в пре-
дыдущей схеме.
• Асфальт, даваемый этой схемой, имеет температуру размягчения около 150 °C,
что позволяет получать его в жидкой фазе и перекачивать по трубопроводу
на установку газификации или на асфальтовый завод.
• Так как деасфальтизат не содержит асфальтенов, его можно направлять на уста-
новку H-Oil в смеси с непереработанным сырьем.
• Существенно сокращается содержание примесей, снижаются плотность
и вязкость.
• Деасфальтизат можно смешивать с непереработанным атмосферным остатком
и в качестве сырья направлять на установку H-Oil. Там его можно подвергнуть
глубокому гидрокрекингу, так как в нем нет асфальтенов, но есть в умеренных
количествах смолы, ароматические и предельные углеводороды.
• Продукты этой схемы требуют менее жестких условий гидроочистки, чем про-
дукты схемы «Н-Oil + коксование», особенно удаление серы и коксового остатка.
Глава 2. Технологии переработки тяжелой нефти
95
4. Гидрокрекинг в кипящем слое плюс газификация. Непревращенный остаток про-
цесса Н-Oil направляется на установку газификации. Основная выгода от объеди-
нения этих двух процессов заключается в получении водорода для самого процесса
Н-Oil или для дальнейшей гидроочистки. Общая глубина превращения сырья состав-
ляет около 83 %, поскольку нет особых причин для ее увеличения до 90 % и выше
[Gragnani, 2001].
5. Гидрокрекинг в фазе суспензии плюс замедленное коксование. Хотя Uniflex, будучи
процессом гидрокрекинга в фазе суспензии, имеет мало общего с замедленным кок-
сованием, эти процессы можно объединить. Интеграцию можно осуществлять путем
избирательного перенаправления выходных потоков установок и использования об-
щей инфраструктуры и систем разделения легких фракций. Переработка вакуумно-
го остатка в процессе Uniflex, осуществляемая перед замедленным коксованием, дает
следующие преимущества [Gillis et al., 2009]:
• существенное увеличение выхода продуктов;
• снижение чистого выхода кокса (степень снижения зависит от выбранной глу-
бины превращения в процессе Uniflex')',
• с увеличением глубины превращения в процессе Uniflex его экономическая
привлекательность по сравнению с замедленным коксованием возрастает.
6. Другие комбинации. Многообещающие варианты облагораживания тяжелой
нефти возникают при комбинировании гидроочистки в неподвижном слое с дру-
гими процессами: коксованием, газификацией, висбрекингом, деасфальтизацией,
гидрокрекингом в кипящем слое и в фазе суспензии. Один из главных недостатков
гидрокрекинга при высоком давлении (например, в кипящем слое и в фазе суспен-
зии) — образование осадка. Остроту этой проблемы снимают процессы, проводимые
в неподвижном слое в умеренно жестких условиях реакций, что увеличивает возмож-
ности их комбинирования с другими вариантами. Другая проблема — это асфальте-
ны, наличие которых чревато деактивацией катализаторов и образованием осадка.
Поэтому асфальтены необходимо удалить (путем деасфальтизации) или крекировать
(в суспензионном реакторе), прежде чем подавать тяжелую нефть в реактор гидро-
очистки в неподвижном слое: это продлит срок службы катализаторов и сэкономит
их расход. Отсутствие асфальтенов (или минимизация их содержания) в сырье реак-
тора гидроочистки в неподвижном слое позволит оптимизировать условия реакций
гидроочистки с тем, чтобы сделать их более избирательными к выходу ценных про-
дуктов: например, бензина и дизельного топлива. В то же время подключение дру-
гих процессов после гидроочистки в неподвижном слое создает стимулы для полного
превращения фракции остатка, что сегодня проявляется в виде тенденции к полно-
му исключению тяжелого нефтяного топлива из номенклатуры продукции нефтепе-
рерабатывающих заводов.
Некоторые схемы комбинирования процессов обеднения углеродом и обогаще-
ния водородом представлены на рис. 2.8 и 2.9. На рис. 2,10 показаны другие возмож-
ные комбинации различных технологий облагораживания.
96
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Облагоро-
женная
нефть
------->
Гидрокрекинг в кипящем слое + замедленное коксование
Рис. 2.8. Комбинированные процессы: гидрокрекинга в кипящем слое; замедленного
атмосферная перегонка; БФ — бензиновая фракция; ВГ — вакуумный газойль; ВО —
Л Г — легкий газойль; ОКГ — отбор кислых газов; СрД — средние дистилляты; ТГ —
Глава 2. Технологии переработки тяжелой нефти
97
Атмосферная
перегонка
Атмосферный
остаток
Дистилляты
Облагоро-
женная
нефть
------->
Деасфаль-
тизация
ВО
Асфальт ф
Гидрокрекинг в кипящем слое + деасфальтизация
Деасфальтизат
коксования и гидрокрекинга в кипящем слое; деасфальтизации. Сокращения: АП —
вакуумный остаток; ВП — вакуумная перегонка; КОА — колонна отпарки асфальта;
тяжелый газойль
98
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Дистилляты
——-.....-—-~>1
Атмо-
Дистилляты
—----------я
"I
I
, Кислый
——_—>
। газ
I Кислая
1вода *
I Бф -
—I--------->
I Вакуумный
। газойль „
----------->1
I
I
I
I
Облагоро-
женная
нефть
Процесс
Н-ОН
1 Синтетический газ
Гидрокрекинг в кипящем слое + газификация
Рис. 2.9. Комбинированные процессы: гидрокрекинга в кипящем слое; газификации
атмосферная перегонка; БФ — бензиновая фракция; ВО — вакуумный остаток;
вакуумный газойль; ЛГ — легкий газойль; ОКГ — отбор кислых газов; ООО —
с суспензионным слоем); СрД — средние дистилляты; ТВГ — тяжелый вакуумный
Глава 2. Технологии переработки тяжелой нефти
99
Атмо-
сферная
перегонка 1
Дистилляты
Вакуумная перегонка Дистилляты
Газойль
ЛВГ
Процесс в фазе
ВО суспензии
Асфальт
Бензиновая фракция
и газойль
Гидрокрекинг в фазе суспензии + замедленное коксование
Облагоро-
женная
нефть
и гидрокрекинга в фазе суспензии; замедленного коксования. Сокращения; АП —
ВП — вакуумная перегонка; КФ — колонна фракционирования; ЛВГ — легкий
отделение отфильтрованного осадка; СР — суспензионные реакторы (реакторы
газойль; ТГ — тяжелый газойль; ТГК — тяжелый газойль коксования
100
Часть 1. Свойства тяжелой нефти и технологии ее переработки
Рис. 2.10. Возможные комбинации различных технологий облагораживания
ЛИТЕРАТУРА
Ancheyta, J., Betancourt, G., Marroquin, G., Centeno, G., Munoz, J.A., Alonso, E 2010a. Process for
the catalytic hydrotreatment of heavy hydrocarbons of petroleum. US Patent 7651204 B2.
Ancheyta, J., Speight, J.G. 2007. Hydroprocessingof Heavy Oils and Residua, CRC Press, Taylor & Francis
Group, Boca Raton, FL.
Ancheyta, J., Trejo, E, Rana, M.S. 2010b. Asphaltenes: Chemical Transformations during Hydroprocessing
of Heavy Oils, CRC Press, Taylor & Francis Group, Boca Raton, FL.
Atkins, L., Higgins, T, Barnes, C. 2010. Heavy crude oil: Global analysis and outlook to 2030. Hart
Energy Consulting report, November.
Bearden, R. 1997. MICROCAT-RC: Technology for hydroconversion upgrading of petroleum residues,
Division of Petroleum Chemistry, American Chemical Society, San Francisco, CA, April 13—17.
Billon, A., Morel, E, Penes, J.P. 1997. Use of solvent deasphalting process in residues conversion schemes.
In 15th World Petroleum Congress, Beijing, China, October 12—17.
Bressan, L., McGrath, M.J. 2007. Heavy crude upgrading: An option for gasification. In Gasification
Technology Council Conference, San Francisco, CA, October.
Castaneda, L.C., Munoz, J.A.D., Ancheyta, J. 2012. Combined process schemes for upgrading of heavy
petroleum. Fuel 100:110—127.
Chevron Lummus. 2010. Converting low-value feed to high-value-CGL. Hydro processing technologies
and catalysts. Brochure.
Chevron Lummus. 2011. RDS/VRDS/OCR/UFR Lummus technology. СВ & I company brochure.
Daniel, M., Lerman, D.B., Peck, L.B. 1988. Amocos LC-flning residue hydrocracker yield
and performance correlations from a commercial unit. In NPRA Annual Meeting, San Antonio, TX,
March.
Глава 2. Технологии переработки тяжелой нефти
101
Dickenson, R.L., Karp, A.D., Johnson, Н.Е. 1988. Heavy oil processing—Progress and outlook. \nFourth
Forum on Advances in the Refining Industry, Pemex Refinacion and Institute Mexicano del Petroleo, Mexico
City, Mexico, August 18-19.
Dickenson, R.L., Schulman, B.L., Biasca, F.E., Johnson, H.E. 1996. The bottom-of-the-barrel: Real
options to avoid fuel oil. In NPRA 1996 Annual Meeting, San Antonio, TX, paper AM-96-57, 14 pp.
Elliott, J.D. 2003. Delayed coking: Recycle to zero recycle concepts & considerations. In NPRA Annual
Meeting, San Antonio, TX, March 23-25, paper AM-03-30.
Elliott, J.D., McGrath, M.J. 2009. Conversion of very heavy residues. Hydrocarbon Engineering. March.
Elliott, J.D., Wedlake, D.A. 2007. Residue upgrading with SYDEC delayed coking: Benefits and
economics. In First Asia Bottom of the Barrel Technology Conference, Kuala Lumpur, Malaysia, June.
Furimsky, E. 2000. Characterization of cokes from fluid/flexi-coking of heavy feeds. Fuel Process. Technol.
6:205-230.
Gillis, D., Van Wees, M., Zimmerman, P. 2001. Upgrading residues to maximize distillate yields. UOP
tech paper, 2009. http://www.uop.com (retrieved on March 4, 2011).
Gragnani, A. 2011. New and advanced technological processes by Axens. http://www.axens.net/upload/
presentations/fichier/interfaces05_hoil.pdf (retrieved on March 3, 2011).
Hamilton, G.L. 2002. Delayed coker design considerations and project execution. In NPRA Annual
Meeting, San Antonio, TX, March 17—19, paper AM-02-06.
Haniford, R.R. 2003. Coke is good, but less is better. In NPRA Annual Meeting, San Antonio, TX, March
23-25, paper AM-03-90.
Houde, E.J., McGrath, M.J. 2006. Residue upgrading. In The Seventh International Downstream
Technology & Catalyst Conference & Exhibition, London, U.K., February.
Houde, E.J., Thompson, G., Marzin, R., Periera, P., McGrath, M., Feintuch, H. 1998. The
Aquaconversion™ process: A new approach to residue processing. In NPRA Annual Meeting, San Francisco,
CA, March 15-17, paper AM-98-09.
HTI. 2011. Headwaters incorporated announces successful commercial implementation of the HCATR
heavy oil upgrading technology at the Neste Oil Porvoo Refinery. Headwaters Incorporated News Bulletin,
January, 1:1.
Kamienski, P, Wright, M., de Wit, M., Wakui, T. 2007. Maximize value of resid conversion with
FLEXICOKING technology maximize technology. In BBI'C Technology Conference, Athens, Greece,
October 10—12.
Kressmann, S., Boyer, C., Colyar, J.J., Schweitzer, J.M., Viguie, J.C. 2000. Improvements of ebullated-
bed technology for upgrading heavy oils. Oil Gas Sci. Technol. Rev. IFP 55(4):397—406.
Kressmann, S., Colyar, J.J., Peer, E., Billon, A., Morel, F. 1998. H-oil process based heavy crudes refining
schemes. In Proceedings of Seventh Unitar Conference on Heavy Crude and Tar Sands, Beijing, China, October
27-30, pp. 857-866.
Lars, S., Andersson, T., Lundin, S.T., Jaras, S., Ottersedt, J.-E. 1984. An ESCA study of metal deposition
on cracking catalysts. Appl. Catal. 9(3):317-325.
Letzsch, WS. 1997. The future of fluid catalytic cracking. In NPRA Annual Meeting, San Antonio, TX,
paper AM-97-65, March 16—18.
Liu, Y., Gao, L., Wen, L., Zong, B. 2009. Recent advances in heavy oil hydroprocessing technologies.
Recent Pat. Chem. Eng. 2:22—36.
Lunin, G., Chambers, L.W, Parsons, B.I. 1985. The commercialization of the CANMET hydrocracking
process, CANMET Branch, Energy, Mines & Resources. In Technical Meeting/Petroleum Conference of the
South Saskatchewan Section, Regina, Saskatchewan, Canada, September 15-17.
Marano, J.J. 2003. Refinery technology profiles: Gasification and supporting technologies. U.S.
Department of Energy, National Energy Technology Laboratory, Pittsburgh, PA. Energy Information
Administration, Washington, DC, June.
Mart, C.J., Ellis, E.G., McCaffrey, Jr. D.S. 2005. ExxonMobil resid upgrading technologies.
In PETROTECH2005, 6th International Petroleum Conference and Exhibition, New Delhi, India.
102
Часть 1. Свойства тяжелой нефти и технологии ее переработки
McGehee. J. 2006. Solvent deasphalting in today's deep conversion refinery R&D—Heavy oil upgrading.
In AIChE-Chicago Symposium, Chicago, IL, October 2.
McGrath, M.J. 2008. Solvent deasphalting—An economic residue upgrading technology. In Third Russia
& CiS Bottom of the Barrel Technolog)’ Conference, Moscow, Russia, April 24-25.
Meyers, R.A. 2004. Handbook of Petroleum Refining Processes, 3rd edn., Chapter 3, McGraw-Hill, New
York.
Morrison, M.E., Billon, A., Stephens, G., Hennico, A., Peries, J.P. 1994. Hyvahl-Solvahl—Key processes
for upgrading of residues. In NPRA Annual Meeting, San Antonio, TX, paper AM-94-25 24P, March 20-22.
Otterstedt, J.E., Gevert, S.B., Jaras, S.G., Menon, P.G. 1986. Fluid catalytic cracking of heavy (residual)
oil fractions: A review. Appl. Catal. 22:159—179.
Parrish, M.R., Hammond, D.G., Citarella, V.A. 1996. Fluid coking: A continuous, flexible and reliable
conversion process. Hydrocarb. Technol. Inf. Spring: 112(4):25-31.
Phillips, G., McGrath, M. 1998, Residue upgrading options for eastern Europe. In World Refining
Association, Budapest, Hungary, October 13—14.
Plain, C., Benazzi, E., Guillaume, D. 2011. Residue desulphurization and conversion. The Refining,
Gas and Petrochemicals Processing Website, http://www.axens.net/upload/news/fichier/ptq_q2_2006.pdf
(retrieved on March 3, 2011).
Pruden, B., Muir, G., Skipek, M. 1993. The CANMET hydrocracking process: Recent developments; oil
sands—Our petroleum future. Alberta Research Council, Edmonton, Alberta, Canada, p. 277.
Rana, M.S., Samano, V., Ancheyta, J., Diaz, J.A.I. 2007. A review of recent advances on process technol-
ogies for upgrading of heavy oils and residua. Fuel 86(9): 1216-1231.
Rispoli, G. 2009. Heavy' oil upgrading. In GE Oil & Gas Annual Meeting, Florence, Italy, January 26-27.
Sanfilippo, D. 2009. Bottom of the barrel technology within refining extracting additional value from oil
feedstocks using EST technology. In WTG Webinar Connected from San Donato Milanese, Milan, Italy, No-
vember 18.
Scheffer, B., van Koten, M.A., Robschlager, K.W., de Boks, EC. 1998. The shell residue hydroconversion
process: Development and achievements. Catal. Today 43(3—4)2:17-224.
Shell. 2011. Shell soaker visbreaking. Technical sheet, http://www.cbi.com/images/uploads/tech_sheets/
Visbreaking.pdf (retrieved on February 21, 2011).
Silva, A.E., Rohrig, H.K., Dufresne, A.R. March 1998. CANMET process going into Montreal refinery.
OHGas J. 26:81-88.
Solari, R.B., Marzin, R., Zbinden, H. 1997. Comparison of carbon rejection and hydrogen addition pro-
cesses in production—Upgrading complexes. In 15th World Petroleum Congress, Beijing, China, October
12-17.
Stiegel, G.J. 2005. Gasification technologies. In Clean, Secure and Affordable Energy Systems, IGCC and
Clean Coal Technologies Conference, Tampa, FL, June 28.
Vartivarian, D., Andrawis, H. 2006. Delayed coking schemes are most economical for heavyoil upgrad-
ing. Oil Gas J. 104(6):52—56.
Wallace, P.S., Anderson, M.K., Rodarte, А.1., Preston, WE. 1998. Heavy oil upgrading by the separa-
tion and gasification of asphaltenes. In The Gasification Technologies Conference, San Francisco, CA, October.
Wenzel, F., Kretschmar, K. 1993. Veba-Combi-Cracking Process—Status and future trends. Oil sands—
Our petroleum future. Alberta Research Council, Edmonton, Alberta, Canada, pp. 268—253.
Yuandong, L., Liang, G., Langyou, W., Baoning, Z. 2009. Recent advances in heavy oil hydroprocessing
technologies. Recent Pat. Chem. Eng. 2:22—36.
Zhang, S., Liu, D., Deng, W., Que, G. 2007. A review of slurry-phase hydrocracking heavy oil technology.
Energy Fuels 21(6):3057—3062.
Часть II
Моделирование
некаталитических процессов
Глава 3. Моделирование
висбрекинга
Эта глава посвящена одному из старейших процессов облагораживания тяжелой
нефти — висбрекингу. Дается описание процесса, раскрываются его общие аспекты.
Рассматриваются типы, параметры и химия процесса, а также (кратко) моделирова-
ние его кинетики. Читатель найдет здесь ряд зависимостей, необходимых для расчета
выхода продуктов и глубины превращения. Прилагаются примеры применения этих
зависимостей для типичного сырья и рабочих условий. Предлагаются модели про-
цессов обоих типов — висбрекинга в змеевике печи и висбрекинга с выносной реак-
ционной камерой.
3.1. ВВЕДЕНИЕ
Со времени своего внедрения в 1939 г. процесс висбрекинга широко применялся для
переработки нефтезаводских остатков (атмосферной или вакуумной перегонки) и дру-
гого тяжелого сырья в газ, бензин, дистилляты и остаток висбрекинга. Само название
висбрекинг происходит от английских слов viscosity (вязкость) и breaking (разрушение).
Висбрекинг — это некаталитический термический процесс, снижающий вязкость
тяжелого сырья. Мягкие условия крекинга способствуют повышенному выходу бен-
зиновой фракции и меньшему выходу газа и кокса. Глубина превращения в процес-
се висбрекинга составляет около 30 %. Во избежание образования кокса пребывание
в условиях реакций должно быть непродолжительным. Основная задача висбрекин-
га — уменьшить концентрацию тяжелых фракций в остатке атмосферной или вакуум-
ной перегонки, увеличивая таким образом выход более ценных продуктов. Иными
словами, висбрекинг снижает вязкость тяжелого сырья при нагревании его в печи
за счет термического расщепления крупных углеводородных молекул.
Спрос на малоценное остаточное нефтяное топливо продолжает снижаться. По-
этому висбрекинг, позволяющий уменьшить выход такого топлива, стал важным не-
фтезаводским процессом. Так как остаток висбрекинга имеет пониженную вязкость,
для доведения ее до требуемого рынком уровня затрачивается меньше разбавителей
(средних дистиллятов), которые можно направить на производство других, более
ценных продуктов.
106
Часть 2. Моделирование некаталитических процессов
Несмотря на то что висбрекинг представляет несомненный интерес как техноло-
гия, при небольших капитальных затратах не требующая жестких условий процесса
и применения катализаторов и позволяющая сэкономить энергоносители (т. е. об-
ходящаяся дешевле других технологий облагораживания), растущие экологические
требования и увеличение спроса на более легкие продукты неуклонно снижают ее
конкурентоспособность. Висбрекинг — относительно зрелый процесс, что затрудня-
ет его включение в новаторские схемы переработки. Там, где спрос на тяжелое не-
фтяное топливо все еще сравнительно высок, висбрекинг продолжает удерживать
позиции, но в будущем значимость этого процесса, несомненно, упадет.
Главный недостаток процесса висбрекинга — наличие в его продуктах непредель-
ных соединений. Например, бензин висбрекинга содержит олефины и диолефи-
ны, которые вступают во вторичные реакции, образуя смолу и нелетучий смолистый
остаток.
3.2. ОПИСАНИЕ ПРОЦЕССА
Процесс висбрекинга относительно прост. Остаток атмосферной или вакуумной
перегонки прокачивают через теплообменники предварительного подогрева, а за-
тем подают в змеевик печи. Там остаток нагревается до температуры, необходимой
для протекания реакций при атмосферном давлении. Конструкция печи рассчитана
на мягкий крекинг сырья в контролируемых условиях. Главные параметры процес-
са в печи (время пребывания, температура и давление) регулируют так, чтобы опти-
мизировать термический крекинг по свободнорадикальному механизму, завершаю-
щийся образованием требуемых продуктов. Затем реакционная смесь проходит через
зону выдержки, расположенную в самой печи или во внешней камере. Поток из печи
быстро охлаждают холодным газойлем, чтобы остановить реакцию и предотвратить
дальнейший крекинг до нежелательных легких продуктов (стадия закалки). Охлаж-
денный поток поступает в зону испарения перегонной колонны (содержащей около
27 тарелок), где разделяется на газ, бензиновую фракцию, дистиллят и остаток. Дис-
тиллят после отпарки соединяют с остатком, отбираемым снизу колонны, и исполь-
зуют для добавления в тяжелое нефтяное топливо [Speight, 2011]. Нарис. 3.1 показана
типичная установка висбрекинга с выносной реакционной камерой, перерабатываю-
щая вакуумный остаток [Stratiev and Nikolaev, 2009].
3.3. СПОСОБЫ ВИСБРЕКИНГА
В промышленных масштабах применяются технологии висбрекинга двух типов.
1. Печной висбрекинг. Превращение достигается высокотемпературным крекин-
гом в течение заранее определенного, сравнительно короткого времени пребывания
в змеевике печи.
2. Висбрекинг с выносной реакционной камерой. Процесс проводится при по-
ниженных температурах в течение более длительного времени. Большая часть
Глава 3. Моделирование висбрекинга
107
превращения происходит в реакционной камере (сокинг-камере), где двухфазный
поток из змеевика печи находится более продолжительное время в условиях более
низких температур.
Типичные схемы процессов в змеевике и с выносной реакционной камерой изо-
бражены соответственно на рис. 3.2 и 3.3.
Перегретый пар
Пар
Пар
Вакуумный
остаток
с установки
вакуумной
перегонки № 2
Пресная
вода
Газойль
Остаток висбрекинга
Пресная
вода
Рис. 3.1. Типичная установка висбрекинга с выносной реакционной камерой
[Stratiev, D. and Nikolaev, N., Petrol. Coal, 51(2), 140, 2009]
Вакуумный
остаток
с установки
вакуумной
перегонки № 1
Закалка
-3 Кислая вода
, Бензин
на установку
парового
крекинга
Сырье
перегонки
Газ
Нестабильная
бензиновая
фракция
Легкий
газойль
Остаток
висбрекинга
Рис. 3.2. Упрощенная схема печного висбрекинга
108
Часть 2. Моделирование некаталитических процессов
Рис. 3.3. Упрощенная схема висбрекинга с выносной реакционной камерой
3.3.1. Висбрекинг в змеевике
Висбрекинг в змеевике (печной висбрекинг) характеризуется высокой температурой
и коротким временем пребывания в условиях реакции. Остаточное сырье проходит
по змеевику печи, где нагревается до высокой температуры, вызывающей частичное
испарение и неглубокий крекинг. Температуру в печи регулируют изменением подачи
воздуха к горелкам печи, а скорость движения сырья в змеевике — изменением пода-
чи насосов. Глубина реакций крекинга зависит от скорости движения сырья. Чтобы
остановить реакции крекинга, поток из змеевика быстро охлаждают теплообменом
с холодным сырьем либо с газойлем или остатком колонны фракционирования.
После охлаждения парожидкостная смесь поступает в колонну фракционирования,
где разделяется на отходящий газ, бензиновую фракцию, газойль и висбрекинг-оста-
ток. Этот остаток смешивают с более легкими разбавителями и получают нефтяное
топливо. В качестве разбавителя часто используется газойль, отбираемый из колон-
ны фракционирования.
3.3.2. Висбрекинг с выносной реакционной камерой
Висбрекинг с выносной реакционной камерой характеризуется пониженными тем-
пературами и продленным временем пребывания. Глубина превращения в змеевике
невелика, так что большая часть реакций протекает в реакционной камере. Там поток
из печи в течение определенного времени выдерживается при определенной темпе-
ратуре, достаточной для протекания реакций крекинга. Смесь из камеры направля-
ется в колонну фракционирования, где разделяется на продукты нескольких типов.
Эта разновидность висбрекинга, протекающего при более низкой температуре, по-
требляет меньше энергии и дает меньше остатка.
Глава 3. Моделирование висбрекинга
109
3.3.3. Различия между процессами
Перечисленные два способа висбрекинга схожи по сути и различаются в основ-
ном лишь температурами и временем пребывания в условиях реакций. Качество
и выход продуктов процессов при заданной глубине превращения практически
одинаковы.
Некоторые другие различия представлены ниже.
• В случае висбрекинга в реакционной камере температура в печи ниже, что по-
зволяет экономить топливо, но зато печь и камеру приходится очищать от кок-
са, что требует дополнительного оборудования. Со снижением рабочей темпе-
ратуры уменьшаются также эксплуатационные затраты.
• Расход топлива на висбрекинг с выносной реакционной камерой составляет
около 70 % от расхода на висбрекинг в змеевике.
• Висбрекинг в змеевике проводится в двухкамерной печи, что обеспечива-
ет повышенную гибкость подведения тепла и лучший контроль нагревания
сырья.
• При печном висбрекинге легче удалять кокс с внутренней поверхности змееви-
ка; для этого применяется паровоздушный выжиг.
• Висбрекинг с реакционной камерой потребляет меньше энергии при той же
глубине превращения.
• Высокие температуры при печном змеевике позволяют извлекать больше тяже-
лого газойля, чего нельзя добиться висбрекингом в реакционной камере без ва-
куумного испарителя.
• При печном висбрекинге в трубах печи быстро отлагается кокс, что вызывает
уменьшение проходного сечения труб, препятствует теплопередаче, уменьша-
ет пропускную способность и снижает производительность и эффективность
процесса. Для компенсации приходится сжигать больше топлива или умень-
шать скорость подачи сырья.
• Благодаря пониженной температуре при висбрекинге с реакционной камерой
кокс отлагается медленнее.
• Кокс при печном висбрекинге необходимо удалять чаще, но при висбрекинге
с реакционной камерой для этого приходится останавливать работу установки.
• Рабочий цикл печного висбрекинга длится от 3 до 6 мес., висбрекинга с реак-
ционной камерой — от 6 до 18 мес.
• Расход топлива на печной висбрекинг составляет 1—1,5 % от массы сырья;
для висбрекинга с реакционной камерой расход на 30—35 % ниже.
3.4. ПАРАМЕТРЫ ПРОЦЕССА
К параметрам процесса висбрекинга относятся свойства сырья, температура, давле-
ние, время пребывания (определяемое скоростью подачи сырья) и параметры закач-
ки водяного пара [Negin and Van Tine, 2004; Quignard and Kressmann, 2011].
110
Часть 2. Моделирование некаталитических процессов
3.4.1. Свойства сырья
На установку висбрекинга обычно подают остатки атмосферной и вакуумной пере-
гонки нефти. Свойства сырья изменяются в широких пределах и зависят от проис-
хождения нефти и степени отгонки дистиллятов. Глубина превращения такого сырья
составляет, в зависимости от его свойств и жесткости условий реакций, от 10 до 50 %.
Мерой жесткости условий висбрекинга является глубина превращения остаточно-
го сырья в газ, бензиновую фракцию и газойль. Глубина превращения определяется
как доля атмосферного (343 °С+) или вакуумного (538 °С+) остатка, превращающе-
гося в более низкокипящие компоненты. Глубину превращения определяют и отно-
сительно других границ кипения: например, относительно 165, 350 и 480 °C [Strati-
ev et al., 2008]. Глубину превращения атмосферного остатка рассчитывают по формуле
Л'343,С+
Доля фракции 343 °С+ в сырье - Доля фракции 343 °С+ в продукте
Доля фракции 343 °С+ в сырье
100. (3.1)
На глубину превращения, достигаемую при заданной жесткости условий, влияют
колебания в качестве сырья. Ниже перечислены свойства сырья, оказывающие глав-
ное влияние на глубину превращения.
• Содержание асфальтенов. Высокое содержание асфальтенов в сырье снижает
глубину его превращения. В умеренно жестких условиях молекулы асфальте-
нов при прохождении через печь изменяются мало. В жестких условиях они
склонны разрушаться и выпадать из реакционной смеси, что ведет к получе-
нию нестабильного остатка, непригодного для приготовления нефтяного то-
плива. Когда это происходит, технологические линии и трубы печи загрязня-
ются, что вызывает неполное сгорание топлива и преждевременную остановку
установки (сокращение длительности цикла).
• Температура размягчения. Сырье с невысокой температурой размягчения и низ-
ким содержанием асфальтенов хорошо поддается висбрекингу. В нем больше не-
асфальтеновых тяжелых дистиллятных фракций, которые крекируются до более
низкокипящих и менее вязких газойлей, что дает общее снижение вязкости.
• Содержание натрия. Высокое содержание натрия в сырье может ускорить за-
коксовывание труб печи.
• Коксовый остаток (коксуемость по Конрадсону, CCR). Высокая коксуемость,
как и высокое содержание натрия, ускоряет закоксовывание труб. Чем меньше
коксуемость и ниже содержание натрия, тем дольше цикл работы установки.
Сырье с высокой коксуемостью и содержанием асфальтенов дает больше газов,
бензина и кокса.
• Вязкость. При увеличении глубины превращения вязкость остатка висбре-
кинга вначале снижается, а затем, когда условия становятся достаточно жест-
кими, резко возрастает. Такое поведение явно говорит том, что при повыше-
нии жесткости условий в определенный момент, зависящий от свойств сырья,
резко усиливается образование предшественников кокса. Точка резкого по-
вышения вязкости ассоциируется с выходом нефтяного топлива из области
стабильности.
Глава 3. Моделирование висбрекинга
111
3.4.2. Температура
Хотя реакции происходят в глубине змеевика, важна температура не внутри змее-
вика, а на выходе из него, которая изменяется в пределах от 430 до 490 °C. Величи-
на этой температуры зависит от свойств сырья и типа процесса (в змеевике или в ре-
акционной камере). Температура на выходе из змеевика выбирается в зависимости
отцелевой глубины превращения.
При печном висбрекинге сырье поступает в печь, имея температуру 300-330 °C,
а реакции протекают в интервале температур 460-480 °C. Продукт охлаждается
до 350 °C.
Температура на выходе из змеевика при печном висбрекинге выше (около 480 °C),
чем при висбрекинге в реакционной камере (около 450 °C).
Температура сырья по длине змеевика непрерывно возрастает. Но ввиду того, что
реакции протекают с поглощением тепла (192 ккал на 1 кг продукта с температурой
кипения до 204 °C), после прохождения определенной точки рост температуры не-
сколько замедляется.
При висбрекинге с реакционной камерой температура сырья удерживается на бо-
лее низком уровне (около 450 °C); после выхода из змеевика она несколько снижает-
ся (примерно на 10-20°С) из-за поглощения теплоты в реакциях крекинга. Нараста-
ние кокса в змеевике происходит значительно медленнее, так как процесс протекает
при менее высокой температуре и глубина превращения в змеевике ниже.
3.4.3. Давление
Для предотвращения испарения сырья при печном висбрекинге достаточно давле-
ния в несколько бар. В случае висбрекинга с реакционной камерой давление выбира-
ют так, чтобы целевые легкие продукты испарялись и быстро покидали реакционную
зону, а тяжелые дозревали в жидкой фазе. Для вакуумного остатка необходимо давле-
ние в 5-8 бар, для атмосферного — от 10 до 12 бар.
Чтобы упростить перемещение сырья при печном висбрекинге, поддержива-
ют несколько повышенное входное давление около 2,5 МПа. Потери давления
по длине реактора (змеевика) обусловлены двухфазным трением и составляют
около 1,7 МПа.
При висбрекинге с реакционной камерой давление на входе в камеру равно при-
мерно 1,3 МПа. На выходе оно несколько ниже из-за гидростатического напора, кре-
кинга и испарения.
3.4.4. Время пребывания в реакционной зоне
Время пребывания сокращают увеличением скорости подачи сырья, но при этом из-
меняются условия потока в змеевике и реакционной камере.
При печном висбрекинге время пребывания в змеевике печи составляет 2-5 мин.
112
Часть 2. Моделирование некаталитических процессов
Газовая фаза на входе состоит преимущественно из водяного пара. Благодаря ре-
акциям крекинга и частичному испарению по длине змеевика доля газовой фазы
возрастает.
При висбрекинге с реакционной камерой сырье 2-3 мин проходит змеевик, затем
15—25 мин выдерживается в камере.
3.4.5. Закачка пара
Водяной пар вместе с сырьем закачивают:
• для улучшения гидродинамики потока;
• регулирования времени пребывания;
• увеличения коэффициента теплопередачи вблизи стенок через турбулизацию
потока;
• предотвращения закоксовывания стенок труб змеевика.
При введении пара в сырье время его пребывания в змеевике сокращается; снижа-
ется также глубина превращения. Для компенсации снижения глубины превращения
необходимо увеличить температуру. Расход пара обычно составляет около 1 % от мас-
совой подачи сырья.
3.4.6. Главные параметры процесса
Из всех параметров процесса висбрекинга наиболее важны температура, давление
и время пребывания. Увеличение любого из них означает возрастание жесткости ус-
ловий реакций. Эти параметры в определенных пределах взаимозаменяемы, что по-
зволяет достигать заданной жесткости условий (глубины превращения). Каким бы
способом ни достигалась данная глубина превращения, выход и качество продуктов
практически одни и те же.
Глубина превращения находится в прямой зависимости от температуры и времени
пребывания; тем не менее она имеет верхний предел, определяемый появлением не-
стабильности остатка с последующей флокуляцией асфальтенов, что в конечном сче-
те ведет к образованию кокса.
На рис. 3.4 показаны типичные кривые изменения температуры, давления и глуби-
ны превращения (выраженной в виде процентного отношения суммы масс газа и бен-
зиновой фракции с конечной температурой кипения 165 °C к массе сырья) по длине
реакционной системы для процессов висбрекинга в змеевике и реакционной камере.
3.5. ХИМИЯ ПРОЦЕССА
Все реакции, происходящие при висбрекинге, инициируются термически и протека-
ют по свободнорадикальному механизму [Quignard and Kressmann, 2011]. Их после-
довательность представлена ниже.
Глава 3. Моделирование висбрекинга
113
• Инициирование: образование свободных радикалов путем разрыва свя-
зей С-С,
• Распространение: свободные радикалы вступают в реакции переноса водорода,
распада и образования новых низкомолекулярных свободных радикалов, изо-
меризации, циклизации, конденсации.
• Рекомбинация: свободные радикалы образуют попарные связи и превращение
замедляется.
Ниже описываются некоторые из этих реакций [Bosworth, 2005].
500
450 -
400 -
350 -
300 -
и 250 “
го
I 200 -
ш 500
Е
S
£
450 -
400 -
350 -
300 -
250 -
200 -
Рис. 3.4. Типичные кривые изменения температуры, давления и глубины превращения
по длине реактора в процессах висбрекинга в змеевике (верхний график) и в реакционной
камере (нижний график): (—) — температура; (—) — давление; (——) — глубина превра-
щения [Akbar, М. and Geelen, Н., Hydrocarb. Process., 60(5), 81, 1981]
114
Часть 2. Моделирование некаталитических процессов
3.5.1. Разрыв связи С-С
В первую очередь отщепляются, образуя предельные и непредельные цепные углеводо-
роды, длинные боковые парафиновые цепи, присоединенные к алкилированным на-
фтеновым или высококипящим ароматическим углеводородам из числа присутствую-
щих в молекулах асфальтенов. Преимущественное удаление или укорачивание боковых
цепей до метиловых и этиловых групп обусловлено большей термической стойкостью
как кольцевых систем алкилированных нафтенов, так и ароматических колец.
3.5.2. Дегидрирование
Дегидрирование парафинов и нафтенов дает соответственно олефины и ароматиче-
ские углеводороды. Дегидрирование представляет собой обратимую реакцию, ско-
рость которой снижается по мере увеличения длины цепи.
3.5.3. Изомеризация
Из-за термодинамически неблагоприятных условий глубина реакций изомеризации
при висбрекинге ограничена, в отличие от разрыва связи С-С.
3.5.4. Полимеризация и конденсация
Полимеризация и конденсация в процессе висбрекинга рассматриваются как по-
бочные реакции. Они происходят, когда часть образовавшихся углеводородных ра-
дикалов рекомбинирует, давая сравнительно стабильные высокомолекулярные
продукты — в том числе полиароматические углеводороды с конденсированной аро-
матической структурой, являющиеся прекурсорами кокса.
Продукты реакций полимеризации дегидрируются и обогащаются углеродом, ста-
новясь более твердыми и образуя кокс.
3.5.5. Реакции с участием гетероатомов
• Азот. Большая часть основного и неосновного азота сырья сконцентрирована
в смолах и асфальтенах остатка как до реакции, так и после нее. Азот труднее
поддается крекингу и остается сосредоточенным в жидкой фазе.
• Сера. По наблюдениям, с увеличением глубины превращения содержание серы
снижается, что можно объяснить десульфуризацией и распределением серы
в газовой фазе (в виде сероводорода) и дистиллятных продуктах.
• Металлы. В остаточную фракцию переходят ванадий и никель, большей частью
присоединенные к азоту порфириновых и непорфириновых структур, а также
относительно небольшие количества натрия и железа.
Глава 3. Моделирование висбрекинга
115
3.6. КИНЕТИКА РЕАКЦИЙ
Как и в случае других основных процессов превращения нефтяных фракций (напри-
мер, каталитического крекинга и гидрокрекинга), для представления кинетики реак-
ций висбрекинга широко используются дискретные модели. Применение дискрет-
ных моделей обусловлено сложностью сырья и продуктов процесса висбрекинга, что
крайне затрудняет описание его кинетики на молекулярном уровне. Реакции висбре-
кинга предлагается описывать степенными выражениями первого порядка. Зависи-
мость констант скоростей реакций первого порядка от температуры можно предста-
вить формулой Аррениуса. При большой глубине превращения начинает проявлять
себя полимеризация и порядок реакций отклоняется от первого.
Опубликовано несколько моделей кинетики, различающихся способом дискре-
тизации и выведенных для разных видов сырья, методик проведения экспериментов
и условий реакций. Эти модели реакций представлены, в частности, параллельны-
ми и параллельно-последовательными видами. По числу дискретных (сосредото-
ченных) элементов кинетические модели можно сгруппировать следующим образом
[Joshi et al., 2008]:
1) 2-элементные модели [Al-Soufi et al., 1988; Krishna et al., 1988; Di Carlo and Ja-
nis, 1992];
2) 3-элементная модель [Benito et al., 1995];
3) 4-элементные модели [Del Bianco et al., 1993; Trauth et al., 1992];
4) 5-элементные модели [Kataria et al., 2004; Singh et al., 2005];
5) 6-элементная модель [Reza et al., 2011];
6) 8-элементная модель [Takatsuka et al., 1989];
7) 12-элементная модель [Xiao et al., 2002].
Схемы реакций для этих кинетических моделей представлены на рис. 3.5.
Значения кинетических параметров (констант скоростей реакций, энергий акти-
вации, предэкспоненциальных множителей), приведенные авторами, были получены
ими опытным путем при различных температурах, давлениях, временах пребывания,
режимах работы реактора (непрерывный или периодический процесс, висбрекинг
в змеевике или в реакционной камере) и типах сырья (атмосферный остаток, вакуум-
ный остаток, асфальтены, отделенные от нефтей различного происхождения).
Далее в качестве примера будет рассмотрена модель с пятью дискретными эле-
ментами, которую описали Сингх и соавторы [Singh et al., 2005]. Целью их иссле-
дования было разработать кинетическую модель термического крекинга в условиях
низкой жесткости. Элементы модели — вакуумный остаток (ВО, 500 °С+), тяжелый
газойль (ТГ, 350-500 °C), легкий газойль (ЛГ, 150-350 °C), бензиновая фракция (БФ,
С5—150 °C) и газы (Г, С, + С2 плюс сжиженный нефтяной газ, то есть С5-). Как можно
видеть из левой части рис. 3.6, модель имеет 10 кинетических параметров (otJc, доА:|0).
Для оценки параметров использовались экспериментальные данные термического
крекинга, проводимого в периодическом реакторе емкостью 400 мл из нержавеющей
стали, четырех разных остатков вакуумной перегонки индийской и ближневосточ-
ной нефтей.
116
Часть 2. Моделирование некаталитических процессов
Остаточное
сырье
Продукты
крекинга
Остаточное
сырье
Облегченные продукты
2-элементные модели
[Al-Soufi et al., 1998;
Di Carlo and Janis, 1992;
Krishna etal., 1998]
Кокс
3-элементная модель
[Benito etal., 1995]
Асфальтены
* Мальтены
Остаточное
4-элементная модель
[Del Bianco et al., 1993]
4-элементная модель
[Trauth et al., 1992]
♦ С
Остаточное
Остаточное
сырье
5-элементная модель
[Kataria et al., 2004]
5-элементная модель
[Sing etal.,2005]
б-элементная модель [Reza et al., 2011]
538.с+370-538 °C 150-370"С 150°с_
Компоненты, —>
растворимые
вн-С,
Компоненты,
нераство- ~*
римыев н-С7
и растворимые
в толуоле
Компоненты,
нераство-_Компоненты,
римые растворимые
в толуоле в хинолине
и растворимые
в хинолине
300-330"С
270-300 °C
240-270 °C
210-240 °C
480-510 °C
Остаточное
сырье
330-360 °C
360-390 °C
390^20 °C
420-450 °C
450^30 °C
12-элементная модель [Takatsuka et al., 1989]
8-элементная модель [Xiao et al., 2002]
Рис. 3.5. Схемы реакций висбрекинга для различных кинетических моделей с сосредото-
ченными элементами: ЛГ — легкий газойль; ВГ — вакуумный газойль; СНГ — сжиженный
нефтяной газ
Глава 3. Моделирование висбрекинга
117
Первоначальная модель
Сингха с соавторами (2005)
Упрощенная модель
Сингха с соавторами (2005)
Рис. 3.6. Кинетические модели, предложенные Сингхом и соавторами
[Singh, J. et al., Chem. Eng. J., 108, 239, 2005]
Исходя из рис. 3.6 и степенного закона первого порядка, можно написать следую-
щие выражения для скоростей реакций:
^В0/(Л) = -(^ + ^2 + ^ + Ш0; (3.2)
^тг/(^) = ^во-(М М^тг; (3-3)
^лг/(Л) = /с2уВ0 + ^тг-(^ + Л9)улг; (3.4)
^Убф / (^) — ^Уво 3" ^Утг 3" ^Улг — ^Убф> (3.5)
rfyr / (rfr) = ^в0 + ^Т[. + Л^ЛГ + Л10уБф, (3.6)
где у. — массовая концентрация различных фракций при времени пребывания т.
(Остальные сокращения см. в конце главы.)
Исходя из величин констант скоростей реакций, полученных оценкой параме-
тров, и используя графический метод определения порядка реакций Delplot {Delaware
Plot — метод, разработанный университетом штата Делавэр), некоторые исследовате-
ли [Bhore et al., 1990; Singh et al., 2005] заметили, что в предлагаемой схеме реакции,
управляемые кинетическими параметрами k7-ki0, не являются доминирующими,
и из этих параметров при температуре 430 °C лишь к& принимает значимую величину.
По этой причине пути реакций через параметры к7, к9 и кю были исключены из рас-
смотрения и предложена упрощенная реалистичная модель с семью константами ско-
ростей реакций термического крекинга, изображенная в правой части рис. 3.6. Выра-
жения скоростей реакций для различных сосредоточенных элементов имеют тот же
вид, что и в уравнениях (3.2)—(3.6), но с исключенными параметрами kv к9 и к10.
В другой работе [Castellanos et al., 1991] предложены следующие выражения для
расчета значений энергий активации и предэкспоненциальных множителей в зави-
симости от молярной массы сырья (MWI) и продуктов (Л/Ж), участвующих в каждом
маршруте реакции (z ->у):
118
Часть 2. Моделирование некаталитических процессов
к. = А к ехр
-М,
RT
(3.7)
- {а0 + aiMWj + а2МИ^)еХр
(3.8)
£4.. = йп + й,Л/Ж+^Л/Ж. (3.9)
Приведенные значения параметров a. vtb. составляли: а0= 1,5072- Ю12,^^ 1,90-10®,
а2 = 2,0561 • 10% а3 = 146,95, аА = 11,349, % = 42894, bt = -4,5 и Ь2 = 3.
Формулы (3.7)—(3.9) позволяют предсказать величины А и ЕА по известным свой-
ствам сырья и продуктов. Чтобы приступить к расчетам, необходимо знать молярные
массы различных элементов модели, которые можно установить анализом состава
сырья и продуктов промышленных установок висбрекинга. Если эти данные неиз-
вестны, приближенные значения молярных масс различных элементов модели мож-
но рассчитать из данных анализа нефти, перегонкой которой было получено сырье
висбрекинга.
3.7. МОДЕЛИРОВАНИЕ РЕАКТОРОВ
3.7.1. Эмпирические зависимости
Фахим и соавторы [Fahim et al., 2009] представили следующие эмпирические зави-
симости для определения выхода продуктов, плотности по шкале API и содержания
серы в продуктах висбрекинга, выведенные изданных [Maples, 1993].
• Выход продуктов, % масс.:
уг = 0,189825%+ 0,677163; (3.10)
уБф = 0,738321%+ 0,260174; (3.11)
уво = —0,146668%2 - 2,203644% + 98,677947; (3.12)
у„ , = 0,02023% + 0,060438,- 0,156; (3.13)
Н2Ь J
у[л = 100 - yf - %Ф -7ВО - J>H2S’ (3-14)
где Г — газ; Гл — газойль, Гл = ЛГ + ТГ; % — глубина превращения.
• Содержание серы в продуктах, %масс.:
SH2S = (32/34)yH2S;
SM = 0,260112SZ;
(3.15)
(3.16)
Глава 3. Моделирование висбрекинга
119
Srj] = 0,539924Sz; (3.17)
SBo = S/-SE(t)-Srn-SH2S, (3.18)
где Sz— содержание серы в сырье.
• Плотность продуктов, ° API'.
APLb = -0,26215* + 0,315121ЛРЛ + 56,83723; (3.19)
АР1Гл = -0,052919* + 0,52228042/17^ + 12,9318914; (3.20)
АР1В0 = -0,7462183*+ 1,2913825ЛР/,-2,6831388, (3.21)
где APIf— плотность сырья, ° API.
Мейплз [Maples, 1993] определил глубину превращения как относительный сум-
марный выход газа и бензиновой фракции:
х=+г++бф100> 22)
+во
где * — глубина превращения.
Для применения этих зависимостей нужно знать глубину превращения, содержа-
ние серы в сырье и его плотность по шкале API. Формулы (3.10)—(3.21) были выведе-
ны на основе 30 массивов данных для глубин превращения от 3,8 до 15,69 %, которые
подготовил Мейплз. То есть для вывода зависимостей использовались глубины пре-
вращения меньшие, чем при промышленном висбрекинге с реакционной камерой.
Иными словами, указанные зависимости можно использовать лишь для быстрых
приближенных расчетов в ограниченном интервале условий, в том числе при огра-
ниченных глубинах превращения.
3.7.2. Моделирование реакторов
3.7.2.1. Общие характеристики реакторов висбрекинга
Печной висбрекинг проводится при повышенной температуре и малом времени
пребывания; из-за этого он получил сокращенное наименование HTST (High-Tem-
perature, Short-residence-Time). Сырье поступает в печь с температурой 300-330 °C
и в течение 2-5 мин подвергается в змеевике крекингу при температуре 460-480 °C.
Реакции крекинга протекают в жидкой фазе с поглощением небольшого количества
теплоты (192 ккал на 1 кг продукта с температурой кипения до 204 °C).
В другом варианте висбрекинга сырье проходит змеевик за более короткое вре-
мя (2—3 мин) и подвергается воздействию более низких температур (около 450 °C).
После этого поток из змеевика поступает в реакционную камеру, которую в опреде-
ленной степени можно считать адиабатической, и выдерживается там относительно
долго (15-25 мин). Пониженная температура в змеевике компенсируется длительным
120
Часть 2. Моделирование некаталитических процессов
пребыванием в реакционной камере, что в итоге дает примерно такую же глубину
превращения, как и при печном висбрекинге. Висбрекинг с реакционной камерой
сокращенно обозначают как процесс LTHT (Low-Temperature, High-residence-Time).
Реакции крекинга протекают в змеевике и в реакционной камере. Они идут с по-
глощением теплоты, поэтому температура на выходе камеры на 10-20 °C ниже, чем
на входе.
3.7.2.2. Моделирование процессов в змеевике
и в реакционной камере
3.7.2.2.1. Обзор современных технологий
Змеевики и реакционные камеры можно отнести к аппаратам с газожидкостны-
ми системами, в которых диспергированная фаза состоит из образовавшихся газов,
испаренных низкомолекулярных углеводородов и водяного пара, а сплошную фазу
представляет остаток висбрекинга. Таким образом, змеевик можно рассматривать
в качестве двухфазного реактора идеального вытеснения (PFR) либо последователь-
ности равнообъемных реакторов идеального смешения (CSTR), а реакционную каме-
ру— как барботажный колонный реактор или реактор идеального смешения. Реакции
висбрекинга протекают в жидкой фазе по свободнорадикальному механизму, причем
время пребывания в реакторах обоих типов определяется скоростью потока сырья.
На время пребывания в жидкой фазе косвенно влияет рабочее давление. Повыше-
ние давления подавляет испарение низкокипящих фракций, тем самым увеличивая
объем жидкой фазы и время пребывания. Помимо этого, соотношение приведенных
скоростей газа и жидкости, составляющее обычно от 2 до 5, может изменяться в зави-
симости от глубины превращения, рабочего давления и количества пара, вводимого
в змеевик. При моделировании реакторов обоих типов учет этих эффектов поможет
точнее рассчитать время пребывания в жидкой фазе.
Разными авторами предложены различные подходы к моделированию реакторов
висбрекинга [Joshi et al., 2008].
• Кастельяно с соавторами [Castellanos et al., 1991] представил сырье и продук-
ты набором дискретных псевдокомпонентов, сгруппированных в соответствии
с числом атомов углерода. Предложена лишь модель змеевикового реактора,
ведущего себя как реактор идеального вытеснения.
• Карбонелл и Жирарделло [Carbonell and Guirardello, 1997] предложили ги-
дродинамическую модель (потери давления, радиальное распределение газа
и суспензии, эффективная турбулентная вязкость и эпюра скоростей жидкой
фазы). Они также смоделировали наложение реакций термического крекин-
га с учетом их распространения в радиальном направлении, чтобы предсказать
глубину превращения тяжелой нефти. Было установлено, что на выход продук-
тов сильно влияет картина рециркуляции (возвратного смешивания) жидкой
фазы. Однако модели экспериментально не проверялись.
• Миттал и соавторы [Mittal et al., 1999] смоделировали висбрекингсреакционной
камерой как совокупность реактора идеального вытеснения и последовательно
Глава 3. Моделирование висбрекинга
121
соединенных с ним реакторов идеального смешения. Потери давления в змее-
вике печи оценивались по уравнениям Беггса и Брилла [Beggs and Brill, 1973].
С большой точностью были рассчитаны потери давления в змеевике, профиль
распределения температур в змеевике и реакционной камере, а также глубина
превращения остаточного сырья. Модель была разработана для однотипного
сырья, а реакции крекинга заменялись одной суммарной реакцией: моделиро-
вался крекинг вакуумного остатка до газа и бензиновой фракции.
• Кулкарни [Kulkami, 2005] смоделировал установку висбрекинга с реакционной
камерой экспериментальных масштабов и попытался устранить расхождения
между данными опытной установки и реактора периодического действия, ум-
ножая константы скоростей реакций в периодическом реакторе на подходящие
коэффициенты масштабирования. Змеевик моделировался сдвоенным реакто-
ром идеального вытеснения; реакционная камера представлялась последова-
тельно соединенными реакторами идеального смешения с промежуточным об-
ратным перемешиванием.
Таким образом, опубликованные модели реакторов висбрекинга в змеевике и в ре-
акционной камере разрабатывались с применением разных подходов. Для их провер-
ки использовались экспериментальные (периодический реактор и опытная установка)
и промышленные данные установок висбрекинга в змеевике и в реакционной камере.
Здесь при иллюстрации моделирования реакторов висбрекинга принимается, что
змеевик ведет себя как реактор идеального вытеснения и может быть смоделирован
уравнением кинетики такого реактора либо представлен в виде последовательности
равнообъемных реакторов идеального смешения (так называемая ячеечная модель).
Реакционная камера ведет себя как адиабатический реактор идеального смешения.
Схемы представления реакторов процессов двух типов изображены на рис. 3.7.
Рис. 3.7. Схематическое представление змеевика и реакционной камеры
в целях моделирования
122
Часть 2. Моделирование некаталитических процессов
3.7.2.2.2. Модель с реактором идеального вытеснения
Змеевик можно моделировать уравнением кинетики реактора идеального вытеснения:
(3.23)
Jo (~^)
Уравнение (3.23) вместе с уравнениями кинетической модели образует систему
обыкновенных дифференциальных уравнений первого порядка, которую можно ре-
шить относительно длины реактора численно методом Рунге—Кутты четвертого по-
рядка. Для случая изотермической работы реактора существует аналитическое реше-
ние системы. Например, для пятиэлементной кинетической модели, предложенной
Сингхом и соавторами [Singh et al., 2005], для начальных условий
т = 0,
^0 = ^00- <3-24)
^тг = 3;лг = ^БФ=3;г = О <3'25)
решение системы уравнений (3.2)—(3.6) и (3.23) для выхода продуктов имеет вид
yBo=wexP(-W; <3'26)
Утг = ^ + Ув°°[схр(-^) - ехр(-ед; (3.27)
&В
Улг = ^вооИехР(-Алт)--5ехР(-Авт) + (Я-Л)ехр(-^т)]; (3.28)
1;БФ=>’вооИехР(~/’мт)+ С]ехр(-Агст) + (jB, — Л, — С\)ехр(—Л10т)]; (3.29)
Уг -^воо
1 С М?
— kt +---!-<—
кА кв-кА
+ ДД0 + Ak9 (1 - ехр(-^т)) -
1 Г /с|/с7
1А\кв-ка
+ В{к{0 +Bkg (1 - ехр(-л:лт))
+ ~ А^к7 + сЛо)х ~ ехр(-АДт)) + (Д + Д - С,) (1 - ехр(-ЛДт))
Кс
(3.30)
Здесь константы КА, Кв, Кс, А, В, Д, В, и С, зависят от значений кинетических
констант k^—k^, определяемых из формул (3.7)—(3.9), и вычисляются следующим
образом:
КА ~ kl + к2 + ^3 + кА
(3.31)
+
Глава 3. Моделирование висбрекинга
123
Кв ~ ks + К + kr
Kc=ks + kv
—!—[k, +.. 1
кс-кА\- к3-кл)'
k,k5
SKB-KAXKC-KB)
/1 — | & + AL ч | -
I KB-KA)kl0-KA
я = Bk, +-к'кб |------
1 I KB-KA)kl0-KB’
c _ (^-Ж
1 k>a-Kc'
(3.32)
(3.33)
(3.34)
(3.35)
(3.36)
(3.37)
(3.38)
Для упрощенной модели Сингха и соавторов [Singh et al., 2005] можно использо-
вать те же уравнения, исключив из них кинетические параметры к7, к., и kw.
3.7.2.2.3. Модель с последовательностью реакторов
идеального смешения
При висбрекинге с реакционной камерой змеевик можно представить как последо-
вательность реакторов идеального смешения (ячеечная модель). Реакционная каме-
ра при этом моделируется одним реактором идеального смешения. Баланс масс /-го
компонента ву-м реакторе имеет вид
КДо ~ У,) - VCSTR(3.39)
где
Fq — массовая скорость сырья на входе в реактор, принятая постоянной;
у — массовая концентрация /-го компонента на входе в реактор;
у — массовая концентрация /-го компонента на выходе из j-ro реактора;
— объем одного реактора (одинаковый для всех реакторов);
р0 — плотность реакционной смеси;
г.. — скорость реакции /-го компонента ву-м реакторе.
Для случая модели Сингха и соавторов скорость реакции г /-го компонента в у-м
реакторе определяется уравнениями (3.2)—(3.6).
В уравнении (3.39) г зависит от констант скоростей реакций и массовых
концентраций:
124
Часть 2. Моделирование некаталитических процессов
<3-40)
Зависимость к. от температуры представляется законом Аррениуса (см. (3.7)).
Уравнения (3.39) и (3.40) применимы к одному реактору идеального смешения,
имитирующему реакционную камеру, и к последовательности таких реакторов, ими-
тирующей змеевик.
3.7.2.2.4. Тепловой баланс для последовательности реакторов
идеального смешения
Тепловой баланс дляу'-го реактора идеального смешения имеет вид
/ N \
Q- f.cjr - Го) - Л»,Л,„| (3.41)
где 1
С — теплоемкость реакционной смеси, вычисляемая как средневзвешенная ве-
личина по величинам теплоемкостей элементов модели [Кет, 1950];
Т. — температура на выходе из /-го реактора;
TQ — температура на входе в/-Й реактор;
\HR — теплота реакции, принятая равной 800 кДж/кг для продуктов с температу-
рой кипения до 204 °C [Filho and Sugaya, 2001];
N — число компонентов модели.
Для каждого реактора необходимо совместно решить уравнения (3.40) и (3.41). Ре-
шение дает массовую концентрацию у и температуру Т. /-го компонента на выходе
из у-го реактора.
3.7.2.2.5. Парожидкостное равновесие
Промышленные реакторы висбрекинга работают в неизотермических условиях, ког-
да температура в реакторе не является постоянной. Колебания температуры вызывают
изменения скоростей потоков, составов и теплофизических свойств паровой и жид-
кой фаз. Для учета испарения углеводородов необходимы расчеты равновесия паровой
и жидкой фаз, выявляющие влияние равновесия на поведение реактора. На рис. 3.8 схе-
матически представлена модель реактора, учитывающая парожидкостное равновесие.
Согласно этой модели, сырье разделяется на входе в реактор на две фазы: жидкую
и паровую, составы и расходы которых определяются расчетом испарения. В реак-
тор поступает лишь жидкая фаза. По уравнениям модели реактора рассчитывают-
ся расходы и составы образовавшихся в нем жидкостей и газов, а также температура
в реакторе. Поток из реактора смешивается с паровой фазой, выделенной на входе
в реактор, после чего определяются новые значения расходов, составов и темпера-
туры. Новая смесь поступает на следующий этап расчета или в следующий реактор.
Процедуры расчетов по модели равновесия фаз и кинетической модели продолжа-
ются до исчерпания длины змеевика, если он моделируется как реактор идеального
вытеснения, или до последнего реактора, если змеевик моделируется как последова-
тельность реакторов идеального смешения.
Глава 3. Моделирование висбрекинга
125
Сырье
Реактор 1
Реактор 2
ТоУ;,о |________ ...
Модель равно-1 ж""'
весия паровой —------
и жидкой фаз
Паровая
фаза
Т1 У/,1
Модель равно-1 фаза
весия паровой--------
и жидкой фаз
Паровая
фаза
Т2У1,2
Жидкая -----------------
-—-—> Модель реактора
Смесь паровой
и жидкой фаз
Жидкая
Модель реактора
Смесь паровой
и жидкой фаз
Рис. 3.8. Модель реактора висбрекинга с учетом парожидкостного равновесия
Испаренная доля и составы жидкой и паровой фаз в расчетах равновесия фаз вы-
числяются методом Рэчфорда-Райса, константы равновесия К. в котором определя-
ются по уравнениям Вильсона с учетом поправок на состав по уравнению состояния
Пенга-Робинсона [McCain, 1990].
3.7.3. Моделирование процесса висбрекинга
3.7.3.1. Характеристики реакторов и рабочие условия
Для иллюстрации описанной выше модели процесса объем реакторов был принят
таким, чтобы время пребывания в змеевике и в реакционной камере составляло со-
ответственно 2,7 и 19,1 мин. Это типичные значения для реакторов промышленных
установок висбрекинга.
126
Часть 2. Моделирование некаталитических процессов
Число, длина и диаметр труб печи, а также диаметр и высота реакционной каме-
ры зависят от конструкции и типа установки висбрекинга. Например, Реза и соавто-
ры [Reza et al., 2011] сообщили о создании модели промышленной установки висбре-
кинга со следующими характеристиками реакционных секций:
Змеевик
Ч исло труб 128 (76 в конвекционной секции, 52 — в радиантной)
Длинатруб, м 18,745
Наружный диаметр труб, м 0,114
Реакционная камера
Наружный диаметр, м 2,405
Длина, м 16,5
Реакционная камера обычно представляет собой цилиндрический вертикаль-
ный аппарат с соотношением высоты и диаметра около 6:1 и объемом, который дол-
жен обеспечить пребывание жидкого потока в течение 15—25 мин. Необходимость
более длительного пребывания в реакционной камере по сравнению со змеевиком
(2—3 мин) становится очевидной, если учесть разность соответствующих температур
реакций.
Температуры на входе и выходе змеевика равны 326,5 и 439 °C, давление — соответ-
ственно 2,62 и 0,90 МПа (потери давления составляют 1,72 МПа). Для массового рас-
хода в целях воспроизведения процесса принимается типичная величина 124 300 кг/ч
(номинальная скорость подачи около 20 000 баррель/сут). В змеевик вместе с сырьем
подается также 1 %масс. водяного пара. Для потерь давления и скорости подачи пара
взяты типичные значения из литературных источников [Joshi et al., 2008]. Змеевик
моделируется как неизотермический реактор, и необходимая скорость подведения
тепла принята за 4,33 106 кДж/ч. Реакционная камера моделируется как адиабатиче-
ский реактор, поэтому температура в ней снижается из-за поглощения тепла в реак-
циях висбрекинга.
Висбрекинг — термический процесс, а выход различных углеводородных продук-
тов и глубина превращения сырья напрямую определяются температурой реакции.
То есть прямой мерой жесткости висбрекинга является температура, до которой на-
гревается сырье в печи.
Не следует забывать, что в данной схеме висбрекинга, т. е. в змеевике и реакцион-
ной камере, основная часть реакций крекинга происходит не в змеевике печи, а в вы-
носной камере, расположенной вне печи (сокинг-камера). Поэтому можно ожидать,
что глубина превращения в змеевике будет меньше, чем в реакционной камере.
3.7.3.2. Свойства сырья и продуктов
Сырьем при расчетном моделировании является остаток перегонки смеси тяже-
лых нефтей «Майя» и «Истмо» (Мексика). Объемное содержание фракций с темпе-
ратурой кипения ниже 538 °C равно 9,25 %. Состав сырья выражается следующим
образом:
Глава 3. Моделирование висбрекинга
127
Легкий газойль (200—400 °C), %об.
Тяжелый газойль (400-538 °C), %об.
Вакуумный остаток (538 °С+), %об.
0,51
8,74
90,75
Данные эксплуатации промышленной установки висбрекинга позволили вычис-
лить некоторые свойства продуктов (такие как плотность, кривая разгонки, состав).
В свою очередь, это позволило рассчитать значения свойств, необходимые для моде-
лирования (плотность, теплоемкость и молярную массу). Расчетные значения неко-
торых свойств продуктов висбрекинга приведены в табл. 3.1.
Свойства продуктов висбрекинга
Таблица 3.1
Продукты Интервал кипения, °C Отн. плотность 60/60 °F Теплоемкость, кДж/кг °C Молярная масса
Газ — 0,364 3,16 34,2
Бензиновая фракция <200 0,745 3,68 123,6
Легкий газойль 200^400 0,854 3,47 231,1
Тяжелый газойль 400-538 0,914 3,28 498,3
Вакуумный остаток >538 1,037 3,01 1052,0
3.7.3.3. Результаты моделирования
Результаты моделирования представлены на рис. 3.9-3.12 в виде графиков из-
менения выхода продуктов, глубины превращения, температуры и соотношения
пар/жидкость по длине змеевика, а также на входе и выходе реакционной камеры.
Для расчетов использовалась упрощенная модель Сингха и соавторов [Singh et al.,
2005], представленная в правой части рис. 3.6.
Глубина превращения х538 ,с+ остатка с температурой кипения выше 538 °C и выхо-
ды продуктов у. вычислялись по следующим формулам:
•^ззв ’С+
Доля фракции 538 °C + в сырье - Доля фракции 538 °C + в продукте
Доля фракции 538 °C + в сырье
100; (3.42)
Масс, расход фракции i в продукте - Масс, расход фракции i в сырье
=------------- ------------------------------------------------- 100 (3 43)
Масс, расход вакуумного остатка в сырье
На рисунках можно видеть зависимость параметров от безразмерной (т. е. относи-
тельной) длины змеевика. Значение 1 соответствует концу змеевика, затем идет от-
резок прямой, отражающий поведение реакционной камеры. Было установлено, что
поведение реактора идеального вытеснения, которым моделируется змеевик, вос-
производят десять последовательно соединенных реакторов идеального смешения
равного объема. Иными словами, каждые 10 % длины змеевика представлены одним
реактором идеального смешения (CSTR).
128
Часть 2. Моделирование некаталитических процессов
Змеевик Реакционная
камера
Рис. 3.9. Расчетные значения температуры и отношения пар/жидкость:
(—) — с учетом равновесия пар-жидкость; (—) — без его учета
Змеевик
Реакционная
камера
Рис. 3.10. Расчетные значения выхода вакуумного остатка и глубины превращения:
(- -) — с учетом равновесия пар-жидкость; (—) — без его учета
Глава 3. Моделирование висбрекинга
129
*-----------------------------------------------►
Змеевик Реакционная
камера
Рис. 3.11. Расчетные значения выходов тяжелого газойля и бензиновой фракции:
(—) — с учетом равновесия пар-жидкость; (—) — без его учета
Рис. 3.12. Расчетные значения выходов газа и легкого газойля:
(—) — с учетом равновесия пар-жидкость; (—) — без его учета
Данные рис. 3.9-3.12 позволяют сделать ряд выводов.
• Выход всех продуктов (газ, бензиновая фракция, легкий и тяжелый газойли)
по длине реакторов возрастает, а выход вакуумного остатка (непревращенного
сырья), как и следует ожидать, снижается.
• По мере прохождения реакционной смеси по реакторной системе глубина пре-
вращения возрастает. В змеевике она низка (менее 0,5 %); большая часть пре-
вращения осуществляется в реакционной камере.
130
Часть 2. Моделирование некаталитических процессов
• На большей части длины змеевика (от 0 до 60 %, где достигается температура
395 ’С) влияние парожидкостного равновесия пренебрежимо мало. Это значит,
что до этой точки реакционная смесь содержит существенно больше жидкости,
чем пара. Известно также, что реакция крекинга обычно не приобретает выра-
женный характер, пока температура не превысит 400 °C.
• В последней части змеевика, занимающей от 60 до 100 % его длины, значе-
ния всех параметров, вычисленные с учетом и без учета равновесия пар—жид-
кость, расходятся, хотя и незначительно. В этой части змеевика соотношение
пар / жидкость возрастает до 0,44. Отсюда можно заключить, что прирост тем-
пературы в данной секции (от 397 до 439 °C) вызывает, во-первых, образова-
ние большего количества паров, во-вторых — усиление реакций, дающих газы
и легкие углеводороды.
• Учет парожидкостного равновесия по-разному влияет на продукты висбрекин-
га. Моделирование с учетом равновесия дает пониженный выход легких про-
дуктов (газа, бензина и легкого газойля), но для тяжелого газойля наблюдается
противоположное. Потоки обоих реакторов представляют собой парожидкост-
ную смесь. На самом деле сырье и остаток висбрекинга находятся полностью
в жидкой фазе, легкий газойль, бензин и газы — преимущественно в паровой
фазе, а тяжелый газойль распределен между двумя фазами. Кроме того, дли-
тельность пребывания фаз в реакторах неодинакова. Соотношение времен пре-
бывания паровой и жидкой фаз в змеевике печи колеблется между 0,2 и 0,5,
а для реакционной камеры составляет лишь 0,1. Эта разница является при-
чиной менее продолжительного пребывания газойля в реакционной камере
по сравнению со змеевиком и, соответственно, замедления его распада на бен-
зин и газы.
• Для завершения реакций висбрекинга поток из змеевика направляется в ре-
акционную камеру. Изменения в выходе продуктов и глубине превращения
в реакционной камере гораздо значительнее, чем в змеевике. Благодаря более
длительному пребыванию большая часть превращения осуществляется в реак-
ционной камере.
• Температура в реакционной камере снижается с 439 °C на входе до 428 °C на вы-
ходе, что обусловлено поглощением тепла в реакциях висбрекинга и адиабати-
ческими условиями.
• Моделирование реакционной камеры с учетом парожидкостного равновесия
сильно изменяет результаты расчета выхода продуктов и глубины превраще-
ния. Сырье (частично превращенный вакуумный остаток и продукты висбре-
кинга), поступающее в этот реактор из змеевика, содержит определенное коли-
чество паров, которое возрастает благодаря длительному времени пребывания
в реакционной камере.
• Различие в выходе продуктов в змеевике и в реакционной камере объясняется
разными значениями энергий активации реакций. В табл. 3.2 приведены значе-
ния энергий активации, вычисленные по уравнениям (3.7)—(3.9). Из таблицы
видно, что энергия активации реакции образования тяжелого газойля из остатка
(39,7 ккал/моль) меньше энергий активации реакций его разложения на легкий
Глава 3. Моделирование висбрекинга
131
газойль (41,3 ккал/моль) и на бензиновую фракцию (41,0 ккал/моль). Поэтому
меньшая температура в реакционной камере вызывает увеличение выхода та-
ких промежуточных продуктов, как тяжелый газойль. Аналогично ведет себя
и легкий газойль.
Таблица 3.2
Параметры уравнения Аррениуса для различных реакций висбрекинга
Реакция Множитель А, ч~' Энергия активации £,, ккал/моль
ВО->ТГ 4,42 10’ 39,7
ВО->ЛГ 1,28 • 10’ 38,9
ВО—>БФ 1,17 • 10“ 38,5
ВО -» газ 3,68-10’° 38,3
ТГ->ЛГ 4,69'10s 41,3
ТГ—>БФ 1,82-10’ 41,0
ЛГ->БФ 1,30-1012 42,2
Сокращения: БФ — бензиновая фракция; ВО — вакуумный остаток, ЛГ и ТГ — соответ-
ственно легкий и тяжелый газойли.
Из числа других выводов по результатам моделирования процесса висбрекинга
в змеевике и реакционной камере можно отметить следующие.
• При высокой температуре происходят загрязнение или закупорка труб змее-
вика (а также реакционной камеры). Поэтому змеевик до определенной тем-
пературы эксплуатируют в неизотермическом режиме; при этом температу-
ра в реакционной камере ниже, чем при изотермической работе змеевика, что
значительно уменьшает скорость загрязнения.
• Жесткость условий (температура реакции и время пребывания) обычно огра-
ничивают из-за необходимости получать остаток висбрекинга, часто исполь-
зуемого как компонент стабильного нефтяного топлива, дающего мало осадка
при хранении. Осадок нежелателен по той причине, что он может быстро за-
бить фильтры насосов, а их очистка требует немало времени.
• Когда температура в реакторе достигает 460 °C, глубина крекинга и выход газа
и бензиновой фракции существенно увеличиваются. Вязкость продукта сни-
жается с одновременным ростом образования отложений кокса в трубах змее-
вика и в реакционной камере. Увеличение жесткости условий сокращает дли-
тельность цикла и приводит к получению нестабильного нефтяного топлива
с осадком.
3.7.3.4. Заключительные замечания и рекомендации
Моделирование реакторов висбрекинга, т. е. змеевика и реакционной камеры, из-за
сложной природы сырья и участвующих в процессе реакций продолжает оставаться
132
Часть 2. Моделирование некаталитических процессов
трудной задачей. Для моделирования реакторов висбрекинга обоих типов применя-
лись разные стратегии. Однако все они учитывали, что реакционная смесь является
газожидкостной системой, состоящей из диспергированной и сплошной фаз, а реак-
ции протекают преимущественно в жидкой фазе. Хотя сообщалось о различных под-
ходах к моделированию, змеевик чаще всего упрощенно представляется как реактор
идеального вытеснения, а реакционная камера — как реактор идеального смешения.
Кинетические модели реакторов висбрекинга основаны большей частью на ме-
тоде дискретизации, в рамках которого важнейшие продукты висбрекинга — газы
(иногда по отдельности газы С(-С2 и С3—Ц), бензиновая фракция, легкий и тяже-
лый газойли и вакуумный остаток — отображаются в модели как сосредоточенные
псевдокомпоненты. Некоторые модели включают в число продуктов кокс, но в боль-
шинстве случаев он исключен из рассмотрения, так как процесс протекает в мягких
условиях (низкая температура, короткое время пребывания и малая глубина превра-
щения в змеевике). Поэтому скорость образования кокса в реакционной камере пре-
небрежимо мала.
Экспериментальные данные различных уровней (периодический реактор, опыт-
ная установка, промышленные установки) обширны, а выведенные из них параме-
тры кинетической модели во многом зависят от свойств сырья. Поэтому примене-
ние для сырья одного типа параметров, выведенных для другого, рискованно и может
увеличить погрешность прогнозирования выхода продуктов. Это один из типичных
недостатков моделей с дискретными элементами. Что интересно, лишь одна модель
содержит расчет энергий активации и факторов частоты соударений молекул в за-
висимости от молярной массы дискретных компонентов реакционной схемы. При-
веденные результаты хорошо согласуются с промышленными данными. Этот под-
ход мог бы быть полезен для моделирования влияния сырья на реактор висбрекинга.
В таком случае молярные массы компонентов становятся параметрами характериза-
ции сырья, правильная оценка которых необходима для получения приемлемых ре-
зультатов расчетов.
Еще один пункт расхождений опубликованных дискретных кинетических моде-
лей — интервал кипения различных продуктов висбрекинга, использованный при
выводе констант скоростей реакций. Например, Сингх и соавторы [Singh et al., 2005]
определяют вакуумный остаток как фракцию с нижней границей кипения 500 °C,
тогда как чаще принимается величина 538 °C. Это обстоятельство усложняет сравне-
ние значений кинетических параметров, полученных из других результатов, и при-
менение их для расчета выхода продуктов с другими интервалами кипения.
Что до остаточного сырья, то его описание должно исходить из общедоступных
сведений и включать в себя все важные особенности. Типичные результаты анали-
за сырья состоят из кривой разгонки и данных о кинематической вязкости, плотно-
сти, содержании серы и асфальтенов и коксуемости по Конрадсону. Для разработки
более детальных кинетических моделей необходимо более подробное описание сы-
рья. Улучшить возможности кинетических моделей могло бы, например, включение
в описание сырья метода SARA (определение содержания предельных углеводородов,
ароматических соединений, смол и асфальтенов) и анализа каждого компонента.
Глава 3. Моделирование висбрекинга
133
Если затронуть вопрос гидродинамики, то здесь следует иметь в виду, что время
пребывания жидкой фазы зависит от давления, так как его рост подавляет испарение
низкокипящих фракций, а также увеличивает объем жидкой фазы — и, следователь-
но, время ее пребывания. В промышленных установках висбрекинга приведенная
скорость газа в 2—5 раз больше приведенной скорости жидкости, причем это соот-
ношение может изменяться в зависимости от глубины превращения, рабочего давле-
ния и количества пара, вводимого в змеевик; соответственно, может изменяться ре-
жим работы реакторов. Поэтому, чтобы точно предсказать время пребывания жидкой
фазы и правильно смоделировать гидродинамику реактора, необходимо учесть все
эти эффекты. Еще один существенный параметр — потери давления.
Желательно, чтобы модель висбрекинга позволяла рассчитать, кроме выхода про-
дуктов, глубины превращения и распределения температур, еще и свойства продук-
тов, которые можно получить из данного сырья, а также жесткость условий реакций
и геометрические характеристики установки. Из свойств остатка висбрекинга важ-
но знать, в частности, вязкость, содержание асфальтенов, коксуемость по Конрадсо-
ну и стабильность. Нужны данные и о прочих свойствах продуктов, таких как плот-
ность, содержание серы, температура текучести и кривая разгонки.
Надежная модель процесса висбрекинга поможет спрогнозировать выход и свой-
ства продуктов, глубину превращения и прочие параметры процесса, когда стоит за-
дача определить:
• наиболее прибыльную рабочую область при данном сырье;
• влияние изменения рабочих условий на выход продуктов, распределение тем-
ператур и потери давления;
• новые рабочие условия, обеспечивающие требуемый прирост глубины
превращения;
• максимальную глубину превращения, при которой продукты висбрекинга со-
храняют стабильность;
• максимально возможное снижение вязкости сырья без существенного ухудше-
ния стабильности нефтяного топлива.
ОБОЗНАЧЕНИЯ
ДЯя Теплота реакции
Ро Плотность реакционной смеси
т Время пребывания
БФ Бензиновая фракция
ВГ Вакуумный газойль
ВО Вакуумный остаток
Г Газ(ы)
лг Легкий газойль
тг Тяжелый газойль
134
Часть 2. Моделирование некаталитических процессов
а, . Константы уравнения (3.8) для расчета факторов частоты соударений
молекул
Ь. Константы уравнения (3.9) для расчета энергий активации
А Константа, определяемая формулой (3.34)
А, Константа, определяемая формулой (3.36)
APIf Плотность сырья по шкале API
А Фактор частоты соударений молекул в реакции i -> j
АР1Бф Плотность бензиновой фракции по шкале API
АР1Ъ0 Плотность вакуумного остатка по шкале API
АР1Гп Плотность газойля по шкале API
В Константа, определяемая формулой (3.35)
Константа, определяемая формулой (3.37)
С] Константа, определяемая формулой (3.38)
С Теплоемкость реакционной смеси
ЕА Энергия активации реакции i —> j
F. Массовый расход /-го компонента
КА Константа, определяемая формулой (3.31)
Кв Константа, определяемая формулой (3.32)
Кс Константа, определяемая формулой (3.33)
к. Константа скорости /-Й реакции
MW] Молярная масса f-го компонента в реакции / —> j
MW} Молярная масса у-го компонента в реакции / -> j
Q Скорость теплопередачи
R Универсальная газовая постоянная
г. Скорость реакции /-го компонента
Sz Содержание серы в сырье
Sj-ьз Содержание серы в сероводороде
5Бф Содержание серы в бензиновой фракции
SB0 Содержание серы в вакуумном остатке
Srji Содержание серы в газойле
Т Температура
К'лтл Объем реактора идеального смешения
Ирлг Объем реактора идеального вытеснения
х Глубина превращения
у,. Выход /-го продукта
Глава 3. Моделирование висбрекинга
135
ЛИТЕРАТУРА
Akbar, М., Geelen, Н. 1981. Visbreaker uses soaker dram. Hydrocarbon Process. 60(5):81—85.
Al-Soufi, H.H., Savaya, Z.F., Mohammed, H.K., Al-Azawi, I.A. 1988. Thermal conversion (visbreaking)
ofheavy Iraqi residue. Fuel 67:1714—1715.
Beggs, H.D., Brill, J.P. 1973. A study of two phase flow in inclined pipes. J. Petrol. Techno!. 17:607—617.
Benito, A.M., Martinez, M.T., Fernandez, I., Miranda, J.L. 1995. Visbreaking of an asphaltenic coal res-
idue. Fuel 74:922-927.
Bhore, N.A., Klein, M.T., Bischoff, K.B. 1990. The Delplot technique: A new method for reaction path-
way analysis. Ind. Eng. Chem. Res. 29:313—316.
Bosworth, D. 2005. Visbreaking. In Refining and Petrochemicals, Encyclopaedia of Hydrocarbons, Vol. II.
Ed. Marches! Grafiche, Rome, Italy, Chapter 5.2.
Carbonell, M.M., Guirardello, R. 1997. The effect of bubbling regime on gas and liquid phase mixing in
bubble column reactors. Chem. Eng. Sci. 52:4179-4185.
Castellanos, J., Cano, J.L., Del Rosal, R., Briones, V.M., Mancilla, R.L. 1991. Kinetic model predicts
visbreaker yields. Oil Gas J. 89(11):76-82.
Del Bianco, A., Panariti, N., Beltrame, P.L., Carniti, P. 1993. Thermal cracking of petroleum residues. 1.
Kinetic analysis of the reaction. Fuel 72:75-80.
Di Carlo, S., Janis, B. 1992. Composition and visbreakability of petroleum residues. Chem. Eng. Sci.
47(9-11):2695—2700.
Fahim, M., Al-Sahhaf, T, Elkilani, A. 2009. Fundamentals of Petroleum Refining, Elsevier, Amsterdam,
the Netherlands.
Filho, R. M., Sugaya, M.F. 2001. A computer aided tool for heavy oil thermal cracking process simulation.
Comput. Chem. Eng. 25:683—692.
Joshi, J.B., Pandit, A.B., Kataria, K.L., Kulkarni, R.P., Sawarkar, A.N., Tandon, D., Ram, Y.,
Kumar, M.M. 2008. Petroleum residue upgradation via visbreaking: A review. Ind. Eng. Chem. Res.
47:8960-8988.
Kataria, K.L., Kulkarni, R.P., Pandit, A.B., Joshi, J.B., Kumar, M.M. 2004. Kinetics of low severity vis-
breaking. Ind. Eng. Chem. Res. 43:1373—1387.
Kern, D.Q. 1950. Process Heat Transfer, McGraw-Hill Kogakusha Ltd., Tokyo, Japan.
Krishna, R., Kuchhal, Y.K., Sama, G.S., Singh, LD. 1988. Visbreaking studies on aghajari long residue.
7W67(3):379-383.
Kulkarni, R.P. 2005. Modelling of multiphase reactors: Visbreaking. PhD thesis, University of Mumbai,
Mumbai, India.
Maples, R.E. 1993. Petroleum Refinery Process Economics, Penn Well Books, Tulsa, OK.
McCain, W. 1990. The Properties of Petroleum Fluids, Penn Well Books, Tulsa, OK.
Mittal, S., Maiti, R.N., Lahiri, R.N., Ram Babu, D. 1999. Modelling and simulation of vis-breaker. In:
Proceedings of Third International Petroleum Conference and Exploration, PETROTECH-33, New Delhi, In-
dia, p. 349.
Negin, K.M., Van Tine, F.M. 2004. FW/UOP visbreaking process. In Handbook of Petroleum Refining
Processes, McGraw Hill, New York, Chapter 12.3.
Quignard, A., Kressmann, S., 2011. Visbreaking. In Heavy Crude Oils. From Geology to Upgrading: An
Overview (A. Hue, Ed.), Editions Technip, Paris, France, Chapter 16.
Reza, S., Mohaddecy, S., Sadighi, S. 2011. Simulation and kinetic modeling of vacuum residue soak-
er-visbreaking. Petrol. Coal 53( 1 ):26-34.
Singh, J., Kumara, M.M., Saxena, A.K., Kumar, S. 2005. Reaction pathways and product yields in mild
thermal cracking of vacuum residues: A multi-lump kinetic model. Chem. Eng. J. 108:239—248.
Speight, J.G. 2011. The Refinery of the Future, Gulf Professional Publishing, Burlington, MA.
Stratiev, D., Kirilov, K., Belchev, Z., Petkov, P. 2008. How do feedstocks affect visbreaker operations? Hy-
drocarbon Process. 6(June): 105—112.
136 Часть 2, Моделирование некаталитических процессов
Stratiev, D., Nikolaev, N. 2009. Dependence of visbreaker residue properties on unit operation severity
and the residual fuel oil specification. Petrol. Coal 51(2): 140-145.
Takatsuka, T, Kajiyama, R., Hashimoto, H., Matsuo, 1., Miwa, S.A. 1989. Practical model of thermal
cracking of residual oil. J. Chem. Eng. Jpn. 22(3):304—310.
Trauth, D.M., Yasar, M., Neurock, M., Nasigam, A., Klein, M., Kukes, S.G. 1992. Asphaltene and resid
pyrolysis: Effect of reaction environment. Fuel Sci. Technol. In!. 10(7): 1161—1179.
Xiao, J., Wang, L., Chen, Q., Wang, D. 2002, Modeling for product distribution in thermal conversion of
heavy oil. Petrol. Sci. Technol. 20(5—6):605—612.
Глава 4. Моделирование процесса
газификации
В этой главе описывается технология газификации углеродсодержащего сырья.
При этом основное внимание уделяется методу газификации в потоке, который под-
ходит для переработки нефтяных остатков. Рассматривается влияние ключевых па-
раметров процесса на состав образующегося синтез-газа. Даются сведения о разра-
ботке равновесной модели, балансах масс и энергии, приводится алгоритм решения
этих балансов. Полученная модель проверяется на опубликованных эксперимен-
тальных данных и затем используется для исследования влияния условий газифика-
ции на состав синтез-газа. В конце главы показано применение модели для исследо-
вания влияния свойств сырья на продукты газификации и для определения условий
максимального выхода водорода.
4.1. ВВЕДЕНИЕ
Газификация — гибкая, проверенная в промышленных масштабах, эффективная тех-
нология получения ценных продуктов из малоценного сырья. Она позволяет преоб-
разовать любой твердый или полужидкий углеродистый материал (промежуточные
побочные продукты, асфальт, нефтяной кокс и гудрон) в синтетический газ (син-
тез-газ). Без каких-либо твердых отходов, требующих утилизации, может быть пре-
вращен почти весь углерод [Furimsky, 1999].
Газификация нефтяных остатков дает возможность исключить тяжелое нефтя-
ное топливо из номенклатуры продукции нефтеперерабатывающих заводов и полнее
удовлетворять растущий спрос на легкие продукты за счет их производства из мало-
ценных остатков без привлечения дополнительных объемов нефти. Процесс гази-
фикации остатков можно построить так, чтобы он давал оптимальные количества
водорода, электроэнергии и пара для внутреннего потребления на нефтеперерабаты-
вающем заводе [Higman and Van der Burgt, 2008].
Газификация сырья подразумевает воздействие на него высоких температур в при-
сутствии регулируемого количества кислорода и/или водяного пара. Продуктом яв-
ляется синтез-газ (или просто сингаз), представляющий собой смесь водорода, мета-
на, оксида углерода и диоксида углерода.
138
Часть 2. Моделирование некаталитических процессов
Синтез-газ, получаемый газификацией, как топливо эффективнее исходного сы-
рья, поскольку его можно не только сжигать, получая более высокие температуры,
но и использовать в топливных элементах. Из синтез-газа можно производить ме-
танол и водород либо посредством процесса Фишера—Тропша перерабатывать его
в синтетическое топливо.
Из всех технологий облагораживания тяжелой нефти только газификация может
полностью исключить выход тяжелых остатков. Технология газификации настоль-
ко гибка, что позволяет перерабатывать нефтезаводские остатки любого типа, в том
числе нефтяной кокс, производя при этом ряд ценных продуктов, электроэнергию,
пар, водород и различное химическое сырье. Никакая другая технология переработ-
ки малоценных нефтезаводских остатков не дает столь низкий уровень вредных для
экологии выбросов, как газификация [Rana et al., 2007].
4.2. ТИПЫ ПРОЦЕССОВ ГАЗИФИКАЦИИ
Установки газификации, осуществляемой в газогенераторах или газификаторах,
обычно классифицируют по типу слоя сырья, в котором проводится процесс. Как
следствие, выделяют три группы процессов газификации: в стационарном слое,
в кипящем слое и в потоке. Сегодня в промышленных масштабах применяются реак-
торы газификации всех этих типов.
4.2.1. Газификация в стационарном слое
Сырье поступает в неподвижный слой с постоянной высотой. В таких газогенерато-
рах реакции газификации чаще всего происходят в неподвижной зоне, обычно под-
держиваемой колосниковой решеткой (колосником). Газифицирующий агент (иначе
называемый дутьем) может подаваться двумя способами: снизу, т. е. противоточно
сырью (восходящее дутье), или сверху — параллельно сырью (нисходящее дутье).
Размеры частиц сырья должны быть в пределах от 2 до 50 мм. Реактор, в зави-
симости от газифицирующего агента, может работать в режиме сухого (темпера-
тура в зоне газификации 800-1050 °C) или жидкого (температура в зоне газифика-
ции около 1400 °C) шлакоудаления. Газогенераторы могут работать под давлением
от 0,1 до 3,0 МПа; время пребывания составляет 1-3 ч. Для газификации нефтеза-
водских остатков газогенераторы стационарного слоя могут оказаться неприемлемы-
ми [Speight, 1980; Schuetze and Hofmann, 1984; Choi et al., 2003; Salam and Bhattacha-
rya, 2006].
4.2.1.1. Противоточные газогенераторы стационарного слоя
Газогенераторы с противоточным, или восходящим, дутьем относятся к старейшим
и простейшим аппаратам. Газифицирующий агент (пар, кислород и/или воздух) пода-
ется снизу; газ выходит сверху противоточно сырью, опускающемуся в реакционный
Глава 4. Моделирование процесса газификации
139
слой. Для реакторов этого типа характерна низкая кратность пара к сырью, что по-
зволяет достигать температур выше точки плавления золы. Пропускная способность
невелика. Термический КПД высок, так как температура выходящего газа относи-
тельно низка.
Внизу протекают реакции окисления, несколько выше — реакции восстанов-
ления, а в верхней секции реактора в результате конвекционного и радиационно-
го переноса тепла из расположенных ниже зон происходит термическое разложе-
ние (пиролиз) сырья. Горячий сингаз из зоны газификации нагревает и пиролизует
опускающееся вниз сырье. Золу удаляют снизу газогенератора. Образующиеся смола
и летучие продукты уносятся газом. Расход кислорода очень мал, а сингаз содержит
продукты пиролиза.
4.2.1.2. Параллельноточные газогенераторы
стационарного слоя
Газифицирующий агент движется параллельно с сырьем и вводится в зоне окисления
или несколько выше. Через сжигание небольшого количества сырья или из внешнего
источника необходимо подводить тепло к верхней части слоя. Большую часть тепло-
ты часто подводят через газифицирующий агент, подавая его в верх слоя; в этом слу-
чае энергетический КПД становится сравнимым с КПД противоточного газогенера-
тора. Образующийся газ, имеющий высокую температуру, отводится с низа аппарата
и движется в одном направлении с сырьем.
Главные недостатки параллельноточных газогенераторов — неспособность рабо-
тать с сырьем низкой плотности, вызванная нестабильностью потока и большими
потерями давления, особые проблемы с высокой зольностью топлива, пониженный
КПД, обусловленный отсутствием внутреннего теплообмена, и низкая теплота сго-
рания получаемого газа.
4.2.2. Газогенераторы с кипящим (псевдоожиженным) слоем
Газифицирующий агент (кислород и пар или воздух) приводит слой сырья во взве-
шенное состояние. Зола отводится в сухом виде или в виде крупных кусков, выпада-
ющих из слоя. В аппаратах с сухим шлакоудалением температуры относительно низ-
ки, поэтому сырье должно иметь высокую химическую активность; в газогенераторах
с кусковым шлакоудалением температуры несколько выше. Пропускная способность
выше, чем у аппаратов со стационарным слоем, но не столь высока, как при газифи-
кации в потоке. Газогенераторы с кипящим слоем лучше других приспособлены для
переработки сырья, дающего золу с высокой коррозионной активностью.
Одна из важных особенностей газогенераторов с кипящим слоем — способ, ко-
торым сырье приводится во взвешенное состояние. Восходящий поток газифициру-
ющего агента придает высокую скорость частицам сырья и вызывает турбулентное
перемешивание реагентов. В результате улучшается тепло- и массоперенос между га-
зовой и твердой фазами и образуется более однородный сингаз.
140
Часть 2. Моделирование некаталитических процессов
Главное преимущество газификации с кипящим слоем — гибкость требований
к сырью. Лучший контроль температуры позволяет постоянно удерживать ее на уровне
ниже температуры плавления золы. Мелкий материал можно перерабатывать без пред-
варительной подготовки. Качественное перемешивание сырья и окислителя улучшает
тепло- и массоперенос и способствует равномерному распределению материала в слое.
В газогенераторах с кипящим слоем воздух продувается через слой твердых частиц
со скоростью, достаточной для удержания их во взвешенном состоянии. Теплота под-
водится от внешнего источника, а сырье подается лишь после подогрева до достаточ-
но высокой температуры. Сырье вводится снизу, перемешивается с материалом слоя
и нагревается до его температуры. В результате сырье пиролизуется очень быстро
и образующийся продукт содержит сравнительно большое количество компонентов
в газовой фазе. Протекающие в дальнейшем процессы газификации и превращения
образовавшихся смол протекают в газовой фазе. Для борьбы с выдуванием угля газо-
генераторы оснащают внутренними циклонами. Циклоны также частично улавлива-
ют и возвращают в слой пылевидную часть сырья, уносимую сингазом.
4.2.3. Факельные газогенераторы
Такие аппараты работают в параллельноточном режиме с коротким временем пребы-
вания (несколько секунд). Сырье (пылевидное твердое или распыленное жидкое) га-
зифицируется кислородом или (значительно реже) воздухом. Реакции газификации
протекают в плотном облаке очень тонких частиц. Малые размеры частиц необходи-
мы для улучшения массопереноса и перемещения в потоке газа. Большая часть золы
удаляется. Факельные газогенераторы работают при высоких температурах (допусти-
мых благодаря короткому времени пребывания), а расстояния между частицами сы-
рья значительно превышают их размеры.
Благодаря повышенным температурам и давлениям пропускная способность ап-
паратов больше, чем у газогенераторов других типов, но расход кислорода выше,
а термический КПД ниже, так как газ перед очисткой приходится охлаждать.
Факельные газогенераторы лучше других подходят для газификации нефтезавод-
ских остатков.
4.2.4. Другие способы газификации
В литературных источниках упоминаются новаторские способы газификации, в том
числе:
' • гидротермическая газификация;
• газификация в воде в сверхкритическом состоянии;
• плазменно-дуговая газификация;
• двухступенчатая газификация;
• газификация с двумя зонами подвода воздуха;
• риформинг в водной фазе.
Глава 4. Моделирование процесса газификации
141
4.3. ПАРАМЕТРЫ ПРОЦЕССА
На показатели процесса газификации (на состав продукта, КПД, теплоту сгорания
синтез-газа и т. д.) влияет ряд параметров, которые в определенной степени зависят
другот друга. Важнейшие из них описываются ниже. В табл. 4.1 [Ni and Williams, 1995;
Graggen et al., 2008] перечислены характерные рабочие условия в газогенераторах раз-
личных типов.
Таблица 4.1
Некоторые рабочие условия процессов газификации
Ъш процесса В стационарном слое В кипящем слое В потоке
Шлакоудаление Сухая зола Шлак Сухая зола Кусковая зола Шлак
Тип сырья Твердое Твердое Твердое и жидкое
Размер частиц сырья, мм 6-50 6-50 6-10 6-10 <0,1
Температура выходящего газа, С Низкая Средняя Высокая
425-650 900-1050 1250-1600
Время пребывания 15—30 мин 5—50 с 1-Юс
Расход окислителя Низкий Средний Высокий
Расход пара Высокий Низкий Средний Низкий
4.3.1. Температура
Скорость процесса сильно зависит от температуры. Реакции обычно обратимы и про-
текают до равновесия, и на состав продукта можно влиять, изменяя температуру.
По сообщениям, при повышении температуры увеличивается концентрация го-
рючего газа и снижается,выход полукокса (с соответственным увеличением глубины
превращения). Кроме того, возрастает выход синтез-газа (Н2, СО) и повышается его
теплота сгорания, а выход жидких и твердых продуктов снижается.
4.3.2. Давление
Давление оказывает значительное влияние на реакции газификации. Сообщалось,
например, что для различных видов сырья константа скорости первого порядка, ско-
рость газификации, молярные соотношения Н2/СО и СО2/СО и выход метана с по-
вышением давления при постоянной температуре возрастают. При более высоких
температурах прирост этих параметров становится еще заметнее. Одно из достоинств
проведения процесса при высоком давлении — высокая скорость реакции, что по-
зволяет уменьшить размеры реактора.
142
Часть 2. Моделирование некаталитических процессов
4.3.3. Скорость начала псевдоожижения
Для газогенераторов с кипящим слоем важна скорость начала псевдоожижения. Чем
она больше, тем выше температура и ниже теплота сгорания производимого газа
(при использовании воздуха для газификации в продуктах увеличивается содержа-
ние О2 и N2).
4.3.4. Соотношение воздух/пар
Когда соотношение воздух / пар увеличивается, возрастает и объемный выход газа.
Растет также теплота сгорания газа, но при определенном соотношении воздух/пар
она достигает максимума. Эффект наиболее выражен при низких соотношениях, по-
скольку заметный вклад в процесс газификации вносит водяной пар. Пар, даже если
его нет в составе газифицирующего агента, образуется на стадии высвобождения ле-
тучих веществ.
4.3.5. Коэффициент избытка окислителя
Сильнее всего на работу газогенераторов влияет коэффициент избытка окислителя,
определяемый как отношение действительного количества окислителя к теоретиче-
ски необходимому для полного сгорания сырья. Этот параметр влияет на температу-
ру слоя, качество газа и термический КПД. Повышение коэффициента избытка вы-
зывает снижение потерь давления, увеличение скорости образования газа в случае
газификации воздухом и повышение температуры. Уменьшение коэффициента из-
бытка приводит к снижению содержания горючих компонентов в производимом газе
и теплоты его сгорания.
4.3.6. Размер частиц
С увеличением размеров частиц скорость распространения тепла в них снижает-
ся, что приводит к снижению скорости их нагревания. При постоянной температуре
с уменьшением размеров частиц возрастают скорость газификации, выход газа и со-
держание горючих компонентов. Теплота сгорания сначала возрастает (если началь-
ный размер был достаточно велик), достигает максимума, а затем снижается. Зависи-
мость скорости газификации от размера частиц сырья установили Эдрич и соавторы
[Edrichetal., 1985].
4.4. ОПИСАНИЕ ПРОЦЕССА
Как было указано выше, для газификации нефтяных остатков предпочтительны фа-
кельные газогенераторы. Поэтому процесс здесь описывается применительно к ним.
Глава 4. Моделирование процесса газификации
143
Сырье подается с верха реактора или сбоку от него. Возможна подача в расплав-
ленном виде. В сухом виде сырье из шлюзовых бункеров подается шнеком к горел-
кам, где его подхватывает газифицирующая среда и со скоростью, превышающей
скорость распространения пламени, перемещает в реактор. Для подачи в виде рас-
плава или пульпы используются насосы. Из частиц, оказавшихся в реакторе, бы-
стро испаряются летучие вещества, так что частицы теряют всякое сходство с ис-
ходным сырьем. Высокая рабочая температура (в факеле газогенератора она может
превышать 1500 °C) эффективно разрушает все углеводороды, которые могли обра-
зоваться на стадии испарения. Тем самым снимаются проблемы загрязнения газа
водным конденсатом и упрощается очистка от примесей. Скорость реакции высо-
ка, и для исключения проблем, возникающих при прерывании подачи сырья, ну-
жен надлежащий контроль избытка кислорода. Время пребывания в зоне реакции
исчисляется секундами. Факельные газогенераторы обычно эксплуатируют при
высоких давлениях.
Есть несколько типов промышленных факельных газогенераторов. Основные
критерии выбора — производительность, экономические показатели и соответ-
ствие экологическим требованиям. Два типа факельных газогенераторов показа-
ны на рис. 4.1.
Угольная пульпа
Вода
Кислород с установки
разделения воздуха
Холодильник
радиантного типа
для сингаза
Сингаз
Очищенная
сточная вода
Пар высокого
давления
Шлак
в котел-
утилизатор
* т
« Топливный газ
разделения
воздуха
Шлаководяная пульпа
Шлак
Газогенератор конструкции Texaco
Газогенератор конструкции E-gas
Рис. 4.1. Факельные газогенераторы
144
Часть 2. Моделирование некаталитических процессов
4.5. ХИМИЯ И ТЕРМОДИНАМИКА ПРОЦЕССА
Ниже перечислены важнейшие из реакций, происходящих при газификации, в чис-
ло которых входят превращения углерода, оксида углерода, диоксида углерода, водо-
рода, воды (или водяного пара), атакжеи метана [Robinson, 2006; Speight, 2007].
♦ Окисление:
C + '/zOj^CO А/7° =-110,60 МДж/кмоль (4.1)
С + О2 «-> СО2 Ай° = -393,78 МДж/кмоль (4.2)
СО + !6О2 СО2 А//0 = -283,18 МДж/кмоль (4.3)
• Метанирование (образование метана):
С + 2Н2^СН4 АД° =-74,86 МДж/кмоль (4.4)
• Конверсия водяного газа:
С + Н,0 «-> СО + Н2 А№ = +131,38 МДж/кмоль (4.5)
• Реакция Будуара:
С + СО, ^2СО АД0 =+172,58 МДж/кмоль (4.6)
• Конверсия окиси углерода СО:
СО + Н2О СО2 + Н2 АЯ° = -41,20 МДж/кмоль (4.7)
• Реакция риформинга метана водяным паром:
СН4 + Н2О «-> СО + ЗН2 Ай° = +206,24 МДж/кмоль (4.8)
• Горение метана:
СН4 + 3/2О2 СО + 2Н2О АЯ° =-519,70 МДж/кмоль (4.9)
СН4 +2О2 ~ СО2 + 2Н2О АЯ° =-802,88 МДж/кмоль (4.10)
• Реакции с участием серы:
H2S + СО2 «-> COS + Н2О АН0 =+34,23 МДж/кмоль (4.11)
2Н.О +COSTCO+ 21^ + 80, АЯ°= +214,76 МДж/кмоль (4.12)
Н2 + COS «-> СО + H2S АД0 =+6,97 МДж/кмоль (4.13)
• Образование оксидов азота:
Глава 4. Моделирование процесса газификации
145
'/2N2 + CO2 NO + CO A//° = +373,49 МДж/кмоль (4.14)
N, + CO2 N,0 + CO AH’ = +365,24 МДж/кмоль (4.15)
• Гидролиз HCN: hcn + h2o~nh3 + co A//° = —49,66 МДж/кмоль (4.16)
Газификация углеродистого или углеводородного сырья может происходить по-
средством реакций окисления (4.1), метанирования (4.4), конверсии водяного
газа (4.5) и Будуара (4.6). В основе большинства процессов газификации лежит ба-
ланс реакций частичного окисления (4.1) и конверсии водяного газа (4.5).
Кроме углерода, водорода и кислорода, нефтяные остатки содержат серу, которая
переходит в H2S и COS и следовые количества S2 и CS2. В типичных условиях газифи-
кации преобладает сероводород, в котором сосредоточено около 93—96 % всей серы;
остальная сера содержится в COS. Азот сырья превращается в элементарный азот,
NH3 и HCN. Из хлорсодержащих продуктов, образующихся при газификации, пре-
обладает НС1. Сера и азот присутствуют в сырье обычно в малых количествах, по-
этому их влиянием на основные компоненты синтез-газа — водород и оксид углеро-
да — можно пренебречь. Из-за высоких температур углеводороды не образуются при
газификации в заметных количествах; единственным исключением является метан.
Реакциям конверсии водяного газа и метанирования благоприятствуют высокие
температуры (выше 600 °C). Состав конечного продукта зависит от достигаемой сте-
пени равновесия между различными реакциями в газовой фазе.
Горение происходит очень быстро и с выделением большого количества тепла.
Реакции горения (С + О, СО, и С + %О2 СО) обеспечивают энергию, необ-
ходимую для поддержания эндотермических реакций газификации. Они потребля-
ют большую часть подаваемого в газогенератор кислорода, а выделяемое ими тепло
позволяет достичь температур, способствующих разрыву химических связей и бла-
гоприятствующих реакциям Будуара и конверсии водяного газа. Экзотермическими
являются все реакции окисления.
4.6. МОДЕЛИРОВАНИЕ РЕАКТОРА ГАЗИФИКАЦИИ
Показано, что равновесная модель позволяет предсказать термодинамические пре-
делы реакций, происходящих в процессе газификации [Sharma, 2008]. Существует
множество литературных источников, предлагающих схожие модели реактора гази-
фикации. Все эти модели основаны на термодинамическом равновесии, даже если
разработаны для разных типов сырья. В основном они различаются реакциями, вы-
бранными для представления равновесия между образующимися газообразными
продуктами, подходом к рассмотрению равновесия в реакциях, учитываемыми фак-
торами и исходными предположениями. Некоторые авторы задаются степенью пре-
вращения углерода в газообразные компоненты или даже принимают ее равной 100 %
[Zainal et al., 2001; Jarungthammachote and Dutta, 2007; Melgar et al., 2007], в то время
146
Часть 2. Моделирование некаталитических процессов
как другие определяют ее расчетным путем [Altafini et al., 2003; Mountouris et al., 2006;
Sharma, 2008; Yoshida et al., 2008]. Одни авторы изучают зоны окисления и восста-
новления по отдельности [Melgar et al., 2007; Sharma, 2008; Yoshida et al., 2008], дру-
гие рассматривают газогенератор как единую систему [Zainal et al., 2001; Mountour-
is et al., 2006; Choi et al., 2007; Jarungthammachote and Dutta, 2007].
4.6.1. Уравнения модели
Для расчета выхода синтез-газа из остатка вакуумной перегонки нефти предлагает-
ся модель, которую описали Йошида и соавторы [Yoshida et al., 2008]. Модель хороша
тем, что степень превращения углерода в компоненты в газовой фазе определяется
расчетным путем. Для этого моделируется зона окисления и затем глобальным моде-
лированием газогенератора определяется состав производимого синтез-газа.
Нефтяные остатки обычно характеризуют методом экспресс-анализа, количе-
ственно определяющего лишь доли компонентов топлива (fuel materials — FM), золы
и влаги, и элементного анализа (определения содержания С, Н, О, N, S). Горючая
фракция при экспресс-анализе обычно подразделяется на связанный углерод (fixed
carbon — FC) и летучие соединения (volatile material — FM). В целях расчета обе фрак-
ции рассматриваются совокупно как сухое беззольное горючее вещество (содержание
которого выражается в процентах по массе). По результатам элементного анализа,
выраженным в процентах по массе, можно найти сокращенную формулу и молярную
массу горючей фракции.
4.6.1.1. Баланс масс
Баланс масс в реакциях газификации нефтяного остатка предлагается представить
уравнением (4.17). Сухой вакуумный остаток (ВО) реагирует с водой (содержащейся
в сырье плюс отдельно подаваемой в газогенератор) и кислородом (чистым или кис-
лородом воздуха), давая главные компоненты, составляющие синтез-газ, и твердый
остаточный углерод (если степень превращения меньше 100 %). Вторичные газообраз-
ные соединения, образующиеся в остаточных количествах, не принимаются в расчет,
так как в противном случае модель стала бы чрезмерно сложной. Если для газифика-
ции используется чистый кислород, то азотом воздуха в левой части уравнения (4.17)
можно пренебречь. Сокращенная формула вакуумного остатка (СН^ОДМ,) в расче-
те на один моль углерода выводится из его беззольного сухого элементного состава.
CH OyS _N + wH2O + m(O2 + 3,762N2) =
= «.H + n CO + «.CO, + «,НО + «.CH, + «,H,S + «,N + «„C (4.17)
Приравнивая числа атомов соответствующих элементов в правой и левой ча-
стях (4.17), можно получить уравнения (4.18)-(4.23). Здесь п-пъ — число молей со-
ответствующих продуктов газификации на один моль вакуумного остатка:
Глава 4. Моделирование процесса газификации
147
• Баланс углерода:
1 = л, + л3 + л5 + ns. (4.18)
• Баланс водорода: 1/2х + w = л, + л4 + 2ns + л6. (4.19)
• Баланс кислорода: f+ w + 2т = л, + 2л, + л.. г 2 3 4 (4.20)
• Баланс серы: г = «€. (4.21)
• Баланс азота: 1/21 + 3,762m = пг (4.22)
• Общий баланс в газовой фазе:
п= л, + л, + л, + л, + л, + л, + л,. 1 1 L 3 4 3 и / (4.23)
Степень превращения углерода определяется как доля углерода, превращенного
в компоненты газовой фазы:
Масса углерода, превращенного в компоненты газовой фазы _ з ~ ,л
'1 Масса углерода в сырье "ц-
Уравнения (4.18)—(4.23) можно переписать, используя мольные доли фазе (у,.) и степень превращения углерода: в газовой
11 = лг(у2+у3+у5); (4.25)
1/2х + w - z = лг(у, +у4 + 2у5); (4.26)
/+ w + 2ти = лг(у2 + 2у3 + у4); (4.27)
(4.28)
X/2t + 3,762m = лгу7; (4.29)
У1+3'2+3'з+^+^+Л+У7= Ь (4.30)
где
л,.
У/ = —
«7
(4.31)
148
Часть 2. Моделирование некаталитических процессов
Введем следующее определение кратности воды к сырью (Явод/В0):
„ _ Масса Н2О, подаваемой в газогенератор
Люд/во Г7 ’ (4,32)
Масса вакуумного остатка
Примем Лвод/Во за рабочий параметр газогенератора и выразим через него число w
участвующих в реакции газификации молей воды на 1 моль горючего материала:
_ Процент влаги в вакуумном остатке+100Лвод/Во МЖВ0 (4 33)
Процент FM MWH?0’
где Л/Жво — молярная масса беззольного сухого вакуумного остатка; MWH — мо-
лярная масса воды; FM= FC + VMв вакуумном остатке (процентное содержание FM,
определяется экспресс-анализом).
Когда в качестве окислителя подается чистый кислород, можно ввести в качестве
рабочего параметра кратность кислорода к сырью Аоуво:
_ Масса О2, подаваемого в газогенератор (4 34)
°2/в° Масса вакуумного остатка
Если величина Л0з/В0 известна, число молей подаваемого кислорода на один моль
вакуумного остатка можно найти по формуле
Ю07?О2/во МЖВО
т = 'И--------’ (4.35)
Процент FM MW0^
где MWQ^ — молярная масса кислорода.
Для случая, когда окислителем является воздух, вводится рабочий параметр 7?возд/В0
кратности воздуха к сырью:
„ _ Масса воздуха, подаваемого в газогенератор ,.
возд/ВО ~ 77 ' (4.30)
Масса вакуумного остатка
В этом случае стехиометрический коэффициент т в уравнении (4.17) равен чис-
лу молей подаваемого воздуха на 1 моль вакуумного остатка и вычисляется следую-
щим образом:
т = Жоэд/во (437)
Процент FM MWtsaa
где (ИЖ — молярная масса воздуха.
4.6.1.2. Термодинамическое равновесие
Константу равновесия К реакции J можно вычислить по изменению свободной
энергии Гиббса при температуре реакции (см. (4.38)). В свою очередь, изменение
свободной энергии Гиббса зависит от динамики энтальпии и энтропии в реакции
(см. (4.39)):
Глава 4. Моделирование процесса газификации
149
где
AG,.
In К =-----L
J RT
ДО. = ДЯ - 7ДД,
j j 1’
(4.38)
(4.39)
f А О’,
ДЯ. = ДЯ.0 + Я[——dT-,
1 1 J R
г ДСп.
ДД. = ДД° + 7?[—,
1 1 I R Т
(4.40)
(4.41)
где R — универсальная газовая постоянная, равная 8,314 кДж/кмоль • К; Т— темпе-
ратура, К; d — дифференциал от Т; ДНй, и ДСр. — соответственно стандартная эн-
тальпия, стандартная энтропия и молярная теплоемкость в у'-й реакции. Подставляя
(4.39)-(4.41) в (4.38), получим следующее уравнение:
ДЯ" 1ггДСр. №°.тсДСр. dT
1пЯ = —^dT + -^[—.
1 RT Т R R J R Т
‘0 h
Из уравнения (4.39) выразим стандартную энтропию ДД° у-й реакции при стан-
дартной температуре TQ = 298,15 К:
ДЯ°-ДО°
20
(4.42)
(4.43)
Подставляя это выражение в (4.42), можно найти зависимость константы равно-
весия К реакции j от стандартной энергии Гиббса, стандартной энтальпии реакции
и температуры системы:
Дб° дя°
1пЯ.= -J + —-
j т т
20 20
Величину Д(7° можно найти по стандартной энергии Гиббса (G°;), а ДЯ° — по стан-
дартной энтальпии (Я°.) образования соединений, участвующих в реакции:
_L_1
Jo т
1 f .it f ЛСО dT
— ------- dT + ---------
T' R J R T
О >0
(4.44)
(4.45)
(4.46)
Таким же способом можно найти коэффициенты для расчета теплоемкостиу-й ре-
акции (ДСру):
AZ)
^Ср. = ДА. + ДВТ + ДС'Т1 + (А АТ)
ДЛу = Хт../4; (4.48)
150
Часть 2. Моделирование некаталитических процессов
= (4.49)
= (4.50)
^D. = Yv,.jDi, (4.51)
где А, В, С, D — соединения; v — стехиометрический коэффициент /-го соедине-
ния в j-й реакции (положительный для продуктов и отрицательный для реагентов).
Термодинамические данные и константы, необходимые для решения этих уравнений
и расчета теплоемкости, представлены в табл. 4.2 и 4.3.
Чтобы равновесная модель давала более-менее точные величины выхода соеди-
нений, образующих синтез-газ, необходимо выбрать наиболее представительные ре-
акции. В таком случае лучше всего сосредоточить внимание на секциях окисления
и восстановления. Так как кислород, подаваемый в газогенератор, полностью расхо-
дуется в секции окисления, а углерод окисляется и переходит в газовую фазу, послед-
няя крайне важна для процесса: именно она в основном определяет степень превра-
щения углерода. Тогда представительными для равновесия, смещенного в сторону
образования компонентов синтез-газа, будут реакции метанирования (4.4) и Буду-
ара (4.6). Основные соединения, образующиеся на этапе окисления, достигают рав-
новесия в секции восстановления по реакциям (4.7) и (4.8). Реакциями (4.9) и (4.10)
из-за недостатка кислорода в системе и низкого выхода метана можно пренебречь.
Не принимаются во внимание также реакции (4.11)—(4.16), поскольку они определя-
ют равновесие только для следовых компонентов синтез-газа.
Выбрав наиболее представительные реакции процесса газификации, по уравне-
ниям (4.40)—(4.51) можно вычислить константы химического равновесия. Зависимо-
сти констант равновесия реакций (4.4)-(4.8) от температуры, показанные на рис. 4.2,
согласуются с результатами, которые получил Шарма [Sharma, 2008]. Видно, что ре-
акция метанирования (см. (4.4)) на этапе окисления лишь слегка смещена в прямом
направлении (причем это смещение наиболее выражено при высоких температурах),
а реакция разложения метана (см. (4.8)) на этапе восстановления смещена сильнее.
Поэтому можно ожидать, что при высоких температурах концентрация метана в син-
тез-газе будет низкой. Аналогично, высокие температуры не благоприятствуют обра-
зованию СО, (см. (4.7)), но способствуют выходу СО и Н2 (уравнения (4.5) и (4.6)).
Чтобы решить систему уравнений для каждой секции, необходимо, чтобы чис-
ло уравнений было равно числу неизвестных. Уравнение общего баланса масс дает
шесть уравнений (4.25)-(4.30), которые справедливы для газогенератора в целом
и поэтому входят как составная часть в системы уравнений всех секций. Число не-
известных для секции окисления равно девяти, но после определения степени пре-
вращения углерода остается восемь неизвестных в уравнении общего баланса. Не-
обходимые уравнения можно получить исходя из равновесия реакций газификации.
Для секции окисления выбраны реакции (4.4)—(4.6). Для глобального баланса, наря-
ду с реакцией (4.7), выбрана реакция (4.4), так как она является лимитирующей для
образования метана.
Таблица 4.2
Термодинамические свойства соединений, участвующих в процессе газификации
Соединение Формула Фаза ? (25 °C), кДж/кмоль Gf “ (25 °C), кДж/кмоль 5) “(25 °C), кДж/кмоль С, к см’/моль Zc СО Дипольный момент
Вода н2о ЖИДК. -286 020 -237 400 70,13 647,3 221,2 57,1 0,235 0,344 1,8
газ. -241 980 -228 760 188,85 647,3 221,2 57,1 0,235 0,344 1,8
Кислород Тоже 0 0 205,17 154,6 50,4 73,4 0,288 0,025 0
Азот N2 » 0 0 191,62 126,2 33,9 89,8 0,29 0,039 0
Водород Н2 » 0 0 130,61 33,19 13 65,1 0,306 -0,218 0
Оксид углерода со » -110 600 -137 240 197,68 132,9 35 93,2 0,295 0,066 0,1
Диоксид углерода со2 » -393 780 -394 640 213,82 304,1 73,8 93,9 0,274 0,239 0
Метан сн4 » -74 860 -50 850 186,44 190,4 46 99,2 0,288 0,011 0
Сероводород H2S » -20 930 -33 830 205,82 373,2 89,4 98,6 0,284 0,097 0,9
Диоксид серы so2 » -297 100 -300 410 248,23 430,8 78,8 122,2 0,269 0,256 1,6
Углерод (графит) с » 0 0 5,74 — — — — — —
Двухатомная сера S2 » 129 787,68 81 002,24 — — — — — — —
Оксид азота NO » 90 310 86 640,00 210,72 180 64,8 57,7 0,25 0,588 0,2
Сероксид углерода cos » -138 500 -165 800,00 — 378,8 63,5 136,3 0,275 0,105 0,7
Цианид водорода HCN » 134 820 124 350 201,85 456,7 53,9 138,8 0,197 0,388 3
Аммиак NH3 » -46 220 -16 720 192,76 405,5 113,5 72,5 0,244 0,25 1,5
Закись азота n2o » 82 060 104180 220,02 309,6 72,4 97,4 0,274 0,165 0,2
Глава 4. Моделирование процесса газификации
Примечание. Тс — температура компонента; р — его парциальное давление; Vc — его мольный объем.
Источник: [Poling, В.Е. et al., The Properties of Gases and Liquids, 5th edn., McGraw-Hill, New York, 2001; Poling, B.E. et al., Perry’s Chemi-
cal Engineer’s Handbook, Section 2: Physical and Chemical Data, 8th edn., McGraw-Hill, New York, 2008].
(Л
Константы для расчета теплоемкости соединений, участвующих в процессе газификации
Таблица 4.3
Соединение Формула Фаза Молярная масса А В С D
Вода Н2О ЖИДК. 34,080 66,581 2,2762-10-2 -2,3946-10-6 2,0325-105
газ. 34,080 28,166 1,4667-10-2 — 1,5433 -10-6 1,0023-105
Кислород О2 To же 31,999 31,119 3,1088-10-4 -3,3884-10-8 -1,6342-105
Азот N2 » 28,013 27,883 2,9838-10-’ -3,1384-IO*7 3,8452-10-6
Водород Н2 » 2,0160 25,310 8,2575-10-’ -8,6850- IO'7 1,0601 - ю5
Оксид углерода со » 28,010 28,448 2,3633'10-’ -2,4877-10'7 4,2919-10’
Диоксид углерода со2 » 44,010 36,299 2,0352 -10"2 -2,1455-10*6 -4,4910-105
Метан сн4 » 16,043 23,607 4,9622- 10-2 -5,2248-10'6 -2,1280-105
Сероводород H2S » 34,080 29,805 1,5288-10-2 -1,6093- IO'6 -5,5732-10’
Диоксид серы so2 » 64,063 42,129 1,2388 10-2 — 1,3078-10'6 5,3787 - 10s
Углерод (графит) с » 12,011 16,336 6,0972-10-’ -6,4762-IO'7 -8,3634-105
Двухатомная сера S2 » 128,12 35,899 0,0012552 -3,30536-10-* 0,0000
Оксид азота NO » 30,006 27,599 0,0064315 -6,7677- IO*7 3,2370-104
Сероксид углерода cos » 60,070 23,570 7,9840 -10-2 -7,0170-10-5 2,4530 10-8
Цианид водорода HCN » 27,026 31,096 2,4898-10-2 -2,6225-10'6 -2,2130 -105
Аммиак NH3 » 17,030 30,519 2,4586-10-’ -2,5893-10'6 -1,8315-105
Закись азота n2o » 44,013 37,741 1,9836-10-’ | -2,0910- 10'6 -4,2985-105
Источник: [De Souza-Santos, M.L., Solid Fuels Combustion and Gasification: Modeling, Simulation, and Equipment Operation, Marcel Dekker,
Inc., New York, 2004; Poling, B.E. et al., The Properties of Gases and Liquids, 5th edn., McGraw-Hill, New York, 2001; Poling, B.E. et al., Perry's
Chemical Engineer’s Handbook, Section 2: Physical and Chemical Data, 8th edn., McGraw-Hill, New York, 2008].
Примечания. I. Ср =A + ВТ + СП + D/П (кДж/кмоль• К) для всех соединений, за исключением HCN. Для HCN Ср = А + ВТ + СП +
+ DV (кДж/кмоль• К).
2. Для жидкой воды данные действительны при температурах от 298 до 500 К, для других соединений — при температурах от 298 до 1500 К.
152 Часть 2. Моделирование некаталитических процессов
Глава 4. Моделирование процесса газификации
153
Рис. 4.2. Константы равновесия основных реакций процесса газификации
Константа равновесия реакции определяется как произведение коэффициентов
активности всех участвующих в ней соединений, возведенных в степени, равные со-
ответствующим стехиометрическим коэффициентам:
V,
<3, '
(4.52)
где для соединений в газовой фазе
Р
аГ^УГ^
(4-53)
Здесь ф; — фугитивность; Р — давление, бар; Рй — стандартное давление, при 25 °C
принимаемое обычно равным Рй = 1 бар.
Применение уравнения (4.52) к реакциям (4.4)-(4.7) дает следующие выражения
для констант равновесия:
2
к - асо . (4.54)
Л1 асасо2
^2 = асоан2 (4.55)
асан2о
*з = . ДСН„ flCflH2 (4.56)
^4 = дсо2 дн. (4.57)
С0иН,0
154
Часть 2. Моделирование некаталитических процессов
Ввиду высоких температур (особенно в случае газификации вакуумного остат-
ка, когда могут быть достигнуты температуры выше 1200 °C) соединения в газовой
фазе можно считать идеальным газом. Кроме того, жесткие условия в газогенерато-
ре приближают фугитивность соединений в газовой фазе к единице (ср. ® 1). Исхо-
дя из этого и принимая идеальное поведение, коэффициенты активности из форму-
лы (4.53) можно принять зависящими лишь от парциального давления соединения
в газовой смеси:
(4.58)
Активность углерода определяется формулой [Yoshida et al., 2008]
Vc^-Z’o)
«с = ехР RT ’ <4'59)
где vc — молярный объем углерода, м3/кмоль, равный РМс/рс, РМС — молярная мас-
са углерода, равная 12,0107; рс — плотность углерода (графит), равная 2260 кг/м3.
4.6.1.3. Баланс энергий
Чтобы рассчитать константы равновесия реакций, необходимо знать температуру
в реакторе. Она определяется притоком кислорода и наличием воды в системе. Ввиду
этого необходимо задаться начальным значением температуры, чтобы получить пер-
вую оценку степени превращения и состава конечных соединений, а затем по обще-
му балансу энергий рассчитать истинную величину температуры.
Предположим, что газогенератор работает в адиабатическом режиме, а все пото-
ки поступают в него при температуре окружающей среды. Примем, кроме того, что
энтальпия образования чистых соединений, таких как О2, N2 и Н2, равна нулю, а для
остальных соединений используется стандартная энтальпия образования при 25 °C,
значения которой приведены в табл. 4.2. Тогда из баланса энтальпий в суммарной ре-
акции (4.17) можно вывести следующее уравнение:
Hf.BO + W^/,H2O(ximk.) — «1ЛН2 + П2^СО + ИЗ^СО2 + И^Н2О(гаэ.) +
+ «5ЛСН4 + + П7^2 + «SAC(Sr <4'60)
где h — энтальпия компонентов; (S) — сера в соединениях с углеродом.
г
h.= Hf? + \Cp,dT. (4.61)
Принимая, что реакция горения вакуумного остатка (на сухой основе) в присут-
ствии стехиометрического количества кислорода протекает до конца (см. (4.62)),
на основании закона Гесса можно вычислить стандартную энтальпию образова-
ния Н/во в кДж/кмоль (уравнение (4.63)) [De Souza-Santos, 2004]. Энтальпия реак-
ции горения вакуумного остатка соответствует низшей теплоте сгорания (low heating
Глава 4. Моделирование процесса газификации
155
value — LHV), определяемой как количество теплоты, выделяющейся при сжигании
топлива стехиометрическим количеством кислорода:
CHO;S N, + (l+x/4 + ?/2 + z —//2)О2 СО2 + х/2Н2О + zSO, + ?NO (4.62)
Я/во = LHVB0 + Я’,С02 + + <ко- (4-63)
Величину LHV, кДж/кмоль, можно вычислить, если известна величина высшей
теплоты сгорания (high heating value — HHV). Высшая теплота сгорания — это сумма
теплоты полного сгорания стехиометрического количества топлива и теплоты кон-
денсации воды, образующейся при сгорании. Если для реакции (4.62) принять, что
при горении весь водород вакуумного остатка переходит в воду, то LHVопределяет-
ся из выражения
LHVm = (HHVao - 9WHh^0)MWB0t (4.64)
где HHV— высшая теплота сгорания, кДж/кг; JVH — массовая доля водорода в ва-
куумном остатке (на сухой основе с учетом золы); h^f0 — скрытая теплота испарения
воды при 25 °C, равная 2259,44 кДж/кг.
Есть множество зависимостей для расчета величины HHVпо элементному соста-
ву топлива. Для вакуумного остатка хорошо подходит следующая формула [Channi-
wala and Parikh, 2002]:
HHVBQ = 0,3491 % (C) + 1,1783 % (H) + 0,1005 % (S) -
- 0,1034 % (O) - 0,0151 % (N) - 0,0211 % (золы). (4.65)
Здесь процент (золы) — зольность топлива. Величина HHVвыражается в МДж/кг,
концентрации берутся на сухой основе в массовых процентах. Формула дает сред-
нюю погрешность ±1,45 % и применима в следующих пределах:
0<С <92,25;
0,43 < Н < 25,15;
0 < О < 50;
5 < N < 5,6;
0 <S <94,08;
0 < Зольность < 71,4;
4,745 <HHV< 55,345;
156
Часть 2. Моделирование некаталитических процессов
На сухой основе сумма концентраций С, Н, О, N, S и золы равна 100 %. На влаж-
ной основе в 100 % войдет и количество влаги.
4.6.1.4. Теплота сгорания синтез-газа и КПД газификации
Теплотворную способность (теплотворность) производимого синтез-газа (на сухой
основе) можно найти суммированием теплотворностей компонентов (Н2, СО, СН4,
и H2S), умноженных на их мольные доли на сухой основе [Melgar et al., 2007]. Вели-
чины удельной теплотворной способности основных компонентов синтез-газа пе-
речислены в табл. 4.4. Тепловой вклад H2S невелик из-за его малой концентрации
в синтез-газе (SG).
HHV% = у%НН7„2 + y%HHVc0 + у^ HHVctij + y^HHV^, (4.66)
LHV% = y^LHV^ + + ЛУн>ЯГсн< + <s^h2s. (4.67)
Таблица 4.4
Значения высшей и низшей теплот сгорания компонентов
синтез-газа
Компонент HHV, МДж/н. м3 LHV, МДж/н. м3
н2 12,759 10,784
СО 12,630 12,628
сн4 39,758 35,807
H2s 25,219 23,225
Источник: [Waldheim, L, and Nilsson, Т, Heating value of gases from biomass gasification, Re-
port prepared for IEA Bioenergy Agreement, Task 20—Thermal Gasification of Biomass, TPS Termis-
ka Processer AB, Sweden, 2001].
Опираясь на первый закон термодинамики, степень использования тепла (т| ), за-
ключенного в топливе (КПД газификации), можно определить как отношение энер-
гии, заключенной в производимом синтез-газе (£„,), к энергии (£й|), заключенной
в сырье, поступающем в газогенератор. При этом необходимо допустить, что син-
тез-газ покидает процесс при стандартной температуре (25 °C), теряя энергию, рав-
ную его физическому теплосодержанию [Melgar et al., 2007]:
Е
% = уЧ00; (4.68)
Ein = LHV30. (4.69)
Энергия синтез-газа (Диг) — это та энергия, которая выделяется при полном сго-
рании компонентов газа (Н2, СО, СН4 и H2S). Реакции горения СН4, Н2, СО и H2S вы-
ражаются соответственно уравнениями (4.10), (4.70), (4.71) и (4.72):
Глава 4. Моделирование процесса газификации
157
Н2 + 1/2О2 Н2О
СО + 1/2О2 СО,
(4.70)
(4.71)
H2S + 3/2O2 ~ SO2 + Н2О
(4.72)
Учитывая, что реакции горения являются экзотермическими, Е можно опреде-
лить из следующих формул, в которых пр nv и, ии6- числа молей компонентов, по-
лученных из 1 моля вакуумного остатка (на сухой основе) в соответствии с уравнени-
ем (4.17):
Ео,„ = (-^H2 ) + «2 (-Д^со ) + «5 (-Д#сн4) + «6 (-Д^Н,_5 ); (4.73)
Ео,„ = Щ [Н^ -^,Н2о(га3)Н (Я°со °.со2
+ П5 f,CO, 2^/,Н2О(газ)) + /? б(^ /,H2S /.SO, /,Н,О(газ)). (4.74)
4.6.2. Решение уравнений модели
Вычислить состав синтез-газа можно только методом итераций. При этом необходи-
мо решить уравнения:
• баланса масс (шесть линейных уравнений, применимых как к зоне окисления,
так и к зоне восстановления);
• термохимического равновесия (четыре нелинейных уравнения);
• баланса энергий (одно нелинейное уравнение).
Должны быть известны следующие начальные условия:
• скорость подачи вакуумного остатка;
♦ скорость подачи воды;
• скорость подачи воздуха или кислорода;
• температура на входе.
Для зоны окисления нужно найти девять неизвестных:
• доли продуктов в газовой фазе (у,-ys);
• степень превращения углерода (т|).
Решаемая система содержит уравнения (4.24)-(4.30) и (4.54)—(4.56). Составы, вы-
числяемые на этом этапе, соответствуют газу, поступающему в зону восстановления,
и в последующих расчетах не участвуют.
Определив величину степени превращения углерода, через глобальное моделиро-
вание газогенератора можно найти состав выходящего из него синтез-газа. Число не-
известных сокращается до восьми, а модель описывается уравнениями (4.24)—(4.30)
и (4.56)-(4.57). В обоих случаях система содержит как линейные, так и нелиней-
ные уравнения, образующие квадратную матрицу. Нелинейные уравнения системы
158
Часть 2. Моделирование некатг!литических процессов
можно решить итерационным методом Ньютона—Рафсона или другим гибридным
методом (например, алгоритмом Левенберга—Марквардта).
Затем вычисляются числа молей компонентов синтез-газа полученных
из одного моля вакуумного остатка. Это дает возможность решить уравнение (4.60)
баланса энтальпий при адиабатической работе, в котором неизвестной остается
лишь температура. Таким образом вычисляется и принимается новое значение тем-
пературы в системе. Описанная процедура повторяется до тех пор, пока расчетное
значение температуры не сойдется с принятым. Окончательно определив состав син-
тез-газа, по уравнениям (4.66)-(4.68) можно вычислить его теплотворную способ-
ность и энергетический КПД процесса. Итерационный метод решения схематически
показан на рис. 4.3.
Рис. 4.3. Итерационная процедура решения уравнений модели газификации
4.7. МОДЕЛИРОВАНИЕ ПРОЦЕССА ГАЗИФИКАЦИИ
4.7.1. Проверка модели
Модель процесса проверялась на взятых из литературных источников эксперимен-
тальных данных газификации вакуумного остатка с плотностью 4,3 ° API [Marion and
Muenger, 1981]. Свойства остатка представлены в табл. 4.5. По данным источника,
эксперимент проводился в следующих интервалах условий: давление от атмосфер-
ного до 85 бар; температура от 1100 до 1400 °C; кратности воды и кислорода к сырью
постоянны и равны в массовом выражении соответственно 0,4065 и 1,0637. При ими-
тации процесса температуру и давление изменяли до получения наименьшего рас-
хождения между экспериментальным и расчетным составами синтез-газа. Наилуч-
шее согласие наблюдалось при давлении 11 бар и температуре 1260 °C.
Глава 4. Моделирование процесса газификации
159
Таблица 4.5
Данные экспресс-анализа и элементного анализа вакуумного остатка
с плотностью 4,3 °АР1, на котором проверялась модель газификации
Показатель Значение
Экспресс-анализ, %масс.
Н2О 0
Связанный углерод + летучие вещества 99,96
Зола 0,04
Элементный анализ, %масс.
С 83,83
Н 9,66
О 0
N 0,31
S 6,20
Высшая теплота сгорания (HHV), кДж/кг 40340
Источник: [Marion, С.Р. and Muenger, J.R., Energy Prog., 1(1—4), 27,1981].
На рис. 4.4 расчетные концентрации компонентов синтез-газа сравниваются
с экспериментальными. Можно отметить хорошее согласие теории с эксперимен-
том. Абсолютная погрешность расчета содержания основных газообразных продук-
тов (СО, Н2и СО2) меньше 1,5 %; для СН4 она составляет 3,3 %. С высокой точностью
предсказан также энергетический КПД процесса: расчетная величина равна 79,06 %,
экспериментальная — 82,9 %. Величины удельного расхода кислорода, выраженные
в кубометрах чистого кислорода на 1000 кубометров произведенного Н, + СО (объе-
мы приведены к нормальным условиям), практически совпадают.
Рис. 4.4. Сравнение расчетного (□) и экспериментального ()
составов синтез-газа (на сухой основе)
160
Часть 2. Моделирование некаталитических процессов
Полученные результаты четко демонстрируют пригодность модели для расчета со-
става синтез-газа и других параметров процесса газификации остатка вакуумной пе-
регонки нефти.
4.7.2. Влияние условий реакций
4.7.2.1. Влияние давления
Влияние давления изучалось при следующих постоянных условиях: Н2О / ВО = 0,4065;
О2/ВО = 1,0637; Т= 1260 °C (1533 К). Как можно видеть из рис. 4.5, рост давления
в интервале от 1 до 70 бар на состав синтез-газа влияет слабо. Концентрации водо-
рода и оксида углерода незначительно снижаются, в то время как концентрации ди-
оксида углерода, воды и метана с увеличением температуры обнаруживают противо-
положное поведение. Сера вакуумного остатка при высоких рабочих температурах
в основном переходит в H2S, и при изменении давления ее общее содержание остает-
ся практически постоянным.
Давление, бар
Рис. 4.5. Влияние давления и температуры на состав синтез-газа
Температура, К
На рис. 4.6 (слева) показаны зависимости энергетического КПД газогенерато-
ра (coldgas efficiency — CGE) и удельного расхода кислорода от давления (SOC). Вид-
но, что увеличение давления снижает энергетический КПД; это ведет к увеличению
удельного расхода кислорода и уменьшению соотношения Н2/ СО в произведенном
сингазе.
Глава 4. Моделирование процесса газификации
161
го
Ч
О
300
£ 150
1125
"5 юо
f 75
50
т-0,8800
0,8738
-0,8675
0,8613
•0,8550
-0,8488
0,8425
0,8363
0,8300
0,8238
0,8175
О 10 20 30 40 50 60 70 80
Температура, К
Давление, бар
Рис. 4.6. Влияние давления и температуры
на энергетический КПД процесса и удельный расход кислорода
На рис. 4.7 показано, что в условиях, выбранных для проверки модели (Р = 11 бар,
Т= 1533 К, Н2О/ВО = 0,4065 и О2/ВО = 1,0637), степень превращения углерода со-
ставляет 100 %. Видно также, что добиться 100%-ной степени превращения при за-
данной величине соотношения Н2О/ВО можно изменением соотношения О2/ВО.
Чем больше величина Н2О/ВО, тем меньшее соотношение О2/ВО потребуется для
достижения 100%-ной степени превращения.
Рис. 4.7. Влияние давления и соотношений О2/ВО и Н2О/ВО на степень превращения
углерода при 7=1533 К
Из полученных результатов видно, что выбор давления газификации представляет
собой вопрос экономического характера и определяется главным образом выбором
целевого продукта и давления, при котором он будет потребляться. Процессы хими-
ческого синтеза низкого давления (например, производство оксопродуктов и уксус-
ной кислоты) обычно потребляют синтез-газ под давлением около 35 бар. В процессе
162
Часть 2. Моделирование некаталитических процессов
синтеза метанола, в некоторых процессах гидрообессеривания и в ряде других син-
тез-газ подается под давлением 62 бар. Для процессов, протекающих при высоких
давлениях, таких как гидрокрекинг и синтез аммиака, необходим газ под давлени-
ем 83 бар.
Важно понимать, что давление незначительно влияет на химизм и показатели эта-
па частичного окисления и мало сказывается на оптимизации всего производства,
если брать в расчет процессы разделения воздуха, конверсии СО (когда необходимо
производить Н,), очистки газа и компримирования (при необходимости) очищен-
ного газа. В целом удобнее производить газ пониженного давления и доставлять его
к месту потребления, не сжимая до требуемого давления.
Высокое давление газификации дает следующие преимущества для завода в целом:
• увеличение объема производства как Н2 + СО, так и пара высокого и среднего
давления;
• более эффективная очистка газа (в особенности если она основана на физиче-
ской адсорбции);
• общее снижение затрат на компримирование (газогенератор производит
в 3-4 раза больше сингаза по объему, чем потребляет кислорода).
4.7.2.2. Влияние температуры
В газогенераторе развиваются температуры порядка 1100-1400 °C. Как правило, про-
цесс газификации рассчитывают на такую температуру, при которой расход кисло-
рода и скорость подачи сырья, а также состав синтез-газа достигают оптимальных
уровней.
В условиях, для которых проверялась модель (Н2О/ВО = 0,4065, О2/ВО = 1,0637
иР= 11 бар), увеличение температуры в интервале 1100-1500 °C (1373-1773 К) на со-
став синтез-газа влияет довольно мало (см. рис. 4.5). Концентрации СО и Н2О незна-
чительно возрастают, концентрации Н2, СО2 и СН4 убывают.
Повышение температуры газификации увеличивает энергетический КПД процес-
са и снижает удельный расход кислорода; соотношение Н2/ СО в синтез-газе тоже
снижается, но не столь значительно, как при увеличении давления.
С повышением температуры степень превращения углерода возрастает (рис. 4.8).
Это объясняется тем, что реакции Будуара и взаимодействия углерода с паром про-
текают с поглощением тепла, поэтому при увеличении температуры равновесие этих
реакций смещается вправо.
4.7.2.3. Влияние кратности кислорода к сырью
На рис. 4.9 показано влияние соотношения О2/ВО в интервале 0,5-1,1 в условиях,
принятых для проверки модели (Р = 11 бар, Т= 1260 °C и Н2О/ВО = 0,4065). При
увеличении О2/ВО степень превращения углерода стремилась к 100 %. Величи-
ну соотношения, при которой она достигала 100 %, указывает вертикальная пун-
ктирная линия на рис. 4.9. В областях слева и справа от этой линии соотношение
Глава 4. Моделирование процесса газификации
163
О2/ВО по-разному влияет на состав синтез-газа. Согласно принципу Ле Шателье,
с увеличением О2 / ВО реакция протекает так: выход Н2 и СО уменьшается, а вы-
ход СО2 и Н2О увеличивается, чтобы компенсировать увеличение количества О,.
Избыток О, реагирует с Н2, давая Н,О. Это явление наблюдается справа от пун-
ктирной линии. Для участка, расположенного слева от этой линии, справедливо
следующее: если в системе имеется твердый углерод, то увеличение количества О2
способствует реакциям Будуара и горения углерода; следовательно, мольная доля
СО возрастает.
н2о/во
60 -I------1------ь
0,4 0,5 0,6
1333
1433
1533
----1633
---- 1733
Ч----------1
1 1,1
Соотношение О2 / ВО
Рис. 4.8. Влияние температуры и соотношений О2/ВО и Н2О/ВО
на степень превращения углерода при Р= 11 бар
Рис. 4.9. Влияние соотношения О2/ВО на степень превращения углерода и состав
синтез-газа (Р= 11 бар, Т = 1260 °C, Н2О/ВО = 0,4065)
Содержание О2 в вакуумном остатке (определяемое химическим анализом) име-
ет большое значение, так как влияет на точку, в которой степень превращения угле-
рода достигает 100 %. С увеличением содержания кислорода эта точка сдвигается
влево.
164
Часть 2. Моделирование некаталитических процессов
4.7.2.4. Влияние кратности воды к сырью
Изучалось влияние соотношения Н2О/ВО в интервале 0,2-1,0 на состав синтез-газа
вусловиях, принятыхдля проверки модели (Р= Пбар, Т= 1260 °C, О2/ВО = 1,0637);
результаты показаны на рис. 4.10. С увеличением соотношения Н2О / ВО выход воды,
не вступающей в реакцию, возрастает, в то время как выход Н2 и СО снижается; в осо-
бенности заметно уменьшение выхода СО.
Соотношение Н2О / ВО
Рис. 4.10. Влияние соотношения Н2О/ВО на состав синтез-газа
(Р= 11 бар, Т= 1260 °C, О2/ВО = 1,0637)
С увеличением соотношения Н2О / ВО возрастает количество воды, что снижает
температуру в реакторе газификации. Для поддержания температуры на требуемом
уровне нужно увеличить полноту горения, подавая дополнительное количество
кислорода. Это вызовет увеличение расхода горючих компонентов вакуумно-
го остатка. Таким образом, с увеличением соотношения Н2О / ВО выход СО2 воз-
растает, так как повышается полнота сгорания, а скорость реакции газификации
(С + Н2О -» СО + Н2), которая является эндотермической, снижается. Выход СО
уменьшается на большую величину, чем выход Н2, что объясняется дополнитель-
ным расходом СО в реакции конверсии водяного газа. Теоретически метан почти
не образуется.
Чтобы четче выявить влияние соотношения Н2О / ВО на состав синтез-газа и сте-
пень превращения углерода, соотношение О2 / ВО было изменено по сравнению
с тем, что использовалось при проверке модели (1,0637) и обеспечивало 100%-ное
превращение углерода, и было принято равным 0,7 с сохранением других рабочих ус-
ловий (Р = 11 бар, Т= 1260 °C). Из результатов, представленных на рис. 4.11, видно,
что вода важна для производства водорода, так как весь получаемый водород образу-
ется из воды.
С увеличением соотношения Н2О / ВО степень превращения углерода возрастает
и благодаря реакции с водяным паром достигает 100%. Когда количество воды увели-
чивается, реакции углерода с паром и конверсии СО протекают в таком направлении,
Глава 4. Моделирование процесса газификации
165
что выход Н2 возрастает. Эти реакции протекают в области слева от пунктирной ли-
нии, так что после исчерпания твердого углерода продолжается лишь реакция кон-
версии СО. Изменение состава синтез-газа в зависимости от соотношения Н2О / ВО
можно объяснить тем, что реакция конверсии СО подчиняется принципу Ле Шателье.
Рис. 4.11. Влияние соотношения Н2О/ВО на степень превращения углерода и состав
синтез-газа (Р = 11 бар, Т = 1260 “С, О2/ВО = 0,7)
Соотношение Н2О / ВО обычно регулируют для того, чтобы ограничить темпера-
туру реакции или посредством реакции конверсии водяного газа изменить соотно-
шение Н2/СО в синтез-газе.
4.7.3. Применение модели
4.7.3.1. Применение модели к вакуумным остаткам
с различными свойствами
Модель газификации была применена к вакуумным остаткам с температурой ки-
пения выше 538 °C (1000 °F), полученным методом разгонки для определения ИТК
из следующих образцов нефти:
• сырой нефти плотностью 10 ° API;
• сырой нефти плотностью 13 ° API;
• сырой нефти плотностью 21 ° API;
• облагороженной нефти плотностью 25 ° API.
Результаты экспресс-анализа и элементного анализа остатков приведены
в табл. 4.6. Первые три остатка получены из сырой нефти, последний — из нефти, об-
лагороженной каталитической гидроочисткой [Ancheyta et al., 2010].
По этим остаткам четырех видов оценивались влияние плотности сырья, а так-
же влияние предварительной гидроочистки на продукты газификации. Результаты
прогонки модели на этих четырех видах сырья в идентичных условиях приведены
в табл. 4.7.
166
Часть 2. Моделирование некаталитических процессов
Таблица 4.6
Данные экспресс-аналнза и элементного анализа различных вакуумных остатков
Показатель Плотность нефти, из которой получен остаток, ‘APJ
10 13 21 25
Экспресс-анализ, %масс.
Н,0 1,12 1,84 0,53 0,21
Связанный углерод + летучие вещества 98,48 97,90 99,34 99,72
Зола 0,40 0,26 0,13 0,07
Элементный анализ, %масс.
С 81,05 81,66 82,86 87,23
Н 9,80 9,87 10,01 10,01
О 0,49 0,34 0,21 0,17
N 0,81 0,88 0,76 0,40
S 7,85 7,25 6,16 2,19
Таблица 4.7
Расчетные результаты газификации различных вакуумных остатков
Показатель Плотность нефти, из которой получен остаток, ° API
10 13 21 25
Рабочие условия
Р, бар 11
Т, °C 1260
Источник О2 Чистый кислород
Соотношение Н2О / ВО 0,4065
Соотношение О,/ВО 1,0637
Результаты расчетов по модели, %мольн. на сухой основе
н, 37,70 37,89 39,18 40,93
СО 42,70 42,58 43,82 46,22
со. 5,52 5,51 4,78 3,80
н,о 11,87 11,95 10,41 8,19
сн4 0,24 0,24 0,26 0,29
H,S 1,76 1,61 1,36 0,47
N, 0,21 0,22 0,19 0,10
CGE, % 79,69 79,18 81,55 83,66
SOC, % 301,49 299,99 285,57 264,96
cc,% 100 100 100 100
Высшая теплота сгорания сухого газа 12,19 12,17 12,26 12,30
Низшая теплота сгорания сухого газа 11,29 11,27 11,35 11,40
Примечание. CGE — энергетический КПД процесса; SOC — удельный расход О2; СС — сте-
пень превращения углерода.
Глава 4. Моделирование процесса газификации
167
Видно, что вакуумный остаток нефти с плотностью 25 °Л/7дает больше СО и Н2
и меньше СО2, Н2О и H2S. Благодаря предварительной гидроочистке эта нефть содер-
жит больше водорода и меньше примесей (серы, металлов и др.), чем сырые нефти,
так что газификация ее остатка дает лучшие результаты. Кроме того, сам процесс по-
требляет меньше кислорода и показывает более высокий энергетический КПД.
4.7.3.2. Оптимизация выхода водорода
На нефтеперерабатывающих заводах процесс газификации проводят так, чтобы по-
лучить максимально возможное количество водорода для установок гидрообессе-
ривания и гидрокрекинга. Как говорилось выше, главными параметрами процесса
являются давление, температура, а также кратности кислорода и воды к сырью. Ис-
следовалось влияние давления на выход водорода при постоянной кратности кис-
лорода и двух значениях кратности воды к сырью. Для каждого значения давления
вычислялись температуры, соответствующие максимальному выходу водорода. Дан-
ные рис. 4.12 показывают, что с уменьшением давления снижается и температура, не-
обходимая для достижения равновесия. Видно, что при обоих значениях кратности
воды к остатку (0,5 и 1 по массе) давление оказывает обратное влияние на выход во-
дорода. Следует упомянуть, что реакции газификации моделируются исходя из тер-
модинамического равновесия. Равновесие реакций конверсии водяного газа и СО
зависит от количества воды, равновесие других реакций (метанирования и Будуа-
ра) — от давления, поскольку число молей продуктов меньше числа молей реаген-
тов в газовой фазе. Таким образом, необходимо найти правильный баланс между ко-
личеством воды и давлением. При высокой кратности воды к сырью максимальный
выход водорода и метана наблюдается при низких давлениях. Если давление высоко,
реакция метанирования потребляет больше водорода, чем дают реакции конверсии
водяного газа и СО, и выход водорода уменьшается.
Рис. 4.12. Изменение выхода водорода в зависимости от давления
при О2 / ВО = 1: (*) — при Н2О / ВО = 1; (♦) — при Н2О / ВО = 0,5
168
Часть 2. Моделирование некаталитических процессов
Изучалось также влияние кратности кислорода к сырью на выход водорода при
постоянной кратности воды к сырью. Результаты моделирования представлены
на рис. 4.13. Область условий реакций разделяется диагональной линией на две зоны:
неприемлемую (слева от линии) и приемлемую (справа от линии). Эту линию опре-
деляет минимальная температура газификации (860 °C), которая ограничивает ми-
нимально допустимую величину кратности кислорода [Gasification Technologies
Council, 2012]. Видно, что при давлениях выше 3 бар наилучшие результаты дости-
гаются при высоких значениях кратности кислорода к сырью. В таких условиях вы-
сокому выходу водорода способствует то обстоятельство, что с кислородом реагирует
больше углерода, так что для реакции с водородом с образованием метана углерода
остается меньше. При низком давлении реакция метанирования может потреблять
водород быстрее, чем его дают реакции конверсии. В этом случае существует компро-
мисс между давлением и количеством кислорода: на линии, соответствующей давле-
нию 1 бар, появляется точка максимума.
Кратность кислорода к сырью (по массе)
Рис. 4.13. Изменение выхода водорода в зависимости от кратности кислорода к сырью
при Н2О/ВО = 1: (♦) Р= 1 бар; () Р= 3 бар; (*) Р = 10 бар; (') Р= 50 бар; (*) Р= 100 бар
На основании полученных результатов можно заключить, что максимальному
выходу водорода способствуют следующие условия: низкое давление (1 бар), высо-
кая кратность воды к сырью (1:1 по массе) и высокая кратность кислорода к сырью
(1:1 по массе),
ЛИТЕРАТУРА
Altafini, C.R., Winder, RR., Barreto, R.M. 2003. Prediction of the working parameters of a wood
waste gasifier though an equilibrium model. Energy Comers. Manage. 44:2763—2777.
Ancheyta, J., Betancourt, G., Marroquin, G., Centeno, G., Munoz, J.A.D., Alonso, E 2010. Process
for the catalytic hydrotreatment of heavy hydrocarbons of petroleum. US7,651,604 B2. January 26,2010.
Channiwala, S.A., Parikh, RR 2002. A unified correlation for estimating HHV of solid, liquid and gaseous
fuels. Fuel 81:1051-1063.
Глава 4. Моделирование процесса газификации
169
Choi,YC., Lee, J.G.,Y>on,S. J., Park, M.H.2007. Experimental and theoretical study on the characteristics
of vacuum residue gasification in an entrained-flow gasifier. Korean J. Chem. Eng. 24( 1 ):60-66.
Choi, YC., Park, TJ., Kim, J.H., Lee, J.G., Hong, J.C., Kim, YG., Na, J.L, Renevier, A. 2003.
Experimental study on the characteristics of vacuum residue gasification in an entrained-flow gasifier. Eneigy
E’wg./. 12(1):49—57.
DeSouza-Santos, M.L. 2004. Solid Fuels Combustion andGasification: Modeling, Simulation, and Equipment
Operation, Marcel Dekker, Inc., NewYirk.
Edrich, R., Bradley, T, Grabboski, M. 1985. The gasification of ponderosa pine charcoal.
In Fundamentals of Thermochemical Biomass Conversion (R.P Overend, TA. Milne, K.L. Mudge, Eds.),
Elsevier Applied Science Publishers, London, U.K.
Furimsky, E. 1999. Gasification in petroleum refinery of 21st century Oil Gas Sci. Technol. Rev. IFP
54(5):597—618.
Gasification Technologies Council. 2012. Gasification. Redefining clean energy. http://www. kentuckynewgas.
com/wp-content/uploads/2008/12/GTC_Gasification_Brochure.pdf (accessed on January 17, 2012).
Graggen, A.Z., Haueter, P, Maag, G., Romero, M., Steinfeld, A. 2008. Hydrogen production by steam-
gasification of carbonaceous materials using concentrated solar energy—IV Reactor experimentation with
vacuum residue. Int. J. Hydrogen Energy 33:679-684.
Higman, C., Vm der Burgt, M. 2008. Gasification, 2nd edn., Gulf Professional Publishing, Burlington,
MA.
Jarungthammachote, S., Dutta, A. 2007. Thermodynamic equilibrium model and second law analysis of
a downdraft waste gasifier. Energy 32:1660-1669.
Marion, C.P, Muenger, J.R. 1981. Partial oxidation syngas can help improve refining econom- ics. Eneigy
Png. 1(1—4):27—32.
Melgar, A., Perez, J.E, Laget, H., Horillo, A. 2007. Thermochemical equilibrium modelling of a gasifying
process. Energy Corners. Manage. 48:59—67.
Mountouris, A., Vnitsas, E., Tassios, D. 2006. Solid waste plasma gasification: Equilibrium model
development and exergy analysis. Energy Comers. Manage. 47:1723-1737.
Ni, Q., Williams, A. 1995. A simulation study on the performance of an entrained-flow coal gasifier. Fuel
74(l):102—110.
Poling, B.E., Prausnitz, J.M., O’Conell, J.P2001. The Properties of Gases and Liquids, 5th edn., McGraw-
Hill, NewYark.
Poling, B.E., Thomson, G.H., Friend, D.G., Rowley R.L., Wilding, WV 2008. Ferry’s Chemical Engineer’s
Handbook, Section 2: Physical and Chemical Data, 8th edn., McGraw-Hill, New York.
Rana, M.S., Samano, V, Ancheyta, J., Diaz, J. 2007. A review of recent advances on process technologies
for upgrading of heavy oils and residua. Fuel 86:1216—1231.
Robinson, PR. 2006. Petroleum processing overview In Practical Advances in Petroleum Processing,
Vol. 1 (C.S. Hsu, PR. Robinson, Eds.), Springer, New York, Chapter 1. Salam, PA., Bhattacharya, S.C. 2006.
A comparative study of charcoal gasification in two types of spouted bed reactors. Energy 31:228-243.
Schuetze, B., Hofmann, H. 1984. How to upgrade heavy feeds. Hydrocarbon Process. 2:75—82. Sharma,
A.K. 2008. Equilibrium modeling of global reduction reactions for a downdraft (biomass) gasifier. Eneigy
Comers. Manage. 49:832—842.
Speight, J.G. 1980. The Chemistry and Technology of Petroleum, 1st edn., Marcel Dekker, Inc., New York.
Speight, J.G. 2007. The Chemistry and Technology of Petroleum, 4th edn., CRC Press, Taylor & Francis
Group, LLC, New York.
Wildheim, L., Nilsson, T 2001. Heating value of gases from biomass gasification. Report prepared for
IEA Bioenergy Agreement, Task 20—Thermal Gasification of Biomass. TPS Termiska Processer AB, Sweden.
Yoshida, H., Kiyono, E, Tajima, H., Yamasaki, A., Ogasawara, K., Masuyama, T 2008. Two-stage equilibrium
model for a coal gasifier to predict the accurate carbon conversion in hydrogen production. Fuel 87:2186—2193.
Zainal, Z.A., Ali, R., Lean, C.H., Seetharamu, K.N. 2001. Prediction of performance of a downdraft
gasifier using equilibrium modeling for different biomass materials. Energy Comers. Manage. 42:1499—1515.
Глава 5. Моделирование
процесса коксования
Эта глава посвящена моделированию коксования как наиболее распространенно-
го процесса превращения вакуумного остатка на нефтеперерабатывающих заводах.
Кратко освещаются основные типы процессов: замедленное коксование и коксова-
ние в псевдоожиженном слое — флюидкокинг и флексикокинг (т. е. флюидкокинг
с газификацией кокса). Замедленное коксование описывается подробно. Рассма-
триваются главные параметры процесса и их влияние на выход и свойства продук-
тов. Кратко излагаются фундаментальные аспекты каждого процесса (химия, кине-
тика и термическое разложение асфальтенов). Подчеркиваются сложность реакций
коксования и трудность задачи построения детальной кинетической модели. Дается
оценка некоторым опубликованным в литературных источниках зависимостям, ис-
пользуемым для прогнозирования выхода и свойств продуктов коксования.
5.1. ВВЕДЕНИЕ
В настоящее время из всех технологий облагораживания тяжелого сырья путем обед-
нения углеродом коксование, в особенности замедленное, применяется наиболее
часто, так как обеспечивает нефтеперерабатывающим заводам наибольшую выго-
ду. Цель коксования — превращение остаточного сырья (остатков атмосферной и ва-
куумной перегонки нефти) в более легкие и ценные продукты, в том числе в газы,
бензиновую фракцию, легкий и тяжелый газойли, а также в твердый продукт —
нефтяной кокс. При коксовании длинные углеводородные молекулы термиче-
ски расщепляются на более короткие. Объемный выход жидких продуктов зависит
от свойств сырья: например, при замедленном коксовании вакуумного остатка неф-
ти плотностью 22 ° API и коксуемостью по Конрадсону (CCR) 10,79 % объемный вы-
ход жидких продуктов составляет около 90 %; для вакуумного остатка нефти с плот-
ностью 12,7 °APIviCCR 16,4 % он равен 80,5 %.
Как уже говорилось, наиболее распространено замедленное коксование, кото-
рое проводится при температуре 450-470 °C. «Замедленным» этот процесс назва-
ли в связи с тем, что реакции крекинга доходят до завершения не в змеевике печи,
где это привело бы к закоксовыванию труб и преждевременному прерыванию рабо-
ты установки, а в камерах коксования, где подогретое сырье пребывает достаточно
Глава 5, Моделирование процесса коксования
171
длительное время. Тепла, сообщаемого сырью в змеевике печи, вполне достаточно
для деструктивной дистилляции, но реакции восстановления до кокса не успевают
начаться до того, как сырье попадет в камеру коксования. Иными словами, подогрев
в печи лишь инициирует реакции крекинга, а завершаются они в камерах коксова-
ния [Sawarkar et al., 2007].
Нефтеперерабатывающие заводы, располагающие установками коксования, ча-
сто называют заводами с безостаточной переработкой нефти. Достоинство коксо-
вания — присущая процессу гибкость превращения разнообразного сырья, позво-
ляющая решать проблему падения спроса на тяжелое нефтяное топливо переходом
на производство более ценных и легких продуктов.
Одной из целей процесса коксования может быть и производство кокса различ-
ных видов. Коксование может давать, в зависимости от свойств сырья, конструкции
установки и рабочих условий, кокс нескольких видов [Speight, 1980]. Эти разновид-
ности представлены ниже.
• Топливный кокс. Производится большинством установок коксования, главная
задача которых заключается в максимальном увеличении количества жидких
продуктов и уменьшении выхода кокса низкой ценности. Такой кокс использу-
ется как топливо для технологических печей и теплоэнергоустановок.
• Анодный кокс. Производится из сырья с низким содержанием серы и металлов.
Из такого кокса изготавливают аноды для алюминиевой промышленности.
• Игольчатый кокс. Производится из сырья с высоким содержанием ароматиче-
ских углеводородов и низким содержанием асфальтенов, серы и золы. Облада-
ет высокой прочностью и низким коэффициентом теплового расширения. Ис-
пользуется для изготовления электродов для сталелитейной промышленности
и производства графита.
Физические и химические свойства топливного, анодного и игольчатого кокса су-
щественно различаются. Именно они определяют, как будет использоваться кокс:
в качестве топлива, в целях прокаливания и изготовления электродов для алюмини-
евой, химической или сталелитейной промышленности, в сфере газификации и вы-
работки пара, электричества либо как газовое сырье для нефтехимической промыш-
ленности. Главной задачей установок коксования нефтеперерабатывающих заводов
остается выпуск жидких продуктов. Кроме того, производимый на заводах топлив-
ный кокс является дополнительным источником прибыли, так как из него путем га-
зификации можно успешно получать синтез-газ [Jacob, 1971; Dymond, 1991].
5.2. ПРОЦЕССЫ КОКСОВАНИЯ
Процессы коксования бывают трех основных видов [Rana et al., 2007].
1. Замедленное, или задержанное, коксование, которое может давать коксовый орех
(разновидность топливного кокса), губчатый кокс (используется для производства
электродного кокса или как топливо) или игольчатый кокс. Дает большую часть про-
изводимого сегодня в мире кокса.
172
Часть 2. Моделирование некаталитических процессов
2. Коксование в псевдоожиженном слое, или непрерывное коксование — флюидко-
кинг. Дает, как правило, топливный кокс.
3. Флексикокинг. Представляет собой комбинацию флюидкокинга и газификации
получаемого кокса. Дает синтез-газ с низкой теплотворностью.
5.2.1. Замедленное коксование
Замедленное коксование — полунепрерывный процесс термического крекинга, при-
меняемый на нефтеперерабатывающих заводах для превращения остатков атмо-
сферной и вакуумной перегонки нефти в жидкие и газообразные продукты. Остат-
ком процесса является твердый углеродистый продукт — нефтяной кокс, ценность
которого зависит от таких свойств, как содержание серы, металлов и т. д. Установ-
ка замедленного коксования производит жирный газ, бензиновую фракцию, легкий
и тяжелый газойли и кокс. В камерах коксования отделяется кокс, что сопровожда-
ется возрастанием соотношения Н/С в остальных продуктах. Последние нестабиль-
ны, не насыщены водородом и требуют дальнейшего гидрирования. Основная часть
асфальтенов, серы и металлов сырья переходит в кокс. Другие продукты представля-
ют собой ненасыщенные газы (олефины) и жидкости с высоким содержанием аро-
матических соединений.
Все жидкие продукты требуют дальнейшей переработки, так как содержат много
олефинов. Из-за этого жидкие продукты химически нестабильны и малопригодны
в качестве компонентов конечных продуктов (бензина и дизельного топлива). Бен-
зиновую фракцию и легкий газойль подвергают гидроочистке, тяжелый газойль на-
правляется на установки гидрокрекинга или каталитического крекинга в псевдоожи-
женном слое (FCC), а горячий остаток накапливается в коксовой камере, пока она
не будет заполнена коксом.
Ниже описаны процессы, происходящие на установке замедленного коксования
[Kruse et al., 1995; Ellis and Paul, 1998; Hughes et al., 2000].
• Сырье проходит через низ колонны фракционирования, где смешивается с жид-
ким рециркулятом, конденсирующимся из потока паров камеры коксования.
• Смесь прокачивается через печь коксования и поступает в одну из двух пере-
ключаемых камер коксования.
• Сразу же начинается крекинг с образованием кокса и продуктов в паровой фазе.
• Кокс остается в камере, а пары поднимаются вверх и после выхода из камеры
направляются в колонну фракционирования продуктов.
• Необходимое число камер коксования в каждом конкретном случае зависит
от качества и количества сырья и выбранной периодичности цикла коксова-
ния. В любом случае необходимы как минимум две камеры: пока одна работает
в режиме коксования, из другой выгружается кокс.
• Для выгрузки кокса открывают верхний и нижний люки камеры. Вращаю-
щимся гидрорезаком, создающим водяные струи высокого давления, с верха
до низа камеры вдоль ее оси вырезают отверстие.
Глава 5. Моделирование процесса коксования
173
• Вода не только вырезает отверстие, но и охлаждает кокс, образуя пар.
Гидрорезак медленно поднимают, разрезая кокс на куски, которые падают
на дно камеры.
Длительность цикла работы камер коксования составляет, как правило, 18-24 ч.
В цикл входят подогрев камеры, заполнение потоком из печи, крекинг с образовани-
ем кокса, газа и жидких продуктов, охлаждение камеры и выгрузка кокса.
Выход кокса может составлять до 30 % от массы сырья. Например, вакуумный
остаток нефти с плотностью 22 ° API к CCR 10,79 % дает примерно 12,2 % кокса; для
вакуумного остатка нефти с плотностью 12,7 ° API и CCR 16,4 % эта цифра доходит
до 22,3 %. Свежий (сырой) кокс, называемый зеленым, вагонетками, грузовиками
или ленточным конвейером доставляют на установку прокаливания, которая перера-
батывает его в различные сорта товарного продукта. Зеленый кокс можно также ис-
пользовать как топливо.
Установку коксования часто проектируют так, чтобы она давала максимум жидких
продуктов и минимум жирного газа и кокса. Превращение осуществляется подогре-
вом сырья до высокой температуры и подачей его в камеру коксования увеличенных
размеров, время пребывания в которой достаточно для завершения основных реак-
ций крекинга.
Многие нефтеперерабатывающие заводы отдают предпочтение процессу за-
медленного коксования из-за присущей ему гибкости в плане переработки остат-
ков любого вида. Процесс обеспечивает частичное превращение остаточного сы-
рья в жидкие продукты (бензиновую и дизельную фракции), практически полностью
очищенные от металлов и коксового остатка.
Избирательность процесса зависит от рабочих условий, главным образом от тем-
пературы и давления. Замедленное коксование дороже деасфальтизации растворите-
лями, но дешевле других термических процессов. Недостатки процесса — большой
выход кокса и низкий выход жидких продуктов. Несмотря на это, замедленное кок-
сование остается преобладающим процессом переработки остатков на нефтепере-
рабатывающих заводах. Совершенствование этой технологии позволило увеличить
выход легких продуктов, снизить рабочее давление, а также уменьшить выход кокса
и количество рециркулята.
5.2.2. Непрерывное коксование (флюидкокинг)
Процесс непрерывного коксования протекает в псевдоожиженном (кипящем) слое.
Подогретое сырье (остаток перегонки нефти) распыляется над псевдоожиженным
слоем горячих мелких частиц кокса. Реакции в псевдоожиженном слое проводятся
при более высоких температурах и в течение меньшего времени, чем при замедлен-
ном коксовании. Такие условия благоприятствуют увеличению выхода жидких про-
дуктов и уменьшению выхода кокса. Используются два технологических аппарата:
реактор и коксонагреватель. В последнем за счет сжигания части кокса вырабатыва-
ется тепло, которое циркулирующими частицами кокса передается в реактор. В реак-
торе поддерживается псевдоожиженный слой частиц, перемешиваемый подаваемым
174
Часть 2. Моделирование некаталитических процессов
снизу паром. Сырье вводится непосредственно в реактор и равномерно распределяет-
ся поверх слоя, где претерпевает крекинг и испарение. Пары сырья, подвергаясь кре-
кингу, обволакивают частицы кокса слоем жидкости. Частицы обрастают до тех пор,
пока не будут выведены из процесса; их место занимают другие зародышевые части-
цы. Кокс является одновременно и продуктом, и теплоносителем [Furimsky, 2000].
Сырье (вакуумный остаток) подогревается до 260 °C и поступает в расположенный
над реактором скруббер (парциальный конденсатор) с рабочей температурой 370 °C.
Тяжелые углеводороды сырья образуют суспензию с тонкими частицами кокса и воз-
вращаются в реактор. Последний работает при температурах 510—566 °C. Возвращае-
мое сырье через форсунки распыляется над псевдоожиженным слоем в реакторе, где
расщепляется на пары и легкие газы. Пары и газы проходят для очистки через скруб-
бер и поступают в перегонную колонну. Образующийся в реакторе кокс наслаивается
на существующие частицы. Снизу в реактор подается водяной пар. Там же находит-
ся скруббер, в котором с поверхности частиц удаляются тяжелые углеводороды. Пар
поддерживает слой в псевдоожиженном состоянии. Часть кокса, отбираемая из ре-
актора, поступает в коксонагреватель. От 15 до 30 % этого кокса сжигается, осталь-
ная часть возвращается в реактор для подведения к нему тепла. Температура в коксо-
нагревателе поддерживается в интервале 593—677 °C. При сжигании кокса образуется
дымовой газе низким теплосодержанием [McKetta, 1992].
5.2.3. Флексикокинг
Флексикокинг — это сочетание технологий непрерывного коксования и газифика-
ции. Кокс перерабатывается в синтез-газ, но температура (1000 °C) недостаточна
для сжигания всего кокса. Флексикокинг дает лишь 2 % кокса в качестве конечного
продукта; большая часть кокса сжигается для подогрева сырья. Процесс непрерыв-
ного коксования дополняется еще одним псевдоожиженным слоем, действующим
как газогенератор. В слой подаются водяной пар и воздух, которые реагируют с кок-
сом, образуя низкокалорийный синтез-газ. Газогенератор работает при температу-
ре 816-982 °C. Излишки горячего кокса, остающегося после сгорания, поступают
в промежуточный теплообменник, где нагревают холодный кокс, выходящий из ре-
актора, а затем возвращаются в коксонагреватель. Реактор работает таким же обра-
зом, как и при непрерывном коксовании [McKetta, 1992; Furimsky, 2000].
Процессы флюидкокинга и флексикокинга разработаны на основе технологии ка-
талитического крекинга в псевдоожиженном слое. В обоих процессах циркулирующий
кокс переносит тепло от коксонагревателя к реактору. Частицы кокса в реакгоре игра-
ют роль центров, на которых происходят реакции крекинга остаточного сырья до лег-
ких продуктов. Флюид- и флексикокинг способны давать больший объемный выход
жидкостей, чем замедленное коксование. Короткое время пребывания способствует
увеличению выхода дистиллятов и уменьшению выхода кокса, но за счет некоторо-
го ухудшения качества продуктов. Благодаря повышенному выходу жидких продуктов,
а также меньшему расходу энергоносителей и топлива флексикокинг обладает некото-
рым преимуществом перед замедленным коксованием [McKetta, 1992; Furimsky, 2000].
Глава 5. Моделирование процесса коксования
175
5.3. ОПИСАНИЕ ПРОЦЕССА
Из всех технологий обеднения углеродом наиболее распространено замедлен-
ное коксование, поэтому оно рассматривается здесь как типичная технология
коксования. На рис. 5.1 показана типичная технологическая схема замедленно-
го коксования, включающая в себя печь, камеры коксования, секции фракцио-
нирования и отпарки и оборудование выгрузки кокса. Фактическая схема зависит
от стратегии проектирования конкретного завода и существующих технологиче-
ских возможностей.
Ниже перечислены основные моменты технологического процесса на установке
замедленного коксования [Hamilton, 2002; Elliot, 2003; Haniford, 2003].
• Вакуумный остаток (первичное сырье) подается в нижнюю часть зоны ис-
парения колонны фракционирования или непосредственно под тарелку от-
бора газойля, где нагревается, а легкие фракции отбираются в виде боковых
погонов.
• Фракции легче тяжелого газойля испаряются, а вакуумный остаток вместе с ре-
циркулирующим потоком тяжелого продукта (вторичное сырье) снизу колон-
ны подается в печь коксования, температура на выходе из которой составляет
от 480 до 515 °C.
• В змеевик печи во избежание преждевременного закоксовывания вводится во-
дяной пар.
• Парожидкостная смесь, выходящая из печи, поступает в одну из двух камер
коксования, где происходят реакции крекинга и накапливается кокс. Другую
камеру в это же время освобождают от кокса, очищают и готовят к следующему
циклу работы.
* Для непрерывной работы нужны две камеры коксования: пока одна камера ра-
ботает в режиме коксования, из другой выгружается кокс.
• Коксование до заполнения камеры (примерно на 4/5 объема) обычно про-
должается около 24 ч. Нарастание слоя кокса происходит со скоростью
около 0,6 м/ч.
• Температура в камере коксования составляет от 415 до 465 °C, давление — от 0,1
до 0,4 МПа.
• Горячие пары, которые состоят из водяного пара и продуктов реакций термиче-
ского крекинга (газ, бензиновая фракция, газойли), поступают из камеры кок-
сования во фракционирующую колонну, где охлаждаются жидким сырьем для
предотвращения образования значительных количеств кокса в колонне и (од-
новременно) конденсации части тяжелых фракций (рециркулята).
• Газы и пары коксования поднимаются через охлаждающие тарелки колонны.
• Под тарелкой отбора газойля выше точки ввода свежего сырья обычно есть две-
три дополнительные тарелки, орошаемые частично охлажденным газойлем.
Они уменьшают унос свежего жидкого сырья и рециркулята отбираемым га-
зойлем и обеспечивают контроль верхней границы его кипения.
Рис. 5.1. Технологическая схема типичной установки замедленного коксования
_С
0J
q
м
Глава 5. Моделирование процесса коксования 177
• Газойль отбирают через обычную отпарную секцию, содержащую 6-8 тарелок.
Под нижнюю тарелку секции подается пар для отгона легких фракций и кон-
троля температуры начала кипения газойля.
• Смесь водяного пара и легких фракций, отбираемую с верха отпарной секции,
возвращают в колонну на 1-2 тарелки выше точки отбора.
• Часть отбираемого газойля через систему циркуляционного орошения воз-
вращают в колонну, чтобы максимально использовать высокоуровневое тепло
и свести к минимуму количество низкоуровневого тепла, отводимого из верх-
него конденсатора, которое обычно невозможно использовать в теплообмен-
никах и приходится выбрасывать через водяные или воздушные холодильники.
• Тарелка отбора газойля обычно располагается на 8—10 тарелок ниже точки от-
бора бензиновой фракции или верха колонны.
• Если бензиновая фракция отбирается сбоку, выше точки ее отбора нужны до-
полнительные тарелки.
• Сырье в одну из двух камер направляют с помощью системы кранов.
• Камеры коксования работают попеременно в одном из двух режимов:
в режиме заполнения, когда происходят реакции коксования, в камере на-
капливается кокс, а газообразные продукты выводятся через верх и на-
правляются в колонну фракционирования;
• режиме выгрузки, при котором камера отсекается от остальной части цир-
куляционной системы и осуществляются гидрорезка и выгрузка кокса.
• Кокс из камеры выгружают следующим образом:
охлаждают кокс водой;
открывают люки камеры и вдоль оси камеры высверливают отверстие
в слое кокса;
в высверленное отверстие опускают гидрорезак с насадками для создания
высоконапорных струй. Медленно поднимают гидрорезак. Куски влажно-
го вырезаемого кокса падают вниз, где выходят через нижний люк и по же-
лобу отводятся на отстойную площадку.
5.4. ПАРАМЕТРЫ ПРОЦЕССА
Через подбор рабочих параметров можно изменять качество и выход продуктов кок-
сования сообразно целям, которые ставит перед собой нефтеперерабатывающий за-
вод. Ниже кратко описываются важнейшие условия процесса на установке коксова-
ния [Speight, 1980; Furimsky, 2000; Hue, 2011].
5.4.1. Температура на выходе из печи и на входе в камеры
коксования
Температура — основной параметр, определяющий жесткость условий коксования.
Температура определяет качество кокса. Ее изменения влекут за собой целый ряд эф-
фектов, которые представлены ниже.
178
Часть 2. Моделирование некаталитических процессов
• Повышение температуры вызывает усиленное испарение летучих веществ,
снижение выхода кокса и увеличение выхода дистиллятов, но регулировать та-
ким образом количество летучих веществ, остающихся в коксе, можно лишь
в узком интервале.
• Изменением температуры также регулируют количество остающихся в коксе
летучих веществ. Чем выше температура, тем меньше их остается и тем тверже
кокс.
• Высокая температура в печи может вызвать закоксовывание змеевика.
• Слишком высокие температуры вызывают образование очень твердого кокса,
который трудно вырезать гидрорезаком.
• Повышение температуры в конце цикла коксования сокращает время, необхо-
димое для переключения камер.
• Если температура слишком низка, реакции коксования протекают не до кон-
ца и усиливается образование кокса и тяжелого недококсованного продукта.
Иными словами, низкая температура ведет к неполному коксованию. Умень-
шение длительности цикла увеличивает пропускную способность, но снижа-
ет выход жидких продуктов и сокращает срок службы камер коксования.
5.4.2. Давление в камере коксования
Изменение давления в камерах коксования влияет на процесс следующим образом:
• при уменьшении давления снижается выход кокса;
• на каждые 0,5 кг/см2 снижения давление в камере коксования:
выход жидких продуктов возрастает на 1,3 %об. (относительно свежего
сырья);
выход кокса по массе снижается на 1 %;
• для получения кокса высокого качества камеры коксования должны ра-
ботать под повышенным давлением (увеличение давления подавляет ис-
парение газойля в камере коксования, а время удержания ароматических
соединений продлевается, что способствует образованию кокса лучшего
качества);
• для получения игольчатого кокса необходимо давление 10,5 кг/см2;
• при повышении давления снижаются коэффициент теплового расширения
и удельное электрическое сопротивление получаемого кокса; а также увели-
чивается объем жидкой фазы, которая остается в камере и принимает участие
в реакциях, ведущих к образованию кокса;
• низкое давление способствует увеличению скорости паровой фазы в камере
коксования и уносу кокса.
Так как экономика нефтепереработки требует минимизации производства кок-
са, новые установки коксования проектируют в расчете на рабочее давление 1 бар
(15psi)', существующие установки работают под давлением 2,4 бар (35psi).
Глава 5. Моделирование процесса коксования 179
5.4.3. Коэффициент рециркуляции сырья
Коэффициент рециркуляции сырья* 1 (CFR) определяется следующим образом:
Скорость подачи вторичного сырья в печь
Скорость подачи первичного сырья в колонну фракционирования
Чтобы увеличить пропускную способность, современные установки коксования
эксплуатируют с коэффициентом рециркуляции 10 % и менее (до 3 %). Коэффици-
ент рециркуляции определяет конечную температуру кипения газойля. Повышение
коэффициента рециркуляции вызывает:
• увеличение выхода кокса и улучшение его свойств;
• снижение содержания смол и асфальтенов во вторичном сырье и рост концен-
трации ароматических соединений в камерах коксования;
• увеличение объемов производства газа и бензина, снижение выхода дистилля-
тов, замедление отложения кокса в трубах змеевика печи;
• улучшение свойств тяжелого газойля коксования, в том числе плотности, кок-
суемости, содержания металлов и верхней границы кипения;
• снижение выхода жидких продуктов;
• продление межремонтного периода работы печи коксования.
5.4.4. Виды сырья
Типичное сырье установок коксования — вакуумный остаток. Можно перерабаты-
вать и сырье других видов:
• остаток атмосферной перегонки;
• остаток висбрекинга;
• нефтяной шлам;
• остатки процессов гидроочистки;
• битуминозную нефть;
• остатки процессов деасфальтизации (асфальт, или битум);
• сырье других видов, которое с годами привыкли перерабатывать в установках
коксования: гильсонит, пек бурого угля, каменноугольный пек, вредные отхо-
ды нефтепереработки и отходы пластмасс;
• тяжелые нефти с высоким содержанием ароматических соединений2 (их пере-
рабатывают для получения высококачественного игольчатого кокса).
1 В действительности здесь имеется в виду не коэффициент рециркуляции (К^), а коэффициент за-
грузки (К1а1р), так как вторичное сырье представляет собой сумму расходов (или скорости подачи) первич-
ного сырья и рециркулята, т. е. всю загрузку установки, а Крти = Кир — 1. — Примеч. науч.ред.
1 Предпочтительным сырьем для получения игольчатого кокса являются малосернистые высокоаро-
магизированные тяжелые фракции и остатки деструктивного происхождения: дистиллятные крекинг-
остатки, декантированный газойль каталитического крекинга и др. [Мейерс Р. А. Основные процессы
нефтепереработки. Справочник: пер. с англ. 3-го изд. / [Р. А. Мейерс и др.]; под ред. Глаголевой О. Ф.,
Лыкова О. П. - СПб.: ЦОП «Профессия», 2011. - 944 с.] — Примеч. науч.ред.
180
Часть 2. Моделирование некаталитических процессов
Ниже перечислены свойства сырья, оказывающие определяющее влияние на ка-
чество дистиллятов и кокса.
• Нижняя граница кипения. Истинные границы кипения остатков атмосферной
и вакуумной перегонки обычно принимаются равными соответственно 343 °C
(649 °F) и 538 °C (1000 °F), но они могут быть другими (это зависит от свойств
перерабатываемой нефти и рабочих параметров перегонных колонн). Со-
держание серы и металлов в остатке и его коксуемость по Конрадсону зави-
сят от нижней границы кипения, поэтому выбор границы кипения влияет как
на выход, так и на качество продуктов.
• Коксовый остаток (коксуемость) по Конрадсону. Это свойство сырья наиболее
важно для процесса коксования, так как оно определяет количество образу-
ющегося кокса. Как следствие, именно оно принято за меру склонности сы-
рья к образованию кокса. Сырье с высокой коксуемостью по Конрадсону дает
больше кокса. Следовательно, для максимального производства жидких про-
дуктов желательно сырье с низким коксовым остатком по Конрадсону.
• Содержание серы и азота. Сера концентрируется в коксе и тяжелых жидких
продуктах. Разнообразные соединения серы и азота по-разному распределены
в получаемых продуктах и отходящем газе.
• Содержание металлов. Большая часть металлов, в особенности никель и вана-
дий, концентрируется в коксе, что ухудшает качество кокса.
5.5. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ПРОЦЕССА
КОКСОВАНИЯ
5.5.1. Химия процесса
Сырье процесса обычно содержит компоненты различных типов, из которых на про-
цесс коксования сильнее всего влияют следующие [Furimsky, 2000; Нис, 2011]:
• асфальтены — самые тяжелые, трудно поддающиеся переработке высокомоле-
кулярные компоненты с относительно высокой ароматичностью, часто с высо-
ким содержанием металлов;
• смолы — длинноцепные тяжелые углеводороды средней ароматичности, в кото-
рых растворены асфальтены. Смолы относятся к тяжелым газойлям, кипящим
выше 427 °C (800 °F), и содержат органические соединения серы, азота и тяже-
лых металлов.
Установка коксования расщепляет эти компоненты на более легкие и ценные жидкие
и газообразные продукты, а наиболее тяжелая часть остается в виде твердого кокса.
Реакции превращения являются эндотермическими, и для их завершения необходи-
мо подводить большое количество теплоты.
В процессе коксования участвуют реакции трех основных типов. Эти реакции
могут протекать как параллельно, так и последовательно; все они представлены ниже.
Глава 5. Моделирование процесса коксования 181
—
1. Частичное испарение и мягкий крекинг (висбрекинг) сырья в змеевике печи. Тепло,
необходимое для инициирования испарения и крекинга, передается сырью в печи.
2. Термический крекинг. В ходе этой реакции высокомолекулярные соединения
расщепляются на более легкие, которые затем разделяются на продукты. Реакция
протекает с поглощением больших количеств теплоты. Тепло, необходимое для ини-
циирования крекинга, подводится внутри печи. Температуру и время пребывания сы-
рья в печи жестко контролируют, чтобы коксование в змеевике было минимальным.
3, Полимеризация. Молекулы углеводородов с небольшими размерами соединя-
ются в более крупные. В результате этой реакции образуется кокс. Полимеризация
начинается в жидкой фазе и продолжается на различных этапах процесса. Реакции
полимеризации занимают много времени, и для их завершения реагенты пребывают
в камере коксования достаточно долго.
В ходе этих реакций смолы и асфальтены расщепляются на более легкие углеводо-
роды и кокс. Легкие промежуточные продукты крекинга подвергаются дальнейше-
му крекингу, превращаясь в низкомолекулярные соединения: водород, легкие газы
и легкие жидкости (бензин и другие дистилляты).
Карбонизация, т. е. термическое превращение органических веществ в углероди-
стый продукт — кокс, состоит из сложной последовательности химических реакций
и диффузионных процессов, которые протекают как в жидкой, так и в твердой фазе.
Цепь реакций включает в себя отщепление замещающих атомов и групп от органиче-
ских молекул и последующий их переход в крупные ароматические структуры,
Нефтяной кокс образуется посредством двух разных механизмов.
1. Под действием высоких температур высокомолекулярные соединения, к ко-
торым относятся асфальтены и смолы, деалкилируются, образуя соединения с от-
крытой цепью и метиленовые группы СН2. Остатком является углерод, т. е. кокс.
2. К образованию кокса ведет также дегидрирование тяжелых продуктов с по-
следующей полимеризацией и конденсацией радикалов высокомолекулярных со-
единений (главным образом ароматических углеводородов).
5.5.2. Кинетика реакций
Кинетика реакций коксования — тема множества опубликованных исследований.
В них подробно описаны механизмы образования кокса, взаимопревращения ком-
понентов разных классов растворимости, роль этих компонентов в образовании кок-
са, влияние структурных свойств на скорость образования и выход кокса и, в числе
прочего, вывод различных зависимостей.
Большинство предложенных моделей основано на структурных изменениях
в остатках, и лишь несколько учитывают псевдокомпоненты промышленной зна-
чимости. Некоторые из этих моделей уже были проиллюстрированы на рис. 3,5
(см. главу 3). Например, модель, которую предложили Дель Бьянко и соавторы [Del
Bianco et al., 1993], содержит четыре дискретных компонента — вакуумный остаток,
фракцию дистиллятов, промежуточные продукты реакций и кокс, но не учитыва-
ет газовой фракции. Хорошо известно, что газы С,-С4 относятся к числу основных
182
Часть 2. Моделирование некаталитических процессов
продуктов процесса коксования и что их выход может быть достаточно высоким в за-
висимости от жесткости условий и свойств сырья. Поэтому кинетическая модель
должна содержать псевдокомпонент газовой фракции. Иначе говоря, кинетическая
модель, предусматривающая подробное описание реакций, происходящих в процес-
се коксования, должным образом еще не рассмотрена. Как уже сообщалось, общий
подход заключается в выборе небольшого числа дискретных компонентов и фор-
мулировании различных параллельных и последовательных реакций между ними
[Zhou et al., 2007]. Главный недостаток такого подхода, как и любых других дискрет-
ных моделей кинетики, состоит в том, что всякое изменение состава и свойств сырья
требует перерасчета параметров модели.
Вследствие сложности, присущей реакциям коксования, аналитическую мо-
дель вывести трудно. Разработка детальной кинетической модели реакций коксо-
вания требует решения двух проблем: 1) трудно описать сырье, т. е. тяжелый оста-
ток; 2) довольно сложно определить константы скоростей реакций и их зависимость
от свойств сырья. Применимость простых, т. е. дискретных, моделей ограничена тем,
что они конкретизируют условия реакций и свойства сырья. Именно по этой причи-
не для моделирования и имитации процессов коксования используют главным обра-
зом эмпирические зависимости. Чтобы такие зависимости можно было использовать
для прогнозирования выхода и качества продуктов в широком спектре условий реак-
ций и типов сырья, их разработка должна опираться на обширную базу эксперимен-
тальных данных.
5.5.3. Термическое разложение асфальтенов
Вопрос термического разложения асфальтенов привлекает к себе внимание по той
причине, что в условиях высоких температур асфальтены склонны к образованию
кокса. Ключ к пониманию процесса переработки нефтяных остатков путем коксо-
вания лежит в выявлении химизма образования кокса при различных температурах
[Goncalves et al., 2001; Douda et al., 2004]. Предложены различные пути реакций тер-
мического разложения асфальтенов, главными продуктами которых являются алка-
ны от С, до С40 и полициклические ароматические углеводороды с числом аромати-
ческих колец от 1 до 4 [Schucker and Keweshan, 1980; Yasar et al., 2001]. При пиролизе
асфальтенов в интервале температур от 350 до 400 °C разрываются связи С—S, тогда
как при температурах выше 400 °C начинает преобладать разрыв связей С-С
[Zhao et al., 2001]. Образование метана и других н-алканов при пиролизе указывает
на то, что наружная поверхность молекул асфальтенов содержит термически нестой-
кие алкильные группы [Ali and Saleem, 1991]. Йошида и соавторы [Yoshida et al., 1984]
методом термогравиметрического анализа (ТГА) исследовали термическое разложе-
ние асфальтенов, полученных из угля, и пришли к выводу, что в интервале темпера-
тур от 300 до 500 °C скорость потери массы асфальтенов достигает наибольшей вели-
чины, а после 500 °C снижается. Кок [Кок, 1993] сообщил, что если ТГА проводится
в атмосфере воздуха, можно выделить три области реакций: низкотемпературного
Глава 5. Моделирование процесса коксования
183
окисления, накопления горючих материалов и высокотемпературного окисления.
Хаузер с соавторами [Hauser et al., 2008] исследовали пиролиз асфальтенов при 412 °C
и нашли, что соотношение чисел атомов углерода в ароматических и алифатических
соединениях возросло примерно с 1 до 3, а соотношение Н/С снизилось с 1,1 до 0,8.
Сведений о термогравиметрическом анализе нефтяных фракций значительно
меньше. В ранних исследованиях [Ciajolo and Barbella, 1984] утверждается, что пи-
ролиз парафиновых, ароматических, полярных и асфальтеновых фракций тяже-
лых нефтей может быть истолкован как испарение парафиновых и ароматических
фракций в низкотемпературной фазе и пиролиз полярных и асфальтеновых соеди-
нений с образованием частиц углеродистого остатка в высокотемпературной фазе.
Дауда и соавторы [Douda et al., 2004] осуществили пиролиз асфальтенов нефти
«Майя» и получили в виде продуктов мальтены (предельные, ароматические и По-
лярные соединения), кокс и газы. После разделения мальтенов методом жидкостной
хроматографии было установлено, что их предельная фракция состоит из тетраци-
клоалканов (18,2 %), алканов (15,9 %) и гексациклоалканов (10,9 %), а ароматиче-
ская — из моноароматических (22,6 %), диароматических (26,5 %), тиофеновых аро-
матических (19,5 %) и пентаароматических (1,3 %) соединений. Дауда с соавторами
[Douda et al., 2008] также установили, что при температурах вплоть до 215 °C мальте-
ны нефти «Майя» стабильны, далее при температурах до 550 °C происходит быстрое
превращение, а при температуре около 1000 °C наблюдается полное разложение. Вы-
ход кокса из асфальтенов изменялся в пределах от 25 до 60 %масс. и более; основным
объектом исследований выступали асфальтеновые компоненты, дававшие выход
термического кокса в интервале от 35 до 65 %масс. Асфальтены обеспечивают высо-
кий выход летучих продуктов, в том числе конденсируемых жидкостей и газов [Shi-
roto et al., 1983; Speight, 2004]. Но в образование кокса они вносят наибольший вклад
среди всех фракций SARA. По наблюдениям Дауды и соавторов [Douda et al., 2004],
термогравиметрический анализ асфальтенов нефти «Майя» показывает значитель-
ное разложение асфальтенов в интервале температур 300-680 °C; максимума ско-
рость разложения достигает при 478 °C.
Считается, что важную роль в термолизе смол и асфальтенов играет гетероцикли-
ческий азот. Сначала от ароматических колец отщепляются алкильные группы.
В ходе вторичных реакций происходят ароматизация нафтенов и конденсация аро-
матических колец, что инициирует образование кокса. Таким образом, на началь-
ном этапе образуются летучие углеводороды и нелетучие гетероатомные соедине-
ния. Получающиеся продукты нерастворимы в реакционной среде, что способствует
карбонизации и образованию в конечном счете кокса [Magaril and Aksenova, 1968;
Dias, 1988]. Ранее было показано [Schuckerand Keweshan, 1980; Savage etal., 1988], что
при термолизе атомное соотношение Н/С в асфальтенах быстро снижается и (когда
время реакции становится достаточно большим) стремится к асимптотическому пре-
делу. При этом крекинг асфальтенов практически не затрагивает их ядра. Экспери-
ментальные данные дают основание предполагать, что термический крекинг асфаль-
тенов и смол — сложный процесс, в ходе которого происходят следующие реакции:
• отрыва алкильных цепей от ароматических групп;
• дегидрирования нафтенов с образованием ароматических соединений;
184
Часть 2. Моделирование некаталитических процессов
• конденсации ароматических колен до высших соединений с сочлененными
кольцами;
• димеризации и олигомеризации.
Термический крекинг всегда сопровождается отрывом алкильных цепей, а реак-
ции дегидрирования и конденсации являются следствием дефицита водорода.
5.6. КИНЕТИКА РЕАКЦИЙ КОКСОВАНИЯ
Определить кинетические параметры реакций коксования и пиролиза можно про-
ведением ТГА с различными скоростями нагревания. Опираясь на изоконверсион-
ные методы, Шукер [Schucker, 1983] вывел ряд значений энергий активации при
пиролизе асфальтенов в зависимости от их испаренной доли. При более высоких
степенях превращения величины энергий активации были больше. Ших и Сон
[Shih and Sohn, 1980], напротив, обнаружили, что при различных значениях степе-
ни превращения энергия активации была почти постоянна. Используя прямую за-
висимость Аррениуса, Парк и соавторы [Park et al., 2009] установили, что в области
разложения углеродистых материалов при 350—600 °C энергия активации долж-
на находиться в интервале 146—246 кДж/моль. В первой области (50—350 °C), где
происходит испарение легких органических веществ, энергии активации меньше
(19-23 кДж/моль).
5.6.1. Разделение атмосферного остатка
Методом хроматографического разделения были выделены различные фракции
остатка атмосферной перегонки тяжелой нефти. Свойства нефти и остатка ее атмо-
сферной перегонки представлены в табл. 5.1. Асфальтены были получены осажде-
нием н-гептаном в атмосфере азота под давлением 25 кг/см2 при постоянном пере-
мешивании. В двух хроматографических колонках с глинистой и алюмооксидной
насадками было осуществлено фракционирование SARA. Более подробные сведения
об этой процедуре можно найти в других источниках [Alvarez et al., 2011].
Таблица 5.1
Свойства тяжелой нефти и остатка ее атмосферной перегонки
Свойство Нефть Атмосферный остаток
Плотность, “API 12 5,6
Содержание серы, %масс. 5,29 6,08
Содержание металлов, мг/кг: N1 V 89 441 97 493
Глава 5. IМоделирование процесса коксования
185
Таблица 5.1, окончание
Свойство Нефть Атмосферный остаток
Элементный состав, %масс.:
С 83,98 82,56
Н 10,28 10,56
N 0,40 0,74
Состав SARA, %масс.:
предельные углеводороды 15,83 11,75
ароматические углеводороды 36,74 23,66
СМОЛЫ 18,61 34,27
асфальтены 28,82 30,32
После выделения фракций предельных и ароматических углеводородов, смол
и асфальтенов был проведен ТГА каждой фракции, а также атмосферного остатка.
Температура изменялась в пределах от комнатной до 800 °C. Для определения кинети-
ческих параметров коксообразования анализ всех фракций и атмосферного остатка
(за исключением предельных углеводородов) проводился при трех разных скоростях
нагревания: 8, 12 и 16 °С/мин. Термогравиметрический анализ фракции предельных
углеводородов, ввиду ее склонности к раннему термическому разложению, прово-
дился лишь при единственном значении скорости нагревания (8 °С/мин).
5.6.2. Неизотермическая кинетика
Неизотермическим методом были найдены кинетические параметры пиролиза ас-
фальтенов. Данные ТГАбыли проанализированы по методике Фридмана [Dias, 1988],
а кинетика была рассчитана для реакции первого порядка. Метод основывается
на сравнении данных, полученных при различных линейных скоростях нагревания.
С его помощью можно найти энергию активации ряда процессов, когда форма кине-
тического уравнения неизвестна. Согласно Шукеру и Кевешану [Schucker and Kewe-
shan, 1980], термическое разложение тяжелых компонентов можно описать следую-
щим образом:
А—> а(кокс) + (1 — а)Г, (5.2)
Где д _ реагент; И — летучая фракция; а — стехиометрический коэффициент для
кокса.
Из этого уравнения можно вывести следующее кинетическое выражение для ле-
тучих продуктов:
186
Часть 2. Моделирование некаталитических процессов
где Vo — суммарное количество испаренных веществ. Линейное преобразование
уравнения (5.3) дает выражение
111 (5.4)
at J К1
где х = V/ Vo — доля летучей фракции, которая при термическом разложении соот-
ветствует степени превращения асфальтенов.
Уравнение (5.4) легко использовать, подставляя различные значения степени
превращения х, изменяющиеся в интервале от 0,1 до 0,8. По первым производным
кривых ТГА были определены величины dx / dt и Т для всех значений степени пре-
вращения, достигаемых при различных скоростях нагревания (8, 12, 16 °С/мин). Ли-
нейной регрессией формулы (5.4) был получен ряд значений энергии активации (£j
и предэкспоненциальных множителей (/<0) в зависимости от степени превраще-
ния асфальтенов. Применение этого метода подробнее описано в другом источни-
ке [Trejo et al., 2010].
5.6.3. Термическое разложение
Ниже изложены выводы, следующие из анализа представленных на рис. 5.2 результа-
тов ТГА атмосферного остатка и его фракций SARA.
• Атмосферный остаток. Масса изменяется в интервале температур от 100
до 500 °C. Вначале испарение идет главным образом за счет отгонки легких ал-
канов и продолжается до 350 °C. При температурах от 350 до 500 °C происходит
быстрое испарение, свидетельствующее о крекинге тяжелых фракций — асфаль-
тенов и смол. В интервале от 500до 800 °C испарение идет с низкой и почти посто-
янной скоростью. Выход кокса из атмосферного остатка составляет 16,3 %масс.
• Асфальтены. Испарение, происходящее при температурах от комнатной
до 350 °C, соответствует почти 10 % превращения. Сообщалось [Karacan and
Kok, 1997], что изменения, обнаруживаемые при температуре около 350 °C, мо-
гут быть объяснены отщеплением алкильных групп от периферийных участков
асфальтеновых структур. Наиболее существенная потеря массы наблюдается
в интервале температур от 430 до 550 °C, где следует ожидать разрыва межмо-
лекулярных и химических связей, в том числе серных мостиков и связей С-С.
На этой стадии асфальтены могут быть превращены в газы и ценные компо-
ненты, т. е. масла. При продолжении пиролиза в интервале от 550 до 800 °C ас-
фальтены вступают в реакции конденсации, образуя кокс в качестве конечного
продукта. В интервале от 600 до 800 °C выход кокса из асфальтеновой фракции
остается практически постоянным (43,1 %масс.).
• Смолы. Эта фракция дает 7 % потери массы в интервале от 50 до 150 °C, что отра-
жает испарение легких алканов. В интервале от 150 до 350 °C отгон продолжает-
ся. Благодаря реакциям крекинга в интервале от 350до 500 °C потеря массы силь-
но ускоряется. В этом интервале термически разлагается почти 80 %масс. смол.
Глава 5. Моделирование процесса коксования
187
Интервал пиролиза смол самый широкий по сравнению с другими фракциями.
Возможное объяснение этому состоит в том, что во время пиролиза смолы спо-
собны принимать участие в реакциях конденсации с образованием более тя-
желых молекул — например, асфальтенов [Phillips et al., 1985]. Из слабо при-
соединенных алкильных групп образуются свободные радикалы, стремящиеся
нейтрализоваться и конденсироваться в более крупные молекулы. После ис-
парения большей части легких соединений на начальных стадиях нагревания
происходит испарение оставшихся крупных молекул смол и асфальтенов, об-
разовавшихся путем конденсации, что и объясняет широкий интервал пироли-
за смол. Выход кокса из смол составляет 4,6 %масс.
• Ароматические углеводороды. При температуре до 100 °C они не испытывают су-
щественных потерь массы. В интервале от 100 до 320 °C наиболее выраженным
изменением является, пожалуй, испарение моно-, ди-, три- и высших поли-
циклических соединений. В интервале от 320 до 480 °C смолы вступают в ре-
акции крекинга. По выходу кокса (3,8 %масс.) ароматические углеводороды
очень близки к смолам.
• Предельные углеводороды. Они дают очень мало кокса (0,3 %масс.), так как все
соединения этой фракции полностью испаряются. Предельные углеводоро-
ды практически не вступают в реакции крекинга: все изменения происходят
на стадии отгонки.
• При температурах ниже 400 °C термограммы асфальтенов и атмосферно-
го остатка обнаруживают схожее поведение; в интервале от 450 до 800 °C они
практически идентичны. Это может указывать на то, что в целом поведение ат-
мосферного остатка в этом интервале определяют реакции с участием тяжелых
фракций вроде асфальтенов.
На рис. 5.2 показаны также скорости изменения массы каждой фракции и атмо-
сферного остатка. Максимальные скорости изменения массы асфальтенов и смол
наблюдаются при температурах соответственно 467 и 455 °C. Точка максимальной
скорости потери массы асфальтенов (467 °C) очень близка к полученной другими ис-
следователями [Ritchie et al., 1979; Khulbe et al., 1984; Karacan and Kok, 1997]. При
сравнительно низких температурах (ниже 350 °C) термическое разложение асфаль-
тенов идет через отщепление периферийных групп. При температурах выше 350 °C
асфальтеновые структуры сильно сокращаются в размерах [Moschopedis et al., 1978].
На начальных стадиях пиролиза идет отгонка низкомолекулярных соединений,
но с повышением температуры возможно также расщепление все более крупных мо-
лекул с образованием летучих фрагментов [Douda et al., 2004]. Асфальтены образу-
ются кольцами полициклических ароматических соединений, связанными с нафте-
новыми и алкильными группами, а также с гетеросоединениями. Основные силы
молекулярного взаимодействия в структурах асфальтенов — это межмолекулярные
силы и слабые химические связи, которые при повышении температуры разрушают-
ся или ослабляются. Вначале газы высвобождаются в результате разрыва алкильных
цепей. Но при достаточно высоких температурах (выше 450 °C) количество высво-
бождаемых газов увеличивается вследствие разложения асфальтенов, когда разрыва-
ются более сильные химические связи и разрушаются скелеты молекул.
188
Часть 2. Моделирование некаталитических процессов
— Предельные
углеводороды
— Ароматические
углеводороды
... Смолы
---Асфальтены
— Атмосферный
остаток
100
90
80
70
60
50
| 40
30
20
10
0
0 100 200 300 400 500 600 700 800
Температура, °C
Рис. 5.2. Термограммы и кривые скорости изменения массы атмосферного остатка
и его фракций при нагревании со скоростью 8 °С/мин в атмосфере азота
Скорость потери массы атмосферного остатка достигает максимума при 431 °C,
а вдоль всей термограммы наблюдаются небольшие изгибы. Это можно объяснить
последовательным испарением компонентов с разными температурами кипения.
Более четко оно выражено у предельных углеводородов с последовательно отгоняе-
мыми легкими компонентами. У таких углеводородов максимальная скорость поте-
ри массы наблюдается при 212 °C. Термограмма ароматических углеводородов тоже
имеет множество изгибов, образуемых последовательным испарением соединений
различных классов, т. е. моно-, ди-, три- и полициклических ароматических соедине-
ний. Скорость потери массы максимальна при 395 °C. В табл. 5.2 для каждой фракции
приведены величины выхода кокса и значения температур, при которых скорость по-
тери массы достигает максимума.
Глава 5. Моделирование процесса коксования
189
Таблица 5.2
Выход кокса из атмосферного остатка н его фракций SARA при скорости
нагревания 8 °С/мин
Атмосферный остаток/ его фракция Выход кокса, %масс. Температура максимума скорости потери массы, °C
Атмосферный остаток 16,3 431
Предельные углеводороды 0,3 212
Ароматические углеводороды 3,8 395
Смолы 4,6 455
Асфальтены 43,1 467
5.6.4. Кинетические параметры
Кинетические параметры оценивают по результатам ТГА, которые дают общие сведе-
ния о кинетике суммарной реакции. Для анализа данных ТГА использовался подход,
не привязанный к конкретной модели и основанный на изоконверсионном методе
Фридмана. Метод позволяет определить величины энергии активации и предэкспо-
ненциального множителя при известном порядке реакции и неизвестной зависимо-
сти скорости от степени превращения.
Для получения кинетических параметров коксообразования анализировались тя-
желые фракции (смолы и асфальтены) и атмосферный остаток. На рис. 5.3 в виде
зависимости степени превращения от температуры показан ход испарения атмо-
сферного остатка, асфальтенов и смол. По сообщениям ряда исследователей [Soni-
bare et al., 2003; Meng et al., 2007; Park et al., 2009], с увеличением скорости нагревания
испарение смещается в сторону высоких температур. При наивысших скоростях на-
гревания реакции протекают очень быстро, так что выделение газов уменьшает мас-
су образца гораздо сильнее, чем при небольших скоростях. По оси ординат отложена
величина испаренной части исходного образца. Видно, что при высоких темпера-
турах кривые асимптотически приближаются к предельному значению. На рис. 5.4
изображены зависимости 1п(</х/dt) от 1 / Т (графики Аррениуса) для атмосферного
остатка, асфальтенов и смол при различных величинах степени превращения в ин-
тервале от 0,1 до 0,8. Для величин степени превращения 0,9 и выше графики не по-
казаны ввиду ненадежности данных, что объясняется трудностью получения точных
значений dx/dt вблизи момента завершения реакций. Как следует из табл. 5.3, во всех
случаях получены высокие значения коэффициента корреляции (г). Значения энер-
гии активации (ЕД в таблице выражены в ккал/моль, предэкспоненциального мно-
жителя (к0) — в мин-1. При степени превращения выше 0,4 в величине энергии ак-
тивации происходят определенные изменения, о чем говорит ее существенный рост.
При малых значениях степени превращения (от 0,1 до 0,4) происходит лишь испаре-
ние легких компонентов. Для увеличения степени превращения необходим крекинг
тяжелых фракций, т. е. смол и асфальтенов, что влечет за собой увеличение энергии
активации. Высказано предположение [Collett and Rand, 1980], что увеличение энер-
гии активации отражает изменения в природе лимитирующей стадии.
190
Часть 2. Моделирование некаталитических процессов
Рис. 5.3. Испаренные доли атмосферного остатка, асфальтенов и смол
при различных скоростях нагревания
|-лаВа 5. Моделирование процесса коксования
191
з,о
2,5
2,0
1,5
1,0
0,5
0,0
-0,5
-1,0
0,0013 0,0014 0,0015 0,0016 0,0017 0,0018 0,0019 0,002 0,0021 0,0022
1 /Г, к-1
Рис. 5.4. Графики Аррениуса для атмосферного остатка, асфальтенов и смол
при различных степенях превращения
192
Часть 2. Моделирование некаталитических процессов
Таблица 5.3
Значения энергии активации и предэкспоненциального множителя в зависимости
от степени превращения
X Атмосферный остаток Асфальтены Смолы Ароматические углеводороды
еа *0 Г Ея ft0 r r ЕЛ ko r
0,1 11,5 4,1 105 0,989 41,0 1,4- Ю” 0,978 10,8 4,4 104 0,992 10,8 6,7 IO7 0,991
0,2 15,3 4,4-106 0,999 54,9 1,8 1018 0,998 14,8 5,9-10s 0,998 14,5 7,8-10’ 0,997
0,3 12,4 1,0' 10s 0,995 56,7 4,2 • 1018 0,996 21,1 3,9 • 107 0,999 14,2 1,7-10’ 0,991
0,4 12,7 4,8 Ю4 0,991 52,8 1,6-IO17 0,993 35,8 1,3 1012 0,999 17,6 4,6-10'° 0,992
0,5 20,1 9,5-106 0,985 58,6 6,7 • 1018 0,995 45,9 1,2 1015 0,996 17,9 1,3 1010 0,990
0,6 24,8 1,2-10s 0,994 52,1 4,6-1016 0,998 49,8 1,3- IO'6 0,998 21,6 1,1-ю11 0,992
0,7 27,8 2,7' 108 0,994 53,1 5,9-1016 0,999 47,5 1,7 1015 0,994 19,8 8,9 • 1010 0,994
0,8 30,0 2,4-108 0,980 58,6 8,1 • 1017 0,987 48,9 2,6-1015 0,999 19,2 1,2-1010 0,994
Наиболее существенное превращение асфальтенов происходит в интервале при-
мерно от 430 до 550 °C. По сравнению с атмосферным остатком и другими фракци-
ями это наиболее узкий интервал, где преобладают реакции крекинга. Выход кокса
при всех скоростях нагревания был один и тот же (43,1 %масс.), а энергия активации
возрастала, достигая пиковых величин при степенях превращения, равных 0,5 и 0,8,
По сравнению с атмосферным остатком и другими фракциями энергия активации
асфальтенов выше, что можно связать с образованием свободных радикалов в реак-
циях крекинга. Атмосферный остаток образуется отделением путем перегонки более
легких компонентов, вследствие чего тяжелые компоненты концентрируются в тя-
желых фракциях; в этих условиях единственной существенной реакцией является
крекинг.
Испарение смол завершается примерно при 500 °C. Выход кокса при всех трех
значениях скорости нагревания один и тот же (4,6 %масс.). С увеличением степени
превращения энергия активации изменяется, достигая наибольших значений в ин-
тервале между 0,4 и 0,8. Такое поведение может быть следствием реакций, происхо-
дящих при крекинге смол с образованием свободных радикалов, конденсирующих-
ся в виде кокса.
Кривая испарения и график Аррениуса для ароматических углеводородов не пред-
ставлены; для них приведены лишь значения энергии активации, предэкспоненци-
ального множителя и коэффициента корреляции в табл. 5.3. Энергия активации этой
фракции меньше, чем у асфальтенов и смол; при малых степенях превращения она
примерно та же, что у атмосферного остатка. Термогравиметрические данные пока-
зывают, что наибольший вклад в образование кокса дают асфальтены; за ними сле-
дуют смолы и ароматические углеводороды. По энергии активации атмосферный
остаток занимает промежуточное положение между ароматическими углеводорода-
ми и смолами. Энергия активации атмосферного остатка приблизительно та же, что
Глава 5. Моделирование процесса коксования 193
у смол и ароматических углеводородов, если степень превращения невелика, но если
последняя выше 0,5, то энергия активации стремится возрасти. Вероятно, это гово-
рит о реакции асфальтенов и смол.
5.6.5. Примечания
Рассмотренные выше результаты свидетельствуют о том, что основной коксообра-
зующей фракцией являются асфальтены. Выход кокса из асфальтенов составлял
43,1 %масс. Следует ожидать, что в реакциях крекинга асфальтены превращают-
ся в газы, масла и смолы. Смолы и ароматические углеводороды дают почти одно
и то же количество кокса, но энергия активации смол несколько выше. Предельные
углеводороды практически полностью испаряются. Атмосферный остаток демон-
стрирует рост энергии активации с увеличением степени превращения. Это говорит
о том, что на ранних этапах нагревания происходит испарение легких компонентов,
после чего начинается крекинг тяжелых фракций — смол и асфальтенов.
Более детальное представление о пиролизе атмосферного остатка, асфальте-
нов, смол и ароматических углеводородов дают кинетические данные, получен-
ные изоконверсионным методом. Во всех случаях коэффициенты корреляции были
близки к единице, а кинетика хорошо отвечала реакциям первого порядка. Значе-
ния энергии активации составляли от 11,5 до 30 ккал/кмоль для атмосферного остат-
ка, от 41,0 до 58,6 ккал/кмоль для асфальтенов, от 10,8 до 49,8 ккал/кмоль для смол
йот 10,8 до 21,6 ккал/кмоль для ароматических углеводородов. Различия в величине
энергии активации могут означать изменения силы связей в ходе реакций.
5.7. ЗАВИСИМОСТИ ДЛЯ ПРОГНОЗИРОВАНИЯ
ВЫХОДА КОКСА
5.7.1. Зависимости
Либерман [Lieberman, 1986] предложил несколько эмпирических правил для расчета
выхода кокса при замедленном коксовании, в том числе следующие:
• на каждые 8 psi снижения давления в камере коксования выход кокса уменьша-
ется на 1,0 %масс.;
• на каждые 10 °F (5,5 °C) увеличения температуры в линии отбора паров из каме-
ры коксования выход кокса снижается на 0,8 %масс.
Помимо этих эмпирических правил, опубликован ряд различных зависимостей
для расчета выхода, плотности и содержания серы в продуктах замедленного коксо-
вания. Большинство из них в качестве входных данных использует лишь коксовый
остаток сырья по Конрадсону (ССЯ); некоторые учитывают также влияние давления
и температуры. Зависимости выведены для широкого спектра видов сырья, свойства
которых можно найти в соответствующей литературе. Ввиду эмпирической природы
194
Часть 2. Моделирование некаталитических процессов
этих зависимостей рискованно применять вне областей свойств сырья, рабочих усло-
вий и выхода продуктов, для которых они были выведены. Расчетные величины в та-
ких случаях можно использовать лишь как приближенные оценки.
5.7.1.1. Зависимости Гзри и Хэндверка
Основываясь на данных, полученных с промышленных и опытных установок, Гэри
и Хэндверк [Gary and Handwerk, 2001] опубликовали следующие эмпирические
зависимости:
• выход продуктов, %масс.:
= 7,8 + 0,144 X CCR ырье; (5.5)
ЛеНзиН=1Ь29+0’343><ССЛеЫ₽Ье; (5.6)
у = 1,6 х CCR ; •'кокс ’ сырье’ (5.7)
v = 100 — у — V — V : •'газойль ^raaCCj) •'беиэин •'кокс’ (5.8)
• выход продуктов, %об.:
Убензин (%об.) = (131,5 + ЛР/сырье)186,5 ху6ензин (%масс.); (5.9)
Лазойль (%06-) = <131’5 +^l/>Ue)155’5X^b (%МаСС.). (5.10)
В промышленном производстве фракции бензина и газойля можно разделить
на легкий и тяжелый погоны и использовать их следующим образом:
• легкая бензиновая фракция коксования (ЛБК): гидроочистка и насыщение оле-
финов, затем изомеризация для повышения октанового числа или непосред-
ственное использование как компонента товарного бензина;
• тяжелая бензиновая фракция коксования (ТБК): гидроочистка и риформинг;
• легкий газойль коксования (ЛГК): сырье для гидрокрекинга или каталитического
крекинга в псевдоожиженном слое;
• тяжелый газойль коксования (ТГК): тяжелое нефтяное топливо, сырье для ката-
литического крекинга после гидроочистки или вакуумная перегонка.
Рассчитав по формулам (5.9) и (5.10) выходы бензина и газойля, соотношение их
легкой и тяжелой фракций можно найти следующим образом:
Злбк(65м«) — 0,351 ху6ензин (%об.); (5.11)
'тбкоомр/) — 0’649 ху6ензин (%об.); (5.12)
'лгк(зо-и/>/)- 0,673 хугиойль (%об.); (5.13)
'тгк(1.глр/) = 0,327 хугаюйль (%об.). (5.14)
Глава 5. Моделирование процесса коксования
195
Для расчета состава газа (молярная масса 22,12) и содержания серы и азота в про-
дуктах можно воспользоваться типичными цифрами, приведенными в табл. 5.4.
Из состава газа исключена сера, а содержание ее и азота в продуктах дано в процен-
тах относительно соответствующей примеси в сырье.
Таблица 5.4
Типичное распределение серы и азота в продуктах и состав газа замедленного коксования
Компонент Сера, % Азот, %
Газ 30,0 —
ЛБК 1,7 —
ТБК 3,3 1
Л ГК 15,4 2
ТГК 19,6 22
Кокс 30 75
Всего 100,0 100
Состав газа, %мольн.: [Maples, 1993] [Gary and Handwerk, 2001]
метан 27,2 51,4
этилен 6,7 1,5
этан 17,8 15,9
пропилен 10,8 3,1
пропан 14,4 8,2
бутилен 11,0 2,4
изобутан 2,4 1,0
«-бутан 8,8 2,6
водород 0,9 13,7
диоксид углерода — 0,2
всего 100,0 100,0
Зависимости даны для следующих условий: давление в камере коксования
35-45 psi (0,25—0,32 МПа), сырье — прямогонный остаток, верхние границы кипе-
ния газойля и бензина равны соответственно 468—496 и 204 °C. Все зависимости Гэри
и Хэндверка основаны на влиянии коксуемости сырья по Конрадсону (ССА) и мо-
гут использоваться для расчета выхода в первом приближении и в предварительных
исследованиях.
5.7.1.2. Зависимости Мейплза
Мейплз [Maples, 1993] вывел аналогичные зависимости, исходя изданных промыш-
ленных и опытных установок, работающих в типичных условиях (сырье — прямо-
гонный остаток с плотностью 1,4—2,5 "API и CCR = 25,5 %масс.). Он разработал
196
Часть 2. Моделирование некаталитических процессов
линейные графики для определения выхода продуктов в массовых процентах, откуда
получил приведенные ниже зависимости. Зависимость для выхода бензиновой фрак-
ции можно вывести из графика, но Мейплз предложил рассчитывать его по разности.
• Выход продуктов:
-^газ(С4) = 4,1264 + 0,2745 х ССА ; (5.15)
У п z газойль = 79,225- 1,9418 хССА ; (5.16)
Укоке -0,3765 + 1,6755 хССЯ ; ’ ’ сырье’ (5.17)
У бензин = 100 — у —V —у zra3(C4) •'газойль •'кокс’ (5.18)
Мейплз разработал аналогичные графики для распределения серы и плотности
(в градусах АРГ) продуктов, из которых вывел ряд линейных зависимостей.
• Распределение серы в продуктах:
Sfi = 0,2002xS ; (5.19)
бензин ’ сырье’ v
S й = 0,7489 X S • (5.20)
газойль ’ сырье’ ' w/
S = 1,395 xS . (5.21)
кокс ’ сырье
• Плотность продуктов, °АРГ.
АРГ = 53,604 + 0,3404 хАРГ . (5.22)
бензин ’ ’ сырье’ v /
API а = 10,356 + 0,9131 хАРГ . (5.23)
газойль ’ сырье v '
Все зависимости даны для процесса замедленного коксования.
Ниже представлен еще один набор зависимостей для процесса флексикокинга
[Maples, 1993].
• Выход продуктов:
Лаз(с4 > = 5,206667 + 0,17943 х CCR- 4 сырье (5.24)
18,594587-0,115234хССЯ ; z бензин ’ ’ сырье’ (5.25)
1 >875742 + 1,037233 xCCR ; z кокс ’ ’ сырье’ (5.26)
^газойль Ю0 •'^газСС^ ) ^бензин У кокс’ (5.27)
Распределение серы в продуктах:
s бензин = 0,193461 xS ; бензин ’ сырье’ (5.28)
Глава 5. Моделирование процесса коксования
197
S . = 0,16921 + 0,91482xS ; (5.29) газойль ’ ’ сырье’ \ ^7 S = 0,18691 + 1,399667x5 ; (5.30) S=S — SK — S , — S (5.31) газ сырье бензин газойль кокс х 7
Плотность продуктов, ° АРЕ
АР7 й = газойль = -0,79976 + 1,264942 к API + 0,506675 х CCR . (5.32) ’ ’ сырье ’ сырье 4 7
Выход газов: щ = 3,200754 -0,028627 х CCR ; (5.33) У(Сз) = 0,456001 + О,О28627(-yj; (5.34) Гсз = ^газ(С<) ~ УС4~ Л С, )• (5.35)
5.7.1.3. Зависимости Шаброна и Спайта
Основываясь на опубликованной ранее работе Спайта [Speight, 1981], содержа-
щей формулы, по которым рассчитывался выход продуктов коксования остат-
ков перегонки нефтей Уилмингтона и Западного Техаса, Шаброн и Спайт [Schab-
ron and Speight, 1997] разработали новые зависимости, применимые к различным
видам сырья со следующими свойствами: плотность от 3,2 до 14,1 °АРГ, соотно-
шение Н / С от 1,4 до 1,7; коксуемость по Конрадсону от 7,5 до 29,2 %масс.; со-
держание никеля от 11 до 176 мг/кг, ванадия — от 5,2 до 612 мг/кг, асфальтенов —
от 4,4 до 32,4 %масс. Исходя из данных по шести видам сырья, Шаброн и Спайт
ввели коэффициент корреляции CF, зависящий от свойств асфальтенов (VPOMW—
молярная масса, определенная методом осмометрии по давлению паров в толуоле)
и их содержания в сырье:
CF=VPOMW . хАсфальтены [1-(Н + С) . ]. (5.36)
асфальтены сырье1- v 'асфальтены-’ 4 7
По этому коэффициенту вычисляется коксуемость сырья по Конрадсону (ССЛ):
ССЛ = 8,19 + 0,18CF. (5.37)
Затем величина CCR, рассчитанная по формуле (5.37), подставляется в зависимо-
сти Гэри и Хэндверка [Gary and Handwerk, 2001], что дает
З’с-i = 8,98 + 0,0259CF; (5.38)
уг = 14,1+0,0617CF; (5.39)
7 бензин ’ ’ ’ v '
198
Часть 2. Моделирование некаталитических процессов
^„= 13,1 + 0,288^; (5.40)
ЛаэоЛль = ЮО - Уг^) - - ^кокс. (5.41)
Таким образом, величины выхода кокса, газа, бензиновой фракции и газойля
можно вычислить в зависимости от содержания асфальтенов в сырье, молярной мас-
сы асфальтенов и содержания гетероэлементов в асфальтенах. Эти зависимости труд-
нее использовать, так как они требуют характеризации асфальтенов.
5.7.1.4. Зависимости Кастильони
Кастильони [Castiglioni, 1983] сообщил о графическом методе прогнозирования вы-
хода продуктов процесса коксования. Исходя из представленных им графиков, Аги-
льер и соавторы [Aguilar et al., 2012] разработали зависимости, основанные на рабо-
чем давлении и температуре. Метод предполагает три этапа («шага»).
1. Расчет выхода кокса при стандартном и фактическом давлениях:
где
А = Ь,хССЯсырк + Ь2; (5.43)
Д,=ЛЛ; (5.44)
Ь=АР). (5.45)
Для расчета выхода остальных продуктов вводится коэффициент приведения CFR-.
CFR = -0Д62152 + 1,28065-0,0072, (5.46)
где
„ Локс ПР11 рабочем давлении
Г) — - - - , (J. 4 /)
Локс И511 стандартном давлении
Стандартное давление принимается равным 0 бар (изб.). Величины выхо-
да газа и бензиновой фракции, а также суммарный выход продуктов вычисляются
по формуле
^ = С1/кокс+С2Локс+С3- (5.48)
2. Расчет коэффициента поправок, уточняющих выход продуктов замедленного
коксования F1;
F\=dpFR2 + d2CFR + dy
(5.49)
Глава 5. Моделирование процесса коксования 199
Этот коэффициент применяется для внесения поправок в величины выхода газа,
кокса, бензиновой фракции и суммарный выход продуктов, вычисленные на 1-м шаге:
Т,(шаг2)=У/(шаг1)ХН,.. <5-50)
3, Расчет коэффициента F1 для вычисления новых значений выхода кокса и бен-
зиновой фракции:
Д = еЛокс + е2’ <5'51)
где /= бензиновая фракция, кокс; е. =flCFR).
УЛшагЗ) — У/(шаг2) Х (5.52)
Конечные значения выхода продуктов вычисляются: угаз — на шаге 2; уиокс — на
шаге 3; у6еиэии - на шаге 3; уо6щий - на шаге 2. Таким образом,
^газойль ^обший У газ(С4~) З’бензин ^кокс’ (5.53)
Параметры зависимостей Кастильони для замедленного коксования представле-
ны в табл. 5.5.
Таблица 5.5
Параметры зависимостей Кастильони
Характеристика Зависимые параметры
Выход кокса «2 «2 Т, °C
3,2351 0,8352 427
2,03 0,8866 438
-3,0232 0,9699 449
Коэффициент А *2 Р, бар (изб.)
0,9433 -0,435 0
0,989 1,0532 1,04
0,9655 2,2898 2,07
Выходы ci С2
Газ -0,0055 0,3722 0,5101
Бензиновая фракция 0,00287 -0,1816752 3,889472
Всего -0,0139 0,7397 101,71
Коэффициент /1 «г
Газ -0,1066 0,9106 0,1965
Кокс -0,2621 1,2806 -0,0072
Бензиновая фракция -0,2419 1,1269 0,1294
Всего -0,8189 4,1057 -2,9785
200
Часть 2. Моделирование некаталитических процессов
Таблица 5.5, окончание
Характеристика Зависимые параметры
Коэффициент FL е, «2 СРЛ
Бензиновая фракция 0,00454545 0,88636364 1,1
То же 0,01193182 0,701704455 1,2
» 0,01988636 0,50284091 1,3
» 0,025 0,375 1,4
Кокс -0,0013 1,0346 1,1
То же -0,0024 1,0614 1,2
» -0,0037 1,0919 1,3
» -0,0053 1,1333 1,4
Источник: [Castiglioni, В.Р., Hydrocarbon Process., 62, 77, 1983].
Исходя из графиков Кастильони, Алберс [Albers, 1996] вывел ряд зависимостей,
которые представлены ниже.
• Выход кокса
= CCR -10,4753 + 4,094 ln( Р+15) - 0,2224 l[ln( Р +15)]2 +10,095 - 8,853 10~3 Т
?кокс 2,373-6,5979 • 10’3Т + 5,837 • 10'6Т~
• Выход других продуктов при Р = 0 и CFR = 1:
Уеух.™ = 0,76366 + 0,28299укокс + 2,0745 • 10-укокс - 1,8178 • 10-укокс; (5.55)
^иэин = -3,297 + 3,5559укокс - 0,16062/кокс + 2,4672 • 10-Укокс; (5.56)
Го6ший = Ю0,37 + 1,0354укокс - 0,033294/кокс + 3,8258 • 10-укок, (5.57)
• Коэффициенты поправки выхода в зависимости от коэффициента приведения
CFR:
Лухпп = Лухгаз + (0,49235 + 0,28789 х CFR + 0,30729 х CFR2 - 0,08715 х CFR3)', (5.58)
бензин= ^е.пин(-°’88234 + 3’1756 х CFR ~ 1,5648 х CFR2 + 0,27514 х CFR2)', (5.59)
Локс= Локс(-°’84217 + 2,9805 х CFR - 1,3648 х CFR2 + 0,22925 х CFR2); (5.60)
З^обЩИЙ 3,общНЙ + (-3,7703 + 5,6364 х CFR - 1,6785 х CFR2 + 0,13387 х CFR2). (5.61)
Выход газойля после внесения поправок:
З'газойль Х,би|Ий ^сух.гм З’бензни 3" Укокс,
(5.62)
Глава 5. Моделирование процесса коксования
201
• Поправка при высоком выходе кокса (если укокс > 25):
у, =К [1+(У — 25)(—0,9968 + 0,13484xCFR- 0,03766хСГТ?2)]; (5.63)
’'бензин z бензин1 '-'кокс ’ ZJI 4 '
= \окс[1 + (укокс- 25)(-0,01627 -0,01779 xCF/? + 1,5027- 10’3 х СГЯ2)]. (5.64)
5.7.1.5. Зависимости Смита и соавторов
Смит и соавторы [Smith et al., 2006] заметили, что предыдущие зависимости не учи-
тывают влияния давления на выход продуктов, и предложили следующие выражения:
УГ!п1С.= 7,4 + 0,10 х СС7? + -0’8^Г--5); (5.65)
к = 10,29 + 0,20 х CCR + 2’5<Р~15). (5.66)
z бензин ’ 7 20
3(Р-15)
у = 1,6 х CCR + -4-—L- (5.67)
Угазойль = 1 °0 - Угаэ(С;) “ У6енэин ~ Укокс- (5-68)
Авторы не сообщают, для какого сырья и рабочих условий выведены предложен-
ные зависимости, но утверждают, что они дают лучшие результаты, чем зависимо-
сти Гэри и Хэндверка, что видно из табл. 5.6. Так как зависимости Гэри и Хэндверка
не учитывают влияния давления, для разных давлений они дают одни и те же резуль-
таты, в то время как зависимости Смита и соавторов с высокой точностью прогнози-
руют выход кокса в процессе замедленного коксования при двух значениях давления.
Авторы применили зависимости для замедленного коксования и других различных
процессов, чтобы через имитацию производственного процесса всего нефтеперера-
батывающего завода подобрать оптимальное нефтяное сырье, переработка которого
дает максимальную прибыль и отвечает рыночному спросу.
Таблица 5.6
Сравнение результатов применения зависимостей Гэри и Хэндверка (1)
и Смита и соавторов (2) к коксованию сырья с CCR = 18,1 %масс.
Сырье Зависимости при давлении 15 psi (изб.) Зависимости при давлении 35 psi (изб.)
Фактическая 1 2 Фактическая 1 2
Газ 9,1 10,4 9,2 9,9 10,4 10,0
Бензиновая фракция 12,5 17,5 13,9 15,0 17,5 16,4
Газойль 51,2 43,1 49,7 44,9 43,1 43,4
Кокс 27,2 29,0 27,2 30,2 29,0 30,2
202
Часть 2. Моделирование некаталитических процессов
5.7.1.6. Зависимости Фолька и соавторов
Сотрудники университета г. Талса [Volk et al., 2002] провели ряд экспериментов по за-
медленному коксованию различного прямогонного остаточного сырья (с содержани-
ем микроуглеродистого остатка (micro-carbon residue, MCR) от 16 до 29 %масс.) в ми-
крореакторе. Условия реакций варьировались в следующих пределах: температура
от 900 до 950 °F (482-510 °C), давление от 6 до 40psi (изб.) (0,04-0,28 МПа) при раз-
личных величинах объемной скорости (liquidspace velocity, LSV). На основании опыт-
ных данных были выведены следующие линейные зависимости:
У = —1П39ССА + 0,04197-0,28977 + 1103,0875К+ 41,59; (5.69)
у = 0,9407СС7? — 0,06097+0,15297—319,759757+ 65,075; (5.70)
у = 0Д729СС7? + 0,01917+0,136467—786,319757— 6,762; (5.71)
У = -0,3086ССЯ + 0,01377 + 0,15717-819,63757+ 16,461; (5.72)
улгк = -0,3339СС/? - 0,026357- 0,03927 + 70,957757+ 50,452; (5.73)
утгк = -0,4714СС7 + 0,05467- 0,40767+ 1851,76757- 25,315. (5.74)
Были выведены также эмпирические формулы для прогнозирования содержа-
ния серы (в массовых процентах) в продуктах коксования различного сырья в зави-
симости от содержания серы в сырье (Sci>lpl>e, %масс.), температуры (°F) и давления
(psi (изб.)). Для одного из видов сырья зависимости имеют следующий вид:
S =—0,0417657+0,1056427- 586,479 х S + 71,4037; (5.75)
S =0,09805857-0,1413957+ 1321,503 xScbi- 38,9963; (5.76)
жидк. прод ’ ’ сырье '
S = -0,05629317+ 0,0357537 - 735,024 xS + 67,5925. (5.77)
По заявлениям авторов, их зависимости не подходят для прогнозирования выхо-
да продуктов промышленной установки замедленного коксования, так как дают для
микрореактора заниженный выход жидких продуктов по сравнению с промышлен-
ным процессом. Наихудший результат наблюдается при наинизших скоростях пода-
чи сырья. Кроме того, объемная скорость LSV, величина которой учитывается в этих
зависимостях и вычисляется исходя из объема реактора, применительно к промыш-
ленным установкам имеет другой смысл, поскольку реакции начинаются до того,
как сырье поступит в камеру коксования. Поэтому авторы предложили поправки для
определения выхода продуктов промышленных процессов. Поправочные коэффи-
циенты определялись так, чтобы давать минимальное расхождение между расчетны-
ми и фактическими данными:
уХМ -0,91укокс; (5.78)
Глава 5. Моделирование процесса коксования
203
ЛПГ =0,82Лаз;
пром = 100 — „пром _ пром.
Z жидк. прод 1VV Лкокс /газ ,
' пром А
У жидк. прод
ч У жидк. прод
' пром
Лжидк.прод ,
1 J
У жидк. прод,
пром _ пром пром _ пром
У ТГК У жидк. прод У бензин У ЛГК "
пром _ Q
Л бензин ’ -бензин
Ж*=0,90^лгк
(5.79)
(5.80)
(5.81)
(5.82)
(5.83)
5.7.2. Применение зависимостей
Описанные выше зависимости (за исключением зависимостей Шаброна и Спайта,
которые требуют знания свойств асфальтенов) были использованы при прогнозиро-
вании выхода продуктов коксования различных вакуумных остатков. Затем расчет-
ные значения сравнивались с фактическими, полученными с промышленных уста-
новок коксования. Сравнения проводились для оценки влияния свойств сырья,
давления и температуры.
5.7.2.1. Влияние свойств сырья
В табл. 5.7 представлены данные, полученные при промышленном коксовании
сырья трех видов (вакуумных остатков различных нефтей с нижней границей кипе-
ния 538 °C) с CCR от 22 до 31 %масс. и плотностью от 2,3 до 7,9 °АР1. Наблюдается ти-
пичное поведение: т. е. повышение CCR сырья вызывает увеличение выхода кокса,
газа и бензиновой фракции и уменьшение выхода газойля.
Таблица 5.7
Свойства сырья и выход продуктов замедленного коксования при
давлении 30 psi (абс.) (0,2 МПа) и температуре 900 °F (482 °C)
Показатель Вид сырья
А В С
Свойства сырья
CCR, %масс. 31 29 22,4
Плотность, °АР1 2,3 3,7 7,9
Выход, %масс.
Газ 9,05 9,00 8,59
Бензиновая фракция 11,21 10,81 10,70
Газойль 39,94 44,68 50,71
Кокс 39,80 35,51 30,00
204
Часть 2. Моделирование некаталитических процессов
На рис. 5.5 выход продуктов коксования, предсказанный зависимостями, срав-
нивается с фактическим. По точности зависимости можно подразделить на две
группы.
1. Менее точные зависимости Гэри и Хэндверка, Мейплза и Смита с соавторами,
дающие общее среднее абсолютное расхождение, превышающее 10 %. Меньшая точ-
ность отчасти объясняется применением зависимостей к сырью со свойствами вне
области их определения.
2. Зависимости Фолька с соавторами и Кастильони, дающие общее среднее аб-
солютное расхождение менее 8 %. В целом все зависимости склонны завышать
выход газа, бензиновой фракции и кокса и занижать выход газойля. Зависимо-
сти Кастильони для всех видов сырья дают наименьшие значения выхода газойля;
для сырья с низкой коксуемостью по Конрадсону расчетный выход кокса меньше
фактического.
Рис. 5.5. Влияние свойств сырья на выход продуктов коксования и сравнение
фактических значений с расчетными: (♦) — Гэри и Хэндверк; () — Фольк и соавторы;
(А) — Мейплз; (х) — Смит и соавторы; (•) — Кастильони
Что удивительно, наиболее широко применяемые и известные зависимости
Гэри и Хэндверка, Мейплза и Смита с соавторами показывают ошибки выхода газа
от 21 до 32 %, бензиновой фракции — от 31 до 62 %, газойля — от 35 до 46 % и кокса —
от 17 до 21 %. Следует иметь в виду, что методы как Гэри и Хэндверка, так и Мейплза
используют только величину CCR, в то время как метод Смита с соавторами учитыва-
ет влияние давления, что несколько улучшает точность, но ненамного.
Зависимости второй группы (Фолька с соавторами и Кастильони) учитывают,
кроме коксового остатка и давления, влияние температуры и скорости подачи сырья.
Глава 5. Моделирование процесса коксования
205
Учет дополнительных факторов значительно повышает точность прогноза. По сооб-
щениям [Depew et al., 1988], зависимости Кастильони достаточно хорошо предсказы-
вают выход продуктов коксования.
Влияние свойств сырья проверялось лишь на трех его видах. Более того, все
эти виды были получены перегонкой схожих сортов нефти (смеси нефтей «Майя»
и «Истмас»), Отсюда следует, что проверка на более обширной выборке сырья
позволит уточнить порядок расположения зависимостей по точности прогноза.
Несмотря на это, можно утверждать, что зависимости, которые учитывают вли-
яние давления и температуры, дают более точные значения выхода продуктов
коксования.
5.7.2.2. Влияние давления
Влияние давления можно проверить только по зависимостям Фолька с соавторами,
Кастильони, а также Смита с соавторами, поскольку зависимости как Гэри и Хэнд-
верка, так и Мейплза его не учитывают.
В табл. 5.8 приведены величины выхода продуктов промышленной установки за-
медленного коксования сырья с CCR = 15,6 %масс. и плотностью 10,3 ° API. Данные
получены для одной и той же температуры (930 °F, или 499 °C) и двух значений давле-
ния (15 и 40psi (абс.), или 0,1 и 0,28 МПа). Как и следует ожидать, с увеличением дав-
ления выход кокса повышается.
Таблица 5.8
Выход продуктов замедленного коксования сырья с CCR = 15,6 %масс.
и плотностью 10,3 °ЛР/при температуре 930 °F (499 °C)
и двух значениях давления
Продукт Выход продуктов при давлении, psi (абс.)
15 40
Газ 8,20 12,77
Бензиновая фракция 17,20 21,20
Газойль 56,83 43,33
Кокс 17,77 22,70
На рис. 5.6 предсказанные значения выхода продуктов сравниваются с фактиче-
скими. В данном случае худшие результаты дают зависимости Кастильони, а подходы
Фолька с соавторами и Смита с соавторами прогнозируют выход продуктов с вы-
сокой точностью. Зависимости Кастильони выведены для более низких давлений
и температур, чем те, для которых были получены реальные промышленные данные,
поэтому расчет по ним требует интерполяции, что вносит дополнительную погреш-
ность. Кроме того, данные для сравнения были взяты из работы Фолька и соавторов
206
Часть 2. Моделирование некаталитических процессов
[Volk et al., 2002], и причина большей точности их зависимостей может крыться имен-
но в этом.
Рис. 5.6. Влияние давления на выход продуктов коксования и сравнение фактических зна-
чений с расчетными: () — Фольк и соавторы; (х) — Смит и соавторы; (•) — Кастильони
5.7.2.3. Влияние температуры
Влияние температуры можно проверить только по зависимостям Фолька с соавтора-
ми и Кастильони.
В табл. 5.9 приведены данные о выходе продуктов промышленной установки за-
медленного коксования сырья с CCR = 19,0 % масс. и плотностью 5,7 ° API. Влияние
температуры исследовалось при двух ее значениях — 900 °F (482 °C) и 930 °F (499 °C) —
и постоянном давлении 30 psi (абс.), или 0,2 МПа. Можно видеть, что повышение ра-
бочей температуры снижает выход кокса.
Таблица 5.9
Выход продуктов замедленного коксования сырья
с CCR = 19 %масс. и плотностью 5,7 °АР1
при давлении 30 psi (абс.) (0,2 МПа) и двух значениях температуры
Продукт Выход продуктов при температуре, °F (°C)
900 (482) 930 (499)
Газ 5,00 6,50
Бензиновая фракция 14,00 15,00
Газойль 59,00 58,50
Кокс 22,00 20,00
Глава 5. Моделирование процесса коксования
207
На рис. 5.7 приводится сравнение расчетных значений выхода с реальными. За-
висимости Фолька и соавторов опять дают лучшие результаты, чем зависимости Ка-
стильони. Как и в случае изучения влияния давления, данные были взяты из работы
Фолька и соавторов [Volk et al., 2002]. Хотя давление и попадало в интервал приме-
нимости зависимостей Кастильони, температура была существенно выше интервала
800-840 °F (427-449 °C).
Рис. 5.7. Влияние температуры на выход продуктов коксования и сравнение фактических
значений с расчетными: () — Фольк и соавторы; (•) — Кастильони
5.7.3. Заключительные положения
В последние годы самой предпочтительной технологией облагораживания остаточ-
ного сырья стало замедленное коксование. Надежные средства моделирования про-
цесса — необходимое условие правильного прогнозирования выхода продуктов.
Предпринималось множество попыток вывести фундаментальные модели, в част-
ности кинетические, которые отображают сложные реакции, происходящие в про-
цессе коксования. Однако главной проблемой остается способность модели адекват-
но описать крупные молекулы и пути реакций, в которых они принимают участие.
По этой причине до сих пор широко применяются эмпирические подходы, т. е. зави-
симости, дающие довольно точные результаты.
Определяющий параметр процесса коксования, влияющий на выход продук-
тов, — качество сырья. Именно поэтому главным параметром любых зависимостей
является CCR. Другие рабочие параметры (температура, давление и т. д.) также очень
важны, поскольку позволяют улучшить точность рассчитываемого выхода продук-
тов. К примеру, Предель [Predel, 1992], сравнив несколько методик по расчету выхо-
да продуктов коксования, подтвердил, что включение в зависимости рабочих усло-
вий в качестве переменных улучшает значимость прогнозирования.
208
Часть 2, Моделирование некаталитических процессов
Рабочие условия промышленных установок коксования, данные с которых ис-
пользовались для вывода первых зависимостей, отличаются от современных, в осо-
бенности давления и температуры. Это основная причина их неприменимости к дру-
гим условиям реакций.
Для более надежного сравнения эмпирических зависимостей необходимы более
обширные промышленные данные о выходе продуктов коксования различного сы-
рья в различных условиях. Объем данных должен быть достаточен для того, чтобы
по ним можно было однозначно установить применимость зависимостей для про-
гнозирования выхода продуктов.
ОБОЗНАЧЕНИЯ
А Реагент. Параметр зависимостей Кастильони [Castiglioni, 1983]
а!’а2 а В Ь Ь2 С. d 1 Параметры зависимостей Кастильони Стехиометрический коэффициент Параметры зависимостей Кастильони Параметры зависимостей Кастильони Параметры зависимостей Кастильони Параметры зависимостей Кастильони
ei CCR CF CFR F\ Параметры зависимостей Кастильони Коксовый остаток (коксуемость) по Конрадсону, %масс. Коэффициент корреляции Коэффициент рециркуляции сырья Энергия активации, ккал/моль Коэффициент поправки значений выхода продуктов замедленного коксования
Fl Коэффициент обновления значений выхода продуктов замедленного коксования
Предэкспоненциальный множитель, или фактор частоты соударений молекул
LSV P R T V Vo VPO Объемная скорость подачи сырья Давление Универсальная газовая постоянная Температура Доля испаренного материала Общее количество испаренного материала Осмометрия давления паров
X Степень превращения асфальтенов
Глава 5. Моделирование процесса коксования
209
у Выход, % масс.
^пр°м Выход на промышленной установке, %масс.
ДБК Легкая бензиновая фракция коксования
ЛГК Легкий газойль коксования
ТБК Тяжелая бензиновая фракция коксования
ТГА Термогравиметрический анализ
ТГК Тяжелый газойль коксования
ЛИТЕРАТУРА
Aguilar, R.A., Ancheyta, J., Trejo, E 2012. Simulation and planning of a petroleum refinery based on
carbon rejection processes. Fuel 100:80-90.
Albers, J.E. 1996. Modeling of a delayed coker. MSc thesis. Texas Tech University, Lubbock, TX.
All, M.F., Saleem, M. 1991. Thermal decomposition of Saudi crude oil asphaltenes. Fuel Sci. Technol. Int.
9:461-484.
Alvarez, E., Marroquin, G., Trejo, E, Centeno, G., Ancheyta, J., Diaz, J.A.L 2011. Pyrolysis kinetics of
atmospheric residue and its SARA fractions. Fuel 90:3602—3607.
Castiglioni, B.P 1983. How to predict coker yield. Hydrocarbon Process. 62:77—79.
Ciajolo, A., Barbella, R. 1984. Pyrolysis and oxidation of heavy fuel oils and their fractions in a
thermogravimetric apparatus. Fuel 63:657-661.
Collett, G.W, Rand, B. 1980. Thermogravimetric investigation of the pyrolysis of pitch materials:
A compensation effect and variation in kinetic parameters with heating rate. Thermochim.Acta 41:153—165.
Del Bianco, A., Panariti, N., Beltrame, PL., Carniti, P 1993. Thermal cracking of petroleum residues 1.
Kinetic analysis of the reaction. Fuel 72:75-80.
Depew, C.A., Hashemi, M.H., Davis, J. 1988. Evaluation of alternative control strategies for delayed
coker. Pnoc. Am. Control Conf. 88( l):240—246.
Dias, J.R. 1988. Handbook of Pblycyclic Hydrocarbons. Part B. Fblycyclic Isomers and Heleroatoms
Analogs of Benzenoid Hydrocarbons, Elsevier, NewYjrk.
Douda, J., Alvarez, R., Navarrete, J. 2008. Characterization of maya asphaltene and maltene by means
of pyrolysis application. Energy Fuels 22:2619—2628.
Douda, J., Llanos, M.E., Alvarez, R., Lopez, C., Montoya, J.A. 2004. Pyrolysis applied to the study of a
maya asphaltene. J. Anal. Appl. Pyrolysis 71:601—612.
Dymond, R.E. 1991. Wbrld marketsforpetroleum coke. Hydrocarbon Process. 9:162-C-162-J.
Elliot, J.D. 2003. Delayed coking: Recycle to zero recycle concepts & considerations. NPRA Annual
Meeting, San Antonio, TX, March 23—25, Paper AM-03-30.
Ellis, PJ., Paul, C.A. 1998. Tutorial: Delayed coking fundamentals. AIChE 1998 Spring National
Meeting, New Orleans, LA, March 8—12.
Furimsky, E. 2000. Characterization of cokes from fluid/flexi-coking of heavy feeds. Fuel Process.
Technol. 67:205—230.
Gary, J.H., Handwerk, G.E. 2001. Petroleum Refining—Technology and Economics, 2nd edn., Marcel-
Dekker, New York.
Goncalves, M.L.A., Teixeira, M.A.G., Pereira, R.C.L., Mercury, R.L.P, Matos, J.R. 2001.
Contribution of thermal analysis for characterization of asphaltenes from Brazilian crude oil. J. Therm.
Anal. Calorim. 64:697—706.
Hamilton, G.L. 2002. Delayed coker design considerations and project execution. NPRA Annual Meeting,
San Antonio, TX, March 17-19, Paper AM-02-06.
210
Часть 2. Моделирование некаталитических процессов
Haniford, R.R. 2003. Coke is good, but less is better. NPRA Annual Meeting, San Antonio, TX, March
23-25, Paper AM-03-90.
Hauser, A., Bahzad, D., Stanislaus, A., Behbahani, M. 2008. Thermogravimetric analysis studies on the
thermal stability of asphaltenes: Pyrolysis behavior of heavy oil asphaltenes. Eneigy Fuels 22:449—454.
Hue, A.-Y 2011. Heavy Crude Oils: From Geology to Upgrading: An Overview, Technip, Paris, France.
Hughes, G., Doerksen, B., Englehom, A., Romero, S. 2000. Tutorial: Delayed coking commercial
technology In: AIChE Spring National Meeting, Atlanta, GA, March.
Jacob, R.R. 1971. Coke quality and how to make it. Hydrocarbon Process. 9:132—136. Karacan, O.,
Kok, M.V 1997. Pyrolysis analysis ofcrude oilsand their fractions. Eneigy Fuels 11:385—391.
Khulbe, K.C., Sachdev, A.K., Mann, R.S., Davis, S. 1984. TGA studies of asphaltenes derived from cold
lake (Canada) bitumen. Fuel Process. Technol. 8:259—266.
Kok, M.V 1993. Use of thermal equipment to evaluate crude oils. Themochim. Acta 214:315-324.
Kruse, C., Ragsdale, R., Roth, J., Hughes, G. 1995. Low pressure coker operation with distillate recycle.
NPRA Annual Meeting, San Francisco, CA, March.
Lieberman, N.R, 1986. Troubleshooting Refinery Processes, PennMfell Publishing Company Tulsa, OK.
Magaril, R.Z., Aksenova, E.I. 1968. Study of the mechanism of coke formation in the cracking of
petroleum. Int. Chem. Eng. 8:727-729.
Maples, R.E. 1993. Petroleum Refinery Process Economics, PennWfcll Publishing Company, Tulsa, OK.
McKetta. 1992. Petroleum Processing Handbook, Marcel Dekker, NewYtrk.
Meng, M., Hu, H., Zhang, Q., Li, X., Wu, B. 2007. Pyrolysis behaviors of Tumuji oil sand by
thermogravimetry (TG) and in a fixed bed reactor. Energy Fuels 21:2245—2249.
Moschopedis, S.E., Parkash, S., Speight, J.G. 1978. Thermal decomposition of asphaltenes. Fuel
57:431-434.
Park, YC., Paek, J.Y, Bae, D.H., Shun, D. 2009. Study of pyrolysis kinetics of Alberta oil sand by
thermogravimetric analysis. Korean J. Chem. Eng. 26:1608—1612.
Phillips, C.R., Haidar, YC., Poo, C. 1985. Kinetic models for the thermal cracking ofAthabasca bitumen.
Fuel 64:678-691.
Predel, H. 1992. Theoretical and practical methods for coke yield calculations and optimization. Light
Metals 92:601-607.
Rana, M.S., Samano, V, Ancheyta, J„ Diaz, J.A.I. 2007. A review of recent advances on process
technologies for upgrading of heavy oils and residua. Fuel 86(9): 1216—1231.
Ritchie, R.G.S., Roche, R.S., Steedman, W 1979. Pyrolysis of Athabasca tar sands: Analysis of the
condensable products from asphaltene. Fuel 58:523—530.
Savage, PE., Klein, M.T, Kukes S.G. 1988. Asphaltene reaction pathways. Ill: Effect of reaction
environment. Energy Fuels 2:619-628.
Sawarkar, A.N., Pandit, A.B., Samant, S.D., Joshi, J.B. 2007. Petroleum residue upgrading via delayed
coking: A review Can. J. Chem. Eng. 85:1-24.
Schabron, J.E, Speight, J.G. 1997. An evaluation of the delayed-coking product yield of heavy feed-
stocks using asphaltene content and carbon residue. Rev. I’Institut Fran ais du Petrole 52(1):73—85.
Schucker, R.C. 1983. Thermogravimetric determination of the coking kinetics of Arab heavy vacuum
residuum. Ind. Eng. Chem. Proc. Des. Dev. 22:615-619.
Schucker, R.C., Keweshan, C.E 1980. Reactivity of cold lake asphaltenes. Prepr. Pap. Am. Chem. Soc.
Div. Fuel Chem. 25:155—165.
Shih, S.M., Sohn, H.Y 1980. Nonisothermal determination of the intrinsic kinetics of oil generation from
oil shale. Ind. Eng. Chem. Proc. Des. Dev. 19:420—426.
Shiroto, Y, Nakata, S., Fukui, Y, Takeuchi, C. 1983. Asphaltene cracking in catalytic hydrotreating
of heavy oils. 3. Characterization of products from catalytic hydroprocessing of Khafji vacuum residue. Ind.
Eng. Chem. Proc. Des. Dev. 22:248-257.
Smith, A., Frow, M., Quddus, J., Howell, D., Reed, T, Landrum, C., Clifton, B. 2006. Delayed coking
model. In Refinery Modeling. http.7/wwwou.edu/class/che-design/a-design/projects-2006/Refinery%20
Planning%20Presentation.pdf [ Retrieved 2012-01-15].
Глава 5. Моделирование процесса коксования
211
Sonibare, О.О., Egashira, R., Adedosu, ТА. 2003. Thermo-oxidative reactions of Nigerian oil sand
bitumen. Thermochim. Acta 405:195-205.
Speight, J.G. 1980. The Chemistry and Technology of Petroleum, Marcel Dekker Inc., New York.
Speight, J.G. 1981. The Desulfurization ofHeavy Oils and Residua,
Speight, J.G. 2004. Petroleum asphaltenes. Part 2. The effect of asphaltenes and resin constituents on
recovery and refining processes. Oil Gas Sci. Technol. 59:479—488.
Trejo, E, Rana, M.S., Ancheyta, J. 2010. Thermogravimetric determination of coke from asphaltenes,
resinsand sediments and coking kinetics of heavy crude asphaltenes. Catal. Today 150:272—278.
Volk, M., Wisecarver, K.D., Sheppard, C.M. 2002. Fundamentals of delayed coking. University of Tulsa,
Tulsa, OK.
Yisar, M., Trauth, D.M., Klein, M.T 2001. Asphaltene and resid pyrolysis. 2. The effect of reaction
environment on pathways and selectivities. Energy Fuels 15:504-509.
Y>shida, R., Takeda, S., Teramoto, S., Matsushita, T, Takeya, G. 1984. Thermal behavior of coal-derived
asphaltenes. Fuel Process. Technol. 9:307—313.
Zhao, Y, Gray, M.R., Chung, K.H. 2001. Molar kinetics and selectivity in cracking of athabasca
asphaltenes. Energy Fuels 15:751—755.
Zhou, X.-L., Chen, S.-Z., Li, C.-L. 2007. A predictive kinetic model for delayed coking. Petrol. Sci.
Есйио/. 25(12):1539-1548.
Глава 6. Некаталитическая
(термическая) гидропереработка
В этой главе рассматриваются некаталитические процессы гидрообессеривания
(НГДО), гидродеметаллизации (НГДМ) и гидрокрекинга (НГДК) тяжелой нефти
и атмосферного остатка. Были проведены опыты на двух разных лабораторных уста-
новках в реакторах с неподвижным слоем, соединенных последовательно и работаю-
щих в адиабатическом и изотермическом режимах. Реакторы загружалисыинертным
материалом (карбидом кремния). Опыты проводились с сырьем различных видов:
с двумя сортами тяжелой нефти плотностью соответственно 13 и 21 ° API и с остат-
ками атмосферной перегонки нефтей этих сортов. Проверялось влияние давления,
времени пребывания в реакторе, температуры и типа сырья на некаталитические ре-
акции и распределение температур вдоль оси реакторов. С применением степенного
закона изучалась кинетика различных некаталитических реакций.
6.1. ВВЕДЕНИЕ
При каталитической гидропереработке тяжелых нефтей одновременно с катализи-
руемыми реакциями неизбежно протекают термические. В зависимости от условий
эксперимента, типа сырья и используемого оборудования, глубина (степень завер-
шенности) термических реакций может быть такой, что влияние катализаторов либо
скрыто, либо неверно интерпретируется. Важную роль в некаталитической гидро-
очистке играют состав сырья и его свойства. Парафиновые соединения легко под-
даются каталитическому или термическому крекингу, в то время как ароматические
соединения превратить труднее: наиболее тяжелая их часть стремится к уплотнению
с образованием кокса [Ancheyta and Speight, 2007].
Все фракции нефти в определенных условиях претерпевают гидрокрекинг. На-
пример, в процессе каталитического риформинга бензиновая фракция крекирует-
ся с образованием легких углеводородных газов, что ведет к потере выхода жидких
продуктов [Prestvik et al., 2004]. Но реакция гидрокрекинга не всегда является неже-
лательной. Например, реакции гидрокрекинга таких тяжелых фракций, как вакуум-
ный газойль и остаток, дают в результате более легкие и ценные продукты — бензин
и средние дистилляты [Sanford, 1994; Speight, 2000]. Таким гидрокрекингом управляют
Глава 6. Некаталитическая (термическая) гидропереработка
213
посредством соответствующих катализаторов и условий реакций. Степень превраще-
ния тяжелого сырья зависит от рабочих условий (температуры, давления и времени
контакта) и, разумеется, свойств сырья и типа катализатора [Gray, 1994]. Большая
часть реакций гидрокрекинга протекает по каталитическому механизму. Разработано
множество промышленных катализаторов с активными центрами и промоторами из-
бирательного действия, способствующими расщеплению крупных углеводородных
молекул и присоединению водорода [Topsoe et al., 1996]. Хорошо известно, каким об-
разом типы и концентрации кислотных центров на поверхности частиц катализатора
влияют на реакции гидрокрекинга [Wade et al., 2009].
Тяжелые нефти и остатки содержат компоненты, характеризующиеся большим
количеством гетероатомов и асфальтеновых структур. Свойства и химическое строе-
ние этих сложных компонентов — еще один фактор, во многом определяющий глу-
бину термических реакций. К примеру, наиболее вероятно превращение асфальтенов
с малым числом ароматических колец и большим числом боковых алкильных цепей
[Ancheyta et al., 2009].
Помимо каталитических, могут протекать и термические (т. е. некаталитиче-
ские) реакции гидрокрекинга, что обусловлено особенностями отдельного процес-
са [Kim et al., 1998; Zhang et al., 2001]. Глубина реакций термического гидрокрекинга
сильно зависит от рабочих условий [Yang et al., 2011]. Поскольку это некаталитиче-
ские реакции, они требуют большей энергии для активации, чем реакции, промоти-
руемые катализаторами, и поэтому им благоприятствуют более высокие температуры,
близкие к температурам в конце цикла каталитического процесса, т. е. в конце сро-
ка службы катализатора. Кажущаяся энергия активации термической реакции выше,
чем каталитической, и это может быть лимитирующим фактором [Yang et al., 2011].
Кроме того, термические реакции могут протекать как в жидкой, так и в газовой
фазе, поскольку не зависят от активных центров какого-либо катализатора. Степень
превращения определяется температурой и разветвленностью углеводородных моле-
кул. Реакция термического гидрокрекинга — цепная и протекает по свободноради-
кальному механизму. Некоторые исследователи установили, что начало реакций ги-
дрокрекинга обуславливает температура [Martinez et al., 1997].
При термической гидроочистке тяжелые молекулы расщепляются на более мел-
кие, что способствует удалению серы и других гетероатомов [Kin et al., 1998]. Если
есть катализатор, термические реакции протекают в пространстве между его части-
цами в жидкой фазе, а также в газовой, и нечувствительны к диффузионным ограни-
чениям [Rahimi and Gentzis, 2003].
Изучением термических реакций в процессах гидропереработки занимались мно-
гие исследователи. Марафи и соавторы [Marafi et al., 2008] провели ряд эксперимен-
тов при различных значениях объемных скоростей подачи сырья и температур. Они
пришли к выводу, что для точного анализа данных гидропереработки необходимо
учитывать термические реакции. Юрайден и соавторы [Juraidan et al., 2006] показали,
что в интервале температур в реакторе от 330 до 410 °C глубина (степень) НГДО мо-
жет изменяться от 0,35 до 21 %. Это означает, что данная термическая реакция край-
не важна и должна приниматься в расчет при изучении каталитической гидроочист-
ки тяжелых нефтей.
214
Часть 2. Моделирование некаталитических процессов
Вследствие большого разнообразия соединений, присутствующих в сырье, гидро-
крекинг тяжелых фракций сопровождается сложными химическими реакция-
ми. Из-за этого чрезвычайно трудно исследовать кинетику и механизм превраще-
ний каждого компонента системы. Некоторые исследователи прибегают к ядерной
магнитно-резонансной (ЯМР) спектроскопии для физико-химической характери-
зации углеводородных групп, к элементному анализу для определения усреднен-
ных значений молекулярных параметров и к широкому спектру методов химиче-
ского разложения для выявления класса соединений — например, к анализу 5ИЛ4
[Sanford, 1994]. Такую характеризацию осуществляют до и после проведения реак-
ций, чтобы можно было изучить различные пути реакций и выявить их механиз-
мы. Так называемые композиционные модели описывают изменения в составе ре-
агентов в зависимости от их расположения в реакторе и от времени [Jia et al., 2009].
Это дает возможность определить (несмотря на участие в процессе тяжелых углево-
дородов) кинетику реакций отщепления отдельных элементов, а также гидрокре-
кинга избранных углеводородных групп, если известны их начальные и конечные
концентрации [Melisa et al., 2004].
Большая часть исследований кинетики гидрокрекинга нефтяного сырья прово-
дится при наличии катализатора и в жестких условиях реакций, а сырьем являет-
ся вакуумный газойль. Некаталитическому гидрокрекингу, в особенности тяжелых
остатков, уделяется мало внимания. Общепризнано, что температура оказывает наи-
большее влияние на глубину гидрокрекинга, но на нее влияют и другие параметры
процесса: например, давление и кратность водорода к сырью. Таким образом, сни-
жение жесткости условий существенно изменяет кинетику реакций [Singh et al., 2005;
Marafi et al., 2008]. Иначе говоря, в жестких и мягких условиях кинетика реакций бу-
дет разной.
6.2. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
6.2.1. Сырье
В качестве сырья для экспериментов выступили два сорта тяжелой нефти плотно-
стью соответственно 13 и 21 “АР/и их атмосферные остатки. Остаток получали пе-
регонкой нефти на одноступенчатой установке по методу ASTM D\ 160. Перегонная
колонна с одной теоретической тарелкой загружалась нефтью в стеклянной колбе.
Колонна работала в ручном режиме, и разделение продукта осуществлялось без рек-
тификации, чтобы имитировать однократную перегонку. Первую ступень разделения
начинали при атмосферном давлении. Температура жидкости в круглодонной пере-
гонной колбе достигала примерно 275 °C. Температуру медленно увеличивали, чтобы
избежать термического крекинга образца. На второй ступени колонна работала под
давлением 100 мм рт. ст., а заключительная разгонка образца проводилась под давле-
нием 2 мм рт. ст. Объемный выход остатка нефтей с плотностями 13 и 21 ° API соста-
вил соответственно 70,5 и 56,1 %. Основные свойства образцов нефти и соответству-
ющих атмосферных остатков представлены в табл. 6.1.
Глава 6. Некаталитическая (термическая) гидропереработка
215
Таблица 6.1
Свойства сырья
Свойства Нефть, 21 "API Нефть, 13 ‘API АО нефти, 21 ‘API АО нефти, 13 ‘API
Относительная плотность, 60/60 °F 0,9260 0,9812 1,0206 1,0505
Плотность, ‘API 21,31 12,71 7,14 3,20
Содержание серы, %масс. 3,57 5,22 4,6 6,21
Коксуемость по Рамсботтому, %масс. 10,87 16,07 17,66 21,48
Компоненты, нерастворимые в н-Сг %масс. 11,32 18,78 17,74 25,10
Содержание Ni, мг/кг 53,4 84,0 86,6 117,4
Содержание V, мг/кг 298,1 423,0 489,0 578,2
Разгонка ИТК, °C:
НК/5 % 23/84 51/136 340/383 374/410
перегоняется 10/15 % 128/170 199/252 410/435 446/475
» 20/25 % 206/242 299/340 454/475 504/529
» 30/35 % 280/317 378/421 497/524 551/-
» 40/45 % 352/386 468/517
» 50/55 % 428/469
» 60% 512
Отгон до 538 °C, %об. 62,9 46,99 36,0 27,3
Характеризующий фактор К 11,30 11,32 15,59 16,61
Из таблицы видно, что более тяжелое сырье содержит больше примесей (серы,
коксового остатка по Рамсботтому и металлов) и дает меньший выход дистил-
лятов при 538 °C. Величины характеризующего фактора К образцов нефти близ-
ки (около 11,3); то же самое верно и для остатков (около 16). Это объясняется
зависимостью К от интервала температур кипения и относительной плотности
(К= Me АВР13 / sg, где МеАВР — среднечисленная температура кипения, англ, mean
average boiling point', sg — относительная плотность). Высокий фактор Ку остатков оз-
начает, что они содержат больше парафинов, чем образцы нефти. Поэтому можно
ожидать, что глубина реакции НГДО остатков будет больше.
6.2.2. Оборудование
Эксперименты проводились на двух лабораторных установках высокого давления,
оборудованных реакторами с неподвижным слоем. На установке с одним реактором
(рис. 6.1) определяли кинетику реакций и изучали влияние температуры и соотноше-
ния FmT/ KSiC (отношения суммарного массового потока на входе реактора к общему
количеству инертного материала — карбида кремния SiC). Реактор загружали 200 мл
216
Часть 2. Моделирование некаталитических процессов
карбида кремния и запускали в изотермическом режиме. Температуру поддержива-
ли на требуемом уровне с помощью электронагревателей. Газ анализировали поточ-
ным газовым хроматографом. Жидкий продукт собирали в коллектор для дальней-
шего анализа.
Рис. 6.1. Упрощенная схема однореакторной лабораторной установки
На установке с двумя реакторами с неподвижным слоем, работающими в адиаба-
тическом режиме, изучали влияние температуры, давления и свойств сырья на НГДО
(рис. 6.2). Водород в реакторы подавался компрессором высокого давления через
массовые расходомеры. Сырье вводилось насосом с высоким давлением нагнетания.
Поток сырья контролировался по массе и измерялся электронными весами. Реак-
торная секция состояла из двух реакторов из нержавеющей стали объемом по 500 мл.
Реакция могла проводиться в неподвижном слое в адиабатическом или изотермиче-
ском режиме. В данном эксперименте реакторы работали в адиабатическом режиме
при нисходящем потоке сырья. В реакторы было загружено по 500 мл инертного ма-
териала (карбида кремния). Карбид кремния имеет плотность 3,2 г/см’ и химически
инертен. Он обладает высокой теплопроводностью и прочностью и низким коэффи-
циентом теплового расширения (4,0-10-6/К) и не испытывает фазовых переходов,
способных вызвать нарушения сплошности при тепловом расширении.
Адиабатический режим работы достигался отключением электронагревателей
в стенках реакторов, затем контролировалась лишь температура на входе в слой.
Во избежание потерь тепла в окружающую среду нагреватели реакторов снабжались
наружной теплоизоляцией.
Установка была оборудована тремя сепараторами: один работал при высокой тем-
пературе и под высоким давлением, другой — под высоким давлением, третий — под
низким давлением, при котором получались легкая фракция и гидроочищенные про-
дукты. Во второй сепаратор (высокого давления) вводилась вода, чтобы нейтрализо-
вать сероводород и аммиак, образующиеся в ходе реакций. Во все реакторы подавал-
ся водород высокой чистоты (99,9 %мольн.).
Глава 6. Некаталитическая (термическая) гидропереработка
217
Рис. 6.2. Упрощенная схема двухреакторной лабораторной установки
6.2.3. Условия реакций
Эксперименты в однореакторной установке проводились при давлении
9,81 МПа и кратности водорода к сырью 890 м3/м3 (5000 фут3/баррель), темпе-
ратура и соотношение FmTl Fs.c изменялись соответственно в интервалах от 380
до 420 °C и от 0,23 до 0,65 г/см3 • ч. Сырьем являлся атмосферный остаток нефти
плотностью 13 ° API.
Влияние типа сырья в двухреакторной установке изучалось в следующих условиях:
давление 6,86 МПа и 9,81 МПа, кратность водорода к сырью 890 м3/м3, соотношение
FtnT/ KSiC = 0,54 г/см3 • ч. Температуру изменяли в интервале от 380 до 420 °C. Резерву-
ар с сырьем, сепараторы и трубы приходилось подогревать до 140 °C, чтобы избежать
закупоривания.
6.2.4. Методы анализа
Газообразный продукт анализировали поточным газовым хроматографом модели
Agilent 6890 с катарометром (детектором теплопроводности).
Относительная плотность (при 60/60 °F) образцов нефти и остатков определя-
ли пикнометром по методу ASTM £>70. Содержание серы измеряли спектрометром
модели 5££И-2100 HORIBA методом энергодисперсионной рентгенофлуоресцент-
ной спектрометрии (ASTM £>4294). Склонность к коксообразованию (коксуемость)
определяли методом Рамсботтома (ASTM £>524), измеряя количество углеродистого
218
Часть 2. Моделирование некаталитических процессов
остатка после выпаривания и пиролиза пробы. Содержание асфальтенов определя-
ли по ASTM D3279 как количество компонентов, нерастворимых в н-гептане. Содер-
жание никеля и ванадия измеряли атомно-абсорбционным спектрометром модели
Solar. Кривую разгонки стабилизированного сырья (разгонка для определения ИТК)
получали по ASTM £>2892.
6.3. РЕЗУЛЬТАТЫ
При анализе влияния условий реакций на удаление примесей и изменение свойств
продуктов первыми в этом разделе рассматриваются результаты, полученные надвух-
реакторной установке. Затем на основании результатов работы однореакторной уста-
новки выводятся кинетические параметры различных некаталитических реакций.
6.3.1. Двухреакторная установка
6.3.1.1. Некаталитическое гидрообессеривание
Глубина реакции НГДО (степень удаления серы) в двухреакторной установке оцени-
валась по следующей формуле:
нгдо,=
I sf )
100,
(6.1)
где Sf и S' — содержание серы, %масс., соответственно в сырье и в продук-
те; I (г = 1,2) — номер реактора; НГДО1 — степень удаления серы в 1-м реакторе,
НГДО2 — суммарная степень удаления серы на выходе из 2-го реактора (формула
не учитывает возможного изменения массового потока жидкости, которое предпо-
лагается минимальным).
6.3.1.1.1. Влияние температуры и давления
На рис. 6.3 показана степень НГДО сырья четырех видов на выходе из второго реак-
тора при рабочих давлениях 9,81 и 6,86 МПа. Можно видеть, что чем выше темпера-
тура, тем больше степень удаления серы. Виды сырья по степени удаления серы рас-
полагаются следующим образом:
Нефть (13 °АРГ) < нефть (21 ‘API) < АО нефти (21 °АР1) < АО нефти (13 °АРГ).
НГДО2 тяжелого сырья больше, чем НГДО легкого. Остатки содержат соединения
серы тех же типов, что и нефти, из которых они получены. Кроме того, нефти и их
остатки содержат одно и то же количество асфальтенов и металлов: после отделения
легкой фракции те и другие концентрируются в остатке. Таким образом, нефть и ее
остаток различаются лишь тем, что первая содержит легкий дистиллят, отделяемый
Глава 6. Некаталитическая (термическая) гидропереработка
219
при перегонке. Степень удаления серы зависит от наличия легких фракций, а также
их состава. Термическому крекингу более подвержены тяжелые парафиновые углево-
дороды, чем легкие. Поскольку сера высвобождается при расщеплении молекул тя-
желых углеводородов, степень ее удаления из остатка больше, чем из соответствую-
щей нефти [Kawai and Kumata, 1998; Yang et al., 1998].
Температура, °C
Рис. 6.3. Суммарная глубина термического гидрообессеривания (НГДО2) тяжелых
нефтей и их атмосферных остатков в зависимости от температуры и давления:
(♦) — нефть (13 °АРГ); (А) — АО нефти (13 ° АРГ);
(и) — нефть (21 °АРГ); (х) — АО нефти (21 °АРГ)
Будучи сравнительно небольшой при температурах ниже 390 “С, глубина НГДО
быстро возрастает при дальнейшем нагревании. Такое поведение объясняется
большими значениями энергий активации реакций термического обессеривания
[Yang et al., 2011]. Результаты согласуются с теми, что получили Юрайден и соавто-
ры [Juraidan et al., 2006] для некаталитической гидротермической реакции боскан-
ской нефти.
Термические реакции происходят по свободнорадикальному механизму
[Yang et al., 2011]. Поэтому можно ожидать, что высокое парциальное давление во-
дорода в реакторе должно благоприятствовать реакции насыщения и водород быстро
нейтрализует свободные радикалы, препятствуя увеличению степени превращения.
220
Часть 2. Моделирование некаталитических процессов
Тем не менее в пределах условий, в которых протекали изучавшиеся реакции, влия-
ние давления на глубину НГДО было незначительным; это говорит о том, что преоб-
ладающее влияние на нее оказывает температура.
6.3.1.1.2. Влияние времени пребывания
Влияние времени пребывания проверяется отбором пробы и анализом продук-
та на выходе из первого реактора. Глубина НГДО, показана на рис. 6.4. В целом на-
блюдается та же картина, что и на выходе из второго реактора. Глубина НГДО лег-
кого сырья почти одинакова; для тяжелого сырья она существенно выше. Далее,
НГДО, < НГДО,, что объясняется более коротким временем пребывания в первом
реакторе. Следует отметить, что в поток первого реактора вводился водород. Целью
было охладить поток и предотвратить чрезмерный перепад температур, а также под-
держать кратность водорода к сырью в реакторах на одном уровне. Это значит, что
кривая НГДО во втором реакторе не является продолжением кривой в первом, а ана-
логична ей, так как температуры на входе в реакторы одинаковы.
Рис. 6.4. Глубина термического гидрообессеривания (НГДО,) тяжелых нефтей и их
атмосферных остатков в 1-м реакторе в зависимости от температуры и давления:
(♦) — нефть (13 °АРГ); (А) — АО нефти (13 °АР1);
() — нефть (21 ° API); (х) — АО нефти (21 ° API)
Глава 6. Некаталитическая (термическая) гидропереработка 221
6.3.1.1.3. Влияние типа сырья
Главное влияние на глубину НГДО оказывает температура, но состав и свойства так-
же важны. Степень удаления серы из наиболее тяжелого сырья (остаток нефти плот-
ностью 13 °АРГ) наиболее высока, что обусловлено значительным содержанием па-
рафинов (наивысшая величина характеризующего фактора К, см. табл. 6.1). Кин
и соавторы [Kin et al., 1988] провели опыты по гидрообессериванию тяжелых би-
тумных нефтей при наличии натриевого катализатора на алюмооксидном носителе
и объяснили высокую степень удаления серы термическими реакциями с участием
алифатических сульфидов, присутствующих в тяжелых нефтях. Авторы сообщили,
что в интервале температур 370-410 °C степень превращения серы составляла от 3
до 15 %. Сэнфорд [Sanford, 1999] предположил, что первый этап превращения за-
ключается главным образом в разрыве слабых углерод-углеродных связей с образо-
ванием дистиллятов. При формировании промежуточных ароматических биради-
калов из гидроароматических структур происходит дополнительное превращение.
Перенос водорода к углеродному центру ароматического радикала останавливает ре-
акцию конденсации, ведущую к образованию кокса. Важную роль в процессе игра-
ет водородный радикал, образующийся в ходе реакции переноса водорода, Реакция
этого радикала с конденсированным ароматическим центром дает в конечном счете
большие количества газа и дистиллятов. Механизм, который предложили Янг и соав-
торы [Yang et al., 1998], предполагает, что первым этапом гидротермических реакций
является превращение парафиновых цепей. Поэтому сырье с большим содержанием
парафинов менее стойко в условиях реакций НГДО (высокая температура и высокое
парциальное давление водорода). Крупные, разветвленные молекулы легче поддают-
ся расщеплению. Нефти содержат больше низкомолекулярных соединений, которые
более стабильны, поэтому глубина НГДО нефтей меньше.
В отличие от каталитического гидрообессеривания, глубина которого увеличива-
ется с ростом давления, удаление серы в процессе НГДО происходит по свободно-
радикальному механизму, и наличие водорода может нейтрализовать свободные ра-
дикалы, участвующие в удалении серы. Более тяжелое и химически активное сырье
(дающее больше свободных радикалов) чувствительнее к повышению давления, так
как способствует насыщению свободных радикалов [Sanford, 1994].
6.3.1.2. Избирательность к некаталитическим процессам
гидрообессеривания и гидродеметаллизации
На рис. 6.5 изображена зависимость соотношения степеней удаления серы и ме-
таллов (НГДО / НГДМ) на выходе из второго реактора от температуры. Видно,
что в интервале 380—420 °C это соотношение возрастает с 2 до 3,5. Вследствие вы-
сокого соотношения FmT/ KSiC, т. е. короткого времени пребывания, глубина ре-
акции НГДО оказывается больше, чем реакции НГДМ. Из этого следует, что для
реакции НГДМ необходимо больше времени. Хорошо известно, что металлы (ни-
кель и ванадий) концентрируются главным образом в тяжелых молекулах (в ас-
фальтенах), которые являются ненасыщенными, в то время как сера содержится
222
Часть 2. Моделирование некаталитических процессов
в виде неасфальтеновых соединений, или мостиков, между молекулами асфальте-
нов. Кроме того, концентрация серы в сырье значительно выше, чем концентра-
ция металлов (5,869 %масс. серы против 0,06924 %масс. металлов). Что касается
влияния температуры, из рис. 6.5 видно, что зависимость соотношения степеней
НГДО / НГДМ от температуры в интервале 380-420 °C носит линейный харак-
тер; это означает, что некаталитические реакции удаления серы и металлов не яв-
ляются избирательными. С повышением температуры скорость обеих реакций,
как и любой другой химической реакции, возрастает, но повышение температуры
не действует избирательно в пользу удаления серы — несмотря на то, что концен-
трация последней в сырье гораздо выше и ее атомы расположены в более доступ-
ных местах молекул асфальтенов.
Температура, °C
Рис. 6.5. Соотношение степеней НГДО и НГДМ: () — FmT/ VSiC = 0,54 r/см3• ч;
(о) — FmJ = 0,43 г/см3 • ч; (□) — FmJ V = 0,33 г/см3 ч
\ / I о|ь ' ' ' I
6.3.1.3. Влияние температуры и соотношения FmT/VSiC
на плотность продукта
На рис. 6.6 показаны изменения плотности продукта по шкале API в зависи-
мости от температуры и соотношения FmT / KSiC. В наиболее жестких условиях
(FmTl KSiC = 0,23 г/см3 • ч, температура 420 °C) суммарный прирост плотности в граду-
сах API относительно плотности сырья (3,2 ° API) составляет около 7 единиц. Видно
также, что температура меньше влияет на плотность в градусах API, чем соотношение
FmTi KSjC, хотя при высоких температурах (400-420 °C) плотность по шкале API воз-
растает быстрее, чем при низких. Наблюдаемые изменения плотности по API прямо
связаны с глубиной гидрокрекинга: чем выше плотность по API, тем больше глуби-
на гидрокрекинга. При всех значениях температуры зависимость плотности в граду-
сах API от соотношения FmT/ Ияс близка к линейной; это говорите том, что кинетику
реакций гидрокрекинга можно представить простой моделью, подчиняющейся сте-
пенному закону.
(Глава
б. Некаталитическая (термическая) гидропереработка
223
Рис. 6.6. Влияние температуры и соотношения FmTl l/Sjc на плотность продукта по шка-
ле API: (*) — 380 °C; (х) — 390 °C; (А) — 400 °C; () — 410 °C; (♦) — 420 °C
6.3.1.4. Влияние температуры и соотношения Fmr/VSiC
на кривую разгонки продукта
На рис. 6.7 показано влияние температуры в реакторе на кривую разгонки продуктов
при FmT/ KSiC = 0,23 г/см3 • ч. Чтобы избежать перекрытия графиков и не загромождать
рисунок, показаны лишь кривые для сырья и для продуктов при значениях темпера-
туры в реакторе, равных 380 и 420 °C. Кривые разгонки сырья и продуктов при тем-
пературе в реакторе 380 °C схожи и расходятся не более чем на 20 °C. Это говорит
отом, что при 380 °C наиболее тяжелые фракции остаются практически неизменны-
ми. Тем не менее, температура в реакторе оказывает значительное влияние на кривую
разгонки продуктов, о чем свидетельствуют данные разгонки продукта, полученно-
го при 420 °C.
Влияние соотношения FmT/ Ksic на кривую разгонки продуктов гидрокрекинга ил-
люстрирует рис. 6.8. Здесь сравниваются сырье и продукты, полученные при темпера-
туре в реакторе 420 °C и величинах соотношения FmT/ KSiC, равных 0,23 и 0,65 г/см3 • ч.
Графики подтверждают, что соотношение FmT/ KSiC в исследуемом интервале влия-
ет на кривую разгонки продуктов сильнее, чем температура, поскольку при большем
значении FmT/ VSiC (0,65 г/см3 • ч) кривая разгонки продуктов довольно сильно сме-
щена вправо от кривой разгонки сырья (см. рис. 6.8), тогда как на рис. 6.7 при мень-
шем значении температуры (380 °C) она близка к кривой разгонки сырья. Такое по-
ведение согласуется с влиянием на плотность продукта по шкале API.
6.3.1.5. Влияние на состав суммарного жидкого продукта
Результаты, полученные при анализе продуктов имитации перегонки, использовались
для расчета относительных количеств бензиновой фракции, средних дистиллятов,
224
Часть 2. Моделирование некаталитических процессов
Отгон, %масс.
Рис. 6.7. Влияние температуры в реакторе на кривую разгонки продуктов
при FmTl l/S]C = 0,23 г/см3 • ч: (-) — кривая разгонки сырья; (—) — кривая разгонки продук-
тов при 380 °C; (•••) — кривая разгонки продуктов при 420 °C
Рис. 6.8. Влияние соотношения FmT/ |/Sic на кривую разгонки продуктов при температуре
в реакторе 420 °C: (-) — кривая разгонки сырья; (—) — кривая разгонки продуктов
при FmTl l/S1C = 0,65 г/см3 • ч; (••) — кривая разгонки продуктов при FmTl l/SiC = 0,23 г/см3 • ч
Глава 6. Некаталитическая (термическая) гидропереработка
225
вакуумного газойля и непревращенного вакуумного остатка. В данном иссле-
довании были приняты следующие интервалы кипения фракций: бензино-
вая фракция — НК—204 °C, средние дистилляты — 204—343 °C, вакуумный га-
зойль — 343—538 °C, вакуумный остаток — выше 538 °C. Состав сырья (остаток
атмосферной перегонки нефти с плотностью 13 ° API) был следующим: бензиновая
фракция — 0 %, средние дистилляты — 6,69 %, вакуумный газойль — 36,13 %, ва-
куумный остаток — 57,18 %.
Исходя из этих данных был вычислен выход каждой фракции:
Масса фракции в продукте - Масса фракции в сырье
' Масса вакуумного остатка в сырье
Сообщалось, что реакции термического и каталитического гидрокрекинга проте-
кают последовательно [Yang et al., 2011], т. е. гидрокрекинг вакуумного остатка дает
вакуумный газойль, гидрокрекинг вакуумного газойля — средние дистилляты и т. д.
В зависимости от условий протекание реакций по некоторым путям может оказаться
затрудненным или вообще не происходить.
На первом этапе через отрыв разветвленных цепей, окружающих крупные нена-
сыщенные ароматические молекулы, происходит крекинг вакуумного остатка с об-
разованием вакуумного газойля, а также незначительных количеств легких продук-
тов — бензина и газов. На последующих этапах непрерывно происходят реакции
гидрокрекинга вакуумного газойля и других более легких фракций. В этих реакциях
участвуют также вакуумный газойль, средние дистилляты и бензиновая фракция,
изначально содержавшиеся в сырье.
На рис. 6.9 показаны зависимости выхода различных фракций от температуры при
термическом гидрокрекинге вакуумного остатка при двух значениях соотношения
FmT/ KSiC (0,23 и 0,65 г/см3 • ч).
Выход всех продуктов с повышением температуры в реакторе возрастает. Когда
реактор работает в мягких условиях (высокое соотношение FmTl KSiC и низкая тем-
пература), выход каждой фракции меньше 2 %масс. Но при FmT/ KSiC = 0,23 г/см’ - ч
выход продуктов гидрокрекинга более чем в два раза превышает выход при
FmTl Ks.c = 0,65 г/см3 • ч. Такое соотношение примерно сохраняется при более вы-
соких температурах в реакторе, особенно в случае средних дистиллятов. По выходу
при FmT/ KSiC = 0,23 г/см3 • ч продукты располагаются в такой последовательности:
средние дистилляты > вакуумный газойль > бензиновая фракция >> газы. В случае
Fm.(/ KSiC = 0,65 г/см3 • ч последовательность другая: средние дистилляты ® вакуум-
ный газойль > бензиновая фракция >> газы. Когда температура в реакторе высока,
изменение соотношения FmT/ KSiC от 0,23 до 0,65 г/см3 • ч сильно отражается на по-
следовательности расположения продуктов по величине выхода. Все это позволяет
предположить, что избирательность реакций к продуктам зависит от жесткости ус-
ловий. Безусловно, такое поведение связано с температурой — параметром, управ-
ляющим реакцией гидрокрекинга. Имеет значение и время контакта, но не в такой
степени, как температура.
226
Часть 2. Моделирование некаталитических процессов
Температура, °C
Рис. 6.9. Зависимость выхода фракций от температуры и величины соотношения
FmT/ l/sjc: (и) — вакуумный газойль; (А) — средние дистилляты; (х) — бензиновая фрак-
ция; (*) — газы
Наблюдаемое поведение выхода средних дистиллятов и вакуумного газойля может
объяснить как повышенный суммарный выход жидкого продукта с температурой ки-
пения выше 300 °C, так и то, что вакуумный газойль крекируется легче, чем средние
дистилляты. В пределах исследованных рабочих условий выход газов не претерпева-
ет серьезных изменений, оставаясь практически на одном и том же низком уровне.
Наиболее подходящее объяснение этому состоит в том, что не достигается энергия
активации гидрокрекинга таких легких фракций, как бензиновая.
Исходя из сказанного, можно утверждать, что не все фракции сырья крекируются
одинаково легко. Труднее всего гидрокрекингу поддается бензиновая фракция.
Глава 6. Некаталитическая (термическая) гидропереработка
227
6.3.1.6. Распределение температур по оси реакторов
На рис. 6.10 показано распределение температур по оси двух реакторов в зависимо-
сти от сырья при различных условиях реакций. Кривые построены для трех разных
совокупностей условий: для низкой температуры и высокого давления, для высо-
кой температуры и высокого давления и для высокой температуры и низкого давле-
ния. Высота слоя в каждом реакторе составляла около 50 см; оба реактора работали
в режиме нисходящего потока (стрелки указывают, что движение сырья направле-
но сверху вниз). Во всех случаях сырье перед входом в первый реактор имело темпе-
ратуру на 3 °C ниже точки уставки. При входе сырье подогревалось до точки устав-
ки, после чего температура во всех точках слоя оставалась постоянной. Наблюдаемое
на выходе из реактора снижение температуры составляет 5—8 °C. Вероятно, оно обу-
словлено тепловыми потерями. Продукт первого реактора смешивался с водородом
и через теплоизолированную трубу подавался во второй реактор.
Рис. 6.10. Распределение температур по оси реакторов в зависимости от сырья, темпера-
туры и давления: (♦) — нефть (13 ° API); (А) — АО нефти (13 ° API); () — нефть (21 ° API);
(х) — АО нефти (21 ° API)
228
Часть 2. Моделирование некаталитических процессов
На входе во второй реактор смесь опять подогревалась до той же температуры, что
и на входе в первый реактор. Температурный профиль во втором реакторе показывает
снижение температуры. Судя по наблюдаемой картине, на участке от входа реактора
до 60-70 % его длины протекают эндотермические реакции, а на остальном участке
реактора происходит выделение теплоты, сопровождающееся превышением темпе-
ратуры уставки примерно на 15-25 °C.
Смена эндотермических реакций экзотермическими объясняется следующим обра-
зом. В термических процессах обычно происходят реакции двух типов: 1) эндотермиче-
ские реакции расщепления, при которых из крупных молекул образуются более мелкие;
2) экзотермические реакции конденсации, в ходе которых мелкие молекулы соединя-
ются в более крупные. В рассмотренном эксперименте в первом реакторе преобладают
реакции расщепления. Образовавшиеся мелкие молекулы вступают во втором реакторе
в реакции конденсации, вследствие чего температура в реакторе повышается.
Результаты, полученные при высоких температурах, свидетельствуют о важных из-
менениях в избирательности термических реакций. Превращение активных веществ,
подверженных гидрокрекингу (гидрообессериванию), способствует реакции деги-
дрирования, протекающей с поглощением тепла. Когда реактор работает при низкой
температуре, степень превращения активных веществ ниже и преобладает некатали-
тическая реакция термического гидрообессеривания (НГДО). Работа реактора при
разных температурах подразумевает изменение константы равновесия реакций ги-
дрирования и дегидрирования ароматических и нафтеновых соединений. Это явление
было продемонстрировано многими исследователями. Например, Столл и соавторы
[Stoll et al., 1998] определили значения констант равновесия реакции гидрирования
для ряда ароматических соединений. В случае гидрирования фенантрена значения
указанных констант в интервале 325-425 °C изменялись от 107 до 10~4. Это предпола-
гает изменение термического поведения реакционных систем, когда при повышении
температуры экзотермические реакции переходят в эндотермические.
Детально исследовать и моделировать не только термические, но и каталитические
реакции гидрокрекинга очень нелегко. Это объясняется сложным составом сырья,
трудностью его характеризации, а также проблемой учета большого числа реакций,
происходящих в процессе термокаталитического гидрокрекинга [Ferreira et al., 2007].
6.3.2. Однореакторная установка
6.3.2.1. Кинетика некаталитической гидроочистки
Для оценки кинетических параметров реакций НГДО и НГДМ использовались зна-
чения общего содержания серы и металлов (никеля и ванадия) при разных величинах
объемной скорости подачи сырья и температуры. Предполагалось, что реактор рабо-
тает в изотермическом режиме идеального вытеснения: это позволяло использовать
соответствующие формулы расчета.
Кинетика реакций НГДО и НГДМ моделировалась степенными уравнениями.
Совместным решением уравнений модели реактора и кинетической модели были
получены следующие выражения для расчета степени превращения серы и металлов:
Глава 6. Некаталитическая (термическая) гидропереработка
229
НГДОрасч = 1 -
НГДМрасч = 1 -
(Fmr/rsiC '
(FmT/V^ f
(6.3)
(6.4)
где НГДСрасч и НГДМрасч — расчетные степени превращения соответственно серы
и металлов; к — константа скорости; п — порядок реакции; Sfn — содержание со-
ответственно серы и металлов в сырье.
Величины к и п разных реакций могут быть различными.
С помощью алгоритма оптимизации, вычисляющего сумму квадратов отклонений
расчетных значений содержания серы и металлов от экспериментальных, был опре-
делен наилучший набор кинетических параметров. На рис. 6.11 и 6.12 сравниваются
значения степеней превращения серы и металлов, полученные расчетным (см. фор-
мулы (6.3) и (6.4)) и экспериментальным путем. Во всех случаях наблюдается хоро-
шее согласие между теорией и экспериментом. Порядок реакции НГДО оказался
равным 1,9, энергия активации — 48,2 ккал/моль. Для реакции НГДМ соответствую-
щие величины составили 1,4 и 42 ккал/моль.
Марафи и соавторы [Marafi et al., 2008] выполнили расчет кинетики некатали-
тических реакций гидроочистки тяжелой босканской нефти с более высоким со-
держанием серы и металлов (S = 5,54 %масс., Ni + V = 1353 мг/кг) в аналогичных
условиях. Порядок реакции НГДО был равен 1,74, а энергия активации составила
25,42 ккал/моль, что примерно в два раза меньше значения, указанного в настоящем
разделе. Порядок реакций НГДМ варьировался от 1,38 до 1,79, энергия активации —
от 43,83 до 50,665 ккал/моль. Наблюдаемые различия можно объяснить использова-
нием в экспериментах сырья разных типов.
Температура, °C
Рис. 6.11. Степень некаталитического превращения серы остатка атмосферной перегонки
нефти с плотностью 13 ° API при различных значениях температуры и соотношения FmTl VEjC:
(•) — FmTl VSiC = 0,65 г/см3 ч; () — FmTl VSiC = 0,54 г/см3 ч; (о) — FmT/ VSiC = 0,43 г/см3 ч;
(□) — FmTl VSiC = 0,33 г/см3 • ч; (A) FmTl VEiC = 0,23 г/см3 ч; (—) — расчетные значения
230
Часть 2. Моделирование некаталитических процессов
Рис. 6.12. Степень некаталитического превращения металлов остатка атмосфер-
ной перегонки нефти с плотностью 13 ° API при различных значениях температуры
и соотношения FmTl \/SiC: () — FmTl l/sjc = 0,54 г/см3 • ч; (о) — FmTl l/SlC = 0,43 г/см3 • ч;
(□) — FmTl VSiC = 0,33 г/см3 • ч; (—) — расчетные значения
На рис. 6.11 и 6.12 также показано влияние величин отношения FmT/ KS|C и темпе-
ратуры на степени НГДО и НГДМ. Как и следует ожидать, при низких температурах
и высоких значениях соотношения FmT/ KSiC степень некаталитического превращения
серы минимальна (меньше 2 %). В более жестких условиях (7тиг/Узк; = 0,2 г/см3-ч,
Т= 420 °C) степень НГДО достигала 25 %, в то время как степень НГДМ не подни-
малась выше 6 %.
6.3.2.2. Кинетика превращения вакуумного остатка
Степень превращения вакуумного остатка вычислялась следующим образом:
xBO=gBa0~gB°, (6.5)
?во,о
где£воо — содержание вакуумного остатка (температура кипения выше 538 °C) в сы-
рье; gBO — содержание вакуумного остатка в продукте.
Влияние температуры на степень превращения вакуумного остатка показано
на рис. 6.13. Видно, что при 420 °C изменения степени превращения наиболее
заметны. Это объясняется тем, что высокие температуры способствуют реакци-
ям некаталитического гидрокрекинга. Что касается зависимости от соотноше-
ния FmT/ Ks c, она почти линейна, а наклон прямой этой зависимости от тем-
пературы в реакторе зависит мало. Степень превращения вакуумного остатка
с повышением температуры увеличивается по той причине, что реакция являет-
ся не каталитической, т. е. энергия активации обеспечивается за счет температуры
[Khorasheh et al., 1989].
Глава 6. Некаталитическая (термическая) гидропереработка
231
Рис. 6.13. Влияние температуры и соотношения FmT! |/SiC на степень превращения
вакуумного остатка с нижней границей кипения 538 °C:
(♦) — 420 °C; () — 410 °C; (А) — 400 °C; (х) — 390 °C; (*) — 380 °C
Каталитические и термические реакции протекают по разным механизмам, по-
этому можно ожидать, что их порядки также будут различаться. Степени превра-
щения вакуумного остатка, достигаемой при каталитическом крекинге, достаточно
хорошо отвечает кинетика реакции второго порядка [Sanchez and Ancheyta, 2007].
Что касается некаталитических реакций гидрокрекинга, можно вывести следующую
взаимосвязь между степенью превращения вакуумного остатка хво, кинетическими
параметрами (порядком реакции п и константой скорости реакции fc0), суммарным
массовым потоком FmTn объемом инертного материала KSiC.
Для и = 1
In
— ^£вао
rsic
FmT j
Для п 1
(1 - -1 = ад0.о(" - 0'
Г Шу
(6.6)
(6.7)
1 '
1— хво>
Предполагалось, что реактор работает в режиме идеального вытеснения, что огра-
ничения массопереноса вследствие отсутствия катализатора не действуют, а кинети-
ка подчиняется степенному закону. Для каждого значения температуры методом наи-
меньшего квадратичного отклонения находили оптимальный набор кинетических
параметров (л и к0), из которого расчетным путем была определена энергия актива-
ции, оказавшаяся равной 42 ккал/моль.
Были опробованы различные значения порядка реакции. На рис. 6.14 представ-
лены степени превращения вакуумного остатка при 420 °C для значений порядка
232
Часть 2. Моделирование некаталитических процессов
реакции, равных п = 1, 1,5 и 2. Величины /(хво), откладываемые по осям ординат
на рис. 6.14, определяли по левым частям уравнений (6.6) и (6.7). Значения коэффи-
циента корреляции в целом были высоки и близки к единице, достигая наибольшей
величины при п = 2. Определялась также сумма квадратов отклонений расчетных зна-
чений степени превращения от экспериментальных; она существенно убывала, ког-
да порядок реакции приближался к 2. Ту же величину порядка реакции термического
гидрокрекинга тяжелых фракций обнаружили Иньсянь и Да [Yingxian and Da, 2011].
Рис. 6.14. Степень термического превращения вакуумного остатка при 420 °C для раз-
личных значений порядка реакции: () — л = 1; (•) — л = 1,5; (А) — л = 2. Обозначения:
SSE — среднеквадратичное отклонение
6.3.2.3. Кинетика некаталитического гидрокрекинга
Каталитический гидрокрекинг тяжелой нефти хорошо описывается пятикомпонент-
ной дискретной моделью, показанной на рис. 6.15 [Sanchez et al., 2005]. Хотя катали-
тические и термические реакции протекают по разным механизмам, та же кинетиче-
ская модель была использована для представления некаталитического гидрокрекинга.
Глава 6. Некаталитическая (термическая) гидропереработка
233
Предполагалось, что реакция гидрокрекинга вакуумного остатка подчиняется урав-
нению кинетики второго порядка; соответственно, для других реакций был принят
первый порядок. Уравнения (6.8)—(6.12) выражают скорости г. реакций компонентов
через их содержание у. в продукте и соответствующие константы скоростей к...
Вакуумный остаток (ВО):
^ВО ^оУ ВО’ (6.8)
где к0 = + к2 + к3 + кА.
Вакуумный газойль (ВГ):
Гивг ~ ^\У ВО — ^5 + к6 + ^Т^ВГ (6-9)
Средние дистилляты (СД):
ГСД = кЛо + ^ВГ - (^8 + ^СД- (6-10)
Бензиновая фракция (БФ):
ГБФ — к1У ВО + кбУВГ + кбУСД _ ^1оУбФ’ (6.11)
Газы (Г):
ГГ ~ ВО + ^уГвг + СД + ^1оУбФ' (6-12)
Рис. 6.15. Общая схема реакций каталитического гидрокрекинга тяжелых нефтей
Концентрации фракций на входе в реактор и выходе из него вычислялись по сле-
дующей формуле:
g,
(6.13)
где#. — суммарная масса z-й фракции (газы, бензин, средние дистилляты и вакуум-
ный газойль); gT — суммарная масса потока, входящего в реактор (вакуумный оста-
ток + водород).
234
Часть 2. Моделирование некаталитических процессов
Кинетическую модель объединяли со следующей моделью реактора:
^.с 7 А
FmT i ~r>
(6.14)
Полученную систему решали методом Рунге—Кутты. Предполагалось, что ограни-
чения массопереноса не действуют из-за отсутствия катализатора, а реактор работает
в режиме идеального вытеснения. Значения параметров определялись методом после-
довательных приближений, описанным в другом источнике [Ancheyta and Sotelo, 2000].
Методом минимизации целевой функции, которая принималась равной сумме квадра-
тов отклонений расчетных значений концентраций фракций в продукте от экспери-
ментальных, выявляли набор кинетических параметров, наилучшим образом согла-
сующийся с экспериментальными данными. Решение находили методом Марквардта
для задач нелинейной регрессии с использованием критерия наименьших квадратов.
Значения кинетических параметров и энергий активаций приведены в табл. 6.2.
Как можно видеть из рис. 6.16, концентрации всех компонентов в продукте пред-
сказываются достаточно точно: средняя абсолютная погрешность не превышает 5 %.
Величины энергий активации, необходимых для отдельных путей реакций гидро-
крекинга вакуумного остатка, находятся в пределах от 30 до 55 ккал/моль. Среднее
арифметическое четырех значений энергии активации (43,2 ккал/моль) довольно
близко к энергии активации суммарной реакции (42,4 ккал/моль). Из этого следует,
что кинетика реакции, в которой компоненты рассматриваются как единый продукт
(продукт = вакуумный газойль + средние дистилляты + бензиновая фракция + газы),
схожа с кинетикой реакций отдельных компонентов.
Таблица 6.2
Кинетические параметры
Пара- метр Путь реакции Температура, °C £(, ккал/моль
380 390 400 410 420
/с« ВО-* продукт 0,046 0,068 0,109 0,177 0,301 42,4
п 2,000 2,000 2,000 2,000 2,000
*1 ВО^ ВГ 0,017 0,024 0,047 0,090 0,162 52,0
во-* СД 0,015 0,022 0,032 0,047 0,074 36,0
во-* БФ 0,013 0,020 0,028 0,035 0,053 30,2
во-* газы 0,001 0,002 0,003 0,005 0,011 54,7
к<: вг^> СД 0,002 0,003 0,006 0,015 0,029 62,7
к6 ВГ-* БФ 0,000 0,000 0,000 0,000 0,000
к1 вг^ газы 0,000 0,000 0,000 0,000 0,000
к> сд-* БФ 2,50 • 10-4 1,02-10-’ 4,12-10-’ 1,17-10-2 2,47 IO2 104,8
к1 сд-* газы 0,000 0,000 0,000 0,000 0,000
БФ- газы 0,000 0,000 0,000 0,000 0,000
Сокращения: Ел — энергия активации, БФ — бензиновая фракция, ВО — вакуумный оста-
ток, ВГ — вакуумный газойль, СД — средние дистилляты.
Глава 6. Некаталитическая (термическая) гидропереработка
235
Концентрация компонента, д./д-
Рис. 6.16. Концентрации компонентов
(значки — экспериментальные значения, линии — расчетные значения):
(•) — 420 °C; (о) — 410 °C; (А) — 400 °C; (А) — 390 °C; () — 380 °C
236
Часть 2. Моделирование некаталитических процессов
По значениям констант скоростей видно, что в исследованном интервале тем-
ператур реакция гидрокрекинга вакуумного остатка имеет большую селективность
по более тяжелым компонентам и что газы образуются только из вакуумного остат-
ка. Это вполне ожидаемо: при каталитическом гидрокрекинге бензиновой фракции
и средних дистиллятов в аналогичных условиях газы тоже не образуются [Sanchez
and Ancheyta, 2007]. Видно также, что гидрокрекинг вакуумного газойля дает толь-
ко средние дистилляты, а гидрокрекинг средних дистиллятов — только бензиновую
фракцию. Все эти факты учитывает упрощенная схема термического гидрокрекинга
тяжелых нефтей (рис. 6.17) с шестью константами скоростей.
Рис. 6.17. Предлагаемая схема реакций термического гидрокрекинга тяжелых нефтей
Ниже приводятся некоторые ключевые особенности некаталитической гидро-
очистки, которые следуют из рассмотренных фактов.
• Термическое гидрообессеривание — важная реакция гидроочистки тяжелых
нефтей.
• Глубина реакций термического гидрообессеривания и гидродеметаллизации
зависит от ряда условий: качества сырья, температуры, времени пребывания
и давления.
• На глубину термических реакций преобладающее влияние оказывает
температура.
• Высокое давление подавляет термическое обессеривание, так как свободные
радикалы быстрее нейтрализуются в обогащенной водородом среде.
• При температуре в реакторе 420 °C и соотношении Fmr/ KSjC = 0,2 г/см3 • ч сте-
пень термического гидрообессеривания может доходить до 25 %масс., в то вре-
мя как степень термической гидродеметаллизации не превышает 6 %масс.
• С увеличением времени пребывания выделение тепла возрастает вследствие
реакций конденсации мелких молекул, образовавшихся на первом этапе, в бо-
лее крупные.
• При некаталитическом гидрокрекинге степень превращения вакуумно-
го остатка может достигать 42 %масс., вызывая увеличение плотности сы-
рья с 3,2 до 9,7 °АР1 (т. е. снижение абсолютной плотности крекинг-остатка
до 1,0021 г/см3 при плотности сырья в 1,0505 г/см3).
ава 6. Некаталитическая (термическая) гидропереработка
237
• Реакции некаталитического гидрокрекинга происходят последовательно
с превращением тяжелых фракций в более легкие. В исследованном интерва-
ле рабочих условий гидрокрекинг бензиновой фракции с образованием газов
не происходит.
’ Эксперимент показывает, что некаталитический гидрокрекинг тяжелых не-
фтей достаточно точно описывается пятикомпонентной кинетической моде-
лью с шестью кинетическими параметрами.
• Реакции термического (некаталитического) гидрокрекинга происходят доста-
точно глубоко даже в сравнительно мягких условиях, поэтому их необходимо
учитывать при расчете каталитических процессов.
ОБОЗНАЧЕНИЯ
Латинские
FmT s. Энергия активации, ккал/моль Суммарный массовый поток на входе в реактор, г/ч Содержание z-й фракции в продукте, г
?i,0 St #во &BO.O К Содержание i-й фракции в сырье, г Суммарная масса потоков на входе в реактор, г Содержание вакуумного остатка (538 °С+) в продукте, г Содержание вакуумного остатка (538 °С+) в сырье, г Характеризующий фактор
к Константа скорости реакции НГДО или НГДМ
к0 Константа скорости суммарной реакции гидрокрекинга вакуумного остатка, г/см3 ч
к< Константа скорости реакции термического гидрокрекинга вакуумного остатка до вакуумного газойля, г/см3 • ч
к2 Константа скорости реакции термического гидрокрекинга вакуумного остатка до средних дистиллятов, г/см3 • ч
кз Константа скорости реакции термического гидрокрекинга вакуумного остатка до бензина, г/см3 • ч
к. Константа скорости реакции термического гидрокрекинга вакуумного остатка до газов, г/см3 • ч
к5 Константа скорости реакции термического гидрокрекинга вакуумного газойля до средних дистиллятов, г/см3 • ч
к6 Константа скорости реакции термического гидрокрекинга вакуумного газойля до бензина, г/см3 • ч
238
Часть 2. Моделирование некаталитических процессов
/<7 Константа скорости реакции термического гидрокрекинга вакуумного
газойля до газов, г/см3 • ч
к3 Константа скорости реакции термического гидрокрекинга средних дис-
тиллятов до бензина, г/см3 ч
Константа скорости реакции термического гидрокрекинга средних дис-
тиллятов до газов, г/см3 • ч
Л10 Константа скорости реакции термического гидрокрекинга бензиновой
фракции да газов, г/см3 • ч
МеАВР Среднечисленная температура кипения, R
Mf Содержание металлов в сырье
п Порядок реакции термического гидрокрекинга, НГДО или НГДМ ва-
куумного остатка
гг Скорость реакции газов, г/см3 • ч
гсд Скорость реакции средних дистиллятов, г/см3 • ч
тБФ Скорость реакции бензиновой фракции, г/см3 ч
гвг Скорость реакции вакуумного газойля, г/см3 • ч
тво Скорость реакции вакуумного остатка, г/см3 • ч
S Содержание серы в сырье, %масс.
sg Удельный вес (относительная плотность) при 60 °F
S Содержание серы в продукте, %масс.
KSiC Суммарный объем инертного материала, см3
хво Степень превращения вакуумного остатка
Y. Выход z-й фракции, %
у. Концентрация z-й фракции в продукте
у. 0 Концентрация z-й фракции в сырье
уБФ Концентрация бензиновой фракции
увг Концентрация вакуумного газойля
уво Концентрация вакумного остатка
уг Концентрация газов
усд Концентрация средних дистиллятов
Нижние индексы
SiC Карбид кремния
БФ Бензиновая фракция
ВГ Вакуумный газойль
Глава 6. Некаталитическая (термическая) гидропереработка
239
ВО Вакуумный остаток
Г Газы
СД Средние дистилляты
ЛИТЕРАТУРА
Ancheyta, J., Sotelo, R. 2000. Estimation of kinetic constants of a five-lump model for fluid catalytic
cracking process using simpler sub-models. Energy Fuels 14:1226—1231.
Ancheyta, J., Speight, J.G. 2007. Hydroprocessing of Heavy Oils and Residua, CRC Press, Taylor &
Francis Group, Boca Raton, FL.
Ancheyta, J., Trejo, E, Rana, M.S. 2009. Asphaltenes: Chemical Transformations during Hydroprocessing
of Heavy Oils, CRC Press, Taylor & Francis Group, Boca Raton, FL.
Ferreira Da Silva, R.M.C., de Medeiro, J.L., Araujo, O.Q.E 2007. A network of chemical reactions
for modeling hydrocracking reactors. In Proceedings of European Congress of Chemical Engineering (ECCE-6),
Copenhagen, Denmark, September 16-20.
Gray M.R. 1994. Upgrading Petroleum Residues and Heavy Oils, Marcel Dekker Inc., Newark.
Jia, N., Moore, R.G., Mehta, S.A., Ursenbach, M.G. 2009. Kinetic modeling of thermal cracking
reactions. Fue/88:1376—1382
Juraidan, M., Al-Shamali, M., Qabazard, H., Kam, E.K.T 2006. A refined hydroprocessing multicatalyst
deactivation and reactor performance models pilot-plant accelerated test applications. Energy Fuels
20:1354-1364.
Kawai, FL, Kumata, E 1998. Free radical behavior in thermal cracking reaction using petroleum heavy oil
and model compounds. Catal. Today 43:281-289.
Khorasheh, E, Rangwala, H.A., Gray M.R., Dalia Lana, LG. 1989. Interactions between thermal and
catalytic reactions in mild hydrocracking of gas oil. Energy Fuels 3:716-722.
Kim, J.W, Longstaff, D.C., Hanson, EV 1998. Catalytic and thermal effects during hydrotreating of
bitumen-derived heavy oils. Fuel 77(15): 1815—1823.
Marafi, A., Kam, E.K.T, Stanislaus, A. 2008. A kinetic study on non-catalytic reactions in
hydroprocessing Boscan crude oil. Fuel 87:2131—2140.
Martinez, M.T, Benito, A.M., Callejas, M.A. 1997. Kinetics of asphaltene hydroconversion 1. Thermal
hydrocracking of a coal residue. Fuel 76( 10) :899—905.
Melis, S., Erby, L., Sassu, L., Baratti, R. 2004. A model for the hydrogenation of aromatic compounds
during gasoil hydroprocessing. Chem. Eng. Sci. .
Prestvik, R., Moljord, K., Grande, K., Holmen, A. 2004. Compositional analysis of naphtha and
reformate. In Catalytic Naphtha Reforming (G.J. Antos, A.M. Aitani, Eds.), 2ndedn., Marcel Dekker Inc.,
New York, pp. 1-34.
Raltimi, PM., Gentzis, T 2003. Thermal hydrocracking of cold lake vacuum bottoms asphaltenes and
their subcomponents. Fuel Process. Technol. 80:69—79.
Ramirez, S., Ancheyta, J., Centeno, G., Marroquin, G. 2011. Non-catalytic hydrodesulfurization and
hydrodemetallization of residua. Fuel 90:3571—3576.
Sanchez, S., Ancheyta, J. 2007. Effect of pressure on the kinetics of moderate hydrocracking of Maya
crude oil. Energy Fuels 21:653—661.
Sanchez, S., Rodriguez, M.A., Ancheyta, J. 2005. Kinetic model for moderate hydrocracking of heavy
oils. Ind. Eng. Chem. Res. 44:9409-9413.
Sanford, E.C. 1994. Molecular approach to understanding residuum conversion. Ind. Eng. Chem. Res.
33:109-117.
Singh, J., Kumar, M.M., Saxena, A.K., Kumar, S. 2005. Reaction pathways and product yields in
mild thermal cracking of vacuum residues: A multi-lump kinetic model. Chem. Eng. J. 108:239-248.
240 Часть 2. Моделирование некаталитических процессов
Speight, J.G. 2000. The Desulfurization of Heavy Oils and Residua, Marcel Dekker Inc., New "fork.
Stoll, P, Demirel, B., Wiser, WH. 1998. Thermodynamic probability of the conversion of multiring
aromatics to isoparaffins and cycloparaffins. Fuel Process. Technol. 55:83-91. Topsoe, H., Clausen, B.S.,
Massoth, EE. 1996. Hydrotreating Catalysis Science and Technology, Springer-Xferlag, New York.
Wide, R., Vislocky, J., Maesen, T, Torchia, D. 2009. Hydrocracking Catalyst Developments and Innovative
Processing Schemes, NPRA, AM-2009-12, San Antonio, TX.
Y ang, M.-G., Nakamura, I., Fujimoto, K. 1998. Hydro-thermal cracking of heavy oils and its model
compound. Catal. Today 43:273—280.
Y ing, C., Zheng, H., Du, E, Xu, C. 2011. Reaction Characteristicsand Mechanism of Residuum Hydrocracking,
University of Petroleum, State Key Laboratory of Heavy Oil Processing, http://www.anl.gov/PCS/acsfiiel/
preprint%20archive/Files/43_3_BOSTON_08-98_0668. pdf (accessed on October 5, 2011).
Y ingxian, Z., Da, L. 2011. Kinetics and selectivity in thermal hydrocracking and catalytic hydrocracking
of asphaltenes. China Petrol. Process. Petrochem. Technol. 13:24—31.
Z hang, C., Lee, C.W., Keogh, R.A., Demirel, B., Davis, B.H. 2001. Thermal and catalytic conversion of
asphaltenes. Fuel 80(10): 1131 -1146.
Часть III
Моделирование каталитических
процессов
Глава 7. Моделирование
каталитической гидроочистки
В этой главе рассматривается каталитическая гидроочистка — один из важней-
ших процессов в нефтеперерабатывающей промышленности, не только улучшаю-
щий качество нефтей тяжелых сортов, но и дающий топливные продукты с низким
содержанием примесей, а также сырье для других процессов переработки. Освеща-
ются основные аспекты этого процесса: его характеристики, параметры и типы ис-
пользуемых реакторов.
Разрабатывается детализированная модель для стационарной и динамической
имитации реактора гидроочистки. Построение модели иллюстрируется на приме-
ре газойля, полученного из тяжелой нефти. Освещаются опыты, проводимые на ла-
бораторном реакторе с целью определить кинетические константы различных реак-
ций гидроочистки и соответствующих гидродинамических параметров. Полученная
модель применялась для имитации поведения изотермического лабораторного и не-
изотермического промышленного реакторов гидроочистки. При моделировании ре-
актора гидроочистки в качестве сырья был выбран тяжелый газойль. Он упрощал
проведение опытов, не создавал трудностей при анализе продуктов и сохранял ак-
тивность катализатора в условиях исследования, что давало возможность адекватно
применить и проверить модель. Последующие главы посвящены ее адаптации и ис-
пользованию для моделирования гидропереработки тяжелой нефти.
7.1. ВВЕДЕНИЕ
Каталитическая гидроочистка (гидропереработка, гидрооблагораживание) — ос-
военная технология, которая уже шесть десятилетий применяется в нефтеперера-
батывающей промышленности для повышения качества углеводородного сырья.
Это важнейший процесс очистки разнообразного сырья — от прямогонного бен-
зина до вакуумного остатка, тяжелых и очень тяжелых сортов нефти. Гидроочистка
дистиллятов преследует две главные цели: 1) удаление таких примесей, как гетеро-
элементы (сера, азот и кислород); 2) насыщение ароматических соединений через
присоединение водорода. В случае тяжелых нефтей и остатков к этим задачам добав-
ляются еще удаление металлов (никеля и ванадия) и увеличение выхода дистиллятов
за счет тяжелых фракций — вакуумного остатка и асфальтенов [Ancheyta et al., 2005].
244
Часть 3. Моделирование каталитических процессов
7.1.1. Значение гидроочистки в нефтепереработке
На первом этапе переработки нефть поступает в колонну атмосферной перегонки,
где отделяются прямогонные дистилляты: бензин, керосин и газойль. По ряду техни-
ческих причин эти сырые продукты нельзя сразу же использовать в качестве мотор-
ных топлив: во-первых, они содержат слишком много примесей (серу, азот), а так-
же ароматических соединений; во-вторых, бензиновая и дизельная фракции имеют
слишком низкие соответственно октановые и цетановые числа. Сырые продукты
превращают в товарные посредством различных процессов облагораживания. В этом
контексте гидроочистка играет главную роль, причем на нескольких этапах нефтепе-
реработки. Это можно видеть на рис. 7.1, где изображена схема типичного нефтепе-
рерабатывающего завода, ориентированного на производство бензина. Переработка
остаточного сырья, т. е. вакуумного остатка, представлена на рисунке коксовани-
ем, хотя это может быть также и гидроочистка с гидрокрекингом. Согласно обзору
мировой нефтеперерабатывающей промышленности [OGJ, 2010], установки гидро-
очистки располагают наибольшими производственными мощностями по сравнению
с другими операциями нефтепереработки (45,43 млн баррель/сут).
Газ
Установка
газофракцио-
нирования
Легкие нефтяные газы
-
Олефины Полимеризация
Атмосферная
перегонка
1
Газы с других установок
Легкий прямогонный
бензин
Алкилиро-
вание
Нефть
И
Обессоливание
1.
г
*
Тяжелый прямогонный
бензин
Реактивное
топливо
Легкий прямогон-
ный газойль
Тяжелый прямогонный
газойль
(
Вакуумная
перегонка
Коксование |
ГО
ГО
ГО
Деасфаль-
Вакуумный
газойль го
ГО
Риформинг
бензина
Изомери-
зация
Экстракция
ароматических
углеводородов Бензин
Гидрокрекинг
Ароматические углеводороды
Реактивное топливо
Дизельное топливо
Каталитический
крекинг в псевдо
ожиженном слое (FCC)
Асфальт
Экстракционная деасфальтизация
Кокс
I *
А '
Рис. 7.1. Значимость гидропереработки на типичном нефтеперерабатывающем заводе
Глава 7. Моделирование каталитической гидроочистки
245
Благодаря росту спроса на моторные топлива и ужесточению законов об охране
природы процесс гидроочистки приобретает все большее значение. Ниже перечис-
лены его основные применения в сфере нефтепереработки.
1. Подготовка сырья для таких процессов превращения, как изомеризация, ка-
талитический риформинг, каталитический крекинг и гидрокрекинг, заключающая-
ся в снижении содержания серы, азота (в особенности азота оснований), металлов
и полициклических ароматических углеводородов. Главная задача — получить обла-
гороженное сырье с минимально возможным содержанием агентов, ухудшающих ак-
тивность кислотных катализаторов в следующих процессах [Dahl et al., 1996; O’Con-
nor et al., 1996; Sau et al.,2005]:
а) удаление серы из прямогонного бензина с целью предотвратить отравление
катализаторов риформинга, изготавливаемых из благородных металлов;
б) удаление азота оснований (для предотвращения обратимого отравления
кислотных катализаторов), серы и металлов (для предотвращения потери ак-
тивности цеолитных катализаторов и нежелательного промотирования реак-
ций дегидрирования, увеличивающих избирательность к сухому газу), а также
насыщение полициклических ароматических углеводородов (способствующих
образованию кокса) вакуумного газойля с целью получить более качественное
сырье для процессов гидрокрекинга и каталитического крекинга в псевдоожи-
женном слое (FCC).
2. Доочистка дистиллятов — заключительный этап производства моторных то-
плив, отвечающих экологическим стандартам (например, бензина и дизельного то-
плива с ультранизким содержанием серы). Это направление применения гидроочист-
ки выглядит весьма перспективным в связи с ужесточением экологических законов.
Необходимость минимизировать уровень выбросов оксидов серы (SOx) и азота (NOx),
а также летучих органических веществ привела к тому, что низкое содержание серы,
азота и ароматических углеводородов стало обязательным свойством современных
автомобильных топлив.
3. Облагораживание тяжелых нефтей. Это сравнительно новое применение про-
цесса, когда гидропереработке подвергается непосредственно тяжелая нефть. Оно
позволяет достичь следующих целей:
а) снизить плотность и вязкость, чтобы облегчить транспортировку;
б) осуществить превращение тяжелых фракций для увеличения выхода дис-
тиллятов, а также снизить содержание примесей, чтобы повысить товарную цен-
ность нефти;
в) улучшить качество нефти, направляемой на заводы, которые были постро-
ены давно с расчетом на переработку обычной нефти и поэтому не располагают
возможностями для переработки тяжелой нефти.
Разнообразие существующих процессов гидропереработки позволяет перера-
батывать нефтезаводское сырье любых типов и решать любую конкретную зада-
чу [Rana et al., 2007]. Процессы различаются конструкциями реакторов, типами
применяемых катализаторов, рабочими условиями и технологическими схемами.
Из всех типов реакторов в гидропроцессах шире всего из-за присущей им гибкости
246
Часть 3, Моделирование каталитических процессов
и простоты применяются реакторы с неподвижным слоем [Furimsky, 1998]. Могут
применяться также реакторы с кипящим слоем, особенно для облагораживания наи-
более тяжелых фракций. Главная особенность таких реакторов — возможность заме-
нить отработанный катализатор без прерывания работы установки. Выбор той или
иной технологии определяется скоростью потери активности катализатора, что зави-
сит от свойств сырья [Sie, 2001].
Помимо упомянутых выше традиционных применений гидроочистки, в дина-
мической среде нефтеперерабатывающей отрасли появляется множество других
технологий, большинство которых используются для облагораживания наиболее
тяжелых видов сырья. К таким технологиям относится процесс обессеривания (де-
сульфуризации) атмосферных и вакуумных остатков, разработанный для реше-
ния множества задач, в числе которых подготовка сырья каталитического крекин-
га и гидрокрекинга продуктов коксования и остатков перегонки [Scheuennan et al.,
1993; Kressmann et al., 1998]. Еще один пример процесса из этой категории — ги-
дрокрекинг остатков в псевдоожиженном слое с одновременными обессерива-
нием и деметаллизацией. Недавно технология гидроочистки стала применяться
по отношению к предварительному (т. е. проводимому до атмосферной перегон-
ки) облагораживанию тяжелых и очень тяжелых нефтей, дающему так называемую
синтетическую нефть [Ancheyta et al., 2010]. По сравнению с традиционными тер-
мическими процессами облагораживания гидропереработка дает больше жидких
продуктов хорошего качества и значительно меньше кокса [Speight, 2004]. Поэто-
му можно ожидать, что из-за роста добычи тяжелой нефти эта группа процессов
займет главное место в технологической цепи нефтеперерабатывающих заводов
будущего.
Гидрооблагораживание проводится в широком спектре рабочих условий.
Жесткость условий регулируют в зависимости от свойств сырья и требуемой струк-
туры целевых продуктов. Главные параметры процесса — давление, температура,
кратность водорода к сырью и удельная (по отношению к катализатору) объемная
скорость подачи сырья. Каждый параметр влияет на любой отдельно взятый аспект
процесса, поэтому для эффективной переработки необходимо тщательно подобрать
совокупность рабочих условий.
Процесс гидроочистки, проводимый в присутствии катализатора в атмосфе-
ре водорода, увеличивает водород-углеродное соотношение. Происходят реак-
ции гидрообессеривания (ГДО), гидродеазотирования (ГДА), гидродеароматиза-
ции (ГДАр), гидродеоксигенации (ГДОк) и гидродеметаллизации (ГДМ). Еще один
тип реакций гидропереработки — гидрокрекинг, при котором высокомолекуляр-
ные соединения расщепляются на низкомолекулярные. Применительно к гидро-
крекингу асфальтеновой фракции процесс часто называют гидродеасфальтизацией
(ГДАс). Химию этих реакций можно представить как процесс, в который посред-
ством различных механизмов гидрогенолиза и гидрирования вовлекается подводи-
мый из внешнего источника водород, замещающий гетероатомы и стабилизирую-
щий ненасыщенные продукты. Реакции ГДО, ГДА, ГДОк и ГДМ рассматриваются
как происходящие по механизму гидрогенолиза, в котором молекула водорода
Глава 7. Моделирование каталитической гидроочистки
247
расщепляет связь углерода с гетероатомом. Молекулы олефинов и ароматических
углеводородов гидрируются непосредственно, без всякого разрыва связей. Напро-
тив, реакции гидрокрекинга и гидродеасфальтизации представляют собой кислот-
но-катализируемый крекинг в сочетании с гидрированием. Но из-за сложности
состава, присущей нефти и ее фракциям, химия процесса гидроочистки в действи-
тельности гораздо сложнее.
Для адекватного расчета и имитирования процесса гидропереработки необходимо
глубоко изучить аспекты моделирования кинетики и реакторов. Это нетривиальная
задача, так как в реакторе одновременно происходит множество физических и хими-
ческих процессов и явлений: достижение равновесия фаз; массоперенос реагентов
и продуктов между газовой, жидкой и твердой фазами; диффузия в частицах катали-
затора; сложная цепь реакций; потеря активности катализатора и др. В идеале модель
работы реактора должна учитывать влияние всех этих явлений. Но в действительно-
сти при выборе уровня сложности модели обычно руководствуются целями прогно-
зирования [Mederos et al., 2009].
7.1.2. Текущая ситуация в нефтеперерабатывающей отрасли
Не секрет, что мировое энергопотребление постоянно растет. Сегодня нефть, буду-
чи наиболее широко используемым источником жидких топлив, обеспечивает око-
ло трети мирового энергоснабжения [International Energy Agency, 2010]. Ожидает-
ся, что такая ситуация сохранится как минимум ближайшие 50 лет [Ancheyta and
Speight, 2007а]. На рис. 7.2 показан рост мирового потребления продуктов нефтепе-
реработки с 1990 по 2010г. Можно видеть, что запоследние Юлетон составил 12,2%.
Данная тенденция обусловлена быстрым ростом спроса на автомобильные и авиаци-
онные топлива, особенно в таких развивающихся странах, как Китай, Россия и госу-
дарства Латинской Америки [Enerdata, 2011].
Между тем структура мировых запасов нефти сдвигается в сторону тяжелых
и очень тяжелых сортов, что означает ухудшение среднего качества нефти. Типичный
пример этой тенденции — Мексика, где на тяжелую нефть приходится более полови-
ны суммарной нефтедобычи (см. рис. 7.2). Более того, если до недавнего времени за-
пасы мексиканской нефти состояли из тяжелой нефти «Майя» (плотностью 21 ° API),
то сейчас их структура сместилась в сторону еще более тяжелых сортов с плотностью
10-16 “API. В результате на нефтеперерабатывающие заводы поставляется нефть все
более худшего качества. Сегодня заводы, традиционно нацеленные на переработ-
ку нефти легких сортов, сталкиваются с коренными изменениями в составе нефти:
им приходится перерабатывать сырье с повышенным содержанием серы, металлов
и асфальтенов.
Переработку тяжелых нефтей сильно затрудняет сложность их состава.
На рис. 7.2 показаны свойства двух сортов тяжелой («Майя» и «Ку-Малоб-Са-
ап») и одного сорта легкой («Истмас») мексиканской нефти, чтобы подчеркнуть
различия между ними. По сравнению с легкими сортами тяжелая нефть содержит
гораздо больше примесей, в том числе серы, азота, металлов (никель и ванадий)
248
Часть 3. Моделирование каталитических процессов
и асфальтенов. Кроме того, велико (более 50 %об.) количество остатка с темпера-
турой кипения выше 343 °C, называемого атмосферным; это говорит о низком вы-
ходе светлых дистиллятов. В этом смысле остаток составляет основную часть тяже-
лых нефтей и поэтому сильно влияет на их свойства, так как в нем концентрируется
основная масса примесей. Эта фракция подлежит тщательному изучению, так как
в будущем она станет основным сырьем для получения ценных жидких продук-
тов. Помимо всего прочего, возникают большие трудности при транспортировке
по трубопроводам тяжелых нефтей: они имеют чрезвычайно низкие реологические
качества, а смешивание с другими сортами нефти сопряжено с проблемами ста-
бильности и несовместимости.
в) Свойства мексиканской нефти различных сортов
Свойства Сорт нефти
«Ку-Малоб-Саап» «Майя» «Истмас»
Относительная плотность при 60 °F (15,5 °C), °АР1 12,9(0,9799) 21,3 (0,9260) 33,2(0,8591)
S, %масс. 5,19 3,57 1,80
N, мг/кг 4771 3200 1446
Ni, мг/кг 83 53 20
V, мг/кг 501 298 79
Компоненты, нерастворимые в С7, %масс. 17,03 11,32 3,06
Выход атмосферного осадка (343 °С+), %об. 73,88 56,4 45,31
Рис. 7.2. Мировое потребление продуктов нефтепереработки (а), добыча нефти в Мек-
сике (6) и свойства мексиканских нефтей (в) [Enerdata, Global Energy Statistical Yearbook],
Доступно на сайте http://www.enerdata.net, 2011. В скобках даны показатели, выраженные
в безразмерных величинах (относительная плотность, т. е. плотность продукта, деленная
на плотность воды)
Глава 7. Моделирование каталитической гидроочистки 249
Современные тенденции в нефтедобыче и на рынке топливных продуктов ста-
вят перед отраслью грандиозные задачи. Чтобы решить их, нефтеперерабатываю-
щие предприятия должны строить свою деятельность с учетом следующих главных
аспектов:
1) увеличение производительности для удовлетворения растущего спроса на то-
пливные продукты;
2) повышение требований к качеству конечных продуктов;
3) значительное ухудшение качества сырья.
Все эти обстоятельства вынуждают предварительно облагораживать тяжелые неф-
ти, чтобы иметь возможность перерабатывать их в больших объемах. Гидроперера-
ботка отлично помогает решить эту сложную задачу.
7.2. ОПИСАНИЕ ПРОЦЕССА
На рис. 7.3 представлена схема типичной установки и типов реакторов гидропере-
работки. Все начинается с подготовки сырья. Нефтяное сырье смешивается с цир-
кулирующим водородом и подогревается в печи до температуры реакции. Затем
газожидкостная смесь поступает в реактор или в нескольких последовательно со-
единенных реакторов. В зависимости от количества выделяемого тепла может ока-
заться необходимым промежуточное охлаждение реагентов между слоями катализа-
тора или между реакторами. Охлаждение обычно осуществляется циркулирующим
газом. Кроме того, реактор должен быть оборудован внутренними устройствами
для распределения и смешивания реагентов на входе и между слоями. Поток из ре-
актора поступает в сепаратор высокого давления, где от жидких продуктов отделя-
ются газы. Чтобы удалить из жидкости остаточные количества растворенных кис-
лых газов (сероводорода и аммиака), ее подают в отпарную колонну. Газ сепаратора
с той же целью отмывается раствором диэтаноламина (ДЭА). Получаемый водо-
род высокой чистоты компримируется и возвращается на вход в реактор. Продукт
гидрообработки может разделяться на несколько дистиллятных фракций, что зави-
сит от глубины процесса.
В реакторах гидропереработки обычно применяют кобальт-молибденовый или
никель-молибденовый катализатор на алюмооксидном носителе; состав и струк-
турные свойства катализатора определяет специфика решаемой задачи. Жесткость
рабочих условий зависит от типа сырья и требований к качеству целевого продук-
та. Процесс, как правило, проводят при высоких давлениях и температурах. Типич-
ные промышленные установки работают при давлениях от 2 до 20 МПа, температу-
рах от 320 до 440 °C, кратности водорода к сырью от 350 до 1800 н. м3/м3 и объемной
скорости подачи сырья от 0,2 до 8 ч-1 [Satterfield, 1975].
Что касается типов реакторов, в настоящее время для гидрообработки применя-
ют реакторы с неподвижным, движущимся и кипящим слоем катализатора (РНС,
РДС и РК.С соответственно), а также реакторы с суспензионным слоем (РСС)
[Furimsky, 1998]. Все эти виды реакторов изображены на рис. 7.3.
250
Часть 3. Моделирование каталитических процессов
Свежий водород
Циркулирующий
водород
* !
На дожит
Нефтяное сырье J
I <
Реактор
с неподвижным
орошаемым
слоем
Сырье
и водород
Продукты
и водород
: Регенерированный ДЭА
Аминовый
Реактор
Насыщен-
ный ДЭА
Т .......... ,
скруббер
Пар
Противоточный
реактор с движу-
щимся слоем
Отпарная
колонна
Прямоточный
реактор с движу-
щимся слоем
Сырье
и водород
Продукты
и водород
«
Кислый газ
Продукт гидро-
процесса
Реактор
с суспензионным
слоем
Продукты, водород
и катализатор
' Катали-
[ затор
Продукты Катали. Сырье
Катали-
иводород 3 иводо- 3
род
Реактор
с кипящим
слоем
Продукты
и водород
Катали-
затор
Катали-
затор
Сырье
и водо- Катали-
род затор
Сырье, водород
и катализатор
Рис. 7.3. Типичная технологическая схема и реакторы установок гидропроцесса
Благодаря своей простоте, гибкости к типу сырья и удобству в эксплуатации наи-
более широкое применение во всех операциях гидропереработки нашли реакторы
с неподвижным слоем. Реакторы с движущимся и кипящим слоем разрабатыва-
лись исключительно для облагораживания сырья наиболее тяжелых типов [Anchey-
ta, 2007]. Реакторы с движущимся слоем вначале применялись для очистки только
бензиновой фракции и газойля, но в конце концов их приспособили и к более тяже-
лому сырью, в том числе к вакуумным газойлям и остаткам. Главный недостаток РДС
при переработке тяжелого сырья — радикальное сокращение цикла службы катали-
затора, вызываемое быстрой потерей активности вследствие накопления металлов
и закоксовывания [Sie, 2001]. Решить эту проблему помогло внедрение многослойных
каталитических систем, позволяющих рациональнее использовать заряд катализато-
ра и существенно продлить цикл его службы [Marafi et al., 2003]. Главная особенность
таких технологий — входной слой макропористого катализатора деметаллизации.
Он способствует разобщению молекул асфальтенов и удалению металлов. Последую-
щие катализаторы гидродеметаллизации и гидрокрекинга перерабатывают уже сырье
с низким содержанием металлов и прекурсоров кокса.
Недостаток короткого срока службы устранен в реакторах с движущимся и кипя-
щим слоем. В РДС и РКС можно заменять отработанный катализатор без прерыва-
ния работы реактора, поэтому это оптимальный вариант для переработки наиболее
Глава 7. Моделирование каталитической гидроочистки
251
проблематичного сырья (с высоким содержанием металлов и асфальтенов). В реак-
торах с движущимся слоем работа в режиме вытеснения сочетается с периодической
(1-2 раза в неделю) заменой катализатора. Эти реакторы особенно хорошо подхо-
дят для входной деметаллизации сырья, проводимой в целях защиты последующих
реакторов гидродеметаллизации и гидрокрекинга в неподвижном слое [Sie, 2001].
Наиболее передовая технология гидропереработки — реакторы с кипящим слоем.
Они рассчитаны на прямое облагораживание сырья тяжелых типов без всякой под-
готовки [Rana et al., 2007]. Технология непрерывной замены позволяет применять
в них обычные высокоактивные катализаторы гидродеметаллизации и гидрокрекин-
га. Эти реакторы имеют высокую приспосабливаемость. Глубина превращения в них
весьма высока (до 90 %об.), а содержание серы, металлов и азота в продуктах низко
[Martinez et al., 2010]. Главные недостатки реакторов с кипящим слоем — повышен-
ная скорость образования осадков и высокий расход катализатора.
7.3. ТИПЫ РЕАКТОРОВ
Существующие технологии реакторов гидропереработки различаются главным об-
разом типом каталитического слоя. По этому признаку они подразделяются на ре-
акторы с неподвижным, движущимся, кипящим и суспензионным слоем. К наибо-
лее распространенным относятся реакторы с неподвижным слоем; реакторы других
типов более сложны и отличаются узкой специализацией. Главным критерием выбо-
ра типа реактора является скорость потери активности катализатора, которая сильно
зависит от содержания металлов и асфальтенов в сырье [Sie, 2001]. Для переработки
сырья наиболее тяжелых типов хорошо подходят реакторы с движущимся, кипящим
и суспензионным слоем, поскольку в них предусмотрена возможность замены ката-
лизатора без прерывания работы установки. Тяжелое сырье в реакторах с неподвиж-
ным слоем можно перерабатывать лишь в том случае, если ожидаемая длительность
срока службы катализатора находится в экономически приемлемых пределах (как
правило, около 6 мес.). Ниже рассматриваются особенности реакторов каждого типа.
7.3.1. Реакторы с неподвижным слоем
Реакторы с неподвижным слоем (РНС) применяются на большинстве промышлен-
ных установок гидроочистки и гидрокрекинга. Изначально РНС предназначались
для переработки бензиновой фракции, керосина и газойля, но со временем их при-
способили и для гидрооблагораживания более тяжелого сырья, в том числе вакуум-
ных газойлей и остатков атмосферной и вакуумной перегонки. Предпочтительность
применения РНС в нефтепереработке обусловлена их простотой, гибкостью и удоб-
ством в эксплуатации.
Реакционная среда в реакторах гидропереработки является, как правило, трехфаз-
ной (газовая, жидкая и твердая фазы). Газовая фаза состоит в основном из водоро-
да, газообразных продуктов реакций и частично испаренных углеводородов, жидкая
252
Часть 3. Моделирование каталитических процессов
фаза представлена углеводородным сырьем, а твердая — каталитическим слоем.
Единственное исключение — реакторы гидроочистки бензина, в которых вследствие
полного испарения углеводородов существуют всего лишь две фазы (газовая и твер-
дая). Сосуществование трех фаз переводит реакторы гидропереработки в неподвиж-
ном слое в особую категорию так называемых реакторов с орошаемым слоем. В таком
реакторе потоки газообразных и жидких реагентов и продуктов движутся совмест-
но вниз через неподвижный слой частиц катализатора [Al-Dahhan et al., 1997]. Схе-
ма орошающего режима течения приведена на рис. 7.4. В таком потоке дисперсной
фазой считается жидкость, сплошной — газ. Жидкость стремится образовать тонкую
пленку, обтекающую частицы катализатора, а газовая фаза движется отдельно, запол-
няя остальное пространство между частицами в слое. Орошающий режим течения
обычно достигается при сравнительно низких скоростях газа и жидкости, когда вза-
имодействие между этими фазами минимально.
При орошающем режиме течения происходит массоперенос реагентов и продук-
тов между тремя фазами. На рис. 7.4 схематически изображены также явления массо-
переноса, происходящие в соответствии с двухпленочной теорией [Korsten and Hoff-
mann, 1996] в реакторе с орошаемым слоем. Водород, будучи основным реагентом
в газовой фазе, должен диффундировать из газовой фазы к границе раздела «газ—жид-
кость», пересечь ее и перейти в жидкую фазу. Сопротивлением в газовой пленке обыч-
но пренебрегают, а концентрации газообразных веществ на границе раздела определя-
ются равновесием с парциальным давлением в газовой фазе. Газообразные реагенты,
органические соединения и углеводороды в жидкой фазе перемещаются к частицам
катализатора через границу раздела «жидкость — твердое тело», чтобы прореагировать
друг с другом. Углеводородные продукты возвращаются в жидкую фазу, а газы, обра-
зовавшиеся в результате химических реакций (в том числе сероводород, аммиак и лег-
кие углеводороды), тем же способом перемещаются в газовую фазу.
Одно из главных достоинств реакторов с орошаемым слоем состоит в том, что ре-
жим течения жидкости в них близок к режиму идеального вытеснения, поэтому они
по характеристикам превосходят другие трехфазные реакторы. Кроме того, у них вы-
сокое соотношение объема загрузки катализатора к объему жидкой фазы. Они име-
ют очень простую конструкцию, не требуют больших капиталовложений и наиболее
гибки в отношении пропускной способности и жесткости условий реакций для полу-
чения различных степеней превращения.
Один из недостатков реакторов орошаемого слоя — ограниченная диффузия реа-
гентов вглубь частиц катализатора. Это обусловлено тем, что на практике в реактор
приходится загружать сравнительно крупный катализатор, чтобы избежать больших
потерь давления. Но главный недостаток реакторов этого типа заключается в том, что
катализатор со временем теряет активность. Кроме того, из-за наличия в сырье твер-
дых примесей (ржавчина, соли, коксовая мелочь и т. д.) и продуктов реакций (кокс
и отложения металлов) реакторы с орошаемым слоем подвержены быстрому загряз-
нению [Chou, 2004]. Это означает, что с определенной периодичностью реактор при-
ходится останавливать, чтобы заменить катализатор. Процедура замены занимает
около месяца и требует полной разборки реактора. Длительность цикла определя-
ется, в зависимости от типа процесса и свойств сырья, нарастанием потерь давле-
ния или потерей активности катализатора до определенного уровня. Цикл работы
ава 7. Моделирование каталитической гидроочистки
253
Сырье и водород
Продукты
Застойная зона
Ароматические
углеводороды
Углеводороды
С*
сн2
Сероводород PH2s
Ph2s
д D cnh;
Аммиак PNH __
р*
С*
Легкие угле- PLH_
водороды р* —j
Орошающий
режим течения
Поток газа
Поток жидкости
Поток газа
Сухая зона
Сопротивление
массопереносу
между газовой
и жидкой фазами
Сопротивление
массопереносу
между жидкой
и газовой фазами
Сера
Азот
Водород Рн.
ci
С
Жидкая фаза
Г s
L NH3
Cllh
cH,s
Clh
Газовая фаза
Твердая фаза
(катализатор)
нс. 7.4. Схема орошающего режима течения и распределения концентраций в реакторе
идропереработки [Mederos, F.S. et al., Catal. Rev. Sci. Eng., 51(4), 485, 2009]. Обозначе-
ния: P— парциальное давление паров; звездочка означает насыщенные пары
254
Часть 3. Моделирование каталитических процессов
большинства установок длится обычно 2 года, но он может сократиться до 6—12 мес.
в случае гидроочистки остатков либо увеличиться до 5 лет при переработке сырья
с относительно небольшим содержанием металлов и асфальтенов [Robinson and Dol-
bear, 2006]. В реакторах с орошаемым слоем нередки нарушения распределения ре-
агентов в слое вследствие неудовлетворительной работы внутреннего оборудования
[Alvarez et al., 2007b], В результате отдельные части слоя работают с перегрузкой, что
может вызвать образование зон перегрева, а другие используются недостаточно пол-
но. Все это ведет к общему снижению эффективности реактора и сокрашению цик-
ла работы.
7.3.1.1. Охлаждение в реакторах с неподвижным слоем
катализатора
Промышленные реакторы с неподвижным слоем (РНС) работают в адиабатическом
режиме, поэтому могут требовать охлаждения в зависимости от тепловыделения. Те-
пловая стабильность нужна для безопасной работы реактора, получения продуктов,
свойства которых отвечают требованиям спецификаций, и обеспечения приемлемой
длительности службы катализатора. Традиционный способ сохранить контроль над
температурой в РНС заключается в охлаждении горячего потока через смешивание
со сравнительно холодным газом. Поток обычно охлаждают водородом, но иногда
для этой цели применяют жидкости. Охлаждение осуществляется в так называемых
секциях охлаждения между слоями. По сути, это просто камеры, в которых горячий
поток смешивается с охлаждающей средой.
Охлаждение водородом типично для большинства процессов гидроочистки и ги-
дрокрекинга с несколькими каталитическими слоями. Охлаждающий газ отбирает-
ся из потока возвращаемого газа и вводится в секции промежуточного охлаждения
между слоями. Охлаждение водородом выгодно тем, что частично восполняет водо-
род, химически связанный в предыдущих слоях. Газовая фаза обогащается водоро-
дом, который подавляет коксообразование и одновременно снижает концентрации
таких ингибиторов реакций, как сероводород и аммиак [Alvarez et al., 2007а]. До-
ступность водорода для охлаждения зависит главным образом от скорости цирку-
ляции газа, которая во многом определяется экономическими соображениями: для
большей скорости циркуляции нужен более мощный компрессор. Установки ги-
дрокрекинга обычно имеют не менее пяти точек ввода охлаждающего газа, так как
рассчитаны на высокую кратность водорода к сырью (около 2000 н. м3/м3). При ма-
лых скоростях циркуляции скорость отбора охлаждающего газа ограничена также
проектной кратностью водорода к сырью в реакторе. Это обусловлено тем, что от-
бор газа уменьшает количество водорода, возвращаемого на вход в реактор, снижая
таким образом среднюю кратность водорода к сырью, особенно в первом катали-
тическом слое. Возвращаемый газ должен быть правильно распределен между вхо-
дом в реактор и точками охлаждения, чтобы избежать работы при кратности ниже
проектной и связанных с этим проблем (таких как повышенное коксообразование
и ухудшение результатов).
Глава 7. Моделирование каталитической гидроочистки
255
Процессы с жидкостным охлаждением распространены не так широко, как с во-
дородным [Alvarez et al., 2007а]. Применение жидкостей для охлаждения может ока-
заться целесообразным ввиду их большей теплоемкости и меньших затрат на ком-
примирование. Такие процессы можно подразделить на две широкие категории:
1) с распределенной подачей сырья и 2) с частичной рециркуляцией продукта. В пер-
вом случае сырье разделяют на ряд фракций и избирательно вводят их в различных
местах по длине реактора (рис. 7.5, а). Наиболее тяжелую фракцию подают, как пра-
вило, на вход в реактор, чтобы она контактировала с полным зарядом катализатора.
Легкие фракции вводят как охлаждающие потоки между нижерасположенными сло-
ями, где они смешиваются с более тяжелыми фракциями из предшествующих сло-
ев. Такой способ подачи сырья и порядок расположения слоев катализатора улучша-
ют избирательность процесса. В патентной литературе последних лет есть описание
нескольких процессов с распределенной подачей сырья, предназначенных для об-
лагораживания легких и средних дистиллятов, бензина каталитического крекинга,
вакуумного газойля и продуктов процесса Фишера—Тропша. Ко второй группе от-
носятся процессы, в которых охлаждение осуществляется возвращаемой частью
жидкого потока. Помимо охлаждения, непрореагировавшие вещества получают воз-
можность еще раз пройти через реактор (рис. 7.5, б).
Рис. 7.5. Общие схемы процессов с жидкостным охлаждением:
а — с распределенной подачей сырья; б — с частичной рециркуляцией потока
[Alvarez, A. et al., Energy Fuels, 21, 1133, 2007a]
Жидкостное охлаждение имеет свои недостатки: из-за увеличения объемной ско-
рости подачи сырья (т. е. снижения жесткости условий) ухудшаются результаты рабо-
ты реактора, снижается парциальное давление водорода (если происходит испарение
жидкости), падают концентрации реагентов, в реакционную смесь поступают но-
вые, более активные вещества. Компенсировать это можно либо увеличением разме-
ров реактора и количества катализатора, либо повышением температуры в реакторе.
Первый вариант явно ведет к увеличению капитальных затрат, а второй может со-
кратить длительность цикла или изменить требования к материалам конструкции
реактора.
256
Часть 3. Моделирование каталитических процессов
7.3.1.2. Внутреннее оборудование реакторов
Работоспособность РНС сильно зависит от конструкции внутреннего оборудования
[Alvarez et al., 2007b], Это оборудование равномерно распределяет реагенты и тепло-
вые потоки в объеме слоя, тем самым улучшая эффективность использования ката-
лизатора и способствуя нормальному протеканию процесса, отвечает за промежуточ-
ное охлаждение и обеспечивает защиту от загрязнения механическими примесями
[Ouwerkerk et al., 1999]. Неправильный выбор внутренней конструкции реактора ве-
дет к неравномерному распределению реагентов, неэффективному использованию
катализатора и ухудшению промежуточного охлаждения.
Внутренние устройства реактора гидропереработки схематически изображены
на рис. 7.6. Оборудование может располагаться на входе, между слоями и на выходе.
Входное оборудование состоит из верхней распределительной тарелки, фильтрующе-
го устройства и отсортированного слоя керамических шаров. Процессы с большим
тепловыделением проводят в многосекционных реакторах с промежуточными сек-
циями охлаждения. Секция охлаждения состоит из тарелки сбора жидкости, устрой-
ства ввода охлаждающей среды, камеры смешения охлаждающей среды с горячими
реагентами и тарелки вторичного распределения. Выходное оборудование включает
в себя устройство вывода, сборник жидкости, опорную решетку и слой керамических
шаров. На рис. 7.6 показаны также осевое и радиальное распределения температур
в реакторе. Распределение по оси соответствует типичному для установок гидропе-
реработки повышению температуры. Радиальный перепад температур вызывается
охлаждением и смешением в промежуточных секциях. Радиальное распределение
отражает эксплуатационные качества внутреннего оборудования реактора. Удов-
летворительной работе оборудования соответствует узкий перепад температур по-
сле охлаждения и распределения. Расширение перепада температур свидетельствует
о неудовлетворительном распределении. Если распределительные тарелки и секции
охлаждения не работают должным образом, негативные последствия, возникающие
при движении реагентов по оси реактора, накапливаются.
С точки зрения эффективности использования катализатора наиболее важный
элемент внутреннего оборудования — это распределительные тарелки, так как они
отвечают за равномерное распределение жидкости в слое. Традиционные распреде-
лительные устройства, такие как тарелки с паровыми патрубками, ситчатые или кол-
пачковые тарелки, в РНС работают неудовлетворительно. Напротив, распределите-
ли современных конструкций (например, высокодисперсные тарелки конструкции
Shell, тарелки VLT конструкции Topsoe, реакторные технологии Spider Vortex компа-
нии Exxon, сдвоенные тарелки Akzo Nobel и вихревые колпачковые тарелки конструк-
ции Fluor) обеспечивают полное орошение каталитического слоя.
На рис. 7.7 сравниваются конструктивные особенности некоторых распредели-
тельных тарелок. Шагом орошения тарелки называется расстояние между центра-
ми двух соседних точек орошения (рис. 7.7, а). Чем меньше шаг орошения, тем бли-
же к верху слоя достигается равномерное распределение, поскольку больше число
источников орошения. Большой шаг орошения уменьшает полезный объем ката-
лизатора, так как равномерное распределение достигается ниже по слою за счет
'Глава 7. Моделирование каталитической гидроочистки
257
Водород
Сырье
“ Охлаж-
дающий
водород
Вход .
Керами че- J
скиешары [
Каталитиче-
ский слой I
Опорная решетка •
Секция охлаждения
Керамические <
шары
Каталитиче-
ский слой
Опорная решетка
Выход
Распределение
и защита от обрастания
Сбор жидкости
Охлаждение
и смешение
Вторичное
распределение
Сбор жидкости
Продукт
Рис. 7.6. Внутренние устройства реактора гидроочистки: (—) — осевое распределение
температур; (••) — радиальное распределение температур. Обозначения: РРТ — ради-
альное распределение температур [Alvarez, A. et al., Energy Fuels, 21,1731,2007b]
в) r
Ситчатая тарелка
с газовыми патрубками
Тарелка современной
конструкции
Рис. 7.7. Конструктивные особенности распределительных тарелок:
а — влияние шага орошения на распределение жидкости; б — сравнение двух тарелок
с разными шагами орошения; е — пятна орошения, образуемые тарелками некоторых
конструкций [Alvarez, A. et al., Energy Fuels, 21, 1731, 2007b]
258 Часть 3. Моделирование каталитических процессов
радиальной дисперсии. На рис. 7.7, б сравнивается шаг орошения тарелки Vapor-Lift
конструкции Topsee и колпачковой тарелки. Очевидно, что у тарелки Topsoe точки
орошения расположены гуще, поэтому она обеспечивает значительно лучшее рас-
пределение жидкости. У тарелок традиционных конструкций из-за больших разме-
ров колпачков шаг орошения значительно шире. Этот недостаток способствует так-
же образованию мертвых зон (где нет источников жидкости) вблизи стенок реактора,
из-за чего значительное количество катализатора не используется и остается уязви-
мым для образования зон перегрева. Возможно, наиболее важная конструктивная
особенность — пятно орошения, так как его размеры определяют смачиваемую часть
катализатора в верхней части слоя. Тарелки традиционных конструкций (рис. 7.7, в)
дают дискообразное пятно орошения, смачивающее лишь поверхность, находящую-
ся непосредственно подточкой орошения (смачивается только 10—30 % площади по-
верхности слоя). Точки орошения тарелок современных конструкций работают как
форсунки с большим углом распыления и дают очень широкие пятна, покрывающие
всю поверхность слоя. В этих точках орошения энергия газа используется для увлече-
ния скопившейся на тарелке жидкости и диспергирования жидкой фазы.
Секция охлаждения представляет собой камеру, в которой горячие реагенты сме-
шиваются с охлаждающей средой. Она должна выполнять следующие функции;
1) вводить среду охлаждения; 2) смешивать ее с горячим потоком предшествую-
щего слоя; 3) вторично распределять жидкие и газообразные реагенты по сечению
последующего слоя. Как правило, секция охлаждения имеет устройство ввода ох-
лаждающей среды и определенные устройства для эффективного смешивания газа
и жидкости. Камера охлаждения обычно оборудуется отбойными лопатками и пе-
регородками для турбулизации и завихрения потоков, что улучшает контакт жид-
кости и газа. Примерами новейших технологий промежуточного охлаждения яв-
ляются секции охлаждения UFQ (Ultra-Flat Quench) конструкции Shell, Spider Vortex
конструкции ExxonMobil и внутриреакторные устройства Nautilus и Isomix кон-
струкции Chevron-Lummus.
7.3.2. Реакторы с движущимся слоем катализатора
В технологиях с движущимся слоем устранена необходимость останавливать работу
реактора вследствие полной потери активности катализатора. Главная особенность
ректоров с движущимся слоем (РДС) состоит в том, что они сочетают работу в режи-
ме вытеснения, присущем реакторам с неподвижным слоем (РНС), с возможностью
периодической замены части отработанного катализатора во время работы установ-
ки [Furimsky, 1998]. Поэтому РДС хорошо приспособлены для переработки тяжелого
сырья с высоким содержанием металлов и асфальтенов.
Название «движущийся слой» обусловлено способом, которым осуществляется
замена отработанного катализатора. Слой катализатора периодически частично вы-
тесняется вниз под действием сил тяжести. В верх реактора поступает свежий ката-
лизатор, а снизу отбирается отработанный. Сырье в реактор может подаваться как
и»
:|Мава 7. Моделирование каталитической гидроочистки
259
навстречу движению катализатора (противоточно), так и в одном направлении с ним
(прямоточно). Периодичность замены катализатора определяется скоростью потери
его активности. Замена обычно осуществляется 1-2 раза в неделю [Gosselink, 1998].
В РДС активность катализатора распределена по длине значительно эффектив-
нее, чем в РНС [Sie, 2001]. Периодическое добавление свежего катализатора уве-
личивает общую глубину гидродеметаллизации и гидродеасфальтизации. В отли-
чие от РНС, значительная часть задержанного металла и кокса покидает РДС вместе
счастицами катализатора. Эта особенность позволяет эксплуатировать РДС при бо-
лее высоких давлениях (20 МПа) и температурах (400—430 °C), чем типичные РНС
[Furimsky, 1998]. Поэтому они допускают большие концентрации металлов и других
примесей, чем РНС, даже при том же типе катализатора и в более жестких условиях.
Катализаторы РДС должны иметь улучшенные механические свойства и противосто-
ять измельчению и истиранию.
Наиболее представительными промышленными разработками, используюшими
технологию движущегося слоя, являются бункерный реактор процесса Нусоп ком-
пании Shell и реактор OCR {On-stream Catalyst Replacement — оперативная замена ка-
тализатора) компании Chevron [Ancheyta, 2007]. Бункерный реактор работает в пря-
моточном режиме, реактор OCR — в противоточном (сырье подается снизу). Схема
реактора OCR изображена на рис. 7.8. Противоточный режим эффективнее прямо-
точного в смысле расхода катализатора, так как свежее сырье сначала контактиру-
ет с отработанной частью катализатора внизу реактора, а затем уже частично деме-
таллизированное сырье контактирует с более свежим катализатором в верхней части
реактора. Реакторы с движущимся слоем работают во входных ступенях установок
гидропереработки остатков, осуществляя деметаллизацию и обеспечивая защиту ка-
тализатора гидрообессеривания и гидрокрекинга последующих реакторов с непод-
вижным слоем [Sie, 2001].
7.3.3. Реакторы с кипящим слоем катализатора
Реакторы с кипящим слоем (РКС) относятся к наиболее сложным технологиям ги-
Дропереработки. Они предназначены для облагораживания тяжелого и очень тяже-
лого неподготовленного сырья [Rana et al., 2007]. В РКС отработанный катализатор
непрерывно заменяется; таким образом, потеря активности катализатора не ограни-
чивает выбор сырья и степень превращения. Эта особенность РКС позволяет исполь-
зовать в них обычные высокоактивные катализаторы гидрообессеривания (ГДО)
и гидрокрекинга вместо входного катализатора гидродеметаллизации (ГДМ), приме-
няемого в реакторах гидроочистки в неподвижном слое. Процессы в кипящем слое
применяются главным образом для ГДМ/ГДО и гидрокрекинга остатков. В числе
примеров такой технологии, имеющих промышленное значение, можно указать про-
цесс Н-Oil (рис. 7.8) компании Axens и процесс LC-Fining компании Chevron Lummus.
Реакторы и рабочие параметры этих процессов весьма схожи, но имеют определен-
ные конструктивные различия.
260
Часть 3. Моделирование каталитических процессов
Реактор ОСЯ
Реактор гидропереработки в фазе суспензии
Катализатор
Рециркуля-
ционный
поддон
Граница
разрыхлен-
ного слоя
Граница
плотного
слоя
i Газожидкостный
4
□ Газ
ПТЗ Газожидкостная смесь
О Катализатор
► Кипящий слой
рующий продукт
РКС Н-ОН
Рис. 7.8. Реакторы с движущимся (РДС), кипящим (РКС) и суспензионным (РСС) слоем.
[Ancheyta, J., Reactors for hydroprocessing, in Hydroprocessing of Heavy Oils and Residua,
Ancheyta, J., Speight, J.G., eds., CRC Press, Taylor & Francis, Boca Raton, FL, 2007, p. 92]
Глава 7. Моделирование каталитической гидроочистки
261
Рабочая среда в реакторе гидропереработки в кипящем слое представляет собой
фехфазную систему, в которой циркулирующий газ всплывает сквозь турбулент-
ную суспензию, образованную смесью углеводородов и частиц катализатора [Rob-
inson, 2006]. Сырье и водород подаются снизу и, поднимаясь вверх, проходят через
разрыхленный («кипящий») слой частиц катализатора, удерживаемых во взвешен-
ном состоянии. Вверху реактора технологическая среда отделяется от катализатора
и поступает в газожидкостный сепаратор. Большинство частиц катализатора возвра-
щается в реактор. Часть продукта, скапливающаяся на рециркуляционном поддоне,
возвращается в низ реактора и смешивается со свежим сырьем. Циркуляция жидкости
в реакторе поддерживается насосом. Из-за больших скоростей рециркуляции можно
считать, что РКС ведет себя почти как реактор идеального смешения. Поэтому усло-
вия реакций близки к изотермическим. Катализатор заменяется периодически. Све-
жий катализатор подается через верх реактора, отработанный отбирается снизу.
Переработка тяжелых нефтей в РКС дает много преимуществ. Эти реакторы очень
гибки в работе, степень гидропревращения может доходить до 90 %об., а уровень со-
держания серы, металлов и азота в конечных продуктах низок. Механические при-
меси могут свободно перемещаться в кипящем слое, что уменьшает его загрязнение
и рост потерь давления [Furimsky, 1998]. Размер частиц не ограничен потерями дав-
ления, поэтому можно использовать более мелкий катализатор. Это значительно ос-
лабляет диффузионные ограничения и снижает склонность к закупорке устьев пор
отложениями металлов. Реактор работает почти в изотермическом режиме, что уве-
личивает избирательность к целевым продуктам, улучшает теплоперенос, сокраща-
ет риск местного перегрева и снижает выход кокса. В отличие от РНС, общая ак-
тивность катализатора, а следовательно, и качество продуктов остаются практически
неизменными в течение всего цикла.
Один из недостатков РКС — чрезмерный расход катализатора. Режим обратно-
го смешения кинетически менее благоприятен, чем режим вытеснения. Катализатор
должен иметь высокую механическую прочность, так как условия в реакторе способ-
ствуют интенсивному истиранию и размыванию частиц. Из-за малого соотношения
объема катализатора и объема жидкости этот реактор должен иметь большой объ-
ем. Высокая конверсия (более 50 %об.) приводит к усиленному образованию осадка.
Расчет и масштабирование РКС затрудняется сложностью их гидродинамики.
7.3.4. Реакторы с суспензионным слоем
Гидропереработка в реакторах с суспензионным слоем (РСС) относится к нетипич-
ным технологиям. В отличие от типичных технологий, в РСС используют дешевые
катализирующие добавки однократного применения, чтобы снизить затраты на ката-
лизатор [Furmisky, 1998]. Такие реакторы предназначены главным образом для пред-
варительного облагораживания тяжелых нефтей и битумов. Процесс обычно про-
водится в жестких условиях (при температуре выше 450 °C и давлении 14-21 МПа),
отвечающих режиму термического крекинга [Robinson and Dolbear, 2006]. Для
262
Часть 3. Моделирование каталитических процессов
подавления коксообразования сырье смешивают с порошкообразной добавкой
в низкой концентрации — обычно с углем или солями железа. Сегодня разработано
несколько технологий гидропревращения в фазе суспензии, но их промышленная ре-
ализация ограничена демонстрационными масштабами (примеры: Veba Combi-Crack-
ing компании KBR, Hydrocracking компании Canmet, Aurabon компании UOP и HDH
Cracking компании Intevep).
Упрощенная схема РСС показана на рис. 7.8. Сначала сырье смешивают с ката-
лизирующей добавкой и нагревают до температуры в реакторе. Затем подогретую су-
спензию смешивают с водородом и подают полученную смесь в низ реактора, кото-
рый представляет собой, по сути, просто емкость. Суспензия порошка в жидкости
благодаря малым размерам частиц ведет себя как однородная фаза, сквозь которую
всплывают пузырьки газа. Так как жидкость и катализатор движутся в одном направ-
лении, РСС можно считать работающим в режиме вытеснения [Speight, 2000]. От-
работанный катализатор покидает реактор в смеси с жидким продуктом, стремясь
в безопасной форме сконцентрироваться в непревращенной части.
В целом, благодаря высокой степени гидропревращения (более 90 %об.), низ-
ким затратам на катализатор и простоте конструкции реактора, процесс гидропре-
вращения в фазе суспензии может оказаться целесообразным для облагораживания
наиболее тяжелых видов сырья. Суспензионная фаза характеризуется улучшенным
теплопереносом и термически более стабильна. Главный недостаток технологии —
крайне низкое качество непревращенной части сырья (очень высокое содержание
серы и металлов).
7.4. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
7.4.1. Реакции гидроочистки
Превращения, которым подвергаются углеводороды в условиях процесса, невозмож-
но понять без знания химии реакций гидроочистки. Упрощенно процесс гидропе-
реработки можно представить как перенос водорода [Ancheyta and Speight, 2007b]:
последний подводится из внешнего источника и замещает гетероатомы, уменьшая
молярную массу углеводородов в исходной смеси посредством различных механиз-
мов гидрирования и гидрогенолиза. Но на деле явления, происходящие на молеку-
лярном уровне, значительно сложнее.
Детально описать реакции гидроочистки крайне трудно из-за сложности состава
промышленного сырья. Нефтяные фракции, в зависимости от интервала их кипения,
могут содержать от нескольких сотен до нескольких тысяч различных соединений.
Это подразумевает огромное множество последовательно и параллельно протекаю-
щих реакций. Ввиду высокой степени полидисперсности углеводородных смесей хи-
мическая активность реагентов, участвующих в таких реакциях, меняется в широких
пределах. Проблематичен и сбор данных о процессе на молекулярном уровне; анали-
тические возможности определения детального состава нефтяных фракций (особен-
но наиболее тяжелых) сильно ограничены.
7. Моделирование каталитической гидроочистки 263
Как было обозначено выше, реакции гидроочистки классифицируют по удаляе-
мым примесям (сера, азот, кислород, металлы и асфальтены). В случае реакций ГДО,
ГДА, ГДОк и ГДМ некоторые различия в преимущественной активности по отно-
шению к определенным реакциям можно объяснить исходя из энергий связи ато-
мов углерода с гетероатомами. Но такой подход не учитывает ни различных конфи-
турационных эффектов, возникающих в типичных для нефти сложных трехмерных
структурах, ни взаимодействий, которые возможны между реагентами и продуктами
в этой сложнейшей реакционной схеме [Speight, 2000]. Отсюда представляется це-
лесообразным описать химию реакций гидропереработки через модельные соедине-
ния и простые смеси. Как заметили Джирджис и Гейтс [Girgis and Gates, 1991], глав-
ным объектом исследований химии гидроочистки должны быть наиболее стойкие
(т. е. трудно реагирующие) соединения, находящиеся в тяжелых фракциях, как наи-
более важные для моделирования процесса и разработки катализаторов.
Природа химических превращений, происходящих при каталитической гидро-
очистке, во многом определяется целями проведения процесса. С утяжелением сы-
рья сложность процесса возрастает, так как соединения становятся более сложными
по структуре и менее активными. Именно по этой причине гидрооблагораживание
легких дистиллятов проводят в менее жестких условиях, чем переработку тяжелого
сырья. Вообще говоря, каталитическая гидроочистка может протекать параллельно
по двум механизмам: гидрогенолиза и гидрирования. Первый предусматривает не-
посредственный разрыв атомом водорода одинарной связи углерода и гетероатома.
К гетероатомам относятся атомы всех элементов, отличных от водорода и углерода,
в том числе атомы серы, азота, кислорода и металлов. При гидрировании водород
присоединяется к молекуле без разрыва каких-либо связей. Во многих случаях ги-
дрогенолизу должно предшествовать гидрирование, что будет описано ниже. Далее
кратко рассматриваются основные реакции гидрогенолиза и гидрирования в процес-
се каталитической гидроочистки.
7.4.1.1. Гидрообессеривание
В результате гидрообессеривания (ГДО) из углеводородов извлекается сера, которая
выделяется в виде H2S. Активность компонентов, содержащих серу, может сильно за-
висеть от структуры молекулы. В целом углеводородные типы по активности в реакци-
ях ГДО располагаются в следующем порядке: парафины > нафтены > ароматические
структуры. Наиболее активны меркаптаны и сульфиды, за которыми идут нафтены
и шестичленные ароматические структуры [Mochida and Choi, 2006]. Более стойки та-
кие пятичленные ароматические соединения, как тиофены; химически наименее ак-
тивны бензотиофены, дибензотиофены и алкилзамещенные дибензотиофены.
Удаление серы происходит непосредственно по механизму гидрогенолиза или
косвенно через предварительное гидрирование. На рис. 7.9 показаны два возможных
маршрута ГДО дибензотиофена. В прямом маршруте атом серы отщепляется и заме-
няется атомом водорода. Другой механизм требует насыщения одного из ароматиче-
ских колец перед удалением серы. Очищенный от серы продукт может подвергаться
дальнейшему гидрированию до полного насыщения молекул.
Фрагменты колец
отложения сульфидов металлов
Рис. 7.9. Примеры маршрутов реакций гидроочистки
264 Часть 3. Моделирование каталитических процессов
Глава 7. Моделирование каталитической гидроочистки
265
Ясно, что предпочтительным был бы прямой маршрут, так как в нем эффектив-
нее расходуется водород, но такой маршрут сильно зависит от структуры молекулы.
Конденсация ароматических колец или замещение алкильной группой вблизи ато-
ма серы стерически затрудняют молекулу и делают ее менее активной. На косвен-
ный маршрут структурные эффекты влияют меньше. Гидрирование ароматического
кольца уменьшает энергию связей атомов серы и углерода, что упрощает их разрыв.
Предполагают, что насыщение структуры облегчает доступ атома серы к активным
центрам катализатора [Mochida and Choi, 2006]. Но этот маршрут термодинамически
менее благоприятен, так как при низких давлениях и высоких температурах реакция
гидрирования ограничена равновесием [Vrinat, 1983]. Еще один аспект, влияющий
на ГДО, — сильное ингибирование адсорбируемыми продуктами реакции, в том
числе H2S, NH3 и органическими соединениями азота. Поэтому преобладание того
или иного механизма реакции будет зависеть от свойств сырья и условий процесса.
7.4.1.2. Гидродеазотирование
Азот в нефти содержится обычно в ароматических кольцах. Азотсодержащие веще-
ства подразделяются на основания (например, хинолин, акридин и пиридин) и неос-
новные соединения (индол, карбазол и пиррол). Для промышленности наибольший
интерес представляют азотсодержащие основания, так как неосновные соединения
быстро гидрируются до основных [Furimsky and Massoth, 2005]. Общепризнано, что
извлечение азота путем гидрогенолиза требует полного предварительного гидриро-
вания ароматических колец для ослабления сильной ароматической связи C-N [Gir-
gis and Gates, 1991]. В целом реакция гидродеазотирования (ГДА) остатков протека-
ет значительно труднее, чем ГДО, и уж в любом случае труднее, чем ГДМ и ГДАс,
поэтому требует более жестких условий. На рис. 7.9 изображен механизм реакции
ГДА хинолина, предложенный Джиойя и Ли [Gioia and Lee, 1986]. Предпочтитель-
ный (быстрый) маршрут реакции лежит через насыщение обоих колец перед разры-
вом связи С—N. Другой маршрут короче и расходует меньше водорода, но реакци-
ей по нему можно пренебречь, так как она протекает очень медленно [Dolbear, 1998].
Конечными продуктами в этом случае являются насыщенный углеводород и NH3.
Кроме термодинамических ограничений реакции ГДА, на активность азотсодер-
жащих соединений сильно влияет их молекулярная структура, мешающая абсор-
бции атома азота активным центром [Но, 1988]. Азотсодержащие основания явля-
ются также сильнейшими ингибиторами центров гидрирования, а следовательно,
и всех реакций, которые могут протекать по этому маршруту (например, ГДО). По-
этому для глубокого обессеривания дизельной фракции важно удалить азотсодержа-
щие ингибиторы.
7.4.1.3. Гидродеоксигенация
Реакции гидрооксигенации (ГДОк) уделяется значительно меньше внимания,
чем реакциям ГДО и ГДА, так как органические соединения кислорода содержат-
ся в нефти в низких концентрациях. Реакция ГДОк состоит в удалении кислорода
266
Часть 3. Моделирование каталитических процессов
из углеводородов и превращении его в воду. Соединения кислорода присутствуют
в виде органических фенолов, нафтола, фурана и их производных [Furimsky, 1983].
Как и в случае реакций ГДО и ГДА, активность кислородсодержащих соединений
уменьшается с молярной массой. Сообщалось, что реакция ГДОк протекает преиму-
щественно через частичное насыщение ароматических колец, а не через прямой ги-
дрогенолиз [Girgis and Gates, 1991].
7.4.1.4. Гидродеметаллизация
Никель и ванадий обычно содержатся в нефти в виде металлопорфиринов. Эти ве-
щества концентрируются в наиболее тяжелых фракциях, особенно в асфальтеновой.
При реакции ГДМ никель и ванадий переходят в соответствующие сульфиды и от-
лагаются на поверхности катализатора, вызывая необратимую потерю активности.
Янссене и соавторы [Janssens et al., 1996] предложили схему реакции ГДМ металло-
порфиринов, согласно которой молекула претерпевает гидрирование пиррольных
колец с последующим разрывом связи Ni-N или V-N путем гидрогенолиза и фраг-
ментацией порфириновой структуры (рис. 7.9). Экспериментальные данные показы-
вают, что ванадий удалить обычно легче, чем никель. Эта разница в активности была
объяснена тем фактом, что ванадиевые порфирины стремятся сконцентрироваться
на периферии молекулы асфальтена, в то время как никелевые в некоторой степе-
ни скрыты внутри ядра и поэтому требуют предварительного расщепления молекулы
[Beuther and Schmid, 1963]. Кроме того, выдвинуто предположение, что атом кисло-
рода, образующий перпендикулярную связь со структурой ванадиевых порфиринов,
улучшает адсорбцию каталитической поверхностью [Kobayashi et al., 1987].
7.4.1.5. Реакции гидрирования
Реакции гидрирования можно разделить на две категории: насыщение олефинов и ги-
дродеароматизация. Олефины гидрируются до их насыщенных гомологов практиче-
ски необратимо. Реакция насыщения ароматических соединений до нафтенов в про-
мышленных рабочих условиях является обратимой. На рис. 7.9 изображены реакции
гидрирования нафталина до тетрагидронафталина и бутенилбензола до н-бутилбензо-
ла. Ароматические соединения присутствуют в нефти в моно-, ди-, три- и полицикли-
ческой формах. Из-за стабильности бензольной молекулы и ее гомологов наиболее
трудно происходит насыщение моноароматических соединений. Полиароматические
соединения гидрируются последовательно кольцо за кольцом, причем равновесие
благоприятствует насыщению первого кольца [АН, 2007]. Гидрирование играет также
определяющую роль в реакциях ГДО и ГДА, о чем уже говорилось выше.
7.4.1.6. Гидрокрекинг
Цель реакций гидрокрекинга заключается в разрыве связей С—С с тем, чтобы
уменьшить молярную массу сырья. Химия реакций напоминает химию каталити-
ческого крекинга, но с одновременным гидрированием продуктов. В катализато-
рах функцию крекинга осуществляют кислотные центры, а функцию гидрирования
Глава 7. Моделирование каталитической гидроочистки
267
и дегидрирования — металлические. На рис. 7.9 изображен механизм гидрокрекин-
га и-парафина на центрах двух типов [Robinson and Dolbear, 2006]. Сначала молекула
парафина хемосорбируется металлическим центром и дегидрируется до соответству-
ющего олефина. Затем молекула олефина мигрирует к кислотному центру Брёнстеда
и протонируется до карбениевого иона. Парафиновый карбениевый ион может под-
вергнуться перегруппировке (например, сдвигу метиловой группы) с образованием
более стабильного карбониевого иона. Этот ион расщепляется в [3-позиции с образо-
ванием олефина и меньшего карбениевого иона. Образующийся олефин может под-
вергнуться дальнейшему крекингу или гидрироваться на металлическом центре.
Одним из важных аспектов гидрокрекинга является то, что концентрации азота
и полициклических ароматических соединений в сырье должны быть низкими, что-
бы избежать отравления катализатора и коксообразования. Известно, что органи-
ческие азотсодержащие основания сильно ингибируют функцию крекинга катали-
затора. Кроме того, для ограничения коксообразования необходимо поддерживать
парциальное давление водорода надостаточно высоком уровне, чтобы стабилизиро-
вать олефиновые продукты.
ТАЛЛ. Гидродеасфальтизация
Асфальтены представляют собой различающиеся по растворимости классы соеди-
нений в остаточных фракциях нефти. В условиях процессов гидропревращения ас-
фальтены подвергаются ряду сложных химических реакций. Механизм этих реакций
зависит от рабочих условий и структуры молекул. К примеру, реакция ГДАс очень
чувствительна к температуре, что можно объяснить изменением избирательности
к реакциям, когда при низких температурах преобладает гидрирование, а при вы-
соких — гидрокрекинг [Ancheyta et al., 2003]. Превращения асфальтенов тесно свя-
заны с удалением серы, азота и металлов, так как асфальтеновая структура включает
в себя множество молекул, содержащих гетероатомы [Mullins, 1995]. Недавно выдви-
галось предположение, что при гидродеасфальтизации крупные асфальтеновые агре-
гаты переходят в более мелкие с меньшей молярной массой [Gauthier et al., 2008].
В зависимости от степени гидропревращения процесс может начинаться с диссоциа-
ции асфальтеновых агрегатов с последующим деалкилированием путем отщепления
мелких боковых цепей и завершается расщеплением алкильных мостиков, соединя-
ющих крупные полиароматические ядра. В условиях высокой степени превращения
(при температуре выше 400 °C) остаются молекулы асфальтенов, сложенные крупны-
ми полиароматическими ядрами. Из-за этих молекул и образуется осадок. Показано,
что при высоких температурах свой вклад в превращение вносят также некаталити-
ческие реакции термического крекинга [Marafi et al., 2008].
7.4.2. Кинетика реакций
Нефтяное сырье состоит из смесей углеводородов, структура и молекулярный со-
став которых довольно сложны. В контексте моделирования это означает громадное
268
Часть 3. Моделирование каталитических процессов
множество взаимосвязанных реакций. Поэтому моделированию процессов превра-
щения углеводородов присущ высокий уровень сложности.
В попытках предсказать, хотя бы частично, структуру выхода продуктов процессов
нефтепереработки разрабатывались модели их кинетики. До сегодняшнего дня боль-
шинство кинетических моделей по практическим причинам по-прежнему представ-
лены дискретными компонентами. Кинетику реакций дискретных компонентов лег-
ко вывести из обычных аналитических сведений. Но достижения в методах анализа,
благодаря которым сегодня можно получать необходимую информацию о молеку-
лярной структуре сложных углеводородов, и прогресс в вычислительной технике по-
зволили разрабатывать детальные модели на молекулярном уровне [Klein et al., 2006].
При дискретном подходе сырье и продукты представляются в виде химических
сущностей, или псевдокомпонентов, определяемых стандартными методами ана-
лиза. Их обычно разграничивают в зависимости от целей моделирования. Напри-
мер, традиционная схема разграничения псевдокомпонентов при каталитическом
крекинге и гидрокрекинге формулируется исходя из так называемой интервальной
дискретизации [Но, 2008]. Она заключается в том, что сырье разбивается по темпе-
ратурам кипения или растворимости на конечное число псевдокомпонентов, кото-
рые состоят из групп молекул с относительно схожими физическими и химически-
ми свойствами. Псевдокомпоненты обычно определяют в соответствии с интервалом
кипения нефтяной фракции, представляющей интерес с практической точки зрения
(например, бензиновая фракция определяется интервалом от точки начала кипе-
ния до 204 °C, средние дистилляты — от 204 до 343 °C, вакуумный газойль — от 343
до 538 °C и вакуумный остаток — температурой выше 538 °C). Другой способ зада-
ния псевдокомпонентов заключается в разграничении их по классам углеводородов
(PIONA — парафины, изопарафины, олефины, нафтены и ароматические углеводо-
роды; SARA — предельные углеводороды, ароматические углеводороды, смолы и ас-
фальтены), иногда с разбиением на субкомпоненты по числу атомов углерода (па-
рафины С5, парафины С6 и т. д.), как при каталитическом риформинге бензиновой
фракции. Результирующие химические сущности связываются сравнительно про-
стой реакционной схемой.
Другой тип дискретизации встречается, когда кинетику реакций группы опреде-
ленных химических веществ формулируют как кинетику единого псевдокомпонен-
та. Такой подход типичен для представления кинетики реакций гидроочистки, где
псевдокомпоненты определяют как общие химические классы (например, как об-
щую серу, азот, ароматические соединения, металлы (Ni + V)), а иногда и как клас-
сы растворимости (например, как асфальтены). В этом случае дискретизация более
радикальна, поскольку каждый агрегат содержит, как правило, полидисперсную вы-
борку молекул. Более детальное представление можно получить объединением двух
упомянутых выше методов дискретизации [Verstraete et al., 2007]: первый набор псев-
докомпонентов определяется методом фракционирования и подразделяется далее
по элементному составу (сера, азот и т. д.). Таким путем можно предсказать динамику
продуктов гидрокрекинга, равно как и их атомный состав.
Моделирование кинетики на молекулярном уровне предлагает описывать процесс
исходя из его реальной химии. Этот подход учитывает превращение молекул сырья
0! а в a 7- Моделирование каталитической гидроочистки
269
каждого отдельного типа. Разработка таких моделей требует знания основополагаю-
щих механизмов реакций и невозможна без детального описания сырья. До сих пор
опубликовано лишь несколько моделей гидроочистки такого характера [Frament, 2004;
Ghosh et al., 2009; Lypez-GarcHa et al., 2010]. Все они сосредоточены на ГДО, ГДА
И ГДАр сырья относительно легких типов, таких как бензиновая фракция, газойль
^легкий рецикловый газойль, о молекулярной структуре которого имеется достаточ-
но сведений. Формулировки этих моделей позволяют отслеживать превращения всех
доступных для наблюдения веществ, но не описывают процесс на механистическом
уровне. В связи с огромным количеством реакций в подобных моделях применяются
разнообразные методы уменьшения числа параметров скоростей, основанные боль-
шей частью на структурном вкладе. В этом смысле моделирование гидрокрекинга идет
на шаг впереди, так как на механистическом уровне он уже смоделирован. Фромен
и соавторы ввели концепцию «единичного события», исходя из которой некоторые
кислотно-катализируемые процессы могут быть представлены элементарными ста-
диями превращения карбениевого или карбониевого иона [Baltanas et al., 1989]. Этот
подход был применен к моделированию кинетики превращений такого промышлен-
ного тяжелого сырья, как вакуумный газойль [Kumar and Froment, 2007].
Эти два подхода к моделированию представляют противоположные крайности.
Различия между ними достаточно очевидны. Безусловно, желательно иметь сред-
ства, позволяющие прогнозировать в деталях выход продуктов процессов нефтепере-
работки. Но это поистине грандиозная задача, так как ее решение требует гигантских
вычислительных мощностей и изощренных аналитических методов для измерения
характеристик сложного сырья на молекулярном уровне. До настоящего времени мо-
делирование на молекулярном уровне остается ограниченным упомянутыми выше
сравнительно легкими типами сырья. «Экстраполяция» этого подхода на тяжелые
нефти и остатки перегонки остается под вопросом и продолжает быть предметом
усилий исследователей. Помимо всего, модели молекулярного уровня, как прави-
ло, не позволяют использовать программные средства моделирования гидродинами-
ки, а также контроля и управления производственным процессом, так как для это-
го необходимы запредельные вычислительные мощности [Но, 2008]. Ситуация ясна:
чем большее число реагентов учитывает модель, тем шире совокупность реакций, тем
большее число параметров скоростей необходимо вычислять и тем выше объем тре-
буемых экспериментальных данных.
В то же время дискретизация — наиболее распространенный подход к моделиро-
ванию кинетики, так как она радикально упрощает задачу. И действительно, дискрет-
ные модели уже несколько десятков лет используются для расчета процессов превра-
щения углеводородов. Многие промышленные установки до сих пор проектируют
на основе этого подхода. Он широко применяется и в других областях, в том числе
при отборе катализаторов, контроле в режиме реального времени, при управлении
процессами, в исследованиях базовых процессов и при динамическом моделирова-
нии. Но дискретным моделям присуща проблема: кинетика реакций в них не опи-
рается на химию процесса. Им сопутствует существенная потеря информации о ка-
ждом из многокомпонентных агрегатов. Коренным недостатком дискретной модели
270
Часть 3. Моделирование каталитических процессов
кинетики является то, что, независимо от числа псевдокомпонентов, параметры ско-
ростей определяются составом сырья. Таким образом, модели этого типа предназна-
чены для частных случаев и не допускают экстраполяцию. Всякий раз, когда меняет-
ся состав сырья или хотя бы схема процесса, модель приходится переформулировать,
проведя новый цикл экспериментов. Несмотря на все эти ограничения, сложность ре-
ального сырья, особенно тяжелого, наталкивает на мысль о том, что применение дис-
кретных моделей в исследованиях кинетики гидроочистки и гидрокрекинга будет про-
должено, по меньшей мере до тех пор, пока прогресс в методах анализа не позволит
напрямую определять структурные свойства столь сложных молекул, как асфальтены.
Что касается уравнений скоростей, большинство исследований кинетики на мо-
дельных соединениях соглашается с тем, что реакции гидропереработки следуют
простой кинетике первого порядка. Однако реакции гидроочистки реального сырья
имеют более высокий или даже дробный порядок. Обобщенное уравнение скорости
данной реакции гидрооблагораживания имеет вид
г=*/7"С£, (7.1)
где kj — Аррениусова константа скорости у'-й реакции; С. и Сн_, — концентрации со-
ответственно /-го псевдокомпонента и водорода; тх — порядок реакции /-го псевдо-
компонента; т2 — порядок реакции водорода.
Кинетическая модель в такой форме явным образом учитывает влияние концен-
трации водорода на общую скорость реакции. Это базовое выражение применяется
ко всем реакциям гидроочистки и к множеству типов сырья.
Для учета разницы активностей, когда сырье состоит из широкого спектра хими-
ческих веществ, псевдокомпоненты можно разбить на активные и стойкие соедине-
ния. Тогда результирующее выражение объединит в себе две параллельные реакции
первого порядка:
г=^лСЛ(1-у)к.2С., (7.2)
где у — доля активных веществ; (1 — у) — доля неактивных веществ; к. к — кон-
станты скоростей реакций каждой из фракций.
Следует отметить, что в этом случае концентрация водорода входит в выражение не-
явно, но его можно видоизменить так, чтобы явным образом учитывать эту переменную.
В других случаях можно учитывать ингибирующий эффект определенных сое-
динений (в том числе H2S, NH3, органических соединений азота и даже асфальте-
нов), обусловленный параллельно протекающей адсорбцией. Выделяя из констан-
ты скорости член, отвечающий за адсорбцию, можно получить следующее уравнение
Ленгмюра-Хиншельвуда:
где К, — коэффициент адсорбции вещества к', С.. — концентрация адсорбируемого ве-
щества к.
Глава
7, Моделирование каталитической гидроочистки
271
Предполагается, что адсорбция происходит на двух центрах; отсюда показатель 2
в знаменателе.
Помимо этих трех базовых форм, в литературе представлен длинный перечень вы-
ражений скоростей реакций, являющихся, по сути, вариантами уравнений (7.1 )-(7.3).
Например, предлагаются упрощенные реакционные схемы для представления, в за-
висимости от доступности сведений о маршрутах реакций, обратимого гидрирования
ароматических колец, последовательного гидрирования полиароматических колец,
параллельных и последовательных реакций гидрокрекинга.
Эффект группирования широкого спектра компонентов в единый псевдокомпо-
нент больше всего отражается на порядке реакции п. В дискретных моделях кинети-
ки наблюдаемые порядки реакций составляют от 0,5 до 2. Порядки реакций выше 1
обычно объясняются существенными различиями в активности молекул в пределах
псевдокомпонентов. На рис. 7.10 показаны константы скоростей первого порядка
гидрообессеривания бензотиофена, дибензотиофена и его алкилзамещенных про-
изводных. Порядок разницы в активности между классами близок к единице. Это
говорит о значительном влиянии структуры на активность молекул. Таким образом,
дискретизированную смесь можно представить набором параллельно протекающих
реакций первого порядка активных и стойких веществ. Тогда второй порядок реак-
ций обусловлен «живучестью» в реакционной системе наиболее стойких веществ
[Но and Aris, 1987].
Рис. 7.10. Значения констант первого порядка скоростей реакций ГДО
различных соединений серы [Ancheyta, 2007]
С другой стороны, для гидроочистки тяжелой нефти и остатков характерна ки-
нетика дробного порядка [Alvarez and Ancheyta, 2008]. Безусловно, дробный поря-
док реакций труднее объяснить. Наиболее убедительное объяснение этому явлению
Til
Часть 3. Моделирование каталитических процессов
можно найти в работе Грея и соавторов [Gray et al., 1995]. Исходя из математическо-
го анализа простой схемы из двух параллельных реакций (прямого гидрогенолиза ти-
офена и гидрирования тиофена с последующей прямой десульфуризацией), авторы
делают вывод о том, что группирование этих двух маршрутов реакций в кинетиче-
ское выражение единого псевдокомпонента может дать в прямом смысле слова лю-
бой порядок реакций, если в одном из маршрутов, каким в данном случае является
обратимое гидрирование, имеет место частичное превращение. Этот вывод можно
напрямую применить к тяжелым фракциям, поскольку всякая сущность химических
превращений обычно объединяет в себе и последовательные, и параллельные реак-
ции (гидрирование, гидрогенолиз и гидротермическую очистку).
При разработке и формулировании дискретных моделей трудно отталкивать-
ся исключительно от кинетических экспериментов. Базовая совокупность экспери-
ментов с переменным временем пребывания и температурой позволяет определить
лишь глобальные значения порядка реакции и энергии активации. Порядок реак-
ции водорода определяется по влиянию изменения общего давления при постоян-
ных значениях других параметров. Коэффициенты адсорбции при учете эффектов
ингибирования (например, H2S или NH3) определяются изменением начальных кон-
центраций этих веществ — опять-таки при постоянных значениях остальных параме-
тров. В идеальном случае для получения истинных значений параметров скоростей
условия эксперимента должны гарантировать отсутствие явлений массоперено-
са (малый размер частиц). Но, как правило, кинетические испытания в целях мас-
штабирования и разработки процессов проводятся с использованием катализаторов
промышленной крупности в реакторах с орошаемым слоем, чтобы эмулировать про-
мышленные условия [Bej, 2002]. Они дают относительные модели кинетики, которые
неявным образом учитывают влияние массопереноса внутри частиц. Однако эти мо-
дели (вследствие гидродинамических эффектов при разных масштабах или величи-
нах производительности процесса) трудно поддаются масштабированию и поэтому
требуют введения определенного рода поправок (например, эффективности смачи-
вания катализатора). Более подробные сведения о кинетике отдельных реакций ги-
дроочистки и об определении сопутствующих параметров излагаются далее (в этой
и последующих главах).
7.4.3. Термодинамика
Для прогнозирования химического равновесия необходимо знать термодинамику ре-
акций гидропроцессов. Подробные термохимические данные об отдельных реакциях
применительно к сложному сырью, как правило, отсутствуют [Girgis and Gates, 1991].
В литературе предлагаются некоторые обобщения о реакциях каждого типа, осно-
ванные на теоретических расчетах и результатах немногочисленных исследований
на модельных соединениях. Приближения констант равновесия в целях моделирова-
ния в большинстве случаев находят с помощью методов вклада групп.
В табл. 7.1 приведены значения стандартной теплоты и констант равновесия от-
дельных реакций гидроочистки. Положительные константы равновесия реакций
Глава 7. Моделирование каталитической гидроочистки 273
ГДО и ГДА указывают на то, что в применяемых на практике интервалах рабочих
условий эти реакции необратимы и при наличии стехиометрических количеств во-
дорода могут продолжаться до завершения. С другой стороны, гидрирование арома-
тических углеводородов в типичных условиях гидропереработки обратимо и полное
превращение недостижимо, так как при высоких температурах равновесие смещает-
ся в сторону дегидрирования. Со снижением температуры реакции значения кон-
стант равновесия в целом склонны уменьшаться, что согласуется с экзотермиче-
ской природой реакции гидроочистки. В случае ГДО величины констант становятся
значительно меньше единицы лишь при температурах, существенно превышающих
применяемые на практике (выше 425 °C) [АП, 2007]. Видно также, что энтальпии ре-
акций существенно разнятся. В пределах каждого класса реакций количество выде-
ляемой теплоты обычно зависит от величины стехиометрического расхода водорода.
Таблица 7.1
Значения стандартных энтальпий и констант равновесия типичных реакций гидроочистки
Резкими ДГР •g*.
200 °C 300 °C 400 °C
Гидрообессеривание
c,h7-sh + h2«->c.hs + h.s -57 6,92 5,87 5,15
Тиофен + ЗН2 <-ч- н-С4Н|0 + H2S -262 14,13 9,33 6,04
Бензотиофен + Н2 <-> этилбензол + H2S -203 16,65 12,85 10,20
Дибензотиофен + 2Н2 бифенил + H2S -148 15,23 12,50 10,61
ГИдродеазотнрование
Индол + ЗН2 <-ч- этилбензол + NH3 -49 — 7,8 5,0
Карбазол + 2Н2 бифенил + NH3 -126 — 6,8 5,1
Пиридин + 5Н2 <-ч- н-пентан + NH3 -362 — 8,9 4,4
Хинолин + 4Н2 <-ч- пропилбензол + NH3 -272 — 7,0 3,3
Пщродеароматизация
Нафталин + 2Н2 <-> тетралин -140 1,26 -1,13 -2,80
Тетралин + ЗН2 <-» транс-декалин -193 0,74 -2,95 -5,56
Циклогекс илбензол + ЗН, <-> циклогексилгексан -295 2,47 -1,86 -4,91
Фенантрен + 4Н2 октагидрофенантрен -251 1,16 -3,64 -7,12
Источник: [Ancheyta, 2011].
9 Стандартная энтальпия реакции, килоджоулей на 1 моль органического реагента.
Реакции гидрирования при переработке углеводородов подразделяются на реак-
ции насыщения при крекинге алканов и разрыве колец, реакции гидродеаромати-
зации и реакции насыщения олефинов [Jaffe, 1974]. Реакции первого типа связаны
с гидрогенолизом сг-связи С-С в нафтеновом кольце или боковой алкильной цепи.
Это наименее экзотермичная из всех реакций гидрирования, так как разрыв о-связи
274
Часть 3. Моделирование каталитических процессов
С-С требует большого количества энергии. Реакция насыщения олефинов проте-
кает быстро, выделяет много теплоты (105-120 кДж на 1 моль Н2) и в практических
условиях необратима. Ароматические кольца вследствие стабилизации структуры
эффектом мезомерии труднее поддаются гидрированию, чем олефины. Реакция их
гидрирования также менее экзотермична, чем насыщение олефинов, поскольку на-
сыщение сопряженных ет-связей С-С ароматических колец до о-связей поглощает
больше энергии, чем насыщение несопряженных от-связей С-С олефинов. Хотя те-
пловыделение при гидрировании ароматических колец возрастает пропорционально
расходу водорода, количество теплоты, выделяемой на 1 моль водорода, остается до-
вольно постоянным (58-70 кДж на 1 моль Н2).
Как уже говорилось, гидрирование полициклических соединений до насыщенных
конечных продуктов осуществляется последовательно кольцо за кольцом. Установле-
но, что равновесие благоприятствует насыщению первого кольца, тогда как послед-
нее кольцо из-за резонансной стабилизации насыщается труднее всего [АН, 2007].
Гидрирование ароматических соединений имеет место также в реакциях ГДО, ГДА
и даже ГДМ сложного сырья. Если в ароматическую структуру встроены гетероато-
мы, реакция обычно протекает по маршруту гидрирования, когда до разрыва связи
углерода с гетероатомом сначала гидрируется по меньшей мере одно соседнее арома-
тическое кольцо. При высоких температурах и низких парциальных давлениях водо-
рода превращение по этому маршруту может тормозиться из-за низкой равновесной
концентрации частично гидрированных промежуточных продуктов. Поэтому на об-
щую скорость этих реакций может неблагоприятно влиять термодинамика гидриро-
вания ароматических соединений.
Обратимость реакций гидрирования ароматических соединений — фактор, важ-
ный для промышленной практики. Максимальная степень превращения арома-
тических соединений сильно зависит от рабочих условий, особенно температуры
реакции, а также от типа и количества присутствующих в сырье ароматических сое-
динений, объемной скорости подачи сырья, парциального давления водорода и типа
катализатора. Повышение температуры реакции с целью увеличить глубину ГДО
и ГДМ уменьшает степень равновесного превращения ароматических соединений.
Это особенно важно, например, при ГДО газойля, так как для приведения продук-
та в соответствие требованиям спецификаций желательно одновременно снизить со-
держание серы, азота и ароматических соединений. Концентрацию этих примесей
необходимо понизить также при подготовке сырья каталитического крекинга в псев-
доожиженном слое (FCC), чтобы предотвратить закоксовывание кислотных катали-
заторов и в то же время получить чистые продукты. Поэтому условия реакций следу-
ет подобрать так, чтобы сбалансировать равновесие гидрирования со степенью ГДО
и других реакций. Общее решение состоит в проведении процесса при повышенном
парциальном давлении водорода, что позволит увеличить равновесные концентра-
ции насыщенных продуктов.
Большинство этих операций (за исключением гидроочистки бензиновой фрак-
ции и керосина) рассматриваются как крайне экзотермичные. Скорость роста тем-
пературы при движении вниз по слою катализатора зависит главным образом
7. Моделирование каталитической гидроочистки
275
да концентрации ароматических углеводородов и соединений серы, а также от глу-
бкны соответствующих реакций. Сообщалось, что прирост температуры в некото-
рых установках гидроочистки может превышать 100 °C [Robinson and Dolbear, 2006].
' Для проектирования безопасного реактора необходимы данные по тепловыде-
ЛСНию. Методом вкладов групп можно получить достаточно хорошие приближе-
ния общего тепловыделения от каждой отдельной реакции, но это требует разработ-
ки сложной модели кинетики на молекулярном уровне. В литературе приводятся,
Как правило, сведения об отдельных реакциях гидроочистки (обычно ГДО, ГДА
И ГДАр), полученные на модельных соединениях. В отдельных работах даются све-
дения о тепловыделении реакций конкретных процессов, которые можно осмотри-
тельно использовать при моделировании реакторов. Суммарная теплота реакции —
ООДгоняемый параметр, выводимый из тепловых балансов нескольких аналогичных
процессов гидроочистки. В нем учитывается вклад всех реакций: ГДО, гидрирова-
ния и т. д. [Dohler and Rupp, 1987]. Для точного воспроизведения промышленных
данных любого конкретного процесса значения этих параметров обычно требуется
Подгонять. Эти значения обычно приводятся в виде количества выделяемой теплоты,
приходящейся на единицу определенного реагента (удаленной серы, крекирован-
ного углеводорода, израсходованного водорода и т. д.). Например, Моэнти и соав-
торы [Mohanty et al., 1991] указывают, что теплота реакции гидрокрекинга вакуум-
ного газойля составляет —42 МДж на 1 кмоль израсходованного водорода, тогда как
Шах и Параскос [Shah and Paraskos, 1975] сообщают о следующих величинах тепло-
выделения в различных процессах: -7820 кДж на 1 кг удаленной серы для ГДО остат-
ков; -582 кДж на 1 кг превращенного газойля для гидрокрекинга газойля; -8147 кДж
на 1 кг удаленного азота для ГДА сланцевой нефти.
7.4.4. Катализаторы
Типичные промышленные катализаторы гидроочистки состоят из активных ме-
таллов в виде оксидов (например, молибдена и вольфрама) с такими промотора-
ми, как кобальт или никель, на подложке (т. е. носителе) из гамма-оксида алюми-
ния (у-А12О3). Оксид алюминия предпочтителен как материал носителя благодаря
гибким структурным свойствам, возможности высокодисперсного распределения
активных металлов, высокой механической прочности и низкой стоимости [Mochi-
da and Choi, 2006]. Уровень кислотности гамма-оксида алюминия сравнительно не-
высок, что снижает глубину реакций крекинга и коксообразование. Активная фаза
образуется распределением активных металлов в порах носителя путем пропитыва-
ния. Активная фаза обычно состоит из молибденового прекурсора (15-20 %масс.)
и, в зависимости от назначения катализатора, никелевого или кобальтового промо-
тора (1—5 %масс.). Основная функция промотора заключается в существенном улуч-
шении активности и избирательности активного металла.
Катализаторы гидрокрекинга характеризуются двойной функциональностью.
Функцию крекинга промотирует высококислая подложка, а за функции гидрирования
276
Часть 3. Моделирование каталитических процессов
и дегидрирования отвечает фаза активного металла. Как правило, подложка изготав-
ливается из аморфных оксидов кремния и алюминия либо из кристаллических це-
олитов (X- и К цеолиты), причем кислотность последних выше [Robinson and Dol-
bear, 2006]. Функцию гидрирования могут катализировать благородные металлы
(палладий и платина) или никель-молибденовый и никель-ванадиевый сульфи-
ды. Благородные металлы обладают наивысшей активностью гидрирования, но они
весьма дороги и легко отравляются сероводородом. Фаза металла отвечает за иници-
ирование крекинга путем дегидрирования насыщенных углеводородов до соответ-
ствующих промежуточных олефинов и стабилизацию конечных продуктов крекин-
га путем гидрирования [Gruia, 2006]. Во избежание чрезмерного коксообразования
нестабильными продуктами необходим компромисс между функциями металла
и крекинга.
Чтобы правильно подобрать катализатор, нужно тщательно проверить химические
свойства сырья и ожидаемых продуктов. Для обычной гидроочистки дистиллятов
(бензиновой фракции, керосина и газойля) наиболее важны удельная поверхность
и состав активной фазы. Для ГДО в промышленности традиционно применяются ко-
бальт-молибденовые катализаторы. Для реакций насыщения и ГДА хорошо подхо-
дят никель-молибденовые катализаторы, характеризуемые лучшей способностью ги-
дрирования. Поэтому если процесс протекает по маршруту гидрирования (т. е. при
очистке газойля, содержащего ароматические соединения с гетероатомами серы
и азота), промотированные никелем катализаторы предпочтительнее кобальт-мо-
либденовых. Заметной способностью гидрирования обладают никель-вольфрамовые
катализаторы, но из-за высокой стоимости они в промышленной практике применя-
ются редко. Реакторы промышленных установок гидрокрекинга вакуумного газойля
обычно имеют входной слой высокоактивных никель-молибденовых катализаторов,
защищающий катализаторы гидрокрекинга от прекурсоров кокса и отравления азо-
том и серой.
Труднее всего выбрать катализатор для тяжелой нефти и остатков. Помимо базо-
вых функций гидрирования и гидрокрекинга, такой катализатор должен обладать
способностью сопротивляться потере активности, вызываемой отложением метал-
лов и кокса. В этом смысле главную роль в обеспечении приемлемой длительности
цикла службы катализатора играют его структурные свойства (пористость). Кислот-
ность носителя должна обеспечивать достаточно умеренный гидрокрекинг, чтобы
избежать чрезмерного закоксовывания и образования осадка. Наивысшую произво-
дительность обычно дает сочетание избирательных катализаторов с разными харак-
теристиками. Типичная сортированная каталитическая система содержит входной
слой макропористого катализатора ГДМ, промежуточный слой сбалансированно-
го катализатора ГДМ и ГДО, а также высокоактивный выходной слой катализатора
ГДО, ГДА и гидрокрекинга. Для процесса наиболее важен входной катализатор ГДМ.
Его задача — разъединить молекулы асфальтенов и удалить никель и ванадий с тем,
чтобы катализаторы последующих слоев могли работать с частично облагороженным
сырьем. Скорость реакции ограничена диффузией крупных молекул внутри частиц,
поэтому размеры пор частиц играют важную роль [Furimsky, 1998]. Если размеры пор
Шва 7. Моделирование каталитической гидроочистки 277
Слишком малы, их устья быстро забиваются, что ведет к преждевременной останов-
ке работы. Общую стабильность и длительность цикла службы катализатора опреде-
ляет его металлоемкость, т. е. способность хранить определенное количество метал-
лов. Средняя секция содержит в себе более активный катализатор, обеспечивающий
частичное ГДО и дополнительное удаление металлов. В выходном слое применяет-
ся типичный высокоактивный катализатор, осуществляющий ГДО, ГДА и гидрокре-
кинг. Как правило, в сортированных системах размеры пор по направлению к выходу
уменьшаются, а активность возрастает.
Чтобы обеспечить оптимальную работу установки, необходимо тщательно вы-
бирать не только состав катализатора и его структурные свойства, но также форму
Ж размеры его частиц. Для гидроочистки дистиллятов хорошо подходит катализатор
с частицами таких традиционных форм, как шары и таблетки. Для гидрооблагора-
Живания тяжелого сырья эти формы не подходят, так как вследствие диффузионных
©граничений крупные молекулы не имеют возможности достаточно глубоко прони-
кать внутрь частиц. Поэтому необходимо сократить диффузионный путь, что дости-
гается уменьшением размеров частиц. Но уменьшение размеров частиц катализатора
в реакторах с неподвижным слоем часто вызывает рост потерь давления. На практике
эту проблему решают использованием катализатора с частицами других форм, в том
числе трилистников и четырехлистников. Эти формы характеризуются увеличенной
площадью поверхности, за счет чего сокращается диффузионный путь.
Общие эксплуатационные качества промышленных катализаторов оценивают
по качеству продуктов, избирательности процесса, начальной активности и стабиль-
ности в течение цикла [Gruia, 2006]. Два последних фактора можно отнести к эксплу-
атационным критериям, так как они определяют график управления рабочей тем-
пературой в течение цикла. От начальной активности катализатора зависит выбор
Начальной температуры цикла, при которой качество продуктов отвечает требовани-
ям, Стабильность катализатора наиболее важна в середине цикла и в его конце, так
как она определяет скорость повышения температуры, при которой качество про-
дуктов остается в пределах требований спецификаций. Стабильность особенно важ-
на при переработке тяжелого сырья, когда необходимо обеспечить приемлемую дли-
тельность цикла, несмотря на быструю потерю активности из-за отложения металлов
йкокса,
7.5. ПАРАМЕТРЫ ПРОЦЕССА
Точный выбор рабочих условий обеспечивает наилучшую производительность про-
цесса. Главные параметры (температуру, давление, объемную скорость подачи сырья
и кратность водорода к сырью) подбирают исходя из целей процесса гидропереработ-
ки. Типичные условия различных процессов показаны в табл. 7.2. Большинство этих
процессов, за исключением гидрокрекинга остатков в кипящем слое катализатора,
проводится в реакторах с неподвижным слоем. Естественно, чем тяжелее сырье, тем
жестче должны быть условия. Дистилляты перерабатывают в более мягких условиях,
чем остаточное сырье. Гидрокрекинг требует более жестких условий и значительно
278 Часть 3. Моделирование каталитических процеа
большего расхода водорода, чем гидроочистка. В последующих разделах кратко рас-
сматривается влияние главных параметров процесса.
Таблица 7.2
Типичные рабочие условия различных нроцессов гидроочистки и гидрокрекинга
Процесс Г,”С Рн,, МПа LHSV. ч-‘ Кратность Н2 к сырью, н. м3/м3 Расход Hj, н. м3/м3
Гидроочистка
Бензиновая фракция 320 1-2 3-8 60 2-10
Керосин 330 2-3 2-5 80 5-15
Газойль 340 2,5-4 1,5-4 140 20-40
Вакуумный газойль 360 5-9 1-2 210 50-80
Остаток 370-410 8-13 0,2-0,5 > 525 100-175
Гидрокрекинг
Вакуумный газойль 380-430 9-20 0,5-1,5 1000-2000 150-300
Остаток 400-440 12-21 0,1-0,5 1000-2000 150-300
Примечание: LHSV— объемная скорость подачи сырья.
Источник: [Kundu, A. et al. Rev. Chem. Eng., 19, 531, 2003; Robinson and Dolbear, Hydrotreat-
ing and hydrocracking: Fundamentals. Practical Advances in Petroleum Processing, Volume 1 (C.S, Hsu,
P.R. Robinson, Eds.), Springer, New York, 2006].
7.5.1. Температура реакций
Температура реакций — безусловно, важнейший параметр процесса. К ней очень
чувствительны глубина и избирательность реакций гидроочистки и гидрокрекинга,
так как с увеличением температуры константы их скоростей возрастают экспоненци-
ально. Поэтому для достижения требуемой избирательности температуру подбирают
в соответствии с химией процесса. Согласно табл. 7.2, гидропереработку легких ди-
стиллятов проводят в интервале температур, достаточно высоких для почти полного
завершения реакций ГДО и ГДА (т. е. при 320—340 °C). При более высоких температу-
рах возможно расщепление легких углеводородов вследствие термического крекинга
и, в случае гидроочистки газойля, неблагоприятного смещения равновесия реакции
гидрирования ароматических соединений. Остатки и вакуумный газойль более стой-
ки по своей природе, поэтому их гидропереработка требует более высоких темпера-
тур. В этом случае для увеличения равновесной концентрации насыщенных колец
важно проводить процесс при повышенных парциальных давлениях водорода. При
наивысших температурах проводятся процессы гидрокрекинга, так как для разрыва
связей С-С требуется больше энергии, чем для реакций гидроочистки.
Повышение температуры дает несколько побочных эффектов, которые нуждаются
в тщательной оценке. Даже небольшое превышение температуры выше приемлемых
значений ведет к потере избирательности и чрезмерной деактивации катализатора.
la 7. Моделирование каталитической гидроочистки 279
Поэтому для каждого отдельного случая существует определенная предельно допу-
стимая температура. Увеличение температуры реакции неизбежно ускоряет коксо-
Образование вследствие роста скорости конденсации нестабильных продуктов кре-
КИНга. Высокие температуры при гидропереработке остатков увеличивают глубину
но в то же время ускоряют необратимую потерю активности из-за осаждения
металлов. Температуры выше 410 °C способствуют термическому крекингу ценных
углеводородных компонентов с образованием значительных количеств низкомоле-
кулярных жидкостей и газов. Кроме того, крекинг остатков в жестких условиях мо-
жет вызвать образование осадков, склонных загрязнять оборудование всех видов.
В промышленных реакторах с неподвижным слоем температура по мере продви-
жения вниз по слою возрастает. По этой причине основную проблему в операциях
Гидропереработки составляет контроль температуры. Обычно для ограничения те-
пловыделения меньшими и безопасными порциями общий объем катализатора рас-
пределяют по нескольким слоям с промежуточным охлаждением между ними [Satter-
field, 1975]. Для гидроочистки бензиновой фракции и керосина обычно достаточно
Одного слоя, так как тепловыделение сравнительно невелико, но в случае более тяже-
лого сырья однослойный реактор может оказаться нецелесообразным ввиду чрезмер-
ного роста температуры. В подобных случаях каталитические слои располагают так,
Чтобы добиться более благоприятного распределения температур. На рис. 7.11 пока-
зан трехслойный реактор с двумя секциями промежуточного охлаждения.
Охлаждение
Рис. 7.11. Трехслойный реактор гидроочистки с промежуточным охлаждением
Число слоев катализатора в реакторе гидроочистки выбирают в зависимости
от общего количества выделяемой теплоты. Высота слоя определяется предельно
280
Часть 3. Моделирование каталитических процессов
допустимой температурой. В идеальном случае для максимально эффективного ис-
пользования заряда катализатора распределение слоев должно быть таким, что-
бы прирост температуры во всех слоях (а следовательно, и средняя температура
в каждом слое) был одинаковым [Gruia, 2006]. Допустимый прирост температуры
в слое лежит, в зависимости от технических характеристик процесса, где-то между
15 и 30 °C. Обычно проектируют многослойные конструкции с возрастающей вы-
сотой слоя, так как скорость тепловыделения постепенно снижается по высоте ре-
актора от входа к выходу. В большинстве случаев реакторы гидроочистки газойля
имеют два слоя, а реакторы гидрокрекинга — от четырех до шести [Robinson and
Dolbear, 2006].
На распределение температур в реакторе влияет также потеря активности ката-
лизатора. В течение цикла ее компенсируют периодическим увеличением темпера-
туры, вследствие чего амплитуда профиля температур постепенно смещается вверх.
Когда верхняя граница температуры достигает предельно допустимой для матери-
ала конструкции реактора, цикл прекращают. Если температура по оси реактора
распределена неправильно, возможна преждевременная вынужденная останов-
ка — особенно при быстрой потере активности, как при гидрообработке остатков.
В таких случаях желательно выбрать минимально возможную величину прироста
температуры в слое, чтобы задержать момент достижения предельно допустимой
температуры. Это означает увеличение числа слоев и соответственно габаритов ре-
актора, что необходимо для размещения дополнительного оборудования промежу-
точного охлаждения.
7.5.2. Парциальное давление водорода
Установки гидрооблагораживания обычно работают в интервале давлений от 1
до 30 МПа. Высокие давления подавляют образование кокса на частицах катали-
затора, увеличивают количество водорода, доступного для жидкой фазы, повыша-
ют степень превращения, улучшают теплоперенос и позволяют увеличить объемную
скорость циркуляции газа [Kundu et al., 2003]. Парциальное давление водорода (Р^
равно произведению общего давления на степень чистоты (в мольных процентах)
циркулирующего газа. Как и в случае температуры, необходимое парциальное дав-
ление водорода возрастает с увеличением плотности и вязкости сырья, а также тре-
буемой степени превращения. В связи с этим процессы гидрокрекинга проводят при
наивысших парциальных давлениях водорода, что позволяет добиться максимальной
степени превращения.
На практике основная функция водорода состоит в том, чтобы способствовать
протеканию реакций гидрирования. В углеводородах растворяется и становится до-
ступной для реакций лишь небольшая часть газообразного водорода, поэтому столь
важно увеличить общее давление. Влияние парциального давления водорода на ре-
акции гидроочистки и гидрокрекинга подтверждено множеством исследований.
В целом высокое парциальное давление водорода способствует благоприятному рав-
новесию реакций гидрирования, что в свою очередь повышает степень насыщения
Глава 7. Моделирование каталитической гидроочистки 281
ароматических углеводородов и существенно улучшает ГДО и ГДА стойких соедине-
ний, протекающие по маршруту предварительного гидрирования. На установках ги-
дрокрекинга высокое парциальное давление водорода необходимо для гидрирования
продуктов крекинга до более стабильных компонентов и удержания контроля над
коксообразованием. Если реактор работает под давлением ниже допустимого, отло-
жения кокса быстро снижают активность катализатора.
Хотя повышение парциального давления водорода играет положительную роль,
для него есть определенные практические ограничения. Допустимое давление огра-
ничено техническими характеристиками оборудования. Поэтому, чтобы максималь-
но приблизить парциальное давление водорода к проектному, важно поддерживать
чистоту циркулирующего водорода на максимально высоком уровне. Другой фак-
тор, ограничивающий давление, — сильное удорожание реактора: чтобы выдержи-
вать высокие давления, он должен иметь толстую стенку.
7.5.3. Скорость подачи сырья
Скорость подачи сырья — это отношение скорости подачи сырья к количеству ката-
лизатора, загруженного в реактор. Она показывает число объемов реактора (имеется
в виду лишь объем, занимаемый катализатором), которые могут быть переработаны
за единицу времени. Скорость подачи может выражаться на объемной {LHSV — объ-
емная скорость подачи сырья) или массовой (WHSV— массовая скорость подачи сы-
рья) основе:
Суммарная объемная скорость подачи сырья в реактор, м3/ч
LHSV-----------------------------------------------5-------[ч ];
Суммарный объем заряда катализатора, м
WHSV~ Суммарная массовая СКОРОСТЬ подачи сырья в реактор, кг/ч
Суммарная масса заряда катализатора, кг
(7.4)
(7.5)
Жесткость процесса находится в обратной зависимости от LHSV. Низкая ве-
личина LHSV означает, что за единицу времени перерабатывается меньше сы-
рья (т. е. сырье дольше контактирует с катализатором). Гидроочистку дистиллятов
обычно проводят при большей величине LHSV(> 1), чем гидроочистку и гидрокре-
кинг остатков (< 1). Объемная скорость подачи сырья выбирается при проектиро-
вании реактора. Она определяет необходимый заряд катализатора, а следовательно,
и емкость реактора, необходимую для обеспечения заданной производительности
установки.
7.5.4. Кратность водорода к сырью и скорость циркуляции
Кратность водорода к сырью — это стандартная мера объемной скорости циркуля-
ции водорода в реакционной системе относительно объемной скорости подачи сы-
рья, выражаемая следующей формулой:
282 Часть 3. Моделирование каталитических процессов
Н /сырье = Суммарный объем Н2, подаваемого в реактор, н. м3 /ч мумз] д ф
2 Суммарный объем сырья, подаваемого в реактор, м3/ч
Чтобы создать в реакторе среду, достаточно обогащенную водородом, необхо-
дима циркуляция газа. Она обеспечивает поддержание парциального давления во-
дорода на нужном уровне и увеличивает количество водорода, контактирующего
с катализатором и молекулами углеводородов. Скорость циркуляции выбирается
исходя из целей процесса и экономических соображений. Если ее величина (рав-
но как и парциального давления водорода) ниже расчетной, усиливается коксо-
образование и уменьшается степень превращения. Скорость циркуляции особен-
но важна для продления цикла службы катализатора при гидрокрекинге, который
по этой причине обычно проводят при очень высоких значениях кратности во-
дорода к сырью (1000-2000 н. м3/м3). Вместе с тем поддержание высокой скоро-
сти циркуляции требует увеличения расхода теплоносителей и более мощного
компрессора.
Циркуляция газа влияет на равновесие жидкой и газовой фаз в реакторе [Hoeks-
tra, 2007]. Большинство установок гидропереработки работает с частично испарен-
ным углеводородным сырьем. Это обстоятельство влияет на состав газа и скорости
реакций. Повышением кратности водорода к сырью можно добиться концентриро-
вания наиболее тяжелых и стойких соединений (например, дибензотиофенов при
переработке газойля) в жидкой фазе и увеличения времени их контакта с катализа-
тором. Но к увеличению скорости циркуляции газа следует подходить осторожно,
иначе может оказаться так, что некоторые вещества в испаренной фракции не будут
иметь доступа к активным центрам частиц катализатора.
Циркуляцию газа на установке гидропереработки иллюстрирует рис. 7.12. Про-
дукты реакции поступают в сепаратор высокого давления, где от газовой смеси отде-
ляются жидкие продукты. Газ из сепаратора направляется в скруббер. Там из него че-
рез контакт с аминовым раствором удаляются сероводород и аммиак. Обогащенный
водородом (около 80-85 %мольн. Н2) газ сжимается в циркуляционном компрессоре
и возвращается на вход в реактор, а также используется для промежуточного охлаж-
дения между слоями. Часть газа (10-15 %), выходящего из скруббера, направляется
в систему газового топлива или на установку очистки водорода (например, в адсор-
бер переменного давления). Газ, нагнетаемый циркуляционным компрессором, сме-
шивается с продуктом установки очистки и добавочным водородом (степень чистоты
96-99,9 %) и подается на вход в реактор. Добавочный водород необходим для ком-
пенсации собственных потерь, складывающихся из химического расхода водорода,
частичного отбора в систему газового топлива, потерь в скруббере и уноса углеводо-
родными продуктами в растворенном виде.
При прохождении каждого слоя в реакторе кратность водорода к сырью снижает-
ся из-за его химического расхода. Промежуточное охлаждение играет роль источника
пополнения водородом на входе в следующий слой. Распределение кратности водо-
рода к сырью вдоль оси реактора обратно пропорционально распределению темпе-
ратуры, что будет видно из имитаций процесса, описанных в следующем подразделе.
Йпава 7. Моделирование каталитической гидроочистки
283
Добавочный водород Н,
________
Нефтяное
сырье
Циркулирующий Отбор на установку
водород ОЧИСТКИ_______►
<----—Т—------1 I
В систему
газового
Очищен-
ный газ
,'становк топл|/|ва
ОЧИСТКИ :
.лдород
*
Реактор
с неподвиж-
ным слоем
Охлаждение
Кислый газ
Циркуляционный
компрессор
Регенерированный
аминовый раствор
Скруббер
Сепаратор
высокого
давления
Продукт гидроочистки
* Насыщенный
аминовый раствор
Рис. 7.12. Циркуляция водорода на установке гидропереработки
7.5.5. Активация катализатора
Промышленные катализаторы гидропереработки производят в виде металлической
фазы, состоящей из соответствующих оксидов (например, СоО/МоО3). Для перевода
металлической фазы в активное состояние оксиды необходимо превратить в сульфи-
ды. Активация, которую называюттакже сульфидированием, осуществляется воздей-
ствием сульфидирующего агента. В современной практике применяют три способа
сульфидирования: 1) углеводородным сырьем, содержащим серу (обычно прямогон-
ным газойлем); 2) в газовой фазе смесью H,/H2S; 3) сульфидированным (с добавкой
сульфидирующего агента) сырьем [Hallie, 1982].
Процесс сульфидирования обычно проводят на месте (т. е. когда металлическая
фаза уже диспергирована на носителе). Однако предварительное сульфидирование
более перспективно с точки зрения улучшения активности катализатора [Marro-
quin et al., 2004]. Суть процесса состоит в пропускании сульфидирующего потока че-
рез каталитический слой в обогащенной водородом атмосфере, с тем чтобы окси-
ды металлов могли превратиться в сульфиды. Общепризнано, что сульфидирование
в жидкой фазе эффективнее и происходит быстрее, чем в газовой фазе. Это обуслов-
лено тем, что поток углеводородов способствует орошению слоя и более однородно-
му распределению сульфидов. Жидкость также поглощает тепло, выделяемое в реак-
циях сульфидирования. Если выбран способ активации сульфидированным сырьем,
в него добавляют такие сульфидирующие агенты, как сероуглерод (CS2), диметил-
сульфид, диметилдисульфид и этилмеркаптан. Наиболее эффективным из них для
лабораторного и промышленного сульфидирования является диметилдисульфид.
284
Часть 3. Моделирование каталитических процессов
Процедуру проводят при температуре разложения сульфидирующего агента.
Для активации сульфидирующими агентами требуются, как правило, значительно
более низкие (от 160 до 260 °C) температуры, чем в случае с несульфидированным
сырьем (300-350 °C) [H-allie, 1982]. Для контроля тепловыделения важна концентра-
ция серы в сырье. Рекомендуемое содержание серы в несульфидированном сырье со-
ставляет 0,5-2,0 %масс., в сульфидированном — ниже 1 %масс.
На рис. 7.13 показан порядок сульфидирования никель-молибденового катализа-
тора гидрообессеривания [Marroquin et al., 2004]. Перед сульфидированием катали-
затор сушат в потоке водорода при низкой температуре, чтобы удалить остатки воды,
представляющей опасность для катализатора при высоких температурах. Затем слой
катализатора полностью пропитывают сырьем, чтобы в нем не осталось сухих зон.
Когда катализатор готов к сульфидированию, через него пропускают сульфидиру-
ющее сырье (например, прямогонный газойль, сульфидированный диметилдисуль-
фидом) и повышают температуру до 260 °C. На данном этапе температура должна
быть достаточно высокой для разложения сульфидирующего агента, но не превы-
шать 260-315 °C, чтобы исключить закоксовывание и восстановление оксида ме-
талла до того, как сульфидирование будет завершено. Это первый этап, называемый
инициированием. По его завершении температуру повышают до следующего уровня
(290-350 °C). Эксперименты показывают, что оптимальная активность катализатора
достигается при температурах, близких к предельно допустимым. Это заключитель-
ный этап, обеспечивающий максимальное поглощение серы металлической фазой.
Следует заметить, что в реакциях сульфидирования выделяется сероводород, что слу-
жит индикатором состояния процесса активации. Появление значительных коли-
честв сероводорода на выходе газа говорит о том, что процесс близок к завершению
и металлическая фаза катализатора насыщена серой [Robinson and Dolbear, 2006].
Сульфидирование
120
Нормальные
условия
30 °С/ч
Н2
260 °C----
Пропитка /зо°С/ч
150 °C <
Сушка
2ч
30 °С/ч
Соотношение
водород/сырье
Второй уровень
290-350 °C 0---—------
Первый уровень /зо °с/ч
Зч У
Соотношение водород /
сульфидирующее сырье
Р = 2,75 МПа
LHSV= 2,0 ч’1
Кратность водорода
к сырью = 356 н. мэ/м3
Рис. 7.13. Условия сульфидирования катализатора ГДО
[Marroquin, G. et al., Catal. Today, 98, 75, 2004]
Глава 7. Моделирование каталитической гидроочистки
285
7.6. МОДЕЛИРОВАНИЕ ГИДРООЧИСТКИ ГАЗОЙЛЯ,
ПОЛУЧЕННОГО ИЗ ТЯЖЕЛОЙ НЕФТИ
Этот раздел иллюстрирует применение кинетических данных лабораторных экспе-
риментов к динамической одномерной модели гетерогенного реактора идеального
вытеснения, а также использование модели для предсказания поведения промыш-
ленного реактора гидроочистки в нестационарном режиме. В качестве сырья выбран
газойль, полученный из тяжелой нефти. Последующие главы посвящены модели-
рованию реактора гидроочистки газойля тяжелой нефти на основе принятых здесь
допущений.
7.6.1. Экспериментальная часть
7.6.1.1. Материалы и оборудование
Все эксперименты проводились на полученном из тяжелой нефти прямогонном
газойле, свойства которого перечислены в табл. 7.3. Применялся промышленный
сульфидированный кобальт-молибденовый катализатор на гамма-алюмооксидном
носителе. В реактор загружали 99,43 г (100 мл) размолотого и просеянного порошка
катализатора, свойства которого также приведены в табл. 7.3. Лабораторный реак-
тор с нисходящим потоком работал в изотермическом режиме с независимым кон-
тролем температуры, обеспечиваемым трехзонной электрической печью. Внутрен-
ний диаметр реактора — 2,54 см. В центре реактора имелось гнездо для термопары
с наружным диаметром 0,635 см. Высота каталитического слоя составляла 25,2 см
(рис. 7.14).
Таблица 7.3
Свойства газойля и катализатора
Показатель Значение
Газойль
Плотность, г/см3:
при 15,6 °C и 586 мм рт. ст. 0,873
» 20 °C и 586 мм рт. ст. 0,869
Вязкость, сП:
при 20 °C и 586 мм рт. ст. 7,2554
» 40 °C и 586 мм рт. ст. 4,0161
» 60 °C и 586 мм рт. ст. 2,5455
Общая сера, %масс. 2,19
286 Часть 3. Моделирование каталитических процессоий|
Таблица 7.3, окончание
Показатель Значение
Азот, мг/кг:
азот оснований 136
неосновный азот 194
Олефины, %масс. 4,83
Ароматические соединения, %масс.: 34,9
полиароматические 6,6
диароматические 7,8
моноароматические 20,5
Предельные соединения, %масс.: 65,1
нафтены 46,17
парафины 18,93
Разгонка при атмосферном давлении, °C:
НК 284,8
выкипает 5 %об. 291,4
» 10 %об. 292,8
» 90%об. 318,8
» 95 %об. 323,7
КК 328,7
Катализатор3
Кобальт, %масс. 3,47
Молибден, %масс. 14,70
у-А12О3, %масс. 81,83
Форма частиц Размолотый Трилистник
Эквивалентный диаметр частиц, см 0,0230 0,2540
Насыпная плотность, г/см3 0,9943 0,9200
а Предварительно сульфидированный. Размолотый катализатор просеивался через сито
крупностью 60/70 меш.
7.6.1.2. Эксперименты
Перед проведением экспериментов катализатор активировали согласно процедуре,
описанной в другом источнике [Rana et al., 2004]. Эксперименты после сульфиди-
рования катализатора проводились при постоянных величинах давления 5,3 МПа
и кратности водорода к сырью 356 н. м3/м3 (2000 ст. куб. фут/баррель). Температуру
изменяли от 340 до 380 °C, объемную скорость подачи сырья — от I до 3 ч_|. Пробы
продуктов отбирали в установившемся режиме работы с периодичностью 6 ч после
успокоения в течение 4 ч между экспериментами.
тмппелироеание каталитической гидроочистки
Газовый
Насосы подачи сырья
10 см
----Е[
Сырье —м
_ хроматограф
Сухой лед а
Сода
Приемник
охладителя
Сепараторы
высокого
давления
V Нейтрали-
» затор Н25
Резервуары
для продуктов
| Продукт гидроочистки
Изотермический
реактор с неподвиж-
ным слоем
Зона -<Q(ass woo|
подогрева 4
У 2 см__
Зона -J 25,2 см
реакции I
г“ 2 см__________
Компенсиру- J Стекло^Х
ющая секция [
Helfy-Pack
1/16"
Опорная
решетка
X
X
©
Рис. 7.14. Лабораторная экспериментальная установка и реактор:
7.6.1.3. Методы анализа
Для характеризации проб атмосферного газойля и жидких продуктов применялись
следующие методы. Содержание серы определялось рентгенофлуоресцентным спек-
трометром (метод ASTMР4294). Перед этим пробы продуктов гидроочистки для отго-
на растворенных сероводорода и аммиака обрабатывали газообразным азотом. Общее
содержание азота определяли методом хемолюминесценции (ASTM £>4629), содер-
жание азота оснований — методом титрования цветовым индикатором (UOP 313).
Сырье и жидкие продукты разделяли на фракции предельных и ароматических
углеводородов методом флуоресцентно-индикаторной адсорбции (ASTM £>1319).
Содержание различных ароматических структур (поли-, ди- и моноароматических
288
Часть 3. Моделирование каталитических процессов
углеводородов) в ароматической части жидких продуктов определяли методом над-
критической жидкостной хроматографии (метод ASTM £>5186). Содержание нафте-
нов в исходном сырье измеряли по количеству предельных углеводородов методом
масс-спектрометрии с полевой десорбцией (метод ASTM Л2425). Глубину гидрокре-
кинга газойля до легких продуктов находили по материальному балансу, используя
данные анализа проб газообразных и жидких продуктов методом газовой хромато-
графии. Содержание олефинов вычисляли по бромному числу, которое находили ме-
тодом ASTM Di 159. Остальные физические свойства сырья и продуктов определя-
ли стандартными методами: плотность жидкости — осцилляционным денсиметром
(метод ASTM Л4052), вязкость — вискозиметром Штабингера (метод ASTM 2)7042),
распределение температур кипения — методом ASTM 2)86. Газообразные продукты
анализировали поточным газовым хроматографом. Физические свойства катализа-
торов — насыпную плотность и суммарный поровый объем — определяли соответ-
ственно методами ASTM С29 и ASTM DA222 на основании теории Брунаэура—Эмме-
та-Теллера (БЭТ).
7.6.2. Построение модели реактора
Реактор орошаемого слоя для гидроочистки нефтяных фракций в лабораторном
и промышленном масштабах имитируется динамической одномерной гетерогенной
моделью, основанной на опубликованной стационарной модели трехфазного реак-
тора [Korsten and Hoffmann, 1996; Rodriguez and Ancheyta, 2004].
7.6.2.1. Допущения, принимаемые в рамках модели
• Реакторы (лабораторный и промышленный) работают в динамическом режиме.
• Как жидкая, так и газовая фазы движутся в режиме вытеснения (в пробковом
режиме).
• Лабораторный реактор работает в изотермических и изобарических условиях.
• Условия в промышленном реакторе изотермичны лишь в радиальном
направлении.
• По высоте слоя не происходят ни испарение жидкости, ни конденсация паров.
• Сопротивлением массопереносу на газовой стороне границы раздела газовой
и жидкой фаз в лабораторном реакторе можно пренебречь.
• В лабораторном реакторе лимитирующим является сопротивление массопере-
носу в пленке на границе жидкой и твердой фаз, в промышленном — сопротив-
ление массопереносу внутри частиц.
• Сопротивление массопереносу внутри частиц оценивается с помощью коэф-
фициентов эффективности.
• Химические реакции происходят лишь в жидкой фазе при контакте с поверх-
ностью твердой фазы.
• Объем жидкости в лабораторном и промышленном реакторах остается
постоянным.
Глава 7, Моделирование каталитической гидроочистки
289
• В промышленном реакторе частицы катализатора смачиваются полностью.
• Так как эксперименты проводятся в течение сравнительно короткого времени
на свежем катализаторе, его активность остается постоянной.
• Внутри частиц нет перепадов температур (изотермия в твердой фазе).
• Баланс энергии для газовой фазы не учитывается, так как она имеет значитель-
но меньшую теплоемкость, чем жидкая.
В связи с предполагаемым отсутствием испарения жидкости и конденсации па-
ров в слое следует отметить крайнюю сложность моделирования испарения сы-
рья в реакторе орошаемого слоя. До настоящего времени известна лишь одна рабо-
та, рассматривающая этот вопрос при моделировании реактора гидропереработки
[Chen et al., 2011]. Авторы этого исследования пришли к выводу, что учет испарения
углеводородов (равновесия паровой и жидкой фаз) приводит лишь к незначитель-
ным изменениям в распределениях температуры и степени деароматизации. Далее,
авторы обнаружили, что хотя степень ГДО в реакторе может быть высокой, влия-
ние испарения при высоких степенях превращения становится меньше. Учет равно-
весия пара и жидкости дает высокие значения степени ГДО, что означает большую
температуру и, соответственно, более сильное испарение. Таким образом, два этих
фактора сильно зависят друг от друга, и их влиянием можно пренебречь. Подобный
вывод согласуется с расчетами Акгермана и соавторов [Akgerman et al., 1985], ука-
завших, что при высоких степенях превращения различия между моделями, учиты-
вающими и не учитывающими испарение, вследствие исчерпания лимитирующего
реагента сокращаются до минимума. В данном исследовании степень ГДО превыша-
ет 95 %, поэтому можно ожидать, что, согласно результатам указанных выше работ,
влияние испарения будет минимальным. Кроме того, в экспериментах использовал-
ся газойль тяжелой нефти, серосодержащие и ароматические соединения которого,
помимо того что они содержатся в повышенных количествах, имеют большую мо-
лярную массу, чем те, что содержатся в обычном газойле. Сложность и большая мо-
лярная масса этих примесей делают сырье менее летучим. В заключение следует ска-
зать, что, хотя учет равновесия паровой и жидкой фаз можно только приветствовать,
гипотезу об их предполагаемом равновесии нельзя считать достаточно обоснованной
[Froment et al., 1994].
7.6.2.2. Баланс масс в нестационарном состоянии
Принимая во внимание перечисленные выше допущения, массоперенос соединений
в реакторе с орошаемым слоем описывается следующей совокупностью уравнений
в частных производных.
Газовая фаза
Нестационарное уравнение баланса масс в каталитическом слое для соединений,
присутствующих в газовой фазе:
п G G ( G
RTl dt RTl dz L' \H!
(7.7)
290
Часть 3. Моделирование каталитических процессов
где i пробегает множество, элементами которого являются Н2, H2S, NH3 и СН4. Знак
«минус» соответствует прямотоку, знак «плюс» — противотоку. (Остальные значе-
ния см. в конце главы.)
Жидкая фаза
Нестационарное уравнение баланса масс в каталитическом слое для газообразных
соединений в жидкой фазе:
е - и dtf . г а (_ cL'\ - ksa (cL-Cs 'l (7 8)
— UL +^~LiaL rj '-'I ('SLih
Ct OZ {H, ) ' '
где i пробегает множество, элементами которого являются Н2, H2S, NH3 и СН4.
Модель предполагает, что органические соединения серы и азота, олефины и аро-
матические соединения, равно как и жидкие углеводороды, нелетучи; следовательно,
нестационарное уравнение баланса масс для жидких соединений имеет вид
(С^ - С), (7.9)
dt dz
где i пробегает множество, элементами которого являются S (соединения серы),
N (соединения азота), Poly (полиароматические соединения), Di (диароматические
соединения), Mono (моноароматические соединения), Naph (нафтены), О (олефи-
ны), GO (газойль) и Naphtha (бензиновая фракция).
Твердая фаза
Компоненты, перемещающиеся между жидкой и твердой фазами, расходуют-
ся или образуются в химической реакции на смоченной поверхности катализатора
в соответствии со следующими обыкновенными дифференциальными уравнениями
первого порядка:
N R1
£s(l-ea)—= -С) + (7.Ю)
(7/
где i пробегает множество, элементами которого являются Н2, H2S, NH3, СН4, S, N,
Poly, Di, Mono, Naph, О, GO и Naphtha', j пробегает множество, элементами которого
являются ГДО (гидродесульфуризация, или гидрообессеривание), ГДА (гидродеазо-
тирование), ГДДрА/ (гидродеароматизация полиароматических соединений), ГДАра
(гидродеароматизация диароматических соединений), ГДАрМотю (гидродеароматиза-
ция моноароматических соединений), ГДАр^а (гидродеароматизация нафтенов),
ГДОл (гидрирование олефинов) и HCR (гидрокрекинг).
7.6.2.3. Баланс теплоты в нестационарном состоянии
С учетом того, что реакции гидрооблагораживания экзотермичны по приро-
де, баланс энергий в промышленном реакторе гидроочистки, работающем в ади-
абатических неизотермических условиях, дается следующими выражениями
[Froment et al., 1994].
Глава 7. Моделирование каталитической гидроочистки 291
Жидкая фаза
' ElPlQ7£ Д£ = ~UlPlCPl “Z ^lLSaS^L ~ Т$ )• (7.11)
tit tiz
Гйзовая фаза
£spsCps^ = h^s(TL-Tss) + fwp^ (7.12)
О/ j=j
7.6.2Л. Гоаничные условия
Поскольку математическая модель представляется системой дифференциальных
уравнений (в том числе в частных производных, независимыми переменными в ко-
торых являются время и пространственные координаты), необходимо задать началь-
ные и граничные условия для газовой, жидкой и твердой фаз в прямоточном режиме.
Начальные условия
„Р^10 ° Р° - ‘ - Нг. H,S, NH, и СИ.;
с‘=(с‘)о;
Сд,.=0, где i=н2, H2s, NH3, СН4,5, N, Poly, Di, Mono, Naph, O.GOvi Naphtha',
T = = T •
7 L 7S 70’
при 0 < z < LB pG = 0, где z. = Нд NH, и CH .
= 0;
C/i(-=0, W i ~ H2, H2S, NH3, CH4, S, N, Poly, Di, Mono, Naph, 0, GOn Naphtha',
T = Ts= 'r.
JL 'S 70>
при Z = LB p° = о, где i = H2, H2S, NH3 и CH4;
C/- = 0;
C^,.=0, где i=H2, H2S, NH3, CH4, S, N, Poly, Di, Mono, Naph, O,GOa Naphtha',
TL = TSS=TO.
Циничные условия
Для?>0 где/=Н,, H,S, NH3hCH4;
лриг=0: C‘=(C‘)J
Сд, - 0, гдеi- н2, H2S, NH3, СН4, S, N, Poly, Di, Mono, Naph, О, GOn Naphtha',
292
Часть 3. Моделирование каталитических процессов
Если вместо рециркуляции газа в реактор подается водород высокой чистоты
(в случае лабораторного реактора) или возвращаемый водород подвергается очист-
ке (промышленные установки гидроочистки), величины парциальных давлений
и молярных концентраций Cf в жидкой фазе для компонентов H2S, NH3 и СНу
на входе слоя (^ = 0) близки к нулю.
7.6.2.5. Метод интегрирования
Модель реактора решалась численно методом прямых [Schiesser, 1991; Walas, 1991].
Уравнения в частных производных, описывающие массо- и теплоперенос в реак-
торе, преобразовывались в обыкновенные дифференциальные уравнения перво-
го порядка. Для этого частные производные по осевой координате дискретизирова-
лись заменой левыми конечно-разностными выражениями, а частные производные
по времени оставлялись без дискретизации. Полученная система обыкновенных
дифференциальных уравнений решалась относительно времени методом Рунге-
Кутты четвертого порядка.
7.6.3. Модели кинетики реакций
7.6.3.1. Гидрообессеривание
Для моделирования реакции ГДО было принято следующее обобщенное
представление:
Аг- S + 2Н2 —/Сгдо > Ar—Н + H2S (7.13)
где Ar-S — серосодержащее соединение; Н2 — водород; Ar—Н — соответствующее
ароматическое соединение, не содержащее серы; H2S — сероводород.
Кинетическая модель, описывающая реакцию ГДО, основывается на опублико-
ванном ранее уравнении
TI ((-iS ул, zyiS ул,
_ лш,ГДО '.'-'S15 > \'~'SLhJ
Ггдо" (1 + jKhsC;lh)2 • (7л4)
Константа равновесия адсорбции H2S (Кн ) зависит от температуры и может быть
вычислена по правилу Вант-Гоффа:
^h,s — ^o.h,s ехР
&Hgds,H2S
I RTSS
(7.15)
7.6.3.2. Гидродеазотирование
Как и в случае реакции ГДА, все органические соединения азота в сырье группиру-
ются в один псевдокомпонент. Для представления всех реакций ГДА использовалось
следующее обобщенное стехиометрическое уравнение:
Глава 7. Моделирование каталитической гидроочистки
293
Ar-N + ЗН2 —А>да ~> Ar—Н + NH3 (7.16)
где Ar-N — азотсодержащее соединение; Ar—Н — соответствующее ароматическое
соединение, не содержащее азот; NH3 — аммиак.
Для реакции ГДА принималась следующая кинетика псевдопервого порядка от-
носительно общей концентрации азота [Bhaskar et al., 2004]:
ГГДА~РяГГДа/ [SS (1 —Ss)] ~^т,ГДА (QsTJV ). (7-17)
Хотя соединения азота вследствие параллельно протекающей адсорбции могут
влиять на активность в реакциях ГДО, здесь это влияние не учитывалось по следую-
щим причинам:
• использовавшийся в экспериментах газойль тяжелой нефти содержал гораздо
меньше азота, чем серы и ароматических соединений;
• кобальт-молибденовый катализатор на носителе, который использовали в экс-
периментах, более избирателен к удалению серы;
• значительное влияние азота на ГДО и другие реакции доказано в исследова-
ниях, посвященных производству дизельного топлива со сверхнизким со-
держанием серы, о чем в данном случае речи не идет. Влияние конкурирую-
щей адсорбции азота на реакции ГДО важно лишь в указанном случае [Но and
Nguyen, 2006].
7.6.3.3. Гидродеароматизация
Совокупность реакций ГДАр моделировалась следующими стехиометрическими
уравнениями [Chowdhury et al., 2002]:
Полиароматические соединения + Н2 <——> Диароматические соединения (7.18)
«То/у-
Диароматические соединения + 2Н < —> Моноароматические соединения (7.19)
2 &DI-
Моноароматические соединения + ЗН2 * Нафтены (7.20)
Скорости реакций ГДАр выражаются следующим образом [Cheng et al., 2004]:
ГГДАрр«/> ~ ^»>,ГДАрМг( Р Н, ) SLPoly ) ~ ^й.ГДАрР«/>-(^' SLDi) ’ (7.21)
^ГДАро. ^'«.ГДАрр^^Т^Н’) ( SLPoly) "лГДАрр,,/,. ( SLDi)
+ ^чгдАрДт’нз) (Суш)_^.гдАрй-(С«шопО)’ (7.22)
294
Часть 3. Моделирование каталитических процессов
ГДАрчърл
If'
;м,ГДАрл&ик
ш^ГДАрд/оло*
•’L = -к
ГДАрл/осо Д1,ГДАр.м>лА
SLMono
SLMono
/иТДАрл/о
М,ГДАрАЛт<>
(7.23)
'/лХДАрл/йяоД SLNaph/’
(7.24)
Так как решить эту систему уравнений довольно трудно даже в случае первого по-
рядка, реакции ГДАр были описаны следующей упрощенной системой, выражаю-
щей скорости реакций первого порядка [Chowdhury et al., 2002; Bhaskar et al., 2004]:
,-’L = L I
ГДАрд,,,. ш.ГДАр/ыЛ
SLPoly
/лГДАр/^
(7.25)
ГДАр.\>А
'L = к
ГДАрл/ (л.ГДАрОг*
'L = к I
ГДАр.Шю /н.ГДАрльлг.’
•'Г = -X |
ГДАрмио /м.ГДАрллп»
Лд
SLMono
ш,ГДАрг»-
inXRkpMc.
SLNaph )4
SLMono
р?,ГДАрл/ов<>-\ SLNaph /
(7.26)
(7.27)
(7.28)
Ввиду того, что условия в лабораторном реакторе являются изобарическими,
а водород подается в избытке, необходимо исключить лимитирование концентра-
ции водорода, растворенного в жидкой фазе. Для этого значения (р% ) ’, (р°, j2 и
V остаются постоянными; тогда уравнения (7.25)—(7.28) сокращаются до
где
r'L — к* (fS )_£' (CS У ГДАрл,,, "ХГДАрлДЛSLPoly) nin,ra^pr^\ SLDIP (7.29)
r'L =k' (cs }-к' (cs У 'ГДАргь л7я,ГДАрп1\^ SLDi / /я,ГДАрй-\ SLMoho)4 (7.30)
r'L -k' (Cs }-k' (Cs У ' ГДАрМвд /»,ГДАрм>™>\ SLMono) /я,ГДАрА4»«-\ SLNaph)' (7.31)
tL if / s-iS + ГДАрЛф» inТДАрлЛшоV SLMono) йТДАрААмм-\ SLNaph /’ (7.32)
^/л.ГДАрР^ - ^ni.rflAppo?j(^H2 ) > (7.33)
^нг.ГДАри“ ^й,ГДАро>(/’н, ) ; (7.34)
к' —к* ( а V'3 ш.ГДАрлнио //;,ГДАрлл,лп\^Н2 ) (7.35)
Глава 7. Моделирование каталитической гидроочистки
295
7.6.3.4. Гидрирование олефинов
Гкдрирование олефинов (ГДОл) представлялось следующим стехиометрическим
уравнением:
R-CH * CH-R' + Н2 —/СгдОл > R-CH2-CH2-R' (7.36)
Для реакции ГДОл тоже была принята кинетика псевдопервого порядка относи-
тельно общей концентрации олефинов, определяемая следующим уравнением ско-
рости реакции [Bhaskaret al., 2004]:
' ГДОл и.ГДОл(<-'.аО
(7.37)
7.6.3.5. Мягкий гидрокрекинг
Реакция гидрокрекинга представлялась трехкомпонентной моделью следующим об-
разом [Bhaskar et al., 2004]:
Легкие газы — Газойль —-—> Бензиновая фракция —~> Легкие газы
(Здесь к легким газам отнесены газообразные углеводороды С,—С4.)
Для реакции гидрокрекинга была принята кинетика псевдопервого порядка:
гво ~ fw?BraoK^-S Ss) ~ (Qv.go) + ^2 (^я.со)> (7.38)
rNaphtha ~ fwP BrNaphlha / ) = —'SLGo) + K^SLNapMia)’ (7'^9)
rLG = fv. pBrLG I(e5 GS ) - ( CsLGO ) - ^3 'SLNaphtha )' (7.40)
7.6.4. Расчет параметров
7.6.4Л . Кинетические параметры
Задавшись допущениями модели и уравнениями реактора и сформулировав выра-
жения скоростей реакций ГДО, ГДА, ГДАр и гидрирования олефинов, по экспери-
ментальным данным, полученным для стационарных состояний, через оптимизацию
по алгоритму нелинейной регрессии Левенберга—Марквардта можно найти величи-
ны коэффициентов адсорбции, констант равновесия, порядков реакций, коэффи-
циентов частоты столкновений и энергий активации. Экспериментальные значения
параметров для стационарных состояний использовались в динамической моде-
ли реактора с орошаемым слоем для перерасчета значений кинетических параме-
тров, которые затем принимались как окончательные. Зависимость от температуры
296
Часть 3. Моделирование каталитических процессов
истинных значений констант скоростей реакций, показанных в табл. 7.4, описывает-
ся законом Аррениуса.
Таблица 7.4
Кинетические и гидродинамические параметры реакций гидроочистки газойля
Реакция,) к u Eaj, кДж/моль. дял., - кДж/моль. кДж/моль-H2
ГДО” 2,639287 - IO17 150,10 5,166888 34,02 34,89r
ГДА 1,553296 1012 172,28 — — 21,62д
ГДАРА„. 2,413720 1010 156,09 9,856780 — 58,6Г
ГДАри 2,855135 • 10s 131,91 66,570899 — 58,6Г
ГДАр^ 11,143786 54,31 438,006631 — 58,6Г
ГДОл 9,463471 • 10s 138,15 — — 101,10*
HCR,.,, „ , (j()~ Naphtha 1,384870-10-' 56,41 — — 29,3 Г
HCRtt0_/6. 2,391640-106 139,46 — — 29,3 Г
HCR Naphtha-I.G 2,212240 -1016 273,78 — — 29,3 Г
Температура, °C: 340 350 360 370 380 A' 0,0401397 0,1759018 0,1963370 1,0277 IO3 1,9970- IO3 b' 1,5552485 0,8613340 0,6933859 2,2143412 2,5912252
Примечание'. HCR — реакция гидрокрекинга.
“ Размерности единиц: для реакций ГДО см. раздел «Обозначения»; для реакций ГДАр
и ГДОл [Ао] = см3/г- с; для реакций ГДА и мягкого гидрокрекинга [&0] = 1/с.
6 Для реакции ГДА i = H2S, единица измерения см3/моль H2S; для реакций ГДАр значе-
ние Ко безразмерно и берется при Го = 340 °C.
”т1 = 1,8; /и, = 0,96.
г [Ueda et al., 1975].
д [Tarhan, 1983].
с [Jaffe, 1974].
* [Chen et al., 2001].
Динамическая модель рассматривалась как краевая задача, так как концентра-
ции реагентов на входе и выходе реактора известны. При решении этой двухточеч-
ной краевой задачи кажущиеся значения констант скоростей реакций определялись
методом «пристрелки» [Bhaskar et al., 2004]. Этот метод сводит решение краевой за-
дачи к итеративному решению задачи с начальными условиями (задача Коши) путем
последовательных приближений. В качестве начальной выбирается точка с наи-
лучшими из известных условиями. Недостающим начальным условиям присваива-
ются какие-либо пробные значения. Затем полученная начальная задача решается
Глава 7. Моделирование каталитической гидроочистки
297
методом Рунге-Кутты четвертого порядка. Полученное решение сравнивается с из-
вестными граничными условиями, по результатам сравнения в принятые значения
начальных условий вносятся поправки и начальная задача решается снова. Для каж-
дого эксперимента процесс повторяется до тех пор, пока расхождение между ре-
зультатом расчета и известными граничными условиями не станет меньше заданной
неотрицательной целевой функции [Carnahan et al., 1969]:
F=
LB,exp •
(7-41)
Lff ,calc
Скорость реакции ГДО предполагалась отвечающей кинетической модели, опи-
сываемой уравнением (7.14), при т, = 1,8 и т2 = 0,96.
Для нахождения констант скоростей реакций ГДАр, протекающих в обратном на-
правлении, определялись константы равновесия:
ГДАрР()/}. , г ’
К/">ГДАр?„6,_
(7.42)
_ Л7п,ГДАри .
ГДАр а — ’
«•Й.ГДАрСг._
__ ^т,ГДАр,№,|а
(7.43)
т/ —________________
ГДАРл/мю 7 '
ет,ГДАр.,
• ** гМоип-
(7.44)
Константы равновесия обратимых реакций для разных температур вычислялись
по формуле Вант-Гоффа:
~ -^О-ГДАй.,,, ,а,м..АТ0 ) еХР
_1_____1_
То Tss
(7.45)
Реакции гидрирования ароматических соединений протекают с выделением от 63
до 71 кДж теплоты на 1 моль Н2. Поэтому средняя теплота реакции ГДАр была приня-
та равной 67 кДж на 1 моль прореагировавшего Н2. Кинетические параметры реакций
гидрирования поли-, ди- и моноароматических соединений приведены в табл. 7.4.
Реакции мягкого гидрокрекинга представляются следующими обыкновенными
дифференциальными уравнениями:
dQ/LHSV)
(7.46)
^Naphtha _ . у — k Y
d(\/LHSV) 1 00 3 W"'M’
(7.47)
--------= k Y + kY
d(VLHSV) 2 00 3
(7.48)
R
298
Часть 3. Моделирование каталитических процессов
Константы скоростей реакций гидрокрекинга вычислялись методом минимиза-
ции разности между экспериментальным и расчетным значениями выхода продук-
тов, Целевая функция для расчета этих параметров задавалась следующим образом:
(7.49)
*=1 (=/
где
IX,.=ь <7'50’
/=/
Здесь N — число опытов, N — число псевдокомпонентов. Задача решается че-
рез оптимизацию Левенберга—Марквардта. Расчетные величины кинетических па-
раметров реакций гидрокрекинга приведены в табл. 7.4.
7.6.4.2. Коэффициент эффективности катализатора
Коэффициент эффективности катализатора можно вычислить в зависимости от ве-
личины обобщенного модуля Тиле (Ф). Согласно [Froment et al., 2010], обобщенный
модуль Тиле необратимой реакции п-го порядка равен
Ф- =-
(7.51)
Для обратимой реакции и-го порядка обобщенный модуль Тиле равен
где
р5=рг/(1 -ед).
(7.55)
Эквивалентный диаметр частиц (dpe) вычислялся по формуле, которую предложи-
ли Купер и соавторы [Cooper et al., 1986]:
, 6<W)
dpe =-----
(7.56)
Ф$
Коэффициент удлинения пробега т колеблется от 2 до 7. В литературе его зна-
чение обычно принимается равным 4 [Bird et al., 2002]. Согласно Саттерфилду
Глава 7. Моделирование каталитической гидроочистки
299
[Satterfield, 1970], в жидкостях кнудсеновская диффузия не наблюдается; следова-
тельно, = 0,
Значения Т| для каждой реакции определяются по следующим формулам [Dudu-
fcovic, 1977; Tarhan, 1983; Froment et al., 2010]:
• дляф^ < 3
(7.57)
• для > 3
(7.58)
7.6.4.3. Гидродинамические параметры
Саттерфилд [Satterfield, 1975] определяет эффективность смачивания катализатора
следующим образом:
k'
k'
пт, ГДО
(7.59)
По сообщениям, эффективность смачивания катализатора в экспериментальных
реакторах составляет от 0,12 до 0,6, в промышленных — от 0,7 до 1,0 [Satterfield, 1975;
Bhaskaret al., 2004].
Согласно модели частичного смачивания, предложенной Бонди [Bondi, 1971]
и модифицированной Саттерфилдом [Satterfield, 1975], кажущееся к' гд0 и истин-
ное k'in гдо значения константы скорости и приведенная массовая скорость жидкости
GmL связаны следующим выражением:
,, 110
^оде.гдо - । А' ' (7.60)
^ш.гдо 10 (G„,l)
Кривая зависимости fw от Re£, определяемая этим уравнением, получается варьи-
рованием приведенной массовой скорости жидкости лишь для одного значения тем-
пературы в реакторе. Значения эмпирических констант Л' и Ь' для разных температур
даны в табл. 7.4. Зависимость истинного значения к'п гдо константы скорости реак-
ции от температуры можно выразить формулой Аррениуса:
К. ГДО
(-F
- к'о гдо ехР -----
0,1 ди ЬЧ
(7.61)
Транспортные и теплофизические свойства рассчитываются в модели по форму-
лам, опубликованным в открытых литературных источниках [Mederos et al., 2009].
300
Часть 3. Моделирование каталитических процессов
7.6.5. Анализ результатов
Для анализа и имитации работы лабораторного реактора применялась модель трех-
фазного изотермического реактора. Решение модели экспериментального реактора
представляет собой задачу с начальными условиями, так как концентрации реаген-
тов и продуктов на входе реактора известны. Модель решалась при значениях кине-
тических параметров, полученных из данных экспериментов.
Начальные молярные концентрации серосодержащих и азотсодержащих соедине-
ний и олефинов можно рассчитать по следующим формулам:
(С»)« = 247, oV
(7.62)
(7.63)
(7.64)
Молярные массы остальных органических соединений принимались равными
молярной массе пробы в целом, так что их молярные концентрации можно рассчи-
тать исходя из массовых долей:
MWL
(7.65)
где i пробегает множество, элементами которого являются Poly, Di, Mono, Naph, GO
и Naphtha. Выходные массовые доли рассчитываются по формуле
(w/)z =
(7.66)
Начальная концентрация газообразных соединений в жидкой фазе оценивается
по выражению
(7.67)
где i пробегает множество, элементами которого являются Н2, H2S, NH3 и СН4. Для
представления кинетики легких углеводородов (С^-С,,) был выбран метан.
7.6.5.1. Имитация динамического режима в изотермическом
лабораторном реакторе гидроочистки
Результаты имитации лабораторного реактора основываются на описанных выше
экспериментальных рабочих условиях и значениях параметров. При имитации по-
ведения лабораторного реактора с орошаемым слоем использовались значения
Глава 7. Моделирование каталитической гидроочистки
301
кинетических параметров, вычисленные ранее методом минимизации разности
между экспериментальным и расчетным значениями выхода продуктов.
На рис. 7.15 представлено распределение концентрации серы в жидкой фазе и на
поверхности катализатора по высоте слоя в разные моменты времени. Профили кон-
центрации получены при температуре в реакторе 340 °C, давлении 5,3 МПа, объем-
ной скорости подачи сырья LHSV= 2,5 ч-1 и кратности водорода к сырью 356 н. м3/м3
(2000 ст. куб. фут/баррель). Расчетное значение концентрации серы на выходе реактора
хорошо согласуется с экспериментальным. Через 60 с сырье проходит лишь 20 % высо-
ты реактора, поэтому количество серы в остальных 80 % высоты реактора равно нулю,
т. е. эта часть реактора пуста. С течением времени реактор полностью заполняется ре-
акционной смесью, и через 1700 с достигается стационарное состояние. На рис. 7.16
показаны распределения парциального давления и концентрации водорода в жидкой
фазе по высоте слоя в разные моменты времени. Имитацией работы в стационарном
состоянии установлено, что форма кривой концентрации водорода в жидкой фазе за-
висит от соотношения между скоростью массопереноса и скоростью реакции. По ре-
зультатам имитации динамического режима это видно гораздо лучше.
На том же рис. 7.16 показаны распределения парциального давления H2S и его мо-
лярной концентрации в жидкой фазе по высоте слоя. Как и в случае Н2, наблюдают-
ся предсказуемые тенденции. Так как в любой момент времени сероводорода на вхо-
де в реактор нет, его парциальное давление и концентрация возрастают по слою.
По прошествии 60 с H2S образуется в определенной части высоты реактора, посколь-
ку здесь имеется сера, которая реагирует с Н2. За этой точкой реакционной смеси нет
и реакция отсутствует.
Результаты имитации ГДО в стационарном состоянии (к моменту времени 1700 с)
были сопоставлены с экспериментальными значениями. Было найдено, что модель
с высокой точностью имитирует работу лабораторного реактора с орошаемым сло-
ем, давая погрешности определения степени превращения серы от -1,13 до +0,56 %.
Имитация других реакций тоже дала хорошее согласие между расчетными и экспери-
ментальными значениями концентрации.
Координата z по высоте реактора, см
Рис. 7.15. Распределение концентрации серы в лабораторном реакторе с орошаемым
слоем в различные моменты времени: (-•-) — через 60 с; (.) — через 400 с;
(—х—) — через 900 с; (-) — через 1700 с; (о) — экспериментальные значения
302
Часть 3. Моделирование каталитических процессов
Координата z по высоте реактора, см
Координата z по высоте реактора, см
Координата z по высоте реактора, см
Рис. 7.16. Распределения парциальных давлений и молярных концентраций в жидкой
фазе Н2 и H2S в лабораторном реакторе с орошаемым слоем в различные моменты
времени: (-•-) — через 60 с; (.................) — через 400 с;
(-Х-) — через 900 с; (--) — через 1700 с
Координата z по высоте реактора, см
7.6.5.2. Имитация динамического режима в изобарическом
неизотермическом промышленном реакторе гидроочистки
Работа промышленного реактора имитировалась трехфазной неизотермической мо-
делью. Решение модели в этом случае опять-таки представляет собой задачу с на-
чальными условиями, так как концентрации реагентов и продуктов и начальные тем-
пературы фаз на входе в реактор известны. Модель решалась при значениях констант
скоростей, вычисленных по результатам экспериментов на лабораторном реакторе.
Учитывая высокие приведенные скорости жидкости в промышленных реакторах, эф-
фективность смачивания катализатора/, была принята равной 1,0 [Satterfield, 1975;
Korsten and Hoffmann, 1996].
На рис. 7.17, а и б показаны динамические распределения концентрации серы
и температуры в промышленном реакторе, построенные по уравнениям баланса
масс и энергий. Для сравнения приводятся также результаты имитации переход-
ного поведения концентраций серы в лабораторном реакторе и эксперименталь-
ное значение температуры. Можно видеть, что стационарное состояние достига-
ется за одно и то же время (1700 с). Это обусловлено тем, что значения объемной
скорости подачи сырья и начальной температуры в реакторах были выбраны одина-
ковыми. Поскольку динамическая модель проверяется лишь в одной стационарной
Глава 7. Моделирование каталитической гидроочистки
303
точке, остается некоторая неопределенность в формах динамических распределе-
ний в промышленном и лабораторном реакторах. Согласно Карберри и Варма [Car-
berry and Varma, 1987], небольшие пики в распределении концентрации серы на вы-
ходе из промышленного реактора объясняются типичным способом реагирования
реакторов гидроочистки на движение жидкости в обход некоторых участков ката-
литического слоя.
Время, с Координата z по высоте реактора, см
Время, с
Координата z по высоте реактора, см
Рис. 7.17. Динамические распределения концентрации серы (а) и температуры
(б) в зависимости от времени в промышленном (—) (z = 853,44 см; uL = 0,72 см/с;
uG = 8,93 см/с) и лабораторном (—) (z = 25,2 см; uL = 1,81-10-2 см/с; uG = 0,24 см/с)
реакторах при 340 °C и 5,3 МПа и по высоте слоя (в и г) в промышленном реакторе:
(-•-) — через 60 с; (.) — через 400 с; (-х-) — через 900 с; (-) — через 1700 с;
(о) — экспериментальные значения
На рис. 7.17 показаны также результаты имитации динамики температуры в про-
мышленном реакторе. Явление так называемого «обращенного» поведения в началь-
ной части слоя, о чем сообщали другие исследователи [Mederos et al., 2006], не было
обнаружено. Это явление можно объяснить быстрым и глубоким превращением реа-
гентов на первых 25 % высоты слоя, что ведет к нехватке реагентов для остальной ча-
сти и переходному понижению температуры.
Кроме того, показаны расчетные динамические распределения молярной кон-
центрации серы по высоте слоя в промышленном реакторе в различные моменты
времени от 60 до 1700 с при температуре на входе 340 °C. Имитация динамического
304
Часть 3. Моделирование каталитических процессов
поведения проводилась при тех же условиях реакций, что и при имитации лаборатор-
ного реактора. Экспериментальные значения концентрации серы на выходе из изо-
термического лабораторного реактора обозначены значком «о». Хименес и соавторы
[Jimenez et al., 2007] уже сообщали о распределениях с резко выраженным снижением
содержания серы в начальной части реактора. Авторы связали их с рассматриваемой
моделью кинетики и имитируемыми рабочими условиями. Далее, из-за повышения
температуры каталитического слоя, наблюдаемого в жидкой фазе в адиабатическом
реакторе, экспериментальные значения концентрации серы в лабораторном реакто-
ре были выше, чем расчетные в промышленном реакторе.
На рис. 7.17 изображены также расчетные изменения температуры жидкой фазы
по высоте каталитического слоя в промышленном реакторе в различные моменты
времени. Профиль температур катализатора (т. е. твердой фазы) не показан, так как
разность температур жидкости и катализатора составляла всего лишь 0,03 °C. Как со-
общалось в другом источнике [Mederos et al., 2006], это обстоятельство оправдывает
имитацию теплового баланса лишь одним уравнением для псевдофазы. Из рисунка
видно, что благодаря большей степени удаления примесей в начальной части реак-
тора прирост температуры здесь выше, о чем сообщали и другие исследователи [Ro-
driguez et al., 2004].
График на рис. 7.18 иллюстрирует переходное поведение концентрации серы
в промышленном реакторе гидроочистки. Видны четкие максимумы в переходных
значениях, наибольший из которых достигается вверху (т. е. на входе) реактора; да-
лее вниз по слою максимумы убывают. Знание переходного поведения концентра-
ций серы важно для настройки уставок контроллеров в системах управления процес-
сами гидроочистки.
1,2-Ю""
1,1 •ПГ’
s ЙОЛО-4
1 9,0-10’5
S 8,0-10-5
3 7,0-10-5
8 6,o io 5
I 5,0-10“5
2. 4,0-10'5
| 3,0-10~s
z 2,0-10s
£ 1.0-10-5
о
О 200 400 600 800 1000 1200 1400 1600 1800
Время, с
Рис. 7.18. Динамика концентраций серы в жидкой фазе в промышленном реакторе
в различных точках слоя: (.....) — z = 213,4 см; (--) — z = 284,5 см;
(-—) — z = 426,7 см; (-—) — z = 640,1 см; (-) — z = 853,4 см; (о) — эксперимен-
тальное значение в лабораторном реакторе
Глава 7. Моделирование каталитической гидроочистки
305
На рис. 7.19 показана динамика расчетных концентраций основных соединений
(соединений серы и азота; поли-, ди- и моноароматических соединений; нафтенов
и олефинов) в жидкой фазе в нескольких точках по высоте слоя в промышленном
реакторе. Можно видеть, что активности содержащихся в сырье моно-, ди- и кон-
денсированных полициклических соединений в реакциях гидрирования суще-
ственно различаются. Так как в гидрировании последнего кольца участвует больше
водорода (3 моля для моноароматических соединений по сравнению с 2 для диаро-
матических и 1 для полициклических), типичные условия реакций гидроочистки
термодинамически менее благоприятны для гидрирования первого кольца поли-
ароматических соединений (меньше значения констант равновесия), чем для ги-
дрирования последнего кольца моноароматических. С другой стороны, согласно
наблюдениям, кинетически наиболее благоприятно гидрирование первого коль-
ца полициклических соединений. Скорости гидрирования последующих колец
склонны к снижению (см. табл. 7.4), и гидрирование последнего кольца (что соот-
ветствует моноароматическим соединениям) по сравнению с начальными этапами
протекает значительно труднее. В этих экспериментах гидрирование моноаромати-
ческих соединений было лимитировано термодинамическим равновесием, благо-
приятствующим обратной реакции, поскольку концентрация моноароматических
соединений по высоте слоя вместо убывания возрастала. Это означает, что обрат-
ная реакция превращения диароматических соединений в моноароматические
протекала значительно быстрее, чем прямая реакция превращения моноаромати-
ческих соединений в нафтены; как следствие, общая концентрация ароматических
соединений снижается незначительно.
На рис. 7.20 изображена динамика концентраций газойля и бензиновой фрак-
ции в промышленном реакторе, а также распределение концентраций жидких сое-
динений, участвующих в реакциях мягкого гидрокрекинга, по высоте слоя в стаци-
онарном состоянии. Из-за низкой кислотности гамма-алюмооксидного носителя
использовавшийся в экспериментах промышленный кобальт-молибденовый ка-
тализатор давал низкий выход бензиновой фракции в реакциях гидрокрекинга.
Из табл. 7.4 можно видеть, что гидрокрекинг газойля до бензиновой фракции про-
текает медленнее, чем до легких газов, что согласуется со значениями предэкспо-
ненциального множителя. Это обусловлено тем, что отщепление боковых цепей
с числом атомов углерода от 1 до 4 происходит легче, чем отщепление длинных бо-
ковых цепей с большим числом атомов углерода, которые соответствуют бензино-
вой фракции.
На рис. 7.20 показано также переходное поведение парциальных давлений водо-
рода, сероводорода и аммиака в газовой фазе в нескольких точках слоя. Как правило,
парциальное давление водорода по высоте слоя снижается из-за химического рас-
хода и увеличения растворимости, а парциальные давления сероводорода и аммиа-
ка вследствие удаления серы и азота ведут себя противоположным образом. Картина
парциальных давлений метана не показана, так как его выход в условиях мягкого ги-
дрокрекинга пренебрежимо мал.
1,8- 10’5
1,7-Ю"5
1,6-10-5
1,5 -10’5
1,4 - IO'5
1,3 - IO’5
1,2-1O’5
1,1 • 1O’S
1,0-10-5
9,0 • 1O'6
8,0 • 10*6
7,0-10-»
6,0-Ю-6
5,0-Ю-6
4,0 • 10-6
3,0-ю-6
2,0-1 О*6
1,0-ю-6
О
О 200 400 600 800 10001200140016001800
б) 2,0-Ю-4
1,8-Ю-4
1,6-10-»
S 1,4-10-»
*
§ | 1,2-10-»
ф о
g = 1,0-10J
| | 8,0-Ю-5
п- Ф
s f 6,0-Ю-5
5 =с
С g 4,0-10’5
2,0-10’5
О
О 200 400 600 800 10001200 140016001800
а) 2,4-10-»
2,2 -1СГ»
„ 2,0-10-»
ф и 1,8-10-*
П 1,6-ю-
S 1 1,4-10-»
н * 1,2 -10-»
| ф 1,0-Ю-4
£ f 8,0 10 s
5 g 6,0 10 s
и 4,0 -10'5
2,0 • 10'5
О
О 200 400 600 800 10001200 140016001800
Время, с Время, с Время, с
8,0-10-»
7,0-10-1
6,0 10-*
5,0-10"»
4,0-10-*
3,0-10-*
2,0-10-»
1,0-10-»
О
О 200 400 600 800 100012001400 1600 1800
Э) 1,7 • 10-3
1,6-10’3
1,5 • 10-э
1,4-10-’
„ 1,3 -10-3
§ 1,2 -Ю-3
> 1,1 -10-3
с 1,0-Ю'3
| 9,0 -10-*
8,0-10-*
5 7,0-10-»
g 6,0 -ю-*
-в- 5,0-10-*
X 4,0-10-»
з,о-ю-»
2,0-10-*
1,0 -1 о-*
О
О 200 400 600 800 10001200 14001600 1800
е) 1,5-10-»
1,4-10-»
1,3-10-»
1,2-10-»
S 1,1 • 10-»
1,ою-»
§ 9,0 -10'5
S 8,0 -10’5
£ 7,0 -10-5
§ 6,0 -1 о-5
g 5,0 -10-5
О 4,0-10'5
3,0 • 10*5
2,0-10*5
1,0- ю-5
О
О 200 400 600 800 10001200 140016001800
Время, с
Время, с
Врем?, с
Рис. 7.19. Динамические концентрации: а — соединений азота; б — полициклических соединений; в — диароматических соедине-
ний; г — моноароматических соединений; д — нафтенов; е — олефинов в жидкой фазе в промышленном реакторе. Обозначения:
(...) — z = 0 см; (———) — z = 213,4 см; (--) — z = 426,7 см; (-) — z = 640,1 см; (-) — z = 853,4 см; (о) — эксперимен-
тальное значение в лабораторном реакторе
306 Часть 3. Моделирование каталитических процессов
Рис. 7.20. Динамические концентрации газойля (а) и бензиновой фракции (б), распре-
деление их концентраций в жидкой фазе в стационарном состоянии (в) и переходное
поведение парциальных давлений Н2 (г), H2S (д') и NH3 (е) в нескольких точках слоя
в промышленном реакторе. Обозначения: ( ) — z = 0 см; (---) — z = 213,4 см;
(---) — z = 426,7 см; (-) — z = 640,1 см; (-) — z = 853,4 см; (о) — эксперименталь-
ное значение в лабораторном реакторе
На рис. 7.21 представлены стационарные распределения парциальных давлений
основных газообразных соединений (Н2, H2S, NH3 и СН4) в модели промышлен-
ного реактора гидроочистки. Видно, что по высоте слоя парциальное давление во-
дорода в газовой фазе постепенно снижается, а сероводорода — возрастает по мере
гидрообессеривания. По мнению Мелиса с соавторами [Melis et al., 2004], практи-
чески постоянная величина парциального давления Н2 по высоте слоя означает, что
гидроочистка проводилась при избытке водорода. Парциальное давление метана
308
Часть 3. Моделирование каталитических процессов
незначительно возрастает из-за реакций мягкого гидрокрекинга (заключающихся
главным образом в отщеплении алкильных групп), вызванных увеличением темпера-
туры по высоте слоя. Выход аммиака пренебрежимо мал по сравнению с выходом се-
роводорода, что можно объяснить низкой степенью превращения соединений азота.
Рис. 7.21. Стационарные распределения парциальных давлений газообразных соедине-
ний и переходная динамика температуры жидкости в нескольких точках слоя в модели
промышленного реактора
На том же рисунке изображена динамика температуры жидкости в нескольких
точках слоя в промышленном реакторе. Приведены расчетные кривые изменения
температуры в жидкой фазе, начинающиеся от входной температуры, равной 340 °C,
за период времени, примерно равный 1700 с. Видно, что устанавливается заметный
градиент температур вдоль оси реактора.
Глава 7. Моделирование каталитической гидроочистки
309
ОБОЗНАЧЕНИЯ
Греческие
^C*,H2S ML Энтальпия адсорбции H2S, Дж/моль Теплота реакции j в жидкой фазе, Дж/моль
es Доля пустот в каталитическом слое, см3/см3 Пористость частиц катализатора, см3/см3 Межчастичное удержание фазы/, см3/см3 Безразмерный коэффициент эффективности катализатора в реакции j в жидкой фазе
Г Доля активных веществ
Рз Р/ Плотность каталитического слоя, г/см3 Плотность фазы/в условиях процесса, г/см3 Безразмерный стехиометрический коэффициент компонента i в реак- ции у в жидкой фазе
£ Коэффициент разбавления слоя, см3/см3 Безразмерный коэффициент формы (отношение площади поверхно- сти шара эквивалентного объема к площади поверхности частицы)
ф£ Безразмерный модуль Тиле реакции j в жидкой фазе
т Коэффициент удлинения пробега в частице катализатора, см/см
Латинские
aL Удельная (на единицу объема реактора) поверхность раздела газовой и жидкой фаз, см2/см3
as Удельная (на единицу объема реактора) поверхность раздела жидкой и твердой фаз, см2/см3
A',b' cPf Cf Безразмерные константы зависимости Бонди Удельная теплоемкость фазы/, Дж/(г- К) Молярная концентрация компонента i в сплошной жидкой фазе, моль/см3
CL Молярная концентрация компонента i на поверхности твердой фазы, покрываемой жидкой фазой, моль/см3
d pe DL et Эквивалентный диаметр частиц, см Коэффициент диффузии соединения i в жидкой фазе в поры ката- лизатора, см3/см • с
310
Часть 3. Моделирование каталитических процессов
At Коэффициент молекулярной диффузии соединения i в жидкой фазе, см3/см-с
А, Л Энергия активации реакции j, Дж/моль Эффективность смачивания катализатора, см’/см2
F G , tnL ^LS Оптимизируемая объективная функция, моль/см3 Приведенная массовая скорость жидкой фазы, г/см2-с Коэффициент теплопереноса в жидкой пленке вокруг частиц катали- затора, Дж/с • см2 К)
н с, Коэффициент Генри компонента z, МПа • см3/моль Истинные значения констант скоростей реакций мягкого гидрокре- кинга, моль/см3•с
к' . appj Кажущееся значение константы скорости гетерогенной реак- ции j, см3/моль • г • с
k'inJ Истинное значение константы скорости гетерогенной реак- ции J, см3/моль • г с
kaj к oj к- Фактор частоты столкновений для реакции j, моль/см3 • с Фактор частоты столкновений д ля гетерогенной реакции/, см’/моль -гс Коэффициент массопереноса соединения i из жидкой фазы в твер- дую, см3/см2 • с
Общий коэффициент массопереноса соединения z из газовой фазы в твердую, см3/см2 • с
К J Безразмерная константа равновесия реакции j (одинаковая для реак- ций ГДАрл// ГДАра. и ГДАрШю)
ПэЬ Константа равновесия адсорбции H2S активными центрами ката- лизатора, см3/моль
г. Oj Безразмерный предэкспоненциальный множитель для константы равновесия реакции j
F" Л0.Н25 Предэкспоненциальный множитель для константы равновесия адсо- рбции H2S, см’/моль
LB тх, mv п MW, р- Высота каталитического слоя, см Безразмерный порядок реакции Молярная масса жидкой фазы, г/моль Безразмерное число реакций в жидкой фазе Парциальное давление компонента z в сплошной газовой фазе, МПа
rJ Скорость реакции j на единицу объема в жидкой фазе, моль/см3 • с
г- Скорость реакции j на единицу массы катализатора в жидкой фазе, моль/гс
Глава 7. Моделирование каталитической гидроочистки 311
7? Sp т Tf uf V р Y 1 Универсальная газовая постоянная, Дж/моль К Общая геометрическая площадь поверхности катализатора, см2 Время, с Температура фазы/, К Приведенная скорость фазы/, см3/см2 • с Общий геометрический объем частицы катализатора, см3 Массовая доля соединения i в жидкой фазе, г/г Массовая доля в реакциях мягкого гидрокрекинга, г/г
Z Координата по высоте реактора, см
Нижние индексы
арр calc сн4 Di Кажущееся значение Расчетное значение Метан Диароматические соединения
exp f Экспериментальное значение Фаза (газ, жидкость, твердое тело); конечное состояние или состоя- ние на выходе
G GO НС HCR Газовая фаза Газойль Углеводород Реакция гидрокрекинга
н2 h2s i Молекулярный водород Сероводород Индекс компонента
in Истинное значение
j к L Mono N Naph NH3 0 Poly Индекс реакции Индекс эксперимента Жидкая фаза Моноароматические соединения Соединения азота Нафтены Аммиак Олефины Полиароматические соединения
312
Часть 3. Моделирование каталитических процессов
5 Соединения серы; твердая фаза; состояние на внешней поверхности
частиц катализатора
О Начальное значение или значение на входе; начальное условие
ГДА Реакция гидродеазотирования
ГДАр Реакция гидродеароматизации
ГДО Реакция гидрообессеривания
ГДОл Реакция гидрирования олефинов
Верхние индексы
G Газовая фаза; газовая сторона границы раздела газа и жидкости
L Жидкая фаза; жидкостная сторона границы раздела газа и жидкости
S Твердая фаза; граница раздела жидкой и твердой фаз
ЛИТЕРАТУРА
Akgerman, A., Collins, G.M., Hook, B.D. 1985. Effect of feed volatility on conversion in trickle-bed
reactors. Ind. Eng, Chem. Fundam. 24:396-401.
Al-Dahhan, M.H., Larachi, E, Dudukovic, M.P, Laurent, A. 1997. High-pressure trickle-bed reactors.
Ind. Eng. Chem. Res. 36:3292—3314.
Ali, S.A. 2007. Thermodynamics of hydroprocessing reactions. In Hydroprocessing of Heavy Oils and
Residua (J. Ancheyta, J.G. Speight, Eds.), CRC Press, Taylor & Francis, Boca Raton, FL, Chapter 4.
Alvarez, A., Ancheyta, J. 2008. Modeling residue hydroprocessing in a multi-fixed-bed reactor system.
Appt. Catal. A 351(2):148—158.
Alvarez, A., Ancheyta, J., Munoz, J.A.D. 2007a. Comparison of quench systems in commercial fixed-bed
hydroprocessing reactors. Eneigy Fuels 21:1133-1144.
Alvarez, A., Ramirez, S., Ancheyta, J., Rodriguez, L.M. 2007b. Key role of reactor internals in
hydroprocessing of oil fractions. Eneigy Fuels 21:1731—1740.
Ancheyta, J. 2007. Reactors for hydroprocessing. In Hydroprocessing of Heavy Oils and Residua
(J. Ancheyta, J.G. Speight, Eds.), CRC Press, Taylor & Francis, Boca Raton, FL, p. 92.
Ancheyta, J., Betancourt, G., Marroquin, G., Centeno, G., Munoz, J.A.D., Alonso, E 2010. Process
for the catalytic hydrotreatment of heavy hydrocarbons of petroleum. U.S. Patent 7651604 B2. January 26.
Ancheyta, J., Centeno, G., Trejo, E, Marroquin, G. 2003. Changes in asphaltene properties during
hydrotreating of heavy crudes. Energy Fuels 17(5):1233—1238.
Ancheyta, J., Rana, M.S., Furimsky, E. 2005. Hydroprocessing of heavy petroleum feeds: Tutorial. Catal.
Today 109:3—15.
Ancheyta, J., Speight, J. 2007a. Heavy oils and residua. In Hydroprocessing of Heavy Oils and Residua
(J. Ancheyta, J.G. Speight, Eds.), CRC Press, Taylor & Francis, Boca Raton, FL, Chapter 1.
Ancheyta, J., Speight, J. 2007b. Hydroprocessing chemistry. In Hydroprocessing of Heavy Oils and Residua
(J. Ancheyta, J.G. Speight, Eds.), CRC Press, Taylor & Francis, Boca Raton, FL, Chapter 3.
Baltanas, M.A., Vanraemdonck, K.K., Froment, G.E, Mohedas, S.R. 1989. Fundamental kinetic
modeling of hydroisomerization and hydrocracking on noble-metal-loaded faujasites. 1. Rate parameters
for hydroisomerization. Ind. Eng. Chem. Res. 28:899—910.
Глава 7. Моделирование каталитической гидроочистки
313
Bej, S.K. 2002. Performance evaluation of hydroprocessing catalysts: A review of experimental techniques.
Energy Fuels 16(3):774—784.
Beuther, H., Schmid, B.K. 1963. Reaction mechanisms and rates in residue hydrodesulfurization.
In Proceedings ofthe 6th World Petroleum Congress, Frankfurt, Germany Section3, pp. 297—310.
Bhaskar, M., Vilavarasu, G., Sairam, B., Balaraman, K.S., Balu, K. 2004. Three-phase reactor model to
simulate the performance of pilot-plant and industrial trickle-bed reactors sustaining hydrotreating reactions.
Ind. Eng. Chem. Res. 43(21):6654—6669.
Bird, R.B., Stewart, WE., Lightfoot, E.N. 2002. Transport Phenomena, John Wiley & Sons, NewWrk.
Bondi, A. 1971. Handling kinetics from trickle-phase reactors. Chem. Tech. l(March):185—188.
Carberry, J., Vurna, A. 1987. Chemical Reaction and Reactor Engineering, Chemical Industries/26, Marcel
Dekker, New Y>rk.
Carnahan, B., Luther, H.A., Wilkes, J.O. 1969. Applied Numerical Methods, John Wiley and Sons, Inc.,
NewWrk.
Chen, J., Mulgundmath, V, Wing, N. 2011. Accounting for vapor-liquid equilibrium in the modeling
and simulation of a commercial hydrotreating reactor. Ind. Eng. Chem. Res. 50:1571-1579.
Cheng, Z.-M., Fang, X.-C., Zeng, R.-H., Han, B.-P, Huang, L., Wan, W-K. 2004. Deep removal
of sulfur and aromatics from diesel through two-stage concurrently and countercurrently operated fixed-bed
reactors. Chem. Eng. Sci. 59(22—23):5465—5472.
Chou, T 2004. Causes of fouling in hydroprocessing units. Petrol. Technol. Q. Q4:79—85. Chowdhury, R,
Pedemera, E., Reimert, R. 2002. Trickle-bed reactor model for desulfurization and dearomatization of diesel.
Л/СЛЕ/. 48(1):126—135.
Cooper, B.H., Donnis, B.B.L., Moyse, B. 1986. Hydroprocessing conditions affect catalyst shape
selection. OG/84(49):39-44.
Dahl, I.M, Tangstad, E., Mostad, H.B., Andersen, K. 1996. Effect of hydrotreating on catalytic cracking
ofa VGO. Energy Fuels 10:85—90.
Ddhler, W, Rupp, M. 1987. Comparison of performance of an industrial VGO-treater with reactor
model predictions. Chem. Eng. Technol. 10(l):349—352.
Dolbear, G.E. 1998. Hydrocracking: Reactions, catalysts, and processes, \n Petroleum Chemistry and Refining
(J.G. Speight, Ed.), Taylor & Francis, Wishington, DC, Chapter 7.
Dudukovic, M.P 1977. Catalyst effectiveness factor and contacting efficiency in trickle-bed reactors.
AIChEJ. 23(6):940—944.
Enerdata. 2011. Global Energy Statistical Yearbook. Available on http://www.enerdata.net [Retrieved
2011-01-15].
Froment, G.E 2004. Modeling in the development of hydrotreatment process. Catal. Today 98:43-54.
Froment, G.E, Bischoff, K.B., De Wilde, J. 2010. Chemical Reactor Analysis and Design, 3rd edn.,
Wiley, NewWrk.
Froment, G.E, Depauw, G.A., Vmrysselberghe, V 1994. Kinetic modeling and reactor simulation in
hydrodesulfurization of oil fractions. Ind. Eng. Chem. Res. 33( 12):2975—2988.
Furimsky, E. 1983. Chemistryofcatalytichydrodeoxygenation. Catal. Rev. Sci. Eng. 25:421—458.
Furimsky, E. 1998. Selection of catalysts and reactors for hydroprocessing. Appl. Catal. A
Furimsky, E., Massoth, EE. 2005. Hydrodenitrogenation of petroleum. Catal. Rev. Sci. Eng. 47:297-489.
Gauthier, T, Danial-Fortain, P, Merdrignac, L, Guibard, I., Quoineaud, A.-A. 2008. Studies
on the evolution of asphaltene structure during hydroconversion of petroleum residues. Catal. Today
130(2-4):429—438.
Ghosh, P, Andrews, A.T, Quann, R.J., Halbert, TR. 2009. Detailed kinetic model for the
hydrodesulfurization of FCC naphtha. Energy Fuels 23:5743—5759.
Gioia, E, Lee, V 1986. Effect ofhydrogen partial pressure on catalytic hydrodenitrogenation of quinoline.
Ind. Eng. Chem. Proc. Des. Dev. 25:918—925.
314
Часть 3. Моделирование каталитических процессов
Girgis, M.J., Gates, В.С. 1991. Reactivities, reaction networks, and kinetics in high-pressure catalytic
hydroprocessing. Ind. Eng. Chem. Sci. 30:2021—2058.
Gosselink, J.W 1998. Sulfide catalysts in refineries. Catlech 2:127-144.
Gray M.R., Ayasse, A.R., Chan, E.W, Vtljkovic, M. 1995. Kinetics of hydrodesulfurization of thiophenic
and sulfide sulfur in athabasca bitumen. Energy Fuels 9(3):500-506.
Gruia, A. 2006. Recent advances in hydrocracking. In Practical Advances in Petroleum Processing,
Volume 1 (C.S. Hsu, PR. Robinson, Eds.), Springer, New York, Chapter 8. Hallie, H. 1982. Experience
reveals best presulfiding techniques for HDS and HDN catalysts. Oil and Gas J. 68:69-74.
Ho,TC. 1988. Hydrodenitrogenation catalysis. Catal. Rev. Sci. Eng. 30:117—160.
Ho, TC. 2008. Kinetic modeling of large-scale reaction systems. Catal. Rev. Sci. Eng. 50(3):287—378.
Ho, TC., Aris, R. 1987. On apparent second-order kinetics. AIChEJ. 33(6): 1050—1051.
Ho, TC., Nguyen, D. 2006. Modeling ofcompetitiveadsorption of nitrogen species in hydrodesulfurization.
Chem. Eng. Comm. 193:460—477.
Hoekstra, G. 2007. The effects of gas-to-oil rate in ultra low sulfur diesel hydrotreating. Catal. Today
127:99-102.
International Energy Agency 2010. Key world energy statistics. Available on http://www iea.org [Retrieved
2010-12-15].
Jaffe, S.B. 1974. Kinetics of heat release in petroleum hydrogenation. Ind. Eng. Chem. Process. Des.
Dev. 13:34-39.
Janssens, J.P, Elst, G., Schrikkema, E.G., van Langeveld, A.D., Sie, S.T, Moulijn, J.A. 1996.
Development of a mechanistic picture of the hydrodemetallization reaction of metallo-tetraphenylporphyrin
on a molecular level. Reel. Trav. Chim. Pay-Bas. 115(11—12):465—473.
Jimenez, E, Ojeda, K., Sanchez, E., Karafov, V, Maciel Filho, R. 2007. Modeling of trickle bed reactor
for hydrotreating of vacuum gas oils: Effect of kinetic type on reactor modeling. In 17th European Symposium
on Computer Aided Process Engineering— ESCAPE 17 (V Plesu, PS. Agachi, Eds.), Elsevier BY Amsterdam, the
Netherlands.
Klein, M.T, Hou, G., Bertolacini, R., Broadbelt, L.J., Kumar, A. 2006. Molecular Modeling in Heavy
Hydrocarbon Conversions, CRC Press, Taylor & Francis, Boca Raton, FL.
Kobayashi, S., Kushiyama, S., Aizawa, R., Koinuma,Y, Inoue, K., Shimizu, Y, Egi, K. 1987.
Kinetic study on the hydrotreating of heavy oil. 2. Effect of catalyst pore size. Ind. Eng. Chem. Res.
26:2245-2250.
Korsten, H., Hoffmann, U. 1996. Three-phase reactor model for hydrotreating in pilot trickle-bed reactors.
AIChEJ. 42(5):1350—1360.
Kressmann, S., Morel, E, Harle, V, Kasztelan, S. 1998. Recent developments in fixed-bed catalytic
residue upgrading. Catal. Today 43:203—215.
Kumar, H., Froment, G. 2007. Mechanistic kinetic modeling of the hydrocracking of complex feedstocks,
such as vacuum gas oils. Ind. Eng. Chem. Res. 46:5881 —5897.
Kundu, A., Nigam, K.D.P, Duquenne, A.M., Delmas, H. 2003. Recent developments in hydroprocessing
reactors. Rev. Chem. Eng. 19:531—605.
Lopez-Garcia, C., Hudebine, D., Schweitzer, J.-M., \6rstraete, J.J., Ferre, D. 2010. In-depth modeling
of gasoil hydrotreating: From feedstock reconstruction to reactor stability analysis. Catal. Today 150:279-299.
Marafi, A., Fukase, S., Al-Marri, M., Stanislaus, A. 2003. A comparative study of the effect of catalyst
type on hydrotreating kinetics of Kuwaiti atmospheric residue. Energy Fuels 17(3):661—668.
Marafi, A., Maruyama, E, Stanislaus, A., Kam, E. 2008. Multicatalyst system testing methodology for
upgrading residual oils. Ind. Eng. Chem. Res. 47(3):724—741.
Marroquin, G., Ancheyta, J., Diaz, J.A.l. 2004. On the effect of reaction conditions on liquid phase
sulfiding of a NiMo HDS catalyst. Catal. Tbday 98:75—81.
Martinez, J., Sanchez, J.L., Ancheyta, J., Ruiz, R.S. 2010. A review of process aspects and modeling of
ebullated bed reactors for hydrocracking of heavy oils. Catal. Rev. Sci. Eng. 52:60-105.
Mederos, ES., Elizalde, L, Ancheyta, J. 2009. Steady-state and dynamic reactor models for
hydrotreatment of fractions: A review Catal. Rev Sci. Eng. 51 (4):485—607.
Глава 7. Моделирование каталитической гидроочистки
315
Mederos, ES., Rodriguez, М.А., Ancheyta, J., Arce, E. 2006. Dynamic modeling and simulation of
catalytic hydrotreating reactors. Energy Fuels 20(3): 936—945.
Melis, S„ Erby, L., Sassu, L., Baratti, R. 2004. A model for the hydrogenation of aromatic compounds
during gasoil hydroprocessing. Chem. Eng. Sci. 59(22—23):5671 —5677.
Mochida, I., Choi, K.-H. 2006. Current progress in catalysts and catalysis for hydrotreating. In Practical
Advances in Petroleum Piocessing, Volume 1 (C.S. Hsu, PR. Robinson, Eds.), Springer, New York, Chapter 9.
Mohanty S., Saraf, D.N., Kunzru, D. 1991. Modeling of a hydrocracking reactor. Fuel Process.
Technol. 29:1—17.
Mullins, O.C. 1995. Sulfur and nitrogen structures in asphaltenes and related materials quantified by
XANES spectroscopy In Asphaltenes: Fundamentals and Applications (E.Y Sheu, O.C. Mullins, Eds.),
Plenum Press, New York, Chapter 2.
O’Connor, P, Brevoord, E., Pouwels, A.C., Wijngaards, N.J. 1996. Catalyst deactivation in fluid catalytic
cracking: A review of mechanisms and testing methods. In Deactivation and Testing of Hydrocarbon-Processing
Catalysts (P O’Connor, T Takatsuka, G.L. Mbolery, Eds.), ACS Symposium Series, Ablume 634, Chapter 10.
OGJ. 2010. Worldwide refinery-capacities as of January 1, 2011. Oil Gas J., Dec. 6, 1:12—13.
Ouwerkerk, C.E.D., Bratland, E.S., Hagan, A.P, Kikkert, B.L.J.P, Zonnevylle, M.C. 1999. Performance
optimization of fixed bed processes. Petrol. Technol. Q. 2:21-30.
Rana, M.S., Ancheyta, J., Rayo, P, Malty, S.K. 2004. Effect of alumina preparation on
hydrodemetallization and hydrodesulfurizationof Maya crude. Catal. Today 98:151-160.
Rana, M.S., Samano, V, Ancheyta, J., Diaz, J.A.I. 2007. A review of recent advances on process
technologies for upgrading heavy oils and residua. Fuel 8:1216-1231.
Robinson, PR. 2006. Petroleum processing overview. In Practical Advances in Petroleum Processing,
Volume 1 (C.S. Hsu, PR. Robinson, Eds.), Springer, New York, Chapter 1.
Robinson, PR., Dolbear, G.E. 2006. Hydrotreating and hydrocracking: Fundamentals. In Practical
Advances in Petroleum Processing, Volume 1 (C.S. Hsu, PR. Robinson, Eds.), Springer, New York, Chapter 7.
Rodriguez, M.A., Ancheyta, J. 2004. Modeling of hydrodesulfurization (HDS), hydrodenitrogenation
(HDN), and the hydrogenation of aromatics (HDA) in a vacuum gas oil hydrotreater. Energy Fuels
18(3):789—794.
Satterfield, C.N. 1970. Mass Transfer in Heterogeneous Catalysis, MIT Press, Cambridge, MA.
Satterfield, C.N. 1975. Trickle bed reactors. AIChEJ. 21 (2):209—228.
Sau, M., Basak, K., Manna, U., Santra, M., \6rma, R.P 2005. Effect of organic nitrogen compounds on
hydrotreating and hydrocracking reactions. Catal. Today 109:112-119.
Scheuerman, G.L., Johnson, D.R., Reynolds, B.E., Bachtel, R.W, Threlkel, R.S. 1993. Advances in
chevron RDS technology for heavy oil upgrading flexibility Fuel Process. Technol. 35:39-54.
Schiesser, WE. 1991. The Numerical Method of Lines—Integration of Partial Differential Equations,
Academic Press, Inc., San Diego, CA.
Shah, YT, Paraskos, J.A. 1975. Criteria for axial dispersion effects in adiabatic trickle bed hydroprocessing
reactors. Chem. Eng. Sci. 30(9): 1169-1176.
Sie, S.T 1991. Scale effects in laboratory and pilot-plant reactors for trickle-flow processes. Rev. Inst. Ft:
dulkt. 46(4):501-515.
Speight, J.G. 2000. The Desulfurization of Heavy Oils and Residua, 2nd edn., Marcel Dekker, New York.
Speight, J.G. 2004. New approaches to hydroprocessing. Catal. Today 98:55-60. Tarhan, M.O. 1983.
Catalytic Reactor Design, McGraw-Hill, New York.
Virstraete, J.J., Le Lannic, K., Guibard, I. 2007. Modeling fixed-bed residue hydrotreating processes.
Chem. Eng. Sci. 62:5402—5408.
Vrinat, M.L. 1983. The kinetics of the hydrodesulfurization process: A review Appl. Catal. 6:137-158.
'Ahlas, S.M. 1991. Modeling with Differential Equations in Chemical Engineering, Butterworth-Heinemann,
Boston, MA.
Глава 8. Моделирование
и имитация гидропереработки
тяжелой нефти
В этой главе описываются модель реактора гидропереработки и ее применение
для расчета и имитации процесса облагораживания тяжелой нефти, разработанного
Мексиканским нефтяным институтом {IMP). Рассматриваются задачи эксперимен-
тальных исследований, их результаты, а также построение математической модели
реактора, его масштабирование и расчет. Приводится анализ результатов моделиро-
вания в различных масштабах.
8.1. ВВЕДЕНИЕ
Базовым этапом разработки химических процессов всегда является моделирование
кинетики и реактора. Главная цель моделирования — построение вычислительного
аппарата, позволяющего предсказывать распределение продуктов и поведение реак-
тора в различных рабочих условиях. В этом смысле моделирование гидропереработ-
ки тяжелой нефти представляет собой крайне трудную задачу, создаваемую не толь-
ко одновременным протеканием множества физических и химических процессов,
но и действием самых разных факторов (таких как равновесие фаз, массоперенос ре-
агентов и продуктов между газовой, жидкой и твердой фазами, диффузия в частицах
катализатора, сложная цепь реакций, потеря активности катализатора и т. д.). В иде-
але для улучшения прогнозирования необходимо учитывать вклад всех этих явлений
и факторов. Но в действительности уровень сложности модели обычно выбирают ис-
ходя из поставленных целей.
Модель реактора гидропереработки тяжелой нефти, если сравнивать ее с разрабо-
танной в главе 6 моделью реактора гидроочистки газойля тяжелой нефти, имеет свои
особенности, связанные с переработкой тяжелого сырья:
• наличие в сырье более сложных соединений, в том числе асфальтенов и соеди-
нений металлов (в основном ванадия и никеля), вступающих в реакции гидро-
деасфальтизации (ГДАс) и гидродеметаллизации (ГДМ);
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
317
• потеря активности катализатора из-за отложения кокса и металлов (причем
кокс отлагается на начальной стадии цикла службы катализатора, а металлы —
в течение всего цикла);
• повышенная глубина каталитического гидрокрекинга вакуумного остатка (тем-
пература кипения выше 538 °C), хотя и не столь большая, как в случае процес-
сов гидрокрекинга под высоким давлением — например, такого как H-OU',
• необходимость охлаждения определенных частей реактора из-за выделения
тепла в экзотермических реакциях и конструкционных ограничений на макси-
мальную температуру;
• необходимость более подробной характеризации сырья и продуктов и проведе-
ния длительных экспериментов для определения кинетики реакций и построе-
ния модели деактивации катализатора.
8.2. ОПИСАНИЕ ПРОЦЕССА ГИДРООБЛАГОРАЖИВАНИЯ
МЕКСИКАНСКОГО НЕФТЯНОГО ИНСТИТУТА
Процесс IMP — это технология предварительного каталитического гидрооблагора-
живания тяжелой нефти с целью улучшить ее транспортные и прочие характери-
стики [Ancheyta et al., 2010]. Облагороженная нефть, называемая иногда синтети-
ческой, имеет те же свойства, что и нефть промежуточной плотности (22-30 ° API),
но содержит меньше серы и других примесей. Главная особенность процесса —
каскад реакторов с неподвижным слоем, загруженных сортированными катализа-
торами, часть которых селективна к гидроочистке, часть — к гидрокрекингу, в со-
четании с ограниченным рабочим давлением для подавления образования шлама
и осадка.
Базовая технологическая схема процесса IMP изображена на рис. 8.1. Начальный
этап предусматривает разделение компонентов во всем интервале кипения тяже-
лой нефти на легкую и тяжелую (обычно это атмосферный остаток) фракции. Тяже-
лая фракция подвергается переработке во входном реакторе с неподвижным слоем,
где удаляются значительные количества металлов и асфальтенов и по меньшей мере
часть серы и азота. Частично превращенные продукты первой ступени поступают во
второй реактор с неподвижным слоем. Там удаляется значительная часть серы и азота
и происходит умеренный гидрокрекинг. Поток второго реактора направляется в се-
паратор высокого давления, где жидкие продукты отделяются от газов. Жидкость
из сепаратора высокого давления отпаривают с целью удалить остатки растворен-
ного сероводорода. Газ поступает в аминовый скруббер, где подвергается улавлива-
нию сероводорода и аммиака. Оставшийся водород высокой чистоты компримирует-
ся и возвращается в реакционную систему. Жидкость смешивают с легкой фракцией
(получая облагороженную нефть) или вместе с ней направляют в перегонную колон-
ну. В первом случае процесс служит для облагораживания нефти в целях повышения
ее товарных качеств и цены, во втором — для предварительной очистки нефти перед
подачей ее в атмосферную перегонную колонну.
318
Часть 3, Моделирование каталитических процессов
Добавочный
водород__________
Легкая фракция
Циркулирующий
водород
Отдув части газа
Тяжелая
нефть
Кислый газ
Облагоро-
женная нефть
давления
Рис. 8.1. Процесс //WP для облагораживания тяжелой нефти
На рис. 8.2 в качестве примера результативности процесса IMP показаны свой-
ства различных облагороженных нефтей и исходных тяжелых нефтей, очисткой ко-
торых первые были получены. В целом наблюдается заметное улучшение физических
и химических свойств нефти. Значительно снижается содержание серы (на 70—80 %),
металлов (на 60—80 %) и асфальтенов (на 60—70 %). В результате большего выхода
легких фракций существенно увеличивается плотность по шкале API. Улучшение
характеристик значительно облегчает трубопроводную транспортировку и перера-
ботку нефти и увеличивает ее рыночную ценность. На рис. 8.2 показаны также кри-
вые разгонки сырья и продукта для случая 1. Величины выхода средних дистиллятов
(фракции реактивного топлива, керосина и легкого прямогонного газойля) и сырья
каталитического крекинга (фракции тяжелого прямогонного и вакуумного газойля)
возрастают соответственно с 14,0 до 20,5 %об., тогда как выход бензиновой фрак-
ции остается практически неизменным, а выход вакуумного остатка резко падает
(примерно на 23,0 %об). То обстоятельство, что условия процесса благоприятству-
ют выходу средних дистиллятов и сырья каталитического крекинга по сравнению
с выходом бензиновой фракции, говорит о том, что процесс ориентирован на мягкий
гидрокрекинг. Это объясняется умеренной жесткостью условий реакций, которые
выбираются так, чтобы достичь оптимального баланса между качеством продукта,
с одной стороны, и скоростью потери активности катализатора и загрязнением слоя
(образованием осадка) — с другой. При этом нужно обеспечить еще и приемлемую
длительность цикла.
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
319
8.3. ЭКСПЕРИМЕНТАЛЬНЫЕ ИССЛЕДОВАНИЯ
На основе результатов серии детальных лабораторных экспериментов была постро-
ена и проверена модель процесса облагораживания тяжелой нефти. Для сбора дан-
ных об общих показателях процесса в целом, кинетике реакций и динамике потери
активности катализатора разработали программу экспериментальных работ. В даль-
нейших подразделах даются описания каждого экспериментального исследования
и полученных результатов.
8.3.1. Получение кинетических данных
Чтобы исследовать кинетику реакций процесса облагораживания тяжелой нефти,
провели серию экспериментов [Alvarez and Ancheyta, 2008]. Установку оснастили
двумя последовательно соединенными реакторами с неподвижным слоем. Каждый
реактор был изготовлен из трубной стали и мог вмещать до 500 см3 катализато-
ра. (Упрощенная схема установки представлена на рис. 6.2, см. главу 6.) Реакторы
на всю высоту загружались тремя слоями экструдированного никель-молибденового
Случай 1 Случай 2 Случай 2
Сырье Продукт Сырье Продукт Сырье Продукт
Плотность, °АР1 21,3 27,5 15,9 25,2 9,89 22,6
Содержание серы, %масс. 3,57 1,08 4,60 1,10 5,72 1,10
Содержание Ni + V, мг/кг 352 152 430 97 588 99
Содержание компонентов, нерас- творимых в С7, %масс. 11,3 3,7 15,7 4,7 20,9 8,3
Рис. 8.2. Типичные свойства нефти, облагороженной
посредством процесса IMP
320
Часть 3. Моделирование каталитических процессов
катализатора: входной слой (30 %) состоял из катализатора, избирательного к ГДМ,
средний слой (30 %) — из катализатора со сбалансированными активностями ГДМ
и гидрообессеривания (ГДО), выходной слой (40 %) — из катализатора, избиратель-
ного к ГДО и гидрокрекингу. Имелась возможность подавать водород во второй реак-
тор, чтобы возместить расход водорода в первом реакторе, а также имитировать водо-
родное охлаждение. Катализаторы сульфидировались в слое прямогонным газойлем
(1,46 %масс. серы) с содержанием 1,0 %масс. диметилдисульфида в следующих рабо-
чих условиях: давление 2,75 МПа, скорость подачи газойля 2000 см3/ч, кратность во-
дорода к сырью 356 н. м’/м3, температура 260 °C в течение первых 3 ч работы и 320 °C
в течение последующих 12 ч.
Сырье приготавливалось разделением мексиканской тяжелой нефти (плотность
13 ° API) на легкую и тяжелую (нижняя граница кипения 343 °C) фракции в соответ-
ствии с технологической схемой процесса 1МР(см. рис. 8.1). Тяжелой фракцией, ко-
торая подавалась в реакционную систему, в данном случае был атмосферный остаток,
а легкой — смесь бензиновой фракции и средних дистиллятов. В табл. 8.1 представле-
ны их физические и химические свойства, а также выход после фракционирования.
Таблица 8.1
Свойства сырья для кинетических исследований
Свойства Сырье
Тяжелая нефть Тяжелая фракция Легкая фракция
Выход жидкостей, %об. 100 73,88 26,12
Плотность при 15,6 °C, г/см3 0,9788 1,0326 0,8253
Плотность, ° API 12,9 5,4 39,8
Кинематическая вязкость, сСт:
при 25 °C 16 555 — 2,07
» 121 °C — 1638 —
Содержание С, %масс. 82,75 82,50 83,80
Содержание Н, %масс. 10,58 9,80 13,48
Молярное соотношение Н/С 1,524 1,410 1,917
Содержание S, %масс. 5,19 5,74 2,05
Содержание N, мг/кг 4771 5960 236
Содержание металлов, мг/кг:
Ni 83 102 0
V 501 620 0
Ni + V 584 722 0
Содержание компонентов, нераствори- мых в «-С7, %масс. 17,03 21,77 0
Разгонка, °C: ASTM D2892 ASTM D\ 160 ASTMD86
НК 45 296 812
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
321
Таблица 8.1, окончание
Свойства Сырье
Тяжелая нефть Тяжелая фракция Легкая фракция
выкипает 5/10 %об. 126/187 372/401 129/137
» 20/30 %об. 281/367 475/521 159/181
» 40/60 %об. 454/- 541/- 204/241
» 90 %об. — — 288
кк — — 318
Отгон при 538 °C, %об. 52,38 35,25 —
Детали программы эксперимента показаны на рис. 8.3. Чтобы провести испыта-
ния при установившейся активности катализатора, свежий катализатор вначале ста-
билизировали низкотемпературной (360 °C) переработкой сырья в течение 100 ч.
После начала периода потери активности опыты проводили при последовательно
увеличивающихся значениях объемной скорости подачи сырья (0,25-1,0 ч-1) и тем-
пературы (380-420 °C). Значения давления и кратности водорода к сырью в тече-
ние всего опыта поддерживались на постоянных уровнях (соответственно 9,81 МПа
и 891 н. м3/м3). Добавочный водород для компенсации химического расхода в пер-
вом реакторе подавался между реакторами в соотношении 151 н. м3/м3. При каждом
испытании в течение 5 ч устанавливались требуемые рабочие условия, затем в тече-
ние 5—12 ч (в зависимости от скорости подачи сырья) после стабилизации процес-
са получали представительные пробы продуктов. В стационарных условиях дважды
отбираются пробы продукта и определяется баланс масс. Опыты проводятся при об-
щей продолжительности работы катализатора, не превышающей 250 ч, чтобы свести
к минимуму влияние потери активности на кинетические данные. Через некоторое
время после окончания опытов проверяют активность катализатора (на рис. 8.3 пер-
вое и проверочное испытания отмечены кружками).
440 -
420 -
Загрузка
и сульфиди-
рование
катализатора
Начальный
период потери
активности
400 -
380 -
360 -
Кинетические эксперименты
Проверочное
Объемная
скорость
подачи сырья
0,25 ч-’
Объемная скорость подачи
испытание
0 50 100 150 200 250 300 350
Время работы, ч
Рис. 8.3. Детали программы эксперимента по формированию кинетических данных
322 Часть 3. Моделирование каталитических процессов
Физические и химические свойства проб сырья и продуктов определяли следу-
ющими стандартными методами: плотность по шкале API — ASTM £>287; кинема-
тическую вязкость — ASTM £>7042 (вискозиметром Штабингера SVM 3000); общее
содержание серы — ASTM £>4294; общее содержание углерода, водорода и азота —
ASTM £>5291; содержание компонентов, нерастворимых в н-С7, — ASTM D3219-, со-
держание металлов — (Ni и V) — ASTM£>5863 (атомно-адсорбционный метод); выход
вакуумного остатка (538 °С+) — разгонкой по методу ASTM D\ 160.
На рис. 8.4 показана динамика содержания металлов и серы в продуктах гидро-
очистки в течение эксперимента. Первые 100 ч работы соответствуют начальному
периоду закоксовывания, который всегда имеет место, после чего активность ка-
тализатора становится сравнительно стабильной. При гидропереработке тяжелой
нефти принято выделять два периода деактивации: 1) потеря активности катали-
затора вследствие отложения кокса на поверхности частиц; 2) потеря активности
из-за отложения металлов. Закоксовывание происходит в течение первых часов
работы слоя, отложение металлов — вплоть до окончания цикла. Потеря актив-
ности катализатора при гидропереработке тяжелой нефти может быть значитель-
ной, что зависит от структурных свойств катализатора, условий реакций и уров-
ня содержания примесей в сырье. Для улучшения стабильности рекомендуется
применять во входном слое катализатор с высокой металлоемкостью, чтобы по-
следующие слои, предназначенные для других целей (например, для ГДО), могли
перерабатывать сырье с низким содержанием металлов. Применяемая в экспери-
ментах каталитическая система была рассчитана на переработку тяжелой неф-
ти и состояла из входного слоя катализатора ГДМ с высокой металлоемкостью,
за которым следовали катализаторы ГДМ/ГДО и ГДО (патентованная каталити-
ческая система IMP). Поэтому ожидалось, что после первого и неизбежного пери-
ода деактивации вследствие закоксовывания активность катализатора останется
более или менее постоянной. Это четко демонстрирует рис. 8.4, на котором на-
блюдается постепенное снижение активности ГДО и ГДМ в течение нескольких
первых часов работы, после чего к концу первых 100 ч она остается сравнитель-
но стабильной.
Кинетические испытания проводились в течение 225 ч работы при четырех значе-
ниях объемной скорости подачи сырья (LHSV) при каждом из трех значений темпе-
ратуры, что дает в общей сложности 12 испытаний. В целом наблюдалось увеличение
глубины ГДО и ГДМ с повышением температуры и снижением LHSV. Провероч-
ное испытание, проведенное после окончания эксперимента, позволило установить,
что в течение эксперимента (на рис. 8.4 первое и проверочное испытания отмечены
кружками) активность катализатора снизилась менее чем на 1 %. На основании этого
был сделан вывод, что в ходе экспериментов с изменением LHSVи температуры по-
теря активности катализатора, обусловленная отложениями металлов, была незначи-
тельной, так что предположение о постоянной активности катализатора можно счи-
тать справедливым.
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
323
Время работы, ч
Рис. 8.4. Кривые содержания металлов и серы в продукте в течение эксперимента:
() — Ni + V; (•) — S; (о) — проверочное испытание; Г, = 380, 400, 420 °C
8.3.2. Исследование влияния тяжелого сырья
на деактивацию катализатора
Основной недостаток гидропереработки тяжелого сырья в реакторах с неподвижным
Слоем — старение катализатора вследствие отложения металлов (Ni + V) и кокса. Не-
сомненно, это главная проблема, так как от скорости потери активности зависят дли-
тельность цикла и экономические показатели процесса. Скорость потери активности
определяется составом сырья — прежде всего, содержанием металлов и асфальтенов.
Типичные величины срока службы катализатора при переработке различного сырья
показаны на рис. 1.14 (см. главу 1). Легкое сырье (например, бензиновая фракция
и газойль) снижает активность катализатора только путем сравнительно медленно-
го процесса отложения кокса, и установка может работать несколько лет без заме-
ны катализатора. В случае тяжелого сырья (тяжелая нефть и остаток перегонки) срок
службы катализатора может сократиться, в зависимости от содержания примесей,
до 6—12 мес.
В литературе можно найти множество подтверждений тому, что деактивация раз-
ного сырья носит индивидуальный характер. Поэтому при разработке всякого про-
цесса важно оценить влияние сырья различных типов на активность катализатора.
В целях оценки влияния состава сырья на деактивацию катализатора проводился ряд
экспериментов [Centeno et al., 2012]. Испытания проводились в лабораторном реак-
торе с неподвижным слоем (см. рис. 6.1 главы 6 и рис. 7.14 главы 7), загруженном
100 см3 катализатора. Использовался промышленный никель-молибденовый катали-
затор на алюмооксидном носителе, содержащий 10,7 %масс. молибдена и 2,9 %масс.
никеля. Катализатор имел следующие характеристики: экструдированные частицы
четырехлепестковой формы с удельной поверхностью 175 м2/г, удельным поровым
324
Часть 3. Моделирование каталитических процессов
объемом 0,56 см’/г и средним диаметром пор 127 А. Катализатор сульфидировался
на месте дизельной фракцией с 2,5 %масс. диметилдисульфида по той же методике
и при тех же рабочих условиях, что были описаны в предыдущем подразделе.
Исследования проводились на тяжелом сырье трех различных вддов: тяжелой
нефти (TH), атмосферном остатке тяжелой нефти (АОТН) и особо тяжелой нефти
(ОТН). Основные физические и химические свойства сырья перечислены в табл. 8.2.
Атмосферный остаток тяжелой нефти получали фракционированием TH в сте-
клянной колонне емкостью 40 л. Видно, что все типы сырья содержат много метал-
лов (350-535 мг/кг никеля и ванадия в сумме) и асфальтенов (10—19 %масс.), из-за
чего хорошо подходят для исследования потери активности вследствие отложения
металлов.
Таблица 8.2
Свойства тяжелого сырья и его асфальтенов
Свойства TH АОТН ОТН
Нефть
Относительная плотность, 15,6 °С/15,6 °C 0,9264 1,0219 0,9845
Плотность, °АР1 21,24 6,97 12,23
Содержание S, %масс. 3,44 4,73 5,25
Содержание компонентов, нерастворимых в С7 ,%масс. 10,91 17,74 19,23
Содержание Ni, мг/кг 54,7 87,6 87,1
Содержание V, мг/кг 298,8 411,5 448,0
Асфальтены
Содержание С, %масс. 83,86 83,22 83,91
Содержание Н, %масс. 7,58 7,52 7,57
Содержание S, %масс. 7,11 7,55 7,15
Атомное соотношение Н/С 1,0770 1,0766 1,0749
Эксперименты на каждом сырье проводились при следующих рабочих услови-
ях: три значения температуры (380, 400 и 420 °C); объемная скорость 0,5 ч-'; давле-
ние 6,86 МПа; кратность водорода к сырью 891 н. м3/м3. Результативность процесса
определяли по пробам, отбирая их через каждые 10 ч в течение 240 ч работы. Про-
дукты описывали перечисленными выше стандартными методами анализа. После за-
вершения эксперимента катализатор был выгружен из сосуда реактора для анализа.
В эксперименте с ОТН пробы катализатора отбирались из всех трех слоев (входного,
среднего и выходного). Содержание Ni и V на отработанном катализаторе определя-
ли атомно-абсорбционным спектрометром Solaar АА. Содержание серы определя-
лось сжиганием в элементном анализаторе Perkin-Elmer-24GG.
На рис. 8.5 представлена динамика изменения глубины реакций ГДМ, ГДО
и ГДАс для трех видов сырья при различных температурах. При наинизшей
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
325
температуре (380 °C) виды сырья по глубине удаления примесей располагались
вследующем порядке: TH > АОТН > ОТН. Этот факт можно объяснить разной ак-
тивностью видов сырья. Очевидно, что для превращения наиболее легкого сырья
достаточно меньших температур. Тяжелое сырье содержит преимущественно более
сложные структуры и поэтому труднее всего поддается облагораживанию. При по-
вышенных температурах (400-420 °C) тенденция изменяется в пользу ОТН: TH >
ОТН > АОТН. Это может говорить об изменении избирательности реакций: она бо-
лее благоприятна для превращения примесей, содержащихся в ОТН, чем в АОТН.
При 380 °C в процессе преобладают реакции гидрирования [Ancheyta et al., 2003].
Такие условия подходят для переработки сырья с относительно низким содержа-
нием асфальтенов. При повышенной температуре начинает преобладать гидрокре-
кинг таких высокомолекулярных соединений, как асфальтены. В целом это спо-
собствует удалению серы, азота и металлов, так как большинство гетероатомов
скрыто внутри асфальтеновых молекул. Именно этим объясняется то обстоятель-
ство, что для любого сырья на рис. 8.5 с увеличением степени ГДАс возрастают сте-
пени и ГДМ, и ГДО.
#
2
р
л
Z
Ф
с
ф
О
100
80
60
40
20
О
100
90
80
70
60
50
100
90
80
70
60
380 °C
□□аааа
400 °C
420 °C
О
°°
В°о°°°ооОп„
••в°пп ООО°°ооооооооо
•••Ва°оп°°%аоапа
80
70
60
50
40
30
20
380 °C
100
90
80
70
60
50
40
30
20
380 °C
I
о
°0о°ооооооооооо
• °°ооо
400 °C
90
g 80
(_
£ 70
ф
ф 60
- 50
400 °C
ВВссВ а
, °BO00BogBnD
100
0s
и 90
Ч 80
л 70
ф 60
<5 50
° 40
30
420 °C
50 I...................... ‘
0 50 100 150 200 250
100
90
80
70
60
50
0 50 100 150 200 250
°В0а
’• °В8воо
•_ О“8°ааааао0
о оо о
........
100
90
80
70
60
50
420 °C
40 I- -..................
0 50 100 150 200 250
Время работы, ч
Время работы, ч
Время работы, ч
Рис. 8.5. Динамика изменения глубины реакций ГДМ, ГДО и ГДАс:
(о) — ТН; (•) — АОТН; (□) — ОТН
326
Часть 3. Моделирование каталитических процессов
Что касается картины деактивации, все кривые показывают довольно хорошо вы-
раженное гиперболическое снижение активности в течение первых 100—120 ч рабо-
ты, после чего происходит линейная и сравнительно медленная потеря активности.
Первый этап вызывается начальным отложением кокса, происходящим в первые
часы работы и затем достигающим установившегося состояния. Второй этап обуслов-
лен отложением металлов, продолжающимся в течение всего цикла. Эксперимен-
тальные данные говорят об отсутствии явного влияния типа сырья на общую картину
деактивации катализатора. Это может объясняться тем, что начальный этап потери
активности определяется удельной коксонасыщенностью катализатора, которая за-
висит не от свойств сырья, а от концентрации кислотных центров. Но с увеличени-
ем времени работы скорость деактивации начинает все больше зависеть от скорости
отложения металлов, которая, в свою очередь, определяется свойствами сырья и ус-
ловиями реакций. Ниже будет представлен более тщательный анализ, опирающийся
на имитацию реактора.
На рис. 8.6 представлены результаты анализа проб отработанного катализатора,
отобранных в различных точках реактора. На входе в реактор в основном отлагает-
ся ванадий, тогда как углерод скапливается преимущественно на выходе. Убывающая
концентрация ванадия говорит о том, что входная часть слоя улавливает больше ме-
таллов. Именно по этой причине в промышленных реакторах применяются сортиро-
ванные каталитические системы, входной катализатор которых более стоек к отложе-
нию металлов и защищает от них нижележащие катализаторы, рассчитанные на другие
реакции (например, на реакции ГДО и гидрокрекинга). Рост концентрации углерода
объясняется тем, что кокс образуется в результате последовательных реакций, проте-
кающих ниже по слою [Furimsky and Massoth, 1999]. Кривые, описывающие влияние
температуры на скорости отложения углерода и ванадия, почти одинаковы.
Содержание V, %масс.
Содержание С, %масс.
Рис. 8.6. Содержание ванадия и углерода в пробах отработанного катализатора,
отобранных в различных точках каталитического слоя после переработки ОТН'
(о) — 380 °C; (•) — 400 °C; (□) — 420 °C
Глава 0. Моделирование и имитация гидропереработки тяжелой нефти 327
8.3.3. Испытание катализатора на долговременную стабильность
Стабильность катализатора крайне важна, так как определяет продолжительность
рабочего цикла. В этом смысле способность входного катализатора накапливать
большие количества металлов также имеет большое значение. Назначение входно-
го катализатора — разъединять молекулы асфальтенов и облегчать удаление никеля
и ванадия с тем, чтобы нижележащие катализаторы могли перерабатывать частично
деметаллизованное сырье. Так как скорость реакций лимитируется диффузией круп-
ных молекул асфальтенов внутри частиц, важнейшей характеристикой катализато-
ров этого типа становится размер пор [Furimsky, 1998]. Если диаметр пор слишком
мал, их устья быстро забиваются, что ведет к преждевременному прерыванию цикла.
Был проведен эксперимент длительностью 5,2 мес. по испытанию многослойной
каталитической системы на стабильность в типичных условиях процесса IMP [Alva-
rez et al., 2011]. Исследование проводилось на другой лабораторной установке, со-
стоящей из двух последовательных реакторов с неподвижным слоем (каждый из ко-
торых вмещал около 900 см3 катализатора); схема установки аналогична той, что
показана на рис. 6.2 (см. главу 6). Весь объем реакторов был загружен такой же трех-
слойной каталитической системой, как и в экспериментах по получению кинетиче-
скихданных (ее активировали идентичным способом). Сырьем служил атмосферный
остаток, описанный в табл. 8.1 (единственное отличие состояло в том, что он был по-
лучен из тяжелой нефти с плотностью 13 ° API).
Немаловажный аспект испытания катализатора на стабильность — выбор режима
его работы [Marafi et al., 2008] (предполагающий либо постоянную производитель-
ность, либо постоянную температуру). Для промышленной эксплуатации наиболее
представителен первый режим, когда температуру периодически повышают с целью
компенсировать потери активности. По этой причине стабильность катализатора
в данном исследовании оценивалась именно для первого режима. В отличие от боль-
шинства процессов облагораживания остатков, работающих при постоянной глуби-
не ГДО, целью было поддерживать постоянную плотность продукта, так как этот па-
раметр лучше всего характеризует нефть.
Исследование проводилось при следующих рабочих условиях: LHSV = 0,25 ч-1;
начальная температура 380 °C; давление 9,81 МПа; кратность водорода к сы-
рью 891 н. м3/м3. Пробы продуктов для определения баланса масс отбирались каж-
дые 12 ч. Чтобы обеспечить слежение за поведением первого реактора, каждые 24 ч
отбирались пробы между реакторами. Объем проб не превышал 2 % от общей массы
сырья: это пресекало возникновение заметных возмущений в системе. Такого коли-
чества вполне достаточно для проведения анализов — в частности, предназначенных
для определения содержания металлов в продуктах и оценки их количества, погло-
щаемого в первом реакторе.
На рис. 8.7 изображены последовательность изменений температуры и поведе-
ние плотности по шкале API облагороженной нефти в течение всего цикла. Темпе-
ратуру повышали периодически (начиная с 380 °C), с тем чтобы поддерживать плот-
ность облагороженной нефти на уровне 23 ° API. Имели место два случая нарушения
непрерывности (один — через 1500 ч работы, другой — через 2500 ч), вызванные
328
Часть 3. Моделирование каталитических процессов
Время работы, ч
Рис. 8.7. Порядок изменений температуры и поведение плотности облагороженной нефти
в течение цикла испытания катализатора на стабильность:
(—) — температура; (о) — плотность, ° API
эксплуатационными проблемами из-за больших перепадов давления между реак-
торами и системой сепарации. В этих случаях снижали температуру и для удале-
ния пробок переходили с тяжелого сырья на легкий газойль. Длительность наруше-
ний нормальной работы составила в общей сложности 297 ч (на рис. 8.7 эти периоды
не показаны). Как только перепады давлений возвращались в приемлемые преде-
лы, подачу сырья вновь переключали с газойля на остаток и повышали температуру
до значения, обеспечивающего целевую величину плотности по шкале API. Испыта-
ние показало, что технология IMP способна поддерживать качество продукта в пре-
делах требований в течение 3440 ч (около 5 мес.) работы. После этого, хотя плотность
по шкале API продукта и оставалась на приемлемом уровне, работу приходилось пре-
кращать из-за слишком сильного повышения температуры (выше 398 °C): подобное
решение позволяло избежать чрезмерного закоксовывания, которое могло привести
к дальнейшему загрязнению.
8.4. МОДЕЛИРОВАНИЕ
Математическая модель процесса строилась на основе модели трехфазного реакто-
ра вытеснения, которую разработали Корстен и Хоффманн [Korsten and Hoffmann,
1996]. Модель учитывает массоперенос на границах раздела жидкости с твердой и га-
зовой фазами. Для оценки коэффициентов массопереноса и свойств сред в условиях
процесса используются эмпирические зависимости. Сырье и продукты представля-
ются шестью химическими псевдокомпонентами (соединения серы, азота, никеля,
ванадия, асфальтены и вакуумный остаток с температурой кипения выше 538 °C), ко-
торые определяются общим элементным и физико-химическим методами анализа.
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти 329
В соответствии с этим модель рассматривает реакции ГДО, ГДА, ГДМ (гидродени-
келизации (ГДН) и гидродеванадизации (ГДВ)), ГДАс и гидрокрекинга вакуумного
остатка. Газовая фаза считается состоящей из водорода, сероводорода и продуктов
крекинга (представленных главным образом метаном СН4). В уравнениях материаль-
ного баланса член, описывающий реакции, представляется кинетическим выраже-
нием с кажущейся величиной константы скорости. Модель формулировалась с тем,
чтобы описать как стационарное, так и динамическое поведение. Уравнения матери-
ального и теплового балансов исходили из следующих допущений:
• реактор работает в режиме вытеснения;
• скорость жидкости постоянна по высоте реактора;
• не происходит испарения жидкости;
• давление постоянно;
• сопротивление теплопереносу между фазами отсутствует;
• в кажущихся значениях констант скоростей учитывается диффузия внутри ча-
стиц катализатора;
• деактивация катализатора коксом происходит в течение первых 100 ч работы,
а затем достигает равновесия;
• металлы отлагаются на катализаторе в течение всего цикла.
8.4.1. Стационарные уравнения баланса масс и теплоты
Уравнения модели опираются на перенос реагентов между фазами, который проис-
ходит в реакторах с орошаемым слоем [Alvarez and Ancheyta, 2008]. Водород, буду-
чи основным газообразным реагентом, первым переносится из газовой фазы в массу
жидкости. Реагенты в жидкой фазе (химические псевдокомпоненты и растворенный
водород) диффундируют к частицам катализатора, где вступают в реакцию. Газо-
образные продукты — H2S и СН4 — проходят через жидкую фазу и переносятся в га-
зовую, а углеводородные продукты возвращаются в жидкость.
Изменение молярного потока газообразных соединений по высоте реактора равно
скорости переноса из газовой фазы в жидкую:
Р N- ri
dN°
dz
= -A,ldaL
(8.1)
где i — множество, элементами которого являются Н2, H,S и СН4.
Изменение концентрации газообразных соединений в жидкой фазе обусловлено
переносом масс из газовой фазы в жидкую и в твердую:
dCf 1 (,L ( Р N°
dz uL[ '
-Cf -к°а8((^-С°)
)
(8.2)
где i — множество, элементами которого являются Н2, H2S и СН4.
330
Часть 3. Моделирование каталитических процессов
Химические псевдокомпоненты переносятся из массы жидкости на поверхность
частиц катализатора:
^L = -±kfas(C‘--Cf), < (8.3)
az uL
где i — множество, элементами которого являются соединения серы (S), азота (N),
никеля (Ni), ванадия (V), асфальтены (Asph) и вакуумный остаток (ВО).
Вещества, переносимые через границу раздела жидкой и твердой фаз, расходуют-
ся или образуются в химической реакции:
^(С^-С^) = ±г.и,0, (8-4)
где i — множество, элементами которого являются Н2, H2S, СН4, S, N, Ni, V, Asph
и ВО; г. представляет локальную скорость реакцииJ в точке z по высоте реактора в мо-
мент времени t, причем j пробегает множество, элементами которого являются реак-
ции ГДО, ГДА, ГДН, ГДВ, ГДАс и гидрокрекинга. Для реагентов берется знак «ми-
нус», для продуктов — знак «плюс».
Для представления адиабатической работы промышленного реактора необходимо
учесть тепловой баланс. Если допустить, что все три фазы имеют одну и ту же темпе-
ратуру, то псевдооднородное уравнение баланса энергии будет следующим:
^- = ((~АД'д)ггдо)-- г (8.5)
dz ' iiLpLCpL+uGpGCpG
Член, соответствующий в энергетическом балансе источнику тепла, определяется
общей теплотой (-7820 кДж на 1 кг серы) реакции ГДО атмосферного остатка [Shah
and Paraskos, 1975]. Общая теплота реакции — подгоняемый параметр, получаемый
из балансов теплоты нескольких схожих процессов гидроочистки, в которых учиты-
вается вклад всех реакций (ГДО, гидрирования и т. д.) [Dohler and Rupp, 1987]. Для
адекватного воспроизведения данных любого отдельно взятого промышленного про-
цесса эти параметры обычно требуют точной подгонки. Их значения обычно указы-
вают как количество теплоты, выделяемое в расчете на единицу количества опреде-
ленного реагента (удаленной серы, крекированного углеводорода, израсходованного
водорода и т. д.).
В промышленных реакторах гидропереработки обычно предусматривается не-
сколько точек охлаждения. Газовое охлаждение снижает температуру реакций и из-
меняет состав газовой фазы, а следовательно, и условия на входе в следующий ката-
литический слой. Охлаждение можно моделировать как смешивание охлаждающего
потока с газожидкостным потоком слоя. Скорость охлаждения q, требуемая для полу-
чения охлажденной смеси с определенной температурой Т.п или наоборот, рассчиты-
валась исходя из следующего уравнения баланса энергии [Alvarez and Ancheyta, 2009]:
Ti„ Tln Tm
J kupPidT + J gou,CpGdT + j qCpQdT = 0. (8.6)
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
331
Фактический поток газа, поступающего в следующий слой, определяется суммой
расхода охлаждающего газа и потока предыдущего слоя. Состав этого газа определя-
ется материальными балансами по отдельным газообразным соединениям.
Обширная подборка полученных из различных литературных источников эмпи-
рических зависимостей, применявшихся в данной модели для определения свойств
нефти, величин растворимости газов и коэффициентов массопереноса на границах
раздела жидкости с газообразной и твердой фазами, приведена в работе Альвареса
и Анчиты [Alvarez and Ancheyta, 2008].
8.4.2. Динамические уравнения баланса масс и тепла
Динамические уравнения модели получаются включением в уравнения материаль-
ного и теплового балансов из предыдущего подраздела члена, описывающего акку-
муляцию соответствующей физической величины. Тогда переходный материальный
баланс химических веществ в газовой фазе можно представить в следующем виде:
ео
acf
dt
(8-7)
где I — множество, элементами которого являются Н2, H2S и СН4.
Из уравнения (8.7) можно заметить, что приведенная скорость газа ис вслед-
ствие расходования Н2, образования H2S и СН4 и термического эффекта изменя-
ется вдоль оси реактора. Дополнительное выражение для оценки этого параметра
можно получить суммированием переходных балансов масс всех газообразных со-
единений [Schweitzer et al., 2010]:
duc RT гсР dT , L C°RT L ио dT
—^ =-----------У^а, —--------С, +-2-—
dz Р RT dt Y ' L Ht ' Т dz'
(8.8)
Так как объем газа вдоль оси реактора изменяется, на каждом шаге интегрирова-
ния необходимо проверять выполнение условия постоянства давления.
Динамика концентраций газообразных соединений в жидкой фазе дается
выражением
L dt dz [ Н. ' ' s\ ' • Р
(8.9)
где / — множество, элементами которого являются Н2, H2S и СН4.
Переходной баланс масс химических псевдокомпонентов в жидкой фазе имеет вид
ас dcL ,е / , -
е£ —- = -и, —!— kfaA (А - С, )
L dt L dz 1 s{ ‘ h
(8.10)
где i— множество, элементами которого являются S, N, Ni, V, Asph и BO.
332
Часть 3. Моделирование каталитических процессов
Динамическое уравнение баланса масс веществ, переносимых между жидкой фа-
зой и частицами катализатора, имеет вид
£р(1-е)^ = Л,Ч(^ -С*)±г.и,0, - (8.11)
где i — множество, элементами которого являются Н2, H2S, СН4, S, N, Ni, V, Asph
и ВО.
Наконец, переходный баланс энергий описывается выражением
„ „ dT ( „ dT „ д(игГ) . . rr . 'l
elPl^Pl + £gPgQ>g ”77 — — ^Pg^Pg д *" (—^sHr )Ддо (8.12)
dt \ dz dz ) ' '
Начальные условия для системы уравнений в частных производных выражаются
следующим образом.
Для t = 0, z = О
C»XC-U
С,с = 0,
где i — множество, элементами которого являются H2S и СН4;
с‘ =(сд.
где i — множество, элементами которого являются Н2, S, N, Ni, V, Asph и ВО;
cf = o,
где i — множество, элементами которого являются H2S и СН4;
С- = О,
где i — множество, элементами которого являются Н2, H2S, СН4, S, N, Ni, V, Asph
и ВО;
Т=Т0. (8.13)
Для t = 0, z > О
где i — множество, элементами которого являются Н2, H,S и СН4;
С- = О,
где i — множество, элементами которого являются Н2, H2S, СН4, S, N, Ni, V, Asph
и ВО;
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
333
С,5 = О,
где i — множество, элементами которого являются Н2, H2S, CH4, S, N, Ni, V, Asph
и ВО;
^По-
граничные условия имеют следующий вид:
Для/>0, z = 0
cl =(CS.L
cG = o,
где i — множество, элементами которого являются H2S и СН4;
где i — множество, элементами которого являются Н2, S, N, Ni, V, Asph и ВО;
С,£ = О,
где i — множество, элементами которого являются H,S и СН4;
С- = О,
где i — множество, элементами которого являются Н2, H2S, CH4, S, N, Ni, V, Asph
и ВО;
Т= То. (8.14)
Доля пустот в слое (е) определялась по эмпирической зависимости, предложен-
ной Хофи и Бевериджем [Haughey and Beveridge, 1969]. Для определения удержания
жидкости el (доли объема реактора, занимаемой жидкостью) была выбрана зависи-
мость Ринга и Миссена [Ring and Missen, 1991], так как они проводили исследование
в схожих условиях (ГДО дибензотиофена в трехслойном реакторе при температурах
от 330 до 370 °C и давлении 10 МПа). Удержание газа е0 было принято равным разно-
сти между долей пустот и удержанием жидкости [Whitaker, 1973]. Пористость катали-
затора Ер определялась по данным его физического описания [Mederos et al., 2009а].
8.4.3. Кинетика реакций
Разработка кинетических моделей молекулярного уровня для реакций гидроперера-
ботки остатков до сих пор остается проблемой ввиду отсутствия аналитических мето-
дов, способных дать молекулярное описание такого сырья. Тяжелые фракции могут
334
Часть 3. Моделирование каталитических процессов
содержать несколько тысяч разных компонентов. Строго говоря, это влечет за собой
великое множество последовательно и параллельно протекающих реакций и широ-
кое распределение активностей соединений, обусловленное высокой степенью по-
лидисперсности. В сложившейся практике принято ранжировать молекулы разных
типов по их глобальным свойствам, таким как элементный состав или температура
кипения, и получать хорошо известные реакционные схемы с дискретной кинети-
кой [Но, 2008].
В уравнениях баланса масс член, описывающий реакции, представляется кинети-
ческим выражением n-го порядка с кажущейся константой скорости:
г.а,о = ^Ф7(2’^(с'Г’ <8Л5)
где kj№ — кажущаяся константа скорости/-й реакции, подчиняющаяся закону Арре-
ниуса; ф — показатель деактивации (снижения активности)/-й реакции в точке с осе-
вой координатой z в момент времени t; С$ — концентрация /-го псевдокомпонента
вблизи частиц катализатора; п. — порядок /-й реакции. В случае ГДО учитываются
также ингибирующие эффекты адсорбции H2S и концентрации Н2:
(сЛ"‘до(Сн )0,5
гГД0(г, 0 = ^0ФГД0(^)\ 1 \-2У <8-16)
(1 + 7С SC„ s)
Что касается газообразных соединений, расход Н2 и образование H2S учитывают-
ся через суммарные молярные стехиометрические коэффициенты. Метан, согласно
допущению, образуется лишь при крекинге вакуумного остатка.
8.4.4. Масштабирование кинетических данных
Хорошо известно, что кинетические данные лабораторного реактора с орошаемым
слоем нельзя напрямую масштабировать и использовать для расчета промышленных
реакторов [Bej, 2002]. Газогидродинамические факторы вызывают отклонение от ре-
жима вытеснения и ухудшают смачивание каталитического слоя, что влияет на об-
щую результативность процесса. Из-за этого кажущиеся величины констант скоро-
стей часто возрастают с увеличением масштаба реактора (т. е. скорости жидкости)
[Satterfield, 1975]. В таком случае кажущееся значение константы скорости можно
определить как истинное значение, искаженное диффузионными ограничениями
внутри частиц катализатора и неполным его смачиванием:
=ЧоЧс^7, (8.17)
где к"’ — истинное значение константы скорости, а р0 и г]с£ представляют соот-
ветственно фактор эффективности катализатора и эффективность его внешнего
смачивания.
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
335
Учитывая, что эксперименты проводились с частицами катализатора промышлен-
ных размеров, фактор эффективности объединяется с к"', что дает значение констан-
ты скорости, отнесенное к частицам (кр):
к1‘рр=ч\СЕк^ (8.18)
Величина к’’ позволяет характеризовать катализатор для промышленных условий
без моделирования внутричастичной диффузии, но лишь в том случае, если в экс-
периментах используется тот же катализатор, что и выбранный для промышленного
применения [Sie, 1991]. Результирующее выражение дает связь простого вида между
характеристиками реакторов лабораторного и промышленного масштабов через эф-
фективность смачивания как параметр масштабирования. Коэффициент рС£ являет-
ся мерой степени использования катализатора, которая в условиях промышленных
масштабов стремится к единице (г]С£ ~ 1) [Mederos et al., 2009b].
Подходящий способ решения этой задачи заключается в измерении величины карр
при разных скоростях потока жидкости (и по возможности при двух масштабах ре-
акторов) и последующей подгонке данных под следующее эмпирическое уравнение
[Bondi, 1971]:
1 _ 1 _А_
карр fa? (j ® • (8d9)
Когда приведенная массовая скорость жидкости в уравнении (8.19) стремит-
ся к бесконечности, кажущееся значение константы скорости приближается к зна-
чению константы скорости, отнесенному к частице; параметры А и В являются под-
гоняемыми. Для ряда систем величина показателя степени В лежит между 0,5 и 0,7;
чаще всего она близка к 2/3. Константа скорости, отнесенная к частицам, получается
в виде 1 /кр экстраполяцией до бесконечности приведенной массовой скорости жид-
кости (определяется точкой пересечения с осью ординат). В этом случае использо-
вались данные экспериментов на установках лабораторного и полупромышленного
(с пропускной способностью около 10 баррель/сут) масштабов.
8.4.5. Деактивация катализатора
Потеря активности катализатора гидропереработки тяжелой нефти происходит
под действием двух факторов: закоксовывания и накопления отложений метал-
лов. Есть много доказательств того, что начальный период быстрой потери актив-
ности обусловлен отложением кокса, образование которого происходит в течение
первых (примерно 100) часов работы, а затем, по всей видимости, достигает рав-
новесия, тогда как металлы примерно с постоянной скоростью накапливаются те-
чение всего цикла [Furimsky and Massoth, 1999; Sie, 2001]. Вклад этих двух про-
цессов в общую скорость деактивации катализатора можно выразить следующей
формулой:
336
Часть 3. Моделирование каталитических процессов
ф.(г)0 = ф^(О + ФГЬ(^О) (8.20)
где ф“г и ф^'"'1 — соответственно функции деактивации коксом в момент времени
t < 100 ч и металлами (Ni + V) в произвольный момент времени t в течение цикла.
Функция деактивации определяется как отношение локальных скоростей у-й реак-
ции в момент времени t и в начальный момент (Г = 0, т. е. свежий катализатор).
Влияние этих двух процессов на активность катализатора моделируется в соответ-
ствии со следующим эмпирическим выражением [Alvarez et al., 2011]:
ф.-Сг, t) ~ Гр (хмос(2’0) • (8-21)
(1+М
Первый член в правой части уравнения (8.21) представляет гиперболическую на-
чальную зависимость активности катализатора ву-й реакции от времени с подгоня-
емыми параметрами а. и р;. В перечне допущений было принято, что быстрое на-
чальное снижение активности происходит в течение первых 100 ч работы, после чего
начальный период деактивации заканчивается, т. е. значение этой функции в осталь-
ной части цикла остается постоянным.
Необходимо подчеркнуть, что правильнее было бы описать процесс путем увязы-
вания этой функции с коксующимся реагентом; к сожалению, экспериментальных
сведений, которые могли бы пролить свет на этот механизм, не было. Как следствие,
не было и других способов представить это поведение. Иными словами, выражение
данного члена как функции от количества катализатора на коксе (COQ потребова-
ло бы проведения экспериментов для каждого значения времени со свежим катали-
затором, что сделало бы их очень дорогостоящими и длительными. Выбор же этой
простой функции времени оправдывается исследованиями, показавшими, что кри-
вая деактивации в начальный период цикла носит один и тот же характер для различ-
ных видов сырья в широком интервале температур.
Второй член в правой части уравнения (8.21) представляет вклад отложения ме-
таллов в деактивацию катализатора в течение всего цикла. Эта функция прямо связа-
на с количеством металлов на катализаторе в точке с осевой координатой z в момент
времени t через подгоняемый параметр дляу-й реакции, величина которого инди-
видуальна для каждого из трех катализаторов. Переменная хмос определяется как от-
ношение локальной концентрации металлов на катализаторе (МОС) в момент време-
ни Г к максимальной, которая также индивидуальна для каждого катализатора. Такая
зависимость позволяет ввести переменное во времени распределение активностей
катализаторов по оси реактора. Количество металлов на катализаторе определяется
интегрированием уравнения баланса масс
dMOC(z,t)
— амосггям (z, (8.22)
где МОС выражается в процентах по массе; ггдм — локальная скорость реакции ги-
дродеметаллизации в момент времени /; амос — коэффициент пересчета единиц
измерения.
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
337
8.4.6. Метод решения
8.4.6.1. Моделирование установившегося процесса
Стационарная модель состоит из системы взаимосвязанных обыкновенных диф-
ференциальных и алгебраических уравнений. Моделирование осуществляется па-
раллельным интегрированием уравнений баланса масс газовой и жидкой фаз в осе-
вом направлении методом Рунге—Кутты четвертого порядка. Уравнения теплового
баланса используются лишь при расчетах промышленных реакторов. Между шага-
ми интегрирования методом Ньютона—Рафсона решаются алгебраические уравне-
ния для твердой фазы. На каждом шаге интегрирования обновляются также значения
физических свойств и коэффициентов массопереноса.
Способом квазистационарного приближения осуществлялось моделирова-
ние процесса с динамически изменяющейся активностью катализатора (Lababi-
di et al., 1998]. Способ основан на том принципе, что если в масштабах длитель-
ности рабочего цикла старение катализатора происходит относительно медленно,
то можно разбить цикл на небольшие временные отрезки и принять, что в тече-
ние каждого такого отрезка процесс остается стабильным. В данном случае дли-
тельность отрезков принята равной периодичности отбора проб для определе-
ния материального баланса в испытаниях катализатора на стабильность (12 ч).
Моделирование начинается с момента времени t = 0, когда катализатор находит-
ся в свежем состоянии (для всего цикла службы катализатора ф = 1,0). По реше-
ниям стационарных уравнений баланса теплоты и масс, получаемым упомяну-
тым выше способом, определяются распределения концентраций и температуры.
Далее по осевым распределениям концентраций металлов определяются локаль-
ные количества металлов на катализаторе МОС и вычисляются функции деакти-
вации для каждой реакции. Затем дают приращение переменной времени и по-
вторяют вычисления.
8.4.6.2. Моделирование динамического процесса
Уравнения в частных производных, входящие в динамическую модель, путем дис-
кретизации по осевой координате заменой левыми конечно-разностными вы-
ражениями сводились к обыкновенным дифференциальным уравнениям. Для
пространственной дискретизации весь слой разбивался на 100 равновеликих сег-
ментов, что давало такое же число обыкновенных дифференциальных уравнений
для каждой зависимой переменной (всего получалось 2200 уравнений баланса).
Для расчетов зон охлаждения вводились дополнительные сегменты, служившие
для обновления значений температуры реакций и состава газовой фазы. Таким об-
разом, общее число сегментов в осевом направлении было равно 100+1 вход реак-
тора + число сегментов зон охлаждения. Система обыкновенных дифференциаль-
ных уравнений параллельно интегрировалась по времени методом Рунге-Кутты
четвертого порядка.
338
Часть 3. Моделирование каталитических процессов
8.5. СОГЛАСОВАНИЕ ДАННЫХ
Модель реактора подгоняли к данным лабораторной установки. Набор параметров
оптимизировали методом Левенберга—Марквардта через минимизацию суммы ква-
дратов расхождений экспериментальных и расчетных данных. Оценки параметров
осуществлялись вычислением отдельных доверительных интервалов по критерию
Стьюдента.
8.5.1. Кинетические параметры
Кинетические параметры перечислены в табл. 8.3. Можно сразу же заметить, что по-
рядок реакций (л) изменяется от 0,55 до 2,0. Это типично для дискретных кинетиче-
ских схем [Marafi et al., 2010]. Такое поведение является результатом группирования
широкого множества различных веществ в единой реакции. Порядки реакций выше
1,0 обычно объясняются большими расхождениями в активности молекул в пределах
псевдокомпонентов. Таким образом, дискретную смесь можно отобразить через мно-
жество конкурирующих реакций первого порядка активных и устойчивых веществ.
Тогда второму порядку реакции отвечает «живучесть» наиболее устойчивых веществ
в реакционной системе [Но and Aris, 1987]. Это означает, что в реакторе с неподвиж-
ным слоем наиболее активные вещества быстро исчерпываются в начальной части
слоя, а наиболее стойкие остаются на всем протяжении высоты реактора. В таком
случае второй порядок реакций ГДА и гидрокрекинга предполагает наличие очень
трудно превращаемых азотсодержащих и высокомолекулярных соединений.
Таблица 8.3
Кинетические параметры реакций гидропереработки
Реакция п Ед, кДж/моль
ГДО 1,17 3,568 109 моль/см3 ч 104,04
ГДА 2,00 1,580 • 103 мг/кг-ч 94,25
ГДН 0,55 8,73 2-106 мг/кг-ч 85,39
гдв 1,56 1,323-106 мг/кг-ч 98,52
ГДАс 0,75 7,178-108 %масс./ч 116,79
Гидрокрекинг 2,00 2,843 108 %масс./ч 141,72
Образование СН4 — 8,439-10“ %масс./ч 198,97
Дробные порядки реакций имеют явно другое происхождение, убедительное объ-
яснение которого можно найти в работе Грея и соавторов [Gray et al., 1995] (см. также
подраздел 6.4.2 настоящей книги). Согласно этому объяснению, дробный поря-
док реакций ГДАс и ГДН может быть результатом наложения механизмов после-
довательных и параллельных реакций. Эту идею поддерживают несколько групп
Главе 8. Моделирование и имитация гидропереработки тяжелой нефти
339
исследователей. Например, Готье и соавторы [Gauthier et al., 2008] выдвигают гипо-
тезу превращения крупных асфальтеновых агрегатов через ряд этапов гидрирования
и гидрокрекинга в низкомолекулярные, а Янссене и соавторы [Janssens et al., 1996]
предлагают схему реакций ГДМ металлопорфиринов, в которой молекула претер-
певает гидрирование пиррольных блоков с последующим разрывом связей Ni-N
или V-N путем гидрогенолиза, что сопровождается фрагментацией порфириновой
структуры.
Кажущиеся величины энергий активации (Ел) составляли от 85 до 142 кДж/моль
(от 20 до 34 ккал/моль), что согласуется с результатами большинства исследований,
посвященных кинетике реакций гидроочистки тяжелой нефти [Marafi et al., 2010].
Для сырья такого типа характерен низкий уровень энергий активации, так как на ско-
рости реакций крупных молекул сильно влияют скорости их внутричастичной диф-
фузии [Marafi et al., 2003].
На рис. 8.8 приведено сравнение экспериментальных и расчетных значений кине-
тических данных. Диаграммы четко свидетельствуют об их хорошем согласии. Боль-
шинство кинетических данных попадают в интервал ±5 %, хотя для реакций ГДА,
гидрокрекинга и образования легких углеводородов наблюдаются небольшие откло-
нения. В целом все точки случайно распределены по обе стороны от прямых линий;
это говорит о том, что модель не дает результатов, систематически завышенных или
заниженных по сравнению с данными экспериментальных наблюдений.
Рис. 8.8. Сравнение экспериментальных и расчетных значений кинетических данных:
(—) — границы отклонения на ±5 %
8.5.2. Параметры деактивации
В табл. 8.4 приведены значения параметров деактивации, рассчитанные по результа-
там испытаний катализатора на долговременную стабильность и исследований вли-
яния тяжелого сырья на его активность. Буквы аир обозначают параметры гипер-
болической функции, описывающей снижение активности катализатора вследствие
340 Часть 3. Моделирование каталитических процессов
отложения кокса, у — показатель степенной зависимости, представляющей медлен-
ную деактивацию отложениями металлов (см. уравнение (8.21)). Все эти параметры
не зависят от температуры; тем не менее, их значения индивидуальны для каждой ре-
акции и для каждого сырья. В случае испытания катализатора на стабильность вели-
чина параметра у зависит также от положения в каталитической системе (входной,
промежуточный или выходной слой), что позволяет выбирать скорость потери ак-
тивности каждого катализатора в определенной реакции. Для реакций ГДН и ГДВ
значения параметра у уменьшались по направлению от входа к выходу, т. е. модель
предсказывает, что по скорости потери активности в реакциях ГДН и ГДВ различ-
ные части каталитического слоя располагаются в следующем порядке: вход < середи-
на < выход. Это согласуется с поведением реальной каталитической системы: вход-
ной катализатор выбирают значительно более стойким к деактивации металлами,
чем промежуточный или выходной. В реакциях ГДО и гидрокрекинга значения па-
раметра у ведут себя в обратном порядке: выход < середина < вход. Это связано с тем,
что высокоактивный выходной катализатор ГДО и гидрокрекинга защищен входным
и средним слоями и перерабатывает частично деметаллизованное сырье.
Таблица 8.4
Параметры функций деактивации
Процесс а Р Y
Входной Промежуточный Выходной
Испытание катализатора иа долговременную стабильность
гдо 3,81 0,06 0,34 0,35 0,46
ГДН 2,70 0,08 0,96 0,75 0,45
ГДВ 4,57-10-1 0,10 3,00 0,52 0,31
Гидрокрекинг 2,80 0,05 1,15 1,49 2,70
Процесс а Р Y
Исследование влияния тяжелого сырья на каталитическую активность
гдо 1,09'Ю-2 0,30 0,21
ГДМ 4,34-10-1 0,04 0,17
ГДАс 2,34-10-' 0,08 0,15
Из рис. 8.9 можно видеть, что результаты, которые дает модель, хорошо согласу-
ются с кривыми деактивации катализатора, построенными на основании данных ис-
пытаний катализатора на долговременную стабильность и исследований влияния
тяжелого сырья на его активность. Большинство экспериментальных значений слу-
чайно распределено в интервале шириной ±5 % вокруг теоретических кривых, что
подтверждает справедливость предложенной функции деактивации. На рис. 8.9, б
видно, насколько хорошо данные снижения активности катализатора в каждой из ре-
акций (ГДО, ГДМ и ГДАс) согласуются с соответствующим единым набором параме-
тров во всем интервале температур.
Йява 8. Моделирование и имитация гидропереработки тяжелой нефти
341
Экспериментальные данные
снижения активности, %
Рис. 8.9. Сравнение экспериментальных и расчетных значений снижения активности:
а — данные испытания катализатора на долговременную стабильность; б — данные
исследований влияния тяжелого сырья на активность катализатора; (—) — границы
интервала шириной ±5 %
б)
40 50 60 70 80 90 100
Экспериментальные данные
снижения активности, %
8.6. МОДЕЛИРОВАНИЕ ПРОЦЕССА
НА ЛАБОРАТОРНОЙ УСТАНОВКЕ
8.6.1. Моделирование реактора при стационарной активности
катализатора
Использовалась модель реактора применительно к лабораторной установке при уста-
новившемся режиме работы катализатора, наступающем после начального периода
потери активности. На рис. 8.10 представлены распределения молярных концентра-
ций серы, азота и металлов по высоте реакторов в жидкой и твердой фазах в обоих ре-
акторах (/?! и 7?,). Для сравнения показаны также экспериментальные значения. Как
видно, модель позволяет отследить эволюцию каждого химического псевдокомпо-
нента и показывает хорошее согласие с экспериментальными данными. Можно за-
метить, что между жидкой и твердой фазами есть перепад концентраций, который
постепенно исчезает по направлению к выходу из реакторов. Такое поведение объ-
ясняется сопротивлением массопереносу на границе раздела фаз. Оно определяет-
ся коэффициентом массопереноса, который зависит главным образом от физиче-
ских свойств (плотности и вязкости) и приведенной массовой скорости жидкости.
На рис. 8.11 показаны распределения по высоте слоя величины коэффициента мас-
сопереноса через межфазную границу и плотности жидкости как главного параметра
для его расчета. По направлению к выходу массоперенос заметно улучшается, так как
сырье в результате гидрокрекинга становится легче, причем этот эффект усиливается
с повышением температуры.
342
Часть 3. Моделирование каталитических процессов
Безразмерная координата по высоте реактора
Рис. 8.10. Распределения молярных концентраций серы, азота и металлов
по высоте реакторов. Расчетные значения:
(—) — в жидкой фазе, (—) — в твердой фазе; () — экспериментальные значения
На рис. 8.12 представлены распределения молярных концентраций Н2 и H2S
в жидкой и твердой фазах при температуре в реакторе 380 °C. Форма кривых опре-
деляется балансом между химическими реакциями, с одной стороны, и массопе-
реносом между газовой и жидкой фазами — с другой. На участке от входа до точки
с координатой 0,05 (5 % суммарной высоты реакторов) концентрация Н2 быстро па-
дает, а концентрация H2S существенно возрастает вследствие высоких скоростей ре-
акций на этом участке. В остальной части реактора Н2 концентрируется в жидкой
фазе, a H2S высвобождается из жидкой фазы в газовую, так что концентрации в этой
части реактора определяются равновесием между газовой и жидкой фазами. Здесь
опять действует сопротивление массопереносу между жидкой и твердой фазами, по-
степенно исчезающее по мере протекания реакции.
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
343
Рис. 8.11. Распределения величины коэффициента массопереноса через межфазную
границу и плотности жидкости в зависимости от координаты по высоте реакторов:
(—) — расчетные значения; () — экспериментальные значения
Безразмерная координата по высоте реактора
Рис. 8.12. Распределения молярных концентраций водорода и сероводорода. Расчетные
значения: (—) — в жидкой фазе; (—) — в твердой фазе
График, представленный на рис. 8.13, иллюстрирует изменение состава газовой
фазы по высоте реакторов при 380 °C. Для подтверждения модели показаны также
экспериментальные данные газохроматографического анализа газовой фазы на вы-
ходе реакционной системы. Как видно, в результате расхода Н2 его парциальное
давление быстро снижается. С другой стороны, вследствие образования H2S и СН4
344
Часть 3. Моделирование каталитических процессов
их парциальные давления по высоте реакторов возрастают. В точке с относительной
координатой 0,5, которая соответствует межреакторной зоне, наблюдается заметный
скачок парциальных давлений Н2, H2S и СН4. Он вызван межреакторным водород-
ным охлаждением. Охлаждающий газ обогащает газовую фазу водородом и разбавля-
ет H2S и NH3, ингибирующие реакцию гидроочистки. Такое распределение состава
газовой фазы типично для промышленных реакторов гидроочистки с несколькими
зонами межреакторного водородного охлаждения.
Безразмерная координата по высоте реактора
Рис. 8.13. Изменение состава газовой фазы по высоте реакторов:
(—) — расчетное; () — экспериментальное
На рис. 8.14 изображены распределения приведенной скорости газа («с) и кратно-
сти водорода ксырью (относительно ее значения на входе, которое равно 890 н. м3/м3).
Для сравнения включены экспериментальные значения кратности водорода к сырью
на выходе реакционной системы, определенные по расходу водорода. В целом пове-
дение обеих переменных похоже на поведение парциального давления Н2. Это обу-
словлено расходом Н2 по высоте реакторов, из-за которого существенно снижаются
кратность водорода к сырью, объем газа и его приведенная скорость. Как и следует
ожидать, при более высоких температурах возрастает расход Н2, поэтому кратность
водорода к сырью и приведенная скорость газа падают быстрее. Вклад H2S и СН„
в общую объемную скорость газа пренебрежимо мал по сравнению с вкладом Н2. Об-
наружить это можно, лишь составив молярный баланс масс в газовой фазе. В боль-
шинстве моделей скорость газа считается постоянной, поэтому любые возможные ее
изменения не принимаются во внимание. В данном случае межреакторное охлажде-
ние скачкообразно увеличивает кратность водорода к сырью, а также приведенную
скорость газа.
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
345
Безразмерная координата по высоте реактора
Рис. 8.14. Распределения кратности водорода к сырью и приведенной скорости газа.
Расчетные значения: (—) — 380 °C; (—) — 400 °C; () — экспериментальные значения
В целом результаты расчета по модели находились в хорошем согласии с экспе-
риментальными наблюдениями. Модель отражает важнейшие химические пре-
вращения и фазовые явления, происходящие по высоте каталитического слоя при
различных температурах.
8.6.2. Моделирование реактора при переменной активности
катализатора
8.6.2.1. Влияние типа сырья и температуры реакций
на деактивацию катализатора
Модель реактора использовалась при анализе процесса деактивации катализато-
ра тяжелым сырьем различного типа. Данные рис. 8.15 демонстрируют динами-
ку активности катализатора в реакции ГДО тяжелой нефти (TH) и ее атмосферно-
го остатка (АОТН). Можно видеть, что в экспериментальном интервале температур
модель достаточно хорошо прогнозирует кривые деактивации. Форма начально-
го участка кривых старения не изменяется при переходе от одного сырья к другому.
На рис. 8.16, а показана динамика изменения функции деактивации (фгдо) ДЛЯ трех
видов сырья. Четко видно, что функция деактивации во всех случаях убывает схожим
образом. Но это справедливо лишь для начальной части цикла, так как длительность
экспериментов мала по сравнению с продолжительностью цикла в промышленной
практике. Безусловно, можно ожидать, что в больших временных масштабах кривые
изменения функции деактивации будут расходиться пропорционально тяжести сы-
рья. Модель способна отразить этот эффект, так как он непосредственно связан с ло-
кальной концентрацией отложений металлов.
346
Часть 3. Моделирование каталитических процессов
Рис. 8.15. Зависимость степени ГДО тяжелой нефти и ее атмосферного остатка от времени
работы катализатора. Значки показывают экспериментальные значения, линии — расчетные
Рис. 8.16. Динамика изменения функции деактивации катализатора в реакции ГДО:
а — влияние типа сырья при 380 °C; б — влияние температуры в случае тяжелой нефти.
Обозначения: ОТН — очвнь тяжелая нефть
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
347
Если затронуть влияние температуры, то можно напомнить, что кривые старения
для каждой реакции и каждого типа сырья отвечают одному и тому же набору пара-
метров во всем интервале температур. Это означает, что кривые активности в период
проведения экспериментов не зависят от времени, о чем свидетельствует их нало-
жение на рис. 8.16, б. Тем не менее, при увеличении длительности работы скорость
деактивации с повышением температуры будет неизменно возрастать, что обуслов-
лено ростом скорости реакции ГДМ. Отсюда был сделан вывод, что выбор простой
функции времени для описания старения катализатора на начальном отрезке рабо-
ты вполне оправдан.
8.G.2.2. Результативность процесса при испытании
катализатора на долговременную стабильность
На рис. 8.17 показана динамика результативности ГДО, ГДН, ГДВ и гидрокре-
кинга вакуумного остатка. Модель достаточно хорошо предсказывает результа-
тивность процесса гидропереработки в течение первых 5 мес. работы. В режиме
постоянной результативности кривые деактивации ведут себя по-разному: напри-
мер, степень ГДО и ГДН с течением времени все больше снижается, тогда как
кривые степени ГДВ и гидрокрекинга лучше реагируют на повышение температу-
ры. Такое поведение обусловлено различными факторами, в том числе свойства-
ми катализаторов, значениями энергий активации, типом сырья и термическим
эффектом.
100 -I
Время работы, ч
Рис. 8.17. Динамика результативности процесса гидропереработки. Значки показывают
экспериментальные значения, линии — расчетные
348
Часть 3. Моделирование каталитических процессов
На рис. 8.18 представлены изменения во времени глубины ГДМ в первом реак-
торе (Л,) и общей глубины ГДМ (Ri + Л2). Как было указано в описании экспери-
ментов, за работой первого реактора следили путем отбора межреакторных проб.
Видно, что данные первого реактора достаточно широко отклоняются от расчет-
ных кривых, но в целом модель способна отражать общую тенденцию. Большие от-
клонения объясняются тем, что межреакторные пробы были невелики по объему
(2 % от всего объема потока) и поэтому недостаточно точно представляли кинети-
ку. Тем не менее объем проб был приемлем для приближенной оценки поглощения
металлов в первом реакторе и проверки возможностей модели. Что касается об-
щей глубины ГДМ, модель работает достаточно хорошо, так как оценка параметров
проводилась по суммарным (по двум реакторам) данным старения катализатора.
На рис. 8.18 показана также динамика изменения количества металлов, отлагаю-
щихся на катализаторе в реакторах. Очевидно, что накопление металлов следует
линейному закону. Налицо хорошее согласие между результатами, которые дает
модель, и наблюдаемым количеством металлов на катализаторе, которое определя-
лось исходя из баланса металлов в сырье и продуктах. Следует отметить, что коли-
чество металлов, поглощенных в первом реакторе, доходит до 120 % (относительно
массы свежего катализатора), а во втором реакторе эта величина составляет лишь
около 20 %. Очевидно, что входной катализатор защищает от металлов последую-
щие слои.
90 т
Время работы, ч
Рис. 8.18. Глубина ГДМ и количество металлов на катализаторе (ЛЮС). Значки показыва-
ют экспериментальные значения, линии — расчетные. Экспериментальные значения ()
ЛЮС в конце цикла определялись исходя из баланса металлов в сырье и продуктах
Время работы, ч
Модель способна также описать динамику осевого распределения МОС, представ-
ленную на рис. 8.19. Видно, что в начальный период (через 500 ч) кривая осевого
распределения МОС относительно полога. С течением времени она приобретает ти-
пичную убывающую форму, особенно в первом реакторе. Очевидно, больше всего
металлов поглощает входной катализатор, занимающий первые 30 % общего объема
реакторов (0-0,3); за ним идет промежуточный катализатор, занимающий остальной
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
349
объем первого реактора и часть объема второго (0,3—0,6). Такое послойное распо-
ложение катализатора существенно снижает содержание металлов в реакционной
смеси, поступающей в слой высокоактивного катализатора ГДО, который занимает
выходную часть второго реактора (0,6—1,0).
Безразмерная координата по высоте реакторов
Рис. 8.19. Расчетные распределения МОС по оси реакторов (в процентах относительно
общей массы катализатора в реакторах)
8.7. МАСШТАБИРОВАНИЕ КИНЕТИЧЕСКИХ ДАННЫХ,
ПОЛУЧЕННЫХ НА ЛАБОРАТОРНОЙ УСТАНОВКЕ
Для процессов, проводимых в реакторах с орошаемым слоем, крайне желательно,
чтобы в превращении участвовал весь объем катализатора. В идеале для этого ка-
ждая частица катализатора должна быть покрыта пленкой, образуемой обтекающей
их жидкостью [Sie, 1991]. Но реальное поведение лабораторных реакторов обычно
не всегда таково, та к как малая скорость жидкости в них вызывает неполное смачи-
вание катализатора. В подобных случаях жидкость движется преимущественно че-
рез определенные части слоя. Некоторые частицы катализатора остаются частично
смачиваемыми, некоторые — совершенно сухими. Это обстоятельство уменьшает
эффективную площадь массопереноса между жидкими реагентами и катализато-
ром и тем самым снижает скорость реакции [Al-Dahhan and Dudukovic, 1995]. При
гидропереработке нефтяных фракций малая величина фактора эффективности ка-
тализатора усугубляет проблему, поскольку жидкость не может достаточно быстро
проникнуть внутрь частиц и реакция происходит лишь в тонкой пленке под сма-
чиваемой поверхностью [Harold and Ng, 1987]. Поэтому для масштабирования ки-
нетических данных, полученных на лабораторном реакторе с орошаемым слоем,
важно оценить эффективность смачивания и определить степень использования
350
Часть 3. Моделирование каталитических процессов
катализатора, чтобы внести соответствующие поправки в кинетические параметры
реакций.
На рис. 8.20 представлен анализ влияния масштаба на эффективность смачива-
ния катализатора. Теоретическая кривая получена по уравнению (8.19) с использо-
ванием данных лабораторного и полупромышленного масштаба при 380 °C. Форма
кривой говорит о том, что в этой системе эффективность смачивания слабо зави-
сит от приведенной массовой скорости жидкости. Действительно, величины эф-
фективности смачивания во всех опытах лежат в области промышленных значений
(0,7—1,0). Значение этого параметра всегда выше 0,7 (используется более 70 % ка-
тализатора) даже при минимальной величине LHSV. При испытании на установ-
ке полупромышленного масштаба оно близко к 1, поскольку такая установка пе-
рерабатывает примерно в 230 больше сырья, чем лабораторная, работающая при
LHSV= 0,25 ч-1. Это явно свидетельствует о том, что в отношении коэффициен-
та использования катализатора реактор полупромышленного масштаба ведет себя
идеально, так что подобное испытание можно считать представительным для про-
мышленного масштаба.
Рис. 8.20. Зависимость эффективности смачивания катализатора от приведенной
массовой скорости жидкости: () — лабораторный масштаб; (□) — полупромышленный
масштаб; (—) — расчетные значения
Величина эффективности смачивания индивидуальна для каждой системы, по-
скольку определяется геометрией реактора и частиц катализатора, свойствами сырья
и рабочими условиями. То, что в данной частной системе значения эффективности
смачивания относительно велики даже при небольших скоростях жидкости, мож-
но объяснить; а) вязкостью сырья; б) рабочими условиями (в особенности темпера-
турой реакции) и кратностью водорода к сырью; в) размерами реактора. Вязкость,
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
351
например, помогает распространению жидкости в пористых материалах, тем самым
улучшая ее распределение. Влияние вязкости видно из формулы так называемого
числа смачивания (1У) [Sie, 1991]:
И/= . (8.23)
Pi^gC
Это безразмерное число определяет соотношение между силами трения и тяже-
сти, причем силы трения выражает числитель дроби, а силы тяжести — знаменатель.
Для равномерного распределения жидкости по насадочному слою силы сопротивле-
ния течению должны превышать силы тяжести. Это вынуждает жидкость проходить
через все доступные каналы и течь более равномерно. В противном случае возника-
ет преимущественное течение жидкости в обход некоторых частей каталитического
слоя. На основании многочисленных наблюдений Гиерман [Gierman, 1988] заклю-
чил, что для равномерного распределения жидкости необходимо выполнение усло-
вия W> 5-10~6. Из этого анализа можно сделать вывод, что сильнее всего на эффек-
тивность смачивания влияют скорость жидкости и вязкость сырья. Именно по этой
причине опытные реакторы, перерабатывающие сравнительно легкое сырье (напри-
мер, прямогонный и даже вакуумный газойль), предрасположены к неполному сма-
чиванию катализатора. В случае более тяжелых фракций, как например атмосферный
остаток, вязкость жидкости в тех же рабочих условиях может быть до двух порядков
выше, что безусловно улучшает смачивание катализатора.
Другие параметры процесса, которые положительно влияют на эффективность
смачивания катализатора и явно не учитываются в модели частичного смачива-
ния, — температура реакции и кратность водорода к сырью. Теоретическая кривая
на рис. 8.20 действительна лишь для определенных значений этих величин. Увели-
чение любой из этих переменных (в частности, температуры реакции) сместит вверх
зону, в которой эффективность смачивания катализатора является жесткой функци-
ей скорости жидкости.
Еще один важный фактор — размеры реактора. Внутренний диаметр и высота
(общая высота каталитического слоя) реакторов лабораторной установки, использо-
вавшейся в этой работе, были больше, чем у типичных лабораторных реакторов. Боль-
шее соотношение диаметра реактора и диаметра частиц снижает воздействие пристен-
ного эффекта и увеличивает коэффициент использования катализатора [Sie, 1996].
Увеличенная высота слоя в экспериментальных реакторах уменьшает разброс массо-
потока вдоль оси реактора и обеспечивает достаточное расстояние для улучшения ра-
диального распределения по мере движения жидкости вниз [Mederos et al., 2009b].
На рис. 8.21 сравнивается результативность процесса на установках полупромыш-
ленного и лабораторного масштабов при LHSV= 0,2 ч'1 и температуре 380 °C. Видно,
что благодаря большим скоростям жидкости и лучшему использованию катализато-
ра полупромышленная установка превосходит лабораторную. Влияние этих факто-
ров на работу реактора позволяет отразить включение параметра масштабирования
в уравнения модели. В целом модель дает достаточно хорошее приближение резуль-
тативности процесса в реакторах разных масштабов.
352
Часть 3. Моделирование каталитических процессов
О 10 20 30 40 50 60 70 80 90 100
Глубина реакции, %
Рис. 8.21. Сравнение результативности процессов
полупромышленного и лабораторного масштабов
8.8. МОДЕЛИРОВАНИЕ ПРОЦЕССА
В ПРОМЫШЛЕННОМ РЕАКТОРЕ
8.8.1. Выбор компоновки и моделирование работы реактора
при постоянной активности катализатора
Хорошо известно, что между реакторами промышленного и лабораторного масшта-
бов есть определенные различия. Важнейшим из них является то, что промышленные
реакторы работают в адиабатическом режиме и возвращаемый газ с целью проме-
жуточного охлаждения распределяется по высоте реактора, в то время как лабора-
торные реакторы работают в изотермическом режиме и газ подается лишь на вход
реактора. В адиабатическом режиме температура в реакторе по мере движения реак-
ционной смеси вниз по слою повышается. Одной из важнейших задач в операциях
гидропереработки является контроль температуры. Обычно суммарное тепловыде-
ление разбивают на более мелкие и безопасные порции, размещая катализатор в не-
сколько слоев и предусматривая промежуточное охлаждение между ними, в то время
как однослойный реактор был бы невыгоден из-за чрезмерного повышения темпе-
ратуры. Таким образом, приемлемая конструкция промышленного реактора должна
отвечать требованиям безопасности и воспроизводить среднюю температуру и крат-
ность водорода к сырью, принятые на основании данных лабораторных экспери-
ментов. Для этого выбирают входную температуру, устанавливают допустимый пере-
пад температур в слое (отсюда же определяются необходимое число каталитических
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
353
слоев и их высота) и находят количества газа, которые необходимо отбирать из кон-
тура циркуляции для промежуточного охлаждения.
Для расчета и моделирования промышленных реакторов облагораживания тяже-
лой нефти по технологии IMP была применена модель реактора, учитывающая упо-
мянутые выше критерии. Было принято, что реакторы работают при следующих
средних условиях: общая объемная скорость подачи сырья LHSV1 * = 0,25 ч_|, темпе-
ратура 380 °C, кратность водорода к сырью 890 н. м3/м3, давление 9,81 МПа. Главной
целью было найти такую компоновку слоев, при которой распределения темпера-
тур и кратностей водорода к сырью давали бы требуемые средние значения. Для это-
го последовательно анализировали несколько компоновок, переходящих одна в дру-
гую. Такой подход позволял выявить недостатки выбранной компоновки и затем
улучшить ее так, чтобы она отвечала поставленным критериям.
На рис. 8.22 представлены одна из возможных компоновок реакторов и получен-
ные в результате имитации распределения средней температуры, кратности водорода
к сырью и степеней превращения химических псевдокомпонентов. Для ограничения
резкого роста температуры, вызываемого реакциями гидропереработки, весь объем
катализатора был разделен на шесть слоев. Для реактора R, ввиду большого тепловы-
деления (около 72 °C) в этой секции потребовалось четыре слоя, в то время как для
реактора А2 оказалось достаточно двух слоев. Значения на входе и приросты темпера-
туры в слоях выбирались более или менее одинаковыми, чтобы средние температуры
в слоях были близки к требуемой (пунктирная линия). Требование одинакового пе-
репада температур дает, как правило, слои возрастающей высоты. Из рис. 8.22 можно
видеть, что в схеме предусмотрено всего две точки охлаждения (обе в R^ и три тепло-
обменника. Выбор такой компоновки был обусловлен необходимостью сбалансиро-
вать количество газа, отбираемого для охлаждения из контура циркуляции, и требу-
емой средней кратности водорода к сырью (пунктирная линия). Более подробный
анализ баланса водорода будет рассмотрен далее [Mucoz et al., 2005].
На рис. 8.23 изображена диаграмма зависимости средней кратности водорода к сы-
рью в реакторах от числа точек водородного охлаждения. Анализ показывает, что тре-
бование поддерживать расчетную кратность водорода к сырью (890 н. м3/м3) допуска-
ет не более двух точек промежуточного водородного охлаждения. Если увеличивать
число точек охлаждения (три, четыре или пять точек), средняя кратность водорода
к сырью падает ниже расчетной, так как каждая дополнительная точка охлаждения
уменьшает количество водорода, поступающего на вход Аг Это нежелательно, осо-
бенно в А,, поскольку уменьшение кратности водорода к сырью отрицательно влияет
на срок службы катализатора. Из всего сказанного можно заключить, что невозмож-
но одновременно отводить всю теплоту путем охлаждения водородом и поддержи-
вать среднюю кратность водорода к сырью в реакторах на расчетном уровне. Именно
по этой причине в предлагаемую компоновку включены три теплообменника. Дру-
гой вариант с поддержанием расчетного значения кратности водорода к сырью в ре-
акторе А, (вариант +5) мог бы заключаться в увеличении на 43 % скорости цирку-
ляции газа. Однако это ухудшает экономические показатели процесса из-за роста
затрат на компримирование газа [Alvarez et al., 2009].
1 LHSV — дословно «часовая объемная скорость по жидкому сырью», но чаще всего слово «часовая»
опускают, так как это следует из размерности. — Примеч. науч. ред.
354
Часть 3. Моделирование каталитических процессов
Базовая
Продукт
Рис. 8.22. Распределения средней температуры, кратности водорода к сырью и степе-
ней превращения химических псевдокомпонентов, полученные в результате имитации
процесса в реакторах с изображенной слева компоновкой: (—) — средние величины
температуры и кратности водорода к сырью. Сокращения: АО — атмосферный остаток;
ВО — вакуумный остаток
Безразмерная
концентрация
0,2 0,4 0,6 0,8 1,0
0,2 0,4 0,6 0,8 1,0
1400 -]
1200 -
1000 -
800 -
600 -
Расчетная величина
400 -I-1----1--1---1--'---1--1—-г---1-----1->---г—
0 1 2 3 4 5 +5
Число точек охлаждения водородом
Рис. 8.23. Зависимость средней кратности водорода к сырью
от числа точек водородного охлаждения
8. Моделирование и имитация гидропереработки тяжелой нефти 355
8.8.2. Моделирование и анализ работы реакторов в течение
рабочего цикла
В промышленной практике постепенное снижение активности катализатора ком-
пенсируют периодическим увеличением температуры реакций. При достижении
в любой точке реактора максимально допустимой температуры, определяемой ха-
рактеристиками материала реактора, цикл приходится прерывать. Активность ката-
лизатора к концу цикла необязательно исчерпывается полностью, так как металлы
накапливаются во входной части реактора, о чем свидетельствует убывающий харак-
тер распределения металлов на катализаторе. Из этого следует, что неверный выбор
компоновки реактора может приводить к преждевременной остановке из-за раннего
появления высокотемпературных зон в каталитических слоях. Таким образом, пра-
вильно выбранная компоновка (т. е. число слоев и их расположение) сильно влияет
на время достижения максимально допустимой температуры.
Проблема, связанная с конструкцией реакторов в системах такого типа, возника-
ет из-за неравномерности распределения активности и температур по оси, которая
ктому же усиливается со временем [Shah et al., 1976]. Для максимального продления
длительности цикла этот фактор необходимо тщательно оценить. С помощью моде-
ли работы реактора анализировалось влияние его компоновки на поведение процес-
са IMP в течение цикла. Главная цель состояла в том, чтобы проверить, обеспечивает
ли данная компоновка такую же длительность цикла, как и достигнутую при испыта-
нии катализатора на стабильность (5 мес.), или же она способна продлить время ра-
боты до достижения максимально допустимой температуры (420 °C). Были проана-
лизированы следующие случаи.
• случай 1: два однослойных реактора равной высоты с одним промежуточным
охлаждением;
• случай 2: первый реактор с двумя слоями равной высоты с последующим од-
нослойным реактором; водородное охлаждение в двух точках;
• случай 3: один реактор с четырьмя слоями возрастающей высоты, после кото-
рого следует реактор с двумя слоями возрастающей высоты; охлаждение водо-
родом в двух точках и путем теплообмена — в трех.
Первая компоновка (случай 1) идентична компоновке лабораторной установки,
а третья (случай 3) представлена на рис. 8.22.
На рис. 8.24 показана динамика изменения во времени распределения темпера-
туры по высоте реакторов. Главная особенность результатов моделирования работы
реакторов состоит в том, что со временем кривая распределения температуры сме-
щается вверх, так как температуру сырья приходится периодически повышать, что-
бы поддерживать плотность продукта на постоянном уровне. Компоновка в первом
случае характеризуется большим и непрерывным тепловыделением в обоих реакто-
рах. Из-за значительного перепада температур в слое на выходе из каждого реактора
образуются высокотемпературные зоны. Прирост температуры во втором реакторе
со временем замедляется, поскольку выходной катализатор менее стоек к отложению
металлов и теряет активность быстрее, в то время как катализаторы в первом реакторе
356
Часть 3. Моделирование каталитических процессов
Безразмерная координата по высоте реакторов
Рис. 8.24. Динамика изменения распределения температуры по высоте реакторов
более стабильны. В конечном счете цикл приходится прерывать раньше, так как тем-
пература на выходе из первого реактора достигает максимально допустимых значе-
ний (420 °C) за 2748 ч (около 3,8 мес., что меньше 5,2). Недостаток такой компо-
новки очевиден: это отсутствие охлаждения в первом реакторе, где оно нужнее.
Во втором случае водородное охлаждение в первом реакторе несколько продлевает
(до 2964 ч, т. е. около 4,1 мес.) время работы до достижения максимально допустимой
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
357
температуры, но теперь высокотемпературная зона смещается к выходу из второ-
го реактора. В обоих случаях «недоиспользуется» значительная часть катализатора,
а длительность цикла меньше, чем при испытании катализатора на стабильность.
Кроме того, высокие температуры на выходе из реакторов могут вызвать термиче-
ский крекинг, чреватый проблемами потерь давления, аналогичными наблюдаемым
при испытании катализатора на старение. В третьем случае эти недостатки реша-
ются применением многослойной конструкции с возрастающей высотой слоев. Та-
кая компоновка дает лучшее распределение температур по высоте. Иными словами,
уровни температур во всех слоях очень схожи, а зоны перегрева отсутствуют. Дли-
тельность цикла до достижения максимально допустимой температуры составляет
теперь 4500 ч (около 6,3 мес.).
На рис. 8.25 показаны картины распределения количества металлов на катализа-
торе через 2700 ч работы во всех трех случаях. В третьем случае кривая распределения
носит типичный убывающий характер. Это означает, что основная часть металлов от-
лагается на входном катализаторе, как и должно быть. В случаях 1 и 2 с неприемле-
мой компоновкой реакторов основная масса отлагаемых металлов сдвигается к сере-
дине и к выходу. В этих случаях температура сырья значительно ниже, чем в третьем
случае, что снижает скорость ГДМ во входной секции. В то же время повышенные
температуры в остальной части высоты реакторов ускоряют отложение металлов,
а следовательно, и деактивацию промежуточного и выходного катализаторов. Полу-
чает объяснение и то обстоятельство, что прирост температуры во втором реакторе
в первых двух случаях замедляется со временем быстрее, чем в третьем случае.
Безразмерная координата по высоте реакторов
Рис. 8.25. Распределение количества металлов на катализаторе
по высоте реакторов через 2700 ч работы
Главное, о чем свидетельствует данный анализ, — это то, что неверный выбор ком-
поновки реакторов не позволит ни максимально продлить цикл службы, ни повы-
сить эффективность катализатора. Требуемую длительность цикла (не менее той,
358 Часть 3. Моделирование каталитических процессов
которая была достигнута при испытании катализатора на стабильность) дает третья
компоновка (см. рис. 8.22). Ключевой момент проектирования процессов с боль-
шим тепловыделением заключается в том, чтобы разбить суммарное тепловыделе-
ние на меньшие порции (т. е. уменьшить перепады температуры в слоях), с тем чтобы
получить более равномерное распределение температуры по высоте реактора. Для
достижения этой цели предпочтительнее распределить катализатор по слоям воз-
растающей, а не одинаковой, высоты, так как по мере прохождения реакционной
смеси через реактор количество выделяемой теплоты уменьшается. Для получения
желательного распределения температур необходимо также правильно выбрать ско-
рость охлаждения. Чтобы получить еще более равномерное распределение темпера-
тур, можно увеличить число слоев, тем самым снизив зависимость от температурных
ограничений. Но тогда возрастает необходимая высота реактора, а следовательно,
и затраты на внутреннее оборудование реакторов.
8.8.3. Переходное поведение реактора при пуске
Без понимания динамики поведения реакторов для экзотермических процессов не-
возможны ни их проектирование, ни безопасная эксплуатация. Это большая про-
блема для гидропереработки, особенно при пуске установок со свежим катализато-
ром, так как местный перегрев может занять буквально считаные минуты [Yan, 1980].
Зоны местного перегрева могут возникать в результате нарушения однородности по-
тока жидкости в каталитическом слое [Agrawal et al., 2007]. Такая ситуация может
привести к термической нестабильности, если скорость теплопередачи недостаточна
для своевременного рассеяния теплоты [Alvarez et al., 2007].
Реакторы гидропереработки следует проектировать и эксплуатировать так, чтобы
не допускать больших колебаний температуры. Кроме того, нужен способ оптималь-
ного пуска со свежим катализатором, чтобы максимально сократить время достиже-
ния требуемых свойств продукта при стабильной работе реактора. Для анализа дина-
мики поведения процесса и выработки правил эффективного пуска установки была
использована динамическая модель реактора [Alvarez and Ancheyta, 2012].
Сначала проверяли непротиворечивость модели. Для этого следовало удостове-
риться, что результаты имитации динамической работы согласуются со стационарны-
ми данными, полученными на лабораторной установке. Баланс энергии при имитации
не рассматривался, так как установка рассчитана на изотермический режим рабо-
ты. На рис. 8.26 представлены переходные распределения концентраций серы и азота
в жидкой и твердой фазах в установившемся режиме работы катализатора. Результаты
иллюстрируют, как углеводородный фронт медленно опускается по каталитическому
слою, постепенно заполняя реакторы. Заметно, что через 100 мин на выходе появляется
небольшое количество продукта. Этот момент соответствует среднему времени пребы-
вания, определяемому скоростью движения жидкости в порах. Через 200 мин система
окончательно стабилизируется. Можно видеть, что в установившемся режиме резуль-
таты моделирования хорошо согласуются с данными экспериментальных наблюдений.
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
359
Безразмерная координата по высоте реактора
Рис. 8.26. Распределения безразмерных концентраций серы и азота при Т = 380 °C,
LHSV= 0,25 ч-1, Р= 9,81 МПа и кратности водорода к сырью 891 н. м3/м3. Расчетные значе-
ния: (—) — для жидкой фазы, (—) — для твердой фазы; () — экспериментальные значения
Затем динамической моделью имитировали работу первого реактора промышлен-
ной установки IMP. Реактор имел четыре слоя и три точки охлаждения (£),, Q2 и Q},
см. рис. 8.22). Так как на данном этапе процесса катализатор контактирует со свежим
сырьем, имеют место резкие перепады концентраций и температур по высоте реак-
тора. Проверялся динамический отклик системы в этих условиях; также анализиро-
валось влияние на нее параметров охлаждения и температуры сырья.
8.8.3.1. Охлаждение
Первый важный параметр — момент начала охлаждения, обеспечивающий достиже-
ние тепловой стабильности. Проверялись следующие варианты: а) после заполне-
ния реактора жидкостью; б) когда фронт углеводородов достигает зоны охлаждения;
в) до того, как фронт углеводородов достигнет зоны охлаждения. Моделирование от-
талкивалось от предпосылки, что катализатор находится в стабильном состоянии
(после первых 100 ч начала работы) и что начальная температура слоя равна темпера-
туре сырья, поступающего в реактор. Рабочие условия были приняты следующими:
360
Часть 3. Моделирование каталитических процессов
температура сырья 371 °C (тогда средняя температура в реакторе составит 380 °C),
LHSV= 0,25 чч, Р= 9,81 МПа, кратность водорода к сырью 891 н. м3/м3.
На рис. 8.27 показаны распределения температуры в каждом из перечисленных
выше случаев. В первом из них (вариант а) начало охлаждения откладывалось до пол-
ного насыщения слоя жидкостью. Прохождение фронта жидкости через реактор за-
нимало около 60 мин. В этот период тепловая волна при прохождении через реактор
чрезвычайно усиливалась. Уже через 13 мин появлялась зона с температурой, близ-
кой к 400 °C. Далее реактор быстро перегревался значительно выше допустимых пре-
делов (выше 420 °C). В этом режиме возможны усиленное закоксовывание, повы-
шенный расход водорода, образование больших количеств сухого газа и разложение
ценных продуктов. Более того, может выйти из-под контроля реактор или оказать-
ся нарушенной целостность его сосуда, что ведет к катастрофическим последствиям.
Теоретически после того, как через 60 мин будет одновременно начато охлаждение во
всех трех точках (Q, Q2 и О3), процесс в конечном итоге через 130 мин должен достиг-
нуть установившегося состояния. Но на практике такой сценарий непрактичен и не-
безопасен, так как реактор слишком долго подвергается перегреву.
Во втором случае (рис. 8.27, б), охлаждение в точках Qt, Q2 и Q.. начинается соот-
ветственно через 10, 20 и 40 мин. Момент начала охлаждения соответствует времени
достижения углеводородным фронтом точки охлаждения. Резкое охлаждение пре-
дотвращает развитие тепловой волны и снижает риск перегрева. Поэтому этот сце-
нарий представляет правильную тактику предотвращения появления зон перегре-
ва. Интересной особенностью моделирования является то, что охлаждение вызывает
значительные возмущения в естественном движении тепловой волны и создает пе-
реходные колебания температуры. Охлаждающая среда, рассеивая тепло, прерыва-
ет процесс его аккумуляции. Образующаяся отрицательная тепловая волна при про-
хождении по реактору быстро рассеивается.
Что касается последнего случая (рис. 8.27, в), одновременное раннее (через
10 мин) начало охлаждения во всех трех точках тоже позволяет удержать тепловой
контроль в реакторе. Так как к этому времени реакционная среда еще не достигла по-
следних двух точек охлаждения, часть слоя, не находящаяся в контакте с жидкостью,
охлаждается. Поэтому моделирование показывает наличие зон, температура в кото-
рых ниже температуры сырья. Через 60 мин температура постепенно выравнивается
и достигает таких же средних значений, как во втором случае (см. рис. 8.27, б).
Модель способна отражать также поведение газовой фазы. На рис. 8.28 сравни-
ваются составы газа через 60 мин в случаях (а) и (б). В случае (а) четко проявляются
симптомы повышенной температуры: сильный рост расхода Н, и образования СН4
за счет целевых продуктов. Для предотвращения коксообразования на катализаторе
важно поддерживать концентрацию Н2 надостаточно высоком уровне.
Главный вывод, следующий из этой части анализа, состоит в том, что охлаждение
следует начинать незадолго до того, как углеводороды достигнут соответствующей
точки охлаждения. Даже небольшая задержка начала охлаждения может вызвать бы-
стрый перегрев и сильно повлиять на работу установки.
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
361
Рис. 8.27. Переходные распределения температур при установившемся состоянии
катализатора: а — охлаждение начинается одновременно во всех точках через 60 мин;
б— охлаждение в точках Q,, Q2 и Q3 начинается соответственно через 10, 20 и 40 мин;
в — охлаждение начинается одновременно во всех точках через 10 мин
362
Часть 3. Моделирование каталитических процессов
Безразмерная координата по высоте реактора
Рис. 8.28. Распределения концентраций Н2 и СН4 через 60 мин:
(—) — случай (а); (—) — случай (6)
8.8.3.2. Температура сырья
Температура сырья — один из параметров, наиболее сильно влияющих на динамиче-
ское поведение реактора. Общее правило пуска установок утверждает, что по сообра-
жениям безопасности пуск реактора должен осуществляться при наименьшей воз-
можной температуре [Yan, 1980]. Такое правило обусловлено тем фактом, что свежий
катализатор больше подвержен образованию зон перегрева.
На рис. 8.29 изображены распределения температуры с учетом свежего состояния
катализатора. Температура сырья была принята равной 360 °C, а охлаждение начина-
лось соответственно через 10, 20 и 40 мин, как в рассмотренном выше случае (б). Хотя
температура сырья была на 11 °C ниже, Чем в предыдущей имитации (см. рис. 8.27),
и охлаждение начиналось своевременно, кривые показывают образование переход-
ных высокотемпературных зон в последних двух слоях. Это означает, что неточный
выбор температуры сырья чреват перегревом реактора.
На рис. 8.30 представлена динамика температуры на выходе слоев (То) для свеже-
го катализатора. Охлаждение опять начиналось так же, как и в случае (б). Зависи-
мость довольно проста: с увеличением температуры сырья возрастает и тепловыде-
ление. Результаты показывают, что с увеличением температуры сырья до 365—370 “С
выходные температуры превышают 400 °C, но затем по мере деактивации катализа-
тора постепенно снижаются. Если температуру сырья увеличить еще больше, реактор
перегревается (температура на выходе превышает 420 °C). Поэтому лучше всего запу-
скать реактор при температуре сырья между 355 и 360 °C, чтобы удержать температуру
в наиболее горячих зонах на приемлемом уровне. Имитация показывает также пере-
ходные возмущения, вызываемые охлаждением. Температура на выходе второго слоя
отражает влияние охлаждения в первой точке, на выходе третьего слоя — суммарное
влияние охлаждения в первой и второй точках.
f ва 8 Моделирование и имитация гидропереработки тяжелой нефти
363
0,0 0,2 0,4 0,6 0,8 1,0
Безразмерная координата по высоте реактора
Рис. 8.29. Переходные распределения температуры для свежего катализатора
Рис. 8.30. Влияние температуры сырья на температуру
на выходе слоев для свежего катализатора
364
Часть 3. Моделирование каталитических процессов
8.8.3.2. Тактика пуска
При пуске установки гидропереработки необходимо стабилизировать катализатор
чтобы его активность достигла квазистационарного состояния. Стабилизация катали-
затора в данном процессе занимает обычно около 100 ч. Для безопасной стабилиза-
ции катализатора нужна эффективная тактика пуска. Предыдущий анализ четко пока-
зывает, что реактор следует запускать при низкой температуре на входе (355—360 °C)
а охлаждение должно начинаться вовремя. Кроме того, чтобы соблюсти требования
к свойствам продуктов реакций, температуру следует повышать постепенно. Исходя
из вышесказанного предлагается следующий порядок пуска: температура сырья в на-
чале пуска устанавливается равной 355 °C, а затем, после первых 20 ч работы, повы-
шается на 2 °C через каждые 10 ч; охлаждение в трех точках начинается через 10, 20
и 40 мин. Конечная цель «температурной программы» — достичь температуры сырья
в 371 °C, чтобы средняя температура в реакторе составила 380 °C. Для сравнения при-
водится другая тактика, по которой начальная температура сразу устанавливается рав-
ной 371 °C с тем, чтобы средняя температура при пуске реактора была равна требуемой,
Сравнение результативности методов приведено на рис. 8.31. Эти методы пред-
ставляют собой два разных способа получения в конечном счете одной и той же сред-
ней температуры в реакторе.
Рис. 8.31. Результативность двух методов пуска
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
365
Безразмерная координата по высоте реактора
Рис. 8.32. Переходные распределения температур при разных способах пуска
В первом случае из-за периодического повышения температуры получается сту-
пенчатая картина пуска. Во втором случае процесс начинается с высоких степеней
превращения, которые затем постепенно выравниваются из-за деактивации катали-
затора. Но первый способ более безопасен в отношении температуры, о чем свиде-
тельствует рис. 8.32. Пуск при менее высокой температуре сырья придает реактору
более приемлемое поведение, чем при пуске по второму способу, когда в первые часы
работы температура превышает 410 °C. Суммируя, можно сказать, что тепловое по-
ведение реактора процесса IMP в первые часы работы при пуске по первому способу
более приемлемо, чем по второму. Такую тактику пуска вполне можно использовать
в целом ряде процессов гидропревращения в неподвижном слое катализатора.
ОБОЗНАЧЕНИЯ
ЛАТИНСКИЕ И РУССКИЕ
А Параметр уравнения (8.19)
амос Коэффициент пересчета единиц измерения в уравнении (8.22)
366
Часть 3. Моделирование каталитических процессов
aL as Л Asph Б С- Площадь границы раздела жидкой и газовой фаз, см-1 Площадь границы раздела жидкой и твердой фаз, см-1 Площадь поперечного сечения реактора, см2 Асфальтены Параметр уравнения (8.19) Молярная концентрация i-ro соединения в газовой фазе, моль/см3 Молярная концентрация i-ro соединения в жидкой фазе, моль/см3 Молярная концентрация z-ro соединения в твердой фазе, моль/см3
сос Ска CpL d i> Еа G Количество кокса на катализаторе Удельная теплоемкость газа, Дж/г- К Удельная теплоемкость жидкости, Дж/ г • К Диаметр частиц катализатора, см Энергия активации, кДж/моль Массовая скорость газа, г/с
Sc gl H. k“pp V 4 k- Ускорение свободного падения, см/с2 Приведенная массовая скорость жидкости, кг/м2 • с Константа закона Генри для i-ro соединения, МПа • см3/моль Кажущееся значение константы скорости/-й реакции Истинное значение константы скорости j-й реакции Значение константы скорости J-й реакции, отнесенное к частицам Коэффициент массопереноса i-ro соединения из газовой фазы в жид- кую, см/с
kf Коэффициент массопереноса z-ro соединения из жидкой фазы в твер- дую, см/с
*H2S L MOC Константа равновесия адсорбции H2S, см3/моль Массовая скорость жидкости, г/с Количество металлов на катализаторе
n. N Ni NJ P Порядок/-й реакции Азот Никель Молярная скорость z-ro газообразного соединения, моль/с Общее давление, МПа
? Q Массовая скорость охлаждающей среды, г/с Охлаждение
r J Скоростьj-й реакции, моль/см3-с
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
367
S t т Первый реактор Второй реактор Сера Время Температура
UG UL N W Приведенная скорость газа, см/с Приведенная скорость жидкости, см/с Ванадий Число смачивания
хмос Z ВО ГДА ГДАс ГДВ ГДМ ГДН гдо Отношение количества металлов на катализаторе к массе катализатора Координата по высоте реактора Вакуумный остаток Гидродеазотирование Гидродеасфальтизация Гидродеванадизация Гидродеметаллизация Гидроденикелизация Гидрообессеривание
ГРЕЧЕСКИЕ
aj ₽, X- Параметр уравнения (8.21) Параметр уравнения (8.21) Параметр уравнения (8.21)
£с £l % 6 Суммарная теплота реакции, кДж/кг серы Удержание газа Удержание жидкости Пористость катализатора Доля пустот в каталитическом слое
Чо Чс£ РС Pi Ф, Коэффициент эффективности катализатора Эффективность внешнего смачивания катализатора Вязкость жидкости Плотность газа в условиях процесса, г/см3 Плотность жидкости в условиях процесса, г/см3 Функция деактивации у-й реакции
368
Часть 3. Моделирование каталитических
пР°Цессов
НИЖНИЕ ИНДЕКСЫ
in Значение на входе следующего каталитического слоя
out Значение на выходе предыдущего каталитического слоя
Q Охлаждающий поток
О Значение на входе реактора
ЛИТЕРАТУРА
Agrawal, R., Wfest, D.H., Balakotaiah, V 2007. Modeling and analysis of local hot spot formation in
down-flow adiabatic packed-bed reactors. Chem. Eng. Sci. 62(18—20):4926—4943.
Al-Dahhan, M.H., Dudukovic, M.P 1995. Catalyst wetting efficiency in trickle-bed reactors at high
pressure. Chem. Eng. Sci. 50(15):2377—2389.
Alvarez, A., Ancheyta, J. 2008. Modeling residue hydroprocessing in a multi-fixed-bed reactor system.
Appl. Ся/я/. Л 351(2):148—158.
Alvarez A., Ancheyta, J. 2009. Effect of liquid quenching on hydroprocessing of heavy crude oils in a
fixed-bed reactor system. Ind. Eng. Chem. Res. 48(3): 1228—1236.
Alvarez, A., Ancheyta, J. 2012. Transient behavior of residual oil fixed-bed hydrodemetallization. Chem.
Eng. J. 197:204-214.
Alvarez, A., Ancheyta, J., Centeno, G., Marroquin, G. 2011. A modeling study on the effect of reactor
configuration on the cycle length of heavy oil fixed-bed hydroprocessing. Fuel 90(12):3551 -3560
Alvarez, A., Ancheyta, J., Munoz, J.A.D. 2007. Comparison of quench systems in commercial fixed-bed
hydroprocessing reactors. Energy Fuels 21 (2): 1133-1144.
Alvarez, A., Ancheyta, J., Munoz, J.A.D. 2009. Modeling, simulation and analysis of heavy oil
hydroprocessing in fixed-bed reactors employing liquid quench streams. Appl. Catal. A 361 (1-2): 1—12.
Ancheyta, J., Centeno, G., Trejo, E, Marroquin, G. 2003. Changes in asphaltene properties during
hydrotreating of heavy crudes. Eneigy Fuels 17(5): 1233-1238.
Ancheyta, J., Betancourt, G., Marroquin, G., Centeno, G., Mufioz, J.A.D., Alonso, E 2010. Process
for the catalytic hydrotreatment of heavy hydrocarbons of petroleum. U.S. Patent 7651604 B2, Jan26.
Bej, S.K. 2002. Performance evaluation of hydroprocessing catalysts: a review of experimen- tai techniques.
Energy Fuels 16(3):774—784.
Bondi, A. 1971. Handling kinetics from trickle-phase reactors. Chem. Technol. l(March): 185-188.
Centeno, G., Ancheyta, J., Alvarez, A., Marroquin, G., Alonso, E, Castillo, A. 2012. Effect of different
heavy feedstocks on the deactivation of a commercial hydrotreating catalyst. Fuel 100:73—79.
Dohler, W, Rupp, M. 1987. Comparison of performance of an industrial VGO-treater with reactor
model predictions. Chem. Eng. Technol. 10(1 ):349—352.
Furimsky, E. 1998. Selection of catalysts and reactors for hydroprocessing. Appl. Catal. A171 (2): 177-206.
Furimsky, E., Massoth, EE. 1999. Deactivation of hydroprocessing catalysts. Catal. TocAvy 52(4): 381 —495.
Gauthier, T, Danial-Fortain, P, Merdrignac, I., Guibard, I., Quoineaud, A.-A. 2008. Studies
on the evolution of asphaltene structure during hydroconversion of petroleum residues. Catal. Today
130(2—4):429—438.
Gierman, H. 1988. Design of laboratory hydrotreating reactors. Scaling-down of trickle-flow reactors.
Appl. Catal. A 43(2):277-286.
Gray, M.R., Ayasse,A.R., Chan, E.W, \feljkovic, M. 1995. Kinetics of hydrodesulfurization of thiophenic
and sulfide sulfur in athabasca bitumen. Energy Fuels 9(3):500—506.
Harold, M.P, Ng, K.M. 1987. Effectiveness enhancement and reactant depletion in a partially wetted
catalyst. AIChE J. 3 3 (9): 1448-1465.
Глава 8. Моделирование и имитация гидропереработки тяжелой нефти
369
Haughey, D.P, Beveridge, G.S. 1969. Structural properties of packed beds: a review. Can. J. Chem. Eng.
47(2):130—140.
Ho, TC. 2008. Kinetic modeling of large-scale reaction systems. Catal. Rev. Sci. Eng. 50(3):287—378.
Ho, TC., Aris, R. 1987. On apparent second-order kinetics. AIChEJ. 33(6): 1050-1051.
Janssens, J.P, Elst, G., Schrikkema, E.G., van Langeveld, A.D., Sie, S.T, Moulijn, J.A. 1996.
Development of a mechanistic picture of the hydrodemetallization reaction of metallo-tetraphenylporphyrin
on a molecular level. Reel. Trav. Chim. Pay Bas. 115(11—12):465—473.
Korsten, H., Hoffmann, U. 1996. Three-phase reactor model for hydrotreating in pilot trickle-bed
reactors.AIChEJ. 42(5). 1350—1360.
Lababidi, H.M.S., Shaban, H.I., Al-Radwan, S., Alper, E. 1998. Simulation of an atmospheric residue
desulfurization unit by quasi-steady state modeling. Chem. Eng. Technol. 21 (2): 193—200.
Marafi, A., Fukase, S., Al-Marri, M., Stanislaus, A. 2003. A comparative study of the effect of catalyst
type on hydrotreating kinetics of Kuwaiti atmospheric residue. Energy Fuels 17(3):661-668.
Marafi, A., Maruyama, E, Stanislaus, A., Kam, E. 2008. Multicatalyst system testing methodology for
upgrading residual oils. Ind. Eng. Chem. Res. 47(3):724—741
Marafi, A., Stanislaus, A., Furimsky, E. 2010. Kinetics and modeling of petroleum residues
hydroprocessing. Catal. Rev. Sci. Eng. 52(2):204—324.
Mederos, FS., Ancheyta, J., Chen, J. 2009b. Review on criteria to ensure ideal behaviors in trickle-bed
reactors. Appl. Catal. A 355(1-2):1-19.
Mederos, FS., Elizalde, I., Ancheyta, J. 2009a. Steady-state and dynamic reactor models for
hydrotreatment offractions: a review Catal. Rev. Sci. Eng. 51(4):485—607.
Munoz, J.A.D., Alvarez, A., Ancheyta, J., Rodriguez, M.A., Marroquin, G. 2005. Process heat integration
of a heavy crude hydrotreatment plant. Catal. Today 109( 1—4):214—218.
Ring, Z.E., Missen, R.W 1991. Trickle-bed reactors: tracer study of liquid holdup and wetting efficiency
at high temperature and pressure. Can. J. Chem. Eng. 69(4): 1016—1020.
Satterfield, C.N. 1975. Trickle bed reactors. AIChEJ. 21 (2):209—228.
Schweitzer, J.-M., Lopez-Garcia, C., Ferre, D. 2010. Thermal runaway analysis of a three-phase
reactor for LCO hydrotreatment. Chem. Eng. Sci. 65(1 ):313—321.
Shah, YT, Mhaskar, R.D., Paraskos, J.A. 1976. Optimum quench location for a hydrodesulfurization
reactor with time varying catalyst activity. Ind. Eng. Chem. Process Des. Dev. 15(3):400—406.
Shah,YT, Paraskos, J.A. 1975. Criteria for axial dispersion effects in adiabatic trickle bed hydroprocessing
reactors. Chem. Eng. Sci. 30(9): 1169—1176.
Sie, S.T 1991. Scale effects in laboratory and pilot-plant reactors for trickle-flow processes. Rev. Inst. Ft:
t/uftr. 46(4):501—515.
Sie, S.T 1996. Miniaturization of hydroprocessing catalyst testing systems: theory and practice. AIChEJ.
42(12)3498-3507.
Sie, S.T 2001. Consequences of catalyst deactivation for process design and operation. Appl. Catal. A
212(1):129—151.
Yin, TY 1980. Dynamics of a trickle-bed hydrocracker with a quenching system. Can. J. Chem. Eng.
58(2):259-266.
Whitaker, S. 1973. The transport equations for multi-phase systems. Chem. Eng. Sci. 28(1): 139-147.
Глава 9. Моделирование
лабораторного реактора
для гидроочистки мексиканской
тяжелой нефти «Майя»
Эта глава иллюстрирует моделирование работы лабораторного реактора гидро-
очистки тяжелой нефти. Гидрообессеривание (ГДО) и гидродеметаллизация (ГДМ)
тяжелой нефти осуществлялись в умеренных условиях реакций. Значения параме-
тров кинетической модели выводились из экспериментальных данных, полученных
при следующих условиях: объемная скорость подачи сырья варьировалась от 0,33
до 1,5 ч_|, температура — от 380 до 420 °C, а давление и кратность водорода к сырью
поддерживались на постоянном уровне (соответственно 6,9 МПа и 5000 ст. фут3/бар-
рель). Лабораторный реактор моделировался как одномерный гетерогенный. При-
водятся подробные сведения об экспериментах и этапах разработки модели, опи-
сывается ее применение для моделирования процессов ГДО и ГДМ мексиканской
нефти «Майя».
9.1. ВВЕДЕНИЕ
Рост добычи тяжелой нефти и необходимость ее переработки побуждают уделять
больше внимания разработке соответствующих моделей кинетики и реакторов. Для
этого обычно выбирают простейшую модель реактора и по данным экспериментов,
проводимых в условиях контроля кинетики, выводят значения кинетических пара-
метров реакций в сплошной жидкой фазе. Из-за сложности состава нефти для пред-
ставления всех однотипных реакций используется один псевдокомпонент, Такой
подход может оказаться успешным при условии, что градиенты концентраций меж-
ду фазами и внутри частиц минимальны, а реактор работает в изотермических ус-
ловиях в режиме вытеснения. Если эти условия не выполняются, для адекватного
моделирования любого реактора нужны надежные зависимости, которые учитыва-
ли бы ограничения массопереноса и другие факторы, влияющие на работу реакто-
ра (такие как распределение жидкости, особенно в маломасштабных экспериментах,
Глава 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя» 371
Я эффективность смачивания катализатора или доли жидкости). Медерос и соавторы
(Mederos et al., 2009] рассмотрели все эти аспекты и перечислили наиболее распро-
страненные методы и зависимости. Были приведены и рекомендации относительно
Того, какой подход следует использовать в отдельных случаях.
В малых реакторах, применяемых в лабораторных масштабах, обычно действуют
ограничения для массопереноса. Если реакторы описываются простыми псевдого-
мофазными моделями, эти эффекты проявляются в величинах констант скоростей
реакций. Чтобы точнее описать работу реактора, используют концепцию долей фаз,
хотя дня описания поведения реактора, судя по сообщениям, лучше подходит кон-
цепция смачивания катализатора.
Концепцию доли жидкости впервые использовали Генри и Гильберт [Нету and
Gilbert, 1973], а другие исследователи применили ее для моделирования работы реак-
торов с орошаемым слоем [Paraskos et al., 1975]. Хотя о применении концепции сма-
чивания катализатора сообщается в целом ряде работ, лишь в нескольких из них при-
водятся величины, связывающие поток жидкости с эффективностью смачивания.
Когда потоки жидкости очень малы, реактор действует в режиме орошаемого слоя
и независимым фактором процесса выступает эффективность смачивания.
Ниже приводится моделирование процесса гидроочистки тяжелой нефти в уме-
ренных условиях в лабораторном реакторе (в частности, для реакций удаления серы,
никеля и ванадия). Также проверяется способность модели адекватно предсказывать
поведение реактора в условиях, отличных от тех, для которых определялись параме-
тры модели. При выводе параметров модели кинетики используются полученные
экспериментальным путем сведения о влиянии температуры, объемной скорости по-
дачи сырья и давления в широких интервалах значений. Особо тщательно освещает-
ся порядок определения параметров разработанной модели.
9.2. МОДЕЛЬ
Надежность модели зависит от способов ее проверки, среди которых — тестирование
с использованием независимых данных, а также массопереносных и термодинами-
ческих свойств в условиях реакций, полученных экспериментальным путем. Реактор
гидроочистки тяжелой нефти содержит три фазы: неиспареиные углеводороды (жид-
кая фаза), испаренные углеводороды плюс водород (газовая фаза) и неподвижный
каталитический слой (твердая фаза). Таким образом, моделируется трехфазный гете-
рогенный реактор с неподвижным слоем катализатора. Для представления реального
экспериментального реактора необходимо принять ряд допущений.
9.2.1. Допущения при разработке модели
В данном исследовании были приняты следующие допущения:
• жидкая и газовая фазы движутся в режиме вытеснения (в пробковом режиме);
• потери давления пренебрежимо малы;
372
Часть 3. Моделирование каталитических процессов
• реактор работает в изотермическом режиме;
• испарением жидкости можно пренебречь;
• существует определенное сопротивление массопереносу между жидкостью
и частицами;
• сопротивления массопереносу между газовой и жидкой фазами со стороны га-
зовой фазы пренебрежимо малы;
• активность катализатора постоянна во всем слое;
• порядки реакций не зависят от концентрации гетероатомных соединений
и температуры;
• размеры и форма частиц катализатора одинаковы;
• газовая фаза на входе в реактор содержит только водород;
• средневзвешенная температура кипения жидкости по мере ее реагирования
не изменяется вследствие умеренных условий реакций;
• уравнения массопереноса справедливы для действующей каталитической си-
стемы и размеров частиц;
• закон Генри справедлив при действующих значениях концентраций водорода
и сероводорода, растворяющихся в жидкой фазе.
В дальнейшем принимаются дополнительные допущения для упрощения обще-
го баланса масс.
9.2.2. Описание модели
Молярный баланс масс по фазам при моделировании многофазных реакторов имеет
вид [Gunjal and Ranade, 2007]
~Ei£kfk,i + УеДОХ = V(£AGAVA,) + et5,.t. (9.1)
at
Уравнение (9.1) учитывает колебания в нестационарном состоянии (d(ECf) / df),
конвективный поток (V(e£/C/)), диффузионный поток (V(eCtZDV/)) и образование
и расходование соединений (еУ). Уравнение позволяет моделировать работу реакто-
ров с неподвижным слоем, в которых катализатор выступает как твердая фаза, а жид-
кость и газ — как подвижные фазы.
В уравнении (9.1) к - любая фаза, i — компоненты смеси, Ск — общая концентра-
ция в фазе k,fki — молярная доля /-го соединения в фазе к, Uk — скорость молекул
фазы к или ее средняя действительная скорость для усредненных моделей, Dk — ко-
эффициент диффузии соединений в фазе к, S.к — член, описывающий источник (об-
разование и расходование) /-го соединения.
Если допустить, что изменения концентраций происходят лишь в осевом направ-
лении (z) в цилиндрических координатах, уравнение (9.1) сокращается до
— Е,и,Ск — £kS.
dz к к к'‘ к '< (9.2)
Ямвва 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя» 373
Если доли и скорости фаз считать постоянными, уравнение (9.2) принимает вид
Uk^ckJ=SiJt. (9-3)
dz
Гунджал и Ранад [Gunjal and Ranade, 2007], моделируя трехфазный реактор гидро-
очистки, представили источник на границе раздела газовой и жидкой фаз выражением
^G.i ~ K-GLiaGL
1 Gi __f*
тт
(9.4)
а в жидкой фазе — выражением
^L,i ~ ^-GLiaGL
1 Gi _ f*
гт
nr
+рв^гЛс^)-
j=>
(9.5)
Скорость расходования реагентов в установившемся состоянии можно прирав-
нять к скорости массопереноса между жидкой и твердой фазами:
Q,) Ps1)7).
(9.6)
Подставляя соотношение (9.6) в уравнение (9.5), с учетом отсутствия сопротивле-
ния массопереносу между газовой и жидкой фазами на стороне газовой фазы мож-
но получить
$L.i ~ кцаь
С.
[н,
+ ^Sias(^Li Cgi\
(9.7)
При таком подходе в уравнениях баланса используется концепция долей фаз;
предполагается, что реакции происходят в жидкой фазе.
Чтобы моделировать в рамках реакции ГДО не только соединения серы в жидкой
фазе, но и распределения водорода и сероводорода в реакторе, уравнение (9.6) видо-
изменяется умножением правой части на стехиометрический коэффициент т>:
ksias CSi) и, Ра Г| ггдо.
(9.8)
9.2.2.1. Стехиометрические коэффициенты реакций ГДО
В жидкой углеводородной смеси содержится большое число различных соединений
серы. По этой причине (а также оттого, что псевдокомпоненты учитывают лишь об-
щие концентрации — например, общее содержание серы) невозможно знать зара-
нее величины стехиометрических коэффициентов в происходящих реакциях. Чтобы
обойти эту проблему, предлагается следующее обобщенное уравнение [Korsten and
Hoffmann, 1996]:
374
Часть 3. Моделирование каталитических процессов
,<НС - S)£ + (Н2)„ -> «„(НС), + «,„ (Н,s)c, (9.9)
где LnG— соответственно жидкая и газовая фазы; HC-S — углеводороды, содержа-
щие серу; НС — углеводороды, не содержащие серы.
Стехиометрический коэффициент для H2S можно найти по балансу масс. По со-
держанию серы в жидкости на входе и выходе из реактора можно определить коли-
чество прореагировавшей серы. Затем, зная содержание сероводорода в жидкой и га-
зовой фазах, можно написать следующее уравнение молярного баланса масс для H2S
в реакции ГДО:
wh,s ~/7h,s.o'1 (whc-s,o х xnc-s)> (9.Ю)
UHC-S
где xHC S — степень превращения, определяемая как
WHC-S,0
В лабораторных опытах на вход реактора принято подавать водород высокой чи-
стоты, так что nH2S 0 = 0; тогда уравнение (9.10) упрощается до
wh,s =------(whc-s,ox whc-s) (9-12)
UHC-S
Число молей соединений серы находится по формуле
WHC-S = ! ^^oih (9.13)
где /иНС 5 — масса жидкости, содержащей соединения серы; xiS — массовая доля серы;
MWojl — средняя молярная масса жидкости.
Таким образом, располагая данными о массе жидкости и концентрации серы
на входе и выходе реактора и количестве сероводорода после разделения фаз, из урав-
нения (9.12) путем линейной регрессии (у = nH2S, т = ,uH,s/'oHC_s, х= (nHC_S0 — nHC_s))
можно вывести величину отношения vH s/vHC_s.
Применить ту же процедуру для нахождения стехиометрического коэффициента
водорода не получится, так как он расходуется в нескольких реакциях. Если протека-
ет только реакция ГДО, величину можно определить, хотя и с некоторой погрешно-
стью, поскольку водород расходуется и на насыщение углеводородов. Если же это не так,
что чаще всего и бывает, величину можно принять несколько выше, чем для серово-
дорода, поскольку Н2 присутствует в избытке и величина его стехиометрического коэф-
фициента не имеет значения для моделирования ГДО [Korsten and Hoffmann, 1996].
Э.2.2.2. Коэффициенты скоростей реакций
Ввиду объединения реагентов в псевдокомпоненты кажущиеся значения констант
скоростей реакций (к ) зависят от типа катализатора, массо- и теплопереноса,
Глава 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя» 375
условий на поверхности катализатора и распределения серы и металлов в сырье.
Установлено, например, что соединения серы при гидроочистке дизельных фракций
реагируют с разными скоростями. Стойкие соединения (например, дибензотиофены
с алкильными радикалами в положениях 4-, 6- и 4-6-, показывают низкие значения
констант скоростей, тогда как у соединений без алкильных заместителей в этих по-
ложениях они выше [Ma et al., 1994].
Рабочие условия на поверхности катализатора связывают кажущиеся значения
констант скоростей с истинными (&.J и долями жидкости (е£) посредством следую-
щего уравнения [Ma et al., 1994]:
к \ьк.гг (9.14)
В определении скорости реакции кинетические данные можно связать с жидкой
фазой либо с объемом или массой катализатора [Levespiel, 1999]. В случае реакторов
с насадочным слоем определение скорости реакции удобно связывать с массой или
объемом катализатора, а вместо физической скорости жидкости можно использовать
приведенную. В уравнениях баланса с приведенной скоростью связь истинного зна-
чения константы скорости с кажущимся более очевидна, если использовать коэффи-
циент эффективности смачивания (fw):
к iik.f. (9.15)
Параскос и соавторы [Paraskos et al., 1975] заметили, что в условиях неполного
смачивания кажущийся порядок реакции изменяется с увеличением степени превра-
щения при постоянной константе скорости реакции. Как было установлено ранее,
Лорр зависит от эффективности смачивания или доли жидкости, причем оба этих фак-
тора сами зависят от скорости жидкости. Кроме того, с изменением LHSVизменяется
также карр [Korsten and Hoffmann, 1996], т. е. кажущаяся константа скорости на самом
деле константой не является. Отчасти это обусловлено разной активностью соедине-
ний серы, проявляемых при ГДО: например, оставшиеся соединения серы при высо-
кой степени превращения более стойки, чем при низкой.
Было найдено, что доля жидкости зависит от ее вязкости и приведенной мас-
совой скорости в степени 1/3. В некоторых исследованиях доля жидкости связы-
вается с отношением приведенной массовой скорости к вязкости, возведенным
в степень 1/3. Во всяком случае, доля жидкости зависит от ее вязкости. Важную
роль играет также размер частиц катализатора: доля жидкости обратно пропор-
циональна размеру частиц в степени 2/3 [Satterfield et al., 1969]. Таким образом,
вполне возможны заметные эффекты при изменении размеров частиц и вязко-
сти жидкости; может быть разным и вид зависимостей, связывающих долю жид-
кости с физическими свойствами сырья, рабочими параметрами и размерами ча-
стиц катализатора.
В опубликованных исследованиях работу псевдогомофазной модели гидро-
очистки рассматривают с использованием концепции доли жидкости в реакторе
[Tarhan, 1983], в то время как в гетерогенных моделях предпочитают концепцию
376
Часть 3. Моделирование каталитических процессов
эффективности смачивания [Levespiel, 1999]. Несмотря на это, можно ожидать, что
эти два подхода будут давать сравнимые значения кинетических параметров (по-
рядка реакции и кажущейся энергии активации), так как доля жидкости связана
с эффективной площадью контакта между жидкостью и твердым катализатором
[Satterfield, 1975].
Модели, основанные на приведенной скорости жидкости, широко используются
различными исследователями для моделирования трехфазных экспериментальных
реакторов [Korsten and Hoffmann, 1996; Chowdhuri et al., 2002; Rodriguez and Anchey-
ta, 2004; Mederos et al., 2006]. В настоящей работе при имитации ГДО и ГДМ тяжелой
нефти применяется тот же подход.
9.2.2.3. Определение кинетических параметров
9.2.2.3.1. Порядок реакции гидрообессеривания
Кинетика реакции ГДО тяжелой нефти обычно моделируется степенным зако-
ном. Порядок реакции удаления серы составляет, судя по сообщениям, от 1 до 3,
причем наименьшие значения относятся к легким дистиллятам, а наибольшие —
к тяжелому сырью. Порядок реакции зависит главным образом от типа и кон-
центрации соединений серы, присутствующих в нефтяном дистилляте. Сведений
о кинетике ГДО тяжелых фракций в литературных источниках приводится мало.
Например, Озаки и соавторы [Ozaki et al., 1975] для интерпретации кинетических
данных, полученных при ГДО атмосферного остатка кувейтской нефти в реакто-
ре с неподвижным слоем с нисходящим потоком, использовали простую кинети-
ческую модель, подчиняющуюся степенному закону. Авторы нашли, что при по-
вышении температуры кажущийся порядок реакции изменяется. При 380 и 410 °C
значения порядка реакции составляли соответственно 2,4 и 1,55. Позже Кам и со-
авторы [Kam et al., 2008], используя процедуру, основанную на статистических ар-
гументах, адаптировали данные Озаки и соавторов ко второму порядку реакции
при всех температурах.
Порядок реакции ГДО моделируют также уравнением Ленгмюра-Хиншельвуда
[Korsten and Hoffmann, 1996]:
. «(eg, Г
ГГДО Kpp , ,2 • (9.16)
(l + ^s^s)
В настоящей работе используется уравнение (9.16): это позволяет учесть ингиби-
рующее влияние H2S на реакцию ГДО. Величина т; определяется методом мини-
мизации расхождения между расчетными и экспериментальными данными удале-
ния серы. Порядок реакции для водорода (т2), согласно некоторым литературным
источникам [Ross, 1965; Mederos, 2010], принят равным 0,5. Такое значения поряд-
ка реакции объясняется диссоциацией Н2 на активных центрах катализатора. Вторая
степень знаменателя в уравнении (9.16) обусловлена тем, что реакция происходит по-
следовательно на двух активных центрах каталитической поверхности.
Глава 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя»
377
9.2.2.3.2. Порядок реакции гидродеметаллизации
Другая важная реакция гидроочистки тяжелой нефти, помимо ГДО, — гидродеметал-
лизация (ГДМ), а именно реакции удаления никеля (ГДН) и ванадия (ГДВ).
Зависимость скоростей реакций ГДМ от концентраций металлов (CN и Cv) обычно
представляют простым степенным законом:
ГГДН “ ^ТДНCn'i’ (9.17)
ггдв — • (9.18)
Скорости реакций ГДМ косвенно зависят от парциального давления водорода че-
рез константы скоростей (£гдн и /сгдв), поскольку их выражения сочетают кажущуюся
скорость реакции с концентрацией водорода.
Для нахождения порядка и кажущихся значений констант реакций ГДМ их внача-
ле определяли, предполагая псевдогомофазную фазу в реакторе, а затем пересчиты-
вали с учетом сопротивления массопереносу между жидкой и твердой фазами.
Э.2.2.4. Оценка транспортных
и термодинамических свойств
Все используемые здесь зависимости выбирались исходя из их области применимо-
сти в зависимости от условий процесса (главным образом температуры, плотности
сырья или размера частиц катализатора). Важность адекватной оценки транспорт-
ных и термодинамических свойств обусловлена их влиянием на работу реактора че-
рез коэффициенты массопереноса и кинетические параметры. Без сомнения, для
моделирования реактора наиболее важны плотность жидкости и газа, вязкость жид-
костей, растворимость и коэффициенты диффузии. С другой стороны, можно ожи-
дать определенных отклонений от зависимостей, поскольку в данном исследовании
экспериментальные реакторы работали в режиме орошаемого слоя. Для учета этого
обстоятельства можно использовать концепции доли жидкости или эффективности
смачивания катализатора. В первом случае важны распределение и размеры частиц
катализатора и диаметр реактора. Эти факторы взаимосвязаны, и путь к их правиль-
ному учету лежит через долю пустот в каталитическом слое. Ввиду отсутствия сведе-
ний о значениях транспортных и термодинамических свойствах в рабочих условиях,
для их оценки использовались эмпирические зависимости из литературных источ-
ников. Ниже описываются эти зависимости, смысл содержащихся в них переменных
и причины их выбора.
9.2.2.4.1. Доля пустот в слое
Опубликован ряд формул для определения доли пустот в слое. Хофи и Беверидж
[Haughey and Beveridge, 1969] сообщили о следующей зависимости для сферической
насадки:
378
Часть 3. Моделирование каталитических процессов
е„ = 0,39 + 0,07(c/p/c/f) + 0,54(rf/cZ()2, (9.19)
где £0 — общая доля пустот в слое, т. е. полученная с учетом межчастичных пустот
в насадочном слое (средней пустотности) и пристеночного эффекта (пустоты между
стенками сосуда реактора и насадкой); d — эквивалентный диаметр частиц; dt — вну-
тренний диаметр сосуда реактора.
Общая доля пустот в слое связана со средней пустотностью е формулой
ео = е + О,34(</,/</,), (9.20)
где в равно 0,36 и 0,39 соответственно для плотной и для рыхлой нерегулярной насад-
ки. Из уравнения (9.20) видно, что £0 -> £ при d /d^ 0, поскольку влияние присте-
ночного эффекта снижается [Foumeny et al., 1991; Sie, 1996].
Диксон и соавторы [Dixon et al., 1988] изменили уравнение (9.19), включив в него
результаты Кармана [Carman, 1937], что дало
е = 0,40 + 0,05(</,Д) + 0,412(rf/</()2, (9.21)
где <//</, <0,5.
Для сферической насадки Беньяхья и О’Нейлл [Benyahia and O’Neill, 2005] пред-
ложили также следующую зависимость:
1 740
е = 0,390 + ----(9.22)
(</,Я+1,140)
где 1,5 < </,/</,< 50.
Хофи и Беверидж предположили также, что при dt / dp > 10 пристеночным эф-
фектом можно пренебречь. Таким образом, можно ожидать, что при очень малых
размерах частиц, выбираемых для межчастичных сопротивлений массопереносу,
значения средней пустотности и общей доли пустот одинаковы и стремятся к пре-
делу, равному 0,69. Это согласуется с экспериментальными наблюдениями Чу и Нг
[ChuandNg, 1989].
Хотя упомянутые зависимости были выведены для непористой насадки, их мож-
но использовать для расчета средней пустотности пористой насадки, так как предпо-
лагается, что последняя заполнена жидкостью и поэтому доля удерживаемой в порах
жидкости является внутренней. Так что, если нужно определить внешнюю долю жид-
кости, можно пользоваться приведенными выше зависимостями.
9.2.2.4.2. Доля жидкости
Для расчета линейных скоростей необходимо знать доли жидкости и газа, по-
скольку они позволяют связать время пребывания и степень превращения [Tar-
han, 1983]. Доли жидкости при малых и больших значениях числа Рейнольдса
должны, по-видимому, вычисляться по разным формулам. Были сопоставлены две
Глава 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя»
379
зависимости с близкими областями применимости: формула для пористых частиц,
опубликованная Ланге и соавторами [Lange et al., 2005], и формула для непористых
частиц, записанная Фу и Таном [Fu and Tan, 1996]. Шварц и Робертс [Schwartz and
Roberts, 1973] показали, что вычисляемые по этим формулам значения внешней
доли жидкости в пористой насадке и общей доли жидкости в непористой доста-
точно близки:
• для пористого катализатора [Lange et al., 2005]
= 0,002(4 /<V28 R<38 ПРИ °’05 b5 ; (9.23)
• для непористого катализатора [Fu and Tan, 1996]
h,, = 1,505 Re?’29 Ga;°’3X°’22 при ReL < 2,3,
(9.24)
4
где -j- =
4
16b3 u,p,dn
——-y; Re' = Ga =
9тг(1-в) цл£
PZ '
Сравнение внешних долей жидкости по этим формулам, проводимое при допу-
щении, что статическая внешняя доля жидкости составляет от 0,02 до 0,05 [Hofmann,
1977], показало, что формула Фу и Тана дает меньшие значения.
Фу и Тан [Fu and Tan, 1996] заметили, что размер частиц насадки и вязкость ока-
зывают сильное влияние на общую долю жидкости и что при уменьшении разме-
ров частиц и/или увеличении вязкости доля жидкости возрастает. Ланге и соавто-
ры соотносили результаты лишь с числом Рейнольдса жидкости (ReJ, т. е. считали
влияние силы тяжести пренебрежимо малым. Они пришли к выводу, что при очень
малых числах Рейнольдса, характерных для маломасштабных экспериментальных
реакторов, доля жидкости прямо пропорциональна приведенной массовой скоро-
сти и обратно пропорциональна вязкости жидкости. Фу и Тан проводили исследова-
ния на жидкостях с вязкостью 0,22 сП и непористых частицах размерами 0,5-1,9 мм,
Ланге и соавторы — на жидкостях с вязкостью 0,795 и 1,22 сП и пористых частицах
размерами от 0,57 до 3 мм. Во втором случае вязкость жидкости близка к той, кото-
рую имеет нефть при температурах и давлениях, использовавшихся в данном иссле-
довании. Поэтому для моделирования лабораторного реактора была выбрана зависи-
мость Ланге с соавторами.
9.2.2.4.3. Эффективность смачивания
Жидкая фаза движется преимущественно через одну часть неподвижного каталити-
ческого слоя, а газовая — через другую. Такое неоднородное распределение фаз назы-
вается неполным смачиванием [Sie, 1996]. Эффективность смачивания обычно мо-
делируют через константу скорости. Наиболее распространенный подход, который
предложили Саттерфилд и Робертс [Satterfield and Roberts, 1973], исходившие из на-
блюдений Бонди [Bondi, 1971], связывает истинное значение константы скорости
с кажущимся посредством следующего уравнения:
380
Часть 3. Моделирование каталитических процессов
где Gl — приведенная массовая скорость, кг/м2 с; А, В, к — константы, эксперимен-
тально определяемые изменением объемной скорости жидкости.
Ввиду отсутствия сведений об истинных значениях констант скоростей реакций
ГДО предлагается следующая модификация уравнения (9.25):
где k" — псевдоистинная константа скорости, зависящая от коэффициента эффек-
тивности. Дудукович [Dudukovic, 1977] вывел соотношение между эффективностью
контактирования и коэффициентом эффективности и заметил, что при большом
диффузионном сопротивлении степень использования катализатора пропорцио-
нальна площади внешней смачиваемой поверхности, а в полностью контролируемом
режиме — площади внутренней смачиваемой поверхности. Отсюда следовало, что
в последнем случае изменения внешней смачиваемой поверхности не влияют на ско-
рость реакций.
9.2.2.4.4. Вязкость жидкости
Большинство зависимостей, используемых при расчете вязкости, в случае тяжелой
нефти дают ошибочные результаты, поскольку разработаны для легких дистилля-
тов. Гласе [Glaso, 1980] сообщил о зависимости, применимой к нефтяным фракци-
ям с плотностью 20 °АР1 я выше [Riazi, 2005]. Это, пожалуй, наиболее подходящая
формула вязкости для нефтяных фракций. Для более тяжелых нефтей погрешности
по данной формуле могут достигать 100 % [Bennison, 1998; Riazi, 2005].
Здесь использовалась выведенная Гйасё формула для вязкости (ц£) нефтей с плот-
ностью от 20 до 45,8 ° АРР.
цЛ = c[log (у^)]", (9.27)
где
с = 3,141 • 10|0Т-3’444; (9.28)
d= 10,313 log Т- 36,447. (9.29)
(Здесь Т— температура, °F; 1АР,— плотность нефти, ° API.)
9.2.2.4.5. Массоперенос
На массоперенос сильно влияют размер частиц и пористость катализатора. Напри-
мер, Гото и Смит [Goto and Smith, 1975] сравнили коэффициенты массопереноса для
катализатора и непористого Р-нафтола с двумя размерами частиц: 0,291 и 0,0541 см.
Глава 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя»
381
Исследователи обнаружили, что можно ожидать сильную зависимость коэффициен-
тов массопереноса от размеров частиц, если они сравнительно малы, и что формулы,
выведенные для крупных непористых частиц, неприменимы для пористых. Что же
касается широко применяемой формулы Ван Кревелена и Крекельса [Van Krevelen
and Krekels, 1948] для расчета массопереноса из жидкой фазы в твердую, то она дей-
ствительна лишь для частиц определенных — достаточно крупных — размеров. Здесь
используется зависимость Гото и Смита, хотя значения параметров (коэффициенты
и показатели степени) приходилось определять экстраполяцией, так как значения
размеров частиц в экспериментах выходили за пределы, для которых была выведе-
на зависимость.
Уравнения Гото и Смита выглядят следующим образом:
• для массопереноса из газовой фазы в жидкую
kLaL z а gl
BLi I М-£ у
Р/А,у
(9.30)
• для массопереноса из жидкой фазы в частицы
ksaL __ а Ч. Pi
dl, 4pJ IpA/J
(9.31)
Значения параметров aL, as, n[ и ns в уравнениях (9.30) и (9.31) приводятся в рабо-
те Гото и Смита [Goto and Smith, 1975].
9.2.2.4.6. Растворимость
В настоящей работе для расчета растворимости водорода в жидкости использует-
ся формула, которую предложили Корстен и Хоффманн [Korsten and Hoffmann,
1996]. Сравнение результатов расчетов по этой формуле с данными Лала с соавтора-
ми [Lal et al., 1999] и Кая с соавторами [Cai et al., 2001] показало хорошее согласие.
Для растворимости H2S зависимость найти трудно из-за отсутствия значений пара-
метров взаимодействия. К тяжелой нефти неприменима формула, которую предло-
жили Кэрролл и Мазер [Carroll and Mather, 1995] для расчета параметров бинарного
взаимодействия H2S с парафинами в зависимости от средневзвешенной температуры
кипения. Корстен и Хоффманн [Korsten and Hoffmann, 1996] указали, что примене-
ние закона Генри для расчета выхода H2S может быть ошибочным, так как коэффи-
циенты Генри ограничены малыми концентрациями растворенного вещества. По-
этому чем выше степень ГДО, а следовательно, и выход H2S, тем больше отклонение
от закона Генри. Уменьшение кратности водорода к сырью увеличивает парциальное
давление H2S, что также вызывает отклонение от закона Генри.
Для коэффициента растворимости сероводорода Хн можно вывести экспоненци-
альное выражение:
XHjS = ехр(а + b х Г).
(9.32)
382 Часть 3. Моделирование каталитических процессов
В настоящей работе коэффициент Генри для H,S вычислялся без параметров вза-
имодействия с использованием модуля фракционного расчета программного паке-
та HYSYS.
9.2.2.4.7. Коэффициенты диффузии
Для расчета коэффициентов диффузии жидких углеводородов при низких дав-
лениях предлагается использовать модифицированную формулу Тина и Калуса
[Reid et al., 1987]:
тгО.267 /т, Л
D°ab- 8,93- р , (9.33)
где V — молярные объемы веществ; Т — температура; р — динамическая вязкость.
Коэффициенты поверхностного натяжения <з берутся для средней температуры ки-
пения (Ть). Значения коэффициентов поверхностного натяжения большинства орга-
нических жидкостей при Ть близки; следовательно, их соотношение при возведении
в степень с небольшим показателем дает примерно единицу. Тогда уравнение (9.33)
принимает вид
Г/-0.267 Т {гу
^8,93-КГ8-^— -М . (9.34)
Молярные объемы рассчитывались по формуле Тина и Калуса [Туп and Calus, 1979]:
V = 0,285V(]':m, (9.35)
где значение критического объема (Кс) для газообразных соединений берется из ли-
тературных источников, а для жидкости вычисляется по формуле Риази и Дауберта
[Riazi and Daubert, 1987].
Для решения уравнения (9.35) необходимо, чтобы была известна величина крити-
ческого объема компонента. Для газов ее найти нетрудно, но для соединений серы
и металлов эту величину приходится принимать такой же, как у углеводородов, в ко-
торых эти соединения содержатся. Разумеется, такое предположение не отражает
действительного положения вещей, поскольку реакционная смесь на самом деле со-
держит ряд соединений металлов и серы. Но из-за сложности углеводородной сме-
си выявление этих соединений и определение их свойств до сих пор остается труд-
ной задачей. Поэтому гипотеза, согласно которой критические объемы соединений
серы и металлов равны критическому объему углеводорода, соответствующего сред-
ней температуре кипения, представляется достаточно приемлемой.
9.2.2.4.8. Плотность
Плотность нефти р(Р, Т) можно вычислить по формуле Стендинга-Каца:
р(Р, Т) = р(Р0, Го) + Дрр + Дрг, (9.36)
Глава 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя» 383
где р(Р0, То) — плотность нефти в стандартных условиях, т. е. при давлении
14,7 psi (абс.) и температуре 60 °F (15,6 °C); Др? — поправка на сжимаемость нефти;
ЛРг — поправка на тепловое расширение нефти.
При этом
Др„ = 0,167+ 16,181 • 1о-°.°425р(ДЛ)_
р 1000
- 0,01Г0,299 + 263- io-°'o6O3p(?»-7’»)"|—(9.37)
L J 1000
Арт = {0,0133 +152,4 [р(РЛ) + Др,] -°'245 ) (Т- 520)
— (8,1х10-6+152,4 ю-°’764[р(Р»-7Ь)+Др"1| (7’-520)2. (9.38)
Плотность водорода в газовой фазе можно рассчитать по хорошо известному урав-
нению состояния Пенга-Робинсона [Peng and Robinson, 1976].
9.2.3. Решение модели
Система уравнений (9.3)—(9.7) решалась в пакете MATLAB® путем расчета всех
транспортных термодинамических свойств в имитируемых условиях. Обыкновенные
дифференциальные уравнения решались методом Рунге-Кутты, а алгебраические
уравнения, «порождаемые» уравнением (9.8) на каждом шаге интегрирования, —
с помощью функции fsolve из пакета MATLAB. Чтобы добиться сходимости, во вто-
ром случае для начальных значений концентраций серы и металлов у поверхности
катализатора принимался тот же порядок величины, что и в жидкости. Граничные ус-
ловия для решения системы дифференциальных уравнений имеют вид
Cs(z = 0) = Cs°,Cs(z = £) = C''; (9.39)
CNi(z = 0) = CCNi(z = £) = CNLi; (9.40)
Cv(z = 0) = C“,Cv(z=£) = CvL; (9.41)
^ = 0) = PhVh2(z = £) = Ph2; (9.42)
PH2S(z = 0) = 0,PH2S(z = Z,) = ^2S. (9.43)
Начальные условия (кроме парциальных давлений) можно определить, приняв,
что соединения серы и металлов имеют ту же среднюю молярную массу, что и вся не-
фтяная смесь в целом. Поэтому для нахождения концентраций в жидкости на входе
в реактор и выходе из него использовались следующие выражения:
384
Часть 3. Моделирование каталитических процессов
Pl „о .
» тг ’
qL -
' MWml
(9.44)
(9.45)
где р£ — плотность жидкой нефти в условиях реакции; xw. — массовая доля металлов
или серы в нефтяной смеси; MWgil — средняя молярная масса нефтяной смеси.
В приведенных уравнениях точка с z = 0 располагается на входе, а с z - L — на вы-
ходе каталитического слоя. При выводе значений кинетических параметров реакций
ГДО и ГДМ использовались все имеющиеся данные для давления 6,9 МПа. По ка-
ждой реакции использовалось восемнадцать экспериментальных точек, т. е. по шесть
точек данных при различных скоростях подачи сырья для каждого из трех значений
температуры. Для нахождения кажущейся константы скорости при каждом значении
объемной скорости подачи сырья уравнения (9.3)-(9.7) решали при условии посто-
янной температуры. Для улучшения точности область изменения приведенной мас-
совой скорости жидкости была разбита на 220 интервалов. После проверки влияния
LHSVна кажущееся значение константы скорости определяли значения параметров
уравнения (9.25). Для этого строилась линейная зависимость вида у = тх + Ь, где
у=]/к , т = А’,х= ]/G.e, b = I / к*..
9.3. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
9.3.1. Характеристики сырья
Сырьем в экспериментах служила нефть «Майя», основные свойства которой приве-
дены в табл. 9.1. Содержание серы в сырье и продуктах определялось анализатором
SLFA-2100 компании HORIBA по стандартному методу ASTM 7)4294. Точки кривой
разгонки находили имитированной дистилляцией по методу ASTM 7)5307.
Характеристики сырья (тяжелая нефть «Майя»)
Таблица 9.1
Свойство Значение
Относительная плотность (</4м) 0,9220
Средняя молярная масса 550
Общая сера, %масс. 3,51
Общий азот, мг/кг 3559
Разгонка, °C:
НК 30,21
испаряется 5 %масс. 136,4
» 10 %масс. 187,8
/Глава 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя» 385
Таблица 9.1, окончание
Свойство Значение
» 30 %масс. 325,9
» 40 %масс. 394,9
» 50 %масс. 461,7
» 60 %масс. 533,8
Содержание металлов, мг/кг: Ni V 53 292
Концентрации никеля и ванадия в сырье определяли атомно-абсорбционным
спектрометром SolaarAA. Для каждого измерения отбирали пробу массой 4 г. Пробу
разбавляли раствором HNO, и для удаления жидкости сжигали. Затем пробу прока-
ливали в течение 4 ч при температуре 500 °C. После прокаливания в пробу добавляли
смесь НС1 и НЬ1О3 и определяли количество металлов.
9.3.2. Экспериментальный реактор
Центральным узлом лабораторной установки являлся реактор с карманом для термо-
пары вдоль оси реактора. Внутренний диаметр реактора составлял 2,54 см, внешний
диаметр кармана термопары — 0,635 см. Электрические нагреватели позволяли ре-
актору обеспечивать изотермическую работу. Реактор работал в режиме нисходящего
потока, сырье и водород подавались вместе. Схемы лабораторной установки и реак-
тора показаны на рис. 6.1 (глава 6) и 7.14 (глава 7).
9.3.3. Изотермическая работа реактора
Для подтверждения того, что экспериментальный реактор работает в изотермическом
режиме, было проведено несколько опытов, заключавшихся в измерении фактиче-
ских значений температуры вдоль слоя и сравнении их с ожидаемыми. Максималь-
ное отклонение от изотермического режима, составившее около 5 °C, наблюдалось
при наиболее жестких условиях, т. е. при температуре 420 °C и объемной скорости
(LHSV) 0,33 ч-1.
9.3.4. Свойства катализатора
Во всех экспериментах применялся промышленный никель-молибденовый катали-
затор на у-алюмооксидном носителе со следующими свойствами: LHSV= 175 м2/г,
386
Часть 3. Моделирование каталитических процессов
средневзвешенный удельный поровый объем 0,56 см3/г, средневзвешенный диаметр
пор 127 А, содержание молибдена 10,66 %масс., содержание никеля 2,88 %масс. Ча-
стицы четырехлепестковой формы имели диаметр 2,54 мм.
9.3.5. Загрузка катализатора
В реактор загружали 100 мл катализатора, просеянного через сита определенной
крупности, обеспечивавшие получение нужного гранулометрического состава. Ко-
нечный средний размер частиц был равен 0,25 мм. Хотя не все частицы имели одни
и те же форму и размеры, различия были небольшими. Уменьшение размера ча-
стиц изменяло характеристики массопереноса внутри них при реакциях; кроме того,
на работу реактора могли влиять характеристики орошения слоя (доля жидкости,
эффективность смачивания). Конечная плотность каталитического слоя была рав-
на 0,7305 г/см3.
9.3.6. Активация катализатора
После загрузки катализатор активировали пропусканием через него в течение 18 ч
бензиновой фракции с добавкой 0,8 %масс. CS2 при давлении 5,3 МПа, кратности
водорода к сырью 2000 ст. фут3/баррель, температуре 230 °C и LHSV= 3,2 ч-1.
9.3.7. Сопротивление массопереносу
Эксперименты на катализаторах различного гранулометрического состава показали,
что при размере частиц 0,25 мм и менее степень ГДМ и ГДО остается практически
постоянной. Таким образом, именно этот размер позволял свести к минимуму кон-
центрации градиентов внутри частиц.
Проводились исследования сопротивления массопереносу между жидкостью
и частицами катализатора при разных значениях соотношения потока жидкости
и количества катализатора [Perego and Paratello, 1999]. Эксперименты выполнялись
при двух значениях объемной скорости подачи сырья (0,33 и 1,5 ч-1) и температу-
ры (360 и 420 °C). Было найдено, что при LHSV= 1,5 ч~' степень превращения су-
щественно не изменялась. Но при LHSV= 0,33 ч_| и том же количестве катализатора
(100 мл) степень превращения с изменением температуры не оставалась постоянной.
Это означает, что при высоких скоростях потока жидкости сопротивление межфаз-
ному массопереносу минимально, а при низких остается значительным. Чтобы уве-
личить поток жидкости при той же объемной скорости подачи сырья, следовало за-
грузить в реактор больше катализатора, но конструкция реактора не позволяла этого
сделать. Таким образом, данные, полученные при низких значениях LHSV, зависят
от сопротивления межфазному массопереносу.
Имва 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя»
387
9.3.8. Влияние условий реакций
Изучалось влияние LHSV, температуры и давления на реакции ГДО, ГДН и ГДВ при
постоянной кратности водорода к сырью, равной 5000 ст. фут3/баррель. Эксперимен-
ты проводили при шести значениях LHSV(0,33, 0,5, 0,75, 1,0, 1,25 и 15 ч_|), трех зна-
чениях температуры (380, 400 и 420 °C) и постоянном общем давлении (6,86 МПа)
на входе в реактор. Так как использовался водород 99,99%-ной чистоты, можно было
считать, что общее давление на входе равно парциальному давлению водорода. Для
проверки работоспособности модели были получены данные при тех же значениях
LHSVи температуры, но с увеличением общего начального давления до 8,3 МПа.
9.4. РЕЗУЛЬТАТЫ
9.4.1. Стехиометрические коэффициенты
Обобщенное стехиометрическое уравнение реакции удаления серы (см. (9.9)) мож-
но разделить на dhcs и использовать все стехиометрические коэффициенты в виде
соотношения o./oHC_s. На рис. 9.1 демонстрируется применение уравнения (9.12)
в целях оценки стехиометрического коэффициента для сероводорода. Наклон пря-
мой на рисунке (соотношение uhs/dhcs) близок к 18. Водород не только содер-
жится в образующемся H2S, но и расходуется в других реакциях (например, насы-
щения углеводородов). Поэтому стехиометрический коэффициент для него больше,
чем для сероводорода. Далее, ввиду значительного избытка водорода в реакционной
смеси (5000 ст. фут3/баррель) его стехиометрический коэффициент влияет на пове-
дение модели не столь значительно, как В данной имитации было принято зна-
чение = 23.
Количество прореагировавшего углеводорода
с содержанием серы, г-моль
рис. 9.1. Определение стехиометрического коэффициента для H2S
388 Часть 3. Моделирование каталитических процессов
Другие исследования, в которых фигурирует баланс водорода, показывают, что
с увеличением молярной массы соединения серы значения иНз возрастают; напри-
мер, для меркаптанов оно равно 1, для сульфидов — 2, дисульфидов — 3,' а для тио-
фенов — 4 [Lee et al., 2008]. В данном случае эксперименты проводились на тяжелой
нефти с высоким содержанием серы (3,5 %масс.) и асфальтенов (11,3 %масс.), при-
чем около 30 % всей серы содержалось в асфальтенах. Поэтому высокие наблюдав-
шиеся значения стехиометрического коэффициента для Н2 и H2S могут объясняться
типом соединений серы в сырье, а также допущением, что органические соединения
серы имеют ту же молярную массу, что и вся жидкость в целом: чем больше средняя
молярная масса, тем выше стехиометрический коэффициент.
Значения стехиометрического коэффициента были приняты постоянными. С дру-
гой стороны, при движении газожидкостной смеси по реактору средние молярные
массы веществ изменяются. Возрастают и молярные массы соединений серы. Число
ароматических и нафтеновых колец в этих соединениях тоже изменяется, что влия-
ет на расход водорода.
9.4.2. Кинетические параметры реакций гидрообессеривания
и гидродеметаллизации
Значения всех кинетических параметров определялись при трех значениях темпера-
туры (380, 400 и 420 °C), шести значениях LHSV(0,33, 0,5, 0,75, 1,0, 1,25 и 1,5 ч_|), по-
стоянной кратности водорода к сырью (5000 фут3/баррель) и постоянном начальном
давлении (6,9 МПа).
Порядок реакции концентрирования серы на каталитической поверхности (т^
оказался близким к 2, что хорошо согласуется с результатами других исследований,
в которых соединения серы представлялись единым псевдокомпонентом [Ozakeetal.,
1975; Но and Aris, 1987; Kam et al., 2008]. Кажущееся значение порядка суммарной
реакции (при степенном законе) можно объяснить широкими различиями активно-
сти соединений серы, присутствующих в нефти. Таким образом, хотя отдельные сое-
динения проявляют первый порядок реакции, группирование ряда этих соединений
в единый псевдокомпонент увеличивает кажущееся значение порядка реакции. Кон-
станта адсорбции была принята равной 40 000 см3/г-моль.
При поддержании постоянной константы скорости для всех значений LHSV
и постоянных значений температуры и давления наблюдалось значительное рас-
хождение в порядке реакции. Было установлено, что применение модели псев-
догомофазного реактора и концепции доли жидкости, помимо прочих преи-
муществ, дает значительно лучшее согласие с экспериментальными данными.
Из-за условий плохого контакта, типичного для маломасштабных эксперимен-
тов, модель реактора была заменена на гетерогенную, а концепция доли жидко-
сти — на эффективность смачивания. Было обнаружено, что изменение массовой
скорости жидкости определенным образом влияет на кажущуюся константу ско-
рости (рис. 9.2). Об этом же сообщили Сильвестр и Питайягульсарн [Sylvester and
Глава 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя» 389
Pitayagulsarn, 1974]. Решением уравнений (9.3)—(9.5) и (9.7)—(9.8) для каждого зна-
чения объемной скорости подачи сырья при постоянной температуре были полу-
чены кажущиеся значения констант скоростей. Та же процедура была выполнена
для всех значений температуры, что дало возможность определить путем линейной
регрессии значения параметров уравнения (9.26). Окончательная подгонка пара-
метров с использованием доли жидкости и эффективности смачивания оказалась
приемлемой. Можно было улучшить эффективность смачивания, используя ката-
лизатор с более широким распределением размеров частиц (например, разбавив его
более тонким инертным материалом), что свело бы к минимуму неравномерность
распределения и другие сопутствующие эффекты, но этого не было сделано. К тому
же нет формул для расчета эффективности смачивания такого разбавленного слоя.
Как следствие, отсутствуют и способы математического выражения доли жидкости
и времени пребывания в этих условиях.
Рис. 9.2. Зависимость между карр и массовой скоростью жидкости при температуре 400 °C
и давлении 6,9 МПа
При наивысшем значении LHSV и температуре 380 °C была получена приведен-
ная массовая скорость жидкости, равная 0,0822 кг/м2 • с, что соответствует эффек-
тивности смачивания 0,11. Саттерфилд [Satterfield, 1975] для близкого (0,08 кг/м2-с)
значения приведенной массовой скорости жидкости при гидроочистке тяжелого га-
зойля сообщает об эффективности смачивания между 0,12 и 0,2. На этом основании
можно заключить, что при низких значениях LHSV эффективность смачивания ка-
тализатора будет сильно влиять на работу реактора. Хотя рабочие условия и свойства
катализатора и сырья в исследовании Саттерфилда были иными, можно усмотреть
определенную схожесть в работе реакторов, а следовательно, и в значениях эффек-
тивности смачивания. Саттерфилд не сообщал о каком-либо влиянии испарения;
аналогичное допущение принимается и здесь, так как сырьем является тяжелая
нефть — а значит, можно ожидать, что в газовую фазу перейдет пренебрежимо малое
количество жидкости.
390
Часть 3. Моделирование каталитических
пР°ЦессОв
В табл. 9.2 представлены значения констант для уравнения (9.26). Была установ
лена линейная зависимость А' и В от температуры, причем значение В близко к еди
нице. Это согласуется с линейной зависимостью данных о ка№ и LHSV (см. рис. 9 2)
Корстен и Хоффманн [Korsten and Hoffmann, 1996] представили в графическом виде
зависимость между числом Рейнольдса жидкости (Re£) и эффективностью смачива
ния. При малых значениях Re£ наблюдается линейная зависимость. Значения кон-
стант А' и В определяются транспортными свойствами жидкого сырья, которые
в свою очередь зависят от температуры. Для всех значений LHSV при трех значени-
ях температуры и одном значении давления (6,9 МПа) были вычислены числа Рей-
нольдса жидкости. Далее экстраполяцией уравнения (9.26) до бесконечности были
определены псевдоистинные значения констант скоростей. По кажущимся и псевдо-
истинным значениям констант скоростей была вычислена эффективность смачива-
ния для каждого значения температуры и LHSV. Результаты этих расчетов представ-
лены на рис. 9.3 в виде зависимости между Re£ и эффективностью смачивания (f).
Видно, что с увеличением числа Рейнольдса возрастает также эффективность смачи-
вания. Эти результаты иллюстрируют рост эффективности смачивания с увеличени-
ем объемной скорости жидкости.
Таблица 9.2
Значения констант для уравнения
эффективности смачивания
Температура,°C .4x10’ В
380 2,43 1,000
400 1,60 1,048
420 1,16 1,079
Ret
Рис. 9.3. Зависимость между эффективностью смачивания
и числом Рейнольдса жидкости: (о) — 380 °C; (□) — 400 °C; (д) — 420 °C
Глава 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя»
391
Определив псевдоистинные значения констант скоростей при каждой температу-
ре, можно воспользоваться уравнением Аррениуса для нахождения значений энер-
гии активации (£л), что отражено на рис. 9.4. Найденные кажущиеся значения ЕА ре-
акции ГДО были небольшими (11,73 ккал/г-моль), что объясняется определенными
ограничениями массопереноса [Vrinat, 1983]. Предэкспоненциальный множитель
оказался равным 3,3 • 108 см3/г- с.
i/г,к -1
Рис. 9.4. График Аррениуса для реакции ГДО
В случае реакции ГДМ вначале была сделана попытка найти кажущееся значение
константы скорости через эффективность смачивания катализатора, но, в отличие
от подхода, в рамках которого концепция эффективности смачивания не использова-
лась, какого-либо влияния объемной скорости подачи сырья на карр не было обнару-
жено. Это обстоятельство было объяснено минимизацией сопротивлений внешнему
массопереносу, о чем говорилось в описании экспериментальной части. Дудукович
[Dudukovic, 1977] указал, что преобладание кинетического режима означает слабое
влияние степени смачивания внешней поверхности, что и подтвердилось на практике.
Порядки реакций удаления никеля и ванадия (ГДН и ГДВ) оказались равными со-
ответственно 1,3 и 2,3, а энергии активации — 31,2 и 37,1 ккал/г-моль. Аналогичные
значения энергий активации и порядков реакций приводятся и в литературе [Mara-
fi et al., 2010]. Предэкспоненциальные множители были равны 4,74- 108 (см3/г • с)
(см3/г-моль)0-3 и 5,27 • 1017 (см3/г- с) (см3/г-моль)‘>3. Хотя имеются сообщения о том,
что для отдельных веществ порядок реакции ГДМ близок к 1 (что справедливо для
реакции ГДО), группирование ряда металлсодержащих веществ в единый псевдо-
компонент изменяет порядок суммарной реакции в сторону более высоких значений.
9.4.3. Моделирование лабораторного реактора
На рис. 9.5 показано распределение содержания серы в жидкой фазе и у поверх-
ности катализатора по высоте реактора. Наблюдается типичное поведение, т. е.
392
Часть 3. Моделирование каталитических процессов
по мере прохождения реакционной смеси через реактор концентрация серы снижа-
ется. Распределение концентраций водорода и сероводорода представлено на рис.
9.6. Видно, что наибольшего значения концентрация H2S в жидкой фазе достига-
ет у входа в реактор, в то время как концентрация водорода здесь наинизшая. Это
объясняется следующим образом: вблизи входа в реактор на границе раздела жид-
кой и твердой фаз происходит более интенсивный массоперенос, чем на границе
раздела газообразной и жидкой фаз, так что практически весь образующийся се-
роводород остается в массе жидкости и вблизи поверхности катализатора. Вместе
с тем быстрый расход содержащегося в жидкости водорода означает снижение его
концентрации из-за массопереноса. При дальнейшем движении реакционной сме-
си перенос сероводорода из жидкой фазы в газовую и концентрирование водорода
в жидкой фазе постепенно усиливаются вследствие действия эффекта равновесия.
Допуская равновесие на последнем участке каталитического слоя, можно ожидать
постоянства концентраций этих газов, но в экспериментальном реакторе этого,
по-видимому, не происходит.
На рис. 9.7 показаны парциальные давления H,S и Н2 в зависимости от положе-
ния в слое. По мере продвижения по слою парциальное давление H2S возрастает,
Н2 — убывает. Рост парциального давления H,S обусловлен его образованием в ре-
акции ГДО, а парциальное давление водорода убывает вследствие его расходования
в реакции.
Данные рис. 9.8 демонстрируют зависимость массовой концентрации серы
на выходе из реактора от объемной скорости подачи сырья (L/AST) и температу-
ры. Наблюдается приемлемое согласие между расчетными и экспериментальными
данными.
Рис. 9.5. Распределение концентрации серы в жидкой фазе и у поверхности
катализатора при температуре 400 °C, давлении 6,9 МПа и LHSV = 1 ч*1:
(•) — экспериментальное значение
Оша 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя»
393
Рис. 9.6 Распределения концентраций H2S и Н2 в массе жидкости (—) и у поверхности
катализатора (—) при температуре 400 °C, давлении 6,9 МПа и LHSV = 1 ч-1
Рис. 9.7. Парциальные давления водорода и сероводорода в зависимости от положения
в слое при температуре 400 °C, давлении 6,9 МПа и LHSV = 1 ч~1
394
Часть 3. Моделирование каталитических процессов
Рис. 9.8. Зависимость концентрации серы на выходе из реактора от величины, обратной
LHSV, при давлении 6,9 МПа и температуре: (о) — 380 °C; (□) — 400 °C; (й) — 420 °C.
Обозначения: линии показывают расчетные значения, значки — экспериментальные
По модели были рассчитаны также концентрации серы при давлении 8,3 МПа,
отличном от того, которое фигурировало при выводе значений кинетических пара-
метров (т. е. расчет давал независимые результаты). Расчет проводился для 12 точек,
соответствующих двум значениям температуры (380 и 400 °C) и шести значениям
LHSV(0,33, 0,5, 0,75, 1, 1,25 и 1,5 ч~‘) при давлении 8,3 МПа. Как видно из рис. 9.9,
результаты также хорошо согласуются с экспериментальными данными. Расчет рас-
пределения концентрации серы по высоте реактора, проведенный для наиболее
жестких условий (т. е. при LHSV— 0,33 ч_|, давлении 8,3 МПа и температуре 420 °C),
также дал приемлемое согласие с экспериментом, что видно из рис. 9.10. Наблюда-
лось определенное повышение степени превращения серы при увеличении общего
давления, что было вполне ожидаемым.
Рис. 9.9. Зависимость концентрации серы на выходе из реактора от величины,
обратной LHSV, при давлении 8,3 МПа и температуре: (о) — 380 °C; (□) — 400 °C.
Обозначения: линии показывают расчетные значения, значки — экспериментальные
Глава 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя» 395
Рис. 9.10. Концентрации серы в жидкой фазе в зависимости от положения
в слое при температуре 420 °C, LHSV = 0,33 ч~’ и давлении 8,3 МПа:
() — экспериментальное значение
На рис. 9.11 показаны концентрации никеля и ванадия в массе жидкости и у
каталитической поверхности в зависимости от положения в слое при температу-
ре 400 °C. На рис. 9.12 изображены зависимости тех же величин от 1 / LHSV. Как
и в случае реакции ГДО, результаты расчета хорошо согласуются с эксперименталь-
ными данными.
Положение по высоте слоя, см
Рис. 9.11. Концентрации никеля и ванадия в массе жидкости (—) и у каталитиче-
ской поверхности (—) в зависимости от положения в слое при температуре 400 °C
и LHSV = 1,5 ч-1. Обозначения: линии показывают расчетные значения,
значки — экспериментальные
396
Часть 3. Моделирование каталитических процессов
1 / LHSV, ч-’
Рис. 9.12. Концентрации никеля и ванадия в зависимости от 1 /LHSV при температуре:
(•) —380 °C; (о) —420 °C
9.4.4. Замечания о допущениях в модели
При моделировании работы лабораторного реактора для ГДО, ГДН и ГДВ был сделан
ряд предположений. Хотя экспериментальные и расчетные результаты хорошо согла-
суются, налицо определенные отклонения: во-первых, система уравнений для оцен-
ки транспортных и термодинамических свойств не позволяет рассчитывать их точ-
ные значения; во-вторых, сделанные предположения, пусть и вполне реалистичные,
могут вызывать ошибки в конечных результатах. Гидрокрекинг не учитывается до-
пущениями в модели, но в наиболее жестких условиях он все же может иметь место.
Далее, считается, что такие кинетические параметры, как порядок реакций с участи-
ем соединений, содержащих серу и металлы, не зависят от изменений концентрации
(а следовательно, и средней молярной массы) этих гетероатомных соединений в ре-
акторе, однако это не всегда верно [Trytten et al., 1990; Ancheyta et al., 2002].
В литературных источниках нет сведений, указывающих на значения эффек-
тивности смачивания катализатора при очень малых числах Рейнольдса, поэтому
Глава 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя»
397
неопределенность в этой области остается. Как можно видеть, есть ряд допущений,
которые создают расхождение между экспериментальными и теоретическими ре-
зультатами. В ряде работ, где описывалась модель реактора с орошаемым слоем для
гидроочистки нефтяных фракций, было получено хорошее согласие. Но воссоздать
описанную там модель невозможно, так как значения параметров не публиковались.
Казалось бы, можно воспроизвести экспериментальные данные подбором неиз-
вестных параметров, однако последние не будут иметь никакого физического смыс-
ла. Каждый параметр, вводимый в простую модель реактора, должен вносить свой
вклад в интерпретацию таких данных, как кинетические и диффузионные ограниче-
ния, и быть действительно необходимым. В отсутствие экспериментальных данных
критерием оценки значений этих параметров должна выступать область применимо-
сти; в противном случае группирование нескольких параметров в единую константу
не может считаться реалистичным подходом и, что еще хуже, распространение мо-
дели на другие масштабы или рабочие условия может оказаться попросту опасным.
Иными словами, оптимизация значений параметров модели глобальной минимиза-
цией расхождений не гарантирует реалистичность подхода, если при разработке мо-
дели сделано достаточно большое число допущений и справедливость внесения по-
правок в значения оцениваемых параметров не проверяется путем расчета по модели.
В данной модели кажущиеся порядки реакций, константы скоростей и некоторые
гидродинамические параметры являются подгоняемыми, так что они действитель-
ны лишь для изучаемой системы; тем не менее процедуру можно легко приспособить
для других условий.
Вероятно, если разумно использовать формулы для оценки термодинамических
и транспортных свойств в гетерогенной модели реактора, можно получить пред-
ставление о свойствах всякой реакционной системы. Конечно, наилучший способ
проверки зависимостей — получение данных об этих свойствах в условиях реак-
ций. Впрочем, из-за отсутствия надлежащего оборудования и разнообразия затра-
гиваемых свойств такое не всегда осуществимо. Вместе с тем желательно, чтобы
предстоящие исследования опирались на определенные экспериментальные све-
дения о важнейших свойствах. Это позволит подобрать наилучшие, т. е. наиболее
надежные формулы, позволяющие воспроизводить экспериментальные данные.
Тогда единственными подгоняемыми параметрами модели будут те, что касают-
ся кинетики реакций. Считается, что группирование соединений серы в единый
псевдокомпонент дает более точное описание кинетики. Тем не менее, ввиду от-
сутствия адекватных формул для оценки физических свойств высокомолекуляр-
ных фракций, должна быть проведена дополнительная экспериментальная ра-
бота. Важно также учесть частичное испарение сырья (главным образом легких
фракций), даже если уравнения для расчета фазового равновесия зависят от типа
сырья. В настоящее время попытки смоделировать поведение таких сложных си-
стем, как реакторы гидроочистки тяжелой нефти, ограничены лишь частны-
ми случаями, характеризуемыми ограниченными областями изменения пара-
метров, конфигурации процесса, типа катализаторов и сырья. Универсальных
моделей нет.
398 Часть 3. Моделирование каталитических процессов
ОБОЗНАЧЕНИЯ
ГРЕЧЕСКИЕ
«г Параметр уравнения массопереноса из газовой фазы в жидкую, см Параметр уравнения массопереноса из жидкой фазы в частицы катали- затора, см
I API е Плотность, ° API Доля пустот в слое, см3/см3
ео Е£ Л X. Средняя пустотность слоя, см3/см3 Доля жидкости, см3/см3 Глубина (коэффициент эффективности) реакции Коэффициент растворимости z-ro соединения, н. л/кг - МПа
р. Pi о Динамическая вязкость жидкости, мПа • с Плотность катализатора, г/см3 Плотность жидкости, г/см3 Коэффициент поверхностного натяжения
1) Стехиометрические коэффициенты
ЛАТИНСКИЕ
А, В Параметры формулы для эффективности смачивания
ai as с, d cGi ск Си cNi cst Удельная поверхность границы раздела газовой и жидкой фаз, см2/см3 Удельная поверхность границы раздела жидкой и твердой фаз, см2/см3 Параметры формулы Гласё Молярная концентрация i-ro компонента в массе газа, моль/см3 Общая молярная концентрация в к-й фазе, моль/см3 Молярная концентрация z-ro компонента в массе жидкости, моль/см3 Молярная концентрация никеля, моль/см3 Молярная концентрация z-ro компонента у каталитической поверхно- сти, моль/см3
Су Du Молярная концентрация ванадия, моль/см3 Коэффициент молекулярной диффузии i-ro компонента в жидкости, см2/с
d р d, Z Эквивалентный диаметр частиц, см Внутренний диаметр реактора, см Мольная доля i-ro соединения, моль/моль
Глава 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя»
399
4 Коэффициент эффективности смачивания, безразмерная величина
g Ускорение свободного падения, см2/с
Ga Число Галилея для жидкой фазы, d3nLg / ц2
Gl Приведенная массовая скорость, кг/м2 • с
Я, Константа закона Генри для /-го соединения, МПа • см’/моль
карр Кажущееся значение константы скорости реакции ГДО (см3/г с)
(см3/моль)0'5
^HjS Константа равновесия адсорбции (см3/моль)
kin Истинное значение константы скорости (см3/г- с) (см3/моль)0 5
kL Коэффициент массопереноса между газом и жидкостью на стороне жид-
кости, см3/см2 • с
Общий коэффициент массопереноса между газом и жидкостью, см3/см2 • с
ks Коэффициент массопереноса из жидкой фазы в твердую, см3/см2 • с
^гдв Константа скорости реакции ГДВ (см3/г • с) (см’/моль)”]!
£Гдн Константа скорости реакции ГДН (см3/г • с) (см3/моль)"21
LHSV Объемная скорость подачи жидкого сырья, см3/см3 с
т Масса жидкости, г
т2 Порядок реакции соответственно для серы и водорода
Средняя молярная масса, г/г-моль
«р п2 Порядок реакции соответственно для никеля и ванадия
и. Число молей /-го соединения, моль
и£ Параметр уравнения массопереноса из газовой фазы в жидкую
ns Параметр уравнения массопереноса из жидкой фазы в частицы катализатора
Р Общее давление, МПа
Ра Парциальное давление /-го компонента в массе газа, МПа
г Скорость реакции, моль/г- с
Rez Число Рейнольдса для жидкой фазы, uLpLdp/pL
S, Образование или расходование /-го соединения
/ Время, ч
Т Температура
U Скорость молекул
uL Приведенная скорость жидкости, см3/см2 с
V. Молярный объем /-го соединения, см3/моль
х Степень превращения или массовая доля в жидкости /-го компонента, г/г
Z Координата по высоте слоя, см
400
Часть 3. Моделирование каталитических процессов
НИЖНИЕ ИНДЕКСЫ
В Каталитический слой
G Газ
Н2 Водород
H2S Сероводород
НС Углеводороды, не содержащие серу
НС—S Углеводороды, содержащие серу
i i-й компонент
к к-я фаза
L Жидкость
Ni Никель
S; S Сера; поверхность катализатора
V Ванадий
ГДВ Гидродеванадизация
ГДН Гидроденикелизация
ГДО Гидрообессеривание
ЛИТЕРАТУРА
Ancheyta, J., Angeles, M.J., Macias, M.J., Marroquin, G., Morales, R. 2002. Changes in apparent
reaction order and activation energy in the hydrodesulfurization of real feed- stocks. Energy Fuels
16(0:189-193.
Bennison, TG. 1998. Prediction of heavy oil viscosity IBC UK Conf. LTD. In: Heavy Oil Field
Development Conference, London, England, December.
Benyahia, E, O’Neill, K.E. 2005. Enhanced voidage correlations for packed beds of various particle
shapes and sizes. Particulate Sci. Technol. 23:169—177.
Bondi, A. 1971. Handling kinetics from trickle-phase reactors. Chem. Technol. 1:185-188. Cai, H.-Y,
Shaw; J.M., Chung, K.H. 2001. Hydrogen solubility measurements in heavy oil and bitumen cuts. Fuel
80:1055-1063.
Carman, PC. 1937. Flow through granular beds. Trans. Inst. Chem. Eng. 15:150-166.
Carroll, J.J., Mather, A.E. 1995. A generalized correlation for the Peng-Robinson interaction coef- ficients
for paraffin-hydrogen sulfide binary systems. Fluid Phase Equilib. 105(2):221—228. Chowdhury, R., Pedemera, E.,
Reimert, R. 2002. Trickle-bed reactor model for desulfurization and dearomatization of diesel. AIChE J.
48(1): 126-135.
Chu, C.E, Ng, K.M. 1989. Flow in packed tubes with a small tube to particle diameter ratio. AIChEJ.
35(1): 148—158.
Dixon, A.G. 1988. Correlations for wall and particle shape effects on fixed bed bulk voidage. Can. J. Chem.
Eng. 66:705—708.
Dudukovic, M.P 1977. Catalyst effectiveness factor and contacting efficiency in trickle-bed reactors.
AIChEJ. 23(6):940—944.
Foumeny, E.A., Moallemi, H.A., McGreavy, C., Castro, J.A.A. 1991. Elucidation of mean voidage in
packed beds. Can. J. Chem. Eng. 69:1010—1015.
Глава 9. Моделирование лабораторного реактора для гидроочистки тяжелой нефти «Майя» 401
Fu, M.S., Tan, C.S. 1996. Liquid holdup and axial-dispersion in trickle-bed reactors. Chem. Eng. Sci.
51:5357-5361.
Glas0, 0. 1980. Generalized pressure-volume-temperature correlations. ./. Petrol. Technol.
32(5):785—795.
Goto, S., Smith, J.M. 1975. Trickle-bed reactor performance. Part I. Holdup and mass transfer effects,
AIChEJ. 21(4):706—713.
Gunjal, PR., Ranade, W 2007. Modeling of laboratory and commercial scale hydro-processing reactors
using CFD. Chem. Eng. Sci. 62(18—20):5512—5526.
Haughey, D.P, Beveridge, G.S. 1969. Structural properties of packed beds: a review. Can. J. Chem. Eng.
47(2):130—140,
Henry, H.C., Gilbert, J.B. 1973. Scale up of pilot plant data for catalytic hydroprocessing. Ind. Eng. Chem.
Proc. Des. Dev. 12(3):328—334.
Ho,TC., Aris, R. 1987. Onapparent second-order kinetics. AIChEJ. 33(6): 1050—1051.
Hofmann, H. 1977. Hydrodynamics, transport phenomena, and mathematical models in trickle-bed
reactors. Ini. Chem. Eng. 17(1): 19—28.
Kam, E.K.T, Al-Bazzaz, H,, Al-Fadhli, J. 2008. Simple procedure for interpreting hydrotreating kinetic
data. Ind. Eng. Chem. Res. 47:8594-8601.
Korsten, H., Hoffmann, U. 1996. Three-phase reactor model for hydrotreating in pilot trickle-bed
reactors. AIChEJ. 42(5): 1350— 1360.
Lal, D., Otto, ED., Mather, A.E. 1999. Solubility of hydrogen in athabasca bitumen. Fuel
78(12):1437—1441.
Lange, R., Schubert, M., Bauer, T 2005. Liquid holdup in trickle-bed reactors at very low liquid
Reynolds number. Ind. Eng. Chem. Res. 44:6504—6508.
Lee, C.K., Magalhaes, C., da Silva, L.E., Osowki, C.A. 2008. Study compares methods that measure
hydrogen use in diesel hydrotreaters. Oil Gas J. 13:58—63.
Levespiel, O. 1999. Chemical Reaction Engineering, 3rd edn., John Wiley & Sons, New York.
Ma, X., Sakanishi, K., Mochida, I. 1994. Hydrodesulfurization reactivities of various sulfur compounds
in diesel fuel. Ind. Eng. Chem. Res. 33:218—222.
Marafi, A., Stanislaus, A., Furimsky, E. 2010. Kinetics and modeling of petroleum residues
hydroprocessing. Calal. Rev. Sci. Eng. 52:204—324.
Mederos, ES. 2010. Dynamic modeling and simulation of a three-phase reactor for hydrotreating of oil
fractions, PhD thesis, IMP Postgraduate School, Mexico.
Mederos, E, Elizalde, 1., Ancheyta, J. 2009. Steady-state and dynamic reactor models for
hydrotreatment of oil fractions: a review Catal. Rev. Sci. Eng. 51:485—607.
Mederos, ES., Rodriguez, M.A., Ancheyta, J., Arce, E. 2006. Dynamic modeling and simulation of
catalytic hydrotreating reactors. Energy Fuels 20(3):936—945.
Paraskos, J.A., Frayer, J.A., Shah,YT 1975. Effect of holdup incomplete catalyst wetting and backmixing
during hydroprocessing in trickle bed reactors. Ind. Eng. Chem. Proc. Des. Dev. 14(3):315-322.
Peng, D.Y, Robinson, D.B. 1976. A new two-constant equation of state. Ind. Eng. Chem. Fundam.
15(1):59—64.
Perego, C., Peratello, S. 1999. Experimental methods in catalytic kinetics. Catal. Today
52(2-3):133—145.
Reid, R.C., Prausnitz, J.M., Foiling, B.E. 1987. The Properties of Gases and Liquids, McGraw Hill, New
York.
Riazi, M.R. 2005. Characterization and Properties of Petroleum Fractions, ASTM International Standards
Worldwide, Philadelphia, PA.
Riazi, M.R., Daubert, ТЕ. 1987. Characterization parameters for petroleum fractions. Ind. Eng. Chem.
Res. 26:755-759.
Rodriguez, M,A., Ancheyta, J. 2004. Modeling of hydrodesulfurization (HDS), hydrodenitrogenation
(HDN), and the hydrogenation of aromatics (HDA) in a vacuum gas oil hydrotreater. Energy Fuels
402
Часть 3. Моделирование каталитических процессов
Ross, L.D. 1965. Performance of trickle bed reactors. Chem. Eng. Ptvg. 61(1O):77—82.
Satterfield, C.N. 1975. Trickle bed reactors. AIChEJ. 21 (2):2O9—228.
Satterfield, C.N., Pelossof, A.A., Sherwood, TK. 1969. Mass transfer limitation in a tricklp bed reactor.
AIChEJ. 15:226-234.
Schwartz, J.G., Roberts, G.W 1973. An evaluation of models for liquid backmixing in trickle-bed reactors.
Ind. Eng. Chem. Proc. Des. Dev. 12(3):262-271.
Sie, S.T 1996. Miniaturization of hydroprocessing catalyst testing systems: theory and practice. AIChEJ.
42(12):3498—3507.
Sylvester, N.D., Pitayagulsam, P 1974. Effect of catalyst wetting on conversion in a trickle-bed reactor
Can. J. Chem. Eng. 52:539-540.
Tarhan, M.O. 1983. Catalytic Reactor Design, McGraw-Hill, New York.
Trytten, L.C., Gray, M.R., Sanford, E.C. 1990. Hydroprocessing of narrow-boiling gas oil fractions:
dependence of reaction kinetics on molecular weight. Ind. Eng. Chem. Res. 29:725-730.
Туп, M.T, Calus, WE 1979. Estimating liquid molal volume. Processing 21 (4).16—17.
\hn Krevelen, D.W, Krekels, J.TC. 1948. Rate of dissolution of solid substances part i. rate of mass
transfer in granular beds (physical dissolution). Rec. Trav. Chim. 67:512-520.
Vrinat, M.L. 1983. The kinetics of the hydrodesulfurization process: a review. App. Catal. 6(2): 137-158.
Глава 10. Моделирование
реакторов с кипящим или
суспензионным слоем
В начале главы дается обзор основных характеристик реакторов с кипящим сло-
ем (РКС). Разъясняются такие ключевые аспекты применения этих реакторов для
гидрокрекинга тяжелой нефти, как образование осадка, износ катализатора и поте-
ря его активности. Математическое представление систем с кипящим слоем орга-
низуется в модели гидродинамики, масштабирования и реактора. Кратко рассма-
тривается моделирование реактора для процессов, протекающих в фазе суспензии
(суспензионного реактора). Моделируется кинетика гидроочистки и гидрокрекинга
атмосферного остатка тяжелой нефти на лабораторной установке с проточным реак-
тором смешения (CSTR), который имитирует реактор с кипящим слоем.
10.1. ВВЕДЕНИЕ
Реакторы с кипящим (взвешенным) слоем катализатора — это многофазные ката-
литические реакторы принципиально новой конструкции, занявшие важное ме-
сто в таких промышленных процессах, как гидрокрекинг и гидроочистка нефтяных
остатков, крекинг углеводородов для получения олефинов и ароматических соеди-
нений путем частичного окисления, производство адипонитрила из адипиновой
кислоты и аммиака, гидрирование каменноугольной смолы (каталитическое ожи-
жение угля), каталитическое гидрирование ненасыщенных жирных масел, метани-
рование в жидкой фазе (превращение окиси углерода в метан), переработка сточных
вод биологической оксигенацией, производство бисульфита кальция для целлюлоз-
но-бумажной промышленности, синтез Фишера—Тропша [Chaudhari et al., 1986;
Martinez et al., 2010].
Значимость РКС для нефтепереработки в последние годы возросла из-за резкого
увеличения объемов поставок тяжелого сырья на НПЗ, а следовательно, и объемов ги-
дрокрекинга. Вследствие высокого содержания серы, азота, металлов (никель и вана-
дий) и асфальтенов, отрицательно влияющих на активность и стабильность катализа-
торов, тяжелое сырье трудно поддается переработке традиционными технологиями.
404
Часть 3. Моделирование каталитических процессов
Новые процессы направлены на превращение тяжелых, высококипящих компо-
нентов в более легкие и низкокипящие продукты через разрыв углеродных связей
с одновременным или последующим гидрированием. Существует ряд промышленно
применяемых или находящихся на стадии разработки технологий гидроочистки и ги-
дрокрекинга нефтяных остатков [Ancheyta et al., 2005а]. В процессах такого назначе-
ния применяются реакторы четырех типов: с неподвижным слоем (РНС), с кипящим
слоем (РКС), с движущимся слоем (РДС) и суспензионным слоем (РСС). Особенно-
сти, преимущества и недостатки реакторов каждого из этих типов рассмотрены в раз-
деле 7.3 (см. главу 7), а также описаны в литературе [Ancheyta and Speight, 2007]. При
переработке тяжелой нефти в реакторах всех типов присутствуют три фазы. Реакторы
с неподвижным слоем обычно работают в режиме нисходящего потока, когда жид-
кость и газ (в основном состоящий из водорода) опускаются вниз через каталитиче-
ский слой (твердая фаза). Основное преимущество реакторов этого типа заключа-
ется в простоте их эксплуатации в промышленных масштабах. Главный недостаток
РНС — накопление металлов (никеля и ванадия) и кокса в устьях пор, перекрыва-
ющее доступ реагентов к их внутренней поверхности. В РКС этот недостаток устра-
няется за счет приведения катализатора во взвешенное состояние. Катализатор до-
бавляется и удаляется из реактора с такой периодичностью, чтобы поддерживать его
среднюю активность на постоянном уровне. В целом технология кипящего слоя луч-
ше остальных подходит для проведения реакций с большим тепловыделением и для
переработки сырья, которое из-за высокого содержания примесей трудно перераба-
тывать в реакторах с неподвижным (стационарным) слоем [Ancheyta et al., 2005b].
Реакторы гидрокрекинга в фазе суспензии предназначены для превращения тя-
желых вакуумных остатков, но они еще не в полной мере применяются в промыш-
ленных масштабах. Сырьем реакторов такого типа служит смесь нефтяного остатка
и твердого носителя (добавки). Назначение добавки — обеспечить поверхность для
осаждения превращенных при гидрокрекинге асфальтенов и металлов. Суспензион-
ные реакторы работают при высоких температурах и давлениях, а степень превраще-
ния остатка в них может превышать 90 % [Kressmann et al., 1998]. К сожалению, эти
реакторы дают низкокачественные дистиллятные и вакуумные продукты с дефици-
том водорода, которые вследствие высокого содержания серы и металлов не подхо-
дят для использования в качестве топлива, если не смешать их, например, с углем или
тяжелым топливным мазутом [Ancheyta and Speight, 2007].
Со временем было предложено множество способов облагораживания остатков
в РКС. Реализована схема с последовательным соединением РКС и промежуточ-
ным разделением газа и жидкости с целью концентрировать жидкость, поступаю-
щую в следующий реактор [Coylar and MacArthur, 1996, 2001; Baldassari et al., 2002].
Иногда сырье, подаваемое во вторую ступень, смешивают с ароматическим раство-
рителем и/или с остатком [Baldassari et al., 2002]. В других случаях непревращенный
остаток, возвращаемый в первый реактор, смешивают с газойлем, чтобы повысить
степень превращения [McDaniel et al., 1989]. При переработке тяжелого сырья при-
меняют и другие подходы: например, предварительно подготавливают сырье, посту-
пающее в РКС, путем частичной деметаллизации с использованием каталитической
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем 405
добавки (гетерополикислота с содержанием молибдена и солей металлов переход-
ной группы, растворенных в кислородсодержащем полярном растворителе) [Johnson
and Brown, 1994], промежуточной экстракции [Kolstad et al., 1993] и деасфальтиза-
ции [Coylar, 2007; Coylar et al., 2007; Lenglet, 2008] растворителями. При нормаль-
ной работе РКС реакционная зона содержит значительные количества паровых и га-
зовых пузырей, состоящих из избыточного водорода, легких углеводородных газов,
сероводорода, аммиака, водяного пара и т. д. Чрезмерный объем пузырей (высокая
доля газа) может ухудшить эффективность процесса и снизить выход продуктов. Пу-
зыри занимают значительную часть общего объема реактора, которую эффективнее
было бы использовать для облагораживания сырья. Высказывалась мнение, что пе-
ренос точки подачи газовой фазы на уровень выше распределительной тарелки мо-
жет уменьшить долю газа и существенно улучшить результативность переработки
[Buttke et al., 1994]. Разработаны также оригинальные устройства для отвода паров
из поддона и линии рециркуляции [Chan and Colvert, 1989; Buttke and Frey, 1990]. По-
казано, что применение катализатора определенного состава на носителе (подлож-
ке) с частицами несферической формы и неоднородных размеров повышает степень
превращения тяжелого сырья [Harle et al., 2000; Roy-Auberger et al., 2007]. Послед-
нее новшество на сегодняшний день — сочетание пористого катализатора на под-
ложке с коллоидным. Последний заполняет свободные зоны, образующиеся между
частицами катализатора на носителе, что улучшает работу реактора гидропереработ-
ки в кипящем слое [Lott and Lee, 2008].
Хотя технология РКС применяется во многих процессах и в последние годы ши-
роко используется при гидрокрекинге тяжелых остатков, сведения о подробностях
моделирования и других аспектах таких реакторов скудны и не раскрываются право-
обладателями. В настоящей главе сделана попытка как можно более полно и исчер-
пывающе осветить аспекты процесса и моделирования РКС на основании сведений,
почерпнутых из открытых литературных источников (в том числе патентной лите-
ратуры, научных трудов и материалов конференций). Рассматриваемые модели ка-
саются РКС в различных приложениях, но наибольший акцент делается на анализе
моделей, применяемых к гидрокрекингу нефтяных остатков. Следует отметить скуд-
ность публикуемых в литературе сведений, что объясняется главным образом слож-
ностью систем РКС. В последних подвижными являются все три фазы (газовая, жид-
кая и твердая), что сильно усложняет их моделирование и имитацию по сравнению
с другими системами.
10.2. РЕАКТОРЫ С КИПЯЩИМ СЛОЕМ
В РКС действует трехфазная реакционная система, газовая фаза которой в случае
гидрокрекинга тяжелых нефтяных фракций состоит из водорода и частично испа-
ренных углеводородов, жидкая — из неиспаренной тяжелой части углеводородного
сырья, а твердая — из особого катализатора, физические свойства которого способ-
ствуют псевдоожижению в реакторе. Схематическое изображение РКС представле-
но на рис. 10.1.
406
Часть 3. Моделирование каталитических процессе
Линия добавления
катализатора
б)
Поток ——
Линия добавления
катализатора *
Штуцер кармана
для термопары
Рециркуля-
ционный
поддон
Уровень
разрыхленного
катализатора
Карман для радио-
активного
источника датчиков
плотности
Датчики
плотности
Уровень
катализатора
нормальной
плотности
Линия подачи
водорода
и сырья
Линия
отбора Линия возврата
катализа- непревращенного
Термопары
корпуса реактора
Распределитель-
ная тарелка
Линия подачи
водорода и
EZj J"a3
j Смесь газа
и жидкости
ГТИ
Катализатор
ющий насос
тора сырья (рециркулята)
Рис. 10.1. Реакторы гидрокрекинга в кипящем слое: а — реактор Н-ОН; б — реактор
LC-Fining [Ancheyta, J. and Speight, J.G., Hydroprocessing of Heavy Oils and Residua, CRC
Press, Taylor & Francis Group, Boca Raton, FL, 2007]
Катализатором в РКС обычно служит экструдат диаметром 0,8 мм, активными ме-
таллами в котором являются молибден с никелем или кобальтом. Катализатор под-
держивается во взвешенном состоянии восходящими потоками жидкости (сырье
и жидкий рециркулят) и газа (свежий водород и циркулирующий газ), которые по-
ступают в реактор и распределяются по его сечению распределительной тарелкой.
Высота взвешенной («разрыхленной») части каталитического слоя контролирует-
ся преимущественно скоростью рециркуляции жидкости. Поток жидкости, отби-
раемой из парожидкостного сепаратора внутри реактора, регулируют изменением
оборотов разрыхляющего насоса (центробежный насос герметизированного испол-
нения). Взвешенное состояние катализатора снижает потери давления в реакторе
и обеспечивает работу слоя в практически изотермическом режиме смешения. Таким
образом, можно считать, что РКС ведет себя как реактор с непрерывным перемеши-
ванием (CSTR). Уровень активности катализатора регулируется добавлением свеже-
го и отбором отработанного катализатора. Разделение жидкости и газа для подачи
в разрыхляющий насос можно осуществлять и вне реактора. При такой схеме рабо-
чие условия в сепараторе должны быть такими же, как и в реакторе. Кроме того, при
внешнем разделении высвобождается объем, занимаемый рециркуляционным под-
доном, и в реакторы можно загрузить больше катализатора.
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
407
Ключевая особенность РКС — возможность периодического добавления не-
больших количеств катализатора, что позволяет поддерживать качество продуктов
на постоянном уровне. Поэтому длительность цикла РКС не зависит от активно-
сти катализатора и определяется контролем образования осадка (шлама), ограни-
чиваемого обычно величиной в 0,8-1,0 %масс. [Kressmann et al., 1998; Ancheyta and
Speight, 2007].
10.2.1. Элементы реактора с кипящим слоем
10.2.1.1. Рециркуляционный поддон
Рециркуляционный поддон рассчитан на максимально возможное разделение жид-
кости и газа, с тем чтобы свести к минимуму количество газа, возвращаемого в реак-
тор [Сох, 1990]. Он состоит из желоба с внутренними спиралевидными элементами,
которые сообщают рабочей среде касательную скорость. Газожидкостная смесь про-
ходит через патрубки разделительного аппарата, нижние концы которых находятся
под уровнем жидкости в реакторе. Газовая часть, пройдя поддон, поднимается в верх
реактора. Часть жидкости стекает по стояку, поступает в рециркуляционный насос
и вместе со свежим жидким сырьем и водородом проходит через распределитель-
ную систему, возвращаясь в зону реакции и поддерживая равномерность восходяще-
го потока реакционной среды через разрыхленный каталитический слой. Остальная
часть жидкости отделяется в циклонах от газа, образуя жидкий продукт [Chan and
Colvert, 1989].
Если уровень жидкости ниже верхнего края рециркуляционного поддона, цирку-
ляция сырья через поддон, стояк и колпачки распределителя в реакционную зону
не происходит. Это может вызвать застой жидкости и возникновение зон перегрева,
что делает работу реактора опасной. Риски потери циркуляции жидкости, поврежде-
ния оборудования и катализатора, вынужденной остановки реактора гидроочистки
и последующего простоя установок может предотвратить рециркуляционный поддон
плавающей конструкции [Сох, 1990].
10.2.1.2. Система распределения потока
Система распределения потока состоит из 1) камеры, образуемой днищем и стенка-
ми реактора в нижней его части; 2) распределительной тарелки, расположенной под
слоем катализатора; 3) трубы для подачи водорода и углеводородной смеси в каме-
ру; 4) смесительной юбки, жестко соединенной с внутренним концом трубы. Юбка
имеет по меньшей мере две разнесенные друг от друга отбойные перегородки, Вну-
тренняя перегородка является сплошной, а другие имеют центральный проход, рас-
положенный над сплошной перегородкой. Юбка смешивает поступающие в камеру
водород и углеводородное сырье и в сочетании с распределительной тарелкой обе-
спечивает достаточно равномерное распределение потока газожидкостной смеси
по сечению слоя катализатора. Неправильное распределение потоков на входе в ре-
актор может вызвать эксплуатационные проблемы, в том числе коксообразование
408
Часть 3. Моделирование каталитических процессов
в камере, неравномерность распределения потока по сечению слоя, местные просе-
дания слоя или образование в нем кокса. Эти проблемы сокращают срок службы ка-
тализатора, вызывают частые остановки реактора и укорачивают периоды' его нор-
мальной работы [Li and Eccles, 1987].
10.2.1.3. Распределительная тарелка
Распределительная тарелка равномерно распределяет поток по сечению каталитическо-
го слоя. Она содержит множество отверстий с патрубками и колпачками [Татра, 1992],
аналогичных тем, которые имеются на колпачковых тарелках перегонных колонн. Не-
обходимое число колпачков определяется расчетом или оценивается исходя из диаметра
колпачков, расстояния между ними, диаметра тарелки и диаметра стояка (рециркуля-
ционной линии) [Sayles, 2006]. Такая конструкция создает гидравлическое сопротив-
ление, вследствие чего на тарелке возникает перепад давлений около 0,02—0,035 МПа.
Такой перепад теоретически достаточен для равномерного прохождения газожид-
костной смеси через все отверстия тарелки [Татра, 1992]. Еще более равномерное
распределение газа и жидкости по сечению слоя обеспечивает двухъярусная тарелка.
Нижний ярус тарелки содержит только патрубки, верхний ярус — патрубки с колпач-
ками [Milligan, 1985]. Однородный поток смеси газа и жидкости достигается также
сочетанием колпачковой конструкции с диафрагменным смесителем [Colvert, 1989].
10.2.1.4. Стояк
Стояк представляет собой длинную возвратную линию постоянного диаметра, кото-
рая расположена внутри реактора и сообщается с разрыхляющим насосом. Если уро-
вень в реакторе падает ниже верхнего конца стояка, жидкость в насос не поступает,
что вызывает вынужденную остановку работы реактора. Для устранения этого недо-
статка можно сделать отверстия вдоль верхней части стояка [Hookham, 1993].
10.2.1.5. Разрыхляющий насос
Разрыхляющий, или рециркуляционный, насос получает через стояк жидкость из ре-
циркуляционного поддона и через распределительную тарелку подает ее в катали-
тический слой. Скорость циркуляции должна быть достаточна для подъема частиц
катализатора и удержания верхней части слоя в стационарном разрыхленном и взве-
шенном состоянии. Насос может быть заключен в корпус реактора (встроенный на-
сос). При таком расположении насос работает в условиях высоких давлений и темпе-
ратур, так что необходимо предусмотреть возможность управлять насосом снаружи
[Lyzinski et al., 1993].
Обычно применяются центробежные насосы с короткозамкнутым асинхронным
двигателем герметизированного исполнения [Kressmann et al., 1998].
К достоинствам таких насосов относятся отсутствие сальников и систем промыв-
ки уплотнений, минимальная потребность в обслуживании, повышенная надеж-
ность, минимальное число узлов, максимальная безопасность и компактная кон-
струкция [Chacchia et al., 2004].
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
409
При внесении некоторых модификаций в систему рециркуляции насосы гермети-
зированного исполнения могут перекачивать жидкости с температурой до 538 °C, вы-
соколетучие жидкости и жидкости с содержанием твердых частиц [Karassik et al., 2001 ].
Крайне важно поддерживать разрыхляющий насос в рабочем состоянии. Это позво-
лит избежать превращения кипящего слоя в неподвижный, что чревато возникнове-
нием зон перегрева и нештатной работой реактора, а в худшем случае — перегревом
металла выше допустимых температур [Mounce and Rubin, 1971].
10.2.2. Достоинства и недостатки реакторов с кипящим слоем
Ниже перечислены основные достоинства и недостатки реакторов гидропереработки
с кипящим слоем [Ancheyta and Speight, 2007].
Достоинства
• Очень гибкая работа (в режимах низкой и высокой степени превращения).
• Возможность периодического отбора отработанного катализатора и добав-
ления свежего без прерывания работы реактора, что позволяет поддерживать
среднюю активность катализатора на требуемом постоянном уровне. Это свой-
ство РКС увеличивает коэффициент использования технологической установ-
ки и снижает затраты на обслуживание, связанные с загрузкой и выгрузкой ка-
тализатора, характерные для РНС.
• Конструкция реактора (разрыхление слоя насосом с увеличением объема
на 30—50 %) обеспечивает между частицами свободное пространство, достаточ-
ное для прохождения механических примесей без их накопления, закупорки
слоя и роста потерь давления в нем.
• Вследствие меньшего диаметра частиц катализатора в значительной мере устра-
няются диффузионные ограничения, что повышает скорость реакций и улуч-
шает эффективность катализатора. Кроме того, устья пор частиц меньших раз-
меров менее склонны к закупорке отложениями металлов.
• Высокие скорости теплопереноса уменьшают перегрев слоя и замедляют
закоксовывание.
• Средняя активность рабочего заряда катализатора устанавливается на постоян-
ном уровне, что означает неизменное качество продуктов в течение всего цик-
ла. В этом заключается главное отличие РКС от РНС, рабочую температуру
в которых приходится периодически повышать, чтобы увеличить избиратель-
ность катализатора и компенсировать падение его активности.
• Реакторы с кипящим слоем работают в режиме, близком к изотермическому
режиму реактора идеального смешения. Теплота реакций рассеивается лучше,
что позволяет РКС работать при более высоких температурах, т. е. с более высо-
кой степенью превращения сырья. Перепад температур в промышленных РКС
не превышает 5 °C, что делает ненужным устройство охлаждения внутри реак-
тора [Mounce and Rubin, 1971]. Теплоту для обеспечения необходимой темпера-
туры реакций в зоне гидрокрекинга может дать смесь тяжелого рециклового га-
зойля с 95 %об. ароматических углеводородов [Sayles et al., 1990].
410
Часть 3. Моделирование каталитических процессов
Недостатки
• Отсутствие режима вытеснения, который кинетически выгоднее режима сме-
шения. Этот недостаток можно частично устранить последовательным соеди-
нением нескольких РКС.
• Износ и эрозия катализатора. Отсюда следует, что катализатор должен быть ме-
ханически прочным и стойким к износу.
♦ Из-за меньшего размера частиц и меньшей доли катализатора (т. е. большей
доли пустот) объем каталитического слоя должен быть выше, чем у реакторов
с неподвижным или движущимся слоем.
• Значительный расход катализатора.
• В реакторе могут развиваться застойные зоны, что ведет к нестабильным и неш-
татным условиям. Для предотвращения роста таких зон необходимо тщатель-
ное наблюдение за температурным полем в реакторе.
• Повышенная интенсивность образования осадка.
• Для масштабирования и расчета РКС по сравнению с реакторами других типов
требуется больше данных и сведений (например, о составе сырья, свойствах ка-
тализатора, аспектах катализа и химической кинетики, гидродинамике, явле-
ниях тепло- и массопереноса в масштабах отдельных частиц и всего слоя и т. д.).
10.2.3 . Рабочий заряд катализатора
Ежесуточные затраты НПЗ на катализатор для РКС выражаются тысячами долла-
ров. Выбор Оптимальная скорость замены катализатора выбирается на основе ком-
промисса между качеством и выходом продуктов и эксплуатационными качествами
слоя, с одной стороны, и затратами — с другой. Ключевой момент при оптимизации
скорости добавления свежего катализатора — расчет равновесных свойств слоя. Од-
ним из основных условий, определяющих кинетику реакций, является количество
катализатора в слое. Равновесный заряд катализатора в кипящем слое не обязатель-
но занимает один и тот же объем. Объем слоя контролируют поддержанием посто-
янного уровня. Но с изменением рабочих условий изменяется и степень разрыхле-
ния слоя, так что рабочий заряд катализатора в течение цикла может изменяться.
На практике определить баланс масс катализатора нелегко из-за погрешностей, до-
пускаемых при измерении чистой массы и анализе отработанного катализатора. Ра-
бочий заряд каталитического слоя определяется как масса свежего катализатора, при
которой устраняется эффект изменения массы, обусловленный отложением метал-
лов и кокса. На практике условно принимают, что количество добавляемого свежего
катализатора равно количеству отбираемого отработанного и, стало быть, что рабо-
чий заряд каталитического слоя остается постоянным. Более того, принимается, что
объемы обоих катализаторов равны несмотря на изменение плотности [Sayles, 2006].
В литературных источниках [Soderberg, 1988; Chan and Strickland, 1990; Sayles, 2006]
можно найти подходы к определению рабочего заряда катализатора, основанные
на изменениях давления и плотности.
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
411
10.2.4 . Образование осадка
Одна из проблем, встречающихся при гидрокрекинге остаточного сырья с высокой
степенью превращения, заключается в образовании мягких коксообразных веществ,
называемых осадком, или шламом. Углеродистый материал осадка часто образует от-
ложения в реакторе и расположенных после него технологических аппаратах, а так-
же на поверхности катализатора, вызывая проблемы работоспособности оборудова-
ния и быстрой потери активности катализатора [Stanislaus et al., 2005; Wandas, 2007].
Образование осадка начинается в секции реактора и происходит особенно быстро
в условиях повышенной жесткости, необходимых для достижения высокой степени
превращения вакуумного остатка (с температурой кипения выше 538 °С+) либо по-
вышенной пропускной способности при неизменной степени превращения. В жест-
ких условиях происходит осаждение асфальтенов в непревращенной вакуумном
остатке. Асфальтены ведут себя как коллоидные вещества, образуя мицеллы, агре-
гаты и хлопья. Интенсивность флокуляции асфальтенов зависит от температуры,
давления и растворяющей способности других углеводородов в остаточном сырье
[Robert et al., 2002]. Из-за прогрессирующего накопления и флокуляции асфальтенов
дестабилизация непревращенного сырья может происходить и в секции улавливания
[Me Namara et al., 2007].
Шлам обычно скапливается в нижерасположенных сепараторах, теплообменни-
ках и колоннах фракционирования и загрязняет линии перекачки, рано или позд-
но вызывая остановку работы установки. Загрязнение оборудования коксообраз-
ным шламом, который образуется в процессе гидропереработки остаточного сырья,
может повлечь за собой большие финансовые расходы, связанные с эксплуатацией,
обслуживанием и простоем установки. На НПЗ часто применяют химические и ме-
ханические средства удаления отложений осадка. Иногда используют химические
добавки, подавляющие образование осадка.
Проблемы образования осадка и загрязнения оборудования приобретают осо-
бую остроту, когда степень превращения остаточного сырья в дистилляты высока
и превышает 50 %. Чтобы снизить остроту этих проблем, НПЗ часто вынуждены
эксплуатировать установки гидрокрекинга остаточного сырья с пониженной сте-
пенью превращения. Таким образом, образование осадка представляет собой кри-
тический фактор, ограничивающий максимально достижимую степень превра-
щения в промышленных установках гидропереработки остаточного сырья. Для
достижения высокой степени превращения крайне важно предотвратить или по-
давить образование осаждающихся материалов [Stanislaus, 2005]. Такие проблемы,
как загрязнение и закупорка змеевика печи подогрева сырья, сепараторов высокого
и низкого давления, вакуумной колонны, рециркуляционных насосов и холодиль-
ников, повышенная частота обслуживания фильтров очистки продуктов, фильтров
всасывания насосов колонн атмосферной и вакуумной перегонки и теплообмен-
ников остатка вакуумной колонны, нестабильность топливного мазута и несоот-
ветствие его спецификациям, вызываются преимущественно образованием осадка
[Me Namara et al., 2007].
412
Часть 3. Моделирование каталитических процессов
10.2.5 . Износ частиц катализатора
Проблему износа частиц катализатора можно охарактеризовать как образование ка-
талитической мелочи вследствие соударений частиц друг с другом и со стенками ре-
актора. Значимость этой проблемы может варьироваться от «чистого» истирания
частиц до полной их фрагментации. Первое подразумевает, что происходит лишь
удаление выступов на поверхности частиц с незначительным изменением исходно-
го гранулометрического состава, тогда как во втором случае частицы распадаются
на фрагменты примерно одинаковых размеров [Werther and Reppenhagen, 1999]. Если
размеры частиц становятся меньше, увеличивается доля взвешенных частиц в верх-
ней части слоя. В результате нарушается однородность слоя и становится возможным
унос катализатора [Galiasso and Caprioli, 2005].
Кам и соавторы [Kam et al., 2001] проанализировали эффект смешивания ката-
лизаторов гидроочистки в реакторе установки Н-ОП на НПЗ «Шуайба» Кувейтской
национальной нефтяной компании. Физические свойства смешиваемых катализато-
ров представлены на рис. 10.2. Хотя существенные различия в свойствах катализа-
торов А и В отсутствуют, их смешение при сохранении той же кратности газа к сы-
рью неожиданно затруднило контроль и стабилизацию разрыхленного слоя в РКС.
Главные эксплуатационные проблемы сводились к высокой скорости износа частиц,
повышенному расходу катализатора и увеличению перепадов температур в реакторе.
Кроме того, был неустойчив уровень разрыхленного катализатора в РКС, а степень
превращения вакуумного остатка была слишком низкой. Наблюдалось также нали-
чие значительного количества катализаторной мелочи в расположенных после реак-
тора циркуляционных контурах и оборудовании. Был испытан дополнительный ка-
тализатор С на алюмооксидном носителе с частицами тех же размеров, что и А или В.
Сведений о его физических свойствах не сообщалось. На рис. 10.2 сравнивается так-
же содержание мелочи катализаторов различных типов при испытаниях РКС с по-
вышенными скоростями подачи газа и жидкого сырья. Хотя все катализаторы име-
ли схожие свойства, катализатор В менее пригоден, чем Л, а катализатор С применять
вообще не рекомендуется.
Образование мелочи при износе катализатора влияет на стабильность работы ре-
актора и изменяет характеристики процесса, вызывая проблемы, схожие с теми, ко-
торые наблюдаются при смешении двух разных катализаторов.
10.2.6 . Деактивация катализатора
Одна из основных проблем гидропереработки тяжелых нефтяных фракций — поте-
ря активности катализатора. От срока его службы очень часто зависят экономиче-
ские показатели процесса. Скорость потери активности катализатора при перера-
ботке легких фракций (бензиновой фракции и средних дистиллятов) минимальна,
но в случае тяжелого сырья (тяжелая нефть и остатки перегонки) она может быть
значительной и существенно сокращать цикл работы. Основная причина деакти-
вации катализатора — потеря активных центров, вызываемая главным образом их
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
413
отравлением сильно адсорбируемыми веществами, перекрытием отложениями кокса
и металлов, закупоркой устьев пор и спеканием активной фазы [Ancheyta et al., 2002].
Свойство Катализатор А Катализатор В
Средняя длина частиц, мм 5,2 4,7
Содержание мелочи (крупностью менее 40 меш), %масс. 0,03 0,03
Диаметр частиц, мм 0,97 1,038
Насыпная плотность, кг/м3 0,447 0,453
Среднее сопротивление раздавливанию, кг/мм 0,95 0,86
Потери на истирание, %масс. 0,9 0,9
Удельная площадь поверхности, м7г 310 289
Удельный поровый объем (по адсорбции Н2О), см3/г 0,81 0,81
Время работы, сут
Рис. 10.2. Зависимость выхода каталитической мелочи от времени работы для катали-
заторов различных типов: (о) — катализатор А; () — катализатор В; (*) — катализатор С
[Kam, Е.К.Т. et at, Catal. Today, 64, 297, 2001]
Гальяссо и Каприоли [Galiasso and Caprioli, 2005] проанализировали деактивацию
никель-молибденового катализатора на алюмооксидном носителе при гидрокрекин-
ге тяжелого остатка в РКС. Главной целью исследователей было продемонстрировать
влияние внешнего слоя частиц катализатора на массоперенос и реакции, происходя-
щие при гидрокрекинге тяжелого остатка.
Свойства и условия работы исследовавшихся катализаторов показаны в табл. 10.1.
Катализатор каждого типа состоял из обычных, насеченных и размолотых гра-
нул (со средним размером менее 150 мкм). Сырьем служил остаток вакуумной пе-
регонки нефти «Джобо-Моричал» из нефтеносного пояса Ориноко в Венесуэле
414
Часть 3. Моделирование каталитических процессов
(плотность 8 ° API) с температурой начала кипения 490 °C и содержанием 2,02 % серы,
0,93 % азота, 22 % коксового остатка по Конрадсону, 21 % компонентов, нераствори-
мых в н-пентане и 714 мг/кг ванадия. Исследования активности проводились в реак-
торе смешения.
Таблица 10.1
Свойства катализаторов, использованных в исследовании деактивации при гидрокрекинге
тяжелого остатка в РКС
Свойства Началь- ные М D2 D3 D4 D5
Рабочая температура, °C — 410 420 430 410 430
Время работы 1 неделя 1 неделя 1 неделя 1 месяц 1 месяц
Мо03, %масс. 12,5
N1O, %масс. 4,2 4,7 4,6 4,7 12,3 9,4
V, %масс. 0 3,7 3,7 3,8 60,48 40,32
Удельный объем макропор, см3/г 0,19 0,16 0,15 0,15 0,09 0,06
Удельный объем мезопор, см3/г 0,25 0,21 0,19 0,17 0,09 0,11
Удельный объем микропор, см3/г 0,15 0,08 0,03 0,01 0 0
С/Н — 0,9 0,88 0,84 0,96 0,94
Компоненты, растворимые в кси- лоле, %масс. — 0,6 0,45 о,з 0,4 0,45
Углерод в гранулах, %масс. — 6,1 6,4 7,1 8,9 9,8
Углерод в насеченных гранулах, %масс. — 5,3 5,4 5,6 6,1 5,9
Сера, %масс. 5,8 6,32 6,44 6,55 10,4 10,1
Эффективный коэффициент диффузии, С5/Не, см2/с 0,08 0,0065 0,0033 0,0028 0,0021 —
Источник: [Galiasso, R. and Caprioli, L., Catal. Today, 109, 185, 2005].
Исследование отработанного катализатора показало, что внешний слой, окру-
жающий частицы катализатора, вначале является проницаемым и состоит главным
образом из кокса с предграфитовой углеродной структурой. Непрерывное наслаи-
вание кокса и накопление металлов делают внешний слой менее проницаемым, по-
этому диффузия в жидкой фазе, активность и избирательность ухудшаются. Кроме
того, из-за уменьшения площади сечения и закупорки пор ограничивается диффузия
в них соединений металлов и серы.
Было установлено, что металлы отлагаются на внешней поверхности мезо- и ма-
кропоровых структур, распределяясь по диаметру гранул в форме буквы U. Влияние
закупорки внешнего слоя усиливается с повышением температуры.
Аль-Далама и Станислаус [Al-Dalama and Stanislaus, 2006] изучали деактивацию
в реакторах с неподвижным и кипящим слоем, исходя из сведений, полученных при
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем 415
анализе отработанных катализаторов. Образцы отработанных катализаторов были
предоставлены НПЗ Кувейтской национальной нефтяной компании.
Установка состояла из РКС, загруженного кобальт-молибденовым катализато-
ром на алюмооксидном носителе. Сырьем служил остаток вакуумной перегонки
кувейтской экспортной нефти, разбавленный 10—15 % рециклового газойля (с со-
держанием 5,2 %масс. серы, 110 мг/кг металлов (V + Ni) и 4400 мг/кг азота). Образ-
цы отбирались из равновесного катализатора реактора, состоявшего из смеси силь-
но загрязненного и частично загрязненного катализаторов. Свойства сильно и слабо
загрязненных частей и их смеси представлены в табл. 10.2. Сильно загрязненный
катализатор содержал значительно больше (13,83 %масс.) ванадия, чем слабо загряз-
ненный (4,37 %масс.). Заметных различий в содержании кокса не было обнаружено.
Таблица 10.2
Свойства сильно и слабо загрязненных частей отработанного никель-молибденового
катализатора из промышленного РКС
Свойства катализатора Смесь Слабо загрязненный Сильно загрязненный
Содержание веществ, %масс.: V 10,58 4,37 13,83
С 16,20 15,80 16,30
Ni 4,03 3,48 5,21
Удельная поверхность, м2/г 68 122 55
Насыпная плотность, г/см3 1,09 0,97 1,21
Боковая прочность на раздавливание, фунт/мм 1,8 2,10 1,78
Удельный поровый объем, см3/г 0,17 0,21 0,11
Распределение длин частиц, %масс.: < 1,5 мм 25,2 14,4 40,0
1,5—3 мм 42,3 23,5 37,0
3,0—6 мм 32,5 61,3 23,0
> 6 мм 0,0 0,8 0,0
Источник: [Al-Dalama, К. and Stanislaus, A., Chem. Eng. J., 120, 33, 2006].
Слабо загрязненный катализатор имел существенно большие удельные значе-
ния поверхности и порового объема, чем сильно загрязненный. Полностью де-
активированный катализатор, вероятно, вообще не отбирался. Отмечались также
существенные изменения физических размеров частиц. Распределение длин заметно
изменилось в сторону увеличения содержания мелких частиц с длиной менее 2 мм.
Полученные результаты свидетельствуют о том, что деактивация катализатора
и нарушение нормальной работы РКС вызываются не только отложением метал-
лов и кокса, но и изменениями физических и механических свойств катализатора
416
Часть 3. Моделирование каталитических процессов
в динамических условиях процесса. Это отмечается прежде всего по изменению на-
сыпной плотности, которая оказывает существенное влияние на разрыхление слоя:
для получения нужной степени его разрыхления при большей насыпной плотности
требуется большая скорость циркуляции жидкости.
Гальяссо [Galiasso, 2007] изучал влияние рециркуляции 10 % не превращенного
остатка с температурой начала кипения 500 °C на деактивацию катализатора в устано-
вившемся режиме работы РКС. Проверялись активность катализатора и степень ее по-
тери в установившихся условиях после прохождения нескольких циклов возврата при
степени превращения 60 % за цикл. Сырьем служил остаток той же нефти, что и в ис-
следовании Гальяссо и Каприоли [Galiasso and Caprioli, 2005], при следующих рабочих
условиях: температура 420 °C, давление 13,6 МПа, объемная скорость подачи 0,5 ч-1.
Превращение (со степенью 60 %) вакуумного остатка (ВО0) осуществлялось с помо-
щью никель-молибденового катализатора на алюмооксидном носителе. Остатку перво-
го превращения присваивалось наименование ВОГ Во второй цикл превращения по-
давалась смесь 90 %масс. свежего (ВО0) прямогонного вакуумного остатка с 10 %масс.
остатка ВОГ Остаток второго цикла именовался ВО2. Затем в третий цикл опять подава-
лась смесь 90 % ВО0 и 10 % ВО2 с получением остатка ВО3 и так далее до получения ВО5.
Затем в РКС емкостью 0,05 л [так в оригинале, но это странно (науч, ред.)], за-
полняемом поочередно каждым из различных остатков ВО., при постоянных рабочих
условиях измеряли активность катализатора в реакциях гидрокрекинга, гидрообес-
серивания (ГДО) и гидродеметаллизации (ГДМ). Полученные результаты показа-
ны в табл. 10.3. Сравнивая активности катализатора, отработанного в течение 6 мес.
на свежем сырье (ВО0), после промывки ксилолом со снятием (активность «б») и без
снятия верхнего слоя (активность «в»), можно убедиться в том, что наружные слои
частиц в определенной степени закупориваются коксом и металлами, так как по-
сле частичного удаления отложений активность повышается, а степень деактивации
снижается. Отложения металлов и кокса, влияя на диффузионные ограничения вну-
три частиц, по-разному изменяют степень деактивации катализатора в реакциях ги-
дрокрекинга, ГДО и ГДМ.
Таблица 10.3
Степень деактивации катализатора в реакциях гцдрокрекинга/ГДО/ГДМ
после переработки рециркулята
Свойства/число проходов Начальные ВО0» ВО1 ВО2 во, во5
МоО3,%масс. 12,5 — — — — —
NiO, %масс. 4,2 — — — — —
Средний диаметр пор, А 112 45 75 60 50 30
С/Н 0,8 0,9 0,84 0,89 0,92 0,95
Компоненты, растворимые в ксилоле, %масс. 0,6 0,1 0,55 0,5 0,45 0,4
Кокс, %масс. 1,1 9 7,1 7,9 8,5 9,0
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем 417
Таблица 10.3, окончание
Свойства/число проходов Начальные во; во, во2 во3 во5
Активность6, %масс. 60/59/55 45/40/38 50/50/45 46/48/40 38/39/30 24/25/16,
Активность’, %масс. — 46/44/43 38/50/30 33/44/28 29/34/26 25/30/22,
Активностьг, % масс. 66/60/44 55/52/50 58/50/40 52/46/36 46/38/30 40/30/26,
Площадь молибденовой поверхности (РФС) 0,22 0,01 0,1 0,06 0,02 0,01
Источник: [Galiasso, R., Fuel Process. Technol., 88, 779, 2007].
3 Через 6 мес. работы без рециркуляции.
6 После промывки ксилолом.
' После промывки ксилолом и снятия верхнего слоя.
г После промывки ксилолом и шлифовки.
РФС — рентгеновская фотоэлектронная спектроскопия.
Как показывает таблица, с увеличением числа проходов от 1 до 5 активность сни-
жается. Эти результаты подтверждают влияние рециркуляции непревращенного сы-
рья на закупорку устьев пор, так как после снятия верхнего слоя (кокса и металлов)
и шлифования активность катализатора во всех реакциях улучшается.
Чем больше число циклов рециркуляции, тем больше металлов и углеродистых
материалов отлагается во внешнем слое частиц и тем менее растворимыми в ксилоле
становятся отложения. Вне всякого сомнения, рециркуляция непревращенного сы-
рья воздействует на внешний слой частиц, ухудшая диффузию молекул в поры и бло-
кируя доступ к поверхностным активным центрам.
10.2.7 . Экономические аспекты процесса
Цены на нефтепродукты зависят от цен на нефть и сезонного спроса на продукты.
Спрос на топливный мазут наиболее высок зимой, когда возрастает потребление те-
пловой энергии в холодных регионах. Спрос на бензин, напротив, максимален в лет-
нее время из-за увеличения числа отдыхающих. Установки гидропереработки в кипя-
щем слое рассчитаны на производство продуктов, пользующихся высоким сезонным
спросом [Nongbri and Clausen, 1992].
Капитальные затраты на установку Н-Oil составляют 6,7-8,9 млн долл. Эксплуа-
тационные затраты в зависимости от перерабатываемого сырья колеблются от 0,33
до 0,65 долл, на баррель (в ценах 1970 г. для условий побережья Мексиканского за-
лива) [Mounce and Rubin, 1971]. Экономические показатели процессов в кипящем
слое зависят от разницы цен на сырье и продукты, а также (в определенной степени)
от местоположения НПЗ. Чем больше ценовой разрыв, тем лучше экономические
показатели. Расчетный чистый доход от эксплуатации установки с пропускной спо-
собностью 51 964 баррелей за рабочие сутки составляет 66,4 млн долл, в год (в ценах
США по состоянию на 1990 г.) [Nongbri and Clausen, 1992].
418 Часть 3. Моделирование каталитических процессов.
10.3. ПРОМЫШЛЕННЫЕ ТЕХНОЛОГИИ КИПЯЩЕГО СЛОЯ
10.3.1. Процесс Н-ОН
Процесс Н-Oil был разработан в конце 1950-х гг. компанией Hydrocarbon Research Inc.
(HRI), чтобы упростить облагораживание остаточного сырья в реакторах с неподвиж-
ным слоем. Начальная буква (Я) в названии процесса символизирует процесс обо-
гащения водородом. Первая демонстрационная установка Н-Oil была введена в дей-
ствие в 1964 г. на НПЗ компании в Лейк-Чарльзе, штат Луизиана, а в 1968 г. на НПЗ
Кувейтской национальной нефтяной компании в Шуайбе вступила в строй первая
промышленная (одноступенчатая) установка. В 1984 г. заработала первая двухступен-
чатая установка, сооруженная HRI на НПЗ компании Motiva (впоследствии Texaco)
в Конвенте, штат Луизиана [McNamara et al., 2007]. В октябре 1994 г. права на техно-
логии HRI, в том числе на процессы кипящего слоя Н-Oil и T-Star, выкупил Француз-
ский нефтяной институт (IFP) [Kressmann et al., 1998].
Процесс H-Oil может работать в широком интервале степеней превращения и под-
ходит, в частности, для переработки тяжелого вакуумного остатка с высоким содержа-
нием металлов и коксуемостью. Еще одно важное преимущество H-Oil— способность
поддерживать постоянное качество продуктов в течение всего цикла. Поскольку ре-
актор H-Oil позволяет уникально сочетать работу в режиме смешения и псевдоожи-
женный слой катализатора, можно полагать, что он приспособлен к проведению эк-
зотермических реакций, к переработке сырья с высоким содержанием механических
примесей и к изменчивости типа сырья и целей переработки (например, к проведе-
нию одно- или двухступенчатого процесса). Типичная технологическая схема про-
цесса H-Oil представлена на рис. 10.3.
Рис. 10.3. Типичная технологическая схема процесса Н-ОН
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем 419
Правильно спроектированную установку Н-OU можно переключить с переработки
остатка на гидрирование газойля, не прерывая ее работу. То же относится и к замене
Катализатора в связи с изменением операции. Равновесный катализатор, оставший-
ся от предыдущей операции, можно хранить до тех пор, пока он не потребуется вновь
[Johnson etal., 1968].
В 1980-х гг. появились так называемые технологии второго поколения, состо-
явшие во внедрении автоматизированного управления и разработке катализаторов
Повышенной активности. Кроме того, улучшения коснулись конфигурации про-
цесса и конструкции реакторов. Усовершенствованные технологии позволили уве-
личить пропускную способность установок при невиданных ранее уровнях обессе-
ривания, деметаллизации и глубине гидрокрекинга [Wisdom and Colyar, 1995; Colyar
and Wisdom, 1997].
В отдельных случаях весьма привлекательным вариантом может оказаться сочета-
ние процесса H-Oil с коксованием или каталитическим крекингом. Установка Н-ОЦ,
работая в сравнительно мягких условиях, может существенно снижать содержание
серы, ванадия и коксуемость и таким образом подготавливать сырье для установки
коксования с ограниченной пропускной способностью и строжайшими требования-
ми к качеству производимого кокса. Двухступенчатая установка Н-ОН может подго-
тавливать сырье с низким содержанием серы и металлов для процесса каталитиче-
ского крекинга [Eccles, 1980; Bechtel and Wisdom, 1991].
10.3.2. Процесс Т-Star
Еще один процесс в кипящем слое катализатора — T-Star (сокращение от Texa-
co Strategic Total Activity Retention), представляющий собой расширение процес-
са H-Oil. Установки T-Star способны поддерживать общую глубину превращения
от 20 до 60 % (в частности, глубину ГДО от 93 до 99 %). Установка может работать
в режиме предварительной очистки сырья для каталитического крекинга в псев-
доожиженном слое (FCC) либо в режиме гидрокрекинга вакуумного газойля. Для
реактора T-Star вполне подходит катализатор процесса H-Oil. Реактор T-Star мож-
но также задействовать вместе с реактором H-Oil, чтобы улучшить качество ди-
стиллятных продуктов последнего, а также прямогонных дистиллятов, легкого
и тяжелого газойлей FCC, а также газойлей коксования. В режиме мягкого гидро-
крекинга процесс T-Star обеспечивает глубину превращения до 60 %об. Преиму-
щество эксплуатации установки T-Star в подобном режиме заключается в том, что
его катализатор нечувствителен к содержанию серы и азота в сырье и обеспечи-
вает постоянство как глубины превращения и выхода, так и качества продуктов.
Стабильная производительность обусловлена возможностью замены катализато-
ра без прерывания работы установки. Демонстрацию процесса T-Star в промыш-
ленном масштабе наряду с пуском установок H-Oil осуществили совместно компа-
ния Husky Oil, Канада, и Французский нефтяной институт (IFP) [Johns et al., 1993;
Rana et al., 2007].
420
Часть 3. Моделирование каталитических процессов
10.3.3. Процесс LC-Fining
Процесс LC-Fining, разработанный компанией АВ В Lummus Crest, представляет ветвь
технологий HRI, промышленная эксплуатация которой началась впервые в 1984 г.
на НПЗ компании ВР в Тексас-Сити. Установка LC-Finingкомпании ВР была первой
трехступенчатой установкой гидропереработки в кипящем слое. Буквы LC в назва-
нии означают Lummus Corporation — компанию-разработчика процесса.
Недавно в процессы H-Oil и LC-Fining внедрили технологии гидропереработки
в кипящем слое следующего поколения, предусматривающие промежуточное раз-
деление потока между реакторами многоступенчатой установки с целью улучшить
кинетику реакций в последнем реакторе и повысить общую результативность про-
цесса. Промежуточное разделение, необходимое для повышения степени превра-
щения или увеличения пропускной способности при той же степени превращения,
можно добавить путем модернизации существующей установки [Colyar et al., 2001;
Me Namara et al., 2007]. Упрощенная схема промежуточного разделения показана
на рис. 10.4.
Рис. 10.4. Промежуточное разделение в процессе кипящего слоя
LC-Fining — это процесс гидрирования, который можно использовать для ГДО,
ГДМ и гидрокрекинга остатка атмосферной или вакуумной перегонки. Процесс
LC-Fining хорошо подходит для гидроочистки особо тяжелых остатков, битумов и ва-
куумных остатков и показал в этих случаях хорошую длительность цикла. К общим
достоинствам процесса LC-Fining относятся малые капиталовложения, большой вы-
ход легких продуктов и низкие эксплуатационные затраты. Процесс дает полный
спектр высококачественных дистиллятов. Тяжелый остаток можно использовать как
топливный мазут, синтетическую нефть или сырье для процессов FCC, коксования,
висбрекинга либо деасфальтизации экстракцией растворителями. Процесс LC-Fining
способен обеспечивать степень ГДО от 60 до 90 %, ГДМ — от 50 до 98 %, сниже-
ния коксового остатка — от 35 до 80 %. Конструкция реактора показана на рис. 10.1
[Rana et al., 2007].
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
421
В табл. 10.4 приведен перечень действующих установок гидропереработки в кипя-
щем слое, а также некоторых планируемых к пуску в предстоящие годы. В табл. 10.5
представлена сводка рабочих условий процессов в кипящем слое (H-Oil и LC-Fining).
Таблица 10.4
Действующие и планируемые к пуску установки гидропереработки в кипящем слое
Компания Местоположение Технология Дата пуска Проектная пропускная способность, тыс. барре- лей в сутки Сырье
Действующие установки
LKNPC Шуайба, Кувейт н-ои 1968 50 Вакуумный остаток
2. РЕМЕХ-' Саламанка, Мексика То же 1972 18,5 То же
3. BP- Amoco Тексас-Сити, США LC-Fining 1984 60 Остатки тяжелой нефти
4. Motiva Конвент, США H-Oil 1984 43 Вакуумный остаток
5. Syncrude Милдред-Лейк, Ка- нада LC-Fining 1988 40 Битум
6. Husky Ллойдминстер, Ка- нада Н-Oil/ H-Oil 1992/ 2006 32/39 Тяжелая нефть
7. Tbnen Кавасаки, Япония Тоже 1997 25/25 Вакуумный остаток
8. РЕМЕР Тула, Мексика H-Oil 1997 50 То же
9.ENL Милаццо, Италия LC-Fining 1998 25 Остаточное сырье
10. PKN Плоцк, Польша H-Oil 1999 34 Вакуумный остаток
11. Slovnaft Братислава, Словакия LC-Fining 2000 23 То же
12. Shell Scotford (линии 1 и 2) Скотфорд, Канада To же 2003 79 Битум
13. Лукойл Пермь, Россия T-Star 2004 70,4 Вакуумный газойль
14. Neste Оу Пурву, Финляндия LC-Fining 2007 40 То же
15. Shenhua Внутренняя Монго- лия, Китай T-Star 2007 69,5 Жидкие продукты газификации угля
Установки, планируемые к пуску в предстоящие годы
16. Petro-Canada Эдмонтон, Канада LC-Fining 2009 50 Битуминозная нефть
17. Shell Scotford (Train 3) Альберта, Канада To же 2009 47 Битум
18. Мозырь Мозырь, Белоруссия H-Oil 2010 60 Вакуумный остаток
19, Northwest Upgrading Калгари, Канада LC-Fining 2011 29 Тяжелая нефть
“ Планируется модернизация для подготовки сырья ТСС.
6 После декабря 2008 г. планируется модернизация для подготовки сырья FCC.
422
Часть 3. Моделирование каталитических процессов
Таблица 10.5
Рабочие условия процессов H-Oil, LC-Fining и T-Star
Параметр Н-ОН LC-Fining T-Star
Правообладатель Ахеп (HRI/IFP) ABB Lummus Amoco Oil (BP) Chevron
Температура, °C 415-440 385-450 300-550
Давление, МПа 16,8-20,7 7,0-18,9 8-11
LHSV, ч-' 0,4-1,3 — —
Скорость замены катализатора, кг на тонну сырья 0,3-2,0 — —
Пропускная способность одной линии, баррель/сут < 34 000 < 35 000
Расход Н2, фут’/баррель 1410 1350 650
Степень превращения, % 45-90 40-97 20-85
Степень ГДО, % 55-92 60-90 93-99
Степень ГДМ, % 65-90 50-98 —
Выход продуктов, %масс.
H-Oil LC-Fining T-Star
Продукт Выход Продукт Выход Продукт Выход
c-c4 3,5 c4 2,35 с,-с5» 10,74
C-204 °C 4 17,6 C5—177 °C 12,6 С,-182 °Ca 6 14,6
204-371 °C 22,1 177-371 °C 30,6 182-343 °C' 35,68
371-565 °C 34,0 371-550 °C 21,5 343-380 °Ca 3,01
565 °C+ 22,8 550 °C+ 32,9 380-566 °C“ 23,72
566 °C+a 11,63
Источники: [Johns, W.F. et al., Texaco T-star process for ebullated bed hydrotreating/hydro-
cracking, в NPRA Annual Meeting, March 21—23, Convention Center San Antonio, TX, 1993;
Pratt, R.E. et al., Heavy oil hydroprocessing, в Proceedings of the Sixth UNITAR International Confer-
ence on Heavy Crude and TarSands, Vol. 2(10), c. 1—9, 1995; Rana, M.S. et al., Fuel, 86, 1216, 2007].
" Выход из деасфальтизованного газойля гидрокрекинга.
10.4. МОДЕЛИРОВАНИЕ РЕАКТОРА С КИПЯЩИМ СЛОЕМ
Выше уже говорилось, что РКС моделировать труднее, чем реакторы других типов.
Это, безусловно, одна из труднейших для моделирования реакторных систем. Про-
блему представляет разработка модели не только РКС, но и реакций в фазах, уча-
ствующих в процессе. В частности, если взять случай гидрокрекинга остатка тяже-
лой нефти, то в системе, состоящей из твердой (катализатора), газовой (главным
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
423
образом водород) и жидкой (углеводородное сырье) фаз по различным маршрутам
одновременно протекает множество различных реакций — ГДО, ГДМ, гидродеазо-
тирования (ГДА), гидродеасфальтизации (ГДАс) и т. д. При этом не стоит забывать,
что катализатор непрерывно движется в реакторе, который ведет себя как система
идеального смешения. В то же время из-за отложения кокса и металлов активность
катализатора снижается, так что приходится добавлять свежий катализатор, чтобы
компенсировать потерю и поддерживать активность на более или менее постоян-
ном уровне.
10.4.1. Исследования гидродинамики
Хотя промышленные РКС чаще всего работают в условиях высоких температур
и давлений, наблюдается явный недостаток исследований, относящихся имен-
но к этим условиям процесса. Опубликовано лишь несколько трудов по гидроди-
намике высоких давлений и температур [Jiang et al., 1992, 1997; Luo et al., 1997;
Ruiz et al., 2005; Sanchez et al., 2008]. Сообщается, что давление и температура
сильно влияют (через характеристики барботажа и свойства жидкости) на гидро-
динамику РКС.
Руис и соавторы [Ruiz et al., 2005] исследовали гидродинамические характеристи-
ки систем кипящего слоя, работающих в условиях высоких давлений и температур.
Целью их работы было исследовать влияние этих параметров на гидродинамические
свойства систем кипящего слоя. Измерялись значения пустотности слоя и мини-
мальной псевдоожижающей скорости жидкости, которые затем сравнивались с ре-
зультатами расчетов по хорошо известным уравнениям и моделям, разработанным
для нормальных условий. Анализировалось также влияние температуры и давления
на пузырьковый режим течения. В результате была предложена эмпирическая фор-
мула для расчета скорости газа, соответствующей переходу между режимами дис-
пергирования и коагуляции пузырьков при высоком давлении. Было замечено, что
при данной скорости газа пустотность кипящего слоя, работающего при нормальной
температуре и нормальном или высоком давлении, увеличивается с ростом скорости
потока жидкости, а также с увеличением ее вязкости. Аналогичные результаты были
получены для слоя, работающего при температуре 100 °C. Пустотность слоя при этой
температуре росла и с увеличением скорости газа (результатом было непрерывное
разрыхление слоя). С другой стороны, при увеличении температуры с 20 до 100 °C пу-
стотность снижалась (рис. 10.5).
Так как влияние давления на пузырьковый режим не учитывалось, при высоких
давлениях формула дает неверные значения переходной скорости потока. Подгон-
кой экспериментальных данных из открытых источников к выражению, аналогич-
ному предложенному Зангом и соавторами [Zhang et al., 1997], была получена эмпи-
рическая зависимость. В использованных данных давление изменялось в интервале
от 5,6 до 15,6 МПа, что соответствует давлениям в реакторах гидропереработки тяже-
лых фракций нефти в кипящем слое.
424
Часть 3. Моделирование каталитических процессов
Рис. 10.5. Влияние скорости газа на пустотность слоя при различных значениях
давления Р, температуры Т и скорости жидкости Ц: () — Ц = 1,4 см/с, Т = 20 °C,
Р= 15 МПа; (□) — Ц = 1,4 см/с, Т= 20 °C, Р= 7,5 МПа; (*) — Ц= 0,7 см/с, 7=20 °C,
Р= 15 МПа; (Д) — Ц = 0,7 см/с, 7 = 100 °C, Р= 15 МПа; (•) — U= 0,7 см/с, 7=20 °C,
Р= 7,5 МПа; (о) — Ц = 0,7 см/с, 7= 100 °C, Р= 7,5 МПа
[Jiang, Р. et al., Ind. Eng. Chem. Res., 31,2322, 1992]
Зависимости были получены для двух размеров частиц.
Для частиц диаметром 2,1 мм
z 4-0.667
— = 0,756Fr9,339 Аг/'8321 — 15,6 МПа >Р> 10,1 МПа, (10.1)
и, ° Ip/J
где
н =-0,22411 — 1-1,1566. (10.2)
UJ
(См. все расшифровки формул в конце главы.)
Для частиц диаметром 3,0 мм
z 4-0,667
— = 3,41 103Fr“'Ai',"1,7259 — 15,6 МПа >Р>Р> 5,62 МПа. (10.3)
и, g IpJ
График на рис. 10.6 демонстрирует хорошее согласие между результатами, полу-
ченными по формуле (10.3), и данными из литературных источников. Показаны так-
же результаты расчета по формуле Занга. Они занижены по сравнению с экспери-
ментальными данными.
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
425
Рис. 10.6. Значения отношения скорости газа к скорости жидкости, полученные по экспе-
риментальным данным из литературных источников, а также путем расчета: (♦) — по фор-
муле Занга; () — по формуле (10.3) [Jiang, Р. et al., Ind. Eng. Chem. Res. 31,2322, 1992]
10.4.2. Исследования обратного масштабирования
Дешпанде и соавторы [Deshpande et al., 1992] изучали обратное масштабирование
РКС, уменьшая высоту реакторов и проверяя идентичность гидродинамических ха-
рактеристик в промышленных и лабораторных установках. Чтобы значение приве-
денной скорости жидкости при уменьшении высоты реактора оставалось тем же са-
мым, необходимо снизить линейную скорость жидкости. Для разрыхления слоя при
пониженной линейной скорости жидкости должен быть уменьшен также размер ча-
стиц катализатора. Уменьшение приведенной скорости жидкости и размера частиц
катализатора должно осуществляться так, чтобы важные гидродинамические харак-
теристики, в том числе доля фаз, оставались неизменными.
Предлагаемая процедура обратного масштабирования гидродинамики реактора
заключается в следующем:
1) уменьшение Ц, что дает увеличение доли твердой фазы е_;
2) уменьшение <7, для восстановления значения
3) корректировка других параметров системы, дающая одинаковые значения до-
лей жидкости е, и газа eg в обоих реакторах.
В число корректируемых параметров входили dp, Dc, Ц и Ug. Полученные результаты
представлены в табл. 10.6. Частицы больших диаметров соответствуют промышлен-
ной установке, меньших диаметров — лабораторной установке. Расчет долей фаз вы-
полнялся согласно обобщенной модели спутного потока по формулам (10.4), (10.5)
[Bhatia and Epstein, 1974].
426
Часть 3. Моделирование каталитических процессов
Доля жидкости:
'g ^oEg) '+ioEg-
(10.4)
Пустотность слоя:
!-es =
~Eg~k^g)' /'+O + ^o)Eg.
(10.5)
Таблица 10.6
Параметры промышленной и лабораторной установок
при обратном масштабировании
Параметры для установок Номер испытания
1 2 3 4 5 6
Промышленная
0,005 0,005 0,005 0,005 0,005 0,005
Ц, м/с 0,0753 0,0752 0,0752 0,0673 0,0673 0,0673
Ue, м/с 0,0753 0,0752 0,0752 0,0673 0,0673 0,0673
е/ 0,5206 0,5174 0,5152 0,4888 0,4979 0,4943
£г 0,0631 0,0714 0,0784 0,0839 0,0573 0,0675
Лабораторная
Ч»м 0,003 0,003 0,003 0,003 0,003 0,003
Ц, м/с 0,0561 0,0561 0,0527 0,0467 0,0466 0,0466
С, м/с 0,1186 0,1483 0,1779 0,1727 0,0858 0,1147
е/ 0,5324 0,5295 0,5272 0,4833 0,4922 0,4889
е * 0,0609 0,0689 0,0758 0,0854 0,0584 0,0686
Источник'. [Deshpande, D.A. et al., Similitude studies in threephase ebullated bed reactors, in East-
ern Oil Shale Symposium, November 17—20, Hyatt Regency, Lexington, KY, 1992].
Видно, что доли фаз в каждом из случаев почти идентичны. При таком подхо-
де к обратному масштабированию трехфазного промышленного РКС до лабора-
торного предполагается, что рабочие условия (давление и температура) в реакторах
одинаковы.
Сафонюк и соавторы [Safoniuk et al., 1999] предложили метод обратного масшта-
бирования трехфазного псевдоожиженного слоя с использованием пи-теоремы Ба-
кингема и следующих безразмерных критериев геометрического и динамического
подобия:
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
427
М = -Ц^; (Ю.6)
= (10.7)
С
Prf = —; (Ю.9)
Р/
3,=^-. (ю.ю)
Для сопоставления упомянутых выше безразмерных комплексов использовались
данные, полученные с промышленной колонны диаметром 0,91 м. Колонна была
оснащена трехфазной системой, состоящей из керосина, водорода и катализатора
на керамическом носителе, и работала при давлении около 3,5 МПа и нормальной
температуре.
Эксперименты проводились при нормальных атмосферных условиях в акриловой
колонне с внутренним диаметром 0,0826 м. Жидкой фазой являлся водный раствор
сульфата магния с концентрацией 20 %масс., газовой фазой — воздух, а твердая фаза
была представлена цилиндрическими частицами алюминия.
Значения наиболее важных параметров перечислены в табл. 10.7. Можно заме-
тить, что выбранные для эксперимента материалы не дают строго соответствия всех
безразмерных критериев. Тем не менее большинство данных довольно хорошо со-
гласуются с теми, которые были получены в колонне диаметром 0,91 м (рис. 10.7).
Значения безразмерных критериев достаточно близки, но для получения оптималь-
ного соответствия долей фаз все же нужны дополнительные исследования на других
материалах.
Таблица 10.7
Физические свойства материалов, использованных в методе масштабирования по критериям
геометрического и динамического подобия
Свойство Колонна диаметром 0,91 м Лабораторная колонна
Диаметр колонны Dc, м 0,914 0,0826
Плотность жидкости р;, кг/м3 800 1183
Вязкость жидкости и,, Па с 1,5 10'3 3,6 • 10-3
Скорость жидкости Ц, м/с 0-0,019 0,007-0,031
428
Часть 3. Моделирование каталитических процессов
Таблица 10. 7, окончание
Свойство Колонна диаметром 0,91 м Лабораторная колонна
Плотность частиц р,, кг/м3 1,68 2,68
Диаметр частиц dn, мм 0,8 1,0
Длина частиц Lf, мм 2,4 2,6
Плотность газа рг, кг/м3 2,72 1,2
Скорость газа Up м/с 0-0,09 0-0,122
Коэффициент поверхностного натяжения на границе раздела газа и жидкости, о, Н/м 2,5-10-2 2,5-10-2
Число Мортона М 4,0 • 10-’ 3,05-10-’
Число Рейнольдса Re 0,20 0,151
Соотношение плотностей 0-8,1 0-10,2
Соотношение скоростей 0—00 0-18
Источник'. [Safoniuk, М. et al., Chem. Eng. Sci., 54, 4961, 1999].
Re9=Re/*₽u(-)
Рис. 10.7. Зависимость кажущейся доли газа в надслоевом пространстве от числа
Рейнольдса газа (Re^ при различных значениях числа Рейнольдса жидкости (Re(). Черные
значки — данные лабораторной колонны, белые — промышленные данные. Обозначе-
ния: (□) — Re, = 0; () Re, = 2,434; (Д) Re, = 2,64; (о) Re, = 4,4; (*) Re, = 6,41; (v) Re, = 6,6;
(♦) Re, = 8,7; (•) Re, = 10,12 [Safoniuk, M. etal., Chem. Eng. Sci., 54, 4961, 1999]
Предложенный Сафонюком и соавторами метод обратного масштабирования
гидродинамики трехфазного псевдоожиженного слоя, основанный на принципах
динамического подобия, опробовали Санчес и соавторы [Sanchez et al., 2008]. Не-
скольким системам, работавшим при давлениях в интервале от 0,79 до 15,6 МПа
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
429
(моделируемые системы, т. е. прототипы), сопоставлялись модельные системы при
атмосферном давлении. Эксперименты проводились для проверки метода через
сравнение общих долей фаз и режима потока в системах, работающих при сильно
различающихся давлениях.
Пустотность слоя в прототипных и модельных системах качественно схожа,
а количественные расхождения не превышают 13 %, но при повышении давле-
ния в прототипной системе расхождения возрастают (рис. 10.8). В целом доля газа
в модельных системах завышена по сравнению с прототипными, что можно объяс-
нить вспениванием используемой в них фракции; это говорит о склонности к ак-
кумуляции небольших пузырьков газа даже при низкой его линейной скорости. Ре-
зультаты расчета приведенной скорости дрейфа газа для прототипных и модельных
систем показывают, что, хотя большинство из них работает в дисперсном пузырь-
ковом режиме, линейные скорости газа, соответствующие переходу между режима-
ми диспергирования и коагуляции пузырьков, в системах высокого давления выше
(рис. 10.9 и 10.10).
Используемые безразмерные критерии недостаточно хорошо подходят для харак-
теризации вспенивающихся фракций. Поэтому в целях расширения области согласо-
вания гидродинамических параметров и значительного сокращения количественных
расхождений рекомендуется использовать более плотные газы и фракции, не склон-
ные к вспениванию.
Рис. 10.8. Зависимости пустотности слоя и долей газа и жидкости от числа Рейнольдса
газа (Re ) для прототипной и модельной систем. Пустотность слоя: () — прототип
(Re,= 0,77,V= 34 °C, Р = 10,10 МПа); (□) — модель (Re, = 0,77, Т= 22 °C, Р= 0,08 МПа).
Доли газа и жидкости, прототип (Re, = 0,77, Т= 34 °C, Р = 10,10 МПа): (Д) — ед; (°) — е,.
Доли газа и жидкости, модель (Re, = 0,77, Т = 34 °C, Р = 10,10 МПа): (*) — ед; (•) — е,
[Sanchez, J.L. et al., Catal. Today, 130, 519, 2008]
430
Часть 3. Моделирование каталитических процессов
Доля газа
Рис. 10.9. Зависимость приведенной скорости дрейфа от доли газа для прототипных
систем, работающих при разных давлениях и температуре Т= 34 °C: (я) — Re, = 1,15,
Р = 0,79 МПа; (•) — Re, = 0,99, Р = 2,86 МПа; (*) — Re, = 0,88, Р = 5,62 МПа;
(Т)_ Re,= о,77, Р = 10,10 МПа; (►) — Re, = 0,70, Р= 15,60 МПа [Sanchez, J.L. et al., Catal.
Today, 130, 519, 2008]
Доля газа
Рис. 10.10. Зависимость приведенной скорости дрейфа от доли газа для соответствую-
щих модельных систем, работающих при разных температурах и давлении Р - 0,08 МПа:
() — Re,= 1,15, 7=35 °C; (•) — Re,= 1,06, 7=32 °C; (*) — Re, = 0,99, 7=30 °C;
(v) — Re, = 0,88, 7=25 °C; (<) — Re,= 0,77, 7=22 °C; (►) - Re, = 0,70, 7= 19°C [San-
chez, J.L. et al., Catal. Today, 130, 519, 2008]
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
431
10.4.3. Моделирование реактора
Число математических моделей, разработанных для описания работы трехфазных
реакторов в переходном и установившемся режимах, довольно велико. Поэтому эти
модели необходимо классифицировать по назначению и с учетом присущих им огра-
ничений. Фромен и соавторы [Froment et al., 2010] предложили общую классифи-
кацию моделей адиабатических и неадиабатических реакторов, которая справедлива
и для РКС (табл. 10.8).
Таблица 10.8
Классификация математических моделей каталитических реакторов
Модели Псевдогомогеиные (Л) (7= 7, С= С) Гетерогенные (В) (Т* 7, С* С)
Одномер- ные АТ Базовая модель идеального вытеснения ЛИ: AI + продольное смешение 51: Л1+ межфазные градиенты 511: ЛИ + 51 + внутричастичные градиенты
Двумерные ЛШ: А1 + ЛИ + поперечное сме- шение 5Ш: В\ + 511 + поперечное смешение
Источник: [Froment, G.F. et al., Chemical Reactor Analysis and Design, 3rd edn., John Wiley &
Sons Inc., New York, 2010].
Предполагается, что явления конвекции, массопереноса и диспергирования про-
исходят в направлении потока. Поэтому большинство предлагаемых моделей реакто-
ров относятся к одномерным.
Если перепад температур из-за экзотермичности реакций не слишком велик, что
и имеет место в РКС, адекватно отобразить поведение реактора может псевдогомо-
генная модель. Такие модели предполагают, что скорость массопереноса через гра-
ницы раздела фаз всегда больше скорости реакции, т, е. массо- и теплоперенос че-
рез границы раздела принимаются незначительными. Из этого следует, что значения
температуры и концентраций у поверхности катализатора и вдали от нее одни и те же.
Маллади и соавторы [Malladi etal., 1982] смоделировали РКС как проточный реак-
тор смешения, чтобы проанализировать последствия прерывания рециркуляции не-
превращенного или подачи свежего сырья и определить запас времени для предот-
вращения значительного повышения температуры. Результаты имитации показали,
что при отказе рециркуляционного насоса значительное повышение температуры
происходит примерно через 15 мин; если же падает скорость подачи свежего сырья,
температура в реакторе поднимается до 538 °C менее чем за 30 с.
Матос и Жирарделло [Matos and Guirardello, 2000] описали поведение концен-
траций тяжелых фракций нефти при гидрокрекинге в РКС кинетической моделью,
основанной на псевдокомпонентах. Система моделировалась как изотермический
трубчатый реактор с продольным диспергированием. Водород и сырье движутся сни-
зу вверх, а твердые частицы катализатора образуют разрыхленный слой, оставаясь
в реакторе. Имитация работы проводилась при различных значениях приведенной
432 Часть 3. Моделирование каталитических процессов
скорости жидкости, высоты реактора и степени перемешивания (числа Пекле). Мо-
дель реактора разрабатывалась при следующих допущениях:
• установившийся режим работы;
• постоянное поперечное сечение реактора;
• однородное температурное поле (работа в изотермическом режиме);
• однородная плотность жидкой фазы;
• прямоугольная эпюра скоростей в поперечном сечении реактора (пробковый
режим потока);
• прямоугольная эпюра концентраций в поперечном сечении реактора, кон-
центрации изменяются лишь в осевом направлении;
• водород в газовой фазе присутствует в избытке и жидкое сырье полностью
насыщено растворенным водородом; скорость реакций от парциального дав-
ления водорода явным образом не зависит, поэтому нет необходимости учи-
тывать баланс масс водорода;
• одинаковый для всех псевдокомпонентов коэффициент продольного
диспергирования.
На основании этих предположений общий баланс масс в жидкой фазе дается
выражением
(10.11)
dz dz j=\
при следующих граничных условиях:
UlCiO=UlC-ElD^.,Z = 0-, (Ю.12)
’ dz
-£,D^ = 0,z = L. (10.13)
dz
Все концентрации выражаются через массовые единицы. Для численного реше-
ния модели массовым долям псевдокомпонентов на входе и долям жидкости в реак-
торе присваивались произвольные начальные значения.
Система обыкновенных дифференциальных уравнений решалась методом орто-
гональной коллокации с безразмерными переменными в балансах масс и граничных
условиях.
Эпюры концентраций рассчитывались последовательно в следующем поряд-
ке: тяжелые ароматические углеводороды, легкие ароматические углеводороды, тя-
желые нафтены, легкие нафтены, тяжелые парафины, легкие парафины. В расчете
концентраций последующего псевдокомпонента участвовала эпюра концентраций
предыдущего.
Результаты показывают, что с увеличением высоты реактора глубина реакции воз-
растает, а с увеличением приведенной скорости жидкости — убывает. Это происходит
оттого, что изменение указанных параметров косвенно влияет на время пребывания,
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
433
а последнее, в свою очередь, прямо влияет на скорость реакции из-за изменения
доли жидкости. Был также сделан вывод, что увеличение числа Пекле приближает
режим течения жидкости в реакторе к пробковому, происходящему без продольной
дисперсии. В пределе твердые частицы подвергаются воздействию жидкости одной
и той же концентрации, что способствует поддержанию постоянной скорости реак-
ции по высоте реактора.
Несмотря на общий характер подхода, модель применима к разному сырью,
перерабатываемому в различных рабочих условиях. Полезность подхода зависит
от надежности методов детального композиционного анализа перерабатываемой
тяжелой нефти.
Чтобы рассчитать поведение индикаторного газа (метана) в реакторе, Сейлс и со-
авторы [Sales et al., 2005] смоделировали динамическое поведение газовой фазы
в трехфазном реакторе псевдоожиженного слоя. Была предложена двухзонная мо-
дель газовой фазы, учитывающая рабочие характеристики системы. Общий объ-
ем И, занимаемый газовой фазой в реакторе, был представлен трубчатым реактором
объема И,, последовательно соединенным с проточным реактором смешения объе-
мом К2. Объемы зон связаны формулами
= (ю-14)
^2 = (1-а)К. (10.15)
Доля газа выражается как
е =a/g(K, + a/s)-'. (10.16)
Газ в зоне 1 (У ) течет в жидкой фазе, и для индикаторного газа предлагается сле-
дующая гомогенная модель с продольной дисперсией:
dt J
d2C т дС„т
= D ------f--U —g—
ax'g dz1 g dz
(10.17)
По предположению, зона 2 (Г2) ведет себя как проточный реактор смешения
со следующим уравнением баланса масс индикаторного газа:
Начальные и граничные условия системы имеют следующий вид:
1 = 0,С =С =0;
’ gT g ’
I ЗС„Г
(10.19)
(10.20)
z=0
434
Часть 3. Моделирование каталитических процессов
V dCT D (дС >
-4Ц1-ар)—^- =——
Qg dt иД dz k-L
(10.21)
где С — концентрация индикаторного газа; С ТЕ — концентрация индикаторного
газа на выходе из зоны 2.
Чтобы найти распределение времени пребывания в реакторе с псевдоожижен-
ным слоем, были проведены эксперименты в инертных условиях. Роль твердой
фазы играл катализатор на гамма-алюмооксидной подложке, газовой фазы — воз-
дух, а жидкой — вода. Поток воды, поднимаясь через слой однонаправленно с пото-
ком воздуха, поддерживал катализатор во взвешенном состоянии. Результаты имита-
ции динамического поведения индикаторного газа показаны на рис. 10.11. Решение
системы уравнений (10.18)—(10.21) позволило найти гидродинамические параметры:
долю газа и число Пекле газовой фазы. Из результатов расчета авторы заключили, что
при работе реактора в гомогенном режиме с низкой линейной скоростью газа доля
газа и число Пекле газовой фазы слабо влияют на трехфазную систему. При увеличе-
нии линейной скорости газа значения числа Пекле и доли газа возрастают. Ослабле-
ние эффектов смешения компенсируется повышенной стойкостью газожидкостной
эмульсии, объясняемой наличием твердых частиц катализатора.
Рис. 10.11. Моделирование динамического поведения индикаторного газа в трехфазном
РКС: (—) — экспериментальные значения на выходе; (•) — теоретические значения
на выходе; (---) — экспериментальные значения на входе [Sales, F.G. et al., Braz. J.
Chem. Eng., 22, 443, 2005]
Швейцер и Крессман [Schweitzer and Kressman, 2004] задались целью описать ги-
дродинамику РКС. Для этого они провели эксперименты в промышленных услови-
ях, используя в качестве радиоактивного индикатора изотоп аргона (‘"Аг). Рабочие
условия были следующими: давление 10-20 МПа, температура 380-430 °C, кратность
циркуляции 10-20 л/л, кратность водорода к сырью 650 л/л, объемная скорость
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
435
подачи сырья 0,74 ч_|. Была предложена работоспособная модель реактора, описы-
вающая согласно показаниям датчиков радиоактивности картину потоков жидкости
и газа, а также массоперенос в жидкой фазе. При расчетах равновесия 4|Аг в газовой
и жидкой фазах коэффициент распределения (К) оценивался по уравнениям состоя-
ния Редлиха—Квонга—Соава и Пенга—Робинсона.
Гидродинамическая модель разбивает реактор на две зоны: каталитическую и бар-
ботажную. Течение жидкости в обеих зонах представляется моделью дисперсного по-
тока. Дисперсия происходит между зоной барботажа и кипящим слоем. Газ и возвра-
щаемая жидкость движутся в пробковом режиме. Учитывается также массоперенос
аргона между газовой и жидкой фазами.
Балансы масс аргона в жидкой фазе в кипящем слое, барботажной зоне и рецир-
куляционной линии представляются уравнениями (10.22)—(10.24).
Кипящий слой:
= (10.22)
dz 1 dz Н J dt
Барботажная зона:
г; С Аг тт дСм л. (с
<|0-23>
Рециркуляционная линия:
Доля жидкости, а также линейные скорости жидкости и газа в каждой зоне реак-
тора принимаются постоянными.
Балансы масс аргона в газовой фазе в кипящем слое и барботажной зоне представ-
ляются выражениями (10.25) и (10.26).
Кипящий слой:
-UgS— + k,aRT ——[с^--^-1 = ^ (1025)
81 dz 1 egi ( * dt
Барботажная зона:
тт ЭРдг । j 4RT^~£s:2)(r ^ArY.^Ar
-Ug2^- + kiARJ---- CAr_77~ (10.26
& £g2 h^j dt
В области рабочих условий коэффициент Генри аргона (TfJ постоянен и равен
0,015 МПа • м3/моль. Гидродинамическая модель содержит всего пять расчетных па-
раметров: коэффициенты продольной дисперсии жидкой фазы в трех- и двухфазных
зонах Daxl и Dax2, доли газа в трех- и двухфазных зонах е , и е 2 и коэффициент массопе-
реноса из газовой фазы в жидкую кр. Оптимизированные значения этих параметров
436 Часть 3. Моделирование каталитических процессов
представлены в табл. 10.9. Швейцер и Крессман [Schweitzer and Kressman, 2004] при-
вели формулы для их расчета. На рис. 10.12 показаны две кинетические схемы для
описания гидрокрекинга и термического крекинга. Эксперименты по определению
кинетических параметров проводились на канадской тяжелой нефти. По кривым ис-
тинных температур кипения были определены четыре псевдокомпонента, основные
свойства которых перечислены в табл. 10.10.
Таблица 10.9
Расчетные гидродинамические параметры РКС при переработке канадской тяжелой нефти
Параметр Оптимизированное значение Доверительная вероятность Ширина 95%-ного до- верительного интервала /-Критерий
Еч 9,3960-10-1 8,9330-10 2 8,8590-10-' 40,6
1,1833-IO’2 2,8384-IO'3 2,6504 -10-' 1,6
Z) ,,m2/c 2,5979 - IO”3 2,2967- JO’1 2,8990-10-' 17,3
D „ m2/c 1,4184-IO-4 4,4459-10-' 2,3921- IO"3 2,9
Лд, c_| 3,9842- IO'2 3,8003-10-' 4,1681- 10-' 43,3
Источник: [Schweitzer, J.-M. and Kressmann, S., Chem. Eng. Sci., 59, 5637, 2004].
Каталитический крекинг
Термический крекинг
Рис. 10.12. Кинетические схемы реакций, описывающие гидрокрекинг и термический
крекинг [Schweitzer, J.-M. and Kressmann, S., Chem. Eng. Sci., 59, 5637, 2004]
Таблица 10.10
Характеристики псевдокомпоиентов, канадская тяжелая нефть
Псевдокомпонент Интервал кипения, °C Молярная масса, г/моль Плотность, кг/м3 Массовая доля
А > 524 903 1073 64,78
В 343-524 393 975 31,18
С 177-343 210 888 4,04
D 80-177 113 799 0
Источник: [Schweitzer, J.-M., Kressmann, S., Chem. Eng. Sci,. 59, 5637, 2004].
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
437
Уравнения кинетики каталитического и термического крекинга (уравне-
ния (10.27) и (10.28)) анализировались при следующих предположениях: скорость
реакции отвечает степенному закону; стехиометрический коэффициент водорода
в суммарной каталитической реакции равен 3; тяжелые фракции могут только пре-
вращаться в легкие; порядки всех реакций одинаковы; реакции гидродеоксигена-
ции (ГДОк), ГДО, ГДА и ГДМ не учитываются. Оптимизированные безразмерные
Значения кинетических констант каталитического процесса в рабочих условиях
равнялись ксАВ = 8 • 10s, кАС = k\D =kcCD = 2 10s, квс = kcBD = 3- 10s. Энергия активации
составила 200 кДж/моль.
Уравнение кинетики каталитического крекинга:
rc =kcCCas
(10.27)
Уравнение кинетики термического крекинга:
(10.28)
где более тяжелый псевдокомпонент/превращается в более легкий псевдокомпоненту.
После подключения кинетической модели к гидродинамической можно написать
следующие уравнения баланса масс z-ro псевдокомпонента и водорода (z = А, В, С,
D, Н2) для каждой зоны:
Трехфазная зона:
( s,H, УдС.. д2С.. dC,,
/ + а-е^-еДЯГ]~дГ ~ dz2 U/i ~дГ1
+ г< РА _±г<----------(10 29)
l-£gl-Es ' (l-egl -es)RT dz
Двухфазная зона:
( s7H. }дС.. d2C,, dC..
(1-еАЯГ dt ' dz2 dz
\ v е2' 7
д(Це2Н^.)
' (1-ек2)/?Г dz
(10.30)
На рис. 10.13 сравниваются расчетные и экспериментальные значения степени
превращения и выходы псевдокомпонентов. Наблюдается хорошее согласие между
теоретическими и экспериментальными результатами. Модель проверялась без учета
термического крекинга. Постоянство парциальных давлений после трехфазной зоны
на рис. 10.14 объясняется именно этим обстоятельством.
Предложенная модель, хотя и не учитывает термический крекинг, дает хорошее
согласие с экспериментальными данными. Тем не менее, нужны экспериментальные
данные в более широкой области рабочих условий, чтобы построить более точную
кинетическую модель.
438
Часть 3. Моделирование каталитических процессов
Экспериментальные значения выхода
псевдокомпонентов и степени превращения
Рис. 10.13. Сравнение модельных и экспериментальных результатов (степень превраще-
ния и выходы псевдокомпонентов): (•) — степень превращения; () — выход А; (л) — вы-
ход В; (х) — выход С; (о) — выход D [Schweitzer, J.-M. and Kressmann, S., Chem. Eng. Sci.,
59, 5637, 2004]
0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,0
Координата по высоте реактора, м
Рис. 10.14. Распределение парциальных давлений в РКС: (—) — А; (—) — В; (•• •) — С;
~ D; ( ) — Н2 [Schweitzer, J.-M. and Kressmann, S., Chem. Eng. Sci., 59, 5637, 2004]
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
439
Экклз (Eccles, 1993) моделировал кинетику гидропереработки остатков в РКС сте-
пенным законом, принимая, что в реакторе происходит обратное смешивание в жид-
кой фазе. Модель состояла из единственного уравнения для расчета фактора частоты^
и включала степень превращения и все переменные, не зависящие от реактора:
x/WHSV) =Ae*p(-EA/RT)C“C^, (10.31)
или
А = xA(WHSV)exp(-EA/RT)C7aC£, (10.32)
где WHSV— удельная массовая скорость подачи сырья; А — массовая доля относи-
тельно массы жидкости в реакторе непревращенного остатка или гудрона либо одной
из их составляющих — коксового остатка, остаточной серы, ванадия или никеля, (Ре-
агентом В является водород.)
Кинетическая модель применялась для слежения за степенью превращения остат-
ка на промышленной установке.
Ежесуточные расчеты значений фактора частоты, должным образом учитываю-
щие любые изменения скорости добавления катализатора и подачи сырья, дают кос-
венную оценку текущей эффективности реактора. Изменения расчетного значения,
зафиксированные после установления нормальной динамики (занимающего не-
сколько суток), могут свидетельствовать о таких проблемах в реакторе, как образова-
ние отложений кокса при нарушениях нормального режима работы или увеличение
доли газа из-за механического отказа внутреннего оборудования реактора либо не-
предвиденного изменения реологических свойств жидкой фазы.
Расчет фактора частоты — полезный инструмент контроля, однако требующий
тщательного измерения рабочих параметров. Из рис. 10.15 можно видеть, что разброс
данных, если они неполны, может оказаться значительным, что затрудняет выявле-
ние нормальной динамики. Модель этого типа была проверена на промышленных
данных; тем не менее, рекомендуется проверить ее на экспериментальных данных
лабораторной установки, чтобы убедиться в корректности принятых значений пара-
метров модели и улучшить даваемые ею результаты.
10.5. МОДЕЛИРОВАНИЕ РЕАКТОРОВ
С СУСПЕНЗИОННЫМ СЛОЕМ
Основой для последующей разработки РКС как аппарата для превращения тяжелой
нефти [Eccles, 1993] явилась технология превращения в фазе суспензии, применяв-
шаяся в Германии в годы Второй мировой войны для гидрирования угля и камен-
ноугольного дегтя (процесс Н-СоаГ) [MacArthur, 1993]. Главное различие между РКС
и суспензионным реактором состоит в том, что в первом частицы катализатора до-
статочно крупны, чтобы рассматривать их как отдельную фазу (твердую), тогда как
440
Часть 3. Моделирование каталитических процессов
Время работы, сут
Рис. 10.15. Слежение за степенью превращения остатка в промышленном РКС с помо-
щью модели косвенной оценки [Eccles, R.M., Fuel Process. Technol., 35, 21-38, 1993]
во втором они малы и считаются частью жидкой фазы, что изменяет динамику пото-
ка. Математические подходы к моделированию обоих реакторов одинаковы; един-
ственная разница заключается в том, что в РКС необходимо учитывать как эпюру
концентрации твердой фазы, так и скорость оседания частиц катализатора, а в су-
спензионном реакторе концентрация твердой фазы принимается постоянной и ско-
рость оседания частиц во внимание не принимается. Ниже перечисляются некото-
рые интересные работы, посвященные математическому моделированию реакторов
с суспензионным слоем (РСС).
Састри и соавторы [Sastri et al., 1983] смоделировали трехфазную некаталитиче-
скую, но химически активную систему для получения промышленного концентрата
гидросульфита цинка (ZnS2O4) в РСС. Ими были предложены три разные математи-
ческие модели режимов реактора — вытеснения, продольной диффузии и идеально-
го смешения. Авторы сравнили численные решения моделей и пришли к выводу, что
согласие расчетных результатов с экспериментальными данными было хорошим для
модели вытеснения с продольной дисперсией (диффузией), приемлемым для модели
вытеснения и неудовлетворительным для модели идеального смешения.
Васко де Толедо и соавторы [Vasco de Toledo et al., 2001] разработали динамические
модели каталитических РСС. Были предложены уравнения баланса масс и теплоты,
а также охлаждающей среды для получения 2-метилциклогексанола гидрированием
ортокрезола на никелевом катализаторе на кремнеоксидном носителе. Сообщалось
о двух гетерогенных моделях. Одна из них учитывала внешние и внутренние сопро-
тивления массо- и теплопереносу в частицы катализатора (динамическая модель I),
другая — лишь внешние сопротивления (динамическая модель II).
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
441
Численное моделирование позволило воспроизвести динамическое поведение
реактора, прежде всего тепловой баланс. Обе модели, несмотря на различия между
ними, почти одинаково воспроизводили динамику температур реактора и охлажда-
ющей среды. Что до выбора той или иной модели, авторы рекомендуют следующее:
если коэффициенты внутреннего тепло- и массопереноса в частицах катализатора
достаточно велики, динамическое поведение реактора лучше представляет модель I;
в случае затруднений в измерении этих параметров следует выбирать модель II. Для
проектных и модельных исследований, не ограниченных временем проведения
и сложностью расчетов, более надежна модель I; если же эти факторы являются огра-
ничивающими, лучшим вариантом будет модель II.
Парулекар и Шах [Parulekar and Shah, 1980] разработали модель гидрокрекин-
га в трехфазном суспензионном реакторе псевдоожиженного слоя в установившем-
ся режиме. Модель опирается на кинетику, отвечающую степенному закону, и может
применяться для расчета таких процессов, как ожижение и ГДО угля. Предполагает-
ся, что приведенные скорости газа и жидкости зависят от скоростей абсорбции реа-
гентов и десорбции продуктов из катализатора. Для расчета изменения приведенных
скоростей в баланс каждой фазы было введено дополнительное уравнение. Модель
можно применять в широких интервалах размеров частиц и содержания твердой
фазы, но при больших значениях этих параметров (или при изменении линейных
скоростей, а также долей жидкости и газа в осевом направлении) ее применимость
ограничена.
В рамках модели распределения твердой фазы была предложена модель для расче-
та скорости оседания твердых частиц в затрудненных условиях, применимая в иссле-
довавшихся интервалах размеров частиц и содержания твердой фазы. Предложенная
модель представляет собой модификацию закона Фика, которая учитывает скорость
оседания частиц. В центре исследования был анализ влияния главных параметров
процессов гидрокрекинга: давления, температуры, линейных скоростей газа и жид-
кости, высоты и диаметра реактора, массовой доли твердых частиц в сырьевой су-
спензии и размера частиц катализатора. По результатам имитации можно видеть, что
увеличение давления, температуры или высоты реактора приводит к повышению вы-
хода продукта, тогда как при увеличении значений остальных анализируемых пара-
метров выход сначала достигает максимума, а затем существенно снижается. Таким
образом, существует оптимальный набор рабочих условий в РСС гидрокрекинга.
Вярна и Салми [Warna and Salmi, 1996] разработали динамические модели трехфаз-
ных реакторов с суспензионным и орошаемым слоями, работающих в неизотермиче-
ских условиях. Фазы газа, жидкости и катализатора описывались параболическими
уравнениями в частных производных относительно переменных, выражающих объ-
емные скорости однонаправленных и противоточных потоков. Для моделирования
РСС была выбрана реакция окисления SO2.
Эта модель РСС не учитывала скорости оседания твердых частиц, поэтому облада-
ла расчетным поведением, характерным для РКС. По модели проверялось лишь рас-
пределение SO2 в газовой фазе. Хотя надежность модели не анализировалась с учетом
влияния других параметров (в том числе длины и диаметра реактора, размеров частиц
и приведенных скоростей газа и жидкости), она может оказаться полезной.
442 Часть 3. Моделирование каталитических процессов
10.6. ИССЛЕДОВАНИЕ КИНЕТИКИ ГИДРОКРЕКИНГА
ТЯЖЕЛОЙ НЕФТИ В ПРОТОЧНОМ РЕАКТОРЕ СМЕШЕНИЯ
Чтобы удовлетворить растущий спрос на легкие продукты (в том числе бензин, ке-
росин, реактивное и дизельное топлива), нефтеперерабатывающие компании вы-
нуждены постепенно переходить на переработку тяжелой и особо тяжелой нефти
с высоким содержанием серы, азота, асфальтенов и металлов. Превращение сырья
такого типа — трудная задача, которая решается разработкой новых процессов и ка-
тализаторов. Параллельно ведутся кинетические исследования с использованием
различных катализаторов, типов сырья, реакторов и условий реакций. В большин-
стве случаев исследуется установившийся процесс, и лишь некоторые исследования
рассматривают начальный период деактивации катализатора. В литературе описаны
различные подходы к моделированию кинетики гидрокрекинга нефтяных фракций.
Это представление: 1) дискретными псевдокомпонентами (агрегирование); 2) непре-
рывными смесями (непрерывное агрегирование); 3) единичными событиями [Qader
and Hill, 1969; Stangeland, 1974; Yui and Sanford, 1989; Martens and Marin, 2001; Ba-
sak et al., 2004; Botchwey et al., 2004; Kumar and Froment, 2007; Sadighi et al., 2010].
Однако лишь немногие из указанных подходов затрагивают гидрокрекинг тяжелой
нефти. Деактивацией катализатора обычно пренебрегают, поскольку сырьем в боль-
шинстве случаев служит вакуумный газойль.
Бенито и Мартинес [Benito and Martinez, 1996], моделируя крекинг тяжелой фрак-
ции с получением легких дистиллятов, придерживались простой кинетики первого по-
рядка. Сырьем служил остаток гидрогенизации угля с высоким содержанием асфаль-
тенов. Гидрокрекинг осуществлялся в трубчатом реакторе на никель-молибденовом
(NiO-MoO3) катализаторе на алюмооксидном носителе. Исследования проводились
в широкой области температур (425—475 °C) при постоянном давлении 153 кг/см2.
Авторы обнаружили, что повышение жесткости условий реакций увеличивает выход
средних дистиллятов и газов, но заметного роста содержания кокса не отмечается.
Аяссе и соавторы [Ayasse et al., 1997] разработали семикомпонентную модель гидро-
крекинга битумов Атабаски в проточном реакторе смешения на никель-молибдено-
вом катализаторе на у-алюмооксидном носителе при температуре 430 °C и давлении
140 кг/см2. Гидрокрекинг моделировался с минимально необходимым числом незави-
симых реакций путем введения концепции стехиометрических коэффициентов для
представления избирательности к продуктам. Деактивация катализатора учитывалась
типичной функцией экспоненциального убывания. Модели могли предсказывать
выход продуктов при умеренно жестких условиях на основе данных многократных
экспериментов (в том числе эксперимента над сырьем с повышенным содержанием
вакуумного остатка). Санчес и соавторы [Sanchez et al., 2005] предложили пятиком-
понентную модель гидрокрекинга тяжелой нефти. Кинетические параметры рассчи-
тывались по данным гидрокрекинга тяжелой нефти «Майя» с высоким содержани-
ем серы (3,51 %масс.) и металлов (345 мг/кг). Эксперименты проводились в реакторе
с неподвижным слоем (РНС) и нисходящим потоком сырья на промышленном ни-
кель-молибденовом катализаторе при температуре 380-420 °C, LHSV= 0,33-1,5 ч-1,
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
443
постоянной кратности водорода к сырью, равной 5000 фут3/баррель и постоянном
давлении 70 кг/см2. Предложенная модель давала хорошее согласие с эксперимен-
тальными данными: средняя погрешность была меньше 5 %. Лориа и соавторы [Lo-
ria et al., 2011] применили пятикомпонентную кинетическую модель Санчеса к ги-
дропереработке битумов Атабаски в трубчатом реакторе на ультрадисперсном
никель-молибден-вольфрамовом катализаторе. Температура реакции и удельная
скорость подачи сырья составляли соответственно 320—380 °C и 0,09—0,2 ч"1 при по-
стоянном давлении 28 кг/см2 и постоянной кратности водорода к сырью 3600 фут3/
баррель. Средняя погрешность прогнозирования выхода каждого псевдокомпонента
не превышала 7 %.
Недавно был опубликован обзор других подходов к моделированию кинетики ги-
дрокрекинга тяжелой нефти [Ancheyta et al., 2005]. Из обзора следует, что основанные
на агрегировании модели будут по-прежнему использоваться в исследованиях кине-
тики гидрокрекинга тяжелой нефти, так как новые, более детальные и сложные под-
ходы не решают всех проблем ввиду сложности нефтяного сырья. С другой стороны,
для надлежащего проектирования реактора и самого процесса важны кинетические
исследования, учитывающие деактивацию катализатора. Основными причинами де-
активации считаются накопление углеродистых и металлических отложений и изме-
нение структуры компонентов катализатора. В течение нескольких первых часов ра-
боты слоя очень интенсивно отлагается кокс, после чего деактивация коксом быстро
достигает квазистационарного уровня, а металлы сырья (главным образом никель
и ванадий) преобразуются в сульфиды, которые отлагаются в порах и необратимо
деактивируют катализатор [Forzatti and Lietti, 1999; Ancheyta et al., 2003; Meng et al.,
2007; Froment, 2008].
Существует мнение, что если убывание активности обусловлено образованием
кокса (а последнее зависит от концентраций реагирующих веществ), то активность
катализатора не может быть простой функцией времени, т. е. деактивацию ката-
лизатора правильнее было бы описывать функцией концентрации агента, кото-
рый действительно отвечает за деактивацию [Meng et al., 2007; Froment, 2008; Fro-
ment et al., 2010]. Однако при гидрокрекинге тяжелой нефти потерю активности
катализатора вызывает отложение не только кокса, но и металлов, которое про-
исходит по другим, сложным механизмам. Это обстоятельство, наряду с необходи-
мостью проведения ряда экспериментов (подготовка установки, загрузка и акти-
вация катализатора, корректирование условий реакций, отбор жидких продуктов
для описания, выгрузка катализатора) для получения различных образцов деакти-
вированного катализатора и дальнейшей их характеризации, может потребовать
больших материальных и временных затрат. В данном случае такой подход нецеле-
сообразен. Именно по этой причине, т. е. для упрощения получения данных, пред-
почтение оказывается выражению деактивации катализатора в виде функции вре-
мени работы слоя.
Вдобавок для выделения правильного вида функции деактивации необходи-
мы более глубокие знания физики и химии процесса. Впрочем, если условия реак-
ций (давление, температура, кратность водорода к сырью, приведенная скорость)
444
Часть 3. Моделирование каталитических процессов
в лаборатории аналогичны условиям полупромышленного или промышленного мас-
штаба, то представление скорости деактивации функцией времени можно считать
подходящим для моделирования кинетики. Однако на другие рабочие условия такую
функцию распространять нельзя, поэтому при изменении рабочих условий должны
быть предложены новые значения параметров функции деактивации.
В данном разделе для представления реакций, происходящих при гидрокрекин-
ге тяжелой нефти, применяются пятикомпонентная модель кинетики и зависимая
от времени неизбирательная функция деактивации.
10.6.1. Экспериментальная часть
10.6.1.1. Оборудование
Все эксперименты проводились в лабораторном проточном реакторе смешения с ка-
талитическим пакетом (CSTBR), имитирующем РКС (рис. 10.16), Тяжелое нефтя-
ное сырье подогревалось, смешивалось с водородом и пропускалось через реактор.
Температура измерялась тремя термопарами, расположенными вверху, в середине
и внизу каталитического пакета. Жидкость и газ разделялись в сепараторах низкого
и высокого давления. С низа сепараторов отбирались жидкие продукты. Газы, отби-
раемые с верха, направлялись для анализа в хроматограф. Установка имела распреде-
ленную систему управления через ПИД-контроллеры и компьютеризованную систе-
му дистанционного телеуправления.
Проточный
реактор
смешения
с каталитическим
пакетом
Низкотемпе-
ратурный
сепаратор
низкого
давления
Жидкие продукты
Рис. 10.16. Проточный реактор смешения с каталитическим пакетом
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
445
10.6.1.2. Загрузка и активация катализатора
Основные свойства применявшегося катализатора перечислены в табл. 10.11. В па-
кет загружали 100 мл катализатора, который активировали сульфидированием га-
зойлем, содержащим 1,6 %масс. диметилдисульфида, в следующих условиях: ско-
рость подачи газойля 2 мл в час на 1 мл катализатора, давление 28 кг/см2, кратность
водорода к сырью 2000 ст. фут3/баррель, 3 ч при температуре 260 °C и 12 ч при тем-
пературе 320 °C.
Таблица 10.11
Свойства катализатора
Свойства Значение
Номинальный размер, дюймы 1/18
NiO/Ni, % по сухой массе 0,73/0,58
МоО3/Мо, % по сухой массе 3,27/2,18
Физические свойства:
средняя насыпная плотность, г/мл 0,45
плотность в уплотненном состоянии, г/мл 0,55
удельная поверхность, м2/г 197,2
удельный поровый объем, мл/г 0,85
средний диаметр пор, А 172,6
Распределение размеров пор, %об.:
< 50 А 2,32
50-100 А 14,86
100-200 А 49,44
200-500 А 30,34
500-1000 А 2,26
>1000 А 0,78
10.6.1.3. Эксперименты и анализ продуктов
Сырьем служил остаток атмосферной перегонки (312 °С+) особо тяжелой (плот-
ность 13 ‘APT) нефти. Основные свойства сырья представлены в табл. 10.12. Все экс-
перименты проводились при постоянном давлении 100 кг/см2 и постоянной кратно-
сти водорода к сырью 5000 ст. фут3/баррель. Объемная скорость подачи сырья (LSHV)
в зависимости от температуры составляла: при 380 и 400 °C — 0,5, 0,75, 1,0 и 1,25 ч_|;
при 410 °C — 0,75 и 1,25 ч_|; при 420 °C — 0,5 и 1,0 ч_|. Сырье и продукты анализиро-
вали имитированной дистилляцией по методу ASTM £>7169.
446
Часть 3. Моделирование каталитических процессов
Таблица 10.12
Свойства сырья
Свойства Значение
Относительная плотность, 60/60 °F 1,028
Плотность, "API 6,15
Элементный анализ, %масс.:
углерод 82,67
водород 10,07
сера 6,17
азот 1,09
Коксуемость по Конрадсону, %масс. 20,83
Содержание металлов, мг/кг:
Ni 106
V 558
Кинематическая вязкость при 130 °C, сСт 330,12
Разгонка по А5ТЛ/2)7169, °C:
НК 206,0
выкипает 10 % 344,7
» 30% 462,3
» 50% 568,3
» 70% 644,7
» 90% 700,9
кк 744,6
10.6.2. Анализ результатов
10.6.2.1. Ограничения массопереноса
Чтобы убедиться в том, что экспериментальные данные представляли истинную ки-
нетику, на катализаторе с частицами промышленных размеров было проверено от-
сутствие градиентов внешнего массопереноса. Условия эксперимента были сле-
дующими: температура 380 °C, давление 100 кг/см2, кратность водорода к сырью
5000 ст. фут’/баррель, скорость подачи сырья 0,5 мл за час на 1 мл катализатора, ча-
стота вращения вала мешалки от 750 до 1250 об/мин. Скорость вращения вала мешал-
ки оптимизировали по степени превращения серы, которую определяли каждые 10 ч.
Результаты представлены на рис. 10.17. Заметен характерный начальный период де-
активации, после которого степень превращения серы стабилизируется. Стабилиза-
ция по изменении скорости вращения происходит в течение 40 ч. При 750 об/мин
степень превращения составляла в среднем 44,5 %. Вслед за ускорением вращения
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
447
до 1250 об/мин степень превращения тоже возросла до 45,9 %. После этого скорость
вращения вала мешалки была установлена равной 1000 об/мин и наблюдалась при-
мерно такая же (45,8 %) степень превращения, как при 1250 об/мин. На основании
этого факта было принято, что внешние сопротивления массопереносу существенно
снижаются при 1000 об/мин.
Рис. 10.17. Изменение степени превращения серы в зависимости
от частоты вращения вала мешалки
Так как все эксперименты проводились на катализаторе с частицами промыш-
ленных размеров, для расчета внутричастичных сопротивлений массопереносу при
каждой паре значений температуры и приведенной скорости в кинетическое выра-
жение был включен коэффициент эффективности.
10.6.2.2. Моделирование кинетики
Использованная кинетическая схема показана на рис. 6.15 (см. главу 6). Она вклю-
чает в себя пять агрегатов (псевдокомпонентов) и была ранее описана в литерату-
ре [Sanchez et al., 2005]. Агрегатами являются газы, бензиновая фракция (темпера-
тура начала кипения 204 °C), средние дистилляты (204-343 °C), вакуумный газойль
(343—538 °C) и непревращенный вакуумный остаток (538 °С+). Как говорилось
выше, кинетика при проведении экспериментов искажалась ввиду наличия вну-
тренних диффузионных ограничений, из-за чего необходимо вводить коэффициент
эффективности (ц).
448
Часть 3. Моделирование каталитических процессов
Скорость деактивации учитывалась следующей функцией:
1
(1 + V)'
(10.33)
Кинетическое выражение (г) для каждой реакции формулируется в виде функции
от концентраций продуктов (у.), кинетической константы и коэффициента эффек-
тивности. Концентрации продуктов определялись по балансам масс в лабораторной
установке и имитированным кривым разгонки. Порядок реакции гидрокрекинга ва-
куумного остатка был принят равным двум, остальных реакций — единице [Sanchez
and Ancheyta, 2007]. Исходя из этих соображений, скорости реакций предлагаемой
модели записываются в следующем виде:
Вакуумный остаток (ВО):
''во =-'PWbo2’ (10-34)
где к0 = к{ + к2 + к3 + к*.
Вакуумный газойль (ВГ):
гш. = (pnt^^ao2 - <k5 + k6 + (10.35)
Средние дистилляты (СД):
гсд = ФП[^во2 + ^вг - <*8 + М^сд]- (10-36)
Бензиновая фракция (БФ):
ГБФ = ФП[^ВО2 + ^ВГ + ^СД - Мвф]’ (10-37)
Газы (Г):
Гг = <рт)[^ВО2 + ^ВГ + ^СД + МбфЬ <10-38)
Концентрации на входе и выходе реактора вычислялись с учетом следующего
уравнения:
g,
у> = ~
ST
(10.39)
Кинетическую модель была подключена к модели реактора. При каждом значе-
нии температуры по набору кинетических констант концентрации продуктов вычис-
лялись согласно следующему уравнению:
_ Л,о - л
"Ч (-^)
(10.40)
Применялся описанный в литературе [Ancheyta and Sotelo, 2000] метод последо-
вательного определения значений параметров. Для упрощения расчета параметров
Глава 10, Моделирование реакторов с кипящим или суспензионным слоем
449
исходная кинетическая модель сокращалась до двух-, трех- и четырехкомпонентных
моделей, как показано на рис. 10.18. При таком подходе можно получить значения
некоторых параметров, остающиеся постоянными при решении пятикомпонентной
модели, что сокращает число одновременно вычисляемых параметров.
2L-M1 4L-M1
к к]
Остаток ------°-----► Продукты Остаток ------------------► БГ
3L-M1
Рис. 10.18. Двух-, трех- и четырехкомпонентные кинетические модели:
БФ — бензиновая фракция; ВГ — вакуумный газойль. Обозначения: к— константы скоро-
стей реакций (последние обозначены стрелками на схемах); 2L, 3L, 4L — соответственно
двух-, трех- и четырехгрупповая модели; М — номер модели реакции
Через минимизацию целевой функции, равной сумме квадратов разностей между
экспериментальными и расчетными значениями концентраций продуктов, опреде-
лялась наилучшая совокупность кинетических параметров и коэффициентов эффек-
тивности. Целевая функция решалась по критерию наименьших квадратов методом
нелинейной регрессии по алгоритму Марквардта.
450
Часть 3. Моделирование каталитических процессов
В табл. 10.13 приведены найденные значения коэффициентов эффективности для
различных рабочих условий. Можно видеть, что с повышением температуры реак-
ции и коэффициент эффективности уменьшается. С ростом температуры кон-
станта скорости возрастает быстрее, чем коэффициент диффузии; лимитирующая
роль диффузии становится более выраженной, поэтому коэффициент эффективно-
сти уменьшается. Уменьшение коэффициента эффективности при увеличении LHSV
и постоянной температуре можно объяснить увеличением вязкости реакционной
смеси. Поскольку вязкость прямо связана с коэффициентом диффузии, ее увеличе-
ние ведет к уменьшению коэффициента эффективности.
Таблица 10.13
Коэффициенты эффективности
LHSV, ч-' ^ЗвО’С ТЪоО’С ТЪпгс ^42О*С
0,50 0,8118 0,6843 0,6470
0,75 0,7646 0,6114 0,5644
1,00 0,7061 0,5403 0,5293
1,25 0,6649 0,4621 0,4077
Значения кинетических параметров, констант деактивации, а также энергий ак-
тивации перечислены в табл. 10.14. На рис. 10.19 показаны графики Аррениуса для
всех кинетических констант. В первых четырех строчках табл. 10.14 приведены рас-
четные параметры суммарной реакции гидрокрекинга вакуумного остатка (вакуум-
ный остаток —> продукты гидрокрекинга). Порядок реакции, как и ожидалось, был
равен 2, что согласуется с опубликованными данными [Sanchez and Ancheyta, 2007].
Порядок деактивации катализатора был близок к 0,22. В расчетах параметров других
псевдокомпонентов это значение оставалось постоянным. Что любопытно, значения
энергий активации и деактивации были близки (50 и 53 ккал/моль соответственно);
в принципе, это означает отсутствие преобладания процесса деактивации над сум-
марной реакцией гидрокрекинга.
Таблица 10.14
Кинетические параметры
Па- ра- метр Маршрут реакции Температура ккал/ моль
380 °C 400 °C 410 °C 420 °C
Вакуумный остаток —> продукты 14,349 49,841 84,237 131,297 50,22
ПН 2,000 2,000 2,000 2,000
3,799 15,117 21,917 42,473 53,32
т 0,222 0,222 0,222 0,222
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
451
Таблица 10.14, окончание
Па- ра- метр Маршрут реакции Температура ккал/ моль
380 °C 400 °C 410 °C 420 °C
Вакуумный остаток —> вакуумный газойль 7,958 25,733 42,215 62,613 46,92
к2 Вакуумный остаток средние дистилляты 4,037 15,097 26,016 42,672 53,34
кз Вакуумный остаток —> бензиновая фракция 1,571 6,052 10,964 16,803 54,10
Вакуумный остаток -* газы 0,783 2,960 5,042 9,209 55,21
^5 Вакуумный газойль средние дистилляты 0,031 0,098 0,120 0,175 38,94
Вакуумный газойль -> бензиновая фракция 0,004 0,013 0,026 0,035 51,17
^7 Вакуумный газойль -> газы 0,003 0,005 0,016 0,024 52,30
^8 Средние дистилляты бензиновая фракция 0,051 0,148 0,200 0,360 42,72
Средние дистилляты -> газы 0,0009 0,0026 0,0036 0,0072 44,89
Бензиновая фракция -> газы 0,054 0,239 0,626 0,978 67,21
Значения энергии активации, присущей отдельным маршрутам гидрокрекинга ва-
куумного остатка, составляли от 46 до 55 ккал/моль. Среднее значение (52 ккал/моль)
довольно близко к энергии активации суммарной реакции. Из этого следует, что ки-
нетика реакции при объединении различных углеводородных компонентов в единый
агрегат (продукт = вакуумный газойль + дистилляты + бензиновая фракция + газы)
схожа с кинетиками реакций отдельных псевдокомпонентов.
Меньшие значения энергий активации вакуумного остатка до вакуумного газой-
ля и вакуумного газойля до средних дистиллятов означают преобладание средних ди-
стиллятов над бензиновой фракцией и газами. Гидрокрекинг средних дистиллятов
до бензиновой фракции и газов происходит быстрее, чем другие реакции. Наименее
благоприятна реакция гидрокрекинга бензиновой фракции до газов.
Можно видеть, что реакция гидрокрекинга вакуумного остатка наиболее изби-
рательна к образованию вакуумного газойля; по избирательности реакции агре-
гаты располагаются в следующем порядке: вакуумный газойль > средние дистил-
ляты > бензиновая фракция > газы. Реакция гидрокрекинга вакуумного газойля
наиболее избирательна к средним дистиллятам; затем следуют бензиновая фракция
и газы. Образование средних дистиллятов происходит примерно в восемь и двенад-
цать раз быстрее, чем соответственно бензиновой фракции и газов, и при более низ-
кой температуре. Реакция гидрокрекинга средних дистиллятов более избирательна
к бензиновой фракции, чем к газам, причем бензиновая фракция образуется прибли-
зительно в 55 раз быстрее, чем газы. Все эти тенденции говорят о том, что реакции
гидрокрекинга протекают последовательно. Наибольший относительный выход газа
дает вакуумный остаток; меньший вклад дают соответственно вакуумный газойль,
средние дистилляты и бензиновая фракция,
452
Часть 3. Моделирование каталитических процессов
Рис. 10.19. Графики Аррениуса различных кинетических параметров
На рис. 10.20 сравниваются значения концентраций псевдокомпонентов
в продукте, полученные экспериментальным путем и расчетом по уравнени-
ям (10.1)—(10.8) при значениях к., приведенных в табл. 10.14. Можно видеть, что со-
став продукта предсказывается довольно точно. На рис. 10.21 показано случайное
распределение разностей между экспериментальными и расчетными значениями
концентраций; видно, что количества положительных и отрицательных отклоне-
ний примерно равны. В табл. 10.15 приведены значения длин отрезков, отсекаемых
на осях координат, и наклона (для линеаризованных соотношений на рис. 10.20),
чисел положительных и отрицательных разностей, а также максимальных абсо-
лютных величин относительных погрешностей. Можно видеть, что значения угла
наклона и длин отсекаемых отрезков близки соответственно к 1 и 0; это говорит
о хорошем согласии между экспериментальными и расчетными значениями кон-
центраций. Наибольшую относительную погрешность (4,8 %) дает расчет концен-
трации бензиновой фракции.
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
453
Экспериментальные значения концентраций (у,)
Рис. 10.20. Сравнение расчетных и экспериментальных значений концентраций агрегатов
в продукте: (+) — вакуумный остаток; (о) — вакуумный газойль; (Д) — средние дистилля-
ты; (□) — бензиновая фракция; (о) — газы
„ 0,008 ;
Е -I
01 I
х 0,006
С
S
2 0,004
ш
J) 0,002
з- 0,000
ГО -г,--У. ~ •'>, >•. J
5 -0,002 \ - ' - . - .
=г . ' ' .. ' '
X J-
О 1 -•
£ -0,004 j ’
х -I " '
х -0,006 I-
I !
о -0,008 1________,-----------------------------------------;________,________
0 20 40 60 80 100 120
Число точек данных
Рис 10 21. Отклонения концентраций псевдокомпонентов: (+) — вакуумный остаток;
__вакуумный газойль; (Д) — средние дистилляты; (□) — бензиновая фракция;
' ' (о) — газы
454
Часть 3. Моделирование каталитических процессов
Таблица 10.15
Статистический анализ
Агрегат Наклон Отсекаемый отрезок Отклонение (+) Отклонение (-) Максимальная погрешность, %
Вакуумный остаток 1,0001 -0,0038 61 57 4,24
Вакуумный газойль 0,9952 0,0021 57 61 1,68
Средние дистилляты 1,0017 0,0002 49 69 2,71
Бензиновая фракция 0,9994 -0,0002 72 46 4,79
Газы 0,9846 0,0013 54 64 1,32
10.6.3. Выводы
Результаты экспериментов, изложенные в предыдущем подразделе, можно свести
к следующему.
• В проточном реакторе смешения с пакетным катализатором в интервалах тем-
ператур от 380 до 420 °C и LHSV от 0,5 до 1,25 мл/мл получены данные для рас-
чета кинетических параметров пятикомпонентной модели гидрокрекинга тя-
желой нефти с учетом деактивации катализатора. Результаты гидрокрекинга
вакуумного остатка, вакуумного газойля и средних дистиллятов говорят о по-
вышенной избирательности реакции к более тяжелым псевдокомпонентам
в исследованном интервале температур.
• Легкие компоненты более чувствительны к изменениям температуры, так как
для них энергия активации выше, тогда как тяжелые компоненты легче подда-
ются гидрокрекингу. Как следствие, условия для реакции гидрокрекинга бен-
зиновой фракции до газов наименее благоприятны.
• Концентрации выбранных для модели псевдокомпонентов могут быть пред-
сказаны с погрешностью менее 5,0 %.
10.7. ЗАКЛЮЧИТЕЛЬНЫЕ ЗАМЕЧАНИЯ
Трудность математического моделирования РКС обусловлена главным образом
сложностью гидродинамики реакционной среды в реакторе. Реакторы с кипящим
слоем моделируется либо как простой трубчатый реактор, либо как трубчатый реак-
тор, соединенный последовательно с проточным реактором смешения с пакетным
катализатором. Предлагаются также ограниченные модели, основанные на косвен-
ной оценке эффективности реактора. Их ограниченность заключается в большой по-
грешности данных, используемых для выявления правильной тенденции, и в приме-
нимости лишь как средства контроля, а не прогнозирования.
При обратном масштабировании РКС необходимо убедиться в том, что гидроди-
намические характеристики идентичны, а доли фаз неизменны. Ключевую роль при
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
455
обратном масштабировании играют диаметр частиц катализатора, диаметр реактора,
а также приведенные скорости жидкости и газа. Для прямого масштабирования от-
лично подходят методы, основанные на геометрическом и динамическом подобии.
Задача значительно усложняется, если смешиваются частицы катализатора раз-
ных размеров и в слое присутствует каталитическая мелочь, образующаяся при ис-
тирании, поскольку в этом случае могут появиться проблемы контроля и стабилиза-
ции уровня слоя в РКС при заданной кратности сырья к газу. Возможны и серьезные
эксплуатационные проблемы, в том числе вынужденное увеличение скорости добав-
ления катализатора, увеличение расхода водорода, повышение температуры в слое
и перепада температур в реакторе.
Деактивация катализатора при гидрокрекинге тяжелого остатка в РКС вызыва-
ется главным образом отложением кокса в начальный период работы и закупоркой
пор вследствие диффузии в них соединений металлов. Накопление металлов в тече-
ние цикла изменяет насыпную плотность катализатора, от которой зависит скорость
жидкости, требуемая для разрыхления слоев. Эффект усиливается с повышением
температуры. Необходимо иметь в виду, что на деактивацию катализатора влияет
также кратность рециркуляции непревращенного сырья; чем она выше, тем больше
углеродистых веществ и металлов отлагается на внешнем слое частиц катализатора.
Одна из задач моделирования РКС — надежный расчет физических параметров.
Широко применяются эмпирические формулы, но для гидрокрекинга тяжелого сы-
рья в жестких условиях нет формул, на которые можно положиться. Довольно высоки
затраты на проведение экспериментов, необходимых для определения кинетических
параметров. Кроме того, применимость некоторых существующих аналитических
методов описания ограничена легкими фракциями нефти. Это говорит о необходи-
мости дополнительных исследований, позволяющих усовершенствовать методы ана-
лиза и методики проведения экспериментов до такой степени, которая позволила бы
точнее определять кинетические параметры реакции гидрокрекинга тяжелого сырья
и успешно моделировать РКС.
ОБОЗНАЧЕНИЯ
Латинские
А Фактор частоты
Ат Число Архимеда, безразмерная величина
Концентрация аргона, моль/м3
С Концентрация индикаторного газа, моль/м3 или моль/см3
CgT Концентрация индикаторного газа в жидкой фазе (зона 1), моль/м3 или
моль/см3
CgTE Концентрация индикаторного газа на выходе зоны 2, моль/м3 или
моль/см3
456
Часть 3. Моделирование каталитических процессов
С Т1 Концентрация индикаторного газа на входе трубчатого реактора, моль/м3
или моль/см3
СНг Концентрация водорода, моль/см3
С. Концентрация /-го псевдокомпонента, моль/м3 или моль/см3
С 0 Концентрация z-го псевдокомпонента на входе, моль/м3 или моль/см3
С Концентрация /-го псевдо компонента в жидкости, моль/м3
С Концентрация на поверхности катализатора, моль/м3
D Коэффициент продольной дисперсии, м2/ч
Dax! Коэффициент продольной дисперсии жидкости в трехфазной зоне, м2/с
D 2 Коэффициент продольной дисперсии жидкости в двухфазной зоне, м2/с
Daxg Коэффициент продольной дисперсии индикаторного газа, м2/с
Dc Диаметр реактора, м
d Диаметр частицы, м
Еа Энергия активации, Дж/моль
Е Число Этвёша, безразмерная величина
Fr Число Фруда, безразмерная величина
g Ускорение свободного падения, 9,8 м/с2
g Содержание /-й фракции в продукте, г
g.o Содержание /-Й фракции в сырье, г
gT Суммарный поток, поступающий в реактор, г
Н. Коэффициент Генри /-го компонента, МПа м3/моль
j Приведенная скорость дрейфа газа, м/с
№ Кинетическая константа в каталитическом процессе, безразмерная
величина
к'ц Кинетическая константа втермическом процессе, безразмерная величина
к:а Коэффициент массопереноса, м/с
kQ Отношение диаметра спутной струи к диаметру пузыря, безразмерная ве-
личина; константа скорости суммарной реакции гидрокрекинга вакуум-
ного остатка, ч-1
к, Константа скорости второго порядка в случае реакции гидрокрекинга
вакуумного остатка до вакуумного газойля, ч_|
к2 Константа скорости второго порядка в случае реакции гидрокрекинга
вакуумного остатка до средних дистиллятов, ч-1
ку Константа скорости второго порядка в случае реакции гидрокрекинга
вакуумного остатка до бензиновой фракции, ч_|
к* Константа скорости второго порядка в случае реакции гидрокрекинга
вакуумного остатка до газов, ч_|
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
457
к5 Константа скорости второго порядка в случае реакции гидрокрекинга
вакуумного газойля до средних дистиллятов, ч~'
к6 Константа скорости второго порядка в случае реакции гидрокрекинга
вакуумного газойля до бензиновой фракции, ч-1
Константа скорости второго порядка в случае реакции гидрокрекинга
вакуумного газойля до газов, ч-1
Лв Константа скорости второго порядка в случае реакции гидрокрекинга
средних дистиллятов до бензиновой фракции, ч*1
к9 Константа скорости второго порядка в случае реакции гидрокрекинга
средних дистиллятов до газов, ч_|
А|0 Константа скорости второго порядка в случае реакции гидрокрекинга
бензиновой фракции до газов, ч_|
клер Кинетические константы для трех- и четырехкомпонентных кинетиче-
ских моделей, ч_|
kd Константа скорости деактивации, Г1
L Высота реактора, м
Lp Длина частицы, м
М Число Мортона, безразмерная величина
т Порядок скорости деактивации
тТо Суммарный массовый поток, г/ч
п Индекс Ричардсона-Заки, безразмерная величина; порядок реакции ги-
дрокрекинга вакуумного остатка
Р Давление, Па
Р Парциальное давление аргона, Па
Q Объемная скорость потока, л/ч
R Универсальная газовая постоянная, Дж/моль • К
Re Число Рейнольдса, безразмерная величина
Г Скорость реакции в каталитическом процессе, моль/кг • с
Скорость реакции в термическом процессе, моль/м3 • с
г Скорость реакции бензиновой фракции, г/г • ч
г Скорость реакции вакуумного газойля, г/г - ч
ВГ
rgo Скорость реакции вакуумного остатка, г/г - ч
гг Скорость реакции газов, г/г - ч
г Скорость реакции средних дистиллятов, г/г • ч
Т Температура, К
t Время, с или ч
т Температура на поверхности катализатора, К
458
Часть 3. Моделирование каталитических процессов
и <4. и, и. и. к cat Приведенная скорость, м/с Скорость газа в трехфазной зоне, м/с Скорость газа в трехфазной зоне, м/с Экстраполированная приведенная скорость жидкости, м/с Приведенная скорость жидкости в трехфазной зоне, м/с Приведенная скорость жидкости в двухфазной зоне, м/с Линейная скорость рециркулирующей жидкости Общий объем, м3 Масса катализатора, г
WHSV Массовая скорость подачи сырья, ч
ХЛ •^БФ •^ВГ У ВО Уг Усд Z Молярная доля компонентах Концентрация бензиновой фракции Концентрация вакуумного газойля Концентрация вакуумного остатка Концентрация газа Концентрация средних дистиллятов Положение по высоте реактора, м
Греческие
а Порядок реакции для псевдокомпонентов
а g ос.. V 3 3, Зд Ар Объемная доля газа Стехиометрический коэффициент, безразмерная величина Порядок реакции для Н2 Соотношение плотностей, безразмерная величина Соотношение приведенных скоростей, безразмерная величина Выталкивающая сила (Др = р; — pg), кг/м3
е Доля, безразмерная величина
Е , gi е > & Л Доля газа в трехфазной зоне, безразмерная величина Доля газа в двухфазной зоне, безразмерная величина Коэффициент эффективности
м Вязкость, Па • с
р Плотность, кг/м3
о Коэффициент поверхностного натяжения, Н/м
<Р Активность катализатора
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
459
Нижние индексы
cat Катализатор
/ Жидкость
s Твердая фаза
БФ Бензиновая фракция
ВГ Вакуумный газойль
ВО Вакуумный остаток
Г Газ
СД Средние дистилляты
ЛИТЕРАТУРА
Al-Dalama, К., Stanislaus, А. 2006. Comparison between deactivation pattern of catalyst in fixed-
bed and ebullating-bed residue hydroprocessing units. Chem. Eng. J. 120:33—42.
Ancheyta, J., Betancourt, G., Centeno, G., Marroquin, G. 2003. Catalyst deactivation during
hydroprocessing of Maya heavy crude oil. (II) Effect of temperature during time-on-stream. Energy Fuels
17:462-467.
Ancheyta, J., Betancourt, G., Centeno, G., Marroquin, G., Alonso, E, Garciafigueroa, E. 2002. Catalyst
deactivation during hydroprocessing of Maya heavy crude oil. I. Evaluation at constant operating conditions.
Energy Fuels 16:1438—1443.
Ancheyta, J., Rana, M.S., Furimsky, E. 2005a. Hydroprocessing of heavy petroleum feeds: Tutorial. Calal.
Today 109:3-15.
Ancheyta, J., Sanchez, S., Rodriguez, M.A. 2005b. Kinetic modeling of hydrocracking of heavy oil
fractions: A review Catal. Today 109:76—92.
Ancheyta, J., Sotelo, R. 2007. Estimation of kinetic constants of a five-lump model for fluid catalytic
cracking process using simpler sub-models. Enetgy Fuels 14:1226-1231.
Ancheyta, J., Speight, J.G. 2007. Hydroprocessing of Heavy Oils and Residua, CRC Press, Taylor & Francis
Group, Boca Raton, FL.
Ayasse,A.R., Nagaishi, H., Chan, E.W, Gray, M.R. 1997. Lumped kinetics of hydrocracking of bitumen.
F«e/76.T025—1033.
Baldassari, M.C., Louie, WS., Mukherjee, U.K. 2002. Multiple stage ebullating bed hydro-cracking
with interstage stripping and separating. U.S. Patent 6,454,932, September 24.
Basak, K., Sau, M., Manna, U., \6rma, R.P 2004. Industrial hydrocracker model based on novel
continuum lumping approach for optimization in petroleum refinery. Catal. Today 98:253-264.
Bechtel, R.R., Wisdom, L.I. 1991. Sequential operation of H-oil and residue FCC. American Institute of
Chemical Engineers. In: Symposium on Resid Upgrading Processes, Houston, TX, April 7-11.
Benito, AM., Martinez, M.T 1996. Catalytic hydrocracking of an asphaltenic coal residue. Energy Fuels
10:1235-1240.
Bhatia, VK., Epstein, N. 1974. Three Phase Fluidization: A Generalized TThke Model. Fluidization and
its Applications (H. Angelino, J.P Couderc, H. Gilbert, C. Laguerue, Eds.), Cepadues Editions, Toulouse,
p. 380.
Botchwey, C., Dalai, A.K., Adjaye, J. 2004. Kinetics of bitumen-derived gas oil upgrading using a
commercial NiMo/AljC^ Catalyst. Can. J. Chem. Eng. 82:478—487.
Buttke, R.D., Frey J.R. 1990. Vtpor collector and process for ebullated bed reactors. U.S. Patent
4,950,459, August 21.
460
Часть 3. Моделирование каталитических процессов
Buttke, R.D., Peck, L.B., Nikolaides, С. 1994. Reduced gas holdup in an ebullated bed reactor U.S. Patent
5,308,476, May 3.
Chacchia, D., Perkins, K., \hlente, N. 2004. Sealless pumps, eliminating the single most failure prone
point of the pump. Pulsafeeder Inc. http://wwwpulsa.com/new/docs/SeallessPumps.pdf (February 16,
2004).
Chan, TY, Colvert, J.H. 1989. Liquid degaser in an ebullated bed process. U.S. Patent 4,886,644,
December 12.
Chan, TY, Strickland, J.C. 1990. Catalyst inventory control in an ebullated bed process. U.S. Patent
4,902,407, February 20.
Chaudhari, R.V, Shah, YT, Foster, N.R. 1986. Novel gas-liquid-solid reactors. Catal. Rev. Sci. Eng.
28(4):431—518.
Colvert, J.H. 1989. Bubble cap assembly in an ebullated bed reactor. U.S. Patent 4,874,583, October 17.
Cox, J.A. 1990. Floating recycle pan for ebullated bed reactors. U.S. Patent 4,911,893, March 27.
Coylar, J. J. 2007. Effective integration of solvent deasphalting and ebullated-bed processing. U.S. Patent
7,214,308, May 8.
Coylar, J.J., Kressmann, S., Gueret, C. 2007. Integrated SDA and ebullated-bed process. U.S. Patent
7,279,090, October 9.
Coylar, J.J., MacArthur, J.B. 1996. Complete catalytic hydroconversion process for heavy petroleum
feedstocks. European Patent Office EP0732389, September 18.
Colyar, J.J., MacArthur, J.B., Peer, E.D. 2001. Catalytic hydrogenation process utilizing multi-stage
ebullated bed reactors. U.S. Patent 6,270,654, August 7.
Colyar, J.J., Wisdom, L.1.1997.TheH-OIL® process: A worldwide leader in vacuum residue hydro-processing.
In: NPRA Annual Meeting, March 16—18, Convention Center, San Antonio, TX.
Deshpande, D.A., Deo, M.D., Hanson, EV 1992. Similitude studies in three-phase ebullated bed re-
actors. In: Eastern Oil Shale Symposium, November 17—20. Hyatt Regency, Lexington, KY
Eccles, R.M. 1980. H-Oil®—Aflexibleprocessformaximumoilyieldfromdifficultfeedstocks. In: SS/h Na-
tional Meeting American Institute of Chemical Engineers, Philadelphia, PA, June 8—12.
Eccles, R.M. 1993. Residue hydroprocessing using ebullated-bed reactors. Fuel Process. Technol. 35:21-38.
Forzatti, P, Lietti, L. 1999. Catalyst deactivation. Catal. Today 52:165-181.
Froment, G.E 2008. Kinetic modeling of hydrocarbon processing and the effect of catalyst deactivation
by coke formation. Catal. Rev. Sci. Eng. 50:1-18.
Froment, G.E, Bischoff, K.B., De Wilde, J. 2010. Chemical Reactor Analysis and Design, 3rd edn., John
Wiley & Sons Inc., Newark.
Galiasso, R. 2007. Effect of recycling the unconverted residue on a hydrocracking catalyst operating in an
ebullated bed reactor. Fuel Process. Technol. 88:779—785.
Galiasso, R., Caprioli, L. 2005. Catalyst pore plugging effects on hydrocracking reactions in an ebullated
bed reactor operation. Catal. Today 109:185—194.
Harle, V, Kazstelan, S., Morel, E, Kressmann, S., Courty, P 2000. Hydrotreating hydrocarbon feeds in an
ebullating bed reactor. U.S. Patent 6,132,597, October 17.
Hookham, D.E. 1993. On-stream time for ebullating bed reactor. U.S. Patent 5,253,403, October 19.
Jiang, P, Arters, D., Fan, L.-S. 1992. Pressure effects on the hydrodynamic behavior of gas-liquid-solid
fluidized beds. Ind. Eng. Chem. Res. 31:2322—2327.
Jiang, P, Luo, X., Tsao-Jen, L., Fan, L.-S. 1997. High temperature and high-pressure three-phase
fluidization-bed expansion phenomena. Powder Technol. 90:103—113.
Johns, WE, Clausen, G., Nongbri, G., Kaufman, H., 1993. Texaco T-star process for ebullated bed
hydrotreating/hydrocracking. In: NPRAAnnualMeeting, Convention Center San Antonio, TX, March 21-23.
Johnson, R., Alpert, S.B., Lehman, L.W 1968. Refinery applications of the H-Oil process. In: 33rd
Midyear Meeting of the American Petroleum Institute’s Division of Refining, Philadelphia, PA, May 16.
Johnson, A.R., Brown, E.C. 1994. Combination process for the pretreatment and hydroconversion of
heavy residual oils. U.S. Patent 5,320,741, June 14.
Глава 10. Моделирование реакторов с кипящим или суспензионным слоем
461
Kam, Е.К.Т., Jasam, Е, Al-Mashan, М. 2001. Catalyst attrition in ebullated-bed hydrotreator
operations. Catal. Today 64:297-308.
Karassik, I.J., Messina, J.R, Cooper, R, Heald, C.C. 2001. Pump Handbook, 3rd edn., McGraw-Hill,
New York.
Kolstad, J.J., Beaton, WL, Taylor, J.L. 1993. Means for and methods of removing heavy bottoms from
an effluent of a high temperature flash drum. U.S. Patent 5,258,117, November 2.
Kressmann, S., Colyar, J.J., Peer, E., Billon, A., More, E 1998. H-Oil process based heavy crudes
refining schemes. In: Proceedings of 7th UNITAR Conference on Heavy Crude and Tar Sands, Beijing, China,
October 27-30.
Kumar, H., Froment, G.E 2007. Mechanistic kinetic modeling of the hydrocracking of complex
feedstocks, such as vacuum gas oils. Ind. Eng. Chem. Res. 46:5881-5897.
Lenglet, E. 2008. Process for pre-refining crude oil with moderate multi-step hydroconversion of virgin
asphalt in the presence of diluent. U.S. Patent Application 20080289999, November 27.
Li, A.S., Eccles, R.M. 1987. Fluid flow distribution system for fluidized bed reactors. U.S. Patent
4,702,891, October 27.
Loria, H., Trujillo-Ferrer, G., Sosa-Stull, C., Pereira-Almao, P. 2011. Kinetic modeling of bitumen
hydroprocessing at in-reservoir conditions employing ultradispersed catalysts. Energy Euels 25:1364—1372.
Lott, R.K., Lee, L.-K. 2008. Ebullated bed hydroprocessing methods and systems and methods of
upgrading an existing ebullated bed system. U.S. Patent 7,449,103, November 11.
Luo, X., Jiang, PT, Fan, L.-S. 1997. High-pressure three-phase fluidization: Hydrodynamics and heat
transfer. AIChEJ. 43( 10):2432—2445.
Lyzinski, D., Buttke, R.D., Taylor, J.L., Hall,W.M. 1993. Laboratory simulator of reactor for a petroleum
refinery. U.S. Patent 5186904, February 16.
MacArthur, J.B. 1993. Evolution of HRI’S coal liquefaction technologies. In: Annual International
Pittsbuigh Coal Conference, University of Pittsburgh School of Engineering Centre of Energy.
Malladi, M., Otero-Schipper, P.H., Gross, B. 1982. Dynamics of ebullated bed reactor following recycle
failure. In: American Institute of Chemical Engineers 1982 Fall Meeting, Los Angeles, CA, November.
Martens, G.G., Marin, G.B. 2001. Kinetics for hydrocracking based on structural classes: Model
development and application. AIChE J. 47:1607—1622.
Martinez, J., Shnchez, J.L., Ancheyta, J., Ruiz, R. 2010. A review of process aspects and modeling of
ebullated bed reactors for hydrocracking of heavy oils. Catal. Rev. 52(l):60—105.
Matos, E.M., Guirardello, R. 2000. Modeling and simulation ofthe hydrocracking ofheavy oil fractions.
Braz. J. Chem. Eng. 17:79—90.
McDaniel, N.K., Vasti, N.C., Woods, N.R., Boening, R.E. 1989. Resid hydrotreating with high
temperature flash drum recycle oil. U.S. Patent 4,808,289, February 28.
Me Namara, D.J., Sherwood, D.E., Ginestra, J.M., Ijlstra, W.D. 2007. Correct catalyst selection leads
to improved on-stream factor in high conversion resid upgrading units. In: Second Russia & CIS Bottom of the
Barrel Technology Conference and Exhibition, Marriot Grand Hotel, Moscow, 18—19 April.
Meng, X., Xu, C., Gao, J. 2007. Coking behavior and catalyst deactivation for catalytic pyrolysis of
heavy oil. Fuel 86:1720—1726.
Milligan, J.D. 1985. Staged flow distribution grid assembly for ebullated bed reactor. UK-IPO
GB2148141, May 30.
Mounce, W., Rubin, R.S. 1971. Desulfurization of light and heavy hydrocarbons—Part I. In: Sixty-
Eighth National Meeting, AIChE. Houston, TX, February 28-March 4.
Nongbri, G., Clausen, G.A. 1992. Commercial application of the ebullated bed technology.
In: Fluidization FIT—Proceedings of the Seventh Engineering Foundation Conference on Fluidization, Brisbane,
Queensland, Australia, May 3—8.
Parulekar, S.J., Shah, Y.T 1980. Steady state behavior of gas-liquid-solid fluidized-bed reactors. Chem.
Eng. J. 20:21-33.
Pratt, R.E., Nongbri, G., Clausen, G.A., Bavarian, E 1995. Heavy oil hydroprocessing. In: Proceedings of
the 6th UNITAR International Conference on Heavy Crude and Tar Sands, \bl. 2(10), pp. 1—9.
462
Часть 3. Моделирование каталитических процессов
Qader, S.A., Hill, G.R. 1969. Hydrocracking of gas oil. Ind. Eng. Chem. Proc. Des. Dev. 8:98-105.
Rana, M.S., Samano, V, Ancheyta, J., Diaz, J.A.I. 2007. A review of recent advances on process
technologies for upgrading of heavy oils and residua. Fuel 86:1216—1231.
Robert, E.C., Mendrignac, I., Kressmann, S., Colyar, J. 2002. H-oil® process. Contributions of ana-
lytical tools for the understanding of sediment formation. In: AICHE Spring National Meeting, New Orleans,
LA, March 10—14.
Roy-Auberger, M., Guillaume, D., Kressmann, S., Le Loarer, J.-L., Chapat, J.-C. 2007. Irregularly
shaped, non-spherical supported catalyst and process for hydroconversion of heavy petroleum fractions.
InternationalApplication No. PCT/FR2006/002765, July 19.
Ruiz, R.S., Alonso, E, Ancheyta, J. 2005. Pressure and temperature effects on the hydrodynamic
characteristics of ebullated-bed systems. Catal. Today 109:205—213.
Sadighi, S., Ahmad, A., Mohaddecyz, S.R.S. 2010. 6-Lump kinetic model for a commercial vacuum gas
oil hydrocracker. Inter. J. Chem. Reactor Eng. 8:1-24.
Safoniuk, M., Grace, J.R., Hackman, L., McKnight, C.A. 1999. Use of dimensional similitude for scale-
up of hydrodynamics in three-phase fluidized beds. Chem. Eng. Sci. 54:4961—4966.
Sales, EG., Maranhao, L.C.A., Pereira, J.A.R.E, Abreu, C.A.M. 2005. Experimental dynamic evaluation
of three-phase reactors. Braz. J. Chem. Eng. 22:443-452.
Sanchez, S., Ancheyta, J. 2007. Effect of pressure on the kinetics of moderate hydrocracking of Maya
crude oil. Eneigy Fuels 21:653—661.
Sanchez, S., Rodriguez, M.A., Ancheyta, J. 2005. Kinetic model for moderate hydrocracking of heavy
oils. Ind. Eng. Chem. Res. 44:9409—9413.
Sanchez, J.L., Ruiz, R.S., Alonso, E, Ancheyta, J. 2008. Evaluation of the hydrodynamics of high-
pressure ebullated beds based on dimensional similitude. Catal. Today 130:519—526.
Sastri, N.VS., Epstein, N., Hirata, A., Koshijima, I., Izumi, M. 1983. Zinc hydrosulphic by three-phase
fluidization: Experiments and model. Can. J. Chem. Eng. 61:635-646.
Sayles, S. 2006. The ebullition factor. Hydrocarbon Eng. 11(3):35—40.
Sayles, S.M., Livingston, WB., Bellinger, M.P 1990. Temperature control in an ebullated bed reactor.
U.S. Patent 4,913,800, April 3.
Scheweitzer, J.-M., Kressmann, S. 2004. Ebullated bed reactor modeling for residue conversion. Chem.
Eng. Sci. 59:5637-5645.
Soderberg, D.J. 1988. Catalyst inventory determination. U.S. Patent 4,750,989, June 14.
Stangeland, B.E. 1974. A kinetic model for the prediction of hydrocracker yields. Ind. Eng. Chem. Proc.
Des. Dev. 13:71—76.
Stanislaus, A, Hauser, A., Marafi, M. 2005. Investigation of the mechanism of sediment formation in
residual oil hydrocracking process through characterization of sediment deposits. Catal. Today 109:167-177.
Tampa, G.E. 1992. Ebullated bed grid plate and skirt to prevent flow maldistribution and catalyst attrition.
U.S. Patent 5,100,629, March 31.
Visco de Toledo, E.C., Leite de Antana, P,W)lfMaciel, M.R., MacielFilho, R. 2001. Dynamic modeling of
a three-phase catalytic slurry reactor. Chem. Eng. Sci. 56:6055—6061.
Wandas, R. 2007. Structural characterization of asphaltenes from raw and desulfurized vacuum residue
and correlation between asphaltene content and the tendency of sediment formation in H-oil heavy products.
Petrol. Sci. Technol. 25:153—168.
Wilma, J., Salmi, T. 1996. Dynamic modeling of catalytic three phase reactors. Comput. Chem. Eng. 20:39-47.
Werther, J., Reppenhagen, J. 1999. Catalyst attrition in fluidized-bed systems. AIChEJ. 45(9):2001—2010.
Wisdom, L.L, Colyar, J.J. 1995. Second generation ebullated-bed technology. In: AICHE 1995 Spring
National Meeting, Session 46—Resid Hydroprocessing Advances, Houston, TX, March 19—23.
Yui, S.M., Sanford, E.C. 1989. Mild hydrocracking ofbitumen-derived coker and hydrocracker heavy gas
oils: Kinetics, product yields, and product properties. Ind. Eng. Chem. Res. 28:1278-1284.
Zhang, J.P, Grace, J.R., Epstein, N„ Lim, K.S. 1997. Flow regime identification in gas-liquid flow and
three-phase fluidized beds. Chem. Eng. Sci. 52:3979-3992.
Глава 11. Моделирование
гидрокрекинга непрерывным
агрегированием кинетики
Основная тема настоящей главы — моделирование кинетики гидрокрекинга тяжелой
нефти с использованием метода непрерывного агрегирования (МНА). Наряду с под-
робным изложением решения модели, обсуждаются главные аспекты МНА. Освеща-
ются вопросы, которые предстоит решить в будущих исследованиях. По эксперимен-
тальным данным, полученным в изотермическом реакторе при различных значениях
температур, объемной скорости подачи сырья (£Я5И) и давлении и постоянной крат-
ности водорода к сырью, вычисляются параметры модели. Предлагаются различные за-
висимости, связывающие значения параметров с давлением и температурой. Описы-
вается моделирование совместно протекающих реакций гидрообессеривания (ГДО)
и гидрокрекинга с использованием данного подхода. Оптимизированные значения
параметров модели используются для прогнозирования результатов при условиях ре-
акций, отличных от тех, для которых они были выведены. Демонстрируется хорошее
согласие между экспериментальными данными и результатами, полученными МНА
кинетики.
11.1. ВВЕДЕНИЕ
Гидрокрекинг — один из наиболее привлекательных путей получения средних дис-
тиллятов из тяжелой нефти и остатков. Гидрокрекинг и раньше был одним из наи-
более востребованных процессов в нефтепереработке, но в последнее время его роль
возросла еще больше, так как все чаще приходится иметь дело с тяжелыми нефтя-
ными фракциями [Ancheyta et al., 2005]. Выбор рабочих условий и катализатора за-
висит от целей процесса. Фундаментальные исследования гидрокрекинга в жестких
и мягких условиях привели к появлению технологий, рассчитанных на два этих раз-
ных варианта процесса [Valarasu et al., 2003; Rana etal., 2007]. Для получения синтети-
ческой нефти мягкий гидрокрекинг предпочтительнее жесткого, так как потребляет
меньше водорода и дает меньший выход легких углеводородов и нежелательных вто-
ричных продуктов. Главным продуктом в этом случае являются средние дистилляты
464
Часть 3. Моделирование каталитических процессов
[Ancheyta et al., 2001; Botchway et al., 2003]. Повышенную избирательность мягко-
го гидрокрекинга к выходу средних дистиллятов на примере тяжелой мексиканской
нефти наблюдали Анчита и соавторы [Ancheyta et al., 2001].
При предварительных расчетах гидрокрекинга тяжелой нефти важно понимать,
каким образом условия реакций влияют на избирательность и глубину процесса. Раз-
умеется, нужно знать заранее и тип перерабатываемого сырья. Эти сведения мож-
но получить из результатов экспериментов либо путем имитации. Проведение экс-
периментов весьма желательно, но требует значительных финансовых и временных
затрат. Имитация же, напротив, недорога и осуществляется быстро. Для нее обыч-
но выбирают упрощенную модель реактора. Например, реактор с неподвижным сло-
ем при определенных допущениях довольно точно описывается моделью идеального
вытеснения. Чтобы достичь приемлемого согласия с экспериментальными данны-
ми, в эту модель можно ввести дополнительные члены и параметры [Mederos et al.,
2009а,Ь]. Но моделирование гидрокрекинга тяжелой нефти осложняется наличием
в ней больших количеств тяжелых углеводородов: это сильно затрудняет детальное
описание сырья и продуктов. Применяемая кинетическая модель должна правильно
отражать работу реактора гидрокрекинга. Существуют разные подходы к моделиро-
ванию кинетики гидрокрекинга. Обзор этих подходов опубликовали Анчита и соав-
торы [Ancheyta et al., 2005].
Кинетику реакций гидрокрекинга нефтяного сырья обычно моделируют методом
дискретного приближения, а именно агрегированием соединений сырья и продуктов
в несколько псевдокомпонентов, часто характеризуемых интервалами кипения при
атмосферном давлении (примерами таких агрегатов являются бензиновая фракция,
средние дистилляты, остаток и т. д.), которые вступают в различные последователь-
ные и параллельные реакции [Ayasse et al., 1997]. Основные преимущества этого ме-
тода — простота вычислений и сравнительно небольшой объем данных, необходи-
мых для расчета параметров. Чем больше число агрегатов, тем точнее приближение,
но вместе с тем увеличивается и число параметров, подлежащих вычислению. Метод
непрерывного агрегирования успешно применяется при моделировании гидрокре-
кинга тяжелой нефти [Ancheyta et al., 2005; Sanchez et al., 2005]. Его главный недо-
статок заключается в том, что на каждом шаге составления балансов масс и энергии
приходится переопределять значения необходимых для этого свойств всех агрегатов
(в том числе значения плотности, вязкости, молярной массы и кривой разгонки), по-
скольку они непрерывно изменяются в реакторе.
Один из других подходов предлагает рассматривать реакции гидрокрекинга как
аналог дисперсии распределения молярных масс реакционной смеси вдоль оси ре-
актора [Krishna and Saxena, 1989]. Данный подход позволяет предсказать всю кривую
разгонки, нуждаясь при этом лишь в нескольких параметрах. Эта модель применя-
лась для выявления корреляционной связи между данными лабораторных и опыт-
ных установок.
Фромен [Froment, 2005] предложил детальное приближение процесса гидрокрекин-
га, известное под названием модели единичных событий. Метод позволяет сохранять
данные молекулярного уровня и точно предсказывать состав суммарного продукта.
Но к моделированию гидрокрекинга тяжелой нефти этот подход еще не применялся.
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
465
Еще один метод предлагает рассматривать нефть как смесь бесконечного чис-
ла компонентов, т. е. представлять ее непрерывной смесью вместо дискретной. По-
добную смесь можно описать функцией распределения концентраций [Aris and Ga-
valas, 1966]. Нефтяную фракцию можно считать непрерывной смесью, если она
содержит очень большое число неопознанных веществ, а различия между смежными
веществами минимальны [Aris, 1989]. Рассматривать смесь как непрерывный ряд ве-
ществ впервые предложил Де Дондер [DeDonder, 1931]. Теория непрерывных смесей
успешно применяется в других областях химии [Aris, 1989]. Арис и Гавалас [Aris and
Gavalas, 1966] первыми экстраполировали идею непрерывных смесей на химические
реакции, описав многие параллельно протекающие реакции гидрокрекинга нефтя-
ных фракций. Даже если можно идентифицировать компоненты смеси, представ-
ление ее в виде непрерывного ряда имеет свои преимущества: кинетическое агре-
гирование в этом случае осуществляется интегрированием вместо суммирования.
Решаемые задачи представляются интегралами, с которыми во многих случаях удоб-
нее иметь дело [Chou and Но, 1988]. При непрерывном кинетическом моделирова-
нии сырье и продукты чаще всего характеризуют углеродным числом или истинны-
ми температурами кипения [Astarita and Ocono, 1988]. Эти свойства можно связать
друг с другом [Maxwell, 1965]. В случае таких сложных смесей, как нефтяные фрак-
ции, предпочтительнее иметь дело с истинными температурами кипения, так как их
можно определить привычными методами ASTM.
После публикации новаторской работы Ариса и Гаваласа стали появляться сооб-
щения о различных аспектах теории непрерывного агрегирования и ее применении
к химическим реакциям. К примеру, Чу и Хо [Chou and Но, 1988] представили про-
цедуру моделирования нелинейной реакции с использованием МНА. Ее ключевой
момент — введение распределения по типам веществ, что обеспечивает кинетиче-
ское согласование с дискретно агрегированной смесью, на базе которой построена
непрерывная смесь. Маккой и Ванг [McCoy and Wang, 1994], рассматривая гидро-
крекинг как процесс фрагментации, получили общее выражение для стехиометри-
ческого коэффициента бинарной фрагментации и показали, что при определенных
условиях она хорошо описывается гауссовым распределением. Баланс масс при мо-
делировании непрерывной смесью дается дифференциально-интегральным уравне-
нием. Зифф [Ziff, 1991] исследовал аналитическое решение дифференциально-инте-
грального уравнения фрагментации, но он рассматривал лишь гипотетические или
весьма ограниченные случаи. О практическом применении непрерывного агрегиро-
вания сообщали Броварзик и Келен [Browarzik and Kehlen, 1994], а также Пейсото
и де Медейрос [Peixoto and de Medeiros, 1999], но в этих исследованиях не использо-
вались такие сложные смеси, как нефть и остатки ее перегонки.
Лаксминарасимхан и соавторы [Laxminarasimhan et al., 1996] представили резуль-
таты применения функции распределения выхода, выведенной на основании опу-
бликованных данных, к описанию выхода продуктов и избирательности гидрокре-
кинга. Функция предполагала асимметричное гауссово распределение. Сообщалось
о хорошем согласии между экспериментальными и расчетными значениями выхода
продуктов гидрокрекинга реакционной смеси. Распаду на фрагменты в процессе ги-
дрокрекинга противопоставлялось дробление частиц: это упрощало представление
466
Часть 3. Моделирование каталитических процессов
компонентов стехиометрического распределения и открывало возможность экстра-
поляции на реакции с порядком, отличным от 1. Кроме того, в формулировку, опи-
рающуюся на аналогию с дроблением, можно ввести несколько индексов. Это позво-
лит моделировать реакции гидрокрекинга, в которых принимают участие соединения
всех классов, а именно парафиновых, нафтеновых и ароматических углеводородов.
Основываясь на работе Лаксминарасимхана, Хорашех с соавторами [Khorasheh et al.,
2001, 2005] и Ашури с соавторами [Ashuri et al., 2007] применили идею непрерывного
моделирования к реакциям гидрокрекинга, гидрообессеривания (ГДО) и гидродеазоти-
рования (ГДА), чтобы рассчитать зависимость параметров от температуры. Используя
МНА, Басак и соавторы [Basak et al., 2004] смоделировали потребление водорода и рас-
пределение температуры в каталитическом слое промышленной установки гидрокре-
кинга. Хорашех и соавторы [Khorasheh et al., 2005] указали, что с помощью дискретной
модели можно достаточно точно предсказать массовую долю тяжелой фракции в сме-
си, но для легких фракций результаты будут неудовлетворительными. В расчетах выхо-
да легких фракций альтернативой этому методу может служить непрерывная модель.
При пользовании моделью Лаксминарасимхана и соавторов [Laxminarasim-
han et al., 1996] необходимо учитывать начальные условия дифференциально-инте-
грального уравнения. В таких случаях используется анализ полидисперсности [Ostrows-
ky and Somette, 1981]. Говиндхаканнан и Риггс [Govindhakannan and Riggs, 2007], исходя
из данных Беннетта и Бурна [Bennett and Bourne, 1972] и опираясь на метод и параме-
тры Лаксминарасимхана и соавторов, предложили способ построения (через алгоритм
оптимизации) непрерывной функции концентрации сырья, применимой для описа-
ния гидрокрекинга сырья любого типа. Были получены успешные результаты.
Главный недостаток метода дискретного моделирования — вычисление кон-
центраций легких и тяжелых соединений в продуктах, требующее обширных экс-
периментальных работ и громоздких расчетов для нахождения ряда констант ско-
ростей. К тому же остается неопределенность, обусловленная множеством путей
возможных реакций, причем формулы последних могут потребовать изменений [Sa-
gighi et al., 2010]. Из других недостатков метода дискретного агрегатного моделирова-
ния следует отметить следующие [Basak et al., 2004]:
• ввиду возможных различий в составах агрегатов в одних и тех же интервалах ки-
пения дискретную модель нельзя распространить на сырье, отличное от того,
дня которого она была построена;
• с изменением степени превращения меняются составы агрегатов и истинные
значения кинетических констант;
• в кинетическую схему не включен механизм реакций (недостаточно количе-
ство агрегатов);
• метод не способен прогнозировать изменение свойств продуктов.
В то же время непрерывная кинетическая модель обладает следующими достоин-
ствами [Laxminarasimhan et al., 1996; Sau et al., 1997; Narasimham et al., 1999; Kho-
rasheh et al., 2005; Elizalde et al., 2009]:
• предсказание всей кривой кипения по небольшому числу параметров;
• строгое следование химии процесса;
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
467
• единый (первый) порядок реакции, имеющий физический смысл;
• легкость расчета любой фракции с произвольно заданными начальными и ко-
нечными температурами кипения;
• предсказание кривой распределения гетероатомных соединений;
• легкость адаптации модели к кинетике реакций тяжелой и особо тяжелой неф-
ти и остатков.
Все эти преимущества и недостатки довольно важны, однако общее превосходство
непрерывной кинетической модели над дискретной точнее всего характеризуется ее
способностью предсказывать динамику изменения концентрации гетероатомных со-
единений в зависимости от времени пребывания в реакционной зоне и распределе-
ния температур кипения.
11.2. НЕПРЕРЫВНОЕ АГРЕГАТНОЕ
МОДЕЛИРОВАНИЕ КИНЕТИКИ
11.2.1. Описание модели
Математическое описание кинетики гидрокрекинга в терминах непрерывности опи-
рается на уравнения баланса масс или количеств, учитывающие изменения во време-
ни и пространстве поведения функций распределения вида C(l, z, х), где х — вектор
переменных, описывающих такие изменяющиеся в процессе свойства, как разме-
ры, молярная масса и т. д., z — вектор, учитывающий пространственную размерность
процесса, I — время. Концентрация таких частиц в области (х, х + г/х) равна С(х)г/х.
Согласно Маккою и Вангу [McCoy and Wang, 1994], в системе идеального вытеснения
(характеризуемой бинарной фрагментацией частицы со свойством х' на две частицы
со свойствами (х' -х) и х), в которой пространственные изменения происходят лишь
вдоль оси z, уравнение динамики функции С(/, х, z) имеет вид
—+v~~~ + |i = 2? Л(х')С(/, z, x')Q(x, х')(/х' - fr(x)C(Z, z, х), (11.1)
dt dz dx '
где v — линейная скорость потока; ц — скорость нарастания; Q(x, х') — стехиометри-
ческий коэффициент, или ядро; к(х) — скорость фрагментации.
Для установившегося процесса при условии, что происходит чистая фрагментация
(т. е. нарастание отсутствует: у = 0), уравнение (11.1) сокращается до
v——- - 2|k(x')C(z,x')Q(x,x’)dx’ -k(x)C(z,x). (П-2)
X
Для реактора идеального вытеснения его удобнее записать в виде
= 2 j&(х')С(т, x')Q(x,x')o'x'-к(х)С(х, х), (11Д)
гдет = ^/р.
468
Часть 3. Моделирование каталитических процессов
В своей работе Маккой и Ванг предложили следующую обобщенную зависимость
для коэффициента скорости к(х'):
к(х) = кхр, (11.4)
где к — константа пропорциональности, а р принимает различные значения в от-
дельных случаях.
Были получены решения уравнения (11.3) для определенных типов ядра и различ-
ных частных значений />; в общем случаер является вещественным числом и уравне-
ние (11.3) может быть решено лишь численным способом.
Чу и Хо [Chou and Но, 1988] предложили способ обеспечить непротиворечивость
методов непрерывного и дискретного агрегирования в случае нелинейной кинетики.
Для этого они ввели функцию распределения по типам D(k), которую можно рассма-
тривать как якобиан преобразования дискретной по своей природе величины х' в не-
прерывную переменную к, так что
С(т, x')dx' = с(к, x)D(k)dk.
(11.5)
В случае гидрокрекинга для непрерывного кинетического агрегирования можно
использовать данные кривой кипения (зависимость между температурой кипения
и отогнанной долей wt, где физическую температуру Т можно преобразовать в без-
размерную 0). Эти данные неявно зависят от величины х', т. е. 0 = 0(х')- Таким обра-
зом, всякое изменение величины х' может вызвать изменение состава (w/(0, т)), опре-
деляемое уравнением
С(т, x')dx' = wt(Q, r)dQ.
Из уравнений (11.5) и (11.6) следует
w/(0, т)с/0 = с(к, x)D(k)dk.
(Н.6)
(И.7)
Согласно Лаксминарасимхану и соавторам [Laxminarasimhan et al., 1996], если
в качестве изменяющейся в реакторе величины выбрать к, то после перехода к дру-
гим координатам (от 0 к к) уравнение (11.3) приобретает вид
= -кс(к, т) + Г p(k,x)xc(xs)D(x)dx, (11.8)
с/т •
где к(х') = х; Q(x, х') = р(к, х); C(z, x)dx' = с(х, T)D(k)dk. Верхний предел интеграла
в данном случае изменяется на &тах, а величина к — активность вещества.
Уравнение (11.8) является более общим, чем уравнение (11.3), так как позволя-
ет через функцию распределения по типам ввести нелинейную кинетику [Chou and
Но, 1988; Laxminarasimhan et al., 1996]. Параметры в уравнении (11.8) подчиняются
следующим зависимостям:
' TBP(h)-TBP(T)
(Н.9)
Глава 11, Моделирование гидрокрекинга непрерывным агрегированием кинетики
469
1 -Г(Ц7К)"“ -0,51/a.T
р(к, К) = е -* - А + В ; (и.ю)
S0d2n
Л = е-(0.5/а1)-; (Н.П)
Б = 5[1-(/с/А)]; (11.12)
j p(x,kj)D{x)dx= 1; 0 (П.13)
Г 1 Г -ГФ/Х)"»-0,5}/а,12 У = —= е Ll ’ J - А + В ° JoV24 D(x)dx; (П-14)
С,
wt. = j с(х, т) £>(х)dx. (11.15)
ь.
Уравнение (11.8) содержит пять подгоночных параметров: а, а0, ар 5 и Лтах. Они
позволяют наилучшим образом согласовать кривую с(к, т) с экспериментальными
данными.
Фактор (якобиан), учитывающий преобразование преобразования х' —> к, дол-
жен зависеть также от экспериментально измеримой величины 0. Следовательно, его
можно записать в виде
dx' dB
d V ак
Лаксминарасимхан и соавторы предложили считать, что при п—»оо величину dx'Id^
можно приблизить к значениям п, а константа скорости к, по аналогии с уравнени-
ем (11.4), равна
£=*max0,/a. (11.17)
Отсюда
на ,а_,
D(k) = —к (11.18)
^гпах
где а и ^тах — вещественные положительно определенные подгоночные параметры
модели; 0 — безразмерная температура, определяемая уравнением (11.9). Величина
к прямо пропорциональна безразмерной температуре; следовательно, чем выше тем-
пература кипения, тем больше активность.
При моделировании гидрокрекинга тяжелой нефти с использованием МНА по-
рядок реакции для всех псевдокомпонентов принимается равным единице. Поэтому
470
Часть 3. Моделирование каталитических процессов
каждый псевдокомпонент должен иметь собственную скорость реакции, т. е. актив-
ность. Этим объясняется наличие ряда скоростей реакций и их распределение, дава-
емое уравне нием (11.17).
11.2.2. Допущения модели для реактора с неподвижным слоем
Моделирование кинетики методом непрерывного агрегирования осуществлялось
при определенных допущениях и предположениях, которые представлены ниже.
• Эксперименты проводились в изотермических условиях в отсутствие внутри-
фазных и межфазных градиентов, т. е. реакции протекали в чисто кинетиче-
ском режиме. Это объяснялось прошлым опытом работы на той же экспери-
ментальной установке.
• Гидрокрекинг достаточно хорошо описывается реакцией первого порядка, по-
скольку водород присутствует в большом избытке.
• При проведении опытов подавался водород высокой чистоты (99,9 %) в режиме
однократного прохождения. Поэтому полное давление на входе в реактор мож-
но считать равным давлению подачи водорода.
• При гидрокрекинге в различных условиях образуются определенные коли-
чества легких газов. Эти газы заложены в модель, так как расчетные кривые
кипения предсказывают состав не только продукта гидрокрекинга, но и га-
зообразного продукта. Поэтому любые изменения времени пребывания и пар-
циального давления водорода уже учтены в балансе масс через кривые кипения.
• Продольная и поперечная дисперсии отсутствуют и реактор работает в режиме
идеального вытеснения.
• Сбор экспериментальных данных осуществляется в установившемся режиме
работы.
• Скорость расщепления псевдокомпонента зависит от его молярной массы, а сле-
довательно, и от температуры его кипения в нормальных условиях; таким обра-
зом, чем выше температура кипения, тем больше значение константы скорости.
На основе этих соображений поведение реактора было представлено режимом
идеального вытеснения.
11.2.3. Решение модели
Дифференциально-интегральное уравнение (11.8) решается в следующей
последовательности.
Так как функция с(к, т) является непрерывной, на малом интервале ее можно при-
близить линейной функцией (рис. 11.1) с угловым коэффициентом
с(£,+1,т)-с(^,т)
—— °'”’
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
471
где т — величина, обратная LHSV, или среднее время пребывания, вычисленное
на входе реактора (влияние т на плотность реакционной смеси принимается несуще-
ственным); к. — активность г-го компонента.
Рис. 11.1. Линеаризация функции с(к, -с) на малом интервале
Линейная интерполяция (приближение полиномом Лагранжа первой степени)
в интервале к. < к < к.+1 для произвольного значения времени пребывания дает
с(к, т) =
к-к;
Д |с(^1,т)+|' -'^1 |с(£,.,т),
*7+1 J \*j *7+1 J
(11.20)
а для нулевого времени пребывания
I к-к 1 ( к-к 1
с(к, 0) = с(/с,+1,0)+ с(^,0),
(И.21)
где значение т = 0 соответствует сырью, а т > 0 — продукту реакции, получаемому при
различных значениях времени пребывания.
Начальной точкой решения дифференциально-интегрального уравнения, соот-
ветствующей начальному распределению, является (с(к, 0)). Говиндхаканнан и Риггс
[Govindhakannan and Riggs, 2007], используя уравнение (11.15), разработали методику
построения непрерывной функции концентрации в нулевой момент времени, чтобы
представить начальное значение для дифференциально-интегрального уравнения.
Предложенный ими метод исходит из того, что массовую долю компонентов с актив-
ностью между к. и к.+} можно вычислить на основании уравнения (11.15) (табл. 11.1).
Как следствие, для сырья можно ввести следующее упрощение:
^1т1
wf.= j c(k,O)D(k)dk,
1с,
(11.22)
где wt. — массовая доля углеводородов с активностью к, лежащей в интервале (к,, к.+]).
472
Часть 3. Моделирование каталитических процессов
Сумма массовых долей, разумеется, равна единице:
п
(U.23)
<=|
где п — общее число компонентов в реакционной смеси.
Таблица 11.1
Состав сырья и продукта гидрокрекинга
Сырье Продукт
Т, °C Массовая доля Безразмерная температура (0) Г, °C Массовая доля Безразмерная температура (0)
Газы
(СН4) 0,00 0,0000 -161,55 0,0023 0,0000
(С2Н6) 0,00 0,0847 -88,55 0,0043 0,0847
(H2S) 0,00 0,1183 -59,65 0,0268 0,1183
(С3Н8) 0,00 0,1387 -42,05 0,0284 0,1387
(С4Н10) 0,00 0,1870 -0,45 0,0293 0,1870
Жидкость
30,21 0,00 0,2226 85,25 0,0587 0,2865
136,40 0,05 0,3458 152,22 0,1072 0,3642
187,80 0,10 0,4055 198,01 0,1557 0,4173
227,10 0,15 0,4511 217,98 0,2043 0,4405
261,10 0,20 0,4906 244,63 0,2528 0,4715
292,90 0,25 0,5275 268,34 0,3013 0,4990
325,87 0,30 0,5657 293,29 0,3499 0,5279
360,54 0,35 0,6060 317,16 0,3984 0,5556
394,92 0,40 0,6459 341,46 0,4469 0,5838
428,12 0,45 0,6844 365,75 0,4955 0,6120
461,69 0,50 0,7234 389,88 0,5440 0,6400
497,08 0,55 0,7645 414,57 0,5925 0,6687
533,84 0,60 0,8071 438,96 0,6411 0,6970
— — — 465,48 0,6896 0,7278
— — — 493,71 0,7381 0,7606
— — — 523,99 0,7867 0,7957
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
473
Подстановка выражения (11.21) в правую часть уравнения (11.22) дает
Г с(Л,0)ад^ = с(Л,.,0) f .LAl £(£)<&+c(£ 0)f (11.24)
I,
Уравнение (11.24) эквивалентно уравнению (11.22), и после упрощения получается
wtf = а^с(к., 0) + a.2c(ki+l, 0).
(11.25)
Значения и ai2 даются выражениями (11.26) и (11.27), представляющими собой
решения интегралов в правой части уравнения (11.24) при допущении справедливо-
сти формулы (11.18) для D(k):
1 па ГГ С1 CY (k:+' , *,“Y
ап=-—7т~ А г ; (Н.26)
ki~kM A Lla+1 aJ la+1 aJ.
1 и-a 17 C , £“7 (ka+' ka+' Y
Таким образом, задачу нахождения начального распределения концентраций
с(к, 0) можно сформулировать следующим образом:
wt(0) = А(к)с(к, 0), (11.28)
где
Wt(ty = (iV] w2 w3 (11.29)
Яц a]2 0 0 •• • 0 0 >
0 a2, a22 0 •• • 0 0
А{к) = 0 0 a3i a32 • 0 0 ; (11.30)
[0 0 0 an,l an,2^
(с(^,0) = с(Л„0) с(£2,0) с(Л3,0) ••• с(Л,„0) с(Л„+1,0))г. (11.31)
А(к) можно получить, присвоив начальные значения к и а. Интервалы активно-
сти вычисляются по уравнению (11.17) по двум значениям безразмерной температуры
0 для сырья и каждого псевдокомпонента. Величина и7(0) определяется по экспери-
ментальной кривой истинных температур кипения интерполяцией по эксперимен-
тальным точкам для различных значений числа п компонентов в смеси и экстраполя-
цией до максимальной точки кипения; при этом полагается, что при 0 = 1 суммарная
массовая доля wt достигает 1. По данным разгонки величины wt. определяются раз-
ностью массовых долей на концах интервала значений безразмерной температуры
(0, 0 + А0):
474
Часть 3. Моделирование каталитических процессов
Ч = Н+Д0-Н- (11.32)
Теперь, зная w/ и А(к), можно найти с(к, 0) путем решения уравнения (11.28) [Aris,
1989] и минимизации целевой функции
Min J(c(k, 0)) = £[c(£„0)-c(£,.+1,0)]2
(11.33)
при следующих условиях:
Р > 0: Р = а, ктх и с(к , 0) > 0.
Получив начальное значение, можно перейти к решению уравнения (11.8). Прежде
всего дискретизируется и решается интегральная часть, для чего в уравнение (11.8)
подставляется выражение с(к, т), обеспечиваемое формулой (11.20). Это дает
^^1 = -к/с(к,.,г) +
ат
п
+ х с(к’т>
/+1
D{x)dxt)
(11.34)
где к а,
** /1+1
к виду
к . После некоторых преобразований уравнение (11.34) приводится
dc(k^,x)
' -с(к.,г)
цт
Г х-к... |
-------- D(x)dx
у — ^7+1 )
»+i 7 [ х-Л.
+ Х ~c(kj,t)D(x)dx +
»+i Ч' ( x - к. 'l
+ Е J Р(ких)-х- у----ctk^D^dx, (11.35)
7='+i kj kj kJ+i J
где i изменяется от 1 до л.
Приводя все это к виду, удобному для вычислений при любом времени пребыва-
ния т., получим
^^ = С(Л,.,т^-(-Л,.+/^+^с(^.,Тг)./2.+ £C(V,.)/3,. (11 36)
для i = 1, 2,..., п + 1 и 0 < т < Ттах, где
^11
А, = J P(ki,x)x
ki
" х~км ^1
D(x)dx‘,
(11.37)
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
475
4~ [/?(£,.,х)х —----------— D(x)dx', (11.38)
? /с .,
^/4-1
4 = J Р(к<,х)
kf
D(x)dx.
(11.39)
При i = и + 1, разумеется, /1Р I2J и 1у обращаются в нуль.
Функции уравнения (11.36), т. е. к., I[p 1у, I, можно представить в виде матрицы
с размерностью (и + 1)х(н + 1):
~к\ +4 4 + 4 • 4+4 • - 4,+4 •4+1
0 —4 + Аг • 4+4 • 4и + Цп 4и+1
0 0 4/;+|
4 + 4 ; . (11.40)
0 0 0 4,+4, 4+1
1 0 0 0 0 — £ S+I 7
Задав значения для параметров а, а0, а(, 5 и ктм непрерывной кинетической мо-
дели, можно легко найти матрицу В(к), так как для этого придется лишь вычислить
интегралы уравнений (11.37)-(11.39) и выполнить некоторые арифметические дей-
ствия. Вычислив В(к), в следующем порядке решаем уравнение (11.36).
Для соединения с наивысшей активностью (£;+1) уравнение (11.36) сокращается до
=-кп+}с(кп+1,тг). (11.41)
ат
Уравнение (11.41) можно решить аналитически как задачу с начальным услови-
ем, что дает
c(4+i Л-) = c(4+i> Vi К*"*14 где 0 < т< т. < ттах. (11.42)
На первом шаге с(/с|+1, т.), т._, = 0; тогда решение уравнения (11.42) можно запи-
сать в виде
с(кп+1, т.) = с(к1+1, 0).
(11.43)
Для соединения с меньшей активностью (кл) уравнение (11.36) приводится к виду
= [~А + 4, ] С(К, тг) + 12пс(к,^ ,т,.). (11.44)
Это уравнение опять-таки можно решить как задачу с начальным условием для
с(кп, т) = c(kn, т_() при т = т._г
Процедура продолжается до тех пор, пока не дойдет до наиболее легкого и наиме-
нее активного соединения (; = 1). Затем все расчеты повторяются для других значений
476
Часть 3. Моделирование каталитических процессов
величины, обратной LHSV (т). При этом кривая с(к, т), полученная для определен-
ного значения т, используется как начальное значение при расчетах для следующего
большего значения времени пребывания.
Найденная для желаемого значения т кривая с(к, т) интегрируется по уравне-
нию (11.15) с использованием формулы (11.28), записанной в виде
w/(t) = А(к)с(к, т). (11.45)
Получается распределение массовой доли продуктов для любого значения вели-
чины, обратной LHSV, при фиксированном интервале безразмерных температур, от-
несенных к сырью. Это дает возможность проверить баланс масс в каждом интервале
времени пребывания. Модификацией уравнения (11.28) через изменение интерва-
лов к. можно выбрать другие интервалы из области изменения 0.
Чтобы сравнить результаты имитации с экспериментальными данными о продук-
тах, необходимо вычислить новую матрицу А(к) уравнения (11.28), выбрав интер-
валы к. и к.+1, соответствующие интервалам безразмерных температур для экспери-
ментальных точек для (п + 1) соединений продуктов, что дает п значений массовых
долей. Так как экспериментальные данные доступны не во всей области изменения
безразмерных температур, можно выбрать иной способ интегрирования кривой с(к,
т). Приемлемым альтернативным способом расчета массовых долей на каждом ин-
тервале желаемой области безразмерных температур является формула трапеций, так
как по ней можно получить линейное приближение кривой с(к, т) при достаточно
большом числе соединений (л).
Вычислив массовые доли в каждом интервале экспериментальных значений без-
размерных температур кипения (wtp,ed), их сравнивают с аналогичными массовы-
ми долями, найденными по экспериментальным данным (wt™1”), чтобы найти опти-
мальный набор параметров непрерывной кинетической модели. Целевой функцией
является сумма квадратов разностей J(w(0)) между расчетными и экспериментальны-
ми значениями массовых долей в одном и том же интервале безразмерных темпера-
тур, даваемая выражением
/м9))=(ц-46)
/=1
при следующих условиях:
Р> 0: Р= а, ап, а,, 8 и к v
0 1 max
и
п
Ylwt'-= L
Последнее условие можно удовлетворить увеличением числа интервалов области
изменения, т. е. выбором по возможности большего числа л,
Описанный алгоритм схематически показан на рис. 11.2. Оптимизация со-
стоит из двух шагов [Govindhakannan and Riggs, 2007]: 1) нахождение начального
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
477
распределения с(к, 0) с двумя параметрами (а и А:тах); 2) нахождение кривой с{к, т)
с пятью параметрами (а, а0, а{, 8 и Лтах).
Рис. 11.2. Процедура нахождения наилучшего набора параметров
непрерывной кинетической модели
11.3. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
11.3.1. Гидрокрекинг нефти «Майя»
Эксперименты проводились на стендовой лабораторной установке в изотермическом
реакторе с неподвижным слоем (см. рис. 6.1 главы 6 и рис. 7.14 главы 7). Подробные
описания экспериментальной установки и порядка проведения опытов приведены
478
Часть 3. Моделирование каталитических процессов
в предыдущих главах. Центром установки является изотермический реактор с вну-
тренним диаметром 2,5 см и общей длиной 143 см. Изотермические условия под-
держивались тремя электронагревателями, распределенными по высоте слоя ката-
лизатора. Температура вдоль слоя измерялась съемной термопарой, расположенной
в кармане.
Во всех экспериментах по гидропереработке сырьем служила нефть «Майя»
(см. ее свойства в табл. 9.1 главы 9). Применялся промышленный никель-молибде-
новый катализатор на гамма-алюмооксидном носителе, имевший следующие свой-
ства: удельная поверхность 175 м2/г, средний удельный поровый объем 0,56 см3/г,
средний диаметр пор 127 А, массовое содержание каталитических металлов —
10,66 % молибдена и 2,88 % никеля. В реактор загружали 100 мл предварительно
измельченного и просеянного катализатора. Конечный размер частиц катализатора
во всех опытах составлял 0,25 мм. Перед проведением опытов катализатор активи-
ровали на месте сульфидированием гидрообессеренной бензиновой фракцией, со-
державшей 0,8 %масс. CS2. Гидрокрекинг нефти «Майя» проводился в следующих
рабочих условиях: давление 9,8 МПа, LHSV= 1,5, 0,5 и 0,33 ч-1, температура в ин-
тервале 380-420 °C, постоянная кратность водорода к сырью 5000 фут’/баррель. Ис-
тинные температуры кипения сырья и продуктов определялись имитированной раз-
гонкой по методу ASTM £>5307.
11.3.2. Влияние давления на гидрокрекинг нефти «Майя»
Сырьем служила та же нефть «Майя». Наиболее подробные сведения о свойствах
этой нефти (таких как содержание металлов и серы, относительная плотность
по шкале API и т. д.) по сравнению с другими сортами сообщили Рана и соавторы
[Rana et al., 2007]. Применялся тот же промышленный катализатор NiMo/Al,O3, те же
процедуры загрузки и сульфидирования и те же методы исследования свойств сырья
и продуктов. Сведения о других подробностях экспериментов можно найти в дру-
гом источнике [Ancheyta et al., 2001]. Эксперименты проводились в той же лабо-
раторной изотермической установке в следующих рабочих условиях: давление 6,9,
8,3 и 9,8 МПа; температура 380-420 °C; кратность водорода к сырью 5000 ст. куб.
фут/баррель; LHSV= 1,5, 0,5 и 0,33 ч-1.
11.3.3. Одновременные гидрообессеривание и гидрокрекинг
тяжелой нефти
Сырьем в опытах по гидропереработке служил остаток атмосферной перегонки тя-
желой нефти с плотностью 13 ° API. Остаточное сырье обладало плотностью 5,4 ° API
и включало в себя следующие вещества: сера — 5,74 %масс.; никель и ванадий —
722 мг/кг; компоненты, нерастворимые в «-гептане, — 21,77 %масс. Сырье последо-
вательно проходило через два экспериментальных реактора с насадкой, состоящей
из никель-молибденового катализатора гидроочистки на гамма-алюмооксидном
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
479
носителе. Катализатор имел следующие свойства: номинальный диаметр частиц
1/18 дюйма, удельная поверхность 205 м2/г, удельный поровый объем 0,825 см3/г,
средний диаметр пор 160,8 А, содержание каталитических металлов — 1,12 %масс.
никеля и 4,36 %масс. молибдена. Опыты проводились в следующих рабочих усло-
виях: общая LHSV= 0,2 ч_|, температура реакции 382 °C, полное давление 9,6 МПа,
кратность водорода к сырью 5800 ст. куб. фут/баррель.
Анализ сырья и продуктов предусматривал измерение содержания серы и постро-
ение кривой разгонки. Содержание серы определяли рентгенофлуоресцентным ана-
лизатором SLFA-2100 компании HORIBA по ASTM £>4294. Коэффициент вариации
этого прибора составляет 0,006 %.
Кривую разгонки строили по истинным температурам кипения. Извлекаемые при
разгонке промежуточные фракции анализировали на содержание серы: таким обра-
зом, помимо кривой кипения, получали также кривую содержания серы.
11.4. ПОШАГОВЫЙ ПРИМЕР ПРИМЕНЕНИЯ МОДЕЛИ
11.4.1. Исходные данные
По описанному выше алгоритму был определен набор параметров непрерывной ки-
нетической модели гидрокрекинга нефти «Майя» в мягких условиях. Эксперимен-
тальные данные получали на стендовом реакторе при следующих рабочих условиях:
полное давление 6,9 МПа, температура 400 °C, LHSV = 0,5 ч~', кратность водорода
к сырью 5000 ст. куб. фут/баррель. Сырье и жидкий продукт анализировали разгон-
кой по методу ASTM£>5307, который, насколько известно, дает надежные результаты
при температурах до 538 °C (Espin osa-Peca et al., 2004]. Данные разгонки сырья сни-
мали через каждые 5 % от начальной (30,21 °C) до конечной (533,84 °C) температу-
ры кипения. Легкие газы в сырье отсутствовали. Отбираемые на выходе из реактора
жидкие и газообразные продукты анализировались отдельно. По известному балансу
масс состав смеси на выходе из реактора можно привести к нормализованному виду.
При нормализации непрореагировавший водород не учитывается.
11.4.2. Учет температуры кипения
По известным величинам ТВР(Г) и TBP(h) по уравнению (11.9) вычисляются безраз-
мерные температуры.
Самый легкий газ, которые дает гидрокрекинг, — метан, являющийся углеводо-
родом с наинизшей температурой кипения ТВР([) = -161,55 °C. Гидрообессеривание
(ГДО) дает также H2S, который необходимо учитывать в составе продукта.
В жидком продукте гидрокрекинга трудно выделить наиболее тяжелый предста-
вительный углеводород для определения наивысшей возможной температуры кипе-
ния (TBP(h)), так как метод экспериментальной разгонки ограничен температурой
538 °C. Поэтому значением TBP(h) необходимо задаться.
480
Часть 3. Моделирование каталитических процессов
На первый взгляд, выбор значения TBP(h) произволен. Внесем некоторые поясне-
ния. Вне всякого сомнения, сырье и продукты содержат углеводороды с температу-
рами кипения до 1000 °C и выше. Например, с помощью имитатора технологических
процессов HYSYS путем экстраполирования были выявлены температуры кипения
до 800—850 °C, но обнаружилась погрешность в воспроизведении данных экспери-
ментальной разгонки. Кроме того, если выбрать более высокое значение TBP(h),
то значения 0., вычисляемые по уравнению (11.9), окажутся меньше, а кривая без-
размерной температуры — более сплюснутой, так что из-за изменения порядка ве-
личин параметров и переменных погрешность дальнейших расчетов окажется выше.
Поэтому значение TBP{h) должно быть выбрано таким, чтобы оно, с одной стороны,
было как можно ближе к концу кипения сырья, а с другой — чтобы при построении
полной кривой кипения (т. е. экстраполяции до выбранной конечной температуры
кипения) экспериментальные данные кипения воспроизводились по возможности
максимально точно. В табл. 11.1 показаны выбранная наивысшая температура кипе-
ния, результаты нормализации точек кипения и соответствующие нарастающие зна-
чения массовых долей.
11.4.3. Численное решение
Чтобы начать процедуру оптимизации, область изменения безразмерной температу-
ры 0 необходимо разбить на определенное число интервалов. Замечено, что при числе
интервалов менее 40 начальное значение с(к, 0) при наложенных ограничениях опти-
мизировать не удается, поэтому рекомендуется большее количество интервалов. Не-
обходимо также выбрать оптимальный шаг приращения времени пребывания, что-
бы обеспечить достаточно низкую погрешность расчетов при приемлемых затратах
времени. Для вычисления интегралов в уравнениях (11.13)—(11.14) и (11.37)—(11.39)
нужен алгоритм квадратуры, так как подынтегральные выражения содержат анали-
тически неинтегрируемую функцию Гаусса р(к, К). В настоящей работе использова-
лась адаптивная квадратура Лобатто, а система обыкновенных дифференциальных
уравнений (см. (11.36)) решалась явным методом Рунге—Кутты 4-5-го порядка. Мас-
совые доли продуктов при максимальном времени пребывания (ттах), необходимые
для расчетов массовых долей на каждом интервале области изменения безразмерной
температуры, вычислялись по формуле трапеций. Численное решение осуществля-
лось с помощью пакета MATLAB!® (процедуры odeM> и quad\ соответственно для мето-
дов Рунге—Кутты и адаптивной квадратуры Лобатто).
11.4.4. Результаты
Перенормировка кривой кипения включением газов в жидкий продукт позволяет
предсказать все псевдосоединения продукта гидрокрекинга — от самых легких до са-
мых тяжелых. В представленном здесь случае исследования в продукте после анализа
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
481
было выявлено лишь пять различных газов, хотя модель предполагала непрерывное
распределение концентраций в продукте гидрокрекинга.
11.4.4.1. Максимальная температура кипения
Значения максимальной температуры кипения сырья и продукта (т. е. температуры,
при которой испаряется вся жидкость) нельзя ни предсказать, ни определить экспе-
риментально. Дело в том, что эмпирические зависимости способны уверенно пред-
сказать температуру испарения лишь 90—95 % жидкости (и это в том случае, если есть
надежные данные для нескольких низкокипящих фракций); экспериментальные
методы тоже дают температуры для аналогичных уровней отгонки. В случае тяже-
лой и особо тяжелой нефти и остатков перегонки предсказание температуры отгон-
ки 90-95 % жидкости посредством эмпирических зависимостей нельзя считать на-
дежным ввиду ограничений, налагаемых на максимальную температуру испарения.
Наиболее точные экспериментальные результаты дает метод разгонки для опреде-
ления истинных температур кипения (метод ИТК), но для него нужны проба боль-
шого объема, значительные затраты времени и дорогостоящая аппаратура, не счи-
тая уже упомянутых выше проблем определения температур начала и конца кипения.
К другим распространенным методам относятся высокотемпературная имитирован-
ная дистилляция и термогравиметрический анализ, позволяющие воспроизвести
кривые истинных температур кипения [Padlo and Kuger, 1996]. Первый из них ис-
пользует хроматограф, и максимально измеримые точки конца кипения ограниче-
ны предельно допустимой рабочей температурой (около 750 °C). Согласно второму
методу, по эталонным чистым углеводородам с известными температурами кипения
строится калибровочная кривая, по которой определяется кривая кипения анализи-
руемой пробы.
Отсюда следует, что не стоит определять экспериментальным методом наивыс-
шую температуру кипения таких смесей, как тяжелая нефть. Целесообразнее оценить
или измерить конечную точку кипения отдельно взятой пробы, а затем принять, что
сырье и продукты гидрокрекинга имеют ту же температуру конца кипения. Иногда
оценить максимальную температуру кипения можно по опубликованным в литера-
туре эмпирическим зависимостям, но применимость таких зависимостей к тяжелым
углеводородам находится под вопросом. Из числа других подходов можно упомянуть
линейную экстраполяцию или выбор наивысшей возможной температуры; в обоих
случаях необходимо, чтобы при соответствующей безразмерной температуре мас-
совая доля достигала единицы. Наилучшим значением максимальной температуры
кипения будет то, которое соответствует минимальному расхождению между экспе-
риментальными данными и результатами имитации. Из этих соображений можно за-
ключить, что параметры модели непрерывного кинетического агрегирования могут
принимать разные численные значения, зависящие от выбранной наивысшей темпе-
ратуры кипения псевдосоединений смеси.
В данном случае температура конца кипения была принята равной 700 °C. Можно
предполагать, что эта величина влияет на значения явных параметров кинетической
модели, хотя сама и не принадлежит к ним.
482
Часть 3. Моделирование каталитических процессов
11.4.4.2. Разбиение области изменения и линейное приближение
функции выхода
По мере увеличения числа интервалов, используемых для линейного приближения
функций непрерывной модели, баланс масс все больше приближается к единице.
Но большое число интервалов может снизить скорость расчета параметров, к тому
же будет накапливаться числовая погрешность от округления значений. Для рас-
чета функций выхода р(к, К) и начального распределения применялась линейная
интерполяция полиномами Лагранжа. Для сокращения приемлемого числа интер-
валов (я) линейное приближение можно заменить полиномами высшего порядка,
но полиномы достаточно высокого порядка могут давать приближения осциллиру-
ющего характера; помимо этого, может потребоваться больше времени для реше-
ния уравнения (Н.28). Так что можно ожидать, что простое линейное приближе-
ние даст хорошие результаты при приемлемом числе интервалов. Для начала можно
выбрать небольшое число п, чтобы предварительно сузить интервалы значений па-
раметров модели. После схождения число интервалов можно увеличить, чтобы по-
лучить лучшую оценку параметров. В данном примере для схождения баланса масс
с погрешностью +0,05 % оказалось достаточным разбиение области изменения 0
на 150 интервалов.
11.4.4.3. Шаг приращения времени пребывания
Хотя на начальном этапе сравнивать экспериментальные данные с результата-
ми имитации требуется лишь для части фракций, можно решить уравнение (11.28)
для промежуточных значений времени пребывания, чтобы избежать потери точно-
сти в балансе масс и обновить начальные значения дифференциально-интегрально-
го уравнения. В настоящей работе величина шага приращения времени пребывания
была принята равной 0,02. Меньшая величина шага в итоге давала почти такие же
значения параметров модели.
11.4.4.4. Значения параметров модели
Можно ожидать, что при расчете параметров модели наиболее трудным для оптими-
зации окажется как экспоненциальный параметр, абсолютная величина которо-
го к тому же больше, чем у других параметров. В данном случае наилучшее значение
было найдено применением представленной на рис. 11.2 итерационной процедуры
при постоянных значениях остальных параметров модели. Было отмечено, что, хотя
алгоритм оптимизации MATLAB способен сходиться близко к оптимальному значе-
нию, отклонения от предложенного значения были очень небольшими, т. е. алгоритм
программы не достигал нужной сходимости.
Применением показанной на рис. 11.2 процедуры и данных выше рекоменда-
ций был получен набор параметров непрерывной кинетической модели гидрокре-
кинга нефти «Майя» для данного случая. Параметры имели следующие значения:
а = 0,245,д = 1,396, a =22,0, S = 4,46-10-5 и к . = 0,537 ч"1.
и I max
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
483
11.4.4.5. Результаты применения модели
С использованием указанных выше значений параметров модели были получены
данные имитации, показанные наряду с экспериментальными данными на рис. 11.3.
Наблюдается хорошее согласие между расчетными и экспериментальными значени-
ями нарастающей массовой доли (иД) в зависимости от безразмерной температуры 0.
График полезен тем, что он включает все вещества в сырье и продуктах — как газо-
образные, так и жидкие. По рис. 11.3 можно воспроизвести кривую кипения. Для
этого по уравнению (11.9) следует вычислить значение физической температуры, со-
ответствующее каждому значению 0;., и перенормировать долю жидкости, предвари-
тельно исключив из общего количества продукта количество газов.
Рис. 11.3. Сравнение экспериментальных данных (значки) и результатов
имитации (линии): (•) — сырье; (*) — продукт гидрокрекинга.
Г = 400 °C, Р = 8,3 МПа, LHSV = 0,5 ч'1
Процедуру можно применять к данным при разных значениях температуры и дру-
гих переменных (таких как тип катализатора, тип сырья, давление и т. д.), чтобы
установить зависимость параметров кинетической модели от этих переменных для
любого отдельно взятого процесса гидрокрекинга. Чисто математически это возмож-
но, хотя полученные зависимости могут оказаться справедливы лишь для тех интер-
валов условий, в пределах которых были выведены значения параметров.
11.5. МОДЕЛИРОВАНИЕ ГИДРОКРЕКИНГА
НЕФТИ «МАЙЯ»
11.5.1. Экспериментальные результаты
На рис. 11.4 показано влияние LHSV на кривые кипения при температуре 420 °C
в реакторе. Хорошо видно, что чем меньше LHSV(т. е. чем больше время контакта),
484
Часть 3. Моделирование каталитических процессов
тем выше глубина гидрокрекинга. Степень (или глубину) гидрокрекинга можно
определить по графику зависимости между температурой и массовой долей сдвигом
вправо от точки с данным значением температуры до пересечения с соответствую-
щей кривой.
600
500
400
и
о
га
300
га
о.
си
i 200
£
100
0
0 20 40 60 80 100
Массовая доля, %
Рис. 11.4. Влияние LHSV на гидрокрекинг нефти «Майя»
при температуре 420 °C и давлении 9,8 МПа: (♦) — сырье; (х) — LHSV =1,5 ч~1;
(о) — LHSV = 0,5 ч-1; (*) — LHSV = 0,33 ч’1
Результаты, представленные на рис. 11.4, заставляют предполагать, что усло-
вия гидрокрекинга не слишком жесткие, так как количество легких дистиллятов
(т. е. углеводородов с температурой кипения ниже 200 °C) остается почти неизмен-
ным. Реакция наиболее избирательна к средним дистиллятам. Об этом говорят
и значения параметров пятикомпонентной дискретной модели [Sanchez et al., 2005].
Значения параметров, описывающих превращение тяжелого компонента (например,
остатка) в средние дистилляты и в легкие фракции (например, в бензин), в первом
случае больше.
На рис. 11.5 показано влияние температуры на кривые разгонки. В отличие
от LHSV, температура оказывает более выраженное влияние на выход легких дис-
тиллятов. Изменение температуры от 380 до 400 °C оказывает слабое влияние на кри-
вую разгонки продукта гидрокрекинга, но по достижении 420 °C кривая разгонки
сильно смещается вправо. Это означает, что при температурах выше 400 °C преоб-
ладает реакция гидрокрекинга, а в остальных случаях более выраженный харак-
тер носит реакция гидрирования. Как бы то ни было, температура заметнее влияет
на превращение высокомолекулярных углеводородов, т. е. углеводородов в интерва-
ле кипения вакуумного газойля и остатка перегонки; на углеводороды с температу-
рами кипения ниже 200 °C оказывают более или менее схожее влияние температуры
в интервале 380-420 °C.
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
485
600
500
У 400
га
| 300
<11
2 200
Ф
100
о
о 20 40 60 80 100
Массовая доля, %
Рис. 11.5. Влияние температуры на гидрокрекинг нефти «Майя» при LHSV = 0,33 ч-1
и Р = 9,8 МПа: (♦) — сырье; (х) — Т = 380 °C; (о) — Т = 400 °C; V) _ т= 420 °C
11.5.2. Расчет параметров
Для определения оптимального набора параметров (8, а, а0, л, и /сти) непрерывной
кинетической модели использовались экспериментальные данные разгонки про-
дуктов гидрокрекинга, полученные при наименьшей объемной скорости подачи сы-
рья (LHSV = 0,33 ч-1) и трех значениях температуры реакции. На рис. 11.6 приведе-
но сравнение расчетных и экспериментальных безразмерных кривых кипения сырья
и продукта гидрокрекинга, полученного при LHSV= 0,33 ч-1 и температуре 420 °C.
Такие безразмерные кривые строит и выводит MATLAB по введенным данным. При
Т= 420 °C оптимальные значения параметров таковы: а = 0,246, л0 = 1,487, а{ = 22,83,
8 = 1,67 • 10-6 и /<тах = 1,33 ч-1. После оптимизации выявлялась зависимость этих па-
раметров от температуры. Четыре из них (8, а, л0 и /<п|ах) показали явную зависимость
от температуры, что видно из рис. 11.7. То же самое наблюдали и другие исследова-
тели [Khorasheh et al., 2001, 2005; Ashuri et al., 2007]. Параметр в исследовавшемся
интервале температур (380—420 °C) оставался постоянным.
Рис. 11.6. Расчетные и экспериментальные безразмерные кривые кипения сырья и про-
дукта гидрокрекинга, полученного при LHSV = 0,33 ч-1 и давлении 9,8 МПа: (♦) — сырье;
(Л) — продукт крекинга при Т = 420 °C
486
Часть 3. Моделирование каталитических процессов
Рис. 11.7. Температурная зависимость параметров
непрерывной кинетической модели
1п(/гтах) = -1,933 3 -104/Г + 28,197
Из рис. 11.6 явствует, что модель дает весьма точные прогнозы. Среднее расхожде-
ние между экспериментальными и расчетными значениями не превышало 5 %.
11.5.3. Проверка модели
После того как значения параметров непрерывной кинетической модели были обна-
ружены, их применили для предсказания кривых кипения продуктов гидрокрекин-
га, полученных при значениях LHSV, отличных от 0,33 ч_|. На рис. 11.8, а представ-
лен график зависимости нарастающей массовой доли от безразмерной температуры,
который можно преобразовать в более привычный график корреляции температуры
и массовой доли отгона (рис. 11.8, б). Очевидно хорошее согласие между экспери-
ментальными и расчетными результатами. Среднее по всему набору рабочих условий
расхождение между ними тоже было меньше 5 %.
Был проведен также анализ регрессионных остатков; результаты представле-
ны на рис. 11.9. Распределение было случайным и не подчинялось каким-либо за-
кономерностям. Остатки колебались в пределах ±5 %; было всего 54 точки с поло-
жительным остатком и 51 точка с отрицательным. Эти статистические результаты
подтверждают, что оптимизированные значения параметров модели, их зависимости
от температуры и непрерывная кинетическая модель в целом достаточно точно отве-
чают экспериментальным данным кривых кипения сырья и продуктов гидрокрекин-
га нефти «Майя».
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
487
Рис. 11.8. Сравнение расчетных и экспериментальных результатов: а — зависимость
между нарастающей массовой долей и безразмерной температурой; б — кривые кипения
сырья и продукта гидрокрекинга при LHSV = 0,5 ч~’ и 9,8 МПа. Обозначения: (—) — сырье;
(х) — Т = 380 °C; (о) — Т = 400 °C; (а) — Т = 420 °C
Рис. 11.9. Диаграмма остатков для непрерывной кинетической модели
488
Часть 3. Моделирование каталитических процессов
11.5.4. Применение модели
Модель МНА, в отличие от дискретной модели, способна «предсказать» всю кри-
вую разгонки продукта гидрокрекинга. Для примера в табл. 11.2 показаны расчет-
ные составы продукта при двух вариантах разбиения (на пять и на девять компо-
нентов), вычисленные по уравнению (11.8) исходя из оптимизированных значений
параметров. Непрерывная кинетическая модель позволяет дать прогноз для все-
го интервала температур кипения. Таким образом, чтобы определить выход лю-
бой фракции, совсем необязательно пересчитывать значения параметров: можно
легко получить результаты для обоих вариантов разбиения продукта. Однако дис-
кретная модель дает их лишь для пятикомпонентного варианта; чтобы получить
аналогичные результаты для девятикомпонентного варианта, необходимо, кроме
пересчета значений параметров, провести дополнительные эксперименты. Поэто-
му сравнить результаты можно лишь для пятикомпонентного варианта разбиения
продукта. Обе модели дают сравнительно небольшие отклонения расчетного соста-
ва от экспериментального.
Таблица 11.2
Составы продукта гидрокрекинга, найденные опытным путем и рассчитанные по дискретной
пятикомпонентной и непрерывной кинетическим моделям (7= 400 °C, LHSV= 0,5 ч*1)
Кол-во компонен- тов Фракция, °C Интервал темпера- тур, °с Состав продукта, %масс.
Опытные значения Пятикомпо- нентная модель Непрерыв- ная модель
Пять компо- нентов Газы С-НЮ 2,71 2,67 2,40
Бензиновая фракция НК-204 11,90 14,17 12,87
Средние дистилляты 204-343 26,86 27,62 24,74
Сырье каталитического крекинга 343-538 36,03 36,80 37,34
Остаток перегонки 538+ 22,50 18,74 22,65
Девять компо- нентов Газы С-НК 2,71 — 2,40
Легкая бензиновая фракция НК-177 7,71 — 9,19
Тяжелая бензиновая фракция 177-204 4,19 — 3,68
Фракция реактивного топлива 204-274 13,36 — 11,88
Керосин 274-316 8,23 — 7,85
Легкий газойль 316-343 5,27 — 5,01
Тяжелый газойль 343-454 21,93 — 21,88
Вакуумный газойль 454-538 14,12 — 15,46
Остаток перегонки 538+ 22,50 — 22,65
“ НК — температура начала кипения жидкого продукта. Здесь она принята равной темпера-
туре кипения «-пентана при атмосферном давлении (35,05 °C).
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
489
Диаграмма на рис. 11.10 сравнивает экспериментальные данные выхода де-
вятикомпонентного набора продуктов гидрокрекинга при различных величинах
температуры и LHSV с расчетными, которые были вычислены по непрерывной
кинетической модели. Показаны также величины коэффициента корреляции г2,
углового коэффициента S полученной прямой и ординаты Ь точки ее пересечения
с осью расчетных значений. Видно, что кинетическая модель достаточно хоро-
шо предсказывает экспериментальные значения, так как г2 и S близки к единице,
а b — к нулю.
Рис. 11.10. Сравнение расчетных и экспериментальных
значений нарастающей массовой доли выхода различных фракций
при различных величинах температуры и LHSV
Главное достоинство непрерывной модели заключается в том, что можно пред-
сказать состав продукта при различных интервалах кипения. Это иллюстрирует
рис. 11.11, где показан выход бензина в зависимости от конечной температуры ки-
пения, выбранной в качестве независимой переменной. Наблюдаемое поведение
соответствует ожидаемому: т. е. чем выше температура реакции, а также чем выше
верхняя граница кипения бензиновой фракции, тем больше выход бензина. Раз-
ница между значениями выхода бензина при температурах реакции 380 и 400 °C
несущественна, тогда как при 420 °C она становится более выраженной. Это об-
стоятельство говорит в пользу того, что при температурах выше 400 °C реакция
гидрокрекинга начинает преобладать над реакцией гидрирования. Возможность
вычислять состав продукта при различных интервалах кипения весьма полезна
в условиях, когда из-за экологических ограничений приходится изменять границы
кипения продуктов.
490
Часть 3. Моделирование каталитических процессов
Верхняя граница кипения бензиновой фракции, "С
Рис. 11.11. Расчетные значения выхода бензина при LHSV = 0,33 ч-1 в зависимости
от температуры реакции и конечной температуры кипения фракции: (—) — сырье;
(х) — КК = 380 °C; (о) — КК = 400 °C; (А) — КК = 420 °C
11.6. МОДЕЛИРОВАНИЕ ВЛИЯНИЯ ДАВЛЕНИЯ
И ТЕМПЕРАТУРЫ НА ГИДРОКРЕКИНГ НЕФТИ «МАЙЯ»
11.6.1. История исследований
11.6.1.1. Обзор литературы
Несколькими исследователями изучалось влияние температуры реакции и скорости
подачи сырья на кинетику гидрокрекинга [Scherzer and Gruia, 1996; Rassev, 2003]. Это
влияние на структуру выхода известно хорошо: чем выше температура, тем выше вы-
ход легких фракций; чем меньше LHSV, тем выше суммарный выход продукта. Изу-
чению влияния давления на кинетику гидрокрекинга и гидроочистки посвящено
лишь небольшое число публикаций [Jenkins and Stephens, 1980; Rassev, 2003; Sanchez
and Ancheyta, 2007]. Согласно утверждениям их авторов, давление по-разному влия-
ет на скорость этих реакций в зависимости от характеристик сырья, типа катализато-
ра и рабочих условий.
К примеру, Куадер и Хилл [Qader and Hill, 1969] наблюдали при полных давле-
ниях до 10,3 МПа линейную зависимость выхода газа гидрокрекинга от давления,
но на выход бензина оно оказывало меньшее влияние, определяемое интервалами
его значений при исследованиях. Самби и соавторы [Sambi et al., 1982] обнаружили
снижение плотности и вязкости продуктов по мере повышения давления, что харак-
терно для высокой степени гидрокрекинга. Санчес и Анчита [Sanchez and Anchey-
ta, 2007] сообщили, что влияние давления носит более выраженный характер при
первичном гидрокрекинге каждого псевдокомпонента. Они также установили экспе-
риментальным путем, что давление влияет на гидрокрекинг слабее, чем LHSVи тем-
пература, что подтверждает опубликованные ранее результаты. Ботчуи и соавторы
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
491
[Botchwey et al., 2003], изучавшие мягкий гидрокрекинг газойля в интервалах темпе-
ратур от 340 до 420 °C, LHSVот 0,5 до 2 ч_| и давлений от 6,5 до 11 МПА, наблюдали
незначительное влияние давления на структуру распределения продуктов и избира-
тельность в исследовавшихся интервалах условий.
11.6.1.2. Влияние давления
Большинство опубликованных исследований не учитывают влияние давления, так
как этот параметр при экспериментах поддерживается постоянным, и представля-
ют скорость реакции в виде простой зависимости от константы скорости и концен-
трации углеводородов. Константа скорости связывается с температурой посредством
уравнения Аррениуса, хотя в сложных системах иногда необходимо учитывать влия-
ние и других параметров реакций. Сообщается [Mapiour et al., 2010], что сложные си-
стемы лучше представлять многопараметрическими моделями, так как они учитыва-
ют большее число параметров процесса и тем самым позволяют выявлять их влияние
на скорость реакции.
В случае дискретного агрегатного моделирования влияние давления и темпера-
туры принято выражать измененным уравнением Аррениуса, имеющим следующий
вид [Jenkins and Stephens, 1980; Dufresne et al., 1987; Yui and Sanford, 1989; Ancheyta
and Villafuerte, 2000; Ancheyta et al., 2002]:
Z \w2
*гдк= V'£rw/*7' -Я • (11.47)
)
Необратимую реакцию гидрокрекинга (ГДК) часто моделируют кинетикой, под-
чиняющейся степенному закону:
_/гдк = ^гдк^'с' ^н,' • (11 -48)
Уравнение (11.48) описывает скорость реакции гидрокрекинга как функцию кон-
станты скорости кТ№, концентрации углеводородов Сс и парциального давления во-
дорода Рп. Парциальное давление водорода вместо его концентрации в жидкости
используется по той причине, что количество газов принято измерять не концентра-
цией, а парциальным давлением. В соответствии с законом Генри, концентрация рас-
творенного водорода в жидкости прямо пропорциональна его парциальному давле-
нию, и в реакторах с орошаемым слоем парциальное давление отслеживать легче,
чем концентрацию.
Когда благодаря большому избытку водорода его парциальное давление поддер-
живается постоянным, вводится псевдоконстанта скорости реакции, учитывающая
парциальное давление водорода и константу скорости реакции:
~ ггдк = ^гдк^'с' ’ (11.49)
^гдк “ ^тдк^н,' • (11.50)
492
Часть 3. Моделирование каталитических процессов
Сравнивая уравнения (11.47) и (11.50), можно заключить, что константа скорости
£гдк в уравнении (11.47) является на самом деле псевдоконстантой (к*Г№).
Величину п2, которая, согласно формуле (11.48), представляет собой порядок ре-
акции водорода, можно считать близкой к 0,5. В пользу этого говорят следующие
аргументы. Реакции гидрирования ароматических углеводородов потребляют раз-
ные количества водорода в расчете на один моль, но расход водорода, приходящий-
ся на один превращенный атом углерода, постоянен и равен 0,5 молям. Таким обра-
зом, в случае одного ароматического атома углерода п2 = 0,5 [Yui and Sanford, 1991].
Санчес и Анчита [Sanchez and Ancheyta, 2007] заметили, что при гидрокрекинге ва-
куумных дистиллятов величина п2 колеблется между 0,5 и 0,8. Для других псевдоком-
понентов величина п2 для первичного гидрокрекинга может быть больше благодаря
высокой концентрации ароматических углеводородов, тогда как для гидрокрекинга
легких фракций она меньше и может быть даже отрицательной, как установили Бал-
танас и соавторы [Baltanas et al., 1983]. Такое поведение наблюдали и другие исследо-
ватели: например, при любых значениях давления и переменной температуре выход
газов оставался практически постоянным, но при самой низкой температуре степень
превращения непрерывно возрастала в условиях переменных парциальных давлений
водорода [Qader and Hill, 1969]. То же явление наблюдалось с увеличением полного
давления при гидрокрекинге нефти «Майя» в умеренных условиях [Sanchez and An-
cheyta, 2007].
11.6.1.3. Значимость влияния давления
Когда происходит реакция гидрокрекинга, количество прекурсоров кокса возрастает.
Вследствие этого поверхность катализатора может стать насыщенной, т. е. покрытой
коксом и углеводородами. При последующем увеличении давления площадь катали-
тической поверхности, доступная для протекания реакции, уже не влияет на степень
превращения [Ancheyta et al., 2009].
Парциальное давление водорода влияет не только на скорость гидрокрекинга,
но и на другие явления, происходящие во время реакции. К примеру, слишком высо-
кое давление может вызвать насыщение катализатора, так как ускоряется образова-
ние кокса, снижающее активность катализатора, и дальнейшее увеличение давления
не дает ожидаемого изменения активности [Speight, 2000]. Кроме того, проводить ре-
акцию при слишком высоких давлениях не позволяют конструктивные ограничения
(в основном со стороны реакторов).
Таким образом, учет влияния давления в кинетической модели гидрокрекинга бо-
лее чем оправдан.
11.6.2. Учет влияния давления
Зависимость параметров непрерывной кинетической модели от температуры ра-
нее обычно представлялась следующим образом [Khorasheh et al., 2001; Pacheco and
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
493
Dassori, 2002]. В коротких интервалах температур значения а, а0, а, и 5 линеаризуют-
ся зависимостью вида
а, а0, at, 5 = b + тТ, (11.51)
а к связывается с температурой зависимостью Аррениуса
1п(^ах) = й + /и/Г. (11.52)
В соответствии с уравнением (11.47), зависимость ктт от температуры и давления
можно выразить следующим образом:
In (£ ) = Л + —+ Clnf—\ (11.53)
4 max7 ут I р I
Остальные параметры модели (а, а0, а, и 5) подчиняются зависимости вида
Величины А и В учитывают влияние температуры на параметры модели [Kho-
rasheh et al., 2001; Elizalde et ah, 2009]. Величины С и D учитывают соответственно
влияние давления и совокупное влияние давления и температуры (эти переменные
могут действовать синергически, изменяя распределение выхода продуктов гидро-
крекинга). Все эти величины — А, В, С и D — зависят от типа катализатора и соста-
ва сырья.
Величины То и Ро в уравнении (11.54) равны наименьшим значениям соответствен-
но температуры и давления; здесь они приняты равными То = 380 °C и Ро = 6,9 МПа.
Уравнение (11.8) решалось для различных условий, и с использованием алгорит-
ма Левенберга-Марквардта был найден оптимальный набор параметров модели. Ре-
шение уравнения (11.8) начинается с наиболее тяжелого псевдокомпонента, так как
для этого нужен лишь первый член с правой стороны уравнения. Затем расчет выпол-
няется для более низкокипящего псевдокомпонента — и так далее до тех пор, пока
не будет достигнут самый легкий из них, который в типичных условиях реакций при-
нимается химически неактивным. В данном случае за наиболее легкое соединение
был принят метан; температура кипения самого тяжелого псевдокомпонента при ат-
мосферном давлении была взята за 700 °C.
Решение уравнения (11.8) относительно с(к, f) позволяет по формуле (11.15) вы-
числить состав смеси (C(t)) в любой момент времени в любом сечении реактора.
В качестве критерия оптимизации была принята сумма расхождений между экс-
периментальными и расчетными значениями. (Подробнее о численном решении
таких уравнений можно узнать в других источниках [Laxminarasimhan et al., 1996;
Elizalde et al., 2009].) Порядок решения был следующим. Данные разгонки сы-
рья и продуктов гидрокрекинга, полученные с шагом в 5 %масс., преобразовыва-
лись в безразмерную температуру (см. (11.9)). Затем по процедуре Говиндхаканнана
494
Часть 3. Моделирование каталитических процессов
и Риггса [Govindhakannan and Riggs, 2007] находили начальное распределение кон-
центраций с(к, 0). Для улучшения точности результатов имитации интервал изме-
нения активности разбивали на 150 интервалов. Наконец, по методике Лаксми-
нарасимхана и соавторов [Laxminarasimhan et al., 1996] (см. (11.8)) модель решали
в пакете MATLAB для каждой пары значений температуры и давления и наимень-
шей LHSV. Оптимизировав для каждой пары значений температуры и давления
значения параметров непрерывной кинетической модели, строили для них зависи-
мости вида (11.53) и (11.54).
11.6.3. Анализ результатов
11.6.3.1. Экспериментальные данные
На рис. 11.12 показаны экспериментальные кривые разгонки жидкого продукта, по-
лученного при различных условиях реакции. Наблюдаемое поведение типично для
гидрокрекинга: чем ниже удельная скорость подачи сырья, тем больше выход лег-
ких фракций, а чем выше температура, тем больше выход жидкости. Видно, что для
отдельно взятого значения давления наибольшая степень превращения достигается
при наименьшей LHSVvi наибольшей температуре.
Давление в реакторе сильнее всего влияет на выход средних дистиллятов и фрак-
ций с более высокой температурой кипения (больше 200 °C). Иными словами, сред-
ние значения давления не благоприятствуют выходу таких легких дистиллятов, как
бензин. Это наблюдение подтверждается тем, что фракции с температурами кипе-
ния от начальной до примерно 200 °C не подвергаются вторичному крекингу. Газы
образуются только вследствие крекинга тяжелых фракций. При наинизшем зна-
чении давления (6,9 МПа) глубина гидрокрекинга была меньше, чем при наивыс-
шем (9,8 МПа). Это дает основание полагать, что при 6,9 МПа преобладает реак-
ция гидрирования, при более высоких давлениях — реакция гидрокрекинга. Такое
поведение обусловлено тем, что промежуточные продукты, образовавшиеся в ре-
зультате реакции гидрирования, затем легко расщепляются. Влияние парциального
давления наиболее заметно при низких температурах и с ее повышением усиливает-
ся незначительно.
11.6.3.2. Зависимость параметров модели от давления
и температуры
На рис. 11.13, а показана зависимость значений параметров непрерывной кине-
тической модели от температуры при трех значениях давления. Можно видеть,
что в исследованном интервале температур (380—420 °C) эта зависимость являет-
ся линейной [Elizalde et al., 2009]. Параметр а, от температуры вообще не зависит.
В табл. 11.3 приведены значения констант зависимостей (11.51) и (11.52) при дав-
лении 6,9 МПа.
Отгон, %масс. Отгон, %масс. Отгон, %масс.
Температура, °C
Рис. 11.12. Кривые кипения продуктов гидрокрекинга, полученных при различных условиях реакции.
Значения LHSV: (—) — 1,5 ч'1; (•) — 0,5 ч~1; (- - -) — 0,33 ч~1
Р = 9,8 МПа
Т =400 °C
Температура, °C
Температура, °C
Глава 11 Моделирование гидрокрекинга непрерывным агрегированием кинетики
496
Часть 3. Моделирование каталитических процессов
□
□
□
ю
Е
1п(Р/Р0)
(1 /7)-103, К-1
Рис. 11.13. Зависимость параметров непрерывной кинетической модели от: а — темпе-
ратуры при (□) — ₽= 6,9 МПа; (о) — Р = 8,3 МПа; (А) — Р = 9,8 МПа; б — температуры
и давления при (□) — Т = 380 °C; (о) — Т = 400 °C; (А) — Т = 420 °C. Расчетные значения
показаны линиями, экспериментальные — значками
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
497
Таблица 11.3
Константы зависимостей (11.51) н (11.52) при давлении 6,9 МПа
Параметр модели (у) Температурно-зависимая переменная (х) у = Ь + шх
bi m.
а Т -0,686 2,32-10“3
ао Т -0,633 5,2 • 10-3
т 22,83 0
5 т 3,72-10-“ -8,84- IO’7
1/Т 23,355 -1,6254-104
Примечание. Твыражается в °C, 1 / Т— в К’1.
На рис. 11.13, б демонстрируется совокупное влияние давления и температуры
на значения параметров модели. Эта зависимость, аналогично температурной зави-
симости (см. рис. 11.13, а, табл. 11.3), для а, а0,5 и Лтах носит линейный характер, в то
время как опять остается независимой от этих двух переменных.
Опытные и промышленные реакторы с орошаемым слоем работают при высоких
температурах, что способствует расширению газа и препятствует растворению га-
зообразных реагентов в жидкости [Al-Dahhan et aL, 1997]. Высокие давления улуч-
шают растворимость газообразных реагентов. Другие выгоды от высоких давле-
ний — улучшение массопереноса, переработка больших объемов газа при меньших
затратах и замедление деактивации катализатора. Давление влияет на такие фи-
зико-химические свойства газов и жидкостей, как плотность, динамическая вяз-
кость, коэффициент молекулярной диффузии, растворимость газов, коэффициент
межфазного натяжения и удельная теплоемкость, хотя при типичных для гидрокре-
кинга значениях основное его влияние заключается в уменьшении коэффициента
молекулярной диффузии газов. Аль-Даххан и соавторы [Al-Dahhan et al., 1997] по-
казали, что растворимость водорода в тетралиновой смеси с увеличением его пар-
циального давления повышается почти линейно. Мапюа и соавторы [Mapiour et al.,
2009] заметили, что с увеличением чистоты (парциального давления) водорода
от 50 до 100 % плотность продуктов гидроочистки изменяется линейно. Все эти на-
блюдения подтверждают линейную зависимость параметров непрерывной кинети-
ческой модели от давления.
Давление при невысоких его значениях оказывает незначительное влияние на па-
раметры модели, за исключением /стх, о чем говорит малая величина коэффициента
наклона кривых в исследованном интервале температур. С повышением температу-
ры влияние давления усиливается, но по достижении наивысшей температуры осла-
бевает. Это согласуется с экспериментальными результатами, свидетельствующими
о том, что при высоких температурах давление влияет на кривую разгонки меньше,
чем при низких.
498
Часть 3. Моделирование каталитических процессов
11.6.3.3. Значения параметров модели как функции давления
В табл. 11.4 приведены значения констант зависимостей (11.53) и (11.54). Величи-
на показателя безразмерного давления для fcmax, полученная путем регрессии, равна
С = 0,7223, что очень близко к опубликованному ранее [Sanchez and Ancheyta, 2007]
подобному значению для образования средних дистиллятов из остатка перегонки
в дискретной кинетической модели (С = 0,69), полученному на основании анало-
гичных экспериментальных результатов. Эти более или менее схожие значения со-
ответствуют гидрокрекингу тяжелой нефти при умеренных условиях реакций, ори-
ентированных на максимальный выход средних дистиллятов [Ancheyta et al., 2001;
Valarasu et al., 2003]. Совпадение значений С в моделях (дискретной и непрерывной)
можно объяснить тем, что оба подхода дают среднее значение показателя для реакци-
онной схемы одного и того же набора псевдокомпонентов. Как можно видеть, с уве-
личением давления реакции увеличивается также значение kmsx. Следовательно, воз-
растает и скорость реакции гидрокрекинга, хотя давление в этом отношении влияет
слабее, чем LHSVи температура. Остальные параметры модели по-разному реагиру-
ют на изменение полного давления. Повышение давления вызывает увеличение зна-
чений а и а0, в то время как значение 5 уменьшается.
Таблица 11.4
Значения констант зависимостей (11.53) и (11.54)
Параметр модели Значения констант в зависимостях (11.53) и (11.54)
А В С D
а -2,635 2,845 1,301 -1,318
°0 -4,534 5,855 2,731 -2,705
22,83 0 0 0
5 1,408-10-3 -1,328-10-3 -7,841-10'" 7,408-10“"
1п(Л ) 4 max' 27,396 -1,8994-10" 0,7223 0
Примечание. Для а, а0, 6 и /стах Ти TQ выражаются в К; Р и PQ выражаются в МПа.
Значение константы Л зависимости (11.54), описывающей влияние температуры,
для трех параметров модели (а, а0 и 5) почти в два раза превышает значение констан-
ты С, отражающей влияние давления. Это обстоятельство говорит в пользу того, что
давление на гидрокрекинг тяжелой нефти влияет слабее, чем температура. С изме-
нением давления меняются и некоторые физико-химические свойства реакционной
системы (такие как плотность, вязкость и т. д.), что в свою очередь влияет на коэф-
фициент молекулярной диффузии газа и жидкости. Образование газа с увеличени-
ем давления ускоряется, но влияние этого обстоятельства на динамику сред отчасти
компенсируется большим избытком водорода, который играет преобладающую роль
в динамике газовой фазы. Таким образом, зависимость параметров непрерывной
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
499
кинетической модели от давления и температуры определяется не только чисто ки-
нетическими аспектами: большое влияние оказывают также термодинамические
и гидрогазодинамические эффекты в газожидкостной смеси.
11.6.3.4. Расчетные кривые разгонки
С использованием зависимостей (11.53), (11.54) и приведенных в табл. 11.4 значений
параметров непрерывной кинетической модели был проведен расчет кривых разгон-
ки продуктов гидрокрекинга, полученных при условиях, отличных от тех, при кото-
рых были вычислены параметры модели, т. е. при других значениях LHSV. Сравне-
ние расчетных и экспериментальных точек показано на рис. 11.14 в виде диаграммы
остатков.
Рис. 11.14. Диаграмма остатков для данных разгонки, полученных
по непрерывной кинетической модели
Остатки вычислялись как разность между экспериментальными и расчетными зна-
чениями данных разгонки. Экспериментальные значения брались через каждые 5 %
отгонки продукта; те же значения массовой доли вычислялись по данным расчет-
ной кривой разгонки. Для определения остатков использовалось в общей сложно-
сти 452 экспериментальных значения. Диаграмма подтверждает точность модельных
расчетов гидрокрекинга тяжелой нефти в умеренных условиях, так как точки слу-
чайно распределены в пределах ±4 %. Кроме того, число положительных остатков
почти совпадает с числом отрицательных; это говорит о том, что модель не завышает
и не занижает результаты относительно экспериментальных значений.
На рис. 11.15 показаны расчетные кривые разгонки продуктов гидрокрекинга, по-
лученных при LHSV= 0,5 ч_), Т= 420 °C и трех значениях давления, а также аналогич-
ные экспериментальные данные. Видно, что в интервале исследованных значений
давления его влияние сравнительно невелико. Можно также видеть, что на высоко-
кипящие (с температурой кипения выше 250 °C) фракции давление влияет заметнее,
500
Часть 3. Моделирование каталитических процессов
чем на легкие: к примеру, в интервале 0—30 % кривые перекрываются, что говорит
о минимальном влиянии давления. Это соответствует результатам других исследо-
ваний, согласно которым значение показателя безразмерного давления для реак-
ций высококипящих псевдокомпонентов больше, чем для реакций низкокипящих.
То есть чем выше температура кипения дистиллятов, тем больше показатель степени
и тем сильнее влияние давления [Sanchez and Ancheyta, 2007].
Рис. 11.15. Расчетные кривые разгонки продуктов гидрокрекинга, полученных при
LHSV = 0,5 ч-1 и Г =420 °C: (□) — Р = 6,9 МПа; (о) — р= 8,3 МПа; (Л) —Р = 9,8 МПа.
Расчетные значения показаны линиями, экспериментальные — значками
На рис. 11.16, а показаны расчетные безразмерные кривые разгонки продуктов
гидрокрекинга, полученных при LHSV= 0,5 ч-1 и различных значениях температур
и давлений; для сравнения представлены также экспериментальные данные. График
представляет все компоненты смеси — от наиболее легких газообразных до самых тя-
желых жидких. Чтобы не загромождать график, показаны лишь три кривые для про-
дуктов, полученных при различных условиях, и кривая для сырья. Можно видеть
хорошее согласие между расчетными и экспериментальными кривыми. По расчет-
ным результатам рис. 11.16, а построены обычные кривые разгонки жидкого про-
дукта, показанные на рис. 11.16, б. К средним дистиллятам на этих кривых отнесены
углеводороды с температурами кипения выше 200 °C и ниже 350 °C. Построена также
кривая разгонки сырья, демонстрирующая влияние процесса гидрокрекинга на вы-
ход жидких продуктов. На рисунке показано также, что в случае бензиновой фракции
(температура кипения ниже 200 °C) происходит небольшое, но заметное изменение
в составе. Если для прогнозирования выхода бензиновой фракции использовать дис-
кретную модель, то конечная температура кипения фиксирована (т. е. равна 200 °C)
и при других ее значениях вычислить выход невозможно, пока не будут пересчита-
ны кинетические параметры дискретной модели. Иначе говоря, эта модель не годит-
ся для прогнозирования выхода продуктов при переменных температурах кипения.
Модель непрерывного агрегирования лишена этого недостатка: она позволяет пред-
сказать всю кривую разгонки и определить выход продукта с любым интервалом
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
501
кипения. Этот факт говорит о малопригодное™ модели дискретного агрегирова-
ния для расчета состава продуктов гидрокрекинга. Погрешности в расчете состава,
в свою очередь, порождают ошибки в расчетах свойств, зависящих от состава.
Отгон, %масс.
Рис. 11.16. Расчетные безразмерные кривые разгонки (а); кривые разгонки продуктов (б),
полученных при LHSV = 0,5 ч'1 и при следующих условиях: (□) — Т = 380 °C, Р = 6,9 МПа;
(о) — т = 400 °C, Р = 8,3 МПа; (А) — Т = 420 °C, Р = 9,8 МПа; (•) — кривая разгонки
сырья. Расчетные значения показаны линиями, экспериментальные — значками
Параметры модели непрерывного кинетического агрегирования для суммарной
реакции гидрокрекинга (т. е. для гидрокрекинга фракций без различения в них аро-
матических, нафтеновых и парафиновых углеводородов) коррелируют с LHSV, тем-
пературой и давлением, так что могут быть использованы для имитации влияния
последних на гидрокрекинг тяжелой нефти. Используя подобные данные и пятиком-
понентную кинетическую модель, Санчес и Анчита [Sanchez and Ancheyta, 2007] по-
казали, что повышение давления при гидрокрекинге в умеренных условиях сильнее
отражается на тяжелых фракциях, чем на легких.
502
Часть 3. Моделирование каталитических процессов
Разработанная непрерывная кинетическая модель и зависимости ее параметров
от температуры и давления были проверены расчетом кривых разгонки таких про-
дуктов гидрокрекинга, что были получены при значениях LHSV, отличных от тех,
при которых были выведены значения параметров. Сравнение кривых разгонки, по-
лученных экспериментальным и расчетным путями, показывает хорошее согласие.
11.7. СОВМЕСТНОЕ МОДЕЛИРОВАНИЕ
ГИДРООБЕССЕРИВАНИЯ ТЯЖЕЛОЙ НЕФТИ
Ранее гидрокрекинг и гидрообессеривание (ГДО) исследовали как отдельные реак-
ции. В переработке тяжелой нефти и остаточного сырья эти реакции считаются важ-
нейшими. В литературе широко обсуждаются различные подходы к моделированию
гидрокрекинга [Scherzer and Gruia, 1996; Froment, 2005; Balasubramanian and Push-
pavanam, 2008; Ho, 2008]. В то же время в последних публикациях появляются обзо-
ры различных моделей кинетики ГДО [Mederos et al., 2009а; Marafi et al., 2010]. Если
не считать множества публикаций, посвященных раздельному рассмотрению этих
двух реакций, то работы, освещающие моделирование совместного протекания про-
цессов гидрокрекинга и ГДО, можно пересчитать по пальцам. К примеру, Верстра-
ете и соавторы [Verstraete et al., 2007] использовали дискретное агрегирование для
моделирования гидрокрекинга вакуумного остатка и расчета концентраций серы,
азота и металлов как функций координаты вдоль оси реактора. Садиги и соавторы
[Sadhighi et al., 2010] смоделировали процесс гидрокрекинга с учетом влияния ги-
дроочистки на его граничные условия, а Хорашех и соавторы [Khorasheh et al., 2005]
успешно воспроизвели распределение серы в битумном сырье как функцию времени
и положения в проточном реакторе идеального смешения (CSTR).
11.7.1. Описание модели
11.7.1.1. Модель гидрокрекинга
Экспериментальная установка моделировалась как одномерный псевдогомогенный
реактор. Реакции гидрокрекинга отображались классической непрерывной кинети-
ческой моделью [Laxminarasimhan et al., 1996]. Таким образом, баланс масс в реакторе
можно представить дифференциально-интегральным уравнением (11.55). Это и по-
следующие уравнения аналогичны формулам (11.8)—(11.15) с подстановкой £гдк вме-
сто k [Elizalde and Ancheyta, 2012]:
^с(^дК,т) = с J ^хс (11.55)
^ГДК
Уравнение утверждает, что скорость изменения концентрации с(/сгдк, т) всякого
псевдосоединения как функции времени пребывания т и активности Лгдк в реакции
гидрокрекинга зависит от двух факторов:
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
503
• скорости расщепления этого соединения (первый член в правой части);
• скорости его образования из более крупных молекул, т. е. из более высококипя-
щих соединений (второй член в правой части).
Подынтегральная функция р(Агдк, А) определяется выражением (11.56):
P(k™’ ® Sa^ е>,-да/хГ-0.5}/о,]2 _A + B , (И.56)
где A = e-(0.5/O,)2. (11.57)
£ = 8[1 -(*raK/A)]. (11.58)
Выражение (11.56), представляющее функцию распределения выхода, учитывает
образование вещества с активностью &гдк из веществ с активностью К> 1сГ№-
К другим свойствам функции р(кгак, К) относятся [Laxminarasimhan et al., 1996]:
а) Х^гдк’ А) = 0 при /сгдк = А;
б) р(/<гдк, А), удовлетворяющее уравнению баланса масс
К
^P(x,K)D(x)dx= \\ (11.59)
о
в) р(/сгдк, А), являющееся положительной функцией в области допустимых зна-
чений параметров модели;
г) р(Л:гдк, А) = 0, если &гдк > А (это означает несущественность эффектов
димеризации).
D как функция активности представляет собой якобиан преобразования дискрет-
ного распределения углеводородов в непрерывное [Chou and Но, 1988]. Математиче-
ски D выражается следующим образом:
Л*гда) = -^да. (11.60)
^гпах
К другим вспомогательным уравнениям модели гидрокрекинга относятся:
g — “ТДК _ gl/a.
^max
^ГДК _ gl/a.
^max
D(x)dx\
k2
C(f) = j c(x, t) D(x)dx-,
k.
(11.61)
(11.62)
(11.63)
(11.64)
504
Часть 3. Моделирование каталитических процессов
С'(?)= j c(x,t)D(x)dx-, (11.65)
О
— j D(x)dx = 1. (11.66)
N о
Параметрами модели являются а, а0, ар 5 и Лтах; они могут принимать только по-
ложительные значения. В непрерывной кинетической модели удовлетворяются и не-
которые другие ограничения, в том числе закон сохранения массы и критерий нор-
мальности распределения по типам веществ (см. (11.65)).
11.7.1.2. Модель гидрообессеривания
В целях моделирования ГДО было принято, что реакционная смесь содержит очень
большое число соединений серы. Сау и соавторы [Sau et al., 1997] получили следую-
щее соотношение между безразмерной температурой кипения 0 и активностью кГ
соединения серы в реакции ГДО:
j/
*гдо = ^-^1п г-(е--1)0₽ . (11.67)
Данное соотношение было получено из результатов экспериментов с модель-
ными соединениями. По сути, оно представляет собой выражение с подгоночны-
ми параметрами [Narasimhan et al., 1999]. Уравнение (11.67) показывает, что актив-
ность соединения серы обратно пропорциональна температуре кипения фракции,
т. е. наиболее активные соединения содержатся в легких фракциях, а наиболее стой-
кие — в тяжелых [Sau et al., 1997]. Непрерывное моделирование кинетики позволяет
представить в виде непрерывного распределения конечное, хотя и неопределенное,
число соединений в нефтяной смеси. В идеале кинетическое поведение всякого со-
единения серы — это инвариант относительно такого представления, так что для вза-
имной непротиворечивости дискретного и непрерывного представлений необходи-
мо ввести некоторый множитель. Этот множитель, представляющий собой якобиан
перехода от координат 0 к координатам к, был принят равным следующему соотно-
шению [Narasimhan et al., 1999]:
Ждо)=^-^-. (11.68)
""тдо
Производную от 0 по коэффициенту активности в реакции ГДО (^гдо) можно най-
ти из уравнения (11.67).
Если считать, что происходят лишь реакции ГДО различных соединений серы, то
для каждого такого соединения можно написать следующий дифференциальный ба-
ланс массы в реакторе идеального вытеснения:
de
= (11.69)
ят
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
505
Согласно приведенным выше публикациям, порядок реакции принимается рав-
ным 1 [Но, 1991]. Общую концентрацию серы в жидкости можно вычислить тем же
способом, что и в случае чистого гидрокрекинга, т. е.
S
w4°'a/W= J С5(^ГД0,т)О(^Д0)^гД0. (11.70)
^min S
Можно заметить, что в подынтегральном выражении уравнения (11.70) функ-
ция -Р(£гдо) играет роль весового множителя, аналогично в случае гидрокрекинга.
Как видно из рис. 11.17 [Khorasheh et al., 2005], реакция гидрообессеривания в ре-
акторе гидроочистки протекает параллельно реакции гидрокрекинга. Тогда, с учетом
показанных маршрутов реакций, материальный баланс можно вместо (11.69) напи-
сать в виде
dc кТ
,5 = — (Лгдо + ^Тдо) £5+ I р(к,х) xc$D(x)dx. (11.71)
Это означает, что изменение концентрации всякого соединения серы как функ-
ции времени пребывания обусловлено двумя процессами:
1) уменьшением вследствие гидрирования и расщепления высокомолекулярных
соединений на низкокипящие (первый член в правой части, описываемый &гдо);
2) увеличением из-за одновременного гидрокрекинга высокомолекулярных
фракций (второй член в правой части, описываемый А: к). Этот член подобен ана-
логичному члену в случае чистого гидрокрекинга — за тем исключением, что вместо
с(АГД0, t), т. е. концентрации высококипящих углеводородов, необходимо подстав-
лять концентрацию серы cs в этих углеводородах.
/сГдк Низкомолекулярные
соединения серы
Соедине-
ния серы
fc-??"'''- Неуглеводородные
I ЦЕ* т П эЭ
соединения серы 1
Рис. 11.17. Превращение соединений серы в реакциях гидрообессеривания
и гидрокрекинга [Khorasheh, F. et al., Petrol. Coa/47, 40-48, 2005]
Следует заметить, что как А:гдк, так и Агдо являются функциями безразмерной тем-
пературы, так что активности ГДО всякого соединения соответствует определенная
активность гидрокрекинга. Таким образом, Агдк и Агдо можно взаимоувязать через
уравнения (11.61) и (11.67), хотя это может потребовать интерполяции. В набор па-
раметров модели ГДО, кроме параметров гидрокрекинга, входят (3, AmaxS и A:minS. Здесь
AmnxS — активность ГДО наиболее низкокипящего, AminS — наиболее высококипящего
соединения серы. Если в уравнении (11.71) к^ принимает нулевое значение, то фор-
мула сокращается до (11.69). То есть ГДО не зависит от гидрокрекинга, так как рас-
пределение температур кипения в продукте остается неизменным.
506
Часть 3. Моделирование каталитических процессов
11.7.2. Решение модели
Распределение серы и концентрации углеводородов в сырье можно найти решением
уравнений (11.55) и (11.71) через процедуру, описанную вдругом источнике [Elizalde
and Ancheyta, 2011].
Сначала решалось уравнение (11.55), чтобы найти параметры а, а0, а,, 8 и ктм ки-
нетической модели гидрокрекинга. Затем с использованием найденных значений ре-
шалось уравнение (11.71) и находились значения параметров р, AmaxS и AminS. Все под-
программы писались в пакете MATLAB. При определении значений всех параметров
модели применялся критерий, обеспечивающий минимизацию суммы квадратов от-
клонений расчетных точек от экспериментальных. Подробнее о численном решении
уравнения (11.55) можно узнать в другом источнике [Elizalde and Ancheyta, 2011].
11.7.3. Анализ результатов
11.7.3.1. Гидронренинг
Параметры модели, отвечающие за гидрокрекинг, определялись по уравнению (11.55)
на основании экспериментальных кривых разгонки, т. е. на основании данных о тем-
пературах кипения и соответствующих массовых долях отгона. Они имели следую-
щие значения: а = 0,35, а0 = 1,50, = 22,0, 6=1,31-10-8 и Атх = 0,18 ч_|. Значение
параметра Атах, являющегося прямым показателем глубины гидрокрекинга, говорит
о том, что благодаря умеренным условиям реакция протекает с невысокой скоро-
стью. На рис. 11.18 сравниваются расчетные и экспериментальные данные безраз-
мерных кривых кипения сырья и продукта гидрокрекинга. Наблюдается хорошее
согласие, величина погрешности по всем точкам меньше 2 %. Видно, что реакция
гидрокрекинга более избирательна к средним дистиллятам, чем к легким фракциям;
последние почти не подвергаются крекингу. Иными словами, жесткость условий ре-
акций в данном исследовании такова, что тяжелые фракции превращаются в легкие
по каскадному механизму: вакуумный остаток превращается в вакуумный газойль,
вакуумный газойль — в средние дистилляты и так далее, но для увеличения выхода
самой легкой фракции (бензиновой) этого недостаточно.
11.7.3.2. Гидрообессеривание
По уравнению (11.71) на основании экспериментальных кривых разгонки соединений
серы (т. е. исходя из данных о температурах кипения и соответствующих массовых до-
лях содержания серы) и с использованием найденных ранее значений параметров для
гидрокрекинга были рассчитаны значения параметров для реакции ГДО. Они имели
следующие значения: kmxS = 7,5 ч~‘, = 0,08 ч-1 и р = 2. Расчетные данные разгон-
ки соединений серы сравниваются на рис. 11.19 с экспериментальными. Каки в случае
реакции гидрокрекинга, наблюдается хорошее согласие при величине погрешности
менее 2 %. Согласие между экспериментальными и расчетными результатами подтвер-
ждают и другие статистические параметры: угловой коэффициент, отсекаемый отре-
зок и квадрат коэффициента корреляции равны соответственно 1,003, 1 10-4 и 0,9996.
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
507
Рис. 11.18. Сравнение экспериментальных (значки) и расчетных (линии) данных разгонки
сырья и продукта гидрокрекинга, полученного при следующих условиях: Т = 382 °C,
Р = 9,8 МПа, LHSV = 0,2 ч*1, кратность водорода к сырью 5800 ст. куб. фут/баррель.
Обозначения-. (•) — сырье; (о) — продукт гидроочистки
Рис. 11.19. Сравнение экспериментальных (значки) и расчетных (линии) данных разгонки
сырья и продукта гидроочистки, полученного при следующих условиях: Т = 382 °C,
Р = 9,8 МПа, LHSV= 0,2 ч-1, кратность водорода к сырью 5800 ст. куб. фут/баррель.
Обозначения: (•) — распределение серы в сырье; (о) — распределение серы в продукте
Следует отметить, что с увеличением температуры кипения молярная масса
и сложность соединений серы, присутствующих в сырье, также возрастают. Актив-
ность соединений серы с увеличением молярной массы в целом снижается. К наи-
более стойким относятся те из них, в которых атом серы окружен алкильными
группами (например, 4,6-диметилдибензотиофен). Несмотря на все эти особенности
распределения серы, применяемая непрерывная кинетическая модель без искаже-
ний улавливает различия в активности и способна правильно имитировать реакции
как гидрокрекинга, так и гидрообессеривания.
508
Часть 3. Моделирование каталитических процессов
Что касается формы кривых распределения серы в сырье и продукте гидроочист-
ки, все они представляют собой типичные восходящие кривые. (То есть чем тяжелее
фракция, тем больше серы она содержит.) Из легких фракций продукта сера практи-
чески полностью удаляется, а в тяжелых фракциях она все еще остается, хотя и в го-
раздо меньших количествах, чем в сырье. Снижение содержания серы в тяжелых
фракциях можно считать следствием гидрокрекинга, так как соединения серы в по-
добных фракциях более стойки и в условиях исследования реагируют труднее, что
подтверждается и малой величиной &mjnS. Это говорит в пользу того, что удаление се-
росодержащих соединений связано с гидрокрекингом, поэтому в кинетических ис-
следованиях необходимо учитывать его влияние на реакцию гидрообессеривания.
Таким образом, кинетическая модель непрерывного агрегирования способна пред-
сказать для типичных условий реакций выход дистиллята с данным интервалом ки-
пения и распределение серы в нем.
11.7.3.3. Заключительные замечания
Появилось несколько публикаций о попытках моделирования совместно протека-
ющих реакций гидрообессеривания и гидрокрекинга. Некоторые из них упускают
из виду факт одновременности реакций. Такое допущение справедливо лишь в том
случае, если степень гидрокрекинга при гидроочистке незначительна [Korsten and
Hoffmann, 1996], что обычно имеет место при переработке дистиллятов — например,
бензиновой фракции, газойля или даже сырья каталитического крекинга (вакуумных
газойлей), — но не при гидроочистке тяжелого сырья.
Типичным параметром зависимостей для транспортных свойств нефтяных фрак-
ций является средняя температура кипения, которую обычно принимают неизмен-
ной. Это верно лишь для случаев, когда гидрокрекинг практически отсутствует. Если
же он имеет место, возможны изменения транспортных свойств, в результате чего
модель реактора будет давать неверные результаты. Это обстоятельство может приве-
сти к введению в модель дополнительных параметров, маскирующих истинное пове-
дение реактора гидроочистки.
Если общее содержание серы имитируется моделью, использующей агрегирова-
ние или усреднение распределения без должного подтверждения экспериментальны-
ми данными, применимость прогнозирования содержания серы в продуктах может
быть ограничена узким интервалом условий реакций, что исключает возможность
получить достоверные результаты. Когда распределение серы принимается по опу-
бликованным в литературных источниках зависимостям между содержанием серы
и температурой кипения, значения параметров модели обязательно должны под-
тверждаться экспериментальными данными распределения серы при разгонке [Chou
and Но, 1988].
Получение надежных данных разгонки и кривых распределения серы — нелегкая
задача. Для определения ИТК и выделения различных фракций, необходимых для
дальнейшего анализа на содержание серы и определения удельного веса, нужна про-
ба объемом не менее 4 л. Если требуется более детальный анализ, необходимый объем
пробы может возрасти до 40 л. Получение пробы такого объема само по себе является
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
509
проблемой. Эксперименты обычно проводятся на малых установках при небольшой
скорости подачи сырья, и получение необходимого количества продукта гидроочист-
ки занимает много времени. Таковы основные причины, по которым исследователь-
ские группы, работающие над гидропереработкой тяжелой нефти, не проводят ис-
следований, подобных описанному. Опубликован ряд сообщений о применении
непрерывного кинетического агрегирования для моделирования ГДО и предсказа-
ния изменений в свойствах продуктов. Но авторы этих публикаций не формулируют
исходных допущений, подтверждаемых экспериментальными данными.
11.8. ЗНАЧИМОСТЬ ПАРАМЕТРОВ
НЕПРЕРЫВНОЙ МОДЕЛИ
11.8.1. О параметрах модели
Параметр а соотносится с зависимостью от размера («ширины») псевдокомпонен-
тов, аа0 и а, учитывают положение максимума кривой р(к, К). При К > к температура
кипения компонента с активностью к ниже, чем у компонентов с активностью К, по-
этому он может образовываться из всех компонентов с активностью К > к. Параметр
5, имеющий сравнительно малую величину, отражает начальную форму безразмер-
ной кривой кипения (зависимость wt от 9) и учитывает тот факт, что при к = 0 функ-
ция р(к, К) имеет малое, но конечное значение. Если к = К, то р(к, К) = 0, поскольку
компонент с активностью к не возникает сам по себе, а образуется из более высоко-
кипящих компонентов или расщепляется на фракции с меньшей температурой кипе-
ния. Наконец, /стах представляет скорость разложения псевдосоединений с наивыс-
шей активностью.
Лаксминарасимхан и соавторы [Laxminarasimhan et al., 1996] утверждают, что па-
раметры непрерывной кинетической модели (а, ад, а{, 5 и кт!о) могут зависеть от ка-
тализатора гидрокрекинга, а также от свойств сырья. В частности, £ти коррелирует
с температурой, подчиняясь закону Аррениуса [Basak et al., 2004]. Другие авторы со-
глашаются с тем, что эти параметры модели являются функциями типа катализато-
ра, а также температуры и давления [Khorasheh et al., 2001; Elizalde et al., 2009, 2010].
До настоящего времени предложены лишь эмпирические полиномиальные зависи-
мости параметров от типа катализатора, свойств сырья и рабочих условий. Слож-
ность планирования соответствующих экспериментов и значительные затраты на их
проведение сильно затрудняют исследование в данной области.
11.8.2. Другие факторы, влияющие на параметры модели
Выше говорилось, что параметры модели определяются активностью катализатора
и свойствами сырья. Но остаются неисследованными другие факторы, влияющие
на процесс гидрокрекинга, в том числе степень термического крекинга, который мо-
жет происходить при гидрокрекинге тяжелой нефти. Необходимо глубже исследовать
510
Часть 3. Моделирование каталитических процессов
влияние этого фактора, чтобы параметры модели гарантированно отражали только
каталитический гидрокрекинг, а не совокупный результат каталитического и терми-
ческого крекинга.
Другое распространенное допущение при применении непрерывного агрегирова-
ния к кинетике гидрокрекинга — то, что реакционная смесь остается жидкой. Опре-
деленные исследования в этом направлении вполне целесообразны, хотя можно
ожидать, что испарение тяжелых фракций будет пренебрежимо малым.
11.8.3. Открытые вопросы
Ниже перечислены вопросы, требующие исследования.
1. Истинная сущность параметров модели и явное выражение их зависимости
от активности и состава катализатора. Приемлемым, пусть и эмпирическим, подхо-
дом может оказаться установление связи параметров модели с рабочими параметрами.
2. Возможность улучшить детализацию модели добавлением новых пиков
в функцию распределения выхода (мультимодальное распределение), что, наря-
ду с непрерывным описанием, может дать дополнительные сведения о механизме
процесса.
3. Необходимо исследовать неоднозначность решений модели и обоснован-
ность экстраполяции при расчете максимальной температуры кипения, хотя послед-
нее может оказаться не особо важным.
4. Нерешенным вопросом остается также гибкость встраивания непрерывного
подхода к кинетике в гетерогенную модель реактора. Главная проблема при модели-
ровании гидроочистки тяжелых фракций — отсутствие надежных зависимостей для
оценки термодинамических и транспортных свойств. Так что если псевдогомогенное
описание гидрокрекинга и может быть приемлемым, переход к истинно гомогенной
модели — нелегкая задача.
5. При описании гидрокрекинга может оказаться целесообразным исследовать
функцию выхода другого типа (например, видоизмененную гамма-функцию), что-
бы сравнить результаты с теми, которые дает детализация функции р(к, К) введени-
ем новых параметров, и выделить решение, наилучшим образом описывающее слож-
ные реакции гидрокрекинга тяжелой нефти различных типов в различных рабочих
условиях.
Поскольку моделирование гидрокрекинга тяжелой нефти методом непрерывного
агрегирования учитывает сразу несколько явлений, лучшей из функций распределе-
ния является та, которая допускает подгонку через параметры модели [Govindhakan-
nan and Riggs, 2007]. Константы скорости к, якобиан преобразования D(k) и функ-
ции распределения выхода непрерывной кинетической модели, описанной в данной
работе, используют подгоночные параметры, рекомендованные Чу и Хо [Chou and
Но, 1988]. Чтобы найти значения этих подгоночных параметров и их зависимость
от условий реакций, типа катализатора и свойств сырья, устранив при этом влияние
термического крекинга и ограничений массопереноса, необходимы детальные и точ-
ные экспериментальные данные.
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
511
ОБОЗНАЧЕНИЯ
ЛАТИНСКИЕ И РУССКИЕ
А, В, С, D Константы многопараметрических выражений, связывающих параме-
ао> Я1> *^0 Ь, т с(к, т) тры модели с температурой и давлением Параметры функции распределения выхода Параметры, учитывающие влияние температуры на параметры модели Концентрация компонента с активностью к при времени пребывания т
с(А:, 0) Концентрация компонента с активностью к при времени пребывания т = 0
Q cs Концентрация углеводорода Концентрация серы в произвольный момент времени, отвечающая со- единению с активностью кгао
C(t, Z, х) D(k) Мдк) Функция распределения вектора переменных х Функция распределения по типам компонентов Функция распределения по типам компонентов для реакции гидрокрекинга
А*Гдо) ^гдк I К к(х) к0 к max Функция распределения по типам компонентов для реакции ГДС Энергия активации реакции гидрокрекинга Интеграл Активность компонента, ч_| Скорость фрагментации Предэкспоненциальный множитель в уравнении Аррениуса Активность компонента смеси с наименьшей температурой кипения, ч_|
к с maxS Активность компонента смеси с наименьшей температурой кипения в реакции гидрообессеривания, ч-1
к с mm3 Активность компонента смеси с наивысшей температурой кипения в реакции гидрообессеривания, ч~‘
Ъ- ГДК ^гдо LHSV Константа скорости для гидрокрекинга; размерность обратна времени Активность компонента в реакции гидрообессеривания, ч-1 Объемная скорость подачи сырья, ч-1
т п, N Коэффициент наклона прямой Общее число типов псевдосоединений в реакционной смеси
П1’П1 Порядки отдельных реакций для углеводородов и водорода; п2 — сте- пенной показатель, учитывающий влияние давления
512
Часть 3. Моделирование каталитических процессов
р р0 Ль Р<к,К) Общее давление, МПа Давление сравнения, МПа Парциальное давление водорода, МПа Выход компонента с активностью к при гидрокрекинге компонентов с активностью К
~rvj№. R Т Скорость реакции гидрокрекинга Универсальная газовая постоянная Температура по абсолютной шкале, К
t TBP(h) Время Наивысшая температура кипения псевдокомпонентов реакционной смеси, К
TBP(l) Наинизшая температура кипения псевдокомпонентов реакционной смеси, К
V Скорость потока (расход жидкости за единицу времени)
X х' Активность любого вида, ч_|; переменная интегрирования Произвольное свойство
X Вектор свойств
wt Массовая доля компонента
z Вектор пространственных координат
НИЖНИЕ ИНДЕКСЫ
г Порядковый номер псевдокомпонента Нижний индекс для обозначения произвольного времени пребывания
ВЕРХНИЕ ИНДЕКСЫ
Р Любой экспоненциальный параметр
ГРЕЧЕСКИЕ
а 3 5 Параметр модели Параметр модели Параметр модели для функции распределения выходар(к, К)
0 Приведенная истинная температура кипения; безразмерная величина
к р Константа пропорциональности
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
513
р Скорость нарастания
т Время пребывания, ч; обратно объемной скорости подачи сырья, ч-1
ттах Максимальное время пребывания, ч
Q (х, х') Стехиометрический коэффициент (ядро)
ЛИТЕРАТУРА
Al-Dahhan, М.Н., Larachi, Е, Dudukovic, M.P, Laurent, А. 1997. High-pressure trickle-bed reactors: a
review, Ind. Eng. Chem. Res. 36:3292—3314.
Ancheyta, J., Betancourt, G., Marroquin, G., Perez, A.M., Maity, S.K., Cortez, M.T, del Rio, R. 2001.
An exploratory study for obtaining synthetic crudes from heavy crude oils via hydrotreating. Energy Fuels
15:120-127.
Ancheyta, J., Sanchez, S., Rodriguez, M.A. 2005. Kinetic modeling of hydrocracking of heavy oil fractions:
a review. Catal. Today 109:76—92.
Ancheyta, J., Trejo, E, Rana, M.S. 2009. Asphaltenes: Chemical Transformation during Hydroprocessing of
Heavyoils (Chemical Industries), CRC Press Taylor & Francis Group, Boca Raton, FL.
Ancheyta, J., Villafuerte, E. 2000. Kinetic modeling of naphtha catalytic reforming reactions. Energy
Fuels 14:1032-1037.
Ancheyta, J., Villafuerte, E., Schacht, P, Aguilar, R., Gonzalez, E. 2002. Simulation ofasemi- regenerative
reforming plant using feedstocks with and without benzene precursors. Chem. Eng. Tecnol. 25:541-546.
Aris, R. 1989. Reactions in continuous mixtures. AIChEJ. 35:539-548.
Aris, R., Gavalas G.R. 1966. On the theory of reactions in continuations mixtures. Phil. Trans. Roy. Soc.
260:351-393.
Ashuri, E., Khorasheh, E, Gray, M.R. 2007. Development of a continuous kinetic model for catalytic
hydrodenitrogenation of bitumen. Scientia Iranica 14:152—160.
Astarita, G., Ocone, R. 1988. Lumping nonlinear kinetics. AIChEJ. 34:1299-1309.
Ayasse, A.R., Nagaishi, H., Chan, E.W, Gray, M.R. 1997. Lumped kinetics of hydrocracking of bitumen.
Fuel 76:1025-1033.
Balasubramanian, B., Pushpavanam, S. 2008. Modeling discrimination in hydrocracking of vacuum gas
oil using discrete lumped kinetics. Fuel 87:1660-1672.
Baltanas, M.A., Vamsina, H., Froment, G.E 1983. Hydroisomerization and hydrocracking. 5. Kinetic
analysis of rate data for n-octane. Ind. Eng. Chem. Prod. Res. Dev. 22:531—539.
Basak, K., Sau, M., Manna, U., \ferma, PR. 2004. Industrial hydrocracker model based on novel
continuum lumping approach for optimization in petroleum refinery. Catal. Today 98:253-264.
Bennett, R.N., Bourne K.H. 1972. A study process reactions and corresponding product yields and
qualities. In: Proceedings of the ACS Symposium on Advances in Distillate and Residual Oil Technology,
New York, August 27-September 1, G45—G62.
Botchwey, C., Dalai, A.K., Adjaye, J. 2003. Product selectivity during hydrotreating and mild hydrocracking
of bitumen-derived gas oil. Energy Fuels 17:1372-1381.
Browarzik, D., Kehlen, H. 1994. Hydrocracking process of n-alkanes by continuous kinetics. Chem. Eng.
Sci. 49:923-926.
Chou, M.Y, Ho, TC. 1988. Continuum theory for lumping nonlinear reactions. /17СЛЕ7.34:1519—1527.
DeDonder,Th. 1931. L’Affinite (secondpartie), Chapter III, Gauthier-Villars, Paris.
Dufresne, P, Bigeard, PH., Billon, A. 1987. New developments in hydrocracking: low pressure high-
conversion hydrocracking. Catal. Today 1:367—384.
Elizalde, L, Ancheyta, J. 2011. On the detailed solution and application of the continuous kinetic
lumping modeling to hydrocracking of heavy oils. Fuel 90:3542-3550.
514
Часть 3. Моделирование каталитических процессов
Elizalde, 1., Ancheyta, J. 2012. Modeling the simultaneous hydrodesulfurization and hydro-crackingof
heavy residue oil by using the continuous kinetic lumping approach. Energy Fuels 26:1999-2004.
Elizalde, I., Rodriguez, M.A., Ancheyta, J. 2009. Application of continuous kinetic lumping modeling
to moderate hydrocracking of heavy oil. Appl. Catal. A 365:237—242.
Elizalde, I., Rodriguez, M.A., Ancheyta J. 2010. Modeling the effect of pressure and temperature on the
hydrocracking of heavy crude oil by the continuous kinetic lumping approach. Appl. Catal. A 382:205—212.
Espinosa-Pena, M., Figueroa-Gomez, Y, Jimdnez-Cruz, E 2004. Simulated distillation yield curves in
heavy crude oils: a comparison of precision between ASTM D-5307 and ASTM D-2892 physical distillation.
Eneigy Fuels 18(6):1832—1840.
Froment, G.F 2005. Single event kinetic modeling of complex catalytic processes. Catal. Rev. 47:83-124.
Govindhakannan, J., Riggs J.B. 2007. On the construction of a continuous concentration-reactivity
function for the continuum lumping approach, [nd. Eng. Chem. Res. 46:1653—1656.
Ho, TC. 1991. Modeling of reaction kinetics for petroleum fractions. In: Kinetic and Thermodynamic
Lumping of Multicomponent Mixtures (G. Astarita, S.I. Sandler, Eds). Proceedings of an ACS Symposium on
kinetic and thermodynamic lumping of multi- component mixtures, Atlanta, GA, April 15.
Ho, TC. 2008. Kinetic modeling of large-scale reaction systems. Catal. Rev.-Sci. £wg. 50:287-378,
Jenkins, J.H., Stephens, TW 1980. Kinetics of catalytic reforming. Hydrocarbon Process. 11:163-168.
Khorasheh, E, Chan, E.C., Gray M.R. 2005. Development of a continuous kinetic model for catalytic
hydrodesulfurization of bitumen. Petrol. Coal 47:40-48.
Khorasheh, F, Zainali, H., Chan, E.C., Gray, M.R. 2001. Kinetic modeling of bitumen hydro-cracking
reactions. Petrol. Coal43:208-218.
Korsten, H., Hoffmann, U. 1996. Three-phase reactor model for hydrotreating in pilot trickle-bed
reactors. AIChEJ. 42:1350—1360.
Krishna, R., Saxena, K. 1989. Use of an axial-dispersion model for kinetic description of hydrocracking.
Chem. Eng. Sci. 44:703—712,
Laxminarasimhan, C.S., Xferma, R.P, Ramachandran, PA. 1996. Continuous lumping model for
simulation of hydrocracking. AIChEJ. 42:2645-2653.
Mapiour, M., Sundaramurthy, V, Dalai, A.K., Adjaye, J. 2009. Effect of hydrogen purity on
hydroprocessing of heavy gas oil derived from oil-sands bitumen. Energy Fuels 23:2129—2135.
Mapiour, M., Sundaramurthy, V, Dalai, A.K., Adjaye, J. 2010. Effects of hydrogen partial pressure
on hydrotreating of heavy gas oil derived from oil-sands bitumen: experimental and kinetics. Eneigy Fuels
24:772-784.
Marafi, A., Stanislaus, A., Furimsky, E. 2010. Kinetics and modeling of petroleum residues
hydroprocessing. Catal. Rev. Sci. Eng. 52:204—324.
Maxwell, J.B. 1965. Data Book on Hydrocarbons: Application to Process Engineering, 8th edn., Vin
Nostrand, NewYrrk.
McCoy, B., Wng, M. 1994. Continuous-mixture fragmentation kinetics: particle size reduction and
molecular cracking. Chem. Eng. Sci. 49:3773—3785.
Mederos, E, Ancheyta, J., Chen, J. 2009b. Review on criteria to ensure ideal behaviors in trickle-bed
reactors. Appl. Catal. A 355:1-19.
Mederos, E, Elizalde, I., Ancheyta, J. 2009a. Steady-state and dynamic reactor models for hydrotreatment
of oil fractions: a review Catal. Rev. Sci. Eng. 51:485—607.
Narasimhan, C.S.L., Sau, M., Verma, R.P 1999. An integrated approach for hydrocracker modeling. In:
Hydrotreatment and Hydrocracking of Oil Fractions (B. Delmon et al., Ed),pp. 297-306, Elsevier, Amsterdam.
Ostrowsky, N., Sornette, D. 1981. Exponential sampling method for light scattering polydispersity
analysis. Optica Acta 28:1059—1070.
Pacheco, M.A., Dassori, C.G. 2002. Hydrocracking: an improved kinetic model and reactor modeling.
Chem. Eng. Commun. 189:1684—1704.
Padlo, D.M., Kuger, E.L. 1996. Simulated distillation of heavy oils using an evaporative light scattering
detector. Eneigy Fuels 10:1031—1035.
Глава 11. Моделирование гидрокрекинга непрерывным агрегированием кинетики
515
Peixoto, ЕС., de Medeiros, J .L. 1999. Modelling and parameter estimation in reactive continuous mixtures:
the catalytic cracking of alkanes, Rartl. Braz.J. Chem. Eng. 16(1):65-81.
Qader, S.A., Hill, G.R. 1969. Hydrocracking of gas oil. Ind. Eng. Chem. Proc. Des. Dev. 8:98—105.
Rana, M.S., Samano, V, Ancheyta, J., Diaz, J.A.I. 2007. A review of recent advances on process
technologies for upgrading of heavy oils and residua. Fuel 86:1216—1231.
Rassev, S. 2003. Thermal and Catalytic Processes in Atroleum Refining, Marcel Decker, Inc.. NewYark.
Sadighi, S., Arshad, A., Rashidzadeh, M. 2010. 4 -Lump kinetic model for vacuum gas oil hydrocracker
involving hydrogen consumption. Kor. J. Chem. Eng. 27(4): 1099—1108.
Sambi, I.S., Khulbe, K.C., Mann, R.S. 1982. Catalytic hydrotreatment of heavy gasoil. Ind. Eng. Chem.
Prod. Res. Dev. 21:575-580.
Sanchez, S., Ancheyta, J. 2007. Effect of pressure on the kinetics of moderate hydrocracking of maya
crude oil. Energy Fuels 21:653-661.
Sanchez, S., Rodriguez, M.A., Ancheyta J. 2005. Kinetic model for moderate hydrocracking of heavy
oils. Energy Fuels 44:9409—9413.
Sau, M., Narasimham, C.S.L., \6rma, R.P 1997. A kinetic model for hydrodesulfurization.
In: Hydrotreatment and Hydrocracking of Oil Fractions {Stud. Sutfi Sci. Catal.) (G.E Froment et al., Ed),
pp. 421-435, Elsevier Science, Amsterdam.
Scherzer, J., Gruia, A.J. 1996. Hydrocracking Science and Technology, Series: Chemical Industries;
vol. 66, Marcel Decker, Inc., New York.
Speight, J.G. 2000. The Desulfurization of Heavy Oils and Residua, Marcel Dekker, NewYark.
Vtlarasu, G., Bhaskar, M., Balaraman, K.S. 2003. Mild hydrocracking: a review of the process, catalysts,
reaction, kinetics and advantages. Petrol. Sci. Technol. 21:1185—1205.
\6rstraete, J.J., Le Lannic, K., Guibard, I. 2007. Modeling fixed-bed residue hydrotreting processes.
Chem. Eng. Sci. 62:5402-5408.
Artii, S.M., Sanford, E.C. 1989. Mild hydrocracking of bitumen-derived coker and hydro-cracker heavy gas
oils: kinetics, product yields, and product properties. Ind. Eng. Chem. Res. 28:1278—1284.
Yti, S.M., Sanford, E.C. 1991. Kinetics of aromatics hydrogenation of bitumen-derived gas oils. Can. J.
Chem. Eng. 69:1087-1095.
Ziff, M.R. 1991. New solutions to the fragmentation equation. J. Phys. A. Math. Gen. 24:2821-2828.
Глава 12. Зависимости
и прочие вопросы
Последняя, но оттого не менее важная глава посвящена тем вопросам, на которые
при моделировании гидрокаталитической переработки часто обращают мало внима-
ния, но без решения которых на различных стадиях экспериментальных исследова-
ний, расчета или имитации реакторов (в том числе масштабирования) не обойтись.
Эти вопросы перечислены ниже.
• Зависимости, позволяющие прогнозировать свойства продуктов и параметры
процесса гидроочистки (обычно публикуются в литературных источниках).
Выводимые для определенного сырья, катализатора, экспериментальной уста-
новки и условий реакций, эти зависимости, тем не менее, полезны при бы-
стрых расчетах.
• Расход водорода. Данный вопрос крайне важен при разработке технологий,
так как от количества потребляемого водорода зависят производительность
водородной установки, мощность компрессоров циркулирующего и добавоч-
ного водорода, внутриреакторное и межреакторное охлаждение и другие па-
раметры. Поэтому без приемлемой методики расчета потребления водорода
не обойтись.
• Степень превращения остатка (фракции с температурой кипения выше 538 °C).
Найти ее нетрудно, но иногда расчет степени превращения остатка услож-
няется тем, что для очистки или в других эксплуатационных целях в реактор
наряду с основным сырьем (т. е. вакуумным остатком) приходится подавать
облегченное.
• Содержание металлов в катализаторе. Этот вопрос особенно актуален при
гидропереработке тяжелой нефти, так как отложение металлов определя-
ет момент времени, когда установку приходится останавливать для замены
катализатора. Анализ содержания металлов может быть отнесен к различ-
ным базам: к свежему, отработанному, регенерированному или равновес-
ному катализатору. Это может вызвать путаницу при расчете срока службы
катализатора.
• Функции распределения вероятностей. В литературе приводится множество фун-
кций с различным числом параметров, но их способность аппроксимировать
Глава 12. Зависимости и прочие вопросы
517
кривые разгонки нефти не установлена. В целях моделирования полезно знать,
какая функция лучше всего подходит для данных разгонки.
12.1. ЗАВИСИМОСТИ ДЛЯ ПРОГНОЗИРОВАНИЯ
СВОЙСТВ ПРОДУКТОВ ГИДРООЧИСТКИ
ТЯЖЕЛОЙ НЕФТИ
В отсутствие подходящих моделей, опирающихся на теоретические принципы, для
представления явлений любого типа не остается ничего иного, как воспользоваться
зависимостями — при условии, что они хотя бы отчасти подкреплены теорией, ко-
торая представляет лежащие в основе этих явлений процессы. Для вывода наилуч-
шей зависимости, способной достаточно точно воспроизводить результаты экспе-
риментов, требуется достаточный объем экспериментальных данных. Тем не менее,
уравнения и значения параметров остаются основанными на весьма узких выборках
данных и справедливы лишь для частного набора экспериментальных условий, уста-
новки, катализатора и свойств сырья, для которых и были выведены. Поэтому мо-
жет потребоваться подгонка значений параметров, если они должны отражать новые
обстоятельства.
В прошлом при расчете целых промышленных установок широко применялись
простые зависимости, что объяснялось ограниченностью вычислительных мощно-
стей. Те, кто нуждается в быстрых расчетах, до сих пор предпочитают соотносить
промышленные данные с определенными рабочими параметрами процесса. Зави-
симостями чаще всего пользуются для выявления влияния условий реакций на вы-
ход продуктов и степень превращения, расчета свойств продуктов и определения
некоторых параметров процессов.
Что касается гидроочистки нефтяных фракций, есть немало публикаций о по-
пытках связать степень удаления примесей со свойствами сырья, параметрами
процесса и т. д. Цаматсулис и соавторы [Tsamatsoulis et al., 1991] одними из пер-
вых обнаружили связь между свойствами сырья и процессом гидроочистки. Це-
лью исследования был вывод уравнений, связывающих степени гидрообессе-
ривания, гидрокрекинга, обессеривания асфальтеновых и неасфальтеновых
фракций и расход водорода со свойствами остатка атмосферной перегонки неф-
ти из месторождения у греческого острова Тасос. В течение всего процесса из-
мерялись плотность и коксуемость. По их значениям вычислялись плотность
по шкале API и коксуемость по Конрадсону, значениям которых, в свою оче-
редь, сопоставлялись степени обессеривания и крекинга асфальтенов. Для опи-
сания связи между переменными авторы вывели степенные выражения с че-
тырьмя подгоночными параметрами. Авторы пришли к выводу, что результаты
их исследования, пусть оно и не было обобщающим, свидетельствуют о том,
что глубокое обессеривание сопровождается радикальным улучшением свойств
сырья.
518
Часть 3. Моделирование каталитических процессов
Нг Сью Хунг [Ng, 1997] представил базу экспериментальных данных, позволяю-
щую оценить пригодность экстракции растворителями с последующим каталити-
ческим крекингом для облагораживания остатка перегонки тяжелой нефти из биту-
минозных песчаников Атабаски. На основании изменений фазового поведения он
вывел уравнения, описывающие связь содержания асфальтенов с содержанием ме-
таллов, общей серы и общего азота, коксуемостью по Конрадсону и плотностью де-
асфальтированной нефти. Зависимости, предложенные Нг, хотя и выведенные для
процесса деасфальтизации остатка перегонки, могут оказаться справедливыми и для
гидрокрекинга, так как в асфальтенах концентрируются существенные количества
таких гетероатомов, как кислород, сера, азот, ванадий и никель. Физическое удале-
ние или каталитическое превращение асфальтенов значительно снижают содержание
примесей в нефти, чем и определяется тесная связь между содержанием асфальтенов
и содержанием некоторых примесей в нефти. Нг обнаружил, что все переменные,
кроме плотности, с увеличением содержания асфальтенов возрастают по кривой, об-
ращенной выпуклостью вниз; фактически это означало экспоненциальную зависи-
мость. Сообщалось о положительной линейной связи между плотностью и содержа-
нием асфальтенов.
Трасобарес и соавторы [Trasobares et al., 1998] проанализировали связь коксо-
вого остатка (коксуемости) по Конрадсону с такими свойствами сырья, как содер-
жание асфальтенов, содержание водорода, соотношение водород /углерод и со-
держание остатка, при каталитической гидроочистке остатка перегонки нефти
«Майя». Была обнаружена линейная связь коксуемости со всеми этими параме-
трами. Выход газа тоже линейно зависел от степени уменьшения содержания кок-
сового остатка.
Калледжас и соавторы [Callejas et al., 1999] исследовали кинетику реакций удале-
ния серы, азота, никеля и ванадия из остатка нефти «Майя» на установке гидроочист-
ки, работающей при высоких температурах (375—415 °C) и парциальном давлении
водорода (10-15 МПа). Эксперименты позволили вывести линейную зависимость
между степенями удаления серы и металлов в процессе гидрирования.
Хо [Но, 2003] применил метод частных наименьших квадратов к многопараметри-
ческой матрице X = {х..} размерности лхр, где п = 13 — число типов сырья, ар = 24 —
число дескрипторов (т. е. таких физических и химических свойств, как содержание
серы, азота, ароматических углеводородов, относительная плотность по шкале API
и содержание дибензотиофенов). Он получил корреляцию свойств с химической ак-
тивностью, что позволило количественно интерпретировать влияние свойств сырья
на гидрообессеривание средних дистиллятов.
Хо и Маркли [Но and Markley, 2004] в поисках компактной и устойчивой (с наи-
меньшим числом подгоночных параметров) корреляции, которая связывала бы
свойства предварительно гидроочищенных дистиллятов с их активностью в реак-
ции гидродесульфуризации, провели серию экспериментов с 13 типами сырья, раз-
личающимися происхождением и предварительной обработкой; сырье каждого
типа характеризовалось 10 свойствами. Перебрав все 10 свойств и их комбинации,
Глава 12. Зависимости и прочие вопросы
519
исследователи получили линейную функцию, которая удовлетворительно описыва-
ла активность. Кроме того, они пришли к выводу, что самым значимым параметром
являлось содержание азота.
Фердоус и соавторы [Ferdous et al., 2006J провели ряд экспериментов и иссле-
дований кинетики в целях оптимизации условий и расчета кинетических параме-
тров процесса гидродеазотирования и гидрообессеривания тяжелого газойля биту-
минозной нефти Атабаски. Регрессионным анализом экспериментальных данных
были получены степенные выражения с шестью подгоночными параметрами,
описывающие связь общего содержания азота и серы с температурой, давлением
и удельной объемной скоростью подачи сырья.
Попытка воспроизвести по приведенным в литературных источниках зависи-
мостям экспериментальные данные гидроочистки в условиях, отличных от тех,
для которых были выведены эти зависимости, будет, скорее всего, безуспешной.
Чтобы расширить область применимости зависимостей, необходимо пересчи-
тать значения корреляционных параметров. Основная задачей этого раздела —
сравнение результатов, даваемых известными зависимостями, с эксперимен-
тальными данными, выходящими за область определения этих зависимостей,
и оптимизация корреляционных параметров с целью улучшить результативность
прогнозирования.
12.1.1. Описание зависимостей
Для сравнения были выбраны 17 зависимостей, найденных в литературных источ-
никах. В табл. 12.1 приведены сведения о рабочих условиях и типах реактора, сы-
рья и катализатора, послужившие основанием для выведения этих зависимостей.
Общий вид зависимостей показан в табл. 12.2, значения соответствующих параме-
тров—в табл. 12.3.
Таблица 12.1
Рабочие условия, в предположении которых были выведены зависимости
Реактор Сырье Катали- затор или раствори- тель Г, °C Р, МПа LHSV Кратность водорода, ст. куб. фут/ баррель Литератур- ный источник
Реактор с орошае- мым слоем Атмосфер- ный оста- ток нефти «Тасос», 315 °С+ СоМо/ А12О3 350-465 5,0 0,16-1,46 ч-1 1200— 10500 Tsamats- oulis et al., 1991
Реактор смешения“ Вакуум- ный оста- ток нефти «Майя», 540 °С+ NiMo/ у-А12О3 375-415 10,0- 15,0 1,4—7,1 л/ч г 6000— 10000 Trasoba- res et al., 1998
520
Часть 3. Моделирование каталитических процессов
Таблица 12.1, окончание
Реактор Сырье Катали- затор или раствори- тель Г, °C Р, МПа LHSV Кратность водорода, ст. куб. фут/ баррель Литератур- ный источник
Реактор смешения'1 Вакуум- ный оста- ток нефти «Майя», 540 °С+ NiMo/ 7-А12О3 375-415 10,0— 15,0 1,4—7,1 л/ч • г 6000— 10000 Callejas and Martinez, 1999
Реактор с орошае- мым слоем Тяжелый вакуум- ный га- зойль би- туминоз- ной нефти «Атабаска» NiMoB/ А12о3 340-420 6,1- 10,2 0,5—2,0 ч-1 3300 Ferdons et al., 2006
Экстрак- ция рас- творителем и катали- тический крекинг (реак- тор для испыта- ния на ми- кроактив- ность) Вакуумный остаток би- тумов «Ата- баска» и тяже- лой нефти «Ллойд- минстер» Цеолит/ А12О3 (катали- тический крекинг) 510 — 20 г/ч г — Ng, 1997
Пропан 75 3,21 — —
«-Бутан 120 2,86 — —
«-Пентан 160 2,52 — —
«-Гептан 75 0,45 — —
Скорость перемешивания 2000—3500 об/мин.
Таблица 12.2
Общий вид зависимостей
Сокращенное обозначение типа Общий вид Номер уравнения
SL У = aa + atx (12.1)
P y = aa + atx + arE + ape1 (12.2)
MNLl у = (a0 + a, T + ajc + a3P + a^2)2 + a5 (12.3)
MNLl y = a0 + atT+ 0^ + ajc2 + ajx (12.4)
El у = aoeia,x> (12.5)
El У= aox“‘ (12.6)
Таблица 12.3
Значения параметров зависимостей
Тйп зависимости Г, °C Р, МПа У «0 «2 a3 «4 «5 X Уравнение
SL 375 10-15 ГДН 5,46 1,35 — — — — ГДО» (12.7)
SL 400, 415 10-15 ГДН 72,68 0,34 — — — — ГДО» (12.8)
SL 375 10-15 ГДВ’ 14,04 1,64 — — — — ГДОа (12.9)
SL 400,415 10-15 ГДВ’ 54,8 0,60 — — — — ГДО’ (12.10)
SL 375-415 10-12,5 Asph6 -0,393 0,657 — — — — CCR6 (12.11)
SL 75 3,45 Р15-С 0,9318 0,0021 — — — — Asph6 (12.12)
Р 350-465 5-18 API 9,6 2,69 10 1 -5,26-10’ 4,58-10 5 — — ГДОа (12.13)
Р 350-465 5-18 API 9,6 5,31 IO'1 -9,33-10"’ 6,17 -10 5 — — ГКа (12.14)
Р 350-465 5-15 CCR- 15,2 —1,76-10-* 4,08-IO-3 -3,82-10-s — — ГДОа (12.15)
Р 350-465 5-15 CCR- 15,2 -3,22-10-' 5,33-10"’ —3,69 10 3 — — гка (12.16)
Р 350-465 18,14 RCR- 14,1 -1,8510-' 3,97-10-’ -3,61-10-5 — — ГДОа (12.17)
Р 350-465 18,14 RCR- 14,1 -3,26-10-' 4,82-10’ -2,97-10 5 — — ГКа (12.18)
MNL1 340-420 6,1-10,2 ГДА" -11,31069 0,054205 -6,38681 2,21-10-’ 1,700813 -0,07 LHSV (12.19)
MNL2 340-420 6,1-10,2 ГДО» 78,99496 0,086721 -121,97487 8,89582 0,23395 — LHSV (12.20)
Е\ 75 3,45 s6 3,107 0,0132 — — — — Asph6 (12.21)
Е1 75 3,45 Met- 0,0032 2,905 — — — — Asph6 (12.22)
Е2 75 3,45 CCR6 0,0815 1,3966 — — — — Asph6 (12.23)
Е1 75 3,45 № 116 0,9795 — — — — Asph6 (12.24)
Глава 12. Зависимости и прочие вопросы
а Значения, %,
6 Значения, %масс.
“ Значения, мг/кг.
Примечания. Асфальтены определяются как фракция, нерастворимая в «-гептане. Сокращения: CCR — коксуемость по Конрадсону;
Met — металлы; RCR — коксуемость по Рамсботтому; ГДА — гидродеазотирование; ГДВ — гидродеванадизация, ГДН — гидроденике-
лизация; ГК — гидрокрекинг;
<л
го
522
Часть 3. Моделирование каталитических процессов
Зависимости были классифицированы по типам как линейные, полиномиальные
полиномиальные от нескольких переменных и показательные. Исходя из вида, кото-
рый можно определить по табл. 12.2 и 12.3, они кратко характеризуются следующим
образом.
• Линейные зависимости (SL). Линейные зависимости, приводимые в литера-
турных источниках, описываются уравнением общего вида (12.1). Каллед-
жас и соавторы [Callejas et al., 1999] вывели линейные зависимости, связыва-
ющие процент удаления серы с процентом удаления металлов (а именно Ni
и V). Они нашли, что связь степени гидрообессеривания (ГДО) со степенью
гидроденикелизации (ГДН) (см. (12.7) и (12.8)) и гидродеванадизации (ГДВ)
(см. (12.9) и (12.10)) является линейной при значениях коэффициента корре-
ляции (г) между 0,88 и 0,99. Трасобарес и соавторы [Trasobares et al., 1998] вы-
вели линейную зависимость между содержанием асфальтенов (фракция, не-
растворимая в н-гептане) (Asph) и коксовым остатком по Конрадсону (CCR,
см. (12.11)) с г» 0,93. Hr[Ng, 1997] нашел линейную зависимость между плот-
ностью и содержанием асфальтенов с коэффициентом корреляции г = 0,9129
(см. (12.12)).
• Однопараметрические полиномиальные зависимости (Р). Все однопараметри-
ческие полиномиальные зависимости для свойств продуктов представля-
лись многочленами третьей степени общего вида (12.2). Цаматсулис и со-
авторы [Tsamatsoulis et al., 1991] вывели полиномиальные выражения,
прогнозирующие свойства продуктов каталитической гидроочистки, в том
числе плотность по шкале API и коксовых остатков по Конрадсону (CCR)
и Рамсботтому (RCR), как функций степеней ГДО и гидрокрекинга (ГК).
Эти зависимости (уравнения (12.13)—(12.18)) справедливы для степеней ГДО
и ГК меньше 97-98 %.
• Многопараметрические полиномиальные зависимости (MNL). Общий вид этих
зависимостей дается уравнениями (12.3) и (12.4). Фердоус и соавторы [Fer-
dous et al., 2006] сообщили о двух таких нелинейных зависимостях, связыва-
ющих степени гидродеазотирования (ГДА) и ГДО с рабочими условиями —
температурой, давлением и LHSV. Указывалось, что зависимости для расчета
ГДА (см. (12.19)) и ГДО (см. (12.20)) имеют значения г1, равные соответственно
0,97 и 0,93.
• Экспоненциальные зависимости (Е). Общий вид этих зависимостей описывает-
ся уравнениями (12.5) и (12.6). Hr[Ng, 1997] вывел различные зависимости для
прогнозирования общего содержания серы, азота, металлов (Ni + V) и кок-
суемости по Конрадсону в деасфальтизате как функции содержания асфаль-
та (определенного автором как суммарное содержание смол и асфальтенов)
в сырье. Зависимость для содержания серы (см. (12.21)) имела сравнитель-
но невысокое значение Р (0,6328); у зависимостей для содержания метал-
лов, коксуемости и азота (см. (12.22)—(12.24)) оно колеблется между 0,8819
и 0,9389.
Глава 12. Зависимости и прочие вопросы
523
12.1.2. Анализ результатов
12.1.2.1. Экспериментальные данные
Экспериментальные данные, использованные в настоящей работе, были полу-
чены гидроочисткой сырья шести различных типов, обозначенных следующим
образом:
1) А — остаток 343 °С+ атмосферной перегонки нефти с плотностью 13 ° API;
2) В — остаток 390 °С+ атмосферной перегонки нефти с плотностью 13 ° API;
3) С — остаток 326 °С+ атмосферной перегонки нефти смешанного сорта;
4) D — остаток вакуумной перегонки нефти смешанного сорта;
5) Е — остаток 538 °С+ вакуумной перегонки нефти «Майя»;
6) F— нефть «Мдйя».
Свойства сырья представлены в табл. 12.4. Они охватывают широкие диапазо-
ны значений плотности по шкале API (от 3,2 до 21), общего содержания серы (от 3,4
до 6,2 %масс.) и суммарного содержания никеля и ванадия (от 353 до 778 мг/кг). Все
виды сырья подвергались гидроочистке на катализаторах, перечисленных в табл. 12.5.
Рабочие условия, каталитическая система и типы реакторов гидроочистки для каж-
дого случая показаны в табл. 12.6.
Таблица 12.4
Свойства сырья различных типов
Свойство Тйп сырья
А В С D Е F
Относительная плотность при 60 Т/60 °F 1,0336 1,0504 1,0053 1,0464 1,0609 0,9284
Плотность, ° API 5,40 3,21 9,25 3,73 1,88 20,91
Общее содержание серы, %масс. 5,74 6,21 3,74 4,51 5,08 3,44
Содержание асфальтенов’, %масс. 21,77 25,1 10,18 17,75 25,46 12,4
Содержание азота, %масс. н/дб н/д6 4400 6100 6200 3700
Содержание металлов, мг/кг:
Ni 102,00 119,00 56,20 84,30 130,00 54,70
V 620,00 637,00 297,30 418,30 647,90 298,80
Ni + V 722,00 756,00 353,50 502,60 777,90 353,50
Коксуемость по Конрадсону, %масс. 20,80 20,34 14,30 22,59 22,83 11,73
Коксуемость по Рамсботтому, %масс. 22,18 21,48 13,39 24,98 25,41 10,50
“ Фракция, нерастворимая в «-гептане.
6 Нет данных.
524
Часть 3. Моделирование каталитических процессов
Таблица 12.5
Катализаторы, применявшиеся в экспериментах по гидроочистке
Обозначение катализатора Тип
I NiMo/Al2O3 для гидродеметаллизации (ГДМ)
II NiMo/Al2O3 для ГДМ/ГДО
III NiMo/Al2O3 для ГДО
IV Равновесный катализатор процесса El-Oil
V Свежий катализатор процесса H-OU
VI Активированный уголь - Fe
VII NiMo/y-Al2O3
VIII NiMo/Y-Al2O3 — Т1О2 для ГДМ
IX CoMo/y-A12O3 - TiO2 для ГДО
Таблица 12.6
Типы реактора и катализаторов и условия гидроочистки сырья каждого типа
Тип сырья Реакторы’ Я./Я2 Катализаторы (Я,/Я2)6 Т,°С Р, МПа Общая величина LHSV, ч* Кратность водорода, ст. куб, футов иа баррель
А TBR/TBR I-II/II-III 350-380 9,8 0,25 5000
EBR/EBR
В TBR/EBR IV-V/IV-V 380 9,8 0,25 5000
EBR/TBR
С EBR/EBR IV-V/IV-V 400 6,9-14,7 0,10-0,20 3000
D TBR/TBR VI/VI 400-415 18,1 0,15 7500
Е CSTR/— VII/- 400-435 9,8 0,12 15 000
VI/- 400-421
F TBR/TBR VIII/IX 360-400 5,3-6,9 0,25-1,25 5000
“ Я, и Я2 означают соответственно реактор 1 и реактор 2.
6 Катализаторы обозначены римскими цифрами.
Примечание. TBR — реактор с орошаемым слоем, EBR — реактор с кипящим слоем, CSTR —
реактор смешивания.
12.1.2.2. Расчет по оригинальным значениям параметров
По зависимостям (12.7)—(12.24) вычислялись свойства продуктов гидроочистки,
в том числе плотность по шкале APT, коксуемость по Конрадсону (CCR) и Рамсботто-
му (RCR), содержание асфальтенов, металлов и серы. Независимыми переменными
в этих формулах выступают другие свойства продуктов и некоторые рабочие условия.
Глава 12. Зависимости и прочие вопросы
525
Большинство зависимостей представлено сравнительно простыми линейными, по-
линомиальными и экспоненциальными функциями одной переменной. Более слож-
ные зависимости выражаются нелинейными функциями, зависящими от двух или
более параметров рабочих условий.
Сначала для каждого типа сырья получали результаты его экспериментальной
гидроочистки. Затем, располагая известными свойствами сырья и условиями его ги-
дроочистки, каждую зависимость проверяли на значениях параметров, заимствован-
ных из литературных источников. Таким путем были получены в общей сложности
84 диаграммы, кривые на которых описывались уравнениями (12.25)—(12.113), пере-
численными в табл. 12.7. Следует подчеркнуть, что приведенные в источниках значе-
ния параметров справедливы лишь для ожидаемых сырья и условий (см. табл. 12.3),
поэтому при других данных экспериментальной гидроочистки значения г2 в боль-
шинстве случаев оказались небольшими. Некоторые из зависимостей требуют дан-
ных, имеющихся не для всех типов сырья, поэтому не все уравнения можно было
проверить на сырье каждого типа. В случае экспоненциальной зависимости в каче-
стве независимой переменной вместо содержания асфальта использовалось содержа-
ние асфальтенов (фракции, нерастворимой в н-гептане).
Таблица 12.7
Зависимости, проверявшиеся на сырье имеющихся типов
Номер уравнения Сырье Номер уравнения Сырье Номер уравнения Сырье
(12.25)“ А 0,5566 (12.55)“ Е 0,8984 (12.85)м В 0,3394
(12.26)“ В 0,365 (12.56)“ F 0,8287 (12.86)“ С 0,0456
(12.27)“ F 0,8409 (12.57)“ (A-F) 0,4313 (12.87)” D 0,8729
(12.28)“ (Л, В, F) 0,4154 (12.58)“ А 0,9380 (12.88)” Е 0,4689
(12.29)“ С 0,8798 (12.59)' В 0,4608 (12.89)” F 0,9101
(12.30)" D 0,2563 (12.60)' С 0,2147 (12.90)” (A-F) 0,4337
(12.31)“ Е 0,8254 (12.61)' D 0,8072 (12.91)" В 0,755
(12.32)" (С-Е) 0,2776 (12.62)' (A-D) 0,1368 (12.92)" С 0,6648
(12.33)° А 0,6685 (12.63)ж С 0,6373 (12.93)" D 0,2437
(12.34)° В 0,2329 (12.64)ж D 0,6938 (12.94)" Е 0,6125
(12.35)° F 0,7975 (12.65)ж Е 0,9078 (12.95)" F 0,7467
(12.36)6 (А, В, F) 0,1422 (12.66)ж F 0,8209 (12.96)" (A-F) 0,2477
(12.37)° С 0,8802 (12.67)Ж {C-F) 0,6881 (12.97)° А 0,7449
(12.38)° D 0,903 (12.68)“ С 0,1839 (12.98)° В 0,8703
(12.39)° Е 0,7322 (12.69)“ D 0,7352 (12.99)° С 0,7064
526
Часть 3. Моделирование каталитических процессов
Таблица 12.7, окончание
Номер уравнения Сырье Номер уравнения Сырье Номер уравнения Сырье
(12.40)6 (С-£) 0,1459 (12.70)” (С,Д) 0,3853 [(12.100)° D 0,315
(12.41)” С 0,3252 (12.71)” С 0,6251 1 (12.101)° Е 0,9351
(12.42)” D 0,2139 (12.72)" D 0,6124 | (12.102)° F 0,8718
(12.43)” Е 0,0018 (12.73)" Е 0,9168 | (12.103)° (A-F) 0,8228
(12.44)” (С-Е) 0,0002 (12.74)" F 0,824 (12.104)” С 0,8809
(12.45)” В 0,8505 (12.75)” (С, Е, F) 0,8843 |(12.105)" D 0,2209
(12.46)” С 0,6626 (12.76)” С 0,1887 (12.106)” Е 0,8968
(12.47)” D 0,2765 (12.77)” D 0,732 (12.107)” F 0,8661
(12.48)” Е 0,8596 (12.78)” (C,D) 0,3656 (12.108)” (C-F) 0,8092
(12.49)” F 0,7988 (12.79)” С 0,8755 | (12.109)р С 0,7494
(12.50)” (B-F) 0,5056 (12.80)” D 0,0442 (12.110)” D 0,0381
(12.51)”* А 0,9228 (12.81)” Е 0,5268 (12.111)” Е 0,8457
(12.52)”” В 0,7323 (12.82)” F 0,8896 (12.112)” F 0,8416
(12.53)”* С 0,9171 (12.83)” (C-F) 0,3300 (12.113)” (C-F) 0,3973
(12.54)”” D 0,8657 (12.84)”' А 0,9787
Уравнения зависимостей:
1) общего вида (12.1):
»ГДН =ДГДО);
6 ГДВ =ДГДО);
“Asph =flCCR);
г Pirc =Л-4уМ);
2) общего вида (12.2):
МР/=ЛГДО);
еЛР/=ДГК);
* CCR =ДГДО);
”ССА=ДГК);
" АСА =ДГДО);
к АСА=ДГК);
3) общего вида (12.3):
ЛГДА=ЛГ,ДЯ5И,Р);
4) общего вида (12.4):
«гдо =дг, гяАИ;
5) общего вида (12.5):
н S =f(Asph),
6) общего вида (12.6):
° Met = flAsphy.
” CCA =f(Asph);
p N =f(Asph).
Точность прогнозирования измерялась суммой квадратов отклонений (SSE) меж-
ду расчетными и экспериментальными значениями. Величины SSE для всех зави-
симостей показаны на рис. 12.1. Для большинства зависимостей величина SSE
превышает 10.
Были построены также диаграммы рассеяния расчетных значений свойств отно-
сительно экспериментальных. Было установлено, что пять зависимостей различных
типов (формулы (12.38), (12.51), (12.58), (12.84) и (12.104)) адекватно воспроизводят
результаты для некоторых типов сырья.
Глава 12. Зависимости и прочие вопросы
527
20 30 40 50 60 70 80 90 100 110
Номер уравнения зависимости
Рис. 12.1. Величины SSE для каждой зависимости, вычисленные с использованием:
(•) — оригинальных значений параметров; (о) — оптимизированных
Отмечалось также, что одни зависимости дают приемлемые результаты для неко-
торых типов сырья, другие — неудовлетворительные. Например, уравнение (12.14)
предсказывает плотность по шкале API продукта гидроочистки сырья А лучше, чем
сырья Е, что видно из рис. 12.2.
Когда точность зависимостей проверялась на всех типах сырья, наилучшее рас-
пределение данных вокруг линии на диаграмме рассеяния давало уравнение (12.103).
Судя по диаграмме для этой зависимости (см. рис. 12.2), расчет содержания метал-
лов по оригинальным значениям параметров на другой выборке эксперименталь-
ных данных дает приемлемые результаты. Это свидетельствует о сильной корреляции
между содержанием асфальтенов и металлов.
528
Часть 3. Моделирование каталитических процессов
Расчетные значения содержания (Ni + V), мг/кг
Рис. 12.2. Плотность по шкале API, вычисленная по оригинальным значениям пара-
метров: (•) — по уравнению (12.58) для сырья А; (о) — по уравнению (12.60) для сы-
рья С (а); общее содержание металлов во всех типах сырья по уравнению (12.103) (б)
12.1.2.3. Расчет по оптимизированным значениям параметров
Расчеты по приведенным в литературных источниках значениям параметров боль-
шей частью были совершенно неудовлетворительными, хотя в некоторых случаях
и давали приемлемые результаты. Это говорит о том, что значения параметров ка-
ждой зависимости можно оптимизировать так, чтобы они предсказывали экспери-
ментальные данные с приемлемой погрешностью.
Методами линейного и нелинейного регрессионного анализа была проведена
оптимизация параметров. При оптимизированных значениях параметров величи-
ны SSE для 26 зависимостей оказались меньше 5 (см. рис. 12.1); большинство этих
зависимостей были полиномиальными. Наименьшими были величины SSE для
Глава 12. Зависимости и прочие вопросы
529
уравнений (12.45)—(12.49), описывающих линейную зависимость плотности от со-
держания асфальтенов в процессе деасфальтизации. Чем меньше была величина
SSE, тем больше были значения г2 и тем лучше распределялись данные вокруг ли-
нии на диаграмме рассеяния (рис. 12.3) по сравнению с результатами расчета по ори-
гинальным значениям параметров. Эти зависимости представляются подходящими
для предсказания плотности по содержанию асфальтенов. Однако следует отметить,
что малые величины SSE в этом случае обусловлены не сильной корреляцией (значе-
ния г2 от 0,2 до 0,86), а малыми значениями плотности по сравнению с расчетными
значениями других свойств.
Плотность, °АР1 (расчетные значения)
Рис. 12.3. Плотность в градусах API, вычисленная по уравнению (12.58):
(•) — по оригинальным значениям параметров; (о) — по оптимизированным
Плотность по шкале API как функцию степени ГДО наилучшим образом опи-
сывали полиномиальные уравнения (12.51)—(12.56) с коэффициентами корреля-
ции от 0,73 до 0,92 (в зависимости от типа сырья). Содержание серы, как функция
от содержания асфальтенов, лучше всего описывалось экспоненциальной функцией
(уравнения (12.91), (12.92), (12.94) и (12.95)). Она давала хорошее согласие с экспе-
риментальными данными для всех типов сырья, кроме Д; значения г2 для нее лежали
в интервале 0,6-0,97 (табл. 12.7).
Худшие результаты давали линейные зависимости и показательная функция,
по которой рассчитывалось содержание азота: для них приводимые в литературе зна-
чения г2 лежали между 0,0018 и 0,8. Многопараметрические полиномиальные зави-
симости для расчета степеней ГДА и ГДО давали значения г2 в интервале 0,87-0,97.
Независимыми параметрами в них выступают давление, температура и объемная
скорость подачи сырья (LHSV). В располагаемом наборе экспериментальных данных
сведения имелись лишь для сырья типа F (для трех значений температуры, двух зна-
чений давления и четырех значений LHSV) и типа С (одного значения температуры,
530
Часть 3. Моделирование каталитических процессов
четырех значений давления и двух значений LHSV). Экспериментальные данные для
сырья других типов были получены при константных значениях давления и LHSV,
непостоянной была лишь температура. Отсюда следует, что уравнение (12.89) позво-
ляет рассчитывать содержание серы в продуктах гидроочистки, но для проверки дава-
емой им погрешности необходим более полный набор данных. Зависимость (12.84),
хотя значения г2 для нее близки к 0,98, а значение SSE — к 1,29, не слишком хороша,
так как оптимизация ее параметров проводилась при постоянном значении LHSV,
что делает результаты расчета зависимыми от одной лишь температуры.
12.1.2.4. Корреляция значений параметров со свойствами сырья
После определения оптимизированных значений параметров для всех зависимостей
была сделана попытка связать эти значения с такими свойствами сырья, как плот-
ность по шкале API, содержание серы, металлов, азота и асфальтенов и коксовый
остаток (коксуемость) по Конрадсону и Рамсботтому. Для этого исследования были
выбраны лишь те зависимости, у которых найденные ранее значения Р были прием-
лемыми (больше 0,8) по меньшей мере для четырех типов сырья. Такими являлись:
1) полиномиальная зависимость для плотности по шкале API(уравнения (12.51),
(12.53)-(12.56)) с г2 > 0,82;
2) многопараметрические полиномиальные зависимости для степени удаления
серы (уравнения (12.84), (12.87) и (12.89)) с г2 > 0,87;
3) показательная зависимость для содержания металлов (уравнения (12.98),
(12.101) и (12.102)) с г2 >0,87;
4) показательная зависимость для коксуемости по Конрадсону (уравне-
ния (12.104), (12.106) и (12.107)) с г2 > 0,86.
Как уже говорилось выше, показательные зависимости были выведены не для
процессов гидроочистки и гидрокрекинга. Однако нет ничего удивительного в том,
что они хорошо предсказывают по содержанию асфальтенов содержание металлов
и коксуемость по Конрадсону: хорошо известно, что металлы концентрируются в ас-
фальтеновой фракции, а образование кокса непосредственно связано с асфальтена-
ми [Ancheyta and Speight, 2007].
Зависимости имели различный вид и варьировались от простых уравнений с од-
ним параметром до сложных с четырьмя и более параметрами. Сначала каждый оп-
тимизируемый параметр связывался с отдельными свойствами сырья, затем вы-
водились зависимости для комбинаций двух свойств. Попытка связать значения
параметров более чем с двумя свойствами не предпринималась, так как повлекла бы
чрезмерное усложнение уравнений. Чтобы сохранить простоту зависимостей, не вво-
дились тригонометрические функции и выбиралось не более двух параметров. В пер-
вом случае, т. е. при корреляции с одним свойством, было найдено наибольшее зна-
чение г2. Во втором случае (корреляция с двумя свойствами) для улучшения точности
рассматривались три параметра.
Воспроизвести с высокой точностью значения параметров одно- и многопараметри-
ческих полиномиальных зависимостей (типов Р и MNL) как функции свойств сырья
не удалось, так как эти зависимости, имея соответственно по четыре и шесть параметров,
Глава 12. Зависимости и прочие вопросы
531
весьма чувствительны к изменениям значений последних. По этим формулам были по-
лучены завышенные и заниженные результаты свойств продуктов гидроочистки сырья
некоторых типов, как показывает рис. 12.4 для случая полиномиальной зависимости.
Показательные зависимости, имеющие лишь два параметра (формулы (12.104), (12.106)
и (12.107)), также давали завышенные и заниженные результаты, хотя и более точные,
чем зависимости типов PvlMNL', об этом свидетельствует тот же рис. 12.4.
CCR (расчетные значения), %масс.
Рис. 12.4. Завышенные и заниженные результаты расчета плотности по шкале API
(уравнение (12.13)) и коксового остатка по Конрадсону (уравнение (12.23)) по значениям
параметров, коррелированным со свойствами сырья: (•) — сырье D; (о) — сырье Е
Корреляция значений параметров со свойствами сырья дала хорошие результа-
ты лишь для зависимостей (12.98), (12.101) и (12.102). Реализация описанного выше
подхода к коррелированию иногда оказывается проблематичной, так как новые па-
раметры вносят новые погрешности в оценку исходных. Поэтому в целях улучшения
оценки в исходное показательное уравнение (12.22) вместо первоначальных пара-
метров были подставлены полученные для них зависимости (см. а0 и о , табл. 12.8).
Затем минимизировали среднеквадратичную ошибку и определили новый набор
532
Часть 3. Моделирование каталитических процессов
параметров. Этот набор улучшил согласие, так как величины остатков были меньше,
чем полученные предыдущим способом (рис. 12.5). Результаты сведены в табл. 12.8.
На рис. 12.6 показано сравнение экспериментальных данных о свойствах продуктов
с расчетными. Очевидна некоторая схожесть результатов расчетов по предсказанным
и исходным значениям параметров. Это свидетельствует о том, что при сокращенном
числе параметров можно получить достаточно сильную корреляцию между свойства-
ми сырья и значениями параметров. Тем не менее, корреляцию такого вида для уве-
ренности необходимо проверить на большем числе типов сырья.
Таблица 12.8
Значения параметров уравнения (12.22), полученные оптимизацией и найденные по зависимостям
Зависимости для расчета параметров Сырье Значения параметров
Исходные Расчетные
ао °! а» «1
_ b&Sy. l + btSF 15,4810 -0,4810 В 21,6820 1,0225 22,8744 0,9984
а[ = b'jAPIyA- b{AsphF 0,0137 0,0380 Е 24,8503 1,0050 25,5933 0,9940
F 45,3056 0,6437 37,1681 0,7571
SF — общее содержание серы, %масс.; Asphf. — содержание асфальтенов, %масс.; APIF —
плотность сырья по шкале API.
100
в
80-
60 -
40-
о
20-
°о-
• <">
о •
Q Л.
оо
о
о
о
8° о 6
-20 -
о
о
о
о
-40 -
О
б©
о
-60
О 5 10 15 20 25 30 35 40 45 50 55
Рис. 12.5. Остатки значений CCR, полученные по показательной зависимости (12.23) для
сырья типов В, Е и F: (•) — подстановкой в исходное уравнение полученных зависимо-
стей для расчета значений параметров; (о) — с применением значений параметров как
функций свойств сырья (табл. 12.8)
Глава 12. Зависимости и прочие вопросы
533
Расчетные значения содержания (Ni + V), мг/кг
Рис. 12.6. Сравнение значений общего содержания металлов для сырья типа В, получен-
ных путем эксперимента и расчета: (•) — по исходным значениям параметров;
(х) — по предсказанным значениям параметров
12.2. РАСХОД ВОДОРОДА ПРИ КАТАЛИТИЧЕСКОЙ
ГИДРООЧИСТКЕ
Водород — один из важнейших продуктов современной нефтехимии. Его обычно
получают риформингом природного газа или (еще чаще) бензиновой фракции. Ос-
новные потребители водорода на НПЗ — процессы гидроочистки и гидрокрекинга.
Проблема водородного баланса вызывает особый интерес и беспокойство у нефте-
переработчиков, так как производственная политика некоторых заводов зависит
от доступности водорода. К примеру, при проблемах на установке каталитическо-
го риформинга бензина или ее остановке на обслуживание установки гидроочист-
ки и гидрокрекинга недополучают водород, что может вызвать перерыв в их ра-
боте. По этой причине водород представляет собой важный ресурс в производстве
чистых топлив, в том числе бензина и дизельного топлива с низким содержанием
серы.
Отдельно взятая промышленная установка гидроочистки в нормальном режиме
работает с катализатором и сырьем, свойства которых более или менее постоянны,
поэтому потребляемое ею количество водорода почти не меняется, Но при заметных
изменениях в свойствах катализатора и сырья, происходящих в новых условиях, нуж-
но точно знать величину расхода водорода, чтобы предвидеть его возможное влияние
на общий баланс водорода на НПЗ. Далее, точное определение расхода водорода не-
обходимо для составления уравнений баланса масс и выполнения других технико-
экономических расчетов, при разработке новых применений процесса гидроочистки
и испытании новых катализаторов в различных масштабах, в том числе в микрореак-
торах, стендовых реакторах и опытных установках.
534
Часть 3. Моделирование каталитических процессов
Распространенный способ расчета потребления водорода заключается в определе-
нии разности между количеством водорода, поступающего в реактор и покидающе-
го его, т. е. метод основан на балансе водорода в газовой фазе. Существуют и другие
подходы, основанные: 1) на подсчете расхода водорода в отдельных реакциях, про-
текающих в процессе гидроочистки; 2) балансе водорода в жидкой фазе (т. е. в жид-
ком сырье и жидком продукте); 3) общем балансе водорода с учетом всех потоков, по-
ступающих в реактор и покидающих его; 4) расчетах химических реакций. Каждый
из этих подходов нуждается в том или ином физико-химическом описании потоков
жидкости и газа, а также в определении других параметров, необходимых для состав-
ления баланса водорода. Погрешности расчета расхода водорода зависят от точно-
сти экспериментальных методик и оборудования. Используя в качестве сырья тяже-
лый газойль, полученный из битумов Атабаски, Мапюа и соавторы [Mapiour et al.,
2010] разработали многопараметрическую модель, которая включает в себя зависи-
мости между парциальным давлением водорода и степенями удаления примесей при
гидроочистке. По их наблюдениям, при возрастании парциального давления водо-
рода усиливается его растворение в жидкости и увеличивается расход. Ли и соавто-
ры [Lee et al., 2008] сравнили три способа расчета расхода водорода: по балансу водо-
рода в газовой фазе, по балансу водорода в жидкой фазе и по потреблению водорода
различными реакциями, происходящими при гидроочистке. Эксперименты этих ав-
торов по получению дизельного топлива с ультранизким содержанием серы путем
ГДО (на опытной установке) сырья, состоящего из 62 %об. прямогонного тяжелого
газойля, 10 %об. легкого газойля коксования и 28 %об. легкого рециклового газойля
каталитического крекинга, показали, что расчет расхода водорода только по резуль-
татам анализа газов дает менее надежные результаты. Стратьев и соавторы [Strati-
ev et al., 2009] установили, что правильный баланс водорода на НПЗ можно найти
исходя из химии процессов, потребляющих водород и производящих водород; к та-
ким процессам относятся гидроочистка нефтяных фракций, а также каталитический
и паровой риформинг. Исследователи вывели уравнения для расчета количества во-
дорода, потребляемого реакциями ГДО, гидродеароматизации (ГДАр), ГДА и гидри-
рования в промышленных установках. Эти уравнения позволяют оптимизировать
расход водорода на НПЗ.
Рамачандран и Менон [Ramachandran and Menon, 1998] представили краткий об-
зор различных применений водорода в промышленности. Они показали, что расход
водорода на современных НПЗ составляет около 1 %масс. перерабатываемой нефти.
В табл. 12.9 представлены типичные данные по расходу водорода в различных про-
цессах НПЗ. Большое потребление водорода в процессе гидрокрекинга вакуумных
дистиллятов объясняется тем, что он расходуется не только на удаление серы, но и на
увеличение соотношения водород/углерод в продуктах по сравнению с сырьем [Ai-
tani, 1996].
В ближайшем будущем практически все фракции сернистой нефти будут перера-
батываться в каталитических процессах при наличии водорода. Общая масса фрак-
ций, подлежащих гидроочистке и гидрокрекингу, достигнет 90 % от массы перераба-
тываемой нефти [Aitani, 1996]. Поэтому без определения расхода водорода с высокой
точностью не обойтись.
Глава 12. Зависимости и прочие вопросы
535
Таблица 12.9
Типичные данные по расходу водорода в различных процессах нефтепереработки
Процесс Расход, %масс.
от сырья от нефти
Гидроочистка: прямогонный бензин бензин крекинга 0,05 0,7-1,0 0,01 0,05-0,1
Гидрообессеривание: малосернистый газойль с содержанием серы менее 0,05 % высокосернистый газойль с содержанием серы более 0,05 % 0,15 0,35 0,04 0,05
Гидрирование рецикловых газойлей 3 0,3
Гидрокрекинг вакуумного газойля 2-3 0,5-0,8
12.2.1. Расход водорода
Расход водорода в процессах гидропереработки определяется свойствами сырья, сте-
пенью его превращения, степенью удаления примесей и свойствами катализатора. Об-
лагораживание тяжелого сырья до заданной степени требует значительного количества
водорода [Ancheyta and Speight, 2007]. Чтобы приступить к исследованиям промышлен-
ного применения нового процесса или катализатора, необходимо располагать деталь-
ным балансом масс. Без него невозможен не только учет всех технологических пото-
ков, но и нахождение распределения водорода в этих потоках. Расход водорода обычно
вычисляют на основании экспериментальных данных по его балансу в газовой фазе
либо по содержанию в жидкой фазе сырья и продуктов. На рис. 12.7 показаны все по-
токи, участвующие в общем балансе масс в реакторе гидроочистки. Здесь GaL! и Lief —
суммарные количества соответственно газа, поступающего в реактор, и жидкого сы-
рья, подлежащего гидроочистке, Gas1 и Liq' — суммарные количества соответственно
газообразного продукта, содержащего непрореагировавший водород, побочный серо-
водород и легкие углеводороды от С! до С4, и гидроочищенного жидкого продукта.
На основании рис. 12.7 можно написать следующие уравнения баланса масс.
Общий баланс масс:
Gas0 + Liq° = Gas1 + Liq1.
Баланс водорода в газовых потоках:
(Н2)° = (Н2)' + ДНш.
Общий баланс водорода:
=н>н;._
(12.114)
(12.115)
(12.116)
536
Часть 3. Моделирование каталитических процессов
Добавочный
водород
' <Н2>°
Реактор
Gas0] <H2S)°
(Ci-C6)°
Газообразный
продукт
GosH <H2S)'
Углеводородное
сырье
Ь<?°
(я) Массовый расходомер
@ Газовый хроматограф
(Н2)’
(Ci-Ce)1
Li q1
Г идроочищенный
жидкий продукт
Сепаратор
низкого
давления
Сепаратор
высокого '
давления
Рис. 12.7. Потоки, участвующие в материальном балансе на установке гидроочистки
Здесь (Н,)° и (Н2)‘ — суммарные количества водорода, соответственно поступаю-
щего в реактор и покидающего его. Если чистоту водорода принять близкой к 100 %,
то = (Н2)°; ДН^ — разность количеств водорода, поступающего в реактор и по-
кидающего его, определяемая по балансу масс в газовых потоках, которую обычно
(и ошибочно) принимают за расход водорода.
Н°ж и Н',„ — суммарные эквивалентные количества водорода (содержащегося
в газовых потоках, соответственно поступающих в реактор и покидающих его), скла-
дывающиеся из чистого водорода, водорода, содержащегося в H2S, и водорода, со-
держащегося в углеводородах С,—С4 (см. рис. 12.7). и Н'1Г/ — суммарные количе-
ства водорода, содержащегося соответственно в жидком сырье и в гидроочищенном
жидком продукте.
Н'? состоит из водорода , содержащегося в жидком веществе, и водорода
растворенного в жидкости:
+HL- (12.117)
Поэтому истинный суммарный расход водорода Нгога равен
Н = ДН + Н^,. (12.118)
cons gas a,ss ' '
Значения Нд и предпочтительнее получать экспериментальным путем —
например, элементным анализом. Однако можно оценить их и по эмпирическим
зависимостям.
Глава 12. Зависимости и прочие вопросы
537
Отталкиваясь от этих уравнений, выведенных из материального баланса, можно
предложить следующие подходы к расчету расхода водорода в процессах гидроочистки.
12.2.1.1. Расчет по балансу водорода в газовых потоках
Наиболее известен способ расчета расхода водорода на установках гидроочистки че-
рез измерение количества водорода на входе (Н2)° и на выходе (Н,)1 реакторной си-
стемы. В соответствии с уравнением (12.115), разность между этими величинами
равна химическому расходу водорода. Хотя данный способ наиболее распростра-
нен на промышленных установках гидроочистки, сообщалось о его неточности [Ai-
tani, 1996; Stratievet al.,2009]. Подобный расчет требует наличия данных о скоростях
газовых потоков на входе и выходе реактора, часто измеряемых в объемных едини-
цах, а также о составе этих потоков, для определения которого необходим поточный
газовый хроматограф.
Согласно уравнению (12.118), истинный расход Н2 вычисляется сложением коли-
чества водорода, растворенного в жидком продукте, и расхода водорода АН , опре-
деляемого по балансу в газовых потоках.
12.2.1.2. Расчет по общему балансу водорода
Более точен способ расчета, основанный на измерении содержания водорода в жид-
ком сырье и в гидроочищенном жидком продукте Н',9. Измерять содержание во-
дорода в жидком сырье и продукте можно различными методами: ASTMD5291 (стан-
дартный метод измерения содержания углерода, водорода и азота в нефтепродуктах),
ASTM 7)4808 (метод измерения содержания водорода ЯМР-спектроскопией низко-
го разрешения) и ASTM7)6733 (метод газовой хроматографии высокого разрешения).
Все эти методы требуют наличия аналитической аппаратуры, которой располага-
ют далеко не все НПЗ. Как вариант, содержание водорода можно оценить по эмпи-
рическим зависимостям, перечисленным в табл. 12.10.
Таблица 12.10
Эмпирические зависимости для оценки содержания водорода в нефтяных фракциях
Источник Зависимость Примечания
[Riazi and Daubert, 1986; Riazi et al. 1999] 100-%S %h=i+c/h Содержание азота, кислорода и ме- таллов пренебрежимо мало по срав- нению с содержанием С, Н и S
[Goossens, 1997] 82,952-65,34n 306 %H = 30,646 + — : p Mw Mw = 84-459, Ть = 60-480 °C, п= 1,38-1,51, %Н= 12,2-15,6 % масс.
[ASTM D3343, 2002] 52,407 + 0,014487] -7,O18x, %H = — — sg -0,901x, ’ A Выведена на основании данных об авиационных топливах 247 типов и чистых углеводородах 84 типов
538
Часть 3. Моделирование каталитических процессов
Таблица 12.10, окончание
Источник Зависимость Примечания
[Dhulesia, 1986] %H = 52,825 - 14,260л - 21,329s#- - 0,0024Mv - 0,0525 + 0,7571n (v) Выведена на основании данных о сырье каталитического крекин- га 33 типов
[Jenkins and Walsh, 1968] %H = 11,7 - 12,89s# + 0.0389ЛР Выведена для реактивных топлив с анилиновыми точками в интер- вале 56—77 °C
[Winn, 1957; Baird, 1981] %H = 26- 15s# Первая из известных и простей- шая зависимость
[Goodger and Vere, 1985] %H= 14,9-6,38%, Выведена на основании данных о реактивных топливах
[Korsten and Hoffman, 1996] , T , 1 Л = a0 + a T + a, — + aj" + a4 — ’ P P' Предназначена для расчета рас- творимости водорода в продуктах гидроочистки
[Mapiour and Sundaramurthy, 2009] Hfe =-23,95817 + + о'б7529(Нр11г/о,)3,56483Рн + 0,00215960- - 0,003948(н’|п.о.)2 + 0,23328PH2 + + 0,11314(H „JPH0,0001697(H ^GO Предназначена для оценки рас- творимости водорода
Обозначения:
%Н — содержание водорода, %масс.;
С/ Н — массовое соотношение углерода к водороду;
р — плотность жидкости при 20 °C;
п — показатель преломления при 20 °C;
Mw — молярная масса;
Ть — средняя температура кипения при 10, 50 и 90 %об. отгона, К;
sg — относительная плотность при 60 °F/60 °F;
хА — доля ароматических углеводородов в смеси;
АР— анилиновая точка, К;
v — вязкость при 98,9 °C, сСт;
Р — давление, МПа;
GO — отношение Н2 к С/Н;
Н — чистота водорода.
Чтобы общий баланс водорода сошелся, необходимо учесть суммарные эквива-
лентные количества водорода, содержащегося в газовых потоках, поступающих в ре-
актор (H“OJ) и покидающих его (Тогда расход водорода по его общему балансу
можно вычислить следующим образом:
Ндаи = +НЯС_Л9 +HH;S. (12.119)
12.2.1.3. Расчет по потреблению водорода в химических реакциях
Химическая характеризация потоков, показанных на рис. 12.7, способна дать све-
дения, по которым можно определить расход водорода в различных реакциях
Глава 12. Зависимости и прочие вопросы
539
гидроочистки. Ли и соавторы [Lee etal., 2008] сообщили о методе, основанном на под-
счете суммарного стехиометрического расхода водорода в реакциях, протекающих
с его поглощением:
Н . — Нгпп + Нгпд + НГПГ) + Нглп + Нг„, ;
chem ГДО ГДА 1ДОк ГДОл ГДАр’
нгдо - 0,0252sgz
Нрдок 0’ ^Sg,
S sg
S —p-^yb ;
sgf
Nsgn
N —Р_&^у
sgf P
°PS£p
(12.120)
(12.121)
(12.122)
(12.123)
^гдол Bty
BVSP r
sgf ”
(12.124)
PNA„sgn
----P-°LYB +3 МА,-
sgf
y
sgf P
Уравнения (12.121) и (12.122) были выведены на основании следующих реакций
общего вида:
НГДАР=3,3^ \PNAf-
(12.125)
Нгда“ °>08^/
С H„S + vraoH2-> С Н + H2S (12.126)
С Н N + vraAH2-> С Н +2|х_3| + NH3 (12.127)
Величины угдо и угда в реакциях ГДО (см. (12.126)) и ГДА (см. (12.127)) представ-
ляют собой стехиометрический расход водорода, т. е. молярное отношение Н, к угле-
водороду. Авторы приняли, что значение угдо равно 1 для меркаптанов, 2 для сульфи-
дов, 3 для дисульфидов, 3 для бензотиофенов, 4 для тиофенов, 2 для дибензотиофенов
и 3,6 для типичных дизельных фракций, а значение угдд — 1 для первичных аминов,
2 для вторичных аминов, 3 для третичных аминов, 1 для анилинов, 4 для пирролов,
6 для индолов, 7 для хинолинов и карбазолов и 5,0 для типичных дизельных фракций.
Для реакции гидродеоксигенации было принято значение уГДОк = 5,0, для реакции ги-
дродеароматизации — УГДАр = 3,3 (для превращения поли ароматических соединений
в моноароматические) и угда = 3,0 (для превращения моноароматических соедине-
ний в циклические предельные) [Lee et al., 2008].
Бром легко присоединяется к двойным углеродным связям, поэтому содержание
олефинов определялось по бромному числу, которое использовалось в дальнейших
расчетах расхода водорода по уравнению (12.124).
540
Часть 3. Моделирование каталитических процессов
Стратьев и соавторы [Stratiev et al., 2009] сообщили об аналогичном способе рас-
чета расхода водорода по результатам химического анализа потоков и основным ре-
акциям процесса гидроочистки. Их исследование, проведенное на установках гидро-
очистки дизельного топлива и газойля на различном сырье и в различных рабочих
условиях, дало следующие формулы:
ПГДАр(Р№4)
и
гдо
1000
о л
32 р
2 PNAf
1000
Mwf
u
n ГДАр(Л+7Н4)
r... 100
DiAf-----
f Mw,
Y„BT
—р- +2
320 )
гт
^ГДОк
10DBT.
-----L-S
32 р
1222-0^22 4-^;
Mwf PMwpj ’ 100
-PNAp
IQ^p
Mw„
2 ( TriAf
-DiAp
H
ГДА
YBT \
р р
320 )
J Ю00
+ 3 MA,----MA,
J Mw, 1
1000 ЮК
-----TnA ----
Mwr Mw
^140
Y„ AO
p____p_
100 140 ?
22,4-^-;
100
22,4—,
100
\ I
+ TriAr -TriA. +
f p 100
22,4-2^-;
100
(12.128)
(12.129)
(12.130)
Pr
22,4-^-.
100
(12.131)
(12.132)
юк/
Mvp p
p
Эти авторы предположили, что количеством водорода, расходуемого в реакции
ГДОк при гидроочистке дизельного топлива, можно пренебречь. По их сообщению,
на химический расход водорода заметно влияет качество сырья, что подтверждает
результаты предшествующих исследований. Разница между экспериментальными
и расчетными значениями расхода водорода составляла 29,2 % (±10 н. м3/м3).
12.2.1.4. Расчет по среднему вкладу отдельных реакций
Когда экспериментальных данных недостаточно или необходимо быстро оценить
расход водорода, можно прибегнуть к статистике. Эдгар [Edgar, 1993] сообщил следу-
ющие цифры среднего вклада каждой реакции гидроочистки в общий расход водоро-
да (в ст. куб. футах на баррель):
ГДО = 95-100 на 1 %масс. обессеривания; (12.133)
ГДА = 300-350 на 1 %масс. деазотирования; (12.134)
ГК = 25 на 1 %об. гидрокрекинга; (12.135)
ГДАр = 27 на 1 %масс. деароматизации; (12.136)
Глава 12. Зависимости и прочие вопросы
541
ГДМ = 26 на 1 мг/кг деметаллизации.
(12.137)
Спайт [Speight, 1999], в дополнение к данным Эдгара, предложил поправки к рас-
ходу водорода на содержание металлов (табл. 12.11).
Таблица 12.11
Поправки к расходу водорода в зависимости от содержания металлов
Ni + V, мг/кг Поправка, % Ni + V, мг/кг Поправка, %
0-100 -2 700 12
200 1 800 16
300 2,5 900 21
400 4 1000 28
500 6,5 1100 38
600 9 1200 50
12.2.1.5. Расчет по кинетической модели
Как правило, расчет расхода водорода по кинетическим данным осуществляется сло-
жением скоростей всех реакций, протекающих с поглощением водорода. Разумеется,
при этом необходимо учитывать стехиометрические коэффициенты. Чем больше ре-
акций с поглощением водорода учитывает кинетическая модель, тем точнее опреде-
ляется расход водорода.
В литературе описываются различные упрошенные подходы, основанные на ки-
нетике реакций. Папайаннакос и Георгиу [Papayannakos and Georgiou, 1988] разрабо-
тали кинетическую модель для расчета расхода водорода при каталитическом гидро-
обессеривании в реакторе с орошаемым слоем. В качестве сырья был принят остаток
атмосферной перегонки греческой нефти из залежей в Эгейском море. Исследовате-
ли установили, что модель дает хорошее согласие с экспериментальными данными
по расходу водорода.
Эти авторы составили дифференциальное уравнение баланса масс для водорода
в реакторе идеального вытеснения, работающего в изотермическом режиме. Сум-
марный расход водорода определяется начальной концентрацией всех связей, спо-
собных присоединять водород в условиях реакции. Наблюдаемая скорость потребле-
ния водорода состоит из суммы скоростей реакций водорода со связями, способными
присоединять водород. Принимая отсутствие внутричастичных диффузионных эф-
фектов, авторы вывели следующее уравнение:
(Сн)‘- - (С^)- = (а - 1)^(1 - е)Л , (12.138)
где
Сн — остаточная потребность в водороде (концентрация связей, которые в усло-
виях реакции должны присоединять водород), выраженная в молях Н2 на 1 м3 сырья;
542
Часть 3. Моделирование каталитических процессов
а — порядок реакции, зависящий от концентрации и активности реагирующих
соединений;
СГат ~ суммарный расход водорода;
4 — относительная остаточная активность катализатора;
е — доля пустот в каталитическом слое;
к — константа скорости с учетом парциального давления водорода (к = кР );
VK — объем каталитического слоя;
Ql — объемная скорость реакционной смеси.
По экспериментальным данным методом регрессионного анализа были опреде-
лены порядок реакции и суммарный расход водорода. Порядок суммарной реакции
водорода оказался равным а = 2, а величина суммарного расхода водорода в интер-
вале температур 350-430 °C для катализаторов двух типов составила 277 н. м3/м3. Ди-
аграммы зависимости от плотности остатка по шкале API, взятые из литературных
источников, дают величину суммарного расхода водорода, равную 207 н. м3/м3 [Nel-
son, 1977].
Бойтер и Шмид [Beuther and Schmid, 1963] измерили расход водорода при ГДО ва-
куумного остатка и нашли, что при степени гидрообессеривания, равной 99,2 %, ис-
комая величина составляет 277 н. м3/м3. Для второго порядка реакции водорода урав-
нение (12.138) принимает вид
1
TJ
UHr_OT„
)^Г-соти
(12.139)
= (1-£)Л
Авторы сообщили о графической зависимости для расчета истинной констан-
ты скорости реакции по экспериментальным данным в интервале температур
350-430 °C, а также о графике Аррениуса для расчета истинных констант скорости
расходования водорода.
12.2.2. Растворимость водорода
Чтобы правильно найти расход водорода по его общему балансу, нужно знать раство-
римость водорода в жидких потоках (см. рис. 12.7). Уравнения баланса масс исходят
из допущения, что равновесие газа и жидкости подчиняется закону Генри:
Н = (12.140)
где Я — константа Генри; T|w — молярный объем газа в нормальных условиях; X — ко-
эффициент растворимости; pz — плотность жидкости в условиях процесса.
Тогда концентрация водорода определяется следующим выражением:
Рн
сн,_ = (12.141)
Глава 12. Зависимости и прочие вопросы
543
Корстен и Хоффман [Korsten and Hoffman, 1996] сообщили о следующей зависи-
мости для расчета растворимости водорода в продукте гидроочистки:
Т 1
~к = а +а.Т+ а2 — + а3Т2+аА—, (12.142)
Р Р
где
а0 = -0,559729;
а1 = —0,42947 • 10-3;
а2 = 3,07539 10-3;
а3= 1,94593-IO*6;
аА = 0,835783;
Т— температура, °C;
р — плотность при 20 °C, г/см3;
X — коэффициент растворимости водорода, н. л/(кг - МПа).
Риази и Руми [Riazi and Roomi, 2007] предложили метод расчета растворимости
водорода в нефтяных фракциях при различных температурах и давлениях. Метод
применим к фракциям с молярной массой от 70 до 650, что соответствует числу ато-
мов углерода от 6 до 46.
Риази и Вера [Riazi and Vera, 2005] предложили композиционную модель пара-
финовых, нафтеновых и ароматических углеводородов для расчета растворимости
легких газов (в том числе водорода, метана, этана и диоксида углерода) в различ-
ных жидких нефтяных и угольных фракциях при разных температурах и давлениях.
На основании предложенной модели получены зависимости для расчета молярной
растворимости газа в жидкой смеси по данным парожидкостного равновесия.
Мапюа и соавторы [Mapiouret al., 2010] вывели следующую зависимость для оцен-
ки растворимости водорода (НЛя):
Н„й1 = -23,95817 + О,67529(Н а^)3,5б483Рн + 0,00215900 - 0,003948(Н +
+ 0,23328Рн2 + 0,11314(Нр„гД9,)Рн х 0,0001697(^^)00, (12.143)
где Рн — парциальное давление водорода, МПа; (Н ) — чистота водорода на входе
в реактор, %масс.; GO — кратность водорода к сырью, мл/мл.
Количество газообразного водорода, растворенного в жидкости, можно рассчи-
тать также с помощью имитатора технологических процессов HYSYS по эксперимен-
тальным данным парожидкостного равновесия при типичных условиях в контейнере
для хранения жидкого продукта гидроочистки. В расчетах равновесия можно приме-
нить уравнение состояния Чао-Сидера, справедливое при температурах ниже 257 °C
[Mapiour et al., 2010].
12.2.3. Анализ результатов
12.2.3.1. Экспериментальные данные
Эффективность разнообразных подходов к определению расхода водорода прове-
рялась на различных данных, собранных в ходе предшествующих экспериментов
544
Часть 3. Моделирование каталитических процессов
на лабораторной установке по гидроочистке бензиновой фракции (HDSN), прямо-
гонного газойля (HDSD), тяжелого газойля (HDSG) и остатков атмосферной (HDAR)
и вакуумной (HDVR) перегонки.
На установке соединялись два потока сырья — водорода и углеводородов. Ско-
рость обоих потоков контролировалась массовыми расходомерами. Водород посту-
пал непосредственно в цилиндр высокого давления через регулятор входного давле-
ния газа. Давление жидкого сырья контролировалось насосом. Температура сырья,
поступающего в реактор, регулировалась электрическими нагревателями.
Реактор содержал неподвижный слой катализатора. Продукт реакции представлял
собой парожидкостную смесь, которая подавалась в сепаратор высокого давления.
Жидкий продукт сепаратора поступал в сепаратор низкого давления, где отделялись
остатки паровой фазы. Газы из сепараторов высокого и низкого давления анализи-
ровались газовым хроматографом и проходили через массовый расходомер. Количе-
ство жидкости из сепаратора низкого давления, представлявшей собой продукт ги-
дроочистки, измерялось расходомером.
Рабочие условия различных экспериментов на лабораторной установке сведе-
ны в табл. 12,12. Смешанное сырье получали смешиванием указанных в табли-
це компонентов. Катализаторы, использовавшиеся в экспериментах, перечисле-
ны в табл. 12.13. Свойства сырья и продукта в каждом эксперименте приведены
в табл. 12.14. Погрешность схождения общего баланса масс во всех экспериментах
не превышала 0,8 %, общего баланса водорода — 1 %. Таким образом, эксперимен-
тальные данные гидроочистки нефтяных фракций можно было уверенно использо-
вать в расчетах расхода водорода.
Таблица 12.12
Рабочие условия в экспериментах на лабораторной установке
Процесс Сырье Условия
ГДО бензиновой фракции (HDSN) Бензиновая фракция /’=54,8 Т=290 LHSV=5 Н2/НС = 664
ГДО газойля (HDSD) Газойль Керосин Легкий газойль после HDSG Р=54 Т=360 LHSV= 2,4 Н2/НС = 2200
ГДО тяжелого газойля (HDSG) Газойль коксования Легкий рецикловый газойль Р = 139 Т= 338 Н2/НС= 2000 LHSV= 6 (ГДМ) LHSV= 0,78 (ГДО)
Глава 12. Зависимости и прочие вопросы
545
Таблица 12.12, окончание
Процесс Сырье Условия
Гидроочистка атмосферного остатка (HDAR) Атмосферный остаток Р= 100 Г= 397 Н2/НС= 5000 LHSV= 0,25
Гидрокрекинг вакуумного остатка (HDVR) Вакуумный остаток Легкий рецикловый газойль Экстракт масляной фракции Р = 175 Г=410 н2/ НС = 3200 LHSV= 0,18
Обозначения: Р — давление, кг/см2; Т— температура, °C; LHSV— объемная скорость пода-
чижидкого сырья, ч_|; Н2/НС — кратность водорода к углеводородному сырью, фут’/баррель.
Таблица 12.13
Катализаторы, использовавшиеся в экспериментах на лабораторной установке
Тип катализатора Применение Размер частиц, дюймы
Со-Мо ГДО бензиновой фракции (HDSN) 1/8
Ni-Mo ГДО газойля (HDSG) 1/8
Со—Мо ГДО тяжелого газойля 1/20
Со—Мо Гидроочистка атмосферного остатка 1/14
Ni-Mo Гидрокрекинг вакуумного остатка 1/32
Выше были описаны все существующие методы расчета расхода водорода при ги-
дроочистке нефтяных фракций, включая кинетические модели. Поскольку для не-
которых моделей нужны данные о термодинамических свойствах и кинетических
параметрах, а также о свойствах катализатора, ввиду отсутствия этих данных расчет
расхода водорода по кинетическим моделям ниже не рассматривается.
12.2.3.2. Расчет по общему балансу водорода
По результатам элементного анализа жидких и хроматографии газовых потоков
(см. рис. 12.7) составлялся общий баланс водорода. Количество водорода в неугле-
водородных (H2S) и легких углеводородных газообразных соединениях вычислялось
по молярному эквиваленту; предполагалось, что все эти соединения содержатся толь-
ко в газообразном продукте (Gas1). На основании этого баланса по уравнению (12.119)
вычислялась величина расхода водорода.
На рис. 12.8 показаны величины расхода водорода, определенные по эксперимен-
тальным данным для различного сырья. В силу общего баланса расход водорода зави-
сит от природы сырья. Этот известный факт объясняется тем, что тяжелые фракции
содержат больше примесей, в том числе серы, азота, асфальтенов, ароматических
Таблица 12.14
Свойства сырья н продуктов в экспериментах по гидроочистке
Свойства Ед. изм. HDSN HDSD HDSG HDAS HDRV
Сырье Продукт Сырье Продукт Сырье Продукт Сырье Продукт Сырье Продукт
Элементный анализ:
углерод %масс. 84,95 84,37 85,37 85,22 85,43 85,27 81,00 86,19 82,16 81,82
водород %масс. 14,86 15,55 13,31 14,14 11,6 12,89 13,23 15,93 13,57 16,26
Относительная плотность при 60/60 °F 0,7303 0,7288 0,8774 0,8625 0,929 0,8882 1,0232 0,9428 1,0199 0,929
Относительная плотность при 20/4 °C 0,7268 0,7253 0,8745 0,8596 0,9261 0,8853 1,0203 0,9399 1,017 0,9261
Показатель преломления 1,4064 1,4055 1,4895 1,4806 1,5196 1,4947 1,5456 1,5146 1,5413 1,5212
Молярная масса г/г моль 104 102,3 225,3 220,8 278,4 258,7 602,6 443,4 625,4 277,2
Средняя температура ки- пения °C 105,8 103,1 275,3 270,4 320,7 301,5 563,1 450,8 576,1 302,9
Вязкость при 99 °C сСт 0,3539 0,33 1,195 1,153 2,202 1,83 1315 164 1560,71 3,126
Бромное число г/100 г 1,87 0,01 3,81 1,17 — — — — — —
Общая сера: %масс. 0,1911 0,0085 1,126 0,0055 2,868 0,405 5,52 0,8725 4,268 1,029
бензотиофены 0,1204 0 1,0608 0 2,5812 0,162 — — — —
дибензотиофены 0,0707 0,0085 0,0652 0,0055 0,2868 0,243 — — — —
Ароматич. углеводороды: %об. 10,230 8,540 41,5 31,2 53,2 48,5 — — — —
полиароматические 4,23 3,54 12,0 Н,7 13,4 10,0 — — — —
моноароматические 6,0 5,0 29,5 19,5 39,8 29,5 — — — —
Асфальтены %масс. — — — — — — 20,57 6,3 14,15 6,2
Общий азот мг/кг 1,4 0,3 246 0,3 1504 447 3200 1500 4400 2210
Олефины %об. 0,889 0,715 2,6 2,4 3,1 2,8 — — — —
Никель мг/кг — — — — — — 104 46 77,21 14,2
Ванадий мг/кг — — — — — — 501 152 412,19 | 13,82 j
546 Часть 3. Моделирование каталитических процессов
Глава 12. Зависимости и прочие вопросы
547
соединений и металлов; соответственно, их удаление требует большего количества
водорода. Различия в величине расхода водорода обусловлены также разными ти-
пами реакций, преобладающими в процессе гидроочистки того или иного сырья.
В процессе гидроочистки легких и средних дистиллятов преобладает реакция гидро-
десульфуризации. В случае тяжелых фракций происходят также гидродеметаллиза-
ция и гидрокрекинг, и в меньшей степени — реакции гидродеазотирования, гидроде-
ароматизации и насыщения олефинов.
Плотность сырья, °АР1
Рис. 12.8. Значения расхода водорода, найденные по экспериментальным данным
исходя из общего баланса водорода
Расчет расхода водорода по его общему балансу требует проведения элемент-
ного анализа, поэтому на НПЗ редко пользуются этим способом, предпочитая ему
быстрый расчет по уравнению (12.115). Можно также воспользоваться одной из за-
висимостей для определения содержания водорода в жидких потоках, перечислен-
ных в табл. 12.10.
На рис. 12.9 приведено сравнение результатов определения расхода водорода
по экспериментальным данным и по общему балансу с использованием зависимо-
стей для расчета его содержания в жидких потоках.
Одни зависимости дают лучшие результаты для тяжелого сырья, другие — для лег-
кого; это объясняется тем, что они выведены для разных условий, поэтому области
их применимости различаются. Как правило, расчет расхода водорода по его обще-
му балансу, проводимый с использованием зависимостей для содержания в жидких
потоках, дает заниженные значения по сравнению с экспериментальными. В случае
гидрообессеривания (ГДО) легких дистиллятов зависимости дают схожие результаты
с наименьшими отклонениями от экспериментальных. Самые большие ошибки ха-
рактерны для ГДО тяжелого сырья.
Наихудшие результаты дают зависимости, учитывающие лишь одно свойство
жидкости (зависимости Бюро стандартов и Гуджера). Большую ошибку дает также за-
висимость ASTM Д3343 — вероятно, потому, что она предназначена для легких и чи-
стых углеводородов.
548
Часть 3. Моделирование каталитических процессов
Наилучшие результаты давал расчет расхода водорода по следующим
зависимостям:
• ГДО бензиновой фракции — зависимость Дженкинса—Уолша;
• ГДО легких и тяжелых газойлей — зависимость Дулезиа;
• гидроочистка атмосферных и вакуумных остатков — зависимость Гуусенса.
Все они давали отклонения от экспериментальных результатов в интервале
от 9 до 15 %, что вполне соответствует областям применимости, указанным их
авторами.
12.2.3.3. Расчет по балансу водорода в газовых потоках
Составлялся баланс количеств водорода в газовых потоках, поступающих в реактор
и покидающих его, а также определялось количество водорода, растворенного в жид-
кости, содержащегося как в H2S, так и в углеводородах С,-С6; последние определя-
лись газовой хроматографией и расчетом молярных эквивалентов.
Для всех фракций количество растворенного водорода было сравнительно неве-
лико, так как условия в контейнере хранения жидкого продукта гидроочистки были
близки к окружающим, а растворимость водорода сильно зависит от давления и тем-
пературы. Количество водорода, растворенного в гидроочищенном жидком потоке,
определялось не экспериментальным путем, а по двум разным зависимостям; под-
робности расчета здесь опущены.
На рис. 12.9 были показаны результаты расчета расхода водорода для всех вариан-
тов гидроочистки по балансу в газовых потоках и количеству растворенного водоро-
да; последнее определялось расчетом по зависимостям и на имитаторе HYSYS,
Зависимости Корстена-Хоффмана и Мапюа-Сундарамурти и имитатор HYSYS
давали для растворенного водорода схожие результаты. В случае гидроочистки лег-
ких и тяжелых газойлей расход водорода по уравнению (12.118) отклонялся от ре-
зультатов расчета по общему балансу менее чем на 0,23 %; для гидроочистки тяжелых
фракций (остатков) отклонения колебались между 1,14 и 2,73 %. В целом можно счи-
тать, что расчеты расхода водорода по балансу в газовых потоках и по общему балан-
су дают близкие результаты.
В балансе водорода в газовых потоках (уравнение (12.115)) количество растворен-
ного водорода составляет 0,1—6 % от его общего расхода; такой вклад можно счи-
тать небольшим. Это наиболее вероятная причина того, что на НПЗ расход водоро-
да предпочитают указывать как разность между его количествами во входящих
и выходящих газовых потоках.
12.2.3.4. Расчет по потреблению водорода
в химических реакциях
Для расчета расхода водорода по свойствам входящих и выходящих жидких пото-
ков использовались зависимости Ли и Стратьева-Цингова. Эти схожие зависимо-
сти, учитывающие свойства сырья и продукта, выведены для гидроочистки легких
газойлей.
Глава 12. Зависимости и прочие вопросы
549
На рис. 12.9 было представлено сравнение результатов расчета по указанным за-
висимостям и расчета по общему балансу водорода с использованием эксперимен-
тальных данных. Величины расхода водорода при ГДО атмосферных и вакуумных
остатков не показаны, так как эти зависимости не учитывают вклад реакций ги-
дродеметаллизации и гидрокрекинга. В целом эти зависимости занижают значе-
ния расхода. Наименьшую погрешность они дают для случая ГДО легких газойлей.
12.2.3.5. Расчет по среднему вкладу отдельных реакций
Проводился также расчет расхода водорода при гидроочистке по методу Эдгара с уче-
том поправок Спайта на содержание металлов в сырье и продуктах.
Результаты расчета по методу Эдгара—Спайта сравниваются на рис. 12.9 с резуль-
татами расчета по общему балансу водорода, включающего в себя использование
экспериментальных данных. Для гидроочистки тяжелого сырья метод дает явно луч-
шие результаты. В случае легких фракций ошибка превышает 50 %.
Результаты расчетов, представленных в подразделе 12.2.3, можно прокомментиро-
вать следующим образом.
• Величину расхода водорода невозможно напрямую определить по эксплуата-
ционным данным установки гидроочистки. Распространенный способ расче-
та по количествам водорода во входящем и выходящем газовых потоках может
занижать результаты на величину до 6 %. Истинную величину расхода водоро-
да лучше всего определять по общему его балансу с учетом скоростей потоков,
данных газовой хроматографии и элементного анализа жидкости и количества
растворенного водорода.
• Для расчета содержания водорода в жидких потоках лучше всего подходят сле-
дующие зависимости: Дженкинса—Уолша для бензиновой фракции; Дуле-
зиа для легкого и тяжелого газойлей; Гуусенса для атмосферного и вакуумного
остатков.
• Зависимости Корстена-Хоффмана и Мапюа—Сундарамурти, как и имитатор
HYSYS, дают схожие результаты для количества растворенного водорода, поэ-
тому выбор среди них не играет важной роли.
• При быстрых расчетах вклад реакций гидроочистки средних фракций в общий
расход водорода можно оценивать по зависимостям Ли и Стратьева—Цингова,
тогда как для тяжелых фракций лучше подходит способ Эдгара-Спайта.
12.3. ИСТИННЫЕ ЗНАЧЕНИЯ СТЕПЕНИ ПРЕВРАЩЕНИЯ
И ВЫХОДА ПРОДУКТА ПРИ ГИДРОПЕРЕРАБОТКЕ
ТЯЖЕЛОЙ НЕФТИ
Расчет степени превращения и выхода — задача, часто встречающаяся при исследо-
вании реакций в различных масштабах (в микрореакторах, лабораторных реакторах,
реакторах опытных и промышленных установок). При экспериментах на модельных
соединениях степень превращения определить легко:
б) 1200г
с
ф
& 1000i
га
<Р
800
VD
600
с
га
о 4001
а.
о
g
£ 200
г> 1400
£
о. 1200
о.
га
о
1000
Экспериментальные данные
□ Метод Эдгара-Спайта
800
600
400
200
HDSD
HDVR
Рис. 12.9. Результаты расчета расхода водорода: a — по общему балансу и зависимостям для содержания водорода в жидких
потоках; б — по балансу водорода в газовых потоках и зависимостям для содержания растворенного водорода; в — по поглоще-
нию водорода в химических реакциях; г — по среднему вкладу отдельных реакций
550 Часть 3. Моделирование каталитических процессов
Глава 12. Зависимости и прочие вопросы
551
Начальное число молей ЛР - Конечное число молей Л Р /
/ Начальное число молей Л Р,
где ЛР — лимитирующий реагент.
Иногда ЛР по какой-либо причине разбавляют другим соединением: напри-
мер, при гидродесульфуризации дибензотиофена часто в качестве внутреннего
эталона используют н-декан. В этом конкретном примере для определения степе-
ни превращения площади пиков на хроматограмме соотносят с начальными кон-
центрациями соединений, предполагая, что н-декан в условиях реакции остается
непревращенным [Rana et al., 2007]. Но когда эксперименты проводятся в про-
мышленном масштабе на реальном сырье, вычисление степени превращения мо-
жет быть нелегкой задачей. Трудность в том, что в некоторых промышленных про-
цессах в реактор вместе с сырьем подаются и другие материалы: это усложняет
сведение баланса масс. Например, при промышленном гидрокрекинге вакуумно-
го остатка в реакторе с кипящим слоем катализатора сырье в целях уменьшения
осадка часто разбавляют легкими жидкостями, обогащенными ароматическими
соединениями, которые производят на другой или на той же установке [Anchey-
ta and Speight, 2007]. Свойства и состав такого нестандартного сырья вносят иска-
жения в расчеты степени превращения и других величин, проводимые на основа-
нии баланса масс.
12.3.1. Экспериментальные данные
В данном подразделе определение истинных значений степени превращения и вы-
хода в случаях, подобных описанному выше, иллюстрируется на примере с экспе-
риментальными данными, полученными при гидрокрекинге остатка на опытной
установке в реакторе с кипящим слоем. Сырье, катализатор и условия реакций те
же, что и на промышленной установке гидрокрекинга. Опытная установка схема-
тически показана на рис. 12.10. Подробнее о ней можно узнать в другом источни-
ке [Ruiz et al., 2005]. Для нормальной работы установки сырье (обычно вакуумный
остаток) необходимо подавать вместе с вакуумным газойлем. Это позволит исклю-
чить закупорку линий и запорно-регулирующих устройств, предотвратить отказ
разрыхляющих (рециркуляционных) насосов и избежать возможного прекращения
работы установки. Гидрокрекинг такого комбинированного сырья дает газы, лег-
кие фракции (бензин, средние дистилляты, вакуумный газойль) и остаток. Среди
продуктов гидрокрекинга, покидающих реактор, есть фракция вакуумного газойля,
аналогичная входящему в состав сырья. Проблема заключается в том, что на прак-
тике невозможно отделить газойль сырья от газойля, образующегося в реакторе, так
как их интервалы кипения близки. Если этот газойль не включить в баланс масс как
отдельную составляющую, расчетные значения выхода и степени превращения бу-
дут отличаться от истинных. Однако существует альтернативный способ «разделе-
ния» газойлей — имитация.
552
Часть 3. Моделирование каталитических процессов
Вода
К' । Сепаратор высокого
давления и низкой
температуры
А Сепаратор высокого
ч ’ давления и высокой
U температуры
f Сепаратор
( (низкого
давления
крекинга вода углеводороды
Продукт
гидро-
Рис. 12.10. Схема опытной установки с реакторами с кипящим слоем
12.3.2. Методика
В вышеупомянутом примере с опытной установкой для различения газойля ги-
дрокрекинга от газойля сырья использовались данные расхода вакуумного газой-
ля (45,97 мл/ч) и остаточного сырья (318,3 мл/ч) на входе и продукта гидрокрекинга
(372,71 мл/ч) на выходе, а также кривые их разгонки (рис. 12.11). Кривые экспери-
ментальной разгонки, показанные на рис. 12.10, соответствуют остаточному сырью
(поток 1, см. рис. 12.10), вакуумному газойлю (поток 3), смеси вакуумного газойля
и остатка (смесь потоков 1 и 3) и продукту гидрокрекинга (вместе с вакуумным га-
зойлем сырьевой смеси, сумма потоков 4 и 5). Первые три определялись методом
ASTM D\ 160, так как они описывают продукты вакуумной перегонки нефти. Послед-
няя кривая определялась методом ASTM£>2892 (метод измерения истинных темпера-
тур кипения), поскольку она описывает и легкие продукты гидрокрекинга.
Технологические потоки представлялись в имитаторе HYSYS псевдокомпонента-
ми в соответствии с их средними нормальными (т. е. при атмосферном давлении)
температурами кипения. Было выбрано в общей сложности 36 псевдокомпонентов;
их данные можно увидеть в табл. 12.15. На основании данных отдельных кривых раз-
гонки (зависимость объемного процента отгонки от температуры, рис. 12.11) была
определена объемная доля каждого псевдокомпонента в вакуумном газойле и про-
дукте гидрокрекинга. Затем по общему объемному расходу и объемной доле псев-
докомпонентов вычислялись их объемные расходы. Затем по разности объемных
расходов псевдокомпонентов в продукте и в сырьевой смеси вычислялись их величи-
ны их расхода, а также их доли в продукте гидрокрекинга за вычетом газойля сырья.
Глава 12. Зависимости и прочие вопросы
553
Например, расход в сырьевой смеси псевдокомпонента со средней температурой ки-
пения 294 °C равен 0,37 мл/ч, в продукте гидрокрекинга — 11,81 мл/ч; истинная ве-
личина расхода равна (11,81 - 0,37) мл/ч = 11,44 мл/ч. Процедура предполагает, что
исходный газойль сырьевой смеси не вступает в реакцию.
Рис. 12.11. Кривые разгонки различных потоков: (—) — вакуумный газойль (ASTM D1160);
(—) — вакуумный остаток (ASTM D1160); (—) — вакуумный газойль плюс вакуумный
остаток (ASTM D1160); (--------) — продукт реактора, содержащий вакуумный
газойль (ASTM D2892)
Таблица 12.15
Объемный расход н состав потоков
Средняя температура кипения псевдокомпонента, °C Вакуумный газойль в сырьевой смеси Продукт гидрокрекинга (с вакуумным газойлем) Продукт гидрокрекинга (без вакуумного газойля)
%об. мл/ч %об. мл/ч мл/ч %об.
84 0,00 0,00 0,83 3,11 3,11 0,95
98 0,00 0,00 0,80 3,00 3,00 0,92
112 0,00 0,00 0,82 3,05 3,05 0,93
126 0,00 0,00 0,87 3,24 3,24 0,99
140 0,00 0,00 1,00 3,73 3,73 1,14
154 0,00 0,00 1,21 4,53 4,53 1,39
168 0,00 0,00 1,50 5,59 5,59 1,71
182 0,00 0,00 1,73 6,45 6,45 1,97
196 0,00 0,00 1,86 6,93 6,93 2,12
210 0,00 0,00 2,19 8,14 8,14 2,49
224 0,00 0,00 2,60 9,68 9,68 2,96
238 0,00 0,00 3,22 11,99 11,99 3,67
554
Часть 3. Моделирование каталитических процессов
Таблица 12.15, окончание
Средняя температура кипения псевдоком- понента, °C Вакуумный газойль в сырьевой смеси Продукт гидрокрекинга (с вакуумным газойлем) Продукт гидрокрекинга (без вакуумного газойля)
%об. мл/ч %об. мл/ч мл/ч %об.
252 0,00 0,00 3,27 12,20 12,20 3,73
266 0,00 0,00 2,90 10,80 10,80 3,31
280 0,00 0,00 2,92 10,90 10,90 3,34
294 0,81 0,37 3,17 11,81 11,44 3,50
308 1,05 0,48 2,98 11,09 10,61 3,25
322 1,27 0,58 3,02 11,26 10,68 3,27
336 1,61 0,74 2,93 10,90 10,16 3,11
350 2,22 1,02 2,95 10,98 9,96 3,05
364 3,18 1,46 2,87 10,72 9,26 2,83
378 3,81 1,75 2,85 10,61 8,86 2,71
392 5,00 2,30 2,75 10,25 7,95 2,43
406 9,02 4,15 3,10 11,56 7,41 2,27
420 9,32 4,28 3,22 12,01 7,73 2,37
440 17,13 7,88 5,77 21,51 13,63 4,17
469 16,50 7,58 6,23 23,23 15,65 4,79
496 14,17 6,52 5,68 21,15 14,64 4,48
523 9,17 4,22 4,81 17,94 13,72 4,20
554 3,59 1,65 3,91 14,56 14,56 3,95
578 2,17 1,00 4,03 15,04 15,04 4,30
605 0,00 0,00 3,12 11,63 11,63 3,56
632 0,00 0,00 2,29 8,53 8,53 2,61
670 0,00 0,00 3,28 12,23 12,23 3,74
739 0,00 0,00 1,58 5,88 5,88 1,80
790 0,00 0,00 1,74 6,50 6,50 1,99
Всего 100,00 45,97 100,00 372,71 326,74 100,00
12.3.3. Результаты
По кривой разгонки, скорректированной вычетом исходного вакуумного газойля
из продукта гидрокрекинга, через интерполяцию с применением функции распре-
деления вероятностей (подробнее см. [Sanchez and Ancheyta, 2007]) был определен
истинный состав продукта. Затем были вычислены объемные выходы компонентов
продукта. Для этого объемный расход компонента делили на объемный расход соот-
ветствующего сырья — т. е. на суммарный расход вакуумного газойля и вакуумного
Глава 12, Зависимости и прочие вопросы
555
остатка, если исходный газойль не вычитался из продукта, и на расход вакуумного
остатка в противном случае. Результаты расчетов представлены в табл. 12.16. Следу-
ет заметить, что в промышленной практике выход продуктов определяют по соответ-
ствующим объемным расходам на выходе перегонной колонны, а в данной работе —
по результатам лабораторной разгонки. Так как при промышленной перегонке всегда
есть перекрывающиеся фракции, значения выхода продуктов могут слегка отличать-
ся от найденных путем лабораторной разгонки.
Таблица 12.16
Степень превращения и выход продуктов при гидрокрекинге
Фракция Интервал ИСТИННЫХ температур кипения, °C Доля в продукте, %об. Выход, %об.
С газойлем сырья Без газойля сырья С газойлем сырья Без газойля сырья
Бензин НК-200 10,1 12,8 10,3 13,1
Керосин 200-260 11,7 14,1 12,0 14,5
Газойль 260-400 29,7 30,2 30,4 31,0
Вакуумный газойль 400-538 28,2 22,9 28,8 23,5
Вакуумный остаток 538+ 20,3 20,0 20,8 20,5
Всего 100,0 100,0 102,3 102,6
Степень превращения — — 67,9 71,5
Степень превращения остатка вычислялась исходя из данных разгонки по методу
ASTM ZJ116O следующим образом:
Степень превращения =
'VFfxf™-VFpxf™''
I J
100%,
(12.144)
где VFf и VFp — объемные расходы сырья и продукта; у^38 и у538 — доли фракции
с температурой кипения выше 538 °C соответственно в сырье и продукте.
Следует упомянуть, что вещество продукта, определяемое массовой или объемной
долей в интервале кипения остатка, по составу и свойствам отличается от вещества
сырья с аналогичной температурой кипения. Расчет степени превращения на прак-
тике предполагает, что между составами обоих веществ нет различий. Это может вы-
глядеть бессмысленным, так как подразумевает рассмотрение одного и того же ком-
понента, но чисто практически расчет соответствует уравнению (12.144).
Совершенно очевидно, что если не вычитать вакуумный газойль из продукта, сте-
пень превращения и выходы продуктов будут вычислены с ошибкой. Ошибка в сте-
пени превращения достигает 3,6 %. Ошибки в долях и выходе бензиновой фракции,
керосина и газойля составляют от 0,5 до 2,8 %об. Выход вакуумного газойля, безус-
ловно, окажется ниже, если вычесть исходный газойль из продукта.
556
Часть 3. Моделирование каталитических процессов
Эти результаты подчеркивают важность моделирования при определении истинных
величин степени превращения и выхода продуктов в операциях гидрокрекинга. Пред-
ложенный подход можно применять не только при гидрокрекинге, но и в других случа-
ях, когда специфичные требования вынуждают подавать в реактор нестандартное сырье.
12.4. ПЕРЕСЧЕТ СОДЕРЖАНИЯ МЕТАЛЛОВ
НА СВЕЖИЙ КАТАЛИЗАТОР
Катализаторы, применяемые в нефтепереработке, в той или иной степени подверже-
ны потере активности. В основе большинства механизмов деактивации лежит отло-
жение кокса и металлов. При гидропереработке тяжелой нефти в течение нескольких
первых часов работы происходит обратимая (временная) потеря активности вслед-
ствие интенсивного отложения кокса. Отложение металлов, вызывающее необра-
тимую (постоянную) потерю активности, происходит в течение всего цикла. В не-
которых процессах — например, при каталитическом крекинге в псевдоожиженном
слое (FCC, иначе каталитическом крекинг-флюиде) — катализатор теряет активность
очень быстро (в течение секунд). Катализатор, пройдя через лифт-реактор, поступает
в регенератор, где подвергается выжиганию кокса с собственной поверхности. Такая
система позволяет проводить процесс непрерывно. В других процессах деактивация
катализатора может происходить очень медленно (годами) — например, вследствие
закоксовывания при ГДО бензиновой фракции, — или сравнительно быстро (в тече-
ние месяцев) из-за отложения кокса и металлов (гидроочистка тяжелой нефти).
В нефтеперерабатывающей промышленности есть общее правило: чем тяжелее
сырье и чем больше примесей оно содержит, тем быстрее катализатор теряет актив-
ность. Скорость деактивации также во многом определяется каталитической систе-
мой, условиями реакций и конструкцией реактора [Ancheyta et al., 2005а].
В промышленной практике катализаторы относят к той или иной категории в за-
висимости от текущей стадии их рабочего цикла. Ниже приведено их описание, чаще
всего используемое в нефетепереработке [Ancheyta and Speight, 2007].
• Свежий катализатор — катализатор в том виде, в каком он поставляется
производителем.
• Отработанный катализатор — катализатор, выгруженный из реактора после
определенного периода отработки.
• Регенерированный катализатор — отработанный катализатор после выжига
кокса с его поверхности.
• Равновесный катализатор — катализатор, средняя активность которого поддер-
живается на постоянном уровне благодаря периодическому отбору из реактора
части отработанного катализатора и подгрузке такого же количества свежего.
Уровень активности такого катализатора достигает установившегося, т. е. рав-
новесного, значения.
Из средств слежения за активностью катализаторов в ходе производственных
процессов на НПЗ чаще всего применяют следующие: анализ структурных свойств
Глава 12. Зависимости и прочие вопросы
557
(площади поверхности, порового объема, среднего диаметра пор и распределе-
ния размеров пор) методом адсорбции и десорбции азота; определение содержа-
ния металлов (главным образом ванадия и никеля) методом атомно-абсорбци-
онной спектроскопии; измерение содержания кокса методом сжигания. Кроме
того, есть и более сложные методы, которыми пользуются большей частью в де-
тальных исследованиях деактивации. В их числе спектроскопия ядерного магнит-
ного резонанса (ЯМР), рентгеновская фотоэлектронная спектроскопия (РФЭС),
просвечивающая (трансмиссионная) электронная микроскопия (ПЭМ), растро-
вая (сканирующая) электронная микроскопия (РЭМ) и термогравиметрический
анализ (ТГА).
В частном случае гидропереработки в неподвижном каталитическом слое поте-
ря катализатором активности обусловлена главным образом образованием на его по-
верхности и в порах отложений кокса и металлов. По этой причине такие процессы
проводят в нескольких каталитических слоях. Входной (защитный) слой, характе-
ризуемый пониженным содержанием активного металла и надлежащим распределе-
нием размеров пор, должен обладать высокой металлоемкостью, что позволяет ему
накапливать большие количества металлов, сохранять активность в течение длитель-
ного времени и предотвращать преждевременную потерю активности других катали-
заторов. Для слоев, расположенных после него, выбирают катализаторы, более из-
бирательные к удалению серы и других примесей и к крекингу тяжелых соединений
до более легких [Rana et al., 2005а,Ь].
Снижение активности катализатора отражается на превращении сырья и удале-
нии примесей. На НПЗ обычно допускают снижение активности до определенного
уровня, а для сохранения производительности и качества продуктов постепенно по-
вышают температуру в реакторе, чтобы компенсировать потерю активности. Степень
деактивации можно найти сравнением активностей рабочего и свежего катализато-
ров. Например, активность катализатора FCC определяют испытанием на микроак-
тивность, проводимым в стандартных условиях на стандартном сырье.
После того как катализатор гидропереработки отработает определенное время,
следует измерить содержание в нем металлов: это даст возможность оценить ско-
рость их отложения и срок службы катализатора. Полученная информация позво-
лит, в частности, решить, какой катализатор лучше всего подходит для переработки
данного сырья. Эти параметры представляют большой интерес не только для про-
изводителей катализаторов, но и для нефтепереработчиков, разработчиков техноло-
гических процессов и исследователей катализаторов, поскольку экономические по-
казатели установки гидроочистки определяются главным образом сроком службы
катализатора.
12.4.1. Постановка задачи
Как было сказано выше, отложение металлов при гидроочистке тяжелого сырья —
одна из основных причин потери активности катализатора, поэтому в первую очередь
558
Часть 3. Моделирование каталитических процессов
необходимо правильно определить содержание металлов. При этом важно не толь-
ко найти его, но и привести результаты анализа к одному и тому же базису. Иными
словами, за измерением содержания металла в отработанном катализаторе (в грам-
мах металла на грамм отработанного катализатора) должно следовать составление ба-
ланса масс, предполагающее пересчет результата в граммы металла на грамм свежего
катализатора. Такой пересчет позволит сравнивать катализаторы независимо от со-
стояния образца, т. е. вне зависимости от того, является ли катализатор свежим, отра-
ботанным или регенерированным. Пересчет несложен и может даже показаться три-
виальным. Однако здесь важна не столько методика расчета сама по себе, сколько
приведение к общей базе публикуемых в работах исследователей результатов, полу-
чаемых исходя из простого баланса масс. Вот несколько примеров, в которых могло
произойти отступление от этого правила.
• Марафи и соавторы [Marafi et al., 2007, 2008] проводили испытания по уско-
ренному старению различных катализаторов ГДО атмосферного остатка с це-
лью определить их максимальную металлоемкость. Для расчета содержания
металлов в катализаторе (МОС) исследователи составили баланс масс метал-
лов, поступающих в реактор и покидающих его. Разность делили на количество
свежего катализатора, загруженного в реактор. Проводился также анализ от-
работанного катализатора, результаты которого выражались в граммах металла
на 100 г свежего катализатора. В данном случае содержание металлов выража-
лось правильно, т. е. приводилось к свежему катализатору.
• Калледжас и соавторы [Callejas et al., 2001а,b] проводили эксперименты по ги-
дроочистке остатка перегонки нефти «Майя». Они позаботились о том, чтобы
привести величину МОС к свежему катализатору, но в качестве элемента с по-
стоянной концентрацией вместо алюминия взяли молибден. Это увеличило
погрешность результатов, так как любой катализатор гидроочистки содержит
меньше молибдена, чем алюминия. Несмотря на это, результаты ясно показы-
вали, насколько большой может быть разница в содержании металлов, если вы-
разить ее в расчете на свежий катализатор вместо отработанного. Например,
содержание ванадия на 100 г свежего катализатора было примерно в два раза
больше, чем на 100 г отработанного,
• Некоторые авторы [Nucez-Isaza et al., 2000; Sun et al., 2001; Valverde et al., 2008]
указывают содержание металлов относительно отработанного катализатора
и не пересчитывают результаты на свежий. Это следует из опубликованных эти-
ми авторами результатов, поскольку содержание молибдена в них было пере-
менным. Если бы результаты были выражены относительно свежего катализа-
тора, то содержание молибдена оставалось постоянным. Это могло не влиять
на сделанные указанными авторами выводы, но значения МОС и даже порядок
расположения катализаторов по металлоемкости могли оказаться другими.
• В известных публикациях этому аспекту не придается особого значения, и ре-
зультаты, опубликованные одними авторами, используются другими без при-
ведения данных к свежему катализатору [Furimsky and Massoth, 1999; Tahur and
Thomas, 1985].
Глава 12. Зависимости и прочие вопросы
559
12.4.2. Пробы катализатора
Пересчет содержания металлов на свежий катализатор иллюстрировался на пробах
отработанного катализатора, отобранных из реактора опытной установки гидроо-
чистки. В реактор были загружены катализаторы трех типов разной функциональ-
ности — ГДМ, ГДМ/ГДО и ГДО, — на которых в течение 155 суток проводилась ги-
дроочистка атмосферного остатка (плотность 5 °АР1, содержание никеля 84 мг/кг,
содержание ванадия 423 мг/кг) тяжелой нефти (плотность 13 °АРГ). Для анализа от-
бирались только пробы катализатора ГДО (удельная поверхность 189 м2/г, удельный
поровый объем 0,69 см3/г, средний диаметр пор 147 ), Условия реакций, характери-
стики опытной установки и порядок проведения экспериментов не имеют большого
значения для целей настоящей работы; при необходимости о них можно подробнее
узнать в другом источнике [Ancheyta et al., 2007]. Что действительно важно — это по-
рядок отбора проб, исключающий контакт с окружающей средой. Его подробно опи-
сали Анчита и соавторы [Ancheyta et al., 2003]. Сразу же после отбора пробы ее пе-
реносят в инертную среду, поместив, например, в сосуд с толуолом или н-гептаном.
Проба хранится в герметически закрытом сосуде вплоть до начала анализа. При про-
ведении анализа с пробой следует обращаться осторожно, не допуская контакта ката-
лизатора с воздухом, иначе свойства первого могут измениться.
Катализатор промывают толуолом в аппарате Сокслета, чтобы удалить углеводо-
роды и другие примеси, адсорбированные на его поверхности, и высушивают. Затем
катализатор в течение 5 ч регенерируют горячим воздухом при температуре 550 °C,
чтобы выжечь большую часть кокса и серы. Удаление оставшегося твердого кокса
требует более жестких условий регенерации.
12.4.3. Результаты
В табл. 12.17 приведены состав и структурные свойства свежего, отработанного
и регенерированного катализаторов. Из таблицы видно, что значения структурных
свойств существенно (примерно на 50 %) меньше, чем часто публикуемые другими
авторами, проводившими эксперименты по гидропереработке тяжелой нефти.
Результаты анализа свежего катализатора не требуют никакой обработки, так как
они уже приведены должным образом (выражаются в грамме металла на 100 г све-
жего катализатора). В 3-м и 4-м столбцах приведены значения содержания металлов
в отработанном и регенерированном катализаторах, не пересчитанные на свежий ка-
тализатор. Видно, что содержание А1 и Мо в отработанном катализаторе уменьшает-
ся по сравнению со свежим, а содержание Ni возрастает. Ванадия, углерода и серы
в свежем катализаторе нет: они появляются только в отработанном. Регенерация
существенно снижает содержание S и С и увеличивает содержание Al, Mo, V и Ni.
Эти заключения могут оказаться неверными, так как результаты анализа не приве-
дены к одной и той же основе. Например, содержание А1 и Мо должно оставаться
более или менее постоянным, так как этих элементов обычно нет в тяжелом сырье,
проходящем через реактор; в данном же случае наблюдается кажущееся снижение
560
Часть 3. Моделирование каталитических процессов
содержания А1 примерно на 17 %. Разумеется, могут быть незначительные отклоне-
ния, обусловленные погрешностью анализа, но столь большое отклонение выходит
за мыслимые пределы погрешностей эксперимента. Поскольку никель содержится
как в свежем катализаторе, так и в сырье, для определения истинного количества ни-
келя, отложившегося на катализаторе, необходимо составить баланс масс.
Таблица 12.17
Структурные свойства и содержание металлов, кокса и серы в катализаторе ГДО
Катализатор
Свойства Свежий Отработанный Регенерированный
Удельная поверхность, м2/г 189 89 93
Удельный поровый объем, см3/г 0,69 0,28 0,35
Средний диаметр пор, А 147 118 138
Свежий, %масс. Отработанный, %масс. Регенерированный, %масс. Изменение3, %масс. Изменение6, %масс.
А1 42,50 25,49 28,14 -17,01 +2,65
Мо 6,97 4,22 4,52 -2,75 +0,30
Ni 2,19 3,1 3,59 +0,91 +0,49
V 0 11,39 12,32 +11,39 +0,93
С 0 9,32 0,63 +9,32 -8,69
S 0 7,6 1,25 +7,6 -6,35
Примечание. Знак «минус» означает уменьшение, «плюс» — увеличение.
“ Относительно свежего катализатора.
6 Относительно отработанного катализатора.
При пересчете результатов анализа на свежий катализатор используется то
обстоятельство, что по меньшей мере один из элементов отсутствует в сырье;
в данном случае оно не содержит ни алюминий, ни молибден. Это значит, что не-
зависимо от состояния катализатора в пробе (свежий, отработанный или регене-
рированный), их количества (в единицах массы) должны оставаться теми же, так
как эти металлы в условиях промывки и регенерации не претерпевают измене-
ний. При составе, указанном в табл. 12.17, в 100 г свежего катализатора содержит-
ся 42,5 г А1 и 6,97 г Мо.
Катализаторы гидроочистки содержат больше всего алюминия, поэтому удобнее
сводить баланс масс по нему, так как вносимая погрешность будет меньше. Для этой
цели можно взять и молибден, но в этом случае погрешность будет больше.
Масса отработанного катализатора, соответствующего 100 г свежего, вследствие
отложения V, Ni, С и S будет больше 100 г. Точную ее величину определить практиче-
ски невозможно из-за потерь при отборе и обработке пробы, но по балансу алюми-
ния можно вычислить следующим образом:
Глава 12. Зависимости и прочие вопросы
561
Для отработанного катализатора
^5 49 %масс
Масса А1 (г) = Масса отработанного катализатора (г) х —-----: = 42,50 г;
100 %масс.
Масса отработанного катализатора = 166,73 г.
Для регенерированного катализатора
28,14 %масс. _
Масса А1 (г) = Масса регенерированного катализатора (г) х ———------42,5 г;
100%масс.
Масса регенерированного катализатора = 151,03 г.
Найдя массы отработанного и регенерированного катализаторов, можно вычис-
лить и массы других элементов. Например, для ванадия в отработанном катализаторе
Масса V (г) = 166,73 г х 11’39%масЕ: = 18;99 г>
100 %масс.
Отнеся это количество к 100 г свежего катализатора, получим содержание вана-
дия в пересчете на свежий катализатор; оно равно 18,99 г ванадия на 100 г свежего
катализатора.
В табл. 12.18 приведены результаты расчетов для всех элементов пробы. В случае
никеля его конечное количество уменьшено на величину начального (2,19 %масс.).
Теперь можно провести более точное сравнение. Во-первых, содержание алюминия
во всех пробах остается постоянным (42,5 %), так как именно по нему сводился ба-
ланс масс. Изменение содержания молибдена меньше, чем по табл. 12.17, и не вы-
ходит за пределы погрешности эксперимента. Во-вторых, значения содержания ва-
надия в отработанном и регенерированном катализаторах почти одинаковы, так как
регенерация не затрагивает ванадия. Эти значения, отнесенные к свежему катализа-
тору, несколько меньше, чем отнесенные к отработанному. Наблюдаемые различия
объясняются погрешностью анализа. По данным табл. 12.17 все эти выводы сделать
невозможно.
По цифрам содержания, отнесенным к отработанному (см. табл. 12.17) и к свеже-
му (см. табл. 12.18) катализаторам, можно оценить общее содержание металлов в от-
работанном катализаторе, скорость их отложения в течение 155 суток работы (пред-
полагается, что отложение происходит по линейному закону) и остаточный срок
службы, приняв указанную производителем максимальную металлоемкость (33 %
от массы свежего катализатора). Результаты расчетов сведены в табл. 12.19. Разница
в результатах (вычисленных относительно свежего и отработанного катализаторов)
весьма значима. Расчет относительно отработанного катализатора завышает остаточ-
ный срок службы примерно на 60 %.
Эти результаты демонстрируют, что содержание металлов должно быть выражено
в пересчете на свежий катализатор. Активности отработанного (с отложениями ме-
таллов) и свежего катализаторов следует сравнивать по скорости данной каталитиче-
ской реакции, проводимой в заданных условиях.
562
Часть 3. Моделирование каталитических процессов
Таблица 12.18
Содержание металлов, углерода и серы в катализаторах ГДО, отнесенное
к 100 г свежего катализатора
Свежий, %масс. Отработанный Регенерированный
%масс. Изменение", %масс. %масс. Изменение8, %масс.
А1 42,5 42,5 0 42,5 0
Мо 6,97 7,04 +0,07 6,83 -0,21
Ni 2,19 1,52" -0,67 2,11" +0,59
V 0 18,99 + 18,99 18,61 -0,38
С 0 15,54 + 15,54 0,95 -14,59
S 0 12,67 + 12,67 1,89 -10,78
Знак «минус» означает уменьшение, знак «плюс» — увеличение.
" Относительно свежего катализатора.
6 Относительно отработанного катализатора.
’ С вычетом начального содержания никеля.
Таблица 12.19
Показатели деактивации катализатора ГДО
Показатели Отработанный катализатор
относительно отработанного относительно свежего
Ni через 155 сут, г 0,91 1,52
V через 155 сут, г 11,39 18,99
Ni + V через 155 сут, г 12,3 20,51
Скорость отложения металлов, г/сут 0,0794 0,1323
Остаточный срок службы, мес. 13,9 8,3
12.5. ПРИМЕНЕНИЕ ФУНКЦИЙ РАСПРЕДЕЛЕНИЯ
ВЕРОЯТНОСТЕЙ ДЛЯ ПРИБЛИЖЕНИЯ КРИВЫХ
КИПЕНИЯ НЕФТИ
Методы характеризации нефти — от хорошо известных и широко применяемых ком-
плексных методов анализа до сложных исследований с использованием современных
лабораторных средств (ЯМР-спектроскопии, масс-спектроскопии, рентгенострук-
турного анализа и т. д.) — предмет целого ряда работ. После 1930-х гг. реализация
более точных методов анализа нефти рассматривалась в нескольких исследованиях
[Watson and Nelson, 1933]. Хотя изощренные современные подходы к характеризации
нефти улучшают понимание ее структуры, продолжают широко применяться тради-
ционные методы. Остаются весьма популярными и эмпирические зависимости для
Глава 12. Зависимости и прочие вопросы
563
оценки массовых свойств нефтяных фракций, основанные главным образом на кри-
вых кипения и плотности. Эти зависимости весьма полезны при расчете процес-
сов, особенно при недостатке имеющихся экспериментальных данных [American Pe-
troleum Institute, 1976]. В случае кривых кипения иногда в распоряжении есть лишь
ограниченное число точек на кривой разгонки, так что для определения требуемого
значения приходится прибегать к интер- и экстраполяции. Существует ряд методов
ASTM для получения данных разгонки: D5307, D2892, D1160, 2)86 и т. д. Все они пред-
полагают применение стандартизованного оборудования; результаты указываются
в виде значений температур, при которых объемная или массовая доля отогнанной
части достигает определенных значений. Кривые кипения строятся по конечным на-
борам пар значений доли отгона и температуры; их можно приблизить различными
функциями, чтобы получить непрерывное представление. Для нахождения проме-
жуточных точек на кривых кипения применяют методы параболической регрессии,
приближения кубическими сплайнами и интерполяции многочленами Лагранжа.
Другой подход, дающий более точные поправки, основан на подгонке функций рас-
пределения вероятностей к данным разгонки методом наименьших квадратов.
Функции распределения вероятностей находят широкое применение. Так, их
используют:
• в программах моделирования технологических процессов, где они применя-
ются совместно с методами численного интегрирования для нахождения опти-
мального числа псевдокомпонентов, которыми, например, представляются не-
фтяные фракции [Whitson, 1983; Whitson et al., 1988];
• расчетах фазового равновесия, когда применяются методы непрерывного тер-
модинамического представления [Willman and Teja, 1986; Kehlen and Ratzsch,
1987; Peng et al., 1987];
• для описания степени химических превращений, происходящих в процессах
нефтепереработки; в этих целях вероятностные функции стали применяться
сравнительно недавно [Bacaud et al., 1986; Krishna and Saxena, 1989].
Из других примеров применения функций распределения можно упомянуть
описание продуктов реакций полимеризации, представляющих собой смеси сое-
динений различной молярной массы; представление распределения размеров ча-
стиц пыли и аэрозолей в экологических исследованиях; описание суточного рас-
пределения осадков, уровня рек и озер и других метеорологических явлений
[Kumaraswamy, 1980].
Эмпирические зависимости, входными величинами в которых чаще всего являют-
ся данные разгонки и плотности, применяются для характеризации нефтяных фрак-
ций (определения молярной массы, критических свойств и т. д.). Эти зависимости
можно также использовать для выделения псевдокомпонентов или агрегатов (бензи-
новой фракции, средних дистиллятов и т. д.) в продуктах реакций некоторых типич-
ных процессов нефтепереработки — гидрокрекинга, каталитического крекинга и др.
[Ancheyta et al., 2005b]. Чтобы иметь точное и достоверное представление данных раз-
гонки непрерывными функциями, необходимое для интерполяции, нужен строгий
анализ других аспектов, помимо традиционных методов интерполирования.
564
Часть 3. Моделирование каталитических процессов
12.5.1. Функции распределения вероятности
Функции распределения вероятности помогают описать вероятность наступления
определенного события. Они могут быть дискретными, когда число таких собы-
тий дискретно (счетно), или непрерывными, если множество событий непрерывно.
В данной работе рассматриваются только непрерывные функции. Функции распре-
деления вероятности определены для небольшого числа параметров, обладают чис-
ловыми характеристиками (такими как математическое ожидание, медиана и дис-
персия) и принимают две основные формы: 1) функции плотности распределения
вероятности (дифференциальная форма) и 2) кумулятивной функции распределения
(интегральная форма). Чаще всего применяется первая форма; хорошо известным
пример такой функции — распределение Гаусса, иначе называемое нормальным рас-
пределением [Evans et al., 1993]. Интегральные функции характеризуются монотон-
ным возрастанием. В качестве их аналога можно указать кривые разгонки. Функции
распределения вероятности благодаря их простоте можно без труда встроить в ком-
пьютерные программы моделирования, оптимизации и управления. Выбор функ-
ции, адекватно представляющей экспериментальные данные, определяется ее харак-
теристиками функций и не должен задаваться заранее.
В расчетах в области нефтепереработки используется ряд функций распределения.
Уитсон и соавторы [Whitson, 1983; Whitson et al., 1988] предложили описывать угле-
водороды С7+ трехпараметрической гамма-функцией. Дулезиа [Dhulesia, 1984] вы-
двинул идею, согласно которой кривые разгонки нефтяных фракций описывались
функцией распределения Вейбулла в интегральной форме. Функция Вейбулла была
опробована на данных разгонки сырья и продуктов каталитического крекинга. При-
ближение кривых разгонки показало хорошее согласие с экспериментальными дан-
ными. В расчетах фазового равновесия при непрерывном представлении термодина-
мики использовалась функция нормального распределения [Kehlen et al., 1987]. При
моделировании поведения реакций гидрокрекинга использовались гауссовы функ-
ции распределения вероятностей и распределения ошибок [Bacaud et al., 1996; Krish-
na et al., 1989]. Уиллман и Тейа [Willman and Teja, 1986] использовали двухмерное ло-
гнормальное распределение ддя описания состава смесей, участвующих в расчетах
фазового равновесия. В расчетах по уравнениям состояния распределение нефтяных
фракций описывалось бета-функцией [Peng et al., 1987]. Функции/2 и показательные
функции распределения, представляющие собой упрощенные варианты гамма-функ-
ции, применялись для описания тяжелых фракций и вывода формул по расчету фазо-
вого равновесия пластовых жидкостей [Behrens and Sandler, 1988; Luks etal., 1990]. Ри-
ази [Riazi, 1989] использовал видоизмененную функцию Вейбулла, чтобы обосновать
метод предсказания полного распределения молярной массы, температур кипения,
плотности и показателя преломления фракций С7+. Семейство функций распреде-
ления экстремальных значений находит применение в исследованиях таких природ-
ных явлений, как атмосферные осадки, наводнения, загрязнение воздушной среды
и коррозия. Функции Гумбеля, Фреше и Вейбулла можно отнести к одному и тому
же семейству обобщенных функций распределения экстремальных значений [Kotz
and Nadarajah, 2000]. Распределение Кумарасвами благодаря своей универсальности
Глава 12. Зависимости и прочие вопросы
565
и двусторонней ограниченности сравнимо с бета-функцией, но в обеих формах (диф-
ференциальной и интегральной) имеет более простой вид [Kumaraswamy, 1980]. Рас-
пределение Кумарасвами, хотя оно изначально предназначалось для моделирования
гидравлики, применяется в строительстве для описания физических величин с нало-
женными на них ограничениями.
В этом разделе из всего множества описанных в литературе функций распределе-
ния для анализа отобрано 25; они перечислены в табл. 12.20. Из рассмотрения выпа-
дает несколько хорошо известных функций, в том числе лямбда-распределение Тью-
ки, распределение Коши и распределение Фишера [Heckert and Filliben, 2003], так
как либо они редко применяются, либо у них отсутствует интегральная форма в ана-
литическом виде. Хотя большинство функций, перечисленных в табл. 12.20, не на-
ходят широкого применения в технических приложениях, все они могут оказаться
полезными для описания данных окружающего мира. В табл. 12.21 приведены опре-
деления функций распределения вероятностей в обеих (дифференциальной и инте-
гральной) формах. По виду функции могут несколько отличаться от тех, что приво-
дятся в литературных источниках, а также существовать под другими названиями.
Таблица 12.20
Функции распределения вероятности
Номер функ- ции Название функции Число параме- тров Формы представления
Дифферен- циальная Инте- гральная
1 Альфа-распределение (нормализованное) 2 ф ф
2 Альфа-распределение 4 ф ф
3 Бета-распределение 4 г 1
4 Распределение Бредфорда 3 — —
5 Распределение Бурра 4 — —
6 Распределение «хи-квадрат» 3 г г
7 Распределение усталостной долговечности 3 — ф
8 Распределение Фиска (лог-логистическое) 3 — —
9 Распределение Фреше 3 — —
10 Свернутое нормальное распределение 2 — ф
11 Гамма-распределение 3 г г
12 Обобщенное распределение экстремальных значений 3 — —
13 Обобщенное логистическое распределение 3 — —
14 Распределение Гумбеля 2 — —
15 55-распределение Джонсона 4 — ф
16 Распределение Кумарасвами 4 — —
17 Полунормальное распределение 2 — —
566
Часть 3. Моделирование каталитических процессов
Таблица 12.20, окончание
Номер функ- ции Название функции Число параме- тров Формы представления
Дифферен- циальная Интеграль- ная
18 Логнормальное распределение 2 — ф
19 Распределение Накагами 3 г г
20 Нормальное распределение 2 — ф
21 Распределение Риази 3 — —
22 /-Распределение Стьюдента 3 г I
23 Распределение Вальда 2 — ф
24 Распределение Вейбулла 3 — —
25 Распределение экстремальных значений Вейбулла 4 — —
Примечание. Ф — интегральная функция нормального распределения; Г — гамма-функция;
I — неполная бета-функция.
12.5.2. Методика
12.5.2.1. Данные
Точность приближения выбранными функциями проверялась на данных из двух
источников. Первый — это работа [Sanchez et al., 2007] и приведенные там ссыл-
ки; второй — данные, полученные в лабораториях Мексиканского института нефти
и университета Альберты. В число отобранных входили: пробы сырой нефти; вакуум-
ного газойля; остатков атмосферной и вакуумной перегонки; атмосферного газой-
ля; легкого рециклового газойля; гидроочищенного легкого рециклового газойля;
сырья каталитического крекинга; сырья и продуктов термических процессов, про-
водимых в умеренных условиях; гидроочистки вакуумного остатка; гидроочистки
газойлей, полученных из битумной нефти; гидроочистки средних дистиллятов. Дан-
ные отгонки были получены в основном на пробах нефти из Кувейта, Саудовской
Аравии, Мексики и Канады. Было проанализировано в общей сложности 137 кри-
вых разгонки, каждая из которых имела по меньшей мере шесть эксперименталь-
ных точек; всего было 1627 пар значений температуры и процента отгона. Стандарт-
ными методами (методы ASTM 7)2892 и ASTM DI 160 физической разгонки, методы
ASTM £>5307 и ASTM 7)6352 имитированной и высокотемпературной имитированной
разгонки) были получены экспериментальные данные, которые затем собрали в еди-
ную базу данных. Данные разгонки не приводились к единой базе, а обрабатывались
в том виде, в каком их давал метод (ASTM 7)2892, ASTM D\ 160, ASTM 7)5307 и т. д.).
Взятые из литературных источников данные температуры и выхода, приведенные
в различных единицах, пересчитывались соответственно в градусы Цельсия и про-
центы по массе.
Глава 12. Зависимости и прочие вопросы
567
12.5.2.2. Иллюстрация расчета параметров на примере
Статистическими методами было проведено сравнение точности приближения
функциями, перечисленными в табл. 12.20, 12.21. Описанный ниже порядок расчета
параметров иллюстрируется четырехпараметрическим бета-распределением на при-
мере выборки данных, полученных имитированной разгонкой гидрокрекированной
нефти «Майя».
1. Данные температуры приводились к безразмерному виду:
Т-Т
Q = (12.145)
-Г “
где 0. — безразмерная температура; Т. — истинная температура кипения; То и Г, —
опорные значения температуры, выбираемые так, чтобы значения 0. находились
между 0 и 1 (в данном примере То = 30 °C и Т[ = 1000 °C).
На рис. 12.12 показаны кривая безразмерных температур кипения и исходные дан-
ные разгонки выбранной пробы. Кривая безразмерных температур начинается пра-
вее нуля и заканчивается левее единицы, так как интервал, охватываемый опорными
значениями То и Т\, шире интервала кипения пробы.
Температура кипения, °C
Рис. 12.12. Данные разгонки гидрокрекированной нефти «Майя», полученные:
(о) — экспериментальным путем; (-) — приближением бета-функцией
2. Вычислялся оптимальный набор параметров функции распределения. Оп-
тимизация проводилась по критерию минимума суммы квадратов остатков (Л55),
определяемому формулой (12.146):
(12.146)
где у ИУ«1Н ~ соответственно экспериментальные и расчетные значения массовых
долей. Оптимальные значения параметров бета-функции, найденные по показанным
Таблица 12.21
Определения непрерывных функций распределения вероятности
Номер функции Распределение Функция распределения плотности вероятности Кумулятивная функция распределения
1 Альфа (нормализ.) (С,Р) D Г 1С О? г=ехР С (2Ф(с)у/2п 2 V у) Ф(С-(Р/у)) Ф(С)
2 Альфа (А, В, С, D) D Г Иг- dY В /2Ф(С)л/2л L 2 к f ) Ф(С-(Р//)) Ф(С)
3 Бета (А, В, С, D) Г(С+Р) (у-я)с-‘ Г(С)Г(Р)(В-Л)с^-' (В-y)'-D / .В-А )
4 Бредфорда (А, В, С) С [С(у-Л) + В-Л]1п(С + 1) 1п[1 + (С(у-Л)/(В-/1))] 1п(С + 1)
5 Бурра (А, В, С, D) —rc-'(i+rc)-D-' в (1 + /'с)"°
6 Хи-квадрат (А, В, С) /с~‘ехр(-(1 / 2)Г) Г -И 21 В Г(С/2) — 1 сч и I СЧ 1
7 Усталостной долго- вечности (А, В, С) у1-а2 Г /7 (у/л)+(А/у)-2\1 2^С2Ву2[УА\ у Ч 2С2 JJ ф \w<s-G4/X>°-51 С J
8 Фиска (А, В, С) с tc-' 5[1 + Л]2 1 1+/‘с
568 Часть 3. Моделирование каталитических процессов
9 Фреше (А, В, С) ехр(-Гс) ехр(-Сс)
10 Свернутое нормальное (А, В) 1 к —J— cosh В Мл ^Ау} С 1 у2+Я2''| — ехр -7— <B2J Д 2 В2 J (—у — А Ф(0-ф| -
11 Гамма (А, В, С) —!—Iе'' ехр(-Г) ВГ(С) Г(С,/)
12 Обобщенное распре- деление экстремаль- ных значений (А, В, С) 1 -1-i Г — —(1 + 0) сехр-(1 + О)с В ех] -(1 + СГр
13 Обобщенное логисти- ческое (А, В, С) С ехр(-/) 1
В [1 + ехр(—г1)]”7*' [1+ехр(-г)]г
14 Гумбеля (А, В) -^ехр(-г)ехр[-ехр(-О] В exp[-exp(-z)]
15 55-распределение Джонсона (А, В, С, D) D(B-A) f [C+Dln((y-А)/(В-у))]2' Г- ехр-1 (у-Л)(В - у)-у2л [ 2 J Ф C + Dln[~--"1
16 Кумарасвами (А, В, С, D) / ЧС-1Г / \С~] CD ZzA 1-И^- ^B-yJ ^В-у) 1- *4—Т D
17 Полунормальное (А, В) to | - гГТгч] й тз 1 2Ф(г)-1
18 Логнормальное (А, В) 1 ByV^exp 1<1п(у)-ЛУ 2< В ) Ф( 1п(у)-ЛМ В )
19 Накагами (А, В, С) ?Сс ес-' <жр(-аг) ВГ(С) г(с,а2)
Глава 12. Зависимости и прочие вопросы 569
Таблица 12.21, окончание
Номер функции Распределение Функция распределения плотности вероятности Кумулятивная функция распределения
20 Нормальное (А, В) 1 /=ехр Ву/2й ' 1 —т < 2 ) Ф(Г)
21 Риази (А, В) В /1-1 ( В в ~У ехр -—у А { А 1 oq| 1 Си X и 1
22 Г- Распределение Стьюдента (А, В, С) 1 Г((С + 1)/2) Г + 7 Т“ Ву/^с Г(С/2) [+cj 7 “17 1-J | — /• X + о '"'‘•j ю | К> I 1?) 1 \ у 1Л О V о
23 Вальда (А, В) wexp В (у~а\ 2 А А ) 4Д— Vv л J (2В\( [В-у-А"} Ч А ) [у у A J
24 Вейбулла (А, В, С) jfc~‘ схр(-Л) 1-ехр(-гс)
25 Распределение экс- тремальных значений Вейбулла (А, В, С, D) CDtC । г г г в ехр( t )[1 ехр( t )] [1 - ехр(-1 )]
570 Часть 3. Моделирование каталитических процессов
Глава 12. Зависимости и прочие вопросы
571
на рис. 12.12 данным, были следующими: А = 0,08962; В = 1,05013; С = 2,49003;
D = 6,34186. Чтобы гарантировать правильность расчетных значений и глобальной
сходимости критерия к минимуму, проводился анализ чувствительности, процедура
которого описана в другом источнике [Alcazar and Ancheyta, 2007].
3. При оптимизированных значениях параметров функции распределения вы-
числялись значения выхода жидкости (см. рис. 12.12).
4. По экспериментальным и полученным расчетным значениям проводились
статистический анализ и анализ остатков, чтобы выявить возможные ошибки мо-
дели. В описываемом примере значения максимальной абсолютной ошибки, сред-
ней абсолютной ошибки, суммы квадратов остатков (RSS) и стандартного отклоне-
ния (SD) оказались равными соответственно 1,19, 1,36, 5,34 и 0,56.
5. В большинстве случаев наибольшие отклонения имели место в экстремаль-
ных точках кривых кипения. Это может объясняться низкой точностью измерений
в указанных частях кривой. Поэтому в практических целях целесообразно исключить
из рассмотрения начальные и конечные точки кривых или даже точки 5 и 95 % отгона.
12.5.2.3. Расчет параметров всех функций распределения
В описанном выше порядке были вычислены параметры всех функций распределе-
ния из табл. 12.21 для каждой из 137 выборок данных разгонки.
На рис. 12.13 сравниваются экспериментальные данные и расчетные значения,
полученные путем приближения бета-функцией. По рисунку можно визуально оце-
нить точность приближения. Статистический анализ диаграммы, на которой пред-
ставлено рассеяние экспериментальных значений выхода жидкости относительно
расчетных, позволил получить значения коэффициента корреляции R2, коэффи-
циента угла наклона, отсекаемого отрезка и стандартного отклонения. В табл. 12.22
представлена сводка статистических параметров, полученных регрессионным анали-
зом диаграммы рассеяния. Таблица позволяет получить количественное представле-
ние о точности приближения распределения бета-функцией.
Экспериментальное значение выхода жидкости, %масс.
Рис. 12.13. Сравнение экспериментальных данных и расчетных значений, полученных
путем приближения экспериментальных кривых бета-функцией
572
Часть 3. Моделирование каталитических процессов
Таблица 12.22
Статистические параметры, найденные регрессионным анализом приближения бета-функцией
Параметры Значения
Индикаторы регрессии
Коэффициент корреляции (Л2) 0,999
Стандартное отклонение. %масс. 0,814
Число наблюдений 1474
Коэффициенты Нижние (95 %) Верхние (95 %)
Коэффициент наклона 1,002 1,0033 1,0002
Отсекаемый отрезок -0,0655 0,0062 -0,1372
Дисперсионный анализ Df Сумма квадратов Средний квадрат Критерий F
Регрессия 1 1,0961-Ю6 1,0961 Ю6 1,6560-10s
Число остатков 1473 973,66 0,6619 —
Всего 1474 1,0971 -106 — —
Функции распределения ранжировались по точности приближения в соответ-
ствии со статистическими индикаторами. Вначале применялась методика, ос-
нованная на регрессионном анализе. В ней главным критерием ранжирования
функций являлось стандартное отклонение SD, вычисляемое по формуле (12.147);
учитывались также коэффициент корреляции R-, коэффициент наклона и отсека-
емый отрезок:
\RSS
SD = J---(12.147)
V п-2
Второй подход учитывает информационные критерии Акаике и Байеса. Инфор-
мационный критерий Акаике (AIC) — критерий выбора моделей, учитывающий как
сложность модели, так и качество приближения [Bumham and Anderson, 1998]. На-
значение критерия — выбор модели, наилучшим образом объясняющей данные при
минимальном числе свободных параметров. При случайном распределении остатков
АТС вычисляется по формуле
, (RSS^
А1С=2к+п\п\------ , (12.148)
\ п )
где к — число параметров; п — число наблюдений; RSS — сумма квадратов остатков.
В формулу AIC входит штрафной член 2к, представляющий собой возрастающую
функцию числа параметров. Эта особенность делает критерий весьма полезным при
сравнении моделей с разным числом параметров. В данном исследовании в качестве
предпочтительной выбиралась функция распределения с наименьшим значением
критерия АТС.
Глава 12. Зависимости и прочие вопросы
573
Выражение для байесовского информационного критерия для моделей со случай-
но распределенными остатками имеет вид
В7С = А:1п(п)+«1п| — (12.149)
(, и )
Байесовский критерий (BIC) по сравнению с AIC строже штрафует за число сво-
бодных параметров. Как и в случае AIC, к предпочтительным относили модели с наи-
меньшим значением В1С. Так как Л/С сильно зависит от размера выборки, рекомен-
дуется пользоваться относительными значениями и выбирать модель по разности
значений А., определяемой уравнением (12.150). Модели, для которых Д. > 10, мож-
но считать не объясняющими существенные отклонения в данных и исключать их
из дальнейшего рассмотрения:
Ь=А1С-А1С . (12.150)
Бернхем и Андерсон [Burnham and Anderson, 1998] предложили использовать для
выбора модели взвешенный критерий Акаике (о>.), учитывающий достоверность
того, что в данной ситуации i-я модель является наилучшей среди остальных. Моде-
ли сравниваются по отношению достоверностей (со,/со), где индекс 1 соответствует
проверяемой модели, a j пробегает множество остальных моделей. Взвешенный кри-
терий AIC и отношение достоверностей вычисляются по формулам
со. = RexP((~1/2)A-) ; (12.151)
£ехр((-1/2)Д,.)
Г=1
( 1
— = ехр ---- . (12.152)
со,. ^2Aj
12.5.3. Результаты
12.5.3.1. Ранжирование функций
Для выбора функции распределения, наилучшим образом описывающей кривые
кипения, вначале осуществлялось приближение экспериментальных данных раз-
личными функциями. Наибольшие ошибки в приближении получались в точках,
близких к концам кривых кипения, следующие по величине — в точках вблизи на-
чала кривых. Эта проблема особенно выражена в случае симметричных распре-
делений — нормального распределения и ^-распределения Стьюдента. Трудность
приближения точек начала и конца кипения функциями распределения усугубля-
ется большими погрешностями измерения на концах кривых разгонки. Эти по-
грешности связаны со снижением чувствительности экспериментальной аппарату-
ры в начале и конце испытаний и наблюдаются независимо от того, как аппаратура
574
Часть 3. Моделирование каталитических процессов
приводится в действие — вручную или автоматически. Погрешность эксперимента
является переменной и зависит от применяемого стандартного метода. Например,
метод ASTM 2)2892 допускает погрешности определения доли отгона до 1,2 % масс.;
в методе ASTMD1160 погрешности (в зависимости от местоположения точки на кри-
вой разгонки) могут составлять от 1,7 до 5,7 %масс.
Выбор наилучшей функции распределения — непростая задача. Выбирать
можно по различным статистическим критериям: по стандартным отклонени-
ям, коэффициентам корреляции, информационным критериям Акаике и Байеса
и даже по занимаемому процессорному времени. Все эти критерии представле-
ны в табл. 12.23. Общепризнано, что коэффициенты корреляции не очень под-
ходят для сравнения моделей. В данном исследовании значения коэффициен-
тов корреляции для всех функций распределения были весьма близки к единице
(от 0,986 до 0,999). Точнее говоря, значение 7?2 меньше 0,99 имело лишь альфа-
распределение.
Таблица 12.23
Ранжирование функций распределения по различным критериям сравнения
Номер фун- КНИИ Я2 Наклон Отсекае- мый отре- зок, %масс. SD, %масс. Ранг по SD Средний AIC Средний BIC Ранг по AIC Процес- сорное время11
1 0,986 1,023 -1,47 3,374 25 31,79 32,57 25 2,267
2 0,998 1,001 0,08 1,276 10 3,62 5,19 12 2,143
3 0,999 1,002 -0,01 0,814 2 -14,77 -13,20 2 3,705
4 0,996 1,007 -0,37 1,778 13 10,66 11,84 17 0,219
5 0,995 1,026 -0,60 1,992 21 -0,41 1,15 9 0,124
6 0,998 1,001 0,06 1,182 8 -4,04 -2,86 6 1,924
7 0,995 1,014 -0,71 1,911 19 19,02 20,19 22 0,724
8 0,998 0,998 0,22 1,264 9 4,76 5,93 13 0,114
9 0,996 1,002 -0,17 1,801 15 7,96 9,14 16 0,143
10 0,995 0,987 0,90 1,876 18 15,34 16,12 18 1,257
И 0,998 1,003 -0,09 1,072 5 -5,87 -4,69 5 3,400
12 0,996 1,002 -0,17 1,797 14 7,67 8,84 14 0,124
13 0,998 0,996 0,37 1,132 7 2,54 3,72 11 0,114
14 0,995 1,000 -0,07 1,84 16 7,78 8,56 15 0,086
15 0,993 1,016 -0,92 2,397 24 21,65 22,43 24 0,657
16 0,998 1,001 0,02 1,126 6 -3,90 -2,34 7 0,771
17 0,999 1,004 -0,05 0,885 4 -13,19 -11,62 3 0,219
Глава 12. Зависимости и прочие вопросы
575
Таблица 12.23, окончание
Номер фун- кции R2 Наклон Отсекае- мый отре- зок, %масс. SD, %масс. Раш- по SD Средний AIC Средний BIC Ранг по AIC Процес- сорное время"
18 0,996 1,012 -0,61 1,849 17 15,55 16,34 19 0,838
19 0,997 1,000 0,22 1,393 11 -0,49 0,69 8 2,086
20 0,994 0,986 0,91 2,093 23 18,34 19,12 21 1,000
21 0,996 0,997 0,45 1,673 12 2,03 3,21 10 0,124
22 0,994 0,984 1,00 2,064 22 19,66 20,83 23 4,381
23 0,995 1,015 -0,79 1,987 20 18,20 18,98 20 1,305
24 0,999 1,002 0,01 0,865 3 -10,44 -9,26 4 0,124
25 1 1,003 -0,06 0,599 1 -16,61 -15,04 1 0,248
“ Относительно времени, требуемого для нормального распределения.
Диаграмма рассеяния на примере бета-распределения показана на рис. 12.13; ко-
эффициенты наклона линий и отсекаемые отрезки на соответствующих диаграммах
для всех моделей указаны в таблице 12.23. Во всех случаях коэффициенты наклона
линии на диаграммах рассеяния практически равны единице (0,984-1,026), а отсе-
каемые отрезки лежат в интервале от 1,47 до 1,00. Отсеивать неподходящие модели
целесообразнее по неслучайному распределению остатков. Тренды в распределении
значений остатков характерны практически для всех двухпараметрических моделей.
Из последних по этому критерию исключаются нормализованное альфа-распределе-
ние, распределение Фреше, свернутое нормальное, полунормальное, логнормальное,
нормальное распределения, Г-распределение Стьюдента и распределение Валь-
да, из трехпараметрических моделей — распределение усталостной долговечности
и обобщенное распределение экстремальных значений. Любопытно, что из двухпа-
раметрических моделей распределение остатков носило случайный характер лишь
у распределения Гумбеля. Четырехпараметрические модели, как и следовало ожи-
дать, приемлемым образом описывали экспериментальные данные. Для сравнения
на рис. 12.14 показаны диаграммы остатков для наихудшей и наилучшей моделей —
двухпараметрического альфа-распределения и четырехпараметрического распреде-
ления экстремальных значений Вейбулла. Очень хорошо видны различия в оценках
и тренд: если во втором случае остатки имеют значения от —5 до +5 и распределены
случайно, то в первом они колеблются в интервале от -15 до +5 и проявляют ярко
выраженный тренд.
Модели можно ранжировать по стандартному отклонению. Поскольку его значе-
ния для рассматриваемых функций рассеяны в интервале от 0,59 до 3,37 %масс., этот
критерий подходит лучше, чем значения R2 или коэффициенты наклона и отсекае-
мые отрезки на диаграммах рассеяния.
576
Часть 3. Моделирование каталитических процессов
Рис. 12.14. Диаграммы остатков: а — для нормализованного альфа-распределения;
б — для распределения экстремальных значений Вейбулла
Исходя из приведенных в табл. 12.23 значений коэффициента корреляции, коэф-
фициента наклона, отсекаемого отрезка и стандартного отклонения, функции рас-
пределения были классифицированы по точности приближения следующим образом.
• Группа 1:
SD< 1,0;
0,999 < R2 <1.
Число функций: 4 (3, 17, 24 и 25).
• Группа 2:
1,0 < SD < 1,992;
0,995 < R2 < 0,998.
Число функций: 11 (2, 4, 5, 6, 8, 9, И, 13, 14, 16, 19 и 21).
• Группа 3: исключены ввиду тренда в распределении остатков.
Число функций: 10 (1, 7, 10, 12, 15, 18, 20, 22 и 23).
Глава 12. Зависимости и прочие вопросы
577
Наиболее точны функции группы 1, а функции группы 3 не описывают надлежа-
щим образом функциональную связь данных разгонки.
При выборе модели необходимо учитывать не только точность приближения,
но и достоверность расчетных параметров. Хорошо известно, что увеличение чис-
ла свободных параметров может улучшить точность приближения, но снизить до-
стоверность оценок параметров модели. Поэтому сравнение моделей с разным
числом параметров, построенных на выборках разных размеров, исключитель-
но по критерию стандартного отклонения может дать неприемлемые результаты.
В данной работе при выборе функций распределения, наилучшим образом опи-
сывающих данные разгонки, различия в числе параметров учитывались критери-
ями AIC и BIC.
Для каждой выборки данных и для каждой функции был вычислен критерий
AIC, а затем функции были повторно ранжированы по его среднему значению. Ана-
логичная процедура была выполнена для критерия В1С. Значения AIC и BIC пред-
ставлены в табл. 12.23. Хотя критерий Байеса штрафует функции с числом параме-
тров более трех, результаты основанного на нем ранжирования очень схожи с теми,
что дает ранжирование по критерию АЮ, единственное исключение состоит в том,
что ранги распределения Гумбеля и обобщенного распределения экстремальных
значений меняются местами. Следует заметить, что четыре первых места при ран-
жировании по критерию AIC занимают те же функции, что и при ранжировании
по критерию стандартного отклонения (распределение экстремальных значений
Вейбулла, бета-распределение, распределение Кумарасвами и распределение Вей-
булла), т. е. функции группы 1. Единственное различие — порядок расположения
функций внутри группы.
Остается вопрос статистической значимости различий в способности функций
адекватно описывать данные разгонки. Показано, что при проверке статистической
значимости на предмет того, насколько выбранная модель описывает эксперимен-
тальные данные лучше других с более низким рангом, не стоит полагаться на крите-
рии А1С и BIC [Burnham and Anderson, 1998]. Модель лучше всего выбирать по отно-
шению достоверностей и распределению остатков. В табл. 12.24 приведены значения
AIC и отношения достоверностей для 10 функций наивысшего ранга по критерию
AIC. Можно видеть, что различия между четырьмя лучшими функциями (значения А
от 1,84 до 6,17) не настолько велики, чтобы четко свидетельствовать о существовании
среди них однозначно лучшей модели. Так как значения А. для этих моделей доста-
точно схожи, при переходе к другой выборке данных лучшей может оказаться любая
другая из четырех функций. В случае данных разгонки априорный выбор из четы-
рех моделей (распределение экстремальных значений Вейбулла, бета-распределение,
распределение Кумарасвами и распределение Вейбулла) не рекомендуется; вместо
этого следует оценить все функции. Анализ данных о значении отношения достовер-
ностей позволяет заключить, что большое его значение для распределения Вейбулла
(w./w, = 21,9) делает крайне маловероятным вероятность того, что эта модель могла
бы быть наилучшей.
578
Часть 3. Моделирование каталитических процессов
Таблица 12.24
Значения Д. и отношения достоверностей для 10 наилучших функций
Ранг по кри- терию AIC Функция (число параметров) Номер функции Среднее значение АТС Д. 1 СО. со./со.
1 Распределение экстремальных значений Вейбулла (4) 25 -16,61 0 0,61213 1,0
2 Бета-распределение (4) 3 -14,77 1,84 0,24387 2,5
3 Распределение Кумарасвами (4) 17 -13,19 3,42 0,11049 5,5
4 Распределение Вейбулла (3) 24 -10,44 6,17 0,02794 21,9
5 Гамма-распределение (3) 11 -5,87 10,75 0,00284 215,5
6 Распределение «хи-квадрат» (3) 6 -4,04 12,58 0,00114 538,1
7 55-распределение Джонсона (4) 16 -3,90 12,71 0,00106 575,2
8 Распределение Накагами (3) 19 -0,49 16,12 0,00019 3,171,9
9 Распределение Бурра (4) 5 -0,41 46,20 0,00019 3,289,8
10 Распределение Риази (3) 21 2,03 18,64 0,00005 11,178,2
Выборки данных в этом исследовании содержали от 6 до 19 эксперименталь-
ных точек. Чтобы проверить, не влияет ли размер выборок на выбор модели, для
каждой функции была построена диаграмма зависимости среднего значения крите-
рия АТС от размера выборки. На рис. 12.15 показаны результаты для распределения
экстремальных значений Вейбулла (ранг 1) и гамма-распределения (ранг 5). Видны
две четко выраженные группы, причем каждая группа для одной функции совпада-
ет с аналогичной группой для другой (размеры выборок в группах от 6 до 10 и от 11
до 19). Для каждой функции были вычислены средние значения АТС в каждой из та-
ких групп, после чего функции были повторно упорядочены по этому критерию.
Ранги функций изменились незначительно. Немаловажно, что состав группы четы-
рех лучших функций не изменился.
Для каждой функции было вычислено процессорное время, затраченное на оп-
тимизацию ее параметров. Результаты представлены в табл. 12.23; за единицу при-
нято время оптимизации параметров нормального распределения. Если смотреть
на функции группы 1 (ранг от 1 до 4), можно заметить, что оптимизация параме-
тров распределений экстремальных значений Вейбулла, Кумарасвами и Вейбул-
ла занимает сравнимые отрезки времени (соответственно 0,248, 0,124 и 0,219);
тогда для бета-распределения (ранг 2) требуется в десять раз больше време-
ни (3,705). Это может объясняться простотой первых трех распределений: они,
в отличие бета-распределения, не содержат каких-либо специальных функций
(см. табл. 12.20).
Глава 12. Зависимости и прочие вопросы
579
Размер выборки
Рис. 12.15. Диаграмма зависимости среднего значения критерия AIC от размера выборки:
белые столбики представляют гамма-распределение, черные — распределение экстре-
мальных значений Вейбулла
Основываясь на числе параметров (2, 3 или 4) функций распределения, можно
сделать ряд наблюдений, представленных ниже.
• Наилучшее приближение дают четырехпараметрические функции. Пять из них
входят в первую десятку, а распределение экстремальных значений Вейбулла,
бета-распределение и распределение Кумарасвами — в первую четверку.
• Некоторые трехпараметрические функции способны достаточно точно при-
ближать данные разгонки: распределение Вейбулла и гамма-распределение
имеют стандартные отклонения, равные соответственно 0,86 и 1,07 %масс.,
и входят в первую пятерку.
• Двухпараметрические функции недостаточно хорошо приближают данные.
Все они, кроме одной, входят в группу 3.
• Следует отметить, что гамма-распределение и нормальное распределение, ко-
торыми чаще всего приближают данные разгонки, занимают соответственно
5-е и 20-е места.
12.5.3.2. Проверка наилучших функций
Точность приближения четырьмя лучшими и одной худшей функциями (распреде-
лением экстремальных значений Вейбулла, распределением Кумарасвами, распре-
делением Вейбулла, бета-распределением и нормализованным альфа-распределе-
нием) проверялась на других наборах данных. Для этого было отобрано тридцать
проб сырой нефти трех сортов и их фракций с различными интервалами кипения,
по которым было получено в общей сложности 346 точек данных. Они охватыва-
ли широкий интервал температур (от 20 до 540 °C). Результаты проверки представ-
лены в табл. 12.25. На рис. 12.16 показаны остатки для распределения экстремаль-
ных значений Вейбулла и нормализованного альфа-распределения. Из таблицы
580
Часть 3. Моделирование каталитических процессов.
можно видеть, что стандартные отклонения и остатки для четырех лучших функ-
ций меньше, чем для нормализованного альфа-распределения (величина SD около
2,5 против 4,1), что значения коэффициента корреляции и коэффициента накло-
на диаграммы рассеяния ближе к единице, а отсекаемые отрезки — ближе к нулю.
Кроме того, абсолютная величина разности между положительными остатками для
нормализованного альфа-распределения более чем в два раза выше по сравнению
с четырьмя лучшими функциями; это говорит о том, что оно завышает результаты
по сравнению с экспериментальными. Проверка значений критериев А1С и В1С, Д.
и отношения достоверностей дает для проверочной выборки тот же порядок ран-
жирования функций, что и для испытательной. Результаты проверки подтвержда-
ют гипотезу, согласно которой распределение экстремальных значений Вейбулла,
распределение Кумарасвами и распределение Вейбулла точнее остальных прибли-
жают данные разгонки.
Таблица 12.25
Статистические параметры регрессионного анализа результатов для проверочной выборки
Параметр Распределение эксперименталь- ных значений Вейбулла Распределение Кумарасвами Распределение Вейбулла Бета-распре- деление Нормализован- ное альфа- распределенне
Номер функции по табл. 12.20 25 17 24 3 1
R2 0,994 0,994 0,994 0,993 0,984
SD 2,38 2,43 2,54 2,75 4,10
Коэффициент наклона 1,004 ±0,016 1,009 ±0,016 1,006 ±0,017 1,008 ± 0,009 1,010 ±0,028
Среднее значе- ние AIC 18,11 18,54 19,34 19,92 36,09
Среднее значе- ние BIC 20,18 20,61 20,89 21,99 37,13
0 0,43 1,23 1,81 17,99
Отношение до- стоверностей 1 1,24 1,85 2,47 8048
Число положи- тельных остат- ков 164 167 172 176 195
Число отрица- тельных остат- ков 182 179 174 170 151
Абсолютная разность0 18 12 2 6 44
3 Абсолютное значение разности между положительными и отрицательными остатками.
Глава 12. Зависимости и прочие вопросы
581
Безразмерная температура (0)
Рис. 12.16. Диаграмма распределения остатков для проверочной выборки данных:
(+) — распределение экстремальных значений Вейбулла;
(о) — нормализованное альфа-распределение
Для иллюстрации приближения экспериментальных данных разгонки функция-
ми распределения из литературы [El-Kady, 1979] была взята выборка данных разгонки
продуктов гидрокрекинга вакуумного газойля (интервал разгонки 380-550 °C, моляр-
ная масса 425 г/моль, плотность при 15 °C 0,931 г/мл), полученных в реакторе с непод-
вижным слоем при давлении 10 МПа и LHSV= 0,5 ч_|. Литературные данные о составе
различных продуктов гидрокрекинга приведены в табл. 12.26. Полные кривые кипения
продуктов не были представлены, но их можно воспроизвести по значениям темпера-
тур и соответствующего выхода жидкого продукта, используя приближение функция-
ми распределения. Начало кипения бензиновой фракции определялось по н-пентану,
так что в общей сложности имелось шесть экспериментальных значений температуры.
Повторением описанной выше процедуры были найдены оптимальные параметры для
распределения экстремальных значений Вейбулла, а затем интерполированием этой
функцией по ограниченному набору данных была воспроизведена полная кривая раз-
гонки. Результаты представлены в той же табл. 12.26. Эта процедура и данные были
успешно применены для моделирования кинетики гидрокрекинга, требовавшего пол-
ных кривых кипения [Ancheyta et al., 2005b], На рис. 12.17 сравниваются результаты
воспроизведения кривых кипения функцией распределения экстремальных значений
Вейбулла и распространенным методом интерполирования (кусочная интерполяция
кубическими эрмитовыми сплайнами). Эрмитова интерполяция выбрана для сравне-
ния по той причине, что она сохраняет кусочную монотонность и форму кривых. Вид-
но, что кривые, воспроизведенные кубической интерполяцией, хотя и проходят через
все экспериментальные точки, имеют локальные «бугры». Экспериментальные кривые
разгонки обычно не имеют таких особенностей. С другой стороны, функция распре-
деления экстремальных значений Вейбулла очень хорошо приближает форму кривой
и экспериментальные точки. Следует отметить, что Дулезиа [Dhnlesia, 1984] ранее уже
рекомендовал эту функцию для описания кривых разгонки сырья и продуктов катали-
тического крекинга.
582
Часть 3. Моделирование каталитических процессов
Таблица 12.26
Состав продуктов гидрокрекинга при Р= 10 МПа и LHSV= 0,5 ч_|
Параметр Значение для продукта
1 2 3
Температура реакции, °C 410 430 450
Значения выхода:
газы (С2—С5) 5,90 10,42 17,23
легкая бензиновая фракция (НК—80 °C) 3,51 6,92 19,03
бензин (80—150 °C) 9,07 15,26 36,03
керосин (150—250 °C) 21,23 22,84 11,15
газойль (250—380 °C) 25,28 28,26 13,75
остаток (380—538 °C) 35,01 16,30 2,81
Расчетная температура отгонки жидкого продукта, °C:
НК 36,0 36,0 36,0
5% 86,4 60,9 46,3
10% 125,6 89,7 56,5
30% 228,1 174,4 87,8
50% 321,9 247,6 122,4
70% 401,9 325,0 174,9
90% 479,9 428,2 307,9
95% 505,7 468,9 394,4
КК 538,0 538,0 538,0
Источник: [El-Kady, F.Y., Indian J. Technol., 17, 176, 1979].
Рис. 12.17. Приближение распределением экстремальных значений Вейбулла (—)
и кубическими эрмитовыми сплайнами (--) экспериментальных данных разгонки продуктов
гидрокрекинга, полученных при различных температурах: (•) — 410 °C, (и) — 430 °C,
(а) —450 °C [El-Kady, F.Y., Indian J. Technol., 17, 176, 1979]
Глава 12. Зависимости и прочие вопросы
583
Ниже представлены выводы, следующие из рассмотренных выше результатов.
• В целом функции распределения вероятностей в интегральной форме очень хо-
рошо подходят для приближения данных разгонки.
• Если следовать статистическому анализу применения 25 различных функций
распределения к 1474 экспериментальным точкам, есть только два способа
расположить функции по точности приближения экспериментальных данных:
1) по критериям стандартного отклонения, коэффициента корреляции, анали-
за остатков; 2) по критериям AIC и BIC.
• Эти способы дают схожее ранжирование, хотя критерий стандартного отклоне-
ния может давать ошибки из-за различий в числе параметров.
• Оказалось возможным найти группу из четырех функций распределения, по-
грешность приближения которыми находится в пределах погрешности измере-
ний. При окончательном выборе наиболее подходящих функций учитывался,
кроме того, критерий занимаемого процессорного времени, зависящий от про-
стоты распределения.
• Для приближения данных разгонки рекомендуются распределение экстре-
мальных значений Вейбулла, распределение Кумарасвами и распределение
Вейбулла, о применении которых для этой цели ранее не сообщалось. Необ-
ходимы дальнейшие исследования для выявления связи между особенностями
этих функций и поведением кривых разгонки нефти.
ОБОЗНАЧЕНИЯ
ЛАТИНСКИЕ
А, В, С, D Параметры распределений
AIC Информационный критерий Акаике
BIC Информационный критерий Байеса
Вг Бромное число, граммов Вг на 100 г продукта
ВТ Содержание бензотиофеновой серы, %масс.
СН Остаточная потребность в водороде, молей Н2 на 1 м3 сырья
сн Концентрация водорода в уравнении растворимости, молей Н2 на 1 м3
сырья
Ст Общий расход водорода, молей Н2 на 1 м3 сырья
DBT Содержание дибензотиофеновой серы, %масс.
DiA Содержание диароматических углеводородов, %масс.
GaT Общее количество газообразного сырья, кг
Gas1 Общее количество газообразного продукта, кг
GO Кратность газа к сырью, мл/мл
Н Коэффициент Генри
584
Часть 3. Моделирование каталитических процессов
(Н2)° Общее количество водорода, поступающего в реактор, кг
(Н2)‘ Общее количество водорода, покидающего реактор, кг
Н°О( Общее количество газообразного водорода, подаваемого в реактор, кг
Н° Общее количество водорода в жидком сырье, кг
Н‘,(К Общее количество водорода в газообразном продукте реактора, кг
Общее количество водорода в гидроочищенном жидком продукте, кг
Н„ s Количество водорода, содержащегося в газообразном H2S, кг
НсЛто/ Стехиометрический расход водорода в химических реакциях, н. м3/м3
Нсии Истинный общий расход водорода, ст. куб. фут/баррель
АНрц Разность между входящими и выходящими количествами водорода,
определенная по балансу масс в газовых потоках, кг
НДя Растворимость водорода, ст. куб. фут/баррель
Количество водорода, растворенного в жидком продукте, кг
Н Чистота водорода, %масс.
НГато Экспериментально измеренный расход водорода, молей Н2 на 1 м3
сырья
НГДА Расход водорода в реакциях гидродеазотирования, ст. куб. фут/бар-
рель или н. м3/м3
НГДАр Расход водорода в реакциях гидрирования ароматических углеводо-
родов, ст. куб. фут/баррель или н. м3/м3
Нгдк Расход водорода в реакциях гидрокрекинга, ст. куб. фут/баррель
Нгдм Расход водорода в реакциях гидродеметаллизации, ст. куб. фут/баррель
Нгдо Расход водорода в реакциях гидрообессеривания, ст. куб. фут/баррель
или н. м3/м3
НГДОк Расход водорода в реакциях гидродеоксигенации, н. м3/м3
НГДОл Расход водорода в реакциях гидрирования олефинов, н. м3/м3
I Неполная бета-функция
К Число свободных параметров
к Истинная константа скорости реакции, (м3 сырья)2 / [(м3 катализ.)
(моль Н2) • с]
LiqQ Общее количество жидкого сырья, кг
Liq' Общее количество жидкого продукта, кг
МА Содержание моноароматических углеводородов, %масс.
Mw Молярная масса
N Общее содержание азота, мг/кг или %масс.
Глава 12. Зависимости и прочие вопросы
585
п Число наблюдений
О Общее содержание кислорода, %масс.
Рн Парциальное давление водорода, МПа
PNA Содержание полиароматических углеводородов, %масс.
Ql Объемный расход жидкости, м3/с
R Ранг модели
R2 Коэффициент корреляции
RSS Сумма квадратов остатков
SD Стандартное отклонение
sg Плотность при 60 °F
S Общее содержание серы, %масс.
Т Независимая переменная, истинная температура кипения, °C
Tri А Содержание триароматических углеводородов, %масс.
7° Т2 Опорные значения температуры
VR Объем каталитического слоя, м3
Y Выход жидкости (объемная доля)
у Расчетное значение массовой доли
у Экспериментальное значение массовой доли
НИЖНИЕ ИНДЕКСЫ
f Сырье
р Продукты
ВЕРХНИЕ ИНДЕКСЫ
0 Значение на входе
1 Значение на выходе
ГРЕЧЕСКИЕ
а Порядок реакции
Г Гамма-функция
А. Разность значений AIC для лучшей модели и z-й модели
£ Доля пустот в каталитическом слое
586
Часть 3. Моделирование каталитических процессов
Т|N Молярная масса газа в стандартных условиях, моль/л
Ф Интегральная форма функции нормального распределения
0 Безразмерная температура
i; Относительная остаточная активность катализатора
X Коэффициент растворимости, (н. л Н,) / [(кг сырья) МПа]
р Плотность при 20 °C, г/мл
р£ Плотность жидкости, г/мл
со; Значение взвешенного критерия Акаике (AIC) для z-й модели
и, / со. Отношение достоверностей для z-й модели относительно 1 -й модели
ЛИТЕРАТУРА
Al-Dahhan, М.Н., Larachi, Е, Dudukovic, M.P, Laurent, А. 1997. High-pressure trickle-bed reactors:
a review, Ind. Eng. Chem. Res. 36:3292—3314.
Ancheyta, J., Betancourt, G., Marroquin, G., Perez, A.M., Maity, S.K., Cortez, M.T, del Rio, R. 2001.
An exploratory study for obtaining synthetic crudes from heavy crude oils via hydrotreating. Energy Fuels
15:120-127.
Ancheyta, J., Sanchez, S., Rodnguez, M.A. 2005. Kinetic modeling of hydrocracking ofheavy oil fractions:
a review Catal. Today 109:76—92.
Ancheyta, J., Trejo, E, Rana, M.S. 2009. Asphaltenes: Chemical Transformation during Hydrop recessing of
Heavyoils (Chemical Industries), CRC Press Taylor & Francis Group, Boca Raton, FL.
Ancheyta, J., Villafuerte, E. 2000. Kinetic modeling of naphtha catalytic reforming reactions. Energy
Fuels 14:1032-1037.
Ancheyta, J., Villafuerte, E., Schacht, P, Aguilar, R., Gonzalez, E. 2002. Simulation of a semi-regenerative
reforming plant using feedstocks with and without benzene precursors. Chem. Eng. Tecnol. 25:541-546.
Aris, R. 1989. Reactions in continuous mixtures. ATChEJ. 35:539—548.
Aris, R., Gavalas G.R. 1966. On the theory of reactions in continuations mixtures. Phil. Trans. Roy. Soc.
260:351-393.
Ashuri, E., Khorasheh, E, Gray, M.R. 2007. Development of a continuous kinetic model for catalytic
hydrodenitrogenation of bitumen. Scientia Iranica 14:152—160.
Astarita, G., Ocone, R. 1988. Lumping nonlinear kinetics. AIChEJ. 34:1299—1309.
Ayasse, A.R., Nagaishi, H., Chan, E.W, Gray, M.R. 1997. Lumped kinetics of hydrocracking of bitumen.
Fue/76:1025—1033.
Balasubramanian, B., Pushpavanam, S. 2008. Modeling discrimination in hydrocracking of vacuum gas
oil using discrete lumped kinetics. /ие/87:1660—1672.
Baltanas, M.A., Vimsina, H., Froment, G.E 1983. Hydroisomerization and hydrocracking. 5. Kinetic
analysis of rate data for n-octane. Ind. Eng. Chem. Prod. Res. Dev. 22:531—539.
Basak, K., Sau, M., Manna, U., \ferma, PR. 2004. Industrial hydrocracker model based on novel
continuum lumping approach for optimization in petroleum refinery Catal. Today 98:253-264.
Bennett, R.N., Bourne K.H. 1972. A study process reactions and corresponding product yields and
qualities. In: Proceedings of the ACS Symposium on Advances in Distillate and Residual Oil Technology
New York, August 27-September 1, G45-G62.
Botchwey, C., Dalai, A.K., Adjaye, J. 2003. Product selectivity during hydrotreating and mild
hydrocracking of bitumen-derived gas oil. Energy Fuels 17:1372-1381.
Глава 12. Зависимости и прочие вопросы
587
Browarzik, D., Kehlen, Н. 1994. Hydrocracking process of n-alkanes by continuous kinetics. Chem. Eng.
Sci. 49:923—926.
Chou, M.Y, Ho, TC. 1988. Continuum theory for lumping nonlinear reactions. AIChE J. 34:1519—1527.
DeDonder, Th. 1931. L’Affinite (secondpartie), Chapter III, Gauthier-Villars, Paris.
Dufresne, P, Bigeard, PH., Billon, A. 1987. New developments in hydrocracking: low pressure high-
conversion hydrocracking. Catal. Today 1:367—384.
Elizalde, I., Ancheyta, J. 2011. On the detailed solution and application of the continuous kinetic
lumping modeling to hydrocracking of heavy oils. Fuel 90:3542—3550.
Elizalde, I., Ancheyta, J. 2012. Modeling the simultaneous hydrodesulfurization and hydrocracking of
heavy residue oil by using the continuous kinetic lumping approach. Energy Fuels 26:1999—2004.
Elizalde, I., Rodriguez, M.A., Ancheyta, J. 2009. Application of continuous kinetic lumping modeling
to moderate hydrocracking of heavy oil. Appl. Catal. A 365:237—242.
Elizalde, I., Rodriguez, M.A., Ancheyta J. 2010. Modeling the effect of pressure and temperature on the
hydrocracking of heavy crude oil by the continuous kinetic lumping approach. Appl. Catal. A 382:205-212.
Espinosa-Pena, M., Figueroa-G6mez, Y, Jim6nez-Cruz, E 2004. Simulated distillation yield curves in
heavy crude oils: a comparison of precision between ASTM D-5307 and ASTM D-2892 physical distillation.
Energy Fuels 18(6): 1832—1840.
Froment, G.E 2005. Single event kinetic modeling of complex catalytic processes. Catal. Rev. 47:83—124.
Govindhakannan, J., Riggs J.B. 2007. On the construction of a continuous concentration-reactivity
function for the continuum lumping approach. Ind. Eng. Chem. Res. 46:1653—1656.
Ho, TC. 1991. Modeling of reaction kinetics for petroleum fractions. In: Kinetic and Thermodynamic
Lumping of Multicomponent Mixtures (G. Astarita, S.I. Sandler, Eds). Proceedings of an ACS Symposium on
kinetic and thermodynamic lumping of multi-component mixtures, Atlanta, GA, April 15.
Ho, TC. 2008. Kinetic modeling of large-scale reaction systems. Catal. Rev.-Sci. Eng. 50:287—378.
Jenkins, J.H., Stephens, TW 1980. Kinetics of catalytic reforming. Hydrocarbon Process. 11:163—168.
Khorasheh, E, Chan, E.C., Gray M.R. 2005. Development of a continuous kinetic model for catalytic
hydrodesulfurization of bitumen. Petrol. Coal 47:40-48.
Khorasheh, E, Zainali, H., Chan, E.C., Gray, M.R. 2001. Kinetic modeling of bitumen hydrocracking
reactions. Petrol. Coal 43:208—218.
Korsten, H., Hoffmann, U. 1996. Three-phase reactor model for hydrotreating in pilot trickle-bed
reactors. AIChEJ. 42:1350—1360.
Krishna, R., Saxena, K. 1989. Use of an axial-dispersion model for kinetic description of hydrocracking.
Chem. Eng. Sci. 44:703—712.
Laxminarasimhan, C.S., lerma. R.P, Ramachandran, PA. 1996. Continuous lumping model for
simulation of hydrocracking. AIChEJ. 42:2645-2653.
Mapiour, M., Sundaramurthy, V, Dalai, A.K., Adjaye, J. 2009. Effect of hydrogen purity on
hydroprocessing of heavy gas oil derived from oil-sands bitumen. Energy Fuels 23:2129-2135.
Mapiour, M., Sundaramurthy, V, Dalai, A.K., Adjaye, J. 2010. Effects of hydrogen partial pressure
on hydrotreating of heavy gas oil derived from oil-sands bitumen: experimental and kinetics. Energy Fuels
24:772-784.
Marafi, A., Stanislaus, A., Furimsky, E. 2010. Kinetics and modeling of petroleum residues
hydroprocessing. Catal. Rev. Sci. Eng. 52:204-324.
Maxwell, J.B. 1965. Data Book on Hydrocarbons: Application to Process Engineering, Sth edn., Vm
Nostrand, New "fork.
McCoy, B., Wing, M. 1994. Continuous-mixture fragmentation kinetics: particle size reduction and
molecular cracking. Chem. Eng. Sci. 49:3773—3785.
Mederos, E, Ancheyta, J., Chen, J. 2009b. Review on criteria to ensure ideal behaviors in trickle-bed
reactors. Appl. Catal. A 355:1-19.
Mederos, E, Elizalde, I., Ancheyta, J. 2009a. Steady-state and dynamic reactor models for hydrotreatment
of oil fractions: a review Catal. Rev. Sci. Eng. 51:485—607
588
Часть 3. Моделирование каталитических процессов
Narasimhan, C.S.L., Sau, М., \ёппа, R.P 1999. An integrated approach for hydrocracker modeling.
In: Hydrotreatment and Hydrocracking of Oil Fractions (B. Delmon et al., Ed), pp. 297—306, Elsevier,
Amsterdam.
Ostrowsky, N., Sornette, D. 1981. Exponential sampling method for light scattering polydispersity
analysis. Optica Acta 28:1059—1070.
Pacheco, M.A., Dassori, C.G. 2002. Hydrocracking: an improved kinetic model and reactor modeling.
Chem. Eng. Commun. 189:1684—1704.
Padlo, D.M., Kuger, E.L. 1996. Simulated distillation of heavy oils using an evaporative light scattering
detector. Energy Fuels 10:1031-1035.
Peixoto, EC., de Medeiros, J.L. 1999. Modelling and parameter estimation in reactive continuous mixtures:
the catalytic cracking of alkanes, Part I. Braz. J. Chem. Eng. 16( 1):65—81.
Qader, S.A., Hill, G.R. 1969. Hydrocracking of gas oil. Ind. Eng. Chem. Five. Des. Dev. 8:98-105.
Rana, M.S., Samano, V, Ancheyta, J., Diaz, J.A.I. 2007. A review of recent advances on process
technologies for upgrading of heavy oils and residua. Fuel 86:1216—1231.
Rassev, S. 2003. Thermal and Catalytic Processes in Petroleum Refining, Marcel Decker, Inc., New York.
Sadighi, S., Arshad, A., Rashidzadeh, M. 2010. 4-Lump kinetic model for vacuum gas oil hydrocracker
involving hydrogen consumption. Kor. J. Chem. Eng. 27(4):1099—1108.
Sambi, I.S., Khulbe, K.C., Mann, R.S. 1982. Catalytic hydrotreatment of heavy gas oil. Ind. Eng. Chem.
Prod. Res. Dev. 21:575-580.
Sanchez, S., Ancheyta, J. 2007. Effect of pressure on the kinetics of moderate hydrocracking of maya
crude oil. Energy Fuels 21:653-661.
Sanchez, S., Rodriguez, M.A., Ancheyta J. 2005. Kinetic model for moderate hydrocracking of heavy
oils. Energy Fuels 44:9409—9413.
Sau, M., Narasimham, C.S.L., \6rma, R.P 1997. A kinetic model for hydrodesulfurization.
In: Hydrotreatment and Hydrocracking of Oil Fractions (Stud. Surf. Sci. Catal.) (G.E Froment et al., Ed),
pp. 421-435, Elsevier Science, Amsterdam.
Scherzer, J., Gruia, A.J. 1996. Hydrocracking Science and Technology, Series: Chemical Industries;
vol. 66, Marcel Decker, Inc., New York.
Speight, J.G. 2000. The Desulfurization of Heavy Oils and Residua, Marcel Dekker, New York.
Vilarasu, G., Bhaskar, M., Balaraman, K.S. 2003. Mild hydrocracking: a review of the process, catalysts,
reaction, kinetics and advantages. Petrol. Sci. Technol. 21:1185-1205.
\6rstraete, J.J., Le Lannic, K., Guibard, I. 2007. Modeling fixed-bed residue hydrotreting processes.
Chem. Eng. Sci. 62:5402—5408.
Yui, S.M., Sanford, E.C. 1989. Mild hydrocracking ofbitumen-derived coker and hydro-cracker heavy gas
oils: kinetics, product yields, and product properties. Ind. Eng. Chem. Res. 28:1278—1284.
Yui, S.M., Sanford, E.C. 1991. Kinetics of aromatics hydrogenation ofbitumen-derived gas oils. Can. J.
Chem. Eng. 69:1087-1095.
Ziff, M.R. 1991. New solutions to the fragmentation equation. J. Phys. A. Math. Gen. 24:2821-2828.
Хорхе Анчита
Переработка тяжелой нефти
Реакторы и моделирование процессов
Перевод с английского языка
под редакцией О. Ф. Глаголевой, В. А. Винокурова
ISBN 978-5-91884-068-9
Ответственный редактор А. А. Стешко
ЦОП «Профессия»
Санкт-Петербург, 190020, а/я 140
Тел./факс: (812) 313-54-14
URL: www.epcprof.ru
E-mail: info@epcprof.ru
Интернет-магазин и онлайн-заказ книг издательства:
www.epcprof.ru
Подписано в печать 14.11.2014
Формат 70x100 1/16. Усл. печ. л. 47,96
Бумага офсетная. Заказ № 6436.
Отпечатано способом ролевой струйной печати
в ОАО «Первая Образцовая типография»
Филиал «Чеховский Печатный Двор»
142300, Московская область, г. Чехов, ул. Полиграфистов, д. 1
Сайт: www.chpd.nl, E-mail: sales@chpd.ra, т/ф. 8(496)726-54-10